

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 11/30/02. The contractual start date was in January 2014. The final report began editorial review in April 2018 and was accepted for publication in August 2018. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Sobha Sivaprasad has received research grants, personal fees and advisory board honoraria from Bayer AG (Leverkusen, Germany), Novartis International AG (Basel, Switzerland) and Allergan plc (Dublin, Republic of Ireland), research grants and advisory board honoraria from F. Hoffman-La Roche AG (Basel, Switzerland) and Boehringer Ingelheim GmbH (Ingelheim am Rhein, Germany) and advisory board honoraria from Heidelberg Engineering GmbH (Heidelberg, Germany). Sobha Sivaprasad is also on the National Institute for Health Research (NIHR) Health Technology Assessment Commissioning Committee. Philip Hykin reports grants, personal fees and non-financial support from Novartis International AG; grants, personal fees, non-financial support and other (including travel expenses and advisory board honoraria) from Bayer; and grants and personal fees from Allergan AG outside the submitted work. Andrew Toby Prevost is a member of the NIHR Public Health Research Funding Committee.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2019. This work was produced by Sivaprasad et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2019 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background
Diabetic retinopathy
Diabetes mellitus is a major public health problem; an estimated 415 million people suffer from this condition globally in 2015, and this number is expected to increase to 642 million in 2040. 1 Diabetic retinopathy is the most common complication of diabetes mellitus. Approximately 30% of people with diabetes mellitus develop diabetic retinopathy. 2 With the increasing prevalence of diabetes mellitus globally, diabetic retinopathy is emerging as the leading cause of avoidable blindness worldwide. 3 The progression and severity of diabetic retinopathy can be delayed by optimal control of medical risk factors such as hyperglycaemia, hypertension and hyperlipidemia. 4,5 However, despite better control of these well-established risk factors, good uptake of established national diabetic retinopathy screening programmes in many countries and improved patient awareness, diabetic retinopathy remains a significant morbidity, which indicates the need for alternate management options for this condition. The two vision-threatening complications of diabetic retinopathy are diabetic macular oedema (DMO) and proliferative diabetic retinopathy. DMO is caused by accumulation of excess extracellular fluid in the macula.
Diabetic macular oedema
This study focuses on participants with DMO. A total of 8–10% of people with diabetes mellitus have clinically significant DMO. 6 DMO is characterised by leakage of fluid-compromised blood vessels in the central retina and is the most common cause of sight-threatening retinopathy. When DMO affects the central few degrees of vision, it causes visual impairment.
Clinically significant oedema may or may not involve the central 1000 µm of the macula. Non-centre-involving macular oedema does not usually affect visual acuity. In the UK, participants with early, non-sight-threatening DMO are referred to the Hospital Eye Service to be monitored more closely for progression to centre-involving DMO. These participants are monitored every 4–6 months in eye clinics for disease progression to the centre using slit-lamp biomicroscopy and optical coherence tomography (OCT). The OCT provides information on the changes in the retinal thickness and morphology of the retina caused by DMO. Treatment is available only when the DMO becomes clinically significant or shows progression to the centre. Laser treatment was the standard of care when the DMO becomes clinically significant. 7 Although laser treatment reduces the risk of moderate visual loss by 50% at this stage, it is not effective in restoring visual acuity and has significant side effects that have an impact on the quality of life of these participants. A newer treatment option [i.e. injections of vascular endothelial growth factor (VEGF) blockers] has replaced laser treatment as the standard of care for centre-involving DMO. 8 Intravitreal steroids may also be used usually as a second-line agent in participants with centre-involving macular oedema not responding to other options. 9,10
The natural history of the disease is to progress from non-central to centre-involving DMO. 11 However, the time to progression to centre varies significantly. There are no treatment options for non-clinically significant DMO except optimal control of diabetes mellitus and hypertension. Laser photocoagulation may be performed for non-central clinically significant macular oedema but most cases are managed conservatively in clinics. Preventative trials such as the Phase III ruboxistaurin (Arxxant; Eli Lilly and Company, Indianapolis, IN, USA) clinical trial12 and the Phase II topical non-steroidal anti-inflammatory eye drops13 for non-centre-involving DMO were not found to be beneficial. Therefore, there is a substantial unmet need for both treatment and prevention of progression of non-centre-involving DMO.
Dark adaptation and role of hypoxia in diabetic macular oedema and diabetic retinopathy
The exact pathogenesis of diabetic retinopathy and DMO is uncertain. In this project we studied the role of hypoxia as a contributory factor to the development and progression of DMO and diabetic retinopathy. The evidence regarding the role of hypoxia in DMO is conflicting. 14 First of all, inhaling 100% oxygen has been shown to alleviate DMO in the short term. 15 Photoreceptors may be responsible for causing the relative hypoxia in the retina and in diabetic eyes, this hypoxia may be sufficient to cause or aggravate microvascular changes. 16 The microvascular changes of diabetic retinopathy occur only in the retina and not in the brain, despite having the same embryologic origin and, therefore, there is a specific retinal factor causing diabetic retinopathy. 17 The obvious difference between the two tissues is the presence of photoreceptors in the retina. A total of 95% of these are rods that are used in night vision only. Rods use more oxygen than any other cell in the body and the maximal consumption is during dark adaptation when the oxygen tension at the level of the rods reduce to zero. 18 If the microvasculature is already compromised in diabetes mellitus, this relative hypoxia can increase the VEGF drive and contribute to worsening of the DMO and diabetic retinopathy. 16
Rationale for a randomised clinical trial and mechanistic evaluation
Arden hypothesised that if dark-adaptation can be prevented, the rod dark current would never become maximal and DMO and diabetic retinopathy could be alleviated by decreasing the oxygen demand. 16 As people only dark adapt at night during sleep, sleeping in an illuminated environment should prevent or reverse the condition. Sufficient light is transmitted by the eyelids to reduce the dark current. Therefore, wearing a lightmask of 500–505 nm to suppress the rod-induced dark adaptation may alleviate DMO and prevent the progression of diabetic retinopathy. The lightmasks ensure the uniform illumination of weak light, to which the eyes adapts quickly owing to the Troxler Effect. However, it is important that the masks are comfortable to wear and do not disturb sleep. A few short trials have supported the proof-of-concept that prevention of dark adaptation by using lightmasks may be safe and effective in reducing the retinal thickness in non-central DMO. 19,20
The design must permit the mask to be worn by people with different head shapes and deliver rod excitation efficiently. The spectral output is important and should be matched as closely as possible to the response spectrum of the rod cells. This has been tested in two clinical trials. The first was a proof-of-concept study in which 12 participants slept in a mask containing a chemiluminescent source that exposed one eye only to light. 19 The trial lasted 3 months and all participants found the masks comfortable and the method of treatment acceptable. There were no reports of adverse effects. Measurements of colour contrast sensitivity and examination of standard fundus photographs showed that in the 10 participants for whom complete records were available, colour vision improved and the number of red dots (microaneurysms and small dot haemorrhages) decreased. These results were significant even though the trial was short and the numbers treated were small. A second study was carried out by the chief investigator of this grant at King’s College Hospital using electronic sources of 505-nm light-emitting diodes to illuminate one eye. 20 The electrical power of the system was < 3 mW. A total of 40 participants were recruited and follow-up visits were at 3 and 6 months. All participants had early bilateral DMO and the eye with more swollen retinal thickness was chosen as the study eye. Thirty-four out of 40 participants completed the study and the rest did not attend their exit visit appointments and so we are not aware of their mask wear-time. Mean baseline OCT macular cube thickness (Cirrus, Carl Zeiss Meditec AG, Jena, Germany) was equivalent for study and fellow eyes. But study eyes had a greater mean thickness in the central subfield zone 1 (282 ± 53 µm) than the fellow eyes (256 ± 19 µm). At baseline, 28 study eyes showed intraretinal cysts compared with nine in the fellow eyes. At 6 months, only 19 study eyes had cysts compared with 20 cysts in fellow eyes. After 6 months, the worst-affected Early Treatment Diabetic Retinopathy Study (ETDRS) zone and the central subfield zone 1 reduced in thickness in study eyes by 12 µm only (p = 0.01). The secondary outcomes of change in visual acuity, achromatic contrast sensitivity, and microperimetric thresholds improved significantly in study eyes and deteriorated in fellow eyes.
Based on this study, Polyphotonix Medical Ltd (Durham, UK) manufactured the Noctura 400 Sleep Masks designed to suppress rod-related dark adaptation. The acceptability of this lightmask has been reported in normal eyes as well as eyes with DMO. This report was published after the CLEOPATRA (clinical efficacy and safety of light-masks at preventing dark-adaptation in the treatment of early diabetic macular oedema) study was initiated21 and showed no serious adverse events (SAEs) from the masks, although 27% of the participants had withdrawn by the end of the study at 3 months. A further 6-month study conducted in 2012 evaluating the lightmask in participants with diabetic retinopathy with central macular thickness of at least 220 µm were included. 22 Thirty-five out of the 45 recruited participants completed the study. The primary outcomes were safety and mask wear-time. The average mask wear-time was 4.96 [standard deviation (SD) 2.97] hours and showed interindividual variations. A total of 7% of participants experienced mask-related adverse events (AEs). However, the effect of this treatment option needs to be investigated in a randomised controlled trial (RCT) powered to answer the research question of whether or not the lightmask has a superior effect to a placebo. In this project, our main study was a multicentre randomised clinical trial to study the effects of wearing a lightmask to suppress dark adaptation during sleep at night versus a non-lightmask arm in participants with non-central DMO over 24 months. 23
There are only very small observational studies that suggest that hypoxia contributes to DMO and diabetic retinopathy. 15 In the mechanistic evaluation, we explored the concept of hypoxia as a contributing factor in early DMO. We studied the effect of inhalation of 100% oxygen and lightmasks on the retinal morphology measured by OCT and visual function in a small sample of 28 participants with non-central DMO to establish whether or not local regions of retinal oedema correspond to areas of hypoxia, and whether or not functional defects of outer and inner retinal layers and corresponding anatomical changes improved with 100% oxygen or the lightmasks.
Hypotheses
The working hypothesis was that offering lightmasks to wear during sleep at night, compared with not doing so, reduces the thickness in the zone identified at baseline to have maximal thickness (caused by DMO, of which there is clinical evidence) in participants with non-centre-involving DMO at 24 months’ follow-up from baseline, as measured by OCT.
The statistical null hypothesis was that wearing lightmasks does not alter the absolute thickness at the zone of maximum thickness as determined by OCT at 24 months when compared with standard care (non-lightmask).
Statistical alternative hypothesis was that wearing lightmasks alters the absolute thickness at the zone of maximum thickness, as determined by OCT at 24 months, when compared with standard care (non-lightmask). If there was a significant reduction in absolute thickness in the zone of maximum thickness in participants wearing the lightmasks compared with the control arm (from the trial sample by two-sided 5% significance level for the test of zero difference in mean thickness between arms), then the null hypothesis would be rejected.
Trial objectives
The primary objective was to explore whether or not wearing lightmasks during sleep at night reduces, relative to the non-lightmask arm, the maximal zone thickness as measured by OCT in the study eye of participants with non-centre-involving DMO at 24 months.
The secondary objectives were:
-
exploration of whether or not wearing lightmasks during sleep at night reduces, relative to the non-lightmask arm, the baseline maximal zone thickness as measured by OCT in the study eye of participants with non-centre-involving DMO at 12 months
-
assessment of the safety profile of the lightmasks, including sleep disturbance, using validated questionnaires at 12 and 24 months
-
evaluation of the compliance of wearing these lightmasks over 24 months
-
evaluation of change in macular thickness in both the parafoveal zones as well as the perifoveal zones by OCT, macular volume, progress to centre-involving DMO and change in morphological characteristics of DMO, proportion of participants requiring treatment for DMO, best corrected visual acuity (BCVA), and the levels of diabetic retinopathy at 12 and 24 months.
The objectives of the mechanistic evaluation were exploration of:
-
the changes induced by supplemental oxygen on multifocal electroretinography (mfERG) and scotopic microperimetry as a function of retinal location at 12 months
-
whether or not supplemental oxygen affects tests of function in regions of anatomical change and in those of adjacent regions without apparent change to a different extent at 12 months
-
whether outer (microperimetry) or inner retinal (mfERG) functional loss is closely associated with structural changes at 12 months
-
whether or not the use of lightmasks affects retinal hypoxia in regions of anatomical disturbance, and whether or not a distinction can be made regarding inner and outer retinal functional changes at 12 months
-
whether or not the long-term changes induced by lightmasks are similar to changes induced by oxygen supplementation at 12 months.
Chapter 2 Methods
Trial design
This is a Phase III, randomised controlled, single-masked, clinical trial that evaluated the clinical effectiveness and safety of lightmasks in treating and preventing the progression of non-centre-involving DMO. A mechanistic evaluation studied the role of hypoxia in early DMO.
Target population
The target population, to which inferences from the end of this trial are intended to generalise, is the population of participants with diabetes mellitus who have non-central, non-clinically significant macular oedema (early DMO).
Trial population
The trial population, from which the study sample was drawn, was defined as participants aged ≥ 18 years who attended the ophthalmology referral centres in the UK (n = 15 clinical sites). In subjects for whom both eyes met the inclusion criteria, the eye with the worse visual acuity was included in the study to be the study eye. The fellow eye (non-study eye) was treated in accordance with the NHS standard of care. In subjects for whom only one eye meets the inclusion criteria, that eye was the study eye and the fellow eye (non-study eye) was also monitored and treated in accordance with the NHS standard of care.
Selection and withdrawal of subjects
Inclusion criteria
-
Subjects of either sex aged ≥ 18 years.
-
Diagnosis of diabetes mellitus (type 1 or 2). Any one of the following will be considered to be sufficient evidence that diabetes mellitus is present:
-
current regular use of insulin for the treatment of diabetes mellitus
-
current regular use of oral anti-hyperglycaemic agents for the treatment of diabetes mellitus
-
documented diabetes mellitus by American Diabetes Association and/or World Health Organization criteria.
-
-
The BCVA in the study eye better than 55 ETDRS letters (Snellen visual acuity 6/24).
-
On clinical examination, retinal thickening caused by early DMO not involving the central 1000 µm of the macula characterised by presence of microaneurysm, exudates or oedema and OCT evidence of increased retinal thickness in at least one non-central ETDRS zone of ≥ 320 µm.
-
Previous macular laser, intravitreal steroids or anti-VEGF treatment is permitted provided the last treatment was done at least 4 months before date of recruitment.
-
Media clarity, pupillary dilatation and subject co-operation sufficient for adequate fundus photographs.
-
Ability to return for study visits.
-
Ability to give informed consent throughout the duration of the study.
Exclusion criteria
The following exclusions applied to the study eye only (i.e. they could be present for the non-study eye):
-
Clinical evidence of centre-involving macular oedema (central subfield on OCT of > 300 µm).
-
Macular oedema is considered to be due to a cause other than DMO.
-
An ocular condition is present (other than diabetes mellitus) that, in the opinion of the investigator, might affect macular oedema or alter visual acuity during the course of the study (e.g. vein occlusion, uveitis or other ocular inflammatory disease, neovascular glaucoma, Irvine–Gass syndrome, etc.).
-
History of treatment for DMO at any time in the past 4 months (e.g. focal/grid macular photocoagulation, intravitreal or peribulbar corticosteroids, anti-VEGF drugs, or any other treatment) in the study eye.
-
History of pan-retinal scatter photocoagulation in the study eye.
-
Active proliferative diabetic retinopathy in the study eye.
-
A condition that, in the opinion of the investigator, would preclude participation in the study.
-
Corneal scarring, vitreous opacities, severe asteroid hyalosis that would inhibit proper visualisation, inability to be positioned in front of the OCT device, inability to understand the requirements of the imaging, and nystagmus.
-
Participants with active insomnia or any other relevant sleep disturbances.
Selection of participants
Participants were identified from diabetic retinopathy screening programmes and medical retina clinics of the trial sites and their satellite clinics. In addition, participants were referred by other medical retina consultants from other hospitals to the principal investigators (Table 1).
| Type of approach | Method of approach | Patient responsea | Reminderb |
|---|---|---|---|
| Approach through medical retina clinics at participating sites | In clinic with PIS, via invitation letter (if eligible participants noted in clinic registers or databases) or via study poster in waiting room or reception area | By telephone | Reminder letter or telephone |
| Approach through named PIC | In clinic with PIS, via invitation letter (if eligible participants noted in clinic registers or databases) or via study poster in waiting room or reception area | By telephone to the PIC, with request to forward contact details to the trial site | |
| Approach through any UK diabetic retinopathy screening programme | Generally in clinic without PIS, via invitation letter (if eligible participants noted in clinic registers or databases) or via study poster in waiting room or reception area | Generally participants will directly confirm their interest to be contacted by one of the trial sites, so that the screener/grader can then forward contact details to the nearest trial centre | Reminder letter sent by PIC |
| Approach through any UK diabetic retinopathy screening programme | Generally in clinic without PIS, via invitation letter (if eligible participants noted in clinic registers or databases) or via study poster in waiting room or reception area | Generally participants will directly confirm their interest to be contacted by one of the trial sites, so that the screener/grader can then forward contact details to the nearest trial centre | Not applicable |
Recruitment strategy
Pre-screening of participants
To prevent participants from being subjected to unnecessary trial procedures, potential participants were pre-screened with an OCT before consenting to the study and being subjected to any trial screening procedures to ensure the exclusion of eyes with centre-involving macular oedema defined as central subfield of > 300 µm.
Rescreening of participants
Participants excluded because of any of the following exclusion criteria were allowed to be screened twice (a total of three screens) at least 1 month apart:
-
Does not meet inclusion criteria of non-central retinal thickness of > 320 µm with morphological evidence of oedema, microaneurysms or exudates.
-
History of any laser treatment carried out 4 months previously or anti-VEGF therapy carried out for DMO more than 2 months previously that has evidence of resolving central macular oedema.
Description of the investigational treatment
The investigational treatment evaluated in the study was the use of lightmasks during sleep at night for 24 months in treating and preventing the progression of non-centre-involving DMO.
The lightmask is manufactured by Polyphotonix Medical Ltd, to deliver 500- to 505-nm light through closed eyelids. The lightmask contains a light-emitting unit of two organic light-emitting diodes (OLEDs) placed within a fabric mask and this lightmask was placed over both of the patient’s eyes.
The fabric mask is made of nylon, polyurethane and polyester. The pod and fabric mask are designed to be thin and flexible and contoured to complement the face and improve comfort for the wearer. The OLEDs are powered by two 3V (CR2450) batteries, which power the device without the need for an external power source or recharging. At the end of the mask’s lifetime of 83 days, a replacement lightmask is required. A new fabric mask was provided with each mask to minimise contamination resulting from continued use.
The mask is time, date and touch sensitive and would only ‘work’ between pre-determined operational windows – typically 20.00 to 08.00 during the lifetime of the lightmask. Within these times the mask can be activated by a light touch. If worn within 3 minutes of activation, sensors on the pod keep the mask illuminated for the night’s therapy. The times at which the mask was worn were logged for compliance analysis. The returned mask contained anonymised data on wear-time that was downloaded to the manufacturer’s portal for producing data for the mask compliance on an individual level.
The lightmask has Conformite Européenne (CE) certification as a 2a medical device and its design and manufacture met the standards of ISO13485. The intensity of the light was approximately six orders of magnitude less than for threshold toxicity and two orders below that which causes a 1% change in the melatonin cycle that drives circadian rhythms. We tested sleep disturbance and daytime drowsiness by the use of validated sleep questionnaires. Compliance with the lightmasks was assumed to be an issue and so all site personnel were informed to stress optimal compliance with all participants and each mask was equipped with a capacitive sensor and memory chip capable of sensing when the mask was worn. These data from the sensor were downloaded and analysed by the company to provide us with an accurate measure of compliance in this trial.
Dosing regimen
The participants were advised to wear the lightmask each night, receiving a maximum of 8 hours’ therapy per night. The optical output of the mask was tuned by the manufacturer to optimise scotopic intensity while minimising photopic intensity. The masks regulate the light output to a constant luminosity x, 60 cd/m2 ≤ × ≤ 100 cd/m2, which is well below toxic levels of luminosity but of sufficient scotopic intensity to prevent dark adaption. Emission of < 470 nm is < 3% of total output posing little or no risk of harm.
If patient compliance was deemed to be low (< 70% of the maximum therapeutic dose), the patient received telephone reminders and/or counselling from the research team at each site.
Concomitant therapy
All concomitant and current and past therapies in the last 12 months were recorded at screening. Any change in concomitant medications were recorded at each visit. If the clinician observed disease progression and opted to treat the DMO, a variety of options were allowed.
Laser photocoagulation could be performed as indicated for clinically significant macular oedema if the investigator decided that the oedema had deteriorated to require laser after considering the risks and benefits of laser therapy. Laser therapy was arranged to be done at the same visit or deferred to the next visit. Laser treatment was avoided between study visits unless a detrimental effect was anticipated if laser was deferred to the next visit. Regardless of laser treatment, the participant continued to wear the mask until the end of the study. Repeat laser treatment was also done at any scheduled visit but the interval between two laser treatment sessions had to be not less than 4 months apart.
As intravitreal anti-VEGF is the first line option for treatment of centre-involving DMO of > 400 µm, this treatment could be offered to participants if the oedema deteriorated in order to meet the eligibility for current standard of care. Intravitreal steroids could also be given to these individuals as per investigator discretion but the masks had to be worn until the end of the study.
The fellow eye could be treated according to standard of care and this included laser photocoagulation, intravitreal anti-VEGF therapy or steroids.
Pan-retinal photocoagulation to either eye was permitted if high-risk retinal or disc neovascularisation was observed in any visit. The patient had to be seen at 2-weekly intervals until sufficient pan-retinal photocoagulation was applied. The participants continued to use the masks until the end of the study.
Changes in medications related to diabetes mellitus were recorded within concomitant medications.
Cataract surgery was avoided during the period of the study and was recorded as a protocol deviation if the surgery was deemed necessary and performed during the study period.
Randomisation procedure
A patient identification number (PIN) was generated by registering the patient on the MACRO electronic case report form (eCRF) system (InferMed MACRO, Elsevier, Oxford, UK) after consent had been signed. This unique PIN was recorded on all source data worksheets (SDWs) and used to identify the patient throughout the study. Randomisation was via a bespoke web-based randomisation system hosted at the King’s Clinical Trials Unit (KCTU).
Authorised site staff were allocated a username and password for the randomisation system. Once a patient was consented, all baseline data collected and eligibility confirmed, the staff member logged into the randomisation system (www.ctu.co.uk) and clicked ‘randomisation – advanced’ and selected CLEOPATRA and entered the participants details using the unique PIN. The ‘help’ section of the system had video demonstrations to aid new staff in using the system. Once randomised, the system automatically generated confirmation e-mails to key staff, with or without treatment allocation information depending on their role in the study.
