

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 12/206/30. The contractual start date was in October 2014. The final report began editorial review in September 2018 and was accepted for publication in March 2019. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Joseph Alsousou reports a grant from the National Institute for Health Research (NIHR) Academic Clinical Fellowship programme during the conduct of the study. David J Keene reports grants from a NIHR postdoctoral fellowship during the conduct of the study. Jacqueline Y Thompson reports that she was in receipt of a grant from Thames Valley Clinical Research Network Contingency Fund for physiotherapist training. Sarah E Lamb reports grants from the NIHR Health Technology Assessment (HTA) programme for the AIM study during the conduct of this study. Sarah E Lamb was also a member of the HTA Additional Capacity Funding Board (ongoing), HTA Clinical Trials Board (2010–15), HTA End of Life Care and Add-on Studies (2015), HTA Funding Boards Policy Group (formerly Clinical Study Group) (2010–15), HTA Maternal, Neonatal and Child Health Methods Group (2013–15), HTA post-board funding teleconference (2010–15), HTA Primary Care Themed Call Board (2013–14), HTA Prioritisation Group (ongoing) and NIHR Clinical Trials Unit Standing Advisory Committee (2012–16) during this study. Sarah E Lamb is Co-director of the Oxford Clinical Trials Research Unit and Professor of Rehabilitation at Warwick Clinical Trials Unit, receiving funding from NIHR for both. Sarah E Lamb also receives funding from the NIHR Collaboration for Leadership in Applied Health Research and Care Oxford at Oxford Health NHS Foundation Trust. Keith Willett declares design royalties from Zimmer Biomet (Warsaw, IN, USA), outside the submitted work, for intramedullary bone fixation implants, and held the role of Medical Director for Acute Care, NHS England, during the conduct of this study.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2019. This work was produced by Alsousou et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2019 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
In this chapter, we provide a background to the current management of Achilles tendon rupture (ATR), summarise the evidence base for the use of platelet-rich plasma (PRP) as a treatment and outline the objectives of the PRP in Achilles Tendon Healing (PATH-2) trial.
Background
Achilles tendon
The Achilles tendon is the largest and strongest tendon in the human body. It is the tendon of the gastrocnemius–soleus calf muscle complex. The Achilles tendon inserts to the calcaneum (heel bone), forming the major plantar flexor and stabiliser of the ankle joint. 1 The tendon is rounded and narrow in shape at its midpoint and finally fans out at the insertion (Figure 1). The tendon fibres rotate around 90° as they insert into the calcaneum. The narrowest part of the tendon is approximately 4 cm above the insertion.
FIGURE 1.
Anatomy of the Achilles tendon. The image on the left shows the left Achilles tendon projection at the posterior aspect of the calf and heel (1, calf muscles; 2, Achilles tendon; 3, Achilles tendon insertion to heel); and the image on the right is an illustration of the muscle–tendon unit (1a and 1b, gastrocnemius muscle medial and lateral heads; 1c, soleus muscle; 2, Achilles tendon fibres; 2a, gastrocnemius Achilles fibres; 2b, soleus Achilles fibres; 3, tendon insertion to the calcaneum).


The structure of the muscle–tendon unit allows for effective force to be generated when lifting the heel during locomotive activities such as walking and running. 1,2 The human Achilles tendon also stores and returns energy. 3 The tendon stretches in proportion to the force applied during the downwards motion of the body and then recoils to release most of the energy stored (74%) during the upwards movement.
Blood supply to the tendon is variable but is least in the middle (mid-portion). The mid-portion is the narrowest and a relatively avascular region within the tendon that corresponds to the most frequently injured area. Degenerative changes, common with advancing age and metabolic and chronic diseases, further compromise tendon vascularity and structure. 4 Degenerative changes in the tendon decrease collagen cross-linking and small repetitive tears weaken the tensile strength of the tendon. The Achilles tendon ruptures when it is subjected to a load that exceeds its mechanical capacity.
Achilles tendon rupture epidemiology
The Achilles tendon is the most commonly injured tendon in the human body, accounting for 20% of all tendon ruptures. 5 The incidence of this injury is rising, due to increased engagement in high-impact recreational sport, common particularly among men in the third and fourth decades of life. 6,7 Other factors that might increase the risk of injury include previous injury to the contralateral leg, direct injection of steroids or administration of systemic corticosteroids and fluoroquinolones. 8 In the UK, 11,000 injuries are reported each year. 9 In Denmark, an increase in incidence from 22.1 ATRs per 100,000 person-years in 1991 to 32.6 per 100,000 person-years in 2002 was reported by Gulati et al. 10 The incidence of ATR is 12–18 cases per 100,000 people per year for sedentary professionals. 11 A Cochrane review12 reported rehabilitation and work absence of 63–108 days, highlighting the socioeconomic burden of the injury.
The injury mechanism of ATR is mostly related to loading the tendon during weight-bearing physical activity. Typical acute injuries result from rapid force shifts to the lower leg during sports. This can result in partial or complete tear from direct (acute) trauma. During clinical assessment, patients often describe a history of feeling a direct blow localised to the posterior aspect of the ankle with a sudden onset of pain, swelling, bruising and difficulty with walking, usually during a sport activity. Physical examination of the area commonly shows indentation at the locations of the tendon rupture, soft-tissue swelling and tenderness, but these signs can be masked at the acute stage.
Spontaneous ruptures without a notable trauma, more common in ageing tendons, are typically associated with pre-existing chronic, pathological changes in the tendon. Degenerative changes, such as poor vascular supply, calcification, tendolipomatosis (replacement of tendon tissue with fat), necrosis (cell or tissue death), hypoxia and mucoid degeneration, have been shown to be present in ruptured tendons not related to sports. 13 Once an acute tear to the tendon occurs, the structural, biochemical and functional properties of the tendon are disrupted. Histopathological changes in the injured tissues have been reported. These include high vascularity, collagen disorganisation and hypercellularity relatively close to the ruptured site. There is a reduction in the number and diameter of type I collagen fibres, which are replaced with larger type III fibres. 8
Clinically, a common method of examination to assess the integrity of the Achilles tendon is to lay the patient prone on a plinth with their foot hanging over the edge. In this position, the tendon gap is usually palpable and easy to observe, and on squeezing the calf muscle bulk, ankle plantarflexion can be anticipated if the tendon is intact (Simmonds–Thompson test). 14–16 These methods of assessment may result in misdiagnosis when the tendon gap is filled with oedema or when a plantarflexion response is generated by other intact muscles that also plantarflex the ankle (i.e. tibialis posterior and the peronei). 17 However, significant plantar flexion weakness on directed push-off, abnormal or poor gait pattern and inability to perform a heel rise on the affected leg are often indicative of a complete tendon tear. 18 Diagnosis is usually made clinically from the history and physical assessments, but some clinical centres use ultrasound and magnetic resonance imaging to support diagnosis. There are also treatment protocols that direct management using imaging findings,10,18,19 but this is not currently routine practice in the NHS.
Healing of the tendon (repair and regeneration)
The healing of an injured tendon takes place in three overlapping, successive stages: inflammation, repair and remodelling. These cellular responses are directed by numerous growth factors that are upregulated and activated at various stages of the healing process. 13,20 In the inflammatory stage, haematoma forms and platelets release growth factors and bioactive proteins to attract inflammatory cells, including neutrophils. This haematoma and the inflammatory cells eventually organise into granulation and scar tissue. 13,20 In the repair stage, recruitment, activation and proliferation of tenocytes and fibroblast begins in the injury site. During this phase, neutrophil levels drop while macrophages continue to release growth factors that direct cell activity. In addition, intrinsic and extrinsic tenocytes synthesise and establish a network of extracellular matrix composed of type III collagen fibres. The remodelling stage starts 1–2 months post injury and persists for ≥ 12 months. Tenocytes and collagen mature and become aligned with the direction of stress. Over time, the synthesis of collagen type III fibres decreases, while type I fibres become dominant and fibrous scarring tissue remodel towards the original tendon tissue. 13,20
Although tendons have the ability to heal after injury, the natural process is slow and incomplete. 20 This limited healing capacity of tendons may result from a combination of relatively poor vascularisation and cellular turnover, changes to the matrix structure and the demanding mechanical environment of this connective tissue. 21–24 Poor vascularisation negatively affects the inflammation that is essential for the restoration of the mechanical and biological properties of tendons during the healing process. 25,26 Ageing tenocytes – the dominant cell type in tendons – tend not to be able to facilitate the proper differentiation of progenitor cells, resulting in ineffective healing. This leads to scar formation rather than tendon tissue regeneration, which results in reduced tensile strength. 13,20
The mechanical properties of the healed tendon do not fully recover, leading to weakness of the muscle tendon unit and risk of re-rupture. Even with the best contemporary management, tendon injuries give rise to substantial morbidity that lasts for many months and poses considerable challenges for clinicians and patients during the lengthy healing and recovery period. 27
Management of Achilles tendon rupture
The current treatment strategies for acute ATR are either surgical repair using sutures or non-surgical management by immobilisation of the ankle using a boot or cast. 19 Whether surgical or non-surgical management of ATR is the optimal treatment approach is uncertain. 7,19 The decision for the type of management is often based on patient-related factors such as timing of presentation, comorbidities, activity demands and previous tendon function. 28 Notably, there is a decline in the proportion of tendons treated surgically worldwide. 7
Open, minimally invasive or percutaneous techniques for operative repair seem to show quicker return to work and greater heel-rise height during physical assessments. 29 However, surgical complications including sural nerve injury, keloid formation, wound breakdown or infection are risks to be considered, although they may be reduced when percutaneous surgical repair is used. 29,30
Non-surgical management involves short periods (6–8 weeks) of ankle immobilisation starting in an equinus (plantarflexed) ankle position in a cast, rigid boot or ankle orthoses. 29 Recent developments of early functional rehabilitation and range of motion protocol with early weight-bearing – using controlled ankle motion boots – during non-surgical treatment have demonstrated similar surgical outcomes in terms of re-rupture rates (3–5%) and function. However, non-surgical management does not increase the rate of re-rupture or other complications compared with surgery. 31–33 The inclusion of functional rehabilitation as an adjunct to non-surgical management has not demonstrated improvement in biomechanical tendon properties or tendon elongation, which influence plantarflexion strength. 29,34 Although different surgical techniques and non-surgical approaches can be used, evidence shows similar outcomes. 35 These approaches are not known to alter the existing biological regenerative pathway of the tendon, so the lengthy rehabilitation, reduced function and re-rupture risk (3–5%) all remain. 36 Functional impairments identified by calf atrophy, heel-raise power compared with the opposite side, poor walking pattern (gait) and lower levels of physical activity are reported as outcomes of these management strategies. 37
Platelet-rich plasma
Although significant advancements in the clinical management of ATR have resulted in some improvements in functional outcomes,38 there has been little progress made with regard to the restoration of the structural and biomechanical integrity of an injured tendon to its original state. Thus, it is unsurprising that novel therapies are being explored to diminish the degree and duration of morbidity. Biological adjuncts such as PRP may augment the regeneration of a torn tendon. For example, platelet-derived growth factor (PDGF), a derivative of PRP, seems to upregulate the synthesis of cells in tendons that direct optimal cellular repair response, which results in less scar tissue formation, stimulates healing and improves biomechanics. 20 Evidence to support its clinical application in the management of musculoskeletal conditions remains inconclusive. 39
Platelet-rich plasma is an autologous derivative of whole blood that contains a higher concentration of platelets and other bioactive compounds. PRP is produced by separating the red blood cells from the platelets and other constituents in whole blood. The method of PRP preparation, such as centrifugation or filtration; the concentration and volume of platelets and white blood cells (leucocytes); the activation method; and growth factor levels have all been implicated in the local effects of PRP. 39–41 Unsurprisingly, the nature of the target tissue – acute, subacute or chronic – seems to affect local or systemic effects of PRP. 42
As part of the normal injury response to an acute tendon rupture, platelets immediately contribute to the blood clot that forms within the injured tissue. That response includes multiple growth factors released from the platelets’ α-granules interacting with neutrophils, macrophages and other cells, working synergistically to provide a wound healing mixture. This is part of a cascade within the injured tissue over a period ranging from minutes to an average of several days. 43,44 Active growth factors in PRP in the rupture site promote cell motility and recruit undifferentiated stem cells and surviving tenocytes, which then proliferate to start the healing process. 45 The upregulation of growth factors such as vascular endothelial growth factors (VEGFs) contained within PRP attracts vascular endothelial cells, which stimulate angiogenesis to restore normal tissue conditions. 46 PDGF, insulin-like growth factor 1 (IGF-1) and fibroblast growth factor (FGF) facilitate proliferation of cells and hasten the start of the remodelling phase. 47 Transforming growth factor beta (TGF-β) promotes the formation of fibrous tissue and the expression, organisation and maturation of collagen fibrils, enhancing the biomechanical strength of the tendon. 48
Although there is evidence suggesting that leucocyte-poor PRP may produce better outcomes than leucocyte-rich PRP (L-PRP), leucocytes contained in PRP seem to interact with the tissue of the tendon, promoting healing by stimulating tenocytes within the injured area to produce their own growth factors. 49 Furthermore, PRP assists the initiation of a cascade of steps that induce the synthesis of fibroblasts, collagen fibrils, matrix-degrading enzymes and extracellular matrix. This sequential release of proteins is amplified to recruit a diverse range of cell types to act on the injured tendon and disrupted blood vessels. So far, conclusions from basic laboratory testing have indicated that PRP could be a novel agent for tendon injury. 44 Augmenting the natural biological healing pathway to improve patient recovery in Achilles tendon injury may therefore have significant clinical implications on the health system at large by reducing demands for surgical repair, accelerating rehabilitation, improving physical activity participation and reducing re-injury rates.
Achilles tendon injury and platelet-rich plasma
Although PRP shows some promise as a treatment for promoting faster healing in tendons,20,50,51 the potential enhancement of healing demonstrated in laboratory studies is yet to be evidenced in the patient setting. The mechanisms of action through which PRP exerts its regenerative effects in humans are not completely understood. A systematic review has summarised the evidence from animal experiments that evaluated the use of platelets in the treatment of ATR. 52 The authors reported a moderate beneficial effect of PRP in the treatment of ATR in animal models. The observed effects were attributed to accelerated and enhanced scar tissue maturation. 52 An early systematic review of clinical trials of PRP in humans by Taylor et al. 50 reported faster healing and better function in the management of tendon and ligament injuries. However, a more recent review by Gholami et al. 51 reported no beneficial effects of PRP in the management of sports injuries for pain or function compared with other management options.
A number of studies using different postoperative protocols have investigated the effect of platelets on the treatment of ATR in humans. Two case reports, one of an injured athlete53 and the other of a complete tear in an active 71-year-old male,54 showed positive effects of quick return to play after non-surgical management. In a case–control study by Sánchez et al.,55 12 athletes with Achilles tendon injury were treated with either PRP and surgery, or surgery alone; the PRP and surgery group (six athletes) achieved significantly quicker recovery and better functional outcome measures of greater range of motion and return to play than those treated with surgery alone. However, this effect was seen only at 22 weeks, and a difference was not found at the 1-year follow-up. 56 Similarly, one small clinical trial (15 participants per group) demonstrated no significant difference in structural and functional outcomes between surgery with PRP and surgery alone. 57 However, Zou et al. 58 reported some improvement in isokinetic muscle strength, ankle range of motion and measures of quality of life in a clinical study of 36 people with ATR comparing surgery and PRP with surgery alone.
A randomised controlled trial (RCT) (n = 30) by Schepull et al. 56 showed no difference for functional outcomes or biomechanical tendon properties at 1-year follow-up when PRP with surgery was compared with surgery alone. 59 However, the authors also stated that a limitation in their platelet preparation technique and storage of up to 20 hours resulted in only a 20% release of growth factors from the platelets. The level of activation to release growth factors may have an impact on the regenerative process and could alter the clinical outcome. We observed good activation rates of 69% as a measure of the quality of PRP used in a clinical pilot study,57 in which 20 patients were randomised to receive non-surgical treatment with or without PRP injection or operative management with PRP gel, with standard rehabilitation for both groups. Achilles tendon Total Rupture Score (ATRS) and the Victoria Institute of Sport Activity for Achilles (VISA-A) score showed significantly better PRP group outcomes starting from week 3 to week 24 of follow-up.
All of these small clinical studies used PRP as an adjunct to surgical repair, which may have influenced the effect of PRP on the tendon as there is additional surgical trauma and the mechanical effects of having a suture across the tendon rupture gap. Overall, the results from RCTs with small numbers of participants that have assessed PRP in ATR highlight that the current evidence is inconclusive.
Rationale for the PATH-2 study
Despite significant advances in the management of musculoskeletal injuries (e.g. novel operating techniques),27 slow recovery, morbidity and complication rates persist. PRP as an alternative, regenerative orthobiological agent has gained popularity on the basis that it could augment the management of traumatic musculoskeletal injuries. It is estimated that PRP is used to treat 86,000 tendon, ligament and muscle disorders annually in the USA and Europe. 60 There is also evidence to suggest that PRP injections are being administered in the NHS and private medical practices in the UK. 59
Platelet-rich plasma was initially used only as an adjunct during orthopaedic surgery, but in recent times it has been applied in other areas of sports medicine. Basic experimental research has provided a biological rationale for PRP, with encouraging results from histological and biomechanical assessments; however, its clinical efficacy is yet to be confirmed by findings from studies with robust designs. Although PRP remains an attractive treatment option, owing to its simplicity, patient acceptability, affordability and practicality, the lack of proven clinical effects means that its use is controversial.
Although PRP has been shown to be relatively safe, preparation of PRP has yet to be standardised. 61 For example, some authors have suggested that administration during the early inflammatory stage of healing may disrupt normal underlying physiological processes. This may result in the downregulation of certain growth factors, such as TGF-β, which may promote the formation of fibrous tissue. 61 Others did not find a link with injection stage. Although injecting PRP concentrates may, in theory, increase the risk of infection or exacerbate the underlying microbial activity within spontaneous ruptured tendons,23 no clinical impact on outcome has been reported. 36,50
There is substantial variation in the validity and type of outcomes measured, as well as inconsistency in the effect size of PRP observed from published studies. The underpowered and inadequately designed studies to date suggest that no definite conclusions can be made on PRP as a superior (or inferior) treatment option over standard care in the management of acute ATR. A recent meta-analysis of PRP for orthopaedic conditions62 stated the need for adequately powered studies using disease-specific and patient-important outcomes to investigate the effect of PRP.
We conducted a multicentre, pragmatic, parallel-group, blinded, placebo-controlled RCT to assess the efficacy of PRP. Patients presenting with acute ATR receiving non-surgical management at orthopaedic trauma surgery departments in the NHS in the UK were randomised to receive PRP or placebo (dry-needle injection). PRP and rehabilitation were standardised. Two substudies of blood and tendon biopsy were integrated into the main trial design to assess the components of PRP and to further explore the mechanism of action on injured Achilles tendons.
Research objectives
-
To evaluate the clinical efficacy of PRP among patients with acute ATR using an objective measure of mechanical muscle–tendon function.
-
To evaluate secondary outcome measures of patient-reported function, pain and quality of life.
-
To determine in an exploratory substudy the key components of PRP that may contribute to its mechanism of action.
-
To identify the tissue-level parameters that PRP may alter to exert its effects in an exploratory biopsy substudy.
Chapter 2 Clinical trial methods
Summary of study design
The PATH-2 trial was a multicentre, parallel-group, participant- and outcome assessor-blinded, randomised, placebo-controlled trial with two embedded mechanistic substudies. In NHS hospitals in England and Wales, patients with acute ATR for non-surgical treatment were randomised 1 : 1 to receive either an injection of PRP into the rupture gap or a placebo injection (dry-needle insertion). In substudy 1, whole blood and PRP were sent to a central specialised laboratory (Institute of Inflammation and Ageing, University of Birmingham) for analysis. In substudy 2, immunohistochemical analysis was carried out on tendon biopsy material from participants at one site (Oxford).
Settings and locations
Nineteen NHS hospital orthopaedic or trauma clinics in England and Wales screened and recruited participants for this study:
-
Oxford University Hospitals NHS Foundation Trust
-
University Hospitals of Leicester NHS Trust
-
Taunton and Somerset NHS Foundation Trust
-
North Bristol NHS Trust
-
Cardiff and Vale University Health Board
-
Shrewsbury and Telford Hospital NHS Trust
-
Barts Health NHS Trust
-
University Hospitals Coventry & Warwickshire NHS Trust
-
Warrington and Halton Hospitals NHS Foundation Trust
-
Basildon and Thurrock University Hospitals NHS Foundation Trust
-
Royal Liverpool and Broadgreen University Hospitals NHS Trust
-
Peterborough and Stamford Hospitals NHS Foundation Trust
-
Abertawe Bro Morgannwg University Health Board
-
Aintree University Hospital NHS Foundation Trust
-
University Hospital of South Manchester NHS Foundation Trust
-
Sheffield Teaching Hospitals NHS Foundation Trust
-
Royal Devon and Exeter NHS Foundation Trust
-
Mid Cheshire Hospitals NHS Foundation Trust
-
Royal Surrey County NHS Foundation Trust.
The principal investigator (PI) at each site was a trauma and orthopaedic surgeon. Sites were selected after completion of a site feasibility questionnaire, signed by the PI, which was used to assess whether or not each site had the appropriate resources to deliver the project and meet recruitment targets. The PI supervised implementation of the trial protocol at the site and co-ordinated with local physiotherapy services to provide a standardised rehabilitation protocol and to arrange a blinded physiotherapist to be the assessor for the primary outcome measurement. Confirmation of collaboration was provided in writing to the PI.
Participants
Participant screening and eligibility assessment
All patients attending orthopaedic or trauma clinics with a suspected acute ATR were screened for inclusion in the trial. Patients were identified for screening for the trial at this clinic visit, normally in the 12 days after the initial visit to hospital, where the attending surgeon (or an extended-scope physiotherapist) confirmed appropriateness for non-surgical treatment and trial eligibility.
Patients with acute ATR were eligible for the trial if they met all of the inclusion criteria and none of the exclusion criteria.
Patients were eligible for inclusion if:
-
they were willing and able to give informed consent for participation in the trial
-
they were aged ≥ 18 years
-
they were ambulatory prior to injury without the use of walking aids or assistance of another person
-
they were diagnosed with an acute, complete ATR
-
they presented and received trial treatment within 12 days post injury
-
the decision had been made for them to receive non-surgical treatment
-
they were able (in the investigator’s opinion) and willing to comply with all trial requirements
-
they were able to attend a PATH-2 trial hospital site for the 24-week follow-up.
Patients were excluded if they:
-
had an Achilles tendon injury at the insertion to the calcaneum or at the musculotendinous junction
-
had a previous major tendon or ankle injury or deformity to either lower leg
-
had a history of diabetes mellitus
-
had a known platelet abnormality or haematological disorder
-
were currently using systemic cortisone or a treatment dose of an anticoagulant (i.e. a prophylactic dose for preventing thrombosis was not an exclusion criterion)
-
had evidence of lower-limb gangrene/ulcers or peripheral vascular disease
-
had a history of hepatic or renal impairment or dialysis
-
were female and pregnant or breastfeeding
-
were receiving, or had received within the previous 3 months, radiotherapy or chemotherapy
-
had inadequate venous access for drawing blood
-
had any other significant disease or disorder that, in the opinion of the investigator, might put the participant at risk because of participation in the trial or might influence the result of the trial or the patient’s ability to participate in the trial.
Members of the local research team informed the patient of the trial and carried out the informed consent process, baseline data collection and randomisation.
Standard treatment for this non-surgical population is usually application of a plaster cast, orthopaedic brace or splint during the clinic visit. The PATH-2 trial treatment options required the intervention to be delivered before the definitive immobilisation method was applied. Therefore, the time frame between the informed consent process and treatment was relatively short. To help raise awareness of the trial while patients waited in the clinic, sites displayed trial posters and distributed participant information sheets (PISs) in clinic waiting areas.
The attending clinician conducted a clinical examination and decided with the patient whether management would be surgical or non-surgical. If non-surgical management was appropriate, and the clinician confirmed the patient’s eligibility for the trial, the patient was informed of the trial and given a PIS. The potential participants were allowed as much time as practically possible in this type of acute injury to consider the information, and had the opportunity to ask questions of the attending clinical team and a member of the research team.
Consent was obtained by a member of the local research team who was trained in Good Clinical Practice (GCP) and authorised by the PI to take consent: this could be a research nurse, physiotherapist or surgeon at the local NHS trust, or a nurse assigned from the local National Institute for Health Research (NIHR) clinical research network. The person taking consent presented the PIS and consent form to the participant by means of a verbal discussion. The PIS detailed the exact nature of the trial, the implications and constraints of the protocol, the known side effects and any risks involved in taking part. It was clearly stated that the participant was free to withdraw from the trial at any time for any reason without prejudice to future care and with no obligation to give the reason for withdrawal. The consent form was personally signed and dated by the participant and by the person who obtained consent.
A copy of the signed consent form was given to the participant, and one copy was sent to the trial co-ordinating team in Oxford to facilitate central monitoring. The original signed consent form was retained in the medical notes, and a copy was held in the investigator site file. Consent forms were held in a secure location separately from trial data. Permission was obtained to inform the participant’s general practitioner (GP) of trial participation.
The PIS specified that a blood sample of up to a maximum of 55 ml may be taken. Only a maximum amount was stated, as detailing the exact amount of blood withdrawn per treatment group would have revealed treatment allocation; the amount drawn varied depending on treatment. The PIS also outlined that the sample remaining after treatment for the PRP injection group and the sample for the placebo group would be dispatched to a member of the central research team in the Institute of Inflammation and Ageing, University of Birmingham, for analysis. Samples were anonymised before dispatch, and identified using only the participant’s unique trial number. No laboratory results were reported back to participants or the recruiting centre.
The PIS stated that name and contact details (including mobile phone number, telephone number and e-mail address) would be collected to facilitate follow-up, full data collection and reporting of results. A copy of the contact details would be sent to the trial co-ordinating team in Oxford. These details were used by the trial team to check contact details through NHS Digital and to provide other basic trial-related information that was needed for follow-up.
Permission was sought to allow access to participant data by responsible members of the University of Oxford or the NHS trust for monitoring or audit of the trial to ensure that regulations were complied with.
The 24-week follow-up visit included the heel-rise endurance test (HRET) (primary outcome data collection). The participant was asked to consent to video-recording (without audio) of their ankle and leg movements at the time of the test. Consent for the video-recording was given on a second consent form so that the participant could consent to participation in the trial without consenting to video-recording. The participant personally signed and dated the consent form for video-recording, followed by the person who obtained consent, and the procedures for handling the form were the same as for the intervention consent form, detailed previously. Filming did not include the participant’s face; it focused on the legs, thus reducing the risk of participants being identifiable from the film. The video file (or any still photographs from it) was labelled with the unique trial number, and no identifying details were used. Permission was sought to send the video file to the trial team in Oxford, where it was held and viewed by members of the research team.
At one site (Oxford) where participants were also asked to consent to the biopsy sample collection for substudy 2, a version of the PIS was used that, in addition to the content described previously, invited the patient to attend a separate trial visit during which a small sample of tendon tissue would be removed from the injured tendon. A separate consent form that included consent to giving a tissue sample was provided for these participants. The participant was under no obligation to give a tissue sample.
Baseline assessments
Following signed consent, baseline data were collected and the participant was randomised. The injection treatment took place during the same visit in most cases. A GCP-trained member of the local research team oversaw the participant’s completion of the paper baseline questionnaire, which included:
-
background information and demographics
-
participant-reported questions including date of injury, which leg was injured, general health, current medication, allergies, smoking status, alcohol use, sport activities, age, date of birth, sex, employment status, type of employment, activities related to standing/walking/driving, any medication taken for pain or inflammation, recreational activities prior to injury, the activity that led to the torn tendon, previous rupture history, height and weight
-
-
the Patient-Specific Functional Scale (PSFS)63,64
-
participant reported – three important activities the participant was having difficulty with as a result of their ATR (range is 0–10; a higher score indicates that the participant is closer to achieving their goal)
-
-
the ATRS
-
participant reported – questions specific to ATR (range is 0–100; a higher score indicates better function)
-
-
the Short Form questionnaire-12 items (SF-12) version 2 acute
-
participant reported – without injury (recall of pre-injury function and health state) (range is 0–100 for each of physical health and mental health; a higher score indicates a higher level of health)
-
-
the SF-12 version 2 acute
-
participant reported – with injury (current function/health state) (range as for pre injury)
-
-
pain visual analogue scale (VAS)
-
participant reported – baseline report of pain prior to treatment using a VAS (range of scores is 0–100; a higher score indicates greater pain).
-
The following events took place in the clinic during the baseline data-collection period:
-
Blood sample – following randomisation, a blood sample was taken and the intervention was prepared by a member of the local team according to allocated treatment.
-
Participant contact details – contact details, including NHS number, were collected with a preferred time to be contacted to organise trial follow-up.
-
Pain diary – a pain diary was provided to the participant before leaving the clinic, to be completed at home during the first 2 weeks following injection treatment, and to be returned to the trial office by post using a Freepost account.
-
General practitioner letter – the participant’s GP was informed of their participation in the PATH-2 trial.
Randomisation
Participants were randomly allocated (1 : 1) to receive either the PRP injection or placebo. Randomisation was carried out by a GCP-trained member of the local research team, either by telephone or via a secure central computer-based system provided by the Oxford Clinical Trials Research Unit (OCTRU) randomisation service. A randomisation method with minimisation algorithm using site and age group (< 55 years and ≥ 55 years) as strata and variable block sizes was used to ensure that sites and age groups were balanced across the treatment groups. A probabilistic element of 0.8 was introduced to reduce predictability of allocation. The service was accessible by telephone (during normal office hours: 08.00 to 17.00) and via a secure randomisation website (24 hours per day/7 days per week). Details of the block sizes and allocations were confidential and were known only by the trial statistician and OCTRU programmer.
Interventions
Once randomisation had taken place and the allocation was known to the research team, a blood sample was taken from the participant and prepared as appropriate for the treatment to be delivered by an unblinded research nurse (Figure 2).
FIGURE 2.
Activities taking place on day of randomisation.

