
Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 10/60/27. The contractual start date was in August 2011. The final report began editorial review in September 2015 and was accepted for publication in February 2016. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Sebastian L Johnston has received institutional funding for a clinical trial and consultant compensation from Centocor, Sanofi Pasteur, GlaxoSmithKline and Synairgen; institutional funding for a research grant and consultant compensation from Chiesi, Boehringer Ingelheim and Novartis; and consultant compensation from Grünenthal. He is also a shareholder in Synairgen and has nine relevant licensed patents and one relevant patent pending. Christopher Brightling has received institutional grants and consultant compensation from GlaxoSmithKline, AstraZeneca, Boehringher Ingleheim, Novartis, Chiesi and Roche/Genentech. Dave Singh has received grants and personal fees from Almirall, AstraZeneca, Boehringher Ingleheim, Chiesi, GlaxoSmithKline, Glenmark, Johnson & Johnson, Merck, NAPP, Novartis, Pfizer, Takeda, Teva, Theravance and Verona, and personal fees from Genentech and Skyepharma. Bernard Higgins has taken the role of local principal investigator for multicentre studies funded by Novartis and Roche. Christopher Corrigan has received a grant and personal fees for attendance at scientific conferences and payments for lectures from Allergy Therapeutics; a grant and personal fees from Novartis for research collaborations and consultancy not connected with the current research; a grant for attendance at scientific conferences from Stallergenes, Boehringer Ingelheim and Diagenics; and personal fees from AstraZeneca for speaking at conferences. Rekha Chaudhuri reports a grant and personal fees for attendance at scientific conferences and advisory board meetings: Novartis Pharmaceuticals, Astra-Zeneca, Teva and GlaxoSmithKline.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2016. This work was produced by Johnston et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Background
Importance of asthma exacerbations
Asthma is the most prevalent respiratory disease; in developed countries it is diagnosed in 5–10% of adults and 10–15% of children, with around 30% of children reporting wheeze in the last year. 1 Most asthma morbidity and mortality results from acute exacerbations: 5–10% of asthmatics have been hospitalised with an exacerbation and an estimated ≈25,000 of Europeans die unnecessarily of asthma each year. Exacerbations also account for ≈50% of total expenditure on asthma care. 2 More than half of asthma patients report having an exacerbation in the last year, with more than one-third of children and more than one-quarter of adults requiring urgent medical care visits as a result. 3
Aetiology of asthma exacerbations
Respiratory viral infections are the major cause of asthma exacerbations in children (80–85%)4,5 and adults (75–80%). 6–8 However, non-viral respiratory pathogens such as Mycoplasma pneumoniae and Chlamydophila pneumoniae have also been associated with wheezing episodes and asthma exacerbations in both adults and children. 9–13 Interestingly, in two of these studies virus detection rates were ≈80%,9,11 whereas serological positivity for atypical bacterial infection/reactivation can be as high as 40–60%,9,13 indicating that viral and atypical bacterial infections probably interact in increasing the risk of asthma exacerbations.
There is little published evidence that standard bacterial infections are important in the aetiology of asthma exacerbations;14 however, patients with asthma have increased susceptibility to respiratory bacterial infections,15–17 increased carriage of pathogenic respiratory bacteria identified by culture18 and molecular techniques,19 and impaired interferon responses to bacterial polysaccharides. 20 In addition, viral infection impairs innate responses that are important in antibacterial immunity21 and increases bacterial adherence to bronchial epithelial cells. 22 There is therefore good evidence that bacterial respiratory infections are both more common and more severe in asthma and that viral infection can increase susceptibility to bacterial infection.
A recent study of 361 children with > 800 stable and exacerbation airway samples collected during the first 3 years of life and analysed for standard bacteria and respiratory viruses has confirmed that acute wheezing episodes were associated with both bacterial infection [odds ratio 2.9, 95% confidence interval (CI) 1.9 to 4.3; p < 0.001] and viral infection (odds ratio 2.8, 95% CI 1.7 to 4.4; p < 0.01). 23 We therefore hypothesise that standard bacterial infections are also likely to be important in the aetiology of asthma exacerbations in adults and aimed to investigate this in this study.
Treatment of asthma exacerbations
When asthma exacerbations occur, treatment options are limited to bronchodilators and (cortico)steroids. Beyond the addition of magnesium in severe exacerbations, treatments have developed very little in the last 50 years. Current therapeutic strategies are of limited efficacy and the development of new approaches addressing the aetiological agents causing the exacerbations is urgently needed. Current asthma guidelines recommend specifically that antibiotic therapy should not be administered routinely in asthma exacerbations. 24
New approaches to treatment of asthma exacerbations
If atypical bacteria are causal or contributory factors in asthma exacerbations, then treatment with antibiotics with activity against mycoplasma and chlamydia species would be expected to be beneficial in asthma exacerbations. Adults with acute exacerbations of asthma and treated with telithromycin (a ketolide antibiotic closely related to macrolides, with both classes being highly active against M. and C. pneumoniae) as a supplement to standard care showed a statistically significantly greater reduction in asthma symptoms (p < 0.005) and improvement in lung function (p = 0.001) and faster recovery (p = 0.03) than those treated with placebo. 13 The magnitude of the treatment effect was also highly clinically significant, with the improvement in symptoms resulting from telithromycin treatment being approximately 50% greater than with standard therapy (plus placebo). Improvement in lung function was almost 100% greater and, importantly, recovery time to a 50% improvement in clinical symptoms 3 days faster in those receiving active treatment. This treatment therefore had a clear therapeutic effect; however, this study requires confirmation in a second similar study before revision of guidelines can be considered. Ideally, confirmation would be with a further study with telithromycin; however, issues with toxicity have limited the use of telithromycin to severe life-threatening infections.
The macrolide antibiotic azithromycin is a safe and well-tolerated alternative that has been used for many years in the treatment of respiratory disease but has thus far not been studied in acute exacerbations of asthma. We therefore hypothesised that treatment with azithromycin might be of benefit in acute asthma exacerbations. The AZALEA (AZithromycin Against pLacebo for acute Exacerbations of Asthma) study investigated the effectiveness of azithromycin as a supplement to standard care for adult patients with acute exacerbations of asthma, following as closely as possible the design of the telithromycin study, with the aim of providing confirmation or otherwise of those results.
Mechanisms of activity of macrolide/ketolide antibiotics in the treatment of asthma exacerbations
Macrolide/ketolide antibiotics might have therapeutic effects/benefits in treating asthma exacerbations through treatment of either standard or atypical bacterial infections or both. In addition, both macrolide and ketolide antibiotics have anti-inflammatory properties that are independent of their antibacterial activity, which may be beneficial in reducing airway inflammation, and is known to be important in the pathogenesis of asthma exacerbations. 7,25 In addition to these three possible mechanisms of action, we also believe that antiviral activity is a fourth possible mechanism.
We have previously reported that impaired type I and type III interferon production by virus-infected bronchial epithelial cells and macrophages is important in the pathogenesis of asthma exacerbations. 20,26 We have also recently shown that azithromycin, but not erythromycin or telithromycin, significantly increased rhinovirus-induced type I and type III interferon and interferon-stimulated antiviral protein production in primary bronchial epithelial cells, as well as significantly reducing rhinovirus replication and release in bronchial epithelial cells. 27 Azithromycin has also been shown to reduce illness severity in a mouse model of viral bronchiolitis. 28 Thus, azithromycin has the potential to have direct antiviral activity by augmenting the production of those interferons that we have already shown to be deficient in asthma exacerbations,20,26 and this activity may make it a better treatment option than telithromycin, which does not appear to have this property. 27 A further mechanistic aim of our study, therefore, was to investigate the frequencies of standard bacterial, atypical bacterial and viral infections in these exacerbations to determine the relative importance of each of these infections, and of possible coinfections with one or more agents, in the aetiology of acute exacerbations of asthma in adult subjects. We have also performed subgroup analyses to determine whether or not any treatment benefit observed is greater in those with evidence of one or more of these infections, with the aim of shedding some light on the possible mechanism(s) of action of azithromycin in this context.
Concerns regarding antimicrobial resistance
This clinical trial is important as there are significant concerns regarding the development of resistance against macrolide antibiotics. Although these concerns are somewhat mitigated by the short course of therapy being studied (relative, for example, to ongoing clinical trials investigating long-term treatment in severe asthma), determining whether or not azithromycin has efficacy in this context will, if the study is negative, help limit the inappropriate use of antibiotics (in a recent study of adult asthma exacerbations, 57% of subjects received antibiotics29).
If the study is positive, then determining the frequencies of detection of standard bacterial, atypical bacterial and viral infections in these exacerbations, combined with the subgroup analyses assessing the efficacy of the intervention in those with evidence of one or more of these infections, would help guide the use of such therapies in subgroups of asthma exacerbations that may respond better to such therapies, as well as guiding future investigation of the efficacy of alternative antibiotics with shorter durations of action or different spectra of antimicrobial/viral/inflammatory activity.
Choice of and duration of therapy
Although the course of azithromycin therapy is only 3 days, azithromycin has a multiple-dose tissue half-life of 68 hours and will therefore persist in the lung at significant concentrations for around 10 days after a 3-day course of therapy. 30 The main aim of this study was to determine whether or not the telithromycin results could be validated in a study with a similar antibiotic, with a similar mechanism and duration of action. Telithromycin was given for 10 days (the standard licensed duration of therapy for other respiratory indications) and the primary outcome variable was assessed at 10 days. 13 As our aim was to determine whether or not the telithromycin results can be validated, we felt that it was important to use the same primary outcome variable and as similar a duration of action as is possible (given that we cannot use telithromycin because of liver toxicity). This was one reason that we chose to study azithromycin rather than other macrolide antibiotics. Other reasons for choosing azithromycin include its antiviral activity, not shared with other macrolides,27 a more favourable drug interaction profile30 and excellent concentration at sites of infection. 30
Are patterns of airway inflammation associated with aetiology and treatment outcomes?
Different patterns of airway inflammation have been identified in both stable asthma and during exacerbations; these have been classified as neutrophilic, eosinophilic, mixed granulocytic or pauci-granulocytic. However, it is not known whether or not these different patterns of inflammation are associated with different aetiologies for the exacerbation, nor whether or not they are related to treatment outcome. Our final aim was, therefore, to characterise the inflammatory cell profiles in sputum at presentation, to determine if exacerbation aetiology as well as any possible treatment benefit was related to the types of airway inflammation present (neutrophilic, eosinophilic, mixed granulocytic or pauci-granulocytic).
Need for the AZALEA study
There are no systematic reviews of and no published reports of clinical trials investigating the efficacy of azithromycin in the treatment of (acute) asthma exacerbations. At the time of protocol development for this study there were no similar studies registered on ClinicalTrials.gov. The only somewhat similar study is the AZMATICS (AZithroMycin/Asthma Trial In Community Settings) study (NCT00266851),31 which planned to enrol 200 adult patients with asthma, either stable persistent or in exacerbation, and treat for 3 months to answer the question, ‘Will a 12-week treatment with the antibiotic, azithromycin, result in a statistically significant and clinically meaningful improvement in overall asthma symptoms and other patient-oriented asthma outcomes 1 year after initiation of treatment of adult primary care patients with asthma?’. Thus, the aims, design, timing of outcome analysis and treatment length are clearly very different from those of the AZALEA study.
Chapter 2 Research objectives
Primary objective
The primary objective was to assess the efficacy of azithromycin using a diary card summary symptom score, with symptoms including wheezing, breathlessness and coughing, assessed at 10 days after randomisation.
Secondary objectives
-
Assessment of the following additional efficacy end points at baseline and 5 and 10 days post randomisation:
-
health status assessed by the Acute Asthma Quality of Life Questionnaire (AQLQ)32
-
health status assessed by the Mini AQLQ33
-
pulmonary function tests [forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC), FEV1 : FVC ratio, peak expiratory flow (PEF), forced mid-expiratory flow rate (FEF25–75%) and forced expiratory flow rate at 50% expiration (FEF50%).
-
-
Primary and secondary outcomes assessed 5 and 10 days post randomisation to permit better estimation of the optimum timing of primary/secondary outcome variables in future similar studies.
-
Time to a 50% reduction in symptom score.
Exploratory analyses
-
Trends in primary and secondary outcomes over the time course of the exacerbation up to 10 days.
-
Assessment of efficacy outcomes in relation to initial C. pneumoniae and/or M. pneumoniae status.
-
Assessment of efficacy outcomes in relation to initial standard bacteriological status.
-
Assessment of efficacy outcomes in relation to initial virological status.
-
Assessment of efficacy outcomes in relation to initial sputum inflammatory cell status.
Chapter 3 Methods
A short version of this extended study has been published in JAMA Internal Medicine. 34 Some text in this report is reproduced with permission from JAMA Internal Medicine. 2016. http://dx.doi.org/10.1001/jamainternmed.2016.5664. Copyright © 2016 American Medical Association. All rights reserved.
Trial design
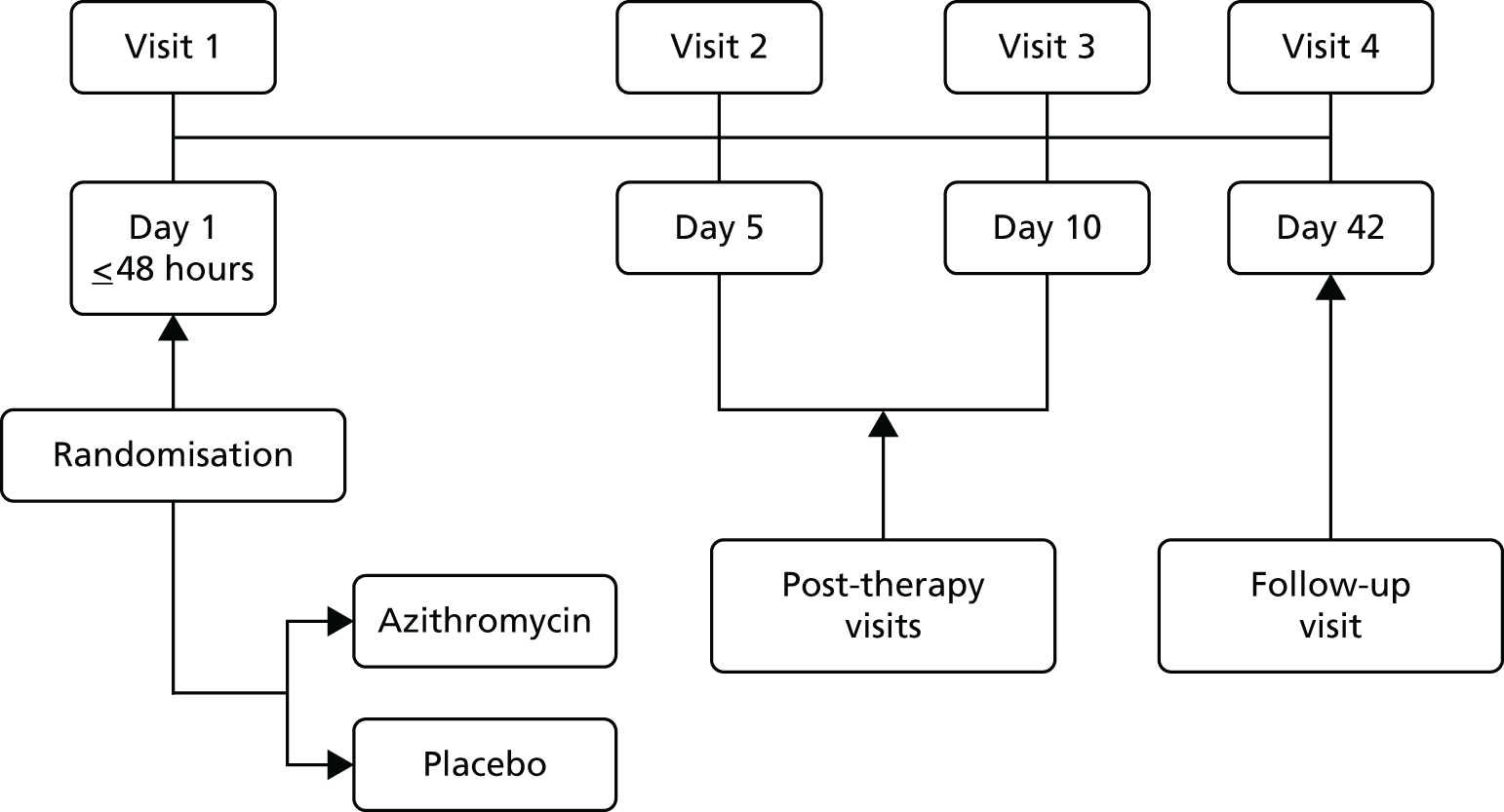
This was a multicentre, randomised, double-blind, placebo-controlled study. Eligible patients were randomised within 48 hours of initial presentation to medical care with an acute deterioration in asthma control and requiring a course of oral steroids. Patients were randomised on a 1 : 1 basis to receive either (1) azithromycin or (2) placebo. The duration of therapy with study medication (active or placebo) was 3 days, with post-therapy assessments/visits up to 10 days and a follow-up visit at 6 weeks.
Figure 1 summarises the design for the study.
FIGURE 1.
Summary of study design.
Participants
Participants in the study were adult patients with a documented history of asthma for > 6 consecutive months and presenting within 48 hours (of initial presentation to medical care) with an acute deterioration in asthma control and requiring a course of oral steroids.
Inclusion criteria
-
Adults of either sex aged 18–55 years or aged 56–65 years with a < 20 pack-year smoking history or aged > 65 years with a < 5 pack-year smoking history.
-
Patients with a documented history of asthma for > 6 months and presenting within 48 hours (of initial presentation to medical care) with an acute deterioration in asthma control (increased wheeze, dyspnoea and/or cough and/or reduced PEF) and requiring a course of oral steroids.
-
Patients with a PEF or FEV1 of < 80% of predicted normal or patient’s best at presentation, at recruitment or in the time elapsed between presentation and recruitment.
-
Patients must be able to complete diaries and quality-of-life questionnaires.
-
Patients must sign and date an informed consent form prior to any study procedures.
Exclusion criteria
-
Patients with known prolongation of the QT interval, with a history of torsades de pointes, congenital long QT syndrome, bradyarrhythmias or uncompensated heart failure, on drugs known to prolong the QT interval, with ongoing proarrhythmic conditions such as uncorrected hypokalaemia or hypomagnesaemia or clinically significant bradycardia and receiving class IA (quinidine, procainamide) or class III (dofetilide, amiodarone, sotalol) antiarrhythmic agents.
-
Smokers aged 56–65 years with a ≥ 20 pack-year history or aged > 65 years with a ≥ 5 pack-year history.
-
Patients requiring immediate transfer/referral to an intensive care unit.
-
Patients who have used oral or systemic antibiotics within 28 days prior to enrolment.
-
Patients with known impaired hepatic function (alanine aminotransferase/aspartate aminotransferase) more than two times the upper limit of normal.
-
Patients with significant lung disease [including chronic obstructive pulmonary disease (COPD)] other than asthma.
-
Patients taking > 20 mg of oral corticosteroids daily as maintenance therapy.
-
Patients requiring other antibiotic therapy.
-
Patients who are receiving other medications or who have other disease conditions or infections that could interfere with the evaluation of drug efficacy or safety.
-
Women who are breastfeeding or who are pregnant, as demonstrated by a urine pregnancy test carried out before exposure to study medication or the start of any study procedure that could pose a risk to the fetus.
-
Patients with suspected or known hypersensitivity to, or suspected serious adverse reaction to, azithromycin or any of the macrolide or ketolide class of antibiotics, erythromycin or any excipients thereof.
-
Patients who have received treatment with any other investigational drug within 1 month prior to study entry or who have such treatment planned for the study period during the treatment or follow-up phase.
-
Patients with a concomitant condition (including clinically relevant cardiovascular, hepatic, neurological, endocrine or other major systemic disease) making implementation of the protocol or interpretation of the study results difficult.
-
Patients with a mental condition rendering them unable to understand the nature, scope and possible consequences of the study.
-
Patients unlikely to comply with the protocol, for example unco-operative attitude or an inability to return for follow-up visits.
-
No patient was allowed to enrol in this study more than once.
Interventions
All patients in the study received treatment with either azithromycin or placebo, as per randomised allocation. The identity of the treatment regimen was blinded by encapsulating active medication in opaque capsules to match the placebo.
Those patients randomised to azithromycin received 500 mg of azithromycin (two 250-mg capsules) once a day for 3 days (this is the routine dose given in clinical care). Those patients randomised to the placebo received two placebo capsules once a day for 3 days. The duration of treatment with the study medications was 3 days. Patients were instructed to take the study medication at least 1 hour before or 2 hours after food; if they were taking antacids they were instructed to take the study medication at least 1 hour before or 2 hours after the antacids.
The time of administration of the study medication and the labelling on the study medication containers was documented on the case report forms for patients throughout the study. The first dose was given in the presence of a member of the research team.
Outcomes
Primary outcome
The primary outcome was the diary card summary symptom score, with symptoms including wheezing, breathlessness and coughing, assessed at 10 days after randomisation.
Secondary outcomes
-
Additional efficacy end points assessed at baseline and 5 and 10 months post randomisation were:
-
health status assessed by Acute AQLQ
-
health status assessed by Mini AQLQ
-
pulmonary function tests (FEV1, FVC, FEV1 : FVC ratio, PEF, FEF25–75%, FEF50%).
-
-
Primary and secondary outcomes assessed 5 and 10 days post randomisation to permit better estimation of the optimum timing of the assessment of primary/secondary outcome variables in future similar studies (the efficacy of telithromycin was assessed at 10 days only).
-
Time to a 50% reduction in symptom score.
Exploratory analyses
-
Trends in primary and secondary outcomes over the time course of the exacerbation up to 10 days.
-
Assessment of efficacy outcomes in relation to initial C. pneumoniae and/or M. pneumoniae status.
-
Assessment of efficacy outcomes in relation to initial standard bacteriological status.
-
Assessment of efficacy outcomes in relation to initial virological status.
-
Assessment of efficacy outcomes in relation to initial sputum inflammatory cell status.
Data collection
Electronic case record form
Data management was carried out using the InForm™ ITM (Integrated Trial Management) System version 4.6, a web-based data entry system that builds an Oracle database for each individual clinical trial (Oracle Corporation, Redwood City, CA, USA). Trial data were captured on a bespoke web-based electronic case report form (eCRF), with built-in validation rules to identify data entry errors in real time and a full audit trail of data entry and changes. All those entering data were trained prior to start-up and given personal login details, with access to forms restricted according to site and role. The eCRF was designed in accordance with the requirements of the trial protocol.
Timescale of trial evaluations
Daily evaluations
Visit 1 (day 1) for each patient occurred within 48 hours of initial presentation to medical care with an acute deterioration in asthma control and requiring a course of oral steroids. Patients were then seen by the research team at visit 2 (day 5 ± 1 day) and visit 3 (day 10 ± 1 day). At visit 1 patients were instructed on the recording of information on the symptom diary cards and were asked to complete the diary at the end of each day for 10 days. Symptom diary cards were reviewed at visits 2 and 3 and recordings were entered onto the eCRF.
Follow-up evaluation
This final follow-up evaluation took place at visit 4 (day 42 ± 2 weeks). At this visit a final serology sample was taken to assess for atypical pathogens and any adverse events (AEs) were recorded.
Schedule of investigations
A summary of the tests and investigations undertaken is provided in Table 1.
| Study procedure | Visit 1 (day 1, within 48 hours of initial presentation) | Visit 2 (day 5 ± 1 day) | Visit 3 (day 10 ±1 day) | Visit 4 (follow-up visit, day 42 ± 2 weeks) |
|---|---|---|---|---|
| Informed consent | ✗ | |||
| Inclusion/exclusion criteria review | ✗ | |||
| Demographics | ✗ | |||
| Medical/surgical history | ✗ | |||
| Record previous and concomitant treatments | ✗ | ✗ | ✗ | |
| Pulmonary function tests (FEV1, FVC, FEV1 : FVC ratio, FEF25–75%, FEF50%, PEF) | ✗ | ✗ | ✗ | |
| Urine pregnancy testa | ✗ | |||
| Serology for atypical pathogens | ✗ | ✗ | ||
| Nose and throat swabs and nasal mucus sample for PCR analysis | ✗ | |||
| Spontaneous/induced sputum for PCR analysis | ✗ | |||
| Culture of sputum for standard bacteria (quantitative) | ✗ | |||
| Sputum for cell differential count | ✗ | |||
| Full blood count | ✗ | |||
| Dispense diary – diary training | ✗ | |||
| Diary review | ✗ | ✗ | ||
| Return diary to investigator | ✗ | ✗ | ||
| Health outcomes assessment – Acute AQLQ | ✗ | ✗ | ✗ | |
| Health outcomes assessment – Mini AQLQ | ✗ | ✗ | ✗ | |
| Randomisation and dispense study medication | ✗ | |||
| Collect and count unused medication | ✗ | |||
| AE review | ✗ | ✗ | ✗ |
Clinical investigations
Pulmonary function tests
A spirometer meeting all American Thoracic Society recommendations was used for the pulmonary function tests, performed at visits 1–3. Pulmonary function tests were measured three times in a consistent position (standing or sitting) throughout the study. The best FEV1, FVC, FEV1 : FVC ratio, FEF25–75%, FEF50% and PEF were recorded on the eCRF as follows:
-
FEV1 in litres
-
FVC in litres
-
FEV1 : FVC ratio
-
FEF25–75% in litres per second
-
FEF50% in litres per second
-
PEF in litres per minute.
Patients’ daily recordings
All patients were supplied with a diary in which to record salbutamol (reliever) use, asthma symptom ratings and number of night-time awakenings because of asthma symptoms. The daily diary included four questions measuring daytime asthma symptoms, with patients rating the frequency and severity of their symptoms on a 7-point scale (with 0 denoting no symptoms and 6 denoting severe symptoms). The summary diary symptom score was calculated as the mean of these four daytime activity scores (the frequency of asthma symptoms, the severity of asthma symptoms, the level of activity performed today and the effect of asthma on activity) recorded at the end of the day. At visit 1, patients were instructed on how to record information in the diary (Box 1) and were asked to complete the diary at the end of each day for 10 days (with the nocturnal questions referring to the previous night). They were reminded of the recording instructions at visits 2 and 3 and to return all of the completed diary cards to the site at visit 4. All diary cards were retained in the participant files for data entry and monitoring.
I. Daytime symptom diary scale questions:
-
How often did you experience asthma symptoms today?
0 (none of the time) 1 2 3 4 5 6 (all of the time)
-
How much did your asthma symptoms bother you today?
0 (not at all bothered) 1 2 3 4 5 6 (severely bothered)
-
How much activity could you do today?
0 (more than usual activity) 1 2 3 4 5 6 (less than usual activity)
-
How often did your asthma affect your activities today?
0 (none of the time) 1 2 3 4 5 6 (all of the time)
II. Nocturnal diary scale question:
-
Did you wake up with asthma symptoms? (This can be awakening in the middle of the night or on awakening in the morning)
□ No □ Once □ More than once □ Awake ‘all night’
III. Number of inhalations of salbutamol will be recorded in the diary. Each patient should be reminded that salbutamol should be used only as needed for symptoms, not on a regular basis or prophylactically.
IV. Study medication will be recorded in the diary. Any concomitant medication use will be recorded in the diary.
V. Adverse events – patients will record all unusual health-related events in the diary regardless of relationship to medication.
Clinical sample collection
Respiratory samples
-
A nasal mucus sample and nasal and throat swabs were taken when possible at visit 1 to enable polymerase chain reaction (PCR) analysis for viruses and atypical bacteria to be undertaken. Nasal mucus samples were taken using clean soft tissues, which were subsequently placed into plastic freezer bags, stored at –80 °C and transferred at intervals to Imperial College London for analysis. Sites were supplied with flocked swabs for nasal and throat sample collection. Swabs were taken and then placed into a bijoux with phosphate-buffered saline or normal saline, frozen at –80 °C and sent to Imperial College London at intervals for analysis.
-
For patients with a productive cough, deep expectorated sputum was collected at visit 1 after rinsing the mouth with sterile water. The deep cough specimen was collected into a sterile Petri dish and patients were instructed not to expectorate saliva or postnasal discharge into the container.
-
In patients unable to produce an adequate sample of spontaneous sputum, sputum was induced in accordance with published protocols using isotonic saline7,35 if the visit took place at the recruiting site.
-
Sputum was sent fresh to the local microbiology laboratory for standard bacteriology using local standard operating procedures. A sputum pellet was frozen at –80 °C in a sterile microfuge tube and sent in batches by sites to Imperial College London to be processed for PCR. Remaining sputum was processed and sent in batches to the University of Leicester for cell differential counts. Those responsible for sputum processing at sites received specific training from the University of Leicester, which was documented by means of either a training certificate or written confirmation that previous training in the area was sufficient and additional training was not required.
-
If sputum was not obtained at visit 1 because of a non-productive cough or for any other reason, this was documented on the case report form. If there was no attempt to collect sputum this was reported as a protocol deviation.
Serology
Acute (visit 1) and convalescent (follow-up visit 4 at day 42) serum samples were obtained and analysed for atypical pathogens at Imperial College London laboratories. At visit 1 and visit 4 10 ml of blood was collected; this was processed on site to obtain serum and the serum was aliquoted and transferred immediately to a –80 °C freezer. At intervals these stored aliquots were sent to Imperial College London for analysis. In addition, at visit 1 an additional 3 ml of blood was collected from patients for a full blood count.
Health outcomes data
Health outcomes were measured to determine overall assessment of symptom resolution during the first 10 days based on global subject diary assessment.
Health status was assessed at visits 1–3 using the Acute AQLQ and Mini AQLQ.
Each site was provided with:
-
an Acute AQLQ
-
a Mini AQLQ
-
background information on the AQLQ, administration information and details on the analysis of the AQLQ
-
AQLQ coloured cards.
All staff at the different sites who were responsible for administering the AQLQs were asked to familiarise themselves with the contents of the different items before administering the questionnaires.
The questionnaires were interviewer administered and not self-administered. The Acute AQLQ contained a response sheet with columns used to record the responses at visits 1, 2 and 3. A new Mini AQLQ was printed for each visit when the Mini AQLQ was administered and patient responses were recorded directly onto the AQLQ. All patient responses/completed AQLQs were kept in the participant files for source data verification (SDV) and were also entered into the InForm eCRF database.
If possible, staff at the different sites were asked to complete the AQLQ first during clinic visits, before any discussion with a health professional, as this may have influenced how patients completed the questionnaires.
Pharmacovigilance definitions and procedures
Definitions
Adverse event
An AE was defined as any untoward medical occurrence in a patient (or clinical trial subject) administered a medicinal product and which does not necessarily have a causal relationship with the treatment.
The AE may have been:
-
a new illness
-
a worsening of a sign or symptom of the condition under treatment or of a concomitant illness
-
an effect of the study medication, including the comparator
-
a combination of two or more of these factors.
If the event met the criteria for ‘serious’, then the event was reported as a serious adverse event (SAE) (see Serious adverse event or serious adverse reaction).
Adverse reaction
All AEs judged by either the reporting investigator or the sponsor as having a reasonable causal relationship to an investigational medicinal product (IMP) were reported as adverse reactions (ARs).
Unexpected adverse reaction
An AR the nature or severity of which was not consistent with the Summary of Product Characteristics for azithromycin was reported as an unexpected AR. Side effects documented in the Summary of Product Characteristics that occurred in a more severe form than anticipated were also considered to be unexpected.
Serious adverse event or serious adverse reaction
A SAE or serious adverse reaction was defined as any untoward medical occurrence or effect that at any dose:
-
results in death
-
is life-threatening – refers to an event in which the subject was at risk of death at the time of the event; it does not refer to an event that hypothetically might have caused death if it were more severe
-
requires hospitalisation or prolongation of an existing inpatient hospitalisation
-
results in persistent or significant disability or incapacity – there is a substantial disruption to a person’s ability to carry out normal life functions
-
is a congenital abnormality or birth defect.
Medical judgement was exercised in deciding whether or not an AE/AR was serious in other situations. Important AEs/ARs that were not immediately life-threatening or which did not result in death or hospitalisation but which may have jeopardised the subject or may have required intervention to prevent one of the other outcomes listed in the definition above were also considered to be serious.
Hospitalisation of the patient as a direct result of the asthma exacerbation was not recorded as a SAE as this was part of routine clinical care and was not related to participation in the trial.
Suspected unexpected serious adverse reaction
A suspected unexpected serious adverse reaction (SUSAR) was any suspected AR related to an IMP that was both unexpected and serious.
Causality
The assignment of the causality of AEs and ARs was made by the investigator responsible for the care of the participant using the definitions in Table 2. If any doubt about causality existed the local investigator would inform the chief investigator.
| Relationship | Description |
|---|---|
| Unrelated | There is no evidence of any causal relationship |
| Unlikely | There is little evidence to suggest that there is a causal relationship (e.g. the event did not occur within a reasonable time after administration of the trial medication). There is another reasonable explanation for the event (e.g. the participant’s clinical condition, other concomitant treatment) |
| Possible | There is some evidence to suggest a causal relationship (e.g. because the event occurs within a reasonable time after administration of the trial medication); however, the influence of other factors may have contributed to the event (e.g. the participant’s clinical condition, other concomitant treatments) |
| Probable | There is evidence to suggest a causal relationship and the influence of other factors is unlikely |
| Definitely | There is clear evidence to suggest a causal relationship and other possible contributing factors can be ruled out |
| Not assessable | There is insufficient or incomplete evidence to make a clinical judgement about the causal relationship |
Period of observation
For the purposes of this study, the period of observation extended from the time that a subject gave informed consent until 7 days after the last dose of study medication.
Reporting procedures
All AEs that occurred after a subject had signed the informed consent were documented on the pages provided on the case report form. The trial eCRF included dedicated forms for reporting SAEs.
Non-serious adverse reactions/adverse events
All such events, whether expected or not, were recorded on the relevant case report form. These were reported to the Medicines and Healthcare products Regulatory Agency (MHRA) and Research Ethics Committee (REC) on the annual safety report form on the anniversary of the date that a favourable opinion for the study was given.
Serious adverse events and suspected unexpected serious adverse reactions
Fatal or life-threatening SAEs and SUSARs were reported to the chief investigator (who reported to the sponsor) on the day that the local site became aware of an event. The SAE form included the nature of the event, the date of onset, severity, corrective therapies given, outcome and causality (i.e. unrelated, unlikely, possible, probably, definitely). Additional information was sent to the chief investigator and sponsor within 5 days if the event had not resolved at the time of reporting.
Serious adverse events
Investigators were advised to report SAEs via the eCRF within 24 hours of becoming aware of an event and to include an assessment of expectedness and causality in the SAE report. Each SAE report was reviewed by the Imperial Clinical Trials Unit (ICTU) and chief investigator. The flow chart provided in Figure 2 shows the reporting procedures used.
FIGURE 2.
Reporting procedure for AEs. CRGO, Clinical Research and Governance Office.

