
Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 10/60/03. The contractual start date was in February 2012. The final report began editorial review in August 2015 and was accepted for publication in September 2016. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Disclaimer
This report contains transcripts of interviews conducted in the course of the research and contains language that may offend some readers.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2017. This work was produced by Robson et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Major congenital anomalies affect 1–1.5% of births and are a leading cause of neonatal death and disability, resulting in substantial emotional and economic burden for families and society. 1 Chromosomal anomalies are responsible for 20–25% of major fetal anomalies. 1 The national Fetal Anomaly Screening Programme (FASP) recommends ultrasound screening for congenital anomalies at 11–14 and 18–20 weeks of pregnancy. 2 Early screening for chromosomal anomalies at 11–14 weeks incorporates ultrasound measurement of nuchal translucency (NT); 2–3% of pregnant women are identified as being at high risk of having a baby with the chromosomal anomaly after screening and are offered chorionic villus sampling (CVS) or amniocentesis. 3 When the NT is ≥ 3.5 mm (equivalent to a 99th centile) the risk of a chromosomal anomaly is > 20% and FASP recommends the offer of invasive prenatal diagnosis irrespective of other screening markers. 4 Fetuses with a NT of ≥ 3.5 mm are also at increased risk of structural anomalies and genetic syndromes. 4
Screening for structural anomalies is recommended at the 18+0 to 20+6 week ultrasound scan,5 although a recent systematic review has shown that approximately 50% of fetal structural anomalies can be detected by an ultrasound scan at 11–14 weeks of pregnancy. 6 Approximately 15% of fetuses with a structural anomaly detected in the second trimester have an underlying chromosomal anomaly although rates vary with the specific structural anomaly and whether or not the anomaly is isolated or one of a number of ultrasound-detected defects;7,8 rates increase from 29%, when two or more structural anomalies are detected, to > 70% with six or more. 7
When women opt for invasive testing because of an increased fetal risk of chromosomal anomaly, laboratories perform quantitative fluorescent polymerase chain reaction (QF-PCR) for rapid aneuploidy detection. This test detects chromosome copy number by amplification of repeat sequences at polymorphic loci. It is efficient (one technician can examine up to 5000 samples per year) and detects trisomies 21, 18 and 13 as well as sex chromosome anomalies and triploidy within 2 days in 5–14% of cases. 9 In addition, G-banded karyotyping of cultured cells detects other aneuploidies and large unbalanced chromosome rearrangements at a resolution of 5–10 Mb in a further 5% of cases. 10–12 However, karyotyping is slow (it can take 2 weeks to receive the results of the test) and labour intensive (one technician can examine ≈250 samples/year). 10 The majority of these other aneuploidies and rearrangements occur in fetuses with multiple anomalies or an increased NT. 7,11,13,14 Furthermore, when a structural anomaly is suggestive of a well-defined microdeletion/duplication syndrome (e.g. del22q11), additional targeted fluorescence in situ hybridisation (FISH) or multiplex ligation-dependent probe amplification (MLPA) may be performed. 12,15 When testing fails to identify a chromosomal anomaly in a fetus with a sonographic anomaly, counselling parents about aetiology and prognosis is difficult because some babies are later found to have other abnormalities and/or learning difficulties.
Array comparative genomic hybridisation (CGH) or chromosomal microarray (CMA) involves the isolation and differential labelling of genomic deoxyribonucleic acid (DNA) from a patient and a reference (control) followed by co-hybridisation to a matrix of DNA fragments (probes) that can be short (25- to 60-mer) oligonucleotides or larger fragments (up to 250 kb). 16 The fluorescence ratio for each probe reflects the average copy number ratio between patient and control DNA. Use of probes with known genome positions allows identification of DNA copy number variants (CNVs).
The first CMA platforms used bacterial artificial chromosome (BAC) clones of 100–150 kb in length, generating an intense hybridisation signal with a high signal-to-noise ratio. 17 BAC arrays have an average resolution of 0.5–1 Mb and multiplexing is limited; therefore, they are suitable only for low throughput. Subsequent oligonucleotide comparative genomic hybridisation (oligoCGH) arrays use shorter probes (typically 50–60 base pairs), providing higher resolution (40–400 kb) but the shorter probes result in less specific hybridisation and lower signal-to-noise ratio. Single nucleotide polymorphism (SNP) arrays use probes that are either 25 or 50 base pairs long and, therefore, tend to have the lowest signal-to-noise ratio. The resolution of oligonucleotide-based arrays is dependent on the number and spacing (density) of the probes and the number of consecutive probes required for a CNV call. Resolution therefore varies with the array design and the calling parameters. 17
Interpretation of CMA results in a clinical setting can be challenging as copy number variation occurs in normal individuals; approximately 12% of the human genome exhibits such variation. 18 Variants can be classified as pathogenic (i.e. causative of the phenotype), benign or novel variants of unknown significance (VOUS). More than 99% of benign CNVs are inherited and the vast majority of these are < 500 kb. 19 Thus, in order to classify novel CNVs, follow-up studies (e.g. parental arrays sometimes followed by FISH, MLPA and QF-PCR on fetal/parental DNA) are often required. Regularly updated online databases that catalogue CMA results from normal individuals and those with phenotypic anomalies provide invaluable information to assist in array classification. One perceived disadvantage of the increased diagnostic yield of smaller CNVs with oligonucleotide-based (either CGH or SNP) arrays is the need for more parental follow-up tests and the increased rate of VOUS. 17,20
Microarray technology has identified novel microdeletion/duplication syndromes, many of which are mediated by specific genomic motifs called segmental duplications. In postnatal life, these account for 25% of all recurring microdeletion/duplication cases. Array CGH has also been used to further characterise visible chromosomal rearrangements and, in some cases, unmask novel Mendelian disorders. When CMA has been applied to children with undiagnosed developmental delay and an apparently normal G-banded karyotype, 10–15% had pathogenic de novo CNVs of 150–15,000 kb. 21,22 The improved detection of pathogenic CNVs, together with reduced cost of arrays, led to the recommendation that CMA should be the first-line test for postnatal referrals with developmental delay and dysmorphism. 23 UK guidelines, published in 2011,24 recommended postnatal CMA for the investigation of developmental delay and dysmorphism. This policy has been implemented in regional genetics laboratories across the country.
Initial prenatal studies employed low-resolution BAC arrays on fetal DNA extracted after fetal loss25 and pathogenic CNVs were detected in ≈10% of cases. Subsequent studies confirmed that CMA could be applied to uncultured and cultured amniocytes and chorionic villi with detection of aneuploidies, known microdeletion syndromes and large unbalanced CNVs. 26–28 Shaffer et al. 29 and Coppinger et al. 30 used BAC arrays in two studies of 151 and 182 prenatal samples with normal karyotypes collected for varying clinical indications. Pathogenic CNVs were detected in 1.3–2.7% of cases and benign CNVs in a further 8.8%. The highest yield of pathogenic CNVs (4.5%) was found in cases with an abnormal ultrasound scan.
Following the introduction of higher resolution oligonucleotide arrays, Van den Veyver et al. 31 used a targeted array in 300 prenatal samples from cases referred primarily for advanced maternal age. They detected 58 (19.3%) CNVs, of which 15 (5.0%) were deemed pathogenic and three (1.0%) of uncertain significance. Subsequent studies used CMA in fetuses with structural anomalies identified on ultrasound but a normal karyotype, and CNVs were reported in 2–15% of cases. 32–35 In 2011, Hillman et al. 36 reported a meta-analysis of eight prenatal studies which showed that overall array CGH detected 12% [95% confidence interval (CI) 8.8% to 16.4%] more chromosomal imbalances when karyotyping was normal. When the analysis was confined to fetuses with a structural anomaly on ultrasound (six studies, 359 cases) CMA detected 11.2% (95% CI 5.7% to 22.1%) more chromosomal imbalances overall. The rate fell to 5.2% (95% CI 1.9% to 13.9%) when benign CNVs were removed. 36 This included 1.9% (95% CI 0.4% to 9.5%) for which the result reported was of unknown significance. 36 The largest of these studies included only 151 cases. In the same year, Leung et al. 37 reported the first study of CMA in a cohort of 48 fetuses with increased NT (> 3.5 mm) at 11+2–14+1 weeks’ gestation and a normal karyotype. CNVs were reported in six (12.5%) cases and considered pathogenic in four (8.3%) cases. Thus, at the time of commencing the Evaluation of Array Comparative genomic Hybridisation (EACH) study, the available literature suggested that CMA may detect more pathogenic CNVs in fetuses with ultrasound-identified structural anomalies and increased NT than karyotyping.
Policy development in NHS-based health care is complex. This is particularly apparent in the area of fetal anomaly, in which health care involves a significant degree of scientific uncertainty about prognoses, alongside difficult decision-making for parents about the care pathways and reproductive choices available to them following prenatal diagnosis. With respect to CMA, there is concern that reporting of VOUS may not only increase parental anxiety but lead parents to choose to terminate a fetus that is potentially at low risk of an adverse outcome. 36 Although technological developments in fetal medicine can help reduce uncertainty, Williams38 noted that sometimes developments have been implemented without due consideration of the ethical and social issues that may arise from the technology for the parents and health professionals involved in care provision. Furthermore, implementation of health policy is influenced by a range of factors, including local level staff views about changes to service provision. 39 Prior to commencing the EACH study, no work had been done on patient or professional views on prenatal CMA or on possible barriers to or facilitators of implementation. In their systematic review of prenatal CMA, Hillman et al. 36 emphasised the need for qualitative research in this area.
In England and Wales, around 35,000 women undergo karyotyping per annum (at a cost of £3.3M). 10 Since CMA was introduced into prenatal diagnosis, when the cost per case was estimated at ≈£600,40 array costs have decreased dramatically and higher-density oligonucleotide arrays are now available for < £150. However, given the reported increased detection rates of pathogenic variants and VOUS with CMA, the associated costs of additional tests, reviewing results and clinical follow-up to explain CMA results are expected to be higher than karyotyping. Thus, at the time of commencing the EACH study, there was an urgent need to assess the clinical and cost efficacy of CMA before introduction into NHS practice, as had already occurred in some US centres. Indeed, despite the limited evidence, an American College of Obstetrics and Gynecology Committee Opinion in 2009 stated that ‘Targeted array CGH, in concert with genetic counselling, can be offered as an adjunct tool in prenatal cases with abnormal anatomic findings and a normal conventional karyotype’. 41
If CMA is shown to detect pathogenic genomic imbalances more effectively than karyotyping and at a lower cost, then array CGH should replace karyotyping, allowing the streamlining of genetic service delivery with a single diagnostic platform across prenatal and postnatal life. CMA could also dramatically further our understanding of fetal anomalies and, in so doing, enhance patient counselling and management. The rationale for the EACH study was, therefore, to evaluate the potential for CMA to replace karyotyping for the investigation of fetuses with ultrasound anomalies after rapid exclusion of common aneuploidies by QF-PCR. The specific research aim was to provide guidance to health service providers on whether CMA should replace karyotyping in the prenatal diagnosis of fetal anomalies by determining whether or not CMA is an acceptable, cost-effective and robust method to detect more clinically significant de novo chromosomal imbalances than conventional karyotyping in fetuses with (1) one or more structural anomalies identified by ultrasound and (2) an isolated NT of ≥ 3.5 mm identified at the routine 11- to 14-week ultrasound scan.
Over recent years, along with developments in CMA, there have also been considerable advances in the development of non-invasive prenatal testing (NIPT) based on cell-free fetal deoxyribonucleic acid (cffDNA) in maternal plasma. By 2011, when the EACH study was funded, several studies had demonstrated the potential for NIPT for common trisomies and even other large CNVs. 42–45 The National Institute for Health Research (NIHR) had funded the Reliable Accurate Prenatal non-Invasive Diagnosis (RAPID) project to (1) develop standards for implementation of NIPT into routine NHS practice for fetal sex determination and single gene disorders, (2) evaluate the potential of NIPT for trisomy 21 and other major trisomies and (3) explore the potential for NIPT of significant unbalanced CNVs using next generating sequencing of cffDNA. 46 As the EACH study would be recruiting a cohort enriched for both aneuploidy and other CNVs, combining studies would allow a large-scale evaluation of NIPT on the EACH cohort. Therefore, a secondary research aim was to contribute maternal blood samples to the RAPID project. The results of the RAPID project will be reported separately. 46
In order to achieve these aims the specific research objectives of the EACH study were as follows.
Laboratory evaluation
-
To compare detection rates of:
-
pathogenic CNVs and VOUS by karyotyping and CMA in two target populations of fetuses with ultrasound-detected anomalies after exclusion of major numerical sex and autosomal chromosomal anomalies by QF-PCR
-
trisomy 21 and other major trisomies by QF-PCR and NIPT (to be reported with the RAPID project).
-
-
To compare turnaround times (TATs) from receipt of sample to issuing of results for:
-
karyotyping and CMA
-
QF-PCR and NIPT (to be reported with the RAPID project).
-
Cost issues
-
To calculate costs of karyotyping and CMA to NHS and Personal Social Services (including DNA extraction, quality metrics, labelling and follow-on tests on fetal/parental DNA).
-
To calculate the cost per additional pathogenic CNV detected by array CGH (and associated follow-on testing) relative to karyotyping.
Public and health professional attitudes
-
To evaluate parent and health professional attitudes to CMA.
-
To determine which factors influence parents’ and health professionals’ choices and decision-making about CMA (and the potential to replace karyotyping).
An economic evaluation of NIPT and an evaluation of public and health professional attitudes to NIPT was already included as part of the RAPID project.
Chapter 2 Methods
The EACH study was designed as a multicentre experimental research cohort study with an additional cost analysis and assessment of patients’, health professionals’ and commissioners’ preferences for array CGH.
Study population
Inclusion criteria
Women with a fetus (singleton or dichorionic twin) undergoing QF-PCR and conventional karyotyping by amniocentesis, CVS or fetal blood sampling for clinical indications with
-
one or more structural anomalies identified on an ultrasound scan at any gestational age or
-
an isolated NT of ≥ 3.5 mm in a fetus with a crown–rump length of 45.0–84.0 mm
were included in the study.
Fetuses with isolated fetal growth restriction (defined as an abdominal circumference of more than two standard deviations below the mean for gestational age according to local standards) were also included in the structural anomaly group. Following feedback from Trial Steering Committee members and site principal investigators, fetuses with a thickened nuchal fold (NF) (defined as a NF of > 6 mm) at 18+0 to 20+6 weeks were also included in the structural anomaly group. 47
Fetuses with a structural anomaly and a NT of ≥ 3.5 mm identified at 11+2 to 14+1 weeks of pregnancy were included in the structural anomaly group. Fetuses diagnosed with an isolated cystic hygroma between 11+2 and 14+1 weeks of pregnancy were included in the NT group.
Only those fetuses with a normal QF-PCR result or a sex chromosome aneuploidy that was unlikely to explain the ultrasound anomaly (e.g. XXX, XXY and XYY) underwent CMA. Cases were recruited from selected fetal medicine units in England and Wales. Sunderland Royal Hospital (City Hospitals Sunderland NHS Foundation Trust) also acted as a patient identification centre for the study; suitable women were given the patient information leaflet and those interested in participating were referred to the Royal Victoria Infirmary Fetal Medicine Unit where they were approached to take part in the study.
Exclusion criteria
Women meeting the following criteria were regarded as ineligible for the study:
-
A fetus with single or multiple ultrasound variants (or markers). In this context fetal cerebral ventriculomegaly (ventricular atrium of ≥ 10 mm) was classed as a structural anomaly.
-
Participant declined to take part in the study.
-
Participant was under the age of 16 years.
-
Participant was unable to read English and understand the study information leaflet.
Women who had consented to the EACH study but who were subsequently found to have a fetus with a common aneuploidy on QF-PCR were also excluded from the study.
Recruitment and consent
Eligible women attending fetal medicine clinics were identified by a member of the medical staff providing care or by a research nurse/midwife screening the medical notes. Those meeting the inclusion criteria were approached to take part in the study and given an information leaflet (see Appendix 1). Informed consent discussions were undertaken by appropriate site staff (as per the delegation log) involved in the study, including medical staff and research nurses/midwives, with opportunity for participants to ask questions.
Those wishing to participate in the study gave informed written consent to use any excess fetal material (villi, amniocytes or fetal blood) available after setting up routine testing (QF-PCR and karyotyping). They also consented to donating a blood sample of up to 20 ml. In addition, women were also asked to give written consent to contact after birth to arrange assessment of their infant (see Appendix 2). When partners were in attendance they were also approached and asked for written informed consent to take a blood sample. Parents were advised that this was a research study designed to evaluate new methods of fetal chromosomal analysis but that in a few cases the research may reveal additional information that may inform pregnancy management. Should this occur, the information would be transmitted to them via the health professionals responsible for their care.
The original signed consent form was retained in the investigator site file, with a copy in the clinical notes and a copy provided to the participant. Women taking part in the study and those who declined were added to the screening and recruitment log to ensure that they were not approached again later in their pregnancy.
Ethics approval was granted by National Research Ethics Service Committee North East – Newcastle and North Tyneside (reference 11/NE/0331) on 4 January 2012. Three subsequent amendments were approved:
-
Amendment 1 (approval date 26 October 2012) to include an updated health economic analysis plan and to amend the definition of increased NT to ≥ 3.5 mm (to be consistent with national guidance from NHS Screening Programmes).
-
Amendment 2 (approval date 14 February 2013) to include Sunderland Royal Hospital as a participant identification centre site, Birmingham Women’s Hospital as a recruitment site and also widening the inclusion criteria to include structural anomalies identified at any gestation.
-
Amendment 3 (approval date 23 December 2013) to include the use of fetal blood samples.
The Newcastle upon Tyne Hospitals NHS Foundation Trust acted as study Sponsor (reference 5878).
Laboratory workflow
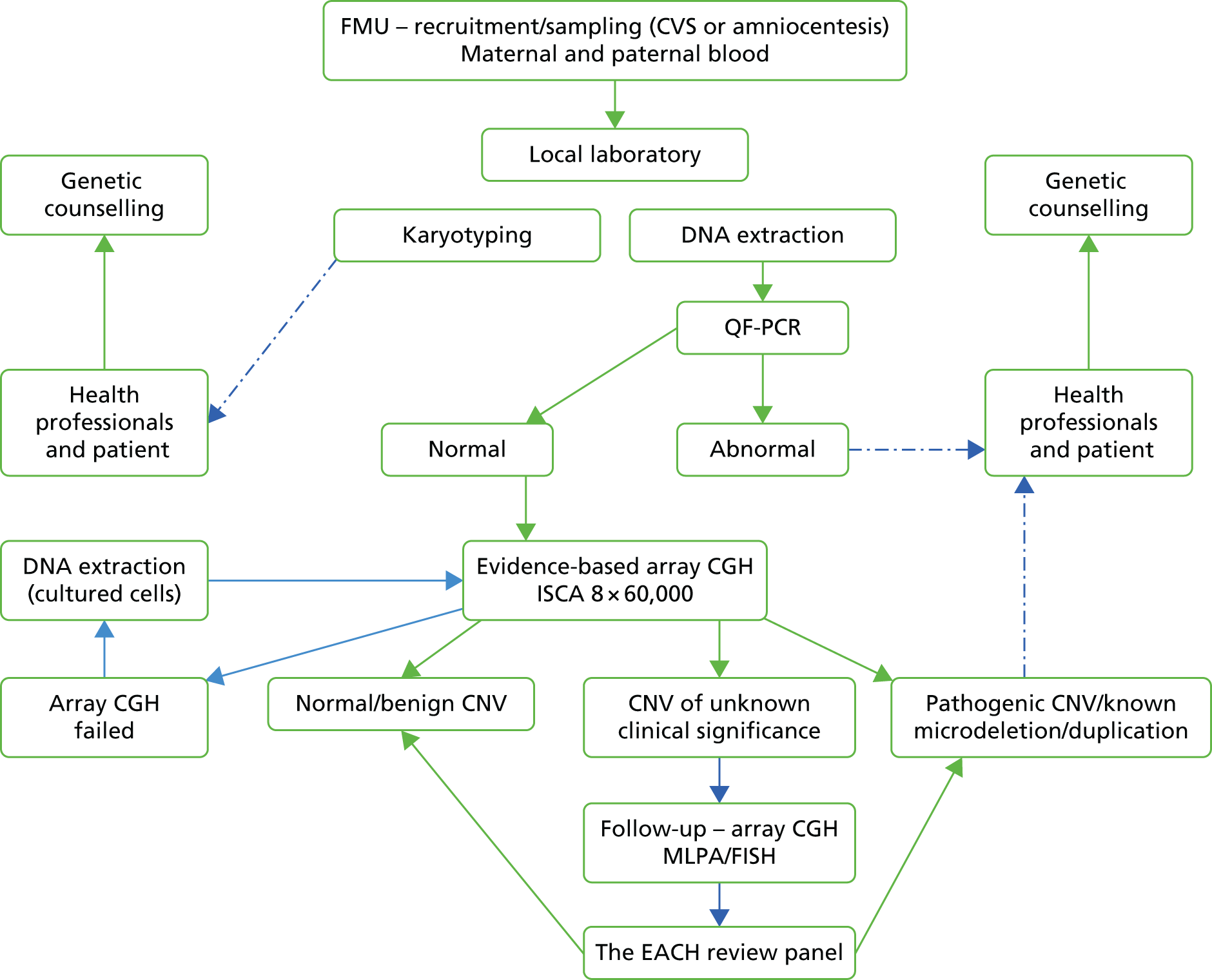
All nine participating cytogenetic laboratories followed their existing clinical pathways for karyotyping and adapted their workflows to incorporate DNA extraction from amniocytes and chorionic villi for both QF-PCR and CMA (Figure 1). In most instances, parental blood samples were routinely stored until the results of the CMA were known and DNA was extracted. Parental chromosome preparations were made only if follow-up parental studies were required (see Figure 1).
FIGURE 1.
Laboratory workflow in the EACH study. FMU, fetal medicine unit; ISCA, International Standards for Cytogenomic Array.
Deoxyribonucleic acid extraction
Deoxyribonucleic acid was extracted from the available uncultured fetal samples, predominantly chorionic villi and amniotic fluid. DNA extraction was achieved in accordance with to either local laboratory protocols or using a method developed specifically for obtaining high-quality DNA from small volumes of source prenatal material. 48 This method was developed during planned preliminary (pre-recruitment) studies undertaken at the Wessex Regional Genetics Laboratory (WRGL) and the North-East Thames Regional Genetics Laboratory (NETRGL). In brief, a collection of commercially available DNA extraction kits and quantification techniques were evaluated and compared using minimal quantities of amniotic fluid (2–4 ml), chorionic villi (2–5 mg) and cultured cells (under 150,000 cells). The work identified the following optimised technical workflow:
-
DNA extraction using the iGENatalTM kit (iLab Tech, Madrid, Spain)
-
Quantification by the Qubit® 2.0 Fluorometer with the Qubit® dsDNA BR assay kit (InvitrogenTM/Life TechnologiesTM, Thermo Fisher Scientific, Waltham, MA, USA).
Subsequent CMA experiments using Oxford Gene Technology’s CytoSure™ (Oxford, UK) International Standards for Cytogenomic Arrays (ISCAs) 8 × 60,000 oligonucleotide array platform demonstrated good-quality array profiles, providing further evidence that the extracted DNA was of high quality. The extractions yielded between 210 ng and 1825 ng of prenatal DNA from 4 ml of amniotic fluid and, using this platform, successful arrays were achieved with as little as 125 ng of prenatal DNA, provided this was matched with an equivalent amount of control DNA in the experiment. 48
Quantitative fluorescent polymerase chain reaction
Quantitative fluorescent polymerase chain reaction was carried out using either commercial kits [QStar plus (Elucigene® QST*R Plus v2 kit, Elucigene Diagnostics, Manchester, UK) or Devyser Complete v2 kit (Devyser AB, Hagersten, Sweden)] or in-house products and protocols. Primers targeting chromosomes 13, 18, 21, X and Y were used. Cases of trisomies 13, 18 and 21, triploidy and 45X were reported to the referring clinicians and patients and no further actions with respect to the EACH study and CMA were performed. When other sex chromosome aneuploidies were detected (47,XXY, 47,XYY, 47,XXX and one case of 49,XXXXX), these were processed by CMA as these chromosomal abnormalities were not expected to be associated with fetal structural abnormalities.
Standard karyotyping
Prenatal samples were processed for standard karyotyping in accordance with the standard operating procedures in operation in each of the participating cytogenetic laboratories. All laboratories retained samples of viable fibroblasts in vitro in case these were needed for either (1) repeat DNA extraction and repeat CMA using fibroblast DNA and/or (2) additional cytogenetic preparations required for FISH follow-up studies.
Array comparative genomic hybridisation
At the outset of the EACH study it was agreed that all of the participating laboratories would use the same array design based on the International Standard for Cytogenomic Array Consortium consensus. 49 The array selected for use consisted of an 8-plex of 60,000 60-mer oligonucleotides with a backbone resolution of ≈75 kb, but with considerably higher coverage over telomeric and pericentromeric chromosomal regions in known microdeletion and microduplication syndromic regions and also for a number of key developmental and haploinsufficient genes. The laboratories obtained their arrays from a number of commercial vendors and in most cases used the analytical software supplied with the arrays. All CMA results are presented in GRch37/hg19 format. 50
All CMA tests were carried out using the protocol provided by the manufacturers and the QF-PCR sex chromosome result was used to (1) detect cases with significant maternal cell contamination (MCC) (in the event of which the DNA was extracted from cultured fibroblasts) and (2) select the sex-matched control for the array CGH experiment. All but one of the laboratories incorporated the EACH samples into their routine postnatal CMA protocols; the exception ran separate prenatal arrays in batches. In cases with a normal result, or when only known benign CNVs were observed, normal CMA and karyotyping results were reported to the referring clinician. When an abnormal array CGH result [e.g. involving one of the known microdeletion syndromes or further quantifying the extent of the imbalance(s) associated with chromosomally visible abnormalities] was received, this was reported to the referring clinician. In cases for which the clinical significance of the CNV was uncertain (VOUS), the laboratory carried out follow-up studies, principally parent versus parent array CGH or FISH, to determine whether the CNV had been inherited or was de novo. Once this information was available, the case was either reported on the basis of the information to hand or referred to the EACH review panel for further consideration. It should be noted that the participating laboratories recorded all cases of pathogenic CNVs and VOUS into a bespoke database designed by Cartagenia Bench™ (version 4.0, Cartagenia N.V., Leuven, Belgium). Known benign CNVs were recorded only locally and not incorporated into the Cartagenia Bench laboratory proforma.
Database of phenotypes, demographics and laboratory results
To accommodate the multicentre design of the EACH study, the recording of all data sets was facilitated by a bespoke online version of Cartagenia Bench. The proformas were designed by SCR and LC (fetal phenotyping and demographics) and JAC (laboratory results). Demographic and fetal phenotypic data were uploaded by research nurses/midwives from the participating fetal medicine units and all identifiable patient information was removed. Clinical scientists at the participating cytogenetic laboratories were then provided access to the demographic and fetal phenotyping information and added all aspects of the laboratory processes via the laboratory proformas. Pregnancy outcome data, when available, were uploaded by research nurses/midwives. Anonymised post-mortem reports and paediatric discharge summaries were uploaded into Cartagenia Bench.
Data queries and outstanding pregnancy outcomes were sent to each participating centre every 4–6 months. In the period between close of recruitment and data closure (for cleaning and analysis). Efforts to get complete outcome data focused on cases with an abnormal karyotype and/or CMA. Data monitoring and analyses were facilitated by downloading all of the data fields into Microsoft Excel® 2010 (Microsoft Corporation, Redmond, WA, USA) spreadsheets.
Follow-up studies
Several laboratory techniques were used to determine the parental origins of CNVs detected by karyotyping or CMA: parental karyotyping, parent versus parent CMA [in which DNA from both parents was run on a single array but only the area of imbalance(s) observed in the fetus was analysed to determine whether or not the CNV was inherited], FISH, MLPA, QF-PCR and, rarely, other molecular cytogenetic techniques.
For cases requiring FISH follow-up studies, the WRGL had an in-house collection of FISH clones consisting of the 30,000 Sanger BACs set, which was acquired by the WRGL in 2007. Since then, ≈2500 of the 30,000 clones have been used for FISH studies and labelled DNA was available for distribution to the participating laboratories, if required. A database of these clones was made available to all participating laboratories. FISH-ready clones were aliquoted within 24 hours and sent by post to the requesting laboratory. TATs for producing FISH-ready clones for other targeted regions of the genome were ≈10 days. All clones were sent to the requesting laboratory in a ‘FISH-ready’ format and use of these FISH clones was contingent on each laboratory verifying the position and how informative the clone(s) were by using the proband’s chromosomes as the positive control.
The EACH study copy number variant exclusion criteria
At the outset of the study, a clinical review group was established to review the available literature and to draw up a list of VOUS with low penetrance and/or low expressivity which would therefore not be reported within the context of prenatal diagnosis (Table 1). However, the final decision whether or not to report CNVs on this list was made by the local clinical scientists and ultimately the referring clinicians.
| Variant | Start (build 37) | End (build 37) | Interval |
|---|---|---|---|
| 1q21.1 microdeletion | 146,512,930 | 147,737,500 | 1.22 |
| 1q21.1 microduplication | 146,512,930 | 147,737,500 | 1.22 |
| 15q11.2 BP1–BP2 | 22,765,637 | 23,217,454 | 0.45 |
| 16p13.11 microdeletion | 15,504,454 | 16,284,248 | 0.78 |
| 16p13.11 microduplication | 15,504,454 | 16,284,248 | 0.78 |
| 22q11 duplication | 18,546,349 | 22,336,469 | 3.79 |
Classification of karyotype and chromosome microarray findings
All structural and numerical abnormalities identified by karyotyping were reported according to standard laboratory protocols. All such cases were included as an abnormal karyotype for the purposes of the EACH study, whether balanced or not (e.g. translocations and inversions); this included cases ultimately found to represent confined placental mosaicism (CPM) and rare, probably benign, variants (e.g. mosaic loss of Y).
The laboratories participating within the EACH study utilised their own standard operating procedures to classify the CNVs detected by their analytical CMA software. These classifications have been divided into the following three categories.
-
Pathogenic CNVs with a clearly defined abnormal phenotype (e.g. known microdeletion syndrome) or a variant with a probability of a phenotypic effect.
-
Variants of unknown significance for which the possible genotype and phenotypic effects were uncertain. In some cases, based on parental origin studies and informed by local and international CNV data sets, a local decision was made not to report the VOUS. In other cases, based on the recommendation of the EACH review panel (see EACH review panel), a decision was made not to report the VOUS.
-
Common variants, or those recognised as benign and not relevant to the presenting phenotype, were not reported. These cases included those on the EACH predetermined list of CNVs (see Table 1).
The EACH review panel
The EACH review panel consisted of five consultant clinical cytogeneticists and five consultant clinical geneticists chaired by DGW. Cases for review were sent to the clinical (DGW) and scientific (JAC) leads for distribution and decision feedback by e-mail. All cases were examined by a quorum of two geneticists and two cytogeneticists. The length of time between referral and decision was recorded as well as the details of the case and the final recommendation of the panel.
At the outset of the study, it was planned that only CNVs with direct relevance to the ongoing clinical fetal phenotype would be reported but this was amended when VOUS with significant clinical risks to other and/or future family members were recorded {e.g. the 455-kb deletion with a maternally inherited Duchenne muscular dystrophy (DMD) gene ascertained in a set of female twins [NCL65675(2)], which was classified as a pathogenic variant}.
Final result groupings
Based on the results of the karyotype and CMA and any follow-up tests, the results of the EACH study were grouped as follows:
-
normal karyotype and normal CMA
-
abnormal karyotype and normal CMA
-
abnormal karyotype and pathogenic CNV on CMA
-
normal karyotype and pathogenic CNV on CMA
-
normal karyotype and VOUS on CMA.
Two more complex scenarios arose when assigning final result groups:
-
The finding of a VOUS on CMA when it was assessed that there was a high probability of a phenotypic effect but one of the parents was unavailable either for analysis or to confirm parental origin and/or parental phenotype. Such cases were assigned to the ‘pathogenic CNV’ group.
-
The finding of a pathogenic CNV on CMA but the karyotype failed and, given the CMA result, the karyotype was not repeated by the laboratory. The two cases in this group (CAN62785 and UCH1372792) were assigned to the ‘abnormal karyotype and pathogenic CNV on CMA’ group.
Evaluation of laboratory turnaround time
Turnaround times for conventional karyotyping and CMA were measured from the date the fetal sample was received in the laboratory to the date the final karyotype or CMA report was issued by the laboratory. This reflects the way in which NHS laboratories collect reporting time data for comparison with Clinical Pathology Accreditation (CPA) standards. This TAT not only includes the technical aspects of the testing but also a robust process of writing, reviewing and authorising the test reports. Laboratories followed their own reporting protocols and the EACH study design had no direct impact on the order in which the karyotyping and CMA results were reported. The EACH laboratory protocol also required that all samples were first tested using QF-PCR. The time taken for QF-PCR was not analysed separately but considered part of the overall reporting process.
The EACH study assessed the use of CMA within the context of prenatal diagnosis of chromosomal anomalies in the UK NHS. The primary responsibility of participating laboratories was therefore to provide the commissioned test (i.e. karyotyping) and CMA was a supplementary technique, although laboratories strove to produce the CMA report as quickly as possible. To try and better understand the actual timescales for undertaking and reporting each test, including the time taken to perform any additional laboratory tests (e.g. parental karyotyping, parent versus parent CMA), laboratories were also asked to record the following on Cartagenia Bench:
-
the dates the karyotype and CMA were set up and reported
-
the dates any follow-up tests were set up and reported.
Given the EACH design, use of the date of CMA set-up and reporting may provide a more reliable estimate of TAT should CMA replace karyotyping and, hence, be carried out as a standalone test.
Exclusions
Karyotype TAT results excluded data from London centres because karyotype testing was rarely performed by the participating laboratory (NETRGL at Great Ormond Street Hospital, London), the majority being undertaken by other non-NHS providers.
Economic analysis
Implementation of CMA into prenatal diagnosis may have significant cost consequences. The costs of the CMA test and associated costs of reviewing the results are expected to be higher than the costs of conventional karyotyping. In addition, women undergoing CMA may need extra clinical visits or additional tests to investigate an abnormality. The EACH economic analysis aimed to investigate the cost-effectiveness of replacing conventional karyotyping with CMA in the prenatal diagnosis pathway of fetal anomalies.
