

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 12/170/45. The contractual start date was in November 2015. The final report began editorial review in July 2019 and was accepted for publication in January 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2020. This work was produced by McCartney et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2020 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Research summary
During the Trial of low-dose adjunctive alTeplase during priMary PCI (T-TIME), the aims and objectives of the study were unchanged. A list of the amendments to the trial protocol is provided in Appendix 1.
To best ensure the safety of the participants, additional exclusion criteria were (1) a requirement for immunosuppressive drug therapy at any time during the past 3 months and (2) active or prophylactic treatment with oral or parenteral antibiotic, antifungal or antiviral therapy to prevent or treat infection. The change was implemented to ensure the safety of the participants.
Finally, on the recommendation of the Data and Safety Monitoring Committee, recruitment was discontinued on 21 December 2017 because a prespecified futility criterion for efficacy was met. Specifically, the conditional power for an analysis on the primary efficacy outcome based on 40% of the randomised population (n = 267) with follow-up to 3 months was < 30% in both treatment groups. The sponsor initially placed a temporary halt on enrolment pending a review by the Trial Steering Committee (TSC) and funder, and no further enrolment occurred following this determination.
Background
Ischaemic heart disease is a major cause of premature disability1 and death2. Acute coronary thrombosis causes ST-elevation myocardial infarction (STEMI). The evidence-based standard of care for STEMI is primary percutaneous coronary intervention (PCI) to emergently reopen the occluded coronary artery and secure vessel patency with a stent. 3 Primary PCI restores normal coronary artery blood flow in more than 9 in 10 patients. 4 However, failed microvascular reperfusion has been estimated to occur in almost half of all treated patients. 5,6 This complication, described as microvascular obstruction, is independently predictive of an unfavourable cardiac prognosis. 7 During primary PCI, displacement of thrombus within the lumen of the main coronary artery and microvascular thrombosis,8–12 notably of fibrin-rich microthrombi,9 are substrates for microvascular obstruction. Clinicians lack the therapeutic tools to treat microvascular obstruction. 3
Fibrinolytic therapy is also effective for acute coronary thrombosis. 13 Facilitated PCI involving half- or full-dose adjunctive fibrinolytic therapy that is given before PCI with stenting improves coronary flow acutely. 14,15 However, this therapeutic strategy16,17 causes paradoxical activation of thrombin, clot formation and bleeding. Sezer et al. 15 implemented a modified, pharmaco-invasive strategy by administering adjunctive low-dose thrombolytic therapy with 250 kU of streptokinase at the end of primary PCI in 41 patients with acute STEMI. This approach appeared to improve myocardial blood flow, as revealed by coronary angiography and invasive measures of coronary microvascular function during repeat coronary angiography 2 days after PCI.
However, this study had some limitations:
-
The improvement in perfusion did not translate into improvements in long-term ventricular size or function.
-
Streptokinase is not selective for fibrin and it has a higher risk of bleeding than fibrin-specific thrombolytics, such as alteplase and tenecteplase. 16
-
Streptokinase was given through the guide catheter rather than through local delivery of the lytic drug directly into the culprit artery. Drug administration into the guide catheter means that the thrombolytic would be dispersed into coronary branches that were not connected to the culprit artery; that is, the drug was delivered to non-relevant coronary arteries, which potentially reduced drug exposure to the fibrin-rich thrombus in the culprit artery microcirculation.
-
Given that streptokinase was given after the end of the PCI procedure, thrombolysis could only treat established microvascular thrombosis rather than prevent its occurrence, as would be the case if it were administered earlier, during PCI (as is proposed in our study).
These problems may explain why Sezer et al. ’s15 findings have not been adopted into clinical practice. Sezer et al. ’s15 findings indicate that intracoronary thrombolysis has the potential to reduce microvascular damage during primary PCI. Our proposal used alteplase, which is a more potent fibrin-specific second-generation lytic drug than streptokinase with comparable potency to third-generation drugs, such as reteplase; accordingly, our trial aimed to determine whether or not alteplase would reduce microembolic thrombi and improve microvascular function when delivered locally.
Rationale
Primary PCI is a multistep procedure. The first step involves coronary angiography to reveal the coronary anatomy and the culprit lesion implicated in the acute myocardial infarction (MI). Second, coronary reperfusion may be achieved using a thin (0.014-inch) coronary guidewire followed by balloon angioplasty and/or selective thrombus removal by transcatheter aspiration thrombectomy. Third, the standard of care procedure involves stenting followed by optimisation of the stent result by high-pressure balloon angioplasty in the stent.
Patients with acute STEMI who present with a blocked coronary artery and/or an artery with a heavy thrombus burden are at an increased risk of developing heart failure. This trial aimed to enrol patients with a heavy coronary thrombus burden at initial angiography to test the hypothesis that a therapeutic strategy involving reduced-dose alteplase, given early after coronary reperfusion as a single dose, would both prevent and treat distal microvascular thrombosis and microvascular obstruction. The trial aimed to determine the lowest dose of alteplase that would be effective in reducing microvascular obstruction.
The T-TIME trial aimed to investigate whether or not a therapeutic strategy involving low-dose intracoronary fibrinolytic therapy with alteplase infused early after coronary reperfusion would both prevent and reduce microvascular obstruction.
Alteplase: prior experience and dose selection
Alteplase is a fibrin-specific, second-generation plasminogen activator and thrombolytic drug. Full standard-dose alteplase (100 mg) is widely used worldwide as a primary reperfusion therapy for STEMI in hospitals where primary PCI is not available. 3
The main difference between alteplase and third-generation lytic drugs is that alteplase has a circulating half-life of approximately 5 minutes (hepatic metabolism), so is administered as an intravenous infusion over 90 minutes, whereas third-generation thrombolytics, such as reteplase and tenecteplase, have longer half-lives (i.e. 15 minutes) and so can be administered as a single-bolus infusion. 18,19 However, although alteplase has a shorter circulating half-life, the deep-tissue beta half-life of alteplase is 40 minutes; this pharmacokinetic property is very relevant for local drug delivery into the coronary microcirculation. The difference in the clearance of alteplase between blood and deep tissues is because alteplase is metabolised in the liver, which means that when alteplase is directly administered into tissues it will persist for a much longer period of time, as it does not initially pass through the liver in the blood. The efficacy of alteplase in restoring normal [Thrombolysis in Myocardial Infarction (TIMI) grade 3] coronary artery blood flow in similar to that of reteplase and tenecteplase. 3 Fibrinolytic drugs also have procoagulant effects;20,21 however, haemostatic effects, such as thrombin activation, are similar between alteplase and reteplase, although procoagulant effects may be lower with tenecteplase owing to lower activation of the kallikrein-factor XII system. 20,21 Concomitant treatment with antithrombotic drugs (e.g. heparin, glycoprotein IIb/IIIa inhibitors and bivalirudin) attenuates the procoagulant effects of thrombolytic drugs. 3
Antithrombotic therapy and thrombolysis
Thrombolytic drugs lyse fibrin in a red thrombus; however, these drugs do not have antiplatelet effects and so have no effect on platelet-rich ‘white’ clots. Thrombolytic drugs are also associated with procoagulant effects, including platelet activation. 3 For these reasons, optimal antiplatelet therapy and anticoagulation during and after thrombolysis are essential. 1 In the ASSENT-IV trial,16 full-dose thrombolysis prior to primary PCI with stents in the absence of glycoprotein IIb/IIIa inhibitors was associated with an increased risk of reinfarction (possibly because of platelet activation) and bleeding. The ASSENT-IV16 investigators concluded that suboptimal antithrombotic therapy, including the absence of heparin after the initial 5000 IU bolus, the lack of an upfront loading dose with clopidogrel and the prohibition of glycoprotein IIb/IIIa inhibitor therapy (except in bailout circumstances) contributed to the early thrombotic complications (i.e. recurrent MI) observed in the tenecteplase-treated patients.
Clinical trials of primary PCI with reperfusion facilitated by administering full-dose thrombolysis during transfer to hospital by ambulance prior to PCI (facilitated primary PCI) have been negative (including microvascular obstruction outcomes in some of the trials), such as ASSENT-IV,16 FINESSE17 and LIPSIA-STEMI. 22 In FINESSE,17 there was an improvement in microcirculation indices in the half-dose reteplase/abciximab group. In this case, any effect of reteplase to enhance platelet activation may have been attenuated by concomitant antiplatelet therapy with abciximab. Overall, the reasons why facilitated PCI trials were unsuccessful may include (1) treatment delays with PCI that are associated with lytic therapy versus usual care (nearly 30-minute difference in LIPSIA, which is a very substantial prolongation in ischaemic time), (2) inadequate anticoagulation and antiplatelet therapy, (3) harmful bleeding in STEMI patients with femoral artery access (at times, a non-compressible puncture site) and (4) the fact that earlier trials used streptokinase (a non-fibrin-specific lytic with ‘off-target’ effects). In LIPSIA,17 only 29% of thrombolysed patients received optimal antiplatelet therapy with glycoprotein IIb/IIIa inhibitors. The LIPSIA investigators recognised that antiplatelet therapy (to minimise bleeding risk in full-dose thrombolysis) had been inadequate in their trial and that thrombolysis prior to PCI had prolonged the ischaemic period in the facilitated group of patients. The LIPSIA investigators specifically referred to the potential value of reduced-dose thrombolysis with optimal antiplatelet therapy, which is what we proposed in the T-TIME trial, and uniquely in a dose-ranging trial with intracoronary lytic administration after reperfusion.
Evidence base in relation to safety (systemic effect) and efficacy for low-dose alteplase ≈10 mg (one-tenth of standard dose, 100 mg)
Sezer et al. 15 used 250,000 units of streptokinase, which is one-sixth of the usual dose given intravenously in acute MI (1.5 million units). Given that streptokinase is much less effective than alteplase, we believe that using a lower equivalent dose of alteplase, such as one-tenth of the standard dose, is an appropriate strategy. The licensed thrombolytic dose for a person > 65 kg is 100 mg of alteplase. We have, therefore, adopted a 10-fold lower dose as the lowest dose of alteplase in this study.
Safety of 10 mg of alteplase
The main safety concern with thrombolytic drugs such as alteplase is bleeding, which may occur at the site of vascular access or systemically. The risk of bleeding is dose dependent20,21 and the usual systemic dose is weight adjusted.
Low-dose alteplase (e.g. 2–4 mg) is widely used in hospitals worldwide to recanalise central venous catheters that are occluded with a thrombus, and the 10-mg loading dose is the standard initial step with systemic thrombolysis for acute MI. 3
Evidence base in relation to safety (systemic effect) and efficacy for low-dose alteplase ≈20 mg (one-fifth of standard dose, 100 mg)
Reduced-dose (20 mg) alteplase has been described in prior PCI studies. In the TIMI 14 A trial, led by Antman et al. ,23 888 STEMI patients who had presented within 12 hours of the onset of symptoms were treated with aspirin and randomised initially to either 100 mg of accelerated-dose alteplase (control) or abciximab (bolus 0.25 mg/kg and 12-hour infusion of 0.125 µg/kg per minute) alone or in combination with reduced doses of alteplase (20–65 mg) or streptokinase (500,000 U to 1.5 MU). Reperfusion treatment regimens consisting of total alteplase doses of 20, 35, 50 or 65 mg produced 90-minute TIMI grade 3 flow rates that were at least comparable to those observed with full-dose alteplase alone. Higher rates of TIMI grade 3 flow at both 60 minutes and 90 minutes were observed with increasing duration of administration of alteplase, progressing from a bolus alone to a bolus followed by either a 30-minute infusion or a 60-minute infusion (p < 0.02). Based on results from the initial dose-finding phase, TIMI grade 3 flow rates were highest in the 50 mg of alteplase plus abciximab group versus the alteplase-only group at both 60 minutes (72% vs. 43%; p = 0.0009) and 90 minutes (77% vs. 62%; p = 0.02). Major haemorrhage occurred in 6% of patients receiving alteplase alone (n = 235), 3% with abciximab alone (n = 532), 10% with streptokinase plus abciximab (n = 143), 7% with 50 mg of alteplase plus abciximab and low-dose heparin (n = 103), and 1% with 50 mg of alteplase plus abciximab with very-low-dose heparin (n = 70).
Standard care with primary PCI does not involve alteplase;3 therefore, the following three-arm design was adopted in which the alteplase or placebo was administered at the start of the PCI procedure:
-
control group – placebo
-
group A – 10 mg of alteplase
-
group B – 20 mg of alteplase.
The rationale for administering low-dose fibrinolytic therapy into the culprit coronary artery at the start of primary PCI (i.e. immediately after coronary reperfusion) is to reduce microvascular obstruction, the infarct size and the future risk of heart failure. Given that alteplase has a ‘deep tissue’ half-life of up to 40 minutes, we anticipate that there should be effective local thrombolysis during the PCI procedure. Therefore, the rationale for giving alteplase at the start of primary PCI is to treat and reduce persistent microvascular obstruction from the earliest time point and to be effective during the course of the procedure.
Our design exploits the timely therapeutic effects with the front-loaded dose approach using alteplase. The standard intravenous loading dose results in a high initial circulating concentration of alteplase. Locally, this is associated with improved initial fibrinolysis (culprit or microcirculatory patency). Our approach aimed to recapitulate this high initial local concentration at the start of primary PCI by direct administration of the drug into the culprit artery, while avoiding the potentially harmful effects of high systemic concentrations of the thrombolytic drug. Impaired intracoronary perfusion during STEMI means that there is a slower ‘washout’ of the drug, which we hypothesised would help to maintain therapeutic concentrations of alteplase locally for targeting and degrading fibrin by plasminogen activation. We hypothesised that sustained local intracoronary perfusion directly into the culprit coronary artery with therapeutic concentrations of alteplase would improve the achievement of therapeutic microvascular concentrations in the volume of tissue being treated.
The hypothesis for the mode of action of our intervention is based on initial prolonged exposure to alteplase caused by the poor perfusion created by the clot; this, in turn, will allow distribution of alteplase into the microvasculature of deep tissue where the half-life of alteplase is ≈40 minutes. Local intracoronary administration should lead to effective plasminogen activation and fibrinolysis within the microvasculature and reduce the complications seen post PCI that may be attributable to distal microvascular obstruction.
Hypothesis
We hypothesised that a therapeutic strategy involving low-dose intracoronary fibrinolytic therapy with alteplase infused early after coronary reperfusion will prevent and reduce microvascular obstruction. To test this hypothesis we enrolled patients with acute STEMI and a large thrombus burden evident at initial coronary angiography. A three-arm design was adopted in which 10 mg or 20 mg of alteplase (representing one-tenth and one-fifth of the standard dose, respectively) or placebo was administered following reperfusion and before stent implantation when residual thrombus burden is at its greatest. The trial aimed to determine the lowest effective dose of alteplase in reducing microvascular obstruction, and multiple surrogate outcomes for efficacy, safety and mechanisms were assessed.
Chapter 2 Research objectives
Objectives
The T-TIME trial was a double-blind, randomised, parallel-group, placebo-controlled clinical trial that was designed to examine the efficacy and safety of reduced-dose intracoronary alteplase in STEMI patients receiving primary PCI.
Primary objective
The primary objective was to determine the lowest dose of alteplase that is effective in reducing microvascular obstruction. More specifically, the primary objective was to determine the safety and efficacy of reduced doses (10 mg and 20 mg) of intracoronary alteplase compared with placebo as an adjunct to PCI in reducing microvascular obstruction, and its consequences in high-risk patients with STEMI. The T-TIME trial was designed as a Phase II clinical trial and was powered to provide definitive information on the primary outcome. Therefore, the results from the T-TIME trial were intended to inform the rationale for undertaking a larger Phase III trial that ultimately would be necessary before this therapeutic strategy could be recommended in practice guidelines. If the primary analysis disclosed evidence of a treatment effect on the primary outcome, then this would support the case for the design of a larger definitive trial. If the results of the primary analysis were not positive, then the case for progressing to a Phase III trial of this intervention, as designed, would not be supported.
Secondary objectives
Mechanistic
To explore mechanisms associated with any beneficial effects of reduced doses of alteplase.
Safety
To determine the rates of adverse events associated with reduced doses of alteplase administered directly into the coronary artery as an adjunct to PCI.
Chapter 3 Methods
Trial design
This was a randomised, double-blind, parallel-group, Phase II clinical trial of low-dose adjunctive alteplase during primary PCI (Figures 1 and 2).
FIGURE 1.
Graphical depiction of the trial protocol. Patients presenting with acute STEMI meeting the study eligibility criteria were enrolled by research staff in the cardiac catheterisation laboratory following reperfusion of the culprit artery. Blood tests were performed acutely (0, 2 and 24 hours after randomisation, which defines baseline) and again at the time of multiparametric magnetic resonance imaging (MRI) at 2–7 days and 3 months after enrolment. An electrocardiogram (ECG) was carried out prior to reperfusion (baseline), at 60 minutes and then again at 3 months. Parametric magnetic resonance imaging, starting from left: cine imaging allows calculation of left ventricular function and volumes; late gadolinium-enhanced MRI allows determination of infarct size (bright white area) and microvascular obstruction (hypointense, black core) within the infarct highlighted with the yellow arrow; T2* magnetic resonance imaging, far right image revealed myocardial haemorrhage (white arrow) within the infarct core. MVO, microvascular obstruction.
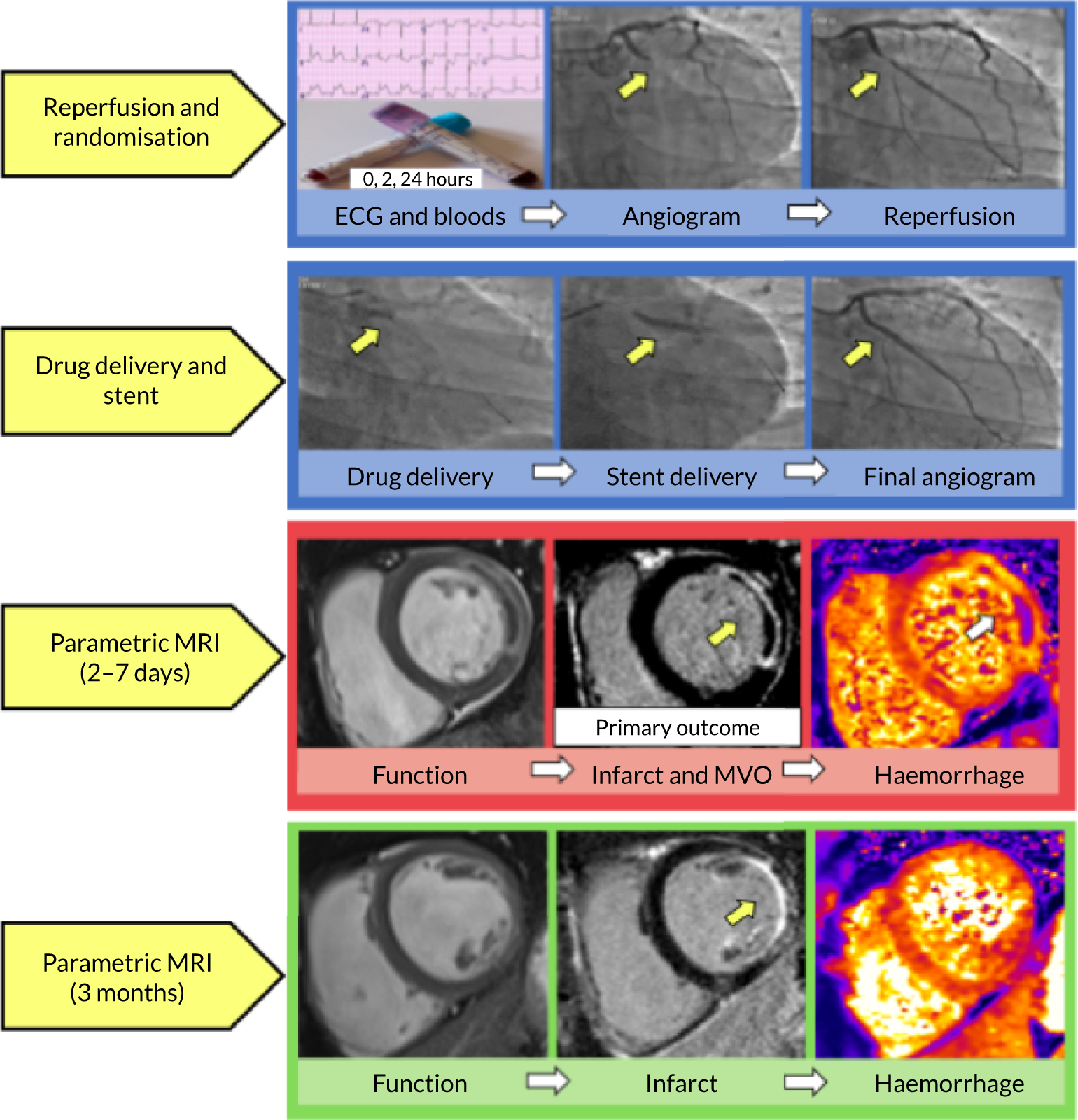
FIGURE 2.
Study procedures. EDTA, ethylenediaminetetraacetic acid; NT-proBNP, N-terminal pro–B-type natriuretic peptide; NYHA, New York Heart Association.