Suspected unexpected serious adverse reactions
If an AR was considered to be serious, unexpected and related to the IMP (possibly, probably or definitely related), this would have met the definition of SUSAR, requiring expedited reporting to the MHRA, REC and sponsor. However, there were no SUSARs in the AZALEA study.
Annual safety reports
Annual safety reports were provided to the REC and MHRA, in accordance with clinical trial regulations, on the anniversary of the clinical trial authorisation each year. A total of three annual safety reports were submitted over the course of the trial.
Statistical considerations
Sample size
The sample size calculation was based on the primary outcome: change from baseline in diary card summary asthma symptom scores at 10 days after randomisation. Our previous study [the Telithromycin, Chlamydophila, and Asthma trial (TELICAST)]13 found a mean decrease in symptom score of 1.3 in the treatment group and 1.0 in the control group, resulting in a difference of –0.3 [standard deviation (SD) 0.783] between the groups at 10 days. In the previous study this was accompanied by a difference in lung function (FEV1) of 290 ml and by a 3-day faster time to a 50% improvement in symptoms, both of which were clinically impressive. We therefore felt that a difference of 0.3 in symptom score was clinically relevant.
Using a two-sided t-test at a 1% significance level with 80% power, 161 patients in each group were needed to detect the same difference in asthma scores between the groups. A significance level of 1% was chosen to provide greater certainty in the assessment of the primary outcome variable, as well as greater power for the subgroup exploratory analyses, as those subgroup analyses that were performed in the 280-patient TELICAST study13 were uninformative.
Taking into account the dropout rate of 15% in our previous study,13 we proposed to recruit 190 patients in each arm of the study. To be able to run the trial within the project timelines, we initially intended to involve 10 centres.
Randomisation
Randomisation was web based via access to a secure Imperial College London server and was performed using the InForm ITM System. Patient allocation was stratified by centre performed in random length blocks. Either the research nurse or the principal investigator (PI) at each site enrolled and then randomised each patient into the study using the InForm database.
The randomisation lists were generated by an ICTU statistician. Details such as the block size were kept confidential and held separately by the ICTU.
Blinding
This was a double-blind trial and so all participants and care providers and therefore those assessing outcomes were blinded to study treatment. Members of the trial team managing and analysing the data were also blind to the treatment received.
The identity of the study medications was blinded and the medications were packaged and supplied by Sharp Clinical Services (Crickhowell, UK) with code-break envelopes. Overencapsulated azithromycin capsules and placebo capsules were placed into child-resistant tamper-evident containers and a randomised label applied to each container.
Emergency identification of study medication/unblinding
If it was medically imperative to know what study medication a subject was receiving, the investigator or an authorised person could contact the on-call pharmacist who could open the relevant code-break envelope that corresponded to the randomisation label on the patient’s study drug container, exposing the blinded information. Clear instructions were provided to sites to ensure that no unnecessary or unintentional unblinding occurred and clear guidelines on when a code-break envelope should be opened were given. Any code break must have been documented in the code-break log and the trial manager notified in writing as soon as possible.
There was no requirement for unblinding during the AZALEA study and therefore no patients were unblinded before the statistical analysis took place.
Definition of day 1 for patient diary scores
Day 1 was defined in the statistical analysis plan (SAP) as follows: ‘the day 1 diary card of each patient will be defined to be the first diary that is completed within 24 hours of randomisation’. This definition was not applicable because the actual time of completing the diary cards was not recorded in InForm (nor the time of randomisation); therefore, if the date of the diary was the day after the randomisation it was not possible to determine whether or not that was within 24 hours.
After careful consideration, the study team agreed to amend the definition of day 1 to the following: ‘the day of the administration of the first dose of study drug, or if it is not available, then the day of the randomisation. If no diary card has been completed on that day, their day 1 diary card will be treated as missing. Diary cards for days 2–10 will be determined in a similar way’.
The date of study drug administration was available for all randomised patients.
This had no effect on the secondary outcomes as both the Acute AQLQ, Mini AQLQ and pulmonary function tests were conducted only three times and the analysis was based on the order of visits not on the days.
Missing data
Before starting data analysis, the level and pattern of missing data in the baseline variables and outcomes were analysed by forming appropriate tables. Additionally, the likely causes of any missingness were investigated. This information was used to determine whether the level and type of missing data had the potential to introduce bias into the analysis or to substantially reduce the precision of estimates related to treatment effects. Missing data in the patient diaries took one of several forms: no patient diary returned for any day (patient missingness), all data missing for ≥ 1 day (day missingness) and data missing for some but not all of the individual questions for a particular day (item missingness). Of these, the level of item missingness was expected to be minimal. According to the SAP, if any item missingness occurred the scores for the missing questions were interpolated from the previous and subsequent day scores. This process was conducted for two missing entries.
If any item missingness occurred in AQLQ scores, the summary score for that day was treated as missing.
Missing data for the pulmonary function tests were expected to occur because of the spirometer not recording some measures. As this was unrelated to patient outcome, it was reasonable to assume that this missingness was uninformative and that multilevel models fitted to all observed data would provide unbiased parameter estimates.
Modelling patient diary scores
The modelling process for patient diary scores was carried out based on the methods outlined in the SAP, which was finalised before the unblinding. All patients who returned at least one diary card (and received the study drug) were included in the analysis but only those diary cards that were collected in the investigated 10-day time frame were included. Clinical efficacy analyses were carried out on an intention-to-treat basis. A data check for outliers was also planned in the SAP, which included a series of longitudinal plots (one for each centre) of diary score for each patient, differentiating between treatment arm; box plots of diary score by treatment arm for each day were also produced as well as a table of summary statistics of diary score by day and treatment arm (including mean, SD, median and lower and upper quartiles).
Multilevel modelling was used to calculate the unbiased estimates of differences in diary scores between the treatment arms for each day. The terms used in the model were set out in the SAP and are explained in the following section. Different relationships between time and diary scores were compared including linear, quadratic and square root relationships. These models differed in their ‘time’ covariate. Fixed and random effects and the use of splines were also investigated. The goodness of fit of these models was assessed by residual plots, which are provided in Appendix 1. The final model includes only time and time × treatment interaction as covariates and diary score as the outcome variable. Treatment by itself was not included in the model, which means that there was not any difference in diary scores assumed at baseline between the treatment arms. Any observed difference could happen only by chance (caused by the randomisation).
The three main components of the model
Let DSid represent the diary score for patient i on day d, d = 1, . . .,10 and t(i) represent the treatment given to individual i (azithromycin or placebo). Then, DSid was modelled as the sum of three components: an intercept term, a change over time term and a residual error term, that is:
Possible choices for each of these components are outlined in the following sections. The options explored for the primary analysis were determined by the results of the exploratory analysis and the final choice was the simplest model that satisfies standard checks of model fit (e.g. residual plots).
Intercept term
The intercept term will estimate the diary score on day 1 (the day of randomisation and start of the study medication). This term will consist of an individual-level random effect, which will be drawn from a distribution parameterised using the associated centre-level random effect. Hence the unexplained variation in the diary scores will be split into three components corresponding to the three levels of the model, that is, the variation attributable to the centre (between-centre variation) and the individual (between-individual variation), as well as the residual variation (within-individual variation).
Additionally, baseline covariates can be incorporated into the model at the individual level. None will be incorporated for the initial analysis unless the baseline characteristics analysis reveals a substantial imbalance. Further analyses will examine the effect of incorporating baseline variables (age, sex, asthma severity, smoking history and asthma exacerbation).
Change over time term
This term will capture the change in the diary score from the start of study medication (day 1) and hence time will enter the model as day 1. The simplest assumption would be a linear change over the 10-day period; however, alternatives such as including a quadratic term or using splines may need to be considered as the rate of change may not be constant over the 10-day period. The covariates in this term were time (in days), which shows the daily change in the reference (placebo) group, and a time × treatment interaction, which shows the daily mean difference between the two groups. The difference at day 10 and its 95% CI were planned to be presented to answer the research question.
Residual error term
We assumed that the residual errors have a normal distribution. An alternative was to assume that these errors follow a heavier tailed distribution such as a t distribution with 4 degrees of freedom, which will provide robustness to outliers. Normality of residual errors was checked graphically.
Modelling Acute Asthma Quality of Life Questionnaire score, Mini Asthma Quality of Life Questionnaire score and pulmonary function
Similar models were used to assess the day 10 differences in change in Acute AQLQ and Mini AQLQ scores and pulmonary function between the two treatment arms.
Statistical analysis plan
A SAP was prepared by the trial investigators and trial statistician and reviewed and agreed by the Trial Steering Committee (TSC) and Data Monitoring and Ethics Committee (DMEC) prior to the end of the recruitment period.
Statistical software
All analyses were performed using Stata 13 (StataCorp LP, College Station, TX, USA).
Trial organisation
Trial management
The UK Clinical Research Collaboration-registered ICTU was responsible for trial management, quality assurance, trial statistics and development and maintenance of the trial database. A dedicated trial manager and clinical trials monitor were appointed through the ICTU to oversee the day-to-day management and monitoring of the project from set-up to close.
Trial sponsor
The sponsor of the trial was Imperial College London. The sponsor’s role is clearly set out in the European Clinical Trials Directive36 and NHS Research Governance Framework37 documents. Imperial College London signed a clinical trial agreement with each of the participating centres prior to the start of recruitment at each centre.
Ethical considerations
The trial was conducted in accordance with the Declaration of Helsinki [see www.wma.net/ (accessed 4 April 2016)] on research involving human subjects. The study protocol, patient information sheet and consent form were submitted to the REC prior to the start of the study and a favourable opinion was obtained on the 15 June 2011.
Consent
Patients were provided with the patient information sheet (see Appendix 2) and were given sufficient time to consider participation and discuss the trial with the research staff prior to consent and enrolment. Full written informed consent was taken using the ethically approved consent form.
Research governance
The trial was carried out in accordance with the NHS Research Governance Framework37 and local NHS permission was granted by the research and development departments at each participating site prior to recruitment commencing.
Regulatory requirements
As a randomised trial of an IMP, the AZALEA trial was conducted in accordance with the European Clinical Trials Directive36 and the Medicines for Human Use (Clinical Trials) Regulations 200438 as well as ICH Good Clinical Practice guidelines. 39,40 The trial received Clinical Trial Authorisation from the MHRA on 21 July 2011 and was registered in the European Community with a EudraCT number of 2011–001093–26.
Trial registration
The trial was registered on the ClinicalTrials.gov clinical trial database with a reference number of NCT01444469.
National Institute for Health Research Clinical Research Network portfolio
The AZALEA trial was adopted on the National Institute for Health Research (NIHR) Clinical Research Network (CRN) portfolio with a UKCRN ID number of 11358. Accrual data were uploaded onto the NIHR CRN database on a monthly basis.
Summary of protocol amendments
The following amendments were made to the trial protocol following approval of the first version of the document by the REC and MHRA.
-
Version 2: in addition to minor typographical clarifications to the wording of the protocol, the following changes were made: addition of a throat swab (in case a sufficient sample was not obtained from the nasal mucus and nasal swab); refinement of inclusion criteria to include FEV1 as well as PEF as a measurement of lung function; refinement of exclusion criteria to clarify the type of antibiotic use that would be excluded; and inclusion of a statement that hospitalisation as a direct result of the asthma exacerbation was not a SAE.
-
Version 3: refinement of inclusion criteria to include patients aged > 65 years with a smoking history of < 5 pack-years.
-
Version 4: refinement of the eligibility criteria to include patients presenting within 48 hours (of initial presentation to medical care) with an acute deterioration of asthma control (instead of 24 hours as in the previous protocol version); recruitment extension to April 2014; and minor administrative changes.
-
Version 5: protocol amendment to introduce participant reimbursements for completing study visits and returning all symptom diaries.
-
Version 6: addition of an extra exclusion criterion to reflect guidelines released from the US Food and Drug Administration on the use of azithromycin.
Trial committees
Trial Steering Committee
A TSC was established to oversee the conduct of the study. The TSC met seven times over the course of the trial (6 January 2012, 5 July 2012, 18 January 2013, 4 April 2013, 31 October 2013, 10 April 2014 and 24 July 2015). Copies of the minutes from each meeting were sent to the funder, the Efficacy and Mechanism Evaluation programme of the NIHR. The TSC approved the trial protocol prior to the start of the study and received regular recruitment reports throughout the duration of the trial.
The TSC membership was as follows:
-
independent members:
-
Professor Wisia Wedzicha – chairperson
-
Professor Peter Calverley – independent member
-
Professor Ratko Djukanovic – independent member
-
Ms Leanne Metcalf, Asthma UK – patient representative, independent member
-
Professor Mike Thomas – independent member.
-
-
non-members in attendance:
-
Professor Deborah Ashby – senior statistician
-
Professor Chris Brightling – PI, Leicester
-
Mrs Mary Cross – operations manager, ICTU
-
Professor Sebastian Johnston – chief investigator
-
Ms Laura Robison – trial manager (until February 2013)
-
Dr Zahid Sattar – trial manager (until April 2015)
-
Dr Jane Warwick – senior statistician (until June 2014)
-
Dr Alexina Mason – junior statistician (until January 2015)
-
Dr Ernie Wong – research fellow, Imperial College London.
-
Data Monitoring and Ethics Committee
An independent DMEC was established to review SAE reports and any ongoing safety issues. The DMEC meetings took place on 31 May 2012, 29 November 2012, 2 December 2013 and 24 July 2015.
The first DMEC meeting to agree the charter outlining operational details and responsibilities took place early in the trial, on 31 May 2012. The DMEC provided feedback reports from each meeting for the chair of the TSC and these were reviewed at subsequent TSC meetings as applicable.
The DMEC membership was as follows:
-
independent members:
-
Professor Jonathan Grigg – chairperson
-
Dr Stephen Bremner – independent statistician
-
Dr Peter Howarth – independent member.
-
Data management
Predefined data ranges were included in the eCRF, which raised automated queries if data outside the expected range were entered. In addition to the automated queries, the trial data were reviewed on a regular basis by the trial monitor to look for discrepancies and errors. In addition to the regular checks performed by the trial monitor, the trial statistician also performed a series of checks on snapshots of data to look for inconsistencies.
Risk assessment and monitoring plan
A risk assessment was performed by the ICTU quality assurance manager prior to the start of the trial. The result of the risk assessment indicated that the study was of medium risk and that 50% of trial data, 100% of consent forms and 100% of SAEs should be source verified. A monitoring plan was prepared in accordance with the risk assessment to specify the frequency of monitoring visits and amount of SDV required.
The requirements of a medium-risk trial for monitoring are:
-
at least two monitoring visits to be performed in total or one to three visits per annum
-
SDV should be carried out for 50% of subjects to check for eligibility, existence, drug delivery (to patients), end points and AEs
-
SDV should be carried out for 100% of consent forms and SAEs
-
at each monitoring visit the monitor will verify research approvals, drug accountability, regulatory documents and archiving.
Monitoring visits
A site initiation visit was performed at all participating centres. Interim monitoring visits were carried out depending on recruitment rates and close-out visits were carried out at all centres following the final follow-up visit for the last patient recruited. The monitoring visits were conducted mainly by the trial monitor.
Investigational medicinal product manufacturer
The overencapsulation of azithromycin capsules and production of matching placebo was undertaken by Sharp Clinical Services, a MHRA-licensed manufacturing unit with expertise in manufacturing and overencapsulating IMPs.
Patient and public involvement
Patient representatives were consulted during preparation of the patient information sheet. The TSC membership included a patient representative from Asthma UK who was invited to attend all TSC meetings and who was included in all relevant correspondence.
In addition, the trial manager attended the National Heart and Lung Institute at the Royal Brompton Hospital, London, on several occasions to meet the respiratory consumer group. At the group meetings an update on study progress was given and any relevant issues were discussed, including the following:
-
Patient leaflet and poster – members of the group were asked for their comments and feedback on the language and appropriateness of the patient leaflet and poster, used as tools to help introduce the study to patients.
-
Ongoing issues affecting recruitment such as whether or not the group felt that patients would be more likely to participate in the study if they were approached by a study doctor rather than a nurse (in looking at why consenting to the study was low) and whether or not introducing patient payments for visits would increase the number of visits attended (in looking at why the level of missing visits was high).
Useful feedback was received from the group and incorporated into study documents and procedures when relevant as the study progressed.
Chapter 4 Results
Participant flow
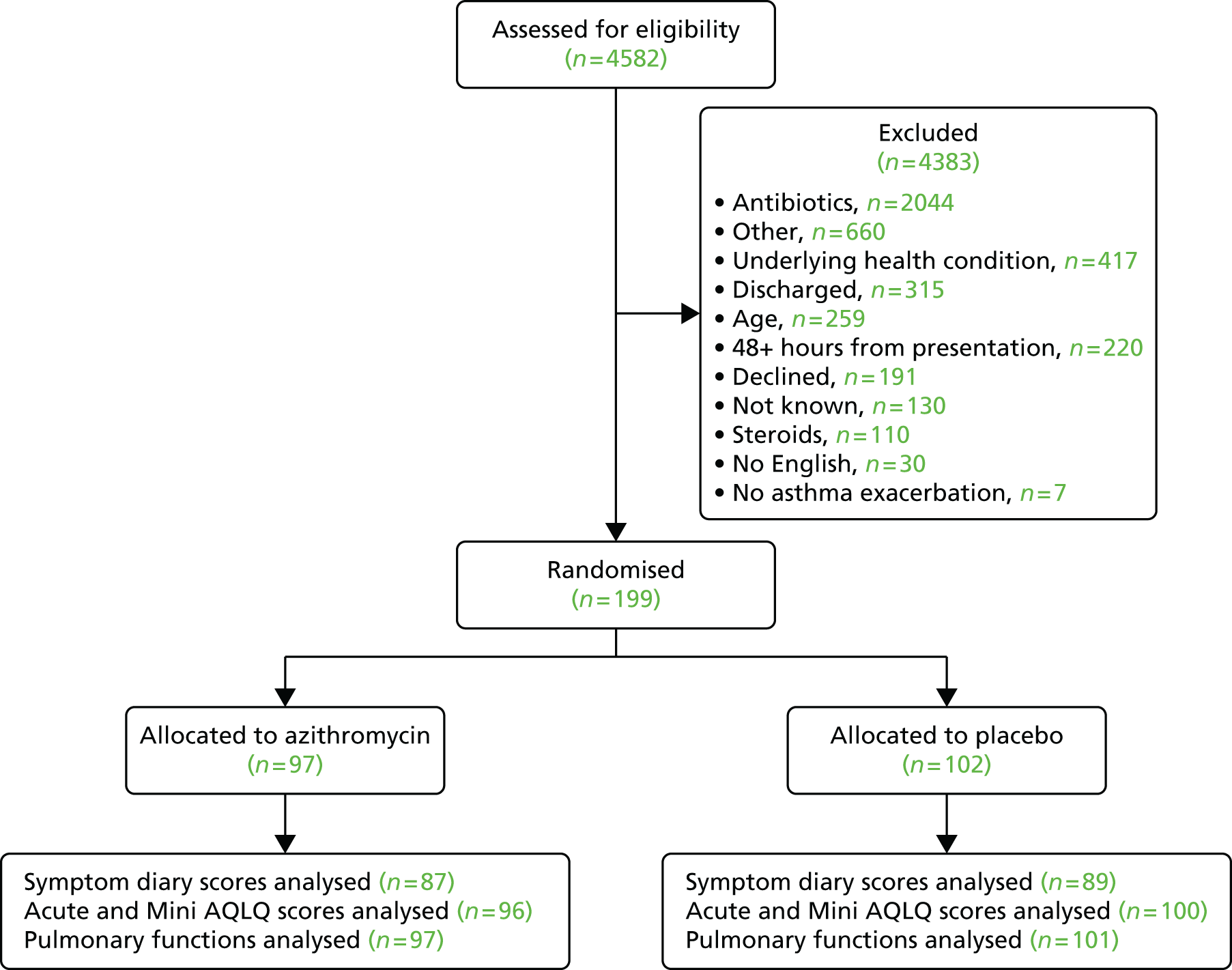
The flow of patients is summarised in Figure 3, including the numbers of patients screened, randomised and completing the trial.
FIGURE 3.
Consolidated Standards of Reporting Trials diagram for the AZALEA trial. Reproduced with permission from JAMA Internal Medicine. 2016. http://dx.doi.org/10.1001/jamainternmed.2016.5664. Copyright © 2016 American Medical Association. All rights reserved.
Screening
In total, 4582 patients were screened at the participating hospitals/centres over the duration of the trial. Of the 390 patients meeting the eligibility criteria, 199 were randomised to the trial as 191 declined to participate. Table 3 summarises the numbers of screened and eligible patients recruited to the trial and Table 4 summarises the reasons for non-recruitment.
| Centrea | Date opened | Patients screenedb | Recruited |
|---|---|---|---|
| Queen Alexandra Hospital, Portsmouth | 5 December 2011 | 298 | 56 |
| Birmingham Heartlands Hospital, Birmingham | 10 January 2012 | 520 | 29 |
| Western & Royal Infirmary, Glasgow | 16 December 2011 | 1333 | 28 |
| St Mary’s Hospital, London | 17 October 2011 | 414 | 11 |
| University Hospital of North Tees, Stockton-on-Tees | 16 October 2012 | 199 | 10 |
| Nottingham City Hospital and Queen’s Medical Centre, Nottingham | 2 April 2012 | 172 | 10 |
| Glenfield Hospital, Leicester | 2 February 2012 | 182 | 10 |
| New Cross Hospital, Wolverhampton | 25 June 2013 | 25 | 8 |
| East Surrey Hospital, Redhill | 27 March 2013 | 84 | 7 |
| Blackpool Victoria Hospital, Blackpool | 14 December 2012 | 78 | 6 |
| Rowden Medical Partnership (GP surgery), Chippenham | 11 March 2013 | 11 | 6 |
| Ipswich Hospital, Ipswich | 24 April 2013 | 108 | 3 |
| St James’s University Hospital, Leeds | 16 January 2013 | 100 | 3 |
| Norfolk and Norwich University Hospital, Norwich | 11 January 2013 | 43 | 3 |
| Worcestershire Acute Hospitals NHS Trust, Worcester | 27 March 2013 | 43 | 2 |
| Countess of Chester Hospital, Chester | 7 February 2013 | 39 | 2 |
| Gloucestershire Royal Hospital, Gloucester | 20 January 2014 | 45 | 1 |
| Princess Royal Hospital, Telford | 9 December 2013 | 32 | 1 |
| Musgrove Park Hospital, Taunton | 25 March 2013 | 85 | 1 |
| Royal Berkshire Hospital, Reading | 7 February 2013 | 136 | 1 |
| Freeman Hospital, Newcastle upon Tyne | 2 April 2012 | 32 | 1 |
| Guy’s and St Thomas’ NHS Foundation Trust, London | 18 April 2012 | 253 | 0 |
| Charing Cross Hospital, Imperial College Healthcare NHS Trust, London | 22 November 2011 | 131 | 0 |
| Derriford Hospital, Plymouth | 21 March 2013 | 96 | 0 |
| University Hospital of South Manchester NHS Foundation Trust, Manchester | 12 June 2012 | 56 | 0 |
| Hammersmith Hospital, Imperial College Healthcare NHS Trust, London | 17 January 2012 | 31 | 0 |
| Sherwood Forest Hospitals NHS Foundation Trust, Sutton in Ashfield | 1 November 2012 | 22 | 0 |
| Great Western Hospitals NHS Foundation Trust, Swindon | 25 March 2013 | – | 0 |
| Barnsley Hospital, Barnsley | 21 October 2013 | – | 0 |
| James Cook University Hospital, Middlesbrough | 12 December 2013 | 14 | 0 |
| Leighton Hospital, Crewe | 4 December 2013 | – | 0 |
| Total | 4582 | 199 |
| Reason | Number of patients | Comments |
|---|---|---|
| No English language | 30 | |
| > 48 hours from presentation | 220 | Includes patients screened > 24 hours from presentation prior to the protocol amendment |
| Already taking antibiotics | 2044 | |
| No asthma exacerbation | 7 | Includes no asthma exacerbation and no exacerbation |
| Underlying health condition | 417 | Includes COPD and comorbidities |
| Declined | 191 | |
| Other | 660 | |
| Steroids | 110 | Patients not requiring steroids for their exacerbation and therefore ineligible |
| Age | 259 | |
| Discharged | 315 | Includes ‘unable to contact’ |
| Not known | 130 | Reason for exclusion and/or time of screening not recorded |
| Total | 4383 |
Recruitment and retention
Recruitment lasted for 2.5 years, from September 2011 to April 2014. The actual recruitment period was longer than the original target of 1 year. The delays in starting the trial and continuing issues with slower recruitment were associated with the following:
-
delays in opening sites to recruitment
-
in the initial year of recruitment we encountered an unusually mild winter with lower than anticipated numbers of asthma exacerbations
-
one of the greatest factors restricting recruitment was patients being prescribed antibiotics by doctors [in accident and emergency (A&E) departments and by general practitioners (GPs)], despite the current British Thoracic Society guidelines41 stating that antibiotics should not be routinely given to treat asthma exacerbations
-
research teams at some recruiting sites were based at a different hospital to the A&E department; consequently, the research nurse and investigator were not always available to travel to the other site to recruit patients.
Because of these reasons, recruitment proved extremely challenging. To counteract this, further recruiting centres were opened, inclusion criteria were relaxed and the recruitment time was extended. However, despite all of these efforts only 199 subjects were recruited (of the original target of 380) by the medication expiry date and, in the absence of any further funding, the study had to be terminated, despite not reaching its recruitment target.
Recruitment rate
The target recruitment rate for the study was three to four patients per month per centre, based on the original 10 centres recruiting and a target recruitment figure of 380.
Accrual of patients during the whole study period is presented in Figure 4. Monthly and cumulative monthly accrual of patients is shown in Figure 5.
FIGURE 4.
Cumulative monthly accrual of patients into the AZALEA trial.