Model structure
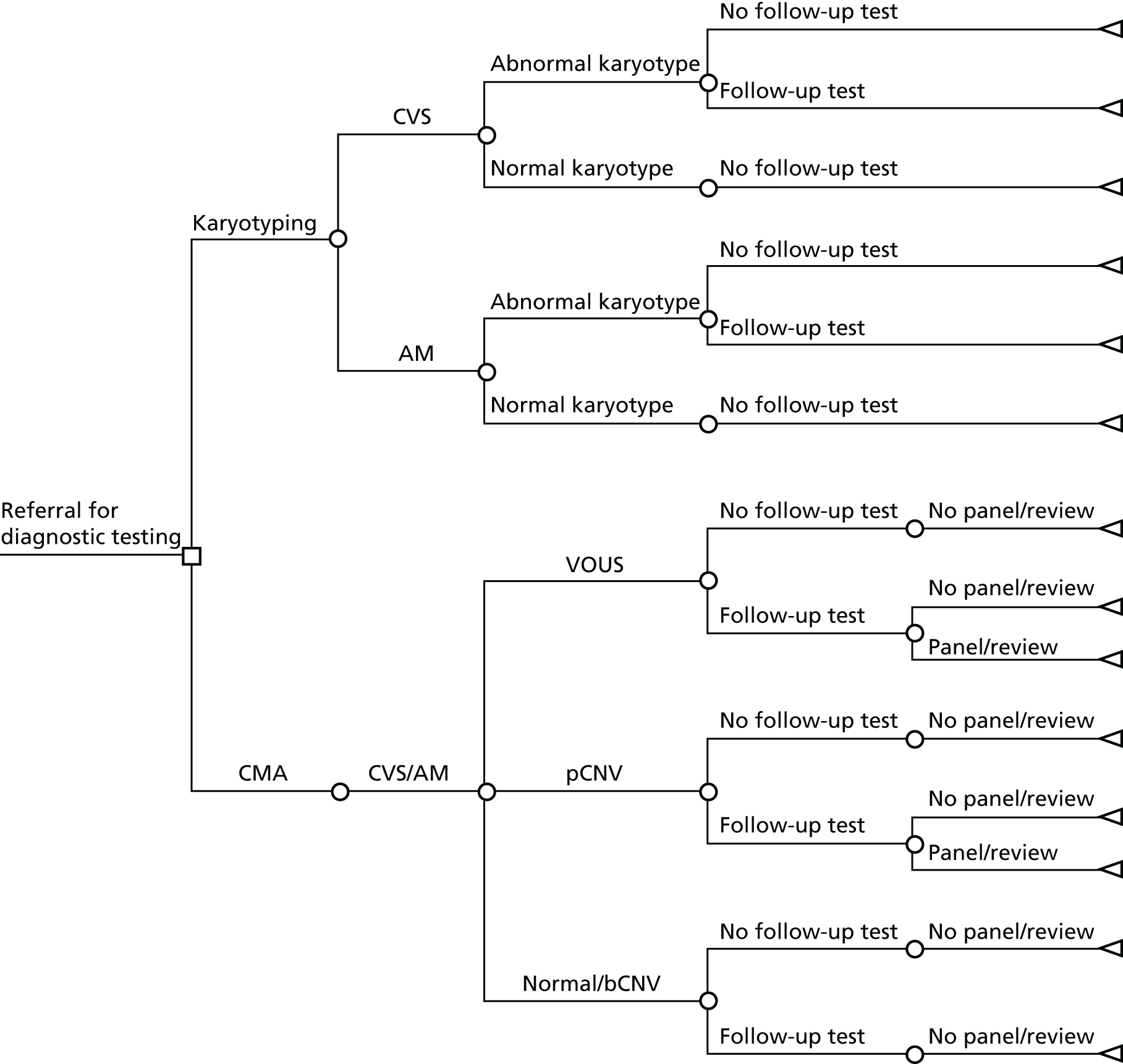
A decision-analytic model was developed describing the two diagnostic options (conventional karyotyping and CMA) and the possible pathways following these. The structure of this model is depicted in Figure 2. The population consists of women referred for diagnostic testing (karyotyping) with a normal QF-PCR result. In the karyotyping pathway, women underwent either CVS or amniocentesis. A small proportion of cases had diagnostic testing on other fetal tissues (e.g. fetal blood) but, for simplification, they were not considered in this model. In the case of an abnormal karyotype, a proportion of women underwent one or more follow-up tests (see Chapter 3). If the karyotype was normal, women did not undergo any follow-up tests. In the CMA pathway, array CGH was performed using extracted DNA. When the result of the test showed a CNV, a proportion of women underwent one or more follow-up tests (see Chapter 3). If these suggested a benign variant, no further action was taken but, if the results suggested a pathogenic CNV or a VOUS, some cases were referred to the EACH review panel, which made a recommendation about whether or not the variant was likely to be clinically significant and should be reported.
FIGURE 2.
Decision tree depicting the current diagnostic pathway and the new pathway using CMA. AM, amniocentesis; bCNV, benign copy number variant; pCNV, pathogenic copy number variant.
Model inputs
Data from the EACH study (collected on Cartagenia Bench) were used to identify the number of women in each branch of the pathway. All women underwent karyotyping as well as CMA and the analysis was run for all women referred for diagnostic testing, but also separately for women in the increased NT and structural anomaly groups. As in the main EACH analysis, cases for which the fetus had a structural anomaly and an increased NT were analysed in the structural anomaly group.
Costs
Data on the costs of karyotyping and CMA were collected from nine cytogenetic laboratories participating in the EACH study. The average costs were used as base-case values in the current model. Costs of laboratory follow-up testing were collected from WRGL, Salisbury. Costs of clinical follow-up (either at a clinical genetics and/or a fetal medicine department) focused specifically on follow-up consultations related to test results (i.e. not just because of the underlying ultrasound finding). These costs were estimated using NHS reference costs. 51 Finally, we estimated the costs of the EACH panel review by looking at the time spent on each case and the number of participants in the panel. The costs per minute for a medical consultant and a senior laboratory scientist were estimated using data from the Personal Social Services Research Unit. 52 We assumed that for each review there was an equal chance that each person involved was a medical consultant or a senior laboratory scientist. All costs are expressed in 2012–13 British pounds. The time horizon in this study was the duration of pregnancy and, therefore, no discounting was necessary.
Model outputs
The main outcome of the model was the incremental costs per pathogenic CNV detected by CMA compared with conventional karyotyping, using the base-case values for every parameter. This summarises how much more it costs to detect one additional significant abnormality with CMA than karyotyping. In the UK, the recommended outcomes for economic evaluations are quality-adjusted life-years (QALYs) and the recommended cost-effectiveness measure is based on the incremental costs per QALY gained. 53 One advantage of using the incremental costs per QALY gained is that there is a published threshold that can be used to judge whether or not an intervention is good value for money (£20,000 to £30,000 per QALY gained53). However, measuring QALYs arising from prenatal testing is challenging. It is problematic to determine the quality of life of a child with an abnormality and of other unaffected children as well as the quality of life of the parents. There is also uncertainty about what happens when a pregnant mother decides to terminate an affected pregnancy. She might get pregnant again and have another (healthy) baby or she might not. Owing to these complexities, the economic consequences of prenatal testing are not usually reported using QALYs.
Uncertainty around our input parameters was taken into account using one-way sensitivity analysis and probabilistic sensitivity analysis. In the one-way sensitivity analysis, parameters were varied one at a time over a plausible range (mostly equal to 95% confidence limits) to assess how the main outcome varied. A scenario analysis was performed excluding the costs of the review panel to investigate the costs per case detected if no review panel would be required or if it were not available. In this scenario, we assumed that the costs of the review panel were £0 but that the outcomes in terms of abnormalities would not change. A probabilistic sensitivity analysis was performed, producing 1000 simulations of the outputs based on drawing random samples from the probability distributions of all input parameters to calculate the chance that CMA would be cost-effective at different levels of willingness to pay for extra cases detected. Beta distributions were used for all probabilities and gamma distributions for all costs.
Statistical analysis
The sample size was based on a comparison of the rates of chromosome imbalances and pathogenic CNVs detected by karyotyping and CMA in each of the two target populations using McNemar’s test for paired binary outcomes (for each fetus each result is either normal or abnormal, giving four possible outcome pairs) and a quantification of the difference in detection rates. A sample of 500 in each target population gave in excess of 90% power to detect a difference in detection rates at the 5% significance level, assuming that karyotyping and CMA detected anomalies in 5% and 10% of fetuses, respectively (nQuery 6.0, Statsols, Cork, Ireland). This increase in diagnostic yield was deemed clinically important by collaborators and was consistent with the meta-analysis reported by Hillman et al. 36 in 2011. Array CGH was assumed to detect all the imbalances detected by karyotyping (i.e. the proportion of discordant outcomes was assumed to be close to 5%). This sample size would give a 95% CI for the difference in detection rates of ±2%.
Continuous variables were described by either means (standard deviations) or medians [interquartile ranges (IQRs)] depending on their distribution and binary and categorical variables were described using numbers (proportions). The proportions of subjects with pathogenic CNVs and VOUS were calculated with exact 95% CIs. Statistical comparisons of pathogenic CNVs/VOUS between karyotyping and CMA were made using McNemar’s test and the exact version of the test was used when numbers were too small. Comparisons between patient subgroups (e.g. NT of ≤/> 6 mm) were made using the chi-squared test, and Fisher’s exact test was used when numbers were too small. A statistical comparison of the differences in TATs between karyotyping and CMA was made using the paired t-test. A comparison of times by laboratory, year and epoch was made using one-way analysis of variance; the Kruskal–Wallis test was used when the distribution of times was skewed.
Qualitative substudy
The qualitative substudy aimed to explore the ways in which CMA is viewed by parents, health professionals and commissioners, and to examine the social and policy issues that result from the experiences of those most closely involved with the intervention.
The sampling approach used was purposive, to enhance the diversity of views represented within the sample (rather than aiming to be statistically representative of some wider population). Women who had participated in the main EACH trial at two of the study sites (Newcastle and Leeds) were asked at the time of consent whether or not they would agree to be interviewed about their experience. The details of those who agreed were passed to the researcher (CL), who posted them a study pack a minimum of 3 months after completion of the pregnancy (birth or termination of pregnancy). The pack contained information about the substudy and a consent to contact form with a reply slip. Those agreeing were contacted and an interview was arranged at a place of their choosing. After 2–4 weeks, if no reply had been received, women were sent a further letter by way of reminder. If no response was received it was assumed that they did not wish to take part and no further contact was made. The aim was to undertake a total of 15–20 parent interviews (with up to 10 in each of the two study groups). Once sufficient numbers of women who had received ‘normal’ CMA results had been interviewed, women with a pathogenic CNV only were purposively sampled. Owing to difficulties recruiting sufficient women with an abnormal result, recruitment was extended to involve a third study site (Birmingham).
Health professionals from two of the study sites (Newcastle and Leeds) were identified by using an initial list of eligible professionals compiled by the principal investigator at each site. From this list, a purposive sample was identified to ensure the inclusion of a range of professional viewpoints. National and local commissioners were identified through a convenience sample through a contact with the Accountable Commissioner for NHS commissioning of national specialised fetal medicine services.
Identified individuals were sent a study pack by post or e-mail (via their work addresses) with a consent to contact form and reply slip. Those agreeing were contacted and an interview was arranged at a place of their choosing. If no response was received from the initial invitation, professionals were sent another invitation 2–4 weeks later by way of reminder. If no response was received, it was assumed that they did not wish to take part and no further contact was made. The aim was to undertake a total of 15–20 professional interviews.
Prior to the planned interviews, the participant was given the opportunity by CL to ask any questions. If the participant was happy to proceed, written consent was obtained. Data collection was via in-depth, semistructured, interviews with the participants. This allowed the researcher to draw on a standard topic guide (see Appendices 3 and 4) to ensure that key topics were covered, but also allowed participants sufficient freedom to describe their experiences in a narrative of their choice. All interviews were audio-recorded, transcribed verbatim and anonymised. All participants were given a study identifier and all identifying names and locations were removed from the transcripts and replaced with identifiers. The anonymised transcripts formed the data for the analysis.
The data analysis was informed by a generative thematic approach,54 drawing on Silverman’s55 approach to the analysis of interview texts to evaluate how participants describe and conceptualise their experiences. The qualitative analysis software package ATLAS.ti (version 7.0, Cleverbridge AG, Cologne, Germany) was used to facilitate the thematic analysis of the data. The parent data and the professional data were analysed as separate groupings initially, before being brought together for the final analysis. Each transcript was coded by the substudy research associate (CL). Another member of the project team (RG) worked with a sample of the transcripts to contribute to the design of the coding framework and to provide an element of inter-rater reliability to the coding of the data. After the data were coded, they were organised into key themes and subthemes.
Chapter 3 Results
Study recruitment
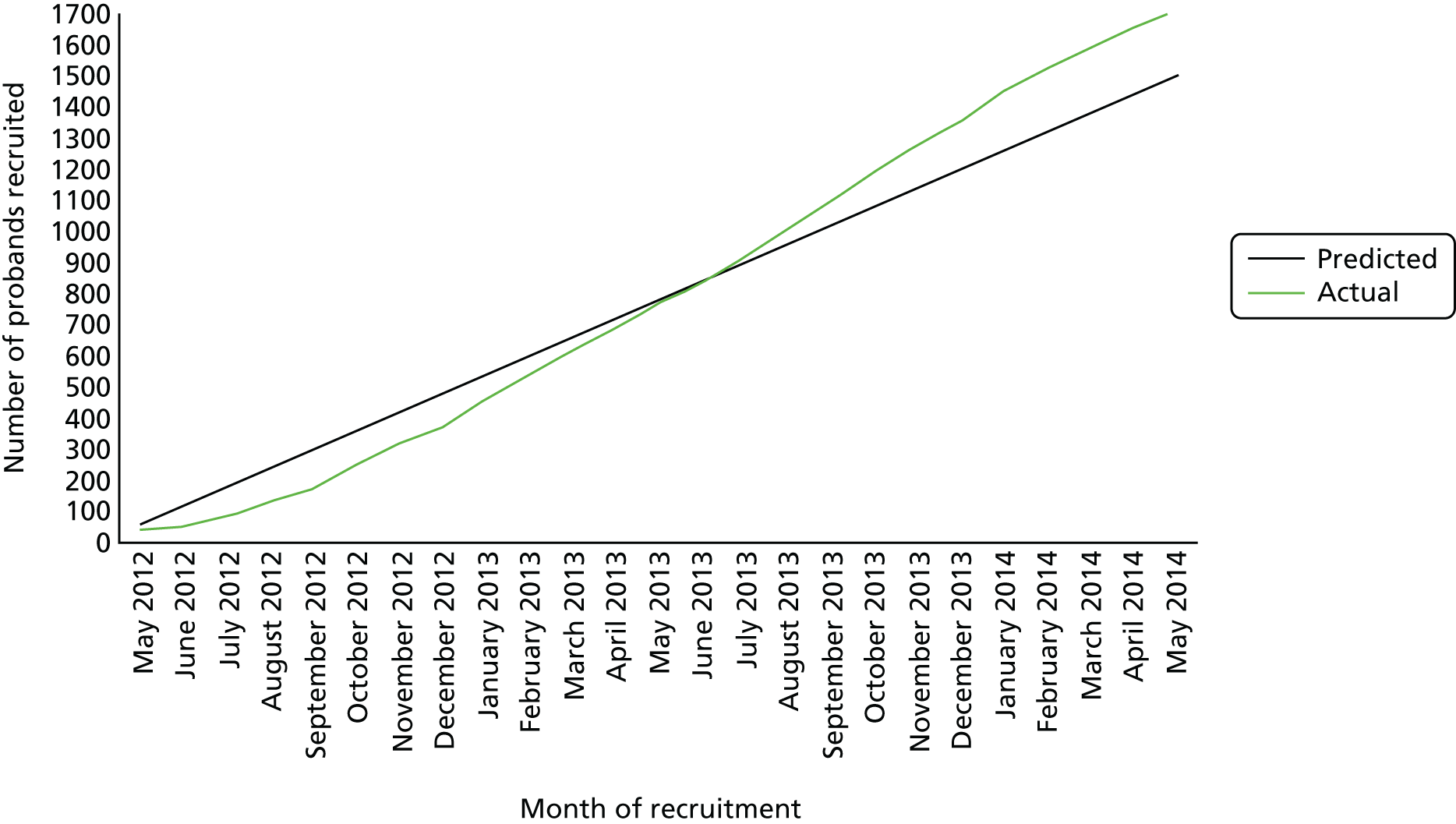
Cases were recruited from 20 fetal medicine units with samples analysed in nine cytogenetic laboratories in England and Wales (Table 2). Two large centres (Cardiff and Birmingham) joined the study after the initial set-up, having secured independent resources (for staff and consumables). In total, 1718 probands were recruited to the EACH study between May 2012 and May 2014 (Figure 3).
| Cytogenetic laboratory | Fetal medicine units | Number of cases |
|---|---|---|
| North East Thames Regional Genetics Service, London | University College Hospital, London | 470 |
| Royal Free Hospital, Londona | 1 | |
| Royal London Hospitala | 2 | |
| Homerton University Hospital, Londona | 3 | |
| Queens Hospital, Romford, Essex | 29 | |
| Queen Charlotte’s and Chelsea Hospital, London | 23 | |
| St George’s Hospital, London | 123 | |
| WRGL, Salisbury | Princess Anne Hospital, Southampton | 105 |
| Frimley Park Hospital, Camberley | 55 | |
| Queen Alexandra Hospital, Portsmouth | 19 | |
| East Anglian Medical Genetics Service, Cambridge | The Rosie Hospital, Cambridge | 59 |
| Norfolk and Norwich University Hospitalb | 16 | |
| Northern Genetics Service, Newcastle | Royal Victoria Infirmary, Newcastle | 168 |
| James Cook University Hospital, Middlesbrough | 37 | |
| Yorkshire Regional Genetics Service, Leeds | Leeds General Infirmary, Leeds | 118 |
| Merseyside and Cheshire Regional Molecular Genetics Laboratory, Liverpool | Liverpool Women’s Crown Street Hospital, Liverpool | 120 |
| Bristol Genetics Laboratory, Bristol | St Michael’s Hospital, Bristol | 131 |
| Royal Devon and Exeter Hospital, Exeter | 53 | |
| All Wales Medical Genetics Service, Cardiffb | University Hospital of Wales, Cardiffb | 57 |
| West Midlands Regional Genetics Laboratory, Birminghamb | Birmingham Women’s Hospital, Birminghamb | 129 |
FIGURE 3.
Number of probands recruited to the EACH study.
A total of 1460 (85.0%) women also gave consent to contact them after birth to arrange assessment of their infant. A paternal blood sample was obtained in 1347 (78.4%) cases. Thus, in total, 3065 subjects consented to the study. The source tissue used for DNA extraction was recorded in 1546 cases (90.0%) (Table 3).
| Source tissue for DNA extraction | Number of cases (%) |
|---|---|
| Chorionic villi | 863 (55.8) |
| Amniotic fluid | 631 (40.8) |
| Fetal blood | 12 (0.8) |
| Fetal tissue | 13 (0.8) |
| Cultured fibroblastsa | 17 (1.1) |
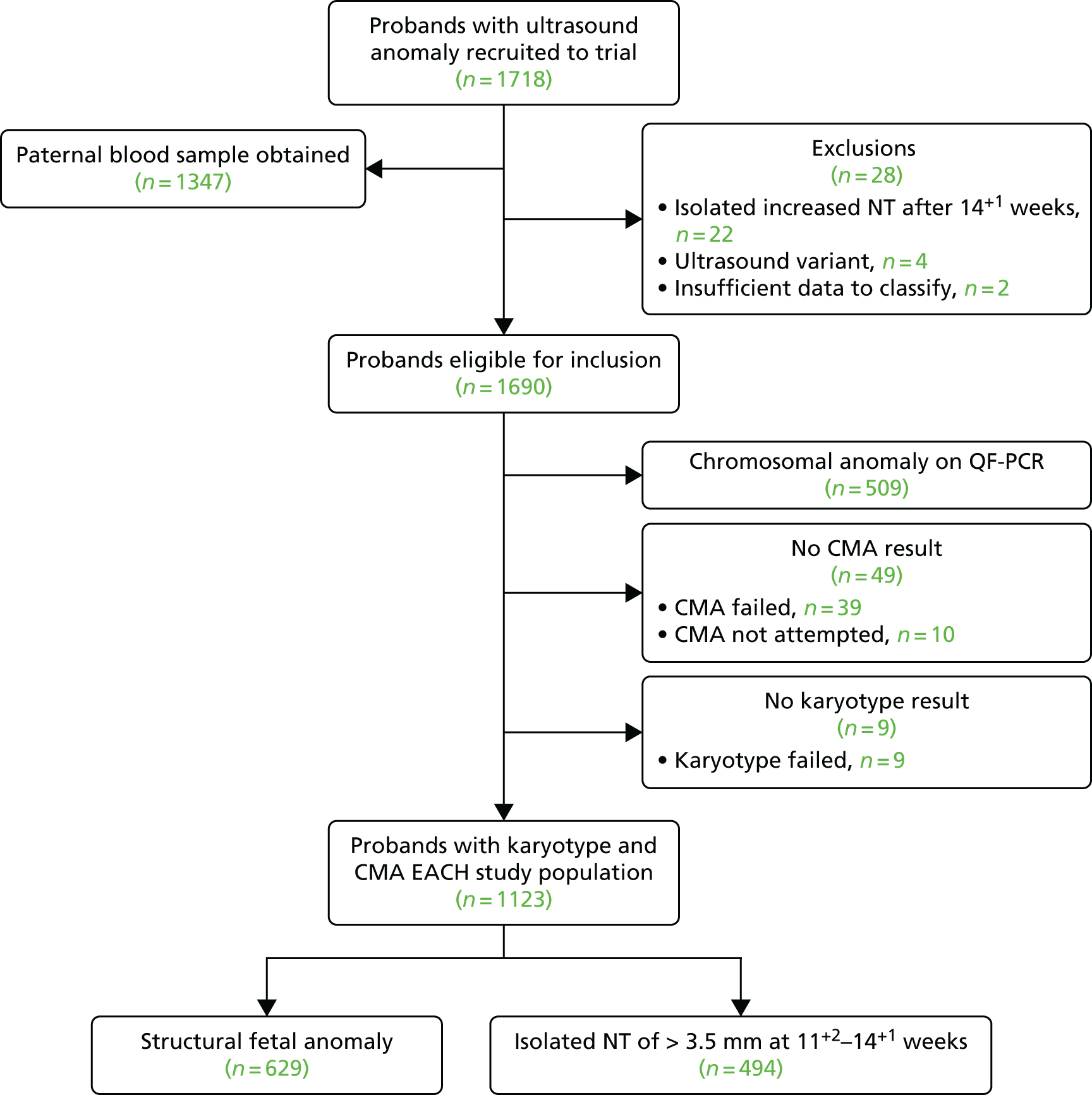
Out of the 1718 probands recruited, 28 were subsequently excluded. The reasons for exclusion are shown in Table 4. No parent requested withdrawal from the study.
| Reason for exclusion | Number of cases |
|---|---|
| Failure to meet ultrasound inclusion criteria | |
| Isolated increased NT identified after 14+1 weeks | 22 |
| Single or multiple ultrasound variants | 4 |
| Insufficient data to classify | 2 |
Out of the 1690 included cases, 509 cases (30.3%) had a common chromosomal anomaly detected by QF-PCR and, therefore, did not proceed to CMA (Table 5). In a further 10 cases, fetal material was not available for DNA extraction and subsequent CMA. In total, karyotyping failed in 9 out of 1181 cases (0.8%) and CMA failed in 39 out of 1171 cases (3.3%).
| Chromosomal anomaly | Number (%) of cases |
|---|---|
| Trisomy 21 | 231 (45.5) |
| Trisomy 18 | 121 (24.0) |
| Trisomy 13 | 43 (8.5) |
| XO | 66 (13.0) |
| Triploidy | 26 (5.0) |
| Aneuploidy with structural chromosome anomaly | 14 (2.5) |
| Autosomal mosaicsa | 8 (1.5) |
Thus, 1123 probands with a karyotype and CMA result were available for analysis and this group constituted the EACH study population (Figure 4).
FIGURE 4.
Trial profile of the EACH study.
Study groups
Increased nuchal translucency group
Of the 1123 cases making up the EACH study population, 494 (44.0%) had an isolated NT identified on a routine scan at 11+2 to 14+1 weeks of pregnancy. The clinical characteristics of this group are shown in Table 6. Pregnancy outcome data were unavailable in 187 (38%) cases but in only 2 out of the 64 (3%) cases with an abnormal karyotype and/or CMA.
| Clinical characteristic | Data | |
|---|---|---|
| Age (years), mean (SD) | 30.7 (5.4) | |
| Primiparous (%) | 197 (47.6%)a | |
| Gestation (weeks), mean (SD) | 13.1 (1.5) | |
| NT of > 5 mm | 161 (32.6%)b | |
| NT of > 6 mm | 82 (17.2%)b | |
| Pregnancy outcome | Normal karyotype and CMA | Abnormal karyotype +/– CMA |
| Live birth | 203 | 33 |
| Stillbirth/neonatal death | 5 | 3 |
| Miscarriage | 6 | 2 |
| Termination of pregnancy | 31 | 24 |
| Unknown | 185 | 2 |
| Total | 430 | 64 |
The distribution of recorded NT measurements is shown in Figure 5. A further 20 cases recruited between 11+2 and 14+1 weeks’ gestation had an ultrasound diagnosis of isolated cystic hygroma but no NT measurement recorded. These cases were included in the increased NT group.
FIGURE 5.
Nuchal translucency measurements in 474 fetuses in the increased NT group.

Structural anomaly group
A total of 629 cases (56%) had a structural anomaly identified. The clinical characteristics of this group are shown in Table 7. Pregnancy outcome data were unavailable in 167 (27%) cases but in only 1 out of the 89 (1%) cases with an abnormal karyotype and/or CMA.
| Clinical characteristic | Data | |
|---|---|---|
| Age (years), mean (SD) | 30.7 (5.7) | |
| Primiparous (%) | 299 (55.8%)a | |
| Gestation (weeks), mean (SD) | 20.2 (4.8) | |
| Number of fetal anomalies | ||
| One | 382 (60.7%) | |
| Two | 156 (24.8%) | |
| Three | 49 (7.8%) | |
| More than three | 42 (6.7%) | |
| Pregnancy outcome | Normal karyotype and CMA | Abnormal karyotype +/– CMA |
| Live birth | 225 | 38 |
| Stillbirth/neonatal death | 31 | 4 |
| Miscarriage | 11 | 0 |
| Termination of pregnancy | 107 | 46 |
| Unknown | 166 | 1 |
| Total | 540 | 89 |
The fetal anomalies detected are shown in Table 8 broken down by the body system. Brain anomalies were the most common (17.6% of all anomalies detected by ultrasound), followed by cardiac (17.5%) and skeletal (14.5%). Overall, 382 cases (60.7%) had an single (isolated) fetal anomaly.
| Structure | Anomaly | Isolated | Multiple | Total |
|---|---|---|---|---|
| Face | Cleft lip and/or palate | 29 | 45 | 74 |
| Micrognathia | 0 | 9 | 9 | |
| Other (including flat face, abnormal nose, macroglossia, hypertelorism and not defined) | 2 | 12 | 14 | |
| Total | 31 | 66 | 97 | |
| Brain | Ventriculomegaly and/or agenesis of corpus callosum and/or posterior fossa anomaly | 65 | 23 | 88 |
| Agenesis of corpus callosum | 5 | 6 | 11 | |
| Posterior fossa anomaly | 10 | 21 | 31 | |
| Holoprosencephaly | 7 | 9 | 16 | |
| Encephalocele | 1 | 3 | 4 | |
| Anencephaly | 1 | 1 | 2 | |
| Cyst | 1 | 2 | 3 | |
| Microcephaly | 5 | 2 | 7 | |
| Other (including abnormal cavum septum pellucidum, choroid plexus cyst, dolichocephaly and not defined) | 1 | 11 | 12 | |
| Total | 96 | 78 | 174 | |
| Spine | Spina bifida and/or ventriculomegaly | 13 | 5 | 18 |
| Kyphoscoliosis (including hemivertebrae) | 0 | 8 | 8 | |
| Sacrococcygeal teratoma | 3 | 1 | 4 | |
| Other (not defined) | 0 | 7 | 7 | |
| Total | 16 | 21 | 37 | |
| Neck | NF of > 6 mm at 18+0 to 20+6 weeks | 7 | 29 | 36 |
| Cystic hygroma | 0 | 9 | 9 | |
| Other (not defined) | 0 | 1 | 1 | |
| Total | 7 | 39 | 46 | |
| Thorax | Diaphragmatic hernia | 14 | 5 | 19 |
| Pleural effusion | 10 | 14 | 24 | |
| Other (including echogenic lungs, short ribs) | 1 | 9 | 10 | |
| Total | 25 | 28 | 53 | |
| Cardiac | Abnormal four chamber (specific anomaly undefined) | 20 | 40 | 60 |
| Abnormal outflow tracts (specific anomaly undefined) | 8 | 1 | 9 | |
| Ventricular septal defect | 7 | 19 | 26 | |
| Atrioventricular septal defect | 9 | 3 | 12 | |
| Tetralogy of Fallot | 11 | 3 | 14 | |
| Hypoplastic left heart | 9 | 4 | 13 | |
| Pulmonary atresia, hypoplastic right heart | 5 | 3 | 8 | |
| Transposition of great arteries | 6 | 1 | 7 | |
| Coarctation of aorta | 4 | 1 | 5 | |
| Pericardial effusion | 3 | 6 | 9 | |
| Other (including total anomalous pulmonary venous drainage, double outlet right ventricle, dextrocardia, Ebstein’s anomaly) | 1 | 9 | 10 | |
| Total | 83 | 90 | 173 | |
| Abdominal wall | Exomphalos | 27 | 12 | 39 |
| Other (including bladder exstrophy, body stalk anomaly) | 2 | 2 | 4 | |
| Total | 29 | 14 | 43 | |
| Gastrointestinal tract | Isolated ascites | 4 | 13 | 17 |
| Echogenic bowel and/or liver | 0 | 31 | 31 | |
| Absent stomach or stomach anomaly undefined | 0 | 20 | 20 | |
| Bowel obstruction (including duodenal atresia) | 3 | 9 | 12 | |
| Cyst | 1 | 2 | 3 | |
| Total | 8 | 75 | 83 | |
| Genitourinary tract | Megacystis | 20 | 6 | 26 |
| Multicystic kidney | 2 | 7 | 9 | |
| Hydronephrosis | 1 | 8 | 9 | |
| Renal agenesis | 0 | 4 | 4 | |
| Enlarged echogenic kidney | 2 | 6 | 8 | |
| Other (including pelvic kidney, horseshoe kidney, duplex kidney) | 1 | 6 | 7 | |
| Abnormal genitalia (including clitoromegaly, microphalus, bifid scrotum) | 0 | 5 | 5 | |
| Total | 26 | 42 | 68 | |
| Skeletal | Talipes | 17 | 27 | 44 |
| Other hand/foot abnormalities (including absent, ectrodactyly, polydactyly) | 5 | 22 | 27 | |
| Absent, short or abnormal long bones (including micromelia and undefined skeletal dysplasia) | 14 | 27 | 41 | |
| Short femur | 0 | 12 | 12 | |
| Flexion/extension anomalies of limbs | 6 | 13 | 19 | |
| Total | 42 | 101 | 143 | |
| Other | Hydrops | 2 | 1 | 3 |
| Fetal growth restriction | 11 | 38 | 49 | |
| Cord anomalies (single umbilical artery, cyst) | 0 | 11 | 11 | |
| Amniotic fluid anomalies (oligohydramnios, hydramnios) | 0 | 7 | 7 | |
| Total | 13 | 57 | 70 | |
| Overall total | 376 | 611 | 987 | |
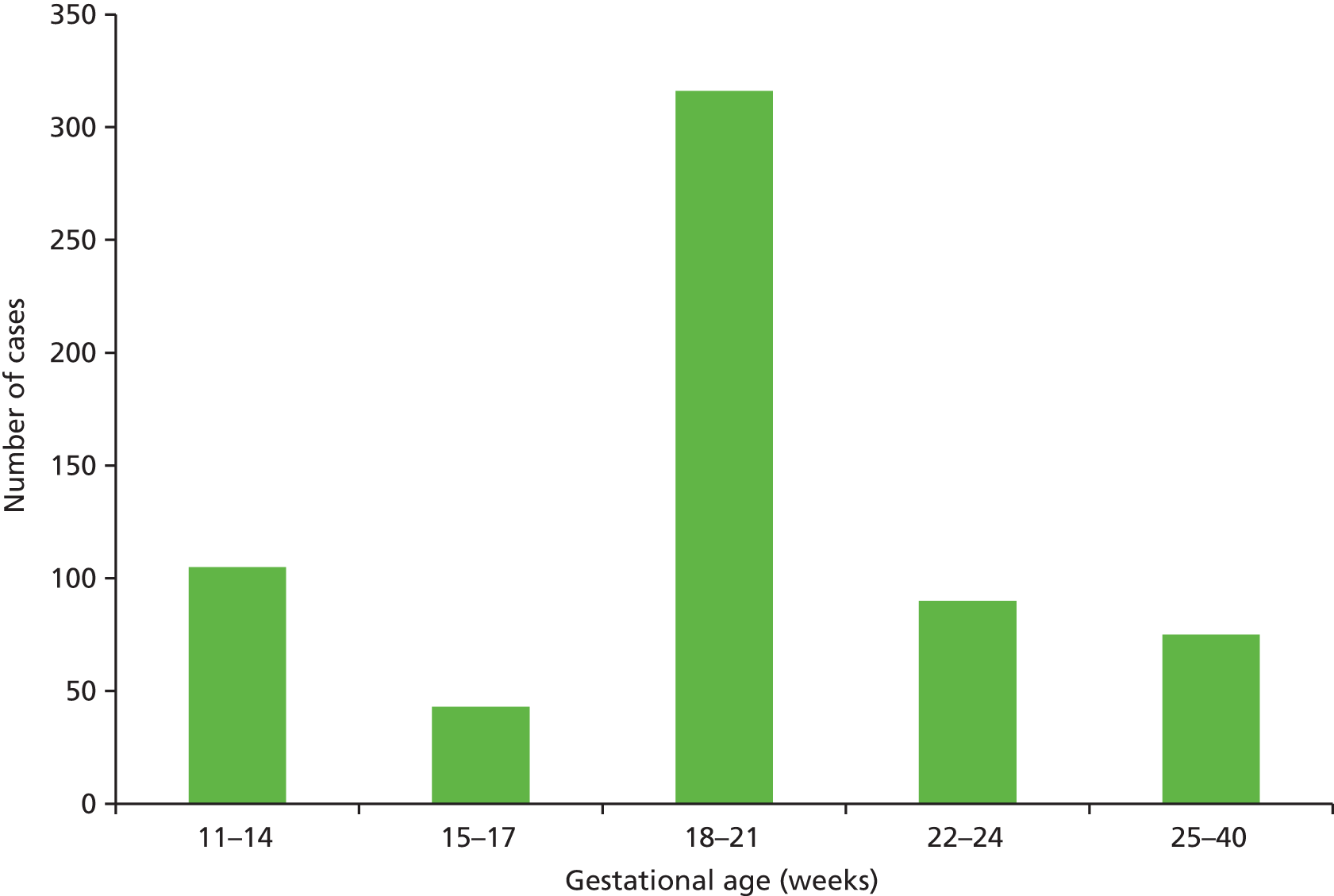
The distribution of gestational age at diagnosis in the structural anomaly group is shown in Figure 6. In total, 105 cases (16.7%) were detected at 11–14 weeks’ gestation and 75 (11.9%) were detected after 24 weeks’ gestation.
