

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 13/158/48. The contractual start date was in November 2015. The final report began editorial review in November 2018 and was accepted for publication in February 2019. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Helen Yates reports personal fees from AbbVie Inc. (Lake Bluff, IL, USA) outside the submitted work.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2019. This work was produced by Yates et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2019 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background
Bronchopulmonary dysplasia (BPD) affects approximately 25% of all babies born at < 30 weeks’ gestation1,2 and represents a significant health-care burden. Few interventions are proven to reduce BPD incidence or severity. 3 Babies who develop BPD are at increased risk of long-term respiratory and neurodevelopmental impairment. 4,5 Natarajan et al. reported that 7% of survivors with BPD had moderate to severe cerebral palsy (CP) and 11.4% had severely abnormal neurodevelopmental outcomes [i.e. composite cognitive scores on Bayley Scales of Infant and Toddler Development III (BSID-III) of < 70 points]. 5 These outcomes are seen in only 2% and 4.6% of matched babies without BPD in the study by Natarajan et al. 5 When these outcomes are combined (to account for comorbidity) the results suggest that 200 babies per annum in the UK develop moderate or severe disability solely attributable to BPD. 5 Therapies like postnatal corticosteroids, which reduce the burden of BPD, will therefore reduce the burden of neurodevelopmental and respiratory impairment that BPD entails.
Since the 1970s, postnatal corticosteroids have been used in a variety of dose regimens to ameliorate the lungs’ inflammatory responses and reduce the incidence of BPD. 6–8 Babies with BPD exhibit a variety of inflammatory biomarkers and numerous cytokines are implicated in the evolution of the disease. 9–11 Postnatal corticosteroids have been found to downregulate many proinflammatory cytokines in these babies12 and have been shown in meta-analyses to facilitate the extubation of preterm babies and reduce the incidence of BPD. 6,7 The most recent Cochrane review of late administration of corticosteroids (in infants > 7 days of age) shows that corticosteroids were associated with a lower rate of failure to extubate by the seventh day of treatment [risk ratio 0.65, 95% confidence interval (CI) 0.59 to 0.72] and reduction of the incidence of BPD (risk ratio 0.77, 95% CI 0.67 to 0.88). 7 As a result of these benefits, postnatal corticosteroid therapy became commonplace. In 1995, 72.5% of all surviving babies born at < 26 weeks’ gestation in the UK received postnatal corticosteroids. 13
Concerns about the long-term neurodevelopmental impact of postnatal corticosteroids were first raised in 1974,8 but it was in 2001 that Barrington’s meta-analysis showed that babies treated with postnatal corticosteroids have an increased risk of long-term adverse neurodevelopmental outcomes (CP: RR 1.92, 95% CI 1.41 to 2.61). 14 In addition to this, studies in animals have shown that postnatal exposure to corticosteroids during the critical period of late neurogenesis can impair myelination and neuronal maturation. 15–17 As a result of these findings, postnatal corticosteroids became less popular with clinicians18 and their use declined dramatically. This in turn resulted in an increased prevalence of BPD. 19
Further investigation of the relationship between postnatal corticosteroids, BPD and neurodevelopmental impairment has shown that the relationship between corticosteroids and neurodevelopmental outcomes is complex and poorly understood. Meta-analyses have revealed that the risk of adverse neurodevelopmental outcome is modified by the corticosteroid used,6 the postnatal age at which the corticosteroids are given,6 7 the cumulative dose used20 and the baby’s underlying risk of developing BPD21 (Table 1). An increased risk of adverse outcomes is seen only in those trials that include babies:
-
treated with dexamethasone at < 7 days of age to prevent BPD (CP: RR 1.75, 95% CI 1.2 to 2.55)6
-
exposed to high doses (i.e. a > 3.0 mg/kg cumulative dose) of dexamethasone [CP: odds ratio (OR) 2.3, 95% CI 1.22 to 4.36]20
-
treated with postnatal corticosteroids who are at low risk of developing BPD. 21
| Administration and dose | Condition | |
|---|---|---|
| CP | BPD at 36 weeks’ PMA | |
| Administration, RR (95% CI) | ||
| Early (< 8 days) administration of dexamethasone6 | 1.75 (1.2 to 2.55) | 0.71 (0.62 to 0.81) |
| Early (< 8 days) administration of hydrocortisone6 | 1.05 (0.66 to 1.66) | 0.91 (0.8 to 1.05) |
| Late (> 7 days) administration of corticosteroid7 | 1.1 (0.79 to 1.54) | 0.77 (0.67 to 0.88) |
| Dose, OR (95% CI) | ||
| Low cumulative dose (i.e. < 3.0 mg/kg) of dexamethasone20 | 0.61 (0.28 to 1.34) | 0.66 (0.54 to 0.8) |
| High cumulative dose (i.e. ≥ 3.0 mg/kg) of dexamethasone20 | 2.3 (1.22 to 4.36) | 0.34 (0.2 to 0.57) |
Hydrocortisone has been used to prevent BPD in babies < 7 days of age and, although it is not associated with an increased risk of cerebral palsy (CP: RR 1.05, 95% CI 0.66 to 1.66), hydrocortisone treatment does not reduce the risk of BPD (BPD: RR 0.91, 95% CI 0.8, 1.05). 6 To date, there has been only one published study giving hydrocortisone to 64 ventilator-dependent babies aged 10–14 days,22 this is combined with 20 trials (1360 babies) with an arm that was randomised to dexamethasone in the meta-analysis results in Table 1.
No increased risk of adverse neurodevelopment or CP was seen in those trials that use corticosteroids in babies:
-
over 7 days of age to treat established or evolving BPD (CP: RR 1.1, 95% CI 0.77 to 1.56)7
-
exposed to lower doses of dexamethasone (CP: OR 0.61, 95% CI 0.28 to 1.34)20
-
at high risk of BPD (control rates of BPD of c. 65%). 21
It was calculated that every 10% increase in babies BPD rate reduces the risk difference of CP by 2.4% (95% CI 0.3% to 4.3%). 21
To reduce the risk of adverse neurodevelopmental outcomes while maintaining the beneficial pulmonary effects, many clinicians across the UK looked to using low-dose (i.e. a ≤ 3.0 mg/kg cumulative dose) and very low-dose (i.e. a ≤ 1.5 mg/kg cumulative dose) regimens of dexamethasone in babies over 8 days of age at high risk of BPD. 23 In Belfast, from 2004, very low-dose dexamethasone was used to treat ventilator-dependent babies with BPD,24 although any reported benefits were not based on randomised trials. Another tertiary unit introduced the Minidex protocol. 25 The Minidex regimen is 50 µg/kg/day dexamethasone every day for 10 days, then on alternate days for 6 days. The regimen was derived from the physiological corticosteroid replacement dose and is lower than the next lowest dose regimen that has been subjected to short-term assessment. 26 Leeds Teaching Hospitals Trust published its results as a retrospective cohort study. 25 This report showed that babies treated with this regimen were extubated significantly faster than control babies, with a hazard ratio (HR) of 6.24 (95% CI 2.34 to 16.63). 25
There is insufficient evidence to support these very low-dose regimens of dexamethasone. 20 No very low-dose regimen of dexamethasone has been subjected to a randomised controlled trial that was adequately powered to detect neurodevelopmental outcomes.
Rationale
There is no definitive evidence of the impact of very low-dose dexamethasone on clinically important outcomes, size of effect or safety. An effectiveness trial aimed at establishing the 2-year neurodevelopmental outcomes of babies receiving the Minidex regimen is needed to inform practice. However, prior to this, a smaller study to determine short-term clinical efficacy, acceptability, recruitment and retention is necessary to allow the definitive trial to be planned. The MINIDEX trial was designed to meet the need for a preliminary efficacy study and, in December 2014, was funded by the National Institute for Health Research (NIHR) Efficacy and Mechanism Evaluation (EME) programme. In this efficacy study, extubation and respiratory morbidity prior to hospital discharge were used as surrogate outcomes for the clinically relevant outcome of long-term respiratory morbidity.
If the treatment ultimately proves to be effective, it will benefit patients by improving pulmonary function and facilitating extubation, thereby reducing both the duration of invasive ventilation and the risk of adverse long-term respiratory outcomes. Clinical trials are justified to assess the benefit–risk balance for this therapy. The trial participants are limited exclusively to those babies deemed to be at high risk of BPD. The meta-regression by Doyle et al. suggests that the risk of neurodevelopmental impairment attributable to a low dose of corticosteroids for the participants in this trial should be minimal. 21 Should the therapy be shown not to facilitate extubation, then it can be recommended that the off-label use of very low-dose dexamethasone for this indication is discontinued. This would save babies across the UK from unnecessary exposure to a potential neurotoxin.
Family involvement in care is important, that is, being actively involved in the baby’s day-to-day care and having contact with their child is an important part of both parental bonding and baby development. 27 It is hypothesised that improved clinical stability of the baby will facilitate increased family involvement in the baby’s care and improve developmental care. Therefore, piloting an assessment tool that could measure the extent of family involvement is wished for.
Collection of cytokine profiles in a subset of the babies will expand scientific knowledge of the processes involved and generate mechanistic hypotheses. One possibility is that cytokine profiles may make it possible to identify babies who are likely to respond to corticosteroids and those who are likely to be non-responders. If non-responders can be identified, it may be possible to target therapy avoiding unnecessary exposure.
Chapter 2 Research objectives
Aim
The aim of the study was to assess the efficacy of very low-dose dexamethasone in facilitating the extubation of ventilator-dependent preterm babies born at < 30 weeks’ gestation who are at high risk of developing BPD.
Hypothesis
Administration of very low-dose dexamethasone to premature babies who are dependent on intermittent positive-pressure respiratory support, via endotracheal intubation, will facilitate extubation.
Research questions
-
How effective is very low-dose dexamethasone at reducing the duration of invasive intermittent positive-pressure ventilation in ventilator-dependent preterm babies born at < 30 weeks’ gestation who are at high risk of developing BPD?
-
What is the safety of very low-dose dexamethasone?
-
– Does very low-dose dexamethasone increase the risk of adverse effects that are seen with high-dose dexamethasone, for example hypertension, hyperglycaemia, confirmed/suspected sepsis, gastrointestinal perforation/necrotising enterocolitis (NEC)?
-
– Does very low-dose dexamethasone therapy result in changes in babies’ cranial ultrasound scans between randomisation and 36 weeks’ postmenstrual age (PMA)?
-
– Does very low-dose dexamethasone therapy result in changes in babies’ neonatal growth?
-
-
Will it be possible to perform a large multicentre randomised controlled trial to assess the 2-year neurodevelopmental outcomes of treatment with very low-dose dexamethasone?
-
Is a novel assessment of the family’s involvement with their baby feasible?
-
Does very low-dose dexamethasone affect changes in babies’ biochemical inflammatory cytokine profile?
Chapter 3 Methods
The main part of the Methods chapter describes, in the interests of transparency, the study as planned.
The final paragraphs of the chapter describe events during the trial and the adaptations that were made to the methods and analysis to respond to these events.
Trial design
This was a multicentre, randomised, masked, parallel-group, placebo-controlled Phase 2b trial to examine the efficacy of very low-dose dexamethasone in facilitating the extubation of ventilator-dependent preterm babies.
The trial was designed as a feasibility study for a subsequent study of the clinical effectiveness of very low-dose dexamethasone in reducing the incidence of BPD at 36 weeks’ PMA. As such, the trial had a surrogate outcome as a primary aim but collected information about a range of clinical outcomes and trial characteristics to support the planning of a subsequent trial.
Babies were randomised in a one-to-one ratio to receive either a very low dose of dexamethasone or a matched placebo. Allocation was masked from the baby’s family, the clinical team, the research team and the statistical analysis team.
The trial was anticipated to take 30 months to complete and aimed to recruit, over 18 months, a total of 94 babies in up to 25 sites across the UK.
Parents of babies recruited at Leeds General Infirmary, Bradford Royal Infirmary and Hull Royal Infirmary were asked to consent to their baby having samples taken for cytokine estimation to allow the modelling of the babies’ inflammatory networks.
In addition, a novel diary to capture family experiences was piloted.
