
Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 09/800/04. The contractual start date was in October 2007. The final report began editorial review in February 2016 and was accepted for publication in March 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Jack Satsangi reports personal fees from Takeda Pharmaceutical Company Ltd, AbbVie Inc. and Dr Falk Pharma UK Ltd, and research funding from the European Commission. Nicholas A Kennedy reports grants from The Wellcome Trust; personal fees from Merck & Co., Inc., Takeda Pharmaceutical Company Ltd, Dr Falk Pharma UK Ltd, Allergan, plc, and Pharmacosmos A/S; non-financial support from Norgine BV, AbbVie Inc., Shire, plc, and Janssen Global Services, LLC; and other support from Merck & Co., Inc., AbbVie Inc. and Takeda Pharmaceutical Company Ltd outside the submitted work. Ian Arnott reports personal fees from Vifor Pharma Management Ltd, Takeda Pharmaceutical Company Ltd and Dr Falk Pharma UK Ltd. Steff Lewis reports membership of the Health Technology Assessment Efficient Study Designs Board and University of Edinburgh grant funding.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2017. This work was produced by Satsangi et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Crohn’s disease (CD) is a chronic, relapsing, inflammatory illness that can involve any part of the gastrointestinal tract, but most commonly involves the terminal ileum. The prevalence in northern England has been estimated to be 144 per population of 100,000. 1 The incidence among young people in Scotland is the highest in the UK, and continues to rise. 2 Although the mortality associated with CD is low, there is significant morbidity, with serious effects on growth, development, education and employment potential.
Crohn’s disease treatment
Treatment algorithms for the medical management of active CD have evolved very rapidly in recent years, and the relative importance of thiopurines in induction of remission, and in maintenance, has been re-evaluated since this study was first designed. Although steroids, biological therapies and combination therapies (biologics used together with thiopurine agents) have an evidence basis for induction of remission, a recent Cochrane review suggested that there was no place for monotherapy with thiopurine in induction of remission. 3
However, for patients entering medically induced remission, immunosuppression with azathioprine or mercaptopurine (MP) is an effective maintenance strategy (odds ratio 3.17, number needed to treat 3.3 favouring 2 mg/kg azathioprine over placebo), offering an acceptable balance of efficacy, tolerability and cost. 4 The relative merits of monotherapy with thiopurines alone, anti-tumour necrosis factor alpha (TNF-α) agents or combination regimes of thiopurines and biological agents remain contentious in this setting of maintenance therapy.
Despite current medical therapy, up to 50% of patients have aggressive disease and require surgery within 5 years of diagnosis. 5 Unfortunately, disease relapse rates are high within 2 years of surgery for both endoscopic (72–98%) and clinical recurrence (37–70%). 6 Re-operation rates cumulate at 5% of patients per year. 7
Evidence for postoperative use of thiopurines
The development of strategies to prevent or delay postoperative recurrence of CD is therefore of major clinical importance; however, there is a paucity of evidence to support any particular drug strategy. 8,9 The thiopurines, azathioprine and MP have an established role in inducing remission in CD and in the maintenance of medically induced remission. These agents have historically been suggested in treatment algorithms for patients at ‘high risk’ of postoperative relapse, but the evidence basis for their use in this context, and indeed the evidence that clinical parameters may predict patients at ‘high risk’ of relapse, is weak. 7 A meta-analysis conducted by Jones et al. 10 found that the efficacy data for thiopurines in the postoperative setting were inconclusive and, aside from smoking, there were no consistent predictors of postoperative relapse. A 2014 Cochrane review also concluded that, although there was evidence to suggest that thiopurines may reduce endoscopic and clinical recurrence, the quality of evidence was degraded by small numbers and flawed study designs. 3 The value of thiopurine metabolites in this situation is unknown. 11 More recently, the role of biological therapies in postoperative prophylaxis has received considerable attention. Following smaller randomised studies of infliximab in postoperative disease, the recent Post-Operative Crohn’s Endoscopic Recurrence (POCER) study demonstrated that targeted escalation of immune-modulatory therapy (thiopurines followed by adalimumab) in patients with early endoscopic evidence of recurrence may delay subsequent endoscopic, although not clinical, recurrence. 11–13
Current practice
Currently no medical therapy is licensed to prevent postoperative recurrence in CD. As well as thiopurine agents and anti-TNF-α agents, 5-aminosalicylates, corticosteroids and metronidazole have all been suggested, but the evidence in favour of these drugs is weak and none is widely used in the UK. 6 Postoperative maintenance therapy with azathioprine or MP is more widely used and is included in a number of clinical algorithms, but trial data from controlled trials available thus far have failed to demonstrate whether or not this is an effective therapy. Existing studies have been underpowered, poorly designed or used an inappropriate drug dose. 6,14–19
For these reasons, we consider that evidence-based strategies to prevent postoperative recurrence are both an unmet clinical need and a research priority.
Controversies in use of thiopurines in postoperative use
As we discussed in our recent review (see Jones et al. 10), there are many unresolved controversies associated with the use of thiopurines in the prevention of postoperative recurrence in CD, and this remains contentious. The definition of recurrence itself, specifically the distinction between endoscopic, clinical and surgical recurrence, the identification of high-risk individuals, the influence of pre-operative therapies on responsiveness, and the relative merits of thiopurines compared with anti-TNF-α therapies are key issues; however, no real evidence-based consensus has emerged. 10 The European Crohn’s and Colitis Organisation (ECCO) 2010 guidelines recommend their use postoperatively in ‘high-risk’ patients (defined as smokers and those with previous intestinal surgery, penetrating disease, perianal disease or extensive small bowel section). 20 The British Society of Gastroenterology’s guidelines are more cautious in emphasising the weak evidence base for risk factors in predicting disease recurrence (with the exception of smoking) and for the use of thiopurines in this setting. 8
The use of thiopurines by women, or the male partners of women, trying to conceive or in pregnancy
With respect to safety, use in patients of childbearing age has been a key issue. When the trial commenced, the published guidelines of both ECCO and the British Society of Gastroenterology supported the use of thiopurines in inflammatory bowel disease (IBD) in women of childbearing potential and during pregnancy on the grounds that the risk to the unborn child from disease activity appeared greater than continued therapy. As part of the initial trial assessment by the Medicines and Healthcare products Regulatory Agency, supporting documentation was reviewed and the inclusion of women of childbearing potential was approved without inclusion criteria of agreement to the use of contraceptives. No comment was made for inclusion of males who could potentially father a child while receiving MP. However, the sponsor, Trial Management Group (TMG) and Trial Steering Committee (TSC) examined this position in more detail in 2010.
The TMG and TSC considered the evidence and decided not to exclude female patients who either were planning to become pregnant or became pregnant, or male patients whose partners were planning to become pregnant. The existing patient information sheets, however, were revised to detail the potential risks to male patients. All enrolled male patients were also reconsented to the new information sheets and consent forms. Pregnancy data were also included in the TSC and Data Monitoring Committee (DMC) standard reports.
Since 2010, several large studies have been published examining thiopurine use for IBD during pregnancy, which provided further support for the initial guidelines. The Cancers Et Surrisque Associé aux Maladies inflammatoires intestinales En France (CESAME) study group examined 215 pregnancies in 204 women and concluded that thiopurine use during pregnancy was not associated with increased risks, including an increased risk of congenital abnormalities. 21 This has been supported by the findings of two further studies published in 2013 and 2014: Casanova et al. 22 included a cohort of 187 pregnancies in participants using thiopurines in IBD and found no evidence of an increased risk of complications, whereas Ban et al. 23 included a cohort of 115 pregnancies in participants using thiopurines for CD and found no evidence of an increased risk of major birth defects.
Potential health-care impact
The present study has an important potential impact on health care in terms of defining, with accuracy, the clinical effectiveness of thiopurine therapy in the prevention of postoperative relapse. The data from the study overall suggest that routine postoperative use of MP in all patients undergoing surgery is not justified as a preventative measure. The subgroup analyses have helped identify smokers as the subgroup of individuals at highest risk of relapse, and helped to demonstrate that this subgroup is most likely to benefit from thiopurine therapy, thereby helping to target immunomodulatory therapy towards patients most likely to benefit. The important secondary objectives allowed comparison of clinical and endoscopic recurrence rates, and analysis of clinical as well as biomarker predictors of disease recurrence. Importantly, we have assessed age, the need for previous surgery, previous thiopurine use and smoking as potential determinants of outcome.
Rationale for research
At present, there is no widely accepted algorithm or management plan in preventing postoperative relapse of CD. Non-evidence-based algorithms have been widely used and the present study will allow clear evidence to be defined in the largest study carried out in the use of thiopurine therapy.
The secondary objectives allowed comparison of clinical and endoscopic recurrence rates, and the effects of thiopurine therapy on each of these aspects. The study design has also allowed examination of drug safety, tolerability and adherence to therapy. The biomarkers in the study allow the relative predictive value of faecal calprotectin levels in assessing recurrence to be defined, as well as the relationship between thiopurine use and calprotectin levels. Quality of life was also measured in the present study. The primary objective of this study was to assess if MP can prevent or delay postoperative recurrence in CD.
Study aims
The primary objective of the Trial Of Prevention of Post operative Crohn’s disease (TOPPIC) was to assess if MP can prevent or delay postoperative recurrence of CD.
Secondary objectives included if MP can prevent or delay endoscopic evidence of recurrence and, finally, whether or not endoscopic recurrence can predict clinical recurrence, whether or not faecal calprotectin levels can be used as a non-invasive marker of disease recurrence that may remove the need for colonoscopy in some patients, whether or not drug metabolite levels relate to clinical efficacy of MP, and whether or not we can predict postoperative recurrence using clinical, genetic or serological data.
Development of TOPPIC
Development of TOPPIC was initiated in 2006 by a team of collaborators based in Scotland, with ethics and clinical trial authorisation sought in 2007. The trial was extended throughout the UK in 2010 to include collaborators from across England and Wales.
Chapter 2 Methods
Trial design
This was a multicentre, parallel-group, double-blind, randomised controlled study conducted in the UK (29 sites). Participants were allocated 1 : 1 to active treatment with MP or a matching placebo.
Following slower than projected recruitment in the first 24 months, the protocol was modified to reduce the number of colonoscopies performed as part of the trial and to allow the inclusion of patients with a previous history of malignancy, providing that they had been in remission for 5 years.
Participants
Participants were adults aged ≥ 16 years in Scotland (≥ 18 years in England and Wales) with a histologically confirmed diagnosis of CD (according to the Lennard-Jones criteria) undergoing ileocolonic or small bowel resection. 24 Patients were approached ≤ 3 months after surgery during the perioperative period and, following consent for screening, underwent a screening assessment consisting of a blood sample to check thiopurine S-methyltransferase (TPMT) activity and a postoperative stool sample to exclude enteric infection.
One week prior to randomisation, eligible participants underwent standard safety blood tests (including checks of liver function, haemoglobin levels, and white blood cell, neutrophil and platelet counts) in addition to a urinary pregnancy test.
Inclusion and exclusion criteria are described in the following sections.
Inclusion criteria
-
Aged ≥ 16 years in Scotland and ≥ 18 years in England and Wales.
-
Established diagnosis of CD confirmed at recent resection.
-
Ileocolonic or small bowel resection ≤ 3 months before screening.
-
Had ≤ 100 cm of fixed small bowel resected in total; a previous ileocolonic resection was accepted.
-
Able to start oral nutrition in the first 2 postoperative weeks.
-
Normal or heterozygous TPMT (activity present or reduced, consistent with carrier status).
-
Able to provide written informed consent prior to screening and comply with the requirements of the study protocol.
-
Not taking antibiotics at least 2 weeks prior to randomisation.
Exclusion criteria
-
Pregnancy at baseline or breastfeeding.
-
Known hypersensitivity or intolerance to MP.
-
Pancreatitis associated with azathioprine.
-
Receiving experimental treatment for CD ≤ 4 weeks prior to study entry.
-
Known to require further surgery at study entry (i.e. for the removal of an abscess developing from the primary surgery).
-
Strictureplasty procedure alone (strictureplasty and resection procedure together will not be considered an exclusion criterion).
-
Presence of a stoma.
Identifying participants
The study aimed to recruit and randomise from participating sites. Potentially eligible patients were identified by clinicians and research/specialist nurses at sites following ileocolonic or small bowel resection.
Settings and locations where the data were collected
The study took place in the following tertiary referral hospitals in the UK: Western General Hospital, Edinburgh; Aberdeen Royal Infirmary, Aberdeen; Ninewells Hospital, Dundee; Glasgow Royal Infirmary and Stobhill Hospital, Glasgow; Royal Devon and Exeter Hospital, Exeter; University Hospital Coventry, Coventry; Royal Liverpool University Hospital, Liverpool; Manchester Royal Infirmary, Manchester; Bristol Royal Infirmary, Bristol; University College Hospital, Royal Free Hospital and Royal London Hospital, London; John Radcliffe Hospital, Oxford; Salford Royal Hospital, Salford; Singleton Hospital, Swansea; Torbay Hospital, Torquay; Southampton General Hospital, Southampton; County Durham and Darlington Memorial Hospitals, Durham and Darlington; Royal Stoke University Hospital, Stoke-on-Trent; Queen’s Medical Centre, Nottingham; St Mark’s Hospital, Harrow; Rotherham General Hospital, Rotherham; Queen Elizabeth Hospital, Birmingham; and Raigmore Hospital, Inverness. Randomisation was open from May 2008 until June 2012 (a total of 49 months).
Interventions
The intervention was a random allocation to MP in 50-mg tablets or placebo in identical 50-mg tablets and matched packaging supplied to sites by the manufacturer.
Outcomes
The primary study outcome was clinical recurrence of CD defined by a Crohn’s Disease Activity Index (CDAI) score of ≥ 150 points together with a 100-point rise in the CDAI score from baseline, with the need for anti-inflammatory rescue therapy or primary surgical intervention. 25
The secondary study outcomes were as follows:
-
clinical recurrence defined by a CDAI score of ≥ 150 points together with a 100-point rise in the CDAI score from baseline, or anti-inflammatory rescue therapy or primary surgical intervention
-
the need for a second operation to remove recurrent CD from the anastomotic site
-
endoscopic recurrence using the Rutgeerts and Crohn’s Disease Endoscopic Index of Severity (CDEIS) scoring systems26,27
-
faecal calprotectin levels
-
thioguanine (TGN) levels
-
changes in self-rated quality-of-life scores.
Measurements
Following patient consent for screening, the trial screening process consisted of analysis of TPMT activity and a postoperative stool sample to exclude enteric infection. One week prior to randomisation, eligible participants underwent blood tests (including liver function tests, haemoglobin concentration tests, and white blood cell, neutrophil and platelet counts) and a urinary pregnancy test. Patients returned to designated clinics run by clinical teams for a baseline assessment including the CDAI, patient-reported outcome measures including the Inflammatory Bowel Disease Questionnaire (IBDQ), a physical examination and a blood sample for assay of drug metabolite level. 28 Additional blood samples for genetic and serological analysis were also taken, and results are reported separately.
Patients were randomised at baseline to receive either MP or placebo. MP was given at a dose of 1–1.5 mg/kg of body weight, rounded to the nearest 25 mg as a single daily dose in the morning. Patients with low TPMT activity were prescribed MP at half the normal dose. Treatment was on a maintenance basis for 3 years, with the dose adjusted up or down for weight changes during the study.
Follow-up data were collected at 10 time points, at weeks 6, 13, 31, 49, 67, 85, 103, 121, 139 and 157 post randomisation. At each visit, the following data were collected: CDAI score, physical characteristics (as measured by examination), concomitant medications taken and patient-reported outcomes including the IBDQ. At weeks 0, 13, 49, 103 and 157 post randomisation, patients were also asked to provide additional stool and blood samples for the central assessment of faecal calprotectin and TGN metabolite levels, with endoscopic assessment at weeks 49 and 157 post randomisation.
Sample processing
Stool samples were collected at participating centres and frozen on site at –80 °C. Samples were then shipped on dry ice in two batches during 2014 and 2015 to a central biochemistry laboratory at NHS Lothian Western General Hospital in Edinburgh. Faecal calprotectin levels were all assessed using the CALPRO® calprotectin enzyme-linked immunosorbent assay (alkaline phosphatase) (NovaTec Immundiagnostica GmbH, Dietzenbach, Germany) and processed by technicians blinded to the treatment allocation. Blood samples for TGN testing were sent ambient to Viapath (formerly GSTS Pathology; London, UK) for analysis in the Purine Research Laboratory. An independent analyst blinded to the treatment allocation then assessed each level.
Therapeutic monitoring
All patients underwent regular safety blood monitoring every week for the initial 6 weeks and thereafter at 6-weekly intervals as long as the patient remained on the study drug. Blood samples were processed at participating site laboratories. An independent central clinician who was blinded to the treatment allocation reviewed results. Prespecified dose reduction or cessation then occurred in the event of abnormal monitoring parameters. In the event of patient intolerance (profound nausea or persistent influenza-like symptoms), protocol-driven dose reduction was also undertaken. If abnormal parameters improved after a temporary stop, treatment was started again at a lower level. The central clinicians, blinded to the treatment allocation, made all of these decisions.
To protect blinding, a programme of sham dose reductions was planned for patients on placebo. On the advice of the DMC these were not undertaken, although the investigators were not informed of this decision, hence protecting the study blinding.
