
Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 11/14/25. The contractual start date was in March 2012. The final report began editorial review in February 2015 and was accepted for publication in October 2015. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
All authors report grants from the National Institute for Health Research, grants from Cystic Fibrosis Trust, grants from Just Gene Therapy, grants from Medicor Foundation and report that Genzyme, a Sanofi company, manufactured and provided Lipid 67. Ian A Pringle also has a patent WO200711062 issued. Ronald K Scheule also reports other funding from Genzyme, a Sanofi company, outside the submitted work and has a patent US5783565 issued, a patent US5840710 issued, and a patent US5935936 issued. Michelle C Manvell also reports grants from Royal Brompton Biomedical Research Unit (National institute for Health Research funds), outside the submitted work. Steve Cunningham also reports personal fees from Gilead, outside the submitted work and is a principle investigator for Vertex Pharmaceuticals studies in children with cystic fibrosis. Nicholas J Simmonds also reports personal fees from cystic fibrosis Adherence Steering Committee, personal fees from Vertex advisory board, personal fees from Pharmaxis advisory board and lecture fee, personal fees from Gilead advisory boards and lecture fee, personal fees from Eumedica lecture fee, personal fees from Forest Laboratories honorarium, outside the submitted work. Eric WFW Alton also has a patent WO2013061091 pending. Christopher Boyd also has a patent WO2013061091 pending. Alexandra L Quittner is a member of the advisory board for Genentech Inc., reports consulting for AbbVie Pharmaceuticals, grants from Cystic Fibrosis Foundation, grants from Vertex Pharmaceuticals and grants from Novartis Pharmaceuticals, outside the submitted work. Uta Griesenbach also has a patent WO2013061091 pending, and a patent European Patent Application Number 12784648.3 pending. Seng H Cheng is an employee of Genzyme, a Sanofi Company and also has a patent US5650096 issued, a patent US5747471 issued, and a patent US5840710 issued. David J Porteous also has a patent WO2013061091 pending. J Alastair Innes also has a patent WO2013061091 pending. Jane C Davies also reports fees paid to employing institution from Vertex for her role as clinical trials lead, educational meetings and advisory board participation, fees paid to employing institution from Novartis for advisory board participation and fees paid to employing institution from Proteostasis for advisory board participation, outside the submitted work, and has a patent WO2013061091 pending. Stephen C Hyde also has a patent WO200711062 issued, and a patent WO2013061091 pending. Lee A Davies has a patent US 20140242690 A1 pending, and a patent WO2013061091 pending. Deborah R Gill also has a patent WO200711062 issued, and a patent WO2013061091 pending. James M Wilson is a founder of, holds equity in, an advisor to and grant recipient from, REGENXBIO and Dimension Therapeutics. He is an advisor to Solid Gene Therapy. He is an inventor on patents licensed to various biopharmaceutical companies.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2016. This work was produced by Alton et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction and background
Cystic fibrosis
Pathophysiology
Cystic fibrosis (CF), a common, genetically inherited disease, is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. Inherited in an autosomal recessive fashion, it has a prevalence of approximately 1 : 2500 and a carrier rate of around 1 : 25 in the Caucasian population. It is estimated that there are approximately 70,000–100,000 people with CF worldwide and over 10,000 on the current UK registry. It is a multisystem disease, primarily affecting the lungs, but also with significant pancreatic, liver and gastrointestinal involvement and infertility in the majority of males. Lung involvement occurs from an early age with intermittent then chronic bacterial infection, inflammation and eventual bronchiectasis, fibrosis and death from respiratory failure. 1 People affected with CF are particularly susceptible to certain bacteria, including Staphylococcus aureus Rosenbach 1884, Pseudomonas aeruginosa (Schroeter 1872) Migula 1900 and Burkholderia cepacia (Palleroni and Holmes 1981) Yabuuchi et al. 1993, although more recently the use of molecular detection techniques has revealed much more diverse bacterial populations present in the lower airway than hitherto suspected. 2
The gene responsible for CF was identified in 1985 and found to be localised to the long arm of chromosome 7, position 7q21–24,3 with the sequence being fully identified in 1989. 4 Over 2000 mutations in the CFTR gene have been described, although not all are disease-causing; some mutations still produce a functional protein and are probably more accurately termed benign polymorphisms. In its normal, wild-type, form, the CFTR gene encodes a 1480-amino-acid protein5 that sits on the cell surface and acts as a cyclic adenosine monophosphate (cAMP)-regulated ion channel. CFTR is a direct conductor of chloride and (probably) bicarbonate ions and interacts closely with the major sodium-absorbing channel, epithelial sodium channel (ENaC), inhibiting its activity. Defects in CFTR lead to reduced chloride secretion and hyperabsorption of sodium and, thus, water, the combined effect of which is dehydration of the cell surface. 6 Mucociliary clearance is impaired and the infective/inflammatory sequelae of this ensues. It is not known if the decreased volume of airway surface liquid alone causes the predisposition to infection, if the immune system is also affected causing a defect in pathogen killing,7 or whether or not other environmental factors within the CF airway prevent the innate immune system functioning as efficiently as in individuals without CF.
Clinical presentation, diagnosis and disease manifestation
The majority of today’s older children and adults, the population included in this trial, will have been diagnosed once they had developed symptoms suggestive of the disease. In many countries, the UK included, this picture has changed dramatically with the introduction of newborn screening; currently, infants are diagnosed around 3–4 weeks of age based on dried-blood-spot screening. 8 The screens differ somewhat globally, but the UK test measures immunoreactive trypsinogen (IRT) and pancreatic enzyme levels, which are increased in CF because of pancreatic duct obstruction. Samples with high levels of IRT are sent for mutation testing. This allows identification and initiation of treatment presymptomatically and before the downstream sequelae have arisen.
The gold standard diagnosis is based on measuring chloride ion levels in sweat samples. Lack of chloride ion reabsorption by CFTR in the sweat gland leads to high levels being lost in the sweat, typically > 100 mmol/l in comparison with values of < 30 mmol/l in healthy individuals. 9 Diagnosis can also be established on CFTR mutation testing, although, because of the large number of mutations, a negative test cannot rule out the diagnosis unless the entire CFTR gene has been sequenced; this is rarely employed as a first-line diagnostic tool and is reserved for difficult diagnoses. However, whereas previously it may have been sufficient to secure a diagnosis on the basis of a sweat test, with the recent development of mutation-specific small-molecule treatments,10 it is becoming increasingly necessary for a patient’s mutation(s) to be defined.
Disease presentation may be respiratory or nutritional and is often both. 1 Respiratory problems present as cough, wheeze, lower respiratory tract infections, sputum production and upper airway disease including nasal polyps and sinusitis. Lung health deteriorates over time, largely driven by infection and the host inflammatory response. Disease is thought to begin in the small airways, although this may just reflect a greater impact of obstruction at this site. Airway obstruction related to mucus accumulation and airway wall inflammation is measurable on physiological testing, the most commonly employed being forced expiratory manoeuvres with spirometry. Over time, and despite optimal treatment at specialist centres, lung function declines. A major contributor to this decline is episodes of pulmonary exacerbation, periods of increased symptoms and acute drop in lung function that is often not fully regained after treatment. 11
The most common manifestation of the pancreatic exocrine disease, present in > 95% of patients, is poor weight gain with steatorrhoea. Later, patients may develop pancreatic endocrine dysfunction with impaired glucose metabolism and CF-related diabetes mellitus; this has respiratory consequences that are poorly understood, but is clearly associated with an increased rate of progression of lung disease.
Conventional management of patients with cystic fibrosis
Cystic fibrosis is a multisystem disease and treatment can be optimally conducted only with the help of a full multidisciplinary team (CF physician, specialist nurse, physiotherapist, dietitian, clinical psychologist, pharmacist) and input from ancillary specialists with expert knowledge of CF (ear, nose and throat surgeon, obstetrician, endocrinologist). CF patients should be seen at least every 3 months by the core CF team. A large number of treatment guidelines have been published. 12–14
The main aims of respiratory care are clearance of mucus, prevention and early eradication of infection and suppression of infection once chronic. Sputum clearance is achieved with regular physiotherapy alongside adjunctive inhaled mucoactive agents: hypertonic saline has been shown to aid clearance,15 most likely be rehydrating the airway surface and stimulating cough. Recombinant human deoxyribonuclease (DNase) (dornase alfa; Pulmozyme®, Roche Products Ltd) breaks down the high levels of deoxyribonucleic acid (DNA) in the CF airway resulting from degradation of neutrophils and decreases mucus viscosity. 16 Regular microbiological surveillance is essential for the early detection of infecting bacteria; sputum samples or cough swabs are collected at every patient contact. P. aeruginosa is an extremely common pathogen, often being acquired first in childhood and becoming chronic in adulthood in around 60–70% of patients. Its presence is associated with a more rapid decline in lung function and, therefore, aggressive eradication is employed when it is first encountered. 17 Once it becomes chronic, long-term nebulised antibiotics such as colistimethate sodium (Colomycin®, Forest Laboratories UK Ltd) or the aminoglycoside, tobramycin for inhaled solution (Tobi®, Novartis Pharmaceuticals UK Ltd), are administered to maintain suppression of bacterial numbers and thereby attempt to limit the host inflammatory response. 18 There are a small number of other organisms that pose particular problems, either because of high transmissibility [e.g. meticillin-resistant Staphylococcus aureus (MRSA) and B. cepacia], or because they are related to a more rapid rate of clinical decline [e.g. Mycobacterium abscessus (Moore and Frerichs 1953) Kusunoki and Ezaki 1992]. These organisms are difficult to treat and, furthermore, such patients often miss out on inclusion in clinical trials of new drugs, either because of infection control issues or concerns over baseline stability. Periods of pulmonary exacerbation are treated with increased attention to airway clearance and antibiotics, often given intravenously and requiring hospital admission.
Therefore, CF management not only poses a huge burden on the patients and their families, but also carries with it substantial health-care system costs. 19 Ultimately, once respiratory failure has ensued, lung transplantation is the only option. There is a shortage of organs in most developed countries and many patients die while on the waiting list. Even after receiving a transplant, complications frequently occur, most importantly the development of bronchiolitis obliterans, although some patients may do well for many years. 20 Currently in the UK, median age at death is around 29 years of age. 21 Thus, despite expensive and burdensome treatments, life expectancy is significantly reduced for CF patients. Progress is being made in several areas of drug development, for example with newer antibiotics, but many feel that a significant improvement in health is likely to be achievable only with novel strategies to tackle the basic defect in the CFTR gene rather than these downstream consequences. The next sections describe the progress that has been made in small-molecule and gene therapy fields to this end.
Novel small-molecule cystic fibrosis transmembrane conductance regulator modulators
An understanding of the various mechanisms by which different mutations lead to CFTR dysfunction has led to the grouping of mutations into classes. 22
Class I mutations lead to a premature ‘stop’ in the messenger ribonucleic acid (mRNA) and a short, non-functioning protein. Mutations of class II encode a structurally abnormal, misfolded protein that does not traffic to its site of action on the apical cell surface but is removed by the endoplasmic reticulum and degraded. Class III–VI mutations reach the cell surface but fail to function appropriately: class III mutations have decreased activation of the channel and remain closed; and class IV mutations cause decreased conductance of ions across the channel. Class V mutations encode proteins with splice-site mutations and result in reduced amounts of CFTR at the cell surface, so some function occurs but at a reduced level. Class VI mutations lead to a shortened half-life because of protein instability and may also impair CFTR’s regulation of neighbouring channels. Many mutations lead to defects in more than one of these classes; for example, F508del (previously ΔF508), the most common mutation worldwide, is clearly a class II misfolding defect, but also opens infrequently and has a short half-life, so also possesses class III and VI features.
Treatments for class I mutations
Class I, or nonsense, mutations account for approximately 5–10% of the worldwide CF population, although the incidence is increased to around 60% among Israeli Jewish CF subjects. 23,24 Based on the initial observation that premature truncation codons could be over-read in the presence of gentamicin (Garamycin®, Schering-Plough)24–27 one synthetic agent, ataluren (previously known as PTC124; Translarna™, PTC Therapeutics), has progressed to clinical trials. 28 This drug was administered orally to CF patients aged ≥ 6 years with at least one class I mutation in a parallel-design randomised controlled trial. Despite encouraging Phase II data, neither the primary end point (change in forced expiratory volume in the first second; FEV1) nor multiple secondary end points were met. A post hoc subgroup analysis demonstrated a treatment effect (TE) in the subgroup who were not receiving nebulised aminoglycoside antibiotics, which may have been interfering with the mechanism of action. A further trial is planned, but to date these patients do not have any other mutation-specific drug approaches in the pipeline.
Treatments for class III mutations
Ivacaftor (Kalydeco®, Vertex Pharmaceuticals, Boston, MA, USA) is the first mutation-specific drug to be approved in the USA, Europe and Australasia. It is a CFTR potentiator, which increases the channel’s open probability. Clinical trial results prove, for the first time, that improving CFTR function can lead to significant benefits.
Class III mutations lead to a protein that remains closed for almost 100% of the time, a so-called ‘gating’ defect. The most common class III mutation is G551D (new nomenclature Gly551Asp). Other class III mutations are rare and together probably account for only a further 1% of patients.
Ivacaftor was identified via high-throughput screening technology,29–31 and progressed rapidly through preclinical testing into CF clinical trials, based on significant financial support from the CF Foundation in the USA. Twice-daily oral administration in patients with a G551D mutation led to a 10% absolute improvement in FEV1, a reduction in pulmonary exacerbations and improved quality-of-life scores; additionally, it seemed to have benefits outside the lungs, with improvement in weight and body mass index (BMI) thought to be related to gut bicarbonate secretion. It was initially licensed (and is now available in the UK) for patients with the G551D mutation, but a subsequent trial demonstrated similar benefit for the small group of patients with other class III mutations,10 for which the drug has now also been licensed and is awaiting funding approval. Furthermore, a trial in patients with the class IV mutation R117H (NCT01614457), which demonstrated smaller benefits, has led, to date, to the US Food and Drug Administration approval and is being considered by the European Medicines Agency.
Treatments for class II mutations
Class II defects are present in the majority of patients with CF worldwide. F508del is carried by approximately 70–80% of the CF population, with 50% of the total CF population being homozygous. 32 As described above, the vast majority of class II CFTR is degraded by intracellular processes and fails to reach the cell surface. The absolute amount that escapes this degradation differs between individuals and, may account, in part, for the phenotypic variation observed. The concept of CFTR ‘correctors’ has been around for many years, although the term has only recently been coined. In the 1980s, an observation was made that low temperatures and the compound glycerol, stabilised misfolded CFTR protein and facilitated trafficking; the term ‘molecular chaperone’ was coined for a drug with these capabilities. 32,33 High-throughput screening was conducted by several commercial and academically funded groups and one of these, Vertex Pharmaceuticals, identified a promising small molecule, named VX-809 (lumacaftor, Vertex Pharmaceuticals). In vitro chloride transport could be achieved at 14% of wild-type CF levels. 34 The results from the first Phase II trial in 89 patients were somewhat disappointing. As the drug is given systemically, sweat chloride is a convenient biomarker and had shown utility in trials of potentiator agents. In this trial, there was a statistically significant reduction in sweat chloride, confirming proof of concept, but the magnitude was small at around 7 mmol/l. In addition, no change was seen in nasal potential difference (PD), a localised bioelectrical read-out of CFTR ion transport, nor clinical parameters within the 4-week treatment period. 35 As well as a trafficking defect, F508del CFTR protein is abnormally gated. It was, therefore, hypothesised that VX-809 could improve the localisation of CFTR protein to the cell surface, but once there it would need additional help to function; this led to the notion of combination trials with potentiators.
Ivacaftor, which had been shown to have dramatic improvement in the movement of ions across the cell membrane in class III mutations,29–31 was considered unlikely to benefit patients homozygous for F508del, and this was borne out in a Phase II study of 140 patients. 35 However, in vitro, combining VX-809 with ivacaftor doubled the chloride transport compared with that of VX-809 alone. A large Phase III global clinical trial programme has recently been reported to demonstrate a statistically significant but relatively modest 2–4% absolute improvement in percentage predicted FEV1 and to reduce the frequency of pulmonary exacerbations. 36 It did not show clinical benefit in patients with only one copy of F508del. Vertex is also developing an alternative corrector, VX-661. It possesses a possible advantage over lumacaftor in that its metabolic pathway does not interact with that of VX-770 and achieving therapeutic levels may be easier. Phase II clinical trials of VX-661 alone, and in combination with ivacaftor, in CF patients homozygous for F508del have been encouraging and a Phase III programme is planned.
Gene therapy
In contrast to the mutation-specific approach described above, gene therapy, described as ‘the introduction or alteration of genetic material within a cell or organism with the intentions of curing or treating a disease’,37 has potential for CF patients with mutations in any class. The first studies of human administration of healthy copies of the CFTR gene in CF patients were developed very soon after the gene was cloned in 1989. 38
Two components are required in a gene therapy product: (1) a normal copy of the CFTR gene (along with required regulatory constructs); and (2) a gene transfer agent, either a viral or non-viral vector. The choice and suitability of such a vector depends on a number of factors, such as the size of the gene to be carried, the requirement for, and tolerability of, repeated administration and the target cells for treatment.
Choice of vector and clinical trials
Clinical trials of CFTR gene transfer to CF subjects have been undertaken using a variety of viral vector systems, including adenovirus and adeno-associated virus (AAV). Encouragingly, adenovirus-based gene transfer led to the correction of the cAMP-stimulated CFTR chloride channel defect in the nose of treated CF subjects. 39,40 However, the efficiency of adenovirus-based gene transfer to human tissues appears to be low,41,42 and is associated with lung inflammation43,44 and the development of neutralising immune responses that, to date, has precluded repeated administration. 45 AAV-based CFTR gene transfer appears less proinflammatory,46–48 although repeated administration is also inhibited by immunological reactivity. 49
Non-viral vectors have also been used in clinical trials of CFTR gene transfer to CF subjects. Ten clinical trials have been performed worldwide to date,50–58 six of which have been in the UK [Gene Therapy Advisory Committee (GTAC) numbers: 002, 007, 008, 009, 015 and 140] and conducted by current groups within the UK CF Gene Therapy Consortium (UK CFGTC). The first of these non-viral-formulation clinical trials assessed the safety and efficacy of a single nasal delivery of plasmid DNA (pDNA) containing the CFTR complementary DNA (cDNA) under the transcriptional control of the simian virus 40 (SV40) promoter complexed with 3beta-[N-(N′,N′-dimethylaminoethane)carbamoyl]cholesterol (DC-Chol)–dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) liposomes (GTAC number: 002). 50 Evidence for pDNA transfer, vector-derived CFTR mRNA expression and partial correction of the CFTR chloride ion channel defect was obtained in a subset of subjects. Similar modest, but positive evidence of CFTR gene transfer and function were obtained after nasal delivery of a pDNA containing the CFTR cDNA under the transcriptional control of the respiratory syncytial virus 3′ long terminal repeat (RSV 3’LTR) promoter complexed with DC-Chol–DOPE liposomes (GTAC number: 007). 51 Alternative formulations in which pDNAs containing the CFTR cDNA under the transcriptional control of the human cytomegalovirus (CMV) immediate early enhancer/promoter were complexed with 1,2-dioleoyl-3-trimethylammoniumpropane liposomes (GTAC number: 008),52 p-ethyl-dimyristoylphosphaditylcholine–cholesterol liposomes,53 or a polycationic peptide consisting of a N-terminal cysteine followed by 30 lysine residues54 were broadly shown to be similarly effective. Importantly, in an additional clinical trial utilising the RSV 3′LTR promoter plasmid complexed with DC-Chol–DOPE liposomes, repeated nasal delivery of a non-viral formulation was shown to be equivalently effective after each of three successive administrations (GTAC number: 015). 55 Crucially, no important safety considerations were raised after nasal application of any of these formulations. Collectively, these clinical data provided proof of principle for CF non-viral gene therapy, but highlighted the need for formulations with enhanced efficacy. Extensive chemical optimisation of the promising DC-Chol formulation by Genzyme Inc. (Cambridge, MA, USA) generated the cationic lipid cholest-5-en-3-ol (3β)-,3-[(3-aminopropyl)[4-[(3-aminopropyl)amino]butyl]carbamate] (GL67) with improved gene transfer potency,59 well-characterised safety parameters60 and desirable stability during aerosolisation. 61 Clinical trials with GL67-based non-viral formulations have been encouraging. A single nasal administration of a pDNA containing the CFTR cDNA under the transcriptional control of the CMV promoter (plasmid pCF1-CFTR) complexed with GL67–DOPE was shown to be safe, to direct vector-derived mRNA expression and to produce an overall ≈ 20% correction of the CFTR chloride ion channel defect. 56 For efficient jet nebuliser-directed aerosol delivery, 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethylene glycol 5000)] (ammonium salt) (DMPE-PEG5000) was added to the GL67–DOPE to generate a formulation termed GL67A that consisted of a mixture of GL67–DOPE–DMPE-PEG5000 at a 1 : 2 : 0.05 molar ratio61 (for further details see GL67A cationic lipid). The GL67A formulation lacking pDNA was shown to be safe and well tolerated after a single lung administration in healthy volunteers. 62 Delivery of pCF1-CFTR complexed with GL67A to the lungs57 or nose and lungs (GTAC number: 009)58 of CF subjects led to an overall ≈ 25% correction of the CFTR chloride ion channel defect. However, CFTR expression in these subjects, and those in all other clinical trials described above, diminished quickly, such that it was essentially undetectable within 1 week of administration. Furthermore, pCF1-CFTR/GL67A lung administration was associated with a mild flu-like syndrome that resolved within 36 hours. 57,58
Further in vivo preclinical studies have identified cytosine–phosphate–guanidine (CpG) dinucleotides in the pCF1-CFTR pDNA as the likely cause of the mild flu-like symptoms following dosing and may also impact on the duration of pDNA-mediated transgene expression. The CpG motifs delivered by the pCF1-CFTR/GL67A formulation appear to be sensed by the innate immune system via the toll-like receptor 9 (TLR9) pathway. 63 Stimulation of the TLR9 pathway led to a host inflammatory reaction typified by the elevation of inflammatory cytokines within the lung milieu and recruitment of lung neutrophils. 64
Thus, proof of principle for non-viral CFTR gene therapy had been demonstrated, but improvements were needed specifically with regard to duration of expression and pro-inflammatory responses.
UK Cystic Fibrosis Gene Therapy Consortium
At this stage, in 2001, and in response to an initiative launched by the UK CF Trust, the three UK sites active in CF gene therapy (University of Edinburgh, Imperial College London/Royal Brompton Hospital and University of Oxford) came together to form the UK CF Gene Therapy Consortium (www.cfgenetherapy.org.uk/). The aim of the consortium is to develop clinically relevant gene therapy for patients with CF. The wave 1 programme was based on the recognition that repeated application would necessitate a non-viral vector and aimed to identify the best, currently available non-viral formulation coupled with a long-lasting plasmid with little or no proinflammatory potential and to take this into a clinical trial designed and powered, for the first time, to detect clinical benefit. The repeated-dose trial is the focus of this report. Work packages in the wave 1 programme leading up to this point have included a study of outcome measures on patients undergoing a pulmonary exacerbation;65 a large, longitudinal study of outcome measures in stable patients (from which power calculations were made for this trial); and a single-application safety and dose-ranging pilot study (see Choice of dose and adjunctive treatments: single-dose pilot study). The consortium is also working on a wave 2 programme developing and assessing a pseudotyped lentiviral vector, which seems to be repeatedly administrable. Although gene transfer efficiency seems greater with this approach than with our non-viral equivalents, the requirement for preclinical development and toxicity testing placed this further in the future and led to the initial focus on wave 1.
Section pGM1/GL67A for clinical trial use describes our chosen clinical trial formulation in detail and the rationale for its development.
pGM1/GL67A for clinical trial use
Composition
We found GL67A (further details in GL67A cationic lipid) to be superior to others in both small and large animal models, having undertaken an extensive preclinical assessment of available non-viral formulations. GL67A has previously been administered to both healthy volunteers and CF subjects, including in a single-dose nebuliser study66 and our recently completed safety study Evaluation of safety and gene expression with a single dose of pGM169/GL67A administered to the nose and lung of individuals with cystic fibrosis (sponsor’s protocol number: cro851; EudraCT reference: 2007-004050-85). 62 For both the single-dose safety study and the trial described in this report, we made substantial changes to the CFTR plasmid, which was specifically designed with the following features:
-
The promoters most commonly used are of viral origin; we had previously incorporated a CMV promoter which, although capable of high-level gene expression, is usually of short duration. The current formulation contains a human elongation factor 1 alpha (EEF1A1) gene promoter. Our preclinical work demonstrated more sustained levels of expression (up to 2 months) with this new promoter; this could translate into a less frequent dosing regimen. The gene expression profile observed in the recent single-dose study supported this hypothesis, with relatively few changes observed at early time points and several subjects demonstrating functional changes in nasal PD several months after dosing.
-
Unmethylated CpG dinucleotides are present in high numbers in pDNA used in gene therapy trials. These bacterially derived components are recognised by humans as foreign and likely responsible for the flu-like responses, reported in our previous trial in the group receiving DNA/lipid but not lipid alone. Although these responses were mild and self-limiting, requiring only treatments with antipyretics, we considered them undesirable in a repeated-dose trial. In an attempt to reduce such an inflammatory response, the new plasmid has been rendered completely CpG free.
Plasmid
pGM169 is a covalently closed, circular, double-stranded pDNA molecule of 6549 base pairs purified from bacteria. It is based on a novel CpG-free plasmid backbone described in the international patent application PCT/GB2007/00110433. A diagrammatic representation of pGM169 is presented in Figure 1. pGM169 contains a CpG-free version of the CFTR coding sequence termed soCFTR2 under the transcriptional control of a novel CpG-free human CMV enhancer/elongation factor 1 alpha (hCEFI) enhancer/promoter. Other plasmid elements include a CpG-free version of the bovine growth hormone polyadenylation sequence, a CpG-free version of the R6K bacterial plasmid origin of replication and a CpG-free version of the kanamycin resistance gene under the transcriptional control of the CpG-free synthetic bacterial promoter sequence termed EM7.
FIGURE 1.
Plasmid pGM169. The basic features of pGM169 (proceeding clockwise from 0 base pairs) are the CpG-free human cytomegalovirus enhancer/elongation factor 1 alpha enhancer/promoter; a CpG-free synthetic intron sequence to enhance mRNA splicing; a CpG-free version of the CFTR coding sequence termed soCFTR2; a CpG-free version of the bovine growth hormone polyadenylation sequence; a CpG-free version of the R6K bacterial plasmid origin of replication; a CpG-free version of the kanamycin resistance gene; and a CpG-free synthetic bacterial promoter sequence termed EM7. bp, base pair; BGH, bovine growth hormone.

Good manufacturing practice (GMP) manufacture of pGM169 was conducted by VGXi Inc. (The Woodlands, TX, USA). Bacteria containing the plasmid were fermented to a high density and harvested. The bacteria were then lysed to release their contents, including the plasmid, into solution. The lysate was subjected to three significant purification steps: (1) solid/liquid separation, (2) ion-exchange chromatography and (3) hydrophobic interaction chromatography. Subsequently, the purified plasmid was concentrated and desalted by ultrafiltration/diafiltration into a sterile 8 mM sodium chloride (NaCl) solution and finally subjected to aseptic filtration to provide the bulk drug substance. This bulk was aseptically filled into single-unit vials and stored at ≤ –70 °C. To prepare the final drug substance, single or multiple pooled lots of bulk drug substance were, if necessary, diluted to 5.3 ± 0.3 mg/ml with sterile 8 mM NaCl and then filled into 10-ml clear glass vials at a fill level of 5.2 ± 0.2 ml. Vials were stored at –80 °C. The material is stable for at least 3 years.
GL67A cationic lipid
The cationic lipid mixture GL67A is an excipient, consisting of a mixture of three components (the structure of which is shown in Figures 2–4; GL67, DOPE, and DMPE-PEG5000) formulated at a 1 : 2 : 0.05 molar ratio.
FIGURE 2.
Structure of GL67. GL67 is included in the GL67A cationic lipid mixture for its DNA-binding properties (i.e. binding of pGM169).

FIGURE 3.
Structure of DOPE. DOPE is included in the GL67A cationic lipid mixture for its endocytosis-inducing properties.

