
Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 08/99/24. The contractual start date was in January 2011. The final report began editorial review in March 2015 and was accepted for publication in October 2015. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2016. This work was produced by Ottensmeier et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Background
Importance of the health problem to the NHS
In the UK, 7279 patients were diagnosed with leukaemia in 2005,1 but despite continuing advances in diagnosis and treatment the majority of these individuals will eventually die from their disease. 2
Summary of current evidence
Chronic myeloid leukaemia
Chronic myeloid leukaemia (CML) is a clonal disease of the haematopoietic stem cells (HSCs) in which a reciprocal translocation, t(9;22)(q34;q11), known as the Philadelphia chromosome, results in a fusion gene, BCR–ABL (breakpoint cluster region–Abelson murine leukaemia viral oncogene homolog 1), which in turn expresses an activated tyrosine kinase, and is regarded as the initiating lesion of CML. 3,4 Until quite recently, the only treatment to offer the possibility of long-term disease-free survival was allogeneic stem cell transplantation (alloSCT), the ‘curative’ effect of which is mediated in large part through the alloimmune graft-versus-leukaemia (GVL) effect. 5 However, alloSCT carries a substantial risk of mortality and is only available to a minority of patients. Tyrosine kinase inhibitors (TKIs), notably imatinib, have replaced alloSCT as first-line therapy for CML owing to their lower toxicity and impressive efficacy. Although > 85% of imatinib-treated patients with chronic-phase CML (CML-CP) achieve a complete cytogenetic response (CCyR), the majority of patients have persisting molecular disease, as assessed by quantitative polymerase chain reaction (qPCR) for BCR–ABL transcripts, and almost all will relapse following imatinib withdrawal. 6,7 Functional leukaemia CD34+ (cluster of differentiation 34-positive) progenitor cells have been identified in such patients within CCyR, suggesting the presence of a reservoir of leukaemia cells resistant to TKIs. 8 Furthermore, the durability of these responses has not yet been established. In contrast, long-term survivors of alloSCT very rarely have any detectable molecular disease, indicating that all leukaemia cells must be susceptible to immune destruction (GVL effect). Therefore, novel strategies to eradicate quiescent CML stem cells are required, especially because these cells provide a reservoir for disease relapse.
The immunological effect of alloSCT and donor lymphocyte infusions (DLIs) suggests that an approach based on the amplification of the patient’s own immune response to the disease could add to the responses seen after treatment with the TKI.
Based on our own data we argue here that vaccinating against the Wilms’ tumour antigen 1 (WT1), using deoxyribonucleic acid (DNA) vaccination, is an attractive choice for delivering this immune attack. 9,10 The validity of WT1, as a target for immunotherapy in CML, was recently shown in work published by Yong et al. [in John Barrett’s group at the National Institutes of Health (USA)]. 11 This group studied the expression of leukaemia-associated antigens, including WT1, within the CD34+ primitive stem cell and committed progenitor cell pools in CML patients. WT1 is significantly overexpressed in all CD34+ subpopulations in CML encompassing the most primitive HSCs to the most mature cells,11 which escape control by imatinib. Taken in the context of these clinical data and that from other groups, which show that even suboptimal vaccination with peptide can have clinical effects,11,12 these data strongly suggest that active immunotherapy other than allotransplantation holds significant promise by the induction of tumour antigen-specific CD8+ (cluster of differentiation 8-positive) T lymphocytes (T cells) without adding toxicity.
Clearly it is critical to choose the best clinical setting in which to vaccinate, as previous data have shown that the effect of TKIs as a drug class on the immune system is variable,13 and can be either suppressive or stimulatory. For imatinib specifically, in vivo data show that it can be immunostimulatory, supporting our proposed trial, both in murine14,15 as well as human16–18 studies. Furthermore, Wang et al. 14 demonstrated that in vivo treatment with imatinib not only prevented the induction of tolerance, while preserving responsiveness to a subsequent immunisation, but, critically, enhanced vaccine efficacy. In patients, low-frequency CD8+ T-cell responses to four leukaemia-associated antigens (ABL kinase, proteinase 3, telomerase and WT1) were detected in CML patients on imatinib and show the immune system’s ability to respond to leukaemia-associated antigens in the presence of imatinib. 19 It is therefore unsurprising that two vaccine studies using BCR–ABL peptides in patients with CML treated with imatinib clearly demonstrated the successful induction of CD8+ and CD4+ (cluster of differentiation 4-positive) T cells against the vaccine, even with a suboptimal peptide vaccine approach. 20,21 Bocchia et al. 20 found that antileukaemia T-cell responses could be stimulated after vaccination in 9 out of 14 patients. In the EPIC trial, T-cell responses to CD4+ T-cell responses against the vaccine were seen in all patients and 14 of 19 patients developed T-cell responses to BCR–ABL peptides. 21 Prospective analysis of immune responses to vaccination against influenza A (H1N1, 2009 strain) and Streptococcus pneumoniae (Klein 1884) Chester 1901 in 50 CML-CP patients treated with imatinib (Glivec®, Novartis Pharmaceuticals UK Ltd), dasatinib (Sprycel®, Bristol-Myers Squibb) or nilotinib (Tasigna®, Novartis Pharmaceuticals UK Ltd) and 15 healthy controls was recently performed. 22 Significant CD8+ and CD4+ T-cell responses against flu were induced in patients with CML-CP on TKIs following vaccination and there was no significant difference in the vaccine-induced T-cell response between CML-CP patients on TKIs and healthy controls (manuscript in preparation). These data strongly support that vaccination of patients on stable doses of imatinib will induce immune responses.
Acute myeloid leukaemia
Acute myeloid leukaemia (AML) is a disease of older adults with a median age of 68 years23 and an incidence of 8–12 per 100,000 population. Advances in the understanding of the pathophysiology of AML have not yet led to major improvements in disease-free and overall survival of adults with this disease. Only about one-third of those aged between 18 and 60 years who are diagnosed with AML can be cured; disease-free survival is rare and current therapy is devastating in older adults. Treatment of AML involves chemotherapy with high remission rates in up to 85% of patients; however, remissions are often short lived and > 70% of patients will progress and die from their disease within 2 years (see Figure 1). 24 Treatment also causes significant morbidity and mortality. AlloSCT from a compatible donor carries a 20–75% chance of long-term disease-free survival depending on whether the transplant is performed in remission or with residual disease. Death from relapse is the most common cause of treatment failure following transplant. At this point, a minority of patients respond to chemotherapy and DLIs, but remission rates are around 15% with only a fraction being durable. 17,18 There is, therefore, a need to devise better treatments for AML.
In AML, WT1 has been established as a marker for minimal residual disease (MRD). 25 Additionally, WT1 gene expression has been suggested to carry adverse prognostic implications in AML based on data from a number of studies. 26,27 A recent trial by the European LeukemiaNet defined and standardised a WT1 real-time qPCR assay as a marker for MRD monitoring and risk stratification in AML. 28 We intend to exploit this for the proposed trial of WT1 vaccination. As in CML, peptide vaccination has been tested with some success12,29–33 and the data support that active immunotherapy other than allotransplantation holds significant promise by the induction of tumour antigen-specific CD8+ T cells without added toxicity.
The purpose of the trial was to build on an established programme of DNA fusion gene vaccination delivered by intramuscular injection and exploiting this unique experience with electroporation, to induce durable immune responses with the aim of controlling the disease by precision attack of the tumour by CD8+ T cells. The aim of the trial was to evaluate an identical vaccine strategy in two parallel settings with the purpose of identifying the most promising context for eventual Phase III testing. The hypothesis was that molecular and clinical responses, induced by T cells can be predicted by increases in the number of CD8+ T cells, specific for the vaccine-encoded T-cell epitopes.
Trialling two patient groups will maximise the knowledge gained from this vaccine trial. Patients with CML will allow a direct and objective assessment of the antileukaemia effect of vaccination at the molecular level by BCR–ABL and WT1 monitoring. Patients with AML offer a difficult challenge to haematologists. The advantage of including this patient group is twofold: (1) the antileukaemia effect of vaccination can be assessed objectively by measuring WT1 gene expression levels; and (2), more importantly, data can be collected on the highly clinically relevant question of whether or not vaccination will prevent relapse in this patient group.
Selection of patients for vaccine therapy
Novel therapies are often first introduced in patient groups who have failed all conventional treatment options and have far advanced or metastatic disease. This strategy is inappropriate for vaccine treatments, which depend upon an intact well-functioning immune system, known to be severely impaired in advanced cancers. The cohort to be studied here has therefore been chosen to reflect this conclusion.
Immunotherapy in haematological malignancies targeting Wilms’ tumour antigen 1
DNA fusion vaccines were initially developed to treat B-cell malignancies,34 which showed that fusion of the microbial sequence, fragment C from the tetanus toxin (FrC) to idiotypic tumour antigen provided the T-cell help required to induce humoral35 and CD4+ T-cell responses in preclinical models. 36 Early clinical testing was undertaken in a Phase I/II dose escalation trial [LIFTT trial; Gene Therapy Advisory Committee (GTAC) 029A], with individual idiotypic DNA fusion vaccines to treat patients with follicular lymphoma. The vaccine was safe and 14 out of 18 patients showed an antibody and/or CD4+ T-cell responses against the FrC portion of the fusion gene. Encouragingly, 6 out of 16 patients showed responses to the tumour-specific idiotypic antigen (manuscript in preparation). There was no evidence of a dose response for doses ranging from 500 µg/dose to 2500 µg/dose. 9 Overall, however, the levels of response were relatively low and improvements were sought.
An important development has been electroporation, which has been shown to dramatically increase DNA vaccine performance in mice37 and rhesus macaques,38 and this method of delivery was used in our clinical trial in patients with prostate cancer. We found clear evidence for amplification of antibody and CD4+ T-cell responses in patients. 39 For induction of CD8+ T-cell responses, the vaccine design was modified by reducing the FrC sequence to a single domain (p.DOM; plasmid domain 1 from FrC). This decreased the potential for peptide competition but retained the major histocompatibility complex class II-restricted peptide p30. 39 An epitope-specific sequence was then inserted at the C-terminus of FrC to aid processing/presentation. In multiple models, this p.DOM epitope design (Figure 1a) was able to induce high levels of epitope-specific CD8+ T cells. 9
Importantly, provision of high levels of T-cell help enables induction of immune responses in tolerant settings. 9,39
The preclinical data appear to predict response in humans. 10 For patients with relapsed prostate cancer, a p.DOM epitope design incorporating a peptide sequence from the prostate-specific membrane antigen (PSMA; GTAC 089) has induced high levels of epitope-specific interferon gamma (IFN-γ), producing CD8+ T-cell responses in 67% (10/15) of patients. 40 Data from the 10 patients in the group receiving the lowest dose levels of DNA and DNA/electroporation are shown in Figure 1b. This was the first ever trial to exploit delivery of DNA by electroporation and it was found this approach was safe and readily accepted by patients. 10 Responses were robust and persistent over many months to the end of follow-up in the trial at 18 months (Figure 1b).
FIGURE 1.
Vaccination of patients with the p.DOM epitope vaccine. (a) The p.DOM epitope vaccine consists of a DNA plasmid backbone incorporating cytosine–phosphate–guanine sites. The first domain of the tetanus toxin (DOM; TT865–1120) provides T-cell help when linked to a tumour-associated nucleotide sequence encoding the human leucocyte antigen class I binding epitope of interest. This format allows the appropriate processing and presentation of the peptide. (b) Spots per million peripheral blood mononuclear cell producing IFN-γ in human leucocyte antigen A2-positive patients treated with three monthly doses of DNA (p.DOM.PSMA27). The figure shows data from the first dose cohort, analysed in a cultured.
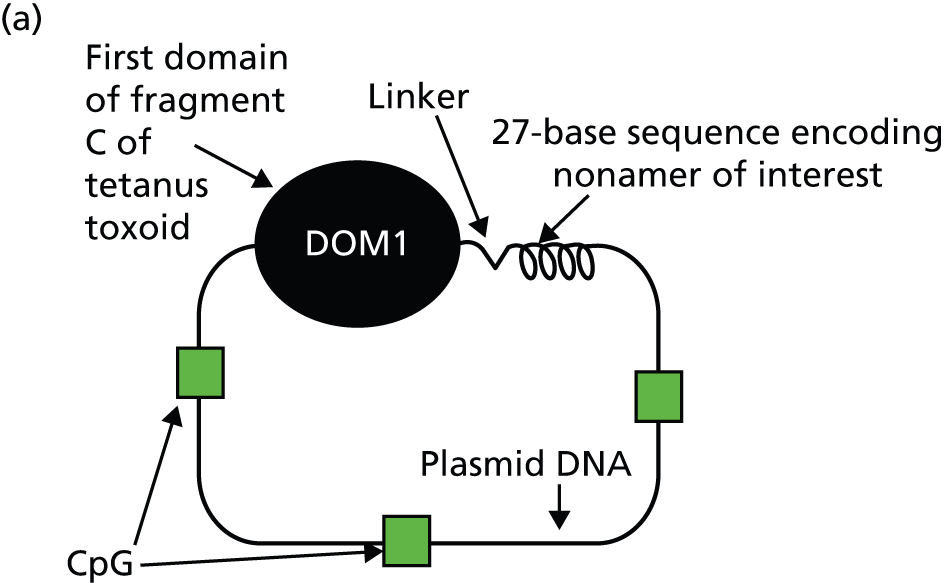
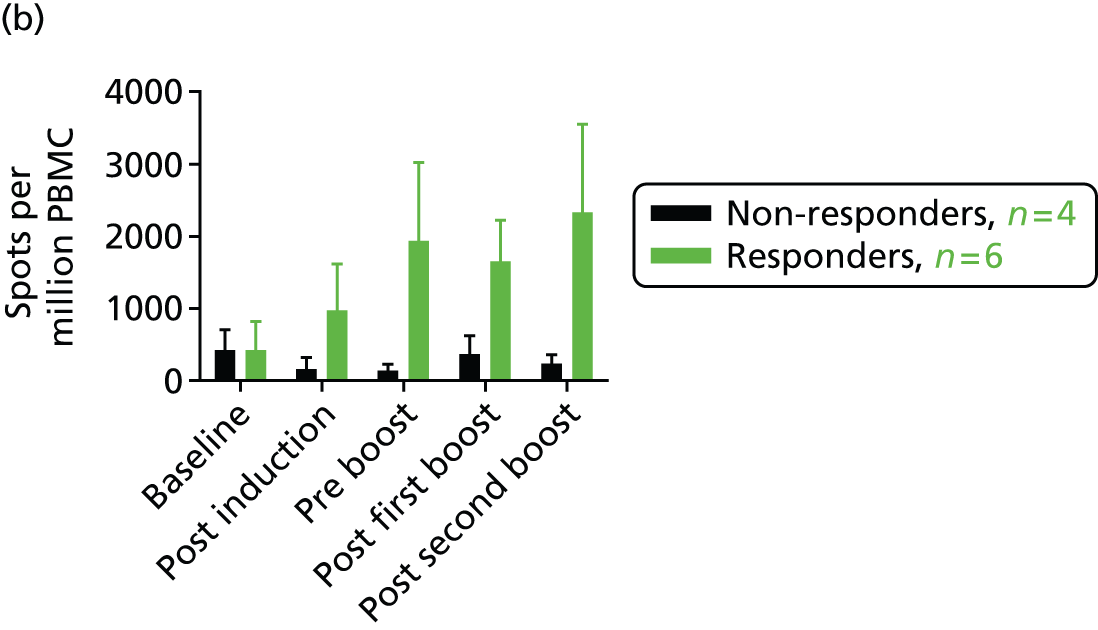
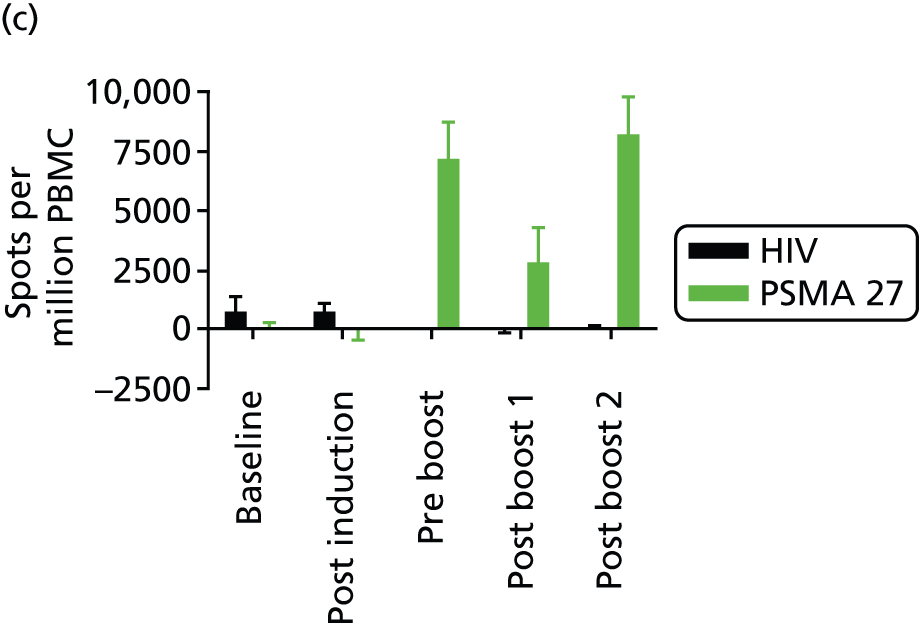
Figure 2 illustrates the CD8+ T-cell analyses in more detail. In Figure 2a and 2b two non-responders are shown, one of whom (Figure 2b) had pre-existing levels of PSMA peptide 27-specific T cells at baseline. It is interesting to note that these cells appear to leave the circulation post vaccination and become visible again after the first booster injection at 6 months. Further data are required to allow interpretation of this observation. In Figure 2c and 2d, two of the six responders at dose level 1 are shown. The patient in Figure 2c was treated with DNA alone followed by DNA delivered by electroporation, whereas the patient in Figure 2d was treated with DNA/electroporation on five occasions.
FIGURE 2.
The CD8+ T-cell responses to DNA vaccination analysed over time by enzyme-linked immunospot. (a) and (b) show data on two out of four non-responders, of which the patient in (b) shows a low-level CD8+ T-cell response to the PSMA peptide 27 at baseline. As there is no significant increase in levels of PMBCs producing IFN-γ above the baseline, this patient has been classified as a non-responder; (c) and (d) show examples of patients that have significantly increased levels of PMBCs producing IFN-γ compared with baseline levels and to the human immunodeficiency virus-negative control. (n = 6 in the first dose cohort.) HIV, human immunodeficiency virus; PBMC, peripheral blood mononuclear cell.



The effectiveness of the p.DOM epitope design for the treatment of myeloid malignancies has been explored based on these clinical results. WT1 has emerged as one of the most promising targets for immunotherapy of haematological malignancies including CML, AML and myelodysplastic syndromes. 29,41–43 It is also a potential target for the treatment of solid tumours. 43–46 Despite its ubiquitous expression during embryogenesis, WT1 expression in normal individuals is limited to renal podocytes, gonadal cells and a small proportion of CD34+ cells,47–50 where expression is significantly lower (10- to 100-fold). 47 This could raise a concern about autoimmunity, but, reassuringly, the available data document selectivity of attack against tumour cells, sparing the CD34+ cells51,52 and without any evidence of renal or other autoimmune toxicity in murine models53–55 or patients. 29,41–43
The WT1 peptide vaccines have been tested both in preclinical models51,52,56 and in clinical trials. 12,41–43 The data from clinical trials document that T-cell responses can be induced in patients and confirm the presence of an expandable CD8+ T-cell repertoire. Importantly, the ability of peptide vaccines to induce measurable clinical responses has been documented. However, a key problem with class I-restricted peptide vaccines is the inability of this approach to provide linked CD4+ T-cell help, which is crucial for the maintenance of tumour antigen-specific CD8+ T-cell populations. In the clinic, this is visible in poor persistence of the detected CD8+ T-cell responses. In contrast, it was that the p.DOM epitope fusion vaccines appeared to be able to deliver CD8+ T-cell responses, which show long-term persistence (see Figures 1b and 2c and 2d).
Recently, three domain 1 from fragment C of tetanus toxin (DOM) epitope vaccines were evaluated, each encoding a different, previously described, WT1-derived, human leucocyte antigen A2 (HLA A2)-restricted peptide. 56 All were able to induce CD8+ T-cell responses in ‘humanised’, and presumably tolerised, mice expressing HLA A2 and these killed human WT1-positive (WT1+), HHD+ leukaemia cells ex vivo. A direct comparison with a WT1 peptide vaccine (plus T-cell help and adjuvant) showed a clear superiority of the DNA fusion vaccine. 56 In parallel, we showed that low numbers of human WT1 peptide-specific T cells could be expanded in vitro to kill WT1+ HLA A2+ leukaemia cells. The WT1 peptide 37 (WT1-37) and WT1 peptide 126 (WT1-126) peptides were selected for current studies. We have already documented clinically the ability of p.DOM epitope vaccines to induce cytotoxic T lymphocytes (CTLs) and anticipate that dual attack against more than one epitope will provide added clinical benefit. 9,10 Vaccination with p.DOM–WT1-37 and p.DOM–WT1-126 into different locations will allow us to avoid antigenic competition. Given the clear effect on the response to the FrC portion of the vaccine in the prostate trial, electroporation was used as a delivery strategy. 9,10,56
Chapter 2 Aims
Recently, three DOM epitope vaccines were developed, each encoding a different, previously described, WT1-derived, HLA A2-restricted peptide. 56 All vaccines were able to induce CD8+ T-cell responses in ‘humanised’, and presumably tolerised, mice expressing HLA A2, and these T cells were capable of killing human WT1+ HHD+ leukaemia cells ex vivo. A direct comparison with a WT1 peptide vaccine (plus T-cell help and adjuvant) showed a clear superiority of the DNA fusion vaccine. 56 In parallel, we showed that low numbers of human WT1 peptide-specific T cells could be expanded in vitro to kill HLA A2+ WT1+ leukaemia cells. 56 Peptides WT1-37 (amino acid sequence: VLDFAPPGA) and WT1-126 (amino acid sequence: RMFPNAPYL) were selected for current studies.