FIGURE 6.
Gestational age at diagnosis in 629 fetuses in the structural anomaly group.
Laboratory karyotype and chromosome microarray results
The breakdown of the karyotype and CMA results according to the final EACH groups is shown in Table 9. In total, 42 cases (3.7%) had a normal karyotype but a pathogenic CNV detected on CMA. A further 38 (3.4%) had a normal karyotype and a VOUS detected on CMA.
| Group | Increased NT group | Structural anomaly group | Total (%) |
|---|---|---|---|
| Normal karyotype and normal CMA | 430 | 540 | 970 (86.4) |
| Abnormal karyotype and normal CMA | 8 | 7 | 15 (1.3) |
| Abnormal karyotype and pathogenic CNV on CMA | 26 | 32 | 58 (5.2) |
| Normal karyotype and pathogenic CNV on CMA | 13 | 29 | 42 (3.7) |
| Normal karyotype and VOUS on CMA | 17 | 21 | 38 (3.4) |
| Total | 494 | 629 | 1123 (100) |
The follow-up tests undertaken in each of the final groups are shown in Table 10. In total, 268 additional investigations were performed and, in some cases, CNVs detected by CMA could be classified as benign (and, hence, the CMA normal) only after parental origin studies were carried out.
| Group | Parental karyotype | FISH | MLPA | PvP | Othera | Total |
|---|---|---|---|---|---|---|
| Normal karyotype and normal CMA | 0 | 12 | 3 | 37 | 7 | 59 |
| Abnormal karyotype and normal CMA | 14 | 0 | 0 | 1 | 0 | 15 |
| Abnormal karyotype and pathogenic CNV on CMA | 46 | 9 | 0 | 10 | 4 | 69 |
| Normal karyotype and pathogenic CNV on CMA | 0 | 42 | 15 | 27 | 4 | 88 |
| Normal karyotype and VOUS on CMA | 0 | 18 | 0 | 16 | 3 | 37 |
| Total | 60 | 81 | 18 | 91 | 18 | 268 |
Abnormal karyotype and normal chromosome microarray group
Fifteen (1.3%) cases were found to have an abnormal karyotype and a normal CMA: eight in the increased NT group (Table 11) and seven in the structural anomaly group (Table 12). Eight cases involved apparently balanced structural rearrangements, two involved inversions and a further two involved low-grade mosaics involving autosomal trisomies. The remaining three cases were shown, using FISH, to be heterochromatic supernumerary marker chromosomes (two derived from either chromosome 14 or 22 and one from chromosome 15), none of which showed an imbalance on CMA. Further molecular studies on the parental origin of one of the marker chromosomes (BIRM2357) revealed paternal uniparental disomy (UPD) for chromosome 14 consistent with a diagnosis of Wang syndrome.
| Primary identifier | Karyotype | CMA result (ISCN) | NT (mm) | Pregnancy outcome | Findings |
|---|---|---|---|---|---|
| LIV22978 | 45,XY,der(13;15)(q10;q10)pat | arr(1–22) × 2,(XY) × 1 | 5.1 | LB (39 weeks), 3820 g | NN: normal. FU: none |
| UCH1325884 | 46,X,t(X;2)(q26.3;p21)dn | arr(1–22,X) × 2 | 4.4 | TOP (17 weeks) | No PM |
| CAM41514 | 46,XX,t(13;20)(p13;p11.1)dn | arr(1–22,X) × 2 | 4.6 | LB (39 weeks), 3180 g | NN: normal (including echo). FU: genetics – normal at 10 weeks |
| JCU72470 | 46,XY,t(4;17)(q27;q23,1)mat | arr(1–22) × 2,(XY) × 1 | 4.0 | LB (38 weeks), 3120 g | NN: VSD, positional talipes right foot. FU: paediatric cardiology |
| BIRM2342 | 47,XX,+16[2]/46,XX[38] | arr(1–22,X) × 2 | 6.9 | TOP (16 weeks) | PM: achondrogenesis, pulmonary hyperplasia |
| EXE43104 | 47,XX,+mar(15) | arr(1–22,X) × 2 | 4.2 | LB (39 weeks), 3390 g | NN: normal. FU: normal (no details) |
| NCL112506 | 47,XY,+mar.ish mar(D14Z1/D22Z1 +)/46,XY | arr(1–22) × 2,(XY) × 1 | 4.0 | LB (40 weeks), 3415 g | NN: normal. FU: none |
| BIRM2357 | 47,XY,+mar(14/22), upd(14)pat[7]/46,XY[3] | arr(1–22) × 2,(XY) × 1 | 3.6 | TOP (21 weeks) | No PM |
| Primary identifier | Karyotype | CMA result (ISCN) | GA (weeks) | Anomalies | Pregnancy outcome | Findings |
|---|---|---|---|---|---|---|
| LEE54389 | 46,XX,t(2;5)(q21.1;q31.3) | arr(1–22,X) × 2 | 20 | CDH | TOP (21 weeks), 310 g | PM: right diaphragmatic hernia. FGR |
| UCH1267463 | 46,XX,t(11;18)(p15.1:q21.3)mat | arr(1–22,X) × 2 | 35 | Ebstein’s anomaly, hydrops | LB (34 weeks), 2700 g | NN: Ebstein’s anomaly. Pleural effusions (drained). FU: paediatric cardiology |
| SOU776398 | 46,XX,t(6;10)(q14.2?2;q24.3)pat | arr(1–22,X) × 2 | 22 | CL + P | LB (39 weeks) | NN: CL + P. FU: cleft team |
| BIRM2349 | 46,XY,t(1;14)(p13.1;q24.3)pat | arr(1–22) × 2,(XY) × 1 | 22 | Pericardial effusion | LB (33 weeks), 2630 g | Slight thickening interventricular septum. No effusion. FU: paediatric cardiology |
| BIRM2729 | 47,XX,+ 20[8]/46,XX[22] | arr(1–22,X) × 2 | 20 | VM, ACC | TOP (23 weeks), 422 g | PM: VM, ACC, mild micrognathia. FGR |
| UCH111464 | 46,XY,inv(15)(p11.2q11.2)mat | arr(1–22) × 2,(XY) × 1 | 13 | Megacystis | SB (35 weeks), 1880 g | PM: ASD, left SVC, pulmonary agenesis (right), TOF, renal agenesis (bilateral), urethral aresia, polysplenia, intestinal malrotation, anal atresia, polymicrogyria and global hypoxic ischaemia |
| UCH1336195 | 46,XY,inv(14)(q24.3q32.3)mat | arr(1–22) × 2,(XY) × 1 | 20 | VM, ACC | TOP (21 weeks) | No details |
Abnormal karyotype and abnormal chromosome microarray group
A total of 58 visible cytogenetic abnormalities were diagnosed, all of which were also identified and further characterised by CMA. These comprised three mosaic marker or ring chromosomes, 25 visible deletions or duplications, 13 numerical abnormalities (not involving chromosomes 13, 18 or 21), 11 unbalanced translocations and six sex chromosome abnormalities (excluding 45X), none of which showed an additional imbalance on CMA. Twenty-six of these variants were found in the increased NT group (Table 13) and 32 were found in the structural anomaly group (Table 14).
| Primary identifier | Karyotype | CMA result (ISCN) | NT (mm) | Pregnancy outcome | Findings |
|---|---|---|---|---|---|
| Mosaic marker or ring chromosomes (n = 1) | |||||
| LEE46083 | 45,X[9]/46,X,+mar[14] | 45,X[9]/46,X,der(X)del(X)(pter-Xp11.22)del(X)(q12-qter)[14]. arrXp22.3p11.22(60726–53616540) × 1,Xq13.1q28(67906874–155232885) × 1 | 13.0 | SM (20 weeks), 110 g | No PM |
| Deletions or duplications (n = 6) | |||||
| SOU830120 | 46,XY,del(13)(q?31) | arr 13q31.1q34(82,221,354–115,109,878) × 1 | 5.8 | TOP (13 weeks) | No PM |
| LIV14267 | 46,XY,i(18)(q10).ish 18q21(BCL2X3)[85]/18q21(BCL2X2)[43] | arr16q23.1q24.3(78,471,999–90,148,364) × 2∼3, 18p11.32p11.21(148,993–14,081,858) × 1,18q11.1q23(18,854,104–78,012,800) × 2∼3 | 7.0 | TOP (17 weeks), 150 g | No PM |
| NCL50426 | 46,XY,dup(21)(q21q22) | arr 21q21.2q22.2(25783769–40747892) × 3 | 6.0 | TOP (16 weeks) | No PM |
| LIV23362 | 46,XX,del(2)(q22.1q23.3) | arr 2q22.1q23.3(141,192,304–153,143,527) × 1dn | CH | NND (28 weeks), 1175 g | NN: hypoplastic left heart. Mowat–Wilson syndrome. No PM |
| NCL108541 | 46,XY,rec(8)dup(8q)inv(p23.2q22.1)mat | arr8p23.3p23.2(191,559–6,880,333) × 1,8q22.1q24.3(96,764,771–146,279,990) × 3 mat | 6.0 | TOP (14 weeks) | No PM. Placental histology: villous dysmorphism |
| QCH132334 | 46,XX,del(9)(q22.33q33) | arr 9q22.33q33.2(100,595,648–124,539,832) × 1 dn | CH | TOP (15 weeks), 53 g | PM: fluid around nape of neck, excess skin oedema. CL + P. Small adrenal glands |
| Numerical abnormalities (not involving chromosomes 13, 18 or 21) (n = 6) | |||||
| SMH7781975 | 47,XY.nuc ish (D3Z1 × 2∼3) | arr(3) × 2∼3 | 4.3 | LB (39 weeks), 3547 g | NN: normal. FU: none |
| CAM62826 | 47,XY,+ 7[21]/46,XY[19] | arr(7) × 2∼3 | 3.7 | LB (40 weeks), 3200 g | NN: normal. Postnatal karyotype: normal (presumed CPM). FU (7 weeks): normal – discharged |
| LIV22831a | 47,XX+ 9[4]/46,XX[26]. nuc ish 9cen(D9Z1 × 3)[23]/9cen(D9z1 × 2)[177] | arr 9p24.3 9q34.3(204,221–141,073,875) × 2∼3 | 4.0 | LB (39 weeks), 4180 g | NN: bilateral hydroureters, cleft palate, talipes. FU: no details |
| UCH331937 | 47,XX,+ 22 | arr(1–21) × 2,(22) × 3 | 4.6 | TOP (16 weeks), 90 g | No PM |
| QHR233174 | 47,XY,+ 22//47,XY,+ 22[10]/46,XY[28] | arr(22) × 3 | 4.5 | U/K | |
| CAM62785 | Fail | arr(22) × 3 | 5.7 | SM (15 weeks) | No PM |
| Unbalanced translocations (n = 8) | |||||
| LIV24321m | 46,XY,der(11)t(11;14)(q25;q23.1)pat | arr11q25(132,058,925–134,868,378) × 1,14q23.1q32.33(58,658,169–107,284,502)X3pat | 4.3 | TOP (17 weeks) | No PM |
| UCH1238714 | 45,XY,der(15)t(15;21)(q?26.2;q22.1),–21 | arr15q26.2q26.3(95,078,659–102,347,831) × 1, 21q11.2q22.11(14,630,740–33,522,228) × 1 dn | 7.0 | TOP (14 weeks) | No PM |
| NCL109148 | 46,XX,der(18)t(18;21)(p11.1;q11.1)dn | arr18p11.32p11.21(149,992–14,081,857) × 1,21q11.2–21q22.3(15,485,037–48,090,287) × 3 dn | 4.5 | TOP (14 weeks) | No PM |
| LEE46556 | 46,XX,der(5)t(5;16)(p15.33;p11.2)mat | arr5p15.33(22,179–2,167,668 × 1, 2,274,718 × 2), arr 16p13.3p11.2(82,437 × 2,93,748–32,066,997 × 3, 32,091,685 × 2)mat | 6.3 | NND (31 weeks), 920 g | NN: no details, no PM |
| LIV22199 | 46,X,der(X;6)(q28;q21)dn | arr6q21q27(106,781,991–170,921,060) × 3, Xq27.1q28(139,886,233–153,790,884) × 3, Xq28(153,791,775–155,232,885) × 1 | 5.4 | TOP (18 weeks) | No PM |
| FPH316572 | 46,XX,der(13)t(8;13)(p12;p11.2)dn | arr 8pterp12(1–35,358,143) × 3 dn | 3.6 | TOP (14 weeks) | No PM |
| SMH7740386 | 46,XX,add(5)(q34) | arr5q34-q35.2(162,790,636–176,217,126) × 1,14q31.3(87,593,423–89,531,709) × 3 | 5.7 | TOP (12 weeks) | No PM |
| NCL122751 | 46,XY,der(13)t(Y;13) pat | arr Yq11.23q12(28609947–59373566) × 2 pat | 4.7 | LB (41 weeks), 2535 g | NN: arthrogryposis. Microcephaly. MRI: severe thinning of CC. FU (1 year): developmental delay, seizures. Pharyngomalacia (tube fed) |
| Sex chromosome anomalies (excluding 45X) (n = 5) | |||||
| UCH1304931 | mos 45,X[5]/46,XY[80] | Mosaic loss of Y | 3.9 | LB (40 weeks), 3270 g | NN: normal. FU: none |
| NCL125013 | 47,XXY/46,XX | arr(Y) × 0∼1 | 4.7 | TOP (20 weeks), 293 g | PM: face; hypertelorism, large mouth, small nose, broad philtrum, low ears. Upper limbs; missing radii, missing ulna (left), single digit on each hand. |
| NCL124643 | 49,XXXXX | arr(X) × 5 | CH | TOP (14 weeks) | No PM |
| UCH1218666 | 47,XYY | arr(X) × 1,(Y) × 2 | 3.7 | LB (40 weeks), 3235 g | NN: normal. FU: none |
| UCH1402900 | 47,XXY | arr (1–22,X) × 2,(Y) × 1 | 4.6 | TOP (13 weeks) | No PM |
| Primary identifier | Karyotype | CMA result (ISCN) | GA (week) | Anomaly | Pregnancy outcome | Findings |
|---|---|---|---|---|---|---|
| Mosaic marker or ring chromosomes (n = 2) | ||||||
| BIRM3448 | 46,XX,r(13)(p11q33.3)dn[24]/45,XX,–13[6] | arr3q33.3q34(108,607,127–115,092,619) × 1 dn | 30 | Microcephaly | LB (35 weeks), 2205 g | NN: microcephaly (HC –4 SDs), prominent nasal bridge, overlapping toes. FU: paediatric |
| SOU800062 | 47,XY,+mar[11]/48,XY,+mar × 2[3]/46,XY[1] | arr5p14.1p11(24,771,971–46,115,173) × 3∼4 dn | 16 | Talipes, hypoplasia cerebellar vermis | TOP (16 weeks) | No PM |
| Deletions or duplications (n = 19) | ||||||
| UCH89103 | mos 47,XX,+i(10)(p10)[10]/46,XX[20] dn | arr10p15.3p11.1(–634,320–38,576,048) × 2∼3 dn | 13 | VSD, dilated IV ventricle, SUA | TOP (16 weeks), 135 g | No PM |
| UCH1237834 | 47,XY,+del(10)(q11.22) | arr10p15.3q11.22(153,760–46,122,249) × 3 | 20 | ACC, unilateral CL + P, pericardial effusion | TOP (21 weeks), 306 g | PM: unilateral CL + P. No other anomalies. Placenta; villous dysmorphism |
| BIRM4047 | 46,XX,del(18)(q22.31)[4]/46,XX[46] | arr15q22.31q26.3(65,238,649–102,383,444) × 2∼3, 18q21.2q23(48,765,742–78,012,800) × 1∼2 | 12 | A4CV, ↑ NT (5.2 mm) | TOP (18 weeks) | No PM |
| UCH887694 | 46,XY,del(18)(p10) | arr18p11.32p11.21(149,089–14,007,195) × 1 dn | 18 | TGA, VSD, hydronephrosis | TOP (19 weeks) | No PM |
| UCH1330425 | 46,XX,del(1)(q41q42) | arr1q41q42.13(216,191,238–227,859,405) × 1 dn | 13 | CDH, talipes | TOP | No PM |
| UCH1330653 | 46,XY,del(2)(p23p25.1)dn | arr2p25.1p23.3(10,840,030–27,554,410) × 1 dn | 24 | Hypoplastic cerebellum, ↑ CM, CPC, umbilical cord cyst | TOP (26 weeks), 890 g | No PM |
| LIV23483 | 46,XX,del(5)(q23.3q31.3) | arr5q23.3q31.3(129,309,631–141,136,904) × 1 dn | 15 | Abnormal cardiac outflow tracts | TOP (16 weeks), 70 g | PM – flat nose, small chin. Nuchal oedema. Bilateral medially placed thumbs. Pre-axial polydactyly feet, talipes. Midline CP. DORV, VSD and pulmonary atresia (Fallot type) |
| UCH1371888 | 46,XX,del(4)(p15.2) | arr 4p16.3p15.2(–329,400–21,995,360) × 1 | 24 | Depressed nasal bridge, abnormal RSCA, short femora and humeri, ↑ NT (8.5 mm) | TOP (27 weeks), 770 g | No PM |
| SOU414862 | 46,XY,del(4)(p15.32)dn | arr 4pterp15.33(1–15,542,084) × 1 dn | 21 | Bilateral CL + P | LB (35 weeks), 1500 g | NN: CL + P. Hypospadias. Neuroblastoma. Hypotonia. Wolf–Hirschorn syndrome. FU: multiple services |
| UCH1308244 | 46,XY,del(6)(p25) | arr 6p25.3p24.3(293,430–8,093,850) × 1 dn | 21 | Talipes, abnormal brain | TOP (24 weeks) | No PM |
| SOU815286 | 46,XX,add(6)(p25).ish del(6)(p25.3p25.3)(CTB-62I11-,RP11–118B18-) | arr 6pterp25.1(1–5,875,603) × 1 | 21 | Partial cerebellar vermian agenesis | TOP (21 weeks), 453 g | PM: hypoplasia posterior and inferior portions cerebellum |
| SOU820778 | 46,XX,del(8)(p23.2) | arr 8p23.1(8,130,572–11,723,203) × 1 | 21 | A4CV, VSD | LB (37 weeks), 2790 g | NN; AVSD, bilateral superior vena cava, coarctation of aorta (repaired). FU: paediatric cardiology |
| UCH1318267 | 46,XY.ish del(8)(q24.3q24.3)(RP11–65A5-)dn | arr 8q24.3(140,186,782–144,969,635) × 1 | 13 | Talipes, polydactyly, ↑ NT (4.0 mm) | TOP (20 weeks) | PM: dysmorphic face. Coarctation of aorta, hypoplastic left ventricle, VSD, persistent left SVC. Talipes, single palmar creases, polydactyly (right foot). Intestinal malrotation, posterior encephalocele |
| UCH1273556 | 47,XX,+del(9)(q11) | arr 9p24.3q13(–516,980–67,298,992) × 3 | 27 | Micrognathia, talipes | TOP | No PM |
| LEE45306 | 46,XX,?dup(9)(q33q31).ish der(9)(D9S325 +) | arr 9q31.1q31.2(106,128,887 × 2, 106,174,653–131,987,296 × 3, 132,043,658 × 2)dn | 28 | Spina bifida, VM | SB (34 weeks), 1690 g | PM: enlarged head, low set ears. CP. Lumbosacral neural tube defect |
| EXE44352 | 46,X,del(X)(q27.1).ish der(X)t(X;3)(q27.1;q29)mat | arr Xq27.1q28(139,384,647–155,226,057) × 1,3q29(197,146,239–197,837,069) × 3 mat | 22 | VM | LB (38 weeks), 2740 g | NN: normal. FU: no details |
| UCH1372792a | Failed karyotype | arr 15q26.2q26.3(91,836,928–102,481,320) × 1, Xp22.12(20,265,130–20,413,100) × 2 | 28 | VSD, hypolastic aorta, echogenic kidneys, talipes, small thorax | TOP (30 weeks), 1735 g | No PM |
| EXE28065 | 46,XX,dup(10)(q23.1q23.2) | arr 10q22.3q23.2(81,962,149–89,065,420) × 3 mat | 23 | A3VV | TOP (23 weeks) | No PM |
| BIRM5007 | 46,XY,trp(14)(q32.1q32.3)dn | arr 14q32.12q32.31(93,211,807–102,565,945) × 4 dn | 20 | VM, absent cerebellar vermis | TOP (23 weeks), 514 g | PM: VM, absent cerebellar vermis. Short humeri and femora, platyspondyly, chondrodysplasia punctata. Skull – wormian bones, small occipital bone. Face – prominent nasal bridge, wide down-turned mouth and low set ears. FGR |
| Numerical abnormalities (not including chromosomes 13, 18 or 21) (n = 7) | ||||||
| LEE45743 | 47,XY,+8[8]/46,XY[12] | arr 8p23.3q24.3(191559–146279990) × ∼3,dn | 13 | Omphalocele | TOP (17 weeks), 200 g | No PM |
| LIV19386(2) | 48,XXX,+8[9]/47,XXX[3] | arr 8p23.3q24.3(191,560–146,279,991) × 2∼3,Xp22.33q28(291,307–155,232,885) × 3 | 14 | Omphalocele | LB (34 weeks), 2260 g | NN: normal (omphalocele resolved prenatally). FU: none |
| SMH6703657 | 47,XX,+9[11]/46,XX[11] | arr(9) × 2∼3 | 22 | FGR | TOP (24 weeks) | No PM |
| BIRM2341 | 47,XX,+14[3]/46,XX[27] | arr 14q11.2q32.33(20,608,246–107,287,476) × 2∼3 | 21 | VSD | TOP (25 weeks) | No PM |
| LIV23880 | 47,XY+22 | arr 22q11.1q13.33(17,397,528–51,178,235) × 3 | 20 | Coarctation of aorta, VSD | TOP (23 weeks) | No PM |
| UCH1371771 | 47,XX,+22[13]/46,XX[37] | arr (1–21,X) × 2,(22) × 2∼3 | 21 | Tetraology of Fallot, ACC, short femurs, FGR, SUA | LB (37 weeks), 2105 g | NN: tetralogy of Fallot. Neurological examination normal. FU: paediatric cardiology |
| BIRM3011 | 47,XY,+2[1]/46,XY[59] | arr 2p25.3q37.3(17,049–243,068,370) × 2∼3 | 15 | Megacystis | LB (39 weeks), 3930 g | NN: renal hypoplasia, chronic renal disease (serum creatinine 138 µmol/l). FU: paediatric nephrology |
| Unbalanced translocations (n = 3) | ||||||
| NCL103473 | 46,XX,der(4)t(4;6)(q34;q2?3) | arr 6q24.1q27(139023538–170921059) × 3,4q34.3q35.3(179762434–190896644) × 1 pat | 20 | Micrognathia, talipes, small stomach | LB (36 weeks), 2200 g | NN: severe micrognathia, tracheostomy at birth. Panhypopituitarism, talipes. Died (day 16) respiratory failure |
| UCH1222910 | 45,XY,der(7)t(7;21)(q36.3;q21.1),–21 | arr 7q36.3(159,030,166–159,123,275) × 1,arr 21q11.2q21.1(14,547,650–20,260,349) × 1 | 23 | VM | TOP | No PM |
| SMH7510833 | 46,XX,der(13)t(2;13)(q37.3;q32.1)pat | arr 2q37.3(238,140,879–243,007,457) × 3,13q32.2q34(98,930,808–115,092,581) × 1 | 15 | Holoprosencephaly | TOP (17 weeks) | No PM |
| Sex chromosome anomalies (excluding 45X) (n = 1) | ||||||
| POR1405022 | 47,XXY | arr(X) × 2,(Y) × 1 | 21 | VM | LB (38 weeks), 3745 g | NN: normal. FU: no details |
Normal karyotype and abnormal chromosome microarray group
A total of 42 cases with a normal karyotype and a CMA finding consistent with pathogenic CNV deemed relevant to clinical findings and prognosis were reported. Thirteen out of the 42 cases were in the increased NT group (Table 15) and the remaining 29 cases were in the structural anomaly group (Table 16). A total of 39 cases had a single CNV and three had two CNVs. The parental origins of 36 out of the 42 CNVs were determined: four were maternal, seven paternal and 25 had arisen de novo.
| Primary identifier | CMA result (ISCN) | NT (mm) | S/U (size)a | Gene(s)/syndrome | Pregnancy outcome | Post-mortem/examination findings |
|---|---|---|---|---|---|---|
| UCH1321680a | arr 1p36.22(11,087,529–12,530,093) × 1 mat | 4.5 | U (1.4 Mb) | U/K | LB (37 weeks), 2884 g | NN: normal. FU: none |
| BIRM4382a | arr 1q25.2(180,230,183–180,235,718) × 1 pat | 3.7 | S (5.5 kb) | LHX4 | LB (39 weeks), 3685 g | NN: normal. FU: none |
| LIV23966(2) | arr 2p25.3q37.3(17,049–243,068,370) × 2∼3 | CH | S (mos + 2) | Cryptic mosaic trisomy 2 | TOP (27 weeks), 600 gb | PM: no details other than ‘complex congenital heart disease’ |
| NCL62560 | arr 6p21.1(40574797–43262906) × 1 dn | 7.1 | U (2.6 Mb) | GUCA1B; TREM2; PRPH2; FEX6; CUL7 | TOP (16 weeks) | No PM |
| QCH132506 | arr 8q23.1(106,386,421–107,440,948) × 3 | 5.9 | S (1 Mb) | ZFPM2 | TOP (18 weeks), 54 g | PM: abnormal-shaped head, retroflexion of head. Small cerebral hemispheres. Facial dysmorphism (hypertelorism, low-set ears, micrognathia, depressed nasal bridge), CP. Absent clavicles, cervical ribs. Arthrogryposis (fixed flexion deformities limbs), overlapping digits. Pterygium (left arm). Scoliosis. Intestinal malrotation. Hydrops. FGR |
| LEE46496a | arr 10q11.22q11.23(46,205,720–51,780,880) × 3 pat | 3.6 | U (5.6 Mb) | U/K | LB (39 weeks), 3040 g | NN: normal. FU: none |
| BIRM4681 | arr 15q25.2(83,283,533–84,812,664) × 1 dn, 16p13.11(14,968,878–16,194,549) × 3 pat | 6.3 | U (1.5 Mb), U (1.2 Mb) | U/K | NND (40 weeks), 2740 g | Left CDH, hypoplasia aortic arch, hydronephrosis. Died day 3 (no PM) |
| SOU835706 | arr 16p13.2(8,221,327–9,218,395) × 1 dn | CH | S (997 kb) | PMM2 | TOP (17 weeks), 179 g | PM: hydrops. Cystic hygromata. Prominent genital tubercle. Mild preductal coarctation. Incomplete intestinal rotation. Focal hepatic calcification. Posteriorly rotated ears |
| SOU404408 | arr 17q12(34,856,305–36,248,926) × 1 dn | 5.6 | S (1.4 Mb) | RCD | LB (40 weeks), 3425 g | NN: normal. FU: no details |
| LIV23984a | arr 19p13.2(13,017,793–13,270,578) × 1 dn,22q11.21(18,661,749–21,540,318) × 3 dn | 9.0 | S (252 kb), S (2.8 Mb) | NFIX Sotos-LIKE, DiGeorge CR | TOP (17 weeks) | No PM |
| LEE49189a | arr Xp21.1(31,646,912–31,763,032) × 3 pat | 4.8 | S (116 kb) | DMD | LB (40 weeks), 3550 g | NN: normal. FU: none |
| UCH85053 | arr 15q21.3q22.1(57,283,540–59,150,660) × 1 dn | 5.6 | U (1.9 Mb) | U/K | TOP (14 weeks) | No PM |
| SGH86084 | arr(1–22) × 2,(X) × 1∼2 | 4.0 | Uc | CPM | LB (40 weeks), 3000 g | NN: normal. FU: none |
| Primary identifier | CMA result (ISCN) | GA (weeks) | Anomaly | Size | Gene(s)/syndrome | Outcome | Post-mortem/examination findings |
|---|---|---|---|---|---|---|---|
| BIRM3010 | arr 1p12(120,409,757–120,520,249) × 1 mat | 22 | DILV, VSD | S (11 kb) | NOTCH 2 | TOP (24 weeks), 670 g | No PM |
| UCH1297290 | arr 1q21.1(145,415,152–145,799,552) × 1 pat | 11 | Absent radius | S (384 kb) | RBM8 A (second hit) | TOP (12 weeks) | No PM |
| BIRM3619a | arr 15q11.2q13.1(23,082,792–29,030,488) × 3 dn | 27 | CL, talipes, nuchal thickening | S (5.9 Mb) | dup(15)mat | TOP (31 weeks), 2120 g | No PM |
| SMH7702525 | arr 1q21.1(145,415,156–145,899,418) × 1 dn | 14 | Absent radius, fixed and flexed wrists | S (484 kb) | RBM8 A (second hit) | TOP (15 weeks), 69 g | PM: bilateral radial aplasia, radial deviation hands. Feet – subtalar inversion. Horseshoe kidney. Small VSD. TAR syndrome |
| SMH7784077 | arr 1q21.1(145,415,156–145,899,418) × 1 | 20 | VM, micrognathia, nuchal thickening; short humerus and radius | S (48 kb) | RBM8 A (second hit) | TOP (21 weeks), 351 g | PM: mild VM. Facial dysmorphism. Horseshoe kidney. Lower limbs – flexion contractures. Upper limbs – phocomelia. TAR syndrome |
| UCH1291069a | arr 1q21.3(154,132,656–154,537,648) × 1 dn | 12 | AVSD, CL + P, omphalocele, talipes | S (404 kb) | IL6R | TOP (no details) | No PM |
| SOU293507a | arr 2q13(111,442,175–113,065,741) × 1 pat | 19 | Small bowel obstruction; pelvic kidney | U (1.6 Mb) | MERTK, RP38 | LB (41 weeks), 4290 g | NN: duplex kidneys. Syndactyly (second/third toes). FU: paediatric nephrology |
| SMH6152191 | arr 3q29(196,686,412–197,837,069) × 1 dn | 21 | Nuchal thickening, oligohydramnios | U (1.15 Mb) | U/K | LB (36 weeks), 1950 g | NN: FGR. Hereditary spherocytosis. Blackfan–Diamond anaemia. Stridor – tracheal narrowing. Normal echo, brain MRI. FU: haematology, ENT |
| LIV12390 | arr 6q22.2q22.31(118,446,046–119,296,210) × 3 dn | 23 | Skeletal dysplasia | U (850 kb) | PLN, ASF1 A, MCM9 | TOP (23 weeks), 210 g | PM: moderate micromelia, mild bowing. Histology: abnormal diaphysis with membranous ossification. Campomelic dysplasia. Pierre Robin sequence (micrognathia and CP) |
| SOU354300a | arr 6q23.3(135,656,228–136,152,953) × 3 pat | 20 | Hypoplastic cerebellum | S (496 kb) | Joubert syndrome | TOP (23 weeks) | No PM |
| NCL123872 | arr 8p23.1(11036904–11725065) × 1 | 22 | A4CV | S (688 kb) | GATA 4 | LB (39 weeks), 2965 g | NN: hypoplastic right heart (tricuspid atresia), VSD, ASD. FU: cardiology – awaiting Glenn procedure |
| FPH310628 | arr 8q12.1q12.2(61,431,781–61,915,254) × 1 dn,17q12(34,856,305–36,248,926) × 3 dn | 21 | Tetralogy of Fallot | S (483 kb), S (1.4 Mb) | CHD7, RCD | TOP (28 weeks) | No PM |
| JCU68514a | arr 7q11.23(72,286,180–74,481,510) × 3 dn | 20 | Dandy–Walker malformation | S (2.2 Mb) | Williams syndrome CR | LB (40 weeks), 3300 g | NN: bilateral polydactyly, MRI: Dandy–Walker variant, FU (4 months): normal |
| SOU824564 | arr 7q36.2q36.3(154,878,001–159,128,555) × 1 | 20 | CL + P | S (4.2 Mb) | LMBR1, SHH | TOP (22 weeks) | No PM |
| UCH1252744a | arr 16p13.11(15,557,250–16,396,490) × 1 dn | 30 | A4CV (?AVSD), dilated PA, bilateral SVC, hydramnios, absent stomach, SUA. unilateral renal agenesis | S (839 kb) | MYH11, AAT4, FAA4 | LB (34 weeks), 1840 g | NN: oesophageal atresia with TOF (repaired), chylothorax. Atrial isomerism, ASD. Renal agenesis (left). Asplenia. Died at 5 weeks (sepsis) |
| CARMU348298a | arr 16q24.3(88,803,492–90,014,642) × 3 dn | 21 | A3VV, dilated PA, talipes | U (1.2 Mb) | KBG syndrome | TOP (25 weeks) | No PM |
| UCH1151793a | arr 16q24.3(89,412,888–89,459,256) × 1 dn | 20 | VM, partial ACC, posterior fossa cyst | S (46 kb) | KBG syndrome | TOP (23 weeks), 608 g | PM: mild facial dysmorphism (tented upper lip, anteverted nares). Cervical rib. No brain abnormality |
| EXE41271 | arr 17p11.2(16,637,872–20,294,010) × 3 dn | 22 | FGR, short radius, fixed flexed wrist | S (3.6 Mb) | Potocki–Lupski syndrome | TOP (23 weeks), 400 g | PM: left arm – mesomelia, contractures elbow and hand. FGR. Potocki–Lupski syndrome |
| EXE35412 | arr 17p12(14,111,754–15,442,178) × 3 | 13 | Omphalocele | S (1.3 Mb) | PMP22 pat | LB (38 weeks), 2890 g | NN: normal (omphalocele resolved prenatally). FU: none |
| BIRM2773 | arr 22q11.2(18,894,865–21,505,388) × 1 dn | 24 | A4CV, echogenic bowel; hydronephrosis | S (2.5 Mb) | DGV | LB (37 weeks), 2818 g | NN: tetralogy of Fallot with right aortic arch. DiGeorge syndrome |
| SOU382387a | arr 22q11.21(18,894,820–21,025,719) × 1 dn | 21 | VSD, ? Truncus arteriosus | S (2.5 Mb) | DGV | TOP (23 weeks) | No PM |
| SOU773161 | arr 22q11.21(18,894,820–21,440,515) × 1 dn | 21 | Tetralogy of Fallot | S (2.5 Mb) | DGV | TOP (23 weeks) | No PM |
| BIRM3449 | arr 22q11.21(18,894,865–21,440,485) × 1 dn | 21 | VSD | S (2.5 Mb) | DGV | TOP (26 weeks), 950 g | No PM |
| BIRM4673 | arr 22q11.21(18,894,865–21,440,485) × 1 dn | 27 | A4CV (VSD, hypoplastic arch), talipes | S (2.5 Mb) | DGV | LB (40 weeks), 3440 g | No details |
| UCH1396696 | arr 22q11.21(18,741,080–21,681,780) × 1 | 28 | Hydrops, cerebral vascular anomaly | S (2.9 Mb) | DGV | SB (28 weeks) | No PM |
| EXE49950 | arr 22q11.21(18,765,102–20,311,733) × 1 | 20 | A4CV (conotruncal anomaly? VSD) | S (1.6 Mb) | CECR, CES | TOP (22 weeks) | No PM |
| NCL65675(2)a | arr Xp21.1(31,646,911–32,102,589) × 1 mat | 20 | VM | S (455 kb) | DMD | LB (34 weeks), 2800 g | NN: postnatal cranial ultrasound scan normal. No other anomalies. FU: neonatology (due to prematurity) |
| UCH1372254a | arr Xq27.1(139,356,336–139,830,384) × 2 mat | 20 | Spina bifida, VM | S (474 kb) | SOX 3 | TOP | No details |
| UCH1285528a | arr Xp22.31(7,223,740–7,555,459) × 0 dn/arr(1–22) × 2,(XY) × 0∼1 dn | 12 | Omphalocele, flexed knees, hypoplastic nasal bone | S (332 kb) | STS, CPM | LB (38 weeks), 2649 g | NN: no details |
In two cases (UCH1285528 and SGH86084), the CMA imbalances were subsequently found to be confined to the placenta (CPM). Both of these cases, which had a normal outcome, represent false positives.