Dose changes
All randomised patients allocated to the treatment group were given 1–1.5 mg/kg body weight, rounded to the nearest 25 mg, to be taken as single daily dose in the morning. Treatment was on a maintenance basis for 3 years or until the study drug was discontinued. Patients heterozygous for TPMT mutations were prescribed MP at half the normal dose. The dose was adjusted up or down for weight changes according to Table 1.
| Body weight (kg) | Initial dose MP (mg) | Amount per body weight (mg/kg) |
|---|---|---|
| < 33 | 50 alternate days | 1–1.5 |
| 33–49.9 | 50 daily | 1–1.5 |
| 50–74.9 | 50/100 alternate days | 1–1.5 |
| 75–99.9 | 100 daily | 1–1.33 |
| 100–150 | 150 daily | 1–1.5 |
| > 150 | 200 daily | > 1 |
The blinded treatment of either MP or placebo could also be reduced or temporarily stopped following patient intolerance (profound nausea or persistent influenza-like symptoms) or abnormal blood safety results. In these cases, the dose was reduced in accordance with Table 2 and not increased again during the course of the trial. If symptoms or blood safety test result abnormalities persisted and were, in the view of the investigator, of sufficient severity, the drug could be stopped. One of the blinded clinicians assessing safety blood test results could also stop the drug.
| Initial dose of MP (mg) | Reduced dose of MP (mg) | Per cent reduction |
|---|---|---|
| 50 alternate days | Stop | 100 |
| 50 daily | 50 alternate days | 50 |
| 50/100 alternate days | 50 daily | 33 |
| 100 daily | 50 daily | 50 |
| 150 daily | 50/100 alternate days | 50 |
| 200 daily | 100 daily | 50 |
Sample size
It was estimated that a study population of 182 evaluable patients would provide the trial with 80% power to detect a reduction in the frequency of recurrence from 50% in the placebo group to 30% in the treatment arm by 3 years at the 5% level of significance. Preliminary data from surgical and pathology databases at the five Scottish recruiting sites indicated that this was an achievable target. In 2006–7, approximately 130 ileocolonic resections were performed annually at the five Scottish recruiting sites initially proposed. We estimated that 60% of the potential patients could be recruited, giving a sample population of 234 patients and allowing for a 15% dropout rate. This would leave an evaluable population of 200 patients over 3 years.
These figures were based on an intention-to-treat analysis, with the number needed to treat in order to prevent one recurrence predicted to be five. It is notable that the treatment effect of 20% was lower than in previously conducted studies (40% reduction in mild CD lesions and 75% reduction in more severe lesions in Hanauer et al. ;18 25% in Ardizzone et al. 19). We also judged a treatment effect of 20% to be appropriate because, given the side-effect profile of MP, it was arguable that a treatment effect of significantly < 20% would be of limited clinical significance.
The sample size chosen had 80% power to detect a reduction in the frequency of recurrence from 50% in the placebo group to 30% in the treatment arm by 3 years at the 5% level of significance.
Interim analyses and stopping guidelines
An independent DMC reviewed trial data throughout the course of the trial, and there were no formal stopping rules. The study recruitment period was extended and additional sites in England and Wales were opened up to address concerns regarding the recruitment rate. Aside from this, the trial recruited according to plan.
Randomisation
Sequence generation
Randomisation was by a central, internet-based, secure password-protected randomisation database. Patient identifiers and some clinical details were entered to confirm eligibility (inclusion and exclusion criteria) and to prevent re-recruitment. The random allocation sequence was generated by the programmers at a UK Clinical Research Collaboration-registered trials unit [Edinburgh Clinical Trials Unit (ECTU)].
Type of randomisation
Randomisation was stratified according to smoking status at baseline (yes/no) and by recruiting site (31 blocks in total).
Implementation
Patients were individually enrolled by clinicians and research/specialist nurses at participating centres (predominantly nursing staff) to one of the two parallel treatment groups. Randomisation had to be within ≤ 3 months of surgery.
Once the randomisation procedure had been completed, a prescription was generated and provided to the participant by a research nurse. The prescription contained details of the number of tablets and bottle codes. The participant was asked to take the prescription to the site pharmacy to receive their allocated blinded treatment. All subsequent prescriptions were generated and processed in the same way.
Blinding
Trial medication and packaging were identical in appearance. All study staff involved in the day-to-day management of the trial, hospital staff and patients were blind to the study intervention. It was not possible to tell which treatment a patient had been allocated from the study numbers, medication packets or prescriptions. To further reduce the opportunity for accidental unblinding, the routine safety blood test results were sent to a blinded central reader for assessment by an independent monitor at each participating site who was not involved in the day-to-day management of the trial or the patient. The clinicians assessing blood results and the study management team based in Edinburgh were blind to the assigned intervention. Blinding was broken only for patients in whom an urgent clinical need was identified in terms of their clinical management, typically following a CD relapse.
Trial data management
Trial data were manually entered into a web-based data collection system by researchers at participating sites. In this system, active server pages supported web-based electronic case report forms and queried a Microsoft SQL Server 2000 database (Microsoft Corporation, Redmond, WA, USA). Data were automatically encrypted (using Secure Sockets Layer protocol V329) when entered into the forms, transferred via the internet, decrypted and stored in a secure maintained network at the University of Edinburgh. The level of encryption was dependent on the client’s web browser and operating system, but University of Edinburgh servers could offer up to 256-bit encryption. In the same network, data were then queried using statistical software (SAS; version 9.4, SAS Institute Inc., Cary, NC, USA) for the purposes of data analysis. All changes to electronic data by site staff were traceable by date and user. A copy of the trial electronic case report form is given in Report Supplementary Material 1.
Statistical methods
An analysis was undertaken using SAS. The full statistical analysis plan is given in Report Supplementary Material 2.
Primary outcomes
The primary outcome variable was postoperative recurrence of CD and its timing if it recurred. Analysis was intention to treat and was based on the application of a Cox proportional hazards model. The primary analysis included terms for treatment, the variables on which the randomisation was stratified (smoking status and recruitment site) and adjusted for baseline values of previous treatment with MP and previous treatment with azathioprine. Adjusted and unadjusted Cox proportional hazard ratios (HRs) are presented as the comparison of MP versus placebo (reference), with a HR of < 1 indicating a treatment effect in favour of MP. The adjusted analysis was considered to be the primary analysis of the primary outcome.
Secondary outcomes
The secondary outcome variable of clinical recurrence of CD (defined by a CDAI score of ≥ 150 points together with a 100-point rise in the CDAI score from baseline) or the need for anti-inflammatory rescue therapy or primary surgical intervention was analysed in the same manner as the primary outcome. For this secondary outcome, as for the primary outcome, the adjusted analysis was considered the primary analysis.
Endoscopic recurrence using both the Rutgeerts and CDEIS scoring systems was summarised by time and treatment group. Colonoscopy results at week 157 post randomisation (study visit 12) were compared between the treatment groups using a chi-squared test to compare the incidence of positive colonoscopies in the MP group and the placebo group. Both adjusted and unadjusted analyses were performed, with the adjusted analysis incorporating the same covariates as for the primary outcome, with odds ratios and 95% confidence intervals (CIs) presented. CDEIS scores at week 157 post randomisation were compared between treatment groups using a t-test. Adjusted and unadjusted analyses were performed, with treatment effects and 95% CIs presented. Faecal calprotectin results were summarised by time and treatment group, both as a continuous measure and categorically.
The use of faecal calprotectin levels as a non-invasive marker of disease recurrence was examined in two ways: first, it was considered as a time-dependent covariate in Cox proportional hazards model and, second, levels were compared descriptively between those with negative and positive colonoscopies (defined as negative if the Rutgeerts score was < i2 and positive if the Rutgeerts score was ≥ i2) at weeks 49 and 157 post randomisation. Similarly, TGN levels of the MP drug metabolite were also considered as a time-dependent covariate in the Cox proportional hazards model.
The quality-of-life variables were analysed using repeated measures analysis of covariance to evaluate treatment and treatment by time interactions. Quality of life, as measured by the IBDQ, was summarised at each visit based on observed scores and change from baseline scores for each of the four IBDQ subscales (bowel symptoms, emotional health, systemic systems and social function). Averages and totals across all subscales were summarised similarly. In addition, the overall average and overall total scores were analysed using a change from baseline repeated measures analysis of covariance to evaluate the effect of treatment over time. Quality of life, as measured by the EuroQol-5 Dimensions (EQ-5D), was summarised by treatment group across study visits.
Subgroup analyses
Subgroup analyses of the primary and secondary outcomes were carried out to assess for a treatment effect in terms of thiopurine naivety, previous treatment with infliximab or methotrexate, previous surgery, smoking status, duration of disease and age at diagnosis. The interaction between subgroup and treatment was included in the Cox regression model to determine if the treatment effect differed by subgroup. The same subgroups analysed for the primary and secondary outcomes were also analysed with respect to colonoscopy results and CDEIS scores.
Publication policy
To safeguard the integrity of the trial, the primary results of the trial were published by the group as a whole in collaboration with local investigators, and local investigators were acknowledged. 30 The success of the trial was dependent on the collaboration of many people and, particularly, the local investigators. The results were, therefore, presented to the trial local investigators first.
Organisation
A TSC and a DMC were established. No formal charter was put in place, but both committees met formally every 6 months throughout the trial in accordance with Medical Research Council (MRC)/Efficacy and Mechanism Evaluation (EME) programme requirements following circulation of a standardised report. Day-to-day management of the trial was overseen by a TMG comprising the chief investigator, the principal investigator at the lead site in Edinburgh, the lead research nurse and the trial manager. Support from the Chief Scientist Officer enabled a dedicated specialist nurse to support the trial at the initial five Scottish sites. The local Comprehensive Local Research Networks supported research nursing time and employed or re-allocated a research nurse to support all aspects of the trial at sites across England and Wales.
Confidentiality
Patients were identified by their trial number to ensure confidentiality. Stringent precautions were taken to ensure confidentiality of names and addresses at ECTU and the sites. The chief investigator and local investigators ensured conservation of records in areas to which access is restricted.
Audit
A risk-based monitoring strategy was implemented for all participating sites with onsite monitoring conducted by either the sponsor’s monitoring team or a member of the TMG at a selected number of sites depending on the issues identified at each site. Site visits were conducted during the course of the trial at University College Hospital, London, on 5 December 2012, Torbay Hospital, Torquay, on 6 November 2013, Ninewells Hospital, Dundee, on 12 March 2014, Aberdeen Royal Infirmary, Aberdeen, on 11 September 2015, Bristol Royal Infirmary, Bristol, on 30 September 2015 and St Marks Hospital, Harrow, on 7 October 2015. A central audit of trial management was conducted on 12 December 2012 at the central co-ordinating site at ECTU. In addition, an investigation was conducted by the sponsor following the lodging of a formal complaint to the Medicines and Healthcare products Regulatory Agency in July 2013 regarding the wording on an information newsletter sent to a site regarding withdrawal procedures. Recommendations were made following the investigation, but no evidence was found of improper conduct regarding withdrawal procedures.
Termination of the study
Before termination of recruitment, ECTU contacted all sites by telephone or e-mail in order to inform sites of the final date for recruitment. Once the recruitment period had expired, the internet-based randomisation database was disabled to prevent further recruitment. All recruited patients received trial medication for a maximum of 3 years (36 months) post randomisation. The final patient’s final visit took place in April 2015 and the database was locked in June 2015. A declaration of the end of trial form was sent to the Multicentre Research Ethics Committee following the formal trial end date of 30 September 2015. The following documents were archived in each site file to be kept for at least 20 years in accordance with MRC policy: original consent forms, data forms, trial-related documents and correspondence. The trial master files at ECTU will be archived for at least 20 years.
Funding
The costs for the study itself were covered by a grant from the MRC. See the MRC/EME programme website for further project information. 31 Additional support was provided by the Chief Scientist Officer.
Indemnity
If there was negligent harm during the clinical trial, then the NHS body owes a duty of care to the person harmed and NHS indemnity covered NHS staff, medical academic staff with honorary contracts and those conducting the trial. NHS indemnity did not offer no-fault compensation. The co-sponsors were responsible for ensuring that proper provision was made for insurance or indemnity to cover their liability and the liability of the chief investigator and staff.
Ethics approval and research governance
Ethics approval for the study was given by Scotland A Research Ethics Committee in August 2007 (reference number 07/MRE00/74). Local NHS management approval and appropriate site-specific assessments were obtained at each participating NHS trust. The trial was registered with the International Standard Randomised Controlled Trial Register under the reference number ISRCTN89489788 along with the European Clinical Trials Database under the reference number 2006-005800-15, and the UK Clinical Research Network Portfolio Database under the reference number 5813. A summary of the changes made to the original protocol is given in Report Supplementary Material 3 and an overview of all participant information and consent forms is given in Report Supplementary Material 4.
Chapter 3 Results
Recruitment
Patients were recruited from the following dates: June 2008 to April 2012 (46 months). The addition of 24 sites in England and Wales was in response to slower than anticipated recruitment over the initial 24 months. Initial study recruitment projection figures were based on surgical data collected from the five original Scottish hospitals in the 5 years prior to the grant application. These data indicated that 60% of patients would proceed from surgery into the trial. In practice, the proportion of patients recruited into the trial following surgery at the initial five sites was 30% as a result of higher than expected contraindications to the trial entry and patients’ greater than anticipated reluctance to receive placebo. An extension to the study recruitment period was granted in 2010, along with approval for the trial to be extended to additional centres across England and Wales. The trial completed recruitment on target according to the revised schedule on 23 April 2012, with the final patient’s final visit completed on schedule in April 2015.
Participant flow
A Consolidated Standards of Reporting Trials (CONSORT) diagram for recruitment is provided in Figure 1. In total, 328 patients were screened and 240 were randomised, thereby meeting and slightly exceeding the recruitment target of 234. Seventy-eight per cent of eligible patients were randomised to the study following screening, with 22% of patients declining to take part.
FIGURE 1.
The CONSORT diagram for TOPPIC.
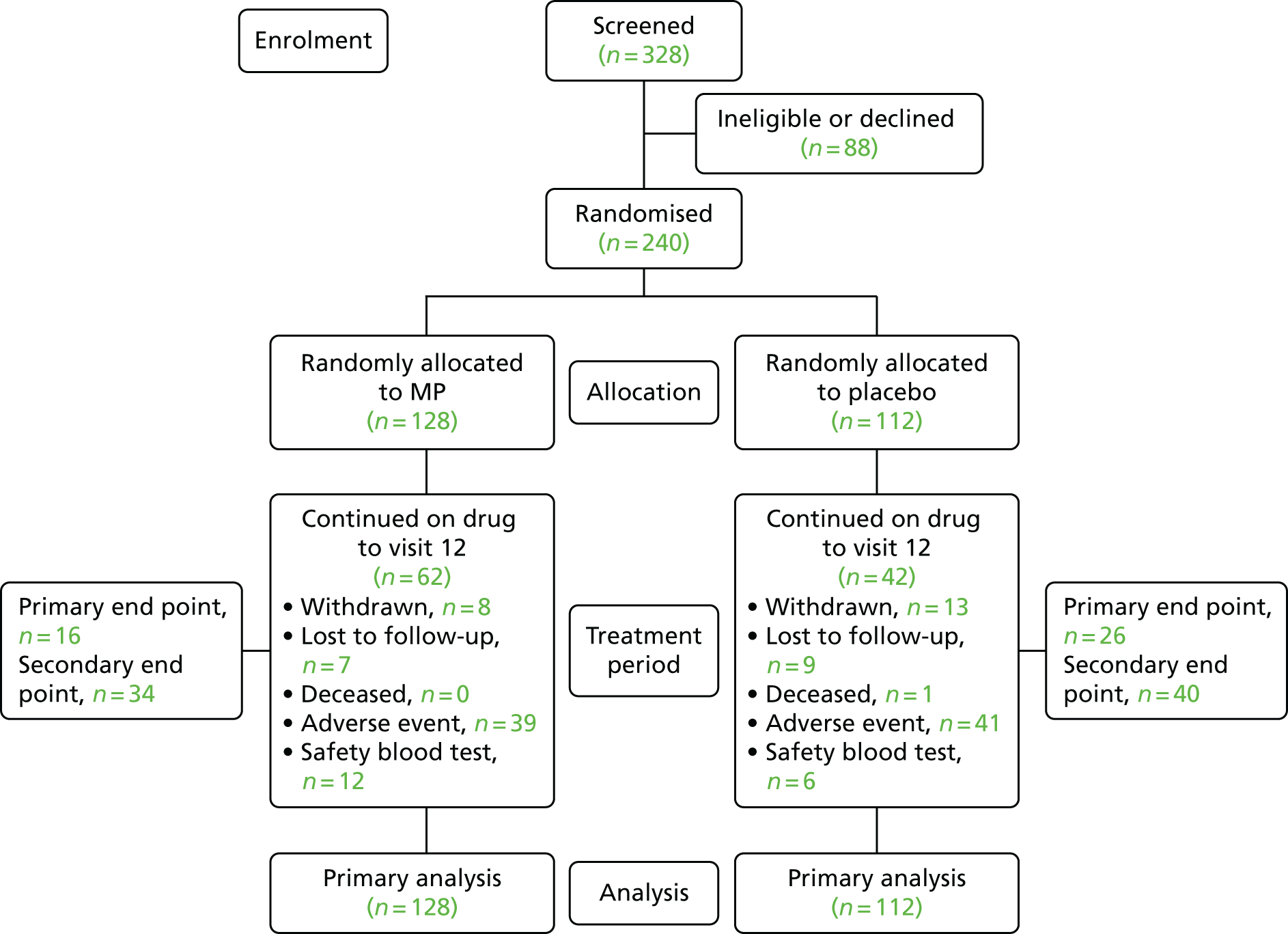
All 240 randomised patients were analysed. A total of 128 (53%) participants were randomised to receive MP and 112 (47%) to placebo. All randomised patients received at least one dose of study drug. A protocol violation was recorded involving a participant who was prescribed a study drug from the wrong treatment arm 6 weeks post randomisation; the error was reported and the correct study drug issued.