Injection treatment was delivered in the outpatient clinic by a surgeon (consultant, registrar or clinical fellow) or an extended-scope physiotherapist who was appropriately qualified, as delegated by the PI (an orthopaedic surgeon). Full training in preparing and administering the intervention was given by central trial office staff and detailed instructions were provided in an illustrated intervention and blood processing manual, which was accessible to local staff when preparing the intervention and managing samples.
For participants in the PRP group, 5 ml of venous blood was drawn off into a 5-ml syringe, followed by 50 ml of venous blood into a 60-ml syringe that contained 8 ml of anticoagulant. The blood was drawn slowly to avoid platelet agitation and early activation. The participant was asked to wait in an adjoining room while the intervention was prepared. The 60-ml syringe was fitted into the trial-specific centrifuge provided to all sites (MAG-200 MAGELLAN® Autologous Platelet Separator, Arteriocyte Medical Systems, Hopkinton, MA, USA). A sterile, disposable PRP kit (MDK 300/MDK 300-1 platelet separation chamber, Arteriocyte Medical Systems, Hopkinton, MA, USA) was placed in the centrifuge, and the centrifuge was set to run to produce 8 ml of PRP, which was fed into a 10-ml syringe.
For participants in the placebo group, 5 ml of venous blood was drawn off into a 5-ml syringe. The participant was asked to wait in an adjoining room while the intervention was prepared. An empty syringe was prepared. The participant was not approached with the placebo intervention until a length of time had passed, similar to the time taken to produce PRP.
For both groups, the participant was asked to return to the treatment room and to lie prone on a treatment bed with the tendon exposed. The tendon gap was palpated clinically to determine the injection site and the area was cleaned. Local anaesthetic (1–2 ml) was administered to the skin.
Participants in the PRP group were given a 4-ml injection of PRP into the tendon rupture gap. Participants in the placebo group received a placebo injection: a dry needle of the same size was introduced via the skin into the tendon tissue, held in the skin briefly and withdrawn without injecting anything so that the biological haematoma was minimally disturbed.
For both groups, the injection area was covered with a dressing and the participant had application of a plaster cast or an orthopaedic boot with the ankle immobilised in the equinus (plantarflexed) ankle position.
If a participant randomised to the PRP group was unable to receive a PRP injection for any technical reason or if blood withdrawal failed, they received a placebo injection. This was recorded, and, whenever possible the participant remained blinded to knowledge of their treatment group. Trusts followed their own policy to manage deep-vein thrombosis (DVT) prophylaxis.
After injection delivery
The centrifuge produced 8 ml of PRP, of which 4 ml was injected into the participant’s Achilles tendon; 4 ml remained for laboratory analysis (see Chapter 4 for further details of the preparation of these samples). Immediately after the intervention, a member of the local team prepared the remaining PRP (PRP group) and the 5-ml whole-blood sample (PRP and placebo groups) for storage or dispatch. Samples were sent to a member of the central team at the University of Birmingham research laboratory. A single, specialised courier (Davies International, Hampshire, UK) was used for this purpose and a pre-paid account was established with the courier to facilitate ease of sample transport.
The 4 ml of PRP that remained after injection (PRP group only) was prepared for dispatch to the laboratory; 1 ml of PRP was transferred to a microtube and stored at the site in a –70 °C freezer within 2 hours of PRP production. These samples were collected by the dedicated courier at the end of recruitment and transported to the central laboratory for analysis for substudy 1.
In the PRP group, 1 ml of PRP was transferred to a microtube and placed in a trial-specific courier pouch; 2 ml of PRP was transferred into activation vials from a proprietary platelet-activation kit (CB kits, Platelet Solutions Ltd, Nottingham, UK) supplied by the central trial office for platelet activation and fixation. The platelet activation and fixation took around 5 minutes and full instructions were given in the intervention and blood processing manual. When the process of platelet activation and fixation was complete, these two tubes were placed in the courier pouch.
For both groups, the 5-ml venous whole-blood sample was transferred to a tube containing ethylenediaminetetraacetic acid, an anticoagulant, and placed in the courier pouch. Thus, the courier pouch contained four tubes if the participant was randomised to PRP (three tubes containing PRP and one tube containing blood) and one tube if the participant was randomised to placebo (blood only).
The courier pouch was packaged in accordance with instructions in the intervention and blood processing manual and the site contacted the courier for collection and next-day delivery service. Until collection, the courier package was stored at room temperature.
Preparation of PRP and samples and courier dispatch were recorded on a blood tracking sheet. The original version of this form was enclosed with the courier pouch for dispatch to the laboratory. A copy was sent separately to the central trial office and a copy was retained at the site. Blood and PRP analysis results were not reported back to the recruiting centre as they did not have an impact on future treatment.
Injection training
Training in the delivery of both the PRP injection and the imitation injection was provided by the trial team and recorded on a PATH-2 trial training form (treatment related), which was signed by the clinician receiving training and retained at the site. Training consisted of the provision of a training manual for all sites. At most sites, a training video or training session by a PATH-2 trial trainer using the trial kit and trial procedures was provided in addition to this manual. The PI at each site identified surgeons (or extended-scope physiotherapists) to be trained, and recorded those who completed training on the site delegation log. Only those individuals who were trial trained and listed on the delegation log were able to carry out trial treatments. When the delegation log was updated, a copy was sent to the trial co-ordinating office in Oxford. The PI at each site identified local staff who were responsible for preparing the blood and PRP samples for treatment and for later dispatch. Only those individuals who were listed on the delegation log as having this responsibility were able to carry out preparation of the blood and PRP and to manage blood and PRP samples.
Monitoring intervention delivery
Intervention delivery was monitored centrally by PATH-2 trial staff. Site staff documented the intervention, including preparation and processing of samples, using a blood sample and treatment case report form (CRF) and a blood tracking sheet. Site staff were trained in completion of these forms by central trial staff. The original blood tracking sheet was sent with the samples to the Birmingham laboratory as supporting documentation for substudy 1. Copies of these completed forms were returned to the trial office and copies were kept at the site.
On receipt of the two forms, trial office staff checked that the forms were fully and correctly completed, that the intervention had been carried out by an individual whom the PI had identified on the delegation log as suitably qualified and that the procedures had been completed within the time frame specified in the intervention and blood processing manual. If any problems had occurred during the preparation of the samples, intervention delivery or sample processing, the site was contacted to investigate whether or not a protocol deviation had occurred and if additional training was required.
Rehabilitation
All participants received standard care for their injury in accordance with local site procedures. Immediately after treatment, the participant’s ankle was placed in a splint, orthopaedic brace or plaster cast. Rehabilitation was prescribed by local members of the clinical and physiotherapy teams. However, it was necessary to standardise key elements of rehabilitation in order to reduce the risk of efficacy interference due to substantial variation in rehabilitation protocols. The standardisation for PATH-2 participants involved:
-
ankle immobilisation in the equinus ankle position in a splint, orthopaedic brace or plaster cast for a minimum of 3 weeks
-
referral to physiotherapy for rehabilitation
-
avoiding more than 6 weeks of rigid full-time immobilisation without ankle motion or weight-bearing.
Adherence to these guidelines was monitored by asking participants questions about their progress with rehabilitation. Standardisation of specific elements of rehabilitation, such as the splinting method, when weight-bearing commenced or specific exercises, was not mandated.
Outcomes
Primary outcome
The primary objective of the trial was to evaluate the clinical efficacy of PRP in acute ATR in terms of muscle–tendon function. The primary outcome measure at 24 weeks post randomisation was the Limb Symmetry Index (LSI) of the work performed by each lower limb in joules (J) during the HRET. The HRET is a validated objective measure of Achilles tendon muscle unit function. 65
The HRET involves repetitive concentric–eccentric muscle actions of the plantar flexors in a single-leg stance until exhaustion, with performance quantified as (1) total work in J, (2) number of repetitions and (3) maximum heel-rise height in cm. 65 Total work was selected as the primary outcome measure because this index provides a measure of plantar flexor muscle endurance and Achilles tendon function as it incorporates the maximum height of each repetition. Total work (J) was computed as the product of body mass (kg), total vertical displacement (m) and the constant 9.807 converting kilopond metres to joules. To quantify performance, a linear displacement sensor (MUSCLELAB™, Ergotest Innovation A.S., Porsgrunn, Norway) was attached to the participant’s heel during the test and recorded on software on a laptop computer. A PATH-2-specific user interface version of the MUSCLELAB software was developed with the manufacturer for ease of use in a multicentre trial. Key features of this software and more background on the HRET development are described in further detail in Appendix 2.
Heel-rise endurance test procedure for PATH-2
At the 24-week follow-up visit, the HRET procedure was explained to each participant by the blinded outcome assessor. Standardised participant information and instructions for the HRET involved the participant watching a video demonstration of the HRET and reading standardised written instructions detailing their expected conduct during the test and the test termination criteria. The standardised warm-up involved 5 minutes of usual-pace walking followed by 10 double-leg heel raises. Before testing each leg, the participants were asked to stand on the box being used and were allowed to familiarise themselves with the expected timing of heel raises by lifting both heels together.
The HRET was first carried out on the uninjured limb and then on the injured limb. The test started with the participant standing on one leg on a 10° incline box (so that the ankle was in a dorsiflexed position) with the cord from the linear encoder strapped to the participant’s heel (Figure 3).
FIGURE 3.
A participant performing the HRET. The participant stands on 10° incline box with a cord attached to their heel and connected to a linear encoder, and raises and lowers their heel repeatedly until fatigued.
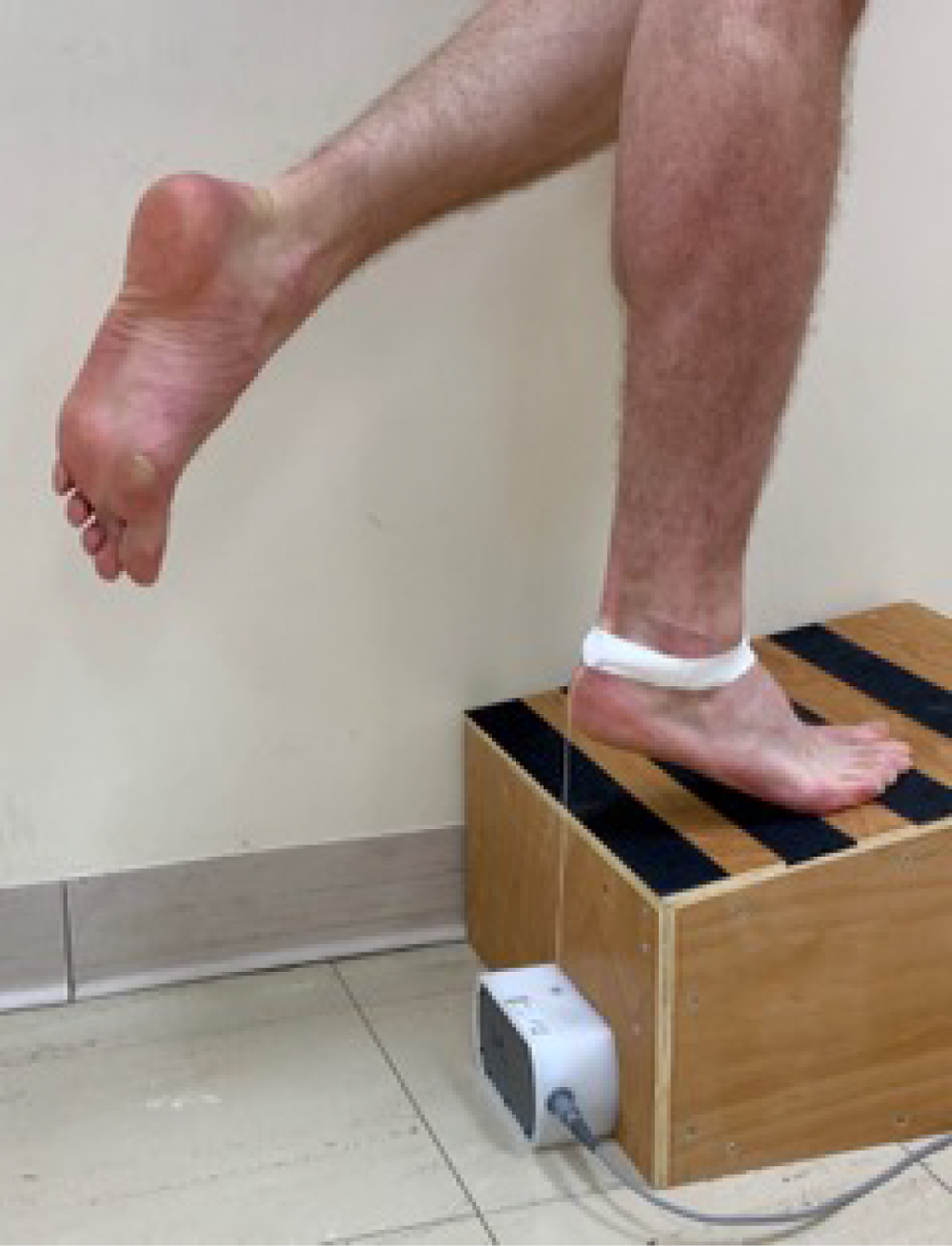
The following standardisation parameters were adopted:
-
Ankle starting position of 10° dorsiflexion, produced by conducting the HRET on a custom-made 10° incline box.
-
Knee starting position of full extension.
-
Height of each repetition to be as high as possible.
-
Pace of 30 raises per minute, guided by a digital metronome.
-
Balance support by the fingertips only.
-
Strictly defined test termination criteria – participants either stopped (i.e. volitional task failure) or were audibly instructed to stop with both feet flat on the box whenever any of the following test termination criteria were observed: (1) inability to keep pace with the metronome, (2) inability to maintain full knee extension of the standing leg or (3) using more than fingertip support. The desired end point was volitional task failure; however, outcome assessors were encouraged to use verbal prompts whenever the termination criteria were observed and to stop the test if the participant did not respond to two consecutive prompts.
Secure data transfer and confidentiality were assured by copying the HRET data from the encrypted laptop to an encrypted Universal Serial Bus (USB) drive that was sent to the trial office. Sites retained a copy of the entire site HRET data set on the dedicated trial laptop.
Assessor training material consisting of high-quality training videos, which were made by the PATH-2 trial team and produced by Oxford Medical Illustration (Oxford University Hospitals NHS Foundation Trust, Oxford, UK), and a training and reference manual. Face-to-face training was delivered by a member of the PATH-2 trial team to each outcome assessor prior to their first participant’s 24-week follow-up appointment.
The linear encoder, a very sensitive device, recorded minimal movements that might not represent actual heel rises; it could also pick up any additional movements of the heel (e.g. participants tend to step off the box or lift the leg up at the end of the test). To dismiss potential measurement errors, two members of the trial team, who were blinded to treatment allocation, independently reviewed videos of all assessments for which participants consented to recording. The invalid heel-raise repetitions in the HRET data were identified so that could they could be dealt with in the analysis (see Statistical methods).
Secondary outcome measures
The secondary outcome measures were as follows:
-
The ATRS,66 a validated patient-reported outcome measure (PROM) incorporating 10 questions relating to muscle strength, fatigue, pain and function at 4, 7, 13 and 24 weeks post treatment. The score range is 0–100.
-
The SF-12 version 2 acute, a health-related quality-of-life PROM67 at 4, 7, 13 and 24 weeks post treatment. The scale consists of a 12-item short questionnaire that assesses eight different health domains reflecting both the physical health (physical functioning, role participation with physical health problems, bodily pain and general health) and the mental health (vitality, social functioning, role participation with emotional health problems and mental health) of the evaluated participants.
-
A daily pain diary, reported by the participant for 14 days beginning on the day of treatment, using a VAS. The participant was asked to place a vertical mark on a 100-mm horizontal line between the extremes of ‘no pain’ and ‘worst pain imaginable’. Trial office staff measured the distance to the vertical mark and converted the distance to a daily measure (0–100) of pain.
-
The pain component of the ATRS at 4, 7, 13 and 24 weeks post treatment. In the ATRS, the participant was asked ‘Are you limited due to pain in the calf/Achilles tendon/foot?’ and invited to circle an integer from 0 to 10 inclusive, 0 indicating major limitations/symptoms and 10 indicating no limitations/symptoms.
-
The PSFS,63 a PROM indicating progress on self-selected recovery goals relating to daily activities affected by the injury on an 11-point scale (0–10), at 4, 7, 13 and 24 weeks post treatment.
-
The maximum number of repetitions (heel rises) recorded during the HRET68 at 24 weeks post treatment.
-
The maximum vertical displacement (cm) recorded during the HRET68 at 24 weeks post treatment.
Adverse events
Complication events reported by participants were explored at two levels: serious adverse events (SAEs) and adverse events (AEs).
Serious adverse events
A SAE is an untoward medical occurrence that:
-
results in death
-
is life-threatening
-
requires inpatient hospitalisation or prolongation of existing hospitalisation
-
results in persistent or significant disability/incapacity
-
is a congenital anomaly/birth defect
-
is another important medical event.
Adverse events
Adverse events were events that were related to the trial treatment or conditions that did not require specific time-critical reporting but were collected as part of standard data collection in the PATH-2 trial. AEs were broken down further into foreseeable and unforeseeable AEs.
Foreseeable AEs:
-
bruising and discomfort at the venesection site
-
mild discomfort or minor bleeding from the ATR site following injection
-
technical complications of the lower-leg casting and splinting
-
consequences of depending on walking aids
-
syncopal (fainting) episode associated with venesection or tendon injection
-
discomfort at the ATR site during rehabilitation
-
swelling or bruising of the lower leg and foot
-
deep-vein thrombosis in a lower limb
-
re-rupture of the treated Achilles tendon (including any surgery on the Achilles tendon treated in the trial).
Unforeseeable AEs:
-
serious infection at the ATR injection site
-
skin breakdown or ulceration of the treated lower leg other than ‘plaster sores’
-
severe pain requiring more than simple analgesia beyond 10 days after injection.
Events that were related to trial treatment and that were either foreseeable or unforeseeable were defined as AEs.
Serious adverse events were reported to the chief investigator by the local research team using a SAE form within 24 hours of their becoming aware of the event, the PI having assessed causality and relatedness.
Adverse events were reported to the central trial office on an AE form by site staff. In addition, participants were asked in follow-ups to report any type of AE that they had experienced.
Blinding
Blinding during treatment preparation and intervention
This was a double-blinded (participant and primary outcome assessor) trial. Participants remained blind to allocation throughout the trial. Those staff involved in treatment delivery were aware of treatment allocation because of the nature of the intervention. The primary outcome assessors were not aware of the allocation or treatment received.
Site staff were trained consistently to maintain blinding. Care was taken not to draw the participant’s attention to the amount of blood taken as this could have indicated the intervention group. To facilitate treatment blinding, preparation of the intervention took place in a location away from the treatment room, or the participant waited outside, as there was a difference in the number of consumables handled depending on treatment allocation. When the injection was prepared, it remained out of the participant’s view.
If a participant randomised to the placebo group was within listening distance of the centrifuge, the centrifuge was run on a dummy cycle. The wait time for the participant was approximately 17 minutes; this was the time taken to run the centrifuge to produce PRP. During the interventions, the participant was requested to lie face down with their foot and ankle slightly off the edge of the bed, and there was an option of placing a pillow on the back of the participant’s shoulders below the neck to help visually shield the intervention procedure. The verbal cues used by site staff at the time of treatment delivery did not refer to allocation and were the same for both groups.
Full guidance on ensuring that the correct amount of blood was withdrawn into the appropriate tube or syringe while ensuring that the participant remained blinded to allocation was provided in the PATH-2 trial intervention and blood processing training materials, which were retained in an area where they were not visible to the public.
Facilitating blinding immediately post treatment
The PATH-2 blood sample and treatment CRF collected confirmation that allocated treatment was received. When a treatment other than that allocated was indicated on the CRF, the trial co-ordinating team contacted the site for further details and recorded the details. Additional training or troubleshooting of any issues was instigated when necessary.
In hospital notes and in the letter to the participant’s GP, it was recorded that an injection was delivered according to the random allocation assigned, but the type of injection was not recorded.
To assess the completeness of participant blinding, after the 24-week trial questionnaire and HRET assessments were completed, participants were asked which treatment they believed they received (‘PRP injection’, ‘imitation injection’ or ‘don’t know’) and why they believed this, along with questions around their experience of taking part in the trial. These data were used to compute two blinding indices with 95% confidence intervals (CIs) (James et al. 69 and Bang et al. 70 blinding indices).
Blinded follow-up assessors
Throughout the trial, follow-up was carried out wherever possible by blinded assessors unaware of treatment allocation. A physiotherapist or assessor who was blind to allocation carried out the 24-week trial follow-up assessment, including the HRET.
Sample size
Sample size
The trial was powered on the work LSI from the HRET at 24 weeks post randomisation and on the ATRS. There were no formal interim analyses of the outcomes planned for the PATH-2 trial. However, the Data and Safety Monitoring Committee (DSMC) agreed to check the sample size assumptions when at least half of the originally planned number of participants had completed the 24-week primary end-point assessment because they were based on the findings of a single previous study conducted in a single centre. 65 This decision was documented in the statistical analysis plan version 1.0.
Original calculation of sample size
It was calculated that 214 participants (107 per treatment group) would provide 90% power to detect a standardised difference of 0.5 in the HRET work measured by the work LSI at 24 weeks post randomisation and with 5% (two-sided) significance allowing for 20% loss to follow-up. This was based on previous data from the non-surgical arm of the 2010 study by Nilsson-Helander et al. ,65 in which a clinically important difference of 10 points with a standard deviation (SD) of 20 points was observed.
This sample size was also to provide 90% power and 5% (two-sided) significance to detect a standardised effect size of 0.5 in the ATRS between the two treatment groups, based on a difference of 11% and a SD of 21.4%.
Sample size review and recalculation
The DSMC agreed at their first meeting that the assumptions for the sample size should be reviewed after approximately half of the participants had been recruited. This review would be a blinded estimate of work LSI variability only. The first variability analysis was undertaken in October 2016 when half of the participants had been recruited (n = 104). It was observed that the SD of the work LSI was smaller than that used in the sample size calculation so no changes to the sample size were recommended (observed SD 17.37, versus sample size SD 20). However, at this time only 27 participants had completed the primary end-point assessment.
As of June 2017, approximately half of the participants had reached their primary end point and recruitment was still ongoing. Therefore, to provide a more robust estimate of the actual variability of the work LSI, a further sample size assumption review was carried out. Using cleaned and validated data from 75 participants, the observed SD was 24, which was higher than that used in the sample size calculation. Based on this observed SD of 24 (being aware that this could go up or down with the addition of further participant data), we recalculated the sample size based on 80% power, which, allowing for 20% loss to follow-up, required a sample size of 226 patients. The DSMC advised the Trial Steering Committee (TSC) and trial management team during this process.
The trial continued to recruit to an agreed minimum of 230 participants (overshooting to account for further minimal fluctuations in sample size assumptions), to ensure that there would be a minimum of 80% power for the primary outcome analysis. These approaches, and the reduction in trial power, were supported by the TSC and by the funder.
Statistical methods
Software employed
All analyses outlined here and reported in Chapter 4 were undertaken using Stata® version 15.0 (StataCorp LP, College Station, TX, USA).
Blinded analysis
Initial exploratory analyses were conducted on a blinded data set (not separated by treatment group) to identify the presence of missing values and to clarify the distribution of continuous variables. These analyses were also used to finalise trial populations and to aid in the identification of key prognostic variables to be included in the adjusted analysis. All subsequent analyses described were conducted on an unblinded data set. Following this blinded analysis, the statistical analysis plan was updated to incorporate any changes.
Data validation
Initial analytical steps assessed the validity of the final, blinded data set. The first of these steps was to manually check, when possible, the use of Stata for importing data and merging data sets for at least 20 participants who were randomly sampled. Once accurate importation was confirmed, data in the data set were validated by checking for duplicate records, checking the values of the range of variables and validating potential outliers against CRFs. Any discrepancies that could not be rectified were referred back to the trial sites. Finally, the production of calculated variables using Stata was manually checked for at least 20 participants who were randomly sampled.
Trial populations
Intention-to-treat population
This required the inclusion of all randomised participants, to be analysed in the groups to which they were allocated. For the primary outcome, analysis included a modified intention-to-treat (mITT) population, defined as all randomised intention-to-treat (ITT) participants with available work LSI data (i.e. at least one valid repetition for each lower limb after HRET data validation by the blinded reviewers of the assessment video files). For the secondary outcome, analysis also included a mITT population, defined as all randomised participants who completed their 24-week follow-up questionnaires, analysed in the groups they were allocated to.
Complier-average causal effect population
Complier-average causal effect (CACE) is an analytic approach that provides a robust estimate of the treatment effect among compliant participants. 71,72 Participants who received a ‘poor-quality’ PRP injection (e.g. received PRP with concentrations of platelets lower than the concentration in their whole blood or did not receive the PRP) in the PRP group were classified as not having received their allocated treatment.
Descriptive analysis
All participants were included in descriptive analyses. Recruitment into the trial was explored, including the numbers of individuals assessed, recruited, randomly assigned to the PRP injection or placebo, receiving treatment, completing the trial protocol and analysed for the primary outcome. Any protocol deviations and violations were investigated and issues with the screening data, including potential incidents of ineligible patients being randomised, were explored. The proportion of unblinded assessors for each assessment was assessed, and the James et al. 69 and Bang et al. 70 indices were calculated to determine whether or not participants were successfully blinded to the intervention they received.
The baseline comparability of participant-level data for each of the treatment groups was summarised. For baseline and follow-up data collected manually through the use of forms or questionnaires, data compliance was explored; all available data collected from these forms were summarised and the proportion of missing items from completed questionnaires was examined. Data-availability patterns for individual variables were assessed both overall and for the two treatment groups separately to explore missing information. Missing values were checked for consistency and the proportion of missing values per variable was assessed. Differentiations were made between partially completed and fully missing outcome data. For measures comprising multiple items, for example SF-12, pro-rata estimation of total and subscale scores was employed for each treatment group, using the appropriate scale-specific scoring guidelines. Imputation of data was utilised for sensitivity checking and is described in further detail in Primary outcome. Imputation was carried out as recommended for each relevant questionnaire, with the imputation value determined by the distribution of the underlying data. Methods utilised for imputation were assessed manually, when possible, for at least 20 participants who were randomly sampled. Sensitivity analyses were conducted to ensure that the missing-at-random assumption for imputation was met.
Comparisons of losses to follow-up were carried out. The proportions of participants defaulting or withdrawing from the trial over the whole period of trial follow-up, and at each analysis time point, were compared between the PRP and placebo groups. The importance of differences identified were assessed using a chi-squared test. For all analyses, a p-value of < 0.05 was considered indicative of a significant difference.
Quality assurance and compliance with the intervention, including any deviations from protocols prior to and during the trial, were assessed. Treatment received was summarised by treatment groups, and time frames for the steps with blood processing were explored to ensure that all participants received their PRP injection within 2 hours of their blood sample being taken. The grade of the health-care professional conducting the procedure was summarised. The importance of apparent differences in compliances was assessed using a chi-squared test.
Finally, data quality and the effect of the treatment received were assessed using CACE analysis in place of the originally planned per-protocol (PP) analysis, as this was deemed the optimal approach. As per the PP analysis, CACE analysis allows for adjustment to account for participants whose received treatment either complied or did not comply with that allocated to them; however, CACE analysis does so without assuming that compliers are the same as non-compliers, as would be the case with a PP analysis. CACE analysis retains information on the original treatment allocation that participants received, which allows us to forgo the core assumption of a PP analysis – that receipt of treatment is random with respect to the outcome predictors – and to enhance our understanding of the effect of PRP on ATR recovery. The decision to use CACE was taken post finalisation of the statistical analysis plan but ahead of the end of follow-up and, therefore, ahead of unblinding.
For the CACE analysis, compliance was defined as participants who received the intervention allocated to them who had sufficient platelets present in their PRP sample to classify it as platelet rich (i.e. higher platelet concentration in PRP than in whole blood). The results from the CACE analysis were reported with standard errors, p-values and 95% CIs, and assessments were made to ensure that the primary outcome estimation was captured within this range.
Standard care for ATR for all trial participants was unaffected by this trial. All participants should therefore have had their injured lower limb immobilised, received a referral for physiotherapy, received delivery of venous thromboembolism prophylaxis and received advice about when to start weight-bearing and ankle-motion exercises. Compliance with these different aspects of standard care was compared between treatment groups using a chi-squared test for categorical data and a Student’s t-test for continuous data.
Analysis of the primary outcome
The primary outcome for this trial was muscle–tendon function, measured by the LSI of maximum work, performed during the HRET (see Primary outcome for more detail). The work LSI was calculated for all trial participants, at 24 weeks (varying by –2 to +8 weeks) post randomisation, as follows:
The proportions of individuals missing data for outcome and explanatory variables were explored, and work LSI data for all participants were assessed for normality. Missing HRET data were defined and handled as follows:
-
Participants with true missing data – participants who were lost to follow-up and participants for whom technical errors were experienced during HRETs. The participants who were lost to follow-up were any participants who either did not attend their 24-week follow-up at all or did not remain at their appointment to complete their HRET. The measurements for these participants were kept as missing.
-
Participants with true zero measurements – participants who attempted to complete their HRET assessment but their attempts were insufficient for the encoder to record any results. Zero measurements for these participants were included for analysis.
-
Participants with potential zero measurements – participants who did not attempt to complete their HRET in at least one leg, despite attending the follow-up appointment. Zero measurements for these participants were included for analysis.
The data followed a near-normal distribution, with a small elevation around zero due to participants defined as having true and potential zero measurements. However, work LSI data did not require transformation prior to inclusion in the final model as post-estimation assessments indicated that the distribution was suitable for the regression technique employed. The impact of zero inflation of the primary outcome measure was assessed using a two-parts model to (1) identify differences between participants with positive work LSI results and participants with zero measures, and (2) identify the impact of the inclusion of participants with zero measures on the primary outcome results. Mean work LSI differences, robust standard errors, p-values and 95% CIs were reported and compared with the core primary outcome results for deviation or interpretation changes.
An unpaired Student’s t-test was used to explore the unadjusted effects of PRP compared with those of placebo.
Multivariate linear regression was used to investigate the effect of PRP on ATR recovery. The base-unadjusted regression model was built using work LSI as the dependent variable and treatment as the key independent variable. The principal analysis model built on this model by adjusting for participant age group and clustering by trial site. Supplementary regression models were built, which adjusted for the effects of sex, body mass index (BMI) and smoking status.
Sensitivity analyses were carried out using imputation of values for missing HRET data to examine the robustness of the conclusions made from the analyses to address the primary aims. Missing HRET data were handled using two approaches. The first was to employ simple imputation of the average concentric displacement (upwards movement); this method was used for missing data associated with specific repetitions of heel rise. The second method to handle missing data was to employ multiple imputation by chained equations; this was used to calculate work LSI for participants with data missing for entire assessments. The multiple imputation using chained equations procedure created a series of complete data sets (observed plus imputed data) in which analyses were carried out individually. The regression parameter estimates plus corresponding standard errors obtained from the analyses of each of the 50 imputed data sets were then combined using the Rubin’s rule approach. When models did not converge during the multiple imputation process, some predictor preselection was carried out based on clinical expertise and subject matter. Sensitivity analyses were employed to assess the assumptions made in preselecting these predictor variables. The method utilised for multiple imputation inclusion was to use the stratification variables already in the principal adjusted model and to further account for any variables with an R2 value of ≥ 0.2 following correlation assessments. This resulted in three additional variables being selected: whether or not the participant jogged or ran, undertook weight training or participated in squash before their injury.
For all analyses, a two-sided p-value of 0.05 (5% significance level) was used to indicate evidence of statistical significance.
Analysis of secondary outcomes
The number of repetitions and the maximum heel-rise height recorded during the HRET assessment were explored as secondary outcome measures. Furthermore, four PROMs [pain as measured by the VAS (daily over the 14 days immediately post treatment), ATRS, PSFS and the SF-12 mental and physical health scores] were explored.
The ITT population was used for secondary outcome analyses.
Repeated-measures linear mixed-effects regression models were used to allow the data collected at all follow-up time points to be taken into account. Time elapsed from the intervention to the outcome measurements was included in the models as a random effect factor, considering that not all participants had their follow-up assessments at exactly the same time. Mean differences and 95% CIs were examined. All analyses were adjusted for the stratification factors included in the adjusted analysis of the primary outcome. As pre-injury scores had been collected for the SF-12 data, the models for this PROM were built so that the principal and supplementary analyses adjusted for these scores.
Sensitivity analyses were carried out on the overall ATRS data sets as this was the key secondary outcome. The first of these analyses used the mITT population used in the primary outcome analysis to assess the impact of selecting the mITT population used in the primary outcome analysis. The second applied multiple imputation by chained equations for those participants with data missing with missing ATRSs. The multiple imputation by chained equations technique that was applied to the ATRSs was the same as for the primary outcome, although the additional variables selected for inclusion to inform the imputation were different. The six additional variables used to build the imputation data sets in this situation were BMI as a continuous variable, the date of the ATR, the date when the treatment was received, the date when a participant started carrying out rehabilitation exercises (as recorded at week 13), whether or not a participant was jogging or running by week 24 and whether or not a participant was carrying out ‘do it yourself’ (DIY), heavy housework or gardening by week 24.
As with the primary outcome analysis, data quality was assessed using CACE on the overall ATRS data, to examine the robustness of the conclusions made from the analyses.
Adverse events
The number of participants experiencing one or more AE of any type over the 24 weeks was explored. The number of participants experiencing each specific type of AE over the 24 weeks was also analysed. The number of separate complication events was not explored owing to the opportunity for re-reporting by participants across the different follow-up time points and the nature of the events of interest.
Patient and public involvement
The Efficacy and Mechanism Evaluation programme recognises the value of active involvement of public contributors in research. In our patient and public involvement (PPI) work, we consulted trauma injury patients at John Radcliffe Hospital. Our aim was to ensure that:
-
interventions were acceptable to patients
-
follow-up procedures reduced the patient burden while obtaining meaningful scientific data
-
patient information sheets and questionnaires were easy to read and user-friendly.
The protocol73 was designed following a small-scale pilot study with the help of Achilles tendon injury patients at the John Radcliffe Hospital and the Oxford Trauma User Group (OTUG), which is composed of patients who have experienced the John Radcliffe Hospital orthopaedic trauma service and who volunteer to engage on clinical and research issues. Recommendations of OTUG members were incorporated in the original research proposal.
A questionnaire was sent to the OTUG members to explore opinions regarding the trial aims and procedures; 75 out of 105 replied to the survey. All responders indicated that the project might benefit patients with ATR. Ninety per cent were happy with the trial design; some felt that the number of follow-up contacts could be inconvenient. Taking that feedback, and experience of the pilot study,74 into account, follow-up procedures were refined to reduce the number of follow-up contacts, thereby reducing participant burden.
Participant satisfaction with the intervention was explored in the pilot study. All 20 patients tolerated the injection well and no side effects were reported. The use of needle biopsy under local anaesthetic, as in substudy 2, was also reported during the pilot study to be acceptable.
A patient representative joined the TSC as a PPI representative to contribute to management of the trial. One of the co-applicants acted as a mentor to the PPI representative, being available by telephone before and after the meetings to help with interpretation of trial briefing documents. This provided an opportunity for the PPI representative to raise issues prior to meetings, which she did, and if necessary the mentor could raise issues on her behalf. All committee members were requested to use plain English in meetings.
The PPI representative was also actively involved in designing the approach to patients and the text of patient-facing materials (questionnaire and letters) and for the 2-year extended follow-up; the latter is ongoing and not reported as part of this trial.
Members of the trial management team attended the INVOLVE conference and PPI research meetings.
Outcomes for patient and public involvement
In the main trial, participants reported satisfaction at the end of the trial in a post-assessment questionnaire. We asked participants to rate their satisfaction with the trial on a five-point scale of ‘very dissatisfied’ to ‘very satisfied’. Eighty-nine per cent of participants declared themselves ‘satisfied’ or ‘very satisfied’. We invited comments on what could have improved their experience; the vast majority who answered (n = 37) said ‘nothing’ and none asked for fewer forms, although five said that some questions were not relevant and three asked for more information.
An adjustment to the statistical analysis related to how the pain score would be reported is an example of how feedback from our PPI representative improved the conduct of the trial.
The PPI representative reviewed a graphical information slide to be used in future public-facing presentations.
Overall, from participant feedback, we believe that our PPI involvement improved trial conduct and participant experience.
Ethics approval and monitoring
Ethics approval
The trial was given a favourable opinion by the South Central – Oxford A Research Ethics Committee on 11 November 2014 (reference number 14/SC/1333) and each site was granted site-specific approval from its NHS trust research and development department before trial commencement.
Data and Safety Monitoring Committee
The DSMC was established to safeguard the interests of trial participants, monitor the main outcome measures including safety and efficacy, and monitor data quality and completeness. In accordance with the DSMC charter, the DSMC received and reviewed information on the progress and data collection of the trial and advised the TSC on the conduct of the trial.
Trial Steering Committee
The TSC provided expert oversight of the trial on behalf of the sponsor and funder. Through its independent chairperson, the TSC also provided advice to the Trial Management Group (TMG), the funder and OCTRU on all aspects of the trial.
Trial Management Group
The day-to-day management of the trial was overseen by a TMG comprising the chief investigator, the lead research nurse at the lead site (John Radcliffe Hospital), the co-applicants, the trial statistician, research physiotherapists and the trial manager.
Trial registration
This trial is registered with the International Standard Randomised Controlled Trial Number (ISRCTN) Registry (ISRCTN54992179) and ClinicalTrials.gov (NCT02302664).
Summary of changes to the trial protocol
The changes to the project protocol are summarised in Table 1.
| Protocol version | Date issued | Details of changes made |
|---|---|---|
| 1.0 | 10 September 2014 | Not applicable as this was the first issue |
| 2.0 | 12 February 2015 |
|
| 3.0 | 8 March 2016 |
|
| 4.0 | 8 March 2016 | Removal of ‘draft’ from filenames, removal of draft watermark and removal of tracked changes from documents for version 3.0 |
| 5.0 | 21 April 2017 |
|
| 6.0 | 24 July 2017 |
|
Chapter 3 Clinical trial results
Trial participants
A total of 230 eligible participants were recruited from July 2015 to September 2017 from 19 NHS hospitals across England and Wales. Participants attended clinic visits at the time of randomisation (baseline) and at 24 weeks post intervention, with the last 24-week visit taking place in March 2018. Participants were also contacted by the trial team via telephone to complete follow-up questionnaires at 4, 7 and 13 weeks post intervention. The trial ended when the recruitment target of 230 was reached.
One participant withdrew from the trial. This participant withdrew following randomisation but prior to receiving any intervention. We therefore do not have baseline or follow-up data relating to this participant. The reason given for withdrawal was that the participant had decided that they would prefer a surgical intervention for their ATR. This participant had been randomised to the PRP injection group.
The number of participants at each stage of the PATH-2 trial, from assessment for eligibility through to analysis, is presented in the Consolidated Standards of Reporting Trials (CONSORT) flow diagram in Figure 4.
FIGURE 4.
The PATH-2 trial CONSORT flow diagram.