Excluding the two cases of CPM, 32 of the remaining 40 cases (80%) had at least one CNV within recognised syndromic regions (e.g. six Di George/velocardiofacial syndrome 22q11.21 deletions, one 17p11.2 Potocki–Lupski syndrome, two 17q12 renal cyst and diabetes imbalances) and eight involved the coding regions of key developmental genes (e.g. NOTCH2, DMD), or the size and gene content resulted in classification as pathogenic.
Within this group, 17 cases were referred to the panel and in 16 cases it was recommended the result was reported. There were two cases for which CMA diagnosed a cryptic chromosome abnormality undetected by karyotyping:
-
an interstitial proximal 15q duplication involving the Prader–Willi/Angelman critical region which had arisen on the maternal homologue (BIRM3619)
-
a mosaic trisomy 2 (LIV23966–2) (not visible cytogenetically from cultured CVS cells).
Normal karyotype and variants of unknown significance on chromosome microarray
In total, 38 cases with a normal karyotype were classified as having VOUS on CMA. This excluded eight cases on the EACH exclusion list (see Table 1), which consisted of six 15q11.2 BP1–BP2 imbalances (ranging in size from 319 to 448 kb), one 1q21.1 duplication (1.9 Mb) and one 16p13.11 duplication (1.6 Mb). Of the remaining 38 VOUS, 17 were in the increased NT group (Table 17) and 21 were in the structural anomaly group (Table 18). Twelve cases were referred to the EACH review panel and in each case the panel recommended not reporting the VOUS.
| Primary identifier | CMA result (ISCN) | NT (mm) | Size | Gene(s) | Pregnancy outcome | Findings |
|---|---|---|---|---|---|---|
| LIV24788 | arr 1q21.1(145,415,217–145,899,369) × 1 mat | 4.3 | 484 kb | RBM8 A, PEX11B | LB (41 weeks), 4105 g | NN: normal. FU: none |
| CAM57558 | arr 4q25(110,601,739–110,915,872) × 1 mat | 3.9 | 314 kb | CFI | LB (40 weeks), 4210 g | NN: normal. FU: none |
| BIRM3453 | arr 7q11.21(66,540,149–66,655,829) × 1 | 4.4 | 115 kb | No genes | LB (38 weeks), 3270 g | NN: normal. FU: none |
| LEE53803 | arr 9p24.3(416,322–500,555) × 3 | 3.6 | 84 kb | DOCK8, KANK1 | LB (41 weeks), 3500 g | NN: normal. FU: none |
| LEE54756 | arr 11q25(134,300,606–134,720,315) × 3 | 4.0 | 420 kb | No genes | LB (40 weeks), 3700 g | NN: normal. FU: none |
| LEE54757 | arr 11q25(134,381,662–134,716,359) × 3 | 4.4 | 420 kb | No genes | LB (40 weeks), 3550 g | NN: normal. FU: none |
| JCU68924 | arr 12p11.23p11.22(26,573,102–28,495,882) × 3 pat | 4.3 | 1.9 Mb | PTHLH, BDE2 | LB (41 weeks), 4320 g | NN: normal. FU: none |
| LIV24307 | arr 15q13.3(32,065,030–32,635,930) × 3 | 4.8 | 570 kb | CHRNA7 | LB (39 weeks), 2760 g | NN: normal. FU: none |
| NCL122058 | arr 15q13.2q13.3(30,491,443–32,899,529) × 3 | 3.6 | 2.4 Mb | CHRNA, FAN1, TRPM1 | LB (39 weeks), 3080 g | NN: diaphragmatic hernia (repaired). Hypolastic transverse aortic arch, VSD. FU: paediatric cardiology/surgery |
| LEE51494 | arr 16p11.2(33,850,611–34,173,701) × 1 | 3.7 | 323 kb/3.4 Mb | No genes/centromere | LB (41 weeks), 4335 g | NN: normal. FU: none |
| LEE53014 | arr Xp22.31(7,491,619–7,744,162) × 1 | 6.2 | 252 kb | No genes | LB (39 weeks), 3300 g | NN: Ebstein’s anomaly. FU: paediatric cardiology |
| PORQ615229 | arr Xq21.31 or Yp11.2(90,740,551–91,377,360 or 4,527,541–6,465,436) × 1.ish Xq21.31(RP11–339M12 +),del(Y)(p11.2p11.2)(RP11–339M12-) | 3.7 | 636 kb | PCDH11X | LB (39 weeks), 4115 g | NN: normal. FU: none |
| SGH80978a | arr 1q21.1(145,258,240–146,002,128) × 3 | 3.8 | 743 kb | 1q21.1 dup syndrome | U/K | Normal anomaly scan/echo at 20 weeks, moved abroad |
| CAM59605a | arr 10q26.3(132,580,596–135,434,178) × 1 mat | 6.4 | 2.8 Mb | No genes | LB (40 weeks), 4360 g | NN: normal. FU: none |
| BIRM4051a | arr 16p13.11(14,910,228–16,194,549) × 1 | 4.1 | 1.28 Mb | NDE1, NUDE, LIS4, MHAC, MYH11 AAT4 FAA4 | LB (38 weeks), 3460 g | NN: normal. FU: none |
| NCL117734a | arr 22q11.22q11.23(23,012,042–25,065,546) × 3 mat | 5.1 | 2.0 Mb | Distal 22q dup | LB (38 weeks), 2810 g | NN: normal. FU: none |
| SMH0111272a | arr Xp11.23(47,330,209–47,335,088) × 1 mat | 3.8 | 4.8 kb | X-linked MR | LB (38 weeks), 2405 g | NN: normal. FU: none |
| Primary identifier | CMA result (ISCN) | GA (weeks) | Anomaly | Size | Gene(s) | Pregnancy outcome | Findings |
|---|---|---|---|---|---|---|---|
| SOU819483 | arr 3q29(196,544,887–196,559,842) × 3 mat, arr 16q23.3(82,685,769–82,926,739) × 1 pat | 13 | Ectopia cordis, asymmetrical skull/brain, CH | 15 kb 240 kb | No genes | TOP (14 weeks) | No PM |
| NCL121723 | arr 4q35.2(187,607,091–188,238,570) × 1 | 21 | Pleural effusion, echogenic bowel/liver | 631 kb | FAT1 | LB (39 weeks), 3175 g | NN: normal. FU: none |
| LEE46232 | arr 5p14.3(20,474,778–21,402,969) × 1 | 14 | Bladder extrophy | 928 kb | No genes | LB (35 weeks), 2230 g | NN: cloacal exstrophy, omphalocele, imperforate anus, pelvic kidney, spinal anomaly. FU: paediatric urology |
| LEE46579 | arr 6p25.3(259,911–866,364) × 1 | 20 | Echogenic bowel, CPC | 173 kb | IRF4, LSIRF | LB (38 weeks), 2315 g | NN: normal (including cranial ultrasound). FU: none |
| LEE52107 | arr 9p24.3(839,182–1,050,031) × 3 | 32 | Short femora | 210 kb | DMRT1–3 | LB (36 weeks), 2250 g | NN: positional talipes. FU: height ninth centile, otherwise normal |
| LEE47093 | arr 14q11.2(22,598,056–22,897,060) × 3 | 15 | Cerebral cyst | 299 kb | No genes | LB (40 weeks), 2800 g | NN: normal. FU: none |
| LEE47304 | arr 14q11.2(22,669,473–22,897,060) × 3 | 21 | A4CV, CL + P | 227 kb | No genes | LB (40 weeks), 3240 g | NN: bilateral CL + P. Atrioventricular septal defect. FU: multiple services |
| CAM63103 | arr 16p11.2(28,786,703–28,992,408) × 1 | 19 | CL, echogenic bowel | 206 kb | 3 OMIM genes | TOP (25 weeks) | No PM |
| BIRM5006 | arr 16q23.3q24.1(83,912,627–84,233,642) × 1 | 21 | Pleural effusion | 321 kb | MLYCD, MCD, SLC38A8, FVH2, LRRC50, ODA7, CILD13 | LB (39 weeks), 3065 g | NN examination: left hydrothorax. Pleuroamniotic drain removed. FU: paediatric |
| JCU67801 | arr 17p13.2p13.3(2,721,952–3,614,612) × 1 pat | 20 | CL + P | 892 kb | ASPA, TRPV3m CTNS | LB (41 weeks), 3540 g | NN: CL + P. FU: cleft team |
| LIV24162 | arr Xq22.2(102,605,762–102,857,832) × 2 mat | 21 | CL, short femora | 252 kb | 1–5 OMIM genes | LB (40 weeks), 3804 g | NN: bilateral CL + P. FU: cleft team |
| UCH1316390 | arr Xp21.2(30,325,822–30,327,478) × 3 | 12 | Short femora, humeri, ulnae, radii, oligodactyly, VM, hypo-ossified skull | 1.6 kb | DAX1 | TOP (13 weeks) | No PM |
| QHR122614a | arr 2p14(66,703,773–68,478,303) × 3 mat | 12 | Omphalocele | 1.7 Mb | No genes | LB (38 weeks), 3215 g | NN: no details |
| SMH7718328a | arr 2q31.1(175,386,410–176,248,574) × 3 mat | 21 | Talipes | 862 kb | WIPF1, CHN1 | LB (39 weeks), 4360 g | NN: bilateral talipes. FU: casts, tenotomy (4 months) |
| SOU773245a | arr 3p12.3p12.2(80,312,677–83,304,547) × 3 pat | 20 | CL | 2.9 Mb | GBE1 | LB (40 weeks) | NN: CL (no further details) |
| CARMX840228a | arr 3p26.3(1492692–2655555) × 3 | 20 | Tetralogy of Fallot | 1.16 Mb | CNTN4 | LB (39 weeks), 2910 g | NN: double outlet RV, VSD, pulmonary stenosis (repaired at 2 weeks). FU: paediatric cardiology |
| SOU755972a | arr 6q23.2(131,643,570–132,082,405) × 1 mat | 20 | Multicystic kidney | 439 kb | MED23 | U/K | |
| SOU343509a | arr 16p11.2(29,423,111–30,289,657) × 3 dn | 21 | CL | 866 kb | KIF22, PRRT2, ALDOA, TBX6, CORO1A | LB (42 weeks), 3515 g | NN: CL + P. FU: cleft team |
| SMH6770372a | arr 16p13.3(6,819,616–6,972,344) × 1 | 19 | Spina bifida, VM, CP, A4CV, multi-cystic kidney, talipes | 152 kb | No genes | NND (36 weeks), 3000 g | No PM |
| UCH1357393a | arr 17q11.2(29,423,410–29,435,610) × 3 pat | 20 | Short femora | 12 kb | NF1 | LB | No details |
| SOU773467a | arr Xq13.3(74,463,844–74,651,264) × 3 pat | 21 | Megacystis, hydronephrosis, ? microcolon | 187 kb | ZDHHC15 | LB (34 weeks), 2165 g | NN: megacystis microcolon, died at 3 months. No PM |
Detection rates of imbalances in each subject group
Increased nuchal translucency group
The percentages of pathogenic CNVs and VOUS in fetuses with a NT of ≥ 3.5 mm are shown in Table 19. There was no difference in the rates of abnormal karyotype and pathogenic CNV on CMA, but the rate of any CNV (pathogenic + VOUS) on CMA was 4.5% (95% CI 1.8% to 7.1%) higher than the rate of abnormal karyotype.
| Group | n | Percentage (95% CI) | Percentage difference (95% CI) | p-value |
|---|---|---|---|---|
| Abnormal karyotype | 34 | 6.9 (4.8 to 9.5) | ||
| Pathogenic CNV on CMA | 39 | 7.9 (5.7 to 10.6) | 1.0 (–1.0 to 3.0) | 0.28 |
| VOUS on CMA | 17 | 3.4 (2.0 to 5.5) | ||
| Any CNV on CMA | 56 | 11.3 (8.7 to 14.5) | 4.5 (1.8 to 7.1) | < 0.001 |
The relationship between karyotypes and CNVs on CMA in fetuses with a NT of > 3.5 mm is shown in Table 20.
| Group | CMA | ||
|---|---|---|---|
| Karyotyping | No CNV | Pathogenic CNV | VOUS |
| Normal | 430 | 13 | 17 |
| Abnormal | 8 | 26 | 0 |
The percentages of pathogenic CNVs and VOUS in the subgroup of 159 fetuses with a NT of > 5 mm and the 83 fetuses with a NT of > 6 mm (including those with cystic hygroma) are shown in Table 21. In all subgroups except those with a NT of > 6 mm the percentages of any CNV on CMA were greater than the percentages of abnormal karyotype.
| Group | n | Percentage (95% CI) | Percentage difference (95% CI) | p-value |
|---|---|---|---|---|
| NT of ≤ 5 mm (n = 335) | ||||
| Abnormal karyotype | 19 | 5.7 (3.5 to 8.8) | ||
| Pathogenic CNV on CMA | 18 | 5.4 (3.2 to 8.4) | –0.3 (–2.5 to 1.9) | 0.76 |
| Any CNV on CMA | 32 | 9.6 (6.6 to 13.2) | 3.9 (0.7 to 7.1) | 0.009 |
| NT of > 5 mm (n = 159) | ||||
| Abnormal karyotype | 15 | 9.4 (5.4 to 15.1) | ||
| Pathogenic CNV on CMA | 21 | 13.2 (8.4 to 19.5) | 3.8 (–0.7 to 8.3) | 0.11 |
| Any CNV on CMA | 24 | 15.1 (9.9 to 21.6) | 5.7 (0.7 to 10.6) | 0.022 |
| NT of ≤ 6 mm (n = 411) | ||||
| Abnormal karyotype | 26 | 6.4 (4.2 to 9.2) | ||
| Pathogenic CNV on CMA | 29 | 7.1 (4.8 to 10.0) | 0.7 (–1.5 to 3.0) | 0.47 |
| Any CNV on CMA | 44 | 10.7 (7.9 to 14.1) | 4.4 (1.5 to 7.3) | 0.001 |
| NT of > 6 mm (n = 83) | ||||
| Abnormal karyotype | 8 | 9.6 (4.3 to 18.1) | ||
| Pathogenic CNV on CMA | 10 | 12.0 (5.9 to 21.0) | 2.4 (–3.4 to 8.1) | 0.63 |
| Any CNV on CMA | 12 | 14.5 (7.7 to 23.9) | 4.8 (–2.1 to 11.7) | 0.22 |
The relationship between imbalances detected by karyotyping and CMA in fetuses with a NT of ≤ 5 mm and of > 5 mm and in fetuses with a NT of ≤ 6 mm and of > 6 mm is shown in Table 22.
| Group | CMA | ||
|---|---|---|---|
| No CNV | pCNV | VOUS | |
| NT of ≤ 5 mm (n = 335) | |||
| Karyotyping | |||
| Normal | 297 | 5 | 14 |
| Abnormal | 6 | 13 | 0 |
| NT of > 5 mm (n = 159) | |||
| Karyotyping | |||
| Normal | 133 | 8 | 3 |
| Abnormal | 2 | 13 | 0 |
| NT of ≤ 6 mm (n = 409) | |||
| Karyotyping | |||
| Normal | 358 | 10 | 15 |
| Abnormal | 7 | 19 | 0 |
| NT of > 6 mm (n = 85) | |||
| Karyotyping | |||
| Normal | 72 | 3 | 2 |
| Abnormal | 1 | 7 | 0 |
Abnormal karyotype was more common in fetuses with a NT of > 5 mm than in those with a NT of ≤ 5 mm (p = 0.003), but there was no difference when 6 mm was used as a cut-off point. Similarly, pathogenic CNVs were more frequent in fetuses with a NT of > 5 mm than in those with a NT of ≤ 5 mm (p < 0.003), but there was no difference when 6 mm was used as a cut-off point. There was no difference in the rates of any CNV using either cut-off point.
Structural anomaly group
The percentages of pathogenic CNVs and VOUS in fetuses with one or more structural anomalies are shown in Table 23. This includes the 36 fetuses with increased NT and one or more structural anomalies. The rate of pathogenic CNVs on CMA was 3.5% (95% CI 1.5% to 5.5%) higher than the rate of abnormal karyotype (p < 0.001). The rate of any CNV (pathogenic + VOUS) was 6.8% (95% CI 4.4% to 9.3%) higher than the rate of abnormal karyotype.
| Group | n | Percentage (95% CI) | Percentage difference (95% CI) | p-value |
|---|---|---|---|---|
| Abnormal karyotype | 39 | 6.2 (4.4 to 8.4) | ||
| Pathogenic CNV on CMA | 61 | 9.7 (7.5 to 12.3) | 3.5 (1.5 to 5.5) | < 0.001 |
| VOUS on CMA | 21 | 3.3 (2.1 to 5.1) | ||
| Any CNV on CMA | 82 | 13.0 (10.5 to 15.9) | 6.8 (4.4 to 9.3) | < 0.001 |
The relationship between imbalances detected by karyotyping and CMA in fetuses with a structural anomaly is shown in Table 24.
| Group | CMA | ||
|---|---|---|---|
| Karyotyping | No CNV | Pathogenic CNV | VOUS |
| Normal | 540 | 29 | 21 |
| Abnormal | 7 | 32 | 0 |
The percentages of pathogenic CNVs and VOUS in the subgroup of 253 fetuses (40.2%) with multiple structural anomalies are shown in Table 25. In both subgroups of fetuses, the percentages of pathogenic CNV and any CNV were greater than the percentages of abnormal karyotype. However, although the rate of pathogenic CNVs on CMA was marginally higher in fetuses with multiple structural anomalies than in those with one structural anomaly, the difference was not statistically significant (p = 0.34).
| Group | n | Percentage (95% CI) | Percentage difference (95% CI) | p-value |
|---|---|---|---|---|
| One structural anomaly (n = 376) | ||||
| Abnormal karyotype | 22 | 5.9 (3.7 to 8.7) | ||
| Pathogenic CNV on CMA | 33 | 8.8 (6.1 to 12.1) | 2.9 (0.2 to 5.7) | 0.022 |
| Any CNV on CMA | 43 | 11.4 (8.3 to 15.1) | 5.3 (2.3 to 8.8) | < 0.001 |
| Multiple structural anomalies (n = 253) | ||||
| Abnormal karyotype | 17 | 6.7 (4.0 to 10.5) | ||
| Pathogenic CNV on CMA | 28 | 11.1 (7.5 to 15.6) | 4.3 (1.2 to 7.5) | 0.003 |
| Any CNV on CMA | 39 | 15.4 (11.2 to 20.5) | 8.7 (4.7 to 12.7) | < 0.001 |
The relationship between imbalances detected by karyotyping and CMA in fetuses with one or more structural anomalies is shown in Table 26.
| Group | CMA | ||
|---|---|---|---|
| No CNV | Pathogenic CNV | VOUS | |
| One structural anomaly (n = 376) | |||
| Karyotyping | |||
| Normal | 327 | 17 | 10 |
| Abnormal | 6 | 16 | 0 |
| More than one structural anomaly (n = 253) | |||
| Karyotyping | |||
| Normal | 213 | 12 | 11 |
| Abnormal | 1 | 16 | 0 |
The percentages of pathogenic CNV and VOUS in fetuses with a cardiac anomaly (whether isolated or associated with other structural anomalies) and without a cardiac anomaly are shown in Table 27. In both subgroups the rates of pathogenic CNVs and any CNV were higher than the rate of abnormal karyotype.
| Group | n | Percentage (95% CI) | Percentage difference (95% CI) | p-value |
|---|---|---|---|---|
| No cardiac anomaly (n = 456) | ||||
| Abnormal karyotype | 25 | 5.5 (3.6 to 8.0) | ||
| Pathogenic CNV on CMA | 36 | 7.9 (5.6 to 10.8) | 2.4 (0.2 to 4.6) | 0.016 |
| Any CNV on CMA | 53 | 11.6 (8.8 to 14.9) | 6.2 (3.3 to 9.0) | < 0.001 |
| Cardiac anomaly (n = 173) | ||||
| Abnormal karyotype | 14 | 8.1 (4.5 to 13.2) | ||
| Pathogenic CNV on CMA | 25 | 14.5 (9.6 to 20.6) | 6.4 (1.5 to 11.2) | < 0.001 |
| Any CNV on CMA | 29 | 16.8 (11.5 to 23.2) | 8.7 (3.3 to 14.0) | 0.001 |
The relationship between imbalances detected by karyotyping and CMA in fetuses with and without a cardiac anomaly is shown in Table 28.
| Group | CMA | ||
|---|---|---|---|
| No CNV | Pathogenic CNV | VOUS | |
| No cardiac anomaly (n = 456) | |||
| Karyotyping | |||
| Normal | 398 | 16 | 17 |
| Abnormal | 5 | 20 | 0 |
| Cardiac anomaly (n = 173) | |||
| Karyotyping | |||
| Normal | 142 | 13 | 4 |
| Abnormal | 2 | 12 | 0 |
There was no difference in the percentage of abnormal karyotypes or any CNV among fetuses with and without a cardiac anomaly. However, the percentage of pathogenic CNVs was greater in the subgroup of fetuses with a cardiac anomaly than in the subgroup without (p = 0.013).
Laboratory turnaround times
For the TAT analysis, the results from the increased NT and structural anomaly groups were combined, as were the results from amniotic fluid and chorionic villus samples.
The TATs for karyotyping and CMA are shown in Table 29. Valid dates for sample receipt and karyotype report were available for 685 cases with a median interval of 12 days (IQR 10–14 days). Valid dates for sample receipt and CMA report were available for 714 cases with a median of 15 days (IQR 12–25 days). The median difference between the karyotype and CMA TAT was 3 days (IQR 0–13 days) and this was statistically significant (p < 0.0001). The median TAT for cases for which additional investigations were required is also shown in Table 29.
| Group | Median (days) | IQR |
|---|---|---|
| Karyotyping | ||
| Sample receipt to report: all cases (n = 685) | 12 | 10–14 |
| Sample receipt to report where parental karyotyping performed (n = 23) | 19.5 | 14–25.5 |
| Set-up to report: all cases (n = 588) | 12 | 9–14 |
| CMA | ||
| Sample receipt to report: all cases (n = 714) | 15 | 12–25 |
| Sample receipt to report: cases where parent vs. parent array performed (n = 61) | 22 | 18–29 |
| Sample receipt to report: cases where FISH performed (n = 17) | 26 | 19–41 |
| Set-up to report: all cases (n = 1105) | 6 | 4–9 |
A further analysis was performed on times from test set-up to report. As expected, given the EACH laboratory protocol, the median time for karyotyping was very similar to the TAT reported above (median 12 days, IQR 9–14 days). However, for CMA, when the test was set up after the QR-PCR result was available, the time was much shorter (median 6 days, IQR 4–9 days). The median difference between karyotype and CMA TAT was –5 days (IQR –8 to –2 days), indicating that TATs from set-up to report were quicker with CMA (p < 0.0001).
Factors affecting chromosome microarray turnaround time
Laboratory
To determine if there were differences between laboratories in terms of CMA TAT, median intervals were compared (Table 30). The differences between laboratories were statistically significant (p < 0.0001).
| Laboratory | Number | Median TAT (days) (IQR) |
|---|---|---|
| West Midlands Regional GL | 105 | 11 (9–14) |
| East Anglian Medical GS | 45 | 18 (14–29) |
| All Wales Medical GS | 30 | 28.5 (22–47) |
| Bristol GL | 113 | 16 (14–26) |
| Merseyside and Cheshire Regional GL | 84 | 15 (13–32) |
| Wessex Regional GL | 116 | 12 (9–15) |
| Northern GS | 99 | 15 (13–19) |
| Yorkshire Regional GS | 66 | 26 (15–32) |
A similar comparison was undertaken for times from CMA set-up to report (Table 31). As with the overall TATs, the differences between the laboratories for these times were statistically significant (p = 0.0001), suggesting that some laboratories were able to perform, interpret and report the CMA faster than others.
| Laboratory | Number | Median TAT (days) (IQR) |
|---|---|---|
| West Midlands Regional GL | 105 | 2 (2–5) |
| East Anglian Medical GS | 49 | 3 (2–5) |
| All Wales Medical GS | 30 | 9 (6–17) |
| Bristol GL | 115 | 5 (2–8) |
| Merseyside and Cheshire Regional GL | 86 | 6 (4–10) |
| Wessex Regional GL | 117 | 6 (6–7) |
| Northern GS | 136 | 7 (6–8) |
| Yorkshire Regional GS | 66 | 6 (3–8) |
| North East Thames Regional GS | 398 | 8 (5–12) |
Duration of study
To determine if increasing laboratory experience with CMA affected TAT, median intervals were compared for three successive time epochs (with approximately equal numbers of cases) (Table 32). The differences between epochs were statistically significant (p = 0.007).
| Epoch | Number | Median TAT (days) (IQR) |
|---|---|---|
| One (November 2011to April 2013) | 242 | 17 (13–28) |
| Two (April 2013 to November 2013) | 239 | 14 (12–22) |
| Three (November 2013 to May 2014) | 233 | 14 (12–22) |
The EACH review panel
In total, 32 cases were referred to the EACH review panel (Table 33). Details of the individual cases are reported in Tables 13–18. In total, the panel recommended that 16 CNVs should be reported, which included one case for which the karyotyping failed but CNV was deemed pathogenic by the panel (UCH1372792). In one case (SOU382387), owing to the complex nature of the CNV, the 22q11.2 deletion was not reported immediately, but after parental follow-up studies had been completed.
| Group | Report | Not report |
|---|---|---|
| Increased NT (n = 11) | 7 | 6 |
| Structural anomaly (n = 21) | 9 | 10 |
When full parental data were available to the panel, the median time to reach a decision was 3 days (range 1–7 days). When full parental data were not initially available (n = 4), the length of time to make a recommendation ranged from 6 to 14 days. In 24 cases, two clinical geneticists and two cytogeneticists reached a decision with at least a 3 out of 4 consensus. In eight cases, additional colleagues were asked for an opinion when consensus could not be reached or when there was considerable uncertainty about whether or not to recommend reporting. The relationship between the size of the CNV and the panel decision is shown in Table 34. It is noteworthy that 9 out of the 16 CNVs (56%) that were recommended for reporting were < 1 Mb in size, whereas 6 out of 16 (38%) of those not reported were > 1 Mb.
| Size of CNV | Reported | Not reported | ||
|---|---|---|---|---|
| Deletions | Duplications | Deletions | Duplications | |
| < 500 kb | 4 | 3 | 4 | 2 |
| 500–1000 kb | 1 | 1 | – | 4 |
| 1–2 Mb | 3 | 1 | 1 | 2 |
| > 2 Mb | 3 | 1 | 2 | |
| Total | 8 | 8 | 6 | 10 |
Health economic analysis
A list of data used in the decision-analytic model is provided in Table 35. The decision tree depicting the current diagnostic pathway and the new pathway using CMA, with corresponding percentages in each branch, is shown in Figure 7. Almost half of the women underwent amniocentesis for karyotyping; this proportion was smaller in the increased NT group (13%) than in the anomaly group (78%). The karyotype was abnormal in 6.5% of cases, of which 56% underwent further follow-up testing (which was most often parental karyotyping). Following CMA, a pathogenic CNV was found in 8.9% of cases, of which 73% underwent follow-up testing, and a VOUS was found in 3.4% of cases, of which than 48% underwent follow-up testing. In the increased NT group, 23% of the pathogenic CNV cases that had a follow-up test were sent to the EACH review panel, compared with 100% of the VOUS cases. In the anomaly group, 28% of the pathogenic CNV cases and 64% of the VOUS cases that had follow-up tests were sent to the panel. Abnormal results (abnormal karyotype or pathogenic CNV) resulted in a genetics consultation more often than a normal result (41% vs. 7%).
| Parameter | All cases (n = 1123) | Increased NT (n = 494) | Structural anomaly (n = 629) |
|---|---|---|---|
| Invasive test | |||
| Chorionic villus sample | 51.4 (48.5 to 54.4) | 87.5 (84.4 to 90.3) | 22.4 (18.9 to 25.6) |
| Amniocentesis | 48.6 (45.6 to 51.5) | 12.5 (9.7 to 15.6) | 77.8 (74.4 to 81.1) |
| Test results | |||
| Karyotype abnormal | 6.5 (5.1 to 8.0) | 6.9 (4.9 to 9.3) | 6.2 (4.5 to 8.2) |
| Pathogenic CNV on CMA | 8.9 (7.3 to 10.6) | 7.9 (5.7 to 10.4) | 9.7 (7.5 to 12.1) |
| VOUS on CMA | 3.4 (2.5 to 4.6) | 3.4 (2.0 to 5.2) | 3.5 (2.2 to 5.1) |
| Cases undergoing follow-up tests | |||
| Karyotype abnormal | 56.2 (44.7 to 67.3) | 52.9 (36.4 to 69.2) | 59.0 (43.4 to 73.7) |
| Pathogenic CNV on CMA | 73.0 (63.9 to 81.2) | 66.7 (51.3 to 80.4) | 77.0 (65.8 to 86.6) |
| VOUS on CMA | 48.7 (33.4 to 64.2) | 29.4 (11.0 to 52.4) | 63.6 (43.0 to 81.9) |
| Other (benign) CNV on CMA | 4.6 (3.4 to 6.1) | 5.4 (3.6 to 7.9) | 3.9 (2.4 to 5.7) |
| Proportion of follow-up tests | |||
| FISH | 18.6 (12.7 to 25.3) | 20.0 (11.0 to 30.9) | 17.6 (10.4 to 26.4) |
| Parental karyotype | 20.7 (14.5 to 27.6) | 21.7 (12.3 to 32.8) | 20.0 (12.3 to 29.1) |
| PvP CMA | 62.8 (54.8 to 70.4) | 65.0 (52.6 to 76.4) | 61.2 (50.7 to 71.2) |
| MLPA | 4.1 (1.5 to 7.9) | 1.7 (0.0 to 6.1) | 5.9 (2.0 to 11.7) |
| QF-PCR | 3.4 (1.1 to 7.0) | 3.3 (0.4 to 9.1) | 3.5 (0.7 to 8.3) |
| Other | 2.1 (0.4 to 4.9) | 3.3 (0.4 to 9.1) | 1.2 (0.0 to 4.3) |
| Cases sent to the EACH review panel | |||
| After pathogenic CNV and follow-up test(s) | 26.0 (16.7 to 36.6) | 23.1 (9.4 to 40.7) | 27.7 (16.0 to 41.1) |
| After VOUS and follow-up test(s) | 73.7 (52.4 to 90.3) | 99.8 (99.0 to 100) | 64.3 (38.6 to 86.1) |
| Follow-up action: after abnormal result | |||
| Genetics consultation | 40.9 (32.1 to 49.9) | 40.4 (27.0 to 54.6) | 41.2 (29.8 to 53.0) |
| Fetal medicine consultation | 67.0 (58.1 to 75.2) | 68.1 (54.2 to 80.5) | 66.2 (54.6 to 78.8) |
| Follow-up action: after normal result | |||
| Genetics consultation | 7.4 (5.9 to 9.1) | 5.4 (3.5 to 7.7) | 9.1 (6.8 to 11.6) |
| Fetal medicine consultation | 47.9 (44.8 to 51.0) | 51.9 (47.3 to 56.5) | 44.8 (40.7 to 48.9) |
FIGURE 7.
Decision tree depicting the current diagnostic pathway and the new pathway using CMA. Percentages of cases in each branch are shown: all cases, increased NT group and anomaly group. AM, amniocentesis; bCNV, benign CNV; pCNV, pathogenic copy number variant.