Summary of ineligible and non-recruited patients
Table 3 summarises the number of ineligible and non-recruited patients at the participating centres. Five centres did not randomise any patients during the recruitment period and were closed early.
| Centre number | Centre name | Pre-screening ineligibles | Number of candidates | Number randomised | Number of ineligibles | Number of non-recruits |
|---|---|---|---|---|---|---|
| 11 | Edinburgh | 150 | 91 | 78a | 1 | 14 |
| 12 | Aberdeen | 62 | 23 | 17 | 1 | 4 |
| 13 | Dundee | 49 | 29 | 21 | 2 | 6 |
| 14 | Glasgow Stobhill | 15 | 25 | 19 | 3 | 3 |
| 15 | Glasgow Royal Infirmary | 29 | 15 | 8 | 1 | 6 |
| 16 | Exeter | 25 | 28 | 11 | 0 | 17 |
| 17 | Coventry | 15 | 6 | 6 | 0 | 0 |
| 18 | Liverpool | 25 | 14 | 8 | 2 | 3 |
| 19 | Manchester | 20 | 5 | 3 | 0 | 2 |
| 20 | Bristol | 3 | 6 | 5 | 0 | 1 |
| 21 | UCLH | 59 | 7 | 5 | 0 | 2 |
| 24 | Oxford | 32 | 12 | 9 | 2 | 1 |
| 25 | Salford | 31 | 4 | 2 | 1 | 1 |
| 26 | Swansea | 11 | 6 | 4 | 1 | 1 |
| 27 | Royal Free | 14 | 2 | 2 | 0 | 0 |
| 28 | Barts | 24 | 1 | 1 | 0 | 0 |
| 29 | Torbay | 11 | 6 | 6 | 0 | 0 |
| 30 | Norfolk & Norwich | 0 | 1 | 0 | 0 | 1 |
| 31 | Stockton | 4 | 1 | 0 | 0 | 1 |
| 32 | Southampton | 18 | 4 | 2 | 1 | 1 |
| 33 | Plymouth | 20 | 3 | 3 | 0 | 0 |
| 34 | Durham | 34 | 8 | 6b | 1 | 2 |
| 35 | Hull | 22 | 3 | 3 | 0 | 0 |
| 36 | Leeds | 36 | 10 | 8 | 1 | 1 |
| 38 | Wolverhampton | 36 | 0 | 0 | 0 | 0 |
| 39 | North Staffordshire | 0 | 2 | 1 | 0 | 1 |
| 40 | Nottingham | 15 | 2 | 2 | 0 | 0 |
| 42 | St Marks | 14 | 1 | 1 | 0 | 0 |
| 43 | Rotherham | 20 | 5 | 4 | 0 | 0 |
| 44 | Birmingham Queen Elizabeth | 0 | 3 | 2 | 1 | 0 |
| 45 | Inverness | 2 | 1 | 1 | 0 | 0 |
| 46 | Darlington | 14 | 4 | 2 | 2 | 0 |
| 47 | Bradford | 5 | 1 | 0 | 0 | 1 |
| 48 | Birmingham City | 11 | 0 | 0 | 0 | 0 |
Characteristics used for stratification of randomisation
Randomised patients were stratified according to centre and smoking status (31 blocks in total). Table 4 shows the numbers of patients randomised per recruiting centre, with the Edinburgh site recruiting the highest proportion of patients (32.5%). Smoking status (defined as those patients smoking more than one cigarette per day) is shown in Table 5.
| Centre name | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Edinburgh | 38 (29.7) | 40 (35.7) | 78 (32.5) |
| Aberdeen | 9 (7.0) | 8 (7.1) | 17 (7.1) |
| Dundee | 11 (8.6) | 10 (8.9) | 21 (8.8) |
| Glasgow Stobhill | 10 (7.8) | 9 (8.0) | 19 (7.9) |
| Glasgow Royal Infirmary | 5 (3.9) | 3 (2.7) | 8 (3.3) |
| Exeter | 6 (4.7) | 5 (4.5) | 11 (4.6) |
| Coventry | 3 (2.3) | 3 (2.7) | 6 (2.5) |
| Liverpool | 6 (4.7) | 2 (1.8) | 8 (3.3) |
| Manchester | 2 (1.6) | 1 (0.9) | 3 (1.3) |
| Bristol | 2 (1.6) | 3 (2.7) | 5 (2.1) |
| UCLH | 3 (2.3) | 2 (1.8) | 5 (2.1) |
| Oxford | 5 (3.9) | 4 (3.6) | 9 (3.8) |
| Salford | 2 (1.6) | 0 (0.0) | 2 (0.8) |
| Swansea | 3 (2.3) | 1 (0.9) | 4 (1.7) |
| Royal Free | 1 (0.8) | 1 (0.9) | 2 (0.8) |
| Barts | 1 (0.8) | 0 (0.0) | 1 (0.4) |
| Torbay | 4 (3.1) | 2 (1.8) | 6 (2.5) |
| Southampton | 1 (0.8) | 1 (0.9) | 2 (0.8) |
| Plymouth | 2 (1.6) | 1 (0.9) | 3 (1.3) |
| Durham | 3 (2.3) | 3 (2.7) | 6 (2.5) |
| Hull | 1 (0.8) | 2 (1.8) | 3 (1.3) |
| Leeds | 4 (3.1) | 4 (3.6) | 8 (3.3) |
| North Staffordshire | 1 (0.8) | 4 (3.6) | 8 (3.3) |
| Nottingham | 1 (0.8) | 1 (0.9) | 2 (0.8) |
| St Marks | 0 (0.0) | 1 (0.9) | 2 (0.8) |
| Rotherham | 2 (1.6) | 2 (1.8) | 4 (1.7) |
| Birmingham Queen Elizabeth | 0 (0.0) | 2 (1.8) | 2 (0.8) |
| Inverness | 1 (0.8) | 0 (0.0) | 1 (0.4) |
| Darlington | 1 (0.8) | 1 (0.9) | 2 (0.8) |
| Smoking status | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Smoker | 29 (22.7) | 26 (23.2) | 55 (22.9) |
| Non-smoker | 99 (77.3) | 86 (76.8) | 185 (77.1) |
Characteristics at pre-assessment
Characteristics of randomised patients at pre-assessment are shown in Table 6. The group randomised to MP had slightly more patients who had previously been treated with MP, azathioprine and immunosuppressant agents, although all other demographics and characteristics were similar.
| Patient characteristic | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Sex | |||
| Female | 79 (61.7) | 67 (59.8) | 146 (60.8) |
| Male | 49 (38.3) | 45 (40.2) | 94 (39.2) |
| Previous treatment with MP | |||
| Yes | 14 (10.9) | 5 (4.5) | 19 (7.9) |
| No | 114 (89.1) | 106 (94.6) | 220 (91.7) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| Previous treatment with azathioprine | |||
| Yes | 80 (62.5) | 47 (42.0) | 127 (52.9) |
| No | 48 (37.5) | 64 (57.1) | 112 (46.7) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| Previous treatment with infliximab | |||
| Yes | 21 (16.4) | 15 (13.4) | 36 (15.0) |
| No | 104 (81.3) | 96 (85.7) | 200 (83.3) |
| Missing | 3 (2.3) | 1 (0.9) | 4 (1.7) |
| Previous treatment with methotrexate | |||
| Yes | 8 (6.3) | 7 (6.3) | 15 (6.3) |
| No | 120 (93.8) | 104 (92.9) | 224 (93.3) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| Previous treatment with other corticosteroids | |||
| Yes | 97 (75.8) | 79 (70.5) | 176 (73.3) |
| No | 31 (24.2) | 32 (28.6) | 63 (26.3) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| Any previous immunosuppressants | |||
| Yes | 112 (87.5) | 86 (76.8) | 198 (82.5) |
| No | 16 (12.5) | 25 (22.3) | 41 (17.1) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| CD location | |||
| Ileal | 54 (42.2) | 39 (34.8) | 93 (38.8) |
| Colonic | 4 (3.1) | 2 (1.8) | 6 (2.5) |
| Ileocolonic | 70 (54.7) | 70 (62.5) | 140 (58.3) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| CD type | |||
| Non-stricturing non-penetrating | 50 (39.1) | 45 (40.2) | 95 (39.6) |
| Stricturing | 63 (49.2) | 46 (41.1) | 109 (45.4) |
| Penetrating | 15 (11.7) | 19 (17.0) | 34 (14.2) |
| Missing | 0 (0.0) | 2 (1.8) | 2 (0.8) |
| Previous surgery | |||
| Yes | 46 (35.9) | 28 (25.0) | 74 (30.8) |
| No | 82 (64.1) | 83 (74.1) | 165 (68.8) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| TPMT | |||
| Normal | 116 (90.6) | 96 (85.7) | 212 (88.3) |
| Heterozygous | 12 (9.4) | 16 (14.3) | 28 (11.7) |
| Age at diagnosis (years) | |||
| ≤ 40 | 103 (80.5) | 87 (77.7) | 190 (79.2) |
| > 40 | 25 (19.5) | 23 (20.5) | 48 (20.0) |
| Unknown | 0 (0.0) | 2 (1.8) | 2 (0.8) |
| Duration of CD (years) | |||
| ≤ 1 | 37 (28.9) | 41 (36.6) | 78 (32.5) |
| > 1 | 91 (71.1) | 69 (61.6) | 160 (66.7) |
| Unknown | 0 (0.0) | 2 (1.8) | 2 (0.8) |
| Age (years) | |||
| Mean | 39.2 | 38.2 | 38.7 |
| Median | 38.0 | 36.0 | 38.0 |
| SD | 12.8 | 13.4 | 13.1 |
| Q1, Q3 | 28, 50 | 28, 48 | 28, 48 |
| Min., max. | 17, 67 | 17, 75 | 17, 75 |
| n (missing) | 128 (0) | 112 (0) | 240 (0) |
| Duration of CD (years) | |||
| Mean | 7.7 | 7.6 | 7.6 |
| Median | 3.0 | 4.0 | 3.0 |
| SD | 9.7 | 9.5 | 9.6 |
| Q1, Q3 | 0,11 | 0, 11 | 0,11 |
| Min., max. | 0, 39 | 0, 47 | 0,47 |
| n (missing) | 128 (0) | 110 (2) | 238 (2) |
Patient characteristics at randomisation
Table 7 shows the baseline characteristics of participants at randomisation (week 0). The two treatment groups were similar across the variables assessed, which included weight, faecal calprotectin levels, and CDAI or IBDQ scores.
| Patient characteristic | Treatment | Overall (N = 240) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Weight (kg) | |||
| Mean | 70.7 | 70.7 | 70.7 |
| Median | 69.9 | 68.8 | 69.4 |
| SD | 14.4 | 13.7 | 14.0 |
| Q1, Q3 | 58.5, 80.1 | 62.0, 76.8 | 60.0, 78.1 |
| Min., max. | 44.3, 111.9 | 43.6, 123.8 | 43.6, 123.8 |
| n (missing) | 128 (0) | 112 (0) | 240 (0) |
| Height (cm) | |||
| Mean | 168 | 169 | 169 |
| Median | 168 | 168 | 168 |
| SD | 9 | 9 | 9 |
| Q1, Q3 | 161, 175 | 162, 177 | 161, 176 |
| Min., max. | 149, 193 | 150, 193 | 149, 193 |
| n (missing) | 128 (0) | 112 (0) | 240 (0) |
| Faecal calprotectin (µg/g of faeces) | |||
| Mean | 124.4 | 160.1 | 141.2 |
| Median | 80.0 | 77.5 | 80.0 |
| SD | 170.9 | 213.9 | 192.7 |
| Q1, Q3 | 30, 125 | 40, 165 | 40, 140 |
| Min., max. | 0, 920 | 0, 1040 | 0, 1040 |
| n (missing) | 108 (20) | 96 (16) | 204 (36) |
| Neutrophil count (109/l) | |||
| Mean | 4.45 | 4.22 | 4.34 |
| Median | 4.20 | 3.88 | 4.10 |
| SD | 1.76 | 1.63 | 1.70 |
| Q1, Q3 | 3.4, 5.2 | 3.0, 5.0 | 3.2, 5.1 |
| Min., max. | 1.5, 13.8 | 1.7, 8.9 | 1.5, 13.8 |
| n (missing) | 127 (1) | 111 (1) | 238 (2) |
| CDAI score | |||
| Mean | 130 | 121 | 125 |
| Median | 111 | 112 | 112 |
| SD | 86 | 72 | 80 |
| Q1, Q3 | 63, 179 | 63, 161 | 63, 169 |
| Min., max. | 2, 459 | 5, 368 | 2, 459 |
| n (missing) | 128 (0) | 112 (0) | 240 (0) |
| IBDQ: overall average | |||
| Mean | 5.19 | 5.31 | 5.24 |
| Median | 5.38 | 5.56 | 5.47 |
| SD | 0.99 | 0.94 | 0.97 |
| Q1, Q3 | 4.4, 5.9 | 4.9, 6.0 | 4.7, 6.0 |
| Min., max. | 1.9, 7.0 | 2.7, 6.7 | 1.9, 7.0 |
| n (missing) | 128 (0) | 111 (1) | 239 (1) |
| IBDQ: overall total | |||
| Mean | 166 | 170 | 168 |
| Median | 172 | 178 | 175 |
| SD | 32 | 30 | 31 |
| Q1, Q3 | 142, 188 | 156, 191 | 150, 191 |
| Min., max. | 61, 223 | 85, 215 | 61, 223 |
| n (missing) | 128 (0) | 111 (1) | 239 (1) |
| SF-36: physical component score | |||
| Mean | 44.2 | 42.6 | 43.5 |
| Median | 45.0 | 43.2 | 44.5 |
| SD | 9.4 | 10.5 | 9.9 |
| Q1, Q3 | 37, 52 | 36, 51 | 36, 52 |
| Min., max. | 22, 60 | 6, 60 | 6, 60 |
| n (missing) | 124 (4) | 110 (2) | 234 (6) |
| SF-36: mental component score | |||
| Mean | 45.3 | 46.4 | 45.8 |
| Median | 47.0 | 49.7 | 47.7 |
| SD | 11.9 | 12.5 | 12.3 |
| Q1, Q3 | 38, 55 | 39, 56 | 38, 56 |
| Min., max. | 13, 66 | 15, 69 | 13, 69 |
| n (missing) | 124 (4) | 110 (2) | 234 (6) |
| EQ-5D: mobility, n (%) | |||
| I have no problems in walking about | 107 (83.6) | 87 (77.7) | 194 (80.8) |
| I have some problems in walking about | 21 (16.4) | 24 (21.4) | 45 (18.8) |
| I am confined to bed | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| EQ-5D: self-care, n (%) | |||
| I have no problems with self-care | 125 (97.7) | 104 (92.9) | 229 (95.4) |
| I have some problems washing or dressing myself | 3 (2.3) | 7 (6.3) | 10 (4.2) |
| I am unable to wash or dress myself | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| EQ-5D: usual activities, n (%) | |||
| I have no problems | 75 (58.6) | 61 (54.4) | 136 (56.7) |
| I have some problems | 47 (36.7) | 41 (36.6) | 88 (36.7) |
| I am unable to perform my usual activities | 6 (4.7) | 9 (8.0) | 15 (6.3) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| EQ-5D: pain/discomfort, n (%) | |||
| I have no pain or discomfort | 64 (50.0) | 56 (50.0) | 120 (50.0) |
| I have moderate pain or discomfort | 61 (47.7) | 54 (48.2) | 115 (47.9) |
| I have extreme pain or discomfort | 3 (2.3) | 1 (0.9) | 4 (1.7) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| EQ-5D: anxiety/depression, n (%) | |||
| I am not anxious or depressed | 93 (72.7) | 74 (66.1) | 167 (69.6) |
| I am moderately anxious or depressed | 33 (25.8) | 34 (30.4) | 67 (27.9) |
| I am extremely anxious or depressed | 2 (1.6) | 3 (2.7) | 5 (2.1) |
| Missing | 0 (0.0) | 1 (0.9) | 1 (0.4) |
Adherence to trial protocol
Adherence to trial medication
A summary of the duration of trial medication is shown in Table 8, with Figure 2 showing a Kaplan–Meier plot of the duration of medication in years. The reasons for non-completion of the full treatment period are detailed in Table 9. One hundred and thirty-six patients (56.7%) did not complete 3 years of trial medication, with 80 (58.8%) patients not completing as the result of adverse events, 21 (15.4%) because of early withdrawal, 18 (13.2%) because of abnormal safety blood results, 16 (11.8%) being lost to follow-up and one (0.7%) patient dying.
| Duration of trial medication (years) | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| ≥ 3 | 62 (48.4) | 42 (37.5) | 104 (43.3) |
| < 3 | 66 (51.6) | 70 (62.5) | 136 (56.7) |
FIGURE 2.
Kaplan–Meier plot showing duration of trial medication in years.

