

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 08/99/08. The contractual start date was in September 2010. The final report began editorial review in April 2016 and was accepted for publication in April 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Daniel F McAuley reports grants from the National Institute for Health Research (NIHR) Efficacy and Mechanism Evaluation (EME) programme, a Medical Research Council and NIHR partnership, Health Research Board, Health and Social Care Research and Development division of the Intensive Care Society of Ireland and REVIVE for the conduct of the study. In addition, Daniel F McAuley reports personal fees from GlaxoSmithKline, Bayer and Peptinnovate Ltd. His institution has received grants from NIHR and other funds were received from GlaxoSmithKline for undertaking bronchoscopy as part of a clinical trial. Daniel F McAuley also holds a patent for a treatment for acute respiratory distress syndrome awarded to Queen’s University Belfast and is a member of the Health Technology Assessment (HTA) Commissioning Board. John G Laffey reports grants from the NIHR EME programme, grants from Health Research Award and from the Health Research Board during the conduct of the study. Cecilia M O’Kane reports grants from the NIHR EME programme during the conduct of the study and her spouse has received consultancy fees from GlaxoSmithKline, Peptinnovate Ltd and Bayer. Cecilia M O’Kane has received funds from GlaxoSmithKline for undertaking bronchoscopy as part of a clinical trial, and has received travel and accommodation funding from AstraZeneca for attending a respiratory conference. Gavin D Perkins reports other funds from the Intensive Care Foundation during the conduct of the study, grants and personal fees from GlaxoSmithKline outside the submitted work, is a member of the Health Services and Delivery Research panel and is a NIHR senior investigator.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2018. This work was produced by McAuley et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2018 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Description of acute respiratory distress syndrome
Acute respiratory distress syndrome (ARDS) is a condition characterised by a failure of pulmonary oxygen exchange due to increased alveolar–capillary permeability and resultant pulmonary oedema. It can be caused by primary lung conditions such as aspiration, pneumonitis or can arise as a complication of non-pulmonary conditions such as severe sepsis. The syndrome was first described by Ashbaugh et al. 1 in 1967 in a group of 12 patients with acute onset on dyspnoea, tachypnoea, refactory hypoxaemia, reduced pulmonary compliance and diffuse alveolar shadowing on their chest radiographs. All the patients required positive and expiratory pressure to maintain arterial oxygenation. The term ‘adult respiratory distress syndrome’ was initially used to describe the condition2 but it was subsequently renamed ARDS because it may also occur in children. 3 The current definition arose from the American–European Consensus Conference in 19944 and recognised two grades of the disease, separated by the degree of hypoxaemia. ARDS was reserved for the more severe grade, with acute lung injury (ALI) being used as the overall term. The definition of ALI/ARDS requires:
-
acute onset of bilateral infiltrates on chest radiographs
-
pulmonary arterial occlusion pressure (PAOP) of < 18 mmHg (if measured) or absence of clinical signs of left atrial hypertension
-
ratio of partial pressure of arterial oxygen to the fraction of inspired oxygen (PaO2 : FiO2) of < 200 mmHg (26.7 kPa) for ARDS, or PaO2 : FiO2 of < 300 mmHg (40 kPa) for ALI.
Incidence and burden of acute lung injury
Acute lung injury is a major cause of morbidity and mortality. It affects all age groups, has a high mortality rate of up to 30–50%5–7 and causes a long-term reduction in quality of life for survivors. 7 ALI has significant resource implications, prolonging intensive care unit (ICU) and hospital stay, and requiring rehabilitation in the community. 8 The cost per ICU bed-day exceeds £1400 and delivery of critical care to patients with ALI accounts for a significant proportion of ICU capacity. Only 54% of survivors are able to return to work 12 months after hospital discharge. 9 The high incidence, mortality, long-term consequences and high economic costs mean that ALI is an extremely important problem.
Rationale for the trial
Prior evidence
There was a large body of evidence from in vitro and animal studies suggesting that statins might be beneficial in ALI. 10 In summary, statins improved epithelial and endothelial function to reduce alveolar capillary permeability and reduced pulmonary oedema. In addition, they modulated the inflammatory cascade; regulated inflammatory cell recruitment, activation and apoptosis; and reduced cytokine and protease activity. This suggested that statins might improve outcomes, as high levels and persistence of inflammatory mediators in ALI are associated with poor outcome. 11
Lack of published randomised controlled trials of statins in acute lung injury
There was a lack of published randomised controlled trials (RCTs) of statins in ALI. We conducted a systematic review, searched registries of ongoing clinical trials and contacted national and international experts in ALI. The National Institutes of Health had conducted a Phase III multicentre trial involving statins, but this involved rosuvastatin (Crestor, AstraZeneca UK Ltd) compared with placebo for up to 28 days in patients with sepsis-induced respiratory failure in the USA. 12 Our trial examined simvastatin and investigated ALI due to all aetiologies, as well as studying the potential mechanism of action by which statins act. In addition, unlike the US trial, an economic evaluation was undertaken in this study.
Observational studies support a clinical trial of a statin in acute lung injury
Acute lung injury is the most common complication of severe sepsis. 13 In patients with sepsis, most observational studies14–17 suggest that statins are associated with better outcomes, as measured by morbidity and mortality. Similarly, most observational studies18–20 have suggested a beneficial effect of statins in patients with pneumonia, supporting a potential role for statins in modulating pulmonary inflammation.
The Irish Critical Care Trials Group (ICCTG) has undertaken a prospective observational study in patients with ALI, which found that mortality was lower in patients receiving statins during their ICU stay. After adjusting for plateau pressure, severity of illness and other relevant covariates in a multiple logistic regression model, patients receiving statins had a much lower probability of death, although this failed to reach significance [odds ratio (OR) 0.27, 95% confidence interval (CI) 0.06 to 1.21; p = 0.09]. 21 Similarly, in a retrospective study, statin usage in patients with ALI was associated with increased ventilator-free days (VFDs) and reduced mortality, although again this was not significant. 22 These observational studies were not powered to examine the effect of statins on mortality.
It was not clear if the association with better outcomes in these studies was due to statins as opposed to statins representing a surrogate marker for improved access to health care. Moreover, these studies did not demonstrate whether or not beneficial effects would occur when statins were commenced after the onset of ALI. Although these data suggested that statins may have a potentially beneficial pharmacological treatment in ALI, a trial powered for important clinical outcomes was required.
Simvastatin reduces lipopolysaccharide-induced pulmonary and systemic inflammation in humans
We had conducted a study to examine if simvastatin modulates pathogenic mechanisms important in the development of lung injury in a model of acute lung inflammation induced by inhaled lipopolysaccharide (LPS) in healthy human volunteers. 23 In this double-blind, placebo-controlled study, participants were randomised to the simvastatin group or the placebo orally for 4 days prior to LPS inhalation group. Pre treatment with simvastatin reduced mediators of early ALI in bronchoalveolar lavage fluid including tumour necrosis factor alpha (TNF-α), neutrophil myeloperoxidase and protease release as measured by neutrophil elastase and matrix metalloproteinase (MMP)-7, -8 and -9. Furthermore, there was a significant reduction in systemic inflammation as measured by plasma C-reactive protein (CRP) levels. These effects were associated with reduced nuclear factor kappa B translocation. These novel findings provided the first proof of the principle that simvastatin has important anti-inflammatory effects in vivo in humans who are challenged with aerosolised endotoxin. These mechanistic findings were supported by a randomised placebo-controlled study that found 80 mg of simvastatin for 4 days reduced systemic cytokine responses induced by low-dose intravenous LPS in healthy subjects. 24 Finally, a randomised placebo-controlled study in patients with acute bacterial infection found that simvastatin, commenced prior to the development of sepsis-induced organ dysfunction, also reduced the levels of systemic inflammatory cytokines [TNF-α and interleukin 6 (IL-6)]. 25
Proof of concept that simvastatin improves pulmonary and non-pulmonary organ dysfunction, reduces inflammation and is well tolerated in patients with acute lung injury
We had completed a single-centre, randomised, double-blind, placebo-controlled Phase II study of simvastatin (80 mg for up to 14 days) in 60 patients with ALI. By day 14 there was a trend to improvement in pulmonary dysfunction, as measured by the oxygenation index (OI), respiratory system compliance and lung injury score in the simvastatin-treated group. Non-pulmonary organ dysfunction, as measured by the sequential organ failure assessment (SOFA) score was significantly lower in the simvastatin-treated group, with improvements in cardiovascular, renal and coagulation function. There was no difference in outcome for patients with sepsis- or non-sepsis-related ALI. Importantly, 80 mg of simvastatin was well tolerated with no increase in adverse events (AEs). In addition, we found that, unlike placebo, simvastatin decreased pulmonary IL-8 2.5-fold by day 3, with a trend to a decrease in IL-6 2.9-fold. In addition, at day 14 plasma CRP was lower with a trend to reduced plasma IL-6 in the simvastatin-treated group. 26
Together these results reflected the beneficial effects seen in previous in vitro and animal studies. 10 The study described above was not designed or powered to show an effect of simvastatin on VFDs or mortality. However, pulmonary and non-pulmonary organ dysfunction, as well as high levels of inflammatory cytokines, were associated with fewer VFDs and higher ICU mortality, which suggested that simvastatin might lead to improved clinical outcomes.
The findings above were supported by two small prospective randomised controlled studies involving the acute use of statins in patients with sepsis and pneumonia. 27,28 Choi et al. 27 studied 10 mg of atorvastatin (Lipitor®, Pfizer) given once daily in 74 patients with sepsis and pneumonia. Hospital mortality was reduced in the atorvastatin group compared with placebo, although this failed to reach significance (47% vs. 53%; p = 0.06). Similarly, Gonzalez et al. 28 conducted a study of 80 mg of simvastatin given once daily or placebo for 14 days in 40 patients with sepsis and found that simvastatin decreased the length of hospital stay.
Rationale for statins in acute lung injury
Statins have been proven to be a well-tolerated class of drugs. An improved mortality rate and no AEs have been reported in observational studies in critically ill patients with sepsis who were receiving statins. 15–21 Importantly, no toxicity was reported when statins were continued throughout the ICU course.
A dose of 80 mg of simvastatin is within the licensed therapeutic range for the treatment of hypercholesterolaemia in the UK. Although a different patient population, there is evidence regarding the safety of 80 mg of simvastatin in patients with cardiovascular disease. In a study in which 2265 patients following an acute coronary syndrome were randomised to receive 80 mg of simvastatin, myopathy [creatine kinase (CK) levels of > 10 times the upper limit of normal associated with muscle symptoms] occurred in only 0.4% of participants and rhabdomyolysis (CK levels of > 10,000 units/l with or without muscle symptoms) occurred in 0.13% of participants receiving 80 mg of simvastatin. 29 Importantly in this study, follow-up was only at months 1, 4 and 8 and every 4 months thereafter for up to 24 months until trial completion. In a further study in which 6031 patients with a history of a previous myocardial infarction were randomised to receive 80 mg of simvastatin, myopathy occurred in 0.9% and rhabdomyolysis in 0.18% of participants. 30 In this study, participants were seen for follow-up only at 2, 4, 8 and 12 months, and then at 6-month intervals with a median follow-up of 6 years. It is important to emphasis the maximum treatment period with 80 mg of simvastatin in this study was 28 days with safety monitoring (CK and liver transaminases) at days 3, 7, 14, 21 and 28.
The data from our proof-of-concept study reassuringly found that 80 mg of simvastatin was well tolerated and not associated with increased AEs compared with placebo. There was no difference in CK levels or numbers of patients with a CK level of > 10 times the upper limit of normal between the groups. There were no differences in creatinine levels between the groups. Reassuringly, there was a trend towards a lower incidence of renal replacement therapy at day 14 in the simvastatin-treated group. Liver transaminases [alanine transaminase (ALT) and aspartate aminotransferase (AST)] were commonly elevated and, although not significant, this was more common in the placebo-treated group. There were no differences in AEs or serious adverse events (SAEs) between the groups. No drug-related SAEs occurred during the study. 26
Rationale for choice of simvastatin
The diverse effects of statins appeared to represent a class effect. As outlined above, in both in vitro and animal experiments statins showed consistent effects regardless of the choice of statin. In addition, retrospective and prospective human studies have included multiple statins and shown beneficial effects. However, as the only statin with proof-of-concept efficacy and safety data in ALI, simvastatin was investigated in this study.
Rationale for simvastatin 28-day duration of treatment
The decision to examine treatment for up to 28 days was based on (1) data from our proof-of-concept study that demonstrated ongoing clinical improvement to day 14,26 (2) data showing that the upper interquartile range for duration of ICU stay in patients with ALI/ARDS is 14–18 days5,7,9 and (3) observational trials that showed benefit with no reported toxicity when statins were continued throughout the ICU stay. 21,22
Rationale for 80 mg of simvastatin dosage
Although there were a large number of data suggesting that statins might be beneficial in animal models of ALI, only a single animal study compared two doses of simvastatin (5 mg/kg or 20 mg/kg given intraperitoneally 24 hours before and concomitantly with LPS to induce lung injury) and only the higher dose was effective in attenuating lung injury. 31
Importantly, a retrospective observational study of statin usage in patients with sepsis found a greater mortality benefit in patients who were receiving a higher dose of statin. 32
A dose of 80 mg of simvastatin is the only dose with proof-of-concept data and is well tolerated in ALI. Therefore, 80 mg of simvastatin compared with placebo once daily was investigated in this study.
Although it is acknowledged that the risk of adverse side effects is dose related, on the basis of available evidence 80 mg of simvastatin is safe, particularly given that the duration of treatment was only up to 28 days and these patients were intensively monitored.
There are no effective pharmacological therapies for acute lung injury
The Cochrane systematic review of pharmacological treatments that included 22 studies of 14 different drugs concluded that ‘effective pharmacotherapy for ALI is extremely limited, with insufficient evidence to support any specific intervention’. 33
The National Heart, Lung and Blood Institute working group considered the future research directions in ALI in 2002 and concluded that clinical trials underpinned by mechanistic investigations were essential to develop new therapies for ALI. 34
Chapter 2 Methods
Trial summary
Hydroxymethylglutaryl-CoA reductase inhibition with simvastatin in Acute lung injury to Reduce Pulmonary dysfunction (HARP-2) was a multicentre, randomised, allocation-concealed, double-blind, placebo-controlled clinical trial of 80 mg of enteral simvastatin or placebo once daily for a maximum of 28 days. Patients were recruited from adult general ICUs in 40 hospitals throughout the UK and Ireland. The study was approved by the Office for Research Ethics Committees Northern Ireland (ORECNI) (10/NIR02/36) and research governance departments at each site in the UK and by the Clinical Research Ethics Committee (CREC) at each site in Ireland (8/10). The study was approved by the Medicines and Healthcare Products Regulatory Agency (MHRA) (32485/0020/001-0001) and Irish Medicines Board (IMB) (CT900/495/1).
The Northern Ireland Clinical Trials Unit (NICTU) co-ordinated the overall trial, with support from the Health Research Board (HRB) Galway Clinical Research Facility for centres in Ireland.
The study was conducted in accordance with the protocol and the statistical analysis plan and reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) guidelines. 35
The primary outcome measure was VFDs up to day 28 (defined as the number of days from the time of initiating unassisted breathing to day 28 after randomisation), with follow-up for mortality and quality of life to 12 months. The target sample size was 540 patients. The trial protocol and the results to 28 days have been published. 36,37
Objectives
The aim was to test the hypothesis that treatment with 80 mg of enteral simvastatin once daily for a maximum of 28 days was of therapeutic value in patients with ALI. The study had two distinct objectives:
-
Objective 1: to conduct a prospective randomised, double-blind, placebo-controlled Phase II multitrial of simvastatin for the treatment of ALI.
-
Objective 2: to study the biological effect of simvastatin treatment on mechanisms implicated in the development of ARDS.
Outcome measures
Primary outcome measure
The primary outcome measure was VFDs to day 28, defined as the number of days from the time of initiating unassisted breathing to day 28 after randomisation, assuming survival for at least 2 consecutive calendar days after initiating unassisted breathing and continued unassisted breathing to day 28. If a patient returned to assisted breathing and subsequently achieved unassisted breathing to day 28, VFDs were counted from the end of the last period of assisted breathing to day 28. A period of assisted breathing lasting < 24 hours and for the purpose of a surgical procedure was not counted against the VFD calculation. If a patient was receiving assisted breathing at day 27 or died prior to day 28, VFDs were counted as zero. Patients transferred to another hospital or other health-care facility were followed to day 28 to assess this end point.
In keeping with previous trials,38,39 unassisted breathing was defined as:
-
extubated with supplemental oxygen or room air or
-
open T-tube breathing or
-
tracheostomy mask breathing or
-
continuous positive airway pressure of ≤ 5 cm H2O without pressure support.
Patients receiving pressure support via non-invasive ventilation were defined as receiving assisted ventilation.
Secondary outcome measures
The secondary outcomes for this clinical trial included clinical outcomes, safety, biological mechanisms and data for the economic evaluation.
Clinical outcomes
-
Change in OI from baseline to day 3, 7, 14 and 28.
-
Change in SOFA score from baseline to day 3, 7, 14 and 28.
-
Non-pulmonary organ failure-free days (defined as the number of days in the first 28 days after randomisation that the patient has none of the following: cardiovascular support, renal support, liver support or neurological support).
-
All-cause mortality 28 days post randomisation.
-
Mortality at (first) discharge from critical care.
-
Mortality at (first) discharge from hospital.
-
Mortality at 12 months post randomisation.
Safety outcomes
-
A CK level of > 10 times the upper limit of normal (measured on days 1, 3, 7, 14, 21 and 28).
-
ALT/AST level of > 8 times the upper limit of normal (measured on days 1, 3, 7, 14, 21 and 28).
-
Need for renal replacement therapy in patients with CK levels elevated > 10-fold.
-
The SAEs and occurrence of suspected unexpected serious adverse reactions (SUSARs), as defined in Chapter 3, Safety outcomes.
Biological mechanisms
The effects of statins on biological mechanisms known to be important in ARDS were investigated as below:
-
Neutrophil activation as measured by plasma MMP-8 concentration.
-
Plasma acute phase inflammatory response as measured by CRP, IL-6 and vitamin D concentration.
-
Alveolar epithelial injury as measured by plasma receptor for advanced glycation end-products (RAGE) concentration and alveolar endothelial injury as measured by plasma angiopoietin 2 (Ang2) concentration.
Data for economic evaluation
-
Health-related quality of life (HRQoL)
-
EuroQol-5 Dimensions (EQ-5D) at discharge, 3, 6 and 12 months post randomisation.
-
-
Resource use
-
length of ICU stay (level 3 care)
-
length of high-dependency unit (HDU) stay (level 2 care).
-
length of hospital stay
-
health service contacts up to 12 months post randomisation.
-
Inclusion/exclusion criteria
Inclusion criteria
-
Patients receiving invasive mechanical ventilation.
-
Patient with ALI4 as defined by:
-
acute onset of hypoxic respiratory failure (PaO2 : FiO2 of ≤ 40 kPa from two arterial blood gas tests > 1 hour apart)
-
bilateral infiltrates on chest radiograph consistent with pulmonary oedema
-
no clinical evidence of left atrial hypertension or, if measured, a PAOP of ≤ 18 mmHg. If a patient has a PAOP of > 18 mmHg, then the other criteria must have persisted for > 12 hours after the PAOP had declined to < 18 mmHg, and still be within the 48-hour enrolment window.
-
Acute onset was defined as follows: the duration of the hypoxia criterion (1) and the chest radiograph criterion (2) must have been < 28 days at the time of randomisation.
Infiltrates considered ‘consistent with pulmonary oedema’ included any patchy or diffuse infiltrates not fully explained by mass, atelectasis, or effusion or opacities known to be chronic (> 28 days). The findings of vascular redistribution, indistinct vessels and indistinct cardiac borders were not considered ‘consistent with pulmonary oedema’.
All ALI criteria (under the second bullet point above) must have occurred within the same 24-hour period. The time of onset of ALI was when the last ALI criterion was met. Patients were enrolled within 48 hours of ALI onset.
Exclusion criteria
-
Aged < 16 years.
-
There had been > 48 hours since the onset of ALI.
-
Patient was known to be pregnant.
-
A CK level of > 10 times the upper limit of the normal range.*
-
Transaminase levels of > 8 times the upper limit of the normal range.*
-
Patients receiving ongoing and sustained treatment with any of the following: itraconazole, ketoconazole, human immunodeficiency virus (HIV) protease inhibitors, nefazodone (Dutonin, Bristol-Myers Squibb), ciclosporin, amiodarone, verapamil or diltiazem.
-
Patients with severe renal impairment (estimated creatinine clearance of < 30 ml/minute) not receiving renal replacement therapy.
-
Severe liver disease (Child–Pugh score of > 12).
-
Current or recent treatment (within 2 weeks) with statins.
-
Physician decision that a statin was required for proven indication.
-
Contraindication to enteral drug administration (e.g. patients with mechanical bowel obstruction). Patients with high gastric aspirates due to an ileus were not excluded.
-
Domiciliary mechanical ventilation except for continuous positive airway pressure/bilevel positive airway pressure used for sleep-disordered breathing.
-
Known participation in other investigational medicinal product trials within 30 days.
-
Consent declined.
-
Treatment withdrawal imminent within 24 hours.
-
Non-English-speaking patients or those who did not adequately understand verbal or written information unless an interpreter was available.
*If CK, ALT and AST values were not available as part of routine care, a blood sample was obtained after informed consent but before randomisation.
The CK, ALT and AST values could be obtained up to 72 hours prior to randomisation.
The following amendments relating to eligibility were made during the study.
-
Protocol version 2.0: the exclusion criteria were amended to allow patients receiving low-dose erythromycin as a prokinetic to be included.
-
Protocol version 3.0: concomitant use of clarithromycin and erythromycin and domicilliary ventilation for sleep-disordered breathing were removed as exclusion criteria.
-
Protocol version 4.0: level of ALT and AST for eligibility and discontinuation of study drug was changed from five times the upper limit of normal to eight times the upper limit of normal.
Consent
The study was conducted in accordance with the ethics principles that have their origin in the Declaration of Helsinki. Eligible patients were included in the trial only after written informed consent was obtained. Informed consent was obtained prior to conducting any trial-specific procedures and the process for obtaining informed consent was documented in the patient’s medical records (source documents that were reviewed at the time of on-site monitoring visits).
Informed consent procedure for the UK
Informed consent forms approved by the Research Ethics Committee (REC) were provided to each trial site. The principal investigator (PI) was responsible for ensuring that informed consent for trial participation was given by each patient or a legal representative. This required that the informed consent form was signed and personally dated by the patient or by the patient’s legally acceptable representative. An appropriately trained doctor or nurse took consent. If no consent was given, then the patient was not randomised into the trial.
The incapacitating nature of the condition precluded obtaining prospective informed consent from participants. In this situation, informed consent was sought from a personal legal representative (PerLR) or professional legal representative (ProfLR) when a PerLR was not available.
Personal legal representative consent: UK
Informed consent was sought from the patient’s PerLR, who was a relative, partner or close friend. The PerLR was informed about the trial by the responsible clinician or a member of the research team and they were provided with a copy of the covering statement for the PerLR with an attached patient information sheet (PIS) and asked to give an opinion on whether or not the patient would object to taking part in such medical research. If the PerLR decided that the patient would have no objection to participating in the trial, then they were asked to sign two copies of the PerLR consent form, which were then countersigned by the person taking consent. A copy of the signed informed consent form was placed in the patients’ medical records, while the originals were retained by the PerLR and by the PI in the investigator site file (ISF).
Professional legal representative consent: UK
If the patient was unable to give informed consent and no PerLR was available, a doctor who was not connected with the conduct of the trial acted as a ProfLR. The doctor was informed about the trial by the responsible clinician or a member of the research team and given a copy of the PIS. If the doctor decided that the patient was suitable for entry into the trial, they were asked to sign two copies of the ProfLR consent form. A copy of the signed informed consent form was placed in the patients’ medical records, while the originals were retained by the doctor ProfLR and by the PI in the ISF.
Retrospective patient consent: UK
Patients were informed of their participation in the trial by the responsible clinician or a member of the research team once they regained capacity to understand the details of the trial. The responsible clinician or a member of the research team discussed the study with the patient and the patient was given a copy of the PIS to keep. The patient was asked for consent to participate in the trial and to sign two copies of the consent to continue form, which were then countersigned by the person taking consent. A copy of the signed consent form was placed in the patient’s medical records while the originals were retained by the patient and by the PI in the ISF. When consent to continue was not obtained, consent from the legal representative remained valid. If the patient refused consent, data collected about the patient were not entered into the analysis.
Informed consent/assent procedure for Ireland
Informed consent/assent forms approved by the CREC were provided to each trial site. The PI was responsible for ensuring that informed consent/assent for trial participation was given by each patient or their representative respectively. This required that the informed consent/assent form be signed and personally dated by the patient or by their representative. An appropriately trained doctor or nurse took consent. If no assent was given, the patient was not randomised into the trial.
The incapacitating nature of the condition precluded obtaining prospective informed consent from participants. In this situation, informed assent was sought from the patient’s representative or from a professional representative if no suitable representative was available.
