

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 13/95/10. The contractual start date was in September 2015. The final report began editorial review in November 2019 and was accepted for publication in June 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2021 Butler et al. This work was produced by Butler et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2021 Butler et al.
Chapter 1 Introduction
Text in this chapter has been reproduced form the published protocol paper by Owen-Jones et al. 1 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Rigorous research is needed to underpin improved care for care home residents
As the population ages, care homes are becoming an increasingly common and important sector for delivering health and social care. Novel interventions that aim to enhance care and quality of life should not be taken up in routine care without efficacy first being established. Thus far, there have been few randomised controlled trials (RCTs) of interventions that aim to improve the quality of care for care home residents (CHRs). As a result, far more is known about effective treatments in hospitals than about what works most effectively to improve care and the experience for older people living in care homes. 2
Because immunity wanes with increasing age and frailty, CHRs are more prone to infections and account for a high volume of antibiotic use. Antibiotic use drives antimicrobial resistance (AMR). Regular probiotics, so-called ‘good bacteria’, have been proposed as a feasible and safe intervention that may reduce the incidence of infections and, thus, antibiotic use. However, regular probiotics for reducing infections and antibiotic use have so far not been evaluated in a rigorous, long-term RCT among CHRs. Furthermore, there are gaps in our understanding of how probiotics might work. Therefore, we evaluated the efficacy and possible mechanism of probiotics in CHRs to prevent infections and thus reduce antibiotic use.
What are probiotics and how might they work?
Probiotics are defined by the World Health Organization as ‘live microorganisms which when administered in adequate amounts, confer a health benefit to the host’. 3 They are present in certain products available in supermarkets as foodstuffs and in formulations used for specific therapeutic purposes. They may prevent infection by blocking pathogenic colonisation and enhancing gut–immune interaction, with influence on mucosal and systemic immunity, leading to enhanced natural killer (NK) cell activity and vaccine response in older people. 4 The gastrointestinal tract may be a major reservoir for antimicrobial-resistant bacteria. This is important because many urinary tract infections (UTIs) are thought to arise from auto-inoculation with organisms from the gut. 5 As people age, the microbiological diversity in their gut reduces because they are less mobile and adopt different eating patterns and their immune systems become less active. By increasing the diversity of microorganisms in the gut, taking probiotics regularly may reduce the prevalence of pathogenic bacteria and stimulate the immune system, adding to overall resilience and general well-being as well as reducing infections and antibiotic use. Ageing is independently associated with reduced immune response to infections. 6
Are probiotics safe and feasible for care home residents?
Although probiotics carry theoretical risks of causing infection beyond the gut and transferring antibiotic-resistant genes, there have been no reports of bacteraemia or fungaemia attributable to the probiotics when used in trials,7–9 and only a very few case reports in patients with immunodeficiency or severe ulceration of the colonic mucosa. 10 Gastrointestinal side effects and rash are generally no more common in patients on probiotics than in those on placebo. 11 Systematic reviews found no serious adverse effects in participants of trials of probiotics for antibiotic-associated diarrhoea (AAD). Some older people already use them regularly, despite an inadequate evidence base supporting their effect on common infectious diseases. Adverse events (AEs) in trials of probiotics have generally not been well reported. 12
However, there is emerging evidence that probiotics after antibiotic use may delay the reconstitution of the faecal and mucosal microbiome, because soluble factors secreted from the probiotic species could inhibit human microbiome growth. 13 Inhibition by probiotics of reconstitution of the human microbiome may be a mechanism that explains a surprise finding from one retrospective hospital-based cohort study of 5209 patients for whom prior probiotic exposure was an independent predictor of Clostridium difficile infection [odds ratio (OR) 1.39, 95% confidence interval (CI) 1.08 to 1.80]. 13
Probiotics are feasible to administer to CHRs in the course of routine care: Carlsson et al. 14 confirmed the feasibility of serving a probiotic intervention for 6 months to people with dementia in care homes. The authors found that the intervention was easy to serve alongside usual diet, there were few side effects and staff were able to complete the processes and measures. In contrast to antibiotic use, long-term probiotic use does not result in resistance in commensal gut organisms. 15
Existing trials and the research gap
Small trials found benefits from probiotic colonisation by multidrug-resistant Gram-negative organisms in critically ill patients16 and by vancomycin-resistant enterococci (VRE) in renal dialysis patients. 17 A Cochrane review18 of probiotics to prevent acute upper respiratory tract infections (URTIs) included 13 RCTs, 10 of which were meta-analysed (including a total of 3720 participants). Probiotics reduced episodes of acute URTI (OR 0.53, 95% CI 0.37 to 0.76), duration of episodes (mean difference –1.89 days, 95% CI –2.03 to –1.75 days; p < 0.001, low-quality evidence) and antibiotic prescribing (OR 0.65, 95% CI 0.45 to 0.94). Side effects of probiotics were minor. The review noted poor allocation concealment, and insufficient heterogeneity in several studies, and recommended that future RCTs should include older people, as few were included in the trials reviewed.
King et al. 19 systematically reviewed studies of probiotics to reduce antibiotic use for common acute infections. The authors included 17 RCTs, all conducted in infants and children. The mean duration of probiotic supplementation ranged from 4 days to 9 months. Children who received probiotics had a lower risk of being prescribed antibiotics than those receiving placebo (pooled relative risk 0.71, 95% CI 0.54 to 0.94); however, trial quality was rated as being variable. The authors recommended evaluating probiotics to reduce antibiotic use in other population groups.
Regarding older adults, Turchet et al. 20 performed a single-centre pilot study of otherwise well ‘free-living’ older people, randomised to receive a probiotic containing Lactobacillus casei or placebo for 3 weeks. The authors found no differences in frequency of URTI in this short study, but did find a reduction in the severity and duration of URTIs. Guillemard et al. 21 randomised otherwise well people living in care homes in France to a probiotic containing L. casei or placebo for 3 months. Probiotics decreased the duration of common infectious diseases, especially URTIs. Makino et al. 22 in Japan considered whether or not the intake of yogurt fermented with L. delbrueckii over 12 weeks had an effect on the common cold, and found that the risk was 2.6 times lower (OR 0.39) and that there was increased NK cell activity in the yoghurt group. However, randomisation and concealment may not have been adequate in this study. Van Puyenbroeck et al. 23 found no difference in the duration of respiratory symptoms or the probability of respiratory symptoms in ‘healthy older people’ in nursing homes in Antwerp from a probiotic containing L. casei Shirota for 176 days. However, people with relevant medical conditions and those with cognitive deficits were excluded, and these people may be most likely to benefit meaningfully from regular probiotics. Antibiotic prescription rates were not reported. The large number of missing self-complete diary data generated analytical challenges.
Thus, overall, the evidence base supporting a recommendation for frailer elderly people either to consume or to not consume probiotics is insufficient to robustly guide care. New trials that are properly designed in terms of allocation and concealment, that focus on antibiotic use for all-cause infections in CHRs (including those without capacity and who are most frail) and with reliable, frequent external ascertainment and with longer-term follow-up are therefore warranted.
Rationale for the PRINCESS trial
Infections are a common cause of suffering for and increased resource use by CHRs. Even so-called ‘minor infections’ such as UTIs and URTIs can have an important negative impact on the health, well-being and dignity of older frail people. The 2010 Adult Social Care Statistics reported that there were 229,900 people in residential care in England, with numbers predicted to steadily increase. 24 The 2011 census reported that there were 291,000 people over the age of 65 years resident in the care home population in England and Wales. 24
A year-long observational study (n = 274) identified 609 infections that led to an antibiotic prescription incidence of 2.16 antibiotic prescriptions per resident year (95% CI 1.90 to 2.46). The most common indications were respiratory tract infections (RTIs) (47% of prescriptions), UTIs (29%) and skin infections (18%). 25 Common infectious diseases in CHRs led to suffering, loss of dignity, hospitalisation, general practitioner (GP) visits and death. A health needs assessment of 240 CHRs in South Tyneside Primary Care Trust (now called South Tyneside Clinical Commissioning Group) identified 167 hospital admissions accounting for 1595 bed-days over 1 year. 26 On average, at least four beds in the acute trust were used for CHRs all year round, costing around £400,000. About 25% of admissions were due to infection. Common infectious diseases in CHRs increase GP and care home burden and costs, affecting opportunities for other aspects of care. Antibiotic prescribing leads to health-care-associated infections, such as C. difficile infection, and drives AMR. Infection with an antimicrobial-resistant organism is commonly associated with recent consumption of antibiotics, even after controlling for age, comorbidity and other risk factors. 27 Antimicrobial-resistant infections are more serious, last longer and are costlier to manage. 28,29 Probiotics have been found to be effective in preventing AAD,9,11,30 although a recent large trial31 found no benefit. However, there are no adequate data on probiotics for the prevention of all-cause common infections and antibiotic prescribing in CHRs.
Health-care-associated infections include common infectious diseases and cause significant debility, hospital admissions and the death of CHRs, burdening both the health service and care home staff. Among CHRs in Norway, the risk of deterioration in general physical condition was twice as high those who developed health-care-associated infections than in matched controls who did not have health-care-associated infection, and the former were nine times more likely to be admitted to hospital. 32
Carriage of antimicrobial-resistant bowel organisms increases the chances of an antimicrobial-resistant UTI. Antimicrobial-resistant Gram-negative septicaemia and antimicrobial-resistant UTI are on the increase in the community,33 especially among older people. 34 Care homes are a reservoir for antimicrobial-resistant organisms that cycle between the community and hospitals. 35,36
There have been calls for ‘diligently planned large-scale randomised and blinded trials, preferably devoid of commercial interests’ with end points being objectively assessed. 37
Rationale for the probiotics selected for evaluation in the PRINCESS trial
The effects of probiotics are thought to vary by strain because of differing resistance to gastric acid and bile, ability to colonise mucosa and susceptibility to antibiotics. The probiotic combination selected for this study comprised two organisms, Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 Lactobacillus rhamnosus GG has been extensively studied in a wide variety of populations including older individuals. 38 This strain has been found to be safe. Although the effect of this probiotic strain on infections in CHRs or older people has not yet been assessed, relevant evidence proving efficacy in other populations supports its use in this trial. A meta-analysis of RCTs involving 1805 children found that Lactobacillus rhamnosus GG was associated with a reduced risk of otitis media, URTIs and antibiotic treatments. 39 This meta-analysis included a double-blind, placebo-controlled RCT of 742 hospitalised children that found that Lactobacillus rhamnosus GG reduced the risk of gastrointestinal infections (GIs) [risk ratio (RR) 0.40, 95% CI 0.25 to 0.70] and RTIs (RR 0.38, 95% CI 0.18 to 0.85), and the duration of these infections. 40 A RCT of 281 children in day care found that Lactobacillus rhamnosus GG reduced the risk of RTIs over 3 months (RR 0.66, 95% CI 0.52 to 8.82, number needed to treat = 5) and reduced the time with a RTI. 41 A double-blind RCT found that Lactobacillus rhamnosus GG acts as an immune adjuvant to influenza vaccination, as measured by levels of protective antibodies to the H3N2 flu strain. This study stressed the need for future studies of probiotics as immune adjuvants, focusing on groups known to have a poor response to influenza vaccination. 42 When administered orally as lozenges, a combination of probiotics has been found to have a beneficial effect on oral and dental health, reducing plaque and gingival inflammation without affecting oral microbiota. 43
Many studies include more than one probiotic strain in the intervention, and, in several studies, Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 have been included in the same probiotic formulation. 44,45 A randomised placebo-controlled trial of 231 college students taking a combination probiotic including Bifidobacterium animalis subsp. lactis BB-12 for 12 weeks found that the duration and severity of URTIs were reduced by the active probiotic combination. 46 A randomised, placebo-controlled, double-blind, parallel-group dose–response study investigated the impact of 4-week commercial yoghurt consumption supplemented with Bifidobacterium animalis subsp. lactis BB-12. The probiotic strain remained active during gut transit and was associated with an increase in beneficial bacteria and a reduction in potentially pathogenic bacteria in faeces. 47 In a double-blind, placebo-controlled study, 109 1-month-old infants receiving Bifidobacterium animalis subsp. lactis BB-12 were reported to have experienced fewer respiratory infections (65% vs. 94%, RR 0.69, 95% CI 0.53 to 0.89; p = 0.014) than the control infants. 48 A randomised, double-blind, placebo-controlled study of infant formula supplemented with the probiotics Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 or placebo was administered daily until the age of 12 months. Those infants receiving the active probiotic had fewer episodes of otitis media, received fewer courses of antibiotics in the course of routine care and had fewer recurrent RTIs. 49 A multispecies probiotic supplementation that included Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 reduced symptoms of irritable bowel syndrome and stabilised the bowel microbiota. 50 A randomised, double-blind, placebo-controlled trial in 12 wards in two nursing homes in Finland involved 209 CHRs who received (1) 109 colony-forming units (CFUs) per day of B. longum strains or (2) 109 CFU per day of Bifidobacterium animalis subsp. lactis BB-12 or (3) placebo for 7 months. There was 85% compliance, and the groups receiving active products had more frequent bowel movements than the placebo group. 51 In a 7-week double-blind crossover study, 36 patients were randomised to receiving yoghurt with Bifidobacterium animalis subsp. lactis BB-12 or placebo. Bifidobacterium animalis subsp. lactis BB-12 was safe. During the Bifidobacterium animalis subsp. lactis BB-12 intake period, the defecation frequency increased compared with the no-intake period for the group, and comfort after defecation improved significantly. 52
Thus, neither proposed probiotic strain has ever been evaluated for the purposes intended in the PRINCESS (Probiotics to Reduce Infections iN CarE home reSidentS) trial and there is evidence from other population groups that these strains have the effect of reducing frequency, severity and duration of infections and stabilising bowel flora. Many infections in older people result in auto-inoculation from the bowel. It is possible that the two strains complement each other in terms of efficacy as a result of different modes of action.
Objectives of the PRINCESS trial
The primary objective of the PRINCESS trial was to evaluate the effect of a dose of daily oral probiotics on cumulative systemic antibiotic administration days for all-cause, acute infections.
Secondary objectives included determining the effect of daily probiotic intake on subcategories of infections, incidence and duration of diarrhoea in CHRs who are being treated with oral antibiotics, incidence and duration of infections, prevalence of C. difficile infection, health utility, well-being, hospitalisation, mortality, gastrointestinal colonisation with antimicrobial-resistant bacteria, and Candida from oral samples.
Mechanistic immunology outcomes aimed to explore the effect of daily probiotic combination intake on influenza vaccine response, cytokine and chemokines, monocyte and neutrophils, probiotic in stool samples, and influence of baseline vitamin D on estimates of AMR.
Chapter 2 Methods for the randomised trial
Text in this chapter has been reproduced form the published protocol paper by Owen-Jones et al. 1 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Trial design
The PRINCESS trial was an individually randomised, double-blind placebo-controlled trial of the clinical efficacy and safety of a combination of two probiotic organisms given daily to CHRs in reducing cumulative antibiotic administration days (CAAD). Trial participants were CHRs from 23 care homes in Wales and England. Following enrolment, and on completion of a baseline assessment, participants were randomised to receive a capsule containing either the probiotic combination (intervention) or a formulation of maltodextrin, microcrystalline cellulose, magnesium stearate and silicon dioxide (placebo control) to be taken once daily for 52 weeks.
An internal pilot was conducted in four care homes (two in Wales and two in England) between December 2016 and February 2017. The aims of the internal pilot included assessing levels of interest expressed by care homes in implementing the trial, estimating the likely CHR recruitment rate, establishing that it would be possible to ascertain adherence to the study product (SP) (probiotic or placebo) and measuring study outcomes for most of the participants. The internal pilot also allowed us to develop a mechanism for capturing antibiotic use while participants were away from the care home or in hospital. A hospital discharge case report form (CRF) was developed for collection of hospital data after the internal pilot and was used in most cases. No other trial procedures were changed as a result of the internal pilot phase.
The trial protocol has been published1 and amendments made to the trial protocol during the conduct of the trial are listed in Appendix 1.
Involving the care home population
We adopted broad inclusion criteria to maximise generalisability of the trials’ findings. In particular, CHRs with mental capacity and CHRs without mental capacity to provide informed consent were eligible. An observational study,25 undertaken by the research team, found that 72% of CHRs included in an observational study in care homes lacked mental capacity to provide informed consent, and that those lacking mental capacity had a higher incidence of infections, which were more likely to be serious and harder to treat. 25
Those lacking capacity to consent might, therefore, potentially receive greater benefit from the active intervention, and make up an important proportion of CHRs. Furthermore, even those who lack capacity should not be denied the opportunity to participate in research that might be relevant to their situation because of age or lack of capacity, unless there are good scientific reasons to justify their exclusion. 53 Interventions should be evaluated in those for whom the interventions, if found to be effective, are intended, and, similarly, mechanisms should be explored across the whole of the relevant population. 54 Therefore, we considered that it was important that those CHRs lacking capacity to consent should be eligible for inclusion in the trial to maximise the applicability and benefit of findings. This decision was supported by research previously undertaken with CHRs, CHRs’ relatives, and health and social care professionals. 55–57 The trial was given a favourable ethics opinion by Wales Research Ethics Committee (REC) 3 (reference number 15/WA/0306; approval received on 13 November 2015).
Patient and public involvement
Two patient and public representatives who had previously contributed to a care home study25 agreed to perform this role for the PRINCESS trial. One of the representatives had been a care home manager (CHM) and the other was a relative of a CHR and had been employed as a carer in a care home. One of the representatives maintained an advisory role throughout the study, attending monthly Trial Management Group meetings, assisting with the preparation of annual newsletters and advising on results dissemination methods and events.
Setting and participants
Involvement and setting up of care homes
Between December 2016 and May 2018, a total of 23 care homes were recruited, set up for the study and opened to CHR recruitment. Care home set-up had to be staggered as training was resource intensive; therefore, not all 23 care homes were open to CHR recruitment simultaneously. CHMs took the role of principal investigator (PI) in 20 of the care homes, and an NHS-employed psychogeriatrician assumed the role of PI in the remaining three care homes. Training was delivered to the PIs in three sessions, with care home staff attending one or two of the relevant sessions. Most of the CHMs (those that were also PIs) did not have any prior research experience, but this was not a barrier following discussion with the REC (who granted the non-NHS site approval).
The training of care home staff and PIs in trial implementation was delivered by either the research nurses (RNs) or the trial managers from Cardiff University or the University of Oxford clinical trials units. Recruitment of CHRs, further training of care home staff, and biological samples and data collection were undertaken by the trained RNs. The trial RNs were employed directly by Cardiff University or the University of Oxford, or were provided by Clinical Research Networks (CRNs) or specific NHS trusts. Owing to the low-risk nature of the trial, a risk-based approach to study monitoring was adopted and outlined in the trial risk assessment document. We planned to monitor the study centrally, to be followed up, if required, by a site visit; there were no preplanned monitoring visits.
Participant recruitment
The recruitment process was undertaken as a collaboration between care home staff and RNs; the process itself varied depending on whether or not the CHR was deemed to have mental capacity to consent.
Eligibility
Inclusion criteria
-
Currently living in a care home setting (residential, nursing or mixed).
-
Willing and able to give informed consent for participation in the trial or, if the participant lacks capacity, a consultee is willing to complete a consultee declaration form for this participant on their behalf.
-
Aged ≥ 65 years.
Exclusion criteria
-
Known to be immunocompromised (requiring immunosuppressants, on long-term, high-dose, oral, intramuscular or intravenous steroids).
-
Currently taking regular probiotics and not willing to adapt to trial protocol.
-
Currently participating in a clinical trial of an investigational medicinal product (CTIMP) or has been a participant in a CTIMP in the last 30 days.
-
A temporary CHR (i.e. < 1 month of planned transitional/respite residential care).
-
Death is thought to be imminent.
-
Is lactose intolerant.
Mental capacity assessment
In accordance with the Mental Capacity Act 2005,58 potential PRINCESS trial participants were assumed to have mental capacity to consent unless there were reasons for doubting this. If there were concerns about a CHR’s capacity to understand the information about the trial and decide for themselves whether or not to take part, a mental capacity assessment was undertaken in accordance with the Mental Capacity Act 2005. 58 The outcome was documented using a mental capacity assessment form designed specifically for the PRINCESS trial.
Mental capacity is decision – and time – specific, because it relates to whether or not a person can make a specific decision at a specific time. Mental capacity assessment in the PRINCESS trial was always undertaken jointly between care home staff and the RNs. Care home staff had important knowledge relating to factors such as (1) CHRs’ medical history, (2) previous mental capacity issues/fluctuation and (3) personal knowledge of how best to communicate with CHRs, all of which had important implications for mental capacity assessment. RNs, on the other hand, had in-depth knowledge of the study, enabling them to answer any questions that CHRs/consultees might have, and of the mental capacity assessment procedure specific to the PRINCESS trial. CHRs’ confidentiality was maintained during the mental capacity assessment process.
Maximising capability to provide informed consent
The recruitment materials for CHRs were designed to take into account age-related sight and hearing loss, conditions affecting mobility or motor skills, or cognitive impairment. These included a pictorial information booklet that the RN could use in place of, or alongside, the participant information sheet during discussions about the trial. The participant information sheet and consent forms were A3 in size as standard with proportionally larger font. The CHR consent forms were designed such that the option of verbal consent (witnessed by an adult independent of the trial) could be provided if CHRs were unable to add their signature to the consent form because of joint or neurological conditions.
Consultee model of recruitment
If a mental capacity assessment indicated that a CHR lacked capacity to make an informed decision about participating in the trial, the involvement of a consultee was required, in accordance with the Mental Capacity Act 2005. 58 In these situations, a member of care home staff would identify a close relative (usually next of kin) or a close friend who could act as a personal consultee to provide advice on behalf of the CHR. The consultee was asked for their advice on whether or not, in their opinion, the CHR would have wished to take part in the trial, were they to have had capacity [section 32(4)];58 this was in conjunction with the RN, who had the final decision about the CHR being recruited. If no personal consultee could be identified, a nominated consultee was suggested by the staff at the care home. The nominated consultee could be a member of the care home team who was prepared to be consulted but had no formal involvement with the trial [section 32(3)]. 58,59 In accordance with the provisions of the Mental Capacity Act 2005, any indication that the CHR objected to participation, or to a trial-related procedure, was respected and taken as withdrawal of consent (section 33). 58
Ongoing process of consent
Consent was considered an ongoing process. If a CHR’s capacity appeared to diminish or increase during their trial participation, a further assessment of their mental capacity in relation to the trial was carried out. If a CHR was assessed to have lost capacity, having previously consented to participate, consultee advice was sought regarding their continuing trial participation. The reverse was also applied: if the CHR gained capacity, informed consent would have been obtained.
Trial interventions
The active and placebo capsules were manufactured by Chr. Hansen A/S (Hørsholm, Denmark) and supplied to the trial team at no cost to the study. Boxes of active SP and boxes of placebo SP were sent from Chr. Hansen directly to Production Services and Clinical Trials, Nottingham University Hospitals NHS Trust, for labelling with trial information and pack identifiers. The boxes were then transported to the Centre for Trials Research, Cardiff University (CTR), for long-term storage in a temperature-monitored fridge. Because the SP was stable at room temperature for 2 years, some packs were stored at room temperature at the CTR before posting to study sites.
During the trial, owing to expiry dates, three batches of SP were sent from Chr. Hansen to Nottingham University Hospitals NHS Trust for labelling (the last two batches were from the same manufacture run).
The SP capsules contained either Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 or placebo (containing maltodextrin, microcrystalline cellulose, magnesium stearate and silicon dioxide). The dates of manufacture of SP were December 2015 (total cell count per capsule 1.6 × 1010) and March 2017 (total cell count per capsule 1.36 × 1010).
There were 31 capsules per tube. SP capsules could be swallowed whole, opened and sprinkled onto warm or cold (but not hot) food or dissolved in warm or cold (but not hot) liquid. Before any use of the probiotics, a sample probiotic and placebo were labelled and sent for analysis. Each capsule was dissolved in 10 ml of sterile water then further diluted 10–3 and 10–6. All three solutions (50 µl) were spiral plated onto two plates each of Columbia blood agar (CBA) and fastidious anaerobe agar (FAA) (both from Thermo Fisher Scientific Oxoid Ltd, Basingstoke, UK) and incubated at 35 ± 1 °C aerobically and in carbon dioxide (CBA plates) anaerobically (FAA plates) for 24–48 hours. All colony types were identified using matrix-assisted laser desorption/ionisation time of flight (MALDI-TOF) mass spectrometry and individual counts were made.
Data collection and randomisation
Data were collected at the care home, using information gathered from the participant, the participant’s relatives, the care home staff, hospital discharge summaries and the participant’s care home records. Data were collected either on paper CRFs or electronically via a Structured Query Language (SQL) online database, which was hosted on a secure password-protected server at the CTR. All data collected on paper were later entered electronically to the database either by the RNs remotely or at the CTR. Data were added to the database using a secure tablet, a laptop or a PC. A trial-specific Data Management Plan was developed outlining in detail the procedures that were put in place to ensure that high-quality data were produced for statistical analysis. The database had in-built validation rules and ranges checks, with incomplete fields and data outliers flagged at the time of entry. Missing data and validation errors were queried periodically with RNs, as necessary. A list of self-evident corrections was iterated during the trial to enable correction of unambiguous data entry errors. Data collected on paper held or returned to the central monitoring site (i.e. CTR) were subject to quality control checks against data held on the database (10% of data collected on paper). All data, including sensitive and personal data, were handled in accordance with the Data Protection Act 199860 and then the General Data Protection Regulation 2016. 61
On completion of data cleaning, the data were extracted from the database and provided to the statistician for analysis. As per Cardiff University’s procedures, data will be retained for 15 years following trial closure.