| Reason | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Adverse event | 39 (59.1) | 41 (58.6) | 80 (58.8) |
| Blood test result | 12 (18.2) | 6 (8.6) | 18 (13.2) |
| Early withdrawal | 8 (12.1) | 13 (18.6) | 21 (15.4) |
| Deceased | 0 (0.0) | 1 (1.4) | 1 (0.7) |
| Lost to follow-up | 7 (10.6) | 9 (12.9) | 16 (11.8) |
Dosage was adjusted throughout the trial in accordance with the protocol (see Tables 1 and 2). Table 10 provides a summary of doses at first and final visits. Table 11 summarises doses at final visit compared with dose first allocated. Only a small proportion of patients continued on the same dosage (n = 63, 26.3%) or at an increased dosage (n = 13, 5.4%); in the majority either the dose was decreased (n = 68, 28.3%) or the trial medication was stopped altogether (n = 96, 40.0%). The dosage was decreased in a higher proportion of patients in the MP group (39.1%) than in the placebo group (16.1%), whereas a higher proportion in the placebo group than in the MP group remained on the same dosage (37.5% compared with 16.4%, respectively) or stopped trial medication (42.9% compared with 37.5, respectively). The dose was increased in a small number of patients, but the proportion was marginally higher in the MP group than in the placebo group (7.0% compared with 3.6%).
| Dosage | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Dose (mg) allocated at first visit | |||
| 50 alternate days | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| 50 daily | 17 (13.3) | 17 (15.2) | 34 (14.2) |
| 50/100 alternate days | 73 (57.0) | 62 (55.4) | 135 (56.3) |
| 100 daily | 34 (26.6) | 27 (24.1) | 61 (25.4) |
| 150 daily | 4 (3.1) | 5 (4.5) | 9 (3.8) |
| Dose allocated at final visit | |||
| Stop | 48 (37.5) | 48 (42.9) | 96 (40.0) |
| 50 alternate days | 17 (13.3) | 5 (4.5) | 22 (9.2) |
| 50 daily | 33 (25.8) | 20 (17.9) | 53 (22.1) |
| 50/100 alternate days | 17 (13.3) | 25 (22.3) | 42 (17.5) |
| 100 daily | 13 (10.2) | 13 (11.6) | 26 (10.8) |
| 150 daily | 0 (0.0) | 1 (0.9) | 1 (0.4) |
| Final dose change | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Dose decreased | 50 (39.1) | 18 (16.1) | 68 (28.3) |
| Dose stayed the same | 21 (16.4) | 42 (37.5) | 63 (26.3) |
| Dose increased | 9 (7.0) | 4 (3.6) | 13 (5.4) |
| Trial medication stopped | 48 (37.5) | 48 (42.9) | 96 (40.0) |
Adherence to the trial visits and withdrawal
Table 12 shows the number of patients who reached the end of the study (week 157 post randomisation; n = 161, 67.1%). Seventy-nine patients did not reach the final study visit, with 52 withdrawn early at their own choice or by their clinician and a further 26 being lost to follow-up or non-attenders; there was one death.
| Parameter | Category | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | |||
| Week 12 (week 157, n = 161) | Patient attended | 89 (69.5) | 72 (64.3) | 161 (67.1) |
| Patient did not attend | 7 (5.5) | 2 (1.8) | 9 (3.8) | |
| Early withdrawal | 24 (18.8) | 26 (23.2) | 50 (20.8) | |
| Withdrawn by clinician | 1 (0.8) | 1 (0.9) | 2 (0.8) | |
| Lost to follow-up | 7 (5.5) | 10 (8.9) | 17 (7.1) | |
| Deceased | 0 (0.0) | 1 (0.9) | 1 (0.4) | |
The length of patient follow-up, in months, is shown in Table 13. The mean follow-up period was 28.6 months overall (maximum follow-up was 48 months), with a slightly longer follow-up in the MP group than in the placebo group (29.3 months vs. 27.8 months).
| Months | Treatment | Overall (N = 240) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Mean | 29.3 | 27.8 | 28.6 |
| Median | 36.0 | 36.0 | 36.0 |
| SD | 12.2 | 12.2 | 12.2 |
| Q1, Q3 | 28, 36 | 20, 36 | 23, 36 |
| Min., max. | 0, 48 | 0, 38 | 0, 48 |
| n (missing) | 128 (0) | 112 (0) | 240 (0) |
Adherence to study blinding
Treatment blinding was broken for 12 patients, and the reasons are listed in Table 14.
| Patient ID | Centre | Date | Explanation | |
|---|---|---|---|---|
| Randomisation | Unblinding | |||
| Treatment group | ||||
| MP | ||||
| 11003 | Edinburgh | 13 June 2008 | 27 January 2009 | Primary end point reached |
| 11030 | Edinburgh | 3 November 2009 | 27 February 2014 | Adverse event post trial |
| 11064 | Edinburgh | 21 February 2011 | 5 April 2012 | Other |
| Placebo | ||||
| 11053 | Edinburgh | 18 August 2010 | 29 February 2012 | Other |
| 11078 | Edinburgh | 5 September 2011 | 31 May 2012 | Other |
| 11082 | Edinburgh | 13 September 2011 | 2 September 2013 | Other |
| 12002 | Aberdeen | 30 October 2008 | 3 February 2009 | SUSAR |
| 12014 | Aberdeen | 31 March 2010 | 17 May 2010 | SUSAR |
| 14010 | Glasgow Stobhill | 6 October 2009 | 26 October 2009 | SUSAR |
| 16004 | Exeter | 17 June 2010 | 25 November 2010 | SUSAR |
| 18007 | Liverpool | 3 May 2011 | 9 July 2013 | SUSAR |
| 44003 | Birmingham Queen Elizabeth | 16 January 2012 | 17 March 2015 | Adverse event post trial |
Adherence to trial procedures
A total of nine protocol violations were recorded during the course of the study: five relating to study sample storage and four relating to study dosage and affecting specific sites and specific patients. A total of 216 deviations were recorded. The deviation categories are provided in Table 15.
| Protocol deviation | Overall (n) |
|---|---|
| Study visit missed or out of window | 49 |
| Safety bloods missed or out of window | 89 |
| Colonoscopy missed or out of window | 22 |
| Questionnaire missed or out of window | 3 |
| Calprotectin sample missed or out of window | 25 |
| Medication miscounted or taken incorrectly | 11 |
| TGN sample missed or out of window | 3 |
| Other | 14 |
Concomitant medication
Concomitant medications are shown in Table 16.
| Parameter | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Concomitant medications taken? | |||
| Yes | 119 (93.0) | 111 (99.1) | 230 (95.8) |
| No | 9 (7.0) | 1 (0.9) | 10 (4.2) |
| Total number of concomitant medications | 1051 | 965 | 2016 |
| Number of prohibited and non-prohibited medications | |||
| Prohibited | 93 (8.8) | 126 (13.1) | 219 (10.9) |
| Non-prohibited | 958 (91.2) | 839 (86.9) | 1797 (89.1) |
| Number of occasions concomitant medications taken | |||
| 1 | 7 (5.9) | 7 (6.3) | 14 (6.1) |
| 2 | 10 (8.4) | 12 (10.8) | 22 (9.6) |
| 3 | 9 (7.6) | 7 (6.3) | 16 (7.0) |
| 4 | 9 (7.6) | 5 (4.5) | 14 (6.1) |
| 5 | 12 (10.1) | 12 (10.8) | 24 (10.4) |
| 6–10 | 31 (26.1) | 33 (29.7) | 64 (27.8) |
| 11–15 | 22 (18.5) | 21 (18.9) | 43 (18.7) |
| > 15 | 19 (16.0) | 14 (12.6) | 33 (14.3) |
| Requiring rescue therapy for CD? | |||
| Yes | 30 (25.2) | 37 (33.3) | 67 (29.1) |
| No | 89 (74.8) | 74 (66.7) | 163 (70.9) |
| Number of occasions rescue therapy taken for CD | |||
| 1 | 10 (33.3) | 10 (27.0) | 20 (29.9) |
| 2 | 3 (10.0) | 11 (29.7) | 14 (20.9) |
| 3 | 8 (26.7) | 6 (16.2) | 14 (20.9) |
| 4 | 4 (13.3) | 4 (10.8) | 8 (11.9) |
| 5 | 1 (3.3) | 2 (5.4) | 3 (4.5) |
| > 5 | 4 (13.3) | 4 (10.8) | 8 (11.9) |
Outcomes and estimation
Primary outcome
The primary outcome was postoperative clinical recurrence of CD, defined as a CDAI score of > 150 points with an increase from baseline of 100 points, together with the need for anti-inflammatory rescue therapy or primary surgical intervention.
As shown in Tables 17 and 18, the primary outcome was reached by 42 patients (17.5%); 16 out of 128 (12.5%) on MP versus 26 out of 112 (23.2%) on placebo (HR 0.535, 95% CI 0.27 to 1.06; adjusted p = 0.073; HR 0.527, 95% CI 0.28 to 0.99; unadjusted p = 0.046). Of the 42 who reached the primary end point, 37 participants (88%) met the CDAI score trigger and had rescue therapy initiated, whereas five (12%) met the CDAI score trigger and had both rescue therapy and primary surgical intervention.
| Primary outcome | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Primary outcome met? | |||
| Yes | 16 (12.5) | 26 (23.2) | 42 (17.5) |
| No | 112 (87.5) | 86 (76.8) | 198 (82.5) |
| Analysis type | HR | 95% CI level | p > χ2 | |
|---|---|---|---|---|
| Lower | Upper | |||
| Adjusted | 0.535 | 0.27 | 1.06 | 0.073 |
| Unadjusted | 0.527 | 0.28 | 0.99 | 0.046 |
The HR of < 1 for both the adjusted and unadjusted analyses indicates a treatment effect in favour of MP. The associated confidence limits for the adjusted and unadjusted results demonstrate borderline statistical significance at the 5% level.
For the 42 patients who reached primary outcome, Table 19 shows the follow-up and recurrence for the 26 (10.8%) participants reaching the primary outcome and completing the full 36-month follow-up, and for the 16 (6.7%) participants reaching the primary outcome with < 36 months of follow-up in the trial.
| Follow-up and recurrence | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Follow-up of ≥ 3 years and primary outcome | 11 (8.6) | 15 (13.4) | 26 (10.8) |
| Follow-up of < 3 years and primary outcome | 5 (3.9) | 11 (9.8) | 16 (6.7) |
| Other | 112 (87.5) | 86 (76.8) | 198 (82.5) |
Secondary outcome: clinical recurrence
The secondary outcome of clinical recurrence was postoperative clinical recurrence of CD, defined as a CDAI score of > 150 points with an increase from baseline of 100 points, or the need for anti-inflammatory rescue therapy or primary surgical intervention.
The secondary outcome events and statistical analysis are summarised in Tables 20 and 21 respectively. The secondary outcome was reached by 74 (30.8%) patients: 34 out of 128 (26.6%) on MP versus 40 out of 112 (35.7%) on placebo (HR 0.737, 95% CI 0.44 to 1.23; adjusted p = 0.243; HR 0.746, 95% CI 0.47 to 1.18; unadjusted p = 0.211). Similar to the primary outcome, the HRs are in favour of MP, but the associated CIs indicate a lack of statistical significance.
| Secondary outcome | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Secondary outcome met? | |||
| Yes | 34 (26.6) | 40 (35.7) | 74 (30.8) |
| No | 94 (73.4) | 72 (64.3) | 166 (69.2) |
| Analysis type | HR | 95% CI limit | p > χ2 | |
|---|---|---|---|---|
| Lower | Upper | |||
| Adjusted | 0.737 | 0.44 | 1.23 | 0.243 |
| Unadjusted | 0.746 | 0.47 | 1.18 | 0.211 |
Crohn’s Disease Activity Index score
The mean score change from baseline in the CDAI for patients is shown in Figure 3 from randomisation at visit 2 (week 0) to the final study visit at visit 12 (104 weeks post randomisation).
FIGURE 3.
Mean change from baseline in CDAI scores per study visit.

Secondary outcome: surgical intervention
Tables 22 and 23 present a summary of the patients who have had surgical intervention and the associated time (in days) to the intervention.
| Days | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Had surgical intervention? | |||
| Yes | 3 (2.3) | 4 (3.6) | 7 (2.9) |
| No | 125 (97.7) | 108 (96.4) | 233 (97.1) |
| Number of occasions that surgery was undertaken | |||
| 1 | 2 (66.7) | 4 (100) | 6 (85.7) |
| 2 | 1 (33.3) | 0 (0.0) | 1 (14.3) |
| Time to surgical intervention (days) | Treatment | Overall (N = 240) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Mean | 460.7 | 582.8 | 530.4 |
| Median | 629.0 | 581.0 | 629.0 |
| SD | 357.6 | 511.7 | 421.7 |
| Q1, Q3 | 50, 703 | 149, 1017 | 60, 924 |
| Min., max. | 50, 703 | 60, 1109 | 50, 1109 |
| n (missing) | 3 (0) | 4 (0) | 7 (0) |
A small number of patients (7, 2.9%) had a secondary surgical intervention during the course of the trial, with similar numbers across the MP and placebo groups. The mean time to surgical intervention was 530.4 days, with a mean time being shorter in the MP group than in the placebo group (460.7 days vs. 582.8 days).
Secondary outcome: endoscopic recurrence
Tables 24 and 25 present a summary of endoscopic recurrence, as defined by Rutgeerts score, at visits 6 (week 49) and 12 (week 157), categorised by overall score and as either negative or positive. Figure 4 presents the number of negative and positive scores as bar charts.
| Time point | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Visit 6 (week 49, n = 208) | |||
| i0: no lesions | 33 (29.7) | 14 (14.4) | 47 (22.6) |
| i1: ≤ 5 aphthous ulcers | 24 (21.6) | 28 (28.9) | 52 (25.0) |
| i2: > 5 aphthous ulcers | 18 (16.2) | 19 (19.6) | 37 (17.8) |
| i3: diffuse aphthous ileitis | 11 (9.9) | 13 (13.4) | 24 (11.5) |
| i4: diffuse ileal inflammation | 5 (4.5) | 3 (3.1) | 8 (3.8) |
| Other | 4 (3.6) | 0 (0.0) | 4 (1.9) |
| Missing | 16 (14.4) | 20 (20.6) | 36 (17.3) |
| Visit 12 (week 157, n = 161) | |||
| i0: no lesions | 20 (22.5) | 9 (12.5) | 29 (18.0) |
| i1: ≤ 5 aphthous ulcers | 18 (20.2) | 20 (27.8) | 38 (23.6) |
| i2: > 5 aphthous ulcers | 18 (20.2) | 11 (15.3) | 29 (18.0) |
| i3: diffuse aphthous ileitis | 4 (4.5) | 5 (6.9) | 9 (5.6) |
| i4: diffuse ileal inflammation | 7 (7.9) | 12 (16.7) | 19 (11.8) |
| Other | 2 (2.2) | 2 (2.8) | 4 (2.5) |
| Missing | 20 (22.5) | 13 (18.1) | 33 (20.5) |
| Time point | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Visit 6 (week 49, n = 208) | |||
| Negative (< i2) | 57 (51.4) | 42 (43.3) | 99 (47.6) |
| Positive (≥ i2) | 34 (30.6) | 35 (36.1) | 69 (33.2) |
| Other | 4 (3.6) | 0 (0.0) | 4 (1.9) |
| Missing | 16 (14.4) | 20 (20.6) | 36 (17.3) |
| Visit 12 (week 157, n = 161) | |||
| Negative (< i2) | 38 (42.7) | 29 (40.3) | 67 (41.6) |
| Positive (≥ i2) | 29 (32.6) | 28 (38.9) | 57 (35.4) |
| Other | 2 (2.2) | 2 (2.8) | 4 (2.5) |
| Missing | 20 (22.5) | 13 (18.1) | 33 (20.5) |
FIGURE 4.
Colonoscopy by study visit (visit 6 was week 49 post randomisation and visit 12 was week 157 post randomisation). (a) Visit 6 and (b) visit 12.

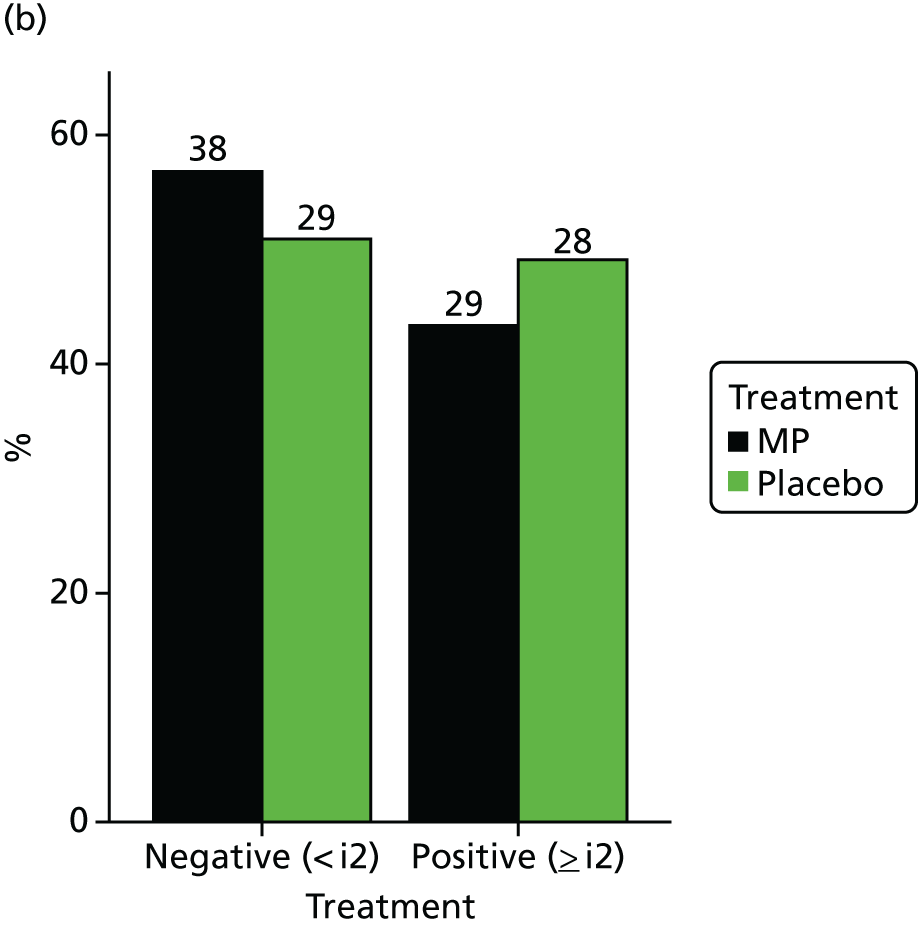
Colonoscopy results (defined as negative or positive using the Rutgeerts score) are also summarised and the results of the statistical analysis of visit 12 colonoscopy results are presented in Table 26.
| Analysis type | Visit 12 odds ratio | 95% CI limit | p-value | |
|---|---|---|---|---|
| Lower | Upper | |||
| Adjusted | 0.66 | 0.26 | 1.67 | 0.382 |
| Unadjusted | 0.79 | 0.39 | 1.61 | 0.516 |
Comparing the incidence of positive colonoscopies at visit 12 for the MP group with the placebo group gave an adjusted odds ratio of 0.66 (95% CI 0.26 to 1.67; p = 0.382) and an unadjusted odds ratio of 0.79 (95% CI 0.39 to 1.61; p = 0.516), indicating no significant difference between treatment arms.