Patient representative assent: Ireland
Informed assent was sought from the patient’s representative who was a relative, partner or close friend. The patient representative was informed about the trial by the responsible clinician or a member of the research team and they were provided with a copy of the covering statement for the representative with an attached PIS and asked to give an opinion on whether or not the patient would object to taking part in such medical research. If the patient representative decided that the patient would have no objection to participating in the trial they were asked to sign two copies of the patient representative assent form, which was then countersigned by the person taking consent. A copy of the signed informed assent form was placed in the patient’s medical records, while the originals were retained by the patient representative and by the PI in the ISF.
Professional representative assent: Ireland
If the patient was unable to give informed consent and no patient representative was available, a doctor who was not connected with the conduct of the trial acted as a professional representative. The doctor was informed about the trial by the responsible clinician or a member of the research team and given a copy of the PIS. If the doctor decided that the patient was suitable for entry into the trial, they were asked to sign two copies of the professional representative assent form. A copy of the signed informed assent form was placed in the patient’s medical records, while the originals were retained by the professional representative and by the PI in the ISF.
Retrospective patient consent: Ireland
Patients were informed of their participation in the trial by the responsible clinician or a member of the research team once they regained the capacity to understand the details of the trial. The responsible clinician or a member of the research team discussed the study with the patient and the patient was given a copy of the PIS to keep. The patient was asked for consent to participate in the trial and to sign two copies of the consent to continue form, which was then countersigned by the person taking consent. A copy of the signed consent form was placed in the patient’s medical records while the originals were retained by the patient and by the PI in the ISF. When consent to continue was not obtained, consent from the patient or professional representative remained valid. If the patient refused consent, data collected about the patient were not entered into the analysis.
Withdrawal of consent: UK and Ireland
Patients could withdraw or be withdrawn (by PerLR or ProfLR) from the trial at any time without prejudice. Data recorded up to the point of withdrawal were included in the trial analysis, unless consent/assent to use the patient’s data had also been withdrawn. If a patient or legal representative requested termination of the trial drug during the treatment period, the drug was stopped but the patient continued to be followed up as part of the trial. If a patient or a PerLR withdrew consent/assent during trial treatment, the trial drug was stopped but permission was sought to access medical records for data related to the trial. If a patient or PerLR wished to withdraw from the trial after completion of trial treatment, permission to access medical records for trial data was sought.
Randomisation
After informed consent, patients were randomised using an automated 24-hour telephone randomisation service provided by the Centre for Healthcare Randomised Trials (University of Aberdeen, UK). Randomisation was stratified by site and by vasopressor requirement (defined as any inotropic requirement except dopamine of < 6 µg per kg per minute). The randomisation service used a computer-generated random number sequence and allocated a numbered treatment pack to each patient.
Each site participating in the study had a unique site number that had to be entered when using the randomisation system. The randomisation service required confirmation that the patient fulfilled the trial entry criteria and requested the data required for stratification. Using the computer-generated random number sequence, the randomisation service allocated a unique trial identification number in accordance with the study randomisation schedule prepared prior to the start of the trial. This identification number was used throughout the trial for purposes of patient identification. The clinician then recorded the unique patient identification number on to the prescription, which was then handed to the pharmacy who handed over the matching pre-numbered drug pack. The randomisation service confirmed randomisation details by e-mail to the Clinical Trials Unit (CTU), chief investigator and to the study site pharmacy.
Trial treatment
The trial drug packs contained 70 × 40-mg tablets of either simvastatin or placebo. This allowed for 80 mg (2 × 40-mg tablets) to be given daily up to a maximum of 28 days with 7 days of study drug over requirement to allow for spillage or spoilage.
Drug pack preparation and supply
Patient drug packs were prepared by Victoria Pharmaceuticals, Belfast, UK. A dose of 40 mg of simvastatin or identical placebo tablets were packaged in a white opaque high-density polyethylene plastic container, which was sealed with a tamper-evident seal and labelled in compliance with applicable regulatory requirements. All trial drugs were packaged identically and identified only by the unique trial identifier.
Drug packs were stored by Victoria Pharmaceuticals and dispatched by them to participating hospital pharmacies under the instruction of the trial manager who monitored recruitment at participating sites. Hospital pharmacies ensured that all study drugs were stored in a secured area that was separate from normal hospital stock under the manufacturer’s recommended storage conditions.
As several sites experienced problems with accessing the pharmacy out of office hours, advice was sought from MHRA on storing the study drug on ICUs. MHRA advised that this was acceptable on the following conditions being met: this must be acceptable to the site pharmacist, the drugs must be received in to the site pharmacy and dispensed out to the ICU, accountability logs must record this action, and study drugs must be kept in a secure temperature-monitored environment. Several sites stored the study drug on the ICU and the location within the ICU was checked at monitoring visits.
Administration of trial drug
The first dose of study drug was administered as soon as possible, ideally within 4 hours of randomisation and subsequent doses were given each morning starting on the following calendar day. If, for any reason, a dose was not administered at the intended time, clinicians were advised to administer it subsequently but not > 12 hours after the intended time of administration. The study drug was most commonly administered via a nasogastric tube; however, if the patient was extubated and receiving oral intake prior to critical care discharge, then the study drug could be administered orally.
If patients received more than a single bolus of amiodarone after randomisation, then the dose was reduced to 40 mg on alternate dates (i.e. one tablet on alternate days for the duration of the treatment period).
Trial drug termination criteria
The trial drug was terminated if any one of the following conditions was met, prior to the maximum treatment period (28 days from randomisation):
-
study drug-related AE
-
CK level of > 10 times the upper limit of normal
-
ALT/AST level of > 8 times the upper limit of normal
-
-
development of a clinical condition requiring immediate treatment with a statin
-
discharge from critical care environment
-
death
-
discontinuation of active medical treatment
-
patient or relative request for withdrawal of patient from the study
-
decision by the attending clinician that the study drug should be discontinued on safety grounds.
Clinical management of patients in the trial
Patients involved in the HARP-2 trial were managed according to best practice established locally on each unit.
Participating ICUs were encouraged to use low tidal volume ventilation at 6–8 ml per kg of predicted body weight and to maintain plateau pressure of < 30 cm H2O. All other treatment was determined by the patients’ physicians.
Serious adverse events and suspected unexpected serious adverse reactions
Assessment of causality
Each AE was clinically assessed for causality based on the information available (i.e. the relationship of the AE to the study drug). For the purposes of this trial, the causality was assessed using the categories presented below. Drug-related AEs were defined as those considered by the PI to have a possible, probable or definite relationship to the study drug. The PI at each site was responsible for evaluating all AEs for causality using the following guide:
-
Unrelated: clinical event with an incompatible time relationship to study drug administration, and that could be explained by underlying disease or other drugs or chemicals.
-
Unlikely: clinical event for which the time relationship to study drug administration makes a causal connection improbable, but that could plausibly be explained by underlying disease or other drugs or chemicals.
-
Possible: clinical event with reasonable time relationship to study drug administration, but that could also be explained by concurrent disease or other drugs or chemicals.
-
Probable: clinical event with a reasonable time relationship to study drug administration, and is unlikely to be attributed to concurrent disease or other drugs or chemicals.
-
Definite: clinical event with plausible time relationship to study drug administration, and that cannot be explained by concurrent disease or other drugs or chemicals.
The AEs were reported and documented on the relevant pages of the case report form (CRF), in accordance with the procedures outlined below. The PI at each site was responsible for evaluating all AEs for expectedness in addition to causality and severity.
Drug-related AEs were defined as those with a possible, probable or definite relationship to the study drug. The site PI was asked to assess causality and record this as a ‘yes’ or ‘no’ in the CRF.
Adverse event reporting period
The AE reporting period for this trial began on enrolment into the trial and ended 30 days following the last administration of the study drug. All AEs assessed by the PI as possibly or probably related to the study drug and all SAEs that occurred during this time were followed until they were resolved or were clearly determined to be due to a patient’s stable or chronic condition or intercurrent illness(es).
Adverse event reporting
Because the HARP-2 trial was recruiting from a population that was already in a life-threatening situation, it was expected that many of the participants would experience AEs. Events that were expected in this population (i.e. events that were in keeping with the patient’s underlying medical condition) were not required to be reported as AEs.
An adverse reaction (AR) is an AE that is related to the administration of the study drug. Any AEs that were related to the study drug were reported on the AE form within the CRF.
The following are ARs that were expected and reported on the AE form within the CRF:
-
CK level of > 10 times the upper limit of normal
-
ALT/AST level of > 8 times the upper limit of normal.
An unexpected adverse reaction (UAR) is an AE that is related to the administration of the study drug and that is unexpected, in that it has not been previously reported in the current summary of product characteristics (SPC). Clinicians were instructed to report all UARs.
Serious adverse event reporting
A SAE was defined as an AE that fulfilled one or more of the criteria for severity:
-
results in death
-
is immediately life-threatening
-
requires hospitalisation or prolongs existing hospitalisation
-
results in persistent or significant disability or incapacity
-
results in congenital abnormality or birth defect
-
requires medical intervention to prevent one of the above, or is otherwise considered medically significant.
Because the HARP-2 trial was recruiting from a population that was already in a life-threatening situation, it was expected that many of the participants would experience SAEs. Events that were expected in this population (i.e. events that were in keeping with the patient’s underlying medical condition) and that were collected as outcomes of the trial, including death and organ failure, were not reported as SAEs.
The SAEs were evaluated by the PI for causality (i.e. their relationship to study drug) and expectedness. All other SAEs were reported using the SAE form in the patient’s CRF and were reported to the CTU within 24 hours of the clinician becoming aware of the event. The CTU was responsible for reporting SAEs to the sponsors, ethics committees, MHRA and IMB within the required timelines as per the regulatory requirements.
A serious adverse reaction (SAR) is a SAE that is related to the administration of the study drug. The following SAR was expected and clinicians were advised that it must be reported on the SAE form within the CRF:
-
need for renal replacement therapy in patients with CK levels of > 10 times the upper limit of normal.
The SUSARs are SAEs that are considered to be caused by the study drug and are unexpected (i.e. their nature or severity is not consistent with the SPC). All SUSARs were the subject of expedited reporting to meet regulatory requirements.
All AEs and SAEs were classified using common terminology criteria for adverse events (CTCAE) version 4 (v4.03, 14 June 2010) apart from those AEs/SAEs that were protocol related. According to CTCAE v4.0, elevated ALT, AST and CK fall under the broad category of investigations. However, these AEs were expected ARs in this trial and, as such, they have been presented separately.
When CK levels were elevated and required renal replacement therapy, this SAR fell under the musculoskeletal category and was also reported separately.
Data collection
Hospital data
The Acute Physiology and Chronic Health Evaluation (APACHE) II scores were used as part of the description of the trial population. For centres that participated in the Intensive Care National Audit and Research Centre (ICNARC) Case Mix Programme (CMP), the APACHE II scores were obtained from ICNARC; therefore, these centres supplied only the CMP number for HARP-2 trial participants. Centres that did not participate in the CMP collected all of the data to allow calculation of the APACHE II score.
At enrolment, the patients’ demographic characteristics, ventilatory and physiological variables and admission APACHE II scores were recorded. The cause of ARDS was identified by the treating clinician. For each day in the ICU, ventilatory and physiological variables as well as data on organ support based on the UK’s critical care minimum data set40 were recorded. Vital status at 28 days was collected but cause of death was not recorded.
Data were collected and recorded on a two-part carbonised CRF by the site research team from the time the patient was considered for entry into the trial through to their discharge from hospital. In the event that a patient was transferred to another hospital, the site research team liaised with the receiving hospital to ensure complete data collection. On completion of the data collection period, the PI signed off the CRF; the top copy of each CRF was returned to the CTU and the bottom copy was retained in the CRF booklet at the recruiting site. Submitted data were reviewed for completeness on receipt at the CTU and entered onto a secure, backed-up custom database. Entries on the CRF that were ambiguous, unintelligible or incomplete were queried with the hospital research staff who completed the CRF.
Discharge and follow-up questionnaires
To provide an economic evaluation, a HRQoL questionnaire was measured using the EuroQol-5 Dimensions, three-level version (EQ-5D-3L) questionnaire administered at hospital discharge by site staff. The CTU followed up surviving patients with a further EQ-5D questionnaire at 3, 6 and 12 months post randomisation. Resource utilisation data were also collected at 6 and 12 months.
To minimise the risk of causing distress by contacting relatives of patients who had since died, the CTU used a NHS central register and/or contacted the patient’s general practitioner (GP) to ascertain the patient’s survival status prior to any contact being made.
If questionnaires were not returned, a maximum of two telephone contacts were made to the patient to check that the questionnaire had been received and that the patient was happy to complete it. In the event that the patient stated non-receipt, a second copy of the questionnaire was sent out. If the second questionnaire was not returned, the patient was contacted again by telephone to follow up. To increase the percentage of questionnaire returns an amendment was submitted to the ethics committee to obtain permission to change our procedures to send out a £5 (€5 for sites in Ireland) gift voucher with the first questionnaire as a thank you gesture for patients taking part in the study.
Methods for assays
The MMP-8, IL-6, Ang 2 and RAGE were measured using Duoset enzyme-linked immunosorbent assay kits from R&D Systems Europe (Oxford, UK). According to the manufacturer’s instructions, samples were run neat or in reagent diluent (phosphate-buffered saline/1% bovine serum albumin) to obtain values within the detection range of the standard curve. When neat values were less than the lowest standard, then they were assigned the value of the lowest standard. The range of detection for the analyses was as follows.
-
MMP-8: 31–4000 pg/ml
-
IL-6: 9–600 pg/ml
-
Ang2: 94–6000 pg/ml
-
RAGE: 62.5–4000 pg/ml.
The CRP was measured by immunoturbidimetric assay performed by Randox Laboratories (Crumlin, Northern Ireland). The range of detection was 0.3–402 mg/l.
Plasma 25-hydroxyvitamin D was measured by liquid chromatography mass spectrometry by colleagues in the laboratory of Barbara Obermayer-Pietsch, Heidelberg, Germany. Values at or below the lower limit of detection were assigned a value of 6.99 ng/ml.
Statistical methods
Analyses were conducted on all outcome data obtained from all participants as randomised and regardless of protocol adherence (i.e. intention-to-treat analysis). All statistical tests were at the two-sided p-value of 0.05 unless adjustment for multiple testing was needed. As VFDs and oscillator frequency-free days (OFFDs) had a bimodal distribution, the groups were initially analysed by t-test with difference in means and 95% CIs presented. A secondary analysis of these outcome measures involving a bootstrapped t-test was also conducted to support the findings of the t-tests as detailed in version 3 of the statistical analysis plan. This differed from the main analysis for VFDs detailed in the protocol but was approved by the Trial Steering Committee and independent Data Monitoring and Ethics Committee (DMEC). The comparison of other continuous outcomes was by analysis of variance, including covariates when appropriate. Statistical diagnostic methods were used to check for violations of the assumption, and transformations were performed when required. A statistical interaction test was used to assess differences in treatment effects between the subgroups. For binary outcome measures, risk ratios and associated 95% CIs were calculated. Time-to-event data were presented using Kaplan–Meier plots. In all time-to-event analyses, patients who did not experience the event in question (e.g. death) were censored on the date last seen or 60 days. Time-to-event data were tested using a log-rank chi-squared test. Hazard ratios (HRs) were calculated to test the difference between the treatment groups using the Cox proportional hazards model. All HRs were presented with a two-sided 95% CI. Median follow-up time was calculated.
Subgroup analyses used a statistical test for interaction and were reported using 99% CI.
Four subgroup analyses were prespecified by:
-
age in quartiles
-
vasopressor requirement (defined as any inotropic requirement except dopamine of < 6 µg per kg per minute) as presence or absence
-
sepsis versus non-sepsis aetiology
-
CRP level at baseline in quartiles.
Every effort was made to minimise missing baseline and outcome data in this trial and imputation was not used.
Exploratory analysis on organ dysfunction was carried out using the Mann–Whitney U-test.
Exploratory analysis on biomarkers was carried out using student t-tests and Fisher’s exact test, presented graphically by day, when applicable, and split by baseline quartiles.
Sample size calculation
Sample size assumptions were based on previously published data. Assuming a mean number of VFDs of 12.7 days [standard deviation (SD) 10.6 days],41 we estimated that a sample of 524 patients would need to be enrolled in order for the study to have 80% power, at a two-tailed significance level of 0.05, to detect a mean between-group difference of 2.6 VFDs. On the basis of data from the Pulmonary Artery Catheters in Management of Patients in Intensive Care (PAC-Man) trial,42 a dropout rate of 3% was estimated and, therefore, a total of 540 patients (270 in each group) was required.
When the primary outcome measure of VFDs was available for 270 patients, a sample size review was undertaken by the DMEC independent statistician. The purpose of this was to check that the within-groups variance was not substantially underestimated, which would mean that the sample size had been underestimated.
No other data were analysed. The group allocation of the patients was not revealed and this review did not compare the two groups to examine treatment effects. In keeping with recommendations on interim sample size review,43 the review would not lead to a reduction of the sample size. The review led to a recommendation that the sample size remained unchanged.
Ethics and regulatory approvals
Ethics approval was given for the study by ORECNI REC B (UK sites: 10/NIR02/36) in September 2010 and by CREC (ROI sites: 8/10) in July 2010. Local approval and permission from the research and development (R&D) department of each participating trust was received prior to sites commencing on the study. This was not applicable to Republic of Ireland-based sites.
The MHRA gave approval for the study in August 2010 (UK sites: 32485/0020/001-0001) [and IMB (now Health Products Regulatory Authority)] gave approval in October 2010 (CT900/495/1).
During the trial, the following amendments were submitted to ethics and regulatory authorities.
Amendment one (main changes)
Protocol v1.0_24.06.10 was submitted to ORECNI in the original application for ethics approval. ORECNI requested some changes that necessitated amending the protocol to v2.0_01.09.10. The major changes included:
-
amending the exclusion criteria to exclude non-English-speaking patients or those who did not adequately understand verbal or written information unless an interpreter was available
-
amending the exclusion criteria to include the wording ‘currently’ and ‘sustained’ in relation to the use of listed concomitant medications. Amiodarone was added to the list of concomitant medications
-
giving clarification that the 80-mg dose was given as two 40-mg tablets
-
adding SOFA score to the schedule of assessments in day 14 and day 28.
Amendment two (main changes)
Protocol v2.0_01.09.10 was amended to v3.0_16.05.11. ORECNI and MHRA approved the following changes.
-
Change of address of the chief investigator.
-
Non-pulmonary organ failure-free days added to secondary outcomes.
-
An additional blood sample was added at day 21 to measure CK and liver function.
-
The exclusion criteria were amended to allow for patients receiving erythromycin as a prokinetic to be included in the study.
-
Change of address of study drug supplier.
-
Other changes: additional sites added.
Amendment three (main changes)
Protocol v3.0_16.05.11 was amended to v4.0_18.07.11 and was approved by ORECNI and MHRA to include the following changes.
-
The exclusion criteria were amended to remove clarithromycin and erythromycin.
-
Clarification was given that domiciliary ventilation used for sleep-disordered breathing would not be included as mechanical ventilation.
-
Scheduling for research samples submission was changed to allow that research samples due on bank holidays or weekends could be collected up to 2 days after the due date (with the exception of day 1).
Amendment four
There was no change to the protocol. Additional sites were added.
Amendment five (main changes)
Protocol v4.0_18.07.11 was amended to v5.0_13.01.12. This amendment was approved by ORECNI and MHRA to include the following change.
-
The exclusion criteria were amended to change the upper limit of normal for ALT and AST from more than five times the upper limit of normal to more than eight times the upper limit of normal.
Amendment six (main changes)
Protocol v5.0_13.01.12 was amended to v6.0_09.01.13. This amendment was approved by ORECNI and MHRA to include the following changes.
-
The protocol was amended to allow NICTU to use a NHS central register and/or contact the patient’s GP to ascertain the patient survival status prior to any contact being made.
-
The PIS and consent form were amended to inform patients and patient representatives of this change.
-
A GP letter was created to advise the patient’s GP of future NICTU contact in relation to patient survival status.
Amendment seven (main changes)
Additional sites added and change of PI at three sites.
Minor amendments included:
-
the inclusion of a £5 or €5 thank-you voucher
-
change of CTU (NICTU) address from Education and Research Centre to Elliott Dynes Building.
Chapter 3 Results
Overview of recruitment
Patients were recruited between 21 December 2010 and 13 March 2014. During the recruiting period to the HARP-2 trial a total of 40 sites participated in the study: five in Ireland, four in Northern Ireland, four in Scotland and 27 in England. One site opened in 2010, 25 in 2011, nine in 2012, four in 2013 and one in 2014 (Table 1).
| Sites | Time period | Total recruited | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 2011 | 2012 | 2013 | January to March 2014 | ||||||
| Screened | Recruited | Screened | Recruited | Screened | Recruited | Screened | Recruited | ||
| Addenbrookes | 26 | 3 | 60 | 9 | 157 | 10 | 45 | 5 | 27 |
| Aintree | 23 | 2 | 13 | 1 | 3 | ||||
| Altnagelvin | 85 | 2 | 74 | 5 | 74 | 3 | 13 | 1 | 11 |
| Antrim | 25 | 3 | 28 | 4 | 45 | 11 | 17 | 3 | 21 |
| Arrowe Park, Wirral | 21 | 2 | 15 | 3 | 5 | ||||
| Berkshire (Royal) | 50 | 3 | 44 | 3 | 69 | 2 | 18 | 0 | 8 |
| Birmingham Heartlands | 105 | 7 | 98 | 20 | 46 | 9 | 9 | 4 | 40 |
| Bristol | 39 | 8 | 43 | 7 | 48 | 5 | 11 | 0 | 20 |
| Coventry | 30 | 2 | 45 | 8 | 78 | 3 | 34 | 1 | 14 |
| Derby (Royal) | 35 | 2 | 3 | 0 | 0 | 0 | 0 | 0 | 2 |
| Dumfries and Galloway | 24 | 4 | 29 | 6 | 28 | 5 | 5 | 2 | 17 |
| Edinburgh (Royal Infirmary) | 123 | 6 | 173 | 6 | 129 | 8 | 27 | 1 | 21 |
| Freemans | 61 | 4 | 85 | 5 | 50 | 3 | 18 | 2 | 14 |
| Frenchay | 27 | 2 | 10 | 2 | 4 | ||||
| Glasgow Victoria | 21 | 3 | 31 | 5 | 19 | 3 | 3 | 1 | 12 |
| Glasgow Western | 47 | 2 | 27 | 0 | 0 | 0 | 0 | 0 | 2 |
| Good Hope | 12 | 0 | 34 | 5 | 27 | 5 | 3 | 0 | 10 |
| Guy’s and St Thomas’ | 58 | 5 | 36 | 8 | 48 | 6 | 5 | 1 | 20 |
| Harefield | 7 | 0 | 16 | 1 | 0 | 0 | 0 | 0 | 1 |
| Hull | 33 | 2 | 58 | 2 | 16 | 2 | 6 | ||
| King’s College London | 33 | 6 | 116 | 27 | 110 | 9 | 16 | 5 | 47 |
| Leeds General Infirmatory and St James’ | 1 | 1 | 46 | 3 | 9 | 0 | 4 | ||
| Norfolk and Norwich | 12 | 2 | 21 | 5 | 3 | 1 | 8 | ||
| Papworth | 45 | 3 | 26 | 2 | 28 | 1 | 11 | 0 | 6 |
| Poole | 29 | 5 | 26 | 9 | 4 | 2 | 16 | ||
| Queen Elizabeth Birmingham | 26 | 6 | 233 | 19 | 27 | 1 | 26 | ||
| Royal Free Hospital | 30 | 1 | 1 | ||||||
| Royal Hospitals Belfast | 156 | 23 | 99 | 17 | 127 | 18 | 23 | 2 | 60 |
| Royal Liverpool | 86 | 5 | 191 | 10 | 32 | 4 | 19 | ||
| Royal Preston | 35 | 1 | 42 | 1 | 5 | 0 | 2 | ||
| Royal Sussex | 68 | 4 | 8 | 0 | 4 | ||||
| St George’s Hospital | 70 | 2 | 35 | 0 | 0 | 0 | 2 | ||
| Ulster Hospital | 56 | 9 | 103 | 6 | 80 | 3 | 15 | 3 | 21 |
| Whiston | 148 | 4 | 143 | 12 | 178 | 4 | 41 | 3 | 23 |
| Worcester (Royal) | 20 | 2 | 27 | 4 | 18 | 3 | 2 | 0 | 9 |
| Beaumont Hospital | 54 | 6 | 33 | 1 | 6 | 0 | 7 | ||
| Cork | 7 | 0 | 33 | 5 | 23 | 4 | 3 | 0 | 9 |
| Galway | 12 | 5 | 99 | 6 | 77 | 4 | 17 | 0 | 15 |
| Mater | 44 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| St Vincent’s Hospital | 18 | 3 | 19 | 0 | 5 | 0 | 3 | ||
| Total | 1225 | 106 | 1880 | 204 | 2302 | 179 | 519 | 51 | 540 |
By July 2011 recruitment was behind target, due to:
-
a longer than anticipated trial start-up, relating in part to delays with local R&D permissions
-
patients being excluded because they were on clarithromycin or erythromycin.
To address this situation, a substantial amendment was submitted and authorised, allowing the removal of clarithromycin and erythromycin from the exclusion criteria. This had a significant effect on recruitment as 10% of patients screened were excluded owing to patients receiving these drugs. In addition, a further amendment was submitted and authorised to change the upper limit of normal for AST and ALT from five times the upper limit of normal to eight times the upper limit of normal.