FIGURE 5.
Monthly and cumulative monthly accrual of patients into the AZALEA trial.

Baseline data
Clinical characteristics
Baseline characteristics of all randomised patients were summarised by treatment group using the median and interquartile range for continuous variables and number and percentage for categorical variables (Table 5). To check for any differences in baseline characteristics between centres, the same variables for centres that recruited at least 10 patients were compared (Table 6). Baseline characteristics were well balanced across treatment arms and centres.
| Factor | Active group, n (%) (N = 97) | Placebo group, n (%) (N = 102) |
|---|---|---|
| Age (years), median (IQR) [min.–max.] | 39.1 (28.9–49.5) [19.5–76.4] | 36.2 (25.4–49.3) [18.2–79.7] |
| Sex | ||
| Female | 64 (66.0) | 75 (73.5) |
| Male | 33 (34.0) | 27 (26.5) |
| Asthma severity (n = 198) | ||
| Step 1: mild intermittent asthma | 7 (7.2) | 13 (12.9) |
| Step 2: regular preventer therapy | 30 (30.9) | 26 (25.7) |
| Step 3: initial add-on therapy | 31 (32.0) | 27 (26.7) |
| Step 4: persistent poor control | 22 (22.7) | 22 (21.8) |
| Step 5: continuous or frequent use of oral steroids | 7 (7.2) | 13 (12.9) |
| Smoking status (n = 198) | ||
| Never smoked | 60 (61.9) | 61 (60.4) |
| Former smoker | 26 (26.8) | 19 (18.8) |
| Current smoker | 11 (11.3) | 21 (20.8) |
| Pack-years, median (IQR) [min.–max.] (n = 75) (current/former smokers) | 5 (1–15) [0–127a] | 5 (2–12) [0–22] |
| Asthma exacerbation (n = 198) | ||
| Mild asthma exacerbation | 5 (5.2) | 3 (3.0) |
| Moderate asthma exacerbation | 26 (26.8) | 35 (34.7) |
| Acute severe asthma | 61 (62.9) | 56 (55.4) |
| Life-threatening asthma | 4 (4.1) | 7 (6.9) |
| Near-fatal asthma | 1 (1.0) | 0 (0.0) |
| Time from presentation to study drug administration, median (IQR) [min.–max.] (n = 192) | 21 (12–29) [1–49] | 22 (14–28) [1–48] |
| Factor | Centre, n (%) | ||||||
|---|---|---|---|---|---|---|---|
| BIR (n = 29) | GLA (n = 28) | LEI (n = 10) | POR (n = 56) | SMH (n = 11) | UNT (n = 10) | OTH (n = 55) | |
| Age (years), median (IQR) | 38.2 (28.3–43.4) | 32.6 (27.45–44.8) | 50.35 (26.7–62.6) | 37.15 (26.25–49.5) | 42.3 (23.2–58.2) | 41.75 (39.1–58.7) | 41.6 (26.1–53.4) |
| Sex | |||||||
| Female | 22 (75.9) | 18 (64.3) | 4 (40.0) | 39 (69.6) | 8 (72.7) | 8 (80.0) | 40 (72.7) |
| Male | 7 (24.1) | 10 (35.7) | 6 (60.0) | 17 (30.4) | 3 (27.3) | 2 (20.0) | 15 (27.3) |
| Asthma severity | |||||||
| Step 1: mild intermittent asthma | 0 (0.0) | 5 (17.9) | 1 (10.0) | 5 (8.9) | 1 (9.1) | 0 (0.0) | 8 (14.8) |
| Step 2: regular preventer therapy | 8 (27.6) | 3 (10.7) | 3 (30.0) | 12 (21.4) | 7 (63.6) | 5 (50.0) | 18 (33.3) |
| Step 3: initial add-on therapy | 15 (51.7) | 8 (28.6) | 3 (30.0) | 13 (23.2) | 1 (9.1) | 4 (40.0) | 14 (25.9) |
| Step 4: persistent poor control | 4 (13.8) | 9 (32.1) | 2 (20.0) | 17 (30.4) | 2 (18.2) | 1 (10.0) | 9 (16.7) |
| Step 5: continuous or frequent use of oral steroids | 2 (6.9) | 3 (10.7) | 1 (10.0) | 9 (16.1) | 0 (0.0) | 0 (0.0) | 5 (9.3) |
| Smoking status | |||||||
| Never smoked | 18 (62.1) | 19 (67.9) | 7 (70.0) | 29 (51.8) | 9 (81.8) | 5 (50.0) | 34 (63.0) |
| Former smoker | 5 (17.2) | 5 (17.9) | 2 (20.0) | 16 (28.6) | 1 (9.1) | 5 (50.0) | 11 (20.4) |
| Current smoker | 6 (20.7) | 4 (14.3) | 1 (10.0) | 11 (19.6) | 1 (9.1) | 0 (0.0) | 9 (16.7) |
| Pack-years, median (IQR) | 5 (2–10) | 2 (2–8) | 8 (1–15) | 7 (2–15) | 2 (1–3) | 10 (10–20) | 4.5 (2–13.5) |
| Asthma exacerbation | |||||||
| Mild asthma exacerbation | 0 (0.0) | 3 (10.7) | 1 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 4 (7.4) |
| Moderate asthma exacerbation | 5 (17.2) | 12 (42.9) | 3 (30.0) | 12 (21.4) | 2 (18.2) | 5 (50.0) | 22 (40.7) |
| Acute severe asthma | 24 (82.8) | 13 (46.4) | 5 (50.0) | 38 (67.9) | 8 (72.7) | 5 (50.0) | 24 (44.4) |
| Life-threatening asthma | 0 (0.0) | 0 (0.0) | 1 (10.0) | 5 (8.9) | 1 (9.1) | 0 (0.0) | 4 (7.4) |
| Near-fatal asthma | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.8) | 0 (0) | 0 (0.0) | 0 (0.0) |
| Time from presentation to study drug administration, median (IQR) | 20 (9–26) | 23 (18–36) | 11.5 (6–19) | 23 (17–30) | 19 (16–21) | 24 (24–27) | 22 (10–28) |
Biological samples
Sputum bacteriology results by treatment arm are shown in Tables 7 and 8. Sputum virology results are shown in Table 9 and nasal virology results are shown in Table 10. The joint sputum and nasal swab virology results are shown in Table 11.
| Factor | Active group, n (%) (N = 97) | Placebo group, n (%) (N = 102) |
|---|---|---|
| Sputum samples collecteda | 52 (53.6) | 53 (52.0) |
| Streptococcus pneumoniae | ||
| Negative | 21 (21.6) | 21 (20.6) |
| Positive | 3 (3.1) | 3 (2.9) |
| Result not available | 73 (75.3) | 78 (76.5) |
| Haemophilus influenzae | ||
| Negative | 24 (24.7) | 20 (19.6) |
| Positive | 0 (0.0) | 4 (3.9) |
| Result not available | 73 (75.3) | 78 (76.5) |
| Moraxella catarrhalis | ||
| Negative | 23 (23.7) | 22 (21.6) |
| Positive | 1 (1.0) | 1 (1.0) |
| Result not available | 73 (75.3) | 79 (77.5) |
| Any bacteria positive including sputum, nasal and serology results | 9 (9.3) | 12 (11.8) |
| Factor | Active group, n (%) (N = 97) | Placebo group, n (%) (N = 102) |
|---|---|---|
| Sputum | ||
| C. pneumoniae negative and M. pneumoniae negative | 40 (41.2) | 36 (35.3) |
| Not available | 57 (58.8) | 66 (64.7) |
| Nasal | ||
| C. pneumoniae negative and M. pneumoniae negative | 95 (97.9) | 96 (94.1) |
| Not available | 2 (2.1) | 6 (5.9) |
| Serology | ||
| C. pneumoniae positive and M. pneumoniae negative | 1 (1.0) | 1 (1.0) |
| C. pneumoniae negative and M. pneumoniae positive | 4 (4.1) | 3 (2.9) |
| C. pneumoniae negative and M. pneumoniae negative | 79 (81.4) | 79 (77.5) |
| Not available | 13 (13.4) | 19 (18.6) |
| C. pneumoniae and M. pneumoniae including sputum, nasal and serology results | ||
| C. pneumoniae positive and M. pneumoniae negative | 1 (1.0) | 1 (1.0) |
| C. pneumoniae negative and M. pneumoniae positive | 4 (4.1) | 3 (2.9) |
| C. pneumoniae negative and M. pneumoniae negative | 79 (81.4) | 79 (77.5) |
| Not available | 13 (13.4) | 19 (18.6) |
| Factor | Active group, n (%) (N = 37) | Placebo group, n (%) (N = 34) |
|---|---|---|
| Rhinovirus | ||
| Negative | 31 (83.8) | 26 (76.5) |
| Positive | 6 (16.2) | 8 (23.5) |
| Other picornaviruses | ||
| Negative | 35 (94.6) | 31 (91.2) |
| Positive | 2 (5.4) | 3 (8.8) |
| Adenoviruses | ||
| Negative | 37 (100.0) | 34 (100.0) |
| Bocavirus | ||
| Negative | 37 (100.0) | 34 (100.0) |
| Respiratory syncytial virus | ||
| Negative | 34 (91.9) | 30 (88.2) |
| Positive | 3 (8.1) | 4 (11.8) |
| Influenza AH1/AH3/B | ||
| Negative | 37 (100.0) | 34 (100.0) |
| Parainfluenza viruses 1–3 | ||
| Negative | 37 (100.0) | 34 (100.0) |
| Human metapneumovirus | ||
| Negative | 37 (100.0) | 34 (100.0) |
| Coronaviruses 229E and/or OC43 | ||
| Negative | 37 (100.0) | 34 (100.0) |
| Any virus | ||
| Negative | 27 (73.0) | 20 (58.8) |
| Positive | 10 (27.0) | 14 (41.2) |
| Factor | Active group, n (%) (N = 95) | Placebo group, n (%) (N = 96) |
|---|---|---|
| Rhinovirus | ||
| Negative | 88 (92.6) | 87 (90.6) |
| Positive | 7 (7.4) | 8 (8.3) |
| Result not available | 0 | 1 (1.0) |
| Other picornaviruses | ||
| Negative | 95 (100.0) | 95 (99.0) |
| Positive | 0 | 0 |
| Result not available | 0 | 1 (1.0) |
| Adenoviruses | ||
| Negative | 95 (100.0) | 95 (99.0) |
| Positive | 0 | 0 |
| Result not available | 0 | 1 (1.0) |
| Bocavirus | ||
| Negative | 95 (100.0) | 95 (99.0) |
| Positive | 0 | 0 |
| Result not available | 0 | 1 (1.0) |
| Respiratory syncytial virus | ||
| Negative | 95 (100.0) | 95 (99.0) |
| Positive | 0 | 0 |
| Result not available | 0 | 1 (1.0) |
| Influenza AH1/AH3/B | ||
| Negative | 95 (100.0) | 95 (99.0) |
| Positive | 0 | 0 |
| Result not available | 0 | 1 (1.0) |
| Parainfluenza viruses 1–3 | ||
| Negative | 95 (100.0) | 95 (99.0) |
| Positive | 0 | 0 |
| Result not available | 0 | 1 (1.0) |
| Human metapneumovirus | ||
| Negative | 95 (100.0) | 95 (99.0) |
| Positive | 0 | 0 |
| Result not available | 0 | 1 (1.0) |
| Coronaviruses 229E and/or OC43 | ||
| Negative | 93 (97.9) | 93 (96.9) |
| Positive | 2 (2.1) | 2 (2.1) |
| Result not available | 0 | 1 (1.0) |
| Any virus | ||
| Negative | 86 (90.5) | 85 (88.5) |
| Positive | 9 (9.5) | 10 (10.4) |
| Result not available | 0 | 1 (1.0) |
| Factor | Active group, n (%) (N = 95) | Placebo group, n (%) (N = 95) |
|---|---|---|
| Rhinovirus | ||
| Negative | 85 (89.5) | 82 (86.3) |
| Positive | 10 (10.5) | 13 (13.7) |
| Other picornaviruses | ||
| Negative | 93 (97.9) | 92 (96.8) |
| Positive | 2 (2.1) | 3 (3.2) |
| Adenoviruses | ||
| Negative | 95 (100.0) | 95 (100.0) |
| Bocavirus | ||
| Negative | 95 (100.0) | 95 (100.0) |
| Respiratory syncytial virus | ||
| Negative | 92 (96.8) | 91 (95.8) |
| Positive | 3 (3.2) | 4 (4.2) |
| Influenza AH1/AH3/B | ||
| Negative | 95 (100.0) | 95 (100.0) |
| Parainfluenza viruses 1–3 | ||
| Negative | 95 (100.0) | 95 (100.0) |
| Human metapneumovirus | ||
| Negative | 95 (100.0) | 95 (100.0) |
| Coronaviruses 229E and/or OC43 | ||
| Negative | 93 (97.9) | 93 (97.9) |
| Positive | 2 (2.1) | 2 (2.1) |
| Any virus | ||
| Negative | 79 (83.2) | 75 (78.9) |
| Positive | 16 (16.8) | 20 (21.1) |
Pulmonary function tests
Pulmonary function test results at baseline (visit 1) are shown in Table 12 by treatment arm and in Table 13 for the centres that recruited ≥ 10 patients.
| Pulmonary function | n | Mean | SD | P25 | Median | P75 | Min. | Max. |
|---|---|---|---|---|---|---|---|---|
| Active group | ||||||||
| FEV1 (l) | 95 | 1.9 | 0.7 | 1.4 | 1.8 | 2.5 | 0.6 | 4.1 |
| FEV1% predicted | 93 | 63.2 | 21.8 | 48 | 63 | 79 | 16 | 113 |
| FVC (l) | 96 | 2.8 | 1.0 | 2.0 | 2.7 | 3.5 | 0.9 | 5.3 |
| FEV1 : FVC ratio | 94 | 69.7 | 13.3 | 62.0 | 70.0 | 79.0 | 35.0 | 93.0 |
| FEF25–75% (l/second) | 80 | 1.6 | 0.9 | 0.9 | 1.4 | 2.1 | 0.3 | 3.9 |
| FEF50% (l/second) | 76 | 1.9 | 1.1 | 1.1 | 1.7 | 2.6 | 0.3 | 4.5 |
| PEF (l/minute) | 93 | 290 | 104 | 215 | Unchanged | 348 | 71 | Unchanged |
| PEF% predicted | 92 | 65.7 | 23.4 | Unchanged | 67.0 | Unchanged | Unchanged | 137 |
| Placebo group | ||||||||
| FEV1 (l) | 96 | 2.1 | 0.8 | 1.5 | 2.0 | 2.6 | 0.6 | 4.5 |
| FEV1% predicted | 96 | 66.3 | 21.0 | 52.5 | 64.0 | 84.0 | 23.0 | 107 |
| FVC (l) | 96 | 3.1 | 1.0 | 2.4 | 3.0 | 3.6 | 1.3 | 6.9 |
| FEV1 : FVC ratio | 96 | 68.8 | 13.7 | 58.0 | 69.0 | 79.5 | 40.0 | 96.0 |
| FEF25–75% (l/second) | 87 | 1.7 | 1.1 | 0.9 | 1.4 | 2.4 | 0.2 | 5.6 |
| FEF50% (l/second) | 84 | 2.0 | 1.3 | 1.1 | 1.7 | 2.8 | 0.2 | 6.1 |
| PEF (l/minute) | 96 | 323 | 98 | 248 | 341 | 390 | 116 | Unchanged |
| PEF% predicted | 95 | 73.0 | 21.5 | 56.0 | 75.0 | Unchanged | Unchanged | Unchanged |
| Factor | Centre, median (IQR) | |||||
|---|---|---|---|---|---|---|
| BIR (n = 29) | GLA (n = 28) | LEI (n = 10) | POR (n = 56) | SMH (n = 11) | UNT (n = 10) | |
| FEV1 (l) | 2.0 (1.5–2.5) | 2.1 (1.8–2.9) | 2.0 (1.4–2.6) | 1.9 (1.4–2.8) | 1.6 (1.3–2.3) | 1.8 (1.2–2.2) |
| FEV1% predicted | 65 (47–78) | 73 (57–93) | 66 (48–82) | 60 (50–79) | 62 (38–77) | 56 (49–68) |
| FVC (l) | 2.8 (2.3–3.6) | 3.2 (2.6–3.9) | 2.9 (2.5–3.2) | 2.9 (2.1–3.6) | 2.7 (1.9–3.7) | 2.4 (2.0–3.0) |
| FEV1 : FVC ratio | 68 (60–81) | 74 (64–85) | 69 (65–79) | 71 (62–84) | 67 (50–74) | 68 (60–73) |
| FEF25–75% (l/second) | 1.3 (1.0–2.2) | 2.0 (1.3–2.5) | 1.5 (0.9–2.3) | 1.6 (0.9–2.4) | 0.9 (0.5–2.0) | 1.1 (0.7–1.7) |
| FEF50% (l/second) | 1.5 (1.2–2.1) | 2.2 (1.7–2.9) | 1.9 (1.1–3.0) | 2.1 (1.1–2.8) | 1.3 (0.7–2.4) | 1.2 (0.8–2.1) |
| PEF (l/minute) | 284 (211–343) | 369 (260–427) | 270 (193–397) | 329 (264–399) | 322 (244–372) | 262 (162–351) |
| PEF% predicted | 63 (46–83) | 78 (67–93) | 65 (43–82) | 75 (56–88) | 78 (45–98) | 62 (41–75) |
Data completeness
Each patient should have completed four visits: a randomisation visit (visit 1) and three follow-up visits (visits 2–4). The timing of these visits and the associated data collection schedule are shown in Table 14.
| Data collection | Visit 1 (day 1) | Visit 2 (day 5 ±1 daya) | Visit 3 (day 10 ±1 daya) | Visit 4 (day 42 ±15 days) |
|---|---|---|---|---|
| Demographics | ✗ | |||
| Pulmonary function tests | ✗ | ✗ | ✗ | |
| Other biological samples | ✗ | ✗ | ||
| Return diary to investigator | ✗ | ✗ | ||
| Acute AQLQ | ✗ | ✗ | ✗ | |
| Mini AQLQ | ✗ | ✗ | ✗ | |
| AE review | ✗ | ✗ | ✗ |
The number of patients missing each visit is shown in Figure 6. Of the 199 patients randomised, all attended visit 1, which coincided with randomisation, but 21 (11%) missed visit 2, 28 (14%) missed visit 3 and 39 (20%) missed visit 4.
FIGURE 6.
Number of missed visits at each time point (n = 199).

