

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 11/14/08. The contractual start date was in July 2013. The final report began editorial review in July 2017 and was accepted for publication in June 2018. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Anthony Gordon reports personal fees and non-financial support from Orion Corporation Orion Pharma, and grants and other from Tenax Therapeutics during the conduct of the study. He also reports personal fees from Ferring Pharmaceuticals, grants from HCA International, personal fees from GlaxoSmithKline plc, personal fees from Baxter Healthcare Ltd and personal fees from Amomed Pharma outside the submitted work. Daniel McAuley reports personal fees from Peptinnovate Ltd, Bayer, Boehringer Ingelheim Ltd and SOBI, personal fees and other from GlaxoSmithKline plc and grants from the National Institute for Health Research (NIHR), Wellcome Trust and other funders outside the submitted work, as well as a patent being issued for the novel treatment for acute respiratory distress syndrome outside the submitted work. Daniel McAuley is a member of the Health Technology Assessment (HTA) General Board. Mervyn Singer reports grants from the NIHR during the conduct of the study and personal fees from Amomed Pharma outside the submitted work. Anthony Gordon, Gavin Perkins and Daniel McAuley are directors of research for the Intensive Care Foundation.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2018. This work was produced by Gordon et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2018 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background
Severe sepsis is responsible for approximately 30% of all admissions to intensive care in the UK, yet, despite improvements in care, the mortality rate from severe sepsis remains high. 1 According to data from the Intensive Care National Audit & Research Centre, the incidence of severe sepsis has increased by 68% over a 9-year period, such that the total number of severe sepsis cases in the UK is in excess of 45,000 per annum, and the hospital mortality rate is approximately 45%. 1 Mortality rates increase with increasing number of organ failures. 2,3 In particular, acute renal failure in severe sepsis is an independent risk factor for death [odds ratio (OR) 2.1]. 4
It was estimated in 2001 that treating critically ill patients who have sepsis costs the NHS more than £700M. 5 As the population ages and receives more complex medical treatments, the incidence of sepsis, and of associated mortality and morbidity, and costs will continue to rise. Therefore, severe sepsis is an extremely important health-care problem.
Levosimendan
Levosimendan (Simdax®; Orion Pharma, Newbury, UK) is a licensed treatment for decompensated heart failure in > 50 countries around the world. It acts by sensitising the myocardium to calcium so that a greater ventricular contraction (and thus stroke volume) can be achieved for the same intracellular calcium concentration, thereby reducing the workload of the failing heart. 6 The drug itself has a short plasma half-life of approximately 1 hour, is around 95% bound to plasma proteins and is fully metabolised in the liver and intestine into both active and inactive metabolites. However, the haemodynamic effects are maintained for up to 7 days after a single 24-hour infusion of levosimendan because of the effects of the active metabolite, OR-1896, which has an elimination half-life of approximately 80 hours. 7
Extensive animal and human investigations have concluded that the mechanism of action of levosimendan also includes vasodilatation, mediated by activation of adenosine triphosphate (ATP)-sensitive sarcolemmal potassium channels and ATP-sensitive mitochondrial potassium channels. 8 This in turn may lead to the maintenance of mitochondrial volume and a reduction in calcium overload seen in ischaemia, thereby preserving mitochondrial function. 9 Levosimendan has also been shown to possess anti-inflammatory properties. 10,11 Post licensing, levosimendan has been extensively investigated in patients with acute heart failure caused by a variety of aetiologies.
As part of the systemic inflammatory response, myocardial dysfunction is seen in > 50% of patients with severe sepsis. 12 The likely mechanism of myocardial dysfunction is a combination of altered calcium trafficking and reduced troponin sensitivity to calcium13 and its presence contributes to multiple organ failure including acute renal failure. The calcium-sensitising and anti-inflammatory actions of levosimendan provide a strong biological rationale for its use in sepsis. In addition, conventional vasoactive support using catecholamines such as noradrenaline and dobutamine may result in sympathetic overstimulation and a range of adverse effects. 14 Evidence of lack of benefits from trials comparing different catecholamine regimens,15 increased mortality in patients exposed to a greater vasopressor load16 and the observation of higher plasma catecholamine levels in non-survivors than in survivors of critical illness17 all provide further evidence of possible harm from conventional catecholamine therapy.
Levosimendan in sepsis: animal studies
The use of levosimendan in severe sepsis has been studied in a range of animal models of sepsis. In an ovine septic shock model, the combination of levosimendan (0.2 µg/kg/minute) and vasopressin as opposed to noradrenaline and vasopressin was associated with improved myocardial, pulmonary and renal function. 18 In a mouse model of septic shock, Zager et al. 19 observed that levosimendan protected against acute renal failure, probably because of vasodilatation in the kidney as a result of levosimendan inducing ATP-sensitive potassium channel activation. Similarly, in a rat model of sepsis, Fries et al. 20 demonstrated an improvement in microvascular perfusion in the buccal mucosa of animals given 0.3 µg/kg/minute of levosimendan. Dubin et al. 21 evaluated higher doses of levosimendan (100-µg loading dose followed by an infusion of 1.6 µg/kg/minute) in an ovine model, demonstrating that levosimendan prevented the reduction in mesenteric oxygen delivery that was seen in the control group animals. In a porcine septic shock model, high-dose levosimendan attenuated the increase in pulmonary vascular resistance and improved both hepatosplanchnic and systemic blood flow compared with control animals. 22 An improved responsiveness to noradrenaline was also seen in the porcine septic shock model study.
In two studies from the same group,23,24 both in porcine models of sepsis, levosimendan failed to show an improvement in hepatosplanchnic perfusion compared with both placebo and dobutamine, as had been seen in other studies. These results were felt, in part, to be the result of the failure to adequately fluid resuscitate the animals and restore mean arterial pressure (MAP) prior to commencing levosimendan. 25
Levosimendan in sepsis: human studies
In humans, evidence of a potential benefit from levosimendan in severe sepsis comes from a combination of case reports and case series,26–29 together with a number of clinical trials. 30–35 In a case series of six patients with refractory septic shock given 0.1–0.2 µg/kg/minute of levosimendan, there was a trend towards improved haemodynamics, associated with a reduction in catecholamine requirements. 29 All but one of these patients survived to hospital discharge despite an Acute Physiology and Chronic Health Evaluation II (APACHE II)-predicted mortality of 60%. 29
Three of the sepsis trials in humans have been led by Dr Morelli. The first trial, published in 2005, compared an infusion of 0.2 µg/kg/minute of levosimendan with an infusion of 5 µg/kg/minute of dobutamine for 24 hours in 28 patients with septic shock and echocardiographically proven acute left ventricular dysfunction. 30 Statistically significant reductions in both pulmonary artery pressure and pulmonary artery occlusion pressure and an increase in left ventricular stroke work index were seen with levosimendan. In particular, levosimendan increased creatinine clearance by 64% while decreasing serum lactate levels compared with dobutamine. 30 In 35 patients with septic shock and the acute respiratory distress syndrome, 0.2 µg/kg/minute of levosimendan increased the cardiac index and reduced the mean pulmonary artery pressure compared with placebo. 31 More recently, in a study of 40 patients who had septic shock, the effects of levosimendan (0.2 µg/kg/minute) on microcirculatory blood flow in the sublingual mucosa were compared with those of dobutamine (5 µg/kg/minute). Blood flow was significantly higher in the levosimendan group (p < 0.001) and there was a trend towards higher central venous oxygen saturation (ScvO2) and arterial pH as well as lower noradrenaline requirements in the levosimendan group. 32 These studies were not of sufficient size to detect any differences in any patient-focused outcomes.
A number of other small trials have been performed. In 42 patients with septic shock, levosimendan compared with dobutamine reduced the number of patients requiring additional catecholamine support with noradrenaline (p < 0.04). 33 In a similar trial, 42 patients who had severe sepsis and a cardiac index of < 2.2 l/minute/m2 received either levosimendan or dobutamine as additional therapy. Cardiac index, ejection fraction and ScvO2 all increased significantly more in the levosimendan group than in the dobutamine group. 34 In a more recent trial of 30 patients with septic shock, patients randomised to receive levosimendan (0.1 µg/kg/minute) had significantly improved splanchnic perfusion as measured by the indocyanine green plasma disappearance rate than those randomised to receive dobutamine (10 µg/kg/minute). 35
Further evidence of a beneficial effect of levosimendan on renal function in sepsis comes from a case–control study of 99 patients with septic shock who received 0.2 µg/kg/minute of levosimendan for 24 hours within 36 hours of admission to the intensive care unit (ICU). Compared with matched control patients, a 24% increase in glomerular filtration rate at 96 hours (p < 0.05) was seen in patients who received levosimendan together with a lower peak serum creatinine concentration (p < 0.05). 36 Similar beneficial effects of levosimendan on renal function have also been demonstrated in patients with acute heart failure. In 88 patients who had acute decompensated heart failure requiring inotropic therapy, levosimendan compared significantly increased the calculated glomerular filtration rate with dobutamine, with an increase of 45% seen at 72 hours after infusion completion. 37
There are no existing systematic reviews of levosimendan in severe sepsis. However, a recent article reviewing the role of levosimendan in sepsis concluded that ‘large-scale multicenter clinical trials are now needed to clarify whether levosimendan improves the overall outcome of patients with sepsis and septic shock’. 38
Risks and benefits
The potential benefits of levosimendan have been reviewed in the previous sections. Levosimendan has been widely used in patients with acute heart failure, has a good safety profile and has no known significant pharmacokinetic drug interactions. According to the levosimendan investigators’ brochure, between September 2000 (when the drug first received a licence in Sweden) and November 2010, an estimated 440,000 patients were treated with levosimendan, with a reported serious adverse drug reaction rate of 791/≈440,000 (0.2%). The most common events reported were hypotension (0.03%) and serious arrhythmias (0.02%).
Levosimendan has been used in > 200 patients with septic shock in published controlled trials and case series without any reported significant adverse effects. Adequate cardiovascular resuscitation with intravenous fluids and noradrenaline, as well as avoiding an initial bolus dose and high-dose infusion (≥ 0.4 µg/kg/minute), help reduce adverse effects when used in sepsis. 38
Levosimendan is currently used in many ICUs within Europe in the treatment of severe sepsis and septic shock and has recently been recommended as an alternative inotrope in the German Sepsis Society guidelines. 39
As highlighted earlier, septic shock is associated with a high mortality rate and many of the drugs required for its treatment, for example high-dose catecholamine infusions, also have significant risks. Available evidence would suggest that levosimendan has a good safety profile and would not add any additional risk in this population. In fact, levosimendan may reduce the risk associated with standard therapy if catecholamine use is reduced.
Toxicology
Conventional studies on general toxicity and genotoxicity have revealed no special hazard for humans with short-term use of levosimendan. In animal studies, levosimendan was not teratogenic, but it caused a generalised reduction in the degree of ossification in rat and rabbit fetuses, with anomalous development of the supraoccipital bone in the rabbit. Pregnant patients were not included in this trial.
Rationale for study
As summarised earlier, there is a substantial body of research that provides proof of concept that levosimendan improves cardiac output, regional perfusion and other physiological end points, including creatinine clearance and glomerular filtration rate, in patients who have septic shock.
We undertook an exploratory trial, designed to identify the important clinical outcome benefits and to explore the mechanism of action of levosimendan in septic shock. Given that multiple organ dysfunction is associated with an increased mortality rate,40 a reduction in the incidence and severity of organ failure would be associated with meaningful benefits to patients and clinicians alike, along with potential reductions in costs to the NHS. The trial protocol41 and the main outcomes of the trial42 have been previously published.
Chapter 2 Research objectives
Objectives
Primary objectives
The main objectives of this trial were to:
-
ascertain if levosimendan reduces the incidence and severity of organ dysfunction compared with placebo in adult patients who have septic shock
-
identify the effect of levosimendan on individual organ function in septic shock
-
establish the safety profile and pharmacokinetics of levosimendan in this group of patients.
Secondary objectives
The secondary objectives were to:
-
identify whether or not levosimendan reduces the need for, and duration of, catecholamine support and thus reduces myocardial injury
-
establish whether or not levosimendan alters the pro- and anti-inflammatory balance in sepsis
-
collect long-term (3- and 6-month) survival data to help inform the appropriate long-term outcome measure for a subsequent effectiveness trial, should the efficacy of levosimendan be confirmed in this trial.
Chapter 3 Methods
Trial design
This was a multicentre, randomised, double-blind, placebo-controlled study. Eligible patients were randomised within 24 hours of meeting all of the inclusion criteria. Patients were randomised to receive either levosimendan or placebo in addition to standard care. The duration of therapy with the study medication (active or placebo) was 24 hours; data were collected daily up to 28 days while in the ICU, with long-term follow-up at 3 and 6 months after hospital admission.
Table 1 summarises the visit and data collection schedule for the study.
| Visit | Day | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8–28 | |
| Screening | ✓ | |||||||
| Informed consent (patient consent/PerLR/ProLR/retrospective patient information and consent) | Patient/PerLR/ProLR will be obtained initially. Retrospective patient consent will be obtained when the patient has recovered | |||||||
| Inclusion/exclusion criteria | ✓ | |||||||
| Randomisation | ✓ | |||||||
| Study drug administration | Study drug infusion for 24 hours | |||||||
| Follow-up | ||||||||
| Blood and urine sampling | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | Patients in the pharmacokinetic study had additional blood sampling at days 8, 10, 13 and 16 | |
| Daily collection of clinical data | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Final visit | On the day of discharge from hospital | |||||||
Randomisation and blinding
Patients were assigned to either levosimendan or placebo in a 1 : 1 ratio. Randomisation was stratified by ICU in permuted blocks of variable sizes of four and six. The randomisation lists were computer generated by a statistician independent of the LeoPARDS (Levosimendan for the Prevention of Acute oRgan Dysfunction in Sepsis) trial team and held separately at the Imperial Clinical Trials Unit (ICTU). The randomisation lists were imported into the InForm system (version 4.6; Oracle Corporation, Red Wood City, CA, USA) to enable web-based randomisation at sites. Study drugs were labelled and packaged identically by Victoria Pharmaceuticals (Belfast, UK) to preserve blinding. Patients, clinical staff and the research team were blinded to drug allocation with the exception of trial statisticians Greg Frazier, Alexina Mason and Shalini Santhakumaran. The senior statistician, Deborah Ashby, remained blinded throughout the trial.
Participants
Participants in the trial were adult patients who had sepsis and cardiovascular failure requiring vasopressors to maintain blood pressure despite adequate fluid resuscitation.
Inclusion criteria
Inclusion criteria were the internationally established consensus definitions of sepsis. In brief:
-
Fulfil two out of four of the criteria of the systemic inflammatory response syndrome (SIRS) because of known or suspected infection within the previous 24 hours. The SIRS criteria are:
-
fever (> 38 °C) or hypothermia (< 36 °C)
-
tachycardia (heart rate > 90 beats per minute)
-
tachypnoea (respiratory rate > 20 breaths per minute or PaCO2 < 4.3 kPa) or need for mechanical ventilation
-
abnormal leucocyte count [> 12,000 cells/mm3, < 4000 cells/mm3 or > 10% immature (band) forms].
-
-
Hypotension, despite adequate intravenous fluid resuscitation, requiring treatment with a vasopressor infusion (e.g. noradrenaline/adrenaline/vasopressin analogue) for ≥ 4 hours and still having an ongoing vasopressor requirement at the time of randomisation.
Exclusion criteria
-
More than 24 hours since meeting all of the inclusion criteria.
-
End-stage renal failure at presentation (previously dialysis dependent).
-
Severe chronic hepatic impairment (Child–Pugh class C).
-
A history of torsades de pointes.
-
Known significant mechanical obstructions affecting ventricular filling or outflow or both.
-
Treatment limitation decision in place [e.g. DNAR (do not attempt resuscitation) or not for ventilation/dialysis].
-
Known or estimated weight of > 135 kg.
-
Known to be pregnant.
-
Previous treatment with levosimendan within 30 days.
-
Known hypersensitivity to levosimendan or any of the excipients.
-
Known to have received another investigational medicinal product (IMP) within 30 days or currently in another interventional trial that might interact with the study drug.
Treatments
Patients were randomised to either the treatment group or the control group.
The study drug was not started until the treating physician was confident that adequate fluid resuscitation had been achieved and the patient had reached the target MAP (suggested target 65–70 mmHg but this could be varied, as detailed below). Adequate fluid resuscitation was achieved using repeated fluid challenges. Examples of appropriate targets included any or all of the following:
-
central venous pressure ≥ 8 mmHg (≥ 12 mmHg in mechanically ventilated patients)
-
good peripheral perfusion on clinical examination
-
other measures of cardiac output/flow (e.g. stroke volume variability, global end-diastolic volume index).
Treatment group
Patients in the treatment group received all normal standard care plus a 24-hour blinded intravenous infusion of levosimendan. The levosimendan infusion started at 0.1 µg/kg/minute and, if tolerated, this was increased after 2–4 hours to 0.2 µg/kg/minute for a further 20–22 hours (total infusion of 24 hours). Levosimendan can cause vasodilatation. Therefore, it was advised that if there was a mild drop in blood pressure an intravenous fluid bolus should be given (e.g. 250–500 ml), fluid status should be reassessed and treatment given as necessary. The vasopressor dose could then be titrated up if needed once any fluid depletion had been corrected. The infusion rate would then be increased after 2–4 hours once the clinician was satisfied that the drug was well tolerated.
If the dose of 0.2 µg/kg/minute was not tolerated (hypotension despite titration of vasopressors, or severe tachycardia), the rate of infusion was reduced back to 0.1 µg/kg/minute. If there was hypotension or tachycardia at an infusion rate of 0.1 µg/kg/minute (either initially or later) then the rate of infusion was reduced to 0.05 µg/kg/minute. If the hypotension or tachycardia continued then the infusion was discontinued (see Figure 1).
FIGURE 1.
LeoPARDS trial drug infusion protocol. HR, heart rate.
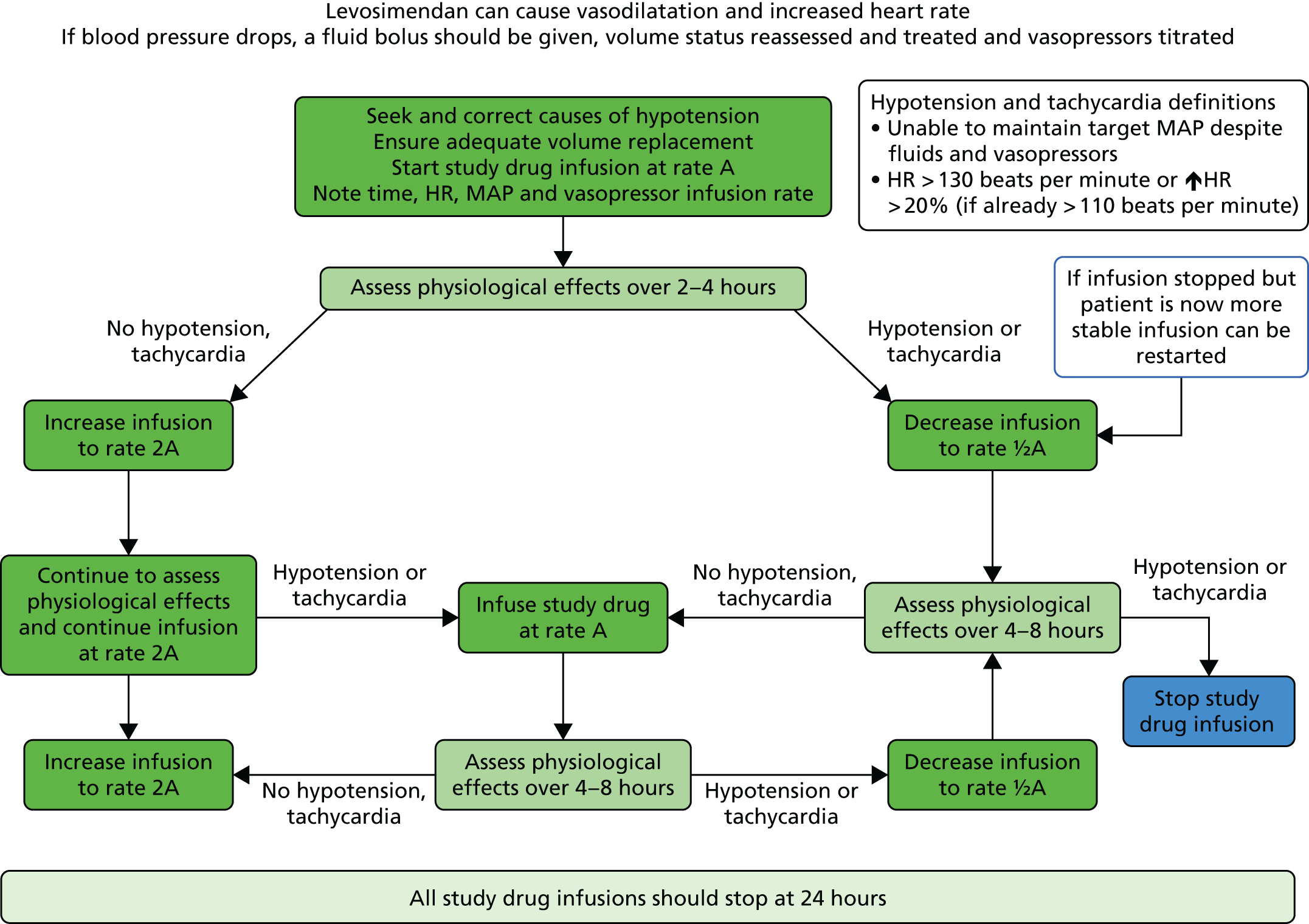
The Summary of Product Characteristics (SmPC) states that an initial bolus of levosimendan should be given followed by a 24-hour infusion of 0.1–0.2 µg/kg/minute (reduced if there is hypotension or tachycardia) when treating acute decompensated heart failure. To avoid hypotension and to maintain safety in this septic shock population during the LeoPARDS trial a bolus dose was never given. The infusion was started at the lower dose to ensure that the drug was well tolerated by patients before being increased to the higher dose. The dose of 0.2 µg/kg/minute has shown clinical benefit in previous septic shock clinical trials and, importantly, has been shown to be safe using detailed global haemodynamic and microcirculatory monitoring. 30–32 The titration of the dose between 0.05 and 0.2 µg/kg/minute helped ensure that patients received an effective dose but that any adverse effects were minimised in each individual patient.
Control group
Patients in the control group received all normal standard care plus a 24-hour blinded intravenous infusion of matching placebo. The placebo infusion rate followed the treatment group regimen.
During the study drug administration period, and especially during the first 6 hours, patients were repeatedly reassessed to ensure adequate fluid resuscitation using any or all of the targets above.
The study drugs were supplied to the ICU by the local pharmacy as specific research study drugs and were stored in separate research stores (e.g. locked boxes/fridges in the ICU). The study drug was drawn up and administered by the bedside critical care nurse (or critical care research nurses). The study drug was prescribed on the patient drug chart by the clinical staff as per the policy of each ICU. Preprinted stickers or preset electronic prescriptions were provided to ensure standardised prescribing, dilution and administration of the drug.
Other treatments
Cardiovascular
Fluids
Crystalloid infusions [e.g. 0.9% saline or compound sodium lactate or Plasma-Lyte® (Baxter Healthcare Ltd, Northampton, UK)] were used for intravenous fluid resuscitation. Starch-containing colloid solutions were not used in view of evidence that they may be associated with adverse outcomes and increased rates of acute kidney injury. 43–45 Gelatin-based solutions and human albumin solutions were allowed as alternative resuscitation fluids.
Fluid resuscitation was given based on repeated assessment of volume status, as detailed above.
Vasoactive drugs: vasopressors
Noradrenaline was the initial vasopressor of choice. After fluid resuscitation it was advised that noradrenaline should be titrated to maintain a target MAP of 65–70 mmHg. In individual patients a higher MAP target could be chosen, for instance if a patient was known to be hypertensive; similarly, in a normotensive patient a lower MAP target could be chosen. However, it was emphasised to investigators that the lowest dose of vasopressor to maintain an acceptable MAP to allow tissue perfusion should always be used.
Vasopressin, or any of its analogues, could also be used as an alternative vasopressor or in addition to noradrenaline.
Vasoactive drugs: inotropes
Additional inotropic agents could be used in either treatment group as clinically indicated (i.e. in the presence of low cardiac output after fluid resuscitation). Dobutamine was the inotropic agent of choice as per the Surviving Sepsis Campaign guidelines,46 but other inotropes, including adrenaline or milrinone, were also allowed. Open-label levosimendan was not to be used in the LeoPARDS trial; however, after it was used at one site, the protocol was amended (v1.3) and it was clarified that open-label levosimendan was not to be used for any patients randomised into the LeoPARDS trial. There was a lack of evidence to recommend a set cardiac output/index target but, in general, an adequate cardiac output to ensure adequate oxygen delivery was to be maintained. It was also advised that a ScvO2 of > 70% should be targeted in the early stages of septic shock management and that dobutamine (or any other inotropes) should be titrated down and patients weaned off once an adequate oxygen delivery was achieved.
Corticosteroids
It was advised that hydrocortisone should be used only for patients who were poorly responsive to vasopressors, that is, who were on high-dose vasopressors, as per the Surviving Sepsis Campaign guidelines. 46 Low doses were used (e.g. 200 mg/day of hydrocortisone in divided doses or as a continuous infusion) and doses were titrated down and patients weaned off once the shock resolved.
Ventilation
A lung-protective ventilation strategy was recommended, that is, 6–8 ml/kg of ideal body weight tidal volume, limiting plateau pressure to ≤ 30 cmH2O, accepting permissive hypercapnia and ensuring adequate levels of positive end-expiratory pressure (PEEP) to prevent extensive lung collapse at the end of expiration.
High-frequency ventilation, neuromuscular blockade, inhaled nitric oxide, prone positioning and extracorporeal membrane oxygenation were all permitted as needed to manage severe hypoxaemia.
Renal support
Continuous venovenous haemo(dia)filtration was the renal replacement therapy (RRT) of choice and was used to treat the recognised complications of renal failure, that is, fluid overload, hyperkalaemia, symptomatic uraemia, drug accumulation and severe acid–base disturbance. High-volume haemofiltration for the management of sepsis (i.e. RRT not to treat kidney failure) was not advised.
Other management
All other general ICU management was based on the latest guidance from the Surviving Sepsis Campaign46 and UK national critical care guidelines [e.g. the ventilator (http://webarchive.nationalarchives.gov.uk/20120118171705/http://hcai.dh.gov.uk/files/2011/03/2011-03-14-HII-Ventilator-Associated-Pneumonia-FINAL.pdf) and central line care bundles (http://webarchive.nationalarchives.gov.uk/20120118171551/http://hcai.dh.gov.uk/files/2011/03/2011-03-14-HII-Central-Venous-Catheter-Care-Bundle-FINAL.pdf)].
Inevitably, there were minor differences in management between different centres but using the Surviving Sepsis Campaign guidelines46 ensured external generalisability of the results. Stratification of randomisation by treating centre also ensured balance of small ICU differences between treatment groups.
Dose modifications for toxicity
The study drug infusion protocol was followed to manage the expected pharmacodynamic effects of levosimendan, in particular vasodilatation and tachycardia.
Follow-up
Participants were followed up daily while on the ICU. Routinely collected clinical data (cardiovascular, respiratory and renal physiological variables as well as haematological, biochemical and microbiological blood test results) were recorded on a daily basis during this time.
Patients were also followed up to ascertain survival status at 28 days post recruitment, at hospital discharge and at 3 and 6 months post recruitment using local hospital clinical records or through the patients’ general practitioners (GPs).
Laboratory evaluations
For blood and urine sampling, 25 ml of blood [≈12 ml in ethylenediaminetetra-acetic acid (EDTA) tubes for plasma, ≈10 ml in plain tubes for serum, ≈2.5 ml in PAXgene tubes for ribonucleic acid) and 10 ml of urine were collected on the day of inclusion (day 1), after 24 hours (day 2) and on days 4 and 6 while still on the ICU. Additional samples for drug-level assays were collected in select patients. Samples were separated locally, frozen according to standardised operating procedures and then sent to the co-ordinating centre in batches for storage and analysis.
End-point management
Outcomes
This section describes the primary and secondary outcomes; further detail of the statistical analysis can be found Statistics and data analysis.
Primary outcome
This trial was designed to fully explore the efficacy and mechanism of action of levosimendan. We therefore examined multiple organ failure, as measured by the Sequential Organ Failure Assessment (SOFA) score, as the primary end point. The SOFA score is the sum of scores relating to six organs. It comprises respiration, coagulation, liver, cardiovascular, renal and central nervous system components, each of which can have values from 0 (normal) to 4 (most abnormal), based on the most extreme values recorded each day. 2 In line with Table S3 in Myburgh et al. ,45 vasopressin treatment (any dose) was assigned a value of 3 for the cardiovascular component. We excluded the central nervous system from the daily calculation after randomisation as sedative drugs prevent an accurate daily assessment in this situation, and it has often been excluded in other septic shock clinical trials. Therefore, the SOFA score ranged from 0 to 20 on each day. Table S1 (see Report Supplementary Material 1) shows the criteria for assigning values. For cardiovascular and renal scores, which have more than one contributing measure, the worst score was used. For the renal component, if urine output was not recorded on a particular day the SOFA component score was not considered missing and was based on the creatinine score only. For data recorded more than once a day on the case report form (MAP and drug dose for the cardiovascular component for the first 4 days), the worst value (i.e. the one leading to the highest SOFA score) was used for each day.
We calculated the SOFA score component attributable to the respiration, coagulation, liver, cardiovascular and renal components for each patient for each day that they were in the ICU, using measurements recorded in the daily data (see Report Supplementary Material 1). We calculated an overall patient SOFA score for each day by adding the five components together. The mean SOFA score during the ICU stay was then calculated by adding the overall patient SOFA scores for all ICU days (up to a maximum of 28 days) and dividing by the number of days. The mean SOFA score in the ICU has been shown to be closely correlated with mortality and its predictive value was similar regardless of the length of stay. 3 This helped solve the ‘truncated by death’ issue as all patients contributed scores while alive in the ICU.
Secondary outcomes
The SOFA score is a composite of several different organ failures and there may be differential effects of levosimendan in different organ systems. Therefore, to gain further insight into the mode of action of levosimendan, we also measured organ-specific outcomes.
Cardiovascular
In all patients we compared oxygen delivery between treatment groups using ScvO2. This was measured and recorded at baseline and 6 and 12 hours and then 12-hourly in all patients with a jugular or subclavian central line for up to 96 hours and then daily to day 5 if the central line remained in situ.
In prespecified ICUs we aimed to measure cardiac output using calibrated devices [e.g. PiCCO (Maquet, Rastalt, Germany), LiDCOplus (LiDCO, London, UK), oesophageal Doppler or pulmonary artery catheter] in all patients included in the study. Cardiac output data were measured and recorded at baseline and 6 and 12 hours and then 12-hourly for up to 96 hours (as long as the device was clinically required).
Renal
In view of the importance of acute kidney injury as an independent determinant of outcome in septic shock and the previous data demonstrating a beneficial effect of levosimendan on kidney function in sepsis we also compared rates of renal failure using the Acute Kidney Injury Network (AKIN) definitions. 47 Each patient was categorised on a daily basis into the increasing stages of renal failure as shown in Table S2 (see Report Supplementary Material 1), with stage 3 defining acute renal failure. The overall score is the worst of the scores from each component, with higher scores indicating poorer renal function.
Abdominal
As poor mesenteric perfusion and bowel ischaemia are believed to be major contributors to the pathogenesis of multiorgan failure in septic shock we analysed bilirubin levels (used to calculate the hepatic SOFA score) over time.
Respiratory
Recent evidence has suggested that levosimendan may also enhance calcium sensitivity in diaphragm muscle and thus improve diaphragm contractility. 48 In patients who required intubation and mechanical ventilation, time to successful liberation from mechanical ventilation was defined as:
-
extubated with face mask, nasal cannulae or room air or
-
T-piece/heat and moisture exchange (HME) filter breathing or
-
tracheostomy mask breathing or
-
continuous positive airway pressure (CPAP) breathing ≤ 5 cmH2O without any pressure support or mandatory ventilation for 48 hours continuously.
We analysed the respiratory end points in three ways:
-
ventilator-free days, defined as:
-
0, for patients who died before extubation
-
days to death minus ventilation days, for patients who died after extubation and before 28 days
-
28 minus the number of ventilation days, for patients who survived to 28 days
-
-
time to extubation, defined as the time to first successful extubation, with deaths treated as still being on the ventilator beyond the end of follow-up (28 days)
-
arterial partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) ratio over time, using data collected at baseline and each day the patient remained ventilated in the ICU (up to 28 days).
Major acute kidney event
A major acute kidney event (MAKE) at a certain time point, for example day 28 or day 90, is a recently suggested alternative renal failure outcome. 49 We applied the version based on 28 days, in line with the daily data collection and other end points.
In the LeoPARDS trial, a patient was defined as having MAKE28 (MAKE 28 days after randomisation) if they experienced any of the following in the 28 days following randomisation:
-
death
-
need for RRT, excluding those who required RRT before randomisation
-
sustained loss of kidney function, defined as acute kidney injury (AKI) stage 2 or 3 at day 28 or on ICU discharge if discharged before day 28.
Other secondary clinical outcomes
Other secondary clinical outcomes were:
-
28-day, ICU and hospital discharge, and 3 and 6-month survival
-
ICU and hospital length of stay, and ICU-free days, defined as the number of days during the 28 days after randomisation that a patient was alive and not in the ICU
-
duration of RRT, defined as the first day of RRT in the ICU to the last day of RRT, including any RRT received post discharge (all days in between were counted, whether or not the patient received RRT)
-
days free from catecholamine therapy, defined as the number of days during the 28 days after randomisation that a patient was alive and did not receive any catecholamine therapy (dobutamine, adrenaline and noradrenaline)
-
organ support data.
Secondary mechanistic outcomes
Serial blood and urine samples were collected from patients, as detailed earlier. A number of assays were considered for analysis including markers of acute kidney injury, myocardial dysfunction, inflammation and intestinal perfusion. Based on the clinical results the final biomarkers assayed included:
-
Markers of myocardial dysfunction:
-
troponin, a widely used marker of myocardial injury and an early prognosticator of poor outcome in sepsis
-
serum brain natriuretic peptide [and N-terminal prohormone of brain natriuretic peptide (NT-proBNP)], demonstrated to be a reliable biomarker of ventricular dysfunction in septic patients and also a prognostic marker for a poor outcome. 50,51
-
-
Biomarkers of systemic inflammation. The original plans had been to use a multiplex inflammatory biomarker bead assay. However, because of discontinuation of one such assay by the manufacturer, no reliable similar alternatives and issues with different dilutions required for different analytes, we selected five key biomarkers to study. These were:
-
the pro-inflammatory cytokine interleukin (IL)-6
-
the chemokines IL-8 and monocyte chemoattractant protein 1 [also known as chemokine ligand 2 (CCL2)] (levosimendan has been shown to reduce levels in sepsis)52
-
the anti-inflammatory cytokine IL-10
-
soluble tumour necrosis factor receptor 1 (sTNFR1), an important soluble receptor that is one of the biomarkers used to characterise a hyperinflammatory phenotype in critical care, which is associated with higher mortality and potentially improved response to anti-inflammatory treatment. 53
-
In addition, samples were stored for subsequent analysis (e.g. genetics/proteomics/metabonomics) in future separately funded projects.
Plasma sample assay methodology
For troponin I, plasma samples were analysed in the clinical laboratories at Imperial College Healthcare NHS Trust. The high sensitive chemiluminescent microparticle immunoassay, Abbott Architect (Abbott Diagnostics, UK), was used. Samples were processed according to normal laboratory procedures.
N-terminal prohormone of brain natriuretic peptide
Plasma samples were quantified using a sandwich enzyme-linked immunosorbent assay (ELISA) kit (Abcam, Cambridge, UK). Samples, on their third freeze–thaw cycle (after measurement of biomarkers; see Inflammatory biomarkers), were run at a 1 : 4 dilution and, when required, were rerun at a 1 : 8 dilution.
Inflammatory biomarkers
Interleukin 6, IL-8, IL-10 and CCL2 were quantified using the ELLA multiplex assay (ProteinSimple™, San Jose, CA, USA). Samples were thawed at room temperature on the first freeze–thaw cycle and were then diluted by 1 : 3 to 1 : 8 in assay buffer and run as per the manufacturer’s instructions. Positive controls of recombinant IL-6, IL-8, IL-10 and CCL2 standards were run alongside to ensure reproducibility. sTNFR1 was assessed using the ELLA Simple Plex assay. Samples were thawed at room temperature on the second freeze–thaw cycle, a minimum of 24 hours after the first cycle. Samples were then diluted 1 : 10 to 1 : 13 and run as specified by the manufacturer’s instructions; positive controls were run alongside.
Pharmacokinetic/pharmacodynamic study
The first 80 patients enrolled in the study (with a full sample set collected) had an additional 3 ml of blood collected while on the ICU on days 2, 4, 6, 8, 10, 13 and 16, for assays of levosimendan and its active metabolites OR-1896 and OR-1855. The assays used liquid chromatography tandem mass spectrometry on a Thermo Finnigan TSQ Quantum™ Ultra device (Thermo Fisher Scientific, Waltham, MA, USA) and were carried out at LGC Group (Fordham, Cambridge, UK); this analysis was paid for by Orion Corporation. A non-compartmental analysis of pooled data was undertaken. The population area under the concentration–time curve (AUC) and the maximum concentration (Cmax) were estimated. The AUC was estimated using the trapezoid rule and the Cmax was estimated from visual inspection. We then compared the AUC and the Cmax between patients with and without acute renal failure, acute liver failure and requirement for RRT and also with previous pharmacokinetic data from other studies. Analysis was performed using GraphPad Prism version 7.00 (GraphPad Software Inc., San Diego, CA, USA).
Data collection
Electronic case record form
Data management was carried out using the InForm ITM (Integrated Trial Management) system, a web-based data entry system that builds an Oracle database for each individual clinical trial. Trial data were captured on a bespoke web-based electronic case record form (eCRF) with built-in validation rules to identify data entry errors in real time and a full audit trail of data entry and changes. All those entering data were trained prior to start-up and given personal login details, with access to forms restricted according to site and role. The eCRF was designed in accordance with the requirements of the trial protocol, and access to the eCRF was password protected and included controlled level of access. A full list of the data collected on each form is provided in Appendix 1.
Pharmacovigilance definitions and procedures
Definitions
An adverse event (AE) was defined as any untoward medical occurrence in a patient or clinical trial subject administered a medicinal product and which does not necessarily have a causal relationship with this treatment. An AE can therefore be any unfavourable and unintended sign (including an abnormal laboratory finding), symptom or disease temporally associated with the use of an IMP, whether or not considered related to the IMP.
An adverse reaction (AR) was defined as an untoward and unintended response to an IMP related to any dose administered. All AEs judged by either the reporting investigator or the sponsor as having a reasonable causal relationship to a medicinal product qualified as adverse reactions. The expression ‘reasonable causal relationship’ means to convey in general that there is evidence or an argument to suggest a causal relationship.
An unexpected AR was defined as an AR, the nature or severity of which is not consistent with the applicable product information (e.g. investigator’s brochure for an unapproved investigational product or SmPC for an authorised product). When the outcome of an AR is not consistent with the applicable product information this AR should be considered as unexpected. Side effects documented in the SmPC that occur in a more severe form than anticipated are also considered to be unexpected.
A serious adverse event (SAE) or serious AR was defined as any untoward medical occurrence or effect that at any dose:
-
results in death
-
is life-threatening – refers to an event in which the subject was at risk of death at the time of the event; it does not refer to an event that hypothetically might have caused death if it were more severe
-
results in hospitalisation or prolongation of an existing inpatient hospitalisation
-
results in persistent or significant disability or incapacity
-
is a congenital anomaly or birth defect.
Medical judgement was exercised in deciding whether or not an AE/AR was serious in other situations. Important AEs/ARs that were not immediately life-threatening or that did not result in death or hospitalisation but may have jeopardised the subject or have required intervention to prevent one of the other outcomes listed in the definition above were also considered serious.
A suspected unexpected serious adverse reaction (SUSAR) was defined as any suspected AR related to an IMP that was both unexpected and serious.
Causality
The assignment of the causality was made by the investigator responsible for the care of a participant using the definitions in Table 2.
| Relationship | Description |
|---|---|
| Unrelated | There is no evidence of any causal relationship |
| Unlikely | There is little evidence to suggest that there is a causal relationship (e.g. the event did not occur within a reasonable time after administration of the trial medication). There is another reasonable explanation for the event (e.g. the participant’s clinical condition, other concomitant treatment) |
| Possible | There is some evidence to suggest a causal relationship (e.g. because the event occurred within a reasonable time after administration of the trial medication). However, the influence of other factors may have contributed to the event (e.g. the participant’s clinical condition, other concomitant treatments) |
| Probable | There is evidence to suggest a causal relationship and the influence of other factors is unlikely |
| Definitely | There is clear evidence to suggest a causal relationship and other possible contributing factors can be ruled out |
| Not assessable | There is insufficient or incomplete evidence to make a clinical judgement of the causal relationship |
If there was any doubt about the causality, the local investigator informed the study co-ordination centre, which notified the chief investigator. In the case of discrepant views on causality between the investigator and others, all parties discussed the case, but the final decision was made by the local investigator.
Reporting procedures
Depending on the nature of the event the subsequent reporting procedures were followed. Any questions concerning AE reporting were directed to the study co-ordination centre in the first instance.
Non-serious adverse reactions/adverse events
All such toxicities, whether expected or not, were required to be recorded on the AE section of the eCRF form within 1 month of the form being due.
Serious adverse reactions/adverse events
Fatal or life-threatening SAEs were required to be reported on the day that the local site became aware of an event. The SAE form asked for the nature of the event, date of onset, severity, corrective therapies given, outcome and causality (i.e. unrelated, unlikely, possible, probably, definitely). The responsible investigator assigned the causality of the event. Additional information was sent to the study co-ordination centre within 5 days of notification if the reaction had not resolved at the time of reporting.
A SAE form was completed and entered into the eCRF, which automatically sent e-mail alerts to the chief investigator, the trial manager and the sponsor. However, relapse, organ failure and death related to sepsis (see following definitions), and hospitalisations for elective treatment of a pre-existing condition, did not need to be reported as a SAE.
Clinical outcomes
Clinical outcomes from sepsis were exempt from AE reporting, unless the investigator deemed an event to be related to the administration of the study drug. The following events were considered clinical outcomes:
-
death related to sepsis
-
cardiovascular failure, including the need for vasopressors/inotropes
-
respiratory failure, including mechanical ventilation and acute lung injury
-
hepatic failure
-
renal failure, including the need for RRT
-
haematological/coagulation failure, including thrombocytopaenia.
Clinical details about these clinical outcomes were routinely collected on the case record form.
In relation to the study drug in this trial the following specific SAEs were recorded on the eCRF:
-
myocardial infarction/acute coronary syndrome
-
life-threatening arrhythmia (e.g. ventricular fibrillation, ventricular tachycardia or atrial fibrillation that leads to hypotension).
Suspected unexpected serious adverse reactions
In the case of SUSARs, the staff at sites were requested to complete the SAE eCRF (within 24 hours). The study co-ordination centre would then be notified by e-mail and would contact the local site for further information.
The study co-ordination centre would then notify the Medicines and Healthcare products Regulatory Agency (MHRA), the Research Ethics Committee (REC) and the sponsor of all SUSARs occurring during the study according to the following timelines: fatal and life-threatening – within 7 days of notification; non-life threatening – within 15 days. All investigators would have been informed of all SUSARs occurring throughout the study.
Local investigators were to report any SUSARs and/or SAEs as required by the protocol and study-specific Standard Operating Procedures (SOPs). There were no SUSARs in the LeoPARDS trial.
Annual safety reports
Annual safety reports were provided to the REC and MHRA, in accordance with clinical trial regulations, on the anniversary of the clinical trial authorisation each year. A total of three annual safety reports were submitted over the course of the trial.
Emergency identification of study medication/unblinding
Prior to any unblinding the site must have made a concerted effort to contact the chief investigator, his deputy or the trial manager.
Sealed envelopes containing the login and password for unblinding from the InForm ITM system were provided to participating ICUs. Sealed envelopes would not be opened under normal circumstances and the integrity of the envelopes was checked at every monitoring visit by the trial monitor.
To unblind a patient, the treating physician/principal investigator (PI) would contact the chief investigator or trial manager to discuss the need for, and obtain approval for, unblinding. If the need for unblinding was agreed, the treating physician/PI would open the sealed envelope. The enclosed login and password would then be used to unblind the patient using the InForm database. A File Note would then be completed detailing the circumstances of the unblinding. After the sealed envelope containing the unblinding instructions had been opened to unblind a patient, a new sealed envelope would be provided to the site by the trial manager/monitor. In the event that the InForm database was inaccessible, once the site and the study co-ordinating centre had agreed that unblinding was the best course of action, the Charing Cross Hospital on-call pharmacist could be contacted. The on-call pharmacist had access to the main trial unblinding list and would be able to expose the unblinded information.
One patient was unblinded in the LeoPARDS trial, after completion of the recruitment phase of the trial but before the statistical analysis of the data took place and just prior to database lock. In this case, the patient had been randomised into the trial, recovered from their episode of septic shock, left the ICU and later that year passed away in hospital. The patient’s family had raised a complaint against the hospital trust and the care and subsequent death of the patient were being reviewed. The independent medical team had requested to know whether the patient had received the active drug or the placebo.