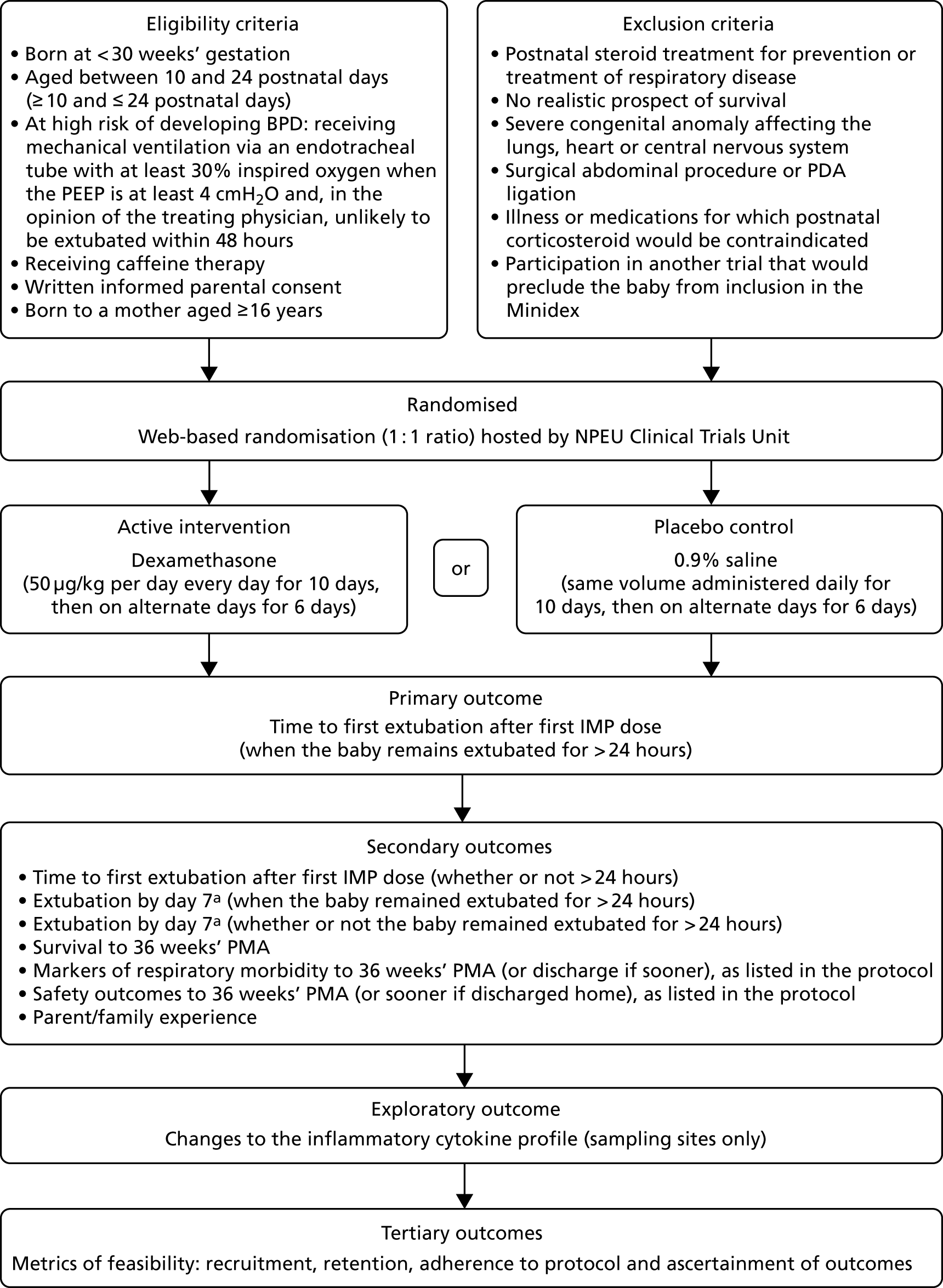
The trial flow diagram is shown in Figure 1.
FIGURE 1.
Trial flow diagram. a, References to ‘day 7’ refer to 7 completed days from the first Investigative Medicinal Product (IMP) dose. NPEU, National Perinatal Epidemiology Unit; PDA, patent ductus arteriosus; PEEP, positive end-expiratory pressure.
Trial registration
The study is registered with the International Standard Randomised Controlled Trial Number (ISRCTN) registry as number ISRCTN81191607.
Ethics and research governance approval
The protocol was reviewed by the North West – Liverpool Central Research Ethics Committee (REC) that gave a favourable opinion (REC reference number 16/NW/0396).
The sponsor was the University of Liverpool (sponsor ID: UoL001206).
Participants
Inclusion criteria
Inclusion criteria included:
-
babies born at < 30 weeks’ gestation
-
babies aged between 10 and 24 postnatal days (i.e. ≥ 10 and ≤ 24 postnatal days)
-
babies at high risk of developing BPD – receiving mechanical ventilation, via an endotracheal tube, receiving at least 30% inspired oxygen when the positive end-expiratory pressure is at least 4 cmH2O and, in the opinion of the treating physician, unlikely to be extubated within 48 hours
-
babies receiving caffeine therapy
-
written informed parental consent
-
babies born to a mother aged ≥ 16 years.
Exclusion criteria
Exclusion criteria included babies:
-
receiving postnatal steroid treatment for prevention or treatment of respiratory disease
-
with no realistic prospect of survival
-
with a severe congenital anomaly affecting the lungs, heart or central nervous system
-
having undergone a surgical abdominal procedure or patent ductus arteriosus (PDA) ligation
-
who are ill or receiving medication for which postnatal corticosteroid would be contraindicated (e.g. an active fungal infection, confirmed or suspected acute sepsis, acute NEC/focal intestinal perforation or cyclo-oxygenase therapy)
-
participating in another trial that would preclude the baby from inclusion in the MINIDEX feasibility trial.
Setting
The trial took place in neonatal units across the UK.
Recruiting sites were tertiary neonatal units that screened potential participants, undertook recruitment, randomised participants, and started trial procedures and administration of Investigative Medicinal Product (IMP) (n = 11; see Appendix 1).
Sites were secondary or tertiary neonatal units that completed trial procedures if participants were transferred from recruiting sites for clinical reasons (n = 26; see Appendix 2).
When more than one trial was open in a recruiting site, the potential for co-recruitment was discussed between the Chief Investigator and Clinical Lead of the MINIDEX study, and the leaders of other trials. Participants could be co-recruited to the following trials: ELFIN (Enteral LactoFerrin In Neonates), PlaNeT-2 (Platelets for Neonatal Transfusion – study 2), PREVAIL (PREVenting infection using Antimicrobial Impregnated Long lines), SPRING (Sustainable PRogramme Incorporating Nutrition and Games (for maximising development, growth and survival) and REACT (Real Time Continuous Glucose Monitoring in the Newborn). Participants could not be co-recruited to Baby-Oscar (Outcome after Selective Early Closure of PDA) and Firefly.
Screening and recruitment
A daily review of the baby’s status allowed the attending clinical team, at recruiting sites, to identify babies who were likely to be eligible for the trial. Trial eligibility was confirmed by a medically qualified clinician who was on the delegation log.
Once a baby had been identified as eligible, a full verbal and written explanation of the trial (via the Parent Information Leaflet) was provided for the parents to consider. Parents who did not speak English were approached only if an unrelated adult interpreter was available.
Following provision of information about the trial, parents had time to consider participation and were given the opportunity to discuss the trial with their family and health-care professionals before they were asked whether or not they would be willing to take part in the trial.
When parents of eligible babies were happy for the babies to participate, the parents provided informed written consent by means of a dated signature.
Randomisation
Randomisation was managed via a secure web-based randomisation facility hosted by the National Perinatal Epidemiology Unit (NPEU) Clinical Trials Unit (CTU), at the University of Oxford, with telephone backup available all at times (i.e. 24/7, 365 days a year). The randomisation program used a minimisation algorithm to ensure balance between the trial groups, with respect to collaborating hospital, sex, multiple births, gestational age at birth and existing diuretic therapy for the 24 hours prior to randomisation. Babies from multiple births were randomised individually.
Cytokines
Parents of babies recruited at specified sites were asked to consent to having opportunistic microsamples of blood and endotracheal tube secretions taken at study entry, and within the 4-hour period prior to the IMP dose on days 4, 7, 10 and 14 after randomisation.
A maximum of 0.5 ml of blood (total blood volume of 1.5 ml, to a maximum of 2.5 ml over the 14 days) was obtained as an ‘extra sample’ by trained staff at the time of a clinically indicated capillary blood sampling episode on these five occasions. Endotracheal tube secretions were obtained at the time of clinically indicated endotracheal tube suction (in ventilated babies only). The endotracheal tube suction was performed by trained staff.
Cytokine profiling
The cytokine analysis has yet to be performed. Cytokines will be profiled in plasma and endotracheal tube secretions by multiplex immunoassay (Bio-Plex Pro™; Bio-Rad, Laboratories, Inc., Hercules, CA, USA) on a Luminex® 200™ System (Luminex Corporation, Austin, TX, USA) cytometer equipped with Bio-Plex Manager software (version 6.11; Bio-Rad, Laboratories, Inc.). 28 The following multiplex immunoassay kits will be used:
-
Bio-Plex Pro Human Cytokine 21-plex Assay (#MF0005KMII; Bio-Rad, Laboratories, Inc.). Analytes to be examined include interleukin (IL) 1 alpha (IL-1α), IL-2 receptor alpha (IL-2Rα), IL-3, IL-12 (specifically the p40 subunit of IL-12), IL-16, IL-18, CTACK (cutaneous T cell-attracting chemokine), growth-related oncogene (GRO) alpha (GRO-α), hepatocyte growth factor (HGF), interferon (IFN) alpha 2 (IFN-α2), leukaemia inhibitory factor (LIF), monocyte chemoattractant protein (MCP) 3 (MCP-3), macrophage colony-stimulating factor (M-CSF), macrophage migration inhibitory factor (MIF), monokine induced by gamma interferon (MIG), β-nerve growth factor (NGF), stem cell factor (SCF), stem cell growth factor (SCGF) beta (SCGF-β), stromal cell-derived factor (SDF) one alpha (SDF-1α), tumour necrosis factor (TNF) beta (TNF-β) and TNF-related apoptosis-inducing ligand (TRAIL).
-
Bio-Plex Pro Human Cytokine 27-plex Assay (#M500KCAF0Y; Bio-Rad, Laboratories, Inc.). Analytes to be examined include basic fibroblast growth factor (bFGF), eotaxin, granulocyte colony-stimulating factor (G-CSF), granulocyte–macrophage colony-stimulating factor (GM-CSF), IFN gamma (IFN-γ), IL 1 beta (IL-1β), IL-1 receptor antagonist (IL-1RA), IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 (specifically the p70 subunit of IL-12), IL-13, IL-15, IL-17, IFN-γ-induced protein 10 (IP-10), MCP 1 (MCP-1), methyl CpG-binding domain protein 1 (MBD1)-containing chromatin-associated factor (MCAF), macrophage inflammatory protein (MIP) one alpha (MIP–1α), MIP 1 beta (MIP-1β), platelet-derived growth factor (PDGF) two B subunits (PDGF-BB), regulated upon activation, normal T cell expressed and secreted (RANTES), TNF alpha (TNF-α) and vascular endothelial growth factor (VEGF).
Plasma samples will be diluted in accordance with the manufacturer’s recommendations. For the endotracheal tube secretions, a small aliquot (i.e. 20 µl) will be set aside for protein concentration determination in order to standardise cytokine concentrations to total protein given the dilutional effect in the collection of these specimens.
Sample size and power
For the primary outcome of time to extubation, assuming 90% power and two-sided 5% level of significance, a log-rank test with a sample size of 70 babies (35 in each trial arm) was required to detect a HR of 2.3 for a control group HR of 1.0. This HR is the lower end of the CI found by the retrospective study (adjusted HR 6.24, 95% CI 2.34 to 16.63). 25 This sample size allowed for the detection of a clinically important absolute risk reduction of 36% for the secondary outcome of failure to extubate by day 7 of the intervention period, with 90% power, a two-sided 5% level of statistical significance, and a control group incidence of 80%. (This is consistent with control group event rates reported in previous trials. 25,26) As other previously published randomised trials of dexamethasone report failure to extubate by day 7 as a primary outcome, it was considered in this study an important secondary outcome to consider in the sample size calculations.
As all centres routinely collect the required data, a negligible loss to follow-up was anticipated. However, as the primary analysis was per protocol, babies were censored for the primary outcome (and other time-to-event outcomes) if the baby either died, if treatment was discontinued before extubation or if the baby remained intubated at day 16 (i.e. the last day of the intervention period). These babies were excluded from the analysis of other secondary outcomes.
Studies and published data sets show that 13.5–18% of babies < 30 weeks’ gestational age have late-onset sepsis;2,29 7% will be treated for NEC;1 and 14% will be treated for PDA with cyclo-oxygenase inhibitors. 30 It was estimated that one-third of late-onset sepsis, half of NEC and one-quarter of treated PDA would occur in the study period. Therefore, it was expected that in up to 14% of babies the study treatment would be permanently discontinued for clinical reasons. It was estimated that 10% of babies would either die before extubation or remain intubated at day 16. Deaths were reported to the Data Monitoring Committee (DMC), which monitored for any safety imbalances or differences in censoring rates between the trial groups. The per-protocol analysis assumed that approximately 23% of all babies would be censored or excluded, giving a final overall sample size target of at least 94 babies.
Cytokine profile sample size
With regard to the secondary outcome of inflammatory network modelling, the samples obtained from specified sites (approximately 16 babies) would be sufficient for detecting differences in absolute cytokine concentrations. This assumption was based on work conducted in the assisted conception setting rather than power calculations, as the principal purpose of measuring cytokines in the current context is not to determine changes in individual cytokines, but rather to understand differences in network evolution in relation to response to dexamethasone and those characterising BPD.