Each participant was randomised to one of two arms: lightmasks (treatment) or non-lightmasks (control). Participants were randomised at the level of the individual, using the method of minimisation incorporating a random element. The minimisation factors were glycated haemoglobin (HbA1c) level (< 8% or ≥ 8%), baseline thickness in excess of 320 µm in parafoveal zones (2 to 5), and study site. As different types of OCT machines were used across sites, having site as a minimisation factor overcame systematic differences between machines.
Participants were randomised into the study only by an authorised member of staff at the study research site, as detailed on the delegation log. Participants could be randomised into the study only once.
Blinding
Control participants were provided with identical non-lightmasks that contain no active light. Primary outcome assessors (optometrists and OCT technicians) remained masked to treatment allocation. The optometrists were the visual acuity examiners and OCT technicians did the OCT scans at all visits and both were masked to the participant study arm. The visual acuity examiners received the participants into the visual acuity lanes with a visual acuity case report form (CRF), study number and detail of study eye and non-study eye to be refracted, but with no previous subject records or CRF by which the subject treatment arm could be identified. Similarly, the OCT technicians received the subjects into the OCT room on a specific CRF that provides details of subject study number and eye to be examined. The subjects were advised at enrolment that they must not discuss the study arm they are in with the OCT or visual acuity examiner. Retinal photographs were graded by masked graders in the independent reading centre at Gloucestershire Eye Unit. This avoided performance and detection bias. We described the completeness of outcome data for each outcome, including reasons for attrition and exclusions from the analysis.
The decision of the Data Monitoring Committee (DMC) was that the trial/senior statisticians would not be blinded to study arm.
Summary of assessments and trial flow chart23
Table 2 and Figure 1 show the summary of assessments and the trial flow chart.
| Study assessments | Screening | Weeka | Montha | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 4 | 8 | 12 | 16 | 20 | 24 | ||
| Medical history | ✗ | |||||||
| Concomitant medications | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| HbA1c levels and BP | ✗ | ✗ | ✗ | |||||
| PIRS (insomnia) and ESS (sleepiness) | ✗ | ✗ | ✗ | ✗ | ||||
| BCVA | ✗ (+R) | ✗ | ✗ | ✗ (+R) | ✗ | ✗ | ✗ (+R) | |
| VA repeated with new refraction | ✗ | |||||||
| OCT – macular thickness | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Repeated OCT – macular thickness | ✗ | ✗ | ||||||
| OCT – morphological characteristics | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Retinal colour photographs 4 | ✗ | ✗ | ✗ | |||||
| Lightmask compliance | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| AEs form | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
FIGURE 1.
Trial pathway showing flow of participants in the trial. Reproduced with permission from Sivaprasad et al. 24 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.
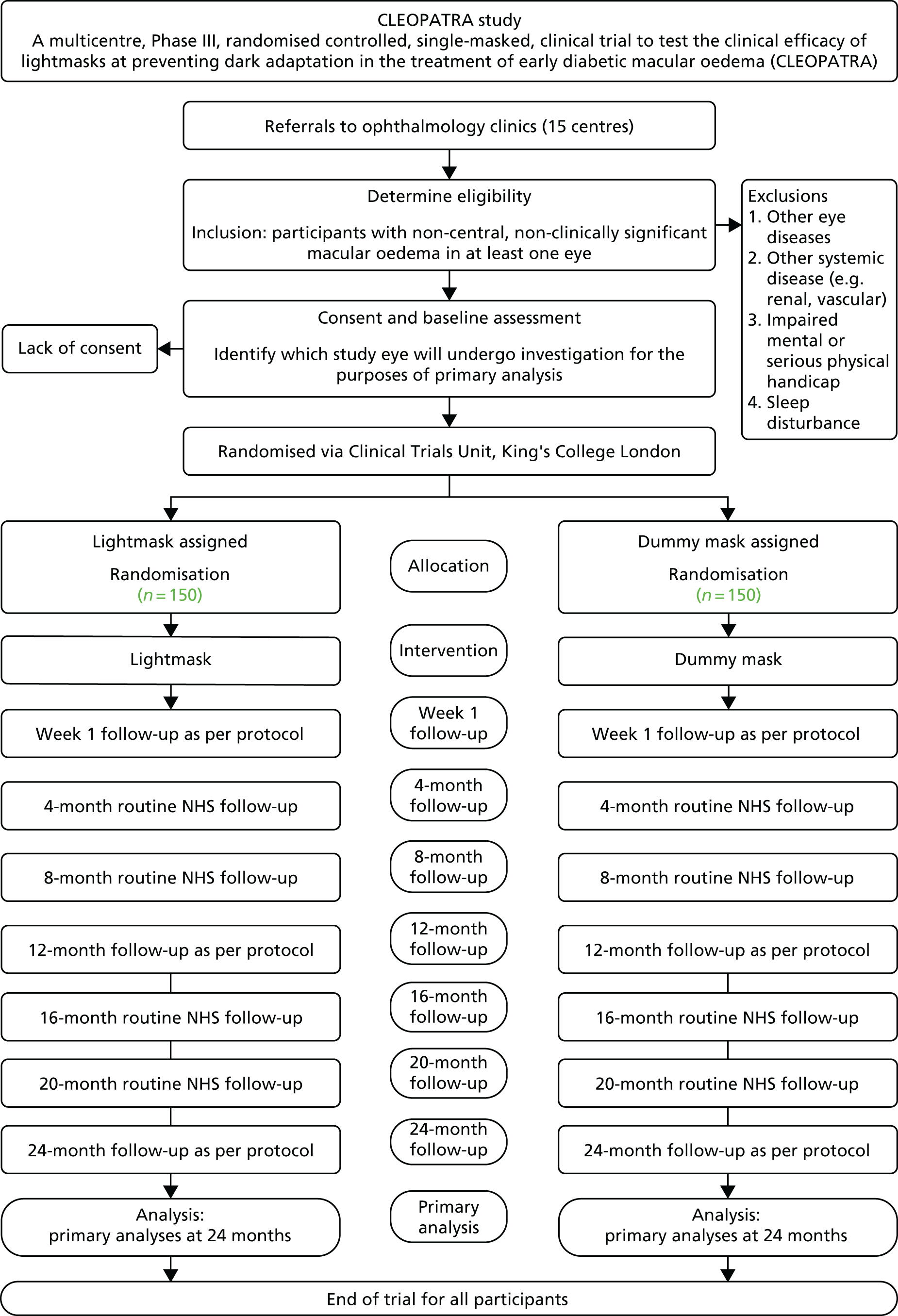
Colour photographs and OCT had to be carried out before commencing on first laser or anti-VEGF therapy. These tests were allowed to be repeated at the investigator’s discretion during the period of the study. The visual acuity tests with new refraction were repeated before dilating the pupils at baseline to measure intertest variability. The baseline tests that include mechanistic tests could be done over 8 days. The last procedure was the issuance of the mask. The week 1 visit could be either in person at the recruiting site or by telephone. If on the telephone, Pittsburgh Insomnia Rating Score and Epworth Sleepiness Scale (ESS) questionnaires were provided to the patient at screening to be completed at home and returned to the recruiting site in a prepaid envelope.
Trial procedures: informed consent
We designed the patient information leaflets and consent forms with service user involvement. We supplied individuals with as much information as they required to make an informed decision about participation in the study. A copy of the informed consent was given to the participant for review before consent was obtained by the investigator. All participants were required to read, sign and date a consent form before participating in the study. All information about participants was collected during the course of the study and was not derived from existing databases.
Optical coherence tomography
The primary outcome was change in the zone of maximal retinal thickness on OCT. OCT is a well-established tool used as an assessment and monitoring tool for DMO. The OCT thickness map is divided into nine zones with the central zone representing the centremost area of the retina (the fovea). The DMO trials to date have used this central zone as the outcome measure because the trials are on centre-involving DMO. However, in non-central DMO the central zone is not affected and disease progresses to the centre over time. Therefore, we used the retinal thickness of the baseline zone of maximal retinal thickness as a measure. We also measured, as a secondary outcome, the thickness of parafoveal zones (zones 2–5) and perifoveal zones (zones 6–9) and macular volume to provide further evidence about changes in other zones at 24 months or at the point of withdrawal. For participants with the same maximum baseline retinal thickness in two zones, the zone located in the parafoveal zone was chosen. When these two zones were in the parafoveal zone, the average retinal thickness was taken in subsequent follow-up measurements. The OCT macular thickness protocol was carried out twice at 12 and 24 months each, to assess test–retest variability. The OCT technicians were masked of the treatment arm. When research appointments had been missed, OCTs performed as part of routine clinical care within the visit time frame were allowed to be used.
Visual acuity tests
The visual acuity tests were carried out using the validated ETDRS vision charts using standard operating procedure (SOPs) for trials in DMO. Refracted visual acuity tests were carried out at baseline and at the point of withdrawal. At baseline, following recording of refracted BCVA, the patient could complete another assessment that did not require pupil dilatation and then return to repeat visual acuity recording with the new refraction to ensure that we accounted for intertest variability. Refraction was not repeated the second time. The optometrists were masked of the treatment arm. At baseline, the optometrist was not allowed to have the recorded visual acuity from the first BCVA test when conducting the second test. The second test could also be done by another visual acuity assessor.
Retinal colour photographs
Colour fundus photographs were carried out at baseline, 12 and 24 months or at the point of withdrawal to explore progression of diabetic retinopathy. The photographs were read by masked graders at the Independent Reading Centre in Gloucestershire Eye Unit.
Sleep and insomnia rating scales
We had additional tests to explore sleep disturbance in this study. We used the validated Pittsburgh Insomnia Score Index questionnaire to assess insomnia. 25 Daytime sleepiness was measured by the ESS, which is another validated self-administered questionnaire. 26 Both questionnaires were administered at baseline, week 1, 12 and 24 months or at the point of withdrawal.
Mechanistic tests
A total of 28 participants who consented to the mechanistic evaluation underwent further tests in the baseline visit. Oximetry, mfERG and scotopic microperimetry were carried out at baseline while breathing either air or oxygen through a face mask. The test began 10 seconds after the gas flows and continued for the length of the test. The tests were repeated at 12 months. Half of the participants used the lightmasks and the other half used the non-lightmasks. Blood pressure and intraocular pressure were also measured in these visits. A within-visit flexibility of 14 days was allowed for these participants to complete all the tests in these visits.
Laboratory tests
Laboratory tests for levels of HbA1c for all participants were carried out at the local laboratories at each site. Results of HbA1c levels from within 3 months of the visit date were also accepted.
Assessment of efficacy
Primary efficacy parameters
The primary efficacy measure was analysed as the difference between arms in the change from baseline in absolute thickness at the zone of maximum thickness as determined by OCT at 24 months.
Secondary efficacy parameters at 12 and 24 months
-
Difference between arms in the change from baseline in absolute thickness at the zone of maximum thickness as determined by OCT at 12 months.
-
Other measures (analysed as difference in arms or within lightmask arm as indicated) included:
-
difference between arms in the change in retinal thickness in the nine ETDRS zones (parafoveal zones 2–5 and perifoveal zones 6–9) and macular volume
-
difference between arms in morphological characteristics of macular thickness
-
difference between arms in the mean change in visual acuity
-
difference between arms in the proportion of centre-involving macular oedema
-
difference between arms in the time to occurrence of centre-involving macular oedema
-
difference between arms in the proportion requiring steroids, macular laser or anti-VEGF treatment
-
difference between arms in the proportion of participants that show progression of retinopathy as measured by the ETDRS severity levels and microaneurysm turnover
-
compliance rates in the lightmask arm.
-
Assessment of safety parameters
-
Difference between arms in the measures of sleep disturbance in terms of daytime sleepiness and insomnia.
-
Difference between arms in ocular and systemic AEs and SAEs.
Assessment of mechanistic parameters
-
Change in P1 (the first positive waveform) and N1 (the first negative waveform) amplitudes and peak time in mfERG after supplemental oxygen.
-
Change in retinal sensitivity in scotopic microperimetry after supplemental oxygen.
-
To determine differences in change in P1 and N1 amplitudes and peak time in mfERG after lightmasks and non-lightmasks at 12 months.
-
To determine differences in change in retinal sensitivity in scotopic microperimetry after lightmasks and non-lightmasks at 12 months.
-
To correlate the changes induced by lightmasks and oxygen supplementation on retinal sensitivity using oximetry.
Assessment of safety
Specification, timing and recording of safety parameters
All AEs and side effects were recorded in the eCRF throughout the study, regardless of their severity or relation to study participation.
Procedures for recording and reporting adverse events
The masks are CE marked. The definitions below are for non-CE marked devices but were useful to classify the AEs in this study. The following definitions were used.
Adverse events
Any untoward medical occurrence which does not necessarily have a causal relationship with the treatment. ‘Treatment’ includes all investigational and non-investigational agents administered during the course of the study. Medical conditions/diseases present before starting study treatment are only considered AEs if they worsen after starting study treatment.
Adverse device effect
Any untoward or unintended responses to the investigational device – all AEs judged by either the reporting investigator or the sponsor as having a reasonable suspected causal relationship to the device (i.e. definitely, probably or possibly related) qualify as adverse reactions (ARs). The expression ‘reasonable causal relationship’ means to convey in general that there is evidence or argument to suggest a causal relationship.
Causality
The assignment of the causality was made by the investigator responsible for the care of the participant using the definitions in Table 3. If any doubt about the causality arose, the investigator informed the chief investigator. In the case of discrepant views on causality between the investigator and others, all parties discussed the case. In the event that no agreement was made, the Medicines and Healthcare products Regulatory Agency (MHRA), main Research Ethics Committee (REC) and other bodies were planned to be informed of both points of view.
| Relationship | Description |
|---|---|
| None | There is no evidence of any causal relationship to study treatment |
| Remote | There is little evidence to suggest there is a causal relationship (e.g. the event did not occur within a reasonable time after administration of the trial intervention). There is another reasonable explanation for the event (e.g. the participant’s clinical condition, other concomitant treatment) |
| Possible | There is some evidence to suggest a causal relationship (e.g. because the event occurs within a reasonable time after administration of the trial intervention). However, the influence of other factors may have contributed to the event (e.g. the participant’s clinical condition, other concomitant treatments) |
| Probable | There is evidence to suggest a causal relationship and the influence of other factors is unlikely |
| Definite | There is clear evidence to suggest a causal relationship and other possible contributing factors can be ruled out |
Serious adverse event
Any untoward medical occurrence or effect that, at any dose:
-
results in death
-
is life-threatening – refers to an event in which the subject was at risk of death at the time of the event; it does not refer to an event which hypothetically might have caused death if it were more severe
-
requires hospitalisation, or prolongation of existing participants’ hospitalisation
-
results in persistent or significant disability or incapacity
-
is a congenital anomaly or birth defect.
Medical judgement was exercised in deciding whether an AE/AR is serious in other situations. Important AE/ARs that were not immediately life-threatening or did not result in death or hospitalisation but may jeopardise the subject or may require intervention to prevent one of the other outcomes listed in the definition above, was considered serious.
Serious adverse device effect
An adverse device effect (ADE) that has resulted in any of the consequences characteristic of a SAE or that might have led to any of these consequences if suitable action had not been taken or intervention had not been made or if circumstances had been less opportune, is defined as a serious adverse device effect (SADE). Note that this definition includes incidents and near incidents.
Serious procedure related adverse event
A SAE that occurs as a result of any procedure specific to the clinical investigation, including modification of the system, is defined as a serious procedure-related adverse event (SPRAE).
Unexpected serious adverse device effect
A SADE that is unexpected in nature is defined as an unexpected serious adverse device effect (USADE).
All AEs and all SAEs were recorded. Depending on the nature of the event, the reporting procedures below were followed. Any questions concerning AE recording/reporting was directed to the trial manager in the first instance. The reporting procedures were as follows:
-
Non-SAEs were recorded on the study CRF. Severity of all AEs were graded on a three-point scale of intensity (mild, moderate, severe):
-
mild – discomfort was noticed, but there is no disruption of normal daily activities
-
moderate – discomfort was sufficient to reduce or affect normal daily activities
-
severe – discomfort was incapacitating, with inability to work or to perform normal daily activities.
-
-
Relationship of an AE to treatment was assessed by the investigator/delegate (must be a clinician) at site, and investigators were responsible for managing all ARs.
-
Serious adverse event (SAE, including SADE): all SAEs, SADEs, SPRAEs and USADEs were recorded and reported on the SAE form to the chief investigator/delegate within 24 hours of learning of its occurrence. The initial report could be made by completing the SAE form, and faxing to the KCTU. A record of this notification (including date of notification) was clearly documented to provide an audit trail. In the case of incomplete information at the time of initial reporting, all appropriate information was provided as follow-up as soon as this became available.
-
Relationship of the SAE to the treatment was assessed by the investigator/delegate (must be a clinician). Treating clinicians reported SAEs in both trial arms which included the assessment of seriousness, and causality. Expectedness was assessed by the confidence interval (CI) once the report was faxed to KCTU.
-
The chief investigator reported all USADEs to the relevant ethics committee within the required time frame, with the support of the KCTU. The sponsor and Polyphotonix Medical Ltd (supplier of the masks) were planned to receive a copy.
-
Onward reporting of all USADEs to the MHRA was the responsibility of Polyphotonix Medical Ltd.
Withdrawal of subjects
Visit windows of ± 10 days promoted prompt visit attendance; non-attendance for study visits prompted follow-up by telephone. However, a delayed visit was entered in the database. An appointment was only defined as missed if the delayed visit was within 10 days of the next predefined trial visit date.
Participants had the right to withdraw from the study at any time for any reason, and without giving a reason. The investigator also had the right to withdraw participants from the study mask in the event of intercurrent illness, AEs, SAEs, suspected unexpected serious adverse reactions, protocol violations, cure, administrative reasons or other reasons. It was understood by all concerned that an excessive rate of withdrawals can render the study uninterpretable; therefore, unnecessary withdrawal of participants should be avoided. Should a patient decide to withdraw from the study, all efforts were made to report the reason for withdrawal as thoroughly as possible. Should a patient withdraw from intervention only, efforts were made to continue to obtain follow-up data, with the permission of the patient.
Participants who wished to withdraw from study mask were asked to confirm whether or not they were willing to provide the following: study-specific data at follow-up month 12 and 24; end of study data as per month 24, at the point of withdrawal; and questionnaire data collected as per routine clinical practice at annual follow up visits. If participants agreed to any of the above, they were asked to complete a confirmation of withdrawal form to document their decision.
Statistics
Sample size
With 300 participants, we anticipated 240 to be followed up (20% dropout). This was sufficient to provide 90% power to detect a 15 µm in mean change of retinal thickness at the zone of maximal thickness from baseline between arms using a two-sided 5% significance level, assuming a SD of 35.7 µm. The chosen detectable effect size (retinal thickness of 15 µm) was both plausible, in terms of being consistent with a CI estimate for this intervention in preceding research,12,20 and also minimally detectable in terms of being distinguishable from test–retest variation. Detectable effect sizes for secondary outcomes based on 240 participants followed up (for 90% power with 5% significance level) would be a between-arm difference in mean outcome of a size that is equivalent to 0.42 of a SD.
The DMC monitored the study power and we regularly provided information such as non-compliance, withdrawal and variability of the primary outcome as increasing proportions of the participants passed each of the 4-monthly measurement points. We followed DMC recommendations and deferred the monitoring of outcome variability within the 6-monthly reports to the DMC.
Justifications for the sample size calculation
Determination of the primary outcome effect size
The 15 µm difference between arms in the mean change in retinal thickness was the clinically relevant difference to be detected. This was plausible as it is consistent with the 95% CI from the previous trial and it is also minimally distinguishable from the 95% range of test–retest variation, which was estimated to be 10.2 µm in the pilot study, with an adequately small test–retest mean change over time of 0.9 µm.
Determination of the primary outcome variability
The SD of the change from baseline in retinal thickness was anticipated to be 35.68 µm, based on the previous pilot trial of this intervention. 19
Clustering of outcomes from eyes within subjects effects
Only one eye per subject was selected for the study despite the fact that the lightmasks covered both eyes. Therefore, clustering did not need to be accounted for in the sample size calculation.
Power to detect effects
The power was chosen to be 90% and was based on a two-sided, unadjusted, unpaired t-test at the 5% level of significance.
Determination of the sample size based on the primary outcome
The sample size was set to be 300 subjects (150 per arm). The target of 240 followed up with data for analysis having the primary outcome allowed for 20% attrition. This was based on the previous study in the same population.
The statistical package used for the calculated sample size was nQuery Advisor (version 4.0; Statistical Solutions, Saugus, MA, USA).
Detectable effects sizes expressed in general standardised form
For secondary outcomes, such as change in visual acuity, a medium effect size difference in means (of size 0.42 of a SD) between the arms, based on the unpaired t-test at the 5% significance level could be detected with 90% power if 240 participants were followed up with data for analysis.
Factors contributing to improved power and precision
It was anticipated that, relative to the methods specified in the sample size calculation, there would be an improvement in power and in the precision of estimated treatment effects on each outcome because the statistical analysis approach would incorporate repeated measures data of each outcome and with adjustment for the baseline corresponding to each outcome.
Analysis
A detailed statistical analysis plan (SAP) was set out to test the study objectives and hypotheses, describe the analytical approaches and procedures necessary to address these for the main trial paper and to provide guidance for further research reported in other papers, promoting consistent approaches and methods. The SAP was updated whenever the DMC recommended any changes based on accruing data or new literature being made available, and the changes were made after approval by the Trial Steering Committee (TSC).
As there can typically be more than one analytical approach to address a hypothesis, there is the potential for different results to be produced from using alternative approaches, alternative methods, alternative outcome definitions and the alternative data that may be involved. These differences can be influential, for example when results are of borderline statistical significance. Therefore, the SAP transparently recorded these decisions that were made about study hypotheses, outcome definitions and statistical procedures, along with their basis and the appropriateness of the assumptions required for their use, in advance of the main trial analysis. Changes within subsequent versions of the SAP prior to analysis were dated, with the basis for the changes reasoned, and recorded as an updated version and reasons justified.
It was also not intended that the strategy set out in the SAP should prohibit sensible practices. However, the principles established in the SAP were planned to be followed as closely as possible when analysing and reporting the trial.
Trial sample
Intention to treat
The achieved trial sample comprised those participants who consented to participate and were actually randomised into this trial. This randomised trial sample was also the trial intention-to-treat (ITT) population. The ITT principle states that every subject will be analysed according to the treatment arm to which they were randomised. In this trial, subjects’ data were analysed according to the ITT strategy, under which at least one of two component analyses is recommended to be based on the ITT population. 27
The trial ITT population comprised all randomised participants, regardless of eligibility (inclusion/exclusion) error, post-randomisation withdrawal and whether the correct randomised treatments were received or other treatments or interventions received.