Data Monitoring and Ethics Committee
An independent Data Monitoring and Ethics Committee (DMEC) was set up to monitor progress, patient safety and any ethical issues involved in this trial. The DMEC reviewed trial progress, recruitment rates, event rates and safety data. A separate charter was drawn up defining its exact remit and criteria for reporting to the Trial Steering Committee (TSC). There were 6-monthly meetings of the independent DMEC.
Early discontinuation
Withdrawal criteria
Patients were free to withdraw at any stage of the study.
If a patient wished to withdraw from the study during the treatment period, the treating physician would no longer follow the trial protocol and the study drug would have been stopped. The patient’s data may or may not have been included in the final analysis, according to the patient’s wishes.
If a patient wished to withdraw from the study after the treatment period, no further data would be collected. The patient’s data may or may not have been included in the final analysis, according to the patient’s wishes.
Loss to follow-up
Patients were followed up post hospital discharge using hospital data provided by each research team and so loss to follow-up was low. If patients could not be traced using this system they were contacted through their GP.
Statistics and data analysis
All patients who were randomised were included in the analysis where possible, unless consent to use the data was withdrawn. For all outcomes, the primary analysis was carried out on an intention-to-treat (ITT) basis. The analysis of the primary outcome and safety data was repeated on an as-treated basis, excluding patients who did not receive any study drug. For all statistical modelling, the validity of the underlying assumptions was checked and any resulting alterations to the model specifications were documented. A full statistical analysis plan (SAP) was developed by the trial investigators and trial statisticians and reviewed and agreed by the TSC (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408).
Missing data
For all measures, the number of patients with missing data was described by drug allocation. We considered whether or not the level and type of missing data had the potential to affect the estimates of interest by introducing bias or reducing precision.
Daily data were recorded as clinically indicated for up to 28 days while patients were in the ICU. A decision not to take a measurement usually reflected the clinical judgement that there had been no change in that variable or the patient was getting better. In the pilot study for the Vasopressin vs Noradrenaline as Initial therapy in Septic Shock (VANISH) trial,54 only a small percentage (4%) of SOFA scores had missing values, and a similar level of missingness was expected for the LeoPARDS trial. For the previous pilot study, missing data were imputed as the last recorded value [last observation carried forward (LOCF)] as it was reasonable to assume that the data were not collected because no change was expected by the clinician. Therefore, for longitudinal outcomes we took the following approach:
-
When there were only 1 or 2 consecutive days of data missing, or when the missing data occurred at the end of follow-up, measurements were imputed using the LOCF.
-
Where there were ≥ 3 days of data missing, the average value of the last available and next available observation was used as the imputed value.
-
If the first day was missing data the value from day 2 was taken.
-
If the values for days 1 and 2 were both missing the baseline value was taken.
If the level of missingness was substantially higher than anticipated or the assumptions governing the approach described earlier were thought to be unreasonable, we planned to conduct sensitivity analyses using Bayesian methods. 55 This was required for the total SOFA score and its components; methods are described further in Primary outcome.
Data management
In addition to the validation rules built into the InForm system (see Data collection) and trial monitoring activities (see Trial management, Monitoring), the trial statistician conducted checks on the data exported from the InForm system. This included checking for discrepancies across forms, inconsistent dates and times, duplicate entries, outliers and missing data. Data and all appropriate documentation will be stored for a minimum of 10 years after the completion of the study, including the follow-up period.
Baseline data and other longitudinal monitoring
Baseline characteristics are described by arm and overall, using the median and interquartile range (IQR) for continuous variables and the number and percentage in each group for categorical variables. The following clinical measures were monitored at varying intervals during the ICU stay:
-
MAP
-
central venous pressure
-
heart rate
-
ScvO2
-
cardiac index
-
platelet count
-
bilirubin
-
lactate
-
arterial oxygen saturation (SaO2)
-
haemoglobin
-
total intravenous fluid administered
-
fluid balance
-
PaO2/FiO2 ratio
-
creatinine
-
total urine output.
These measures are described longitudinally by treatment group using box plots (see Figures 3–14). Note that day 1 is slightly different from the other days as it ran from the time of randomisation to when the next ICU chart was competed, so day 1 is almost always < 24 hours. Subsequent days are all 24-hour periods until the day of discharge. Cardiovascular measures are additionally presented by subgroup as follows. The cardiac index is presented for two groups according to baseline cardiac output (lowest tertile and middle and highest tertiles combined) and ScvO2 is presented for three groups according to baseline ScvO2 [low (< 70%), normal (70–85%) and high (> 85%)]. We planned to investigate treatment differences over time for cardiac output, ScvO2, bilirubin and PaO2/FiO2 ratio (see Primary outcome, Subgroup analysis and Secondary outcomes).
To assess how well the study drug was tolerated, the number of subjects receiving the study drug at 6, 12 and 24 hours was tabulated by dose level and treatment group. The number of subjects receiving noradrenaline and dobutamine, and the median and IQR of the dose received, are described by treatment group.
Safety data
Adverse events were summarised by seriousness, relationship to study medication and treatment group. This information was shown as the number of AEs (subjects could contribute more than one AE) and the number of subjects (each subject is shown only once, using the AE with the most serious classification and the highest level of causality).
Using the descriptions recorded on the AE and SAE report forms, each SAE was assigned to one of the following categories:
-
myocardial infarction/acute coronary syndrome
-
life-threatening arrhythmia (e.g. ventricular fibrillation, ventricular tachycardia or atrial fibrillation that leads to hypotension)
-
other.
Furthermore, SAEs were grouped and tabulated by treatment according to organ system:
-
cardiovascular/circulatory
-
digestive/gastrointestinal
-
nervous
-
respiratory
-
urinary/excretory
-
musculoskeletal
-
skin/hair/nails
-
other.
Adverse events by category and organ system were reported for each treatment group.
Primary outcome
The primary outcome was the mean SOFA score on the ICU up to 28 days after randomisation. The score was calculated as described in End-point management, Primary outcome.
Exploratory analysis
To explore the distribution of the primary outcome, box plots of the total SOFA score by day and treatment arm were produced, along with bar plots by component up to day 7.
The calculation of the mean SOFA score requires a total SOFA score for each day that the patient is in the ICU. To assess the level of missingness, we tabulated the percentage of subjects with at least one missing score during the ICU stay and the percentage of daily scores that were missing. This was carried out for the total SOFA score and for each component separately.
Main analysis of the primary outcome
The mean total SOFA score and its components are presented as means and standard deviations (SDs) and medians and IQRs by treatment group. The treatment difference was the unadjusted mean difference in the total mean SOFA score between the levosimendan arm and the placebo arm, with a 95% confidence interval (CI). Although randomisation was stratified by site, as there were a lot of sites, many of which recruited only a handful of patients, including site as a covariate did not appear to be analytically sensible. Instead, we allowed for any correlation of outcomes between patients at the same site by including a random intercept in the secondary analysis (see Adjusted analysis). Histograms and Q–Q plots were used to check the assumption that the distribution of the total SOFA score was approximately normal. As this was not the case, the CIs for the means in the primary analysis were calculated using bootstrapping, applying the percentile method with 100,000 samples. In addition, the difference in medians is also presented and for the total SOFA score the square root transform was applied, although lack of a back transform meant that this result was less clinically meaningful. To provide quality assurance, the main analysis of the primary outcome was repeated by another statistician at the ICTU using different software (Stata, version 13.1; StataCorp LLC, College Station, TX, USA), including calculation of the SOFA scores from the raw data.
Sensitivity analysis of the primary outcome
We conducted a series of sensitivity analyses, as detailed in the following sections.
Omitting the cardiovascular component
The main analysis was repeated but omitting the cardiovascular component from the SOFA score. The cardiovascular component of the SOFA score is based largely on the dose of catecholamine given to treat shock. Levosimendan has a similar mode of action (improving cardiovascular function) but is not included within the cardiovascular SOFA score. It was expected that the patients treated with levosimendan would need less catecholamine therapy. As the levosimendan therapy is not recorded within the SOFA score this might have resulted in a spurious lowering of the cardiovascular component of the SOFA score.
Adjusted analysis
Regression models were used to check whether or not the main analysis result was sensitive to adjustment for age and severity of illness (as indicated by the APACHE II score at baseline). Adjustment for age and APACHE II score is typically included in the analysis of data from critical care trials. A random intercept model was used to check whether or not the results were sensitive to clustering by ICU.
Bayesian analysis
The overall level of missingness across SOFA components was 6.2%, higher than the 4% expected. LOCF methods may underestimate uncertainty so, as specified in the SAP (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408), to check the robustness of the primary result a sensitivity analysis was conducted, implementing Bayesian models using Markov chain Monte Carlo (MCMC) methods that differed from the main analysis in two ways. First, the imputed values varied with each MCMC iteration, reflecting the additional uncertainty of missing values. Second, the assumptions governing the imputed values differed. An autoregressive process resulting in imputed values similar to the adjacent values was used and assumptions reflecting the clinical expectation that missing values were more likely to be normal were incorporated. Similar to the main analysis, a bootstrap approach was used because of the non-normal distribution of the daily total SOFA scores, with a separate bootstrap sample taken at each MCMC iteration. Further details of the methods are provided in Report Supplementary Material 1.
Post hoc sensitivity analysis
Any differential effect of treatment on ICU discharge or death could affect the comparison of mean SOFA scores between groups. Two further imputation approaches were conducted to investigate this. First, the last recorded score was carried forward to all days after ICU discharge or death, up to day 28. Second, all days alive outside the ICU were assigned the minimum possible score of zero and all days dead were assigned the maximum score obtained by the patient in question. Note that these analyses were conducted post hoc in response to comments from reviewers at publication stage.
Subgroup analysis
The following a priori-defined subgroup analyses were carried out for the primary outcome:
-
cardiac index – using the baseline cardiac index, we split patients into two groups: lowest tertile and middle and highest tertiles combined
-
ScvO2 – using baseline ScvO2, we split patients into three groups: low (< 70%), normal (70–85%) and high (> 85%)
-
lactate – using baseline lactate level, we split patients into two groups: normal (≤ 2 mmol/l) and raised (> 2 mmol/l)
-
noradrenaline – using baseline noradrenaline level, we split patients into two groups: above and below the median.
The results are presented in the same way as for the main analysis of the primary outcome. In addition, a forest plot is used to display the results and, as a post hoc analysis, a permutation test was used to investigate subgroup heterogeneity. 56
Secondary outcomes
Cardiovascular
For both ScvO2 and the cardiac index, we used hierarchical regression models to take into account the structure in the data, incorporating random effects to allow for repeated measurements from individuals. To allow the effect of treatment to vary over time, treatment, time and the interaction between treatment and time terms were included. Mathematical and implementation details are provided in the SAP (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408).
For the cardiac index the difference between groups was summarised as the difference between the AUCs on the log scale. We also calculated the ratio comparing levosimendan and placebo on the original scale, at each time point and averaged over all time points using the geometric mean. In the SAP (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408) we planned two analysis populations: first, patients in the subgroup of ICUs in which ≥ 75% of the trial participants were monitored and, second, all patients, with the latter yielding a larger sample although possibly not representative of the whole population. As only two ICUs contributing 19 patients met the criterion for the first analysis, only the second analysis was carried out.
Renal
For the renal assessment, we chose to focus on renal failure on day 14. This choice was based on simulation with the aim of maximising power to detect a treatment difference (see Power calculations). Full details of the simulation study were included in the SAP (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408). On day 14, each patient was categorised into increasing stages of renal failure according to the international AKIN definitions (0, 1, 2, 3) (see Table S2, Report Supplementary Material 1). If the intervention reduced death, excluding those who had died by day 14 might lead to the appearance of increased illness severity in the treatment group as more patients would have been alive but likely to have had a high AKIN score. Patients who died on or before day 14 were included and classified as stage 4, as death is a worse outcome than renal failure. Patients who were discharged alive from the ICU were assumed to have a score of 0 unless they received RRT post discharge.
This created five-group ordinal categorical data, that is, 4 is worse than 2 but not necessarily twice as bad. Rather than modelling the (log) odds of having the event, as for binary data, we modelled the (log) odds of being a category j or higher, with j = 1, 2, 3 or 4 for the renal failure data. The result of interest is the ratio of these odds in the levosimendan and placebo arms. We can assume that the OR is constant across values of j (a proportional odds model) or independent across j (a non-proportional odds model). Alternatively, we can constrain the ORs to be constant over some but not all values of j (a constrained proportional odds model). This approach was consistent with our expectation that levosimendan would have a greater impact on the transition between the different levels of renal injury/failure (scores 0–3) than between these stages and death (score 4). This assumption was explored using simulation as documented in the SAP (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408) and was found to give the best fit using simulated data with parameters derived from published data and data from the three ICUs within the Imperial College Healthcare NHS Trust. Therefore, the ordinal categorical data for renal failure on day 14 were modelled using a constrained non-proportional odds model, such that the log ORs are assumed to be constant across all of the cumulative probabilities except the last, death. We additionally present results from the proportional and non-proportional models for comparison.
We also present the number and proportion of patients receiving any RRT and the median and IQR for the duration of RRT, separately for all patients and for those receiving some RRT.
Respiratory
Ventilator-free days
Because of the non-parametric distribution of ventilator-free days, we present the number and proportion of patients in each treatment group with no days free and with 28 days free, as well as the absolute difference in proportions and 95% CI. As a summary of the whole population, we present the medians and IQRs, testing the difference between groups using a Mann–Whitney test and presenting the absolute difference in medians with the bootstrapped 95% CI.
Time to extubation
Time to extubation was illustrated using Kaplan–Meier plots and analysed using a Cox proportional hazards model. We present both an unadjusted model and a model adjusted for age, APACHE II score and ICU effects, similar to the regression analysis for the primary outcome.
Arterial partial pressure of oxygen/fraction of inspired oxygen ratio
An appropriate model for the PaO2/FiO2 ratio is a patient-specific random-effects model to allow for repeated measurements per patient. It is plausible that respiratory function is correlated with dropout as a result of death or discharge, potentially leading to biased estimates from the longitudinal model. Therefore, we chose to model the longitudinal respiratory data jointly with survival in order to explicitly acknowledge the underlying relationships and minimise the risk of bias. Following the proposal of Henderson et al. ,57 the patient-specific random effects are linked to the survival model using three parameters modelling the association between the survival time and (1) the intercept, (2) the slope and (3) the current subject-specific random effects. Based on data from the VACS (Vasopressin And Corticosteroids in Septic Shock)58 and VANISH54 trials, for the PaO2/FiO2 ratio we chose a hierarchical model with correlated intercepts and slopes, assuming a linear relationship with time, and for the survival data we chose a Weibull model. Full methods are provided in the SAP (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408).
A number of amendments were made in order to achieve model convergence: PaO2/FiO2 ratio measurements, including at baseline, were mean-centred and standardised, the effect of the baseline measurement was included as a separate parameter, and the N(0,100) prior was used for the parameters linking the longitudinal and survival models and for the intercept and treatment effect in the survival model. This prior has a narrower range than the N(0,10000) prior proposed in the SAP (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408), but it still supports a wide range of reasonable values.
We report:
-
the probability that the PaO2/FiO2 ratio improves more quickly for patients receiving levosimendan than for patients receiving placebo during their stay in the ICU
-
the mean change per day in PaO2/FiO2 ratio for patients receiving levosimendan
-
the mean change per day in PaO2/FiO2 ratio for patients receiving placebo
-
the difference in the mean change in PaO2/FiO2 ratio between patients receiving levosimendan and those receiving placebo over (1) 1 day, (2) 7 days (the length of time that the active metabolite of levosimendan is present in plasma) and (3) the mean length of stay (calculated across both arms).
A series of sensitivity analyses were carried out by repeating the above analysis as follows:
-
restricting the PaO2/FiO2 ratio data to (1) the first 7 days, (2) the first 14 days and (3) the first 21 days
-
incorporating age and APACHE II score at baseline into the measurement and survival models as explanatory variables
-
adjusting for ICU effects by adding a third level to the measurement model (PaO2/FiO2 ratios modelled within patients within ICUs).
Liver
Analogous to the analysis of the PaO2/FiO2 ratio, we jointly modelled bilirubin level as a patient-specific random-effects model and death using a survival model. However, unlike the PaO2/FiO2 ratio, bilirubin has a markedly skewed distribution, so we used a logarithmic transform to better comply with the assumption of normal errors. Similar modifications were made to achieve convergence: the log baseline measurements were mean-centred and standardised, the effect of the baseline measurement was included as a separate parameter and, in the three sensitivity analyses restricted to data from the first 7, 14 and 21 days, a N(0,100) prior was used for the parameters linking the longitudinal and survival models.
Other secondary clinical outcomes
For the outcome of MAKE28, we present the number and proportion of patients in each arm experiencing the event. The treatment difference was described using the risk difference and 95% CI and using logistic regression adjusted for age, APACHE II score and ICU effects.
For survival outcomes we present Kaplan–Meier plots and describe the treatment difference using the hazard ratio from Cox regression models, both unadjusted and adjusted for age, APACHE II score and ICU effects. We also present the number and proportion of patients in each arm experiencing the event, together with the risk difference and 95% CI. This was repeated for the subgroups for the primary outcome (see Primary outcomes, Subgroup analysis) as a post hoc analysis.
For ICU-free days, we took a similar approach as for ventilator-free days, presenting the number and proportion of patients in each treatment group with no free days and the absolute difference in proportions with the 95% CI. As a summary of the whole population, we present the medians and IQRs, testing the difference between groups using a Mann–Whitney test and presenting the absolute difference in medians with the bootstrapped 95% CI. We analysed days free of catecholamine therapy in a similar manner but did not plan a formal hypothesis test.
The median and IQR for length of stay and organ support days are described by treatment arm and for the whole trial population. Length of stay was analysed separately for survivors and non-survivors. No formal statistical comparisons or tests were conducted.
Biomarker data
Seven biomarkers were analysed: one marker of myocardial dysfunction (NT-proBNP), one marker of myocardial injury (troponin I) and five markers of the systemic inflammatory and anti-inflammatory response (IL-6, IL-8, IL-10 and CCL2 and sTNFr1). Samples were taken at baseline (date of study entry) and on days 2, 4 and 6. The number of measurements at each time point was described and box plots produced over time.
The mean total SOFA score (both including and excluding the cardiovascular component), the mean SOFA component scores and 28-day mortality were analysed by subgroups of NT-proBNP and troponin values. The study population was split in two ways:
-
normal compared with high values [upper limit of normal values: 34 ng/l for troponin (Imperial College Healthcare NHS Trust) and 2000 pg/ml for NT-proBNP, according to NICE guidelines59]
-
below and above the median value in the study population.
For all biomarkers, we used Bayesian hierarchical regression models to investigate changes in biomarker levels over time and whether or not trajectories differed for levosimendan and placebo patients. A random intercept term was used to allow for the correlation of multiple measures per patient, with a treatment × time interaction to model differing trajectories in the treatment groups, and adjustment was made for the baseline values of the biomarker. All biomarkers, including baseline values, were log transformed to better comply with the assumption of normal error terms. As this analysis was exploratory we present models both with and without the treatment × time interaction. Sensitivity analysis was performed adjusting for age and APACHE II score at baseline and allowing for clustering by ICU with a further level of random effects. Full details can be found in the SAP (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408) for biomarker analysis.
To describe the effects of levosimendan we present:
-
the estimated change in biomarker levels per day for levosimendan and placebo patients
-
the probability of a faster reduction in biomarker levels in the levosimendan group than in the placebo group
-
the estimated treatment difference on days 2, 4 and 6.
Power calculations
Our sample size was 500 patients. This would provide > 90% power to detect a 0.5-point difference in mean SOFA score assuming a SD of 1.5. 3 In this previous validation study a 1-point rise in mean SOFA score was associated with a significant mortality increase (mean SOFA score 2.1–3.0 = 20% mortality rate, 3.1–4.0 = 36.1% mortality rate and 4.1–5.0 = 73.1% mortality rate; OR 3.06, 95% CI 2.36 to 3.97). We recruited an additional 3% (16 patients) to account for the potential loss to follow-up and withdrawal of consent, as seen in previous UK ICU trials. 60 Hence, 516 patients were recruited, with the goal of obtaining approximately 250 evaluable patients per treatment arm.
Renal outcome
Because of the non-parametric nature of the renal outcome data, calculation of the power for the renal outcome was undertaken using simulation with multistate modelling. The event rates and frequencies of moving between the states of no renal failure, developing renal failure, recovery and death were informed from mortality and organ failure data from the VASST (Vasopressin And Septic Shock Trial),61–63 the CORTICUS (Corticosteroid Therapy of Septic Shock) trial,64 local data from the three adult ICUs within Imperial College Healthcare NHS Trust and the recently completed septic shock study by Gordon et al. 58
Patients were classified according to the three stages of renal dysfunction as per the AKIN definitions47 and data were analysed as ordinal categorical data. Death, which is clearly the worst outcome, was taken into account by classifying it as stage 4, as detailed earlier. We compared the ordinal categorical data for death with acute renal failure at day 14. Based on a range of different transition probabilities we have 65–90% power to detect a 25–35% improvement with levosimendan (previous studies have demonstrated a 24–64% improvement in renal function).
Cardiovascular outcome
For oxygen delivery, 500 patients would provide > 95% power to detect a 5% difference in ScvO2, assuming a SD of 15%. 63
Respiratory outcome
More than 90% of patients who have septic shock will require intubation and mechanical ventilation. Time to final successful liberation from mechanical ventilation was compared between the two treatment groups using survival analysis. A total of 450 patients (90% of 500) would provide 80% power to detect a hazard ratio of 1.4, based on the assumption that, overall, 63% of patients will be successfully liberated from mechanical ventilation by day 28.
The DMEC, including an independent statistician, was instructed within its charter to check these planning assumptions and undertake an interim sample size review.
Treatment
Investigational medicinal product details
Orion Corporation Orion Pharma supplied the study drugs for this trial (Table 3).
| Drug name | Dosage | Description |
|---|---|---|
| Levosimendan | 0.05–0.20 µg/kg/minute | Supplied as 8-ml vials with a nominal filling volume of 5 ml containing 12.5 mg of levosimendan and inactive ingredients (povidone, anhydrous citric acid and anhydrous ethanol) |
| Placebo | 0.05–0.20 µg/kg/minute (equivalent) | Supplied as 8-ml vials with a nominal filling volume of 5 ml containing riboflavin, sodium phosphate, anhydrous ethanol and water |
Labelling, storage and dispensing
Orion Corporation were responsible for assuring that the quality of all IMPs was adequate for the duration of the trial and in compliance with the Good Manufacturing Practice standards (www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001205.jsp&mid=WC0b01ac0580027088).
Levosimendan and the matched placebo were imported from Finland. All study drugs were packaged, labelled to meet the MHRA requirements and distributed to sites by Victoria Pharmaceuticals. The study co-ordination centre kept accurate records of supply to trial centres and destruction of unused IMPs at the end of the trial.
It was each trial centre’s responsibility to ensure that accurate records of IMPs dispensed and returned were maintained and reported to the study co-ordination centre. It was the PI’s responsibility to ensure that accurate records of IMP prescriptions were maintained. The study co-ordination centre tracked supplies of IMPs via information from Victoria Pharmaceuticals and site IMP tracking documents. At the completion of the trial, the study co-ordination centre, via the trial monitor, ensured the destruction of all returned dispensed IMPs (after close-out and before archiving).
Accountability
Hospital pharmacies were responsible for recording study drugs dispensed to the ICU. Preparation of all drug infusions was recorded on the ICU drug accountability form and drug administration was recorded on each patient’s prescription chart. The study pharmacies included a sheet on which the fate of all ampoules was recorded (infused, opened but not infused, unused). At the end of the study all remaining unused drugs were returned to the hospital pharmacies for recording and destruction.
Administrative matters
Source data
Source documents included original documents related to the trial, medical treatment and the history of the participants.
Electronic data capture
The principal means of data collection from participant visits was electronic data capture (EDC) via the internet. Data were entered into the EDC system by site personnel. All source data recorded on the eCRF were signed by the PI or his or her appropriate designee. All changes made following the electronic signing had an electronic audit trail with a signature and date. Specific instructions and further details were outlined in the CRF manual.
Trial management
The UK Clinical Research Collaboration (UKCRC)-registered ICTU was responsible for trial management, quality assurance, trial statistics and development and maintenance of the trial database. A dedicated trial manager and clinical trial monitors were appointed through the ICTU to oversee the day-to-day management and monitoring of the project from set-up to close.
Trial sponsor
The sponsor of the trial was Imperial College London. Imperial College London signed a clinical trial agreement with each of the participating centres prior to the start of recruitment at each centre.
Ethics considerations
The trial was conducted in accordance with principles of the Declaration of Helsinki65 on research involving human subjects. The study protocol, patient information sheet and consent form were submitted to the REC prior to the start of the study and a favourable opinion was obtained on 26 April 2013.
Consent
Patients, personal legal representatives and professional legal representatives were given the patient information sheet and sufficient time to consider participation, and discussed the trial with the research staff prior to consent and enrolment. Full written informed consent was taken using the ethics-approved consent form.
Research governance
The trial was carried out in accordance with the NHS Research Governance Framework (www.hra.nhs.uk/planning-and-improving-research/policies-standards-legislation/uk-policy-framework-health-social-care-research/) and local NHS permission was granted by the research and development departments at each participating site prior to recruitment commencing.
Regulatory requirements
As a randomised trial of an IMP, the LeoPARDS trial was conducted in accordance with the European Clinical Trials Directive (https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-/dir_2001_20/dir_2001_20_en.pdf) and the Medicines for Human Use (Clinical Trials) Regulations 2004 (www.legislation.gov.uk/uksi/2004/1031/pdfs/uksi_20041031_en.pdf), as well as the International Conference of Harmonisation (ICH) Good Clinical Practice (GCP) guidelines (www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf). The trial received clinical trial authorisation from the MHRA on 28 August 2013 and was registered in the European Community with a EudraCT number of 2012-005159-18.
Trial registration
The trial was registered on the ClinicalTrials.gov clinical trial database with the following reference: ISRCTN12776039.
National Institute for Health Research Clinical Research Network portfolio
The LeoPARDS trial was adopted on the National Institute for Health Research (NIHR) Clinical Research Network (CRN) portfolio/Central Portfolio Management System with a UKCRN ID number of 15139. Accrual data were uploaded onto the NIHR CRN database on a monthly basis.
Summary of protocol amendments
A summary of the amendments made to the trial protocol following approval of the first version of the document by the REC and MHRA is provided in Table 4.
| Protocol version | Amendment | Description | Location |
|---|---|---|---|
| 1.1 | Substantial amendment 08 | Clarification of exclusion criteria: previous: Severe hepatic impairment (Child–Pugh class C)New: Severe chronic hepatic impairment (Child–Pugh class C) | p. 12, 5.1.3 |
| Clarification of follow-up: previous: Patients will also be followed up to ascertain survival status at 28 days post recruitment, at hospital discharge, and at 3 and 6 months post recruitment using The Health and Social Care Information Centre [NHS Digital]New:Patients will also be followed up to ascertain survival status at 28 days post recruitment, at hospital discharge, and at 3 and 6 months post recruitment using The Health and Social Care Information Centre [NHS Digital] or via their GP | p. 17, 5.2.5 | ||
| Improve consistency in secondary outcomes, cardiovascular: previous: This should be measured and recorded at baseline, 6 and 12 hours and then 12 hourly in all patients with a jugular or subclavian central line for up to 96 hours and then daily to day 7 if the central line remains in situNew: This should be measured and recorded at baseline, 6 and 12 hours and then 12 hourly in all patients with a jugular or subclavian central line for up to 96 hours and then daily up to and including day 4 if the central line remains in situ | p. 17, 5.3.2 | ||
| Typo, clarification in appendix infusion guideline: previous – diagram did not display possibility of restarting drug; new – diagram now displays possibility of restarting the study drug if the patient has stabilised | p. 33, 5.2.5 13 | ||
| Clarification: 14 appendix 2 – echocardiographic substudy: new – 14 appendix – echocardiographic substudy | pp. 35 and 36 | ||
| 1.2 | Minor amendment 03 | Restart of pharmacokinetic substudy to collect 80 full sample sets | p. 18, 5.3.2 |
| 1.3 | Minor amendment 04 | Clarification that open-label levosimendan is not permitted | p. 16, 5.2.4 |
| 1.4 | Minor clarification to the protocol | Additional wording to clarify the collection of follow-up data from LeoPARDS trial patients | p. 17; p. 33, 13; p. 23, 7.3 |
Trial committees
Trial Management Group
The Trial Management Group (TMG) was set up by the chief investigator and was established to discuss recruitment and other practical aspects of the trial. The TMG met 22 times over the course of the trial, on 5 October 2012, 12 November 2012, 6 December 2012, 14 January 2013, 11 February 2013, 11 March 2013, 8 April 2013, 13 May 2013, 10 June 2013, 12 August 2013, 9 September 2013, 14 October 2013, 11 November 2013, 9 December 2013, 19 May 2014, 28 July 2014, 13 October 2014, 5 December 2014, 11 March 2015, 22 June 2015, 13 October 2015 and 19 May 2016.
The TMG membership is listed below:
-
Professor Anthony Gordon
-
Professor Danny McAuley
-
Professor Mervyn Singer
-
Professor Deborah Ashby
-
Professor Gavin Perkins
-
Dr Rob Orme
-
Dr Shalini Santhakumaran
-
Dr Alexina Mason
-
Dr Farah Al-Beidh
-
Dr Jonas Lexow
-
Mrs Mary Cross
-
Mrs Janis Best-Lane
-
Miss Ashley Campbell.
Trial Steering Committee
A TSC was established to oversee the conduct of the study. The TSC met five times over the course of the trial, on 11 October 2013, 3 June 2014, 29 January 2015, 9 October 2015 and 15 June 2016. Copies of the minutes from each meeting were sent to the funder, the Efficacy and Mechanism Evaluation (EME) programme of the NIHR. The TSC approved the trial protocol prior to the start of the study and received regular recruitment reports throughout the duration of the trial.
The TSC membership is listed below:
-
independent members:
-
Dr Simon Baudouin – chairperson
-
Dr Andrew Rhodes – independent member
-
Dr Phil Hopkins – independent member
-
Mrs Catherine White, ICU Steps – patient representative
-
Mr Simon Denegri – independent member
-
-
non-independent members:
-
Professor Anthony Gordon – chief investigator
-
Mrs Mary Cross – operations manager, ICTU
-
Dr Rob Orme – member
-
Professor Deborah Ashby – co-investigator and statistician
-
Dr Alexina Mason – trial statistician
-
Ms Nabila Youssouf – trial sponsor representative.
-
Data Monitoring and Ethics Committee
An independent DMEC was established to review SAE reports and any ongoing safety issues. The DMEC meetings took place on 26 November 2013, 28 April 2014, 9 December 2014, 26 March 2015, 22 September 2015 and 15 June 2016.
The first DMEC meeting to agree the charter outlining operational details and responsibilities took place early in the trial, on 26 November 2013. The DMEC provided feedback reports for each meeting to the chairperson of the TSC and these were reviewed at subsequent TSC meetings as applicable.
The DMEC membership is listed below:
-
independent members:
-
Professor Julian Bion – independent member chairperson
-
Dr Duncan Wyncoll – independent member
-
Dr Graeme MacLennan – independent statistician
-
-
non-independent attendees (in attendance for open sessions only):
-
Professor Anthony Gordon – chief investigator
-
Mrs Mary Cross – operations manager, ICTU
-
Professor Deborah Ashby – co-investigator and statistician
-
Dr Alexina Mason – senior trial statistician (in attendance for closed sessions)
-
Dr Shalini Santhakumaran – trial statistician (in attendance for closed sessions).
-
Monitoring
A monitoring plan was devised based on risk analysis and was described in detail in the monitoring manual by the trial manager. Trial monitors visited all sites and facilities where the trial took place to ensure compliance with the protocol, GCP and local regulatory compliance, and patient safety and accurate data collection and reporting. Monitoring visits were dependent on the rate of recruitment and number of participants recruited per site. Regular communication with sites by telephone, mail and e-mail was undertaken. Training sessions were organised for the investigators and all site staff at the beginning of the trial and then as appropriate. Initiation visits were completed at all trial centres prior to the recruitment of participants, and consisted of a review of protocol and trial documents, training with respect to trial procedures (informed consent, SAE reporting, inclusion and exclusion criteria), and a review of the recruitment strategy, site facilities and equipment, and essential document receipt, collection and filing, and archiving and inspection. Copies of the trial-specific procedure manuals and related documents were given to the investigators and research team. The approved version of the protocol was followed at all times and any significant protocol deviations were documented on a protocol violation form and submitted to the study co-ordination centre as soon as possible. The investigators allowed the monitors to:
-
inspect the site, facilities, IMP management and materials used for the trial
-
meet all members of the team involved in the trial and ensure that all staff working on the trial were experienced, appropriately trained and had access to review all of the documents relevant to the trial
-
have access to the eCRFs and source data
-
discuss trial progress and any issues with the investigator and site staff on a regular basis.
The monitor ensured that:
-
all participant records were inspected for confirmation of existence, eligibility and informed consent
-
there was adherence to the protocol, including consistency with inclusion/exclusion criteria
-
there was GCP and regulatory compliance
-
trial documentation was complete and up-to-date (e.g. correct versions of documents being used, source data captured) and relevant documents were collected for the Trial Master File (TMF)
-
the eCRFs had been completed correctly and accurately and all entries corresponded to data captured in source documents
-
the IMP accountability records were in order (receipt, dispensing and destruction), storage was under appropriate conditions and secure expiry dates were being checked and adhered to, and dispensing was carried out according to the protocol and trial procedures.
All information dealt with during such visits was treated as strictly confidential. At the end of the trial, close-out visits were performed by the monitors after the final participant visit had been completed and prior to database lock. During this visit the monitors verified that all trial close-out activities had been completed: all queries had been resolved, missing data had been completed, monitoring had been completed, archiving arrangements were in place, IMP accountability was complete and all used and unused IMPs had been destroyed, the Investigator Site File (ISF) was completed and TMF documents had been collected and the end-of-trial notification had been completed.
Patient and public involvement
Two patient and public involvement representatives sat on the TSC and provided input from a patient perspective at trial meetings. Both representatives reviewed and provided feedback on all of the project documents prior to the ethics and regulatory submissions, and their comments were incorporated.
Quality control and quality assurance
Quality control was performed according to ICTU internal procedures. The study was audited by a quality assurance representative of the sponsor. All necessary data and documents were made available for inspection.
Audits and inspections
The study may be subject to inspection and audit by Imperial College London under its remit as sponsor, the study co-ordination centre and other regulatory bodies to ensure adherence to GCP.
Chapter 4 Results
Screening and participant flow
In total, 2382 patients were screened at the participating hospitals/centres over the duration of the trial. Of the 824 patients meeting the eligibility criteria, 308 declined to participate and 516 were randomised to the trial. The flow of patients is summarised in Figure 2, including the numbers of patients screened, randomised and completing the trial. Recruitment ended when the required sample of 516 patients had been enrolled. One patient withdrew consent before the study drug was administered and no data were collected for this patient; this patient was excluded from all analyses. Four patients died before the study drug was administered, three before any post-randomisation data on the primary outcome could be collected. These patients have been included in the ITT analysis using data recorded at baseline. Two patients did not receive the study drug for other reasons, but data were collected and they too have been included in the ITT analysis.
FIGURE 2.
Participant flow in the LeoPARDS trial: Consolidated Standards of Reporting Trials (CONSORT) flow diagram. a, Patients may have more than one reason for exclusion. b, Patients met all criteria, but could not be randomised because of staff availability. c, Other known reasons for exclusion were unavailability of study drug, patient mental health concerns and language barriers; reason not known for 78 patients.
Appendix 1 (see Table 20) provides a list of the study centres with details of the numbers of patients screened and randomised into each group, along with the number of protocol deviations. There were a total of 100 protocol deviations, all of which were minor (they did not significantly affect patient safety or the scientific value of the trial).
Recruitment and retention
Recruitment lasted for 23 months, from January 2014 to December 2015. There was a temporary halt to recruitment between July and September 2014 when the study drug was recalled and repackaged because of moisture damage to some outer cardboard storage boxes.
The actual recruitment period was shorter than the original target of 2.5 years.
Recruitment rate
The target recruitment rate for the study was one to two patients per month per centre, with a target recruitment figure of 516.
The accrual of patients during the whole study period is presented in Appendix 1 (see Figure 28).
Baseline data and longitudinal monitoring
Baseline data
Participant characteristics at baseline are shown in Table 5, with the number of missing values (if any) shown in Appendix 1 (see Table 21).
| Characteristic | Treatment group | Total | |
|---|---|---|---|
| Levosimendan | Placebo | ||
| Age (years) | 67 (58, 75) | 69 (58, 77) | 68 (58, 76) |
| Sex (male) | 145 (56) | 144 (56) | 289 (56) |
| Weight (kg) | 76 (65, 90) | 80 (68, 91) | 79 (66, 90) |
| BMI (kg/m2) | 27 (23, 30) | 28 (24, 32) | 27 (23, 31) |
| Ethnicity | |||
| Asian | 11 (4) | 10 (4) | 21 (4) |
| Black | 4 (2) | 6 (2) | 10 (2) |
| Caucasian | 240 (93) | 240 (93) | 480 (93) |
| Other | 3 (1) | 1 (0) | 4 (1) |
| Recent surgical history | 94 (36) | 95 (37) | 189 (37) |
| APACHE II score | 25 (21, 31) | 25 (21, 30) | 25 (21, 30) |
| Pre-existing conditions | |||
| Ischaemic heart disease | 46 (18) | 31 (12) | 77 (15) |
| Congestive heart failure | 1 (0) | 4 (2) | 5 (1) |
| Cardiac failure | 23 (9) | 26 (10) | 49 (10) |
| Severe COPD | 16 (6) | 11 (4) | 27 (5) |
| Chronic renal failure | 19 (7) | 18 (7) | 37 (7) |
| Cirrhosis | 4 (2) | 6 (2) | 10 (2) |
| Immunocompromised | 23 (9) | 24 (9) | 47 (9) |
| Diabetes | 59 (23) | 51 (20) | 110 (21) |
| Beta-blockers normally taken | 54 (21) | 45 (18) | 99 (19) |
| Organ failureb | |||
| Respiratory | 99 (39) | 101 (39) | 200 (39) |
| Renal | 77 (30) | 74 (29) | 151 (29) |
| Liver | 6 (2) | 8 (3) | 14 (3) |
| Haematological | 16 (6) | 13 (5) | 29 (6) |
| Neurological | 117 (52) | 111 (52) | 228 (52) |
| Source of infection | |||
| Lung | 98 (38) | 103 (40) | 201 (39) |
| Abdomen | 95 (37) | 96 (37) | 191 (37) |
| Urine | 12 (5) | 17 (7) | 29 (6) |
| Primary bacteraemia | 7 (3) | 3 (1) | 10 (2) |
| Neurological | 4 (2) | 1 (0) | 5 (1) |
| Soft tissue or line | 16 (6) | 10 (4) | 26 (5) |
| Other | 25 (10) | 27 (11) | 52 (10) |
| Mechanical ventilation | 207 (80) | 210 (82) | 417 (81) |
| RRT | 44 (17) | 45 (18) | 89 (17) |
| Moderate or severe ARDS | 72 (28) | 59 (23) | 131 (25) |
| Heart rhythm | |||
| Sinus rhythm | 201 (78) | 218 (85) | 419 (82) |
| Atrial fibrillation | 32 (12) | 21 (8) | 53 (10) |
| Paced | 3 (1) | 2 (1) | 5 (1) |
| Other irregular rhythm | 22 (9) | 14 (5) | 36 (7) |
| Physiological variables | |||
| MAP (mmHg) | 74 (68, 80) | 73 (67, 79) | 74 (68, 79) |
| Heart rate (beats/minute) | 97 (82, 111) | 94 (80, 110) | 95 (80, 110) |
| Central venous pressure (mmHg) | 11 (9, 15) | 12 (8, 16) | 11 (8, 15) |
| Cardiac index (l/minute/m2)c | 2.7 (2.2, 3.7) | 3.3 (2.2, 4.0) | 3 (2.2, 3.8) |
| SaO2 (%) | 97 (95, 98) | 97 (95, 98) | 97 (95, 98) |
| ScvO2 (%) | 75 (69, 81) | 76 (70, 81) | 76 (69, 81) |
| Lactate (mmol/l) | 2.2 (1.4, 3.5) | 2.3 (1.5, 3.9) | 2.3 (1.4, 3.6) |
| PaO2/FiO2 (kPa) | 29 (20, 39) | 29 (20, 39) | 29 (20, 39) |
| Creatinine (µmol/l) | 140 (89, 216) | 137 (93, 208) | 138 (91, 213) |
| Bilirubin (µmol/l) | 14 (8, 25) | 15 (9, 27) | 14 (8, 26) |
| Haemoglobin (g/l) | 108 (94, 123) | 108 (93, 125) | 108 (94, 124) |
| Platelets (×109/l) | 212 (134, 299) | 216 (144, 308) | 215 (140, 307) |
| GCS score | 9 (3, 15) | 8 (3, 15) | 9 (3, 15) |
| Time from shock to randomisation (hours)d | 16 (10, 21) | 15 (10, 20) | 16 (10, 21) |
| Vasoactive drug dosage at randomisation | |||
| Noradrenaline (µg/kg/minute) | 0.29 (0.16, 0.52), n = 255 | 0.27 (0.15, 0.44), n = 253 | 0.28 (0.16, 0.47), n = 508 |
| Adrenaline (µg/kg/minute) | 0.14 (0.07, 0.28), n = 21 | 0.13 (0.08, 0.38), n = 21 | 0.14 (0.07, 0.3), n = 42 |
| Vasopressin (units/minute) | 0.03 (0.02, 0.04), n = 33 | 0.03 (0.02, 0.04), n = 37 | 0.03 (0.02, 0.04), n = 70 |
| Dobutamine (µg/kg/minute) | 5.7 (3.5, 8.8), n = 18 | 5 (4.4, 6.2), n = 22 | 5.2 (4.4, 6.5), n = 40 |
Longitudinal measurements
The box plots in Figures 3–14 show the clinical measures monitored at varying intervals throughout the ICU stay. All group allocations are on an ITT basis.
FIGURE 3.
Box plot for MAP, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.
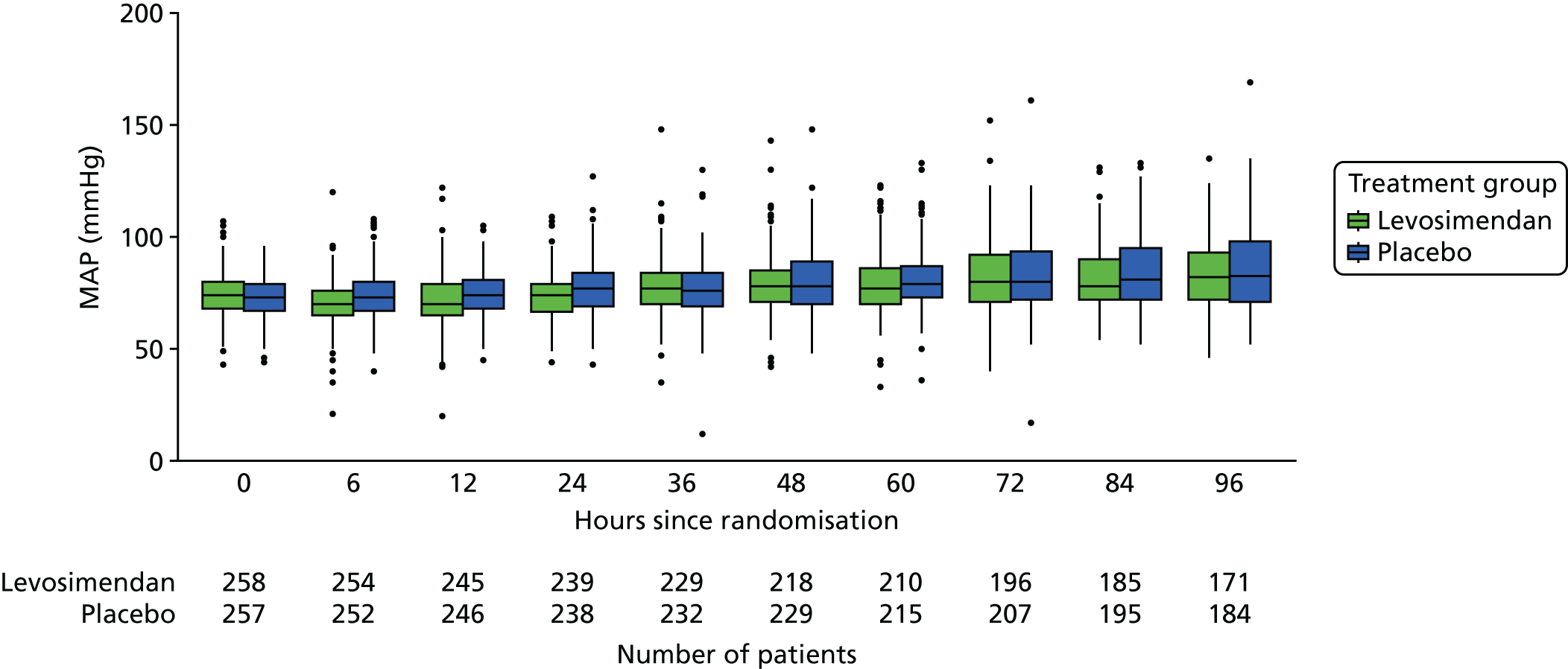
FIGURE 4.
Box plot for heart rate, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.
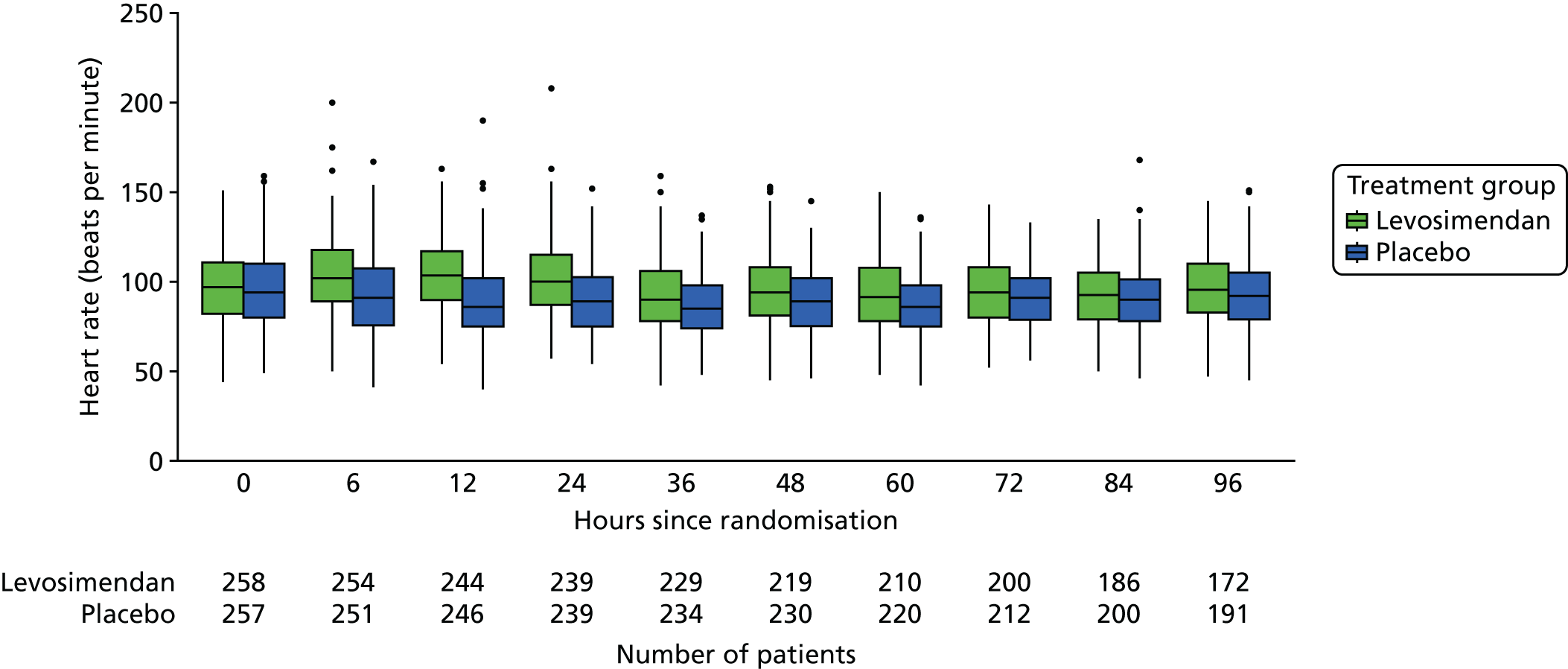
FIGURE 5.
Box plot for ScvO2, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.