FIGURE 4.
Structure of DMPE-PEG5000. DMPE-PEG5000 is included in the GL67A cationic lipid mixture for its charge-shielding properties that facilitate final preparation of the pGM169/GL67A drug product and allow efficient nebulisation.
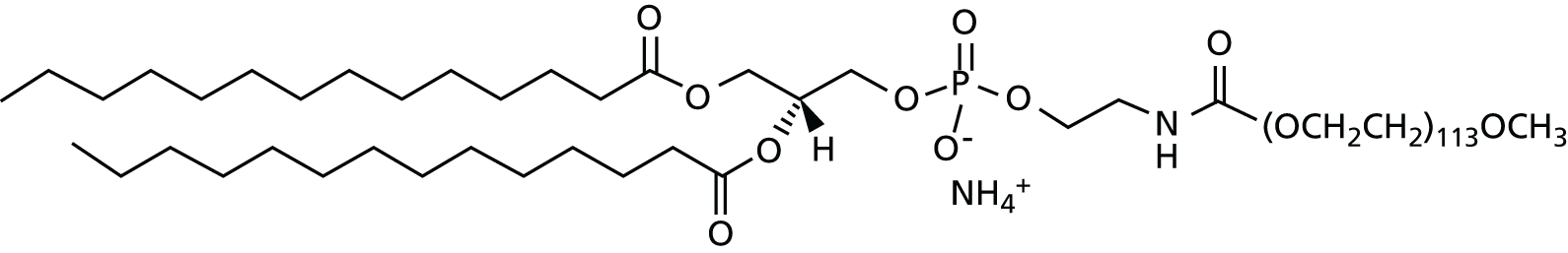
The final formulation containing GL67, DOPE and DMPE-PEG5000 at a 1 : 2 : 0.05 molar ratio and a GL67-to-DNA ratio of 0.75 : 1 was termed GL67A.
Good manufacturing practice-grade GL67 was manufactured by Sanofi-Genzyme (Haverhill, UK). GMP-grade DOPE and DMPE-PEG5000 were purchased from Avanti Polar Lipids (Alabaster, AL, USA). GL67A was formulated from GMP-grade GL67, DOPE and DMPE-PEG5000 by OctoPlus N.V. (Leiden, Netherlands). Briefly, the individual constituents were first dissolved in 2-methylpropan-2-ol (9 : 1 weight-to-weight ratio of 2-methylpropan-2-ol to water) and then mixed in appropriate quantities to obtain a GL67–DOPE–DMPE-PEG5000 molar ratio of 1 : 2 : 0.05. After sterile filtration the lipid mixture was dispensed into individual 10-ml glass lyophilisation vials such that each vial contained 38.8 mg of GL67, 93.7 mg of DOPE and 17.8 g of DMPE-PEG5000. The vials were freeze-dried under nitrogen gas for approximately 94 hours at temperatures ranging from –50 °C to +10 °C. The vials were capped with aluminium crimp caps, coded and stored at –80 °C. The material is stable for at least 3 years.
Preclinical toxicology of the selected formulation: murine
To comply with the Medicines and Healthcare products Regulatory Agency (MHRA) regulations, a multidose toxicology study in rodents (mice) was performed under good laboratory practice and the study was outsourced to a clinical research organisation (CRO).
Protocol development and selection of clinical research organisations
The protocol was developed with the help of a toxicology consultant (Professor Anthony Dayan, Professor of Toxicology at Barts Hospital, London, UK). In addition to standard toxicology, assessment of DNA and mRNA biodistribution into organs other than the lung was assessed using molecular assays. The study was tendered to six national and international CROs, and selection was based on expertise in conducting inhalation toxicology studies and molecular analysis, costs and flexibility. The inhalation study and toxicology assessment were placed with Charles River (Edinburgh, UK) and the molecular analysis at BioReliance (Glasgow, UK). The studies were performed in several stages, detailed in Inhalation study and toxicology assessment at Charles River and Molecular analysis at BioReliance.
Inhalation study and toxicology assessment at Charles River
This was performed in three phases: (1) analytical development; (2) a feasibility study; and (3) the main toxicology study.
Analytical development
This phase was used to develop methodologies, validation and specifications for the analysis of pDNA and liposome in inhalation toxicology studies. An important aspect was to show that the AeroEclipse® II (Trudell Medical International Europe Ltd, Nottingham, UK) nebuliser to be used in the trial was able to generate droplet sizes suitable for mouse inhalation studies.
Feasibility study
This showed that mice tolerated the prolonged (6 hour = maximum feasible dose) exposure in the nebulisation chamber and that gene transfer and gene expression could be achieved using the inhalation chambers used at Charles River.
Main toxicology study
This consisted of four dosing cohorts (low, medium, high and air control), corresponding to approximately 5-, 20- and 60-fold the anticipated human dose (mg/kg). Mice were exposed on 12 occasions over a 6-month period to mimic the human trial protocol (consistent with relevant guidelines). Controls were exposed to air for 6 hours. The study involved approximately 700 mice (including animals dosed for assessment of gene expression and biodistribution; see Molecular analysis at BioReliance). Standard non-invasive assessments were made at regular intervals and post-mortem toxicology undertaken 2 weeks or 3 months after the last dose. Additionally, molecular analyses to assess gene expression and biodistribution were performed. The treatment was well tolerated by all mice. In the high-dose group, small increases in lung weight and circulating neutrophils were seen 2 days after the last dose (dose 12); this was not observed in the cohort sacrificed 14 days after the last dose. Two weeks after administration of the last dose scattered alveolar macrophages were seen on histology in the low- and medium-dose groups. Multifocal alveolar foamy macrophage accumulation and occasional inflammatory changes were recorded in the high-dose group. Fat-laden macrophages were observed by Oil Red O (ORO; VWR International Ltd, Lutterworth, UK) staining. There was no evidence of structural remodelling. All other organs were unremarkable. Three months after administration, findings were no different from baseline in the low-dose group, but were still observed in the medium- and high-dose animals.
Molecular analysis at BioReliance
The study was again performed in three phases: (1) assay transfer; (2) validation of extraction methods; and (3) the main biodistribution study.
Assay transfer
Molecular assays [quantitative polymerase chain reaction (qPCR) and quantitative reverse transcription PCR reaction (qRT-PCR) for the detection of pGM169 were developed by the UK CFGTC and the know-how and experimental details were transferred to BioReliance where the protocols were validated to ensure that sensitivity and reproducibility of the assays were compliant with regulatory requirements.
Validation of extraction
The validation of extraction study demonstrated that the clinical trial plasmid could be successfully retrieved from all tissue selected for the biodistribution and gene expression studies (lung, gonad, gut, spleen, kidney, liver, lymph nodes and blood), as well as determining the detection limits of the assays.
Main biodistribution study
The main biodistribution study was designed to assess the level and persistence of DNA and pGM169-specific mRNA in the above organs. Male and female mice (n = 10 per treated group) were sacrificed after one, six and 12 doses and organs analysed using qPCR. A dose–response relationship was observed between duration of inhalation and the quantity of pDNA present in the lungs 1 day after delivery of 1, 6 and 12 doses (p < 0.0001; Spearman's rank correlation coefficient). pDNA remained detectable in the lungs of animals for up to 21 weeks after the final (high) dose. Levels of pDNA in non-target organs were several orders of magnitude lower than the lungs at day 1 and generally did not persist for > 2 months. pGM169-specific mRNA was not detectable in any organs other than the lung. When CFTR mRNA was measured in the lungs, low levels were detected after a single dose in the low- and medium-dose groups, with increased signal in the high-dose group (p < 0.001; equivalent to ≥ 100% endogenous levels). Robust levels of CFTR mRNA remained in the lung for at least 21 weeks after the final exposure.
In conclusion, all animals tolerated the treatment well. The transient and dose-related systemic inflammatory responses and drop in lung function observed in the single-dose pilot study (see Choice of dose and adjunctive treatments: single-dose pilot study) were not replicated in non-CF mice. Mice only developed mild systemic inflammation at the highest dose (≈50-fold of the human dose) possibly highlighting species differences or an increased response to the lipid–DNA complexes in the inflamed human CF lung. The mouse multidose toxicology study supports progression into a multidose CF gene therapy trial. Key data are summarised in Alton et al. 67
Preclinical toxicology of the selected formulation: ovine
Following MHRA discussions, the UK CFGTC also used its in-house sheep core facility to undertake a study of nine monthly administrations of 10 ml of the therapeutic product administered via the inhalation route. Repeated gene delivery was very well tolerated by the animals, with no obvious clinical signs throughout the study. The majority of changes observed were mild and were observed both in anaesthetic controls (CONs) and gene therapy-treated (GT) animals. Some mild changes in animal body weight and haematological parameters were observed at day 1 in both groups, and are likely to be a consequence of the anaesthetic procedure; for example, food was withheld from animals the night before an anaesthetic procedure to avoid bloating as a result of rumen gas accumulation. Specifically, the absence of a gene therapy-related increase in the acute-phase protein haptoglobin contrasts with previous studies that demonstrated a significant rise following a 20-ml dose of the same pGM169/GL67A complex. Although an upwards trend in the serum creatinine levels was observed in the GT group, the increase was small, all values were within the normal reference range and final values after the 4-week recovery period were lower than baseline levels.
A gene therapy-specific effect was seen in bronchoalveolar lavage (BAL), which was assessed after doses 1, 5 and 9. This was manifested by increases in total BAL cell number, neutrophils, macrophages and lymphocytes. These changes are consistent with the previously observed changes in the sheep lung at day 1 after a 20-ml dose. However, encouragingly, rather than being exacerbated by repeated delivery, the response was less pronounced after dose 9 and the observed changes had always fully resolved by day 15 after delivery. The increase in BAL macrophage and lymphocyte numbers was only observed after dose 1. There were no significant changes in any aspect of lung function attributable to the gene therapy administration.
In the lungs, common to all three groups (GT, CON and untreated animals), was minimal/mild lymphoplasmacytic, and sometimes eosinophilic, inflammation around bronchioles and some blood vessels. This was present in most samples, and is consistent with background immunosurveillance and/or indicative of previous parasite exposure. Alveolar histiocytosis was also common to all three groups, and is not unusual in the ovine lung, especially if sheep are not specific pathogen free or in climate- or pathogen-controlled housing. There were no notable changes that suggest accumulation of lipid related to gene therapy treatment. No adverse effect of the test article was observed on lung morphometry, indicating no changes in alveolar septal thickness or alveolar size after nine doses. In the additional organs assessed, there was nothing to point to an adverse effect of treatment. Lesions present were consistent with background inflammation or immunosurveillance, were incidental and present in all three groups.
Choice of nebuliser for clinical trial programme
In total, five commercially available, Conformité Européenne marked jet nebuliser devices were compared (four PARI Medical Ltd breath-enhanced design nebulisers and one inherently breath-actuated nebuliser: the AeroEclipse® II, Trudell Medical International Europe Ltd, Nottingham, UK).
The different investigations focused on:
-
Median mass aerodynamic diameter (MMAD) aerosol droplet size and fine particle fraction (FPF; that proportion of the aerosol droplets ≤ 5 µm, which are those thought likely to be deposited within the airways) with two commercially available nebuliser compressor devices, or with continuous gas supplies operating at either 29 psi or 50 psi.
-
Aerosol delivery rate, and inhalation efficiency (the proportion of the produced aerosol that can be retained within the lung rather than immediately exhaled) in breath actuated and non-breath actuated modes at an operating gas pressure of 50 psi.
-
pDNA stability during the aerosolisation process. Jet nebulisation results in a modest degree of non-specific pDNA damage. 68 The damage is associated with loss of supercoiled pDNA forms, an increase in linear forms and the appearance of degraded DNA after agarose gel electrophoresis.
All test nebuliser device and operating gas supply combinations resulted in the generation of respirable pCIKLux/GL67A aerosol droplets. A reduction in aerosol MMAD and an increase in aerosol FPF were generally observed with increasing operating pressures. Operating the test nebuliser devices under breath actuation conditions reduced the rate of aerosol delivery, but increased the inhalation efficiency. Both Pari LC+ (PARI Medical Ltd, Surrey, UK) and AeroEclipse® II devices impart a modest degree of pDNA damage during aerosolisation. Of the five nebuliser devices evaluated, only the AeroEclipse® II device is capable of operating in a breath-actuated mode. Given the considerable enhancement in inhalation efficiency of pCIKLux/GL67A aerosol observed and the lack of inferiority in the other parameters measured, the AeroEclipse II was selected as the preferred nebuliser device for clinical studies of pGM169/GL67A. The measured MMAD of pGM169/GL67A aerosols using the AeroEclipse® II was 3.4 ± 0.1 µm, with a FPF of 71.4% ± 1.5%. 69 To confirm that nebulisation does not alter the ability of pGM169/GL67A to generate functional chloride channels, HEK-293T cells [American Type Culture Collection (ATCC), Teddington, UK] were transfected with pGM169/GL67A. The cells were collected prior to nebulisation and at the end of nebulisation (residual volumes in the nebuliser) and an iodide efflux assay, commonly used to assess CFTR function, was performed as previously described. 70 Levels of cAMP-mediated iodide efflux were similar when using pGM169/GL67A before and after nebulisation (data not shown).
Choice of dose and adjunctive treatments: single-dose pilot study
Dose ranging was undertaken in the single-dose trial (manuscript in preparation). In brief, we observed that, despite depletion of pro-inflammatory CpG motifs, 20-ml nebulised doses led to systemic inflammatory responses and an acute reduction in pulmonary function; although these were largely well tolerated, we considered them unacceptable for repeated application. The side effects were clearly dose related. The group receiving 5-ml doses had no, or very low-level, fever, minimal rises in systemic inflammatory markers and small, self-limiting reductions in lung function after dosing; 10 ml was somewhere in between and, although probably acceptable, we had concerns that blinding, of either patients or staff, could be compromised. A 5-ml dose was therefore chosen for the current trial. In addition, standard doses of the antipyretic agent paracetamol post dosing appeared to further reduce both symptoms and inflammatory markers and was administered within 2 hours of dosing and again 6 hours later, with a specific aim to ensure patient blinding.
Summary
Cystic fibrosis is a life-limiting disease; currently, there is no treatment targeted at the basic defect in the majority of patients. The UK CFGTC considers that gene therapy has the potential for broad, mutation-independent applicability and has set out to design a clinical trial of the optimal non-viral formulation powered to detect clinical benefit.
In Chapter 2, the specific design of the trial, along with background and rationale for the choice of outcome measures, is discussed.
Chapter 2 Methods: clinical trial design and outcome measures
This trial was a placebo-controlled, parallel trial of 12 monthly doses of the gene therapy product or placebo. Given the chronic nature of the disease, this time period was judged to be an adequate one over which to observe potential improvements in clinical outcomes, while not overburdening patients. It is a time period chosen in similarly designed trials of other agents.
Trial formulation
-
Active: GL67A nebulised and applied via nasal spray.
-
Placebo: 0.9% saline.
Trial objectives
-
To assess the clinical benefit of the gene therapy formulation when given on a monthly basis over a period of 1 year.
-
To assess the safety and tolerability of the formulation over the same period.
-
To assess gene expression of the formulation over the same period.
Additional research questions
-
Will markers of molecular efficacy (mRNA and PD) measured in the lung correlate with changes in clinical end points?
-
Will markers of molecular efficacy measured in the nose correlate with these markers measured in the lungs in the same patients?
-
Will molecular efficacy increase with repeated administration when measured serially in the same patient?
-
Will it be possible to identify the phenotype of responders or non-responders to allow stratification of patients for future gene therapy trials?
-
Will it be possible to identify stratifiable biomarkers from the secondary outcomes, based on disease severity?
-
Will one (or more) of the secondary outcome measures provide different or better information than FEV1, allowing it to be proposed as a novel registrable biomarker for future trials?
-
Will the extensive preclinical molecular efficacy data produced in support of this study correlate with similar data in humans?
-
Will the extensive two species toxicology package produced in support of this study correlate with the findings in humans?
Trial sponsorship, oversight and approvals
The protocol was developed by the UK CFGTC Strategy Group members and trial statistician (GM) and sponsored by Imperial College London, London, UK.
The protocol was submitted to the GTAC on 7 October 2011 and received approval on 8 March 2012 (GTAC number 184). Clinical trials authorisation was received from the MHRA on 2 February 2012 (EudraCT number 2011-004761-33) and approvals from research and development at all clinical sites was obtained prior to commencing. The trial is registered on clinicaltrials.gov (NCT01621867) and ISRCTN (Current Controlled Trials ISRCTN71164341).
Trial outcome
For a detailed description see Detailed description of outcome measures and assays.
Relative change from baseline in percentage predicted FEV1.
Major secondary outcomes
These include:
-
change in other spirometric values [forced vital capacity (FVC), mid-expiratory flow 25–75% (MEF25–75%)]
-
lung clearance index (LCI)
-
structural parameters on computed tomography (CT) scans of the chest
-
a validated quality-of-life score (as measured by the Cystic Fibrosis Questionnaire – Revised; CFQ-R). 71
Other secondary outcomes
Other measures of lung physiology including:
-
exercise capacity [maximal oxygen uptake (VO2max)] and activity monitoring
-
gas transfer
-
Inflammatory markers:
-
blood – white blood cell count and differential count, C-reactive protein (CRP), serum calprotectin
-
sputum – soluble inflammatory markers, cellular inflammation, 24-hour weight, solid content, DNA content, microbiology.
-
Adverse events (AEs) and other safety measures including:
-
blood biochemistry
-
haematology
-
liver and renal function
-
urinary markers
-
histology
-
fat-laden airway cells.
Mechanistic outcomes
-
Transgene-specific DNA and mRNA on bronchial and nasal brushings.
-
Nasal and lower airway PD measurements (CFTR protein function).
Statistical considerations in design and analysis
Proposed sample and effect sizes
The sample size derivation was based on outcomes of previous clinical trials in CF and available data on what is widely considered the minimum clinically meaningful difference. Over the last two decades, a relatively small number of new drugs have been licensed for the treatment of patients with CF and adopted internationally as standards of care. Those of particular note include the mucolytic agent, recombinant human DNase [rhDNase; dornase alpha (Pulmozyme®, Roche)]; tobramycin for the inhaled solution; and the macrolide, azithromycin.
-
Recombinant DNase acts by breaking down neutrophil-derived DNA, which contributes significantly to mucus viscosity. In the most widely cited double-blind, placebo-controlled trial, the drug led to a relative improvement in FEV1 of 5.8%; this was accompanied by a significant reduction in the frequency of infective exacerbations. 72 This agent is now a licensed product in routine clinical use worldwide.
-
Tobramycin for inhalation solution (TOBI) was designed as an antipseudomonal antibiotic to be administered on alternate months, in contrast to other agents that are administered continuously. At the end of a 6-month trial period, the relative improvement in FEV1 was around 7%. 73 This agent is now considered the gold standard by the regulatory agencies both in Europe and in the USA, to the extent that all other inhaled antibiotics are required to undergo head-to-head testing with it.
-
Azithromycin is an orally bioavailable macrolide that was first considered as a useful therapy in CF after significant success in Japanese panbronchiolitis sufferers. Its mechanism of action is unknown, but is thought likely to relate to its known anti-inflammatory actions. Several trials have been conducted in CF, the largest of which reported a 6.2% TE on FEV1 and a significant reduction in the frequency of exacerbations. 74 Azithromycin is now in widespread clinical use for CF.
Our decision to power for a relative change of 6% in FEV1 was made, in part based on these data, and in part on pragmatic considerations of patient recruitment feasibility and cost of materials. We suggest that the current consensus supports this change as being a clinically meaningful improvement. Based on run-in study data [longitudinal assessment of clinical measurements in patients with cystic fibrosis in preparation for a clinical trial of CFTR gene therapy (unpublished data, Gene Therapy Consortium)], and using the mean of two measurements at baseline and the mean of two measurements at 12 months, we estimate the standard deviation (SD) of the percentage change in percentage predicted FEV1 over 12 months to be 10.0%. The use of duplicate measurements reduces variability substantially, with resultant increases in power. The corresponding SD using only single measurements is 12.2%. We also demonstrated, using data from the run-in study, that analysis of percentage predicted FEV1 is more sensitive than analysis of absolute FEV1; the corresponding SD for percentage change from baseline based on duplicate measurements was 11.6% for absolute FEV1. With the SD of 10.0%, a total sample of 120 evaluable patients would provide 90% power at the 5% significance level (2-sided) to detect a difference of 6% between the randomised groups in the mean change from baseline. With a plan to recruit 130 patients, we would allow a safety margin for subjects leaving the study prior to completion. This number also allows us to be powered at ≥ 80% to detect changes in the secondary outcomes LCI and CT scan parameters, which were smaller than we had previously observed in a study of intravenous antibiotics. 65 The above power calculation was conservative in that covariate adjustment can be anticipated to increase the statistical power.
Statistical analysis
We reviewed the approach taken to the analysis of FEV1 in over 40 published randomised trials of interventions in CF. There is no consistent approach, with roughly half of the trials using absolute changes from baseline (for either absolute FEV1, measured in litres, or percentage predicted FEV1) and the other half using relative changes (i.e. percentage change from baseline in terms of either absolute FEV1 or percentage predicted FEV1). Analysis of absolute changes is typically based on an analysis of covariance (ANCOVA) with baseline FEV1 as a covariate, and in some instances relative changes were also analysed with an ANCOVA to adjust for baseline FEV1. 75 The clinical investigators were of the opinion that in the context of this trial and, in particular, given the age range of the recruits, the clinically relevant measure of TE is the relative (percentage) change from baseline in the percentage predicted FEV1 and not the absolute change. The primary analysis was designed to compare the two randomised groups in terms of the mean percentage change in percentage predicted FEV1 from baseline to end of treatment.
An ANCOVA model was designed to include baseline percentage predicted FEV1, together with the other variables used in the randomisation algorithm, as covariates. ‘Baseline’ will be defined as the mean of the FEV1 values from the two pretreatment assessments. ‘End of treatment’ will be taken as the mean of the values obtained at 14 days and 28 days following the final treatment. The TE will be presented as an adjusted difference in mean percentage change along with its corresponding 95% confidence interval (CI). No interim efficacy analyses were planned. As a sensitivity analysis the above analysis would be repeated, but with the logarithm of the end of treatment percentage predicted FEV1 taken as the response variable, and the logarithm of the baseline percentage predicted FEV1 included as a covariate in place of its raw value. An exploratory analysis compared the two randomised groups in terms of the evolution of FEV1 over the 12 months of treatment. The study was not adequately powered to explore subgroup effects for the primary outcome measure, although we did plan to look at the stability of the TE over subgroups defined by the covariates included in the ANCOVA model. The formal analyses were performed by including interaction terms in the model. A similar approach could be used with certain secondary outcome measures that are closer to the direct mechanism of action of the study intervention, as there is likely to be more statistical power with such variables to explore subgroup effects, which could support a ‘stratified medicine’ approach to the use of gene therapy.
The intention-to-treat (ITT) group was defined as patients randomised and having any subsequent follow-up data. The per-protocol (PP) population was defined as patients receiving ≥ 9 doses. Demographic and safety data are presented for both populations. All efficacy data are presented for the PP population.
Subject identification, recruitment and randomisation
Inclusion criteria
-
Cystic fibrosis confirmed by sweat testing or genetic analysis.
-
Males and females aged ≥ 12 years.
-
FEV1 between 50% and 90% predicted inclusive (Global Lung Function Initiative reference ranges; www.ers-education.org/guidelines/global-lungfunction-initiative.aspx).
-
Clinical stability at screening defined by:
-
not on any additional antibiotics (excluding routine, long-term treatments) for the previous 2 weeks
-
no increase in symptoms such as change in sputum production/colour, increased wheeze or breathlessness over the previous 2 weeks
-
no change in regular respiratory treatments over the previous 4 weeks
-
if any of these apply, entry into the study can be deferred.
-
-
Prepared to take effective contraceptive precautions for the duration of their participation in the study and for 3 months thereafter (as stated in GTAC guidelines)
-
If taking regular rhDNase and is willing, and considered able by independent medical carers, to withhold treatment for 24 hours before and 24 hours after the gene therapy dose
-
Written informed consent obtained.
-
Permission to inform general practitioner of participation in study.
Exclusion criteria
-
Infection with Burkholderia cepacia complex organisms, MRSA or M. abscessus.
-
Significant nasal pathology including polyps, clinically significant rhinosinusitis, or recurrent severe epistaxis (nose bleeds; nasal cohort only).
-
Acute upper respiratory tract infection within the last 2 weeks (entry can be deferred).
-
Previous spontaneous pneumothorax without pleurodesis (bronchoscopic subgroup only).
-
Recurrent severe haemoptysis (bronchoscopic subgroup only).
-
Current smoker.
-
Significant comorbidity including:
-
moderate/severe CF liver disease (varices or significant, sustained elevation of transaminases: alanine transaminase/aspartate transaminase levels of > 100 IU/l)
-
significant renal impairment (serum creatinine levels of > 150 µmol/l)
-
significant coagulopathy (bronchoscopic group only)
-
-
Receiving second-line immunosuppressant drugs, such as methotrexate, ciclosporin and intravenous immunoglobulin preparations.
-
Pregnant or breastfeeding.
Patient recruitment
We had originally anticipated that the majority of trial participants would be recruited from an earlier study (the Gene Therapy Run-in) assessing outcome measures at periods of stability (unpublised data, Gene Therapy Consortuim). However, delays because of funding issues and the resultant drop-off in patient interest meant that recruitment had to be spread more widely. Initial patients were recruited by study staff from the adult and paediatric clinics at the Royal Brompton Hospital and from across Scottish CF centres. Once it was apparent that the pace of recruitment was slower than required, we activated participant identification centres. Local CF centres were activated in November 2012 and nationwide CF centres followed in January 2013; in total, 16 outside centres referred patients. These patients attended one of the three sites for trial-related visits, but continued their clinical care at their referring hospital. All patients had read a detailed, age-specific information sheet and underwent a thorough briefing before signing consent. Parents provided consent for participation of paediatric subjects, who were also asked for their assent to take part. Patient information sheets are available in Appendix 1.
Randomisation
Randomisation was performed by trial staff following a successful screening visit using the electronic InForm™, version 4.6 (Phase Forward Incorporated, Oracle, CA, USA) system. Patients consenting to participate in one or more of the mechanistic subgroups were randomised 2 : 1 to enrich for subjects receiving active treatment; all others were randomised on a 1 : 1 basis. The data required for randomisation and stratification [centre (London or Edinburgh), age (< 18/≥ 18 years), FEV1 (< 70/≥ 70%) and inclusion in the mechanistic subgroups] were entered by a member of the trial team, which led to the generation of a unique patient number corresponding to a blinded randomised arm of the trial. In the event of computer system failure, a prearranged manual randomisation method was available from the InForm team. The unique patient number was entered onto the subject’s prescription sheet and submitted to the trial pharmacists who had access to the unblinding code and prepared the active or placebo product as appropriate.
Preparation of dose and blinding
Assembly of pGM169/GL67A complexes
The pGM169 drug substance and GL67A cationic lipid mixture are imported into the UK by The Clinical Biotechnology Centre of the University of Bristol, Bristol, UK, by a qualified person acting on behalf of the UK National Blood Service, a division of the NHS Blood and Transplant Special Health Authority. The dose preparation is described in full in Appendix 2. Briefly, immediately prior to use, pGM169 and GL67A are thawed to room temperature and GL67A is hydrated in water for injection. pGM169/GL67A complexes are prepared by rapidly mixing 5 ml of pGM169 and 5 ml of GL67A using a disposable syringe-based static mixer device [Plas Pak Industries, Norwich, CT, USA (Figure 5)]. The final dosage form of pGM169/GL67A contains nominally 2.6 mg/ml of pGM169, 3.7 mg/ml of GL67, 8.9 mg/ml of DOPE and 1.7 mg/ml of DMPE-PEG5000. The shelf life of the final formulation was 6 hours.
FIGURE 5.
Mixing of the trial formulation. The pGM169 and GL67A were mixed in a dual-barrel mixing device by loading the components into the syringe (a) and using a fixed-rate plunge mechanism (b) to deliver this into the receiving container.


Blinding
pGM169/GL67A or placebo (0.9% saline) were then filled into AeroEclipse II breath-actuated nebulisers by unblinded trial pharmacists. To avoid any inadvertent unblinding by clinical staff, nebulisers were taped and a tamper-proof seal was attached (Figure 6 and see Appendix 3 for details). Clinical trial staff, participants and trial analysts were blind to allocation until database lock. The 10-ml volumes were placed in opaque nasal spray devices (GlaxoSmithKline parts number AR5989 30 ml bottle/AR9488 30 ml actuator) and the device was primed.
FIGURE 6.
Masking of the nebuliser pots to ensure blinding. AeroEclipse II nebuliser pots are transparent (a) as the gene therapy formulation has a milky appearance that contrasts clearly with that of the 0.9% saline (clear, colourless liquid), it was necessary to mask the pots, which was achieved with tape and (b) a tamper-proof seal was applied to the lid, which also prevented any unintentional spillages.


Administration of doses to trial participants
Patients were pretreated with 200–400 µg of salbutamol via a metered-dose inhaler and spacer device approximately 20 minutes prior to receiving the trial treatment to prevent bronchospasm in response to the hypotonic nature of the trial preparation. Nebulisation took place in sealed negative-pressure cubicles, with external venting to limit spread within the hospital setting; patients were observed and communicated with through a glass window by the trial team. The 5-ml aliquot was nebulised during cycles of tidal breathing while patients were wearing a nose clip (Figure 7) for 3 minutes, following which the nebuliser air supply (8 l/minute) was turned off for a 2-minute rest period. Nebulisation continued for 40 minutes, which was predetermined as a period after which delivery was complete. Patients in the nasal subgroup administered one actuation of the nasal spray device to each nostril during the 2-minute ‘off-nebuliser’ period, which resulted in an approximate 1-ml dose to each nostril (see Figure 7). After dosing, patients either remained in the cubicle for 40 minutes, or were able to leave wearing a mask to limit spread of exhaled trial product in the unit. They were observed for 1–2 hours, but no spirometry or temperature measurements were made, to limit the potential for unblinding related to transient dose-related fever or drop in lung function. Patients were administered paracetamol (dose adjusted for body weight if < 40 kg) within 2 hours of dosing and asked to take another dose 6 hours later, once at home. This was again to prevent unblinding of the active group.
FIGURE 7.
Dose administration. Nebulisation of the trial product in a sealed, negative-pressure cubicle while subject (a) wears a nose clip and; (b) receives delivery of nasal dose. A member of the study team illustrates the technique.