Comparisons of losses to follow-up
A total of 27 participants (27/229, 11.8%) were lost to follow-up by the point of completing their HRET assessment; these participants therefore did not contribute to the primary outcome analysis. Of these participants, 24 (24/229, 10.5%) were true losses to follow-up, with 12 in the placebo group and 12 in the PRP group. Of the 24 participants who failed to complete their HRET assessment:
-
Eleven participants (11/24, 45.8%) completed all follow-up questionnaires up to and including their 24-week questionnaire but failed to remain to complete the assessment.
-
Two participants (2/24, 8.3%) completed their 4-, 7- and 13-week follow-up questionnaires but not their 24-week questionnaire.
-
Four participants (4/24, 16.7%) completed their 4- and 7-week follow-up questionnaires but did not complete their 13- and 24-week questionnaires.
-
Two participants (2/24, 8.3%) completed only their 4-week follow-up questionnaires.
-
One participant (1/24, 4.2%) completed only their 24-week questionnaire.
-
Four participants (4/24, 16.7%) completed no follow-up past the baseline questionnaire.
These participants were spread across 10 sites and there were no discernible differences between them in relation to their age, sex, BMI or smoking status.
In addition to these 24 participants, there were three participants (3/229, 1.3%) who were missing data for their HRET assessments following a technical error. These participants therefore do not contribute to the primary outcome analysis. However, it is worth noting that, although one of these participants did not complete their 13-week follow-up questionnaire, all other questionnaires were completed.
There were no differences between treatment groups in whether or not a participant was lost to follow-up, irrespective of whether participants who experienced a technical error were included in this cohort [PRP injection, 12/113 (10.6%), vs. placebo, 15/116 (12.9%), difference –2.3%; p = 0.588] or not [PRP injection, 12/113 (10.6%), vs. placebo, 12/116 (10.3%), difference 0.3%; p = 1.000].
Trial populations
A total of 229 participants are included in the descriptive statistics, 113 of whom were in the PRP group and 116 of whom were in the placebo group.
Intention-to-treat population
-
Primary outcome (work LSI): a total of 201 participants were included, 100 of whom were in the PRP group and 101 of whom were in the placebo group.
-
Key secondary outcome (ATRS): a total of 216 participants were included, 107 of whom were in the PRP group and 109 of whom were in the placebo group.
Compliance
Baseline, randomisation and treatment delivery forms
There were no differences between treatment groups in compliance with baseline, randomisation and treatment delivery forms. Completion rates for all of these forms were 100% for both treatment groups.
Follow-up diaries and questionnaires
There were no differences between treatment groups in compliance with follow-up questionnaires and diaries. A total of 180 out of 229 participants (78.6%) returned their pain diary for the first 2 weeks post intervention. Completion of all follow-up questionnaires at weeks 4, 7, 13 and 24 was ≥ 90% in both intervention groups.
Treatment allocation compliance
Nine per cent (10/113) of PRP group participants did not receive their allocated treatment, whereas all placebo group participants received the intervention assigned to them. Treatment for participants in the placebo group was dependent on the availability of the trial kit for this intervention. However, treatment for participants in the PRP group was further dependent on the ability to withdraw sufficient whole blood to produce the required 8-ml PRP sample. Therefore, it is not unexpected that a higher number of participants in the PRP group did not receive their allocated treatment.
The reasons for participants in the PRP group not receiving their allocated treatment were:
-
site staff being unable to take a blood sample from the participant (3/10, 30.0%)
-
site staff not being able to take a blood sample of sufficient volume to produce PRP (2/10, 20.0%)
-
technical issues encountered with centrifuge (2/10, 20.0%)
-
participant not remaining on site to receive treatment (1/10, 10.0%)
-
in-date PRP disposable kit not available (1/10, 10.0%)
-
site staff unable to obtain PRP disposable kit (1/10, 10.0%).
In all PRP injection participants who were able to have blood samples drawn (103/113, 91.15%), the mean time from taking the blood sample to their PRP injection was 42 minutes (range 12–105 minutes). This breaks down to a mean of 28 minutes from taking the blood sample to producing the PRP sample (range 1–80 minutes) and a mean of 14 minutes from producing the PRP sample to receiving the injection (range 0–45 minutes). All PRP intervention participants who were able to have blood samples drawn received their injection within the expected 2-hour time frame.
Grade of the clinician administering the treatment
A total of 75.5% of participants [total, 173/229; PRP group, 86/113 (76.1%), vs. placebo group, 87/116 (75.0%)] had their treatment administered by a consultant surgeon; 19.2% had their treatment administered by a surgical registrar or research fellow [total, 44/229; PRP group, 19/113 (16.8%), vs. placebo group, 25/116 (21.5%)], 1.3% had it administered by an extended-scope physiotherapist [total, 3/229; PRP group, 1/113 (0.9%), vs. placebo group, 2/116 (1.7%)] and 1.7% had it administered by any other grade of physiotherapist [total, 4/229; PRP group, 2/113 (1.8%), vs. placebo group, 2/116 (1.7%)]. Although we do not have any information for 2.2% of participants (5/229), there were no discernible differences between groups in relation to the grade of clinician administering the treatment (p = 0.706).
Compliance with standard care
Compliance with standard care is reported in Table 2. There were no discernible differences between treatment groups in compliance or in the use of venous thromboembolism prophylaxis [PRP group, 56/113 (49.5%), vs. placebo group, 58/116 (50.0%), difference –0.4%, p = 0.891]; the use of a below-knee cast [PRP group, 47/113 (41.6%), vs. placebo group, 55/116 (47.4%)] or splint/brace [PRP group, 64/113 (56.6%), vs. placebo group, 61/116 (52.6%)] to immobilise the ankle (p = 0.443); or in the receipt of a physiotherapy consultation within 7 weeks [PRP group, 64/113 (56.6%), vs. placebo group, 68/116 (58.6%)] or 13 weeks [PRP group, 100/113 (88.5%), vs. placebo group, 100/116 (86.2%)] (p = 1.000).
| Treatment received | Intervention group | Total (N = 229) | |
|---|---|---|---|
| PRP (N = 113) | Placebo (N = 116) | ||
| Venous thromboembolism prophylaxis use, n (%) | |||
| No | 54 (47.79) | 58 (50.00) | 112 (48.91) |
| Yes | 56 (49.56) | 58 (50.00) | 114 (49.78) |
| Missing | 3 (2.65) | 0 (0) | 3 (1.31) |
| Method of ankle immobilisation, n (%) | |||
| Below-knee cast | 47 (41.59) | 55 (47.41) | 102 (44.54) |
| Splint/brace | 64 (56.64) | 61 (52.59) | 125 (54.59) |
| Missing | 2 (1.77) | 0 (0) | 2 (0.87) |
| Physiotherapy consultation received, n (%) | |||
| Within 7 weeks of injury | 64 (56.64) | 68 (58.62) | 132 (57.64) |
| Within 13 weeks of injury | 100 (88.50) | 100 (86.21) | 200 (87.34) |
| None reported by 13 weeks | 4 (3.54) | 4 (3.45) | 8 (3.49) |
| Missing at 13 weeks | 9 (7.96) | 12 (10.34) | 21 (9.17) |
| Time to starting weight-bearing (days), n; mean (SD) | 96; 27.04 (24.12) | 93; 27.70 (16.10) | 189; 27.36 (15.24) |
| Time to starting ankle-motion exercises (days), n; mean (SD) | 104; 47.40 (19.57) | 103; 48.48 (19.97) | 207; 47.94 (19.73) |
The date a participant started weight-bearing was recorded at the 4-week and 7-week follow-ups, and the date they started ankle-motion exercises was recorded at 7 and 13 weeks. It was assumed that participants remembered the date they were weight-bearing or exercising their ankle better if the time point was closer to it. The time to start weight-bearing and the time to start ankle exercises was calculated as the date reported minus the date the ATR occurred. The date reported was determined to be the earliest date reported by the participant that this activity took place. To allow for the fact that participants may have stopped either weight-bearing or exercising their ankle between questionnaires, participant binary responses as to whether or not they were carrying out an activity were taken into account, and if participants answered ‘yes’ in the earlier questionnaire and ‘no’ in the later questionnaire, their reported date was not included.
Following from the creation of the described time to weight-bearing and time to ankle-exercising variables, assessments of their distributions demonstrated that they did not significantly differ from normal. Therefore, differences in time taken to complete these tasks between treatment groups were assessed using Student’s t-test; there were no differences in the time to weight-bearing between the PRP group (mean 27.0 days, 95% CI 24.1 to 30.0 days) and the placebo group (mean 27.7 days, 95% CI 24.4 to 31.0 days) (PRP group vs. placebo group mean difference –0.7 days, 95% CI –3.7 to 5.0 days; p = 0.768) or in the time taken to start ankle-motion exercises between the PRP group (mean 47.4 days, 95% CI 43.6 to 51.1 days) and placebo group (mean 48.5 days, 95% CI 44.7 to 52.4 days) (PRP group vs. placebo group mean difference –1.1 days, 95% CI –4.2 to 6.5 days; p = 0.672).
Descriptive analyses
The baseline characteristics of participants according to their intervention group are presented in Table 3. Participants in both intervention groups are remarkably similar in all characteristics, with no observable between-group differences.
| Baseline characteristics | Intervention group | Total (N = 229) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PRP (N = 113) | Placebo (N = 116) | ||||||||
| n | Mean | SD | n | Mean | SD | n | Mean | SD | |
| BMI (kg/m2)a | 113 | 27.69 | 5.29 | 114 | 27.25 | 4.22 | 227 | 27.47 | 4.78 |
| Age (years) | 113 | 45.90 | 13.74 | 116 | 45.16 | 12.43 | 229 | 45.53 | 13.07 |
| Alcohol consumption (units) | 113 | 9.90 | 11.33 | 116 | 10.47 | 10.37 | 229 | 10.19 | 10.83 |
| Days since injury | 113 | 5.35 | 2.95 | 116 | 5.20 | 3.08 | 229 | 5.27 | 3.01 |
Information on the stratification factors (age group and trial site) and other prognostic factors of interest by intervention group are presented in Table 4. This table demonstrates that there are clear differences between the levels of the subgroups; for example, more participants in this trial were aged < 55 years than ≥ 55 years and there were notably more male participants than female participants in this trial. However, there are no noteworthy differences between the intervention groups.
| Participant characteristics | Intervention group, n (%) | Total (N = 229), n (%) | |
|---|---|---|---|
| PRP (N = 113) | Placebo (N = 116) | ||
| Centrea,b | |||
| John Radcliffe Hospital, Oxford | 21 (18.58) | 22 (18.97) | 43 (18.78) |
| Musgrove Park Hospital, Taunton | 9 (7.96) | 8 (6.90) | 17 (7.42) |
| Southmead Hospital, Bristol | 7 (6.19) | 9 (7.76) | 16 (6.99) |
| Llandough Hospital, Cardiff | 0 (0) | 1 (0.86) | 1 (0.44) |
| Royal London Hospital | 7 (6.19) | 8 (6.90) | 15 (6.55) |
| Leicester Royal Infirmary | 22 (19.47) | 21 (18.10) | 43 (18.78) |
| University Hospital Coventry | 5 (4.42) | 5 (4.31) | 10 (4.37) |
| Warrington Hospital | 3 (2.65) | 2 (1.72) | 5 (2.18) |
| Basildon University Hospital | 9 (7.96) | 9 (7.76) | 18 (7.86) |
| Royal Liverpool Hospital | 6 (5.31) | 7 (6.03) | 13 (5.68) |
| Peterborough City Hospital | 6 (5.31) | 5 (4.31) | 11 (4.80) |
| Morriston Hospital, Swansea | 3 (2.65) | 4 (3.45) | 7 (3.06) |
| University Hospital Aintree | 2 (1.77) | 1 (0.86) | 3 (1.31) |
| Wythenshawe Hospital, Manchester | 3 (2.65) | 5 (4.31) | 8 (3.49) |
| Northern General Hospital, Sheffield | 3 (2.65) | 4 (3.45) | 7 (3.06) |
| Royal Devon and Exeter Hospital, Exeter | 1 (0.88) | 1 (0.86) | 2 (0.87) |
| Royal Surrey Hospital, Guildford | 1 (0.88) | 1 (0.86) | 2 (0.87) |
| Leighton Hospital, Crewe | 5 (4.42) | 3 (2.59) | 8 (3.49) |
| Age (years)a,b | |||
| < 55 | 86 (76.11) | 88 (75.86) | 174 (75.98) |
| ≥ 55 | 27 (23.89) | 28 (24.14) | 55 (24.02) |
| Sexb | |||
| Female | 25 (22.12) | 32 (27.59) | 57 (24.89) |
| Male | 88 (77.88) | 84 (72.41) | 172 (75.11) |
| Lower limb injured | |||
| Right | 49 (43.36) | 48 (41.38) | 97 (42.36) |
| Left | 64 (56.64) | 68 (58.62) | 132 (57.64) |
| BMI (kg/m2)c | |||
| Normal weight (18.5–24.99) | 40 (35.40) | 39 (33.62) | 79 (34.50) |
| Overweight (25–29.99) | 43 (38.05) | 46 (39.66) | 89 (38.86) |
| Obese (30–39.99) | 30 (26.55) | 31 (26.72) | 61 (26.64) |
| Smoking statusb | |||
| Smoker | 14 (12.39) | 13 (11.21) | 27 (11.79) |
| Ex-smoker | 28 (24.78) | 38 (32.76) | 66 (28.82) |
| Never smoked | 71 (62.83) | 65 (56.03) | 136 (59.39) |
| Use of antiplatelet medicationd | |||
| Yes | 8 (7.08) | 5 (4.31) | 13 (5.68) |
| No | 78 (69.03) | 86 (73.27) | 163 (71.18) |
| Not askede | 27 (23.89) | 26 (22.42) | 53 (23.14) |
On exploration, it was found that only around 8.0% (13/163) of participants questioned were regularly taking medications that could have an impact on their platelet function, with similar low proportions of participants taking such medications in each intervention group [PRP group, 8/86 (9.3%)a, vs. placebo group, 5/91 (5.5%)].
Pain levels at their injection site as reported by participants – relating to the need for more than simple pain relief – were explored. Only 2.6% of participants (6/229) reported experiencing severe pain at their injection site, with too-small numbers of participants to detect any difference between the PRP group (4/113, 3.5%) and the placebo group (2/116, 1.7%). For more details, see Appendix 3.
The academic and employment characteristics of participants prior to their injuries were explored. These explorations related to participants’ employment or academic status, the type of work undertaken and the time participants spent on their feet or driving. There were no differences between the two intervention groups in relation to any of the academic or employment-related characteristics investigated. For more information, see Appendix 3.
The activity levels of participants before they experienced their rupture and the activity being undertaken when the rupture occurred were explored (Table 5). The majority of participants (157/229, 68.6%) injured themselves during sport. As with previous baseline variables, there were no clear differences between intervention groups in relation to their pre-injury activity levels or the activity being undertaken when the injury occurred.
| Activity type | Intervention group, n (%) | Total (N = 229), n (%) | |
|---|---|---|---|
| PRP (N = 113) | Placebo (N = 116) | ||
| Pre-injury activities | |||
| Fitnessa | |||
| More than once/week | 100 (88.50) | 105 (90.52) | 205 (89.52) |
| Less than once/week | 10 (8.85) | 10 (8.62) | 20 (8.73) |
| Never | 3 (2.65) | 1 (0.86) | 4 (1.75) |
| Ball sportsb | |||
| More than once/week | 24 (21.24) | 25 (21.55) | 49 (21.40) |
| Less than once/week | 22 (19.47) | 21 (18.10) | 43 (18.78) |
| Never | 67 (59.29) | 70 (60.34) | 137 (59.83) |
| Racquet sportsc | |||
| More than once/week | 8 (7.08) | 15 (12.93) | 23 (10.04) |
| Less than once/week | 21 (18.58) | 20 (17.24) | 41 (17.90) |
| Never | 84 (74.34) | 81 (69.83) | 165 (72.05) |
| Non-sporting activityd | |||
| More than once/week | 65 (57.52) | 67 (57.76) | 132 (57.64) |
| Less than once/week | 28 (24.78) | 34 (29.31) | 62 (27.07) |
| Never | 20 (17.70) | 15 (12.93) | 35 (15.28) |
| Mechanism of injury | |||
| During sports | 81 (71.68) | 76 (65.52) | 157 (68.56) |
| Heavy DIY, housework, gardening, etc. | 1 (0.88) | 3 (2.59) | 4 (1.75) |
| Other | 31 (27.43) | 37 (31.90) | 68 (29.69) |
Analyses to address primary aims
Missing data assessments
Missing data explorations were carried out to maximise inclusion in the final analysis. The final classification of participants with missing data following these explorations is given in Table 6. (For information on how missing data were classified, see Chapter 2, Analysis of the primary outcome.)
| Missing data classification | Intervention group, n (%) | Total, n (%) | |
|---|---|---|---|
| PRP | Placebo | ||
| True missing data | 12 (10.62) | 15 (12.93) | 27 (11.79) |
| Loss to follow-up | 12 (10.62) | 12 (10.34) | 24 (10.48) |
| Technical errors | 0 (0) | 3 (2.59) | 3 (1.31) |
| Participants with true zero measurements | 9 (7.96) | 11 (9.48) | 20 (8.73) |
| Participants with potential zero measurements | 4 (3.54) | 1 (0.86) | 5 (2.18) |
Primary analysis outputs
There were three data sets available for the analysis of the primary outcome:
-
all available HRET assessments
-
all available HRET assessments that were deemed valid on review of video footage (see Chapter 2, Primary outcome, for more information on the validation review)
-
all available HRET assessments that were deemed valid on review and included only participants with assessments available in both legs.
The primary outcome analysis focuses on data set 3 (all valid HRETs available on both legs).
Explorations of the mean work exhibited during the HRET assessment in participants’ injured and uninjured legs on all three data sets were carried out. Basic explorations of the subsequent measure for work LSI in participants with measurements in both legs were undertaken. These explorations demonstrated that the mean work LSI and subsequent HRET measurements for both intervention groups were very similar.
Focusing on the third data set, the mean work LSI in the injured leg was lower in both the PRP group (675.2, SD. 622.3) and the placebo group (748.2, SD 630.3) than the mean work LSI in the uninjured legs in the PRP (1783.8, SD 838.8) and placebo (1825.5, SD 796.2) groups. Comparing the subsequent work LSI measurement in the PRP group (mean 34.9) with the work LSI measurement in the placebo group (mean 38.3) using the Student’s t-test demonstrated no difference between these two groups (unadjusted PRP group vs. placebo group difference –3.467, 95% CI –10.979 to 4.246; p = 0.384). These explorations demonstrate that the work LSI measurements for both groups are very similar.
Analyses using multivariate linear regression adjusting for stratification factors (adjusted analysis and principal analysis) and other confounding factors (fully adjusted analysis) were undertaken to assess the robustness of conclusions based on these results (Table 7). It is clear from these analyses that there is no evidence of any difference between the intervention groups in relation to work LSI, recorded at 24 weeks post recovery. Taking account of stratification factors and the defined prognostic variables has no impact on the results attained. Sensitivity analyses were also undertaken accounting for participants with zero measurements (two-parts model), with individual missing HRET repetitions (simple imputation) and missing entire HRET data sets (multiple imputation) and for compliance (CACE), and these showed that the results were robust and, therefore, they had no impact on the interpretation of the results.
| Analysis | n | Intervention group, LSI | Mean difference | Robust standard error | p-value | 95% CI | |||
|---|---|---|---|---|---|---|---|---|---|
| PRP | Placebo | ||||||||
| Mean | 95% CI | Mean | 95% CI | ||||||
| Unadjusteda | |||||||||
| Primary analysis | 201 | 34.925 | 29.528 to 40.321 | 38.292 | 32.922 to 43.661 | –3.367 | 3.860 | 0.384 | –10.979 to 4.246 |
| Sensitivity: two-parts model | 170 | 42.078 | 36.786 to 47.371 | 44.454 | 39.284 to 49.623 | –2.375 | 3.747 | 0.527 | –9.773 to 5.023 |
| Sensitivity: simple imputation | 201 | 34.930 | 29.536 to 40.325 | 38.299 | 32.931 to 43.667 | –3.369 | 3.859 | 0.384 | –10.979 to 4.241 |
| Sensitivity: MICE | 229 | 34.596 | 29.132 to 40.060 | 38.457 | 33.128 to 43.787 | –3.861 | 3.897 | 0.323 | –11.549 to 3.826 |
| Sensitivity: CACE | 201 | 34.551 | 28.599 to 40.502 | 38.292 | 32.988 to 43.595 | –3.741 | 4.642 | 0.380 | –12.095 to 4.614 |
| Adjustedb | |||||||||
| Primary analysis | 201 | 34.671 | 30.945 to 38.397 | 38.543 | 33.753 to 43.333 | –3.872 | 3.120 | 0.231 | –10.454 to 2.710 |
| Sensitivity: two-parts model | 170 | 41.464 | 37.754 to 45.174 | 45.040 | 41.224 to 48.855 | –3.576 | 2.741 | 0.209 | –9.348 to 2.206 |
| Sensitivity: simple imputation | 201 | 34.676 | 30.946 to 38.407 | 38.551 | 33.768 to 43.334 | –3.875 | 3.122 | 0.231 | –10.462 to 2.712 |
| Sensitivity: MICE | 229 | 34.583 | 30.016 to 39.150 | 38.526 | 33.362 to 43.690 | –3.943 | 3.312 | 0.256 | –11.120 to 3.234 |
| Sensitivity: CACE | 201 | 34.235 | 30.242 to 38.228 | 38.548 | 34.241 to 42.854 | –4.313 | 3.413 | 0.206 | –11.003 to 2.377 |
| Fully adjustedc | |||||||||
| Primary analysis | 201 | 34.360 | 30.438 to 38.282 | 38.851 | 34.161 to 43.541 | –4.491 | 3.249 | 0.185 | –11.345 to 2.364 |
| Sensitivity: two-parts model | 170 | 41.614 | 37.552 to 45.675 | 44.897 | 41.177 to 48.617 | –3.283 | 2.886 | 0.271 | –9.372 to 2.805 |
| Sensitivity: simple imputation | 201 | 34.365 | 30.438 to 38.293 | 38.859 | 34.176 to 43.541 | –4.493 | 3.254 | 0.185 | –11.358 to 2.371 |
| Sensitivity: MICE | 229 | 34.378 | 29.740 to 39.017 | 38.594 | 33.361 to 43.827 | –4.216 | 3.400 | 0.237 | –11.572 to 3.140 |
| Sensitivity: CACE | 201 | 33.862 | 29.725 to 37.999 | 38.850 | 34.673 to 43.027 | –4.988 | 3.515 | 0.156 | –11.877 to 1.901 |
Post-estimation assessments of the fully adjusted primary outcome regression were carried out to ensure that the results reported by the regression analyses were valid. There were no major issues identified during these assessments and, therefore, the model was kept as simple as possible.
No formal subgroup analyses were carried out; however, results were explored visually using forest plots to examine whether or not the effect of PRP compared with placebo was consistent across the stratification factors (age group and trial site) and other prognostic variables (BMI categories, smoking status and sex) (Figure 5). Although the variability was wide for some categories, such as trial site, as was expected owing to the low number of participants randomised in some sites, the treatment effect was consistent across subgroups, showing that the results are robust.
FIGURE 5.
Forest plot of work LSI effect demonstrating the effect of the intervention on subgroups of defined stratification and prognostic factors. a, Number in the placebo arm vs. number in the PRP arm. The Cardiff and Royal Devon and Exeter sites were excluded as neither had patients in the treatment arm completing HRET assessment.

Analysis to address secondary outcomes
Overall Achilles tendon Total Rupture Score
An overall ATRS of 0 is the lowest (i.e. poorest) score a participant can attribute to their recovery and an overall ATRS of 100 is the highest. At baseline, the unadjusted mean overall ATRS in the PRP group was 14.0, compared with 11.7 in the placebo group. By week 24, this had risen to 64.9 in the PRP group, compared with 65.6 in the placebo group (Table 8).
| Analysis | n | Intervention group, ATRS | Mean difference | Robust standard error | p-value | 95% CI | |||
|---|---|---|---|---|---|---|---|---|---|
| PRP | Placebo | ||||||||
| Mean | 95% CI | Mean | 95% CI | ||||||
| Baseline | |||||||||
| Adjusted analysis | 216 | 14.090 | 10.974 to 17.205 | 11.668 | 8.637 to 14.699 | 2.422 | 2.218 | 0.275 | –1.925 to 6.769 |
| Sensitivity: HRET mITT | 201 | 14.082 | 10.904 to 17.260 | 11.309 | 8.192 to 14.426 | 2.773 | 2.273 | 0.222 | –1.682 to 7.227 |
| Sensitivity: MICE | 229 | 14.899 | 11.566 to 18.232 | 13.085 | 9.781 to 16.389 | –1.814 | 2.401 | 0.450 | –2.892 to 6.723 |
| Sensitivity: CACE | 216 | 14.480 | 12.139 to 16.821 | 11.918 | 9.789 to 14.047 | 2.562 | 1.534 | 0.095 | –0.446 to 5.569 |
| Week 4 | |||||||||
| Adjusted analysis | 216 | 28.461 | 25.286 to 31.637 | 30.609 | 27.561 to 33.657 | –2.148 | 2.246 | 0.339 | –6.550 to 2.255 |
| Sensitivity: HRET mITT | 201 | 28.441 | 25.212 to 31.670 | 31.304 | 28.168 to 34.441 | –2.864 | 2.298 | 0.213 | –7.369 to 1.641 |
| Sensitivity: MICE | 229 | 28.849 | 27.688 to 34.498 | 31.093 | 27.688 to 34.498 | –2.244 | 2.476 | 0.365 | –7.098 to 2.609 |
| Sensitivity: CACE | 216 | 28.175 | 24.881 to 31.469 | 30.499 | 27.527 to 33.471 | –2.325 | 2.177 | 0.286 | –6.591 to 1.942 |
| Week 7 | |||||||||
| Primary analysis | 216 | 37.579 | 34.432 to 40.727 | 38.619 | 35.537 to 41.702 | –1.040 | 2.248 | 0.644 | –5.446 to 3.366 |
| Sensitivity: HRET mITT | 201 | 37.369 | 34.170 to 40.568 | 38.381 | 35.207 to 41.555 | –1.012 | 2.301 | 0.660 | –5.521 to 3.497 |
| Sensitivity: MICE | 229 | 37.454 | 34.014 to 40.894 | 38.523 | 35.083 to 41.962 | –1.069 | 2.490 | 0.668 | –5.951 to 3.813 |
| Sensitivity: CACE | 216 | 37.407 | 33.671 to 41.144 | 38.832 | 35.491 to 42.173 | –1.424 | 2.640 | 0.589 | –6.598 to 3.749 |
| Week 13 | |||||||||
| Primary analysis | 216 | 51.663 | 48.483 to 54.844 | 53.114 | 50.016 to 56.213 | –1.451 | 2.266 | 0.522 | –5.892 to 2.990 |
| Sensitivity: HRET mITT | 201 | 53.091 | 49.858 to 56.324 | 53.432 | 50.254 to 56.611 | –0.342 | 2.315 | 0.883 | –4.878 to 4.195 |
| Sensitivity: MICE | 229 | 50.344 | 46.846 to 53.842 | 51.656 | 48.135 to 55.178 | –1.312 | 2.498 | 0.599 | –6.210 |
| Sensitivity: CACE | 216 | 51.635 | 47.801 to 55.468 | 53.236 | 49.855 to 56.618 | –1.601 | 2.723 | 0.557 | –6.939 to 3.736 |
| Week 24 | |||||||||
| Primary analysis | 216 | 64.987 | 61.864 to 68.111 | 65.530 | 62.495 to 68.565 | –0.543 | 2.222 | 0.807 | –4.899 to 3.813 |
| Sensitivity: HRET mITT | 201 | 66.321 | 63.135 to 69.507 | 65.815 | 62.694 to 68.936 | 0.506 | 2.277 | 0.824 | –3.956 to 4.968 |
| Sensitivity: MICE | 229 | 62.697 | 59.303 to 66.092 | 63.890 | 60.520 to 67.261 | –1.193 | 2.434 | 0.624 | –5.963 to 3.577 |
| Sensitivity: CACE | 216 | 65.143 | 60.681 to 69.604 | 65.738 | 61.769 to 69.708 | –0.595 | 3.013 | 0.843 | –6.500 to 5.310 |
A repeated-measures mixed-effects regression analysis accounting for the time elapsed since the intervention was received was undertaken (see Table 8 and Figure 6). As with the primary outcome analysis, sensitivity analyses were conducted to assess the robustness of conclusions based on these results. These analyses found no evidence of any difference between the intervention groups in relation to average overall ATRS recorded throughout the recovery period. Taking account of stratification factors had no impact on the results attained. In addition, accounting for participants missing ATRS data (multiple imputation) and for compliance (CACE) had no impact on the interpretation of the results. These analyses have shown that participants’ overall ATRS increased from baseline to 24 weeks post treatment at an almost identical rate in the PRP and the placebo groups, irrespective of adjustment for stratification variables.
FIGURE 6.
The ATRS principal adjusted repeated-measures mixed-effects regression model demonstrating the change in ATRS in PRP and placebo group participants over time. 0 = totally limited because of ATR; 100 = not limited at all because of ATR.