A full-time trial co-ordinator commenced in post in December 2011. This allowed the trial manager to commit more time to progressing the opening of new sites. An expression of interest was sent out and attracted > 20 applications. Twelve of these sites were invited to apply on a competitive basis, which resulted in seven new sites joining the study.
In addition, a contract variation was submitted to extend the study for a further 1 year. This extension was approved and allowed a longer period for recruitment (Figure 1). As a result of these amendments recruitment to the study increased steadily and the recruitment target was achieved. There was no evidence of recruitment fatigue over the course of the trial.
FIGURE 1.
The HARP-2 trial monthly accrual.

An ongoing review of sites not meeting the recruitment target was carried out and as a result of this a total of five sites were closed to recruitment over the recruitment period of the study: one in Ireland (closed October 2012), one in Scotland (closed July 2012) and three in England (July 2012, September 2013 and December 2013) (see Table 1).
An extension of 1 year was granted in April 2012, at which point the timeline for target recruitment was extended by 1 year. Recruitment was completed in March 2014 when the target of 540 was met.
Participants
Out of the 5926 patients who were assessed for eligibility, 540 (9%) underwent randomisation. Four patients who did not fulfil the eligibility criteria were randomised in each group and are included in the analysis. Five patients allocated to simvastatin and three patients in the placebo group did not receive study drug. One patient in the simvastatin group was lost to follow-up. No data on the primary outcome were available for one patient in the simvastatin group and two patients in the placebo group (Figure 2). 37
FIGURE 2.
Flow chart of 12-month mortality BIPAP, bilevel positive airway pressure; CPAP, continuous positive airway pressure; IMP, investigational medicinal product. From The New England Journal of Medicine, McAuley DF, Laffey MD, O’Kane CM, Perkins GD, Mullan B, Trinder J, et al. Simvastatin in the acute respiratory distress syndrome, vol. 371, pp. 1695–703. 37 Copyright © 2014 Massachusetts Medical Society. Reprinted with permission.

Data collection and procedures
To ensure that accurate, complete and reliable data were collected, the CTU provided training to site staff in the format of investigator meetings and/or site initiation visits. The CTU provided the PI and research staff with training on good clinical practice, the study protocol, completion of the CRF and trial procedures including standard operating procedures.
Baseline characteristics
The baseline characteristics of the patients at randomisation were similar in the two study groups. The main causes of ARDS were pneumonia and sepsis. On day 3, the tidal volume in the simvastatin group did not differ significantly from that in the placebo group; the mean difference was 0.05 ml per kg of predicted body weight (95% CI −0.61 to 0.71 ml per kg; p = 0.89) (Table 2). 37
| Characteristic | Treatment group | |
|---|---|---|
| Simvastatin (n = 259) | Placebo (n = 280) | |
| Age (years), mean (SD) | 53.2 (16.1) | 54.4 (16.7) |
| Gender, n (%) | ||
| Male | 137 (52.9) | 170 (60.7) |
| Female | 122 (47.1) | 110 (39.3) |
| Sepsis, n (%) | 189 (73.0) | 218 (77.9) |
| Non-sepsis, n (%) | 70 (27.0) | 62 (22.1) |
| Vasopressor requirement, n (%) | ||
| Yes | 169 (65.2) | 187 (66.8) |
| No | 90 (34.8) | 93 (33.2) |
| Plateau pressure (cm H2O), mean (SD) | 23.55 (6.07) | 23.64 (6.03) |
| APACHE II score, mean (SD) | 19.4 (6.9) | 18.3 (6.2) |
| PaO2 : FiO2 ratio, mean (SD) | 16.4 (7.3) | 17.6 (7.4) |
| Tidal volume per ideal body weight (ml/kg), mean (SD) | 8.1 (2.8) | 8.1 (2.6) |
| Aetiology of ARDS | ||
| Direct, n (%) | ||
| Smoke/toxin inhalation | 1 (0.4) | 2 (0.7) |
| Gastric content aspiration | 21 (8.1) | 29 (10.4) |
| Near drowning | 0 (0) | 0 (0) |
| Thoracic trauma | 22 (8.5) | 10 (3.6) |
| Pneumonia | 161 (62.2) | 154 (55.0) |
| Other | 15 (5.8) | 19 (6.8) |
| Indirect, n (%) | ||
| Sepsis | 106 (40.9) | 118 (42.1) |
| Cardiopulmonary bypass | 1 (0.4) | 0 (0) |
| Pancreatitis | 5 (1.9) | 17 (6.1) |
| Non-thoracic trauma | 4 (1.5) | 8 (2.9) |
| Other | 14 (5.4) | 1 9 (6.8) |
| SOFA score, mean (SD) | 8.60 (3.2) | 8.97 (2.9) |
| OI, mean (SD) | 112.8 (87.3) | 112.0 (89.0) |
| Lowest mean arterial pressure (mmHg), mean (SD) | 65.4 (9.3) | 64.9 (8.4) |
Treatment with study drug
Patients received the study drug for a mean of 10.2 days (SD 7.1 days) in the simvastatin group and 11.0 days (SD 7.9 days) in the placebo group (p = 0.23). The most common reasons for discontinuation of the study drug were discharge from critical care, death and an AE that was considered to be related to the study drug. A total of five patients assigned to the simvastatin group and three assigned to the placebo group received treatment with non-trial statins.
Table 3 presents the mean (SD) for continuous data and the number (%) for categorical data for treatment after trial entry, reasons for termination of study drug and protocol violations. 37
| Study drug administration | Treatment group | |
|---|---|---|
| Simvastatin (n = 259) | Placebo (n = 280) | |
| Study drug given | 254 | 278 |
| Number of days on treatment, mean (SD) | 10.2 (7.1) | 11 (7.9) |
| Reason for termination of study drug, n (%) | ||
| 28 days after randomisation | 20 (7.7) | 28 (10.0) |
| Discharge from critical care | 141 (54.4) | 147 (52.3) |
| Liver transaminases levels of > 5/8 times upper limit | 20 (7.7) | 16 (5.7) |
| CK levels of > 10 times upper limit | 21 (8.1) | 14 (5.0) |
| Request for discontinuation of trial drug by patient or legal representative | 2 (0.8) | 3 (1.1) |
| Discontinuation of active treatment | 9 (3.5) | 8 (2.8) |
| Development of a condition requiring immediate treatment with statin | 2 (0.8) | 3 (1.1) |
| Decision by a physician on safety ground | 3 (1.2) | 4 (1.4) |
| Death | 31 (12.0) | 46 (16.4) |
| Other | 10 (3.9) | 12 (4.3) |
| Non-trial statins | 5 (–) | 3 (–) |
| Days of non-trial statins, mean (SD) | 6 (8) | 3 (2) |
| Protocol violations, n | ||
| Post-randomisation withdrawal | ||
| Refused use of data already collected | 0 | 1 |
| Refused data collection from NHS records | 0 | 1 |
| Withdrew from follow-up | 1 | 4 |
| Ineligible patient | 4 | 4 |
| Did not receive allocated treatment | 5 | 3 |
| Received treatment of other group | 0 | 0 |
| Study drug administered in error | 27 | 24 |
| Study drug omitted in error | 21 | 31 |
As a marker of compliance and absorption, simvastatin concentrations were measured in plasma samples used for biomarker analyses. Simvastatin or simvastatin acid were detectable in plasma in 216 out of 226 (96%) samples from simvastatin-treated patients at day 3. Of the 10 participants in whom simvastatin was not detected at day 3, five had detectable concentrations of simvastatin at day 7. One subject had documented withholding of medication at day 3 and 7.
Because this was not a true pharmacokinetic measurement (i.e. the samples not taken in relation to timing of drug administration), the levels were highly variable and potentially less than the limit of detection of the assay in subjects assigned to simvastatin in the four remaining subjects in whom it was not detectable at days 3 or 7.
Outcomes
Primary outcome
The number of VFDs did not differ significantly between the two study groups (Table 4) [12.6 days (SD 9.9 days) with simvastatin and 11.5 days (SD 10.4 days) with placebo; mean difference 1.1 days, 95% CI −0.6 to 2.8 days; p = 0.21]. 37 There was also no significant between-group difference in the number of VFDs after adjustment for the baseline PaO2 : FiO2 ratio (mean difference 1.4 days, 95% CI −0.3 to 3.2 days; p = 0.10).
| Outcome | Treatment group | |||
|---|---|---|---|---|
| Simvastatin | Placebo | Difference (95% CI); results from bootstrapped t-test | p-value; results from bootstrapped t-test | |
| Primary outcome; VFDs to 28 days post randomisationa | ||||
| n | 258 | 279 | ||
| Mean (SD) | 12.6 (9.9) | 11.5 (10.4) | 1.1 (–0.6 to 2.8); 1.1 (–0.7 to 2.8) | 0.21; 0.22a |
| Non-pulmonary OFFDs in first 28 days (N = 539) | ||||
| n | 257 | 279 | ||
| Mean (SD) | 19.4 (11.1) | 17.8 (11.7) | 1.6 (–0.4 to 3.5); 1.6 (–0.3 to 3.5) | 0.11; 0.10a |
| Change in OI from baseline (N = 404) | ||||
| Day 3 (n = 329) | ||||
| n | 167 | 162 | ||
| Mean (SD) | –25.3 (59.7) | –8.5 (75.1) | –16.8 (–31.5 to –2.1) | 0.02 |
| Day 7 (n = 204) | ||||
| n | 93 | 111 | ||
| Mean (SD) | –33.0 (83.9) | –30.1 (78.5) | –2.9 (–25.4 to 19.5) | 0.80 |
| Day 14 (n = 100) | ||||
| n | 43 | 57 | ||
| Mean (SD) | –37.5 (111.3) | –24.6 (61.8) | –13.0 (–47.7 to 21.7) | 0.46 |
| Day 28 (n = 19) | ||||
| n | 4 | 15 | ||
| Mean (SD) | 20.7 (125.4) | –54.0 (43.6) | 74.7 (–3.5 to 153.0) | 0.06 |
| Change in SOFA from baseline (N = 472) | ||||
| Day 3 (n = 430) | ||||
| n | 205 | 225 | ||
| Mean (SD) | –0.9 (2.2) | –0.8 (2.3) | –0.1 (–0.5 to 0.3) | 0.67 |
| Day 7 (n = 307) | ||||
| n | 152 | 155 | ||
| Mean (SD) | –2.5 (3.0) | –2.5 (2.7) | –0.1 (–0.7 to 0.6) | 0.86 |
| Day 14 (n = 151) | ||||
| n | 70 | 81 | ||
| Mean (SD) | –3.4 (3.3) | –2.4 (3.2) | –1.1 (–2.1 to –0.01) | 0.047 |
| Day 28 (n = 38) | ||||
| n | 15 | 23 | ||
| Mean (SD) | –4.1 (3.9) | –2.7 (4.3) | –1.5 (–4.3 to 1.3) | 0.29 |
| All-cause mortality 28 days post randomisationb | 57/259 (22.0) | 75/280 (26.8) | 0.8 (0.6 to 1.1) | 0.23 |
| Death before discharge from critical careb | 56/259 (21.6) | 70/280 (25.0) | 0.9 (0.6 to 1.2) | 0.36 |
| Death before discharge from hospitalb | 67/259 (25.9) | 90/280 (32.1) | 0.8 (0.6 to 1.1) | 0.13 |
Short-term secondary outcomes
The change from baseline to day 28 in the OI (Figure 3 and see Table 4) did not differ significantly between the two groups, nor did the SOFA score (Figure 4 and see Table 4). 37 There were no significant differences in the number of days free of non-pulmonary organ failure or in mortality at 28 days (see Table 4). 37 Mortality at ICU discharge or hospital discharge (see Table 4) was also not significantly different between the two groups. 37 Among survivors, the mean duration of the ICU stay was 13.9 days (SD 14.4 days) in the simvastatin group and 14.4 days (SD 13.3 days) in the placebo group (mean difference −0.5 days, 95% CI −3.2 to 2.2 days; p = 0.71). The mean duration of the hospital stay was 37.7 days (SD 64.5 days) and 35.4 days (SD 31.1 days) for the simvastatin group and the placebo group, respectively (mean difference 2.3 days, 95% CI −8.0 to 12.6 days; p = 0.66). From randomisation to day 28, there were no significant differences between the two groups in the probability of breathing without assistance or the probability of survival (Figure 5). 37
FIGURE 3.
The OI and 95% CI at days 1, 3, 7, 14 and 28.

FIGURE 4.
Mean SOFA score and 95% CI at days 1, 3, 7, 14 and 28.

FIGURE 5.
Kaplan–Meier plot for probabilities of survival at 28 days and breathing without assistance, from the day of randomisation (day 0) to day 28, according to whether patients received simvastatin or placebo. (a) Unassisted breathing; and (b) survival. From The New England Journal of Medicine, McAuley DF, Laffey MD, O’Kane CM, Perkins GD, Mullan B, Trinder J, et al. Simvastatin in the acute respiratory distress syndrome, vol. 371, pp. 1695–703. 37 Copyright © 2014 Massachusetts Medical Society. Reprinted with permission.


Bootstrapped t-test for non-pulmonary OFFDs was not statistically significant (mean difference 1.6 days, 95% CI –0.3 to 3.5 days; p = 0.10). To adjust for the multiple testing for the change in OI and total SOFA score, a p-value of 0.0125 is considered to be statistically significant.
Exploratory analyses
The SOFA is a secondary outcome for this study (Table 5). 37 Further exploratory analysis was carried out for non-pulmonary dysfunction. There was no significant between-group difference in the proportion of patients with non-pulmonary organ dysfunction, as measured by a SOFA score of < 2 for each organ.
| Variable | Day, n/N | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | p-value | 3 | p-value | 7 | p-value | 14 | p-value | 28 | p-value | ||||||
| Simvastatin | Placebo | Simvastatin | Placebo | Simvastatin | Placebo | Simvastatin | Placebo | Simvastatin | Placebo | ||||||
| Renal | 220/247 | 235/271 | 0.4 | 220/249 | 224/264 | 0.3 | 184/202 | 180/205 | 0.3 | 96/106 | 101/118 | 0.3 | 32/34 | 35/37 | 1.0 |
| Hepatic | 206/240 | 223/269 | 0.4 | 215/247 | 216/261 | 0.2 | 183/198 | 177/202 | 0.1 | 93/102 | 102/113 | 1.0 | 31/32 | 33/36 | 0.6 |
| Cardiovascular | 81/259 | 80/280 | 0.5 | 123/253 | 131/267 | 0.9 | 160/203 | 169/209 | 0.6 | 92/110 | 98/123 | 0.5 | 34/36 | 36/39 | 1.0 |
| Haematological | 188/247 | 210/270 | 0.7 | 183/249 | 200/261 | 0.5 | 172/201 | 175/204 | 1.0 | 97/106 | 109/118 | 0.8 | 30/34 | 33/37 | 1.0 |
| Neurological | 166/241 | 182/263 | 1.0 | 159/237 | 174/256 | 0.8 | 127/188 | 132/202 | 0.7 | 65/103 | 67/115 | 0.5 | 17/32 | 17/37 | 0.6 |
Exploratory biomarker analysis
Neutrophil activation
Neutrophil activation, as measured by plasma MMP-8 concentrations, was compared at baseline and day 3 between the statin and placebo treatment groups. There was no baseline difference between the two groups (difference 4352.00 pg/ml, 95% CI –3410.15 to 12,114.15 pg/ml). Simvastatin did not reduce MMP-8 at day 3 (Figure 6) (difference –1138.12 pg/ml, 95% CI –6895.97 to 4619.74 pg/ml).
FIGURE 6.
Mean (SD) plasma MMP-8 (pg/ml) levels in simvastatin- and placebo-treated groups at baseline and day 3.

Patients were stratified into quartiles according to degree of neutrophil activation at baseline to investigate whether or not those with greater baseline inflammation had a greater response to simvastatin. We found no evidence that high neutrophil activation at baseline predicted a response to simvastatin in terms of VFDs or 28-day mortality (Tables 6 and 7).
| VFDs to 28 days post randomisation | Treatment group, mean (SD) | Difference (99% CI) | |
|---|---|---|---|
| Simvastatin | Placebo | ||
| MMP-8 (pg/ml) | |||
| ≤ 5191 (n = 124) | 15.54 (9.54) | 13.12 (10.96) | 2.41 (–2.40 to 7.23) |
| 5191–13,792 (n = 123) | 11.07 (10.15) | 12.32 (10.81) | –1.25 (–6.22 to 3.72) |
| 13,792–34,631 (n = 124) | 11.87 (9.81) | 10.06 (9.88) | 1.81 (–2.87 to 6.49) |
| > 34,631 (n = 124) | 11.35 (9.44) | 10.44 (10.18) | 0.91 (–3.71 to 5.53) |
| Mortality at 28 days post randomisation | Treatment group, n (%) | p-value | |||
|---|---|---|---|---|---|
| Simvastatin | Placebo | ||||
| Dead | Alive | Dead | Alive | ||
| MMP-8 (pg/ml) | |||||
| ≤ 5191 | 10 (14.71) | 58 (85.29) | 13 (22.81) | 44 (77.19) | 0.26 |
| 5191–13,792 | 16 (28.07) | 41 (71.93) | 16 (23.88) | 51 (76.12) | 0.68 |
| 13,792–34,631 | 9 (16.98) | 44 (83.02) | 21 (29.58) | 50 (70.42) | 0.14 |
| > 34,631 | 14 (23.33) | 46 (76.67) | 20 (31.25) | 44 (68.75) | 0.42 |
We found no evidence that higher neutrophil activation at baseline predicted a reduction in neutrophil activation at day 3 with simvastatin treatment (Table 8).
| MMP-8 (pg/ml) level at baseline quartiles | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Simvastatin | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| ≤ 5191 | 7121.02 (9869.97) | 59 | 7402.07 (14,578.1) | 46 | –281.05 (–5022.03 to 4459.92) | 0.907 |
| > 5191 and ≤ 13,792 | 12,316.53 (27,271.85) | 52 | 18,072.60 (48,578.34) | 61 | –5756.07 (–20,797.17 to 9285.03) | 0.450 |
| > 13,792 and ≤ 34,631 | 17,620.51 (32,966.06) | 44 | 17,527.61 (24,428.20) | 55 | 92.90 (–11,359.92 to 11,545.72) | 0.987 |
| > 34,631 | 24,806.60 (30,740.25) | 49 | 21,661.31 (33,229.70) | 48 | 3145.29 (–9754.41 to 16,044.99) | 0.630 |
Systemic inflammation and acute phase response
Previously in the HARP study26 simvastatin was shown to reduce systemic inflammation in patients with ARDS. We assessed the effect of simvastatin on systemic inflammation and acute phase response, as measured by IL-6 and CRP levels.
Baseline CRP level was the same in the statin and placebo-treated groups (difference –4.8 mg/l, 95% CI –24.0 to 14.5 mg/l). Although CRP levels fell over time, there was no difference between placebo and simvastatin-treated cohorts at day 3 (difference –5.5 mg/l, 95% CI –23.0 to 12.0 mg/l) (Figure 7 shows mean and SD CRP for simvastatin- and placebo-treated groups).
FIGURE 7.
Systemic inflammation: mean (SD) plasma CRP levels in simvastatin- and placebo-treated groups at baseline and day 3.
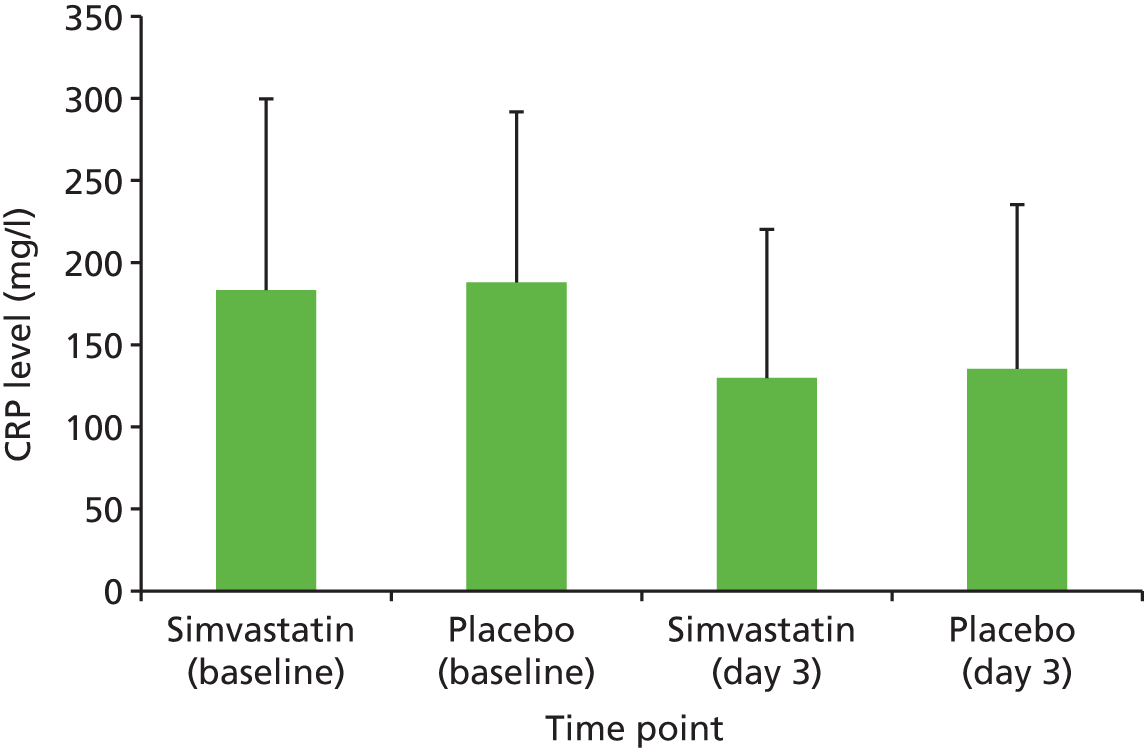
We tested the hypothesis that simvastatin would have a greater effect in those with higher baseline acute phase response, reflecting increased inflammation. Patients were stratified into quartiles according to baseline CRP level. There was no evidence that higher baseline CRP level predicted a greater response to simvastatin in terms of either reducing VFDs or 28-day mortality (Tables 9 and 10).
| VFDs to 28 days post randomisation | Treatment group | Difference (99% CI) | |
|---|---|---|---|
| Simvastatin | Placebo | ||
| CRP (mg/l) level at baseline, mean (SD) | |||
| ≤ 100 (n = 125) | 11.8 (10.4) | 9.0 (10.7) | 2.8 (–2.1 to 7.8) |
| 100–175 (n = 122) | 13.5 (10.5) | 13.2 (10.2) | 0.3 (–4.6 to 5.2) |
| 175–250 (n = 134) | 12.9 (9.8) | 11.2 (10.9) | 1.7 (–3.1 to 6.5) |
| > 250 (n = 126) | 12.7 (9.0) | 12.0 (9.6) | 0.7 (–3.7 to 5.0) |
| Mortality at 28 days post randomisation | Treatment group, n (%) | p-value | |||
|---|---|---|---|---|---|
| Simvastatin | Placebo | ||||
| Dead | Alive | Dead | Alive | ||
| CRP (mg/l) level | |||||
| ≤ 100 (N = 126) | 18 (26.09) | 51 (73.91) | 17 (29.82) | 40 (70.18) | 0.692 |
| 100–175 (N = 124) | 15 (25.42) | 44 (74.58) | 14 (21.54) | 51 (78.46) | 0.674 |
| 175–250 (N = 134) | 10 (17.54) | 47 (82.46) | 27 (35.06) | 50 (64.94) | 0.032 |
| > 250 (N = 126) | 9 (14.52) | 53 (85.48) | 13 (20.31) | 51 (79.69) | 0.484 |
There was no evidence that a greater degree of baseline inflammation predicted a greater response to simvastatin as measured by systemic CRP at day 3 (Table 11).
| CRP (mg/l) level at baseline quartiles | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Simvastatin | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| ≤ 100 | 65.38 (49.94) | 48 | 79.70 (64.21) | 63 | –14.33 (–36.53 to 7.88) | 0.204 |
| > 100 and ≤ 175 | 112.08 (79.90) | 60 | 105.82 (62.05) | 53 | 6.26 (–20.67 to 33.19) | 0.646 |
| > 175 and ≤ 250 | 160.75 (95.04) | 65 | 149.06 (85.00) | 53 | 11.69 (–21.55 to 44.93) | 0.487 |
| > 250 | 186.99 (117.35) | 58 | 184.83 (98.29) | 56 | 2.16 (–38.08 to 42.40) | 0.916 |
Plasma IL-6, a marker of systemic inflammation, was measured in patients at baseline (difference –45.03 pg/ml, 95% CI –342.61 to 252.54 pg/ml) and day 3 (difference 14.10 pg/ml, 95% CI –33.22 to 61.42 pg/ml). There was no difference in baseline level nor day 3 IL-6 level between the two groups (Figure 8).
FIGURE 8.
Systemic inflammation: mean (SD) plasma IL-6 (pg/ml) level in simvastatin- and placebo-treated groups at baseline and day 3.