Study visits and interventions
Early patient safety cohort adaptive design
An adaptive early recruitment phase was designed to permit the early identification of any cumulative side effects: 20 subjects (10 active treatment and 10 placebo) would receive three doses at 4-weekly intervals before any further subjects were dosed. In addition to the visits described below undertaken by the entire cohort, they were also seen on day 2 post each dose. Clinical examination findings, lung function, gas transfer and systemic inflammatory markers would then be reviewed in an unblinded fashion by the Data Monitoring and Ethics Committee (DMEC).
-
Should data prove satisfactory, these subjects would continue with subsequent visits and the remainder of the cohort will begin dosing.
-
Should the data be of sufficient concern to the DMEC that progression was unacceptable, a second cohort of 20 patients would receive three doses of the formulation at a 2.5-ml dose, followed by DMEC review in a similar fashion.
-
If these data were acceptable, the trial would continue with all subjects receiving a 2.5-ml dose of either gene therapy or placebo. In this instance, the initial cohort would be discontinued and an additional 20 ‘naive’ subjects recruited.
-
Should the DMEC have considered these data unacceptable, the trial would have been halted.
-
The adaptive design was not in fact required as there were no concerning safety signals. Safety data from this cohort are presented in Chapter 5.
Schedule of trial visits
Scheduled study visits and interventions are shown in Table 1.
| Summary of study assessments | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Week | 0 | 4 | 8 | 12 | 16 | 20 | 24 | 28 | 32 | 36 | 40 | 44 | 46 | 48 | 49 | ||||||||||
| Visit name | E&C | Intro | Screen | nPD1a | nPD2a | nPD3a | Broncha | Dose 1 | D2a | Dose 2 | D2b | Dose 3 | D2c | Dose 4 | Dose 5 | Dose 6 | Dose 7 | Dose 8 | Dose 9 | Dose 10 | Dose 11 | Dose 12 | F/U1 | F/U2 | Bronch |
| Consent/history/examination | |||||||||||||||||||||||||
| Informed consent | ✗b | ✗b | ✗b | ✗c | |||||||||||||||||||||
| Medical history | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Physical examination | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Quality-of-life questionnaire (CFQ-R) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||||||||||||||||
| Vital signs | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Pulse oximetry | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Prior medications | ✗b | ✗b | ✗b | ||||||||||||||||||||||
| Concomitant medications | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||
| Check diary comments and reissue diary | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||||
| AEs | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||
| Download PiKo-6 device (nSpire Health Ltd, Hertford, UK) | ✗d | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||
| Sweat teste | ✗e | ✗e | ✗e | ||||||||||||||||||||||
| Samplesf | |||||||||||||||||||||||||
| Blood sampling | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| Urine sampling | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| Sputumg | ✗h,i | ✗h,i | ✗h | ✗ | ✗j | ✗ | ✗ | ✗ | ✗ | ✗i | ✗i | ||||||||||||||
| Sputum for NTM | ✗k | ✗k | ✗k | ✗k | ✗k | ✗k | |||||||||||||||||||
| 24-hour sputum weight | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||||||||||
| Investigationsf | |||||||||||||||||||||||||
| LCI | ✗h | ✗h | ✗h | ✗ | ✗ | ✗ | ✗ | ✗ | |||||||||||||||||
| Exercise bike test | ✗k | ✗k | ✗k | ✗k | ✗ | ✗ | |||||||||||||||||||
| Activity monitor | ✗ | ✗ | ✗ | ||||||||||||||||||||||
| CT scan | ✗ | ✗l | ✗ | ||||||||||||||||||||||
| Gas transfer | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||||||||||||
| Bronchial blood flow measurementm | ✗ | ✗ | ✗ | ||||||||||||||||||||||
| Spirometry | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Dosing | |||||||||||||||||||||||||
| Randomisationn | ✗ | ||||||||||||||||||||||||
| Study dose administration | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||||||||||
| Nasal/bronchoscopy subgroups only | |||||||||||||||||||||||||
| Nasal PD | ✗o | ✗o | ✗o | ✗o | ✗o | ✗o | ✗p | ✗m | ✗p | ✗ | ✗p | ✗m | ✗h | ✗h | ✗h | ||||||||||
| Nasal brushing | ✗q | ✗q | ✗k | ✗k | |||||||||||||||||||||
| Bronchoscopy | ✗ | ✗ | |||||||||||||||||||||||
| Predischarge administration | |||||||||||||||||||||||||
| Instructions regarding home spirometry (PiKo-6) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Instructions regarding use of symptom diary | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Schedule next study appointment | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||||
Detailed description of outcome measures and assays
Clinical examination
Clinical examination included recording pulse rate, blood pressure (BP), respiratory rate, temperature, pulse oximetry, height, weight and lung auscultation. Patients were assessed by a team comprising medical, nursing and respiratory physiology personnel.
Spirometry
Forced expiratory manoeuvres from which the FEV1 and the total volume exhaled (FVC) are conventional measures of airway patency and lung health, and are widely used both clinically and in trial settings for CF. The MEF (flow at varying percentages of vital capacity) are thought to more closely reflect small, distal airway disease, but are recognised as being more inherently variable. In CF, FEV1 is closely linked to survival and is the most widely used primary outcome in clinical trials of respiratory interventions. It is recognised by regulatory agencies as a surrogate outcome and for this reason, alongside our longitudinal data demonstrating good power, it was chosen as the primary outcome for this trial. Spirometry was performed on the EasyOne spirometer (New Diagnostic Design Technologies, Zurich, Switzerland) with disposable mouthpiece and filter according to the American Thoracic Society (ATS)/European Respiratory Society (ERS) criteria. 76 A minimum of three measurements and the best FEV1, FVC and MEF25–75% were recorded in absolute values. These values were converted to percentage predicted values based on the Stanojevic references. 77
Lung clearance index
The LCI is a measure derived from a multiple breath washout (MBW), which provides a global measurement of ventilation inhomogeneity. It can be performed either with inhalation of an inert tracer gas, such as sulphur hexafluoride (SF6), or by using 100% oxygen to wash out resident nitrogen. In the case of an exogenous tracer, the gas is inspired until equilibrium is reached between the inhaled and exhaled air at which point the gas source is removed. The number of lung volume turnovers required until the expired tracer gas concentration falls to an arbitrary 1/40th of its starting value is used to calculate the LCI. Individuals with lung disease and greater ventilation inhomogeneity require longer to clear the tracer gas and, therefore, will have a higher (more abnormal) LCI.
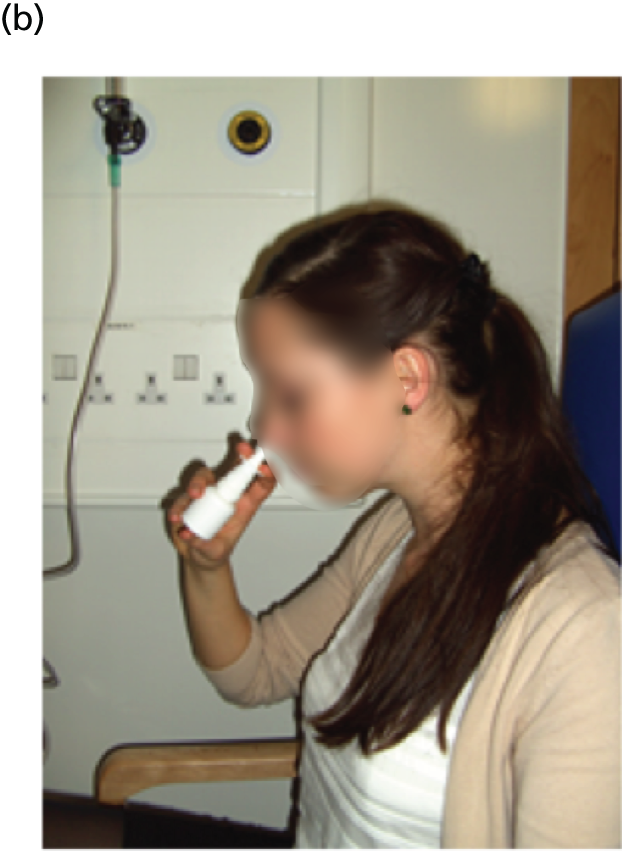
Many different systems have been used to measure MBW in clinical trials in CF and these have been summarised in a recent ATS/ERS consensus document. 78 Although the mass spectrometer is considered the gold standard gas analyser, it is very expensive, custom built for MBW and, therefore, not suitable for widespread use. 78 The method adopted in this trial used the tracer gas, SF6, detected by the Innocor™ photoacoustic gas analyser (Innovision, Glamsbjerg, Denmark). Methodology was as described in a recent trial of the small molecule, ivacaftor. 79 In brief, the procedure is performed with a mouthpiece during tidal breathing while patients wear a nose clip (Figure 8). Triplicate measures are undertaken, with each test taking approximately 15 minutes. Data are subsequently analysed off-line according to strict standard operating procedures (SOPs) for quality control using software developed within the consortium. Every seventh trace was analysed in duplicate by a corresponding team member from the other site.
FIGURE 8.
Lung clearance index. (a) A child performs a LCI on the Innocor gas analyser; (b) wash-in of the tracer gas, SF6 (in green) is undertaken until equilibrium; and (c) the gas supply is disconnected and concentration falls with each subsequent breath as washout occurs (bottom). The black trace denotes flow.
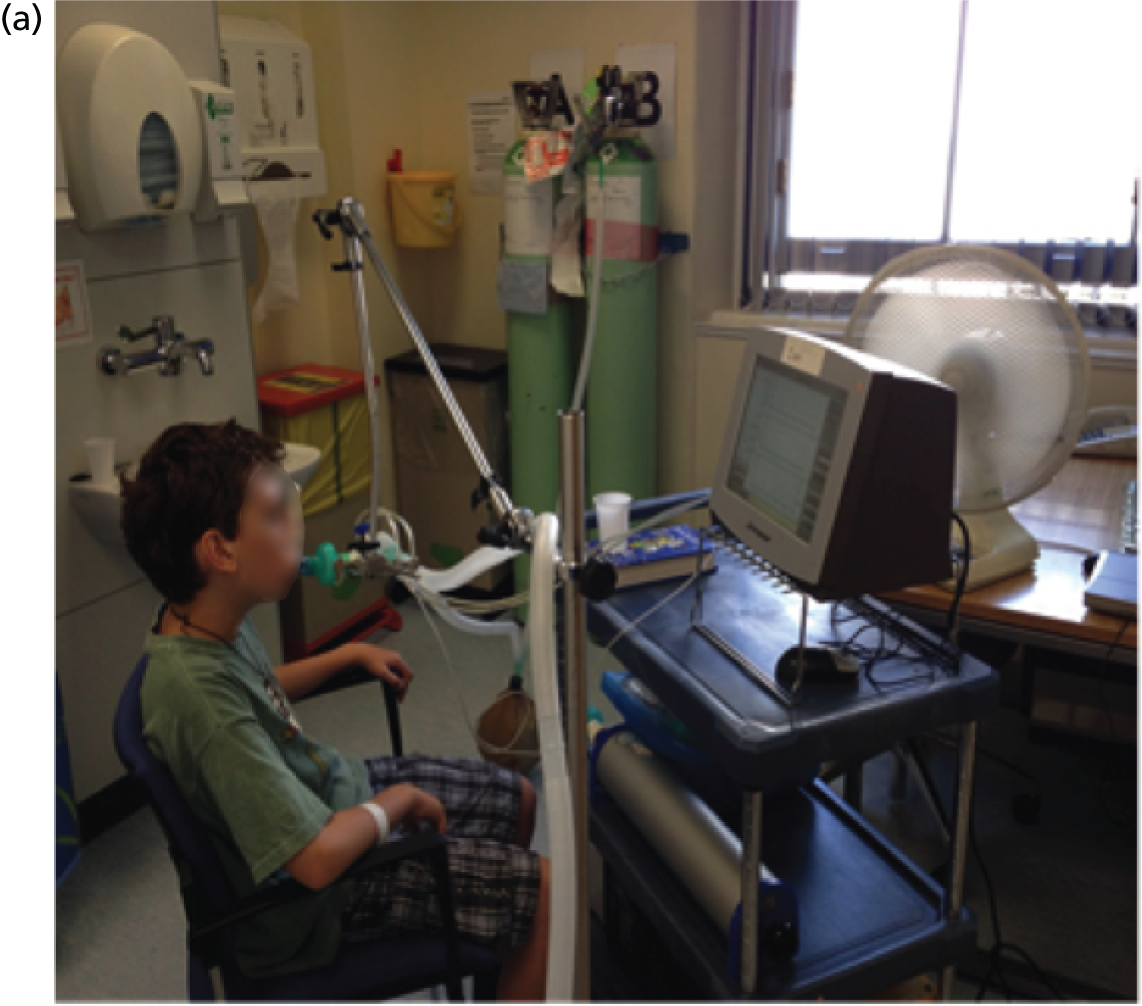
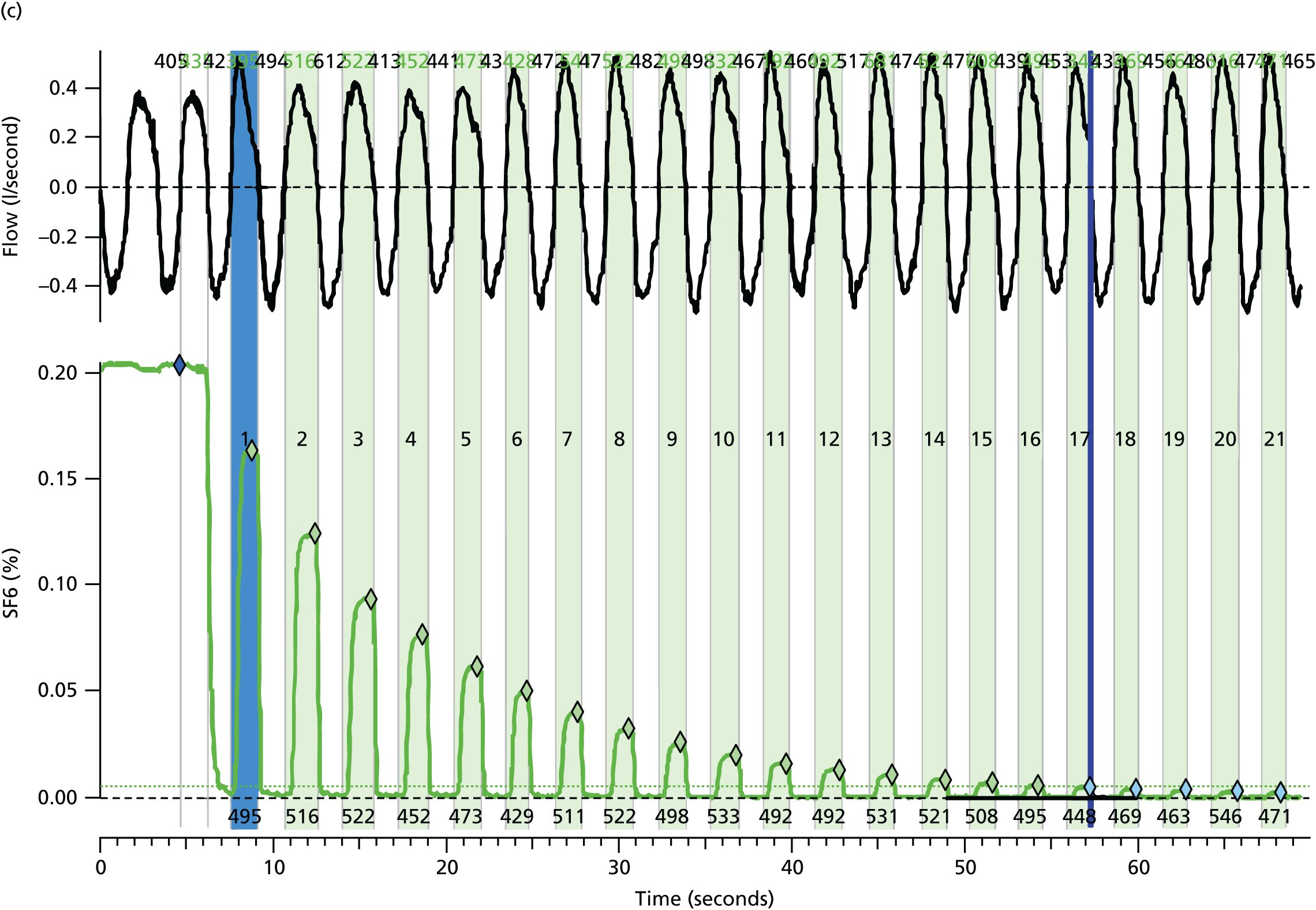
Cycle ergometry
This test of exercise capacity was performed on a stationary exercise bike with breath-by-breath analysis on the Innocor photoacoustic gas analyser and was used to calculate VO2max. Subjects were asked to pedal at a set speed of 70 rpm and maintain this speed throughout the test while wearing a nose clip and breathing via a mouthpiece attached to a gas analyser. The resistance increased automatically each minute for as long as pedalling could be sustained. The starting workload and increase in workload was dependent on the patient’s height, and calculated using the Godfrey protocol.
-
Patients < 120 cm use 10 W starting resistance and 10-W increments.
-
Patients 120–150 cm use 15 W starting resistance and 15-W increments.
-
Patients > 150 cm use 20 W starting resistance and 25-W increments.
The subject’s heart rate, oxygen saturations and Borg scale were measured at rest and at each minute during exercise. The test was stopped by the operator if the patient’s oxygen saturation fell below 80%, if they were unable to maintain the required pedalling rate or if there was any concern about the patient’s condition. Once the test was complete, patients had a 2-minute cool-down period and were monitored until their oxygen saturation returned to baseline. The Innocor system was validated against standard calibrated VO2 equipment using paired exercise tests in 12 CF patients. For VO2max, the mean difference (reference equipment – Innocor) was –0.026 l/minute and the 95% CI was –0.27 to 0.22 l/minute.
Body-worn activity monitoring
Patients were asked to wear the SenseWear® body monitoring system™ (BodyMedia, Pittsburgh, PA, USA) for at least 7 whole days at time points throughout the trial. The device uses accelerometry, heat flux, skin temperature and galvanic skin response sensors to gather physiological data on movement and daily physical activity patterns. These armbands have been validated in normal subjects and a number of patient groups. 80 Data from activity monitors were downloaded and analysed using proprietary software. Final data were analysed when there were at least 4 days of data, at least 1 day was on a weekend and there were at least 10 hours of data for each day.
This analysis yields data for the proportion of time that wearers spend at various levels of energy expenditure. For the purposes of summarising these data in the present trial, a comparison was made (between active and placebo groups) of the percentage of time spent in vigorous exercise (defined as > 3 metabolic equivalents; METs) compared with the percentage of time spent at ≤ 3 METs of activity, as well as the mean number of steps taken per day.
Gas transfer
The transfer factor of the lung for carbon monoxide (TLCO) was measured using a single-breath technique (London site: Jaeger Masterscreen, CareFusion, Germany; Edinburgh site: CPL, nSpire Health, England). Subjects, who were seated and wearing a nose clip, were asked to exhale to residual volume, then take a maximal inspiration (required to be > 90% vital capacity) of the test gas (medical air with 0.28% carbon monoxide and 9% helium) prior to a breath hold of 10 seconds’ duration and a smooth exhalation. This procedure was repeated at least twice on each test day (minimal interval between tests was 4 minutes). Duplicate estimates of TLCO were required to be within 10% or 1.0 mmol/minute/kPa (whichever was greater) to meet requirements for inclusion. At least two technically acceptable tests from up to 10 attempts were obtained and the mean values were used for analysis. The following values were recorded for each test: single-breath TLCO, alveolar volume (VA), transfer coefficient (KCO; derived from TLCO/VA) and inspiratory vital capacity. TLCO and KCO were corrected for most recent haemoglobin (usually obtained on the same day) and expressed as corrected TLCO (TLCOc) and corrected KCO (KCOc) using standard formulae.
Quality-of-life questionnaire
The validated CFQ-R for either adults or children was completed by subjects at time points throughout the trial, always at the start of a visit before any other study investigations. The questionnaire is available in Appendix 4.
Lung computed tomography scanning
High-resolution CT scans were obtained on 64-channel multidetector scanners (SOMATOM Sensation 16 or 64; Siemens Medical Solutions, Erlangen, Germany). On two occasions (pre-dosing and at 28 ± 5 days after dose 12) patients underwent a full scan comprising contiguous thin sections (1.25 mm) through the entire volume of the lungs obtained during inspiration and also interspaced (1-mm sections at 10-mm increments) at end-expiration. A high-spatial-resolution algorithm was used for image reconstruction. All CT scans were scored independently by the same two radiologists specialising in thoracic imaging; scans had been anonymised and scores were assigned with the radiologists blinded to both subject identification and whether pre- or post-dosing. All scoring was performed directly from workstations with access to image manipulation including window settings (default: width 1500 Hounsfield units, centre 500 Hounsfield units). The presence and severity of specific CT scan features were recorded for each lobe as previously described. 81 The features studied were extent of bronchiectasis, severity of bronchiectasis, bronchial wall thickness, small and large airway mucus plugs (all scored per lobe on a range of 0–4 and the results meaned) and gas trapping (scored on a percentage basis) (features illustrated on examples in Figure 9). In addition, as a safety outcome, all subjects underwent a limited-cut inspiratory CT scan pre-dosing at the dose 4 visit; these scans were not formally scored, but were reviewed in real time by the clinical radiology team for any acute changes and the reports were provided to the DMEC.
FIGURE 9.
Computed tomography parameters. (a) Examples of bronchiectasis; (b) gas trapping (areas of mosaic attenuation highlighted in yellow where gas has failed to empty from areas of lung on expiration); and (c) an area of dense small mucus plugs in the left-lower lobe highlighted in yellow.

Venepuncture for blood sampling
This was performed by an experienced practitioner after the application of topical anaesthetic cream, if desired by the patient, via a peripheral vein. The use of central indwelling intravenous catheters was discouraged but not prohibited.
Urine sampling
Urine was collected into a sterile container and tested immediately by dipstick.
Sputum sampling
Sputum was collected after spontaneous expectoration whenever possible. In patients unable to expectorate, induction was considered at screening and follow-up visits only; all other visits included administration of study drug and it was considered undesirable to expose the patients to a procedure known to induce a degree of bronchospasm prior to dosing. The induction protocol followed clinical guidelines and included pretreatment with an inhaled bronchodilator (200 µg of salbutamol or equivalent, 15 minutes before test). Hypertonic saline (7%) was administered via a deVilbiss 2000 (DeVilbiss Healthcare Ltd, Tipton, UK) or equivalent ultrasonic nebuliser. Attempts to induce sputum were made during and after nebulisation and FEV1 was monitored carefully throughout. Prior to dose 1, sputum was expectorated in 72 out of 116 (62%) of patients [active group, 37/62 (60%); placebo group, 35/54 (65%)]. After the last dose, sputum was expectorated in 57 out 116 (49%) of patients [active group: 27/62 (44%); placebo group: 30/54 (56%)].
At certain visits, patients were asked to collect all sputum expectorated over the previous 24 hours into 50-ml Falcon tubes (VWR International Ltd, Leicestershire, UK) and to bring these with them to the study visit.
Nasal potential difference
The characteristic CF ion transport defects, involving the charged ions chloride and sodium, result in an altered PD across the respiratory epithelium, measurable in millivolts. Techniques to measure this for both clinical diagnostic purposes and in the context of trials have been developed. 82,83 When compared with healthy cells expressing CFTR, in CF the absent chloride secretion and sodium hyperabsorption result in an apical cell surface that is electrically more negative under basal or resting (non-stimulated) conditions. Amiloride, as a sodium channel blocker, results in a greater reduction (depolarisation) in nasal PD for CF than non-CF subjects and subsequent ionic and pharmacological challenges to stimulate chloride secretion (via chemical gradients and cAMP stimulation) are largely ineffective in CF but lead to pronounced hyperpolarisation in the presence of functional CFTR (Figure 10).
FIGURE 10.
Representative nasal PD traces from (a) a CF patient; and (b) a healthy volunteer. The CF basal (Ringer’s, R) measures are higher (more negative) than non-CF largely related to sodium hyperabsorption. For the same reason, there is a larger drop (depolarisation) to the sodium channel blocker, amiloride (RA). There is little chloride secretion in response to either a zero chloride solution (ZC, passive gradient) or the cAMP stimulator, isoproterenol (ZCAI), in contrast to the hyperpolarisation observed in the healthy volunteer.

There have been over 20 previous clinical trials of CF gene therapy, which have involved dosing to the nose, lung or both. 84 In trials involving nasal dosing, nasal PD has been used as a main outcome for efficacy. 39,42,50–52,55,58,84–86 However, these tests are inherently variable and gene therapy-related results have been similarly so; correlation of any change in nasal PD with molecular evidence of CFTR has also often been lacking. 50,52,58 Where changes in PD have been reported following gene therapy, these have almost uniquely been restricted to the chloride secretion phase rather than correcting sodium hyperabsorption phases. This likely reflects the higher proportion of cells required to be corrected before changes in sodium hyperabsorption can be corrected (> 90% vs. < 10% for restoration of chloride transport). 87–89 Use is not limited to gene therapy evaluation, and recent studies have included trials of the small-molecule drugs ataluren and ivacaftor. Interestingly, while the latter small-molecule potentiator demonstrated significant and large-magnitude clinical changes, changes in nasal PD were much more modest.
For reasons of variability, patients electing to participate in the nasal dosing substudy underwent up to three nasal PDs on different days prior to dosing and two at follow-up appointments. A minority of subjects also underwent on-treatment recordings at other time points, but these are not included in the analysis owing to the small number.
At the time of the first measurement in any subject, a single-use, disposable, dual-lumen catheter (Marquat, France) was fixed in place at the site of maximal unstimulated PD in one nostril; for all subsequent measurements, the same nostril and the same distance into the nasal cavity was used. Solutions comprised chemicals listed in the Cystic Fibrosis Foundation Therapeutic Development Network (CFF TDN)’s SOP90 and were perfused at room temperature at a rate of 4 ml/minute in the following sequence: Ringer’s solution, Ringer’s plus amiloride (0.1 mM), Ringer’s plus amiloride plus zero chloride and Ringer’s plus amiloride plus zero chloride containing isoproterenol (0.01 mM) (Figure 11). Post-dosing measurements were obtained after 14 ± 2 days and 28 ± 5 days.
FIGURE 11.
Nasal PD. The subject has a double-lumen catheter inserted in one nostril. Solutions are pumped through one lumen while the other is connected to an electrode via conducting cream. There is a reference electrode placed subcutaneously on the forearm.

Traces were obtained using a high-impedance, low-resistance voltmeter (LR-4; Logan Research UK Ltd, Rochester, UK) onto a laptop computer and were scored by four investigators experienced in the technique (EWFWA, JCD, SNS and MDW). The four components (baseline, amiloride, zero chloride and isoproterenol responses) were scored individually using standard criteria.
Nasal brushings
Nasal brushings were performed in subjects in the nasal subgroup both pre-dosing and at 28 ± 5 days post final dose from under the middle or inferior turbinate of both nostrils using interdental brushes (Dent-o-care, London, UK). Cells were suspended in 800 µl of phosphate-buffered saline (PBS) and processed for transgene DNA/mRNA analysis (see Nasal and bronchial brushings).
Bronchoscopy and associated procedures
Bronchoscopy was undertaken in a subgroup of patients for both efficacy (gene transfer assessed by mRNA on brushings and PD) and safety assays (remodelling or lipid deposition in endobronchial biopsies).
Potential difference
Cystic fibrosis transmembrane conductance regulator-mediated ion transport is clearly of more relevance in the lower airway than the nose, but is more complex and invasive to measure. No standardised technique has been developed and only a few research groups perform this procedure. General anaesthesia is required to overcome the challenges of interference from patient movement or coughing, and to avoid local anaesthetic agents that affect ion channel function. The single operating channel within most fibreoptic bronchoscopes limits the size of the PD catheters used. Although some systems allow for a double-lumen PD catheter to pass through this port, our group has designed a single-lumen technique. 91 The technique permits basal and stimulated measurements, although pooling of solutions which then dilute subsequent perfusate needs to be carefully avoided; for this reason, perfusion rate is slower than for nasal measurements and an amiloride phase is omitted. We have previously shown clear separation between CF and non-CF basal PD in the proximal airways, although in both groups PD falls with distal progression and a disease-specific difference is not seen in the most peripheral sites measured. As expected, chloride secretory responses are significantly different between CF and non-CF (Figure 12). This technique was used in our previous trial of GL67-mediated CFTR (with a previous version of the CFTR plasmid incorporating a CMV promoter) and showed significant improvement in chloride secretion on day 2 post dosing. 58
FIGURE 12.
Chloride secretory responses on lower airway PD. Summary data demonstrating the increase in millivolts in lower airway PD in non-CF patients following perfusion with a zero-chloride solution containing isoprenaline (indicating chloride secretion). The effect is absent in patients with CF. The pre–post design of measurements in this trial is in an attempt to detect a signal over and above pre-dosing traces in the actively treated group. 91

In the current trial, all bronchoscopies and associated procedures were performed by a single operator (JCD), with the same senior anaesthetist (BK). Solutions (Ringer’s and a zero-chloride solution) were manufactured by the pharmacy at Eastbourne Hospital, UK and constituted the chemicals in the CFF TDN SOP,90 with the exception that amiloride was omitted from the latter. They were warmed prior to use to reduce the formation of microbubbles, but were used at room temperature and perfused at 100 ml/hour (Figure 13). Basal (non-stimulated) PD was measured on each wall of the distal trachea with sterile Ringer’s solution. Where possible, a stable period (< 1 mV change over 30 seconds) was recorded. Subsequently, at three sites more distally (approximately fifth-generation airways; all in one lung), following a second period with Ringer’s solution, the solution was switched to a zero-chloride equivalent containing isoproterenol (10 µM) for 5 minutes. The catheter was removed and reprimed between measurements. Hardware and software were as previously described. 91 Measurements were completed before the samples described below were obtained.
FIGURE 13.
Bronchial PD measurement. Under video control, the PD catheter is advanced via the bronchoscope onto the lower airway epithelial surface and can be observed on the screen towards the top of the picture. Solutions are perfused via pumps to the right and recordings made via a millivoltmeter connected to a laptop computer.
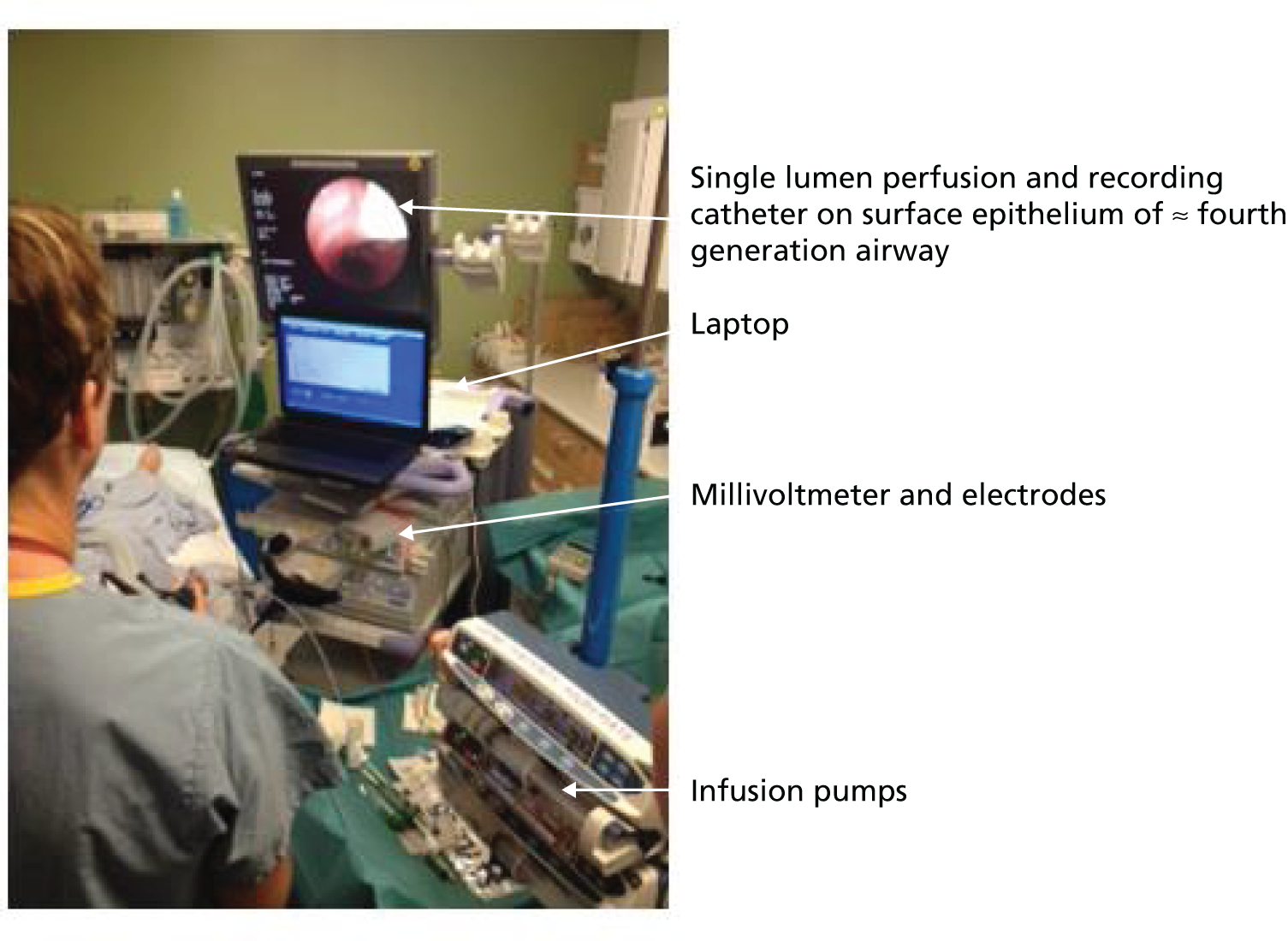
For analysis, recordings (1–3) from the same patient at the same time point were pre-grouped and scored blinded both for acceptability and response. Recordings were only accepted for scoring if (a) the time of any inflexion owing to the zero chloride/isoproterenol solution was approximately 50–80 seconds after onset of perfusion, this duration having previously been shown to represent clearing of the dead space within the catheter (≈50 seconds) and allowing ≈30 seconds for the initiation of any isoproterenol response; and (b) the response fell into one of three categories namely no response, continuous upwards (hyperpolarisation) or continuous downwards (depolarisation) deflection. Recordings were excluded if (a) the response at the 50- to 80-second time point was unstable (rising or falling); or (b) the deflection was characterised by both a depolarisation and a hyperpolarisation. Traces were scored by four investigators with experience in the technique (EWFWA, JCD, KH and SNS).
Bronchial brushing
Ten bronchial brushing samples were obtained from throughout the airways of the same lung using disposable cytology brushes (BC-202D-5010; Olympus UK, Southend-on-Sea, UK); to prevent cell loss that occurs while resheathing the brush, the entire bronchoscope was removed each time and the brush cut while still exposed at the distal end of the scope. Brushed cells were placed into two aliquots of PBS (five brushings in each) and processed for DNA/mRNA analysis (see Nasal and bronchial brushings).
Endobronchial biopsy
Two endobronchial biopsies were obtained from approximately fifth-generation subcarinae with disposable forceps (BC-202D-5010, Olympus UK, Southend-on-Sea, UK; Figure 14) and processed as described in Endobronchial biopsies. Bronchoalveolar lavage was not undertaken, but, where possible, samples of secretions were sent for microbiological culture.
FIGURE 14.
Endobronchial biopsy. (a) The biopsy forceps were advanced under direct vision to a subcarina of an approximate third- to fifth-generation airway where two biopsies were obtained; and (b) representative morphology on a haematoxylin and eosin (H&E)-stained section of an endobronchial biopsy demonstrating pseudostratified columnar epithelium and a submucosa containing smooth muscle, glandular tissue and an inflammatory cell infiltrate.

