
Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 14/187/01. The contractual start date was in September 2015. The final report began editorial review in October 2018 and was accepted for publication in March 2019. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Vaneesha Monk, Caroline Hartley, Jennifer L Bell, Ed Juszczak, Jane Norman, Richard Rogers, Chetan Patel, Eleri Adams and Rebeccah Slater report funding from the National Institute for Health Research (NIHR) Efficacy and Mechanism Evaluation (EME) programme. Fiona Moultrie, Amy Hoskin and Rebeccah Slater report funding from the Wellcome Trust (London, UK). Fiona Moultrie reports funding from the NIHR Biomedical Research Centre (BRC; Oxford University Hospitals Trust, Oxford, UK). Gabrielle Green reports funding from the NIHR Oxford BRC. Jane Norman reports grants from the UK government and charities outside the submitted work and consultancy payments from GlaxoSmithKline plc (Brentford, UK), Dilafor AB (Solna, Sweden) and the Wellcome Trust outside the submitted work. Jane Norman was a member of the Health Technology Assessment and EME Editorial Board from 2012 to 2014. Ed Juszczak is currently a member of the Clinical Trials Units funded by the NIHR committee.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2019. This work was produced by Monk et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2019 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Infant pain
Pain is a subjective experience, of which we all have an intuitive and personal understanding. In nursing, pain is regarded as ‘whatever the experiencing person says it is, existing whenever he says it does’. 1 Self-reporting is therefore central to accurate pain assessment, but the inability of infants to communicate through language renders assessment in this clinical population challenging. Nevertheless, the measurement of pain in infancy is as important as, if not more so than, in adulthood, because it occurs at a time when the brain is vulnerable and plastic, and early pain experiences can be destabilising in the short term and have lasting neurodevelopmental effects. Infants can develop physiological instability, including pauses in their breathing (apnoeas), in the 24–48 hours following painful clinical procedures. 2 In the longer term, cumulative pain exposure has now been associated with altered brain microstructure, corticospinal tract development, cortical thinning, stress hormone expression and impaired cognitive development, in both infancy and at school age. 3
Despite increasing evidence of these immediate and long-term consequences, pain remains substantially under-treated in infancy. Although surgery is no longer performed on infants without analgesia, the historical misconception that infants do not feel pain still affects routine clinical practice. Very few clinical trials have been conducted to test the safety, efficacy and dosing of pharmacological analgesics in this population. Consequently, comfort measures (such as non-nutritive sucking and swaddling) are frequently recommended as an alternative to pharmacological analgesia. Given that infants requiring intensive care experience an average of 12 painful procedures per day, and the youngest and sickest infants can be exposed to up to 50 procedures per day, this is a serious clinical issue, particularly considering that analgesia is frequently not provided. 4 Although efforts can be made to minimise the number of painful procedures performed in intensive care, these repeated procedures are in many cases unavoidable and essential to the treatment and survival of the infants. It is therefore critical that safe and effective analgesics are identified.
Analgesia for infants
The provision of analgesia on neonatal units is heavily dominated by non-pharmacological comfort techniques (such as non-nutritive sucking and sucrose). Evidence of the efficacy of pharmacological analgesics is sparse and contradictory, and general anaesthetics are avoided where possible because of the potential for systemic adverse effects and complications. Retinopathy of prematurity (ROP) screening is an example of a routine invasive painful procedure that is repeatedly performed in premature infants without adequate pain relief. Topical anaesthetics, oral sucrose, breast milk and various combinations of these interventions are often used but have been shown to provide inadequate analgesia for this stressful and painful procedure. 5,6 A recent Cochrane review emphasised the inadequacy of current analgesic protocols for ROP screening and the need for high-quality randomised controlled trials (RCTs) of pharmacological interventions. 7
Although morphine is a potent analgesic that provides effective pain relief for procedural pain in adults,8 developmental pharmacokinetic and pharmacodynamic differences means that we cannot assume that it provides analgesia in infants. The efficacy of morphine in this population currently remains controversial because of contradictory results from a few RCTs. 9–11 Morphine, however, continues to be frequently administered intravenously to ventilated infants for comfort and sedation, despite a Cochrane review concluding that there is insufficient evidence to recommend its routine use for procedural pain relief in infants. 12
Given the compelling need for analgesics in the neonatal population, it is essential that the efficacy of morphine be rigorously assessed. In the PrOcedural Pain in Premature Infants (Poppi) trial, it was investigated whether or not oral morphine could provide pain relief for ROP screening using multiple validated methods of pain assessment. To date, one previous trial has attempted to test the efficacy of morphine during ROP screening;13 however, it was unfortunately stopped early as a result of changes in research regulations from the Medicines and Healthcare products Regulatory Agency (MHRA). Having recruited only 18 infants of a planned sample size of 63 (six of whom received morphine), the authors of the study were unable to make any conclusions on the efficacy of morphine.
Quantifying pain in the neonatal population
Infant pain is usually inferred from observations of behavioural and autonomic activity; changes in facial expression, heart rate and oxygen saturation form the basis of most pain scales. In premature infants, the most validated pain assessment tool is the Premature Infant Pain Profile (PIPP). 14,15 The PIPP is a composite multimodal measure, incorporating measures of heart rate, oxygen saturation and facial expression change, which captures different aspects of the infant pain experience. It has been widely used as the primary outcome measure for assessing infant pain in clinical trials. 16–18 However, in the developing central nervous system where pain pathways are maturing and connectivity is developing, the correspondence between pain perception and behavioural responses is unknown.
Cortical processing is a prerequisite for pain perception; therefore, recording nociceptive brain activity may provide the best surrogate measure of pain in infants. 19 In adults, patterns of electroencephalographic (EEG) activity evoked by noxious stimulation have been extensively studied, and noxious-evoked patterns of brain activity have been characterised in response to various noxious stimuli. Noxious inputs transmitted by Aδ and C nerve fibres are detected in the brain as long-latency (100–400 milliseconds) and ultralong-latency (800–1500 milliseconds) patterns of activity comprising a negative and a positive deflection that is maximal at the vertex, and differing in latency and morphology according to stimulus modality, intensity and location. 20 In adults, the amplitude of noxious-evoked brain activity and verbal reports of pain are significantly reduced by the administration of opiates. 21
Noxious-evoked brain activity has been well characterised in infants in response to heel lancing (a procedure for blood sampling), which has provided an objective, quantifiable and validated neurophysiological measure of acute procedural pain that can be used as an outcome in clinical trials. 22 Studies combining EEG and behavioural measures have additionally identified the limitations of relying on observational behavioural measures to assess pain in infants. For example, nociceptive information can be processed in the infant brain without a concomitant behavioural response. 23 Adopting a multimodal approach in clinical trials, through using behavioural pain scores, EEG and electromyography (EMG), can provide a greater mechanistic understanding of the effects of analgesic drugs by assessing their ability to effectively reduce clinical pain scores, nociceptive brain activity and spinal cord activity evoked by an acute painful procedure. 24
Retinopathy of prematurity screening
Retinopathy of prematurity is a condition of the retinal vascular system that affects premature and very low-birthweight infants and, if untreated, can lead to permanent blindness. ROP screening is a painful eye examination procedure that is repeatedly performed on premature infants to diagnose and stage ROP, and to identify any infants who would benefit from treatment. During the procedure, a speculum is inserted to maintain exposure of the eye and the retina is examined using an ophthalmoscope. A scleral indenter is used to depress and reposition the eye, as necessary, to ensure visualisation of the extremes and thorough examination of the retina. Unfortunately, ROP screening is distressing for parents and both very painful and stressful for infants,25 resulting in high pain scores, increased salivary cortisol and increased rates of apnoeic episodes in the 24 hours after the procedure. 2 Unfortunately, pain management strategies are at present inadequate26. In the Poppi trial, it was investigated whether or not the administration of morphine prior to ROP screening could significantly reduce the pain caused by the procedure, and whether or not the physiological instability that follows the examination could be alleviated through effective pain relief.
Study rationale
Identifying better pain management strategies for common painful procedures performed in neonatal care is a current clinical priority. ROP screening is an essential painful and destabilising procedure that is repeatedly performed in premature infants, for which adequate analgesia cannot be provided because of a lack of evidence-based analgesics. Morphine is an effective analgesic in children and adults, which is commonly used intravenously for sedation and comfort in ventilated infants despite controversial evidence of its analgesic efficacy. The primary objective of the Poppi trial was to investigate the analgesic efficacy of oral morphine using a well-validated clinical pain measure following ROP screening. However, given the development of the objective (i.e. validated neurophysiological measures of pain in infants), the trial also provided an opportunity to gain a mechanistic insight into how morphine affects nociceptive brain and spinal cord activity in this population. Patterns of nociceptive brain activity evoked by ROP screening have unfortunately not yet been characterised or validated. However, by studying infants whose routine blood tests coincided with the morning of ROP screening, EEG and EMG responses to heel lancing could also be recorded and noxious-evoked brain activity could be used as a co-primary outcome measure assessing the analgesic efficacy of morphine. Given the destabilising effects of ROP screening, a comprehensive approach was also devised to assess changes in oxygen saturation, respiratory rate, heart rate and ventilation requirement in the 24 hours before and after the clinical procedures to determine whether or not morphine analgesia could further improve an infant’s clinical stability.
In this study, we chose to administer an oral dose of 100 µg/kg of morphine prior to clinical heel lancing and ROP screening, based on extrapolation from guidance in the BNF for Children 2015. 27 Many neonatal formularies include oral morphine as a treatment option for pain in infants with doses ranging from 50 to 200 µg/kg. However, no RCTs have previously been completed investigating the efficacy and safety of oral morphine for acutely painful procedures in healthy non-ventilated premature infants. A single dose of 200 µg/kg of oral morphine (double the dose in this trial) had previously been used in an incomplete trial of pain relief for ROP screening,13 and the authors reported in their publication that no adverse effects were observed at this dose.
To ensure that all infants received the same baseline standard of care, morphine or placebo was administered to infants in addition to local pain management practices, which included administration of local anaesthetic eye drops prior to ROP screening and swaddling of the infant during painful procedures.
If a bolus dose of oral morphine were to provide safe and effective analgesia for procedural pain that resulted in improved clinical stability, this would have a significant impact on clinical practice for ROP screening and, potentially, provide an analgesic that could be used for other acutely painful procedures that premature infants must undergo, such as laser eye surgery.
Chapter 2 Trial description
This single-centre, double-blind, placebo-controlled randomised clinical trial aimed to determine whether or not a single dose of morphine sulphate (100 µg/kg) administered orally prior to painful clinical procedures could provide safe and effective analgesia for premature infants.
Infants were randomised to receive either oral morphine or an equivalent volume of placebo solution prior to a clinically required heel lance and ROP screening, performed consecutively on the same test occasion. The co-primary outcomes of the trial (i.e. assessing the analgesic efficacy of morphine) were the magnitude of noxious-evoked brain activity in response to heel lancing and a behavioural pain score [as measured using the Premature Infant Pain Profile – Revised (PIPP-R)] after ROP screening. Secondary outcomes assessed the behavioural pain score and limb reflex withdrawal activity to heel lancing, and the physiological stability of the infants over the 24 hours before and after the trial. Finally, the rate of apnoeic episodes requiring emergency resuscitative intervention [non-invasive positive-pressure ventilation (NIPPV)] was assessed as a safety outcome.
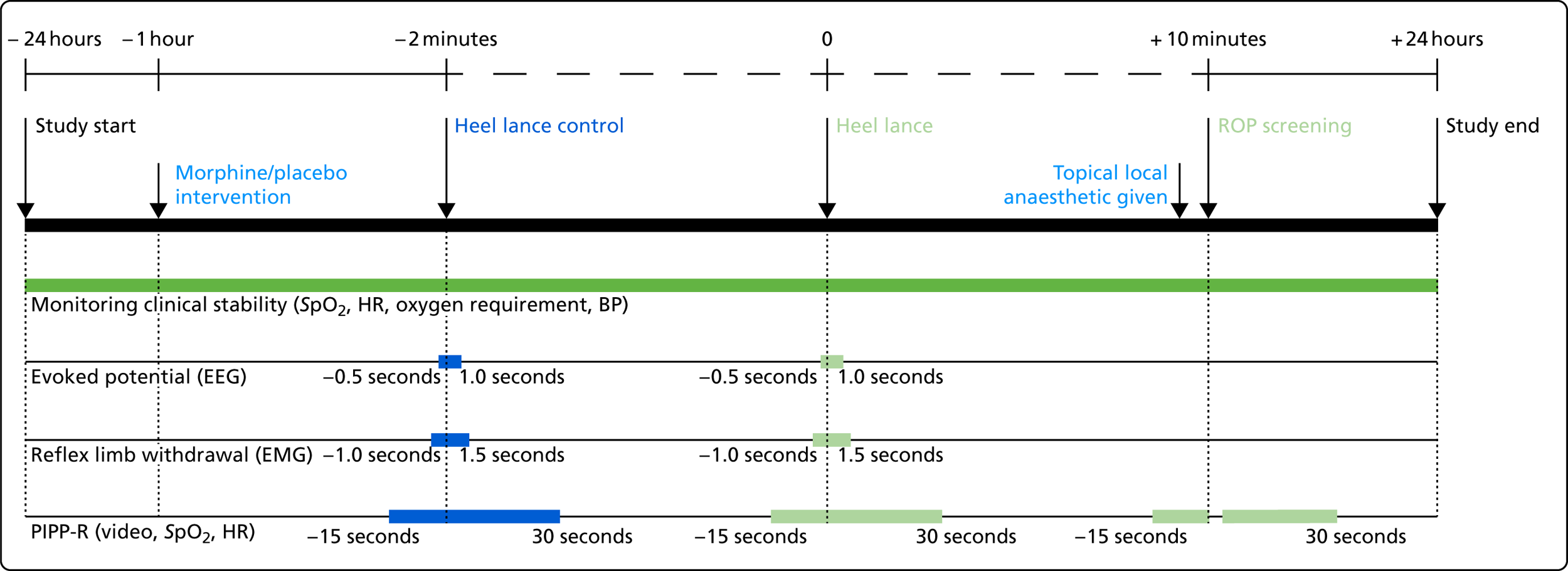
Infants were recruited to the trial from September 2016 to November 2017, and were studied when they required ROP screening and a clinical heel lance on a single test occasion. No noxious procedures were performed solely for the purpose of the study. Infants were included in the study for a 48-hour period; physiological monitoring began 24 hours before the start of the clinical intervention (defined as the heel lance followed by the ROP screening) and ended 24 hours after. Morphine or placebo was administered to infants 1 hour prior to the clinical intervention. EEG and EMG recordings, and data for PIPP-R scoring, were collected only during the clinical intervention. An overview of the experimental protocol is presented in Figure 1, the trial objectives and outcome measures are listed in Box 1, and the trial flow chart is displayed in Figure 2.
FIGURE 1.
Experimental protocol overview. BP, blood pressure; HR, heart rate; SpO2, peripheral capillary oxygen saturation.
-
To test whether or not administration of morphine reduces clinical pain scores (as measured via the PIPP-R) during the 30-second period after ROP screening compared with a placebo (inactive solution).
-
To test whether or not administration of morphine reduces noxious-evoked brain activity following a clinically essential heel lance compared with a placebo (inactive solution).
-
To test whether or not administration of morphine improves clinical stability in the 6-hour and 24-hour periods following the start of the clinical intervention. The clinical intervention is defined as the heel lance followed by ROP screening.
-
To test whether or not administration of morphine reduces clinical pain scores (PIPP-R) and reflex withdrawal activity following a clinically essential heel lance compared with a placebo (inactive solution).
-
To test whether or not administration of morphine is safe by determining whether it results in episodes of respiratory depression or hypotension that require intervention.
-
PIPP-R score during the 30-second period after ROP screening.
-
Magnitude of noxious-evoked brain activity evoked by heel lance.
-
Clinical stability in the 6-hour and 24-hour periods following the start of the clinical intervention. Clinical stability is assessed from pulse oximetry recordings and the need for increased respiratory support.
-
PIPP-R score and magnitude of reflex withdrawal following heel lance.
-
Drug safety is assessed by calculating the number of incidents of apnoea that require intervention using Neopuff™ (Fisher & Paykel Healthcare Limited, Auckland, New Zealand) or bag-and-mask ventilation and the number of incidents of hypotension that require treatment with inotropes in the 24-hour period following the administration of the IMP or placebo.
IMP, Investigational Medicinal Product.
FIGURE 2.
Trial flow chart. IMP, Investigational Medicinal Product; NPEU, National Perinatal Epidemiology Unit.

Chapter 3 Methods
Some of the information in this section has been reproduced from published work by Slater et al. 28 © 2016 Slater et al. 28 This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Recruitment
Screening
Infants were considered eligible for the trial if they met the following inclusion criteria:
-
were inpatients on the neonatal unit at the John Radcliffe Hospital, Oxford
-
were born at < 32 weeks’ gestation or at a birthweight of < 1501 g
-
at the time of the study, were 34–42 weeks gestational age
-
required a clinical heel lance and ROP screening on the same test occasion
-
their parents/guardians had given written informed consent for inclusion in the trial
-
a senior clinician considered inclusion in trial to be medically appropriate.
Infants were considered ineligible for the trial if they met any of the following exclusion criteria:
-
had an intraventricular haemorrhage (IVH) > grade II
-
had short-bowel syndrome
-
were receiving nil by mouth because of documented gut pathology
-
had received opiates in the last 72 hours
-
had received other analgesics or sedatives in the last 24 hours
-
had a previously documented episode of morphine sensitivity
-
had congenital malformation or a genetic condition known to affect neurological development
-
were born to mothers who regularly used opiates during pregnancy or while breastfeeding or while expressing breast milk.
Training in the protocol and eligibility criteria was provided at the site initiation visit, and the secure randomisation website included eligibility checks. This system also highlighted ineligible infants at the point of randomisation, ensuring that staff reviewed this information and could not continue with the randomisation process unless correct.
A screening log (see Report Supplementary Material 8) was maintained throughout the duration of the trial, recording all infants born at < 32 weeks’ gestation or with a birthweight of < 1501 g. The screening log was updated at least once a week by the research staff, documenting information about the infant’s medical record number, date of birth, initials, gestational age at birth, sex, expected date of first ROP examination, eligibility status for the trial, the date the parent information leaflet (PIL; see Report Supplementary Material 6) was given, consent provided, and eligibility status at the proposed time of randomisation. The sponsor reviewed the screening log (see Report Supplementary Material 8) and participant eligibility during monitoring visits; any concerns were stated in the monitoring report and issued to the research team for resolution.
Parental approach
Recruitment was carried out by the research team and health-care professionals who had received Poppi trial training and Good Clinical Practice (GCP) training (as documented on the training log; see Report Supplementary Material 11 and 12), and were signed off to take consent on the delegation log (see Delegation; see Report Supplementary Material 4). To ensure that potentially eligible infants were not missed, Neonatal Intensive Care Unit (NICU) staff were informed about the Poppi trial to enable them to help with identification and recruitment. Promotional materials, such as roll banners, stating the trial eligibility criteria were on display around the NICU (see Improving consent rate).
The research team tried to identify eligible infants as early as possible during their hospitalisation. Following discussion with the clinical team in charge of the infant’s care, parents were approached by the research team and provided with a copy of the PIL for reference (see Appendix 4 and Report Supplementary Material 6). The eligibility of the infant was also documented in the infant’s medical notes at this time by a senior clinician. The research team ensured that parents were given plenty of time to consider participation in the trial, and arranged follow-up meetings for further discussion and to answer any questions, to ensure that parents gained a thorough understanding of the trial prior to consent.
During these meetings, the researchers introduced themselves and described their role within paediatric research at the John Radcliffe Hospital, Oxford. The researchers explained that the research team investigates brain development and how infants respond to sensory stimuli, in particular pain, and discussed the importance of this work to enable the identification of drugs that can relieve pain caused by essential clinical procedures required in premature infants in neonatal intensive care. The researchers informed the parents of the eligibility of their infant to participate in the trial and enquired whether or not the parents would be interested in receiving further details. If parents declined, the researchers did not provide further information, and the screening log (see Report Supplementary Material 8) was updated to confirm that the parents had declined. However, if parents were interested the researchers clarified that participation was voluntary, without obligation, and withdrawal from the trial was possible at any time. The parents were informed that the trial had been reviewed by an ethics committee, was funded by the Wellcome Trust and the National Institute for Health Research (NIHR) Efficacy and Mechanism Evaluation (EME) programme, and sponsored by the University of Oxford.
The parents were given an overview of the eligibility criteria and an explanation of the rationale behind the trial. The researcher explained that the trial was investigating whether or not morphine could provide pain relief for premature infants. They discussed the widespread use of morphine for pain relief in adults and older children, and its intravenous use in ventilated preterm infants. Furthermore, the researcher explained that morphine is not currently used for short-term pain relief for procedures in infants. The researcher stated two aims of the trial: to identify whether or not morphine reduces infant pain scores following ROP screening, and whether or not morphine reduces pain-specific brain responses to heel lancing.
The researcher explained that the trial was a RCT and, therefore, half of the infants taking part would be randomly allocated to receive morphine and half would receive an inactive placebo of the same volume (containing the additive ingredients of the morphine solution only). The researcher explained that the infants would be allocated to a treatment using a computer system and that the trial was blinded, which meant that nobody would know whether the infant had received morphine or placebo, to prevent bias in the analysis. The researcher always clarified, however, that in an emergency the clinical staff could find out which treatment the infant had been given.
Parents were informed that the trial involved monitoring the infants for 24 hours before and 24 hours after their ROP screen, using the same type of leads and machine used clinically to monitor their infant’s heart rate, breathing rate and oxygen saturations. The infant would be given the study drug by mouth or via their feeding tube approximately 1 hour before their planned heel lance and ROP screen. After receiving the study drug, the research team would set up EEG monitoring, which measures brain activity, by placing 10 electrodes (small discs) on the infant’s head using a soluble paste. Sticker electrodes would also be placed on the infant’s thighs to measure their leg reflex away from the stimulus. The researcher explained that these measures would record the infant’s brain activity and leg activity during heel lancing and ROP screening. The researcher described the blood sampling procedure, which would be conducted using a heel lance as standard on the unit, around 5–10 minutes before the ROP screen. Parents were also told that before the heel lance for blood sampling, their infant would also have a control heel lance, which involved the lancet being held against the heel of the foot with the end pointing away from the foot so that when the device was depressed the blade did not pierce the skin. This provided a non-painful comparison with the actual heel lance.
The researcher told the parents that the team would video their infant during the procedures to score the infant’s pain using recordings of their facial expression, and changes in their heart rate and oxygen saturations. The infant would then have their routine ROP screen conducted as usual by one of the ophthalmologists. The EEG and EMG recording would carry on throughout this, and the infant’s face would be videoed again for approximately 2 minutes in order to score the pain after the ROP screen. At this point, the team would settle the infant if needed and take the EEG off. The researcher then explained that the infant would be monitored for a further 24 hours for any effects of the drug or ROP screen (such as an increase in desaturations or bradycardias). Parents were told that the research team would communicate with the clinical team if any problems were identified on the recordings. The researcher also explained that the infant would be randomised only once and studied on one occasion. Parents were told that the team aimed to recruit 156 participants over a 3-year period.
Advantages and disadvantages of participating were discussed. The disadvantages outlined included potential reactions to morphine. The researcher explained that the team would monitor the infant closely for reactions and would work with the clinical staff on the ward to ensure the infant’s safety. There were no direct advantages to taking part, as the team did not know whether or not morphine reduced the effects of the ROP screen and blood test, but the results would help improve understanding of how infants respond to pain and morphine, and demonstrate whether or not morphine is effective at reducing pain in infants. The researcher explained that the team would publish the results of the trial so that they could be used to inform clinical practice and further research. Parents were told that they would be sent a copy of the results once available.
Finally, the research team also explained to parents that the team would review their infant’s medical notes in order to record their birthweight, gestation, age and relevant medical history on a secure online system. Parents were informed that other members of the research team, trial sponsor, regulatory authorities and Oxford University Hospitals (OUH) NHS Foundation Trust may also review relevant sections of the medical notes. Each infant would be referred in documents using an allocated study number, to preserve anonymity. All relevant medical information and video footage would be kept securely, and personal identifiable data would be treated confidentially and according to UK legislation. Parents were also told that if they decided not to enter the trial, their infant would receive the same care that other infants on the neonatal unit receive.
Informed consent
Written consent was obtained before each infant was randomised. Only the mother, father, or person designated by legal process, could sign the consent form (see Appendix 4 and Report Supplementary Material 2). Fathers with parental responsibility could sign a consent form if married to the mother, named on the birth certificate, or if granted parental responsibility through a court order or parental responsibility agreement.
In the case of twins or triplets, each infant had a separate signed consent form. The researcher taking informed consent verified that the parents had read and understood the PIL (see Report Supplementary Material 6), and asked the parents to outline their understanding of what the study involved. The researcher clarified any missing or inaccurate details, before reiterating each item on the consent form (see Report Supplementary Material 2). Parents initialled each box of the consent form and the full names of both the parent and the health professional were clearly recorded with the date.
Following randomisation (see Randomisation), the consent form (see Report Supplementary Material 2) was annotated, and the screening log (see Report Supplementary Material 8) and enrolment log (see Report Supplementary Material 5) were updated. The four copies of the consent form were distributed as follows: the original was sent to the co-ordinating centre [i.e. the National Perinatal Epidemiology Unit Clinical Trials Unit (NPEU CTU), University of Oxford, Oxford, UK], one copy was given to a parent, one copy was filed in the investigator site file (ISF) and one copy was filed in the infant’s medical notes together with a copy of the PIL (see Report Supplementary Material 6) and randomisation details. The recruitment process is outlined in Figure 3.
FIGURE 3.
The recruitment process.

Training and reference documentation for the process of obtaining informed consent, and ensuring accurate formal patient identification, were provided for the site team. This was to ensure that consent forms (see Report Supplementary Material 2) were completed accurately and according to GCP. Consent forms were also checked by the trial manager before filing in the trial master file (TMF), to ensure that they were completed accurately by parents and health professionals. The trial manager also checked that all individuals taking consent had been delegated responsibility by the chief investigator to do so. Copies of delegation logs (see Report Supplementary Material 4) and training logs (see Report Supplementary Material 11 and 12) were supplied to the trial manager on a regular basis. Language barriers were not a concern because the neonatal unit had access to face-to-face interpreters and a telephone interpretation line if required.
Randomisation
Infants were randomised as close as possible to the start of the 48-hour monitoring period (24 hours prior to a clinical heel lance). Eligibility was re-assessed at this time and documented in the medical notes.
Randomisation of participants to morphine sulphate or placebo was managed via a secure web-based randomisation facility hosted by the co-ordinating centre. Participants had an equal chance of receiving morphine sulphate or placebo, and a minimisation algorithm was used to ensure that there was an approximate balance between the groups with respect to gestational age at the time of randomisation, gestational age at birth, number of days on a ventilator, presence of a gastric tube at the time of randomisation, number of days since morphine had been given, intrauterine growth restriction and previous surgery. The minimisation algorithm used a probabilistic mechanism to increase unpredictability and the users of the system had no insight into the next allocation. The balance between the groups for each of the minimisation variables was reviewed by the Data Monitoring Committee (DMC; see Data Monitoring Committee) and reported in the trial publication.
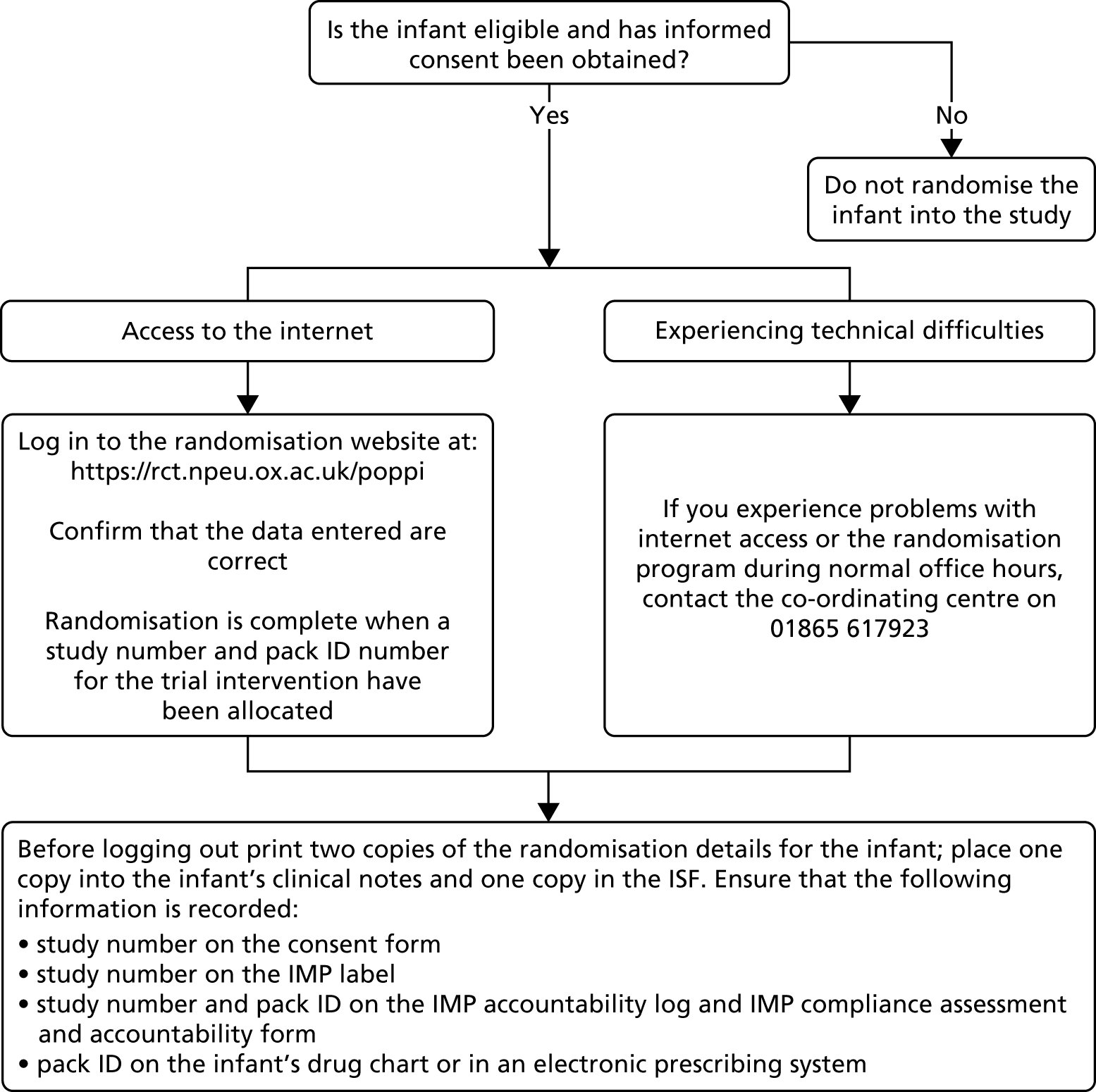
Following confirmation of eligibility and parental informed consent, a researcher randomised the infant by logging into the randomisation program with a username and password at https://rct.npeu.ox.ac.uk/poppi or via the Poppi trial website (www.npeu.ox.ac.uk/poppi) using the ‘randomisation’ link. The researcher verified the inclusion or exclusion criteria to confirm eligibility. The infant was recruited and randomised into the trial only following confirmation that the entered data were correct. The randomisation program provided a five-digit numeric study number and five-character alphanumeric pack identifier (ID) (relating to morphine or placebo). The researcher subsequently checked that the correct pack ID was available and unopened in the temperature-controlled cupboard in the NICU (see Stock management and storage). If it could not be located, or the package was open, the researcher could select ‘Allocate other pack’ from the main menu and the randomisation program allocated a new pack ID of the same type as the original allocation (i.e. active or placebo). The researcher printed two copies of the randomisation details, filing one in the infant’s medical notes and one in the ISF. The researcher added the study number to the consent form (see Report Supplementary Material 2) and the Investigational Medicinal Product (IMP) label; the study number and pack ID to the IMP accountability log (see Report Supplementary Material 1) and the IMP compliance assessment and accountability form (see Stock management and storage); and the pack ID to the infant’s drug chart or electronic prescribing system.