Post-estimation assessments of the fully adjusted primary outcome regression were carried out to ensure that the results reported by the regression analyses were valid. There were no major issues identified during these assessments and, therefore, the model was kept as simple as possible.
As before, no formal subgroup analyses were carried out; however, results for the principal secondary outcome (ATRS) were explored visually using forest plots to examine whether or not the effect of PRP compared with placebo differed based on stratification factors (age group and trial site) and other prognostic variables (BMI categories, smoking status and sex) (Figure 7).
FIGURE 7.
Forest plot of ATRS demonstrating the effect of the intervention on overall ATRS in subgroups of defined stratification and prognostic factors. a, Number in the placebo arm vs. number in the PRP arm. The Cardiff and Royal Devon and Exeter sites were excluded as neither had patients in the treatment arm completing the assessment.
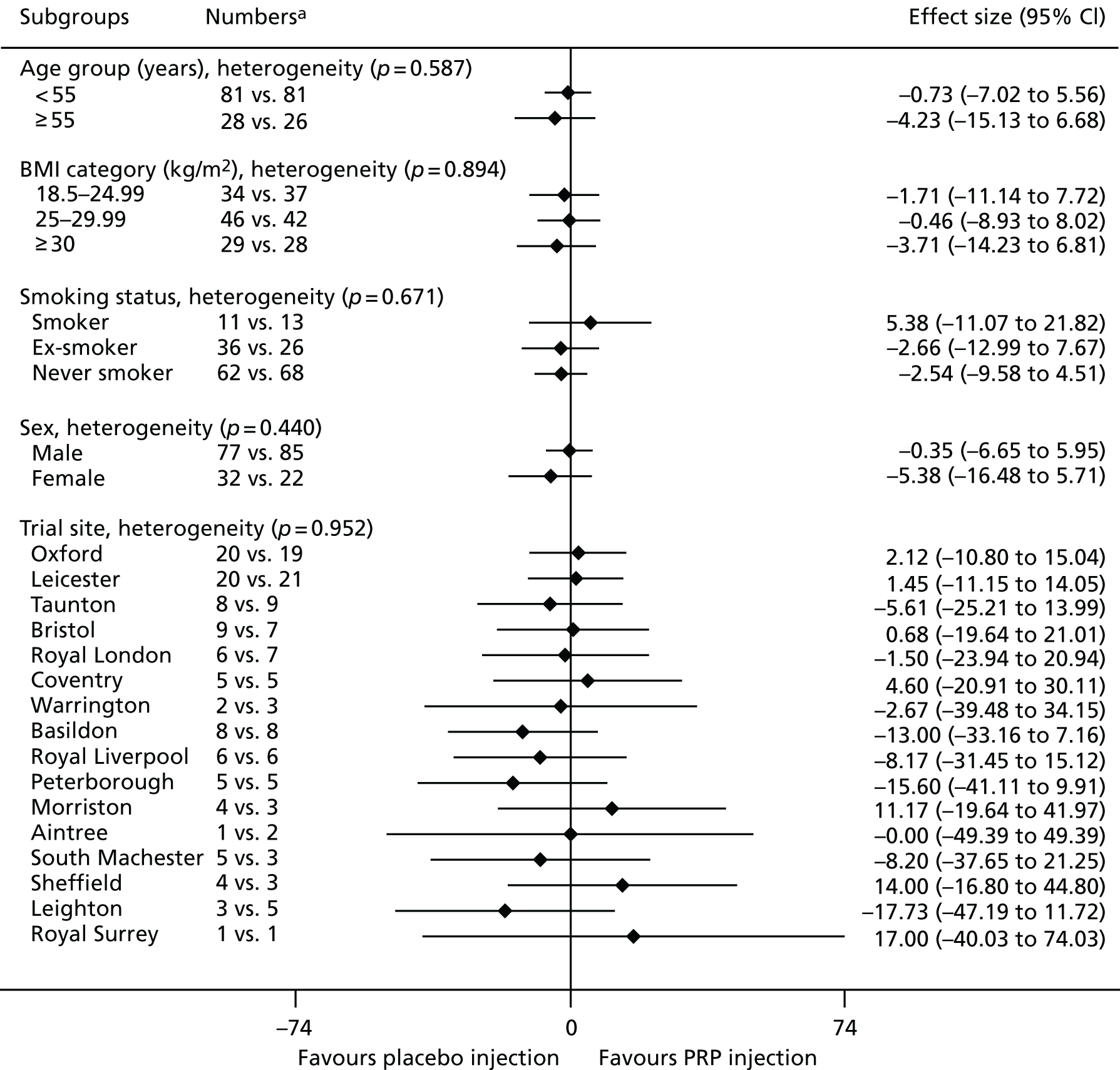
As for the work LSI, this shows that the treatment effect was consistent across subgroups, showing that the results are robust.
Pain-related Achilles tendon Total Rupture Score
Participants’ pain-related ATRSs, a subset of the overall ATRS, increased at a relatively steady rate from baseline to 24 weeks post treatment, with similar rates in the PRP and placebo groups, irrespective of adjusting for stratification variables (Table 9 and Figure 8).
| Time point | n | Intervention group, ATRS pain | Mean difference | Standard error | p-value | 95% CI | |||
|---|---|---|---|---|---|---|---|---|---|
| PRP | Placebo | ||||||||
| Mean | 95% CI | Mean | 95% CI | ||||||
| Baseline | 216 | 3.678 | 3.124 to 4.233 | 3.051 | 2.511 to 3.590 | 0.628 | 0.395 | 0.112 | –0.146 to 1.401 |
| Week 4 | 216 | 6.255 | 5.690 to 6.819 | 6.277 | 5.734 to 6.821 | –0.023 | 0.400 | 0.954 | –0.806 to 0.761 |
| Week 7 | 216 | 7.006 | 6.445 to 7.568 | 6.679 | 6.129 to 7.230 | 0.327 | 0.401 | 0.415 | –0.459 to 1.113 |
| Week 13 | 216 | 7.489 | 6.921 to 8.057 | 7.235 | 6.681 to 7.788 | 0.255 | 0.405 | 0.529 | –0.538 to 1.048 |
| Week 24 | 216 | 7.661 | 7.106 to 8.217 | 7.449 | 6.909 to 7.990 | 0.212 | 0.395 | 0.592 | –0.563 to 0.987 |
FIGURE 8.
The ATRS pain mixed-effect regression model. Results from the adjusted repeated-measures mixed-effects regression model demonstrating the change in ATRS pain measure in PRP and placebo group participants over time. 0 = totally limited because of pain; 10 = not limited at all because of pain.
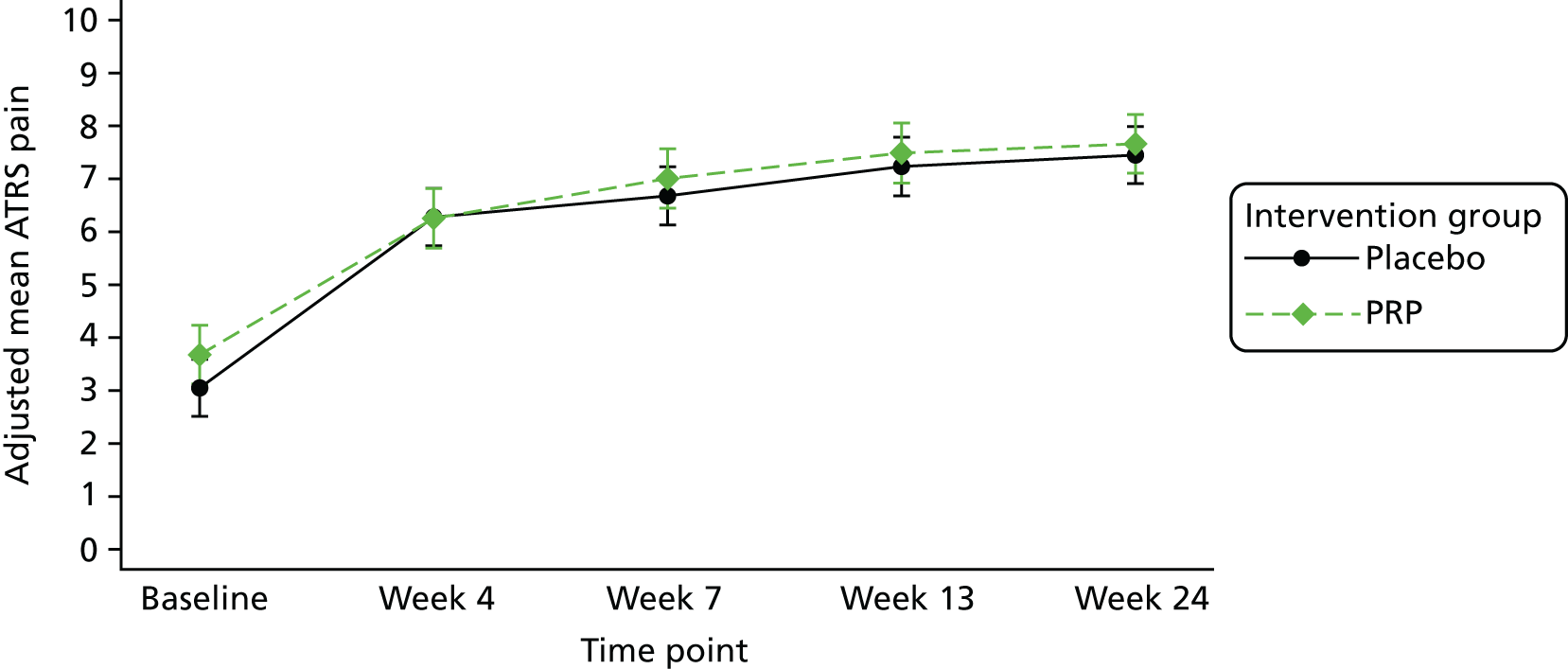
Patient-specific functional scale
Participants’ overall PSFS score increased from baseline to 24 weeks post treatment at an almost identical rate in the PRP and placebo groups, irrespective of adjusting for stratification variables (Table 10 and Figure 9).
| Time point | n | Intervention group, overall PSFS | Mean difference | Standard error | p-value | 95% CI | |||
|---|---|---|---|---|---|---|---|---|---|
| PRP | Placebo | ||||||||
| Mean | 95% CI | Mean | 95% CI | ||||||
| Baseline | 216 | 0.833 | 0.425 to 1.241 | 0.846 | 0.448 to 1.244 | –0.013 | 0.291 | 0.965 | –0.583 to 0.557 |
| Week 4 | 216 | 2.021 | 1.608 to 2.433 | 2.030 | 1.632 to 2.429 | –0.009 | 0.293 | 0.975 | –0.583 to 0.564 |
| Week 7 | 216 | 3.128 | 2.717 to 3.539 | 3.357 | 2.955 to 3.759 | –0.229 | 0.293 | 0.435 | –0.804 to 0.346 |
| Week 13 | 216 | 5.810 | 5.395 to 6.225 | 5.776 | 5.372 to 6.180 | 0.034 | 0.296 | 0.909 | –0.546 to 0.614 |
| Week 24 | 216 | 7.198 | 6.788 to 7.608 | 7.495 | 7.097 to 7.893 | –0.297 | 0.291 | 0.308 | –0.868 to 0.274 |
FIGURE 9.
The PSFS mixed-effects regression model. Results from the adjusted repeated-measures mixed-effects regression model demonstrating the change in PSFS in PRP and placebo group participants over time. 0 = unable to carry out activities; 10 = able to carry out activities at same levels as before ATR.

Maximum concentric displacement and maximum number of repetitions
As with the primary outcome analysis, there was no difference between the intervention groups in the maximum concentric displacement LSI achieved and the maximum number of repetitions achieved (Table 11). After adjusting for all stratification variables, the average maximum concentric displacement LSI [the LSI calculated as (maximum displacement in the injured leg/maximum displacement in the uninjured leg) × 100, to account for potential ability differences] achieved by participants in the PRP group was 55.1 (95% CI 51.4 to 58.8), compared with 55.4 (95% CI 49.6 to 61.3) in the placebo group. This difference was not significant (mean difference –0.3, 95% CI –6.1 to 5.4; p = 0.898). Similarly, after adjusting for stratification variables, the average maximum number of repetitions proportion [calculated as (maximum number of repetitions inthe injured leg/maximum number of repetitions in the uninjured leg) × 100, to account for potential ability differences] achieved by participants in the PRP group was 50.1% (95% CI 43.7% to 56.4%), compared with 60.7% (95% CI 52.8% to 68.7%) in the placebo group. This difference was not statistically significant (mean difference –10.7, 95% CI –21.9 to 0.6; p = 0.061).
| HRET measure | n | Intervention group, maximum displacement | Mean difference | Robust standard error | p-value | 95% CI | |||
|---|---|---|---|---|---|---|---|---|---|
| PRP | Placebo | ||||||||
| Average | 95% CI | Average | 95% CI | ||||||
| Maximum displacement | 201 | 55.099 | 51.436 to 58.762 | 55.453 | 49.610 to 61.296 | –0.354 | 2.717 | 0.898 | –6.087 to 5.378 |
| Maximum number of repetitions | 201 | 50.080 | 43.735 to 56.417 | 60.752 | 52.841 to 68.662 | –10.671 | 5.325 | 0.061 | –21.906 to 0.564 |
Visual analogue scale
Participants’ pain levels as recorded in their pain diaries in the first 2 weeks post intervention demonstrate that the pain experienced by participants decreased at a relatively steady rate from day 1 to day 14 (Figure 10).
FIGURE 10.
Pain VAS mixed-effects regression model. Results from the adjusted repeated-measures mixed-effects regression model demonstrating the change in VAS pain measure in PRP and placebo group participants in the first 2 weeks post intervention. 0 = no pain; 100 = severe pain.
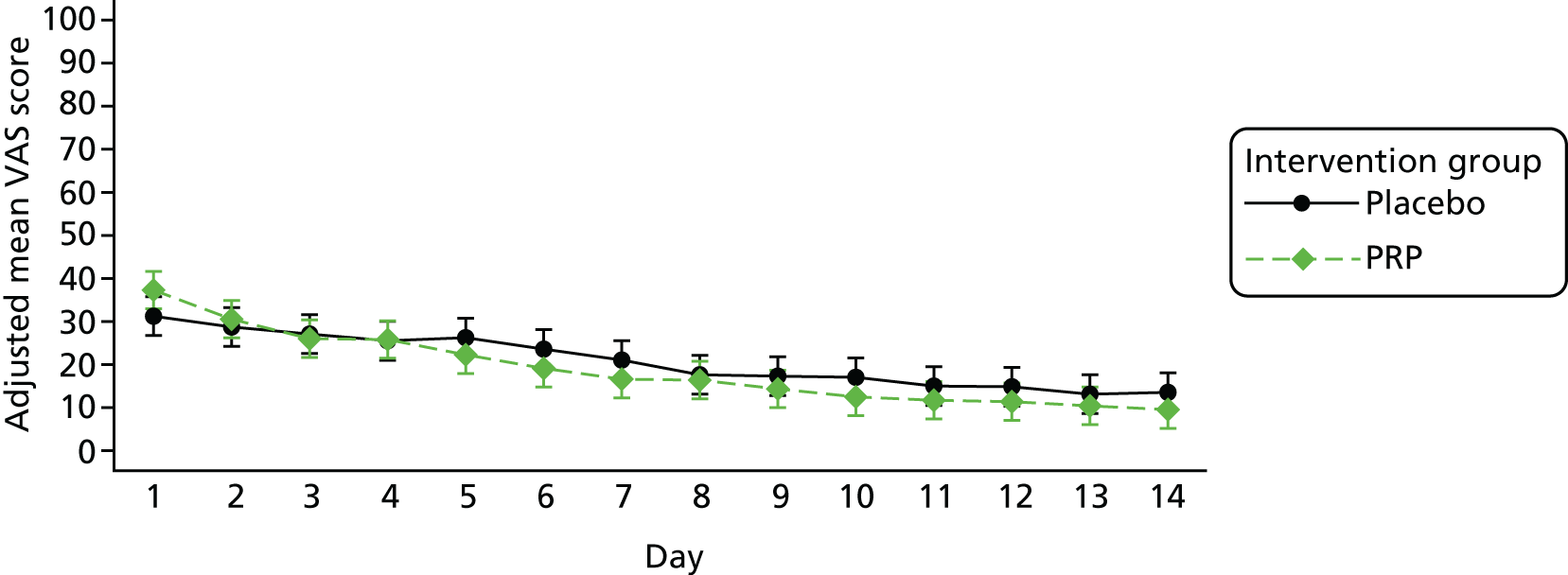
Accounting for the stratification factors, participants in the PRP group reported a mean overall VAS score of 37.3 (95% CI 33.0 to 41.7) at day 1, with the placebo group reporting a similar level of 31.2 (95% CI 26.7 to 35.7). By day 14, participants in the PRP group were reporting a mean VAS score of 9.5 (95% CI 5.2 to 13.9) and participants in the placebo group were reporting a mean VAS score of 13.6 (95% CI 9.0 to 18.1). This difference at day 14 was not significant (mean difference –4.0, 95% CI –10.3 to 2.3; p = 0.210). This demonstrates that the decline in reported pain was at similar rates in the PRP and placebo groups.
Short Form questionnaire-12 items quality of life
Participants’ SF-12 physical component scores (PCSs) increased gradually over time to near-normal levels at 24 weeks post treatment (Figure 11) at an almost identical rate in the PRP and placebo groups, irrespective of stratification variable and accounting for participants’ pre-injury PCSs (Table 12).
FIGURE 11.
The SF-12 PCS mixed-effects regression model. Results from the adjusted repeated-measures mixed-effects regression model demonstrating the change in PCS in PRP and placebo group participants over time, with mean pre-injury score (with 95% CIs) shown.
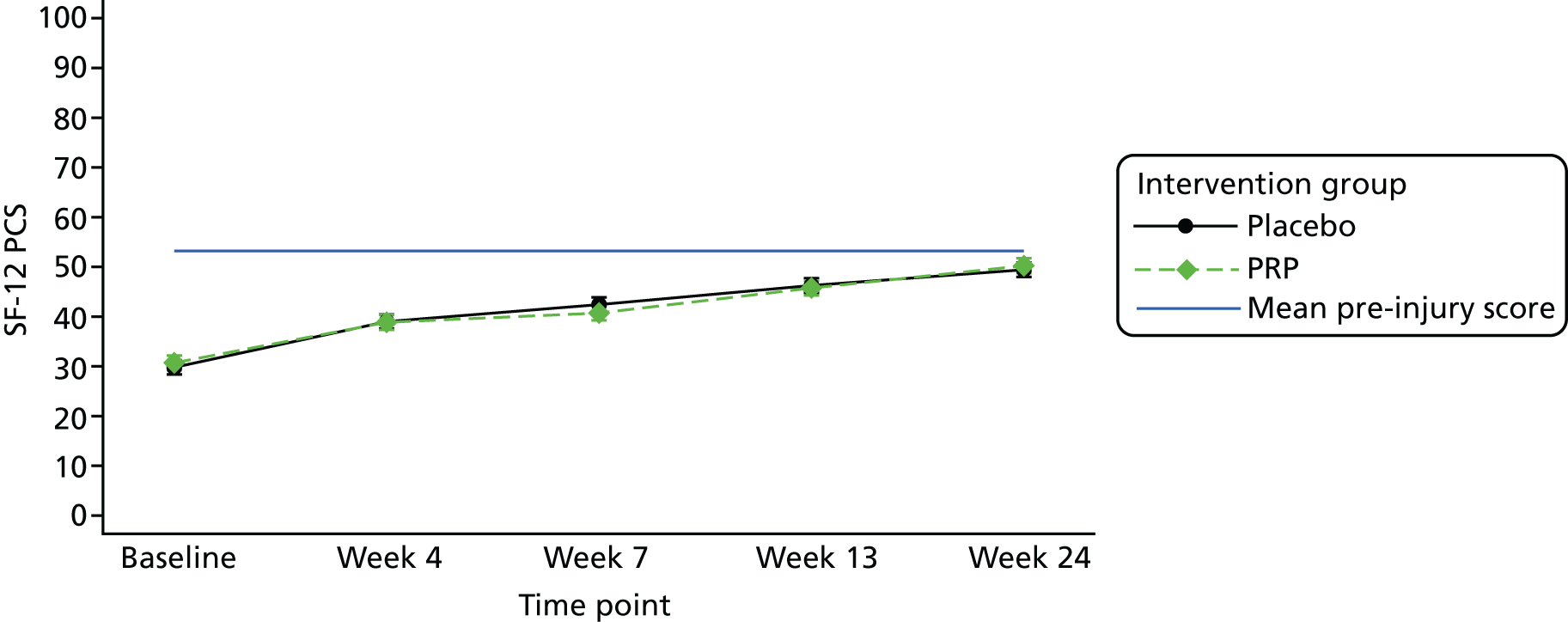
| Time point | n | Intervention group, overall PCS | Mean difference | Standard error | p-value | 95% CI | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| PRP | Placebo | |||||||||
| Mean | 95% CI | Mean | 95% CI | |||||||
| Baseline: post injury | 213 | 30.720 | 29.250 to 32.191 | 29.799 | 28.367 to 31.230 | 0.922 | 1.048 | 0.379 | –1.133 to 2.976 | |
| Week 4 | 213 | 38.865 | 37.376 to 40.354 | 39.001 | 37.565 to 40.438 | –0.136 | 1.057 | 0.897 | –2.208 to 1.935 | |
| Week 7 | 213 | 40.731 | 39.249 to 42.214 | 42.422 | 40.971 to 43.872 | –1.691 | 1.059 | 0.110 | –3.767 to 0.386 | |
| Week 13 | 213 | 45.760 | 44.260 to 47.259 | 46.272 | 44.812 to 47.731 | –0.512 | 1.069 | 0.632 | –2.606 to 1.584 | |
| Week 24 | 213 | 50.240 | 48.751 to 51.729 | 49.436 | 47.996 | 50.876 | 0.805 | 1.058 | 0.447 | –1.269 to 2.879 |
Participants’ SF-12 mental component scores (MCSs) remained relatively stable and close to their pre-injury scores over the entire trial follow-up period (Figure 12). At 24 weeks post treatment, participants in the PRP group had a MCS that was nearly 3 points lower than that of the MCS in the placebo group after adjusting for stratification variables and accounting for participants’ pre-injury MCSs (p = 0.035) (Table 13).
FIGURE 12.
The SF-12 MCS mixed-effects regression model. Results from the adjusted repeated-measures mixed-effects regression model demonstrating the change in MCS in PRP and placebo group participants over time.
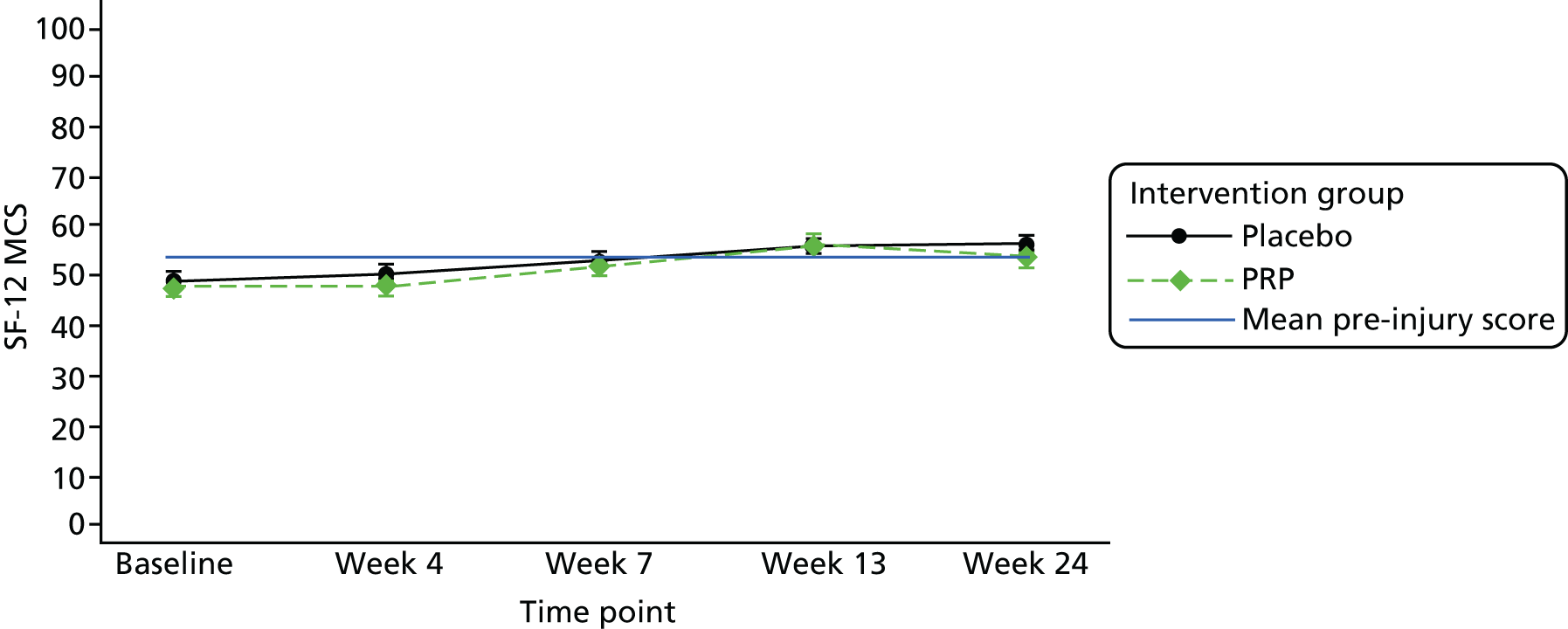
| Time point | n | Intervention group, overall MCS | Mean difference | Standard error | p-value | 95% CI | |||
|---|---|---|---|---|---|---|---|---|---|
| PRP | Placebo | ||||||||
| Mean | 95% CI | Mean | 95% CI | ||||||
| Baseline: post injury | 213 | 47.802 | 46.003 to 49.601 | 49.105 | 47.356 to 50.855 | –1.303 | 1.281 | 0.309 | –3.813 to 1.207 |
| Week 4 | 213 | 48.287 | 46.460 to 50.114 | 50.686 | 48.925 to 52.446 | –2.398 | 1.295 | 0.064 | –4.936 to 0.139 |
| Week 7 | 213 | 52.054 | 50.235 to 53.873 | 53.420 | 51.639 to 55.200 | –1.365 | 1.299 | 0.293 | –3.911 to 1.180 |
| Week 13 | 213 | 56.415 | 54.580 to 58.251 | 55.673 | 53.885 to 57.462 | 0.742 | 1.308 | 0.571 | –1.822 to 3.305 |
| Week 24 | 213 | 53.788 | 51.972 to 55.604 | 56.503 | 54.745 to 58.260 | –2.714 | 1.289 | 0.035 | –5.242 to –0.187 |
Success of blinding
Participants were questioned following their 24-week HRET assessment as to which treatment they believed they received; 210 participants responded to this question (Table 14).
| Participant prediction of treatment | Intervention group, n (%) | Total, n (%) | |
|---|---|---|---|
| PRP | Placebo | ||
| PRP injection | 32 (30.77) | 28 (26.41) | 60 (28.57) |
| Imitation injection | 17 (16.35) | 9 (8.49) | 26 (12.38) |
| Don’t know | 55 (52.88) | 69 (65.09) | 124 (59.05) |
| Total | 104 (100.00) | 106 (100.00) | 210 (100.00) |
Using participant responses to this question, assessments of blinding were carried using the James et al. 69 and Bang et al. 70 blinding indices to determine whether or not participants were successfully blinded to the intervention they received. Using the information generated from both of these assessments, we can be confident that there is no evidence to suggest that blinding in this trial was unsuccessful. For further details on the outcome of these assessments, see Appendix 3.
Adverse events
Adverse events are reported in Table 15. Only one participant (1/229, 0.4%) reported a SAE during the study. There were similar AE rates in the treatment groups [PRP, 84/113 (74%); placebo, 90/116 (78%)]. The overall re-rupture rate among study participants was 4.4% (10/229), with similar rates in the PRP group (6/113, 5.3%) and the placebo group (4/116, 3.5%). Nine of the ten participants with a re-rupture were then treated with surgery. DVT rates were also similar (PRP group: 6/113, 5.3%; placebo group: 5/116, 4.3%).
| Complication type | Intervention group, n (%) | |
|---|---|---|
| PRP (N = 113) | Placebo (N = 116) | |
| Participants experiencing at least one AE of any typea | 84 (74.34) | 90 (77.59) |
| SAEsa | 1 (0.88) | 0 |
| ST elevation myocardial infarction | 1 (0.88) | 0 |
| AEsa | 84 (74.34) | 90 (77.59) |
| Foreseeable AEsa | 83 (73.45) | 87 (75.00) |
| Mild discomfort or minor bleeding following injection | 22 (19.47) | 8 (6.90) |
| Technical complications of lower leg casting and splinting | 38 (33.63) | 28 (24.14) |
| Consequences of depending on walking aids | 1 (0.88) | 1 (0.86) |
| Syncopal episode related to venesection or tendon injection | 0 | 0 |
| Discomfort at rupture site during rehabilitation | 9 (7.96) | 10 (8.62) |
| Swelling or bruising of the lower leg and foot | 68 (60.18) | 77 (66.38) |
| DVT in a lower limb | 6 (5.31) | 5 (4.31) |
| Re-rupture of treated Achilles tendon | 6 (5.31) | 4 (3.45) |
| Unforeseeable AEsa | 16 (14.16) | 19 (16.38) |
| Serious infection of injection site of ATR | 0 | 0 |
| Skin breakdown or ulceration of treated lower leg | 13 (11.50) | 13 (11.21) |
| Severe pain (more than simple analgesia) > 10 days after injection | 6 (5.31) | 6 (5.17) |
| Other AEs related to treatment or ATRa | 13 (11.50) | 13 (11.21) |
| Frequent discomfort at injection site | 5 (4.42) | 6 (5.17) |
| Infection at injection site confirmed by doctor | 0 | 3 (2.59) |
| Infection at non-injection siteb | 0 | 3 (2.59) |
| Other problemc | 9 (7.96) | 6 (5.17) |
There were some complications at the injection site, with 11 out of 229 (5%) participants reporting frequent discomfort and 3 out of 229 (1%) participants reporting clinically diagnosed infections. However, no clinically serious infections were reported.
Summary of key findings
-
The results of the analyses of the primary end point allow us to conclude that there was no evidence of a difference between PRP injection and placebo for the treatment of acute ATR in adults in terms of muscle–tendon function at 24 weeks.
-
There was no evidence of any differences between the intervention groups in the PROMs (ATRS, PSFS, pain and SF-12 physical function) across all time points assessed. There was a small statistically significant difference in SF-12 mental function, with the PRP group doing worse than the placebo group. This was probably due to chance.
-
There was no difference in complications and AEs between the two groups.
-
The results were robust to adjustment for stratification factors (age and centre) with/without other confounding factors and for the sensitivity analyses.
-
The overall re-rupture rate was low, at 4.37%, and no significant difference was seen between the groups.
Chapter 4 Substudy 1: platelet-rich plasma and blood analysis
Introduction
Autologous platelet preparations are increasingly being used in many areas of regenerative medicine. However, there are few properly controlled RCTs and the preparation, content and definitions of many platelet preparations are generally poorly defined, standardised and controlled. Most platelet products therefore have varying concentrations of blood cells [platelets, white blood cells (leucocytes) and red blood cells (erythrocytes)], plasma and fibrin. Studying the potential role(s) of all the cells and their bioactive factors is, therefore, essential to understand the variables associated with the biological activity and efficacy of any platelet preparation.
Although our pilot study demonstrated efficacy signal of a PRP (L-PRP) preparation in ATR healing, as a pilot study it was underpowered to study the variables within PRP preparations that may be associated with efficacy of healing. The PATH-2 trial, powered on the work LSI as the primary outcome, randomised a total of 230 participants, 103 of whom received the PRP product. This provided the opportunity to not only study the variation in cellular and bioactive components within the PRP but also to determine the reliability of using a single device to prepare an autologous sterile product for ATR healing across 19 trial centres.
The aim of substudy 1 was to determine the cellular content of the PRP product (platelets, erythrocytes and leucocytes) and the quality and growth factor content of the PRP preparations used. This provided one of the most comprehensive studies of the within- and between-centre performance of a single preparation device of PRP in a clinical trial setting. The data also provided insight into the key variables within PRP that were associated with the biological efficacy of the product as determined by the primary and secondary outcomes.
Methods
Blood and platelet-rich plasma samples
Venous blood (1–5 ml) was taken from all participants recruited into the trial and anticoagulated within ethylenediaminetetraacetic acid vacutainers (Becton, Dickinson and Company, Plymouth, UK). In the PRP treatment group, an additional 50 ml of venous blood was taken for automated sterile PRP preparation in the centrifuge; 8 ml of sterile PRP was then harvested and 4 ml was used for injection into the Achilles tendon, and the remaining 4 ml was divided into four 1-ml aliquots. One microtube was immediately frozen at –70 °C for storage until the end of the trial when it was used for batch measurement of growth factor levels (see below). The remaining PRP aliquots (each 1 ml) were stimulated at room temperature for 5 minutes by addition to two tubes (Platelet Solutions Ltd, Nottingham, UK) containing either (tube B) saline alone, to provide an unstimulated baseline, or (tube C) adenosine diphosphate and U46619, to fully activate the platelets. Both samples were then fixed using 1 ml of PAMFix (Platelet Solutions Ltd, Nottingham, UK). The whole blood, unfixed PRP and tubes B and C were then transported at room temperature by courier to the Institute of Inflammation and Ageing at the University of Birmingham and processed as soon as possible on arrival.
Cell counting
Whole-blood and unfixed PRP cell counts were conducted using the Sysmex XN-1000 haematology analyser (Sysmex UK, Milton Keynes, UK). The analyser utilises three primary analysis principles: fluorescence flow cytometry, direct current detection with hydrodynamic focusing and sodium lauryl sulfate haemoglobin detection. The WNR (white cell nucleated) channel evaluates white blood cell count, basophils and nucleated red blood cells. An impedance red blood cell count is also reported. The WDF (white blood cell differential) channel also provides a differential count of the neutrophils, lymphocytes, eosinophils, monocytes and immature granulocytes. The platelet fluorescence (PLT-F) channel is a specialised fluorescence optical analysis exclusively for platelets. The PLT-F parameter utilises traditional fluorescence flow cytometry in which platelets are stained with oxazine (a ribonucleic acid binding dye that eliminates any interference mediated by cellular debris). A measurement of platelet production (immature platelet fraction) is also in this channel. The analyser thus reports three platelet counts (platelet impedance, platelet optical and PLT-F), mean platelet volume and the immature platelet fraction. Quality control material (XN Check™, Sysmex UK, Milton Keynes, UK) was tested on a daily basis to ensure instrument performance throughout the trial. The instrument was also enrolled in a national external quality assurance scheme (UK NEQAS, Watford, UK) and maintained on a service contract (Sysmex UK). To determine normal ranges for each cellular parameter, blood samples from 40 healthy volunteers (control cohort) were analysed and normal ranges are represented as the mean value ± 2 SDs.
Measurement of platelet quality
Tubes B and C containing fixed resting and activated platelets, respectively, were analysed for the expression of a platelet-specific activation marker P-selectin (CD62P) by flow cytometry as a measure of platelet quality. After gentle mixing, 5 µl of fixed PRP from tubes B and C was incubated with 5 µl of test antibody [CD62P conjugated to flourescein isothiocyanate (FITC); Beckman Coulter Inc., Brea, CA, USA] or its corresponding isotype control (IgG conjugated to FITC; Beckman Coulter Inc.) with 40 µl of 0.2-µm filtered phosphate buffered saline (pH 7.4) for 20 minutes at room temperature. The samples were then further diluted by the addition of 4 ml of phosphate-buffered saline. The platelets were then analysed by flow cytometry (Accuri™ Flow cytometer, Becton, Dickinson and Company). Platelets were identified according to their characteristic size and granularity (using log-forward scatter and log-side scatter), with a forward-scatter threshold of 25,000 to eliminate noise. An amorphous gate was placed around the platelet population and 10,000 events were collected. Analysis regions were set using the isotype control so that 0.5% of the platelet population was positive. CD62P positivity was then measured and both percentage expression and median fluorescent intensity of all platelet events were recorded.
Measurement of growth factor levels within platelet-rich plasma
At the end of participant recruitment, all frozen aliquots of PRP were shipped to Birmingham for analysis of growth factor levels. Appropriate batches of samples were thawed at 37 °C for 10 minutes. One-fortieth volume of 20% Triton™ X-100 (Roche Diagnostics, Mannhein, Germany) was added to ensure full lysis prior to a further incubation at 37 °C for 10 minutes. Samples were mixed then centrifuged at 1500 g for 10 minutes at room temperature. Supernatants were then removed and simultaneously assayed for five different growth factors (TGF-β1, FGF, VEGF, IGF-1 and PDGF-AB) by commercial enzyme-linked immunosorbent assay (ELISA) kits (Bio-Techne Ltd, Abingdon, UK). Optimal sample dilutions for each growth factor were predetermined by assaying L-PRP samples prepared from control volunteers using the identical method of preparation as in the trial. Diluted samples, blanks and standards were pipetted in duplicate into 96 well plates coated with capture antibodies and incubated as instructed within each growth factor ELISA kit. Plates were then washed four times with the washing buffers provided and the peroxidase-conjugated secondary antibodies provided within each kit were added to each well and incubated as instructed. Plates were then washed four times with washing buffer before addition of substrate solution to each well. The plates were then incubated for 20–30 minutes at room temperature in the dark before the addition of the stop solution. The optical density of each well was then measured immediately using a microplate reader and readings taken at 450 nm. A further reading was taken at 540 nm and the values subtracted from the readings at 450 nm to correct for optical imperfections in the plate. The duplicate readings were averaged for each standard, control and sample, and the zero standard optical density was subtracted. Standard curves were generated by plotting the mean absorbance for each standard on the y-axis against the concentration on the x-axis and the best-fit line generated by regression analysis. Unknown sample values were then read off the regression line and final concentrations were determined by multiplying using the dilution factors used in each assay. The concentration for each growth factor per ml of lysate was reported.
Statistical analysis
The comparability of key blood parameters and bioactive factors for each treatment group was summarised with means and SDs. For whole-blood parameters, a Student’s t-test was used to explore differences between treatment arms.
Correlation between the primary outcome and both key blood parameters and bioactive factors was explored using Pearson’s correlation.
As per analyses in the main study, a p-value of < 0.05 was seen as indicative of a statistically significant difference.
Results
Cell counts within whole blood and platelet-rich plasma
Summary statistics for key blood parameters in whole-blood and PRP samples are given in Table 16. A comparison of the whole-blood counts in the PRP and placebo groups is shown in Figure 13. There was no significant difference in the whole-blood samples between the treatment groups for red blood cell count (mean difference –0.002, 95% CI –0.144 to 0.139; p = 0.974), white blood cell count (mean difference –0.259, 95% CI –0.784 to 0.264; p = 0.330) or platelet count (as PLT-F) (mean difference –19.045, 95% CI –38.310 to 0.040; p = 0.051). Figure 14 illustrates the cell counts within the PRP samples showing the variation in platelet, red blood cell and white blood cell counts, with the whole-blood sample summaries given in the same figure for reference. The mean platelet count (PLT-F) in the PRP sample was 852.5 × 109/l, but with a wide range (6.0–2903.0 × 109/l). Although a small number of PRP platelet counts were lower than original whole-blood counts due to some unforeseen centrifuge problems, the overall mean increase in platelets was 4.1-fold. The mean red blood cell count was 0.9 × 1012/l (range 0.1–9.0 × 1012/l). As expected, the red blood cell count decreased on average in the PRP by a factor of 5.3 of the original count in whole blood. The mean white blood cell count was 15.1 × 109/l (range 1.7–65.3 × 109/l). As this preparation is a L-PRP, the mean white blood cell count increased by a factor of 2.2, as expected. The white blood cell differential count was 14.6% monocytes (mean 2.2 × 109/l, range 0–21.1 × 109/l), 45.2% lymphocytes (mean 6.8 × 109/l, range 0.1–18.7 × 109/l), 39.2% neutrophils (mean 5.9 × 109/l, range 0.2–30.4 × 109/l), 0.1% basophils (mean 0.02 × 109/l, range 0–0.5 × 109/l) and 0.82% eosinophils (mean 0.1 × 109/l, range 0–1.5 × 109/l).
| Parameter | Intervention group | Total (N = 229) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PRP (N = 113) | Placebo (N = 116) | ||||||||
| n | Mean | SD | n | Mean | SD | n | Mean | SD | |
| Whole-blood | |||||||||
| Erythrocytes (1012/l) | 107 | 4.825 | 0.589 | 114 | 4.827 | 0.475 | 221 | 4.826 | 0.532 |
| Leucocytes (109/l) | 107 | 6.742 | 2.049 | 114 | 7.001 | 1.904 | 221 | 6.875 | 1.975 |
| Platelets (109/l)a | 104 | 208.183 | 77.729 | 110 | 227.227 | 63.537 | 214 | 217.972 | 71.264 |
| PRP | |||||||||
| Erythrocytes (1012/l) | 101 | 0.907 | 1.489 | – | – | – | – | – | – |
| Leucocytes (109/l) | 101 | 15.130 | 10.275 | – | – | – | – | – | – |
| Platelets (109/l)a | 98 | 852.551 | 438.958 | – | – | – | – | – | – |
| IGF-1 (ng/ml) | 103 | 78.178 | 23.180 | – | – | – | – | – | – |
| TGF-β1 (ng/ml)b | 97 | 131.915 | 74.372 | – | – | – | – | – | – |
| PDGF-AB (ng/ml)c | 100 | 55.339 | 27.640 | – | – | – | – | – | – |
| VEGF (ng/ml)d | 103 | 0.981 | 0.721 | – | – | – | – | – | – |
| bFGF (pg/ml)e,f | 103 | 111.038 | 76.970 | – | – | – | – | – | – |
FIGURE 13.
Cell counts in whole-blood samples, by intervention group. (a) Erythrocytes; (b) leucocytes; and (c) platelets. a, Using the PLT-F measure.