FIGURE 6.
Box plot for cardiac index, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.
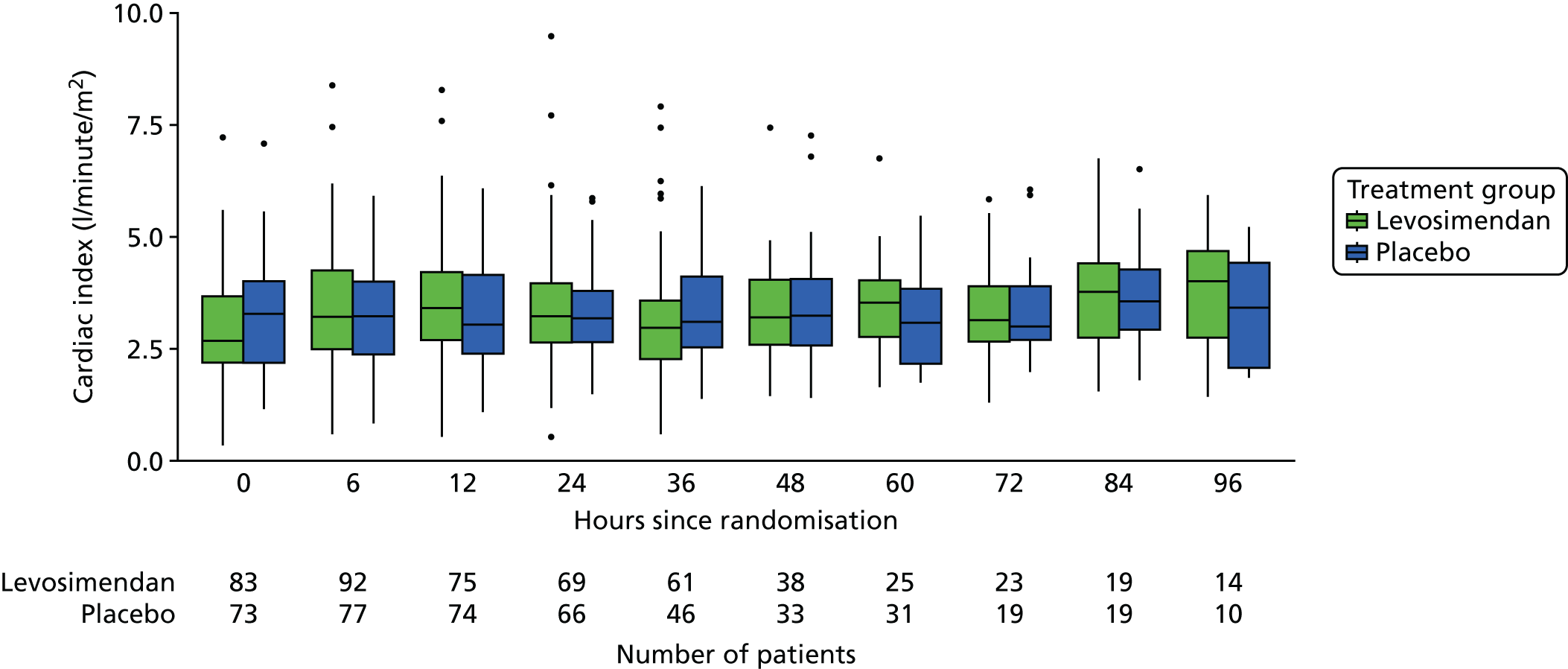
FIGURE 7.
Box plot for platelet count, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.
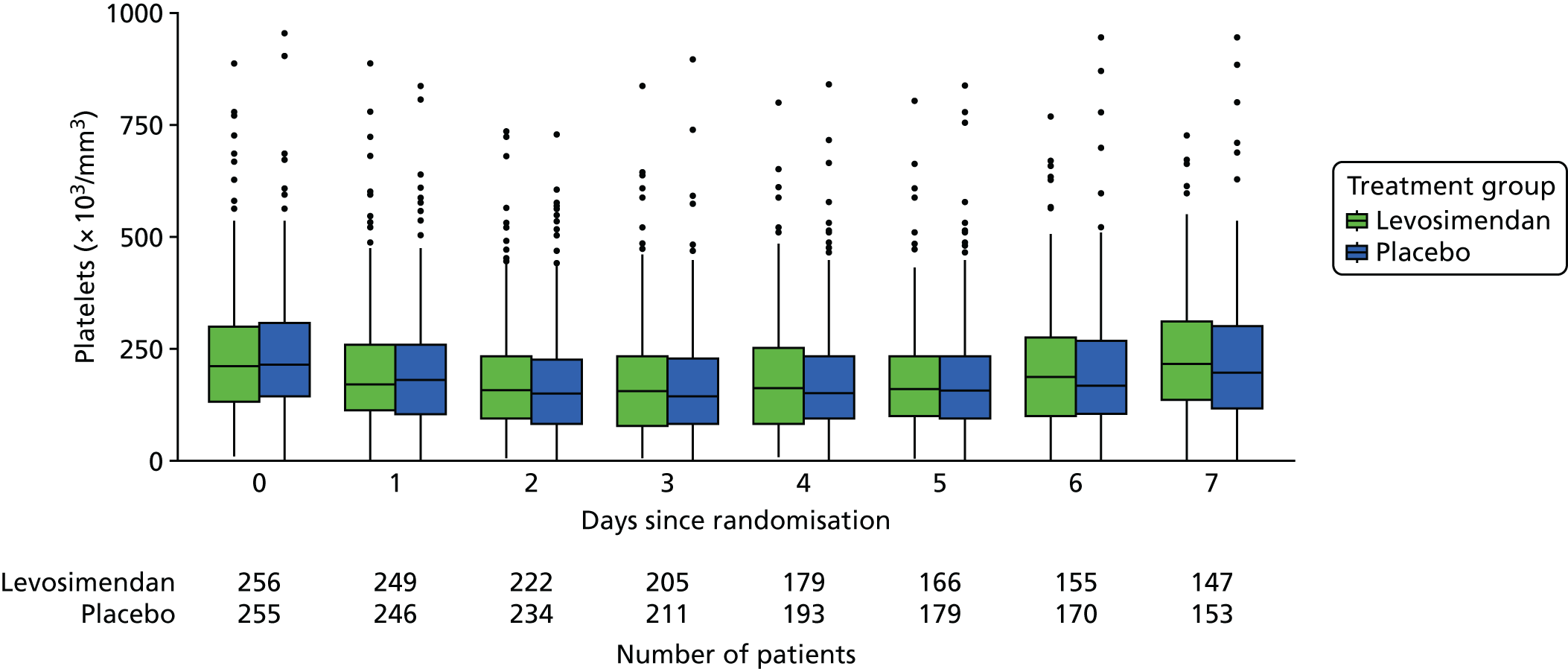
FIGURE 8.
Box plot for bilirubin level, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.
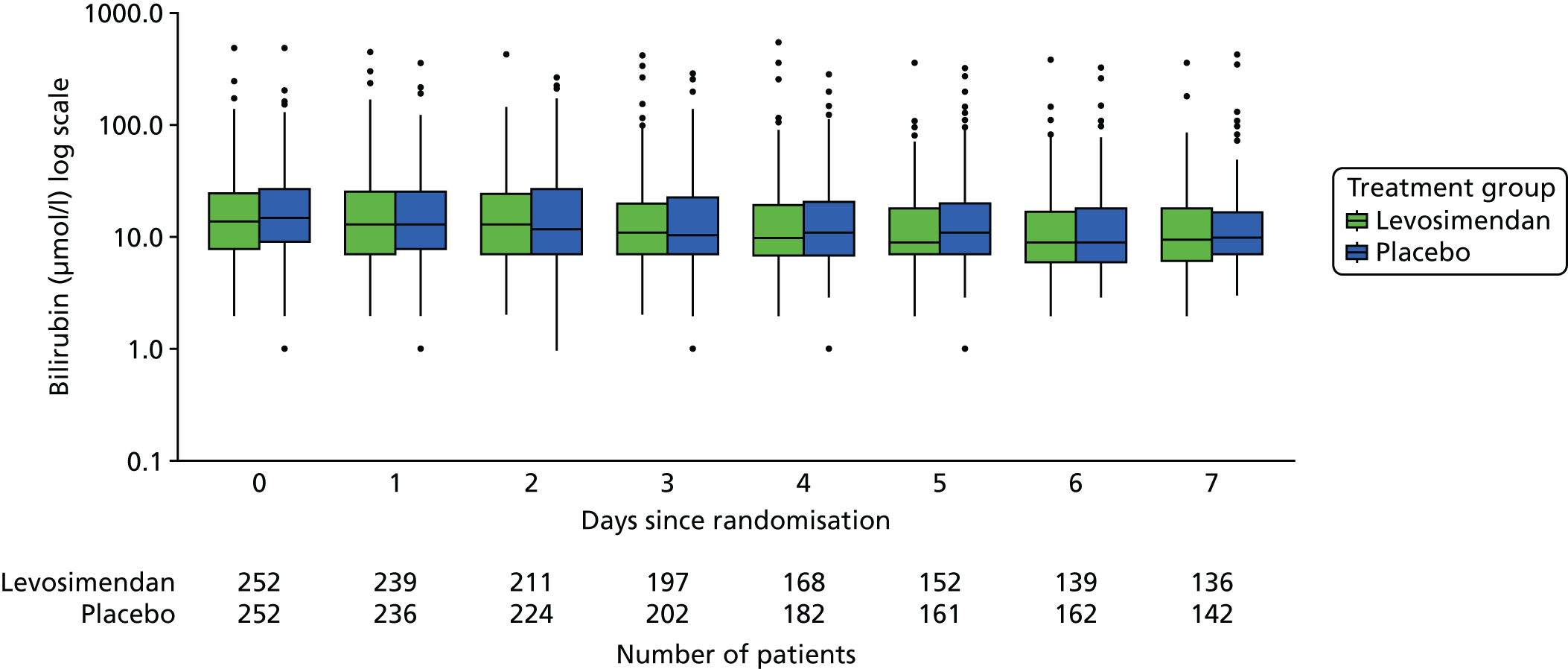
FIGURE 9.
Box plot for lactate level, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.