Endoscopic recurrence was also assessed using CDEIS score. Figure 5 shows CDEIS scores in a box plot, with a higher CDEIS score indicating greater severity of disease. The results of the statistical analysis are shown in Table 27, with an adjusted treatment effect of –0.612 (95% CI –1.92 to 0.69; p = 0.354), where an effect < 0 indicates a treatment effect in favour of MP. Higher CDEIS scores were recorded for the placebo group than for those patients allocated to MP at visits 6 and 12, although the difference was not statistically significant.
FIGURE 5.
Crohn’s Disease Endoscopic Index of severity score by study visit (visit 6 was 49 weeks post randomisation and visit 12 was 157 weeks post randomisation). (a) Visit 6 and (b) visit 12. Mean CDEIS scores are indicated by a diamond in the box plot. The minimum is indicated by the bottom of the lower whisker, the maximum is indicated by the top of the upper whisker. The lower quartile is indicated by the horizontal line at the bottom of the box and the upper quartile is indicated by the horizontal line at the top of the box. Outliers are shown as small circles, identified as being 1.5 times outside the interquartile range. A higher CDEIS score indicates greater severity of the disease.


| Analysis type | Visit 12 treatment effect | 95% CI limit | p-value | |
|---|---|---|---|---|
| Lower | Upper | |||
| Adjusted | –0.612 | –1.92 | 0.69 | 0.354 |
| Unadjusted | –0.661 | –1.89 | 0.57 | 0.289 |
Secondary outcome: faecal calprotectin levels
Faecal calprotectin results were summarised by time and treatment group both as a continuous measure and categorically. The continuous change from baseline to each time point was also summarised and is presented in Figure 6, indicating an increase from baseline across all visits in the placebo group, in contrast to a decrease from baseline across all visits in the MP group. Figure 7 shows a categorical summary of faecal calprotectin levels, indicating the proportions in both MP and placebo groups.
FIGURE 6.
Mean change from baseline in faecal calprotectin level. Faecal calprotectin results of < 20µg/g faeces have been included in the summary with a value of 0.

FIGURE 7.
Categorical summary of faecal calprotectin levels (µg/g) in faeces. (a) MP group and (b) placebo group. Percentages are determined on non-missing data only.
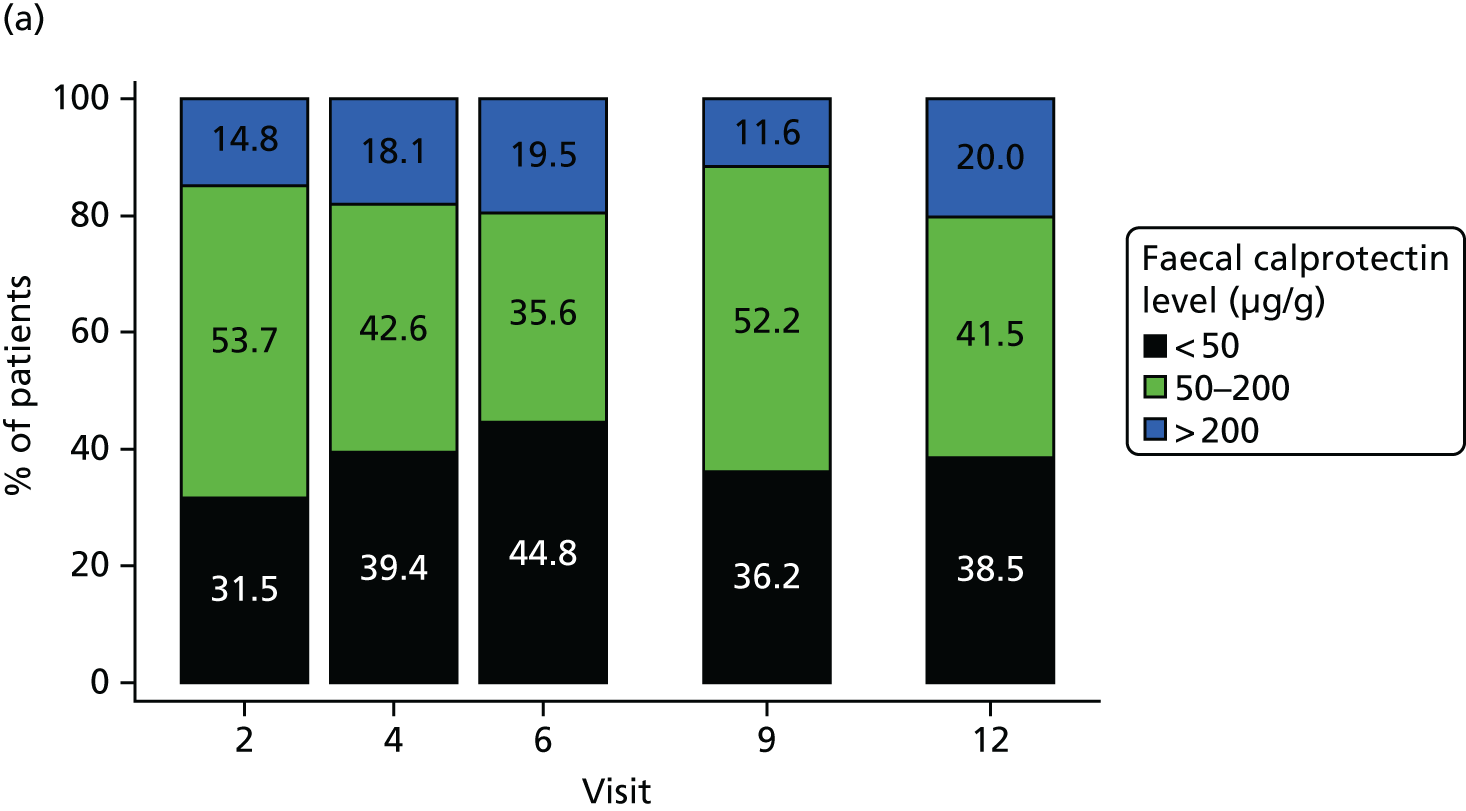
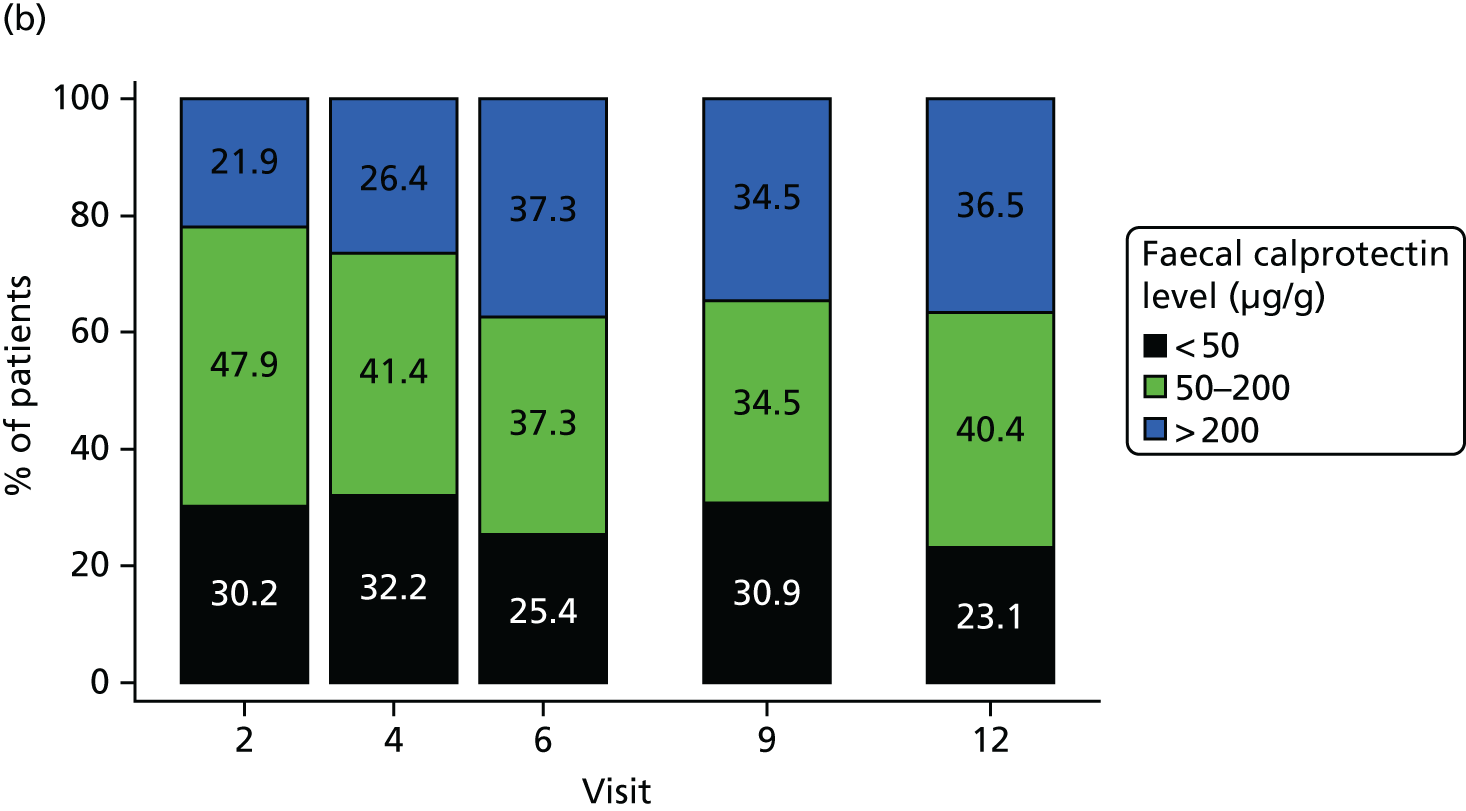
Faecal calprotectin levels were also assessed as a non-invasive marker of clinical recurrence of CD by incorporating it as a time-varying covariate in the Cox proportional hazards model. Tables 28 and 29 summarise faecal calprotectin levels by colonoscopy result at visits 6 and 12. The results of the statistical analysis are presented in Table 30 and indicate that faecal calprotectin levels are a significant predictor of the primary outcome. For each 100 µg/g increase in faecal calprotectin, the probability of reaching the primary outcome increased by 17.7%. These results should, however, be treated with caution as only 31 out of 42 events have been included in this analysis as a result of missing data.
| Colonoscopy result | Treatment | Overall (N = 240) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Negative (< i2) (n = 99, 47.6%) | |||
| Mean | 98 | 181 | 133 |
| Median | 45 | 110 | 60 |
| SD | 148 | 243 | 197 |
| Q1, Q3 | 20, 90 | 30, 180 | 20, 130 |
| Min., max. | 20, 810 | 20, 1080 | 20, 1080 |
| n (missing) | 44 (13) | 33 (9) | 77 (22) |
| Positive (≥ i2) (n = 57, 35.4%) | |||
| Mean | 240 | 353 | 291 |
| Median | 170 | 220 | 170 |
| SD | 305 | 308 | 308 |
| Q1, Q3 | 50, 260 | 120, 590 | 90, 350 |
| Min., max. | 20, 1200 | 20, 1130 | 20, 1200 |
| n (missing) | 27 (7) | 22 (13) | 49 (20) |
| Colonoscopy result | Treatment | Overall (N = 240) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Negative (< i2) (n = 99, 47.6%) | |||
| Mean | 141 | 133 | 137 |
| Median | 45 | 70 | 60 |
| SD | 174 | 170 | 170 |
| Q1, Q3 | 20, 260 | 20,200 | 20, 200 |
| Min., max. | 20, 660 | 20,750 | 20, 750 |
| n (missing) | 24 (14) | 23 (6) | 47 (20) |
| Positive (≥ i2) (n = 57, 35.4%) | |||
| Mean | 146 | 262 | 200 |
| Median | 110 | 200 | 140 |
| SD | 143 | 203 | 181 |
| Q1, Q3 | 50, 180 | 130, 270 | 80, 240 |
| Min., max. | 20, 530 | 30, 750 | 20, 750 |
| n (missing) | 22 (7) | 19 (9) | 41 (16) |
| Model | HR | 95% CI limit | p > χ2 | Number of primary events | |
|---|---|---|---|---|---|
| Lower | Upper | ||||
| 100-unit change in calprotectin | 1.177 | 1.082 | 1.282 | 0.0002 | 31 |
Secondary outcome: thioguanine nucleotide concentration levels
Thioguanine levels were summarised by time and treatment group both as a continuous measure and categorically following independent blind review. Figure 8 shows the mean change from baseline in TGN levels, with a slight and unexplained rise in TGN levels in the placebo arm. Figure 9 presents a categorical summary of TGN levels in the MP group at each of the five time points measured throughout the trial.
FIGURE 8.
Mean change from baseline in TGN levels. RBC, red blood cell.

FIGURE 9.
Categorical summary of TGN levels (pmol/8 × 108 red blood cells). Percentages are determined on non-missing data only. RBC, red blood cell.

In a similar manner to faecal calprotectin levels, TGN was assessed as a non-invasive marker of clinical recurrence of CD by incorporating results from MP patients only as a time-varying covariate in the Cox proportional hazards model. As shown in Table 31, the corresponding time-varying covariate analysis of TGN concentrations (data available for 14 of the 16 patients receiving MP only) indicated that, for every 100-pmol/8 × 108 red blood cell increase in TGN, the hazard of reaching the primary out come decreased by 20% (HR 0.800, 95% CI 0.565 to 1.132; p = 0.207).
| Model | HR | 95% CI limit | p > χ2 | Number of primary events | |
|---|---|---|---|---|---|
| Lower | Upper | ||||
| 100-pmol/8 × 108 red blood cells change in TGN | 0.800 | 0.565 | 1.132 | 0.207 | 14 |
Secondary outcome: changes in self-rated quality-of-life scores
Patient-reported outcome measures (as measured by IBDQ) were summarised at each visit based on observed scores and change from baseline scores for each of the four IBDQ subscales (bowel symptoms, emotional health, systemic systems and social function). Averages and totals across all subscales were summarised similarly.
In addition, the overall average IBDQ score and overall total IBDQ score were statistically analysed using a repeated measures analysis of covariance, modelling change from baseline, fitting terms for treatment, visit and the interaction between treatment and time.
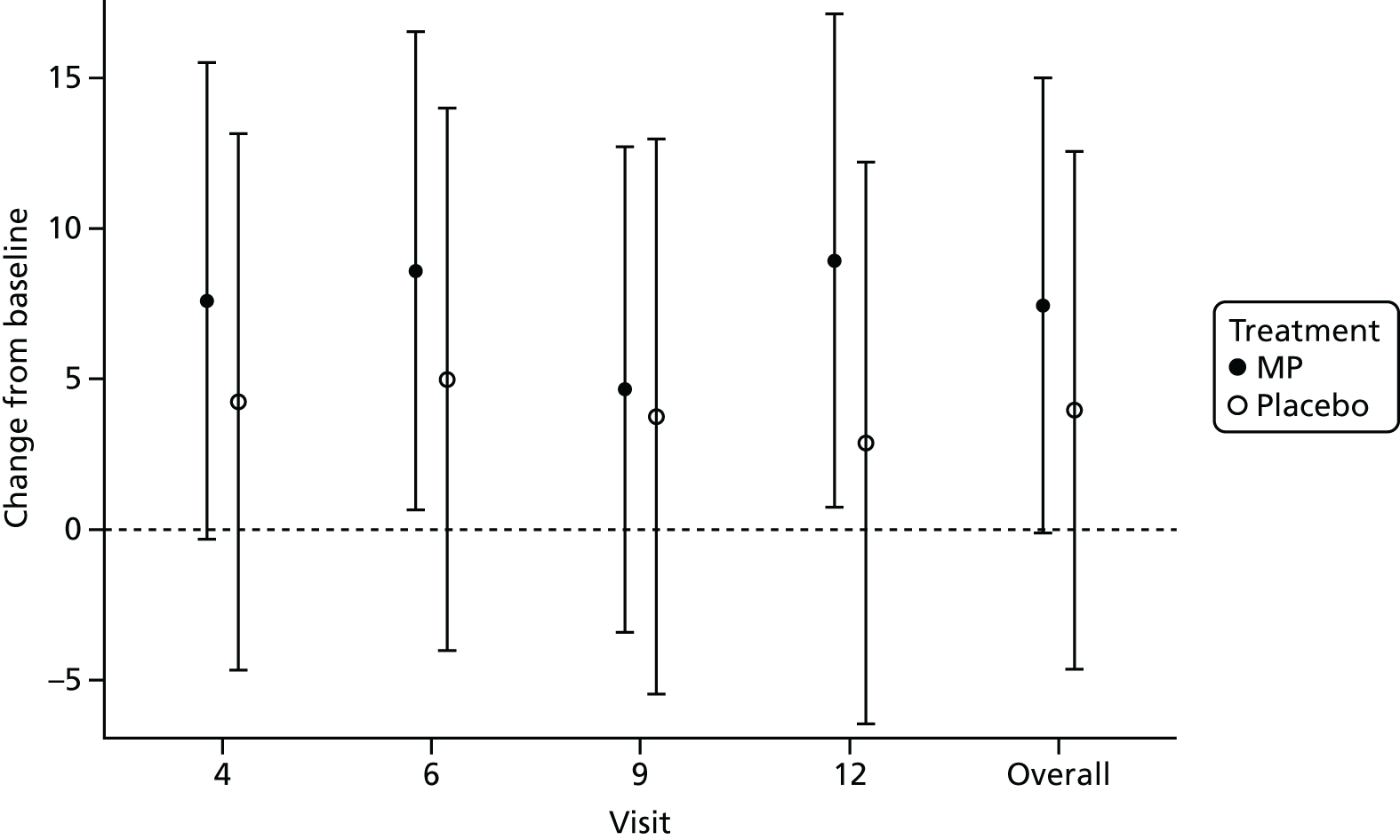
For both the overall average and overall total, there were no significant differences between treatment and placebo groups across all study visits and the study overall. However, both treatment groups showed an improvement in quality of life across all study visits in comparison with baseline, with the MP group demonstrating the greater change across all visits. Figures 10 and 11 depict the results of the statistical analysis for the overall average IBDQ and overall total IBDQ, respectively.