The ability of p.DOM epitope vaccines to induce CTLs has been documented clinically and we anticipate that dual attack against more than one epitope will provide added clinical benefit. Vaccination with p.DOM–WT1-37 and p.DOM–WT1-126 into different locations will allow us to avoid antigenic competition. Given the clear effect on the response to the FrC portion of the vaccine in the prostate trial, electroporation was chosen as the vaccine delivery strategy. 10
The aim of this trial was to bring together substantial preclinical and clinical expertise to exploit the advantages of DNA fusion vaccines to form the basis for larger, randomised studies.
The objectives were to evaluate the:
-
molecular response in patients with CML (i.e. transcript levels of BCR–ABL and WT1) and AML (i.e. transcript level of WT1)
-
time to disease progression, 2-year survival rate (patients with AML)
-
correlation of molecular responses with immunological responses.
Chapter 3 Trial design and methods
Trial design
This was a non-randomised, open-label, single-dose-level Phase II trial in two patient groups (CML and AML) based on HLA A2 genotype. HLA A2+ patients were vaccinated with two DNA vaccines: (1) p.DOM–WT1-37 (epitope sequence: VLDFAPPGA); and (2) p.DOM–WT1-126 (epitope sequence: RMFPNAPYL). Patients with HLA A2-negative (HLA A2–) genotype were not vaccinated and formed the control group. Patients were tested for the presence of the human immunodeficiency virus (HIV), hepatitis B, hepatitis C and syphilis to protect the laboratory personnel, and because these infections may have a significant impact on the immunocompetence of the patient.
Significant change to trial design
The original trial design used Simon’s optimal Phase II trial design. 57,58 This allowed us to undertake a ‘start/stop’ evaluation once 12 patients had been enrolled into each vaccination group of the study and had been evaluated to 6 months (molecular monitoring). An interim analysis of these CML patients’ molecular data (BCR–ABL and WT1) was to be carried out including up to 6 months of data. If a molecular response (change in transcript level of BCR–ABL or WT1) was observed the CML arm may recruit an additional 25 participants. It was designed that the AML arm would also be opened to recruitment at this point.
It became evident early on in the trial that recruitment of the CML patients was significantly behind target. At the same time, new data emerged which illustrated that immune responses detected in the blood appear to evolve by 6 months post vaccination in most patients. 56 These data were not available when the WT1 Immunity via DNA (WIN) trial protocol was developed. Clinical responses (reduction in BCR–ABL transcript levels) are therefore also expected to happen late which would necessitate a halt in recruitment to ‘observe’ outcomes in the first stage prior to initiating the second stage.
This new information manifested to protocol amendment six. The following changes were included in this amendment to increase the available eligible CML patient population and help speed up recruitment:
-
The AML arm was to be opened, independent of the CML result, as a result of the new data that had emerged illustrating that immune responses detected in the blood appear to evolve by 6 months in most patients.
-
Patients on any TKI became eligible (rather than just those on imatinib).
-
The trial design was changed from Simon’s two-stage design58 to A’Hern’s single-stage design. 59
-
Amendment to the sample size for the trial to observe a minimum of 4 out of 32 CML patients who are molecular responders in order to provide evidence that the vaccine warrants further investigation. This is an increase proportionally from 10.8% (4/37) to 12.5% (4/32) and allowed all patients to receive all 12 vaccinations rather than only receive an additional six based on response being observed.
Early closure of the trial
The CML arm of the trial was terminated early by the funders because of poor recruitment. This meant that recruitment of the AML arm could not be implemented until an assessment was done to secure funding to continue with this arm of the trial. The outcome of this assessment was to close the trial. The Research Ethics Committee and the Medicines and Healthcare products Regulatory Agency (MHRA) were notified on the 3 April 2014.
The study met its primary decision-making target with one major molecular response in BCR–ABL transcript levels and, in parallel, new data emerged which illustrated that immune responses detected in the blood appear to evolve by 6 months post vaccination in most patients. 56 The early onset of the major molecular response suggests that vaccination has achieved this molecular event. In parallel, a WT1 molecular response was observed.
A significant amount of time and effort was utilised in an attempt to overcome multiple hurdles that prevented successful recruitment, but despite this the study did not complete recruitment. The failure to achieve the recruitment target was exclusively driven by the lack of recruitment of CML patients.
Some of the major hurdles which contributed to the failure of the studies are as follows:
-
The agreement and enthusiasm of the principal investigator (PI) for the centre at Hammersmith to participate in the study was not matched by a main member of the team to recruit to the study, leading to a lack of recruitment from the centre where the largest number of CML patients was expected.
-
At the feasibility stage, the expert haemato-oncologist (PI) at Hammersmith assessed the cohort size of eligible patients to be 500 patients with CML who would fulfil the entry criteria for the study. This meant that 70% of the recruitment would come from the cohort size at this centre. Unfortunately, neither the cohort size nor recruitment materialised and this was key to the success of the study.
-
Apparent lack of clinical efficacy by the vaccination and, therefore, less interest in the study.
-
The unexpected loss of the PI to a US centre, leaving the study unsupervised in the key centre.
The following key reasons contributed to the failure to achieve the recruitment target:
-
Extensive attempts were made by the chief investigator via the National Institute for Health Research (NIHR) clinical studies group and by visiting national key centres, which mange CML, to recruit new centres to the study. Although there was significant academic interest for this study, it competed directly with an ongoing study in CML, precluding their participation.
-
As highlighted above, the NIHR Efficacy and Mechanism Evaluation (EME) programme did not support recruitment of AML patients. Therefore, recruitment of this study arm was stopped and no patients were vaccinated; it was deemed unethical to recruit one or two patients to the AML arm and then stop the trial.
This was disappointing as the robust induction of FrC responses (10/10 evaluable patients) and WT1 T-cell responses in 7 out of 10 evaluable patients support that the preclinical data link to immunological outcomes as predicted. Overall, the data confirmed the immunogenicity and safety of the vaccine.
Ethical approval and research governance
Ethical approval for the trial was given by the GTAC, National Research Ethics Service National Patient Safety Agency (24 September 2010, reference number GTAC 173). The trial was registered with the International Standard Randomised Controlled Trial Register (ISRCTN) under the reference number ISRCTN 62678383.
Changes to original protocol
A summary of the changes to the original protocol is given in Table 1.
| Change in protocol | REC/MHRA approvals |
|---|---|
| Amendment 1 | |
| Protocol version 1 (20 June 2010): MHRA non-acceptance | 28 September 2010 |
| Protocol version 1 (7 July 2010): conditional approval only | |
| Protocol version 2 (9 September 2010): conditions addressed and protocol approved | |
| Addition of electrocardiography at baseline; logistical changes made for supply and return of IMP; and clarification on monitoring | |
| Amendment 2 | |
| Protocol version 3 (7 October 2010): used to open sites | 1 November 2010 |
| Update in the IMPD included in the protocol | |
| Amendment 3 | |
| Protocol version 4 (29 June 2011): clarifications | 12 August 2011 |
| Clarification of patient pathway throughout the trial from consent; clarification on the schedule of observations and procedure for HLA A2– participants; clarification on inclusion criteria for AML patients with regards to WT1 status; clarification determination of bone status for trial inclusion; clarification on resupply of IMP to sites; clarification of local and central laboratory responsibilities and shipment of samples; amendment of pain assessment case report form to remove patient identifiers; and addition of delayed-type hypersensitivity reaction to be carried out wherever feasible | |
| Amendment 4 | |
| Protocol version 5 (18 October 2011): eligibility broadened | 16 January 2012 |
| Eligibility criteria amended to allow patients with a 6-month history of lymphocyte counts of just below 1 to be included in the trial | |
| Amendment 5 | |
| Protocol version 5 (18 October 2011): changes to the IMPD; no changes to the protocol | 17 May 2012 |
| Stability data for p.DOM–WT1 DNA vaccines to support the proposed expiry date extension plan at the predetermined 18-month time point | |
| Amendment 6 | |
| Protocol version 6 (31 July 2012): eligibility broadened and trial design changed | 8 November 2012 |
| Eligibility criteria widened to include all TKIs to increase recruitment; change in trial design from a two-stage design to a single-stage design; sample size adjusted to 32 CML patients and 37 AML patients; all HLA A2+ patients to receive all 12 vaccinations instead of receiving the second six only if a response is observed | |
| Amendment 8 | |
| Protocol version 6 (31 July 2012): no change to the protocol; temporary halt to the trial | 11 April 2013 |
| CML arm of the trial stopped and temporary halt on trial for AML arm | |
| Amendment 10 | |
| Protocol version 7 (1 October 2013): reduction in follow-up visits | 7 November 2013 |
| Patients last follow-up 12 months post final vaccination. The enzyme-linked immunospot (ELISPOT) assay removed from end-point analysis (replaced by tetramer staining). The 36-month follow-up visit removed | |
| End of trial notification | 3 April 2014 |
Trial setting and sample
Three hospitals (Southampton General Hospital, University Hospital Southampton NHS Foundation Trust; Hammersmith Hospital, Imperial College Healthcare NHS Trust; and Royal Devon and Exeter Hospital, The Royal Devon and Exeter NHS Foundation Trust) were selected to undertake the trial based on their interest, clinical experience and the number of patients with AML and CML patients who may be suitable to participate in the trial. It was anticipated that the Hammersmith Hospital would contribute the majority of the CML patients based on the cohort of about 500 patients treated with imatinib with stable disease. Smaller numbers were expected from Exeter and Southampton.
Inclusion criteria
Chronic lymphocytic leukaemia patients
Those CML patients:
-
who have Philadelphia chromosome-positive CML in chronic phase
-
who are in CCyR but with detectable BCR–ABL transcripts and maintain the CCyR on TKI monotherapy for a minimum of 24 months.
Acute myeloid leukaemia patients
Those AML patients who have either:
-
WT1-positive AML in complete remission (CR) post chemotherapy
-
or AML in morphological CR with incomplete blood count recovery defined as patients who fulfil all of the criteria for CR except for residual neutropenia (< 1000/µl) or thrombocytopenia (< 100,000/µl).
All patients
-
Aged ≥ 18 years.
-
Written informed consent.
-
World Health Organization (WHO) performance status of 0 or 1.
-
For vaccination groups: HLA A2+ in at least one allele.
-
For control groups: HLA A2– in both alleles.
-
Renal function and liver function (creatinine < 1.5 × upper limit of normal, liver function tests < 1.5 × upper limit of normal); lymphocyte count ≥ 1.0 × 109/l (if the lymphocyte count was < 1.0 × 109/l at the time of entry into the trial but had been > 1.0 × 109/l in the last 6 months and had also not declined rapidly in the days and weeks preceding entry, then the patient was eligible); and normal clotting.
-
Haemoglobin level of > 100 g/l.
-
Adequate venous access for repeated blood sampling according to the protocol schedule.
-
If sexually active and possibly fertile, patients must have agreed to use appropriate contraceptive methods during the trial and for 6 months afterwards.
Eligibility criteria were widened to include all TKIs (previously imatinib only) to increase recruitment (amendment 6, 8 November 2012; see Table 1).
Exclusion criteria
Chronic lymphocytic leukaemia patients
-
Chronic lymphocytic leukaemia in accelerated phase or blast crisis or having achieved complete molecular response (CMR) at any point during TKI therapy.
-
A TKI change or dose modification in the previous year, therapy interruption for > 15 days in the 6 months prior to enrolment.
-
Prior interferon alpha therapy.
-
Hypocellular bone marrow (< 20%; indicated by blood counts and most recent bone marrow, where available).
-
A CMR.
Acute myeloid leukaemia patients
-
Acute myeloid leukaemia in haematological relapse or eligible for alloSCT.
-
Hypocellular bone marrow (< 20%).
-
Acute myeloid leukaemia patients with the ‘good-risk’ abnormalities comprising by the core-binding factor leukaemias [i.e. AML with the translocation (8;21) and inversion of chromosome 16, and acute promyelocytic leukaemia with the translocation (15;17)].
All patients
-
Systemic steroids or other drugs with a likely effect on immune competence were forbidden during the trial. The predictable need of their use precluded the patient from trial entry. Inhaled steroids were allowed.
-
Major surgery in the preceding 3–4 weeks from which the patient had not yet recovered.
-
Patients who were of high medical risk because of non-malignant systemic disease, as well as those with active uncontrolled infection.
-
Patients with any other condition which, in the investigator’s opinion, would not make the patient a good candidate for the clinical trial, such as concurrent congestive heart failure or prior history of New York Heart Association class III or IV cardiac disease.
-
Current malignancies at other sites, with the exception of adequately treated basal or squamous cell carcinoma of the skin. Cancer survivors, who had undergone potentially curative therapy for a prior malignancy, had no evidence of that disease for 5 years and were deemed at low risk of recurrence, were eligible for the trial.
-
Patients who are serologically positive for, or are known to suffer from, hepatitis B or C, syphilis or HIV. Counselling was offered to all patients prior to testing.
Trial interventions
The trial was an open-label trial with two groups for both the CML and AML arms:
-
intervention group: all eligible and consenting patients who were HLA A2+
-
control group: all eligible and consenting patients who were HLA A2–.
For the intervention group, the DNA vaccine was administered six times every 4 weeks followed by a further six vaccinations every 3 months to maximum of 24 months at the following dosing amounts:
-
p.DOM–WT1-37: 1 mg/dose/vaccine
-
p.DOM–WT1-126: 1 mg/dose/vaccine.
The original protocol planned that patients would be given vaccines six times, at 4-weekly intervals, and only if a response was observed would they go on to receive the remaining six vaccinations at 3-monthly intervals up to a maximum of 24 months. New data emerged which illustrated that immune responses detected in the blood appear to evolve by 6 months in most patients;58 therefore, the protocol was amended to allow all HLA A2+ patients to receive all 12 vaccinations instead of receiving the second six only if a response was observed.
The vaccine was manufactured at the MHRA-approved Clinical Biotechnology Centre at the Bristol Institute for Transfusion Science (Bristol, UK) in accordance with good manufacturing practice.
The amount of DNA used was 1 mg/dose for p.DOM–WT1-37 and 1 mg/dose for p.DOM–WT1-126 (at a final concentration of 1 mg/0.8 ml). The vaccine was supplied in standard phosphate-buffered saline. The DNA for injection was divided into aliquots for storage at –70 °C in sterile glass vials, and aliquots for sterility and stability testing. The testing was based on the guidelines for injectables described in the European Pharmacopoeia (http://edpm.eu). The most likely contaminant is protein, which was expected to be < 1%. The material was confirmed as pyrogen free by using a Limulus test (BioWhittaker UK Ltd, Wokingham, UK). After delivery to the hospital pharmacy, the vaccine was stored at –70 °C.
The vaccine was thawed for approximately 5 minutes so that it was at room temperature before administration. The vaccines were injected by deep intramuscular injection into separate sites followed by electroporation.
The electroporation device (Elgen1000, Inovio Biomedical corporation, San Diego, CA, USA) was a system specifically designed for the delivery of electrical pulses to selected tissues, including muscle, to facilitate the intracellular uptake of plasmid DNA. The device locally applies controlled, short-duration electric pulses to target tissues to create an electric field that temporarily increases cellular membrane permeability allowing the plasmid DNA to enter the cells.
Operators underwent formal training before being considered competent in the use of the device. Training was provided by Inovio Pharmaceuticals, Inc. (San Diego, CA, USA).
Vaccination schedule
The DNA vaccine was administered 12 times. Patients received the vaccine at 4-weekly intervals into separate sites for the first 6 months, followed by vaccinations every 3 months up to a maximum of 24 months. Vaccines were injected intramuscularly and followed by intramuscular electroporation. Pain assessments were conducted immediately after vaccination and at 48 hours post vaccination.
The first HLA A2+ patient recruited at each site was evaluated 48 hours after administration of the first vaccination, before additional doses were given or before additional patients were vaccinated at that site.
Trial procedures
Recruitment and informed consent
The PIs identified potential eligible patients from their existing patient population either during routine consultation or from a database search. Patients identified from their database were approached for the trial at their next clinic appointment. Patients who were interested in participating in the trial were provided with the patient information leaflet and signed the informed consent form prior to enrolment into the trial.
Registration
After written informed consent was obtained from the patient and before screening commenced, sites registered the patient with the Southampton Clinical Trials Unit (SCTU) to obtain the unique patient identification number. The patient’s eligibility was checked during the registration process to ensure that only patients fulfilling the eligibility criteria were registered. Subsequently, the patient identification number was assigned.
Data collection and management
Sites entered trial-specific data, as specified in the protocol, onto paper case report forms (pCRFs). Completed pCRFs were sent to the SCTU, which was responsible for the data management of the trial. Data were transcribed from pCRFs into an InForm database (InForm version 5.0, ORACLE) at the SCTU. A range of data validation checks were carried out within both InForm and SAS version 9.3 (SAS Institute Inc., Cary, NC, USA) or above to minimise incorrect or missing data.
Molecular samples were sent to Haematology department at Hammersmith Hospital (a clinical pathology-accredited MRD laboratory) for molecular monitoring (qPCR for BCR–ABL/WT1 in CML and WT1 in AML). This is an accepted and routine test for monitoring of this disease. Results from the analysed samples were regularly sent back to SCTU for statistical analysis.
Immunological analyses for vaccine responses, including leukapheresis samples and bone marrow samples, were processed and frozen locally according to an agreed standard operating procedure and stored in liquid nitrogen. Samples were transported to the Cancer Sciences Division (Southampton General Hospital, Southampton, UK) in dry ice and using a temperature logger once a sufficient number or samples had been collected locally (Hammersmith Hospital, Exeter, UK).
Source data verification was undertaken during site monitoring visits, in accordance with the SCTU’s trial monitoring plan. Sites were visited at least once during the trial. At least one monitoring visit was undertaken for each participating site. A total of three planned monitoring visits and one triggered monitoring visit were carried out.
Baseline
The baseline investigations/evaluations that were performed on patients before vaccination included bone marrow aspiration, electrocardiogram (ECG), chest X-ray, HLA A2 status verification, vital signs, WHO performance status, urinalysis, full blood counts (FBCs), blood clotting test, biochemistry (levels of sodium, potassium, calcium, phosphorus, urea and creatinine; total protein, albumin, bilirubin, alkaline phosphate, alanine transaminase, aspartate transaminase mad gamma-glutamyl transferase levels), the creatine kinase (CK) test (for HLA A2+ patients), tests for the presence of syphilis, hepatitis B, hepatitis C and HIV, qPCR (for BCR–ABL and WT1 in CML patients and for WT1 in AML patients), leukapheresis for immunological studies (HLA A2+ patients only), autoimmune profiling and checks for concomitant diseases/treatments.
Follow-up
Patients were followed up as outpatients from the start of treatment at the time points outlined below. Each visit was done from baseline ± 14 days.
-
Visit 1: week 0, within 7 days of baseline.
-
Visit 2: week 2.
-
Visit 3: week 4.
-
Visit 4: week 8.
-
Visit 5: week 10.
-
Visit 6: week 12.
-
Visit 7: week 16.
-
Visit 8: week 20.
-
Visit 9: week 22.
-
Visit 10: week 24.
-
Visit 11: week 32.
-
Visit 12: week 34.
-
From visit 13 onwards, patients were seen at the following time points from baseline ± 14 days: months 11, 14, 17, 17 + 14 days, 20, 23, 24, 27, 30, 33 and 36.
Patients were observed in hospital for 2 hours post vaccination for adverse events (AEs). It was planned that if no AEs were observed after 12 patients completed six doses of vaccination, the 2-hour hospital stay may be discontinued; however, because of the early termination of the study, this was not implemented.
Time points were adjusted if visits were delayed to keep visits as close as possible to the 14-day post-vaccination visits. Many of the baseline investigations were repeated throughout the follow-up period at varying time points. The repeated tests included FBCs and differential blood counts; biochemistry including the CK test and urinalysis; immunological monitoring [65 ml of anticoagulated blood (in lithium heparin tubes) and 5 ml of clotted blood for serum would be taken for immunological monitoring]; ECG; echocardiogram (ECHO), if clinically indicated; bone marrow for immunological (CML and AML) and disease (AML) evaluation; leukapheresis; and molecular analysis of BCR–ABL and WT1 transcripts in CML patients and WT1 transcripts in AML patients (20 ml of anticoagulated blood was taken for qPCR).
All patients were asked to consent to information about their health status being held and maintained by the Health and Social Care Information Centre and the NHS Central Register, to enable long-term follow-up.
Outcome measures
Molecular response
Definition of the BCR–ABL response
For patients with a baseline BCR–ABL transcript level of > 11 transcripts/mg of ribonucleic acid (RNA):
-
Complete molecular response: a BCR–ABL transcript level of 0 transcripts/mg of RNA with an Abelson murine leukaemia viral oncogene homologue 1 (ABL) control level of ≥ 32,000 transcripts/mg of RNA in two consecutive tests. These patients cannot be assessed for a major or minor response as defined below.
For patients with a baseline BCR–ABL transcript level of ≥ 11 transcripts/mg of RNA:
-
Complete molecular response: a BCR–ABL transcript level of 0 transcripts/mg of RNA, with an ABL control level of ≥ 32,000 transcripts/mg of RNA in two consecutive tests.