Post procedure, patients were observed for 4–6 hours prior to discharge.
Sample handling, preparation and assays
Blood
Routine clinical
Blood samples for electrolytes, renal function, CRP, full blood count and coagulation screen were processed and reported by the clinical laboratories in accordance with usual routine hospital practice.
Inflammatory markers
Serum was frozen and stored for subsequent cytokine analysis. Serum interleukin 6 was measured on the Beckman Access 2 immunoassay analyser (Beckman Coulter, High Wycombe, Buckinghamshire, UK). An in-house calprotectin enzyme-linked immunosorbent assay (ELISA) was used (intra-assay coefficient of variation = 5.6%; unpublished observations). Calprotectin antibodies were kind gifts of Erling Sundrehagen, Oslo, Norway. Incubations were carried out in a damp box to prevent potential evaporation during the ELISA protocol. Microtitre plates (Immulux HB, Dynex Technologies, Chantilly, VA, USA) were coated overnight at 4 °C with 100 µl of anticalprotectin (mouse antihuman) (Axis-Shield Diagnostics Ltd, Dundee, UK; Diatec mAb Clone Cp5 B1252, Product number 9790) monoclonal antibody at 2.5 µg/ml in coating buffer (KPL/Insight Biotechnology). Plates were blocked with 1% bovine serum albumin (BSA; Sigma-Aldrich Company Ltd, Gillingham, UK) for 1 hour at 37 °C and washed four times with 0.05% Tween-20 (Sigma-Aldrich Company Ltd, Gillingham, UK). Duplicate samples of 100 µl were added to the plate in 1 : 60,000 dilutions for sputum in PBS and 1 : 500 for serum [50% fetal calf serum (Sigma-Aldrich Company Ltd, Gillingham, UK) in PBS]. Positive controls of calprotectin (Immundiagnostik, Oxford Biosystems, Oxford, UK) were included. Human recombinant calprotectin (Cambridge Biosystems, Cambridge, UK) was used to produce 1.56–100 ng/ml standard curves. The samples: anti-calprotectin [chicken antihuman (Axis-Shield Diagnostics, Ltd, Dundee, UK; lot number: 4095804)] polyclonal antibody at 1 : 5000 dilution; 100 µl of alkaline phosphatase-conjugated donkey anti-chicken immunoglobulin G (Jackson Product Code 703-055-155, Jackson ImmunoResearch, Suffolk, UK) at 1 : 5000 dilution were added to wells in three cycles of incubation at room temperature for 1 hour on a platform vibrator (450 rpm) followed by washings. Finally, 100 µl of BluePhos® Microwell Phosphatase Substrate System (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD, USA) was added to each well. Plates were incubated in the dark at 600 rpm until the blue colour developed, and reached an absorption of 620 nm on a BioTek plate reader (BioTek UK, Swindon, UK). Sample concentrations were calculated using the five-parameter logistic (5PL) non-linear regression curve-fitting model. 92
Antibodies against double-stranded DNA (dsDNA) (immunoglobulin G) were processed by the clinical laboratories using a commercial dsDNA assay was performed on the Immunocap 250 analyser (ThermoScientific, ThermoFisher Diagnostics, Milton Keynes, UK) in accordance with routine hospital practice. Out of 232 samples, 218 (94%) were available for analysis. Peripheral blood mononuclear cells (PBMCs) were extracted and human interferon-γ ELISA for the analysis of CFTR-specific T cells was performed as previously described. 93 Out of 232 samples, 170 (73%) were suitable for analysis. In the remaining 27% of samples, cell viability was too low to perform the assay. Cell viability was likely affected by storage of PBMCs in liquid nitrogen and shipment of samples from the UK to the USA, where the assays were performed.
Sputum
Samples collected into Falcon tubes for 24 hours prior to a visit were weighed. Freshly expectorated sputum obtained during the trial visit was stored on ice for a maximum of 2 hours and processed as previously described. 65 If patients did not produce sufficient sputum to perform all sputum assays, the following priority for assays was assigned: (1) clinical microbiology (generated from cough swabs if patients were completely non-productive); (2) soluble sputum that generated samples for quantification of interleukin 8 (IL-8), calprotectin and extracellular DNA as well as sputum cells; and (3) sputum solid content. Samples to prepare soluble sputum were available from 130 out of 232 samples (56%).
Microbiology
Samples were processed according to CF-specific protocols in the microbiology laboratories at the clinical centres. Semiquantitative reports of cultured bacteria and fungi (and non-tuberculous mycobacteria pre-dose 1 and at follow-up) were provided. Completely non-productive patients underwent cough swabs for microbiological surveillance following clinical protocols.
Soluble inflammatory mediators
Sputum calprotectin was measured as described above for serum. Sputum IL-8 assays were performed using a commercial kit (Human IL-8 Ultra Sensitive Kit, kit number KAC1301; Biosource/Invitrogen, CA, USA), following the manufacturer’s instructions. Plates were read using a Biotek plate reader, and standard curves produced using the 5PL non-linear regression curve-fitting model. Extracellular DNA was quantified in the soluble sputum fraction using the Quant-iT™ PicoGreen® dsDNA Assay Kit (Invitrogen, Paisley, UK) according to the manufacturer’s recommendations. Sputum solid content was determined as previously described. 65 Out of the expected 232 samples, 169 (63%) were not available because patients did not expectorate sufficient sputum.
Cell counts
Cells were isolated from solubilised sputum as described (Tracking65) and approximately 105 sputum cells were cytospun (5 minutes at 500 rpm) onto cytoslides (Thermo Shandon Ltd, Cheshire, UK) (six slides per subject). Slides were air dried and fixed for 10 minutes in 4% formalin (Sigma-Aldrich, St Louis, M0, USA) and subsequently stored at –20 °C until further use. For total and differential cell counts, cells were fixed for 10 minutes in cold methanol (Fisher Scientific, Loughborough, UK), dried and stained with May–Grünwald–Giemsa (MGG quick stain; Bio-Optica, Milan, Italy) using routine histological procedures and neutrophils, lymphocytes, macrophages and eosinophils quantified. Cells were counted on random fields until 300 neutrophils were counted. Total and differential cell counting could be performed on 129 out of 232 (56%) and 111 out of 232 (48%) samples, respectively.
Quantification of intracellular lipid droplets could be performed on 121 out of 232 (52%) of samples and was performed on slides stained with ORO using a protocol adapted from Lian et al. 94 Briefly, defrosted slides were washed in distilled water, dried and placed for 3 minutes in 100% propylene glycol (Sigma-Aldrich, St Louis, M0, USA). Slides were then transferred into pre-warmed (60 °C) ORO solution (0.5% weight to volume in propylene glycol) and stained for 10 minutes in a 60 °C oven. Subsequently slides were placed into 85% (v/v) propylene glycol for 3 minutes at room temperature and rinsed three times in distilled water, counterstained with Harris’s haematoxylin (VWR International Ltd, Lutterworth, UK) and mounted in Aquatex mounting medium (VWR International Ltd, Lutterworth, UK). Quantification of lipid staining was performed blinded on each slide. On each slide 300 neutrophils, 100 macrophages and 100 squamous cells were evaluated for the presence of cytoplasmic lipid droplets. The data are presented as a percentage of lipid-containing cells.
Urine
Samples were dipsticked routinely for glucose, protein and blood. At the start of the trial and on visits including CT scans, post-menarche female patients underwent urinary pregnancy testing.
Nasal and bronchial brushings
Deoxyribonucleic acid and ribonucleic acid (RNA) were simultaneously isolated from nasal and bronchial brushing samples using the AllPrep DNA/RNA Mini Kit (QIAGEN, Manchester, UK). Levels of pGM169-specific and endogenous CFTR DNA and mRNA were quantified using TaqMan® (Life Technologies, Paisley, UK) qRT-PCR instruments and supplies, following reverse transcription, when appropriate, as described by Rose et al. 95 Absolute DNA and in vitro transcribed RNA calibration standards allowed precise copy number quantification. pGM169 DNA levels were determined using a qPCR primer set specific for the soCFTR2 cDNA:
-
50 nM forward primer: 5′-GGAACAGCTCCAAGTGCAAGA-3′
-
900 nM reverse primer: 5′-CCTGGTGTCCTGCACTTCCT-3′
-
100 nM probe: 5′-FAM-CAAGCCCCAGATTGCTGCCCTG-TAMRA-3′.
Levels were normalised to total genomic DNA, as determined using a qPCR primer set specific for the endogenous CFTR gene:
-
300 nM forward primer: 5′-CTTCCCCCATCTTGGTTGTTC-3′
-
300 nM reverse primer: 5′-TGACAGTTGACAATGAAGATAAAGATGA-3′
-
100 nM probe: 5′-VIC-TGTCCCCATTCCAGCCATTTGTATCCT-TAMRA-3′.
pGM169-derived mRNA was determined using qRT-PCR. Reverse transcription reactions (20 µl) contained ≤ 5 µl total RNA, 1.25 units of MultiScribe™ Reverse Transcriptase (Life Technologies) and 0.4 units RNAse inhibitor (Life Technologies) and 400 nM of the appropriate primer:
-
soCFTR2 mRNA: 5′-CCAGCTGAAGAACAGCTTGCT-3′
-
human CFTR (hCFTR) mRNA: 5′-CCAGGCGCTGTCTGTATCCT-3′.
pGM169-derived cDNA levels were determined using a qPCR primer set specific for the correctly spliced exon 1–2 soCFTR2 mRNA:
-
300 nM forward primer: 5’-TCTCCCTCCTGTGAGTTTGGTT-3′
-
300 nM reverse primer: 5’-GCTCACCACAGAGGCCTTCT-3′
-
100 nM probe: 5’-FAM-CTAGCCACCATGCAGAGAAGCCCTCTG-TAMRA-3′.
Levels were normalised to endogenous hCFTR mRNA as determined using a qPCR primer set specific for the correctly spliced exon 1–2 endogenous CFTR mRNA:
-
300 nM forward primer: 5’-GGAAAAGGCCAGCGTTGTC-3′
-
300 nM reverse primer: 5’-CCAGGCGCTGTCTGTATCCT-3′
-
100 nM probe: 5’-VIC-CCAAACTTTTTTTCAGCTGGACCAGACCAA-TAMRA-3′.
The qPCR reactions (10 µl) contained 0.6–6 ng of total DNA or 2 µl of cDNA and 1 × TaqMan® Universal MasterMix (Life Technologies Manchester, UK). Thermocycling was performed in 384-well plates, on a 7900HT cycler (Applied Biosystems, Thermofisher, Foster City, CA, USA), with SDS 2.2 software, and the following cycling conditions: 50 °C for 2 minutes; 95 °C for 10 minutes; then 40 cycles of 95 °C for 15 seconds and 60 °C for 1 minute. Data were calculated as the percentage of pGM169 specific to endogenous CFTR copy number. When samples were positive for pGM169-specific signal, but either quantification fell below the linear range of the standards (limit of quantification; LOQ) or quantification of endogenous CFTR signal was negative, they were scored as positive but not quantifiable (PBNQ). Remaining samples that were negative for endogenous CFTR signal were scored as not determined. Samples negative for the pGM169-specific signal but positive for endogenous CFTR signal were scored as zero. When data are presented as post–pretreatment difference, a conservative approach was adopted for samples scored with the nominal value of PBNQ by substituting a value of either zero or the LOQ as appropriate to maximise the post–pre difference for placebo samples and minimise the post–pre difference for active samples. For bronchial brushings, two nominally identical samples were typically available (each pools of five independent bronchial brushings) and the maximum score was reported.
Endobronchial biopsies
Two biopsies were collected from each patient. One was frozen in 2-methylbutane-cooled liquid nitrogen at –196 °C and the other was immediately fixed in 10% formal saline. The formalin-fixed samples were processed using a Tissue Tek Sakura VIP processor (Sakura Finetek UK Ltd, Thatcham, UK) and embedded in paraffin wax blocks. Three micron sections were then cut from these blocks for haematoxylin and eosin (H&E) staining. For frozen samples, 6- to 8-µm sections were cut for H&E and ORO staining. For H&E staining, slides were thawed at room temperature for 30 minutes, and then placed in Harris’s haematoxylin for 1 minute. Subsequently, they were differentiated in acid alcohol for 5 seconds and left to blue in tap water for 5 minutes. They were then counterstained with eosin for 30 seconds and cover-slipped using an aqueous mountant.
For ORO staining, frozen sections were thawed and air dried at room temperature for 30 minutes. Slides were rinsed in 60% 2-propanol then placed in filtered ORO working solution for 15 minutes. Subsequently, the slides were taken out of the ORO solution and excess dye was washed off with 60% 2-propanol, before rinsing in three changes of distilled water. The sections were counterstained using Harris’s haematoxylin for 30 seconds in order to visualise the nuclei. The sections were left to blue in running tap water for 5 minutes and mounted with a coverslip using Aquatex (an aqueous mountant). H&E slides were scored independently by two blinded pathologists using a semiquantitative scoring system for goblet cell hyperplasia, basement membrane thickening, presence of chronic inflammatory cells (lymphocytes and plasma cells), neutrophils, eosinophils and seromucinous gland hyperplasia. ORO slides were scored for the presence of lipid-laden macrophages using a semiquantitative scoring system.
Data collection and analysis
Trial database, monitoring and audit
The Imperial College Trials Unit (ICTU) built an InForm database specifically for the trial and the staff working on the trial entered the data from source. The InForm database was monitored by both ICTU on a regular basis and the Imperial College Research Office undertook on-site source data verification. Two patients had 100% source data verification and the remainder had 10%. Any queries were flagged on the database as well as in the case report forms and dealt with by the study team.
Data analysis
A detailed statistical analysis plan (SAP) was drafted by the trial statistician (GDM) and was further refined with input from the Trial Management Group (see Appendix 5). The SAP was approved by the Trial Steering Committee (TSC) and finalised ahead of the database being locked and the trial unblinded. Data management was undertaken using Microsoft Excel® (version 14.4.6 for Mac OSX; Microsoft Corporation, Redmond, WA, USA). Descriptive statistics and standard analyses were performed using Prism (Version 5.0c for Mac OSX, Graph Pad Software Inc., San Diego, CA, USA) and or IBM SPSS (version 22.0, IBM Corporation, Armonk, NY, USA). There was no exploration of the impact of missing values in the primary analysis as data were available for 114 out of the 116 PP patients.
Patient and public involvement
Since 2000, when the UK CFGTC started working together, we have had a close relationship with patients and their families, the ethos of the consortium being to take the patients with us along the gene therapy pathway. As well as having a formal patient representative on the TSC and input from the National Institute for Health Research (NIHR) Respiratory Biomedical Research Unit patient group, the UK CFGTC has attended numerous patient and family meetings to discuss the trial programme as well as future research projects. Patient input enabled us to refine the trial design in a number of ways, including the length of time for which a nebuliser may be tolerated and which side effects in an eventual clinical product would be tolerated.
Owing to the length of the study, one of the biggest challenges we had during the trial was patient recruitment. Being so involved with the patient groups enabled us to take advantage of social media and local networks across the UK ensuring that when we opened the patient identification centres patients were already aware of the study and wanted to participate. Importantly, having patients involved at every stage of the research process enabled us to design a product that would be tolerated by the patients if it becomes a licensable product.
In 2013 the patient and public involvement for this project was written up as an INVOLVE case study. 96
Role of the funder
This project was supported by the Efficacy and Mechanism Evaluation (EME) programme, a Medical Research Council and NIHR partnership. The EME programme covered the costs specifically relating to the clinical trial, including staff, patient visits, assays and data analysis and management. In addition funding was received from Medicor Foundation and Cystic Fibrosis Trust to cover the production costs of pGM169/GL67A. The NIHR Clinical Research Network, initially through the Medicines for Children Network and later the North West London Local Clinical Research Network, provided funding for a paediatric nurse. Further funding was provided by Just Gene Therapy.
Chapter 3 Results of clinical efficacy outcomes
Results are reported according to Consolidated Standards of Reporting Trials (CONSORT) guidelines.
Overall timelines
The first patient was screened on 6 June 2012 and the first dose was administered on 13 June 2012. The last patient was screened on 24 June 2013, the last dose was administered on 1 May 2014 and the last follow-up visit was performed on 30 May 2014.
Screening and recruitment
Following prescreening of clinic databases, 191 patients were considered likely to be suitable and agreed to a screening visit. Forty of these were not enrolled, as they failed to fulfil inclusion criteria (see Figure 15). Of the 151 who passed screening, 11 patients subsequently withdrew, either because of a change of mind or because of the development of an exclusion criterion. One hundred and forty patients were randomised: 62 (44%) to placebo and 78 (56%) to active treatment. Two patients in each group withdrew post randomisation and were not able to reattend for any further visits; therefore, this left 136 in the ITT cohort, predefined as being randomised and having any follow-up data available. The PP population was predefined as those patients receiving ≥ 9 doses and comprised 116 patients (placebo, n = 54; active treatment, n = 62). The reasons for 20 patients discontinuing are shown in Figure 15. Of note, the first small-molecule CFTR modulator, ivacaftor, was licensed and approved in UK countries during the trial for patients with the G551D mutation. Three of our subjects elected to leave the trial to enable them to receive this treatment; others were prepared to wait until the end of the trial and continued.
FIGURE 15.
The CONSORT diagram showing screening, randomisation and patients dosed. The ITT and PP groups are identified.
Subject demographics
Subjects randomised to active-treatment and placebo groups were well matched at baseline for age (and proportion of paediatric patients aged < 18 years), sex, centre and CFTR gene mutation class (F508del/F508del vs. other) (Table 2). The two groups were also similar with regard to clinical characteristics including lung function (FEV1%) and BMI, used here as a general measure of nutritional status.
| Characteristic | ITT placebo (n = 60) | ITT active treatment (n = 76) | ITT total (n = 136) | PP placebo (n = 54) | PP active treatment (n = 62) | PP total (n = 116) |
|---|---|---|---|---|---|---|
| Age (years) | ||||||
| Mean | 25.9 | 25.0 | 25.4 | 26.0 | 23.6 | 24.7 |
| Range | 12–64 | 12–57 | 12–64 | 12–64 | 12–57 | 12–64 |
| Age distribution, n (%) | ||||||
| < 18 years | 19 (32) | 25 (33) | 44 (32) | 17 (31) | 23 (37) | 40 (34) |
| ≥ 18 years | 41 (68) | 51 (67) | 92 (68) | 37 (69) | 39 (63) | 76 (66) |
| Sex, n (%) | ||||||
| Female | 29 (48) | 37 (49) | 66 (49) | 25 (46) | 31 (50) | 56 (48) |
| Male | 31 (52) | 39 (51) | 70 (51) | 29 (54) | 31 (50) | 60 (52) |
| Centre distribution, n (%) | ||||||
| Edinburgh | 28 (47) | 27 (36) | 55 (40) | 24 (44) | 22 (36) | 46 (40) |
| London | 32 (53) | 49 (64) | 81 (60) | 30 (56) | 40 (64) | 70 (60) |
| Height (cm) | ||||||
| Mean | 164.5 | 164.3 | 164.4 | 165.0 | 163.6 | 164.3 |
| Range | 146.6–191.3 | 133.8–186.2 | 133.8–191.3 | 147.4–191.3 | 133.8–186.2 | 133.8–191.3 |
| Weight (kg) | ||||||
| Mean | 60.9 | 61.1 | 61.0 | 61.6 | 61.0 | 61.3 |
| Range | 37.0–120.7 | 33.6–105.4 | 33.6–120.7 | 37–120.7 | 33.6–105.4 | 33.6–120.7 |
| Percentage predicted FEV1 | ||||||
| Mean | 67.9 | 69.2 | 68.6 | 69.0 | 69.9 | 69.5 |
| Range | 48.4–89.9 | 50.3–89.6 | 48.4–89.9 | 49.6–89.9 | 50.7–89.6 | 49.6–89.9 |
| BMI | ||||||
| Mean | 22.3 | 22.3 | 22.3 | 22.4 | 22.4 | 22.4 |
| Range | 16.2–41.0 | 16.7–40.7 | 16.2–41.0 | 16.2–41.0 | 16.7–40.7 | 16.2–41.0 |
| Mutation class | ||||||
| F508del/F508del | 29 | 37 | 66 | 26 | 31 | 57 |
| F508del/class 1–6 | 23 | 28 | 51 | 22 | 23 | 45 |
| Not F508del/class 1 | 2 | 5 | 7 | 1 | 3 | 4 |
| Heterozygous/homozygous class 3–6 | 3 | 2 | 5 | 2 | 2 | 4 |
| F508del/unknown class | 3 | 4 | 7 | 3 | 3 | 6 |
Primary outcome: relative change in FEV1 percentage
Out of 116 PP subjects, 114 (placebo, n = 54; active treatment, n = 60) had paired pre–post-treatment measurements of percentage predicted FEV1. Two patients, despite fulfilling the PP definition of receiving ≥ 9 doses, were excluded from this analysis as they did not have spirometry performed at follow-up visits; in one case, this test was contraindicated owing to a recent surgically induced pneumothorax. The other patient had recently withdrawn from the trial because of time commitments and was unable to return for follow-up measurements. There was a significant (p = 0.046; Figure 16) TE in the primary outcome, percentage predicted FEV1, with the active-treatment group having an ANCOVA-adjusted 3.7% (95% CI 0.1% to 7.3%) greater change in FEV1 than the placebo group at 12 months’ follow-up. The effect was seen from month 1 onwards, with a sustained divergence of the two groups (see Figure 16).
FIGURE 16.
Primary end point (relative change in percentage predicted FEV1). Time course of the response of the primary outcome to either placebo (green) or active treatment (black). ‘Pre’ and ‘post’ indicate the mean of two measurements at the respective time points. Error bars indicate SEM. There was a significant (p = 0.046) TE, with the active-treatment group having an ANCOVA-adjusted improvement of 3.7% (95% CI 0.1% to 7.3%) than placebo at 12 months’ follow-up. The effect was seen from month 1 onwards, with a sustained divergence of the two groups. SEM, standard error of the mean.

Changes in FEV1 over the course of the trial were variable between subjects. Figure 17 illustrates individual relative improvements from pretreatment to follow-up in a waterfall plot in which improvement is indicated by a positive value. Post hoc analysis showed that 21 subjects [placebo, n = 6 (11%); active treatment, n = 15 (25%)] demonstrated a change in percentage predicted FEV1 of ≥ 5%.
FIGURE 17.
The distribution of FEV1 changes in individual patients, shown separately for the two groups. (a) Active treatment; and (b) placebo. Positive values indicate an improvement.

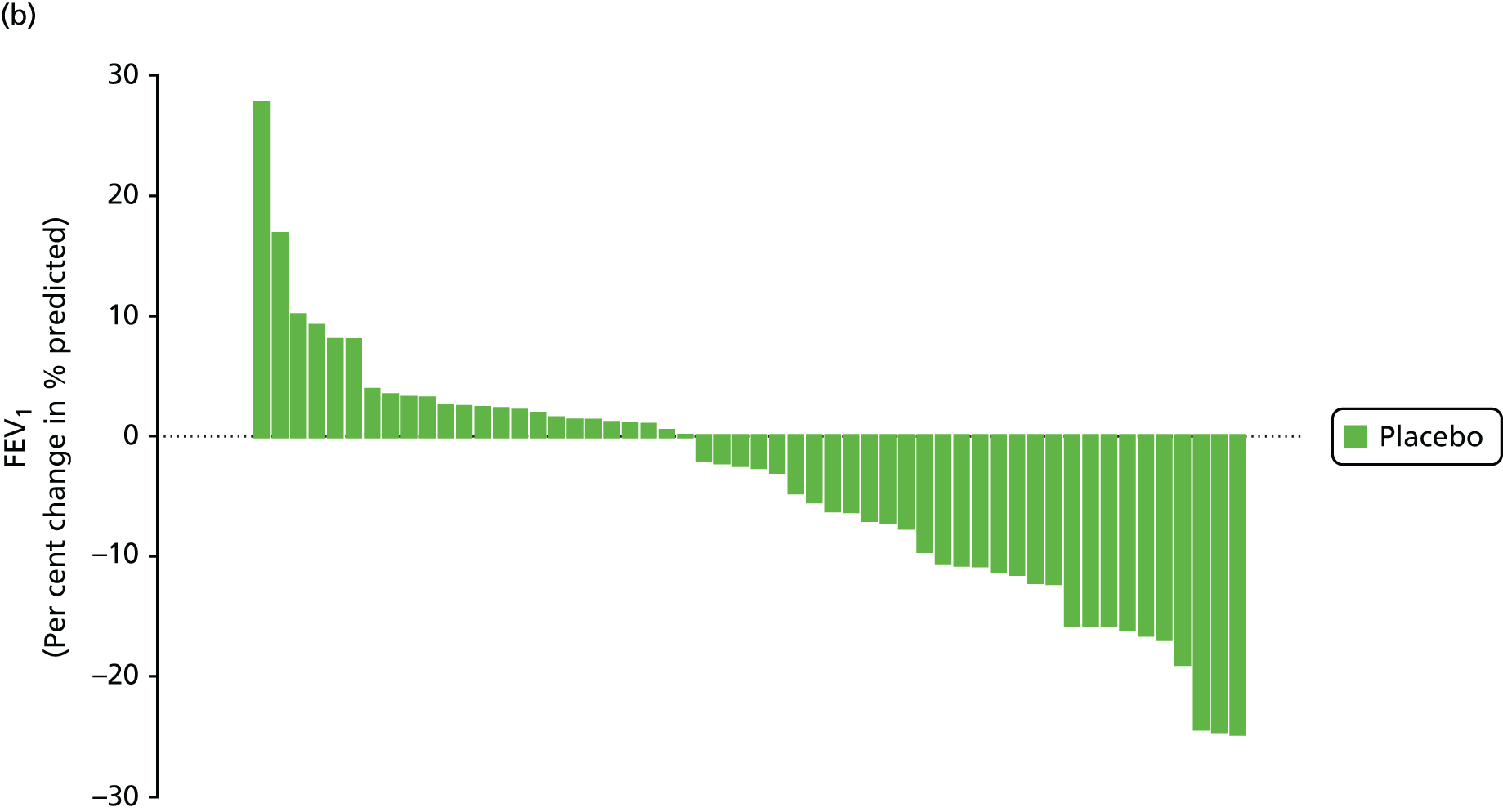
The TE in the ITT population with spirometry measurements both pre-dosing and within the protocol-defined window after their final dose (placebo, n = 56; active treatment, n = 65) was 3.6% (95% CI 0.2% to 7.0%; p = 0.039). The SAP prespecified an additional sensitivity analysis based on the area under the curve for the percentage predicted FEV1. With this analysis, using the PP population, the estimated TE (in units of percentage predicted FEV1, rather than the relative change) was 1.32% (95% CI –0.48% to 3.12%; p = 0.15), consistent with the relative TE observed in the primary analysis.
Major secondary outcomes
Physiology
There was also a significant TE in FVC (p = 0.031; Figure 18; see also Figures 25 and 26). No difference was observed in the LCI at the follow-up period, although the serial time point graph (Figure 19) demonstrates that the active-treatment group appear to have a better-preserved LCI in the earlier stages of the trial.
FIGURE 18.
Forced vital capacity. Time course of the response of FVC to either placebo or active treatment. ‘Pre’ and ‘post’ indicate the mean of two measurements at the respective time points. Error bars indicate SEM. There was a significant (p = 0.031) TE, with the active group having an ANCOVA-adjusted improvement of 3.0% (95% CI 0.3% to 5.8%) compared with placebo at 12 months’ follow-up. SEM, standard error of mean.
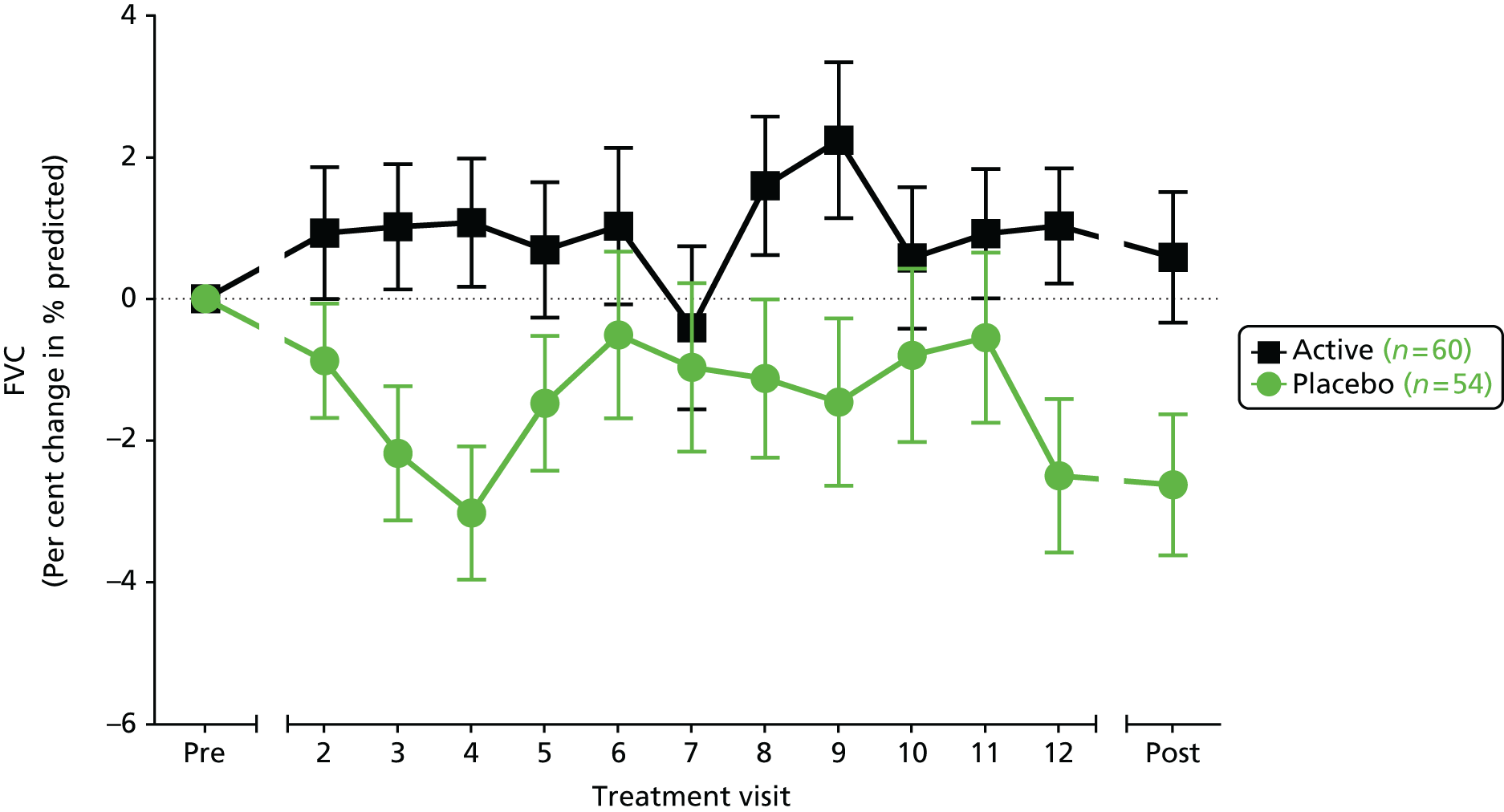
FIGURE 19.
Change in LCI. There was no significant TE in LCI, both groups increasing (worsening) slightly over the year of the trial.
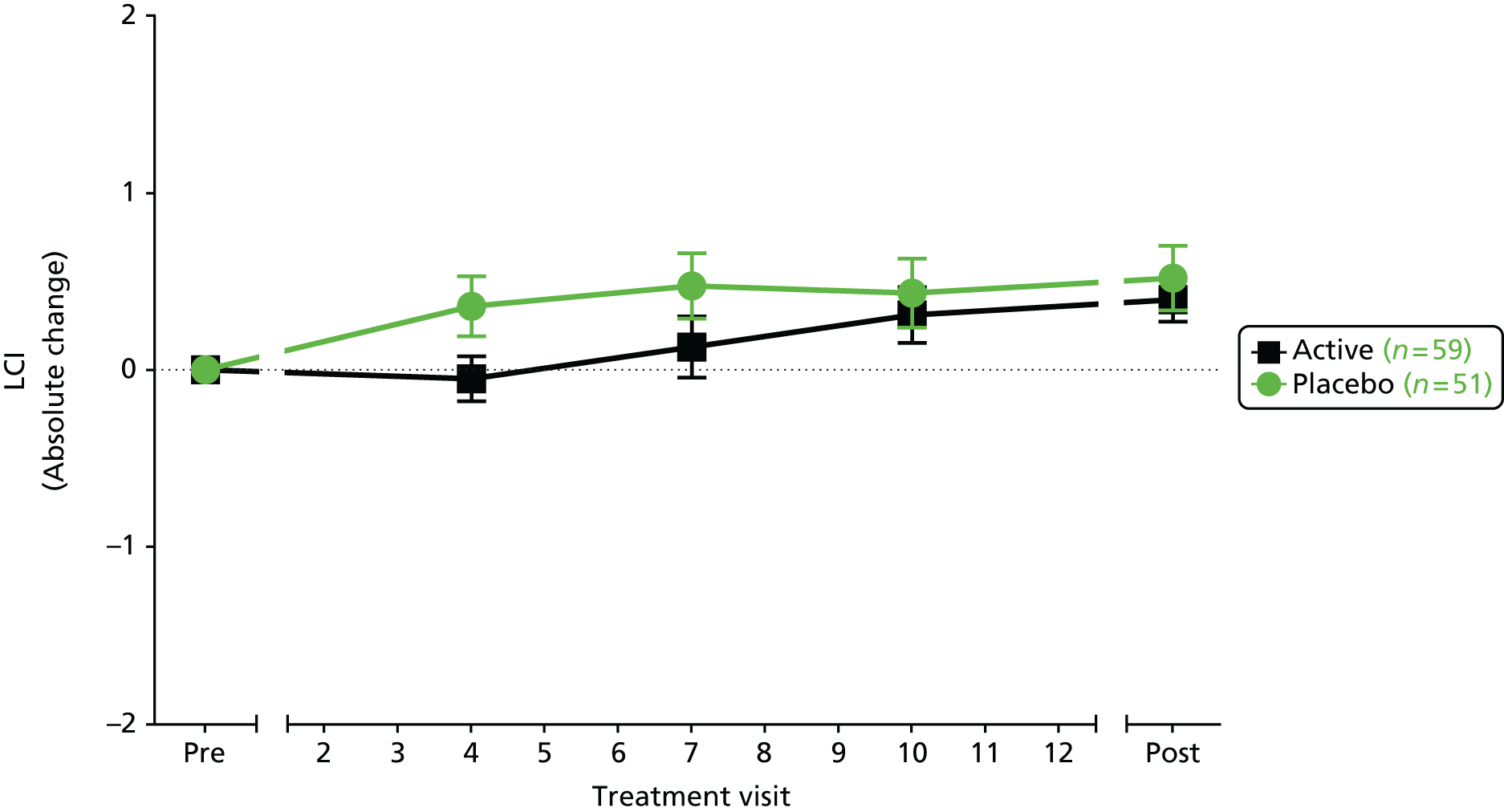
Computed tomography scans
Paired pre and post follow-up high-resolution CT scans were available for 115 patients; the one PP patient missing is the same patient also missing from the analysis of the primary end point as she had been lost to follow-up. The other patient missing from the primary outcome is represented here, as CT scanning did not pose the same risks following her pneumothorax as a forced expiratory manoeuvre would have done.
Bronchiectasis is defined as dilatation and thickening of the airways. It is considered irreversible. An intervention applied for this period of time would not therefore be expected to have any impact on the extent of bronchiectasis and this was the case in this cohort (Figure 20). Although it was not statistically significant, it was interesting to note a trend for less worsening in the severity of bronchiectasis in the active-treatment group than in the placebo.
FIGURE 20.
Change in CT scores for bronchiectasis (a) extent; and (b) severity.
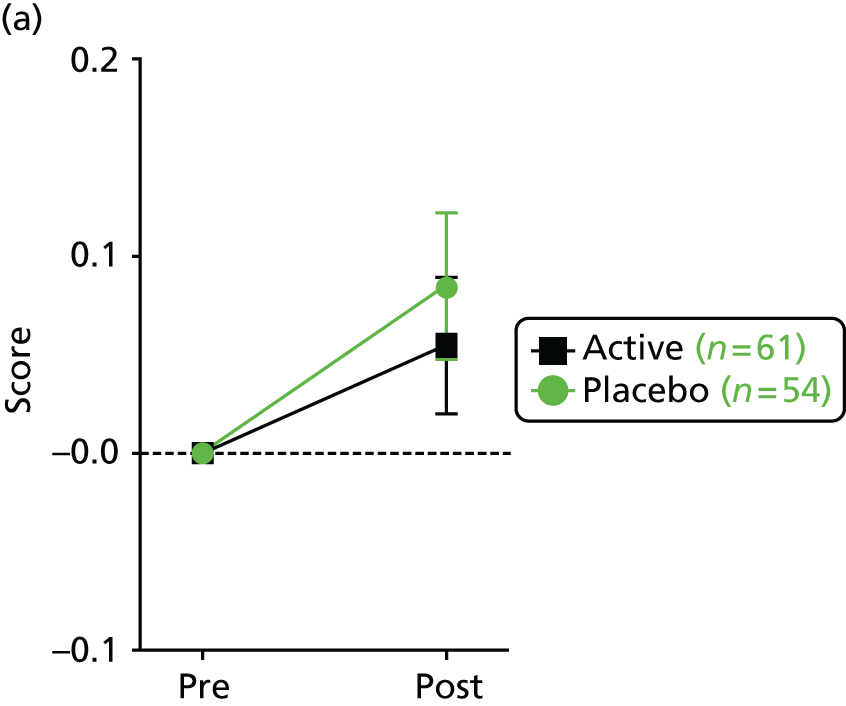

Gas trapping is a feature visible on expiratory films when areas of lung fail to empty properly because of airway obstruction. There were increases in the percentage of gas trapping in the placebo group that were not demonstrated in the active-treatment group and which led to a significant TE (p = 0.048; Figure 21).
FIGURE 21.
Gas trapping on CT scan. Over the course of the study, gas trapping indicative of small airways disease, increased in the placebo group (worsened) but did not in the active-treatment group leading to a statistically significant TE (p = 0.048).