Interventions
Dexamethasone (Hospira, Lake Forest, IL, USA; 3.3 mg/ml solution of dexamethasone for injection) was supplied as a clear sterile solution at a concentration of 3.3 mg per 1 ml in 2-ml vials. Cartons containing 14 single-use vials were provided.
Doses of 50 µg/kg (0.015 ml/kg of 3.3 mg/ml solution) dexamethasone were administered daily on days 1–10 after randomisation (i.e. 10 doses), then on alternate days (i.e. on days 12, 14 and 16), making a total of 13 doses. Doses were calculated on the current working weight of the baby at the time of randomisation (but could be increased in line with subsequent working weight as per local practice) and diluted to the appropriate volume with dextrose or saline. The current working weight was designated by individual units as the weight to manage IMPs with. While the baby had an intravenous line in situ (peripheral or central), each dose was given as a short intravenous infusion over 10–15 minutes. Once intravenous access had been removed, the IMP could be given via the nasogastric tube or orally. The doses were prepared and diluted in the same manner for oral, nasogastric and intravenous administration.
All 13 doses were given unless there was an adverse effect necessitating stoppage, as described below.
The placebo was supplied as a clear sterile solution of 0.9% saline solution for injection. Cartons identical to those for dexamethasone, each containing 14 identical single-use vials, were provided. The volume of IMP to be withdrawn from the vial was calculated following the calculations for dexamethasone dosing and then diluted with dextrose or normal saline for administration.
Supplies of the active product and placebo were prepared in a Medicines and Healthcare products Regulatory Agency (MHRA)-recognised manufacturing facility at Guy’s and St Thomas’s NHS Foundation Trust (London, UK). The active product and placebo were released by the qualified person (i.e. QP released).
Open-label treatment
If the clinical condition of a baby warranted intervention, treatment with open-label corticosteroids after the intervention period could be given and was recorded.
Stopping trial medications
Trial medications could be stopped under the following circumstances:
-
parental wish
-
intolerable adverse reaction to study drug, as determined by the treating clinician (e.g. anaphylaxis)
-
requirement for cyclo-oxygenase therapy
-
if deemed by the treating clinician to be in the baby’s best interest (e.g. overwhelming sepsis or need for open-label corticosteroid).
If the trial medication was stopped, data were collected for the remainder of the trial. No further blood and bronchoalveolar samples were taken if the baby was participating in the cytokine substudy. Samples that were taken inadvertently after the IMP was stopped were analysed.
When parents chose to withdraw their baby from the trial, permission was sought to continue data collection and/or use data up to the point of withdrawal from the trial.
Parent diary
Being actively involved in the baby’s day-to-day care and having contact with their child is an important part of both parental bonding and baby development. The parent diary was developed in conjunction with the parents who provided patient and public involvement (PPI) to the trial. It was designed to capture details about the amount of contact and interaction the family were able to have with their baby during administration of the IMP. The way the family felt the baby reacted to their contact was recorded. It was hoped that this would allow an assessment of the impact of the treatment on the family and their experience of the baby’s neonatal journey to be made. The diaries were trialled by a group of parents whose babies were receiving intensive care, and were found to be easy to use.
Parents were invited to complete a daily log of their interactions with their baby. Interactions were divided into ‘cares’ and ‘contact and cuddles’. ‘Cares’ was the amount of parental involvement in the day-to-day care of a preterm baby, such as nappy changes and mouth care. Contact and cuddles were the amount of time the family were able to spend holding the baby. The degree of contact the baby could tolerate was graded from the least contact of hand-holding to the full contact of skin-to-skin, when the baby is placed inside the parent’s clothing for a cuddle.
Outcomes
The objectives and outcomes are described in Table 2. Further details of the severity criteria used to determine the secondary outcome of BPD are included in Appendices 3 and 4.
| Objectives | Outcome measures | Time point(s) of evaluation of this outcome measure (if applicable) |
|---|---|---|
| Primary objective | ||
| To determine if treatment with very low-dose dexamethasone facilitates the extubation (for > 24 hours) of ventilator-dependent preterm babies of < 30 weeks’ gestation who are at high risk of developing BPD | Time to first extubation after first IMP dose when the baby remains extubated for > 24 hours | |
| Secondary objectives | ||
| To determine if the treatment of ventilator-dependent preterm babies of < 30 weeks’ gestation who are at high risk of developing BPD with very low-dose dexamethasone impacts upon: | ||
| 1. Time to first extubation (whether or not > 24 hours) | Time to first extubation after first IMP dose (whether or not > 24 hours) | |
| 2. Extubation by day 7 |
|
Day 7 of the intervention period |
| 3. Survival to 36 weeks’ PMA | Alive at 36 weeks’ PMA (or discharge home if sooner) | 36 weeks’ PMA (or discharge home if sooner) |
| 4. Respiratory morbidity to 36 weeks’ PMA (or discharge home if sooner) |
|
36 weeks’ PMA (or discharge home if sooner) |
| 5. Safety outcomes |
|
During administration of IMP and for 14 days after last dose of intervention (a–d) CrUSS performed prior to randomisation (or when occurring according to standard clinical practice) and at 36 weeks’ PMA ( ± 2 weeks or discharge home if sooner) Weight measured at trial entry, on day 14 (after randomisation) and 36 weeks’ PMA ( ± 2 weeks or discharge home if sooner) Head circumference measured at trial entry, and at 36 weeks’ PMA ( ± 2 weeks or discharge home if sooner) |
| 6. Parent/family experience | Parent/family experience:
|
Log recorded during 16-day intervention period. Although part of the consent process, this is not part of the source data set and is optional for parents and staff to complete |
| Exploratory objective | ||
| Cytokine profile (consenting babies from specified sites only) | Changes to the inflammatory cytokine profile in blood and endotracheal tube secretion fluid (a mechanistic marker of response) | Endotracheal tube secretions and blood samples taken at trial entry, and on days 4, 7, 10 and 14 of the IMP administration schedule |
| Tertiary objectives | ||
| To assess the acceptability and feasibility of the trial in order to inform the design of the subsequent effectiveness trial by reporting metrics related to: | ||
| 1. Recruitment | Recruitment rate | Trial completion |
| 2. Retention | Retention rate | Trial completion |
| 3. Adherence to protocol | Use of open-label therapy and completion of course | Trial completion |
| 4. Completeness of data | Ascertainment of outcomes | Trial completion |
The trial schedule is shown in Table 3.
| Procedure | Time point | 36 weeks PMA | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre-randomisation | Post-consent pre-IMP | Days after randomisation | ||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17–30 | ||||
| Eligibility assessment | ||||||||||||||||||||
| Consent | ||||||||||||||||||||
| Randomisation | ||||||||||||||||||||
| Baseline clinical data | ||||||||||||||||||||
| IMP administration | ||||||||||||||||||||
| Clinical data collection | ||||||||||||||||||||
| Measurement of occipitofrontal circumference | ||||||||||||||||||||
| Measurement of weight | ||||||||||||||||||||
| Blood sample and endotracheal secretion collectiona | ||||||||||||||||||||
| Oxygen reduction test | ||||||||||||||||||||
| Safety reporting | ||||||||||||||||||||
Hypothesis to be tested
To determine if the treatment with very low-dose dexamethasone of ventilator-dependent preterm babies of < 30 weeks’ gestation who are at high risk of developing BPD impacts upon:
-
time to extubation (where the baby remains extubated for > 24 hours)
-
time to extubation (whether or not the baby remains extubated for > 24 hours)
-
rates of extubation by day 7 of the intervention period (where the baby remains extubated for > 24 hours)
-
rates of extubation by day 7 of the intervention period (whether or not the baby remains extubated for > 24 hours)
-
survival to 36 weeks’ PMA (or discharge home if sooner)
-
respiratory morbidity to 36 weeks’ PMA (or discharge home if sooner)
-
cytokine profile
-
safety outcomes
-
the parent/family experience.
Adverse reactions and adverse events
The Reference Safety Information for pharmacovigilance purposes was the Summary of Product Characteristics for the 3.3 mg/ml dexamethasone product, which was manufactured by Hameln Pharmaceuticals Ltd (Gloucester, UK).
Serious adverse events and serious adverse reactions that were anticipated in the trial population were reported on the case report form (CRF).
Other serious adverse events (SAEs) and serious adverse reactions were reported in an expedited manner to NPEU.
The NPEU CTU reviewed each report, requested any additional information and ensured that it was assessed by the Chief Investigator (or their delegate) within the reporting time frame for potential suspected unexpected serious adverse reactions.
Statistical analysis
Demographic factors, clinical characteristics, and service utilisation were summarised with counts (percentages) for categorical variables, mean [standard deviation (SD)] for normally distributed variables and median [interquartile range (IQR) or other percentile range] for other continuous variables.
The original statistical analysis plan entailed calculating the HR plus 95% CIs for the primary outcome, the risk ratio (plus 95% CI) for dichotomous outcomes, and the mean or median difference (plus 95% CI) for other continuous variables. As this was an efficacy study, the plan was to base the primary inference on a per-protocol analysis, excluding babies who were discontinued from the study treatment. For time-to-event analyses, babies who discontinued or died before extubation or who remained intubated at day 16 would have been censored.
The plan was to conduct a secondary analysis of all outcomes on an intention-to-treat basis. There was also a preplanned subgroup analysis to examine the consistency of the effect of dexamethasone on the primary outcome using the statistical test of interaction across the following subgroups: (1) gestational age, (2) singleton versus multiple pregnancy and (3) diuretic use at trial outset.
Analysis of cytokine data
Basic cytokine data analysis would be largely conducted using a combination of Kruskal–Wallis and post hoc Dunn’s tests. Significance levels would be corrected for multiple comparisons by applying Holm’s sequentially selective Bonferroni method. Thereafter, correlation, principal components and factor analyses would be used as recognised tools to investigate mediator inter-relationships. More informative network-based approaches would be used to model inflammatory cytokine–cytokine network interactions using Bayesian methods refined in-house. These networks offer a framework combining uncertainty (probabilities) and logical structure (independence constraints) to represent complex biological phenomena by generating acyclic graph-based models of joint probability distributions representing the conditional independence between related variables. 31 The key element of this approach is that this allows investigators to capture the complex biological features of cytokines, such as antagonism, synergy and functional redundancy, as recently shown by Beal et al. [rather than relying on the increasingly inadequate T helper (Th) cell type 1/2 (Th1/Th2) paradigm]. 31
Two dichotomisations would be applied: one in response to dexamethasone therapy versus placebo and one relating to the time to extubation (groupings for the latter will be determined empirically based on the data emerging from the study), independent of treatment. Both will incorporate a dynamic aspect to the Bayesian model to account for changes in cytokine profiles and inter-relationships over time. Data from each group will also be used to develop modified variational Bayesian state–space models (in which the human-specific text-learning algorithm will be used; https://compbio.dfci.harvard.edu/predictivenetworks/) to generate prior networks. Initialisation fields will be set to 10 seeds and 1000 iterations, and the maximum hidden-state dimension limited to 20. A z-score equivalent to a 98% significance threshold will be used to define the number of edges reaching significance. Networks will be visualised using either the open-source platform Cytoscape (https://cytoscape.org; 24 May 2019) or the open-graph Viz platform, Gephi (https://gephi.org; 24 May 2019). The spatialisation technique applied will be the force-directed Yifan Hu algorithm, whereas a community detection algorithm will be used to detect the underlying graph topology modular structure. It is also planned to use machine-learning-based methods in an attempt to identify the value of specific small groups of cytokines as predictors of outcome by using a support vector machine and a random forest-based combinatorial approach developed in-house. The main focus of these, that is the cytokines, was to offer a starting point for an iterative stepwise empirical identification of outcome predictors based on feature importance ranking.
Given the discontinuation of the trial, and the comparatively limited number of samples collected, it may now not be possible to conduct a Bayesian-based analysis of the results.
Trial management
Project Management Group
The Project Management Group (PMG) supervised the trial on a day-to-day basis. The PMG comprised the Chief Investigator, Lead Clinician, Trial Manager, Trial Statistician, Trials Programming Team, Senior Trials Managers and CTU Director. PMG meetings were held every 6–8 weeks to provide regular central monitoring of the trial, key items reviewed were recruitment, data monitoring reports, safety, incidents, programming and statistical concerns, IMP stock management, and any other relevant topics such as upcoming reports, oversight committee meetings, and amendments.