FIGURE 10.
Box plot for SaO2, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.

FIGURE 11.
Box plot for total intravenous (i.v.) fluid, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.

FIGURE 12.
Box plot for PaO2/FiO2 ratio, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.
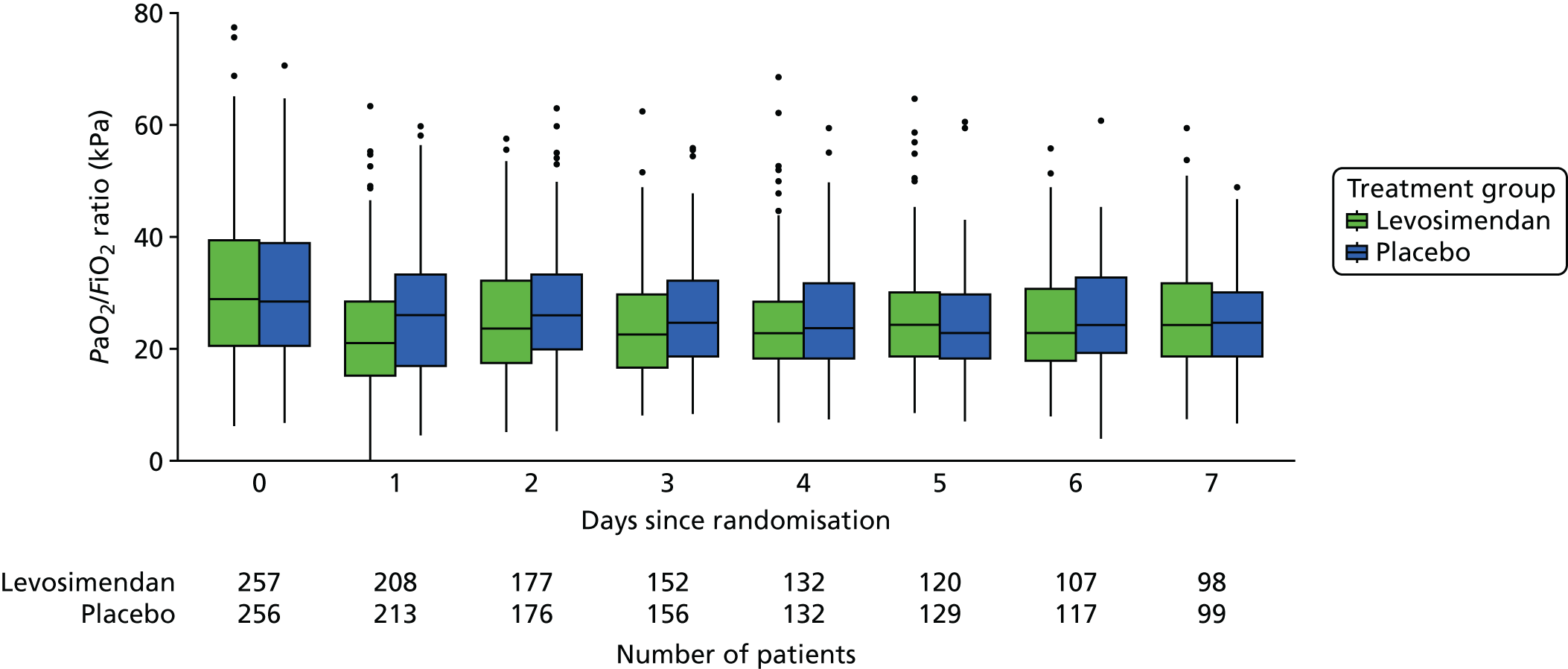
FIGURE 13.
Box plot for creatinine level, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.

FIGURE 14.
Box plot for total urine output, by treatment group. Line = median; box = IQR; whiskers = extremes of the data (1.5 × the IQR); circles = very extreme outliers. The data shown include all patients still alive and still in the ICU for each time point since randomisation.
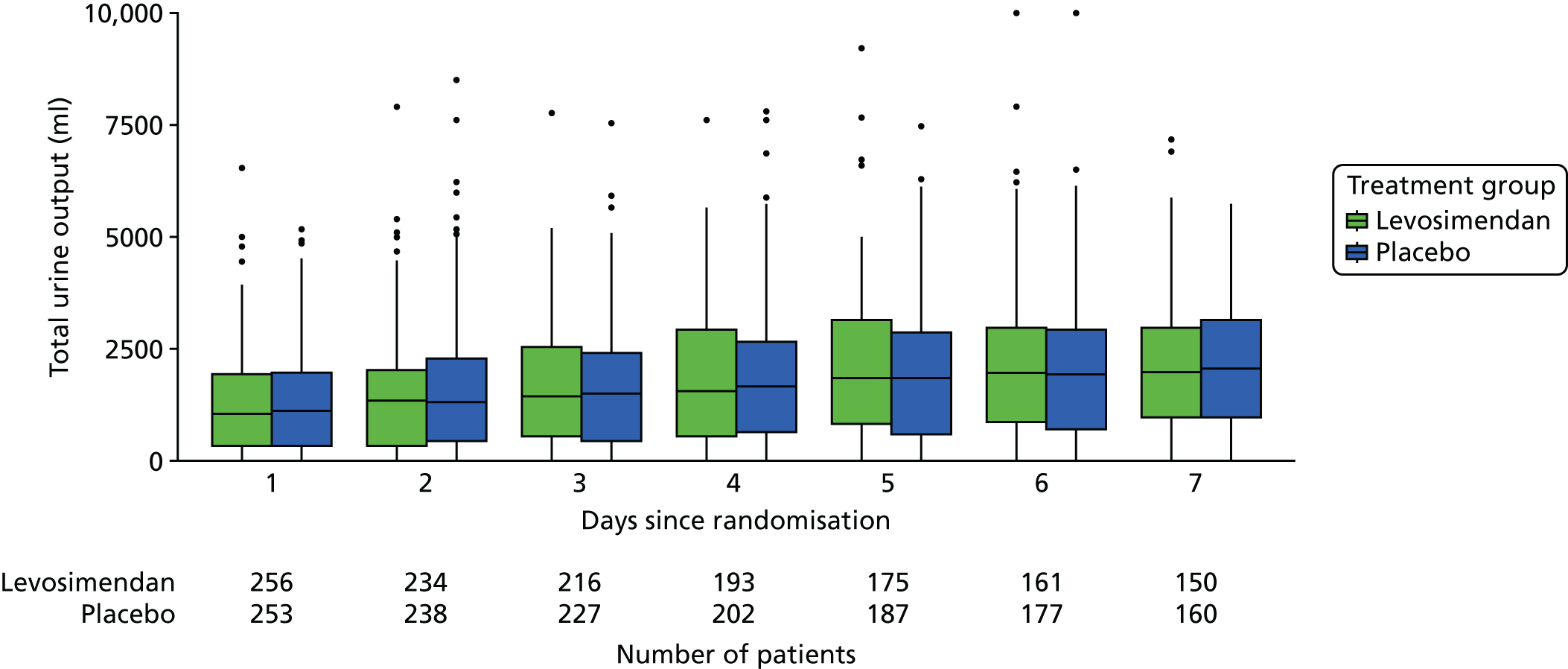
Study drug infusion
Table 6 shows the numbers of patients receiving the study drug by dose at 6, 12 and 24 hours in the levosimendan and placebo groups.
| Dose (µg/kg/minute) | Treatment group, n (%) | Total, n (%) | |
|---|---|---|---|
| Levosimendan | Placebo | ||
| 6 hours | |||
| 0 | 20 (7.8) | 8 (3.1) | 28 (5.4) |
| 0.05 | 36 (14.0) | 11 (4.3) | 47 (9.1) |
| 0.1 | 43 (16.7) | 22 (8.6) | 65 (12.6) |
| 0.2 | 159 (61.6) | 216 (84.0) | 375 (72.8) |
| Total | 258 (100) | 257 (100) | 515 (100) |
| 12 hours | |||
| 0 | 28 (11.3) | 7 (2.8) | 35 (7.0) |
| 0.05 | 32 (12.9) | 9 (3.6) | 41 (8.2) |
| 0.1 | 41 (16.5) | 22 (8.8) | 63 (12.7) |
| 0.2 | 147 (59.3) | 212 (84.8) | 359 (72.1) |
| Total | 248 (100) | 250 (100) | 498 (100) |
| 24 hours | |||
| 0 | 33 (13.5) | 19 (7.7) | 52 (10.6) |
| 0.05 | 26 (10.7) | 6 (2.4) | 32 (6.5) |
| 0.1 | 38 (15.6) | 8 (3.2) | 46 (9.3) |
| 0.2 | 147 (60.2) | 215 (86.7) | 362 (73.6) |
| Total | 244 (100) | 248 (100) | 492 (100) |
Other vasoactive drugs
Tables 7 and 8 show the numbers of patients receiving noradrenaline and dobutamine, respectively, by group and the median dose received at each time point.
| Follow-up (hours) | Treatment group | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | ||||||||
| n (%) | Median dose | LQ, UQ | n (%) | Median dose | LQ, UQ | n (%) | Median dose | LQ, UQ | |
| 6 | 246 (95.3) | 0.32 | 0.17, 0.53 | 240 (93.4) | 0.24 | 0.13, 0.44 | 486 (94.4) | 0.29 | 0.15, 0.50 |
| 12 | 238 (96.0) | 0.30 | 0.16, 0.54 | 232 (92.8) | 0.20 | 0.10, 0.39 | 470 (94.4) | 0.26 | 0.12, 0.47 |
| 24 | 210 (86.1) | 0.28 | 0.14, 0.46 | 195 (78.6) | 0.18 | 0.07, 0.33 | 405 (82.3) | 0.22 | 0.11, 0.40 |
| 36 | 185 (80.8) | 0.18 | 0.09, 0.32 | 167 (71.4) | 0.15 | 0.07, 0.29 | 352 (76.0) | 0.16 | 0.08, 0.31 |
| 48 | 145 (63.9) | 0.13 | 0.07, 0.26 | 124 (53.2) | 0.12 | 0.07, 0.24 | 269 (58.5) | 0.13 | 0.07, 0.25 |
| 60 | 121 (57.3) | 0.11 | 0.05, 0.25 | 107 (48.9) | 0.12 | 0.06, 0.20 | 228 (53.0) | 0.12 | 0.05, 0.24 |
| 72 | 92 (44.4) | 0.11 | 0.06, 0.21 | 78 (36.8) | 0.09 | 0.05, 0.16 | 170 (40.6) | 0.10 | 0.06, 0.20 |
| 84 | 76 (40.6) | 0.10 | 0.06, 0.22 | 66 (33.7) | 0.09 | 0.05, 0.17 | 142 (37.1) | 0.10 | 0.05, 0.21 |
| 96 | 58 (32.0) | 0.11 | 0.06, 0.24 | 61 (31.8) | 0.08 | 0.03, 0.17 | 119 (31.9) | 0.10 | 0.04, 0.20 |
| Follow-up (hours) | Treatment group | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | ||||||||
| n (%) | Median dose | LQ, UQ | n (%) | Median dose | LQ, UQ | n (%) | Median dose | LQ, UQ | |
| 6 | 13 (5.0) | 5.00 | 4.00, 9.10 | 24 (9.3) | 5.00 | 4.40, 5.65 | 37 (7.2) | 5.00 | 4.40, 6.40 |
| 12 | 10 (4.0) | 6.10 | 5.00, 8.80 | 23 (9.2) | 4.40 | 3.50, 5.25 | 33 (6.6) | 4.90 | 3.70, 6.40 |
| 24 | 10 (4.1) | 5.65 | 2.70, 7.00 | 22 (8.9) | 4.55 | 3.40, 5.50 | 32 (6.5) | 4.75 | 3.30, 6.32 |
| 36 | 7 (3.1) | 5.50 | 1.95, 7.25 | 20 (8.5) | 4.55 | 3.70, 6.40 | 27 (5.8) | 4.60 | 2.70, 6.85 |
| 48 | 4 (1.8) | 5.95 | 4.45, 7.93 | 18 (7.7) | 4.20 | 3.08, 4.90 | 22 (4.8) | 4.20 | 3.08, 5.57 |
| 60 | 1 (0.5) | 1.30 | 1.30, 1.30 | 18 (8.2) | 3.70 | 2.05, 6.05 | 19 (4.4) | 3.30 | 1.65, 5.80 |
| 72 | 2 (1.0) | 7.20 | 4.55, 9.85 | 16 (7.5) | 3.90 | 1.73, 5.55 | 18 (4.3) | 3.90 | 1.83, 6.05 |
| 84 | 1 (0.5) | 1.90 | 1.90, 1.90 | 11 (5.6) | 4.60 | 1.65, 6.60 | 12 (3.1) | 4.40 | 1.73, 6.45 |
| 96 | 3 (1.7) | 2.30 | 2.10, 3.75 | 10 (5.2) | 4.40 | 2.00, 6.05 | 13 (3.5) | 4.20 | 1.90, 5.30 |
Safety data
Table 9 shows the numbers of AEs by SAE classification and their relationship to the study medication.
| Classification | Relationshipa | Treatment group | |||
|---|---|---|---|---|---|
| Number of AEsb | Number of subjectsc | ||||
| Levosimendan | Placebo | Levosimendan | Placebo | ||
| Not serious | Definite | 1 | 0 | 1 | 0 |
| Not assessable | 0 | 1 | 0 | 1 | |
| Not related | 26 | 13 | 7 | 4 | |
| Possible | 24 | 16 | 19 | 13 | |
| Probable | 6 | 0 | 5 | 0 | |
| Unlikely | 16 | 16 | 8 | 10 | |
| Serious | Definite | 0 | 0 | 0 | 0 |
| Not assessable | 0 | 0 | 0 | 0 | |
| Not related | 9 | 11 | 8 | 10 | |
| Possible | 15 | 3 | 12 | 1 | |
| Probable | 1 | 0 | 1 | 0 | |
| Unlikely | 13 | 11 | 11 | 11 | |
| Total | All | 111 | 71 | 72 | 50 |
Primary outcome analysis
Exploratory analysis
Figure 15 provides a box plot of the total SOFA score by group and day since randomisation.
FIGURE 15.
Box plot of total SOFA scores, by day and treatment group.