Table 15 shows the pattern of the missed visits; in total, 80% of the patients attended all of the follow-up visits.
| Visits missing | n | % |
|---|---|---|
| None | 159 | 80.0 |
| Visit 2 only | 1 | 0.5 |
| Visits 2 and 4 | 1 | 0.5 |
| Visits 2–4 | 19 | 9.5 |
| Visits 3 and 4 | 9 | 4.5 |
| Visit 4 only | 10 | 5.0 |
| Total | 199 | 100 |
Missing diary cards
Figure 7 shows the extent of the missingness for the diary cards. Day 1 was defined as the day of administration of the study drug (see Chapter 3). The highest level of missingness was observed on day 10 (30%), although the second highest was observed on day 1 (26%).
FIGURE 7.
Number and percentage of missing diary cards by day (n = 199).

A breakdown of diary card missingness for centres with at least 10 recruited patients is shown in Table 16. A relatively high rate of missingness was observed in the largest centre (Queen Alexandra Hospital, Portsmouth), where 41% of the diary cards were missing on the last 2 days.
| Site | Recruited | Day (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| POR | 56 | 39 | 23 | 21 | 29 | 29 | 36 | 32 | 34 | 41 | 41 |
| BIR | 29 | 28 | 14 | 14 | 14 | 14 | 17 | 14 | 14 | 14 | 21 |
| GLA | 28 | 21 | 7 | 7 | 14 | 18 | 18 | 18 | 18 | 18 | 21 |
| SMH | 11 | 27 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 |
| LEI | 10 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 10 | 10 | 20 |
| UNT | 10 | 20 | 10 | 10 | 10 | 20 | 10 | 10 | 10 | 20 | 20 |
| OTH | 55 | 16 | 7 | 9 | 9 | 13 | 11 | 13 | 15 | 15 | 31 |
Table 17 shows the most common patterns of diary card missingness. Regardless of whether day 1 was defined as the day of randomisation or the day of administration of the study drug, 10% of the patients did not complete their diary card on the first day. Apart from that, the missingness of the diary score records can be considered as standard dropout.
| Frequency | % | Day | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| 111 | 63 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| 17 | 10 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| 13 | 7 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| 4 | 2 | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 4 | 2 | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ |
| 3 | 2 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ |
| 2 | 1 | ✗ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 2 | 1 | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 2 | 1 | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 18 | 10 | Other patterns | |||||||||
| Total = 176 | 100 | ||||||||||
We identified two frequent types of missingness in our study data, related to the time of the study drug admission. Some patients (potentially 17) could have received their study drug late in the day and so they started to complete the diary cards on the following day, which means that their first completed diary card was for day 2 and their last diary card was for day 11 by definition. There were some patients (potentially 13) who could have completed their first diary card by assessing their status on the previous day and so they completed diary cards for days 0–9 by definition.
There were also 23 patients who were randomised but who did not return any diary cards.
Missing asthma questionnaires
Asthma questionnaires were missing if a patient did not attend visits 2 or 3. In addition, there were 18 missing overall Acute AQLQ scores (three at visit 1, seven at visit 2 and eight at visit 3) and 18 missing overall Mini AQLQ scores (three at visit 1, seven at visit 2 and eight at visit 3) because one or more items were missing. The missingness was balanced between the treatment arms.
Missing pulmonary function test results
Results from the pulmonary function tests were missing for unattended visits; in addition, some test results were missing for other patients. There were complete results for 160, 150 and 142 patients at visits 1, 2 and 3, respectively (out of a possible 199, 171 and 163 patients who attended visits 1, 2 and 3, respectively). Most of the missing results were for FEF25−75% and FEF50%, which were missing in five cases out of the six recruited patients at Rowden Medical Partnership (GP surgery), Chippenham, and in five cases out of the eight recruited patients at New Cross Hospital, Wolverhampton.
Primary outcome analysis
Exploratory analysis of the primary outcome
As a check for outliers and imbalances, a series of longitudinal plots (one for each centre) of diary score for each patient, differentiating between treatment arms, were produced (see Appendix 1). Figure 8 shows the mean diary score and standard error (SE) for each treatment arm on each day. Box plots of diary scores by treatment arm for each day were also produced to show the distribution of the observed scores graphically (Figure 9). Table 18 shows the observed mean diary scores and SDs for each treatment arm by day and the numbers of observations. Additionally, a table of summary statistics of the diary scores by day and treatment arm was produced, including the numbers of observations, means, SDs, medians and lower and upper quartiles (Table 19).
FIGURE 8.
Observed symptom diary scores by day (with SEs). Reproduced with permission from JAMA Internal Medicine. 2016. http://dx.doi.org/10.1001/jamainternmed.2016.5664. Copyright © 2016 American Medical Association. All rights reserved.

FIGURE 9.
Box plots of observed symptom diary scores. Vertical bars represent the highest/lowest value within 1.5× the interquartile range from the upper/lower quartile. Closed circle represents value(s) outside 1.5× the interquartile range from the upper/lower quartile. Reproduced with permission from JAMA Internal Medicine. 2016. http://dx.doi.org/10.1001/jamainternmed.2016.5664. Copyright © 2016 American Medical Association. All rights reserved.

| Group | Day | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Placebo, mean (SD) | 4.18 (1.48) | 3.45 (1.62) | 3.12 (1.47) | 3.04 (1.57) | 2.87 (1.58) | 2.79 (1.56) | 2.80 (1.69) | 2.43 (1.53) | 2.32 (1.55) | 2.20 (1.51) |
| n | 77 | 86 | 85 | 81 | 81 | 80 | 79 | 77 | 74 | 68 |
| Active, mean (SD) | 4.14 (1.38) | 3.51 (1.42) | 3.09 (1.45) | 2.78 (1.58) | 2.63 (1.51) | 2.44 (1.54) | 2.19 (1.53) | 2.24 (1.61) | 2.22 (1.71) | 2.09 (1.71) |
| n | 71 | 85 | 86 | 84 | 80 | 78 | 81 | 80 | 78 | 71 |
| Day | Placebo group | Active group | ||||
|---|---|---|---|---|---|---|
| n | Diary score, mean (SD) | Diary score, median (IQR) | n | Diary score, mean (SD) | Diary score, median (IQR) | |
| 1 | 77 | 4.18 (1.48) | 4.25 (3.00–5.50) | 71 | 4.14 (1.38) | 4.25 (3.25–5.00) |
| 2 | 86 | 3.45 (1.62) | 3.50 (2.25–5.00) | 85 | 3.51 (1.42) | 3.75 (2.50–4.75) |
| 3 | 85 | 3.12 (1.47) | 3.00 (2.00–4.25) | 86 | 3.09 (1.45) | 3.00 (2.00–4.25) |
| 4 | 81 | 3.04 (1.57) | 3.25 (1.75–4.25) | 84 | 2.78 (1.58) | 2.50 (1.50–4.00) |
| 5 | 81 | 2.87 (1.58) | 3.00 (1.50–4.25) | 80 | 2.63 (1.51) | 2.50 (1.38–3.75) |
| 6 | 80 | 2.79 (1.56) | 2.63 (1.50–4.00) | 78 | 2.44 (1.54) | 2.25 (1.25–3.75) |
| 7 | 79 | 2.80 (1.69) | 3.00 (1.25–4.00) | 81 | 2.19 (1.53) | 2.25 (1.00–3.25) |
| 8 | 77 | 2.43 (1.53) | 2.50 (1.00–3.50) | 80 | 2.24 (1.61) | 2.00 (1.00–3.50) |
| 9 | 74 | 2.32 (1.55) | 2.38 (1.00–3.25) | 78 | 2.22 (1.71) | 2.00 (0.75–3.25) |
| 10 | 68 | 2.20 (1.51) | 2.25 (0.88–3.25) | 71 | 2.09 (1.71) | 1.75 (0.50–3.50) |
Results
A linear change was assumed in the model for the diary score over time, with different slopes for the two treatment arms. Additionally, equal mean scores were assumed at baseline for the two groups as any inequality could have occurred only by chance, because of randomisation. To reduce bias caused by the observed difference at baseline, the main effect of the interaction term was not included in the model as an independent covariate. Sensitivity analysis with the inclusion of this covariate was conducted. The estimated mean diary score at baseline (day 1) in the whole study population was 3.66 (95% CI 3.41 to 3.90). The decrease in diary score observed in the azithromycin group was slightly greater than the decrease in diary score observed in the placebo group. On average the difference in change in diary scores from baseline for the azithromycin group compared with the placebo group was –0.018 per day (95% CI –0.074 to 0.037). The estimated differences with their 95% CIs for each day are provided in Table 20. The mean ‘natural’ background daily decrease in diary score (decrease in the placebo group) was –0.18 (95% CI for the first day alone –0.22 to 0.14). On day 10, the difference between the two groups was not statistically significant, with the estimated mean diary score being lower in the azithromycin group by –0.166 (95% CI –0.670 to 0.337). On day 5 the difference between the groups was –0.074 (95% CI –0.298 to 0.150).
| Day | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Difference in change from baseline (95% CI) | 0 | –0.018 (–0.074 to 0.037) | –0.037 (–0.149 to 0.075) | –0.055 (–0.223 to 0.112) | –0.074 (–0.298 to 0.150) | –0.092 (–0.372 to 0.187) | –0.111 (–0.446 to 0.224) | –0.129 (–0.521 to 0.262) | –0.148 (–0.595 to 0.299) | –0.166 (–0.670 to 0.337) |
Secondary outcome analysis
For all secondary outcomes, an exploratory analysis and assessment of missing data were completed prior to the main analysis. This was analogous to that outlined for the primary outcome. Multilevel models, similar to those specified for the primary outcome, were used to analyse Acute AQLQ and Mini AQLQ scores and also the pulmonary function tests. Details of the models used can be found in Appendix 1. The assessment of missingness for the Acute AQLQs and Mini AQLQs and the pulmonary function tests has been described earlier in this chapter. All presented secondary outcome analyses were prespecified in the SAP.
Acute Asthma Quality of Life Questionnaire and Mini Asthma Quality of Life Questionnaire analysis
Figure 10 shows the mean Acute AQLQ scores and SEs for each treatment arm at each visit. Box plots of observed Acute AQLQ scores by treatment arm for each visit are shown in Figure 11.
FIGURE 10.
Observed mean Acute AQLQ scores and SEs by treatment arm and visit. Reproduced with permission from JAMA Internal Medicine. 2016. http://dx.doi.org/10.1001/jamainternmed.2016.5664. Copyright © 2016 American Medical Association. All rights reserved.

FIGURE 11.
Box plots of observed Acute AQLQ scores by treatment arm and visit. Vertical bars represent the highest/lowest value within 1.5× the interquartile range from the upper/lower quartile. Closed circle represents value(s) outside 1.5× the interquartile range from the upper/lower quartile (circles for active group, diamonds for placebo group). Reproduced with permission from JAMA Internal Medicine. 2016. http://dx.doi.org/10.1001/jamainternmed.2016.5664. Copyright © 2016 American Medical Association. All rights reserved.
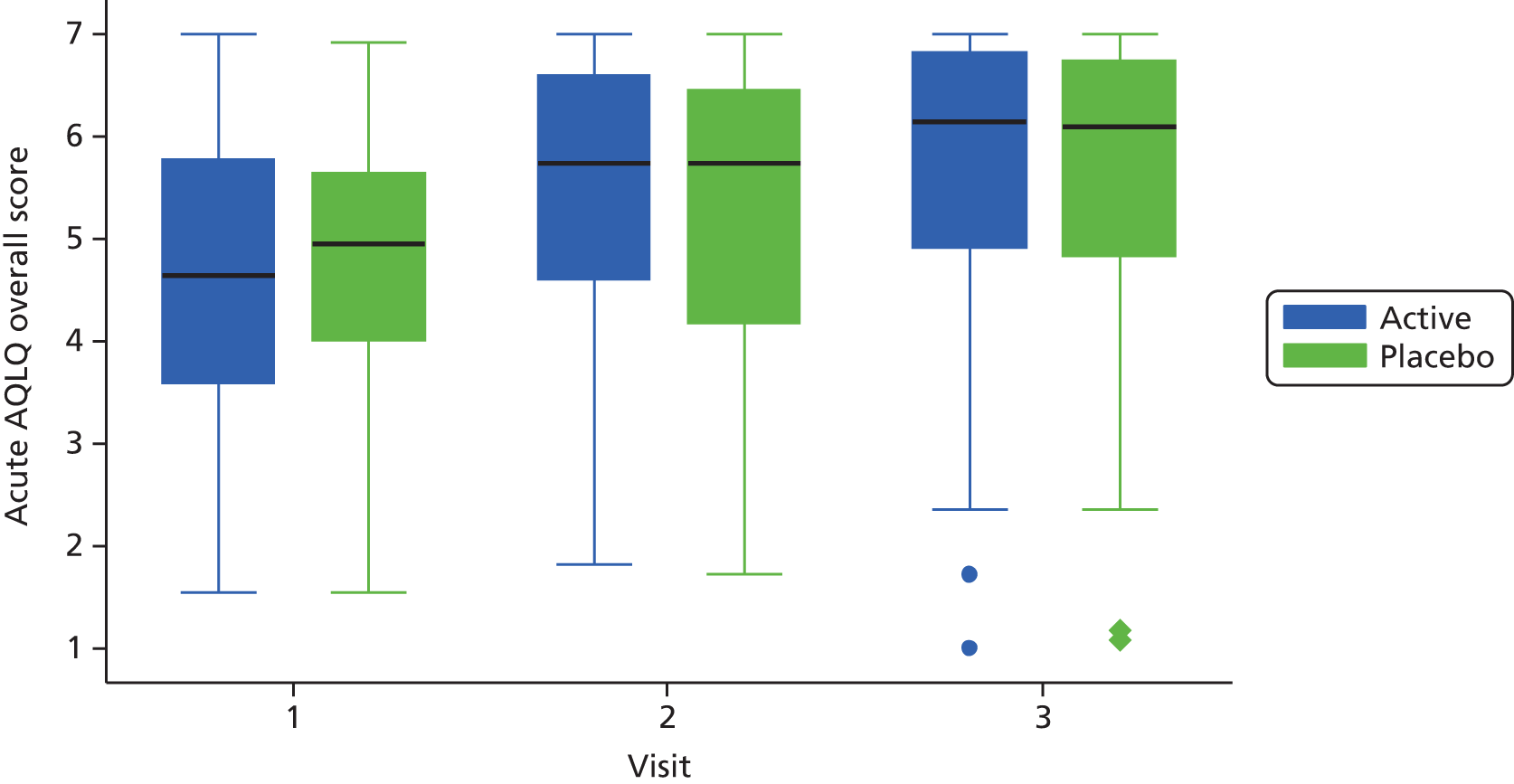
Table 21 shows the statistics of the Acute AQLQ scores for each treatment arm by visit and the numbers of observations.
| Visit | n | Acute AQLQ score | ||||
|---|---|---|---|---|---|---|
| Mean | SD | Median | P25 | P75 | ||
| Placebo group | ||||||
| 1 | 100 | 4.8 | 1.3 | 5.0 | 4.0 | 5.6 |
| 2 | 87 | 5.3 | 1.4 | 5.7 | 4.2 | 6.4 |
| 3 | 83 | 5.6 | 1.5 | 6.1 | 4.8 | 6.7 |
| Active group | ||||||
| 1 | 96 | 4.6 | 1.4 | 4.6 | 3.6 | 5.8 |
| 2 | 84 | 5.4 | 1.3 | 5.7 | 4.6 | 6.6 |
| 3 | 80 | 5.6 | 1.5 | 6.1 | 4.9 | 6.8 |
As for the primary outcome, multilevel modelling was carried out assuming equal mean scores at baseline and a linear change for the Acute AQLQ and Mini AQLQ scores over time, with different slopes for the two treatment arms. Differences in the change in Acute AQLQ scores from baseline for each visit for the azithromycin group compared with the placebo group are provided in Table 22. At visit 3 (day 10) there was no statistically significant difference between the two groups. According to the model, at visit 3 the AQLQ score was higher by 0.130 (95% CI –0.276 to 0.539) in the azithromycin group than in the placebo group. Details of the model can be found in Appendix 1.
| Acute AQLQ score | Visit 1 (day 1) | Visit 2 (day 5) | Visit 3 (day 10) |
|---|---|---|---|
| Difference in change from baseline (95% CI) | 0 | 0.065 (–0.138 to 0.269) | 0.130 (–0.276 to 0.539) |
The same analyses that were conducted for the Acute AQLQ scores were conducted for the Mini AQLQ scores. Figure 12 shows the mean Mini AQLQ scores and SEs for each treatment arm and visit. Box plots of Mini AQLQ scores by treatment arm and visit are shown in Figure 13. Table 23 shows the statistics of the Mini AQLQ scores for each treatment arm and visit.
FIGURE 12.
Observed mean Mini AQLQ scores and SEs by treatment arm and visit. Reproduced with permission from JAMA Internal Medicine. 2016. http://dx.doi.org/10.1001/jamainternmed.2016.5664. Copyright © 2016 American Medical Association. All rights reserved.
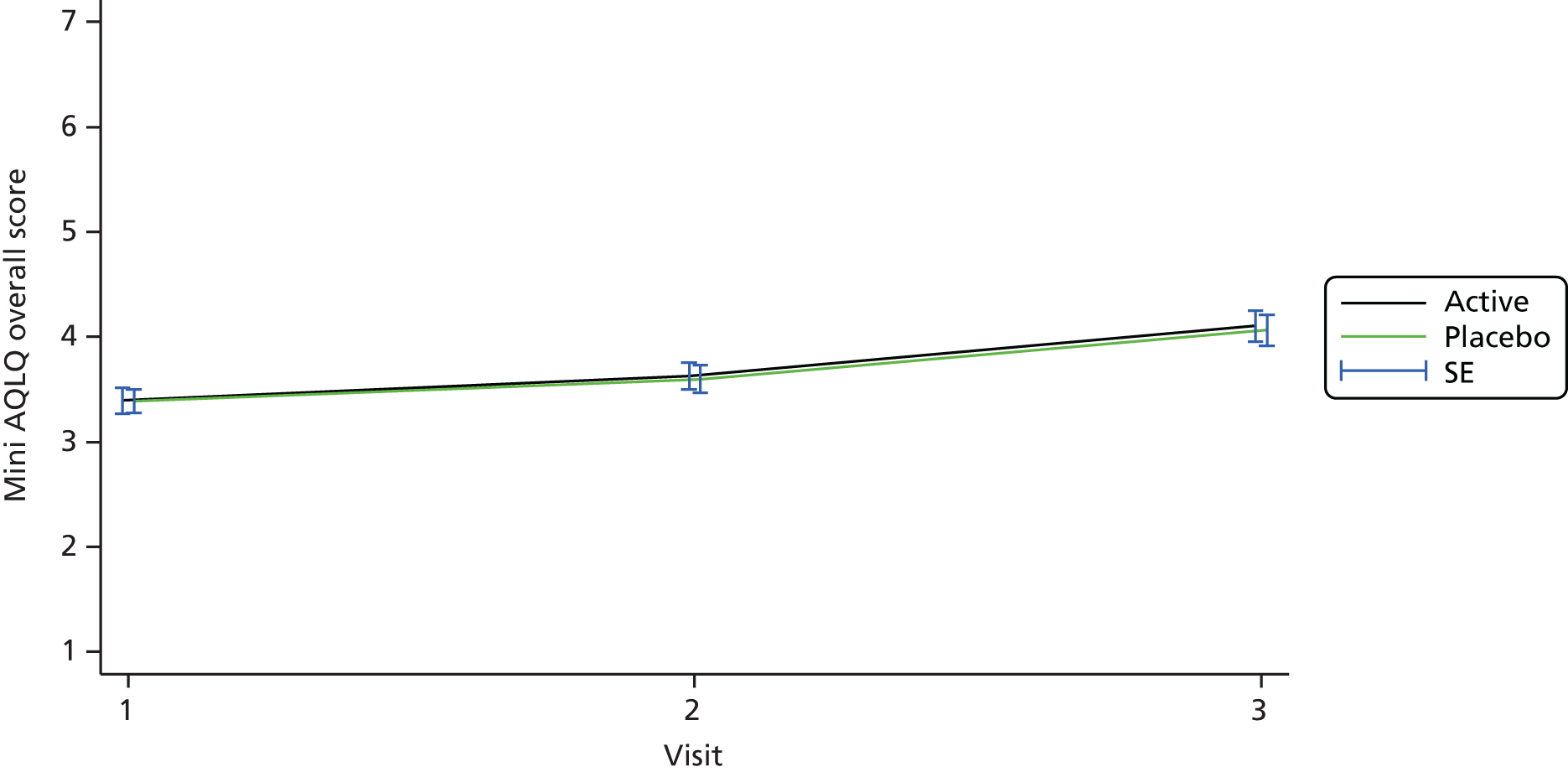
FIGURE 13.
Box plots of observed Mini AQLQ scores by treatment arm and visit. Vertical bars represent the highest/lowest value within 1.5× the interquartile range from the upper/lower quartile. Closed circle represents value(s) outside 1.5× the interquartile range from the upper/lower quartile (circles for active group, diamonds for placebo group). Reproduced with permission from JAMA Internal Medicine. 2016. http://dx.doi.org/10.1001/jamainternmed.2016.5664. Copyright © 2016 American Medical Association. All rights reserved.
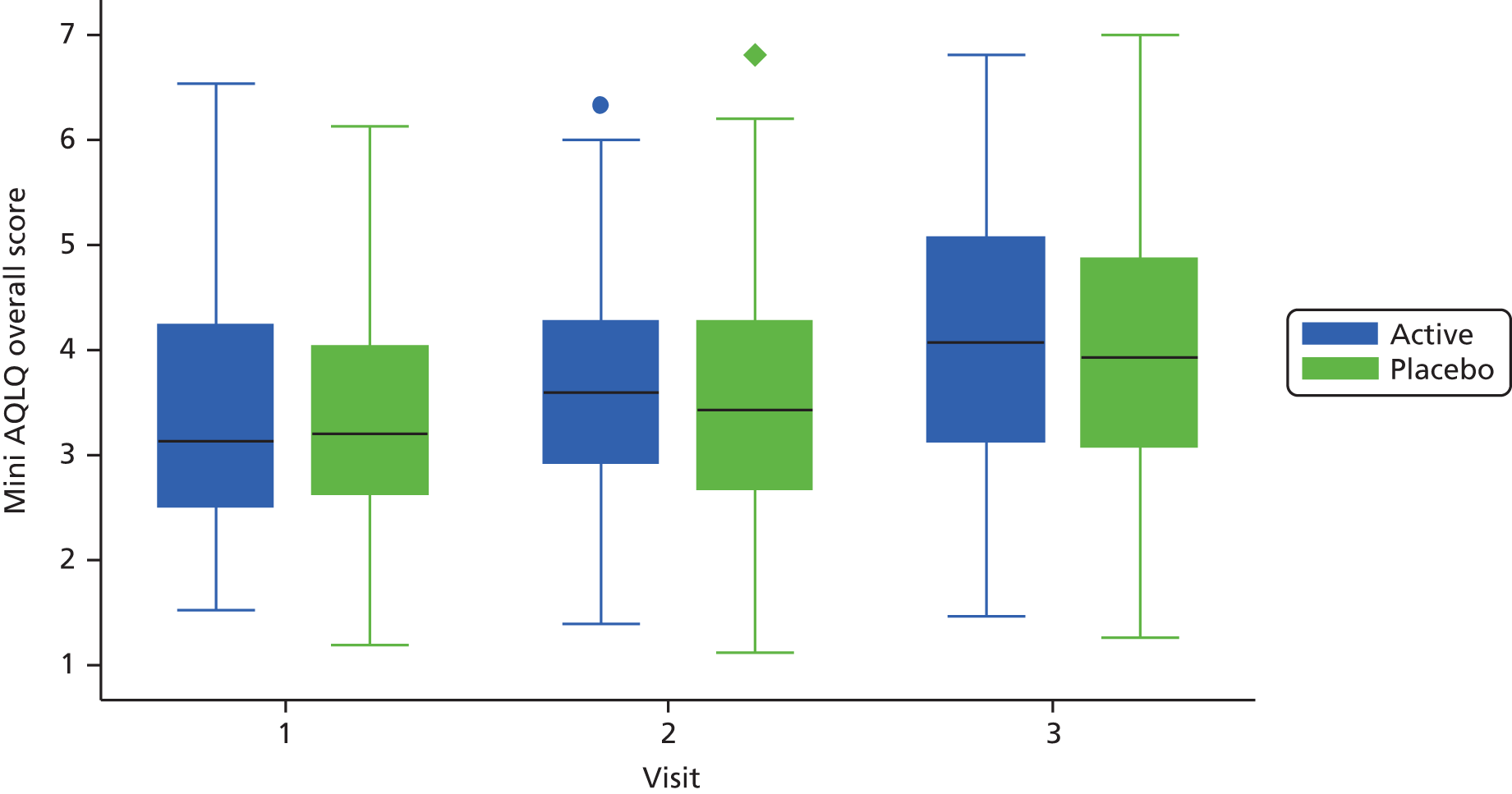
| Visit | Mini AQLQ score | ||||
|---|---|---|---|---|---|
| Mean | SD | Median | P25 | P75 | |
| Placebo group | |||||
| 1 | 3.4 | 1.1 | 3.2 | 2.6 | 4.0 |
| 2 | 3.6 | 1.2 | 3.4 | 2.7 | 4.3 |
| 3 | 4.1 | 1.3 | 3.9 | 3.1 | 4.9 |
| Active group | |||||
| 1 | 3.4 | 1.2 | 3.1 | 2.5 | 4.2 |
| 2 | 3.6 | 1.1 | 3.6 | 2.9 | 4.3 |
| 3 | 4.1 | 1.3 | 4.1 | 3.1 | 5.1 |
Differences in the change in Mini AQLQ scores from baseline for each visit with 95% CIs for azithromycin compared with placebo are shown in Table 24. At visit 3 (day 10) there was no statistically significant difference between the two groups. According to the model, at visit 3 the Mini AQLQ score was lower by –0.042 (95% CI –0.409 to 0.325) in the azithromycin group than in the placebo group. Details of the model can be found in Appendix 1.
| Mini AQLQ score | Visit 1 (day 1) | Visit 2 (day 5) | Visit 3 (day 10) |
|---|---|---|---|
| Difference in change from baseline (95% CI) | 0 | –0.020 (–0.204 to 0.163) | –0.042 (–0.409 to 0.325) |
Pulmonary function test analysis
For the pulmonary function tests, similar exploratory analyses and multilevel modelling were conducted as for the AQLQ scores. Details of the models used can be found in Appendix 1. Table 25 shows the observed pulmonary function test values (mean and SE) for each visit by treatment arm.
| Test | Active groupa | Placebo groupa | ||||
|---|---|---|---|---|---|---|
| Visit 1 day(n = 97) | Visit 2 day (n = 85) | Visit 3 day (n = 80) | Visit 1 day (n = 101) | Visit 2 day (n = 90) | Visit 3 day (n = 83) | |
| FEV1 (l), mean (SD) | 1.94 (0.74) | 2.23 (0.77) | 2.30 (0.83) | 2.11 (0.79) | 2.34 (0.83) | 2.38 (0.91) |
| FVC (l), mean (SD) | 2.80 (1.03) | 3.13 (1.00) | 3.25 (1.08) | 3.09 (1.05) | 3.40 (1.10) | 3.38 (1.09) |
| FEV1 : FVC ratio, mean (SD) | 69.66 (13.33) | 71.71 (12.02) | 71.00 (12.38) | 68.83 (13.71) | 69.28 (12.24) | 70.02 (12.71) |
| FEF25–75% (l/second), mean (SD) | 1.59 (0.89) | 1.85 (0.94) | 1.77 (0.92) | 1.74 (1.14) | 1.83 (1.08) | 1.94 (1.20) |
| FEF50% (l/second), mean (SD) | 1.92 (1.06) | 2.12 (1.05) | 2.19 (1.08) | 2.04 (1.26) | 2.15 (1.24) | 2.32 (1.35) |
| PEF (l/minute), mean (SD) | 289.9 (104.0) | 349.6 (103.5) | 364.8 (108.3) | 323.5 (97.9) | 353.5 (104.2) | 361.2 (111.9) |
Table 26 shows the estimated differences in change in pulmonary function from baseline with 95% CIs for azithromycin compared with placebo.
| Test | Difference in change vs. placebo at visit 3 (day 10) (95% CI) | Difference in change vs. placebo at visit 2 (day 5) (95% CI) | Per-visit change in placebo (95% CI) | Baseline average (95% CI) |
|---|---|---|---|---|
| FEV1 (l), mean (SD) | 0.050 (–0.132 to 0.231) | 0.024 (–0.067 to 0.116) | 0.164 (0.099 to 0.228) | 2.011 (1.875 to 2.146) |
| FVC (l), mean (SD) | 0.038 (–0.166 to 0.243) | 0.019 (–0.083 to 0.122) | 0.200 (0.127 to 0.272) | 2.959 (2.809 to 3.110) |
| FEV1 : FVC ratio, mean (SD) | 1.379 (–1.559 to 4.316) | 0.689 (–0.779 to 2.158) | 0.365 (–0.732 to 1.463) | 69.5 (67.7 to 71.4) |
| FEF25–75% (l/second), mean (SD) | 0.036 (–0.192 to 0.265) | 0.018 (–0.096 to 0.132) | 0.116 (0.035 to 0.197) | 1.631 (1.470 to 1.792) |
| FEF50% (l/second), mean (SD) | 0.045 (–0.234 to 0.324) | 0.022 (–0.117 to 0.162) | 0.161 (0.062 to 0.260) | 1.931 (1.750 to 2.112) |
| PEF (l/minute), mean (SD) | 19.57 (–6.81 to 45.94) | 9.784 (–3.404 to 22.97) | 25.07 (15.49 to 34.65) | 304.8 (285.3 to 324.3) |
Time to a 50% reduction in symptom score analysis
Time from day 1 to a 50% reduction in the initial symptom score was calculated using the mean daytime scores from the diary cards. This variable was calculated as the number of days until the first occasion on which the score was at least 50% lower than the day 1 score (it is possible for this score to come down but then to increase above 50% again). Patients who reached the 50% reduction were considered to have had this event; all other patients were treated as censored at the day of their last recorded diary score (usually day 10 but earlier for patients with missing diary card scores). The observed median time to a 50% reduction in symptom score was 7 days in the placebo group and 6 days in the active group.
Kaplan–Meier curves of time to a 50% reduction in symptom score for each treatment arm (truncated at 10 days) are shown in Figure 14.
FIGURE 14.
Kaplan–Meier curves of time to a 50% reduction in symptom score for each treatment arm (truncated at 10 days). Reproduced with permission from JAMA Internal Medicine. 2016. http://dx.doi.org/10.1001/jamainternmed.2016.5664. Copyright © 2016 American Medical Association. All rights reserved.