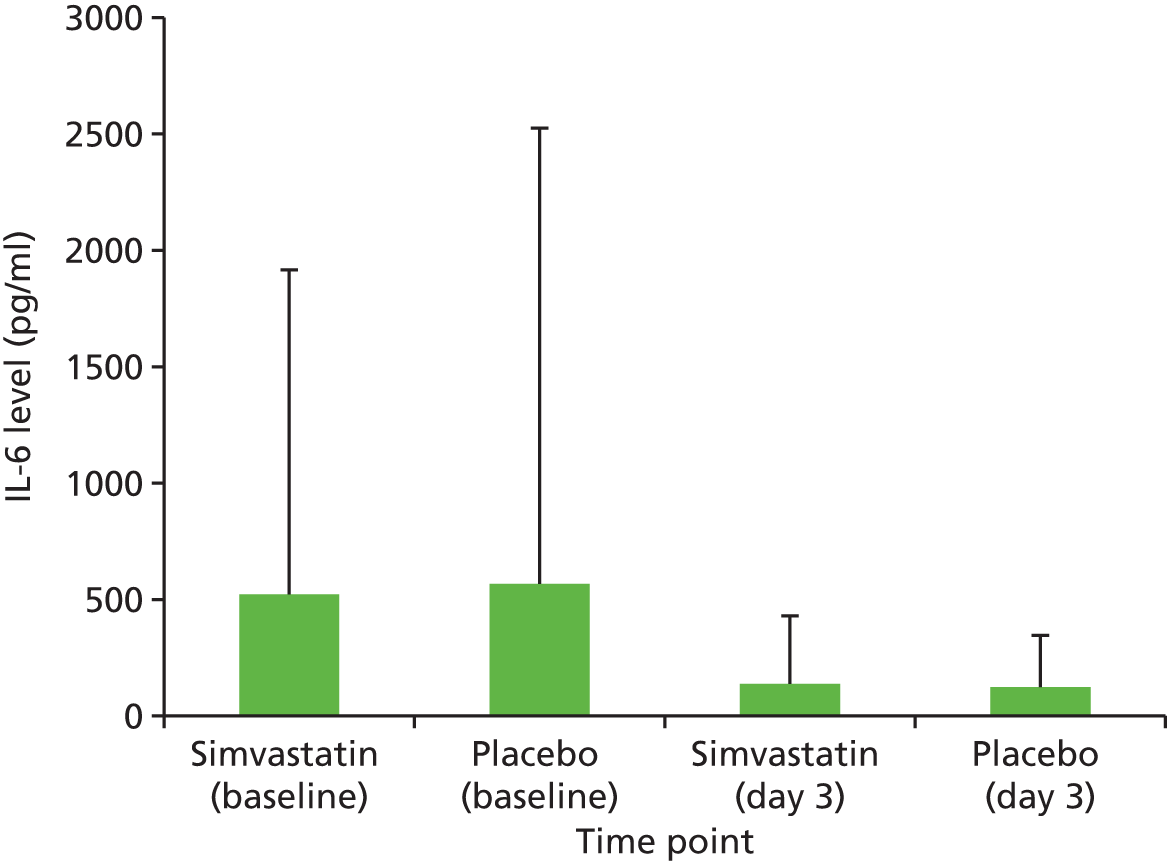
Patients were stratified into quartiles according to baseline IL-6 level to test the hypothesis that higher inflammation at recruitment, as measured by plasma IL-6 level, predicted a greater response to simvastatin. Higher baseline IL-6 level did not predict a greater response to simvastatin in terms of VFDs (Table 12) or 28-day mortality (Table 13).
| VFDs to 28 days post randomisation | Treatment group, mean (SD) | Difference (99% CI) | |
|---|---|---|---|
| Simvastatin | Placebo | ||
| IL-6 (pg/ml) level | |||
| ≤ 52.89 (n = 127) | 15.59 (10.11) | 11.86 (10.42) | 3.73 (–1.04 to 8.49) |
| 52.89–135.15 (n = 126) | 13.49 (9.20) | 13.02 (10.72) | 0.48 (–4.17 to 5.12) |
| 135.15–349.15 (n = 128) | 11.51 (9.88) | 11.92 (10.75) | –0.41 (–5.29 to 4.47) |
| > 349.15 (n = 127) | 9.66 (9.66) | 8.95 (9.64) | 0.71 (–3.77 to 5.19) |
| Mortality at 28 days post randomisation | Treatment group, n (%) | p-value | |||
|---|---|---|---|---|---|
| Simvastatin | Placebo | ||||
| Dead | Alive | Dead | Alive | ||
| IL-6 (pg/ml) level | |||||
| ≤ 53 | 9 (14.29) | 54 (85.71) | 11 (16.92) | 54 (83.08) | 0.81 |
| 53–135 | 13 (19.12) | 55 (80.88) | 9 (15.25) | 50 (84.75) | 0.64 |
| 135–349 | 12 (22.64) | 41 (77.36) | 26 (34.67) | 49 (65.33) | 0.17 |
| > 349 | 18 (29.03) | 44 (70.97) | 26 (40.00) | 39 (60.00) | 0.26 |
We did not find that higher baseline levels of IL-6 predicted a greater reduction in systemic inflammation as measured by IL-6 by day 3 (Table 14).
| IL-6 (pg/ml)-level baseline quartiles | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Simvastatin | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| ≤ 53 | 59.59 (160.41) | 56 | 47.90 (60.73) | 60 | 11.68 (–32.37 to 55.73) | 0.60 |
| > 53 and ≤ 135 | 67.96 (74.0) | 65 | 57.93 (65.35) | 53 | 10.04 (–15.72 to 35.79) | 0.442 |
| > 135 and ≤ 349 | 115.87 (137.02) | 47 | 132.15 (214.82) | 68 | –16.28 (–86.59 to 54.03) | 0.647 |
| > 349 | 312.57 (504.44) | 57 | 227.94 (291.99) | 56 | 84.63 (–69.37 to 238.63) | 0.279 |
As a sterol-based hormone (steroid), vitamin D rises during the acute inflammatory response. We investigated whether or not simvastatin affected circulating plasma 25-hydroxyvitamin D during the course of ARDS. There was no baseline difference between the two groups (difference –0.14, 95% CI –1.47 to 1.19) and no evidence that simvastatin altered plasma vitamin D during the course of ALI (Figure 9).
FIGURE 9.
Acute phase response: mean (SD) circulating plasma vitamin D level in simvastatin- and placebo-treated groups at baseline and days 3, 7 and 14.
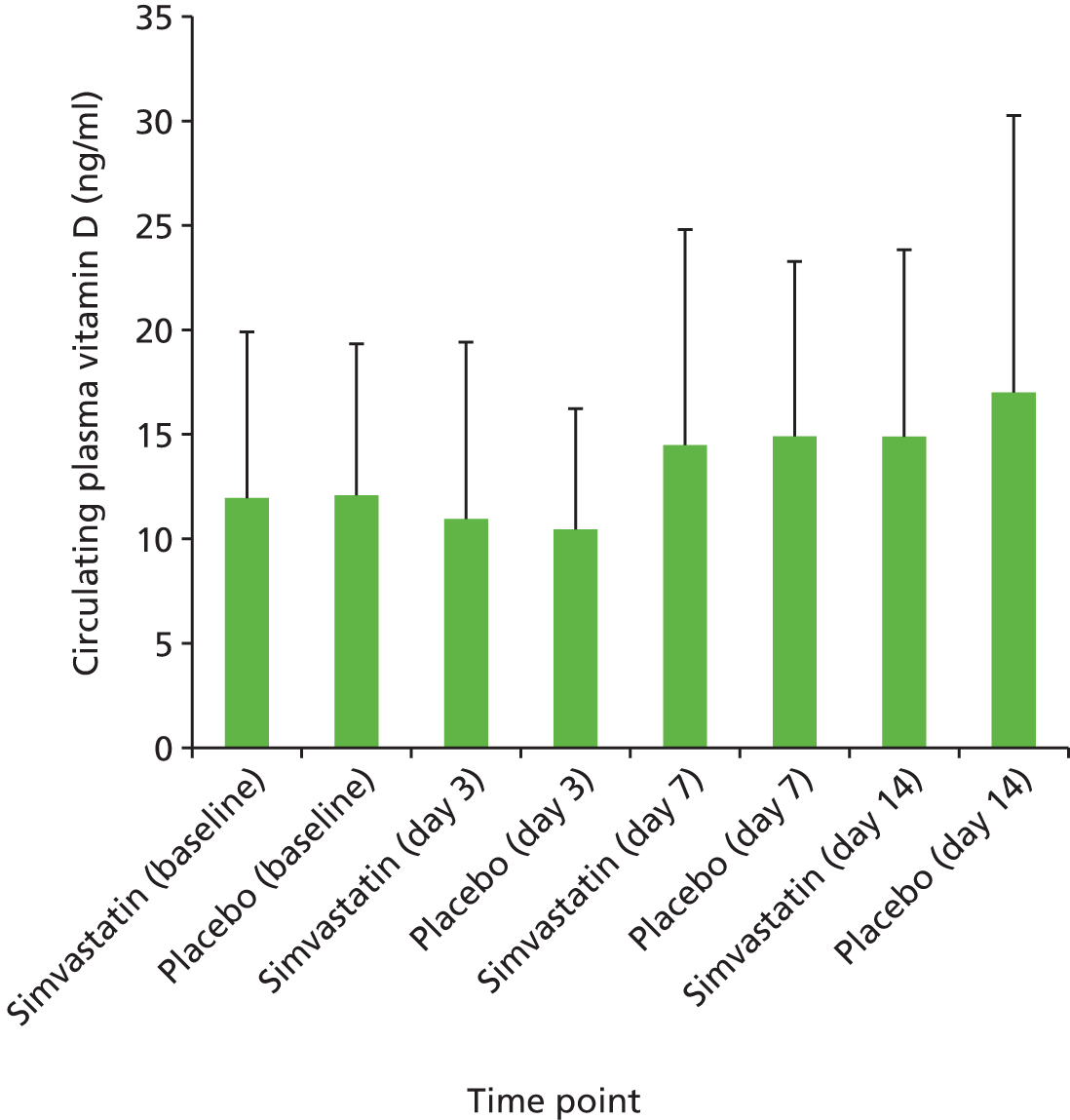
Patients were stratified into quartiles according to baseline vitamin D. There was no evidence that those with higher vitamin D at baseline who received simvastatin had a greater improvement in VFDs or reduction in mortality than those receiving placebo (Tables 15 and 16).
| Primary outcome | Treatment group, mean (SD) | Difference (99% CI) | |
|---|---|---|---|
| Simvastatin | Placebo | ||
| 25-hydroxyvitamin D level (ng/ml) | |||
| < 7a (n = 198) | 12.63 (9.95) | 11.65 (10.64) | 0.98 (–2.85 to 4.81) |
| ≥ 7 and ≤ 9 (n = 55) | 14.63 (8.84) | 7.60 (9.85) | 7.03 (0.30 to 13.77) |
| > 9 and ≤ 14 (n = 125) | 11.92 (9.84) | 11.85 (9.95) | 0.07 (–4.57 to 4.70) |
| > 14 (n = 126) | 12.32 (10.49) | 11.89 (10.81) | 0.42 (–4.55 to 5.40) |
| Secondary outcome | Treatment group, n (%) | p-value | |||
|---|---|---|---|---|---|
| Simvastatin | Placebo | ||||
| Dead | Alive | Dead | Alive | ||
| 25-hydroxyvitamin D level (ng/ml) | |||||
| < 7 (N = 198) | 19 (20.43) | 74 (79.57) | 32 (30.19) | 74 (69.81) | 0.143 |
| ≥ 7 and ≤ 9 (N = 55) | 2 (6.67) | 28 (93.33) | 9 (36.00) | 16 (64.00) | 0.015 |
| > 9 and ≤ 14 (N = 126) | 16 (25.40) | 47 (74.60) | 16 (25.40) | 47 (74.60) | 1.000 |
| > 14 (N = 126) | 16 (26.67) | 44(73.33) | 15 (22.73) | 51 (77.27) | 0.681 |
We did not find that higher baseline levels of vitamin D predicted a greater reduction in acute phase response as measured by vitamin D on day 3 (Table 17).
| 25-hydroxyvitamin D level at baseline quartiles | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Simvastatin | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 25-hydroxyvitamin D level (ng/ml) | ||||||
| ≤ 6.99a | 8.03 (2.25) | 83 | 7.57 (1.57) | 96 | 0.46 (–0.11 to 1.02) | 0.115 |
| > 6.99 and ≤ 8.9 | 8.83 (4.03) | 29 | 8.72 (3.67) | 23 | 0.10 (–2.07 to 2.28) | 0.924 |
| > 8.9 and ≤ 14.325 | 9.36 (4.29) | 56 | 9.80 (4.19) | 54 | –0.44 (–2.04 to 1.17) | 0.591 |
| > 14.325 | 17.40 (13.36) | 56 | 16.89 (7.47) | 55 | 0.51 (–3.57 to 4.59) | 0.805 |
Endothelial injury
Endothelial injury as measured by plasma Ang2 concentrations was compared at baseline and day 3 between the statin and placebo treatment groups. There was no difference in baseline (difference 1153.44 pg/ml, 95% CI –4644.16 to 6951.03 pg/ml) or day 3 (difference 541.09 pg/ml, 95% CI –498.46 to 1580.65 pg/ml) between the two groups (Figure 10).
FIGURE 10.
Endothelial injury: mean (SD) plasma Ang2 (pg/ml) level in simvastatin- and placebo-treated groups at baseline and day 3.

Patients were stratified into quartiles according to degree of endothelial injury at baseline to investigate whether or not those with greater baseline endothelial injury had a greater response to simvastatin. We found no evidence that higher endothelial injury at baseline predicted a response to simvastatin in terms of VFDs or 28-day mortality (Tables 18 and 19).
| VFDs to 28 days post randomisation | Treatment group, mean (SD) | Difference (99% CI) | |
|---|---|---|---|
| Simvastatin | Placebo | ||
| Ang2 (pg/ml) level | |||
| ≤ 4369 (n = 127) | 13.82 (9.78) | 11.88 (11.00) | 1.94 (–2.89 to 6.76) |
| 4369–10,308 (n = 126) | 13.65 (9.81) | 11.55 (10.16) | 2.10 (–2.56 to 6.77) |
| 10,308–23,761 (n = 127) | 12.91 (9.90) | 12.69 (9.95) | 0.22 (–4.42 to 4.86) |
| > 23,761 (n = 127) | 10.30 (9.92) | 9.06 (10.55) | 1.24 (–3.51 to 5.99) |
| Mortality at 28 days post randomisation | Treatment group, n (%) | p-value | |||
|---|---|---|---|---|---|
| Simvastatin | Placebo | ||||
| Dead | Alive | Dead | Alive | ||
| Ang2 (pg/ml) level | |||||
| ≤ 4369 | 12 (17.65) | 56 (82.35) | 16 (26.67) | 44 (73.33) | 0.29 |
| 4369–10,308 | 13 (21.67) | 47 (78.33) | 17 (25.37) | 50 (74.63) | 0.68 |
| 10,308–23,76 | 10 (17.86) | 46 (82.14) | 17 (23.94) | 54 (76.06) | 0.51 |
| > 23,761 | 17 (26.98) | 46 (73.02) | 22 (34.38) | 42 (65.63) | 0.44 |
We did not demonstrate that higher Ang2 at baseline predicted a response to simvastatin in terms of showing reduced endothelial injury at day 3 (Table 20).
| Ang2 (pg/ml)-level baseline quartiles | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Simvastatin | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| ≤ 4369 | 2890.95 (2330.86) | 63 | 3122.72 (2835.92) | 52 | –231.78 (–1186.16 to 722.61) | 0.631 |
| > 4369 and ≤ 10,308 | 4302.81 (6989.11) | 54 | 3769.17 (3067.72) | 56 | 533.64 (–1493.92 to 2561.19) | 0.603 |
| > 10,308 and ≤ 23,761 | 5393.56 (5071.44) | 51 | 4675.61 (4621.78) | 68 | 717.95 (–1049.96 to 2485.87) | 0.423 |
| > 23,761 | 7831.79 (8899.61) | 58 | 6364.04 (6913.79) | 59 | 1467.75 (–1447.80 to 4383.30) | 0.321 |
Alveolar epithelial injury
Type I alveolar epithelial cell injury as measured by plasma RAGE concentrations was compared at baseline (difference 31.83 pg/ml, 95% CI –102.96 to 166.62 pg/ml) and day 3 (difference 103.43 pg/ml, 95% CI –71.54 to 278.40 pg/ml) between the statin and placebo treatment groups (Figure 11). Simvastatin had no effect on plasma RAGE.
FIGURE 11.
Epithelial injury: mean (SD) plasma RAGE (pg/ml) level in simvastatin- and placebo-treated groups at baseline and day 3.
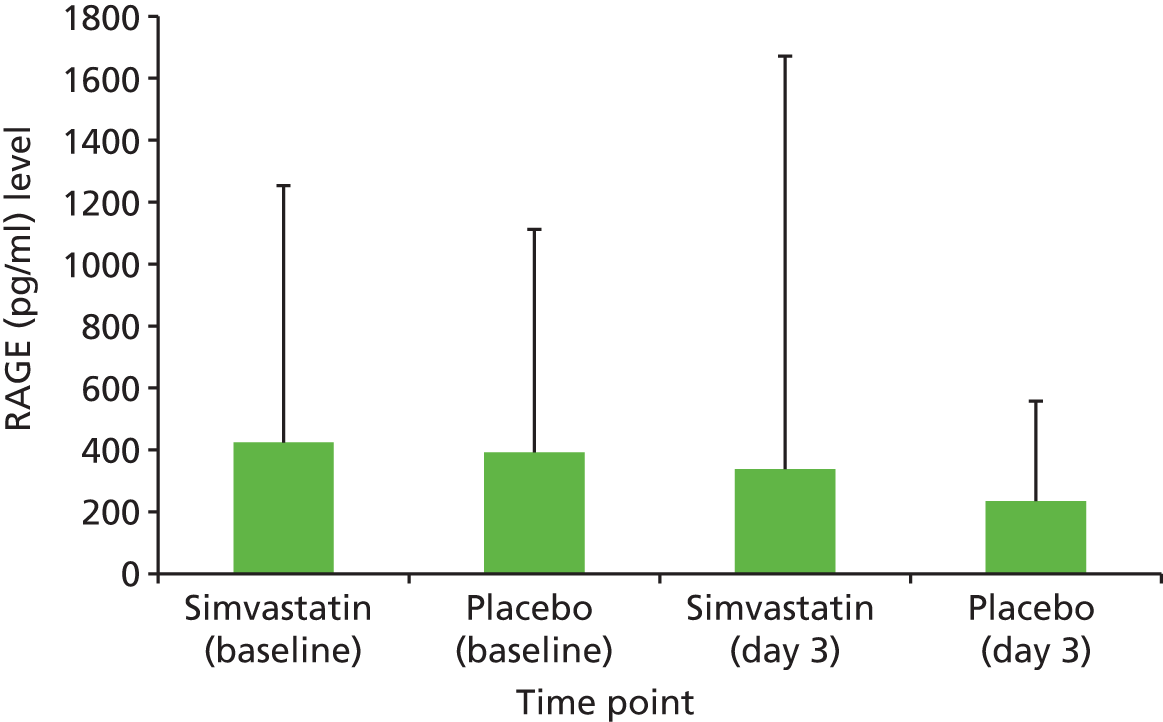
Patients were stratified into quartiles according to degree of epithelial injury at baseline to investigate whether or not those with evidence of greater epithelial injury at recruitment had a differential response to simvastatin. We found no evidence that worse epithelial injury at baseline predicted a response to simvastatin in terms of VFDs (Table 21) or 28-day mortality (Table 22).
| VFDs to 28 days post randomisation | Treatment group, mean (SD) | Difference (99% CI) | |
|---|---|---|---|
| Simvastatin | Placebo | ||
| RAGE (pg/ml) level | |||
| ≤ 63 (n = 167) | 12.70 (9.36) | 12.69 (10.38) | 0.01 (–3.98 to 4.00) |
| > 63 and ≤ 124 (n = 87) | 12.82 (10.69) | 9.83 (10.21) | 2.99 (–2.93 to 8.91) |
| > 124 and ≤ 369 (n = 128) | 14.29 (10.04) | 11.68 (10.54) | 2.61 (–2.16 to 7.37) |
| > 369 (n = 127) | 10.92 (9.88) | 10.68 (10.62) | 0.24 (–4.52 to 5.01) |
| Mortality at 28 days post randomisation | Treatment group, n (%) | p-value | |||
|---|---|---|---|---|---|
| Simvastatin | Placebo | ||||
| Dead | Alive | Dead | Alive | ||
| RAGE (pg/ml) level | |||||
| ≤ 63 (N = 168) | 11 (13.10) | 73 (86.90) | 25 (29.76) | 59 (70.24) | 0.014a |
| > 63 and ≤ 124 (N = 88) | 9 (23.08) | 30 (76.92) | 10 (20.41) | 39 (79.59) | 0.799 |
| > 124 and ≤ 369 (N = 128) | 13 (20.97) | 49 (79.03) | 18 (27.27) | 48 (72.73) | 0.419 |
| > 369 (N = 127) | 19 (30.65) | 43 (69.35) | 19 (29.23) | 46 (70.77) | 1.000 |
In addition, we did not find that higher epithelial injury at baseline predicted a greater response to simvastatin in terms of reducing plasma RAGE at day 3 (Table 23).
| RAGE (pg/ml)-level baseline quartiles | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Simvastatin | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| ≤ 63 | 147.30 (235.06) | 79 | 160.77 (213.85) | 76 | –13.47 (–84.87 to 57.92) | 0.710 |
| > 63 and ≤ 124 | 79.12 (36.70) | 38 | 208.72 (442.53) | 44 | –129.59 (–273.00 to 13.81) | 0.076 |
| > 124 and ≤ 369 | 167.41 (167.68) | 54 | 273.32 (375.67) | 58 | –105.92 (–216.25 to 4.42) | 0.060 |
| > 369 | 948.16 (2587.01) | 56 | 305.42 (257.06) | 59 | 642.74 (–27.83 to 1313.30) | 0.060 |
In summary, the biomarker exploratory analyses did not indicate that simvastatin treatment had any effect on neutrophilic inflammation, systemic inflammation or the acute phase response, nor on alveolar epithelial or endothelial injury in this cohort of patients. We were not able to identify a subgroup of patients with ALI as determined by levels of baseline systemic inflammation or epithelial or endothelial injury who had any beneficial effect from simvastatin.
Subgroup analysis
Subgroup analyses did not suggest that the effects of simvastatin were modified by any of the variables investigated. There was no significant interaction between treatment and age (p = 0.62), vasopressor requirement (p = 0.17), presence or absence of sepsis (p = 0.50) (Table 24)37 or baseline CRP level (p = 0.77) (see Table 9).
| Primary outcome: VFDs to 28 days post randomisation | Treatment group, mean (SD) | Difference (99% CI) | |
|---|---|---|---|
| Simvastatin | Placebo | ||
| Age (years) | |||
| ≤ 42.3 (n = 133) | 15.8 (9.2) | 13.4 (9.8) | 2.4 (–1.9 to 6.7) |
| 42.3–54.3 (n = 134) | 13.5 (9.7) | 14.3 (10.3) | –0.8 (–5.3 to 3.7) |
| 54.3–66.3 (n = 134) | 11.5 (9.9) | 10.3 (10.3) | 1.2 (–3.4 to 5.7) |
| > 66.3 (n = 134) | 9.3 (10.1) | 8.1 (10.2) | 1.1 (–3.5 to 5.7) |
| Vasopressor requirement | |||
| Absent (n = 182) | 14.7 (10.1) | 11.9 (10.4) | 2.8 (–1.2 to 6.8) |
| Present (n = 353) | 11.5 (9.7) | 11.2 (10.4) | 0.3 (–2.5 to 3.1) |
| Sepsis/non-sepsis | |||
| Sepsis (n = 404) | 12.2 (10.1) | 10.8 (10.3) | 1.4 (–1.2 to 4.0) |
| Non-sepsis (n = 131) | 13.7 (9.6) | 13.6 (10.7) | 0.05 (–4.6 to 4.7) |
Long-term outcomes
Mortality at 12 months post randomisation was a long-term clinical outcome for the trial and was not statistically significantly different between the two groups (p = 0.20; Table 25 and Figure 1244).
| Outcome | Treatment group | Risk ratio (95% CI) | p-value (Fisher’s exact test) | |
|---|---|---|---|---|
| Simvastatin | Placebo | |||
| Mortality 12 months post randomisation, n/N (%) | 82/258 (31.8) | 103/276 (37.3) | 0.9 (0.7 to 1.1) | 0.20 |
FIGURE 12.