FIGURE 10.
Change from baseline final model treatment effects and 95% CIs for overall average IBDQ.
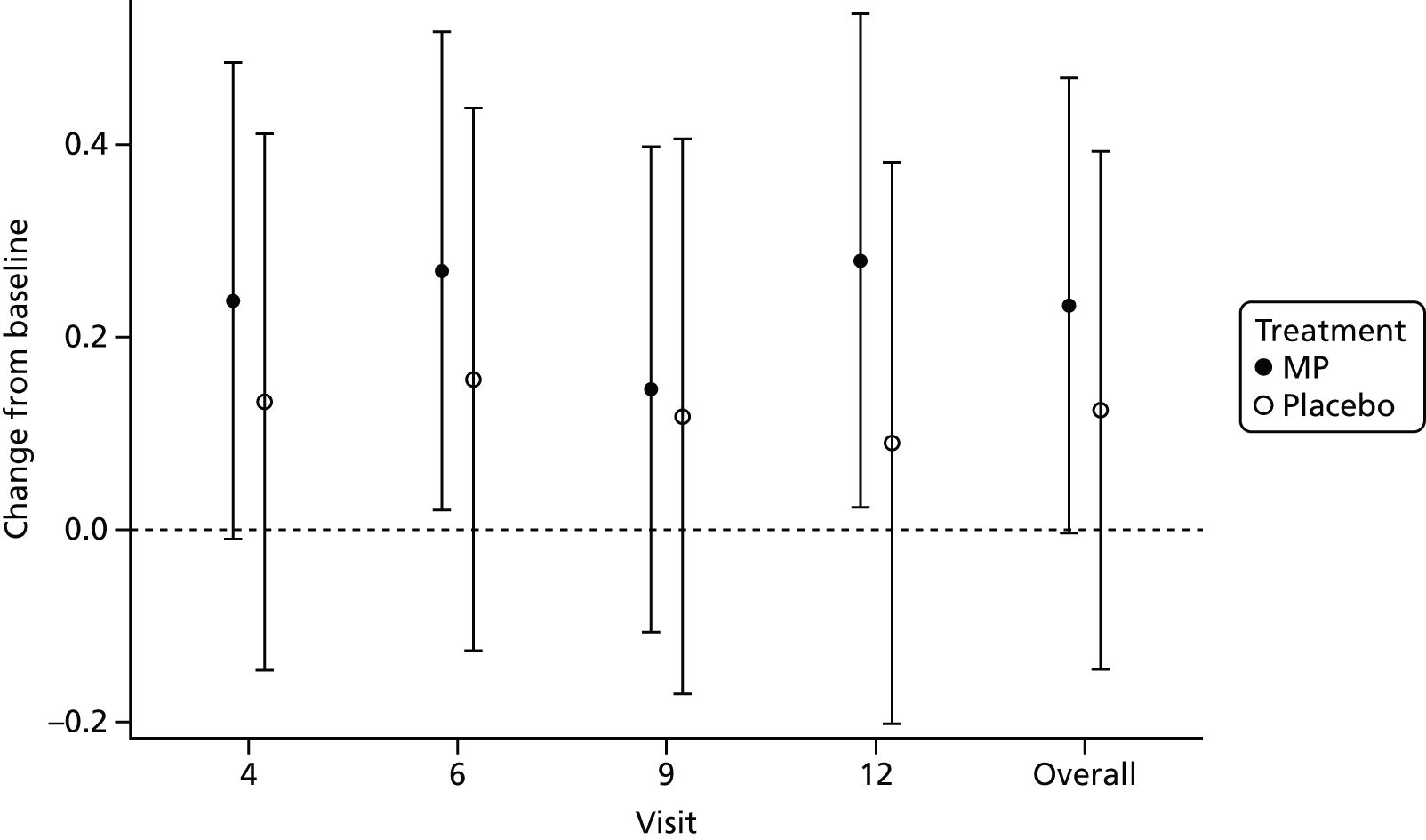
FIGURE 11.
Change from baseline final model treatment effects and 95% CIs for overall total IBDQ.
Safety outcomes
Adverse events
Of 1747 reported adverse events, 355 (20.3%) were infections, although only seven (0.4%) necessitated hospitalisation, with higher rates in the group allocated to placebo. The majority of adverse events were classed as either mild or moderate in severity [868 (91.6%) in the MP group and 728 (91.1%) in the placebo group]. Adverse events caused discontinuation of treatment in 80 patients overall (33%): 39 of the 128 (30%) patients in the MP group versus 41 of the 112 (36.6%) patients in the placebo group. Adverse events in the trial cohort overall are shown in Table 32, with serious adverse events in Table 33. There were two cases of pancreatitis (0.1%, one in the MP group and one in the placebo group) and four malignancies (0.2%, three in the MP group and one in the placebo group): basal cell carcinoma, breast cancer and two cases of lentigo maligna. One participant died of coronary heart disease in the placebo group.
| Parameter | Treatment, n (%) | Overall (N = 240), n (%) | |
|---|---|---|---|
| MP (N = 128) | Placebo (N = 112) | ||
| Had adverse event | |||
| Yes | 121 (94.5) | 105 (93.8) | 226 (94.2) |
| No | 7 (5.5) | 7 (6.3) | 14 (5.8) |
| Severity of worst event | |||
| Mild | 14 (11.6) | 8 (7.6) | 22 (9.7) |
| Moderate | 65 (53.7) | 59 (56.2) | 124 (54.9) |
| Severe | 41 (33.9) | 38 (36.2) | 79 (35.0) |
| Missing | 1 (0.8) | 0 (0.0) | 1 (0.4) |
| Total number of adverse events | 948 | 799 | 1747 |
| All adverse events split by category | |||
| Uncoded | 1 (0.1) | 1 (0.1) | 2 (0.1) |
| GI symptoms: nausea/vomiting | 78 (8.2) | 41 (5.1) | 119 (6.8) |
| GI symptoms: abdominal pain | 132 (13.9) | 141 (17.6) | 273 (15.6) |
| GI symptoms: constipation/diarrhoea | 54 (5.7) | 56 (7.0) | 110 (6.3) |
| GI symptoms: other | 53 (5.6) | 40 (5.0) | 93 (5.3) |
| Worsening CD | 41 (4.3) | 37 (4.6) | 78 (4.5) |
| Rash | 35 (3.7) | 17 (2.1) | 52 (3.0) |
| Headache | 61 (6.4) | 38 (4.8) | 99 (5.7) |
| Infections | 171 (18.0) | 184 (23.0) | 355 (20.3) |
| Pain | 30 (3.2) | 19 (2.4) | 49 (2.8) |
| Cancers | 3 (0.3) | 1 (0.1) | 4 (0.2) |
| Deranged LFT | 4 (0.4) | 5 (0.6) | 9 (0.5) |
| Pancreatitis | 1 (0.1) | 1 (0.1) | 2 (0.1) |
| Joint pain/arthralgia | 72 (7.6) | 65 (8.1) | 137 (7.8) |
| Other | 212 (22.4) | 153 (19.1) | 365 (20.9) |
| System Organ Class (SOC1) | Treatment, number of | Overall, number of | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MP | Placebo | ||||||||
| Patients | Events | Related events | Patients | Events | Related events | Patients | Events | Related events | |
| Cardiac disorders | 3 | 3 | 0 | 2 | 2 | 0 | 5 | 5 | 0 |
| Congenital, familial and genetic disorders | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| Eye disorders | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| GI disorders | 23 | 26 | 2 | 29 | 33 | 4 | 52 | 59 | 6 |
| General disorders and administration site conditions | 1 | 1 | 0 | 1 | 1 | 0 | 2 | 2 | 0 |
| Hepatobiliary disorders | 1 | 1 | 1 | 1 | 1 | 0 | 2 | 2 | 1 |
| Infections and infestations | 4 | 7 | 0 | 3 | 3 | 1 | 7 | 10 | 1 |
| Injury, poisoning and procedural complications | 1 | 1 | 0 | 1 | 1 | 0 | 2 | 2 | 0 |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 1 | 1 | 0 | 1 | 1 | 0 | 2 | 2 | 0 |
| Nervous system disorders | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| Renal and urinary disorders | 4 | 6 | 0 | 2 | 2 | 0 | 6 | 8 | 0 |
| Reproductive system and breast disorders | 2 | 2 | 0 | 0 | 0 | 0 | 2 | 2 | 0 |
| Skin and subcutaneous tissue disorders | 1 | 1 | 0 | 1 | 2 | 1 | 2 | 3 | 1 |
| Surgical and medical procedures | 9 | 9 | 0 | 5 | 5 | 0 | 14 | 14 | 0 |
| Vascular disorders | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 |
| Total | 51 | 59 | 3 | 49 | 54 | 8 | 100 | 113 | 11 |
Deaths
One death was recorded during the study period (coronary heart disease). This was in the placebo group and was considered unrelated to the study intervention.
Pregnancy
Of the 240 randomised patients, 14 pregnancies were reported (Table 34) during the course of the trial [nine female patients (one of whom had two pregnancies) and in the partners of four male patients] with 12 normal children and maternal outcomes. We observed one spontaneous abortion at approximately 21 weeks’ gestation and one congenital anomaly (heart murmur, septal defect and hydrocephalus) in the infant of a patient in the placebo group.
| Patient ID | Centre | Treatment | Sex | Estimated date of delivery | Delivery date | Maternal outcome | Child outcome |
|---|---|---|---|---|---|---|---|
| 11008 | Edinburgh | MP | Malea | 26 July 2009 | Unknown | Unknown | Normal |
| 11012 | Edinburgh | Placebo | Malea | 11 August 2010 | 21 August 2012 | Normal | Normal |
| 11047 | Edinburgh | MP | Female | 12 November 2013 | 4 November 2013 | Unknown | Normal |
| 11054 | Edinburgh | MP | Female | 17 June 2012 | 30 May 2012 | Caesarean | Normal |
| 11067 | Edinburgh | MP | Female | 16 March 2012 | 5 March 2012 | Caesarean | Normal |
| 11075 | Edinburgh | Placebo | Female | 29 March 2013 | 13 March 2013 | Induced (IUGR) | Normal |
| 11075 | Edinburgh | Placebo | Female | 3 May 2014 | 7 April 2014 | Normal | Normal |
| 12017 | Aberdeen | MP | Female | 22 April 2013 | Unknown | Abortion – spontaneous 5 October 2012 | N/A |
| 13024 | Dundee | Placebo | Female | 22 February 2012 | 22 February 2012 | Normal | Normal |
| 14010 | Glasgow Stobhill | Placebo | Female | 15 October 2010 | 22 October 2010 | Forceps/ventouse | Normal |
| 14024b | Glasgow Stobhill | MP | Malea | Unknown | 28 September 2015 | Normal | Normal |
| 15015 | Glasgow Royal Infirmary | MP | Female | 12 June 2013 | 31 May 2013 | Normal | Normal |
| 18007 | Liverpool | Placebo | Female | 11 October 2012 | 26 September 2012 | Normal | Abnormal |
| 34005 | Durham | MP | Malea | Unknown | 3 December 2012 | Normal | Normal |
Safety blood monitoring
Blood safety monitoring tests of interest were summarised by treatment and visit. Abnormal laboratory results outside the normal reference range were also summarised and are presented for each visit in Table 35. The numbers highlighted in italics indicate a higher value than in the other treatment group. Alanine aminotransferase and lymphocyte values were consistently higher across all visits in the MP group, whereas the majority of C-reactive protein values were higher in the placebo group. Albumin and haemoglobin values were higher in the placebo group from visits 6 and 5, respectively, until the end of the trial.
| Visit | Blood safety monitoring test | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALT | ALP | Albumin | C-reactive protein | WBC count | Haemoglobin | Neutrophils | Lymphocytes | |||||||||
| MP | Placebo | MP | Placebo | MP | Placebo | MP | Placebo | MP | Placebo | MP | Placebo | MP | Placebo | MP | Placebo | |
| 2 | 21.9 | 8 | 9 | 11.4 | 4.7 | 5.4 | 21.9 | 20.5 | 8.6 | 8.9 | 48.4 | 42.9 | 7.0 | 5.4 | 37.5 | 35.7 |
| 3 | 24.2 | 6.5 | 6.6 | 13.1 | 2.4 | 7.4 | 26.8 | 29.6 | 17.1 | 9.3 | 42.3 | 28.7 | 5.7 | 6.5 | 49.6 | 42.6 |
| 4 | 17.1 | 5.6 | 6.2 | 8.9 | 8.5 | 5.6 | 18.8 | 27.1 | 13.7 | 8.4 | 26.5 | 23.4 | 4.3 | 3.7 | 47.9 | 31.8 |
| 5 | 16.7 | 11.1 | 4.5 | 4.3 | 6.1 | 6.1 | 12.3 | 23.2 | 7.9 | 5.1 | 22.8 | 26.3 | 6.1 | 5.1 | 50.9 | 30.3 |
| 6 | 13.5 | 7.2 | 6.5 | 4.4 | 3.6 | 7.2 | 15.3 | 21.6 | 5.4 | 7.2 | 17.1 | 23.7 | 2.7 | 6.2 | 51.4 | 25.8 |
| 7 | 17.0 | 9.2 | 2.9 | 3.8 | 2.8 | 5.7 | 14.2 | 18.4 | 9.4 | 8.0 | 13.2 | 20.7 | 4.7 | 5.7 | 44.3 | 19.5 |
| 8 | 18.0 | 7.2 | 3.1 | 5.0 | 3.0 | 7.2 | 15.0 | 16.9 | 12.0 | 7.2 | 13.0 | 22.9 | 8.0 | 7.2 | 49.0 | 19.3 |
| 9 | 13.1 | 3.8 | 2.1 | 0.0 | 4.0 | 5.1 | 10.1 | 11.5 | 5.1 | 7.7 | 14.1 | 16.7 | 6.1 | 6.4 | 46.5 | 19.2 |
| 10 | 13.7 | 6.8 | 1.1 | 1.4 | 4.2 | 5.4 | 11.6 | 10.8 | 7.4 | 6.8 | 6.3 | 13.5 | 3.2 | 0.0 | 36.8 | 18.9 |
| 11 | 9.9 | 2.8 | 1.1 | 1.4 | 2.2 | 4.2 | 7.7 | 11.1 | 4.4 | 2.8 | 8.8 | 15.3 | 2.2 | 1.4 | 46.2 | 15.3 |
| 12 | 16.9 | 5.6 | 2.3 | 1.4 | 3.4 | 5.6 | 9.0 | 5.6 | 4.5 | 2.8 | 7.9 | 8.3 | 2.2 | 1.4 | 47.2 | 15.3 |
Prespecified subgroup analyses of primary and secondary outcomes
Subgroup analyses of the primary and secondary outcomes (clinical recurrence of CD) were analysed similarly to the main analysis of primary and secondary outcomes with the addition of an interaction term for treatment and subgroup in the Cox proportional hazards model.
Primary outcome
The results show that, in the entire study cohort, smokers had a higher chance of reaching the primary outcome than non-smokers [15/55 (27.3%) vs. 27/185 (14.6%); p = 0.018]. Among smokers, 3 out of 29 on MP (10.3%) had a clinical recurrence, compared with 12 out of 26 (46.2%) on placebo, demonstrating that MP was effective at preventing postoperative recurrence in smokers (unadjusted HR 0.127, 95% CI 0.04 to 0.46) but not in non-smokers (unadjusted HR 0.898, 95% CI 0.42 to 1.94). The number needed to treat was calculated as 3 in smokers and 31 in non-smokers. Other subgroup analyses assessing previous TPMT exposure, prior methotrexate use, prior infliximab use, previous surgery and age at diagnosis did not identify any differences between the groups.
Figure 12 shows the results of the overall unadjusted analysis of the primary outcome together with the results of the individual subgroup analyses.
FIGURE 12.
Forest plot of primary outcome (unadjusted). The subgroup p-values are from a test for an interaction between the treatment and subgroup variables.