Informed consent and study protocol
Screening, witnessed verbal informed consent, study drug administration and acute assessments of efficacy took place during the standard of care emergency primary PCI procedure in the cardiac catheterisation laboratory. A screening log was prospectively completed. Only patients who were sufficiently well to understand the information about the study, as described by the attending cardiologist, were eligible to participate. The decision of whether or not a patient was eligible to be included was made and documented by the cardiologist. The study information sheet that had been approved by the research ethics committee was subsequently provided to each participant on the ward, where written informed consent was obtained. The participants were followed up unless consent was withdrawn.
The protocol and statistical analysis plan are provided in Appendix 2. The trial was reviewed and approved by an ethics committee of the West of Scotland Research Ethics Service (reference 13-WS-0119), adhered to Guidelines for Good Clinical Practice in Clinical Trials24 and complied with the Declaration of Helsinki. 25
Participants and eligibility criteria
Patients with a clinical diagnosis of acute STEMI were eligible for randomisation according to the following eligibility criteria.
Inclusion criteria
-
Acute MI (symptom onset ≤ 6 hours) with persistent ST segment elevation or recent left bundle branch block.
-
Coronary artery occlusion (TIMI coronary flow grade of 0 or 1) or impaired coronary flow (TIMI flow grade of 2: slow but complete filling) in the presence of definite angiographic evidence of a thrombus (TIMI grade of 2 or more).
-
Proximal to middle culprit lesion location in a major coronary artery (i.e. the right, left anterior descending, intermediate or circumflex coronary artery).
-
Radial artery access.
Exclusion criteria
Clinical criteria that would exclude the patient from the trial were evaluated by medical, research and nursing staff when the patient arrived in the catheter laboratory.
Coronary
-
Normal flow in the culprit coronary artery at initial angiography (TIMI flow grade of 3).
-
Functional coronary collateral supply (Rentrop grade of 2 or 3) to the culprit artery.
-
Previous infarction in the culprit artery (known or suspected clinically, e.g. wall motion abnormality revealed by echocardiography).
Clinical
-
Cardiogenic shock (Killip class IV).
-
Multivessel PCI intended before cardiac MRI intended for days 2–7.
-
Previous infarction in the culprit artery (known or suspected clinically).
-
Body weight estimated to be < 60 kg.
-
Non-cardiac comorbidity with expected survival < 1 year.
-
Contraindication to contrast-enhanced MRI.
Pacemaker
-
Implantable defibrillator.
-
Known impaired renal function [estimated glomerular filtration rate (eGFR) of < 30 ml/minute/1.73 m2].
-
Significant bleeding disorder either at present or within the past 6 months.
-
Patients with current concomitant oral anticoagulant therapy (international normalised ratio > 1.3), including apixaban, dabigatran and rivaroxaban.
-
Any history of central nervous system damage (i.e. neoplasm, aneurysm, intracranial or spinal surgery).
-
Known haemorrhagic diathesis.
-
Severe hypertension (blood pressure of > 180/110 mmHg) not controlled by medical therapy.
-
Major surgery, biopsy of a parenchymal organ or significant trauma within the past 3 months (this includes any trauma associated with the current acute MI).
-
Recent trauma to the head or cranium (< 2 months).
-
Prolonged cardiopulmonary resuscitation (> 2 minutes) within the past 2 weeks.
-
Acute pericarditis and/or subacute bacterial endocarditis (e.g. valve mass or vegetation revealed by echocardiography).
-
Acute pancreatitis.
-
Severe hepatic dysfunction, including hepatic failure, cirrhosis, portal hypertension (oesophageal varices) and active hepatitis.
-
Active peptic ulceration.
-
Arterial aneurysm and known arterial/venous malformation.
-
Neoplasm with increased bleeding risk.
-
Any known history of haemorrhagic stroke or stroke of unknown origin.
-
Known history of ischaemic stroke or transient ischaemic attack in the preceding 6 months.
-
Dementia.
-
Hypersensitivity to gentamicin or natural rubber.
-
Incapacity or inability to provide informed consent.
-
Previous randomisation to this study or participation in a study with an investigational drug or medical device within 90 days prior to randomisation.
-
Women of child-bearing potential (i.e. premenopause) or breast feeding.
-
Requirement for immunosuppressive drug therapy at any time during the past 3 months, whether administered orally, subcutaneously, intramuscularly or intravenously. This would include corticosteroids (but not inhaled or topical), drugs used following transplantation (e.g. tacrolimus and cyclosporine), antimetabolite therapies [e.g. mycophenolic acid (Myfortic®), azathioprine and leflunomide (Arava®)], and immunomodulators including biologics [e.g. adalimumab (Humira®), etanercept (Enbrel®), aldesleukin and disease-modifying antirheumatic drugs (e.g. cyclophosphamide and methotrexate)]. Please note that this list is not exhaustive and a requirement for other immunosuppressive drugs not listed would also exclude the patient.
-
Active or prophylactic treatment with oral or parenteral antibiotic, antifungal or antiviral therapy to prevent or treat infection.
-
Any anticancer treatment (excluding surgery as this is covered above) at any time during the past 3 months, including chemotherapy, radiotherapy and treatment with biologics such as vascular endothelial growth factor receptor inhibitors (e.g. bevacizumab and pazopanib).
-
Any significant concurrent or recent condition(s) not listed above that, in the opinion of the treating clinician, would pose an additional risk for the patient.
Race/ethnicity was designated by the patient and recorded by the local investigator to provide information on the participation of individuals with different ethnicity.
Setting
The participants were enrolled in 11 hospitals in the UK, and guideline-based medical and invasive management was recommended. 3 Enrolment started on 17 March 2016.
Randomisation, implementation and blinding
Participants were randomised by staff in the catheter laboratory using an interactive voice response-based randomisation system. The randomisation sequence was computer generated, using the method of randomised permuted blocks of length 6, with stratification by location of STEMI (anterior vs. non-anterior) and study site. The allocation sequence was on a 1 : 1 : 1 basis between the placebo, the 10 mg of alteplase and the 20 mg of alteplase groups, and the sequence was concealed electronically. The participants, staff and researchers were blinded to the treatment group allocation.
Standard care
Primary PCI followed contemporary practice guidelines. 3 Standard care for coronary reperfusion was recommended according to contemporary practice guidelines3 using either balloon angioplasty or aspiration thrombectomy for thrombus-containing lesions. A coronary balloon diameter (mm) versus lumen diameter (mm) relationship of < 1 : 1 and a low inflation pressure were recommended to minimise thrombus embolisation. The balloon angioplasty was intended to stabilise the thrombotic lesion and prevent vessel reocclusion prior to stent implantation. Antithrombotic therapy included oral antiplatelet drugs and intravenous heparin (5000 IU or as per standard practice) at the first medical contact. The target activated clotting time was 250 seconds.
Interventions
After successful reperfusion (TIMI flow grade of 2 or above) the participants received the allocated intervention immediately in the catheter laboratory. The study drug (placebo or 10 mg or 20 mg of alteplase) was manually infused before stent implantation. The drug was reconstituted by the clinical staff using 20 ml of sterile water for injection. The cardiologist then infused the solubilised drug over 5–10 minutes directly into the infarct-related artery proximal to the culprit lesion using either an intracoronary catheter or the guiding catheter if selectively engaged.
Outcomes
The methods for the assessments of the primary and secondary outcomes are described in Appendix 3.
Central laboratory analyses
The central laboratory analyses of the primary and secondary outcomes were determined blind to treatment allocation.
Primary outcome
The primary outcome was the amount of microvascular obstruction (percentage of left ventricular mass) demonstrated by late gadolinium-enhanced MRI, 10–15 minutes after administration of contrast medium. Cardiac MRI at 1.5 T was scheduled during the index hospitalisation for 2–7 days after enrolment.
Secondary outcomes
Cardiac magnetic resonance imaging
Magnetic resonance imaging secondary outcomes included microvascular obstruction (presence/absence), myocardial haemorrhage (presence/absence) and the amount of myocardial haemorrhage expressed as a percentage of left ventricular mass on MRI at 2–7 days. Infarct size expressed as a percentage of left ventricular mass, myocardial salvage index, left ventricular end-diastolic volume, left ventricular end-systolic volume and left ventricular ejection fraction were obtained at 2–7 days and 3 months.
Angiography
Angiographic measures of reperfusion (TIMI coronary flow grade, TIMI myocardial perfusion grade and TIMI frame count) and TIMI thrombus grade at the end of PCI were predefined secondary outcomes.
Electrocardiogram
The percentage ST-segment resolution on ECG performed 60 minutes post reperfusion versus pre reperfusion and final infarct size revealed by the Selvester QRS score at 3 months were also calculated.
Biochemistry
The area under the curve (AUC) for troponin T (ng/ml) was measured from blood samples that were obtained immediately before reperfusion (0 hours) and then again at 2 and 24 hours. N-terminal pro–B-type natriuretic peptide (NT-proBNP) concentration (pg/ml) was measured at 2–7 days and 3 months post reperfusion, scheduled at the time of MRI.
Health status
Health-related quality of life (HRQoL) [EuroQol-5 Dimensions, three-level version (EQ-5D-3L)] was recorded at 2–7 days and 3 months post MI. The EQ-5D-3L is a standardised instrument that is used as a measure of health outcome, which is made up of the following two components. The health utility score is a descriptive system composed of five dimensions: mobility, self-care, usual activities, pain/discomfort and anxiety/depression. Scores for each are combined to give a maximum value of 1. Each dimension has three levels: no problems, some problems and extreme problems. The visual analogue scale reports the patient’s self-rated health from 0 (worst imaginable) to 100 (best health imaginable).
Bleeding and coagulation
Fibrinogen and other parameters of coagulation and haemostasis served as surrogate measures of bleeding risk. 20,21 These parameters were measured in blood samples when site logistics permitted blood sample collection. The sampling time points were at baseline before reperfusion (0 hours) and 2 and 24 hours post reperfusion. The parameters included fibrinogen and plasminogen (both measures of coagulation and systemic fibrinolysis), fibrin D-dimer (a measure of fibrin lysis), tissue plasminogen activator (tPA) (a measure of endogenous tPA and any circulating alteplase) and prothrombin fragment 1 + 2 (from here referred to as prothrombin F1 + 2) (a measure of thrombin activation).
Adverse events
Classification of health outcomes
-
Major adverse cardiovascular and cardiac events: cardiovascular death, non-fatal MI, unplanned hospitalisation for transient ischaemic attack or stroke.
-
Major adverse cardiac events (MACEs): cardiac death, non-fatal MI or unplanned hospitalisation for heart failure.
-
Spontaneous MACE: MACE, excluding MI associated with revascularisation procedures (type 4 or 5 MI).
-
MI associated with revascularisation procedures (type 4 or 5 MI).
-
All-cause mortality.
-
Unplanned hospitalisation for heart failure.
-
All-cause death and unplanned hospitalisation for heart failure.
-
Bleeding Academic Research Consortium (BARC) type 3, type 4 and type 5 bleeding events.
A MACE was defined as cardiovascular death, non-fatal MI or unplanned hospitalisation for heart failure. Acute cerebrovascular and systemic bleeds were defined using the BARC criteria. 26
All of these events were adjudicated by a Clinical Event Committee (CEC), which was made up of three cardiologists who were independent of the trial and blinded to the treatment allocation. The CEC charter was established before enrolment began. Coronary revascularisation, including PCI and coronary artery bypass grafting, was prospectively recorded in the clinical report form. Information on serious adverse events during follow-up was obtained by contacting the patients by telephone and reviewing their medical records. Complications that were potentially related to the study procedure were prospectively recorded.
Trial management
The trial was conducted in line with Good Clinical Practice in Clinical Trials24 guidelines and the study complies with the Declaration of Helsinki. 25 There was a trial management group for operational activity, an independent CEC to adjudicate on serious adverse events for safety and efficacy outcomes, an independent Data and Safety Monitoring Committee and a TSC to co-ordinate the trial and liaise with the sponsor and trials unit. Each committee had a charter that was established before enrolment started and an independent chairperson who had relevant expertise. The TSC also included two lay members representing patient and public involvement (PPI). The PPI representatives contributed actively to the meetings. The PPI representatives provided the patient voice when enrolment had been halted after December 2016.
The independent Data and Safety Monitoring Committee met before the enrolment began, and twice again during the active phase of the trial. This committee had responsibility for potentially recommending early discontinuation of the entire study or an individual group because of safety concerns or owing to futility (see Prespecified futility analysis).
The Robertson Centre for Biostatistics within the Glasgow Clinical Trials Unit provided the trial-specific electronic data collection system, acted as an independent co-ordinating centre for randomisation and data management, and conducted the statistical analyses. The trial was approved by the National Research Ethics Service (reference 13/WS/0119). The clinical trial registration number is NCT02257294, and the trial was co-sponsored by the University of Glasgow and Greater Glasgow and Clyde Health Board, NHS Scotland. The sponsors undertook feasibility assessments at each site. The sponsors prospectively monitored the study for safety and monitoring; visits were undertaken in all of the sites. All serious adverse events were prospectively reported to the Pharmacovigilance Unit.
Sample size and statistical methods
The target sample size was 618 patients, which was based on obtaining 186 patients per group with MRI at 2–7 days, allowing for approximately 10% missing data. This was designed to give 90% power at a 5% significance level to detect a difference between two groups of 1.72%, assuming a mean [standard deviation (SD)] of 3.2% (5.1%) for the extent of microvascular obstruction in the comparator group. This calculation was based on the amount of microvascular obstruction demonstrated in the subgroup of patients enrolled into the magnetic resonance in myocardial infarction (MR-MI) cohort study27 who fulfilled the enrolment criteria for the T-TIME trial.
Efficacy analyses were analysed according to randomisation group, that is in relation to randomised treatment allocation regardless of the treatment received. Safety data were analysed in relation to the treatment received. The primary outcome (extent of microvascular obstruction on MRI at 2–7 days post enrolment, as percentage of left ventricular mass) was compared between groups using a stratified Wilcoxon test (van Elteren test), stratified by the location of the MI. The 95% confidence intervals (CIs) for between-group differences in the mean extent of microvascular obstruction were derived by bootstrap resampling (10,000 replicates), stratified by location of MI; percentile CIs are reported. The primary analysis was to compare the 20 mg of alteplase group with the placebo group; if this was significant at a 5% level, the 10 mg of alteplase group would then be compared with the placebo group as a primary analysis. This hierarchical approach was used to preserve the overall type I error rate at 5%. However, if the 20 mg of alteplase group versus the placebo group comparison was not significant, the 10 mg of alteplase group versus the placebo group comparison would be considered as a secondary analysis. Primary and secondary outcomes were also analysed using linear regression (continuous outcomes), logistic regression (binary outcomes) or proportional odds logistic regression (ordinal outcomes). All models were adjusted for the location of the MI. In linear regression models for continuous outcome measures, data were transformed where necessary to improve model residual distributions and were further adjusted for the baseline value of the outcome (where appropriate). For the primary outcome, a post hoc analysis was performed with multiple imputation for the missing outcomes. Regression models were used to assess treatment effects within prespecified subgroups through the use of treatment-by-subgroup interactions (further details are provided in Appendix 3). All tests were two-tailed and assessed at the 5% significance level. Missing secondary outcome data were not imputed. Owing to the potential for type 1 error in the analyses of secondary end points, these end points should be interpreted as exploratory. All statistical analyses were carried out with R studio version 3.2.4 (R Development Core Team 2015, The R Foundation for Statistical Computing, Vienna, Austria) according to a prespecified statistical analysis plan (see Appendix 2).
Prespecified futility analysis
The funder, the EME programme of NIHR, required an interim analysis for futility and specified the criteria before the start of the trial. This analysis was scheduled for when approximately 40% of patients had been randomised and followed up to 3 months. Considering the primary outcome, each active treatment group was compared with the placebo group and if the conditional power for showing a benefit over placebo based on the current trend was less than 30%, then a recommendation would be made to halt that group.
Chapter 4 Results
On the recommendation of the Data and Safety Monitoring Committee, recruitment was discontinued on 21 December 2017 because a prespecified futility criterion for efficacy was met. Specifically, the conditional power for an analysis on the primary efficacy outcome based on 40% of the randomised population (n = 267) with follow-up to 3 months was < 30% in both treatment groups. The median value of microvascular obstruction was based on 251 patients, of whom 124 had a value of zero. The median value was 0.05% of left ventricular mass (0.00–3.19% of left ventricular mass) and the range was 0.00% to 28.75% of left ventricular mass. Overall, microvascular obstruction affected only 20% of the trial participants included in this analysis. The Data and Safety Monitoring Committee also noted that there were no safety concerns. The Data and Safety Monitoring Committee recommended that all randomised participants should be followed up for 3 months and that follow-up data collection should be as complete as possible. The sponsor implemented a temporary halt to enrolment, and following a review by the TSC and sponsor the halt became permanent on 2 February 2018.
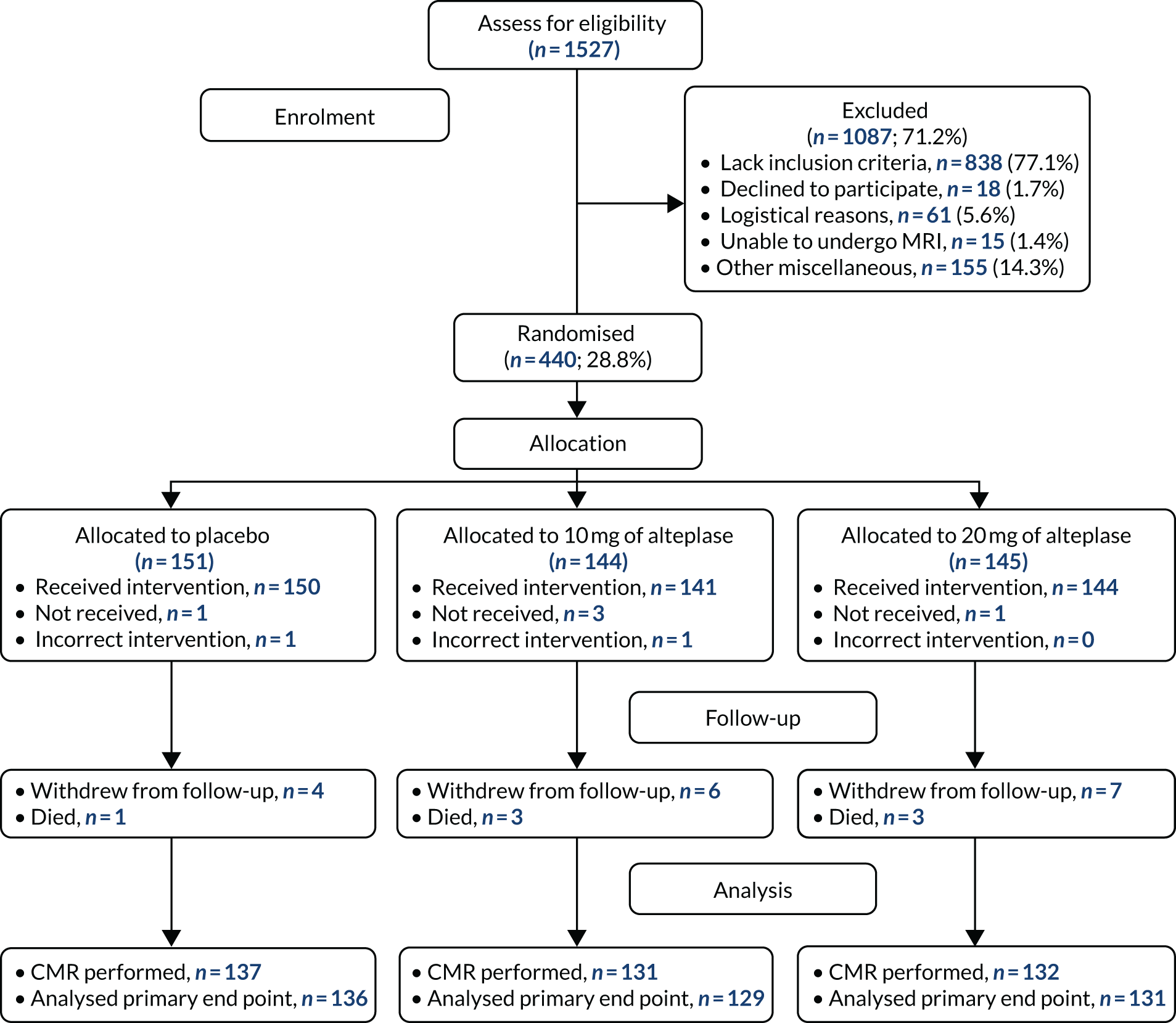
By that time, 1527 patients undergoing primary PCI for acute STEMI had been screened (Figure 3) and 440 patients (mean age 60.5 years, 15% female) had been randomised (placebo group, n = 151; 10 mg of alteplase group, n = 144; 20 mg of alteplase group, n = 145) (Table 1). Seventeen (3.9%) patients withdrew from the study during follow-up. All of the other patients were followed up for 3 months; final follow-up took place on 12 April 2018. Procedure characteristics and outcomes for all randomised patients are shown in Table 2.
FIGURE 3.