Participant tracking through the trial was assisted through database-generated automated e-mails, which were sent when data collection for a particular time point was complete. A summary of data collection by time point is provided in Table 1.
| Outcome/assessment | Time point | ||||||
|---|---|---|---|---|---|---|---|
| Registration | Baseline review | Weekly review (prior to SP initiation) | Weekly review (after commencing SP) | 3-month review | 12-month/second review | Up to 10 days before and after 28 days post influenza vaccinationa | |
| Confirmation of eligibility | ✗ | ✗ | |||||
| Assessment of mental capacity (if applicable) | ✗ | ✗ | ✗ | ||||
| Contact details | ✗ | ||||||
| Written informed consent/consultee declaration form | ✗ | ||||||
| Ethnicity | ✗ | ||||||
| Weight | ✗ | ||||||
| All antimicrobial use | ✗ | ✗ | ✗ | ||||
| Proton pump inhibitor use | ✗ | ||||||
| Use of laxatives | ✗ | ||||||
| Consumption of vitamin D supplements | ✗ | ||||||
| Antibiotic use | ✗ | ✗ | |||||
| Infection | ✗ | ✗ | |||||
| Hospitalisation | ✗ | ✗ | |||||
| Diarrhoea | ✗ | ✗ | |||||
| Dose and mode of SP | ✗ | ||||||
| Well-being and health–utility assessments (ICECAP-O/proxy ICECAP-O/EQ-5D-5L/proxy EQ-5D-5L) | ✗ | ✗ | ✗ | ||||
| Serious AEs (including participant mortality) | ✗ | ✗ | |||||
| Full blood count, serum vitamin D, immunophenotyping (blood sampleb) | ✗ | ✗ | |||||
| Oral microbiology (saliva sample) | ✗ | ✗ | ✗ | ||||
| AMR and gut microbiology (stool sample) | ✗ | ✗ | ✗ | ||||
| Influenza vaccine efficacy (blood sample) | ✗ | ||||||
Baseline review
Following enrolment and registration, data were collected for the baseline review. Data collected at baseline included ethnicity, physical characteristics [height, weight, ulna length, mid-upper arm circumference and frailty (assessed by using the Clinical Frailty Scale score)], medical history (proton pump inhibitor use, use of antimicrobials in the previous 4 weeks, laxative use, vitamin D use). CHRs with capacity were asked to self-complete the EuroQol-5 Dimensions, five-level version (EQ-5D-5L),62,63 and the Investigating Choice Experiments for the Preferences of Older People CAPability measure for Older people (ICECAP-O). 64,65 Care home staff were asked to complete proxy versions of the EQ-5D-5L and ICECAP-O for all participants, including participants who had capacity. The EQ-5D-5L used throughout the trial was amended (with permission from EuroQol Research Foundation) to remove the examples given in the ‘usual activities’ item (‘e.g. work, study, housework, family or leisure activities’), as these were not applicable to the trial population.
With the appropriate documented consent/advice, plus assent at the time of sampling, a saliva swab, stool sample and blood sample were obtained from the participant.
Data collection and samples for the baseline review were collected within 4 weeks of study enrolment, except in cases where data collection was delayed or on hold because of participant illness or hospitalisation.
Randomisation procedure and study product allocation
Care home residents were individually randomised in a 1 : 1 ratio using minimisation. To achieve a balance of probiotic and placebo allocations in care homes, the care home from which a CHR was recruited and the sex of the CHR were used as minimisation variables. The Sortition (Oxford University Innovation Ltd, Oxford, UK) system, developed by the Primary Care Clinical Trials Unit, University of Oxford, was used to assign the CHR allocations.
A list of SP pack identifiers (one per month’s supply of SP) was uploaded to the Sortition system, where the SP identifiers were randomly allocated to probiotic or placebo using random permuted blocks of size 6.
The randomisation procedure, and subsequent SP allocation, was undertaken by the CTR. An automated e-mail was sent to the trial team when data collection for the baseline review was complete. The automated e-mail notification prompted the trial team to randomise the participant. The Sortition system was used to assign the participant to the probiotic or placebo arm by generating the appropriate SP pack identifier. The Sortition system was programmed to initially allocate 2 months’ worth of SP at the time of randomisation. From then on, an automated e-mail notification was sent to the trial team on a monthly basis as a reminder to allocate a subsequent SP pack identifier.
The SP was stored according to the manufacturer’s requirements (i.e. Chr. Hansen A/S) at the CTR. Pack identifiers generated during the randomisation and subsequent SP allocation were matched up with the correct SP packs. A quality control process was carried out to ensure that the SP pack corresponded to the correct pack identifier and participant. The SP was then sent from the CTR to the RNs’ places of work, for them to deliver to the care home.
The only people who had access to the randomisation code were a study-independent statistician and those in the Quality Assurance and Pharmacovigilance Team within the CTR, an independent staff member who developed the Sortition software (based at the Primary Care Clinical Trials Unit, University of Oxford) and the person who labelled the SP at Nottingham University Hospitals NHS Trust. For any unblinding needed during the trial, and at the end of the trial for revealing the allocation, an independent statistician from the CTR (and the CTR Quality Assurance and Pharmacovigilance Team as back-up) was trained how to do this. All members of the PRINCESS trial team, including trial statisticians, were blinded until the database had been locked and the statistical report (with trial arms labelled A and B) delivered.
Follow-up data collection
All baseline and outcome assessments were conducted by people blind to study allocation. Participants were visited each week by the study RNs from the date of randomisation for data collection.
Weekly data collection
Data were collected weekly for up to 52 weeks following randomisation. Participants were reviewed weekly for signs of infection, incidence of diarrhoea, antibiotic use, hospitalisations and AEs, and changes to capacity status. Diarrhoea was defined as ‘the abnormal passing of loose or liquid stools, with increased frequency and/or increased volume’ [National Institute for Health and Care Excellence (NICE) clinical knowledge summaries. 66 © NICE 2018. Diarrhoea – Adult’s Assessment. 2018. Available from URL: https://cks.nice.org.uk/topics/diarrhoea-adults-assessment/. All rights reserved. Subject to Notice of rights.]. The SP dose (full or partial) and method of ingestion (capsule/in food/in liquid/unknown) were also recorded once the participant had commenced taking the SP.
Follow-up reviews
The RNs conducted two follow-up reviews, one at 3 months and a second between 6 and 12 months after the date of randomisation. The time window for collection of 3-month review data was 3 months –14/+28 days, and the window for the 12-month review was 11 months ± 1 month. Owing to the limited time available for CHR follow-up, data for the second review for a minority of participants were collected between 6 and 11 months after randomisation. Every effort was made by the RNs and the trial team to maximise the time between the 3-month review and the second review.
Biological samples
During the enrolment process, consent (or consultee advice) to collect blood, saliva and stool samples was sought. Stool samples were cultured to assess accuracy of randomisation and confirm adherence to the probiotic combination. Two mechanistic substudies requiring blood samples were embedded within the main trial: an immunology substudy and an influenza vaccine substudy. Separate consent was sought for the influenza vaccine substudy (on the main consent form). All samples were voluntary, and the participant could still take part in the trial if they did not wish to have any samples taken.
Blood samples were collected during the baseline review. Participants who were part of the immunology substudy were asked for a repeat blood sample at the 12-month (or final) review. Blood samples for the influenza substudy were scheduled around the participant’s influenza vaccine. Saliva and stool samples were collected during the baseline review and at the 3- and 12-month (or truncated) reviews. Samples taken during the baseline review were taken before randomisation and SP initiation.
Results from sample investigations were not used for routine health monitoring.
Blood samples
Baseline blood sampling and immunology substudy
Blood samples were taken by the RN during the baseline review if consent (or consultee advice) had been obtained for blood samples. The first 155 blood samples collected by the RN from these participants were included in an immunology substudy. A follow-up blood sample taken during the 12-month (or final) review was required only if participants had provided a blood sample at baseline that was included in the immunology substudy.
The procedure for all blood sampling in the trial adhered to the World Health Organization’s Guidelines on Drawing Blood: Best Practices in Phlebotomy67 by trained RNs who were sensitive to the needs of CHRs. In the event of difficulty in drawing blood, the procedure was repeated to a maximum of three times, provided the participant assented to this and did not show any signs of appearing to object. In accordance with the Mental Capacity Act 2005,58 no sampling or procedures were conducted in the event of any indication of distress or discomfort from CHRs.
For participants in the immunology substudy, blood was collected in one ethylenediaminetetraacetic acid (EDTA) (purple) tube and one heparin (green) tube. For those CHRs not in the substudy, blood was collected in one EDTA (purple) tube and one serum separator (gold) tube.
Influenza vaccine substudy
Two blood samples, one pre vaccination and another post vaccination, were required for the influenza vaccine substudy. These were scheduled around the participant’s annual flu vaccination.
Two vaccines were available to use for adults, a trivalent and quadrivalent vaccine for the 2017/2018 flu season (northern hemisphere winter). The trivalent vaccines contained an A/Michigan/45/2015 (H1N1)pdm09-like virus, a A/Hong Kong/4801/2014 (H3N2)-like virus and a B/Brisbane/60/2008-like virus. The quadrivalent vaccines contained two influenza B viruses and contained the above three viruses and a B/Phuket/3073/2013-like virus. 68
Criteria required to participate
To participate in the vaccine substudy the participant had to have been taking the SP for at least 8 weeks prior to receiving their annual flu vaccination. If the participant met this criterion, the first of the two blood samples was taken by the RN immediately before (or up to 10 days prior to) the participant had their flu vaccination using one clot activator (red) tube to collect the sample. The second sample was taken by the RN between 21 and 42 days after the participant had received their vaccination using one clot activator (red) tube to collect the sample.
Laboratory assessments
All blood samples were placed in SpeciSafe® (Alpha Laboratories, Eastleigh, UK) packaging, along with the relevant requisition form, into prelabelled envelopes, and posted by the RN using the Royal Mail (Royal Mail Group plc, London, UK) next-day delivery service to the Department of Human Development and Health, Faculty of Medicine, University of Southampton, Southampton, UK. Blood samples in the heparin tube for the immunology substudy were processed by the laboratory based at this location for immune cell phenotypes, plasma cytokine and chemokine concentrations, cytokine and chemokine responses in whole-blood samples stimulated ex vivo by toll-like receptor (TLR) 2 and TLR 4 agonists, and leucocyte phagocytosis of Escherichia coli.
All other blood samples, including those from the flu vaccine substudy, were forwarded from the University of Southampton’s Department of Human Development and Health to Southampton General Hospital Pathology Care Group. The serum was extracted from the flu vaccine substudy samples, transferred to aliquots, frozen and forwarded to the laboratory at Public Health England (London, UK) to undertake pre- and post-vaccination haemagglutination inhibition assays (HAIs) and measure antibody titres.
The EDTA blood sample was processed at the Southampton General Hospital Pathology Care Group to determine levels of serum vitamin D and carry out a full blood count (FBC). A copy of the results of the FBC and serum vitamin D results was sent to the CTR for data input.
Saliva samples (baseline and 3- and 12-month reviews)
Saliva samples were collected during the baseline review and at the 3- and 12-month (or truncated) reviews. Levels of Candida species were investigated by semiquantitative analysis of oral rinse or saliva samples.
Procedure for obtaining saliva sample
The participant was asked to refrain from eating or drinking for at least 30 minutes prior to taking the saliva sample. The participant was instructed that saliva production could be helped by placing the tip of the tongue behind the front teeth. The preferred method of sampling was to provide the participant with a universal container and ask them to spit into it. At least one teaspoon of saliva was required. Sampling via oral swab was more appropriate if participants could not understand instructions on how to use a universal container. The oral swab was removed from the swab storage tube by twisting the cap and pulling out the integrated cap and oral swab. The gum line was swabbed for up to 2 minutes and, if possible, a sample was taken from any lesions or inflamed areas. The oral swab was inserted into the swab storage tube. Samples were placed in SpeciSafe packaging, with a sample requisition form, in a prelabelled envelope and posted via the Royal Mail next-day delivery service to the Specialist Antimicrobial Chemotherapy Unit, Public Health Wales Microbiology, Cardiff, UK.
Laboratory procedures
Processing saliva samples
Saliva samples were centrifuged at 1147 g for 10 minutes, then the supernatant was discarded. The sediment was resuspended in 1 ml of phosphate-buffered saline, and 50 µl was inoculated on to Brilliance™ Candida agar (Thermo Fisher Scientific Oxoid Ltd) and spread for single colonies. The plates were incubated in air at 30 °C for 48 hours.
Processing oral swabs
Oral swabs were inoculated on to Brilliance Candida agar (Thermo Fisher Scientific Oxoid Ltd) and spread for single colonies. The plates were incubated at 30 °C for 48 hours and colonies identified by MALDI-TOF.
Stool samples (baseline and 3- and 12-month reviews)
Stool samples were collected at baseline and at the 3- and 12-month (or truncated) reviews. Samples were analysed for evidence of the presence of C. difficile and clinical data were collected about symptomatic infection to investigate gastrointestinal colonisation with antimicrobial-resistant bacteria by culture and antibiotic sensitivity of Enterobacterales and VRE, to quantify the number of Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 in stools, to determine if the level of serum vitamin D at baseline correlated with colonisation of antimicrobial-resistant bacteria in faecal isolates by the level of serum vitamin D and antimicrobial-resistant colonisation in the stool sample.
Procedure for obtaining stool sample
The RN or trained care home staff member provided information to the participant as necessary, and reassurance as required. Hand hygiene was performed using soap and water/alcohol hand rub. Gloves and apron were worn. The participant was asked to defecate into a clinically clean bedpan or receiver, with assistance provided as necessary. If the patient had been incontinent, a sample could be obtained from bedlinen or pads, avoiding contamination with urine. Using a ‘Universal Container and spoon unit’, enough stool was scooped to fill approximately one-third of the container (usually > 10 ml). Participant hygiene was attended to as required. If the specimen was being obtained in a residential care home that did not have bedpans or appropriate receivers to catch the sample, cling film could be used as a ‘trap’ placed strategically over the toilet to gather it. Emphasis was given to ensuring good hand hygiene, disposal of waste and decontamination following the procedure. Samples were placed in SpeciSafe packaging, with a sample requisition form, in a prelabelled envelope and posted via the Royal Mail next day-delivery service to the Specialist Antimicrobial Chemotherapy Unit, Public Health Wales Microbiology, Cardiff, UK.
Laboratory procedures
Culture of faecal samples for antimicrobial-resistant and probiotic organisms
A small ball (5 mm in diameter) sample of faeces was used to inoculate 3 ml of saline, then 50 µl was spiral plated onto selective agar to isolate relevant bacteria. Non-selective media used were CBA and FAA. Selective media used were UTI to provide identification of Gram-negative bacteria and some Gram-positive bacteria; VRE to detect VRE; extended-spectrum beta-lactamase (ESBL) plates to detect Gram-negative bacteria resistant to third-generation cephalosporins; carbapenem resistance detection media [carbapenem-resistant Enterobacterales (CRE)] to detect Gram-negative bacteria resistant to carbapenemase (all media from Thermo Fisher Scientific Oxoid Ltd); and Lactobacillus selective agar and Bifidobacterium agar for probiotic detection (Becton, Dickinson Limited, Wokingham, UK). CBA, UTI, VRE, ESBL and CRE plates were incubated in air at 35 ± 1 °C for 18 hours. FAA plates were incubated at 35 ± 1 °C in anaerobic conditions for 18–24 hours. Lactobacillus selective agar plates were incubated at 35 ± 1 °C in carbon dioxide for 24–72 hours and Bifidobacterium agar plates were incubated anaerobically at 35 ± 1 °C for 24–72 hours. A quantitative count (i.e. CFU/ml) of all bacteria was performed and bacteria identified by a Bruker MALDI-TOF and up-to-date database/library. Any of the targeted antimicrobial-resistant bacteria (Gram-negative bacteria possessing resistance to third-generation cephalosporins or carbapenems or VRE) were confirmed using phenotypic and genotypic methods. Combination disc methods (ROSCO Diagnostica, Taastrup, Denmark) were used for the detection of ESBL-, AmpC beta-lactamase- and carbapenemase-producing bacteria. Minimum inhibitory concentrations were determined using agar dilution for clinically relevant antimicrobial agents, amoxicillin, co-amoxiclav, trimethoprim, nitrofurantoin and ciprofloxacin for Gram-negative bacteria and vancomycin for enterococci. Minimum inhibitory concentrations were interpreted using the European Committee on Antimicrobial Susceptibility Testing guidelines version 9. 69
Culture of faeces samples of Clostridium difficile
Stool samples were investigated for C. difficile by suspending a small ball (5 mm in diameter) sample of faeces in 70% methanol then culturing on cycloserine–cefoxitin egg yolk agar (Thermo Fisher Scientific Oxoid Ltd) at 35 ± 1 °C for 72 hours under anaerobic conditions. Colonies were identified and confirmed as C. difficile using colony morphology, fluorescence under long-wave ultraviolet light and MALDI-TOF.
Outcome measures
A table of all objectives and outcome measures is shown in Table 2.
| Objective and outcome measure domain | Objectives | Outcome measures | Time point(s)a of evaluation of this outcome measure |
|---|---|---|---|
| Primary | To evaluate the effect of a daily dose of oral probiotics on CAAD for all-cause, acute infections | CAAD for all-cause infections; total number of days of systemic antibiotic administration as recorded in care home medical records and discharge summaries if the participant is admitted to hospital | Over a 12-montha period (data recorded at regular intervals by RN from care home records) |
| Secondary | To determine the effect of daily probiotic intake | ||
| Infection |
On CAAD for the following subcategories of infection:On incidence and duration of diarrhoea in CHR who are being treated with oral antibiotics The site, incidence and duration of infection (RTI, UTI, skin infection, GI and unexplained fever) On prevalence of C. difficile infection (clinical and bacteriological evidence of C. difficile colonisation) |
Total number of days of antibiotic administration for each infection type as recorded in care home medical records (collect generic name and mode of delivery) (to be recorded as defined daily dose multiples) Number, site and duration (mean and cumulative) of infection Estimation of incidence and duration of all-cause diarrhoea in CHR when taking (and also not taking) oral antibiotic treatment Estimation of incidence and duration of AAD in CHR when taking oral antibiotic treatment Stool sample laboratory analysis |
Over a 12-montha period (data recorded at regular intervals by RN from care home records) Question asked by RN and recorded on weekly record sheet Question asked by RN and recorded on weekly record sheet Over a 12-montha period (data recorded at regular intervals by RN from care home records) |
| Health utility | On participant’s health utility | Self- and/or proxy-reported health-related quality-of-life measurement (EQ-5D-5L) | At baseline, 3 monthsa and up to 12a months |
| Well-being | On participants’ well-being | Self- and/or proxy-reported ICECAP-O | At baseline, 3 monthsa and up to 12a months |
| Hospitalisation | NHS service use | In relation to infections, number of hospital stays for all-cause hospitalisation (as recorded in care home records and discharge summaries) | In a 12-montha period (data recorded at regular intervals by RN from care home records) |
| Mortality | Mortality rates | Total number of deaths of trial participants (collected from care home records) | In a 12-montha period (data recorded at regular intervals by RN from care home records) |
| AMR | Gastrointestinal colonisation with antimicrobial-resistant bacteria | Culture and antibiotic sensitivity of Gram-negative Enterobacterales and VRE from stool samples | Samples taken at baseline, 3a and 12a months |
| Oral microbiology | Levels of Candida species from oral samples | Semiquantitative analysis of oral rinse or saliva samples | Samples taken at baseline, 3 monthsa and up to 12a months |
| Influenza vaccine response (for those who have received influenza vaccine only) | Influenza vaccine efficacy | Blood sample: HAI and antibody titres | Sample taken on the day of (or up to 10 days prior to) and 4 weeks (28 days) post influenza vaccination (trial participants need to have been on SP for at least 2 months prior to influenza vaccination to take part in this substudy) |
| Immunology (ex vivo responses to pathogenic challenge) (n ≈ 100) |
Participant’s cytokine and chemokine response in whole-blood samples stimulated ex vivo by TLR 2 and TLR 4 agonists Participant’s plasma cytokines and chemokines Participant’s monocyte and neutrophil phagocytosis of E. coli |
Response measured by laboratory analysis of whole blood (stimulated ex vivo by TLR 2 and TLR 4 agonists) Measurement of plasma cytokines and chemokines in plasma and whole blood Measurement of monocyte and neutrophil phagocytosis of E. coli |
Baseline and up to 12a months Baseline and up to 12a months Baseline and up to 12a months |
| Gut microbiology | Quantify the amount of probiotic in stool samples | Investigative work to analyse level of Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 from stool sample | Samples taken at baseline, 3 monthsa and up to 12a months |
| Haematology and biochemistry | Haematology; biochemistry | FBC including immune phenotyping; vitamin D level measurement | FBC and vitamin D level measurement at baseline (all participants); FBC on n ≈ 100 (immunology participants only) and up to 12 months |
| Tertiary | To determine if the level of serum vitamin D at baseline correlates with colonisation of antimicrobial-resistant bacteria in faecal isolates | Level of serum vitamin D and antimicrobial-resistant colonisation in faecal sample | Baseline |
| Additional baseline measurements | Demographic information (to include age); Clinical Frailty Scale score; use of proton pump inhibitors, laxatives and dose of vitamin D | Baseline | |
| Other | Record whether or not trial participant refused to give blood/saliva/stool sample | Over 12a months from CHR record | |
| Ask care home staff what arm they think the trial participant is in | 12-month CRF | ||
| Record most common method of delivery of SP (e.g. swallowed with water or sprinkled on food/drink) | Over 12a months from medication administration record sheet | ||
| Qualitative research | To understand how the PRINCESS trial was implemented | Semistructured interviews with key personnel involved in the trial | After follow-up was completed |
Primary outcome
The trial primary outcome measure was CAAD for all-cause infections. This was ascertained from the total number of days of systemic antibiotic administration as recorded in care home medical records and discharge summaries if the participant was admitted to hospital, collected retrospectively by the RNs during weekly visits to CHRs.
Secondary outcomes
Secondary outcomes were investigated, under the following headings.
Infection
-
Total number of days of antibiotic administration for each infection type, as recorded in care home medical records (collected by RNs).
-
Number, site and duration (mean and cumulative) of infection, as recorded in care home medical records (collected by RNs).
-
Estimation of incidence and duration of diarrhoea when taking (and not taking) oral antibiotic treatment, and AAD (question asked by RNs). [Diarrhoea was defined as ‘the abnormal passing of loose or liquid stools, with increased frequency and/or increased volume’ (NICE clinical knowledge summaries).]
-
Stool sample laboratory analysis –
-
prevalence of C. difficile infection
-
culture and antibiotic sensitivity of Gram-negative Enterobacterales and VRE
-
levels of Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12.
-
Oral microbiology
-
Semiquantitative analysis of oral rinse or saliva samples for Candida spp.
Health and well-being
-
Self- and/or proxy-reported standardised measure of health utility (assessed using the EQ-5D-5L). Scale ranged from –0.28 to 1, where higher scores indicate a better health state.
-
Self- and/or proxy-reported health status (assessed using the EQ-5D-5L). Scale ranged from 0 to 100, where higher scores indicate better health status.
-
Self- and/or proxy-reported ICECAP-O. Scale ranged from 0 to 100 points, where higher scores indicate better well-being.
Hospitalisations
-
Number and duration of hospital stays for all-cause hospitalisation (as recorded in care home records and discharge summaries, collected by RNs).
Mortality
-
Deaths (from care home records, collected by RNs).
Mechanistic immunology outcomes
-
Influenza vaccine efficacy (HAI and antibody titres).
-
FBC.
-
Immune cell phenotypes, plasma cytokine and chemokine concentrations, cytokine and chemokine responses in whole-blood samples stimulated ex vivo by TLR 2 and TLR 4 agonists, and leucocyte phagocytosis of E. coli.
Tertiary outcomes/aims
-
To determine if the level of serum vitamin D correlated with colonisation of antimicrobial-resistant bacteria in faecal isolates.
-
Qualitative substudy to understand how the PRINCESS trial was implemented in order to identify opportunities for improving processes that might be relevant for future, similar studies.
Collection of adverse events
An extensive review of published literature and product dossiers provided by Chr. Hansen A/S, the SP manufacturer, led to the conclusion that probiotics, including the strains used in the PRINCESS trial, are generally well tolerated by elderly people and the risks to CHRs associated with the probiotics used in the PRINCESS trial were very low. A review by Doron and Snydman70 summarised the risks. Infections (e.g. septicaemia) caused by the probiotic bacteria had not been reported in trials, and theoretical risks of gene transfer of antibiotic resistance and overstimulation of the immune response had not been reported. Although deleterious metabolic activities (e.g. bowel ischaemia and D-lactic acidosis in patients with pancreatitis) had been reported, the mode of probiotic delivery was different from that in the PRINCESS trial. Therefore, if any of the above events were to occur in the PRINCESS trial, they would have been unexpected.
Minor gastrointestinal symptoms, such as abdominal cramping, nausea, soft stools, flatulence (passing wind) and taste disturbance, had been reported. It is also possible that a minor allergic reaction could have occurred. If these events were to have occurred, they would be classed as expected AEs, but would be classed as unexpected if they fulfilled the definition of serious. Therefore, the patient information sheet stated that side effects from probiotics are very rare and have never been serious, but that flatulence and bloating have been reported to occur in a minority of people.
The study population was expected to have a vast number of health events in the normal course of their care at their stage of life. Given the potential frailty of the trial population and the high incidence of hospitalisation and death in the course of routine care, we aimed to not cloud any true emerging safety profile by collection of unrelated data; the AE reporting procedure was developed to reflect this. AEs that were both (1) not serious and (2) unlikely to be related to the SP or study procedures (e.g. when a participant had a minor fall that did not require hospitalisation) were not recorded.
Non-serious AEs that were, or could be, related to either the SP or the study procedures were recorded on the weekly record form and weekly further information CRF. The AE was described in a free-text box on the CRF.
Serious adverse events (SAEs) were recorded on the weekly record in the further information section of the CRF. This required the RN who was completing the form to consider if any SAEs were related to the study intervention or study procedures and subsequently indicated this using the causality classification (definitely, probably, possibly, unlikely, not related). All SAEs were discussed (in person, by telephone or by e-mail) by the RN with a second reviewer (another PRINCESS trial RN or a PRINCESS trial clinician involved in the study and trained to do this task) to confirm the causality classification (definitely, probably, possibly, unlikely, not related). Details of the second reviewer, date and classification were recorded on the PRINCESS trial weekly record in the further information section of the CRF. Where there was a difference in classification between the two reviewers, the highest category of causality (most likely to be related) was selected. If the main and second reviewer classified the AE as definitely or probably related to study participation or study intervention, a serious adverse reaction (SAR) form was completed. This SAR form was then passed to a clinical reviewer, usually the trial’s chief investigator, who then allocated expectedness. If the SAE was classified as unexpected (and definitely or probably related to study procedure or study intervention), this would require further reporting to the sponsor, the REC and the Independent Data Monitoring Committee. Unblinding would occur if it was classified as related to the study intervention.