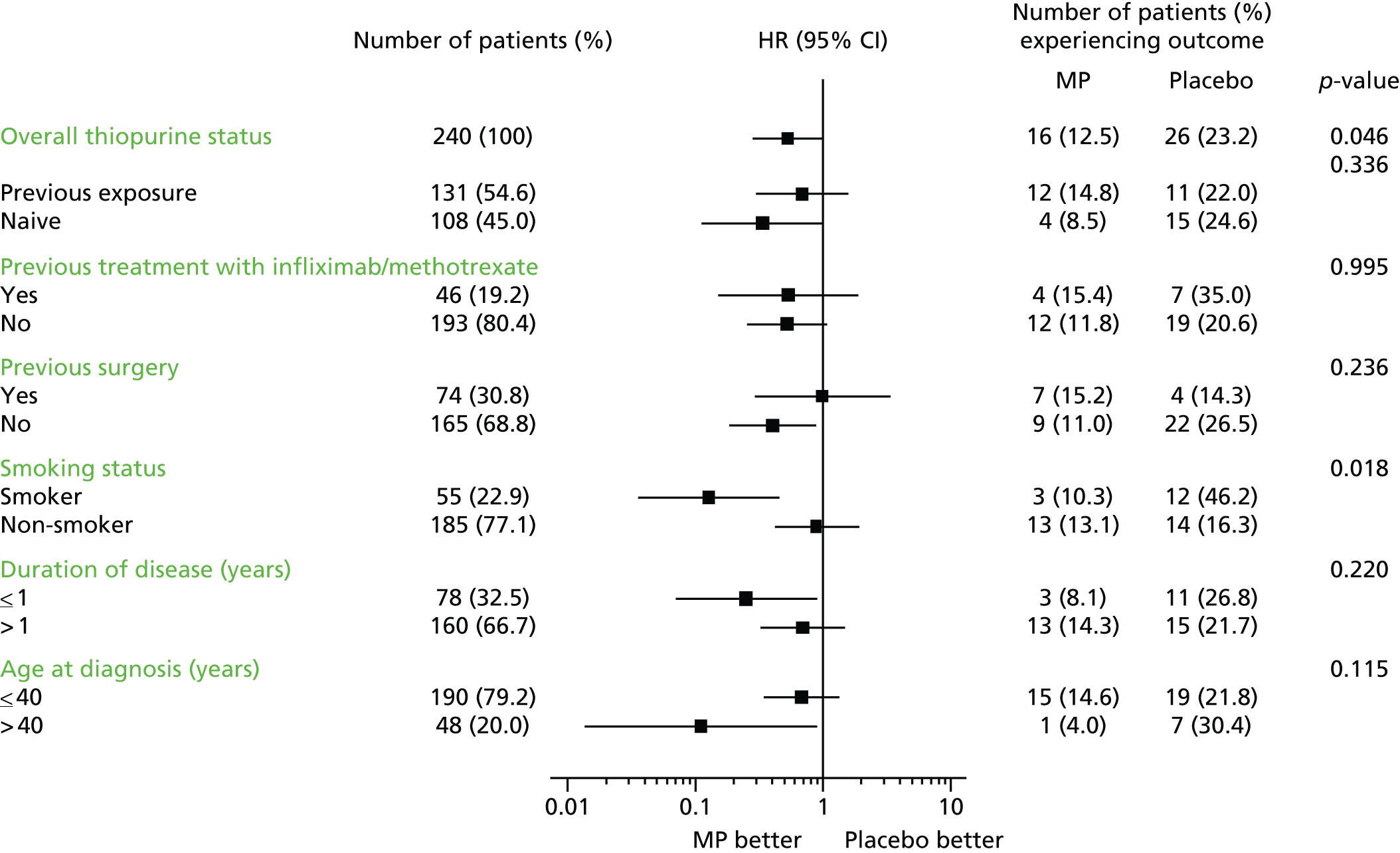
Secondary outcome
Clinical recurrence
Similar to the subgroup analyses of the primary outcome, subgroup analyses for the secondary outcome demonstrated that smokers had a higher chance of reaching the secondary outcome than non-smokers [19/55 (34.5%) vs. 55/185 (29.7%); p = 0.033]. Among smokers, 6 out of 29 on MP (20.7%) had a secondary outcome, compared with 13 out of 26 (50.0%) on placebo, demonstrating that MP was effective at preventing postoperative recurrence in smokers (unadjusted HR 0.270, 95% CI 0.10 to 0.72) but not in non-smokers (unadjusted HR 0.995, 95% CI 0.58 to 1.70). There were no significant differences in the analyses of the other subgroups. Figure 13 shows the results of the overall unadjusted analysis of the secondary outcome together with the results of the individual subgroup analyses.
FIGURE 13.
Forest plot of secondary outcome (unadjusted). The subgroup p-values are from a test for an interaction between the treatment and subgroup variables.

Endoscopic recurrence
There were no significant differences in endoscopic recurrence, as defined by the Rutgeerts score, by treatment group on subgroup analyses except in the thiopurine-naive group (38 in the MP group vs. 29 in the placebo group; p = 0.516).
Figures 14 and 15 show the results of the overall unadjusted analysis of visit 12 colonoscopy results and CDEIS scores, together with the results of the individual subgroup analyses.
FIGURE 14.
Forest plot of colonoscopy results at visit 12 (unadjusted). The percentage of patients with a positive colonoscopy is based on positive (≥ i2) and negative (< i2) colonoscopy results only. Missing and other colonoscopy results have been excluded. The subgroup p-values are from a test for an interaction between the treatment and subgroup variables. i2: ≥ 5 aphthous lesions with normal mucosa between lesions, or skip areas of larger lesions or lesions confined to the ileocolonic anastomosis.

FIGURE 15.
Forest plot of CDEIS score at visit 12 (unadjusted). The subgroup p-values are from a test for an interaction between the treatment and subgroup variables.
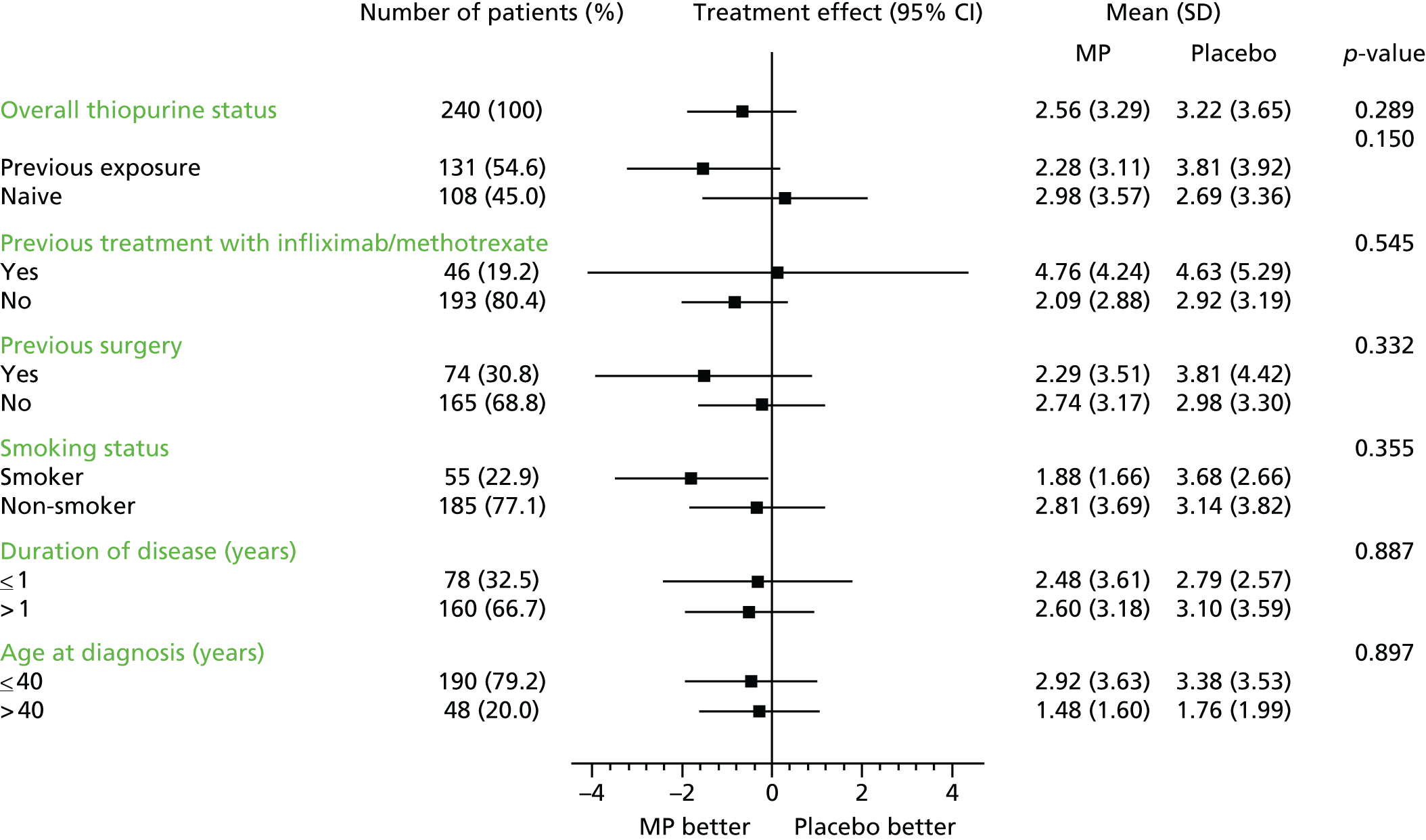
Chapter 4 Discussion
Context
This study is the largest randomised, double-blind study in postoperative CD reported to date. In the study cohort, analysed on an intention-to-treat basis, there is evidence of a reduction of clinical recurrence in patients on MP compared with placebo, although this finding was not statistically significant. It is notable that the treatment effect of 20% is lower than in previously conducted studies (Hanauer et al. :18 40% reduction in mild CD lesions and 75% reduction in more severe lesions; Ardizzone et al. :19 25% reduction). We judged a treatment effect of 20% appropriate because, given the side-effect profile of MP, it is arguable that a treatment effect of significantly < 20% is of limited clinical significance.
Endoscopic recurrence is an important secondary outcome that other studies have prioritised over clinical outcomes. No overall benefit of MP therapy on endoscopic outcome was identified in the present study using the prespecified criteria of a Rutgeerts score of ≥ i2. However, on post hoc analysis, fewer patients on placebo than on MP had endoscopically normal appearances at the anastomosis. The endoscopic subgroup analysis showed an intriguing difference in treatment effect between thiopurine-naive and non-naive groups, with patients previously exposed to thiopurine having greatest evidence of benefit.
The apparent discrepancy between clinical outcome measures and endoscopic outcomes is noteworthy. There are several possible explanations for this, and clearly no consensus regarding whether or not to prioritise clinical outcomes over endoscopic outcomes has emerged from previous trial data. The stringency of our primary outcome and the internal correlation with faecal calprotectin assessment and with thiopurine metabolite concentrations are arguments in favour of prioritising and relying on the clinical primary outcome. Indeed, the validity and reproducibility of a Rutgeerts score of ≥ i2 – defined in the primary study from Rutgeerts et al. ,26 but not subject to detailed replication as a predictor of clinical recurrence – has been debated.
With respect to clinical practice, the data emerging from the trial strongly highlight the importance of cigarette smoking in the pathogenesis and progression of CD, and as a target for therapeutic intervention. Not only do the data show that the effect of MP on primary outcome was of statistical as well as clinical significance in smokers, with a number needed to treat of only 3, but the analysis of factors predictive of primary outcome is also noteworthy. Only smoking habit was predictive of outcome, whereas age, sex or need for previous surgery were not implicated as risk factors for recurrence. The data underline the importance of smoking cessation in disease management, and prioritising this aspect using all available methodologies is of undeniable importance in clinical research. Previous studies have demonstrated that surgical recurrence increases with the number of cigarettes smoked each day, and that smoking cessation reduces clinical and surgical recurrence. 32,33 To date, the constituents responsible in cigarette smoke, and pathogenic mechanisms involved in this association, have not been identified.
Limitations
The present study does have a number of limitations. There was a lower primary event rate than initially anticipated: the actual primary outcome rates in the treatment group were 12.5% in the MP group compared with 23.2% in the placebo group as opposed to expected rates of 30% and 50%. Of the 240 patients recruited into the study, 37 (15%) were also withdrawn or lost to follow-up. This compares favourably to the rates reported in earlier trials. 10,18,19,34 In the present study, 80 (33%) patients discontinued therapy as a result of an adverse event [39 (30%) in the MP group vs. 41 (37%) in placebo group] and an additional 18 (8%) patients withdrew as a result of an abnormal blood test result picked up on safety monitoring [12 (9.4%) in the MP group vs. 6 (5.4%) in the placebo group]. Ardizzone et al. 19 reported treatment discontinuation rates attributable to adverse events of 15 (10.7%) in the azathioprine group and 6 (4%) in the mesalazine group, although figures on treatment discontinuation in other trials are more difficult to gauge from the published figures.
The number of discontinuations does raise questions in terms of adherence in routine clinical practice and, together with the numbers lost to follow-up, raises tantalising questions about the impact on the significance of the primary analysis. In this sense, however, the present study may reflect the real-life use of thiopurines in a postoperative population. We would argue that this trial has considerable value as a pragmatic trial in which the results and experiences of participating patients are highly generalisable to the clinic population seen not only in the UK but in much of Western Europe and North America.
Another limitation was the number of missing values in the analysis of the secondary outcomes, which could affect the overall results. However, the proportion of missing values between treatment groups was assessed and found to be similar.
There were no worrying signals with respect to adverse events in the present study. Indeed, lower than expected rates of pancreatitis and malignancies were reported. Fourteen pregnancies were reported, with 12 healthy births, one miscarriage and one abnormal birth to a mother in the placebo group. The stringent precautions for blood monitoring may well be pertinent to the safety profile demonstrated in the trial.
Strengths
The strengths of this study include the placebo arm, the assessment of both clinical and endoscopic outcomes, dose adjustment by TPMT status, the stringency of the primary end point and the attention to maintenance of blinding throughout, as well as its generalisability to routine clinical practice.
A further issue worthy of discussion and of exploration in further studies is whether or not a stronger treatment effect would have been seen if the TPMT metabolite profiles were available for dose adjustment during the trial, as is now routine in clinical practice. The data presented suggest that a significant proportion of patients may have benefited from higher doses of active drug; indeed, almost 60% had levels below the accepted lower limit of the therapeutic range.
Applicability in UK hospitals and beyond
The study population and the overall findings of the study are highly generalisable in clinical practice in the UK and indeed across Western Europe and North America.
Taken with other recent data from the POCER study investigators,13 the present study helps make progress towards a treatment algorithm in postoperative CD that may prove to be generalisable in clinical practice. We suggest that cessation of cigarette smoking emerges as the key intervention to emphasise ahead of drug therapy. In those who continue to smoke, but not non-smokers, thiopurine therapy appears to be justified in the early postoperative period, and we would now suggest the need to explore dose optimisation by metabolite testing. In thiopurine-intolerant patients, anti-TNF-α therapy may be justified. In all patients, reassessment of clinical status, endoscopic appearances and calprotectin levels in 6 months will determine need for escalation of therapy.
Reducing variance in clinical practice
The present study provides useful insights by identifying size of effect of thiopurine therapy overall, and the subgroup in which it is most effective. The identification of smoking habit as the key risk factor for recurrence is also of considerable importance in practice.
All these represent important points generalisable to clinical practice and intervention.
The present data are suggestive of utility of calprotectin levels in predicting clinical recurrence, and utility of metabolites in optimising thiopurine therapy. These data will need to be explored in further studies but both represent potential biomarkers that may help optimise intervention.
Potential risks not explored in this study
The study had a 3-year treatment period but did not have a follow-up period for recruited patients. The longer-term management of this patient population and the duration of therapy remain unexplored.
Unanticipated findings
The magnitude of effect of smoking habit on primary outcome was unexpected, and represents a key finding.
Patient public involvement
Patient public involvement in TOPPIC was limited. A lay representative formed part of the TSC and, as such, was invited to all formal meetings and provided with the TSC reports and updates with the initial meetings attended.
Chapter 5 Conclusions
Implications for health care
TOPPIC was designed in 2003–4 and completed in 2015 as the largest single study of the efficacy of MP in reducing postoperative recurrence in CD. This has since remained an important question in the context of disease management over time, although the introduction of new biological therapies and the widespread use of biomarkers and therapeutic drug monitoring need to be taken into account in assessing current impact and implications.
We have defined the rate of clinical and endoscopic disease recurrence in the UK population, and have characterised the key risk factor amenable to public health intervention as smoking, while also demonstrating that other proposed risk factors do not have a significant effect in this study. The last finding is not to be underestimated – age, disease behaviour and previous exposure to thiopurines do not appear to influence outcome adversely.
From the clinical perspective, the need to educate and re-educate patients and clinicians regarding the deleterious effects of smoking on outcome is critical, as is the need to put in place all available strategies to help with smoking cessation.
From the trial itself, MP was not effective in reducing the frequency of clinical postoperative recurrence of CD overall, but the data suggest that it has clinically meaningful effect among the subgroup of patients who continue to smoke after surgery. Importantly, with the benefit of the results of therapeutic drug monitoring of TPMT levels available after the trial, it appears that many patients were underdosed per protocol, and the data generated in the trial may underestimate the potential benefit in the clinic.
Taking these data in the context of other published data in this field, we suggest that risk stratification of relapse after surgery is principally influenced by smoking habit, and would offer prophylaxis with thiopurine therapy to those individuals who continue to smoke, if the drug is tolerated. In all patients, these data, and the data from the POCER study, make the case for monitoring faecal calprotectin levels and offering endoscopic assessment within 6–12 months of surgery to those with elevated calprotectin values, or clinical concern of recurrence. At this stage, the introduction of thiopurine therapy or biological therapy needs to be assessed in terms of the presence or absence of symptoms, and endoscopic features.
Recommendations for research
A number of areas in which further research is required emerge from the present study.
-
Although the present study defines the role of MP in postoperative prevention, we also provide evidence that the effect might be underestimated in the present study.
-
A further trial designed to incorporate concomitant real-time measurements of faecal calprotectin levels, TPMT metabolites and other biomarkers in the clinic, and using the combination of these parameters to optimise and target therapy on appropriate individuals, is likely to provide a more accurate estimate of the full therapeutic potential of thiopurines in the postoperative setting.
-
Most importantly, there is a research need to explore the efficacy of other agents as monotherapy or co-therapy with thiopurines – anti-TNF-α therapies, vedolizumab and ustekinumab will all be available in this context in 2017, with other new oral agents in advanced stages of analysis. This area is likely to require specific trials, incorporating the understanding generated in the present study.
-
The trial has also demonstrated the need for research to identify the optimum biologically meaningful outcome measure in studies of postoperative recurrence, with discrepancies between the results when clinical, biochemical and endoscopic end points are considered.
-
With TOPPIC strongly highlighting the effect of smoking on recurrence rates, the data demonstrate the need for directed intervention studies to assess the relative success of alternative strategies in smoking cessation in patients with CD.
-
The trial also highlights the need for mechanistic studies to explore the very well-documented association between smoking and development, with epigenetic, immune-mediated mechanisms currently of particular interest.
-
Exploration of efficacy of other agents in preventing postoperative relapse – these will include anti-TNF-α therapies and anti-adhesion strategies.