Missing data
Appendix 1 (see Table 22) shows the level of missingness across subjects and across daily scores by treatment group. In total, 66% of patients had at least one total SOFA score missing and hence an incomplete set of scores on which to base the primary outcome of mean total SOFA score. Although this is high, each patient should have five component scores recorded for each day in the ICU and missing only one of these would result in the patient having at least one missing total SOFA score. Of the 26,775 scores expected (one for each of the five components for each day that each patient was in the ICU), 1773 (6.2%) were missing, only slightly higher than the 4% anticipated. In the LeoPARDS trial, there was an expectation that scores might not be recorded for 1 or 2 days when there had been no change or when the patient was getting better and approaching discharge. There were 60 instances (across all components) when ≥ 3 consecutive days were missing that did not occur at the end of the ICU stay.
For the main analysis we used a version of LOCF, as described in Chapter 3, Missing data, with a Bayesian sensitivity analysis to check where the results were sensitive to the underestimation of uncertainty in the LOCF approach.
Main analysis of the primary outcome
Table 10 shows the main analysis of the primary outcome (mean total SOFA score averaged over all days that a patient was in the ICU), along with each of the SOFA components separately. Three subjects died soon after randomisation and before any data could be collected; they were assigned baseline values in line with the LOCF imputation. A further two subjects who both had only one daily measurement taken on the ICU before they died were missing the liver component for this day, with no baseline measurement; these patients were assumed to have a normal SOFA score based on our assumptions about missing data and with the majority of these scores being normal. Appendix 1 (see Table 23) shows the corresponding ‘as-treated’ analysis, excluding seven participants who did not receive the study drug and one placebo participant who received open-label levosimendan.
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)b | |
| Respiration | 1.70 | 1.18 | 1.6 (0.76, 2.68) | 1.56 | 1.15 | 1.5 (0.50, 2.36) | 0.14 (–0.06 to 0.34) | 0.10 (–0.20 to 0.54) |
| Coagulation | 0.75 | 1.05 | 0.14 (0.00, 1.19) | 0.75 | 1.02 | 0.3 (0.00, 1.08) | 0.00 (–0.18 to 0.17) | –0.16 (–0.36 to 0.11) |
| Liver | 0.51 | 0.84 | 0.00 (0.00, 0.74) | 0.45 | 0.77 | 0.00 (0.00, 0.64) | 0.06 (–0.08 to 0.19) | 0.00 (0.00 to 0.05) |
| Cardiovascular | 2.27 | 1.20 | 2.00 (1.22, 3.52) | 2.02 | 1.20 | 1.67 (1.00, 3.04) | 0.25 (0.04 to 0.46) | 0.33 (0.07 to 0.62) |
| Renal | 1.46 | 1.49 | 1.00 (0.00, 2.88) | 1.28 | 1.38 | 0.79 (0.00, 2.25) | 0.18 (–0.07 to 0.42) | 0.21 (–0.29 to 0.63) |
| Total | 6.68 | 3.96 | 5.63 (3.75, 9.00) | 6.06 | 3.89 | 5.13 (3.33, 7.86) | 0.61 (–0.07 to 1.29) | 0.50 (–0.28 to 1.25) |
Sensitivity analysis of the primary outcome
Table 11 shows the results of the sensitivity analysis of the primary outcome.
| Variable | Model | |
|---|---|---|
| 1a | 2a | |
| Intercept | 0.63 (–1.16 to 2.42) | 1.19 (0.84 to 1.54) |
| Treatment differenceb | 0.59 (–0.02 to 1.20) | 0.13 (0.02 to 0.25) |
| Age (years) | –0.01 (–0.03 to 0.01) | 0.00 (–0.01 to 0.00) |
| APACHE II score | 0.24 (0.20 to 0.28) | 0.05 (0.04 to 0.06) |
Subgroup analysis of the primary outcome
Figure 16 shows the results of the subgroup analysis of the primary outcome.
FIGURE 16.
Forest plot showing the difference in mean SOFA score, by predefined subgroup.

Secondary outcomes
Time to extubation
Participants in the levosimendan group were less likely than those in the placebo group to be successfully weaned from mechanical ventilation over 28 days (hazard ratio 0.77, 95% CI 0.60 to 0.97; p = 0.03) (Figure 17 and Table 12).
FIGURE 17.
Kaplan–Meier plot for time to extubation. The hazard ratio in the levosimendan group compared with the placebo group was 0.77 (95% CI 0.60 to 0.97; p = 0.03).

| Variable | Analysis | |
|---|---|---|
| Unadjusted | Adjusted | |
| Treatment differencea | 0.78 (0.61 to 0.99) | 0.77 (0.60 to 0.97) |
| Age (years) | 1.00 (0.99 to 1.01) | |
| APACHE II score | 0.94 (0.92 to 0.96) | |
Survival
Figures S4 (see Report Supplementary Material 1) and Figure 18 show the Kaplan–Meier plots for survival to 28 days and 6 months, respectively. Three participants were censored before 6 months. One participant in the placebo group was censored at ICU discharge as post-discharge follow-up was declined. Two participants were censored at 28 days: one participant in the placebo group who had no hospital or primary care data recorded and one participant in the levosimendan group who left the UK.
FIGURE 18.
Kaplan–Meier plot for survival to 6 months.
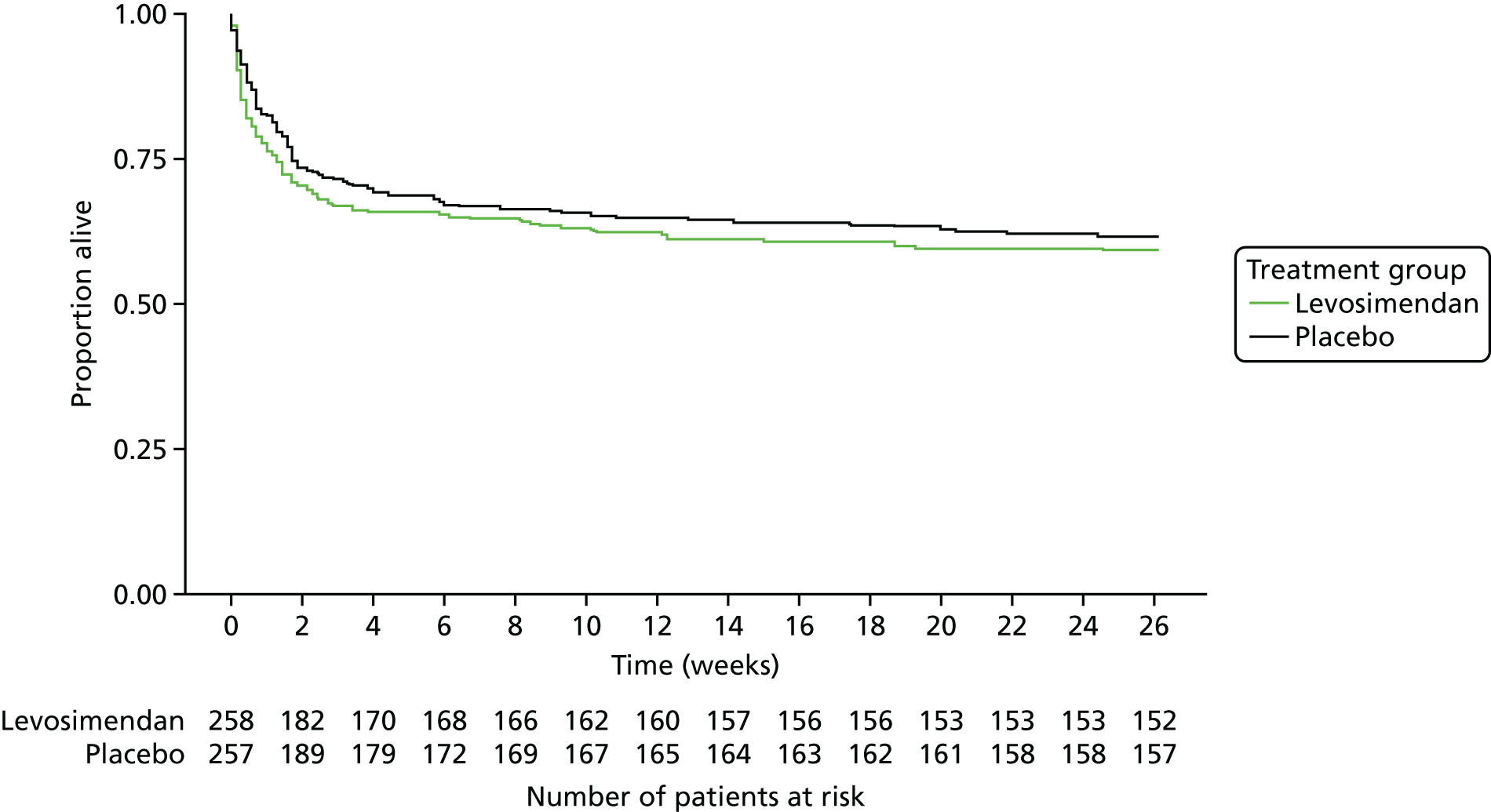
Table S14 (see Report Supplementary Material 1) shows 28-day mortality, mortality before discharge from the ICU and from hospital, and 3- and 6-month mortality established from hospital readmission and GP data. Results from the Cox regression, both unadjusted and adjusted for age, APACHE II score and ICU effects, are shown in Tables S15 and S16 (see Report Supplementary Material 1) for 28-day and 6-month survival, respectively.
Figure 19 shows the results of the subgroup analysis of survival at 28 days.
FIGURE 19.
Forest plot showing the difference in survival at day 28, by subgroup.

A summary of the clinical outcomes is provided in Table 13.
| Outcome | Treatment group, n(%) | Absolute difference (95% CI) | p-valuea | |
|---|---|---|---|---|
| Levosimendan (N = 258) | Placebo (N = 257) | |||
| Primary outcome | ||||
| Mean daily total SOFA score | 6.68 (3.96) | 6.06 (3.89) | 0.61 (–0.07 to 1.29) | 0.053 |
| Mean daily respiratory SOFA score | 1.70 (1.18) | 1.56 (1.15) | 0.14 (–0.06 to 0.34) | 0.23 |
| Mean daily coagulation SOFA score | 0.75 (1.05) | 0.75 (1.02) | 0.00 (–0.18 to 0.17) | 0.55 |
| Mean daily hepatic SOFA score | 0.51 (0.84) | 0.45 (0.77) | 0.06 (–0.08 to 0.19) | 0.65 |
| Mean daily cardiovascular SOFA score | 2.27 (1.20) | 2.02 (1.20) | 0.25 (0.04 to 0.46) | 0.01 |
| Mean daily renal SOFA score | 1.46 (1.49) | 1.28 (1.38) | 0.18 (–0.07 to 0.42) | 0.32 |
| Mean daily SOFA score excluding cardiovascular score | 4.41 (3.13) | 4.05 (3.07) | 0.36 (–0.17 to 0.90) | 0.12 |
| Mean daily total SOFA score, sensitivity analysisb | 7.19 (3.72) | 6.78 (3.74) | 0.41 (–0.24 to 1.06c) | – |
| Secondary outcomes | ||||
| Mortality | ||||
| At 28 days | 89 (34.5) | 79 (30.9)d | 3.6 (–4.5 to 11.7) | 0.43 |
| At ICU discharge | 83 (32.2) | 76 (29.6) | 2.6 (–5.4 to 10.6) | 0.59 |
| At hospital discharge | 97 (37.6) | 84 (32.8)d | 4.8 (–3.5 to 13.0) | 0.30 |
| Number of catecholamine-free days | 22 (0, 26) | 23 (0, 26) | –1.0 (–4.5 to 1.0) | 0.09 |
| Number of ventilation-free days | 16 (0, 25) | 19 (0, 25) | –3.0 (–9.5 to 1.0) | 0.14 |
| MAKE28 | 148 (57.4) | 139 (54.3) | 3.1 (–5.5 to 11.6) | 0.54 |
| Need for new RRT | 62 (24.1) | 62 (24.1) | 0.0 (–7.4 to 7.4) | > 0.99 |
| Sustained renal failure at day 28 or ICU discharge if before 28 days | 118 (45.7) | 108 (42.0) | 3.7 (–4.9 to 12.3) | 0.45 |
| Duration of RRT (days) | 3.0 (1.0, 8.0) | 5.0 (2.0, 9.0) | –2.0 (–3.0 to 0.0) | 0.24 |
| Length of ICU stays (days) | ||||
| All participants | 7.3 (3.2, 14.8) | 8.3 (3.9, 13.5) | –1.0 (–2.6 to 0.8) | 0.66 |
| Survivors | 9.1 (5.0, 16.1) | 9.0 (4.9, 14.1) | 0.2 (–2.0 to 2.7) | 0.31 |
| Non-survivors | 3.2 (1.4, 8.9) | 5.7 (2.2, 11.7) | –2.6 (–5.7 to –0.8) | 0.09 |
| Length of hospital stay (days) | ||||
| All participants | 19.6 (10.1, 40.4) | 22.7 (11.7, 42.3) | –3.1 (–7.0 to 2.2) | 0.24 |
| Survivors | 30.1 (16.8, 48) | 27.7 (18, 52.3) | 2.5 (–5.9 to 8.2) | 0.81 |
| Non-survivors | 8.2 (3.4, 18.6) | 11.3 (5.1, 25.7) | –3.1 (–6.5 to 0.7) | 0.25 |
| Safety outcomes | ||||
| Participants with a SAE (any) | 32 (12.4) | 23 (8.9) | 3.5 (–2.3 to 9.2) | 0.26 |
| Subcategories of SAEs | ||||
| Any life-threatening arrhythmias | 15 (5.8) | 6 (2.3) | 3.5 (–0.3 to 7.3) | 0.08 |
| Atrial fibrillation/supraventricular tachycardia | 8 (3.1) | 1 (0.4) | 2.7 (0.1 to 5.3) | 0.04 |
| Bradycardia | 0 (0.0) | 2 (0.8) | –0.8 (–2.2 to 0.7) | 0.48 |
| Ventricular fibrillation/tachycardia | 7 (2.7) | 3 (1.2) | 1.5 (–1.2 to 4.3) | 0.34 |
| Myocardial infarction/acute coronary syndrome | 3 (1.2) | 1 (0.4) | 0.8 (–1.1 to 2.7) | 0.62 |
| Other | 18 (7.0) | 17 (6.6) | –0.4 (–4.3 to 5.1) | > 0.99 |
Biomarker data
Figures S5–S11 (see Report Supplementary Material 1) show the box plots for each biomarker by treatment group and sample day, including baseline samples. All measures are shown on the log scale. The analysis of the change in biomarkers over time between the two treatment groups is reported in Hierarchical regression models for change in biomarkers over time.
Subgroup analysis of mean SOFA scores and 28-day mortality by cardiovascular markers
The mean total SOFA score (both including and excluding the cardiovascular component), the mean SOFA component scores and 28-day mortality were analysed by subgroups of NT-proBNP and troponin values, categorising the study population based on normal and raised values and values below and above the median value in the study population (Figures 20 and 21 and see Report Supplementary Material 1, Tables S25–S32).
FIGURE 20.
Forest plot showing the difference in mean total SOFA score, by subgroup based on cardiovascular biomarkers at baseline.

FIGURE 21.
Forest plot showing the difference in 28-day mortality, by subgroup based on cardiovascular biomarkers at baseline.
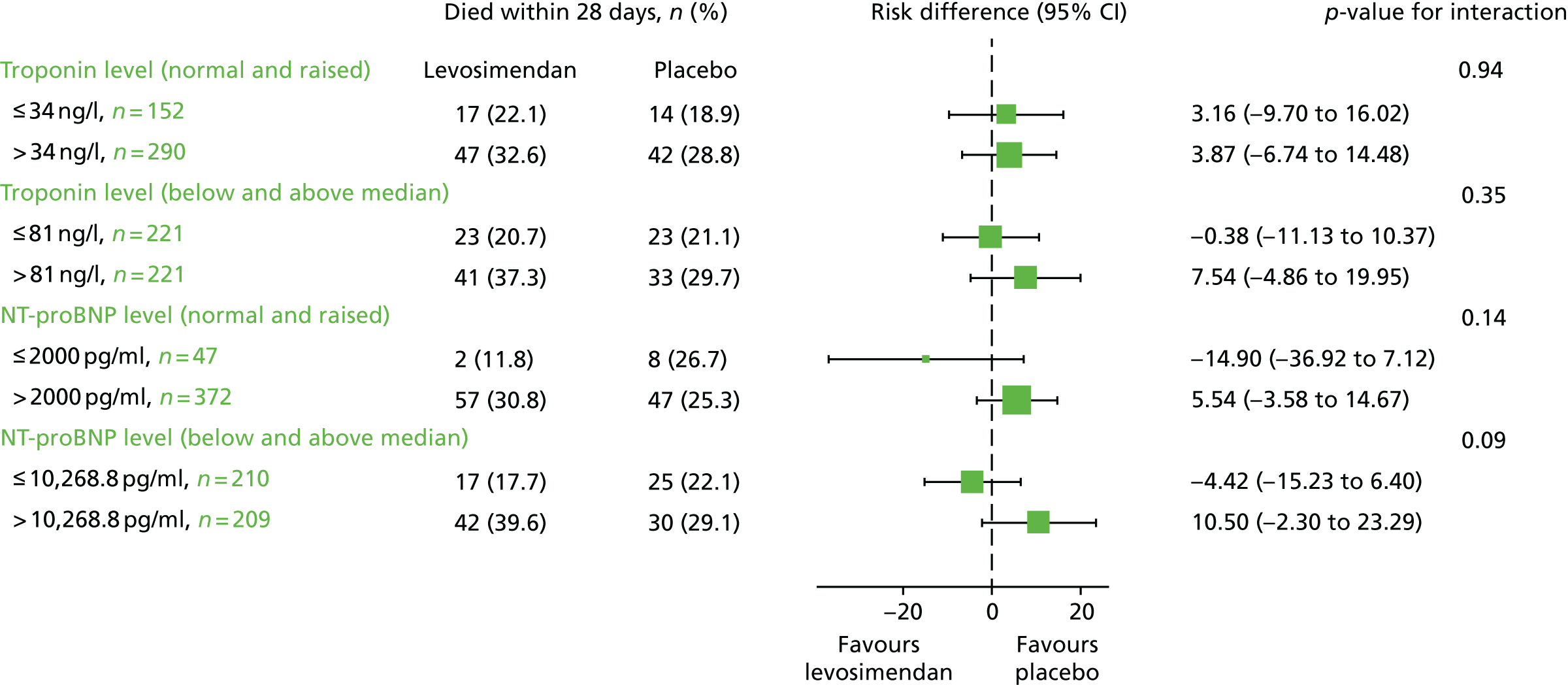
Hierarchical regression models for change in biomarkers over time
To assess if levosimendan alters the pro- and anti-inflammatory balance in sepsis the measured inflammatory biomarkers were analysed using Bayesian hierarchical regression models.
Tables 14 and 15 show the main model as specified in the supplementary SAP (see www.journalslibrary.nihr.ac.uk/programmes/eme/111408) for biomarker data, along with sensitivity analysis of a simpler model assuming that the treatment difference is constant over time (i.e. omitting the treatment*time interaction). For the model with no interaction, the estimated change over time applies to both the levosimendan arm and the placebo arm, and treatment differences are assumed to be constant over time. The fit of the models was compared using the deviance information criterion, values of which are provided in Report Supplementary Material 1 (Tables S37–43).
| Variable | Biomarker | |||
|---|---|---|---|---|
| NT-proBNP | Troponin | |||
| Main model | No interaction | Main model | No interaction | |
| Change per day – levosimendan | 1.09 (1.00 to 1.19) | 1.02 (0.96 to 1.09) | 0.87 (0.74 to 1.01) | 0.80 (0.72 to 0.89) |
| Change per day – placebo | 0.97 (0.90 to 1.05) | 0.75 (0.65 to 0.86) | ||
| Pr(faster reduction in levosimendan) | 0.032 | – | 0.082 | – |
| Treatment difference on day 2 | 1.00 (0.84 to 1.19) | 1.10 (0.94 to 1.27) | 1.12 (0.79 to 1.55) | 1.26 (0.92 to 1.67) |
| Treatment difference on day 4 | 1.12 (0.96 to 1.30) | 1.30 (0.95 to 1.73) | ||
| Treatment difference on day 6 | 1.26 (1.02 to 1.54) | 1.52 (1.01 to 2.19) | ||
| Variable | Biomarker | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CCL2 | IL-6 | IL-8 | IL-10 | sTNFr1 | ||||||
| Main model | No interaction | Main model | No interaction | Main model | No interaction | Main model | No interaction | Main model | No interaction | |
| Change per day – levosimendan | 0.78 (0.73, 0.83) | 0.74 (0.71, 0.77) | 0.50 (0.44, 0.56) | 0.50 (0.46, 0.54) | 0.85 (0.80, 0.91) | 0.82 (0.79, 0.86) | 0.68 (0.63, 0.73) | 0.68 (0.65, 0.72) | 0.90 (0.86, 0.93) | 0.90 (0.87, 0.92) |
| Change per day – placebo | 0.71 (0.66, 0.75) | 0.50 (0.45, 0.56) | 0.80 (0.75, 0.84) | 0.69 (0.65, 0.74) | 0.90 (0.86, 0.93) | |||||
| Pr(faster reduction in levosimendan) | 0.019 | – | 0.536 | – | 0.046 | – | 0.662 | – | 0.500 | – |
| Treatment difference on day 2 | 0.89 (0.78, 1.02) | 0.96 (0.86, 1.07) | 1.00 (0.79, 1.25) | 0.99 (0.82, 1.20) | 1.00 (0.85, 1.17) | 1.06 (0.91, 1.22) | 1.06 (0.90, 1.25) | 1.05 (0.91, 1.20) | 1.02 (0.93, 1.12) | 1.02 (0.94, 1.11) |
| Treatment difference on day 4 | 0.98 (0.87, 1.10) | 0.99 (0.81, 1.20) | 1.08 (0.93, 1.24) | 1.04 (0.90, 1.19) | 1.02 (0.94, 1.11) | |||||
| Treatment difference on day 6 | 1.08 (0.92, 1.26) | 0.99 (0.74, 1.29) | 1.16 (0.97, 1.38) | 1.02 (0.85, 1.23) | 1.02 (0.92, 1.13) | |||||
As the assumption of linear change with time seemed reasonable for all measures, and given the lack of evidence that treatment effects varied over time, we did not consider the treatment differences at each time point separately. As the biomarkers were all log-transformed, the results are displayed as ratios in Tables 14 and 15 and expressed as percentage differences in the accompanying text. For all biomarkers, the results were not sensitive to adjustment for age and APACHE II score at baseline or to ICU effects. These results can be found in Report Supplementary Material 1 (Tables S37–43).
N-terminal prohormone of brain natriuretic peptide
The results for NT-proBNP are shown in Table 14. NT-proBNP increased on average by 9% [95% credible interval (CrI) 0% to 19%] per day in the levosimendan group and decreased by 3% (95% CrI –10% to 5%) in the placebo group. The CrIs include 1, indicating that there is little evidence of longitudinal trends in either arm. The probability that NT-proBNP decreased faster in the levosimendan arm was 3% and the simpler model removing the treatment*time interaction fitted the data equally well (see Report Supplementary Material 1, Tables S37). There was some evidence of a treatment difference only on day 6 (26% difference, 95% CrI 2% to 54%). There were 113 NT-proBNP values below the lower limit of detection (32 of which occurred at baseline) and three above the upper limit of detection (two at baseline).
Troponin
The results for troponin are shown in Table 14. Troponin decreased over time in both arms: by 13% per day (95% CrI –26% to 1%) in the levosimendan arm and by 25% (95% CrI –35% to –14%) in the placebo arm. The probability that troponin decreased faster in the levosimendan arm was 8% and the model without the treatment*time interaction gave a similar fit. There was some evidence of a treatment difference only on day 6, but the CrI was wide (52% difference, 95% CrI 1% to 219%). There were no values outside the limits of detection for troponin.
Chemokine ligand 2
The results for CCL2 are shown in Table 15. CCL2 decreased over time in both arms: by 22% per day (95% CrI –27% to –17%) in the levosimendan arm and by 29% (95% CrI –34% to –25%) in the placebo arm. The probability that CCL2 decreased faster in the levosimendan arm was 2%, but again the simpler model with no interaction fitted the data equally well. There was little evidence of a treatment difference on any day. There were four values above the upper limit of detection, all of which were at baseline, and no values below the lower limit of detection.
Interleukin 6
The results for IL-6 are shown in Table 15. IL-6 decreased over time in both arms: by 50% (95% CrI –56% to –44%) per day in the levosimendan arm and also by 50% (95% CrI –55% to –44%) per day in the placebo arm. The probability that IL-6 decreased faster in the levosimendan arm was 54%. Removing the interaction term slightly improved the fit of the model and there was little evidence of a treatment difference on any day. There were 30 values above the upper limit of detection, 26 of which were at baseline.
Interleukin 8
The results for IL-8 are shown in Table 15. IL-8 decreased over time by 15% per day (95% CrI –20% to –9%) in the levosimendan arm and by 20% per day (95% CrI –25% to –16%) in the placebo arm. Removing the interaction term gave a similar model fit and there was little evidence of a treatment difference on any day. There were four values above the upper limit of detection, two of which were at baseline.
Interleukin 10
The results for IL-10 are shown in Table 15. IL-10 decreased over time by a similar amount in both arms (levosimendan: –32% per day, 95% CrI –37% to –27%; placebo: 31% per day, 95% CrI –35% to –26%). Removing the interaction term slightly improved the fit of the model. There was little evidence of a treatment difference on any day. There was one value above the upper limit of detection and one value below the lower limit of detection.
Soluble tumour necrosis factor receptor 1
The results for sTNFr1 are shown in Table 15. sTNFr1 decreased by the same amount per day in the placebo arm and the levosimendan arm (–10%, 95% CrI –14% to –7%) and the probability of a faster decrease in the placebo arm was therefore 50%. The simpler model with no treatment*time interaction had a slightly better fit. There was little evidence of a treatment effect on any day. There was one value above the upper limit of detection.
Pharmacokinetic analysis
Forty-one participants in the levosimendan group were included in the pharmacokinetic analysis. The mean age of participants was 63.0 years and mean weight was 76.7 kg (Table 16).
| Characteristic | Mean | Median | SD |
|---|---|---|---|
| Age (years) | 63.0 | 64.0 | 14.26 |
| Weight (kg) | 76.7 | 76.0 | 20.24 |
| APACHE II score | 23.3 | 23.5 | 7.29 |
In total, levosimendan and the two metabolites OR-1855 and OR-1896 were measured in 53, 148 and 103 plasma samples respectively (Table 17).
| Time post dose (hours) | Levosimendan, n | Metabolites, n | |
|---|---|---|---|
| OR-1855 | OR-1896 | ||
| Total number of samples | |||
| 22 | 44 | 44 | 44 |
| 66 | 34 | 34 | 34 |
| 144 | 28 | 28 | 28 |
| 192 | 28 | 28 | 28 |
| 240 | 21 | 21 | 21 |
| 312 | 14 | 14 | 14 |
| 384 | 17 | 17 | 17 |
| Total | 186 | 186 | 186 |
| Measurable | |||
| 22 | 40 | 38 | 15 |
| 66 | 11 | 31 | 27 |
| 144 | 1 | 25 | 19 |
| 192 | 0 | 24 | 18 |
| 240 | 1 | 15 | 12 |
| 312 | 0 | 8 | 4 |
| 384 | 0 | 7 | 8 |
| Total | 53 | 148 | 103 |
| Below limit of detection or not reportable | |||
| 22 | 4 | 6 | 29 |
| 66 | 23 | 3 | 7 |
| 144 | 27 | 3 | 9 |
| 192 | 28 | 4 | 10 |
| 240 | 20 | 6 | 9 |
| 312 | 14 | 6 | 10 |
| 384 | 17 | 10 | 9 |
| Total | 133 | 38 | 83 |
Dosage adjustment occurred in line with the trial protocol. The mean patient dose of levosimendan was 18.2 mg during the 24-hour administration period (Table 18).
| Levosimendan dose | Mean | Median | SD |
|---|---|---|---|
| mg/24 hours | 18.200 | 18.600 | 8.10 |
| µg/kg/minute | 0.167 | 0.198 | 0.06 |
The AUC was calculated for individual patients using the trapezoid rule; it was calculated only for patients with at least three concentration measurements (Table 19). Data relating to assay results below the limit of quantification were excluded from the analyses (Figures 22 and 23).
| Parameter | Mean | Median | IQR |
|---|---|---|---|
| Maximum levosimendan concentration (mg/l) | 30.9 | 28.4 | 21.0 |
| Maximum OR-1855 concentration (mg/l) | 2.6 | 1.1 | 1.9 |
| OR-1855 AUC (mg × hour/l) | 397.4 | 126.6 | 327.4 |
| Maximum OR-1896 concentration (mg/l) | 4.0 | 1.6 | 5.1 |
| OR-1896 AUC (mg × hour/l) | 679.1 | 394.6 | 1005.9 |
FIGURE 22.
Relationship between total levosimendan dose (mg/kg/24 hours) and maximum levosimendan concentration (mg/l) (a), maximum OR-1855 concentration (mg/l) (b) and maximum OR-1896 concentration (mg/l) (c).