If the CHR died, the cause of death was sought. If the CHR was admitted to hospital, confirmation of infection and any routine microbiology results were sought. SAEs that were deemed to be not related to the SP or procedures did not require any further action and were managed in accordance with the care home’s routine procedures. These SAEs were reported annually in the safety report to the Independent Data Monitoring Committee.
Statistical methods
Sample size
The original target for the PRINCESS trial was to randomise 330 participants from around 20 care homes in Wales and England for 90% power at the 5% level to demonstrate a 10% relative reduction in CAAD (assuming an average CAAD of 17.4 days and an absolute reduction in the probiotic combination arm to 15.6 days per resident-year). We considered that a 10% reduction was feasible and would be clinically important.
This sample size accounted for 30% of participants being lost to follow-up as a result of withdrawal or death during the study (i.e. 365 days of data from 231 CHRs, and, therefore, an average of 256 days across all 330 randomised CHRs).
An interim assessment conducted after 3 months of recruitment (33 participants) to determine if we met the stop–go contractual criteria of primary outcome ascertainment revealed that the mean percentage of days for which there were valid antibiotic administration data (i.e. either no antibiotics administered or the number of days on which antibiotics had been administered in each week) was 77.5%, or, on average, 283 days out of a possible 365 days. This percentage varied depending on the length of time participants had been in the study; however, it remained high for participants who had been in the study for > 6 months.
Given the slower than anticipated recruitment, and this new information regarding the trade-off between numbers of participants required and the average length of follow-up, we aimed to randomise between 258 and 270 participants. Assuming a mean number of days for which primary outcome data could be available (i.e. accounting for follow-up time and missing data) of approximately 250 days, this would provide at least 82% power to detect a 10% relative reduction in CAAD.
Descriptive data
Care home residents’ characteristics and clinical measures were summarised using frequencies and percentages, means and standard deviations (SDs), or medians and interquartile ranges (IQRs), as appropriate. All analyses have been presented as estimates of treatment effects [adjusted incidence rate ratio (IRR), mean differences or ORs, as appropriate], with associated 95% CIs and p-values.
Analysis populations
All primary and secondary comparative analyses were based primarily on the intention-to-treat population, which included all randomised participants who provided outcome data, without imputation of missing values and regardless of protocol deviations or intervention received.
We conducted several sensitivity analyses for our primary outcome analysis, including all CHRs who initiated treatment, with missing diary data imputed under four different scenarios:
-
worst-case scenario: on all missing diary days, CHRs were assumed to be taking an antibiotic
-
best-case scenario: on all missing diary days, CHRs were assumed to not be taking an antibiotic
-
extreme-case scenario: to investigate the worst-possible scenario for our experimental arm, we assumed that all CHRs randomised to placebo had not taken antibiotics on missing diary days and that all CHRs randomised to probiotic combination had taken antibiotics on missing diary days
-
hypothetical scenario incorporating reason for death: in this scenario, we assumed that any CHR who died because of infection had been taking antibiotics on all missing diary days, with the remainder having their rate imputed for the remaining missing days (i.e. their rate on missing days was assumed to reflect their rate on observed days).
We also considered the extent to which CHRs not fully or partially taking SP on a given day may affect the conclusions drawn on the primary outcome. To investigate this, we modelled the CAAD rate as a continuous outcome and fitted a two-stage least squares instrumental variable regression model to CAAD, with randomisation used as the instrument and the exposure being the percentage of SP fully or partially taken (with this set to zero in the placebo arm). We fitted the model adjusting for CHR sex and using cluster-robust standard errors (SEs) to account for residents in care homes. The interpretation from the model coefficients is the adjusted mean difference in CAAD per percentage point increase in the percentage of SP fully/partially taken. For presentation purposes, we multiplied this coefficient (and associated 95% CI) by 100 to estimate the effect of probiotic combination under the scenario whereby participants took their SP fully/partially 100% of the time.
Primary outcome analysis
The mean CAAD per resident-year was compared between trial arms by fitting a two-level negative binomial regression model, accounting for participants nested within care homes, the length of time observed and the sex of the CHR.
Secondary outcome analyses
Similar to the analysis of the primary outcome, the majority of secondary outcome analyses (CAAD by infection type, rates of infections, rates of diarrhoea) involved the between-arm comparison of rate variables using two-level Poisson or negative binomial regression (depending on the presence of overdispersion). Where rates were low, single-level zero-inflated negative binomial regression models were fitted. Robust SEs were used to account for clustering of CHRs within care homes. All models were adjusted for CHR sex.
The mean duration of infection and mean duration of diarrhoea episodes were compared between arms by fitting a two-stage hurdle model, whereby the presence/absence of at least one episode was compared between arms by fitting a two-level logistic regression model (CHRs nested within care homes and CHR sex were included in the model) and, in those with at least one episode, the mean episode was compared between arms by fitting a two-level linear regression model.
The mean differences for the EQ-5D-5L and ICECAP-O measures were compared between arms by fitting two-level linear regression models, adjusting for CHR sex. Any transformations required to fulfil modelling assumptions are described in table footnotes.
The differences between arms in the proportion of CHRs with Enterobacterales present in stools, Enterobacterales present in stools resistant to at least one of the tested antibiotics, the presence of VRE in stools, candidiasis in saliva, and amount of candidiasis in saliva, at 3 months and at the second follow-up time point, were investigated by fitting two-level logistic regression models [two-level ordinal regression models for the amount of candidiasis (colony count) from oral samples], adjusting for CHR sex.
Subgroup analyses
We explored the extent to which there were any differential treatment effects on CAAD by several prespecified subgroups (CHR sex, capacity to provide informed consent for the trial at baseline and level of clinical frailty at baseline) by extending the primary analysis and fitting a subgroup by trial arm interaction.
Sensitivity analyses
We investigated the consistency of the conclusions drawn on our primary outcome by:
-
including prophylactic antibiotic use in our definition of CAAD
-
ignoring periods of hospitalisation from consideration from both the numerator and the denominator of our outcome.
Statistical software
All planned analyses were conducted using Stata® (version 13.0) (StataCorp LP, College Station, TX, USA) described in detail in a statistical analysis plan, which was finalised prior to database lock.
Secondary mechanistic outcomes
Previous research found a 40% prevalence of multidrug-resistant E. coli in faecal samples from UK nursing home residents36 and a 37% prevalence of Candida species in oral samples from hospitalised older patients. 26 A meta-analysis of six trials of probiotics in critically ill patients reported that probiotics reduced colonisation with multidrug-resistant Gram-negative organisms (OR 0.39, 95% CI 0.16 to 0.95). 7 Despite high prevalence of antimicrobial-resistant colonisation in CHRs, few studies have measured the effect of probiotics on this outcome. Hatakka et al. 71 found that probiotics reduced the risk of Candida species being detected in oral samples in 276 older people by 75% (OR 0.25, 95% CI 0.10 to 0.65). Stool and saliva samples at 6–12 months will provide 90% power at the 5% level to detect a 19% absolute reduction in antimicrobial-resistant bacteria and Candida species, assuming a 30% drop-out rate.
Chapter 3 Results of the randomised trial
Recruitment
We screened 1309 CHRs, of whom 332 were consented into the study. Consultee involvement was required for 220 residents (66.3% of those consented). Baseline data collection was completed for 318 CHRs (seven died and a further seven withdrew between consent and completion of baseline data collection), and a further eight were not randomised (following completion of baseline data collection but prior to randomisation, two CHRs died and six withdrew completely from the study). This is reported in the Consolidated Standards of Reporting Trials (CONSORT) trial flow diagram (Figure 1).
FIGURE 1.
A CONSORT flow diagram. a, Participants withdrew for clinical reasons; b, probiotic arm: four participants withdrew for clinical reasons, two moved care home and in one case the reason for withdrawal was unknown. Placebo arm: five withdrew for clinical reasons, one withdrew as a result of a participant/family request and one moved care home; c, completion of the second follow-up assessments occurred between 144 and 364 days post randomisation and depended on the date of randomisation. The median second review day is the same in both arms (placebo arm: median 319, IQR 219–356; probiotic arm: median 321, IQR 244.5–355.5). Fifty-three participants in each arm had at least one follow-up truncated; and d, probiotic arm: one participant withdrew for clinical reasons, two reasons were not known and one participant had moved care home. Placebo arm: five participants withdrew for clinical reasons, in one case the reason for withdrawal was not known and one participant had moved care home.

Baseline characteristics
Care home residents’ clinical characteristics
A total of 310 CHRs were randomised from 23 care homes between December 2016 and May 2018. The number of randomised CHRs per care home ranged from 2 to 30, with an average of 13.5 CHRs. Of those randomised, and with a coefficient of variation of 0.5, 155 were randomised to daily oral probiotic combination and 155 were randomised to daily oral placebo. The mean age was 85.3 (SD 7.39) years, 33.2% (103/310) were men and 65.8% (204/310) lacked capacity to consent. CHRs in both trial arms were well matched for these and other characteristics at baseline (Tables 3 and 4). See Appendix 5, Table 31, and Appendix 5, Figures 15–17, for baseline characteristic by care homes.
| Variable | Trial arm | Overall | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | |||||||||||
| Number of CHRs | Mean (SD) | Median (IQR) | Minimum to maximum | Number of CHRs | Mean (SD) | Median (IQR) | Minimum to maximum | Number of CHRs | Mean (SD) | Median (IQR) | Minimum to maximum | |
| Age (years) | 155 | 85.6 (7.21) | 87 (82.0–90.5) | 65 to 97 | 155 | 85.1 (7.58) | 86 (80.0–91.0) | 65 to 102 | 310 | 85.3 (7.39) | 86 (81.0–91.0) | 65 to 102 |
| Duration of residence in care home (years) | 154 | 1.8 (2.26) | 1 (0–3) | 0 to 11 | 153 | 1.6 (2.46) | 1 (0–2) | 0 to 15 | 307 | 1.7 (2.36) | 1 (0–2) | 0 to 15 |
| Self-reported overall health (EQ-5D-5L) score | 51 | 64.0 (21.20) | 60 (50–80) | 10 to 100 | 59 | 66.1 (21.30) | 65 (50–80) | 0 to 100 | 110 | 65.1 (21.18) | 62.5 (50–80) | 0 to 100 |
| Proxy-reported overall health (EQ-5D-5L) score | 154 | 66.2 (21.23) | 70 (50–85) | 15 to 100 | 151 | 65.8 (20.69) | 70 (50–80) | 0 to 100 | 305 | 66.0 (20.93) | 70 (50–80) | 0 to 100 |
| Self-reported health utility (EQ-5D-5L) score | 53 | 0.60 (0.244) | 0.63 (0.419–0.801) | 0.058 to 1 | 64 | 0.61 (0.275) | 0.64 (0.367–0.851) | –0.059 to 1 | 117 | 0.61 (0.260) | 0.63 (0.401–0.812) | –0.059 to 1 |
| Proxy-reported health utility (EQ-5D-5L) score | 152 | 0.50 (0.261) | 0.50 (0.282–0.724) | –0.027 to 1 | 152 | 0.53 (0.265) | 0.55 (0.313–0.758) | –0.213 to 1 | 304 | 0.51 (0.263) | 0.52 (0.284–0.745) | –0.213 to 1 |
| Self-report well-being (ICECAP-O) score (points) | 49 | 0.70 (2.000) | 0.70 (0.540–0.868) | 0.306 to 1 | 60 | 0.72 (0.218) | 0.74 (0.669–0.886) | 0.034 to 1 | 109 | 0.71 (0.210) | 0.72 (0.581–0.877) | 0.034 to 1 |
| Proxy-reported well-being (ICECAP-O) score (points) | 145 | 0.69 (0.200) | 0.72 (0.562–0.863) | 0 to 1 | 142 | 0.71 (0.185) | 0.74 (0.583–0.852) | 0 to 1 | 287 | 0.70 (0.193) | 0.74 (0.575–0.857) | 0 to 1 |
| Variable | Trial arm | Overall | ||||
|---|---|---|---|---|---|---|
| Placebo | Probiotic | |||||
| Frequency | % | Frequency | % | Frequency | % | |
| Sex | ||||||
| Male | 51 | 32.9 | 52 | 33.5 | 103 | 33.2 |
| Female | 104 | 67.1 | 103 | 66.5 | 207 | 66.8 |
| Capacity | ||||||
| Lacks | 106 | 68.4 | 98 | 63.2 | 204 | 65.8 |
| Has | 49 | 31.6 | 57 | 36.8 | 106 | 34.2 |
| Clinical Frailty Scale | ||||||
| Very fit | 2 | 1.3 | 2 | 1.3 | 4 | 1.3 |
| Well | 4 | 2.6 | 4 | 2.6 | 8 | 2.6 |
| Managing well | 12 | 7.7 | 7 | 4.5 | 19 | 6.1 |
| Vulnerable | 5 | 3.2 | 6 | 3.9 | 11 | 3.5 |
| Mildly frail | 8 | 5.2 | 12 | 7.7 | 20 | 6.5 |
| Moderately frail | 38 | 24.5 | 46 | 29.7 | 84 | 27.1 |
| Severely frail | 84 | 54.2 | 74 | 47.7 | 158 | 51.0 |
| Very severely frail | 2 | 1.3 | 4 | 2.6 | 6 | 1.9 |
| Terminally ill | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| Prescribed in the last 4 weeks | ||||||
| Antimicrobials | 37 | 23.9 | 45 | 29.0 | 82 | 26.5 |
| Used in the last 4 weeks | ||||||
| Proton pump inhibitor | 52 | 33.5 | 61 | 39.4 | 113 | 36.5 |
| Laxative | 85 | 54.8 | 75 | 48.4 | 160 | 51.6 |
| Vitamin D | 44 | 28.4 | 50 | 32.3 | 94 | 30.3 |
| Growth on selective agar plates | ||||||
| UTI | 75 | 100.0 | 83 | 100.0 | 158 | 100.0 |
| ESBL | 26 | 34.7 | 36 | 43.4 | 62 | 39.2 |
| CRO | 6 | 8.0 | 8 | 9.6 | 14 | 8.9 |
| VRE | 1 | 1.3 | 2 | 2.4 | 3 | 1.9 |
| Growth on non-selective agar plates | ||||||
| FAA | 75 | 100.0 | 83 | 100.0 | 158 | 100.0 |
| CBA | 75 | 100.0 | 83 | 100.0 | 158 | 100.0 |
| Growth of probiotic combination organisms | ||||||
| Lactobacillus spp. | 58 | 77.3 | 67 | 80.7 | 125 | 79.1 |
| L. rhamnosus | 19 | 32.8 | 28 | 41.8 | 47 | 37.6 |
| Bifidobacterium spp. | 74 | 98.7 | 82 | 98.8 | 156 | 98.7 |
| B. animalis subsp. lactis | 4 | 5.4 | 3 | 3.7 | 7 | 4.5 |
| Growth of other organisms | ||||||
| C. difficile | 0 | 0.0 | 6 | 7.2 | 6 | 3.8 |
| Candida spp. | 99 | 78.6 | 96 | 76.8 | 195 | 77.7 |
Baseline stool characteristics
In total, 159 baseline stool samples were received at the laboratory (51.3% of randomised participants), 76 from participants allocated to placebo (49% of participants in this group) and 83 from those allocated to probiotic combination (53.5% of participants in this group). Owing to the method of collecting stool samples in a care home, the RNs were not always able to be present at the collection of samples; therefore, the ‘Universal Container with spoon’ was often left in the care home for the care home staff to collect a sample at a convenient time. This may have contributed to the lower collection rate.
All but one of the stool samples received was processed (one specimen leaked in transit), and there was growth on the non-selective growth medium from all the samples processed. A total of 402 bacterial isolates were cultured from 158 stool samples (i.e. some stool samples contained more than one type of bacteria, e.g. E. coli and Klebsiella pneumoniae), with 189 cultured from 75 samples from participants in the placebo arm and 213 cultured from 83 samples from participants in the probiotic combination arm. The mean number of species cultured was 2.5 (SD 0.88) per sample in the placebo arm and 2.6 (SD 0.93) per sample in the probiotic combination arm. A total of 29 species were cultured (Figure 2).
FIGURE 2.
Bacterial species cultured from stool samples at baseline.
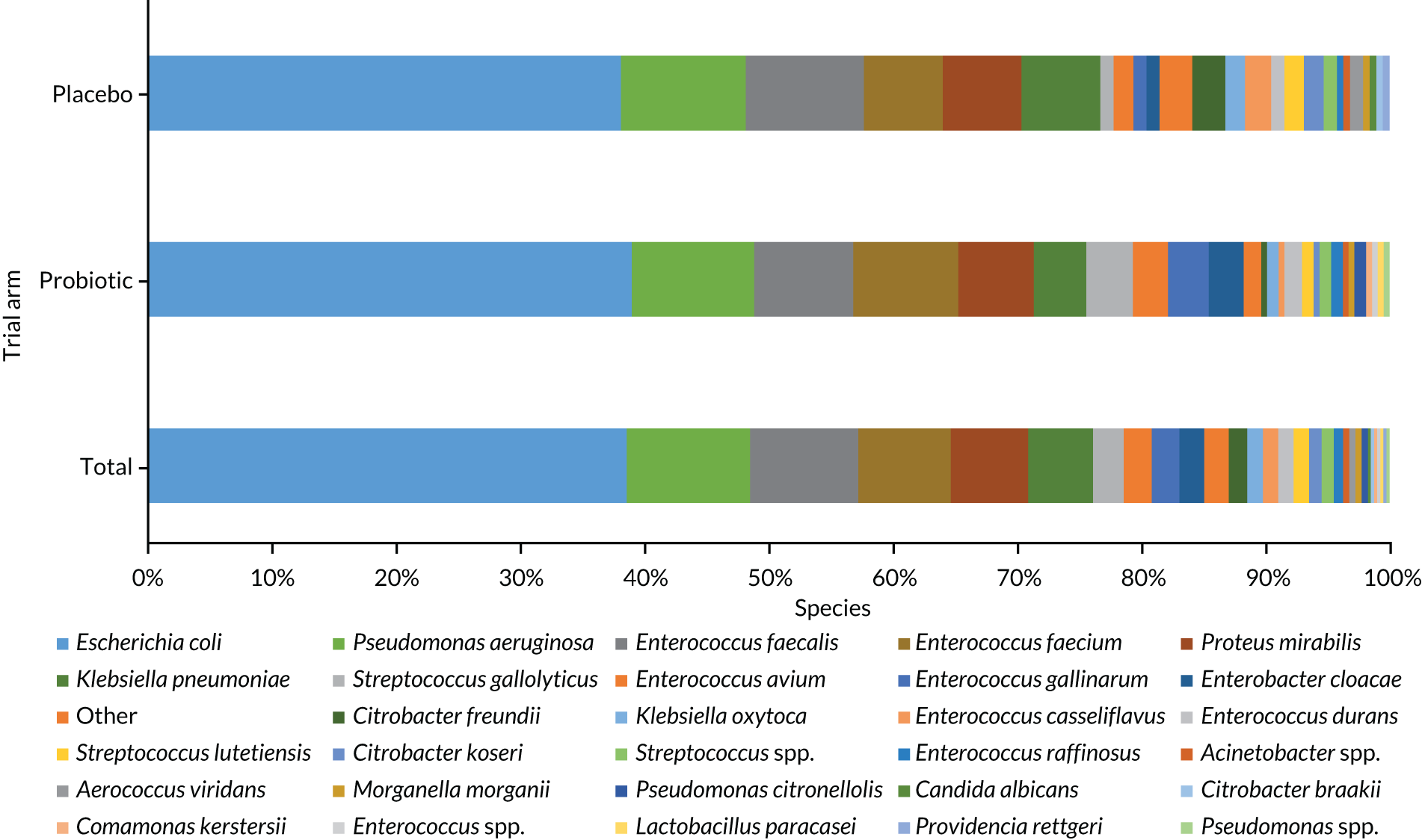
Enterobacterales isolates were cultured from 157 samples (99.4%), and resistance to at least one of the tested antimicrobials was found in 119 of these (75.8%), 57 (77.0%) from participants in the placebo arm and 62 (74.7%) from participants in the probiotic combination arm. AMR in E. coli isolated from baseline samples can be seen in Figure 3.
FIGURE 3.
Antimicrobial susceptibility of E. coli cultured from stool samples at baseline (n = 154).

Three samples were confirmed positive for VRE at baseline.
Sixty-eight samples were found to contain bacteria that grew on ESBL-selective media, suggesting resistance to third-generation cephalosporins. Thirty-one of these were confirmed to harbour an ESBL, with the rest harbouring a native AmpC enzyme conferring third-generation cephalosporin resistance. All ESBL-positive bacteria were identified as E. coli. These enzymes are predominantly carried on plasmids and so are able to transfer to different species; when causing infections, they precipitate treatment with a carbapenem.
Seven samples contained isolates that grew on CRE plates, suggesting carbapenem resistance, mainly Pseudomonas aeruginosa; however, no transferable resistance determinant was confirmed (i.e. carbapenemase).
Stool sample culture for study probiotic combination organisms
Culture of baseline stool samples on Lactobacillus selection agar led to bacterial growth in 125 out of 158 cases (79.1%), in 47 of which (37.6%) the growth of L. rhamnosus was confirmed. Eleven other Lactobacillus species were also confirmed, with the two most frequently recorded other Lactobacillus species (encompassing 80/124, 64.5%) being L. paracasei and L. gasseri. Forty-three samples had two Lactobacillus species and six had three Lactobacillus species.
Culture on Bifidobacterium selection agar led to bacterial growth in 156 out of 158 samples (98.7%). B. animalis subsp. lactis was confirmed in seven samples (7/156, 4.5%) using colony morphology and MALDI-TOF. Seven other Bifidobacterium species were identified, with the two most frequently recorded other Bifidobacterium species (encompassing 73/111, 65.8%) being B. longum and B. adolescentis. Twenty-three samples had two Bifidobacterium species and three had three Bifidobacterium species.
Six samples, from CHRs in different care homes, contained C. difficile (6/158, 3.8%).
Baseline saliva microbiology
Two-hundred and fifty-one saliva samples were processed, of which 195 grew on the Candida selection plate (195/251, 77.7%). The number of recorded organisms ranged from one to four, with more than two-thirds of samples growing one organism (135/195, 69.2%). The mean number of isolates cultured was 1.0 (SD 0.73) per sample in the placebo arm and 1.1 (SD 0.78) per sample in the probiotic combination arm. In total, 11 organisms were cultured (see Appendix 5, Figure 18).
Outcomes ascertainment
A total of 305 of the 310 randomised CHRs contributed to the primary analysis of CAAD for all-cause, acute infections (98.4%). A total of 305 CHRs contributed to the analysis of secondary outcomes relating to infections and diarrhoea (98.4%). The percentage of CHRs included in the secondary outcome analysis of the health-utility and well-being domains, which were collected at 3 months and at the second follow-up via questionnaires, ranged between 20.0% for the self-reported ICECAP-O at the second follow-up (62/310 CHRs) and 83.5% for proxy-reported EQ-5D-5L health utility at 3 months (259/310 CHRs).