The Consolidated Standards of Reporting Trials flow diagram of patient recruitment, randomisation and flow through the study. Two patients (one randomised to the placebo group and one randomised to the 10 mg of alteplase group) received 20 mg of alteplase because an incorrect treatment pack had been selected. Four patients were unable to complete the cardiac magnetic resonance imaging examination, which meant that evaluable data for the primary outcome were not available: placebo group (n = 1), 10 mg of alteplase group (n = 2) and 20 mg of alteplase group (n = 1). CMR, cardiac magnetic resonance imaging.
| Baseline characteristic | Treatment group, n (%)a | ||
|---|---|---|---|
| Placebo (n = 151) | 10 mg of alteplase (n = 144) | 20 mg of alteplase (n = 145) | |
| Clinical | |||
| Age (years), mean (SD) | 60.7 (11.0) | 59.6 (10.3) | 61.2 (9.7) |
| Sex | |||
| Male | 127 (84.1) | 124 (86.1) | 123 (84.8) |
| Female | 24 (15.9) | 20 (13.9) | 22 (15.2) |
| Ethnicity | |||
| White | 143 (94.7) | 134 (93.1) | 136 (93.8) |
| Asian | 7 (4.6) | 9 (6.3) | 8 (5.5) |
| Body mass index (kg/m2), mean (SD) | 28.4 (5.3) | 28.5 (4.8) | 27.8 (4.4) |
| Presenting characteristics, mean (SD) | |||
| Heart rate (b.p.m.) | 73.3 (22.5) | 71.8 (15.9) | 73.5 (17.6) |
| Systolic blood pressure (mmHg) | 132 (26) | 135 (25) | 134 (25) |
| Diastolic blood pressure (mmHg) | 79 (17) | 80 (15) | 81 (15) |
| Infarct location | |||
| Anterior | 65 (43.0) | 62 (43.1) | 64 (44.1) |
| Inferior | 70 (46.4) | 67 (46.5) | 70 (48.3) |
| Lateral | 1 (0.7) | 2 (1.4) | 0 (0.0) |
| Posterior | 14 (9.3) | 11 (7.6) | 8 (5.5) |
| Other | 1 (0.7) | 2 (1.4) | 3 (2.1) |
| Medical history | |||
| Hypertensionb | 47 (31.1) | 45 (31.2) | 49 (33.8) |
| Renal impairmentc | 2 (1.3) | 3 (2.1) | 1 (0.7) |
| Diabetes mellitusb,d | 19 (12.6) | 19 (13.2) | 18 (12.4) |
| Hypercholesterolemiab | 42 (27.8) | 28 (19.4) | 32 (22.1) |
| Percutaneous coronary intervention | 8 (5.3) | 5 (3.5) | 7 (4.8) |
| Coronary artery bypass graft surgery | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Angina | 6 (4.0) | 7 (4.9) | 4 (2.8) |
| Myocardial infarction | 6 (4.0) | 6 (4.2) | 8 (5.5) |
| Stroke or transient ischaemic attackb | 2 (1.3) | 1 (0.7) | 2 (1.4) |
| Peripheral vascular diseaseb | 3 (2.0) | 3 (2.1) | 6 (4.1) |
| Smokingb | |||
| Current | 75 (49.7) | 72 (50.0) | 62 (42.8) |
| Former (stopped > 3 months ago) | 27 (17.9) | 22 (15.3) | 35 (24.1) |
| Never | 49 (32.5) | 50 (34.7) | 48 (33.1) |
| Pre-existing maintenance medication | |||
| Aspirin | 27 (17.9) | 17 (11.8) | 22 (15.2) |
| P2Y12 inhibitor | |||
| Clopidogrel | 1 (0.7) | 0 (0.0) | 1 (0.7) |
| Ticagrelor or prasugrel | 9 (6.0) | 4 (2.8) | 7 (4.8) |
| Statins | 40 (26.5) | 29 (20.1) | 28 (19.3) |
| Beta-blocker | 17 (11.3) | 15 (10.4) | 10 (6.9) |
| ACE-I or ARB | 23 (15.2) | 28 (19.5) | 27 (18.6) |
| Mineralocorticoid receptor antagonist | 1 (0.7) | 2 (1.4) | 1 (0.7) |
| Time frame | |||
| Symptom onset to arrival at primary PCI centre, hours:minutes | 2:05 (1:34, 3:01) | 2:11 (1:31, 3:26) | 2:15 (1:32, 3:15) |
| Arrival at primary PCI centre to reperfusion (minutes) | 24 (19, 35) | 23 (19, 37) | 25 (19, 34) |
| Symptom onset to reperfusion, hours:minutes | 2:36 (2:03, 3:36) | 2:50 (1:55, 4:06) | 2:44 (2:01, 3:49) |
| Initial blood results on admission | |||
| Haemoglobin (g/dl), mean | 14.4 (1.4) | 14.6 (1.2) | 14.7 (1.3) |
| Platelet count (× 103/µl), mean (SD) | 253.7 (59.8) | 267.9 (72.0) | 260.4 (53.3) |
| Creatinine (µmol/l), mean (SD) | 80 (18) | 80 (17) | 80 (18) |
| Troponin T (ng/ml), median (IQR) | 0.06 (0.03–0.13) | 0.06 (0.03–0.10) | 0.06 (0.03–0.12) |
| Characteristic | Treatment group, n (%)a | ||
|---|---|---|---|
| Placebo (n = 151)b | 10 mg of alteplase (n = 144)b | 20 mg alteplase (n = 145)b | |
| Infarct-related artery | |||
| Left anterior descending coronary | 67 (44.4) | 65 (45.1) | 65 (44.8) |
| Circumflex | 20 (13.2) | 18 (12.5) | 18 (12.4) |
| Right | 64 (42.4) | 61 (42.4) | 62 (42.8) |
| Infarct artery diameter (mm), mean (SD) | 3.2 (0.4) | 3.3 (0.5) | 3.2 (0.4) |
| Mode of reperfusion | |||
| Aspiration thrombectomy, n/N (%) | 39/151 (25.8) | 44/143 (30.8) | 42/145 (29.0) |
| Balloon angioplasty, n/N (%) | 112/151 (74.2) | 99/143 (69.2) | 102/145 (70.3) |
| Primary stent, n/N (%) | 0/151 (0.0) | 0/143 (0.0) | 1/145 (0.7) |
| Balloon angioplasty pre-stent deployment | 143 (94.7) | 138 (95.8) | 136 (93.8) |
| PCI with stent implantation, n/N (%) | 149/151 (98.7) | 141/143 (98.6) | 143/144 (99.3) |
| Total number of stents deployed | |||
| 0 | 2 (1.3) | 2 (1.4) | 1 (0.7) |
| 1 | 102 (67.5) | 98 (68.1) | 106 (73.1) |
| 2 | 43 (28.5) | 30 (20.8) | 33 (22.8) |
| ≥ 3 | 4 (2.6) | 14 (9.7) | 5 (3.4) |
| Total length of stents deployed (mm) (n = 435), mean (SD) | 33.4 (13.5) | 35.0 (15.1) | 32.3 (14.6) |
| Post-stent dilatation | 129 (85.4) | 124 (86.1) | 125 (86.2) |
| TIMI flow grade at initial angiographyc | |||
| 0 (no flow) | 130 (86.1) | 113 (78.5) | 111 (76.6) |
| 1 (minimal flow) | 3 (2.0) | 14 (9.7) | 15 (10.3) |
| 2 or 3 (2, slow but complete; 3, normal flow) | 18 (11.9) | 17 (11.8) | 19 (13.1) |
| TIMI thrombus grade at initial angiographyd | |||
| 0–2 (0, no thrombus; 2, definite thrombus; < 1/2 vessel diameter) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 3 (definite, > 1/2 but < 2 vessel diameters) | 4 (2.6) | 2 (1.4) | 5 (3.4) |
| 4 (definite thrombus ≥ 2 vessel diameters) | 18 (11.9) | 28 (19.4) | 29 (20.0) |
| 5 (total occlusion) | 129 (85.4) | 114 (79.2) | 111 (76.6) |
| Acute therapy following the first medical contact | |||
| Aspirin | 130 (86.1) | 128 (88.9) | 125 (86.2) |
| Loading dose of aspirin (mg), n/N (%) | |||
| 300 | 124/130 (95.4) | 123/128 (96.1) | 121/125 (96.8) |
| > 300 | 6/130 (4.6) | 5/128 (3.9) | 4/125 (3.2) |
| Additional antiplatelet medication | |||
| None | 18 (11.9) | 14 (9.7) | 18 (12.4) |
| Clopidogrel | 46 (30.5) | 49 (34.0) | 51 (35.2) |
| Ticagrelor | 84 (55.6) | 76 (52.8) | 70 (48.3) |
| Prasugrel | 3 (2.0) | 5 (3.5) | 6 (4.1) |
| Unfractionated heparin (U), median (IQR) | 10,000 (7000–12,250) | 10,000 (7500–13,000) | 10,000 (7000–13,000) |
| Inhaled oxygen, n/N (%) | 23/148 (15.5) | 25/140 (17.9) | 16/140 (11.4) |
| Intravenous morphine | 105 (69.5) | 109 (75.7) | 112 (77.2) |
| Intravenous or intracoronary glycoprotein IIb/IIIa antagonist, n/N (%) | 17/148 (11.5) | 31/140 (22.1) | 25/140 (17.9) |
| Study drug treatment | |||
| Drug administered | 150 (99.3) | 141 (97.9) | 144 (99.3) |
| Study drug given according to protocol, n/N (%) | 149/150 (99.3) | 139/141 (98.6) | 141/144 (97.9) |
| Duration of study drug infusion (minutes), mean (SD) | 6.4 (1.9) | 6.6 (2.0) | 6.6 (2.0) |
Study intervention
The standard of care procedure and study intervention are illustrated in Appendix 3 and summarised in Table 3. Adjunctive study drug therapy was administered to 435 (98.9%) patients; five patients did not receive any drug (see Figure 3). Two patients (one randomised to the placebo group and one randomised to the 10 mg of alteplase group) received 20 mg of alteplase because an incorrect treatment pack had been selected.
| Characteristic | Treatment group, n (%)a | ||
|---|---|---|---|
| Placebo (n = 137)b | 10 mg of alteplase (n = 131)b | 20 mg of alteplase (n = 132)b | |
| Infarct-related artery | |||
| Left anterior descending coronary | 61 (44.5) | 61 (46.6) | 60 (45.5) |
| Circumflex | 18 (13.1) | 15 (11.5) | 13 (9.8) |
| Right | 58 (42.3) | 55 (42.0) | 59 (44.7) |
| Infarct artery diameter (mm), mean (SD) | 3.2 (0.4) | 3.2 (0.5) | 3.2 (0.4) |
| Mode of reperfusion | |||
| Aspiration thrombectomy | 37 (27.0) | 42 (32.3) | 40 (30.3) |
| Balloon angioplasty | 100 (73.0) | 88 (67.7) | 91 (68.9) |
| Primary stent | 0 (0.0) | 0 (0.0) | 1 (0.8) |
| Balloon angioplasty pre-stent deployment | 129 (94.2) | 126 (96.2) | 125 (94.7) |
| PCI with stent implantation, n/N (%) | 136/137 (99.3) | 128/130 (98.5) | 130/131 (99.2) |
| Total number of stents deployed | |||
| 0 | 1 (0.7) | 2 (1.4) | 1 (0.8) |
| 1 | 92 (67.2) | 88 (67.2) | 99 (75.0) |
| 2 | 40 (29.2) | 28 (21.4) | 28 (21.2) |
| ≥ 3 | 4 (2.9) | 10 (7.6) | 4 (3.0) |
| Total length of stents deployed (n = 396) (mm), mean (SD) | 33.5 (13.8) | 35.7 (15.3) | 32.0 (14.0) |
| Post-stent dilatation | 119 (86.9) | 116 (88.5) | 115 (87.1) |
| TIMI flow grade at initial angiographyc | |||
| 0 (no flow) | 117 (85.4) | 103 (78.6) | 103 (78.0) |
| 1 (minimal flow) | 3 (2.2) | 13 (9.9) | 14 (10.6) |
| 2 or 3 (2, slow but complete; 3, normal flow) | 17 (12.4) | 15 (11.5) | 15 (11.4) |
| TIMI thrombus grade at initial angiographyd | |||
| 0–2 (0, no thrombus; 2, definite thrombus; < 1/2 vessel diameter) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 3 (definite, > 1/2 but < 2 vessel diameters) | 4 (2.6) | 2 (1.5) | 5 (3.8) |
| 4 (definite thrombus, ≥ 2 vessel diameters) | 17 (12.4) | 25 (19.1) | 24 (18.2) |
| 5 (total occlusion) | 116 (84.7) | 104 (79.4) | 103 (78.0) |
| Acute therapy following the first medical contact | |||
| Aspirin | 119 (86.9) | 118 (90.1) | 112 (84.8) |
| Loading dose of aspirin (mg), n/N (%) | |||
| 300 | 114/119 (95.4) | 113/118 (95.8) | 109/112 (97.3) |
| > 300 | 5/119 (4.2) | 5/118 (4.2) | 3/112 (2.7) |
| Additional antiplatelet medication | |||
| None | 15 (10.9) | 10 (7.6) | 17 (12.9) |
| Clopidogrel | 43 (31.4) | 48 (36.6) | 51 (38.6) |
| Ticagrelor | 76 (55.5) | 68 (51.9) | 60 (45.5) |
| Prasugrel | 3 (2.2) | 5 (3.8) | 4 (3.0) |
| Unfractionated heparin (U), median (IQR) | 10,000 (7000–12,250) | 10,000 (7500–13,000) | 10,000 (7000–13,000) |
| Inhaled oxygen, n/N (%) | 21/134 (15.5) | 22/127 (17.3) | 13/129 (10.1) |
| Intravenous morphine, n/N (%) | 93 (67.9) | 100 (76.3) | 104 (78.8) |
| Intravenous or intracoronary glycoprotein IIb/IIIa antagonist, n/N (%) | 13/134 (9.7) | 27/127 (21.3) | 21/129 (16.3) |
| Study drug treatment | |||
| Drug administered | 137 (100.0) | 129 (98.5) | 132 (100.0) |
| Study drug given according to protocol | 136/137 (99.3) | 127/129 (98.4) | 131/132 (99.2) |
| Duration of study drug infusion (minutes), mean (SD) | 6.4 (1.9) | 6.6 (2.0) | 6.6 (2.0) |
Primary and secondary outcomes
Magnetic resonance imaging was performed in 400 (90.9%) patients at 2–7 days and in 367 (83.4%) patients at 3 months post enrolment. The primary end point was available in 396 patients, which meant that data were missing for the primary end point in 10% of patients. The median time to MRI was 4 days [interquartile range (IQR) 3–6 days]: placebo, median 4 days (IQR 3–5 days); 10 mg of alteplase, median 5 days (IQR 3–6 days); 20 mg of alteplase, median 4 days (IQR 3–6 days); and median 91 days (IQR 86–97 days) for the second MRI scan for the study population. Microvascular obstruction was observed in 176 (44.4%) patients and the amount of microvascular obstruction, expressed as the mean percentage of left ventricular mass, was 2.80%. Clinical case examples are illustrated in Figure 4.
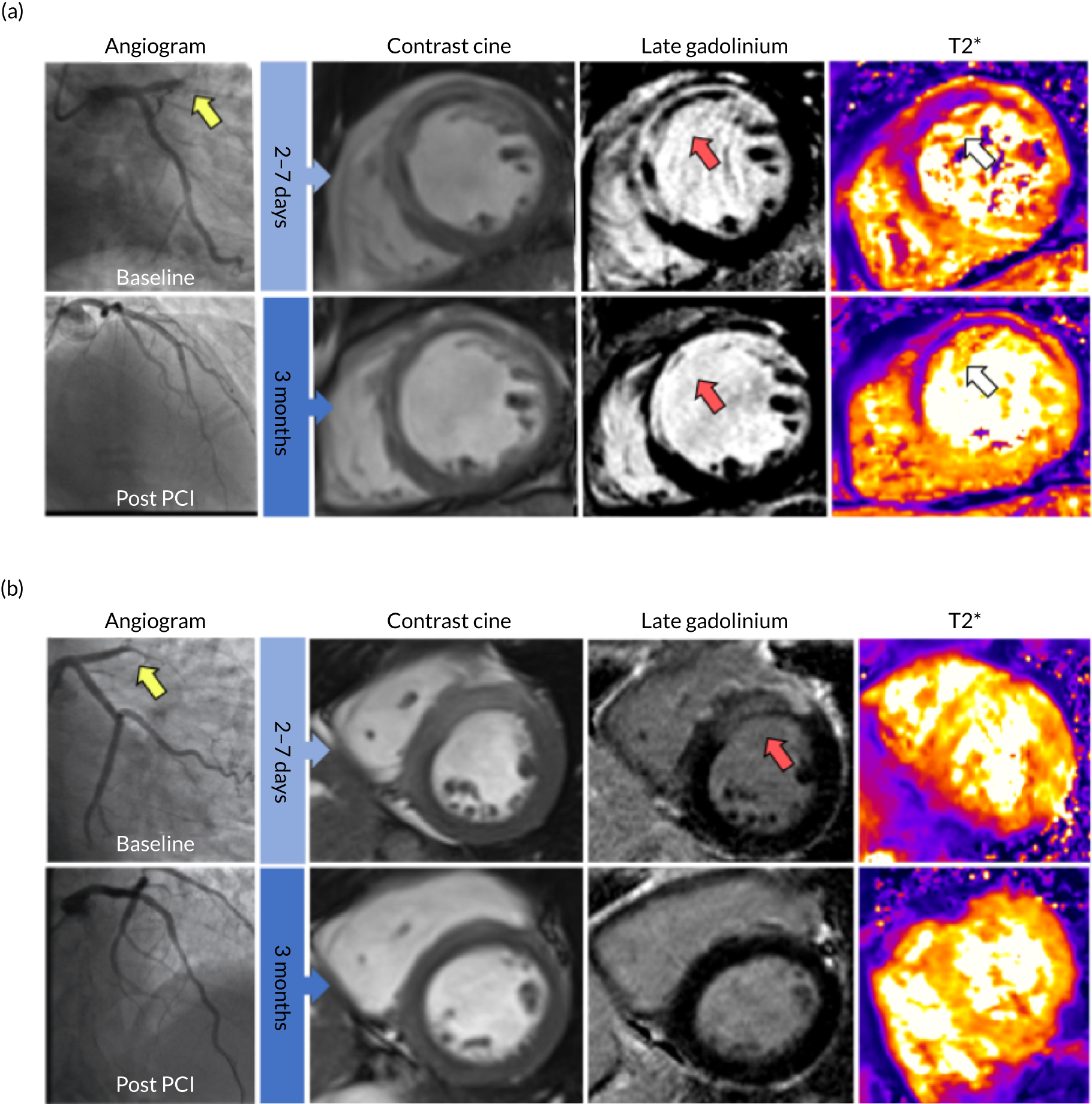
FIGURE 4.
Clinical case examples. Two patients, both with acute anterior STEMI treated successfully with primary PCI within 4 hours of symptom onset. Each patient had thrombolysis in MI (TIMI) grade 3 flow at the end of PCI. MRI was performed at 5 days and 3 days post reperfusion, in a and b respectively. a, Patient with haemorrhagic infarction on MRI. Diagnostic angiogram (top far left image, yellow arrow) revealed an occluded left anterior descending artery with a TIMI 0 flow. T2*-MRI (top far right image) revealed myocardial haemorrhage (white arrow) within the infarct core. Late gadolinium-enhanced MRI revealed microvascular obstruction (top second from right image, red arrow) within the bright area of infarction. The microvascular obstruction within the infarct core spatially corresponded with the myocardial haemorrhage. There was evidence of persistent haemoglobin breakdown products (bottom far right image, white arrow), a reduction in ejection fraction, increased left ventricular end-diastolic volumes and thinned myocardium in the infarct territory on follow-up MRI at 3 months compared to baseline. This represents a case of failed myocardial reperfusion despite successful PCI. b, Patient with an anterior infarct but no MRI evidence of reperfusion injury. Diagnostic angiography (top, far left image, yellow arrow) revealed an occluded left anterior descending artery with a TIMI 0 flow. Late gadolinium-enhanced MRI revealed an anterior infarct (top second from right image, red arrow) with no evidence of microvascular obstruction and no evidence of haemorrhagic transformation on T2*-MRI. There was an improved ejection fraction and a reduction in infarct mass on follow-up MRI at 3 months. This represents a case of successful myocardial reperfusion.
Primary outcome
In the primary analysis, the amount of microvascular obstruction revealed by MRI did not differ between the 20 mg of alteplase group and the placebo group [mean 3.5% vs. 2.3%, estimated difference 1.16%, 95% CI –0.08% to 2.41%; Wilcoxon test (van Elteren test) p = 0.32]. The comparison of the 10 mg of alteplase group with the placebo group then became a secondary outcome [mean 2.6% vs. 2.3%, estimated difference 0.29%, 95% CI –0.76% to 1.35%; Wilcoxon test (van Elteren test) p = 0.74]. Similar results were obtained using a linear regression model, with no evidence of a difference in the primary outcome between all patients who were randomised to the alteplase group and those randomised to the placebo group (mean difference on square-root scale 0.15, 95% CI –0.12 to 0.42; p = 0.28).
Post hoc analysis
A post hoc analysis of the primary outcome including multiple imputation for the missing values was performed, which produced similar results to the primary analysis.