Trial Steering Committee
The members of the Trial Steering Committee (TSC) are included in Table 4.
| Role | Salutation and name | Position of employment and institution |
|---|---|---|
| Independent chairperson | Professor Henry Halliday | Consultant Neonatologist, retired, Belfast, UK |
| Independent vice-chairperson | Dr Gopi Menon | Consultant Neonatologist, Simpson Centre for Reproductive Health, Royal Infirmary of Edinburgh, Edinburgh, UK |
| Independent | Professor Howard Clark | Consultant Neonatologist, Southampton General Hospital, Southampton, UK |
| Independent | Dr Paul Mannix | Consultant Neonatologist, North Bristol NHS Trust, Southmead Hospital, Bristol, UK |
| Independent | Mrs Vanessa Lett | PPI representative, Leicester, UK |
| Independent | Mrs Nicola Kitchin | PPI representative, Ashton-Under-Lyne, UK |
| Independent | Ms Trish Hepburn | Senior Statistician, Faculty of Medicine and Health Sciences, Queen’s Medical Centre, Nottingham, UK |
| Non-independent | Dr Helen Yates | Clinical Lead, Consultant Neonatologist, Hull Royal Infirmary, Hull, UK |
| Non-independent | Dr Mark Turner | Chief Investigator, Consultant Neonatologist, University Department of Women’s and Child Health, Liverpool Women’s and Children’s Hospital, Liverpool, UK |
| Non-independent | Associate Professor Ed Juszczak | NPEU Clinical Trials Unit Director, NPEU CTU, University of Oxford, Oxford, UK |
| Observer | Mrs Ursula Bowler | Senior Trials Manager, NPEU CTU, University of Oxford, Oxford, UK |
| Observer | Dr Vaneesha Short | MINIDEX Trial Manager, National Perinatal Epidemiology Unit CTU, University of Oxford, Oxford, UK |
| Observer | Ms Karen Wilding | Research Integrity and Governance Manager, University of Liverpool/Liverpool Joint Research Office, Liverpool, UK |
Data Monitoring Committee
The members of the DMC are included in Table 5.
| Role | Salutation and name | Position of employment and institution |
|---|---|---|
| Independent chairperson | Professor Ben Stenson | Consultant Neonatologist, Senior Lecturer, Neonatal Unit, Simpson Centre for Reproductive Health, Royal Infirmary of Edinburgh, Edinburgh, UK |
| Independent | Professor Catherine Hewitt | Deputy Director, Statistician, York Trials Unit, University of York, York, UK |
| Independent | Dr Chris Gale | Senior Lecturer in Neonatal Medicine, Imperial College London; Consultant Neonatologist, Chelsea and Westminster NHS Foundation Trust, London, UK |
| Independent | Professor Sailesh Kotecha | Consultant in Paediatrics, Department of Health, Cardiff University School of Medicine, Cardiff, UK |
Public and patient involvement
A focus group of parents of ex-premature babies provided PPI input on the trial structure and assisted with the development of the family-centred study outcomes. One member of this group was a trial co-applicant. The member contributed to the development of the bespoke parent-facing trial materials, including the diary of care.
Data management
All trial data were collected using bespoke CRFs and processed in line with the NPEU CTU standard operating procedures.
Case report form entries were considered source data if the CRF is the site of the original recording (e.g. there is no other written or electronic record of data, for example medical records). All documents are stored safely in confidential conditions.
Minimal personal data were collected and stored at NPEU CTU. The infant’s name and any other identifying details were stored separately and linked by the trial number. Data were stored in an electronic database in which the participant was identified by a trial-specific number. This information will be stored for a period not less than 25 years in order to follow up health-related issues that may become relevant in the future. After the trial has been completed and the reports published, the data will be archived in a secure physical or an electronic location with controlled access.
Circumstances during the trial
The grant award start date was November 2015, with a planned recruitment start date of March 2016 and recruitment completion date of August 2017. Trial set up took longer than anticipated and recruitment did not commence until 21 July 2017.
The reasons for this 16-month delay were:
-
Change in trial sponsor.
Originally the MINIDEX trial was to be sponsored by Leeds Teaching Hospitals NHS Trust. All documentation was prepared and submitted to Leeds Teaching Hospitals NHS Trust for review in this role. The original contract between Leeds Teaching Hospitals NHS Trust and the Department of Health and Social Care was signed on 28 August 2015. However, after a MHRA inspection it became clear to Leeds Teaching Hospitals NHS Trust that it would not be in a position to continue with sponsorship. After negotiations with the funder, the University of Liverpool agreed to take on sponsorship if the Chief Investigator was changed to Dr Mark Turner, who is employed by the University of Liverpool. The change of sponsor took a long time to facilitate and, ultimately, it was 21 October 2016 before the Deed of Novation to transfer sponsorship to the University of Liverpool was sent to all parties. Research agreements with recruiting sites could not be arranged and signed until the transfer of sponsorship was complete, resulting in a significant delay to site initiation.
-
Sourcing matched components for the placebo.
Sourcing and preparing IMP supplies for release by the QP at Guy’s and St Thomas’ NHS Foundation Trust Pharmacy Manufacturing Unit took over 18 months, rather than the 4 months in the study plan. This delay arose from difficulties with sourcing placebo. The manufacturers of the active agent were not able to supply empty vials because of fears about counterfeit medicines. When suitable vials were located after a global search there were extensive difficulties with sourcing an appropriate seal for the vial. After a global search a matching seal was identified, but it was not possible to sterilise the vials with this seal. A seal that could be sterilised was found, but this seal did not provide a perfect match to the seal used with the active agent. The study team conducted a risk analysis and decided the risk of unblinding was sufficiently low to justify using an imperfectly matched seal. The sponsor agreed to this method of working. The funder was informed, and the placebo was QP released on 20 July 2017.
Once the trial opened, the recruitment rate was lower than expected. The trial team contacted sites frequently to identify issues and facilitate their work on recruitment, including protocol amendments.
The proportion of discontinuations was 55%; much higher than the 23% estimated in the sample size calculation. This meant that the number of participants who could contribute to a time-to-event analysis was reduced substantially. The rate of discontinuation could not be reduced partway through the trial because the key reasons for discontinuation were the requirement of open-label steroids (in the opinion of the treating clinician) and overwhelming sepsis (for which antibiotics would be contraindicated with the active drug).
Towards the end of the contracted funding period the study team approached the funders for a no-cost extension.
In April 2018, the funders notified the study team that an extension was not appropriate. The funders reported that the study team had obtained a sufficient number of data sets to conduct the final analysis of the primary outcome. Given the rate of recruitment it would not be a good use of resources to continue recruitment and generate sufficient data sets to conduct the other study analyses.
The batches of trial supplies expired on 30 April 2018. In the absence of further funding it was not possible to order new batches. Recruitment closed on 14 April 2018. The last evaluation of the final participant took place on 24 June 2018. The database was locked on 4 September 2018.
Adaptations made during the trial
The study team made strenuous efforts to identify, and when possible, address the issues relating to trial supplies, the recruitment rate and the discontinuation rate.
Revised statistical analysis
The statistical plan was revised because of the much reduced sample size. Although 22 babies were recruited, the number of events for the primary outcome was much lower than this. A total of 14 participants were censored (mainly as a result of a high rate of discontinuations of the study drug), so only eight participants presented the event of interest for the time-to-event analysis. The main deviations from the analysis described in the protocol were as follows:
-
No comparative analyses were performed for any outcomes. Only descriptive statistics were to be reported by trial arm.
-
Outcomes were reported for all babies randomised minus post-randomisation exclusions.
-
No confidence intervals or p-values were reported.
-
There were no adjustments made for the minimisation factors used at randomisation or clustering owing to multiple births.
-
Subgroup analyses were not carried out.
-
Exploratory analyses on the use of diuretics and oxygen in relation to treatment effect were not performed.
Time to first extubation outcomes were summarised with medians and IQRs. The number of babies in whom the intervention was discontinued or who died before the event of interest occurred, and the number of babies still intubated at day 16 was reported but not included in this summary measure. Extubation by day 7 outcomes were reported with counts and percentages. The numbers of babies who died or in whom the intervention was discontinued before day 7 was reported and excluded from the denominator. Outcomes recorded at 36 weeks’ PMA or discharge (if sooner), and safety outcomes, were reported for all babies randomised and still alive at 36 weeks’ PMA or discharge. These outcomes were summarised with counts (percentages) for categorical variables, mean (SD) for normally distributed variables and median (IQR) for other non-normally distributed continuous variables.
Serious adverse events are listed, by allocation, for all babies randomised.
Chapter 4 Results
The database was locked on 4 September 2018. The grant closed on 31 October 2018. Recruitment stopped on 14 April 2018 after 22 babies, out of the planned sample size of 94, had been randomised to the trial over a period of 9 months. This was a decision made by the funder owing to poor recruitment. It was agreed that the final report should provide a descriptive analysis only because of the small number of babies recruited to the trial.
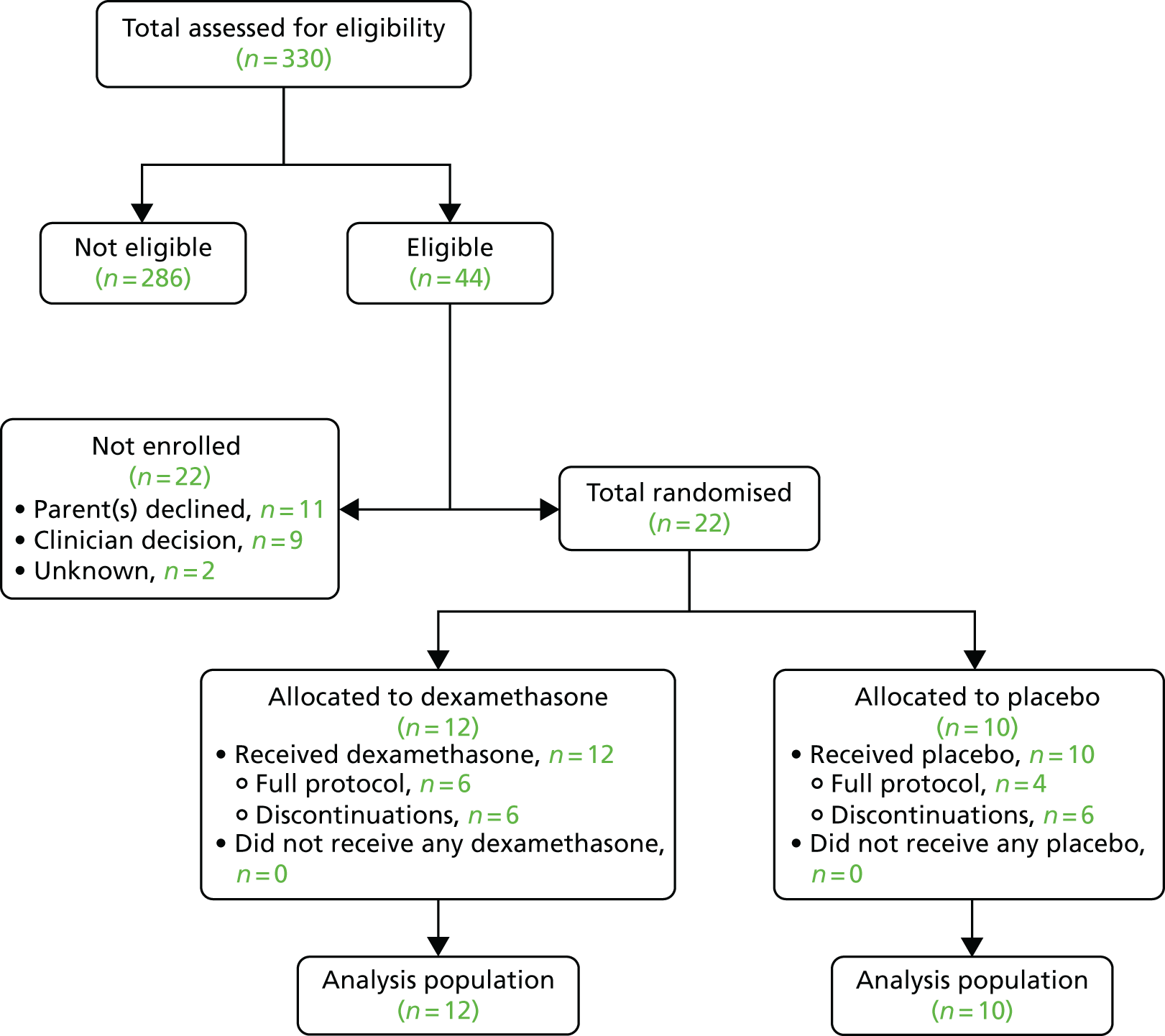
Figure 2 presents the flow of participants in the trial. Among the 22 babies randomised, the intervention was discontinued in 12 (see Table 13) and none was withdrawn from the trial. Maternal and neonatal characteristics and all the outcomes are reported for all babies randomised. There were no post-randomisation exclusions.
FIGURE 2.
Flow of trial participants in the trial.