Per protocol
A per-protocol (PP) analysis was also included. This included all randomised participants but excluded all data from the point that any participant was given intravitreal steroids, anti-VEGF agents or laser treatment. This PP analysis was approved by the TSC and added into version 5.2 of the SAP, because these treatments began to occur and they could substantially improve visual outcomes when given to those who have deteriorated to have poor visual acuity.
This PP analysis was carried out/considered as a secondary analysis and ITT maintained as primary.
Primary outcome
The purpose of the CLEOPATRA trial was to evaluate whether or not prevention of dark adaptation using lightmasks is safe and prevents the progression of early DMO. The principal research question was as follows: does wearing lightmasks during sleep at night decrease or slow the progression of early (non-central, non-clinically significant) DMO? Therefore, the primary efficacy measure was the change from baseline to 24 months in retinal thickness at the zone of maximum thickness at baseline in the study eye. It was measured using standard OCT at baseline and 24 months. For the 12- (secondary) and 24- (primary) month measurements, OCT tests were each done twice to obtain a more precise measurement of retinal thickness.
The trial had two arms, with equal allocation of participants in a 1 : 1 ratio to lightmask treatment or no treatment. After baseline, assessments were carried out at months 4, 8, 12, 16, 20 and 24 (when the last follow-up visit was carried out).
Trial secondary measures
Secondary efficacy outcome measures
The secondary efficacy outcome measures are listed below according to the type of the variable.
Continuous outcome variables
The change in retinal thickness of the baseline zone of maximum thickness at 12 months from baseline.
The change in BCVA at 12 and 24 months was measured using validated ETDRS vision charts. The BCVA test was carried out twice at baseline to reduce the intertest variability and the average BCVA was used in the analysis.
Three secondary continuous outcomes that complement the primary outcome at 12 and 24 months were as follows:
-
Retinal thickness in the central zone (zone 1) of the macula (Figure 2).
-
Retinal thickness in centre and parafoveal zones as measured by the sum of the five regions (1–5) of the macula (see Figure 2). This outcome captured thickness not just in the important central zone but also across the other four neighbouring parafoveal zones.
-
Retinal thickness as measured by the sum of the nine regions (1–9) of the macula. This outcome captured thickness across the macula.
FIGURE 2.
Retinal thickness zones numbered 1–9 in the right and left eye. Reproduced with permission from Sivaprasad et al. 24 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.

Time-to-event outcome variables
Time in months to occurrence of centre-involving macular oedema as defined by > 300 µm on OCT over the 24 months of the study.
Categorical outcome variables
The categorical outcome variables included:
-
Progression to clinically significant centre-involving macular oedema as defined as thickness of > 300 µm on OCT occurring within 12 and 24 months.
-
Requirement for macular laser treatment, anti-VEGF or steroids treatment at 12 and 24 months.
-
Progression of retinopathy as assessed by the independent reading centre as changes in severity of ETDRS scale at 12 and 24 months.
-
Microaneurysm turnover of six or more in a ratio of appearance versus disappearance of microaneurysms, using computerised software at 12 and 24 months. The software was not available for this analysis.
-
Change in status (yes/no) of each morphological characteristic of macular thickening on OCT from baseline to follow-up at 12 and 24 months.
Other secondary outcome measures
-
Change in HbA1c level at 12 and 24 months.
As those developing high levels of HbA1c may have an increase in retinal thickness, it was important to make sure that there was not an unequal number of participants with high HbA1c levels between the arms. Some participants with high levels of HbA1c were treated by their general practitioners, and these medications and other concomitant medications were recorded in each arm.
-
Compliance measures.
Compliance with the lightmasks was determined at 12 and 24 months using an electronic device embedded in the mask, which quantified the time (in minutes) the masks were used each night. As the number of sleep hours generally varied quite considerably from person to person and within person, and the duration of light varied by season, a pragmatic decision of 6 hours (360 minutes) in a day was taken to be sufficient to represent the 100% compliance within a single day and, therefore, also represented the level at, and above which, maximal benefit is derived. An observed duration of > 6 hours (> 360 minutes) on a single day was taken to be 100% compliance for that day.
For each participant, compliance was measured primarily over the 2 years that the mask was offered to the patient provided the masks were returned at their next 4-monthly visit and the anonymised data in each mask was sent to Polyphotonix Medical Ltd to convert into the number of minutes worn per day. A compliant participant was defined for this study (and accepted in the grant application) as masks that were worn 70% of the time. In practice, this means that over 2 years, the mean of the daily percentages (as defined above, being truncated to the 6-hour maximum of 100% on a day where necessary) needs to be ≥ 70%.
This pragmatic approach (i.e. an upper truncated value for 100% and the mean across days) has been adopted in smoking cessation trials where there is daily measurement of compliance to prescribed nicotine replacement therapy. 28
As an illustration, if a participant wore the mask for at least 6 hours on ≥ 70% of the days, then this participant would be compliant. An alternative participant could wear the mask for as little as 70% of 6 hours (i.e. 4.2 hours, every day) and would also be compliant. More specifically and realistically, a participant who accumulates ≥ 70% (in whatever way over time) of the truncated (or available to be counted) 6 hours per day across the 2-year period, was defined to be compliant. Six hours per day over 2 years (i.e. 730 days) is 4380 hours, and 70% of this is 3066 hours.
For the purpose of DMC monitoring, compliance was also calculated in each of the six successive 4-month periods in the trial to match the timing of the outcome measures, and using the same approach, based on the mean of the daily (truncated) percentages within the period, and applying a 70% threshold. If compliance data were missing on a day, then it was assumed that there had been no compliance.
Safety outcome measures
Sleep disturbance was measured as the change in:
-
daytime sleepiness as measured by the ESS score at 12 and 24 months
-
insomnia score measured using the Pittsburgh Insomnia Rating Scale 20-item version (PIRS_20) at 12 and 24 months.
Safety monitoring measures
Safety outcome measures (ocular and systemic AEs) were graded and reported by arm to the DMC as follows:
-
Presence of AEs, ADEs, SAEs, SADEs, SPRAEs and USADEs. All AEs were also graded by intensity (mild, moderate, severe) and by relationship to treatment (none, remote, possible, probable and definite).
Timing of measures
Retinal thickness was measured by OCT at baseline, 4, 8, 12, 16, 20 and 24 months.
Refracted BCVA as well as retinal colour photographs were measured at baseline, 12 and 24 months. The measures of sleep disturbance in terms of daytime sleepiness and insomnia were to be evaluated at baseline, 12 and 24 months.
Participant duration in the study
Each subject was to participate in the trial for 24 months.
Final assessment
The final study assessment was done after the last study subject completed their 24-month assessment.
Subgroup variables
Subgroup analysis was carried out by baseline HbA1c level (< 8% or ≥ 8%).
Outcomes requiring derivation
The source of derivation codes for the list of outcomes on sleep-related questionnaires are shown in the following sections.
Pittsburgh insomnia rating scale – 20-item version25
Each item is scored on a 4-point scale (0–3), with one missing item allowed. If one item is missing, then pro-rating was done [i.e. (sum/19) × 20].
Minimum score = 0 (good), and maximum score = 60 (bad).
Epworth sleepiness scale26
Each item is scored on a 4-point scale (0–3). If one or more item scores is missing, then the ESS is invalid. It is not permitted to interpolate missing item scores.
The minimum score was equal to 0 (low level of daytime sleepiness), and the maximum score was equal to 24 (high level of daytime sleepiness).
Missing items in scale and subscales
The number (%) of participants with complete data for each scale was to be reported. The missing value guidance given by ESS and PIRS_20 scales was used.
Use of data transformation
It was not anticipated that consideration for transformation of any continuous outcomes would be needed because of the reasonably large sample size for group comparisons in the main trial analyses. Assumptions of normality and constant variance required by the models were examined using residual and other diagnostic plots. If relevant, and necessary, a log-transformation would be considered because this retains a sensible interpretation for inferences between arms. This was anticipated to be more relevant in the mechanistic evaluation, in which sample size was smaller, and for measures of areas and volumes. However, if an absolute interpretation was important, then rather than considering data transformation, a non-parametric bootstrap method for obtaining CIs was considered. 29 For the mechanistic evaluation, the analysis plan included consideration of non-parametric methods.
Defining outliers
Outliers are observations that have extreme values relative to other observations observed under the same conditions. An outlier was defined here as a data point being at least four SDs from the mean of its distribution of values observed across other participants. This definition would apply to the transformed scale for those outcomes that may have been log-transformed.
Handling outliers
Sensitivity analysis would be undertaken to check whether or not the outlier is influential by obtaining results with and then without inclusion of the outlier. If the conclusions are changed, then this will be noted. Outliers in key outcomes were reported back to the study team after DMC analysis reporting.
Comparison of rates of adherence and follow-up
Compliance was assessed in the lightmask arm.
Non-attendance at any visit and withdrawal rates were to be calculated per arm and compared between arms using Fisher’s exact test.
Analysis covariates
Stratifiers
It is important to consider which, if any, baseline covariates are to be adjusted for in the analyses. The ICH E9 guideline30 recommends careful consideration is given to those factors on which randomisation may be minimised. These factors tend to be predictive of outcome, as these will tend to improve the precision of the estimated treatment effects and are also therefore those covariates that may introduce chance confounding due to chance imbalances arising between arms. Levels of HbA1c, baseline thickness in excess of 320 µm in parafoveal zones (2 to 5) and study site were chosen as minimisation factors. Levels of HbA1c and study site were included in the analysis as covariates. The baseline thickness in excess of 320 µm in parafoveal zones (2 to 5) was removed as an analysis covariate from version 5.2 of the SAP because the great majority of participants had baseline thickness in excess of 320 µm in parafoveal zones.
Baseline
The corresponding baseline measure for a continuous outcome is also often predictive of the outcome at follow-up, whereas standard errors (SEs) of statistics derived from binary outcomes vary little with the prevalence to offer gain in precision while expending degrees of freedom. Therefore, ‘baseline’ was included as an additional covariate when modelling only continuous outcomes wherever data were available (e.g. for retinal thickness and visual acuity measures).
Participants with missing data in the baseline of a continuous outcome were accommodated in the analysis using the missing indicator method of White and Thompson. 31
Primary outcome analysis
Statistical model
The primary efficacy measure was the change from baseline in retinal thickness at the zone of maximum thickness in the study eye, determined by OCT at 24 months (or ‘retinal thickness’ for short). The primary time point for evaluation of the outcome was 24 months after baseline. As the analysis approach took advantage of covariate adjustment for the baseline retinal thickness, the primary end point could equivalently be regarded to be each participant’s 24-month measurement of retinal thickness. This is convenient because then those with a 24-month outcome but whose baseline measurement was missing were not regarded to be missing on the primary end point, with the same principle to be applied to secondary continuous outcomes. The primary outcome may therefore be referred to below as the 24-month retinal thickness, rather than the change in this from baseline to 24 months.
The primary outcome was analysed using a linear mixed-effect (LME) model, which incorporated the six 4-monthly post-baseline measurements of the outcome over time to 24 months. The model allowed for within-participant correlation in the 4-monthly outcomes measured over time. As the correlation may be stronger for those measurements of retinal thickness taken closer together in time for a participant, the model was specified to have an unstructured variance–covariance matrix, but a simpler correlation structure, such as exchangeable, was planned to have been used if the model with the unstructured matrix did not fit. 32 The fixed effects in the mixed-effects model were the main effect terms for arm, the categorical minimisation factors HbA1c level and study site contrasts, and also ‘time’ contrast terms. Other fixed effects included in the model were the interaction between ‘time’ terms and level of HbA1c, study site and the baseline of the outcome (and as it turned out only for secondary outcomes requiring this, the missing indicator required for the missing indicator method). 31 If the model would not have been able to accommodate as many terms, the interaction between time and study site was planned to have been removed, leaving just a main effect for study site to apply equally across time.
The aim of the primary outcome analysis was to provide an adjusted difference in the mean change score in the 24-month retinal thickness between arms (treatment minus control), as an estimate of the population effect of the lightmask intervention compared with the control dummy-mask. Differences were considered significant if the p-value was < 0.05, and differences between the groups were estimated with 95% CIs.
Missing data assumptions required to be made to interpret the model
This analysis involved fitting a suitable statistical model to all the data that had been observed over the repeated (4-monthly) time points recorded for the outcome, and the valid interpretation of the results from this model implicitly assumes that outcome data that are missing from participants are so-called ‘missing at random’ (MAR). As the outcome was measured repeatedly over time, this means that participants with ‘missing data’ were assumed to have outcomes equal in distribution to participants with ‘observed data’, conditional on the (i.e. for the same given) baseline and 4- to 24-month follow-up measurements of the outcome variable (and controlling for the same impacts over time of minimisation factors and outcome correlation) estimated from the participant data that were included in the analysis. 33
Reasoning behind the chosen model versus alternatives
The use of interim measures in the model (linear mixed effects versus analysis of covariance)
An alternative modelling approach to the likelihood-based LME model approach is the analysis of covariance (ANCOVA) model, where the primary outcome is analysed adjusting for baseline but without representing the available interim measurements within the model. Use of this model also assumes missing outcome data are MAR, but the assumption is needlessly stronger, ignoring the data observed at interim points in those subjects who are missing the primary outcome at 24 months. The LME model better captures any alteration in interim distribution (of observed data) of those participants with missing primary outcome data at 24 months than in the distribution of those with complete observed primary outcome at 24 months, primarily through the within-participant correlation over time.
It was accepted that the MAR assumption associated with the LME model above is more plausible than the alternative MAR assumption associated with ANCOVA because the LME model includes repeated measures of the outcome (rather than ignoring this available interim observed data), and is more plausible than the consequences of the alternative last observation carried forwards (LOCF) approach. The LOCF approach assumes no further change from the last observed value in any participants who have dropped out, whatever the reason for dropout or whatever the outcome values before the one carried forwards, and assumes no uncertainty surrounding the imputed value.
It is plausible that the recent observed interim data on retinal thickness prior to dropout conveys information predictive of future unobserved retinal thickness outcome. The LME model included this, and carried this through via the within-participant correlation, whereas ANCOVA does not. This made a difference when subjects whose observed retinal thickness prior to dropout differs in distribution (given the covariates) from the corresponding retinal thickness in subjects who continue to 24 months.
We anticipated that those dropping out would have been more likely to follow the future mean trend over time of the completers (given with the same covariates and follow-up data to that point) than to be personally stable from point of dropout. The MAR assumption for the LME model more reasonably captured this than the LOCF assumption. This is described further, graphically, in Figure 5. 33
We believe that the MAR assumption relating to the LME model is reasonably plausible in this clinical context of ophthalmology. This is based on considering whether the reasons for dropout in this context are relatively ‘innocent’ or ‘unconnected with outcome or future outcome’ (which would favour the MAR assumption).
Two such situations are as follows: (1) participants may not see improvements in their vision immediately as their retinal thickness improves (and so dropout is less connected with underlying retinal thickness); and (2) wearing masks may disturb sleep (unconnected with underyling retinal thickness).
Such ‘innocent’ reasons mean that dropout may not depend on retinal thickness (having accounted in the model for covariates including retinal thickness itself up to the point of dropout).
Nevertheless, a sensitivity analysis was undertaken to assess the possibility of alternative plausible values of treatment effect arising from potential mishandling of missing data in the primary analysis model. As disease severity and sleepiness disturbance data were collected, it was possible to explore the association between these and dropout to improve the interpretation of the sensitivity analysis results.
The use of the missing indicator method
The use of the missing indicator method31 enabled those with missing baseline data to contribute to the estimate, and for a gain in precision through adjustment for baseline. The inclusion of interim 4-monthly measurements enabled those without the primary outcome but with at least some follow-up data to be able to contribute to the estimate. Those with no post-baseline data for the primary outcome, however, were not included in the main LME model analysis of the primary outcome. An ideal is to include all randomised subjects in this ITT analysis without having to consider a reduced set of participants unless strictly necessary.
The use of linear mixed effects rather than multiple imputation
The LME model for the primary outcome analysis was the first of a two-part approach called the ITT strategy27 in which a second analysis (outlined in Sensitivity analysis for missing data) examines the sensitivity of the results to missing data in the full randomised, ITT, population. This meets the ideal of ITT. The approach to missing data taken for the trial follows the recently published implementation paper of the ITT strategy. 33 In this paper, the similarity was identified between the LME approach and the multiple imputation having corresponding covariates, both in terms of the similarity of the MAR assumption and in terms of the resulting estimates. This underlies the decision for our trial to take a LME approach rather than a multiple imputation approach. Sensitivity analysis (to explore departure from the MAR assumption) may then be added to the LME approach in the same way as it is added within the multiple imputation approach.
As this decision for LME (rather than multiple imputation of missing 24-month measure) is a change from the funded grant application, further discussion is given. A small disadvantage of the multiple imputation approach is that there may be inaccuracy as a result of the use of a finite number of imputations; if the number of imputations is too small then the Monte Carlo error remains large. 33 A potential advantage of the multiple imputation is the ability to incorporate further covariates. However, for the CLEOPATRA trial, the predictive elements of primary outcome are mostly the interim measurements of the primary outcome prior to dropout and the minimisation factors (potential chance confounders). In addition, the multiple imputation model is defined often at a late stage (when prevalence and predictors of dropout can be established), notably after prespecification is possible, which may outweigh any negligible net efficiency advantage. Conversely, the LME approach is defined a priori, above (see Statistical model), and with additional sensitivity analysis below (see Sensitivity analysis for missing data).
Sensitivity analysis for missing data
An expert missing-data group concluded that rather than statisticians reacting to missing data at the end of a trial, there should be comprehensive, proactive planning for handling missing data at the stage of designing trials. 34 The group recommended that there should be consideration of missing data mechanisms (e.g. MAR) and, if the missing data may be informative, that appropriate sensitivity analyses should be undertaken to investigate the robustness of the inferences to the different assumptions made by the main analysis. It has also been recommended that analyses allowing for non-response and low intervention uptake (or compliance) are best specified in advance and included in the analysis plan. 35 In this section we consider the handling of missing data, and in Sensitivity analysis for non-compliance we consider handling low compliance.
In the funded grant application for our trial, a sensitivity analysis was specified, which would be additional to, and nested within, the multiple imputation. Given the change from multiple imputation to the LME model, the sensitivity analysis was instead applied to examine robustness of the LME results to the MAR assumption, with the aim of adequately exploring the impact of departures from the MAR assumption33 on the primary outcome results.
For the sensitivity analysis, we prespecified a range for retinal thickness from –20 µm to +20 µm over which the mean of the ‘unobserved outcome data’ might depart (or be different) from the mean of the ‘observed outcome data’. 33 In other words, this range could be defined as how much a typical subject with missing data may, on average, have had a different estimated treatment effect compared with the corresponding subject with the outcome data observed (given the same baseline covariates and follow-up data in the LME model). The range (–20 µm to + 20 µm) is chosen to represent both negative and positive departures that could potentially arise as the ‘net effect’ of alternative reasons, which may be unknown, such as dropout caused by no anticipated further improvement or dropout caused by no improvement so far, together with no anticipated achievable improvement.
This range of 40 µm (from –20 µm to +20 µm) is generously wide for exploring sensitivity of the main results to departures from the MAR assumption, because 20 µm (as the maximum departure in either direction) is larger than the detectable between-arm treatment effect (difference in means) of 15 µm that is set in the sample size calculation, and because it represents over half of the SD of 35.68 µm, which is a sizeable shift in the mean of the distribution for dropouts compared with completers. The generous width would also cover the situation that there may be some bias in the estimated treatment effect from having chosen this LME model for the main analysis.
At the end of the trial, the fractions of individuals with missing data for retinal thickness at 24 months was available in each arm fi (for intervention) and fc (for control). The parameter representing excess retinal thickness in those missing compared with those observed, δ, included values across the range –20 µm to +20 µm. Three scenarios were undertaken within the sensitivity analysis. 33,35 These reflect whether departures from the MAR assumption apply within the intervention arm only (lightmasks), within the control arm only (non-lightmasks) or within both arms equally and in the same direction (thereby potentially cancelling out across the sensitivity range, if the dropout rate were to be the same in both arms).
Scenario 1: the treatment effect from the LME model will be increased by fiδ.
Scenario 2: the treatment effect from the LME model will be increased by –fcδ.
Scenario 3: the treatment effect from the LME model will be increased by (fi–fc)δ.
Sensitivity analysis for non-compliance
Incomplete uptake of trial interventions often means that randomised groups have more similar experience than the investigators had intended, which usually causes the difference in outcomes to be smaller than it would have been with better uptake.
White et al. 35
For this trial, this means that low compliance with the active lightmasks would bring about two groups that have more similar intervention experience to each other than was planned (i.e. intervention more similar to control through low compliance).
We could use compliance to make the MAR assumption more plausible by jointly modelling the continuous (percentage) compliance variable as an additional outcome variable in the LME model alongside the post-baseline 4-monthly retinal thickness outcome measures, where compliance has a natural zero (equivalent to intervention uptake in the control arm) and would need to be standardised to have the same SD as the 24-month retinal thickness outcome. The model was extended with an extra parameter such that the residual SD in the model was allowed to be different in each arm of the trial. 35
However, the purpose of incorporating compliance is to estimate a causal effect of treatment among compliers. This purpose arises from the CLEOPATRA trial being funded by the Efficacy and Mechanism Evaluation programme of the National Institute for Health Research (NIHR), where there is a need to understand what the treatment effect might be if compliance were adequate. ‘Efficacy’ is of interest from this additional analysis, rather than ‘effectiveness incorporating low compliance’. In the application, a complier average causal effect (CACE) of treatment analysis, under a MAR assumption, was specified, as described in Dunn et al. 36
Further to the previous sensitivity analysis, we conducted an analysis estimating the effect of lightmasks versus non-lightmasks on the primary outcome (retinal thickness) in a complier population, while respecting randomisation. This approach provided a better estimate of the true effect without suffering from potential biases seen in a per-protocol analysis.
A CACE analysis was carried out as recommended and outlined by Dunn et al. 36 The CACE estimate is the comparison of the average outcome of the compliers in the lightmasks arm with the average outcome of the comparable group of ‘would-be compliers’ in the non-lightmasks arm (not requiring measurement of compliance in the control arm). A participant was assumed to be a complier, if he/she wore the mask for at least 70% of the time over the 2-year period, where a maximum of 6 hours is taken to be 100% on a single night. A ‘would-be complier’ is then a person in the control arm who would have complied with their treatment allocation had they been allocated to the intervention arm. To determine the CACE estimate, the ‘exclusion restriction’ criteria was anticipated, which assumes that the mean retinal thickness for non-compliers in the control arm is the same as that for those in the treatment arm (i.e. there is no effect of the ‘label’ of one’s group on outcome among non-compliers).