The imputation analysis involved 10 imputed data sets and two predictive models: one to predict the presence of any microvascular obstruction and one to predict the extent of microvascular obstruction, if any. For the presence of microvascular obstruction, a logistic regression model was used that included age, sex, location of MI and time from symptom onset to reperfusion as predictors; for the extent of microvascular obstruction (where present), a linear model for the log of microvascular obstruction extent was used and the same set of predictors were included. From these models, the presence of microvascular obstruction was randomly imputed along with the extent (if present) for those patients with missing data for microvascular obstruction at 2–7 days. The analysis was repeated 10 times.
The primary analyses were conducted on the imputed data sets and the results were combined using Rubin’s rules. 28 For the van Elteren test results, the results using multiple imputation data were as follows: 20 mg of alteplase versus placebo, p = 0.279; 10 mg of alteplase versus placebo, p = 0.794; active treatment (20 mg and 10 mg of alteplase) versus placebo, p = 0.422. For the linear regression of the square root of microvascular obstruction, the multiple imputation results were as follows: 20 mg of alteplase versus placebo, p = 0.148; 10 mg of alteplase versus placebo, p = 0.700; active treatment (20 mg and 10 mg of alteplase) versus placebo, p = 0.283.
Prespecified subgroup analyses of primary outcome
Treatment effect differences on the primary outcome between prespecified subgroups that were defined by baseline characteristics were assessed. None of the interaction tests was statistically significant (Tables 4 and 5). In the subgroup of patients who presented > 4 hours after symptom onset, the estimated mean difference in the square root of the amount of microvascular obstruction between the 20 mg of alteplase group (n = 27) and the placebo group (n = 26) was 1.12 (95% CI 0.42 to 1.82; p = 0.002); however, the test for interaction was not statistically significant (p = 0.06) so this subgroup finding should not be interpreted as different from the overall effect.
| Outcome | Primary outcome analysed, mean (SD)a | Between-group comparison | Interaction p-value | |||||
|---|---|---|---|---|---|---|---|---|
| Placebo (n = 136) | 10 mg of alteplase (n = 129) | 20 mg of alteplase (n = 131) | 20 mg of alteplase vs. placebo, p-value | 20 mg of alteplase vs. placebo (95% CI) | 10 mg of alteplase vs. placebo, p-valueb | 10 mg of alteplase vs. placebo (95% CI)b | ||
| Primary outcomec | ||||||||
| Microvascular obstruction at 2–7 days post enrolment (% of left ventricular mass) | 2.3 (4.3) | 2.6 (4.5) | 3.5 (5.8) | 0.32d | 1.16 (–0.08 to 2.41) | 0.74d | 0.29 (–0.76 to 1.35) | |
| Interquartile range | 0.0–3.2 | 0.0–3.8 | 0.0–4.8 | |||||
| Range, min–max | 0–28.8 | 0–27.0 | 0–26.1 | |||||
| Secondary analysis: comparing the square root of microvascular obstruction, 2–7 days post enrolment | 0.15e | 0.23 (–0.09 to 0.55) | 0.68e | 0.07 (–0.25 to 0.39) | ||||
| Prespecified analysis of primary outcome (microvascular obstruction), by characteristics at baseline | ||||||||
| Ischaemic time | ||||||||
| < 2 hours (n = 98, 24.7%) | 1.4 (2.7) | 1.5 (2.7) | 2.7 (5.0) | 0.48e | 0.25 (–0.43 to 0.92) | 0.78e | 0.09 (–0.55 to 0.73) | 0.09f |
| 2–4 hours (n = 215, 54.3%) | 3.0 (5.0) | 3.1 (5.3) | 3.2 (5.7) | 0.79e | –0.06 (–0.47 to 0.35) | 0.95e | –0.01 (–0.45 to 0.42) | |
| ≥ 4 hours (n = 83, 21.0%) | 1.1 (2.6) | 3.1 (4.6) | 5.2 (6.9) | 0.002e | 1.12 (0.42 to 1.82) | 0.13f | 0.53 (–0.15 to 1.22) | |
| Sex | ||||||||
| Male (n = 338, 85.4%) | 2.4 (4.5) | 2.8 (4.6) | 4.0 (6.2) | 0.05e | 0.34 (0.00 to 0.68) | 0.51e | 0.11 (–0.23 to 0.45) | 0.27f |
| Female (n = 58, 14.6%) | 1.8 (2.8) | 1.7 (4.0) | 0.8 (1.6) | 0.35e | –0.39 (–1.20 to 0.43) | 0.60e | –0.22 (–1.06 to 0.61) | |
| Age | ||||||||
| < 55 years (n = 113, 28.5%) | 1.8 (2.9) | 3.2 (5.2) | 3.1 (4.7) | 0.51e | 0.21 (–0.42 to 0.85) | 0.23e | 0.34 (–0.22 to 0.91) | 0.68f |
| 55–65 years (n = 168, 42.4%) | 3.0 (5.6) | 2.3 (3.8) | 3.6 (6.4) | 0.58e | 0.14 (–0.35 to 0.63) | 0.47e | –0.19 (–0.71 to 0.33) | |
| ≥ 65 years (n = 115, 29.0%) | 2.1 (3.8) | 2.5 (4.5) | 3.5 (5.5) | 0.27e | 0.33 (–0.25 to 0.91) | 0.75e | 0.10 (–0.50 to 0.69) | |
| MI location | ||||||||
| Anterior (n = 178, 44.9%) | 3.4 (5.6) | 2.8 (4.0) | 4.7 (6.9) | 0.28e | 0.26 (–0.21 to 0.74) | 0.74e | –0.08 (–0.55 to 0.39) | 0.57f |
| Non-anterior (n = 221, 55.1%) | 1.5 (2.7) | 2.5 (4.8) | 2.5 (4.6) | 0.35e | 0.20 (–0.22 to 0.63) | 0.38e | 0.19 (–0.24 to 0.62) | |
| Smoking status | ||||||||
| Never (n = 137, 34.6%) | 2.4 (4.0) | 1.9 (3.7) | 4.7 (7.5) | 0.19e | 0.36 (–0.18 to 0.90) | 0.38e | –0.25 (–0.79 to 0.30) | 0.22f |
| Former (n = 74, 18.7%) | 1.3 (3.4) | 1.8 (2.9) | 3.2 (5.7) | 0.10e | 0.57 (–0.12 to 1.27) | 0.50e | 0.28 (–0.52 to 1.07) | |
| Current (n = 185, 46.7%) | 2.6 (4.8) | 3.3 (5.2) | 2.6 (3.8) | 0.99e | 0.00 (–0.48 to 0.47) | 0.41e | 0.19 (–0.26 to 0.64) | |
| Initial TIMI coronary flow grade | ||||||||
| 0 (no flow) (n = 320, 80.8%) | 2.6 (4.5) | 3.1 (4.9) | 3.9 (6.2) | 0.23e | 0.21 (–0.13 to 0.56) | 0.73e | 0.06 (–0.28 to 0.41) | 0.71f |
| 1 (minimal flow), (n = 30, 7.6%) | 0.0 (0.0) | 1.4 (2.3) | 3.5 (4.8) | 0.10e | 1.37 (–0.24 to 2.98) | 0.29e | 0.87 (–0.76 to 2.50) | |
| 2 or more (2, slow but complete; 3, normal flow) (n = 47, 11.6%) | 0.7 (2.9) | 0.5 (1.0) | 0.8 (2.9) | 0.88e | 0.07 (–0.83 to 0.97) | 0.90e | 0.06 (–0.86 to 0.97) | |
| Pre-existing antiplatelet medication | ||||||||
| Yes (n = 58, 14.6%) | 2.4 (3.5) | 1.4 (4.0) | 3.9 (6.3) | 0.44e | 0.31 (–0.48 to 1.11) | 0.30e | –0.46 (–1.32 to 0.40) | 0.31f |
| No (n = 341, 85.4%) | 2.3 (4.5) | 2.8 (4.5) | 3.4 (5.8) | 0.22e | 0.22 (–0.13 to 0.56) | 0.41e | 0.14 (–0.20 to 0.49) | |
| Outcome | Alteplase groups vs. placebo group, p-value | Alteplase groups vs. placebo group, effect estimates (95% CI) | Interaction, p-value |
|---|---|---|---|
| Secondary analysis of primary outcome, microvascular obstruction at 2–7 days | 0.28a | 0.15 (–0.12 to 0.42) | |
| Prespecified analysis of primary outcome by characteristics at baseline | |||
| Ischaemic time | |||
| < 2 hours | 0.59a | 0.16 (–0.42 to 0.74) | 0.06b |
| 2–4 hours | 0.84a | –0.04 (–0.40 to 0.32) | |
| ≥ 4 hours | 0.009a | 0.81 (0.21 to 1.42) | |
| Sex | |||
| Male | 0.13a | 0.23 (–0.07 to 0.52) | 0.17b |
| Female | 0.39a | –0.31 (–1.02 to 0.40) | |
| Age | |||
| < 55 years | 0.25a | 0.29 (–0.21 to 0.79) | 0.65b |
| 55–65 years | 0.98a | –0.01 (–0.45 to 0.44) | |
| ≥ 65 years | 0.40a | 0.21 (–0.29 to 0.71) | |
| MI location | |||
| Anterior | 0.66a | 0.09 (–0.32 to 0.50) | 0.70b |
| Non-anterior | 0.29a | 0.20 (–0.17 to 0.57) | |
| Smoking status | |||
| Never | 0.80a | 0.06 (–0.41 to 0.53) | 0.57b |
| Former | 0.15a | 0.46 (–0.17 to 1.10) | |
| Current | 0.61a | 0.10 (–0.29 to 0.50) | |
| Initial TIMI coronary flow grade | |||
| 0 (no flow) | 0.37a | 0.14 (–0.16 to 0.43) | 0.45b |
| 1 (minimal flow) | 0.15a | 1.13 (–0.41 to 2.67) | |
| 2 or 3 (2, slow but complete; 3, normal flow) | 0.87a | 0.06 (–0.71 to 0.84) | |
| Pre-existing antiplatelet medication | |||
| Yes | 0.96a | –0.02 (–0.71 to 0.68) | 0.61b |
| No | 0.24a | 0.18 (–0.12 to 0.48) | |
Secondary outcomes
The seconday outcomes for efficacy and health outomes for alteplase are shown in Tables 6 and 7. The secondary outcomes for safety are shown in Tables 8 and 9.
| Outcome | Treatment group | Between-group comparison | |||||
|---|---|---|---|---|---|---|---|
| Placebo (n = 151) | 10 mg of alteplase (n = 144)a | 20 mg of alteplase (n = 145)a | 20 mg of alteplase vs. placebo (95% CI) | 20 mg of alteplase vs. placebo, p-value | 10 mg of alteplase vs. placebo (95% CI) | 10 mg of alteplase vs. placebo, p-value | |
| Coronary angiogram | |||||||
| TIMI flow grade after PCI | |||||||
| 0 (no flow) | 0 (0.0) | 1 (0.7) | 1 (0.7) | 0.73 (0.41 to 1.29) | 0.28b | 0.83 (0.46 to 1.49) | 0.52b |
| 1 (minimal flow) | 6 (4.0) | 6 (4.2) | 3 (2.1) | ||||
| 2 (slow but complete) | 20 (13.2) | 22 (15.3) | 29 (20.0) | ||||
| 3 (normal flow) | 125 (82.8) | 115 (79.9) | 112 (77.2) | ||||
| TIMI myocardial perfusion grade after PCIc | |||||||
| 0 (no blush) | 46 (30.5) | 62 (43.1) | 49 (33.8) | 0.93 (0.61 to 1.41) | 0.73b | 0.70 (0.46 to 1.08) | 0.11b |
| 1 (incomplete clearance) | 11 (7.3) | 2 (1.4) | 9 (6.2) | ||||
| 2 (persistent) | 51 (33.8) | 48 (33.3) | 49 (33.8) | ||||
| 3 (normal blush and clearance) | 43 (28.5) | 32 (22.2) | 38 (26.2) | ||||
| Corrected TIMI frame count after PCId (n = 438) | 20 (14, 27) | 20 (15, 28) | 22 (14, 30) | 1.07 (0.94 to 1.23) | 0.31e | 1.10 (0.96 to 1.25) | 0.19e |
| TIMI thrombus grade after PCI | |||||||
| 0 (no thrombus) | 147 (97.4) | 137 (95.1) | 141 (97.2) | 1.04 (0.25 to 4.23) | 0.96f | 1.87 (0.53 to 6.53) | 0.33f |
| 1 (possible thrombus) | 2 (1.3) | 4 (2.8) | 3 (2.1) | ||||
| 2 (definite, < 1/2 vessel diameter) | 1 (0.7) | 2 (1.4) | 0 (0.0) | ||||
| 3 (definite, > 1/2 but < 2 vessel diameters) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||||
| 4 (definite, > 2 vessel diameters) | 1 (0.7) | 0 (0.0) | 0 (0.0) | ||||
| 5 (occluded artery) | 0 (0.0) | 1 (0.7) | 1 (0.7) | ||||
| Electrocardiogram | |||||||
| Acute | |||||||
| ST-segment resolution 60 minutes (%) (n = 396), mean (SD) | 49.6 (38.9) | 43.7 (45.0) | 44.2 (46.0) | –5.0 (–15.1 to 5.2) | 0.34e | –5.8 (–16.2 to 4.6) | 0.27e |
| 3 months | |||||||
| Final infarct size (Selvester score),g % of left ventricular mass (n = 368), mean (SD) | 10.3 (9.0) | 12.1 (10.6) | 12.1 (9.9) | 1.70 (–0.64 to 4.03) | 0.15e | 1.69 (–0.69 to 4.08) | 0.16e |
| Biochemistry | |||||||
| Acute | |||||||
| Troponin T AUC (ng/l) (n = 317), mean (SD) | 2.80 (1.10, 5.32) | 2.94 (1.57, 5.98) | 3.80 (1.56, 6.63) | 1.53 (1.12 to 2.11) | 0.008e | 1.52 (1.10 to 2.09) | 0.01e |
| NT-proBNP (pg/ml) at 2–7 days post enrolment (n = 394), mean (SD) | 784 (386, 1350) | 849 (417, 1586) | 791 (412, 1355) | 1.00 (0.78 to 1.27) | 0.98e | 1.12 (0.88 to 1.42) | 0.35e |
| 3 months | |||||||
| NT-proBNP (n = 372), pg/ml | 228 (94, 521) | 260 (100, 442) | 239 (119, 528) | 0.99 (0.76 to 1.28) | 0.93e | 0.98 (0.75 to 1.27) | 0.88e |
| Ratio of NT-proBNP from 3 months to 2–7 days (n = 354) | 0.32 (0.20, 0.58) | 0.33 (0.18, 0.51) | 0.34 (0.22, 0.49) | 0.95 (0.79 to 1.15) | 0.63e | 8.69 (0.77 to 1.13) | 0.45e |
| Cardiac MRI | |||||||
| 2–7 days | |||||||
| Microvascular obstruction (n = 396), n/N (%) | 59/136 (43.4) | 58/129 (45.0) | 59/131 (45.0) | 1.07 (0.66 to 1.73) | 0.80f | 1.06 (0.65 to 1.73) | 0.81f |
| Myocardial haemorrhage (n = 378), n/N (%) | 52/128 (40.6) | 54/121 (44.6) | 56/129 (43.4) | 1.12 (0.68 to 1.84) | 0.65f | 1.17 (0.71 to 1.94) | 0.54f |
| Myocardial haemorrhage (% of left ventricular mass), mean (SD) (n = 360) | 1.56 (3.78) | 1.98 (3.68) | 2.45 (4.80) | 0.86 (–0.17 to 1.88) | 0.10e | 0.36 (–0.69 to 1.41) | 0.50e |
| Infarct size (% of left ventricular mass), mean (SD) (n = 396) | 26.3 (13.7) | 27.3 (12.4) | 26.6 (13.4) | 0.23 (–2.64 to 3.11) | 0.87e | 0.80 (–2.08 to 3.69) | 0.58e |
| Extent of myocardial oedema, % of left ventricular mass, mean (SD) (n = 397) | 40.4 (11.4) | 41.9 (11.4) | 41.0 (11.5) | 0.44 (–1.72 to 2.60) | 0.69e | 1.17 (–0.99 to 3.33) | 0.29e |
| Myocardial salvage index (n = 396), mean (SD) | 0.37 (0.25) | 0.36 (0.23) | 0.36 (0.24) | –0.01 (–0.07 to 0.05) | 0.68e | –0.01 (–0.07 to 0.04) | 0.64e |
| Left ventricular end-diastolic volume (ml) (n = 400) | 166 (144, 195) | 177 (157, 208) | 166 (143, 195) | 1.00 (0.95 to 1.05) | 0.94e | 1.06 (1.01 to 1.12) | 0.02e |
| Left ventricular end-systolic volume (ml) (n = 400) | 90 (77, 112) | 96 (80, 124) | 95 (79, 110) | 1.00 (0.94 to 1.08) | 0.93e | 1.08 (1.01 to 1.16) | 0.03e |
| Left ventricular ejection fraction (n = 400), % mean (SD) | 44.5 (8.8) | 43.6 (8.1) | 44.2 (8.4) | –0.27 (–2.16 to 1.63) | 0.78e | –0.76 (–2.66 to 1.14) | 0.44e |
| 3 months | |||||||
| Infarct size, % left ventricular mass (n = 364), mean (SD) | 18.7 (12.5) | 18.5 (11.2) | 19.3 (12.2) | 0.79 (–2.00 to 3.58) | 0.58e | –0.21 (–3.02 to 2.61) | 0.89e |
| Myocardial salvage indexh (n = 358), mean (SD) | 0.56 (0.24) | 0.58 (0.21) | 0.55 (0.23) | –0.02 (–0.08 to 0.04) | 0.52e | 0.01 (–0.05 to 0.07) | 0.67e |
| Left ventricular end-diastolic volume (ml) (n = 367), mean (SD) | 172 (42) | 183 (49) | 171 (40) | 1.00 (0.94 to 1.06) | 0.87e | 1.06 (1.00 to 1.12) | 0.06e |
| Left ventricular end-systolic volume (ml) (n = 367), mean (SD) | 89 (38) | 96 (41) | 91 (35) | 1.03 (0.94 to 1.12) | 0.57e | 1.08 (0.99 to 1.18) | 0.09e |
| Left ventricular ejection fraction (n = 367), % mean (SD) | 49.8 (8.8) | 48.5 (8.0) | 48.6 (8.5) | –1.25 (–3.30 to 0.80) | 0.23e | –1.30 (–3.38 to 0.77) | 0.22e |
| Health status and quality of life | |||||||
| Health-related quality of life:i health utility score at 2–7 days (n = 391), mean (SD) | 0.81 (0.19) | 0.82 (0.19) | 0.81 (0.20) | –0.01 (–0.06 to 0.03) | 0.60e | –0.002 (–0.049 to 0.044) | 0.92e |
| Health-related quality of life: EQ-5D-3Li health utility score at 3 months (n = 391), mean (SD) | 0.88 (0.16) | 0.89 (0.15) | 0.88 (0.16) | –0.002 (–0.04 to 0.04) | 0.93e | 0.008 (–0.03 to 0.05) | 0.68e |
| Change from baseline health status at 3 months (n = 378), mean (SD) | 0.07 (0.17) | 0.08 (0.23) | 0.06 (0.18) | –0.005 (–0.05 to 0.04) | 0.83e | 0.014 (–0.03 to 0.06) | 0.55e |
| Health-related quality of life: EQ-5D-3Li visual analogue score (n = 396), mean (SD) | 78.0 (17.5) | 79.8 (15.2) | 81.8 (15.1) | 3.89 (0.07 to 7.71) | 0.05e | 1.89 (–1.97 to 5.75) | 0.34e |
| Change from baseline health status to 3 months (n = 392), mean (SD) | 5.6 (19.9) | 8.2 (17.2) | 6.7 (18.4) | 1.0 (–3.45 to 5.59) | 0.66e | 2.50 (–2.00 to 7.01) | 0.28e |
| Outcome | Alteplase groups vs. placebo group, p-value | Alteplase groups vs. placebo group, effect estimates (95% CI) |
|---|---|---|
| Coronary angiogram | ||
| TIMI flow grade after PCI | ||
| 0–3 | 0.32a | 0.77 (0.47 to 1.29) |
| TIMI myocardial perfusion grade after PCIb | ||
| 0–3 | 0.26a | 0.81 (0.57 to 1.17) |
| Corrected TIMI frame count after PCIc | 0.17d | 1.08 (0.97 to 1.22) |
| TIMI thrombus grade after PCI | ||
| 0–5 | 0.53e | 1.45 (0.45 to 4.63) |
| Electrocardiogram (acute) | ||
| ST-segment resolution at 60 minutes, % | 0.23d | –5.4 (–14.2 to 3.5) |
| Biochemistry (acute) | ||
| Troponin T AUC | 0.002d | 1.53 (1.16 to 2.01) |
| NT-proBNP at 2–7 days (pg/ml) | 0.59d | 1.06 (0.86 to 1.30) |
| Cardiac MRI (2–7 days) | ||
| Microvascular obstruction | 0.77e | 1.06 (0.70 to 1.62) |
| Myocardial haemorrhage | 0.54e | 1.15 (0.74 to 1.77) |
| Myocardial haemorrhage, % left ventricular mass | 0.18d | 0.62 (–0.28 to 1.52) |
| Infarct size, % left ventricular mass | 0.68d | 0.52 (–1.96 to 3.00) |
| Extent of myocardial oedema, % left ventricular mass | 0.40d | 0.81 (–1.06 to 2.67) |
| Myocardial salvage index | 0.61d | –0.01 (–0.06 to 0.04) |
| Left ventricular end-diastolic volume (ml) | 0.21d | 1.03 (0.98 to 1.08) |
| Left ventricular end-systolic volume (ml) | 0.20d | 1.04 (0.98 to 1.11) |
| Left ventricular ejection fraction, % | 0.54d | –0.51 (–2.15 to 1.13) |
| Electrocardiogram (3 months) | ||
| Final infarct size (Selvester score)f | 0.10d | 1.69 (–0.33 to 3.72) |
| Biochemistry (3 months) | ||
| NT-proBNP (pg/ml) | 0.89d | 0.98 (0.79 to 1.23) |
| Change in NT-proBNP at 3 months from days 2–7 post enrolment (pg/ml) | 0.91d | 16.30 (–263.50 to 296.10) |
| Cardiac MRI (3 months) | ||
| Infarct size, % left ventricular mass | 0.81d | 0.30 (–2.13 to 2.73) |
| Myocardial salvage indexg | 0.89d | 0.00 (–0.05 to 0.05) |
| Left ventricular end-diastolic volume (ml) | 0.34d | 1.03 (0.97 to 1.08) |
| Left ventricular end-systolic volume (ml) | 0.20d | 1.05 (0.97 to 1.14) |
| Left ventricular ejection fraction, % | 0.16d | –1.28 (–3.06 to 0.51) |
| Health status and quality of life | ||
| Health-related quality of life: EQ-5D-3Lh health utility score | 0.85d | 0.003 (–0.03 to 0.04) |
| Change from baseline health status at 3 months | 0.83d | 0.005 (–0.04 to 0.05) |
| Health-related quality of life: EQ-5D-3Lh visual analogue score | 0.09d | 2.91 (–0.40 to 6.22) |
| Change from baseline health status to 3 months | 0.38d | 1.73 (–2.12 to 5.59) |
| Outcome | Treatment groupa | Between-group comparison p-valueb | ||||
|---|---|---|---|---|---|---|
| Placebo group (n = 149) | 10 mg of alteplase (n = 140) | 20 mg of alteplase group (n = 146) | 20 mg of alteplase vs. placebo | 10 mg of alteplase vs. placebo | Alteplase vs. placebo | |
| Coronary angiogram, n (%) | ||||||
| No re-flow | 26 (17.4) | 19 (13.6) | 18 (12.3) | 0.88 | 0.48 | 0.63 |
| Slow re-flow | 6 (4.0) | 7 (5.0) | 14 (9.6) | |||
| Normal flow | 117 (78.5) | 114 (81.4) | 114 (78.1) | |||
| Haematology and coagulation | ||||||
| Activated clotting time (seconds) at 2 hours from baseline | 256 (218–300) | 275 (229–350) | 250 (219–310) | 0.67 | 0.05 | 0.37 |
| Ratio of fibrinogen (mg/dl) at 2 hours from baseline | 1 (0.95–1.1) | 1 (0.92–1.12) | 1 (0.91–1.08) | 0.09 | 0.68 | 0.22 |
| Change in plasminogen (U/dl) at 2 hours from baseline | 1.7 (7.5) | –3.6 (8.9) | –9.7 (9.9) | < 0.001 | < 0.001 | < 0.001 |
| Ratio of fibrin D-dimer (ng/ml) at 2 hours from baseline | 1.08 (0.95–1.34) | 3.37 (2.23–4.79) | 4.56 (2.91–7.36) | < 0.001 | < 0.001 | < 0.001 |
| Ratio of prothrombin F1 + 2 (pmol/l) at 2 hours from baseline | 1.05 (0.89–1.34) | 1.21 (1.03–1.52) | 1.27 (1.06–1.57) | 0.002 | < 0.001 | < 0.001 |
| Ratio of tPA (ng/ml) at 2 hours from baseline | 1.1 (1.0–1.3) | 1.3 (1.2–1.7) | 1.5 (1.3–2.0) | < 0.001 | 0.05 | 0.002 |
| Haemoglobin at 24 hours (g/l) | 143.1 (11.3) | 141.8 (12.9) | 143.3 (13.5) | 0.95 | 0.40 | 0.66 |
| Change in haemoglobin (g/l) at 24 hours from baseline | –1.8 (9.5) | –3.9 (9.2) | –3.6 (10.0) | 0.13 | 0.08 | 0.06 |