FIGURE 23.
Relationship between total levosimendan dose (mg/kg/24 hours) and OR-1855 AUC (mg × hour/l) (a) and OR-1896 AUC (mg × hour/l) (b).

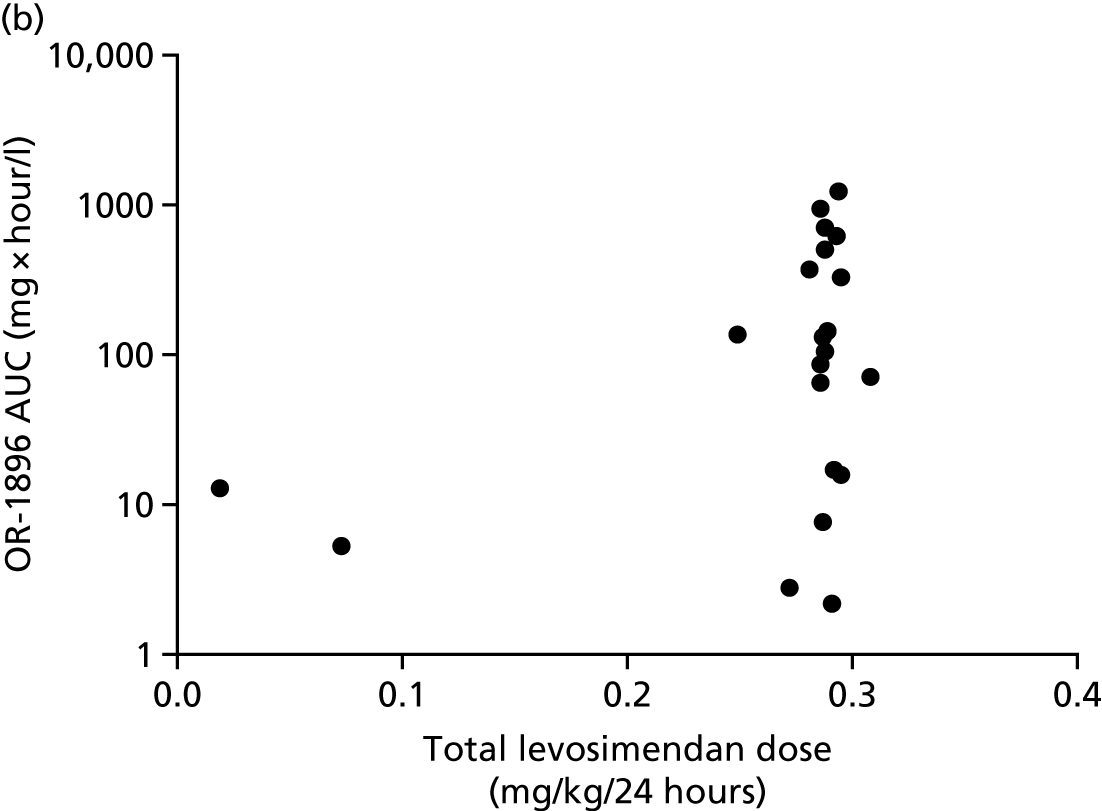
The concentration–time profiles for levosimendan and both metabolites (OR-1855 and OR-1896) are shown in Figures 24–26, respectively. Levosimendan undergoes rapid clearance in the first 48 hours. The peak concentration of both metabolites occurs at 6 days, with both metabolites detectable at 16 days. The mean maximum OR-1855 concentration was 2.8 mg/l, with an AUC of 615.9 mg × hour/l (see Figure 25). The mean maximum OR-1896 concentration was 4.0 mg/l, with an AUC of 962.9 mg × hour/l (see Figure 26).
FIGURE 24.
Mean levosimendan concentration following infusion. The range is the standard error of the mean; n is the number of observations at each time point.
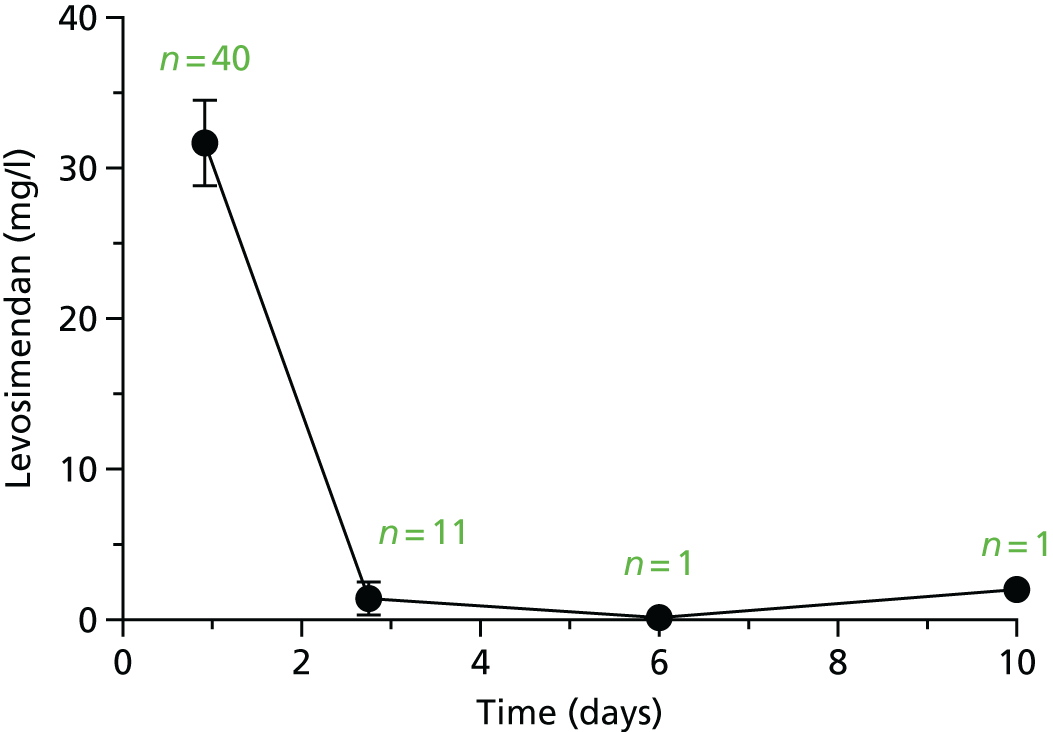
FIGURE 25.
Mean OR-1855 concentration following levosimendan infusion. The range is the standard error of the mean; n is the number of observations at each time point.