If there were problems accessing the online randomisation program during normal working hours, the research team contacted the co-ordinating centre by telephone (the co-ordinating centre could carry out the randomisation on behalf of the research team). The trial manager would report system failures to the programming team, and they would be investigated by the senior trials programmer. Following randomisation, the researcher placed a cot card (see Report Supplementary Material 3) on the infant’s cot and a notes sticker on the infant’s medical notes, and ensured that the clinical team were aware that the infant was enrolled in the Poppi trial. The trial’s randomisation process is outlined in Figure 4.
FIGURE 4.
Randomisation process.
Recruitment challenges
Throughout the trial, the research team encountered a number of challenges to recruitment. A summary of recruitment is presented in Chapter 4, Results, but overall the recruitment rate was approximately 50% of the target rate.
Barrier nursing (December 2016–January 2017)
No infants were recruited from mid-December 2016 to mid-January 2017, as the neonatal unit at the John Radcliffe Hospital had an outbreak of an infectious strain of Serratia marcescens. This bacterium was isolated in a number of infants who were potentially eligible for inclusion in the Poppi trial. It was not clinically appropriate to approach these infants because of the highly infectious nature of this organism and the need for strict barrier nursing.
Retinopathy of prematurity screening
Following the start of recruitment, the research team realised that some parents were not aware that their infant was due to undergo, or had already undergone, ROP screening. This represented an unmet clinical need and complicated recruitment to the trial, as a full explanation of the clinical procedure was necessary prior to explaining why the trial was taking place. The amount of information being conveyed was overburdening parents, resulting in a negative impact on recruitment. The team decided that information provision about ROP screening should be improved. The team identified a previous ROP screening PIL that was no longer being distributed to parents as it provided out-of-date information regarding the procedure. The clinical members of the research team revised and re-designed the ROP screening PIL, which was subsequently approved by the trust, endorsed by the Support for the Sick Newborn and their Parents (SSNAP) charity, and is now distributed on the neonatal unit as standard practice. The research team agreed that this PIL rapidly increased parents’ awareness of the procedure, which improved the parental approach for the trial and recruitment.
Promotion of early discharge from the neonatal unit also affected recruitment. Infants could participate in the trial only if undergoing ROP screening as inpatients at the John Radcliffe Hospital (as the trial procedures involved 24-hour monitoring of the infant before and after the clinical intervention); therefore, infants undergoing ROP screening in outpatient clinics could not be recruited. Many infants whose parents were approached about trial participation were discharged prior to ROP screening and underwent the procedure as outpatients. The rates of early discharge increased because of the success of the home feeding programme. The Project Management Group (PMG; comprising the chief investigator, principal investigator, core team at site, trial manager, trial statistician, co-ordinating centre programming team and co-ordinating centre senior management team) considered the possibility of providing a clinical area where infants could return for their ROP screens as inpatients. This would have potentially increased the mean age of participants. Nevertheless, the logistics and financial implications were not fully explored by the PMG or the Trial Steering Committee (TSC; see Trial Steering Committee), as the trial was closed early (see Trial closure).
Improving consent rate
The research team incorporated a number of strategies to improve the consent rate (initially at approximately 40%), by raising awareness of the trial and by maximising the information resources available to staff and parents. This included improving trial visibility among staff by investing in branded lanyards and badges for the research team to wear, providing branded mugs in the staff areas, and distributing branded pens and hand sanitisers for use on the neonatal unit. Trial posters (see Report Supplementary Material 7) and roll banners were also displayed around the neonatal unit, and the trial was presented at grand rounds by members of the research team. The research team also liaised with SSNAP (see Patient and public involvement) to increase its involvement in the trial for further promotion. SSNAP teddy bears were purchased and Poppi trial badges were attached, and these were provided to parents by the SSNAP team. The SSNAP team also identified a number of volunteers to act as Poppi trial champions, who promoted the trial and discussed it with parents. The research team also attended SSNAP coffee mornings to build relationships with the parents and answer questions regarding the Poppi trial, neonatal pain or their infant’s care. The group also created contact cards with the core site team’s pictures and roles, so that members of the Poppi trial team could easily be identified on the neonatal unit.
These attempts to increase the trial’s visibility among staff and parents were very successful in improving the consent rate over the last few months of recruitment. The consent rate increased from 40% to approximately 75%. The research team also considered recruiting existing clinical nurses to act as Poppi trial champions. The Poppi trial research nurse identified five or six interested nurses and a training programme was devised with the trial manager. Unfortunately, the Poppi trial champion training was not implemented prior to the trial terminating early.
Re-randomisation
The PMG considered randomising participants more than once to increase the likelihood of reaching the recruitment target. Trial participants could potentially have been randomised into the trial on more than one occasion as they often required multiple ROP screens. Each ROP screen would have constituted an individual test occasion, with separate informed consent and an independent randomisation process. ROP screening is repeated with a minimum interval of approximately 1 week; therefore, there would have been no overlap between test occasions. Repeated exposure to morphine or placebo would have been unlikely to impact patient safety or the validity of outcome data considering that all infants would have experienced prior pain and many would have previously been exposed to morphine. Approximately 60% of the infants who participated in the trial could have been studied on a second occasion if re-randomisation had been originally implemented. The PMG concluded that introducing re-randomisation would contribute to an increased recruitment rate, and would not compromise patient safety or the scientific integrity of the Poppi trial. The TSC was consulted and planned to discuss the proposal at the next oversight meeting. However, the trial was stopped before this could be considered further.
Reassessment of power calculation and eligibility criteria
Prior to the early closure of the trial, the PMG considered re-assessment of the power calculation to address difficulties with recruitment. The original power calculation was based on pre-existing data, but new data were available from a recent publication from the research team. 22 The repeat power calculation suggested that the sample size required was lower than initially calculated. The original calculation had also accounted for a broad age range, but infants recruited to the trial were aged 34–36 weeks’ gestation. Therefore, the age could be restricted to an upper limit of 38 weeks’ gestation (without affecting potential re-randomisation in the event of a second inpatient ROP screen). The PMG agreed that the power calculation would need to consider both co-primary outcome measures, but a re-calculation could be justified given the new data available. The PMG also considered extending the lower age limit to 32 weeks’ gestation, the minimum age at first ROP screening. However, the pattern of noxious-evoked brain activity used as the co-primary outcome in the trial is not often observed in infants aged < 34 weeks’ gestation19 and the published measures were validated only for infants aged ≥ 34 weeks’ gestation. However, the Poppi trial was stopped before a decision was reached.
Additional research sites
The PMG also discussed the option of opening more research sites to address the low recruitment rate. However, this would require considerable additional funding for a full-time research nurse and specialist equipment. The recording and measurement techniques used in the trial were developed by the research group, so intensive training at additional sites would be required to ensure that there was confidence and competency in study set-up, collection of data and identification of key values (e.g. measures of noxious-specific brain activity). Alternatively, the original research team could have visited other sites when infants were recruited. However, as a result of the labour-intensiveness of the trial and expertise required this was not a feasible option.
Investigational Medicinal Product management
Participants received an oral dose of either morphine or an inactive placebo. The active IMP comprised morphine sulphate suspended in a cherry-flavoured oral suspending base (SyrSpend®; Fagron BV, Rotterdam, The Netherlands) and the placebo was SyrSpend in isolation. A pre-prepared solution at a standard concentration, as specified in the protocol, was used. As the Poppi trial was blinded, staff and parents did not know the treatment allocation of the infants. Infants had a 50% chance of receiving placebo or morphine. The IMP was provided in 10-ml amber-coloured glass bottles, with an individual sterile oral/enteral 3-ml syringe. On randomisation, infants were allocated an IMP pack ID. Issues with IMP manufacturing were the key source of delays to the trial opening for recruitment. Further information is provided in Appendix 1. All IMP stock was held in a temperature-controlled cupboard on the neonatal unit; further information about temperature control and monitoring is presented in Appendix 2.
Research staff were aware of the potential side effects of morphine, which include vomiting, constipation, drowsiness, hypersensitivity reactions, bradycardia, tachycardia, hypothermia, muscle rigidity, urinary retention, antidiuretic effect and respiratory depression. The temperature of the IMP was managed at all times to ensure that it remained below the safe limit of 25 °C.
Stock management and storage
Electronic stock management
The trial used an online stock management system, which was developed by the co-ordinating centre and accessed through the randomisation website. The status (i.e. in transit to hospital, in the pharmacy, in the neonatal unit, allocated, destroyed) for all IMP packs was available on this online stock management system. Once pack numbers had been generated by the head of trials programming, these appeared in the system. The trial manager then ‘assigned’ to the site the pack IDs that were being manufactured and an automatic alert e-mail was sent to the trial manager, pharmacy, research nurse and Stockport Pharmaceuticals (Stockport NHS Foundation Trust, Stockport, UK). Once manufacture and qualified professional (QP) release were complete, the trial manager ‘sent’ the packs to the hospital on the online system (an automatic alert e-mail was sent to the trial manager, pharmacy and research nurse). On receipt of the packs at pharmacy, the pharmacy team used the online system to register packs as ‘in pharmacy’ and indicate any damaged packs (an automatic alert e-mail was sent to the trial manager, pharmacy and research nurse). The research nurse collected the undamaged IMP packs from the pharmacy and transferred them to the temperature-controlled cupboard on the neonatal unit, and registered them ‘in NICU’ on the online system, triggering an automatic alert e-mail to the trial manager, pharmacy and research nurse. The randomisation system could then be used to allocate packs to participants at randomisation. Throughout the trial, the trial manager monitored stock levels.
Paper stock management
When an IMP batch was received and transferred to the neonatal unit, the research nurse completed a stock transfer form documenting the pack IDs and date of transfer. An accountability log (see Report Supplementary Material 1) was maintained by the research nurse indicating pack ID, date of dispense from the pharmacy, the study number of the participant to whom the pack had been allocated and the eventual outcome of the pack, that is, either returned to the pharmacy or used and/or destroyed (and the corresponding date). Copies of these documents were forwarded to the trial manager for storage in the TMF.
Investigational Medicinal Product prescription and administration
Only individuals signed off on the delegation log (see Report Supplementary Material 4) were prescribed the IMP, using an IMP compliance assessment and accountability form (a dedicated prescription chart; see Figure 5) and the drug monograph for the IMP. Doses were oral/nasogastric and prescribed at 100 µg/kg. Once the IMP compliance assessment and accountability form was completed, the same prescription was written on the infant’s drug chart as ‘Poppi IMP Morphine or Placebo Pack ID XXXX’. The working weight was used to calculate the IMP prescription; an example dose calculation is presented in Figure 6.
FIGURE 5.
Investigational Medicinal Product compliance assessment and accountability form.

FIGURE 6.
Investigational Medicinal Product prescription calculation.

The dose was also double checked by qualified clinical staff and training was provided to include discarding excess solution prior to administration to the infant. Members of staff prescribing, preparing and administering the IMP were listed on delegation log (see Report Supplementary Material 4) with their designated duties. Any qualified member of the neonatal nursing staff was able to check the IMP dose; staff did not need to be listed on the delegation log (see Report Supplementary Material 4) because checking doses forms part of standard neonatal care. The IMP was administered after the first set of ROP eye drops; this was approximately 1 hour before the heel lance and ROP screen, allowing sufficient time for the morphine to be absorbed. The IMP could be administered by orogastric tube, nasogastric tube or directly into the mouth. The IMP was never added to a bottle feed in case the infant did not complete the feed. Following administration, the second part of the IMP compliance assessment and accountability form and the infant’s drug chart were signed to indicate administration (Figure 7). The form was returned to the co-ordinating centre each time the IMP was administered and checked by the trial manager to ensure that accountability had been demonstrated. Any errors identified regarding drug administration would have been reported as incidents and assessed, before being reviewed by the PMG. The trial sponsor also reviewed drug accountability records during site monitoring.
FIGURE 7.
The IMP compliance assessment and accountability form.

Accountability and destruction
Once the IMP had been administered, any IMP remaining in the vial was disposed of as per trust procedure (excess controlled drugs were disposed of in a sharps bin along with the bottle, syringe and packaging).
Any quarantined packs, packs that could not be allocated (e.g. for participants who had withdrawn after randomisation and pack allocation, but before IMP administration) and expired packs were returned to the pharmacy for destruction. A stock transfer form was completed documenting the transfer of IMP packs from the neonatal unit to the pharmacy, and the accountability log (see Report Supplementary Material 1) was updated to reflect that these packs had been returned to pharmacy. Stock transfer forms and updated accountability logs (see Report Supplementary Material 1) were e-mailed to the co-ordinating centre for storage in the TMF. In the case of expired or quarantined packs, the head of trials programming updated the online stock management system to prevent these packs from being allocated on randomisation.
The pharmacy contacted the trial manager to request authorisation for the destruction of transferred IMP packs. Following authorisation and destruction, the pharmacy team e-mailed the co-ordinating centre a copy of the trust accountability and destruction log to confirm that all relevant IMP packs had been destroyed. The trial manager marked each IMP pack on the online stock management system as ‘destroyed’.
Study processes
Recording techniques
Approximately 1 hour before the heel lance (and after randomisation), the IMP was administered. The research team then set up the EEG, EMG and video monitoring. When the drug reached maximum efficacy (i.e. approximately 1 hour post administration) a heel lance control stimulus and clinical heel lance were performed. During each procedure noxious-evoked brain activity (as measured using EEG), reflex withdrawal activity (as measured using EMG on the biceps femoris of each leg), physiological activity (including oxygen saturation and heart rate) and facial expression change (using video monitoring) were recorded.
After the heel lance was complete, routine ROP screening was carried out by an ophthalmologist or an appropriately trained ophthalmology trainee. The time taken for the screening to be performed was recorded. Pulse oximetry, respiratory monitoring and video monitoring were recorded during and immediately after the ROP screen.
Once the ROP screen was complete, pulse oximetry, oxygen requirement and blood pressure continued to be monitored and recorded by the data-logging equipment for 24 hours after the start of the clinical intervention. Clinical stability was assessed throughout the 48-hour trial period. These measures were calculated from pulse oximetry recordings and requirement for respiratory support. Pulse oximetry data were monitored and downloaded to the data-logging equipment for 24 hours before and 24 hours after the start of the clinical intervention. Throughout the 48-hour trial period, blood pressure was monitored every 6 hours and changes in respiratory support (including type of respiratory support modality and oxygen requirement) were recorded. Any identification of abnormal clinical findings would have been reported to the clinical team. The recording measures used in the trial are summarised in Table 1.
| Recording technique | Measurement |
|---|---|
| EEG | Noxious-evoked brain activity |
| EMG | Reflex withdrawal |
| Pulse oximetry | Heart rate |
| Pulse oximetry | Oxygen saturation |
| Video recording | Facial expression change |
| Blood pressure monitor | Blood pressure |
| Oxygen flow meter (if required) | Oxygen requirement |
Electroencephalography
Electrophysiological activity was recorded using the ™SynAmpsRT 64-channel headbox (Compumedics Neuroscan, Abbotsford, VIC, Australia) and Compumedics Neuroscan amplifiers (bandwidth from direct current – 400 Hz, sampling rate 2 kHz), and recorded using ®CURRYSCAN7 Neuroimaging Suite (Compumedics Neuroscan). All equipment conformed to the electrical safety standard for medical devices, IEC 60601-1. Eight EEG recording electrodes were positioned on the scalp at Cz, CPz, C3, C4, FCz, T3, T4 and Oz according to the modified international 10–20 system. Reference and ground electrodes were placed at Fz and the forehead, respectively. EEG conductive paste was used to optimise contact with the scalp. All impedances were reduced to approximately 5 kΩ by rubbing the skin with the EEG preparation gel prior to electrode placement. An ECG electrode was also placed on the left clavicle and a movement transducer on the abdomen to record respiration.
Electromyography
Bipolar EMG electrodes (Ambu® Neuroline 700 solid-gel surface electrodes; Ambu A/S, Ballerup, Denmark) were placed on the biceps femoris of each leg to measure reflex withdrawal. EMG was recorded before and after the control heel lance and the heel lance.
Clinical pain scores
Clinical pain scores were calculated using the validated PIPP-R score. PIPP-R is a composite measure of behavioural, physiological and contextual factors associated with the pain response: gestational age, behavioural state, heart rate, oxygen saturation, and duration of brow bulge, eye squeeze and nasolabial furrow. Each indicator is rated on a four-point scale (0–3) and summed (to a maximum possible score of 21). In the PIPP-R, gestational age and behavioural state (contextual indicators) are included only if either the physiological or the behavioural variables are scored > 0. 15
Videos of facial expression were recorded throughout the procedures and scored offline from single frames using the PIPP-R facial coding system. Changes in heart rate and oxygen saturation were downloaded from the pulse oximeter and used to calculate the PIPP-R score. Heart rate, oxygen saturation and facial expression were recorded in the 15-second period before and the 30-second period after the heel lance and the heel lance control. These measures were recorded in the 15-second period before ROP screening and in the 30-second period after ROP screening was complete. 14,15
Pulse oximetry
Heart rate and oxygen saturation were measured throughout the 48-hour study period and downloaded to a computer. These data were used to calculate the clinical pain scores following the heel lance and ROP screening, and to assess clinical stability during the 6- and 24-hour periods after the start of the clinical intervention. Data were analysed offline and the numbers of clinically relevant episodes were calculated based on the following definitions:
-
bradycardic episode – pulse rate of < 100 beats per minute (b.p.m.) for at least 15 seconds
-
tachycardic episode – pulse rate of > 200 b.p.m. for at least 15 seconds
-
desaturation episode – oxygen saturation of < 80% for at least 10 seconds
-
apnoea episode – cessation of respiratory air flow for at least 20 seconds.
Episodes of artefact (i.e. infant handling or essential clinical procedures) were documented throughout the recordings.
Blood pressure monitoring
Blood pressure was monitored using a standard blood pressure cuff approximately every 6 hours during the 48-hour study period.
Increased respiratory support
Any change in respiratory support modality or a significant change in oxygen requirement was documented during the 48-hour study period. Increased respiratory support was defined as a significant increase in oxygen requirement or an increase in ‘respiratory support modality’. Respiratory support modality was classified on a graded 1–4 scale: grade 1 = self-ventilating in air; grade 2 = low flow (0.01–0.35 l/minute; 100% oxygen); grade 3 = high flow (1–8 l/minute), continuous positive airway pressure, or duoPAP (21–100% oxygen); and grade 4 = ventilator (21–100% oxygen). If an infant had a change in ‘respiratory support modality’ that resulted in an increase in grade, this was considered an increase in respiratory support. A significant increase in oxygen requirement was defined as an increase in oxygen supply by more than 10%, a flow rate change of more than 1 l/minute (if receiving high-flow therapy) or a flow rate change of more than 0.04 l/minute (if receiving low-flow oxygen).
Clinical stability recording
Clinical stability was assessed by the number of episodes of bradycardia, number of episodes of tachycardia, number of episodes of desaturation, number of episodes of apnoea, and requirement for an increase in respiratory support at the 6- and 24-hour time points before and after the clinical intervention.
Outcome measures
Premature Infant Pain Profile – Revised
Pain scores were calculated using the PIPP-R score (to a maximum value of 21; higher score implies greater pain). PIPP-R scores were calculated during the control heel lance, during the heel lance and during the 30-second period after the ROP screening (it was not possible to accurately calculate PIPP-R scores during ROP screening because the eyes were held open by a speculum). If the trial had not been stopped early (see Trial closure), an interim analysis was planned for once data had been collected from 50% of trial participants. In this planned analysis, 20% of PIPP-R scores would have been re-calculated by two investigators to ensure that there was inter-rater and intra-rater reliability. Inter-rater and intra-rater reliability would also have been calculated in 20% of trial participants at the end of the trial.
As the trial stopped after 31 participants had been recruited (one infant was withdrawn, so data were available only for 30 participants), the PMG agreed that all PIPP-R scores would be re-calculated. The research nurse was the primary scorer and a researcher from the site team was the second scorer. Inter-rater reliability was then assessed. The research nurse then calculated all PIPP-R scores again; these were compared with the original scores and assessed for intra-rater reliability.
The research nurse and researcher were both well experienced in calculating PIPP-R scores and further training was not necessary. No other individuals calculated PIPP-R scores in this trial, but had the trial not stopped early there may have been other less experienced scorers. In preparation for this, a guidance sheet was available for reference. This guidance sheet provided step-by-step instructions for how to calculate PIPP-R scores. Before calculating any PIPP-R scores for the trial, any inexperienced researchers would have been required to practise PIPP-R scoring using a training set of at least 30 infant videos and accompanying relevant data. The scores calculated by an experienced PIPP-R scorer for these videos were available; the researcher would then compare their calculated scores for this training set with the pre-calculated scores, and conduct an inter-rater reliability assessment.
Noxious-evoked brain activity
Electroencephalography data were high-pass filtered at 0.5 Hz and low-pass filtered at 70 Hz, with a notch filter at 50 Hz and harmonics. Noxious-evoked brain activity evoked by a clinical heel lance has been well characterised in previous studies. 19,20,29 A template based on the principal component, which reflects the noxious-evoked activity in infants, was defined in an independent data set. 22 This noxious-evoked template was projected onto the EEG data recorded in the 400- to 700-millisecond period following each heel lance and control heel lance, and the relative weight of the component was calculated for each infant to determine the magnitude of the noxious-evoked brain activity. A greater weight indicated a stronger noxious-specific-evoked response. Prior to projecting the template, the data were first Woody filtered in the region of 400–700 milliseconds after the stimulus with a maximum jitter of ± 50 milliseconds to allow for individual variation in latency to the response. Although the brain activity characterised was directly related to nociceptive input, it did not reflect all nociceptive activity that took place across the brain or all aspects of the pain experience. 22 The response to the control heel lance was recorded to confirm that the brain activity evoked by the heel lance was noxious. This formed an important data quality control check. 19
Reflex withdrawal
The reflex withdrawal response was characterised using the EMG data. EMG data were high-pass filtered at 10 Hz and low-pass filtered at 500 Hz, with a notch filter at 50 Hz and harmonics. The data were baseline corrected to the pre-stimulus mean and the root-mean-square (RMS) of the signal calculated in 250-millisecond windows. 30 The average RMS value was calculated in the 1000 milliseconds post stimulation. 29,30 Higher values imply greater pain.
Clinical stability measures
Clinical stability was assessed using five clinical stability measures calculated in the 6-hour and 24-hour periods before and after the start of the clinical intervention. The following outcome measures were calculated: the number of episodes of bradycardia; the number of episodes of tachycardia; the number of episodes of desaturation; and the number of episodes of apnoea. Episodes of bradycardia, tachycardia and oxygen desaturation were presented as the standardised difference in number of episodes between the 24- or 6-hour period before and after the clinical intervention. The standardised difference in number of episodes in the periods before and after the clinical procedure was defined as the difference in number of episodes, as a proportion of the total number of episodes, for every infant, symmetrically in both the 6- and the 24-hour periods (e.g. in the 24-hour period post procedure relative to the 24-hour period pre procedure); therefore, results could range from –1.0 to 1.0. A positive value indicated more episodes of the outcome measure in the time period after the clinical intervention than in the period before; a negative value indicated more episodes of the outcome measure in the time period before the clinical intervention than in the period after. A value of zero indicated no difference in the number of episodes of the outcome before and after the clinical intervention.
The episodes of apnoea outcome was calculated as a composite of infants who either developed new-onset apnoeas or had an increased number of apnoeas in the 6- and 24-hour periods after the clinical intervention.
Requirement of an increase in respiratory support was also calculated as a binary outcome.
Safety
The safety of the intervention was established by calculating the number of incidents of apnoea that required intervention using Neopuff or bag-and-mask ventilation and the number of incidents of hypotension that required treatment with inotropes in the 24-hour period following the administration of the IMP.
Other
The trial team were aware that any changes to local practice and/or national guidelines for the use of sucrose or a pacifier as a standard comfort technique during clinical procedures might impact outcome data. To combat this, the PMG decided that all comfort techniques would be recorded on case report forms (CRFs) and monitored by the DMC.
Clinical procedures
Heel lance
Infants were given a heel lance during routine investigations when blood samples were clinically required. Given the frequency with which blood samples are required in premature infants, the heel lance could be timed to occur immediately before the ROP screening. The heel lance was linked electronically to the recording equipment, using methods that have been used in previous studies. 19,20,29 This methodology provided an opportunity to record the precise timing of when the heel lance occurred without interfering with clinical practice. Each infant also received a control heel lance, in which the lancet was rotated such that on release the blade did not touch the foot.
Retinopathy of prematurity screen
All infants born at < 32 weeks’ gestation or with a birthweight of < 1501 g are screened for ROP. 25 The decision to conduct ROP screening was for clinical reasons and independent of the trial. National and local policy guidelines were followed to determine when screening would be performed. 25 The research team was aware that ROP screening for infants eligible for the trial may change weekly and, therefore, checked timings each week. The ophthalmology team were made aware that timings were critical and needed to be adhered to at all times as per the protocol, and the ophthalmology team was asked to alert the research team as soon as possible if there were any problems with timings (i.e. at least 24 hours before the planned test occasion).
The ophthalmology team was also asked to contact the team a minimum of 1 hour before the test occasion in the case of an emergency change in timings, to allow the team to delay IMP administration. The team also emphasised that changes in timings could reduce possible morphine effect and influence the results of the trial.
An ophthalmologist or suitably experienced ophthalmology trainee carried out screening as per standard practice. The ophthalmologist/ophthalmology trainee was reminded that EEG would be carried out during the ROP screen to measure brain activity prior to, throughout and following the procedure. The ophthalmologist/ophthalmology trainee was asked to avoid contact with the leads and to not hold the infant’s forehead, as too much contact or disturbance would result in artefact and make the EEG for that time frame unusable. The screening procedure involved dilatation of both pupils using mydriatic eye drops (i.e. 1% tropicamide and 2.5% phenylephrine) approximately 1 hour prior to ROP screening. At the time of ROP screening, an assistant supported the infant’s head. Topical local anaesthetic (i.e. 0.5% proxymetacaine) instillation was followed 10 seconds later by insertion of an eyelid speculum, which was used to hold the eyelid open during the examination. The eye was intermittently lubricated using sterile saline drops. A Flynn-style indenter was used to stabilise the eye, allowing for a standardised-intensity light beam to enter the eye through a condensing lens as part of the binocular ophthalmoscopic fundus examination. The right eye was examined first, following which the speculum was removed and inserted into the left eye to repeat the process.
Information technology and data collection
All data were stored in line with the co-ordinating centre data protection standard operating procedure (SOP). Personal data were not shared outside the research team. The co-ordinating centre believes that data produced by publicly funded research are a public resource and that as many data as possible should be made available to the scientific community with as few restrictions as possible insofar as this is compatible with the legal requirements for the protection of personal information and within the financial resources available.
Administrative database
For the collection and management of non-clinical data, a web-based trial administration database application (TADA) was used (developed in-house at the co-ordinating centre). TADA allowed the study team to manage all non-clinical data relating to the trial, such as participant contact details and site staff details, and helped co-ordinate the collection of clinical data collection forms (including chasing missing forms, helping co-ordinate the management and resolution of clinical data queries, and generating reports for PMG meetings and other trial overview metrics).
OpenClinica
OpenClinica set-up
The trial required a database in which clinical data could be entered, stored and managed securely in accordance with regulatory requirements. To meet these requirements an OpenClinica database (OpenClinica LLC, Waltham, MA, USA), managed by the co-ordinating centre, was used.
Before data entry began, the trial was created in OpenClinica using a validated web browser automation script; the study involved a number of data collection points, each associated with an electronic CRF (eCRF). Each eCRF was validated to ensure that it was fit for purpose. Only when an eCRF was validated and signed off by a trials programmer was it uploaded into OpenClinica and made available for data entry.