Kaplan–Meier plot for 12-month survival. Amended from Agus et al. 44 This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
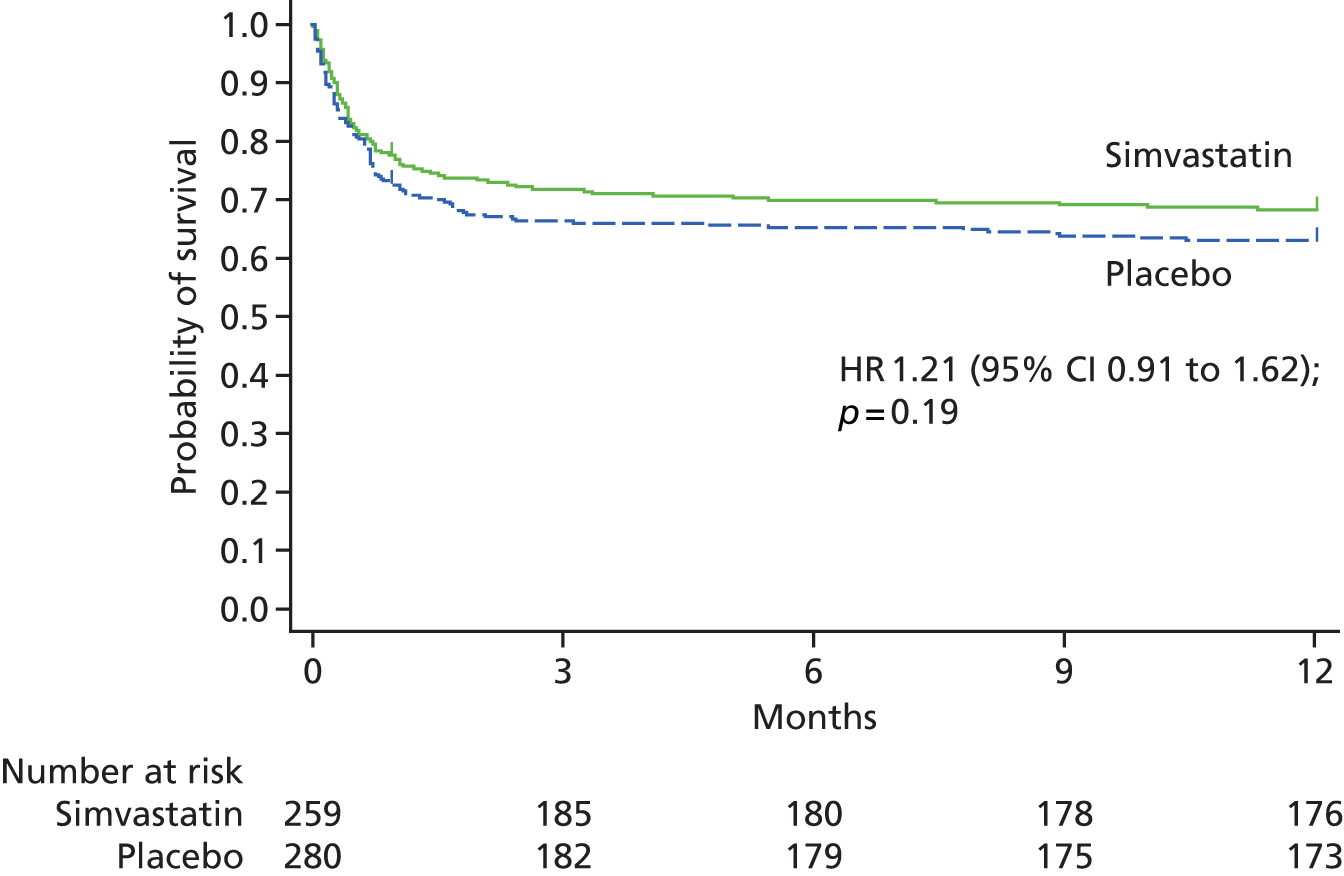
Adverse events
Safety outcomes
A total of 121 patients experienced AEs and a total of 170 AEs were reported in the study. The SAEs were reported in 25 patients (11 patients in the simvastatin group and 14 patients in the placebo group). In total, 28 SAEs were reported (12 in the simvastatin group and the 16 in the placebo group), with one patient in the simvastatin group having two SAEs and two patients in the placebo group each having two SAEs. In the simvastatin group one SAE was assessed to be related to study drug and this was not assessed to be an unexpected SAE. In the placebo group three SAEs were assessed to be related to study drug and, of these, two were thought to be unexpected SAEs.
There were two SUSARs reported during the HARP-2 study, neither of which was fatal. Both SUSARs were reported to the relevant competent authorities and ethics committee within the applicable 15-day window.
Table 26 summarises AEs, SAEs, SUSARs, ARs and SARs by treatment group. 37 Table 27 shows that the absolute values of AST and CK were significantly elevated at day 14 in the simvastatin-treated group, and that the elevation in CK persisted to day 21, reflecting the increased incidence of AEs related to CK and elevated transaminases. Table 27 presents the mean (SD) for highest ALT (units per litre), AST (units per litre) and CK (units per litre) collected over the course of the trial.
| Outcome | Treatment group, n | OR (95% CI) | p-value | |
|---|---|---|---|---|
| Simvastatin | Placebo | |||
| AEs, SAEs and SUSARs | ||||
| Total number of SAEs; patients | 12; 11 | 16; 14 | 0.8 (0.3 to 2.0) | 0.84 |
| Related to study druga | 1 | 3 | 0.4 (0.0 to 5.9) | 0.61; 0.42b |
| Related to study drug and unexpectedc | 0 | 2 | 0.0 (0.0 to 2.6) | 0.49; 0.32b |
| Total number of AEs; patients | 84; 59 | 86; 62 | 1.0 (0.7 to 1.6) | 0.92 |
| Related to study druga | 45 | 30 | 2.2 (1.1 to 4.2) | 0.02; 0.01b |
| SAEs | ||||
| Cardiac disorders | 3 | 5 | 0.7 (0.1 to 5.1) | 1.00 |
| Gastrointestinal disorders | 3 | 0 | d | 0.07 |
| Hepatobiliary disorders | 0 | 1 | 0e | 1.00 |
| Infections and infestations | 1 | 2 | 0.6 (0.0 to 14.0) | 1.00 |
| Injury, poisoning and procedural complications | 1 | 1 | 1.4 (0.0 to 144.1) | 1.00 |
| Musculoskeletal and connective tissue disorders/elevated CK levels | 1 | 1 | 1.4 (0.0 to 144.1) | 1.00 |
| Nervous system disorders | 1 | 2 | 0.6 (0.0 to 14.0) | 1.00 |
| Renal and urinary disorders | 1 | 0 | d | 0.43 |
| Respiratory, thoracic and mediastinal disorders | 1 | 4 | 0.3 (0.0 to 3.5) | 0.36 |
| AEs | ||||
| ALT levels of > 8 times the upper limit of normal and/or AST levels of > 8 times the upper limit of normalf | 34 | 24 | 1.8 (0.9 to 3.5) | 0.11; 0.06b |
| Blood and lymphatic system disorders | 1 | 1 | 1.0 (0.0 to 81.3) | 1.00 |
| Cardiac disorders | 6 | 11 | 0.5 (0.2 to 1.6) | 0.31 |
| CK levels of > 10 times the upper limit of normalf | 24 | 16 | 1.8 (0.8 to 3.9) | 0.15; 0.09b |
| Gastrointestinal disorders | 6 | 4 | 1.6 (0.4 to 7.9) | 0.53 |
| General disorders and administration site conditions | 1 | 3 | 0.3 (0.0 to 4.3) | 0.62 |
| Infections and infestations | 2 | 6 | 0.3 (0.0 to 1.9) | 0.28 |
| Injury, poisoning and procedural complications | 1 | 3 | 0.3 (0.0 to 4.3) | 0.62 |
| Investigations | 2 | 2 | 1.0 (0.1 to 14.4) | 1.00 |
| Nervous system disorders | 1 | 3 | 0.3 (0.0 to 4.3) | 0.62 |
| Renal and urinary disorders | 2 | 2 | 1.0 (0.1 to 14.4) | 1.00 |
| Respiratory, thoracic and mediastinal disorders | 4 | 5 | 0.8 (0.2 to 3.9) | 1.00 |
| Skin and subcutaneous tissue disorders | 0 | 6 | 0.0 (0.0 to 0.7) | 0.03 |
| ARsa | ||||
| CK levels of > 10 times the upper limit of normal | 17 | 8 | 2.5 (0.9 to 7.0) | 0.05; 0.04b |
| ALT levels of > 8 times the upper limit of normal and/or AST levels of > 8 times the upper limit of normal | 26 | 16 | 2.0 (0.9 to 4.3) | 0.08; 0.05b |
| SARsa | ||||
| Need for renal replacement therapy in patients with CK elevated > 10-fold | 1 | 1 | 1.4 (0.0 to 114.1) | 1.00 |
| Day | Treatment group, mean (SD) | p-valuea | |
|---|---|---|---|
| Simvastatin | Placebo | ||
| 1 | |||
| Highest ALT (U/L) | 45.5 (47.1) | 45.8 (43.2) | |
| Highest AST (U/L) | 59.9 (49.4) | 65.3 (63.9) | |
| Highest CK (U/L) | 327.2 (499.3) | 298.3 (487.7) | |
| 3 | |||
| Highest ALT (U/L) | 48.1 (54.9) | 65.8 (215.2) | 0.47 |
| Highest AST (U/L) | 71.3 (103.1) | 102.1 (454.1) | 0.37 |
| Highest CK (U/L) | 414.4 (1001.8) | 350.4 (941.5) | 0.54 |
| 7 | |||
| Highest ALT (U/L) | 60.0 (63.0) | 76.7 (155.7) | 0.15 |
| Highest AST (U/L) | 65.1 (57.6) | 76.9 (153.1) | 0.34 |
| Highest CK (U/L) | 170.5 (293.4) | 183.8 (450.3) | 0.70 |
| 14 | |||
| Highest ALT (U/L) | 84.8 (142.1) | 74.3 (115.2) | 0.59 |
| Highest AST (U/L) | 110.7 (218.3) | 58.5 (52.5) | 0.03 |
| Highest CK (U/L) | 409.7 (1350.9) | 104.4 (311.9) | 0.03 |
| 21 | |||
| Highest ALT (U/L) | 96.2 (110.9) | 72.3 (67.9) | 0.21 |
| Highest AST (U/L) | 64.2 (74.1) | 75.5 (126.7) | 0.81 |
| Highest CK (U/L) | 364.4 (912.2) | 77.2 (174.5) | 0.03 |
| 28 | |||
| Highest ALT (U/L) | 57.7 (46.9) | 105.6 (230.0) | 0.30 |
| Highest AST (U/L) | 47.0 (54.8) | 64.8 (111.5) | 0.98 |
| Highest CK (U/L) | 219.9 (631.7) | 131.3 (317.2) | 0.59 |
Discussion
In this large, multicentre, double-blind, randomised, placebo-controlled clinical trial in patients with ARDS, simvastatin did not improve short- or long-term clinical outcomes. VFDs is a composite measure that includes death. We analysed VFDs overall as well as the composites of duration of ventilation (see Table 29) and mortality (see Table 4 and Figure 537) separately, and both were in the same direction. Mortality rates were lower in the simvastatin group, although this was not statistically significant. Owing to the central limit theorem and the large sample size of 540 in the trial, we used a t-test (parametric technique) that assumes normality; however, we backed this up with a bootstrapped t-test, which is a non-parametric technique and makes no assumptions on the distribution of the data, which in fact was bimodal. Simvastatin was associated with an increase in AEs, but there was no increase in SAEs.
Our biomarker study findings indicated that simvastatin did not modulate pathogenic mechanisms implicated in the development of ALI. ALI is characterised by neutrophilic inflammation and alveolar epithelial and endothelial cell dysfunction. It is usually accompanied by evidence of systemic inflammation and systemic endothelial dysfunction. Measurement of neutrophil activation by plasma MMP-8 was chosen as this marker is neutrophil-specific, reflects their activation status and is easily and reproducibly measurable in highly inflamed samples. In addition, MMP-8 has been shown to be modulated by simvastatin in our healthy volunteer study of lung injury. 23 CRP is the gold-standard marker of systemic inflammation in clinical care and has been shown to fall in statin-treated ARDS patients in our previous Phase IIa study. 26 IL-6 has been shown in multiple studies to have important baseline predictive value and to correlate better with degree of lung injury and progress of disease in ARDS than many other biomarkers. In addition, we had shown a trend to reduction in IL-6 the statin-treated group in our Phase IIa HARP study. 26
We have previously identified (McAuley D, O’Kane C. Queen’s University Belfast, 2015; unpublished data) a rise in vitamin D concentrations over the course of ARDS, suggesting that it is synthesised as part of a generalised sterol-increase response in stress. We hypothesised that simvastatin would inhibit inflammation and the stress response and that this would be reflected by a reduction in neutrophil activation (MMP-8) and markers of inflammation/acute phase (CRP, vitamin D and IL-6). RAGE is a type I alveolar epithelial cell injury marker, accounting for the majority of the alveolar epithelial surface. Higher baseline levels of RAGE, indicating greater alveolar injury, are predictive of death in ARDS.
Angiopoietin 2 is a biomarker of endothelial inflammation and has been shown in several studies to be an independent prognostic marker for death in patients with ARDS. Although it is possible that the biomarkers chosen were not the ‘best’ biomarkers to show the effect of simvastatin, we did choose biomarkers that have been shown to be prognostically and mechanistically important in previous studies in ARDS and that, in the case of IL-6 and CRP, have previously shown a response to simvastatin in an ARDS population (albeit in a small study). 26 The absence of even a trend across this range of markers that reflect the different key pathophysiological mechanisms of ARDS (inflammation, endothelial and epithelial injury and neutrophil activation) correlates with the absence of effect of simvastatin on clinical outcome measures, and supports our conclusion that simvastatin did not modulate these pathophysiological mechanisms. Potential explanations for the absence of effect include potential significant variability in the tissue levels of drug achieved, with levels being inadequate to impair inflammation, or a true absence of these ‘off-target’ statin effects in patients with disease. It is also possible that by the time ALI has presented clinically, the cascade of inflammation and systemic inflammatory response is already too far advanced to allow statins to have an effect. It is possible that statins are potentially more useful as a pre-treatment strategy to inhibit these mechanisms at the time of onset.
The study was a well-conducted, large, multicentre, allocation-concealed, randomised, double-blind, parallel-group trial. This is the gold standard to determine the potential benefits of a therapeutic intervention and, therefore, it is highly unlikely that deficiencies in the trial design accounted for the lack of difference between the groups.
Statins can modulate mechanisms important in the pathogenesis of ALI. 10 Observational studies support a clinical trial of a statin in ALI. 21 Furthermore, simvastatin reduced LPS-induced pulmonary and systemic inflammation in humans23 and a proof-of-concept study found that simvastatin improved pulmonary and non-pulmonary organ dysfunction, reduced inflammation and was well tolerated in patients with ALI. 26 Therefore, the rationale for the use of simvastatin was well justified.
One consideration is that the study may have been underpowered. The sample size calculation was based on a between-group difference of 2.6 VFDs. The 95% CI for VFDs was –0.6 to 2.8 days; therefore, it is possible that the study concluded incorrectly that there was no difference between the groups.
A further possibility for the lack of efficacy relates to the statin and dosage used. The choice of 80 mg of simvastatin used was based on our previous data23,36 that indicated 80 mg of simvastatin improved surrogate clinical outcomes and biological mechanisms believed to be important in the development of ALI. Although the majority of data indicate that the pleotropic effects of statins are likely to be a class effect, we used simvastatin as it was the only statin with proof-of-concept human data. There are also more recent data to indicate that simvastatin may be more effective than atorvastatin,45 supporting the use of simvastatin in this study. An 80-mg dose of simvastatin is a high dosage; this was selected based not only on our pilot data but also on data from a study in which simvastatin 5 mg/kg and 20 mg/kg were investigated in a murine LPS model of ALI and only the higher dose was effective in attenuating lung injury. 31 Furthermore, observational studies of statin usage in critically ill patients found a greater mortality benefit in patients who were receiving a higher dose of statin. 32 It is possible that adverse effects at the simvastatin dosage used outweighed a beneficial effect. Our data would suggest that this is unlikely given that, although there was a higher incidence of AEs related to simvastatin, the numbers of SAEs were similar in both groups. Furthermore, the higher incidence of AEs did not lead to simvastatin being stopped earlier or more frequently.
Although plasma CRP fell in both groups, there was no difference between the groups. This raises the possibility that the lack of efficacy could mean that the dosage used was not sufficient, or not adequately absorbed to reach therapeutic simvastatin concentrations and we acknowledge that a potential limitation is that a formal pharmacokinetic/pharmacodynamics study was not undertaken to confirm adequate absorption and drug exposure. However, we demonstrated that simvastatin was present in the patients randomised to simvastatin. We believe that it is likely that an adequate simvastatin concentration was achieved for several reasons. Prior studies in the critically ill indicate that 80 mg of enterally daily administered simvastatin produces systemic drug concentrations that are in the high therapeutic range, and that drug absorption is seen even in patients with high nasogastric aspirates. Furthermore, given that patients in the simvastatin treatment group received the therapy for a mean of 10 days, it would be expected that this would be sufficient to see a treatment effect if one was present. Finally, the increased incidence of expected statin-related AEs would provide support that sufficient simvastatin concentrations were achieved. We believe that the lack of an effect on plasma CRP suggests that statins in ARDS are unable to modulate inflammation sufficiently to translate into a beneficial clinical effect. One potential reason for the absence of an effect may be related to the fact that 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase may already be significantly inhibited as reflected by the low cholesterol levels seen in critically ill patients.
Following on from our previous Phase II study36 we recruited a heterogenous cohort of patients with ARDS due to any aetiology, which was an attempt to ensure that our findings would be applicable to the overall population of patients with ARDS. The observed mortality in the trial was lower than that described in unselected observational studies. It is recognised that patients who were recruited to clinical trials may represent a selected population owing to the study eligibility criteria, which is further indicated by the fact that only approximately 9% of those meeting the inclusion criteria were subsequently recruited. However, the results of the trial are still generalisable to those patients fulfilling the trial eligibility criteria.
Recent data have suggested that it may be possible to identify subphenotypes within the overall population of patients with ARDS. 46 Although we failed to identify improvements in biological mechanisms implicated in ARDS in the overall cohort, further work could explore if it is possible to identify a population that might be more responsive to simvastatin.
There are frequently reasons, independent of pulmonary status, such as neurological impairment, that may affect the decision to extubate and that may prolong the duration of ventilation. These non-pulmonary factors may not be modifiable regardless of an improvement in pulmonary function. Therefore, it is possible that such factors could have accounted for the absence of any difference in VFDs. However, given the absence of any difference in pulmonary organ dysfunction between the groups, it is less likely that this was a major confounding issue in the finding of no difference in VFDs.
Although we recommended best practice management for ARDS, including lung protective ventilation, we did not measure details of clinical management. At randomisation, the tidal volume was 8.1 ml/kg of predicted body weight and it is possible that this level of tidal volume may have confounded the potential effects of simvastatin. However, this is unlikely given the similar absence of benefit for rosuvastatin in the SAILS study,12 in which the mean tidal volume was 6.6–6.8 ml/kg of predicted body weight. Our data for tidal volume and plateau pressure are consistent with those seen in other clinical trials in critical care in which ventilation is not strictly protocolised. 47
Conclusion
In conclusion, our study showed that high dose enteral simvastatin, while safe and with minimal adverse effects, is not effective at improving clinical outcomes in patients with ARDS.
Chapter 4 Economic evaluation
Methods
Aim and perspective
The aim of the economic evaluation was to assess the cost-effectiveness of simvastatin compared with placebo at 12 months. The study was a cost–utility analysis undertaken alongside the main trial. Incremental cost-effectiveness ratios (ICERs) were used to present cost per quality-adjusted life-year (QALY). This ratio represents the difference in mean health service cost of the simvastatin and placebo groups divided by the difference in mean QALYs between groups to establish the cost per QALY gained, in keeping with the recommendations of National Institute for Health and Care Excellence (NICE). 48 The perspective of the analysis was the NHS and Personal Social Services. In addition, data of private and informal care were collected but these were not included in the cost-effectiveness analysis.
Health and social care service use and costs
Data on the health and social care services used by all patients were collected 12 months post randomisation. Data relating to patients’ primary hospital admission were collected prospectively via the CRF until primary hospital discharge or death. Data on levels of care and organ support were obtained from the daily data collection section of the CRF. These data (on levels of care and organ support) were only available for a maximum of 28 days or until discharge to a separate HDU or ward. It was assumed that patients were discharged to a rehabilitation ward for respiratory disorders. Other ICU days included the remaining time spent in the ICU after 28 days (if applicable) and any other stay in the ICU during a patient’s primary admission, but level of care was not recorded for these ICU days. It was anticipated that some patients would move multiple times between ICU, HDU and the ward during their primary admission, thus, the CRF tracked this movement so that length of stay could be calculated for each. Patients’ use of medications other than the study drug was not included in the economic analysis.
Use of health and social care services after hospital discharge was collected retrospectively via questionnaires posted out to surviving patients at 6 and 12 months post randomisation. Mortality status was established prior to posting via contact with the research sites, engagement with the Health and Social Care Information Service (now known as NHS Digital), and contact with GPs. The questionnaire asked patients to report their use of primary and secondary care services, and private and informal care.
Individual-level resource use was combined with unit costs to estimate total costs of health and social care use for each participant in the trial. Unit costs were obtained from publicly available sources and set at 2013–14 prices.
These were NHS Reference Costs49 for hospital resources, the Unit Costs of Health and Social Care 201450 for general practice services and the NHS Electronic Drug Tariff51 for the cost of simvastatin (Table 28).
| Resource item | Unit cost (£) | Source |
|---|---|---|
| Primary admission | ||
| Intensive care level 1 day | 696.12 | NHS Reference Costs 2013–1449 (XC07Z) adult critical care |
| Intensive care level 2 day | 932.10 | NHS Reference Costs 2013–1449 (XC06Z) adult critical care |
| Intensive care level 3 day | 1440.64 | NHS Reference Costs 2013–1449 (XC01Z-XC05Z weighted average) adult critical care |
| Other ICU day | 1228.65 | NHS Reference Costs 2013–1449 (XC01Z-XC07Z weighted average) adult critical care |
| HDU day | 932.10 | NHS Reference Costs 2013–1449 (XC06Z) adult critical care |
| Ward bed-day | 437.00 | NHS Reference Costs 2013–1449 (VC40Z) rehabilitation for respiratory disorders |
| 80-mg simvastatin tablets, 28 tablets per pack | 2.02 | NHS Electronic Drug Tariff 51 |
| Other hospital services | ||
| Non-specific ward days | 483.04 | NHS Reference Costs 2013–1449 (weighted average length of stay and cost of non-elective long stays) |
| Outpatient attendance | 109.00 | Unit Costs of Health and Social Care 2014,50 p. 111 |
| Attendance at A&E | 233.00 | Unit Costs of Health and Social Care 2014,50 p. 111 (see and treat and convey) |
| Community health services | ||
| GP surgery consultation | 46.00 | Unit Costs of Health and Social Care 2014,50 p. 195 |
| GP telephone consultation | 28.00 | Unit Costs of Health and Social Care 2014,50 p. 195 |
| GP home consultation | 115.00 | Unit Costs of Health and Social Care 2013,52 p. 191 (inflated using the hospital and community health services index) |
| GP out-of-hours consultation | 115.00 | Unit Costs of Health and Social Care 2013,52 p. 191 (home visit unit cost assumed as above) |
| GP nurse surgery consultation | 13.70 | Unit Costs of Health and Social Care 2014,50 p. 192 (per 15.5-minute surgery consultation) |
| GP nurse telephone consultation | 4.85 | Unit Costs of Health and Social Care 2014,50 p. 192 (per 7.1-minute telephone consultation)a |
| GP nurse home visit | 24.29 | Unit Costs of Health and Social Care 2014,50 p. 192 (per 15.5-minute consultation and 12-minute travel assumeda) |
| District nurse home visit | 39.00 | Unit Costs of Health and Social Care 2014,50 p. 187 |
| Social worker visit | 79.00 | Unit Costs of Health and Social Care 2014,50 p. 206 (per 1-hour cost assumed to include travel) |
| Physiotherapist visit | 51.00 | Unit Costs of Health and Social Care 2014,50 p. 179 |
| Occupational therapist visit | 77.00 | Unit Costs of Health and Social Care 2014,50 p. 180 |
| Dietitian visit | 37.00 | Unit Costs of Health and Social Care 2014,50 p. 238 (with qualifications) |
| Nurse specialist visit | 74.00 | Unit Costs of Health and Social Care 2014,50 p. 190 (per 1-hour cost assumed to include travel) |
| Rapid response/acute care episode | 182.00 | Unit Costs of Health and Social Care 2013,52 p. 111 (inflated using the hospital and community health services index) |
| Psychotherapy/counselling | 50.00 | Unit Costs of Health and Social Care 2014,50 p. 51 |
| Day centre | 38.00 | Unit Costs of Health and Social Care 2014,50 p. 38 (per client session) |
| Care services | ||
| Home help/care worker | 17.00 | Unit Costs of Health and Social Care 2014,50 p. 210 (per 1-hour cost assumed to include travel) |
| Delivered meals | 6.60 | Unit Costs of Health and Social Care 2014,50 p. 127 (per meal) |
| Nursing home | 511.00 | Unit Costs of Health and Social Care 2014,50 p. 33 (per week) |
| Respite | 511.00 | Nursing home cost assumed as above |
| Residential care home | 493.00 | Unit Costs of Health and Social Care 2014,50 p. 34 (weekly) |
| Sheltered housing | 443.00 | Unit Costs of Health and Social Care 2014,50 p. 39 (extra care housing for older people, weekly) |
Health outcomes
The economic analysis used the QALY as the measurement of health outcome. The QALY reflects the impact of an intervention on both the quality and quantity of a patient’s life and is recommended by NICE in economic evaluations. 48 QALYs were derived using the EQ-5D-3L. 53 This generic HRQoL instrument, which has been used frequently in the critically ill,54–56 provides a description of health using five dimensions (mobility, self-care, usual activities, pain/discomfort and anxiety/depression), each with three levels of severity. Patients also place their health on a visual analogue scale (VAS), for which 0 represents the worst imaginable health state and 100 the best imaginable health state. The UK social preference weights for EQ-5D-3L health states57 were used to obtain single utility values from the responses.