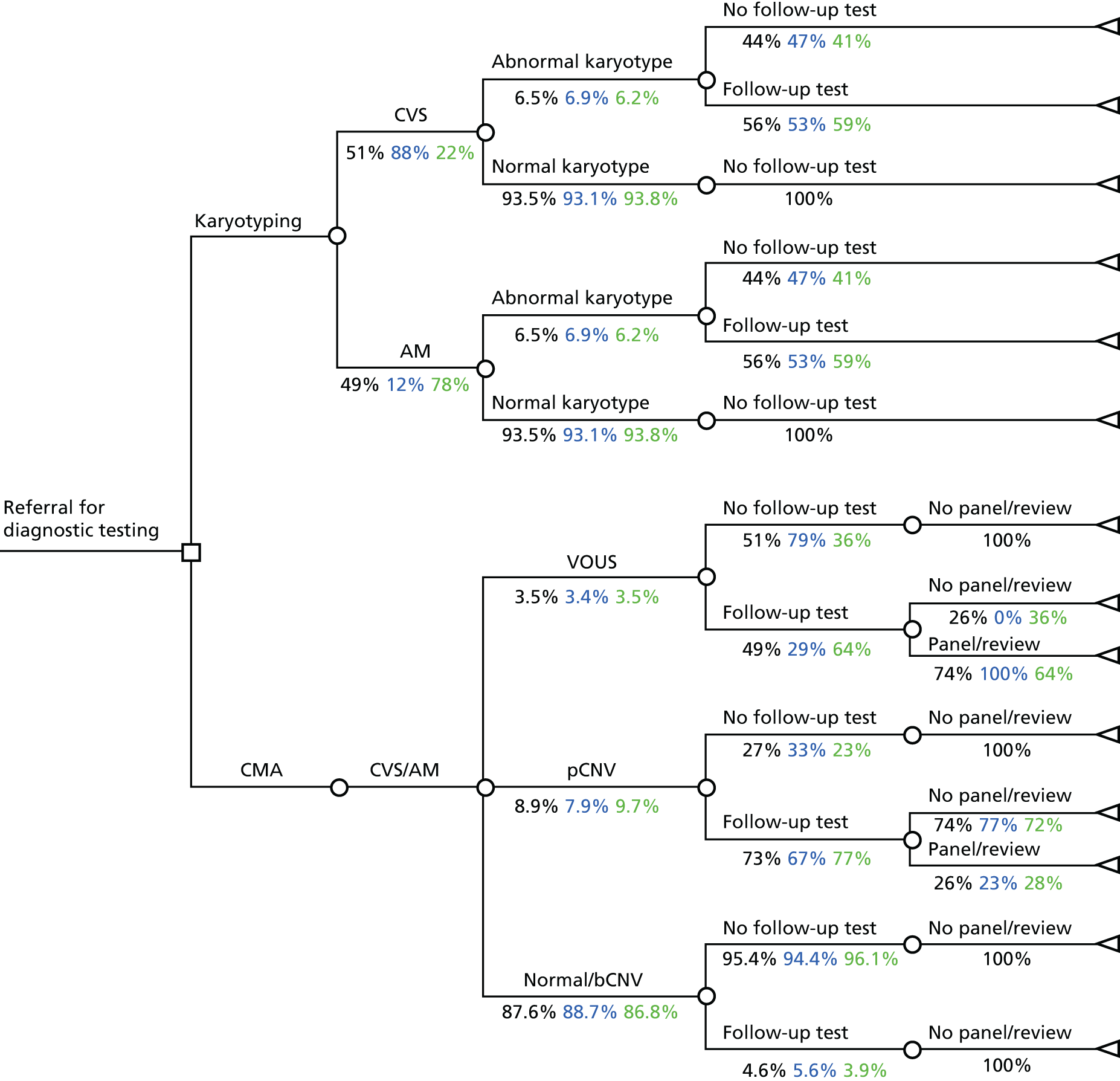
The cost input parameters are summarised in Table 36. There were minimal differences in follow-up laboratory costs between the two study groups: £390 in the increased NT group compared with £383 in the structural anomaly group (average £386). There were also minimal cost differences between the increased NT and structural anomaly groups in terms of the costs of clinical follow-up actions in abnormal cases [£225 vs. £226, respectively (average £226)] and in normal cases [£79 vs. £85, respectively (average £82)]. Estimated EACH panel costs are shown in Table 37. All costs are expressed in 2012–13 British pounds. The time horizon in this study was the duration of pregnancy and, therefore, no discounting was necessary.
| Parameter | Base-case value (range) (£) | Source |
|---|---|---|
| Laboratory primary test | ||
| Chorionic villus karyotyping | 272 (240–307) | The EACH study laboratories |
| Amniotic fluid karyotyping | 234 (212–258) | The EACH study laboratories |
| CMA | 322 (280–367) | The EACH study laboratories |
| Laboratory follow-up test | ||
| FISH | 393 (225–608) | The EACH study laboratories (3 × £131a) |
| Parental karyotype | 508 (453–566) | Costs of karyotyping (2 × £254b) |
| PvP CMA | 352 (298–413) | One × CMA + two additional DNA extractions (2 × £15) |
| MLPA | 300 (171–465) | The EACH study laboratories (3 × £100a) |
| Other | 100 | Assumed similar to one MLPA |
| Clinical follow-up action | ||
| Clinical genetics consultation | 363 (207–561) | NHS reference costs 2012–1352 |
| Fetal medicine consultation | 116 (66–179) | NHS reference costs 2012–1352 |
| Case type | Number of cases | Resource use | Unit costs (per minute)53 (£) | Total | ||||
|---|---|---|---|---|---|---|---|---|
| People involved | Minutes/person | Total minutes/case | Medical consultant | Laboratory scientist | Average | Cost (£)/case | ||
| Brief | 1 | 3 | 15 | 45 | 2.32 | 2.13 | 2.22 | 100.08 |
| Average | 22 | 4.27 | 25 | 107 | 2.32 | 2.13 | 2.22 | 237.57 |
| Difficult | 8 | 5 | 75 | 375 | 2.32 | 2.13 | 2.22 | 834.01 |
| Weighted average | 387 | |||||||
Table 38 shows the total costs of the karyotyping pathway and the CMA pathway, as well as the number of abnormal karyotypes/pathogenic CNVs found in each pathway. If all cases were considered, the costs per case in the CMA pathway were £113 higher than in the karyotyping pathway, increasing the number of abnormalities found by 0.024. Therefore, the incremental cost per additional abnormality found was £4703. The incremental costs and the number of additional abnormalities found were lower in the increased NT group than in the anomaly group (£96 vs. £127 and 0.01 vs. 0.035, respectively). The incremental costs per additional abnormality found were £9439 in the increased NT group and £3635 in the anomaly group.
| Parameter | Total costs (£) | Abnormality found | Incremental costs per extra case (pCNV) detected (£) |
|---|---|---|---|
| All cases | |||
| Karyotyping | 485.90 | 0.065 | |
| CMA | 598.97 | 0.089 | |
| Increment | 113.07 | 0.024 | 4703 |
| Increased NT | |||
| Karyotyping | 499.09 | 0.069 | |
| CMA | 594.63 | 0.079 | |
| Increment | 95.54 | 0.010 | 9439 |
| Anomaly | |||
| Karyotyping | 475.17 | 0.062 | |
| CMA | 602.30 | 0.097 | |
| Increment | 127.13 | 0.035 | 3635 |
Sensitivity analysis
In the one-way sensitivity analysis, only a few parameters were found to affect the results appreciably. The largest influence was seen for the number of pathogenic CNV results found by CMA. The costs per extra pathogenic CNV detected would be £13,690 if a pathogenic CNV result was seen in 7.3% of the cases, compared with £2851 if a pathogenic CNV result was seen in 10.6% of the cases. A similar influence was seen for the number of abnormal karyotypes. In addition, the costs of CMA influenced the results appreciably (costs per extra case detected varied from £2872 to £6686 when the costs of the test were varied from £280 to £367). A smaller, but still relevant, influence was seen for the costs of karyotyping and the costs of the panel review (Figure 8). Similar results were found for the increased NT group and the structural anomaly group. In the increased NT group, the number of abnormal results found had a very large influence on the results. If the percentage of pathogenic CNV results was low (lower limit 5.7%) or the percentage of abnormal karyotype results was high (upper limit 9.3%), CMA was less effective and more costly than karyotyping (dominated by karyotyping). The costs of chorionic villus karyotyping had a larger influence in the increased NT group (because the majority of women in this group underwent CVS instead of amniocentesis) and the costs of amniotic fluid karyotyping had a larger influence in the anomaly group (because the majority of women in this group underwent amniocentesis).
FIGURE 8.
Tornado diagram of the incremental costs per extra pathogenic CNV detected by CMA vs. conventional karyotyping thresholds in all cases and by subgroup. (a) All cases; (b) increased NT; and (c) anomaly. aCGH, array CGH; AM, amniocentesis.
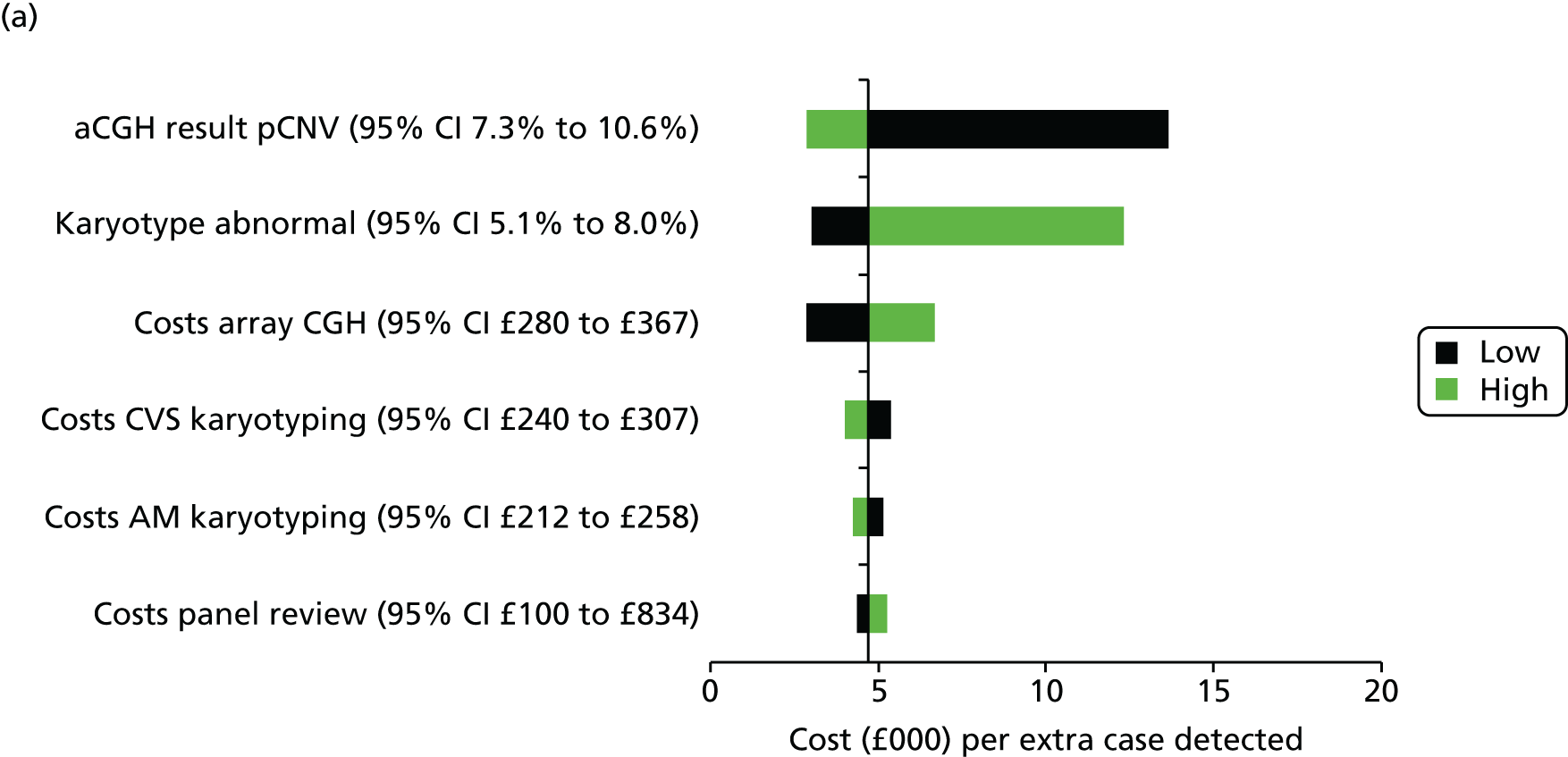
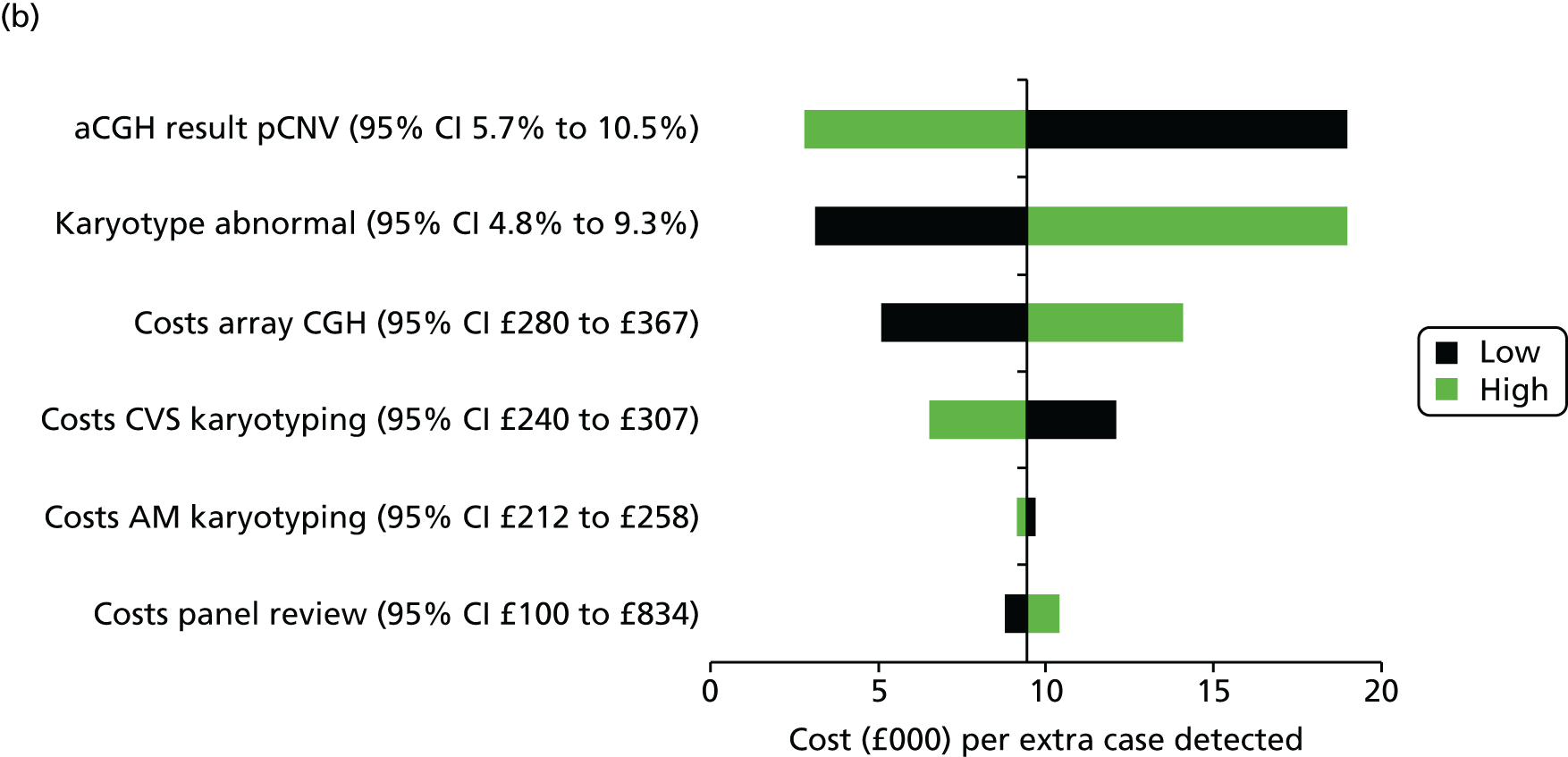
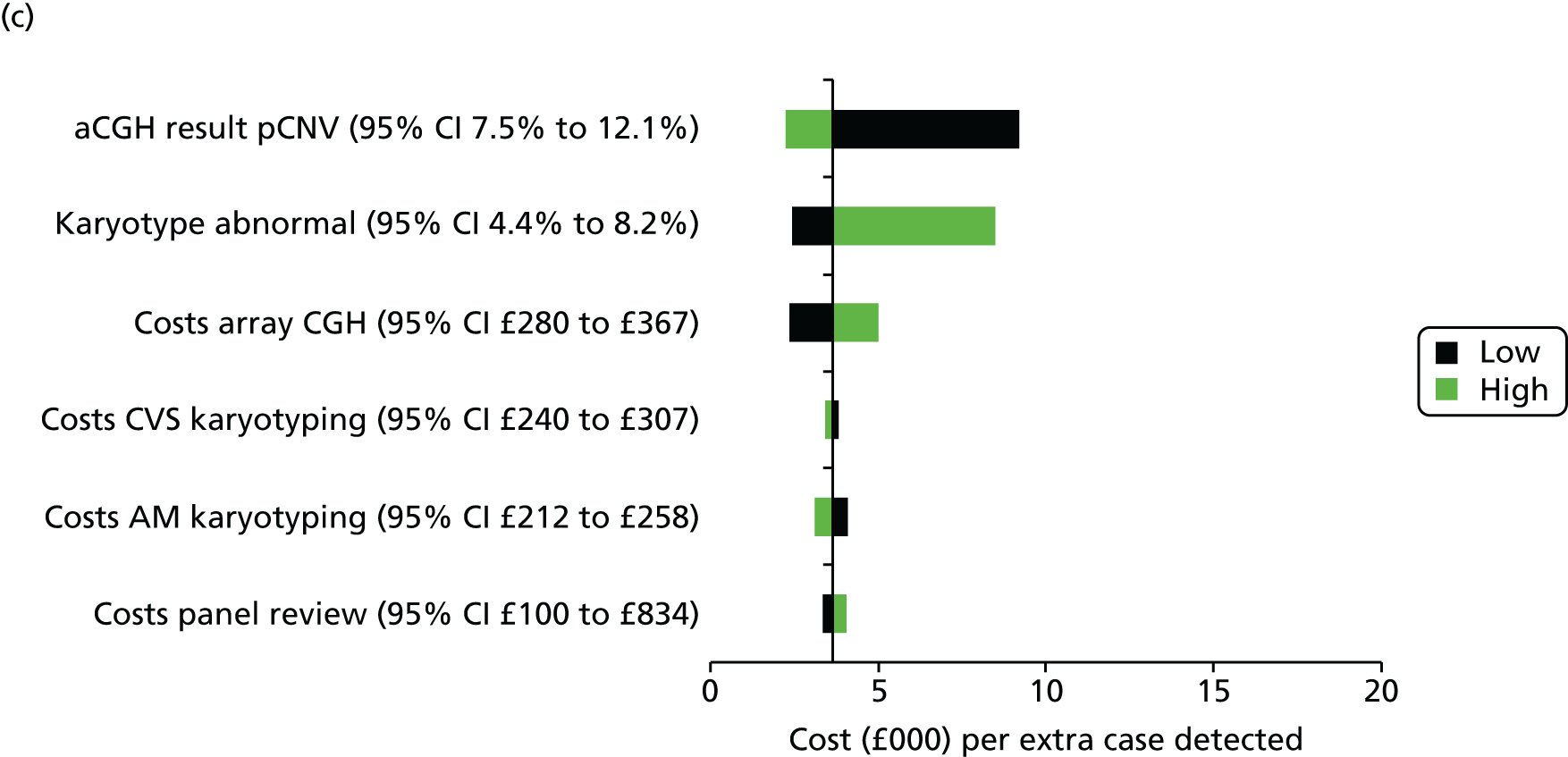
In the scenario analysis in which the EACH review panel was excluded, CMA increased costs by £102 (costs per extra case detected was £4230) if all cases were considered, by £87 (costs per extra case detected was £8588) for the increased NT group and by £114 (costs per extra case detected was £3248) for the anomaly group.
In the probabilistic sensitivity analysis, 0% of the simulations were found to be cost-effective if the willingness to pay per incremental abnormality found was £1000 and 87% of the simulations were found to be cost-effective if the willingness to pay was £10,000. Figure 9 shows the chance that CMA would be cost-effective at different levels of willingness to pay. For cases with an increased NT, the probability that CMA would be cost-effective was lower (52%) than for cases with a structural anomaly (93%) at a willingness to pay of £10,000 per incremental abnormality found.
FIGURE 9.
Cost-effectiveness acceptability curves showing the chance that CMA would be cost-effective given different willingness to pay thresholds in all cases and by subgroup. (a) All cases; (b) increased NT; and (c) structural anomaly.

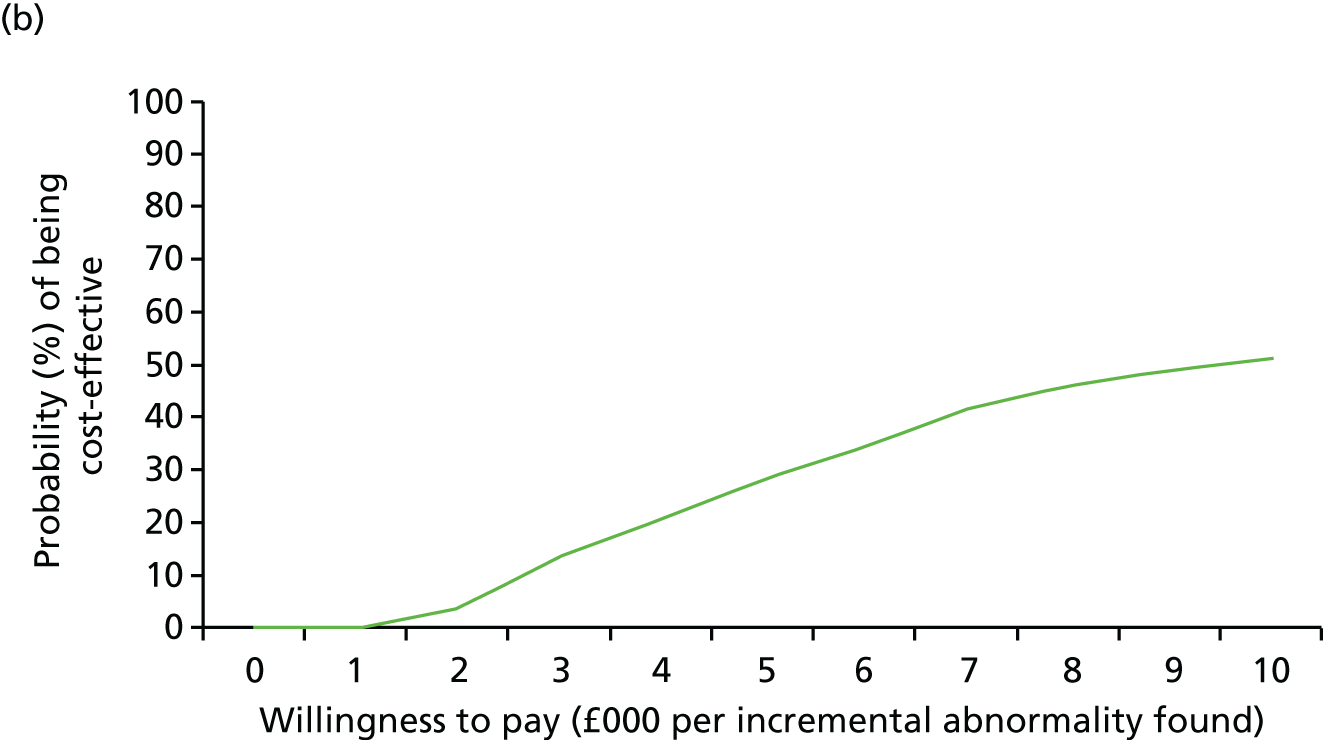
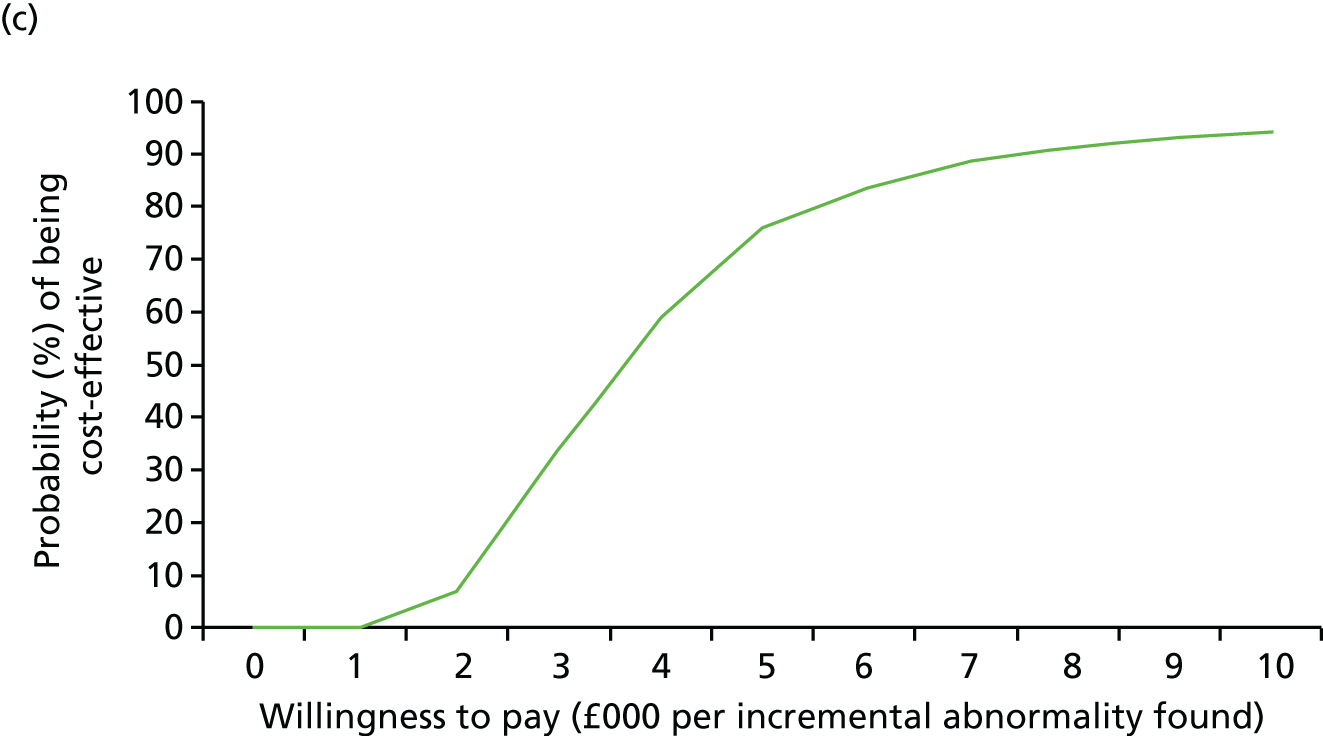
Qualitative substudy
Out of a total of 98 parents, health professionals and commissioners contacted and invited to participate in the substudy, 48 respondents were interviewed (Table 39): 16 women (five with their partners present) from the three study sites, 18 health professionals from two study sites and six commissioners. Two women and three health professionals declined to participate, one woman was not present when the researcher arrived at her home, one woman agreed to be interviewed but was thereafter not contactable and the remaining 24 women and 20 health professionals never responded. Thus, the response rates for women and health professionals were 40% and 58%, respectively. The recruitment rate for commissioner participants was 50%.
| Category | SiteA | SiteB | SiteC | Total |
|---|---|---|---|---|
| Cytogeneticist | 2 | 0 | – | 2 |
| Fetal medicine midwife | 1 | 3 | – | 4 |
| Fetal medicine consultant | 5 | 3 | – | 8 |
| Consultant clinical geneticist | 3 | 2 | – | 5 |
| Genetic counsellor | 2 | 0 | – | 2 |
| Women (partner) | 5 (2) | 9 (2) | 2 (1) | 16 (5) |
| Commissioners | – | – | – | 6 |
| Total | 20 | 19 | 3 | 48 |
Out of the six commissioners interviewed, three were involved with commissioning national specialised services (e.g. fetal medicine and medical genetics) and three with commissioning local services (as part of the Area Teams within NHS England North). All women and partners were interviewed in their home and all health professional and commissioners were interviewed at their place of work. The case details of the parent participants are shown in Table 40.
| Participant site/number | Age (years) | Gestation+ (weeks) | Study group | Karyotype result | CMA result | Pregnancy outcome |
|---|---|---|---|---|---|---|
| SiteA, WOM1 | 41 | Term | Increased NT | Normal | VOUS | LB |
| SiteA, WOM2 | 42 | 12+6 | Increased NT | Abnormal | Normal | TOP |
| SiteA, WOM3 | 35 | 21+2 | SA | Normal | Normal | TOP |
| SiteA, WOM4 | 44 | 39+6 | Increased NT | Normal | VOUS | LB |
| SiteA, WOM5a | 34 | 16+1 | Increased NT | Abnormal | pCNV | TOP |
| SiteB, WOM1 | 29 | 13+3 | Increased NT + SA | Normal | Normal | TOP |
| SiteB, WOM2 | 36 | 22 | SA | Normal | Normal | SpA |
| SiteB, WOM3 | 37 | 24 | SA | Abnormal | pCNV | TOP |
| SiteB, WOM4a | 35 | 21+3 | SA | Normal | Normal | TOP |
| SiteB, WOM5 | 31 | 37+4 | Increased NT | Normal | Normal | LB |
| SiteB, WOM6 | 27 | 39+4 | Increased NT | Normal | pCNV | LB |
| SiteB, WOM7 | 32 | 35+6 | SA | Normal | pCNV | LB |
| SiteB, WOM8 | 33 | 18+1 | SA | Abnormal | pCNV | TOP |
| SiteB, WOM9a | 35 | Term | SA | Normal | VOUS | LB |
| SiteC, WOM1 | 32 | 32+3 | SA | Normal | pCNV | TOP |
| SiteC, WOM2a | 41 | 30+2 | SA | Normal | pCNV | TOP |
The generative thematic analysis resulted in the identification of four key themes emerging from the data: (1) the functionality of CMA, (2) introducing CMA into the clinical consultation, (3) decision-making and (4) embedding CMA into standard clinical practice.
Theme 1: the functionality of chromosome microarray
Referral pathway for chromosome microarray
Women recruited into the EACH study were referred from hospitals with and without a specialised fetal medicine centre. Referrals were from obstetric consultants and sonographers. Some women had discussed karyotyping with their consultant prior to referral. The health professionals’ accounts implied that there were clear guidelines for referral that were generally understood and followed. Exceptions to this occurred when a referral to a consultant with a special interest in fetal medicine had taken place before a referral to the specialised centre. Clearly this pathway is appropriate in some cases, potentially preventing women attending a fetal medicine centre unnecessarily. However, it did have implications for the care of women whose referral was delayed:
. . . sometimes that’s entirely appropriate but it can occasionally mean that we get a slightly delayed referral, erm, which . . . depending on gestation can mean that we’re coming up to pretty time sensitive areas in terms of whether people are approached in 24 weeks by the time they come to see us with an abnormality.
SiteA, FM2
The parents interviewed recalled how the time between the initial identification of the ultrasound anomaly and having the diagnostic testing appeared to move slowly: ‘A minute feels like a day’ (SiteB, WOM4). The time frames for referral to a fetal medicine centre varied from 1 to 10 days, depending on the availability of appointments, and all parents highlighted how difficult this waiting time was for them. Once referred, the parents were seen by a fetal medicine consultant and counselled regarding their options for pregnancy management. Expectations around invasive testing differed between parent participants; those seen in specialised fetal medicine centres expected to have access to invasive testing on the same day:
I just wished I could just hopped on that bed that day and got that needle through me. I wanted it done instantly but I know it doesn’t work that way . . . if people want the test I think it should be available very quickly if possible.
SiteA, WOM4
The feasibility of array as a replacement for karyotype
The health professionals discussed the advantages and disadvantages of CMA as a clinical test when compared with karyotyping. Overall, CMA was seen as the better option in most cases owing to the greater information provided, but there were cautions. Some comparisons were made in terms of time taken for the test result and the reporting process:
To do a karyotype somebody physically has to look down a microscope, it’s not something you can rush. Array CGH, it takes a long time to interpret the results but you’ve got machinery that’s doing it for you, erm, and so it’s, the detail that you get surpasses what you can get from a karyotype.
SiteA, GC1
A fetal medicine consultant commented that arrays were taking longer from their perspective but acknowledged that this was likely to improve as the laboratory staff became more experienced with the technology and the reporting procedures. The main issue for the health professionals was the degree of uncertainty associated with CMA. It was acknowledged that the array was more likely to detect an abnormality than karyotyping and if this abnormality was a known deletion or duplication, then the test was perceived to be extremely useful. However, if it was a variant of unknown clinical significance, this made counselling far more challenging. Despite this, the majority of the health professional group still stated that they would rather have more information than less. The implications of this uncertainty for counselling are covered in greater detail in Theme 3: decision-making.
Some clinicians felt there would still be situations in which they would want karyotyping instead of CMA, for example following a high-risk screening test for trisomy 21. However, it was more generally felt that CMA, as a clinical test, could feasibly replace the standard karyotype altogether:
Karyotypes are obsolete . . . I think karyotypes are yesterday’s. Arrays are today’s, just . . . next gen sequencing is tomorrow and you’ve got to move with the times.
SiteB, CG2
Theme 2: introducing chromosome microarray into the clinical consultation
Understandings of the EACH research
The parents interviewed had varied understandings of the EACH research and the CMA test. Even when parents received an abnormal result, their comprehension of the information given to them at the time of enrolment was not always optimal. There were four main areas where patient understanding seemed problematic: (1) awareness (or lack of awareness) that the test could potentially have an impact on their own pregnancy, (2) mistaking the EACH research for concurrent research on non-invasive testing, (3) mistaking the EACH test for whole-genome sequencing and (4) recollection (or lack of recollection) of any information regarding the array.
The lack of awareness of the potential impact on their own pregnancy was found in the parents’ narratives around reasons for taking part in the research. They spoke of wishing to give something back, participating in research to help others and/or their immediate family or taking part in research to support scientific endeavour. Many of the women who had normal results (or who never received a result confirming normality) spoke of how they were keen to contribute to research that would enable the clinicians to test women in the future using non-invasive procedures:
And if it is a case of being able to take blood and stuff from mum and dad and, erm, you know work out if there is a, a problem there, erm, then it’s got to be easier than having the risk of miscarriage hasn’t it really.
SiteB, WOM9
Two of the women interviewed confused the array with whole-genome sequencing and felt reassured by the normal result on aspects not actually covered by the test:
We know that we’re healthy. [Laughter.] That we haven’t got like cancer or owt like that. One thing that we could say that we are all perfectly healthy and it were, we were both like oh that’s a good thing we went through all that. At least we know we’re fine.
SiteB, WOM6
Mistaking the array for whole-genome sequencing was also something that some of the health professionals expressed concerns about. They warned of the importance of ensuring that parents fully understood the limitations of the array test.
Many of the parents interviewed had no or poor recollection of being spoken to about the array test or the EACH research:
I just remember the doctor asking if they could, for a study to take extra, they would have to take extra cells but it wouldn’t, you know it wasn’t going in again with another needle, it was just like a bit extra at the time that she took the cells that they needed, erm, I can’t really remember to be quite honest.
SiteA, WOM2
The majority of parents interviewed in both the normal and abnormal (pathogenic copy number variant) CMA result groups expressed this sort of recollection. Those who received an abnormal test result subsequently developed a better understanding of the specific condition in their case, but admitted to not understanding the initial implications of the array test. Only one couple were able to describe their understanding in a way that suggested a good grasp of the processes involved:
They talked about my blood and X’s blood, to sort of see what genetic problems we might have and then using that to compare with the, erm, baby’s blood and then looking for any differences that may or may not be there and the genetic information, erm, some of which would be, erm, sort of known problems, so kind of a known 22q11, but obviously there were other genetic type problems that aren’t necessarily diagnosable that easily and it was part of that bigger picture. I, that’s not a very description of it really.
SiteC, WOM1
Knowledge is power
Despite the apparent lack of understanding among the parent participants around CMA, there was a strong theme of parents believing that more information was always a good thing. They felt that additional information would enable them to make better choices and would give them definitive answers, with comments such as ‘I would rather be overloaded with information and, than not have anything at all’ (SiteB, WOM2) and ‘Knowledge is power, it’s all about choice isn’t it?’ (SiteB, WOM3).