-
Major response: a fall of > 1-log in the breakpoint cluster region (BCR) to ABL transcript level ratio. Confirmed in an ABL control level of ≥ 32,000 transcripts/mg of RNA in two consecutive samples at any time during follow-up.
-
Minor response: a fall of > 0.5-log in the BCR to ABL transcript level ratio. Confirmed in an ABL control copy transcript level of ≥ 32,000 transcripts/mg of RNA in two consecutive samples at any time during follow-up.
Definition of WT1 response
For patients with a baseline WT1 to β-glucuronidase (GUS) transcript level ratio of < 0.1%:
-
Complete molecular response: 0% WT1 to GUS transcript level ratio, with a GUS control level of ≥ 32,000 transcripts/mg of RNA; in two consecutive tests these patients cannot be assessed for a major or minor response as defined below.
For patients with a baseline WT1 to GUS transcript level ratio of ≥ 0.1%:
-
Complete molecular response: 0% WT1 to GUS transcript level ratio, with a GUS control level ≥ 32,000 transcripts/mg of RNA; in two consecutive tests.
-
Major response: a fall of > 1-log in the WT1 to GUS transcript level ratio. Confirmed in a GUS control level of ≥ 32,000 transcripts/mg of RNA in two consecutive samples at any time during follow-up.
-
Minor response: a fall of > 0.5-log in the WT1 to GUS transcript level ratio. Confirmed in a GUS control level of ≥ 32,000 transcripts/mg of RNA in two consecutive samples at any time during follow-up.
Immunological response
Definition of validated assay by tetramer
A positive response using the validated assays by tetramer will be determined by the following criteria:
-
tetramer staining at the post-vaccination time point is over the cut-off value for the specific tetramer when the pre-vaccination baseline time point (visit number 00 or 01) is below this cut-off value or
-
tetramer staining at the post-vaccination time point is more than twofold above the baseline time point when the baseline time point measures over the cut-off value for the specific tetramer
-
tetramer staining is reviewed and confirmed by at least two independent, flow cytometric-experienced scientists.
Immunological responses may also be assessed by other research assays such as intracellular cytokine staining and enzyme-linked immunospot. Only results produced by validated end-point assays will be reported on.
All patients who are removed from the study for reasons other than progressive disease will be re-evaluated at the time of treatment discontinuation.
Primary outcome
Chronic myeloid leukaemia
The primary outcome for the CML treatment group was a molecular BCR–ABL response (major or minor response or CMR), as defined in Definition of BCR–ABL response.
Acute myeloid leukaemia
The primary outcome for the AML treatment group was time to disease progression. Disease progression in AML is defined as disease relapse.
Secondary outcomes
Chronic myeloid leukaemia
The secondary outcomes for the CML treatment group are as follows:.
-
A BCR–ABL molecular response (major response or CMR), as defined in Definition of BCR–ABL response.
-
A WT1 molecular response of (major or minor response or CMR), as defined in Definition of WT1 response.
-
Time to disease progression. Disease progression for CML patients is defined as a loss in complete haematological response, where at least one factor falls out of the following ranges:60
-
a white blood cell count of < 10 × 109/l
-
basophils levels of < 5%
-
no myelocytes, promyelocytes or myeloblasts in the differential blood count
-
a platelet count of < 450 × 109/l
-
a non-palpable spleen.
-
-
Time to next treatment. A next treatment is defined as the first drug taken during the course of the study with an indication to treat CML.
-
Time to molecular response (for both BCR–ABL and WT1):
-
measured from the beginning of TKI treatment
-
measured from the time of obtaining informed consent.
-
-
Overall survival.
-
Toxicity assessed according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. 1
-
Pain assessment immediately after vaccination (see Appendix 1).
-
Pain assessment at 48 hours post vaccination (see Appendix 2).
Acute myeloid leukaemia
The secondary outcomes for the AML treatment group are as follows:
-
2-year overall survival.
-
2-year progression-free survival (PFS).
-
Overall survival.
-
A WT1 molecular response (major or minor response or CMR), as defined in Definition of WT1 response.
-
Toxicity assessed according to the National Cancer Institute’s CTCAE, version 4.0.
-
Pain assessment immediately after vaccination.
-
Pain assessment at 48 hours post vaccination.
Human leucocyte antigen A2-positive patients only (chronic myeloid leukaemia and acute myeloid leukaemia)
The secondary outcomes for only the HLA A2+ patients are as follows:
-
Immune response to WT1 epitope-specific T cells in blood and/or bone marrow, using validated assays by tetramer staining. A positive response using the validated assays by tetramer staining is defined in Definition of validated assay by tetramer.
-
Number of WT1-specific T-cells after peptide challenge to the skin (wherever assessment is feasible).
-
Immune response to FrC, evaluated using validated assays by enzyme-linked immunosorbent assay (ELISA).
-
Number of humoral responses (B lymphocytes) to the vaccine components evaluated using ELISA.
Sample size
Original sample size
For sample size calculation, we used Simon’s optimal Phase-II trial design for clinical development of therapeutic cancer vaccines. 60,61 This would allow us to undertake a ‘start/stop’ evaluation once 12 patients had been enrolled into each vaccination group of the study and had been evaluated to 6 months (molecular monitoring).
If one or more responders were observed in the 12 initial patients, this particular vaccination group would be extended to 37 patients. This would allow the study to distinguish between p0 = 5% (standard response) and p1 = 20% (expected response) with α = 0.1% (two-sided) and β = 0.10, where p0 is the probability of a clinically uninteresting true response rate and p1 is the probability of a sufficiently promising true response rate. This gives a less than 10% chance of rejecting a useful vaccine, with error probability limits of α 0.10 and β < 0.10, even if the true response rate were to be p1 = 20%.
If no molecular responses were seen in the first 12 CML or AML patients, recruitment would have ceased for this patient group, as there will not be sufficient clinical interest to pursue the trial further. This optimal design had an expected sample size under H0 (the null hypothesis) of 24 and a maximum sample size of 37 in each patient group (AML and CML).
Revised sample size
The sample size was revised in line with the change in trial design from Simon’s two-stage60 to A’Hern’s one-stage trial design. 59 This was approved in version 6 of the protocol on 8 November 2012 (see Table 1).
Chronic myeloid leukaemia
The sample size for the HLA A2+ group had been determined using A’Hern’s single-stage trial design59 and the primary outcome measure of molecular response (BCR–ABL or WT1 transcript level) for CML patients.
A molecular response (BCR–ABL or WT1 transcript level) rate of 20% would imply that the vaccine clearly warrants further investigation. A molecular response rate of ≤ 5% would be unacceptable and would indicate that the vaccine does not warrant further investigation. The probability of obtaining a false-positive result, α (i.e. incorrectly accepting for further trial a treatment that has a true response rate of 5%), was set at 10%. The probability of a false-negative result, β (i.e. incorrectly rejecting for further trial a treatment with a true response rate of 20%), was set at 10%. Using these parameters (α = 0.1, β = 0.1, p0 = 5%, p1 = 20%), 32 HLA A2+ patients needed to be recruited to the trial. The drop-out rate was expected to be < 10% and hence a total of 36 patients would be recruited. The control arm consisted of all eligible and consenting patients who were HLA A2–.
This trial needed to observe a minimum of four molecular responders out of 32 patients in order to provide evidence that the vaccine warrants further investigation.
Acute myeloid leukaemia
The sample size calculation for the AML treatment group was based on A’Hern’s single-stage design59 and the primary outcome measure of PFS at 2 years post vaccination.
A PFS rate of 50% at 2 years post vaccination would indicate that the vaccine warrants further investigation. A PFS rate of ≥ 30% at 2 years post vaccination would be unacceptable and would indicate the vaccine does not warrant further investigation. The probability of obtaining a false-positive result, α (i.e. incorrectly accepting for further trial a treatment that has a true PFS rate of 30%), was set at 10%. The probability of a false-negative result, β (i.e. incorrectly rejecting for further trial a treatment with a true PFS rate of 50%) was set at 10%. Using these parameters, α = 0.1, β = 0.1, p0 = 30%, p1 = 50%, 39 HLA A2+ patients needed to be recruited to the trial. The dropout rate was expected to be > 10%. The control arm consisted of all eligible and consenting patients who were HLA A2–.
This trial needed to observe a minimum of 16 out of 39 patients who are progression free and alive at 2 years in order to provide evidence that the vaccine warrants further investigation.
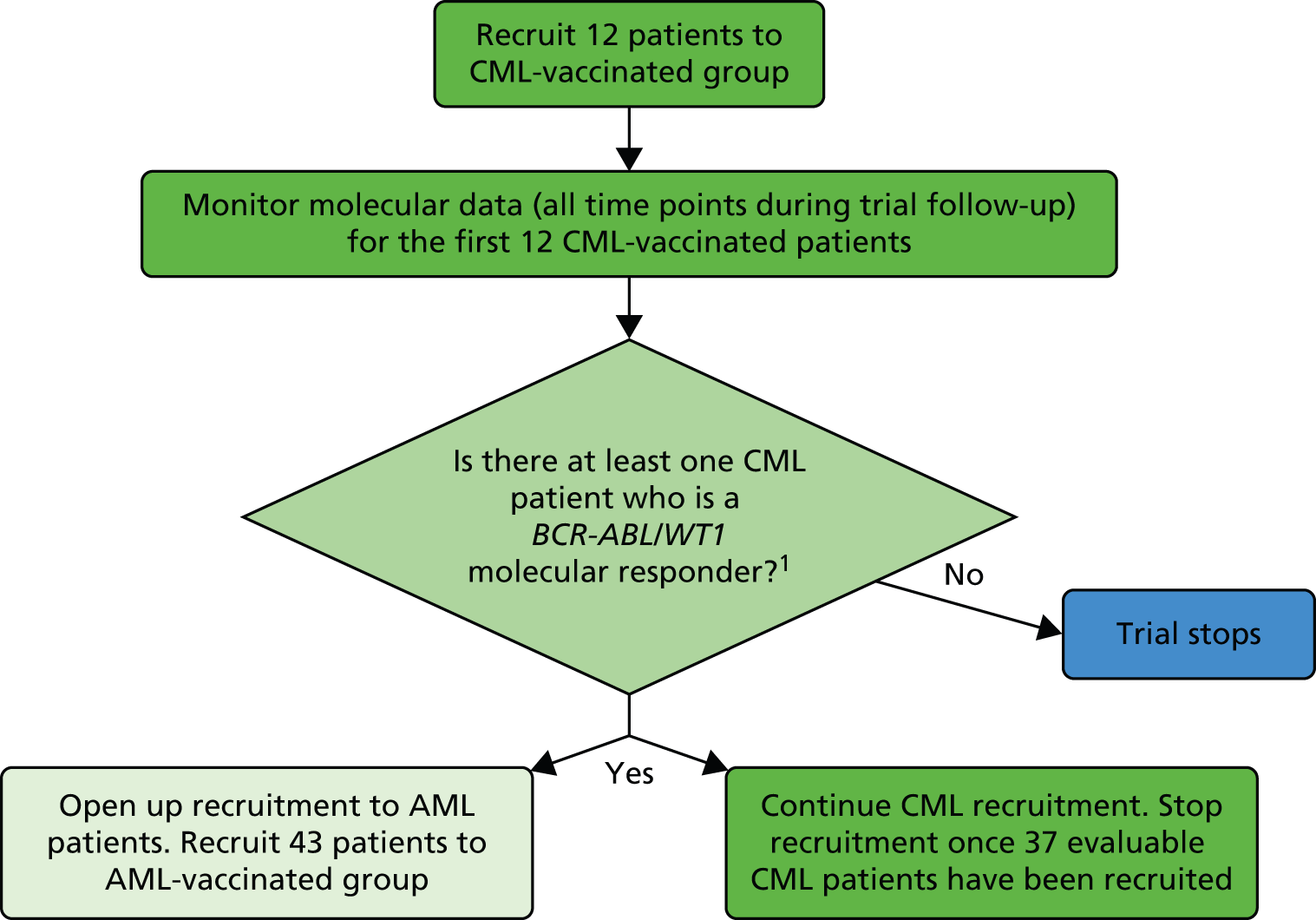
Interim analyses and stopping guidelines
Following the original trial design of Simon’s optimal phase II trial design,60 there was a ‘start/stop’ evaluation once 12 patients had been enrolled into the trial (Figure 3). The molecular data for these patients will be evaluated (molecular monitoring). If no molecular responses are seen in the first 12 vaccinated patients with CML, recruitment will cease for CML and will not begin for AML, as there will not be sufficient clinical interest to pursue further. After the change in trial design to A’Hern’s one-stage design,59 this stopping evaluation was no longer in required.
FIGURE 3.
Flow diagram showing the original two-stage design for the CML patient group.
A Data Monitoring and Ethics Committee (DMEC) was convened on behalf of, and with input from, the Trial Management Group. The committee’s remit was to meet at regular intervals to review the safety listings and recruitment and to make a recommendation on whether or not to continue beyond 12 patients in the CML and AML cohorts. Unplanned DMEC meetings were to be called if required during the study.
In addition, the DMEC would review any cases of CTCAE grade 3 adverse reactions, possibly or likely to be related to vaccination to assess if there were sufficient reason to suspend or terminate the study.
Statistical analysis
All trial analyses and reporting were carried out following the Consolidated Standards of Reporting Trials (CONSORT) guidelines. Statistical analysis was carried out in SAS, version 9.3, following a predefined statistical analysis plan.
Analysis populations
Molecular analysis
The analyses of the molecular responses were performed on all patients with molecular data at a minimum of two post-baseline time points (HLA A2+ patients must also have received at least one dose of the vaccine).
Immunological analysis
To be evaluable for an immunological response, HLA A2+ patients must have received at least one dose of the vaccine and the immunological testing must have been available until at least week 8 post first dose.
Safety analysis
For safety analyses, all HLA A2+ patients who received at least one trial drug administration were evaluable for toxicity. All controls were included in the safety analyses, where relevant.
Other analysis (intention-to-treat population)
For all other analyses, an intention-to-treat (ITT) principle (i.e. the planned treatment regimen) was used, which included all registered patients who obtained HLA A2+ status.
Preliminary analyses
Disposition of patients and follow-up information on the ITT population were analysed separately for HLA A2+ and HLA A2– patients (see Figure 4 and Table 2). Major protocol deviations were also listed for the ITT population (see Box 1).
Patient characteristics recorded at baseline, including the patient’s age, gender, ECHO result and WHO performance status, were summarised by HLA A2 status for the ITT population (see Table 3). In addition, baseline BCR–ABL and WT1 molecular data were analysed separately for the molecular analysis population and summarised by HLA A2 status (see Tables 4 and 5).
Dosing information on pre-trial imatinib treatment was summarised by HLA A2 status for the ITT population (see Table 6).
Treatment analyses
Vaccination administration and electroporation failure summarises were presented for the ITT population (HLA A2+ patients only), together with details of any reasons for premature withdrawals from treatment (see Tables 7–9).
Safety analyses
Serious adverse events (SAEs), including a SAE summary and listing of SAEs by system organ class and CTCAE term, were presented by HLA A2 status for the safety analysis population (Tables 10 and 11). In addition, a complete SAE listing was provided for the safety analysis population (see Table 12).
Information on all AEs was also presented by HLA A2 status for the safety analysis population, which comprised a summary by worst CTCAE grade recorded across the following type of AEs:
-
all AEs (see Table 13)
-
cardio-related AEs (see Table 14)
-
renal-related AEs (see Table 15)
-
bone marrow-related AEs (see Table 16)
-
other-related AEs (see Table 17).
Fisher’s exact test was used to compare differences in rates of toxicity in CML patients with controls for the safety population. No adjustment was made for multiple testing and, therefore, borderline significance should not be overinterpreted.
A list of all AEs was also presented for the safety analysis population (see Table 18). In addition, any concomitant medications taken because of pain within 48 hours following vaccination were summarised for HLA A2+ patients only in the safety analysis population (see Table 19).
Primary analyses
The primary outcome is a BCR–ABL molecular response (major or minor response or CMR), which was summarised for the molecular analysis population by HLA A2 status (see Table 20).
Secondary analyses
Secondary outcomes carried out on the molecular analysis population included:
-
BCR–ABL (major response or CMR), summarised by HLA A2 status (see Table 21)
-
WT1 (major or minor response or CMR), summarised by HLA A2 status (see Table 22).
In addition, the following time-to-event secondary outcomes were also analysed by HLA A2 status for the ITT population:
In each case, the median time to event was presented along with time to event listings, a log-rank test, results from an unadjusted Cox proportional hazards model with alpha set at the 10% two-sided level and a Kaplan–Meier plot.
Time to a BCR–ABL response (major or minor or CMR) and time to a WT1 response (major or minor or CMR) from the beginning of inhibitor treatment and also from the date of informed consent were also analysed using a similar approach for the molecular analysis population, together with the duration of BCR–ABL and WT1 responses (see Table 27 and Figure 9, and Table 28 and Figure 10, respectively).
Pain assessment information recorded immediately after vaccination and recorded 48 hours post vaccination were summarised in terms of the median pain score recorded and the worst pain score recorded for HLA A2+ patients in the ITT population (see Tables 29 and 30, respectively).
Immunological analyses
The immunological analyses carried out on vaccinated HLA A2+ patients included:
-
anti-FrC antibody in serum, as evaluated by a validated ELISA
-
WT1-specific T-cell responses in blood and bone marrow, as evaluated by a validated WT1 tetramer assay.
The ELISA acceptance criteria included a post-vaccination time point level > twofold increase, as well as significantly greater levels of anti-FrC antibody compared with pre-vaccination baseline (p < 0.05, Dunnett’s multiple comparisons test).
The WT1 tetramer acceptance criteria included a post-vaccination time point level > twofold over baseline and confirmed by three independent flow cytometric experts.
Chapter 4 Trial results
The intention to recruit 39 AML patients was not realised because of the early termination of the trial. The information included in this section only relates to patients in the CML treatment group.
Recruitment
Below is a summary of the recruitment milestones in the WIN trial:
-
Southampton (first site) opened: 1 February 2011.
-
Hammersmith opened: 9 May 2011.
-
Exeter opened: 21 November 2011.
-
First patient registered: 1 June 2011.
-
Chronic myeloid leukaemia arm closed: 26 February 3013.
-
Last patient, last visit: 31 July 2014.
-
Trial termination: 25 March 2014.
Flow of participants through the trial
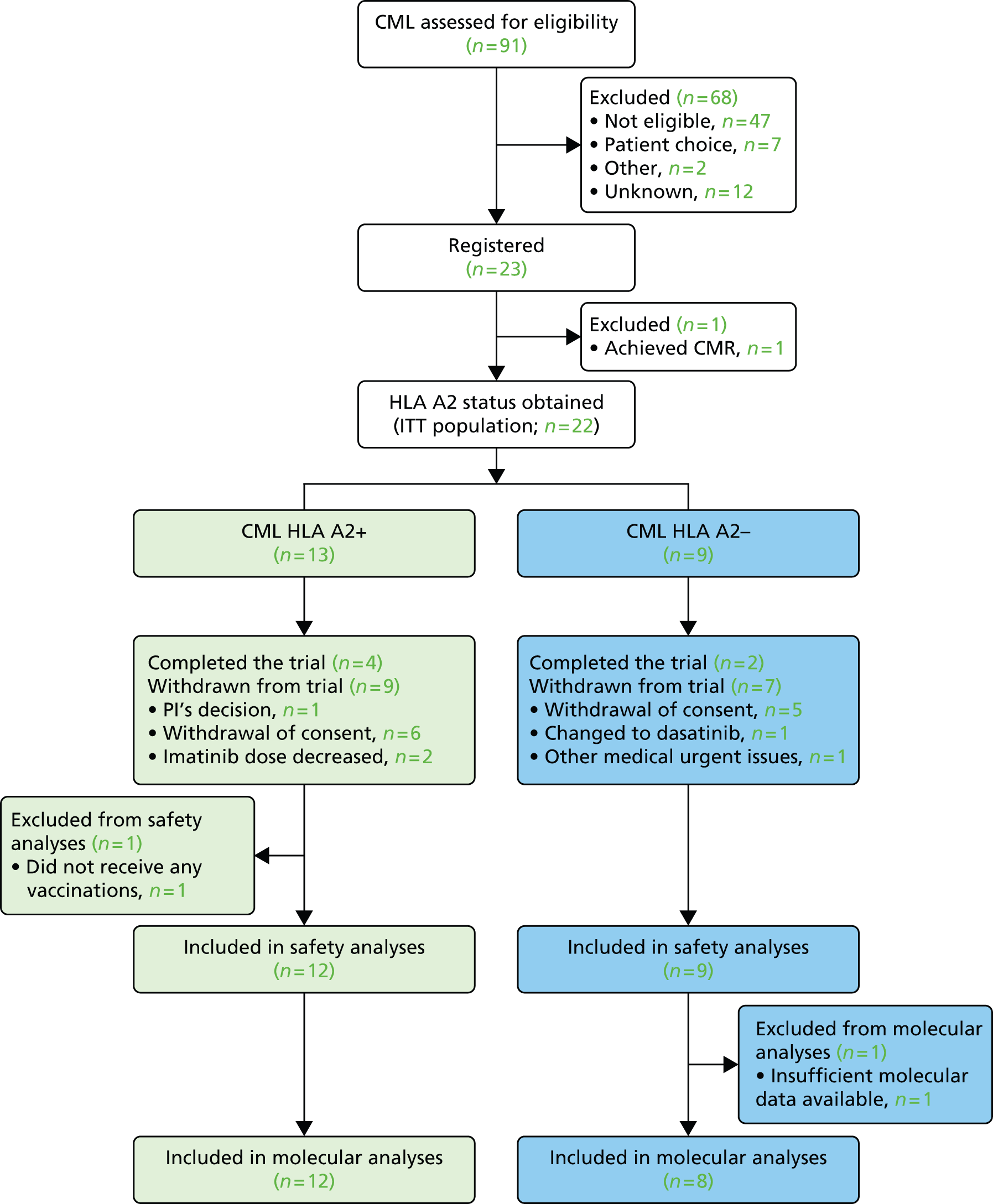
A total of 91 CML patients were assessed for eligibility for entry into the WIN trial and 23 CML patients were registered. Of these, 13 patients were HLA A2+ and 12 out of 13 began vaccination with a control group of nine HLA A2– patients.