The area under the curve method was used to estimate patient-specific QALYs accrued over the 12-month period. This method involves multiplying the utility of the patients’ health state at a certain time point by the duration of the health state and then summing these over the study period. It was assumed that changes in utility over the time points were linear.
The EQ-5D-3L was administered at discharge, and at 3, 6 and 12 months post randomisation. All questionnaires were posted out to survivors, except for at discharge when it was administered face to face. As patients were unconscious and receiving invasive mechanical ventilation at the time of randomisation, baseline utility could not be measured. Instead we used the utility value for an unconscious state (–0.402) from the tariff. 57 This approach has been used previously in economic evaluations of therapies for patients with ARDS. 54,55
Over the course of the trial it became apparent that many patients were still in hospital at 3 months and were then not administered the EQ-5D-3L at this time point. Furthermore, the timing of the discharge questionnaire varied from patient to patient. For these reasons QALYs were calculated using only baseline, 6- and 12-month utility scores in the first instance as these captured the complete study period. Discharge and 3 months were used in the QALY calculation in a sensitivity analysis.
To maximise the return rate of postal questionnaires, the following evidence-based strategies were introduced.
-
Franked, addressed return envelopes were provided with each 3-, 6- and 12-month questionnaire.
-
A ‘thank-you’ voucher to the value of £5 (or €5 for Ireland) was enclosed with the first questionnaire sent out.
-
A covering letter from the trial manager was included with each questionnaire.
-
A follow-up telephone call was made if the questionnaire was not returned with the offer to resend a copy of the questionnaire if needed.
-
A second and final follow-up telephone contact was made as a final reminder.
-
To minimise the risk of causing distress to the relation of a patient who died after leaving the hospital, the CTU contacted the patient’s GP and NHS Digital to ascertain the patient’s survival status prior to sending out any questionnaires.
Analysis and reporting
We undertook the following:
-
analysis of service use and costs
-
analysis of health outcomes
-
cost–utility analysis.
Analysis of service use and costs
The analysis included all patients and their available data. We used mean imputation to impute missing data in cases for which the patient reported using a care service (e.g. carer, home help, delivered meals) but did not provide the number of contacts per week.
Death was not considered a censoring event in the primary analysis and periods after death were counted as observations with known outcome. 58 In practice, this meant that for patients who had died in hospital, costs after hospital discharge until 12-month follow-up were considered to be zero. For patients who were discharged from hospital but had died by 28 days, we also assumed that their costs after hospital discharge until 12 months were zero. This was an acceptable assumption because, out of the patients who were dead at 28 days (24.5%; 132/539), only two patients were discharged from hospital and they both subsequently died within 2 weeks of discharge. For patients who had died by 6 months, costs from 6 to 12 months were considered to be zero. In some cases, we could not assign zero costs to all periods after death. For patients who were discharged from hospital, alive at 28 days but had died by 6 months, costs from discharge to 6 months were considered to be missing as no information was available on their use of resources in the period up to their death. The same was true for patients alive at 6 months but had died by 12 months; costs from 6 to 12 months were treated as missing.
Descriptive statistics were used to summarise the health service resource use and associated costs for (1) primary admission, (2) discharge to 6 months and (3) 6 to 12 months. Differences in mean service use between groups were analysed using two-tailed independent t-tests and differences in proportions were analysed using two-tailed Fisher’s exact test. Non-parametric bootstrapping was used to calculate 95% bootstrap CIs of differential mean costs, drawing 1000 samples of the same size as the original sample separately for each group with replacement.
Analysis of health outcomes
The analysis included all patients and their available data. Descriptive statistics were used to summarise EQ-5D-3L utilities, VAS scores and QALYs at discharge, 3, 6 and 12 months. Differences in utilities and VAS scores between groups were tested using two-tailed independent t-tests. Non-parametric bootstrapping was used to calculate 95% bootstrap CIs of differential mean QALYs. A utility of zero was assigned to patients who were dead at the relevant EQ-5D-3L collection time point (3, 6 and 12 months).
Cost-utility analysis
The analysis included only patients with complete cost and QALY data. The ICER was calculated as the difference in mean health service cost of the simvastatin and placebo groups divided by the difference in mean QALYs between the groups to establish the cost per QALY gained. To account for sampling uncertainty in the total cost and QALY data, non-parametric bootstrapping was performed to generate 1000 bootstrap replicates of the ICERs, which were then plotted on the cost-effectiveness plane. The resulting scatter plot was used to derive the cost-effectiveness acceptability curve (CEAC) by calculating the proportion of the replicates that would be considered cost-effective at various willingness-to-pay (WTP) thresholds for an additional QALY. In general, NICE59 considers interventions with an ICER of < £20,000 to be cost-effective.
Cost-effectiveness acceptability curves were also constructed for the following sensitivity analyses:
-
Multiple regression used to estimate the mean difference between groups for total health service costs and QALYs after adjusting for the baseline variables of age, APACHE II score and vasopressor requirement.
-
Multiple imputation for missing data – missing total cost and QALY data points were filled simultaneously using imputation by chained equations and predictive mean matching to generate five imputed data sets. Treatment group, baseline APACHE II score, age, vasopressor requirement at baseline, mortality at 28 days, mortality at 12 months and primary admission costs were entered into the model as predictors of missing data.
-
Multiple imputation and adjustment for baseline variables simultaneously.
-
Death as a censored event (i.e. data were considered to be missing for patients who had died during the study period).
-
Not using mean imputation for missing care service data (i.e. treated as missing).
-
Using discharge and 3-month EQ-5D-3L data in the calculation of QALYs when available.
All curves were constructed regardless of whether or not the cost and effect differences were statistically significant, in keeping with current health economic practice.
All analyses were performed using Stata® (version 12.0; StataCorp LP, College Station, TX, USA) Intercooled (IC) for Windows® (Microsoft Corporation, Redmond, WA, USA). Costs and QALYs were not discounted as the time horizon of the study was 12 months. Significance (p < 0.05) was judged when the CI of differential means or QALYs excluded zero.
Results
A total of 540 patients were randomised. Five patients withdrew consent during the study but only one did not give permission for the use of their anonymised data collected prior to withdrawal. Thus, 539 patients were included in the health economics analysis: 259 in the simvastatin group and 280 in the placebo group. A total of 266 patients completed the discharge EQ-5D-3L: 131 (49%) in the simvastatin group and 135 (51%) in the placebo group. Out of the 367 patients alive at 3 months, 222 (60%) returned the questionnaire [114 (51%) in the simvastatin group and 108 (49%) in the placebo group]. Out of the 359 patients alive at 6 months, 213 (59%) returned the 6-month questionnaire [114 (32%) in the simvastatin group and 99 (27%) in the placebo group]. Out of the 349 patients alive at 12 months, 185 (53%) returned the 12-month questionnaire [91 (26%) in the simvastatin group and 94 (27%) in the placebo group]. A total of 158 (45%) patients completed both the 6- and 12-month questionnaires: 82 (23%) in the simvastatin group and 76 (22%) in the placebo group.
Analysis of service use and costs
Patients’ use of services during their primary admission to hospital, including simvastatin, is presented in Table 29. As level of care was not recorded for days spent in ICU after 28 days, these data are presented separately. No differences were statistically significant.
| Primary admission (baseline to discharge) | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Simvastatin | Placebo | ||||
| n | Mean (SD) | n | Mean (SD) | ||
| Primary ICU stay days | 259 | 15 (13.41) | 280 | 15 (12.5) | 0.997 |
| Intensive care level 1 days | 259 | 0.53 (1.2) | 280 | 0.46 (1.14) | 0.451 |
| Intensive care level 2 days | 259 | 2.32 (2.85) | 280 | 2.31 (3.1) | 0.958 |
| Intensive care level 3 days | 259 | 10.66 (7.56) | 280 | 10.77 (8.24) | 0.866 |
| Other ICU daysa | 257 | 1.00 (4.84) | 275 | 1.19 (6.33) | 0.695 |
| HDU daysa | 257 | 0.82 (4.28) | 275 | 1.75 (8.05) | 0.098 |
| Ward daysa | 257 | 13.91 (26.10) | 275 | 12.21 (18.61) | 0.285 |
| Advanced respiratory support days | 259 | 10.71 (7.55) | 280 | 10.9 (8.33) | 0.774 |
| Advanced cardiovascular support days | 259 | 2.03 (3.57) | 280 | 2.31 (3.88) | 0.379 |
| Liver support days | 259 | 0.01 (0.11) | 280 | 0 (0) | 0.071 |
| Neurological support days | 259 | 0.64 (2.56) | 280 | 0.71 (2.64) | 0.769 |
| Renal support days | 259 | 1.74 (4.1) | 280 | 2.09 (4.56) | 0.348 |
| 80-mg simvastatin tablets | 259 | 19.93 (14.24) | – | – | – |
Patients’ use of other hospital services (hospital inpatient stay, outpatient attendances and accident and emergency visits) following discharge is presented in Table 30. Patients in the simvastatin group reported fewer hospital inpatient days but more outpatient and accident and emergency visits than the placebo group at both 6 and 12 months; however, these differences were not statistically significant between groups.
| Other hospital service | Discharge to 6 months | 6–12 months | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Simvastatin (N = 179) | Placebo (N = 184) | p-value | Simvastatin (N = 168) | Placebo (N = 190) | p-value | |||||
| n (%) | Mean (SD) | n (%) | Mean (SD) | n (%) | Mean (SD) | n (%) | Mean (SD) | |||
| Hospital inpatient days | 18 (10.06) | 0.99 (4.72) | 21 (11.41) | 1.28 (7.92) | 0.675 | 18 (10.71) | 0.89 (4.36) | 20 (10.53) | 0.99 (6.15) | 0.857 |
| Hospital outpatient appointment | 73 (40.78) | 1.72 (5.49) | 54 (29.35) | 1.36 (4.86) | 0.506 | 51 (30.36) | 1.03 (2.29) | 43 (22.63) | 0.84 (2.68) | 0.467 |
| Hospital accident and emergency visit | 22 (12.29) | 0.3 (1.43) | 17 (9.24) | 0.12 (0.4) | 0.097 | 17 (10.12) | 0.17 (0.65) | 16 (8.42) | 0.15 (0.59) | 0.769 |
Patients’ use of community health services from baseline until 12 months is presented in Table 31. There were no statistically significant differences between the groups.
| Service | Discharge to 6 months | 6–12 months | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Simvastatin (N = 179) | Placebo (N = 184) | p-value | Simvastatin (N = 168) | Placebo (N = 190) | p-value | |||||
| n (%) | Mean (SD) | n (%) | Mean (SD) | n (%) | Mean (SD) | n (%) | Mean (SD) | |||
| GP surgery consultation | 79 (44.13) | 2.41 (4.57) | 65 (35.33) | 2 (4.46) | 0.383 | 61 (36.31) | 2.11 (6.35) | 62 (32.63) | 1.42 (2.84) | 0.176 |
| GP telephone consultation | 28 (15.64) | 0.72 (2.98) | 25 (13.59) | 0.74 (3.1) | 0.927 | 18 (10.71) | 0.93 (8.09) | 17 (8.95) | 0.44 (2.09) | 0.414 |
| GP home consultation | 19 (10.61) | 0.26 (1.45) | 23 (12.5) | 0.33 (1.29) | 0.66 | 9 (5.36) | 0.11 (0.54) | 17 (8.95) | 0.23 (0.93) | 0.149 |
| GP out-of-hours consultation | 6 (3.35) | 0.15 (1.51) | 10 (5.43) | 0.07 (0.33) | 0.513 | 11 (6.55) | 0.08 (0.31) | 9 (4.74) | 0.08 (0.41) | 0.97 |
| GP nurse surgery consultation | 34 (18.99) | 0.73 (2.56) | 29 (15.76) | 1.22 (5.3) | 0.27 | 32 (19.05) | 0.57 (1.74) | 18 (9.47) | 0.93 (7.36) | 0.542 |
| GP nurse telephone consultation | 3 (1.68) | 0.11 (1.14) | 2 (1.09) | 0.05 (0.61) | 0.59 | 2 (1.19) | 0.06 (0.59) | 1 (0.53) | 0.01 (0.07) | 0.207 |
| GP nurse home consultation | 16 (8.94) | 1.13 (5.56) | 18 (9.78) | 1.79 (12.32) | 0.51 | 3 (1.79) | 0.04 (0.29) | 3 (1.58) | 1.44 (9.82) | 0.065 |
| District nurse | 1 (0.56) | 0.02 (0.22) | 5 (2.72) | 0.23 (1.74) | 0.107 | 1 (0.6) | 0.14 (1.85) | 2 (91.05) | 0.04 (0.46) | 0.469 |
| Social worker | 12 (6.7) | 0.14 (0.73) | 11 (5.98) | 0.12 (0.52) | 0.763 | 3 (1.79) | 0.07 (0.64) | 13 (6.84) | 0.21 (1.19) | 0.159 |
| Physiotherapist | 39 (21.79) | 1.87 (5.25) | 25 (13.59) | 1.72 (8.41) | 0.846 | 17 (10.12) | 0.53 (2.16) | 20 (10.53) | 1.28 (6.5) | 0.155 |
| Occupational therapist | 23 (12.85) | 0.67 (2.78) | 14 (7.61) | 0.92 (8.42) | 0.708 | 9 (5.36) | 0.36 (2.74) | 17 (8.95) | 0.32 (1.66) | 0.861 |
| Dietitian | 3 (1.68) | 0.04 (0.32) | 2 (1.09) | 0.04 (0.43) | 0.913 | – | – | – | – | – |
| Nurse specialist | 2 (1.12) | 0.21 (2.62) | 1 (0.54) | 0.02 (0.22) | 0.314 | – | – | – | – | – |
| Rapid response | 4 (2.23) | 0.02 (0.15) | 4 (2.17) | 0.12 (1.41) | 0.36 | 3 (1.79) | 0.02 (0.13) | 3 (1.58) | 0.02 (0.18) | 0.848 |
| Psychotherapy/counselling | 0 (0) | 0 (0) | 2 (1.09) | 0.02 (0.16) | 0.186 | 1 (0.6) | 0.01 (0.08) | 1 (0.53) | 0.24 (3.26) | 0.36 |
| Day centre | – | – | – | – | – | 3 (1.79) | 0.17 (1.52) | 1 (0.53) | 0.05 (0.73) | 0.357 |
Patients’ use of care services is presented in Tables 32 and 33. In the period from discharge to 6 months, mean imputation was used for three patients who used a home help, three patients who used a carer paid for by the health service and one patient who used delivered meals. For the patient who used delivered meals, the mean value from the 6- to 12-month period was imputed as only one patient reported using the service from discharge to 6 months. In the period from 6 to 12 months, mean imputation was used for four patients who used a carer paid for by the health service. Patients in the placebo group received statistically significantly more carer visits paid for by the health service between 6 and 12 months than those in the simvastatin group. All other differences were not significant.
| Care service | Discharge to 6 months | 6–12 months | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Simvastatin (N = 179) | Placebo (N = 184) | p-value | Simvastatin (N = 168) | Placebo (N = 190) | p-value | |||||
| n (%) | Mean (SD) | n (%) | Mean (SD) | n (%) | Mean (SD) | n (%) | Mean (SD) | |||
| Home help visits | 5 (2.79) | 2.46 (16.16) | 3 (1.63) | 1.94 (15.53) | 0.756 | 0 (0) | 0 (0) | 3 (1.58) | 4.93 (54.39) | 0.241 |
| Delivered meals visits | 1 (0.56) | 0.77 (10.31) | 0 (0) | 0 (0) | 0.311 | 0 (0) | 0 (0) | 3 (1.58) | 2.19 (17.81) | 0.112 |
| Carer visits (paid by health-care provider) | 10 (5.59) | 7.52 (32.76) | 10 (5.43) | 13.05 (74.53) | 0.363 | 6 (3.57) | 5.42 (29.41) | 14 (7.37) | 24.46 (104.05) | 0.023 |
| Residential care home (days) | 2 (1.12) | 0.08 (1.05) | 0 (0) | 0 (0) | 0.311 | 1 (0.6) | 1.07 (13.89) | 0 (0) | 0 (0) | 0.288 |
| Nursing home (days) | 4 (2.23) | 1.36 (11.92) | 4 (2.17) | 0.62 (4.23) | 0.427 | 1 (0.6) | 1.07 (13.89) | 0 (0) | 0 (0) | 0.288 |
| Shelter (days) | 1 (0.56) | 0.04 (0.52) | 2 (1.09) | 2.63 (26.94) | 0.199 | 1 (0.6) | 0 (0) | 3 (1.58) | 1.89 (18.42) | 0.183 |
| Respite care (days) | 2 (1.12) | 0.3 (2.86) | 1 (0.54) | 0.15 (2.06) | 0.568 | 0 (0) | 0 (0) | 1 (0.53) | 0.07 (1.02) | 0.348 |
| Service | Discharge to 6 months | 6–12 months | ||||
|---|---|---|---|---|---|---|
| Treatment group | p-value | Treatment group | p-value | |||
| Simvastatin (n = 179) | Placebo (n = 184) | Simvastatin (n = 168) | Placebo (n = 190) | |||
| n (%) | n (%) | n (%) | n (%) | |||
| Paid carer | 6 (3.35) | 0 (0) | 0.638 | 1 (0.60) | 4 (2.11) | 0.376 |
| Unpaid carer | 21 (11.73) | 25 (13.59) | 0.014 | 15 (8.93) | 17 (8.95) | 1.00 |
Patients’ use of private and informal carers is presented in Table 33. Private paid carers represent a cost to the patient rather than the health or social care provider and although they are not included in the subsequent ICER calculation, as they are outside the health-care provider perspective, they are presented for completeness. At 6 months, there was a statistically significant association between treatment and the use of unpaid carers, with a larger proportion of patients in the placebo group using unpaid carers.
Costs for individual resource components were grouped together to estimate mean costs for the primary hospital admission, other hospital services, community services and care-related services over the study period (Table 34). These total costs were calculated only for patients with complete data on each of the cost components. Lower mean costs were observed in the simvastatin group for all service types, with the exception of the primary hospital admission. The total mean cost difference at 12 months was statistically significant (–£4987, 95% CI –£10,060 to –£390). No other difference was statistically significant.
| Service costs | Treatment group | Cost difference (95% CI):a intervention – control | |||
|---|---|---|---|---|---|
| Simvastatin | Placebo | ||||
| n | Mean cost (SD) (£) | n | Mean cost (SD) (£) | ||
| Baseline to 6 months | |||||
| Primary admission | 257 | 26,017.93 (19,825.31) | 275 | 26,311.24 (20,162.46) | –293.31 (–3653.94 to 3161.23) |
| Other hospital services | 179 | 738.19 (2517.87) | 184 | 795.51 (3926.37) | –57.32 (–820.81 to 573.50) |
| Community health services | 179 | 395.58 (776.44) | 184 | 421.30 (1085.47) | –25.72 (–232.82 to 158.40) |
| Care-related services | 179 | 304.29 (1164.08) | 184 | 477.75 (2150.14) | –173.46 (–559.12 to 181.97) |
| 6–12 months | |||||
| Other hospital services | 168 | 579.49 (2215.12) | 190 | 603.51 (3211.87) | –24.02 (–669.03 to 519.74) |
| Community health services | 168 | 223.70 (682.05) | 190 | 284.46 (671.16) | –60.76 (–207.01 to 80.69) |
| Care-related services | 168 | 245.76 (1481.22) | 190 | 639.32 (2810.66) | –393.56 (–870.93 to 36.91) |
| Total: baseline to 6 months | 179 | 25,228.57 (16,640.52) | 183 | 27,989.23 (21,313.84) | –2760.53 (–6694.82 to 1186.83) |
| Total: 6–12 months | 168 | 1048.94 (3282.70) | 190 | 1527.28 (5197.55) | –478.34 (–1472.50 to 377.68) |
| Total: 12-month health service costs | 147 | 24,183.96 (16,969.87) | 160 | 29,171.08 (24,967.77) | –4987.12 (–10,059.91 to –390.40) |
Health outcomes analysis
Mean EQ-5D-3L utilities and VAS scores at discharge, 3, 6 and 12 months, and QALYs at 12 months are presented in Table 35. Patients in the simvastatin group had higher HRQoL at each time point than those in the placebo group, with the most notable difference at 6 months. No differences between the groups were statistically significant. Both groups experienced an overall decline between discharge and 12 months.
| Health outcome | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Simvastatin | Placebo | ||||
| n | Mean score (SD) | n | Mean score (SD) | ||
| EQ-5D-3L utilities | |||||
| Dischargea | 128 | 0.46 (0.38) | 129 | 0.44 (0.37) | 0.630 |
| 3 months | 182 | 0.28 (0.38) | 194 | 0.25 (0.37) | 0.457 |
| 6 months | 184 | 0.31 (0.38) | 188 | 0.23 (0.36) | 0.059 |
| 12 months | 167 | 0.29 (0.37) | 193 | 0.24 (0.37) | 0.143 |
| VAS scores | |||||
| Dischargea | 126 | 61.09 (18.25) | 133 | 56.73 (19.33) | 0.064 |
| 3 months | 183 | 37.03 (0.04) | 200 | 32.25 (33.64) | 0.168 |
| 6 months | 191 | 38.03 (35.59) | 192 | 31.76 (34.94) | 0.083 |
| 12 months | 172 | 34.01 (36.05) | 197 | 29.60 (34.59) | 0.233 |
| QALYs | 152 | 0.17 (0.27) | 166 | 0.06 (0.26) | 0.037 |
Cost–utility analysis
Results from the cost–utility analysis are presented in Table 36. Simvastatin is both less costly and more effective than placebo; thus, it is the dominant strategy with a mean difference in costs of –£3601 and a mean difference in QALYs of 0.064. In this situation, the ICER is not calculated as its magnitude does not convey any meaning. 58 Sampling uncertainty is represented by the joint distribution of the bootstrapped differences in cost and QALY on the cost-effectiveness plane for the primary analysis (Figure 13). The majority of the points lie below the x-axis (indicating that simvastatin is cost saving) and to the right of the y-axis (indicating simvastatin produces more QALYs than placebo). The small number of points lying outside of this area indicate a small degree of variability surrounding the presence and magnitude of cost savings and effectiveness.
| Analysis | Incremental total health service costs (95% CIa) (£) | Incremental QALY gain (95% CI) | Probability of cost-effectiveness at £20,000 per QALY (%) |
|---|---|---|---|
| n = 139 for simvastatin, n = 153 for placebob | |||
| Primary analysis (unadjusted) | –3600.91 (–8061.10 to 859.28) | 0.064 (0.002 to 0.127) | 99 |
| Adjusted for baseline variables | –2661.03 (–7842.76 to 2520.70) | 0.089 (0.025 to 0.151) | 95 |
| n = 259 for simvastatin, n = 280 for placeboc | |||
| Multiply imputed total costs and QALYs | –2132.69 (–5629.21 to 1363.83) | 0.042 (–0.001 to 0.086) | 96 |
| Multiply imputed total costs and adjusted | –1290.35 (–5000.61 to 2419.91) | 0.048 (0.005 to 0.091) | 90 |
| n = 74 for simvastatin, n = 68 for placebob | |||
| Death as a censoring event | –8532.48 (–16,107.75 to –957.21) | 0.056 (–0.022 to 0.135) | 99 |
| n = 137 for simvastatin, n = 151 for placebob | |||
| No mean imputation of care data | –3966.00 (–8503.11 to 571.10) | 0.06 (0.00 to 0.13) | 99 |
| n = 138 for simvastatin, n = 150 for placebob | |||
| QALY calculation using discharge and 3-month EQ-5D-3L | –3559.00 (–8241.41 to 1123.42) | 0.084 (0.005 to 0.162) | 99 |
FIGURE 13.