The outline of the approach to be taken is given here
Sample sizes and means were deduced for the non-lightmasks would-be compliers and would-be-non-compliers in Table 4 by assuming that the proportion of intervention group compliers and control group would-be compliers is the same under randomisation, and that would-be non-compliers in the control group would have the same mean outcome as non-compliers in the intervention group (the exclusion restriction assumption). The sample sizes refer to those followed up with primary outcome data (retinal thickness at 24 months).
| Treatment arm | Compliance | All | |
|---|---|---|---|
| Compliers (≥ 70%) | Non-compliers (< 70%) | ||
| Lightmask |
NI1 MI1 |
NI2 MI2 |
NI MI |
| Non-lightmask |
= NC – (NI2/NI) × NC = (MC – (NI2/NI) × MI2)/(NI1/NI) |
= (NI2/NI) × NC = MI2 |
NC MC |
This gives a simple unadjusted CACE estimate as the ITT estimate (MI – MC) divided by the proportion compliant in the intervention group (NI1/NI). The method has been adapted to a more plausible MAR assumption by replacing the sample sizes at follow-up by those at baseline. In the presence of missing compliance (i.e. insufficient data over time to be able to deduce if compliance is at least, vs. below, 70%), it was primarily assumed that the participant is a non-complier. This was a consequence of coding non-compliant days as 0% compliant days. The required dose to have an effect is not well studied; alternative levels of compliance, 60% and 50%, were also considered.
As planned, we reported an alternative CACE estimator, was a priori preferred because it was obtained from the primary analysis LME model, which adjusted for baseline and accounted for the primary outcome measured at all of the 4-monthly visits. It was calculated as the ratio of the estimated treatment effect to the proportion compliant, following the recommended rule of thumb. 37
Sensitivity analysis to the requirement for treatment of centre-involving macular oedema
Some participants were anticipated, during the 2-year study period, to develop centre-involving macular oedema at the level of 300 µm. If this reached 400 µm they would become eligible for treatment according to National Institute for Health and Care Excellence (NICE) technology appraisal guidance although some may be treated at a higher level depending on clinician discretion. After treatment, the retinal thickness and the primary outcome may be reduced for this reason rather than being due to the randomised treatment alone.
Therefore, we undertook two sensitivity analyses to examine the robustness of the results of the outcome findings. In these sensitivity analyses, we used 400 µm as a treatment-related value because it is not dependent on the clinician’s decision but on an external fixed value from NICE.
First, for a patient receiving treatment before the 24-month end point, the retinal thickness measurement taken just after the patient first reached 400 µm was considered in the analysis to carry forward to be their final measurement in this sensitivity analysis. For all other participants, the retinal thickness reached at the 24-month visit were retained.
Second, we undertook a time-to-event analysis for the time to requirement of treatment (> 400 µm) using stratified Cox proportional hazards regression incorporating the minimisation factor HbA1c. ‘Study site’ was not included because there would be too many strata for the number of expected events to be reliably modelled.
Sensitivity analysis for large changes (i.e. > 50 µm) in the zone of maximum baseline retinal thickness
Given the observation that a subject’s retinal thickness may suffer drops or increases of > 50 µm, the variability in retinal thickness could be different across time, and potentially between treatment arms, and so the non-parametric Mann–Whitney U-test was made available in this case to be used as a sensitivity analysis to compare retinal thickness between arms at 24 months. This was applied to the primary outcome in the SAP version 5.2.
Interim analysis
There was planned to be no formal interim analysis.
Secondary outcomes analysis
Analysis of continuous outcomes
Analysis of the secondary continuous outcomes, such as the change in the central, inner five-zone, or full nine-zone retinal thickness, and changes in the measures of sleep disturbance used the same initial method as that used for the primary outcome (the LME model), but without sensitivity analyses (e.g. for missing data or compliance).
Analysis of binary outcomes
For the binary outcomes, such as the proportion of participants progressing to clinically significant centre-involving macular oedema at 24 months, a difference in proportions was used to compare arms, with 95% CIs and Pearson’s chi-squared tests, or Fisher’s exact tests and Wilson’s exact CIs when any expected table counts were < 5. When required (e.g. small overall numerator), exact unadjusted CIs were estimated for proportions and differences between proportions.
Exploratory analysis of the morphological characteristics
Within each arm the change in the proportion with a morphological characteristic from baseline to 12 and 24 months was summarised using the difference in paired proportions and McNemar’s SE to calculate the CI. These changes in proportions were compared between arms using a Z-test by subtracting the two changes in proportions by their SE.
Analysis methods for secondary outcomes
For the secondary outcomes, the analyses in Table 5 were planned.
| Types of variables | Outcomes | Methods |
|---|---|---|
| Continuous | Change in the baseline zone of maximal retinal thickness at 12 months | LME model (primary outcome analysis model) |
| Change in retinal thickness in the central zone of the macula at 12 and 24 months | LME model | |
| Change in retinal thickness in the centre and parafoveal zones at 12 and 24 months | LME model | |
| Change in retinal thickness over the nine regions of the macula at 12 and 24 months | LME model | |
| Change in BCVA refracted visual acuity at 12 and 24 months | LME model | |
| Changes in daytime sleepiness (ESS) at 12 and 24 months | LME model | |
| Changes in insomnia (PIRS_20) at 12 and 24 months | LME model | |
| Change in macular volume at 12 and 24 months | LME model | |
| Change in HbA1c level at 12 and 24 months | LME model | |
| Time to event | Time to occurrence of centre-involving macular oedema (> 300 µm) | Stratified Cox regression |
| Categorical | Progress to clinically significant centre-involving macular oedema at 12 and 24 months | Difference between proportions with 95% CI |
| Request for macular laser treatment, anti-VEGF treatment or steroids at 12 and 24 months | Difference between proportions with 95% CI | |
| Proportion of participants that show progression of retinopathy (ETDRS scale) at 12 and 24 months | Difference between proportions with 95% CI | |
| Difference in the proportion with a morphological characteristic present from baseline to 12 and 24 months | Z-test based on McNemar’s test SE in each arm | |
| Lightmask compliance every 4 months | One-sample proportion |
Substudy on mechanistic evaluation
The concept of hypoxia as a contributing factor in early DMO was explored in this substudy. The main objective of this substudy was to evaluate the effect of inhalation of oxygen and lightmasks on the retinal morphology and visual function. It was originally planned for this substudy to be carried out with 30 participants of the main study in Moorfields Eye Hospital. However, as the tests were time-consuming, it was difficult to consent participants for the substudy. Therefore, the study was carried out by conducting these time-consuming tests on 28 participants to establish whether or not local regions of retinal oedema corresponded to hypoxia, and whether or not functional defects of outer and inner retinal layers are associated with corresponding anatomical changes. The additional tests included oximetry, mfERG and scotopic microperimetry at baseline and at 12 months. At baseline, these tests were carried out twice: either with breathing air (normal situation) first, followed by participants having their measurement taken while breathing 100% oxygen through a face mask, or vice versa to ensure that the tests were completely done in random order.
Of the 28 participants, half were from the group randomised to use the lightmasks, and the other half from those randomised to use the non-lightmasks.
Comparison of the two baseline tests in all of the 28 participants, were prior to use of any masks, and addressed questions relating to the effect of inhalation of oxygen relative to normal conditions.
Mechanistic evaluation outcomes and correspondent analysis
For each continuous outcome assessed in the mechanistic evaluation substudy, means (SDs) or medians [interquartile ranges (IQRs)] of changes from baseline to 12 months were used to describe all participants. Table 6 shows the outcomes and corresponding analysis.
| Analysis | Outcome measures | Analysis methodsa |
|---|---|---|
| Air vs. oxygen at baseline |
Change in P1 and N1 amplitudes (average within each of the 9 ETDRS zones) and P1 and N1 peak time (average within each of the nine ETDRS zones) in mfERG after supplemental oxygen Y1: mfERG measurements at baseline (air) Y2: mfERG measurements at baseline (oxygen) Comparison: Y1 (after breathing air) vs. Y2 (after oxygen) Participants: all 30 participants |
Paired t-test of each summary measure,b based on aggregating the zones |
| Air vs. oxygen at baseline |
Change in retinal sensitivity in scotopic microperimetry (average within each of the nine ETDRS zones) after supplemental oxygen Y1: scotopic microperimetry measurements at baseline (air) Y2: scotopic microperimetry measurements at baseline (oxygen) Comparison: Y1 (after breathing air) vs. Y2 (after oxygen) Participants: all 30 participants |
Paired t-test of each summary measure,b based on aggregating the zones |
| Change over time by arm in air and/or oxygen |
To determine differences in change in P1 and N1 amplitudes and P1 and N1 peak time in mfERG after lightmasks and non-lightmasks at 12 monthsY: 12 month minus baseline of P1 or N1 X: arm = lightmask vs. dummy mask |
Unpaired t-test of each summary measure,b based on aggregating the zones Graph: 95% limits of agreement for change under oxygen vs. change under air with symbol for arm |
| Change over time by arm in air and/or oxygen |
To determine differences in changes in retinal sensitivity in scotopic perimetry measurements after lightmasks and non-lightmasks at 12 monthsY: 12 month minus baseline of P1 or N1 X: arm = lightmask vs. dummy mask |
Unpaired t-test of each summary measure,b based on aggregating the zones Graph: 95% limits of agreement for change under oxygen vs. change under air with symbol for arm |
| Graph to compare the effect of oxygenation and lightmask |
To correlate the changes induced by lightmasks and oxygen supplementation on retinal sensitivity using oximetry Y1: retinal sensitivity at 12 months in lightmask arm air minus the corresponding air at baseline (lightmask effect) Y2: retinal sensitivity at baseline in lightmask arm oxygen minus the corresponding air at baseline (oxygen effect) Difference = Y1 – Y2 (how well the lightmasks do relative to oxygen) Comparison: Y1 vs. Y2 as a paired t-test (tests for mean difference) Participants: in 15 randomised to receive lightmask Bland–Altman limits of agreement to display the range of differences: plot the difference against the average (of Y1 and Y2) |
95% limits of agreement and paired t-test |
Handling multiple comparisons
All study analyses were based on tests that are two-sided, including the two-sided 95% CIs.
Significance tests were used sparingly and restricted when possible to addressing stated hypotheses. Secondary outcomes, as well as the primary outcome, were summarised using an effect size with a 95% CI. Interpretation for those secondary outcomes that did not directly address the stated study hypotheses were more cautious.
Software
The principal software package was IBM SPSS statistics 23 (IBM Corporation, Armonk, NY, USA) with the R software (The R Foundation for Statistical Computing, Vienna, Austria) planned to be available.
Data Monitoring Committee monitoring
The DMC monitored study power and the study statisticians regularly provided the DMC members (see Appendix 2) with information such as non-compliance, withdrawal and variability of the primary outcome as an increasing proportion of the participants pass each of the 4-monthly measurement points.
Data handling
The chief investigator is the custodian for the trial data. Personal data were regarded as strictly confidential. To preserve anonymity, any data leaving the site identified participants by their initials and a unique study identification code only. No identifiable patient data left the study site. The study complied with the Data Protection Act 1998. 38 All study records and investigator site files (ISFs) were kept at site in a locked filing cabinet with restricted access. Any breach of confidentiality was minimised by adherence to the Data Protection Act 1998,38 with reassurance stated on the consent form to minimise any potential distress.
Data collection
Each participant was assigned a sequential identification number via the InferMed MACRO web-based data entry system. This number, rather than the participant’s name, was used to collect, store and report participant information.
Data management
Data management was consistent with Medical Research Council Guidelines for Good Clinical Practice in Clinical Trials39 and the Data Protection Act. 38 Centre principal investigators ensured that all personnel were familiar with and complied with these guidelines. Data management procedures for the trial were developed and overseen by KCTU.
Electronic case report form
All baseline and follow-up data were entered on the online InferMed MACRO electronic data capture system (www.infermed.com). This system is regulatory compliant [Good Clinical Practice (GCP) and the European Commission Clinical Trial Directive]. An eCRF using the MACRO electronic data capture was programmed by the Clinical Trials Unit in collaboration with the trial manager and trial statistician, and was hosted on a dedicated secure server within King’s College London. The eCRF system had a full audit trail, data discrepancy functionality, database lock functionality and supported real-time data cleaning and reporting. The Clinical Trials Unit provided training, essential documentation and user support to the study centres, as well as on-site audit and monitoring. A detailed SOP covered data recording, online entry, checking, central backup and storage. A regularly updated coding manual was developed to accompany the study database. Each research worker and centre principal investigator had a unique username and password provided by the Clinical Trials Unit for the eCRF. The trial manager provided usernames and passwords to any new researchers. Only those authorised by the trial manager were able to use the system.
Data collection and recording
Baseline data were collected and entered by researchers in each study site prior to randomisation. Each participant was assigned a unique trial PIN at the start of the assessment process. This number was written on all clinical assessment forms, data sheets and databases used to record participant data. Trial data were first entered on to paper source data sheets provided to each centre during the preparation phase. We endeavoured to minimise the use of paper at all times. The data sheets were immediately checked for completeness and accuracy. If data queries arose, these were logged and followed up locally before data were entered online. A hard copy of a record sheet linking patient identity, contact details and trial PIN for all participants was kept at each site. These were placed securely in a locked filing cabinet separate from data sheets. All data were kept secure at all times and maintained in accordance with the requirements of the Data Protection Act 199838 and archived locally according to clinical trial GCP regulations and the host institutions additional procedures.
Quality assurance
The study incorporated a range of data management quality assurance functions. As the data were entered online, the data manager logged any queries generated and fed back to the centre research workers in a timely manner. Maintaining a single point of contact between each centre and the Clinical Trials Unit, the trial manager conducted regular monitoring visits at each centre, checking 10% of entered data for consistency with the written data worksheet. Any necessary alterations to entered data were indicated clearly with an audit trail from the original point of data entry, to ensure that any such amendments, and the reasons for them, could be inspected and tracked.
The study was managed through the KCTU. The Trial Management Group included the chief investigator, operational director, trial manager, data management strategic lead, sponsor representative and other members of the trial team when applicable.
The KCTU provided day-to-day support for the site and provide training through investigator meetings, site initiation visit and routine monitoring visits. The principal investigator was responsible for the day-to-day study conduct at site. Quality control was maintained through adherence to Clinical Trials Unit SOPs, study SOPs, study protocol, the principles of GCP, research governance and clinical trial regulations.
Database lock
After written recording, each research worker transcribed data onto the eCRF within 1 working week of a participant assessment. After completion of all follow-ups and prompt entry of data, the trial manager reviewed the data and issued queries. The research worker answered these queries before the participants’ data were ‘frozen’ within the database. After that time, changes were not made to the database by the centres unless specifically requested by the study office in response to statistician data checks. At the end of the trial, the centre principal investigator reviewed all the data for each participant and provided electronic sign off to verify that all the data are complete and correct. At this point, all data were formally locked for analysis. At the end of the trial, each centre was supplied with a compact disc read-only memory (CD-ROM) containing the eCRF data for their centre. This was filed locally for any future regulatory or internal audit.
Amendments to statistical analysis plan versions
Version 1 was written by the trial statistician on 31 July 2013 and was verified by the senior statistician on 23 August 2013, leading to version 2.
Version 3 was produced on 24 October 2013 after comments from the chief investigator and the trial team.
Version 4 was produced on 31 March 2014 and accounts for comments made at a meeting on 14 February 2014 with the DMC statistician. The main changes were to record that the statisticians would be unblinded in reporting at DMC meetings, that two measurements would be taken of ETDRS at baseline and two of retinal thickness at 12 and 24 months, and that the correlation structure of the analysis of repeated measures continuous outcomes would have an unstructured covariance matrix with covariate contrasts interacting at each follow-up time point. In addition, the principal investigator added a stratifier (whether or not retinal thickness is 320 µm already in any of the inner zones 2–5 at baseline).
Version 5 was produced on 22 May 2014 and this was the first SAP version formally signed off by the DMC chairperson and statistician and TSC chairperson. The method of stratified randomisation was replaced for minimisation because there were too many strata for a reliable stratification.
Version 5.2 was produced in August 2016 after discussions with the trial management team, DMC and TSC in June 2016.
-
A per-protocol analysis was added as a secondary analysis because some subjects take steroids, anti-VEGF or laser treatments, which makes retinal thickness improve substantially.
-
The minimisation stratifier ‘Baseline thickness in excess of 320 µm in zones 2–5 or not’ was to be removed from the planned analysis model and as a subgroup analysis variable because all but a handful of participants had baseline retinal thickness > 320 µm in zones 2–5.
-
A sensitivity analysis to large retinal thickness departures (≥ 50 µm) was added, which incorporates a non-parametric test to the primary outcome.
-
The sensitivity analysis for non-compliance would also take into account the 60% and 50% levels of compliance.
-
The use of anti-VEGF, steroids and laser at 12 and 24 months was added as a secondary outcome.
-
SPSS statistics 23 would be used instead of version 21.
Summary of changes made to protocol
When CLEOPATRA protocol version 1.0, dated 1 February 2012, was first submitted to the REC, the device that was being used was non-CE marked from Keepsight Ltd and, therefore, the trial required MHRA approval. This was applied for in version 2.0, dated 14 February 2013, but the company could not provide the required documents.
We then purchased a device from Polyphotonix Medical Ltd. This device was CE marked and, therefore, no longer required MHRA approval. The protocol was amended to version 3.0, dated 1 August 2013, (substantial amendment 1) and was submitted and approved by the REC because of this and notice was given to the MHRA that the submission for this study was withdrawn.
The changes in protocol included the following.
The lightmasks for this study were changed to be supplied by Polyphotonix Medical Ltd. These masks were already CE marked as a class 2a medical device. These masks have inbuilt software that assessed compliance so a separate compliance data logger was not required. The protocol was amended to incorporate the input from the company. The compliance section was changed to be in line with how compliance was measured for these masks.
The representative of Keepsight Ltd was removed from the protocol and the new suppliers were inserted. The patient information sheet showed the photograph of the new mask and details of the new mask and compliance and the name of the new supplier of the mask. The consent form was amended to show the new version number of the patient information sheet.
A second substantial amendment (substantial amendment 2) was then submitted to the REC for changes in the protocol version 4.0, dated 1 November 2013, that included the addition of five new sites, clarification of the secondary outcomes and altering the participants’ assessments. A DMC member was also removed because she became a principal investigator at an additional site. REC approval was requested and granted for this amendment.
We had received a non-invasive CE-approved oximetry machine for measuring oxygenation within retinal vessels. It gave a more direct measure of oxygenation of the retinal vessels than our originally planned indirect estimation from fluorescein angiography. Therefore, we replaced the original invasive fluorescein angiography test with oximetry. The outcome measures in the mechanistic part were changed to reflect these new measurements. As part of oximetry measurement we also added measurement of blood pressure and ocular examination in the table on summary of assessments. We also replaced the word ‘early’ DMO with ‘non-central’ DMO as ‘early’ is a subjective term while ‘non-central’ is an objective measure of the oedema. This ensured that all sites recruited similar participants into the trial and we could maintain an audit trail on the inclusion criteria. Different sites were using different OCT machines for the outcome measure, so we added ‘macular volume’ as a secondary outcome measure to substantiate or better qualify our primary outcome. We also included patient identification centre sites to enable identification of participants from these diabetic retinopathy screening units. The junior statistician and the research fellow for the mechanistic studies were added. We also formatted the protocol and removed all the typographical errors. These changes were also added to the participant information sheet and informed consent form so the versions were amended to version 4.0 for these documents.
A non-substantial amendment was then submitted to the REC (amendment 3) to remove a sentence from the protocol version 5.0, dated 7 February 2014, that should have been removed previously, to accurately address the TSC (see Appendix 2), to include the company’s video demonstration for participants and instructions for use of the masks provided by the company. A correction of the contents page and typographical errors led to a non-substantial change in protocol to version 5.1, dated 27 February 2014.
A non-substantial amendment (amendment 4) was then submitted to the REC for changes in the protocol version 6.0, dated 16 July 2014, that included the correction of typographical errors, inclusion of an invitation letter and poster to help with patient recruitment.
A final substantial amendment of the protocol version 7.0, dated 3 August 2016 (amendment 5), was submitted and approved by REC and included a change to the address of statistics team, and the addition of per-protocol analysis as a secondary analysis to exclude subjects from the ITT sample who were treated with steroids, anti-VEGF or laser treatments, which makes retinal thickness improve substantially. The baseline stratifier on baseline thickness in excess of 320 µm in zones 2–5 was removed, a sensitivity analysis to large retinal thickness departures (i.e. ≥ 50 µm) was added and a further sensitivity analysis for non-compliance also took into account the 60% and 50% levels of compliance to understand the dose effect of the lightmasks. The use of anti-VEGF, steroids and laser at 12 and 24 months was added as a secondary outcome. The version of the statistical software was amended to SPSS statistics 23.
Chapter 3 Clinical results
Reproduced with permission from Sivaprasad et al. 24 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Between April 2014 and May 2015, 349 participants were assessed for eligibility and 308 were randomly assigned to receive lightmask (n = 154) or non-lightmask or standard care (n = 154).
Setting and locations
The study was conducted at 15 NHS clinical sites (Table 7). The sites were chosen based on previous clinical trial experience or by the estimated volume of potentially eligible participants. Interested site investigators completed a site feasibility questionnaire.
| Site number | Site |
|---|---|
| 01 | Central Middlesex Hospital, London North West University Healthcare NHS Trust, London, UK |
| 02 | Heartlands Hospital, Heart of England NHS Foundation Trust, Bordesley Green East, Birmingham, UK |
| 03 | Stoke Mandeville Hospital, Buckinghamshire Healthcare NHS Trust, Amersham, UK |
| 04 | Maidstone and Tunbridge Wells NHS Trust, Kent, UK |
| 05 | Moorfields Eye Hospital NHS Foundation Trust, London, UK |
| 06 | Bristol Eye Hospital, University Hospitals Bristol NHS Foundation Trust, Bristol, UK |
| 07 | Frimley Park Hospital NHS Foundation Trust, Surrey, UK |
| 08 | City Hospitals Sunderland NHS Foundation Trust, Sunderland Royal Hospital, Sunderland, UK |
| 09 | William Harvey Hospital, East Kent Hospitals University NHS Foundation Trust, Kent, UK |
| 10 | Site code not in use. Site 10 withdrew before recruitment began |
| 11 | Princess Alexandra Hospital NHS Foundation Trust, Essex, UK |
| 12 | Hillingdon Hospital, Hillingdon Hospitals NHS Foundation Trust, Uxbridge, UK |
| 13 | King’s College Hospital NHS Foundation Trust, London, UK |
| 14 | Brighton and Sussex University Hospitals NHS Trust, Sussex Eye Hospital, Brighton, UK |
| 15 | The Royal Wolverhampton NHS Trust, Wolverhampton and Midland Counties Eye Infirmary, West Midlands, UK |
| 16 | Birmingham Midland Eye Centre, City Hospital NHS Trust, Birmingham, UK |
The actual recruitment period was on target to the planned original target of 12 months. Figure 3 shows the actual monthly recruitment compared with the original target.