Twelve HLA A2+ patients were vaccinated and 12 out of 12 as well as nine out of nine control patients were included in both the safety and molecular analyses. Four vaccinated and two control patients completed the study protocol treatment. The other patients either withdrew consent (n = 11) or underwent modification of their TKI treatment, thus requiring study termination.
Only one AML patient was assessed for eligibility for entry into the WIN trial, but decided not to enter into the trial.
Figure 4 shows the CONSORT diagram for the CML group only.
FIGURE 4.
Consolidated Standards of Reporting Trials flow chart (CML group only).
| Characteristic | HLA A2+ (n = 13), n (%) | HLA A2– (n = 9), n (%) | Total (n = 22), n (%) |
|---|---|---|---|
| Number who completed the trial | 4 (30.8) | 2 (22.2) | 6 (27.3) |
| Number who discontinued the trial | 9 (69.2) | 7 (77.8) | 16 (72.7) |
| PI’s decisiona | 1 (11.1) | 0 | 1 (6.3) |
| Withdrawal of consenta | 6 (66.7) | 5 (71.4) | 11 (68.8) |
| Difficulty in complying with study visits because of work commitments and because patient found vaccinations distressingb | 1 (16.7) | 0 | 1 (9.1) |
| As there are going to be no more vaccinationsb | 1 (16.7) | 0 | 1 (9.1) |
| Good response, not keen on the number of visitsb | 1 (16.7) | 0 | 1 (9.1) |
| Patient choiceb | 3 (50.0) | 5 (100) | 8 (72.7) |
| Othera | 2 (22.2) | 2 (28.6) | 4 (25.0) |
| Changed to dasatinibc | 0 | 1 (50.0) | 1 (25.0) |
| Imatinib dose decreased to 300 mg o.d.c | 1 (50.0) | 0 | 1 (25.0) |
| Imatinib dose reduced to 400 mg o.d.c | 1 (50.0) | 0 | 1 (25.0) |
| Other medical urgent issuesc | 0 | 1 (50.0) | 1 (25.0) |
| Time from consent date to end of study (months) | |||
| n | 9 | 7 | 16 |
| Median (IQR) | 18.27 (14.23–26.68) | 14.95 (14.46–18.69) | 14.98 (14.34–23.00) |
| Range | 5.42–33.48 | 3.94–23.00 | 3.94–33.48 |
Major protocol deviations
There were no major protocol deviations. Box 1 summarises the protocol deviations that occurred during the trial.
Missed patient visits:
-
2-C-008: visit 7 and visit 8.
-
2-C-015: visit 3 and visit 4.
-
2-C-016: visit 1 and visit 2.
-
2-C-020: visit 6.
Immunological blood sample volumes below the protocol requirements.
1-C-023: baseline molecular sample taken, but not sent to central laboratory.
Characteristics of the trial sample
Tables 3–5 detail the baseline characteristics of the trial sample. The distribution of patient baseline characteristics was fully consistent with the expected distribution. The arms appear well balanced, within the limits of a small sample size.
As expected from the entry criteria of the trial, the BCR–ABL transcript levels were low. WT1 transcripts were detected in all but one patient, in line with published data sets.
| Characteristic | HLA A2+ (n = 13) | HLA A2– (n = 9) | Total (n = 22) |
|---|---|---|---|
| Gender, n (%)a | |||
| Male | 6 (46.2) | 4 (50.0) | 10 (47.6) |
| Female | 7 (53.8) | 4 (50.0) | 11 (52.4) |
| Missing | 0 (0) | 1 (11.1)b | 1 (4.5) |
| WHO performance status, n (%)a | |||
| 0: Asymptomatic | 13 (100) | 8 (100%) | 21 (100) |
| 1: Symptomatic but completely ambulatory | 0 (0) | 0 (0) | 0 (0) |
| Missing | 0 (0) | 1 (11.1)b | 1 (4.5) |
| ECHO result, n (%)a | |||
| Normal | 10 (76.9) | NAc | 10 (76.9) |
| Abnormal | 0 (0) | NAc | 0 (0) |
| Not done | 3 (23.1) | NAc | 3 (23.1) |
| Age at baseline (years) | |||
| n | 13 | 9 | 22 |
| Median (IQR) | 52 (42–60) | 56 (51–63) | 53 (46–63) |
| Range | 23–66 | 42–73 | 23–73 |
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 8) | Total (n = 20) |
|---|---|---|---|
| Transcript type, n (%) | |||
| e13/e14a2 | 0 (0) | 1 (12.5) | 1 (5.0) |
| e13a2 | 2 (16.7) | 3 (37.5) | 5 (25.0) |
| e13a2/e14a2 | 0 (0) | 1 (12.5) | 1 (5.0) |
| e14a2 | 7 (58.3) | 2 (25.0) | 9 (45.0) |
| Unknown | 3 (25.0) | 1 (12.5) | 4 (20.0) |
| Result, n (%) | |||
| Positive | 12 (100) | 8 (100) | 20 (100) |
| Undetectable | 0 (0) | 0 (0) | 0 (0) |
| Failed | 0 (0) | 0 (0) | 0 (0) |
| Transcript level (transcripts/mg of RNA) | |||
| n | 12 | 8 | 20 |
| Median (IQR) | 6.5 (2.5–12.5) | 16.5 (7.0–29.5) | 7.0 (4.0–23.5) |
| Range | 2–42 | 1–68 | 1–68 |
| ABL control (transcripts/mg of RNA) | |||
| n | 12 | 8 | 20 |
| Median (IQR) | 28,100 (15,900–46,100) | 30,950 (20,450–69,300) | 29,550 (18,150–47,450) |
| Range | 14,300–61,800 | 14,300–97,900 | 14,300–97,900 |
| BCR to ABL transcript level ratio | |||
| n | 12 | 8 | 20 |
| Median (IQR) | 0.021 (0.013–0.056) | 0.036 (0.023–0.079) | 0.027 (0.015–0.061) |
| Range | 0.003–0.098 | 0.005–0.117 | 0.003–0.117 |
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 8) | Total (n = 20) |
|---|---|---|---|
| Result, n (%) | |||
| Positive | 12 (100) | 7 (87.5) | 19 (95.0) |
| Undetectable | 0 (0) | 1 (12.5) | 1 (5.0) |
| Failed | 0 (0) | 0 (0) | 0 (0) |
| Transcript level (transcripts/mg of RNA) | |||
| n | 12 | 8 | 20 |
| Median (IQR) | 5.9 (2.0–9.5) | 3.0 (1.5–7.5) | 4.5 (2.0–9.5) |
| Range | 1–83 | 0–74 | 0–83 |
| GUS control (transcripts/mg of RNA) | |||
| n | 12 | 8 | 20 |
| Median (IQR) | 40,027.2 (23,402.5–59,431.5) | 45,856.9 (36,828.5–59,618.8) | 41,656.9 (31,270.0–59,431.5) |
| Range | 16,828.8–99,100.0 | 22,644.4–12,3530.8 | 16,828.8–123,530.8 |
| WT1 to GUS transcript level ratio | |||
| n | 12 | 8 | 20 |
| Median (IQR) | 0.011 (0.006–0.048) | 0.004 (0.003–0.019) | 0.010 (0.003–0.022) |
| Range | 0.002–0.294 | 0.000–0.025 | 0.000–0.294 |
Treatment adherence
Pre-trial information is described in Table 6 and treatment adherence is described in Tables 7–9.
| Characteristic | HLA A2+ (n = 13) | HLA A2– (n = 9) | Total (n = 22) |
|---|---|---|---|
| Received pre-trial imatinib,a n (%) | 12 (92.3) | 8 (88.9) | 20 (90.9) |
| Maximum imatinib dose receivedb | |||
| 300 mg o.d. | 1 (8.3) | 0 | 1 (5.0) |
| 400 mg o.d. | 6 (50.0) | 4 (50.0) | 10 (50.0) |
| 500 mg o.d. | 0 | 1 (12.5) | 1 (5.0) |
| 600 mg o.d. | 4 (33.3) | 1 (12.5) | 5 (25.0) |
| 800 mg o.d. | 1 (8.3) | 2 (25.0) | 3 (15.0) |
| Imatinib duration (months)c | |||
| n | 12 | 8 | 20 |
| Median (IQR) | 89.1 (69.8–101.7) | 79.1 (67.9–98.0) | 85.0 (69.8–100.8) |
| Range | 28.1–110.1 | 27.8–105.6 | 27.8–110.1 |
| Imatinib duration (years)d | |||
| n | 12 | 8 | 20 |
| Median (IQR) | 7.4 (5.8–8.5) | 6.6 (5.7–8.2) | 7.1 (5.8–8.4) |
| Range | 2.3–9.2 | 2.3–8.8 | 2.3–9.2 |
| Characteristic | HLA A2+ (n = 13) |
|---|---|
| Received treatment,a n (%) | 12 (92.3) |
| Initial p.DOM–WT1-37 or p.DOM–WT1-126 vaccination administration | |
| No vaccines administeredb | 0 (0) |
| At least one vaccine administeredb | 12 (100) |
| At least one p.DOM–WT1-37 vaccination administered where no p.DOM–WT1-126 vaccination administered (or vice versa)b | 0 (0) |
| Number of initial p.DOM–WT1-37 or p.DOM–WT1-126 vaccinations received | |
| n | 12 |
| Median (IQR) | 6 (6–6) |
| Range | 3–6 |
| Proceeded onto additional vaccinations,a n (%) | 1 (7.7) |
| Additional p.DOM–WT1-37 or p.DOM–WT1-126 vaccinations | |
| No vaccines administeredc | 0 (0) |
| At least one vaccine administeredc | 1 (100) |
| At least one p.DOM–WT1-37 vaccination administered where no p.DOM–WT1-126 vaccination administered (or vice versa)c | 0 (0) |
| Number of additional p.DOM–WT1-37 or p.DOM–WT1-126 vaccinations received | |
| n | 1 |
| Median (IQR) | 1 (1–1) |
| Range | 1–1 |
| Received treatment,a n (%) | 12 (92.3) |
| Any (initial or additional) p.DOM–WT1-37 or p.DOM–WT1-126 vaccinations | |
| No vaccines administeredb | 0 (0) |
| At least one vaccine administeredb | 12 (100) |
| At least one p.DOM–WT1-37 vaccination administered where no p.DOM–WT1-126 vaccination administered (or vice versa)b | 0 (0) |
| Number of any (initial or additional) p.DOM–WT1-37 or p.DOM–WT1-126 vaccinations received | |
| n | 12 |
| Median (IQR) | 6 (6–6) |
| Range | 3–7 |
| Characteristic | HLA A2+ (n = 13) |
|---|---|
| Received treatment,a n (%) | 12 (92.3) |
| Initial p.DOM–WT1-37 or WT1-126 electroporation failures | |
| No electroporation failures | 10 (83.3) |
| At least one electroporation failure | 2 (16.7) |
| Electroporation failure details | |
| Electroporation failure in initial p.DOM–WT1-37 vaccinationb | 1 (50.0) |
| Electroporation failure in initial p.DOM–WT1-126 vaccinationc | 1 (50.0) |
| Initial p.DOM–WT1-37 or WT1-126 electroporation failures | |
| n | 12 |
| Median (IQR) | 0 (0–0) |
| Range | 0–1 |
| Proceeded onto additional vaccinations,d n (%) | 1 (7.7) |
| Additional p.DOM–WT1-37 or p.DOM–WT1-126 electroporation failure | |
| No electroporation failures | 1 (100) |
| At least one electroporation failure | 0 (0) |
| Additional p.DOM–WT1-37 or 126 electroporation failures | |
| n | 0 |
| Median (IQR) | 0 (0–0) |
| Range | 0–0 |
| Received treatment,e n (%) | 12 (92.3) |
| Any (initial or additional) p.DOM–WT1-37 or WT1-126 electroporation failures | |
| No electroporation failures | 10 (83.3) |
| At least one electroporation failure | 2 (16.7) |
| Electroporation failure details: | |
| Electroporation failure in initial p.DOM–WT1-37 vaccinationb | 1 (50.0) |
| Electroporation failure in initial p.DOM–WT1-126 vaccinationc | 1 (50.0) |
| Any (initial or additional) p.DOM–WT1-37 or WT1-126 electroporation failures | |
| n | 12 |
| Median (IQR) | 0 (0–0) |
| Range | 0–1 |
| Characteristic | HLA A2+ (n = 13), n (%) |
|---|---|
| Received treatmenta | 12 (92.3) |
| Patients who received all six initial vaccinationsb | 10 (83.3) |
| Patients who did not receive all six initial vaccinationsb | 2 (16.7) |
| Withdrew consent (from treatment only)c | 2 (100) |
| Patients who proceeded to receive additional vaccinationsb | 1 (7.7) |
| Patients who received all six additional vaccinationsd | 0 (0) |
| Patients who did not receive all six additional vaccinations | 1 (100) |
| Withdrew consent (from trial)e | 1 (100) |
Maintenance imatinib treatment was given throughout the trial (see Table 6). Table 6 also describes the duration of pre-trial treatment with imatinib. A course of six pairs of vaccinations (with p.DOM–WT1-37 and p.DOM–WT1-126, respectively) were planned for weeks 0, 4, 8, 12, 16 and 20, followed by maintenance vaccinations. Median vaccination was the expected 12 doses, with a range of 3–6 pairs of vaccinations delivered. Only one patient went on to receive one further set of maintenance vaccinations.
There were two technical failures of electroporation delivery in the same patient at one vaccination time point (Table 8). Ten out of 12 patients completed all six pairs of vaccination to week 20. Two patients withdrew consent for further injections after three doses of the vaccine (see Table 9).
Safety
Serious adverse events
Details of SAEs are provided in Tables 10–12. No vaccine-related SAEs were observed. Three SAEs were observed in the control group and are not vaccine related.
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 9) | Total (n = 21) |
|---|---|---|---|
| Total number of SAEs, n (%) | 0 (0) | 3 | 3 |
| Number of patients experiencing at least one SAE,a n (%) | 0 (0) | 1 (11.1) | 1 (4.8) |
| PI assessment,b n (%) | |||
| SUSAR | 0 (0) | 0 (0) | 0 (0) |
| SAR | 0 (0) | 0 (0) | 0 (0) |
| SAE | 0 (0) | 3 (100) | 3 (100) |
| Clinical reviewer assessment,b n (%) | |||
| SUSAR | 0 (0) | 0 (0) | 0 (0) |
| SAR | 0 (0) | 0 (0) | 0 (0) |
| SAE | 0 (0) | 3 (100) | 3 (100) |
| Grade,b n (%) | |||
| Mild | 0 (0) | 0 (0) | 0 (0) |
| Moderate | 0 (0) | 2 (66.7) | 2 (66.7) |
| Severe | 0 (0) | 1 (33.3) | 1 (33.3) |
| Life-threatening | 0 (0) | 0 (0) | 0 (0) |
| Death related to AE | 0 (0) | 0 (0) | 0 (0) |
| Why was the event serious?,b n (%) | |||
| Resulted in death | 0 (0) | 0 (0) | 0 (0) |
| Life-threatening | 0 (0) | 0 (0) | 0 (0) |
| Required hospitalisation or prolongation of existing hospitalisation | 0 (0) | 3 (100) | 3 (100) |
| Persistent or significant disability/incapacity | 0 (0) | 0 (0) | 0 (0) |
| Congenital anomaly/birth defect | 0 (0) | 0 (0) | 0 (0) |
| CTCAE, version 4, system organ class/terma | HLA A2+ (n = 12) | HLA A2– (n = 9) | Total (n = 21) |
|---|---|---|---|
| Gastrointestinal disorders,b n (%) | 0 (0) | 3 (100) | 3 (100) |
| Abdominal pain,c n (%) | 0 (0) | 3 (100) | 3 (100) |
| HLA A2 status | PI assessment | Clinical reviewer assessment | CTCAE version 4 system organ class | CTCAE version 4 term | Main symptom | Patient ID | Date of informed consent | Date of onset of SAE | Grade | Why was event serious? | Was the vaccine given?a | Last administration of vaccine |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Negative | SAE | SAE | Gastrointestinal disorders | Abdominal pain | Abdominal pain (cause unknown) | 1-C-003 | 3 August 2011 | 30 November 2011b | 3 = severe | 3 = hospitalisation or prolongation of hospitalisation | No/no | NA |
| Negative | SAE | SAE | Gastrointestinal disorders | Abdominal pain | Abdominal pain | 1-C-003 | 3 August 2011 | 09 December 2011 | 2 = moderate | 3 = hospitalisation or prolongation of hospitalisation | No/no | NA |
| Negative | SAE | SAE | Gastrointestinal disorders | Abdominal pain | Abdominal pain | 1-C-003 | 3 August 2011 | 23 December 2011 | 2 = moderate | 3 = hospitalisation or prolongation of hospitalisation | No/no | NA |
Adverse events
In the vaccine group, 11 grade 1–3 AEs were observed, compared with nine grade 1–3 AEs in the control group (Table 13). Of these AEs, palpitations in one vaccinated patient were of special interest (Table 14). In one patient, 12 grade 1–3 renal AEs were documented (pre-existing grade 1 renal dysfunction), with no such AEs observed in the control group (Table 15). No haematological toxicities were observed (Table 16).
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 9) | Total (n = 21) |
|---|---|---|---|
| Worst CTCAE grade experienced, n (%) | |||
| None | 1 (8.3) | 0 (0) | 1 (4.8) |
| Grade 1: mild | 6 (50.0) | 4 (44.4) | 10 (47.6) |
| Grade 2: moderate | 5 (41.7) | 4 (44.4) | 9 (42.9) |
| Grade 3: severe | 0 (0) | 1 (11.1) | 1 (4.8) |
| Grade 4: life-threatening | 0 (0) | 0 (0) | 0 (0) |
| Grade 5: death related to an AE | 0 (0) | 0 (0) | 0 (0) |
| At least one severe (CTCAE grade 3 or above) AE | 0 (0) | 1 (11.1) | 1 (4.8) |
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 9) | Total (n = 21) | p-valued |
|---|---|---|---|---|
| Worst CTCAE grade for cardio-related AEs experienced, n (%) | ||||
| None | 11 (91.7) | 9 (100) | 20 (95.2) | – |
| Grade 1: mild | 0 (0) | 0 (0) | 0 (0) | – |
| Grade 2: moderate | 1 (8.3) | 0 (0) | 1 (4.8) | – |
| Grade 3: severe | 0 (0) | 0 (0) | 0 (0) | – |
| Grade 4: life-threatening | 0 (0) | 0 (0) | 0 (0) | – |
| Grade 5: death related to AE | 0 (0) | 0 (0) | 0 (0) | – |
| At least one severe (CTCAE grade 3 or above) cardio-related AE | 0 (0) | 0 (0) | 0 (0) | – |
| CTCAE, version 4, term, n (%) | ||||
| Palpitations | 1 (8.3) | 0 | 1 (4.8) | > 0.999 |
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 9) | Total (n = 21) | p-valued |
|---|---|---|---|---|
| Worst CTCAE grade for renal-related AEs experienced, n (%) | ||||
| None | 8 (66.7) | 9 (100) | 17 (81.0) | – |
| Grade 1: mild | 3 (25.0) | 0 (0) | 3 (14.3) | – |
| Grade 2: moderate | 1 (8.3) | 0 (0) | 1 (4.8) | – |
| Grade 3: severe | 0 (0) | 0 (0) | 0 (0) | – |
| Grade 4: life-threatening | 0 (0) | 0 (0) | 0 (0) | – |
| Grade 5: death related to AE | 0 (0) | 0 (0) | 0 (0) | – |
| At least one severe (CTCAE grade 3 or above) renal-related AE | 0 (0) | 0 (0) | 0 (0) | – |
| CTCAE, version 4, term, n (%) | ||||
| CK increased | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Renal and urinary disorders, other | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Renal calculi | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Urinary tract pain | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 9) | Total (n = 21) |
|---|---|---|---|
| Worst CTCAE grade for bone marrow-related AEs experienced, n (%) | |||
| None | 12 (100) | 9 (100) | 21 (100) |
| Grade 1: mild | 0 (0) | 0 (0) | 0 (0) |
| Grade 2: moderate | 0 (0) | 0 (0) | 0 (0) |
| Grade 3: severe | 0 (0) | 0 (0) | 0 (0) |
| Grade 4: life-threatening | 0 (0) | 0 (0) | 0 (0) |
| Grade 5: death related to AE | 0 (0) | 0 (0) | 0 (0) |
| At least one severe (CTCAE grade 3 or above) bone marrow-related AE | 0 (0) | 0 (0) | 0 (0) |
A range of mild to moderate AEs was observed (Table 17). No detectable significance was observed between the vaccination and observation arms.