The participating units are shown in Table 6.
| Neonatal unit | Trial arm, n (%) | |
|---|---|---|
| Dexamethasone | Placebo | |
| Hull Royal Infirmary | 2 (16.7) | 3 (30.0) |
| Leicester Royal Infirmary | 1 (8.3) | 2 (20.0) |
| Liverpool Women’s Hospital | 3 (25.0) | 0 (0.0) |
| Birmingham Women’s and Children’s NHS Foundation Trust | 2 (16.7) | 0 (0.0) |
| University Hospital Coventry | 1 (8.3) | 1 (10.0) |
| Bradford Royal Infirmary | 1 (8.3) | 1 (10.0) |
| Leeds General Infirmary | 2 (16.7) | 0 (0.0) |
| Royal Preston Hospital | 0 (0.0) | 2 (20.0) |
| Royal Victoria Infirmary (Newcastle upon Tyne) | 0 (0.0) | 1 (10.0) |
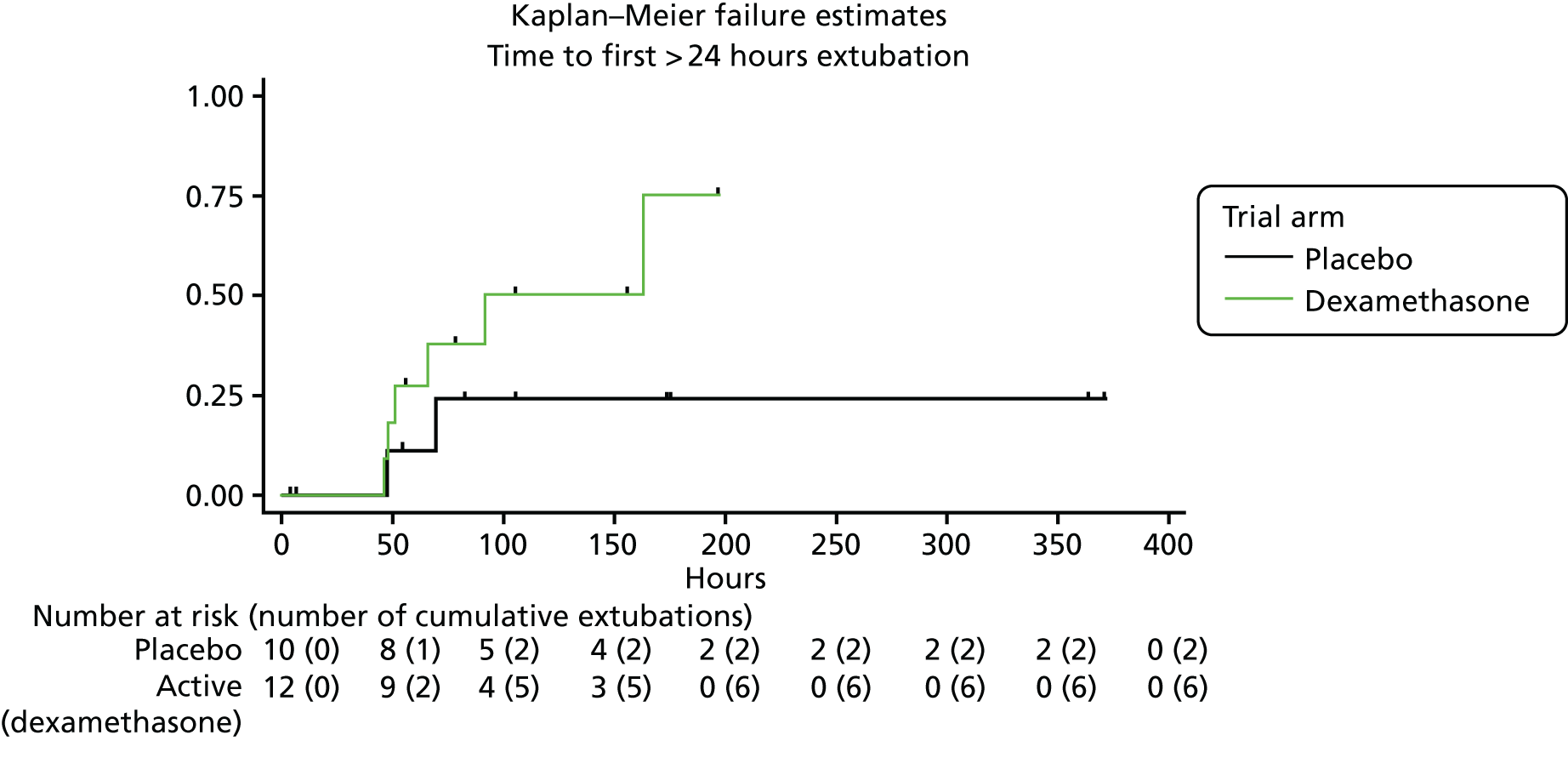
The maternal and neonatal characteristics of the participating babies are shown in Table 7. The primary outcome is shown in Table 8 and Figure 3.
| Characteristic | Trial arm | |
|---|---|---|
| Dexamethasone | Placebo | |
| Total number of babies | 12 | 10 |
| Maternal characteristics | ||
| Maternal age (years) | ||
| Median (IQR) | 26 (21–31) | 28 (23–34) |
| Mother’s ethnic group, n (%) | ||
| White | 11 (91.7) | 9 (90.0) |
| Mixed | 0 (0.0) | 0 (0.0) |
| Asian | 1 (8.3) | 0 (0.0) |
| Black | 0 (0.0) | 1 (10.0) |
| Arab | 0 (0.0) | 0 (0.0) |
| Other | 0 (0.0) | 0 (0.0) |
| Unknown | 0 (0.0) | 0 (0.0) |
| Neonatal characteristics | ||
| Gestational age at birth (completed weeks)a | ||
| Median (IQR) | 25 (24–25) | 25 (23–26) |
| ≤ 25, n (%) | 10 (83.3) | 7 (70.0) |
| 26–27, n (%) | 2 (16.7) | 1 (10.0) |
| 28–29, n (%) | 0 (0.0) | 2 (20.0) |
| Received antenatal steroids | ||
| Number (%) of babies | 10 (83.3) | 7 (70.0) |
| < 4 hours before birth | 2 (20.0) | 1 (14.3) |
| ≥ 4 hours before birth | 8 (80.0) | 6 (85.7) |
| 1 dose | 1 (12.5) | 1 (16.7) |
| 2 doses | 4 (50.0) | 4 (66.7) |
| > 2 doses | 0 (0.0) | 0 (0.0) |
| Not known | 3 (37.5) | 1 (16.7) |
| Time between rupture of membranes and birth, n (%) | ||
| < 24 hours | 6 (50.0) | 7 (70.0) |
| ≥ 24 hours | 4 (33.3) | 3 (30.0) |
| Missing | 2 | 0 |
| Clinical chorioamnionitis evidence | ||
| Number (%) of babies | 0 (0.0) | 1 (10.0) |
| Level of neonatal unit at site of birth, n (%) | ||
| I | 0 (0.0) | 0 (0.0) |
| II | 2 (16.7) | 0 (0.0) |
| III | 10 (83.3) | 10 (100.0) |
| Other | 0 (0.0) | 0 (0.0) |
| Received diuretics for > 24 hours at randomisation, n (%)a | ||
| Number (%) of babies | 3 (25.0) | 2 (20.0) |
| Actual mode of delivery, n (%) | ||
| Vaginal delivery | 10 (83.3) | 6 (60.0) |
| Caesarean section | 2 (16.7) | 4 (40.0) |
| Baby’s sex, n (%)a | ||
| Male | 8 (66.7) | 4 (40.0) |
| Female | 4 (33.3) | 6 (60.0) |
| Indeterminate | 0 (0.0) | 0 (0.0) |
| Birth weight (g) | ||
| Mean (SD) | 730.4 (175.1) | 756.5 (191.1) |
| Birth weight centile | ||
| Mean (SD) | 43.5 (32.5) | 45.6 (20.1) |
| Birth weight centile cut-off points, n (%) | ||
| < Second centile | 1 (8.3) | 0 (0.0) |
| < Ninth centile | 2 (16.7) | 1 (10.0) |
| Postnatal age (days) at trial entry | ||
| Median (IQR) | 13 (11–20) | 16 (14–18) |
| 10–14, n (%) | 7 (58.3) | 3 (30.0) |
| 15–19, n (%) | 2 (16.7) | 6 (60.0) |
| 20–24, n (%) | 3 (25.0) | 1 (10.0) |
| Multiple pregnancy, n (%)a | ||
| Singleton | 11 (91.7) | 8 (80.0) |
| Multiple | 1 (8.3) | 2 (20.0) |
| Birth order (only for multiple births), n (%) | ||
| First born | 1 (100.0) | 1 (50.0) |
| Second born | 0 (0.0) | 1 (50.0) |
| Third born | 0 (0.0) | 0 (0.0) |
| Temperature (°C) at admission to neonatal unit | ||
| Mean (SD) | 36.8 (1.1) | 37.4 (1.1) |
| Not recorded, n (%) | 0 (0.0) | 2 (20.0) |
| Worst base deficit in first 24 hours after birth | ||
| Mean (SD) | 10.9 (5.7) | 9.6 (5.1) |
| Apgar score at 5 minutes | ||
| Median (IQR) | 5 (0–8) | 5 (2–8) |
| Not recorded, n (%) | 3 (25.0) | 1 (10.0) |
| CRIB II score | ||
| Median (IQR) | 15 (12–16) | 15 (9–16) |
| Missing, n | 0 | 2 |
| CRIB score level I (1–5), n (%) | 0 (0.0) | 0 (0.0) |
| CRIB score level II (6–10), n (%) | 1 (8.3) | 3 (37.5) |
| CRIB score level III (11–15), n (%) | 8 (66.7) | 3 (37.5) |
| CRIB score level IV (> 15), n (%) | 3 (25.0) | 2 (25.0) |
| Ibuprofen treatment received before trial entry | ||
| Number (%) of babies | 4 (33.3) | 2 (20.0) |
| Air leak requiring chest drain before trial entry | ||
| Number (%) of babies | 0 (0.0) | 0 (0.0) |
| Hydrocortisone treatment received before trial entry | ||
| Number (%) of babies | 2 (16.7) | 0 (0.0) |
| Baby’s actual weight at trial entry (g) | ||
| Mean (SD) | 862.1 (252.7) | 851.1 (227.5) |
| Head circumference (cm) at trial entryb | ||
| Mean (SD) | 22.9 (1.5) | 23.9 (2.6) |
| Missing, n | 2 | 4 |
| Ventilation method at trial entry, n (%) | ||
| IPPV | 8 (66.7) | 10 (100.0) |
| HFOV | 4 (33.3) | 0 (0.0) |
| FiO2 at trial entry (%) | ||
| Mean (SD) | 49.8 (12.9) | 41.2 (8.0) |
| Mean airway pressure (cmH20) at trial entry | ||
| Mean (SD) | 8.9 (3.9) | 11.5 (3.4) |
| Most recent cranial ultrasound results at trial entry (non-exclusive), n (%) | ||
| Cranial ultrasound not performed | 1 (8.3) | 0 (0.0) |
| No abnormality seen | 4 (36.4) | 7 (70.0) |
| IVH (worst on either side) | ||
| Grade I or II without ventricular dilatation | 5 (45.5) | 2 (20.0) |
| Severe IVH with ventricular dilatation (grade III) | 0 (0.0) | 1 (10.0) |
| IVH causing intraparenchymal bleeding (grade IV) | 1 (9.1) | 0 (0.0) |
| Hydrocephalus | 0 (0.0) | 1 (10.0) |
| PVL | 0 (0.0) | 0 (0.0) |
| Other white-matter injury | 1 (9.1) | 0 (0.0) |
| Outcome | Trial arm | |
|---|---|---|
| Dexamethasone | Placebo | |
| Total number of babies | 12 | 10 |
| Censored (n) | 6 | 8 |
| Discontinuations (before/without extubation) | 6 | 6 |
| Did not have extubation criteria at day 16 | 0 | 2 |
| Not censored/number of events | 6 | 2 |
| Time (hours) to first extubation after first IMP dose (if extubated for > 24 hours) | ||
| Median (IQR) | 58.5 (47.9–91.5) | 58.4 (47.3–69.5) |
FIGURE 3.
Kaplan–Meier failure estimates of the primary outcome.