FIGURE 26.
Mean OR-1896 concentration following levosimendan infusion. The range is the standard error of the mean; n is the number of observations at each time point.
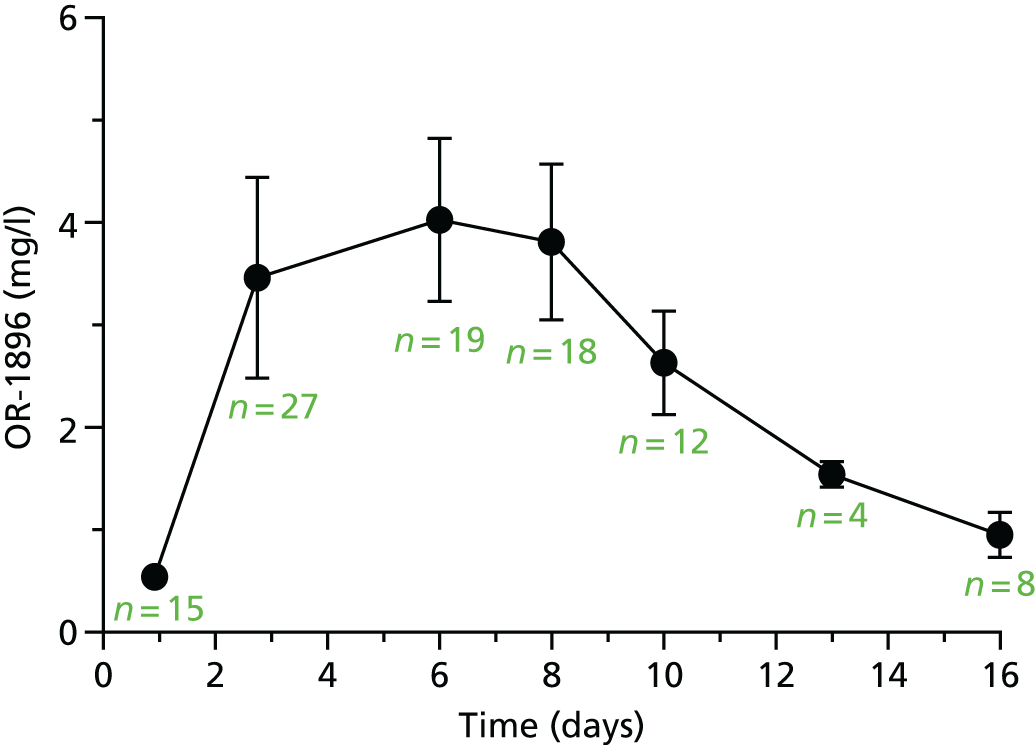
There was a linear relationship between the maximum OR-1855 and OR-1896 concentrations for each patient (Pearson’s r = 0.60; p = 0.0004) and the OR-1855 and OR-1896 AUC for each patient (Pearson’s r = 0.58; p = 0.0035) (Figure 27).
FIGURE 27.
Relationship between the maximum OR-1855 and OR-1896 concentrations for each patient (a) and the maximum OR-1855 AUC and OR-1896 AUC for each patient (b).
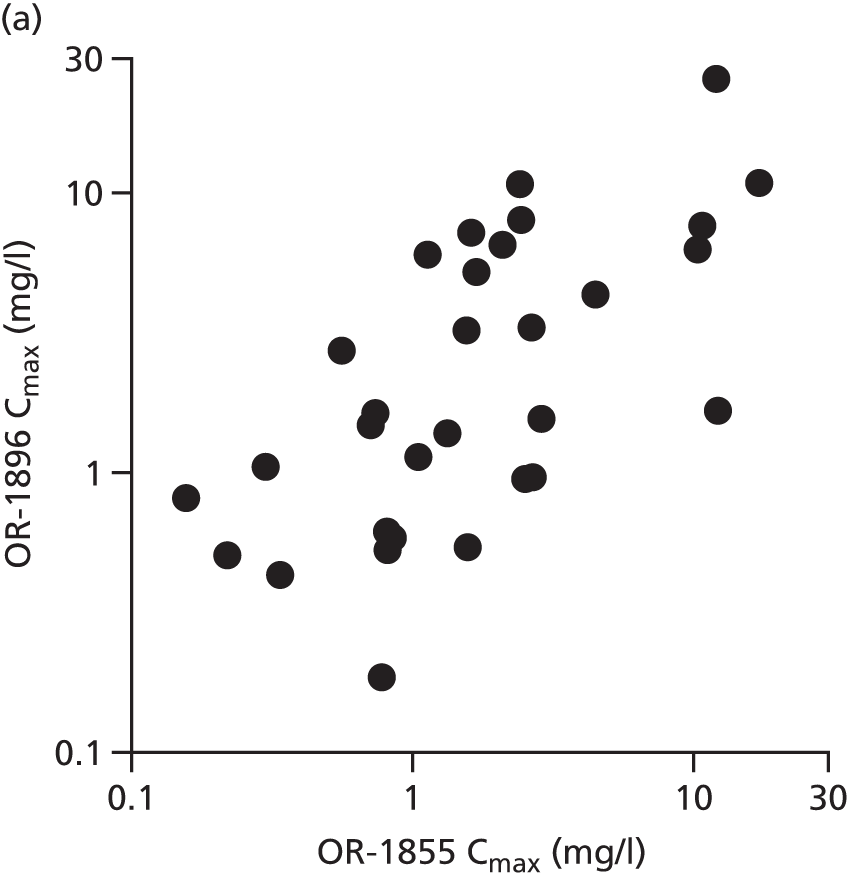
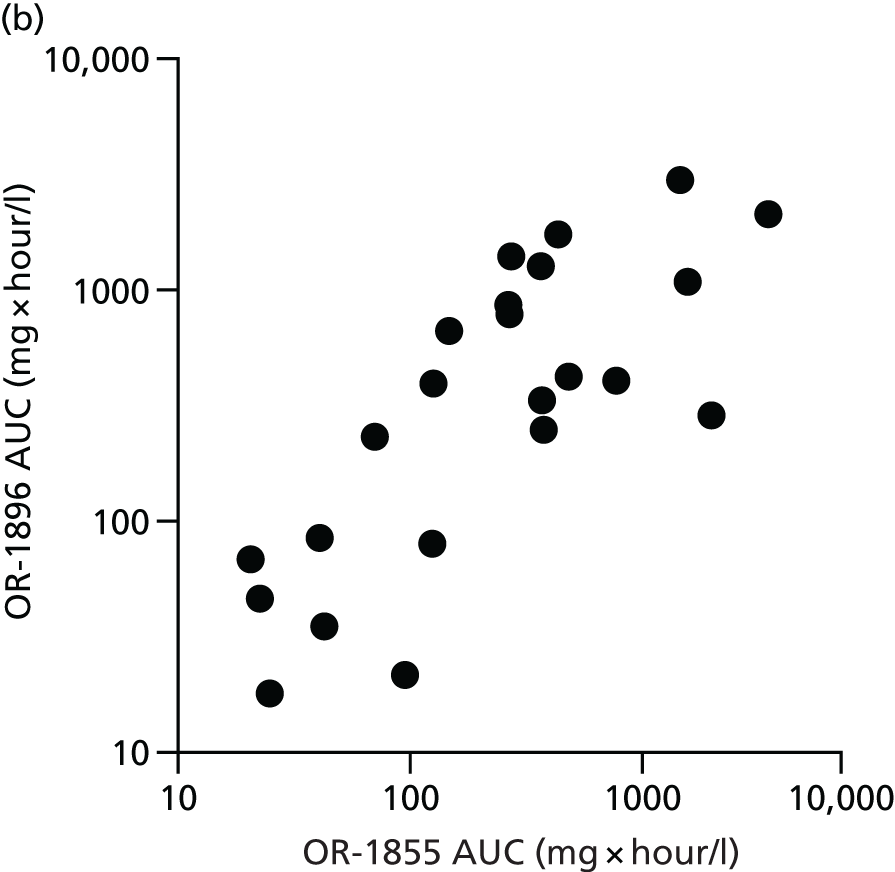
The impact of renal function (peak creatinine level) and RRT is shown in Figures S12–S16 (see Report Supplementary Material 1). No significant differences in metabolite exposure and the use of RRT were found. The influence of liver function (liver component of the SOFA score) on peak metabolite concentration and the AUC is shown in Figures S17 and S18 (see Report Supplementary Material 1); no effects on metabolite levels were found.
Chapter 5 Discussion
In this multicentre, double-blind randomised clinical trial levosimendan did not reduce organ dysfunction or mortality when added to standard care for adult patients who had septic shock. Patients treated with levosimendan required more noradrenaline, had a higher heart rate and required mechanical ventilation for longer. There was also a higher rate of supraventricular tachycardias.
Cardiovascular resuscitation is an essential element of sepsis management. 46 However, there is ever-increasing evidence that high doses of catecholamine infusions16,66 and high circulating levels of catecholamines are associated with worse outcomes and more AEs. 17 Alternative non-catecholamine vasopressor and inotrope options are thus being investigated. 66 Levosimendan produces its inotropic action through different mechanisms from the mechanism of action of catecholamines. By sensitising cardiomyocytes to existing levels of intracellular calcium, an increased myocardial contraction is achieved with a minimal increase in myocardial oxygen demand. This is in contrast to catecholamines, which increase the myocardial oxygen demand. 67 As calcium levels fall in diastole, relaxation of the myocardium is not impaired with levosimendan, and this lusitropic effect may be an additional benefit over catecholamines. 68 Although levosimendan has a half-life of about 1 hour, its active metabolite OR-1896 has a long half-life, and a single 24-hour infusion of levosimendan should provide haemodynamic effects over a week,69 long enough to cover the majority of cases of septic shock. 70
Levosimendan may have other important non-inotropic effects. It opens ATP-sensitive potassium channels in vascular smooth muscle leading to vasodilatation. It may also be protective to the heart and other organs, especially in ischaemia/reperfusion injury. 9,71 Additional potential properties of levosimendan, including anti-inflammatory,52 antioxidative10 and anti-apoptotic effects,72 have all been described.
In view of these pleiotropic effects beyond just inotropy, combined with the fact that myocardial dysfunction is present in > 50% of the septic shock population,12 it may not be evident at the onset of shock and it may also go unrecognised even using cardiac output monitoring,73 we decided to recruit all patients who had septic shock. However, four subgroup analyses were planned to examine the effect of levosimendan in higher risk patients, including those with a low cardiac output, those with impaired oxygen delivery to the tissues and those on high doses of catecholamines. There was no evidence of a beneficial effect of levosimendan in any of these prespecified subgroups with regard to either mean SOFA scores or mortality.
A number of preclinical and small clinical trials have demonstrated a potential benefit of levosimendan for renal,30 liver35 and pulmonary31 function in sepsis. Therefore, the mean daily SOFA score was chosen in this study as the primary outcome to assess the effect of levosimendan on multiple organ function. However, there was no evidence of any beneficial effect on the total SOFA score, either in the whole study population or in any of the predefined subgroups. No benefit was seen for any individual component of the SOFA score nor for any other clinical outcome. The cardiovascular SOFA score was higher in the levosimendan group, reflecting the higher doses of noradrenaline required to maintain MAP.
The detailed analyses of physiological variables over time also did not demonstrate any clear evidence of clinical benefit with regard to individual organ function. Not surprisingly, the cardiac index and central venous saturations tended to be higher in the levosimendan group, reflecting its known inotropic effect. However, this did not lead to any benefits for other organ function, such as liver or kidney function. Although levosimendan does not stimulate beta-adrenoreceptors, we saw a significantly higher heart rate in the levosimendan group, most likely because of vasodilatation but possibly related to the increased requirement for noradrenaline. 63 Although noradrenaline is an alpha-adrenoreceptor agonist causing vasoconstriction, it does have some beta-adrenoreceptor agonist effects and may increase the heart rate compared with non-catecholamine vasoconstrictors. 63 Similarly, there was a higher rate of tachyarrhythmias in levosimendan-treated patients. These may have contributed to the lack of overall clinical benefit, which is consistent with data suggesting a potential benefit of treating persistent tachycardia in sepsis using beta-blockers. 74 This hypothesis is currently being investigated in the STRESS-L [STudy into the REversal of Septic Shock with Landiolol (Beta Blockade)] trial (NIHR EME reference no. 14/150/85, EudraCT number 2017-001785-14; ISRCTN12600919).
Interestingly, the levosimendan group was less likely to be successfully weaned from mechanical ventilation over 28 days. Levosimendan has been reported to sensitise diaphragmatic muscle to calcium, improve contractility and reverse the development of fatigue after muscle loading. 48 Combined with the prolonged inotropic effect of levosimendan and its active metabolite, levosimendan might have been expected to improve weaning from mechanical ventilation. Why the opposite effect was observed remains unclear, especially as there was no difference in intravenous fluid administration nor fluid balance between treatment groups. It is possible that the increased use of noradrenaline in the levosimendan group may have contributed to further catecholamine-induced myocardial dysfunction. The PaO2/FiO2 ratio was also lower in the levosimendan participants on day 1 but there was then a more rapid improvement over time after this. It is possible that any vasodilatation in the first 24 hours may have contributed to an early ventilation/perfusion mismatch.
We also carried out analyses on inflammatory biomarkers over time to assess levosimendan’s anti-inflammatory effects, which have previously been described. 52 Despite measuring a number of both pro- and anti-inflammatory markers, we found no clear effect of levosimendan on these circulating biomarkers.
Limitations
There were limitations to this study that must be considered. This was a trial of levosimendan added to standard care rather than a comparison of levosimendan against an alternative inotrope such as dobutamine. Fewer than 10% of patients in the standard care group received dobutamine. However, there was no difference in outcome (mean SOFA score or mortality) in the prespecified subgroup analysis of patients with a low cardiac index. It should be noted that the number of patients with a measured low cardiac index was small (n = 52) and therefore the study lacks power to fully assess the effect of levosimendan in this subgroup of patients. Similarly, no echocardiographic analyses were performed to provide more detailed information about changes in myocardial function with levosimendan treatment. Therefore, this trial cannot provide guidance as to which inotrope is best for use in the management of sepsis if a low cardiac index is present and an inotrope is being considered. However, we did measure troponin as a marker of myocardial injury and NT-proBNP as a marker of ventricular dysfunction. Surprisingly, those patients with the highest troponin and NT-proBNP levels tended to have higher mean SOFA scores and mortality when treated with levosimendan. Similar to the tachycardia effect described earlier, it is not clear if this is caused by levosimendan directly or is the consequence of the increased noradrenaline use, but it does provide further support for the concept that low cardiac output states in sepsis should not be routinely treated with inotropic drugs.
The dose of levosimendan in this trial was titrated between 0.05 and 0.2 µg/kg/minute in an attempt to administer the maximum tolerated dose. However, it has been reported that a dose of 0.1 µg/kg/minute may be as effective as 0.2 µg/kg/minute and may avoid excessive hypotension. 75 The pharmacokinetic analysis revealed that plasma levels of the parent drug and its active metabolites were very similar to levels seen in other patient cohorts and, importantly, there was no effect of acute kidney or liver failure on these drug levels. This suggests that the dosing regimen used in this trial was appropriate.
It is also worth noting that the target MAP of 65–70 mmHg recommended in the protocol and reiterated regularly at all investigator meetings was frequently exceeded. This is commonly seen in other shock trials61,68 and suggests that the noradrenaline doses administered could have been reduced in both treatment groups.
Future research work
Although this study found no evidence of any clinically meaningful benefits of levosimendan for patients with septic shock, it is still unclear if there is any role for inotropic therapy in the management of septic shock in patients with an extremely low cardiac output. Future studies could compare levosimendan against dobutamine and against placebo to test if there is any benefit from either inotrope for this specific but limited indication.
Although the use of the mean SOFA score as the primary outcome in this study has provided very clear answers about the effects of levosimendan, it is not a perfect outcome measure for a Phase II trial. It is not a patient-centred outcome. However, there are limitations to all currently used outcomes in Phase II trials in sepsis. Future work should investigate the different possible outcomes and highlight their relative strengths and weaknesses.
Conclusions
Among adult patients with septic shock levosimendan when added to standard care does not reduce organ dysfunction or mortality. Patients allocated to the levosimendan group required more noradrenaline, were less likely to be successfully weaned from mechanical ventilation and had more tachycardia and a higher rate of supraventricular arrhythmias, but there were no overall differences in SAEs between the groups.
Acknowledgements
Funding and support
This project was funded by the Efficacy and Mechanism Evaluation (EME) programme (11/14/08), a Medical Research Council (MRC) and NIHR partnership, and by a NIHR Clinician Scientist Award held by Professor Gordon (NIHR/CS/009/007). Study drugs and matching placebo were provided free of charge by Orion Pharmaceuticals and additional research funds were provided by Tenax Therapeutics.
The study, including the ICTU core staff and the InForm team, was supported by the NIHR Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London and also by the UK Intensive Care Foundation.
LeoPARDS trial investigators
Imperial Clinical Trials Unit: Deborah Ashby, Sandra Griffiths, Mary Cross, Jonas Lexow, Farah Al-Beidh, Ashley Campbell, Janis Best-Lane, Alexina Mason, Gregory Frazier and Shalini Santhakumaran.
Trial Steering Committee: Simon Baudouin (Independent Chairperson), Anthony Gordon, Mary Cross, Alexina Mason, Nabila Youssouf, Robert Orme, Deborah Ashby, Andrew Rhodes (Independent Member), Phil Hopkins, (Independent Member), Simon Denegri (Independent Lay Member) and Catherine White (Independent Lay Member).
Data Monitoring Ethics Committee: Julian Bion (Chairperson), Duncan Wyncoll and Graeme MacLennan.
Site investigators
Charing Cross Hospital: Maie Templeton, Kirsty Gladas, Adaeze Ochelli-Okpue and Anthony Gordon (PI).
Hammersmith Hospital: Dorota Banach, Rosemary Matthew-Thomas and Stephen Brett (PI).
Cheltenham General Hospital and Gloucester Royal Infirmary: Phillipa Rusher, Kelly Matthews, Natalie Bynorth and Robert Orme (PI).
Antrim Area Hospital: Orla O’Neill, Emma McKay and Christopher Nutt (PI).
Royal Cornwall Hospital: Karen Burt, Anna Fouracres and Jonathan Paddle (PI).
Royal Victoria Hospital, Belfast: Vanessa Quinn, Leona Bannon, Lia McNamee, Grianiae White and James McNamee (PI).
Birmingham Heartlands Hospital: Teresa Melody, Joanne Gresty, Anna Dennis, Lucie Linhartova and Gavin Perkins (PI).
Bradford Royal Infirmary: Martin Northey and Stephen Fletcher (PI).
Altnagelvin Hospital, Derry: Sinead O’Kane and Noel Hemmings (PI).
James Paget University Hospital, Great Yarmouth: Lynn Everett, Christian Hacon and Hazel Stuart (PI).
Hull Royal Infirmary: Neil Smith, Caroline Abernathy, Vicky Martinson and Andrew Gratix (PI).
Queen Elizabeth Hospital, Kings Lynn: Kate Wong, Leilani Cabreros and Darcy Pearson (PI).
St James’s Hospital, Leeds: Stuart Elliot, Zoe Beardow, Elizabeth Wilby and Andrew Breen (PI).
University College London Hospital: Georgia Bercades, Jung Ryu, Magda Pinto-Rocha and David Brealey (PI).
Manchester Royal Infirmary: Richard Clark, Rachel Pearson, Jon Bannard Smith, Douglas Atkinson and Daniel Conway (PI).
Whythenshawe Hospital, South Manchester: Christine Bowyer, Peter Alexander, Timothy Felton, Jon Hopper and Andrew Bentley (PI).
James Cook University Hospital, Middlesbrough: Keith Hugill, Emanuel Cirstea and Jost Mullenheim (PI).
Norfolk and Norwich University Hospital: Melissa Rosbergen and Jurgens Nortje (PI).
Queen’s Medical Centre, Nottingham: Claudia Woodford, Lucy Ryan, Marc Chikani, Andrew Sharman and Daniel Harvey (PI).
Derriford Hospital, Plymouth: Susan Tyson and Peter MacNaugton (PI).
Poole Hospital: Julie Camasooksai, Sarah Patch, Sarah Jenkins, Lee Tbaily, Helena Bancroft-Barnes, Nicola Venner and Henrik Reschreiter (PI).
Queen Alexandra Hospital, Portsmouth: Johanna Mouland, Steve Rose, Lindsey Roberts, Nicola Lamb, Zoe Daly and David Pogson (PI).
Whiston Hospital, Prescot: Susan Dowling, Amanda McCairn and Kevin Sim (PI).
Royal Preston Hospital: Jacqueline Baldwin, Alexandra Williams, Mark Verlander and Shondipon Laha (PI).
City General Hospital, Stoke-on-Trent: Ruth Salt, Minerva Gellamucho and Nick Coleman (PI).
Musgrove Park Hospital, Taunton: Patricia Doble, Moira Tait and Richard Innes (PI).
Medway Maritime Hospital, Gillingham: James Cullinane, Claire Pegg and Nandita Divekar (PI).
Ipswich Hospital: Stephanie Bell, Heather Baylock and Richard Howard-Griffin (PI).
Kettering General Hospital: Parizade Raymode, Laslo Hollos, Dumisani Ncomanzi and Phil Watt (PI).
Leicester Royal Infirmary: Dawn Hales, Natalie Rich, Neil Flint and Simon Scott (PI).
Royal Free Hospital, London: Sarah James and Daniel Martin (PI).
Pinderfields Hospital, Wakefield: Sarah Buckley and Alistair Rose (PI).
Contributions of authors
Professor Anthony Gordon and Dr Shalini Santhakumaran had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Anthony C Gordon (Professor of Critical Care, Imperial College London and Imperial College Healthcare NHS Trust) (Chief Investigator) was responsible for the study concept and design; acquisition, analysis and interpretation of the data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; and statistical analysis. He also provided administrative, technical or material support and study supervision at the different sites.
Shalini Santhakumaran (Research Statistician, ICTU) was the trial statistician. She was responsible for the acquisition, analysis and interpretation of the data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; and statistical analysis.
Farah Al-Beidh (Trial Manager, Imperial College London) oversaw the management of the trial. She was responsible for the acquisition, analysis, or interpretation of the data; drafting of the manuscript; and critical revision of the manuscript for important intellectual content. She also provided administrative, technical and material support at the different sites.
Robert ML Orme (Consultant of Intensive Care Medcine, Cheltenham General Hospital) (Co-investigator) was responsible for the study concept and design; acquisition, analysis, or interpretation of the data; and critical revision of the manuscript for important intellectual content. He also provided study supervision at the different sites.
Gavin D Perkins (Professor of Intensive Care Medicine, University of Warwick and Heart of England NHS Foundation Trust) (Co-investigator) was responsible for the study concept and design; acquisition, analysis and interpretation of the data; and critical revision of the manuscript for important intellectual content. He also provided study supervision at the different sites.
Mervyn Singer (Professor of Intensive Care Medcine, University College London) (Co-investigator) was responsible for the study concept and design; acquisition, analysis and interpretation of the data; and critical revision of the manuscript for important intellectual content. He also provided study supervision at the different sites.
Daniel F McAuley (Professor of Intensive Care Medicine, Royal Victoria and Queen’s University, Belfast) (Co-investigator) was responsible for the study concept and design; acquisition, analysis and interpretation of the data; and critical revision of the manuscript for important intellectual content. He also provided study supervision at the different sites.
Alexina J Mason (Assistant Professor of Medical Statistics, London School of Hygiene & Tropical Medicine) was the trial statistician. She was responsible for the acquisition, analysis and interpretation of the data; critical revision of the manuscript for important intellectual content; and statistical analysis.
Josie K Ward (Research Assistant, Department of Surgery and Cancer, Imperial College London) was responsible for the biomarker assays; acquisition, analysis and interpretation of the data; and critical revision of the manuscript for important intellectual content.
Kieran P O’Dea (Senior Research Fellow in the Department Surgery and Cancer at Imperial College London) was responsible for the biomarker assays; acquisition, analysis and interpretation of the data; and critical revision of the manuscript for important intellectual content.
Timothy Felton (Consultant of Intensive Care Medicine, University of Manchester) was the pharmacokinetic analysis lead. He was responsible for the acquisition, analysis and interpretation of data and critical revision of the manuscript for important intellectual content.
Mary Cross (Operations Manager, Imperial College London) oversaw the management of the trial. She was responsible for the study concept and design; acquisition, analysis and interpretation of the data; and critical revision of the manuscript for important intellectual content. She also provided administrative, technical or material support and study supervision at the different sites.
Janis Best-Lane (Trial Monitor, Imperial College London) oversaw the monitoring of the trial. She was responsible for the acquisition, analysis and interpretation of the data and critical revision of the manuscript for important intellectual content. She also provided administrative, technical and material support at the different sites.
Jonas Lexow (Trial Manager, Imperial College London) oversaw the management of the trial. He was responsible for the acquisition, analysis and interpretation of the data and critical revision of the manuscript for important intellectual content. He also provided administrative, technical and material support at the different sites.
Ashley Campbell (Trial Monitor, Imperial College London) oversaw the monitoring of the trial. She was responsible for the acquisition, analysis and interpretation of the data and critical revision of the manuscript for important intellectual content. He also provided administrative, technical and material support at the different sites.
Deborah Ashby (Chair in Medical Statistics and Clinical Trials, Co-Director of ICTU and Deputy Head of Imperial School of Public Health) was the senior statistician for the trial. She was responsible for the study concept and design; acquisition, analysis and interpretation of the data; critical revision of the manuscript for important intellectual content; and statistical analysis.
Publications
Orme RM, Perkins GD, McAuley DF, Liu KD, Mason AJ, Morelli A, et al. An efficacy and mechanism evaluation study of Levosimendan for the Prevention of Acute oRgan Dysfunction in Sepsis (LeoPARDS): protocol for a randomized controlled trial. Trials 2014;15:199.
Gordon AC, Perkins GD, Singer M, McAuley DF, Orme RM, Santhakumaran S, et al. Levosimendan for the prevention of acute organ dysfunction in sepsis. New Engl J Med 2016;375:1638–48.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Harrison DA, Welch CA, Eddleston JM. The epidemiology of severe sepsis in England, Wales and Northern Ireland, 1996 to 2004: secondary analysis of a high quality clinical database, the ICNARC Case Mix Programme Database. Crit Care 2006;10. https://doi.org/10.1186/cc4854.
- Vincent JL, Moreno R, Takala J, Willatts S, De Mendonça A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med 1996;22:707-10. https://doi.org/10.1007/BF01709751.
- Ferreira FL, Bota DP, Bross A, Mélot C, Vincent JL. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA 2001;286:1754-8. https://doi.org/10.1001/jama.286.14.1754.
- Oppert M, Engel C, Brunkhorst FM, Bogatsch H, Reinhart K, Frei U, et al. Acute renal failure in patients with severe sepsis and septic shock – a significant independent risk factor for mortality: results from the German Prevalence Study. Nephrol Dial Transplant 2008;23:904-9. https://doi.org/10.1093/ndt/gfm610.
- Davies A, Green C, Hutton J, Chinn C. Severe sepsis: a European estimate of the burden of disease in ICU. Intensive Care Med 2001;27.
- Gheorghiade M, Teerlink JR, Mebazaa A. Pharmacology of new agents for acute heart failure syndromes. Am J Cardiol 2005;96:68G-73G. https://doi.org/10.1016/j.amjcard.2005.07.023.
- Sorsa T, Pollesello P, Solaro RJ. The contractile apparatus as a target for drugs against heart failure: interaction of levosimendan, a calcium sensitiser, with cardiac troponin c. Mol Cell Biochem 2004;266:87-107. https://doi.org/10.1023/B:MCBI.0000049141.37823.19.
- Papp Z, Edes I, Fruhwald S, De Hert SG, Salmenpera M, Leppikangas H, et al. Levosimendan: molecular mechanisms and clinical implications: consensus of experts on the mechanisms of action of levosimendan. Int J Cardiol 2012;159:82-7. https://doi.org/10.1016/j.ijcard.2011.07.022.
- Buckley JF, Singer M, Clapp LH. Role of KATP channels in sepsis. Cardiovasc Res 2006;72:220-30. https://doi.org/10.1016/j.cardiores.2006.07.011.
- Hasslacher J, Bijuklic K, Bertocchi C, Kountchev J, Bellmann R, Dunzendorfer S, et al. Levosimendan inhibits release of reactive oxygen species in polymorphonuclear leukocytes in vitro and in patients with acute heart failure and septic shock: a prospective observational study. Crit Care 2011;15. https://doi.org/10.1186/cc10307.
- Kankaanranta H, Zhang X, Tumelius R, Ruotsalainen M, Haikala H, Nissinen E, et al. Antieosinophilic activity of simendans. J Pharmacol Exp Ther 2007;323:31-8. https://doi.org/10.1124/jpet.107.124057.
- Vieillard-Baron A, Caille V, Charron C, Belliard G, Page B, Jardin F. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med 2008;36:1701-6. https://doi.org/10.1097/CCM.0b013e318174db05.
- Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med 2007;35:1599-608. https://doi.org/10.1097/01.CCM.0000266683.64081.02.
- Dünser MW, Hasibeder WR. Sympathetic overstimulation during critical illness: adverse effects of adrenergic stress. J Intensive Care Med 2009;24:293-316. https://doi.org/10.1177/0885066609340519.
- Annane D, Vignon P, Renault A, Bollaert PE, Charpentier C, Martin C, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007;370:676-84. https://doi.org/10.1016/S0140-6736(07)61344-0.
- Dünser MW, Ruokonen E, Pettilä V, Ulmer H, Torgersen C, Schmittinger CA, et al. Association of arterial blood pressure and vasopressor load with septic shock mortality: a post hoc analysis of a multicenter trial. Crit Care 2009;13. https://doi.org/10.1186/cc8167.
- Boldt J, Menges T, Kuhn D, Diridis C, Hempelmann G. Alterations in circulating vasoactive substances in the critically ill – a comparison between survivors and non-survivors. Intensive Care Med 1995;21:218-25. https://doi.org/10.1007/BF01701475.
- Rehberg S, Ertmer C, Vincent JL, Spiegel HU, Köhler G, Erren M, et al. Effects of combined arginine vasopressin and levosimendan on organ function in ovine septic shock. Crit Care Med 2010;38:2016-23. https://doi.org/10.1097/CCM.0b013e3181ef4694.
- Zager RA, Johnson AC, Lund S, Hanson SY, Abrass CK. Levosimendan protects against experimental endotoxemic acute renal failure. Am J Physiol Renal Physiol 2006;290:F1453-62. https://doi.org/10.1152/ajprenal.00485.2005.
- Fries M, Ince C, Rossaint R, Bleilevens C, Bickenbach J, Rex S, et al. Levosimendan but not norepinephrine improves microvascular oxygenation during experimental septic shock. Crit Care Med 2008;36:1886-91. https://doi.org/10.1097/CCM.0b013e31817cede9.
- Dubin A, Murias G, Sottile JP, Pozo MO, Barán M, Edul VS, et al. Effects of levosimendan and dobutamine in experimental acute endotoxemia: a preliminary controlled study. Intensive Care Med 2007;33:485-94. https://doi.org/10.1007/s00134-006-0519-5.
- García-Septien J, Lorente JA, Delgado MA, de Paula M, Nin N, Moscoso A, et al. Levosimendan increases portal blood flow and attenuates intestinal intramucosal acidosis in experimental septic shock. Shock 2010;34:275-80. https://doi.org/10.1097/SHK.0b013e3181cd8c5b.
- Cunha-Goncalves D, Perez-de-Sa V, Grins E, Dahm PL, Thörne J, Blomquist S. Inotropic support during experimental endotoxemic shock: part I. The effects of levosimendan on splanchnic perfusion. Anesth Analg 2009;109:1568-75. https://doi.org/10.1213/ane.0b013e3181af3fe3.
- Cunha-Goncalves D, Perez-de-Sa V, Larsson A, Thörne J, Blomquist S. Inotropic support during experimental endotoxemic shock: part II. A comparison of levosimendan with dobutamine. Anesth Analg 2009;109:1576-83. https://doi.org/10.1213/ane.0b013e3181af40e0.
- De Backer D, Bracht H. Levosimendan in early sepsis: when good ideas give poor results. Anesth Analg 2009;109:1367-9. https://doi.org/10.1213/ANE.0b013e3181b9e91a.
- Noto A, Giacomini M, Palandi A, Stabile L, Reali-Forster C, Iapichino G. Levosimendan in septic cardiac failure. Intensive Care Med 2005;31:164-5. https://doi.org/10.1007/s00134-004-2502-3.
- Matejovic M, Krouzecky A, Radej J, Novak I. Successful reversal of resistant hypodynamic septic shock with levosimendan. Acta Anaesthesiol Scand 2005;49:127-8. https://doi.org/10.1111/j.1399-6576.2005.00541.x.
- Ramaswamykanive H, Bihari D, Solano TR. Myocardial depression associated with pneumococcal septic shock reversed by levosimendan. Anaesth Intensive Care 2007;35:409-13.
- Powell BP, De Keulenaer BL. Levosimendan in septic shock: a case series. Br J Anaesth 2007;99:447-8. https://doi.org/10.1093/bja/aem225.
- Morelli A, De Castro S, Teboul JL, Singer M, Rocco M, Conti G, et al. Effects of levosimendan on systemic and regional hemodynamics in septic myocardial depression. Intensive Care Med 2005;31:638-44. https://doi.org/10.1007/s00134-005-2619-z.
- Morelli A, Teboul JL, Maggiore SM, Vieillard-Baron A, Rocco M, Conti G, et al. Effects of levosimendan on right ventricular afterload in patients with acute respiratory distress syndrome: a pilot study. Crit Care Med 2006;34:2287-93. https://doi.org/10.1097/01.CCM.0000230244.17174.4F.
- Morelli A, Donati A, Ertmer C, Rehberg S, Lange M, Orecchioni A, et al. Levosimendan for resuscitating the microcirculation in patients with septic shock: a randomized controlled study. Crit Care 2010;14. https://doi.org/10.1186/cc9387.
- Alhashemi JA, Alotaibi QA, Abdullah GM, Shalabi SA. Levosimendan vs dobutamine in septic shock. J Crit Care 2009;24:e14-5. https://doi.org/10.1016/j.jcrc.2009.06.006.
- Vaitsis J, Michalopoulou H, Thomopoulos C. Use of levosimendan in myocardial dysfunction due to sepsis. Crit Care 2009;13. https://doi.org/10.1186/cc7329.
- Memis D, Inal MT, Sut N. The effects of levosimendan vs dobutamine added to dopamine on liver functions assessed with noninvasive liver function monitoring in patients with septic shock. J Crit Care 2012;27:318.e1-6. https://doi.org/10.1016/j.jcrc.2011.06.008.
- Morelli A, Ertmer C, Rehberg S. Effects of levosimendan on renal function in septic shock: a case control study. Crit Care 2009;13. https://doi.org/10.1186/cc7334.
- Yilmaz MB, Yalta K, Yontar C, Karadas F, Erdem A, Turgut OO, et al. Levosimendan improves renal function in patients with acute decompensated heart failure: comparison with dobutamine. Cardiovasc Drugs Ther 2007;21:431-5. https://doi.org/10.1007/s10557-007-6066-7.
- Pinto BB, Rehberg S, Ertmer C, Westphal M. Role of levosimendan in sepsis and septic shock. Curr Opin Anaesthesiol 2008;21:168-77. https://doi.org/10.1097/ACO.0b013e3282f43c56.
- Reinhart K, Brunkhorst FM, Bone HG, Bardutzky J, Dempfle CE, Forst H, et al. Prevention, diagnosis, therapy and follow-up care of sepsis: 1st revision of S-2k guidelines of the German Sepsis Society (Deutsche Sepsis-Gesellschaft e.V. (DSG)) and the German Interdisciplinary Association of Intensive Care and Emergency Medicine (Deutsche Interdisziplinäre Vereinigung für Intensiv- und Notfallmedizin (DIVI)). Ger Med Sci 2010;8. https://doi.org/10.3205/000103.
- Alberti C, Brun-Buisson C, Goodman SV, Guidici D, Granton J, Moreno R, et al. Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med 2003;168:77-84. https://doi.org/10.1164/rccm.200208-785OC.
- Orme RM, Perkins GD, McAuley DF, Liu KD, Mason AJ, Morelli A, et al. An efficacy and mechanism evaluation study of Levosimendan for the Prevention of Acute oRgan Dysfunction in Sepsis (LeoPARDS): protocol for a randomized controlled trial. Trials 2014;15. https://doi.org/10.1186/1745-6215-15-199.
- Gordon AC, Perkins GD, Singer M, McAuley DF, Orme RM, Santhakumaran S, et al. Levosimendan for the prevention of acute organ dysfunction in sepsis. N Engl J Med 2016;375:1638-48. https://doi.org/10.1056/NEJMoa1609409.
- Brunkhorst FM, Engel C, Bloos F, Meier-Hellmann A, Ragaller M, Weiler N, et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008;358:125-39. https://doi.org/10.1056/NEJMoa070716.
- Perner A, Haase N, Guttormsen AB, Tenhunen J, Klemenzson G, Åneman A, et al. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012;367:124-34. https://doi.org/10.1056/NEJMoa1204242.
- Myburgh JA, Finfer S, Bellomo R, Billot L, Cass A, Gattas D, et al. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012;367:1901-11. https://doi.org/10.1056/NEJMoa1209759.
- Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013;41:580-637. https://doi.org/10.1097/CCM.0b013e31827e83af.
- Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, et al. Acute Kidney Injury Network . Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 2007;11. https://doi.org/10.1186/cc5713.
- Doorduin J, Sinderby CA, Beck J, Stegeman DF, van Hees HW, van der Hoeven JG, et al. The calcium sensitizer levosimendan improves human diaphragm function. Am J Respir Crit Care Med 2012;185:90-5. https://doi.org/10.1164/rccm.201107-1268OC.
- Palevsky PM, Molitoris BA, Okusa MD, Levin A, Waikar SS, Wald R, et al. Design of clinical trials in acute kidney injury: report from an NIDDK workshop on trial methodology. Clin J Am Soc Nephrol 2012;7:844-50. https://doi.org/10.2215/CJN.12791211.
- Charpentier J, Luyt CE, Fulla Y, Vinsonneau C, Cariou A, Grabar S, et al. Brain natriuretic peptide: a marker of myocardial dysfunction and prognosis during severe sepsis. Crit Care Med 2004;32:660-5. https://doi.org/10.1097/01.CCM.0000114827.93410.D8.
- Post F, Weilemann LS, Messow CM, Sinning C, Münzel T. B-type natriuretic peptide as a marker for sepsis-induced myocardial depression in intensive care patients. Crit Care Med 2008;36:3030-7. https://doi.org/10.1097/CCM.0b013e31818b9153.
- Wang Q, Yokoo H, Takashina M, Sakata K, Ohashi W, Abedelzaher LA, et al. Anti-inflammatory profile of levosimendan in cecal ligation-induced septic mice and in lipopolysaccharide-stimulated macrophages. Crit Care Med 2015;43:e508-20. https://doi.org/10.1097/CCM.0000000000001269.
- Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, et al. ARDS Network . Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med 2017;195:331-8. https://doi.org/10.1164/rccm.201603-0645OC.
- Gordon AC, Mason AJ, Thirunavukkarasu N, Perkins GD, Cecconi M, Cepkova M, et al. Early vasopressin vs norepinephrine on kidney failure in patients with septic shock: the VANISH randomized clinical trial. JAMA 2016;316:509-18. https://doi.org/10.1001/jama.2016.10485.
- Daniels MJ, Hogan JW. Missing Data in Longitudinal Studies: Strategies for Bayesian Modeling and Sensitivity Analysis. New York, NY: Chapman & Hall/CRC; 2008.
- Anderson MJ, Ter Braak CJF. Permutation tests for multi-factorial analysis of variance. J Stat Comput Sim 2003;73:85-113. https://doi.org/10.1080/00949650215733.
- Henderson R, Diggle P, Dobson A. Joint modelling of longitudinal measurements and event time data. Biostatistics 2000;1:465-80. https://doi.org/10.1093/biostatistics/1.4.465.
- Gordon AC, Mason AJ, Perkins GD, Stotz M, Terblanche M, Ashby D, et al. The interaction of vasopressin and corticosteroids in septic shock: a pilot randomized controlled trial. Crit Care Med 2014;42:1325-33. https://doi.org/10.1097/CCM.0000000000000212.
- Al-Mohammad A, Mant J, Laramee P, Swain S. Chronic Heart Failure Guideline Development Group . Diagnosis and management of adults with chronic heart failure: summary of updated NICE guidance. BMJ 2010;341. https://doi.org/10.1136/bmj.c4130.
- Harvey S, Harrison DA, Singer M, Ashcroft J, Jones CM, Elbourne D, et al. Assessment of the clinical effectiveness of pulmonary artery catheters in management of patients in intensive care (PAC-Man): a randomised controlled trial. Lancet 2005;366:472-7. https://doi.org/10.1016/S0140-6736(05)67061-4.
- Russell JA, Walley KR, Singer J, Gordon AC, Hébert PC, Cooper DJ, et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008;358:877-87. https://doi.org/10.1056/NEJMoa067373.
- Gordon AC, Russell JA, Walley KR, Singer J, Ayers D, Storms MM, et al. The effects of vasopressin on acute kidney injury in septic shock. Intensive Care Med 2010;36:83-91. https://doi.org/10.1007/s00134-009-1687-x.
- Gordon AC, Wang N, Walley KR, Ashby D, Russell JA. The cardio-pulmonary effects of vasopressin compared with norepinephrine in septic shock. Chest 2012;142:593-605. https://doi.org/10.1378/chest.11-2604.
- Moreno R, Sprung CL, Annane D, Chevret S, Briegel J, Keh D, et al. Time course of organ failure in patients with septic shock treated with hydrocortisone: results of the Corticus study. Intensive Care Med 2011;37:1765-72. https://doi.org/10.1007/s00134-011-2334-x.
- World Medical Association Declaration of Helsinki . Ethical principles for medical research involving human subjects. JAMA 2013;310:2191-4. https://doi.org/10.1001/jama.2013.281053.
- Singer M. Catecholamine treatment for shock – equally good or bad?. Lancet 2007;370:636-7. https://doi.org/10.1016/S0140-6736(07)61317-8.
- Ukkonen H, Saraste M, Akkila J, Knuuti MJ, Lehikoinen P, Någren K, et al. Myocardial efficiency during calcium sensitization with levosimendan: a noninvasive study with positron emission tomography and echocardiography in healthy volunteers. Clin Pharmacol Ther 1997;61:596-607. https://doi.org/10.1016/S0009-9236(97)90139-9.
- Zangrillo A, Putzu A, Monaco F, Oriani A, Frau G, De Luca M, et al. Levosimendan reduces mortality in patients with severe sepsis and septic shock: a meta-analysis of randomized trials. J Crit Care 2015;30:908-13. https://doi.org/10.1016/j.jcrc.2015.05.017.
- Kivikko M, Antila S, Eha J, Lehtonen L, Pentikäinen PJ. Pharmacokinetics of levosimendan and its metabolites during and after a 24-hour continuous infusion in patients with severe heart failure. Int J Clin Pharmacol Ther 2002;40:465-71. https://doi.org/10.5414/CPP40465.
- Jörgensen K, Bech-Hanssen O, Houltz E, Ricksten SE. Effects of levosimendan on left ventricular relaxation and early filling at maintained preload and afterload conditions after aortic valve replacement for aortic stenosis. Circulation 2008;117:1075-81. https://doi.org/10.1161/CIRCULATIONAHA.107.722868.
- du Toit EF, Genis A, Opie LH, Pollesello P, Lochner A. A role for the RISK pathway and K(ATP) channels in pre- and post-conditioning induced by levosimendan in the isolated guinea pig heart. Br J Pharmacol 2008;154:41-50. https://doi.org/10.1038/bjp.2008.52.
- Parissis JT, Adamopoulos S, Antoniades C, Kostakis G, Rigas A, Kyrzopoulos S, et al. Effects of levosimendan on circulating pro-inflammatory cytokines and soluble apoptosis mediators in patients with decompensated advanced heart failure. Am J Cardiol 2004;93:1309-12. https://doi.org/10.1016/j.amjcard.2004.01.073.
- Shahul S, Gulati G, Hacker MR, Mahmood F, Canelli R, Nizamuddin J, et al. Detection of myocardial dysfunction in septic shock: a speckle-tracking echocardiography study. Anesth Analg 2015;121:1547-54. https://doi.org/10.1213/ANE.0000000000000943.
- Morelli A, Ertmer C, Westphal M, Rehberg S, Kampmeier T, Ligges S, et al. Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: a randomized clinical trial. JAMA 2013;310:1683-91. https://doi.org/10.1001/jama.2013.278477.
- de Geus HR, Bakker J, Lesaffre EM, le Noble JL. Neutrophil gelatinase-associated lipocalin at ICU admission predicts for acute kidney injury in adult patients. Am J Respir Crit Care Med 2011;183:907-14. https://doi.org/10.1164/rccm.200908-1214OC.
Appendix 1 Additional information
Screening data
| Site | Screened patients (n) | Treatment group, number of randomised patients | Protocol deviations (n) | |
|---|---|---|---|---|
| Levosimendan | Placebo | |||
| Altnagelvin Hospital | 55 | 5 | 5 | 1 |
| Antrim Area Hospital | 51 | 17 | 17 | 3 |
| Birmingham Heartlands | 48 | 19 | 17 | 10 |
| Bradford Royal Infirmary | 19 | 6 | 8 | 6 |
| Charing Cross Hospital | 131 | 13 | 13 | 5 |
| Cheltenham General | 26 | 3 | 3 | 2 |
| City General Hospital | 17 | 2 | 4 | 1 |
| Derriford Hospital | 56 | 4 | 2 | 0 |
| Gloucestershire Royal | 3 | 0 | 1 | 0 |
| Hammersmith Hospital | 100 | 7 | 7 | 2 |
| Hull Royal Infirmary | 91 | 8 | 7 | 5 |
| Ipswich Hospital | 59 | 3 | 3 | 3 |
| James Cook University Hospital | 234 | 3 | 3 | 3 |
| James Paget University Hospital | 28 | 0 | 2 | 0 |
| Kettering General Hospital | 35 | 9 | 9 | 5 |
| Leicester Royal Infirmary | 43 | 8 | 8 | 0 |
| Manchester Royal Infirmary | 33 | 11 | 10 | 4 |
| Medway Maritime Hospital | 54 | 4 | 5 | 2 |
| Musgrove Park Hospital | 27 | 5 | 5 | 1 |
| Norfolk and Norwich University Hospital | 39 | 8 | 6 | 5 |
| Pinderfields Hospital | 18 | 2 | 2 | 0 |
| Poole Hospital | 118 | 13 | 13 | 4 |
| Queen’s Medical Centre | 147 | 9 | 11 | 6 |
| Queen Alexandra Hospital | 160 | 7 | 7 | 1 |
| Queen Elizabeth Hospital | 60 | 9 | 9 | 1 |
| Royal Cornwall Hospital | 55 | 5 | 7 | 2 |
| Royal Free Hospital | 24 | 0 | 1 | 0 |
| Royal Preston Hospital | 36 | 6 | 3 | 0 |
| Royal Victoria Hospital | 59 | 17 | 16 | 3 |
| St James’s Hospital | 38 | 13 | 12 | 7 |
| York Hospital | 46 | 7 | 6 | 1 |
| University College Hospital | 268 | 24 | 24 | 9 |
| University Hospital of South Manchester | 17 | 4 | 3 | 2 |
| Whiston Hospital | 187 | 7 | 8 | 6 |
| Total | 2382 | 258 | 257 | 100 |
Accrual data
FIGURE 28.
Recruitment of patients into the LeoPARDS trial.