Most parents felt that more information would enable them to prepare adequately, again assuming that the information that they would gain would always offer definitive answers. There appeared to be a lack of recognition that the additional information gained from the CMA could introduce greater uncertainty. Only one parent felt that too much information could be worse than none at all in cases where the information was uncertain:
Erm, but I think the tests, I think it’s good to know that you’re constantly improving it and, but sometimes I think too much information’s bad as well, so it would have to be a fine line of how much information . . . I suppose some people, if they knew you were picking up [uncertainties] would wanna know, but I would probably not wanna know, yeah, just different people.
SiteA WOM4
The health professional participants expressed concern about the desire parents have for more information. They recognised that parents tend to prefer more information but, as clinicians, they were keen to ensure that the counselling prior to testing ensured/facilitated an adequate understanding of how an uncertain result could cause parents greater confusion and distress:
I’m not convinced yet that we’ve got this business of a potential uncertainty, erm, over to couples because we’re seeing lots of families where that, that hasn’t been understood. That doesn’t necessarily mean it’s not been explained, it just means it hasn’t been understood.
SiteA, CG4
The issue of ‘true’ informed consent was discussed by many of the health professionals. One fetal medicine consultant in particular was keen to get across that parents did not need to avail themselves of CMA just because the technology was available. Conversely, health professional participants also discussed how parents who had apparently fully understood the test at the time of consent could still be surprised by an abnormal result:
I think that if people are aware that there’s a significant chance that we’ll find something that we can’t explain, then they stop and think, but most of them will still go ahead with that and they’re still surprised when you come back to them and you say ‘Ah guess what, we’ve found something that we can’t explain’, erm, and I’m not sure there’s an easy way of tackling that because that’s just life.
SiteA, CG5
Many of the health professionals felt that even when there was an abnormal result or a VOUS, parents would still prefer to be given as much information as was available. This belief was matched by the data from parents who had been in this situation. Overwhelmingly, parents wanted as much information as possible and, even when that information had led them to make a difficult choice, their preference was to have been informed:
Ultimately, yeah, yeah ‘cos I think it did give us that choice, erm, and we wouldn’t have had that choice you know. ‘Cos even if we decided to continue with the pregnancy and he needed to have the heart surgery, it would all be very planned. I would be up in [city] for 3 months or however long it’s gonna take, erm, so either way, you know, that choice and the ability to prepare yourself is, is so valuable really, and without that choice you don’t have any options.
SiteC WOM1
Despite the issues expressed by parents around understandings of the array, the health professionals were confident that they always fully informed parents of all the issues around the array. Many offered an almost verbatim rendition of the verbal information they would give to parents when consenting for an array and these accounts consistently highlighted the potential for introducing more uncertainty. One of the midwives interviewed offered a possible explanation for the disparity between parents’ understandings and the health professionals’ certainty that informed consent had taken place. She was concerned not only about the complexity of information but also about the timing:
. . . we also have massive concerns about how much information they are actually taking in at that time. They’re given a very complicated, it doesn’t matter how easy you’ve tried to make the information leaflets for any of the studies, it’s very complicated for them to understand and you know they’re willing to help, you know, for any reason so, so we do have massive reservations, so that’s a big thing about it.
SiteB, MW1
Overall, the data from parents and health professionals around the theme of introducing CMA into the clinical consultation pointed to differing perspectives of how well the array had been introduced and how much understanding parents had been able to absorb and retain. Their accounts highlighted the need for ensuring that parents fully understood the implications of having CMA.
Theme 3: decision-making
Dealing with uncertainty
The health professional data suggest that health professionals grapple with variants where the clinical significance is uncertain. Evident in the data overall, it was particularly important to their views on clinical decision-making. Given this context, the multidisciplinary team was identified as important to health professionals in making decisions about whether or not to report a VOUS to parents. In a similar way, the Cartagenia Bench database was seen as having a role in assisting with such decisions in the future. In the context of this study in particular, the function of the expert panel was important.
The health professionals interviewed worked across two sites (A and B). Site A had a formal multidisciplinary team meeting each week whereas Site B made no formal provision but included wider team members more informally. At both sites, participants felt it was important to involve colleagues in the decision-making around uncertain results. Although the clinicians advocated always being completely honest with parents, they admitted that telling parents that they did not know the clinical significance of a test result made them feel uneasy. Involving colleagues in their decisions was seen as a way of being ‘as sure as we possibly can of our own understanding of the position and then being honest with people’ (SiteA, CG4).
A common concern expressed was the possibility of decisions about pregnancy management being made based on an uncertain result, which may have been a normal variant. For example, a clinical geneticist explained:
. . . so it’s possible to be completely normal but to have a chunk of DNA missing, er, and that’s just a normal variant if you like. Er, and some of the things that we pick up on arrays may well be those and we don’t know that yet, if you see what I mean, and that makes me worry about basing, erm, basing pregnancy management decisions on this kind of uncertainty . . .
SiteA, CG5
Some of this discussion focused on the issue of reporting only clinically ‘significant’ results. In particular, a commissioner participant felt quite strongly that there was little benefit in giving a woman a result for which the implications were uncertain. Some professionals considered the possibility of only looking for specific abnormalities so there would be no conflict around reporting VOUS. Others suggested that changing the criteria for having a CMA would reduce the number of VOUS to manage (e.g. increasing the threshold for the raised NT):
. . . it would appear that in some centres, if there’s multiple abnormalities on a scan and the karyotype’s normal then they then go on to do an array and I was thinking that might sort of make more sense because the raised nuchals are just so non-descript and so I personally think that the abnormalities must be more substantial . . . I just think either a very increased nuchal you know it needed a bigger cut-off or some structural abnormalities . . . and then there’s also the suggestion of not doing the whole array and just doing more common micro deletion or duplication syndromes, I don’t . . . we’re, we’re getting lots of positive arrays for not particularly well . . . erm, well known syndromes but I think there has to be something where, erm, there’s sort of substantive evidence that, erm, that you can actually go in to them and say you know ‘80% of these people have whatever or’ . . .
SiteB CG2
However, most participants felt that, despite concerns in the early stages of the study, the reporting of VOUS was appropriate. A clinician with direct experience of managing two cases with pathogenic results stated:
I was worried that we would be reporting every copy number variation even if we didn’t think it was important but that people would be very worried about that and I think the fact that that isn’t how it’s happening is, is the way to do it.
SiteA, FM2
The role of long-term follow-up
The health professionals pointed to the importance of addressing the uncertainties through long-term follow-up of children with VOUS, specifically through a database. Such a resource was seen as fundamental to ensuring that the degree of uncertainty currently faced would decrease over time:
. . . we are building up a database of array results and so it is getting easier. We are then able to actually have a closer look at which genes are involved. We still don’t know what all the genes do, we don’t know the complete picture of those genes but we are building that information up and I suspect that in another 10 years’ time, we will have a lot more information than we do now.
SiteA, CG2
In order for any database to have maximum effect, the importance of continually and universally updating the information stored was stressed. In order to avoid a fragmented approach and ensure that individual laboratories could access the relevant information, there needed to be ownership of the database, but with collaboration in mind:
. . . as more and more information comes in about what is abnormal and what is normal, that needs to be fed back into the database or somebody needs to hold the database so that each individual lab can get access to the information and that’s gonna be incredibly important otherwise if it’s fragmented round the country, every individual unit is going to go through the same hassle every time they find something that they don’t know what the significance is.
SiteB, FM2
The panel
As part of the EACH study an expert panel was set up to support cytogeneticists and clinical geneticists in decision-making around the reporting of VOUS during the study and the discussion around the panel came predominantly from these professional groups. There were differing opinions as to when the panel should be consulted. It was felt by two clinical geneticists that the panel should be a last resort and that they should make as much of the decision as possible, deferring to the panel only when they were absolutely unsure of the significance of the result. Others from across the health professional group saw the panel as an extension of the multidisciplinary team whose expertise should be called on wherever there was doubt to gain a consensus. However, there was some uncertainty among clinical geneticists as well as fetal medicine consultants at both sites about what they would do if the panel made a decision that they did not agree with, and on one occasion this did actually occur:
. . . in the end they [the panel] said don’t report it, it’s not relevant, but they also said, this is our advice but the final decision is down to you as a team. Thanks for that! We all decided they [the parents] have to know this. The whole thing was a bloody nightmare, erm, but we were all completely happy in the decision that we had to tell this family this information because we would all want to know it.
SiteB FM1
Two participants (clinical geneticists) had consulted the panel about two separate cases and were less than satisfied with the outcomes because they were advised to report something that had only once been reported in a paper as being linked with learning difficulties. The clinical team involved with one case recognised that the panel had to maintain a degree of objectivity but felt that they ought to have considered both the indication for testing (a slightly raised NT) as well as the social circumstances (which were difficult). In addition, the clinical team did not feel that the panel had reached a decision quickly enough and that the length of time they had to wait for a response was inappropriate.
There was some discussion among the health professionals about the extent to which the panel decision should be seen as final and which expertise should be represented on the panel. It was felt that there should be a broad range of experts, including cytogeneticists, midwives, genetic counsellors and lay representatives. One of the health professionals suggested that the panel should also be tested against a set of abnormal variants of known significance to be certain they were consistent. But, overall, there was support for a panel to help clinical decision-making among the fetal medicine consultants as well as clinical geneticists. One consultant in particular felt that the experience of the panel had been positive and that the EACH panel was a success:
. . . my understanding is that, you know, erm, laboratories in consultation with their clinical geneticists are pretty clear what would benefit from going to panel, erm, and the panel has been very successful.
SiteA, FM4
Reporting array results to parents
One cytogeneticist interviewed early in the study expressed concern about only informing parents of abnormal results. They felt that normal results should also be reported in order to reassure parents. Although it was originally decided that laboratories would not release formal reports of a normal CMA result, this decision was changed early in the study because of increasing pressure from cytogeneticists. Clinicians then had the option of reporting the normal result to parents. Reporting emerged as a common theme among the parents who had not been informed and these parents felt that it was inappropriate to have heard nothing at all following the test and would have liked to have received some official notification of the normal result. In some cases, this lack of notification left them uneasy about whether or not the results were in fact normal. This concern about whether or not there was actually an anomaly extended beyond the pregnancy and into the early stages of the child’s life. Some parents remained anxious about the health of their child.
Reporting of an ‘abnormal’ result
Health professionals were concerned that parents would be surprised by an abnormal result. The parent data confirmed that they often felt they had been ‘blindsided’ by an abnormal result because previous tests had given false reassurance:
There’s the initial 2 or 3 days waiting for the three big ones and then there’s this 2-week period where they test, and we didn’t about that part because nobody has any problems with that, and this was when we were lured into a false sense of security ‘cos the woman who, from [tertiary unit], that phoned said that when that result comes back [polymerase chain reaction] and those are all clear [it’s] very, very rare, you know the rest is usually OK.
SiteC, WOM2
This couple received an abnormal array result just a few days after the QF-PCR but were grateful to have been involved in the EACH study because it had given them additional information on which they could base their decision on whether or not to continue the pregnancy.
More generally, when the CMA offered a definitive answer, the parents were pleased to have had the array and felt that they gained a lot from the process. Two sets of parents interviewed had been given a result with a known deletion/duplication, which had a direct impact on their decision to terminate the pregnancy. They were able to gather detailed information on the conditions and became extremely knowledgeable about the difficulties faced by affected children. The risks of disability were taken into account when making these choices:
. . . sort of mental health problems, you know the job I do I work in mental health . . . the kind of schizophrenia that I think apparently comes with [condition] is sort of quite treatment resistant . . . they very often can’t sleep at night because they’ve got leg cramps and you know it’s a small medical issue but in terms of quality of life it’s really important.
SiteC, WOM1
Health professional participants speculated about the factors that influenced parent decision-making and these were generally consistent with the factors described by the parent participants. Although it was generally acknowledged that decision-making was multifactorial, there were a number of important common issues, such as the impact of an affected child on the family and particularly on the other children. In the short term, this centred on the amount of parental time that would need to be dedicated to the affected child, and, in the longer term, this centred on the perceived burden on other children of caring for a disabled child:
. . . if they’ve got unaffected children, erm, then sometimes they think well will it be a burden on the children that I’ve got, I won’t be able to look after the children and take them about and do things that, because I’m going to be in and out of hospital with this child etc., etc.
SiteA, GC1
. . . and we decided not to continue with the pregnancy, erm, just the thought of the newborn having heart surgery and then going on with what, I don’t know, erm, yeah, I didn’t feel . . . and obviously already having one child you know, impacts greatly on them really as well so it’s never an easy decision really.
SiteB, WOM1
The type of disability was discussed by the parents and health professionals as being important. Developmental delay was seen as a major factor in decision-making and more likely than physical disability to lead parents to decide to terminate their pregnancy. As one fetal medicine consultant put it:
. . . parents are far more likely to choose to continue with the pregnancy or at least in my experience they’re far more with a structural abnormality than a risk of developmental delay.
SiteB, FM2
One set of parents reflected the thoughts of many of the couples interviewed. They had made the decision to continue with the pregnancy despite significant structural abnormalities. They stated:
For us it’s more if they’re mentally going to be OK we can cope, physically you just, you just adapt. But I didn’t know how I’d cope with a mentally handicapped child . . . we’re not particularly bothered about a physical disability but we were really, really worried about mental disability.
SiteB, WOM9
In addition to the quality of life for the child, additional influencing factors included the law, the number of existing children, the family circumstances and religious beliefs. Clinicians alluded to the influence of social pressure and how parents often asked what others in the same situation generally did. Perhaps unsurprisingly, the influence of social pressure was not discussed by parents as directly having an impact on decision-making, but was evident in their accounts. For example, one woman spoke of how one of her friends had stopped speaking to her after she terminated her pregnancy. However, she was keen to stress that this friend, as well as others who were quick to judge, did not have a disabled child. Some women experienced feelings of selfishness or guilt for ending a pregnancy; however, for most parents, their motives had been the reverse:
I think you’d have to think very carefully whether that’s the life you’d want for yourself and whether you can watch your child go through that knowing that you made that decision for them to do it, because maybe you weren’t quite brave enough to make the other decision.
SiteC, WOM1
In addition, a woman who had decided to terminate stated:
I almost felt a bit selfish wanting to bring this little boy into the world just for us to have this baby in our arms who was only ever going to have a total struggle for the rest of his life.
SiteA, WOM3
Knowledge of the condition gained through CMA armed the parents with relevant information on the impact the condition would have on these influencing factors. They seemed to base their choices around the risks of the worst possible scenario for the given condition. The data suggest that parent decision-making was multifactorial and that the health professionals involved had a good understanding of the issues that influence parents in their choices. The array was an important part of the decision-making process for parents and enabled them to feel they were making more informed choices.
Theme 4: embedding chromosome microarray into standard clinical practice
The introduction of new technologies
The health professionals discussed the process of developing and introducing new technologies. Using historical examples as a means of illustrating the benefits and potential pitfalls, they cautioned against technology for technology’s sake. The commissioner group in particular were careful to place patients at the centre of decisions regarding the implementation of new technologies. The inevitability of new technologies was seen as something that clinicians would continually face. They discussed examples of technology that were now fully embedded in clinical care but which took time to be widely embraced (such as ultrasound). It was noted that although health professionals are not necessarily requesting new technologies, technologies are continually being developed and patients expect those technologies will become available to them. The commissioner group accepted that clinicians wanted to be at the forefront of technology and that this would ultimately enable them to deliver better care. However, they were keen to ensure that the new technology could offer additional and useful information.
Is it gonna improve the care of the patient? ‘Cos at the end of the day, in theory that’s what we’re trying to do, we’re trying to improve the care and the outcomes that patients get, not to kind of so they can go and meet their mates down in London and brag about this lovely shiny machine that they’ve tricked this gullible commissioner to pay them lots of money to do it, you know.
COMM6
The clinicians interviewed were very positive about the potential impact of CMA for parents, despite their cautions about new technologies in general. Although the use of CMA was seen as ‘new’ in the context of fetal diagnosis, arrays were established in paediatrics. Some of the clinicians had direct experience with introducing CMA in the paediatric setting and, drawing from this experience, discussed some of the perceived benefits. The availability of CMA in the paediatric setting was seen as a success in terms of the diagnostic yield and the ability to give answers to parents:
Arrays have taken our detection rate, erm, of chromosomal abnormality from about 3% of the people that we see, erm, of the kids that we see, erm, in that category of children with, with developmental abnormalities, to 20% with 15% being clinically significant, so that’s a huge sea change in terms of our diagnostic capability.
SiteA, CG4
One commissioner commented that it was important to understand the diagnostic yield of a test as this demonstrated the direct impact. In relation to the paediatric population with unexplained developmental delay, clinicians described CMA as a ‘game changer’, giving answers to families who, for years, had not had a diagnosis. This had not only been a huge relief to families but had, in some cases, enabled them to seek support by way of benefits or through the education system for their child.
Support for the use of chromosome microarray in fetal diagnosis
The majority of the health professional participants welcomed CMA and were very keen to see the success of arrays in the paediatric setting repeated in the prenatal setting. They tended to equate the additional information gleaned with offering better choices to parents. However, there was a sense among some that the array would present counselling dilemmas that they felt ill-prepared for. Overall, there was a sense of inevitability around the introduction of the test and it was felt that there would soon be demand from the general public and consumer groups for the NHS to offer the test:
I’m probably more in favour of it rather than not, erm, because I think it is about, I think we’ve got to be . . . well that’s why we do big studies to get, to get the information really so I’m very yeah, let’s wait and see, but I think you know, because it’s being offered, sometimes in the private sector or in America, one of things is that people go on the internet and they read and they will find out about it and I think we’re gonna start, people are gonna start to ask about it, erm, and we’ve got to be able to justify if, why we’re not offering it.
SiteA, FM3
At the heart of discussion around the support for CMA were the implications for the parents and the necessity of ensuring that they were fully informed prior to testing:
I think there is a significant onus on those who are er helping couples make decisions about whether or not they want to apply the technique to give them all of that information in the way that it’s accessible, erm, but I think provided you do that, then it’s a very positive thing.
SiteA, CG4
Interviewing the commissioners provided valuable insight into how commissioning works following the changes to the commissioning process in April 2013. The quality of the research evidence available was seen as central to ensuring that national recommendations were appropriate:
What we’d be looking for is a recognised, they’re either an RC, randomised control study or a recognised trial that’s published. We’d look at the amount of research that’s being done and we’d look at the quality ….. and we’d use our either our public health colleagues predominantly to help us who are experts in evaluating research evidence to tell us whether they felt that actually there was enough evidence in terms of the type of trial that was undertaken and whether the quality of the research recommendations were such that they felt that we could use it for evidence and obviously if NICE [National Institute for Health and Care Excellence] are also supporting, erm, then we would likely support it.
COMM4
One national commissioner summarised her role:
The only thing is that different clinicians have different ideas, so my job is to get them to agree, so if Joe Bloggs down the road says you know ‘We need to implement micro arrays’ but, erm, Mr Jones down the other end says ‘oh no, no, we’re into new sequencing’, erm, and me in the middle will say ‘Well I want this testing done, I don’t mind what technology you use, however, you’ve got to be the same price and the same outcome and that’s what you’ve got to tell me’.
COMM2
However, participants identified some problems in commissioning arrangements. One area of concern for clinicians was the perceived lack of meaningful communication with the commissioning groups:
Well I think it’s much easier when, when a genetic service has a good relationship with its Commissioners, erm, and that’s gonna be a fundamental stumbling block, erm, because our, my, and my Directorate Manager relationship with the 13 CCGs [Clinical Commissioning Groups] within this region is very limited at the moment because we don’t really know how to contact them.
SiteA, CG5
Some participants identified NHS England specialised services clinical reference groups (CRGs) as a vehicle for introducing CMA into NHS practice. CRGs bring together clinicians, commissioners and public health experts with the patients who use the relevant services; they are responsible for preparing national specialised service-level strategy and developing specialised service contract products, such as specifications and policies. One of the local commissioners stated that both the fetal medicine and medical genetics CRG were supportive of CMA but another local commissioner expressed concern around the role of the CRG. One local commissioner felt that recommendations were made that were unrealistic:
At a national level they don’t get involved in the funding elements of that so one criticism, I suppose, that has been made by some area team commissioners about the national process is that well it’s all very well and good for clinical reference groups to sit in a darkened room and write, gold plated service specification that no one can deliver.
COMM4
Many of the commissioners related their roles back to patient care and delivery. For example, they expressed concerns about equity of access to tests as well as the need for patient feedback regarding aspects of care. One commissioner in particular explained their involvement with the Genetic Alliance and how this had been pivotal to the development of a care pathway for a particular disorder:
. . . for example we did a conference on, er, we wanted to review the care pathways for Marfan’s syndrome, erm, so we invited the specific Marfan’s Patient Association to come along to the whole meeting and participate. Present, erm, give us feedback and we’d go to report at the end which has to be signed off. We tend to work very much with patients because they bring a different perspective.
COMM6
Cost and resource implications of funding chromosome microarray in fetal diagnosis
In terms of the cost of CMA in comparison with conventional karyotyping, there was general agreement that, over time, array CGH would become less expensive. It was acknowledged that the initial outlay would be for equipment. However, as the test would be performed only in regional cytogenetic laboratories that were already performing arrays in the paediatric setting, the equipment was already in place so the additional cost would be negligible. Furthermore, it was pointed out by a cytogeneticist that:
. . . we as a, as a lab now are doing CGH follow-up on fetal loss so some of these EACH samples have actually again have had the array carried out and you know we, we’re able to use that information to you know to sort of say well OK we’ve done this analysis already, we don’t need to do it on this fetal loss material so, I mean it’s actually saving you know.
SiteA, CG2
Health professional participants felt that the time taken to interpret an array was comparable to a karyotype. Many pointed to how the array technology would become less expensive and offer a better diagnostic yield whereas the cost of karyotyping would increase:
It’s not the actual cost of the machinery, etc., but if you look at how many karyotypes you do and how many abnormal results you, because that’s what you’re looking for. I mean it sounds awful but you’re not looking for normal you’re looking for an abnormal.
SiteA, GC1
Professional participants envisaged that CMA would replace karyotyping and this would reduce diagnostic costs. Clinicians directly involved in patient care were mindful of cost issues, but the commissioners in particular focused on the broader issue of negotiating cost priorities. Commissioner participants tended to focus on the difficulties faced by key decision-makers in deciding where efficiencies could be made. Although it was agreed that, in the context of CMA, cost savings would be made in the long term, the savings were seen as quite difficult to actually quantify:
. . . it’s a difficult line to tread if you’re talking about terminating pregnancy and that’s you know, so well if say 5% of cases would, erm, would discontinue, it’s a very difficult line to tread and say ‘Well these are our, you know we won’t have the impact on the NHS for the next 5, 10, 15, however many years’ so, it’s a tricky one.
COMM1
. . . in genetics it’s a diagnosis and a testing service that we provide. That’s all we provide in genetics. It’s quite an expensive, erm, service, because some tests are very expensive . . . if you had the test pre-conception and you make that decision whether you want to ‘cos, erm, then if you want to go ahead and try for a child, and then you can’t quantify, it’s just not feasible.
COMM2
The professionals’ accounts of parents’ reproductive choices were framed with reference to ethical questions and concerns regarding the possibility of eugenic effects. The idea of potentially eugenic effects was an uncomfortable one for health professionals and commissioners. However, they felt that, even if these were set to one side momentarily, from a purely economic perspective it was impossible to quantify the benefit of arrays against the potential long-term cost of not having the test available. Given the various care pathways involved and the different support services that may be involved throughout the entirety of the child’s life, even a basic economic quantification of benefit was seen as problematic. Given the nature of antenatal screening and testing and the inevitability of further development of such technologies, the professionals questioned the value of attempting to quantify economic benefit in simple terms.
Despite widespread support for the introduction of CMA in fetal diagnosis, commissioners used a more corporate language of efficiencies and savings. They discussed how they look for evidence of savings in terms of what it would replace, or what would no longer be paid for, because ‘. . . we don’t have any new money. We have to move it around’ (COMM2). Issues of investment were an area of disparity between the commissioners’ and clinicians’ views. The commissioners were clear that their role was to ensure that patients received the best possible care in the here and now. When asked about CMA and the ongoing information that arrays would provide to clinicians as more and more were performed, one commissioner said:
Well is this not research? And we don’t pay for research. We pay for treatments. We’re paying for diagnostics and treatments. That’s what we pay basically. To get people better, either improve their outcomes or the quality of their life for a longer period.
COMM5
Commissioners seemed less inclined to equate long-term initiatives, such as long-term follow-up and maintaining a central database of information about this, with these patient-focused objectives. In contrast, a clinician explained how advances such as CMA were more of a quality improvement issue:
. . . this is actually a quality improvement, erm, development, so we’re actually inevitably wanting to introduce better and better tests and this is just the next in a series of better tests.
SiteA, CG5
Overall, the discussions focused on balancing the needs of patients in the present with the requirement for developing technologies to continually improve patient care and outcomes in the future. The negotiation of cost priorities was not seen as straightforward, but it was largely understood (among the professional participants) that, as time went on, CMA would become less expensive and less time-consuming and would be a feasible replacement for karyotyping.
Considerations for national policy to roll out chromosome microarray in prenatal diagnosis
Health professionals felt that rolling out CMA on a national basis into standard care pathways would be feasible. There was some variability in the estimated time frames for roll out across the UK, which ranged from 6 months to 10 years, with the average estimation being 2 years for parents undergoing invasive testing for fetal anomaly to have access to CMA.
Professional participants felt that in the units already undertaking arrays, either prenatally or postnatally, there would be a seamless shift from the research to standard care. In units not already performing CMA the shift was seen as more of a challenge in terms of needing to draw on expertise from a range of clinical specialties. There was some caution about the potential for a piecemeal approach to implementation and it was felt that appropriate resources should be invested to ensure equity of access:
The problem is, is, how you fund that and how you make sure equity of access across all fetal medicine centres and you know that’s a challenge for national commissioning really, erm, you know, erm, the only way you can ensure equity of access, erm, is to ensure that it’s commissioned as a service for all patients. Introducing arrays in major fetal medicine centres will be relatively straightforward, erm, it’s likely to happen anyway to a greater or lesser extent, erm, so you will get therapeutic creep because it will be fairly obvious that it’s a cost-effective intervention once EACH is published. How quickly you can get that on to a national platform is gonna come down to specialist national commissioning.
SiteA, FM4
The national commissioners were certainly keen to stress that they too needed to ensure equity of access. One in particular felt that the best way to achieve this was to centralise services:
It should be available, it should be available geographically, I think rather than be everywhere and some, I mean some people who might not have arrays don’t necessarily have the expertise to use them or report on them so it would seem sensible to me that you know, you have a mechanism by which you have some sort of centralisation.
COMM6
In addition to the key issue of equitable access to services, health professionals placed heavy emphasis on the importance of education around arrays in relation to adequacy of services provided. A programme of education was seen as essential to train health professionals who would encounter CMAs, in order for them to be able to convey the information appropriately to parents:
I think they need to have a teaching process to teach us properly . . . it could spiral out of control so there has to be, as well as money being thrown to it from a national perspective, there has to be money thrown to on the education of health professionals, on the education of society per se.
SiteB, MW1
It was also emphasised that written information in different languages should be available for women to take away and consider, and that if health professionals were unsure about CMA then they should know whom the most appropriate person to speak to would be.
Overall, the feeling among the health professionals was that CMA was something that would be welcomed by the majority of stakeholders. They were reassured that the test was being properly evaluated prior to roll-out and felt that the issues they were currently facing (around uncertainty, education, information giving and equity of access) were eminently surmountable. One clinician summarised:
We plot out across the next decade and I know that we will be able to refine the information that comes out of the laboratory to different types of clinicians and those clinicians will get used to it and they’ll work out how to integrate those into their workflows, erm, and in 10 years’ time everyone will be comfortable and happy about this and they’ll look at this time and say ‘Blimey do you remember when we started doing arrays?’ and they’ll rock quietly in their chairs on their verandas and they won’t quite believe how difficult it was because now it’s easy peasy.
SiteA, CG5
The future of prenatal testing
Across the health professional group, there was discussion around what the future of prenatal testing would hold. This discussion focused on NIPT and whole-genome sequencing.
Comparisons were made between CMA and NIPT methods. One fetal medicine consultant pointed to how it is possible to ‘screen the whole human genome from free fetal DNA’ (SiteB, FM2). It was acknowledged that NIPT would be more acceptable to parents, who would negotiate their choice around the potential for more, and possibly uncertain, information and the risk of miscarriage following an invasive procedure. The necessity for an invasive procedure was seen by health professional participants as the greatest risk to the future of array technology:
I think the key difference is between an invasive and a non-invasive and I think the thing most threatening to EACH is not people’s perception of the dilemmas it may put them in, I think it’s purely if it’s an invasive test and I think the future will be much more to do with non-invasive testing.
SiteA, FM3
In the medium term, professionals felt that NIPT would be used for routine aneuploidy screening, whereas CMA would be utilised as the diagnostic test in fetuses with structural anomalies and increased NT undergoing invasive testing. Parents would be balancing the risk of miscarriage following an invasive test with the potential for a false-negative result from a non-invasive test. One fetal medicine consultant explained:
. . . at the moment, non-invasive prenatal diagnosis is essentially focused on Trisomy 21 and specific Mendelian genetic disorders. I don’t think it will make any difference to the group of patients that we’re talking about in the medium term, so I think you know for the next 5 years patients who have a major fetal abnormality detected on ultrasound you know are not going to opt for non-invasive prenatal diagnosis because their risks of having an underlying chromosomal abnormality which are not diagnosed by non-invasive are going to be so high.
SiteA, FM4 (participant’s emphasis)
Fetal medicine consultants and clinical geneticists recognised the additional information provided by whole-genome or exome sequencing but highlighted that the techniques were extremely costly. As a result, they felt that sequencing was unlikely to become widely available in clinical practice in the near future. Health professionals also commented that there would be similar challenges to surmount around the interpretation of results and counselling parents:
. . . but the growth area in the future is going to be non-invasive prenatal testing and CGH array, erm, or, or whole-genome sequencing at some point in the future which almost certainly will become available as it becomes cheaper and cheaper, erm, and then we might identify more and more of, of the funny anomalies, erm, is that good or bad? I don’t know, you know do we really want to remove every child with an abnormality off the face of the planet?
SiteB FM2
. . . what’s on the horizon, erm, is whole-genome analysis and in theory you could replace, erm, chromosome testing and arrays completely with whole-genome analysis and that means you sequencing the entire genome. You would reveal every tiny variant within that person so you can’t really start using this until you understand what those variants mean. If you think array is generating confusion at the moment then the whole-genome analysis in a fetus will generate even more, so I don’t see that array will be the final resting place.
SiteA CG5
The Reliable Accurate Prenatal non-Invasive Diagnosis study
A secondary aim of the EACH study was to contribute maternal blood samples to the RAPID study to evaluate NIPT for aneuploidy funded by NIHR Programme Grants for Applied Research (RP-PG-0707-10107). The results of this programme of work will be reported separately but a brief report on cell-free DNA aspects of the EACH study is provided in Appendix 5.
Chapter 4 Discussion
The EACH study was designed to evaluate the clinical effectiveness and cost-effectiveness of prenatal CMA compared with karyotyping within the UK’s NHS. The study involved 8 of the 17 NHS regional genetics laboratories (representing six of the nine NHS Genetics Services in England) as well as the Welsh Genetics Service. All the participating laboratories had experience of running CMA on postnatal samples and, therefore, prenatal samples were integrated into existing postnatal laboratory array workflows. Through the NHS FASP all women in the UK are offered combined ultrasound and biochemical screening for common aneuploidies at 11–14 weeks’ gestation and ultrasound screening for structural anomalies at 18–20 weeks of pregnancy. 2 Women at a high risk of chromosomal anomalies are offered invasive testing with karyotyping of chorionic villus or amniotic fluid samples in QF-PCR-negative cases. 2 Invasive testing and assessment of structural anomalies is undertaken by fetal medicine specialists working in large district hospitals or regional specialised fetal medicine centres. Cases for the EACH study were recruited from 10 of the largest specialised centres in England and Wales as well as 10 smaller units, reflecting current UK practice. The design of the EACH study was therefore predicated both by the existing national prenatal screening/diagnosis programmes and pathways but also by the published prenatal CMA data20 that confirmed that targeting fetuses with ultrasound anomalies would optimise the yield of clinically significant cryptic chromosomal anomalies.
The study has shown that, within this UK context, prenatal CMA in fetuses with a structural anomaly and a normal QF-PCR detects 3.5% more pathogenic CNVs than karyotyping. This rate is consistent with the early meta-analysis of Hillman et al. ,36 which informed the EACH study, and their subsequent BAC array study conducted in Birmingham, which reported pathogenic CNVs in 4.4% of 243 fetuses referred with a structural anomaly on ultrasound. 56 It is also consistent with the large National Institute of Child Health and Human Development (NICHD)-sponsored trial which performed oligonucleotide- or SNP-based arrays in 755 cases with an ultrasound anomaly; 21 (2.8%) were classified as having a pathogenic CNV. 57 However, this is lower than the most recent systematic review of 18 studies (3359 cases), published in 2014, which reported a pooled prevalence of pathogenic CNVs in fetuses with one or more structural anomalies of 6.8% (95% CI 6.0% to 7.7%). 58 This review was dominated by the experience from a single laboratory in the USA which reported pathogenic CNVs in 159 out of 2184 (7.3%) fetuses with ultrasound anomalies. 59 Of note, a recent study from Belgium, which utilised 60,000 or 105,000 oligonucleotide arrays in 383 cases with undefined ‘ultrasound abnormalities’, identified submicroscopic ‘causal’ CNVs in 2.6% of cases, while CMA added valuable information over conventional karyotyping in another 3.9% of cases. 60
Currently, the NHS FASP recommends the offer of invasive prenatal diagnosis when ultrasound screening identifies a NT of ≥ 3.5 mm. 4 The EACH study therefore sought to address the effectiveness of CMA in this specific group. Although CMA detected 4.5% more CNVs than karyotyping in fetuses with an increased NT, there was no difference in the rate of pathogenic variants. This finding was consistent when the NT cut-off point was increased to 5 mm or 6 mm. However, the rate of pathogenic CNVs on CMA was higher in fetuses with a NT of > 5 mm than in those with a NT of 3.5–5 mm. We are not aware of any other studies that have looked at the effectiveness of CMA at different NT cut-off points, although De Wit et al. 58 found no difference in the rates of pathogenic submicroscopic CNVs in fetuses with a NT of > 3.5 mm and those with a cystic hygroma. Grande et al. 61 have recently published a meta-analysis of 17 studies (1696 cases) addressing the incremental yield of CMA over karyotyping in fetuses with an increased NT. When a NT cut-off point was defined, most authors used 3.5 mm and included cystic hygroma, as in the current study. Overall, 5% more pathogenic CNVs were detected by CMA. In the subgroup of fetuses with isolated increased NT (1403 cases with no additional structural anomaly), the pathogenic CNV detection rate was 4% (95% CI 2.0% to 7.0%) higher with CMA. In the largest single study to date which reported CMA results in 215 fetuses with a NT of 3.5–9.0 mm, CMA detected CNVs in 1.4% (95% CI 0.5% to 4.0%), but all three cases were classified as VOUS. 62 These findings are also consistent with the NICHD trial, which included 187 cases with a NT of ≥ 3.5 mm or a cystic hygroma or a NF of > 6 mm; the rate of CNVs not detected by karyotyping (3.8%) was no different to that in the control group (for which CMA was performed for advanced maternal age in the absence of any detected fetal anomaly). 63 Taken together, the results suggest that the added diagnostic yield of CMA over karyotyping is significantly lower in fetuses with an isolated increased NT than in fetuses with a structural anomaly (with or without an increased NT).