Users were provided with an OpenClinica user account and access to the study database only after they had completed the Poppi trial OpenClinica training. Further information about eCRF design and validation is presented in Appendix 3.
Statistics
Power calculation
The primary objective was to determine whether or not morphine administration reduced clinical pain scores (as measured using the PIPP-R) during the 30-second period after ROP screening, in comparison with placebo. The co-primary objective was to determine whether or not morphine administration reduced noxious-evoked brain activity in response to a heel lance, in comparison to placebo. Although the trial closed early (see Trial closure), the trial was designed to be powered to show meaningful differences in the co-primary outcomes.
A two-point reduction in PIPP scores is considered to be a clinically meaningful reduction in pain,31 and scores below seven are considered to reflect minimal pain. 14 Three studies have used PIPP scores to assess analgesic efficacy following ROP screening. 32–34 Using the most conservative data, where the mean PIPP score post ROP screening was 8.3 [standard deviation (SD) 3.5], it was calculated that the trial would require 66 infants per group to observe a two-point reduction in PIPP-R scores, with a power of 90% at a two-sided significance level of 0.05.
The research team also considered the sample size required to observe a significant difference between the groups in the other co-primary outcome measure (noxious-evoked brain activity evoked by a heel lance). In previous analyses,35 the noxious-evoked brain activity evoked by a heel lance had a mean magnitude of 0.2 (SD 0.14). In the Poppi trial, the magnitude of the noxious-evoked brain activity was calculated and compared between the two groups. Studies in adults with chronic pain indicate that morphine treatment attenuates the amplitude of laser-evoked potentials by 33.1%. 36 Furthermore, adults administered tramadol (an alternative opioid treatment) showed up to a 50% reduction in the amplitude of laser-evoked potentials. 2 This reduction in nociceptive brain activity is coupled with the verbal report that tramadol is providing effective pain relief. Tramadol is widely used as an analgesic in adults and trials have shown that it effectively reduces neuropathic pain. 37,38 In the Poppi trial, the investigators, therefore, assumed that a 40% reduction in the magnitude of the noxious-evoked brain activity represented a clinically meaningful reduction in brain activity. A 40% reduction would lead to the noxious components having a post-treatment magnitude of approximately 0.12 (SD 0.14), down from a mean of 0.20. This would also require 66 infants per group for a power of 90%, at a two-sided significance level of 5%.
Allowing for a loss to follow-up rate of approximately 10% (e.g. as a result of technical difficulties during physiological monitoring), a total of 148 participants were required. Moreover, the research team anticipated that the proportion of twins eligible for the trial would be approximately 25% based on past studies [e.g. the National Perinatal Epidemiology Unit (NPEU)-run trials BOOST-II UK (a study investigating which oxygen saturation range should be maintained in very premature infants), I2S2 (a RCT of iodine supplementation in extremely preterm infants with follow-up at 2 years), PiPS (early administration to preterm infants of the probiotic Bifidobacterium breve strain BBG to prevent infection and necrotising enterocolitis) and SIFT (a multicentre RCT of two speeds of daily increment of milk feeding in very preterm or very low-weight infants)]. There is evidence that the correlation in pain outcomes between twins is 0.5. 39 Hence, the effect of clustering was calculated to be 1.06. This inflated the total sample size required (90% power, 5% two-sided significance level, 10% loss to follow-up and accounting for multiple births) from 148 to 156 participants (i.e. 78 per group). This was the recruitment target for the Poppi trial.
Significance levels
In the original statistical analysis plan (SAP; prepared for the trial to continue until reaching the target sample size), a p-value of 0.05 (two-sided 5% significance level) was planned to indicate statistical significance for the analysis of the co-primary outcome measures. The multiplicity issue would be addressed using Hochberg’s procedures for multiple testing to control the family-wise error rate. 40 Therefore, the research team planned to reject the null hypothesis for both outcomes if the p-value was < 0.05 for both outcomes. If the p-value was > 0.05 for one of the outcomes, then the other outcome would be tested at the 2.5% significance level. This method was less stringent than the Bonferroni adjustment, while preserving the original power of the study.
Comparisons of all other outcomes would be reported in full for completeness and transparency. For all other analyses a p-value of 0.01 (two-sided 1% significance level) would be used to indicate statistical significance, in order to take account of the number of comparisons. Two-sided statistical tests and corresponding p-values would be presented throughout; however, for the purposes of interpretation of results, confidence intervals (CIs) would dominate, rather than p-values.
Missing data
There were minimal numbers of missing data in this trial. Where this was the case, in some instances it may have been a result of equipment failure or artefacts within the EEG recording. Missing data occurred at random and so the collected data were representative of the population. Prospectively during trial design, the sample size was inflated by 10% to account for missing data.
Analysis
A detailed SAP was developed in-house and agreed by the TSC before the analysis was undertaken. Owing to the smaller sample size and associated data set at the early trial closure, the SAP was significantly revised to accommodate this. Overall revisions to the SAP are described in Revisions to the statistical analysis plan, and were reviewed by the DMC and approved by the TSC before the analysis began. The original planned analyses for this trial, and the revisions made to these in the light of early trial cessation, are detailed below in Primary and co-primary outcome measures analysis and Secondary outcome measures analysis. The analysis and presentation of results followed the most up-to-date recommendations of the Consolidated Standards of Reporting Trials (CONSORT) group. All comparative analyses were originally planned to adjust for important prognostic factors where possible. All statistical analyses were conducted using Stata®/SE 13.1 (StataCorp LP, College Station, TX, USA) for Windows.
Revisions to the statistical analysis plan
Prior to database lock and analysis of the results, the study team further developed the SAP and discussed each analysis in detail to ensure that the data were presented accurately and appropriately. Although the SAP would usually be completed before recruitment closure, as a result of the early suspension of the trial this document was still in draft form. Furthermore, the draft SAP required substantial revisions to accommodate the lower sample size (i.e. 31 participants from a target of 156). The following deviations from the original planned analyses described in the protocol were considered necessary:
-
Significance levels – 95% CIs should be reported for all comparisons of outcomes, instead of reporting 99% CIs for secondary outcomes as specified in the protocol.
-
Models should not adjust for minimisation factors because of the small sample size.
-
Clinical stability analysis should not use Poisson or linear regression for count outcomes (i.e. the number of episodes of bradycardia, tachycardia, desaturation and apnoea). Instead, the Wilcoxon rank-sum test was performed on the standardised difference in the number of episodes of bradycardia, tachycardia and desaturation before and after the clinical intervention. The median differences between the groups with CIs were calculated for these standardised differences.
-
Increased respiratory support was to be analysed using crude risk ratios, instead of logistic regression.
-
Episodes of apnoea was treated as a binary composite of new-onset apnoeas or an increase in the number of apnoeas, analysed using crude risk ratios.
-
The safety analysis did not use logistic regression, but instead calculated risk differences with CIs.
-
Additional exploratory analyses were added (see Exploratory analyses).
The trial statisticians checked the data for inconsistencies and updated the dummy tables according to the revised SAP. It was ensured that the SAP provided a comprehensive report of the planned analyses and the document was reviewed thoroughly by the investigators and oversight committees. A joint TSC and DMC meeting was held on 6 April 2018 for the oversight committees to review the updated SAP (version 1.0; 6 April 2018). Minor edits to the document were suggested, but overall the attendees approved the document, which was signed off by the chief investigator, principal investigator, senior statistician and TSC chairperson.
However, following sign-off of SAP version 1.0 four errors/omissions were identified:
-
Stopping boundaries had been stated incorrectly (this matched an error identified in the protocol – the stopping boundaries are stated correctly in two other places in the protocol).
-
A typographical error occurred in the criteria for participants randomised in error – ‘born ≤ 32 weeks’ gestation or birthweight ≥ 1501 g’ should be stated instead of ‘born ≥ 32 weeks’ gestation and birthweight ≥ 1501 g’.
-
Postnatal age should have been included in the characteristics at the time of study. This was discussed during trial design and agreed that it could be calculated from the eCRF data. The PMG agreed that this should be incorporated into the new version of the SAP.
The PMG agreed that errors 1 and 2 were typographical errors and error 3 was an oversight, all of which should be corrected in SAP version 2.0. The co-ordinating centre quality assurance (QA) associate agreed that this was an appropriate course of action. The errors corrected were clearly explained in the ‘document history’, and stated to have occurred after unblinding of the investigators. The changes were incorporated into SAP version 2.0 (6 June 2018), which was re-signed by the chief investigator, principal investigator, senior statistician and TSC chairperson.
Primary and co-primary outcome measures analysis
PIPP-R score during the 30-second period after retinopathy of prematurity screening
The original plan was to compare PIPP-R scores (during the 30-second period after ROP screening) in the morphine and placebo groups using linear regression so as to estimate the adjusted mean difference with a 95% CI. If the PIPP-R scores were skewed, the median and interquartile range (IQR) (or entire range, whichever was appropriate) would be presented for each group, and the median difference between groups estimated with a corresponding 95% CI.
In the final analysis, a mean difference was calculated using a t-test, as it was not appropriate to adjust for minimisation factors as a result of the small sample size.
Noxious-evoked brain activity
In the original planned analyses the magnitude of the noxious-evoked brain activity for the morphine and placebo groups would be compared using linear regression, depending on the mean and variance of the data. If appropriate, the mean and SD would be presented for each group and the adjusted mean difference with 95% CI would be estimated. If the outcome data were skewed, the median and IQR (or entire range, whichever is appropriate) would be presented for each group and the median difference estimated between groups with a corresponding 95% CI.
In the final analysis, an unadjusted median difference was estimated using the Hodges–Lehmann estimator and a Wilcoxon rank-sum test, as the data were skewed, and it was not appropriate to adjust for minimisation factors because of the small sample size.
Secondary outcome measures analysis
Premature Infant Pain Profile – Revised score following heel lance
In the original planned analyses, PIPP-R scores following heel lance in the morphine and placebo groups would be compared using linear regression to estimate the adjusted mean difference with 99% CIs (and p-values). If the PIPP-R scores were skewed, the median and IQR (or entire range, whichever was appropriate) would be presented for each group, and the median difference between groups with corresponding 99% CIs (and p-values) estimated.
In the final analysis, a mean difference was calculated using a t-test, as it was not appropriate to adjust for minimisation factors because of the small sample size. As per the updated analysis plan, 95% CIs were estimated instead of 99% CIs.
Reflex withdrawal
In the original planned analyses, the average RMS activity in the 1000 milliseconds post heel lance for the morphine and placebo groups would be compared using linear regression, depending on the mean and variance of the data. If appropriate, the mean and SD for each group would be presented and the adjusted mean difference plus 99% CIs (and p-values) estimated. If the outcome data were skewed, the median and IQR (or entire range, whichever was appropriate) would be presented for each group, and the median difference between groups with corresponding 99% CIs (and p-values) estimated.
In the final analysis, as the outcome data were skewed, an unadjusted median difference was estimated using the Hodges–Lehmann estimator and a Wilcoxon rank-sum test. As per the updated analysis plan, 95% CIs were estimated instead of 99% CIs.
Clinical stability analysis
The number of episodes of bradycardia, tachycardia, desaturation, and apnoea, and the requirement for an increase in respiratory support that occurred in the 24-hour period after the start of the clinical intervention, would be compared in the morphine and placebo groups. In the original planned analyses, depending on the distribution of these counts, either Poisson or linear regression would be used for these analyses to calculate the adjusted effect estimates plus 99% CIs (and p-values). If appropriate, adjustment for the 24-hour baseline period would also be made. Alternatively, the medians and IQRs (or entire range, whichever is appropriate) for each group would be presented and the median differences between groups with corresponding 99% CIs (and p-values) estimated. In the event of occurrences being very infrequent or the outcome of interest being the occurrence of increased respiratory support, logistic regression could have been used as an alternative.
In the final analysis, it was not appropriate to use Poisson or linear regression for these outcomes because of the small sample size (which contributed to the number of zero counts and the distribution of the data). Instead, the Wilcoxon rank-sum test was performed on the standardised difference in the number of episodes of bradycardia, tachycardia and desaturation before and after the clinical intervention, to adjust for the number of pre-procedure episodes. A negligible constant (0.01) was added to each observed value and the difference in the number of episodes in the 6-hour period before and the 6-hour period after the clinical intervention as a proportion of the total number of episodes in the 12-hour period was calculated. Similarly, the difference in the number of episodes in the 24-hour period before and the 24-hour period after the clinical intervention as a proportion of the total number of episodes in the 48-hour period was calculated. The median differences between the groups with 95% CIs were calculated for these standardised differences using the Hodges–Lehmann estimator, and compared using a Wilcoxon rank-sum test.
Increase in respiratory support was analysed using unadjusted risk ratios, because of the small sample size, with 95% CIs. Episodes of apnoea, a composite of infants who either developed new-onset apnoeas or had an increased number of apnoeas in the 6- and 24-hour periods after the clinical intervention, was treated as a binary outcome. These comparisons were also symmetrical, comparing the 6-hour period after the clinical intervention with the 6-hour period before, and the 24-hour period after with the 24-hour period before. The number and proportion of infants who had this composite outcome were presented and risk ratios estimated with 95% CIs.
Safety analysis
Drug safety would be assessed by measuring the number of occurrences of apnoea that required intervention via Neopuff or bag-and-mask ventilation and the number of episodes of hypotension that required treatment with inotropes in the 24-hour period after drug administration. The number of occurrences would be compared in the morphine and placebo groups. Depending on the distribution of these counts, either Poisson or linear regression would be used for these analyses to calculate the adjusted effect estimate plus 99% CIs (and p-values). Alternatively, the median and IQR (or entire range, whichever was appropriate) would be presented for each group and the median difference between groups with corresponding 99% CIs (and p-values) estimated. In the event of occurrences being very infrequent, logistic regression could have been used as an alternative.
In the final analysis, risk differences were calculated with 95% CIs instead.
Exploratory analyses
In addition to the planned analyses, exploratory analyses were performed (see Exploratory analyses).
Planned interim analyses
An interim statistical analysis of the primary and secondary outcomes was planned when 50% of trial participants (78 infants) had completed data collection. In the light of the interim data analysis, and other evidence from relevant studies (including updated overviews of relevant RCTs), the role of the DMC was to inform the TSC if in its view there was proof beyond reasonable doubt that the data indicate that any part of the protocol under investigation was either clearly indicated or contraindicated. Guidelines for early cessation owing to safety concerns were agreed with the DMC and documented. Unless modification or cessation of the trial was recommended by the DMC, the TSC, investigators, collaborators and administrative staff (except those who supply the confidential information) would remain ignorant of the results of the safety analyses.
Trial stopping rules
Drug safety was assessed by measuring the number of occurrences of apnoea that required intervention using Neopuff or bag-and-mask ventilation, or hypotension that required treatment with inotropes, in the 24-hour period after drug administration. The DMC planned to review trial safety outcomes after every 25 participants had been randomised and safety data had been collected (i.e. n = 25, 50, 75, 100 and 125). In addition, the chief investigator (or suitably trained delegate) was notified of every such occurrence.
A formal sequential safety procedure was applied and presented to the DMC for occurrences of apnoea that required intervention using Neopuff or bag-and-mask ventilation. A stopping boundary using group sequential methods with a boundary agreed by the DMC and specified in the DMC charter was employed.
Specifically, a one-sided gamma spending function, with gamma equal to 4.5, was used. This is a type of α-spending function, that is a stopping boundary where the type I error rate is spread across a number of analyses. The boundary is a function of the amount of information collected (in this case, the number of infants with safety outcome data available). A type I error rate of 0.2 was used, which was estimated to result in power of 0.79. This boundary was based on the assumption of a control group event rate of 7% and a difference between the group event rates of 12%.
At the first DMC safety review of 25 infants and the subsequent requested review of 31 infants, a graph showing evidence of the relative safety of the treatments was provided as a guide, in addition to trial data summarised by trial arm, clinical summaries for infants who had experienced safety events and the completed CRFs for all infants. The DMC charter stated that, if the prespecified stopping boundary was crossed, then the DMC should consider terminating the trial, taking into account other considerations (e.g. implications on future infants/clinical practice and follow-up, how convincing the evidence was). It is important to note that the stopping boundary was intended as an aid to the decision to stop the trial, and not as a definitive stopping rule. There was no stopping rule or plan to stop early as a result of benefits from the treatment.
Adherence to the intervention
Adherence to the intervention was reported in a process outcomes table. The median, IQR and range of the volume of IMP given, and the number and percentage of infants receiving more than the correct dose for their weight (i.e. 100 µg/kg), plus a 5% tolerance to account for measurement error, were reported.
Timing of the study treatment in relation to the clinical intervention was also reported in this table. The time between study treatment administration and the clinical intervention was summarised by trial arm using median and IQR, and ranges.
Analysis populations
Post-randomisation exclusions
As this was an efficacy study, the analysis was per protocol (as specified in the protocol). However, as all infants received the drug to which they had been randomised (except for one infant, withdrawn from the placebo arm prior to study), the per-protocol analysis was equivalent to intention-to-treat analysis and the safety analysis was based on the safety population, according to the treatment received.
The following infants would be excluded from the per-protocol analysis population post randomisation:
-
Those who did not receive the study treatment.
-
Those who did not receive the clinical intervention (i.e. heel lance followed by ROP screening).
-
Those randomised in error.
-
Those for whom full consent was not obtained.
-
Those for whom consent to use their data was withdrawn by the parent(s). (Parents could specify whether or not data collected up to the point of withdrawal could be used. If the response was ‘no’, then the infant was considered as a post-randomisation exclusion. If the response was ‘yes’, then the infant was reported as ‘missing’ for any data not collected after withdrawal.)
-
Those for whom an entire record of fraudulent data was detected.
The numbers of post-randomisation exclusions were reported by trial arm, and reasons summarised, in the CONSORT flow diagram.
Population definitions
Per-protocol population
The per-protocol population was all infants randomised who received both the study treatment and the clinical intervention (i.e. heel lance followed by ROP screening), excluding post-randomisation exclusions. Comparative analyses and other descriptive analyses would be presented for this population only.
Baseline characteristics at time of trial entry and time of study were reported for all infants randomised for whom data were available, excluding post-randomisation exclusions.
Safety analysis population
Safety analyses would include all infants randomised who received the study treatment.
Descriptive analyses
Representativeness of trial population and participant throughput
The flow of participants through each stage of the trial was summarised using a CONSORT flow diagram. This described the numbers of participants who were:
-
randomised:
-
randomised in error
-
-
allocated to each intervention:
-
who received the allocated intervention
-
who did not receive the allocated intervention:
-
who received the other intervention
-
who received no study treatment
-
-
-
withdrawn from the trial:
-
data already collected can be used
-
clinical data can be collected from infant’s notes for remaining study period
-
vital signs monitor data can be recorded for remaining study period
-
reason for withdrawal:
-
clinical decision
-
parental request
-
other reasons
-
-
-
included in the analysis
-
excluded post-randomisation (with reasons).
Baseline comparability of randomised groups
The per-protocol population groups were described by allocation group with respect to their demographic and clinical characteristics at trial entry and at the time of study. The time of study was defined as the time of the start of the clinical intervention (i.e. heel lance followed by ROP screening).
Characteristics at trial entry and at time of study are listed in the following sections. Note that * denotes minimisation criteria.
Characteristics at trial entry
-
Gestational age at birth.*
-
Gestational age at randomisation.*
-
Birthweight.
-
Birthweight z-score (adjusted for gestational age and gender at birth).
-
Weight at randomisation.
-
Apgar score at 10 minutes of age.
-
Mode of delivery.
-
Sex.
-
Days ventilated.*
-
Days since morphine was given.*
-
Presence of gastric tube.*
-
Intrauterine growth restriction.*
-
Infant has had surgery.*
-
IVH grade I or II.
-
Infant is one of a multiple pregnancy.
-
Conceived using IVF treatment.
Characteristics at time of study (clinical intervention)
-
Gestational age at time of study.
-
Postnatal age at time of study.
-
Weight at time of study.
-
Level of care received:
-
intensive care unit
-
high-dependency unit (HDU)
-
low-dependency unit (LDU)
-
maternity ward.
-
-
Respiratory support modality:
-
self-ventilating
-
low-flow therapy
-
high-flow therapy.
-
-
Time since last feed.
-
Comfort measures at heel lance:
-
kangaroo care
-
swaddling, containment, facilitated tucking
-
rocking/holding
-
non-nutritive sucking
-
oral sucrose.
-
-
Duration of ROP screening examination.
-
Diagnosis of ROP.
The number and percentage were presented for binary and categorical variables. The mean and SD, or the median and the IQR, and the range if appropriate, were presented for continuous variables. There were no tests of statistical significance performed nor CIs calculated for differences between randomised groups on any baseline variable.
Other descriptive analysis
Other data collected during the trial and the 48-hour observation period were summarised by allocation group for descriptive purposes, as follows:
-
PIPP-R score 30 seconds after the control heel lance
-
magnitude of noxious-evoked brain activity evoked by the control heel lance
-
magnitude of the reflex withdrawal following the control heel lance
-
mean blood pressures (i.e. systolic, diastolic and mean) during, and in the 6- and 24-hour periods before and after, the clinical intervention.
The number and percentage of the total were presented for binary and categorical variables. The mean and SD, or the median and the IQR, and the range if appropriate, were presented for continuous variables. There were no tests of statistical significance performed, nor CIs calculated for differences between randomised groups on any of these end points.
Additional analyses
Additional objectives not prespecified in the protocol were added, in the light of the early suspension of the trial recruitment, as detailed in the following sections.
Additional objectives
The objectives aimed to (1) quantify whether or not, within the study population, the magnitude of the noxious-evoked brain activity, reflex withdrawal activity and PIPP-R scores could discriminate between noxious and non-noxious input and (2) further understand how heart rate, oxygen saturation and respiratory rate were influenced by the clinical intervention (i.e. heel lance and ROP screening) and the administration of morphine.
Investigations explored whether or not:
-
noxious-evoked brain activity and reflex withdrawal activity are different following the heel lance compared with the background activity and control heel lance, independent of the intervention allocation
-
PIPP-R scores are different following the heel lance and the ROP screening compared with control heel lance, independent of the intervention allocation
-
average time courses for heart rate, oxygen saturation and respiratory rate following the clinical intervention differ between the morphine group and the placebo group (through the use of continuous electronic data capture of heart rate, oxygen saturation and respiratory rate for the 24 hours after the clinical intervention).
Analysis of additional objectives
Validation of pain measures
Noxious-evoked brain activity and reflex withdrawal activity were summarised at background, control heel lance and heel lance using means with standard error of the mean and ranges, or medians with standard error of the median (using bootstrap estimation) and ranges if the data were skewed. These summary statistics were also presented graphically.
The PIPP-R scores were summarised at control heel lance, heel lance and ROP screening using means with standard error of the mean and ranges, or medians with standard error of the median (using bootstrap estimation) and ranges if the data were skewed. These summary statistics were also presented graphically.
Extended exploratory analysis of physiological stability
The average changes in heart rate, oxygen saturation and respiratory rate were presented graphically for the 24 hours pre and post clinical intervention, and separately for the morphine and placebo groups. The time courses (which describe the variation in these measures with time) were described and compared between the two groups using non-parametric cluster analysis. 41 Time periods with differences between the morphine and placebo groups were identified by first calculating a t-statistic at each time point. Significant differences in the time courses were defined to have occurred when the t-statistic was above a threshold set at the 97.5th percentile of the t-distribution. Time periods (clusters) of significant activity were identified based on temporal adjacency of the above threshold points. A cluster-based test statistic was calculated from 1000 random permutations of the data.
Trial management
Study documentation
The TMF was created at the beginning of the trial by the trial manager. This was organised to allow efficient maintenance and review of trial-related documentation using a template contents document from the co-ordinating centre. The trial manager, trial sponsor, co-ordinating centre, QA team, MHRA (should an inspection have taken place) and the archivist had access to the TMF; maintenance of the TMF was the responsibility of the trial manager. All documentation stored separately from the TMF had a signed and dated file note within the relevant section of the TMF indicating its location. All essential documents, trial-related documents and important correspondence were stored in the TMF, which was updated regularly throughout the trial. Previous versions of documents were retained in the TMF, but clearly labelled as superseded.
The trial manager provided the site team with an ISF, and the pharmacy contact with a pharmacy folder, containing all relevant documentation for the trial. Throughout the trial, updated documentation was sent to the research team and the pharmacy contact to keep the ISF and pharmacy folder up to date. Details of the protocol, PIL, consent form and other parent-facing materials are presented in Appendix 4. Final versions of the documents approved by the regulatory bodies are listed in Table 2.
| Study document | Current version | Date |
|---|---|---|
| Protocol | 6 | 24 January 2017 |
| PIL (see Report Supplementary Material 6) | 4 | 12 April 2016 |
| Consent form (see Report Supplementary Material 2) | 4 | 12 April 2016 |
| SmPc: 10 mg/5 ml of Oramorph® (Boehringer Ingelheim Limited, Bracknell, UK) oral solution | N/A | 1 April 2014 |
| Promotional items | ||
| Cot cards (see Report Supplementary Material 3) | 1 | 12 August 2015 |
| Thank-you poster (see Report Supplementary Material 10) | 1 | 9 September 2015 |
| Roll banner | 1 | 9 September 2015 |
| Recruitment poster (see Report Supplementary Material 7) | 1 | 9 September 2016 |
| Insert/poster | 1 | 28 September 2017 |
Details of regulatory reporting and communication are presented in Appendix 5.
Training and delegation
Training
A set of training packages and guidance sheets were written and reviewed by the PMG. These documents were used to train research staff and clinical staff in trial procedures. Training packages in the form of presentations were:
-
package 1 (for research staff) – screening, consent and enrolment, IMP management, computer and monitor use, and safety and incident reporting
-
package 2 (for medical staff) – trial outline, prescribing the IMP, and safety and incident reporting
-
package 3 (for nursing staff) – trial outline, IMP administration, IMP storage, monitor and computer use, and safety and incident reporting
-
package 4 (for ophthalmology staff) – trial outline and ROP considerations.
A set of guidance sheets for quick-reference information about study processes (more detailed than in the protocol) for the research team was provided, as follows:
-
recruitment
-
consent checklist
-
randomisation
-
trial checklist
-
cupboard temperature and thermometer management
-
monitor use and computer set-up
-
IMP management
-
unblinding
-
withdrawals
-
safety and incident reporting
-
setting up EEG software
-
exporting from CURRY
-
analysis of EEG and EMG data
-
video management
-
PIPP-R scoring
-
emergency queries
-
trial overview.
A set of guidance sheets for the neonatal unit staff was also provided, as follows:
-
IMP prescribing
-
IMP administration
-
IMP storage
-
monitor and computer
-
ROP considerations
-
unblinding
-
safety and incident reporting
-
emergency queries
-
withdrawals
-
monograph for the Poppi trial IMP
-
quick-guide PC use
-
quick-guide thermometer use.
These training packages and guidance sheets provided the core training for all staff involved in the Poppi trial. Two training logs were maintained throughout the trial (see Report Supplementary Material 11 and 12). The first training log was used to record all training relating to specific guidance sheets and the second was used to record all other types of training, for example OpenClinica training provided by the co-ordinating centre and training given by other equipment providers (see Report Supplementary Material 11 and 12).
Prior to opening for recruitment, trial procedures during ROP screening were piloted (without IMP) on approximately 10 infants. This helped the team prepare for the trial, while also collecting important information about brain activity during ROP examinations. These pilot studies without the IMP and the associated data collection were conducted within the study ‘Investigating pain in the developing human brain’ [Research Ethics Committee (REC) reference number 12/SC/0447].
Delegation
A delegation log (see Report Supplementary Material 4) was maintained throughout the study to record all trial duties that had been delegated to staff by the chief investigator. The recorded duties included screening patients, clinical assessment of eligibility (medically qualified staff only), obtaining informed consent (only if GCP training was complete), randomisation, prescribing IMP (medically qualified staff only), preparing and administering IMP (clinician or registered nurse only), set-up of the physiological monitor, nociceptive monitoring, heel lance (clinician or registered nurse only), ROP screening (ophthalmologist only), data collection, data entry/resolution of data queries (only those individuals who had received OpenClinica training), post-acquisition analysis (i.e. video, for PIPP-R scoring), post-acquisition analysis (electrophysiology/physiology), serious adverse event (SAE) clinical review/causality assessment and sign-off (medically qualified staff only), IMP supply/management (pharmacy staff) and completing IMP accountability logs (see Report Supplementary Material 1). The name of the individual, role, delegated duties, signature, e-mail address, date of duty period, chief investigator signature and date of signature were recorded on the delegation log (see Report Supplementary Material 4). All delegation logs (see Report Supplementary Material 4) were signed off again by the chief investigator, and the principal investigator, prior to site close-out (see Appendix 6, Site close-out).
Risk assessment
A risk assessment was completed annually by the PMG using the co-ordinating centre SOP. The risk assessment identified, evaluated and estimated the level of risk involved in potential situations. A risk assessment procedure was followed that identified potential trial-specific risks (including risks to participants and staff), provided a basis for trial-specific monitoring and management, and provided a complete and transparent record of key decisions made in relation to the risk-adapted trial monitoring and management plans. The purpose of the embedded monitoring plan was to help verify that the rights and well-being of the participants were protected, that the reported trial data were accurate, complete, reliable and verifiable from source documents, and that the conduct of the trial was in compliance with the protocol, trial processes and GCP. Monitoring was an integral process in the quality control of this clinical trial, and the monitoring plan was designed to verify the quality and integrity of the research.
The initial trial risk assessment, monitoring and management plans were finalised on 12 October 2015. These were reviewed and updated yearly by the PMG, using a template document from the co-ordinating centre. Risk areas were prespecified on the template, and the PMG completed the following information for each potential risk: particular risk identified, specific concerns, likelihood (low/medium/high), impact (low/medium/high), how the risk would be minimised, if the minimisation required monitoring. This was supplemented with a monitoring strategy reviewed through central or site visit monitoring, actions and person(s) responsible, frequency of monitoring, and trigger points and escalation route. This was then followed by documented management procedures: actions and person(s) responsible, frequency of management, trigger points and escalation route.