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 9) | Total (n = 21) | p-valuee |
|---|---|---|---|---|
| Worst CTCAE grade for other-related AEs experienced, n (%) | ||||
| None | 1 (8.3) | 0 | 1 (4.8) | – |
| Grade 1: mild | 7 (58.3) | 4 (44.4) | 11 (52.4) | – |
| Grade 2: moderate | 4 (33.3) | 4 (44.4) | 8 (38.1) | – |
| Grade 3: severe | 0 (0) | 1 (11.1) | 1 (4.8) | – |
| Grade 4: life-threatening | 0 (0) | 0 (0) | 0 (0) | – |
| Grade 5: death related to AE | 0 (0) | 0 (0) | 0 (0) | – |
| At least one severe (CTCAE grade 3 or above) other-related AE | 0 (0) | 1 (11.1) | 1 (4.8) | – |
| CTCAE, version 4, term, n (%) | ||||
| Abdominal pain | 0 (0) | 1 (11.1) | 1 (4.8) | 0.429 |
| Anorexia | 0 (0) | 1 (11.1) | 1 (4.8) | 0.429 |
| Bloating | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Bone pain | 0 (0) | 1 (11.1) | 1 (4.8) | 0.429 |
| Bruising | 2 (16.7) | 0 (0) | 2 (9.5) | 0.486 |
| Cough | 1 (8.3) | 2 (22.2) | 3 (14.3) | 0.553 |
| Depression | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Diarrhoea | 1 (8.3) | 1 (11.1) | 2 (9.5) | > 0.999 |
| Dyspepsia | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Ear and labyrinth disorders, other | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Oedema limbs | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Epistaxis | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Eye disorders, other | 2 (16.7) | 1 (11.1) | 3 (14.3) | > 0.999 |
| Eye infection | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Fatigue | 4 (33.3) | 0 (0) | 4 (19.0) | 0.104 |
| Headache | 0 (0) | 2 (22.2) | 2 (9.5) | 0.171 |
| Hypotension | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Injury, poisoning and procedural complications, other | 2 (16.7) | 0 (0) | 2 (9.5) | 0.486 |
| Musculoskeletal and connective tissue disorders, other | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Myalgia | 3 (25.0) | 0 (0) | 3 (14.3) | 0.229 |
| Nail infection | 0 (0) | 1 (11.1) | 1 (4.8) | 0.429 |
| Nausea | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Nervous system disorders, other | 1 (8.3) | 2 (22.2) | 3 (14.3) | 0.553 |
| Pain in extremity | 1 (8.3) | 0 (0) | 1 (4.8) | > 0.999 |
| Respiratory, thoracic and mediastinal disorders, other | 0 (0) | 1 (11.1) | 1 (4.8) | 0.429 |
| Skin and subcutaneous tissue disorders, other | 2 (16.7) | 1 (11.1) | 3 (14.3) | > 0.999 |
| Skin ulceration | 0 (0) | 1 (11.1) | 1 (4.8) | 0.429 |
| Tooth infection | 0 (0) | 1 (11.1) | 1 (4.8) | 0.429 |
| Upper respiratory infection | 2 (16.7) | 5 (55.6) | 7 (33.3) | 0.159 |
| Vomiting | 1 (8.3) | 1 (11.1) | 2 (9.5) | > 0.999 |
| Watering eyes | 0 (0) | 1 (11.1) | 1 (4.8) | 0.429 |
Table 18 provides a complete list of all AEs for the safety analysis population.
| AE category | CTCAE version 4 system organ class | CTCAE version 4 term | Grade | HLA A2 status | Patient ID | Was the event serious? | Relationship | Outcome | AE start date | AE end date |
|---|---|---|---|---|---|---|---|---|---|---|
| Cardio | Cardiac disorders | Palpitations | Moderate | Positive | 2-C-004 | No | Unrelated | Ongoing | March 2013 | Ongoing |
| Cardio | Cardiac disorders | Palpitations | Mild | Positive | 2-C-004 | No | Unrelated | Resolved | September 2012 | September 2012 |
| Other | Ear and labyrinth disorders | Ear and labyrinth disorders, other | Mild | Positive | 3-C-017 | No | Unlikely to be related | Unknown | 12 September 2012 | Unknown |
| Other | Eye disorders | Eye disorders, other | Mild | Negative | 2-C-016 | No | Unrelated | Resolved | 2 August 2012 | 20 August 2012 |
| Other | Eye disorders | Eye disorders, other | Mild | Positive | 1-C-005 | No | Unrelated | Resolved | 27 September 2011 | 13 October 2011 |
| Other | Eye disorders | Eye disorders, other | Mild | Positive | 2-C-013 | No | Unrelated | Ongoing | 4 June 2012 | Ongoing |
| Other | Eye disorders | Watering eyes | Mild | Negative | 3-C-011 | No | Unrelated | Unknown | 14 March 2012 | Unknown |
| Other | Gastrointestinal disorders | Abdominal pain | Severe | Negative | 1-C-003 | Yes | Unrelated | Resolved | 1 December 2011 | 6 December 2011 |
| Other | Gastrointestinal disorders | Abdominal pain | Moderate | Negative | 1-C-003 | No | Unrelated | Resolved | 22 November 2011 | 22 November 2011 |
| Other | Gastrointestinal disorders | Abdominal pain | Moderate | Negative | 1-C-003 | Yes | Unrelated | Resolved | 9 December 2011 | 21 December 2011 |
| Other | Gastrointestinal disorders | Abdominal pain | Moderate | Negative | 1-C-003 | Yes | Unrelated | Resolved | 23 December 2011 | 29 December 2011 |
| Other | Gastrointestinal disorders | Bloating | Moderate | Positive | 3-C-017 | No | Unlikely to be related | Unknown | 12 September 2012 | Unknown |
| Other | Gastrointestinal disorders | Diarrhoea | Mild | Negative | 3-C-018 | No | Unrelated | Unknown | 8 February 2013 | Unknown |
| Other | Gastrointestinal disorders | Diarrhoea | Mild | Positive | 3-C-017 | No | Unlikely to be related | Unknown | 12 September 2012 | Unknown |
| Other | Gastrointestinal disorders | Dyspepsia | Mild | Positive | 3-C-017 | No | Unlikely to be related | Unknown | 12 September 2012 | Unknown |
| Other | Gastrointestinal disorders | Nausea | Moderate | Positive | 2-C-012 | No | Unrelated | Resolved | 16 August 2012 | 18 August 2012 |
| Other | Gastrointestinal disorders | Vomiting | Moderate | Positive | 2-C-012 | No | Unrelated | Resolved | 16 August 2012 | 18 August 2012 |
| Other | Gastrointestinal disorders | Vomiting | Mild | Negative | 3-C-011 | No | Unrelated | Resolved | 12 March 2012 | 14 March 2012 |
| Other | General disorders and administration site conditions | Oedema limbs | Mild | Positive | 1-C-005 | No | Unrelated | Resolved | 5 May 2012 | 8 May 2012 |
| Other | General disorders and administration site conditions | Fatigue | Mild | Positive | 1-C-005 | No | Unrelated | Unknown | 20 June 2012 | Unknown |
| Other | General disorders and administration site conditions | Fatigue | Mild | Positive | 1-C-023 | No | Unlikely to be related | Resolved | 31 January 2013 | 31 January 2013 |
| Other | General disorders and administration site conditions | Fatigue | Mild | Positive | 2-C-014 | No | Unlikely to be related | Resolved | 11 July 2012 | 18 July 2012 |
| Other | General disorders and administration site conditions | Fatigue | Mild | Positive | 2-C-014 | No | Unlikely to be related | Resolved | 11 July 2012 | 18 July 2012 |
| Other | General disorders and administration site conditions | Fatigue | Mild | Positive | 3-C-017 | No | Possibly related | Resolved | 12 June 2012 | 27 June 2012 |
| Other | Infections and infestations | Eye infection | Mild | Positive | 2-C-004 | No | Unrelated | Resolved | 14 October 2012 | 19 October 2012 |
| Other | Infections and infestations | Nail infection | Moderate | Negative | 3-C-011 | No | Unrelated | Resolved | November 2013 | 12 March 2014 |
| Other | Infections and infestations | Tooth infection | Moderate | Negative | 2-C-009 | No | Unrelated | Ongoing | 18 August 2012 | Ongoing |
| Other | Infections and infestations | Upper respiratory infection | Moderate | Negative | 2-C-008 | No | Unrelated | Resolved | 10 December 2011 | 9 January 2012 |
| Other | Infections and infestations | Upper respiratory infection | Moderate | Negative | 2-C-009 | No | Unrelated | Resolved | 12 April 2012 | May 2012 |
| Other | Infections and infestations | Upper respiratory infection | Mild | Negative | 2-C-006 | No | Unrelated | Resolved | 23 December 2011 | 25 December 2011 |
| Other | Infections and infestations | Upper respiratory infection | Mild | Negative | 2-C-008 | No | Unrelated | Resolved | 14 November 2011 | 18 November 2011 |
| Other | Infections and infestations | Upper respiratory infection | Mild | Negative | 2-C-020 | No | Unrelated | Ongoing | 7 October 2012 | Ongoing |
| Other | Infections and infestations | Upper respiratory infection | Mild | Negative | 3-C-011 | No | Unrelated | Resolved | 6 March 2013 | 20 March 2013 |
| Other | Infections and infestations | Upper respiratory infection | Mild | Positive | 2-C-010 | No | Unrelated | Resolved | 8 February 2012 | 18 February 2012 |
| Other | Infections and infestations | Upper respiratory infection | Mild | Positive | 2-C-013 | No | Unrelated | Ongoing | 21 May 2012 | Ongoing |
| Other | Injury, poisoning and procedural complications | Bruising | Mild | Positive | 1-C-022 | No | Unrelated | Ongoing | 2013 | Ongoing |
| Other | Injury, poisoning and procedural complications | Bruising | Mild | Positive | 2-C-002 | No | Possibly related | Resolved | 24 June 2011 | 25 June 2011 |
| Other | Injury, poisoning and procedural complications | Injury, poisoning and procedural complications, other | Moderate | Positive | 1-C-005 | No | Unrelated | Resolved | 21 September 2011 | 21 September 2011 |
| Other | Injury, poisoning and procedural complications | Injury, poisoning and procedural complications, other | Mild | Positive | 2-C-010 | No | Unrelated | Resolved | January 2012 | February 2012 |
| Renal | Investigations | CK increased | Mild | Positive | 3-C-017 | No | Possibly related | Resolved | 30 May 2012 | 26 June 2012 |
| Other | Metabolism and nutrition disorders | Anorexia | Mild | Negative | 3-C-011 | No | Unrelated | Unknown | 28 April 2012 | Unknown |
| Other | Musculoskeletal and connective tissue disorders | Bone pain | Mild | Negative | 2-C-008 | No | Unrelated | Resolved | 6 December 2011 | 12 December 2011 |
| Other | Musculoskeletal and connective tissue disorders | Bone pain | Mild | Negative | 2-C-008 | No | Unrelated | Ongoing | 20 June 2012 | Ongoing |
| Other | Musculoskeletal and connective tissue disorders | Musculoskeletal and connective tissue disorders, other | Mild | Positive | 1-C-005 | No | Probably related | Resolved | 28 September 2011 | 29 September 2011 |
| Other | Musculoskeletal and connective tissue disorders | Myalgia | Moderate | Positive | 2-C-013 | No | Unrelated | Resolved | 1 June 2012 | 1 June 2012 |
| Other | Musculoskeletal and connective tissue disorders | Myalgia | Mild | Positive | 2-C-007 | No | Unrelated | Ongoing | 21 November 2011 | Ongoing |
| Other | Musculoskeletal and connective tissue disorders | Myalgia | Mild | Positive | 2-C-013 | No | Unrelated | Resolved | 1 June 2011 | 1 June 2012 |
| Other | Musculoskeletal and connective tissue disorders | Myalgia | Mild | Positive | 2-C-013 | No | Unrelated | Ongoing | 12 August 2012 | Ongoing |
| Other | Musculoskeletal and connective tissue disorders | Myalgia | Mild | Positive | 2-C-013 | No | Unrelated | Resolved | 15 June 2012 | 15 June 2012 |
| Other | Musculoskeletal and connective tissue disorders | Myalgia | Mild | Positive | 3-C-017 | No | Possibly related | Resolved | 12 June 2012 | 27 June 2012 |
| Other | Musculoskeletal and connective tissue disorders | Pain in extremity | Mild | Positive | 2-C-010 | No | Unrelated | Resolved | 19 April 2012 | 20 April 2012 |
| Other | Nervous system disorders | Headache | Moderate | Negative | 2-C-008 | No | Unrelated | Ongoing | 14 September 2012 | Ongoing |
| Other | Nervous system disorders | Headache | Moderate | Negative | 2-C-015 | No | Unrelated | Resolved | 9 June 2012 | 9 June 2012 |
| Other | Nervous system disorders | Headache | Mild | Negative | 2-C-008 | No | Unrelated | Ongoing | 6 April 2012 | Ongoing |
| Other | Nervous system disorders | Headache | Mild | Negative | 2-C-015 | No | Unrelated | Resolved | 7 October 2012 | 11 October 2012 |
| Other | Nervous system disorders | Nervous system disorders, other | Moderate | Negative | 2-C-015 | No | Unrelated | Resolved | July 2012 | September 2012 |
| Other | Nervous system disorders | Nervous system disorders, other | Mild | Negative | 3-C-011 | No | Unrelated | Resolved | 2011 | 12 September 2012 |
| Other | Nervous system disorders | Nervous system disorders, other | Mild | Positive | 2-C-002 | No | Unrelated | Ongoing | June 2013 | Ongoing |
| Other | Psychiatric disorders | Depression | Mild | Positive | 3-C-017 | No | Possibly related | Unknown | 12 September 2012 | Unknown |
| Renal | Renal and urinary disorders | Renal and urinary disorders, other | Mild | Positive | 2-C-013 | No | Unrelated | Ongoing | 16 May 2012 | Ongoing |
| Renal | Renal and urinary disorders | Renal calculi | Mild | Positive | 2-C-010 | No | Unrelated | Ongoing | October 2013 | Ongoing |
| Renal | Renal and urinary disorders | Urinary tract pain | Moderate | Positive | 2-C-012 | No | Unrelated | Resolved | April 2013 | April 2013 |
| Other | Respiratory, thoracic and mediastinal disorders | Cough | Moderate | Negative | 2-C-009 | No | Unrelated | Resolved | 12 April 2012 | May 2012 |
| Other | Respiratory, thoracic and mediastinal disorders | Cough | Mild | Negative | 2-C-020 | No | Unrelated | Ongoing | 7 October 2012 | Ongoing |
| Other | Respiratory, thoracic and mediastinal disorders | Cough | Mild | Positive | 2-C-010 | No | Unrelated | Resolved | 8 February 2012 | 18 February 2012 |
| Other | Respiratory, thoracic and mediastinal disorders | Epistaxis | Mild | Positive | 1-C-005 | No | Unrelated | Resolved with sequelae | 28 September 2011 | 28 September 2011 |
| Other | Respiratory, thoracic and mediastinal disorders | Respiratory, thoracic and mediastinal disorders, other | Mild | Negative | 3-C-011 | No | Unrelated | Unknown | 28 April 2012 | 12 March 2014 |
| Other | Skin and subcutaneous tissue disorders | Skin and subcutaneous tissue disorders, other | Mild | Negative | 2-C-009 | No | Unrelated | Ongoing | 12 April 2012 | Ongoing |
| Other | Skin and subcutaneous tissue disorders | Skin and subcutaneous tissue disorders, other | Mild | Positive | 1-C-005 | No | Unrelated | Resolved | 5 May 2012 | 8 May 2012 |
| Other | Skin and subcutaneous tissue disorders | Skin and subcutaneous tissue disorders, other | Mild | Positive | 3-C-017 | No | Possibly related | Unknown | 12 September 2012 | Unknown |
| Other | Skin and subcutaneous tissue disorders | Skin ulceration | Moderate | Negative | 2-C-009 | No | Unrelated | Ongoing | 15 June 2012 | Ongoing |
| Other | Vascular disorders | Hypotension | Mild | Positive | 1-C-005 | No | Unrelated | Resolved | 24 November 2011 | 24 November 2011 |
Concomitant medications taken because of pain within 48 hours of vaccination
Two patients used painkillers after vaccination/electroporation, with good symptomatic relief (Table 19). No new clinical insights beyond the pre-trial data on electroporation were observed.
| Characteristic | HLA A2+ (N = 12), n (%) |
|---|---|
| Did the patient receive any concomitant medications due to pain within 48 hours of vaccinations? | |
| Yesa | 2 (18.2) |
| Noa | 9 (81.8) |
| Missing | 1 (8.3) |
| Types of concomitant medications taken for pain within 48 hours of vaccinationsb | |
| Paracetamol | 1 (14.3) |
| Paracetamol and ibuprofen | 2 (28.6) |
| Tramadol for arthritis | 4 (57.1) |
| Total | 7 (100) |
| Within 48 hours of vaccination, how much relief have pain treatments or medications provided?c | |
| Median scored | |
| n | 2 |
| Median (IQR) | 80% (80–80%) |
| Range | 80–80% |
| Worst scoree | |
| n | 2 |
| Median (IQR) | 25% (10–40%) |
| Range | 10–40% |
Primary outcome
The primary outcome was BCR–ABL response (CMR or minor or major response) observed during the trial. There were two BCR–ABL responses, both of which were defined as a major response, one in each HLA A2 group. Hence, there were no differences observed between the groups (Table 20).
| Characteristic | HLA A2+ | HLA A2– | Total |
|---|---|---|---|
| BCR–ABL response (CMR or minor or major), n (%) | |||
| No | 11 (91.7) | 7 (87.5) | 18 (90.0) |
| Yes | 1 (8.3) | 1 (12.5) | 2 (10.0) |
| Total | 12 (100) | 8 (100) | 20 (100) |
| Fisher’s exact test p-valueb ≥ 0.999 | |||
Secondary outcomes
Breakpoint cluster region–Abelson response (major or complete molecular response)
The BCR–ABL major and CMR responses were evaluated, with no change in results to those seen in the primary end point (Table 21).
| Characteristic | HLA A2+ | HLA A2– | Total |
|---|---|---|---|
| BCR–ABL response (CMR or minor or major), n (%) | |||
| No | 11 (91.7) | 7 (87.5) | 18 (90.0) |
| Yes | 1 (8.3) | 1 (12.5) | 2 (10.0) |
| Total | 12 (100) | 8 (100) | 20 (100) |
| Fisher’s exact test p-valueb ≥ 0.999 | |||
WT1 response
There were three WT1 responses observed, all of which were defined as CMR. Two responses were observed in the HLA A2+ group and one in the HLA A2– group. There were no differences observed between the groups (Table 22).
| Characteristic | HLA A2+ | HLA A2– | Total |
|---|---|---|---|
| WT1 response (CMR or minor or major), n (%) | |||
| No | 10 (83.3) | 7 (87.5) | 17 (85.0) |
| Yes | 2 (16.7) | 1 (12.5) | 3 (15.0) |
| Total | 12 (100) | 8 (100) | 20 (100) |
| Fisher’s exact test p-valueb ≥ 0.999 | |||
Follow-up
Median follow-up time was 18 months for HLA A2+ patients and 15 months for HLA A2– patients. No significant differences between the study arms were detected (Table 23).
| Characteristic | HLA A2+ (n = 13) | HLA A2– (n = 9) | Total (n = 22) |
|---|---|---|---|
| Time to last follow-up from date of consent (months)a | |||
| Median (90% CI) | 18.3 (14.6 to 26.7) | 15.0 (14.5 to 23.0) | 18.1 (14.6 to 23.0) |
| Follow-up at 6 months, % (90% CI) | 92.3 (66.0 to 98.5) | 88.9 (54.3 to 97.8) | 90.9 (73.7 to 97.1) |
| Follow-up at 12 months, % (90% CI) | 84.6 (58.6 to 94.9) | 88.9 (54.3 to 97.8) | 86.4 (68.4 to 94.5) |
| Follow-up at 18 months, % (90% CI) | 53.8 (29.4 to 73.1) | 44.4 (17.8 to 68.3) | 50.0 (31.7 to 65.8) |
| Follow-up at 24 months, % (90% CI) | 30.8 (12.2 to 51.7) | 22.2 (5.1 to 46.7) | 27.3 (13.3 to 43.3) |
| Log-rank test | |||
| Number of events observed, n (%) | 13 (100) | 9 (100) | 22 (100) |
| p-value | 0.463 | ||
| Cox’s proportional hazards model | |||
| Hazard ratio (90% CI)b | 0.721 (0.343 to 1.516) | ||
| p-value | 0.469 | ||
A Kaplan–Meier plot for time to last follow-up in the ITT population is presented in Figure 5.
FIGURE 5.
Kaplan–Meier for time to last follow-up: ITT population.

Progression-free survival
There were no disease progression or deaths observed in this trial (Table 24).
| Characteristic | HLA A2+ (n = 13) | HLA A2– (n = 9) | Total (n = 22) |
|---|---|---|---|
| Time to progression or death (PFS) from date of consent (months)a | |||
| Median (90% CI) | NR (NR to NR) | NR (NR to NR) | NR (NR to NR) |
| PFS at 6 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| PFS at 12 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| PFS at 18 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| PFS at 24 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| Log-rank test | |||
| Number of events observed | 0 | 0 | 0 |
| p-value | NA | ||
| Cox’s proportional hazards model | |||
| Hazard ratio (90% CI)b | NA | ||
| p-value | NA | ||
A Kaplan–Meier plot for time to disease progression or death in the ITT population is presented in Figure 6.
FIGURE 6.
Kaplan–Meier for time to disease progression or death: ITT population.