The secondary outcomes are shown in Table 9.
| Outcome | Trial arm | |
|---|---|---|
| Dexamethasone | Placebo | |
| Total number of babies | 12 | 10 |
| Censored, n | 5 | 4 |
| Discontinuations (before/without extubation) | 5 | 4 |
| Not been extubated by day 16 | 0 | 0 |
| Not censored/number of events, n | 7 | 6 |
| Time (hours) to first extubation after first IMP dose (whether or not > 24 hours) | ||
| Median (IQR) | 65.7 (47.9–91.5) | 58.4 (18.3–123.4) |
| Extubation outcomes, n | 8 | 6 |
| Extubation by day 7 (extubated for > 24 hours), n (%)a | 5 (62.5) | 2 (33.3) |
| Extubation by day 7 (whether or not > 24 hours), n (%)a | 5 (62.5) | 4 (66.7) |
| Alive at 36 weeks’ PMA or discharge if sooner, n (%) | 10 (83.3) | 9 (90.0) |
| Total duration (days) of invasive ventilation through ET | ||
| Median (IQR) | 23 (20–27) | 31 (20–47) |
| Total duration (days) of non-invasive respiratory support nasal CPAP, nasal ventilation or high-flow oxygen therapy | ||
| Median (IQR) | 40 (27–50) | 40 (28–46) |
| Total duration (days) of supplemental oxygen | ||
| Median (IQR) | 15 (2 to 19) | 4 (0 to 8) |
| Open-label treatment with corticosteroids received after randomisation (cumulative dose), n (%) | 5 (41.7) | 8 (80.0) |
| < 1000 µg/kg | 2 (40.0) | 4 (50.0) |
| ≥ 1000 µg/kg | 3 (60.0) | 4 (50.0) |
| Diuretics received for > 48 hours after randomisation, n (%) | 8 (66.7) | 9 (90.0) |
| Additionally received diuretics before randomisation | 7 (87.5) | 7 (77.8) |
| Did not receive diuretics before randomisation | 1 (12.5) | 2 (22.2) |
| BPD assessment at 36 weeks’ PMA or discharge if sooner, n (%) | 10 | 9 |
| No BPD | 0 (0.0) | 0 (0.0) |
| BPD | 10 (100.0) | 9 (100.0) |
| Mild | 2 (20.0) | 0 (0.0) |
| Moderate | 2 (20.0) | 2 (22.2) |
| Severe | 6 (60.0) | 7 (77.8) |
The outcomes of the diary of care are shown in Table 10.
| Outcome | Trial arm | |
|---|---|---|
| Dexamethasone | Placebo | |
| Total number of diaries of care returned | 7 | 5 |
| Time (days) to first cuddle | ||
| Median (IQR) | 5 (4–6) | 12 (2–22) |
| Missing | 5 | 3 |
| Time (days) to first skin-to-skin | ||
| Median (IQR) | 4 (3–4) | 2 (0–3) |
| Missing | 5 | 3 |
| Duration of contact (hours) a family member has with baby | ||
| Cuddle | ||
| Median (IQR) | 13 (13–13) | 2 (1–3) |
| Missing | 6 | 3 |
| Skin-to-skin | ||
| Median (IQR) | 15 (9–21) | 9 (3–15) |
| Missing | 5 | 3 |
| Still touch | ||
| Median (IQR) | 1 (0–1) | 2 (0–3) |
| Missing | 5 | 3 |
| Containment | ||
| Median (IQR) | 1 (0–11) | 1 (0–3) |
| Missing | 4 | 2 |
| Frequency of contact a family member has with baby (number of contacts) | ||
| Cuddle | ||
| Median (IQR) | 0 (0–1) | 0 (0–1) |
| Missing | 0 | 0 |
| Skin-to-skin | ||
| Median (IQR) | 0 (0–5) | 0 (0–2) |
| Missing | 0 | 0 |
| Still touch | ||
| Median (IQR) | 0 (0–1) | 1 (1–1) |
| Missing | 0 | 0 |
| Containment | ||
| Median (IQR) | 1 (0–2) | 1 (0–2) |
| Missing | 0 | 0 |
| Average proportion of baby’s response to contact and cuddles | ||
| Well | 81.0 | 47.0 |
| Neutral | 8.0 | 42.0 |
| Not well | 0.0 | 2.0 |
| Missing | 1 | 0 |
| Time (days) to first family care | ||
| Median (IQR) | 1 (1–4) | 4 (1–15) |
| Missing | 4 | 1 |
| Frequency of family care in first week (number of contacts) | ||
| Median (IQR) | 0 (0–6) | 1 (0–5) |
| Missing | 0 | 0 |
| Average proportion of baby’s response to contact and cuddles | ||
| Nappy changes | 40.0 | 59.0 |
| Skin cares | 25.0 | 30.0 |
| Mouth care | 37.0 | 58.0 |
| Missing | 0 | 0 |
| Average proportion of baby’s response to family care | ||
| Well | 38.0 | 51.0 |
| Neutral | 3.0 | 17.0 |
| Not well | 2.0 | 12.0 |
Safety outcomes are shown in Table 11, with SAEs presented in Table 12.
| Outcome | Trial arm | |
|---|---|---|
| Dexamethasone | Placebo | |
| Total number of babies | 12 | 10 |
| Hypertension, n (%) | 0 (0.0) | 1 (10.0) |
| Hyperglycaemia, n (%) | 3 (25.0) | 2 (20.0) |
| Sepsis, n (%) | ||
| Suspected | 11 (91.7) | 9 (90.0) |
| Received antibiotics for ≤ 72 hours | 2 (18.2) | 0 (0.0) |
| Received antibiotics for > 72 hours | 9 (81.8) | 9 (100.0) |
| Culture-positive sepsis | 2 (22.2) | 4 (44.4) |
| Did not receive antibiotics | 0 (0.0) | 0 (0.0) |
| Spontaneous gastrointestinal perforation or NEC | ||
| Total number (%) of babies | 0 (0.0) | 3 (30.0) |
| Spontaneous gastrointestinal perforation, n (%) | 0 (0.0) | 0 (0.0) |
| NEC, n (%) | 0 (0.0) | 1 (10.0) |
| Unclear, n (%) | 0 (0.0) | 2 (20.0) |
| Gastrointestinal haemorrhage (not associated with gastrointestinal perforation or NEC) | ||
| Total number (%) of babies | 0 (0.0) | 0 (0.0) |
| Clinically important deterioration in cranial ultrasound | ||
| Total number (%) of babies | 1 (9.1) | 1 (10.0) |
| Cranial ultrasound not carried out | 1 | 0 |
| New grade III for IVH | 0 (0.0) | 0 (0.0) |
| New grade IV for IVH | 1 (100.0) | 0 (0.0) |
| New PVL | 0 (0.0) | 1 (100.0) |
| New hydrocephalus | 0 (0.0) | 0 (0.0) |
| SDS for weight (g) at study entrya | ||
| Mean (SD) | –1.1 (1.0) | –1.4 (0.6) |
| Missing | 0 | 0 |
| SDS for weight (g) at day 14 after randomisationa | ||
| Mean (SD) | –1.4 (1.1) | –1.4 (0.8) |
| Missing | 1 | 0 |
| SDS for weight (g) at 36 weeks’ PMA (or sooner if discharged home)a | ||
| Mean (SD) | –1.1 (1.0) | –1.7 (1.0) |
| Missing | 1 | 0 |
| SDS for head circumference (cm) at study entrya | ||
| Mean (SD) | –2.1 (0.9) | –1.4 (0.8) |
| Missing | 4 | 7 |
| SDS for head circumference (cm) at 36 weeks’ PMA (or sooner if discharged home)a | ||
| Mean (SD)b | –1.6 (1.0) | –1.9 (1.1) |
| Missing | 2 | 0 |
| Participant ID | SAE number | Allocation (i.e. trial arm) | Centre ID | Description | Severity | Outcome | Causality |
|---|---|---|---|---|---|---|---|
| MI_10031 | 1 | Active (dexamethasone) | 112 | Hypoglycaemia | Mild | Resolved | Not related |
Details of the babies who discontinued the intervention are included in Table 13.
| Participant ID | Allocation (i.e. trial arm) | Centre ID | Randomisation date | Discontinuation date | Description | Reason |
|---|---|---|---|---|---|---|
| MI_10015 | Active (dexamethasone) | 230 | 2 August 2017 | 11 August 2017 | Suspected/confirmed infection | Clinical decision |
| MI_10111 | Active (dexamethasone) | 230 | 6 December 2017 | 11 December 2017 | Suspected/confirmed infection | Clinical decision |
| MI_10120 | Active (dexamethasone) | 160 | 15 December 2017 | 18 December 2017 | Deterioration, given open-label steroids | Clinical decision |
| MI_10168 | Active (dexamethasone) | 224 | 20 January 2018 | 24 January 2018 | Deterioration, given open-label dexamethasone | Clinical decision |
| MI_10202 | Active (dexamethasone) | 230 | 13 March 2018 | 17 March 2018 | Deterioration, given open-label dexamethasone | Clinical decision |
| MI_10216 | Active (dexamethasone) | 224 | 26 March 2018 | 3 April 2018 | Baby accidentally given two doses of the ward stock of dexamethasone instead of the trial-specific dose | Other |
| MI_10048 | Placebo | 167 | 8 September 2017 | 12 September 2017 | Suspected/confirmed infection | Clinical decision |
| MI_10057 | Placebo | 160 | 8 September 2017 | 16 September 2017 | Suspected/confirmed infection | Clinical decision |
| MI_10142 | Placebo | 160 | 16 January 2018 | 19 January 2018 | Deterioration, given open-label dexamethasone | Clinical decision |
| MI_10179 | Placebo | 288 | 27 January 2018 | 4 February 2018 | Deterioration, given open-label dexamethasone | Clinical decision |
| MI_10195 | Placebo | 145 | 2 March 2018 | 7 March 2018 | Deterioration, given open-label dexamethasone | Clinical decision |
| MI_10225 | Placebo | 160 | 11 April 2018 | 12 April 2018 | Suspected/confirmed infection | Clinical decision |
Chapter 5 Discussion
Overview
This trial was not able to identify sufficient babies with the primary outcome to evaluate the study objectives. The main metric of feasibility, recruitment, proved difficult. There was a tendency for open-label medication and a higher than predicted rate of suspected/confirmed sepsis among babies. Recruitment was halted following discussions with the TSC and the funder after 22 babies had been enrolled.
Findings
Baseline characteristics
The study groups were well balanced with regard to maternal and infant characteristics and no clinically important imbalances were observed. There was a preponderance of babies born at < 25 weeks in both arms of the trial and a lower than expected number of babies received antenatal steroids (77% as opposed to the 86% predicted from national data sets). 33 This appears to be indicative that the babies at highest risk of remaining ventilator dependent, and being assessed as being at high risk of developing BPD, are the most extremely preterm infants who do not receive protective antenatal interventions.
Efficacy
Owing to the low number of trial participants and the higher than predicted discontinuation rate it is possible to comment only on possible trends seen at data analysis. It appears that very low-dose dexamethasone may improve lung function and facilitate extubation. 24,25 When compared with placebo, there was a higher proportion of babies extubated at day 7 of life (62.5% in the dexamethasone group vs. 33.3% in the control group) in addition to a reduced duration of invasive ventilation (median of 23 days in the dexamethasone group vs. median of 31 days in the control group) in those babies treated with very low-dose dexamethasone. In support of this finding is the trend to double the requirement for open-label rescue steroids in control group babies (41.7% in the dexamethasone group vs. 80% in the control group).
Safety
Dexamethasone has been extensively researched in this population and its side effect profile is well identified. 6,7 There were no unexpected safety concerns and no clinically important imbalances were observed.
Feasibility
All the clinical outcomes of the trial were ascertained appropriately; however, the trial was difficult to recruit to as there was a lower than expected number of eligible babies. This may be due to a lack of clinician equipoise, the inability to co-recruit to other key neonatal trials or changing neonatal practices.
The rate of discontinuation of the IMP for open-label steroids was higher than expected by the trial team during trial planning, as was the rate of suspected sepsis, with 23% discontinuing for suspected/confirmed sepsis as opposed to the predicted 6%. Discontinuation in favour of open-label steroids seems to represent a loss of clinician equipoise regarding steroid therapy. The discrepancy in the predicted and actual rates of sepsis is likely to reflect the fact that:
-
The published data in this cohort are for confirmed culture-positive late-onset sepsis. Clinicians were discontinuing on the grounds of suspected infection that, although the baby may manifest clinical signs, may not result in positive blood cultures and so will be a more prevalent outcome.
-
The babies selected for the trial are the most at-risk subset of babies and will consequently have a higher than average risk of this outcome.
Family diaries
Only 12 families (55%) completed and returned a diary of care. The completed diaries were incomplete with large numbers of missing data for time to first cuddle, time to first skin-to-skin contact and duration of contact a family member has with their baby.
Among the returned diaries it was notable that there were low values of frequency of contact a family member had with the baby. It is uncertain whether this represents a true finding or incomplete data collection.
Cytokines
The cytokine samples are yet to be analysed. Analysis will take place after the lifespan of the NIHR EME programme funding.
Issues raised by this study
This study informs a number of discussion points relating to the evaluation of medicines to treat evolving BPD.
Preparation of placebos for marketed products
The preparation of placebos for marketed products is a well-recognised problem. In the absence of placebos for marketed products it will be impossible to evaluate existing treatments, many of which have entered routine practice without appropriate evaluation. The unjustified use of legacy products is common in paediatrics, particularly in neonates, and leads to the use of medicines that may not be effective, that may have unrecognised safety concerns and that may not be an appropriate use of resources.
One way forward is to set up a centralised service to commission trial supplies. This could speed up the process or pool expertise in making relevant judgements about the suitability of possible placebo preparations.