Confirmation and distribution of study product
A total of 97.4% (302/310) of the randomised CHRs initiated at least one dose of SP: 98.1% (152/155) in the placebo arm and 96.8% (150/155) in the probiotic combination arm. Of the remaining eight CHRs, five withdrew following randomisation and three died soon after randomisation. Among the 302 CHRs who initiated at least one SP, the median percentage of taken SP either in full dose or in partial dose was 97.8% (IQR 93.56–99.45%), and 89.4% (65,525/77,772) of SPs were taken in capsule form (Tables 5 and 6).
| Variable | Trial arm | Overall | ||||
|---|---|---|---|---|---|---|
| Placebo | Probiotic | |||||
| Number of CHRs | Mean (SD) | Number of CHRs | Mean (SD) | Number of CHRs | Mean (SD) | |
| Antibiotic administration days | 153 | 7.5 (10.61) | 152 | 8.8 (10.87) | 305 | 8.2 (10.74) |
| Incidence of | ||||||
| Any infection | 153 | 2.4 (2.72) | 152 | 2.5 (2.51) | 305 | 2.4 (2.62) |
| UTI | 153 | 0.78 (1.40) | 152 | 0.78 (1.40) | 305 | 0.78 (1.40) |
| GI | 153 | 0.04 (0.20) | 152 | 0.03 (0.18) | 305 | 0.04 (0.19) |
| URTI | 153 | 0.45 (0.90) | 152 | 0.37 (0.75) | 305 | 0.4 (0.83) |
| LRTI | 153 | 0.5 (0.89) | 152 | 0.6 (1.03) | 305 | 0.6 (0.96) |
| Skin infection | 153 | 0.5 (1.10) | 152 | 0.6 (1.22) | 305 | 0.6 (1.16) |
| Cumulative number of infection days | 153 | 20.8 (40.66) | 152 | 21.8 (30.79) | 305 | 21.3 (36.0) |
| Mean duration of infection (days) | 153 | 4.0 (4.90) | 152 | 5.1 (5.03) | 305 | 4.6 (5.0) |
| CAAD | 153 | 12.0 (18.57) | 152 | 12.9 (18.36) | 305 | 12.4 (18.44) |
| Incidence of all-cause diarrhoea | 153 | 1.6 (3.46) | 152 | 1.8 (3.88) | 305 | 1.7 (3.67) |
| Cumulative days of all-cause diarrhoea | 153 | 4.4 (10.84) | 152 | 4.4 (10.16) | 305 | 4.4 (10.49) |
| Mean duration of all-cause diarrhoea episodes (days) | 153 | 0.5 (0.76) | 152 | 0.6 (0.80) | 305 | 0.6 (0.79) |
| Incidence of AAD | 153 | 0.6 (1.83) | 152 | 0.8 (1.97) | 305 | 0.7 (1.90) |
| Cumulative days of AAD | 153 | 21.1 (30.04) | 152 | 25.2 (37.42) | 305 | 23.4 (34.21) |
| Number of hospital stays | 153 | 0.3 (0.57) | 152 | 0.4 (0.70) | 305 | 0.32 (0.64) |
| Variable | Trial arm | Overall | ||||
|---|---|---|---|---|---|---|
| Placebo | Probiotic | |||||
| Frequency | % | Frequency | % | Frequency | % | |
| Initiated at least one | ||||||
| SP | 152 | 98.1 | 150 | 96.8 | 302 | 97.4 |
| Prescribed at least one | ||||||
| Antibiotic | 103 | 67.3 | 111 | 73.0 | 214 | 70.2 |
| Non-prophylactic antibiotic | 97 | 63.4 | 105 | 69.1 | 202 | 66.2 |
| Five most frequently prescribed non-prophylactic antibiotic | ||||||
| Amoxicillin | 72 | 25.1 | 78 | 23.2 | 150 | 24.1 |
| Nitrofurantoin | 65 | 22.6 | 50 | 14.9 | 115 | 18.5 |
| Trimethoprim | 25 | 8.7 | 42 | 12.5 | 67 | 10.8 |
| Flucloxacillin | 28 | 9.8 | 34 | 10.1 | 62 | 10.0 |
| Doxycycline | 26 | 9.1 | 33 | 9.8 | 59 | 9.5 |
| Othera | 71 | 24.7 | 99 | 29.5 | 170 | 27.3 |
| At least one of | ||||||
| Suspected infection | 102 | 66.7 | 111 | 73.0 | 213 | 69.8 |
| UTI | 62 | 40.5 | 57 | 37.5 | 119 | 39.0 |
| GI | 6 | 3.9 | 5 | 3.3 | 11 | 3.6 |
| URTI | 43 | 28.1 | 38 | 25.0 | 81 | 26.6 |
| LRTI | 48 | 31.4 | 54 | 35.5 | 102 | 33.4 |
| Skin infection | 37 | 24.2 | 46 | 31.3 | 83 | 27.2 |
| Other infection | 11 | 7.2 | 10 | 6.6 | 21 | 6.9 |
| All-cause diarrhoea | 61 | 39.9 | 64 | 42.1 | 125 | 41.0 |
| AAD | 32 | 20.9 | 41 | 27.0 | 73 | 23.9 |
| Ever hospitalised | 36 | 23.5 | 42 | 27.6 | 78 | 25.6 |
| Death | 32 | 20.6 | 33 | 21.3 | 65 | 21.0 |
Primary outcome analysis
Care home residents randomised to probiotic combination had a mean CAAD of 12.9 days (SE 1.49 days) and CHRs randomised to placebo had a mean CAAD of 12.0 days (SE 1.50 days). The adjusted CAAD rate in those allocated to probiotic combination compared with placebo was 1.13 days (adjusted IRR 1.13, 95% CI 0.79 to 1.63; p = 0.495) (Table 7).
| Outcome measure | Trial arm | Adjusted IRRa | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||
| Number of CHRs | Mean (SE) | Median (IQR) | Number of CHRs | Mean (SE) | Median (IQR) | ||||
| CAAD | 153 | 12.0 (1.50) | 4.8 (0–16.95) | 152 | 12.9 (1.49) | 7.0 (0–18.05) | 1.13 | 0.79 to 1.63 | 0.495 |
Secondary outcomes analyses
There was evidence that CHRs allocated to probiotic combination were administered more antibiotics for lower respiratory tract infections (LRTIs) (adjusted IRR 1.43, 95% CI 1.05 to 1.93; p = 0.022). We found no evidence of differences between trial arms in antibiotic use for UTIs, URTIs and skin infections (Table 8). No evidence of between-arm differences was found for duration of infection, the number of infection days (Table 9, and see Appendix 5, Figures 19 and 20) or any of our prespecified diarrhoea outcomes (see Appendix 5, Table 32, and Appendix 5, Figure 21).
| Outcome measure | Trial arm | Adjusted IRRa | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||
| Number of CHRs | Mean | SE | Number of CHRs | Mean | SE | ||||
| CAAD for | |||||||||
| UTIs | 153 | 6.7 | 1.10 | 152 | 7.1 | 1.22 | 1.17 | 0.75 to 1.84 | 0.479 |
| URTIs | 153 | 3.4 | 0.82 | 152 | 3.3 | 0.76 | 1.13 | 0.71 to 1.78 | 0.610 |
| LRTIs | 153 | 4.0 | 0.61 | 152 | 6.2 | 1.18 | 1.42 | 1.05 to 1.93 | 0.023 |
| Skin infections | 153 | 3.7 | 1.06 | 152 | 3.4 | 0.70 | 0.92 | 0.54 to 1.57 | 0.759 |
| Outcome measure | Trial arm | Adjusted IRRa | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||
| Number of CHRs | Mean | SE | Number of CHRs | Mean | SE | ||||
| Incidence of | |||||||||
| Any infection | 153 | 2.4 | 0.22 | 152 | 2.5 | 0.20 | 0.99 | 0.79 to 1.24 | 0.915 |
| UTIs | 153 | 0.8 | 0.11 | 152 | 0.8 | 0.11 | 1.14 | 0.62 to 2.09 | 0.675 |
| GIs | 153 | 0.04 | 0.02 | 152 | 0.03 | 0.01 | 0.78 | 0.24 to 2.57 | 0.683 |
| URTIs | 153 | 0.5 | 0.07 | 152 | 0.4 | 0.06 | 0.81 | 0.53 to 1.22 | 0.310 |
| LRTIs | 153 | 0.5 | 0.07 | 152 | 0.6 | 0.08 | 1.17 | 0.81 to 1.67 | 0.407 |
| Skin infections | 153 | 0.4 | 0.07 | 152 | 0.5 | 0.08 | 1.19 | 0.72 to 1.96 | 0.493 |
| Mean duration of infectionb | |||||||||
| At least one infection | 102 | 66.7% | 111 | 70.3% | 1.41c | 0.83 to 2.38 | 0.203 | ||
| Non-zero duration | 102 | 6.0 | 0.48 | 113 | 6.9 | 0.44 | 0.08 | –0.001 to 0.16 | 0.050 |
| Cumulative number of infection days | 153 | 20.8 | 3.29 | 152 | 21.8 | 2.50 | 1.08 | 0.77 to 1.51 | 0.674 |
There was a difference between trial arms in self-reported generic well-being/capability measured using the ICECAP-O scale at the 3-month follow-up, with CHRs allocated to the probiotic combination arm reporting an ICECAP-O value 0.06 points lower (i.e. worse well-being) than those allocated to the placebo arm (adjusted mean difference –0.06 points, 95% CI –0.11 to –0.001 points; p = 0.047). There was no evidence of any between-arm differences for other self- and proxy-reported well-being and quality-of-life outcomes (Tables 10 and 11).
| Outcome measure | Trial arm | Adjusted mean differencea | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||
| Number of CHRs | Mean | SE | Number of CHRs | Mean | SE | ||||
| EQ-5D-5L | |||||||||
| 3 months | |||||||||
| Index value | 43 | 0.64 | 0.03 | 49 | 0.60 | 0.04 | –0.05 | –0.12 to 0.01 | 0.126 |
| Health status | 42 | 65.1 | 3.17 | 44 | 65.0 | 2.73 | –0.26 | –7.97 to 7.45 | 0.947 |
| Second follow-up | |||||||||
| Index value | 31 | 0.61 | 0.05 | 38 | 0.61 | 0.06 | –0.01 | –0.10 to 0.09 | 0.915 |
| Health statusb | 29 | 65.7 | 3.99 | 34 | 65.2 | 3.67 | 24.39 | –1267.86 to 1316.63 | 0.970 |
| ICECAP-O | |||||||||
| 3 months | |||||||||
| Value (points) | 40 | 0.7 | 0.03 | 47 | 0.7 | 0.03 | –0.06 | –0.11 to –0.001 | 0.047 |
| Second follow-up | |||||||||
| Value (points) | 27 | 0.7 | 0.03 | 35 | 0.7 | 0.05 | –0.07 | –0.17 to 0.003 | 0.151 |
| Outcome measure | Trial arm | Adjusted mean differencea | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||
| Number of CHRs | Mean | SE | Number of CHRs | Mean | SE | ||||
| EQ-5D-5L | |||||||||
| 3 months | |||||||||
| Index value | 129 | 0.5 | 0.02 | 130 | 0.5 | 0.02 | –0.01 | –0.05 to 0.03 | 0.655 |
| Health status | 130 | 70.2 | 1.81 | 128 | 70.7 | 1.68 | 0.36 | –4.07 to 4.79 | 0.873 |
| Second follow-up | |||||||||
| Index value | 95 | 0.5 | 0.03 | 97 | 0.5 | 0.03 | 0.01 | –0.05 to 0.06 | 0.793 |
| Health status | 96 | 64.1 | 2.15 | 98 | 64.7 | 2.20 | 0.63 | –4.89 to 6.15 | 0.824 |
| ICECAP-O | |||||||||
| 3 months | |||||||||
| Value (points) | 118 | 0.7 | 0.02 | 117 | 0.7 | 0.02 | 0.004 | –0.04 to 0.04 | 0.852 |
| Second follow-up | |||||||||
| Value (points) | 90 | 0.7 | 0.02 | 84 | 0.7 | 0.02 | –0.01 | –0.05 to 0.03 | 0.688 |
Table 12 describes the findings for hospital stay and death. There was no evidence of any difference between trial arms in terms of CHRs being hospitalised at least once during the post-randomisation study period (adjusted OR 1.25, 95% CI 0.74 to 2.11; p = 0.407), number of hospital stays (adjusted IRR 1.17, 95% CI 0.72 to 1.90; p = 0.530), cumulative number of hospital days (adjusted IRR 1.00, 95% CI 0.43 to 2.29; p = 0.997), death (adjusted OR 1.03, 95% CI 0.59 to 1.80; p = 0.904) or time from randomisation to death (hazard ratio 0.99, 95% CI 0.67 to 1.46; p = 0.958).
| Outcome measure | Trial arm | Adjusted ORa | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||
| Number of CHRs | % | Number of CHRs | % | ||||||
| Ever been hospitalised | 36 | 23.5 | 42 | 27.6 | 1.25 | 0.74 to 2.11 | 0.407 | ||
| Death | 32 | 20.6 | 33 | 21.3 | 1.03 | 0.59 to 1.80 | 0.904 | ||
| Outcome measure | Number of CHRs | Mean | SE | Number of CHRs | Mean | SE | Adjusted IRR b | 95% CI | p-value |
| Hospital stays | |||||||||
| Number | 153 | 0.3 | 0.05 | 152 | 0.4 | 0.06 | 1.17 | 0.72 to 1.90 | 0.530 |
| Cumulative number of hospital days | 153 | 5.4 | 1.57 | 152 | 4.5 | 1.01 | 1.00 | 0.43 to 2.29 | 0.997 |
Secondary microbiology outcomes analyses
Stool analysis
At 3 months post randomisation, 108 stool samples were received at the laboratory (34.8% of randomised participants), 52 from CHRs allocated to placebo (33.5% of participants in this group) and 56 from those allocated to the probiotic combination (36.1% of participants in this group). All samples were processed and there was growth on the UTI selective agar plate in all instances.
A total of 288 isolates were cultured from 108 stool samples: 139 isolates were cultured from 52 samples from participants in the placebo arm and 149 isolates were cultured from 56 samples from participants in the probiotic combination arm. The mean number of isolates cultured per sample was 2.67 in the placebo arm and 2.66 in the probiotic arm. The five most common named isolates that were cultured, accounting for 208 (72.2%), were E. coli, P. aeruginosa, Enterococcus faecalis, E. faecium and K. pneumoniae. In total, 34 different organisms were cultured (Figure 4).
FIGURE 4.
Organisms cultured from stool samples at 3 months post randomisation.
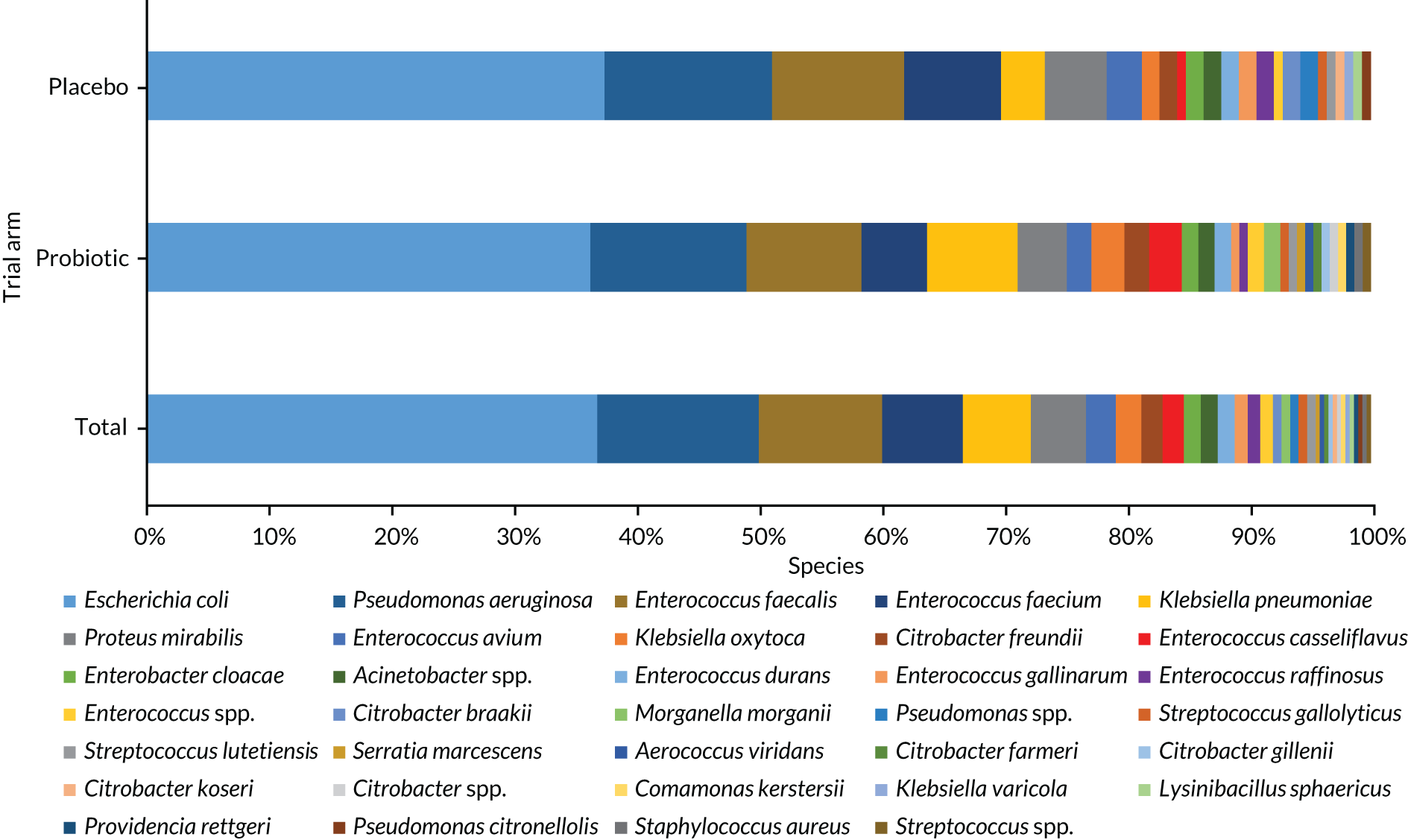
Enterobacterales isolates were cultured from 107 samples (99.1%), and resistance to at least one of the tested antimicrobials was found in 76 of these (71.0%), 39 from participants in the placebo arm (75.0% of participants in this group) and 37 from participants in the probiotic combination arm (67.3% of participants in this group). AMR in E. coli at 3 months can be seen in Figure 5.
FIGURE 5.
Antimicrobial susceptibility of E. coli cultured from stool samples at 3 months (n = 106).

Three samples were confirmed positive for the presence of VRE at 3 months.
At 3 months, 45 samples were found to contain isolates (mainly E. coli and K. pneumoniae) with resistance to third-generation cephalosporins, 16 of which harboured an ESBL-producing bacterium.
Six samples contained carbapenem-resistant isolates, mainly P. aeruginosa; however, no transferable resistance determinant was confirmed (i.e. carbapenemase gene).
At the second follow-up time point, 67 stool samples were received at the laboratory (21.6% of randomised participants), 30 from CHRs allocated to placebo (19.4% of participants in this group) and 37 from those allocated to probiotic combination (23.9% of participants in this group). All but one of the samples were processed (one specimen was too scant) and there was growth on the UTI selective agar plate in all samples processed.
A total of 180 isolates were cultured from 66 stool samples: 75 isolates were cultured from 29 samples from CHRs in the placebo arm and 105 isolates were cultured from 37 samples from CHRs in the probiotic combination arm. The mean number of isolates per sample was 2.59 in the placebo arm and 2.84 in the probiotic arm. The five most common named isolates that were cultured, accounting for 120 (66.7%) isolates were E. coli, E. faecalis, P. aeruginosa, E. faecium and Proteus mirabilis. In total, 30 species were cultured (Figure 6).
FIGURE 6.
Organisms cultured from stool samples at the second follow-up time point.

Enterobacterales were cultured from 65 samples (98.5%), of which 60 were tested for resistance to one or more antimicrobials. Antimicrobial-resistant Enterobacterales isolates were found in 42 (70.0%) samples, of which 19 (70.4%) were from CHRs allocated to placebo and 23 (69.7%) were from those allocated to the probiotic combination. AMR in E. coli isolates at the second follow-up time point can be seen in Figure 7.
FIGURE 7.
Antimicrobial susceptibility of E. coli cultured from stool samples at the second follow-up time point (n = 58).
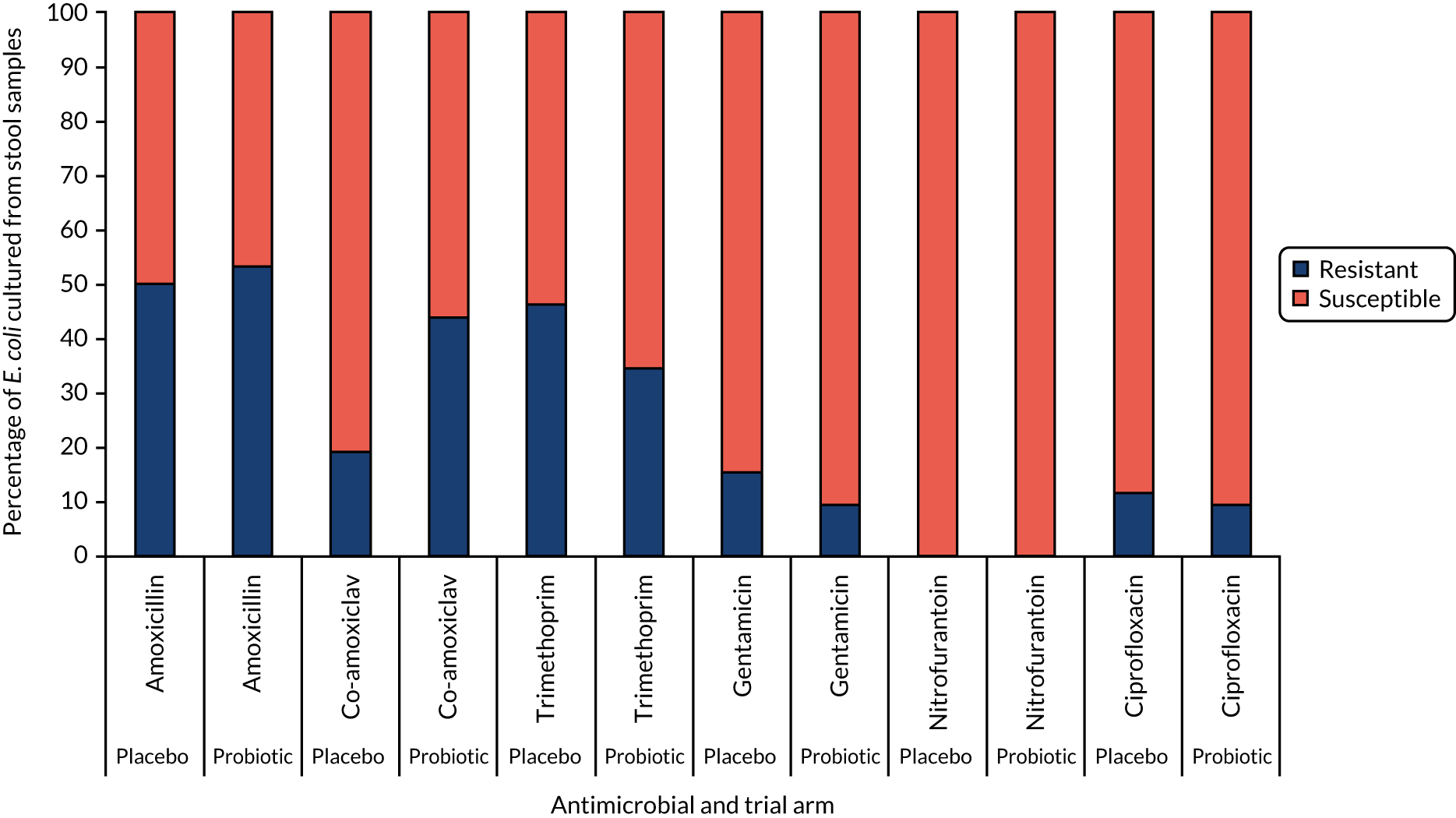
Similar to baseline and 3 months, three samples were confirmed positive for VRE at the second follow-up time point. Only one patient was positive for VRE at baseline, and at 3 and 12 months.
There was no evidence of differences between the trial arms in the presence of Enterobacterales isolates resistant to at least one of the tested antimicrobials at either 3 months or the second follow-up time point (Table 13).
| Outcome | Time point | Trial arm | Adjusted ORa | 95% CI | p-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||||
| Number of CHRs | Frequency | % | Number of CHRs | Frequency | % | Lower limit | Upper limit | ||||
| Enterobacterales in stools | 3 months | 52 | 52 | 100.0 | 55 | 56 | 98.2 | Not analysable | |||
| Second follow-up | 29 | 29 | 100.0 | 36 | 37 | 97.3 | Not analysable | ||||
| Enterobacterales in stools resistant to at least one of the tested antimicrobials | 3 months | 39 | 52 | 75.0 | 37 | 55 | 67.3 | 0.61 | 0.24 | 1.56 | 0.303 |
| Second follow-up | 19 | 27 | 70.0 | 23 | 33 | 69.7 | 0.76 | 0.20 | 2.89 | 0.683 | |
| VRE in stools | 3 months | 0 | 0 | 0.0 | 3 | 3 | 100.0 | Not analysable | |||
| Second follow-up | 0 | 0 | 0.0 | 3 | 3 | 100.0 | Not analysable | ||||
At 3 months post randomisation, 7 out of 107 samples tested (6.5%) were found to contain C. difficile, the majority from CHRs who were randomised to the probiotic combination [6/55 (10.9%) vs. 1/52 (1.9%) in the placebo trial arm; adjusted OR 6.51, 95% CI 0.75 to 56.57; p = 0.090]. At the second follow-up, 2 out of 64 (3.1%) of the samples tested contained C. difficile. Both samples were from CHRs randomised to the probiotic combination.
Stool culture for study probiotic combination organisms
Of the 125 samples that resulted in growth on the Lactobacillus selection agar (125/158, 79.1%), 58 were from CHRs randomised to placebo. The number of samples found to contain L. rhamnosus 3 months post randomisation was higher in the probiotic combination group than in the placebo group [47/56 (83.9%) vs. 19/52 (36.5%); adjusted OR 9.19, 95% CI 3.51 to 24.07; p < 0.001]. This finding persisted at the second follow-up time point [27/37 (73.0%) vs. 9/29 (31.0%); adjusted OR 6.41, 95% CI 2.14 to 19.20; p = 0.001].
Similarly, B. animalis subsp. lactis was cultured more frequently from samples from CHRs randomised to the probiotic combination than from those randomised to placebo at both 3 months [29/56 (51.8%) vs. 2/52 (3.8%); adjusted OR 26.90, 95% CI 5.94 to 121.66; p < 0.001] and the second follow-up time point [21/37 (56.8%) vs. 2/29 (6.9%); adjusted OR 21.96, 95% CI 2.97 to 162.43; p = 0.002]. Study probiotic combination organisms were cultured from stool samples from those CHRs randomised to the probiotic combination intervention in all study sites and were checked against random allocation codes after the database lock.
At both 3 months and the second follow-up time point, the mean colony counts (‘weight of growth’) for both L. rhamnosus and B. animalis subsp. lactis were higher in CHRs allocated to the probiotic combination arm than in those allocated to placebo (Tables 14 and 15). Colonisation with these probiotic organisms is not uncommon; therefore, we expected to see some of the placebo group with these organisms, but the numbers of patients in the placebo trial arm with positive colonisation were small.
| L. rhamnosus GG in stoolsa | Trial arm | Total | |
|---|---|---|---|
| Placebo | Probiotic | ||
| Time point | |||
| Baseline | |||
| Samples with no growth, n (%) | 56/75 (74.7) | 55/83 (66.3) | 111/158 (70.3) |
| Bacterial counts (CFU/ml), mean (SD) | 14,427 (52,666) | 20,020 (77,864) | 17,365 (66,946) |
| Bacterial counts (CFU/ml), median (IQR) | 0 (0–10) | 0 (0–270) | 0 (0–180) |
| 3 months | |||
| Samples with no growth, n (%) | 33/52 (63.5) | 9/56 (16.1) | 42/108 (38.9) |
| Bacterial counts (CFU/ml), mean (SD) | 46,729 (2,772,633.46) | 704,478 (3,048,311) | 387,784 (2,218,564) |
| Bacterial counts (CFU/ml), median (IQR) | 0 (0–2100) | 5550 (240–130,000) | 0 (680–26,000) |
| Second follow-up | |||
| Samples with no growth, n (%) | 20/29 (69.0) | 10/37 (27.0) | 30/66 (45.5) |
| Bacterial counts (CFU/ml), mean (SD) | 13,970 (43,139) | 151,527 (526,728) | 91,086 (39,899) |
| Bacterial counts (CFU/ml), median (IQR) | 0 (0–100) | 29,000 (0–100,000) | 240 (0–40,000) |
| B. animalis subsp. lactis in stoolsa | Trial arm | Total | |
|---|---|---|---|
| Placebo | Probiotic | ||
| Time point | |||
| Baseline | |||
| Samples with no growth, n (%) | 71/75 (97.7) | 80/83 (96.4) | 151/158 (95.6) |
| Bacterial counts (CFU/ml), mean (SD) | 17,800 (106,559) | 2922 (18,773) | 9984 (74,777) |
| Bacterial counts (CFU/ml), median (IQR) | 0 (0–0) | 0 (0–0) | 0 (0–0) |
| 3 months | |||
| Samples with no growth, n (%) | 50/52 (96.2) | 27/56 (48.2) | 77/108 (71.3) |
| Bacterial counts (CFU/ml), mean (SD) | 28,846 (170,738) | 1,717,750 (5,109,484) | 904,574 (3,761,923) |
| Bacterial counts (CFU/ml), median (IQR) | 0 (0–0) | 5000 (0–415,000) | 0 (0–27,500) |
| Second follow-up | |||
| Sample with no growth, n (%) | 27/29 (93.1) | 16/37 (43.2) | 43/66 (65.2) |
| Bacterial counts (CFU/ml), mean (SD) | 362 (1856) | 214,824 (444,510) | 120,591 (347,763) |
| Bacterial counts (CFU/ml), median (IQR) | 0 (0–0) | 2500 (0–180,000) | 0 (0–80,000) |
Saliva analysis
At 3 months post randomisation, 218 saliva samples were received at the laboratory (from 70.3% of randomised CHRs), 105 from participants allocated to placebo (67.7% of participants in this group) and 113 from those allocated to the probiotic combination (72.9% of participants in this group). All but two of the saliva samples received were processed (one snapped and consent was not given for the other).