The results of the EACH study add to the growing evidence of the improved diagnostic accuracy of CMA compared with karyotyping for the detection of numerical and structural chromosomal imbalances in prenatal diagnosis. In a recent systematic review, using the combined rate of pathogenic imbalances detected by karyotyping and CMA as the reference standard, Saldarriaga et al. 64 reported a higher sensitivity (0.942) for CMA than for karyotyping (0.673) with the same specificity (0.999). Negative and positive likelihood ratios were also improved (0.049 vs. 0.291 and 1340 vs. 860, respectively).
The two recent meta-analyses discussed above both showed statistically significant heterogeneity between studies. 56,61 This is likely to reflect the inclusion of smaller cohorts with an artificially high detection rate. 56 It is noteworthy that the results from the EACH study agree more closely with more recent, much larger, studies. However, differences in anomaly case selection, CNV calling strategy and array platform may also have contributed to the lower rates of pathogenic CNVs, than in the published meta-analyses, found in the current study.
The rate of pathogenic CNVs is known to be higher in fetuses with multiple sonographic anomalies. In De Wits’ recent systematic review of studies in fetuses with structural anomalies and a normal karyotype,58 9.1% with multiple anomalies had a pathogenic CNV, compared with 5.6% of those with an isolated anomaly. In the EACH study, 11.1% of fetuses with multiple anomalies had a pathogenic CNV on CMA compared with 8.8% of those with one anomaly, although this difference was not statistically significant. A similar pattern was found in the NICHD study, in which CNVs (both pathogenic and VOUS) undetected by karyotype were more common in fetuses with multiple (n = 206) than isolated (n = 312) anomalies (13.6% vs. 6.7%). 63 We also confirmed the specific benefit of CMA in fetuses with a cardiac anomaly. 63,65,66 Other CMA studies have reported pathogenic CNVs in 2.0–6.6% of fetuses with a cardiac anomaly, normal karyotype and negative or no FISH results for 22q11.2 deletion syndrome. 65,66 Yan et al. 66 found the rate of pathogenic CNVs in this group was not affected by the presence of additional structural anomalies.
Considerable effort went into optimising the laboratory protocols before recruitment to the EACH study commenced. A detailed technical evaluation was performed by scientists at two of the participating laboratories (WRGL and NETRGL), aimed at developing an agreed technical workflow for performing CMA using minimal quantities of amniotic fluid (2–4 ml), chorionic villi (2–5 mg) and cultured cells (< 150,000 cells). Guidance was provided about optimal methods of DNA extraction (using the iGENatalTM kit) and DNA quantitation (using Qubit® 2.0 Fluorometer and Quibit® dsDNA BR assay kit)48 to use with the agreed array design. As a result, 98.9% of arrays in the EACH study were performed on uncultured samples. This is a substantial improvement on the NICHD study in which the CMA result was derived from cultured cells in 21.5% of cases. 57 The array design chosen was based on the ISCA Consortium’s consensus49 and consisted of an 8-plex of 60,000 60-mer oligonucleotides with a backbone resolution of ≈75 kb but with considerably higher coverage over telomeric and pericentromeric chromosomal regions, in known microdeletion/duplication syndromic regions and also for a number of developmental and haploinsufficent genes. Thus, the array design was felt to be optimal to detect known microdeletion and duplication syndrome regions as well as other chromosomal imbalances. The laboratories obtained their arrays from a number of commercial vendors and, in most cases, used the analytical software supplied with the arrays. With the described laboratory workflow, 3.3% of arrays failed (vs. 0.8% of karyotypes). This is higher than in other recent studies56,57 and, in part, may reflect the introduction of prenatal arrays in addition to karyotyping in busy NHS genetics laboratories but also may be due to the relatively small amounts of amniotic fluid and chorionic villi made available for DNA extraction (e.g. ≈5 ml of amniotic fluid in EACH vs. 20 ml exclusively available for DNA extraction in the NICHD study57). In fact, the preliminary work carried out by Callaway et al. 48 was undertaken in anticipation of this problem and almost certainly contributed to the relatively high success rate with less then optimal volumes of starting fetal material for CMA.
The interpretation of CMAs and the strategy for calling CNVs is challenging. The identification of novel VOUS, and the perceived difficulties these cause for clinical management, is seen as the most significant disadvantage of prenatal CMA. To facilitate accurate and timely interpretation of prenatal CMA, the EACH study was designed so laboratories had ready access to parental DNA; blood samples were collected at the time of prenatal diagnostic procedure from 100% of mothers and 78% of fathers. Thus, for most identified CNVs, parental inheritance could be determined from a single parent versus parent array targeted at the area of imbalance. When necessary, FISH-ready clones were made rapidly available by WRGL. In-house interpretation of CNVs has been facilitated by the development and evolution of online international databases with results from normal individuals (e.g. Database of Genomic Variation67) and those with phenotypic anomalies (e.g. DECIPHER68). As laboratories have increased their experience of prenatal CMA many have also developed databases cataloguing CNVs. Through continuous updating of clinical CMA databases (e.g. ISCA Consortium database69), understanding of genotype/phenotype correlations and the clinical significance of novel CNVs is expanding rapidly with an emphasis on penetrance of key phenotypic traits associated with an increasing number of CNVs. 70 To reflect current laboratory practice, local laboratory scientists used their standard in-house interpretation protocols including minimum size cut-off points for deletions and duplications. In this context, deletions as small as 5.5 kb and 11 kb were detected and included in the reported cases (see Tables 15 and 16). The exception to this was the agreed list of low-penetrance CNVs excluded from reporting. Furthermore, to provide additional support to local genetics services in making CNV calls and reporting decisions, an expert panel made up of five consultant clinical cytogeneticists and five consultant clinical geneticists, was available to offer advice when requested. This strategy was chosen to reflect how CMA may be introduced into the NHS. It differs markedly from the NICHD trial, for which all CMA results were interpreted at a single centre and all VOUS were referred to an independent advisory panel.
Variants for which the possible genotype and phenotypic effects were uncertain (VOUS) were not reported in the EACH study. Overall, 38 (3.4%) cases were called as VOUS and the rate was similar in the two study groups. In the majority of cases (58%), the decision was made in-house by the local cytogenetics/clinical genetics team while, of the 32 cases referred to the EACH panel because of local uncertainty, half were not reported (i.e. regarded as a VOUS). The VOUS rate in the EACH study is very similar to the rate reported in the NICHD trial, in which 24 out of 755 (3.2%) arrays performed for an ultrasound anomaly were interpreted as ‘potential for clinical significance’. 57 These VOUS rates are higher than both the rates reported by Hillman et al. 56 in their meta-analysis of studies for which the clinical indication for CMA was a structural anomaly (2.1%, 95% CI 1.3% to 3.3%) and the rates reported in the Leuven study (1.6%) of oligonucleotide arrays in fetuses with ultrasound anomalies. 60 It is also higher than the rate reported from the meta-analysis of increased NT studies (0.8%, 95% CI 0.4% to 1.3%). 61 This is likely to reflect the 60,000 oligonucleotide array design used in the EACH study in comparison with several studies included in both meta-analyses that used BAC arrays. VOUS rates are known to increase as the resolution of the array increases. Hillman et al. 71 compared CNV detection rate in 62 fetal samples using two array platforms (1 Mb targeted BAC array and 60,000 oligonucleotide array). The 60,000 array detected 4.8% (95% CI 1.6% to 13.3%) more pathogenic CNVs but 8% (95% CI 1.3% to 14.8%) more VOUS. 71 Using information on CMA genome coverage (rather than a direct comparison in the same fetal samples), Shaffer et al. 70 estimated that use of a 55,000 array would reduce VOUS rates by 32% compared with a higher resolution 135,000 array while still detecting all but one pathogenic variant (98%). Thus, we believe that the array design employed in the EACH study represents a sensible compromise between pathogenic and VOUS detection rates. It is expected that, as experience of prenatal CMA increases, the number of VOUS calls will reduce. 61
The second part of the laboratory evaluation within the EACH study was to compare TATs. To reflect NHS laboratory practice, we adopted the definition used for CPA (which is one of the services within the UK Accreditation Service), that is, from the date of sample receipt to the date of the final report. Based on this definition, result times for karyotyping were consistent with the UK average (12 days) and times for CMA were consistent with previous research studies. 72 CMA results took a median of 3 days longer than karyotyping (15 days vs. 12 days), although there was a large variation between laboratories. When further testing was required to interpret the array, median TATs were increased to 22–26 days, depending on the test; however, we acknowledge that this may not reflect TAT if CMA were to replace karyotyping. The TAT, as defined for CPA, is also predicated on whether or not rapid aneuploidy detection by QF-PCR is performed prior to setting up the array. We therefore also compared times from set-up to report; in this analysis CMA was 5 days quicker than karyotyping with a median time of 6 days. There remained substantial differences between laboratories (with median times ranging from 2 to 9 days) suggesting that, with greater clinical experience, a median TAT of 7 days is achievable.
There was complete concordance between the detection rate of cytogenetically visible euchromatic unbalanced chromosomal abnormalities and the results obtained by CMA. As expected, array CGH failed to detect 15 visible cytogenetic abnormalities, the majority of which were apparently balanced rearrangements and, therefore, no euchromatic gains or losses were observed on the matched array. 73 The proportion of cases with an abnormal karyotype but normal CMA (1.3%) is comparable to the Hillman et al. 56 series from the UK (2%) and the NICHD study (0.9%). 57 Balanced translocations and inversions are reported in < 0.1% of prenatal diagnostic samples,74 suggesting that such changes are more common in prenatal series selected for fetal anomaly. Although an inherited rearrangement with a normal CMA would not explain ultrasound findings in the index pregnancy, the finding is of relevance to future reproductive counselling. Counselling parents with a de novo apparently balanced rearrangement is more challenging, especially when the indication for testing is an increased NT with no structural anomaly. In a large cytogenetic series, in which amniocentesis was performed primarily for maternal age, the risk of a serious anomaly in fetuses with an apparently balanced de novo rearrangement varied from 3.7% for Robertsonian translocations to 9.4% for inversions. 75 Although many such cases will have an imbalance detected on CMA, either at one or more breakpoints or unrelated to the translocation/inversion,76 the residual risk in those with a normal CMA is unclear.
Confined placental mosaicism, in which the karyotypes of the placental and fetal tissues are divergent, has been extensively reported and reviewed. 77 It occurs in up to 2% of chorionic villus samples (compared with < 0.5% of amniotic fluid samples78). Overall, within the EACH cohort there were three cases with CPM (UCH312964, Birm3011 and SGH 86084) that were recognised prenatally and not reported. There were two further cases in which the CPM was only finally recognised postnatally. The first was a Xp22.31 steroid sulfatase enzyme deletion (UCH1285528) and was reported prenatally. Follow-up studies after delivery showed that the Xp22.31 deletion was confined to the placenta. 79 The case prompted an observation by Kooper and Faas80 that similar false positives could be avoided if the source villi were dissociated into two separate cell suspensions of cytotrophoblast and mesenchymal cells using an adapted version of the protocol of Mann et al. 81 This allows testing of DNA from both fractions whenever mosaicism is suspected. The second case (CAM62826) was reported prenatally to be a trisomy 7 mosaic on karyotyping and non-mosaic trisomy 7 using CMA. Postnatal karyotyping of peripheral blood found no evidence of trisomy 7, confirming CPM. These two cases, both with a normal outcome, represent the only false positives within the EACH study giving a frequency of 2 out of 1123 (0.18%). Aside from these CPM cases, there were three additional cases in the EACH study in which an apparent anomaly was identified by CMA (SGH 85534, QCH130141 and UCH1176628) but follow-up studies found to be cultural or technical artefacts. In addition, there was one case with a benign heterochromatic variant, t(Y;15) (NCL12275). All four cases were reported as normal.
An increasing number of prenatal studies are using SNP-based arrays. Srebniak et al. 82 summarised the advantages of SNP-based arrays over non-SNP arrays in the prenatal setting. SNP arrays can detect triploidy and MCC, although this is less important in the current UK context, where rapid aneuploidy detection with QF-PCR is recommended. With male fetal samples interfering levels of MCC can be excluded when the sex chromosome plots are normal, but this is not possible with a female fetus and MCC testing is recommended to be confident that the results are reflective of the fetal DNA. 83 However, depending on the array and software design, small CNVs can be detected with varying levels of MCC. 81 SNP arrays are also reported to be able to detect levels of mosaicism as low as 5% depending on origin. 82 However, recent studies have confirmed that oligoCGH arrays can detect mosaicism at a level as low as 10%. 84,85 Finally, in contrast to oligoCGH arrays, SNP arrays also offer the advantage of detecting loss of heterozygosity; large regions of homozygosity throughout the genome can suggest consanguinity while regions of homozygosity involving chromosomes 6, 7, 11, 14, 15 and 20 can suggest clinically relevant UPD. 82 In one of the 15 abnormal karyotype/normal CMA cases, additional molecular studies demonstrated paternal UPD(14) of the chromosome 14 homologues in association with mosaicism supernumerary heterochromatic marker 14 chromosome86 and represented the one case in the EACH study for which use of a SNP array would have provided a more comprehensive diagnostic utility. 87,88
The health economic analysis indicated that the mean cost per test was higher for CMA than for karyotyping (£322 vs. £234–272), as was the total cost of the pathway, including laboratory follow-up tests and clinical follow-up (£599 vs. £486). CMA detected more pathogenic CNVs than karyotyping in the structural anomaly group than the NT group (0.035 vs. 0.010 more per pregnancy) and, hence, the incremental cost per pathogenic CNV detected by CMA compared with karyotyping was lower in the structural anomaly group (£3635 vs. £9439). Results were sensitive to the number of pathogenic CNV results found by CMA, the number of abnormal karyotypes and the cost of CMA. If the NHS was willing to pay an extra £10,000 to detect an extra pCNV, then CMA would be cost-effective on 87% of occasions. Few economic evaluations of CMA are available for comparison. Hillman et al. 89 evaluated the cost-effectiveness of CMA compared with nine combinations of conventional techniques in the NHS using data from a cohort of women with a structural anomaly. Testing strategies including CMA detected the most CNVs. Using a base-case unit cost for a 1-Mb BAC array of £405, the authors calculated that, compared with QF-PCR followed by karyotype [if the polymerase chain reaction (PCR) was negative] followed by FISH for 22q11.2 deletion syndrome (if the karyotype was negative and the fetal anomaly was cardiac related), CMA cost an extra £24,600 for every additional CNV. Compared with karyotyping alone, CMA cost an extra £33,300 per pathogenic CNV detected, which fell to £9768 if the unit cost of CMA was £360. The authors concluded that CMA was not cost-effective, although they acknowledged that it may become so in future if the costs of CMA fall; the EACH results support this (in the EACH study, the unit cost of CMA was £322). Hillman et al. 89 included both pathogenic CNVs and VOUS in their definition of additional cases detected. When VOUS were treated as false positives, CMA was dominated by all other strategies (i.e. it was more costly and less effective). The authors noted that treating all VOUS or false positives would underestimate the specificity of CMA because some VOUS would in time be determined to be pathogenic. Definitive judgements about the cost-effectiveness of CMA are problematic because it is unclear what the NHS is willing to pay to detect an additional CNV.
The qualitative substudy aimed to inform the development of any policy product from the main study by providing an evaluation of the acceptability of CMA to major stakeholders. It brings together parents’ perspectives with those of health-care providers (i.e. cytogeneticists, clinical geneticists, genetic counsellors, midwives, fetal medicine consultants and commissioners). No previous studies have evaluated CMA across this range of experiences. Two prior studies have reported parents’ experience of prenatal CMA; Bernhardt et al. 90 conducted 23 telephone interviews with women in the USA who received positive array results. Sixteen of the group had prenatal testing for maternal age or positive screening results. In the UK, Hillman et al. 91 conducted 25 interviews with women after receiving the results of CMA, most of which were normal. In contrast, the women in our study were interviewed at least 3 months after the outcome of the pregnancy, allowing participants to comment on their experience of decision-making after a longer period of reflection. Participants were generally supportive of the process of prenatal diagnosis in general and the use of CMA in particular. However, in each of the participant groups, actual and potential problems were raised for discussion, which clustered around three key issues.
The first was around achieving integration of CMA into the existing care pathways. Accessing CMA involves referral to a fetal medicine centre. Health professional participants were concerned that delays in referral could reduce options for women in terms of termination of pregnancy for fetal anomaly at later gestations. The time between identification of a problem and referral to a specialist centre is a time of great stress for parents and a time when appropriate support is needed. 92 Participants highlighted many advantages of CMA already reported in the literature, particularly the availability of more detailed genetic information. 36 Furthermore, CMA was perceived to be a quicker test than karyotyping. Even when a novel variant was identified, the reporting time was felt to be no slower than karyotyping. Moreover, several health professionals acknowledged that, as more information became available about CNVs, reporting time for arrays was likely to reduce. 25,93,94 The management of VOUS was seen by health professionals as the main challenge associated with implementation of CMA, which is consistent with previous literature. 95–97 Professionals were primarily concerned with ensuring that parents who take up the offer of an array are well informed about the test, particularly the potential for a VOUS. Education programmes for health professionals and appropriate literature for parents were recommended, reinforcing recommendations from other studies of CMA in prenatal diagnosis. 90,91 Commissioners provided a unique insight into the factors that influence commissioning decisions around new technologies. Evidence of clinical utility, improved diagnostic yield and the actual cost featured high on their list of priorities. However, they were also keen to ensure that there was equity of access to care and patients appeared central to their commissioning decisions in line with current policy. 98 Finally, many participants highlighted the future impact of NIPT on care pathways, acknowledging the increased risk of miscarriage associated with invasive prenatal diagnosis. Although NIPT was seen as an appropriate part of the aneuploidy screening pathway,99 several professional participants commented that the higher diagnostic yield of CMA in cases with an ultrasound anomaly meant that this technology was more appropriate for informing pregnancy management decisions.
The second issue was around informed consent. Parent participants had variable understandings of the EACH study. Consistent with prior research,100 some parents participated in the research for altruistic reasons and there was some lack of recognition that the results could potentially have an impact on their own pregnancy. Many parents recalled being told about a concurrent study on NIPT and, although some parents’ accounts demonstrated understanding of CMA as a genetic test, they mistakenly thought it would screen for all genetic abnormalities. These sorts of possible confusion related to CMA have been reported previously90,99 and were raised as concerns by health professionals. Even with apparent understanding at the time of consent, parents who opt for the test can still be surprised by an abnormal result and disappointed when health professionals are unable to interpret the results. 90 Similarly, parents interviewed for the EACH study described feeling lured into a false sense of security by a normal karyotype result and, hence, were shocked by the array result. 89 This highlights the difficulties of offering a test that is additional to standard care in a research study. Parents are unprepared for receiving such news92,101 and, when parents have not fully understood the implications of their participation in research, this can be more problematic.
Despite many parents not fully understanding the array test, and some experiencing shock after receiving an abnormal array result, they tended to accept it on the basis that CMA offered additional information. The potential for uncertain information was either not an immediate concern for parents in the EACH study or was a risk they were prepared to take in seeking clarification/reassurance. Many described gaining a better understanding of potential problems which directly informed their decisions about pregnancy, which was experienced as empowering. The premise that ‘knowledge is power’ prevailed, which was consistent with previous studies, even though this knowledge may, in the end, be seen as ‘toxic’. 90,91 Our findings suggest that informed consent for CMA is challenging because the information is just one facet of a complex scenario, which itself is a time of emotional distress for parents. Distress can have an impact on parents’ recollection of what was said to them92,101 and this effect seems likely in this study.
The management of uncertain prognosis underpins much of the literature on prenatal diagnosis and it featured in discussions about test integration and informed consent. CMA does not necessarily offer certainty in terms of diagnosis or prognosis, raising questions about how much uncertainty can be tolerated given the potential decision to end a pregnancy. Professionals in particular were concerned about these issues; adopting an inclusive, interprofessional team approach to shared clinical decision-making was an important mechanism for managing this concern. This approach is associated with more collaborative team behaviour,102 leading to improved continuity of care and patient satisfaction. 103 Our study suggests that clinicians, especially fetal medicine consultants, value this collaboration more when they are faced with uncertainty. Collaborative working was important to them in ensuring that parents received the best possible information. Other strategies for managing uncertainty were raised. A few health professionals who had faced dilemmas over VOUS saw the use of targeted arrays as a way to reconcile the ambivalence around uncertain results while acknowledging that this strategy did not remove all reporting uncertainty. 85 The majority of the health professionals were keen to see CMA develop in the longer term and to gain more information to offer to parents. Developing collaborative databases of information and ensuring long-term follow-up were seen as fundamental requirements. The EACH study included a review panel to assist in decisions about reporting CNVs. Clinician experience of, and satisfaction with, the panel was variable and there was some ambiguity about the role of the panel in relation to decision-making. Although, in principle, the panel is an excellent resource for clinicians faced with uncertainty about the clinical significance of a CNV and whether or not to report it, clear guidance on what role the panel has in the final decision needs to be developed to increase the professional user experience.
Finally, linked to the issue of managing uncertain prognosis, both parents and health professionals identified the importance of reporting of results. CMA results were reported only if it was felt that the CNV had a high probability of a phenotypic effect. This left some women with a ‘normal’ result unsure; they had understood that there would be longer-term results from the study, albeit that many seemed to have misunderstood the intricacies of the test. However, women who received prenatal testing often remained anxious about the health of their child104,105 and not reporting a result has the potential to add to the concerns of an already worried parent.
The geneticisation of health and disease has been identified as both dominant and problematic in how the social categories normal and abnormal are defined. 106 Much of the literature on user experiences of CMA stems from its use in those with an established personhood status, for example children with developmental delay and/or congenital anomalies. In this context, the concept of informed consent is of critical importance to the understandings of the ethical and social acceptability of genetic testing as a medical practice. The issue of informed consent recurs in the literature on parent perspectives of prenatal diagnosis. 88,91 The dominant biomedical narratives for prenatal diagnosis utilise notions of control, choice and reassurance, but these narratives have been criticised for being too simple. 106 Factors such as strong social support and religious beliefs have influence regardless of social context while class and ethnicity are affected by social structures. The notion of choice in prenatal diagnosis has been problematised because, following the diagnosis of a serious fetal anomaly, parents have a time-limited decision about whether to continue or end the pregnancy. For some parents, this is problematic because it is conceptualised as an ‘involuntary choice’101 and, for others, the problem lies with the lack of true alternatives to termination. Decisions after prenatal diagnosis of fetal anomaly are, therefore, dependent on social context as well as individual views. Underpinning these debates is the idea that the social context in which western populations live make it difficult to say ‘no’ to new developments38,101 because the goal of a ‘healthy baby’ is perceived to be a self-evident good. 107 As genetic technology has progressed, so the options for prenatal diagnosis have multiplied. Therefore, CMA is best conceptualised as a recent innovation in this dynamic sphere of activity. Many of the issues raised in the analysis of the EACH data are best understood as a part of a much bigger social and medical phenomenon. However, their occurrence in this novel situation provides opportunities to understand how those existing dilemmas have shaped the acceptability of the intervention in the study and how that may, in turn, shape policy in the future.
The EACH study has a number of strengths. It was designed to detect a clinically important difference in cryptic chromosomal imbalances in two distinct groups of fetuses with ultrasound anomalies after exclusion of a common aneuploidy by QF-PCR. This large cohort of women was recruited from 20 fetal medicine units with samples analysed in nine cytogenetic laboratories across England and Wales. Pilot work optimised laboratory workflows and a single oligoCGH array design was chosen, based on ISCA consensus, which optimised detection of clinically relevant imbalances while minimising VOUS rates. CNV calls and reporting decisions were made by local genetic teams but advice was available from an expert panel for challenging cases. The results are therefore likely to genuinely reflect the impact of replacing karyotyping with CMA in clinical practice. The study also included an economic analysis which investigated the cost-effectiveness of such a replacement strategy using main unit costs averaged from all nine participating laboratories. The EACH study also included an embedded qualitative study to determine the factors that influence parents’ and health professionals’ choice and decision-making about CMA in this context. Importantly, the sample included professionals making commissioning decisions about prenatal testing.
The study also has a number of limitations. Prenatal arrays were undertaken in addition to karyotyping on ‘routine’ clinical samples in busy NHS laboratories. As a result, limited amounts of fetal material were available for DNA extraction, which accounts for the relatively high CMA failure rate (3.4%) in the EACH study. Furthermore, TATs were calculated using the CPA standard for NHS laboratories even though arrays may not have been set up as soon as possible after sample receipt. Therefore, a more clinically meaningful time was calculated from the time of array set-up to final report. Unlike many prenatal array studies, we attempted to collect pregnancy outcome for all cases but this was not available in 32% of the cohort. However, we were able to get delivery details and some paediatric information from > 95% of the cases with an abnormal karyotype and/or CMA. Understandably, termination of pregnancy rates were very high but surprisingly, given the indications for testing, many parents opted not to have post-mortem examination, limiting the phenotypic information available for a significant number of infants. The main limitation of the health economic analysis was that cost-effectiveness was measured in terms of the incremental cost per pCNV detected. Although this is an appropriate measure for evaluating CMA, it means that it is difficult to make a firm judgement about whether or not CMA is cost-effective because it is unclear how much money the NHS is willing to pay to detect an additional pathogenic CNV. Further research to identify how much the NHS is willing to pay to detect an additional pCNV would be beneficial. In addition, we considered only costs during pregnancy, not following childbirth and, therefore, we did not capture all the potential costs and benefits associated with detected additional pCNVs. Moreover, the study was designed so that participating women underwent both karyotyping and CMA. This is an appropriate study design to investigate the diagnostic accuracy of CMA, but it meant that when the results of CMA and karyotyping were discordant it was difficult to attribute the subsequent laboratory follow-up tests and clinical follow-up to each option. It is acknowledged that, although the sample size for the qualitative study was sufficiently diverse for the purpose, few parents with a ‘normal karyotype and abnormal CMA’ were interviewed. The narratives of the two couples that did fall into this category offered a unique insight into the benefits of CMA as the test directly informed their decision to discontinue the pregnancy.
The EACH study began recruiting in May 2012. Since then there have been significant advances in prenatal testing for chromosomal anomalies which will have an impact on future implementation of CMA as well as future research. Numerous studies summarised in a recent meta-analysis,108 as well as the NHS RAPID project,47 have demonstrated the effectiveness of cell-free DNA in maternal blood to screen for trisomies 21, 18 and 13 using either sequencing (massively parallel or chromosome specific) or SNP-based methodology. As a result, in November 2016, the Department of Health announced the introduction of cell-free DNA as a second stage non-invasive screen for these trisomies. 109 Benachi et al. 110 studied the potential impact of cell-free DNA in 387 cases of structural anomaly (which included 193 cases with increased NT) and out of the 290 cases with a normal cell-free DNA analysis, 23 (7.9%) had additional pathogenic karyotypes. Unfortunately, CMA was not performed. The application of cell-free DNA is already being extended to sex chromosome aneuploidies111 and common microdeletion syndromes. 112 Introduction of cell-free DNA testing is anticipated to dramatically reduce the number of women undergoing invasive testing for chromosomal anomalies. In 2012, the first report of whole-genome sequencing of amniocytes from a fetus with multiple anomalies was reported113 and, since then, further cases of prenatal exome sequencing have been reported, including a series of 30 prenatal and neonatal samples from cases with structural anomalies from Birmingham. 114 Targeting exons has the advantage that only 1–2% of the genome is sequenced, but these key protein-coding regions contain up to 85% of mutations known to cause genetic disorders. In 2013, the Prenatal Assessment of Genome and Exomes (PAGE) project was funded with the aim of analysing 1000 exome sequences (with whole-genome sequencing in a smaller cohort) from a fetus undergoing invasive testing because of a structural anomaly. 114 Samples will be collected from fetal medicine centres throughout the UK, building on the EACH collaboration, and PAGE will identify what additional information exome sequencing provides over CMA. Further studies will be needed to determine whether or not sequencing should replace CMA and, ultimately, if this technology can be applied to detect other chromosomal imbalances in cell-free fetal DNA in maternal blood. 115
Conclusions
The EACH study was designed to provide guidance to health service providers on whether or not CMA should replace karyotyping in the prenatal diagnosis of fetal anomalies. The laboratory evaluation, conducted in nine cytogenetic laboratories across England and Wales, showed that, in the group of fetuses with a structural anomaly, array CGH detected 3.5% more clinically significant CNVs than karyotyping. Although CMA also detected more CNVs in fetuses with increased NT, the increase in pathogenic variants did not achieve statistical significance. Laboratory TATs were slightly longer for CMA than for karyotyping owing to the design of the study. However, analysis of actual set-up to reporting times indicated that, in clinical practice, CMA results would be available on average within 7 days. Cost calculations indicated that, per patient, CMA is on average £113 more costly than karyotyping. Whether or not CMA is cost-effective depends on how much money the NHS would be willing to pay to detect an additional pathogenic CNV. CMA is likely to be more cost-effective in cases with a structural anomaly than in those with increased NT. The qualitative evaluation of attitudes to CMA suggest that parents find the technology acceptable, despite the uncertainties it may introduce, and that, generally, it is acceptable to health professionals and commissioners. However, it will be important to ensure health professionals are able to better inform parents about CMA in order to enhance patient understanding of the implications of the test. The sharing of information on detected variants and the associated phenotypes and outcomes is seen as important to ensure consistent interpretation of CMA results.
Chapter 5 Implications of research
Summary of implications for practice
-
The evidence from the EACH study suggests that CMA should replace karyotyping in care pathways when the indication for fetal testing is one or more structural anomalies or an isolated NT of ≥ 3.5 mm on ultrasound scan after a normal QF-PCR result. Replacement for both indications will ensure there is a consistent prenatal (and postnatal) diagnostic technology.
-
In order to achieve a detection rate greater than when using karyotyping, the experience from the EACH study suggests that a minimum array-CGH resolution of ≈400 kb throughout the genome is optimal.
-
The findings of the EACH study suggest that any variant that can be linked to a potential phenotype of the future child on the basis of the genes involved should be reported, regardless of the size of the imbalance. This should include high-penetrance neurosusceptibility loci. Other (incidental) variants should not be reported.
-
The evidence suggests that until more national (and international) information on prenatal variants has been collected, specifically linking clinical phenotypic and molecular data, there may be benefit in a national advisory group that can provide expert advice to local health-care professionals about variants of possible pathogenic significance. Such a group could also identify incidental findings not to be reported. The operating model of the EACH review panel could serve as a template for such a group.
-
To ensure that consistent, high-quality, information is provided to parents and to help support shared decision-making, consideration should be given to producing a national information sheet and consent form.
Since completion of the EACH study and taking account of the findings, the Joint Committee on Genomics in Medicine published recommendations for the use of CMA in pregnancy in June 2015. 116 The recommendations were endorsed by The Royal College of Pathologists in collaboration with The British Society for Genetic Medicine, The Royal College of Obstetricians and Gynaecologists and The British Maternal and Fetal Medicine Society. The implications for practice from the EACH study are consistent with the recommendations produced by the Joint Committee.
Implications for future research
The evidence supporting the introduction of CMA in the prenatal diagnosis of fetal anomalies is robust. It is anticipated that NIPT for trisomies will be introduced into UK clinical practice very soon. Future research should focus on the role of next-generation sequencing not only on fetal DNA acquired by invasive testing, but also on cffDNA in maternal blood. A particular concern is the rate of false-positive results generated by high-depth sequencing. In addition, following on from the EACH health economic study and relevant to future decisions around new genetic technologies, further research is needed to decide how much money the NHS is willing to pay to detect an additional pathogenic CNV. Finally, following on from the qualitative substudy, and relevant to the introduction of next generation sequencing, further research is needed on methods of enhancing patient understanding and supporting collaborative decision-making when a CNV is detected.
Acknowledgements
The authors would like to thank all the staff from each of the contributing fetal medicine units and cytogenetic laboratories. Special thanks go to the Trial Steering Committee (Natalie Teich, Sara Wynn, Katherine Payne and Alan Cameron), particularly Lorraine Gaunt (chairperson) and the Data Monitoring and Ethics Committee (Eddie Maher and Jenny Barrett), particularly Ruth Newbury-Ecob (chairperson). The authors would also like to thank the Newcastle Clinical Trials Unit, particularly Joanne Morrison (Trial Manager) and all the parents who participated in the study.
Contributions of authors
The first and final draft of the report was prepared by Stephen C Robson.
The report on the RAPID study was prepared by Lyn S Chitty.
The health economic methods, analysis and discussion were prepared by Stephen Morris and Talitha Verhoef.
The statistical analysis was prepared by Gareth Ambler.
The genetic methods, results and discussion were prepared by Diana G Wellesley and John A Crolla.
The qualitative substudy methods, analysis and discussion were prepared by Ruth Graham and Claire Leader.
The lay summary was prepared by Jane Fisher.
Publications
Callaway JLA, Shaffer LG, Chitty LS, Rosenfeld JA, Crolla JA. The clinical utility of microarray technologies applied to prenatal cytogenetics in the presence of a normal conventional karyotype: a review of the literature. Prenat Diagn 2013;33:1–5.
Karampetsou E, Morrogh D, Ballard T, Waters JJ, Lench N, Chitty LS. Confined placental mosaicism: implications for fetal chromosomal analysis using microarray comparative genomic hybridization. Prenat Diagn 2014;34:98–101.
Callaway JLA, Huang S, Karampetsou E, Crolla JA. Perspective on the technical challenges involved in the implementation of array-CGH in prenatal diagnostic testing. Mol Biotechnol 2014;56:312–18.
Robson SC, Crolla J, Chitty L, Wellesley D, Ambler G. Evaluation of array comparative genomic hybridisation in prenatal diagnosis of fetal anomalies (EACH study). BJOG 2015;122(Suppl. 2):3.
Data sharing statement
All available data can be obtained from the corresponding author.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health.
References
- Rankin J, Pattenden S, Abramsky L, Boyd P, Jordan H, Stone D, et al. Prevalence of congenital anomalies in five British regions, 1991-99. Arch Dis Child Fetal Neonatal Ed 2005;90:F374-9. http://dx.doi.org/10.1136/adc.2003.047902.
- NHS FASP . Service Specification n.d. http://fetalanomaly.screening.nhs.uk/specification (accessed May 2015).
- Wright D, Syngelaki A, Bradbury I, Akolekar R, Nicolaides KH. First-trimester screening for trisomies 21, 18 and 13 by ultrasound and biochemical testing. Fetal Diagn Ther 2014;35:118-26. http://dx.doi.org/10.1159/000357430.