The risk associated with the IMP for this trial was considered to be ‘type B’, that is higher than the risk of standard medical care. The justification for this risk level was that morphine is used in neonates for pain relief and sedation, but is not currently used as a pre-emptive analgesic prior to ROP screening. Morphine is commonly used on neonatal units as a sedative. During mechanical ventilation morphine has been shown to reduce physiological instability, reduce behavioural and hormonal stress responses, and improve ventilator synchrony. Morphine is used locally as a treatment for pain relief during ROP laser surgery at the same dose, in combination with other medications. All infants in the trial were monitored as per standard practice. In the event that the infant exhibited side effects, such as reduced respiratory drive, it was possible to reverse the effects of the morphine with naloxone (also used as standard care).
The potential side effects of morphine were risk assessed, with the mitigation being the 24-hour monitoring post IMP administration. Other processes to mitigate risks to participants’ safety included the trial oversight by an independent TSC and DMC. The aims of the DMC included the identification of any trends, such as increases in the number of unexpected or expected events, and taking appropriate action, seeking additional advice or information from investigators where required, and evaluating the risk of the trial continuing. The DMC monitored safety outcomes closely. Drug safety was assessed by measuring the number of occurrences of apnoea that resulted in increases in respiratory support modality or the requirement of intervention via Neopuff or bag-and-mask ventilation in the 24-hour period after ROP screening. The DMC reviewed trial safety outcomes after every 25 infants were randomised and safety data collected (i.e. n = 25, 50, 75, 100 and 125). A formal sequential safety procedure was applied and presented to the DMC. A graph showing evidence of the relative safety of the treatments was provided to act as a guide to the DMC members. If the prespecified boundary was crossed, then the DMC would consider terminating the trial, taking into account other factors (e.g. implications on future patients/clinical practice and follow-up, how convincing the evidence was). The DMC provided advice on the conduct of the trial to the TSC. Pharmacovigilance and serious breaches were continually monitored in accordance with the co-ordinating centre’s SOPs.
Monitoring and oversight
Information about the site initiation and close-out visits from the trial manager is presented in Appendix 6. Details about the sponsor-led monitoring of the trial are provided in Appendix 7 and PMG-led monitoring in Appendix 8.
Oversight of the trial was maintained by the DMC and TSC, the members of which were approved by the trial funder.
Data Monitoring Committee
An independent DMC was appointed, the terms of reference of which were governed by a signed DMC charter. The DMC comprised an independent chairperson (Professor of Neonatal Medicine, Consultant Neonatologist), another independent clinical expert (Consultant Neonatologist, Senior Lecturer) and an independent statistician (Professor of Medical Statistics). The purpose of the DMC was to monitor efficacy and safety end points. Only the independent DMC members and the unblinded trial statisticians had access to unblinded study data (until the closure of the trial; see Trial closure, Oversight). The committee met six times during the trial.
Members of the DMC were nominated by the investigators to the funder for approval. The NIHR EME programme director vetted the nominees and appointed the chairperson and members. The members of the DMC were independent of the trial (were not involved with the trial in any other way and did not have competing interests that could impact on the trial). Any competing interests, both real and potential, were declared at the beginning of each DMC meeting. The trial statistician produced closed reports for the DMC, participated in DMC meetings and took minutes for the closed sessions. The trial manager was responsible for providing input into the open sessions at the DMC meetings, and producing non-confidential trial summaries and reports for the DMC. In accordance with the NIHR research governance guidelines, the DMC was advisory to the TSC.
The DMC was due to review trial safety outcomes after approximately every 25 infants had been randomised and safety data collected (i.e. n = 25, 50, 75, 100 and 125). It also planned to meet when data had been collected from 50% of trial participants (i.e. 78 infants) for an interim analysis of primary and secondary outcomes. Meetings were arranged so that reports could be fed into the TSC meeting. At the request of the DMC, interim meetings, in person or by teleconference, could be organised by the trial manager or trial statistician. DMC members were prepared for such instances where major trial issues needed to be dealt with between meetings, by phone or by e-mail.
The standard format for DMC meetings was a mixture of open and closed sessions. Only DMC members and others who were specifically invited, for example the trial statistician, attended the closed sessions. In open sessions, all those attending the closed session were joined by the chief investigator, principal investigator and trial manager. The meeting format was:
-
open session – introduction and any ‘open’ parts of the report
-
closed session – DMC discussion of the ‘closed’ parts of the report.
If necessary:
-
open session – discussion with other attendees on any matters arising from the previous session(s)
-
closed session – extra closed session.
The aim of the open sessions was to accumulate information relating to recruitment and data quality (e.g. data return rates and compliance). Safety details based on pooled data from the two comparison groups combined were presented with the total number of events for the primary outcome measures and other outcome measures (if relevant), at the discretion of the DMC. The closed session additionally included a review of the efficacy and safety data analyses conducted by the treatment group.
Decisions open to the DMC included no action needed, that is, trial to continue as planned; early stopping as a result of, for example, a clear benefit or harm of one of the treatments (overall or within a subgroup) or external evidence (e.g. published results from another trial); and approving requests for early release of (subsets) of data.
The process of decision-making aimed for consensus. The method of deliberation was not revealed to the TSC to avoid revealing information about the status of the trial’s data. The chairperson summarised discussions and encouraged consensus, and gave their own opinion last. Every effort was made by the DMC to reach a unanimous decision. If the DMC could not achieve this, a vote was taken, although details of the vote were not routinely included in the report to the TSC to avoid accidentally conveying information about the state of the trial data. Implications (e.g. ethical, statistical, practical and financial) for the trial were considered before any recommendation was made. Members of the DMC made every effort to attend all meetings. If any DMC member could not attend, in person or by teleconference, then the DMC could still meet if the chairperson and one other member were present. If the DMC was considering recommending major action after such a meeting, the DMC chairperson would speak with the absent member as soon after the meeting as possible to assess whether or not they agreed. If they did not, a further teleconference would be arranged with the full DMC. The DMC reported its recommendations in writing to the TSC. These were copied to the trial statistician and sent, via the trial manager, in time for consideration at the next TSC meeting. Details of the DMC meetings are presented in Appendix 9.
Trial Steering Committee
A TSC was established to provide overall supervision of the study, the terms of reference of which were governed by a signed TSC charter. The TSC comprised an independent chairperson (Professor of Neonatal Medicine, Neonatal Consultant), an independent vice-chairperson (Consultant Senior Lecturer, Consultant Paediatric Ophthalmologist), an independent clinician (Consultant Neurologist), an independent senior statistician, an independent patient and public involvement (PPI) representative, and the chief investigator and principal investigator (non-independent members). Members of the PMG also attended meetings as observers. The committee met five times during the trial.
The chairperson had previous experience of serving on trial committees and experience of chairing meetings. The chairperson and members of the TSC were nominated by the investigators to the trial funder for approval. The NIHR EME programme director vetted the nominees and appointed the chairperson and members. The vice-chairperson was identified by the TSC at the first meeting. The chairperson’s responsibilities included:
-
chairing the TSC meetings and summarising discussions
-
establishing clear reporting lines to the trial funder, trial sponsor, etc.
-
being familiar with relevant guidance documents and with the role of the DMC
-
providing an independent, experienced opinion if conflicts arose between the needs of the research team, the trial funder, the trial sponsor, the participating organisations and/or any other agencies
-
leading the TSC to provide regular, impartial oversight of the trial, especially to identify and pre-empt problems, ensuring that changes to the protocol were debated and endorsed by the TSC
-
making letters of endorsement available to the project team when requesting approval from the trial funder and trial sponsor for matters, such as changes to protocol
-
being available to provide independent advice as required (not just when the TSC meetings scheduled)
-
commenting on any extension requests and, where appropriate, providing a letter of recommendation to accompany such a request
-
commenting in detail (and when appropriate) regarding the continuation or termination of the trial.
The chairperson of the TSC was directly answerable to the NIHR EME programme.
The trial manager was responsible for arranging the TSC meetings, co-ordinating reports, and producing and circulating minutes and action points. The trial manager was the central point for all communications between the TSC and other bodies, was copied into all correspondence among the TSC members and was kept aware of trial issues as they arose. The TSC aimed to meet, in person or by teleconference, at least annually. At the request of the TSC, interim meetings, in person or by teleconference, could be organised. Meetings were scheduled to follow shortly after the DMC meetings so that the DMC reports could be considered. Prior to the meeting, the trial manager prepared a detailed report providing information on accrual and any matters affecting the trial. Additionally, the material could include a report from the DMC, requests from the PMG or draft publications. No data on trial outcomes were presented by trial arm unless explicitly authorised by the DMC.
Based on recommendations from the DMC, possible decisions from the TSC included:
-
no action needed, that is, trial to continue as planned
-
early stopping owing to, for example, a clear benefit or harm of a treatment or external evidence (this would generally involve a recommendation from the DMC to unblind the TSC to these data)
-
modifying target recruitment, or pre-analysis follow-up, based on any change to the assumptions underlying the original trial sample size calculation (but not on any emerging differences)
-
sanctioning and/or proposing protocol changes
-
approving proposed protocol amendments or new trial substudies
-
approving the presentation of results during the trial or soon after closure on the advice of the DMC
-
approving strategies to improve recruitment.
The TSC quorum for decision-making was a minimum of three independent members (including the chairperson or vice-chairperson, unless otherwise agreed), with either the chief investigator or the principal investigator, and a member of the PMG present. The TSC reported its decisions (through the trial manager) to the PMG and was responsible for implementing any agreed actions. The TSC also provided feedback to the DMC and to the trial funder. Copies of communications passed through the trial manager. Details of the TSC meetings are presented in Appendix 9.
Collaborators
A collaborator agreement dated 9 February 2016 was signed between the University of Oxford and the sites of the trial collaborators (i.e. OUHs NHS Trust, Great Ormond Street Hospital for Children NHS Foundation Trust and the University of Edinburgh). The collaborators on the grant application remained involved in the progress of the trial. They were informed of the early trial closure, invited to review the results which were published in The Lancet42 [see Chapter 4, Results (The Lancet)], and to attend the TSC/DMC/collaborator meeting on 11 June 2018 to discuss the trial closure, and were invited to a thank-you event for trial stakeholders on 12 September 2018 (see Staff and stakeholders).
Safety reporting and unblinding
Throughout the trial any staff could report safety events at any time. The safety reporting process is detailed in Appendix 10. There were two reported SAEs in the trial (see Chapter 4, Serious adverse events) and no suspected unexpected serious adverse reactions (SUSARs).
Unblinding
Treatment allocation for any participant could be unblinded by the clinical team using the randomisation website. The requirements were that the treating clinician felt that it was a genuine emergency and that knowledge of the treatment allocation (i.e. morphine or placebo) was necessary to guide the appropriate clinical management of the infant. The document box, provided at the site initiation visit (see Appendix 6, Site initiation), included a sealed envelope containing the randomisation website address, login details and password, and a single-use access code. These details were entered onto the randomisation website, alongside the study number, details about who was carrying out unblinding and information regarding why unblinding was necessary. The system would then reveal the arm allocation of the infant. The co-ordinating centre would be automatically informed of any cases of unblinding. The unblinding process is detailed in Figure 8.
FIGURE 8.
Unblinding process.

Unblinding reports
The treatment allocation of one infant receiving low-dependency care was unblinded on 30 January 2017, approximately 6 hours after IMP administration, and a SAE was reported (described in Chapter 4, Serious adverse events). The decision to unblind was made by two consultants to guide appropriate clinical management of intermittent poor respiratory effort. The chief investigator was unavailable for guidance, and there were no named safety delegates documented in the TMF. The PMG agreed that two safety delegates should be identified in the event of the chief investigator’s absence.
Incidents
Any deviations from the protocol, trial procedures, GCP or regulatory requirements were reported as incidents. Incidents were reported in writing using an incident and deviation reporting form (of which spare copies were kept in the document box) and faxed or e-mailed to the co-ordinating centre as soon as was practical. Any member of staff could report incidents, and all incidents were investigated and assessed by the trial manager using the co-ordinating centre incident, deviation and serious breach assessment form. The location (e.g. pharmacy, ward, laboratory, co-ordinating centre), classification (i.e. protocol deviation, breach of GCP, deviation from other trial procedures or breach of national legislation) and key area defining the incident (i.e. consent, eligibility, randomisation, IMP) were documented on the form. This form also queried whether the event threatened the safety, health or mental integrity of a trial participant or if it affected the scientific integrity of the trial. The assessment was also completed by a second trial manager or senior member of the co-ordinating centre.
If an event did not fulfil these criteria, it was considered to be an incident. The trial manager provided updates on incidents at the PMG and central monitoring meetings. A number of protocol deviations were anticipated as incidents. These included problems with recording PIPP-R scores because of the use of a pacifier or sucrose, loss of data during recording or at export, accidental unblinding and overdose of the IMP. None of these incidents occurred during the trial. If the event was considered to threaten the safety, health or mental integrity of a trial participant, or affect the scientific integrity of the trial, it would have been considered a serious breach and the trial manager would have contacted the trial sponsor within 1 working day. The trial sponsor would work with the chief investigator and trial team to assess the serious breach and, if appropriate, report it to the REC, regulatory authority and research and development (R&D) department within 7 calendar days.
The incidents/serious breaches were also reviewed to ensure that they did not meet trigger points on the risk assessment and monitoring/management plan (see Risk assessment). If so, a documented escalation plan would be followed. A summary of improvements to processes made to eliminate causes of non-conformities was also documented during the assessment, in the form of corrective action (i.e. implemented to prevent recurrence of non-conformity) and preventative action (i.e. a prediction of a problem and actions made to prevent occurrence) recorded on an assessment form. The assessment form was finalised once the corrective and preventative actions were completed, and signed off by the trial manager. Information from the initial report and assessment form was entered by the trial manager onto a central incident database at the co-ordinating centre.
Incident reports
During the trial, a total of eight incidents were reported. None was considered a serious breach.
-
Temperature monitoring was not recorded by hand every day. The trial process was altered as temperature excursions were clearly identified from the monthly download and maximum/minimum thermometer observation; and missed records on the temperature log (see Report Supplementary Material 9) would not affect the trial or the stability of the IMP. The temperature was to still be recorded by hand whenever an IMP pack was removed from the temperature-controlled cupboard.
-
An infant was not withdrawn from the trial following a period of clinical instability during the 24-hour monitoring before the planned IMP administration (as had been specified in the protocol); instead the infant was studied at the next ROP screen. The participant was not re-randomised, but received the original IMP pack that had been allocated. Monitoring re-commenced 24 hours ahead of the second planned IMP administration. All procedures were completed to protocol. The protocol and trial processes were updated to allow infants to be included in the trial during subsequent ROP screens without re-randomisation, should the infant be clinically unstable during the original monitoring period pre-IMP administration and, therefore, not receive the IMP on the original test occasion.
-
A participant experienced a SAE and the clinical decision was made to unblind the participant and then treat with naloxone. Prior to unblinding, the research team outlined the implications of unblinding to the clinical team. The chief investigator was not on clinical duty at the time to review the SAE and advise immediately. The research team agreed that there would be named safety delegates who could review SAEs in the chief investigator’s absence.
-
On a single consent form (see Report Supplementary Material 2) a parent signed and wrote their name in the wrong box. The signature was written in the name box and the name was written in the signature box. This was identified by the trial manager and site staff were reminded to double-check that the correct boxes were completed when taking consent. The trial manager reviewed each signed consent form (see Report Supplementary Material 2) on receipt.
-
A parent entered their own date of birth in the ‘date of consent’ section of the consent form (see Report Supplementary Material 2), and the health professional corrected an error in their own date of signature. Neither errors were GCP corrected. Staff were reminded that all errors on trial documents should be GCP corrected.
-
Clinical information recorded on the paper CRFs was not transcribed into the medical notes. The member of staff involved was reminded that any clinically relevant information arising from trial processes should be recorded in the medical notes to inform future medical decisions. Trial procedures were changed to ensure that a photocopy of the trial document was placed in the medical notes for future participants.
-
The hand-written daily temperature log for August 2017 was misplaced. Unit staff were reminded to leave completed logs in place for the Poppi trial team to collect. There was no risk to the IMP, as the continuous thermometer recording indicated no temperature excursions.
-
A chief investigator assessment form was not requested on receipt of the first follow-up information report for SAE 002. Follow-up information included blood results for several days after the SAE started, but the medical causality assessment remained the same. A second follow-up information report for SAE 002 had already been submitted before the error was identified, and a chief investigator assessment form had been completed using this information. Following QA advice, an incident was logged. The trial manager reviewed the co-ordinating centre SOP for safety reporting in clinical trials using IMPs, and requested that this be updated to clarify that a chief investigator assessment form should be completed on receipt of any follow-up information forms (as well as the initial report).
Withdrawals
Any staff member could withdraw a participant from the trial if parents wished to withdraw or if it was deemed necessary for clinical care. Parents did not need to give a reason for withdrawing a participant. When withdrawing a participant, staff completed a paper withdrawal form to record the time, date, reason for withdrawal, and permissions from the parents to use data already collected. These data were then entered onto OpenClinica. The time and date of withdrawal were then documented in the infant’s medical notes, together with any other necessary information.
There was one withdrawal from the trial. At the time of study set-up, the clinical team informed the parents that the infant could be discharged on the day of the ROP screen. The parents, therefore, chose to withdraw their infant before monitoring began, in order to prevent delay to discharge (if they had participated, the infant would have been monitored for a further 24 hours after the ROP screen, delaying discharge by 1 day). No data were collected for this infant and, therefore, the infant was excluded from analyses.
Trial closure
Oversight
The DMC met on 4 December 2017 to review the safety outcomes of the first 25 participants. The safety analysis indicated that the stopping boundary had been crossed for the first 25 infants. In order to make a fully informed decision, the independent DMC arranged to meet again on 7 December 2017 to review the data for the co-primary outcomes and re-review the safety data with additional data from the six participants who had been recruited (one of whom was not studied). The stopping boundary for the safety outcome was still crossed with inclusion of the additional participant data, and the efficacy outcomes did not suggest that there was pain relief. Therefore, the DMC recommended that the TSC have access to the unblinded safety report and consider suspending the trial.
The independent TSC members were granted access to the unblinded safety report by the trial statistician and the recommendation of trial suspension was discussed at the TSC meeting on 13 December 2017. At this meeting, the TSC reviewed the unblinded safety report and agreed with the DMC’s view that the stopping boundary had been crossed with little evidence of analgesic efficacy. The TSC recommended suspension rather than cessation of the trial, as it considered that this would perpetuate a serious uncertainty in clinical practice and could lead to the continued infliction of a painful and destabilising procedure on preterm infants without evidenced-based analgesia. It considered that a revised trial design had the potential to resolve a major uncertainty in newborn care, which could provide robust evidence to guide pain management and improve clinical outcomes for infants. Although the TSC considered that the trial was conducted to an extremely high standard with strong methodological rigour, it could not recommend that the trial continue in its current form. The decision was primarily based on concerns about drug safety and a lack of evidence that morphine (at the current dose) provided adequate relief for procedural pain. The TSC recommended that significant changes be made to the trial design and that investigators review the unblinded data in order to guide the development of a revised protocol. These changes would provide insight into (1) the safety issues that resulted in the stopping boundary being crossed; (2) the lack of analgesic efficacy identified from the primary outcome data; and (3) the effect of both morphine and the ROP screening procedure on infant physiology, which could guide future clinical practice. As the investigators would be unblinded, data collected could not be included in the proposed future data set and, instead, should be reported as a standalone study. The trial sponsor and funder were in agreement with these recommendations.
The randomisation system was therefore disabled and recruitment was suspended on 13 December 2017. A substantial amendment for temporary suspension was submitted and approved by the REC and regulatory bodies. Key stakeholders were informed of the decision (including the trust, drug manufacturers and relevant research staff). The trial team met in January 2018 to develop a plan. In mid-February the team provided the trial funder with a detailed breakdown of the required actions before review of the data, protocol development and publication of results. A progress report was submitted to the trial funder that further explained the steps required before database lock and the complexities of the changes required for the SAP (see Revisions to the statistical analysis plan). The team planned to submit an options document to the funder in early May to outline the possible protocol changes following review of the data.
In March, the team received a letter from the funder stating that funding would be withdrawn, and following review of the TSC chairperson’s letter and documentation, submitted in February, it had decided that the suggested trial revisions required submission of a new trial proposal, rather than protocol amendments. The funder also reiterated that the pilot data should be used to guide developments for the new trial proposal. Therefore, the central monitoring team stopped the trial on 15 March 2018 and the data from the 31 participants were analysed.
Regulatory
Following the TSC’s decision on 13 December 2017, recruitment to the trial was suspended immediately and a substantial amendment submitted to the Health Research Authority (HRA), REC and MHRA informing them of the temporary halt. The investigators planned to review the data once the database was locked and, consequently, devise a plan for the development of the trial in the light of the TSC’s recommendations.
Publicity
Following recommendations from the DMC following the safety analysis meeting on 7 December 2017, and those from the TSC on 13 December 2017, the trial was no longer publicised. The Poppi trial website was updated to inform viewers that recruitment to the trial had been paused while the trial data were reviewed. Key stakeholders were also informed of the trial’s current status.
Completion of data entry
Following the decision to suspend the trial, the site team then entered any outstanding CRFs and missing data onto OpenClinica and resolved discrepancy notes in which queries in the data had been raised. The protocol stated that the inter-rater and intra-rater reliability would be assessed for the co-primary outcome (i.e. PIPP-R score), so duplicate CRFs were created for this outcome and validated by the programming team. The PIPP-R score was then recalculated and re-entered by the site team. The programming team also needed to create new CRFs (and validate these) for a number of data items that were recorded during the trial but not transposed onto CRFs (e.g. noxious brain activity at baseline). Given the sudden suspension of recruitment, the PMG agreed that these additional data should be reported in the publication of the results to provide context. The site team then provided the programming team with the additional data, and the programming team uploaded these to the CRFs and completed validation. A sample of the original data within OpenClinica was also cross-checked with the paper CRFs for validation purposes.
Database lock
The senior trials programmer provisionally locked the database in order for any checks to be carried out, by removing permissions to edit the database from all personnel. These checks were signed off by the trial manager, the head of trials programming, the senior statistician and the trial statistician, and included:
-
A sample of 10% of all participants, which was generated randomly, ensuring that at least 10% of each CRF was included. All data on the OpenClinica eCRFs, for each of these participants, were compared with the data in the Stata data set to be used for analysis. In the case of the SAE eCRF, the data in the Stata data set were compared with the data on the paper CRFs. All data on the paper SAE chief investigator assessment CRFs were compared with the data in the Stata data set to be used for analysis.
-
Checking that all forms received at the co-ordinating centre had been entered onto the corresponding eCRF and marked as complete in OpenClinica.
-
Checking that all discrepancy notes in OpenClinica were closed.
-
Checking that the reports generated from the administrative database for consent form issues, CRF signature and delegation log checks.
-
Validation of the process for importing extra details from an Microsoft Excel® (Microsoft Corporation, Redmond, WA, USA) spreadsheet into OpenClinica.
-
Sign-off of the SAP.
Authorisation to lock the database was provided by the chief investigator, the principal investigator, the director of the co-ordinating centre, the senior statistician, the head of trials programming and the trial manager, and the senior trials programmer locked the database. The data set was exported to the trial statistician and the site research team for analysis (the principal investigator signed a memorandum of understanding for the transfer of the data). The trial statistician used this data set to create the final report detailing the results of the trial.
Two differences between the clinical stability source data and OpenClinica data set were discovered on 14 May 2018 by the site team. The error was identified when the raw data were analysed in MATLAB (MathWorks, Natick, MA, USA) rather than the exported data set from OpenClinica, and the outputs differed from the final report (provided by the trial statistician using the OpenClinica data set). Although correction of these errors would not significantly alter the results, the values in the final report at that time were inaccurate.
The PMG met on 17 May 2018 and agreed that the database should be unlocked, these values corrected, then the database relocked. The PMG was attended by the co-ordinating centre QA associate, who provided QA input into the decisions made by the group. The PMG decided that, beforehand, a complete comparison should be made of the clinical stability raw data from MATLAB and data entered onto OpenClinica.
A further meeting of the PMG was held on 22 May 2018. The PMG agreed that the following should be checked against their source data and were selected because of their importance in the final results:
-
co-primary outcome
-
magnitude of noxious-evoked specific brain activity evoked by heel lance (source data from MATLAB – value for each infant)
-
-
important secondary outcomes
-
increase in respiratory support before and after the clinical intervention (source data on cot-side log – yes/no for each infant)
-
-
safety outcomes
-
at least one incident of apnoea that required an intervention using Neopuff or bag-and-mask ventilation in the 24-hour period following drug administration (source data on the cot-side log – yes/no for each infant)
-
at least one incident of hypotension that required treatment with inotropes in the 24-hour period following drug administration (source data on cot-side log – yes/no for each infant).
-
The PIPP-R scores (at ROP screening, heel lance and control heel lance) were already validated by the trial statistician and postdoctoral researcher on 27 March 2018, when the source data were checked. These data, therefore, did not need to be re-checked.
These further outcome data were entered by the postdoctoral researcher and research nurse onto a separate spreadsheet on 24 May 2018. The head of trials programming ran the comparison with the OpenClinica data set and confirmed that there were no further inconsistencies.
The database unlock documentation was then prepared by the co-ordinating centre, documenting any required changes to the database. Following approval from all individuals named on the unlock form (co-ordinating centre director, chief investigator and senior statistician), the database was unlocked on 4 June 2018 by the senior trials programmer, and the trial manager and postdoctoral researcher together entered the correct values. The co-ordinating centre programming team then checked the audit log and verified that only the agreed changes had been made to the data set. The relock was then authorised by the senior trials programmer, head of trials programming, senior statistician, chief investigator, principal investigator, co-ordinating centre director and trial manager. The database was relocked on 6 June 2018 by the senior trials programmer, and the new data set exported for the trial statistician to revise the relevant tables and update the final report. The new data set was transferred to the principal investigator and postdoctoral researcher from the site team to run the additional analyses.
Analysis
The final analysis was completed by the trial statistician according to the SAP, version 2.0 and reviewed by the senior statistician. One deviation was made from the SAP: where risk ratios were not estimable because of no events in one trial arm, risk differences were calculated. This applied to the outcome ‘required an increase in respiratory support’ at both 6 and 24 hours following the clinical intervention. Additional post-hoc exploratory analyses were performed by the on-site Poppi trial team (see Exploratory analyses for more detail).
The co-primary outcomes were validated by an independent statistician at the co-ordinating centre prior to the analysis being finalised.
Patient and public involvement
The research team worked closely with the on-site charity SSNAP during the design, conduct and dissemination of the trial. The charity’s remit is to provide family support for parents with infants on the newborn care unit and to support neonatal medical research.
Trial design
Patient and public involvement input was essential in designing the trial and was considered critical to its success. To ensure that the parents’ views and opinions were considered from the beginning of the trial, the SSNAP parent help team leader was an applicant on the grant application. In collaboration with SSNAP, the research team organised a workshop in which parents of premature infants discussed the proposed clinical trial with the research team. This workshop provided the parents with an opportunity to discuss their ideas about the trial and explore the ethical, practical and emotional issues associated with the research. Furthermore, parents were actively involved in the application for ethical and regulatory approvals. The parents suggested amendments to the research design based on their experiences and edited the proposed PIL (see Report Supplementary Material 6). Parents advised on timing and manner of approach to potential families, and suggested ideas regarding dissemination of information about the trial. Importantly, parents were able to advise on how their expectations about pain management differed from actual clinical practice, and made suggestions about how the team could sensitively approach this with families.
Trial conduct
Parents of prematurely born infants are often unaware and shocked when they realise their infants will experience painful procedures without the provision of pain relief. Raising awareness of this and explaining why so few pain-relieving drugs are available for infants highlighted the importance of the trial and encouraged parent participation. This is important in neonatal research, as families can be in the neonatal unit for extended periods and provide a support network for each other. The SSNAP parent help team leader was the primary support contact for parents and clinical staff, and provided key information to parents about the trial. This endorsement by SSNAP was critical to ensure that parents felt that the trial had been designed and was conducted with the care of their infant, and their own concerns, in mind.
The parent help team leader identified a volunteer from SSNAP to act as a PPI representative on the trial TSC. All SSNAP volunteers are individuals who have had a child or grandchild cared for in the neonatal unit of the John Radcliffe Hospital and, therefore, have the experience to both support and promote the charity. The PPI representative provided invaluable insight into the parents’ perspective and considerations for parents’ views at each stage of the trial, by attending and inputting into every TSC meeting that was held.
The parent help team leader also identified volunteers from SSNAP to raise awareness of the trial on the neonatal unit. The parent help team leader co-ordinated an information session where the research team met with the volunteers and gave a presentation about pain in premature infants and the trial. This provided the volunteers with the opportunity to ask questions about the trial and consider how they might be able to promote the trial. These volunteers acted as a further source of information for parents who were considering participating. The parent help team leader and the SSNAP volunteers provided information about the trial at SSNAP coffee mornings, where parents of premature infants meet for support. The research team attended these meetings, where possible, in order to build further relationships with the parents and answer any questions they may have had about their infant and pain management.
Partway through the trial, the Poppi trial team adapted the existing trust ROP screening PIL to improve and clarify the information provided to parents about the procedure (see Chapter 1, Retinopathy of prematurity screening). The SSNAP team reviewed the document to ensure that it was parent-friendly and provided all relevant information in a suitable format. SSNAP approved the wording and endorsed the document.