A total of 235 isolates were cultured from 168 saliva samples: 119 isolates from cultured from 80 samples from participants in the placebo arm and 116 isolates were cultured from 88 samples from participants in the probiotic combination arm. The five most common named organisms that were cultured, accounting for 229 (97.4%) isolates, were C. albicans, C. glabrata, C. dubliniensis, C. tropicalis and C. parapsilosis. In total, nine different organisms were cultured (Figure 8).
FIGURE 8.
Candida species detected from saliva samples of CHRs at 3 months post randomisation.
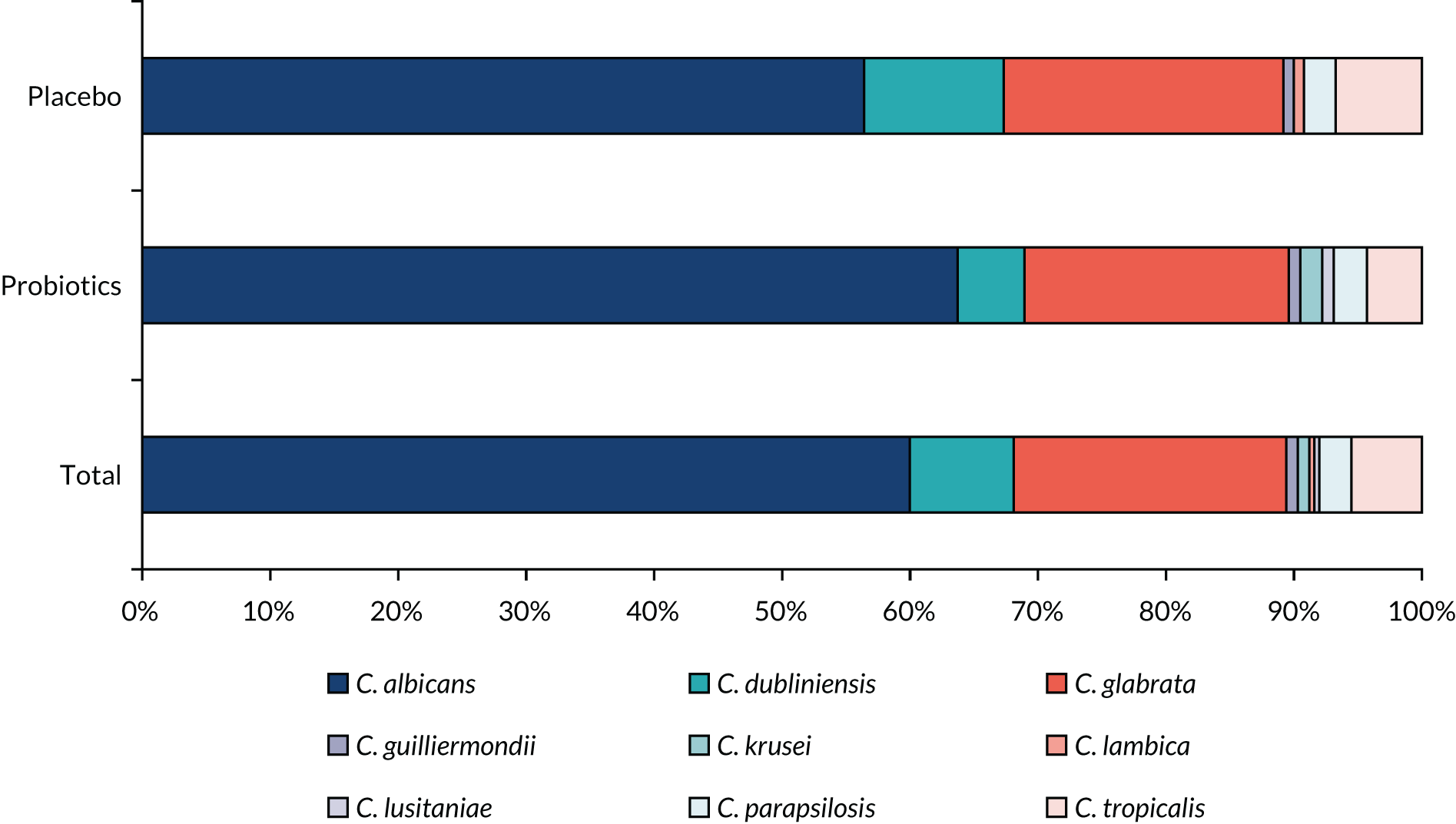
At the second follow-up time point, 161 saliva samples were received at the laboratory (from 51.9% of randomised participants), 76 from CHRs allocated to placebo (49.0% of participants in this group) and 85 from those allocated to the probiotic combination (54.8% of participants in this group). All saliva samples received were processed.
A total of 179 organisms were cultured from 127 saliva samples: 119 organisms were cultured from 57 samples from CHRs in the placebo arm and 116 organisms were cultured from 70 samples from CHRs in the probiotic combination arm. The four most common named organisms that were cultured, accounting for 172 (96.1%) of isolates, were C. albicans, C. glabrata, C. dubliniensis and C. tropicalis. In total, nine different organisms were cultured (see Appendix 5, Figure 22).
There was no evidence of any between-arm differences in terms of presence or amount of candidiasis from oral samples at either follow-up time point (Tables 16 and 17).
| Outcome | Trial arm | Adjusted ORa | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||
| Number of CHRs | Frequency | % | Number of CHRs | Frequency | % | ||||
| Presence of candidiasis from oral samples | |||||||||
| 3 months | 105 | 80 | 76.2 | 113 | 88 | 77.9 | 1.23 | 0.54 to 2.83 | 0.623 |
| Second follow-up | 76 | 57 | 75.0 | 85 | 70 | 82.4 | 1.27 | 0.50 to 3.21 | 0.620 |
| Outcome | Trial arm | Adjusted ORa | 95% CI | p-value | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||||
| Number of CHRs | Weight | Number of organisms | % | Number of CHRs | Weight | Number of organisms | % | ||||
| Amount of candidiasis present in saliva | |||||||||||
| 3 months | 80 | (–/+) | 20 | 16.8 | 88 | (–/+) | 21 | 18.1 | 0.66 | 0.20 to 2.17 | 0.489 |
| (+) | 19 | 16.0 | (+) | 26 | 22.4 | ||||||
| (++) | 38 | 31.9 | (++) | 20 | 17.2 | ||||||
| (+++) | 42 | 35.3 | (+++) | 49 | 42.2 | ||||||
| Second follow-up | 57 | (–/+) | 14 | 17.3 | 70 | (–/+) | 15 | 15.3 | 0.50 | 0.12 to 2.16 | 0.356 |
| (+) | 11 | 13.6 | (+) | 11 | 11.2 | ||||||
| (++) | 23 | 28.4 | (++) | 31 | 31.6 | ||||||
| (+++) | 33 | 40.7 | (+++) | 41 | 41.8 | ||||||
Level of serum vitamin D
Table 18 summarises the findings from the statistical analyses of the AMR at baseline. In total, 158 participants provided a blood sample for the vitamin D test, 14 samples were not processed as a result of an insufficient sample (n = 6), haemolysed specimen (n = 4) or delay/staffing issue (n = 4). Among the 144 participants with an analysable vitamin D sample, 77 also had resistance test data available. The mean serum vitamin D level was 21.2 ng/l higher in participants who provided stool samples containing an Enterobacterales isolate resistant to at least one of the tested antibiotics (95% CI of mean difference 0.03 to 42.43 ng/l). Similarly, the mean serum vitamin D levels were 19.0 ng/l higher in participants who provided samples containing bacteria of any kind that were resistant to at least one of the tested antibiotics (95% CI –2.93 to 40.92 ng/l).
| Outcome | Number of CHRs | Level of serum vitamin D (ng/l) | p-value | ||
|---|---|---|---|---|---|
| Mean (SE) | Mean difference | 95% CI | |||
| Enterobacterales isolates resistant to at least one of the tested antibiotics | |||||
| Not resistant | 23 | 45.6 (6.27) | 21.2 | 0.03 to 42.43 | 0.050 |
| Resistant | 54 | 66.6 (6.40) | |||
| Enterobacterales isolates resistant to third-generation cephalosporins or carbapenems | |||||
| Not resistant | 0 | 0.0 | Not analysable | ||
| Resistant | 17 | 62.7 (10.18) | |||
| Resistant to at least one of the tested antibiotics | |||||
| Not resistant | 21 | 46.8 (6.81) | 19.0 | –2.93 to 40.92 | 0.089 |
| Resistant | 56 | 65.4 (6.23) | |||
Sensitivity analysis
Including prophylactic antibiotic use in the definition of cumulative antibiotic administration days
The mean CAAD including prophylactic antibiotics was 12.7 (SE 1.51) days for CHRs allocated to the placebo arm and 14.6 (SE 1.62) days for CHRs allocated to the probiotic combination arm. The adjusted IRR was 18% (IRR 1.18, 95% CI 0.83 to 1.67) higher in CHRs allocated to the probiotic combination arm than in those allocated to the placebo arm. The null hypothesis was not rejected, and we can conclude that there was no evidence of any difference between trial arms in terms of CAAD including prophylactic antibiotics (Table 19).
| Outcome measure | Trial arm | Adjusted IRRa | 95% CI | p-value | |||
|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||
| Number of CHRs | Mean (SE) (days) | Number of CHRs | Mean (SE) (days) | ||||
| CAAD | 153 | 12.7 (1.51) | 152 | 14.6 (1.62) | 1.18 | 0.83 to 1.67 | 0.363 |
Removing periods of hospitalisation from the primary analysis definition
Ignoring periods of hospitalisation from both the numerator and the denominator of the primary outcome did not alter the conclusions drawn (Table 20).
| Outcome measure | Trial arm | Adjusted IRRa | 95% CI | p-value | |||
|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||
| Number of CHRs | Days, mean (SE) | Number of CHRs | Days, mean (SE) | ||||
| CAAD | 153 | 12.1 (1.69) | 152 | 11.9 (1.36) | 1.07 | 0.74 to 1.54 | 0.726 |
Additional analysis
Missing data adjustments
Of the 310 randomised CHRs, 305 contributed to the primary analysis, with the remaining five contributing no data. Among the 305 who contributed data up until their point of study exit (i.e. completed study, died or withdrew from the study), there were 1245 diary days missing from 30 CHRs in total (373 days from 17 CHRs in the placebo arm and 872 days from 13 CHRs in the probiotic combination arm). In total (including missing days due to withdrawal or death before study completion), there were 21,555 missing diary days from 109 CHRs (11,460 days from 57 CHRs in the placebo arm and 10,095 days from 52 CHRs in the probiotic combination arm). Overall, the main reason for missing diary days was death (15,229 missing days).
Care home residents with complete primary outcome data were more likely to have the capacity to consent and be classified as ‘very fit to managing well’ on the Clinical Frailty Scale (see Appendix 5, Table 33).
In total, 65 CHRs died during the study: 32 in the placebo arm (32/155, 20.6%) and 33 in the probiotic combination arm (33/155, 21.3%). The number of deaths that were infection related was higher in the probiotic combination arm than in the placebo arm (12/33, 26.4%, vs. 6/32, 18.8%) (Table 21).
| Reason for death | Trial arm | Total | ||||
|---|---|---|---|---|---|---|
| Placebo | Probiotic | |||||
| Number of CHRs | % | Number of CHRs | % | Number of CHRs | % | |
| Not infection related | 21 | 65.6 | 20 | 60.6 | 41 | 63.1 |
| Infection related | 6 | 18.8 | 12 | 26.4 | 18 | 27.7 |
| Unknown | 5 | 15.6 | 1 | 3.0 | 6 | 9.2 |
| Total | 32 | 33 | 65 | |||
Four sensitivity analyses were conducted to explore the impact of missing data on our conclusions:
-
worst-case scenario – on all missing diary days, CHRs were assumed to be taking an antibiotic
-
best-case scenario – on all missing diary days, CHRs were assumed to not be taking an antibiotic
-
extreme-case scenario – to investigate the worst possible scenario for our experimental arm, we assumed that all CHRs randomised to placebo had not taken antibiotics on missing diary days and that all CHRs randomised to probiotic combination had taken antibiotics on missing diary days
-
hypothetical scenario incorporating reason for death – in this scenario, we assumed that any CHR who died because of infection had been taking antibiotics on all missing diary days, with the remainder having their rate imputed for the remaining missing days (i.e. their rate on missing days was assumed to reflect their rate on observed days).
Table 22 demonstrates that for assumptions analyses 3 and 4 there is evidence to suggest that CHRs allocated to probiotic combination had a higher rate of CAAD than those allocated to placebo, with the most extreme-case scenario yielding the greatest difference.
| Assumption | Adjusted IRR | 95% CI | p-value |
|---|---|---|---|
| 1. Worst-case scenario | 0.94 | 0.60 to 1.47 | 0.780 |
| 2. Best-case scenario | 1.11 | 0.77 to 1.60 | 0.582 |
| 3. Extreme-case scenario | 5.17 | 3.38 to 7.89 | < 0.001 |
| 4. Hypothetical scenario incorporating reason for death | 1.62 | 1.03 to 2.57 | 0.039 |
Adherence adjusted analyses
Given the high levels of adherence to SP (median percentage of SP taken either in full dose or in partial dose was 97.8%, IQR 93.56–99.45%), there was minimal impact on our study findings when accounting for SP non-adherence [adjusted mean difference in CAAD at 100% fully or partially taken SP is 1.01 days higher in the probiotic combination arm, 95% CI –2.04 (lower) to 4.06 days (higher)].
Subgroup analysis
Table 23 provides model estimates for prespecified subgroup analyses conducted for the primary CAAD.
There was no evidence of a differential intervention effect for CHR sex or baseline capacity to consent to the trial or Clinical Frailty Scale score at baseline.
| Subgroup analysis (n = 305) | Variable | Adjusted IRRa | 95% CI | p-value |
|---|---|---|---|---|
| CHR sex | Placebo | Reference category for trial arm main effect (i.e. effect of trial arm for female subgroup) | 0.952 | |
| Probiotic | 1.01 | 0.65 to 1.59 | ||
| Female | Reference category for sex main effect (i.e. effect of female CHRs allocated to the placebo arm) | 0.763 | ||
| Male | 0.92 | 0.52 to 1.62 | ||
| Probiotic × female | Reference category for trial arm × sex | 0.406 | ||
| Probiotic × male | 1.40 | 0.63 to 3.08 | ||
| Baseline capacity to consent to the trial | Placebo | Reference category for trial arm main effect (i.e. effect of trial arm for lack of capacity subgroup) | 0.411 | |
| Probiotic | 1.21 | 0.77 to 1.91 | ||
| Lack of capacity | Reference category for capacity to consent to the trial (i.e. the effect of with capacity to consent for CHRs allocated to placebo arm) | 0.378 | ||
| With capacity | 1.29 | 0.73 to 2.28 | ||
| Probiotic × lack of capacity | Reference category for trial arm × capacity at consent | 0.636 | ||
| Probiotic × with capacity | 0.83 | 0.38 to 1.80 | ||
| Baseline Clinical Frailty Scale | Placebo | Reference category for trial arm main effect (i.e. effect of trial arm for severely frail to terminally ill subgroup) | 0.215 | |
| Probiotic | 1.38 | 0.83 to 2.85 | ||
| Severely frail to terminally ill | Reference category for Clinical Frailty Scale main effect (i.e. effect of severely frail to terminally ill CHRs allocated to the placebo arm) | 0.204 | ||
| Very fit to managing well | 0.50 | 0.20 to 1.23 | ||
| Vulnerable to moderately frail | 1.17 | 0.66 to 2.07 | ||
| Probiotic × severely frail to terminally ill | 0.312 | |||
| Probiotic × very fit to managing well | 1.24 | 0.34 to 4.55 | ||
| Probiotic × vulnerable to moderately frail | 0.57 | 0.26 to 1.25 | ||
Adverse events
Data about AEs were collected during the study period following randomisation. During this period, 283 events in 120 CHRs were recorded. A total of 60.8% of events were hospitalisations (172/283); in 22.9% of events, CHRs had died (65/283) (Table 24). Three AEs were trial related and occurred in the placebo arm: one participant experienced an increase in difficulty swallowing and the speech and language team recommended that the SP should be discontinued because of a choking risk; one participant experienced frequent episodes of diarrhoea that the care home staff felt had been worse since he started the SP; and one participant reported feeling bloated.
| AE | Trial arm | Total | ||||
|---|---|---|---|---|---|---|
| Placebo | Probiotic | |||||
| Number of CHRs | % | Number of CHRs | % | Number of CHRs | % | |
| At least one AE (percentage of all participants) | ||||||
| Yes | 59 | 38.1 | 61 | 39.4 | 120 | 38.7 |
| AE typea (percentage of AEs) | ||||||
| Resulted in death | 32 | 24.1 | 33 | 22.0 | 65 | 23.0 |
| Life-threatening | 11 | 8.3 | 12 | 8.0 | 23 | 8.1 |
| Hospitalisation | 78 | 58.6 | 94 | 62.7 | 172 | 60.8 |
| Persistent or significant disability or incapacity | 8 | 6.0 | 5 | 3.3 | 13 | 4.6 |
| Congenital anomaly or birth defect | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| Another medically important condition | 4 | 3.0 | 6 | 4.0 | 10 | 3.5 |
| Total | 133 | 150 | 283 | |||
| Trial-related event (percentage of those assessed for relatedness) | ||||||
| No | 105 | 97.2 | 124 | 99.2 | 229 | 98.3 |
| Yes | 3 | 2.8 | 0 | 0.0 | 3 | 1.3 |
| Missing | 0 | 0.0 | 1 | 0.8 | 1 | 0.4 |
| Totalb | 108 | 125 | 233 | |||
EuroQol-5 Dimensions
Self- and proxy-reported EQ-5D-5L data were collected at baseline, 3 months and the second follow-up. The individual domains, split by time point and trial arms, are provided in Appendix 5 (see Tables 34–36 and Figures 23–25). The findings appear to be largely consistent across the domains of the EQ-5D-5L.
Chapter 4 Immunology substudy
Background
Common chronic conditions are linked to an altered immune functionality, which comprises excessive inflammation and declined immunoregulatory and immunostimulatory responses. These changes have been described as biomarkers of ageing, but tend to be heterogeneous between individuals. 72,73 The reduced intestinal microbial diversity in many older people compared with healthy young individuals, as well as healthy old individuals, indicates that gut microbiota might be a target to decrease the progressive immune decline74 and, subsequently, the rate of infections in CHRs. Immunophenotypes provide useful information about functionality of B cells, which have been linked to a decreased antibody production and, therefore, poor response towards influenza vaccination.
Altering the gut microbiota may have the potential to reduce frailty in the aged because it may modulate different aspects of innate75,76 and acquired immunity,77,78 including inflammation. It is therefore relevant to examine strategies tailored towards modifications of gut bacteria in CHRs to improve gut microbial diversity in order to enhance the immune response to exposure to pathogens. In terms of quantifying the immune system, there is a clear consensus around the parameters of the FBC and reference values suggestive of ongoing clinical conditions such as infection. With regard to more detailed immune phenotyping, there are guidelines and a general agreement that age-related alterations include a decreased de novo generation of naive T and B cells, and accumulation of memory cells. 9 In addition, there are many individual immune components and responses that can be measured,11,12 usually by sampling blood, although it is acknowledged that none of these individual immune assessments represent the breadth of the immune response. It is recognised that response to vaccination represents an integrated readout of the ability of the immune system to respond to challenge in vivo. 11,12
Research on the effects of probiotics in CHRs is currently lacking and, hence, we aimed to perform detailed mechanistic analysis of participants’ immune responses to daily probiotics versus placebo in the PRINCESS trial. These findings have been described using FBCs, blood immunophenotypes and various ex vivo immune measures in the trial population. The null hypotheses for our mechanistic analysis were that there would be no difference between groups in FBCs, immunophenotypes, influenza vaccine response, ex vivo cytokine response to TLR agonists, and haematology and biochemistry. The alternative hypothesis was that there would be a difference in these outcomes between the two trial groups.
Aims
The objectives of the mechanistic analyses were to determine the effect of daily probiotic intake on:
-
influenza vaccine response
-
immunology measures (ex vivo responses to immune challenge)
-
haematology and biochemistry measures (FBC) –
-
neutrophils, lymphocytes, monocytes, white blood cells (WBCs), eosinophils, basophils identified through a full blood cell count
-
lymphocyte and monocyte subsets identified through immune cell phenotyping.
-
Methods
The trial design and randomisation procedures were described in Chapter 2. The mechanistic analyses included all participants from the main PRINCESS trial who consented to provide blood samples at baseline and at the end of the trial. We aimed to obtain 150 samples at baseline with a view to achieving between 70 and 100 pairs of samples (allowing for attrition and participant refusal, moving out of care home, etc.). Participants from the main PRINCESS trial were characterised at baseline, and at the end of the study samples were paired according to data availability at both baseline and the end of the intervention period. Influenza vaccine efficacy analysis aimed to include all participants who consented to provide blood samples (or whose consultees had provided agreement on their behalf) and had been taking the SP for at least 2 months prior to receiving the seasonal influenza vaccination.
The schedule of study procedures is reiterated below:
-
screening – screening was conducted prior to recruitment to assess participant eligibility and obtain consent
-
baseline – a baseline blood sample was taken following consent but prior to randomisation
-
second follow-up – blood sample collected at least 6 and up to 12 months post randomisation.
Owing to slower than anticipated recruitment, we truncated the second face-to-face follow-up for some participants. The minimum period until the second face-to-face follow-up was 6 months post randomisation. The time at which this second follow-up visit took place was accounted for in any analysis involving this time point. The majority of analyses were conducted for all participants with available data provided they initiated SP.
The influenza vaccine substudy analysis was conducted for those who consented to blood sample collection (or whose consultees had provided agreement on their behalf), had had a blood sample collected on the day of their routine seasonal influenza vaccination (or up to 10 days prior to this) and another collected 4 weeks (28 days) after they had received their vaccination.
Participants who were enrolled in this study received the most recent recommended seasonal influenza vaccine for the year 2017. The vaccines for the 2017–18 northern hemisphere consisted of the strains A/Michigan/45/2015 (H1N1)pdm09-like virus, A/Hong Kong/4801/2014 (H3N2)-like virus and B/Brisbane/60/2008-like virus. A quadrivalent product was to be used in this study; therefore, the vaccine included B/Phuket/3073/2013.
The blood sample handling, processing and analysis methods are detailed elsewhere. 79
Analysis
Analysis included:
-
Full blood count. Sample: whole blood was collected into EDTA to establish absolute counts for neutrophils, lymphocytes, monocytes, WBCs, platelets, eosinophils and basophils (109/l). The counts were collected using an automated UniCel Beckman Coulter Dxl 800 (Beckman Coulter, High Wycombe, UK).
-
Immune cell phenotypes. Sample: whole blood was collected into lithium heparin. Immune cell phenotypes were established by flow cytometry [as absolute counts using BD Trucount™ tubes (BD Pharmingen, Oxford, UK)]. All antibodies were sourced from BD Pharmingen. T cells [cluster of differentiation 45+ (CD45+)/CD3+], helper T cells (CD45+/CD3+/CD4+), regulatory T cells (CD45+/CD3+/CD4+/CD25+/CD127+LO), cytotoxic T cells (CD45+/CD3+/CD8+), activated cytotoxic T cells (CD45+/CD3+/CD8+/CD25+), CD4+/CD8+ ratio, NK cells (CD45+/CD3–/CD16+), B cells (CD45+/CD3–/CD19+), activated B cells (CD45+/CD3–/CD19+/CD80+ and CD45+/CD3–/CD19+/CD86+), monocytes (CD45+/CD14+) and activated monocytes (CD45+/CD14+/CD80+ and CD45+/CD14+/CD86+) were evaluated. Samples were stained, red blood cells lysed and cells fixed. Tubes were kept at room temperature in the dark overnight and were analysed within 18 hours on a BD FACS LSRF Fortessa™ X-20 special order (BD Biosciences, San Jose, CA, USA).
-
Monocyte and neutrophil phagocytosis. This was assessed in whole blood (lithium heparin) using the Phagotest™ kit (Glycotope Biotechnology GmbH, Heidelberg, Germany) following the manufacturer’s instructions. This is a flow cytometry-based technique (BD FACSCalibur™; BD Biosciences, San Jose, CA, USA) that measures the proportion and fluorescence intensity of monocyte and neutrophil phagocytosis of labelled bacteria.