Scores of mucus plugging (large or small; Figure 22) and airway wall thickness (Figure 23) did not differ significantly between the two groups, although, for every parameter, the change was smaller in and, therefore, favoured the active-treatment group.
FIGURE 22.
Mucus plugging on CT scan. (a) Large mucus plugging; and (b) small mucus plugging (sometimes termed ‘tree in bud’).

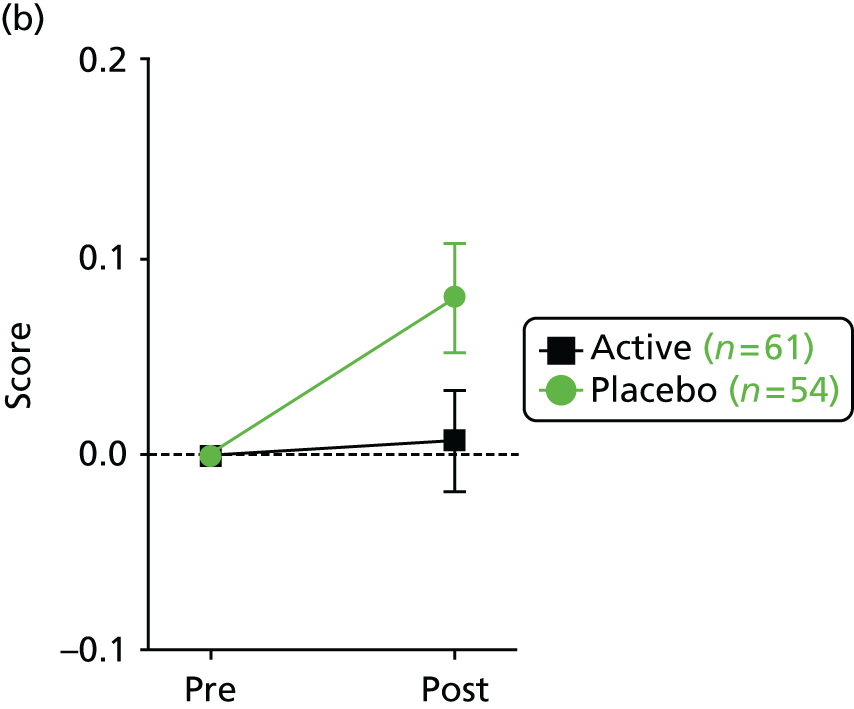
FIGURE 23.
Airway wall thickness on CT scan.

Quality-of-life scores
Baseline quality-of-life scores for the domains of major interest in a trial of respiratory treatment (respiratory and physical) were high for the group as a whole (median 87.5%). There were no statistically significant TEs at the end of the trial, although, again, the TE appeared to favour the active-treatment group (Figure 24).
FIGURE 24.
Quality-of-life (QoL) scores. Change in the (a) respiratory and (b) physical domains on the validated CF quality-of-life questionnaire CFQ-R.
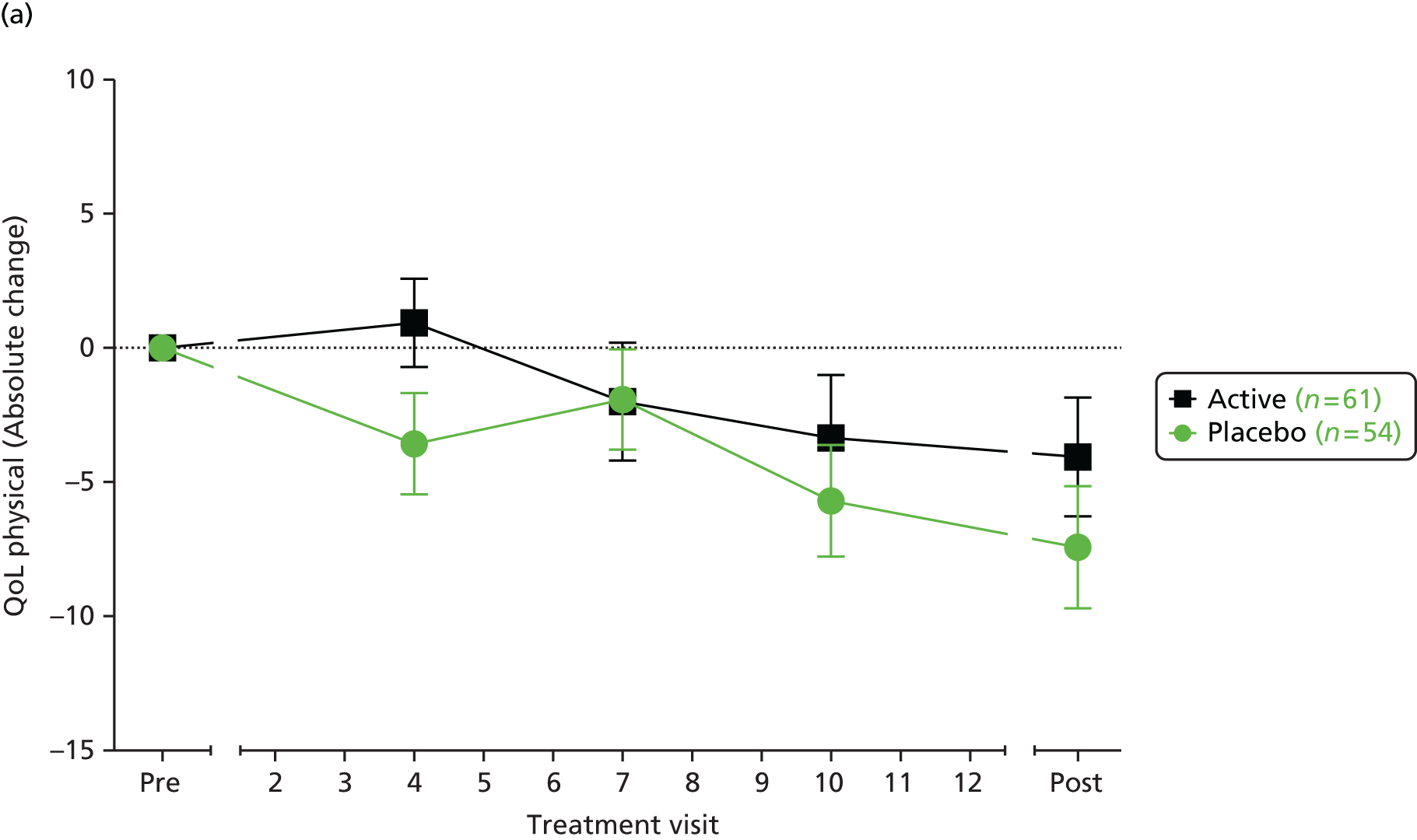

Other secondary outcomes
Figures 25 and 26 show that for all the assays, even those which did not reach significance, the standardised TE favoured the active-treatment group.
FIGURE 25.
Secondary outcome measures. Forest plot showing the responses of secondary outcome measures to placebo or active treatment. To allow results from different end points to be plotted on a common scale, the estimated TEs were standardised to be presented as multiples of the underlying SD (standardised TE). The size of the circle is proportional to the number of subjects represented and the bars indicate 95% CI. *, statistically significant; ESR, erythrocyte sedimentation rate; WBC, white blood cell.
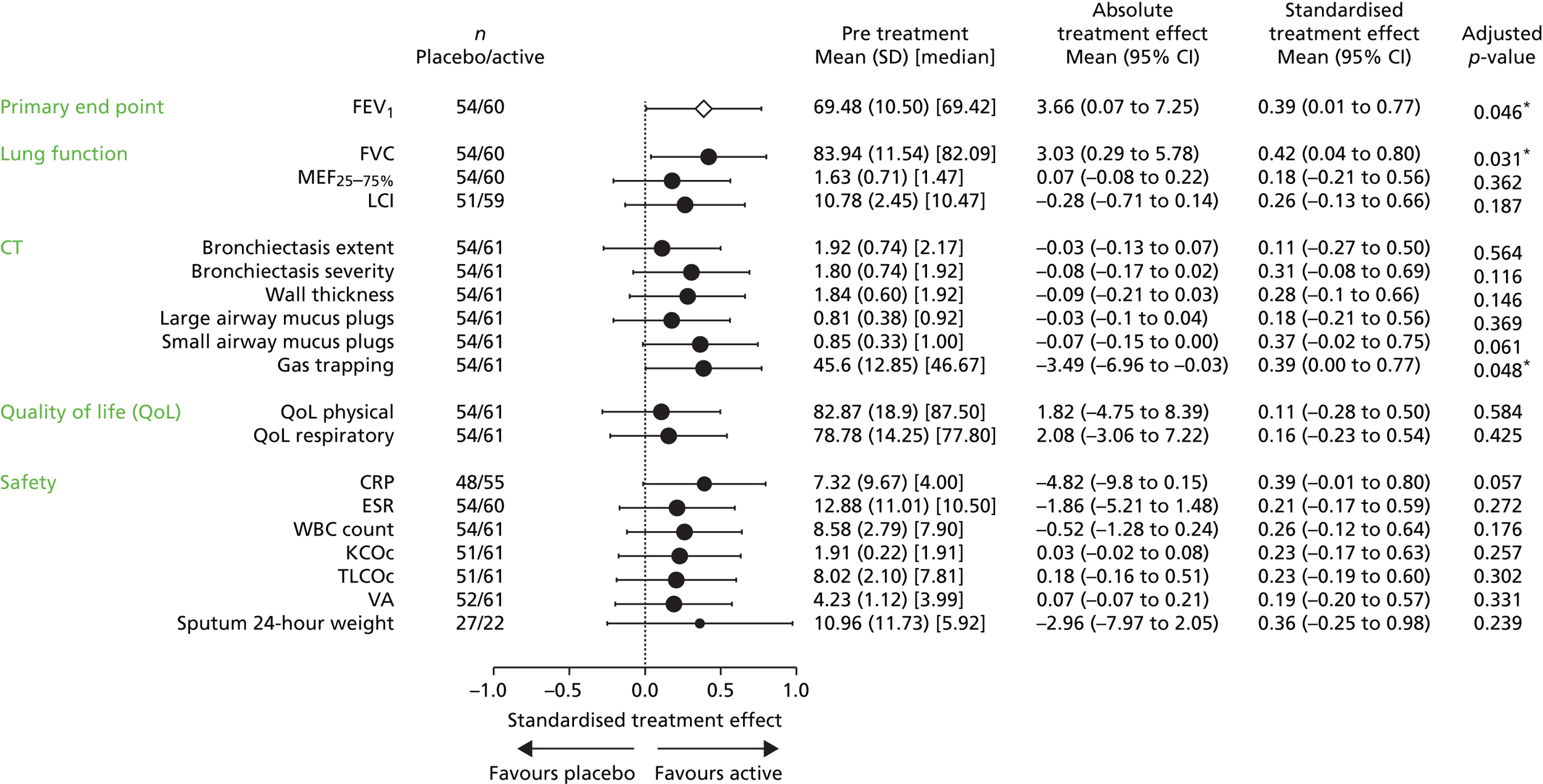
FIGURE 26.
Responses of active and placebo groups separately for secondary outcomes. Forest plot showing the responses of the placebo or active-treatment arms when assessed by the predefined subgroup values shown. To allow results from different endpoints to be plotted on a common scale, the estimated TEs were standardised to be presented as multiples of the underlying SD. The size of the circle is proportional to the number of subjects represented and the bars indicate 95% CI. *, statistically significant; Con meds, concurrent medication; MP, mucus plugs; Pa, P. aeruginosa; TA AE, treatment-associated AE.

Influence of baseline parameters on primary outcome
We had predefined that the influence of baseline parameters on TE of the primary outcome would be explored in an attempt to define a ‘responder’ group. We reasoned that differences between responders and non-responders could be based on any of the following:
-
Severity of underlying lung disease: patients with milder disease could respond more because the inhaled medication reached further into the airways, or less because they were already so well. We examined this using FEV1, LCI and CT parameters.
-
Age.
-
Sex.
-
CFTR mutation class: although we expect gene transfer to be mutation independent, this seemed necessary to confirm.
-
The presence or absence of P. aeruginosa: gene transfer could be affected by the presence of this organism either because of the greater levels of inflammation likely associated with this chronic infection or because of an interaction with the wide array of exoproducts it produces. We did not consider that we had sufficient power to break this down further on the basis of other infections.
-
Concomitant medications. Those considered to be particularly important to examine were:
-
the anti-inflammatory agents, azithromycin and corticosteroids, either of which could affect the host response to the medication
-
DNase, which could degrade the DNA in the trial product (of note, patients receiving this drug were asked to withhold it for 24 hours before and after each dose)
-
hypertonic saline, which by influencing mucus clearance could potentially reduce contact time.
-
-
Side effects caused by the trial medication: we considered the hypotheses that those patients who developed either lower respiratory or systemic side effects acutely after dosing could have either a higher (side effects resulting from successful delivery) or smaller (host response limited success of transfection) chance of being a responder. To examine this, we classified patients on the basis of having (1) lower respiratory or (2) flu-like reactions within 48 hours of at least four doses. There were no subjects in the latter group and, therefore, only the lower airway symptoms were examined in this analysis.
For each of the continuous variables, the whole PP group was divided based on median values for that measure. The other variables were divided into two dichotomous groups.
Figure 27 illustrates that TE was independent of age, sex, mutation class, concomitant treatments and response to dosing. However, stratification by baseline disease severity on the basis of percentage predicted FEV1 suggested a difference in TE between the lower half (49.6–69.2% predicted FEV1) who had a TE of 6.4% (95% CI 0.8% to 12.1%) and the upper half (69.6–89.9% predicted) of TE 0.2% (95% CI –4.6% to 4.9%) (interaction analysis p = 0.065).
FIGURE 27.
Stratification of primary outcome measure. Forest plot showing stratification of the primary outcome response by prespecified variables. To allow results from different end points to be plotted on a common scale, the estimated TEs were standardised to be presented as multiples of the underlying SD (standardised TE). The size of the circle is proportional to the number of subjects represented and the bars indicate 95% CI. For all CT values and LCI, lower numbers indicate less severe disease. Con Meds, Concurrent Medication; MP, mucus plugs; Pa, P. aeruginosa; TA AE, treatment-associated AE.

This is further illustrated in Figure 28, which shows the evolution of FEV1 over time during the trial in (a) the half of patients with more severe disease and (b) the half with milder disease. The greater TE was not because of the use of ‘relative change’ magnifying an effect, as this was also apparent when absolute change in FEV1 percentage was examined (data not shown).
FIGURE 28.
Primary outcome stratified by baseline FEV1. Time course of the primary outcome response stratified by baseline FEV1 at trial entry. (a) Severe group, baseline FEV1 49.6–69.2% predicted; and (b) milder group, baseline FEV1 69.6–89.9% predicted. Error bars indicate SEM. SEM, standard error of the mean.
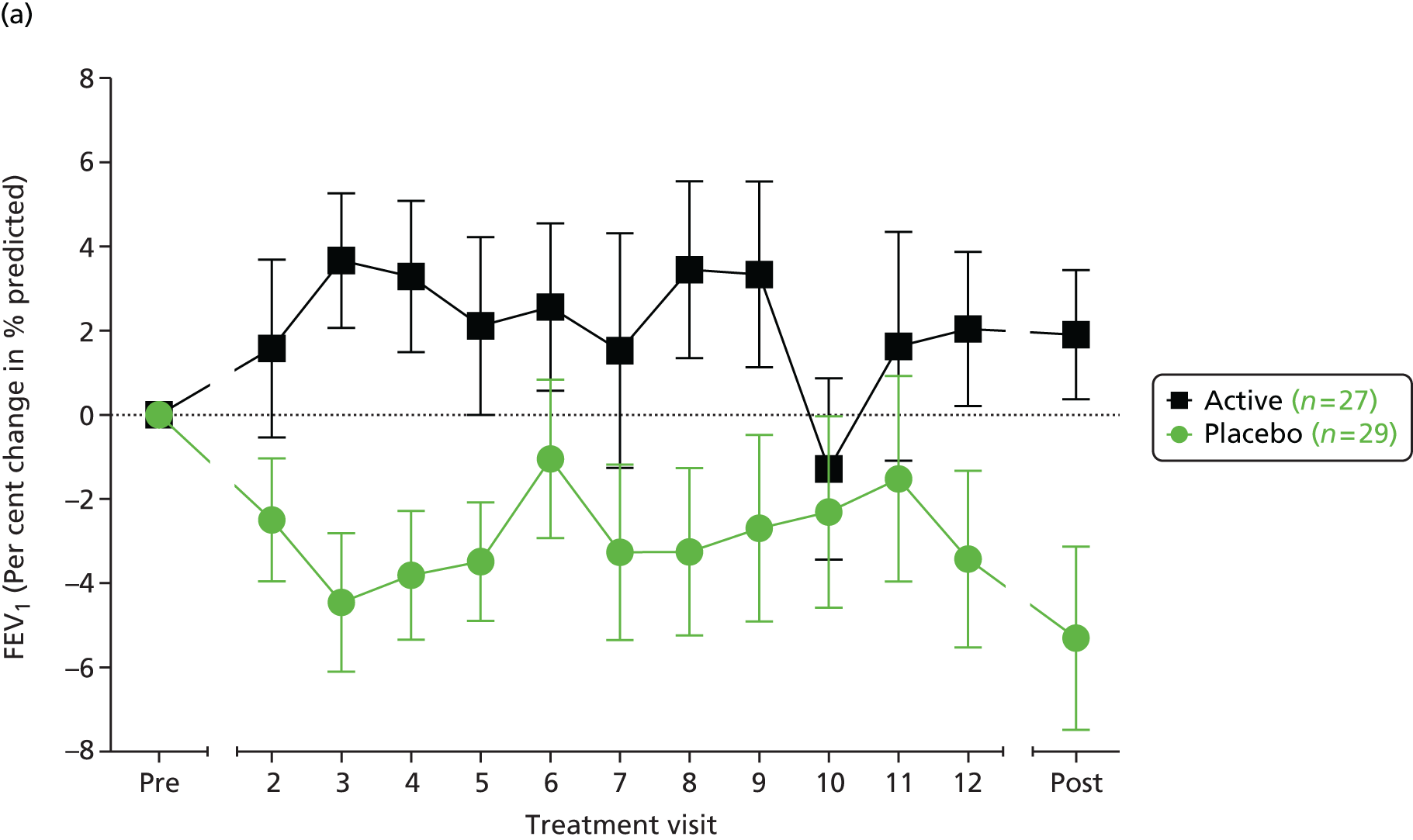
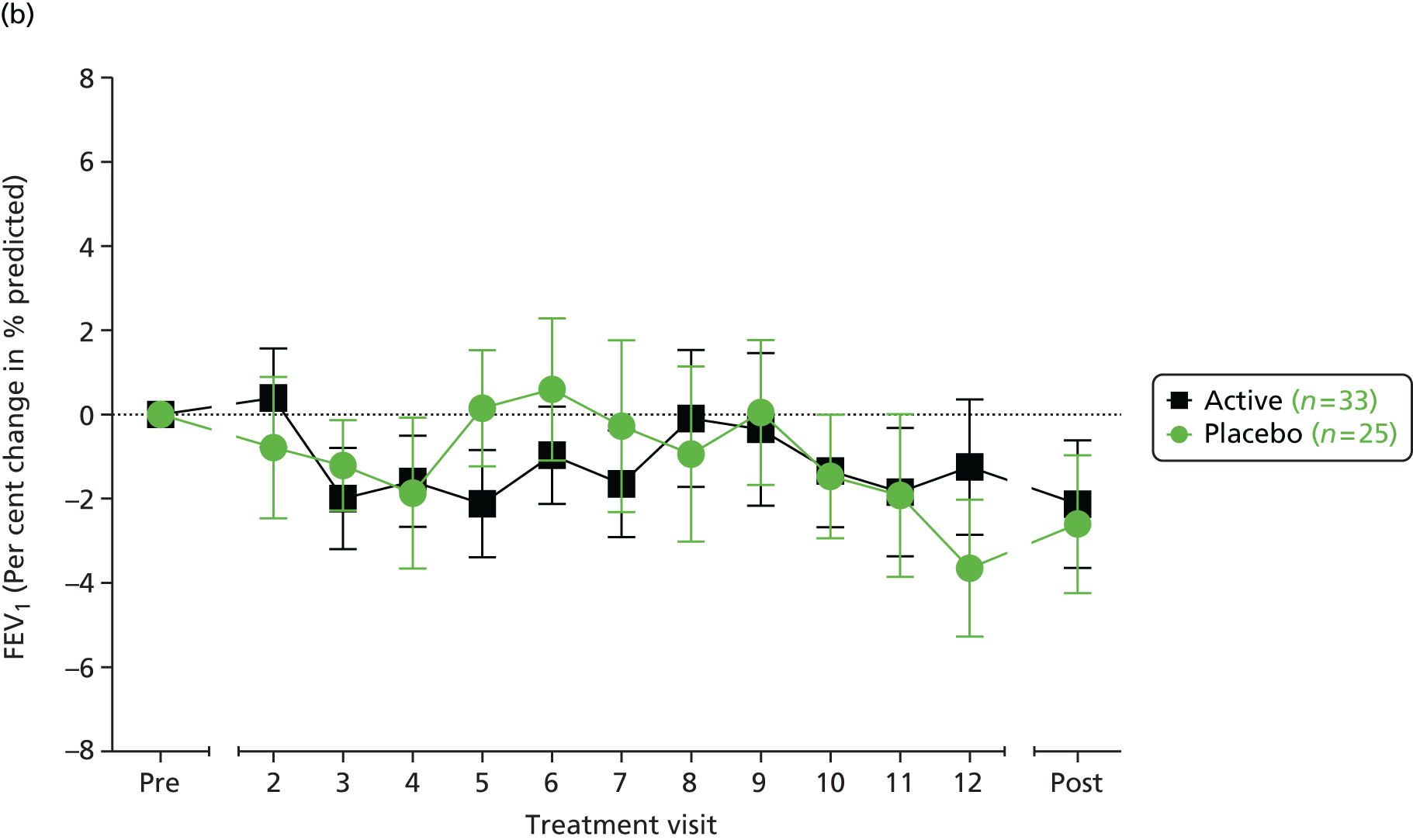
Influence of baseline FEV1 on other outcomes
Having observed that baseline FEV1 appeared to have an important influence in the magnitude of the TE based on the primary outcome, we next examined other outcome measures for this effect. Subjects were divided into upper and lower halves based on the median value for the entire group, as previously, and TEs were compared. Figure 29 shows that many of the assays mirrored the effect which had been seen for the primary outcome, namely the more severe group of patients showing an approximate doubling of the TE compared with the values in the whole, unstratified group. This resulted in an absolute improvement (vs. stabilisation) in many of the assays. Of note, in those with less severe FEV1 at trial entry, biomarkers associated with smaller airways, in particular LCI, still showed a TE favouring the active-treatment group.
FIGURE 29.
Stratification of secondary outcomes. Forest plot showing stratification of secondary outcomes by the severity of baseline FEV1 at trial entry. To allow results from different end points to be plotted on a common scale, the estimated TEs were standardised to be presented as multiples of the underlying SD (standardised TE). The size of the circle is proportional to the number of subjects represented and the bars indicate 95% CI. ESR, erythrocyte sedimentation rate; WBC, white blood cell.
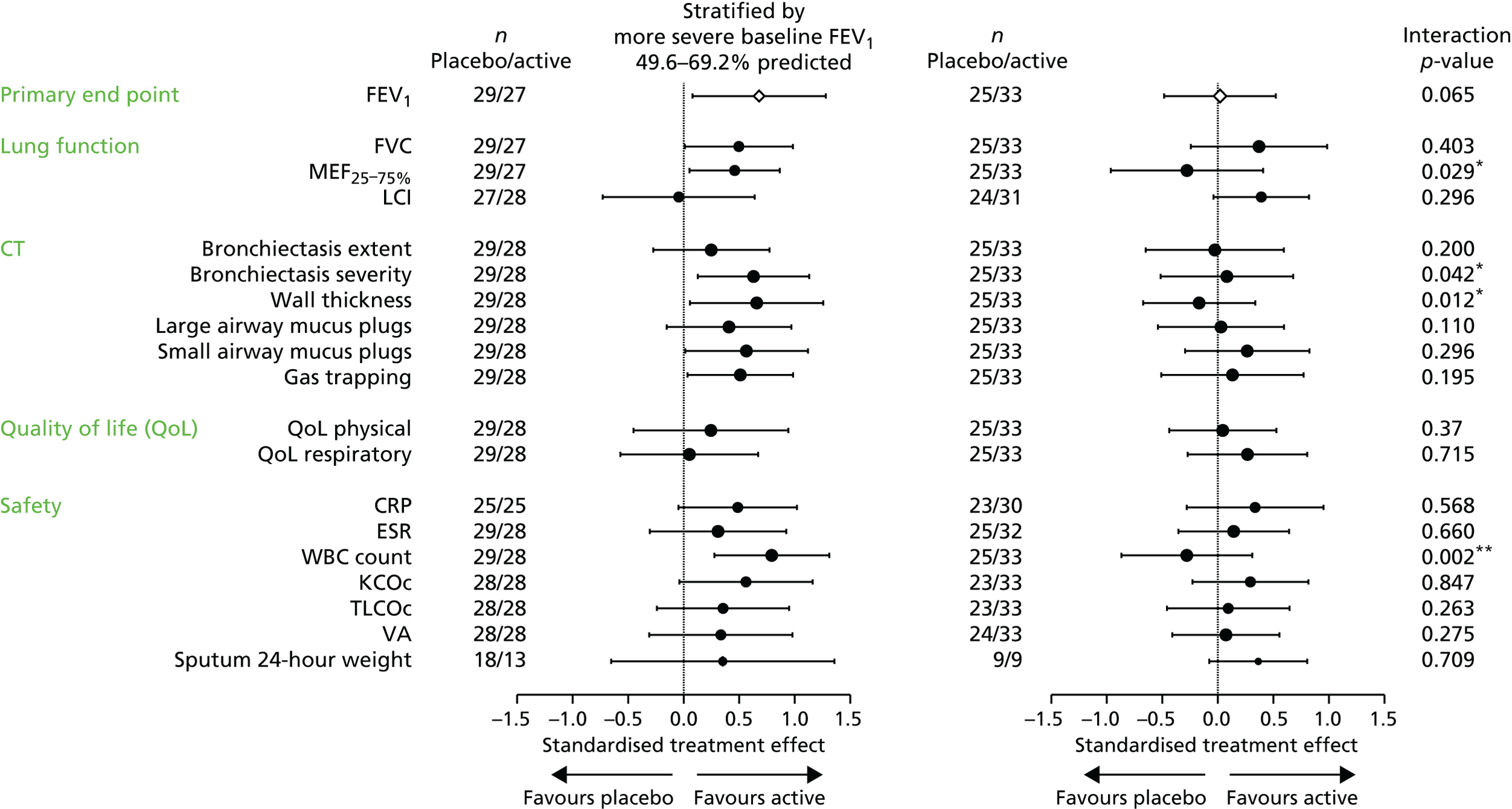
We confirmed that these benefits seen in the more severe subgroup were not related to an increased number of antibiotic courses during the trial. Both active-treatment and placebo groups received a median of three courses of oral or intravenous antibiotics. In the half with more severe disease, stratified by FEV1, both placebo and active-treatment groups received three courses, while among those with less severe disease, the placebo group received three courses and the active-treatment group received two. Thus, the observed TEs were independent of concurrent antibiotic courses.
Summary of efficacy outcomes
The trial achieved its primary outcome confirming a statistically significant TE favouring the active-treatment group on relative change in FEV1. This was underscored by statistically significant TEs in the secondary outcomes, FVC and gas trapping on CT. Furthermore, for all other secondary outcomes, although not statistically significant, the standardised TE analysis favoured active treatment. In general, these improvements were seen across the group and were independent of sex, age and mutation class. In contrast, the magnitude of TE was influenced by the severity of baseline lung disease assessed by FEV1: patients in the more severe half at baseline experienced larger improvements not only in FEV1 but also in several other outcomes. Changes in the small airway measure, LCI, were still seen in patients with less severe baseline FEV1, suggesting that these patients can still benefit.
Chapter 4 Results of mechanistic substudy
Transgene-specific DNA and mRNA
Nasal
In the nasal arm of the substudy, the assay could quantify a dosing-dependent increase in vector-specific DNA in 15 out of 15 active-dose subjects post treatment, although in four of these modest levels of DNA were detected prior to dosing. In the placebo patients, DNA could also be detected in samples from three out of four subjects post dosing (none prior to dosing); no vector-specific mRNA was quantifiable in either group.
Lower airway
In the bronchial arm of the substudy, the assay quantified vector-specific DNA in 10 out of 10 active-treatment patients and zero out of seven placebo subjects post dosing (Figure 30); no vector-specific mRNA was quantifiable in either group.
FIGURE 30.
Assessment of DNA from bronchial brushings in the placebo (n = 7) and active-treatment (n = 10) subgroups. Each circle represents an individual patient.
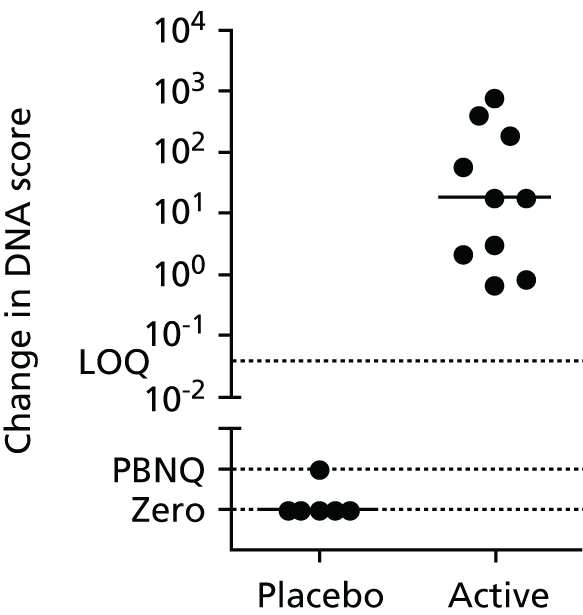
Potential difference measurements
Nasal
The nasal arm comprised 24 patients in the ITT group, 20 of whom were in the PP. One patient had a mean total chloride secretory response ≥ 5 mV pre-dosing and was, therefore, excluded as prespecified in the protocol. Day 28 post-treatment recordings for two active-treatment patients had to be delayed beyond a window of > 7 days of the prespecified interval and were therefore excluded; both patients had usable values at the day 14 post treatment time point. Overall, 75 out of 106 (70.8%) of zero chloride, and 70 out of 106 (66.0%) of isoproterenol recordings were interpretable.
There were no significant changes in baseline values or amiloride responses in either the placebo or active-treatment groups. No significant changes in either the zero chloride or isoproterenol responses were seen. For the zero chloride component 10 out of 14 active-treatment patients and three out of six placebo patients showed net secretion (i.e. a more negative value post treatment than pretreatment); 4 out of 14 active-treatment patients showed mean pre–post-treatment responses (ranging from –3.4 mV to –7.0 mV), which were more negative than the largest placebo response. For the isoproterenol component, 11 out of 12 active-treatment patients and three out of six placebo patients showed net secretion (Figure 31).
FIGURE 31.
The response of the nasal epithelium to perfusion with (a) a zero chloride solution; and (b) a zero chloride solution containing isoproterenol (10 µM). Each symbol indicates the change in this response from trial start to finish for the relevant treatment in an individual patient. The plotted value is the most negative value obtained from all interpretable recordings (range 1–3) at each time point for that patient. A more negative value is in the non-CF direction. The placebo group had a median pre–post trial change of + 0.1 mV (range +1.1 to –2.3 mV) and the active-treatment group –0.6 mV (range +3.5 to –7.0 mV) (p = 0.509). The response of the nasal epithelium to perfusion with a zero chloride solution containing isoproterenol (10 µM). The placebo group showed a median pre–post trial change of +0.3 mV (range +2.9 to –2.6 mV) and the active-treatment group –1.0 mV (range +0.1 to –3.8 mV) (p = 0.424).
Bronchial
In the bronchial subgroup, one active-treatment patient withdrew after six doses, but consented to post-dosing bronchoscopy within the prespecified timing (28 ± 5 days) after the last dose; values for this patient are included in the analysis. Post-dosing bronchoscopy was performed on two placebo patients 58 and 62 days after their last doses, and on two active-treatment patients 49 and 111 days after their last dose. The data for the two placebo patients are included in the final analysis, on the basis that the natural history of bronchial electrophysiology is unlikely to be influenced by a dose of saline approximately 60 days previously. Values for the two active-treatment patients were omitted from the final analysis given the monthly dosing schedule in the trial based on previously generated gene expression data, although their samples were included in the safety analysis. Overall, 66 out of 102 (64.7%) recordings fulfilled the acceptability criteria.
There were no significant changes in basal values in either the placebo or active-treatment groups. Figure 32 shows bronchial chloride responses using the mean of all available interpretable tracings for each patient; a negative value indicates a non-CF direction. The placebo group (n = 7) had a median pre–post trial change of +3.1 mV (range +9.3 to –1.2 mV) and the active-treatment group (n = 10) –1.3 mV (range +4.0 to –5.8 mV) (p = 0.032). Five out of 10 active-treatment patients had values more negative than the largest placebo response. Figure 32b shows the same analysis with only the most negative value recorded for each patient at any time point. The placebo group showed a median pre–post trial change of +2.6 mV (range +9.3 to –1.2 mV) and the active group –2.8 mV (range + 4.0 to –16.8 mV) (p = 0.087). Six out of 10 active-treatment patients had values more negative than the largest placebo response.
FIGURE 32.
Change from pretreatment in the response of the bronchial epithelium to perfusion with a zero chloride solution containing isoproterenol (10 µM). (a) The response of the bronchial epithelium to perfusion with a zero chloride solution containing isoproterenol (10 µM). Each symbol indicates the change in this response from trial start to finish for the relevant treatment in an individual patient. The plotted value is the mean of all interpretable recordings (range 1–3) at each time point for that patient. A more negative value is in the non-CF direction. The placebo group had a median pre–post trial change of +3.1 mV (range +9.3 to –1.2 mV) and the active group –1.0 mV (range +4.0 to –5.8 mV) (p = 0.032). (b) The response of the bronchial epithelium to perfusion with a zero chloride solution containing isoproterenol (10 µM). Each symbol indicates the change in this response from trial start to finish for the relevant treatment in an individual patient. The plotted value is the most negative value obtained from all interpretable recordings (range 1–3) at each time point for that patient. A more negative value is in the non-CF direction. The placebo group showed a median pre–post trial change of +2.6 mV (range +9.3 to –1.2 mV) and the active group –2.8 mV (range + 4.0 to –15.6 mV) (p = 0.087).