FIGURE 3.
Cumulative monthly accrual of participants into the CLEOPATRA trial.

Recruitment was completed in 13 months. Table 8 shows the total number of participants recruited from each site.
| Site number | Site name | Total number of participants recruited |
|---|---|---|
| 01 | Central Middlesex | 4 |
| 02 | Birmingham Heartlands | 11 |
| 03 | Stoke Mandeville | 10 |
| 04 | Maidstone and Tunbridge Wells | 12 |
| 05 | Moorfields | 51 |
| 06 | Bristol Eye Hospital | 12 |
| 07 | Frimley Park | 32 |
| 08 | City Hospital, Sunderland | 25 |
| 09 | William Harvey Hospital | 21 |
| 11 | Princess Alexandra Hospital | 11 |
| 12 | Hillingdon Hospital | 54 |
| 13 | King’s College Hospital | 35 |
| 14 | Brighton | 3 |
| 15 | Wolverhampton | 21 |
| 16 | Birmingham Midlands | 6 |
| Total | 308 |
Study set-up
Site initiation visit
The site initiation visit (SIV) provided the local study team members with a clear understanding of the trial and provided a forum for staff to ask questions. It also provided an opportunity for staff to be trained in the electronic systems and databases required to conduct the trial. The sites were also provided with the necessary study equipment at the SIV.
The principal investigator, research nurses, optometrist, photographer, OCT technician and research co-ordinators/administrators were required to attend this meeting. Other relevant trust members were also invited to join if required by the site. Attendance to the SIV was recorded on a SIV attendance log.
The following documents were reviewed at the visit:
-
site file
-
site initiation attendance record
-
site green-light document
-
site initiation report template
-
user guide for data entry
-
instructions for mask programming
-
demo masks (for demonstration purposes)
-
delegation log.
Key activities
The SIV meeting consisted of training on the protocol, AE reporting, MACRO and randomisation and data entry, checking and monitoring training and an outline of GCP. Sites were given a practical session (when possible) on the information technology (IT) systems being used in the study. The ISF was reviewed with the research team during the SIV. Attention was drawn to key sections including SDWs completion, mask accountability forms and mark order forms. All relevant documentation was also provided electronically to all sites [either on compact disc (CD) or via e-mail]. Training was conducted with staff members who had been identified by the principal investigator as being responsible for programming the masks, database entry and randomisation.
The staff were also shown screen shots of the randomisation system and given guidance on how to use this system. They were given an explanation of the randomisation e-mails and given information on how to effectively randomise and minimise errors.
A demonstration of the MACRO V4 system was also given. The staff members were shown the training site and steps on how to generate a patient PIN. The MACRO V4 user guide was provided in the site file and this document was highlighted to staff during this section of the training.
Site staff were also given access to the dummy database for practice while waiting for the green light.
Mask programming
A demonstration of how to programme the lightmasks was given. The local team was provided with a demo lightmask, which was programmed during this training session. A training version of the PPX works website (Polyphotonix, Durham, UK) was reviewed and the demonstration pod was programmed. Instructions were provided in the site file and these documents were highlighted during this section of the training. Training was also provided on the study laptop, using the dongle provided to gain internet access, on how the compliance data of each returned mask was collected and downloaded. It was highlighted to the site staff that the compliance data are dependent on the mask being returned so that the site staff could be extra vigilant about requesting the return of the used masks.
At the end of the SIV, the following documents were copied for holding in the trial master file (TMF):
-
completed delegation of authority log
-
curricula vitae (CVs) signed and dated by all staff, evidence of GCP training, if not previously sent (site may send these after the SIV if necessary)
-
SIV attendance record.
All clinical procedures were also detailed in an operation manual. The optometrists were certified by the clinical research trial optometrists at Moorfields Eye Hospital. The trial photographers were certified by Moorfields reading centre. These were done outside the SIV meeting.
Each site received a report on the SIV produced by the trial manager. The report was distributed to local staff alongside the greenlight letter. Principal investigators reviewed and signed the report. Copies of the signed report were stored in the TMF and ISF. The trial manager ensured that all relevant documentation and certifications were complete before a greenlight letter was sent to each site to initiate the study access to the IT systems for the trial.
Trial milestones
The study start date was April 2014 and was planned to involve 10 recruiting sites around the UK. The sample size was 300 participants. The eligible participants were identified from the medical retina clinics. New participants were also referred in from the diabetic retinopathy screening programmes. As these participants were asymptomatic, it was necessary for the research teams at each site to communicate well with the participants that the lightmask is evaluated to prevent deterioration of the disease and progression to the central retina when the participants may experience visual impairment. It was challenging to recruit and retain participants and emphasise the importance of using the lightmask in this asymptomatic group of participants.
Success in trial recruitment and management
The NIHR recognises the need for good trial management and recommends the collaboration of a clinical trials unit to conduct the study. A multidisciplinary team at KCTU was funded for the study. As a collaborator for the CLEOPATRA study, the unit was delegated by the sponsor to conduct the trial and the team ensured the appointment of a dedicated trial manager. This was a crucial step for the success of this study. The KCTU also has a trial methodologist, data manager, operations manager and statisticians who met regularly with the chief investigator, other clinical investigators, the sponsor representative and the trial manager for initial brainstorming sessions and then to ensure the smooth running of the study. This group approach and shared ownership were very useful for the successful delivery of the study. During the early part of the set-up phase of the study, the team met very often to develop the protocol, obtain all the regulatory approvals and worked well with Polyphotonix Medical Ltd for timely delivery of the lightmask, training the staff to upload the anonymised compliance data and replace defective masks. The study source documents and CRFs were user-friendly and we paid significant attention to detail to capture all the required outcome measures. Following the approvals, meetings were focused on targeting and solving the barriers to site set-up, identifying and managing recruitment hurdles and monitoring recruitment. During these meetings, we evaluated reasons for initial delay in recruitment.
The start-up delays at some sites were associated with the following:
-
a delay in completion of training and certification of the staff for the study
-
a delay in site approval process
-
participants referred from diabetic retinopathy screening programmes not being targeted
-
other retinal consultants preferred standard of care (no treatment) to intervention
-
several site principal investigators found it challenging to convince participants about the importance of the trial and the potential success of a simple device that could prevent visual impairment due to DMO if proven to be effective and safe
-
the research and clinical team being not in the same area and so participants were being missed.
Recruitment strategies used
The target recruitment rate for the study was one to two participants per month per centre, based on the original 10 centres recruiting and a target recruitment figure of 300. To counteract this initial slow recruitment, five further recruiting centres were opened. No changes were made to the inclusion criteria and the recruitment time was not extended.
The trial management meetings enabled us to pay attention to the trial pathway and ensured that all barriers were tackled in time as we progressed. The trial manager and the chief investigator were always available to provide guidance and support to the clinical sites. The sites were chosen based on previous clinical trial experience or by the estimated volume of potentially eligible participants. Interested sites completed a site feasibility questionnaire. Some sites required support from the UK Clinical Research Network to be able to recruit participants for the study.
Recruitment was monitored closely. We sent out a recruitment league table in a newsletter to all sites every month initially so that the sites were aware of the progress of the study. The chief investigator also e-mailed or telephoned the principal investigator of clinical sites that were recruiting below target. The trial manager had to travel around to all sites to monitor the study at the sites. Although we were behind in recruitment, in March 2015 a late-to-start site (Wolverhampton NHS Trust) started recruiting very well and helped the study meet its target. Two other sites that recruited 30% of the participants were Hillingdon NHS Trust and Moorfields Eye Hospital.
Screening and eligibility
A total of 349 participants were assessed for eligibility. Of these participants, 41 were excluded because they either did not meet the inclusion exclusion criteria or they were not eligible or not keen. The remaining eligible participants (n = 308) were randomised into the trial.
Randomisation
Randomisation was balanced across the treatment groups and hospital sites, and within strata.
On-site monitoring visits were conducted routinely; more monitoring visits were arranged if problems were identified (e.g. poor data collection or under-reporting of primary end-point data) or when a site requested a visit to discuss specific issues (including data collection, screening participants, recruitment and staff training). The main areas of focus included consent, SAEs, essential documents in study site files, and source data verification on selected outcomes, including the primary outcome measure. A monitoring report was prepared after each monitoring visit and the points raised in the monitoring report were then addressed with sites remotely or at the next onsite monitoring visit.
In addition, all AEs and SAEs were monitored at each DMC meeting. The SAE data were collected on paper SAE report forms and faxed to KCTU. Summary details of SAEs were also transcribed to the AE section of the eCRF.
The principal investigator provided an electronic signature for each patient case record form once all queries were resolved and immediately prior to database lock.
Multiple systematic approaches were therefore instituted to ensure that all outcome data were as accurate and complete as possible.
Monitoring visits were conducted at least once annually. The frequency of monitoring increased or decreased at a site during the life cycle of the trial dependent on recruitment and where issues of compliance had been raised.
The first monitoring visit was conducted within 6 months of the first patient being recruited.
The site co-ordinator or lead research nurse was contacted in advance to arrange a visit, requesting that all medical notes, SDWs and the ISFs be provided for the visit. The current mask supply was also to be made available. A meeting at the end of the visit was to be arranged with the co-ordinator or lead research nurse and, if possible, the principal investigator to summarise any findings and to discuss overall trial progress, including patient retention, compliance issues and trial IT issues.
Off-site monitoring via the electronic case report form
A monthly data management report was generated by the data manager in accordance with the data monitoring plan. The trial manager resolved the issues identified in the report with the site. General reviews of all the data entered were also conducted by the trial manager, with particular attention being paid to the AEs reported and full completion of the patient questionnaires. Reminders were sent to sites that had outstanding data to enter, or if queries were flagged that had not been resolved previously.
Off-site monitoring beyond the electronic case report form
The trial monitor used the PPX website to monitor patient compliance (on a patient by patient basis) and generated information that could be cross-checked with the eCRF to ensure accurate data reporting. Each mask had a unique serial number (radio frequency identification) that needed to be entered into the eCRF and recorded on the SDW. Reports on allocated radio frequency identifications were generated from the PPX website and these numbers were cross-checked on the eCRF.
A report was generated monthly to assess the number of returned lightmasks, with missing/lost masks being highlighted to the site for follow-up. Masks that had been returned were reviewed for compliance by downloading the mask to the PPX Works. The company produced an electronic chart on mask wear-time. Participants with low compliance were highlighted to sites for follow-up discussion with the patient to improve compliance.
Mask levels at site were monitored routinely. The trial monitor cross-checked stock sent to site and compared this with current available stock in relation to the number of participants at site and usage of masks by those participants. The trial monitor liaised with PPX to monitor expiring stock at site and instructed sites to return expired stock to PPX on an ongoing basis.
Monthly status update spreadsheets provided by the reading centre at Moorfield’s Eye Hospital were reviewed and cross-checked against the eCRF to assess for missing OCT and colour photograph images at screening, and at months 12 and 24. Any sites with outstanding images were promptly followed up.
The trial monitor telephoned the site co-ordinator or lead research nurse every 8 weeks as a minimum. This call included discussions on retention of participants and collection of primary and secondary outcome data at 12 and 24 months, AE/SAE follow-up, general mask compliance, timelines for data entry and resolution of queries, completion of data in the eCRF, issues with the trial IT systems and site staff.
Updates to the trial (e.g. amendments) were sent to the site for filing as and when needed and confirmation of receipt obtained. When documents were identified as missing from the TMF (e.g. CVs, training records and updated delegation logs), the trial monitor requested these documents from the site.
On-site monitoring via the electronic case report form
The SDWs were compared with the eCRF to confirm that the data were accurate. The following were performed to ensure that the trial outcomes were robust:
-
Eligibility was checked for transcription errors using the inclusion/exclusion, medical history and ophthalmic history forms.
-
SAEs were cross-checked using the tracking list in the office against all SAEs reported at site. When there were outstanding queries or ongoing SAEs, the medical notes were checked for further available information. The AE log and medical notes were also to be checked for any unreported SAEs.
-
A minimum of 80% OCT image printouts at screening, months 12 and 24 (primary outcome measure) were checked for transcription errors. A proportion were within patient assessment checks, which were determined on a site-by-site basis, taking into account the number of participants at that particular site.
-
A minimum of 25% per sleep questionnaire (Epworth and Pittsburgh Insomnia Rating) at screening and at months 12 and 24 were checked for transcription errors.
-
A minimum of 25% of refracted BCVA assessments at screening and at months 12 and 24 were checked for transcription errors.
-
A minimum of 80% of OCT assessor treatment guess forms were checked for transcription errors at the 24-month time point or at the point of withdrawal.
Any discrepancies were highlighted to the site via flagged data in the eCRF and in the monitoring visit report. Reviewed data were confirmed and noted on the eCRF. Once identified queries had been resolved by the site, the responses were reviewed and closed. Any source data that had been verified in the notes were noted on MACRO via the source data verification function. Transcription errors made by sites or information on MACRO that differed from the source data were raised as a data clarification request in MACRO for the site to resolve.
On-site monitoring beyond the electronic case report form
The SDWs were monitored alongside the patient’s notes to ensure that all relevant information had been captured and information had been accurately transcribed. Evidence of consent was also checked in the medical notes for all participants. Any discrepancies were raised with the site, and noted in the monitoring report. When applicable, data were flagged in the eCRF for sites to update.
The ISFs were reviewed for completeness, including filing of all original signed consent forms, CVs, GCP and assessor certifications, and completion of the delegation and enrolment logs. Copies of documents found to be missing during the visit were taken for the TMF.
Discussions with the site co-ordinator or lead research nurse and, when possible, the masked outcome assessors, were conducted to ensure the masking process had worked. The trial monitor also checked that only delegated and appropriately masked outcome assessors were performing these roles via the OCT and photographer record sheet, the delegation log and the certification records in the ISF.
Mask accountability logs were checked in the case of discrepancies identified during off-site monitoring.
Baseline characteristics
The clinical study results have been published in Sivaprasad et al. 24
When we consider the whole study population, the average age of the participants was 57 years (SD 11 years) and 63% (194/308) were males. Mean baseline maximum retinal thickness was 348 µm (SD 28 µm). A total of 286 (93%) participants had baseline maximal retinal thickness in zones 2–5 while 22 (7%) had maximal retinal thickness in the parafoveal zones 6–9. A total of 183 out of 308 (59%) participants were measured on Heidelberg Spectralis (Heidelberg Engineering GmbH, Heidelberg, Germany), 54 (18%) were measured on Cirrus (Carl Zeiss Meditec, Cambridge, UK), 61 (20%) were measured on Topcon 2000 (Topcon Great Britain Medical Ltd, Newbury, UK) and 10 (3%) were measured on RS3000 (Nidek Co. Ltd, Aichi, Japan). Mean baseline macular volume was 8.89 µm3 (SD 0.86 µm3). The first measurement of BCVA was 84.1 (7.5) and second was 84.4 (SD 7.5) and the average of the two means was 84.3 (SD 7.3) ETDRS letters, which was equivalent to 6/6 Snellen. The mean HbA1c level was 68.0 mmol/mol (SD 18.8 mmol/mol), and a total of 154 participants (50%) had a HbA1c concentration of < 8% or 63.9 mmol/l at baseline.
Baseline characteristics were well balanced between treatment arms (Table 9).
| Characteristics | Treatment arm | |
|---|---|---|
| Non-lightmask (N = 153) | Lightmask (N = 155) | |
| Age (years), mean (SD) | 57.9 (11.8) | 56.9 (11.0) |
| Women, % (n) | 40 (61) | 34 (53) |
| Ethnicity, % (n) | ||
| White | 61 (94) | 65 (100) |
| Black | 19 (29) | 17 (27) |
| Asian | 18 (28) | 15 (24) |
| Other | 1 (2) | 3 (4) |
| Smoker, % (n) | 7 (10) | 8 (13) |
| Site, % (n) | ||
| 01 | 2 (3) | 1 (1) |
| 02 | 3 (4) | 5 (7) |
| 03 | 3 (5) | 3 (5) |
| 04 | 4 (6) | 4 (6) |
| 05 | 16 (25) | 17 (26) |
| 06 | 4 (6) | 4 (6) |
| 07 | 10 (16) | 10 (16) |
| 08 | 8 (13) | 8 (12) |
| 09 | 7 (10) | 7 (11) |
| 11 | 4 (6) | 3 (5) |
| 12 | 18 (27) | 17 (27) |
| 13 | 11 (17) | 12 (18) |
| 14 | 1 (1) | 1 (2) |
| 15 | 7 (11) | 6 (10) |
| 16 | 2 (3) | 2 (3) |
| Blood pressure (mmHg), mean (SD) | ||
| Systolic | 140.3 (18.9)a | 137.2 (16.5)a |
| Diastolic | 81.0 (10.2)a | 80.2 (9.4)a |
| Diabetes mellitus, % (n) | ||
| Type 1 | 13 (20) | 20 (31) |
| Type 2 | 87 (133) | 80 (124) |
| Medication, % (n) | ||
| Insulin only | 14 (22) | 28 (43) |
| Oral hypoglycaemic agents only | 49 (75) | 46 (72) |
| Insulin and oral hypoglycaemic agents | 37 (56) | 25 (39) |
| Diet controlled | 0 (0) | 1 (1) |
| BCVA (letters), mean (SD) | 84.3 (7.3) | 84.3 (7.4) |
| Maximal retinal thickness (µm), mean (SD) | 348.8 (24.3) | 345.9 (21.6) |
| Total volume (mm3), mean (SD) | 8.9 (0.9) | 8.8 (0.8) |
| HbA1c level, % (n) | ||
| < 8% (< 63.89 mmol/mol) | 50 (77) | 50 (77) |
| ≥ 8% (≥ 63.90 mmol/mol) | 50 (76) | 50 (78) |
| Severity level (study eye),b % (n) | ||
| 10 | 1 (2) | 1 (2) |
| 20 | 17 (25) | 23 (35) |
| 35 | 69 (101) | 60 (93) |
| 43–47 | 7 (11) | 10 (15) |
| 53 | 0 (0) | 1 (1) |
| 61c | 0 (0) | 1 (2) |
| 65c | 1 (1) | 1 (1) |
| 71–75c | 0 (0) | 1 (1) |
| 81–85c | 0 (0) | 0 (0) |
| 90c | 5 (7) | 3 (4) |
| Intraocular pressure (mmHg), mean (SD) | 16.2 (2.9)a | 16.4 (2.9)d |
There were 62 participants without primary outcome data at 24 months and Table 10 shows that there were no significant differences in baseline characteristics between those participants who dropped out and those that did not except for site, which was already adjusted for in the analysis. This was largely because one site had a high drop-out rate.
| Characteristics | Without 24-month data (N = 62) | With 24-month data (N = 246) | p-value |
|---|---|---|---|
| Age (years), mean (SD) | 57.2 (12.9) | 57.5 (11.0) | 0.87a |
| Women, % (n) | 40 (25) | 36 (89) | 0.55b |
| Ethnicity, % (n) | 0.07c | ||
| White | 76 (47) | 60 (147) | |
| Black | 16 (10) | 19 (46) | |
| Asian | 8 (5) | 19 (47) | |
| Other | 0 (0) | 2 (6) | |
| Smoker, % (n) | 7 (4) | 8 (19) | 0.73b |
| Baseline maximal zone retinal thickness, mean (SD) | 348.1 (22.4) | 347.2 (23.2) | 0.77a |
| Baseline BCVA (letters), mean (SD) | 83.8 (7.6) | 84.4 (7.3) | 0.59a |
| Site, % (n) | 0.0143c | ||
| 01 | 3 (2) | 1 (2) | |
| 02 | 3 (2) | 4 (9) | |
| 03 | 2 (1) | 4 (9) | |
| 04 | 3 (2) | 4 (10) | |
| 05 | 10 (6) | 18 (45) | |
| 06 | 7 (4) | 3 (8) | |
| 07 | 23 (14) | 7 (18) | |
| 08 | 11 (7) | 7 (18) | |
| 09 | 11 (7) | 6 (14) | |
| 11 | 3 (2) | 4 (9) | |
| 12 | 8 (5) | 20 (49) | |
| 13 | 8 (5) | 12 (30) | |
| 14 | 0 (0) | 1 (3) | |
| 15 | 5 (3) | 7 (18) | |
| 16 | 3 (2) | 2 (4) | |
Withdrawals
Participants who withdrew from the trial but were still reviewed in clinics with OCT were also included in the primary outcome analysis. Table 11 shows the number of participants included in the primary outcome data.
| Time point | Number of participants followed up | Number of participants followed up with primary outcomea | ||||
|---|---|---|---|---|---|---|
| Treatment arm | Total | Treatment arm | Total | |||
| Non-lightmask | Lightmask | Non-lightmask | Lightmask | |||
| Baseline | 153 | 155 | 308 | 153 | 155 | 308 |
| 4 months | 121 | 135 | 256 | 119 | 132 | 251 |
| 8 months | 112 | 113 | 225 | 113 | 113 | 226 |
| 12 months | 121 | 130 | 251 | 121 | 130 | 251 |
| 16 months | 102 | 111 | 213 | 103 | 111 | 214 |
| 20 months | 102 | 100 | 202 | 104 | 100 | 204 |
| 24 months | 115 | 122 | 237 | 119 | 127 | 246 |
The flow of patients are shown in the Consolidated Standards of Reporting Trials (CONSORT) flow diagram (Figure 4). 24
FIGURE 4.
The CONSORT flow diagram for the CLEOPATRA trial. Reproduced with permission from Sivaprasad et al. 24 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.

Derivation of the intention-to-treat model population
Participants included in the prespecified ITT LME model were derived as follows: (1) the OCT data were available for 246 participants of 308 randomly assigned participants (lightmask arm, n = 127; and non-lightmask arm, n = 119) at 24 months, and (2) these data included OCT data from routine clinical care obtained from five participants in the lightmask arm and four participants in the non-lightmask arm who attended their clinic appointment but did not attend an intervening research visit. A further 17 participants in the lightmask arm and 14 participants in the non-lightmask arm were included in the model with data from previous time points. Therefore, a total of 277 participants were included in the primary outcome model, 133 in the non-lightmask arm and 144 in the lightmask arm (see Table 11).