-
Circulating plasma immune mediators. Sample: plasma was derived from whole blood (lithium heparin). Multiplex assays (Bio-Techne Magnetic Luminex assays, R&D Systems, Abingdon, UK) were used to measure immune mediators in plasma: tumour necrosis factor alpha (TNF-α), interleukin 6 (IL-6), interleukin 10 (IL-10), IL-12p70, IL-17A, IL-1 receptor antagonist (IL-1Ra/IL-1F3), tumour necrosis factor receptor 2 (TNFR-2), monocyte chemoattractant protein 1 [MCP-1, also known as the chemokine (C–C motif) ligand 2 (CCL2)], interferon gamma (IFN-γ)-inducible protein 10 (CXCL10, previously called IP-10), regulated on activation normal T cell expressed and secreted (RANTES/CCL5), soluble intracellular adhesion molecule 1 (ICAM-1/CD54), vascular cell adhesion molecule 1 (VCAM-1/CD106) and E-selectin (CD62E). Multiplex assays were used in accordance with the manufacturer’s instructions. Plates were read on a Bio-plex 200 Analyser (Bio-Rad Laboratories Inc., Watford, UK).
-
Immune mediator production in stimulated whole-blood cultures. Sample: whole blood was collected into lithium heparin. Whole blood was diluted 1 : 10 in Roswell Park Memorial Institute (RPMI) 1640 culture medium supplemented with penicillin (50 U/ml), streptomycin (50 µg/ml) and L-glutamine (2 mM) (Sigma-Aldrich Company Limited, Gillingham, UK). Cultures were stimulated with lipopolysaccharide (LPS) (10 µg/ml), peptidoglycan (PGN) (5 µg/ml) or phytohaemagglutinin (PHA) (5 µg/ml), respectively. Cultures were incubated for 24 hours at 37 °C in an atmosphere of 95% air and 5% carbon dioxide. Supernatants were collected by centrifuging the plates at 2000 revolutions per minute for 5 minutes and were then stored at –80 °C for analysis. LPS- and PGN-stimulated cultures (both monocyte stimulants) were analysed for IL-1β, IL-6, IL-10 and TNF-α, and PHA-stimulated cultures (T cell mitogen) were analysed for IFN-γ and TNF-α. Multiplex assays were obtained from Bio-Techne Magnetic Luminex assays (R&D Systems, Abingdon, UK) and used in accordance with the manufacturer’s instructions. Plates were read on a Bio-plex 200 Analyser (Bio-Rad, Watford, UK).
-
Anti-seasonal influenza vaccine antibody. Anti-seasonal influenza vaccine antibody titres were measured using the HAI at Public Health England Colindale (London, UK).
Statistical analyses
No imputation was performed. Analyses of normality were conducted by applying the Kolmogorov–Smirnov test and by identification of the Gaussian distribution in the histogram plot. Significant effects of the post-intervention outcome were defined as p-values < 0.05. In non-normally distributed parameters, it was necessary to log-transform the variable to fit a regression model in which analyses were adjusted by allocation (either placebo or probiotic combination), sex and baseline measurement through the analysis of covariance (ANCOVA). Significant effects of the intervention for variables that did not fit the model assumptions were analysed using the Mann–Whitney U-test or the Kruskal–Wallis test if the variable was numerical; the Kruskal–Wallis test was used when samples were numerical and ordinal. Descriptive statistics for all of the variables are presented as median, mean, and percentiles 10 and 90.
Results
A CONSORT flow diagram (Figure 9) depicts the progress of the immunology trial participants through to the end of trial sample collection (obtained for 60 participants). Thirty participants were randomised to the daily oral probiotic combination group and 30 participants to the placebo group. Preliminary analyses testing the influence of delayed processing on the parameters examined in blood indicated the maximum time frame in which these parameters can be analysed for each variable under assessment. To control this, the time of sample collection directly from the participant was recorded and posted with relevant information. This time was noted when the sample was delivered to the specialised laboratory, and analyses were carried out taking account of the time delays. Immune mediators measured in plasma and in supernatants from whole-cell cultures were analysed up to 24 hours after sample collection. Other than those parameters, the great majority of parameters could be analysed up to 72 hours after sample collection, with the exception of specific variables within the FBC, immune phenotypes and phagocytic function, in which analyses had to be conducted within 48 hours after collection (see Figure 9).
FIGURE 9.
A CONSORT flow diagram for the immunology substudy.

Where paired samples for a patient were not available because one or both of the samples arrived at the specialised laboratory outside the predetermined time, the patient was not included in the data set for any analyses affected by the time delay. The analyses conducted are shown in Figure 10.
FIGURE 10.
Analyses conducted in the immunology substudy. ImMed, immune mediators; IMPh, immunophenotypes; PhFx, phagocytic function.

Paired samples, representing sample availability at both baseline and post intervention for a patient, were available for analysis for 30 patients.
Characterisation of participants
A total of 184 participants were randomised and recruited to the immunology substudy at baseline. The mean age was 85.4 years (SD ±7.45 years). The general characteristics of study participants at baseline are shown in Table 25 and frailty characteristics are depicted in Figure 11.
| Variable | Mean | SE | SD |
|---|---|---|---|
| Age (years) | 85.4 | 0.6 | 7.5 |
| Time at care home (years) | 1.9 | 0.2 | 2.2 |
| Height (cm) | 1.6 | 0.0 | 0.1 |
| Weight at study baseline (kg) | 66.6 | 1.4 | 15.7 |
| Middle upper arm circumference (cm) | 27.3 | 0.3 | 3.9 |
FIGURE 11.
Relative distribution of participants according to frailty status. The scale is based on the frailty score, ranked as follows: 1 = fittest category for their age (active and energetic); 2 = well (absent symptomatology of disease but less active); 3 = managing well (medical problems under control but not active); 4 = vulnerable (symptoms that limit activities); 5 = mildly frail (impairment of daily activities); 6 = moderately frail (progressive impairment and decline in activities); and 7 = severely frail (completely dependent cognitively or physically; not terminally ill).

Full blood count
The FBC results are presented for 60 participants. There was no evidence of a difference between participants allocated to the probiotic combination group and participants allocated to the placebo group in numbers of neutrophils, lymphocytes, monocytes, WBC, platelets, eosinophils and basophils in whole blood at the end of the study. Table 26 summarises the outcomes considered for FBC.
| Descriptive statistics categorised according to trial arm | ANCOVA | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter and reference value (109/l) | Trial arm | Adjustment | Adjusted mean difference | 95% CI | p-value | |||
| Placebo (n = 30) | Probiotic (n = 30) | |||||||
| Mean | SE | Mean | SE | |||||
| Neutrophils (2.0–7.5) | ||||||||
| Baseline | 4.8 | 0.4 | 4.6 | 0.4 | Trial arm | 0.006 | –0.058 to 0.069 | 0.861 |
| Post intervention | 4.4 | 0.3 | 4.7 | 0.3 | Sex | 0.017 | –0.047 to 0.081 | 0.593 |
| Baseline | 0.52 | 0.348 to 0.691 | < 0.001 | |||||
| Lymphocytes (1.5–5.0) | ||||||||
| Baseline | 1.4 | 0.1 | 2.4 | 0.5 | Trial arm | 0.018 | –0.047 to 0.082 | 0.583 |
| Post intervention | 1.5 | 0.1 | 2.5 | 0.6 | Sex | 0.007 | –0.055 to 0.068 | 0.826 |
| Baseline | 0.796 | 0.651 to 0.94 | < 0.001 | |||||
| Monocytes (0.2–1.0) | ||||||||
| Baseline | 0.5 | 0.0 | 0.6 | 0.0 | Trial arm | –0.023 | –0.104 to 0.058 | 0.566 |
| Post intervention | 0.5 | 0.0 | 0.6 | 0.1 | Sex | –0.02 | –0.099 to 0.06 | 0.623 |
| Baseline | 0.665 | 0.403 to 0.928 | < 0.001 | |||||
| WBC (4–11) | ||||||||
| Baseline | 6.9 | 0.4 | 8 | 0.6 | Trial arm | 0.014 | –0.035 to 0.064 | 0.563 |
| Post intervention | 7.1 | 0.4 | 8.2 | 0.7 | Sex | 0.006 | –0.044 to 0.055 | 0.824 |
| Baseline | 0.702 | 0.535 to 0.868 | < 0.001 | |||||
| Platelets (140–400) | ||||||||
| Baseline | 281.6 | 15.5 | 291.7 | 22.5 | Trial arm | 0.019 | –0.021 to 0.06 | 0.347 |
| Post intervention | 267.4 | 21.2 | 284 | 15.4 | Sex | –0.008 | –0.054 to 0.037 | 0.718 |
| Baseline | 0.832 | 0.668 to 0.995 | < 0.001 | |||||
| Mann–Whitney U -test: factor (trial arm) | ||||||||
| Eosinophils (0.0–0.5) | ||||||||
| 0.2 | 0.0 | 0.2 | 0.0 | p-value | ||||
| 0.2 | 0.0 | 0.3 | 0.0 | Post intervention | 0.816 | |||
| Basophils (0.0–0.1) | ||||||||
| Baseline | 0 | 0.0 | 0.1 | 0.0 | p-value | |||
| Post intervention | 0.1 | 0.0 | 0.1 | 0.1 | Post intervention | 0.688 | ||
Immunophenotypes
Immune cell phenotypes were determined as absolute cell counts in whole blood for the 60 participants. Data are expressed as total counts and are presented in Table 27. There was no evidence of a difference between participants allocated to the probiotic combination group and participants allocated to the placebo group in T cells (CD45+/CD3+), helper T cells (CD45+/CD3+/CD4+), regulatory T cells (CD45+/CD3+/CD4+/CD25+/CD127+LO), cytotoxic T cells (CD45+/CD3+/CD8+), activated cytotoxic T cells (CD45+/CD3+/CD8+/CD25+), ratio CD4+ : CD8+, NK cells (CD45+/CD3–/CD16+), B cells (CD45+/CD3–CD19+), activated B cells (CD45+/CD3–CD19+/CD80+ and CD45+/CD3–CD19+/CD86+), monocytes (CD45+/CD14+) and activated monocytes (CD45+/CD14+/CD80+ and CD45+/CD3+/CD86+).
| Descriptive statistics categorised according to trial arm | ANCOVA | |||||||
|---|---|---|---|---|---|---|---|---|
| Phagocytic function | Trial arm | Adjustment | Adjusted mean difference | 95% CI | p-value | |||
| Placebo (n = 30) | Probiotic (n = 30) | |||||||
| Mean | SE | Mean | SE | |||||
| T cells | ||||||||
| Baseline | 1298.3 | 50.8 | 1337.8 | 78.5 | Trial arm | 0.03 | –0.02 to 0.08 | 0.26 |
| Post intervention | 1387.7 | 64.9 | 1539.1 | 89.4 | Sex | 0.04 | –0.01 to 0.1 | 0.12 |
| Baseline | 0.65 | 0.41 to 0.9 | < 0.01 | |||||
| Helper T cells | ||||||||
| Baseline | 886.3 | 64.3 | 912.4 | 82.4 | Trial arm | 0.01 | –0.06 to 0.07 | 0.79 |
| Post intervention | 985.8 | 72.6 | 1014.5 | 79.8 | Sex | 0.05 | –0.02 to 0.11 | 0.17 |
| Baseline | 0.79 | 0.66 to 0.93 | < 0.01 | |||||
| Regulatory T cells | ||||||||
| Baseline | 87.2 | 15.4 | 85.7 | 14.3 | Trial arm | 0.02 | –0.08 to 0.12 | 0.71 |
| Post intervention | 80.1 | 13.5 | 84.1 | 13.4 | Sex | –0.05 | –0.16 to 0.06 | 0.34 |
| Baseline | 0.79 | 0.67 to 0.92 | < 0.01 | |||||
| Cytotoxic T cells | ||||||||
| Baseline | 631.2 | 35.2 | 767.0 | 33.3 | Trial arm | –0.01 | –0.06 to 0.04 | 0.67 |
| Post intervention | 717.3 | 38.8 | 803.1 | 37.9 | Sex | –0.04 | –0.08 to 0 | 0.07 |
| Baseline | 0.74 | 0.58 to 0.9 | < 0.01 | |||||
| Activated cytotoxic T cells | ||||||||
| Baseline | 230.9 | 18.2 | 270.0 | 19.2 | Trial arm | –0.02 | –0.09 to 0.05 | 0.59 |
| Post intervention | 278.5 | 14.1 | 282.3 | 20.5 | Sex | –0.02 | –0.1 to 0.05 | 0.48 |
| Baseline | 0.1 | –0.05 to 0.26 | 0.19 | |||||
| Ratio CD4:CD8 | ||||||||
| Baseline | 1.4 | 0.1 | 1.2 | 0.1 | Trial arm | 0.02 | –0.05 to 0.1 | 0.56 |
| Post intervention | 1.4 | 0.1 | 1.3 | 0.1 | Sex | 0.09 | 0.02 to 0.16 | 0.02 |
| Baseline | 0.76 | 0.56 to 0.95 | < 0.01 | |||||
| NK cells | ||||||||
| Baseline | 80.8 | 5.3 | 82.4 | 5.8 | Trial arm | 0.04 | –0.04 to 0.12 | 0.33 |
| Post intervention | 72.6 | 4.9 | 79.2 | 4.2 | Sex | 0.03 | –0.05 to 0.11 | 0.46 |
| Baseline | 0.33 | 0.04 to 0.63 | 0.03 | |||||
| B cells | ||||||||
| Baseline | 221.2 | 20.1 | 240.1 | 19.8 | Trial arm | –0.04 | –0.09 to 0.01 | 0.1 |
| Post intervention | 224.3 | 21.4 | 232.3 | 20.1 | Sex | 0.02 | –0.02 to 0.07 | 0.34 |
| Baseline | 0.84 | 0.73 to 0.95 | < 0.01 | |||||
| B cells – CD80 | ||||||||
| Baseline | 137.8 | 11.2 | 148.5 | 16.3 | Trial arm | 0 | –0.05 to 0.05 | 0.96 |
| Post intervention | 126.9 | 11.6 | 142.7 | 17.1 | Sex | –0.01 | –0.05 to 0.04 | 0.81 |
| Baseline | 0.86 | 0.74 to 0.98 | < 0.01 | |||||
| B cells – CD86 | ||||||||
| Baseline | 141.7 | 12.0 | 155.8 | 17.1 | Trial arm | –0.02 | –0.07 to 0.03 | 0.52 |
| Post intervention | 140.1 | 14.0 | 151.6 | 17.8 | Sex | –0.03 | –0.08 to 0.02 | 0.23 |
| Baseline | 0.88 | 0.76 to 1.01 | < 0.01 | |||||
| Monocytes | ||||||||
| Baseline | 447.6 | 31.5 | 577.6 | 37.7 | Trial arm | –0.02 | –0.11 to 0.07 | 0.6 |
| Post intervention | 504.1 | 41.4 | 569.9 | 40.0 | Sex | 0 | –0.09 to 0.08 | 0.92 |
| Baseline | 0.8 | 0.57 to 1.02 | < 0.01 | |||||
| Monocytes – CD80 | ||||||||
| Baseline | 154.8 | 20.5 | 164.2 | 25.9 | Trial arm | 0.02 | –0.11 to 0.16 | 0.75 |
| Post intervention | 112.9 | 18.3 | 120.4 | 23.1 | Sex | –0.07 | –0.21 to 0.07 | 0.3 |
| Baseline | 0.71 | 0.52 to 0.91 | < 0.01 | |||||
| Monocytes – CD86 | ||||||||
| Baseline | 130.8 | 17.9 | 122.0 | 19.5 | Trial arm | 0.07 | –0.14 to 0.28 | 0.5 |
| Post intervention | 110.9 | 19.5 | 124.4 | 22.3 | Sex | 0.04 | –0.17 to 0.25 | 0.71 |
| Baseline | 0.87 | 0.65 to 1.08 | < 0.01 | |||||
Phagocytic function
Phagocytic function is presented as the percentage of phagocytic activity in monocytes and neutrophils, and the fluorescence activity is a surrogate marker of the number of pathogenic bacteria engulfed. There was no evidence of a difference between participants allocated to probiotic combination and placebo in the phagocytic activity measured in monocytes and neutrophils. Table 28 summarises the outcomes considered within the assessment of the phagocytic activity.
| Descriptive statistics categorised according to trial arm | ANCOVA | |||||||
|---|---|---|---|---|---|---|---|---|
| Phagocytic function | Trial arm | Adjustment | Adjusted mean difference | 95% CI | p-value | |||
| Placebo (n = 30) | Probiotic (n = 30) | |||||||
| Mean | SE | Mean | SE | |||||
| Phagocytic activity neutrophils (%) | ||||||||
| Baseline | 78.7 | 1.8 | 81.2 | 1.4 | Trial arm | 0.006 | –0.028 to 0.039 | 0.727 |
| Post intervention | 81.8 | 2.1 | 83.1 | 2.5 | Sex | 0.024 | –0.01 to 0.058 | 0.158 |
| Baseline | 0.207 | 0.028 to 0.386 | 0.025 | |||||
| Phagocytic activity neutrophils (GMFL) | ||||||||
| Baseline | 272.8 | 10.2 | 243.4 | 9.5 | Trial arm | 0.014 | –0.052 to 0.081 | 0.666 |
| Post intervention | 247.7 | 11.4 | 245.2 | 14.8 | Sex | 0.057 | –0.008 to 0.123 | 0.084 |
| Baseline | 0.398 | 0.145 to 0.65 | 0.003 | |||||
| Phagocytic activity monocytes (%) | ||||||||
| Baseline | 33.9 | 1.6 | 28.3 | 1.4 | Trial arm | 0.032 | –0.069 to 0.134 | 0.524 |
| Post intervention | 29.6 | 1.9 | 29.5 | 2.1 | Sex | 0.001 | –0.098 to 0.1 | 0.979 |
| Baseline | 0.333 | 0.1 to 0.566 | 0.006 | |||||
| Phagocytic activity monocytes (GMFL) | ||||||||
| Baseline | 182.3 | 8.7 | 184.3 | 11.0 | Trial arm | 0.028 | –0.048 to 0.104 | 0.465 |
| Post intervention | 174 | 9.5 | 184.9 | 11.6 | Sex | –0.013 | –0.089 to 0.063 | 0.738 |
| Baseline | 0.039 | –0.176 to 0.254 | 0.070 | |||||
Plasma immune mediators
Immune mediators were analysed in plasma. There was no evidence of a difference between participants allocated to probiotic combination and participants allocated to placebo on these outcomes. Appendix 5, Table 37, summarises the outcomes considered within the assessment of the immune mediators.
Whole-blood cultures stimulated by PGN, LPS or PHA
Immune mediators were analysed in whole-blood cultures by collecting the supernatant after LPS, PGN or PHA stimulation of immune cells for 36 participants in the immunology substudy. No evidence of between-arm differences was found. The results are presented in Appendix 5, Tables 38–40.
Anti-vaccine antibody response (vaccine strain-specific response)
A total of 39 participants were recruited into the anti-vaccine antibody response work during the 2017/18 vaccination schedule (19 participants were randomised to daily oral probiotic and 20 participants were randomised to placebo).
Antibody titres at baseline (pre-influenza vaccination)
Influenza vaccine-specific antibodies were measured in serum. Protection against the viruses is analysed through two outcomes: seroprotection and seroconversion. Seroprotection is defined as an antibody titre ≥ 40 haemagglutination units. At baseline (i.e. pre-influenza vaccination), a large proportion of the population was already seroprotected. The antibody titres for the strain A/Michigan/45/2015 indicated that 41% of the population were seroprotected before the commencement of the study. The proportion of the population seroprotected against the strain A/Hong Kong/4801/2014 was 76.9%. The quadrivalent vaccine included the strains B/Brisbane/60/2008 and B/Phuket/3073/2013, for which the percentages of seroprotection were 94.9% and 84.6%, respectively. Seroprotection status before receiving the quadrivalent version of the influenza vaccine is reported in Figure 12.
FIGURE 12.
Pre-vaccination seroprotection status for quadrivalent influenza vaccine in elderly participants in the PRINCESS immunology substudy.

Titres status and seroprotection post vaccination
No statistical differences were observed in the anti-influenza titres when the analyses from the placebo and probiotic combination groups were compared (see Appendix 5, Figures 26–29).
As expected, there was an increase in the proportion of participants seroprotected, as measured by the presence of anti-vaccine antibody titres in the post-vaccination period. Descriptively, it was observed that the proportion of participants who were seroprotected regardless of allocation of SP (either placebo or probiotic combination) increased. There was an increase in the percentage of seroprotection of 28.2% for the strain A/Michigan/2015, 12.8% for the strain A/Hong Kong/2014, 2.5% for the strain B/Brisbane/2008 and 12.5% for the strain B/Phuket/2013, as described by the frequency of cases presented in Table 29.
| Quadrivalent influenza vaccine strain | Seroprotection status | Time period | |
|---|---|---|---|
| Pre vaccination (n) | Post vaccination (n) | ||
| HAI1_A/Michigan/2015 | No | 23 | 12 |
| Yes | 16 | 27 | |
| Total | 39 | ||
| HAI1_A/Hong Kong/2014 | No | 9 | 4 |
| Yes | 30 | 35 | |
| Total | 39 | ||
| HAI1_B/Brisbane/2008 | No | 2 | 1 |
| Yes | 37 | 38 | |
| Total | 39 | ||
| HAI1_B/Phuket/2013 | No | 6 | 1 |
| Yes | 33 | 38 | |
| Total | 39 | ||
Subsequently, a detailed analysis was conducted of the proportion of seroprotected participants in the post-vaccine period according to group (either placebo or probiotic combination). It was found that the probiotic did not exert any statistically significant effect on the antibody response to the virus strains used in the quadrivalent influenza vaccine. It was observed that the percentage of positive seroprotection was higher, but not significantly so, in the probiotic combination group than in the placebo group for the strains A/Michigan/2015 and A/Hong Kong/2014. Figure 13 illustrates the percentage of seroprotection by group.
FIGURE 13.
Anti-vaccine antibody seroprotective response according to strain of analysis and allocation group (placebo or probiotic combination).

Subjects were defined as experiencing seroconversion when their post-vaccine sample demonstrated a fourfold increase in antibody titres compared with their pre-vaccine sample. The results of the seroconversion assessment are shown in Figure 14 and are presented as titres of antibodies in the post-vaccine period, analysed per strain composing the quadrivalent influenza vaccine. It was found that the probiotic intervention did not exert a significant effect on the seroconversion rate compared with the placebo combination group.
FIGURE 14.
Anti-vaccine antibody seroconversion response according to strain of analysis and allocation group (placebo or probiotic combination).
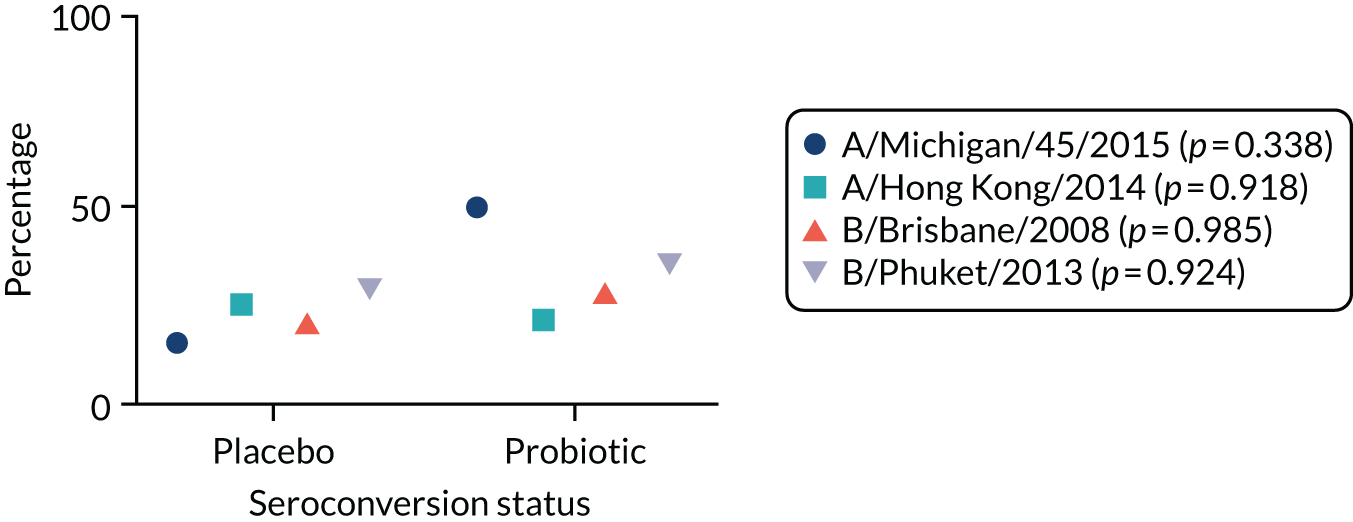
Discussion
There was no evidence to suggest that the effect of a probiotic combination consisting of Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 on blood immune cell numbers or subtypes, or on responses to seasonal influenza vaccination, was different from that of placebo. These results are in keeping with the clinical outcomes of the trial, which showed no evidence of an effect of the probiotics on antibiotic consumption or on the duration or number of infections. Systematic reviews have indicated positive effects of other strains of probiotics in healthy older people14 and, hence, our findings may not be applicable to other populations and probiotic species. Limitations include high losses in the follow-up period, which significantly reduced available data and, hence, statistical power. The relatively raised mortality rate, which is comparable to other studies in older participants, contributed to data loss. 80 Moreover, this multicentre study was exposed to other variables affecting the sample size, including that the clinical settings (care homes) were a considerable distance from the laboratory where immunological and inflammatory analyses would take place. This meant that the time between sample collection and arrival at the laboratory was variable and, when considerable delays occurred, it was not possible to carry out experiments on the sample. This significantly affected the data available for immune mediators, cytokine production and chemokines assessed in this study.
Chapter 5 Qualitative evaluation of the implementation of the PRINCESS trial
Aim
The aim of the qualitative evaluation was to achieve a deep understanding of how the PRINCESS trial was implemented in order to identify aspects that went well and opportunities for improving processes that might be relevant for future, similar studies.
Methods
Setting and participants
The PRINCESS qualitative evaluation study was approved in November 2018 by the Cardiff University School of Medicine REC (reference number 18/69) and a substantial amendment was submitted to the Wales REC 3.
Eligibility to participate in the qualitative evaluation was based on advice from key groups that participated in the trial itself, provided advice on behalf of a CHR about participation (consultee), were family members/friends who experienced the CHR’s participation in the trial, involvement in the research activities required to recruit CHRs, or collection of research data for the trial.