| Outcome | Effect estimates and 95% CIs | ||
|---|---|---|---|
| 20 mg of alteplase vs. placebo | 10 mg of alteplase vs. placebo | Alteplase vs. placebo | |
| Coronary angiogram | |||
| No re-flow | 1.04 (0.60–1.81) | 1.23 (0.69–2.19) | 1.13 (0.69–1.83) |
| Slow re-flow | |||
| Normal flow | |||
| Haematology and coagulation | |||
| Activated clotting time (seconds) at 2 hours | 1.0 (0.9–1.1) | 1.1 (1.0–1.2) | 1.0 (1.0–1.1) |
| Ratio of fibrinogen (mg/dl) at 2 hours from baseline | 0.97 (0.93–1.01) | 0.99 (0.95–1.03) | 0.98 (0.95–1.01) |
| Change in plasminogen (U/dl) at 2 hours from baseline | –11.41 (–13.64 to –9.19) | –5.34 (–7.60 to –3.08) | –8.47 (–10.47 to –6.47) |
| Ratio of fibrin D-dimer (ng/ml) at 2 hours from baseline | 3.94 (3.36–4.62) | 3.11 (2.64–3.65) | 3.51 (3.06–4.04) |
| Ratio of prothrombin F1 + 2 (pmol/l) at 2 hours from baseline | 1.24 (1.08–1.42) | 1.29 (1.13–1.48) | 1.26 (1.13–1.42) |
| Ratio of tPA (ng/ml) at 2 hours from baseline | 1.35 (1.14–1.60) | 1.19 (1.00–1.41) | 1.27 (1.10–1.47) |
| Haemoglobin at 24 hours (g/l) | 0.10 (–2.95–3.15) | –1.31 (–4.38–1.76) | –0.60 (–3.23–2.04) |
| Change in haemoglobin (g/l) at 24 hours from baseline | –1.83 (–4.17–0.51) | –2.11 (–4.45–0.23) | –1.97 (–3.98–0.04) |
Blood chemistry
The AUC for troponin T (ng/ml), which was measured at baseline and at 2 and 24 hours post reperfusion in 317 patients, was increased in both treatment groups compared with placebo (relative difference 1.53, 95% CI 1.16 to 2.01, p = 0.002, for both alteplase groups combined vs. the placebo group) (Figure 5). The AUC for troponin T was 35% higher in patients who were treated with 20 mg of alteplase than those in the placebo group (relative ratio 1.53, 95% CI 1.12 to 2.11; p = 0.008).
FIGURE 5.
Plot of troponin T AUC. The AUC for troponin T (ng/l) was measured from blood samples obtained immediately before reperfusion (0 hours) and then again at 2 and 24 hours. Analysed samples at all three time points were available in 317 patients.

Health-related quality of life
In unadjusted analyses, HRQoL scores were not significantly different between the groups at 3 months. The EQ-5D-3L health utility score was 0.88 in both the 20 mg alteplase group and the placebo group (mean difference –0.002, 95% CI –0.04 to 0.04; p = 0.93) (see Table 6).
Adverse events
Haematology and coagulation
Compared with placebo, there was a dose-related increase in the systemic concentrations of fibrin D-dimer and prothrombin F1 + 2, and a slight reduction in plasminogen, in the alteplase groups (Table 10). The systemic concentrations of fibrinogen and haemoglobin were numerically similar between the groups.
| Baseline values | Treatment group | ||
|---|---|---|---|
| Placebo | 10 mg of alteplase | 20 mg of alteplase | |
| Coagulation | |||
| Fibrinogen (mg/dl), median (IQR) | 339 (280–390) | 331 (260–390) | 327 (270–380) |
| Plasminogen (U/dl), mean (SD) | 93 (14) | 93 (14) | 94 (14) |
| Fibrin D-dimer (µg/ml), median (IQR) | 0.09 (0.07–0.16) | 0.11 (0.07–0.17) | 0.10 (0.07–0.16) |
| Prothrombin F1 + 2 (pmol/l), median (IQR) | 164 (120–220) | 156 (122–234) | 156 (115–242) |
| tPA (ng/ml), median (IQR) | 10 (7.5–12) | 10 (8–12) | 9 (7.5–12) |
Clinical events
The adverse events are described in Table 11. MACE occurred in 15 patients (10.1%) in the placebo, 18 (12.9%) in the 10 mg of alteplase and 12 (8.2%) in the 20 mg of alteplase groups. Two patients experienced a stroke. Aspiration thrombectomy was used in one of these patients, who developed a homonymous hemianopia after the procedure.
| Outcome | Treatment group (as treated), n (%) | p-value | ||
|---|---|---|---|---|
| Placebo (n = 149) | 10 mg of alteplase (n = 140) | 20 mg of alteplase (n = 146) | ||
| CV death, non-fatal MI, unplanned hospitalisation for stroke or TIA (MACCE) | 5 (3.4) | 6 (4.3) | 6 (4.1) | 0.91 |
| CV death, non-fatal MI or unplanned hospitalisation for heart failure (MACE) | 15 (10.1) | 18 (12.9) | 12 (8.2) | 0.43 |
| All-cause death | 1 (0.7) | 3 (2.1) | 3 (2.1) | 0.58 |
| Cardiac death | 1 (0.7) | 3 (2.1) | 2 (1.4) | 0.45 |
| Myocardial infarction associated with revascularisation procedures (type 4 or 5) | 2 (1.3) | 3 (2.1) | 1 (0.7) | 0.54 |
| Fatal or non-fatal MI | 3 (2.0) | 6 (4.3) | 5 (3.4) | 0.51 |
| Stroke or TIA | 0 (0.0) | 0 (0.0) | 2 (1.4) | 0.22 |
| Heart failure requiring unplanned hospitalisation | 13 (8.7) | 13 (9.3) | 8 (5.5) | 0.43 |
| All-cause death or heart failure | 14 (9.4) | 15 (10.7) | 11 (7.5) | 0.64 |
| All bleeding | ||||
| Minor bleeds (BARC 1–2) | 4 (2.6) | 3 (2.1) | 3 (2.1) | 0.32 |
| Major bleeds (BARC 3–5) | 0 (0.0) | 1 (0.7) | 1 (0.7) | 0.55 |
| Bleeding on day of procedure | ||||
| Minor bleeds (BARC 1–2) | 2 (1.3) | 2 (1.4) | 3 (2.1) | 0.90 |
| Major bleeds (BARC 3–5) | 0 (0.0) | 1 (0.7) | 1 (0.7) | 0.55 |
Bleeding events on the day of the procedure
Overall, major bleeds were rare, occurring in one patient in each of the 10 mg and 20 mg of alteplase groups. One patient who presented with acute inferior STEMI experienced recurrent intracranial bleeding and died 8 days after hospital admission. The final diagnosis was infective emboli to the right coronary artery and cerebral circulation secondary to Gram-positive cocci endocarditis of the mitral valve. In addition to standard antithrombotic therapy with ticagrelor, this patient received 20 mg of intracoronary alteplase. One other patient who received 10 mg of alteplase experienced a fatal myocardial rupture. This patient had Q-waves on the initial ECG and a history of chest infection treated with oral steroids.
Chapter 5 Discussion
Among patients with acute STEMI presenting within 6 hours of symptom onset, adjunctive low-dose intracoronary alteplase given during the primary PCI compared with placebo did not reduce microvascular obstruction. Among patients presenting > 4 hours after symptom onset, microvascular obstruction appeared to be greater in patients treated with 20 mg of alteplase than in patients treated with placebo.
This trial has several strengths. The design of our trial and the methods employed may be considered as robust. Therefore, the negative finding was not a result of limitations in the study design or inappropriate patient selection. Comparable in scale to other pivotal trials involving cardiac MRI,22 such as the INFUSE-AMI trial,4 the trial design selected patients with presenting characteristics that increase infarct size (e.g. proximal occlusion of a thrombus-laden coronary artery). Mean infarct size (27% of left ventricular mass) was almost two-fold larger than that observed in an unselected population of STEMI patients. 5,6 By limiting eligibility to an ischaemic time of ≤ 6 hours, the aim was to include participants with salvageable myocardium. 18 Alteplase was used within its licensed indication and at doses that are available in the clinic. Bias was minimised through a double-blind design and the use of core laboratory analyses. The increase in systemic concentrations of fibrin D-dimers without any change in fibrinogen indicates that fibrinolysis and/or fibrin generation were localised to the heart.
Safety
The potential for harm with facilitated PCI was highlighted in the ASSENT-416 and FINESSE17 trials. In ASSENT-4,16 compared with primary PCI (standard care), full-dose tenecteplase combined with PCI was associated with an increase in the primary end point of death, congestive heart failure or shock within 90 days. In those trials, ischaemic cardiac complications and ischaemic stroke were also increased in the intervention group. Despite more initial patency in the infarct-related artery, residual thrombus burden was higher in the facilitated PCI group, and tissue reperfusion and clinical outcomes were worse. 29 These results may be explained by comparatively inadequate anticoagulation and, potentially, the formation of fibrin and thrombus in the group that was treated with tenecteplase. 29,30 The importance of effective anticoagulation to mitigate the prothrombotic effects of fibrinolytic therapy with alteplase has been reported previously. 30
The targeted intracoronary infusion of the study drug was intended to minimise the systemic release of alteplase and minimise bleeding events. Alteplase was selected because it is a fibrin-specific fibrinolytic drug with a brief circulating half-life (≈5 minutes). To further reduce the possibility of harmful remote bleeds, patients with risk factors for bleeding were excluded and the PCI procedures were performed via the radial artery. The rates of bleeding events were within the expected range for primary PCI. 31
Safety assessments were a primary objective of our trial. Circulating fibrinogen concentrations were not different between the groups, indicating that there was no evidence of systemic fibrinolysis or fibrinogenolysis. There were no between-group differences in the haemoglobin concentration at 24 hours or bleeding events. We observed no short-term effect on NT-proBNP, making any compensatory natriuretic effect due to volume overload unlikely. The clinical significance of small increases in left ventricular end-diastolic volume and left ventricular end-systolic volume in the 10 mg of alteplase group at 2–7 days but not at 3 months is uncertain.
The increase in troponin T in the alteplase groups may represent a false-positive result within a series of multiple secondary outcome tests. Alternatively, it may be a true result and, therefore, mechanistically relevant. We think that this scenario is plausible. The AUC for troponin T is distinct from the other measures of infarct size, which were obtained at single time points. An alternative explanation, such as biomarker washout after fibrinolysis, may explain why this rise in troponin T was not associated with an increase in MRI measures of infarction. The dose-related increase in the systemic concentrations of fibrin D-dimer indicates that clot lysis had occurred. An increase in prothrombin F1 + 2 concentrations was observed in the alteplase groups, despite achieving therapeutic anticoagulation with unfractionated heparin. The undesired procoagulant effect of fibrinolytic therapy through thrombin activation30 may have led to microvascular thrombosis, limiting the efficacy of the intervention.
Statistical analysis of the primary outcome
As expected, around half of the participants did not have evidence of microvascular obstruction on the 2- to 7-day MRI scan. This creates zero values and a non-normal distribution in the data set. Different statistical approaches can be considered, each with strengths and limitations. We used a square-root transformation, which may not fully normalise the distribution of the data, back-transformation may be unclear and CIs on a square-root scale may not be easily interpreted. A log-transformation can be back-transformed to produce results as a ratio; however, zero values remain problematic with this approach. A boot-strapping approach is an alternative option.
Overview of current clinical trials
Contemporary practice guidelines call for more research to identify new treatments for microvascular obstruction. 3 There is a widespread, growing interest in the potential efficacy of adjunctive intracoronary fibrinolytic therapy during primary PCI. In our trial, the intervention was scheduled to be carried out at an early stage during primary PCI. The rationale was to infuse intracoronary fibrinolytic therapy upstream of the culprit lesion when thrombus burden was at its greatest and fibrinolytic therapy might be most effective. Intracoronary fibrinolytic therapy was infused before stent implantation to reduce mechanical distal embolisation of a thrombus. An alternative approach proposed by Sezer et al. 15 was to target the administration of adjunctive lytic therapy at the end of primary PCI. At this point, antegrade coronary flow is much improved compared with at the start of the procedure, implying the potential for improved delivery of fibrinolytic therapy to the microcirculation. Based on the new knowledge from the T-TIME trial, future trials should schedule adjunctive intracoronary antithrombotic therapy for the end of the PCI.
The T-TIME trial was designed as a Phase II clinical trial and it was powered to provide definitive information on the primary outcome. The trial was designed to inform the rationale for undertaking a larger Phase III trial that would ultimately be necessary to determine whether or not this strategy could be recommended in practice guidelines. Given that the results of this trial were not positive, there is no case for progressing to a Phase III trial of this intervention, as designed. The intervention should evolve in the light of the new knowledge arising from the T-TIME trial, and any future trial should reflect this.
Three trials are currently investigating this strategy (OPTIMAL, NCT02894138; ACTRN12618000778280; and STRIVE, NCT03335839). Two international, Phase III RCTs are investigating the efficacy of reduced doses of either alteplase or tenecteplase. The Adjunctive, Low-dose tPA in primary PCI for STEMI trial (STRIVE, NCT03335839) will enrol 4000 patients with acute STEMI who present < 6 hours after symptom onset with a large thrombus burden at angiography (TIMI thrombus grade of > 3). The intervention involves open-label intracoronary administration of alteplase (10 mg or 20 mg) or placebo during primary PCI, and the primary composite outcome is MACEs at 180 days. The Restoring Microcirculatory Perfusion in STEMI (RESTORE-MI; ACTRN12618000778280) trial will enrol 1666 patients undergoing primary PCI at < 12 hours after symptom onset. In this double-blind trial, the efficacy and safety of intracoronary administration of tenecteplase (one-third of the weight-adjusted systemic dose) will be compared with placebo in reperfused STEMI patients who have evidence of microvascular dysfunction that is reflected by an index of microcirculatory resistance (IMR) value of > 32 at the end of PCI. The primary outcome is cardiovascular mortality at 24 months. Finally, the Optimal Coronary Flow After PCI for Myocardial Infarction (OPTIMAL) study is a single-centre pilot. The intervention involves the selection of patients with an increased index of microvascular resistance (IMR of > 30) in the infarct-related artery at the end of PCI, who will receive an intracoronary infusion (10 ml) of either 20 mg of alteplase or sodium chloride. The primary outcome is the ratio of myocardial infarct size to area at risk, which is assessed by MRI early after enrolment (days 2–6) to assess the area at risk and again at 3 months to assess the final infarct size. The overall sample size is 90 patients (n = 40 per group), including 10 patients assigned to an observational group with no intervention.
The new knowledge from the T-TIME trial serves as a pause for thought and is relevant to the design of these trials. For example, we have identified a potential risk of intracoronary alteplase to promote myocardial haemorrhage in patients presenting with a prolonged ischaemic time.
Microvascular obstruction persists as an unmet therapeutic need, affecting around half of patients following an acute STEMI. We recommend further research towards therapy development for microvascular obstruction. Novel therapies could be designed to prevent the early occurrence of microvascular obstruction by limiting endothelial damage or by limiting the progression of microvascular obstruction by restoring reflow within the microcirculation. Based on the results of our trial, we support further research involving intracoronary antithrombotic therapy, including fibrinolytic drugs, when targeted only to appropriately selected patient groups who have a low likelihood of developing myocardial haemorrhage. In this regard, the designs of the RESTORE-AMI and STRIVE trials should be reconsidered in the light of the results from the T-TIME trial.
Discontinuation of the trial: the case to continue enrolment
The idea for the T-TIME trial was originally proposed in 2010 and, to date, the case to develop a treatment for failed microvascular reperfusion in patients with acute MI has grown even stronger. Ischaemic heart disease is the number one cause of death and disability in the UK and worldwide (as reported by The Global Burden of Disease Survey). 2 Myocardial infarction is the major cause of death from ischaemic heart disease and the dominant cause of heart failure in developed and developing countries. An increase in the rate of survival from acute MI has led to more patients surviving with persistent heart injury; it is these patients who are at risk of heart failure in the longer term. Despite advances in secondary prevention, the incidence of heart failure has remained persistently high in the past three decades. Failed myocardial reperfusion (or microvascular obstruction) affects half of all-comers with acute MI and has no known treatment, and microvascular obstruction is a driver of heart failure in the longer term. The lack of any available treatment to reduce infarct size in post-MI patients remains an unmet need.
The T-TIME trial was an exploratory Phase II trial that was designed and powered to gather evidence on the safety, efficacy and mechanisms of a potential new treatment approach for patients with acute MI. Given the knowledge gaps for therapy development, the NIHR EME board supported funding of a substantive clinical trial. The T-TIME trial intended to provide definitive results, and, in terms of the primary efficacy outcome, this objective was met. During meetings of the TSC (19 and 27 December 2017), members, including two lay representatives, affirmed that conclusive results, be they ‘positive’ or ‘negative’, would be in the public interest and inconclusive results would be undesirable.