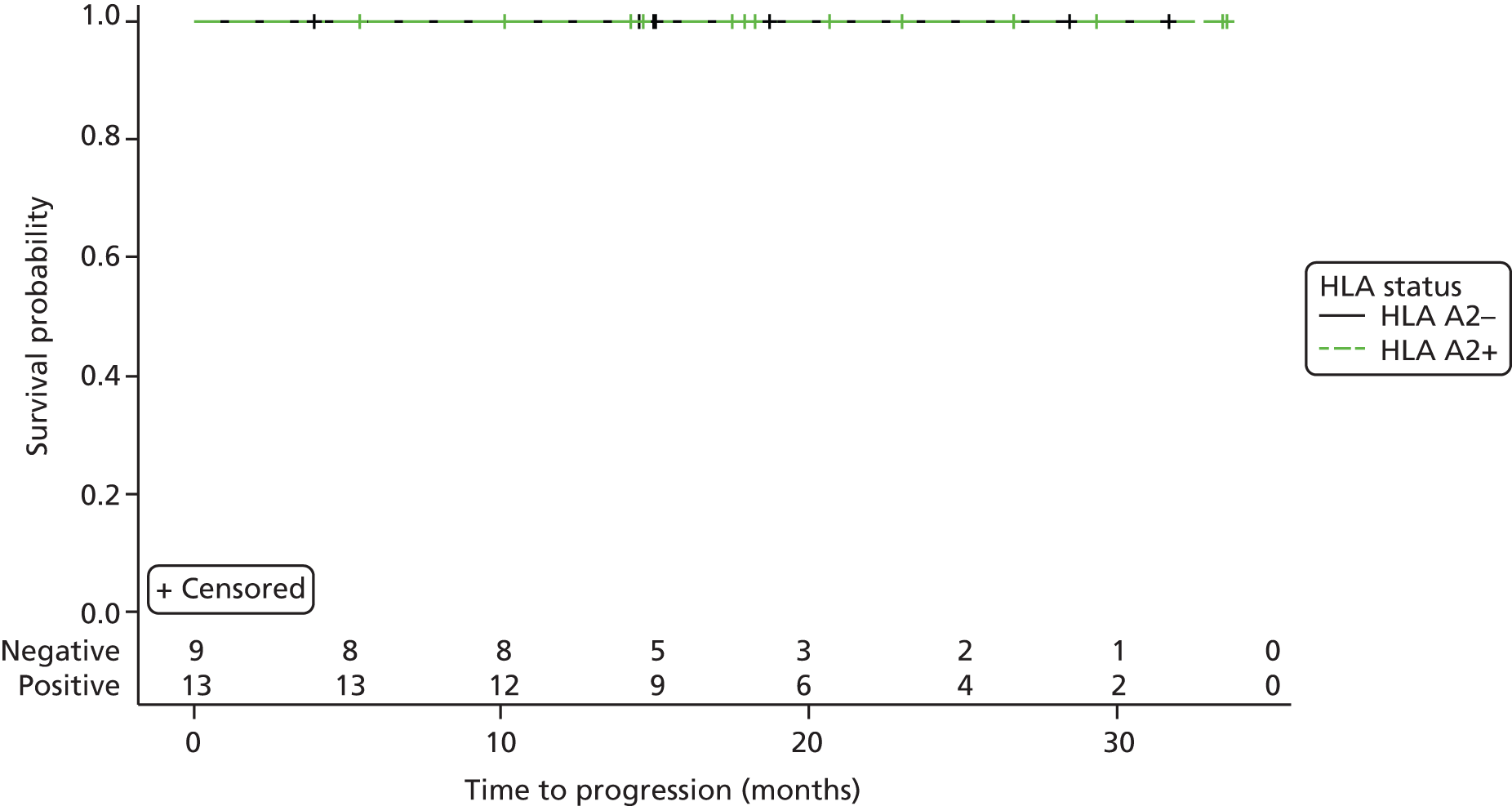
Overall survival
There were no deaths observed in this trial (Table 25).
| Characteristic | HLA A2+ (n = 13) | HLA A2– (n = 9) | Total (n = 22) |
|---|---|---|---|
| Time to death (OS) from date of consent (months)a | |||
| Median (90% CI) | NR (NR to NR) | NR (NR to NR) | NR (NR to NR) |
| OS at 6 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| OS at 12 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| OS at 18 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| OS at 24 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| Log-rank test | |||
| Number of events observed | 0 | 0 | 0 |
| p-value | NA | ||
| Cox’s proportional hazards model | |||
| Hazard ratio (90% CI)b | NA (NA) | ||
| p-value | NA | ||
A Kaplan–Meier plot for time to death in the ITT population is presented in Figure 7.
FIGURE 7.
Kaplan–Meier for time to death: ITT population.
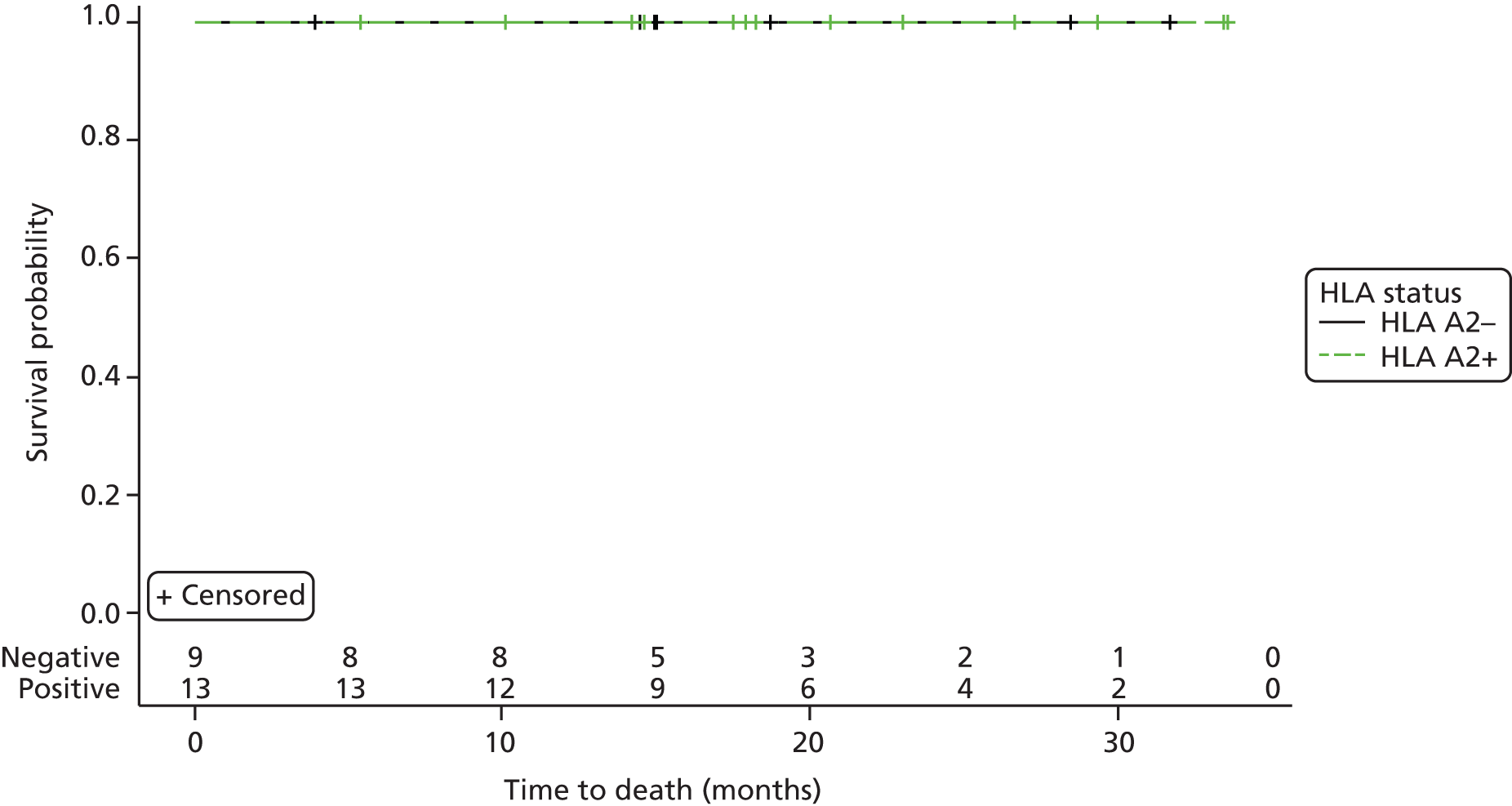
Time to next treatment
Time to next treatment is defined as time from date of consent to the date of next CML treatment or date of last follow-up (whichever occurs first). No additional CML treatment was administered to any patients within this trial (Table 26).
| Characteristic | HLA A2+ (n = 13) | HLA A2– (n = 9) | Total (n = 22) |
|---|---|---|---|
| Time to next treatment from date of consent (months)a | |||
| Median (90% CI) | NR (NR to NR) | NR (NR to NR) | NR (NR to NR) |
| Time to next treatment at 6 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| Time to next treatment at 12 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| Time to next treatment at 18 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| Time to next treatment at 24 months, % (90% CI) | 100 (NA) | 100 (NA) | 100 (NA) |
| Log-rank test | |||
| Number of events observed | 0 | 0 | 0 |
| p-value | NA | ||
| Cox’s proportional hazards model | |||
| Hazard ratio (90% CI)b | NA (NA) | ||
| p-value | NA | ||
A Kaplan–Meier plot for time to next treatment in the ITT population is presented in Figure 8.
FIGURE 8.
Kaplan–Meier for time to next treatment: ITT population.
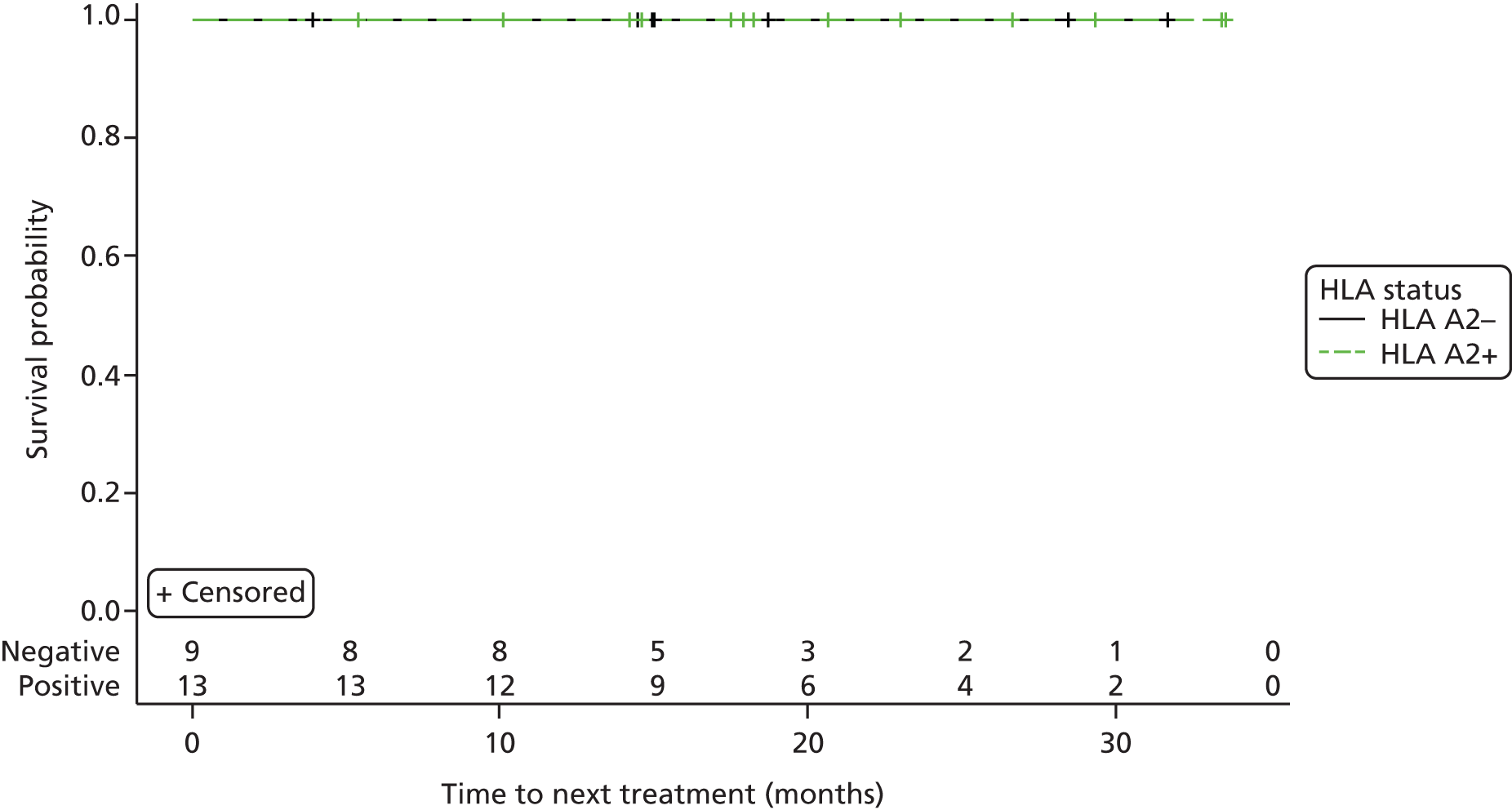
Time to response
Two BCR–ABL responses were observed. One patient in the HLA A2+ group had a response at week 8 and one patient in the HLA A2– group had a response at month 23. The data suggest a vaccine response in the HLA A2+ patient and a late imatinib response in the HLA A2– patient (Table 27).
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 8) | Total (n = 20) |
|---|---|---|---|
| Time to BCR–ABL response from beginning of inhibitor treatment (years)a | |||
| Median time (90% CI) | NR (NR to NR) | NR (NR to NR) | NR (NR to NR) |
| Log-rank test | |||
| Number of events observed, n (%) | 1 (8.3) | 1 (12.5) | 2 (10.0) |
| p-value | 0.768 | – | |
| Cox’s proportional hazards model | |||
| Hazard ratio (90% CI)b | 0.661 (0.064 to 6.782) | – | |
| p-value | 0.770 | – | |
| Time to BCR–ABL response from date of informed consent (months)c | |||
| Median time (90% CI) | NR (NR to NR) | NR (22.8 to NR) | NR (22.8 to NR) |
| Log-rank test | |||
| Number of events observed, n (%) | 1 (8.3) | 1 (12.5) | 2 (10.0) |
| p-value | 0.695 | – | |
| Cox’s proportional hazards model | |||
| Hazard ratio (90% CI)b | 0.577 (0.056 to 5.947) | – | |
| p-value | 0.699 | – | |
| Duration of BCR–ABL response (weeks)d | |||
| n | 1e | 1 | 2 |
| Median (IQR) | 23.1 (23.1–23.1) | 4.1 (4.1–4.1) | 13.6 (4.1–23.1) |
| Range | 23.1–23.1 | 4.1–4.1 | 4.1–23.1 |
A Kaplan–Meier plot for time to for time to BCR–ABL molecular response in the molecular analysis population is presented in Figure 9.
FIGURE 9.
Kaplan–Meier for time to BCR–ABL molecular response: molecular analysis population. (a) From beginning of TKI treatment (in years); and (b) from informed consent (in months).

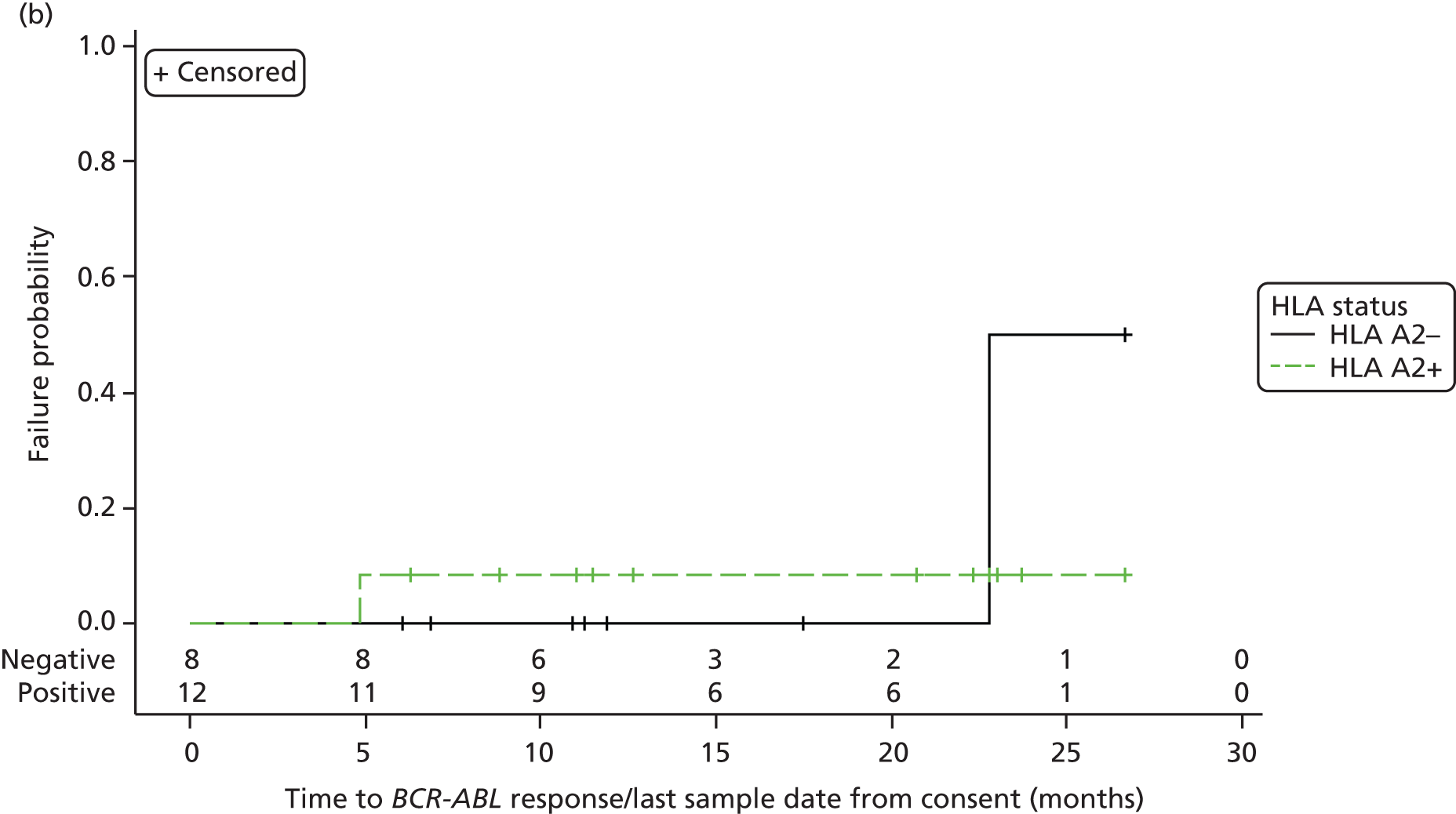
There were three WT1 responses observed. HLA A2+ patients experienced responses at week 20 and week 32. The patient in the HLA A2– group had a response at week 8 (Table 28).
| Characteristic | HLA A2+ (n = 12) | HLA A2– (n = 8) | Total (n = 20) |
|---|---|---|---|
| Time to WT1 response from beginning of inhibitor treatment (years)a | |||
| Median time (90% CI) | NR (8.0 to NR) | NR (8.7 to NR) | NR (8.7 to NR) |
| Log-rank test | |||
| Number of events observed, n (%) | 2 (16.7) | 1 (12.5) | 3 (15.0) |
| p-value | 0.764 | ||
| Cox’s proportional hazards model | |||
| Hazard ratio (90% CI)b | 0.688 (0.088 to 5.397) | ||
| p-value | 0.765 | ||
| Time to WT1 response from date of informed consent (months)c | |||
| Median time (90% CI) | NR (NR to NR) | NR (NR to NR) | NR (NR to NR) |
| Log-rank test | |||
| Number of events observed, n (%) | 2 (16.7) | 1 (12.5) | 3 (15.0) |
| p-value | 0.879 | ||
| Cox’s proportional hazards model | |||
| Hazard ratio (90% CI)b | 1.205 (0.161 to 9.050) | ||
| p-value | 0.879 | ||
| Duration of WT1 response (weeks)d | |||
| n | 2 | 1 | 3 |
| Median (IQR) | 2.0 (1.7– 2.3) | 5.0 (5.0–5.0) | 2.3 (1.7–5.0) |
| Range | 1.7–2.3 | 5.0–5.0 | 1.7–5.0 |
A Kaplan–Meier plot for time to for time to WT1 molecular response in the molecular analysis population is provided in Figure 10.
FIGURE 10.
Kaplan–Meier for time to WT1 molecular response: molecular analysis population. (a) From beginning of inhibitor treatment (in years); and (b) from informed consent (in months).