Substudies
The same model as outlined for the primary outcome was used for the subgroup analyses, which included the following:
-
bacterial culture positive or negative in sputum (Table 27)
-
viral tests positive or negative on nasal swab, throat swab or sputum (Table 28)
-
atypical bacteria positive or negative on nasal swab, throat swab, sputum or serological testing (Table 29)
-
no subgroup analyses defined on sputum cell count characteristics were performed as the numbers per group were too low to be meaningful.
-
the observed mean daily diary scores and error bars during the follow-up by treatment are presented for the any bacterial test-positive subgroup (Figure 15), the atypical bacterial test-positive subgroup (Figure 16) and the bacterial culture positive in sputum subgroup (Figure 17).
| Group | Whole study population (n = 176) | Bacterial testing in sputum missing (n = 93) | Bacterial culture in sputum positive (n = 12) | Bacterial culture in sputum negative (n = 71) |
|---|---|---|---|---|
| Day 10 difference in change from baseline (95% CI) | –0.166 (–0.670 to 0.337) | –0.114 (–0.821 to 0.594) | 1.178 (–0.497 to 2.853) | –0.410 (–1.183 to 0.364) |
| Group | Whole study population (n = 176) | Viral testing positive (n = 31) | Viral testing negative (n = 138) |
|---|---|---|---|
| Day 10 difference in change from baseline (95% CI) | –0.166 (–0.670 to 0.337) | –0.100 (–1.170 to 0.969) | –0.106 (–0.683 to 0.472) |
| Group | Whole study population (n = 176) | Atypicala bacteria positive (n = 8b) | Atypicala bacteria negative (n = 157) | Any bacterial test positive (n = 20) |
|---|---|---|---|---|
| Day 10 difference in change from baseline (95% CI) | –0.166 (–0.670 to 0.337) | 1.391 (–1.214 to 3.996) | 0.044 (–0.465 to 0.554) | 0.198 (–1.546 to 1.942) |
FIGURE 15.
Observed mean diary scores and SEs for the any bacterial test-positive subgroup (n = 20) by treatment arm.