Trial dissemination
Both the SSNAP parent help team leader and the TSC PPI representative attended the TSC/DMC/collaborator meeting to discuss the draft results manuscript for The Lancet [see Results (The Lancet)]. They also attended the family event held on 11 July 2018 to thank the families who participated, and to feedback the results and explain the early trial closure (see Participants). Their involvement in this event was critical to its success, as they had established relationships with many of the families. They were able to provide further support and information from SSNAP, and encouraged families to complete a feedback survey about their experiences of the trial (see Family feedback).
Personnel
The trial manager changed in February 2017. The senior statistician changed in August 2017. An ophthalmology specialist trainee joined the research team in November 2017.
A co-ordinating centre delegation of authority and signature log was maintained in the TMF, documenting all members of staff involved with the trial at the co-ordinating centre and signed off by the chief investigator. The co-ordinating centre also held a delegation document stating the responsibility division between the chief investigator/principal investigator and the co-ordinating centre.
Dissemination
Summary protocol publication (The Lancet)
The summary protocol for the trial was published in The Lancet. 43 This was accepted for publication on 22 January 2016.
Protocol publication
The protocol was published with Wellcome Open Research on 15 November 2016 and the revised protocol was published on 26 January 2017. 28
Results (The Lancet)
A joint TSC, DMC and collaborator meeting was held on 11 June 2018 to review the draft The Lancet manuscript detailing the results of the trial. All invited attendees had the opportunity to review and comment on the draft manuscript electronically prior to the meeting. The attendees largely approved the manuscript and the underlying messages, and provided suggested minor edits throughout the text. The revised manuscript was published in The Lancet in November 2018. 42
Commentary (The Lancet Child & Adolescent Health)
A commentary discussing the ethical considerations within the trial and the regulatory and oversight procedures in place was co-published in The Lancet Child & Adolescent Health in November 2018. 44
Other relevant publications
Members of the Poppi trial team published a paper in Science Translational Medicine22 that described and validated the methods that were used to quantify the primary outcome measure (i.e. noxious-evoked brain activity) in the trial.
Participants
The parents/guardians of the participants of the trial were invited by telephone and by post to a family event on 11 July 2018. The families of 10 participants attended, including parents, grandparents, siblings and aunts. The research team used the event to thank the participants and their families for their contribution and support for the trial. An animation explaining the challenges of assessing pain in infants, and exploring the purpose of the research, was shown to the families. The principal investigator thanked the families, disclosed the results of the analysis and explained the sequence of events leading up to trial closure. The research team were keen that families clearly understood the decision to stop the trial and had the opportunity to discuss any queries or requests for further information with the team in person. Parents of five participants also requested (in writing) to know the treatment allocation of their infant. The event was extremely successful and greatly supported by representatives from SSNAP. A letter was also sent to the parents/guardians of all participants in the post in November 2018, summarising the trial results and explaining that these would soon be published.
Family feedback
The families that attended the Poppi trial family event also completed a feedback survey. The survey was designed by the research team, and reviewed and adapted on the advice of SSNAP. The SSNAP members present at the event helped families to complete the survey, which covered their experiences in the trial and their attitudes to neonatal research. Families were asked about their involvement in research before the trial, the information they were provided about pain, ROP screening, the trial, their decision to participate, their understanding of blinding and random allocation, and the dissemination of results. Families were also asked if they would consider taking part in future research, their concerns about their infant’s discomfort/pain, measures that they felt had helped with their infant’s discomfort/pain, and their infant’s response to discomfort/pain.
The majority of families had not taken part in research prior to the Poppi trial. The amount of information about pain control that had been provided to families varied and approximately half the families had received some information about ROP screening before being approached about the Poppi trial. All families felt that they had received the right amount of information about the trial and all reported that they had understood that their infant might have received either morphine or placebo. The families most consistently reported that finding new medicines to treat pain in infants, and the potential benefit to future infants, were very important reasons for participating in the trial. All families reported that they were pleased that their infant had taken part in the trial and pleased to be invited to the event to be informed about the results. Eight of the nine families reported that they would consider taking part in future research and one family stated that they had already participated in another research study.
Staff and stakeholders
An end-of-trial event was held on 12 September 2018 to thank all the Poppi trial staff and stakeholders for their efforts and input into the trial.
Publicity
Website
A trial website was maintained on the co-ordinating centre website (www.npeu.ox.ac.uk/poppi; accessed 15 June 2019). This resource provided a link to the randomisation and stock management website, and also provided a source of up-to-date information about the trial. The home page provided a short summary of the trial, a recruitment count, videos showing animations about infant pain and the group’s research, and general information about the trial (including eligibility criteria and trial registration details). The home page also included acknowledgement of the funding bodies (i.e. the NIHR EME programme and the Wellcome Trust), and endorsement from Bliss (a charity for babies born premature or sick) and SSNAP. The Poppi trial website provided contact details for core members of the team, information and links about the team’s involvement with the media, information for parents (including a flow diagram of study processes and the PIL), the protocol, relevant publications and a recruitment summary by month. News items were also added regularly, which included updates on recruitment, media coverage and publications.
Media
The publication of the Science Translational Medicine paper (see Other relevant publications) generated a large amount of interest from the media. Articles about the trial, and the impact of this work on the measurement of pain in premature infants were widely published and included articles in the New Scientist (www.newscientist.com/article/2129744-electrode-can-tell-you-if-a-baby-is-really-experiencing-pain/; accessed 15 June 2019), The Guardian and IEEE Spectrum (http://spectrum.ieee.org/the-human-os/biomedical/diagnostics/brain-activity-detector-helps-study-pain-relief-for-babies; accessed 15 June 2019), as well as many other news outlets. Professor Slater took part in an interview about the work and this is available on the International Business Times UK website (www.ibtimes.co.uk/your--pain-brain-scan-can-detect-physical-suffering-infants-1619859; accessed 15 June 2019). Professor Slater and Dr Moultrie also featured in a BBC Radio 4 programme entitled ‘From Agony to Analgesia’, which was broadcast on 16 August 2017. This programme focused on specific research areas in the field of pain, during which Professor Slater and Dr Moultrie discussed novel methods to measure pain in premature infants and the objectives of the trial.
In November 2017, the Poppi trial team joined the #startedinoxford initiative with a video introducing its work to explore how infants feel pain. The #startedinoxford initiative has a website (http://innovation.ox.ac.uk/innovation-news/startedinoxford/; accessed 15 June 2019 ) and YouTube (YouTube, LLC, San Bruno, CA, USA) channel (https://www.youtube.com/results?searchquery=%23StartedinOxford; accessed 15 June 2019) publicising the innovative research being conducted at the University of Oxford.
The clinical doctoral fellow from the Poppi trial team (Dr Fiona Moultrie) collaborated with a professional animator (Miss Charlotte Moultrie) to create an animation explaining the importance of understanding pain in infants, the difficulties of assessment, and new imaging and recording techniques that are being used to understand infant brain development. This animation also discussed the aims of the trial, and the video appealed to parents of infants on the neonatal unit, adults and school children, as well as scientists, doctors and nurses. The video was available on the Poppi trial website, shown to parents at the family event, and exhibited at various public engagement events and public venues including the Oxford Natural History Museum, the Ashmolean Museum and the Cheltenham Science Festival. The work was also recently recognised with an honourable mention at the OxTalent 2018 awards in the digital media category.
Public engagement events
The Poppi trial team presented information about the trial alongside interactive games and displays about neonatal brain development at the ‘FrightFriday’ public engagement event on 25 November 2016 at the Ashmolean Museum in Oxford, which was supported by the Wellcome Trust and the national Festival Finale for the Being Human Festival. The event comprised a late-night opening of the museum to explore the art and science of hope and fear, with performances, workshops and stalls run by researchers across the University of Oxford. The Poppi trial team had a large display, which included Poppi trial posters, an interactive three-dimensional (3D) brain model to demonstrate the similarities between the experience of pain in adults and infants, 3D printed brains and a video animation (https://www.youtube.com/results?search_query=%23startedinoxford; accessed 25 July 2019). The event was extremely successful, and the team’s display was featured in the University of Oxford’s Medical Sciences newsletter (December 2016). The SSNAP parent help team leader (also a grant applicant) was also involved with the Poppi trial and paediatrics department activities at this event.
Conferences
Dr Moultrie gave a presentation entitled ‘Neonatal Pain: Improving our Understanding of the Assessment and Treatment of Pain in Infants’ at the Congenital Cardiac Anaesthesia Network Meeting on 13 May 2016 in Southampton, UK. During the presentation, Dr Moultrie described the definition of pain, methods to measure pain in the newborn, how to investigate the effects of early pain and current research to test analgesics in this population, specifically the Poppi trial.
Dr Hartley also gave a presentation entitled ‘Using Noxious-Evoked Brain Activity to Measure Analgesic Efficacy in Infants’ at the London Pain Consortium Research Update Meeting on 12 May 2016. Dr Hartley provided information about infant pain, surrogate measures of infant pain, the balance of reflex withdrawal and brain activity in early development in response to noxious stimuli, the potential use of brain activity to explore analgesic efficacy and the plans for the Poppi trial.
Dr Moultrie presented a talk entitled ‘Determining Infant Pain Indicators to Support New Approaches to Pain Assessment and Effective Pain Management Strategies’ in a workshop at the International Symposium for Paediatric Pain, in Malaysia, in July 2017. Dr Moultrie explained how quantitative neurophysiological techniques can be used to evaluate nociceptive responses in infants and how these measures can be used as sensitive objective end points in clinical trials to assess analgesic efficacy. She outlined the Poppi trial protocol, which generated substantial interest and discussion. Following the early cessation of the trial and final analysis, Dr Moultrie also described the trial results at the Neonatal Society Autumn Meeting in November 2018 in London, UK, where she won the prize for best presentation.
Other
Literature searches
The research team searched PubMed (National Centre for Biotechnology Information, Bethesda, MD, USA) and Google Scholar (Google Inc., Mountain View, CA, USA) at least fortnightly for any relevant literature using a range of search terms such as ‘infant’, ‘pain’, ‘analgesia’ and ‘morphine’. The research team are based within the Paediatric Neuroimaging Group at the University of Oxford; when any member identified relevant literature, this was circulated to the full research team.
Emergencies
Trial documentation was provided to the neonatal unit, including contact numbers of trial staff who should be contacted in an emergency. A clinical telephone cascade included four members of staff, who gave permission for their contact details to be distributed, and the trial enquiries cascade included three members of staff.
Complaints
All parents provided written informed consent for their infant taking part in the trial. If parents were anxious, members of the clinical and research team answered any questions and discussed concerns with them. If the parents had any complaints during the Poppi trial these would have been documented, any action taken registered in their medical notes, and an incident form completed. In the PIL (see Report Supplementary Material 6), parents were given the contact details of the trial manager, chief investigator and principal investigator if they wanted more information, alongside the contact information for the trial sponsor and the Patient Advice and Liaison Service should they have wished to report a complaint or concern. No complaints were received about the Poppi trial.
Network support
The trial was supported by the Thames Valley and South Midlands Clinical Research Network (CRN). This provided funding to support the full-time salary for a dedicated neonatal research nurse, working exclusively at the newborn care unit at the John Radcliffe Hospital, Oxford. The research team liaised with the neonatal research nurse throughout the set-up of the trial to ensure that clinical staff had up-to-date GCP training. The team continued to collaborate with the neonatal research nurse about establishing effective methods for training neonatal staff members about trial processes during set-up. Once recruitment started, the research team continued to work with the neonatal research nurse to ensure that there was staff training and protocol compliance.
Trial funder
The trial funder was regularly provided with trial updates using the NIHR Evaluation, Trials and Studies Coordinating Centre (NETSCC)’s online management information system. Recruitment updates were submitted on a monthly basis and biannual progress reports were prepared providing information on all aspects of research activity in the trial. Minutes from oversight committee meetings (with actions for the research team) were uploaded periodically, as well as outputs for all public engagement activities, publicity resources and publications. The trial funder was made aware of any trial amendments, and trial documents (both draft and approved versions) were similarly uploaded to the online management system. Partway through recruitment, the Poppi team also met with the EME programme director to discuss the progress and the future of the trial. The programme director enquired about the practicalities of the trial, including information about the live birth rate at the site, standard practice for ROP screening, assessment of trial outcomes, appropriateness of morphine and the proposed plans to increase recruitment. The principal investigator provided detailed information about the trial, trial processes and future research plans. The meeting concluded with all attendees in agreement regarding the importance of the research area and the critical need for continued research into the prevention of pain in infants.
Chapter 4 Results
The results of this trial have now been published by the authors in The Lancet;42 some of the information and figures in this section have been reproduced from this published work. © 2018 The Author(s). 42 Published by Elsevier Ltd. This is an Open Access article distributed in accordance with the terms of the Attribution 4.0 International (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text, figures and tables below include minor additions and formatting changes to the original text.
Recruitment
Recruitment to the trial began on 16 September 2016 and the first participant was recruited on 30 October 2016. A total of 276 infants were screened for eligibility and 95 were approached for participation (Figure 9). Thirty-six infants were enrolled in the trial, of whom 31 had been randomised at the point of recruitment suspensions and, subsequently, trial closure (see Chapter 3, Trial closure). Data were available for only 30 participants, as one infant was withdrawn before being studied (see Chapter 3, Incident reports).
FIGURE 9.
Participant flow.
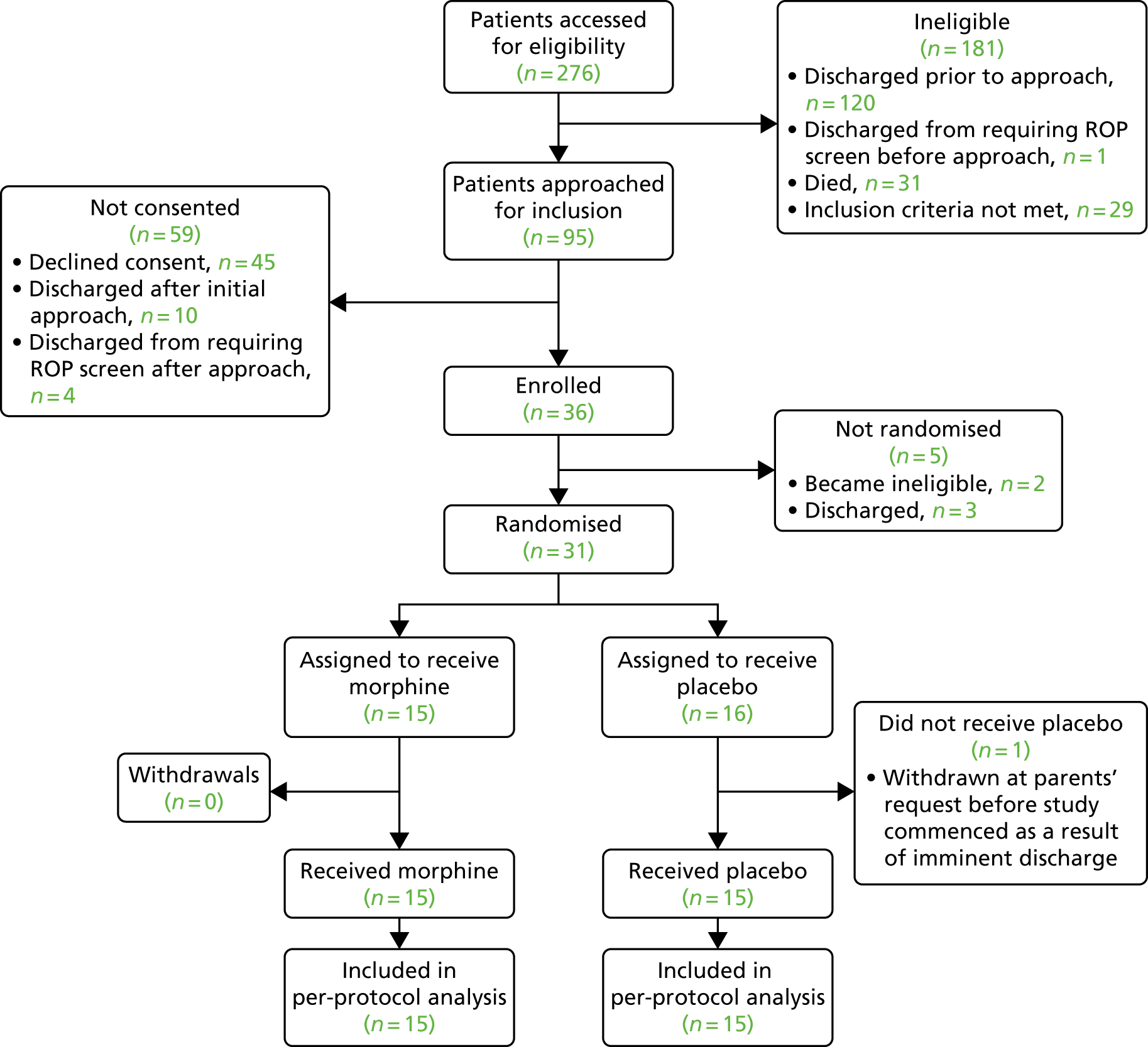
Throughout the trial, approximately two infants were recruited per month. This was below the target rate of four or five infants per month. Issues affecting recruitment, and strategies that were either implemented or suggested to improve recruitment, are described in Recruitment challenges and resulted in substantial increases in recruitment figures in July and September 2017 (six and nine recruits, respectively). The target and actual recruitment rates for the duration of the trial are presented in Figure 10.
FIGURE 10.
Target and actual recruitment.
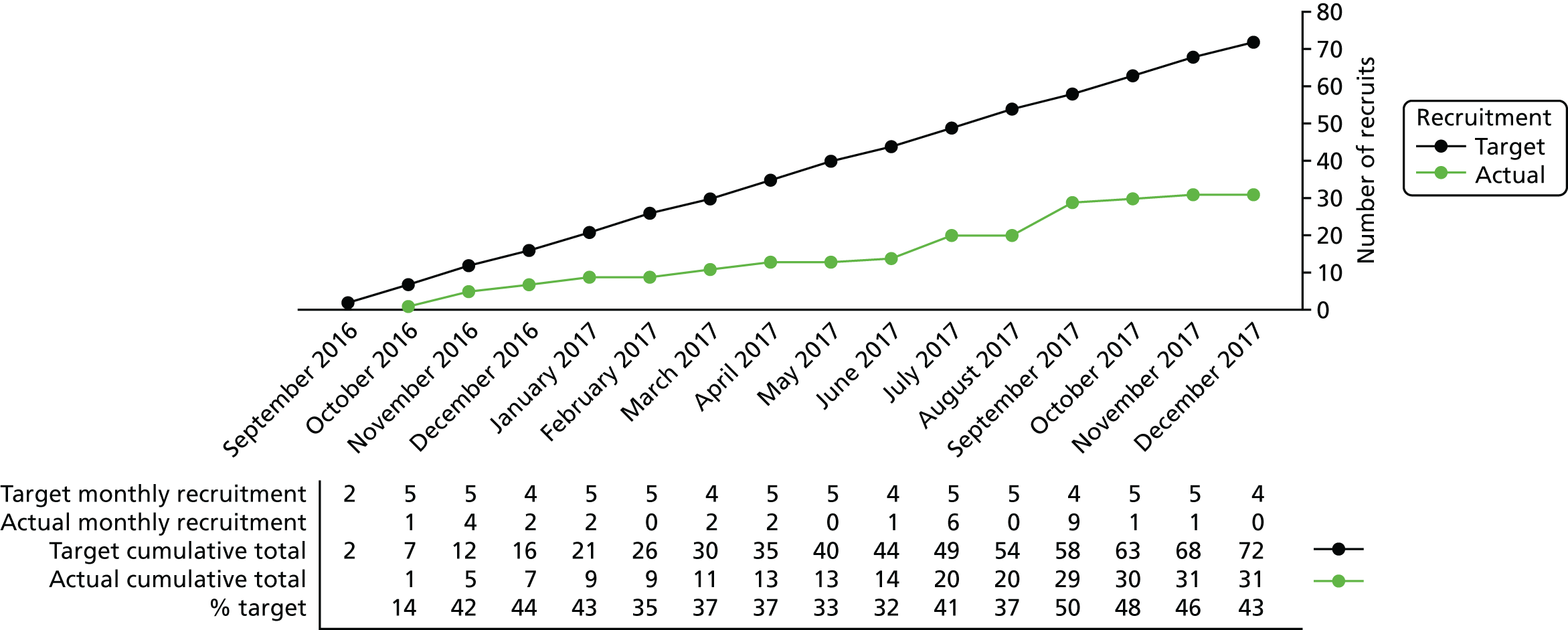
Baseline data
There were no deviations from the exclusion or inclusion criteria. Infant demographics and clinical characteristics at birth and the time of intervention are reported in Table 3 according to group allocation.
| Characteristics | Trial arm | |
|---|---|---|
| Morphine (N = 15) | Placebo (N = 15) | |
| At birth | ||
| Gestational age (weeks),a median (IQR) | 28.1 (26.3–30.1) | 28.6 (27.9–29.7) |
| Birthweight (g), mean (SD) | 1107 (329) | 1173 (350) |
| Birthweight z-score, mean (SD) | –0.4 (0.9) | –0.2 (1.0) |
| Intrauterine growth restriction, n (%) | 2 (13.3) | 3 (20.0) |
| Apgar score at 10 minutes,b median (IQR) | 10.0 (9.0–10.0) | 10.0 (8.0–10.0) |
| Mode of delivery, n (%) | ||
| Spontaneous vaginal delivery | 8 (53.3) | 5 (33.3) |
| Caesarean section | 7 (46.7) | 10 (66.7) |
| Sex (male), n (%) | 12 (80.0) | 8 (53.3) |
| Multiple pregnancy, n (%) | 4 (26.7) | 4 (26.7) |
| At time of randomisation | ||
| Gestational age (weeks)a | 34.7 (34.1–35.1) | 34.7 (34.1–35.1) |
| Days ventilated,a,c median (IQR) | 8.0 (1.0–20.0) | 3.5 (2.0–19.5) |
| Days since morphine last given,a,d median (IQR) | 46.5 (33.5–49.0) | 19.0 (15.0–39.0) |
| Presence of gastric tube,a n (%) | 14 (93.3%) | 15 (100.0%) |
| Infants with IVH (grade I or II), n (%) | 3 (20.0%) | 2 (13.3%) |
| Infants with history of surgery,a n (%) | 0 (0%) | 1 (6.7%) |
| At time of clinical procedure | ||
| Gestational age (weeks), median (IQR) | 35.0 (34.3–35.4) | 34.9 (34.3–36.3) |
| Postnatal age (days), median (IQR) | 50 (28–58) | 49 (43–59) |
| Weight (g), mean (SD) | 2049 (426) | 2128 (331) |
| Duration of ROP screening (seconds), median (IQR) | 97 (82–108) | 91 (83–110) |
| Diagnosis of ROP, n (%) | 2 (13.3%) | 2 (13.3%) |
| Level of care, n (%) | ||
| Intensive care unit | 1 (6.7%) | 1 (6.7%) |
| HDU | 5 (33.3%) | 9 (60.0%) |
| LDU | 9 (60.0%) | 5 (33.3%) |
| Respiratory support modality, n (%) | ||
| Self-ventilating | 9 (60.0%) | 8 (53.3%) |
| Low-flow oxygen therapy | 2 (13.3%) | 1 (6.7%) |
| High-flow oxygen therapy | 4 (26.7%) | 6 (40.0%) |
| Time (minutes) between IMP administration and heel lance, median (IQR) | 61 (57–66) | 63 (58–70) |
Comparison between trial arms: Premature Infant Pain Profile – Revised scores, noxious-evoked brain activity and reflex withdrawal
The co-primary outcome measures used to assess morphine analgesic efficacy were the PIPP-R score following ROP screening and the magnitude of noxious-evoked brain activity following heel lance.
The PIPP-R score following ROP screening was not significantly different between the morphine-treated infants and those who received placebo [morphine: mean PIPP-R score 11.1 (SD 3.2); placebo: mean PIPP-R score 10.5 (SD 3.4); mean difference 0.5, 95% CI –2.0 to 3.0; p = 0.66 (t-test)] (Figure 11). Similarly, the magnitude of the noxious-evoked brain activity following heel lancing did not significantly differ between infants in the two trial arms [morphine: n = 15, median 0.99, IQR 0.40–1.56; placebo: n = 14, median 0.75, IQR 0.33–1.22; median difference 0.25, 95% CI –0.16 to 0.80, p = 0.25 (Wilcoxon rank-sum test)] (Figure 12). Consistent with this, the PIPP-R score and the magnitude of the reflex withdrawal activity evoked by the heel lance did not significantly differ between the two groups [morphine: mean PIPP-R score 7.9 (SD 3.4); placebo: mean PIPP-R score 8.5 (SD 3.9); mean difference –0.6, 95% CI –3.3 to 2.1, p = 0.66; morphine: reflex withdrawal, n = 15, median 24.8, IQR 19.7–44.8; placebo: n = 14, median 12.4, IQR 6.1–46.3; median difference 8.9, 95% CI –12.0 to 22.4, p = 0.48 (Wilcoxon rank-sum test)] (Figures 13 and 14).
FIGURE 11.
Co-primary outcome: mean (SE) PIPP-R score following ROP screening in the infants who received morphine (black) compared with placebo (green). SE, standard error.
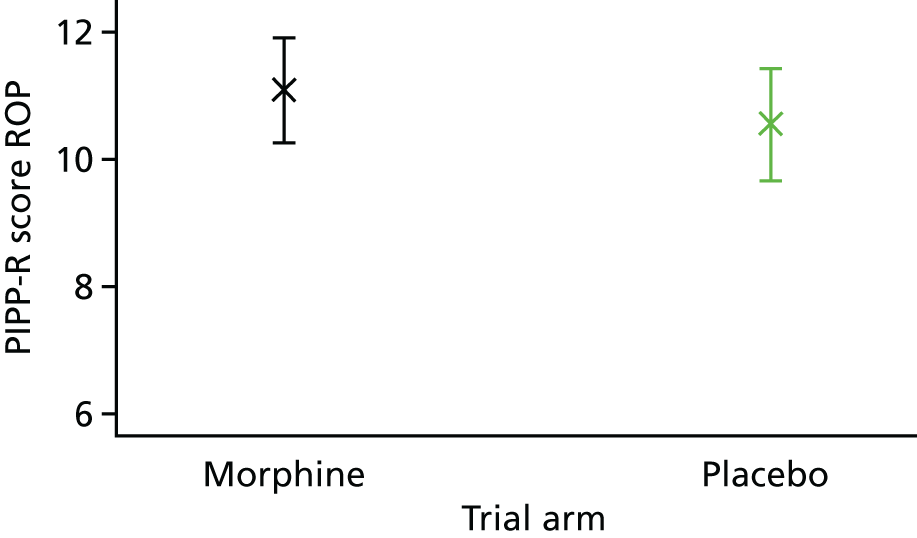
FIGURE 12.
Co-primary outcome: average noxious-evoked brain activity following heel lancing in infants who received morphine compared with placebo. The Woody-filtered EEG is shown overlaid with the template of noxious-evoked brain activity (in blue). The median (SE) magnitude of the noxious-evoked brain activity, quantified using the template, is shown for the two groups. SE, standard error.

FIGURE 13.
Secondary outcome: mean PIPP-R score following heel lance in the infants who received morphine compared with placebo. SE, standard error.

FIGURE 14.
Secondary outcome: median (SE) magnitude of the reflex withdrawal activity following heel lancing in infants who received morphine compared with placebo. The magnitude is quantified using the RMS in 250-millisecond windows. SE, standard error.
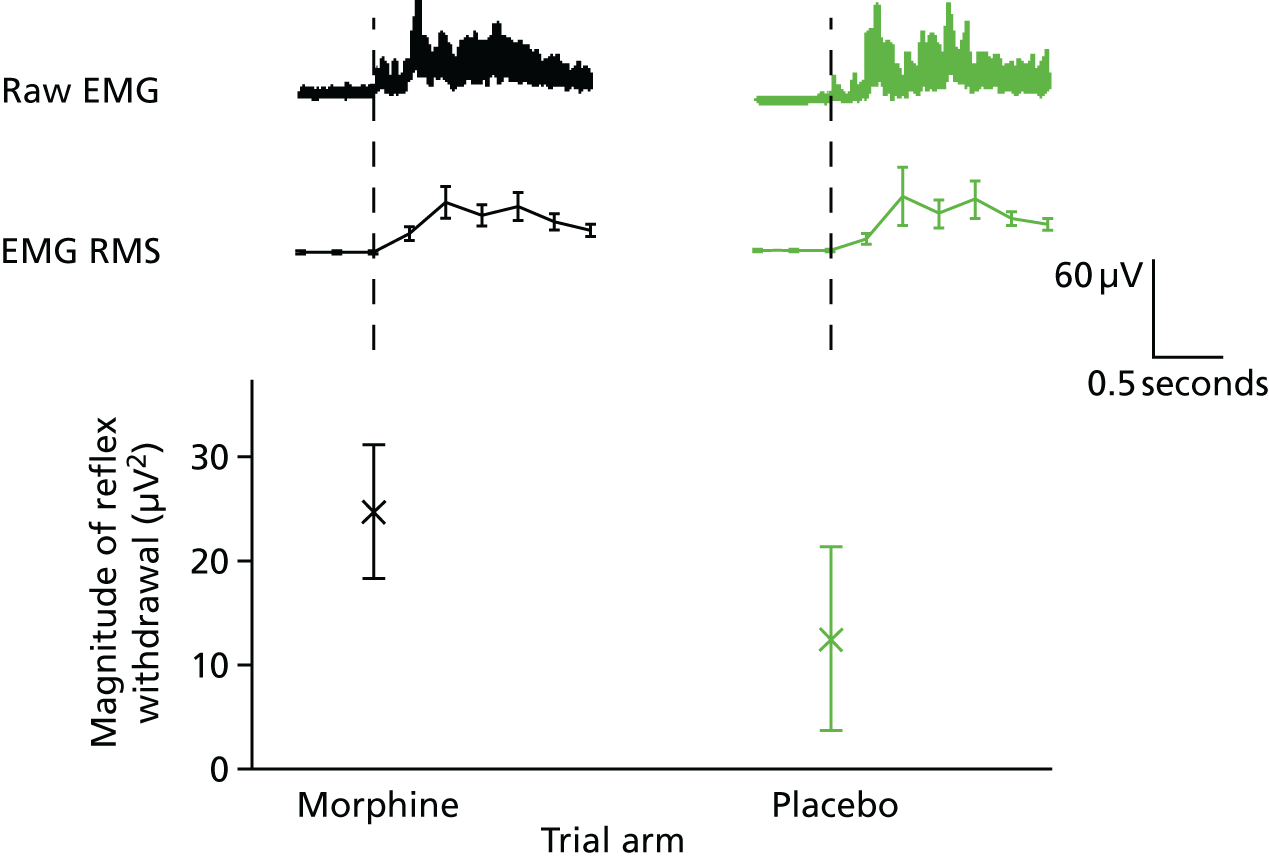
Comparison between background, control lance, heel lance and retinopathy of prematurity screening: Premature Infant Pain Profile – Revised, noxious-evoked brain activity and reflex withdrawal
The magnitude of each outcome measure used to assess analgesic efficacy significantly increased following the noxious procedures compared with non-noxious control stimuli and background activity (Figure 15), demonstrating that the measures were discriminative and appropriate for assessing analgesic efficacy in this population. The average PIPP-R score for all 30 infants was 4.6 (SD 3.2) following the non-noxious control stimulus, 8.2 (SD 3.6) following the heel lance and 10.8 (SD 3.3) following ROP screening (Figure 15), where a score of < 7 is considered indicative of minimal or no pain. 14 The intra-rater reliability was 0.98 (95% CI 0.97 to 0.99) for heel lance PIPP-R scores and 0.97 (95% CI 0.94 to 0.99) for ROP screening. The inter-rater reliability was 0.98 (95% CI 0.95 to 0.99) for heel lance and 0.89 (95% CI 0.79 to 0.95) for ROP screening. In addition, the magnitudes of the noxious-evoked brain activity and reflex withdrawal activity were greater following heel lancing than following the non-noxious control stimulus and background activity [noxious-evoked brain activity: background median 0.09 (IQR 0.02–0.18), control median 0.15 (IQR 0.04–0.24) and heel lance median 0.93 (IQR 0.34–1.25); reflex withdrawal: background median 2.3 µV2 (IQR 1.2–5.2 µV2), control median 4.3 µV2 (IQR 2.5–7.1 µV2) and heel lance median 23.3 µV2 (IQR 9.2–44.8 µV2)] (Figures 16 and 17).