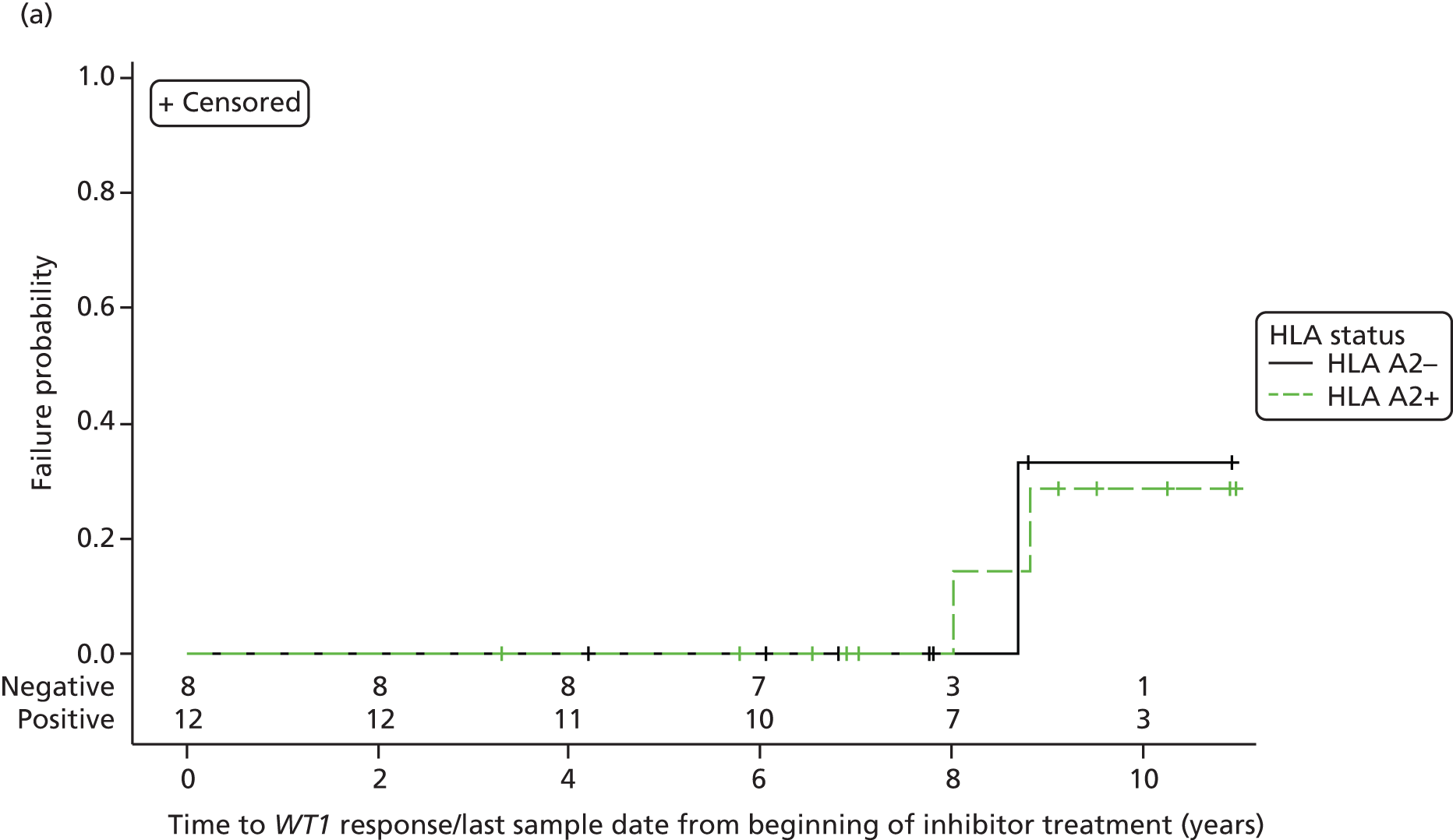

Pain assessment immediately after vaccination
Patient 2-C-021 did not receive any treatment, as outlined by the missing pain assessment data values described in Table 29.
| Characteristic | HLA A2+ (n = 13) |
|---|---|
| Median pain score recordedb | |
| How severe is your pain or discomfort now? | |
| n | 12 |
| Median (IQR) | 1.0 (1.0–2.3) |
| Range | 0.0–5.0 |
| How severe was your pain or discomfort during and immediately after the injection? | |
| n | 12 |
| Median (IQR) | 3.0 (2.0–5.0) |
| Range | 1.0 to 9.0 |
| How distressing is your pain or discomfort now? | |
| n | 12 |
| Median (IQR) | 1.0 (0.3–1.0) |
| Range | 0.0–3.0 |
| How distressing was your pain or discomfort during and immediately after the injection? | |
| n | 12 |
| Median (IQR) | 2.3 (1.0–4.0) |
| Range | 0.0–8.5 |
| Worst pain score recordedc | |
| How severe is your pain or discomfort now? | |
| n | 12 |
| Median (IQR) | 3 (1.50–5.0) |
| Range | 1.0–6.0 |
| How severe was your pain or discomfort during and immediately after the injection? | |
| n | 12 |
| Median (IQR) | 5.0 (3.5–7.0) |
| Range | 1.0–9.0 |
| How distressing is your pain or discomfort now? | |
| n | 12 |
| Median (IQR) | 2.0 (1.0–2.0) |
| Range | 1.0–5.0 |
| How distressing was your pain or discomfort during and immediately after the injection? | |
| n | 12 |
| Median (IQR) | 4.0 (2.0–7.0) |
| Range | 1.0–9.0 |
Pain assessment 48 hours post vaccination
Pain and discomfort data were measured on a visual analogue scale ranging from 0 (no pain/discomfort) to 10 (worst ever pain/discomfort). Electroporation was well tolerated, with short-lived clinical effects on the patient. No new safety information emerged on electroporation.
Patient 2-C-021 did not receive any treatment, as outlined by the missing pain assessment data values described in Table 30.
| Characteristic | HLA A2+ (n = 13) |
|---|---|
| Median pain score recordedb | |
| Pain at its worst in the last 48 hours? | |
| n | 12 |
| Median (IQR) | 0.5 (0.0–1.0) |
| Range | 0.0–4.0 |
| Pain at its least in the last 48 hours? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–1.0 |
| Pain at its average in the last 48 hours? | |
| n | 12 |
| Median (IQR) | 0.3 (0.0–1.0) |
| Range | 0.0–1.0 |
| How much pain do you have right now? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–1.0 |
| During the past 48 hours, has pain interfered with your general activity? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–1.0 |
| During the past 48 hours, has pain interfered with your mood? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–1.0 |
| During the past 48 hours, has pain interfered with your walking ability? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–0.0 |
| During the past 48 hours, has pain interfered with your normal work? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–1.0 |
| During the past 48 hours, has pain interfered with your relations with other people? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–0.0 |
| During the past 48 hours, has pain interfered with your sleep? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–2.5 |
| During the past 48 hours, has pain interfered with your enjoyment of life? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.5) |
| Range | 0.0–1.5 |
| Worst pain score recordedc | |
| Pain at its worst in the last 48 hours? | |
| n | 12 |
| Median (IQR) | 1.0 (1.0–2.0) |
| Range | 0.0–8.0 |
| Pain at its least in the last 48 hours? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–1.0) |
| Range | 0.0–1.0 |
| Pain at its average in the last 48 hours? | |
| n | 12 |
| Median (IQR) | 1.0 (0.5–1.0) |
| Range | 0.0–4.0 |
| How much pain do you have right now? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.5) |
| Range | 0.0–1.0 |
| During the past 48 hours, has pain interfered with your general activity? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.5) |
| Range | 0.0–3.0 |
| During the past 48 hours, has pain interfered with your mood? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–1.0) |
| Range | 0.0–6.0 |
| During the past 48 hours, has pain interfered with your walking ability? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–0.0 |
| During the past 48 hours, has pain interfered with your normal work? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.5) |
| Range | 0.0–2.0 |
| During the past 48 hours, has pain interfered with your relations with other people? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.0) |
| Range | 0.0–1.0 |
| During the past 48 hours, has pain interfered with your sleep? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–0.5) |
| Range | 0.0–4.0 |
| During the past 48 hours, has pain interfered with your enjoyment of life? | |
| n | 12 |
| Median (IQR) | 0.0 (0.0–1.0) |
| Range | 0.0–4.0 |
Immunological analysis
Immunological humoral (antibody) and cellular responses to the vaccine were assessed, using validated assays, in blood and/or bone marrow of 10 out of 12 evaluable vaccinated patients. Results are reported for 10 out of 12 vaccinated patients. Samples for patient 3-C-019 were lost in a freezer thaw at a site prior to the analysis, as were baseline and multiple later time points for serum and cellular samples for patient 3-C-017. Patients 3-C-019 and 3-C-017 were therefore not immunologically evaluable. Immunological analyses demonstrated that the p.DOM–WT1 vaccine can stimulate measurable immune responses against both the DOM and the WT1 components in CML patients in chronic phase on imatinib.
Positive responses to the tetanus-derived component of the p.DOM–WT1 vaccine were assessed by measuring levels of anti-FrC antibody in the patient serum by validated ELISAs. A positive response was assigned for a post-vaccination time point with a greater than twofold increase as well as significantly greater levels of anti-FrC antibody compared with pre-vaccination baseline (p < 0.05, Dunnett’s multiple comparisons test). Positive FrC-antibody responses were detected in a total of 10 out of 10 (100%) evaluable vaccinated patients, providing confirmation of successful vaccine delivery (see Appendix 3).
Immunological responses to the antigen-specific (WT1) component of the vaccine were measured using the validated WT1 tetramer assay. A positive response was assigned when tetramer staining for a time point was greater than twofold over baseline and confirmed by three out of three members of an independent flow cytometry expert panel. WT1-specific T cells were detected for total of 7 out of 10 (70%) evaluable vaccinated patients (see Appendix 4). This included responses to the WT1-37 peptide in 6 out of 10 (60%) and to WT1-126 peptide in 2 out of 10 (20%) evaluable patients.
Chapter 5 Discussion and summary of findings
The study met its primary decision-making target with one major molecular response in BCR–ABL transcript levels. This was the decision-making point for opening the study to the full cohort as well as the AML part of the trial (funded by Bloodwise). The early onset of the major molecular response suggests that vaccination has achieved this molecular event. Two WT1 molecular responses were observed and one WT1 response was seen in the HLA A2– group. One of the control patients developed a BCR–ABL response at 23 weeks.
The vaccine and electroporation appeared safe, with no new toxicities observed or noted. The evaluation of patients for AEs of special interest did not reveal safety concerns. One patient with pre-existing compensated renal failure had fluctuating renal function, but no significant change from baseline was observed. No significant cardiac toxicity or bone marrow toxicities were observed. One event of palpitation was most likely related to anxiety pre-vaccination/pre-electroporation.
No significant differences in frequency of AEs between the HLA A2+ or HLA A2– groups were seen. AEs related to vaccination site reaction were numerically more frequent in the HLA A2+ group; this did not reach significance – a reflection of small sample size.
At an immunological level the vaccine performed as expected. Positive responses to the tetanus-derived component of the p.DOM–WT1 vaccine were assessed by measuring levels of anti-FrC antibody in the patient serum by validated ELISA. A positive response was assigned for a post-vaccination time point with greater than twofold increase as well as significantly greater levels of anti-FrC antibody compared with pre-vaccination baseline (p < 0.05, Dunnett’s multiple comparisons test). Positive FrC-antibody responses were detected in a total of 10 out of 10 (100%) evaluable vaccinated patients, providing confirmation of successful vaccine delivery.
Immunological responses to the antigen-specific (WT1) component of the vaccine were measured using the validated WT1 tetramer assay. A positive response was assigned when tetramer staining for a time point was greater than twofold over baseline and confirmed by three independent flow cytometry experts. Overall, WT1-specific T cells were detected for total of 7 out of 10 (70%) evaluable vaccinated patients. This included responses to WT1-37 peptide in 6 out of 10 (60%) and to WT1-126 peptide in 2 out of 10 (20%) evaluable patients.
The immunological analyses for the WIN trial provide evidence to show that the p.DOM–WT1 vaccine can stimulate measurable immune responses against both the DOM and the WT1 components in chronic-phase CML patients on imatinib.
Recruitment could not be completed for multiple reasons. The key study centre had access to only a fraction of the predicted clinical cohort, severely limiting recruitment of CML patients. Unexpectedly, there was no access to AML patients in this study centre. The principal clinical investigator moved out of the country, and this resulted in loss of oversight and recruitment in the key centre. Attempts to recruit other centres were hampered by directly competing studies and, therefore, the CML extension cohort and the AML cohorts could not be pursued.
This is particularly disappointing as the robust induction of FrC responses (10/10 evaluable patients) and WT1 T-cell responses in 7 out of 10 evaluable patients support that the preclinical data link to immunological outcomes as predicted.
The observation that only 1 out of 12 patients exhibited amolecular response suggests that the rate of clinical benefit in patients on imatinib in chronic-phase CML is low, although if it were possible to withdraw imatinib in 10% of patients this would be clinically significant. Although immunogenicity supports effective delivery of the vaccine and stimulation of the expected immune response, we do not intend to further assess the vaccine approach in this clinical context.
Evaluation in AML remains attractive clinically, but is unlikely to be feasible at this time. A combination of the DNA vaccine approach with strategies to expand T-cell responses with immunomodulatory antibodies is currently being developed.
Acknowledgements
Funding
Chronic myeloid leukaemia arm – NIHR EME programme.
Acute myeloid leukaemia arm – Bloodwise.
Sponsor
University Hospital Southampton NHS Foundation Trust.
Clinical Trials Unit
Joshua Caddy, Mary Ellis, Alex Allen, Nicole Vaughan-Spickers, Emily Moore, Elizabeth Dixon, Jo Musgrove and Kirsty Rogers.
Data Monitoring and Ethics Committee
Professor Alan Melcher, Dr Christy Ralph and Dr Emma Hall.
Trial Steering Committee
Dr Stephen Falk, Dr Andrew Protheroe, Professor Sally Stenning and Professor Diana Eccles.
Investigational medicinal product manufacturer
Bristol Institute of Transfusion Sciences Clinical Biotechnology Centre (BITS), University of Bristol.
Inovio
Provision of the Electroporation Device and training of site staff.
Laboratory
Experimental Cancer Medicine Centre (ECMC) Imperial College Healthcare NHS Trust.
National Cancer Research Network
National Cancer Research Network (NCRN).
Contributions of authors
Christian Ottensmeier, chief investigator.
Megan Bowers, senior clinical trial statistician.
Debbie Hamid, clinical trials manager.
Tom Maishman, trial statistician.
Scott Regan, trial co-ordinator.
Wendy Wood, senior clinical trials manager.
Angelica Cazaly, immunological specialist.
Louise Stanton, senior clinical trial statistician.
Data sharing statement
The available data can be obtained from the SCTU. The results will also be published on the European Clinical Trials Database (EudraCT).
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health.
References
- Cancer Research UK . Cancer Statistics for the UK n.d. http://info.cancerresearchuk.org/cancerstats (accessed 13 February 2016).
- Goldstone AH, Burnett AK, Wheatley K, Smith AG, Hutchinson RM, Clark RE. Attempts to improve treatment outcomes in acute myeloid leukemia (AML) in older patients: the results of the United Kingdom Medical Research Council AML11 trial. Blood 2001;98:1302-11. http://dx.doi.org/10.1182/blood.V98.5.1302.
- Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of BCR–ABL oncogene products. Science 1990;247:1079-82. http://dx.doi.org/10.1126/science.2408149.
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210BCR/ABL gene of the Philadelphia chromosome. Science 1990;247:824-30. http://dx.doi.org/10.1126/science.2406902.
- Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood 2004;103:767-76. http://dx.doi.org/10.1182/blood-2003-02-0342.
- Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006;355:2408-17. http://dx.doi.org/10.1056/NEJMoa062867.
- Hughes T, Branford S. Molecular monitoring of chronic myeloid leukemia. Semin Hematol 2003;40:62-6. http://dx.doi.org/10.1053/shem.2003.50044.
- Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL, et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003;101:4701-7. http://dx.doi.org/10.1182/blood-2002-09-2780.
- Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer 2008;8:108-20. http://dx.doi.org/10.1038/nrc2326.
- Low L, Mander A, McCann K, Dearnaley D, Tjelle T, Mathiesen I, et al. DNA vaccination with electroporation induces increased antibody responses in patients with prostate cancer. Hum Gene Ther 2009;20:1269-78. http://dx.doi.org/10.1089/hum.2009.067.
- Yong AS, Keyvanfar K, Eniafe R, Savani BN, Rezvani K, Sloand EM, et al. Hematopoietic stem cells and progenitors of chronic myeloid leukemia express leukemia-associated antigens: implications for the graft-versus-leukemia effect and peptide vaccine-based immunotherapy. Leukemia 2008;22:1721-7. http://dx.doi.org/10.1038/leu.2008.161.
- Oka Y, Tsuboi A, Taguchi T, Osaki T, Kyo T, Nakajima H, et al. Induction of WT1 (Wilms’ tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc Natl Acad Sci USA 2004;101:13-885. http://dx.doi.org/10.1073/pnas.0405884101.
- Hipp MM, Hilf N, Walter S, Werth D, Brauer KM, Radsak MP, et al. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood 2008;111:5610-20. http://dx.doi.org/10.1182/blood-2007-02-075945.
- Wang H, Cheng F, Cuenca A, Horna P, Zheng Z, Bhalla K, et al. Imatinib mesylate (STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood 2005;105:1135-43. http://dx.doi.org/10.1182/blood-2004-01-0027.
- Mumprecht S, Matter M, Pavelic V, Ochsenbein AF. Imatinib mesylate selectively impairs expansion of memory cytotoxic T cells without affecting the control of primary viral infections. Blood 2006;108:3406-13. http://dx.doi.org/10.1182/blood-2006-04-018705.
- Larmonier N, Janikashvili N, LaCasse CJ, Larmonier CB, Cantrell J, Situ E, et al. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR–ABL tumors. J Immunol 2008;181:6955-63. http://dx.doi.org/10.4049/jimmunol.181.10.6955.
- Collins RH, Shpilberg O, Drobyski WR, Porter DL, Giralt S, Champlin R, et al. Donor leukocyte infusions in 140 patients with relapsed malignancy after allogeneic bone marrow transplantation. J Clin Oncol 1997;15:433-44.
- Vettenranta K, Hovi L, Saarinen-Pihkala UM. Adoptive immunotherapy as consolidation of remission in pediatric AML relapsing post-transplant. Pediatr Transplant 2003;7:446-9. http://dx.doi.org/10.1046/j.1399-3046.2003.00082.x.
- Gannage M, Abel M, Michallet AS, Delluc S, Lambert M, Giraudier S, et al. Ex vivo characterization of multiepitopic tumor-specific CD8 T cells in patients with chronic myeloid leukemia: implications for vaccine development and adoptive cellular immunotherapy. J Immunol 2005;174:8210-18. http://dx.doi.org/10.4049/jimmunol.174.12.8210.
- Bocchia M, Gentili S, Abruzzese E, Fanelli A, Iuliano F, Tabilio A, et al. Effect of a p210 multipeptide vaccine associated with imatinib or interferon in patients with chronic myeloid leukaemia and persistent residual disease: a multicentre observational trial. Lancet 2005;365:657-62. http://dx.doi.org/10.1016/S0140-6736(05)70931-4.
- Rojas JM, Knight K, Wang L, Clark RE. Clinical evaluation of BCR–ABL peptide immunisation in chronic myeloid leukaemia: results of the EPIC study. Leukemia 2007;21:2287-95. http://dx.doi.org/10.1038/sj.leu.2404858.
- de Lavallade H, Garland P, Sekine T, Hoschler K, Marin D, Stringaris K, et al. Repeated vaccination is required to optimize seroprotection against H1N1 in the immunocompromised host. Haematologica 2011;96:307-14. http://dx.doi.org/10.3324/haematol.2010.032664.
- Jemal A, Clegg LX, Ward E, Ries LA, Wu X, Jamison PM. Annual report to the nation on the status of cancer, 1975–2001, with a special feature regarding survival. Cancer 2004;101:3-27. http://dx.doi.org/10.1002/cncr.20288.
- Grimwade D, Walker H, Harrison G, Oliver F, Chatters S, Harrison CJ. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood 2001;98:1312-20. http://dx.doi.org/10.1182/blood.V98.5.1312.
- Brieger J, Weidmann E, Maurer U, Hoelzer D, Mitrou PS, Bergmann L. The Wilms’ tumor gene is frequently expressed in acute myeloblastic leukemias and may provide a marker for residual blast cells detectable by PCR. Ann Oncol 1995;6:811-16.
- Karakas T, Miething CC, Maurer U, Weidmann E, Ackermann H, Hoelzer D, et al. The coexpression of the apoptosis-related genes BCL-2 and WT1 in predicting survival in adult acute myeloid leukemia. Leukemia 2002;16:846-54. http://dx.doi.org/10.1038/sj.leu.2402434.
- Bergmann L, Miething C, Maurer U, Brieger J, Karakas T, Weidmann E, et al. High levels of Wilms’ tumor gene (WT1) mRNA in acute myeloid leukemias are associated with a worse long-term outcome. Blood 1997;90:1217-25.
- Cilloni D, Renneville A, Hermitte F, Hills RK, Daly S, Jovanovic JV, et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: a European LeukemiaNet study. J Clin Oncol 2009;27:5195-201. http://dx.doi.org/10.1200/JCO.2009.22.4865.
- Rezvani K, Yong AS, Mielke S, Savani BN, Musse L, Superata J, et al. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood 2008;111:236-42. http://dx.doi.org/10.1182/blood-2007-08-108241.
- Mailander V, Scheibenbogen C, Thiel E, Letsch A, Blau IW, Keilholz U. Complete remission in a patient with recurrent acute myeloid leukemia induced by vaccination with WT1 peptide in the absence of hematological or renal toxicity. Leukemia 2004;18:165-6. http://dx.doi.org/10.1038/sj.leu.2403186.
- Oka Y, Tsuboi A, Elisseeva OA, Nakajima H, Fujiki F, Kawakami M, et al. WT1 peptide cancer vaccine for patients with hematopoietic malignancies and solid cancers. Sci World J 2007;7:649-65. http://dx.doi.org/10.1100/tsw.2007.119.
- Molldrem J, Dermime S, Parker K, Jiang YZ, Mavroudis D, Hensel N, et al. Targeted T-cell therapy for human leukemia: cytotoxic T lymphocytes specific for a peptide derived from proteinase 3 preferentially lyse human myeloid leukemia cells. Blood 1996;88:2450-7.