Considering some of the secondary outcomes, premature discontinuation of the trial has the potential to lead to inconclusive results through lack of statistical power. This is not the case for all of the secondary outcomes. For example, the higher AUCs with troponin T in the alteplase groups, compared with placebo, present a clear result, albeit one that was not expected. Moreover, the explanation for this result is not fully understood, and more than one mechanism (or explanation) may be operative. If alteplase enhances microvascular perfusion then the higher AUC for troponin may reflect a ‘wash-out’ phenomenon. On the other hand, alteplase may increase the biochemical size of infarction through enhanced thrombosis. Indeed, both mechanisms may be operative. In the absence of any excess of adverse clinical events, allowing the trial to fully recruit would have conferred more power for the statistical analyses of the secondary, mechanistic outcomes, allowing a conclusive examination of the mechanisms and the effects of alteplase. Furthermore, the futility analysis on the primary outcome presented some issues that may not have been fully considered in the design of the analysis. Because microvascular obstruction is a binary outcome, it would occur in around half the participants, which would substantially reduce the power of an interim analysis. On reflection, we are not aware of any previous trial that has undertaken a futility analysis using microvascular obstruction. This gap increased uncertainty for the oversight committees at a crucial time in the determination of whether or not to discontinue enrolment in the trial. Representing a learning point from this experience, if futility analyses are required for clinical trials involving cardiovascular magnetic resonance imaging in patients following acute myocardial infarction, we advocate adoption of continuous surrogate outcome measures with a normal distribution, such as infarct size, rather than microvascular obstruction.
For the futility analysis, the median value of microvascular obstruction was based on 251 patients, and 124 had a value of zero. The median value was 0.05% of left ventricular mass (0–3.19% of left ventricular mass) and the range was 0.00–28.75% of left ventricular mass. The skewed, as expected, distribution with a wide spread may render a futility analysis based on conditional power highly likely to fail a 30% criterion. A calculation could be performed using a non-parametric test, but it is likely to give similar results to the binary conditional power calculation, given that half of the population have a zero value. Moreover, this scenario could be anticipated in other trials in which microvascular obstruction might be adopted, regardless of what the intervention may be. Therefore, this observation is relevant to investigators involved in the design of future trials that might consider adopting microvascular obstruction as a primary outcome measure. In this trial, the conditional power analysis based on 40% of the trial participants was consistent with the final analysis, including microvascular obstruction and other secondary outcome measurements, such as infarct size. Of note, the investigators had originally proposed infarct size (percentage of left ventricular mass) as the primary outcome in the trial and based on feedback during peer review the outcome measure had changed from infarct size to microvascular obstruction.
To our knowledge, this is the first report of a futility analysis based on microvascular obstruction. There is no other example that could serve as a reference to inform the situation that developed in our trial situation. Given that there were no prior studies to draw on, there was uncertainty relating to the validity of the futility analysis based on microvascular obstruction. In the end, there were no time-varying effects of the intervention and the futility analysis was subsequently proven to be valid.
The T-TIME trial was designed and powered to assess for between-group differences in predefined secondary outcome measures that had been identified as distinct measures of efficacy. These outcomes were measured by a range of methods, including (1) coronary angiography for TIMI flow grade, TIMI blush grade and TIMI thrombus grade, (2) ECG, for example the ST-elevation score at 60 minutes post reperfusion and the per cent ST-segment resolution on the 12-lead ECG at 60 minutes, and (3) biochemistry [troponin T (AUC) and NT-proBNP at 3 months]. These outcomes have comparable prognostic significance but reflect different aspects of cardiovascular function and pathobiology. These outcomes were purposefully selected to provide a broad range of information about efficacy and the evaluation of mechanisms, in line with the mission of the EME programme. Each of the secondary outcomes may be considered to have distinct importance.
Our intention a priori had been to analyse and interpret all of these outcomes to form an overall strategic view on the efficacy (and safety) of the intervention. Although there are inevitable correlations between these efficacy outcomes, they are nonetheless distinct. It is entirely possible that for one outcome there may be no obvious treatment effect on one or more of the secondary outcomes and a distinct effect on another. The statistically significant finding with the AUC for troponin T is a case in point. However, several of the other secondary outcomes exhibited directional trends that were not statistically significant. As an exploratory Phase II trial, the T-TIME trial was designed to have sufficient power to definitively answer questions on efficacy, safety and mechanisms across the full range of secondary outcomes. However, statistical power for the secondary outcome analyses was diminished when enrolment was discontinued with a sample size of 440 patients rather than 618. The Data and Safety Monitoring Committee noted ‘no safety concerns’. Nonetheless, given that the primary question had been answered, the funder and sponsor did not support continuing the study.
The T-TIME trial has provided clinically relevant results. Despite being discontinued prematurely, it is one of the largest Phase II therapeutic trials in acute STEMI. Experts, such as Herson et al. ,19 point out that a futility analysis may lead to premature discontinuation and equivocal results. Because the trial was discontinued with 72% of the population enrolled, there is an inevitable loss of statistical power. This means that prespecified secondary analyses, such as of the treatment interactions with key predefined presenting characteristics, for example the duration of ischaemia from symptom onset to first treatment to achieve coronary reperfusion, have reduced statistical power, limiting the strength of any conclusions. Given that other clinical trials are scheduled, there is community interest in learning as much as possible from the T-TIME trial to optimally inform the design of those other trials. Interestingly, Herson et al. 19 also suggest that different thresholds for conditional power on futility may be adopted. They seem to support a less stringent threshold (e.g. 15–25%) than the 30% level of conditional power that was designated during the peer review process in our trial, pointing out that a futility analysis may lead to premature discontinuation and equivocal results due to lack of statistical power.
Patient and public involvement in the decision to discontinue the trial
The TSC included two patient representatives. One of the representatives highlighted that > 400 patients had accepted a risk by agreeing to participate in the trial. These patients had given a personal commitment to the study; therefore, early termination of the study, leading to inconclusive answers, would not best represent their support. Given that the Data and Safety Monitoring Committee had reported ‘no safety concerns’, the patient representatives on the TSC described premature discontinuation of the trial as being highly undesirable. The patient representatives felt that either ‘negative’ or ‘positive’ results would be of value, whereas equivocal results would be unsatisfactory.
Overall, the discontinuation of enrolment in the T-TIME trial introduced an unexpected situation for the investigators, participants and stakeholders. In some respects, the situation was unprecedented. The experience has provided new knowledge and learning that will be relevant to the wider community involved in the design and conduct of clinical trials in patients with acute MI. There is evidence of good communication, shared timely decision-making, research governance and effective implementation of the decision to place a halt on enrolment without delay following receipt of the letter from the Data and Safety Monitoring Committee. There is also evidence that the trial management process between stakeholders was effective in forming a pragmatic decision, and the safety of the participants was assured.
Limitations
The study had several limitations. First, the study presents short-term findings up to 3 months. Second, the trial was discontinued when prespecified futility criteria were met. The interim analysis and related stopping criteria had been required and specified by the funder. The objectives of this Phase II trial included evidence synthesis for evaluation of mechanisms as well as efficacy. The unblinded results are consistent with those in the interim analysis. There was a linear trend indicating that, even if the trial had enrolled the target sample size of 558 participants, the main results would have been the same. To an extent, premature discontinuation limits the evaluation of mechanisms, as some of the analyses for the secondary outcomes, for example myocardial haemorrhage, may be considered underpowered. Furthermore, the sample size was reduced from 618 to 440 patients. This reduction has limited the power for the evaluation of safety outcomes, notably bleeding events. Nonetheless, follow-up to 3 months continued, in line with the Data and Safety Monitoring Committee recommendation, and missing data were minimal. The longer-term implications of adjunctive intracoronary alteplase for health outcomes will be assessed at 12 months. Third, because of the large number of secondary end points and the potential for type I error, all of these findings should be interpreted as exploratory only. Fourth, study drug administration was focused at a single time point before stent implantation when coronary blood flow was variable. Alternatively, in STRIVE and RESTORE-MI, the intervention is scheduled at the end of primary PCI, after stent implantation. Finally, for logistical reasons, platelet-poor plasma was not collected and information on platelet activation is not available.
Conclusions
Among patients with acute STEMI presenting within 6 hours of symptoms, adjunctive low-dose intracoronary alteplase given during the primary PCI compared with placebo did not reduce microvascular obstruction. The study findings do not support this treatment.
Chapter 6 Patient and public involvement
Aim
The aim of the PPI in the T-TIME trial was to involve the public and service users to ensure that the trial was carried out ‘with’ and/or ‘by’ them.
Methods
The methods used for PPI in the study included involvement in the study design, implementation and conduct of the trial, and reporting the results. The study design involved informal and formal feedback from patients and the public. Informal feedback was gained during 2010–12 when the original idea for the intervention was conceived. The chief investigator (CB) sought the views of patients in his care during and after treatment for a heart attack (MI). Following favourable informal feedback, formal input was sought from members of the public who have designated roles as PPI representatives in the Golden Jubilee National Hospital, including Mr Gordon Baird and Mrs Sandra Pairman. PPI feedback was also provided through the ethics committee reviews of the research idea, trial design, patient information sheet and consent form. The idea for the current trial had been preceded by an earlier proposal involving tenecteplase, a third-generation lytic drug, rather than alteplase, a second-generation lytic drug. Our current trial was preceded by an earlier grant from the NIHR EME programme (10/90/12): ‘A randomised parallel group double blind placebo-controlled Trial of very low dose and low dose adjunctive Tenecteplase during priMary PCI’. The award was withdrawn for a number of reasons, for example delays in research and development (R&D) set-up. The design of that trial was developed with the involvement of Ms Brenda Rankin, who was a lay member of the R&D Steering Group in the Golden Jubilee National Hospital and a member of the NHS Board’s Quality Reference Group and Lay Research Group. The T-TIME trial was presented for review at PPI meetings outside the lead site. PPI review took place in Leeds General Infirmary and the Liverpool Heart and Chest Hospital. These meetings involved presentation of the trial to the local PPI group by the local principal investigator (Professor John Greenwood) or research lead (Professor Rod Stables). These PPI presentations formed an integral part of the site review process leading up to the approval of a new study in these hospitals. Therefore, the T-TIME trial has benefited from PPI feedback in multiple forums over an extended period of time.
The conduct of the T-TIME trial was overseen by a TSC. The responsibility of the TSC was to supervise the trial and ensure that it was conducted according to the Medical Research Council Guidelines for Good Clinical Practice. The TSC had two PPI representatives, Mr Gordon Baird and Mrs Sandra Pairman. Mr Baird is a former patient, based in Stranraer, and Mrs Pairman has a designated role as a lay adviser in the Golden Jubilee National Hospital. They participated in the TSC meetings, including one before the start of the trial and twice-yearly during the course of the trial. The meetings had an agenda and minutes. Finally, these PPI representatives received the plain English summary of the report and provided feedback.
During the past 3 years, the T-TIME trial has been presented at a number of PPI events hosted by the sponsor in Glasgow, the lead site in Clydebank and in Leeds.
Study results
The overall results of the study are reported in the Plain English summary. The T-TIME trial benefited from PPI input before, during and after the end of the trial. In general, service users recognised that blocked microvessels in the distribution of the ‘culprit’ heart artery represented a problem of unmet need. They supported the rationale to dissolve the microthrombi using low doses of the ‘clot buster’, alteplase, while placing patient safety as a paramount consideration. They positively contributed to optimise the plain English summary content of the patient information sheet and consent forms. Their feedback on these documents was included in the final, approved versions that were used in the trial. PPI representatives positively contributed to the TSC meetings. In December 2016, following a recommendation from the chairperson of the independent Data Monitoring Committee (IDMC), it was advised that the trial be discontinued based on preliminary evidence of futility. In an urgently convened meeting of the TSC held on 28 December 2017, the PPI members strongly advocated that the trial should continue. The IDMC had noted that there were no safety concerns, and on this basis the PPI representatives voiced concern that, by discontinuing the trial prematurely, all of the trial’s objectives may not be met, reducing the value of the trial’s results. In this regard, they were particularly concerned that by discontinuing the trial the contribution of those patients who had already been enrolled (n = 440) would be undermined. The sponsor took account of this feedback while enrolment was temporarily suspended. Nonetheless, EME programme management required that the recommendation of the IDMC be implemented; therefore, the trial was discontinued.
The PPI representatives and service users contributed to PPI meetings that focused on the trial. Dates of PPI meetings include 24 March 2017 and 25 April 2019 (both in the Queen Elizabeth University Hospital, Glasgow) and 20 May 2019, for the SofTMech Patient Participation Day in the Golden Jubilee National Hospital (URL: www.softmech.org/events/headline_596271_en.html; accessed 29 June 2020).
Discussion and conclusions
In the SoftMech PPI event that took place in the Golden Jubilee National Hospital (20 May 2019), in the light of the T-TIME trial’s neutral results that were presented by Professor Colin Berry, the service user representatives very much supported the case for more research into therapy development for microvascular obstruction. They also supported the role of mathematics and biostatistics for computational heart modelling to better understand the nature of heart injury, to create computational models to predict the effects of novel therapies and, finally, to enhance the efficiency of randomised trials, for example through efficient trial design to reduce the sample size of clinical trials, wherever possible.
On reflection, PPI very much influenced the study design across a broad range of areas. PPI brought added value to the trial. The PPI representatives reviewed the Plain English summary of the trial and provided feedback on the text. There were no negative effects of PPI input.
Reflections/critical perspective
The PPI input enhanced the case for support and helped make the case for funding stronger. Prior PPI input may have facilitated the ethics review, which also benefited from lay/service user appraisal. PPI input enhanced the discussions during the TSC meetings and positively supported the trial when enrolment was suspended. PPI user feedback continues to advocate for more research into microvascular obstruction, which affects half of all patients affected by STEMI (heart attack) and, in the absence of any effective therapy, represents an unmet therapeutic need.
Acknowledgements
Responsible individual at the data co-ordinating centre: Alex McConnachie.
Data co-ordinating centre(s): Robertson Centre for Biostatistics, Glasgow Clinical Trials Unit, University of Glasgow, Glasgow, UK.
Boehringer Ingelheim (Ingelheim am Rhein, Germany) provided the study drugs.
Role of sponsor statement
The University of Glasgow and Greater Glasgow and Clyde Health Board were independent co-sponsors of the trial. The co-sponsors shared oversight and responsibility for the design and conduct of the study, data collection and management, research governance, analysis, and final approval of the manuscript. The sponsor did not have the right to preclude submission of the manuscript.
Access to data and data analysis
Colin Berry, who is the principal investigator for the trial, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Peter J McCartney MRCP; British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Hany Eteiba FRCP; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
Annette M Maznyczka MRCP; British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Margaret McEntegart FRCP; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
John P Greenwood FRCP; Leeds University and Leeds Teaching Hospitals NHS Trust, Leeds, UK; no personal compensation; salaried, no personal compensation.
Douglas F Muir FRCP; The James Cook University Hospital, South Tees Hospitals NHS Foundation Trust, Middlesbrough, UK; salaried, no personal compensation.
Saqib Chowdhary FRCP; Wythenshawe Hospital, Manchester University NHS Foundation Trust, Manchester, UK; salaried, no personal compensation.
Anthony H Gershlick FRCP; University of Leicester, Leicester Biomedical Research Centre, University Hospitals of Leicester NHS Trust, Leicester, UK; salaried, no personal compensation.
Clare Appleby FRCP; Liverpool Heart and Chest Hospital NHS Foundation Trust, Liverpool, UK; salaried, no personal compensation.
James M Cotton FRCP; Wolverhampton University Hospital NHS Trust, Wolverhampton, UK; salaried, no personal compensation.
Andrew Wragg FRCP; Barts Health NHS Trust, London, UK; salaried, no personal compensation.
Nick Curzen FRCP; University Hospital Southampton NHS Foundation Trust, Southampton, UK; salaried, no personal compensation.
Keith G Oldroyd MD; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
Mitchell Lindsay FRCP; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
J Paul Rocchiccioli FRCP; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
Aadil Shaukat FRCP; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
Richard Good FRCP; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
Stuart Watkins FRCP; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
Keith Robertson FRCP; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
Christopher Malkin FRCP; Leeds University and Leeds Teaching Hospitals NHS Trust, Leeds, UK; salaried, no personal compensation.
Lynn Martin BN; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
Lynsey Gillespie PhD; Greater Glasgow and Clyde Health Board, Glasgow, UK; salaried, no personal compensation.
Thomas J Ford FRACP; British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Mark C Petrie FRCP; British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Peter W Macfarlane DSc; Electrocardiology Core Laboratory, University of Glasgow, Glasgow, UK; unsalaried (honorary staff), no personal compensation.
R Campbell Tait FRCPath; Department of Haematology, Royal Infirmary, Glasgow, UK; salaried, no personal compensation.
Paul Welsh PhD; British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Naveed Sattar FMedSci; British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Robin A Weir MD; University Hospital Hairmyres, East Kilbride, UK; salaried, no personal compensation.
Keith A Fox FRCP; University of Edinburgh, Edinburgh, UK; salaried, no personal compensation.
Ian Ford PhD; Robertson Centre for Biostatistics, Institute of Health and Wellbeing, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Alex McConnachie PhD; Robertson Centre for Biostatistics, Glasgow Clinical Trials Unit, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Colin Berry FRCP; British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Contribution and disclosure of the research staff in the participating sites
West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank
David Corcoran MRCP; salaried, no personal compensation.
Andrew Davie FRCP; salaried, no personal compensation.
Stuart Hood FRCP; salaried, no personal compensation.
Joanne Kelly RN; salaried, no personal compensation.
Tom Krysztofiak MRCP; salaried, no personal compensation.
Victoria McNulty MPharm (Hons); salaried, no personal compensation.
Vanessa Orchard HCPC; salaried, no personal compensation.
Jackie Wales FIBMS; salaried, no personal compensation.
Liverpool Heart and Chest Hospital NHS Foundation Trust
Suneil Aggarwal FRCP; salaried, no personal compensation.
Turab Ali FRCP; salaried, no personal compensation.
Fiona Andrews BN; salaried, no personal compensation.
Nicola Browning PhD; salaried, no personal compensation.
Timothy A Fairbairn FRCP; salaried, no personal compensation.
Ruth Hardwick MPharm; salaried, no personal compensation.
J Hasleton FRCP; salaried, no personal compensation.
Babu Kunadian FRCP; salaried, no personal compensation.
Pradeep Magapu FRCP; salaried, no personal compensation.
Nick Palmer FRCP; salaried, no personal compensation.
Rodney Stables FRCP; salaried, no personal compensation.
Clive Taylerson; salaried, no personal compensation.
Hoi Tong HCPC; salaried, no personal compensation.
Sophie Twiss BN; salaried, no personal compensation.
Periaswamy Velavan FRCP; salaried, no personal compensation.
University Hospital Southampton NHS Foundation Trust
Karen Banks RN; salaried, no personal compensation.
Simon Corbett FRCP; salaried, no personal compensation.
Michael Mahmoudi FRCP; salaried, no personal compensation.
Zoe Nicholas BSc; salaried, no personal compensation.
James Wilkinson FRCP; salaried, no personal compensation.
Leeds University and Leeds Teaching Hospitals NHS Trust
Matthew Allan MRCP; salaried, no personal compensation.
Michelle Anderson RN; salaried, no personal compensation.
Daniel J Blackman MD MRCP; salaried, no personal compensation.
Jonathan Blaxill FRCP; salaried, no personal compensation.
Michael Cunnington MD; salaried, no personal compensation.
Vivek Kodoth FRCP; salaried, no personal compensation.
John Kurian FRCP; unsalaried, no personal compensation.
Steven Lindsay FRCP; unsalaried, no personal compensation.
Christopher Malkin FRCP; salaried, no personal compensation.
Jim Mclenachan FRCP; salaried, no personal compensation.
Kathryn Somers RN; salaried, no personal compensation.
Murgu Veersamy FRCP; salaried, no personal compensation.
Leicester Biomedical Research Centre, University Hospitals of Leicester NHS Trust
Reenamol Abraham RN; salaried, no personal compensation.
David Adlam FRCP; salaried, no personal compensation.
Stephen Coleman BPharm; salaried, no personal compensation.
Ian Hudson FRCP; salaried, no personal compensation.
Andrew Ladwiniec MRCP; salaried, no personal compensation.
Gerald P McCann MD; salaried, no personal compensation.
Emma Parker RN; salaried, no personal compensation.
Elved Roberts MD; salaried, no personal compensation.
Joanne Wormleighton DCR (R); salaried, no personal compensation.
Wythenshawe Hospital, Manchester University NHS Foundation Trust
Eltigani Abdelaal MRCP; salaried, no personal compensation.
Hussain Contractor FRCP; salaried, no personal compensation.
Beatris Duran BS Pharm; salaried, no personal compensation.
Robin Egdell FRCP; salaried, no personal compensation.
Susan Ferguson BN; salaried, no personal compensation.
Sarra Giannopoulou BN; salaried, no personal compensation.
Sanjay Sastry FRCP; salaried, no personal compensation.
Jaydeep Sarma FRCP; salaried, no personal compensation.
Matthias Schmitt FRCP; salaried, no personal compensation.