Primary outcome: intention to treat
Primary outcome at 24 months showed that there was no difference in mean change in maximal retinal thickness between the lightmask arm and the non-lightmask arm (Table 12). The adjusted difference between arms was –0.65 µm (95% CI –6.90 to 5.59 µm; p = 0.84).
| Time point | Primary outcome: OCT central subfield thickness (µm) | p-value | ||||
|---|---|---|---|---|---|---|
| Treatment arm, mean (SD); n | Treatment arm, change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | ||||
| Non-lightmask | Lightmask | Non-lightmask | Lightmask | |||
| Baseline | 348.8 (24.3); 153 | 345.9 (21.6); 155 | – | – | – | – |
| 12 months | 339.1 (35.9); 121 | 341.3 (29.7); 132 | –9.5 (3.1) | –4.6 (2.5) | 1.73 (–5.31 to 8.77) | 0.63 |
| 24 months | 336.3 (29.7); 119 | 336.0 (25.5); 127 | –12.9 (2.9) | –9.2 (2.5) | –0.65 (–6.90 to 5.59) | 0.84 |
Table 13 shows that there was no difference in mean change in maximal retinal thickness between arms at all time points, including at the 4- and 8-month time points at which compliance was highest.
| Outcome | Month | |||||
|---|---|---|---|---|---|---|
| 4 | 8 | 12 | 16 | 20 | 24 | |
| Adjusted mean difference in retinal thickness (µm) | 4.64 | 2.35 | 1.73 | –0.27 | 2.46 | –0.65 |
| 95% CI | –1.02 to 10.30 | –3.28 to 7.99 | –5.31 to 8.77 | –6.67 to 6.13 | –3.95 to 8.87 | –6.90 to 5.59 |
Primary outcome: per-protocol secondary analysis
The per-protocol secondary analysis after excluding participants from point of initiating treatment for DMO with laser, steroids or anti-VEGF agents included 266 participants (lightmask arm, n = 140; non-lightmask arm, n = 126) in the linear mixed models. Table 14 shows the number of participants requiring treatment with anti-VEGF, steroids or laser at each follow-up visit. A total of 56 participants required treatment (lightmask arm, n = 23; non-lightmask arm, n = 33).
| Month visit | Number of participants requiring treatment | Number of participants with primary outcome at each follow-up visit after removing those requiring treatment | ||||
|---|---|---|---|---|---|---|
| Treatment arm | Total | Treatment arm | Total (n = 308) | |||
| Non-lightmask | Lightmask | Non-lightmask (n = 153) | Lightmask (n = 155) | |||
| 4 months | 7 | 4 | 11 | 112 | 128 | 240 |
| 8 months | 6 | 3 | 9 | 102 | 106 | 208 |
| 12 months | 9 | 5 | 14 | 102 | 120 | 222 |
| 16 months | 6 | 4 | 10 | 81 | 97 | 178 |
| 20 months | 1 | 6 | 7 | 80 | 82 | 162 |
| 24 months | 4 | 1 | 5 | 90 | 105 | 195 |
The difference in the cumulative proportion of participants requiring treatment between arms was 8% (95% CI 0% to 16%) at 12 months, and 9% (95% CI 1% to 18%) at 24 months. This secondary analysis also showed no significant difference in change in maximal retinal thickness between arms at 12 and 24 months. The adjusted difference at 24 months was 3.23 µm (95% CI –2.11 to 8.58 µm; p = 0.23) (Table 15).
| Time point | Primary outcome: maximum retinal thickness using OCT (µm) | p-value | ||||
|---|---|---|---|---|---|---|
| Treatment arm, mean (SD); n | Treatment arm, change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | ||||
| Non-lightmask | Lightmask | Non-lightmask | Lightmask | |||
| Baseline | 348.8 (24.3); 153 | 347.7 (31.2); 155 | – | – | – | – |
| 12 months | 333.4 (26.6); 102 | 339 (18.6); 120 | –13.1 (2.9) | –5.3 (1.8) | 4.50 (–0.72 to 9.73) | 0.09 |
| 24 months | 330.5 (21.6); 90 | 335.9 (21.9); 105 | –14.6 (2.8) | –8.5 (2.1) | 3.23 (–2.11 to 8.58) | 0.23 |
Lightmask compliance over time
Table 16 shows that the median level and IQR of compliance of using the lightmask was 39.5% (IQR 9.8–78.2%) at 4 months and this reduced to 19.5% (IQR 1.9–51.6%) at 24 months. This decreasing level of compliance was observed at all three definitions of levels of non-compliance.
| Measure | Month | |||||
|---|---|---|---|---|---|---|
| 4 | 8 | 12 | 16 | 20 | 24 | |
| Median | 39.5 | 32.0 | 25.7 | 25.1 | 20.5 | 19.5 |
| IQR | 9.8–78.2 | 5.4–68.6 | 3.8–64 | 2.8–60.7 | 2.3–55.6 | 1.9–51.6 |
| Levels of compliance | ||||||
| 70% | 31% (48/155) | 23% (36/155) | 21% (32/155) | 19% (30/155) | 19% (29/155) | 16% (24/155) |
| 60% | 38% (59/155) | 32% (49/155) | 28% (44/155) | 26% (40/155) | 23% (35/155) | 21% (33/155) |
| 50% | 45% (69/155) | 39% (61/155) | 36% (56/155) | 33% (51/155) | 30% (47/155) | 26% (40/155) |
Sensitivity analysis
Sensitivity analyses for missing data were carried out to represent three possible scenarios to reflect whether departures from the MAR assumption apply within the lightmask arm only, within the non-lightmask arm only and within both arms equally and in the same direction. All three sensitivity analysis scenarios for departures from MAR assumption showed a non-significant effect in the change in maximal retinal thickness of the lightmask arm over the non-lightmask arm (Table 17).
| Three scenarios reflecting whether or not departures from the MAR assumption apply within: | Retinal thickness range over which the mean of the ‘unobserved outcome data’ might depart from the mean of the ‘observed outcome data’ | |
|---|---|---|
| –20 µm | + 20 µm | |
| Scenario 1. The intervention arm only (lightmasks) | –4.3 (–10.5 to 2.0) | 3.0 (–3.3 to 9.2) |
| Scenario 2. The control arm only (non-lightmasks) | –5.1 (–11.3 to 1.1) | 3.8 (–2.5 to 10.0) |
| Scenario 3. Both arms equally and in the same direction | 0.2 (–6.1 to 6.4) | –1.5 (–7.7 to 4.8) |
Assuming those with unobserved outcome data in one, or other, or both arms would take values as much as a prespecified 20 µm either side of the adjusted observed effect, all of the 95% CIs included 0 and excluded –15 µm, thus confirming that the finding of an absence of a clinically important lightmask effect is robust to missing data (Figure 5).
FIGURE 5.
Figure showing the robustness of the lightmask effect to missing data. Reproduced with permission from Sivaprasad et al. 24 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.
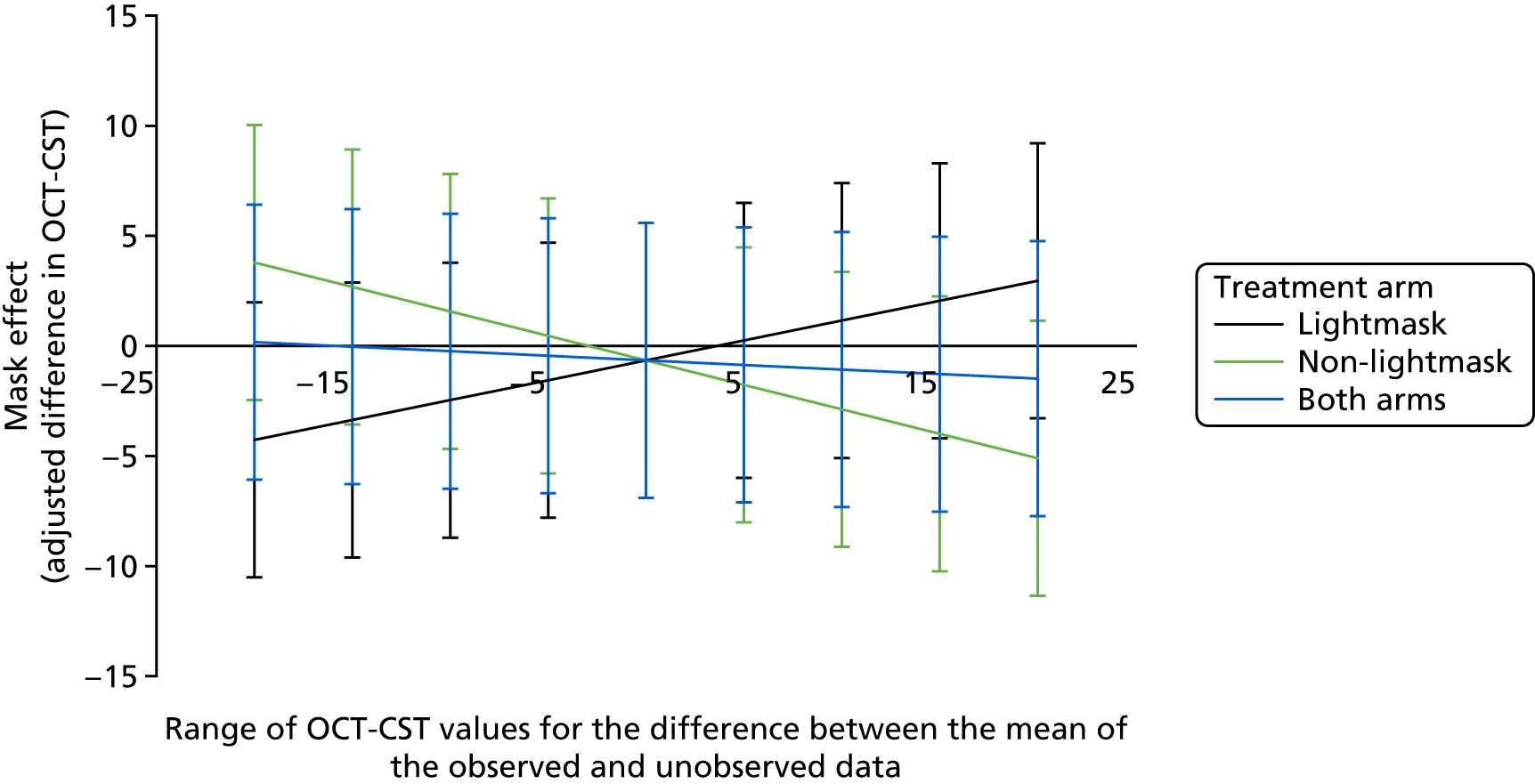
We also performed sensitivity analysis for non-compliance by CACE analysis. The CACE estimate of the treatment effect for compliers defined by 70% compliance was –4.2 (95% CI –44.6 to 36.1), by 60% compliance was –3.1 (95% CI –32.4 to 26.3) and by 50% compliance was –2.5 (95% CI –26.7 to 21.7). This was based on 277 participants (lightmask arm, n = 144; non-lightmask arm, n = 133). Across these three definitions of compliers, the results were consistent in estimating a small non-significant intervention effect, which was not close to the detectable effect of 15 µm retinal thickness.
For the sensitivity analysis on participants who met the requirement for treatment of centre-involving DMO because the retinal thickness reached 400 µm before the 24-month end point, the retinal thickness measurement taken just after the participant first reached 400 µm was considered in the analysis and carried forward to be their final measurement. There were nine participants achieving 400 µm in the central macula: three in the non-lightmask arm who achieved this point at 24 months, and six in the lightmask arm (two achieving it at 24 months, two at 20 months and two at 12 months). A total of 161 participants were included in this LME model and the adjusted difference between arms was –0.22 µm (95% CI –8.36 to 7.92 µm; p = 0.96). Using stratified Cox proportional hazards regression stratified by the minimisation factor HbA1c level, the time-to-event analysis (time to requirement of treatment for retinal thickness to be 400 µm or before the 24-month end point) included 259 participants. The hazard ratio was 2.0 (95% CI 0.5 to 8.0; p = 0.33). For the sensitivity analysis for potential differential variability between arms over time in the zone of maximum baseline retinal thickness, there was no significant difference between arms in the change in retinal thickness from baseline to 24 months using the Mann–Whitney U-test; p = 0.38 (n = 246). The sensitivity analysis on participants that had their OCT scans on the Spectralis OCT at baseline also showed no significant difference between arms (Table 18).
| Time point | Primary outcome: maximum retinal thickness using OCT (µm) | p-value | ||||
|---|---|---|---|---|---|---|
| Treatment arm, mean (SD); n | Treatment arm, change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | ||||
| Non-lightmask | Lightmask | Non-lightmask | Lightmask | |||
| Baseline | 353.8 (26.0); 92 | 349.7 (20.6); 91 | – | – | – | – |
| 12 months | 344.2 (35.2); 73 | 345.1 (32.7); 74 | –9.4 (4.1) | –5.0 (3.8) | 0.15 (–9.83 to 10.13) | 0.98 |
| 24 months | 340.3 (28.4);70 | 339.8 (23.3); 72 | –13.9 (4.0) | –9.3 (2.9) | –0.22 (–8.36 to 7.92) | 0.96 |
Secondary outcomes on retinal thickness and volume
Table 19 shows that there were no significant differences between arms in change from baseline in central subfield thickness, total thickness of central and parafoveal zones, total retinal thickness measured over all nine zones and total macular volume at 12 and 24 months.
| Outcome | Treatment arm, mean (SD); n | Treatment arm, change from baseline, mean (SE) | Adjusted difference between lightmask and non-lightmask (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| Non-lightmask | Lightmask | Non-lightmask | Lightmask | |||
| Retinal thickness in the central zone | ||||||
| Baseline | 271.9 (18.9); 153 | 269.3 (18.9); 155 | – | – | – | – |
| 12 months | 273.8 (34.3); 121 | 274.5 (36.7); 132 | 2.9 (2.6) | 4.9 (2.8) | 1.59 (–5.80 to 8.98) | 0.67 |
| 24 months | 275.1 (42.3); 119 | 274.6 (37.4); 127 | 2.7 (3.4) | 5.2 (3.2) | 2.74 (–6.39 to 11.88) | 0.55 |
| Retinal thickness in the central and parafoveal zones | ||||||
| Baseline | 1604 (83); 153 | 1591 (82); 155 | – | – | – | – |
| 12 months | 1599 (127); 121 | 1600 (121); 132 | –1.1 (7.9) | 7.8 (7.9) | 5.36 (–16.17 to 26.90) | 0.62 |
| 24 months | 1597 (137); 119 | 1589 (124); 127 | –6.1 (9.9) | –0.2 (8.4) | 4.72 (–20.3 to 29.74) | 0.71 |
| Retinal thickness in over the nine zones | ||||||
| Baseline | 2789 (157); 153 | 2764 (142); 155 | – | – | – | – |
| 12 months | 2781 (213); 121 | 2774 (187); 132 | 2.0 (11.9) | 7.5 (11.3) | –0.26 (–31.68 to 31.15) | 0.99 |
| 24 months | 2772 (221); 119 | 2756 (196); 126 | –11.2 (14.7) | –6.9 (12.4) | 1.52 (–34.78 to 37.83) | 0.93 |
| Macular volume | ||||||
| Baseline | 8.9 (0.9); 153 | 8.8 (0.8); 155 | – | – | – | – |
| 12 months | 8.9 (0.9); 122 | 8.8 (0.9); 132 | 0.0 (0.0) | –0.1 (0.0) | –0.06 (–0.19 to 0.06) | 0.34 |
| 24 months | 8.9 (1.2); 121 | 8.9 (0.9); 126 | 0.0 (0.1) | 0.0 (0.0) | –0.01 (–0.15 to 0.14) | 0.92 |
| BCVA refracted | ||||||
| Baseline | 84.3 (7.3); 153 | 84.3 (7.4); 155 | – | – | – | – |
| 12 months | 83.5 (9.1); 120 | 83.6 (8.2); 131 | –0.6 (0.5) | –0.7 (0.4) | –0.07 (–1.38 to 1.23) | 0.91 |
| 24 months | 82.4 (10.1); 115 | 83.0 (8.8); 122 | –1.8 (0.7) | –1.6 (0.5) | 0.13 (–1.45 to 1.71) | 0.87 |
Visual acuity
The adjusted difference in BCVA between arms was also not significant at –0.07 ETDRS letters (95% CI –1.38 to 1.23 letters; p = 0.91) at 12 months and at 0.13 ETDRS letters (95% CI –1.45 to 1.71 letters; p = 0.87 at 24 months) (see Table 1).
Secondary outcomes on morphological characteristics of diabetic macular oedema on optical coherence tomography
Significantly more participants in the lightmask arm showed a resolution of diffuse DMO at 12 months than those in the non-lightmask arm, but this effect was lost at 24 months. There was also a trend in reduction in foveal cysts at 12 and 24 months in the lightmask arm compared with the non-lightmask arm. Changes in visible cysts in the inner ETDRS zones did not differ between arms, but the proportion of participants with visible cysts in the outer ETDRS zones reduced significantly more in the lightmask arm than in the non-lightmask arm (Table 20).
| Characteristic | Treatment arm, % (n/N) | Difference between arms in change from baseline (95% CI) | p-value | |
|---|---|---|---|---|
| Non-lightmask | Lightmask | |||
| Hyporeflectivity of outer nuclear layer without cysts (diffuse) | ||||
| Baseline | 18 (27/153) | 19 (30/155) | ||
| 12 months | 21 (26/121) | 11 (14/132) | ||
| Change from baseline to 12 months | 5 (6/121) | –8 (–10/132) | –13 (–23 to –2) | 0.0246 |
| 24 months | 12 (14/118) | 13 (16/123) | ||
| Change from baseline to 24 months | –6 (–7/118) | –4 (–5/123) | 2 (–10 to 14) | 0.75 |
| Visible cysts in foveal area | ||||
| Baseline | 16 (24/153) | 24 (37/155) | ||
| 12 months | 26 (32/121) | 23 (30/132) | ||
| Change from baseline to 12 months | 10 (12/121) | –2 (–3/132) | –12 (–25 to 0) | 0.052 |
| 24 months | 29 (34/118) | 25 (31/123) | ||
| Change from baseline to 24 months | 13 (15/118) | –1 (–1/123) | –14 (–27 to 0) | 0.054 |
| Visible cysts in inner ETDRS zones (< 3 mm) | ||||
| Baseline | 47 (72/153) | 44 (68/155) | ||
| 12 months | 38 (46/120) | 40 (53/132) | ||
| Change from baseline to 12 months | –12 (–14/120) | –2 (–3/132) | 9 (–4 to 23) | 0.18 |
| 24 months | 39 (46/118) | 33 (40/122) | ||
| Change from baseline to 24 months | –10 (–12/118) | –11 (–14/122) | –1 (–16 to 13) | 0.86 |
| Visible cysts in outer ETDRS zones (3–6 mm) | ||||
| Baseline | 38 (58/152) | 41 (64/155) | ||
| 12 months | 38 (46/120) | 26 (34/132) | ||
| Change from baseline to 12 months | 0 (0/120) | –14 (–18/132) | –14 (–27 to –1) | 0.0398 |
| 24 months | 37 (44/118) | 33 (41/123) | ||
| Change from baseline to 24 months | –1 (–1/118) | –5 (–6/123) | –4 (–17 to 9) | 0.55 |
| Vitreo macular traction | ||||
| Baseline | 0 (0/153) | 2 (3/155) | ||
| 12 months | 0 (0/121) | 2 (3/132) | ||
| Change from baseline to 12 months | 0 (0/121) | 0 (0/132) | 0 (–3 to 3) | 1.00 |
| 24 months | 3 (4/118) | 1 (1/123) | ||
| Change from baseline to 24 months | 3 (4/118) | –2 (–2/123) | –5 (–9 to –1) | 0.0130 |
| Subretinal fluid | ||||
| Baseline | 0 (0/153) | 1 (1/155) | ||
| 12 months | 1 (1/121) | 0 (0/132) | ||
| Change from baseline to 12 months | 1 (1/121) | –1 (–1/132) | –2 (–4 to 1) | 0.16 |
| 24 months | 0 (0/118) | 2 (2/123) | ||
| Change from baseline to 24 months | 0 (0/118) | 1 (1/123) | 1 (–2 to 4) | 0.56 |
| Epiretinal membrane | ||||
| Baseline | 1 (2/153) | 2 (3/155) | ||
| 12 months | 2 (3/121) | 3 (4/132) | ||
| Change from baseline to 12 months | 1 (1/121) | 1 (1/132) | 0 (–4 to 4) | 0.97 |
| 24 months | 7 (8/118) | 4 (5/123) | ||
| Change from baseline to 24 months | 5 (6/118) | 2 (2/123) | –3 (–10 to 3) | 0.29 |
Changes in levels of severity of diabetic retinopathy
The diabetic retinopathy severity grade was defined according to the ETDRS severity grade shown in Table 21.
| ETDRS level | ETDRS severity | ETDRS definition |
|---|---|---|
| 10 | No retinopathy | Diabetic retinopathy absent |
| 20 | Very mild NPDR | Microaneurysms only |
| 35 | Mild NPDR | Hard exudates, cotton wool spots, and/or mild retinal haemorrhages |
| 43 | Moderate NPDR |
43A: retinal haemorrhages moderate (> photograph 1A) in four quadrants or severe (≥ photograph 2A) in one quadrant 43B: mild IRMA (< photograph 8A) in 1–3 quadrants |
| 47 | Moderate NPDR |
47A: both level 43 characteristics 47B: mild IRMA in four quadrants 47C: severe retinal haemorrhage in 2–3 quadrants 47D: venous beading in one quadrant |
| 53 | Severe NPDR |
53A: ≥ 2 level 47 characteristics 53B: severe retinal haemorrhages in four quadrants 53C: moderate to severe IRMA (≥ photograph 8A) in at least one quadrant 53D: venous beading in at least two quadrants 53E: ≥ 2 level 53A-D characteristics |
| 61 | Mild PDR | NVE < 0.5 disk area in ≥ 1 quadrants |
| 65 | Moderate PDR |
65A: NVE ≥ 0.5 disk area in ≥ 1 quadrants 65B: NVD < photograph 10A (0.25–0.33 disk area) |
| 71 and 75 | High-risk PDR | NVD ≥ photograph 10A, or NVD < photograph 10A or NVE ≥ 0.5 disk area plus VH or PRH, or VH or PRH obscuring ≥ 1 disk area |
| 81 and 85 | Advanced PDR | Fundus partially obscured by VH and either new vessels ungradable or retina detached at the centre of the macula |
| 90 | Ungradable | Poor image quality |
The proportion of participants showing progression of retinopathy was low. There was no difference between arms at 12 and 24 months (Table 22).
| Progression of retinopathy | Treatment arm, % (n/N) | Difference in proportions (95% CI) | |
|---|---|---|---|
| Non-lightmask | Lightmask | ||
| 12 months (≥ 2 ETDRS level improvement) | 1 (1/84) | 1 (1/87) | –0.04 (–5.4 to 5.4) |
| 24 months (≥ 2 ETDRS level improvement) | 3 (3/96) | 3 (3/99) | –0.09 (–6.5 to 6.5) |
Treatment outcomes guess form by masked optical coherence tomography technicians and optometrists
The treatment allocation guess form, which measured the success of masking of primary assessors to treatment allocation, was reported for 248 participants by OCT technicians and 246 optometrists. In line with chance, OCT technicians guessed correctly for 55% (137/248) of participants and optometrists for 52% (129/246). Seventy-three per cent (180/248) and 94% (231/246) of the OCT technicians and optometrists, respectively, responded based on a random choice and 27% (68/248) and 4% (15/246) made an educated guess based on a clinical response or adverse event.