Acknowledgements
Use of the IBDQ, authored by Dr Jan Irvine et al. ,35 was made under licence from McMaster University, Hamilton, ON, Canada.
We would like to thank all the participants who took part in the study. We gratefully acknowledge the help of the following people.
The TOPPIC Collaborators Group.
Trial Steering Committee: Chris Probert, John Mansfield, Stuart Ralston, Ruth Slater, Craig Mowat and Steff Lewis.
Data Monitoring Committee: Stephen JW Evans, Huw Roddie and Helen Gillett.
Data Management Group: Jack Satsangi, Ian Arnott and Debbie Kerr.
Safety blood review clinicians: Jack Satsangi, Ian Arnott, Craig Mowat, Ashley Mowat, Aidan Cahill, Mhairi Collie, Jack Winter, Malcolm Smith, Aileen McKinley, Daniel Gaya, John Morris, John Todd, John Thomson and Graham Naismith.
Recruiting centres (collaborating doctors and nurse; the principal investigator is listed first):
-
Western General Hospital, Edinburgh – Dr Ian Arnott, Debbie Kerr and Gail Scott
-
Ninewells Hospital, Dundee – Dr Craig Mowat and Shirley Cleary
-
Aberdeen Royal Infirmary, Aberdeen – Dr Malcolm Smith and Jenny Robb
-
Glasgow Royal Infirmary, Glasgow – Dr Daniel Gaya and Joanna Munro
-
Stobhill Hospital, Glasgow – Dr Aidan Cahill and Liz Lightbody
-
Raigmore Hospital, Inverness – Dr Lindsay Potts and Ian Shread
-
Queen Elizabeth Hospital, Birmingham – Dr Jason Goh, Sian Davis and Vishy Veerana
-
Bristol Royal Infirmary, Bristol – Dr Tom Creed, Jane Bowles and Andrew Parker
-
University Hospital Coventry, Coventry – Dr Chuka Nwokolo and Emily Archer
-
County Durham and Darlington Memorial Hospitals, Durham and Darlington – Dr Anjan Dhar and Claire Shaw
-
Royal Devon and Exeter Hospital, Exeter – Dr Tariq Ahmad, Dr Kenji So, Dr Ollie Waters and Suzie Marriott
-
Hull Royal Infirmary, Hull – Dr Shaji Sebastian and Bronwen Williams
-
Leeds General Infirmary, Leeds – Dr John Hamlin, Dr Alex Ford and Doris Quartey
-
Royal Liverpool University Hospital, Liverpool – Dr Shreedhar Subramanian, Dr Keith Leiper and Kate Martin
-
Manchester Royal Infirmary, Manchester – Dr Scott Levison, Dr Simon Campbell and Jane Taylor
-
University Hospital of North Staffordshire NHS Trust, Stoke-on-Trent – Dr Sandip Sen and Sue Gallagher
-
Nottingham University Hospital, Nottingham – Professor Chris Hawkey, Jenny Salmon and Michelle Mieszek
-
Rotherham NHS Foundation Trust Hospital, Rotherham – Dr Mohamed Yousif, Meredyth Harris and Rachel Walker
-
Royal Free Hospital, London – Dr Charles Murray and Orla Brady
-
Salford Royal Hospital, Salford – Dr Simon Lal and Anne Keen
-
Barts Health NHS Trust St Bartholomew’s Hospital, London – Dr James Lindsay, Louise Langmead, Jacqueline Nichols and Abu Karim
-
St Mark’s Hospital, Harrow – Dr Naila Arebi and Sherrill Tripoli
-
Abertawe Bro Morgannwg University Health Board Singleton Hospital, Swansea – Dr Linzi Thomas and Caradog Thomas
-
Torbay Hospital, Torquay – Dr Cathryn Edwards and Jan Palmer
-
University College Hospital, London – Dr Stuart Bloom, Belinda Theis, Elen Witness and Roman Jastrub.
Edinburgh Clinical Trials Unit: Audrey Duncan, Garry Milne, Allan Walker, Lynsey Milne, Samantha Thomas, Ashma Krishnan, Debbie Kerr, Gail Scott and Helen Watters.
Academic and Clinical Central Office for Research & Development: Marise Bucukoglu, Anne Langston, Ray French and Vikki Young.
Wellcome Trust Clinical Trial Facility Genetics Core: Lee Murphy and Mark Blandford.
Viapath laboratory, Guy’s and St Thomas’ Hospital: Tony Marinaki.
Histopathology laboratory: Ann Jarvie and Susan A Walker.
Pathology laboratory: David Worrall.
Cedars Sinai: Dermot McGovern and Carol J Landers.
We also wish to thank the MRC and National Institute for Health Research (NIHR)’s EME programme, the Scottish Government’s Chief Scientist Office and the NIHR National Portfolio.
Contributions of authors
Professor Jack Satsangi (Consultant in Gastroenterology, Division of Gastroenterology) was the chief investigator. He devised the study, was involved in the design and conduct of research, and was a member of the trial development group, project management group and the TSC. He was also involved in the development and refinement of the protocol, the conduct of the research interpretation and the reporting of results, and he revised the manuscript for important intellectual content.
Dr Nicholas A Kennedy (Consultant Gastroenterologist and Honorary Clinical Senior Lecturer) was a research fellow at the Edinburgh site. He was involved in the conduct of the research interpretation and the reporting of results, including the analysis of the predictive data and samples, and he revised the manuscript for important intellectual content.
Dr Craig Mowat (Consultant in Gastroenterology, Gastroenterology Department) was the principal investigator at the Dundee site. He was involved in the design and conduct of research, and was a member of the trial development group and the TSC. He was also involved in the development and refinement of the protocol, the conduct of the research interpretation and the reporting of results, and he revised the manuscript for important intellectual content.
Dr Ian Arnott (Consultant in Gastroenterology, Division of Gastroenterology) was the principal investigator at the Edinburgh site. He was involved in the design and conduct of research, and was a member of the trial development group, project management group and the TSC. He was also involved in the development and refinement of the protocol, the conduct of the research interpretation and the reporting of results, and he revised the manuscript for important intellectual content.
Mrs Catriona Keerie (Statistician) was the study statistician who wrote the statistical analysis plan and undertook the end-of-study analysis. She prepared the final study statistical report and was involved in the interpretation and reporting of results, and the drafting and revision of this manuscript for important intellectual content.
Dr Steff Lewis (Reader in Medical Statistics) was the lead study statistician and was involved in the design and conduct of research, the TSC, the interpretation and reporting of results, and the drafting and revision of the manuscript for important intellectual content.
Dr Holly Ennis (Trial Manager from August 2013) was involved in the day-to-day trial management, data management and was a member of the TMG.
Publication
Mowat C, Arnott I, Cahill A, Smith M, Ahmad T, Subramanian S, et al. Mercaptopurine versus placebo to prevent recurrence of Crohn’s disease after surgical resection (TOPPIC): a multicentre, double-blind, randomised controlled trial. Lancet Gastroenterol Hepatol 2016;1:273–82.
Data sharing statement
All available data can be obtained from the corresponding author via the ECTU in accordance with the ECTU Data Sharing Policy. This policy is currently under development, but will be based on the MRC Hubs for Trials Methodology Research ‘Good Practice Principles for Sharing Individual Participant Data from Publicly Funded Clinical Trials’, published in April 2015. 36
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health.
References
- Rubin GP, Hungin AP, Kelly PJ, Ling J. Inflammatory bowel disease: epidemiology and management in an English general practice population. Aliment Pharmacol Ther 2000;14:1553-9. https://doi.org/10.1046/j.1365-2036.2000.00886.x.
- Sandborn W, Sutherland L, Pearson D, May G, Modigliani R, Prantera C. Azathioprine or 6-mercaptopurine for inducing remission of Crohn’s disease. Cochrane Database Syst Rev 2000;2.
- Gordon M, Taylor K, Akobeng AK, Thomas AG. Azathioprine and 6-mercaptopurine for maintenance of surgically-induced remission in Crohn’s disease. Cochrane Database Syst Rev 2014;8. https://doi.org/10.1002/14651858.CD010233.pub2.
- Pearson DC, May GR, Fick G, Sutherland LR. Azathioprine for maintaining remission of Crohn’s disease. Cochrane Database Syst Rev 2000;2.
- Michelassi F, Balestracci T, Chappell R, Block GE. Primary and recurrent Crohn’s disease. Experience with 1379 patients. Ann Surg 1991;214:230-8. https://doi.org/10.1097/00000658-199109000-00006.
- Van Assche G, Rutgeerts P. Medical management of postoperative recurrence in Crohn’s disease. Gastroenterol Clin North Am 2004;33:347-60. https://doi.org/10.1016/j.gtc.2004.02.012.
- Post S, Herfarth C, Böhm E, Timmermanns G, Schumacher H, Schürmann G, et al. The impact of disease pattern, surgical management, and individual surgeons on the risk for relaparotomy for recurrent Crohn’s disease. Ann Surg 1996;223:253-60. https://doi.org/10.1097/00000658-199603000-00005.
- Mowat C, Cole A, Windsor A, Ahmad T, Arnott I, Driscoll R, et al. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011;60:571-607. https://doi.org/10.1136/gut.2010.224154.
- Singh S, Garg SK, Pardi DS, Wang Z, Murad MH, Loftus EV. Comparative efficacy of pharmacologic interventions in preventing relapse of Crohn’s disease after surgery: a systematic review and network meta-analysis. Gastroenterol 2015;148:64-76. https://doi.org/10.1053/j.gastro.2014.09.031.
- Jones GR, Kennedy NA, Lees CW, Arnott ID, Satsangi J. Systematic review: The use of thiopurines or anti-TNF in post-operative Crohn’s disease maintenance – progress and prospects. Aliment Pharmacol Ther 2014;39:1253-65. https://doi.org/10.1111/apt.12743.
- Regueiro M, Schraut W, Baidoo L, Kip KE, Sepulveda AR, Pesci M, et al. Infliximab prevents Crohn’s disease recurrence after ileal resection. Gastroenterology 2009;136:441-50. https://doi.org/10.1053/j.gastro.2008.10.051.
- Yoshida K, Fukunaga K, Ikeuchi H, Kamikozuru K, Hida N, Ohda Y, et al. Scheduled infliximab monotherapy to prevent recurrence of Crohn’s disease following ileocolic or ileal resection: a 3-year prospective randomized open trial. Inflamm Bowel Dis 2012;18:1617-23. https://doi.org/10.1002/ibd.21928.
- De Cruz P, Kamm MA, Hamilton AL, Ritchie KJ, Krejany EO, Gorelik A, et al. Crohn’s disease management after intestinal resection: a randomised trial. Lancet 2015;385:1406-17. https://doi.org/10.1016/S0140-6736(14)61908-5.
- Cuillerier E, Lémann M, Bouhnik Y, Allez M, Rambaud JC, Modigliani R. Azathioprine for prevention of postoperative recurrence in Crohn’s disease: a retrospective study. Eur J Gastroenterol Hepatol 2001;13:1291-6. https://doi.org/10.1097/00042737-200111000-00005.
- Domènech E, Scala L, Bernal I, García-Planella E, Casalots A, Piñol M, et al. Azathioprine and mesalazine in the prevention of postsurgical recurrence of Crohn’s disease: a retrospective study. Gastroenterol Hepatol 2004;27:563-7. https://doi.org/10.1016/S0210-5705(03)70533-0.
- Nos P, Hinojosa J, Aguilera V, Molés JR, Pastor M, Ponce J, et al. Azathioprine and 5-ASA in the prevention of postoperative recurrence of Crohn’s disease. Gastroenterol Hepatol 2000;23:374-8.
- Korelitz B, Hanauer S, Rutgeerts P, Present D, Peppercorn M. Post-operative prophylaxis with 6-MP, 5-ASA or placebo in Crohn's disease. A two-year multicenter trial. Gastroenterology 1998;114. https://doi.org/10.1016/S0016-5085(98)84115-2.
- Hanauer SB, Korelitz BI, Rutgeerts P, Peppercorn MA, Thisted RA, Cohen RD, et al. Postoperative maintenance of Crohn’s disease remission with 6-mercaptopurine, mesalamine, or placebo: a 2-year trial. Gastroenterology 2004;127:723-9. https://doi.org/10.1053/j.gastro.2004.06.002.
- Ardizzone S, Maconi G, Sampietro GM, Russo A, Radice E, Colombo E, et al. Azathioprine and mesalamine for prevention of relapse after conservative surgery for Crohn’s disease. Gastroenterology 2004;127:730-40. https://doi.org/10.1053/j.gastro.2004.06.051.
- Dignass A, Van Assche G, Lindsay JO, Lémann M, Söderholm J, Colombel JF, et al. The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: current management. J Crohns Colitis 2010;4:28-62. https://doi.org/10.1016/j.crohns.2009.12.002.
- Coelho J, Beaugerie L, Colombel JF, Hébuterne X, Lerebours E, Lémann M, et al. Pregnancy outcome in patients with inflammatory bowel disease treated with thiopurines: cohort from the CESAME Study. Gut 2011;60:198-203. https://doi.org/10.1136/gut.2010.222893.
- Casanova MJ, Chaparro M, Domènech E, Barreiro-de Acosta M, Bermejo F, Iglesias E, et al. Safety of thiopurines and anti-TNF-α drugs during pregnancy in patients with inflammatory bowel disease. Am J Gastroenterol 2013;108:433-40. https://doi.org/10.1038/ajg.2012.430.
- Ban L, Tata LJ, Fiaschi L, Card T. Limited risks of major congenital anomalies in children of mothers with IBD and effects of medications. Gastroenterology 2014;146:76-84. https://doi.org/10.1053/j.gastro.2013.09.061.
- Lennard-Jones JE. Classification of inflammatory bowel disease. Scand J Gastroenterol Suppl 1989;170:2-6. https://doi.org/10.3109/00365528909091339.
- Best WR, Becktel JM, Singleton JW, Kern F. Development of a Crohn’s disease activity index. National Cooperative Crohn’s Disease Study. Gastroenterology 1976;70:439-44.
- Rutgeerts P, Geboes K, Vantrappen G, Kerremans R, Coenegrachts JL, Coremans G. Natural history of recurrent Crohn’s disease at the ileocolonic anastomosis after curative surgery. Gut 1984;25:665-72. https://doi.org/10.1136/gut.25.6.665.
- Mary JY, Modigliani R. Development and validation of an endoscopic index of the severity for Crohn’s disease: a prospective multicentre study. Groupe d’Etudes Thérapeutiques des Affections Inflammatoires du Tube Digestif (GETAID). Gut 1989;30:983-9. https://doi.org/10.1136/gut.30.7.983.
- Guyatt G, Mitchell A, Irvine EJ, Singer J, Williams N, Goodacre R, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology 1989;96:804-10. https://doi.org/10.1016/S0016-5085(89)80080-0.
- Freier A, Karlton P, Kocher P. The Secure Sockets Layer (SSL) Protocol Version 3.0 2011. https://tools.ietf.org/html/rfc6101 (accessed 6 June 2017).
- Mowat C, Arnott I, Cahill A, Smith M, Ahmad T, Subramanian S, et al. Mercaptopurine versus placebo to prevent recurrence of Crohn’s disease after surgical resection (TOPPIC): a multicentre, double-blind, randomised controlled trial. Lancet Gastroenterol Hepatol 2016;1:273-82. https://doi.org/10.1016/S2468-1253(16)30078-4.
- National Institute for Health Research . Efficacy and Mechanism Evaluation n.d. www.nihr.ac.uk/funding-and-support/funding-for-research-studies/funding-programmes/efficacy-and-mechanism-evaluation/ (accessed 6 June 2017).
- Yamamoto T, Keighley MR. Smoking and disease recurrence after operation for Crohn’s disease. Br J Surg 2000;87:398-404. https://doi.org/10.1046/j.1365-2168.2000.01443.x.
- Reese GE, Nanidis T, Borysiewicz C, Yamamoto T, Orchard T, Tekkis PP. The effect of smoking after surgery for Crohn’s disease: a meta-analysis of observational studies. Int J Colorectal Dis 2008;23:1213-21. https://doi.org/10.1007/s00384-008-0542-9.
- D’Haens GR, Vermeire S, Van Assche G. Therapy of metronidazole with azathioprine to prevent postoperative recurrence of Crohn’s disease: a controlled randomized trial. Gastroenterol 2008;135:1123-9. https://doi.org/10.1053/j.gastro.2008.07.010.
- Irvine EJ. Development and subsequent refinement of the inflammatory bowel disease questionnaire: a quality-of-life instrument for adult patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 1999;28:S23-7. http://dx.doi.org/10.1097/00005176-199904001-00003.
- Smith CT, Hopkins C, Sydes M, Woolfall K, Clarke M, Murray G, et al. Good Practice Principles for Sharing Individual Participant Data from Publicly Funded Clinical Trials 2015. www.methodologyhubs.mrc.ac.uk/pdf/Datasharingguidance2015.pdf (accessed 4 September 2017).
List of abbreviations
- CD
- Crohn’s disease
- CDAI
- Crohn’s Disease Activity Index
- CDEIS
- Crohn’s Disease Endoscopic Index of Severity
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- DMC
- Data Monitoring Committee
- ECCO
- European Crohn’s and Colitis Organisation
- ECTU
- Edinburgh Clinical Trials Unit
- EME
- Efficacy and Mechanism Evaluation
- EQ-5D
- EuroQol-5 Dimensions
- HR
- hazard ratio
- IBD
- inflammatory bowel disease
- IBDQ
- Inflammatory Bowel Disease Questionnaire
- MP
- mercaptopurine
- MRC
- Medical Research Council
- POCER
- Post-Operative Crohn’s Endoscopic Recurrence study
- TGN
- thioguanine
- TMG
- Trial Management Group
- TNF-α
- tumour necrosis factor alpha
- TOPPIC
- Trial Of Prevention of Post operative Crohn’s disease
- TPMT
- thiopurine S-methyltransferase
- TSC
- Trial Steering Committee