Chapter 5 Safety and adverse events
The trial was monitored at intervals by the independent DMEC, who scrutinised the unblinded group data and provided written confirmation to the TSC that the trial could continue with no modifications. Overall, the drug was well tolerated and AEs were of the nature expected in CF trials. In this section we describe the findings from the early safety cohort, AEs including a detailed description of serious AEs (SAEs) and present results of safety assays throughout the study.
Early safety cohort
The early safety cohort was designed to include 20 patients receiving three doses under intensified monitoring conditions ahead of enrolment of the complete cohort. Two patients who had been successfully screened became unwell before their planned first dose and could not be included without delaying timelines, so were excluded from this subgroup. The data presented to the DMEC came from 18 patients (eight of whom received placebo), 17 of whom had received three doses and one two doses. In addition to the procedures outlined in the general protocol, they had been seen for symptom review, lung function and blood tests (inflammatory markers, liver and renal function) at day 2 following each dose. The data from this cohort were reviewed by the Data Monitoring Safety Board at a meeting in September 2012.
Relevant data have been extracted from the closed report, to which the investigators have been unblinded only since database lock; any amendments to the original text have been made only for typographic or language errors, otherwise text and tables were as presented.
Throughout this section, the placebo group is referred to as group A and the active-treatment group as group B.
Adverse events
The AEs are presented as an aggregate number of events and also are listed by site and treatment group. The total numbers of AEs are presented in Tables 3 and 4.
Table 3 presents the total number of AEs that were observed initially in the trial, we observed that that there was only one severe AE.
| Total | London | Edinburgh | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Overall | Group A | Group B | Overall | Group A | Group B | Overall | Group A | Group B | |
| Number of AEs | 130 | 38 | 92 | 103 | 33 | 70 | 27 | 5 | 22 |
| Frequency | |||||||||
| Single episode | 108 | 32 | 76 | 87 | 27 | 60 | 21 | 5 | |
| Intermittent | 17 | 6 | 11 | 11 | 6 | 5 | 6 | 0 | |
| Severity | |||||||||
| Mild | 104 | 23 | 72 | 82 | 24 | 55 | 22 | 5 | 17 |
| Moderate | 12 | 4 | 8 | 7 | 4 | 3 | 5 | 0 | 5 |
| Severe | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| Life-threatening | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Number of expected AEs | 84 | 22 | 62 | 66 | 18 | 48 | 18 | 4 | 14 |
| Expected for study drug | 41 | 16 | 25 | 32 | 12 | 20 | 9 | 4 | 5 |
| Action taken | |||||||||
| None | 121 | 35 | 86 | 94 | 30 | 64 | 27 | 5 | 22 |
| Dose adjusted | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dose interrupted | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| Discontinued | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Medication used | |||||||||
| Yes | 24 | 10 | 14 | 18 | 10 | 8 | 6 | 0 | 6 |
| No | 98 | 28 | 70 | 77 | 23 | 54 | 21 | 5 | 16 |
| Outcome | |||||||||
| Resolved | 105 | 30 | 75 | 83 | 26 | 57 | 22 | 4 | 18 |
| Unresolved | 9 | 5 | 4 | 5 | 5 | 0 | 4 | 0 | 0 |
| Fatal | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Relation to study? | |||||||||
| Unrelated | 19 | 8 | 11 | 11 | 7 | 4 | 8 | 1 | 7 |
| Unlike | 25 | 8 | 17 | 23 | 8 | 15 | 2 | 0 | 2 |
| Possible | 39 | 11 | 28 | 25 | 7 | 18 | 14 | 4 | 10 |
| Probable | 35 | 6 | 29 | 32 | 6 | 26 | 3 | 0 | 3 |
| Almost certain | 9 | 5 | 4 | 9 | 5 | 4 | 0 | 0 | 0 |
Table 4 presents the number of AEs that were observed 2 days after dosing the subjects. No SAEs were observed 2 days after dosing the subjects. Table 5 presents changes in spirometric indices.
| Total | London | Edinburgh | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Overall | Group A | Group B | Overall | Group A | Group B | Overall | Group A | Group B | |
| Number of AEs | 10 | 4 | 6 | 10 | 4 | 6 | 0 | 0 | 0 |
| Frequency | |||||||||
| Single episode | 10 | 4 | 6 | 10 | 4 | 6 | 0 | 0 | 0 |
| Intermittent | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severity | |||||||||
| Mild | 9 | 4 | 5 | 9 | 4 | 5 | 0 | 0 | 0 |
| Moderate | 1 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Life-threatening | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Number of expected AEs | 6 | 3 | 3 | 6 | 3 | 3 | 0 | 0 | 0 |
| Expected for study drug | 4 | 2 | 2 | 4 | 2 | 2 | 0 | 0 | 0 |
| Action taken | |||||||||
| None | 10 | 4 | 6 | 10 | 4 | 6 | 0 | 0 | 0 |
| Dose adjusted | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dose interrupted | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Discontinued | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Medication used | |||||||||
| Yes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| No | 10 | 4 | 6 | 10 | 4 | 6 | 0 | 0 | 0 |
| Outcome | |||||||||
| Resolved | 5 | 1 | 4 | 5 | 1 | 4 | 0 | 0 | 0 |
| Unresolved | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Fatal | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Relation to study? | |||||||||
| Unrelated | 5 | 1 | 4 | 5 | 1 | 4 | 0 | 0 | 0 |
| Unlike | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Possible | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| Probable | 2 | 0 | 2 | 2 | 0 | 2 | 0 | 0 | 0 |
| Almost certain | 2 | 2 | 0 | 2 | 2 | 0 | 0 | 0 | 0 |
We observe that, overall, there were no SAEs occurring during the trial.
Spirometry data
| Dosing period 1 | Dosing period 2 | Dosing period 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Group A | Group B | All | Group A | Group B | All | Group A | Group B | |
| FEV1 (l) | 0.05 (–0.20 to 0.29) n = 17 | 0.00 (–0.12 to 0.17) n = 8 | 0.14 (–0.20 to 0.29) n = 9 | 0.00 (–0.28 to 0.55) n = 18 | –0.07 (–0.22 to 0.06) n = 8 | 0.22 (–0.28 to 0.55) n = 10 | –0.04 (–0.49 to 0.43) n = 18 | –0.10 (–0.31 to 0.04) n = 8 | 0.27 (–0.49 to 0.43) n = 10 |
| Percentage predicted FEV1 | 1.15 (–7.01 to 7.57) n = 18 | –0.03 (–3.87 to 6.24) n = 8 | 3.91 (–7.01 to 7.57) n = 10 | –0.12 (–8.79 to 15.06) n = 18 | –2.26 (–4.99 to 1.62) n = 8 | 6.46 (–8.79 to 15.06) n = 10 | –0.92 (–11.28 to 12.47) n = 18 | –2.92 (–6.65 to 0.42) n = 8 | 7.42 (–11.28 to 12.47) n = 10 |
| FVC (l) | 0.03 (–0.23 to 0.42) n = 18 | –0.04 (–0.23 to 0.25) n = 8 | 0.18 (–0.17 to 0.42) n = 10 | 0.05 (–0.28 to 0.89) n = 18 | 0.02 (–0.28 to 0.57) n = 8 | 0.32 (–0.25 to 0.89) n = 10 | –0.01 (–0.44 to 0.37) n = 17 | –0.11 (–0.44 to 0.10) n = 7 | 0.21 (–0.32 to 0.37) n = 10 |
| MEF25 (l) | 0.07 (–0.13 to 0.45) n = 18 | 0.06 (–0.13 to 0.42) n = 8 | 0.16 (–0.10 to 0.45) n = 10 | 0.03 (–0.18 to 0.42) n = 18 | –0.03 (–0.13 to 0.03) n = 8 | 0.18 (–0.18 to 0.42) n = 10 | –0.04 (–0.42 to 0.21) n = 18 | –0.04 (–0.17 to 0.10) n = 8 | 0.20 (–0.42 to 0.21) n = 10 |
| MEF50 (l) | 0.15 (–0.26 to 0.52) n = 18 | 0.14 (–0.14 to 0.47) n = 8 | 0.25 (–0.26 to 0.52) n = 10 | –0.12 (–0.98 to 0.75) n = 18 | –0.33 (–0.98 to 0.10) n = 8 | 0.39 (–0.61 to 0.75) n = 10 | –0.05 (–0.87 to 1.14) n = 18 | –0.14 (–0.37 to 0.17) n = 8 | 0.64 (–0.87 to 1.14) n = 10 |
| MEF75 (l) | 0.07 (–0.62 to 0.63) n = 17 | 0.05 (–0.62 to 0.60) n = 8 | 0.26 (–0.19 to 0.63) n = 9 | 0.17 (–1.29 to 1.84) n = 17 | –0.10 (–1.29 to 0.69) n = 8 | 0.88 (–0.86 to 1.84) n = 9 | –0.33 (–2.16 to 1.00) n = 17 | –0.32 (–0.95 to 0.08) n = 8 | 1.01 (–2.16 to 1.00) n = 9 |
Clinical examination
Table 6 presents the change in clinical examination parameters from screening day to dosing day. The changes in the clinical examination are presented as a number of cases that change from a positive result to a negative result; change from a negative result to a positive result; and do not change.
| Change in clinical findings | Dosing period 1 | Dosing period 2 | Dosing period 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Group A | Group B | All | Group A | Group B | All | Group A | Group B | |
| Ears | |||||||||
| Normal to abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 8 (100.0) | 9 (100.0) |
| Abnormal to normal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Eyes | |||||||||
| Normal to abnormal | 1 (5.6) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 17 (94.4) | 8 (100.0) | 9 (90.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 8 (100.0) | 9 (100.0) |
| Abnormal to normal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Nose | |||||||||
| Normal to abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (17.7) | 1 (12.5) | 2 (22.2) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 14 (82.4) | 7 (87.5) | 7 (77.8) |
| Abnormal to normal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Throat | |||||||||
| Normal to abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 1 (12.5) | 0 (0.0) | 2 (11.8) | 1 (12.5) | 1 (11.1) |
| No change | 17 (94.4) | 8 (100.0) | 9 (90.0) | 16 (88.9) | 7 (87.5) | 9 (90.0) | 14 (82.4) | 7 (87.5) | 7 (77.8) |
| Abnormal to normal | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.9) | 0 (0.0) | 1 (11.1) |
| Skin | |||||||||
| Normal to abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (11.1) | 0 (0.0) | 2 (20.0) | 2 (11.8) | 1 (12.5) | 1 (11.1) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 14 (77.8) | 8 (100.0) | 6 (60.0) | 13 (76.5) | 7 (87.5) | 6 (66.7) |
| Abnormal to normal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (11.1) | 0 (0.0) | 2 (20.0) | 2 (11.8) | 0 (0.0) | 2 (22.2) |
| Heart | |||||||||
| Normal to abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 17 (94.4) | 7 (87.5) | 10 (100.0) | 17 (94.4) | 7 (87.5) | 10 (100.0) | 16 (94.1) | 7 (87.5) | 9 (100.0) |
| Abnormal to normal | 1 (5.6) | 1 (12.5) | 0 (0.0) | 1 (5.6) | 1 (12.5) | 0 (0.0) | 1 (5.9) | 1 (12.5) | 0 (0.0) |
| Breath sound left lung | |||||||||
| Normal to abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 8 (100.0) | 9 (100.0) |
| Abnormal to normal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Breath sound right lung | |||||||||
| Normal to abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 8 (100.0) | 9 (100.0) |
| Abnormal to normal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Crackles left lung | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.9) | 1 (12.5) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 16 (88.9) | 8 (100.0) | 8 (80.0) | 15 (88.2) | 7 (87.5) | 8 (88.9) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.9) | 0 (0.0) | 1 (11.1) |
| Crackles right lung | |||||||||
| Absent to present | 2 (11.1) | 1 (12.5) | 1 (10.0) | 3 (16.7) | 2 (25.0) | 1 (10.0) | 1 (5.9) | 0 (0.0) | 1 (11.1) |
| No change | 15 (83.3) | 6 (75.0) | 9 (90.0) | 15 (83.3) | 6 (75.0) | 9 (90.0) | 15 (88.2) | 8 (100.0) | 7 (77.8) |
| Present to absent | 1 (5.6) | 1 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.9) | 0 (0.0) | 1 (11.1) |
| Wheeze left lung | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Not change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 8 (100.0) | 9 (100.0) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Wheeze right lung | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 8 (100.0) | 9 (100.0) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Other respiratory problems | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (11.8) | 1 (12.5) | 1 (11.1) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 15 (88.2) | 7 (87.5) | 8 (88.9) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Abdomen | |||||||||
| Normal to abnormal | 1 (5.6) | 1 (12.5) | 0 (0.0) | 1 (5.6) | 1 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 16 (88.9) | 7 (87.5) | 9 (90.0) | 16 (88.9) | 7 (87.5) | 9 (90.0) | 16 (94.1) | 8 (100.0) | 8 (88.9) |
| Abnormal to normal | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.9) | 0 (0.0) | 1 (11.1) |
| Liver | |||||||||
| Normal to abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 8 (100.0) | 9 (100.0) |
| Abnormal to normal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Spleen | |||||||||
| Normal to abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 8 (100.0) | 9 (100.0) |
| Abnormal to normal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Current symptoms
Table 7 presents the symptoms expressed as change from the dosing day to 2 days after dosing.
| Symptom | Dosing period 1 | Dosing period 2 | Dosing period 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Group A | Group B | All | Group A | Group B | All | Group A | Group B | |
| Breathlessness | |||||||||
| Absent to present | 1 (5.6) | 0 (0.0) | 1 (10.0) | 2 (11.1) | 0 (0.0) | 2 (20.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 16 (88.9) | 8 (100.0) | 8 (80.0) | 13 (72.2) | 8 (100.0) | 5 (50.0) | 15 (88.2) | 7 (100.0) | 8 (80.0) |
| Present to absent | 1 (5.6) | 0 (0.0) | 1 (10.0) | 3 (16.7) | 0 (0.0) | 3 (30.0) | 2 (11.8) | 0 (0.0) | 2 (20.0) |
| Increase cough | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 4 (23.5) | 0 (0.0) | 4 (40.0) |
| No change | 17 (94.4) | 8 (100.0) | 9 (90.0) | 12 (66.7) | 7 (87.5) | 5 (50.0) | 9 (52.9) | 6 (85.7) | 3 (30.0) |
| Present to absent | 1 (5.6) | 0 (0.0) | 1 (10.0) | 6 (33.3) | 1 (12.5) | 5 (50.0) | 4 (23.5) | 1 (14.3) | 3 (30.0) |
| Wheeze | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 16 (88.9) | 8 (100.0) | 8 (80.0) | 17 (100.0) | 7 (100.0) | 10 (100.0) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Haemoptysis | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 7 (100.0) | 10 (100.0) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Chest pain | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 7 (100.0) | 10 (100.0) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Cardiovascular | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.9) | 0 (0.0) | 1 (10.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (94.4) | 7 (87.5) | 10 (100.0) | 15 (88.2) | 7 (100.0) | 8 (80.0) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.6) | 1 (12.5) | 0 (0.0) | 1 (5.9) | 0 (0.0) | 1 (10.0) |
| Gastrointestinal | |||||||||
| Absent to present | 1 (5.6) | 1 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (11.8) | 0 (0.0) | 2 (20.0) |
| No change | 16 (88.9) | 7 (87.5) | 9 (90.0) | 17 (94.4) | 8 (100.0) | 9 (90.0) | 12 (70.6) | 6 (85.7) | 6 (60.0) |
| Present to absent | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 3 (17.7) | 1 (14.3) | 2 (20.0) |
| Throat | |||||||||
| Absent to present | 2 (11.1) | 0 (0.0) | 2 (20.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.9) | 0 (0.0) | 1 (10.0) |
| No change | 13 (72.2) | 7 (87.5) | 6 (60.0) | 17 (94.4) | 8 (100.0) | 9 (90.0) | 14 (82.4) | 6 (85.7) | 8 (80.0) |
| Present to absent | 3 (16.7) | 1 (12.5) | 2 (20.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (11.8) | 1 (14.3) | 1 (10.0) |
| Eye | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 7 (100.0) | 10 (100.0) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Ears | |||||||||
| Absent to present | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 18 (100.0) | 8 (100.0) | 10 (100.0) | 18 (100.0) | 8 (100.0) | 10 (100.0) | 17 (100.0) | 7 (100.0) | 10 (100.0) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Nose | |||||||||
| Absent to present | 2 (11.1) | 0 (0.0) | 2 (20.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No change | 15 (83.3) | 8 (100.0) | 7 (70.0) | 16 (88.9) | 7 (87.5) | 9 (90.0) | 17 (100.0) | 7 (100.0) | 10 (100.0) |
| Present to absent | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.6) | 1 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Neurological examination | |||||||||
| Absent to present | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.6) | 1 (12.5) | 0 (0.0) | 3 (17.7) | 1 (14.3) | 2 (20.0) |
| No change | 14 (77.8) | 7 (87.5) | 7 (70.0) | 13 (72.2) | 5 (62.5) | 8 (80.0) | 13 (76.5) | 6 (85.7) | 7 (70.0) |
| Present to absent | 3 (16.7) | 1 (12.5) | 2 (20.0) | 4 (22.2) | 2 (25.0) | 2 (20.0) | 1 (5.9) | 0 (0.0) | 1 (10.0) |
| Skin | |||||||||
| Absent to present | 2 (11.1) | 0 (0.0) | 2 (20.0) | 1 (5.6) | 0 (0.0) | 1 (10.0) | 1 (5.9) | 0 (0.0) | 1 (10.0) |
| No change | 16 (88.9) | 8 (100.0) | 8 (80.0) | 17 (94.4) | 8 (100.0) | 9 (90.0) | 15 (88.2) | 6 (85.7) | 9 (90.0) |
| Present to absent | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.9) | 1 (14.3) | 0 (0.0) |
Vital signs
Table 8 presents the change from screening day to dosing day. A negative change indicates a decrease in vital sign parameters.
| Sign | Dosing period 1 | Dosing period 2 | Dosing period 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Group A | Group B | All | Group A | Group B | All | Group A | Group B | |
| Diastolic BP (mmHg) | 3.83 (–8.00 to 21.00) n = 18 | 5.88 (–6.00 to 20.00) n = 8 | 2.20 (–8.00 to 21.00) n = 10 | 3.44 (–13.00 to 22.00) n = 18 | 3.13 (–4.00 to 12.00) n = 8 | 3.70 (–13.00 to 22.00) n = 10 | 4.82 (–15.00 to 31.00) n = 17 | 5.63 (–4.00 to 17.00) n = 8 | 4.11 (–15.00 to 31.00) n = 9 |
| Systolic BP (mmHg) | –0.56 (–13.00 to 14.00) n = 18 | 1.00 (–13.00 to 14.00) n = 8 | –1.80 (–13.00 to 8.00) n = 10 | 1.78 (–10.00 to 16.00) n = 18 | 3.25 (–10.00 to 16.00) n = 8 | 0.60 (–7.00 to 13.00) n = 10 | 0.24 (–15.00 to 16.00) n = 17 | 2.50 (–9.00 to 16.00) n = 8 | –1.78 (–15.00 to 8.00) n = 9 |
| Pulse (b.p.m.) | 2.11 (–16.00 to 24.00) n = 18 | 4.63 (–8.00 to 24.00) n = 8 | 0.10 (–16.00 to 14.00) n = 10 | 5.94 (–27.00 to 28.00) n = 18 | 4.38 (–27.00 to 23.00) n = 8 | 7.20 (–18.00 to 28.00) n = 10 | 12.06 (–6.00 to 29.00) n = 17 | 6.88 (–6.00 to 25.00) n = 8 | 16.67 (0.00 to 29.00) n = 9 |
| Respiratory rate (b.p.m.) | –0.50 (–12.00 to 6.00) n = 16 | –2.86 (–12.00 to 2.00) n = 7 | 1.33 (–2.00 to 6.00) n = 9 | 0.13 (–10.00 to 8.00) n = 16 | –1.71 (–10.00 to 8.00) n = 7 | 1.56 (–4.00 to 6.00) n = 9 | 1.56 (–6.00 to 10.00) n = 16 | 0.25 (–6.00 to 10.00) n = 8 | 2.88 (–3.00 to 8.00) n = 8 |
| Transcutaneous oxygen saturation (%) | 0.22 (–2.00 to 4.00) n = 18 | –0.13 (–2.00 to 3.00) n = 8 | 0.50 (–1.00 to 4.00) n = 10 | 0.06 (–3.00 to 4.00) n = 18 | 0.63 (–2.00 to 4.00) n = 8 | –0.40 (–3.00 to 2.00) n = 10 | 0.35 (–2.00 to 3.00) n = 17 | 0.63 (–2.00 to 3.00) n = 8 | 0.11 (–1.00 to 2.00) n = 9 |
| Temperature (°C) | –0.01 (–0.60 to 1.00) n = 16 | –0.14 (–0.60 to 0.70) n = 7 | 0.09 (–0.50 to 1.00) n = 9 | 0.04 (–0.70 to 1.10) n = 16 | –0.24 (–0.70 to 0.40) n = 7 | 0.26 (–0.60 to 1.10) n = 9 | 0.13 (–0.60 to 0.90) n = 16 | 0.02 (–0.40 to 0.70) n = 8 | 0.23 (–0.60 to 0.90) n = 8 |
Biochemistry parameters
Table 9 presents the change from screening day to dosing day. A negative change indicates a decrease in the biochemistry parameters.
| Variable | Dosing period 1 | Dosing period 2 | Dosing period 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Group A | Group B | All | Group A | Group B | All | Group A | Group B | |
| Alanine transaminase (units/l) | 2.44 (–2.00 to 14.00) n = 18 | 1.50 (–2.00 to 5.00) n = 8 | 3.20 (0.00 to 14.00) n = 10 | 1.83 (–7.00 to 36.00) n = 18 | 3.50 (–7.00 to 36.00) n = 8 | 0.50 (–3.00 to 5.00) n = 10 | 1.47 (–14.00 to 43.00) n = 17 | 3.38 (–14.00 to 43.00) n = 8 | –0.22 (–9.00 to 9.00) n = 9 |
| Albumin (mmol/l) | –0.78 (–4.00 to 3.00) n = 18 | –0.88 (–4.00 to 3.00) n = 8 | –0.70 (–3.00 to 2.00) n = 10 | –2.06 (–7.00 to 3.00) n = 18 | –2.50 (–7.00 to 3.00) n = 8 | –1.70 (–3.00 to –1.00) n = 10 | –2.18 (–5.00 to 1.00) n = 17 | –1.63 (–5.00 to 1.00) n = 8 | –2.67 (–5.00 to –1.00) n = 9 |
| Alkaline phosphatise (units/l) | –4.08 (–22.00 to 2.00) n = 12 | –1.75 (–7.00 to 2.00) n = 4 | –5.25 (–22.00 to 2.00) n = 8 | –5.67 (–26.00 to 12.00) n = 15 | 2.20 (–9.00 to 12.00) n = 5 | –9.60 (–26.00 to 5.00) n = 10 | –1.93 (–32.00 to 41.00) n = 14 | –0.40 (–9.00 to 12.00) n = 5 | –2.78 (–32.00 to 41.00) n = 9 |
| Aminotransferase (units/l) | 1.38 (–2.00 to 5.00) n = 16 | 1.25 (–2.00 to 3.00) n = 8 | 1.50 (–1.00 to 5.00) n = 8 | 2.41 (–6.00 to 32.00) n = 17 | 3.63 (–6.00 to 32.00) n = 8 | 1.33 (–5.00 to 4.00) n = 9 | 0.88 (–12.00 to 10.00) n = 17 | 0.13 (–12.00 to 10.00) n = 8 | 1.56 (–4.00 to 7.00) n = 9 |
| Amylase (units/l) | –2.94 (–28.00 to 12.00) n = 16 | –4.75 (–28.00 to 12.00) n = 8 | –1.13 (–10.00 to 7.00) n = 8 | –2.88 (–19.00 to 44.00) n = 17 | 0.88 (–19.00 to 44.00) n = 8 | –6.22 (–16.00 to 7.00) n = 9 | –1.63 (–24.00 to 26.00) n = 16 | –1.00 (–24.00 to 26.00) n = 7 | –2.11 (–18.00 to 23.00) n = 9 |
| Bilirubin (g/l) | 0.92 (–2.00 to 6.00) n = 13 | 1.20 (–2.00 to 6.00) n = 5 | 0.75 (–2.00 to 5.00) n = 8 | 0.27 (–6.00 to 6.00) n = 15 | 2.20 (–2.00 to 6.00) n = 5 | –0.70 (–6.00 to 3.00) n = 10 | 0.36 (–7.00 to 8.00) n = 14 | 3.20 (–1.00 to 8.00) n = 5 | –1.22 (–7.00 to 8.00) n = 9 |
| Calcium (mmol/l) | –0.03 (–0.20 to 0.10) n = 18 | –0.04 (–0.20 to 0.00) n = 8 | –0.03 (–0.20 to 0.10) n = 10 | –0.06 (–0.30 to 0.10) n = 17 | –0.06 (–0.20 to 0.10) n = 7 | –0.06 (–0.30 to 0.00) n = 10 | –0.04 (–0.20 to 0.10) n = 16 | –0.03 (–0.20 to 0.10) n = 8 | –0.05 (–0.20 to 0.10) n = 8 |
| Corrected calcium (mmol/l) | –0.03 (–0.20 to 0.10) n = 18 | –0.04 (–0.20 to 0.10) n = 8 | –0.02 (–0.20 to 0.10) n = 10 | –0.03 (–0.20 to 0.10) n = 16 | –0.05 (–0.20 to 0.10) n = 6 | –0.01 (–0.20 to 0.10) n = 10 | –0.01 (–0.20 to 0.10) n = 16 | –0.01 (–0.10 to 0.10) n = 8 | –0.01 (–0.20 to 0.10) n = 8 |
| Creatinine (µmol/l) | 2.00 (–10.00 to 10.00) n = 18 | –0.88 (–10.00 to 9.00) n = 8 | 4.30 (–3.00 to 10.00) n = 10 | 0.44 (–18.00 to 14.00) n = 18 | –2.25 (–18.00 to 6.00) n = 8 | 2.60 (–5.00 to 14.00) n = 10 | 0.65 (–11.00 to 13.00) n = 17 | –1.75 (–11.00 to 5.00) n = 8 | 2.78 (–9.00 to 13.00) n = 9 |
| CRP (mg/l) | –0.94 (–12.00 to 6.00) n = 16 | 0.43 (–5.00 to 6.00) n = 7 | –2.00 (–12.00 to 1.00) n = 9 | –0.29 (–14.00 to 6.00) n = 17 | 0.86 (–1.00 to 5.00) n = 7 | –1.10 (–14.00 to 6.00) n = 10 | 0.06 (–14.00 to 13.00) n = 16 | 1.71 (–6.00 to 13.00) n = 7 | –1.22 (–14.00 to 4.00) n = 9 |
| Glucose (mmol/l) | 0.10 (–1.60 to 4.00) n = 9 | 0.70 (–1.60 to 4.00) n = 4 | –0.38 (–1.60 to 1.00) n = 5 | –0.48 (–2.00 to 0.70) n = 9 | –0.22 (–2.00 to 0.70) n = 4 | –0.68 (–1.70 to 0.50) n = 5 | 0.36 (–2.00 to 4.10) n = 9 | –0.03 (–2.00 to 2.00) n = 4 | 0.66 (–1.20 to 4.10) n = 5 |
| γ-Glutamyl transferase (units/l) | –0.35 (–4.00 to 3.00) n = 17 | –1.00 (–4.00 to 1.00) n = 7 | 0.10 (–2.00 to 3.00) n = 10 | –0.44 (–4.00 to 3.00) n = 18 | –0.63 (–3.00 to 3.00) n = 8 | –0.30 (–4.00 to 3.00) n = 10 | 1.06 (–5.00 to 21.00) n = 17 | 0.63 (–3.00 to 10.00) n = 8 | 1.44 (–5.00 to 21.00) n = 9 |
| Inorganic phosphate (mmol/l) | –0.02 (–0.40 to 0.50) n = 18 | –0.04 (–0.40 to 0.50) n = 8 | 0.00 (–0.40 to 0.20) n = 10 | –0.03 (–0.20 to 0.20) n = 16 | –0.01 (–0.20 to 0.20) n = 7 | –0.04 (–0.20 to 0.20) n = 9 | –0.08 (–0.50 to 0.30) n = 15 | –0.18 (–0.50 to 0.10) n = 8 | 0.03 (–0.20 to 0.30) n = 7 |
| Magnesium (mmol/l) | 0.01 (–0.10 to 0.10) n = 14 | 0.03 (–0.10 to 0.10) n = 7 | –0.01 (–0.10 to 0.00) n = 7 | 0.01 (–0.20 to 0.20) n = 13 | 0.03 (–0.20 to 0.20) n = 6 | –0.01 (–0.10 to 0.00) n = 7 | 0.04 (–0.20 to 0.20) n = 11 | 0.04 (–0.20 to 0.20) n = 7 | 0.03 (–0.10 to 0.10) n = 4 |
| Potassium (mmol/l) | 0.09 (–0.10 to 0.40) n = 17 | 0.18 (–0.10 to 0.40) n = 8 | 0.02 (–0.10 to 0.30) n = 9 | 0.03 (–0.40 to 0.50) n = 18 | 0.04 (–0.40 to 0.40) n = 8 | 0.03 (–0.20 to 0.50) n = 10 | 0.06 (–0.50 to 0.30) n = 17 | 0.09 (–0.20 to 0.30) n = 8 | 0.03 (–0.50 to 0.30) n = 9 |
| Total protein (g/l) | –1.55 (–8.00 to 2.00) n = 11 | –2.75 (–8.00 to 0.00) n = 4 | –0.86 (–4.00 to 2.00) n = 7 | –2.43 (–7.00 to 3.00) n = 14 | –3.80 (–7.00 to 1.00) n = 5 | –1.67 (–7.00 to 3.00) n = 9 | –1.77 (–4.00 to 1.00) n = 13 | –2.20 (–4.00 to 1.00) n = 5 | –1.50 (–4.00 to 1.00) n = 8 |
| Sodium (mmol/l) | –0.28 (–5.00 to 3.00) n = 18 | –0.63 (–4.00 to 2.00) n = 8 | 0.00 (–5.00 to 3.00) n = 10 | –0.28 (–7.00 to 3.00) n = 18 | 0.13 (–2.00 to 3.00) n = 8 | –0.60 (–7.00 to 3.00) n = 10 | 1.00 (–2.00 to 4.00) n = 17 | 1.50 (–1.00 to 4.00) n = 8 | 0.56 (–2.00 to 4.00) n = 9 |
| Urea (mmol/l) | –0.13 (–1.20 to 2.70) n = 18 | –0.53 (–1.20 to 0.60) n = 8 | 0.19 (–1.00 to 2.70) n = 10 | –0.44 (–2.00 to 0.70) n = 18 | –0.69 (–2.00 to 0.70) n = 8 | –0.24 (–1.40 to 0.50) n = 10 | –0.15 (–3.30 to 3.00) n = 17 | –0.44 (–1.70 to 1.30) n = 8 | 0.11 (–3.30 to 3.00) n = 9 |
Haematology parameters
Table 10 presents the change from screening day to dosing day. A negative change indicates a decrease in the haematology parameters.
| Variable | Dosing period 1 | Dosing period 2 | Dosing period 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Group A | Group B | All | Group A | Group B | All | Group A | Group B | |
| Basophils | 0.01 (–0.10 to 0.10) n = 17 | 0.00 (–0.10 to 0.10) n = 8 | 0.01 (–0.10 to 0.10) n = 9 | 0.02 (–0.10 to 0.10) n = 17 | 0.03 (0.00 to 0.10) n = 8 | 0.02 (–0.10 to 0.10) n = 9 | 0.01 (–0.10 to 0.10) n = 17 | 0.01 (0.00 to 0.10) n = 8 | 0.00 (–0.10 to 0.10) n = 9 |
| Eosinophils | 0.01 (–0.12 to 0.11) n = 17 | –0.03 (–0.12 to 0.11) n = 8 | 0.04 (–0.01 to 0.10) n = 9 | 0.07 (–0.10 to 0.61) n = 17 | 0.12 (–0.10 to 0.61) n = 8 | 0.02 (–0.10 to 0.10) n = 9 | 0.06 (–0.12 to 0.36) n = 17 | 0.09 (–0.10 to 0.36) n = 8 | 0.04 (–0.12 to 0.17) n = 9 |
| Erythrocyte sedimentation rate (mm/hour) | –1.93 (–10.00 to 4.00) n = 15 | –2.14 (–10.00 to 4.00) n = 7 | –1.75 (–7.00 to 3.00) n = 8 | –1.60 (–12.00 to 7.00) n = 15 | –1.14 (–12.00 to 7.00) n = 7 | –2.00 (–12.00 to 4.00) n = 8 | 0.40 (–15.00 to 20.00) n = 15 | –1.86 (–15.00 to 5.00) n = 7 | 2.38 (–9.00 to 20.00) n = 8 |
| Lymphocytes | –0.07 (–0.90 to 0.70) n = 17 | –0.01 (–0.90 to 0.70) n = 8 | –0.12 (–0.40 to 0.40) n = 9 | 0.00 (–0.60 to 0.60) n = 17 | 0.09 (–0.60 to 0.60) n = 8 | –0.08 (–0.40 to 0.50) n = 9 | 0.10 (–0.80 to 2.40) n = 17 | 0.03 (–0.70 to 0.70) n = 8 | 0.17 (–0.80 to 2.40) n = 9 |
| Mean corpuscular haemoglobin (pg) | –0.19 (–1.40 to 1.50) n = 17 | –0.45 (–1.40 to 0.30) n = 8 | 0.03 (–0.80 to 1.50) n = 9 | –0.46 (–1.40 to 1.10) n = 17 | –0.68 (–1.20 to –0.10) n = 8 | –0.28 (–1.40 to 1.10) n = 9 | 3.12 (–1.80 to 61.30) n = 17 | –0.67 (–1.80 to 0.50) n = 8 | 6.49 (–1.20 to 61.30) n = 9 |
| Mean corpuscular haemoglobin concentration (g/dl) | –0.50 (–2.00 to 2.00) n = 10 | –1.00 (–2.00 to 0.00) n = 5 | 0.00 (–2.00 to 2.00) n = 5 | –0.10 (–2.00 to 2.00) n = 10 | –0.60 (–2.00 to 0.00) n = 5 | 0.40 (–2.00 to 2.00) n = 5 | 0.25 (–2.00 to 2.00) n = 8 | –0.25 (–2.00 to 1.00) n = 4 | 0.75 (–1.00 to 2.00) n = 4 |
| Mean corpuscular volume (fl) | 0.29 (–3.00 to 3.00) n = 17 | 0.25 (–2.00 to 3.00) n = 8 | 0.33 (–3.00 to 3.00) n = 9 | –1.12 (–4.00 to 3.00) n = 17 | –1.00 (–3.00 to 3.00) n = 8 | –1.22 (–4.00 to 1.00) n = 9 | –1.31 (–6.00 to 1.00) n = 16 | –1.25 (–4.00 to 1.00) n = 8 | –1.38 (–6.00 to 1.00) n = 8 |
| Monocytes | 0.01 (–0.20 to 0.20) n = 17 | 0.00 (–0.20 to 0.14) n = 8 | 0.01 (–0.16 to 0.20) n = 9 | 0.00 (–0.30 to 0.30) n = 17 | –0.02 (–0.30 to 0.14) n = 8 | 0.01 (–0.30 to 0.30) n = 9 | 0.01 (–0.20 to 0.40) n = 17 | –0.01 (–0.10 to 0.10) n = 8 | 0.03 (–0.20 to 0.40) n = 9 |
| Neutrophils | –0.53 (–4.70 to 2.30) n = 17 | –0.65 (–3.30 to 2.20) n = 8 | –0.42 (–4.70 to 2.30) n = 9 | –0.84 (–5.80 to 2.00) n = 17 | –0.85 (–5.80 to 2.00) n = 8 | –0.83 (–3.30 to 0.20) n = 9 | –0.58 (–4.90 to 2.10) n = 17 | –1.34 (–4.20 to 0.70) n = 8 | 0.10 (–4.90 to 2.10) n = 9 |
| Platelets | –4.53 (–74.00 to 32.00) n = 17 | –9.13 (–74.00 to 32.00) n = 8 | –0.44 (–31.00 to 31.00) n = 9 | –10.94 (–93.00 to 84.00) n = 17 | –14.50 (–93.00 to 84.00) n = 8 | –7.78 (–58.00 to 39.00) n = 9 | –5.53 (–97.00 to 59.00) n = 17 | –6.00 (–63.00 to 59.00) n = 8 | –5.11 (–97.00 to 35.00) n = 9 |
Gas transfer
Table 11 presents the change from the screening day to 2 days after dosing. A negative change indicates a decrease in the gas transfer.
| Variable | Dosing period 1 | Dosing period 2 | Dosing period 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Group A | Group B | All | Group A | Group B | All | Group A | Group B | |
| KCO (mmol) | –0.02 (–0.15 to 0.18) n = 18 | 0.01 (–0.06 to 0.13) n = 8 | –0.05 (–0.15 to 0.18) n = 10 | 0.00 (–0.14 to 0.16) n = 17 | 0.00 (–0.11 to 0.16) n = 8 | –0.01 (–0.14 to 0.12) n = 9 | 0.00 (–0.38 to 0.21) n = 16 | 0.04 (–0.06 to 0.21) n = 6 | –0.03 (–0.38 to 0.13) n = 10 |
| KCOc | 0.00 (–0.12 to 0.16) n = 18 | 0.03 (–0.06 to 0.14) n = 8 | –0.03 (–0.12 to 0.16) n = 10 | 0.00 (–0.12 to 0.16) n = 18 | 0.03 (–0.06 to 0.14) n = 8 | –0.03 (–0.12 to 0.16) n = 10 | 0.03 (–0.35 to 0.25) n = 16 | 0.08 (–0.02 to 0.25) n = 6 | 0.00 (–0.35 to 0.17) n = 10 |
| TLCO (mmol) | –0.06 (–1.19 to 1.48) n = 18 | 0.15 (–0.29 to 0.53) n = 8 | –0.22 (–1.19 to 1.48) n = 10 | 0.03 (–1.09 to 1.60) n = 17 | 0.04 (–0.75 to 1.01) n = 8 | 0.03 (–1.09 to 1.60) n = 9 | 0.03 (–1.90 to 1.48) n = 16 | 0.13 (–1.04 to 1.14) n = 6 | –0.02 (–1.90 to 1.48) n = 10 |
| TLCOc | 0.02 (–1.10 to 1.52) n = 18 | 0.24 (0.03 to 0.56) n = 8 | –0.15 (–1.10 to 1.52) n = 10 | –0.49 (–8.90 to 1.68) n = 18 | 0.02 (–0.92 to 0.93) n = 8 | –0.89 (–8.90 to 1.68) n = 10 | –0.29 (–7.31 to 1.77) n = 17 | –0.77 (–7.31 to 1.29) n = 7 | 0.04 (–1.75 to 1.77) n = 10 |
| VA (l) | 0.04 (–0.39 to 1.02) n = 18 | 0.08 (–0.17 to 0.39) n = 8 | 0.02 (–0.39 to 1.02) n = 10 | 0.02 (–0.57 to 0.95) n = 17 | 0.01 (–0.25 to 0.32) n = 8 | 0.03 (–0.57 to 0.95) n = 9 | 0.02 (–0.64 to 0.65) n = 16 | –0.01 (–0.45 to 0.37) n = 6 | 0.04 (–0.64 to 0.65) n = 10 |
In summary, the DMEC did not consider there was a safety signal on reviewing these results and wrote approving continuation of full recruitment into the trial at the 5-ml dose.
Adverse events and serious adverse events
General adverse events
All recorded AEs were categorised as in Table 12, and the frequency of their occurrence expressed proportional to the number of patients in each group. All patients in both groups reported AEs, as would be expected in a population of CF patients monitored for 12 months. However, the majority of AEs were of a nature expected in this clinical context and frequencies were similar between groups; there were no statistically significant differences in number overall or once they were broken down into the categories as tabulated.
| Category | ITT population | PP population | ||
|---|---|---|---|---|
| Placebo (n = 60) | Active treatment (n = 76) | Placebo (n = 54) | Active treatment (n = 62) | |
| Lower airway respiratory symptoms | 7.6 | 8.0 | 7.9 | 9.0 |
| Gastrointestinal symptoms | 2.0 | 1.7 | 2.1 | 1.8 |
| Fever or flu-like symptoms | 1.1 | 1.4 | 1.1 | 1.4 |
| Headache | 1.2 | 1.1 | 1.2 | 1.1 |
| Upper airway symptoms | 2.2 | 3.1 | 2.3 | 3.4 |
| Elevated liver function tests | 0.3 | 0.4 | 0.3 | 0.4 |
| Haematuria | 0.2 | 0.2 | 0.2 | 0.2 |
| Isolated raised inflammatory markers | 0.8 | 0.6 | 0.8 | 0.7 |
| Other | 3.0 | 3.0 | 3.2 | 3.3 |
| All | 18.2 | 19.6 | 19.1 | 21.2 |
One patient in the placebo group (fatigue and increased respiratory symptoms) and one in the active-treatment group (flu-like symptoms) discontinued study treatment because of AEs.
Serious adverse events
There were no deaths during the study. Six SAEs were documented, all in the active-treatment group (Table 13). Neither the DMEC nor the TSC considered any SAE was related to the trial medication, but that one (case 2) was probably related to a trial procedure (bronchoscopy).
| Case | Description |
|---|---|
| 1 | Admission to hospital with acute pancreatitis: 47-year-old pancreatic-sufficient male, with multiple previous episodes of acute, non-alcohol-related pancreatitis. Managed conservatively. 4 weeks after last dose |
| 2 | Admission to hospital with severe headache, fever, pulmonary exacerbation and new isolate of MRSA: 28-year-old male with flu-like illness within 24 hours of trial bronchoscopy. Hospital admission for intravenous antibiotics. 5 weeks after last dose |
| 3 | Pneumothorax after removal of indwelling intravenous access device: 23-year-old female with semi-elective admission for removal and replacement of malfunctioning portacath. Small pneumothorax induced during surgery requiring no intervention. 4 days after last dose |
| 4 | Post-surgical infection: 46-year-old female undergoing laparoscopic Nissen’s fundoplication for severe reflux. 10 days post surgery, admitted with pain and fever secondary to surgical site abscess, treated 20 days after last dose |
| 5 | Admission to hospital with headache, vomiting and viral upper respiratory tract infection symptoms: 25-year-old male/female. Overnight admission with conservative management. 11 days after previous dose |
| 6 | Admission to hospital with minor vomiting illness: 14-year-old male with diabetes. Admitted for assistance with diabetic control. 16 days after previous dose |
Other safety assays
There were no clinically relevant changes in haematology (Figure 33), biochemistry (Figures 34 and 35), urinary markers (Table 14), histology (Figure 36), or lipid staining in sputum (Table 15) or biopsy cells (Table 16) during the trial. There was no immunological evidence to suggest the development of anti-DNA antibodies (Table 17) or anti-CFTR T cells (Table 18). There were no treatment-related differences in change in weight or BMI (Figure 37). Figures 33–37 present longitudinal values as mean (standard error of the mean; SEM), although in many cases error bars are small and not visible outside the mean point.
FIGURE 33.
Haematological parameters.