Changes in sleep questionnaire scores
The ESS scores and PIRS_20 scores did not show significant differences between arms (Table 23).
| Visit | Treatment arm, mean scores (SD); n | Treatment arm, change in scores from baseline, mean (SE); n | Adjusted difference in scores between lightmask and non-lightmask (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| Non-lightmask | Lightmask | Non-lightmask | Lightmask | |||
| Daytime sleepiness (ESS) | ||||||
| Baseline | 6.2 (4.0); 143 | 5.7 (3.7); 148 | ||||
| 12 months | 6.1 (4.2); 112 | 5.5 (4.1); 120 | –0.3 (0.4); 109 | 0.2 (0.3); 116 | –0.11 (–1.05 to 0.83) | 0.82 |
| 24 months | 5.8 (4.7); 107 | 5.7 (4.4); 118 | –0.3 (0.4); 102 | 0.4 (0.3); 112 | 0.51 (–0.42 to 1.43) | 0.28 |
| Insomnia (PIRS_20) | ||||||
| Baseline | 13.6 (10.4); 146 | 12.7 (10.3); 152 | ||||
| 12 months | 13.6 (10.1); 114 | 14.5 (11.7); 121 | –0.2 (0.8); 110 | 2.0 (0.9); 119 | 1.56 (–0.62 to 3.74) | 0.16 |
| 24 months | 13.2 (9.5); 111 | 14.2 (12.4); 116 | –0.4 (1.0); 106 | 1.4 (1.0); 114 | 1.41 (–1.02 to 3.84) | 0.25 |
Safety outcomes
There were 58 SAEs in total (lightmask arm, n = 32; non-lightmask arm, n = 26), but none related to the intervention (Table 24).
| Body system | Treatment arm (n) | Total (n) | |
|---|---|---|---|
| Non-lightmask | Lightmask | ||
| Cardiovascular | 8 | 4 | 12 |
| Respiratory | 3 | 1 | 4 |
| Hepatic | 1 | 1 | 2 |
| Gastrointestinal | 2 | 6 | 8 |
| Genitourinary | 2 | 0 | 2 |
| Endocrine | 3 | 2 | 5 |
| Musculoskeletal | 3 | 6 | 9 |
| Neurological | 2 | 6 | 8 |
| Psychiatric | 0 | 1 | 1 |
| Dermatological | 2 | 1 | 3 |
| Other | 0 | 4 | 4 |
| Total | 26 | 32 | 58 |
There were a total of 340 AEs not related to the intervention (lightmask arm, n = 172; non-lightmask arm, n = 168) (Table 25).
| Body system | Treatment arm (n) | Total (n) | |
|---|---|---|---|
| Non-lightmask | Lightmask | ||
| Eyes | 51 | 40 | 91 |
| Cardiovascular | 5 | 5 | 10 |
| Respiratory | 23 | 13 | 36 |
| Gastrointestinal | 8 | 17 | 25 |
| Genitourinary | 12 | 9 | 21 |
| Endocrine | 6 | 2 | 8 |
| Haematological | 4 | 2 | 6 |
| Musculoskeletal | 16 | 24 | 40 |
| Neurological | 5 | 16 | 21 |
| Psychiatric | 3 | 5 | 8 |
| Immunological | 3 | 2 | 5 |
| Dermatological | 11 | 6 | 17 |
| Allergies | 2 | 1 | 3 |
| Ear, nose, throat | 13 | 14 | 27 |
| Other | 6 | 16 | 22 |
| Total | 168 | 172 | 340 |
There were 72 AEs that were reported as related to the intervention (lightmask arm, n = 50; non-lightmask arm, n = 22) (Table 26).
| Body system | Treatment arm (n) | Total (n) | |
|---|---|---|---|
| Non-lightmask | Lightmask | ||
| Eyes | |||
| Corneal abrasion, corneal ulcer | 3 | 0 | 3 |
| Mask causing pressure on eyes, pain on eyes, uncomfortable masks | 7 | 14 | 21 |
| Sore eyebrows, sore eyelids | 0 | 2 | 2 |
| Subconjunctival haemorrhage | 0 | 1 | 1 |
| Vision deterioration, disturbance | 1 | 2 | 3 |
| Watery eyes, sore eyes, sticky eyes, painful eyes, conjunctivitis | 6 | 14 | 20 |
| Other body systems | |||
| Neurological | |||
| Severe persistent headache | 0 | 2 | 2 |
| Psychiatric | |||
| Insomnia | 0 | 1 | 1 |
| Musculoskeletal | |||
| Left sided neck and skull pain | 0 | 1 | 1 |
| Dermatological | |||
| Scratched face on two occasions getting mask off during sleep | 1 | 0 | 4 |
| Sore skin/small lump side of right eye | 0 | 1 | |
| Pod moving around in mask when turns in bed | 0 | 1 | |
| Wart | 0 | 1 | |
| Other | |||
| Mask slipping off head | 3 | 3 | 6 |
| Sleep disturbance, bad dreams | 1 | 7 | 8 |
| Total | 22 | 50 | 72 |
Mechanistic substudy
A total of 28 participants consented to the mechanistic substudy. Table 27 shows the baseline characteristics of these participants, with more females than males in the lightmask arm than in the non-lightmask arm.
| Characteristic | Treatment arm | |
|---|---|---|
| Lightmask (N = 13) | Non-lightmask (N = 15) | |
| Age (years), median (IQR) | 60.0 (55.5–64.0) | 56.0 (50.0–61.0) |
| Sex (female), % (n/N) | 62% (8/13) | 33% (5/15) |
| HbA1c level (nmol/mol), median (IQR) | 65.0 (56.3–86.9) | 65.0 (55.2–82.5) |
| Maximal retinal thickness (µm), median (IQR) | 349.0 (335.0–355.5) | 353.0 (335.0–368.0) |
| BCVA ETDRS (letters), median (IQR) | 84.5 (78.5–89.3) | 87.0 (82.5–91.0) |
| Macular volume (mm3), median (IQR) | 8.7 (8.3–9.0) | 8.7 (8.5–9.1) |
The median level and IQR of compliance for the substudy participants using the lightmask was 67.7% (IQR 19.1–92.0%) at 4 months, 60.3% (IQR 18.7–81.8%) at 8 months, 61.3% (IQR 18.0–80.9%) at 12 months, 55.1% (IQR 13.9–79.1%) at 16 months, 53.8% (IQR 16.8–75.5%) at 20 months, and 49.1% (IQR 18.0–64.3%) at 24 months. Their compliance was generally higher than the other lightmask participants not included in the substudy.
Complete data for both baseline and 12 months were available for 24 participants with oximetry, 26 for mfERG and 28 for microperimetry. Table 28 shows the changes in pulse oximetry and retinal oximetry after supplemental oxygen.
| Parameter | Breathing air, mean (SD) | After supplemental oxygen, mean (SD) | Mean difference (oxygen – air) (95% CI) | p-value (paired t-test) |
|---|---|---|---|---|
| Baseline (n = 28) | ||||
| Oximetry | N = 24 | N = 24 | ||
| Mean arterial SO2 (%) | 96.7 (5.5) | 96.0 (5.3) | 0.7 (–0.8 to 2.3) | 0.33 |
| Mean venous SO2 (%) | 63.1 (8.5) | 64.3 (8.1) | –1.1 (–3.2 to 0.9) | 0.27 |
| Mean AV SO2 (%) | 36.0 (8.4) | 34.1 (6.5) | 1.8 (–0.9 to 4.6) | 0.18 |
| Mean arterial diameter (µm) | 117.8 (10.2) | 115.9 (10.3) | 1.9 (–1.5 to 5.2) | 0.26 |
| Mean venous diameter (µm) | 147.0 (14.9) | 145.2 (15) | 1.7 (–1.2 to 4.7) | 0.24 |
| mfERG | N = 26 | N = 26 | ||
| Mean N1 (ms) | 16.4 (1.1) | 16.2 (0.7) | –0.2 (–0.6 to 0.2) | 0.30 |
| Mean N1 (nV) | –396 (116) | –408 (93) | –12.5 (–48.1 to 23.2) | 0.48 |
| Mean P2 (ms) | 31.9 (1.7) | 31.8 (1.4) | –0.03 (–0.3 to 0.3) | 0.85 |
| Mean P2 (nV) | 1111 (244) | 1146 (228) | 34.5 (–46.1 to 115.1) | 0.39 |
| Microperimetry | N = 28 | N = 28 | ||
| Mean retinal sensitivity (db) | 16.7 (3.3) | 16.9 (2.8) | 0.2 (–0.3 to 0.8) | 0.45 |
Table 29 shows that there was no difference in retinal function with and without oxygen inhalation in both the Noctura 400 Sleep Masks over non-lightmask. The arterial and venous diameter decreased with oxygen supplementation.
| Parameter | Treatment arm | Difference between arms in the change from baseline when breathing air (t-test) (95% CI) | Difference between arms in the change from baseline after supplemental oxygen (t-test) (95% CI) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lightmask (N = 13) | Non-lightmask (N = 15) | |||||||||||||
| Baseline breathing air, mean (SD) | 12 months breathing air, mean (SD) | Change air, mean (SE) | Baseline after supplemental oxygen, mean (SD) | 12 months breathing oxygen, mean (SD) | Change oxygen, mean (SE) | Baseline breathing air, mean (SD) | 12 months breathing air, mean (SD) | Change air, mean (SE) | Baseline after supplemental oxygen, mean (SD) | 12 months breathing oxygen, mean (SD) | Change oxygen, mean (SE) | |||
| Oximetry | n = 12 | n = 12 | n = 12 | n = 12 | n = 12 | n = 12 | n = 12 | n = 12 | n = 12 | n = 12 | n = 12 | n = 12 | ||
| Mean arterial SO2 (%) | 96.8 (7.0) | 95.4 (9.7) | –1.4 (1.5) | 96.6 (6.5) | 97.0 (7.3) | 0.4 (1.6) | 96.7 (3.9) | 95.3 (5.4) | –1.4 (1.4) | 95.4 (4.0) | 96.9 (6.2) | 1.5 (1.5) | 0.002 (–4.19 to 4.19) p = 0.999 | –1.1 (–5.7 to 3.5) p = 0.62 |
| Mean venous SO2 (%) | 63.4 (8.3) | 59.3 (8.0) | –4.0 (1.8) | 64.5 (8.9) | 66.2 (6.1) | 1.8 (2.3) | 62.9 (9.1) | 63.5 (7.5) | 0.6 (2.7) | 64.0 (7.7) | 66.9 (9.4) | 2.9 (2.1) | –4.6 (–11.3 to 2.1) p = 0.17 | –1.1 (–7.6 to 5.4) p = 0.72 |
| Mean AV SO2 (%) | 34.5 (8.4) | 37.9 (5.2) | 3.4 (1.5) | 33.4 (6.9) | 33.5 (6.5) | 0.1 (1.6) | 37.5 (8.6) | 35.4 (7) | –2.0 (2.7) | 34.9 (6.4) | 36.4 (8.6) | 1.5 (2.0) | 5.5 (–0.9 to 11.8) | –1.4 (–6.8 to 3.9) p = 0.59 |
| Mean arterial diameter (µm) | 118.6 (8.8) | 120.0 (11.8) | 1.4 (2.3) | 117.4 (8.7) | 122.5 (14.3) | 5.1 (3.9) | 116.9 (11.8) | 113.7 (10.3) | –3.2 (2.3) | 114.4 (11.9) | 108.4 (21.1) | –6.0 (4.2) | 4.6 (–2.1 to 11.4) p = 0.17 | 11.1 (–0.9 to 23.1) p = 0.07 |
| Mean venous diameter (µm) | 143.4 (17.1) | 147.1 (17.7) | 3.8 (3.7) | 142.3 (17.9) | 149.3 (22.7) | 7.0 (4.1) | 150.5 (11.9) | 148 (13.9) | –2.6 (3.5) | 148.1 (11.5) | 143.3 (18.9) | –4.8 (3.9) | 6.3 (–4.3 to 17) p = 0.23 | 11.8 (0.04 to 23.6) p = 0.049a |
| mfERG | n = 11 | n = 10 | n = 10 | n = 11 | n = 10 | n = 10 | n = 15 | n = 15 | n = 15 | n = 15 | n = 15 | n = 15 | ||
| Mean N1 (ms) | 16.5 (0.7) | 16.9 (1.2) | 0.5 (0.4) | 16.2 (0.5) | 17.0 (1.3) | 0.7 (0.3) | 16.3 (1.3) | 16.5 (1.2) | 0.2 (0.2) | 16.2 (0.9) | 16.4 (1.3) | 0.2 (0.2) | 0.3 (–0.5 to 1.2) p = 0.46 | 0.5 (–0.3 to 1.4) p = 0.21 |
| Mean N1 (nV) | –453 (99) | –434 (133) | 27 (33) | –420 (73) | –439 (133) | –15 (44) | –354 (112) | –351 (90) | 2 (30) | –399 (107) | –386 (126) | 13 (38) | 25 (–69 to 118) p = 0.59 | –28 (–149 to 93) p = 0.64 |
| Mean P2 (ms) | 32.1 (1.5) | 32.5 (1.5) | 0.6 (0.4) | 32.4 (1.0) | 32.5 (1.6) | 0.2 (0.3) | 31.7 (1.9) | 31.8 (1.4) | 0.1 (0.3) | 31.5 (1.6) | 31.7 (1.6) | 0.3 (0.3) | 0.5 (–0.4 to 1.4) p = 0.26 | –0.1 (–0.9 to 0.8) p = 0.90 |
| Mean P2 (nV) | 1219 (242) | 1214 (256) | –17 (76) | 1165 (195) | 1200 (324) | 54 (109) | 1032 (221) | 1026 (294) | –7 (78) | 1132 (255) | 1084 (373) | –47 (97) | –11 (–246 to 225) p = 0.92 | 101 (–207 to 409) p = 0.50 |
| Microperimetry | n = 13 | n = 12 | n = 12 | n = 13 | n = 12 | n = 12 | n = 15 | n = 15 | n = 15 | n = 15 | n = 15 | n = 15 | ||
| Mean retinal sensitivity (db) | 17.7 (1.3) | 17.7 (1.6) | –0.1 (0.6) | 17.7 (1.8) | 18 (1.5) | 0.4 (0.5) | 15.7 (4.2) | 14.9 (4.4) | –0.8 (0.7) | 16.1 (3.3) | 15.4 (4.8) | –0.7 (0.6) | 0.8 (–1.2 to 2.7) p = 0.42 | 1.1 (–0.7 to 2.9) p = 0.21 |
Chapter 4 Discussion
Summary of findings
The CLEOPATRA trial is the first Phase III RCT that evaluated the Noctura 400 Sleep Mask as an intervention to treat and prevent non-central DMO in a multicentre setting. The results of this study show that offering a Noctura 400 Sleep Mask to wear at night during sleep is not an effective option in the treatment or prevention of progression of non-centre-involving DMO. Although objective assessment of reduction of maximal retinal thickness was our primary outcome, we have made our conclusion based on the primary outcome, per-protocol secondary analysis and five prespecified sensitivity analyses of the primary outcome, as none of these showed any significant therapeutic benefit to wearing the lightmask. In addition, because of the dynamic nature of DMO, we considered several secondary outcomes including reduction in total retinal thickness, macular volume, progression of central subfield thickness to ≥ 300 µm, the proportion of participants requiring treatment for new-onset centre-involving DMO, and the number of participants who were treated with standard therapy during the trial because of worsening DMO. None of the changes in these parameters was significant between arms substantiating the results of the primary outcome. There was also no effect of the lightmask on diabetic retinopathy severity levels.
Both arms showed a gradual mean reduction in the zone of maximal retinal thickness over 24 months. The reduction is within the SD that we used for the sample size calculation. Participants do pay more attention to their diabetic control when they participate in clinical trials. The event rate of progression to centre-involving DMO was also similar to previous reports.
Mechanistic evaluation
This study is the first study to evaluate the effect of oxygen supplementation on retinal function in participants with early DMO. Mean pulse oximetry increased by 2% during 100% oxygen breathing. However, mean retinal arteriolar oxygen saturation was unchanged. This finding suggests that the higher systemic oxygen saturation was not conveyed to the retinal arterioles because of a countercurrent exchange between the central retinal artery and vein, a possible calibration error of retinal oximetry or the arteries are already carrying maximum possible oxygen saturation. 40 Palkovits et al. 41 measured the effect of hyperoxia with a different type of oximeter (from Imedos Systems GmbH, Jena, Germany) and similarly found that the retinal arterioles did not reach 100% oxygen saturation during hyperoxia.
Retinal oxygenation is dependent on the rate of retinal blood flow and the oxygen concentrations in arterial and venous blood. Blood flow, in turn, is dependent on the diameter of the retinal arteries and veins. 41 The diameter of both the arteries and veins did not show a significant change with supplemental oxygen at baseline; however, there was a difference in diameter change of the arteries and veins at 12 months. The mean arterial and venous diameters increased in eyes that used the lightmask, while they decreased in those not wearing the lightmask. Loss of autoregulation of retinal blood vessels is an early change in participants with diabetes mellitus. These study results are in the opposite direction to that predicted, as an increasing blood vessel diameter is typically a sign of increasing severity of diabetic retinopathy and loss of autoregulation. Although these findings may be attributed to a small sample size and technical difficulties with performing these tests, they may also suggest that the inner retinal tissue oxygenation may not be compromised in early DMO. We have not measured the choroidal blood flow or the choroidal oxygen saturation but the fact that there was no change in retinal function with oxygen supplementation suggests that the outer retina may not be significantly affected by hypoxia in early DMO. However, animal experiments have shown significant oxidative stress at the photoreceptor layer,42 although a recent study by Lau and Linsenmeier showed that hypoxia is not a contributory factor for diabetic retinopathy in rodents. 43,44 Therefore, our study results together with the current evidence from laboratory work indicate that further work is required to understand the role of photoreceptors and hypoxia in the development and progression of DMO.
Strengths and limitations
The main strength of this study was that we ensured that the primary outcome was corroborated by predefined sensitivity analysis and secondary analysis to reduce potential systemic biases in this dynamic condition. Other strengths included clear definition of objective end points, publication of the protocol, significant public and patient involvement throughout the study, and strict assessment assured high-quality data and pre-planned analysis for expected non-compliance. The baseline characteristics of the trial population were typical for the intended patient population. The representative multiethnic patient population, together with the multicentre trial design, permit wide generalisability of our results. The primary outcome of retinal thickness was measured twice at the 12 and 24 months time points and the average taken to analyse the outcomes. Similarly, we took the average of two BCVA measurements at baseline to ensure that test–retest variability did not influence the result as the expected differences between arms were small. We took several prespecified steps to increase the robustness of our analysis. We buttressed our primary outcome analysis with several sensitivity analyses. In a study in which we are reliant on asymptomatic participants using an intervention, missing data, suboptimal compliance and drop-out rates can significantly affect the study outcomes. We expected compliance with lightmasks to be an issue based on the phase II study19 and because non-centre-involving DMO is asymptomatic. Therefore, we made multiple efforts to tackle compliance-related issues in this study. We calculated the sample size with a 20% attrition rate, which is higher than most ophthalmic trials. In addition, we allowed for OCTs from clinic appointments to be used in cases for which participants attended a clinic visit and not a clinical trial visit appointment. We had also carefully considered the influence of non-compliance on the potential therapeutic effect of the lightmasks by incorporating a predefined CACE analysis for non-compliance at three levels: –70%, 60% and 50% compliance. The non-compliance was observed as early as 4 months into the trial and across all three definitions of compliers highlighting that offering these lightmasks to wear during sleep is not an effective option for this condition. This study illustrates the importance of the study statisticians planning for all possible outcomes and likely challenges with the analysis. The statisticians were also expertly advised by a very experienced DMC statistician ensuring the robustness of the study outcomes. A full explanation of how the statistical model was designed is provided in Chapter 2. We expect more interventions to be developed to prevent the progression of diabetic retinopathy and DMO and we recommend that considerable planning of anticipated challenges be considered in this area as was done in this study.
We are also grateful to the reviewers of our grant application as it strengthened our study by defining our primary outcome at 24 months rather than 12 months. This 24-month study has clearly shown several outcomes that may not have come to light with a shorter study.
Nevertheless, some limitations should be considered when interpreting the results. The study only shows that offering Noctura 400 Sleep Masks to wear during sleep at night to suppress rod function is not an effective option to treat DMO. It is possible that the retinal illumination achieved with these devices as used in this protocol did not reduce the dark current sufficiently to alter the hypoxic state. The study also showed that offering the lightmasks to participants to use during sleep is associated with suboptimal compliance by 24 months. The electronic recording of compliance was possible only if the participants returned the lightmasks at each 4-monthly clinic visit, and so compliance may have been better than the recorded compliance data suggests.
The study also questions whether or not non-central DMO is the correct patient group in which to test this device. The role of hypoxia is better understood in severe retinopathy. Recent literature on oxygen metabolism in early stages of retinopathy is conflicting. 42,43,45 Oxygen metabolism in different stages of diabetic retinopathy has to be studied further in both animals and humans. Currently, this is challenging because of the technical difficulties of measuring regional variations in oxygen saturation in the retina.
Comparison with existing literature
The studies on the lightmask have all been short-term studies. 19–22 In diabetic retinopathy trials, all interventions should be evaluated for at least 2 years to understand the holistic effect of the intervention on the patient. On average, diabetic retinopathy affects participants for > 20 years and most eyes with mild to moderate retinopathy or non-central DMO deteriorate very slowly. Therefore, clinical trials in this area should be long-acting, with clearly predefined objective end points and should have a control arm. This is the first RCT that has evaluated lightmasks over 2 years, to our knowledge, and we have highlighted that the lightmasks did not show any clinically meaningful benefit at any time point in the study compared with the non-lightmask.