FIGURE 14.
Cell counts in PRP samples, represented against counts in whole-blood samples. (a) Erythrocytes; (b) leucocytes; and (c) platelets. a, Using the PLT-F measure.
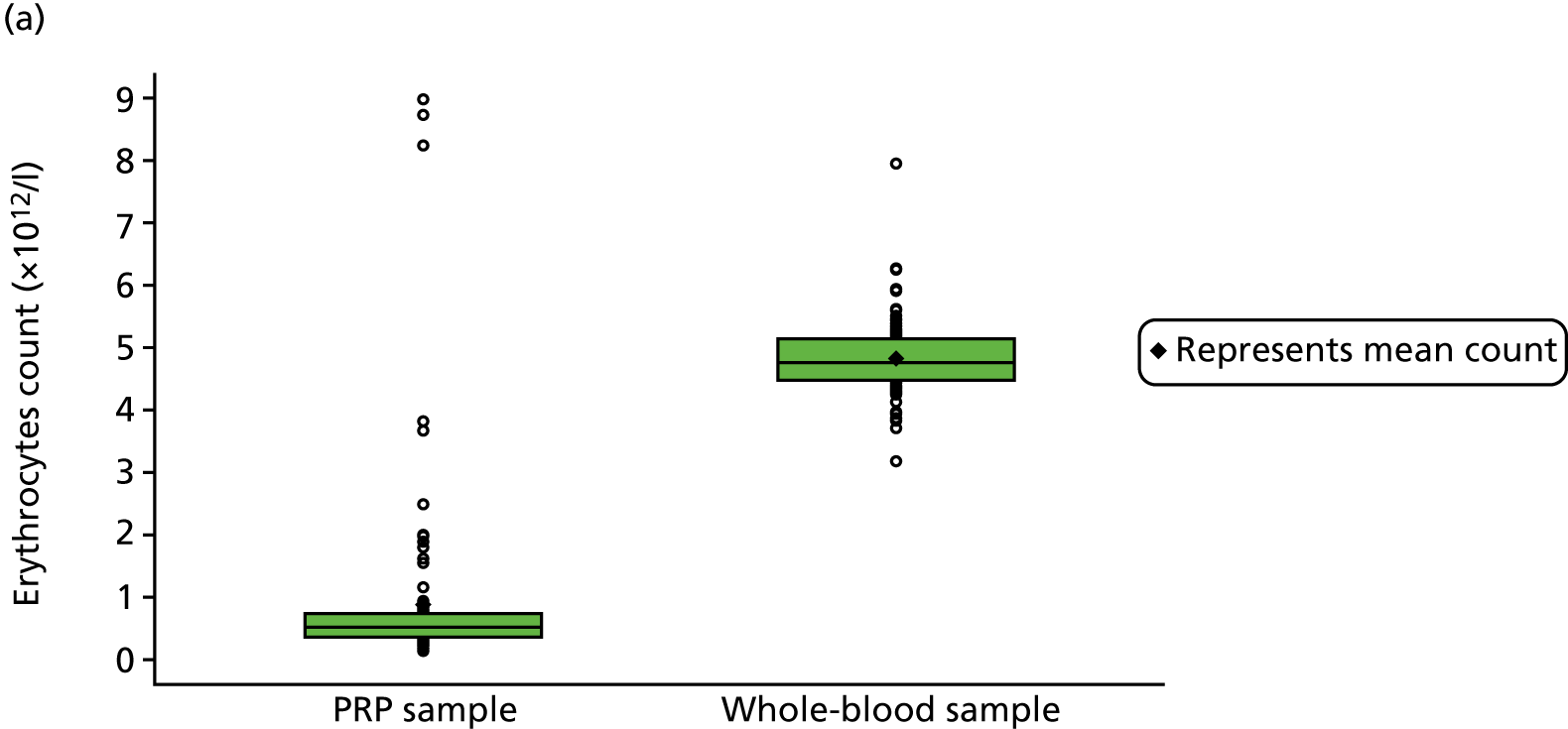

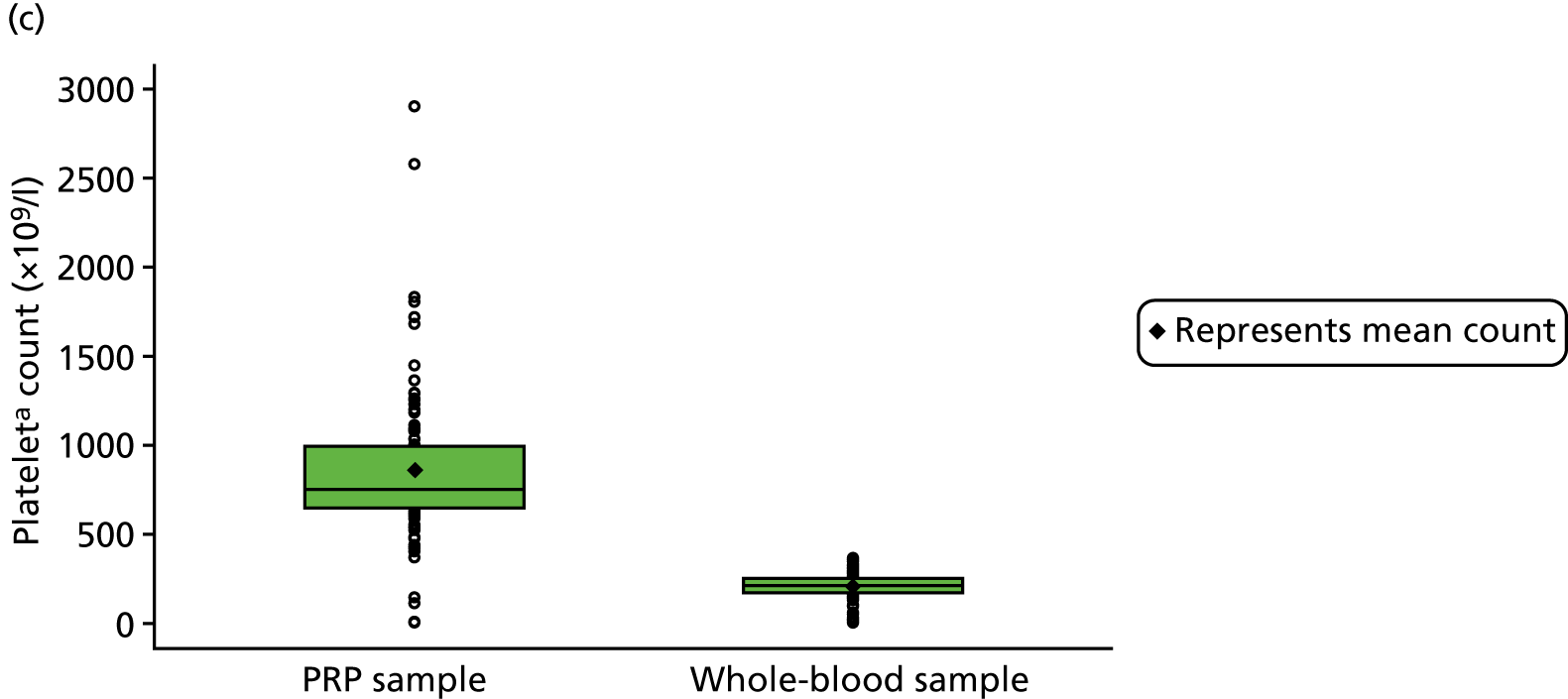
Platelet quality
The basal levels of CD62P expression [percentage and mean fluorescence intensity (MFI)] within the platelets of resting basal PRP and activated PRP samples are shown in Table 17 and Figure 15. In resting PRP, the mean CD62P expression was 4.3% with a MFI of 248.6. The majority of PRP samples were therefore of good quality with low levels of activation, with a few exceptions. In the activated PRP samples, the mean CD62P expression was 60.1% with a MFI of 1208.0. The majority of PRP samples were functional, with the majority of platelets in the PRP preparations being shown to be capable of activation and degranulation, with a few exceptions.
| Parameter | PRP samples | |||||
|---|---|---|---|---|---|---|
| Activated (N = 103) | Resting (N = 103) | |||||
| n | Mean | SD | n | Mean | SD | |
| CD62P expression (%) | 102 | 60.092 | 22.264 | 103 | 4.274 | 5.046 |
| MFI | 102 | 1208.046 | 556.599 | 103 | 248.568 | 50.941 |
FIGURE 15.
Platelet quality in PRP samples, demonstrating both (a) CD62P expression and (b) MFI in activated and resting (unactivated) PRP.


Growth factor levels within platelet-rich plasma
The levels of each growth factor are shown in Table 16 and Figure 16.
FIGURE 16.
Growth factor concentrations in PRP samples.

Blood and platelet-rich plasma variables and clinical outcomes
Baseline blood and the primary outcome measure
Parameters of baseline whole blood taken before the intervention did not correlate with the primary outcome measure at 24 weeks (Figure 17 and Table 18), irrespective of intervention group.
FIGURE 17.
Correlation between whole-blood sample blood cell counts and the primary outcome (work LSI) at 24 weeks. (a) Erythrocytes; (b) leucocytes; and (c) platelets. a, Using the PLT-F measure.
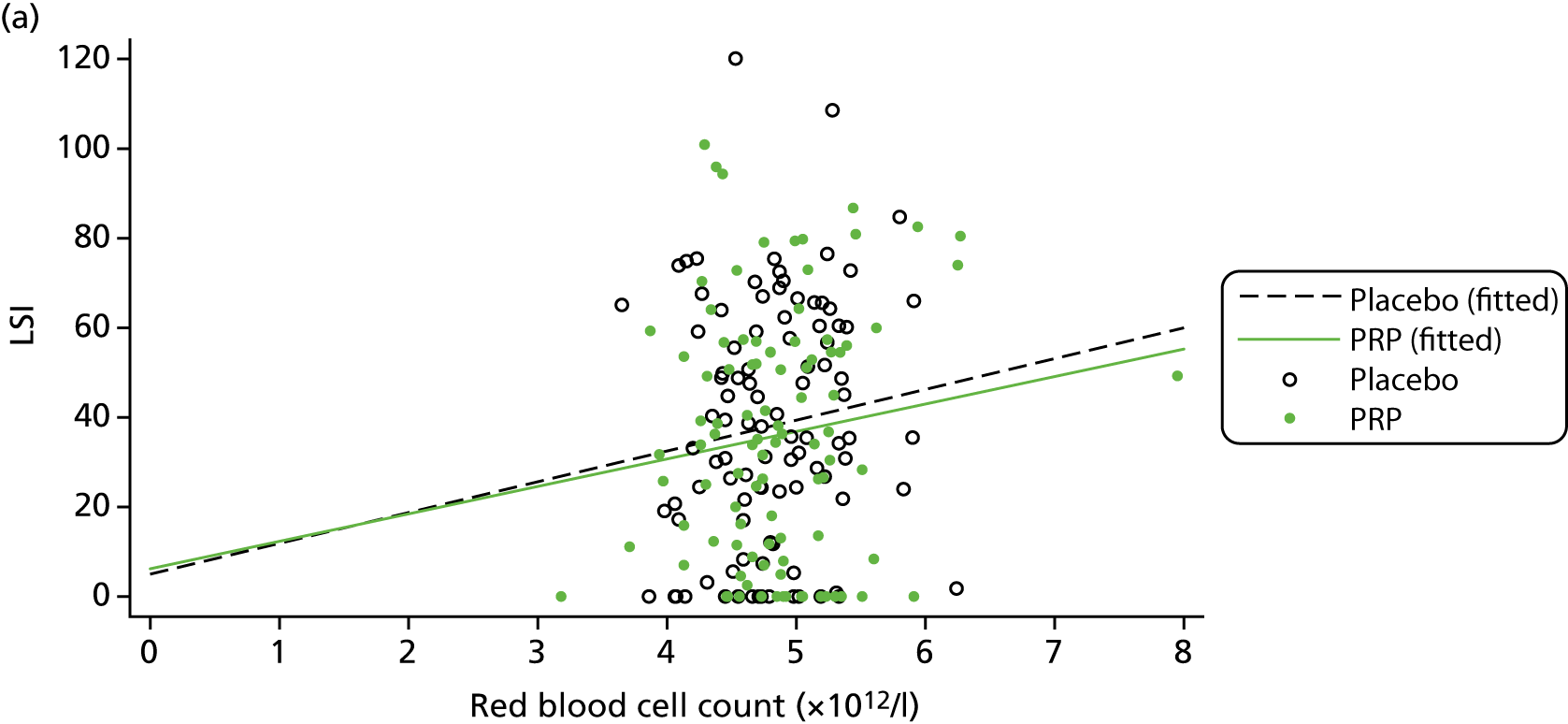
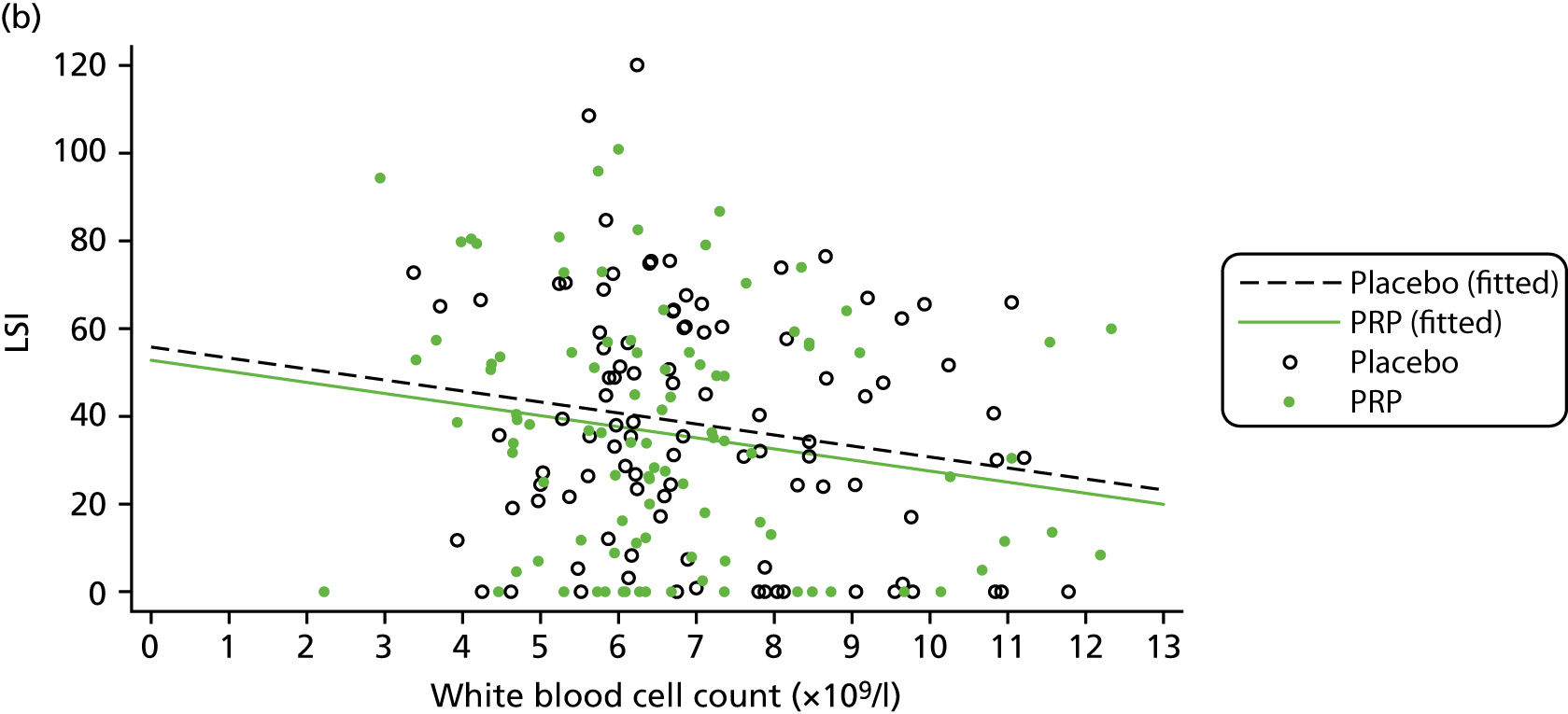
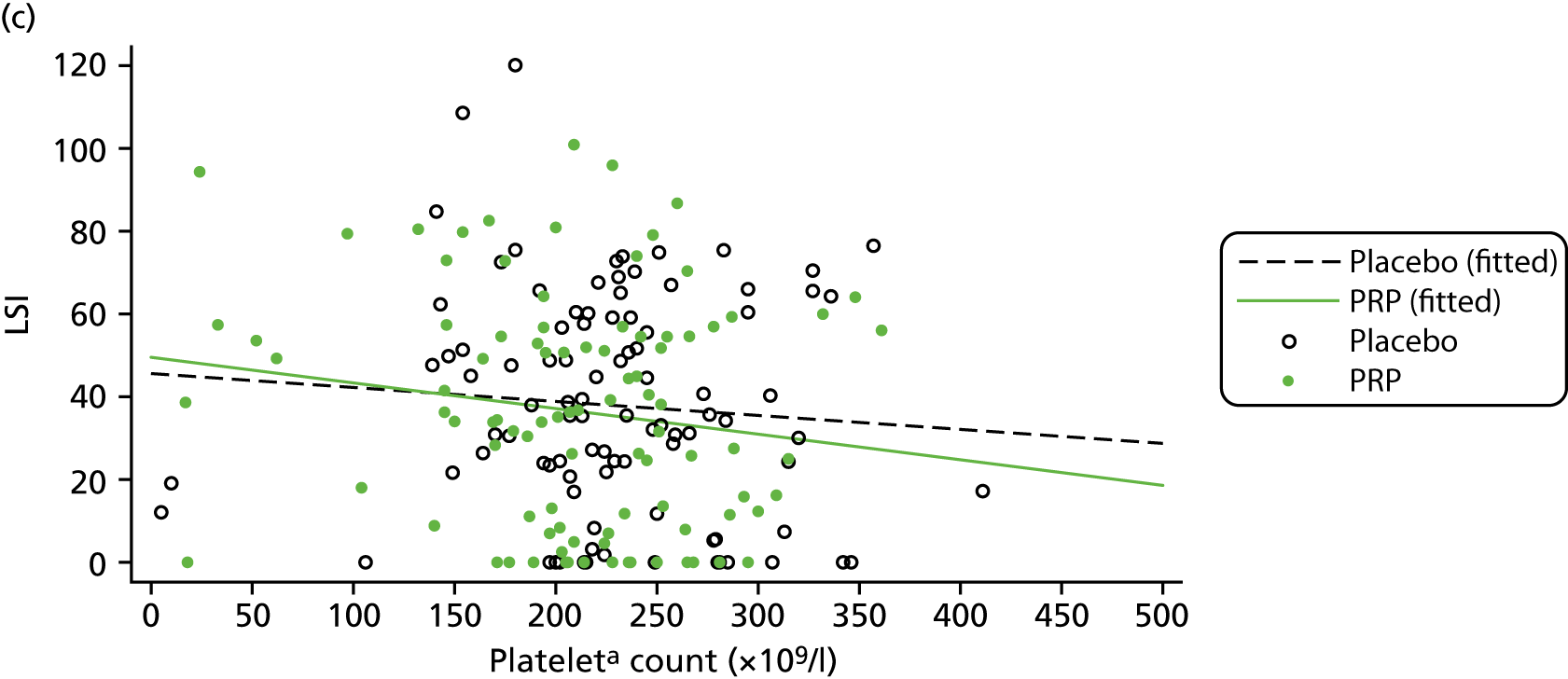
| Parameter by group | n | r | % variancea | p-value |
|---|---|---|---|---|
| Erythrocytes | ||||
| PRP | 97 | 0.133 | 1.780 | 0.193 |
| Placebo | 99 | 0.122 | 1.496 | 0.228 |
| Leucocytes | ||||
| PRP | 97 | –0.184 | 3.393 | 0.071 |
| Placebo | 99 | –0.172 | 2.976 | 0.088 |
| Plateletsb | ||||
| PRP | 94 | –0.153 | 2.329 | 0.142 |
| Placebo | 96 | –0.080 | 0.634 | 0.440 |
Platelet-rich plasma and the primary outcome measure
The PRP cell count and growth factor concentration correlation with the primary outcome measure were analysed to assess if the variability in PRP had any effect on the outcome. The results are displayed in Figures 18–20 and Table 19.
FIGURE 18.
Correlation between PRP sample blood cell counts and the primary outcome (work LSI) at 24 weeks. (a) Erythrocytes; (b) leucocytes; and (c) platelets. a, Using the PLT-F measure.

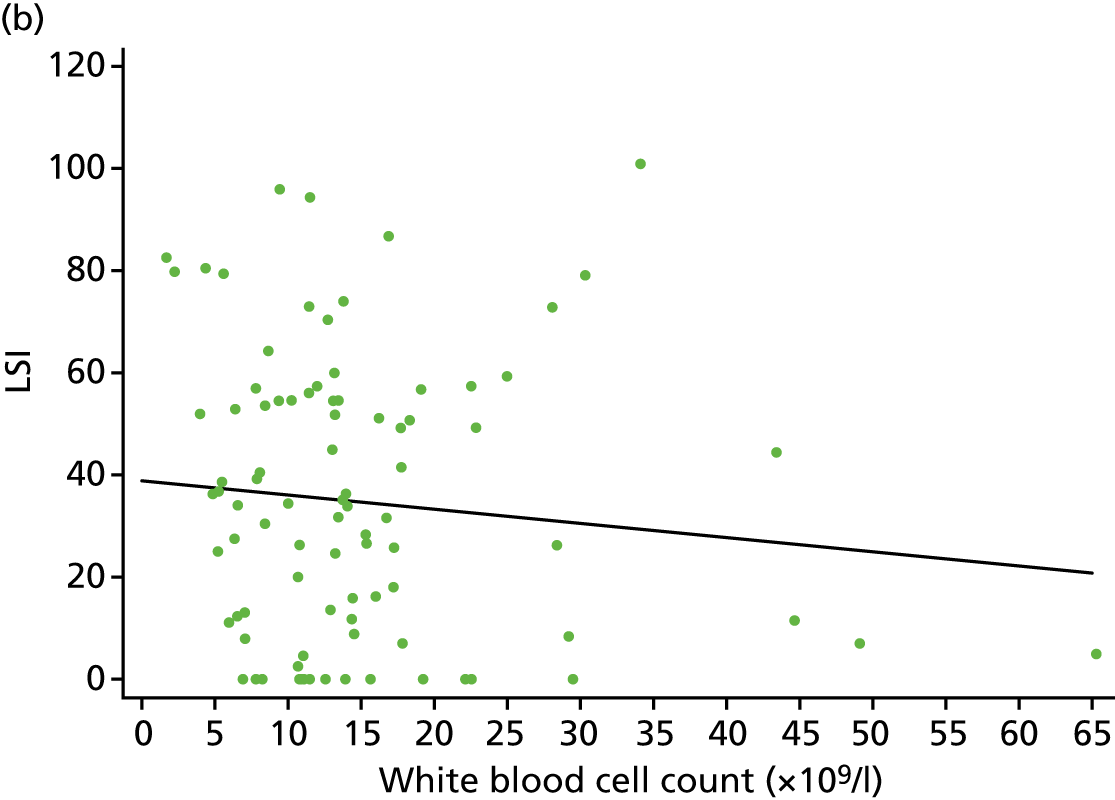
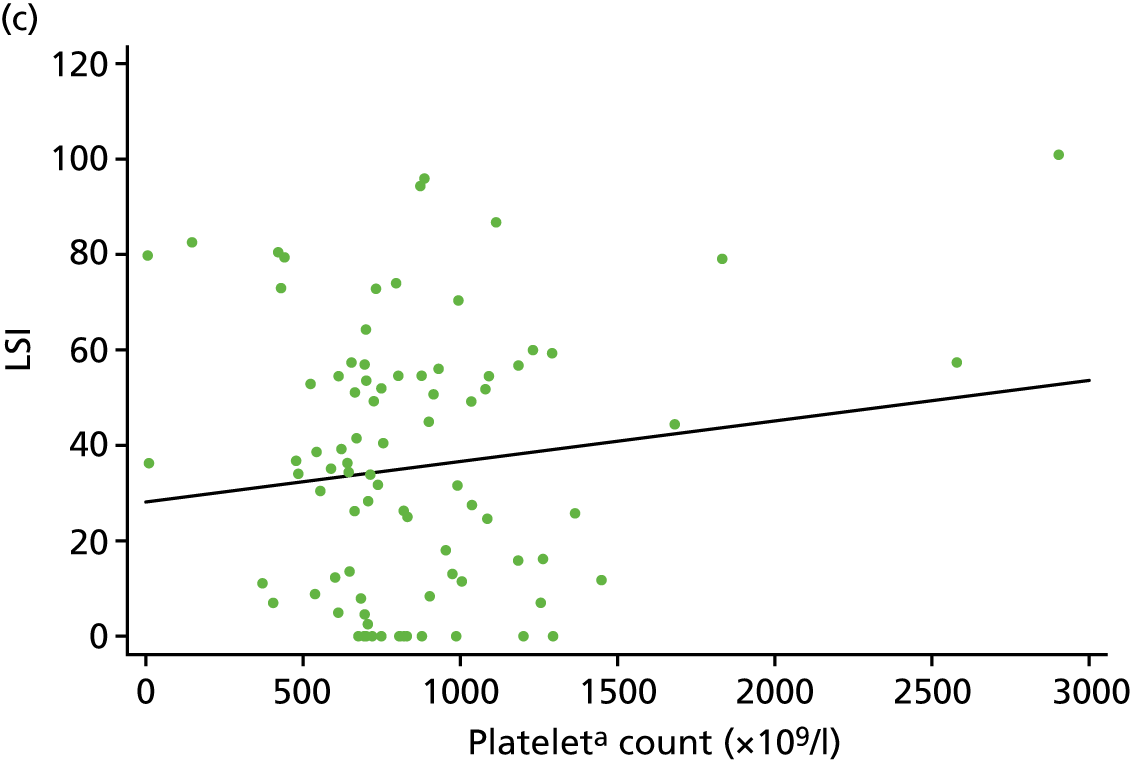
FIGURE 19.
Correlation between platelet quality and the primary outcome (work LSI) at 24 weeks. (a) Activated CD62P expression (%); and (b) activated CD62P MFI expression (%).