- Keilholz U, Letsch A, Aseminssen A, Hofmann WK, Uharek L, Blau W, et al. Clinical and immune responses of WT1-peptide vaccination in patients with acute myeloid leukemia. J Clin Oncol 2006;24.
- Stevenson FK, Zhu D, King CA, Ashworth LJ, Kumar S, Hawkins RE. Idiotypic DNA vaccines against B-cell lymphoma. Immunol Rev 1995;145:211-28. http://dx.doi.org/10.1111/j.1600-065X.1995.tb00083.x.
- Spellerberg MB, Zhu D, Thompsett A, King CA, Hamblin TJ, Stevenson FK. DNA vaccines against lymphoma: promotion of anti-idiotypic antibody responses induced by single chain Fv genes by fusion to tetanus toxin fragment C. J Immunol 1997;159:1885-92.
- King CA, Spellerberg MB, Zhu D, Rice J, Sahota SS, Thompsett AR, et al. DNA vaccines with single-chain Fv fused to fragment C of tetanus toxin induce protective immunity against lymphoma and myeloma. Nat Med 1998;4:1281-6. http://dx.doi.org/10.1038/3266.
- Buchan S, Gronevik E, Mathiesen I, King CA, Stevenson RK, Rice J. Electroporation as a ‘prime/boost’ strategy for naked DNA vaccination against a tumor antigen. J Immunol 2005;174:6292-8. http://dx.doi.org/10.4049/jimmunol.174.10.6292.
- Otten G, Schaefer M, Doe B, Liu H, Srivastava I, . Enhancement of DNA vaccine potency in rhesus macaques by electroporation. Vaccine 2004;22:2489-93. http://dx.doi.org/10.1016/j.vaccine.2003.11.073.
- Rice J, Buchan S, Dewchand H, Simpson E, Stevenson FK. DNA fusion vaccines induce targeted epitope-specific CTLs against minor histocompatibility antigens from a normal or tolerized repertoire. J Immunol 2004;173:4492-9. http://dx.doi.org/10.4049/jimmunol.173.7.4492.
- Ottensmeier C, Low L, Mander A, Williams A, Tjelle T, Campos-Perez J, et al. DNA Fusion Gene Vaccination, Delivered With or Without in Vivo Electroporation – A Potent and Safe Strategy for Inducing Anti-Tumor Immune Responses in Prostate Cancer n.d.
- Kawakami M, Oka Y, Tsuboi A, Harada Y, Elisseeva OA, Furukawa Y, et al. Sugiyama, Clinical and immunologic responses to very low-dose vaccination with WT1 peptide (5 microg/body) in a patient with chronic myelomonocytic leukemia. Int J Hematol 2007;85:426-9. http://dx.doi.org/10.1532/IJH97.06194.
- Oka Y, Tsuboi A, Murakami M, Hirai M, Tominaga N, Nakajima H, et al. Wilms tumor gene peptide-based immunotherapy for patients with overt leukemia from myelodysplastic syndrome (MDS) or MDS with myelofibrosis. Int J Hematol 2003;78. http://dx.doi.org/10.1007/BF02983241.
- Tsuboi A, Oka Y, Udaka K, Murakami M, Masuda T, Nakano A, et al. Enhanced induction of human WT1-specific cytotoxic T lymphocytes with a 9-mer WT1 peptide modified at HLA-A*2402-binding residues. Cancer Immunol Immunother 2002;51:614-20. http://dx.doi.org/10.1007/s00262-002-0328-9.
- Oji Y, Yano M, Nakano Y, Abeno S, Nakatsuka S, Ikeba A, et al. Overexpression of the Wilms’ tumor gene WT1 in esophageal cancer. Anticancer Res 2004;24:3103-8.
- Iiyama T, Udaka K, Takeda S, Takeuchi T, Adachi YC, Ohtsuki Y, et al. WT1 (Wilms’ tumor 1) peptide immunotherapy for renal cell carcinoma. Microbiol Immunol 2007;51:519-30. http://dx.doi.org/10.1111/j.1348-0421.2007.tb03940.x.
- Hosen N, Shirakata T, Nishida S, Yanagihara M, Tsuboi A, Kawakami M, et al. The Wilms’ tumor gene WT1-GFP knock-in mouse reveals the dynamic regulation of WT1 expression in normal and leukemic hematopoiesis. Leukemia 2007;21:1783-91. http://dx.doi.org/10.1038/sj.leu.2404752.
- Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, Oka Y, et al. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood 1997;89:1405-12.
- Lee SB, Haber DA. Wilms tumor and the WT1 gene. Exp Cell Res 2001;264:74-99. http://dx.doi.org/10.1006/excr.2000.5131.
- Rauscher FJ. The WT1 Wilms tumor gene product: a developmentally regulated transcription factor in the kidney that functions as a tumor suppressor. FASEB J 1993;7:896-903.
- Hosen N, Sonoda Y, Oji Y, Kimura T, Minamiguchi H, Tamaki H, et al. Very low frequencies of human normal CD34+ haematopoietic progenitor cells express the Wilms’ tumour gene WT1 at levels similar to those in leukaemia cells. Br J Haematol 2002;116:409-20. http://dx.doi.org/10.1046/j.1365-2141.2002.03261.x.
- Gao L, Bellantuono I, Elsasser A, Marley SB, Gordon MY, Goldman JM, et al. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood 2000;95:2198-203.
- Oka Y, Elisseeva OA, Tsuboi A, Ogawa H, Tamaki H, Li H, et al. Human cytotoxic T-lymphocyte responses specific for peptides of the wild-type Wilms’ tumor gene (WT1) product. Immunogenetics 2000;51:99-107. http://dx.doi.org/10.1007/s002510050018.
- Gaiger A, Reese V, Disis ML, Cheever MA. Immunity to WT1 in the animal model and in patients with acute myeloid leukemia. Blood 2000;96:1480-9.
- Gao L, Xue SA, Hasserjian R, Cotter F, Kaeda J, Goldman JM, et al. Human cytotoxic T lymphocytes specific for Wilms’ tumor antigen-1 inhibit engraftment of leukemia-initiating stem cells in non-obese diabetic-severe combined immunodeficient recipients. Transplantation 2003;75:1429-36. http://dx.doi.org/10.1097/01.TP.0000061516.57346.E8.
- Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood 2000;95:286-93.
- Chaise C, Buchan SL, Rice J, Marquet J, Rouard H, Kuentz M, et al. DNA vaccination induces WT1-specific T-cell responses with potential clinical relevance. Blood 2008;112:2956-64. http://dx.doi.org/10.1182/blood-2008-02-137695.
- Chudley L, McCann K, Mander A, Tjelle T, Campos-Perez J, Godeseth R, et al. DNA fusion-gene vaccination in patients with prostate cancer induces high-frequency CD8(+) T-cell responses and increases PSA doubling time. Cancer Immunol Immunother 2012;61:2161-70. http://dx.doi.org/10.1007/s00262-012-1270-0.
- Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials 1989;10:1-10. http://dx.doi.org/10.1016/0197-2456(89)90015-9.
- A’Hern RP. Sample size tables for exact single-stage phase II designs. Stat Med 2001;20:859-66. http://dx.doi.org/10.1002/sim.721.
- Simon RM, Steinberg SM, Hamilton M, Hildesheim A, Khleif S, Kwak LW, et al. Clinical trial designs for the early clinical development of therapeutic cancer vaccines. J Clin Oncol 2001;19:1848-54.
- Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol 2009;27:6041-51. http://dx.doi.org/10.1200/JCO.2009.25.0779.
Appendix 1 Pain assessment tool: immediately after vaccination
Appendix 2 Pain assessment tool: 48 hours after vaccination
Appendix 3 Anti-fragment C from tetanus toxin antibody enzyme-linked immunosorbent assay results
Anti-FrC antibody ELISA results are shown as mean relative antibody units at each time point (0–23 weeks). Positive responses (shown highlighted in green) represent a greater than twofold, and significantly greater than pre-vaccination, baseline (p< 0.05, Dunnett’s multiple comparisons test). Blue shading represents no sample. The final column shows overall response, with a maximum fold increase relative to baseline.
| FrC | Time point (weeks) | Overall result | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 00 | 01 | 02 | 03 | 04 | 05 | 06 | 07 | 08 | 09 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 23 | ||
| 1-C-005 | 0.4215 | 0.4151 | 0.6618 | 0.6491 | 0.8572 | 0.8491 | 0.7884 | 0.7357 | 0.8395 | 0.8159 | 0.8159 | 0.7603 | 0.7135 | 0.5409 | 0.5194 | 0.4560 | 0.6307 | 0.5164 | 0.5923 | 2.0 | |||
| 1-C-022 | 0.0517 | 0.0624 | 0.0899 | 0.4052 | 1.1441 | 1.4248 | 1.6481 | 1.7005 | 1.7878 | 1.7795 | 1.8024 | 1.2027 | 0.9147 | 0.7652 | 34.9 | ||||||||
| 1-C-023 | 0.6330 | 0.5645 | 1.0376 | 1.0156 | 1.2872 | 1.2920 | 1.2381 | 1.8903 | 1.6056 | 1.7529 | 1.4973 | 1.4630 | 1.3148 | 1.0102 | 0.9034 | 3.0 | |||||||
| 2-C-002 | 1.5938 | 2.2106 | 4.4236 | 2.9914 | 3.6068 | 4.7370 | 3.1848 | 3.2868 | 2.8729 | 5.1475 | 4.5656 | 2.3297 | 4.5745 | 3.0819 | 3.1852 | 2.7010 | 3.3740 | 3.2 | |||||
| 2-C-004 | 0.1043 | 0.1389 | 0.1340 | 0.0573 | 0.1130 | 0.2688 | 0.2195 | 0.2440 | 0.2212 | 0.4068 | 0.3736 | 0.1449 | 0.1633 | 0.0702 | 0.0691 | 3.9 | |||||||
| 2-C-007 | 0.0854 | 0.1092 | 0.1098 | 0.1417 | 0.3030 | 0.4984 | 0.6170 | 0.7719 | 0.4890 | 0.8262 | 0.7443 | 0.3447 | 9.7 | ||||||||||
| 2-C-010 | 0.4321 | 0.5565 | 1.0132 | 1.2200 | 0.9896 | 1.4916 | 1.4232 | 1.4452 | 1.3271 | 1.3318 | 1.2191 | 0.8340 | 0.7065 | 0.6568 | 0.6074 | 0.7267 | 3.5 | ||||||
| 2-C-012 | 0.6950 | 0.7023 | 1.2529 | 1.1530 | 1.1180 | 1.0267 | 1.1176 | 1.3059 | 1.6607 | 1.6134 | 1.5593 | 1.2250 | 1.8651 | 1.0415 | 2.7 | ||||||||
| 2-C-013 | 0.1304 | 0.1458 | 0.3887 | 0.3314 | 0.3412 | 0.4077 | 0.4102 | 0.4713 | 0.4341 | 0.4839 | 0.2557 | 3.7 | |||||||||||
| 2-C-014 | 0.3324 | 0.3583 | 1.0392 | 1.0211 | 1.1308 | 1.0485 | 1.0133 | 1.2261 | 1.2952 | 1.2738 | 0.5880 | 3.9 | |||||||||||
| 3-C-017 | NA | 0.5324 | 0.4440 | 0.4829 | 0.3703 | NA | |||||||||||||||||
| Response frequency | 0 | 0 | 4 | 4 | 8 | 8 | 6 | 8 | 8 | 9 | 6 | 3 | 2 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 10/11 |
Appendix 4 Tetramer analysis results for WT1-37 and WT1-126 peptides
Tetramer analysis results for WT1-37 and WT1-126 peptides are shown as percentage of tetramer-positive cells within the CD8+ T-cell population. Positive responses (shown highlighted in green) represent a post-vaccination time point greater than twofold increase over baseline and confirmed as real by three independent flow cytometrists. Blue shading represents no sample. The final two columns show overall response, with a maximum fold increase relative to baseline.
| Peptide | Time point (weeks) | Overall result | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 00 | 01 | 02 | 03 | 04 | 05 | 06 | 07 | 08 | 09 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 23 | ||
| WT1-37 | |||||||||||||||||||||||
| 1-C-005 | 0.0152 | 0.0152 | 0.0210 | 0.0120 | 0.0117 | 0.0088 | 0.0120 | 0.0183 | 0.0093 | 0.0125 | 0.0135 | 0.0160 | 0.0155 | 0.0090 | 0.0074 | 0.0075 | 0.0086 | 0.0149 | 0.0085 | ||||
| 1-C-022 | 0.0025 | 0.0013 | 0.0036 | 0.0039 | 0.0016 | 0.0024 | 0.0012 | 0.0021 | 0.0022 | 0.0004 | 0.0035 | 0.0017 | 0.0012 | 0.0017 | 0.0009 | 0.0015 | |||||||
| 1-C-023 | 0.0074 | 0.0115 | 0.0110 | 0.0186 | 0.0243 | 0.0267 | 0.0277 | 0.0119 | 0.0276 | 0.0162 | 0.0181 | 0.0230 | 0.0223 | 0.0225 | 0.0099 | 3.7 | |||||||
| 2-C-002 | 0.0378 | 0.0455 | 0.0667 | 0.0547 | 0.0844 | 0.0760 | 0.0663 | 0.0794 | 0.0775 | 0.0612 | 0.0631 | 0.0696 | 0.0490 | 0.0483 | 0.0633 | 0.0648 | 0.0592 | 2.3 | |||||
| 2-C-004 | 0.0026 | 0.0008 | 0.0046 | 0.0034 | 0.0122 | 0.0042 | 0.0063 | 0.0023 | 0.0040 | 0.0057 | 0.0054 | 0.0051 | 0.0045 | 0.0069 | 0.0060 | 4.7 | |||||||
| 2-C-007 | 0.0032 | 0.0027 | 0.0074 | 0.0031 | 0.0079 | 0.0036 | 0.0038 | 0.0035 | 0.0074 | 0.0037 | 0.0020 | 0.0024 | 2.3 | ||||||||||
| 2-C-010 | 0.0031 | 0.0016 | 0.0037 | 0.0047 | 0.0037 | 0.0037 | 0.0036 | 0.0052 | 0.0021 | 0.0031 | 0.0047 | 0.0036 | 0.0026 | 0.0042 | 0.0030 | ||||||||
| 2-C-012 | 0.0028 | 0.0041 | 0.0027 | 0.0158 | 0.0274 | 0.0227 | 0.0182 | 0.0147 | 0.0256 | 0.0162 | 0.0093 | 0.0143 | 0.0149 | 0.0120 | 9.8 | ||||||||
| 2-C-013 | 0.0136 | 0.0117 | 0.0097 | 0.0175 | 0.0148 | 0.0117 | 0.0147 | 0.0132 | 0.0091 | 0.0117 | 0.0077 | ||||||||||||
| 2-C-014 | 0.0056 | 0.0041 | 0.0521 | 0.0096 | 0.0126 | 0.0144 | 0.0093 | 0.0128 | 0.0184 | 0.0148 | 0.0116 | 9.3 | |||||||||||
| 3-C-017 | 0.0121 | 0.0184 | 0.0195 | 0.0176 | 0.0128 | ||||||||||||||||||
| Response frequency | 0 | 0 | 2 | 2 | 5 | 4 | 2 | 3 | 5 | 3 | 0 | 2 | 2 | 1 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 6/11 |
| WT1-126 | |||||||||||||||||||||||
| 1-C-005 | 0.0040 | 0.0040 | 0.0035 | 0.0055 | 0.0082 | 0.0090 | 0.0085 | 0.0085 | 0.0067 | 0.0043 | 0.0062 | 0.0048 | 0.0065 | 0.0030 | 0.0050 | 0.0045 | 0.0050 | 0.0045 | 0.0065 | 2.2 | |||
| 1-C-022 | 0.0005 | 0.0008 | 0.0016 | 0.0004 | 0.0008 | 0.0008 | 0.0013 | 0.0026 | 0.0009 | 0.0004 | 0.0018 | 0.0004 | 0.0009 | 0.0008 | 0.0005 | 0.0010 | |||||||
| 1-C-023 | 0.0050 | 0.0055 | 0.0060 | 0.0070 | 0.0068 | 0.0064 | 0.0085 | 0.0076 | 0.0140 | 0.0112 | 0.0120 | 0.0065 | 0.0123 | 0.0120 | 0.0097 | 2.8 | |||||||
| 2-C-002 | 0.0021 | 0.0028 | 0.0031 | 0.0035 | 0.0018 | 0.0028 | 0.0036 | 0.0030 | 0.0027 | 0.0022 | 0.0021 | 0.0023 | 0.0035 | 0.0093 | 0.0102 | 0.0035 | 0.0064 | ||||||
| 2-C-004 | 0.0007 | 0.0007 | 0.0008 | 0.0000 | 0.0032 | 0.0010 | 0.0011 | 0.0006 | 0.0017 | 0.0025 | 0.0024 | 0.0007 | 0.0020 | 0.0000 | 0.0007 | ||||||||
| 2-C-007 | 0.0012 | 0.0035 | 0.0023 | 0.0040 | 0.0037 | 0.0026 | 0.0019 | 0.0013 | 0.0006 | 0.0029 | 0.0013 | 0.0006 | |||||||||||
| 2-C-010 | 0.0021 | 0.0021 | 0.0000 | 0.0010 | 0.0005 | 0.0010 | 0.0031 | 0.0026 | 0.0016 | 0.0021 | 0.0032 | 0.0042 | 0.0047 | 0.0031 | 0.0029 | ||||||||
| 2-C-012 | 0.0020 | 0.0015 | 0.0006 | 0.0005 | 0.0015 | 0.0015 | 0.0010 | 0.0000 | 0.0019 | 0.0017 | 0.0023 | 0.0005 | 0.0020 | 0.0000 | |||||||||
| 2-C-013 | 0.0015 | 0.0026 | 0.0000 | 0.0015 | 0.0015 | 0.0017 | 0.0036 | 0.0036 | 0.0035 | 0.0046 | 0.0025 | ||||||||||||
| 2-C-014 | 0.0023 | 0.0000 | 0.0009 | 0.0045 | 0.0038 | 0.0013 | 0.0012 | 0.0031 | 0.0021 | 0.0036 | 0.0022 | ||||||||||||
| 3-C-017 | 0.0020 | 0.0048 | 0.0010 | 0.0073 | 0.0035 | ||||||||||||||||||
| Response frequency | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2/11 |
Glossary
- Plasmid domain 1 from fragment C of tetanus toxin
- Used in the vaccine construct as an immune alert signal.
- Wilms’ tumour antigen 1 peptide 37
- Peptide from Wilms’ tumour antigen 1 with the amino acid sequence VLDFAPPGA.
- Wilms’ tumour antigen 1 peptide 126
- Peptide from Wilms’ tumour antigen 1 with the amino acid sequence RMFPNAPYL.
List of abbreviations
- ABL
- Abelson murine leukaemia viral oncogene homolog 1
- AE
- adverse event
- alloSCT
- allogeneic stem cell transplantation
- AML
- acute myeloid leukaemia
- BCR
- breakpoint cluster region
- BCR–ABL
- breakpoint cluster region–Abelson murine leukaemia viral oncogene homolog 1
- CCyR
- complete cytogenetic response
- CD34+
- cluster of differentiation 34 positive
- CD4+
- cluster of differentiation 4 positive
- CD8+
- cluster of differentiation 8 positive
- CK
- creatine kinase
- CML
- chronic myeloid leukaemia
- CML-CP
- chronic-phase chronic myeloid leukaemia
- CMR
- complete molecular response
- CONSORT
- Consolidated Standards Of Reporting Trials
- CR
- complete remission
- CTCAE
- Common Terminology Criteria for Adverse Events
- CTL
- cytotoxic T lymphocyte
- DLI
- donor lymphocyte infusion
- DMEC
- Data Monitoring and Ethics Committee
- DNA
- deoxyribonucleic acid
- DOM
- domain 1 from fragment C of tetanus toxin
- ECG
- electrocardiogram
- ECHO
- echocardiogram
- ELISA
- enzyme-linked immunosorbent assay
- EME
- Efficacy and Mechanism Evaluation
- FBC
- full blood count
- FrC
- fragment C from tetanus toxin
- GTAC
- Gene Therapy Advisory Committee
- GUS
- β-glucuronidase
- GVL
- graft versus leukaemia
- HIV
- human immunodeficiency virus
- HLA A2
- human leucocyte antigen A2
- HLA A2–
- human leucocyte antigen A2 negative
- HLA A2+
- human leucocyte antigen A2 positive
- HSC
- haematopoietic stem cell
- IFN-γ
- interferon gamma
- ISRCTN
- International Standard Randomised Controlled Trial Register
- ITT
- intention to treat
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MRD
- minimal residual disease
- NIHR
- National Institute for Health Research
- pCRF
- paper case report form
- p.DOM
- plasmid domain 1 from fragment C of tetanus toxin
- PFS
- progression-free survival
- PI
- principal investigator
- PSMA
- prostate-specific membrane antigen
- qPCR
- quantitative polymerase chain reaction
- RNA
- ribonucleic acid
- SAE
- serious adverse event
- SCTU
- Southampton Clinical Trials Unit
- T cell
- T lymphocyte
- TKI
- tyrosine kinase inhibitor
- WHO
- World Health Organization
- WIN
- Wilms’ tumour antigen 1 (WT1) Immunity via DNA trial
- WT1
- Wilms’ tumour antigen 1
- WT1-126
- Wilms’ tumour antigen 1 peptide 126
- WT1-37
- Wilms’ tumour antigen 1 peptide 37
- WT1+
- Wilms’ tumour antigen 1 positive