Cost-effectiveness plane for the primary cost-effectiveness analysis (showing bootstrapped replications of mean incremental costs and QALY gain and the WTP threshold of £20,000 per QALY). 44
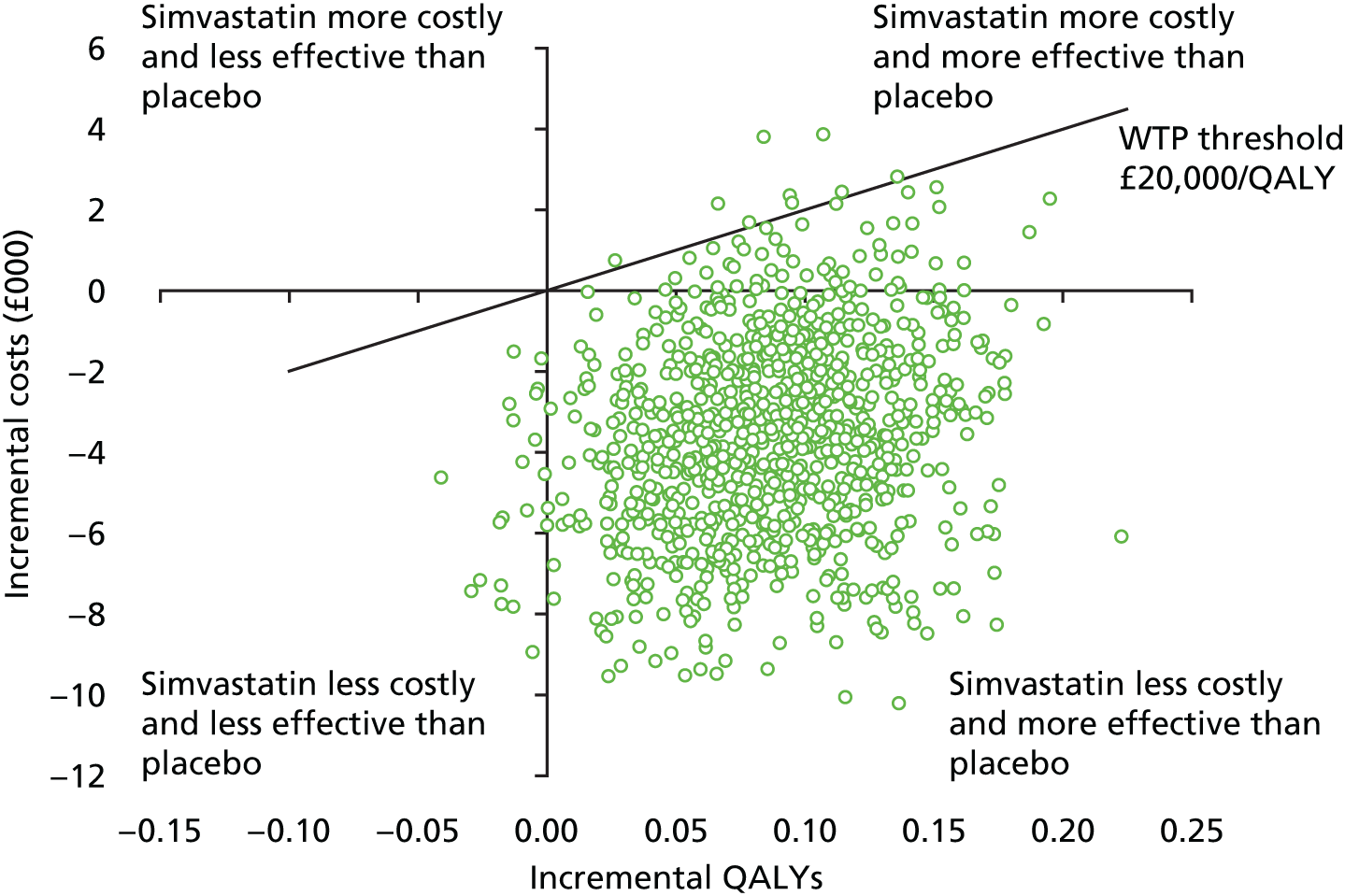
The CEAC for the primary analysis presented in Figure 14 summarises this uncertainty for the decision-maker and presents the probability of simvastatin being cost-effective compared with placebo at different thresholds of WTP per QALY gain for the primary and sensitivity analyses. The CEAC for the primary analysis indicates that, at a WTP threshold of £20,000 per QALY gain, the probability of simvastatin being cost-effective is 99%.
FIGURE 14.
Cost-effectiveness acceptability curve (showing the probability of simvastatin being cost-effective compared with placebo for the primary and sensitivity analyses). Amended from Agus et al. 44 This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.

Sensitivity analyses were conducted to determine the impact of changing particular assumptions on the cost-effectiveness (see Table 36). Although there are some notable effects on differential mean costs and effects, the CEACs for the sensitivity analyses (see Figure 14) indicate that cost-effectiveness of simvastatin was robust to these changes in the assumptions, with the probability of it being cost-effective at £20,000 per QALY never dropping below 90%.
Health economic discussion
The principal finding of the economic evaluation is that simvastatin compared with placebo was associated with lower costs and a significant QALY gain. Although the gain in QALYs was small (0.064; equivalent to 23 days of full health), it was associated with a cost saving equating to £3601 over the 12-month period. These findings were robust to changes in the methodological assumptions. Thus, simvastatin has a very high probability of being cost-effective at 1 year.
There is currently no consensus on what constitutes a minimally important difference in mean QALYs between groups;60 however, 0.05 has been suggested previously. 60,61 The difference observed in our analysis exceeds this, suggesting that the small difference is still meaningful. The small gain in QALYs is consistent with the modest and non-significant benefits observed in the clinical outcomes reported in Chapter 3. As all patients were assigned the same utility score at baseline, the difference in QALYs is due to the HRQoL of simvastatin patients being higher than placebo patients at both 6 and 12 months.
The majority of differences in individual resource use components were not statistically significant. However, when costs were categorised as primary admission, other hospital health services, community health services and care-related services, differences were more apparent. All other costs after discharge were lower in the simvastatin group, with the most notable cost savings associated with care-related services. The results corroborate with the HRQoL analysis. Patients in the placebo group experienced poorer health than those in the simvastatin group over the study period and this had an impact on their use of health services, including some services not paid for by the health services that were not included in the analyses.
There were a number of limitations to the economic evaluation. The study was powered to detect statistically significant differences in the primary outcome and not in costs, QALYs or cost-effectiveness. However, this is typically the case and significance rules are not typically relied on in the interpretation of cost-effectiveness analyses,62 as greater emphasis is placed on the joint distribution of cost and effects. Nonetheless, having a sufficiently powered study would have led to more conclusive results60 and allowed decision-makers to be more confident in the value claim. 58 There are limitations to this study related to the inevitable difficulties of collecting follow-up data from patients recovering from a stay in critical care. A substantial proportion of patients was lost to follow-up in the economic analysis. The cost–utility analysis included only patients with complete cost and QALY data but, of those alive at 12 months, only 45% completed both the 6- and 12-month follow-up questionnaires and some data were missing from these completed questionnaires. Previous intensive care trials54,55 have also found it difficult to achieve high rates of long-term data collection and the experience of the HARP-2 trial confirms that this is a difficult population to follow up. Furthermore, economic data are particularly likely to be missing owing to the reliance on multiple components within HRQoL and resource use questionnaires required for the calculation of QALYs and costs. As a result, the cost–utility analysis was performed on a subgroup of patients with complete cost and QALY data. The utilities derived from the discharge and 3-month EQ-5D-3L were not used in the QALY calculation for the primary analysis. This was due to the variable timing of the discharge questionnaire and the fact that the 3-month questionnaire was not consistently administered to survivors if they were still hospital inpatients. A sensitivity analysis included them in the QALY calculation when they were available and there was a minimal impact on the overall results. The probability of simvastatin being cost-effective at a WTP threshold of £20,000 remained 99%.
Conclusion
Simvastatin was found to be cost-effective at 1 year compared with placebo for the treatment of ARDS, being associated with both a significant QALY gain and a cost saving. The cost-effectiveness remained robust to changes in methodological assumptions. However, given that the health economic analysis was performed on a subgroup of patients and the QALY gain was relatively small, there are currently insufficient data to support the treatment of patients with ARDS with simvastatin in the NHS.
Chapter 5 Overall discussion and conclusion
Despite promising findings for statins in ARDS in early-phase clinical trials, this large, multicentre, double-blind, randomised, placebo-controlled clinical trial found that high-dose enteral simvastatin was not effective at improving clinical outcomes in patients. Mortality rates were lower in the simvastatin group although this difference was not statistically significant. Simvastatin was associated with an increase in AEs, but there was no increase in SAEs. Simvastatin did not modulate mechanisms implicated in the development of ARDS. Simvastatin was found to be cost-effective at 1 year compared with placebo for the treatment of ARDS, being associated with a cost saving and small QALY gain. However, these were secondary outcomes and the cost–utility analysis was performed in a subgroup of patients.
The results do highlight the importance of undertaking a health economic analysis in the setting in which the primary clinical outcome is not significantly different between the trial arms. In addition, it flags important issues regarding the use of short-term clinical outcomes, such as VFDs, which have been shown to poorly correlate with long-term patient-centred outcomes such as long-term mortality63 and QALYs. 63,64 The HARP-2 trial is considered a negative trial to the absence of a significant difference in the primary outcome (VFDs) at 28 days. However, it did achieve a non-significant 5% reduction in mortality and the cost–utility analysis found a significant QALY gain with a non-significant cost saving at 12 months. In the setting of no significant difference in mortality or other clinical outcomes, it is unlikely that the results of the cost-effectiveness analysis will be sufficient to change clinical practice. Had the trial been powered sufficiently for long-term mortality or QALYs, a different conclusion may have been reached. QALYs may be a feasible patient-centred primary outcome for critical care studies as they combine both morbidity and mortality, and have potential gains in statistical power owing to being a continuous variable. 61 Undoubtedly, more work is required to identify valid surrogate outcomes that more closely track patient outcomes.
The definition of ARDS uses a trade-off between feasibility and reliability. 65 Although the ARDS definition identifies clinical phenotypes with predictive validity,66 owing to the underlying biological differences within the overall clinical phenotype, pharmacological interventions may only be effective in a subpopulation of the overall cohort of patients with ARDS in a RCT. 46,67 In support of our hypothesis, using a combination of eight plasma biomarkers and clinical data, Calfee et al. 46 recently identified ARDS subphenotypes that respond differently to differing ventilator strategies using latent class analysis methodology. This study prompted a call for further confirmatory research of this concept and other methods that explore ways to increase the signal-to-noise ratio. 68
Our study was powered to detect a VFD of 2.6 days as the primary outcome. The mean VFD difference between the simvastatin and placebo groups was only 1.1 days, although the upper 95% CI was 2.8 days. Therefore, it is possible that a treatment effect may be present and that a subpopulation within the overall cohort may exist that has a more marked response to statins. It is possible that there is heterogeneity in treatment effect by baseline risk. 67 Thus, identifying ARDS endotypes (subsets with distinct pathophysiological mechanisms) that are more likely to benefit from targeted interventions informed by endotype characteristics is a research priority.
There are many challenges in undertaking a large clinical trial of a pharmacological treatment in patients with ARDS. Given the number of exclusion criteria that exist in any clinical trial, which are designed to include a study population that may benefit from the therapeutic intervention but exclude patients who are unlikely to benefit owing to their underlying condition or who are at increased risk of a complication from the intervention, there is often a high number of patients who are screened who are not eligible. This was approximately 10 : 1 in this study, which is similar to other ARDS trials. 12 This represents a considerable workload and should be considered when setting up such trials.
Surprisingly, the appropriate clinical outcome or set of clinical outcomes to use in a trial recruiting patients with ARDS requiring mechanical ventilation in ICU to date has not been defined. It may be that the VFDs was not the best outcome given that it is a composite outcome combining death and duration of ventilation, and gives similar weight in terms of the outcome to death and being ventilated for 28 days.
Several challenges were identified in relation to progressing sites through the contract completion stage prior to opening as a HARP-2 trial site; a number of sites would not accept the standardised version of the model Clinical Trial Agreement (mCTA) and requested several amendments, which slowed the contracting process and delayed the opening of new sites. In addition, it was also noted that a number of R&D offices completed the review of the mCTA to sign-off stage before they commenced the R&D review, rather than progressing both elements in tandem to speed up the approval process and allow sites to open quicker. To speed up the mCTA sign-off and R&D process, the trial manager telephoned the contract signatory and R&D office on a weekly basis for an update on progress. In addition, the trial co-ordinator hand-delivered the mCTA, Material Transfer Agreement and financial agreement to local signatories for signature, to speed up the local signature process.
To maintain enthusiasm for the study and combat trial fatigue, the following measures were introduced during the lifetime of the study.
-
A review of site recruitment took place on a regular basis to identify under-recruiting sites. These sites were contacted and offered full support and assistance from the HARP-2 trial team to help them meet their target recruitment. This review led to the closure and replacement of five under-recruiting sites.
-
A HARP-2 trial newsletter was created and circulated to sites on a quarterly basis.
-
A research nurse phone-in was organised on a weekly basis to allow a one-to-one discussion with the trial co-ordinator.
A key strength of this study is the successful long-term follow-up of patients who have been discharged from critical care to assess their survival, HRQoL and resource use. However, we did find it difficult to achieve high rates of long-term follow-up data collection, in keeping with previous intensive care trials,54,55 and a considerable amount of staff resource was involved in delivering this aspect of the trial. It is important to identify barriers to follow-up in these patients to inform future critical care studies.
Another potential barrier is the reliance on recall questionnaires at 6 and 12 months, without the provision of a service-use log or aide memoire at hospital discharge. Logs not only help patients keep track of their service use, making recall less daunting or onerous, but they may also help the patient continue to feel engaged in the trial after hospital discharge.
Chapter 6 Implications for health care
Simvastatin is not effective in improving clinical outcomes in patients with ARDS, although the use of simvastatin in critically ill patients does not appear to be associated with serious adverse effects. These results do not provide support for the use of simvastatin in the management of ARDS. However, these data indicate that simvastatin may be used in critically ill patients with a coexisting condition in which a statin is normally indicated (e.g. coronary heart disease).
This study has informed guidance in the Scandinavian clinical practice guideline on fluid and drug therapy in adults with ARDS, which recommended that statins are not used in the treatment of ARDS. 69
Furthermore, this work has been included in two systematic reviews of randomised clinical trials in critically ill patients with severe sepsis, which concluded that statin therapy should not be recommended in the management of ARDS or severe sepsis in critically ill patients. 70,71
Chapter 7 Implications for research
This work has the following implications for research.
-
There is a need to define and validate surrogate outcomes used in early-phase critical care studies as well as their statistical analysis, which can accurately predict responses in patient-centred outcomes. This work is ongoing: www.comet-initiative.org/studies/details/709 (accessed 9 February 2017). 72
-
There is a need to confirm if ARDS endotypes (subsets with distinct pathophysiological mechanisms) that are more likely to benefit from targeted treatment with simvastatin (or indeed other interventions) exist.
-
There is a need to define a core set of clinical outcomes to use in trials recruiting patients with ARDS requiring mechanical ventilation in an ICU. Furthermore, it is important that outcome measures are defined and standardised. This work is ongoing: www.comet-initiative.org/studies/details/709 (accessed 9 February 2017). 72,73
-
The framework for making health-care decisions based on health economic analyses, particularly in the absence of effects on clinical outcomes, in studies of patients with ARDS needs to be developed.
-
The potential role of simvastatin in the prevention of ARDS in patients at high risk of developing ARDS (e.g. in surgical cohorts) has not yet been evaluated.
Acknowledgements
This project was supported by the EME programme, a Medical Research Council (MRC) and NIHR partnership (08/99/08). The EME programme is funded by the MRC and NIHR, with contributions from the Chief Scientist Office in Scotland, National Institute for Social Care and Health Research in Wales and the Health and Social Care R&D division of the Public Health Agency in Northern Ireland.
Daniel F McAuley and Gavin D Perkins are Directors of Research for the Intensive Care Foundation and the support of the UK Intensive Care Foundation is gratefully acknowledged. We thank the HARP-2 trial investigators on behalf of the ICCTG, all patients and their legal representatives who participated in the trial, all of the research nurses and pharmacists in all participating centres for their help, and medical and nursing staff in participating centres who cared for patients and collected data.
We thank Regina Verghis, Evie Gardner and all the staff of the NICTU for their support in delivering this trial. We thank Michael Faherty, Emma Deenihan, Veronica McInerney and Lisa Daly from the HRB Galway Clinical Research Facility, Galway, Ireland, for their help in conducting the study in Ireland.
We are grateful for the support of Margaret McFarland and staff at Victoria Pharmaceuticals (Belfast, UK) for management of the study drug. We are grateful for the support of the ICNARC for providing APACHE II data for sites that participate in ICNARC’s CMP.
The authors acknowledge the support of the Northern Ireland Clinical Research Network and the NIHR Clinical Research Network.
Contributions of authors
Daniel F McAuley conceived the study.
Daniel F McAuley, John G Laffey and Cecilia M O’Kane designed and obtained the funding for the study.
Daniel F McAuley and John G Laffey managed the undertaking of the trial with the CTU staff.
Daniel F McAuley, John G Laffey, Gavin D Perkins, Brian Mullan, Thomas J Trinder, Paul Johnston, Phillip A Hopkins and Andrew J Johnston contributed substantially to patient recruitment and data collection.
Lynn Murphy, Christine McNally and Colette Jackson managed the study and contributed to the review of the manuscript.
Clíona McDowell was the study statistician, Ashley M Agus was the health economist and Daniel F McAuley, John G Laffey and Cecilia M O’Kane interpreted the data.
Daniel F McAuley wrote the first draft of the manuscript and all authors reviewed and approved this final version of the manuscript.
All authors made a substantial contribution to the protocol development.
HARP-2 study group
Trial Management Group
Daniel McAuley (Chief Investigator, UK), John Laffey (Chief Investigator, Ireland), Christine McNally (Trial Manager), Clíona McDowell (Statistician), Ashley Agus (Health Economist), Colette Jackson, Jim O’ Neill (Trial Co-ordinator), Angela Toner (Data Manager), Susan McMullan (Data Manager), Patricia Rafferty (Senior Monitor), Roisin Boyle (Monitor), Emma Deenihan, Michael Scully and Veronica McInerney (HRB Galway Clinical Research Facility, Galway, Ireland).
Trial Steering Committee
Duncan Young (Independent Chairperson), John Radcliffe Hospital, Oxford, UK.
Rupert Pearse, Barts and the London School of Medicine and Dentistry, Royal London Hospital, London, UK.
Kathy Rowan, ICNARC, Tavistock House, London, UK.
Barry Williams, Critical Care Patient Liaison Committee (CritPaL), Dorset, UK.
Daniel F McAuley, Wellcome–Wolfson Institute for Experimental Medicine, Queen’s University of Belfast, Belfast, UK.
John Laffey, Department of Anaesthesia and Intensive Care, Clinical Sciences Institute, National University of Ireland Galway, Galway, Ireland.
Lynn Murphy, NICTU, The Royal Hospitals, Belfast, UK.
Data Monitoring and Ethics Committee
Geoff Bellingan (Chairperson), University College London, London, UK.
David Harrison, ICNARC, Tavistock House, London, UK.
Anthony Gordon, Imperial College/Charing Cross Hospital, London, UK.
Clinical collaborators (* denotes principal investigator)
Addenbrooke’s Hospital, Cambridge: Andrew J Johnston,* Archana Paikray, Cat Yates, Petra Polgarova, Esther Price, Amy McInerney and Katarzyna Zamoscik.
Aintree University Hospital, Liverpool: Ged Dempsey* and Colette Seasman.
Altnagelvin Area Hospital, Londonderry: Lynn Gilfeather,* Noel Hemmings and Sinead O’Kane.
Antrim Area Hospital, Antrim: Paul Johnston,* Lukas Pokorny, Chris Nutt and Orla O’Neill.
Arrowe Park Hospital, Wirral: Prashast Prashast,* Chris Smalley and Reni Jacob.
Beaumont Hospital, Dublin: James O’Rourke,* Syed Farjad Sultan and Carole Schilling.
Birmingham Heartlands Hospital, Birmingham: Gavin D Perkins,* Teresa Melody, Keith Couper, Ron Daniels, Fang Gao and Julian Hull.
Bristol Royal Infirmary, Bristol: Timothy Gould,* Matthew Thomas and Katie Sweet.
Cork University Hospital, Cork: Dorothy Breen* and Emer Neau.
Dumfries and Galloway Royal Infirmary, Dumfries: Willis J Peel,* Catherine Jardine and Paul Jefferson.
Freeman Hospital, Newcastle upon Tyne: Stephen E Wright* and Kayla Harris.
Frenchay Hospital, Bristol: Matthew Thomas* and Sarah Hierons.
Galway University Hospital, Galway: John Laffey* and Veronica McInerney.
Good Hope Hospital, Birmingham: Gavin Perkins.*
Guy’s and St Thomas’ Hospital, London: Luigi Camporota* and Katie Lei.
Harefield Hospital, Harefield: Sundeep Kaul* and Molly Chivburi.
Hull Royal Infirmary, Hull: Andrew Gratix,* Rachael Bennett, Victoria Martinson, Lisa Sleight and Neil Smith.
King’s College Hospital, London: Phillip A Hopkins,* Daniel Hadfield, Sarah Casboult, Fiona Wade-Smith, Julie Dawson, Clare Mellis, Clair Harris, Georgina Parsons and Sinead Helyar.
Leeds Teaching Hospitals NHS Trust, Leeds: Andrew R Bodenham,* Stuart Elliott, Zoe Beardow and Sian Birch.
Mater Misericordiae University Hospital, Dublin: Brian Marsh* and Teresa Martin.
Norfolk and Norwich University Hospitals, Norwich: Akesh Dhrampal* and Melissa Rosbergen.
Papworth Hospital, Cambridge: Stephen Webb* and Fiona Bottrill.
Poole Hospital, Poole: Henrik Reschreiter,* Helena Barcraft-Barnes and Julie Camsooksai.
Queen Elizabeth Hospital Birmingham, Birmingham: Andrew Johnston,* Aisling Clarkson, Conor Bentley, Lauren Cooper, Yongyan Qui, Natalie Mitchell, Ronald Carrera and Arlo Whitehouse.
Royal Berkshire Hospital, Reading: Christopher M Danbury,* Nicola Jacques and Abby Brown.
Royal Derby Hospital, Derby: David Rogerson* and Craig Morris.
Royal Infirmary of Edinburgh, Edinburgh: Timothy Walsh,* Mike Gillies, Grant Price, Kallirroi Kefala, Neil Young, David Hope, Corrienne McCulloch, Jean Antonelli, Pam Ramsay, Kirsty Everingham, Louise Boardman, Heidi Dawson, Fiona Pollock and Joanne Thompson.
Royal Liverpool University Hospital, Liverpool: Ingeborg D Welters,* Lee Poole, Peter Hampshire, Alison Hall, Karen Williams, Anna Walker, Laura Youds, Samantha Hendry, Victoria Waugh, Julie Patrick-Heselton and David Shaw.
Royal Preston Hospital, Preston: Irfan Chaudry* and Jacqueline Baldwin.
Royal Sussex County Hospital, Brighton: Stephen Drage* and Laura Ortiz-Ruiz de Gordoa.
Royal Victoria Hospital, Belfast: Daniel McAuley,* Leona Bannon, Vanessa Quinn, Lia McNamee and Griania White.
St George’s Hospital, London: Maurizio Cecconi* and Johannes Mellinghoff.
St Vincent’s University Hospital, Dublin: Donal Ryan* and Alistair Nichol.
The Royal Free Hospital, London: Banwari Agarwal,* Paula Meale, Sarah James, Kulwant Dhadwal, Daniel Martin, Agnieszka Walecka and Stephen Ward.
Ulster Hospital, Dundonald: Thomas J Trinder,* Samantha Hagan, Janice Montgomery, Catherine Leonard, Elizabeth Lemon and Tom Trinick.
University Hospitals Coventry and Warwickshire, Coventry: Murthy Buddhavarapu,* Geraldine Ward and Christopher Bassford.
Victoria Infirmary, Glasgow: Alan Davidson,* Kate McGuigan, Anissa Benchiheub and Naomi Hickey.
Western Infirmary, Glasgow: Alexander Binning* and Steven Henderson.
Whiston Hospital, Liverpool: Julie A Wood.*
Worcester Royal Hospital, Worcester: Andrew J Burtenshaw,* Dawn Kelly, Terry Martin, Jessica Thrush, Julie Wollaston, Stephen Graystone, Gavin Nicol and Gareth Sellors.
Publications
McAuley DF, Laffey JG, O’Kane CM, Cross M, Perkins GD, Murphy L, et al. Hydroxymethylglutaryl-CoA reductase inhibition with simvastatin in acute lung injury to reduce pulmonary dysfunction (HARP-2) trial: study protocol for a randomized controlled trial. Trials 2012;13:170.
McAuley DF, Laffey JG, O’Kane CM, Perkins GD, Mullan B, Trinder TJ, et al. Simvastatin in acute respiratory distress syndrome. N Engl J Med 2014;371:1695–703.
Agus A, Hulme C, Verghis RM, McDowell C, Jackson C, O’Kane CM, et al. Simvastatin for patients with acute respiratory distress syndrome: long term outcomes and cost-effectiveness from a randomised controlled trial. Crit Care 2017;21:108.
Nagendran M, McAuley DF, Kruger PS, Papazian L, Truwit JD, Laffey JG, et al. Statin therapy for acute respiratory distress syndrome: an individual patient data meta-analysis of randomised clinical trials. Intensive Care Med 2017;43:663–71.
Poole JD, McDowell C, Lall R, Perkins G, McAuley DF, Gao F et al. Individual patient data analysis of tidal volumes used in three large randomized control trials involving patients with acute respiratory distress syndrome. Br J Anaesthes 2017;118:570–5.
Data sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health.
References
- Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet 1967;2:319-23. https://doi.org/10.1016/S0140-6736(67)90168-7.
- Fowler AA, Hamman RF, Good JT, Benson KN, Baird M, Eberle DK, et al. Adult respiratory distress syndrome: risk with common predispositions. Ann Intern Med 1983;98:593-7. https://doi.org/10.7326/0003-4819-98-5-593.
- Petty TL. Acute respiratory distress syndrome (ARDS). Dis Mon 1990;36:1-58.
- Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 1994;149:818-24. https://doi.org/10.1164/ajrccm.149.3.7509706.