FIGURE 20.
Correlation between growth factors and the primary outcome (work LSI) at 24 weeks. (a) IGF-1; (b) TGF-β1; (c) PDGF-AB; (d) VEGF; and (e) bFGF. bFGF, basic fibroblast growth factor.
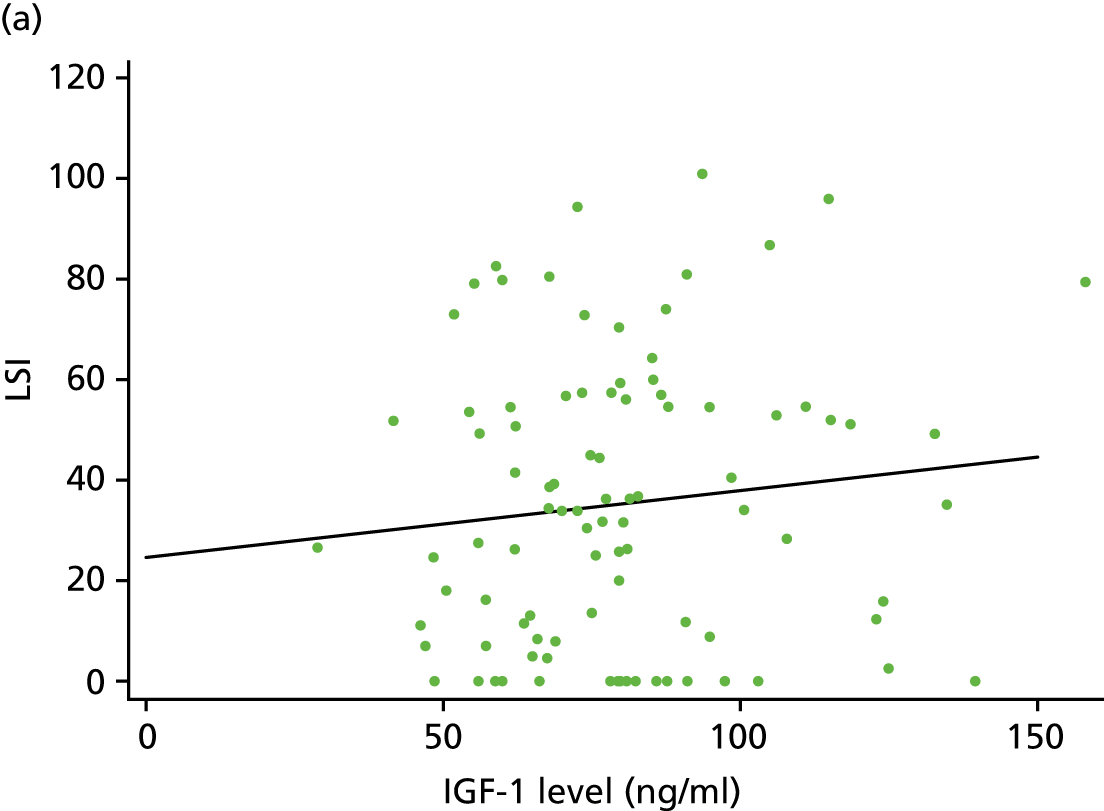
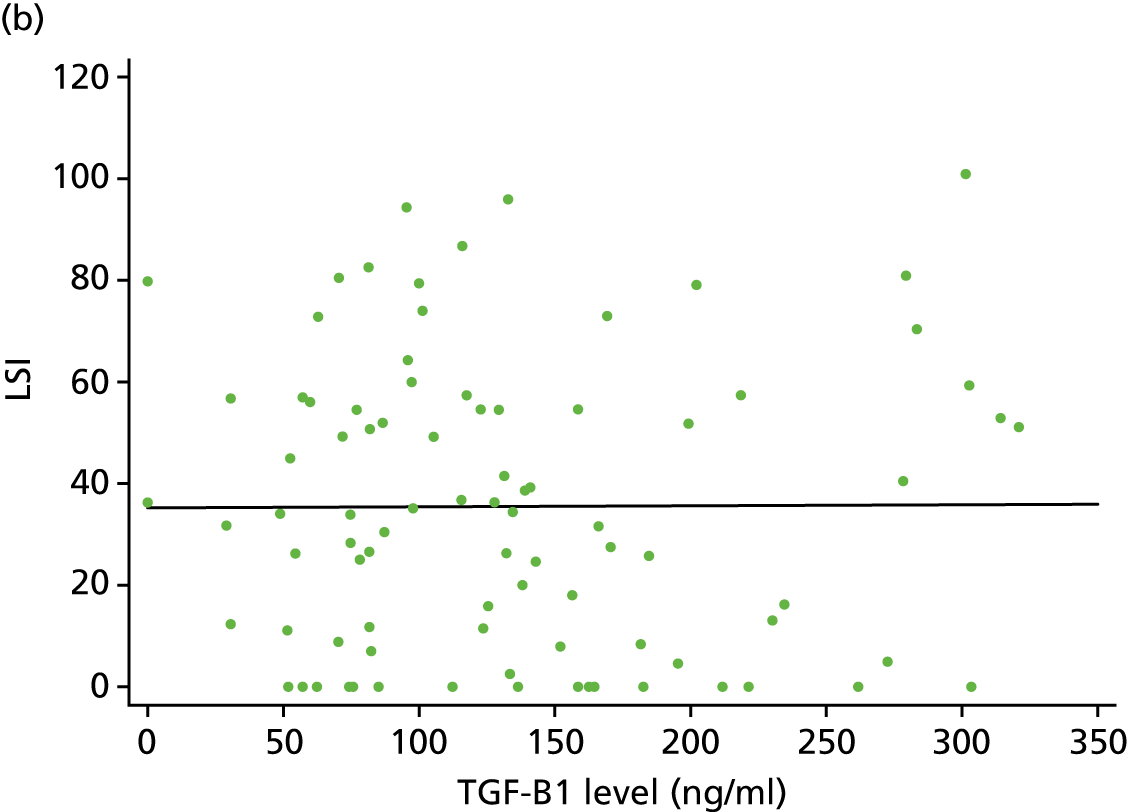