FIGURE 16.
Observed mean diary scores and SEs for the atypical bacterial test-positive subgroup (n = 8) by treatment arm.
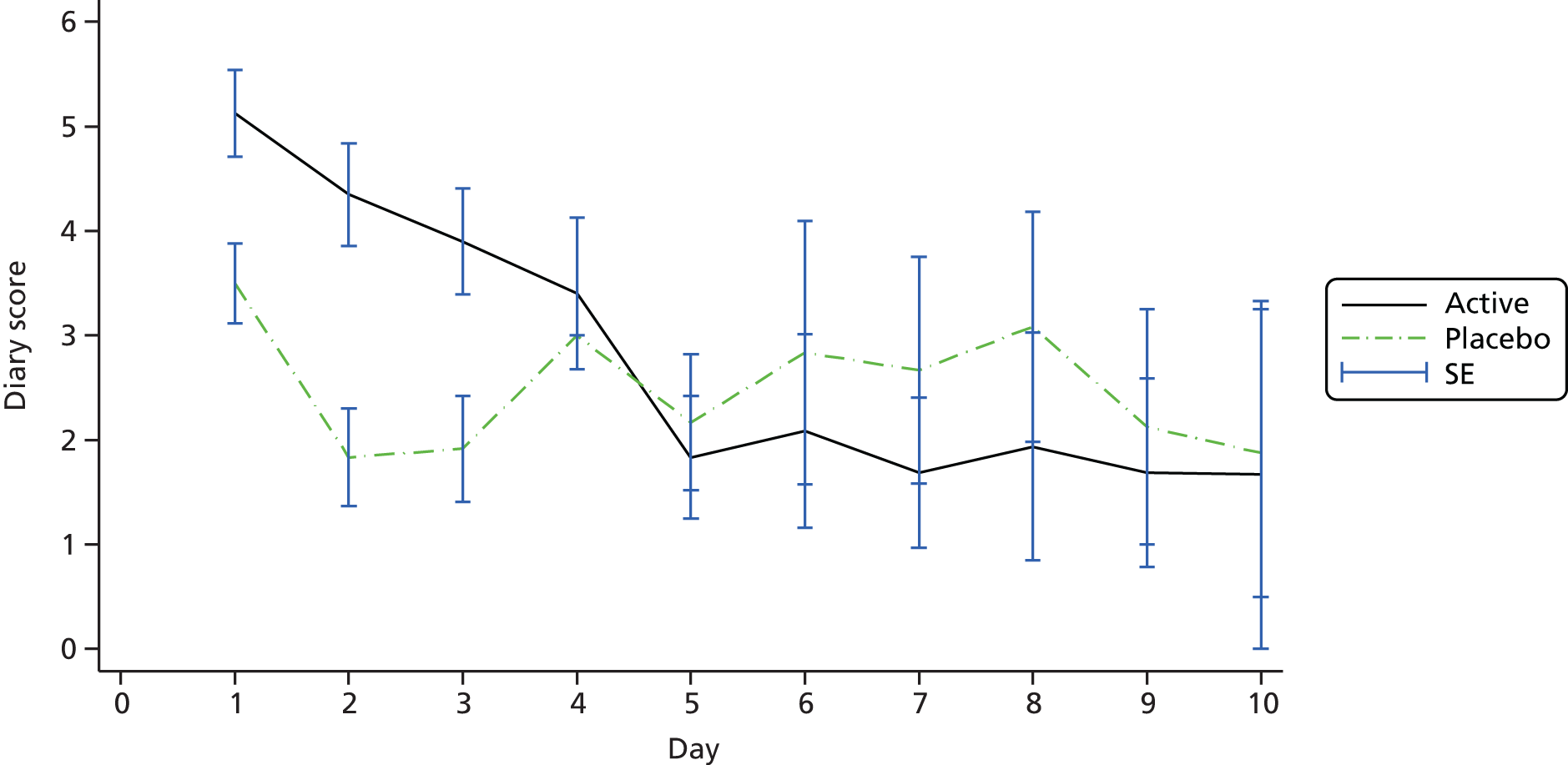
FIGURE 17.
Observed mean diary scores and SEs for the bacterial culture positive in sputum subgroup (n = 12) by treatment arm.
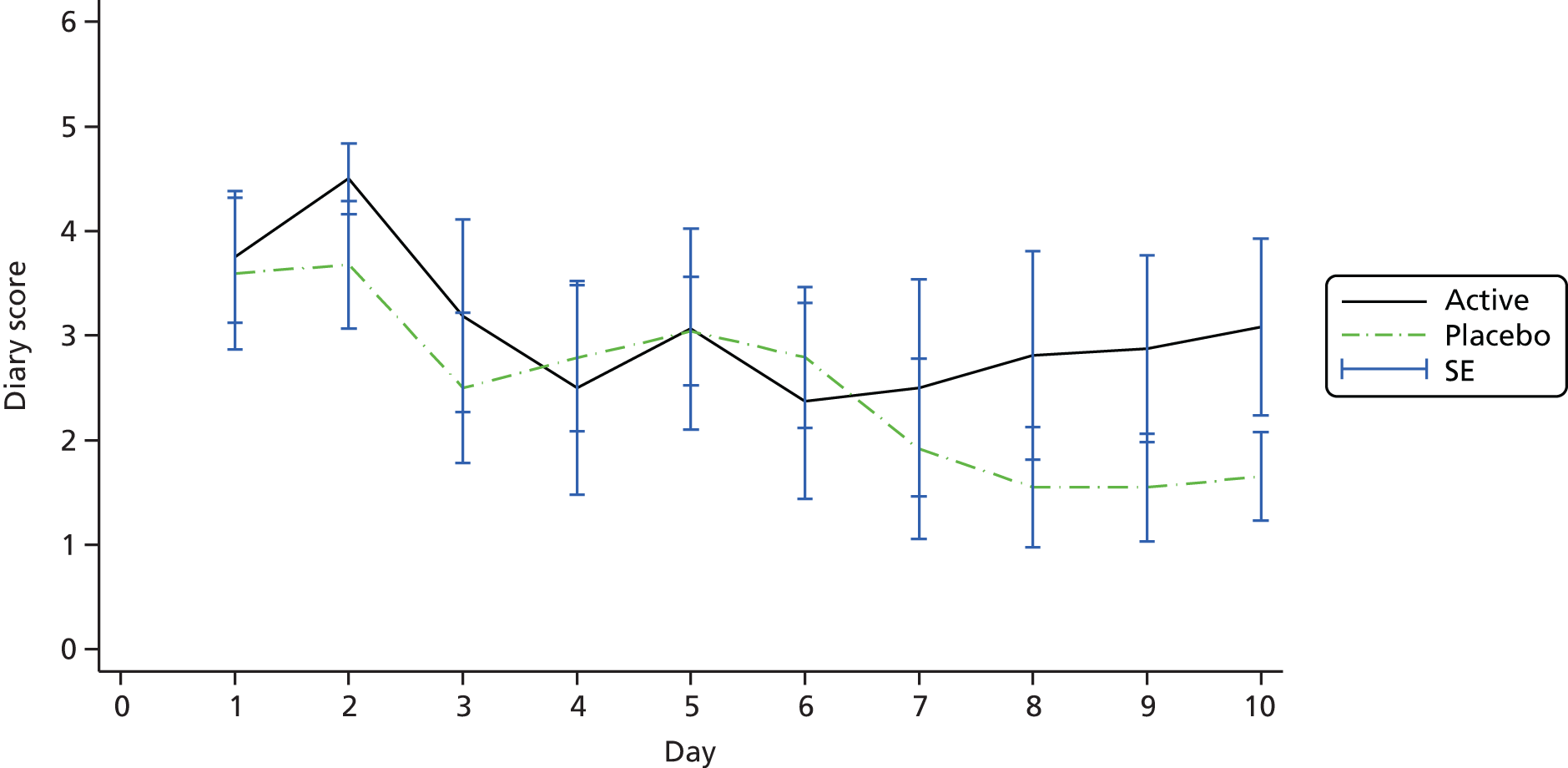
Protocol deviations
Protocol deviations as recorded on the protocol deviation form in the InForm database are summarised by site and category in Table 30.
| Categoryb | Centre | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A23 | A24 | A26 | A30 | BIR | BVH | GLA | LEI | NNU | NOC | NOT | POR | SMH | STJ | UNT | Total | |
| DNA visit | 0 | 0 | 0 | 0 | 8 | 2 | 6 | 1 | 0 | 0 | 0 | 4 | 1 | 0 | 0 | 22 |
| Incomplete Acute AQLQ | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Incomplete Mini AQLQ | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Incomplete set of symptom diaries | 0 | 0 | 0 | 0 | 1 | 0 | 7 | 2 | 0 | 0 | 1 | 16 | 0 | 0 | 1 | 28 |
| Nasal mucus not collected | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| No haematology | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| Prohibited medication | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Spirometry | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 3 | 0 | 0 | 1 | 6 |
| Sputum sample | 1 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 |
| Study drug compliance | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Visit outside protocol schedule | 0 | 1 | 2 | 0 | 0 | 2 | 0 | 4 | 0 | 2 | 0 | 0 | 1 | 1 | 4 | 17 |
| Vital signs not performed | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| Withdrawn/ineligible | 3 | 1 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 10 |
| Total | 4 | 2 | 4 | 3 | 14 | 4 | 18 | 7 | 2 | 4 | 1 | 25 | 2 | 1 | 7 | 98 |
Safety data analysis
Protocol reporting of AEs took place from the time that patients gave informed consent until 7 days after the last dose of study medication. Using the information recorded on the AE eCRF, each AE was categorised using MedDRA (Medical Dictionary for Regulatory Activities) coding system organ class (SOC) terms by a designee or the chief investigator. The numbers of AEs and numbers of patients affected in each category by treatment arm are provided in Tables 31 and 32, respectively.
| AE categorya | Active group, n | Placebo group, n | Total, n |
|---|---|---|---|
| Cardiac disorders | 4 | 2 | 6 |
| Eye disorders | 2 | 1 | 3 |
| Gastrointestinal disorders | 35 | 24 | 59 |
| General disorders | 18 | 25 | 43 |
| Infections and infestations | 0 | 1 | 1 |
| Musculoskeletal and connective tissue disorders | 4 | 6 | 10 |
| Nervous system disorders | 15 | 14 | 29 |
| Psychiatric disorders | 1 | 2 | 3 |
| Reproductive system and breast disorders | 0 | 1 | 1 |
| Respiratory, thoracic and mediastinal disorders | 27 | 37 | 64 |
| Skin and subcutaneous disorders | 0 | 1 | 1 |
| Total | 106 | 114 | 220 |
| AE categoryb | Active group, n | Placebo group, n | Total, n |
|---|---|---|---|
| Cardiac disorders | 4 | 2 | 6 |
| Eye disorders | 2 | 1 | 3 |
| Gastrointestinal disorders | 25 | 20 | 45 |
| General disorders | 16 | 19 | 35 |
| Infections and infestations | 0 | 1 | 1 |
| Musculoskeletal and connective tissue disorders | 3 | 4 | 7 |
| Nervous system disorders | 14 | 13 | 27 |
| Psychiatric disorders | 1 | 2 | 3 |
| Reproductive system and breast disorders | 0 | 1 | 1 |
| Respiratory, thoracic and mediastinal disorders | 20 | 28 | 48 |
| Skin and subcutaneous disorders | 0 | 1 | 1 |
| Total number of adverse eventsc (number of patients affected) | 85 (51) | 92 (52) | 177 (103) |
Table 33 shows the numbers of AEs by category and relationship to study medication. The relationship is missing for four AEs and these are shown as ‘unknown’. No AEs were definitely related to the study medication.
| AE categorya | Relationship to study medication, n | |||||
|---|---|---|---|---|---|---|
| Not related | Unlikely | Possible | Probable | Unknown | Total | |
| Cardiac disorders | 3 | 2 | 1 | 0 | 0 | 6 |
| Eye disorders | 1 | 2 | 0 | 0 | 0 | 3 |
| Gastrointestinal disorders | 9 | 5 | 36 | 7 | 2 | 59 |
| General disorders | 20 | 11 | 11 | 0 | 1 | 43 |
| Infections and infestations | 1 | 0 | 0 | 0 | 0 | 1 |
| Musculoskeletal and connective tissue disorders | 6 | 3 | 1 | 0 | 0 | 10 |
| Nervous system disorders | 8 | 13 | 8 | 0 | 0 | 29 |
| Psychiatric disorders | 0 | 3 | 0 | 0 | 0 | 3 |
| Reproductive system and breast disorders | 1 | 0 | 0 | 0 | 0 | 1 |
| Respiratory, thoracic and mediastinal disorders | 49 | 14 | 0 | 0 | 1 | 64 |
| Skin and subcutaneous disorders | 0 | 0 | 1 | 0 | 0 | 1 |
| Total | 98 | 53 | 58 | 7 | 4 | 220 |
Multiple AEs were reported for some patients, with 51 patients (just less than half of those with AEs) reporting more than one AE. Ten AEs were reported for one patient. Table 34 provides further detail about the distribution of the 220 AEs between the 103 patients who reported AEs.
| Number of AEs | Active group, n | Placebo group, n | Total, n |
|---|---|---|---|
| 1 | 24 | 28 | 52 |
| 2 | 12 | 9 | 21 |
| 3 | 7 | 6 | 13 |
| 4 | 4 | 4 | 8 |
| 5 | 3 | 2 | 5 |
| 6 | 1 | 1 | 2 |
| 8 | 0 | 1 | 1 |
| 10 | 0 | 1 | 1 |
| Total | 51 | 52 | 103 |
Details of the AEs classified as cardiac disorders are given in Table 35. None of these was classified as a SAE. SAEs reported are described in Tables 36 and 37.
| Age (years) | Subject | Arm | Description | Site | Relation | Severity | Outcome | Actiona | Duration |
|---|---|---|---|---|---|---|---|---|---|
| 26 | NOC072 | Placebo | Chest pain | NOC | Not related | Moderate | Recovered | None | Intermittent |
| 36 | NOC077 | Active | Chest pain | NOC | Not related | Mild | Not yet recovered | None | Continuous |
| 22 | POR086 | Active | Palpitations | POR | Unlikely | Mild | Recovered | None | Intermittent |
| 38 | POR120 | Active | Chest pain and pain under left arm pit | POR | Unlikely | Mild | Recovered | None | Single episode |
| 55 | POR195 | Active | Chest pain | POR | Not related | Mild | Recovered | None | Single episode |
| 42 | SMH058 | Placebo | Feeling of tachycardia | SMH | Possible | Mild | Recovered | None | Single episode |
| Age (years) | Subject | Arm | Classification | Action taken | Event description | Site | Relation to study drug | Severity |
|---|---|---|---|---|---|---|---|---|
| 18 | GLA052 | Placebo | Serious | Hospitalisation required | Patient became wheezy and short of breath on 13 October 2012, presented to A&E on 14 October 2012 and was admitted overnight. Diagnosis was exacerbation of asthma | GLA | Unlikely | Moderate |
| 22 | NNU152 | Placebo | Serious | Hospitalisation required | Exacerbation of underlying asthma. Admitted to hospital at 0900 on 7 October 2013 with extreme symptoms of breathlessness | NNU | Not related | Severe |
| 47 | A32197 | Placebo | Serious | Hospitalisation required | Acute exacerbation of asthma | A32 | Not related | Moderate |
| 49 | A29207 | Active | Serious | Hospitalisation required | Shortness of breath and wheeze – non-infective exacerbation of asthma | A29 | Not related | Moderate |
| Subject | Frequency | Comments | Ongoing | Outcome | Category |
|---|---|---|---|---|---|
| GLA052 | Single episode | No | Recovered | Respiratory, thoracic and mediastinal disorders | |
| NNU152 | Unknown | Continuation of patient’s existing underlying condition. Classed as an AE | No | Recovered | Respiratory, thoracic and mediastinal disorders |
| A32197 | Single episode | Admitted to hospital in Chester for 3 nights with asthma exacerbation | No | Recovered | Respiratory, thoracic and mediastinal disorders |
| A29207 | Single episode | Patient was admitted with shortness of breath and kept in overnight | Yes | Not yet recovered | Respiratory, thoracic and mediastinal disorders |
Compliance with study drug schedule
Any unused drug was collected during the end of therapy visit (visit 2, day 5) and study medication compliance was assessed by counting unused capsules. Using this information, further details are provided in Table 38 on the protocol deviations associated with study drug compliance, including the level of compliance and additional comments by treatment arm.
| Subject | Quantity of study treatment returned | Participant | Treatment arm | Protocol deviation | Comment |
|---|---|---|---|---|---|
| A26119 | 3 | Partially compliant | Active | Acute AQLQ missing data at visit 2 | Has only four diary cards |
| NOC050 | 4 | Partially compliant | Placebo | Patient withdrawn from study after first dose | |
| Recorded as a form comment, not protocol deviation | |||||
| POR051 | 4 | Compliant | Placebo | Patient withdrawn from study by investigator, compliant with study medication up to withdrawal. Patient was not uncompliant | |
| Recorded as form comment, not protocol deviation | |||||
| POR041 | 2 | Compliant | Active | – | – |
Study limitations
Recruitment proved extremely challenging because of a milder than expected winter during the recruitment period, the challenges of recruiting from A&E departments that were often distant from the respiratory research teams and the number of patients already receiving antibiotics. To counteract this, further recruiting centres were opened, the inclusion criteria were relaxed and the recruitment time was extended. However, despite all of these efforts only 199 subjects were recruited by the medication expiry date and, in the absence of any further funding, the study had to be terminated despite not reaching its recruitment target. It is therefore possible that the study was underpowered to detect a therapeutic benefit.
It is also possible that patients who might potentially have benefited from antibiotic therapy for their asthma exacerbation were excluded from the study because they had already received them. The population remaining to be randomised could then theoretically have been selected against for antibiotic responsiveness, through having no clinical indication that antibiotic therapy might be of benefit. It is likely, therefore, that those not prescribed antibiotics were strongly negatively selected against for suitability for antibiotics.
This study required all randomised patients to be treated with oral or systemic steroids, with the intention of strengthening the conclusions by reducing the number of milder exacerbations. However, if the population randomised to the present study included largely non-bacterially infected subjects (as the results suggest), this could have resulted in us studying possible anti-inflammatory effects of our drug over a very short period in the face of the powerful anti-inflammatory effects of steroids, with predictably negative results.
Chapter 5 Discussion
This study has found that, in the population of patients with acute severe asthma randomised to treatment, the addition of azithromycin to standard medical care resulted in no statistically or clinically significant therapeutic benefit. The findings were consistently negative across three different symptom scoring tools, including one that was used in a previous study reporting statistically and clinically significant therapeutic benefit with the ketolide antibiotic telithromycin. 13 The findings were also consistently negative for all measures of lung function, including FEV1, which was significantly improved in the previous study with telithromycin. 13 Furthermore, time to a 50% reduction in asthma symptoms, which in a post hoc analysis was also significantly improved in the previous study with telithromycin,13 was not improved with azithromycin.
The different outcomes of the present study and the previous study,13 which employed closely related therapies and very similar designs, requires some interpretation.
Clearly, the two antibiotics studied are different, albeit related. The different outcomes could be explained by differences in their biological properties, including antibacterial, anti-inflammatory and antiviral activities, and possibly other properties, for example pharmacokinetics. 42 We have reported that azithromycin but not telithromycin has antiviral activity27 and so this is an unlikely explanation for the different outcomes in the two studies. In terms of antibacterial activity against relevant respiratory bacteria, telithromycin is reportedly more active than azithromycin against Streptococcus pneumoniae but has similar in vitro activity to azithromycin against both Moraxella catarrhalis and Haemophilus influenzae. 43–45 As the present study detected only three S. pneumoniae infections, one M. catarrhalis infection and no H. influenzae infections in the active treatment arm, differences in activity against these organisms seem unlikely to provide an explanation for the differing outcomes. In terms of anti-inflammatory activities, both drugs reportedly have such properties and have similar activities when compared. 46
A remarkable finding of this study was the number of patients (n = 2044) who had to be excluded as they were already receiving antibiotic therapy for their asthma exacerbation, despite such therapy not being routinely recommended in treatment guidelines. Indeed, for each patient randomised, approximately 10 had to be excluded because they had already received antibiotic therapy for their asthma exacerbation. It is therefore possible that patients who might potentially have benefited from antibiotic therapy for their asthma exacerbation (through having sputum production, sputum purulence, fever, etc.) were excluded from the study through already having received it. The population remaining to be randomised could theoretically have been selected against for antibiotic responsiveness, through having no clinical indication that antibiotic therapy might be of benefit, that is, no sputum production, no sputum purulence or no fever. This is possible as, in the experience of many investigators in this trial, patients being screened had often been seen by their primary care practitioner in the days preceding their emergency room attendance; they had also been seen by emergency room medical staff and many subsequently had also been seen by a member of the on-call respiratory/medical team so that, for many, three independent doctors/teams had assessed them for treatment, including their suitability for antibiotics. It is likely, therefore, that those not prescribed antibiotics were strongly negatively selected against for suitability for antibiotics.
It is also possible that the population randomised was in other ways not representative of the larger population screened, as 10 times more patients (> 2000 other patients) were excluded from the study for other reasons (see Figure 3). The telithromycin study13 did not report the number of patients screened and so it is not possible to determine to what extent these caveats may also have applied to that study.
Recruitment proved extremely challenging; initially there were 10 centres each aiming to recruit 38 subjects over one winter season to meet the power calculation aim of recruiting 380 patients (note that the telithromycin study successfully randomised 270 patients13). Our power calculation deliberately aimed to recruit a larger number of patients to provide statistically robust data to settle this important clinical question regarding efficacy. We also required larger numbers of patients to enable subgroup analyses aimed at potentially important mechanistic questions. Once the recruitment obstacles became clear, including the widespread antibiotic usage, more centres were enrolled, giving a final total of 31. The inclusion criteria were relaxed, including changing the eligibility criteria to include patients presenting within 48 hours (of initial presentation to medical care) with an acute deterioration of asthma control (instead of 24 hours), older subjects and those with low-level smoking histories (to exclude COPD) and the recruitment time was extended to further winter seasons, totalling 2 years and 7 months. However, despite all of these efforts only 199 subjects were recruited by the medication expiry date and, in the absence of any further funding, the study had to be terminated, despite not reaching its recruitment target. It is therefore quite possible that the study was underpowered to detect a therapeutic benefit of the study medication.
A possible trend in favour of active treatment for the primary outcome, the symptom scoring diary, was noted as both the mean and the median symptom scores tended to favour active treatment over several days (see Figures 8 and 9). If true, this might favour a type II error. However, the possibility that the study reached negative conclusions because of being underpowered is less likely given the spread of the data around these possible trends and the consistent nature of all of the other outcomes including the other symptom scoring tools, all measures of lung function and time to a 50% recovery of symptoms.
A further difference between our study and the telithromycin study was that all randomised patients were required to receive oral or systemic steroid treatment, whereas in the telithromycin study only 34.1% of patients randomised to active treatment required steroid therapy/treatment. 13 This requirement for steroid treatment in our study was designed to strengthen the conclusions, by reducing the number of milder exacerbations. However, if the population randomised to the present study included largely non-bacterially infected subjects, as argued earlier, this could have resulted in us studying possible anti-inflammatory effects of our drug, over a very short period in the face of the powerful anti-inflammatory effects of steroids, with predictably negative results. This difference in design could be one explanation for the difference in outcome between the two studies.
The clinical characteristics of the patients in our study were quite similar to the clinical characteristics of the patients in the telithromycin study13 in terms of mean age (39.9 years in our study vs. 39.5 years in the telithromycin study), sex (30.2% male vs. 32% male), mean smoking status (3.44 pack-years vs. 2.15 pack-years), symptom score severity at baseline (exacerbation) (4.16 vs. 2.9) and lung function at baseline (exacerbation) (PEF% predicted 69.4% vs. 55.2%, FEV1% predicted 64.8% vs. 67.2%, FEV1 : FVC ratio 69.2% vs. 72%). Overall, any differences in clinical characteristics do not seem to provide a likely explanation for the difference in outcome between the two studies.
However, the two studies differed strikingly in one important aspect: 61% of telithromycin-treated patients had a positive test for C. pneumoniae and/or M. pneumoniae,13 whereas in the present study only 4.5% of patients tested positive for one or both of these two organisms. Both studies used similar methods of sampling, including sputum (when collected), nasopharyngeal swabs for PCR analysis, acute serology for immunoglobulin M (IgM) and acute and convalescent serology for immunoglobulin G and immunoglobulin A. The serological testing methodologies were very similar, although different PCR assays were used and the laboratories performing the analyses differed (GR Micro, London, UK for the telithromycin study;13 National Heart and Lung Institute, Imperial College London, London, UK for this study). The different detection methods may not have yielded directly comparable results. Of note, in both studies PCR detection rates were very low (three positives for C. pneumoniae and/or M. pneumoniae in the telithromycin study13 and no positives in this study). In contrast, serological positive results were very high in the telithromycin study13 but low in this study. The telithromycin positive results were mostly IgM positives for C. pneumoniae, whereas in our study IgM positivity for this organism was low with only a single positive sample. Both studies used the same assay (C. pneumoniae IgM sandwich enzyme-linked immunosorbent assay; Medac, Hamburg, Germany) and so the discrepancy between the results of this assay is difficult to explain. It might, however, contribute to the difference in clinical outcomes between the two studies.
Sputum culture for standard bacteria was not performed in the telithromycin study and no sputum sample cultured C. pneumoniae and/or M. pneumoniae. 13 In the present study, 105 (52.8%) subjects provided sputum samples for standard bacterial culture and bacterial culture positivity was observed in 6.0% of subjects (4.1% active, 7.8% placebo). These results, together with the negative outcomes in relation to therapy, suggest that the role of bacterial infection in the population studied was unlikely to be important.
Adverse events were infrequent, with a slight preponderance of gastrointestinal AEs in the azithromycin group (35 events) compared with the placebo group (24 events), as is common in antibiotic studies. Interestingly, there was a similar reduction in respiratory, thoracic and mediastinal AEs in the azithromycin group (27 events) as in the placebo group (37 events). A total of 63 of these 64 events proved to be respiratory (the other was backache), suggesting that antibiotic therapy possibly reduced the level of respiratory AEs in this population.
Chapter 6 Conclusions and recommendations
In the population of patients with acute severe asthma randomised to treatment, the addition of azithromycin to standard medical care resulted in no statistically or clinically significant therapeutic benefit, although we could not rule out a –0.3 difference in the primary outcome based on the magnitude of the CIs. For each patient randomised, approximately 10 were excluded because they had already received antibiotic therapy, despite guideline recommendations that such therapy should not be routinely used. The study may therefore have been underpowered to detect therapeutic benefit in the selected minority of patients randomised to treatment.
In the face of such widespread use of antibiotics in naturally occurring asthma exacerbations, without convincing evidence that they are beneficial, it is difficult to come up with coherent recommendations in practice, as a randomised placebo-controlled study of an antibiotic added to therapy in those already receiving an antibiotic appears meaningless, whereas performing an adequately powered study in representative numbers of patients not receiving antibiotic therapy has proved extremely challenging.
Further scientific studies, including human experimentally induced asthma exacerbation studies, should be performed to determine whether there is evidence that bacteria do or do not contribute to the pathogenesis of asthma exacerbations. Such studies should include standard culture techniques as well as modern molecular techniques to study the microbiome to help identify potential pathogens that are difficult to culture. If bacteria do appear to contribute to asthma exacerbation pathogenesis in the human experimentally induced asthma exacerbation setting, a randomised placebo-controlled study in this controlled experimental setting would be the logical next step. Further study of azithromycin in acute exacerbations of asthma in adults and children in settings of low antibiotic usage is required.
Acknowledgements
We would like to thank the following.
-
Trial funder: NIHR Efficacy and Mechanism Evaluation programme.
-
Participating sites:
-
Barnsley Hospital NHS Foundation Trust – Barnsley Hospital, Barnsley
-
Blackpool Teaching Hospitals NHS Foundation Trust – Blackpool Victoria Hospital, Blackpool
-
Countess of Chester Hospital NHS Foundation Trust – Countess of Chester Hospital, Chester
-
Gloucestershire Hospitals NHS Foundation Trust – Gloucestershire Royal Hospital
-
Great Western Hospitals NHS Foundation Trust, Swindon
-
Guy’s and St Thomas’ NHS Foundation Trust
-
Heart of England NHS Foundation Trust – Birmingham Heartlands Hospital, Birmingham
-
Imperial College Healthcare NHS Trust – St Mary’s Hospital, Charing Cross Hospital and Hammersmith Hospital, London
-
Ipswich Hospital NHS Trust – Ipswich Hospital, Ipswich
-
Leeds Teaching Hospitals NHS Trust – St James’s University Hospital, Leeds
-
Mid Cheshire Hospitals NHS Foundation Trust – Leighton Hospital, Crewe
-
Newcastle upon Tyne Hospitals NHS Foundation Trust – Freeman Hospital, Newcastle upon Tyne
-
NHS Greater Glasgow and Clyde – Western & Royal Infirmary, Glasgow
-
NHS Wiltshire – Rowden Medical Partnership, Chippenham
-
Norfolk and Norwich University Hospitals NHS Foundation Trust – Norfolk and Norwich University Hospital
-
North Tees and Hartlepool Hospitals NHS Foundation Trust – University Hospital of North Tees
-
Nottingham University Hospitals NHS Trust – Nottingham City Hospital and Queen’s Medical Centre
-
Plymouth Hospitals NHS Trust – Derriford Hospital, Plymouth
-
Portsmouth Hospitals NHS Trust – Queen Alexandra Hospital
-
Royal Berkshire NHS Foundation Trust – Royal Berkshire Hospital, Reading
-
Royal Wolverhampton Hospitals NHS Trust – New Cross Hospital, Wolverhampton
-
Sherwood Forest Hospitals NHS Foundation Trust
-
Shrewsbury and Telford Hospital NHS Trust – Princess Royal Hospital, Haywards Health
-
South Tees Hospitals NHS Foundation Trust – James Cook Hospital, Middleborough
-
Surrey and Sussex Healthcare NHS Trust – East Surrey Hospital
-
Taunton and Somerset NHS Foundation Trust – Musgrove Park Hospital, Taunton
-
University Hospital of South Manchester NHS Foundation Trust
-
University Hospitals of Leicester NHS Trust – Glenfield Hospital, Leicester
-
Worcestershire Acute Hospitals NHS Trust.
-
-
Patients participating in the trial.
-
Imperial Clinical Trials Unit: ICTU core staff and the InForm team, and therefore the research, were supported by the NIHR Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London.
-
Trial Steering Committee members.
-
Data Monitoring and Ethics Committee members.
-
The AZALEA trial team:
-
Miss Maria-Belen Trujillo-Torralbo, Research Nurse, Imperial College London
-
Mr Ajerico del Rosario, Research Nurse, Imperial College London
-
Elena Kulinskaya, Senior Statistician, Imperial College London
-
Dr Alexina Mason, Statistician, ICTU, School of Public Health, Imperial College London
-
Dr Jane Warwick, Senior Statistician, ICTU, School of Public Health, Imperial College London
-
Dr James Griffiths, PI, Barnsley Hospital, Barnsley
-
Josephine Marange, Research Nurse, Birmingham Heartlands Hospital, Birmingham
-
Dr Tarek Saba, PI, Blackpool Victoria Hospital, Blackpool
-
Sister Judith Saba, Research Nurse, Blackpool Victoria Hospital, Blackpool
-
Professor Stephen Scott, PI, Countess of Chester Hospital, Chester
-
Jillian Andrews, Senior Respiratory Nurse, Countess of Chester Hospital, Chester
-
Dr Duncan Fullerton, Leighton Hospital, Crewe
-
Dr Edward Cetti, PI, East Surrey Hospital, Redhill
-
Patricia Clark, Research Nurse, Western & Royal Infirmary, Glasgow
-
Dr Simon Message, PI, Gloucestershire Royal Hospital, Gloucester
-
Dr Jonathan Douse, PI, Ipswich Hospital, Ipswich
-
Dr Ian Clifton, PI, St James’s University Hospital, Leeds
-
Dr Richard Harrison, PI, University Hospital of North Tees, Stockton-on-Tees
-
Dr Andrew Wilson, PI, Norfolk and Norwich University Hospital
-
Janet Oborne, Research Nurse, King’s Mill Hospital, Sutton in Ashfield
-
Dr Matthew Masoli, PI, Derriford Hospital, Plymouth
-
Dr Thomas Brown, Consultant Respiratory Physician, Queen Alexandra Hospital, Portsmouth
-
Dr Jonathan Owen, Consultant Respiratory Physician, Queen Alexandra Hospital, Portsmouth
-
Dr Dominic Reynish, Clinical Research Fellow, Queen Alexandra Hospital, Portsmouth
-
Dr Heena Mistry, Clinical Research Fellow, Queen Alexandra Hospital, Portsmouth
-
Dr Gavin Durrant, PI, Rowden Medical Partnership, Chippenham
-
Dr Grace Robinson, PI, Royal Berkshire Hospital, Reading
-
Dr Jonathan Mann, PI, Royal Wolverhampton Hospitals NHS Trust, Wolverhampton
-
Dr Ramamurthy Sathyamurthy, PI, James Cook University Hospital, Middlesbrough
-
Dr Andrew Stanton, PI, Great Western Hospital, Swindon
-
Dr Justin Pepperill, PI, Musgrove Park Hospital, Taunton
-
Dr Harmesh Moudgil, PI, Princess Royal Hospital, Telford
-
Dr Steve O’Hickey, PI, Worcestershire Royal Hospital, Worcester.
-
Contributions of authors
Sebastian L Johnston (Chief Investigator, Professor of Respiratory Medicine and Allergy) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Matyas Szigeti (Statistician) contributed to the analysis and interpretation of data, drafting/revising of the report and approval of the report.
Mary Cross (Operations Manager, ICTU) contributed to the design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Christopher Brightling (coapplicant on grant, PI at Leicester, Professor of Respiratory Medicine) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Rekha Chaudhuri (coapplicant on grant, PI at Glasgow, Associate Specialist and Honorary Clinical Associate Professor in Respiratory Medicine) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Timothy Harrison (coapplicant on grant, PI at Nottingham, Associate Professor of Respiratory Medicine) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Adel Mansur (coapplicant on grant, PI at Birmingham, Consultant Physician and Honorary Senior Lecturer in Severe and Brittle Asthma) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Laura Robison (Clinical Trial Manager, ICTU) contributed to the analysis and interpretation of data, drafting/revising of the report and approval of the report.
Zahid Sattar (Clinical Trial Manager, ICTU) contributed to the analysis and interpretation of data, drafting/revising of the report and approval of the report.
David Jackson (Honorary Senior Lecturer, Respiratory Medicine) contributed to the analysis and interpretation of data, drafting/revising of the report and approval of the report.
Patrick Mallia (Clinical Senior Lecturer, Respiratory Medicine) contributed to the analysis and interpretation of data, drafting/revising of the report and approval of the report.
Ernie Wong (Clinical Research Fellow, Airway Disease) contributed to the analysis and interpretation of data, drafting/revising of the report and approval of the report.
Christopher Corrigan (coapplicant on grant, PI at Guy’s and St Thomas’, Professor of Asthma, Allergy and Respiratory Science) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Bernard Higgins (coapplicant on grant, PI at Newcastle, Consultant Respiratory Physician) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Philip Ind (coapplicant on grant, PI at Hammersmith Hospital, Honorary Senior Lecturer in Airway Disease) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Dave Singh (coapplicant on grant, PI at Manchester, Professor of Clinical Pharmacology and Respiratory Medicine) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Neil Thomson (coapplicant on grant, Professor of Respiratory Medicine) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Deborah Ashby (Senior Statistician, Chair in Medical Statistics and Clinical Trials) contributed to the analysis of data, drafting/revising of the report and approval of the report.
Anoop Chauhan (coapplicant on grant, PI at Portsmouth, Consultant Respiratory Physician) contributed to the conception and design of the study, analysis and interpretation of data, drafting/revising of the report and approval of the report.
Publication
Johnston SL, Szigeti M, Cross M, Brightling C, Chaudhuri R, Harrison T, et al. Azithromycin for Acute Exacerbations of Asthma: the AZALEA randomized clinical trial [published online ahead of print 19 September 2016]. JAMA Intern Med 2016.
Data sharing statement
This trial complies with the Imperial College London records management policy, which includes the retention schedule for data by type. When transferring records to the College archive facility (records include both paper and electronic records), there is a specific section relating to clinical trials transfer of records; retrieval of records and access records is also included in the policy. Data are kept for a minimum of 10 years to comply with Imperial College London policy. All available trial data can be obtained from the corresponding author.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health.
References
- Asher MI, Montefort S, Bjorksten B, Lai CK, Strachan DP, Weiland SK, et al. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet 2006;368:733-43. http://dx.doi.org/10.1016/S0140-6736(06)69283-0.
- Weiss KB, Sullivan SD. The health economics of asthma and rhinitis. I. Assessing the economic impact. J Allergy Clin Immunol 2001;107:3-8. http://dx.doi.org/10.1067/mai.2001.112262.
- Rabe KF, Vermeire PA, Soriano JB, Maier WC. Clinical management of asthma in 1999: the Asthma Insights and Reality in Europe (AIRE) study. Eur Respir J 2000;16:802-7. http://dx.doi.org/10.1183/09031936.00.16580200.
- Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ 1995;310:1225-9. http://dx.doi.org/10.1136/bmj.310.6989.1225.
- Chauhan AJ, Inskip HM, Linaker CH, Smith S, Schreiber J, Johnston SL, et al. Personal exposure to nitrogen dioxide (NO2) and the severity of virus-induced asthma in children. Lancet 2003;361:1939-44. http://dx.doi.org/10.1016/S0140-6736(03)13582-9.
- Johnston SL, Pattemore PK, Sanderson G, Smith S, Campbell MJ, Josephs LK, et al. The relationship between upper respiratory infections and hospital admissions for asthma: a time-trend analysis. Am J Respir Crit Care Med 1996;154:654-60. http://dx.doi.org/10.1164/ajrccm.154.3.8810601.
- Wark PA, Johnston SL, Moric I, Simpson JL, Hensley MJ, Gibson PG. Neutrophil degranulation and cell lysis is associated with clinical severity in virus-induced asthma. Eur Respir J 2002;19:68-75. http://dx.doi.org/10.1183/09031936.02.00226302.
- Grissell TV, Powell H, Shafren DR, Boyle MJ, Hensley MJ, Jones PD, et al. Interleukin-10 gene expression in acute virus-induced asthma. Am J Respir Crit Care Med 2005;172:433-9. http://dx.doi.org/10.1164/rccm.200412-1621OC.
- Wark PA, Johnston SL, Simpson JL, Hensley MJ, Gibson PG. Chlamydia pneumoniae immunoglobulin A reactivation and airway inflammation in acute asthma. Eur Respir J 2002;20:834-40. http://dx.doi.org/10.1183/09031936.02.00192002.
- Esposito S, Blasi F, Arosio C, Fioravanti L, Fagetti L, Droghetti R, et al. Importance of acute Mycoplasma pneumoniae and Chlamydia pneumoniae infections in children with wheezing. Eur Respir J 2000;16:1142-6. http://dx.doi.org/10.1034/j.1399-3003.2000.16f21.x.
- Cunningham AF, Johnston SL, Julious SA, Lampe FC, Ward ME. Chronic Chlamydia pneumoniae infection and asthma exacerbations in children. Eur Respir J 1998;11:345-9. http://dx.doi.org/10.1183/09031936.98.11020345.
- Johnston SL, Martin RJ. Chlamydophila pneumoniae and Mycoplasma pneumoniae: a role in asthma pathogenesis?. Am J Respir Crit Care Med 2005;172:1078-89. http://dx.doi.org/10.1164/rccm.200412-1743PP.
- Johnston SL, Blasi F, Black PN, Martin RJ, Farrell DJ, Nieman RB. The effect of telithromycin in acute exacerbations of asthma. N Engl J Med 2006;354:1589-600. http://dx.doi.org/10.1056/NEJMoa044080.
- Pattemore PK, Johnston SL, Bardin PG. Viruses as precipitants of asthma symptoms. I. Epidemiology. Clin Exp Allergy 1992;22:325-36. http://dx.doi.org/10.1111/j.1365-2222.1992.tb03094.x.
- Talbot TR, Hartert TV, Mitchel E, Halasa NB, Arbogast PG, Poehling KA, et al. Asthma as a risk factor for invasive pneumococcal disease. N Engl J Med 2005;352:2082-90. http://dx.doi.org/10.1056/NEJMoa044113.
- Klemets P, Lyytikainen O, Ruutu P, Ollgren J, Kaijalainen T, Leinonen M, et al. Risk of invasive pneumococcal infections among working age adults with asthma. Thorax 2010;65:698-702. http://dx.doi.org/10.1136/thx.2009.132670.
- Pilishvili T, Zell ER, Farley MM, Schaffner W, Lynfield R, Nyquist AC, et al. Risk factors for invasive pneumococcal disease in children in the era of conjugate vaccine use. Pediatrics 2010;126:e9-17. http://dx.doi.org/10.1542/peds.2009-2150.
- Jounio U, Juvonen R, Bloigu A, Silvennoinen-Kassinen S, Kaijalainen T, Kauma H, et al. Pneumococcal carriage is more common in asthmatic than in non-asthmatic young men. Clin Respir J 2010;4:222-9. http://dx.doi.org/10.1111/j.1752-699X.2009.00179.x.
- Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLOS ONE 2010;5. http://dx.doi.org/10.1371/journal.pone.0008578.
- Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, Bartlett NW, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med 2006;12:1023-6. http://dx.doi.org/10.1038/nm1462.
- Oliver BG, Lim S, Wark P, Laza-Stanca V, King N, Black JL, et al. Rhinovirus exposure impairs immune responses to bacterial products in human alveolar macrophages. Thorax 2008;63:519-25. http://dx.doi.org/10.1136/thx.2007.081752.
- Avadhanula V, Rodriguez CA, Devincenzo JP, Wang Y, Webby RJ, Ulett GC, et al. Respiratory viruses augment the adhesion of bacterial pathogens to respiratory epithelium in a viral species- and cell type-dependent manner. J Virol 2006;80:1629-36. http://dx.doi.org/10.1128/JVI.80.4.1629-1636.2006.
- Bisgaard H, Hermansen MN, Bonnelykke K, Stokholm J, Baty F, Skytt NL, et al. Association of bacteria and viruses with wheezy episodes in young children: prospective birth cohort study. BMJ 2010;341. http://dx.doi.org/10.1136/bmj.c4978.
- British Thoracic Society Scottish Intercollegiate Guidelines Network . British Guideline on the Management of Asthma. Thorax 2008;63:1-121. http://dx.doi.org/10.1136/thx.2008.097741.
- Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, et al. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc Natl Acad Sci USA 2008;105:13562-7. http://dx.doi.org/10.1073/pnas.0804181105.
- Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med 2005;201:937-47. http://dx.doi.org/10.1084/jem.20041901.
- Gielen V, Johnston SL, Edwards MR. Azithromycin induces anti-viral responses in bronchial epithelial cells. Eur Respir J 2010;36:646-54. http://dx.doi.org/10.1183/09031936.00095809.
- Beigelman A, Mikols CL, Gunsten SP, Cannon CL, Brody SL, Walter MJ. Azithromycin attenuates airway inflammation in a mouse model of viral bronchiolitis. Respir Res 2010;11. http://dx.doi.org/10.1186/1465-9921-11-90.
- Bafadhel M, Clark TW, Reid C, Medina MJ, Batham S, Barer MR, et al. Procalcitonin and C reactive protein in hospitalised adult patients with community acquired pneumonia, exacerbation of asthma and chronic obstructive pulmonary disease. Chest 2011;139:1410-18. http://dx.doi.org/10.1378/chest.10-1747.
- Amsden GW. Erythromycin, clarithromycin, and azithromycin: are the differences real?. Clin Ther 1996;18:56-72. http://dx.doi.org/10.1016/S0149-2918(96)80179-2.
- Hahn DL, Grasmick M, Hetzel S, Yale S. AZMATICS (AZithroMycin-Asthma Trial In Community Settings) Study Group . Azithromycin for bronchial asthma in adults: an effectiveness trial. J Am Board Fam Med 2012;25:442-59.
- Juniper EF, Svensson K, Mörk AC, Ståhl E. Measuring health-related quality of life in adults during an acute asthma exacerbation. Chest 2004;125:93-7. http://dx.doi.org/10.1378/chest.125.1.93.
- Juniper EF, Guyatt GH, Cox FM, Ferrie PJ, King DR. Development and validation of the Mini Asthma Quality of Life Questionnaire. Eur Respir J 1999;14:32-8. http://dx.doi.org/10.1034/j.1399-3003.1999.14a08.x.
- Johnston SL, Szigeti M, Cross M, Brightling C, Chaudhuri R, Harrison T, et al. Azithromycin for Acute Exacerbations of Asthma: the AZALEA randomized clinical trial. JAMA Intern Med 2016.
- Wark PA, Simpson JL, Hensley MJ, Gibson PG. Safety of sputum induction with isotonic saline in adults with acute severe asthma. Clin Exp Allergy 2001;31:1745-53. http://dx.doi.org/10.1046/j.1365-2222.2001.01230.x.
- Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the Approximation of Laws, Regulations and Administrative Provisions of the Member States Relating to the Implementation of Good Clinical Practice in the Conduct of Clinical Trials on Medicinal Products for Human Use. Brussels: European Commission; 2001.
- Research Governance Framework for Health and Social Care. London: Department of Health; 2005.
- The Medicines for Human Use (Clinical Trials) Regulations (SI 2004 1031), as Amended. London: The Stationery Office; 2004.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) . ICH Topic E6 (R1) Guideline for Good Clinical Practice. Step 5 Note for Guidance on Good Clinical Practice (CPMP ICH 135 95), July 2002 2002.
- Commission Directive 2005/28/EC of 8 April 2005 Laying Down the Principles and Detailed Guidelines for Good Clinical Practice as Regards to Investigational Medicinal Products for Human Use, as Well as the Requirements for Authorisation of the Manufacturing or Importation of Such Products. Brussels: European Commission; 2005.
- British Thoracic Society; Scottish Intercollegiate Guidelines Network . British guideline on the management of asthma. Thorax 2014;69:1-192.
- Zeitlinger M, Wagner CC, Heinisch B. Ketolides – the modern relatives of macrolides: the pharmacokinetic perspective. Clin Pharmacokinet 2009;48:23-38. http://dx.doi.org/10.2165/0003088-200948010-00002.
- Walsh F, Carnegy F, Willcock J, Amyes S. Comparative in vitro activity of telithromycin against macrolide-resistant and -susceptible Streptococcus pneumoniae, Moraxella catarrhalis and Haemophilus influenzae. J Antimicrob Chemother 2004;53:793-6. http://dx.doi.org/10.1093/jac/dkh178.
- Kosowska K, Credito K, Pankuch GA, Hoellman D, Lin G, Clark C, et al. Activities of two novel macrolides, GW 773546 and GW 708408, compared with those of telithromycin, erythromycin, azithromycin, and clarithromycin against Haemophilus influenzae. Antimicrob Agents Chemother 2004;48:4113-19. http://dx.doi.org/10.1128/AAC.48.11.4113-4119.2004.
- De Vecchi E, Nicola L, Larosa M, Drago L. In vitro activity of telithromycin against Haemophilus influenzae at epithelial lining fluid concentrations. BMC Microbiol 2008;8. http://dx.doi.org/10.1186/1471-2180-8-23.
- Kobayashi Y, Wada H, Rossios C, Takagi D, Higaki M, Mikura S, et al. A novel macrolide solithromycin exerts superior anti-inflammatory effect via NF-kappaB inhibition. J Pharmacol Exp Ther 2013;345:76-84. http://dx.doi.org/10.1124/jpet.112.200733.
Appendix 1 Statistical details
Observed diary scores for each centre by treatment arm
FIGURE 18.
Observed diary scores for each centre by treatment arm. A21, Royal Berkshire Hospital, Reading; A22, Rowden Medical Partnership (GP surgery) Chippenham; A23, East Surrey Hospital, Redhill; A24, Countess of Chester Hospital, Chester; A25, Musgrove Park Hospital, Taunton; A26, Worcestershire Acute Hospitals NHS Trust; A29, New Cross Hospital, Wolverhampton; A30, Ipswich Hospital, Ipswich; A32, Princess Royal Hospital, Telford; A35, Gloucestershire Royal Hospital, Gloucester; BIR, Birmingham Heartlands Hospital, Birmingham; BVH, Blackpool Victoria Hospital, Blackpool; GLA, Western & Royal Infirmary, Glasgow; LEI, Glenfield Hospital, Leicester; NEW, Freeman Hospital, Newcastle upon Tyne; NNU, Norfolk and Norwich University Hospital, Norwich; NOC, Nottingham City Hospital, Nottingham; NOT, Queen’s Medical Centre, Nottingham; POR, Queen Alexandra Hospital, Portsmouth; SMH, St Mary’s Hospital, Imperial College Healthcare NHS Trust; STJ, St James’s University Hospital, Leeds; UNT, University Hospital of North Tees, Stockton-on-Tees.
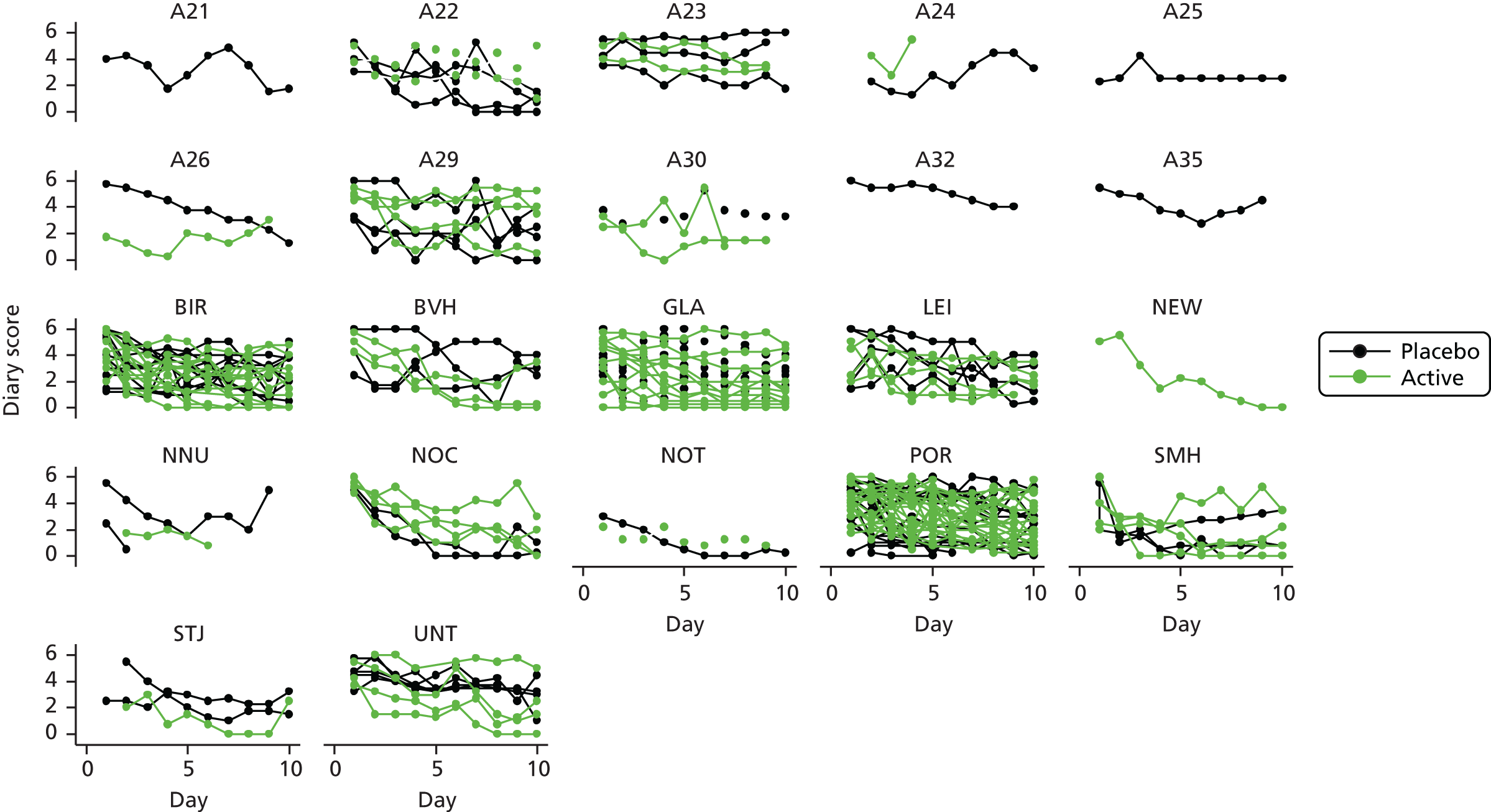
Plots used for model selection
The plots of level 1 and 2 residuals (where appropriate) including the model with splines at day 3 and 7 and the fitted and observed values for some randomly selected patients were also investigated graphically. These plots are presented in Figures 19–37. As can be seen, the more complex alternative models gave more flexibility than the standard linear model, but, overall, the residuals were just barely lower and the pattern of residuals remained the same, so in order of simplicity a linear model was chosen to calculate the estimated scores.
FIGURE 19.
Box plot of residuals for linear and quadratic models. (a) Linear, random intercept; (b) linear, random intercept, slope; (c) quadratic, random intercept, slope; and (d) quadratic, random intercept, slope, quadratic term. Closed circles represent value(s) outside 1.5x the interquartile range from the upper/lower quartile.
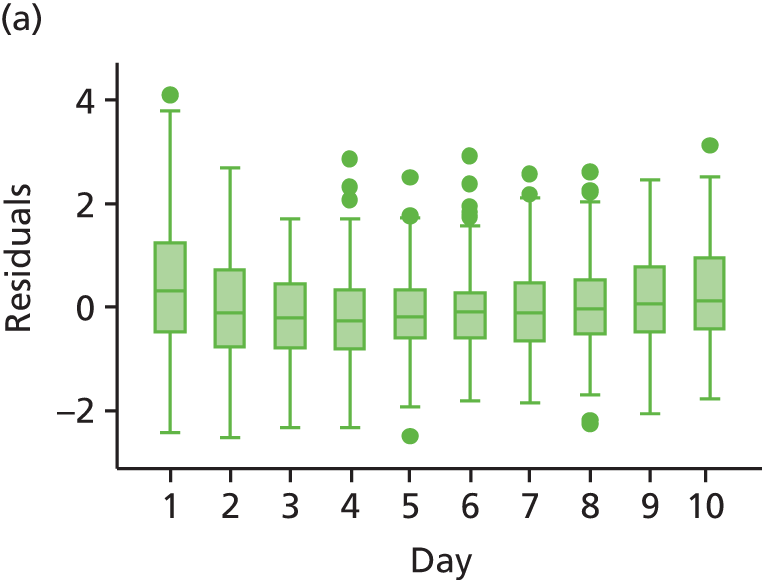
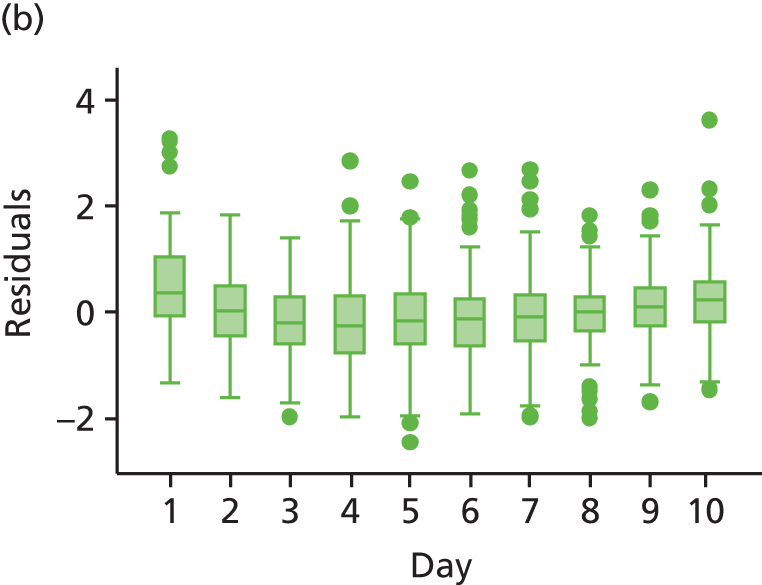