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med 2005;353:1685-93. https://doi.org/10.1056/NEJMoa050333.
- Brun-Buisson C, Minelli C, Bertolini G, Brazzi L, Pimentel J, Lewandowski K, et al. Epidemiology and outcome of acute lung injury in European intensive care units. Results from the ALIVE study. Intensive Care Med 2004;30:51-6. https://doi.org/10.1007/s00134-003-2022-6.
- Dowdy DW, Eid MP, Dennison CR, Mendez-Tellez PA, Herridge MS, Guallar E, et al. Quality of life after acute respiratory distress syndrome: a meta-analysis. Intensive Care Med 2006;32:1115-24. https://doi.org/10.1007/s00134-006-0217-3.
- Rossi C, Simini B, Brazzi L, Rossi G, Radrizzani D, Iapichino G, et al. Variable costs of ICU patients: a multicenter prospective study. Intensive Care Med 2006;32:545-52. https://doi.org/10.1007/s00134-006-0080-2.
- Cheung AM, Tansey CM, Tomlinson G, Diaz-Granados N, Matté A, Barr A, et al. Two-year outcomes, health care use, and costs of survivors of acute respiratory distress syndrome. Am J Respir Crit Care Med 2006;174:538-44. https://doi.org/10.1164/rccm.200505-693OC.
- Craig T, O’Kane CM, McAuley DF, Vincent JL. 27th Yearbook of Intensive Care and Emergency Medicine. Berlin: Springer-Verlag; 2007.
- Frank JA, Parsons PE, Matthay MA. Pathogenetic significance of biological markers of ventilator-associated lung injury in experimental and clinical studies. Chest 2006;130:1906-14. https://doi.org/10.1378/chest.130.6.1906.
- The National Heart, Lung, and Blood Institute ARDS Clinical Trials Network . Rosuvastatin for sepsis-associated Acute Respiratory Distress Syndrome. N Engl J Med 2014;370:2191-200. https://doi.org/10.1056/NEJMoa1401520.
- Monchi M, Bellenfant F, Cariou A, Joly LM, Thebert D, Laurent I, et al. Early predictive factors of survival in the acute respiratory distress syndrome. A multivariate analysis. Am J Respir Crit Care Med 1998;158:1076-81. https://doi.org/10.1164/ajrccm.158.4.9802009.
- Liappis AP, Kan VL, Rochester CG, Simon GL. The effect of statins on mortality in patients with bacteremia. Clin Infect Dis 2001;33:1352-7. https://doi.org/10.1086/323334.
- Almog Y, Shefer A, Novack V, Maimon N, Barski L, Eizinger M, et al. Prior statin therapy is associated with a decreased rate of severe sepsis. Circulation 2004;110:880-5. https://doi.org/10.1161/01.CIR.0000138932.17956.F1.
- Hackam DG, Mamdani M, Li P, Redelmeier DA. Statins and sepsis in patients with cardiovascular disease: a population-based cohort analysis. Lancet 2006;367:413-18. https://doi.org/10.1016/S0140-6736(06)68041-0.
- Kruger P, Fitzsimmons K, Cook D, Jones M, Nimmo G. Statin therapy is associated with fewer deaths in patients with bacteraemia. Intensive Care Med 2006;32:75-9. https://doi.org/10.1007/s00134-005-2859-y.
- Thomsen RW, Riis A, Kornum JB, Christensen S, Johnsen SP, Sørensen HT. Preadmission use of statins and outcomes after hospitalization with pneumonia: population-based cohort study of 29,900 patients. Arch Intern Med 2008;168:2081-7. https://doi.org/10.1001/archinte.168.19.2081.
- Chalmers JD, Singanayagam A, Murray MP, Hill AT. Prior statin use is associated with improved outcomes in community-acquired pneumonia. Am J Med 2008;121:1002-7. https://doi.org/10.1016/j.amjmed.2008.06.030.
- Mortensen EM, Restrepo MI, Anzueto A, Pugh J. The effect of prior statin use on 30-day mortality for patients hospitalized with community-acquired pneumonia. Respir Res 2005;6. https://doi.org/10.1186/1465-9921-6-82.
- Irish Critical Care Trials Group . Acute lung injury and the acute respiratory distress syndrome in Ireland: a prospective audit of epidemiology and management. Crit Care 2008;12. https://doi.org/10.1186/cc6808.
- Kor DJ, Iscimen R, Yilmaz M, Brown MJ, Brown DR, Gajic O. Statin administration did not influence the progression of lung injury or associated organ failures in a cohort of patients with acute lung injury. Intensive Care Med 2009;35:1039-46. https://doi.org/10.1007/s00134-009-1421-8.
- Shyamsundar M, McKeown ST, O’Kane CM, Craig TR, Brown V, Thickett DR, et al. Simvastatin decreases lipopolysaccharide-induced pulmonary inflammation in healthy volunteers. Am J Respir Crit Care Med 2009;179:1107-14. https://doi.org/10.1164/rccm.200810-1584OC.
- Steiner S, Speidl WS, Pleiner J, Seidinger D, Zorn G, Kaun C, et al. Simvastatin blunts endotoxin-induced tissue factor in vivo. Circulation 2005;111:1841-6. https://doi.org/10.1161/01.CIR.0000158665.27783.0C.
- Novack V, Eisinger M, Frenkel A, Terblanche M, Adhikari NK, Douvdevani A, et al. The effects of statin therapy on inflammatory cytokines in patients with bacterial infections: a randomized double-blind placebo controlled clinical trial. Intens Care Med 2009;35:1255-60. https://doi.org/10.1007/s00134-009-1429-0.
- Craig TR, Duffy MJ, Shyamsundar M, McDowell M, O’Kane CM, Elborn JS, et al. A randomized clinical trial of hydroxymethylglutaryl–coenzyme A reductase inhibition for acute lung injury (the HARP study). Am J Respir Crit Care Med 2011;183:620-6. https://doi.org/10.1164/rccm.201003-0423OC.
- Choi HS, Park MJ, Kang HM, Kim YJ, Choi CW, You JH, et al. Statin use in sepsis due to pneumonia. Am J Respir Crit Care Med 2008;177.
- Gonzalez C, Luna A, Morales M, Sanchez J, Guzman C, Granillo J. Statin anti-inflammatory therapy in septic patients. Crit Care 2008;12. https://doi.org/10.1186/cc7059.
- The A to Z Investigators . Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes. JAMA 2004;292:1307-16. https://doi.org/10.1001/jama.292.11.1307.
- Bowman L, Armitage J, Bulbulia R, Parish S, Collins R. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12,064 myocardial infarction survivors. Am Heart J 2007;154:815-23. https://doi.org/10.1016/j.ahj.2007.06.034.
- Jacobson JR, Barnard JW, Grigoryev DN, Ma SF, Tuder RM, Garcia JG. Simvastatin attenuates vascular leak and inflammation in murine inflammatory lung injury. Am J Physiol Lung Cell Mol Physiol 2005;288:L1026-32. https://doi.org/10.1152/ajplung.00354.2004.
- Shah AI, Sobnosky S, Shen AY, Jorgensen MB. High dose statins are associated with a reduced all cause mortality in patients hospitalized with sepsis and severe sepsis. Crit Care Med 2009;36.
- Adhikari N, Burns KE, Meade MO. Pharmacologic therapies for adults with acute lung injury and acute respiratory distress syndrome. Cochrane Database Syst Rev 2004;4. https://doi.org/10.1002/14651858.CD004477.pub2.
- Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, et al. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med 2003;167:1027-35. https://doi.org/10.1164/rccm.200208-966WS.
- Schulz KF, Altman DG, Moher D. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. Trials 2010;11. https://doi.org/10.1186/1745-6215-11-32.
- McAuley DF, Laffey JG, O’Kane CM, Cross M, Perkins GD, Murphy L, et al. Hydroxymethylglutaryl-CoA reductase inhibition with simvastatin in acute lung injury to reduce pulmonary dysfunction (HARP-2) trial: study protocol for a randomized controlled trial. Trials 2012;13. https://doi.org/10.1186/1745-6215-13-170.
- McAuley DF, Laffey JG, O’Kane CM, Perkins GD, Mullan B, Trinder TJ, et al. Simvastatin in the acute respiratory distress syndrome. N Engl J Med 2014;371:1695-703. https://doi.org/10.1056/NEJMoa1403285.
- Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006;354:2564-75. https://doi.org/10.1056/NEJMoa062200.
- Matthay MA, Brower R, Thompson BT, Schoenfeld D, Eisner MD, Carson S, et al. Randomized, placebo-controlled trial of an aerosolized beta–2 adrenergic agonist (albuterol) for the treatment of acute lung injury. Am J Respir Crit Care Med 2009;179. https://doi.org/10.1164/ajrccm-conference.2009.179.1_MeetingAbstracts.A2166.
- Felton TW, Sander R, Al-Aloul M, Dark P, Bentley AM. Can a score derived from the Critical Care Minimum Data Set be used as a marker of organ dysfunction? – a pilot study. BMC Res Notes 2009;2. https://doi.org/10.1186/1756-0500-2-77.
- Brower RG, Hopkins J, Matthay MA, Morris A, Schoenfeld D, Thompson T, et al. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 2000;342:1301-8. https://doi.org/10.1056/NEJM200005043421801.
- Harvey S, Harrison DA, Singer M, Ashcroft J, Jones CM, Elbourne D, et al. Assessment of the clinical effectiveness of pulmonary artery catheters in management of patients in intensive care (PAC-Man): a randomised controlled trial. Lancet 2005;366:472-7. https://doi.org/10.1016/S0140-6736(05)67061-4.
- Machin D, Campbell MJ. Design of Studies for Medical Research. Chichester: Wiley Online Library; 2005.
- Agus A, Hulme C, Verghis RM, McDowell C, Jackson C, O’Kane CM, et al. Simvastatin for patients with acute respiratory distress syndrome: long term outcomes and cost-effectiveness from a randomised controlled trial. Crit Care 2017;21. https://doi.org/10.1186/s13054-017-1695-0.
- Al Harbi SA, Tamim HM, Arabi YM. Association between statin therapy and outcomes in critically ill patients: a nested cohort study. BMC Clin Pharmacol 2011;11. https://doi.org/10.1186/1472-6904-11-12.
- Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2014;2:611-20. https://doi.org/10.1016/S2213-2600(14)70097-9.
- Jaswal DS, Leung JM, Sun J, Cui X, Li Y, Kern S, et al. Tidal volume and plateau pressure use for acute lung injury from 2000 to present: a systematic literature review. Crit Care Med 2014;42:2278-89. https://doi.org/10.1097/CCM.0000000000000504.
- Guide to the Methods of Technology Appraisal 2013. London: NICE; 2013.
- NHS Reference Costs 2013–14. London: Department of Health; 2014.
- Curtis L. Unit Costs of Health and Social Care 2014. Canterbury: PSSRU, University of Kent; 2014.
- Department of Health . NHS Electronic Drug Tariff 2014. www.ppa.org.uk/edt/January_2014/mindex.htm (accessed 22 June 2015).
- Curtis L. Unit Costs of Health and Social Care 2013. Canterbury: PSSRU, University of Kent; 2013.
- EuroQol Group . EuroQol – a new facility for the measurement of health-related quality of life. Health Policy 1990;16:199-208. https://doi.org/10.1016/0168-8510(90)90421-9.
- Gates S, Perkins GD, Lamb SE, Kelly C, Thickett DR, Young JD, et al. Beta-Agonist Lung injury TrIal-2 (BALTI-2): a multicentre, randomised, double-blind, placebo-controlled trial and economic evaluation of intravenous infusion of salbutamol versus placebo in patients with acute respiratory distress syndrome. Health Technol Assess 2013;17. https://doi.org/10.3310/hta17380.
- Lall R, Hamilton P, Young D, Hulme C, Hall P, Shah S, et al. A randomised controlled trial and cost-effectiveness analysis of high-frequency oscillatory ventilation against conventional artificial ventilation for adults with acute respiratory distress syndrome. The OSCAR (OSCillation in ARDS) study. Health Technol Assess 2015;19. https://doi.org/10.3310/hta19230.
- Peek GJ, Elbourne D, Mugford M, Tiruvoipati R, Wilson A, Allen E, et al. Randomised controlled trial and parallel economic evaluation of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR). Health Technol Assess 2010;14. https://doi.org/10.3310/hta14350.
- Dolan P. Modelling valuations for EuroQol health states. Med Care 1997;35:1095-108. https://doi.org/10.1097/00005650-199711000-00002.
- Glick HA, Doshi JA, Sonnad SS, Polsky D. Economic Evaluation in Clinical Trials. Oxford: Oxford University Press; 2007.
- Social Value Judgements: Principles for the Development of NICE Guidance. London: NICE; 2008.
- Hollingworth W, McKell-Redwood D, Hampson L, Metcalfe C. Cost–utility analysis conducted alongside randomized controlled trials: are economic end points considered in sample size calculations and does it matter?. Clin Trials 2013;10:43-5. https://doi.org/10.1177/1740774512465358.
- Ferguson ND, Scales DC, Pinto R, Wilcox ME, Cook DJ, Guyatt GH, et al. Integrating mortality and morbidity outcomes: using quality-adjusted life years in critical care trials. Am J Respir Crit Care Med 2013;187:256-61. https://doi.org/10.1164/rccm.201206-1057OC.
- Claxton K. The irrelevance of inference: a decision-making approach to the stochastic evaluation of health care technologies. J Health Econ 1999;18:341-64. https://doi.org/10.1016/S0167-6296(98)00039-3.
- Karir V, Angus DC, Clermont G, Kong L, Bernard GR, Rubenfeld GD, et al. Relationship Between Ventilator-Free-Days And Patient-Centered Outcomes In Patients With Acute Lung Injury n.d. https://doi.org/10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A3183.
- Spragg RG, Bernard GR, Checkley W, Curtis JR, Gajic O, Guyatt G, et al. Beyond mortality: future clinical research in acute lung injury. Am J Respir Crit Care Med 2010;181:1121-7. https://doi.org/10.1164/rccm.201001-0024WS.
- Rubenfeld GD. Epidemiology of acute lung injury. Crit Care Med 2003;31:276-84. https://doi.org/10.1097/01.CCM.0000057904.62683.2B.
- Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA 2012;307:2526-33. https://doi.org/10.1001/jama.2012.5669.
- Iwashyna TJ, Burke JF, Sussman JB, Prescott HC, Hayward RA, Angus DC. Implications of heterogeneity of treatment effect for reporting and analysis of randomized trials in critical care. Am J Respir Crit Care Med 2015;192:1045-51. https://doi.org/10.1164/rccm.201411-2125CP.
- Rubenfeld GD. Confronting the frustrations of negative clinical trials in acute respiratory distress syndrome. Ann Am Thorac Soc 2015;12:58-63. https://doi.org/10.1513/AnnalsATS.201409-414MG.
- Claesson J, Freundlich M, Gunnarsson I, Laake JH, Møller MH, Vandvik PO, et al. Scandinavian clinical practice guideline on fluid and drug therapy in adults with acute respiratory distress syndrome. Acta Anaesthesiol Scand 2016;60:697-709. https://doi.org/10.1111/aas.12713.
- Thomas G, Hraiech S, Loundou A, Truwit J, Kruger P, Mcauley DF, et al. Statin therapy in critically-ill patients with severe sepsis: a review and meta-analysis of randomized clinical trials. Minerva Anestesiol 2015;81:921-30.
- Nagendran M, McAuley DF, Kruger P, Papazian L, Truwit J, Thompson BT, et al. Statin therapy for acute respiratory distress syndrome: an individual patient data meta-analysis of randomised clinical trials. Intensive Care Med 2017;43:663-71. https://doi.org/10.1007/s00134-016-4649-0.
- COMET Initiative n.d. www.comet-initiative.org/studies/details/709 (accessed 9 February 2017).
- Blackwood B, Ringrow S, Clarke M, Marshall J, Rose L, Williamson P, et al. Core Outcomes in Ventilation Trials (COVenT): protocol for a core outcome set using a Delphi survey with a nested randomised trial and observational cohort study. Trials n.d.;16.
Appendix 1 Serious adverse event listing
| Treatment | Site | Subject number | SAE ID | Date of report | Date of onset | Date of resolution | Ongoing | SAE expected | Causality | SerAdvClass | Date drug started | If discontinued, date stopped | Patient withdrawn as a result | Study drug administered according to protocol |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Simvastatin | s01 | 11,015 | 1 | 13 May 2011 | 13 May 2011 | 9 June 2011 | No | No | Unrelated | Cardiac disorders | 30 April 2011 | 3 May 2011 | No | Yes |
| Simvastatin | s01 | 12,493 | 1 | 10 December 2013 | 9 December 2013 | UNOB | Yes | No | Unrelated | Gastrointestinal | 6 December 2013 | 9 December 2013 | No | Yes |
| Simvastatin | s07 | 11,618 | 1 | 27 June 2012 | 14 June 2012 | UNOB | Yes | No | Unrelated | Renal and urinary disorders | 22 May 2012 | 28 May 2012 | No | Yes |
| Simvastatin | s13 | 11,291 | 1 | 14 October 2011 | 14 October 2011 | 14 October 2011 | No | No | Unlikely | Cardiac disorders | 20 September 2011 | 17 October 2011 | No | Yes |
| Simvastatin | s45 | 11,270 | 1 | 17 November 2011 | 16 November 2011 | UNOB | Yes | No | Unrelated | Cardiac disorders | UNOB | No | No | |
| Simvastatin | s45 | 11,458 | 1 | 6 February 2012 | 3 February 2012 | 3 February 2012 | No | No | Unrelated | Gastrointestinal disorders | 31 January 2012 | 26 February 2012 | No | Yes |
| Simvastatin | s45 | 11,458 | 1 | 7 March 2012 | 5 March 2012 | 7 March 2012 | No | No | Unrelated | Infections and infestations | 31 January 2012 | No | No | |
| Simvastatin | s45 | 11,460 | 1 | 2 April 2012 | 31 March 2012 | 18 April 2012 | No | No | Unrelated | Injury, poisoning and procedural complications | 30 March 2012 | No | Yes | |
| Simvastatin | s45 | 11,844 | 1 | 29 October 2012 | 26 October 2012 | UNOB | Yes | No | Unrelated | Nervous system disorders | 10 October 2012 | 17 October 2012 | No | Yes |
| Simvastatin | s51 | 11,033 | 1 | 14 March 2011 | 9 March 2011 | 21 March 2011 | No | Yes | Probable | Musculoskeletal and connective tissue disorders/elevated creatinine kinase | 15 February 2011 | 9 March 2011 | Yes | Yes |
| Simvastatin | s58 | 11,516 | 1 | 10 May 2012 | 9 May 2012 | 18 July 2012 | No | Yes | Unlikely | Respiratory, thoracic and mediastinal disorders | 27 April 2012 | 10 May 2012 | Yes | Yes |
| Simvastatin | s60 | 12,354 | 1 | 2 August 2013 | 1 August 2013 | 1 August 2013 | No | No | Unrelated | Gastrointestinal disorders | 25 July 2013 | UNOB | No | Yes |
| Placebo | s01 | 11,016 | 1 | 18 May 2011 | 17 May 2011 | 19 May 2011 | No | No | Unrelated | Cardiac disorders | 26 April 2011 | 24 May 2011 | No | Yes |
| Placebo | s01 | 11,434 | 1 | 13 March 2012 | 13 March 2012 | UNOB | Yes | No | Unrelated | Respiratory, thoracic and mediastinal disorders | 3 February 2012 | No | Yes | |
| Placebo | s01 | 11,434 | 1 | 9 February 2012 | 8 February 2012 | 17 February 2012 | No | No | Unrelated | Respiratory, thoracic and mediastinal disorders | 3 February 2012 | 21 February 2012 | No | No |
| Placebo | s01 | 11,436 | 1 | 22 February 2012 | 21 February 2012 | 22 February 2012 | No | No | Unrelated | Cardiac disorders | 21 February 2012 | 22 February 2012 | No | Yes |
| Placebo | s38 | 12,586 | 1 | 3 October 2013 | 2 October 2013 | 2 October 2013 | No | No | Unlikely | Cardiac disorders | 1 October 2013 | No | Yes | |
| Placebo | s43 | 12,745 | 1 | 19 March 2014 | 8 March 2014 | 8 March 2014 | No | No | Unrelated | Injury, poisoning and procedural complications | 6 March 2014 | No | Yes | |
| Placebo | s43 | 12,745 | 1 | 11 April 2014 | 2 April 2014 | 3 April 2014 | No | No | Possible | Nervous system disorders | 6 March 2014 | 29 March 2014 | No | Yes |
| Placebo | s45 | 11,730 | 1 | 4 May 2012 | 4 May 2012 | UNOB | Yes | Yes | Unlikely | Hepatobiliary disorders | 3 May 2012 | 4 May 2012 | No | Yes |
| Placebo | s45 | 11,733 | 1 | 16 July 2012 | 15 July 2012 | 23 July 2012 | No | No | Unrelated | Respiratory, thoracic and mediastinal disorders | 20 June 2012 | 10 July 2012 | No | Yes |
| Placebo | s47 | 11,578 | 1 | 31 May 2012 | 27 May 2012 | UNOB | Yes | Yes | Possible | Musculoskeletal and connective tissue disorders/elevated creatinine kinase | 18 May 2012 | 27 May 2012 | Yes | Yes |
| Placebo | s48 | 12,442 | 1 | 5 November 2013 | 4 November 2013 | 5 November 2013 | No | Unlikely | Cardiac disorders | 4 November 2013 | No | Yes | ||
| Placebo | s58 | 11,519 | 1 | 11 July 2012 | 6 July 2012 | 12 July 2012 | No | No | Unrelated | Infections and infestations | 16 June 2012 | 27 June 2012 | No | Yes |
| Placebo | s59 | 11,699 | 1 | 25 July 2012 | 22 July 2012 | 27 July 2012 | No | No | Possible | Nervous system disorders | 20 July 2012 | 26 July 2012 | No | Yes |
| Placebo | s60 | 11,572 | 1 | 28 May 2012 | 25 May 2012 | UNOB | Yes | No | Unrelated | Cardiac disorders | 16 May 2012 | No | Yes | |
| Placebo | s61 | 11,089 | 1 | 20 June 2011 | 11 June 2011 | 02 July 2011 | No | No | Unrelated | Infections and infestations | 19 May 2011 | 4 June 2011 | No | Yes |
| Placebo | s70 | 12,023 | 1 | 1 February 2013 | 31 January 2013 | UNOB | Yes | No | Unlikely | Respiratory, thoracic and mediastinal disorders | 14 January 2013 | 22 January 2013 | No | Yes |
List of abbreviations
- AE
- adverse event
- ALI
- acute lung injury
- ALT
- alanine transaminase
- Ang2
- angiopoietin 2
- APACHE II
- Acute Physiology And Chronic Health Evaluation
- AR
- adverse reaction
- ARDS
- acute respiratory distress syndrome
- AST
- aspartate aminotransferase
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CK
- creatine kinase
- CMP
- Case Mix Programme
- CONSORT
- Consolidated Standards of Reporting Trials
- CREC
- Clinical Research Ethics Committee
- CRF
- case report form
- CRP
- C-reactive protein
- CTCAE
- common terminology criteria for adverse events
- CTU
- Clinical Trials Unit
- DMEC
- Data Monitoring and Ethics Committee
- EME
- Efficacy and Mechanism Evaluation
- EQ-5D
- EuroQol-5 Dimensions
- EQ-5D-3L
- EuroQol-5 Dimensions, three-level version
- GP
- general practitioner
- HARP-2
- Hydroxymethylglutaryl-CoA reductase inhibition with simvastatin in Acute lung injury to Reduce Pulmonary dysfunction
- HDU
- high-dependency unit
- HR
- hazard ratio
- HRB
- health research board
- HRQoL
- health-related quality of life
- ICCTG
- Irish Critical Care Trials Group
- ICER
- incremental cost-effectiveness ratio
- ICNARC
- Intensive Care National Audit and Research Centre
- ICU
- intensive care unit
- IL
- interleukin
- IMB
- Irish Medicines Board
- ISF
- investigator site file
- LPS
- lipopolysaccharide
- mCTA
- model Clinical Trial Agreement
- MHRA
- Medicines and Healthcare Products Regulatory Agency
- MMP
- matrix metalloproteinase
- MRC
- Medical Research Council
- NICE
- National Institute for Health and Care Excellence
- NICTU
- Northern Ireland Clinical Trials Unit
- NIHR
- National Institute for Health Research
- OFFD
- oscillator frequency-free day
- OI
- oxygenation index
- ORECNI
- Office for Research Ethics Committees Northern Ireland
- PAOP
- pulmonary arterial occlusion pressure
- PerLR
- personal legal representative
- PI
- principal investigator
- PIS
- patient information sheet
- ProfLR
- professional legal representative
- QALY
- quality-adjusted life-year
- R&D
- research and development
- RAGE
- receptor for advanced glycation end-products
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- SAE
- serious adverse event
- SAR
- serious adverse reaction
- SD
- standard deviation
- SOFA
- sequential organ failure assessment
- SPC
- summary of product characteristics
- SUSAR
- suspected unexpected serious adverse reaction
- TNF-α
- tumour necrosis factor alpha
- UAR
- unexpected adverse reaction
- VAS
- visual analogue scale
- VFD
- ventilator-free day
- WTP
- willingness to pay