Missing baseline data
| Characteristic | Treatment group (n) | Total | |
|---|---|---|---|
| Levosimendan | Placebo | ||
| BMI | 6 | 3 | 9 |
| Organ failure | |||
| Respiratory | 1 | 1 | 2 |
| Haematological | 2 | 2 | 4 |
| Liver | 6 | 5 | 11 |
| Neurological | 34 | 45 | 79 |
| Renal | 0 | 1 | 1 |
| Source or infection | 1 | 0 | 1 |
| Heart rhythm | 0 | 2 | 2 |
| Physiological variables | |||
| Central venous pressure | 66 | 77 | 143 |
| SaO2 | 1 | 5 | 6 |
| ScvO2 | 85 | 87 | 172 |
| Lactate | 3 | 2 | 5 |
| PaO2/FiO2 | 1 | 1 | 2 |
| Creatinine | 0 | 2 | 2 |
| Bilirubin | 6 | 5 | 11 |
| Platelets | 2 | 2 | 4 |
| GCS score | 34 | 45 | 79 |
| SOFA component | Treatment group (n) | |||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Subjects | Scores | Subjects | Scores | |||||
| n/N | % | n/N | % | n/N | % | n/N | % | |
| Respiration | 53/258 | 20.5 | 104/2662 | 3.9 | 46/257 | 17.9 | 115/2693 | 4.3 |
| Coagulation | 116/258 | 45.0 | 182/2662 | 6.8 | 114/257 | 44.4 | 223/2693 | 8.3 |
| Liver | 151/258 | 58.5 | 340/2662 | 12.8 | 132/257 | 51.4 | 336/2693 | 13.6 |
| Cardiovascular | 51/258 | 19.8 | 60/2662 | 2.3 | 47/257 | 18.3 | 72/2693 | 2.7 |
| Renal | 92/258 | 35.7 | 141/2662 | 5.3 | 83/257 | 32.3 | 170/2693 | 6.3 |
| Total SOFA score | 175/258 | 67.8 | 441/2662 | 16.6 | 166/257 | 64.6 | 511/2693 | 19.0 |
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)a | |
| Respiration | 1.69 | 1.18 | 1.57 (0.75, 2.67) | 1.55 | 1.14 | 1.5 (0.50, 2.29) | 0.14 (–0.07 to 0.34) | 0.07 (–0.22 to 0.51) |
| Coagulation | 0.75 | 1.06 | 0.14 (0.00, 1.18) | 0.74 | 1.02 | 0.30 (0.00, 1.08) | 0.01 (–0.17 to 0.19) | –0.16 (–0.33 to 0.12) |
| Liver | 0.50 | 0.84 | 0.00 (0.00, 0.74) | 0.44 | 0.76 | 0.00 (0.00, 0.58) | 0.06 (–0.08 to 0.20) | 0.00 (0.00 to 0.00) |
| Cardiovascular | 2.26 | 1.19 | 2.00 (1.23, 3.44) | 2.00 | 1.19 | 1.67 (1.00, 3.00) | 0.26 (0.06 to 0.47) | 0.33 (0.07 to 0.62) |
| Renal | 1.46 | 1.49 | 1.00 (0.00, 2.86) | 1.26 | 1.37 | 0.70 (0.00, 2.25) | 0.19 (–0.05 to 0.44) | 0.30 (–0.28 to 0.68) |
| Total | 6.66 | 3.96 | 5.62 (3.75, 9.00) | 5.99 | 3.84 | 4.89 (3.33, 7.67) | 0.67 (–0.01 to 1.35) | 0.72 (–0.25 to 1.25) |
| SOFA score | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)a | |
| Total | 4.41 | 3.13 | 3.52 (2.15, 5.99) | 4.05 | 3.07 | 3.36 (2, 5.29) | 0.36 (–0.17 to 0.90) | 0.17 (–0.36 to 0.78) |
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | |||
|---|---|---|---|---|---|
| Levosimendan | Placebo | ||||
| Mean | SD | Mean | SD | Mean (95% CrI)a | |
| Respiration | 1.88 | 1.23 | 1.78 | 1.18 | 0.10 (–0.11 to 0.31) |
| Coagulation | 0.83 | 1.08 | 0.86 | 1.09 | –0.03 (–0.22 to 0.16) |
| Liver | 0.49 | 0.82 | 0.48 | 0.81 | 0.01 (–0.13 to 0.15) |
| Cardiovascular | 2.41 | 1.14 | 2.21 | 1.16 | 0.20 (0.00 to 0.39) |
| Renal | 1.58 | 1.54 | 1.44 | 1.44 | 0.14 (–0.12 to 0.40) |
| Total | 7.19 | 3.72 | 6.78 | 3.74 | 0.41 (–0.24 to 1.06) |
| Analysis | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)a | |
| Sensitivity 1b | 5.79 | 4.69 | 4.25 (2.07, 9.00) | 5.21 | 4.48 | 3.86 (1.71, 7.68) | 0.58 (–0.21 to 1.37) | 0.39 (–0.57 to 1.41) |
| Sensitivity 2b | 5.34 | 5.08 | 3.16 (1.07, 9.05) | 4.65 | 4.92 | 2.36 (0.89, 7.75) | 0.69 (–0.18 to 1.56) | 0.80 (–0.32 to 2.25) |
Primary outcome data
Exploratory analysis
FIGURE 29.
Bar plot of respiratory SOFA scores, by day and treatment group.
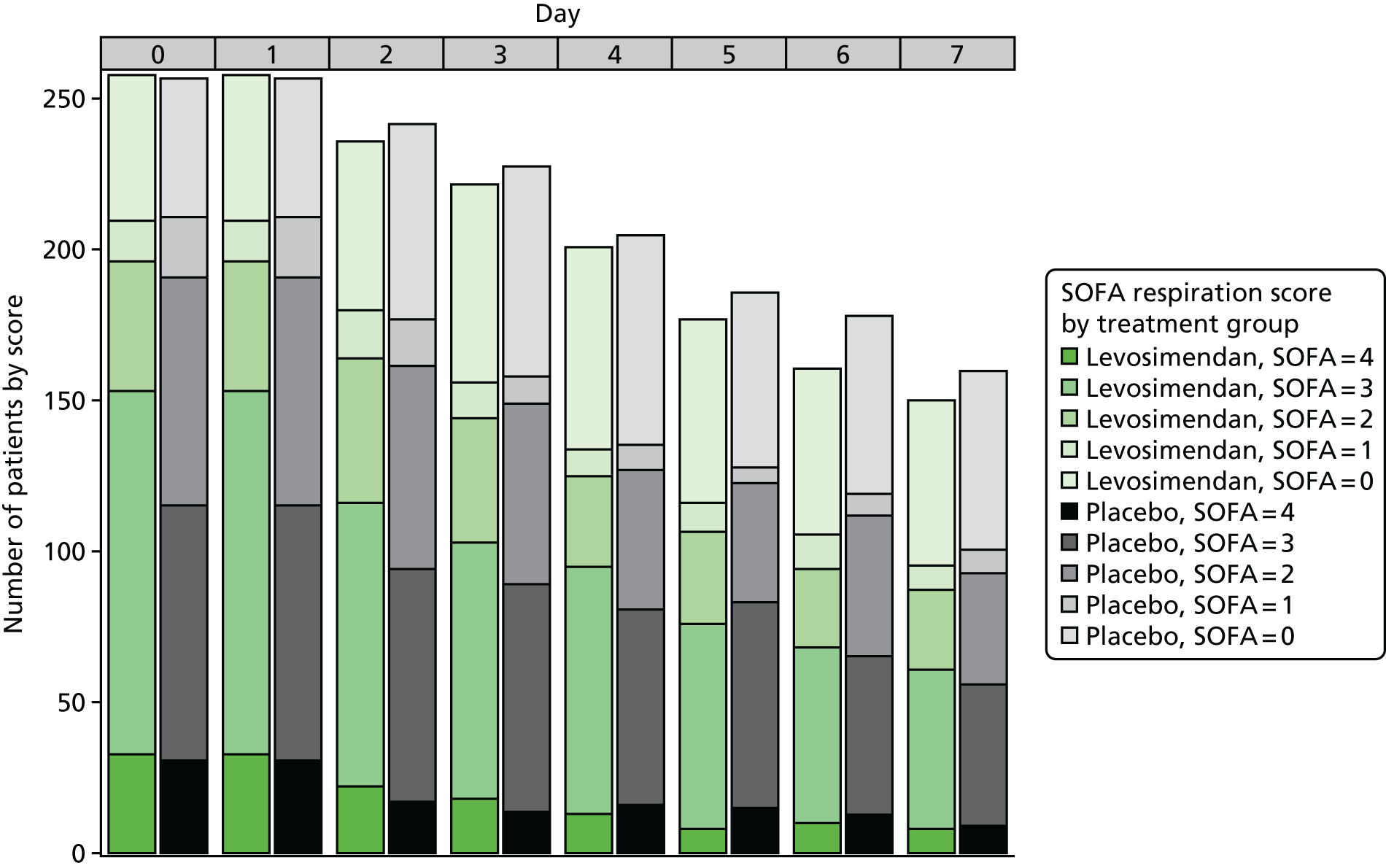
FIGURE 30.
Bar plot of coagulation SOFA scores, by day and treatment group.
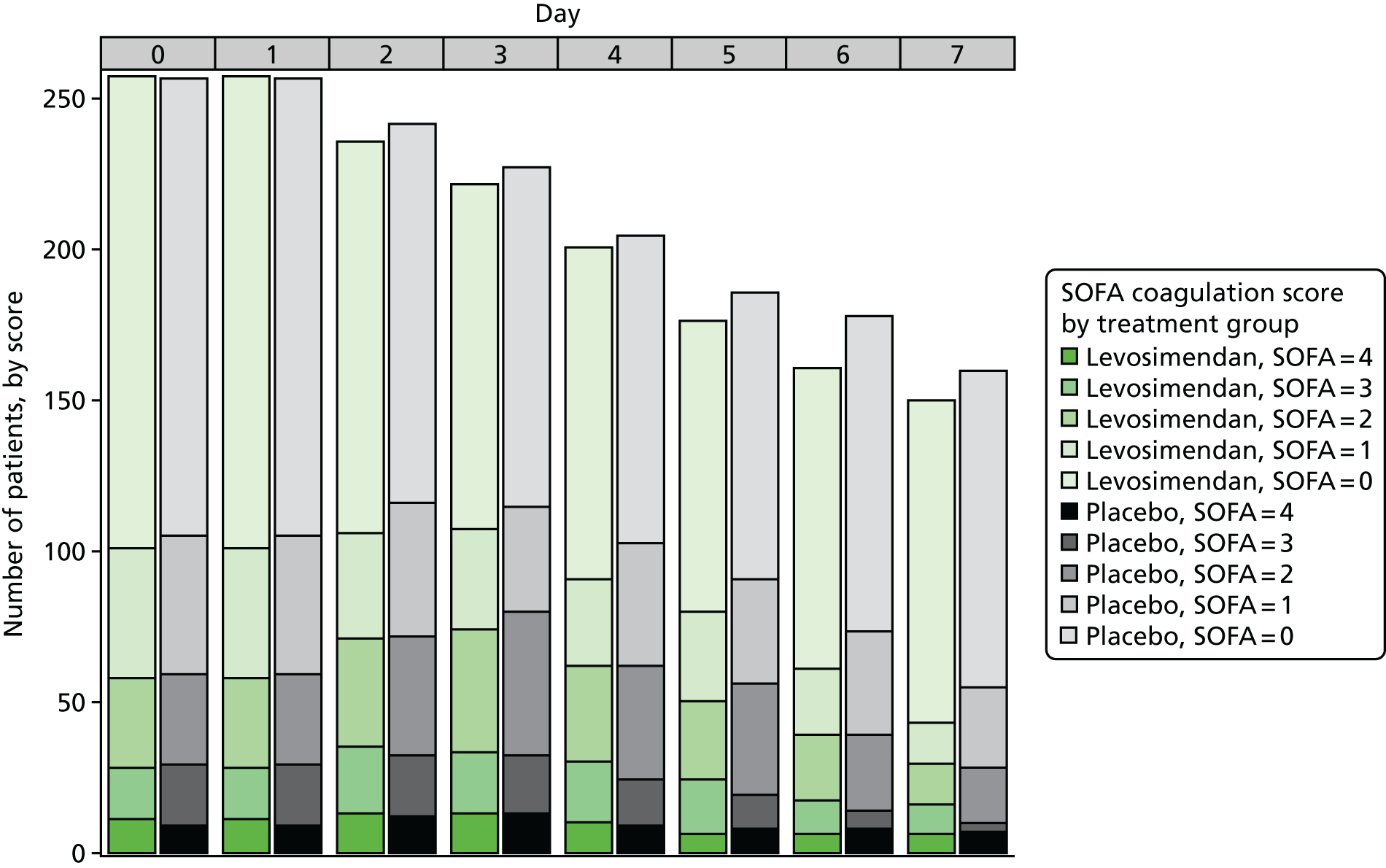
FIGURE 31.
Bar plot of liver SOFA scores, by day and treatment group.
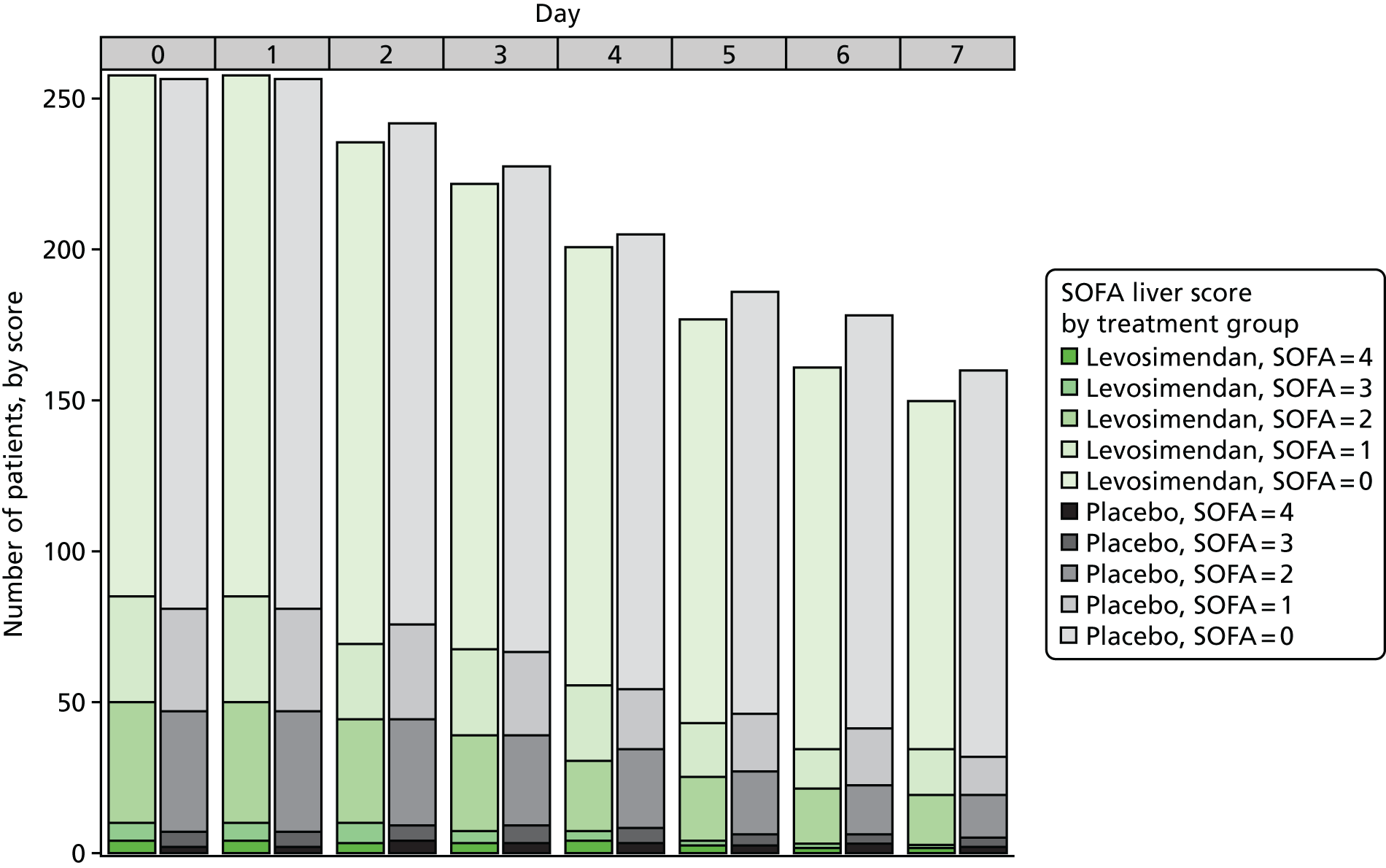
FIGURE 32.
Bar plot of cardiovascular SOFA scores, by day and treatment group.

FIGURE 33.
Bar plot of renal SOFA scores, by day and treatment group.

Subgroup analysis
Cardiac index
Patients were split into the lowest tertile for baseline cardiac index (≤ 2.44 l/minute/m2, 52 patients) and the middle and highest tertiles combined (> 2.44 l/minute/m2, 102 patients).
Tables 27 and 28 show the mean total SOFA scores for the lowest tertile and the middle and highest tertiles combined, respectively.
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)b | |
| Respiration | 2.16 | 1.05 | 2.12 (1.21, 3) | 2.02 | 1.01 | 2.47 (1.07, 2.97) | 0.13 (–0.42 to 0.69) | –0.36 (–0.89 to 1.00) |
| Coagulation | 0.98 | 1.28 | 0.38 (0.00, 1.65) | 1.23 | 1.30 | 1.00 (0.18, 1.71) | –0.25 (–0.96 to 0.44) | –0.62 (–1.09 to 0.45) |
| Liver | 0.65 | 0.91 | 0.00 (0.00, 1.21) | 0.71 | 0.94 | 0.00 (0.00, 1.75) | –0.06 (–0.56 to 0.44) | 0.00 (–1.05 to 0.75) |
| Cardiovascular | 2.87 | 1.31 | 3.81 (1.5, 4.00) | 2.49 | 1.31 | 2.88 (1.27, 3.89) | 0.39 (–0.32 to 1.09) | 0.94 (–1.18 to 2.58) |
| Renal | 2.00 | 1.64 | 1.88 (0.35, 3.74) | 2.25 | 1.24 | 2.16 (1.47, 3.27) | –0.26 (–1.02 to 0.51) | –0.28 (–1.75 to 1.40) |
| Total | 8.66 | 4.86 | 7.88 (4.12, 12.27) | 8.71 | 4.11 | 7.25 (5.83, 10.88) | –0.04 (–2.45 to 2.33) | 0.62 (–4.27 to 4.21) |
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)b | |
| Respiration | 1.95 | 1.20 | 2.05 (1.00, 3.00) | 1.76 | 1.14 | 1.60 (1, 2.39) | 0.19 (–0.25 to 0.63) | 0.45 (–0.35 to 1.29) |
| Coagulation | 0.79 | 0.95 | 0.50 (0.00, 1.22) | 0.66 | 0.91 | 0.17 (0, 1.04) | 0.13 (–0.23 to 0.48) | 0.33 (–0.25 to 0.64) |
| Liver | 0.63 | 0.92 | 0.00 (0.00, 1.25) | 0.54 | 0.73 | 0.18 (0, 1) | 0.09 (–0.22 to 0.41) | –0.18 (–0.44 to 0.17) |
| Cardiovascular | 2.39 | 1.22 | 2.17 (1.25, 4.00) | 1.98 | 1.18 | 1.59 (1, 2.95) | 0.41 (–0.05 to 0.86) | 0.57 (–0.02 to 1.50) |
| Renal | 1.65 | 1.57 | 1.45 (0.00, 3.20) | 1.13 | 1.39 | 0.50 (0.09, 1.57) | 0.52 (–0.05 to 1.08) | 0.95 (–0.44 to 2.12) |
| Total | 7.42 | 3.86 | 6.25 (4.43, 10.5) | 6.08 | 3.74 | 5.36 (3.69, 7.21) | 1.34 (–0.11 to 2.77) | 0.89 (–0.27 to 3.00) |
Central venous oxygen saturation
Patients were categorised according to ScvO2 measured at baseline as low (< 70%, 86 patients), normal (70−85%, 233 patients) and high (> 85%, 21 patients).
Tables 29–31 show the mean total SOFA scores for the low, normal and high ScvO2 groups, respectively.
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)b | |
| Respiration | 1.42 | 1.32 | 1.25 (0.00, 2.42) | 1.65 | 1.45 | 1.55 (0.03, 3.00) | –0.23 (–0.80 to 0.33) | –0.30 (–1.29 to 0.80) |
| Coagulation | 0.95 | 1.18 | 0.50 (0.00, 1.86) | 0.75 | 0.93 | 0.36 (0.00, 1.00) | 0.20 (–0.23 to 0.63) | 0.14 (–0.65 to 0.67) |
| Liver | 0.49 | 0.81 | 0.00 (0.00, 0.67) | 0.67 | 0.83 | 0.09 (0.00, 1.00) | –0.18 (–0.51 to 0.16) | –0.09 (–0.95 to 0.39) |
| Cardiovascular | 2.02 | 1.12 | 1.90 (1.05, 2.83) | 2.33 | 1.37 | 1.81 (1.35, 4.00) | –0.31 (–0.83 to 0.21) | 0.09 (–1.67 to 0.69) |
| Renal | 1.47 | 1.43 | 1.00 (0.09, 2.49) | 1.75 | 1.55 | 1.92 (0.05, 3.07) | –0.28 (–0.89 to 0.33) | –0.92 (–1.67 to 0.86) |
| Total | 6.34 | 3.55 | 5.44 (3.78, 7.64) | 7.15 | 4.76 | 5.67 (3.34, 11.94) | –0.80 (–2.53 to 0.93) | –0.23 (–3.53 to 1.47) |
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)a | |
| Respiration | 1.71 | 1.07 | 1.56 (1.00, 2.64) | 1.56 | 1.00 | 1.52 (0.78, 2.2) | 0.15 (–0.12 to 0.41) | 0.04 (–0.33 to 0.58) |
| Coagulation | 0.66 | 0.89 | 0.14 (0.00, 1.00) | 0.68 | 0.98 | 0.29 (0.00, 1.00) | –0.02 (–0.26 to 0.22) | –0.15 (–0.31 to 0.27) |
| Liver | 0.54 | 0.87 | 0.00 (0, 1) | 0.45 | 0.80 | 0.00 (0, 0.56) | 0.09 (–0.12 to 0.30) | 0.00 (–0.12 to 0.07) |
| Cardiovascular | 2.21 | 1.15 | 2.00 (1.28, 3.24) | 1.94 | 1.10 | 1.61 (1.07, 2.81) | 0.27 (–0.02 to 0.56) | 0.39 (–0.01 to 0.71) |
| Renal | 1.42 | 1.53 | 0.75 (0, 3) | 1.17 | 1.24 | 0.79 (0.05, 1.97) | 0.26 (–0.10 to 0.62) | –0.04 (–0.66 to 0.75) |
| Total | 6.53 | 3.94 | 5.67 (3.67, 8.56) | 5.79 | 3.62 | 4.86 (3.33, 7.04) | 0.74 (–0.22 to 1.71) | 0.81 (–0.55 to 1.69) |
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)a | |
| Respiration | 1.91 | 1.26 | 2.00 (1.33, 3) | 2.23 | 0.94 | 2.12 (1.69, 2.68) | –0.32 (–1.27 to 0.57) | –0.12 (–1.90 to 1.23) |
| Coagulation | 0.61 | 0.80 | 0.00 (0.00, 1.00) | 0.89 | 1.35 | 0.13 (0.00, 1.19) | –0.28 (–1.19 to 0.57) | –0.13 (–1.29 to 1.00) |
| Liver | 0.22 | 0.52 | 0.00 (0.00, 0.00) | 0.47 | 0.84 | 0.00 (0.00, 0.58) | –0.25 (–0.83 to 0.29) | 0.00 (–0.72 to 0.17) |
| Cardiovascular | 2.18 | 1.26 | 2.11 (1.57, 2.50) | 2.32 | 1.31 | 1.67 (1.3, 4.00) | –0.13 (–1.19 to 0.91) | 0.44 (–2.18 to 1.50) |
| Renal | 1.12 | 1.69 | 0.04 (0.00, 2.43) | 1.90 | 1.70 | 1.55 (0.40, 3.83) | –0.79 (–2.14 to 0.64) | –1.51 (–3.85 to 1.95) |
| Total | 6.04 | 3.20 | 4.93 (3.83, 8.07) | 7.81 | 4.17 | 6.09 (5.16, 11.12) | –1.77 (–4.78 to 1.19) | –1.16 (–7.00 to 3.18) |
Lactate
Patients were categorised according to baseline lactate level as normal (≤ 2 mmol/l, 223 patients) and raised (> 2 mmol/l, 282 patients).
Tables 32 and 33 show the mean total SOFA scores for the normal and raised lactate level groups, respectively.
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)a | |
| Respiration | 1.62 | 1.19 | 1.73 (0.45, 2.55) | 1.49 | 1.12 | 1.36 (0.5, 2.19) | 0.13 (–0.17 to 0.43) | 0.36 (–0.09 to 0.73) |
| Coagulation | 0.50 | 0.88 | 0.00 (0.00, 0.65) | 0.44 | 0.81 | 0.00 (0.00, 0.67) | 0.06 (–0.16 to 0.28) | 0.00 (–0.12 to 0.06) |
| Liver | 0.32 | 0.68 | 0.00 (0.00, 0.19) | 0.28 | 0.64 | 0.00 (0.00, 0.13) | 0.04 (–0.13 to 0.22) | 0.00 (0.00 to 0.00) |
| Cardiovascular | 2.12 | 1.13 | 1.86 (1.22, 3.00) | 1.81 | 1.07 | 1.52 (1.00, 2.33) | 0.31 (0.02 to 0.60) | 0.34 (0.03 to 0.70) |
| Renal | 1.19 | 1.38 | 0.54 (0.00, 2.42) | 0.91 | 1.16 | 0.25 (0.00, 1.53) | 0.28 (–0.05 to 0.61) | 0.29 (–0.31 to 0.75) |
| Total | 5.76 | 3.41 | 5.00 (3.41, 7.25) | 4.93 | 3.05 | 4.06 (2.99, 6.34) | 0.83 (–0.01 to 1.67) | 0.94 (0.23 to 1.62) |
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)a | |
| Respiration | 1.76 | 1.19 | 1.46 (1.00, 2.97) | 1.63 | 1.17 | 1.60 (0.63, 2.55) | 0.13 (–0.14 to 0.41) | –0.14 (–0.58 to 0.47) |
| Coagulation | 0.96 | 1.15 | 0.53 (0.00, 1.78) | 0.99 | 1.10 | 0.64 (0.00, 1.50) | –0.03 (–0.29 to 0.23) | –0.11 (–0.56 to 0.28) |
| Liver | 0.67 | 0.93 | 0.11 (0.00, 1.03) | 0.58 | 0.84 | 0.14 (0.00, 1.00) | 0.08 (–0.12 to 0.29) | –0.03 (–0.25 to 0.31) |
| Cardiovascular | 2.40 | 1.25 | 2.15 (1.25, 4.00) | 2.19 | 1.25 | 1.88 (1.05, 3.37) | 0.20 (–0.08 to 0.49) | 0.28 (–0.23 to 0.92) |
| Renal | 1.71 | 1.55 | 1.39 (0.12, 3.08) | 1.57 | 1.46 | 1.33 (0.13, 2.85) | 0.14 (–0.21 to 0.48) | 0.06 (–0.63 to 0.95) |
| Total | 7.50 | 4.23 | 6.64 (4.06, 10.53) | 6.97 | 4.19 | 5.86 (4.12, 9.30) | 0.52 (–0.45 to 1.50) | 0.79 (–0.08 to 1.75) |
Noradrenaline
Patients were categorised into two groups according to baseline noradrenaline level: below the median level (≤ 0.28 µg/kg/minute, 259 patients) and above the median level (> 0.28 µg/kg/minute, 244 patients).
Tables 34 and 35 show the mean total SOFA scores for the groups with a noradrenaline level below and above the median level, respectively.
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)a | |
| Respiration | 1.41 | 1.12 | 1.29 (0.33, 2.44) | 1.34 | 1.12 | 1.2 (0.35, 2.11) | 0.07 (–0.20 to 0.34) | 0.09 (–0.25 to 0.46) |
| Coagulation | 0.57 | 0.99 | 0.00 (0.00, 0.71) | 0.62 | 0.95 | 0.10 (0.00, 1.00) | –0.05 (–0.28 to 0.19) | –0.10 (–0.30 to 0.14) |
| Liver | 0.44 | 0.79 | 0.00 (0.00, 0.56) | 0.39 | 0.71 | 0.00 (0.00, 0.43) | 0.05 (–0.13 to 0.23) | 0.00 (0.00 to 0.00) |
| Cardiovascular | 1.96 | 1.13 | 1.67 (1.00, 3.00) | 1.80 | 1.13 | 1.50 (1.00, 2.50) | 0.16 (–0.11 to 0.43) | 0.17 (–0.20 to 0.50) |
| Renal | 1.13 | 1.38 | 0.31 (0.00, 2.40) | 1.01 | 1.17 | 0.57 (0.00, 1.77) | 0.12 (–0.19 to 0.43) | –0.26 (–0.59 to 0.33) |
| Total | 5.51 | 3.42 | 4.57 (3.25, 7.00) | 5.16 | 3.21 | 4.29 (3.00, 6.65) | 0.35 (–0.45 to 1.16) | 0.29 (–0.84 to 1.00) |
| SOFA component | Treatment group | Absolute difference (levosimendan – placebo) | ||||||
|---|---|---|---|---|---|---|---|---|
| Levosimendan | Placebo | |||||||
| Mean | SD | Median (LQ, UQ) | Mean | SD | Median (LQ, UQ) | Mean (95% CI)a | Median (95% CI)a | |
| Respiration | 1.94 | 1.18 | 1.93 (1.09, 3.00) | 1.78 | 1.15 | 1.85 (0.77, 2.74) | 0.16 (–0.13 to 0.45) | 0.08 (–0.31 to 0.52) |
| Coagulation | 0.90 | 1.08 | 0.53 (0.00, 1.73) | 0.89 | 1.06 | 0.54 (0.00, 1.19) | 0.01 (–0.26 to 0.27) | –0.01 (–0.56 to 0.36) |
| Liver | 0.55 | 0.85 | 0.00 (0.00, 0.83) | 0.52 | 0.84 | 0.00 (0.00, 0.89) | 0.03 (–0.18 to 0.24) | 0.00 (–0.16 to 0.17) |
| Cardiovascular | 2.52 | 1.19 | 2.34 (1.43, 4.00) | 2.23 | 1.22 | 1.83 (1.15, 3.66) | 0.30 (–0.01 to 0.59) | 0.51 (–0.02 to 1.00) |
| Renal | 1.72 | 1.52 | 1.38 (0.16, 3.20) | 1.57 | 1.52 | 1.15 (0.07, 3.00) | 0.15 (–0.22 to 0.53) | 0.23 (–0.62 to 0.99) |
| Total | 7.64 | 4.04 | 6.48 (4.29, 10.65) | 6.99 | 4.29 | 5.63 (3.78, 9.88) | 0.65 (–0.39 to 1.68) | 0.85 (–0.04 to 2.40) |
List of abbreviations
- AE
- adverse event
- AKIN
- Acute Kidney Injury Network
- APACHE II
- Acute Physiology and Chronic Health Evaluation II
- AR
- adverse reaction
- ATP
- adenosine triphosphate
- AUC
- area under the concentration–time curve
- CCL2
- chemokine ligand 2
- CI
- confidence interval
- Cmax
- maximum concentration
- CRF
- case report form
- CrI
- credible interval
- CRN
- Clinical Research Network
- DMEC
- Data Monitoring and Ethics Committee
- eCRF
- electronic case record form
- EDC
- electronic data capture
- EME
- Efficacy and Mechanism Evaluation
- FiO2
- fraction of inspired oxygen
- GCP
- Good Clinical Practice
- GP
- general practitioner
- HME
- heat and moisture exchange
- ICTU
- Imperial Clinical Trials Unit
- ICU
- intensive care unit
- IL
- interleukin
- IMP
- investigational medicinal product
- IQR
- interquartile range
- ITT
- intention to treat
- LeoPARDS
- Levosimendan for the Prevention of Acute oRgan Dysfunction in Sepsis
- LOCF
- last observation carried forward
- MAKE
- major acute kidney event
- MAP
- mean arterial pressure
- MCMC
- Markov chain Monte Carlo
- MHRA
- Medicines and Healthcare products Regulatory Agency
- NIHR
- National Institute for Health Research
- NT-proBNP
- N-terminal prohormone of brain natriuretic peptide
- OR
- odds ratio
- PaO2
- arterial partial pressure of oxygen
- PI
- principal investigator
- REC
- Research Ethics Committee
- RRT
- renal replacement therapy
- SAE
- serious adverse event
- SaO2
- arterial oxygen saturation
- SAP
- statistical analysis plan
- ScvO2
- central venous oxygen saturation
- SD
- standard deviation
- SIRS
- systemic inflammatory response syndrome
- SmPC
- Summary of Product Characteristics
- SOFA
- Sequential Organ Failure Assessment
- sTNFR1
- soluble tumour necrosis factor receptor 1
- SUSAR
- suspected unexpected serious adverse reaction
- TMF
- Trial Master File
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
- VANISH
- Vasopressin vs Noradrenaline as Initial therapy in Septic Shock