- NHS FASP . Programme Statements: Nuchal Translucency Greater Than or Equal to 3.5 Mm n.d. www.fetalanomaly.screening.nhs.uk/programmestatements (accessed May 2015).
- NHS FASP . Standards: 18+0 to 20+6 Weeks Fetal Anomaly Scan – National Standards and Guidance for England 2010 n.d. www.fetalanomaly.screening.nhs.uk/standards (accessed May 2015).
- Rossi AC, Prefumo F. Accuracy of ultrasonography at 11-14 weeks of gestation for detection of fetal structural anomalies: a systematic review. Obstet Gynecol 2013;122:1160-7. http://dx.doi.org/10.1097/AOG.0000000000000015.
- Nicolaides KH, Snijders RJ, Gosden CM, Berry C, Campbell S. Ultrasonographically detectable markers of fetal chromosomal abnormalities. Lancet 1992;340:704-7. https://doi.org/10.1016/0140-6736(92)92240-G.
- Rizzo N, Pittalis MC, Pilu G, Perolo A, Banzi C, Visentin A, et al. Distribution of abnormal karyotypes among malformed fetuses detected by ultrasound throughout gestation. Prenat Diagn 1996;16:159-63. https://doi.org/10.1002/(SICI)1097-0223(199602)16:2<159::AID-PD831>3.0.CO;2-H.
- Mann K, Hills A, Donaghue C, Thomas H, Ogilvie CM. Quantitative fluorescence PCR analysis of > 40,000 prenatal samples for the rapid diagnosis of trisomies 13, 18 and 21 and monosomy X. Prenat Diagn 2012;32:1197-204. http://dx.doi.org/10.1002/pd.3986.
- Grimshaw GM, Szczepura A, Hultén M, MacDonald F, Nevin NC, Sutton F, et al. Evaluation of molecular tests for prenatal diagnosis of chromosome abnormalities. Health Technol Assess 2003;7. https://doi.org/10.3310/hta7100.
- Kagan KO, Chitty LS, Cicero S, Eleftheriades M, Nicolaides KH. Ultrasound findings before amniocentesis in selecting the method of analysing the sample. Prenat Diagn 2007;27:34-9. https://doi.org/10.1002/pd.1615.
- Chitty LS, Kistler J, Akolekar R, Liddle S, Nicolaides K, Levett L. Multiplex ligation-dependent proble amplification (MLPA): a reliable alternative for fetal chromosome analysis. J Mat Fet Neonat Med 2012;25:1383-6. https://doi.org/10.3109/14767058.2011.636093.
- Chitty LS, Kagan KO, Molina FS, Waters JJ, Nicolaides KH. Fetal nuchal translucency scan and early prenatal diagnosis of chromosomal abnormalities by rapid aneuploidy screening: observational study. BMJ 2006;332:452-5. http://dx.doi.org/10.1136/bmj.38730.655197.AE.
- Kagan KO, Avgidou K, Molina FS, Gajewska K, Nicolaides KH. Relation between increased fetal nuchal translucency thickness and chromosomal defects. Obstet Gynecol 2006;107:6-10. https://doi.org/10.1097/01.AOG.0000191301.63871.c6.
- Bellucco FT, Belangero SI, Farah LM, Machado MV, Cruz AP, Lopes LM, et al. Investigating 22q11.2 deletion and other chromosomal aberrations in fetuses with heart defects detected by prenatal echocardiography. Pediatr Cardiol 2010;31:1146-50. http://dx.doi.org/10.1007/s00246-010-9763-0.
- Shaffer LG, Beaudet AL, Brothman AR, Hirsch B, Levy B, Martin CL, et al. Microarray analysis for constitutional cytogenetic abnormalities. Genet Med 2007;9:654-62. https://doi.org/10.1097/GIM.0b013e31814ce3d9.
- Karampetsou E, Morrogh D, Chitty L. Microarray technology for the diagnosis of fetal chromosomal aberrations: which platform should we use?. J Clin Med 2014;3:663-78. http://dx.doi.org/10.3390/jcm3020663.
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature 2006;444:444-54. http://dx.doi.org/10.1038/nature05329.
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet 2008;40:1166-74. http://dx.doi.org/10.1038/ng.238.
- Hillman SC, McMullan DJ, Williams D, Maher ER, Kilby MD. Microarray comparative genomic hybridization in prenatal diagnosis: a review. Ultrasound Obstet Gynecol 2012;40:385-91. http://dx.doi.org/10.1002/uog.11180.
- Shaffer LG, Kashork CD, Saleki R, Rorem E, Sundin K, Ballif BC, et al. Targeted genomic microarray analysis for identification of chromosome abnormalities in 1500 consecutive clinical cases. J Pediatr 2006;149:98-102. http://dx.doi.org/10.1016/j.jpeds.2006.02.006.
- Mefford HC, Batshaw ML, Hoffman EP. Genomics, intellectual disability, and autism. N Engl J Med 2012;366:733-43. http://dx.doi.org/10.1056/NEJMra1114194.
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749-64. http://dx.doi.org/10.1016/j.ajhg.2010.04.006.
- Association of Clinical Cytogenetics . Professional Guidelines for Clinical Cytogenetics n.d. www.acgs.uk.com/media/765587/acc_array_bp_dec2011_2.00.pdf (accessed May 2015).
- Schaeffer AJ, Chung J, Heretis K, Wong A, Ledbetter DH, Lese Martin C. Comparative genomic hybridization-array analysis enhances the detection of aneuploidies and submicroscopic imbalances in spontaneous miscarriages. Am J Hum Genet 2004;74:1168-74. http://dx.doi.org/10.1086/421250.
- Miura S, Miura K, Masuzaki H, Miyake N, Yoshiura K, Sosonkina N, et al. Microarray comparative genomic hybridization (CGH)-based prenatal diagnosis for chromosome abnormalities using cell-free fetal DNA in amniotic fluid. J Hum Genet 2006;51:412-17. https://doi.org/10.1007/s10038-006-0376-7.
- Lapaire O, Lu XY, Johnson KL, Jarrah Z, Stroh H, Cowan JM, et al. Array-CGH analysis of cell-free fetal DNA in 10 mL of amniotic fluid supernatant. Prenat Diagn 2007;27:616-21. https://doi.org/10.1002/pd.1752.
- Sahoo T, Cheung SW, Ward P, Darilek S, Patel A, del Gaudio D, et al. Prenatal diagnosis of chromosomal abnormalities using array-based comparative genomic hybridization. Genet Med 2006;8:719-27. https://doi.org/10.1097/01.gim.0000245576.47154.63.
- Shaffer LG, Coppinger J, Alliman S, Torchia BA, Theisen A, Ballif BC, et al. Comparison of microarray-based detection rates for cytogenetic abnormalities in prenatal and neonatal specimens. Prenat Diagn 2008;28:789-95. http://dx.doi.org/10.1002/pd.2053.
- Coppinger J, Alliman S, Lamb AN, Torchia BS, Bejjani BA, Shaffer LG. Whole-genome microarray analysis in prenatal specimens identifies clinically significant chromosome alterations without increase in results of unclear significance compared to targeted microarray. Prenat Diagn 2009;29:1156-66. http://dx.doi.org/10.1002/pd.2371.
- Van den Veyver IB, Patel A, Shaw CA, Pursley AN, Kang SH, Simovich MJ, et al. Clinical use of array comparative genomic hybridization (aCGH) for prenatal diagnosis in 300 cases. Prenat Diagn 2009;29:29-3. http://dx.doi.org/10.1002/pd.2127.
- Vialard F, Molina Gomes M, Leroy B, Quarello E, Escalona A, Le Sciellour C, et al. Array comparative genomic hybridization in prenatal diagnosis: another experience. Fetal Diagn Ther 2009;25:277-84. https://doi.org/10.1159/000224112.
- Kleeman L, Bianchi DW, Shaffer LG, Rorem E, Cowan J, Craigo SD, et al. Use of array comparative genomic hybridization for prenatal diagnosis of fetuses with sonographic anomalies and normal metaphase karyotype. Prenat Diagn 2009;29:1213-17. http://dx.doi.org/10.1002/pd.2367.
- Tyreman M, Abbott KM, Willatt LR, Nash R, Lees C, Whittaker J, et al. High resolution array analysis: diagnosing pregnancies with abnormal ultrasound findings. J Med Genet 2009;46:531-41. http://dx.doi.org/10.1136/jmg.2008.065482.
- Valduga M, Philippe C, Bach Segura P, Thiebaugeorges O, Miton A, Beri M, et al. A retrospective study by oligonucleotide array-CGH analysis in 50 fetuses with multiple malformations. Prenat Diagn 2010;30:333-41. http://dx.doi.org/10.1002/pd.2460.
- Hillman SC, Pretlove S, Coomarasamy A, McMullan DJ, Davison EV, Maher ER, et al. Additional information from array comparative genomic hybridization technology over conventional karyotyping in prenatal diagnosis: a systematic review and meta-analysis. Ultrasound Obstet Gynecol 2011;37:6-14. http://dx.doi.org/10.1002/uog.7754.
- Leung TY, Vogel I, Lau TK, Chong W, Hyett JA, Petersen OB, et al. Identification of submicroscopic chromosomal aberrations in fetuses with increased nuchal translucency and apparently normal karyotype. Ultrasound Obstet Gynecol 2011;38:314-19. https://doi.org/10.1002/uog.8988.
- Williams C. Dilemmas in fetal medicine: premature application of technology or responding to women’s choice?. Sociol Health Illn 2006;28:1-20. http://dx.doi.org/10.1111/j.1467-9566.2006.00480.x.
- Ham C. Health Policy in Britain. Basingstoke: Palgrave Macmillan; 2009.
- Newman WG, Hamilton S, Ayres J, Sanghera N, Smith A, Gaunt L, et al. Array comparative genomic hybridization for diagnosis of developmental delay: an exploratory cost-consequences analysis. Clin Genet 2007;71:254-9. http://dx.doi.org/10.1111/j.1399-0004.2007.00756.x.
- ACOG Committee Opinion Number 446 . Array comparative genomic hybridization in prenatal diagnosis. Obstet Gynecol 2009;114:1161-3. https://doi.org/10.1097/AOG.0b013e3181c33cad.
- Chiu RW, Akolekar R, Zheng YW, Leung TY, Sun H, Chan KC, et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ 2011;342. http://dx.doi.org/10.1136/bmj.c7401.
- Ehrich M, Deciu C, Zwiefelhofer T, Tynan JA, Cagasan L, Tim R, et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: a study in a clinical setting. Am J Obstet Gynecol 2011;204:205.e1-11. http://dx.doi.org/10.1016/j.ajog.2010.12.060.
- Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE, Neveux LM, Ehrich M, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med 2011;13:913-20. http://dx.doi.org/10.1097/GIM.0b013e3182368a0e.
- Sehnert AJ, Rhees B, Comstock D, de Feo E, Heilek G, Burke J, et al. Optimal detection of fetal chromosomal abnormalities by massively parallel DNA sequencing of cell-free fetal DNA from maternal blood. Clin Chem 2011;57:1042-9. http://dx.doi.org/10.1373/clinchem.2011.165910.
- The NHS Rapid Project . What Is the RAPID Project? 2014. www.rapid.nhs.uk/about-rapid/ (accessed June 2015).
- NHS Fetal Anomaly Screening Programme . Isolated and Increased Nuchal Fold n.d. http://fetalanomaly.screening.nhs.uk/leafletsforprofessionals#fileid11679 (accessed June 2015).
- Callaway JL, Huang S, Karampetsou E, Crolla JA. Perspective on the technical challenges involved in the implementation of array-CGH in prenatal diagnostic testing. Mol Biotechnol 2014;56:312-8. http://dx.doi.org/10.1007/s12033-013-9710-4.
- Baldwin EL, Lee JY, Blake DM, Bunke BP, Alexander CR, Kogan AL, et al. Enhanced detection of clinically relevant genomic imbalances using a targeted plus whole genome oligonucleotide microarray. Genet Med 2008;10:415-29. http://dx.doi.org/10.1097/GIM.0b013e318177015c.
- Human (Homo sapiens) Genome Browser Gateway . Human Genome Browser – hg19 Assembly (Sequences) n.d. http://moma.ki.au.dk/genome-mirror/cgi-bin/hgGateway?db=hg19 (accessed June 2015).
- Department of Health . National Schedule of Reference Costs 2012 13 n.d. www.gov.uk/government/publications/nhs-reference-costs-2012-t0-2013 (accessed August 2015).
- Curtis L. Unit Costs of Health and Social Care 2013. Canterbury: Personal Social Services Research Unit, University of Kent; 2013.
- National Institute for Health and Care Excellence . Guide to the Methods of Technology Appraisal 2013 n.d. www.nice.org.uk/media/D45/1E/GuideToMethodsTechnologyAppraisal/2013 (accessed August 2015).
- Pope C, Ziebland S, Mays N, Pope C, Mays N. Qualitative Research in Health Care. Oxford: Blackwell Science; 2007.
- Silverman D. Interpreting Qualitative Data. London: Sage; 2006.
- Hillman SC, McMullan DJ, Hall G, Togneri FS, James N, Maher EJ, et al. Use of prenatal chromosomal microarray: prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet Gynecol 2013;41:610-20. https://doi.org/10.1002/uog.12464.
- Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med 2012;367:2175-84. http://dx.doi.org/10.1056/NEJMoa1203382.
- De Wit MC, Srebnaik MI, Govaerts LCP, Van Opstal D, Galjaard RJH, Go ATJI. Additional value of prenatal genomic array testing in fetuses with isolated structural ultrasound abnormalities and a normal karyotype: a systematic review of the literature. Ultrasound Obstet Gynecol 2014;43:139-46. https://doi.org/10.1002/uog.12575.
- Shaffer LG, Rosenfeld JA, Dabell MP, Coppinger J, Bandholz AM, Ellison JW, et al. Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound. Prenat Diagn 2012;32:986-95. http://dx.doi.org/10.1002/pd.3943.
- Brady PD, Chiaie BD, Christenhusz G, Dierickx K, Van Den Bogaert K, Menten B, et al. A prospective study of the clinical utility of prenatal chromosomal microarray analysis in fetuses with ultrasound abnormalities and an exploration of a framework for reporting unclassified variants and risk factors. Genet Med 2014;16:469-76. https://doi.org/10.1038/gim.2013.168.
- Grande M, Jansen FA, Blumenfeld YJ, Fisher A, Odibo AO, Haak MC, et al. Genomic microarray in fetuses with increased nuchal translucency and normal karyotype: a systematic review and meta-analysis. Ultrasound Obstet Gynecol 2015;46:650-8. http://dx.doi.org/10.1002/uog.14880.
- Huang J, Poon LC, Akolekar R, Choy KW, Leung TY, Nicolaides KH. Is high fetal nuchal translucency associated with submicroscopic chromosomal abnormalities on array CGH?. Ultrasound Obstet Gynecol 2014;43:620-4. http://dx.doi.org/10.1002/uog.13384.
- Donnelly JC, Platt LD, Rebarber A, Zachary J, Grobman WA, Wapner RJ. Association of copy number variants with specific ultrasonographically detected fetal anomalies. Obstet Gynecol 2014;124:83-90. http://dx.doi.org/10.1097/AOG.0000000000000336.
- Saldarriaga W, Garcia-Perdomo HA, Arango-Pineda J, Fonseca J. Karyotype versus genomic hybridization for the prenatal diagnosis of chromosomal abnormalities: a metaanalysis. Am J Obstet Gynecol 2015;212:330.e1-10. https://doi.org/10.1016/j.ajog.2014.10.011.
- Mademont-Soler I, Morales C, Soler A, Nartinez-Crespo JM, Shen Y, Mararit E, et al. Prenatal diagnosis of chromosomal abnormalities in fetuses with abnormal cardiac ultrasound findings: evaluation of chromosomal microarray-based analysis. Ultrasound Obstet Gynecol 2013;41:375-82. https://doi.org/10.1002/uog.12372.
- Yan Y, Wu Q, Zhang L, Wang X, Dan S, Deng D, et al. Detection of submicroscopic chromosomal aberrations by array-based comparative genomic hybridization in fetuses with congenital heart disease. Ultrasound Obstet Gynecol 2014;43:404-12. https://doi.org/10.1002/uog.13236.
- MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014;42:D986-92. http://dx.doi.org/10.1093/nar/gkt958.
- Bragin E, Chatzimichali EA, Wright CF, Hurles ME, Firth HV, Bevan AP, et al. DECIPHER: database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res 2014;42:D993-D1000. http://dx.doi.org/10.1093/nar/gkt937.
- ClinGen . Clinical Genome Resource n.d. http://clinicalgenome.org/ (accessed June 2015).
- Shaffer LG, Dabell MP, Rosenfeld JA, Neill NJ, Ballif BC, Coppinger J, et al. Referral patterns for microarray testing in prenatal diagnosis. Prenat Diagn 2012;32:344-50. https://doi.org/10.1002/pd.3856.
- Hillman SC, McMullan DJ, Silcock L, Maher ER, Kilby MD. How does altering the resolution of chromosomal microarray analysis in the prenatal setting affect the rates of pathological and uncertain findings?. J Matern Fetal Neonatal Med 2014;27:649-57. http://dx.doi.org/10.3109/14767058.2013.825601.
- Professional Standards Committee . Association for Clinical Cytogenetics n.d. www.cytogenetics.org.uk/ (accessed June 2015).
- Baptista J, Mercer C, Prigmore E, Gribble SM, Carter NP, Maloney V, et al. Breakpoint mapping and array CGH in translocations: comparison of a phenotypically normal and an abnormal cohort. Am J Hum Genet 2008;82:927-36. http://dx.doi.org/10.1016/j.ajhg.2008.02.012.
- Giardino D, Corti C, Ballarati L, Colombo D, Sala E, Villa N, et al. De novo balanced chromosome rearrangements in prenatal diagnosis. Prenat Diagn 2009;29:257-65. http://dx.doi.org/10.1002/pd.2215.
- Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet 1991;49:995-1013.
- Gribble SM, Prigmore E, Burford DC, Porter KM, Ng BL, Douglas EJ, et al. The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J Med Genet 2005;42:8-16. http://dx.doi.org/10.1136/jmg.2004.024141.
- Wolstenholme J. Confined placental mosaicism for trisomies 2, 3, 7, 8, 9, 16, and 22: their incidence, likely origins, and mechanisms for cell lineage compartmentalization. Prenat Diagn 1996;16:511-24. https://doi.org/10.1002/(SICI)1097-0223(199606)16:6<511::AID-PD904>3.0.CO;2-8.
- Carey L, Scott F, Murphy K, Mansfield N, Barahona P, Leigh D, et al. Prenatal diagnosis of chromosomal mosaicism in over 1600 cases using array comparative genomic hybridization as a first line test. Prenat Diagn 2014;34:478-86. http://dx.doi.org/10.1002/pd.4332.
- Karampetsou E, Morrogh D, Ballard T, Waters JJ, Lench N, Chitty LS. Confined placental mosaicism: implications for fetal chromosomal analysis using microarray comparative genomic hybridization. Prenat Diagn 2014;34:98-101. https://doi.org/10.1002/pd.4255.
- Kooper AJ, Faas BH. Comment on ‘confined placental mosaicism: implications for fetal chromosomal analysis using microarray comparative genomic hybridization’. Prenat Diagn 2014;34:815-16. http://dx.doi.org/10.1002/pd.4356.
- Mann K, Kabba M, Donaghue C, Hills A, Ogilvie CM. Analysis of a chromosomally mosaic placenta to assess the cell populations in dissociated chorionic villi: implications for QF-PCR aneuploidy testing. Prenat Diagn 2007;27:287-9. https://doi.org/10.1002/pd.1663.
- Srebniak MI, Van Opstal D, Joosten M, Diderich KE, de Vries FA, Riedijk S, et al. Whole-genome array as a first-line cytogenetic test in prenatal diagnosis. Ultrasound Obstet Gynecol 2015;45:363-72. http://dx.doi.org/10.1002/uog.14745.
- Lamb AN, Rosenfeld JA, Coppinger J, Dodge ET, Dabell MP, Torchia BS, et al. Defining the impact of maternal cell contamination on the interpretation of prenatal microarray analysis. Genet Med 2012;14:914-21. http://dx.doi.org/10.1038/gim.2012.77.
- Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet 2010;19:1263-75. http://dx.doi.org/10.1093/hmg/ddq003.
- Fiorentino F, Caiazzo F, Napolitano S, Spizzichino L, Bono S, Sessa M, et al. Introducing array comparative genomic hybridization into routine prenatal diagnosis practice: a prospective study on over 1000 consecutive clinical cases. Prenat Diagn 2011;31:1270-82. http://dx.doi.org/10.1002/pd.2884.
- Mattes J, Whitehead B, Liehr T, Wilkinson I, Bear J, Fagan K, et al. Paternal uniparental isodisomy for chromosome 14 with mosaicism for a supernumerary marker chromosome 14. Am J Med Genet A 2007;143A:2165-71. https://doi.org/10.1002/ajmg.a.31896.
- Faas BH, Feenstra I, Eggink AJ, Kooper AJ, Pfundt R, van Vugt JM, et al. Non-targeted whole genome 250K SNP array analysis as replacement for karyotyping in fetuses with structural ultrasound anomalies: evaluation of a one-year experience. Prenat Diagn 2012;32:362-70. http://dx.doi.org/10.1002/pd.2948.
- Liao C, Fu F, Li R, Xie GE, Zhang YL, Li J, et al. Implementation of high-resolution SNP arrays in the investigation of fetuses with ultrasound malformations: 5 years of clinical experience. Clin Genet 2014;86:264-9. http://dx.doi.org/10.1111/cge.12271.
- Hillman SC, Barton PM, Roberts TE, Maher ER, McMullan DM, Kilby MD. BAC chromosomal microarray for prenatal detection of chromosome anomalies in fetal ultrasound anomalies: an economic evaluation. Fetal Diagn Ther 2014;36:49-58. http://dx.doi.org/10.1159/000358387.
- Bernhardt BA, Soucier D, Hanson K, Savage MS, Jackson L, Wapner RJ. Women’s experiences receiving abnormal prenatal chromosomal microarray testing results. Genet Med 2013;15:139-45. http://dx.doi.org/10.1038/gim.2012.113.
- Hillman SC, Skelton J, Quinlan-Jones E, Wilson A, Kilby MD. ‘If it helps’ the use of microarray technology in prenatal testing: patient and partners reflections. Am J Med Genet A 2013;161A:1619-27. http://dx.doi.org/10.1002/ajmg.a.35981.
- Statham H, Solomou W, Chitty L. Prenatal diagnosis of fetal abnormality: psychological effects on women in low-risk pregnancies. Baillieres Best Pract Res Clin Obstet Gynaecol 2000;14:731-47. http://dx.doi.org/10.1053/beog.2000.0108.
- Rickman L, Fiegler H, Carter NP, Bobrow M. Prenatal diagnosis by array-CGH. Eur J Med Genet 2005;48:232-40. http://dx.doi.org/10.1016/j.ejmg.2005.03.003.
- American College of Obstetricians and Gynecologists Committee on Genetics . Committee Opinion 581. The use of chromosomal microarray analysis in prenatal diagnosis. Obstet Gynecol 2013;122:1374-7. https://doi.org/10.1097/00006250-201312000-00042.
- Friedman JM. High-resolution array genomic hybridization in prenatal diagnosis. Prenat Diagn 2009;29:20-8. http://dx.doi.org/10.1002/pd.2129.
- Bui T-H, Raymind FL, Van den Veyver IB. Current controversies in prenatal diagnosis 2: should incidental findings arising from prenatal testing always be reported to patients?. Prenat Diagn 2014;34:12-7. https://doi.org/10.1002/pd.4275.
- Vetro A, Bouman K, Hastings R, McMullan DJ, Vermeesch JR, Miller K, et al. The introduction of arrays in prenatal diagnosis: a special challenge. Hum Mutat 2012;33:923-9. http://dx.doi.org/10.1002/humu.22050.
- NHS England . Putting Patients First: The NHS England Business Plan for 2014 15–2016 17 n.d. www.england.nhs.uk/pp-1314-1516/ (accessed June 2015).
- Lichtenbelt KD, Knoers NV, Schuring-Blom GH. From karyotyping to array-CGH in prenatal diagnosis. Cytogenet Genome Res 2011;135:241-50. http://dx.doi.org/10.1159/000334065.
- Heaven B, Murtagh M, Rapley T, May C, Graham R, Kaner E, et al. Patients or research subjects? A qualitative study of participation in a randomised controlled trial of a complex intervention. Patient Educ Couns 2006;62:260-70. https://doi.org/10.1016/j.pec.2005.07.013.
- Sandelowski M, Barroso J. The travesty of choosing after positive prenatal diagnosis. J Obstet Gynecol Neonatal Nurs 2005;34:307-18. http://dx.doi.org/10.1177/0884217505276291.
- Reeves S, Perrier L, Goldman J, Freeth D, Zwarenstein M. Interprofessional education: effects on professional practice and healthcare outcomes (update). Cochrane Database Syst Rev 2013;3. http://dx.doi.org/10.1002/14651858.CD002213.pub3.
- Howell L. Creating a state of the art centre for fetal diagnosis. The importance of a multidisciplinary approach. Prog Pediatr Cardiol 2006;22:121-7. https://doi.org/10.1016/j.ppedcard.2006.01.012.
- Tluczek A, Chevalier McKechnie A, Lynam PA. When the cystic fibrosis label does not fit: a modified uncertainty theory. Qual Heath Res 2010;20:209-23. https://doi.org/10.1177/1049732309356285.
- DeLuca JM, Kearney MH, Norton SA, Arnold GL. Parents’ experiences of expanded newborn screening evaluations. Pediatrics 2011;128:53-61. http://dx.doi.org/10.1542/peds.2010-3413.
- Lippman A. Prenatal genetic testing and screening: constructing needs and reinforcing inequities. Am J Law Med 1991;17:15-50.
- Löwy I. Prenatal diagnosis: the irresistible rise of the ‘visible fetus’. Stud Hist Philos Biol Biomed Sci 2014;47:290-9. http://dx.doi.org/10.1016/j.shpsc.2013.12.003.
- Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: updated meta-analysis. Ultrasound Obstet Gynecol 2015;45:249-66. http://dx.doi.org/10.1002/uog.14791.
- Gov.uk . Safer Screening Test For Pregnant Women n.d. www.gov.uk/government/news/safer-screening-test-for-pregnant-women (accessed November 2016).
- Benachi A, Letourneau A, Kleinfinger P, Senat MV, Gautier E, Favre R, et al. Cell-free DNA analysis in maternal plasma in cases of fetal abnormalities detected on ultrasound examination. Obstet Gynecol 2015;125:1330-7. http://dx.doi.org/10.1097/AOG.0000000000000874.
- Bianchi DW, Parsa S, Bhatt S, Halks-Miller M, Kurtzman K, Sehnert AJ, et al. Fetal sex chromosome testing by maternal plasma DNA sequencing: clinical laboratory experience and biology. Obstet Gynecol 2015;125:375-82. http://dx.doi.org/10.1097/AOG.0000000000000637.
- Wapner RJ, Babiarz JE, Levy B, Stosic M, Zimmermann B, Sigurjonsson S, et al. Expanding the scope of noninvasive prenatal testing: detection of fetal microdeletion syndromes. Am J Obstet Gynecol 2015;212. http://dx.doi.org/10.1016/j.ajog.2014.11.041.
- Talkowski ME, Ordulu Z, Pillalamarri V, Benson CB, Blumenthal I, Connolly S, et al. Clinical diagnosis by whole-genome sequencing of a prenatal sample. N Engl J Med 2012;367:2226-32. http://dx.doi.org/10.1056/NEJMoa1208594.
- Hillman SC, Willams D, Carss KJ, McMullan DJ, Hurles ME, Kilby MD. Prenatal exome sequencing for fetuses with structural abnormalities: the next step. Ultrasound Obstet Gynecol 2015;45:4-9. http://dx.doi.org/10.1002/uog.14653.
- Everett TR, Chitty LS. Cell-free fetal DNA: the new tool in fetal medicine. Ultrasound Obstet Gynecol 2015;45:499-507. http://dx.doi.org/10.1002/uog.14746.
- Gardiner C, Wellesley D, Kilby MD, Kerr B. behalf of the Joint Committee on Genomics in Medicine . Recommendations for the Use of Chromosome Microarray in Pregnancy n.d. www.bsgm.org.uk/media/956141/g144_useofcmapregnancy_jun15.pdf (accessed July 2015).
Appendix 1 The EACH study patient information leaflet
Appendix 2 The EACH study patient consent form
Appendix 3 The EACH study health professional interview topic guide
Appendix 4 The EACH study patient interview topic guide
Appendix 5 Report on cell-free deoxyribonucleic acid aspects of the EACH study
Provided by Professor Lyn Chitty (Institute of Child Health, University College London, 2015, personal communication).
Background
The presence in maternal plasma of cell-free fetal DNA, which is pregnancy specific and represents the whole of the fetal genome, offers enormous promise for the development of a range of prenatal diagnostic tests. Fetal sex can be determined by analysis of cell-free DNA and several other tests are available for definitive prenatal diagnosis of several monogenic disorders. However, it is the development of NIPT for aneuploidy that stands to have the greatest impact on maternity care. The first proof of principle studies using next-generation sequencing were published by academic units in 2008. Larger demonstration projects, led by industry, reporting high sensitivities and specificities (98% and 99%, respectively) for the detection of trisomy 21 in high-risk pregnancies were followed by the commercial launch of NIPT in Asia and the USA in 2011, and in the UK in 2012. It is clear that women and health professionals welcome NIPT but concerns have been raised that ease of access to NIPT may undermine informed consent as there is a risk of this test being seen as routine. These concerns may be overcome by careful pre- and post-test counselling, but this will require education of health professionals and adequate provision of counselling. In the UK, the limitation of NIPT services to private sector provision (at a cost of £400–900) is resulting in inequality of access with increasing demands from women for implementation in public sector maternity care. However, this has significant implications for service provision. Should the test be developed in NHS laboratories or be outsourced to the commercial sector? How should we manage the 1–6% of pregnancies where NIPT fails? The current cost of NIPT is likely to prohibit implementation in the NHS as a replacement for traditional trisomy 21 screening: should we therefore offer it to high-risk women as an intermediate test?
Development of non-invasive prenatal testing for aneuploidy
The funding provided by Efficacy and Mechanism Evaluation assisted the RAPID team with the evaluation of NIPT for aneuploidy by providing consumables for the sequencing of cell-free DNA extracted from ≈1100 maternal blood samples, collected as part of the EACH study, and the development of the bioinformatics algorithm used for the massively parallel sequencing of maternal plasma. This has allowed the RAPID team to deliver an evaluation of NIPT for aneuploidy in the NHS through the NIHR Programme Grants for Applied Research RAPID programme (RP-PG-0707-10107) using sequencing performed at the local regional genetics laboratory at Great Ormond Street Hospital NHS Foundation Trust. The overall aim of this part of the RAPID programme was to deliver a report on all aspects of NIPT implementation (laboratory evaluation, health professional and patient education, and evaluation of the care pathway and a detailed health economic analysis) to the National Screening Committee to inform the decisions on if, how and when to introduce NIPT into the NHS maternity care pathway. The final report has been submitted to the national Screening Committee for comment in May 2015. In November 2016, the Department of Health announced the introduction of NIPT for new, non-invasive, prenatal Down, Edwards and Patau syndromes. 109
Evaluation of non-invasive prenatal testing for the detection of other chromosomal imbalances
Recently, the scope of NIPT has been increased to include selected subchromosomal abnormalities, but the number of samples reported has been small, leading to considerable uncertainty in test sensitivity and specificity. As part of the RAPID programme, a novel calling pipeline based on a segmentation algorithm for the detection of fetal microdeletion and microduplication syndrome in maternal cell-free DNA has been developed. The algorithm has been tested on 31 samples with known subchromosomal abnormalities and many of those from whom these samples were taken were recruited as part of the EACH study.
With the same read depth used in our NIPT for aneuploidy pipeline, our algorithm detected 15 out of 18 samples with a CNV larger than 6 Mb (sensitivity 83%) and three out of three samples with maternally derived abnormalities (sensitivity 100%). There were two false-positive calls in 534 samples with no known subchromosomal abnormalities (specificity 99.6%). Using funding from the Great Ormond Street Biomedical Research Centre, deeper sequencing has been performed. With the higher read depth, 26 out of the 28 samples with fetal subchromosomal abnormalities were detected.
We concluded that test sensitivity is a function of the fetal fraction, read depth and the fetal CNV size. Fetal fraction is a strong determinant of sensitivity and at least one of the two false negatives was due to low fetal fraction. The lack of an independent method for determining fetal fraction, especially for female fetuses, leads to uncertainty in the test sensitivity, which has implications for the future of this technique as a clinical diagnostic test.
Appendix 6 Summary of patient and public involvement
Patients and public were involved throughout the EACH study, as follows.
-
Design: the application had a lay coapplicant (Jane Fisher, Antenatal Results and Choices, London, UK) who helped design the study and contributed to the study application and each version of the protocol.
-
Methods: Jane Fisher, along with the two lay members of the Trial Steering Committee and two lay members of the Reproductive Health Research Group (Newcastle University, Newcastle, UK) contributed to the content of the patient information leaflet and the patient consent form. Jane Fisher also contributed to the interview guide used in the qualitative substudy.
Report: Jane Fisher contributed to the content of final report and prepared the lay summary.
List of abbreviations
- BAC
- bacterial artificial chromosome
- cffDNA
- cell-free fetal deoxyribonucleic acid
- CGH
- comparative genomic hybridisation
- CI
- confidence interval
- CMA
- chromosomal microarray
- CNV
- copy number variant
- CPA
- Clinical Pathology Accreditation
- CPM
- confined placental mosaicism
- CRG
- clinical reference group
- CVS
- chorionic villus sampling
- DMD
- Duchenne muscular dystrophy
- DNA
- deoxyribonucleic acid
- EACH
- Evaluation of Array Comparative genomic Hybridisation
- FASP
- Fetal Anomaly Screening Programme
- FISH
- fluorescence in situ hybridisation
- IQR
- interquartile range
- ISCA
- International Standards for Cytogenomic Array
- MCC
- maternal cell contamination
- MLPA
- multiplex ligation-dependent probe amplification
- NETRGL
- North-East Thames Regional Genetics Laboratory
- NF
- nuchal fold
- NICHD
- National Institute of Child Health and Human Development
- NIHR
- National Institute for Health Research
- NIPT
- non-invasive prenatal testing
- NT
- nuchal translucency
- oligoCGH
- oligonucleotide comparative genomic hybridisation
- PAGE
- Prenatal Assessment of Genome and Exomes
- pCNV
- pathogenic copy number variant
- PCR
- polymerase chain reaction
- QALY
- quality-adjusted life-year
- QF-PCR
- quantitative fluorescent polymerase chain reaction
- RAPID
- Reliable Accurate Prenatal non-Invasive Diagnosis
- SNP
- single nucleotide polymorphism
- TAT
- turnaround time
- UPD
- uniparental disomy
- VOUS
- variant(s) of unknown significance
- WRGL
- Wessex Regional Genetics Laboratory