Linear model with random intercept and slope
FIGURE 20.
Observed and fitted values for randomly selected examples.
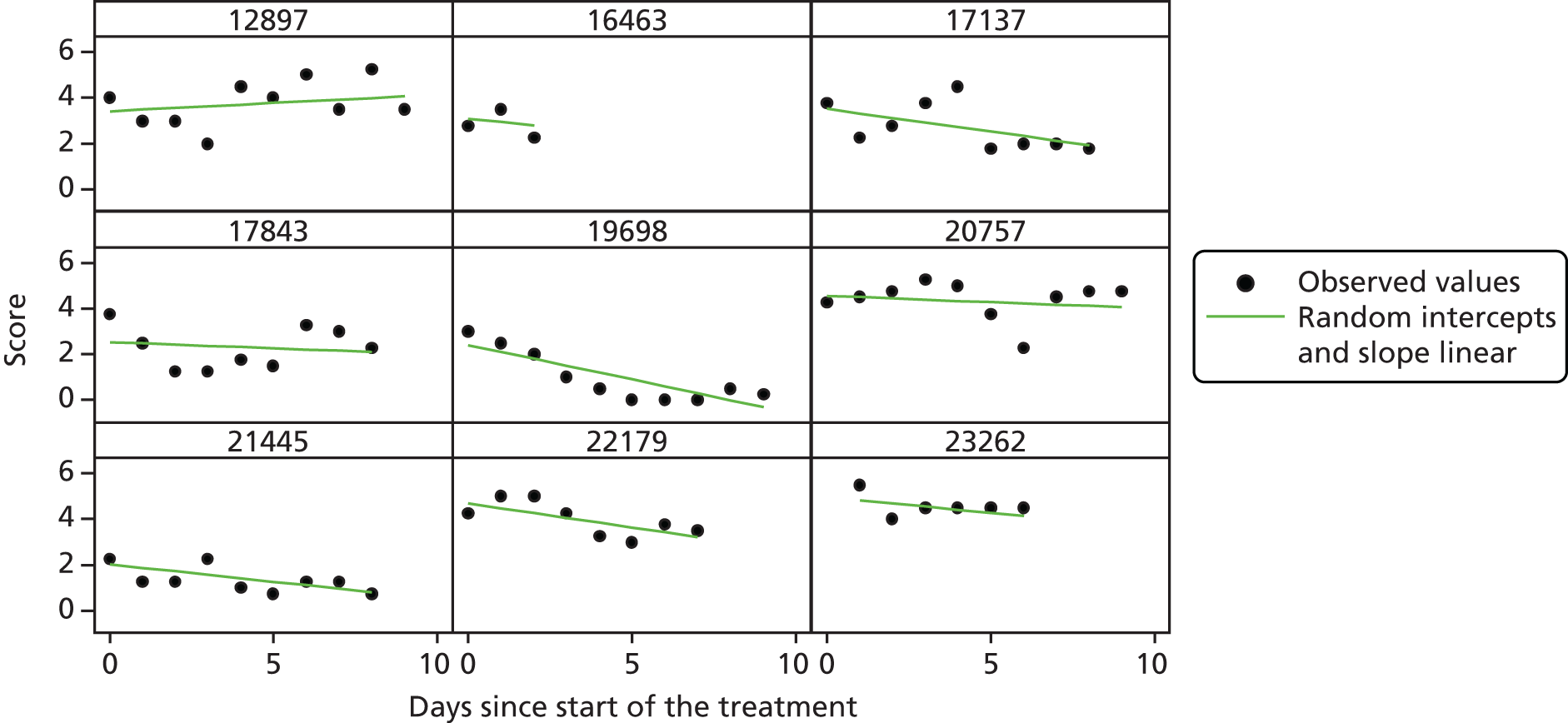
FIGURE 21.
Observed and fitted values for randomly selected examples.
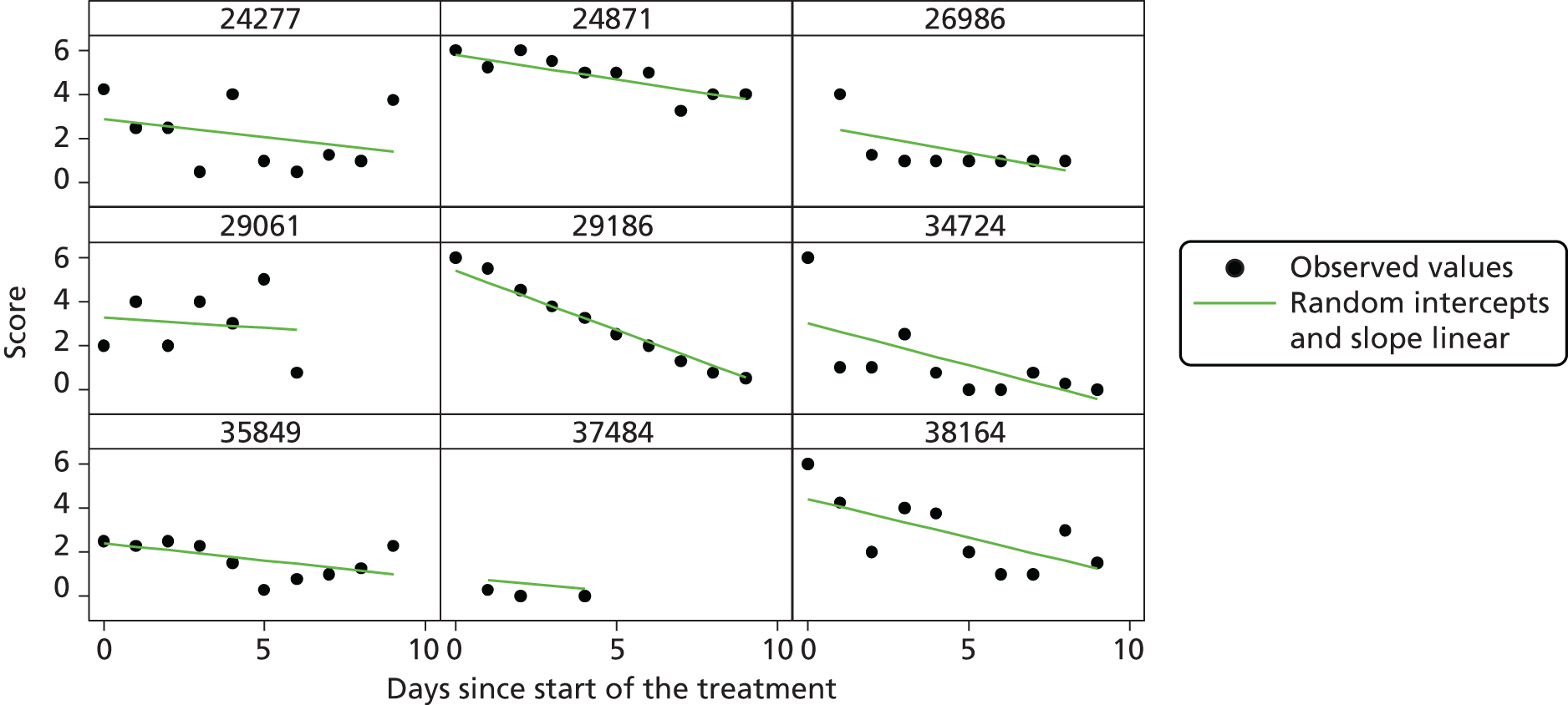
FIGURE 22.
Residuals by day for linear model with random intercept and slope.
FIGURE 23.
Level 2 residuals (slope) for linear model with random intercept and slope (by definition it is constant over time).
FIGURE 24.
Level 2 residuals (intercept) for linear model with random intercept and slope (by definition it is constant over time).

Quadratic model with random intercept and slope
FIGURE 25.
Observed and fitted values for randomly selected examples.

FIGURE 26.
Observed and fitted values for randomly selected examples.
FIGURE 27.
Residuals by day for quadratic model with random intercept and slope.

FIGURE 28.
Level 2 residuals (slope) for quadratic model with random intercept and slope (by definition it is constant over time).
FIGURE 29.
Level 2 residuals (intercept) for quadratic model with random intercept and slope (by definition it is constant over time).
Square root model
FIGURE 30.
Observed and fitted values for randomly selected examples.
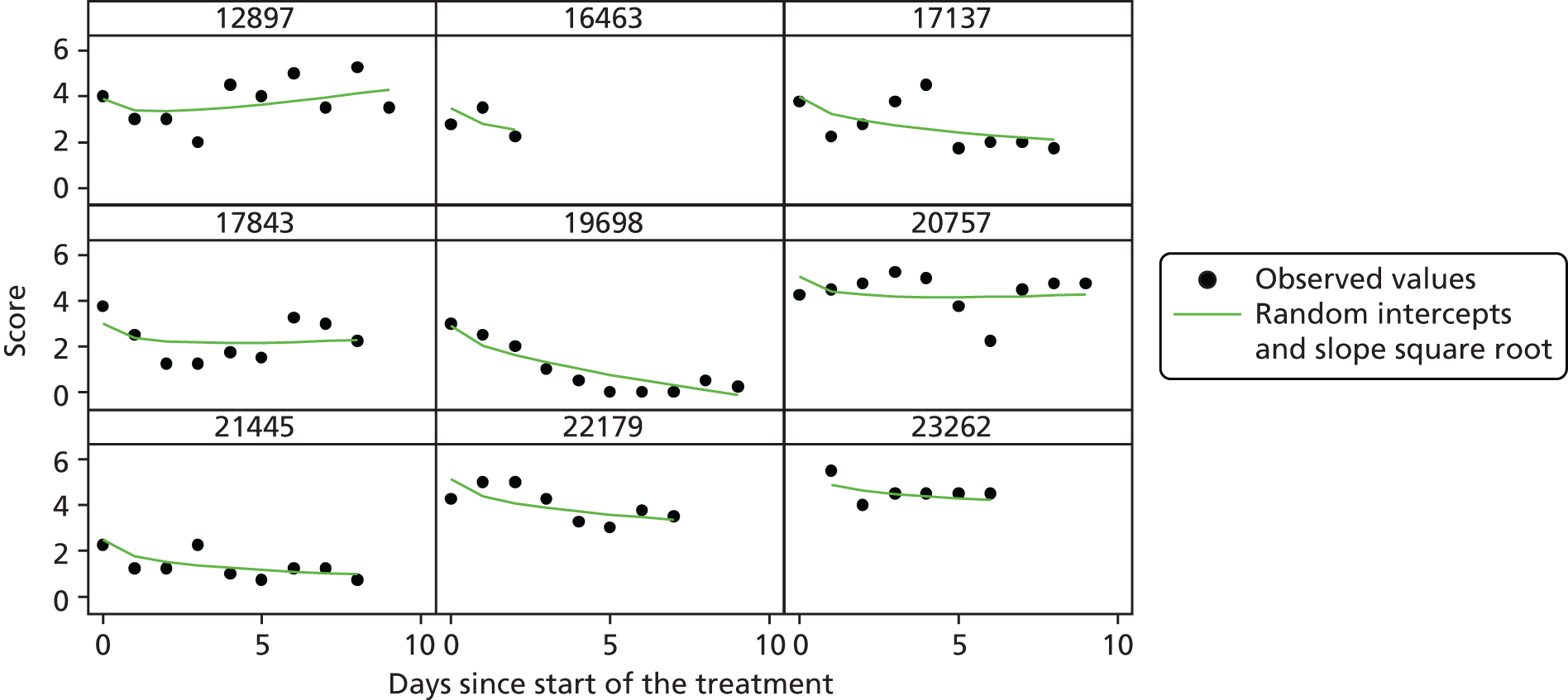
FIGURE 31.
Observed and fitted values for randomly selected examples.

FIGURE 32.
Residuals by day for quadratic model with random intercept and slope.
FIGURE 33.
Level 2 residuals (slope) for square root model with random intercept and slope (by definition it is constant over time).
FIGURE 34.
Level 2 residuals (intercept) for square root model with random intercept and slope (by definition it is constant over time).