FIGURE 15.
Mean (SE) PIPP-R score in all infants following the control heel lance, heel lance, and the ROP screening. SE, standard error.
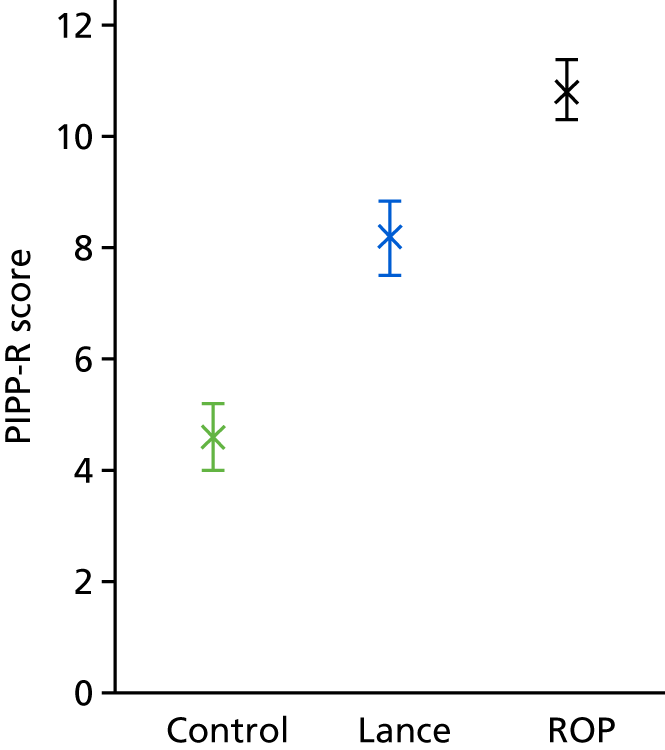
FIGURE 16.
Average EEG in background activity and following the control heel lance and heel lance in all infants. The Woody-filtered EEG is shown overlaid with the noxious-evoked brain activity template (in blue) and the median (SE) magnitude of the noxious-evoked brain activity is shown in all three conditions. SE, standard error.

FIGURE 17.
Reflex withdrawal activity during a background period and following the control heel lance and heel lance in all infants. The RMS in 250-millisecond windows is used to calculate the magnitude of the reflex withdrawal in all three conditions. SE, standard error.

Comparison between arms: clinical stability
The clinical stability of each infant was assessed over 48 hours (24 hours before and after the clinical procedure, defined as the heel lance followed by the ROP screening) by counting the number of episodes of apnoea, bradycardia, tachycardia and oxygen desaturation before and after the clinical procedure, and identifying infants with a significant increase in respiratory support. The standardised differences in the number of episodes of bradycardia, tachycardia and oxygen desaturation were calculated. Infants who received morphine experienced significantly more episodes of oxygen desaturations in the 6- and 24-hour periods following the clinical procedure, and significantly more episodes of bradycardia in the 24-hour period following the clinical procedure, than did infants in the placebo group (Table 4 and Figures 18–21).
| Time period pre and post the clinical procedure | Trial arm, median (IQR) | Median difference (95% CI) | p-value | |
|---|---|---|---|---|
| Morphine | Placebo | |||
| 6 hours | ||||
| Oxygen desaturation | 0.57 (0.00–0.99) | –0.06 (–0.65–0.00) | 0.66 (0.36 to 1.00) | 0.0007*** |
| Bradycardia | 0.50 (0.00–0.99) | 0.00 (0.00–0.98) | 0.33 (0.00 to 0.98) | 0.07 |
| Tachycardia | 0.00 (0.00–0.00) | 0.00 (0.00–0.00) | 0.00 (–0.16 to 0.00) | 0.32 |
| 24 hours | ||||
| Oxygen desaturation | 0.22 (–0.02–0.98) | 0.00 (–0.25–0.08) | 0.33 (0.03 to 0.75) | 0.019* |
| Bradycardia | 0.43 (0.00–1.00) | 0.00 (–0.50–0.60) | 0.43 (0.00 to 1.00) | 0.019* |
| Tachycardia | 0.00 (–0.50–0.98) | 0.00 (0.00–0.00) | 0.00 (–0.38 to 0.98) | 0.57 |
FIGURE 18.
Standardised difference in number of episodes of desaturations in the 6 hours pre and post clinical procedure. Median (SE) of the standardised difference in the number of episodes of desaturation in the 6 hours after the clinical procedure compared with the 6 hours before (the standardised difference is defined as the difference in number of episodes after the clinical procedure compared with before, as a proportion of the total number of episodes – see Chapter 3). SE, standard error. ***p < 0.001.
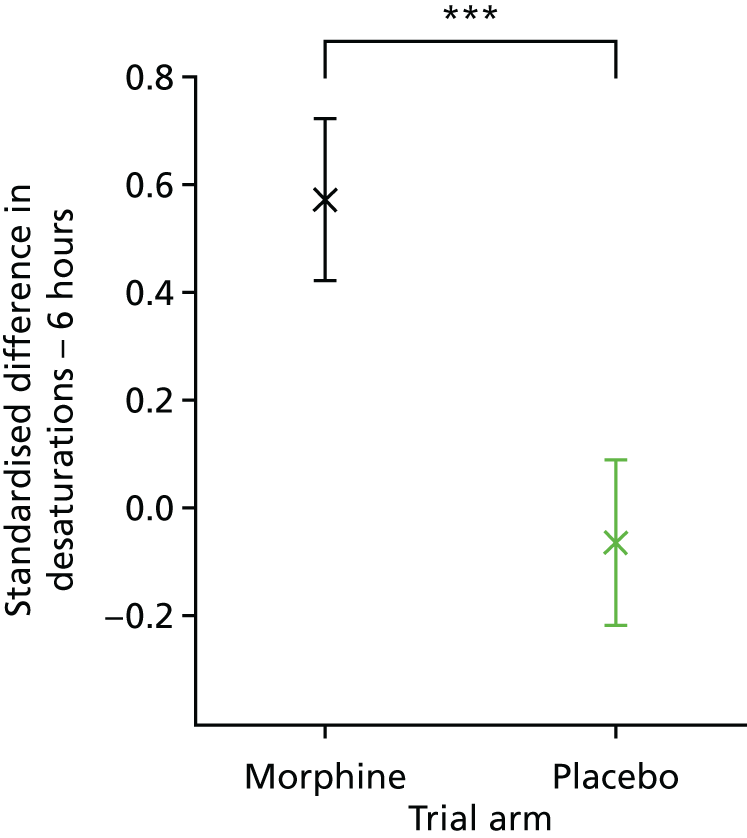
FIGURE 19.
Standardised difference in episodes of desaturations in the 24 hours pre and post clinical procedure. Median (SE) of the standardised difference in the number of episodes of desaturation in the 24 hours following the clinical procedure compared with the 24 hours before, in the morphine (black) and placebo (green) groups. SE, standard error. *p < 0.05.

FIGURE 20.
Standardised difference in episodes of bradycardia in the 6 hours pre and post clinical procedure. Median (SE) of the standardised difference in the number of episodes of bradycardia in the 6 hours after the clinical procedure compared with the 6 hours before, in the morphine (black) and placebo (green) groups. SE, standard error.
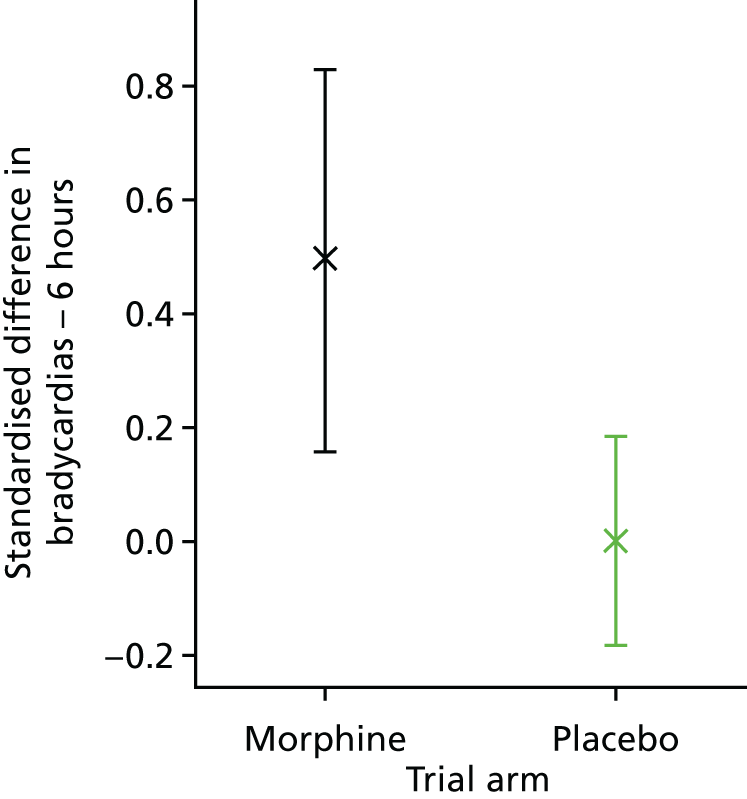
FIGURE 21.
Standardised difference in episodes of bradycardia in the 24 hours pre and post clinical procedure. Median (SE) of the standardised difference in the number of episodes of bradycardia in the 24 hours following the clinical procedure compared with the 24 hours before, in the morphine (black) and placebo (green) groups. SE, standard error. *p < 0.05.
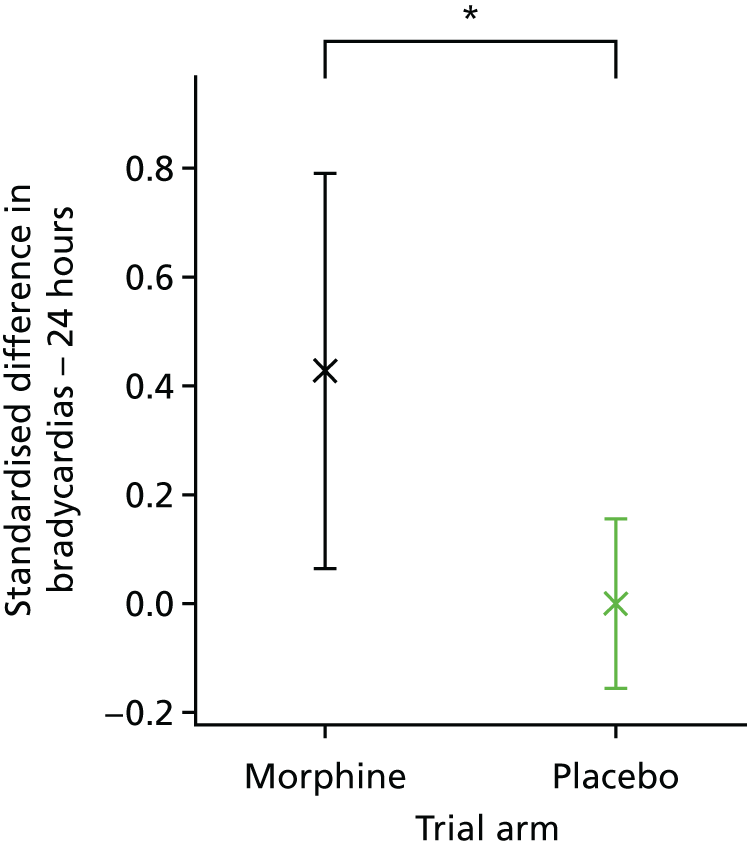
The number of infants who developed new-onset apnoeas or an increase in the number of apnoeic episodes and the number of infants who required a significant increase in respiratory support (see Chapter 3) in the 24-hour period following the clinical procedure were calculated. Eight of the 15 infants (53%) who received morphine developed new-onset apnoeas or experienced an increase in the number of apnoeic episodes in the 24-hour period following the clinical procedure, compared with 3 out of 15 infants (20%) who received placebo (risk ratio 2.7, 95% CI 0.9 to 8.2; p = 0.085). There were also more infants who experienced new-onset or increased apnoeas in the 6-hour period following the clinical procedure in the morphine-treated group (morphine: 7 out of 15 infants; placebo: 3 out of 15 infants; risk ratio 2.3, 95% CI 0.7 to 7.4; p = 0.15). Significantly more infants who received morphine required an increase in respiratory support in the 6 hours following the clinical procedure (morphine: 4 out of 15 infants; placebo: 0 out of 15 infants; risk difference 0.3, 95% CI 0.0 to 0.5; p = 0.020) and over the 24 hours following the clinical procedure (morphine: 5 out of 15 infants; placebo: 0 out of 15 infants; risk difference 0.3, 95% CI 0.1 to 0.6; p = 0.006). There were no differences in the number of episodes of tachycardia in the 6- or 24-hour period following the clinical procedure, or in the number of episodes of bradycardia in the 6-hour period following the clinical procedure (see Table 4).
Drug safety was assessed by considering the number of infants who had apnoeas that required resuscitation with NIPPV and incidences of hypotension that required treatment with inotropes in the 24-hour period following drug administration. The predefined safety stopping boundary was passed, as 3 of the 15 (20%) infants who received morphine had apnoeas requiring resuscitation with NIPPV in the 24 hours after drug administration, compared with 0 of 15 (0%) infants in the placebo arm [difference in proportion 0.2, 80% CI (adjusted to allow for planned multiple analyses) 0.05 to 1.00, p = 0.085; significant at the 20% level allowing for the prespecified stopping boundary]. None of the infants required inotropes and the blood pressure was similar in the two treatment groups (Figure 22).
FIGURE 22.
Blood pressure monitoring. The average mean blood pressure is shown across the 48-hour monitoring period for infants who received placebo (green) or morphine (black). Time 0 is the time of the clinical procedure. Error bars indicate mean ± standard error of the mean.

Comparison between arms: adverse events
In total, 11 of the 30 infants studied experienced adverse events (AEs), two of which had unforeseeable SAEs (Table 5). Eight of the 15 infants who received morphine had respiratory AEs attributed as possibly or probably related to drug administration Two infants had mild events, four infants had moderately severe events and two had moderately severe SAEs, one of which led to unmasking by the clinical team and the administration of two doses of naloxone. Of the 15 infants who received placebo, one had a mild respiratory AE. Two infants in each treatment arm were diagnosed with ROP, an expected SAE.
| Events | Onset of event post IMP administration | Treatment | Grade | Attribution | Allocation |
|---|---|---|---|---|---|
| AEs | |||||
| Nasal congestion | 11 hours 56 minutes | Saline drops | Mild | Not related | Placebo |
| Rash | 4 hours 4 minutes | Cream | Mild | Not related | Placebo |
| Profound desaturations | 17 hours 59 minutes | Facial oxygen | Mild | Not related | Placebo |
| Recurrent desaturations | 8 hours 9 minutes | Stimulation | Mild | Possibly | Morphine |
| Recurrent desaturations | 1 hours 58 minutes | Facial oxygen | Mild | Possibly | Morphine |
| Apnoea | 2 hours 13 minutes | NIPPV, increase high-flow oxygen | Moderate | Possibly | Morphine |
| Recurrent apnoeic episodes | 2 hours 39 minutes | Stimulation, increase low-flow oxygen | Moderate | Possibly | Morphine |
| Recurrent apnoeas | 1 hours 28 minutes | Stimulation × 3, NIPPV × 3 | Moderate | Possibly | Morphine |
| Recurrent desaturation/bradycardias/apnoea | 2 hours 3 minutes | Commenced high-flow oxygen, feed volume reduction | Moderate | Possibly | Morphine |
| SAEs | |||||
| Persistent hypoventilation/desaturation | 6 hours 0 minutes | Moved to HDU, commenced high-flow oxygen | Moderate | Possibly | Morphine |
| Recurrent apnoeas | 6 hours 24 minutes | Moved to HDU, commenced high-flow oxygen, naloxone × 2 | Moderate | Probably | Morphine |
Serious adverse events
There were two reported SAEs, as detailed below.
The first serious adverse event
The infant experienced respiratory problems at the time of a bottle feed and profound apnoea approximately 6 hours after IMP administration. The participant became apnoeic and required bag-and-mask ventilation. It was unclear at the time if this was associated with the IMP. The decision was made to unblind this participant because of intermittently poor respiratory effort. The participant had received the active IMP (i.e. morphine sulphate). The infant was transferred to the HDU and started on high-flow oxygen treatment. The infant was given naloxone at 8 hours and at 9 hours post IMP. The SAE was considered ‘expected’ and the initial report was that it was ‘probably’ related to the IMP.
The infant’s oxygen saturation recording was reviewed after the incident. These data highlighted that in the 24 hours prior to IMP administration, the infant had frequent episodes of physiological instability at the time of feeds. Following the ROP screen, the infant did not have any episodes of desaturation (< 80%) for a period of almost 4 hours, until the bottle feed (which was started at approximately 5 hours 15 minutes post IMP administration). This suggested that the SAE was related to the bottle feed. However, on review of the impedance pneumogram and CRFs it was identified that the infant had four apnoeas between the time of IMP administration and the bottle feed. This suggested that the original assessment, that the SAE was ‘probably’ related to the IMP, was correct.
The second serious adverse event
During a feed (≈4 hours post IMP), the infant had profound desaturations and bradycardias, requiring brief application of facial oxygen and only half the feed volume was given. The infant continued to have recurrent episodes of desaturation and low-flow oxygen treatment was started 5.5 hours post IMP. The infant was transferred from the LDU to the HDU for a partial septic screen and intravenous antibiotics. The infant displayed signs of respiratory distress (grunting and increased work of breathing) and was started on high-flow oxygen at a rate of 4 l/minute. The infant stabilised and high-flow oxygen treatment was discontinued the following day. Chest radiography revealed right middle lobe pneumonia, potentially consistent with aspiration, and the C-reactive protein level rose to a maximum of 13.9 mg/l. The infant was treated with antibiotics for 7 days. The infant remained on the HDU and received a red cell transfusion for anaemia 2 days later. The infant continued to have feed-related desaturations/bradycardias and was commenced on omeprazole 1 week after the study.
The vital signs data were reviewed retrospectively. In the 24 hours pre IMP administration the infant had four apnoeic episodes (as identified on the impedance pneumogram), 12 episodes of desaturation and five bradycardias. In the 24 hours post IMP, the infant had four apnoeic episodes, 14 episodes of desaturation and eight bradycardias. The SAE was considered ‘possibly’ related to the IMP, as morphine could increase the likelihood of aspiration.
Exploratory analyses
Post hoc exploratory analyses revealed that the administration of morphine to infants resulted in a significant reduction in the heart rate and respiratory rate, compared with the placebo group. Morphine administration reduced the group average heart rate by a maximum of 13.9 b.p.m. 1.5 hours after the clinical procedure (i.e. approximately 2.5 hours after drug administration) (Figure 23). The heart rate in the morphine-treated infants was significantly lower than in the placebo group for 6 hours (from 0.5 to 6.5 hours following the clinical procedure, p = 0.0001, cluster-corrected non-parametric analysis). The respiratory rate dropped by an average of 8 breaths per minute 1.5 hours after the clinical procedure and was significantly lower in the morphine group than in the placebo group for 7.5 hours (from 1 to 8.5 hours following the clinical procedure; p = 0.003, cluster-corrected non-parametric analysis; Figure 24). Similarly, 1.5 hours after the clinical procedure the oxygen saturation dropped by an average of 2% in the morphine-treated infants, although this was not significantly lower than in the placebo group (Figure 25). More than 12 hours after the clinical procedure, the oxygen saturation of infants who received placebo dropped by 1.2%, and was significantly different from morphine-treated infants for 2 hours (from 13 to 15 hours following the clinical procedure; p = 0.022).
FIGURE 23.
Average heart rate (b.p.m.) in the morphine and placebo groups during the 48-hour monitoring period. Individual infant traces are baseline corrected to the average baseline across all infants. Time 0 is the point of clinical procedure. Grey boxes indicate time periods when the trial groups are significantly different. Solid lines indicate the mean and shaded areas the standard error.
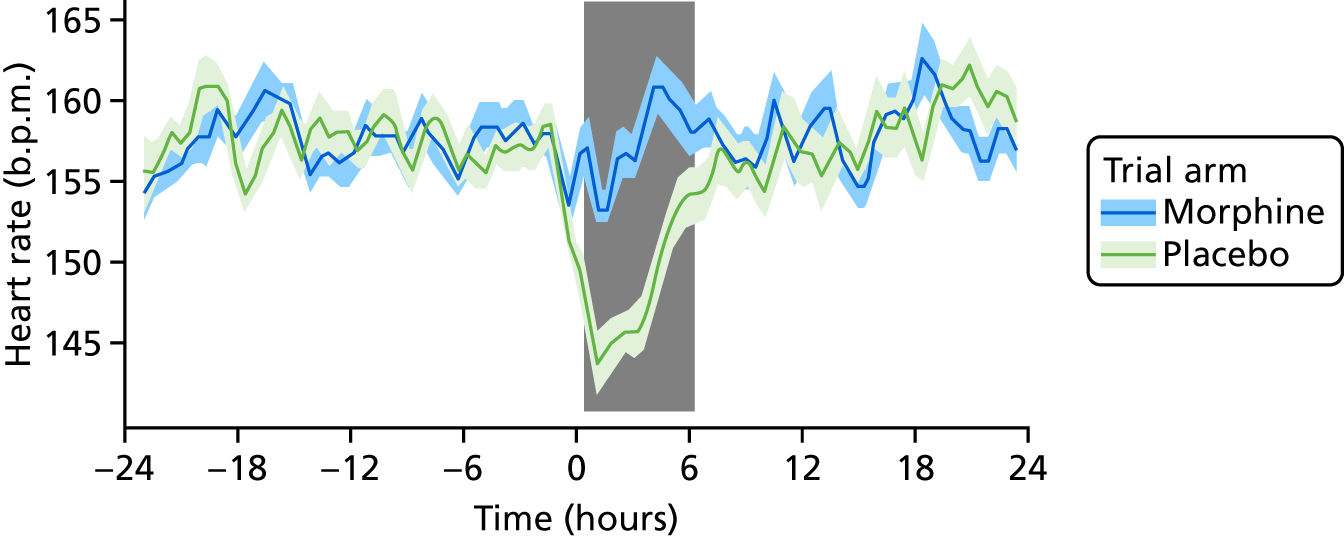
FIGURE 24.
Average respiratory rate in the morphine and placebo groups during the 48-hour monitoring period. Individual infant traces are baseline corrected to the average baseline across all infants. Time 0 is the point of clinical procedure. Grey boxes indicate time periods when the groups are significantly different. Solid lines indicate the mean and shaded areas the standard error.

FIGURE 25.
Average oxygen saturation in the morphine and placebo groups during the 48-hour monitoring period. Individual infant traces are baseline corrected to the average baseline across all infants. Time 0 is the point of clinical procedure. Grey boxes indicate time periods when the groups are significantly different. Solid lines indicate the mean and shaded areas the standard error.

Chapter 5 Discussion
This trial demonstrates that 100 µg/kg oral morphine causes significant cardiorespiratory effects, with no evidence of analgesia, in infants aged 34–39 weeks’ gestational age undergoing an acute painful procedure. Infants who received morphine experienced more episodes of oxygen desaturation, bradycardia and apnoea in the 24 hours following the clinical procedure. The study data suggest that, in non-ventilated premature infants of 1–2 months postnatal age, oral morphine doses recommended by the British National Formulary for Children could cause significant respiratory effects. 27 Intravenous morphine at 10–30 µg/kg has been reported to provide effective analgesia in infants receiving continuous positive airway pressure, and at a dose of 100 µg/kg effective pain relief for central line placement in ventilated preterm infants. 45,46 However, similar AEs, including apnoeas and increased ventilation requirements, were reported. Although these respiratory effects of morphine can be expertly managed in neonatal care, increased requirements for ventilatory support can be expensive, increase the length of hospital stay and cause parental distress.
The timing of the cardiorespiratory effects observed in this study was consistent with the half-life of morphine reported in premature infants, which is approximately 6–8 hours. 47–50 Considering the timing and severity of the AEs, the lack of observable analgesia in this trial is unlikely to be a result of inappropriate timing of the intervention in relation to drug administration or poor absorption of the drug. The study was underpowered for the co-primary outcome measures, as a result of early cessation, and it therefore cannot be concluded whether or not oral morphine provided effective analgesia at this dose. The results suggest that oral morphine at a dose of 100 µg/kg for ROP screening in non-ventilated prematurely born infants is not recommended, and we advise caution for use for other painful procedures. The potential benefits of morphine administration in other contexts should be considered in the light of these findings.
This trial used a multimodal approach to assess both analgesic efficacy and drug safety, providing an overview of the pain experience and a comprehensive time course of the physiological effects of the procedure and drug. Future neonatal clinical trials would benefit from adopting a similar rigorous methodology. Although the study was underpowered to assess the primary outcome measures as a result of early cessation, there was a trend towards infants who received morphine having increased noxious-evoked responses. It is, therefore, perhaps unlikely that an analgesic benefit would have been observed had the trial continued to completion, attaining a sample size of 156 participants.
Chapter 6 Conclusions
The administration of oral morphine at a dose of 100 µg/kg to non-ventilated prematurely born infants undergoing ROP screening caused significant cardiorespiratory effects, without suggestion of analgesic benefit. Therefore, its provision cannot be recommended and we would urge caution when considering its use in other clinical contexts in this population. Identifying effective pain-relieving strategies for this vulnerable patient group is a clinical priority. Future trials should use multimodal approaches to assess the analgesic efficacy and adverse effects of other candidate drugs or interventions, and this could lead to a greater mechanistic understanding of these drug effects. Although this multimodal approach is labour-intensive, it is feasible with a dedicated research team and necessary for future research to ensure that analgesic efficacy is assessed in this population rigorously and appropriately.
Acknowledgements
We thank the National Perinatal Epidemiology Unit (clinical trials unit) for the central management of the trial, specifically Ursula Bowler (senior trials manager), Kayleigh Stanbury (core trials support and previous trial manager), Louise Linsell (senior statistician), Andy King (head of trials programming), David Murray (senior trials programmer), Pollyanna Hardy (previous senior statistician) and Omar Omar (previous trial statistician).
We also thank Alan Worley for building and maintaining the specialised equipment used in this study; Gabriela Schmidt Mellado for help with trial pilot studies and maintenance of EEG equipment; Miranda Buckle, Sezgi Goksan, Marianne van der Vaart and Deniz Gursul for their contribution to data collection; SSNAP for facilitating PPI, review of trial materials, discussion of trial progress, and trial promotion; and Noa Fernandez-Ledo for performing ROP screens. We thank the TSC (chairperson: Neena Modi; vice-chairperson: Cathy Williams; and other members: Simon Farmer, Joanne Gribben and Simon Bond) and the DMC (chairperson: Andy Ewer; and other members: Nigel Stallard and Elaine Boyle) for their advice and support throughout the trial. In particular, we thank them for their dedication to maintaining the safety of participants and their guidance during the early termination of this trial. We thank all the participants and their families for taking part in this study, and the consultant and nursing staff at the Newborn Care Unit at the John Radcliffe Hospital for their tremendous support during the trial recruitment period.
Contributions of authors
Vaneesha Monk (Clinical Research Director, Trial Manager) contributed to the first draft of the manuscript and subsequent revisions, and the critical review of the manuscript.
Fiona Moultrie (Clinical Doctoral Fellow) contributed to the study design, recruitment, data collection, clinical care for the participants during the trial, data entry, data analyses, assessment of SAE causality, the first draft of the manuscript and the critical review of the manuscript.