Sophie Quinn BN; salaried, no personal compensation.
Barts Health NHS Trust, London
Andrew Archbold FRCP; salaried, no personal compensation.
Mervyn Andiapen DN; salaried, no personal compensation.
Charity Evwierhoma BN; salaried, no personal compensation.
Ajay Jain FRCP; salaried, no personal compensation.
Dan Jones FRCP; salaried, no personal compensation.
Simon Kennon FRCP; salaried, no personal compensation.
James Moon FRCP; salaried, no personal compensation.
Anthony Mathur FRCP; salaried, no personal compensation.
Amy Richards BSc; salaried, no personal compensation.
Alexander Sirker FRCP; salaried, no personal compensation.
The James Cook University Hospital, South Tees Hospitals NHS Foundation Trust, Middlesbrough
David Austin FRCP; salaried, no personal compensation.
Mark de Belder FRCP; salaried, no personal compensation.
Justin Carter MRCP; salaried, no personal compensation.
Nicola Cunningham BSc (Hons); salaried, no personal compensation.
Rachel Dale HCPC; salaried, no personal compensation.
Jim Hall FRCP; salaried, no personal compensation.
Stephanie Mack BSc (Hons); salaried, no personal compensation.
Tracy Manders RN; salaried, no personal compensation.
Neil Maredia MD; salaried, no personal compensation.
Karen McLeod BSc (Hons); salaried, no personal compensation.
Luca Settimo PhD Pharm; salaried, no personal compensation.
Andrew Sutton FRCP; salaried, no personal compensation.
Neil Swanson FRCP; salaried, no personal compensation.
Paul Williams FRCP; salaried, no personal compensation.
Robert Wright FRCP; salaried, no personal compensation.
New Cross Hospital, Wolverhampton University Hospital NHS Trust
Victoria Cottam BN; salaried, no personal compensation.
Elisabeth Radford; salaried, no personal compensation.
Ben Wrigley FRCP; salaried, no personal compensation.
Freeman Hospital, The Newcastle Upon Tyne Hospitals NHS Foundation Trust
Rajiv Das FRCP; salaried, no personal compensation.
Samantha Jones BN; salaried, no personal compensation.
Vera Wealleans BN; salaried, no personal compensation.
Royal Infirmary of Edinburgh, NHS Lothian
Nick Cruden FRCP; salaried, no personal compensation.
Laura Flint BN; salaried, no personal compensation.
Ruaridh Buchan MPharm; salaried, no personal compensation.
University Hospitals Bristol NHS Foundation Trust
Chiara Bucciarelli-Ducci FRCP; salaried, no personal compensation.
Tom Johnson FRCP; salaried, no personal compensation.
Vincy Johny BN; salaried, no personal compensation.
Lis McCullagh B.Pharm; salaried, no personal compensation.
Clinical Event Committee
Robin A Weir MRCP; University Hospital Hairmyres, NHS Lanarkshire, East Kilbride, UK; salaried, no personal compensation.
Aengus Murphy MD; University Hospital Monklands, NHS Lanarkshire, East Kilbride, UK; salaried, no personal compensation.
Colin J Petrie PhD; University Hospital Monklands, NHS Lanarkshire, East Kilbride, UK; salaried, no personal compensation.
Eleanor Dinnett MBChB; Robertson Centre for Biostatistics, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Independent Data and Safety Monitoring Committee
Gary A Ford MB BChir; University of Oxford, Oxford, UK; unsalaried, no personal compensation.
Chim C Lang FRCP, University of Dundee, Dundee, UK; salaried, no personal compensation.
John Norrie MSc; University of Edinburgh, Edinburgh, UK; salaried, no personal compensation.
Trial Steering Committee
Keith A Fox FRCP; University of Edinburgh, Edinburgh, UK; salaried, no personal compensation.
Colin Berry FRCP; British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Rajesh Kharbanda FRCP; Oxford University Hospitals NHS Foundation Trust, Oxford, UK; salaried, no personal compensation.
Catherine Labinjoh FRCP; Forth Valley Royal Hospital, NHS Forth Valley, Larbert, UK; salaried, no personal compensation.
Gordon Baird MRCGP; Stranraer; salaried, no personal compensation.
Sandra Pairman BN; Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
David Jamieson PhD; Robertson Centre for Biostatistics, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Sarah Weeden PhD; Robertson Centre for Biostatistics, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Jurgen Van-Melckebeke BSc; Project Management Unit, Glasgow Clinical Research Facility, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Elisabeth Douglas PhD; Clinical Trials Pharmacy, Clinical Research and Development, Greater Glasgow and Clyde Health Board UK; salaried, no personal compensation.
Pamela Surtees; Clinical Trials Pharmacy, Clinical Research and Development, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Barbara Ross BSc (Hons); Clinical Research and Development, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Gemma Brindley PhD; Clinical Research and Development, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Sheila McGowan PhD; Clinical Research and Development, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Emma Jane Gault MA (Hons); College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Debra Stuart PhD; College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Maureen Travers PhD; R&D Office, Clinical Research and Development, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Marc Jones PhD; Pharmacovigilance Office, Clinical Research and Development, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Sharon Kean; Robertson Centre for Biostatistics, Institute of Health and Wellbeing, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Caroline Watson PhD; R&D Governance, Clinical Research and Development, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Emma Miller; Project Management Unit, Glasgow Clinical Research Facility, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Eamonn Bolger BSc, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Gillian Thomson, Project Management Unit, Glasgow Clinical Research Facility, Greater Glasgow and Clyde Health Board, UK; salaried, no personal compensation.
Elaine Butler; Institute of Cardiovascular and Medical Sciences, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Josephine Cooney; Institute of Cardiovascular and Medical Sciences, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Julie Kennedy MSc; Electrocardiology Core Laboratory, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
Emma Leishman BSc; Department of Haematology, Royal Infirmary, Glasgow, UK; salaried, no personal compensation.
Jaclyn Carberry MBChB (Hons); Institute of Cardiovascular and Medical Sciences, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
David Corcoran MRCP; West of Scotland Heart and Lung Centre, Golden Jubilee National Hospital, Clydebank, UK; salaried, no personal compensation.
Aleksandra Radjenovic PhD; Institute of Cardiovascular and Medical Sciences, University of Glasgow, Glasgow, UK; salaried, no personal compensation.
We acknowledge all of the other clinical staff and the patients who participated in this project.
Contributions of authors
Peter J McCartney (https://orcid.org/0000-0001-7482-1811) (Cardiology Specialty Trainee) contributed to screening and enrolment, collected the data, undertook data analyses, interpreted the data and contributed to the manuscript.
Hany Eteiba (Consultant Cardiologist) was a local principal investigator and obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Annette M Maznyczka (https://orcid.org/0000-0002-0118-5153) (Cardiology Specialty Trainee) contributed to screening and enrolment, collected the data, undertook data analyses, interpreted the data and contributed to the manuscript.
Margaret McEntegart (https://orcid.org/0000-0001-5135-3322) (Consultant Cardiologist) obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
John P Greenwood (https://orcid.org/0000-0002-2861-0914) (Consultant Cardiologist) was a local principal investigator and obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Douglas F Muir (https://orcid.org/0000-0003-1871-2869) (Consultant Cardiologist) was a local principal investigator and obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Saqib Chowdhary (Consultant Cardiologist) was a local principal investigator and obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Anthony H Gershlick (https://orcid.org/0000-0002-8044-9048) (Consultant Cardiologist) was a local principal investigator and obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Clare Appleby (https://orcid.org/0000-0002-3838-1174) (Consultant Cardiologist) was a local principal investigator and obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
James M Cotton (https://orcid.org/0000-0001-6548-2723) (Consultant Cardiologist) was a local principal investigator and obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Andrew Wragg (https://orcid.org/0000-0003-0067-1771) (Consultant Cardiologist) was a local principal investigator and obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Nick Curzen (https://orcid.org/0000-0001-9651-7829) (Consultant Cardiologist) was a local principal investigator and obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Keith G Oldroyd (https://orcid.org/0000-0002-7842-3463) (Consultant Cardiologist) obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Mitchell Lindsay (https://orcid.org/0000-0002-4654-712X) (Consultant Cardiologist) obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
J Paul Rocchiccioli (https://orcid.org/0000-0003-1350-1544) (Consultant Cardiologist) obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Aadil Shaukat (Consultant Cardiologist) obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Richard Good (https://orcid.org/0000-0002-3906-1025) (Consultant Cardiologist) obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Stuart Watkins (https://orcid.org/0000-0003-1579-6294) (Consultant Cardiologist) obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Keith Robertson (https://orcid.org/0000-0002-6557-8298) (Consultant Cardiologist) obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Christopher Malkin (https://orcid.org/0000-0002-6643-6830) (Consultant Cardiologist) obtained informed consent, randomised patients, collected data, interpreted the results and contributed to the manuscript.
Lynn Martin (Research Nurse) supported screening and enrolment, data collection and event reporting.
Lynsey Gillespie was project manager for the trial.
Thomas J Ford (https://orcid.org/0000-0003-4009-6652) (Research Fellow) was a co-ordinator for the Clinical Event Committee.
Mark C Petrie (https://orcid.org/0000-0002-6333-9496) (Consultant Cardiologist) was a co-ordinator for the Clinical Event Committee.
Peter W Macfarlane (https://orcid.org/0000-0002-5390-1596) directed and participated in the ECG core laboratory analyses.
R Campbell Tait (https://orcid.org/0000-0003-4842-6783) directed the haematology core laboratory analyses.
Paul Welsh (https://orcid.org/0000-0002-7970-3643) directed the biochemical analyses and contributed to interpretation and to the manuscript.
Naveed Sattar (https://orcid.org/0000-0002-1604-2593) directed the biochemical analyses and contributed to interpretation and to the manuscript.
Robin A Weir (https://orcid.org/0000-0001-9895-2795) (Consultant Cardiologist) chaired the Clinical Event Committee.
Keith A Fox (https://orcid.org/0000-0002-0140-2752) (Emeritus Professor of Cardiology) chaired the Trial Steering Committee and contributed to the interpretation of the data and the manuscript.
Ian Ford (https://orcid.org/0000-0001-5927-1823) (Emeritus Professor of Statistics) contributed to the study design, data analyses and interpretation, and contributed to the manuscript.
Alex McConnachie (https://orcid.org/0000-0002-7262-7000) (Biostatistician) analysed and interpreted the data, and contributed to the manuscript.
Colin Berry (https://orcid.org/0000-0002-4547-8636) (Consultant Cardiologist) conceived the study, obtained the funding, recruited and randomised patients, collected and assessed data blind to treatment group assignment, interpreted the results and wrote the manuscript.
Colin Berry is chief investigator and Saqib Chowdhary, Raj Das, Ian Ford, John P Greenwood, Peter W Macfarlane, Douglas Muir, Keith G Oldroyd, Naveed Sattar and Campbell Tait were co-applicants on the grant from the NIHR EME programme (reference 12/170/45).
Publications
McCartney PJ, Eteiba H, Maznyczka AM, McEntegart M, Greenwood JP, Muir DF, et al. Effect of low-dose intracoronary alteplase during primary percutaneous coronary intervention on microvascular obstruction in patients with acute myocardial infarction: a randomised clinical trial. JAMA 2019;321:56–68.
Berry C, McCartney PJ, Maznyczka AM, T-TIME Group. Fibrinolytic therapy to reduce microvascular obstruction after myocardial infarction-reply. JAMA 2019;321:2033–34.
Berry C, Maznyczka AM, McCartney P. Failed myocardial reperfusion during primary PCI: an unmet therapeutic need. EuroIntervention 2019;14:1628–30.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- HALE Collaborators . Global, regional, and national disability-adjusted life-years (DALYs) for 333 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017;390:1260-344. https://doi.org/10.1016/S0140-6736(17)32130-X.
- GBD 2016 Causes of Death Collaborators . Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017;390:1151-210. https://doi.org/10.1016/S0140-6736(17)32152-9.
- Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, et al. 2017 ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the task force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 2018;39:119-77. https://doi.org/10.1093/eurheartj/ehx393.
- Stone GW, Maehara A, Witzenbichler B, Godlewski J, Parise H, Dambrink JH, et al. Intracoronary abciximab and aspiration thrombectomy in patients with large anterior myocardial infarction: the INFUSE-AMI randomised trial. JAMA 2012;307:1817-26. https://doi.org/10.1001/jama.2012.421.
- Wu KC, Zerhouni EA, Judd RM, Lugo-Olivieri CH, Barouch LA, Schulman SP, et al. Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 1998;97:765-72. https://doi.org/10.1161/01.cir.97.8.765.
- Carrick D, Haig C, Ahmed N, McEntegart M, Petrie MC, Eteiba H, et al. Myocardial hemorrhage after acute reperfused ST-segment-elevation myocardial infarction: relation to microvascular obstruction and prognostic significance. Circ Cardiovasc Imaging 2016;9. https://doi.org/10.1161/CIRCIMAGING.115.004148.
- de Waha S, Patel MR, Granger CB, Ohman EM, Maehara A, Eitel I, et al. Relationship between microvascular obstruction and adverse events following primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: an individual patient data pooled analysis from seven randomised trials. Eur Heart J 2017;38:3502-10. https://doi.org/10.1093/eurheartj/ehx414.
- Schwartz RS, Burke A, Farb A, Kaye D, Lesser JR, Henry TD, et al. Microemboli and microvascular obstruction in acute coronary thrombosis and sudden coronary death: relation to epicardial plaque histopathology. J Am Coll Cardiol 2009;54:2167-73. https://doi.org/10.1016/j.jacc.2009.07.042.
- Zalewski J, Undas A, Godlewski J, Stepien E, Zmudka K. No-reflow phenomenon after acute myocardial infarction is associated with reduced clot permeability and susceptibility to lysis. Arterioscler Thromb Vasc Biol 2007;27:2258-65. https://doi.org/10.1161/ATVBAHA.107.149633.
- Kloner RA, Ganote CE, Jennings RB. The ‘no-reflow’ phenomenon after temporary coronary occlusion in the dog. J Clin Invest 1974;54:1496-508. https://doi.org/10.1172/JCI107898.
- Henriques JPS, Zijlstra F, Ottervanger JP, de Boer M-J, van’t Hof AWJ, Hoorntje JCA, et al. Incidence and clinical significance of distal embolisation during primary angioplasty for acute myocardial infarction. Eur Heart J 2002;23:1112-17. https://doi.org/10.1053/euhj.2001.3035.
- Robbers LF, Eerenberg ES, Teunissen PF, Jansen MF, Hollander MR, Horrevoets AJ, et al. Magnetic resonance imaging-defined areas of microvascular obstruction after acute myocardial infarction represent microvascular destruction and haemorrhage. Eur Heart J 2013;34:2346-53. https://doi.org/10.1093/eurheartj/eht100.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group . Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988;2:349-60. https://doi.org/10.1016/S0140-6736(88)92833-4.
- Hillis WS, Jones CR, Been M, Campbell BC, Fulton WF. Intracoronary thrombolytic therapy performed within a coronary care unit: one year’s experience. Scott Med J 1986;31:25-9. https://doi.org/10.1177/003693308603100106.
- Sezer M, Oflaz H, Gören T, Okçular I, Umman B, Nişanci Y, et al. Intracoronary streptokinase after primary percutaneous coronary intervention. N Engl J Med 2007;356:1823-34. https://doi.org/10.1056/NEJMoa054374.
- Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention (ASSENT-4 PCI) investigators . Primary versus tenecteplase-facilitated percutaneous coronary intervention in patients with ST-segment elevation acute myocardial infarction (ASSENT-4 PCI): randomised trial. Lancet 2006;367:569-78. https://doi.org/10.1016/S0140-6736(06)68147-6.
- Ellis SG, Tendera M, de Belder MA, van Boven AJ, Widimsky P, Janssens L, et al. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008;358:2205-17. https://doi.org/10.1056/NEJMoa0706816.
- Eitel I, Desch S, Fuernau G, Hildebrand L, Gutberlet M, Schuler G, et al. Prognostic significance and determinants of myocardial salvage assessed by cardiovascular magnetic resonance in acute reperfused myocardial infarction. J Am Coll Cardiol 2010;55:2470-9. https://doi.org/10.1016/j.jacc.2010.01.049.
- Herson J, Buyse M, Wittes JT, Harrington D. Designs for Clinical Trials. New York, NY: Springer; 2011.
- Huang X, Moreton FC, Kalladka D, Cheripelli BK, MacIsaac R, Tait RC, et al. Coagulation and fibrinolytic activity of tenecteplase and alteplase in acute ischemic stroke. Stroke 2015;46:3543-6. https://doi.org/10.1161/STROKEAHA.115.011290.
- Rao AK, Pratt C, Berke A, Jaffe A, Ockene I, Schreiber TL, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial – phase I: haemorrhagic manifestations and changes in plasma fibrinogen and the fibrinolytic system in patients treated with recombinant tissue plasminogen activator and streptokinase. J Am Coll Cardiol 1988;11:1-11. https://doi.org/10.1016/0735-1097(88)90158-1.
- Thiele H, Eitel I, Meinberg C, Desch S, Leuschner A, Pfeiffer D, et al. Randomized comparison of pre-hospital-initiated facilitated percutaneous coronary intervention versus primary percutaneous coronary intervention in acute myocardial infarction very early after symptom onset: the LIPSIA-STEMI trial (Leipzig immediate prehospital facilitated angioplasty in ST-segment myocardial infarction). JACC Cardiovasc Interv 2011;4:605-14. https://doi.org/10.1016/j.jcin.2011.01.013.
- Antman EM, Giugliano RP, Gibson CM, McCabe CH, Coussement P, Kleiman NS, et al. Abciximab facilitates the rate and extent of thrombolysis: results of the thrombolysis in myocardial infarction (TIMI) 14 trial. The TIMI 14 Investigators. Circulation 1999;99:2720-32. https://doi.org/10.1161/01.cir.99.21.2720.
- Medicines and Healthcare products Regulatory Agency . Good Clinical Practice for Clinical Trials 2014. www.gov.uk/guidance/good-clinical-practice-for-clinical-trials (accessed 22 October 2018).
- World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013;310:2191-4. https://doi.org/10.1001/jama.2013.281053.
- Mehran R, Rao SV, Bhatt DL, Gibson CM, Caixeta A, Eikelboom J, et al. Standardised bleeding definitions for cardiovascular clinical trials: a consensus report from the Bleeding Academic Research Consortium. Circulation 2011;123:2736-47. https://doi.org/10.1161/CIRCULATIONAHA.110.009449.
- U.S. National Library of Medicine . Detection and Significance of Heart Injury in ST Elevation Myocardial Infarction 2014. https://clinicaltrials.gov/ct2/show/NCT02072850 (accessed 22 October 2018).
- Rubin DB. Inference and missing data. Biometrika 1976;63:581-92.
- Zalewski J, Bogaerts K, Desmet W, Sinnaeve P, Berger P, Grines C, et al. Intraluminal thrombus in facilitated versus primary percutaneous coronary intervention: an angiographic substudy of the ASSENT-4 PCI (Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention) trial. J Am Coll Cardiol 2011;57:1867-73. https://doi.org/10.1016/j.jacc.2010.10.061.
- Rapold HJ. Promotion of thrombin activity by thrombolytic therapy without simultaneous anticoagulation. Lancet 1990;335:481-2. https://doi.org/10.1016/0140-6736(90)90720-P.
- Kwok CS, Khan MA, Rao SV, Kinnaird T, Sperrin M, Buchan I, et al. Access and non-access site bleeding after percutaneous coronary intervention and risk of subsequent mortality and major adverse cardiovascular events: systematic review and meta-analysis. Circ Cardiovasc Interv 2015;8. https://doi.org/10.1161/CIRCINTERVENTIONS.114.001645.
- Clauss A. Gerinnungsphysiologische schnellmethode zur bestimmung des fibrinogens. Acta Haematol 1957;17:237-46.
Appendix 1 Protocol amendments
Amendment 1 (prior to regulatory green light)
Date: 18 July 2014.
Protocol v2.0 and associated documents (short patient information sheet v2.0, patient information sheet v2.0, patient alert card).
Amendment 2 (prior to regulatory green light)
Date: 6 October 2015.
Protocol v3.2 and associated documents (short patient information sheet v3.0, full patient information sheet v3.0, EQ5D questionnaire, patient alert card v2.0).
Amendment 3 (prior to regulatory green light)
Date: 21 January 2016.
Changes to participating sites.
Amendment 4 (prior to regulatory green light)
Date: 16 February 2016.
Revised general practitioner letter (v2.0) and clinical information sheet (v2.0).
Amendment 5
Date: 5 July 2016.
Protocol v4.0 and associated documents (short patient information sheet v3.1, full patient information sheet v3.1). Changes to participating sites.
Amendment 6
Date: 31 August 2016.
Addition of participating site.
Amendment 7
Date: 8 November 2016.
Addition of participating sites.
Amendment 8
Date: 13 December 2016.
Protocol v5.0 and associated documents (short patient information sheet v4.0, full patient information sheet v4.0). Following medical advice from the MHRA (Medicines and Healthcare products Regulatory Agency), the amendment was submitted as an urgent safety measure.
Amendment 9
Date: 22 February 2017.
Addition of participating site.
Amendment 10
Date: 21 March 2017.