An emergent sampling strategy was used, with the aim of capturing a view of the trial from a variety of perspectives. Recruitment of participants for interview varied according to participant group. Those in the care home environment (managers, staff, consultees, relatives and CHRs) were ‘snowball sampled’ (i.e. managers were approached initially). The intention was then for managers to approach staff, who, in turn, could approach CHRs, relatives and personal consultees (i.e. family member or friend who acted as consultee). 81 RNs were approached directly by a qualitative researcher (Helen Stanton) via e-mail and were invited to participate.
Procedure
Interview participants were provided with written information about the qualitative evaluation and were given an opportunity to discuss the interview. Written informed consent was obtained prior to undertaking the interview.
Semistructured interviews were carried out either by telephone or face to face with a qualitative researcher (Helen Stanton). Topic guides [see Appendix 2 (care home management and staff), Appendix 3 (research professionals) and Appendix 4 (CHRs and consultees)] focused on various aspects of the research process of the trial and aimed to elucidate relevant contextual issues and barriers to and facilitators of the implementation of the trial. 82
Analysis
Interviews were digitally audio-recorded and transcribed verbatim by staff within the CTR. Transcripts were checked for accuracy and completeness against the source data and anonymised (Helen Stanton). NVivo 12 (QSR International, Warrington, UK) qualitative analysis software was used to support data management and facilitate coding.
Data were coded iteratively into domains emerging from the data, and those identified a priori. Data were analysed using framework analysis. The organisation and mapping of data using this approach enables within- and between-case comparisons, and enables clear linking of developed categories and themes back to the raw data. 83 Framework analysis involves five stages: (1) familiarisation with the data, (2) development of a thematic framework, (3) applying thematic codes to all of the data (indexing), (4) retrieving and summarising coded data in a chart and (5) interpreting the data by drawing inferences and pulling together relevant themes. 84 This method of analysis is useful if a number of well-defined research aims have been used to guide the topics covered in the interviews, while also allowing themes to emerge from the data that are relevant to the research aims. 82,85
Data coding and analysis was led and conducted by Helen Stanton. After Helen Stanton had coded approximately half of the total interview transcripts, a member of the research team (VS) with experience in this research topic area and expertise in qualitative analysis discussed the coding, framework and themes with Helen Stanton. This helped establish the validity of the coding framework for the analysis to be completed. Victoria Shepherd also coded two of the interview transcripts to assess coding validity.
Results
Semistructured interviews were carried out with nine RNs who implemented the trial in care homes across England and south Wales, seven CHMs from care homes that participated in the trial from across England and south Wales and two relatives who had a parent that participated in the trial. The interviews were carried out between February and May 2019. The interviews lasted between 13 to 71 minutes. Regrettably, one of the audio-recordings of an interview with a CHM was corrupted and could not be used in the analysis; therefore, only six of the seven CHM interviews were included in the analysis. Of the six interviews with CHMs included in the analysis, three were from larger (≥ 50 beds) homes providing residential and nursing care and three were from moderate-sized (20–40 beds) homes providing residential care. Two of the homes (both residential care homes) were privately owned, standalone companies. The remaining four homes were all part of privately owned care home groups. Of the three homes providing residential and nursing care, two were identified as higher recruiters (≥ 12 participants recruited), with one identified as a lower recruiter (fewer than five participants). Two of the three homes providing residential care were moderate recruiters (between 5 and 12 participants) and one was a higher recruiter. Care home size did not appear to be associated with numbers of participants recruited.
The key themes identified through the framework analysis are summarised in Table 30.
| Main theme | Subtheme | Key points |
|---|---|---|
| Acceptability of the trial | Participation in the trial, receiving trial intervention and obtaining samples | RNs and CHMs perceived the intervention as very low risk, with important potential benefits for CHRs. There was additional reassurance that CHRs were in an environment where any trial-related issues could be quickly identified. RNs felt less comfortable taking blood samples than stool and saliva samples from CHRs without capacity |
| Assessing capacity and consultee process | RNs worked with the care home staff to assess CHRs’ capacity. Enrolling CHRs via consultee advice was acceptable because the intervention was perceived as low risk and CHRs could be closely monitored in the care home. Involving those without capacity was acceptable as long as the CHR was amenable to taking the SP | |
| Implementation of research activities | Initial engagement with trial and gaining ‘entry’ to care home | Being mindful of the priorities of care home staff was key to being accepted into the home. Planning ahead and communication prior to visits were important to prevent time being wasted. CHM leadership and a good working management–staff relationship was key in creating an atmosphere where the RNs felt accepted into the care home environment. CHMs indicated that RNs took the lead on various trial activities and played a supportive role in the home |
| Recruitment and consultee process | RNs screened CHRs for suitability for the trial with the CHM and care home staff. Staff at the home approached relatives to act as consultees for CHRs without capacity. Enrolling the CHR via the consultee process was challenging. Engaging relatives via letter was of limited success. Direct communication with relatives was preferred and was a more efficacious method of introducing the trial. The majority of care homes enrolled both CHRs with capacity and CHRs without capacity | |
| Data collection and division of trial-related activities | Data were collected through discussion with the CHR and care home staff, and from care home records and MAR charts. Antibiotic use was well documented but recording adherence to SP was more variable. Where care home records were paper based, RNs had to visit the home often enough to ensure information recall, but not too often as to overburden the care home staff | |
| Obtaining stool samples | RNs frequently reported issues with obtaining stool samples. Obtaining stool samples was delegated to carers who attended to the CHRs’ personal care, but effective communication with these staff to obtain samples was compromised by shift patterns and other competing tasks | |
| Sustainability and routinisation | Some RNs reported that some research activities (stool sampling, monitoring diarrhoea, proxy questionnaires) fell by the wayside when they were not present in the home to remind the care home staff. There was evidence that a longer break between RN visits could lead to care home staff disengaging from the trial activities. The trial was resource intensive, and CHMs and RNs voiced concerns that they would have struggled to maintain the trial activities if they had recruited more CHRs at their care home/site | |
| Mechanisms of implementation | Potential benefit of probiotic, motives for participation and perception of trial ‘value’ | The potential for the active SP to reduce infections and hospitalisations in the older population (because of the severe and lasting impact infections can have) was a driving force behind the home participating in the trial and ensuring successful trial implementation. CHMs’ recognition of the complex set of potential benefits offered by running the trial at the home accompanied effective prioritisation and routinisation of trial activities in the home |
| Leadership, expectations and responsibility | CHM co-ordination and leadership was important during the set-up of the trial, particularly organising care home staff training. Research-enthusiastic CHMs who were often present at the home were able to communicate their expectations with staff. CHMs who took responsibility and ownership for the trial and supporting the research activities created a model for the staff to follow | |
| Understanding the routines and ‘rhythm’ of the care home | Working with the routines and ‘rhythms’ of the homes was key to effective time management and helped to foster good relationships with RNs and care home staff. Planning short visits in advance avoided overburdening very busy staff, and was sufficient for data collection. CHMs appreciated the input and support RNs could offer to the home | |
| Contextual factors | RNs relied more on the nursing staff for some data collection tasks, as care staff did not have capacity to undertake the extra activity. RNs felt that, compared with carers, trained nurses were more adept at documenting SP adherence and medical events like diarrhoea |
To provide context, quotations are captioned by interviewee group [i.e. RN, CHM or relative of a trial participant (relative)].
Acceptability
Participation in the trial, receiving the trial intervention and obtaining samples
Research nurses felt that the intervention being trialled was not problematic for those without capacity because the experimental intervention was perceived as very low risk and could be of benefit. RNs perceived that there was a short-term potential value to taking probiotics for the individual themselves, and, longer term, they saw a prospective societal value in probiotic use, particularly for this trial population.
Additional assurance stemmed from the view that the intervention was also being administered in an environment where CHRs are closely monitored, and where changes in the well-being of the CHRs would be quickly recognised.
The willingness to take on the trial at a particular care home appeared to hinge on the potential benefits of the intervention, and the hope that it would improve the health and well-being of their CHRs. CHMs acknowledged that the active SP was a food supplement, rather than a medication, and that the risk of side effects from the probiotic was extremely low. As the trial progressed, CHMs described some potential adverse effects of the SP; however, this did not seem to affect the overall acceptability of probiotics in this population:
We did have one person we had to stop it, because he’s quite sensitive to things and he started [to have] very loose stools so we did stop him and as soon as he stopped it, that problem stopped . . . everybody else that took part in it there was no issues.
CHM14
Research nurses reported that the actual act of taking another ‘tablet’ (the SP was delivered in capsule form) was the biggest issue with the intervention, particularly for those without capacity. This aspect of the intervention needed to be discussed with CHMs and family members to assess suitability for the trial. CHRs’ concerns about the safety of the SP as communicated in the RN interviews were scarce, with one reporting that a CHR was concerned about the SP interacting with her current medication:
One lady [said] . . . I’ve already got heart problems, I don’t want anything that’s going to jeopardise that . . .
RN11
Research nurses felt that it was not problematic to involve those without capacity as long as the CHR was amenable to taking the SP. This was discussed during the screening process with the CHR’s family and the care home staff. RNs reported that for some relatives the thought of their loved one consuming the SP was problematic; their relative was settled, and they did not want to take part in anything that might jeopardise that. However, there is some indication that consultees were agreeable to their relative taking part, despite acknowledging that their relative may not have been completely adherent to the SP:
. . . I don’t know how compliant [the CHR] was with any medication . . . I couldn’t be 100% sure whether the probiotics were having an impact or not, because I don’t really know how, as I say, compliant [the CHR] was . . .
Relative18
Once CHRs were enrolled on the trial, they were not perturbed by taking the SP in capsule form, and would take the capsule at the same time as their other medication:
I think the project worked really well for the home and the clients, you know, [they] didn’t mind taking the product at all . . .
CHM15
Where samples were concerned, RNs felt less comfortable taking blood samples than stool and saliva samples from CHRs without capacity. Relatives of participating CHRs found it comforting that RNs would not try to take blood at all if CHRs appeared unwilling or if the RN judged it to be inappropriate for any reason. Taking blood from those with capacity was less problematic, with one relative commenting that the CHR would not have distinguished between a sample taken for medical requirements and one taken for research:
. . . I don’t think [my relative] would have noticed, [my relative] never said ‘Oh they came to take bloods today’ . . .
Relative17
One RN described taking the saliva sample as per protocol (i.e. swabbing the gum line for between 1 and 2 minutes) as being too long for CHRs to tolerate if they did not have capacity. Some RNs reported that CHRs with capacity were very keen that all the samples needed were obtained:
. . . The residents who had capacity were very keen to give you everything that you wanted. If you needed blood, if you needed saliva, they were really keen that you took all the samples . . .
RN9
Assessing capacity and consultee process
Research nurses indicated that they worked with the care home staff to initially assess CHRs’ capacity to consent to the trial. Some RNs reported having sometimes disagreed with staff's assessment that a CHR had capacity; however, the RNs knew that the decision lay with the RNs as to whether or not to get consultee advice. However, care home staff could also play an important role in any assessment of a CHR’s capacity to consent, particularly if the CHR had specific communication needs:
. . . as the health-care assistant was explaining, I said can you just ask, you know them really well. Could you ask the three questions with me here? I just want to check, you know, I’m not certain and she said ‘oh, you’re talking in the wrong ear’ and her hearing aid had fallen out.
RN5
There was some indication that RNs found it somewhat saddening when they assessed CHRs who were exhibiting signs of deteriorating mental capacity. Although, overall, RNs were not daunted by the capacity assessment process, and attributed their preparedness to their professional background in mental health or community nursing:
. . . I sort of have done it before with my background in the community. So, as I say, these are the settings that I’m used to . . . I didn’t find it daunting in any way.
RN3
Owing to the perceived low risk of the intervention, and the fact that the CHRs were monitored daily, the process of recruiting CHRs via consultee advice was acceptable to RNs. Using a professional from the care home to act as the nominated consultee was viewed as less acceptable because it was felt that advice should come from someone who had known the CHR before they had lost capacity:
. . . my gut feeling is that it ought to be a family member. Who knew the person when they had capacity because they had a problem with a staff member is that they’ve probably only known the person since they’ve lost capacity.
RN2
Research nurses indicated that one of the motivations for relatives may have been the belief that the care home staff and research team would keep a closer eye on their relative for the duration of the trial. Consultees’ reasons for advising their relative to join the trial, or introducing the trial to their relative, included beliefs about the benefits of probiotics, and for societal benefit or altruistic reasons. Some relatives were very confident that the CHR would have wanted to take part had they been able to decide for themselves:
. . . it seemed to be such a good idea and such a simple idea and [my relative] has always been interested in helping others so when I explained, you know, that the project might help him and help other people he was more than willing to take part . . .
Relative17
If the CHR had not been keen on taking medication before losing capacity, the trial was rejected. Feedback from the CHRs with capacity indicated that most had no objections to taking the SP. CHMs had no misgivings about the acceptability of the consultee process or recruiting CHRs without capacity.
Implementation of research activities
Initial engagement with trial and ‘gaining entry’ to the care home
Care home managers were introduced to the trial via the central trial team, or through other care homes in their commercial group. Interest in participating in the trial also came from the CHM to the trial team. RNs mostly became involved in the trial once initial training and set-up had been undertaken, but a minority were involved in setting up and training care home staff.
Research nurses described how some had previously worked in care homes and were aware of the challenges that they might face in engaging staff, and being welcomed into the environment. Being mindful of the priorities of the care home staff was key to being accepted in to the home to get started with the trial tasks. RNs were sometimes compelled to adjust the time at which they visited, and to extend the time they set aside to engage with staff. Planning ahead, co-ordinating short meetings, agreeing tasks and timelines with the staff and communicating prior to visits were important to prevent time being wasted.
Research nurses felt that their professional identity as nurses was recognised by CHMs and staff, and felt that it carried weight regarding the trust and confidence they were afforded by the CHMs and staff when they were present in the home, or carrying out the trial activities. There was some intimation from the RNs that there was a reciprocal professional understanding between them and the CHMs and staff because of their training and experience in clinical and health-care environments. Where the CHM had a clinical background in nursing, RNs felt that the manager’s knowledge and experience lent itself to an appreciation of the trial and the potential impact it could have on the CHRs.
Care home managers were generally very positive about the RNs carrying out the research tasks for the trial, and were described as being very personable with the CHRs, relatives and staff. CHMs indicated that RNs took the lead on various trial activities and that they also played a supportive role in the home, both practically and in an advisory capacity:
. . . She even helped us. . . . somebody arrested and she had to help me at that time.
CHM14
Care home managers’ leadership and a good management–staff working relationship also appeared key in creating an atmosphere where the RNs felt accepted into the care home environment. There was some indication that a lack of effective leadership and management–staff communication about the trial undermined the RN’s role in the care home:
. . . whilst the management teams generally seemed to recognise the importance of what we were doing, that didn’t always reflect with the staff on the front line. . . . if there was nobody there to sort of say to people, you know, you need to go to this training, they didn’t independently recognise the importance of it enough to come along . . .
RN7s
It could also prove difficult for a new RN to gain entry to the care home midway through the trial after there had been a hiatus of trial activities at the home. A break in research activities could lead to staff disengagement from the trial. For this reason, it was particularly challenging for RNs when trying to familiarise themselves with the rhythm of the care home and build relationships from scratch while also trying to re-engage the staff with the trial:
. . . Different staff and them not knowing me . . . if you go in at the beginning and you set the study up, I think because there was a lot of participants. So, there was a lot that needed doing . . .
RN9
Recruitment and consultee process
Screening the CHRs for suitability for the trial was generally done with the CHM or the nursing staff, and information about the CHR’s eligibility for the trial was forthcoming from staff.
Research nurses took responsibility for the recruitment of CHRs with capacity, and, for reasons of data protection, staff at the home co-ordinated the process of approaching relatives to provide advice for CHRs assessed as lacking capacity. CHMs welcomed the guidance and support from RNs around recruitment. CHMs acknowledged that CHRs’ and relatives’ lack of familiarity with the RN might be a barrier to engaging potential participants, so at times they stepped in to assist:
But because [RN] was new to this place and the residents didn’t know her it was very difficult for her to get any sort of participation from their side so then we spoke together and I said OK let me try and I will try to do that . . .
CHM16
The process of enrolling the CHR via the consultee process was described by some as more challenging and less successful than expected. Relatives were introduced to the trial via letters sent by staff at the care home. The tracking of letters and responses was felt to be hit and miss, with the lack of control over the process being frustrating for some RNs. The lack of face-to-face communication about the trial with relatives who were more remote from the care home could also be frustrating:
It was easier when the consultee came to the home and you could do it face to face with the consultee. I found that an awful lot easier.
RN9
Care home managers also felt that engaging relatives via letter was of limited success. Direct communication with relatives, during visits or through meetings set up to discuss the trial, was a more efficacious method of introducing the trial before the consultee letters arrived. Trial posters that were put up in lifts and communal areas in the home also offered some success in raising awareness of the trial when relatives came in to visit the CHR:
. . . there was an advert, a notice up in the office, and on the window facing outwards for the people to see and they asked for volunteers and it seemed to be such a good idea.
Relative17
One CHM felt that the consultee process and the poor health of the CHRs who required consultee input required a lot of resources, particularly when the CHR may have limited life expectancy and so may not complete the follow-up period. For these reasons this CHM decided not to enrol CHRs who lacked capacity to make the decision to take part for themselves:
. . . instead of trying to figure out how to manage this medication with this person I would go to somebody who was lighter in dependency and who understands more and would take this medication as well.
CHM16
Data collection and division of trial-related activities
Data were collected from the CHR themselves, or through discussion with care home staff. RNs also collected data from the care home records, and medication administration record (MAR) charts, with assistance from the staff. Collecting data about CHRs via the care home staff could be challenging, as staff were often very busy.
Data pertaining to SP adherence and antibiotic use were collected on MAR charts, and then transferred on to the trial weekly records by the RNs. Although antibiotic use was generally well documented, adherence to SP was sometimes not recorded, although nursing staff appeared to be more reliable than carers in this respect. Similarly, the reporting of cases of diarrhoea was seen as far more convincing if it came from the nursing staff:
I would ask [the carers] about various things and sometimes they don’t know how. But they were definitely 100% sure or certain on the nursing side. . .
RN6
Research nurses felt that there was a fine balance between not attending the home too frequently to collect data (to avoid overburdening the care home staff) and not leaving it more than 2–3 weeks, which might lead to issues with staff recall and archiving of paper records. If the home had electronic records this was less of a concern, but there were some frustrations among RNs that they had to access the information about the CHRs via the staff:
. . . But the staffing side of it was frustrating sometimes. A little bit, it felt a little bit out of your control . . . with other research that we do . . . We’re not relying on other people to necessarily provide us, like we always felt like there was a middle man.
RN7
ICECAP-O and EQ-5D-5L data from CHRs with capacity were collected by the RN, CHM or care home staff. RNs tended to leave the proxy versions of the questionnaire for carers to complete, which had a variable success rate. This was attributed to the time pressures on carers and their duty to prioritise other tasks, but there was also an impression that carers were anxious about completing it because they were worried that they might get the answer ‘wrong’. RNs speculated that carers may not be familiar with being asked their opinion on matters such as perceived capability and well-being. Offering their thoughts on such measures was outside the scope of their usual care role, which may have affected their confidence in completing them.
Obtaining stool samples
Obtaining stool samples was delegated to the care home staff. The majority of RNs had issues getting the staff to obtain stool samples. Efforts to facilitate the process, which included leaving labelled pots, writing instructions, leaving phone numbers and pre-preparing envelopes, did little to improve the collection and mailing of stool samples. RNs intimated that having an intermittent presence at the home meant that they could not always be there at the necessary time to remind carers to collect a sample from CHRs. Effective communication about sample collection was also complicated by shift patterns, and because the senior care staff (who were the main point of contact for the RNs) were not necessarily undertaking the CHRs’ personal care:
. . . And even those who had that sort of care, because we weren’t there to remind the carers every day. And they would, you know, they’d have a handover maybe from the night before to the day staff. But it wasn’t part of the normal care so, often didn’t get handed over.
RN9
One RN indicated that CHRs with capacity were happy for their stool samples to be taken, but often needed assistance. Some felt that success with stool samples would improve if a member of the care team who was providing their personal care was specifically delegated that particular responsibility. However, it was noted that obtaining trial samples competed with other priorities for the care staff:
. . . It was probably, I think if you need a sample, you need one person to take responsibility for it. And it’s not top of their agenda.
RN2
Sustainability and routinisation
Although some RNs found that research activities and the collection of samples were maintained between their visits to the home, others felt some that some activities, particularly stool sampling, monitoring diarrhoea and proxy questionnaires, fell by the wayside. This was put down to the fact that they were not present in the home to remind the staff:
. . . You needed to tell them the night before if they go, here’s the pot and explain what to do, but because we were only there once a week, we would be leaving the stuff, asking them . . . if they can collect it whenever they go and you’d come back a week later and it was completely forgotten about. So, that was really difficult to get because we weren’t there . . . to give any reminders.
RN9
There was some feedback from CHMs and RNs that, had there been more CHRs recruited at the site, the research activities would have been very difficult for the care home staff to carry out without extra resource and support in the home:
. . . I think if the take-up had have been that many, I might have had to say we’ll have to drop so many off, because unless somebody else could do it . . .
CHM10
Similarly, taking over research activities in a home when there were a large number of CHRs (≥ 20) enrolled was very challenging for RNs, particularly if the care home environment was unfamiliar and relationships hadn’t yet been built with staff:
. . . I found it a lot easier going into the home where there wasn’t as many people enrolled and starting afresh . . . the regularity of somebody attending was lost, probably for about a month . . . made a massive difference to somebody coming to back in again. I think whenever I came, it was a chore almost for them.
RN9
Mechanisms of implementation
Three themes were identified as facilitating the implementation of the trial in the care homes: (1) the potential benefit of the probiotic, motives for participation and perception of trial ‘value’; (2) leadership and responsibility within the care home; and (3) familiarity with the routines and ‘rhythm’ of the care home.
Potential benefit of probiotic, motives for participation and perception of trial ‘value’
The active SP was recognised as a food supplement and not as a medication to treat or correct a condition. Therefore, being allocated the placebo SP was not viewed as undesirable. Some CHMs, despite being blind to SP allocation, expected the probiotic to confer therapeutic benefits and attributed the improved health of some CHRs to them receiving the ‘active’ SP:
. . . then we looked to join as well, really interested to see would we see any positive benefits . . . [another home] were feeling some positive results, we were really keen that you know we would join as well.
CHM12
The PRINCESS trial was contrasted with a drug trial, where the risks to participants were seen as far higher. The active SP was presented by interviewees as a food supplement, from which the risks to CHRs were negligible, particularly as CHRs can be closely monitored in the care home environment. Reported adverse effects potentially related to the SP were rare, but the contained setting of the care homes maximised the chance of side effects being noticed and reported:
. . . I wasn’t worried about because, because the residents are in a safe setting. They are obviously monitored, they’re looked after . . . I don’t think from a risk point a view that there isn’t much there, no.
RN3
There was collective mention of the positive impact that the probiotic could have on infections and hospitalisations, particularly in the older population, because of the severe and lasting impact that infections can have. All groups of interviewees also mentioned this in the context of the need for greater antibiotic stewardship:
. . . I think that [probiotics are] worthwhile, that’s my personal thoughts are, they are worthwhile, because I don’t think that continual use of antibiotic is doing anybody any good.
Relative18
Research nurses indicated that they formed good relationships with CHMs, who were motivated by their CHRs to improve quality of life, reduce infections, and provide new and novel experiences, and who wanted CHRs to continue to feel part of wider society:
. . . it’d be nice to be involved and involve our residents, I mean two of the residents were just so excited you know to be a part of this study.
CHM10
Care home managers and RNs discussed a range of motivating factors for delivering the research within the care home environment. As well as the potential health impact of the probiotic to the individual, participating in the trial also provided an opportunity for social interaction between the CHRs and the RNs. CHMs and RNs indicated that involving CHRs in research was valuable in and of itself. The perception of CHMs and RNs was that the opportunity to participate in the PRINCESS trial conferred a sense of serving society and making a difference, which enhanced CHRs’ well-being. CHMs’ recognition of the complex set of potential benefits offered by running the trial at the home appeared to accompany effective prioritisation and routinisation of trial activities in the home:
[CHM] was a nurse. Extremely keen on seeing more research in the care home. Couldn’t be more helpful and encouraging and you know, so that, that made it very easy and that was obviously brilliant.
RN3
Leadership, expectations and responsibility
Care home managers discussed their expectations and encouraged feedback from staff about the trial. They recognised the number of activities the staff were already expected to undertake as part of their core role. Sometimes CHMs took responsibility for particular tasks (e.g. gathering MAR charts for the RNs so that they could assess adherence to SP). Although the majority of research activities were carried out by the RNs, some CHMs stepped in to support RNs. This was helpful during the recruitment period because the CHM was already known and trusted by relatives and CHRs:
. . . [my relative] was living in was one of the homes that were taking part in it, CHM contacted me, asked me what I thought . . .
Relative18
Care home managers delegated tasks to the staff who had received training on the trial, but also recognised that the staff needed feedback (from either themselves or the RNs) and supervisory oversight. CHMs praised their staff and acknowledged that a lot is expected of them. CHMs felt that involving the home in the trial could offer an opportunity for their professional development by enabling them to experience research first-hand and do something different from their routine work:
. . . research doesn’t really happen in this sort of setting and it was valuable for the staff really to see how a research project is done in this sort of setting so I do feel that it benefited the staff, the clients and the home really.
CHM15
From the RNs’ perspective, CHMs who were motivated and engaged with the research appeared to set a precedent, which their staff followed. Research-enthusiastic CHMs, who were often present at the home, were able to communicate their expectations with staff. CHMs who were seen to take responsibility and ownership for hosting the trial and supporting the research activities created a model for the staff to follow:
I think it was down to [the CHM’s] leadership . . . she just made it fit into the home and I think the staff have a good relationship with [CHM] . . . I think that makes a difference . . . with some studies, you go into homes and the manager’s like, ‘yes, yes, we’ll do this, and we’ll do that’ . . . and the care staff are like ‘eugh’.
RN4
Care home managers’ co-ordination and leadership was particularly important during the set-up of the trial. CHMs and RNs could agree on slots of time to undertake staff training, which CHMs could instruct their staff to attend. Effective communication between CHMs and RNs about staffing was vital to ensure that new members of staff could be trained prior to giving SP to CHRs:
. . . I think it’s really important to get the managers on board . . . So that the enthusiasm comes from the top . . . Because they’ve got to keep trying to motivate the staff as well . . . It’s not easy.