Caroline Hartley (Senior Postdoctoral Researcher) developed funding proposals, contributed to the study design, recruitment, data collection, data entry, data analyses and the first draft of the manuscript and the critical review of the manuscript.
Amy Hoskin (Research Nurse) contributed to the study design, recruitment, data collection, clinical care for the participants during the trial, data entry, data analyses and the critical review of the manuscript.
Gabrielle Green (Clinical Academic Research Fellow) contributed to the study design, recruitment, data collection, clinical care for the participants during the trial, data entry, assessment of SAE causality and the critical review of the manuscript.
Jennifer L Bell (Trial Statistician) contributed to the data analyses, first draft of the manuscript and the critical review of the manuscript.
Caz Stokes (Family Care Team Leader, PPI representative) facilitated PPI, review of trial materials, discussion of trial progress and trial promotion.
Ed Juszczak (Director of the NPEU CTU) developed funding proposals, contributed to the study design and the critical review of the manuscript.
Jane Norman (Professor of Maternal and Fetal Health, Director of the Edinburgh Tommy’s Centre) developed funding proposals, contributed to the study design and the critical review of the manuscript.
Richard Rogers (Consultant Aneasthetist) developed funding proposals and contributed to the study design and the critical review of the manuscript.
Chetan Patel (Consultant Ophthalmic Surgeon) collected data, conducted ROP screening and contributed to the critical review of the manuscript.
Eleri Adams (Consultant Neonatologist and Director of OUH Newborn Care Services, Chief Investigator) developed the funding proposal, and contributed to the study design, clinical care for the participants during the trial, assessment of SAE causality and the critical review of the manuscript.
Rebeccah Slater (Professor of Paediatric Neuroscience, Principal Investigator) conceived the study, was the grant holder, and contributed to the study design, recruitment, data collection, data entry, data analyses, first draft of the manuscript and the critical review of the manuscript.
Publications
Hartley C, Moultrie F, Juszczak E, Adams E, Slater R. Protocol 15PRT/5747: a blinded randomised placebo-controlled trial investigating the efficacy of morphine analgesia for procedural pain in infants – ISRCTN82342359. Lancet 2016. URL: www.thelancet.com/protocol-reviews/15PRT-5747 (accessed 8 July 2019).
Slater R, Hartley C, Moultrie F, Adams E, Juszczak E, Rogers R, et al. A blinded randomised placebo-controlled trial investigating the efficacy of morphine analgesia for procedural pain in infants: trial protocol. Wellcome Open Res 2016;1:7.
Hartley C, Moultrie F, Hoskin A, Green G, Monk V, Bell JL, et al. Analgesic efficacy and safety of morphine in the PrOcedural Pain in Premature Infants (Poppi) study: randomised placebo-controlled trial. Lancet 2018;392:2595–605.
Moultrie F, Shriver A, Hartley C, Wilkinson D, Ewer AK, Rogers R, et al. A universal right to pain relief-balancing the risks in a vulnerable patient population. Lancet Child Adolesc Health 2018;3:62–4.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- McCaffery M. Nursing Practice Theories Related to Cognition, Bodily Pain, and Man–Environment Interactions. Los Angeles, CA: University of California Print Office; 1968.
- Mitchell AJ, Green A, Jeffs DA, Roberson PK. Physiologic effects of retinopathy of prematurity screening examinations. Adv Neonatal Care 2011;11:291-7. https://doi.org/10.1097/ANC.0b013e318225a332.
- Ranger M, Grunau RE. Early repetitive pain in preterm infants in relation to the developing brain. Pain Manag 2014;4:57-6. https://doi.org/10.2217/pmt.13.61.
- Carbajal R, Rousset A, Danan C, Coquery S, Nolent P, Ducrocq S, et al. Epidemiology and treatment of painful procedures in neonates in intensive care units. JAMA 2008;300:60-7. https://doi.org/10.1001/jama.300.1.60.
- Kandasamy Y, Smith R, Wright IM, Hartley L. Pain relief for premature infants during ophthalmology assessment. J AAPOS 2011;15:276-80. https://doi.org/10.1016/j.jaapos.2011.03.009.
- Sun X, Lemyre B, Barrowman N, O’Connor M. Pain management during eye examinations for retinopathy of prematurity in preterm infants: a systematic review. Acta Paediatr 2010;99:329-34. https://doi.org/10.1111/j.1651-2227.2009.01612.x.
- Dempsey E, McCreery K. Local anaesthetic eye drops for prevention of pain in preterm infants undergoing screening for retinopathy of prematurity. Cochrane Database Syst Rev 2011;9. https://doi.org/10.1002/14651858.CD007645.pub2.
- Ahlers SJ, van Gulik L, van Dongen EP, Bruins P, van de Garde EM, van Boven WJ, et al. Efficacy of an intravenous bolus of morphine 2.5 versus morphine 7.5 mg for procedural pain relief in postoperative cardiothoracic patients in the intensive care unit: a randomised double-blind controlled trial. Anaesth Intensive Care 2012;40:417-26. https://doi.org/10.1177/0310057X1204000306.
- Dyke MP, Kohan R, Evans S. Morphine increases synchronous ventilation in preterm infants. J Paediatr Child Health 1995;31:176-9. https://doi.org/10.1111/j.1440-1754.1995.tb00780.x.
- Anand KJ, Anderson BJ, Holford NH, Hall RW, Young T, Shephard B, et al. Morphine pharmacokinetics and pharmacodynamics in preterm and term neonates: secondary results from the NEOPAIN trial. Br J Anaesth 2008;101:680-9. https://doi.org/10.1093/bja/aen248.
- Anand KJ, Hall RW, Desai N, Shephard B, Bergqvist LL, Young TE, et al. Effects of morphine analgesia in ventilated preterm neonates: primary outcomes from the NEOPAIN randomised trial. Lancet 2004;363:1673-82. https://doi.org/10.1016/S0140-6736(04)16251-X.
- Carbajal R, Lenclen R, Jugie M, Paupe A, Barton BA, Anand KJ. Morphine does not provide adequate analgesia for acute procedural pain among preterm neonates. Pediatrics 2005;115:1494-500. https://doi.org/10.1542/peds.2004-1425.
- Manjunatha CM, Ibhanesebhor SE, Rennix C, Fisher H, Abara R. Pain control during retinopathy of prematurity screening: double-blind, randomised, placebo-controlled study. Infant 2009;5:155-8.
- Stevens B, Johnston C, Petryshen P, Taddio A. Premature Infant Pain Profile: development and initial validation. Clin J Pain 1996;12:13-22. https://doi.org/10.1097/00002508-199603000-00004.
- Stevens BJ, Gibbins S, Yamada J, Dionne K, Lee G, Johnston C, et al. The premature infant pain profile-revised (PIPP-R): initial validation and feasibility. Clin J Pain 2014;30:238-43. https://doi.org/10.1097/AJP.0b013e3182906aed.
- Simonse E, Mulder PG, van Beek RH. Analgesic effect of breast milk versus sucrose for analgesia during heel lance in late preterm infants. Pediatrics 2012;129:657-63. https://doi.org/10.1542/peds.2011-2173.
- Campbell-Yeo ML, Johnston CC, Joseph KS, Feeley N, Chambers CT, Barrington KJ. Cobedding and recovery time after heel lance in preterm twins: results of a randomized trial. Pediatrics 2012;130:500-6. https://doi.org/10.1542/peds.2012-0010.
- Taddio A, Shah V, Hancock R, Smith RW, Stephens D, Atenafu E, et al. Effectiveness of sucrose analgesia in newborns undergoing painful medical procedures. CMAJ 2008;179:37-43. https://doi.org/10.1503/cmaj.071734.
- Slater R, Worley A, Fabrizi L, Roberts S, Meek J, Boyd S, et al. Evoked potentials generated by noxious stimulation in the human infant brain. Eur J Pain 2010;14:321-6. https://doi.org/10.1016/j.ejpain.2009.05.005.
- Worley A, Fabrizi L, Boyd S, Slater R. Multi-modal pain measurements in infants. J Neurosci Methods 2012;205:252-7. https://doi.org/10.1016/j.jneumeth.2012.01.009.
- Truini A, Panuccio G, Galeotti F, Maluccio MR, Sartucci F, Avoli M, et al. Laser-evoked potentials as a tool for assessing the efficacy of antinociceptive drugs. Eur J Pain 2010;14:222-5. https://doi.org/10.1016/j.ejpain.2009.05.001.
- Hartley C, Duff EP, Green G, Mellado GS, Worley A, Rogers R, et al. Nociceptive brain activity as a measure of analgesic efficacy in infants. Sci Transl Med 2017;9. https://doi.org/10.1126/scitranslmed.aah6122.
- Slater R, Cantarella A, Franck L, Meek J, Fitzgerald M. How well do clinical pain assessment tools reflect pain in infants?. PLOS Med 2008;5. https://doi.org/10.1371/journal.pmed.0050129.
- Moultrie F, Slater R, Hartley C. Improving the treatment of infant pain. Curr Opin Support Palliat Care 2017;11:112-17. https://doi.org/10.1097/SPC.0000000000000270.
- BLISS . Guideline for the Screening and Treatment of Retinopathy of Prematurity 2008.
- Disher T, Cameron C, Mitra S, Cathcart K, Campbell-Yeo M. Pain-relieving interventions for retinopathy of prematurity: a meta-analysis. Pediatrics 2018;142. https://doi.org/10.1542/peds.2018-0401.
- Paediatric Formulary Committee . BNF for Children 2015 2015.
- Slater R, Hartley C, Moultrie F, Adams E, Juszczak E, Rogers R, et al. A blinded randomised placebo-controlled trial investigating the efficacy of morphine analgesia for procedural pain in infants: trial protocol. Wellcome Open Res 2016;1. https://doi.org/10.12688/wellcomeopenres.10005.2.
- Slater R, Cornelissen L, Fabrizi L, Patten D, Yoxen J, Worley A, et al. Oral sucrose as an analgesic drug for procedural pain in newborn infants: a randomised controlled trial. Lancet 2010;376:1225-32. https://doi.org/10.1016/S0140-6736(10)61303-7.
- Cornelissen L, Fabrizi L, Patten D, Worley A, Meek J, Boyd S, et al. Postnatal temporal, spatial and modality tuning of nociceptive cutaneous flexion reflexes in human infants. PLOS ONE 2013;8. https://doi.org/10.1371/journal.pone.0076470.
- Lemyre B, Hogan DL, Gaboury I, Sherlock R, Blanchard C, Moher D. How effective is tetracaine 4% gel, before a venepuncture, in reducing procedural pain in infants: a randomized double-blind placebo controlled trial. BMC Pediatr 2007;7. https://doi.org/10.1186/1471-2431-7-7.
- Gal P, Kissling GE, Young WO, Dunaway KK, Marsh VA, Jones SM, et al. Efficacy of sucrose to reduce pain in premature infants during eye examinations for retinopathy of prematurity. Ann Pharmacother 2005;39:1029-33. https://doi.org/10.1345/aph.1E477.
- Marsh VA, Young WO, Dunaway KK, Kissling GE, Carlos RQ, Jones SM, et al. Efficacy of topical anesthetics to reduce pain in premature infants during eye examinations for retinopathy of prematurity. Ann Pharmacother 2005;39:829-33. https://doi.org/10.1345/aph.1E476.
- Mehta M, Mansfield T, VanderVeen DK. Effect of topical anesthesia and age on pain scores during retinopathy of prematurity screening. J Perinatol 2010;30:731-5. https://doi.org/10.1038/jp.2010.36.
- Slater R, Fabrizi L, Worley A, Meek J, Boyd S, Fitzgerald M. Premature infants display increased noxious-evoked neuronal activity in the brain compared to healthy age-matched term-born infants. Neuroimage 2010;52:583-9. https://doi.org/10.1016/j.neuroimage.2010.04.253.
- Lorenz J, Beck H, Bromm B. Cognitive performance, mood and experimental pain before and during morphine-induced analgesia in patients with chronic non-malignant pain. Pain 1997;73:369-75. https://doi.org/10.1016/S0304-3959(97)00123-1.
- Attal N, Cruccu G, Haanpää M, Hansson P, Jensen TS, Nurmikko T, et al. EFNS guidelines on pharmacological treatment of neuropathic pain. Eur J Neurol 2006;13:1153-69. https://doi.org/10.1111/j.1468-1331.2006.01511.x.
- Hollingshead J, Dühmke RM, Cornblath DR. Tramadol for neuropathic pain. Cochrane Database Syst Rev 2006;3. https://doi.org/10.1002/14651858.CD003726.pub3.
- Norbury TA, MacGregor AJ, Urwin J, Spector TD, McMahon SB. Heritability of responses to painful stimuli in women: a classical twin study. Brain 2007;130:3041-9. https://doi.org/10.1093/brain/awm233.
- Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika 1988;75:800-2. https://doi.org/10.1093/biomet/75.4.800.
- Maris E, Oostenveld R. Nonparametric statistical testing of EEG- and MEG-data. J Neurosci Methods 2007;164:177-90. https://doi.org/10.1016/j.jneumeth.2007.03.024.
- Hartley C, Moultrie F, Hoskin A, Green G, Monk V, Bell JL, et al. Analgesic efficacy and safety of morphine in the PrOcedural Pain in Premature Infants (Poppi) study: randomised placebo-controlled trial. Lancet 2018;392:2595-605. https://doi.org/10.1016/S0140-6736(18)31813-0.
- Hartley C, Moultrie F, Juszczak E, Adams E, Slater R. Protocol 15PRT/5747: a blinded randomised placebo-controlled trial investigating the efficacy of morphine analgesia for procedural pain in infants – ISRCTN82342359. Lancet 2016. www.thelancet.com/protocol-reviews/15PRT-5747 (accessed 8 July 2019).
- Moultrie F, Shriver A, Hartley C, Wilkinson D, Ewer AK, Rogers R, et al. A universal right to pain relief-balancing the risks in a vulnerable patient population. Lancet Child Adolesc Health 2018;3:62-4. https://doi.org/10.1016/S2352-4642(18)30269-4.
- Enders J, Gebauer C, Pulzer F, Robel-Tillig E, Knüpfer M. Morphine-related apnoea in CPAP-treated preterm neonates. Acta Paediatr 2006;95:1087-92. https://doi.org/10.1080/08035250600577871.
- Taddio A, Lee C, Yip A, Parvez B, McNamara PJ, Shah V. Intravenous morphine and topical tetracaine for treatment of pain in preterm neonates undergoing central line placement. JAMA 2006;295:793-800. https://doi.org/10.1001/jama.295.7.793.
- Bhat R, Chari G, Gulati A, Aldana O, Velamati R, Bhargava H. Pharmacokinetics of a single dose of morphine in preterm infants during the first week of life. J Pediatr 1990;117:477-81. https://doi.org/10.1016/S0022-3476(05)81102-3.
- Hartley R, Green M, Quinn M, Levene MI. Pharmacokinetics of morphine infusion in premature neonates. Arch Dis Child 1993;69:55-8. https://doi.org/10.1136/adc.69.1_Spec_No.55.
- Pacifici GM. Metabolism and pharmacokinetics of morphine in neonates: a review. Clinics 2016;71:474-80. https://doi.org/10.6061/clinics/2016(08)11.
- Scott CS, Riggs KW, Ling EW, Fitzgerald CE, Hill ML, Grunau RV, et al. Morphine pharmacokinetics and pain assessment in premature newborns. J Pediatr 1999;135:423-9. https://doi.org/10.1016/S0022-3476(99)70163-0.
- Bolland K, Whitehead J. Formal approaches to safety monitoring of clinical trials in life-threatening conditions. Stat Med 2000;19:2899-917. https://doi.org/10.1002/1097-0258(20001115)19:21<2899::AID-SIM597>3.0.CO;2-O.
Appendix 1 Investigational Medicinal Product manufacturing issues
The Royal Free Hospital’s manufacturing unit was initially contracted to manufacture the IMP; however, as a result of unforeseen manufacturing difficulties, the contract was transferred to Stockport Pharmaceuticals in January 2016. Stockport Pharmaceuticals developed the IMP and provided the supporting trial documentation. The active ingredient, dose and concentration remained unchanged [i.e. 200 µg/ml (100 µg/kg) of morphine sulphate] and only the ingredients in the carrier solution and packaging were changed. From June 2016, Stockport Pharmaceuticals ran stability testing on a test IMP batch. This information formed part of the amended documentation (protocol, IMP dossier, labels and clinical trial application form), which was approved by the MHRA and the REC on 15 August 2016 and 23 August 2016, respectively.
The process for the manufacturing of each IMP batch with Stockport Pharmaceuticals was:
-
The trial manager used a shipment request form to inform the manufacturer of the number of packs and date of delivery required.
-
The co-ordinating centre head of trials programming generated the pack IDs and associated allocations and sent these to Stockport Pharmaceuticals.
-
Stockport Pharmaceuticals manufactured the required number of IMP packs and labelled these, as appropriate.
-
The QP ensured that the placebo packs:
-
matched the requirements for appearance
-
had a pH of 4.0–5.0
-
were absent of morphine
-
had a limit of sodium benzoate of > 0.02%
-
had a viable count at 20–25 °C of 0–10 colony-forming unit (CFU)/ml
-
had a viable count at 30–35 °C of 0–100 CFU/ml
-
were absent of Escherichia coli.
-
-
The QP ensured that the morphine packs:
-
matched the requirements for appearance
-
were identical
-
had a pH of 4.0–5.0
-
were checked for weight per ml (no stated limits)
-
had a limit of sodium benzoate of > 0.02%
-
had a morphine assay of 95.0–1–5.0% stated strength
-
had a viable count at 20–25 °C of 0–10 CFU/ml
-
had a viable count at 30–35 °C of 0–100 CFU/ml
-
were absent of E. coli.
-
-
The QP completed the stage 1 batch release on the certificates of analysis.
-
The trial manager (or other approved members of the research team) reviewed the information and completed the stage 2 batch release on the certificates of analysis.
-
Stockport Pharmaceuticals completed the next section of the shipment request form, and sent this with the IMP to the hospital pharmacy. The IMP batch was fitted with temperature monitoring equipment (TempTale®; Sensitech, Sassenheim, the Netherlands) to record temperature in transit (the temperature limit was set at 25 °C to protect the IMP from chemical deterioration).
-
The consignee in the pharmacy completed the final section of the shipment request form to confirm receipt and returned copies of this to Stockport Pharmaceuticals and the trial manager.
-
The pharmacy staff downloaded the TempTale data, ensured that there were no temperature excursions, e-mailed the data to the trial manager and returned the TempTale equipment to Stockport Pharmaceuticals.
Batch 1
Batch 1 of the IMP (33 packs) was manufactured on 6 September 2016 and delivered to the site on 15 September 2016. The expiry date was originally 29 November 2016, and was later extended to 3 March 2017.
Temperature excursion
For the first batch of IMP, a lower limit of 15 °C was set on the TempTale for monitoring the IMP temperature in transit. On receipt at the pharmacy, the team observed that the data logger had alarmed during transit. On review of the data, the excursion was < 2 °C below the lower limit for a time period of less than 3 hours. The quality control team at Stockport Pharmaceuticals confirmed that this minor temperature excursion placed insignificant risk of product deterioration. Chemical deterioration (such as hydrolysis or oxidation) is a temperature-dependent process, which slows (rather than accelerates) with a lower temperature, so this was not a cause for concern in this instance. There was a theoretical possibility that some components may have become insoluble at lower temperatures, but all the components were freely soluble at the concentrations found in the product and would endure far greater temperature reductions before precipitating. It was identified that a lower temperature limit was not necessary for this IMP, as per standard practice. No lower temperature limit was applied to future batches of IMP.
Overlabelling
After the manufacture of IMP batch 1, further stability data were made available from Stockport Pharmaceuticals permitting a shelf life of 6 months. The QP at Stockport Pharmaceuticals confirmed that the stability data supported the extension of the expiry date and that another QP release certificate was not required. These stability data were filed in the product specification file held at Stockport Pharmaceuticals.
The extension conformed to the MHRA approval conditions, which state that ‘the initial shelf life of the drug product will not exceed twice the time period for which there are satisfactory stability data at 25 degrees/60% room humidity’. The remaining 29 IMP packs from batch 1 were therefore over-labelled to extend the IMP expiry date from 29 November 2016 to 3 March 2017. The expiry date was also updated on the co-ordinating centre pack management system (see Chapter 3, Electronic stock management).
Batch 2
Batch 2 of the IMP (29 packs) was manufactured on 16 February 2017 and delivered to site on 2 March 2017. This second batch of IMP was short-dated to 31 May 2017 to match the expiry of the remaining SyrSpend stock held by the manufacturers. The temperature limit was not exceeded while in transit.
Change in SyrSpend supplier
The active IMP comprised morphine sulphate in a cherry-flavoured oral suspending base (SyrSpend). The placebo was SyrSpend in isolation. Stockport Pharmaceuticals sourced SyrSpend from Fagron BV. In December 2016, Stockport Pharmaceuticals was alerted that Fagron BV had changed its supplier of SyrSpend to Tiofarma BV (Oud Beijerland, the Netherlands). In January 2016, Stockport Pharmaceuticals was made aware that the new SyrSpend supplier did not have an IMP licence and, therefore, considered that its product could not be used for future IMP batches. The IMP batch at the time was due to expire 6 March 2017 (IMP batch 1).
Stockport Pharmaceuticals had some remaining stock of SyrSpend from the previous licensed supplier, Terra Pharma (London, UK), which expired on 31 May 2017. A request for a new IMP batch was placed using this stock as a short-term solution (batch 2), but another IMP batch with a different SyrSpend source was required to be manufactured before batch 2 expired. The PMG discussed the following potential long-term solutions:
-
Further stability data were expected at the end of March 2017 for the original IMP. This might have provided stability data for extending the expiry date of the recently requested IMP batch and allow a greater time period to identify another long-term solution.
-
Contact Terra Pharma directly to request a personal batch of SyrSpend. Terra Pharma had not yet confirmed if this was feasible, but had suggested that a minimum order of 1000 bottles would be required. Potential costs of this were explored by Stockport Pharmaceuticals.
-
Stockport Pharmaceuticals had a large supply of unflavoured SyrSpend which could be used (expiry March 2018). This would require an update to the IMP dossier and a substantial amendment to the MHRA. Additionally, as the recruitment period was intended to be 33 months, another longer-term solution would be required for the remaining recruitment period after this SyrSpend supply expired.
-
Use a completely different suspending agent. This would require an update to the IMP dossier and a substantial amendment to the MHRA. However, many other suspending agents were not suitable for neonatal use because of the sugar content or other ingredients.
However, the MHRA confirmed that as the SyrSpend is classed as an ‘excipient’ there was no requirement for the new supplier to hold an IMP licence. A substantial amendment was therefore submitted in March 2017 to change the SyrSpend supplier to Tiofarma BV in the IMP dossier. This amendment was approved by the MHRA, REC and HRA in March/April 2017. IMP batches 3 and 4 were manufactured using SyrSpend from the new supplier.
Batch 3
Batch 3 of the IMP (60 packs) was manufactured on 8 May 2017 and delivered to site on 26 May 2017. This third batch of IMP was dated to 31 December 2017.
Batch 4
Batch 4 of the IMP (60 packs) was manufactured in early December 2017, with a 2019 expiry, in preparation for replacing the IMP batch 3, which was due to expire on 31 December 2017. However, batch 4 was not delivered to site following the trial suspension recommended by the TSC on 13 December 2017 (see Chapter 3, Trial closure, Oversight).
Appendix 2 Temperature control and monitoring
The temperature of the IMP was managed at all times to ensure that it remained below the safe limit of 25 °C. The temperature of the cupboard where the IMP was stored was continuously monitored using a thermometer with USB-downloadable data. The temperature was set to 18 °C as a safety back-up. The temperature was hand-recorded on the temperature record (see Report Supplementary Material 9) whenever an IMP pack was taken from the cupboard, and temperature excursions were checked for (on the thermometer screen) and noted.
Continual temperature monitoring
The thermometer continually recorded and stored temperature information, which was downloaded monthly as a portable document format (PDF) file and sent to the pharmacy and the co-ordinating centre (for storage in the TMF). Any temperature excursions would raise an alarm and would be dealt with as per the protocol. During the download, temperature management was maintained using a maximum–minimum thermometer. This was checked before removing the monitor for downloading information. After the PDF file was generated, the temperature logger was reconfigured to clear the data already recorded and collected. The thermometer was then reconnected. The maximum–minimum thermometer was then checked again to ensure that the temperature of the cupboard had not gone above 25 °C during the download period.
Interval temperature monitoring
The research team recorded the temperature of the cupboard every time an IMP pack was removed and the thermometer was checked for temperature excursions. The temperature and occurrence of excursions were recorded on the temperature record (see Supplementary Material 9). A new record form was completed monthly and a copy provided to the co-ordinating centre.
Temperature excursions
Trial processes were set up to ensure that all temperature excursions were reported as soon as possible. No temperature excursions (i.e. above 25 °C) were reported during the trial. In the event of an excursion, the trial team would have immediately contacted a member of the pharmacy and the affected IMP stock would have been quarantined and not used until authorised. A notice would have been placed on the cupboard and the trial team members would have been informed in person or via e-mail. The IMP would have been moved to the pharmacy or to another secure area where storage requirements could be met, until the trial team and co-ordinating centre could take advice from the QP at the manufacturing site on whether the IMP should be used or destroyed. Any planned studies would have been postponed until a decision was reached.
Appendix 3 Electronic case report form design and validation
Electronic case report form design
In this trial, all data were entered either directly into the eCRF in the database, or first recorded on a paper CRF and then transposed into the appropriate eCRF.
| eCRF name | Paper CRF | Other sources of data used |
|---|---|---|
| Entry | No – eCRF only | Medical notes |
| Test occasion: heel lance | No – eCRF only | Medical notes |
| Clinical stability | Yes (in part) – cotside log | Medical notes |
| PIPP-R score | Yes (in part) – cotside log | Clinical PIPP-R scoring sheet – using data from video recordings, vital signs monitoring on cot-side log and medical notes |
| Neurophysiology | No – eCRF only | EEG data – output from MATLAB |
| Respiratory support | Yes (in part) – cotside log | Medical notes |
| Safety data | Yes (in part) – cotside log | Medical notes |
| Foreseeable AE | No – eCRF only | Medical notes |
| AE | Yes | Medical notes |
| SAE | Yes | All data available (e.g. medical notes, cot-side log) |
| SAE chief investigator assessment | Yes | SAE report form and any other data available |
| Withdrawal | Yes | N/A |
Draft CRFs were created initially by the PMG, which addressed the data collection requirements in the protocol. The CRFs were designed to follow on from each other in an easy to understand and logical sequence. The trial statistician then reviewed the draft CRFs relative to the protocol, drafted dummy tables and drafted a data derivation document. The completed drafts were then reviewed again by the PMG to focus on collection of the correct data to address the objectives stated in the protocol, the unambiguous identification of participants’ CRFs, logical flow of data collection, legibility and ease of comprehension, use of unambiguous and closed questions, avoidance of leading questions, administrative burden on those completing CRFs, avoidance of collection of unnecessary data, and addressing any relevant risks on the risk assessment (see Chapter 3, Risk assessment). The QA team reviewed all CRFs against the co-ordinating centre SOPs.
Electronic case report form validation
Before each eCRF was uploaded to OpenClinica, it was validated in accordance with the co-ordinating centre’s SOP. The validation process was divided into the following processes: naming variables, eCRF design, data verification and validation (including checking data were within an expected range and logic tests), test data entry and verifying data export. Each individual process was completed, reviewed and signed off by the trials programmer before the eCRF was uploaded to OpenClinica and made available for data entry.
When new eCRFs were required, these were validated and added manually using a script by a trials programmer. During the trial, the following minor eCRF amendments were made:
-
AE form updated to allow selection of multiple systems
-
heading corrected in the clinical stability form
-
PIPP-R questions re-ordered to match the order in the clinical paper scoring sheet
-
the respiratory support form updated to allow documentation of up to 20 significant changes in respiratory support.
Appendix 4 Documentation
Protocol
There were six versions of the protocol over the course of the trial.
Version 1
This was the original version of the protocol submitted for approvals. This version of the protocol was approved by the REC on 23 July 2015 and MHRA on 21 August 2015.
Version 2
Version 2 of the protocol incorporated the following updates:
-
Change of measure used to assess clinical pain scores: change from PIPP to PIPP-R.
-
Change to timing of primary outcome measure: change from PIPP score 1 minute after ROP screening to PIPP-R score 30 seconds after ROP screening.
-
Definition of clinical intervention: where applicable, the timing of objectives and outcomes were clarified to state that the measures were assessed ‘following the start of the clinical intervention’. The clinical intervention was defined as the ‘heel lance followed by ROP screening’.
-
Change to eligibility criteria: requirement for a senior clinician to assess suitability of the infant prior to enrolment in the trial changed from an exclusion to an inclusion criterion.
-
Unblinding procedure: changed to allow medical staff to unblind participants immediately without input from the chief investigator or trial team.
-
Changes to procedure for recording AEs: only AEs identified as serious would be recorded.
-
Reporting procedures for foreseeable SAEs: increased respiratory support with a significant increase in oxygen requirement or an increase in support modality of grades 1, 2 and 3 added to the list of foreseeable SAEs.
-
Reporting procedures for SAEs: addition of electronic SAE reporting.
-
Analysis section: simplified, and the implied emphasis switched from significance testing to estimating the magnitude and direction of treatment effects with corresponding CIs.
-
PIPP-R analysis: separated into the 30 seconds post ROP screening (co-primary outcome) and post heel lance (secondary outcome). The analysis method for both was changed from repeated measures analysis of variance (ANOVA) to linear regression, to explicitly mention calculating the mean difference and corresponding CI at a single time point and to facilitate adjustment for minimisation factors (only if required and technically possible). For skewed data, the median difference and corresponding CI would be calculated; this was changed from using Friedman’s test.
-
Independent two-sample t-tests: changed to linear regression (to explicitly mention calculating the mean difference and corresponding CI).