FIGURE 34.
Blood biochemistry and renal markers.



FIGURE 35.
Liver and pancreatic function.


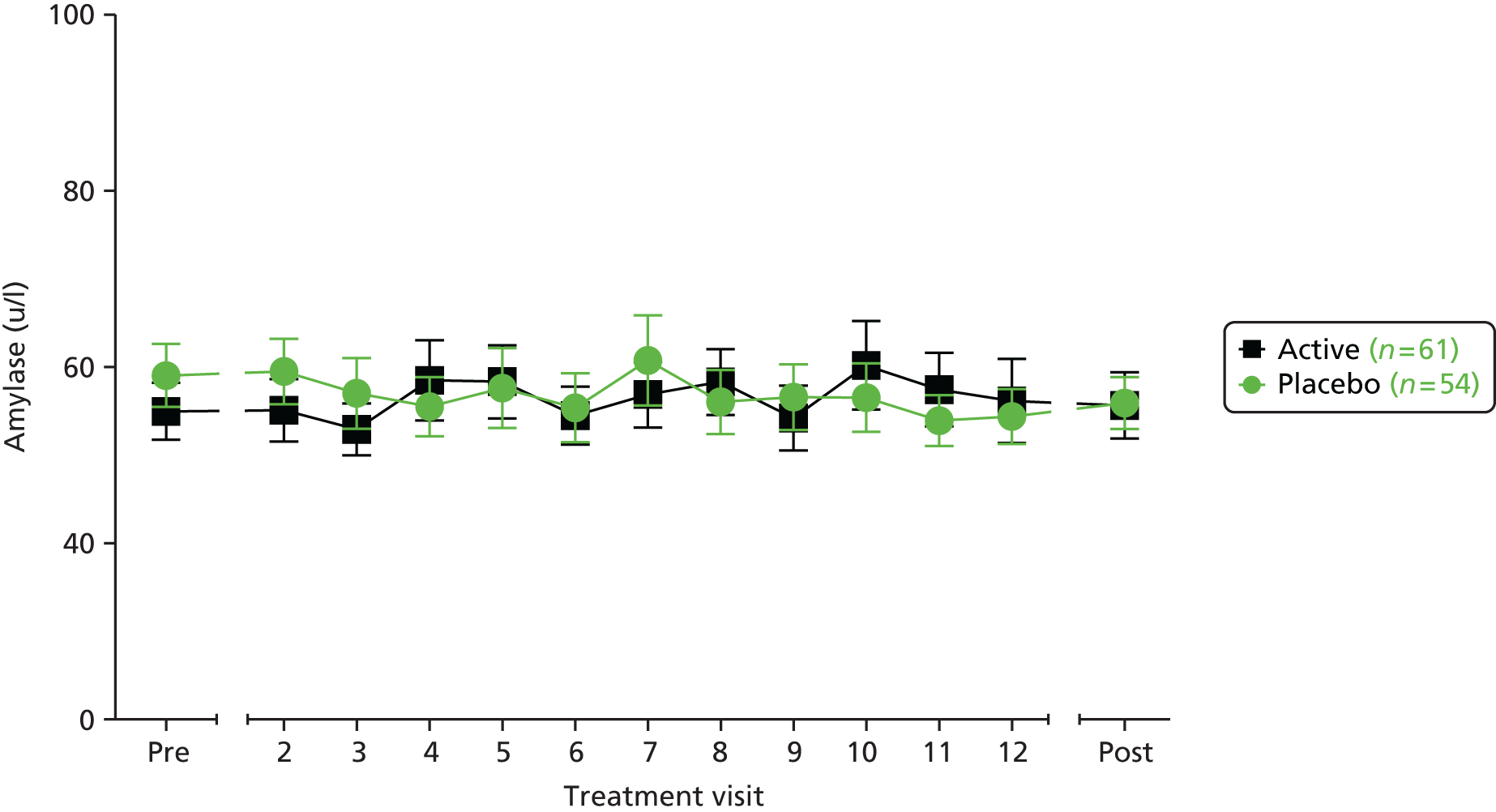
| Urinalysisa | Placebo (n = 54) | Active (n = 62) | ||
|---|---|---|---|---|
| Pretreatment | Post-treatment | Pretreatment | Post-treatment | |
| Urine proteinb | ||||
| Negative | 52 | 47 | 62 | 56 |
| Trace | 2 | 6 | 0 | 5 |
| + Positive | 0 | 0 | 0 | 0 |
| ++ Positive | 0 | 1 | 0 | 0 |
| +++ Positive | 0 | 0 | 0 | 0 |
| ++++ Positive | 0 | 0 | 0 | 0 |
| No result | 0/54 | 0/54 | 0/62 | 1/62 |
| Negative then positive | 2 | 5 | 0 | 4 |
| Positive then negative | 0 | 4 | 0 | 2 |
| Urine bloodc | ||||
| Negative | 48 | 47 | 57 | 49 |
| Trace | 3 | 1 | 1 | 3 |
| + Positive | 0 | 2 | 1 | 1 |
| ++ Positive | 0 | 1 | 3 | 4 |
| +++ Positive | 3 | 3 | 0 | 4 |
| ++++ Positive | 0 | 0 | 0 | 0 |
| No result | 0/54 | 0/54 | 0/62 | 1/62 |
| Negative then positive | 2 | 3 | 2 | 4 |
| Positive then negative | 4 | 6 | 4 | 5 |
| Urine glucosed | ||||
| Negative | 50 | 51 | 55 | 54 |
| Trace | 0 | 0 | 1 | 0 |
| + Positive | 0 | 1 | 1 | 1 |
| ++ Positive | 1 | 0 | 0 | 0 |
| +++ Positive | 1 | 2 | 1 | 2 |
| ++++ Positive | 2 | 0 | 4 | 4 |
| No result | 0/54 | 0/54 | 0/62 | 1/62 |
| Negative then positive | 1 | 1 | 3 | 2 |
| Positive then negative | 0 | 1 | 3 | 1 |
FIGURE 36.
Histological assessment of bronchial biopsies. Bronchial biopsies were collected before the first dose (pre) and 28 ± 5 days after the last dose (post). Sections were stained with H&E. Out of the 50 possible biopsies, 39 (78%) were analysed. The remaining 11 biopsies were not analysable because of poor sample quality (n = 10) or because the patient withdrew from the trial (n = 1). Sections were scored semiquantitatively using a scale from 0 to 6, except for seromucinous gland hyperplasia, which was scored as absent (0)/present (1). All data are expressed as the difference between pre and post treatment, with a positive score indicating an increase in that parameter following treatment. Dots represent scores for individual biopsies. The horizontal bar indicates the median. (a) Goblet cell hyperplasia; (b) basement membrane thickening; (c) numbers of lymphocytes; (d) numbers of neutrophils; (e) numbers of eosinophils; and (f) seromucinous gland hyperplasia.
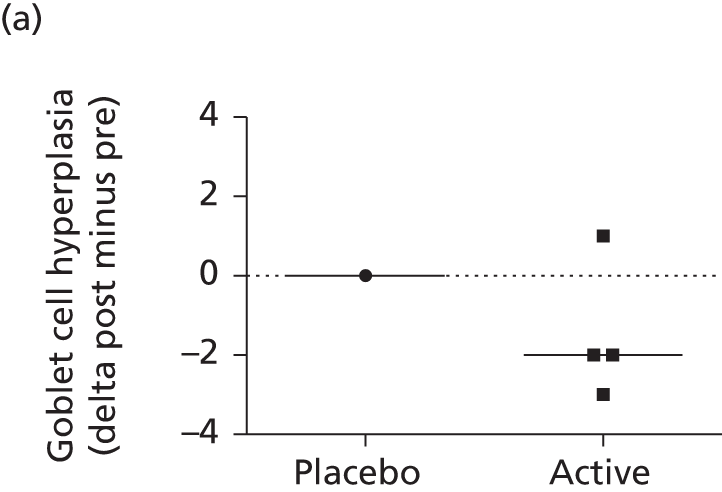

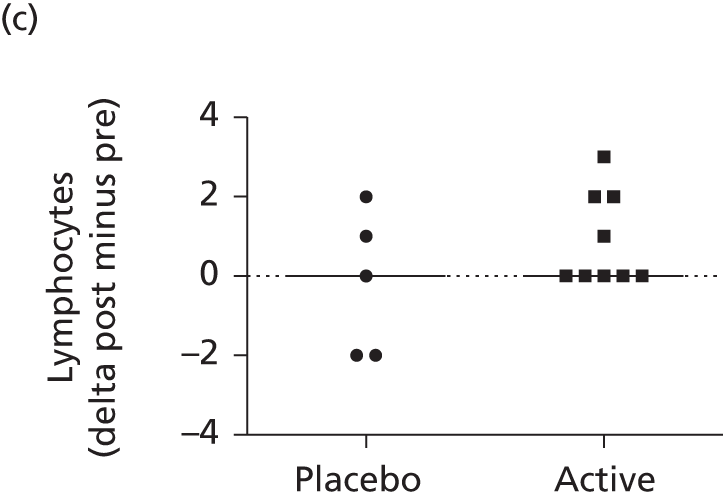
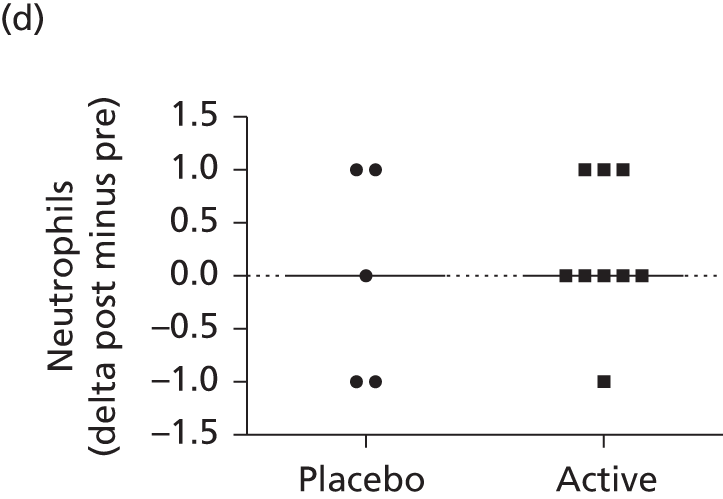
| Lipid-staining sputum cells | Placebo, meana (95% CI), n | Active treatment, meana (95% CI), n | TE, meana (95% CI) | p-value |
|---|---|---|---|---|
| Macrophages | –3.27 (–6.60 to –0.05), 24 | –2.13 (–5.45 to 1.20), 24 | 1.14 (–3.63 to 5.92) | 0.631 |
| Neutrophils | –0.23 (–1.62 to 1.16), 19 | 0.45 (–0.87 to 1.77), 21 | 0.69 (–1.27 to 2.64) | 0.480 |
| Squamous cells | –18.24 (–44.26 to 7.78), 5 | –3.49 (–21.23 to 14.25), 9 | 14.75 (–20.87 to 50.37) | 0.360 |
| Lipid-stained macrophages | Placebo | Active treatment | ||
|---|---|---|---|---|
| Pretreatment | Post-treatment | Pretreatment | Post-treatment | |
| 0 | 3 | 3 | 9 | 6 |
| 1–6 | 2 | 2 | 1 | 6 |
| 7–14 | 2 | |||
| ≥ 15 | 1 | 1 | ||
| Result | Placebo | Active | ||
|---|---|---|---|---|
| Pretreatment | Post-treatment | Pretreatment | Post-treatment | |
| Nuclei anti-nuclear antibodya | ||||
| Negative | 45 | 43 | 51 | 45 |
| + Diffuse | 1 | |||
| ++ Diffuse | 1 | 1 | ||
| +++ Diffuse | ||||
| + Speckled | 5 | 7 | 5 | 7 |
| ++ Speckled | 1 | 1 | 4 | 3 |
| +++ Speckled | ||||
| No result | 3 | 2 | 2 | 5 |
| Nucleoli anti-nuclear antibodyb | ||||
| Negative | 50 | 50 | 60 | 56 |
| + Nucleolar | 1 | 2 | 1 | |
| ++ Nucleolar | ||||
| +++ Nucleolar | ||||
| + Centromere | ||||
| ++ Centromere | ||||
| +++ Centromere | ||||
| No result | 3 | 2 | 2 | 5 |
| CFTR-specific T cells | PP population | |||
|---|---|---|---|---|
| Placebo (n = 54) | Active (n = 62) | |||
| Pre dose | Follow-up | Pre dose | Follow-up | |
| Negative | 33 | 36 | 37 | 49 |
| Positive | 4 | 8 | 1 | 2 |
| Not determined | 17 | 10 | 24 | 11 |
| T-cell conversion | 1b | 1c | ||
FIGURE 37.
Change from baseline: (a) weight; and (b) BMI.