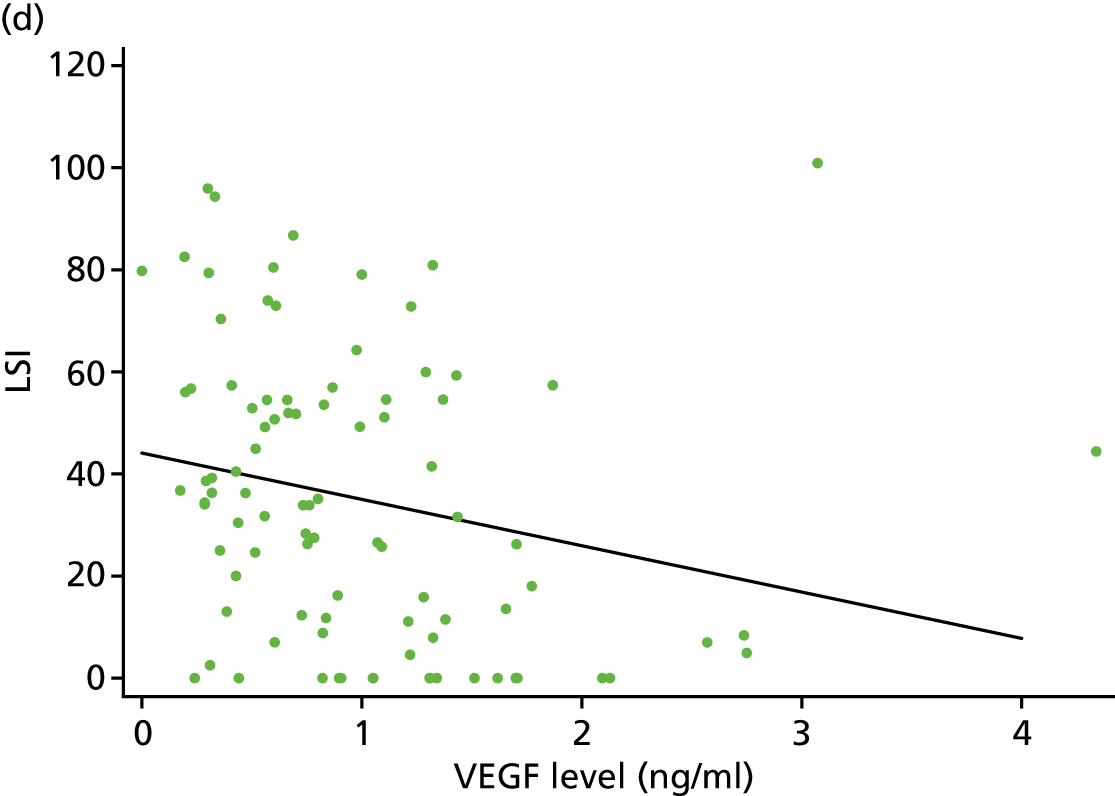

| Parameter | n | r | % variancea | p-value |
|---|---|---|---|---|
| Blood cell counts | ||||
| Erythrocytes | 91 | 0.126 | 1.590 | 0.234 |
| Leucocytes | 91 | –0.103 | 1.053 | 0.333 |
| Plateletsb | 88 | 0.128 | 1.649 | 0.233 |
| Platelet quality | ||||
| Activated CD62P expression (%) | 92 | 0.038 | 0.142 | 0.721 |
| MFI | 92 | –0.004 | 0.002 | 0.966 |
| Growth factors | ||||
| IGF-1 | 93 | 0.124 | 1.248 | 0.287 |
| TGF-β1 | 88 | 0.005 | 0.003 | 0.960 |
| PDGF-AB | 90 | < 0.001 | < 0.001 | 0.998 |
| VEGF | 93 | –0.231 | 5.355 | 0.026 |
| bFGF | 93 | –0.102 | 1.049 | 0.329 |
The red blood cell, white blood cell and platelet (as PLT-F) counts and work LSI at 24 weeks linear regression analysis showed no significant correlation (see Figure 18). This was also the case for the platelet-quality measures of interest (see Figure 19) and for all growth factors (see Figure 20) with the exception of VEGF, for which the evidence suggests a negative correlation between PRP VEGF concentration and week 24 LSI performance (p = 0.026), with around 5% of the variance in LSI being explained by this growth factor.
Discussion
The results showed that the whole-blood parameters in participants’ venous blood samples were identical in both groups immediately prior to injection of PRP or placebo. As it is possible that variation in platelets and leucocytes in whole blood may play a role in the healing process of tendons, these results confirmed that there was no baseline blood variation between the groups.
To our knowledge, this is the largest analysis of PRP in a multicentre RCT in a musculoskeletal disease or injury setting. The results of this substudy showed that the centrifuging method in this population produced L-PRP. The average platelet count in PRP was > 4 times that in whole blood. The leucocyte concentration was 2.2 times the whole-blood leucocyte concentration. This may have an influence on the healing process as both cells play a crucial role in the initial tissue response to an injury.
The growth factors IGF-1, TGF-β1, PDGF-AB and VEGF are reportedly the greatest contributors to tissue healing in PRP. Previous work has identified a need for growth factor analysis so that the interpatient variation in different parameters can be properly explored. Here, the results of such an analysis on 103 samples are presented. The hypothesis that there would be a significant positive relationship between growth factor concentrations and platelet and leucocyte concentrations was mostly supported in this study. Most of the growth factors had a significant or moderate association with platelet concentrations. The lack of association with IGF-1 supports the opinion that platelets are not the major contributors of that factor.
An important finding of the growth factors analysis was the large degree of variation exhibited between participants, even when corrected for platelet concentration. Using correlation and regression analysis, statistically significant associations have been identified between the concentrations of TGF-β1, PDGF-AB, VEGF, basic fibroblast growth factor (bFGF), platelets and leucocytes. These relationships may contextualise the understanding of the biological mechanisms at work in PRP tissue response.
The relationship between product-variable biological content and clinical effect was presented. The key finding was that there was no association of either the cellular or growth factors’ content with the primary outcome measure. The level of platelet activation as a measure of platelet quality at baseline also did not correlate with the clinical outcome. Baseline blood was also assessed to make sure that there was no variation between the two groups. There was no correlation of the cellular component of whole blood on the primary outcome.
The results of substudy 1 confirm that variability in this biological product did not change the outcome of Achilles tendon injury management.
Chapter 5 Substudy 2: immunohistochemistry study of needle biopsies from Achilles tendon rupture lesion site
Introduction
Torn and healing tendons progress through stages of scar tissue formation and remodelling, in which processes of cell death, inflammation, matrix breakdown, stem cell migration, proliferation, revascularisation, tenocyte differentiation, matrix synthesis and matrix remodelling are evident. 76,77 Although increased cellularity and vascular invasion are essential features of initial scar tissue formation, successful remodelling requires a marked reduction in both to levels that are normal for healthy tendon. Critically, the collagen matrix must resolve from small, randomly aligned fibres to large-diameter fibres with high packing density, aligned with tensile loading forces. In addition, existing undamaged collagen bundles must be knitted back into the new tissue. At the same time, a return to normal loading is essential in restoring normal structure and, therefore, mechanical function. 34,78 Why some tendons heal fully but many remain permanently in a scar tissue or fibrotic stage is the necessary focus of much current research, as the dosing and timing of potentially beneficial growth factor, stem cell and mechanical therapies require considerable optimisation. 79 In this needle biopsy substudy, the effects of PRP were investigated 6 weeks after rupture and treatment using relevant cellular markers.
Methods
Sixteen participants (nine in the PRP group and seven in the placebo group) were enrolled from a single selected trial site and gave consent to undergo the sample collection procedure at 6 weeks post rupture. Both the ultrasound radiologists and the tissue investigators were blinded to treatment allocation until analysis was completed. A biopsy from the mid-lesion site was taken under guided ultrasound using a 14-gauge needle (Temno Evolution soft tissue biopsy needle; Merit Medical Systems, South Jordan, UT, USA) slanted diagonally through the mid-line of the Achilles tendon, as previously developed by the trial team. 57 Tissue was collected into ice-cold phosphate-buffered saline and dissected immediately. Two-thirds of the biopsy was fixed in 10% formalin, embedded in paraffin wax and sectioned longitudinally at 4-µm thickness. One-third of the biopsy was snap frozen at –80 °C for follow-up studies.
Sections were stained by hand with routine haematoxylin and eosin or with picrosirius red (10 minutes in 0.1% sirius red dissolved in 1% picric acid). Blood vessels were stained using a Dako Autostainer Link 48 for an angiogenesis marker [i.e. CD31 (Abcam clone C31.3; ab187377)] (Dako Agilent, Santa Clara, CA, USA). A relevant mouse isotype control was undertaken and the detection system was horseradish peroxidase/DAB (3,3′-diaminobenzidine) with haematoxylin counterstain (Envisage FLEX; Dako Agilent, Santa Clara, CA, USA).
Immunohistochemistry sections were imaged to include most of the biopsy area for CD31 (six viewing fields at 200× magnification). Total cell count was obtained from haematoxylin-stained sections, using automatic nuclear counts from six random fields at 400× magnification (ImageJ version 1.51s; http://imagej.nih.gov/ij; accessed February 2019). Collagen density was assessed in four fields of view per section. In all fields of view, large blood vessels and adipose tissue were avoided. Images were scored by a blinded assessor. A modified Bonar scale81 was used and included analysis of the following components relevant to healing: (1) cellularity, (2) collagen fibre density and (3) vascularity. Each variable was scored on a 4-point scale of 0–3 (0, normal; 1, slightly abnormal; 2, abnormal; and 3, markedly abnormal). The samples were scored according to whether or not there was an abnormal appearance (i.e. hypercellular, hypervascular, low collagen). The total score varied between 0 (normal tendon) and 9 (severe abnormality). 80,81 Treatment groups were compared using two-tailed Mann–Whitney U-tests, with significance taken as a p-value of < 0.05.
Results
The bulk of tissue in all biopsies comprised abnormal tendon tissue in the process of active remodelling. There were very few intact mature collagen bundles, while cellularity and vascularity were typically high compared with normal tissue. One of the biopsies was severely compromised in cellularity and vascularity, with minimal intact collagen. It is not possible to assess whether this was prevalent ahead of the rupture injury or developed subsequently, but tissue repair could be challenging with few matrix-secreting cells and absence of vascular remodelling, and this biopsy was scored as 3 (or abnormal) for all three Bonar categories, as previously suggested. 81 The participant in question did not have a re-rupture, suggesting that the biopsy was not representative of the whole lesion.
In exploratory analysis, there were no detectable differences between placebo and PRP-treated groups in either cellularity or vascularity (Table 20). However, the PRP group had lower collagen fibre density. Overall, the combined Bonar score was higher for the PRP group.
| Bonar score | Intervention group, Bonar score | p-value | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | PRP | ||||||||
| Mean (SD) | Median (IQR) | Frequency (n = 7) | Relative frequency | Mean (SD) | Median (IQR) | Frequency (n = 9) | Relative frequency | ||
| Cellularitya | 1.71 (0.49) | 2 (1–2) | 5 | 0.71 | 1.89 (0.60) | 2 (1.5–2) | 6 | 0.67 | 0.665 |
| Collagen stainb | 1.14 (0.38) | 1 (1–1) | 6 | 0.86 | 2.00 (0.50) | 2 (2–2) | 7 | 0.78 | 0.007 |
| Vascularityc | 0.42 (0.53) | 0 (0–1) | 4 | 0.57 | 1.11 (1.05) | 1 (0–2) | 3 | 0.33 | 0.201 |
| Bonar score totald | 3.28 (0.76) | 3 (3–4) | 3 | 0.43 | 5.00 (1.87) | 5 (3.5–6) | 2 | 0.22 | 0.038 |
Platelet-rich plasma quality was measured in substudy 1 for all participants randomised to receive PRP injection. The nine participants from whom biopsies were obtained all received good-quality PRP with low platelet activation on collection and containing high growth factor levels after activation.
Discussion
Biopsies were obtained from 16 participants from one centre who consented to this component of the study. This substudy was intentionally exploratory so the sample size was accordingly limited and not powered for detecting between-group differences in specific measurements.
Tendon healing at 6 weeks after an acute rupture should have significantly progressed but will not be complete. Marked increases in both cellularity and vascularity must happen in order for healing to occur but should then reduce to normal tendon levels. In humans, collagen synthesis and remodelling can be expected to continue for months after the injury. Although Bonar and Movin scores have been developed to assess tendinopathy rather than recovery from injury, they are frequently used for both. 81,82 It is, in fact, difficult to separate the two processes in a biopsy as many tendons will have ruptured because of prior tendinopathy. 8 In this study, the centre of the rupture lesion was biopsied and the main difference in interpretation of scores is that an injured tendon in the process of healing has high cellularity, reduced and more-diffuse collagen and elevated vascularity, which are also common signs of tendinopathy in an uninjured tendinopathic biopsy. A badly compromised injured tendon will have very few cells (low score), massive collagen loss (high score) and few blood vessels (low score), which, overall, could score quite low or ‘good’ on a tendinopathy scale, but is clearly abnormal. It is therefore useful to consider relative differences between groups rather than absolute score.
Although all biopsies except one showed evidence of healing at 6 weeks, there was less collagen fibre density in the PRP group. This did not correlate with differences in cellularity or vascularity, as these parameters were similar in both groups, suggesting equivalent healing processes. There are several possible explanations. The collagen may be forming more slowly in the PRP-treated group or may have been subjected to an early phase of breakdown and is still catching up at 6 weeks. This was a small study of 16 biopsies, which might be prone to chance allocation. Ultimately, the speed of return to full function and the long-term re-rupture rate will indicate whether this difference in healing process is beneficial, negative or unimportant.
Chapter 6 Discussion
In this chapter, we summarise the main findings of the clinical trial and both substudies before considering the internal and external validity of the trial. Interpretation of the results and their relationship with other literature are discussed, followed by consideration of the implications for clinical practice and future research.
Aim and overview of clinical trial findings
Our primary objective was to evaluate the clinical efficacy of PRP among patients with acute ATR using an objective measure of muscle–tendon function, the LSI of work performed during the HRET at 24 weeks post randomisation. PROMs were used to study function, symptoms, pain and quality of life. An analysis of whole-blood and PRP biological components was embedded in the trial in order to determine key components of PRP that may contribute to its mechanism of action and to enhance quality assurance during the trial. A second substudy to explore the possible tissue and immunohistochemical effects of PRP on the healing tendon tissue compared with those of the control was also embedded in the trial and included 16 participants. Investigating efficacy and mechanism together in the present trial provides robust information on PRP application in ATR and offers a rigorous platform for future research into PRP applications in musculoskeletal problems.
This was a prospective, multicentre, parallel-group, participant- and outcome assessor-blinded, randomised, placebo-controlled superiority trial. A total of 230 patients presenting with acute ATR within 12 days of injury were recruited in 19 NHS hospitals in England and Wales.
We found that PRP was not effective in improving muscle–tendon function 24 weeks after acute ATR. There were also no differences in the secondary outcome measures of patient-reported function, symptoms, quality of life and pain at 24 weeks. There were no statistical differences in all secondary outcome measures between the PRP group and the placebo group throughout the follow-up period, indicating that there was no early acceleration of recovery. Including stratification factors and other predefined prognostic variables in the analysis, when relevant, had no impact on the attained primary and secondary outcome results. CACE and sensitivity analyses demonstrated that the conclusions were robust under a range of assumptions.
The PRP group and the control group had nearly the same rate of re-rupture, surgical repairs, swelling, DVT, skin breakdown or ulceration, swelling and casting-related complications. Secondary surgical intervention to repair the Achilles tendon was required by 4% (9/229) of participants; this was carried out on participants who failed non-surgical treatment and had re-rupture of the tendon.
Blood and platelet-rich plasma substudy findings
An analysis of whole-blood cellular composition showed that the two groups had similar baseline blood parameters. PRP analysis showed that the preparation method resulted in a mean 4.1-fold increase in platelet count and a 2.2-fold increase in white blood cell count compared with whole blood. The majority of platelets in the PRP were of good quality with low levels of activation. The PRP produced using the trial procedures was therefore functional, with the majority of platelets in the PRP preparations being shown to be capable of activation and degranulation, with a few exceptions. As would be expected, there was correlation between platelet and leucocyte counts and some growth factor concentrations.
Despite using a standardised preparation method to manage operator error, there was wide variation in platelet count, white blood cell count and growth factor concentrations; this indicates the variable nature of this biological product. However, this did not have an effect on the clinical outcome.
Clinical outcomes and platelet-rich plasma analysis correlation
Parameters of baseline whole blood taken before intervention in both groups did not correlate with the primary outcome measure at 24 weeks.
Overall, there was no association between PRP cellular or growth factor content on the primary outcome measure. The level of platelet activation and white blood cell concentration at baseline also did not correlate with the primary outcome. The results of this mechanistic analysis confirmed that any variability in this biological product did not change the outcome of ATR management.
Immunohistochemistry study findings
In substudy 2, with 16 participants, all except one biopsy showed evidence of healing at 6 weeks. There was less collagen fibre density in the PRP group. This did not correlate with differences in cellularity or vascularity, as these parameters were similar in both groups, suggesting equivalent healing processes.
The findings of this substudy could indicate that collagen was forming more slowly in the PRP group or was subjected to early breakdown at this early 6-week stage. However, this was a small-scale study, meaning that cautious interpretation is required.
The findings of this study could not be linked to the clinical outcome because of the small number of biopsies undertaken.
Internal validity and methodology
The trial was powered to be a definitive superiority trial and had lower than expected loss to follow-up. Blinding of participants and outcome assessors and low levels of missing data across the outcome measures strengthen the trial results. Allocation concealment was ensured through random computer allocation, stratified by site and age, on participant registration via a remote computer randomisation service. Analyses were pre-planned and agreed by the DSMC.
Platelet-rich plasma quality control and measurements in substudy 1 enabled ongoing monitoring during the trial so that any issues could be resolved. Measuring PRP activation and viability allowed us to ensure that viable PRP was injected and also allowed the correlation with the clinical primary outcome measure. During the initial phase of the trial, four PRP samples showed very low platelet counts. The trial was temporarily halted and preparation methods, devices and kit were tested. This was thought to be due to faulty sensors in some devices. Those devices and kits were replaced and the trial continued without any further issues of that nature.
Platelet-rich plasma products vary according to preparation techniques and devices. 83 In this trial, standardising the preparation method and providing the same kit and PRP preparation devices to all centres ensured that any significant variability due to different preparation techniques and devices was eliminated. The results of substudy 1 still show variability in PRP content and activation; however, this is probably due to the variable nature of this biological product rather than preparation methods.
Complier-average causal effect analysis was used to estimate the treatment effect while accounting for whether participants complied or did not comply with the treatment allocated to them (without assuming that compliers were the same as non-compliers as would be the case with PP analysis). The consistency between the conclusions of the mITT findings and CACE analysis improves the confidence in the findings of the PATH-2 trial.
The validation checks on the HRET (see Appendix 3) identified invalid data on a small number of participants and it was reassuring that the conclusions from the estimates with raw HRET data and those with valid data after the checks were consistent. To dismiss potential measurement errors, two members of the trial team who were blinded to treatment allocation reviewed the videos of all assessments independently and any invalid heel-raise repetitions included in the HRET data were discounted. The test was standardised through training the blinded assessors and by offering trial-specific guidance notes and videos.
In the context of the PATH-2 trial, the conclusions drawn from the work LSI are supported by the high precision of estimates and the consistency with the secondary outcomes (ATRS, PSFS, SF-12 and VAS).
On formal analysis, the success of the outcome assessor blinding at the end point was adequate. Both the James et al. 69 and Bang et al. 70 blinding indices gave evidence to support the assumption that, overall, participants in this trial were blinded. This demonstrates that the findings of this trial are extremely unlikely to be due to participants’ expectations of treatment received.
External validity and generalisability
The trial was conducted in acute hospitals, from major trauma centres through to smaller district general hospitals, and, therefore, is representative of the range of settings for ATR management in the UK. The PRP and placebo interventions were delivered by NHS trauma and orthopaedic teams with relatively little trial-specific training. Rehabilitation guidelines were set to augment some standardisation of care for trial participants. All participants should therefore have had their injured lower limb immobilised for at least 3 weeks after injection, avoided more than 6 weeks of rigid full-time immobilisation without ankle motion or weight-bearing and received a referral for physiotherapy. Compliance with these different aspects was measured and was good, with no difference between treatment groups.
The demographics of the trial participants were representative of the age and sex profile expected of adults with ATR,9 with the majority being male and aged < 55 years. Excluding patients with rupture of the Achilles tendon at the insertion or musculotendinous junction limits generalisability. However, the healing in these areas is usually better than that of mid-substance rupture. 8
The primary end point at 24 weeks could be considered too early for definitive assessment of outcome. However, a difference in speed of healing should have been seen when the tendon is in the recovery phase, and our PATH-2 trial primary outcome measure at 24 weeks post injury is timed to capture this. In addition, the theoretical mode of action of PRP by enhancement of cellular response in the early inflammatory healing phase would be expected to peak in the early stages of Achilles tendon repair. A meta-analysis32 demonstrated that improvement of patient outcomes from previously published studies of ATR showed that most improvement occurs in the first 6 months, which, therefore, supports this as the definitive assessment point.
There were no discernible differences between groups in relation to the grade of the clinician administering treatment. Notably, the clinicians were mostly consultant surgeons (75%) and surgical registrars (17%). A number of extended-scope physiotherapists and research fellows have applied the intervention, reflecting the range of clinicians in contemporary orthopaedic clinical practice.
Implications for clinical practice
Achilles tendon rupture leads to a major impairment in function, irrespective of the treatment received. The findings demonstrate that participants recovered < 40% of the muscle–tendon unit function at 24 weeks. This observation is also supported by the secondary outcome measures ATRS and PSFS. The majority of participants continued to experience pain at the end point, although of significantly less severity than at baseline. This underlines the impact of this injury on individuals in terms of day-to-day activities, as well as the wider socioeconomic impact.
The PATH-2 trial results are important for clinicians practising in this field who are aware of PRP use and who may receive requests from patients to administer PRP by injection. These results challenge the growing use of PRP in ATR. For patients, who are increasingly being involved in decision-making about their treatments, these findings are an important contribution to that discussion. The similarity in the primary outcome findings at 24 weeks and for the patient-reported secondary outcomes throughout follow-up strongly support the validity of the conclusion that PRP does not improve the outcome of ATR management. Therefore, the application of PRP for this indication appears unjustified and without benefit.
The trial results also showed that injection is associated with some AEs at the injection site, with 11 out of 229 (5%) participants reporting frequent discomfort and 3 out of 229 (1%) participants reporting clinically diagnosed infections. However, no clinically serious infections were reported.
Re-rupture rates were low in both groups, but it should be noted that the trial was not powered to detect a between-group difference for this outcome.
Platelet-rich plasma application in clinical settings has a significant cost implication for health-care providers. Although we did not carry out a health economic analysis, applying PRP in ATR management will add to the cost of standard care. The cost of applying PRP is variable, but includes (1) a centrifuge, (2) a PRP disposable kit per patient, (3) an injection kit, (4) blood withdrawal and preparation time (15–30 minutes) and (5) surgeon or practitioner injection time (10 minutes).
In addition, PRP injection is usually administered in a fracture or trauma clinic in NHS hospitals, which are busy, with limited time and space allocation for each patient. The time taken to withdraw blood and prepare the PRP and the injection may have an impact on patient flow in those clinics. The injection is usually administered in a clinic room or cubicle and adds to the burden on clinic staff.
A recent report estimated that the value of the PRP market was US$214.3M in 2016, which is expected to reach US$625M globally by 2025. 84 This market estimate does not include the cost to health-care providers in terms of staff and clinic space; however, it does include PRP applications in all specialties. PRP application in sport injuries in Europe is rapidly increasing. 85 Current PRP cost in ATR is unknown but likely to be growing. 85 The findings of this trial will help inform clinical decision-makers, commissioners and payers for health-care services and may limit the cost burden of PRP in ATR management.
Limitations
In the PATH-2 trial, we used and assessed a single PRP preparation system based on a previous study86 to standardise the procedure. There are three methods to prepare PRP: (1) gravitational platelet sequestration or centrifugation, (2) standard cell separators and (3) selective filtration technology or plateletpheresis. Clinically approved devices use one of these methods to produce PRP from autologous blood with varying concentrations and platelet yields. We selected a fully automated system that utilises infrared sensors to detect platelet layers in the centrifuged blood and automatically load it to a syringe, ready to be injected. Our tests of this device and the PRP method in the pilot study importantly demonstrated a viable product and a reliable preparation system. All PRP preparation devices produce varying concentrations of platelets and white blood cells; however, we have shown in this trial that there was no link between platelet count or white blood cell count and the clinical effect.
Nine per cent of participants in the PRP group did not receive the allocated treatment. The allocated intervention for participants in the PRP group was dependent on the ability to withdraw sufficient whole blood to produce the required 8-ml PRP sample, a fully working centrifuge, an available in-date PRP preparation kit and patients willing to wait the time for the preparation. This reflects the reality of this intervention in usual acute NHS hospital settings.
Standardising the rehabilitation protocol across sites can hinder the pragmatic approach of the trial. Within an agreed common approach, we allowed rehabilitation to be tailored to the participant and local provision. All participants had to be referred for physiotherapy but then it was the responsibility of the therapy service to provide what was their usual care. This pragmatic approach may have resulted in some site-to-site variation in provision, but we stratified randomisation and adjusted primary treatment estimates on centre to help manage this.
Documentation of medication that may have an effect on platelet function, such as aspirin or clopidogrel, was not requested until after the trial had started; therefore, information on this was available for only 65% of patients. Only 5.7% of patients who were asked about this were on antiplatelet medication, and this was similar between the two treatment arms; therefore, it was unlikely to have an influence on outcomes.
The timing of the primary end point (24 weeks) may be considered early for final functional recovery of the tendon. However, the results indicate no effect at this early stage when PRP was expected to exert its maximum effect in enhancing recovery. A 2-year follow-up of patient-reported data is being conducted and the results will be reported in due course.
The number of histological study biopsies was too small to secure statistically significant results of the effect of PRP on the tendon tissues. The substudy findings were intentionally considered exploratory and indicative rather than confirmative of the changes in the healing tendon. The limited number of biopsies is within the generally accepted range for similar studies on human participants.
Further research
The PATH-2 trial found no evidence of efficacy for PRP in the treatment of acute ATR. The implications are that similarly robust clinical trials are recommended for investigating the efficacy of PRP applications for other musculoskeletal disorders.
The extent of muscle–tendon impairment identified in the HRET in the injured limb, irrespective of treatment group, highlights the degree of ongoing musculotendinous unit compromise 6 months after tendon rupture. Although musculotendinous impairment has been found in other ATR cohorts,35 the extent of asymmetry between injured and uninjured legs in this trial was substantial. Optimising recovery of neuromuscular function during rehabilitation is therefore a recommended area of future investigation.
There were some variations in the non-surgical management of acute ATRs within the range permitted in the standardised rehabilitation guidelines for the PATH-2 trial. An ongoing clinical trial funded by the NIHR Health Technology Assessment programme (project number 13/115/62) will provide additional evidence regarding the clinical effectiveness and cost-effectiveness of two commonly used protocols: plaster casting versus a rigid functional brace with immediate weight-bearing. 87
Acknowledgements
Thank you to the team at the Institute of Inflammation and Ageing Laboratory, University of Birmingham, that conducted the following: whole-blood and PRP analysis (Dr Paul Harrison, Ms Marie Didembourg, Miss Amarpreet Devi, Dr Rob Dinsdale, Dr Jon Hazeldine, Miss Hema Chahal, Ms Kirsty McGee), and PRP growth factor measurements (Mr Alexander Wood and Dr Paul Harrison). Special thanks to Dr James Teh (Consultant Radiologist) for conducting the guided biopsies for substudy 2, and Mrs Lesley Morgan and Miss Emma Roberts, who facilitated the PATH-2 trial set-up phase.
Contributions of authors
Joseph Alsousou (NIHR Academic Clinical Fellow in Orthopaedics) developed the trial conception, and intervention, designed the trial, developed substudies 1 and 2 and wrote and reviewed the report.
David J Keene (NIHR Postdoctoral Research Fellow, University of Oxford) developed the trial conception, and intervention, was clinical co-ordinator, designed the trial, delivered intervention training and wrote and reviewed the report.
Paul Harrison (Senior Lecturer, University of Birmingham) designed and led substudy 1, conducted the blood and PRP analysis and wrote the report.
Philippa Hulley (Associate Professor, University of Oxford) designed and led substudy 2, conducted the immunohistochemistry analysis and wrote the report.
Susan Wagland (Clinical Trials Manager, University of Oxford) managed and supervised the trial, and wrote and reviewed the report.
Jacqueline Y Thompson (Research Physiotherapist, University of Oxford) delivered the HRET training and wrote the report.
Scott R Parsons (Trial Co-ordinator, University of Oxford) provided administrative and project co-ordination support, and reviewed the report.
Christopher Byrne (Lecturer in Physiotherapy, University of Plymouth) developed the HRET assessment, delivered the HRET training and wrote the report.
Michael M Schlüssel (Medical Statistician, University of Oxford) was trial statistician, conducted the statistical analysis and wrote the report.
Heather M O’Connor (Medical Statistician, University of Oxford) was trial statistician, conducted the statistical analysis and wrote the report.
Susan J Dutton (University Lecturer and OCTRU Lead Statistician, University of Oxford) was senior trial statistician, conducted the statistical analysis and wrote the report.
Sarah E Lamb (Professor in Rehabilitation and Director of Centre for Statistics in Medicine, University of Oxford) developed the trial conception and design, and reviewed the report.
Keith Willett (Professor in Orthopaedic Trauma Surgery, University of Oxford; Medical Director for Acute Care, NHS England) was chief investigator, led the funding application, trial conception and design, and development of interventions, provided overall trial supervision and wrote and reviewed the report.
The PATH-2 trial team
The following members of staff were members of the central trial team:
-
Ramona Barbu, Clinical Trial Support Officer
-
Christopher Byrne, Research Physiotherapist
-
Daryl Hagan, Clinical Trial Administrative Co-ordinator
-
David Keene, Research Physiotherapist, Research Fellow
-
Lesley Morgan, Clinical Trial Manager
-
Scott Parsons, Clinical Trial Administrative Co-ordinator
-
Emma Roberts, Clinical Trial Administrative Co-ordinator
-
Jacqueline Y Thompson, Research Physiotherapist
-
Susan Wagland, Clinical Trial Manager
-
Keith Willett, Chief Investigator.
Trial Management Group
-
Chief Investigator: Professor Keith Willett.
-
Trial Co-investigators: Mr Joseph Alsousou, Dr David J Keene, Dr Paul Harrison, Dr Philippa Hulley, Mrs Susan J Dutton, Professor Sarah E Lamb.
-
Trial Manager: Dr Susan Wagland.
-
Trial Co-ordinator and Administrator: Dr Scott Parsons.
-
Trial Statisticians: Heather M O’Connor, Dr Michael M Schlüssel, Mrs Susan J Dutton.
Principal investigators and research associates by centre
| PI | Research associates |
|---|---|
| Oxford University Hospitals NHS Foundation Trust | |
| Robert Handley | Kathryn Lewis |
| Louise Spoors | |
| Yuhan Zhang | |
| Maria Mestre | |
| Doreeen Muller | |
| Sangeetha Prasath | |
| Martin Austin | |
| University Hospitals of Leicester NHS Trust | |
| Maneesh Bhatia | Manjit Attwal |
| Carla Christie | |
| Tracy Brear | |
| Taunton and Somerset NHS Foundation Trust | |
| Andrew Kelly | Keira Beacham |
| Sarah Brown | |
| James Allen | |
| Alison Whitcher | |
| Kate James | |
| Eliza Foster | |
| North Bristol NHS Trust | |
| Steve Hepple | Steven Barnfield |
| Katherine Coates | |
| Ruth Halliday | |
| Lucille Hooper | |
| Cardiff and Vale University Health Board | |
| Paul Hodgson | Helen Hodgson |
| Cheryl Cleary | |
| Claire Nott | |
| Jessica Whiteman | |
| Shrewsbury and Telford Hospital NHS Trust | |
| Michael Carmont | Louise Tonks |
| Barts Health NHS Trust | |
| Nima Heidari | Jamila Kassam |
| Catherine Hilton | |
| Shanaz Ahmad | |
| Sophie Young | |
| University Hospitals Coventry and Warwickshire NHS Trust | |
| Jonathan Young | Louise Clarkson |
| Sylvia Turner | |
| Kerri McGowan | |
| Janet Lowe | |
| Warrington and Halton Hospitals NHS Foundation Trust | |
| Gareth Stables | Kerry Bunworth |
| Pascal de Feyter | |
| Basildon and Thurrock University Hospitals NHS Foundation Trust | |
| Ranjith Ravindran | Mark Vertue |
| Royal Liverpool and Broadgreen University Hospitals NHS Trust | |
| Simon Frostick (co-PI: Joseph Alsousou) | Chris Walker |
| Sivia Sirikonda | |
| Anjani Sighn | |
| Joanne Stokes-Denson | |
| Lisa Watson | |
| Deborah Scanlon | |
| Philip Walker | |
| Peterborough and Stamford Hospitals NHS Foundation Trust | |
| James Carmichael | Susie O’Sullivan |
| Deborah Butcher | |
| Abertawe Bro Morgannwg University Health Board | |
| Claire Topliss (co-PI: Anne-Marie Hutchison) | Sharon Storton |
| Amanda Cook | |
| Leanne Quinn | |
| Aintree University Hospital NHS Foundation Trust | |
| Lyndon Mason (co-PI: Joseph Alsousou) | Phil Ellison |
| Andrew Molly | |
| Joanne Earley | |
| University Hospital of South Manchester NHS Foundation Trust | |
| Moez Ballal | Joanne Bradley-Potts |
| Georgina Williamson | |
| Richard McCormick | |
| Sheffield Teaching Hospitals NHS Foundation Trust | |
| Mark Davies | Julie Walker |
| Joanne Badloe | |
| Royal Devon and Exeter NHS Foundation Trust | |
| Adrian Hughes | Anna Grice |
| Jayne Dark | |
| Mid Cheshire Hospitals NHS Foundation Trust | |
| Simon Barnes (co-PI: Jagan Velpula) | Kelly Amor |
| Janet Brown | |
| Royal Surrey County NHS Foundation Trust | |
| Matthew Solan | |
Trial Steering Committee
Professor Roger Smith (Chairperson), Dr Catriona Graham, Dr Dylan Morrissey, Professor John O’Byrne, Dr Gustaaf Reurink and Mrs Sarah Webb.
Data and Safety Monitoring Committee
Dr Chao Huang (Chairperson), Mr Andy Goldberg and Dr Carey McClellan.
Copyright acknowledgements
Paper version of SF-1992–2002 by Optum, Inc. SF-12, and SF-12v2 are trademarks of the Medical Outcomes Trust and are used under license. The SF-12v2 Health Survey is copyrighted by Optum, Inc.
Interviewer script for SF-12 1992, 2002, 2011 by Optum, Inc. SF-12, and SF-12v2 are trademarks of the Medical Outcomes Trust and are used under license. The SF-12v2 Health Survey is copyrighted by Optum, Inc.
Publications
Keene DJ, Alsousou J, Willett K. How effective are platelet rich plasma injections in treating musculoskeletal soft tissue injuries? BMJ 2016;352:i517.
Alsousou J, Keene DJ, Hulley PA, Harrison P, Wagland S, Byrne C, et al. Platelet rich plasma in Achilles Tendon Healing 2 (PATH-2) trial: protocol for a multicentre, participant and assessor-blinded, parallel-group randomised clinical trial comparing platelet-rich plasma (PRP) injection versus placebo injection for Achilles tendon rupture. BMJ Open 2017;7:e018135.
Byrne C, Keene DJ, Lamb SE, Willett K. Intrarater reliability and agreement of linear encoder derived heel-rise endurance test outcome measures in healthy adults. J Electromyogr Kinesiol 2017;36:34–9.
Schlüssel MM, Keene DJ, Wagland S, Alsousou J, Lamb SE, Willett K, et al. Platelet-rich plasma in Achilles tendon healing 2 (PATH-2) trial: statistical analysis plan for a multicentre, double-blinded, parallel-group, placebo-controlled randomised clinical trial. Trials 2018;19:464.
Keene DJ, Alsousou J, Harrison P, Hulley P, Wagland S, Parsons SR, et al. Platelet rich plasma injection for acute Achilles tendon rupture: PATH-2 randomised, placebo controlled, superiority trial. BMJ 2019;367:l6132.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Cummins EJ AB, Carr BW, Wrigh RR. The structure of calcaneal tendon (of Achilles) in relation to orthopaedic surgery with additional observations on the plantaris muscle. Surg Gynecol Obstet 1946;83:107-16.
- Canale ST, Beaty J. Campbell’s Operative Orthopeadics. Maryland Heights, MO: Elsevier Mosby; 2013.
- Lichtwark GA, Wilson AM. In vivo mechanical properties of the human Achilles tendon during one-legged hopping. J Exp Biol 2005;208:4715-25. https://doi.org/10.1242/jeb.01950.
- Huttunen TT, Kannus P, Rolf C, Felländer-Tsai L, Mattila VM. Acute Achilles tendon ruptures: incidence of injury and surgery in Sweden between 2001 and 2012. Am J Sports Med 2014;42:2419-23. https://doi.org/10.1177/0363546514540599.
- Oda H, Sano K, Kunimasa Y, Komi PV, Ishikawa M. Neuromechanical modulation of the Achilles tendon during bilateral hopping in patients with unilateral Achilles tendon rupture, over 1 year after surgical repair. Sports Med 2017;47:1221-30. https://doi.org/10.1007/s40279-016-0629-3.
- Clayton RA, Court-Brown CM. The epidemiology of musculoskeletal tendinous and ligamentous injuries. Injury 2008;39:1338-44. https://doi.org/10.1016/j.injury.2008.06.021.
- Mattila VM, Huttunen TT, Haapasalo H, Sillanpää P, Malmivaara A, Pihlajamäki H. Declining incidence of surgery for Achilles tendon rupture follows publication of major RCTs: evidence-influenced change evident using the Finnish registry study. Br J Sports Med 2015;49:1084-6. https://doi.org/10.1136/bjsports-2013-092756.
- Maffulli N, Longo UG, Maffulli GD, Rabitti C, Khanna A, Denaro V. Marked pathological changes proximal and distal to the site of rupture in acute Achilles tendon ruptures. Knee Surg Sports Traumatol Arthrosc 2011;19:680-7. https://doi.org/10.1007/s00167-010-1193-2.
- Maffulli N, Waterston SW, Squair J, Reaper J, Douglas AS. Changing incidence of Achilles tendon rupture in Scotland: a 15-year study. Clin J Sport Med 1999;9:157-60. https://doi.org/10.1097/00042752-199907000-00007.
- Gulati V, Jaggard M, Al-Nammari SS, Uzoigwe C, Gulati P, Ismail N, et al. Management of Achilles tendon injury: a current concepts systematic review. World J Orthop 2015;6:380-6. https://doi.org/10.5312/wjo.v6.i4.380.
- Leppilahti J, Puranen J, Orava S. Incidence of Achilles tendon rupture. Acta Orthop Scand 1996;67:277-9. https://doi.org/10.3109/17453679608994688.
- Khan RJ, Fick D, Brammar TJ, Crawford J, Parker MJ. Interventions for treating acute Achilles tendon ruptures. Cochrane Database Syst Rev 2004;3. https://doi.org/10.1002/14651858.CD003674.pub2.
- Yang G, Rothrauff BB, Tuan RS. Tendon and ligament regeneration and repair: clinical relevance and developmental paradigm. Birth Defects Res C Embryo Today 2013;99:203-22. https://doi.org/10.1002/bdrc.21041.
- Pierre-Jerome C, Moncayo V, Terk MR. MRI of the Achilles tendon: a comprehensive review of the anatomy, biomechanics, and imaging of overuse tendinopathies. Acta Radiol 2010;51:438-54. https://doi.org/10.3109/02841851003627809.
- Schwieterman B, Haas D, Columber K, Knupp D, Cook C. Diagnostic accuracy of physical examination tests of the ankle/foot complex: a systematic review. Int J Sports Phys Ther 2013;8:416-26.
- Amlang MH, Zwipp H, Friedrich A, Peaden A, Bunk A, Rammelt S. Ultrasonographic classification of achilles tendon ruptures as a rationale for individual treatment selection. ISRN Orthop 2011;2011. https://doi.org/10.5402/2011/869703.
- Voleti PB, Buckley MR, Soslowsky LJ. Tendon healing: repair and regeneration. Annu Rev Biomed Eng 2012;14:47-71. https://doi.org/10.1146/annurev-bioeng-071811-150122.
- Fenwick SA, Hazleman BL, Riley GP. The vasculature and its role in the damaged and healing tendon. Arthritis Res 2002;4:252-60. https://doi.org/10.1186/ar416.
- Ahmed IM, Lagopoulos M, McConnell P, Soames RW, Sefton GK. Blood supply of the Achilles tendon. J Orthop Res 1998;16:591-6. https://doi.org/10.1002/jor.1100160511.
- Brockis JG. The blood supply of the flexor and extensor tendons of the fingers in man. J Bone Joint Surg Br 1953;35–B:131-8. https://doi.org/10.1302/0301-620X.35B1.131.
- Kjaer M, Langberg H, Miller BF, Boushel R, Crameri R, Koskinen S, et al. Metabolic activity and collagen turnover in human tendon in response to physical activity. J Musculoskelet Neuronal Interact 2005;5:41-52.
- Thermann H, Frerichs O, Biewener A, Krettek C, Schandelmaier P. Biomechanical studies of human Achilles tendon rupture. Unfallchirurg 1995;98:570-5.
- Rolf CG, Fu SC, Hopkins C, Luan J, Ip M, Yung SH, et al. Presence of bacteria in spontaneous Achilles tendon ruptures. Am J Sports Med 2017;45:2061-7. https://doi.org/10.1177/0363546517696315.
- Metz R, Kerkhoffs GM, Verleisdonk EJ, van der Heijden GJ. Acute Achilles tendon rupture: minimally invasive surgery versus non operative treatment, with immediate full weight bearing. Design of a randomized controlled trial. BMC Musculoskelet Disord 2007;8. https://doi.org/10.1186/1471-2474-8-108.
- Frankewycz B, Krutsch W, Weber A, Arnstberger M, Nerlich M, Pfeifer CG. Rehabilitation of Achilles tendon ruptures: is early functional rehabilitation daily routine?. Arch Orthop Trauma Surg 2017;137:333-40. https://doi.org/10.1007/s00402-017-2627-9.
- Kadakia AR, Dekker RG, Ho BS. Acute Achilles tendon ruptures: an update on treatment. J Am Acad Orthop Surg 2017;25:23-31. https://doi.org/10.5435/JAAOS-D-15-00187.
- Byrne PA, Hopper GP, Wilson WT, Mackay GM. Knotless repair of Achilles tendon rupture in an elite athlete: return to competition in 18 weeks. J Foot Ankle Surg 2017;56:121-4. https://doi.org/10.1053/j.jfas.2016.07.007.
- Costa ML, MacMillan K, Halliday D, Chester R, Shepstone L, Robinson AH, et al. Randomised controlled trials of immediate weight-bearing mobilisation for rupture of the tendo Achillis. J Bone Joint Surg Br 2006;88:69-77. https://doi.org/10.1302/0301-620X.88B1.16549.
- Mark-Christensen T, Troelsen A, Kallemose T, Barfod KW. Functional rehabilitation of patients with acute Achilles tendon rupture: a meta-analysis of current evidence. Knee Surg Sports Traumatol Arthrosc 2016;24:1852-9. https://doi.org/10.1007/s00167-014-3180-5.
- McCormack R, Bovard J. Early functional rehabilitation or cast immobilisation for the postoperative management of acute Achilles tendon rupture? A systematic review and meta-analysis of randomised controlled trials. Br J Sports Med 2015;49. https://doi.org/10.1136/bjsports-2015-094935.
- Heikkinen J, Lantto I, Flinkkila T, Ohtonen P, Niinimaki J, Siira P, et al. Soleus Atrophy is common after the nonsurgical treatment of acute Achilles tendon ruptures: a randomized clinical trial comparing surgical and nonsurgical functional treatments. Am J Sports Med 2017;45:1395-404. https://doi.org/10.1177/0363546517694610.
- Khan RJ, Fick D, Keogh A, Crawford J, Brammar T, Parker M. Treatment of acute achilles tendon ruptures. A meta-analysis of randomized, controlled trials. J Bone Joint Surg Am 2005;87:2202-10. https://doi.org/10.2106/00004623-200510000-00008.
- Kearney RS, McGuinness KR, Achten J, Costa ML. A systematic review of early rehabilitation methods following a rupture of the Achilles tendon. Physiotherapy 2012;98:24-32. https://doi.org/10.1016/j.physio.2011.04.349.
- Zhao JG, Meng XH, Liu L, Zeng XT, Kan SL. Early functional rehabilitation versus traditional immobilization for surgical Achilles tendon repair after acute rupture: a systematic review of overlapping meta-analyses. Sci Rep 2017;7. https://doi.org/10.1038/srep39871.
- Brorsson A, Grävare Silbernagel K, Olsson N, Nilsson Helander K. Calf muscle performance deficits remain 7 years after an Achilles tendon rupture. Am J Sports Med 2018;46:470-7. https://doi.org/10.1177/0363546517737055.
- Keene DJ, Alsousou J, Willett K. How effective are platelet-rich plasma injections in treating musculoskeletal soft tissue injuries?. BMJ 2016;352. https://doi.org/10.1136/bmj.i517.
- Fox G, Gabbe BJ, Richardson M, Oppy A, Page R, Edwards ER, et al. Twelve-month outcomes following surgical repair of the Achilles tendon. Injury 2016;47:2370-4. https://doi.org/10.1016/j.injury.2016.07.013.
- Oudelaar BW, Peerbooms JC, Huis In‘t Veld R, Vochteloo AJH. Concentrations of blood components in commercial platelet-rich plasma separation systems: a review of the literature. Am J Sports Med 2019;47:479-87. https://doi.org/10.1177/0363546517746112.
- Nguyen RT, Borg-Stein J, McInnis K. Applications of platelet-rich plasma in musculoskeletal and sports medicine: an evidence-based approach. PM R 2011;3:226-50. https://doi.org/10.1016/j.pmrj.2010.11.007.
- Alsousou J, Ali A, Willett K, Harrison P. The role of platelet-rich plasma in tissue regeneration. Platelets 2013;24:173-82. https://doi.org/10.3109/09537104.2012.684730.
- Engebretsen L, Steffen K, Alsousou J, Anitua E, Bachl N, Devilee R, et al. IOC consensus paper on the use of platelet-rich plasma in sports medicine. Br J Sports Med 2010;44:1072-81. https://doi.org/10.1136/bjsm.2010.079822.
- Akeda K, An HS, Okuma M, Attawia M, Miyamoto K, Thonar EJ, et al. Platelet-rich plasma stimulates porcine articular chondrocyte proliferation and matrix biosynthesis. Osteoarthr Cartil 2006;14:1272-80. https://doi.org/10.1016/j.joca.2006.05.008.
- Anitua E, Andía I, Sanchez M, Azofra J, del Mar Zalduendo M, de la Fuente M, et al. Autologous preparations rich in growth factors promote proliferation and induce VEGF and HGF production by human tendon cells in culture. J Orthop Res 2005;23:281-6. https://doi.org/10.1016/j.orthres.2004.08.015.
- Anitua E, Sánchez M, Zalduendo MM, de la Fuente M, Prado R, Orive G, et al. Fibroblastic response to treatment with different preparations rich in growth factors. Cell Prolif 2009;42:162-70. https://doi.org/10.1111/j.1365-2184.2009.00583.x.
- Lyras DN, Kazakos K, Tryfonidis M, Agrogiannis G, Botaitis S, Kokka A, et al. Temporal and spatial expression of TGF-beta1 in an Achilles tendon section model after application of platelet-rich plasma. Foot Ankle Surg 2010;16:137-41. https://doi.org/10.1016/j.fas.2009.09.002.
- Bielecki T, Dohan Ehrenfest DM, Everts PA, Wiczkowski A. The role of leukocytes from L-PRP/L-PRF in wound healing and immune defense: new perspectives. Curr Pharm Biotechnol 2012;13:1153-62. https://doi.org/10.2174/138920112800624373.
- de Mos M, van der Windt AE, Jahr H, van Schie HT, Weinans H, Verhaar JA, et al. Can platelet-rich plasma enhance tendon repair? A cell culture study. Am J Sports Med 2008;36:1171-8. https://doi.org/10.1177/0363546508314430.
- Runesson E, Ackermann P, Karlsson J, Eriksson BI. Nucleostemin- and Oct 3/4-positive stem/progenitor cells exhibit disparate anatomical and temporal expression during rat Achilles tendon healing. BMC Musculoskelet Disord 2015;16. https://doi.org/10.1186/s12891-015-0658-3.
- Sadoghi P, Rosso C, Valderrabano V, Leithner A, Vavken P. The role of platelets in the treatment of Achilles tendon injuries. J Orthop Res 2013;31:111-18. https://doi.org/10.1002/jor.22199.
- Taylor DW, Petrera M, Hendry M, Theodoropoulos JS. A systematic review of the use of platelet-rich plasma in sports medicine as a new treatment for tendon and ligament injuries. Clin J Sport Med 2011;21:344-52. https://doi.org/10.1097/JSM.0b013e31821d0f65.
- Gholami M, Ravaghi H, Salehi M, Yekta AA, Doaee S, Jaafaripooyan E. A systematic review and meta-analysis of the application of platelet rich plasma in sports medicine. Electron Physician 2016;8:2325-32. https://doi.org/10.19082/2325.
- Filardo G, Presti ML, Kon E, Marcacci M. Nonoperative biological treatment approach for partial Achilles tendon lesion. Orthopedics 2010;33:120-3. https://doi.org/10.3928/01477447-20100104-31.
- Sampson S, Aufiero D, Meng M, Bledin A, Gillette T, Zall M. Platelet-rich plasma therapy as a first-line treatment for severe Achilles tendon tear: a case report. Int J Ther Rehabil 2011;18:101-5. https://doi.org/10.12968/ijtr.2011.18.2.101.
- De Carli A, Lanzetti RM, Ciompi A, Lupariello D, Vandalà A, Argento G, et al. Can platelet-rich plasma have a role in Achilles tendon surgical repair?. Knee Surg Sports Traumatol Arthrosc 2016;24:2231-7. https://doi.org/10.1007/s00167-015-3580-1.
- Sánchez M, Anitua E, Azofra J, Andía I, Padilla S, Mujika I. Comparison of surgically repaired Achilles tendon tears using platelet-rich fibrin matrices. Am J Sports Med 2007;35:245-51. https://doi.org/10.1177/0363546506294078.
- Schepull T, Kvist J, Norrman H, Trinks M, Berlin G, Aspenberg P. Autologous platelets have no effect on the healing of human achilles tendon ruptures: a randomized single-blind study. Am J Sports Med 2011;39:38-47. https://doi.org/10.1177/0363546510383515.
- Alsousou J, Thompson M, Harrison P, Willett K, Franklin S. Effect of platelet-rich plasma on healing tissues in acute ruptured Achilles tendon: a human immunohistochemistry study. Lancet 2015;385. https://doi.org/10.1016/S0140-6736(15)60334-8.
- Zou J, Mo X, Shi Z, Li T, Xue J, Mei G, et al. A Prospective study of platelet-rich plasma as biological augmentation for Acute achilles tendon rupture repair. Biomed Res Int 2016;2016. https://doi.org/10.1155/2016/9364170.
- NICE . Interventional Procedures Guidance [IPG438] n.d. www.nice.org.uk/guidance/IPG438 (accessed 8 August 2019).
- Wasterlain AS, Braun HJ, Harris AH, Kim HJ, Dragoo JL. The systemic effects of platelet-rich plasma injection. Am J Sports Med 2013;41:186-93. https://doi.org/10.1177/0363546512466383.
- Grassi A, Napoli F, Romandini I, Samuelsson K, Zaffagnini S, Candrian C, et al. Is platelet-rich plasma (PRP) effective in the treatment of acute muscle injuries? A systematic review and meta-analysis. Sports Med 2018;48:971-89. https://doi.org/10.1007/s40279-018-0860-1.
- Sheth U, Simunovic N, Klein G, Fu F, Einhorn TA, Schemitsch E, et al. Efficacy of autologous platelet-rich plasma use for orthopaedic indications: a meta-analysis. J Bone Joint Surg Am 2012;94:298-307. https://doi.org/10.2106/JBJS.K.00154.
- Horn KK, Jennings S, Richardson G, Vliet DV, Hefford C, Abbott JH. The patient-specific functional scale: psychometrics, clinimetrics, and application as a clinical outcome measure. J Orthop Sports Phys Ther 2012;42:30-42. https://doi.org/10.2519/jospt.2012.3727.
- Stratford P, Gill C, Westaway M, Binkley J. Assessing disability and change on individual patients: a report of a patient specific measure. Physiother Can 1995;47:258-63. https://doi.org/10.3138/ptc.47.4.258.
- Nilsson-Helander K, Silbernagel KG, Thomeé R, Faxén E, Olsson N, Eriksson BI, et al. Acute achilles tendon rupture: a randomized, controlled study comparing surgical and nonsurgical treatments using validated outcome measures. Am J Sports Med 2010;38:2186-93. https://doi.org/10.1177/0363546510376052.
- Nilsson-Helander K, Thomeé R, Silbernagel KG, Grävare-Silbernagel K, Thomeé P, Faxén E, et al. The Achilles tendon Total Rupture Score (ATRS): development and validation. Am J Sports Med 2007;35:421-6. https://doi.org/10.1177/0363546506294856.
- Ware J, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care 1996;34:220-33. https://doi.org/10.1097/00005650-199603000-00003.
- Häggmark T, Liedberg H, Eriksson E, Wredmark T. Calf muscle atrophy and muscle function after non-operative vs operative treatment of achilles tendon ruptures. Orthopedics 1986;9:160-4.
- James KE, Bloch DA, Lee KK, Kraemer HC, Fuller RK. An index for assessing blindness in a multi-centre clinical trial: disulfiram for alcohol cessation -- a VA cooperative study. Stat Med 1996;15:1421-34. https://doi.org/10.1002/(SICI)1097-0258(19960715)15:13<1421::AID-SIM266>3.0.CO;2-H.
- Bang H, Ni L, Davis CE. Assessment of blinding in clinical trials. Control Clin Trials 2004;25:143-56. https://doi.org/10.1016/j.cct.2003.10.016.
- Knox CR, Lall R, Hansen Z, Lamb SE. Treatment compliance and effectiveness of a cognitive behavioural intervention for low back pain: a complier average causal effect approach to the BeST data set. BMC Musculoskelet Disord 2014;15. https://doi.org/10.1186/1471-2474-15-17.
- Imbens GW, Rubin DB. Bayesian inference for causal effects in randomized experiments with noncompliance. Ann Statist 1997;25:305-27. https://doi.org/10.1214/aos/1034276631.
- Alsousou J, Keene DJ, Hulley PA, Harrison P, Wagland S, Byrne C, et al. Platelet rich plasma in Achilles Tendon Healing 2 (PATH-2) trial: protocol for a multicentre, participant and assessor-blinded, parallel-group randomised clinical trial comparing platelet-rich plasma (PRP) injection versus placebo injection for Achilles tendon rupture. BMJ Open 2017;7.
- Alsousou J, Handley R, Hulley P, Thompson M, McNally E, Harrison P, et al. Platelet rich plasma in accelerated Achilles tendon regeneration: a randomized controlled trial. Bone Joint J 2012;94–B. https://doi.org/10.1186/ISRCTN93608625.
- Keene DJ, Alsousou J, Harrison P, Hulley P, Wagland S, Parsons SR, et al. Platelet rich plasma injection for acute Achilles tendon rupture: PATH-2 randomised, placebo controlled, superiority trial. BMJ 2019;367.
- Breidenbach AP, Gilday SD, Lalley AL, Dyment NA, Gooch C, Shearn JT, et al. Functional tissue engineering of tendon: establishing biological success criteria for improving tendon repair. J Biomech 2014;47:1941-8. https://doi.org/10.1016/j.jbiomech.2013.10.023.
- Nourissat G, Berenbaum F, Duprez D. Tendon injury: from biology to tendon repair. Nat Rev Rheumatol 2015;11:223-33. https://doi.org/10.1038/nrrheum.2015.26.
- Khan RJ, Carey Smith RL. Surgical interventions for treating acute Achilles tendon ruptures. Cochrane Database Syst Rev 2010;9. https://doi.org/10.1002/14651858.CD003674.pub4.
- Paredes JJ, Andarawis-Puri N. Therapeutics for tendon regeneration: a multidisciplinary review of tendon research for improved healing. Ann N Y Acad Sci 2016;1383:125-38. https://doi.org/10.1111/nyas.13228.
- Khan KM, Cook JL, Bonar F, Harcourt P, Astrom M. Histopathology of common tendinopathies. Update and implications for clinical management. Sports Med 1999;27:393-408. https://doi.org/10.2165/00007256-199927060-00004.
- Fearon A, Dahlstrom JE, Twin J, Cook J, Scott A. The Bonar score revisited: region of evaluation significantly influences the standardized assessment of tendon degeneration. J Sci Med Sport 2014;17:346-50. https://doi.org/10.1016/j.jsams.2013.07.008.
- Maffulli N, Longo UG, Franceschi F, Rabitti C, Denaro V. Movin and Bonar scores assess the same characteristics of tendon histology. Clin Orthop Relat Res 2008;466:1605-11. https://doi.org/10.1007/s11999-008-0261-0.
- Alsousou J. 2013.
- Grand View Research . Platelet Rich Plasma (PRP) Market Analysis by Product (Pure, Leukocyte-Rich, Leukocyte-Rich Fibrin), by Application (Orthopedics, Cosmetic Surgery, Ophthalmic Surgery, Neurosurgery), and Segment Forecasts, 2018–2025 2017. www.grandviewresearch.com/industry-analysis/platelet-rich-plasma-prp-market.
- Insighs FM . Platelet Rich Plasma (PRP) Market: Increased Application of PRP Therapy in Sports Injuries Is Expected to Boost the Growth of PRP Market: Europe Industry Analysis and Opportunity Assessment, 2016–2024 n.d. www.prnewswire.com/news-releases/platelet-rich-plasma-prp-market-europe-industry-analysis-and-opportunity-assessment-2016---2024-300308838.html (accessed 1 August 2019).
- Bostick GP, Jomha NM, Suchak AA, Beaupré LA. Factors associated with calf muscle endurance recovery 1 year after achilles tendon rupture repair. J Orthop Sports Phys Ther 2010;40:345-51. https://doi.org/10.2519/jospt.2010.3204.
- Achten J, Parsons NR, Kearney RL, Maia Schlüssel M, Liew AS, Dutton S, et al. Cast versus functional brace in the rehabilitation of patients treated non-operatively for a rupture of the Achilles tendon: protocol for the UK study of tendo achilles rehabilitation (UK STAR) multi-centre randomised trial. BMJ Open 2017;7. https://doi.org/10.1136/bmjopen-2017-019628.
- Keene DJ, Mistry D, Nam J, Tutton E, Handley R, Morgan L, et al. The Ankle Injury Management (AIM) trial: a pragmatic, multicentre, equivalence randomised controlled trial and economic evaluation comparing close contact casting with open surgical reduction and internal fixation in the treatment of unstable ankle fractures in patients aged over 60 years. Health Technol Assess 2016;20.
- Hébert-Losier K, Newsham-West RJ, Schneiders AG, Sullivan SJ. Raising the standards of the calf-raise test: a systematic review. J Sci Med Sport 2009;12:594-602. https://doi.org/10.1016/j.jsams.2008.12.628.
- Buchgraber A, Pässler HH. Percutaneous repair of Achilles tendon rupture. Immobilization versus functional postoperative treatment. Clin Orthop Relat Res 1997;341:113-22. https://doi.org/10.1097/00003086-199708000-00018.
- Möller M, Lind K, Movin T, Karlsson J. Calf muscle function after Achilles tendon rupture. A prospective, randomised study comparing surgical and non-surgical treatment. Scand J Med Sci Sports 2002;12:9-16. https://doi.org/10.1034/j.1600-0838.2002.120103.x.
- Weber M, Niemann M, Lanz R, Müller T. Nonoperative treatment of acute rupture of the achilles tendon: results of a new protocol and comparison with operative treatment. Am J Sports Med 2003;31:685-91. https://doi.org/10.1177/03635465030310050901.
- Schepull T, Kvist J, Andersson C, Aspenberg P. Mechanical properties during healing of Achilles tendon ruptures to predict final outcome: a pilot Roentgen stereophotogrammetric analysis in 10 patients. BMC Musculoskelet Disord 2007;8. https://doi.org/10.1186/1471-2474-8-116.
- Schepull T, Kvist J, Aspenberg P. Early E-modulus of healing Achilles tendons correlates with late function: similar results with or without surgery. Scand J Med Sci Sports 2012;22:18-23. https://doi.org/10.1111/j.1600-0838.2010.01154.x.
- Suchak AA, Bostick GP, Beaupré LA, Durand DC, Jomha NM. The influence of early weight-bearing compared with non-weight-bearing after surgical repair of the Achilles tendon. J Bone Joint Surg Am 2008;90:1876-83. https://doi.org/10.2106/JBJS.G.01242.
- Silbernagel KG, Steele R, Manal K. Deficits in heel-rise height and achilles tendon elongation occur in patients recovering from an Achilles tendon rupture. Am J Sports Med 2012;40:1564-71. https://doi.org/10.1177/0363546512447926.
- Olsson N, Nilsson-Helander K, Karlsson J, Eriksson BI, Thomée R, Faxén E, et al. Major functional deficits persist 2 years after acute Achilles tendon rupture. Knee Surg Sports Traumatol Arthrosc 2011;19:1385-93. https://doi.org/10.1007/s00167-011-1511-3.
- Brorsson A, Willy RW, Tranberg R, Grävare Silbernagel K. Heel-rise height deficit 1 year after Achilles tendon rupture relates to changes in ankle biomechanics 6 years after injury. Am J Sports Med 2017;45:3060-8. https://doi.org/10.1177/0363546517717698.
- Kottner J, Audigé L, Brorson S, Donner A, Gajewski BJ, Hróbjartsson A, et al. Guidelines for Reporting Reliability and Agreement Studies (GRRAS) were proposed. J Clin Epidemiol 2011;64:96-106. https://doi.org/10.1016/j.jclinepi.2010.03.002.
- Byrne C, Keene DJ, Lamb SE, Willett K. Intrarater reliability and agreement of linear encoder derived heel-rise endurance test outcome measures in healthy adults. J Electromyogr Kinesiol 2017;36:34-9. https://doi.org/10.1016/j.jelekin.2017.07.004.
Appendix 1 The PATH-2 trial: trial management
Introduction
The PATH-2 trial completed recruitment on schedule against the plans agreed with the funder (Figure 21) and also obtained very good levels of follow-up and a relatively low number of missing outcome data. We believe that an experienced, dedicated, trial management team that could apply the same depth for both the scientific elements and the clinical elements of the study in addition to the dedication of the clinicians and researchers at the recruiting hospitals, was critical to the success of the PATH-2 trial.
FIGURE 21.
The PATH-2 trial recruitment against the original and revised targets.