-
Mann–Whitney rank-based test: the test was replaced with estimating the median difference between groups with corresponding CI.
-
Clinical stability and safety analyses: independent two-sample t-tests were changed to either Poisson or linear regression, dependent on the distribution of data or logistic regression for very infrequent occurrences (to explicitly mention calculating the mean difference and corresponding CI). Adjusting for the 24-hour baseline period, if appropriate, was also added for the clinical stability analysis.
-
Analysis: the CI used for reporting secondary outcome measures was changed from 95% to 99%.
-
Interim analysis details: was added for primary and secondary outcome measures.
-
Additional information: was added to provide further justification for the trial, trial procedures, and elaborated definitions.
This version of the protocol was approved by the REC on 6 November 2015; however, the MHRA provided grounds for non-acceptance (see Appendix 5, Research Ethics Committee, Medicines and Healthcare products Regulatory Agency and Health Research Authority). As a result, this version of the protocol was not issued full approvals.
Version 3
Following feedback from the MHRA, it was decided that changes in respiratory support should be removed from the list of foreseeable SAEs. The research team would therefore report any change in respiratory support as a SAE if it met the definition. As more detailed data regarding the potential risks of morphine in the neonatal population would be valuable to clinical practice, following MHRA advice, the research team updated the protocol to indicate that AEs (with the exception of those already listed in the protocol) would be recorded. The protocol was also updated to confirm that the number of infants requiring an increase in respiratory support would be tested using logistic regression as a result of the binary nature of the data, and typographical errors in reference numbers were also amended. This version of the protocol was approved by the REC on 4 January 2016 and MHRA on 2 February 2016.
Version 4
Updates to version 4 of the protocol involved changes of IMP details to reflect the change in IMP supplier. The sequential safety procedure presented to the DMC was also added to the protocol. This would no longer be using the continuous stopping methods, as described by Bolland and Whitehead. 51 There had been recent developments in sequential stopping methods that were supported by commercially available and validated software. The details of the stopping boundaries using group sequential methods would therefore be agreed by the DMC and documented within the DMC charter. This version of the protocol was approved by the REC on 23 August 2016, MHRA on 15 August 2016 and HRA on 31 August 2016.
Version 5
Version 5 of the protocol incorporated the following updates:
-
typographical and grammatical corrections
-
removal of the specification that the syringe used for drug administration would be a 3-ml syringe
-
specific details relating to the two-tailed t-test in the statistical analysis section were removed.
This version of the protocol was approved by the HRA on 25 October 2016. REC and MHRA approval were not required for these non-substantial amendments.
Version 6
Version 6 of the protocol incorporated the following updates:
-
Clarification that the primary outcome (i.e. PIPP-R score) would be recorded during the 30-second period after ROP screening.
-
Typographical and grammatical corrections.
-
Clarification that should exclusion criteria manifest in the first 24 hours of clinical stability monitoring, the IMP or placebo would not be administered at this time. If subsequent ROP screening was required and the infant met all eligibility criteria, the IMP or placebo would be administered at the required time point for this subsequent ROP screen.
-
Clarification that once data analyses were complete, the randomisation code would be released to the primary researchers only (as the statistician would be unblinded to provide analyses to the DMC).
-
Clarification of the approach to summarising and reporting measures of clinical stability, which would include a requirement of an increase in respiratory support.
-
Clarification of the relationship between brain activity and nociceptive activity.
-
Clarification of current practice in the neonatal unit where the study was conducted.
-
Clarification that further detail for the planned analyses for this trial would be presented in a separate SAP, and the adjustments planned for comparative analyses.
-
Clarification that p-values would be obtained alongside CIs for the outcomes of interest.
This version of the protocol was approved by the REC on 15 February 2017, MHRA on 2 March 2017 and HRA on 16 March 2017.
Parent Information Leaflet
Mrs Caz Stokes, Parent Help Team Leader for the on-site charity SSNAP, reviewed and provided PPI input into the PIL (see Report Supplementary Material 6) and consent form (see Report Supplementary Material 2). The suggested edits were implemented into the first version of the document, which was submitted for ethics approval. Throughout the trial there were four versions of the PIL (see Report Supplementary Material 6).
Version 1
Version 1 was original submitted to the REC. The recommendations from the REC were incorporated into version 2.
Version 2
Amendments were made following REC review. The changes include:
-
Typographical and grammatical errors corrected.
-
Under the section entitled ‘Who has reviewed the study?’, the Northampton REC was added.
-
‘What are the benefits?’ was updated to begin ‘We cannot guarantee any direct benefits . . .’
-
The REC reference was inserted.
-
‘What is the purpose of the study’ was updated to provide details regarding current comfort techniques used in practice.
-
The acronym ‘NPEU’ was spelt out in full at least once.
Version 2 of the PIL was approved by the REC on 23 July 2015 and MHRA on 21 August 2015.
Version 3
The wording in section ‘What is the purpose of the study’ was clarified to reassure parents that anaesthetic eye drops were used as standard routine practice. The word ‘novel’ was removed. This version of the PIL was approved by the REC on 4 January 2016 and MHRA on 2 February 2016.
Version 4
Version 4 was corrected for typographical errors. This version of the PIL was approved by the REC on 23 August 2016, MHRA on 15 August 2016, and HRA on 31 August 2016.
Consent form
Throughout the trial there were four versions of the consent form (see Report Supplementary Material 2).
Version 1
The original was submitted to the REC. The recommendations from the REC were incorporated into version 2.
Version 2
Version 2 included changes requested by the REC. The REC requested typographical updates to ensure that there was consistency throughout the document. This version of the consent form was approved by the REC on 23 July 2015 and MHRA on 21 August 2015.
Version 3
The infant’s name was added to version 3 to identify the infant during recruitment (this was originally omitted in error). The colour coding specified in the footer was also removed in case copies were printed on different-coloured paper. This version of the consent form was approved by the REC on 4 January 2016 and MHRA on 2 February 2016.
Version 4
Version 4 was updated to refer to the revised PIL. This version of the consent form was approved by the REC on 23 August 2016, by the MHRA on 15 August 2016 and by the HRA on 31 August 2016.
Other parent-facing materials
Cot cards (version 1.0; 12 August 2015; see Report Supplementary Material 3) were created to help the neonatal unit staff identify trial participants. A recruitment roll banner and poster (version 1.0; 9 September 2015; see Report Supplementary Material 7) was created for display on the neonatal unit to promote the trial. A thank-you poster (version 1.0; 9 September 2015; see Report Supplementary Material 10) was created to give to infants and their parents after taking part in the trial. An insert/poster (version 1.0; 28 September 2017) was also created for attachment to the new standard-practice ROP screening PIL distributed routinely to parents (see Chapter 1, Retinopathy of prematurity screening).
Appendix 5 Regulatory processes
Research Ethics Committee, Medicines and Healthcare products Regulatory Agency and Health Research Authority
Initial approval for the trial, protocol version 1.0, PIL version 2.0 and consent form version 2.0, was issued from the REC on 23 July 2015 and the MHRA on 21 August 2015. The REC approved a substantial amendment on 6 November 2015 for updates to the protocol (version 2.0), PIL (version 3.0), consent form (version 3.0), recruitment poster (version 1.0), recruitment banner (version 1.0) and thank-you poster (version 1.0). However, the MHRA issued grounds for non-acceptance. The MHRA felt that omitting recording of AEs was not acceptable as data regarding the benefit–risk balance in this population were limited; therefore, data recording was important and the plan for a blanket exemption from expedited reporting as a SAE for the requirement for respiratory support was also not appropriate. The research team updated the protocol accordingly in substantial amendment (2), which was approved by the REC (4 January 2016) and the MHRA (2 February 2016).
The HRA became an additional regulatory body in spring 2016 and, therefore, reviewed any subsequent amendments alongside the REC and MHRA. The REC (on 11 May 2016), MHRA (on 16 May 2016) and HRA (on 1 June 2016) approved a substantial amendment (3) to notify a temporary halt to the trial owing to delays in IMP manufacture. The REC (on 23 August 2016), MHRA (on 15 August 2016) and HRA (on 31 August 2016) approved a substantial amendment (4) for updates to the protocol (version 4.0), PIL (version 4.0) and consent form (version 4.0). This amendment also involved an update to the IMP dossier (version 3.0) and IMP labels (version 2.0) following the change in IMP manufacturer from the Royal Free Manufacturing Unit to Stockport Pharmaceuticals.
On 25 October 2016, the HRA approved a non-substantial amendment (5) for updates to the protocol (version 5.0). The REC (on 1 February 2017), MHRA (on 2 March 2017) and HRA (on 16 March 2017) approved a substantial amendment (6) for updates to the protocol (version 6.0). The REC (on 30 March 2017), MHRA (on 19 April 2017) and HRA (on 26 April 2017) approved a substantial amendment (7) to update the IMP dossier to reflect the new SyrSpend supplier (version 4.0); see Appendix 1, Batch 2. On 8 November 2017, the REC and HRA approved a substantial amendment (8) for a new parent-facing information insert to be attached to the ROP screening PIL routinely provided to parents of premature infants on the neonatal unit. MHRA approval was not required for this amendment. The MHRA (on 26 January 2018), REC (3 January 2019) and HRA (on 3 January 2018) also approved a substantial amendment (9) for the temporary halt to the trial, as recommended by the TSC.
Annual reports
The first annual REC report was submitted in July 2016. The second annual REC report was submitted in August 2017.
Development Safety Update Reports
The development safety update report (DSUR) for the IMP morphine sulphate (reporting period from 21 August 2015 to 20 August 2016) was submitted to the MHRA on 19 October 2016. A copy was also forwarded to the trial funder. The DSUR for the IMP morphine sulphate (reporting period from 21 August 2016 to 20 August 2017) was submitted on 19 October 2017.
Trial sponsor
The trial sponsor for the trial was the University of Oxford; acceptance of sponsorship was confirmed in writing on 24 June 2016. The trial sponsor held a division of accountabilities documenting the responsibilities allocated to the trial sponsor and the chief investigator.
The trial sponsor also reviewed and approved all amendments prior to submission for regulatory approvals. Throughout the trial, the sponsor was provided updates about study progress when requested, copied into relevant correspondence with the funder, trust and regulatory bodies, and was provided with copies of all amendment approval letters and reports. The trial sponsor provided annual insurance letters that formed part of the TMF.
Trust research and development
The John Radcliffe Hospital, OUHs NHS Trust, was chosen as the most suitable site for this trial, as this was the location of the chief investigator and the principal investigator (grant holder). This trial also involved use of commissioned equipment and specialised knowledge in the conduct of the trial and handling and analysis of data; the research group who held the equipment and had the appropriate specialist knowledge were also based at the John Radcliffe Hospital (led by the principal investigator). Before trust approval for the trial was issued, a trust feasibility questionnaire was completed documenting the core research team, and planned recruitment information and capacity information. Full trust management approval for the trial was granted on 8 March 2016.
An ‘agreement for a participating site in a clinical study sponsored by the University of Oxford’ was signed between the University of Oxford and the OUHs NHS Trust on 26 August 2015, confirming the trust’s agreement to conduct the trial. Throughout the trial, the trust R&D team was provided with regular recruitment updates and fully informed of any amendments. Amendments were not implemented at the site until they were approved by the R&D team.
Portfolio adoption and International Standard Randomised Controlled Trial Number registration
This trial was considered eligible to receive NIHR CRN support, and a record was created on the NIHR CRN Portfolio. Throughout the trial, the trial manager amended the study record as appropriate, and updated monthly accrual data documenting recruitment. The CRN were made aware of any amendments to the trial. The trial was also prospectively registered with the International Standard Randomised Controlled Trial Number (ISRTCN) registry as ISRCTN82342359.
Appendix 6 Site visits: trial manager
Site initiation
Site initiation visits were conducted by the trial manager on 3 December 2015 (at the same time as the trial sponsor, with the chief investigator, pharmacist and clinical researcher) and 7 December 2015 (with the research nurse and postdoctoral researcher). Topics that were discussed included the approvals process and required contracts, trial objectives, trial design, eligibility criteria, study-specific procedures, the ISF and document box (which contained all the necessary documents for the trial), GCP and other training, the green-light process, screening, enrolment, randomisation, the OpenClinica database, consent, IMP compliance/storage/accountability, incident reporting, withdrawals, safety reporting, unblinding, administration, staffing issues/contact details, conflicting studies, monitoring plan, archiving requirements, the protocol, and pharmacy arrangements. This was documented on a site initiation report form, signed by the chief investigator and trial manager, and a copy placed in the ISF.
Green-light release
The co-ordinating centre used a green-light release form to authorise the site to begin recruitment. This form documented the following information: confirmation of trial sponsor approval, REC favourable opinion, MHRA approval, batch certificate and QP release for IMP, completion of site initiation/training, completed site agreement, trust R&D approval, REC approval for site and receipt of the ISF/document box/pharmacy folder. Once these were confirmed, the trial manager provided, and documented, authorisation of release of the IMP to site. The trial manager confirmed that the required checks had been completed and IMP received at site, and the senior trials programmer activated the site on the online randomisation system and documented this action. The completed green-light release form was issued to site on 16 September 2016 to confirm approval to start recruitment.
Site close-out
Site close-out was conducted remotely by the trial manager after all findings from the trial sponsor close-out visit report had been completed. The co-ordinating centre QA team conducted an internal audit of the TMF, and the trial manager resolved all outstanding items requiring correspondence with the pharmacy and site team. The trial manager then sent the pharmacy contact a detailed pharmacy folder contents checklist. Once the pharmacy contact confirmed that all documentation was present by initialling each checklist item, and signing and dating the checklist, the trial manager then authorised closure of all pharmacy activity for the trial and approved the documentation for archiving as per trust regulations (29 June 2018). Similarly, the trial manager also sent the research nurse a detailed ISF contents checklist. On confirmation from the research nurse that all documentation was present, the trial manager authorised closure of site team activities and approved the documentation for archiving as per trust regulations (5 July 2018).
The TMF and research data were to be archived as per the co-ordinating centre SOPs. The ISF and pharmacy folder were to be archived at site in line with trust policy.
Appendix 7 Monitoring: trial sponsor
Monitoring plan
The trial sponsor provided site monitoring throughout the trial. The purpose of this monitoring was to ensure that the rights and well-being of the participants were protected, and the conduct of the study was in compliance with the protocol, regulatory and GCP requirements. A monitoring plan detailing the schedule for monitoring visits was prepared. The trial sponsor noted that a risk assessment had been produced by the co-ordinating centre, which was specific for this trial and had identified key areas of risk: consent, safety reporting, eligibility and IMP management (see Chapter 3, Risk assessment). The trial sponsor also noted that the principal investigator had limited experience in conducting clinical trials, but did have the support of an experienced clinical trials unit (the co-ordinating centre), that each infant would be included in the trial for 48 hours, and that safety data would be collected for 24 hours after the IMP or placebo was administered.
The monitoring plan indicated that there would be source data in the medical records with a record of gestational age, and a paper cot-side log maintained for the 48-hour trial period for each participant as a back-up to the computer download of vital signs. The trial sponsor would complete source data verification (SDV) on at least 10% of these data at the appropriate site visits. The trial sponsor noted in the monitoring plan that the vital signs, oxygen saturations and brain activity monitor data would be transferred from the monitor (equipment) to another computer for storage and then into the OpenClinica database, but that the verification for these data would be outside the scope of the monitor.
In the initial trial sponsor monitoring plan (finalised on 23 November 2015), four visits were scheduled: initiation (prior to trial start), after enrolment of the fifth participant, after enrolment of the 75th participant (with the aim of focusing on IMP management) and at close-out (within 60 days of study end at the site). However, partway through the trial the trial sponsor decided that another monitoring visit after enrolment of the 125th participant would be beneficial, and this was incorporated into the revised monitoring plan and signed off in October 2017.
Site initiation
The trial sponsor site initiation visit was carried out on 3 December 2015, after all approvals had been obtained and before starting recruitment. Attendees included representatives from the research team, co-ordinating centre and trial sponsor. The visit was conducted in the knowledge that some trial documents were outstanding as a result of limited staff availability. The trial sponsor reported that the ISF was well structured and organised. The missing documentation included the technical agreement with the IMP manufacturer, collaborator agreement and some guidance sheets. The delegation log (see Report Supplementary Material 4) was also unsigned at this point and the co-ordinating centre programming team were still testing the OpenClinica database. The report was completed by the monitor, reviewed internally by the trial sponsor and forwarded to the site to action outstanding items.
Monitoring visits
A routine site monitoring visit from the trial sponsor was carried out on 6 January 2017 after seven participants had been recruited. The ISF, participants’ medical notes, temperature logs and drug accountability logs were reviewed. Participant enrolment status and overall progress of the study were reviewed and discussed with study staff. Where appropriate, all SDV was performed through the SDV portal of the OpenClinica database, with queries opened to the appropriate investigator with e-mail notification. The trial sponsor QA team confirmed that the site visit went well and that the report stated a number of minor findings. The report indicated that the findings from the previous visit had all been resolved. New findings related to the recording of individuals on the delegation log (see Report Supplementary Material 4), missing signatures on the training log (see Report Supplementary Material 11 and 12), a missing signature on the accountability log (see Report Supplementary Material 1), the screening log (see Report Supplementary Material 8) was not up to date, an error which was not GCP corrected, outstanding queries requiring resolution in the OpenClinica database and a number of file notes that had not been marked as superseded. All outstanding actions were completed before site close-out. As the trial stopped after 31 participants were enrolled, the monitoring visits scheduled for after enrolment of the 75th and 125th participants did not take place.
Site close-out
The close-out visit from the trial sponsor took place on 27 April 2018. Outstanding findings from the previous visit were missing signatures on the training log (see Report Supplementary Material 11 and 12) and the lack of a countersignature on the accountability log (see Report Supplementary Material 1). New findings were an individual from pharmacy missing from the delegation log (see Report Supplementary Material 4), and missing end dates and sign-off from the chief investigator and principal investigator on the delegation log. All actions were completed and reported back to the trial sponsor. The trial sponsor confirmed that from a monitoring perspective, the site and the trial were closed on 28 June 2018.
Appendix 8 Monitoring: project management group
Project management group meetings (including the full PMG team, comprising the chief investigator, principal investigator, core site staff, trial manager, statisticians, programmers and co-ordinating centre senior management team) were held every 6–8 weeks. The PMG meetings were interspersed with central monitoring meetings (including the trial manager and co-ordinating centre senior management team) to provide regular central monitoring of the trial.
Project management meetings
The PMG meetings were chaired by the trial manager, who provided an update to the group on recruitment, data monitoring reports, safety, incidents, programming and statistical concerns, IMP stock management, and any other relevant topics such as upcoming reports, oversight committee meetings, and amendments. Key decisions requiring input from all core members of the trial team were discussed at the PMG meetings.
Central monitoring meetings
Items on the risk assessment identified as requiring monitoring were reviewed at each central monitoring meeting. These items included recruitment rates, parental consent, loss of data during export from monitor to computer, data entry and completeness, SAEs, incidents, possible overdose, temperature excursions, completion of drug accountability records, technical challenges in recording data issues, instances where trials staff were uncontactable and instances of unblinding. Ongoing and resolved items were highlighted in each report.
Finance project management group meetings
Finance PMG meetings were held between the central monitoring team (including co-ordinating centre senior management and trial manager) and the co-ordinating centre finance team at quarterly intervals. The trial expenditure and grant monitoring overview were reviewed, and budgeting concerns and projections discussed at these meetings.
Trial master file audits
Internal audits of the TMF were conducted by the co-ordinating centre QA team soon after recruitment began and, again, after the trial had closed. The first audit was conducted on 16 January 2017, and the second audit was conducted on 30 April 2018. The QA team confirmed that there were no key issues with the TMF identified in either audit, and created reports detailing the minor findings for the trial manager to resolve.
Appendix 9 Data Monitoring Committee and Trial Steering Committee meetings
Data Monitoring Committee meetings
Meeting 1: initial meeting
The first meeting of the DMC was held on 23 November 2015, at which the trial was introduced and the charter reviewed. The first meeting was face to face to facilitate full discussion and allow members to get to know each other.
Meeting 2: stopping boundaries meeting
A DMC meeting was held on 1 November 2016 to discuss options for group sequential stopping boundaries for safety. Of three possible options, the DMC agreed that a gamma spending function should be used, which specified a probability of incorrectly stopping for harm (type I error rate) equal to 0.20, resulting in a probability of correctly detecting a safety issue (power) of 0.790. The DMC confirmed that it would like to be alerted to safety outcomes approaching or crossing the stopping boundary and would like further clinical information to review each case individually. The DMC agreed that the next meeting would be scheduled once 25 infants had been recruited to the trial.
Meetings 3 and 4
Meetings 3 and 4 of the DMC were held on 4 and 7 December 2017, respectively, in which the committee recommended that the trial be stopped.
Meeting 5
Meeting 5 of the DMC was a joint committee meeting with the TSC to review the SAP.
Meeting 6
Meeting 6 of the DMC was a joint committee meeting with the TSC and collaborators to review the draft manuscript reporting the trial findings.
Trial Steering Committee meetings
Meeting 1: initial meeting
The first meeting of the TSC was held on 14 December 2015, at which the trial was introduced and the charter reviewed. The TSC advised that the PMG should carry out pilot work to obtain baseline data on clinical stability, carry out a power calculation on the secondary clinical stability outcome measure, investigate use of the Medical Dictionary for Regulatory Activities (the team investigated this, but chose not to pilot this coding for the trial), and contact the trial ophthalmologist about the use of RetCam® (a wide-field digital imaging system; Natus Medical Inc., Pleasanton, CA, USA) and timing of eye drops to ensure that there was consistency across participants (this was discussed at the collaborators meeting in March 2016). The TSC also requested a factual and objective summary outlining reasons for recruitment delays, the PIL and consent form, and the NPEU data-sharing policy to the TSC.
Meeting 2
A second TSC meeting took place on 16 November 2016, in which the trial progress update report and stopping guideline agreed by the DMC were discussed.
Meeting 3
Meeting 3 of the TSC was held on 13 December 2017, in which the decision to temporarily suspend the trial was made.
Meeting 4
Meeting 4 of the TSC was a joint committee meeting with the DMC to review the SAP.
Meeting 5
Meeting 5 of the TSC was a joint committee meeting with the DMC and collaborators to review the draft manuscript reporting the trial findings.
Appendix 10 Safety reporting
Throughout the trial, any staff member could report safety events at any time. Staff on the neonatal unit could complete a paper form (kept in the document box). Trial staff who were trained in OpenClinica could use this database to electronically record and report safety events. Safety data were recorded for 24 hours following the administration of the IMP. Events were defined as AEs or adverse reactions (ARs), SAEs or serious adverse reactions (SARs), and SUSARs.
An AE was any untoward medical occurrence in a participant to whom a medicinal product had been administered, including occurrences that were not necessarily caused by, or related to, that product. An AR was an untoward and unintended response in a participant to an IMP that was related to any dose administered to that participant. A SAE is any untoward medical occurrence that resulted in death, was life-threatening, required inpatient hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability/incapacity, comprised a congenital anomaly or birth defect, or involved any other important medical event. A SAR was an AE that is both serious and, in the opinion of the reporting investigator, believed with reasonable probability to be as a result of one of the trial treatments, based on the information provided. A SUSAR was a SAR for which the nature and severity was not consistent with the information about the medicinal product in question set out in the reference safety information.
The reference safety information used throughout the trial was the Summary of Product Characteristics (SmPC) for Oramorph oral solution, dated 1 April 2014. The appropriateness of using this document as the continued reference safety information for the trial, in comparison to other variations of the SmPC, was reassessed annually by the chief investigator and principal investigator.
All other AEs occurring during the 24-hour period following the administration of the IMP and which were observed by the research team, were recorded on the AE CRF, whether or not the AE was attributed to trial medication. A 24-hour period post IMP administration was chosen because the reference safety information for morphine sulphate indicated that, when administered orally to humans, the levels of plasma-conjugated morphine peaked at ≈ 3 hours and decreased over the following 24 hours. Thus, AEs associated with the IMP were considered likely to occur in the 24-hour period post IMP administration.
Adverse events and adverse reactions
Adverse events that could be foreseen in this population and did not need recording were suspected sepsis (requiring up to 36 hours of antibiotics), anaemia, minor changes in oxygen requirement [i.e. an increase in oxygen supply by < 10%, a flow rate change of < 1 l/minute (high-flow machine) or a flow rate change of < 0.04 l/minute (low-flow machine)] and electrolyte disturbances. All other AEs were recorded on the AE form by the research team, with information taken directly from the infant’s medical notes including a description of the event, time and date of onset, severity (assessed on the following scale: 1 = mild, 2 = moderate, 3 = severe), assessment of relatedness to trial medication (judged by a medically qualified investigator with delegated duty), other suspect drugs or devices and action taken, and follow-up information as necessary. Any AE suspected to be a SAE would be reviewed by a medically qualified investigator. All AEs considered related to the trial medication would be followed up either until resolution or until the event was considered stable. Clinical judgement would be used to decide whether or not an AE was of sufficient severity to require intervention and/or removal from the trial. If so, the participant would be given medical supervision until symptoms ceased or the condition became stable.
Serious adverse events and serious adverse reactions
Foreseeable SAEs were those that could be reasonably foreseen to occur in this population of infants and required recording, but not expedited reporting, as SAEs. Foreseeable SAEs included death (unless unforeseen), necrotising enterocolitis or focal intestinal perforation, intracranial abnormality (haemorrhage, parenchymal infarction or focal white matter damage) on cranial ultrasound scan or other imaging, microbiologically confirmed or clinically suspected late-onset invasive infection, ROP, patent ductus arteriosus (requiring treatment) or congenital abnormalities.
Any unforeseeable SAEs were reported to the co-ordinating centre immediately, but at least within 24 hours of the research site becoming aware of the event. SAEs could be reported electronically using the OpenClinica database, only by research staff with access to this. If reported electronically, the form also had to be printed, with information and a signature obtained from a study clinician carrying out the causality assessment and the completed form e-mailed or faxed to the co-ordinating centre. If completing a paper SAE form (which were kept in the document box), this also had to be e-mailed or faxed to the co-ordinating centre. In both cases, the causality assessment was completed by a medically qualified investigator with delegated duty on the delegation log (see Report Supplementary Material 4):
-
unrelated – where an event is not considered to be related to the IMP
-
possibly – although a relationship to the IMP cannot be completely ruled out, the nature of the event, the underlying disease, concomitant medication or temporal relationship make other explanations possible
-
probably – the temporal relationship and absence of a more likely explanation suggest that the event could be related to the IMP
-
definitely – the known effects of the IMP, its therapeutic class or based on challenge testing, suggest that the IMP is the most likely cause.
The person reporting the SAE was required to ensure, via telephone, that the co-ordinating centre was aware of the SAE report. Any new, updated or corrected information on previously reported SAEs was also reported to the co-ordinating centre, and the outcome of events ‘Resolving’ or ‘Not Resolved’ were followed up until the status of the SAE changed.
The chief investigator or safety delegate also assessed all reported SAEs. The recorded causality assessment could not be downgraded. This was arranged by the co-ordinating centre, using trial-specific SAE assessment forms.
Suspected unexpected serious adverse reactions
The trial process for SUSARs was that if the SAE was related to the IMP and unexpected (i.e. not consistent with the SmPC), then this was considered to be a SUSAR. SUSARs were reported using the SAE form (via the OpenClinica database or paper form) to the co-ordinating centre immediately, but at least within 24 hours of the research site becoming aware of the event. Review would be timely taking into account reporting time for potential SUSARs: all SUSARs would have been reported by the co-ordinating centre to the relevant competent authority and to the REC and other parties, as applicable. For fatal and life-threatening SUSARs, this reporting would be done within 7 calendar days after the trial sponsor or delegate was first aware of the reaction. Any additional relevant information would be reported within 8 calendar days of the initial report. All other SUSARs would be reported to the relevant bodies within 15 calendar days.
List of abbreviations
- 3D
- three-dimensional
- AE
- adverse event
- AR
- adverse reaction
- b.p.m.
- beats per minute
- CFU
- colony-forming unit
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- CRN
- Clinical Research Network
- DMC
- Data Monitoring Committee
- DSUR
- development safety update report
- eCRF
- electronic case report form
- EEG
- electroencephalography
- EME
- Efficacy and Mechanism Evaluation
- EMG
- electromyography
- GCP
- Good Clinical Practice
- HDU
- high-dependency unit
- HRA
- Health Research Authority
- ID
- identifier
- IMP
- Investigational Medicinal Product
- IQR
- interquartile range
- ISF
- investigator site file
- ISRTCN
- International Standard Randomised Controlled Trial Number
- IVH
- intraventricular haemorrhage
- LDU
- low-dependency unit
- MHRA
- Medicines and Healthcare products Regulatory Agency
- NICU
- neonatal intensive care unit
- NIHR
- National Institute for Health Research
- NIPPV
- non-invasive positive-pressure ventilation
- NPEU
- National Perinatal Epidemiology Unit
- NPEU CTU
- National Perinatal Epidemiology Unit Clinical Trials Unit
- OUH
- Oxford University Hospitals
- portable document format
- PIL
- parent information leaflet
- PIPP
- Premature Infant Pain Profile
- PIPP-R
- Premature Infant Pain Profile – Revised
- PMG
- Project Management Group
- Poppi
- PrOcedural Pain in Premature Infants
- PPI
- patient and public involvement
- QA
- quality assurance
- QP
- qualified professional
- R&D
- research and development
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- RMS
- root-mean-square
- ROP
- retinopathy of prematurity
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SAR
- serious adverse reaction
- SD
- standard deviation
- SDV
- source data verification
- SmPC
- Summary of Product Characteristics
- SOP
- standard operating procedure
- SSNAP
- Support for the Sick Newborn and their Parents
- SUSAR
- suspected unexpected serious adverse reaction
- TADA
- trial administration database application
- TMF
- trial master file
- TSC
- Trial Steering Committee