Implications for practice and research recommendations
The CLEOPATRA study shows that the lightmask to prevent dark adaptation is not recommended in clinical practice for DMO or diabetic retinopathy. As recent laboratory experiments have shown conflicting results on the role of oxygen metabolism on microvascular changes in diabetic retinopathy, we do not recommend more clinical trials on lightmasks suppressing rod function until there is very convincing laboratory experiments to suggest that the retinal oxygen consumption for dark adaptation has a detrimental effect on microvascular changes in the retina. We also do not recommend the use of the lightmask to prevent dark adaptation in participants for diabetic retinopathy based on our study results, in which we found no change in diabetic retinopathy severity scale between the study arms. However, our study results encourage more research on interventions for non-central DMO. Currently, several treatment options for early or non-central oedema have not shown clinical benefit. Research in this area should accelerate to tackle the increasing numbers of participants presenting with non-central diabetic oedema given the exponential increase in the worldwide diabetic population. The research should focus on easy to use and cheap options to global benefit. Until new preventative options are made available, control of systemic risk factors remain crucially important as the only effective option for this cohort of participants.
Chapter 5 Conclusion
To our knowledge, this is the first randomised controlled clinical trial that has evaluated the role of a lightmask designed to prevent dark adaptation as a treatment and preventative option for non-central DMO over 24 months. The robust study design and conduct and the study period of over 24 months show that a lightmask to prevent dark adaptation is not a management option for non-central DMO and diabetic retinopathy. The mechanistic study although exploratory in nature also did not support the role of 100% oxygen or the lightmask in improving visual function or decreasing macular thickness suggesting that hypoxia may not be a significant contributory factor to the development and progression of the early stages of DMO and retinopathy. As laboratory experiments have shown that photoreceptor induced oxidative stress and abnormalities in oxygen metabolism may contribute to the development and progression of diabetic retinopathy, we recommend that further laboratory and clinical studies should be performed to evaluate the mechanisms and time points at which rod photoreceptors may contribute to the pathogenesis of microvascular changes in the retina in diabetes mellitus. As the natural course of diabetic retinopathy is dynamic, with the majority of participants showing an overall tendency to progress in severity of retinopathy over several years, interventions targeting rod function, if proven to be of value, should be evaluated for at least 24 months in robust clinical trials so that patient acceptability, safety, effects on quality of life and costs of the intervention can be taken into account.
Acknowledgements
The authors would like to thank the late Geoffrey Arden (Professor Emeritus, Moorfields Eye Hospital and University College London) for his invaluable help. Geoffrey Arden was a co-applicant and was involved in the design of the study. He was the originator of this concept of prevention of rod mediated dark adaptation as a treatment option for diabetic retinopathy and macular oedema.
We would also like to express thanks to the NIHR Local Clinical Research Networks.
Essential support and guidance was provided by the KCTU, the service user group, the TSC (chairperson Gillian Hood) and the DMC (chairperson Sarah Walker). We would also like to thank members of the Gloucester Reading Centre for their support in the trial. We would also like to thank the lay panel in the North East Diabetes Research Network who helped us with the design of the protocol, the patient information sheet and the consent form.
We would like to thank Polyphotonix Medical Ltd for providing the lightmasks and the non-lightmasks at a discounted rate and for providing the electronic compliance data from the returned lightmasks as part of the service agreement.
Finally, and most importantly, we would like to thank all of the participants who took part in the study.
Support
The study was sponsored by NIHR Moorfields Biomedical Research Centre and supported by the UK Clinical Research Network. The research was supported by the NIHR Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and University College London Institute of Ophthalmology, the NIHR Moorfields Clinical Research Facility and the UK Clinical Research Collaboration-registered KCTU at King’s Health Partners, which is part funded by the NIHR Biomedical Research Centre for Mental Health at South London and Maudsley, NHS Foundation Trust and King’s College London. We would also like to thank Maggie Shergill, the Research Manager (Monitoring) and the other team members of the NIHR Evaluation Trials and Studies Coordinating Centre.
Contributions of authors
Sobha Sivaprasad (Professor, Consultant Ophthalmologist) was the chief investigator.
Joana Vasconcelos (Research Fellow, Medical Statistics) was involved in the design and statistical analysis of the study.
Helen Holmes (Trial Manager) was involved in all aspects of the study’s management and monitoring.
Caroline Murphy (King’s Clinical Trial Unit) was a co-applicant and was involved in the design and trial management.
Joanna Kelly (King’s Clinical Trial Unit) was a co-applicant and was involved in the design and data management.
Philip Hykin (Consultant Ophthalmic Surgeon) was a co-applicant and was involved in the design and recruitment of the study.
Andrew Toby Prevost (Professor, Medical Statistics) was a co-applicant and was involved in the design and statistical analysis of the study.
All authors contributed to the first draft, and reviewed, revised and approved the final version of the manuscript.
Publications
Sivaprasad S, Arden G, Prevost AT, Crosby-Nwaobi R, Holmes H, Kelly J, et al. A multicentre Phase III randomised controlled single-masked clinical trial evaluating the clinical efficacy and safety of light-masks at preventing dark-adaptation in the treatment of early diabetic macular oedema (CLEOPATRA): study protocol for a randomised controlled trial. Trials 2014;15:458.
Sivaprasad S, Vasconcelos JC, Prevost AT, Holmes H, Hykin P, George S, et al. Clinical efficacy and safety of a lightmask for prevention of dark adaptation in treating and preventing progression of early diabetic macular oedema at 24 months (CLEOPATRA): a multicentre, Phase III, randomised controlled trial. Lancet Diabetes Endocrinol 2018;6:382–91.
Data-sharing statement
Data are archived at Moorfield Eye Hospital. All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al. IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract 2017;128:40-5. https://doi.org/10.1016/j.diabres.2017.03.024.
- Sivaprasad S, Gupta B, Gulliford MC, Dodhia H, Mohamed M, Nagi D, et al. Ethnic variations in the prevalence of diabetic retinopathy in people with diabetes attending screening in the United Kingdom (DRIVE UK). PLOS ONE 2012;7. https://doi.org/10.1371/journal.pone.0032182.
- Leasher JL, Bourne RR, Flaxman SR, Jonas JB, Keeffe J, Naidoo K, et al. Global estimates on the number of people blind or visually impaired by diabetic retinopathy: a meta-analysis from 1990 to 2010. Diabetes Care 2016;39:1643-9. https://doi.org/10.2337/dc15-2171.
- Zoungas S, Arima H, Gerstein HC, Holman RR, Woodward M, Reaven P, et al. Collaborators on Trials of Lowering Glucose (CONTROL) group . Effects of intensive glucose control on microvascular outcomes in participants with type 2 diabetes: a meta-analysis of individual participant data from randomised controlled trials. Lancet Diabetes Endocrinol 2017;5:431-7. https://doi.org/10.1016/S2213-8587(17)30104-3.
- Lee R, Wong TY, Sabanayagam C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis 2015;2. https://doi.org/10.1186/s40662-015-0026-2.
- Keenan TD, Johnston RL, Donachie PH, Sparrow JM, Stratton IM, Scanlon P. United Kingdom National Ophthalmology Database Study: Diabetic Retinopathy; Report 1: prevalence of centre-involving diabetic macular oedema and other grades of maculopathy and retinopathy in hospital eye services. Eye 2013;27:1397-404. https://doi.org/10.1038/eye.2013.196.
- Photocoagulation for diabetic macular edema . Early Treatment Diabetic Retinopathy Study report number 1. Early Treatment Diabetic Retinopathy Study research group. Arch Ophthalmol 1985;103:1796-806. https://doi.org/10.1001/archopht.1985.01050120030015.
- Virgili G, Parravano M, Evans JR, Gordon I, Lucenteforte E. Anti-vascular endothelial growth factor for diabetic macular oedema: a network meta-analysis. Cochrane Database Syst Rev 2017;6. https://doi.org/10.1002/14651858.CD007419.pub5.
- Veritti D, Sarao V, Diplotti L, Samassa F, Lanzetta P. Fluocinolone acetonide for the treatment of diabetic macular edema. Expert Opin Pharmacother 2017;18:1507-16. https://doi.org/10.1080/14656566.2017.1363182.
- Al-Khersan H, Hariprasad SM, Chhablani J. Dex Implant Study Group . Early response to intravitreal dexamethasone implant therapy in diabetic macular edema may predict visual outcome. Am J Ophthalmol 2017;184:121-8. https://doi.org/10.1016/j.ajo.2017.10.004.
- Gardner TW, Larsen M, Girach A, Zhi X. Protein Kinase C Diabetic Retinopathy Study (PKC-DRS2) Study Group . Diabetic macular oedema and visual loss: relationship to location, severity and duration. Acta Ophthalmol 2009;87:709-13. https://doi.org/10.1111/j.1755-3768.2009.01545.x.
- PKC-DMES Study Group . Effect of ruboxistaurin in participants with diabetic macular edema: thirty-month results of the randomized PKC-DMES clinical trial. Arch Ophthalmol 2007;125:318-24. https://doi.org/10.1001/archopht.125.3.318.
- Friedman SM, Almukhtar TH, Baker CW, Glassman AR, Elman MJ, Bressler NM, et al. Topical nepafenec in eyes with noncentral diabetic macular edema. Retina 2015;35:944-56. https://doi.org/10.1097/IAE.0000000000000403.
- Arden GB, Sivaprasad S. Hypoxia and oxidative stress in the causation of diabetic retinopathy. Curr Diabetes Rev 2011;7:291-304. https://doi.org/10.2174/157339911797415620.
- Nguyen QD, Shah SM, Van Anden E, Sung JU, Vitale S, Campochiaro PA. Supplemental oxygen improves diabetic macular edema: a pilot study. Invest Ophthalmol Vis Sci 2004;45:617-24. https://doi.org/10.1167/iovs.03-0557.
- Sivaprasad S, Arden G. Spare the rods and spoil the retina: revisited. Eye 2016;30:189-92. https://doi.org/10.1038/eye.2015.254.
- Kern TS, Engerman RL. Capillary lesions develop in retina rather than cerebral cortex in diabetes and experimental galactosemia. Arch Ophthalmol 1996;114:306-10. https://doi.org/10.1001/archopht.1996.01100130302013.
- Linsenmeier RA. Effects of light and darkness on oxygen distribution and consumption in the cat retina. J Gen Physiol 1986;88:521-42. https://doi.org/10.1085/jgp.88.4.521.
- Arden GB, Gündüz MK, Kurtenbach A, Völker M, Zrenner E, Gündüz SB, et al. A preliminary trial to determine whether prevention of dark adaptation affects the course of early diabetic retinopathy. Eye 2010;24:1149-55. https://doi.org/10.1038/eye.2009.328.
- Arden GB, Jyothi S, Hogg CH, Lee YF, Sivaprasad S. Regression of early diabetic macular oedema is associated with prevention of dark adaptation. Eye 2011;25:1546-54. https://doi.org/10.1038/eye.2011.264.
- Sahni JN, Czanner G, Gutu T, Taylor SA, Bennett KM, Wuerger SM, et al. Safety and acceptability of an organic light-emitting diode sleep mask as a potential therapy for retinal disease. Eye 2017;31:97-106. https://doi.org/10.1038/eye.2016.259.
- Kuchynka P, Grierson I, Veith M, Hill D. Participants with diabetic eye disease using a potentially therapeutic mask. Do sufficient participants wear the mask and for how long?. Adv Ophthalmol Vis Syst 2017;7.
- Sivaprasad S, Arden G, Prevost AT, Crosby-Nwaobi R, Holmes H, Kelly J, et al. A multicentre phase III randomised controlled single-masked clinical trial evaluating the clinical efficacy and safety of light-masks at preventing dark-adaptation in the treatment of early diabetic macular oedema (CLEOPATRA): study protocol for a randomised controlled trial. Trials 2014;15. https://doi.org/10.1186/1745-6215-15-458.
- Sivaprasad S, Vasconcelos JC, Prevost AT, Holmes H, Hykin P, George S, et al. Clinical efficacy and safety of a light mask for prevention of dark adaptation in treating and preventing progression of early diabetic macular oedema at 24 months (CLEOPATRA): a multicentre, Phase III, randomised controlled trial. Lancet Diabetes Endocrinol 2018;6:382-91. https://doi.org/10.1016/S2213-8587(18)30036-6.
- Moul D, Pilkonis P, Miewald J, Carey T, Buysse DJ. Preliminary study of the test-retest reliability and concurrent validities of the Pittsburgh Insomnia Rating Scale (PIRS). Sleep 2002;25:A246-7.
- Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991;14:540-5. https://doi.org/10.1093/sleep/14.6.540.
- White IR, Horton NJ, Carpenter J, Pocock SJ. Strategy for intention to treat analysis in randomised trials with missing outcome data. BMJ 2011;342. https://doi.org/10.1136/bmj.d40.
- Marteau TM, Aveyard P, Munafò MR, Prevost AT, Hollands GJ, Armstrong D, et al. Effect on adherence to nicotine replacement therapy of informing smokers their dose is determined by their genotype: a randomised controlled trial. PLOS ONE 2012;7. https://doi.org/10.1371/journal.pone.0035249.
- Carpenter J, Bithell J. Bootstrap confidence intervals: when, which, what? A practical guide for medical statisticians. Stat Med 2000;19:1141-64. https://doi.org/10.1002/(SICI)1097-0258(20000515)19:9<1141::AID-SIM479>3.0.CO;2-F.
- International Conference on Harmonisation. Guidance on statistical principles for clinical trials; availability – FDA. Notice Fed Regist 1998;63:49-98.
- White IR, Thompson SG. Adjusting for partially missing baseline measurements in randomized trials. Stat Med 2005;24:993-1007. https://doi.org/10.1002/sim.1981.
- Mallinckrod CH, Lane PW, Schnell D, Peng Y, Mancuso JP. Recommendations for the primary analysis of continuous endpoints in longitudinal clinical trials. Drug Inf J 2008;42:303-19. https://doi.org/10.1177/009286150804200402.
- White IR, Carpenter J, Horton NJ. Including all individuals is not enough: lessons for intention-to-treat analysis. Clin Trials 2012;9:396-407. https://doi.org/10.1177/1740774512450098.
- Burzykowski T, Carpenter J, Coens C, Evans D, France L, Kenward M, et al. Missing data: discussion points from the PSI missing data expert group. Pharm Stat 2010;9:288-97. https://doi.org/10.1002/pst.391.
- White IR, Kalaitzaki E, Thompson SG. Allowing for missing outcome data and incomplete uptake of randomised interventions, with application to an internet-based alcohol trial. Stat Med 2011;30:3192-207. https://doi.org/10.1002/sim.4360.
- Dunn G, Maracy M, Dowrick C, Ayuso-Mateos JL, Dalgard OS, Page H, et al. Estimating psychological treatment effects from a randomised controlled trial with both non-compliance and loss to follow-up. Br J Psychiatry 2003;183:323-31. https://doi.org/10.1192/bjp.183.4.323.
- White IR. Uses and limitations of randomization-based efficacy estimators. Stat Methods Med Res 2005;14:327-47. https://doi.org/10.1191/0962280205sm406oa.
- Data Protection Act 1998: Elizabeth II. Chapter 19. London: The Stationery Office; 1990.
- MRC Guidelines for Good Clinical Practice. London: External Communications; 1998.
- Olafsdottir OB, Eliasdottir TS, Kristjansdottir JV, Hardarson SH, Stefánsson E. Retinal vessel oxygen saturation during 100% oxygen breathing in healthy individuals. PLOS ONE 2015;10. https://doi.org/10.1371/journal.pone.0128780.
- Palkovits S, Lasta M, Told R, Schmidl D, Werkmeister R, Cherecheanu AP, et al. Relation of retinal blood flow and retinal oxygen extraction during stimulation with diffuse luminance flicker. Sci Rep 2015;5. https://doi.org/10.1038/srep18291.
- Liu H, Tang J, Du Y, Saadane A, Tonade D, Samuels I, et al. Photoreceptor cells influence retinal vascular degeneration in mouse models of retinal degeneration and diabetes. Invest Ophthalmol Vis Sci 2016;57:4272-81. https://doi.org/10.1167/iovs.16-19415.
- Tonade D, Liu H, Palczewski K, Kern TS. Photoreceptor cells produce inflammatory products that contribute to retinal vascular permeability in a mouse model of diabetes. Diabetologia 2017;60:2111-20. https://doi.org/10.1007/s00125-017-4381-5.
- Lau JC, Linsenmeier RA. Increased intraretinal PO2 in short-term diabetic rats. Diabetes 2014;63:4338-42.
- Liu W, Wang S, Soetikno B, Yi J, Zhang K, Chen S, et al. Increased retinal oxygen metabolism precedes microvascular alterations in type 1 diabetic mice. Invest Ophthalmol Vis Sci 2017;58:981-9. https://doi.org/10.1167/iovs.16-20600.
Appendix 1 The CLEOPATRA study group and resource centres
The CLEOPATRA study group thanks all of the participants who participated in the study, and all site investigators and research teams.
| Site number | Site |
|---|---|
| 01 | Central Middlesex Hospital, London North West University Healthcare NHS Trust, London, UK |
| 02 | Heartlands Hospital, Heart of England NHS Foundation Trust, Bordesley Green East, Birmingham, UK |
| 03 | Stoke Mandeville Hospital, Buckinghamshire Healthcare NHS Trust, Amersham, UK |
| 04 | Maidstone and Tunbridge Wells NHS Trust, Kent, UK |
| 05 | Moorfields Eye Hospital NHS Foundation Trust, London, UK |
| 06 | Bristol Eye Hospital, University Hospitals Bristol NHS Foundation Trust, Bristol, UK |
| 07 | Frimley Park Hospital NHS Foundation Trust, Surrey, UK |
| 08 | City Hospitals Sunderland NHS Foundation Trust, Sunderland Royal Hospital, Sunderland, UK |
| 09 | William Harvey Hospital, East Kent Hospitals University NHS Foundation Trust, Kent, UK |
| 10 | Site code not in use. Site 10 withdrew before recruitment began |
| 11 | Princess Alexandra Hospital NHS Foundation Trust, Essex, UK |
| 12 | Hillingdon Hospital, Hillingdon Hospitals NHS Foundation Trust, Uxbridge, UK |
| 13 | King’s College Hospital NHS Foundation Trust, London, UK |
| 14 | Brighton and Sussex University Hospitals NHS Trust, Sussex Eye Hospital, Brighton, UK |
| 15 | The Royal Wolverhampton NHS Trust, Wolverhampton and Midland Counties Eye Infirmary, West Midlands, UK |
| 16 | Birmingham Midland Eye Centre, City Hospital NHS Trust, Birmingham, UK |
Resource centres
We would also like to thank the following resource centres for their support of the trial:
-
King’s College Clinical Trial team: Gill Lambert, Beverley White-Alao, Oliver Pressey, Negin Sarafraz-Shekary, Evangelos Georgiou and Janice Jimenez.
-
The Royal Free London NHS Foundation Trust Pharmacists team: Nicky Heath and Sabina Melander.
-
The North East Diabetes Research Network’s Lay Panel.
-
Patient-reported outcomes consultant to the study: Clare Bradley, Royal Holloway, University of London.
-
Mechanistic evaluation team: Luke Nicholson, Roxanne Crosby-Nwaobi and Lauren Leitch-Devlin at the NIHR Moorfields Clinical Research Facility.
-
Certifications for visual acuity and contrast sensitivity: Catherine Grigg and Katherine Binsted at Moorfields Eye Hospital, London.
-
Members of the Networc UK: Usha Chakravarthy, Tunde Peto, Simon Harding, Pauline Lenfestey, Savitha Madhusudhan, Clare Newell, Michelle McGaughey, Vittorio Silvestri, Karleigh Kelso, Barbra Hamill, Graham Young, Irene Leung, Peter Blows, Frank Picton, David Parry and Sophie Leach.
Appendix 2 The CLEOPATRA study committees
Trial Steering Committee
Gillian Hood (independent chairperson; Queen Mary University of London, London, UK); Graham A Hitman (Barts and The London School of Medicine and Dentistry, London, UK); David Crabb (City University, London, UK); Alaistair Denniston (Queen Elizabeth Hospital, Birmingham, UK); Reverend Douglas Lewin (lay member); and Ian Grierson (University of Liverpool, Liverpool, and Polyphotonix Medical Ltd).
Data Monitoring Committee
Sarah Walker (chairperson; Medical Research Council, Clinical Trials Unit, University College London, London, UK), Jackie Sturt (King’s College London, London, UK) and Debendra Sahu (Southampton NHS Trust, Southampton, UK).
Trial Management Group members
Sobha Sivaprasad, Philip Hykin, Andrew Toby Prevost, Joana Vasconcelos, Helen Holmes, Beverley White-Alao, Caroline Murphy, Joanna Kelly and the sponsor representative (Moorfields Eye Hospital, London, UK).
Appendix 3 Summary of patient and public involvement
Participants and the public were involved throughout the CLEOPATRA study, as follows. The North East London lay member panel of the diabetes research network: five lay members of the panel contributed to the design of the study, design and content of the patient information sheets and the consent form. They also contributed to the contents of the letter to the participants informing them of the results.
List of abbreviations
- ADE
- adverse device effect
- AE
- adverse event
- ANCOVA
- analysis of covariance
- AR
- adverse reaction
- BCVA
- best corrected visual acuity
- CACE
- complier average causal effect
- CE
- Conformite Européenne
- CI
- confidence interval
- CLEOPATRA
- clinical efficacy and safety of light-masks at preventing dark-adaptation in the treatment of early diabetic macular oedema
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- CV
- curriculum vitae
- DMC
- Data Monitoring Committee
- DMO
- diabetic macular oedema
- eCRF
- electronic case report form
- ESS
- Epworth Sleepiness Scale
- ETDRS
- Early Treatment Diabetic Retinopathy Study
- GCP
- good clinical practice
- HbA1c
- glycated haemoglobin
- IQR
- interquartile range
- ISF
- investigator site file
- IT
- information technology
- ITT
- intention to treat
- KCTU
- King’s Clinical Trials Unit
- LME
- linear mixed effect
- LOCF
- last observation carried forward
- MAR
- missing at random
- mfERG
- multifocal electroretinography
- MHRA
- Medicines and Healthcare products Regulatory Agency
- N1
- the first negative waveform
- NICE
- National Institute For Health and Care Excellence
- NIHR
- National Institute for Health Research
- OCT
- optical coherence tomography
- OLED
- organic light-emitting diode
- P1
- the first positive waveform
- PIN
- patient identification number
- PIRS_20
- Pittsburgh Insomnia Rating Scale 20-item version
- PP
- per protocol
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- SADE
- serious adverse device effect
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SDW
- source data worksheet
- SE
- standard error
- SIV
- site initiation visit
- SOP
- standard operating procedure
- SPRAE
- serious procedure-related adverse event
- TMF
- trial master file
- TSC
- Trial Steering Committee
- USADE
- unexpected serious adverse device effect
- VEGF
- vascular endothelial growth factor