Recruitment of neonates with evolving bronchopulmonary dysplasia to clinical trials
Although very low-dose dexamethasone had been in use in several centres throughout the UK, the cohort study that formed the basis for planning the study was carried out in a single centre. That centre did not have rates of BPD that differed from national norms. 31 Since the cohort study was carried out, the rate of BPD has not reduced markedly; however, clinicians’ attitudes to postnatal dexamethasone treatment are changing and the ‘best practice’ care of extremely preterm babies is evolving as a result of the alterations in the use of invasive and non-invasive respiratory support. 34,35 The combination of these factors, in addition to the inability to co-recruit to other concurrent neonatal trials, made it significantly more difficult to recruit than anticipated. The proportion of parents of eligible babies approached regarding the trial who gave consent was 66%, which is in line with experience in similar studies. 36
It is speculated that the eligibility criteria do not match all the contemporary predictors of BPD. The patterns of respiratory support currently given to neonates differ from past practice with respect to modes of support during the initial stabilisation period after birth [i.e. nasal continuous positive airway pressure (CPAP); less invasive surfactant administration] and/or subsequently (move to ‘early extubation’ with greater use of volume-guaranteed ventilation or modes of non-invasive ventilation)35 facilitated by advances in technology respiratory support. These changes in ventilation strategies have created a situation whereby babies at high risk of developing BPD are not necessarily subjected to prolonged mechanical ventilation but may instead be managed with non-invasive ventilation. This is a change since the cohort that informed the design of this trial.
To identify a cohort at high risk of developing BPD, it would now be necessary to adjust the threshold for ‘severity’ of respiratory support. The group of babies currently at risk of BPD is likely to include babies who are receiving non-invasive ventilation. The wide variety of strategies and technology for respiratory support of premature babies used throughout the UK poses difficulties in identifying both a homogeneous trial group and a suitable surrogate outcome measure.
In the future, other novel ways of identifying ‘at-risk’ babies and surrogate outcomes, such as biomarkers, may be useful, but need considerable development and are not yet ready for inclusion in clinical trials.
Discontinuation rates among neonates that participate in a trial of evolving bronchopulmonary dysplasia
In total, treatment was discontinued in 12 babies (55%). This is more than double the 23% predicted at trial planning.
Discontinuation related to clinician choice of open-label steroids occurred in six participants (27%). This finding suggests that equipoise was not maintained with this dose. This may reflect changes in clinicians’ attitudes to postnatal steroid use.
Suspicion of sepsis that warranted discontinuation of steroids prompted discontinuation in trial participation in five babies (23%). This is consistent with clinical practice in many centres and raises two issues for the design of clinical trials about BPD:
-
Recruiting enough babies who are evaluable, that is, participants whose steroid treatment is not discontinued because of clinical suspicion of infection.
-
How to study steroids in babies who start steroids more than once. This group needs to be studied because most of the babies in whom steroids were discontinued will have had a subsequent course. Indeed, a proportion of babies will start steroids three or four times. The effects of steroids may differ according to the presence or absence of an infection (e.g. through a disturbance of the cytokine milieu). This group of babies receiving ‘stop–start’ steroids could be studied through a preplanned subgroup analysis of a trial that is powered according to the number of babies whose steroids are not discontinued as a result of concerns about sepsis. An alternative approach would be a trial that is powered to look at all babies who receive steroids. Planning a trial that includes ‘stop–start’ babies would require careful consideration of the management of trial supplies.
Analysis of event rates in trials of evolving bronchopulmonary dysplasia
The very small number of events eligible for assessment of the primary outcome means that the trial was not powered to detect a difference between the groups at the proposed HR of 6. Furthermore, although this HR was quoted in the original application from a previous cohort study, it was considered by the study team to be an extremely large effect size for a trial setting. The lower bound of the 95% CI for that HR (2.3) was therefore used as a more realistic estimate to calculate the sample size of 94, as reported in the MINIDEX trial protocol. Therefore, even if the trial continued to recruit until full data on ≈ 20 events had been recorded (potentially ≈ 40–45 participants might need to be recruited for this because of discontinuations and other exclusions), the trial would still only be powered to detect an implausibly high HR of 6. The team are of the opinion that, even if recruitment continued to this number (≈ 40–45 participants), there would not be enough power to detect a realistic difference in the primary outcome between the two trial arms.
Description and partial validation of a novel parent reported outcome measurement
There was limited uptake of the parent-reported outcomes contained within the diary of care. It is unclear whether this is a reflection of the design of the trial materials or the promotion of the diary by the sites or whether or not it is a result of the variation seen in family engagement with trial processes.
Although the diaries were incomplete, the data provided by those families who did engage with the process suggest that it may be possible, with further refinements, to capture quantifiable data of the way that clinical trials affect the family’s ability to parent and bond with their child. This is an important family-centred outcome, championed by PPI, and ought to be part of the outcomes of other clinical trials.
Conclusions
Ultimately the trial addressed a particular question with a surrogate outcome in order to facilitate the design of a definitive large pragmatic trial using substantive outcomes. The MINIDEX trial has served its purpose and made clear that, in the contemporary clinical context, this approach to the research question is not feasible. The research question remains open and will not be answered by the MINIDEX trial.
Other researchers are continuing to investigate other regimens of postnatal corticosteroid administration. Following the PREMILOC (i.e. early administration of intravenous prophylactic hydrocortisone for the prevention of BPD in extremely preterm babies) trial,37 Laboratoire Aguettant (Lyon, France) is developing a formulation of low-dose hydrocortisone for neonates, with the intent to submit an application to the European Medicines Agency, the US Food and Drug Administration, and Health Canada (Olivier Baud, Hôpital universitaire Robert-Debré, Paris, YEAR, personal communication, 2018). The Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) neonatal network is currently conducting a trial of a higher dose of hydrocortisone tapered over 10 days starting between 14 and 28 days in intubated babies born at < 30 weeks’ gestation. 38 A trial recently published by the STOP-BPD study group randomised 372 ventilator-dependent babies born at < 30 weeks’ gestation or with a birth weight under 1250 g to hydrocortisone or placebo at 7–14 days of life. Hydrocortisone was given as a 22-day tapering course (cumulative dose of 72.5 mg/kg). 39
The STOP-BPD trial found no differences between the groups for the either the composite outcome of BPD and death (OR 0.87, 95% CI 0.54 to 1.38) or BPD (OR 1.24, 95% CI 0.82 to 1.86), leading the researchers to conclude that hydrocortisone should not be used for this indication in these babies. 39 The cumulative dose of hydrocortisone used in the STOP-BPD trial had higher glucocorticoid potency than the cumulative dose of dexamethasone used in MINIDEX trial.
As the incidence of BPD in the active treatment arm of the PREMILOC trial was 22% [as opposed to 26% in the placebo arm, giving a RR of BPD 0.82 (95% CI 0.58 to 1.16)],37 there will continue to be a significant number of babies who require ‘rescue corticosteroids’. The high dose of hydrocortisone on the NICHD neonatal network trial may lead to a side effect profile that may be ameliorated by a lower glucocorticoid dose. The dosage regimen used in STOP-BPD trial did not have an effect on outcomes.
Something needs to be done to optimise the use of steroids in evolving BPD.
Options include:
-
adjust recruitment to current practice – appropriate inclusion criteria
-
minimise rescue treatment – address equipoise among clinicians.
A better understanding of the biology and natural history of the condition will help. Routinely collected data were not sufficient to adequately plan this study. Given the dynamic nature of neonatal care, novel approaches are needed. These novel approaches could include registry-based trials or the use of enhanced routine data collection to support trial planning.
Acknowledgements
The study team would like to thank the staff, patients and parents at the study’s contributing centres.
Dedication
The trial team would like to dedicate this report to the memory of the late Simon Newell.
Contributions of authors
Dr Helen Yates (Consultant Neonatologist, Hull Royal Infirmary) was a co-applicant for the grant submission, involved in developing the trial protocol and trial-related literature and drafted the report.
Virginia Chiocchia (MINIDEX Trial Statistician, NPEU Clinical Trials Unit) was involved in developing the trial protocol and trial-related literature, analysed the data for the trial, and reviewed and revised the report.
Dr Louise Linsell (Lead Medical Statistician, NPEU Clinical Trials Unit) was involved in developing the trial protocol and trial-related literature, analysed the data for the trial, and reviewed and revised the report.
Dr Nicolas Orsi (Clinical Lecturer in Histopathology, Leeds Institute of Cancer and Pathology) was a co-applicant for the grant submission, involved in developing the trial protocol and trial-related literature, and reviewed and revised the report.
Edmund Juszczak (Director, NPEU Clinical Trials Unit) was a co-applicant for the grant submission, involved in developing the trial protocol and trial-related literature, and reviewed and revised the report.
Dr Kathryn Johnson (Consultant Neonatologist, Leeds Neonatal Service) was a co-applicant for the grant submission, involved in developing the trial protocol and trial-related literature, and reviewed and revised the report.
Dr Philip Chetcuti (Consultant Neonatologist, Leeds Neonatal Service) was a co-applicant for the grant submission, involved in developing the trial protocol and trial-related literature, and reviewed and revised the report.
Claire Illingworth (PPI representative) was a co-applicant for the grant submission and was involved in developing the trial protocol and trial-related literature.
Pollyanna Hardy (Statistician, Birmingham Clinical Trials Unit) was a co-applicant for the grant submission and was involved in developing the trial protocol and trial-related literature.
Dr Vaneesha Monk (Trial Manager, Department of Paediatrics, University of Oxford) was involved in developing the trial protocol and trial-related literature.
Dr Simon Newell (Deceased) was a co-applicant for the grant submission, and was involved in developing the trial protocol and trial-related literature.
Dr Mark Turner (Consultant Neonatologist, Liverpool Women’s Hospital) was a co-applicant for the grant submission, involved in developing the trial protocol and trial-related literature and drafted the report.
Publication
Yates HL, Johnson K, Turner MA. The Minidex trial: does very low-dose dexamethasone improve lung function in preterm infants? Infant 2016;12:49–51.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Please note exclusive use will be retained until the publication of major outputs. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Boyle EM, Manktelow BN, Field DJ, Oddie S, Draper ES. The Neonatal Survey Report 2012 n.d. www2.le.ac.uk/departments/health-sciences/research/timms/documents (accessed January 2016).
- The Canadian Neonatal Network (CNN) . CNN 2012 Annual Report 2012. www.canadianneonatalnetwork.org/Portal (accessed January 2016).
- Kennedy KA, Cotten CM, Watterberg KL, Carlo WA. Prevention and management of bronchopulmonary dysplasia: lessons learned from the neonatal research network. Semin Perinatol 2016;40:348-55. https://doi.org/10.1053/j.semperi.2016.05.010.
- Greenough A, Alexander J, Burgess S, Chetcuti PA, Cox S, Lenney W, et al. Home oxygen status and rehospitalisation and primary care requirements of infants with chronic lung disease. Arch Dis Child 2002;86:40-3. https://doi.org/10.1136/adc.86.1.40.
- Natarajan G, Pappas A, Shankaran S, Kendrick DE, Das A, Higgins RD, et al. Outcomes of extremely low birth weight infants with bronchopulmonary dysplasia: impact of the physiologic definition. Early Hum Dev 2012;88:509-15. https://doi.org/10.1016/j.earlhumdev.2011.12.013.
- Doyle LW, Cheong JL, Ehrenkranz RA, Halliday HL. Early (> 8 days) systemic postnatal corticosteroids for prevention of bronchopulmonary dysplasia in preterm infants. Cochrane Database Syst Rev 2017;10. https://doi.org/10.1002/14651858.CD001146.pub5.
- Doyle LW, Cheong JL, Ehrenkranz RA, Halliday HL. Late (> 7 days) systemic postnatal corticosteroids for prevention of bronchopulmonary dysplasia in preterm infants. Cochrane Database Syst Rev 2017;10. https://doi.org/10.1002/14651858.CD001145.pub4.
- Fitzhardinge PM, Eisen A, Lejtenyi C, Metrakos K, Ramsay M. Sequelae of early steroid administration to the newborn infant. Pediatrics 1974;53:877-83.
- Bose CL, Dammann CE, Laughon MM. Bronchopulmonary dysplasia and inflammatory biomarkers in the premature neonate. Arch Dis Child Fetal Neonatal Ed 2008;93:F455-61. https://doi.org/10.1136/adc.2007.121327.
- Tullus K, Noack GW, Burman LG, Nilsson R, Wretlind B, Brauner A. Elevated cytokine levels in tracheobronchial aspirate fluids from ventilator treated neonates with bronchopulmonary dysplasia. Eur J Pediatr 1996;155:112-16. https://doi.org/10.1007/BF02075762.
- Ambalavanan N, Carlo WA, D’Angio CT, McDonald SA, Das A, Schendel D, et al. Cytokines associated with bronchopulmonary dysplasia or death in extremely low birth weight infants. Pediatrics 2009;123:1132-41. https://doi.org/10.1542/peds.2008-0526.
- Li Y, Brauner A, Jonsson B, Soder O, Jensen J, Lagercrantz H, et al. Inhibition of macrophage proinflammatory cytokines by steroids and recombinant IL-10. Biol Neonate 2001;80:124-32. https://doi.org/10.1159/000047131.