Splines (at day 3 and 7)
FIGURE 35.
Observed and fitted values for randomly selected examples.
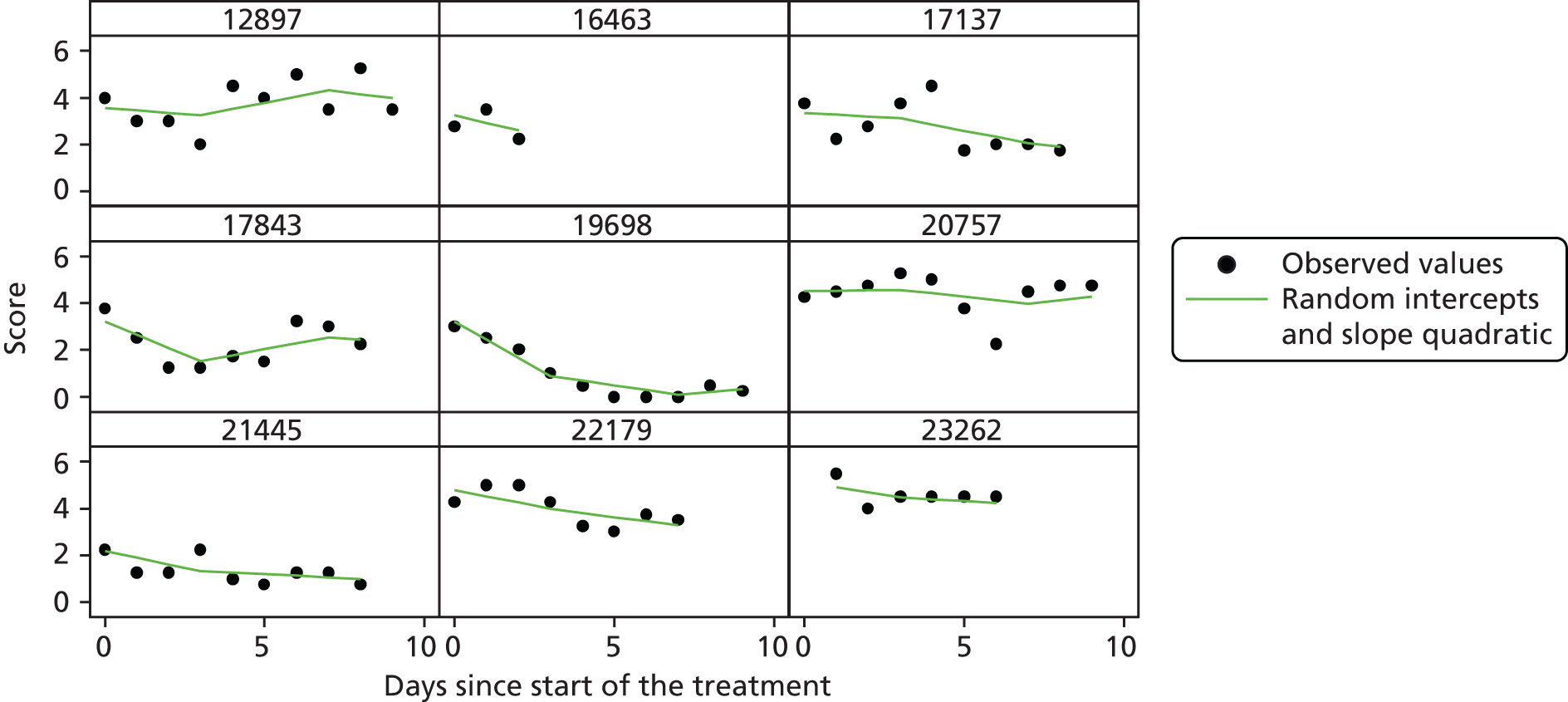
FIGURE 36.
Observed and fitted values for randomly selected examples.
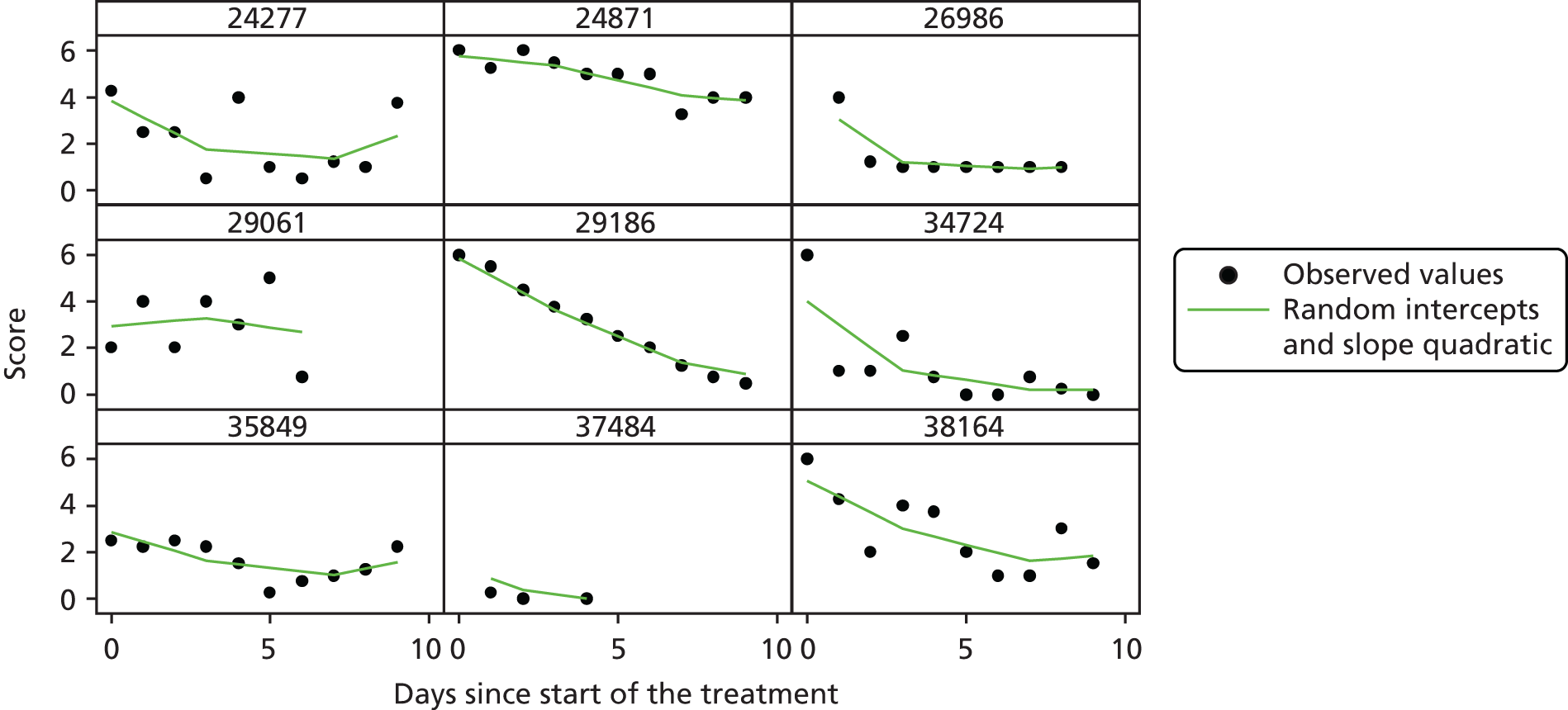
FIGURE 37.
Residuals by day for splines model with random intercept and slope.
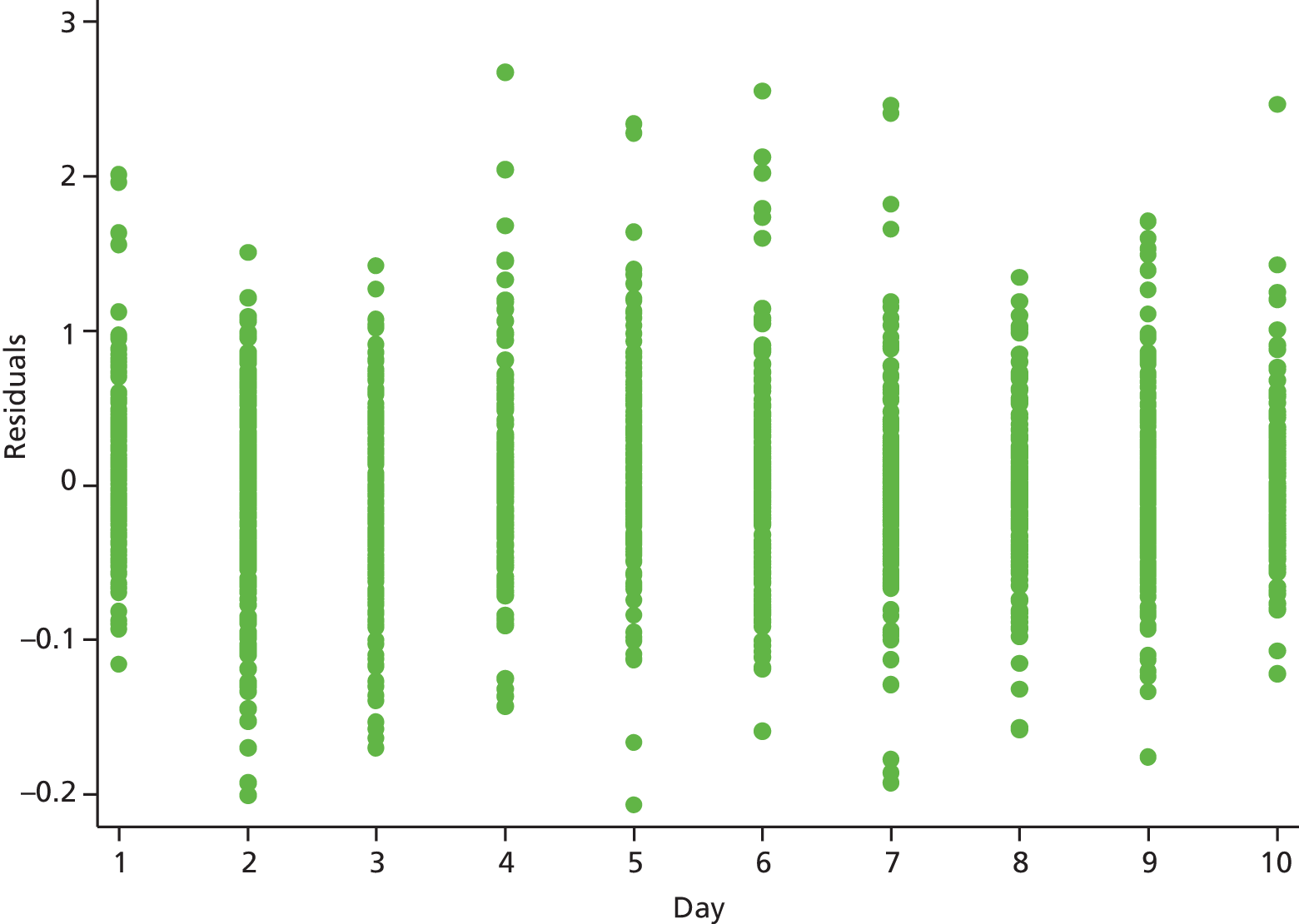
Details of the models for diary and Asthma Quality of Life Questionnaire scores
| Covariates | Factor | Coefficient | 95% CI | p-value |
|---|---|---|---|---|
| Constant | Mean score at baseline in the placebo group | 3.6595 | 3.4169 to 3.9022 | 0.000 |
| Days (centred) | Daily change in placebo group | –0.1792 | –0.2217 to –0.1367 | 0.000 |
| Treatment × day (interaction) | (Treatment effect) Difference in daily change compared with the placebo group | –0.0185 | –0.0744 to 0.0374 | 0.517 |
| Level | Variance | Estimate | 95% CIa |
|---|---|---|---|
| Site | Constant (intercept) | 0.0412 | 0.0012 to 1.4372 |
| Subject | Constant (intercept) | 1.6863 | 1.3063 to 2.1769 |
| Days (slope) | 0.0334 | 0.0251 to 0.0443 | |
| Covariance days – constant | –0.0957 | –0.1461 to –0.0453 | |
| Residuals | 0.6941 | 0.6415 to 0.7510 |
| Covariates | Factor | Coefficient | 95% CI | p-value |
|---|---|---|---|---|
| Constant | Mean score at baseline in the placebo group | 4.727 | 4.491 to 4.962 | 0.000 |
| Visits (centred) | Per-visit change in placebo group | 0.429 | 0.275 to 0.583 | 0.000 |
| Treatment*visit (interaction) | (Treatment effect) | 0.065 | –0.138 to 0.269 | 0.530 |
| Level | Variance | Estimate | 95% CIa |
|---|---|---|---|
| Site | Constant (intercept) | 0.063 | 0.009 to 0.450 |
| Subject | Constant (intercept) | 0.888 | 0.583 to 1.353 |
| Visits (slope) | 0.165 | 0.059 to 0.464 | |
| Covariance visits – constant | –0.074 | –0.272 to 0.125 | |
| Residuals | 0.903 | 0.727 to 1.123 |
| Covariates | Factor | Coefficient | 95% CI | p-value |
|---|---|---|---|---|
| Constant | Mean score at baseline in the placebo group | 3.355 | 3.196 to 3.514 | 0.000 |
| Visits (centred) | Per-visit change in placebo group | 0.350 | 0.214 to 0.486 | 0.000 |
| Treatment*visit (interaction) | (Treatment effect) | –0.021 | –0.204 to 0.163 | 0.823 |
| Level | Variance | Estimate | 95% CIa |
|---|---|---|---|
| Site | Constant (intercept) | 0.000 | 0.000 to 0.000 |
| Subject | Constant (intercept) | 0.803 | 0.569 to 1.133 |
| Visits (slope) | 0.185 | 0.097 to 0.350 | |
| Covariance visits – constant | –0.076 | –0.220 to 0.069 | |
| Residuals | 0.566 | 0.457 to 0.703 |
Appendix 2 Patient information sheet
The use of the antibiotic azithromycin in treatment of patients following acute asthma attacks

Participant information sheet
We would like to invite you to take part in a research study. Before you decide you need to understand why the research is being done and what it would involve for you. Please take time to read the following information carefully. Talk to others about the study if you wish and ask us if there is anything that is not clear or if you would like more information.
Part 1 tells you the purpose of this study and what will happen to you if you take part.
Part 2 gives you more detailed information about the conduct of the study.
Part 1
What is the study about?
Acute attacks (exacerbations) of asthma are common and cause a great deal of suffering in asthmatic patients. Current treatments for asthma attacks are not completely effective and new and better treatments are needed. Viruses often cause asthma attacks and bacterial lung infections have also been associated with asthma attacks. However, the role for bacteria is uncertain. Current asthma guidelines for doctors treating asthma exacerbations do not recommend the routine use of antibiotics. We would like to investigate whether or not azithromycin, which is a safe and well-tolerated antibiotic (an antibacterial) that has been used for many years in the treatment of respiratory disease, might be of benefit in asthma attacks. As there is some evidence that azithromycin has antiviral properties this may add to its benefits (antibiotics do not usually affect viruses). By looking at the effect of azithromycin on asthma attacks this will help us to show whether or not azithromycin should be recommended during an acute asthma attack in addition to the usual care that is provided to these patients as it may help them recover quicker from the exacerbation. We will also be able to look at why azithromycin may be effective – if it is having an antibacterial and/or antiviral effect.
Why have I been invited?
You have been invited to take part in this research study because you have presented with an exacerbation of your asthma, which is the condition we are looking at. We are planning to enrol 380 participants into this study who present to medical care with an asthma exacerbation across different AZALEA research sites across the UK.
Do I have to take part?
It is up to you to decide to join the study. We will describe the study and go through this information sheet. If you agree to take part, we will then ask you to sign a consent form. You are free to withdraw at any time, without giving a reason, and this would not affect the standard of care you receive.
What will happen to me if I agree to take part?
In addition to your current visit we will ask you to attend an additional three visits during which we will take some samples from you (see below for details) and ask you to complete some questionnaires. These visits will be at 5 and 10 days from your current visit and then a final follow-up visit in 6 weeks time; the total time you will be in the research will be 6 weeks.
The study will involve a brief interview and medical examination to find out if you are suitable for the study. A member of the research team will review your medical records and ask some questions about you and any medicines you might be taking. These data will be entered into a central study database and be given a code so that you cannot be identified from the information in the database. Only the study team will have access to identifying information about you and this will be kept confidential as described below.
A summary of the research procedures at each of the study visits is shown in the table below. The research procedures that will occur as part of your current visit should take no longer than 2 hours and all subsequent visits will take around 1 hour.
| Study procedure | Visit 1 (day 1) (current visit) | Visit 2 (day 5) | Visit 3 (day 10) | Visit 4 (follow-up visit, 6 weeks time) |
|---|---|---|---|---|
| Informed consent | ✗ | |||
| Review of medical notes and brief medical examination | ✗ | |||
| Breathing tests | ✗ | ✗ | ✗ | |
| Blood sample | ✗ | ✗ | ||
| Nose swab, throat swab and collection of nasal mucus | ✗ | |||
| Sputum (spit) sample | ✗ | |||
| Complete symptom diary | ✗ | ✗ | ✗ | |
| Complete two asthma questionnaires | ✗ | ✗ | ✗ | |
| Dispense study drug or placebo (‘dummy’ treatment) | ✗ |
Study procedures
Informed consent
At your current visit you will be seen by the study doctor/nurse who will discuss the details of the study and answer any questions you may have. If you wish to take part, the study doctor or nurse will then ask you to sign a consent form and you will be given a signed copy of the form to keep.
Study visits
We will be in contact with you by telephone to co-ordinate the visits and follow-up with you.
Review of medical notes/brief medical examination
The study doctor or nurse will ask you about any current or previous medical conditions and any medications that you take. There is some information we will need to take from your medical notes to help with our research and we will keep this confidential.
Breathing tests
You will be asked to perform a few breathing tests that involve blowing into a tube (spirometry). You will be familiar with these from routine appointments to assess your asthma.
Blood sample
We will collect 30 ml of blood (2 tablespoons worth) today and 30 ml from you in 6 weeks time.
Nose and throat swabs and collection of nasal secretion
A sample of nasal mucus will be taken using a nose swab and a throat swab will be taken. You will also be asked to blow into a tissue to collect a sample of nasal mucus.
Sputum (spit) sample
You will be asked to produce a deep cough into a sterile dish. If there is not enough spit we may also induce a sputum sample from you, which will involve breathing in saline (a salty solution) through a nebuliser (a device used to breath in a salty mist/spray through a mouthpiece) until there is enough sputum available. This should take no longer than 20 minutes and we will check your breathing throughout with a spirometer (described above). You are free to ask to stop the procedure at any time or it may be stopped by the study doctor or nurse if they feel that this is necessary.
Symptom diaries and asthma questionnaires
You will be given a symptom diary to complete at your current visit and will be asked to complete it at home each day for the next 10 days. It should take you less than 5 minutes to complete. You will also be given two asthma questionnaires to complete at your current visit and in 5 and 10 days time when you return for visits. These should take up to 20 minutes in total for you to complete.
Study drug/placebo
Sometimes we do not know which way of treating patients is best. To find out, we need to compare different treatments. We put people into groups and give each group a different treatment. The results are compared to see if one is better. To try to make sure the groups are the same to start with, each patient is put into a group by chance (randomly). In this study 50% of participants will receive the study drug (azithromycin) and 50% will receive a placebo (‘dummy’ treatment). As the study is a ‘double-blind trial’, this means that neither you nor your doctor will know which treatment group you are in (although if your doctor needs to find out he/she can do so).
You will be given either the study drug or the placebo after all other procedures during visit 1, as shown in the above table. You will be asked to take two capsules once a day for 3 days. This is the routine dose given if the drug is prescribed as part of routine care. You should take the study drugs at least 1 hour before or 2 hours after food. If you are taking antacids you should take the study drug at least 1 hour before or 2 hours after antacids. Your study doctor will check the other medication that you may be taking and any medical condition you may have to make sure that this does not conflict with taking azithromycin.
On your next visit in 5 days time we will ask you to return the drug containers and any unused drugs and confirm if you have been able to take the drug as prescribed.
Pregnancy test
If you are female and of childbearing potential we will ask you to undertake a urine pregnancy test before starting study procedures to ensure that you are not pregnant.
What are the alternatives for diagnosis or treatment?
The alternative to participating in this research is to continue through the routine process for treatment of your exacerbation and not participate in the study or take the study drug/placebo.
Are there any disadvantages to taking part in this study?
The disadvantages of taking part in this study are those associated with collecting the samples as well as the time inconvenience to you of attending the AZALEA research site for further visits. We will try to arrange the time of your study visits to be convenient for you.
Blood samples
Blood samples will be taken from a vein in your arm using a sterile needle. Taking blood samples may cause a little discomfort and bruising to your arm but this will resolve in a few days.
Nose and throat swabs and collection of nasal secretion
The nose swab may be associated with mild discomfort and may tickle or make your eyes water slightly but should be painless. The throat swab is not painful but you may find it briefly uncomfortable. Collection of nasal secretion will only involve you blowing into a tissue.
Sputum (spit) sample
Induced sputum as described above may lead to some shortness of breath but we will monitor this with breathing tests. You are free to ask to stop the procedure at any time or it may be stopped by the study doctor or nurse if they feel that this is necessary.
What are the side effects of the study drug?
-
Azithromycin is a licensed antibiotic with a good safety record that has been widely used for a long time. Side effects and allergic reactions are rare. The most common side effects that you may experience are gastrointestinal, that is, nausea, diarrhoea, vomiting and abdominal pain/cramps.
-
There is a low risk of cardiovascular side effects. In rare incidences azithromycin can cause abnormal changes in the electrical activity of the heart, particularly in patients who already have abnormal heart rhythms (arrhythmias) or who are taking medication for these conditions. These patients will therefore be excluded from taking part in the study.
-
If you suspect that you have these or any other symptoms you must inform the study doctor. If you experience severe side effects or an allergic reaction please contact the study doctor immediately on XXXX or attend A&E or phone for an ambulance if you are concerned.
-
If you have reacted badly in the past to azithromycin or related antibiotics and their ingredients then you will not be eligible to participate in this study. Similarly, if you are taking any other medications or have other disease conditions or infections that could interfere with the way that the drug works or with our monitoring of how it works then you will not be included in the study.
Harm to an unborn child
We do not know if the study drug is safe for an unborn child so, to protect the unborn child, pregnant women must not take part in this study and neither should women who plan to become pregnant during the study. Women who are breastfeeding will also not be allowed to take part in the study. Women who are at risk of pregnancy will be asked to have a pregnancy test before taking part in the study to exclude the possibility of pregnancy.
If you are female and find out that you have become pregnant while taking part in the study, you should immediately tell your study doctor.
Are there any benefits to taking part in this study?
You may benefit from this study if you are randomised to receive azithromycin and it is found that this is effective in improving recovery from an asthma exacerbation. However, we do not know whether or not it is effective so we cannot promise that the study will help you. Although this study will not necessarily benefit you directly, it may help us to understand more about the best ways of treating asthma exacerbations in future patients with this condition.
What happens when the research study stops?
You will return to the care of your local doctor (GP) and the hospital doctors you would see routinely.
What if there is a problem?
Any complaint about the way you have been dealt with during the study or any possible harm you might suffer will be addressed. The detailed information on this is given in Part 2.
Will my taking part in the study be kept confidential?
Yes. The details are included in Part 2.
This completes Part 1.
If the information in Part 1 has interested you and you are considering participation, please read the additional information in Part 2 before making any decision.
Part 2
What if relevant new information becomes available?
Sometimes new information about a treatment being used in a study becomes available during the course of the study. If this happens, the study doctor will let you know. You can then make a decision about whether you want to continue in the study. If you decide not to continue in the study, the study doctor will ensure that your ‘normal’ care continues. If you decide to continue in the study, you will be asked to sign an updated consent form.
After receiving new information, the study doctor might want you to withdraw from the study. He may feel it is in your best interest to do this. If this happens, the doctor will explain the reason for this and arrange for your ‘normal’ care to continue. Your participation in the study may be stopped for any of the following reasons:
-
failure to comply with the study instructions
-
a serious reaction, which may require treatment or observation
-
the doctor decides it is in the best interest of your health and welfare to discontinue.
What will happen to the samples I give?
-
The above research samples that we take from you will be used to find out the following information:
-
whether azithromycin treatment is effective during an acute exacerbation of asthma
-
the type of viral and/or bacterial infection present in participants during an exacerbation
-
why azithromycin might be an effective treatment, that is, how it works
-
whether there are ‘markers’ in participants’ blood or spit that would be able to tell us in advance about the severity of and recovery from the acute exacerbation.
-
-
The results from testing your research samples will not affect the care that you receive as part of your routine treatment as we will not know the best treatment until the outcome of the study.
-
All of the samples will be labelled with a code with no identifying information about you. The samples will then be tested in designated laboratories but the laboratory scientists will not be able to identify them as your samples. Your identifying information will be accessible only to authorised members of the research team.
-
If you agree, we would also like to keep any of your samples that are left over after our research. These would be stored and used for future ethically approved research and would be accessible only to authorised members of the research team or regulatory bodies. At any time you want you could ask for these samples to be destroyed.
Can I withdraw from participation in this study?
Your participation in this study is voluntary and you have the right to refuse to participate. You are free to withdraw at any time and do not have to give a reason for this, even after you have agreed to take part. Being part of this study will not affect your normal medical care, either now or in the future.
What will happen if I don’t want to carry on with the study?
You are free to withdraw from the study at any time, without needing to give a reason why. If you withdraw from the study, we would like to keep the samples and data collected up to your withdrawal for our analysis. Any stored blood or tissue samples that can still be identified as yours can be destroyed though if you wish.
What if something goes wrong?
Imperial College London holds insurance policies that apply to this study. If you experience serious and enduring harm or injury as a result of taking part in this study, you may be eligible to claim compensation without having to prove that Imperial College is at fault. This does not affect your legal rights to seek compensation.
If you are harmed because of someone’s negligence, then you may have grounds for a legal action. Regardless of this, if you wish to complain, or have any concerns about any aspect of the way you have been treated during the course of this study, then you should immediately inform the investigator on the contact details below. The normal National Health Service complaints mechanisms are also available to you. If you are still not satisfied with the response, you may contact the Imperial AHSC Joint Research Office.
Who is organising and funding the research?
The research is co-ordinated by Imperial College London and has been funded by the National Institute for Health Research (NIHR) and Medical Research Council. It has been reviewed and given a favourable opinion by the London – Bloomsbury Research Ethics Committee and has been given a favourable opinion by the Medicines and Healthcare products Regulatory Agency (MHRA), the agency that reviews and approves drug studies.
The study is being carried out by experts in asthma from around the country as a collaboration between NHS trusts and universities (a list of these collaborators is available to you should you wish). None of the investigators performing the research or participants taking part will benefit financially from the study.
Will the information on me be kept confidential?
Yes, all personal information will be kept confidential and secure. Only people involved in the study will have access to your personal information. When we send your samples for analysis in designated laboratories they will be labelled with a code and have no identifying information on them. This study will be published in medical journals but it will not be possible to identify you from what is written. With your permission we would also like to retain any of your samples not used in this study for future research projects.
Involvement of the general practitioner/family doctor
With your permission we will inform your GP of your participation in this study.
Expenses and payments
A maximum payment of £50 will be available to you at visit 4 if you have completed all study visits and completed and returned all 10 symptom diaries (this payment is made up of £10 for attending visit 1, £10 for visit 2, £10 for visit 3, £10 for visit 4 and £10 for returning the symptom diaries).
We will also reimburse the cost of your travel expenses for the additional three visits you will be asked to attend for research purposes.
What if I have any problems or would like further information about the study?
If you have a concern about any aspect of this study, you should ask to speak to the researchers on XXXX, who will do their best to answer your questions. If you remain unhappy and wish to complain formally, you can do this through the normal NHS complaints procedure.
Thank you for taking the time to consider participating in our research.
List of abbreviations
- A&E
- accident and emergency
- AE
- adverse event
- AQLQ
- Asthma Quality of Life Questionnaire
- AR
- adverse reaction
- AZALEA
- AZithromycin Against pLacebo for acute Exacerbations of Asthma
- CI
- confidence interval
- COPD
- chronic obstructive pulmonary disease
- CRN
- Clinical Research Network
- DMEC
- Data Monitoring and Ethics Committee
- eCRF
- electronic case report form
- FEF25–75%
- forced mid-expiratory flow rate
- FEF50%
- forced expiratory flow rate at 50% expiration
- FEV1
- forced expiratory volume in 1 second
- FEV1 : FVC
- ratio of forced expiratory volume in 1 second to forced vital capacity
- FVC
- forced vital capacity
- GP
- general practitioner
- ICTU
- Imperial Clinical Trials Unit
- IgM
- immunoglobulin M
- IMP
- investigational medicinal product
- ITM
- Integrated Trial Management
- MHRA
- Medicines and Healthcare products Regulatory Agency
- NIHR
- National Institute for Health Research
- PCR
- polymerase chain reaction
- PEF
- peak expiratory flow
- PI
- principal investigator
- REC
- Research Ethics Committee
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SDV
- source data verification
- SE
- standard error
- SOC
- system organ class
- SUSAR
- suspected unexpected serious adverse reaction
- TELICAST
- Telithromycin, Chlamydophila, and Asthma trial
- TSC
- Trial Steering Committee