Addition of participating site.
Amendment 11
Date: 27 July 2017.
Protocol v6.0 and associated documents (short patient information sheet v5.0, full patient information sheet v5.0, general practitioner letter v3.0, clinical information sheet v3.0).
Amendment 12
Date: 27 July 2017.
Addition of participating site.
Amendment 13
Withdrawn on advice of Research Ethics Committee: removal of participating site.
Amendment 14
Date: 21 December 2017.
Temporary halt to recruitment.
Amendment 15
Date: 30 March 2018.
Protocol v7.0: clarification of the designation of outcome variables as secondary or exploratory.
Appendix 2 Statistical analysis plan
Appendix 3 Supplementary methods
Secondary outcomes
Central laboratory analyses
The central laboratory analyses of the primary and secondary outcomes were determined blind to treatment allocation.
Cardiac magnetic resonance acquisition and analysis
We used cardiac MRI to assess left ventricular dimensions, function and pathology 2–7 days and 3 months post MI. MRI was performed using 1.5-T platforms [MAGNETOM® Avanto (Siemens Healthineers AG, Erlangen Germany) and Philips Intera, Best, the Netherlands]. The imaging protocol followed a standard operating procedure that included planning and localisers, T1-mapping, T2*-mapping, cine MRI with steady-state free precession (SSFP) and late-gadolinium enhancement imaging 10–15 minutes after administration of contrast medium. The scan acquisitions were spatially co-registered and included different slice orientations to enhance diagnostic confidence. The cardiac magnetic resonance imaging protocol is shown in Figure 6.
FIGURE 6.
Flow diagram of the cardiac MRI protocol. FLASH, fast low-angle shot; GRA, gradient recalled echo; i.v. intravenous; LAx, long axis; LV, left ventricular; MOLLI, modified Look–Locker imaging; SAx, short axis.
Typical T2* imaging parameters
The T2* multiecho gradient recalled echo (preferred) or T2*-map bandwidth ≈814 (× 8) Hz/pixel, flip angle 18°, matrix 256 × 115 pixels, spatial resolution 2.6 × 1.6 × 10 mm and a slice thickness of 8 mm with a 2-mm gap.
Contrast medium administration
The intravenous contrast agent used in this study was gadobutrol (Gadovist®, Bayer, Leverkusen, Germany) (1.5 mmol/ml solution for injection), which was administered in two doses in a weight-adjusted contrast volume. This was to obtain information on myocardial perfusion.
The first-dose injection (dose 1 = 0.05 mmol/kg) was given to initiate the first pass of contrast and the second dose (dose 2 = 0.1 mmol/kg ‘top-up’ injection) was given immediately after the first pass. Therefore, the total dose of gadobutrol was 0.15 mmol/kg.
An automated pump injector was used for intravenous injection of gadolinium. The injection rate was 4 ml/second. A three-of-five MRI acquisition protocol for short-axis (SAx) imaging of the first pass of gadolinium contrast perfusion was acquired at the same slice positions as the T2* scans. First-pass perfusion at rest for ‘wash-in’ microvascular obstruction quantification was performed with a fast low-angle shot (FLASH) sequence run simultaneously with the contrast injection. ‘Normal’ standard sequence, that is non-work-in-progress implementation, for three SAx per R-R interval over 1.5 minutes was used.
Typical first-pass imaging parameters: saturation recovery with inversion pulse. T1 101 milliseconds; TR/TE 194/0.98 milliseconds; acquisition window 1000 milliseconds; one concatenation; three SAx slices. If three slices could not be acquired within the R-R cycle, then two concatenations were used.
Cardiac mass and function incorporating area-at-risk imaging with contrast cine-SSFP was acquired 2–3 minutes following the second dose of contrast medium to enable contrast equilibration. The cine-MRI for left ventricular mass and function was collected during the interval between the 3-minute scan and the late enhancement imaging. Cine-MRI was acquired with a SAx left ventricular stack, slices aligned to T2* maps, from the mitral valve to the left ventricular apex (usually 10 slices in total). Extra slices were acquired basally to the mitral valve that incorporate left ventricular outflow tract and potentially to the apex.
SSFP cine breath-hold sequences (with parallel imaging acceleration) were used. The heart was imaged in multiple parallel SAx planes that were 8 mm thick separated by 2-mm gaps, equating to approximately 10 slices and 30 cardiac phases.
Typical SSFP imaging parameters: voxel size 2.0 × 2.0 × 8.0 mm; TR/TE 39.6/1.12 milliseconds; flip angle 55°, matrix 192 × 192 pixels; and slice thickness 8 mm, with a 2-mm gap.
Late enhancement
Late microvascular obstruction and scar was imaged 10–15 minutes after intravenous Gadovist contrast administration using, in general, a motion-corrected T1-weighted phase-sensitive inversion recovery radiofrequency pulse sequence. A full stack, aligned to T2* scans (or cines), and three long-axis views (vertical long axis, horizontal long axis and three-chamber view) were acquired. Phase-sensitive inversion recovery MRI techniques reduce variability relating to myocardial nulling, which is required for late gadolinium-enhancement imaging of infarct versus unaffected myocardium. If a phase-sensitive protocol was not used, a modified Look–Locker inversion time scout was performed prior to using an inversion recovery turbo gradient echo sequence. Phase swaps were performed where appropriate to rule out artefact.
Poor breath-holding: a single shot technique or navigated late gadolinium enhancement imaging was used as an option for poor breath holders.
Typical late gadolinium enhancement and microvascular obstruction imaging parameters with phase-sensitive inversion recovery: matrix 192 × 256 pixels; flip angle 25°; TE 3.36 milliseconds; bandwidth 130 Hz/pixel; echo spacing 8.7 milliseconds and trigger pulse 2. The voxel size is 1.8 × 1.3 × 8 mm. Inversion times individually adjusted to optimise nulling of apparently normal myocardium (typical values, 200–300 milliseconds).
Post-contrast T1 mapping
Three SAx T1-maps (basal, mid and apical positions) that were spatially matched to the same slice positions as the precontrast T1-map scans > 15 minutes after the top-up injection of contrast medium.
Cardiac magnetic resonance imaging at 3 months
Cardiac MRI at 3-month follow-up involved the same scan protocol as was used in the baseline MRI scan. Renal function was known or checked. A full blood count was checked to provide haematocrit for the extracellular volume analysis from the post-contrast T1 map.
Magnetic resonance imaging analysis
The MRI analyses were undertaken using Medis® Suite MR (Medis, Leiden, the Netherlands), which is vendor-independent post-processing software. Peter J McCartney undertook the primary analyses of the scans and related analyses and they were reviewed by Colin Berry (second observer). The research staff were blinded to treatment allocation.
Infarct definition and size
The presence of acute infarction was established based on abnormalities in cine wall motion, rest first-pass myocardial perfusion and late gadolinium-enhancement imaging in two imaging planes. The myocardial mass of late gadolinium (grams) was quantified using computer-assisted planimetry and the territory of infarction was delineated using a five-SD method and expressed as a percentage of total left ventricular mass.
Microvascular obstruction
Microvascular obstruction was defined as a dark zone on early gadolinium enhancement imaging 1-, 3-, 5- and 7-minutes post-contrast injection that remained present within an area of late gadolinium enhancement at 15 minutes. The myocardial mass (grams) of the dark zone was quantified by manual delineation and expressed as a percentage of left ventricular mass.
Myocardial oedema
The presence of myocardial oedema was established based on an area of increased signal intensity on the steady-state free-precession cine images (acquired 2 minutes after gadolinium contrast injection). The myocardial mass was calculated by manual delineation in end-diastole and end-systole. The values were averaged and expressed as a percentage of left ventricular mass.
Myocardial salvage
Myocardial salvage was calculated by subtraction of per cent infarct size from per cent area at risk, as reflected by the extent of oedema. The myocardial salvage index was calculated by dividing the myocardial salvage area by the initial area at risk.
Myocardial haemorrhage
On the T2* parametric maps, a threshold of 20 milliseconds was applied. A region of reduced signal intensity within the infarcted area, with a T2* value of < 20 milliseconds was considered to confirm the presence of myocardial haemorrhage. The area was manually delineated and expressed as a percentage of left ventricular mass.
Coronary angiogram acquisition and analyses
The acquisition and analyses of the coronary angiograms were performed in accordance with the standard operating procedures. Coronary angiograms were acquired during emergency care with cardiac catheter laboratory X-ray and information technology equipment. Feedback was provided to sites on the quality and completeness of the angiograms.
The angiograms were analysed by trained observers (AM, CB and MMcE) using imaging post-processing software (QAngio® XA Medis, Leiden, the Netherlands). The observers were blinded to treatment assignment. Catheter calibration was performed using the catheter calibration function on QAngio. For each lesion, a view perpendicular to the long axis of the vessel was used to avoid foreshortening and overlap of branches. Therefore, the single-plane projection showing the best opacified and most severe lesion with minimal foreshortening and minimal branch overlap was selected.
The TIMI coronary flow grade and thrombus grade were assessed following initial angiography and at the end of the procedure. TIMI myocardial perfusion grade and TIMI frame count were assessed at the end of the procedure. The TIMI frame count and perfusion grade are angiographic measures of microvascular function.
TIMI coronary flow grade
The TIMI coronary flow grade of the infarct-related artery is independently predictive of prognosis.
Classification of TIMI coronary flow grade
-
0: no flow.
-
1: minimal flow past obstruction.
-
2: slow (but complete) filling and slow clearance.
-
3: normal flow and clearance.
TIMI myocardial perfusion grade
The TIMI myocardial perfusion grade provides a score for ground-glass appearance ('blush') of contrast entering the microvasculature and contrast washout, it is predictive of prognosis. 8
Classification of TIMI myocardial perfusion grade
-
0: minimal or no myocardial blush in the distribution of the infarct-related artery.
-
1: myocardial blush is present in the distribution of the infarct-related artery, but there is incomplete clearance of dye between injections (with ≈30 seconds between injections).
-
2: myocardial blush is present in the distribution of the infarct-related artery, but there is slow contrast entry into the microvasculature and slow clearance of contrast. Specifically, blush is strongly persistent (i.e. either does not or only minimally diminishes in intensity) beyond three cardiac cycles after injection.
-
3: myocardial blush is present in the distribution of the infarct-related artery, with normal entry and exit of dye (mild/moderate persistence of dye beyond three cardiac cycles, but notably reduced after three cardiac cycles). Blush that is only mild intensity throughout three cardiac cycles after injection (washout phase) but fades minimally is also classified as grade 3.
Corrected TIMI frame count
The TIMI frame count is an objective continuous variable index of coronary blood flow, representing the amount of time (in frames) for contrast dye to reach a standardised distal landmark, corrected for vessel length (corrected TIMI frame count, normal < 27 frames).
Method
The corrected TIMI frame count is the number of cine frames required for contrast to first reach standardised distal coronary landmarks in the culprit artery and is measured with a frame counter on a cine viewer. A frame rate of 30 frames per second was used in the original description of the method. The common use of slower frame rates will require the count to be adjusted (e.g. 15 frames per second will require the frame count to be doubled). If the culprit vessel is the left anterior descending artery, the frame count needs to be corrected by dividing by 1.7 (correcting for longer vessel length).
To calculate the transit time (seconds) for dye to traverse the length of the artery to the distal landmark, the corrected TIMI frame count is divided by 30 and multiplied by 1000 to convert the time to milliseconds. To calculate the fraction of a cardiac cycle required for dye to traverse the artery (normalising the corrected TIMI frame count for heart rate): fraction of cardiac cycle = (corrected TIMI frame count/30 seconds)/(60/heart rate). A frame count of 100 (a value that is the 99th percentile of patent vessels) was imputed to an occluded vessel.
Classification of TIMI coronary thrombus grade
Thrombus burden revealed during coronary angiography can be classified according to the TIMI thrombus grade:
-
0 – no angiographic characteristics of a thrombus are present.
-
1 – a possible thrombus is present with reduced contrast density, haziness, irregular lesion contour or a smooth convex ‘meniscus’ at the site of total occlusion suggestive but not diagnostic of thrombus.
-
2 – definite thrombus, with greatest dimensions < 1/2 the vessel diameter.
-
3 – definite thrombus but with greatest long axis dimension > 1/2 but < 2 vessel diameters.
-
4 – definite thrombus, with the largest dimension > 2 vessel diameters.
-
5 – total occlusion.
Electrocardiogram acquisition and analysis
Twelve-lead ECG was carried out before coronary reperfusion and 60 minutes afterwards, and again at 3 months. The ECGs were analysed in the University of Glasgow ECG core laboratory, Glasgow, UK.
Electrocardiograms
Twelve-lead ECG was performed in accordance with standard methods by trained cardiology staff using standard ECG recorders available in the cardiology department. ECG records were anonymised by inserting the patient ID number.
Anonymised ECGs were prospectively collected and uploaded to the electronic case report from portal for electronic transfer to the University of Glasgow core laboratory during the course of the study. The ECG core laboratory is certified to ISO 9001:2015 standards by a UKAS accredited organisation. The intention was that ECGs would be analysed using the University of Glasgow ECG analysis program, but this approach proved not to be possible in most participating centres and so copies of all ECGs were uploaded to the electronic case report form portal in PDF format. ECGs were checked for completeness and quality, and feedback was provided to local sites.
The ECG outcomes include the change in summative ST-elevation score on the 60-minute ECG post MI versus pre reperfusion, and the Selvester QRS score was taken as a surrogate ECG measure of infarct size at 3 months. The Selvester score translates subtle changes in ventricular depolarisation on ECG to a surrogate measure of infarct size. There are a maximum of 32 points, with 1 point corresponding to 3% of the left ventricle.
Safety
Parameters of haemostasis and coagulation, including fibrinogen concentration, plasminogen activity, fibrin D-dimer and prothrombin F1 + 2, served as a surrogate measure of bleeding risk and safety. tPA antigen levels were assessed as a potential measure of the systemic overflow of the alteplase administered into the culprit coronary artery. Haemostasis and coagulation parameters were measured in blood samples, when site logistics permitted. The sampling time points were 0, 2 and 24 hours post reperfusion.
A depletion of fibrinogen and plasminogen following thrombolysis correlates with systemic fibrinolysis and may correlate with bleeding risk. Fibrin D-dimer concentration represents a specific measure of fibrinolysis. Fibrin D-dimer concentrations may correlate with the amount of clot lysis and may therefore represent a measure of residual clot burden. Fibrin D-dimer concentrations have the potential to correlate with efficacy and outcome. Prothrombin F1 + 2 is a measure of thrombin activation and correlates with the (undesired) procoagulant effect of thrombolysis. Prothrombin F1 + 2 is depressed by anticoagulants administered before and during PCI. Prothrombin F1 + 2 concentrations may be associated with the dose of alteplase and placebo, and potentially could correlate with adverse thrombotic events.
Local hospital blood sample handling
Blood samples collected into 0.109 M sodium citrate (for haemostasis assays) or ethylenediaminetetraacetic acid (EDTA) (for troponin T and NT-proBNP) were handled in accordance with a sample handling manual that was provided to all sites. Research blood tests at 0, 2 and 24 hours were preferred but not compulsory. The blood samples were centrifuged locally and plasma separated and frozen within 2 hours of sampling. Frozen plasma samples were subsequently transported on dry ice for central laboratory analysis in the Department of Haematology, Glasgow Royal Infirmary. Plasma samples were stored at –80 °C until analysis, with residual samples being transferred to the Glasgow Biorepository for storage at the end of the study.
Central laboratory analyses
Blood samples
Research blood tests at 0, 2 and 24 hours were preferred but not compulsory. In this way blood sample handling out of hours did not become a barrier to enrolment.
Troponin T and NT-proBNP
The EDTA plasma samples were stored at –80 °C in the Glasgow Royal Infirmary until batch analysis at the end of the study. The biochemical analyses were performed in the British Heart Foundation Glasgow Cardiovascular Research Centre.
The EDTA plasma samples were stored to analyse high-sensitivity cardiac troponin T and NT-proBNP on first thaw. Serial measurements of troponin T using Roche high-sensitivity assay were used to provide a biochemical measurement of infarct size (AUC). 13,14 Troponin T (ng/ml) was measured in blood samples collected at baseline (before study drug administration), and 2 ± 1 hours, 24 ± 12 hours, 2–7 days and 12 weeks (± 2 weeks) post MI. NT-pro BNP (pg/ml) was measured in blood samples collected at baseline, 2–7 days and 12 ± 2 weeks to provide a biochemical measurement of left ventricular remodelling (within-subject change in NT-proBNP at follow-up from baseline).
For measurement of both NT-proBNP and high-sensitivity cardiac troponin T, we used an automated method (cobas e411; Roche Holding AG, Basel, Switzerland) calibrated and quality controlled using the manufacturer’s reagents. We also participated in the National External Quality Assurance Scheme. The lower limit of detection of troponin T is 0.003 ng/ml and the 99th percentile value in a healthy subpopulation is 0.0014 ng/ml (Roche Holding AG, data on file). The between-assay coefficient of variations were 2.2% and 4.2% for control materials with mean troponin T concentrations of 2.098 ng/ml and 0.0027 ng/ml, respectively.
For NT-proBNP, the coefficient of variation was 2.6% and 2.4% for control materials with mean NT-proBNP levels of 4418 pg/ml and 142 pg/ml, respectively. The troponin T and NT-proBNP results were provided to the Robertson Centre for Biostatistics, University of Glasgow.
Haemostasis and coagulation laboratory methods
Fibrinogen and other haemostasis parameters served as a surrogate measure of bleeding and safety. Haemostasis and coagulation parameters were measured in blood samples when site logistics permitted. The sampling time points were 0, 2 and 24 hours post reperfusion. The parameters included fibrinogen, fibrin D-dimer and plasminogen activity, tPA antigen and prothrombin F1 + 2.
Sample handling
All plasma samples were processed in a non-standard manner using anonymised bar-coded samples by a trained member of staff.
Assays
Standard laboratory assays (fibrinogen by Clauss method,32 high-sensitivity fibrin D-dimer by latex immunoassay and plasminogen activity by chromogenic assay were performed on an IL Top 700 analyser (Diamond Diagnostics Inc., Holliston, MA, USA) using HemosIL® reagents (Instrumentation Laboratory Company, Bedford, MA, USA). The fibrinogen Clauss assay had a normal reference range of 170–400 mg/dl (internally derived) and an interassay coefficient of variation of 5.8% and 7.7% for low-control samples with mean concentrations of 292 mg/dl and 222 mg/dl, respectively. The fibrin D-dimer assay had a normal reference range of < 0.230 g/ml (manufacturer derived) and an interassay coefficient of variation of 11.7% and 5.2% for control samples with mean concentrations of 0.343 g/ml and 0.770 g/ml, respectively. The plasminogen activity assay had a normal reference range 80–133 U/dl (manufacturer derived), and an interassay coefficient of variation of 2.1% and 1.8% for control samples with mean concentrations of 95.4 U/dl and 29.6 U/dl, respectively.
Non-standard laboratory enzyme-linked immunosorbent assay (ELISA) tests (tPA and prothrombin F1 + 2 antigen levels) were performed on a TECAN Sunrise spectrophotometer (Labtech International Ltd, Heathfield, UK) using Zymutest tPA Antigen (Hyphen BioMed, Neuville-sur-Oise, France) and Enzygnost F1 + 2 Mono (Siemens, Marburg, Germany) commercial kits, respectively. The tPA antigen assay had a normal reference range of < 10 ng/ml (manufacturer derived) and an interassay coefficient of variation of 4.7% and 11% for control samples with mean concentrations of 11.0 ng/ml and 3.1 ng/ml, respectively. The F1 + 2 assay had a normal reference range of 69–229 pmol/l (manufacturer derived) and an interassay coefficient of variation of 7.9% for a normal control sample, with a mean concentration of 97.6 pmol/l.
List of abbreviations
- AUC
- area under the curve
- BARC
- Bleeding Academic Research Consortium
- BN
- Bachelor of Nursing
- CEC
- Clinical Event Committee
- CI
- confidence interval
- ECG
- electrocardiogram
- EDTA
- ethylenediaminetetraacetic acid
- eGFR
- estimated glomerular filtration rate
- EME
- Efficacy and Mechanism Evaluation
- EQ-5D-3L
- EuroQol 5-Dimensions, three-level version
- FRCP
- Fellow of the Royal College of Physicians
- HCPC
- Health and Care Professions Council
- HRQoL
- health-related quality of life
- IDMC
- independent Data Monitoring Committee
- IQR
- interquartile range
- MACE
- major adverse cardiac event
- MD
- Doctor of Medicine
- MI
- myocardial infarction
- MRCP
- Member of the Royal College of Physicians
- MRI
- magnetic resonance imaging
- NIHR
- National Institute for Health Research
- NT-proBNP
- N-terminal pro–B-type natriuretic peptide
- PCI
- percutaneous coronary intervention
- PPI
- patient and public involvement
- R&D
- research and development
- RCT
- randomised controlled trial
- RN
- Registered Nurse
- SD
- standard deviation
- SSFP
- steady-state free precession
- STEMI
- ST-segment elevation myocardial infarction
- TIMI
- Thrombolysis in Myocardial Infarction
- tPA
- tissue plasminogen activator
- TSC
- Trial Steering Committee
- T-TIME
- Trial of low-dose adjunctive alTeplase during priMary PCI