- Costeloe K, Hennessy E, Gibson AT, Marlow N, Wilkinson AR. The EPICure study: outcomes to discharge from hospital for infants born at the threshold of viability. Pediatrics 2000;106:659-71. https://doi.org/10.1542/peds.106.4.659.
- Barrington KJ. The adverse neuro-developmental effects of postnatal steroids in the preterm infant: a systematic review of RCT. Biomed Cent Ped 2001;1. https://doi.org/10.1186/1471-2431-1-1.
- Tsuneishi S, Takada S, Motoike T, Ohashi T, Sano K, Nakamura H. Effects of dexamethasone on the expression of myelin basic protein, proteolipid protein, and glial fibrillary acidic protein genes in developing rat brain. Brain Res Dev Brain Res 1991;61:117-23. https://doi.org/10.1016/0165-3806(91)90121-X.
- Shi B, Rabin SJ, Brandoli C, Mocchetti I. Dexamethasone induces hypertrophy of developing medial septum cholinergic neurons: potential role of nerve growth factor. J Neurosci 1998;18:9326-34. https://doi.org/10.1523/JNEUROSCI.18-22-09326.1998.
- Flagel SB, Vázquez DM, Watson SJ, Neal CR. Effects of tapering neonatal dexamethasone on rat growth, neurodevelopment, and stress response. Am J Physiol Regul Integr Comp Physiol 2002;282:R55-63. https://doi.org/10.1152/ajpregu.2002.282.1.R55.
- Wilson-Costello D, Friedman H, Minich N, Siner B, Taylor G, Schluchter M, et al. Improved neurodevelopmental outcomes for extremely low birth weight infants in 2000–2002. Pediatrics 2007;119:37-45. https://doi.org/10.1542/peds.2006-1416.
- Yoder NA, Harrison M, Clark RH. Time related changes in steroid use and bronchopulmonary dysplasia in preterm infant. Pediatrics 2008;124:673-9. https://doi.org/10.1542/peds.2008-2793.
- Zeng L, Tian J, Song F, Li W, Jiang L, Gui G, et al. Corticosteroids for the prevention of bronchopulmonary dysplasia in preterm infants: a network meta-analysis. Arch Dis Child Fetal Neonatal Edn 2018;103:F506-11. https://doi.org/10.1136/archdischild-2017-313759.
- Doyle LW, Halliday HL, Ehrenkranz RA, Davis PG, Sinclair JC. Impact of postnatal systemic corticosteroids on mortality and cerebral palsy in preterm infants: effect modification by risk for chronic lung disease. Pediatrics 2005;115:655-61. https://doi.org/10.1542/peds.2004-1238.
- Parikh NA, Kennedy KA, Lasky RE, Tyson JE. Neurodevelopmental outcomes of extremely preterm infants randomized to stress dose hydrocortisone. PLOS ONE 2015;10. https://doi.org/10.1371/journal.pone.0137051.
- Job S, Clarke P. Current UK practices in steroid treatment of chronic lung disease. Arch Dis Child Fetal Neonatal Edn 2015;100. https://doi.org/10.1136/archdischild-2014-308060.
- Tanney K, Davis J, Halliday HL, Sweet DG. Extremely low-dose dexamethasone to facilitate extubation in mechanically ventilated preterm babies. Neonatology 2011;100:285-9. https://doi.org/10.1159/000326273.
- Yates HL, Newell SJ. Minidex: very low dose dexamethasone (0.05 mg/kg/day) in chronic lung disease. Arch Dis Child Fetal Neonatal Ed 2011;96:F190-4. https://doi.org/10.1136/adc.2010.187203.
- Doyle LW, Davis PG, Morley CJ, McPhee A, Carlin JB. DART Study Investigators . Low-dose dexamethasone facilitates extubation among chronically ventilator-dependent infants: a multicenter, international, randomized, controlled trial. Pediatrics 2006;117:75-83. https://doi.org/10.1542/peds.2004-2843.
- Feldman R, Eidelman AI, Sirota L, Weller A. Comparison of skin-to-skin (kangaroo) and traditional care: parenting outcomes and pre-term infant development. Pediatrics 2002;110:16-2. https://doi.org/10.1542/peds.110.1.16.
- Baskind NE, Orsi NM, Sharma V. Follicular-phase ovarian follicular fluid and plasma cytokine profiling of natural cycle in vitro fertilization patients. Fertil Steril 2014;102:410-18. https://doi.org/10.1016/j.fertnstert.2014.04.032.
- Wirtschafter DD, Powers RJ, Pettit JS, Lee HC, Boscardin WJ, Ahmad Subeh M, et al. Nosocomial infection reduction in VLBW infants with a statewide quality-improvement model. Pediatrics 2011;127:419-26. https://doi.org/10.1542/peds.2010-1449.
- Herrman K, Bose C, Lewis K, Laughon M. Spontaneous closure of the patent ductus arteriosus in very low birth weight infants following discharge from the neonatal unit. Arch Dis Child Fetal Neonatal Ed 2009;94:F48-50. https://doi.org/10.1136/adc.2007.129270.
- Beal MJ, Falciani F, Ghahramani Z, Rangel C, Wild DL. A Bayesian approach to reconstructing genetic regulatory networks with hidden factors. Bioinformatics 2005;21:349-56. https://doi.org/10.1093/bioinformatics/bti014.
- Scientific Advisory Committee on Nutrition and RCPCH Expert Group . Application of WHO Growth Standards in the UK 2007.
- Royal College of Paediatrics and Child Health . National Neonatal Audit Programme 2017 Annual Report on 2016 Data 2017. www.rcpch.ac.uk/sites/default/files/NNAP_National_Annual_report_2017.pdf (accessed 20 October 2018).
- Dunn MS, Kaempf J, de Klerk A, de Klerk R, Reilly M, Howard D, et al. Randomized trial comparing 3 approaches to the initial respiratory management of preterm neonates. Pediatrics 2011;128:e1069-76. https://doi.org/10.1542/peds.2010-3848.
- Fischer H, Bruher C. Avoiding endotracheal ventilation to prevent bronchopulmonary dysplasia: a meta-analysis. Pediatrics 2013;132:D1-6. https://doi.org/10.1542/peds.2013-1880.
- Zhao W, Hill H, Le Guellec C, Neal T, Mahoney S, Paulas S, et al. Population pharmacokinetics of ciprofloxacin in neonates and young infants less than 3 months of age. Antimicrob Agents Chemother 2014;58:6572-80. https://doi.org/10.1128/AAC.03568-14.
- Baud O, Maury L, Lebail F, Ramful D, El Moussawi F, Nicaise C, et al. Effect of early low-dose hydrocortisone on survival without bronchopulmonary dysplasia in extremely preterm infants (PREMILOC): a double-blind, placebo-controlled, multicentre, randomised trial. Lancet 2016;387:1827-36. https://doi.org/10.1016/S0140-6736(16)00202-6.
- Walsh MC, Shankaran S, Laptook AR, Cotton CM, Carlton D, Sokol G, et al. Hydrocortisone for BPD n.d. https://clinicaltrials.gov/ct2/show/NCT01353313 (accessed 20 October 2018).
- Onland W, Cools F, Kroon A, Rademaker K, Merkus MP, Dijk PH, et al. Effect of hydrocortisone therapy initiated 7 to 14 days after birth on mortality or bronchopulmonary dysplasia among very Preterm infants receiving mechanical ventilation: a randomized clinical trial. JAMA 2019;321:354-63. https://doi.org/10.1001/jama.2018.21443.
Appendix 1 Recruiting sites
The recruiting sites in this feasibility trial were:
-
Birmingham Women’s Hospital, Birmingham, UK
-
Bradford Royal Infirmary, Bradford, UK
-
Hull Royal Infirmary, Hull, UK
-
Leeds General Infirmary, Leeds, UK
-
Leicester Royal Infirmary, Leicester, UK
-
Liverpool Women’s Hospital, Liverpool, UK
-
Royal Hallamshire Hospital, Sheffield, UK
-
Royal Preston Hospital, Preston, UK
-
Royal Victoria Infirmary, Newcastle, UK
-
University Hospital Coventry, Coventry, UK
-
University Hospital of North Tees, Stockton-on-Tees, UK.
Appendix 2 Continuing-care sites
The continuing-care sites in this feasibility trial were:
-
Birmingham City Hospital, Birmingham, UK
-
Birmingham Heartlands Hospital, Birmingham, UK
-
Burnley General Hospital, Burnley, UK
-
Calderdale Royal Hospital, Halifax, UK
-
Chesterfield Royal Hospital, Chesterfield, UK
-
Countess of Chester Hospital, Chester, UK
-
Dewsbury and District Hospital, Dewsbury, UK
-
Diana Princess of Wales Hospital, Grimsby, UK
-
George Eliot Hospital, Nuneaton, UK
-
Glenfield Hospital, Leicester, UK
-
Harrogate District Hospital, Harrogate, UK
-
James Cook University Hospital, Middlesbrough, UK
-
Leicester General Hospital, Leicester, UK
-
Lincoln County Hospital, Lincoln, UK
-
Milton Keynes General Hospital, Milton Keynes, UK
-
New Cross Hospital, Wolverhampton, UK
-
Norfolk and Norwich University Hospital, Norwich, UK
-
North Manchester General Hospital, Manchester, UK
-
Pilgrim Hospital Boston, Boston, UK
-
Pinderfields General Hospital, Wakefield, UK
-
Pontefract Hospital, Pontefract, UK
-
Queen’s Hospital, Burton on Trent, UK
-
Royal Albert Edward Infirmary, Wigan, UK
-
Royal Bolton Hospital, Bolton, UK
-
Royal Oldham Hospital, Oldham, UK
-
Wirral Women and Children’s Hospital, Wirral, UK.
Appendix 3 Severity-based diagnostic criteria for bronchopulmonary dysplasia
| Time point of assessment: 36 weeks’ PMA | |
|---|---|
|
Therapy with oxygen > 21% and/or respiratory support for ≥ 28 days cumulatively and the following levels of oxygen requirement as determined by the oxygen reduction test: |
|
| Mild BPD | Baby is breathing room air |
| Moderate BPD | Baby is in 22–29% oxygen, or 0.01–1.0 l/minute |
| Severe BPD | Baby is in FiO2 ≥ 30% or low-flow oxygen ≥ 1.1 l/minute or the baby is receiving respiratory support (invasive or non-invasive) to achieve saturations of ≥ 91% |
The need for oxygen will be determined by the oxygen reduction test. This is based on the minimum inspired oxygen threshold at which the baby can maintain saturations of ≥ 91%. Babies requiring inspired oxygen to maintain saturations of ≥ 91% are considered to be oxygen dependent.
The oxygen reduction test applies only to babies in < 0.3 inspired oxygen, or a low-flow oxygen of < 1.1 l/minute, and who have not had respiratory support in the previous 24 hours. Those babies not meeting these criteria will not be tested, but their oxygen requirements will be captured on the relevant data form.
Appendix 4 Oxygen reduction test

List of abbreviations
- bFGF
- basic fibroblast growth factor
- BPD
- bronchopulmonary dysplasia
- CI
- confidence interval
- CP
- cerebral palsy
- CPAP
- continuous positive-airway pressure
- CRF
- case report form
- CTACK
- cutaneous T cell-attracting chemokine
- CTU
- Clinical Trials Unit
- DMC
- Data Monitoring Committee
- G-CSF
- granulocyte colony-stimulating factor
- GM-CSF
- granulocyte macrophage colony-stimulating factor
- GRO
- growth-related oncogene
- HGF
- hepatocyte growth factor
- HR
- hazard ratio
- IFN
- interferon
- IFN-γ
- interferon gamma
- IL
- interleukin
- IMP
- Investigative Medicinal Product
- IQR
- interquartile range
- ISRCTN
- International Standard Randomised Controlled Trial Number
- LIF
- leukaemia inhibitory factor
- MBD1
- methyl CpG-binding domain protein one
- MCAF
- methyl CpG-binding domain protein one (MBD1)-containing chromatin-associated factor
- MCP
- monocyte chemotactic protein
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MIF
- macrophage migration inhibitory factor
- MIG
- monokine induced by gamma interferon
- MIP
- macrophage inflammatory protein
- NEC
- necrotising enterocolitis
- NGF
- nerve growth factor
- NICHD
- Eunice Kennedy Shriver National Institute of Child Health and Human Development
- NPEU
- National Perinatal Epidemiology Unit
- OR
- odds ratio
- PDA
- patent ductus arteriosus
- PMA
- postmenstrual age
- PMG
- Project Management Group
- PPI
- patient and public involvement
- QP
- qualified person
- REC
- Research Ethics Committee
- RR
- relative risk
- SAE
- serious adverse event
- SCF
- stem cell factor
- SCGF
- stem cell growth factor
- SD
- standard deviation
- SDF
- stromal cell-derived factor
- TNF
- tumour necrosis factor
- TRAIL
- TNF-related apoptosis-inducing ligand
- TSC
- Trial Steering Committee
- VEGF
- vascular endothelial growth factor