Chapter 6 Discussion and conclusions
In this, the first trial of non-viral-based gene therapy for CF powered to detect clinically relevant changes, the primary outcome was met. Following 12 monthly nebulised doses of pGM169/G67A, there was a significant but modest (3.7%, 95% CI 0.1% to 7.3%; p < 0.05) TE in FEV1 favouring active treatment over placebo. Changes in this outcome were supported by trends in all the secondary outcomes, reaching statistical significance for FVC and gas trapping on CT scan. Underscoring the mechanism of action, there was a significant difference in lower airway CFTR-mediated chloride secretion assessed by PD. No clinically important AEs attributable to the active treatment were seen.
Although these findings are encouraging, it is important to place them in perspective. The active-treatment group demonstrated stabilisation of FEV1 rather than an improvement, while the placebo group declined. The reduction in FEV1 in the placebo group was within the range seen in other trials, but greater than has been reported from registry data. 21 We consider that there are two factors which likely explain this. First, there was a requirement for patients to (a) be clinically stable and (b) have a FEV1 > 50% predicted at screening. The enthusiasm of patients to pass screening may have led to their being in optimal health at that time and, thereafter, regressing to the mean. Second, registry data are most commonly obtained from annual assessment visits, when patients are well; in our clinic, as in most of the UK, annual assessments are deferred if patients are unwell or experiencing an exacerbation. In this trial, we captured data from all patients irrespective of whether they were stable or exacerbating; this could lead to an apparent steeper rate of decline in the placebo group than has been published from registry data sets.
We acknowledge the relatively modest TE in the primary outcome, but would caution against the bar being set too high for novel drugs in CF, based on comparison with the large response in FEV1 to ivacaftor in patients with class 3 mutations. Correction of misfolded CFTR is recognised to be a much more difficult task, as suggested by the recent ivacaftor/lumacaftor trials, with FEV1 outcomes similar to those reported here. Disease stabilisation would be a worthy addition to the CF armoury, particularly if this could be extended to disease prevention if treatment was started early on.
The response observed was heterogeneous, with obvious responders and non-responders. Encouragingly, the data suggest that an approximate doubling of TE (6.4%) could be achieved in the more severe half of patients stratified by FEV1 and reproduced across a wide range of clinically relevant assays. We consider the simplest hypothesis to relate to deposition: in patients with lower baseline FEV1 the relatively more obstructed smaller airways will lead to a larger proportion of the administered dose being deposited proximally, in the larger airways. In preparation for this trial we assessed deposition in CF patients of varying FEV1 severity using technetium-99m-labelled human serum albumin delivered via a different nebuliser system, which has a similar droplet size (3–4 µm) to the pGM169/GL67A formulation. Of the delivered dose deposited in airway generations 2–8, 2.9% (SEM 0.2%) was in those with 70–90% predicted FEV1 (n = 33), whereas approximately twice as much (6.0%, SEM 1.0%) was delivered to this region in patients with 50–70% predicted FEV1 (n = 23). These data are in keeping with the approximately twofold increase in efficacy we noted in the respective severity groups.
There may also be an influence of the increased mitotic rate of cells in more severely inflamed tissues,97 thereby decreasing the proportion of time that the nuclear membrane is intact, the latter acting as a barrier to pDNA entry to the nucleus. It will be important to verify or refute these data in a larger trial with a stratified trial entry design, powered to assess subgroups and that addresses the mechanisms of response heterogeneity.
The change in lower airway PD supports our clinical outcomes and suggests that the changes observed relate to CFTR expression. However, we have also considered the possibility that such clinical changes could be the result of a non-specific response to the active-treatment formulation. This is difficult to rule out for two reasons. First, the placebo was 0.9% saline rather than the ideal comparator of a scrambled CFTR plasmid/liposome complex. Saline was chosen in part based on ethical considerations (not wishing to expose well-controlled CF patients to first-in-man repeated pulmonary dosing of an untested product as a placebo), in part on pragmatic financial consideration, and finally a wish to compare therapy against the natural history of the disease. We know of no evidence suggesting that monthly nebulisation of 0.9% saline is deleterious to lung function, liposome alone has shown no evidence for producing physiological improvements in either non-CF (Chadwick et al. 62) or CF subjects (Alton et al. 58), and pDNA is generally associated with deleterious rather than non-specific beneficial effects. Furthermore, it was neither noted evidence for pathophysiological changes in the airways, such as inflammation, remodelling or lipid accumulation, nor changes in bacterial species, that might explain the changes. Nevertheless, it cannot be excluded that DNA–liposome complexes induce a previously undocumented augmentation of host defences, stimulate mucus clearance by non-specific mechanisms or produce bacterial killing not detected on semiquantitative routine culture. Second, it would have been reassuring additional evidence to demonstrate more robust evidence for molecular CFTR surrogates. We have repeatedly noted the limited sensitivity of assays assessing vector-specific mRNA from human samples in vivo. 55,58 The ratio of area sampled to area dosed, is extremely small, and in ovine studies we have shown that a 20-ml nebulised dose, fourfold greater than that used in the current study, represents the lower threshold for reproducible detection with this assay in airway tissue samples. 98 We have consistently noted that electrophysiological changes are more readily detectable than mRNA, supported by findings in this study. Although we saw significant chloride secretory changes in the bronchial but not in the nasal epithelium, we caution against placing undue weight on either observation. The mechanistic subgroups were opportunistically recruited to, and were not powered to, detect significant changes at the level of correction observed. Rather we would conclude that modest, inconsistent changes can be demonstrated in assays that remain suboptimal to detect low levels of CFTR function when assessed in vivo in man; improvements in these, or other assays, are needed.
Although we are encouraged by the first demonstration of a significant change in lung function associated with gene therapy in CF patients, the improvement was modest and at the lower end of the spectrum seen in clinical trials resulting in changes in patient-related care. Although we did not formally assess infective exacerbations given the relatively small numbers in our study, using antibiotic courses as a surrogate, we saw no obvious changes. It is unlikely, therefore, that, without further improvement in efficacy and response consistency, the current formulation will be suitable for immediate clinical care given the already high treatment cost in CF. Rather, we believe that a follow-on study should examine dose escalation, frequency of dosing, and the potentially attractive combination of gene therapy with a CFTR potentiator. Furthermore, our current study should encourage the introduction of more potent viral vectors into early-phase trials. We suggest the data reported here provide proof of concept that repeated administration of CFTR gene therapy can alter clinically relevant parameters, providing another step along the path of translational CF gene therapy.
Implications for practice
Although encouraging proof of principle that CFTR gene therapy can lead to changes in a clinically relevant outcome, the modest effect of these to date means that further work is necessary before this becomes a clinical therapy.
Recommendations for areas requiring future research
-
Seeking further understanding of responders/non-responders.
-
Seeking to maximise benefit by:
-
increasing dose
-
increasing dosing frequency
-
co-administration of a potentiator drug
-
-
The development of safe and efficacious viral vectors.
Acknowledgements
The TSC was chaired by Professor Ashley Woodcock, University of Manchester, UK, and comprised the following individuals:
-
Professor Pierre Lehn, University of Brest, France
-
Professor Brandon Wainwright, University of Queensland, QLD, Australia
-
Dr Janet Allen, Cystic Fibrosis Trust
-
Dr Lucy Knight, EME, National Institute of Health Research
-
Dr Vicky Knight, EME, National Institute of Health Research
-
Mrs Nikki Samsa, lay member.
The Data Safety and Monitoring Committee was chaired by Dr Colin Wallis, Great Ormond Street Hospital, London, UK, and comprised the following individuals:
-
Dr Caroline Elston, King’s College Hospital, London, UK
-
Professor George Dickson, University of London, UK
-
Dr Julie Morris (statistician), University of Manchester, UK.
We would like to thank the patients, carers and families who gave so much to this trial, those who so generously supported the programme of work that led to this trial by giving to the Cystic Fibrosis Trust; the Medicor Foundation and the Cystic Fibrosis Trust for part-funding the cost of the pDNA and lipid; George Bouliotis, Sandra Griffiths, Nayan Das, Amanda Bravery, Juan Gonzales-Maffe and Ginny Picot, from the ICTU; Jermaine Wright, Amareen Bhambra, Fabricio Ghiraldi, Stephen Man, Judith Foy and Viba Teli from the Royal Brompton Hospital Pharmacy; Peter Brown from the Royal Marsden Pharmacy; Leticia Brown and Michelle Watson-Dotchin from Imperial College London for administrative support; the EME programme team, including Lucy Knight and Dani Preedy who guided us throughout the trial; Tony Hickson and Gursharan Randhawa for help with the intellectual property and commercialisation aspects; and the DMEC (Colin Wallace, Caroline Elston, George Dickson and Julie Morris) and the TSC (Ashley Woodcock, Nikki Samsa, Brandon Wainwright, Pierre Lehn and Janet Allen) for their wise and helpful advice.
In addition, we would like to thank the teams at the referring sites who gave so generously of their time:
-
Oxford Churchill
-
Cathy Mckenny
-
Jeremy Hull
-
Lesley Bennett
-
-
Bristol
-
Nick Bell
-
Kathryn Bateman
-
-
Papworth
-
Judy Ryan
-
Jane Elliott
-
-
Birmingham
-
Maya Desai
-
Michelle Taberner
-
Edward Nash
-
Joanna Whitehouse
-
-
Great Ormond Street Hospital
-
Paul Aurora
-
Ammani Prasad
-
-
Frimley
-
Judith Duguid
-
Alexandra Higton
-
Timothy Ho
-
-
Royal London
-
Dr Siobhan Carr
-
Catherine Lambert
-
-
Southampton
-
Thomas Daniels
-
Victoria Brown
-
-
Leeds
-
Daniel Peckham
-
Amy Driffill
-
-
Kings
-
Hilary Wyatt
-
Felicity Perrin
-
Mike Waller
-
-
Glasgow
-
Gordon MacGregor
-
Steve Bicknell
-
Ewen Ross
-
Anne Devenny
-
-
Wishaw
-
Carol Dryden
-
-
Aberdeen
-
Richard Brooker
-
Graham Devereux
-
Owen Dempsey
-
-
Dundee
-
Helen Rodgers
-
Jonathon McCormick
-
-
Newcastle
-
Steven Bourke
-
David Spencer
-
Chris O’Brien.
-
The research was supported by the NIHR Respiratory Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London and the Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London. The UK CFGTC would not have come together, and this trial would not have happened, without the vision and leadership of Rosie Barnes OBE during her tenure as Chief Executive of the Cystic Fibrosis Trust.
Contributions of authors
Eric WFW Alton, A Christopher Boyd, Steve Cunningham, Jane C Davies, Duncan M Geddes, Deborah R Gill, Andrew P Greening, Uta Griesenbach, Tracy E Higgins, Stephen C Hyde, J Alastair Innes, Gordon D Murray and David J Porteous conceived, designed and analysed the overall study.
David K Armstrong, Katie J Bayfield, Diana Bilton, Paula Carvelli, Gwyneth Davies, Maria H Dewar, Natalie S Dwyer, Hala I Elgmati, Rosanna F Featherstone, Jemyr Gavino, James SR Gibson, David M Hansell, Katharine Harman, Samantha L Hodges, Joseph Jacob, Brian F Keogh, Michael McGovern, Emma K Punch, Alexandra L Quittner, Clare J Saunders, Sarah Sheard, Nicholas J Simmonds, Stephen N Smith, Najwa Soussi, Emma J Spearing, Rosa P Ureta and Michael D Waller assessed patient outcomes and/or undertook and analysed individual in vivo assays.
Deborah Ashby and Gordon D Murray designed and co-ordinated data collection and statistical analysis.
Emily V Bloomfield, MD, and Samia Soussi co-ordinated and undertook the administration of the trial.
Ruaridh Buchan, Nancy Jones, Paul Lloyd-Evans, Gina Rivellini and Keith Smith oversaw trial drug receipt, preparation and dispensing.
June Brand, Roberto Calcedo, Mario Chan, Heather E Davidson, Ann Doherty, Jackie Donovan, Sabrina Gea-Sorli, Laura Hyndman, Maria P Limberis, Alan W Maclean, Michelle C Manvell, Dominique McCormick, Cuixiang Meng, M Angeles Montero, Hazel Milligan, Laura J Moyce, Andrew G Nicholson, Tina Osadolor, Javier Parra-Leiton, Ian A Pringle, Kamila M Pytel, Barbara J Stevenson, Stephanie G Sumner-Jones, Minna Turkkila, Marguerite Y Wasowicz and James M Wilson designed, undertook and analysed in vitro assays.
Seng H Cheng, Ronald K Scheule and Paul Wolstenholme-Hogg co-ordinated the production of lipid 67A.
David S Collie, Lee A Davies and Gerry McLachlan designed, undertook and analysed trial drug delivery studies.
Publications
Alton EW, Boyd AC, Cheng SH, Cunningham S, Davies JC, Gill DR, et al. A randomised, double-blind, placebo-controlled phase IIB clinical trial of repeated application of gene therapy in patients with cystic fibrosis. Thorax 2013;68:1075–7.
Alton EW, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med 2015;3:684–91.
Data sharing statement
Data are available from Professor Eric Alton.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health.
References
- Davies JC, Alton EW, Bush A. Cystic fibrosis. BMJ 2007;335:1255-9. http://dx.doi.org/10.1136/bmj.39391.713229.AD.
- Rabin HR, Surette MG. The cystic fibrosis airway microbiome. Curr Opin Pulm Med 2012;18:622-7. http://dx.doi.org/10.1097/MCP.0b013e328358d49a.
- Knowlton RG, Cohen-Haguenauer O, Van CN, Frezal J, Brown VA, Barker D, et al. A polymorphic DNA marker linked to cystic fibrosis is located on chromosome 7. Nature 1985;318:380-2. http://dx.doi.org/10.1038/318380a0.
- Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989;245:1059-65. http://dx.doi.org/10.1126/science.2772657.
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989;245:1066-73. http://dx.doi.org/10.1126/science.2475911.
- Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 1998;95:1005-15. http://dx.doi.org/10.1016/S0092-8674(00)81724-9.
- Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MH, Ramachandran S, Moninger TO, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012;487:109-13. http://dx.doi.org/10.1038/nature11130.
- Southern KW, Merelle MM, Dankert-Roelse JE, Nagelkerke AD. Newborn screening for cystic fibrosis. Cochrane Database Syst Rev 2009;1. http://dx.doi.org/10.1002/14651858.cd001402.pub2.
- Collie JT, Massie RJ, Jones OA, LeGrys VA, Greaves RF. Sixty-five years since the New York heat wave: advances in sweat testing for cystic fibrosis. Pediatr Pulmonol 2014;49:106-17. http://dx.doi.org/10.1002/ppul.22945.
- De BK, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros 2014;13:674-80. http://dx.doi.org/10.1016/j.jcf.2014.09.005.
- Sanders DB, Bittner RC, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med 2010;182:627-32. http://dx.doi.org/10.1164/rccm.200909-1421OC.
- Stern M, Bertrand DP, Bignamini E, Corey M, Dembski B, Goss CH, et al. European Cystic Fibrosis Society standards of care: quality management in cystic fibrosis. J Cyst Fibros 2014;13:43-59. http://dx.doi.org/10.1016/j.jcf.2014.03.011.
- Conway S, Balfour-Lynn IM, De Rijcke K, Drevinek P, Foweraker J, Havermans T, et al. European Cystic Fibrosis Society standards of care: framework for the Cystic Fibrosis Centre. J Cyst Fibros 2014;13:3-22. http://dx.doi.org/10.1016/j.jcf.2014.03.009.
- Smyth AR, Bell SC, Bojcin S, Bryon M, Duff A, Flume P, et al. European Cystic Fibrosis Society standards of care: best practice guidelines. J Cyst Fibros 2014;13:23-42. http://dx.doi.org/10.1016/j.jcf.2014.03.010.
- Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med 2006;354:241-50. http://dx.doi.org/10.1056/NEJMoa043891.
- Jones AP, Wallis C. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev 2010;3. http://dx.doi.org/10.1002/14651858.cd001127.pub2.
- Ciofu O, Hansen CR, Hoiby N. Respiratory bacterial infections in cystic fibrosis. Curr Opin Pulm Med 2013;3:251-8. http://dx.doi.org/10.1097/MCP.0b013e32835f1afc.
- Quon BS, Goss CH, Ramsey BW. Inhaled antibiotics for lower airway infections. Ann Am Thorac Soc 2014;11:425-34. http://dx.doi.org/10.1513/AnnalsATS.201311-395FR.
- Weiner JR, Toy EL, Sacco P, Duh MS. Costs, quality of life and treatment compliance associated with antibiotic therapies in patients with cystic fibrosis: a review of the literature. Expert Opin Pharmacother 2008;9:751-66. http://dx.doi.org/10.1517/14656566.9.5.751.
- Corris PA. Lung transplantation for cystic fibrosis and bronchiectasis. Semin Respir Crit Care Med 2013;34:297-304. http://dx.doi.org/10.1055/s-0033-1348469.
- Cystic Fibrosis Trust . UK Cystic Fibrosis Registry Annual Data Report 2013–July 2014 n.d. www.cysticfibrosis.org.uk/media/598466/annual-data-report-2013-jul14.pdf (accessed 15 December 2015).
- Boyle MP, De BK. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med 2013;1:158-63. http://dx.doi.org/10.1016/S2213-2600(12)70057-7.
- Wilschanski M, Miller LL, Shoseyov D, Blau H, Rivlin J, Aviram M, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J 2011;38:59-6. http://dx.doi.org/10.1183/09031936.00120910.
- Wilschanski M, Kerem E. New drugs for cystic fibrosis. Expert Opin Investig Drugs 2011;20:1285-92. http://dx.doi.org/10.1517/13543784.2011.600304.
- Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med 1996;2:467-9. http://dx.doi.org/10.1038/nm0496-467.
- Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med 1997;3:1280-4. http://dx.doi.org/10.1038/nm1197-1280.
- Wilschanski M, Famini C, Blau H, Rivlin J, Augarten A, Avital A, et al. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Respir Crit Care Med 2000;161:860-5. http://dx.doi.org/10.1164/ajrccm.161.3.9904116.
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007;447:87-91. http://dx.doi.org/10.1038/nature05756.
- Van GF, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 2006;290:L1117-30. http://dx.doi.org/10.1152/ajplung.00169.2005.
- Van GF, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009;106:18825-30. http://dx.doi.org/10.1073/pnas.0904709106.
- Pasyk S, Li C, Ramjeesingh M, Bear CE. Direct interaction of a small-molecule modulator with G551D-CFTR, a cystic fibrosis-causing mutation associated with severe disease. Biochem J 2009;418:185-90. http://dx.doi.org/10.1042/BJ20081424.
- Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem 1996;271:635-8. http://dx.doi.org/10.1074/jbc.271.2.635.
- Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1996;1:117-25.
- Van GF, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011;108:18843-8. http://dx.doi.org/10.1073/pnas.1105787108.
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012;67:12-8. http://dx.doi.org/10.1136/thoraxjnl-2011-200393.
- Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 2015;373:220-31. http://dx.doi.org/10.1056/NEJMoa1409547.
- Boucher RC. Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med 2007;261:5-16. http://dx.doi.org/10.1111/j.1365-2796.2006.01744.x.
- Trapnell BC, Chu CS, Paakko PK, Banks TC, Yoshimura K, Ferrans VJ, et al. Expression of the cystic fibrosis transmembrane conductance regulator gene in the respiratory tract of normal individuals and individuals with cystic fibrosis. Proc Natl Acad Sci U S A 1991;88:6565-9. http://dx.doi.org/10.1073/pnas.88.15.6565.
- Zabner J, Couture LA, Gregory RJ, Graham SM, Smith AE, Welsh MJ. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 1993;75:207-16. http://dx.doi.org/10.1016/0092-8674(93)80063-K.
- Hay JG, McElvaney NG, Herena J, Crystal RG. Modification of nasal epithelial potential differences of individuals with cystic fibrosis consequent to local administration of a normal CFTR cDNA adenovirus gene transfer vector. Hum Gene Ther 1995;6:1487-96. http://dx.doi.org/10.1089/hum.1995.6.11-1487.
- Grubb BR, Pickles RJ, Ye H, Yankaskas JR, Vick RN, Engelhardt JF, et al. Inefficient gene transfer by adenovirus vector to cystic fibrosis airway epithelia of mice and humans. Nature 1994;371:802-6. http://dx.doi.org/10.1038/371802a0.
- Knowles MR, Hohneker KW, Zhou Z, Olsen JC, Noah TL, Hu PC, et al. A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N Engl J Med 1995;333:823-31. http://dx.doi.org/10.1056/NEJM199509283331302.
- Joseph PM, O’Sullivan BP, Lapey A, Dorkin H, Oren J, Balfour R, et al. Aerosol and lobar administration of a recombinant adenovirus to individuals with cystic fibrosis. I. Methods, safety, and clinical implications. Hum Gene Ther 2001;12:1369-82. http://dx.doi.org/10.1089/104303401750298535.
- Perricone MA, Morris JE, Pavelka K, Plog MS, O’Sullivan BP, Joseph PM, et al. Aerosol and lobar administration of a recombinant adenovirus to individuals with cystic fibrosis. II. Transfection efficiency in airway epithelium. Hum Gene Ther 2001;12:1383-94. http://dx.doi.org/10.1089/104303401750298544.
- Harvey BG, Leopold PL, Hackett NR, Grasso TM, Williams PM, Tucker AL, et al. Airway epithelial CFTR mRNA expression in cystic fibrosis patients after repetitive administration of a recombinant adenovirus. J Clin Invest 1999;104:1245-55. http://dx.doi.org/10.1172/JCI7935.
- Wagner JA, Reynolds T, Moran ML, Moss RB, Wine JJ, Flotte TR, et al. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet 1998;351:1702-3. http://dx.doi.org/10.1016/S0140-6736(05)77740-0.
- Wagner JA, Messner AH, Moran ML, Daifuku R, Kouyama K, Desch JK, et al. Safety and biological efficacy of an adeno-associated virus vector-cystic fibrosis transmembrane regulator (AAV-CFTR) in the cystic fibrosis maxillary sinus. Laryngoscope 1999;109:266-74. http://dx.doi.org/10.1097/00005537-199902000-00017.
- Aitken ML, Moss RB, Waltz DA, Dovey ME, Tonelli MR, McNamara SC, et al. A phase I study of aerosolized administration of tgAAVCF to cystic fibrosis subjects with mild lung disease. Hum Gene Ther 2001;12:1907-16. http://dx.doi.org/10.1089/104303401753153956.
- Wagner JA, Nepomuceno IB, Messner AH, Moran ML, Batson EP, Dimiceli S, et al. A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum Gene Ther 2002;13:1349-59. http://dx.doi.org/10.1089/104303402760128577.
- Caplen NJ, Alton EW, Middleton PG, Dorin JR, Stevenson BJ, Gao X, et al. Liposome-mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Nat Med 1995;1:39-46. http://dx.doi.org/10.1038/nm0195-39.
- Gill DR, Southern KW, Mofford KA, Seddon T, Huang L, Sorgi F, et al. A placebo-controlled study of liposome-mediated gene transfer to the nasal epithelium of patients with cystic fibrosis. Gene Ther 1997;4:199-20. http://dx.doi.org/10.1038/sj.gt.3300391.
- Porteous DJ, Dorin JR, McLachlan G, vidson-Smith H, Davidson H, Stevenson BJ, et al. Evidence for safety and efficacy of DOTAP cationic liposome mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Gene Ther 1997;4:210-18. http://dx.doi.org/10.1038/sj.gt.3300390.
- Noone PG, Hohneker KW, Zhou Z, Johnson LG, Foy C, Gipson C, et al. Safety and biological efficacy of a lipid–CFTR complex for gene transfer in the nasal epithelium of adult patients with cystic fibrosis. Mol Ther 2000;1:105-14. http://dx.doi.org/10.1006/mthe.1999.0009.
- Konstan MW, Davis PB, Wagener JS, Hilliard KA, Stern RC, Milgram LJ, et al. Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum Gene Ther 2004;15:1255-69. http://dx.doi.org/10.1089/hum.2004.15.1255.
- Hyde SC, Southern KW, Gileadi U, Fitzjohn EM, Mofford KA, Waddell BE, et al. Repeat administration of DNA/liposomes to the nasal epithelium of patients with cystic fibrosis. Gene Ther 2000;7:1156-65. http://dx.doi.org/10.1038/sj.gt.3301212.
- Zabner J, Cheng SH, Meeker D, Launspach J, Balfour R, Perricone MA, et al. Comparison of DNA–lipid complexes and DNA alone for gene transfer to cystic fibrosis airway epithelia in vivo. J Clin Invest 1997;100:1529-37. http://dx.doi.org/10.1172/JCI119676.
- Ruiz FE, Clancy JP, Perricone MA, Bebok Z, Hong JS, Cheng SH, et al. A clinical inflammatory syndrome attributable to aerosolized lipid-DNA administration in cystic fibrosis. Hum Gene Ther n.d.;12:751-61. http://dx.doi.org/10.1089/104303401750148667.
- Alton EW, Stern M, Farley R, Jaffe A, Chadwick SL, Phillips J, et al. Cationic lipid-mediated CFTR gene transfer to the lungs and nose of patients with cystic fibrosis: a double-blind placebo-controlled trial. Lancet 1999;353:947-54. http://dx.doi.org/10.1016/S0140-6736(98)06532-5.
- Lee ER, Marshall J, Siegel CS, Jiang C, Yew NS, Nichols MR, et al. Detailed analysis of structures and formulations of cationic lipids for efficient gene transfer to the lung. Hum Gene Ther 1996;7:1701-17. http://dx.doi.org/10.1089/hum.1996.7.14-1701.
- Scheule RK, St George JA, Bagley RG, Marshall J, Kaplan JM, Akita GY, et al. Basis of pulmonary toxicity associated with cationic lipid-mediated gene transfer to the mammalian lung. Hum Gene Ther 1997;8:689-707. http://dx.doi.org/10.1089/hum.1997.8.6-689.
- Eastman SJ, Lukason MJ, Tousignant JD, Murray H, Lane MD, St George JA, et al. A concentrated and stable aerosol formulation of cationic lipid: DNA complexes giving high-level gene expression in mouse lung. Hum Gene Ther 1997;8:765-73. http://dx.doi.org/10.1089/hum.1997.8.6-765.
- Chadwick SL, Kingston HD, Stern M, Cook RM, O’Connor BJ, Lukasson M, et al. Safety of a single aerosol administration of escalating doses of the cationic lipid GL-67/DOPE/DMPE-PEG5000 formulation to the lungs of normal volunteers. Gene Ther 1997;4:937-42. http://dx.doi.org/10.1038/sj.gt.3300481.
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000;408:740-5. http://dx.doi.org/10.1038/35047123.
- Yew NS, Wang KX, Przybylska M, Bagley RG, Stedman M, Marshall J, et al. Contribution of plasmid DNA to inflammation in the lung after administration of cationic lipid: pDNA complexes. Hum Gene Ther 1999;10:223-34. http://dx.doi.org/10.1089/10430349950019011.
- Horsley AR, Davies JC, Gray RD, Macleod KA, Donovan J, Aziz ZA, et al. Changes in physiological, functional and structural markers of cystic fibrosis lung disease with treatment of a pulmonary exacerbation. Thorax 2013;68:532-9. http://dx.doi.org/10.1136/thoraxjnl-2012-202538.
- Alton EW, Boyd AC, Porteous DJ, Davies G, Davies JC, Griesenbach U, et al. A Phase I/IIa safety and efficacy study of nebulized liposome-mediated gene therapy for cystic fibrosis supports a multidose trial. Am J Respir Crit Care Med 2015;192. http://dx.doi.org/10.1164/rccm.201506-1193LE.
- Alton EW, Boyd AC, Cheng SH, Davies JC, Davies LA, Dayan A, et al. Toxicology study assessing efficacy and safety of repeated administration of lipid/DNA complexes to mouse lung. Gene Ther 2014;21:89-95. http://dx.doi.org/10.1038/gt.2013.61.
- Eastman SJ, Tousignant JD, Lukason MJ, Murray H, Siegel CS, Constantino P, et al. Optimization of formulations and conditions for the aerosol delivery of functional cationic lipid:DNA complexes. Hum Gene Ther 1997;8:313-22. http://dx.doi.org/10.1089/hum.1997.8.3-313.
- Davies LA, Nunez-Alonso GA, McLachlan G, Hyde SC, Gill DR. Aerosol delivery of DNA/liposomes to the lung for cystic fibrosis gene therapy. Hum Gene Ther Clin Dev 2014;25:97-107. http://dx.doi.org/10.1089/humc.2014.019.
- Derand R, Bulteau-Pignoux L, Becq F. Comparative pharmacology of the activity of wild-type and G551D mutated CFTR chloride channel: effect of the benzimidazolone derivative NS004. J Membr Biol 2003;194:109-17. http://dx.doi.org/10.1007/s00232-003-2030-z.
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135:1610-18. http://dx.doi.org/10.1378/chest.08-1190.
- Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 1994;331:637-42. http://dx.doi.org/10.1056/NEJM199409083311003.
- Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999;340:23-30. http://dx.doi.org/10.1056/NEJM199901073400104.
- Saiman L, Mayer-Hamblett N, Campbell P, Marshall BC. Heterogeneity of treatment response to azithromycin in patients with cystic fibrosis. Am J Respir Crit Care Med 2005;172:1008-12. http://dx.doi.org/10.1164/rccm.200502-218OC.
- McCoy KS, Quittner AL, Oermann CM, Gibson RL, Retsch-Bogart GZ, Montgomery AB. Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. Am J Respir Crit Care Med 2008;178:921-8. http://dx.doi.org/10.1164/rccm.200712-1804OC.
- Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Eur Respir J 2005;26:319-38. http://dx.doi.org/10.1183/09031936.05.00034805.
- Stanojevic S, Wade A, Stocks J. Reference values for lung function: past, present and future. Eur Respir J 2010;36:12-9. http://dx.doi.org/10.1183/09031936.00143209.
- Robinson PD, Latzin P, Verbanck S, Hall GL, Horsley A, Gappa M, et al. Consensus statement for inert gas washout measurement using multiple- and single-breath tests. Eur Respir J 2013;41:507-22. http://dx.doi.org/10.1183/09031936.00069712.
- Davies J, Sheridan H, Bell N, Cunningham S, Davis SD, Elborn JS, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir Med 2013;1:630-8. http://dx.doi.org/10.1016/S2213-2600(13)70182-6.
- Johannsen DL, Calabro MA, Stewart J, Franke W, Rood JC, Welk GJ. Accuracy of armband monitors for measuring daily energy expenditure in healthy adults. Med Sci Sports Exerc 2010;42:2134-40. http://dx.doi.org/10.1249/MSS.0b013e3181e0b3ff.
- Irving SJ, Ives A, Davies G, Donovan J, Edey AJ, Gill SS, et al. Lung clearance index and high-resolution computed tomography scores in primary ciliary dyskinesia. Am J Respir Crit Care Med 2013;188:545-9. http://dx.doi.org/10.1164/rccm.201304-0800OC.
- Knowles M, Gatzy J, Boucher R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med 1981;305:1489-95. http://dx.doi.org/10.1056/NEJM198112173052502.
- Middleton PG, Geddes DM, Alton EW. Protocols for in vivo measurement of the ion transport defects in cystic fibrosis nasal epithelium. Eur Respir J 1994;7:2050-6.
- Davies JC, Alton EW. Gene therapy for cystic fibrosis. Proc Am Thorac Soc 2010;7:408-14. http://dx.doi.org/10.1513/pats.201004-029AW.
- Flotte TR, Zeitlin PL, Reynolds TC, Heald AE, Pedersen P, Beck S, et al. Phase I trial of intranasal and endobronchial administration of a recombinant adeno-associated virus serotype 2 (rAAV2)-CFTR vector in adult cystic fibrosis patients: a two-part clinical study. Hum Gene Ther 2003;14:1079-88. http://dx.doi.org/10.1089/104303403322124792.
- Zabner J, Ramsey BW, Meeker DP, Aitken ML, Balfour RP, Gibson RL, et al. Repeat administration of an adenovirus vector encoding cystic fibrosis transmembrane conductance regulator to the nasal epithelium of patients with cystic fibrosis. J Clin Invest 1996;97:1504-11. http://dx.doi.org/10.1172/JCI118573.
- Johnson LG, Olsen JC, Sarkadi B, Moore KL, Swanstrom R, Boucher RC. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet 1992;2:21-5. http://dx.doi.org/10.1038/ng0992-21.
- Johnson LG, Boyles SE, Wilson J, Boucher RC. Normalization of raised sodium absorption and raised calcium-mediated chloride secretion by adenovirus-mediated expression of cystic fibrosis transmembrane conductance regulator in primary human cystic fibrosis airway epithelial cells. J Clin Invest 1995;95:1377-82. http://dx.doi.org/10.1172/JCI117789.
- Goldman MJ, Yang Y, Wilson JM. Gene therapy in a xenograft model of cystic fibrosis lung corrects chloride transport more effectively than the sodium defect. Nat Genet 1995;9:126-31. http://dx.doi.org/10.1038/ng0295-126.
- Rowe SM, Clancy JP, Wilschanski M. Nasal potential difference measurements to assess CFTR ion channel activity. Methods Mol Biol 2011;741:69-86. http://dx.doi.org/10.1007/978-1-61779-117-8_6.
- Davies JC, Davies M, McShane D, Smith S, Chadwick S, Jaffe A, et al. Potential difference measurements in the lower airway of children with and without cystic fibrosis. Am J Respir Crit Care Med 2005;171:1015-19. http://dx.doi.org/10.1164/rccm.200408-1116OC.
- Davis D, Zhang A, Etienne C, Huang I, Malit M. Principles of Curve Fitting for Multiplex Sandwich Immunoassays. n.d.
- Calcedo R, Griesenbach U, Dorgan DJ, Soussi S, Boyd AC, Davies JC, et al. Self-reactive CFTR T cells in humans: implications for gene therapy. Hum Gene Ther Clin Dev 2013;24:108-15. http://dx.doi.org/10.1089/humc.2012.249.
- Lian X, Yan C, Yang L, Xu Y, Du H. Lysosomal acid lipase deficiency causes respiratory inflammation and destruction in the lung. Am J Physiol Lung Cell Mol Physiol 2004;286:L801-7. http://dx.doi.org/10.1152/ajplung.00335.2003.
- Rose AC, Goddard CA, Colledge WH, Cheng SH, Gill DR, Hyde SC. Optimisation of real-time quantitative RT-PCR for the evaluation of non-viral mediated gene transfer to the airways. Gene Ther 2002;9:1312-20. http://dx.doi.org/10.1038/sj.gt.3301792.
- INVOLVE . Example 5: A Randomised Double-Blind Placebo Controlled Phase 2B Clinical Trial of Repeated Application of Gene Therapy in Patients With Cystic Fibrosis 2013. http://www.invo.org.uk/wp-content/uploads/2013/10/Example-5-publicinvolvement-in-funding-apps-2013.pdf.
- Leigh MW, Kylander JE, Yankaskas JR, Boucher RC. Cell proliferation in bronchial epithelium and submucosal glands of cystic fibrosis patients. Am J Respir Cell Mol Biol 1995;12:605-12. http://dx.doi.org/10.1165/ajrcmb.12.6.7766425.
- Alton EW, Baker A, Baker E, Boyd AC, Cheng SC, Coles RL, et al. The safety profile of a cationic lipid-mediated cystic fibrosist gene transfer agent following repeated monthly aerosol administration to sheep. Biomaterials 2013;34:10267-77. http://dx.doi.org/10.1016/j.biomaterials.2013.09.023.
Appendix 1 Patient information sheets
Adult patient information sheet
Parent information sheet
Paediatric information sheet
Appendix 2 Dose preparation sheet
Appendix 3 Blinding protocol
Appendix 4 Cystic Fibrosis Questionnaire – Revised quality-of-life questionnaire
© 2000, Quittner, Buu, Watrous and Davis. Revised 2002. CFQ Teen/Adult, English Version 1.0© 2005, Bryon and Stramik. CFQ-UK Teen/Adult, UK-English Language Version 1.0
Appendix 5 Statistical Analysis Plan
List of abbreviations
- 5PL
- five-parameter logistic
- AAV
- adeno-associated virus
- AE
- adverse event
- ANCOVA
- analysis of covariance
- ATS
- American Thoracic Society
- BAL
- bronchoalveolar lavage
- BMI
- body mass index
- BP
- blood pressure
- BSA
- bovine serum albumin
- cAMP
- cyclic adenosine monophosphate
- cDNA
- complementary deoxyribonucleic acid
- CF
- cystic fibrosis
- CFF TDN
- Cystic Fibrosis Foundation Therapeutic Development Network
- CFQ-R
- Cystic Fibrosis Questionnaire – Revised
- CFTR
- cystic fibrosis transmembrane conductance regulator (gene)
- CI
- confidence interval
- CMV
- cytomegalovirus
- CON
- anaesthetic control
- CONSORT
- Consolidated Standards of Reporting Trials
- CpG
- cytosine–phosphate–guanidine
- CRO
- clinical research organisation
- CRP
- C-reactive protein
- CT
- computed tomography
- DC-Chol
- 3beta-[N-(N′,N′-dimethylaminoethane)carbamoyl]cholesterol
- DMEC
- Data Monitoring and Ethics Committee
- DMPE-PEG5000
- 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethylene glycol 5000)] (ammonium salt)
- DNA
- deoxyribonucleic acid
- DNase
- deoxyribonuclease
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- dsDNA
- double-stranded DNA
- ELISA
- enzyme-linked immunosorbent assay
- EME
- Efficacy and Mechanism Evaluation
- ERS
- European Respiratory Society
- FEV1
- forced expiratory volume in the first second
- FPF
- fine particle fraction
- FVC
- forced vital capacity
- GL67
- cholest-5-en-3-ol (3β)-,3-[(3-aminopropyl)[4-[(3-aminopropyl)amino]butyl]carbamate]
- GMP
- good manufacturing practice
- GT
- gene therapy treated
- GTAC
- Gene Therapy Advisory Committee
- H&E
- haematoxylin and eosin
- ICTU
- Imperial College Trials Unit
- IL-8
- interleukin 8
- IRT
- immunoreactive trypsinogen
- ITT
- intention to treat
- KCO
- transfer coefficient
- KCOc
- corrected transfer coefficient
- LCI
- lung clearance index
- LOQ
- limit of quantification
- MBW
- multiple breath washout
- MEF
- mid-expiratory flow
- MET
- metabolic equivalent
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MMAD
- median mass aerodynamic diameter
- mRNA
- messenger ribonucleic acid
- MRSA
- meticillin-resistant Staphylococcus aureus
- NIHR
- National Institute for Health Research
- ORO
- Oil Red O dye
- PBMC
- peripheral blood mononuclear cell
- PBNQ
- positive but not quantifiable
- PBS
- phosphate-buffered saline
- PD
- potential difference
- pDNA
- plasmid DNA
- PP
- per protocol
- qPCR
- quantitative polymerase chain reaction
- qRT-PCR
- quantitative reverse transcription PCR
- rhDNase
- recombinant human DNase
- RNA
- ribonucleic acid
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SEM
- standard error of the mean
- SF6
- sulphur hexafluoride
- soCFTR2
- CpG-free version of the CFTR coding sequence
- SOP
- standard operating procedure
- TE
- treatment effect
- TLCO
- transfer factor of the lung for carbon monoxide
- TLCOc
- corrected transfer factor of the lung for carbon monoxide
- TSC
- Trial Steering Committee
- UK CFGTC
- UK Cystic Fibrosis Gene Therapy Consortium
- VA
- alveolar volume
- VO2max
- maximal oxygen uptake