Management milestones
| Planned activity to be completed | Project month in project management plan agreed with funder, version 2.0, August 2014 | Project month in revised project management plan version 4.0, September 2017 | Actual month or date |
|---|---|---|---|
| First TMG meeting, finalise protocol | 1 (October 2014) | 21 March 2014 | |
| Obtain ethics approval | 3 (December 2014) | 11 November 2014 | |
| First DSMC meeting | 6 (March 2015) | First DSMC/TSC meeting, 31 July 2015 | |
| Second DSMC meeting | 16 (January 2016) | First DSMC meeting, 22 January 2016 | |
| Second TSC meeting | 17 (February 2016) | First TSC meeting, 4 March 2016 | |
| 15 sites open to recruitment (sites recruitment ends) | 18 (March 2016) | Superseded; recruitment target changed | |
| Second DSMC meeting, 22 July 2016 | |||
| 214 patients recruited (recruitment closes) | 26 (November 2016) | Superseded; recruitment target changed | |
| Substudy 2: 16 biopsies taken place (recruitment to substudy 2 closes) | 26 (November 2016) | October 2016 | 11 August 2016 |
| Substudy 1: 107/214 PRP/blood samples collected for analysis | 27 (December 2016) | Superseded; recruitment target changed | |
| Substudy 1: platelet analysis finalised | 27 (December 2017) | November 2017 | March 2018 |
| Third DSMC meeting | 28 (January 2017) | January 2017 | Third DSMC meeting, 3 February 2017 |
| Third TSC meeting | 29 (February 2017) | October 2016 | Second TSC meeting, 14 October 2016 |
| 214 24-week follow-ups completed (primary outcome data collection ends) | 32 (May 2017) | Superseded; recruitment target changed | |
| Substudy 2: biopsy analysis finalised | 32 (May 2017) | May 2017 | April 2018 |
| Final data cleaning | 32 (May 2017) | March 2018 | June 2018 |
| Fourth TSC meeting | July 2017 | Third TSC meeting, 21 July 2017 | |
| Substudy 1: growth factor analysis finalised | 34 (July 2017) | August 2017 | March 2018 |
| 230 patients recruited (recruitment closes) | September 2017 | 18 September 2017 | |
| Substudy 1: 115/230 PRP/blood samples collected for analysis | September 2017 | September 2017 | |
| Second DSMC/TSC meeting, 29 June 2018 | |||
| Site close-out completed | June 2018 | Ongoing | |
| Data analysis | 34 (July 2017) | July 2018 | July 2018 |
| Write-up of draft final report and publications | 35 (August 2017) | April 2018 | 14 August 2018 |
| Dissemination of findings | 35 (August 2017) | July 2018 | August 2019 |
Recruitment target and site selection
The original application predicted a recruitment rate of one patient per participating centre per month, which required a total of 15 sites to be opened at the rate of one per month.
Some sites expressed interest in the trial after seeing a notice on the NIHR website or information disseminated through the clinical research networks. Sites that had participated in our previous AIM (Ankle Injury Management) trial88 and sites with a surgeon who was a member of the British Orthopaedic Foot and Ankle Society were approached if it was thought that their catchment area was large enough. Sites then completed a site feasibility questionnaire, which included their predicted recruitment rate. We continued negotiation with sites that predicted at least one participant per month after allowing for patients opting for surgery and patients who declined to participate. Site initiation visits were held, mostly in person, although some via teleconference, at which the remaining procedures were discussed and any potential problems were raised.
Establishing collaborating sites was hampered due to their inability to obtain excess treatment costs (ETCs), which were required to purchase trial-specific centrifuges for trial treatment. Sites could not obtain local NHS trust research and development approval until funds for the equipment were in place. The chief investigator purchased a small number of centrifuges, allowing two sites to open to recruitment, while addressing the ETC issue, and in December 2015 the Department of Health and Social Care agreed that the PATH-2 trial could apply for a subvention to purchase the equipment. There was further delay while the centrifuges were shipped from the US manufacturer. The start of patient recruitment was delayed from April 2015 to mid-July 2015. The funder was informed of every development throughout.
In February 2016, the recruitment forecast was re-profiled based on the delay to the start of recruitment and the rate of recruitment at that time (0.75 participants per site per month) and the go/no-go (G/NG) decision point 2 was pushed back from 1 May 2016 to 1 July 2016. One site closed early by mutual agreement with the PI as they had not recruited any participants and all patients were choosing surgery, which made them ineligible for the PATH-2 trial. It was agreed that four more sites would be opened to guard against lower than expected recruitment in some newly opened sites. Finally, 18 sites were opened to recruitment.
In July 2016, after the stop/go decision point, a 9-month extension was agreed with the funder, moving the expected end of recruitment to July 2017.
In June 2017, with approximately half of the sample having reached their 24-week follow-up point, variability was looked at for 75 participants with valid cleaned data (SD 24), and found to be greater than that assumed for the sample size calculation (SD 20). To maintain the integrity of the trial data, we proposed continuing recruitment to 229 participants, which would give 80% power to detect a 10% difference (standardised effect size of 0.5) with 20% loss to follow-up (although this was running at 15%). This meant extending recruitment into September 2017, instead of finishing at the end of July 2017 as planned. The DSMC assisted with the proposal and recommended this course of action, and it was supported by the TSC. The funder approved a 2-month no-cost extension and the recruitment target was raised to 230, which was reached in September 2017.
Go/no-go assessment
Go/no-go decision points formed part of the contract with the Department of Health and Social Care for the deliverability of the PATH-2 trial. The purpose of G/NG decision points is for the Efficacy and Mechanism Evaluation programme to review whether or not funding should continue at each identified stage of the project. Two G/NG decision points were specified as follows:
-
Go/no-go decision point 1 (G/NG 1). The milestone for G/NG 1 was ethics approval being sought and approved within 2 months of the project start date. As the project start date was 1 October 2014 and favourable ethics opinion was granted on 11 November 2014, this milestone was achieved.
-
Go/no-go decision point 2 (G/NG 2). The milestones for G/NG 2 were to be assessed in May 2016, 10 months into the recruitment stage, when approximately 50 participants were to have been recruited.
The DSMC was requested to report on the following milestones for G/NG 2:
-
safety of the intervention
-
site recruitment (consider stopping if seven out of a planned 15 sites were not recruited)
-
participant recruitment (consider stopping if < 20% of eligible patients who were approached had consented)
-
intervention compliance (consider stopping if < 80% of participants who started their allocated treatment were compliant)
-
follow-up compliance (consider stopping if loss to follow-up at week 24 was > 30%).
Data on safety, trial conduct and feasibility were reported to the DSMC, which made recommendations about the continuation or otherwise of the trial to the TSC, which made the final decision about the continuation or otherwise of the trial.
In October 2016, the funder approved an amendment to extend recruitment by 9 months and the G/NG position was moved to 1 July 2016. The reasons for the extension were as follows:
-
Delay in site recruitment – establishing collaborating sites was hampered due to sites’ inability to obtain ETCs, which were required to purchase upfront trial-specific centrifuges for trial treatment. In January 2016, we received formal notice that a subvention would be provided by the Department of Health and Social Care to cover the ETCs (to purchase the centrifuges). We proceeded to order centrifuges for trial sites from the supplier.
-
Sites halting recruitment for 1 month because of a quality issue with PRP samples that were produced in the trial-specific centrifuges. The issue was investigated with the help of the supplier and did not recur.
Following a database lock on 4 July 2016, the DSMC chairperson wrote to the funder to confirm that the milestones had been met as follows:
-
Number of sites open to recruitment (seven expected) – 12.
-
Proportion of patients consented/eligible (at least 20%) – 65% (75/116).
-
Number of participants randomised (50 minimum) – 75.
-
Compliance with intervention (80% minimum) – 96%.
-
Losses to follow-up (< 30%) – 0%.
-
Safety – no SAEs were reported by the randomised participants up to the stop/go point. Six AEs were reported.
The DSMC confirmed that the trial met the G/NG milestones and that the trial should continue.
Monitoring of trial recruitment
Sites completed a monthly screening log, declaring all patients who had presented to the emergency department or specialist fracture clinic or foot and ankle clinic with a suspected ATR. Reasons for not recruiting were monitored centrally and trends were identified. In response, certain eligibility criteria were revisited early in the trial: sites with a single weekly fracture clinic could not always randomise participants within the required 7 days from injury, so the criterion was changed to within 12 days from injury; some otherwise-healthy patients were excluded because they were aged > 70 years, so the age criterion was removed and a criterion of ‘ambulatory prior to injury’ was implemented. These two changes to eligibility criteria were implemented in a change to the protocol in March 2016.
We were alert to repeated reports at certain sites, such as patients being missed because they were treated at the weekend when there were no research staff present, or higher than expected rates of patients choosing surgery. This led to discussions with the sites to explore possible solutions, and identification of one site where all patients were having surgery and no patients were recruited. Once this was recognised, it was decided to close the site, by agreement with the PI. A minority of patients declined participation because they were uncomfortable with injections or blood being taken, and it was discussed with sites how this could be introduced to the patients in a less concerning fashion.
Data management
According to the standard operating procedures of the OCTRU, data management procedures were defined in a data management plan. This covered trial databases and data handling, definition of critical data fields, forms and questionnaires used, how protocol deviations were recorded, data rulings, handling data deviations, data security and confidentiality, data set closure, archiving and data sharing. Each data management plan version was signed off by the chief investigator and trial statistician.
The monitoring plan determined the need for central and on-site data monitoring. All sites were monitored centrally, and 12 sites underwent a site monitoring visit, either routine (n = 8) or triggered by a concern (n = 4).
Statistics on data collection, data entry and query management were presented to each TMG meeting for oversight.
Trial promotion
Promotion to patients and the public
Each recruiting centre displayed an ethics-approved patient poster in the fracture clinic, to make patients aware of the trial before they were approached to participate.
The PATH-2 trial has a public-facing web page on the OCTRU trials website (https://path2.octru.ox.ac.uk/; accessed August 2019), where trial information, current recruitment figures and news are publicised. An additional page called Reach Target Event was created in April 2018 with the aim of encouraging site staff to screen and recruit all eligible patients. This was promoted as an alternative to a study day for site staff, and included videos about the trial and the scientific objectives, discussion forums, competitions and cartoons. The Reach Target Event page is now available to the public and was entered into NIHR’s ‘Let’s Get Digital’ competition in June 2017.
Promotion within the trauma and scientific communities
The PATH-2 trial design has been presented at:
-
the fifth NIHR Musculoskeletal Trauma Trials Annual Meeting, Warwick, 11 January 2017
-
the Trauma Orthopaedic Research Collaboration, Oxford, 6 October 2017
-
the sixth NIHR Musculoskeletal Trauma Trials Annual Meeting, Bristol, 10 January 2018
-
the fourth Annual Celebrating Trauma Research in the Thames Valley, Reading, 28 February 2018
-
Tendon UK, Oxford, 11–12 April 2018.
Appendix 2 Development of heel-rise endurance test outcome measures
Background
The HRET was introduced over 30 years ago as an objective outcome measure for assessing functional status following ATR. 68 The HRET involves repetitive concentric–eccentric muscle actions of the plantar flexors in a single-leg stance until exhaustion, with performance quantified by the number of raises (repetitions) performed. 89 Subsequently, the HRET has been consistently employed as an outcome measure in studies of ATR rehabilitation. 86,90–95 However, reporting of test standardisation procedures has been poor and, to our knowledge, no universally acceptable test parameters have been published to date. 89 A systematic review of the test identified six key testing parameters required for adequate standardisation of the test that have been reported inconsistently: (1) ankle starting position, (2) knee starting position, (3) height of heel raise, (4) pace (raises per minute), (5) balance support and (6) test termination criteria. 89 In addition, the use of repetitions as the primary outcome measure in the majority of studies represents a crude and insensitive measure of function as this outcome provides little information on the quality (height) of movements performed. This is important as tendon elongation and disproportionate strength loss at the end of plantar flexor range are two impairments in ATR that are not adequately assessed by a simple measure of repetitions. Therefore, standardisation of testing parameters and selection of outcome measures that are sensitive to ATR functional impairments were the two key areas identified for test development.
Nilsson-Helander et al. 65 advanced HRET methodology by introducing a measuring device in the form of a linear displacement sensor attached to the participant’s heel, which enables the height of each repetition to be measured and three outcome measures quantified: (1) number of repetitions, (2) total work in J and (3) maximum heel-rise height in cm. Their results revealed that work as an endurance measure and maximum height as a measure of tendon function are more sensitive impairment outcome measures than repetitions following ATR. For example, repetitions classified the percentage of patients having normal function (defined as injured limb performance of ≥ 90% of the uninjured limb) at 6 and 12 months after ATR as 38% and 63%, respectively. This compared with 9% and 23% for work and 6% and 22% for maximum height at 6 and 12 months, respectively. Work and maximum height (but not repetitions) also demonstrated significant positive associations at 6 months post ATR with the Achilles tendon Total Rupture Score, a validated PROM being employed in the PATH-2 trial. Deficits at the 6- and 12-month follow-ups were 39% and 24% for endurance (work), 28% and 20% for maximum height and 26% and 5% for repetitions, respectively. 65 In addition, the deficit in maximum height was significantly and highly correlated with Achilles tendon elongation at 6 months (r = –0.94; p = 0.002) and 12 months (r = –0.74; p = 0.037). 96 Further studies have demonstrated that work and maximum height are sensitive to, and predictive of, long-term residual impairments. 97 Both remain significantly impaired at 2 years post ATR, with the majority of recovery occurring in the first 6 to 12 months,97 and deficits at 12 months correlate with abnormal ankle movement and force during walking, jogging and jumping activities at 6 years post ATR. 98 On the basis of current evidence, the use of a linear encoder measurement device during the HRET to provide outcome measures of work and maximum height represents the best available method of assessing ATR rehabilitation in a clinical setting and was adopted for the PATH-2 trial.
Heel-rise endurance test methodology development
The aims of the HRET development work were to (1) develop standardisation procedures for the HRET, (2) develop clinician training material and participant information, (3) develop easy-to-use HRET software and secure data-transfer procedures for clinician outcome assessors and (4) determine the test–retest reliability of HRET outcome measures when applying our procedures.
Standardisation procedures
A linear encoder, consisting of a spring-loaded cord connected to a linear displacement sensor with a measurement resolution of 0.019 mm and a sample rate of 200 Hz, and data-collection software were loaned for pilot testing from a commercial supplier (Encoder and MUSCLELAB™ software, Ergotest Innovation A.S., Porsgrunn, Norway). The key testing parameters identified by Hébert-Losier et al. 89 were used to aid standardisation of the HRET. In addition, we developed a standardised warm-up for participants that was suitable for a clinical environment, consisting of 5 minutes of usual-pace walking followed by 10 double-leg heel raises. The following standardisation parameters were adopted: (1) ankle starting position of 10° dorsiflexion produced by conducting the HRET on a custom-made 10° incline box, (2) knee starting position of full extension, (3) height of each repetition to be as high as possible, (4) pace of 30 raises per minute guided by a digital metronome, (5) balance support by the fingertips only and (6) strictly defined test termination criteria. The criteria for the end of the HRET were that participants either stopped (i.e. volitional task failure) or were audibly instructed to stop with both feet flat on the box whenever any of the following test termination criteria were observed: (1) inability to keep pace with the metronome, (2) inability to maintain full knee extension of the standing leg or (3) using more than fingertip support. The desired end point was volitional task failure; however, outcome assessors were encouraged to use verbal prompts whenever the termination criteria were observed and to stop the test if the participant did not respond to two consecutive prompts. In accordance with Nilsson-Helander et al.,65 three outcome measures were established: (1) total work in J, (2) number of repetitions and (3) maximum heel-rise height in cm. Each could be expressed as a LSI, with the injured limb performance as a percentag of the uninjured limb performance. Total work LSI was selected as the primary outcome measure as this indexe provides a measure of plantar flexor muscle endurance and Achilles tendon function as it incorporates the maximum height of each repetition. Total work (J) was computed as the product of body mass (kg), total vertical displacement (m) and the constant 9.807, converting kilopond metres to J.
Software and training materials
The MUSCLELAB™ software was deemed too complex for use in a clinical environment and a simpler clinician-facing software was designed by the PATH-2 trial team incorporating our own standardised procedures and produced by a commercial partner (PATH-2, MUSCLELAB™, Ergotest Innovation A.S.). Key features of this software were step-by-step instructions for the conduct of the test and the addition and integration of a video-recording of each test, which enabled a post-HRET evaluation of the integrity of each test by the PATH-2 trial team. Standardised participant information and instructions for the HRET involved the participant watching a video demonstration of the HRET and reading standardised written instructions detailing their expected conduct during the test and the test termination criteria. At test completion, the software also guides the clinician on secure participant HRET data transfer by encrypted USB. The HRET equipment and software were packaged with clinician training material, consisting of high-quality training videos created by the PATH-2 trial team and produced by Oxford Medical Illustration, and a training and reference manual. Face-to-face training would be delivered by a member of the PATH-2 trial team to each clinician outcome assessor prior to their first participant’s 24-week follow-up appointment.
Heel-rise endurance test reliability study
Following the establishment of our standardisation procedures, a reliability study was conducted on 38 healthy participants [18 males and 20 females; mean age 36 years (SD 9 years)], mean body mass 71.5 kg (SD 15.3 kg). The findings were reported in accordance with guidelines for reporting reliability and agreement studies,99 and were published in 2017. 100 Reliability was assessed by the intraclass correlation coefficient (ICC) and agreement was assessed by a range of measures including the standard error of measurement (SEM), coefficient of variation (CV) and 95% limits of agreement (LoA). Reliability for repetitions (ICC 0.77, 95% CI 0.66 to 0.85) was equivalent to work (ICC 0.84, 95% CI 0.76 to 0.89) and maximum height (ICC 0.85, 95% CI 0.77 to 0.90). Agreement for repetitions (SEM 6.7, 95% CI 5.8 to 7.9; CV 13.9%, 95% CI 11.9% to 16.8%; 95% LoA –1.9% ± 37.2%) was equivalent to work (SEM 419 J, 95% CI 361 J to 499 J; CV 13.1%, 95% CI 11.2% to 15.8%; 95% LoA 0.1% ± 34.8%), with maximum height being superior (SEM 0.8 cm, 95% CI 0.6 cm to 1.0 cm; CV 6.6%, 95% CI 5.7% to 7.9%; 95% LoA 1.3% ± 17.1%). We found that work and maximum height demonstrated acceptable reliability and agreement that were at least equivalent to those of the traditional repetitions measure.
Appendix 3 Additional results
Participant reporting of pain
One potential issue of injecting PRP into an ATR site is that, by introducing fluid into this area, increased pain may be experienced immediately after. However, there is a possibility that, over time, any pain experienced by participants may reduce faster if PRP has been received, due to the assistance in healing that is offered. As a result, any severe pain experienced by participants was explored, alongside the information relating to pain as reported in their pain diaries, completed daily for the first 2 weeks. The results from these explorations are given in Table 22 and offer no evidence to suggest any difference between intervention groups in pain experienced.
| Pain measure | Intervention group, n (%) | Total (N = 229), n (%) | |
|---|---|---|---|
| PRP (N = 113) | Placebo (N = 116) | ||
| Severe pain at the injection site requiring more than simple pain relief | |||
| No | 102 (90.27) | 106 (91.38) | 208 (90.83) |
| Yes | 4 (3.54) | 2 (1.72) | 6 (2.62) |
| Missing | 7 (6.19) | 8 (6.90) | 15 (6.55) |
| Pain reduction in first 2 weeksa | |||
| No | 18 (15.93) | 21 (18.10) | 39 (17.03) |
| Yes | 74 (65.49) | 63 (54.31) | 137 (59.83) |
| Missing | 21 (18.58) | 32 (27.59) | 53 (23.14) |
Participant employment and academic characteristics
The academic and employment characteristics of participants, as recorded at baseline, are presented in Table 23.
| Academic/employment status | Intervention group, n (%) | Total (N = 229), n (%) | |
|---|---|---|---|
| PRP (N = 113) | Placebo (N = 116) | ||
| Academic status | |||
| Full-time student | 0 (0) | 4 (3.45) | 4 (1.75) |
| Part-time student | 2 (1.77) | 0 (0) | 2 (0.87) |
| Not studying | 111 (98.23) | 112 (96.55) | 223 (91.38) |
| Employment statusa | |||
| > 40 hours/week | 40 (36.04) | 39 (34.82) | 79 (35.43) |
| 25–40 hours/week | 46 (41.44) | 49 (43.75) | 95 (42.60) |
| 10–25 hours/week | 5 (4.50) | 9 (8.04) | 14 (6.28) |
| < 10 hours/week | 2 (1.80) | 2 (1.79) | 4 (1.79) |
| Unemployed, looking for work | 3 (2.70) | 1 (0.89) | 4 (1.79) |
| At home/not looking for paid employment | 2 (1.80) | 4 (3.57) | 6 (2.69) |
| Unable to work owing to illness or disability | 1 (0.90) | 0 (0) | 1 (0.45) |
| Fully retired (no paid work) | 12 (10.81) | 8 (7.14) | 20 (8.97) |
| Type of workb | |||
| Office based | 42 (45.16) | 47 (47.47) | 89 (46.35) |
| Shop work or similar | 5 (5.38) | 4 (4.04) | 9 (4.69) |
| Classroom or equivalent | 11 (11.83) | 11 (11.11) | 22 (11.46) |
| Physical outside work | 6 (6.45) | 10 (10.10) | 16 (8.33) |
| Physical indoor work | 8 (8.60) | 15 (15.15) | 23 (11.98) |
| Mainly travel/on the road | 6 (6.45) | 3 (3.03) | 9 (4.69) |
| Other | 15 (16.13) | 9 (9.09) | 24 (12.50) |
| Childcare | 0 (0) | 1 (1.01) | 1 (0.52) |
| Driving | 4 (4.30) | 0 (0) | 4 (2.08) |
| Manual | 1 (1.08) | 3 (3.03) | 4 (2.08) |
| Office or standing | 10 (10.75) | 5 (5.05) | 15 (7.81) |
| Time spent on feet | |||
| Most of the day | 37 (32.74) | 42 (36.21) | 79 (34.5) |
| > 4 hours/day | 29 (25.66) | 26 (22.41) | 55 (24.02) |
| < 4 hours/day | 24 (21.24) | 24 (20.69) | 48 (20.96) |
| Not much time (mostly sitting) | 23 (20.35) | 24 (20.69) | 47 (20.52) |
| Time spent driving | |||
| Most of the day | 5 (4.42) | 3 (2.59) | 8 (3.49) |
| > 4 hours/day | 7 (6.19) | 3 (2.59) | 10 (4.37) |
| < 4 hours/day | 49 (43.36) | 47 (40.52) | 96 (41.92) |
| Just to/from work | 46 (40.71) | 47 (40.52) | 93 (40.61) |
| Do not drive | 6 (5.31) | 16 (13.79) | 22 (9.61) |
Participants’ pre-injury clinical characteristics
The health of an individual not only affects their risk of experiencing an injury but can also affect their healing. At recruitment, participants were excluded if they were identified to have any medical conditions that would make them unsuitable for this trial. However, at baseline, information on additional medical conditions and medications taken by participants was collected. The summary of this information by intervention group is given in Table 24.
| Clinical characteristics | Intervention group, n (%) | Total (N = 229), n (%) | |
|---|---|---|---|
| PRP (N = 113) | Placebo (N = 116) | ||
| Medical condition in addition to tendon rupture | |||
| Heart disease | 2 (1.77) | 3 (2.59) | 5 (2.18) |
| Hypertension | 10 (8.85) | 14 (12.07) | 24 (10.48) |
| Asthma/COPD | 22 (19.47) | 13 (11.21) | 35 (15.28) |
| Parkinson’s disease | 0 (0) | 0 (0) | 0 (0) |
| Epilepsy | 1 (0.88) | 1 (0.86) | 2 (0.87) |
| Liver disease | 0 (0) | 0 (0) | 0 (0) |
| Stroke/mini-stroke (TIA) | 3 (2.65) | 0 (0) | 3 (1.31) |
| Peptic ulcer | 1 (0.88) | 4 (3.45) | 5 (2.18) |
| Cancer | 6 (5.31) | 3 (2.59) | 9 (3.93) |
| DVT/pulmonary embolism | 0 (0) | 0 (0) | 0 (0) |
| Osteoarthritis | 7 (6.19) | 4 (3.45) | 11 (4.80) |
| Rheumatoid arthritis | 3 (2.65) | 4 (3.45) | 7 (3.06) |
| Allergies | 33 (29.20) | 25 (21.55) | 58 (25.33) |
| Othera | 31 (27.43) | 36 (31.03) | 67 (29.26) |
| Number of comorbidities | |||
| 0 | 44 (38.94) | 56 (48.28) | 100 (43.67) |
| 1 | 36 (31.86) | 28 (24.14) | 64 (27.95) |
| 2 | 20 (17.7) | 23 (19.83) | 43 (18.78) |
| > 2 | 13 (11.50) | 9 (7.76) | 22 (9.61) |
| Taking medication for pain or inflammation before injury | |||
| Yes | 7 (6.19) | 8 (6.90) | 15 (6.55) |
| Nothing regular | 19 (16.81) | 17 (14.66) | 36 (15.72) |
| No | 87 (76.99) | 90 (77.59) | 177 (77.29) |
| Missing | 0 (0) | 1 (0.86) | 1 (0.44) |
Success of blinding
The results from the James et al. 69 blinding index are given in Table 25 and indicate that, overall, participants in this trial were completely blinded. However, the James et al. 69 index automatically assumes that participants reporting that they do not know their treatment allocation are reporting so honestly rather than to give a response they perceive to be desired by their interviewer or to simply avoid making a judgement. Furthermore, this index does not distinguish between treatments. 69 To determine the effect of each treatment group and ensure that ‘don’t know’ responses are taken into account, the Bang et al. 70 index is also used to understand blinding. The Bang et al. 70 blinding index for the PRP group is positive, indicating that participants were more likely to guess that they received a PRP injection than that they received the placebo treatment. However, the absolute value for this is < 0.2, indicating that blinding was likely to have been successful in this treatment group. Conversely, the Bang et al. 70 blinding index for the placebo group is negative, indicating that participants were more likely than those in the control group to guess that they received the PRP injection. However, as with the PRP group, the absolute value for this is < 0.2, indicating that blinding was likely to have been successful in this group. Taking the information from both blinding indices into account, there is no evidence to suggest that blinding in this trial was unsuccessful.
List of abbreviations
- AE
- adverse event
- ATR
- Achilles tendon rupture
- ATRS
- Achilles tendon Total Rupture Score
- bFGF
- basic fibroblast growth factor
- BMI
- body mass index
- CACE
- complier-average causal effect
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- CV
- coefficient of variation
- DSMC
- Data and Safety Monitoring Committee
- DVT
- deep-vein thrombosis
- ELISA
- enzyme-linked immunosorbent assay
- ETC
- excess treatment cost
- FGF
- fibroblast growth factor
- GCP
- Good Clinical Practice
- G/NG
- go/no-go
- G/NG 1
- go/no-go decision point 1
- G/NG 2
- go/no-go decision point 2
- GP
- general practitioner
- HRET
- heel-rise endurance test
- ICC
- intraclass correlation coefficient
- IGF
- insulin-like growth factor
- IGF-1
- insulin-like growth factor 1
- ISRCTN
- International Standard Randomised Controlled Trial Number
- ITT
- intention to treat
- LoA
- limits of agreement
- L-PRP
- leucocyte-rich platelet-rich plasma
- LSI
- Limb Symmetry Index
- MCS
- mental component score
- MFI
- mean fluorescence intensity
- mITT
- modified intention to treat
- NIHR
- National Institute for Health Research
- OCTRU
- Oxford Clinical Trials Research Unit
- OTUG
- Oxford Trauma User Group
- PATH-2
- PRP in Achilles Tendon Healing
- PCS
- physical component score
- PDGF
- platelet-derived growth factor
- PI
- principal investigator
- PIS
- participant information sheet
- PLT-F
- platelet fluorescence
- PP
- per protocol
- PPI
- patient and public involvement
- PROM
- patient-reported outcome measure
- PRP
- platelet-rich plasma
- PSFS
- Patient-Specific Functional Scale
- RCT
- randomised controlled trial
- SAE
- serious adverse event
- SD
- standard deviation
- SEM
- standard error of measurement
- SF-12
- Short Form questionnaire-12 items
- TGF-β
- transforming growth factor beta
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
- USB
- Universal Serial Bus
- VAS
- visual analogue scale
- VEGF
- vascular endothelial growth factor