RN2
Research nurses felt that delegation of research duties to staff (e.g. taking stool samples and asking staff to take responsibility for trial tasks) had to come from the CHM. RNs were concerned that asking for tasks to be done repeatedly could damage the relationship that they had built up with the staff:
. . . it was really tricky getting those stool samples. Very frustrating and it was very difficult for us to know how to motivate people to do it without changing the relationship that we had with the staff in the home . . . that’s the care home manager’s role . . . you can [only] ask for something to be done so many times, can’t you?
RN7
Understanding the routines and ‘rhythm’ of the care home
Becoming familiar with the routines and general workings of the homes facilitated relationship building and helped the RNs to plan trial activities in advance. CHMs appreciated the input and support that RNs could offer to the home:
. . . the RNs that were coming back and fore were very forthcoming, very personable towards the staff and residents and their families . . . they sat in the environment and you couldn’t tell they were here, became part of the team.
CHM15
Research nurses explained that as they spent time at the care home they became familiar with the internal machinery and rhythm of the home. Certain times of day could be very busy, so RNs had to assess this and adjust their schedule accordingly. RNs acknowledged that it could be challenging for staff to carve out a relatively small amount of time (e.g. 30 minutes) to discuss CHRs with them, so planning a small amount of time at regular intervals (e.g. weekly) was more productive. RNs also indicated that arranging their meetings with administrative and managerial staff in advance of attending the care home meant that they could use their time in the home more effectively:
I think maybe with those, with those training dates when nobody turned up, I think maybe we could have just call, called the home a couple of days in advance and made sure it was still OK to go.
RN7
Contextual factors
As the trial progressed some RNs indicated that they relied more on the nursing staff to undertake certain tasks, such as completing EQ-5D-5L and ICECAP-O questionnaires, rather than the carers because the care staff did not have the capacity to undertake the extra activity. This was attributed to the fact that carers were responsible for the CHRs’ personal care, which was very physical, and took up a great deal of their time. CHMs recognised the heavy workload of their caring staff, and relied on feedback from them to understand how they were managing the processes:
Interestingly, as time went on . . . the carers became less keen to do the questionnaires and I tended to rely on the qualified staff to do the questionnaires, which was fine because they were happy to do them. They, they, the carers, it was always a little bit difficult to get them to do them because of time pressures on them.
RN2
Gathering data from nursing staff about the participating CHRs appeared to inspire greater confidence around the validity of the data compared with data gathered from carers. RNs felt that the medical background of the nursing staff meant that they better understood the value of the research, and the importance of communicating accurate information about the CHRs:
. . . sometimes, if they’d had diarrhoea, that wasn’t actually recorded. So, I think that again is possibly due to them not being trained nurses and that sort of thing.
RN1
Research nurses felt that trained nurses were more accustomed to completing medication charts, and recognised the importance of recording medical events, such as diarrhoea. They felt that nurses understood the importance of documenting the SP even though it was not a medication; however, these views were not expressed by CHMs.
Organisational readiness for implementing research was driven by the interests and priorities of the CHMs. CHMs who had an interest in probiotics and were interested in reducing infections and improving the lives of their CHRs were able to implement the research at their homes with the support of the RNs. Factors such as electronic versus paper record-keeping appeared to have less of a bearing on effective data collection. It was important that records were available and accessible, so locking records away, or filing them in a room that was often used for meetings, was sometimes a barrier to data collection. Similarly, if electronic records were kept, it was necessary for a member of staff to be available to access them on behalf of the RN:
. . . they’ve got a computerised [system] so, they can go on each resident and sort of click and see but, they certainly weren’t going to sit with me with the system up for every single person and go through.
RN11
Qualitative evaluation summary
Research nurses, CHMs and relatives of CHRs perceived the trial intervention as very low risk, with important potential benefits. The data presented here echo the findings of previous research, that is that the acceptability of research studies (including interventional trials) with CHRs is dependent on the research being perceived as low risk and absolutely relevant to the care home population. 56 The PRINCESS trial was implemented in a setting where CHRs were closely monitored by the care staff and RNs, which made the involvement of vulnerable older people acceptable. From the perspective of CHRs’ relatives, any intervention that had the potential to reduce the frequency and impact of infections in their loved one was highly desirable. Despite there being an evidence gap concerning the effectiveness of the probiotics used in the PRINCESS trial to prevent all-cause common infections and antibiotic prescribing in this population,1 there was an expectation from some interviewees that participating in the trial would provide those randomised to the active SP with access to a beneficial substance. 86 Therefore, the potential impact of the intervention was likely to have played a key role in the acceptability of the trial with this population. 86
Despite the perceived low risk of the intervention, the most frequently discussed barrier, particularly regarding those without capacity, was taking the SP capsule alongside their regular medication. This feedback was not surprising, given the level of polypharmacy in CHRs with complex needs. 87–89 There was a need to discuss this particular aspect of the trial with the CHR, relatives and staff at the care home during the screening process. There were some concerns (from CHRs and relatives) that the addition of the SP may have a negative impact on health or well-being. An unintentional consequence of this may have been that CHRs with less complex health needs were identified as being suitable to take part in the trial. RNs and CHMs reported that, once CHRs with capacity enrolled on the trial, they generally did not have any issues taking the SP with their other medication.
Implementing the recruitment process was far more administratively complex when CHRs lacked the capacity to provide consent. Engaging with consultees by letter was difficult to monitor effectively and RNs felt more comfortable if they were able to speak face to face with the relative about the trial. Posters and group meetings with relatives were effective methods of engaging those who attended their relative on a more regular basis.
The consultee model of asking a relative to provide advice on behalf of the CHRs was viewed as more acceptable than approaching a professional (i.e. a staff member from the care home) to act as a nominated consultee; it was important that the consultee had known the CHRs before they lost capacity. This is somewhat in keeping with previous exploratory studies around the acceptability of utilising the advice of personal consultees for CHRs who have lost capacity. 56 ‘Gatekeeping’ by practitioners in control of recruitment (via the rejection of one of the approved approaches to recruitment) is a common research phenomenon, particularly when accessing more vulnerable populations. 90–92 This inauspicious view of the nominated consultee enrolment route does, however, leave those CHRs without family unable to participate in research, and risks further underserving an already under-represented population. 93
Interviewees observed that relatives acting as potential consultees utilised a ‘best interest’ perspective (e.g. they felt that participation might unsettle their relative) to inform their decision to give advice for or against participation, or felt bound by a moral imperative for or against participation. 94 The latter basis for decision-making was evidenced when consultees spoke about the type of person their relative was before they lost capacity, indicating, for example, that they disliked taking any unnecessary medication (aligned with advising against trial participation) or that ‘this was right up Dad’s street’ (aligned with advising for participation). The Mental Capacity Act 200558 requires consultees’ decisions to be based on the person’s wishes and feelings about participation; however, decision-making is more complex, as reported by the consultees in this study.
The ability to build relationships and gain entry to care homes, and thus undertake research activities, was heavily facilitated by the leadership, motivation and presence of the CHM. From a research perspective, CHMs are often described as ‘gatekeepers’ of the care home environment. The trial held a great deal of value from the perspectives of the CHMs and, thus, RNs were valued and welcomed into the care home setting. In addition, there was some evidence that consultation between the CHM and care home staff about the trial, and communication around expectations of their required involvement, promoted entry, RN–staff relationships and trial progress. 57 However, despite high levels of CHM and staff motivation, and a good working relationship between the CHMs and RNs, it could be difficult to maintain or sustain research activities when the RNs were absent from the home for longer periods of time (e.g. staff on annual leave or leaving their role). Therefore, it is likely that RN support, and regular care home engagement and presence, is still required when undertaking trials like PRINCESS in care homes. 95
Although not reported by all RNs interviewed, there was a perception by some that the presence and involvement of nursing staff (i.e. staff with qualifications in nursing recognised by the Nursing and Midwifery Council in the UK) in the home better facilitated the gathering of valid information about the CHR and the reliable undertaking of particular tasks (e.g. recording SP adherence, questionnaire completion). Nurses working in the care home context were also contrasted with ‘carers’ (i.e. staff trained in providing CHRs’ personal care) with regard to their perceived confidence, how busy they were and how much they valued the importance of the research. This contrast was not particularly mentioned by CHMs or relatives of participating CHRs. It is possible that there were some qualitative differences between the homes where nursing staff were more involved in the research than carers; however, without an interhome comparison of data quality and quantity, it is difficult to determine if this is indeed one of the many variables that can predict good-quality data collection. 95 Literature around how carers, nursing staff and research professionals work together to undertake research in long-term care facilities is still in its infancy, particularly from the perspective of care assistants, so there is little with which to compare this study’s findings. However, variables such as previous care home experience, relationships with CHRs, interstaff relationships and staff inclusion do appear to play a part in work-related identities and relationships, and may influence the implementation of research activities in care homes. 96,97
Limitations
Although this qualitative evaluation aimed to gather in-depth views from groups of individuals (CHMs, RNs, care home staff, CHRs and relatives of CHRs) involved in the PRINCESS trial, no care home staff (aside from managers) or CHRs volunteered to be interviewed. Despite two relatives of PRINCESS CHRs kindly providing valuable insight into their experience of the trial, from their own perspective and that of their relative, data saturation was not reached in this group of interviewees and, thus, a broad and thorough exploration of the trial from the viewpoint of loved ones was not achieved. The absence of staff’s and CHRs’ views is unfortunate but not unexpected considering the recruitment strategy used. Undertaking research in care homes can be very time-consuming, and the effectiveness of the strategy used to recruit interviewees for this qualitative evaluation serves as a timely reminder that a great deal of time and resource is still required to support research in the care home environment. 57
Furthermore, the views expressed in this evaluation may be biased in favour of the trial, and the associated processes involved with its implementation. Although the objectives of the evaluation were to explore the implementation of the trial, CHMs who agreed to carry out the research in their care home may have been characteristically different, in terms of infrastructure and readiness for research, from other, non-participating, CHMs. Similarly, many of the RNs who were interviewed were experienced researchers, several of whom had a professional background in particular settings, which may have eased the implementation of research in the homes. It is unclear how many staff, CHRs and relatives were approached to take part in this qualitative evaluation and declined. Therefore, the findings reflect the experiences of the relatively small numbers of CHMs, relatives and RNs who participated.
Conclusions
To our knowledge, this is the first qualitative exploration of a double-blind, randomised, placebo-controlled trial carried out in the care home context. The acceptability of the trial, and the readiness to enrol CHRs without capacity, was justified by the low risk and vigilance of the care home setting. The implementation of a placebo-controlled trial in care homes was facilitated by a present and available CHM or clinical lead who demonstrated strong leadership by taking responsibility for trial activities and demonstrated a robust appreciation of the compound benefits that the trial could have for the residents of their home. Nevertheless, conducting this placebo-controlled trial with the care home population undoubtedly required the sustained engagement and activity of trained researchers to set up and maintain research activities in the homes. PRINCESS trial researchers were trained health professionals and had a good understanding and awareness of how care homes function. Their professional identity and experience in a caring profession facilitated the development of good care home staff–researcher relationships, which was key in the effective implementation of the trial in the care home setting.
Chapter 6 Discussion and conclusion
Summary of clinical efficacy findings
The PRINCESS trial found that administration of a daily dose of the probiotics Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 did not reduce CAAD for all-cause, acute infections in CHRs. All secondary outcomes were consistent with the main finding in that we found no evidence of a beneficial effect of the probiotic combination we studied on antibiotic use overall or on the main categories of common infections that affect the population we studied.
Similarly, the intervention did not favourably affect duration of infections and hospitalisations, death, AAD, immune function measurements or health status, capability and quality of life. Our study population consisted of CHRs in the UK with an average age of > 85 years; two-thirds of participants were women and two-thirds of participants lacked mental capacity to consent. Approximately 21% of participants died during follow-up. We found that those randomised to the probiotic combination arm were administered antibiotics on 10% more days than those in the placebo arm. A 10% difference was prespecified as a clinically meaningful difference, but was not statistically significant, because the power of the study was lower than anticipated as a result of fewer events overall than predicted in our power calculations.
Mechanistic findings
In the mechanistic substudies, we found no evidence that the probiotic combination we studied had a significant effect on blood immune cell numbers or subtypes, or on responses to seasonal influenza vaccination.
Qualitative evaluation
The qualitative interviews found that conducting a randomised placebo-controlled trial in a care home was acceptable to care home staff, family members of CHRs and RNs. The implementation of the trial was facilitated by engaged CHMs; however, the sustained involvement of trained researchers to set up and maintain research activities in the homes was required. The relationship between the RNs and care homes was seen as key to the effective implementation of the trial in the care home setting.
Strengths and limitations
Studies of probiotics often produce contradictory findings and have been criticised for poor design, selective reporting and selective inclusion in systematic reviews, poorly described and verified outcomes, inadequate reporting of harms, and reliance on surrogate outcomes. 98
The PRINCESS trial was a double-blind, placebo-controlled trial that recruited successfully to its revised power target.
Randomisation using minimisation according to care home and CHR sex resulted in comparable intervention groups in terms of medical history (including previous antibiotic use); demographic, haematological and microbiological parameters; health status; capability; and quality-of-life measures (i.e. very similar). To maximise the generalisability of the findings of this trial to the wider care home population, an inclusive approach to recruitment was adopted. It is important that populations included in clinical trials should adequately represent the population relevant to the proposed clinical practice94,99 and, thus, adults lacking capacity to provide consent to participate in clinical trials for themselves should be included in relevant health research, unless there are good scientific reasons justifying their exclusion. 53
There is no established minimal clinically important difference for ICECAP-O score. However, a study100 has previously compared the measure in an older, post-acute patient population with the measure in the general population and found a mean difference on the ICECAP-O of 0.04 points. Although this cannot be used to confirm an important difference, our difference of 0.06 points (in the direction of worse well-being for participants allocated to probiotic) was greater, and may be indicative of a distinctive difference between arms.
The probiotic combination was quality assured by the manufacturer and probiotic and placebo were cultured after labelling to ensure correct labelling. SP pack identification numbers were allocated using randomly permuted blocks.
The SP was given to participants by care home staff using existing, routine care home medication administration processes. Administration of SP was recorded on the standard MAR sheets, and the responsible person reported how the SP was given, and whether it was fully, partially or not consumed. Relevant documentation was signed as correct by the responsible care home staff member. We captured daily SP usage data for a total of well over 70,000 days, with > 90% of the SP recorded as having been taken at the full dose. We are able, therefore, to verify adherence by a large proportion of study participants, which is likely to have been higher than a study of ‘free-living’ older people.
Stool samples pre and post probiotic from participants who volunteered for this additional aspect of the study confirmed that the probiotic combination was correctly produced, labelled, taken by participants and entered the gut.
The probiotic and placebo were provided to the study by the manufacturer as an unconditional grant, and the team delivering this publicly funded study was fully independent of the manufacturers of the probiotic combination or free from any other potentially relevant financial conflicts.
Studies of probiotics in older people have suffered from potential bias from poor ascertainment of outcomes. 23 The PRINCESS trial was able to guard against this by ensuring that participants were visited each week by a registered nurse blind to the participant’s allocation. Outcome data were prospectively obtained from the MAR sheets, care home clinical records, hospital records, observation of the participant and discussion with the participant (where possible) and the participants’ friends and family and care home staff. Weekly participant diary data were available for 97.4% of participants randomised to placebo and 98.7% of participants randomised to the probiotic combination. There were missing days for only 1.3% of the days initially scheduled for data capture using the study weekly diary.
Analysis was conducted only once the database had been locked. Analysis was done according to a statistical analysis plan signed off by an Independent Data Monitoring Committee and checked by a statistician independent of the study team. The allocation code was broken only once that statistical analysis report was finalised.
However, study progress and participant recruitment were slower than anticipated, owing mainly to longer than anticipated contracting processes, and difficulty in engaging with consultees. The latter problem was addressed by obtaining approval for a broader range of consultees who could be consulted on behalf of CHRs. We had planned to follow up all participating CHRs for a full year whenever possible, but these delays meant that follow-up had to be truncated for some. A total of 36.5% of participants were followed up for a full year as planned. However, as expected, about one-fifth of study participants died during follow-up and we were not able to follow up a minority (34.2%) of participants for a full year because of study delays. Nevertheless, every participant could have been followed up for at least 6 months. Fewer events, overall, than predicted in our power calculations meant that the study was underpowered to detect a statistically significant clinically meaningful difference in the primary outcome.
Our findings apply only to the probiotic combination studies for CAAD in CHR. Probiotics and probiotic combinations are likely to differ in their efficacy by setting, study population and outcomes measured.
Interpretation of results
These findings show that daily oral administration of the probiotics Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 does not reduce CAAD for all-cause acute infections in this population, or duration of infections, and does not have a measurable benefit on quality of life or a range of infection-related secondary outcomes. Although evidence of no benefit is a robust finding across all the outcomes that we measured, there was a trend towards harm with many of the outcomes, with better evidence for some analyses; for example, more people receiving probiotics experienced LRTIs, and CHRs randomised to the probiotics arm experienced a greater burden of AAD in one measure. Although such findings should be treated with caution, given multiple testing, it is noteworthy that findings in 21 out of 33 secondary outcomes trend away from benefit of the probiotic combination we studied.
Implications for health and social care
Based on these findings, CHRs should not be advised to consume a combination of the probiotics Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 each day to reduce antibiotic administration. It is important to highlight that the conclusions are relevant only for these specific strains of probiotic; we cannot conclude that no probiotic would be helpful in this population. We are not able to make recommendations about other probiotics or probiotic combinations because certain effects of probiotics may be strain specific. Neither are we able to indicate that these findings are applicable to other populations in different settings. It is also important to highlight that probiotic supplementation may vary according to immune status and age; we cannot conclude that a probiotic would be helpful in an enriched subgroup of the population (e.g. those who are immunocompromised, those aged > 80 years or those with the highest level of frailty).
Future research implications
Global sales of probiotics have been valued at over $40B and are projected to reach over $64B by 2023. 101 Some of these probiotics are marketed on the basis of health claims (although no such claims are currently permitted in the European Union), so rigorous research is needed to support evidence-based decision-making about probiotic use. AMR is a public health threat of global significance, and there is widespread interest in interventions that can replace or reduce antibiotic use, such as through the prevention of infections. 102
A systematic review19 of probiotics to reduce antibiotic use for common infections included 17 RCTs, all conducted in infants and children, and covered 13 different probiotic formulations, all comprising single or combination Lactobacillus and Bifidobacterium strains. The review found that probiotics reduced the risk of antibiotic prescription relative to placebo. Mean duration of probiotic supplementation ranged from 4 days to 9 months. The review found significant statistical heterogeneity among study results and variable study quality, with many studies being criticised for poor reporting of outcomes. 19 A further systematic review103 that included 20 RCTs explored the effect of probiotics, specifically Lactobacillus and Bifidobacterium strains, on the duration of acute respiratory infections in otherwise healthy children and adults, and found that these probiotics reduced the duration of respiratory illness. 103
Despite these systematic reviews of trials finding that probiotics had a favourable impact on antibiotic use in children, and on the duration of infections in children and healthy adults, we found evidence of no benefit from the probiotics we studied in a frail, care home population. Our findings are congruent with a more recent meta-analysis on the effectiveness of probiotics in preventing infections in older people, which included 15 studies covering 5916 participants with a mean age of 75.21 years. The authors concluded that the quality of this evidence was poor, that it did not support the use of probiotics for reducing infections in older adults, that safety outcomes were similar between probiotics and placebo and that more research was needed. 104 However, our study does not suffer from many of the potential biases of the studies included in the reviews, or in the study by Van Puyenbroeck et al. ;23 their trial included largely healthy older people, and it found no benefit in terms of antibiotic use or duration of common infections from the probiotic L. casei Shirota, but there was a high risk of ascertainment bias because of missing data.
Regarding mechanisms and potential harms, recent research has suggested that certain probiotics may delay return of the host gut microbiome to its normal state. 105 A retrospective, single-centre study found probiotics to be a risk factor for C. difficile infection in hospitalised patients. 13 Our trial and associated mechanistic findings could not rule out potential harm from this probiotic combination; however, SAEs were similar in our study groups.
Our mechanistic substudy results are in keeping with the clinical outcomes of the trial. Systematic reviews have indicated positive effects of other strains of probiotics in healthy elderly subjects106 and, hence, our findings may not be applicable to other populations and probiotic species.
Because probiotics are a feasible and cheap potential intervention, further rigorous efficacy, mechanisms and effectiveness trials of other probiotics in other population groups and settings may be indicated regarding antibiotic use and susceptibility to and recovery from common infections. Potential harms should be carefully studied in such trials.
Acknowledgements
Acknowledgement of contributions
Other members of the trial team
We would like to acknowledge the late Professor George Lewith (deceased 17 March 2017) for contributing to the development of the research question, study design, obtaining funding and implementation of the study protocol.
In addition to the authors, the PRINCESS trial team comprised other colleagues whom we would like to thank and acknowledge for their contribution to the trial implementation and management: Ms Katy Addison, Ms Cathy Lisles, Ms Charlotte Scoble and Ms Rachael Lee.
Patient and public involvement
The authors would like to acknowledge the contribution of Mr Alun Toghill and Mr James Downs as the public and patient involvement representatives in the Trial Management Group.
Clinical trial units
Two UK Clinical Research Collaboration-registered clinical trials units were involved in the study. The Centre for Trials Research receives funding from Health and Care Research Wales and Cancer Research UK, Cardiff University and University of Oxford Primary Care Clinical Trials Unit.
Independent members of the Trial Steering Committee and Independent Data Monitoring Committee
We would like to acknowledge the contribution of the Trial Steering Committee members for their advice and support – namely Professor Steve Iliffe (University College London), Professor Glenn R Gibson (University of Reading), Dr Jonathan Sandoe (University of Leeds), Dr Meena Rafiq (University College London), Mr Robin Willmott (General Manager, Millbrook Lodge Care Home) – and the members of the Independent Data Monitoring Committee – namely Dr Stephen Bremner (Brighton and Sussex Medical School), Dr Neil Haslam (Royal Liverpool and Broadgreen University Hospitals NHS Trust) and Dr Matthew Ridd (University of Bristol).
Specialist Antimicrobial Chemotherapy Unit, Public Health Wales at the University Hospital of Wales
We would like to acknowledge and thank Ms Jennifer Richards and Ms Leanne Davies of the Specialist Antimicrobial Chemotherapy Unit, Public Health Wales at the University Hospital of Wales, for their dedicated help in processing and analysing the microbiology samples.
Human Development and Health, Faculty of Medicine, University of Southampton
We would like to acknowledge and thank Ms Vivian Castro Herrera and Dr Elizabeth A Miles for their dedicated help in processing and analysing the blood samples for serum cytokine analysis, and for preparing the serum samples for Public Health England.
Southampton General Hospital Pathology Care Group, Southampton General Hospital, University Hospital Southampton NHS Foundation Trust
We would like to acknowledge and thank Ms Wendy Skelton for help in processing and analysing the blood samples for FBC and vitamin D.
Public Health England
We would like to acknowledge and thank Katja Hoschler for help in analysing the flu substudy serum samples for flu subtype/classification at the Public Health Laboratory in London.
Nottingham University Hospitals NHS Trust
We would like to acknowledge and thank Adam Millington, Specialist Pharmacist, Production Services and Clinical Trials, for labelling the SP.
Recruitment sites
We are grateful to all of the CHMs, staff and CHRs who contributed towards this trial, and our thanks go to all of the care homes who participated in this study for their commitment in recruiting participants and engaging in study processes.
Research nurses employed by Cardiff University covered the following care homes: Shire Hall Care Home (Cardiff), Ty Enfys Care Home (Cardiff), Greenhill Manor Care Home (Merthyr Tydfil), Millbrook Care Home (Pontllanfraith), Ysguborwen Care Home (Aberdare) and Hill View Care Home (Bargoed).
Research nurses employed by the University of Oxford covered the following care homes: Homestead Care Home (Oxford), Freeland Care Home (Oxford), Lake House Care Home (Oxford), Ferendune Court Care Home (Oxford), Leafield Care Home (Oxford), Sandown Park Care Home (Oxford), Rosebank Care Home (Oxford) and Churchfields Care Home (Oxford).
Research networks and NHS trusts
We would like to acknowledge and thank the following CRNs and NHS trusts for their support in identifying care homes, recruiting participants and carrying out data collection at these sites: Yorkshire and Humber CRN (Magdalen Park Care Home and Westfield Park Care Home), West Midlands CRN (Berwood Court Care Home and Southcrest Nursing Home), Thames Valley and South Midlands CRN (Rosebank Care Home and Churchfields Care Home), Northumbria Healthcare NHS Foundation Trust (Ashington Grange Care Home and Castleview Care Home) and Derbyshire Healthcare NHS Foundation Trust (Ashbourne Lodge Care Home and Milford House Care Home). A RN from Cwm Taf University Health Board carried out data collection in Greenhill Manor Care Home and Ysguborwen Care Home.
Participants
Finally, and most importantly, we would like to thank all those who participated in the trial, and their consultees (if applicable), without whom this trial would not have been possible.
Contributions of authors
Professor Christopher C Butler (https://orcid.org/0000-0002-0102-3453) was the chief investigator for the study. He contributed to the conception and overall design of the study, obtaining funding and the implementation and interpretation of the work. He led the drafting of the report.
Dr Eleri Owen-Jones (https://orcid.org/0000-0003-0850-4724) (Research Associate) was the trial manager, contributing to the trial design and implementation, acquisition of data and the writing of the Abstract and Chapters 1 and 2 of the report. She also co-ordinated the compilation, formatting, proofreading and final approval of the report.
Mandy Lau (https://orcid.org/0000-0001-5894-570X) (Research Associate) was the trial statistician, conducting the statistical data analysis and contributing to the writing of Chapters 2 and 3, critical review of the report and final approval of the report.
Dr David Gillespie (https://orcid.org/0000-0002-6934-2928) (Deputy Director of Infection, Inflammation and Immunity Trials, Senior Research Fellow) was a co-investigator and contributed to the trial design, conducting the statistical data analysis, the writing of Chapters 2 and 3, critical review of the report and final approval of the report.
Dr Mark Lown (https://orcid.org/0000-0001-8309-568X) (GP and Clinical Lecturer) was a co-investigator and contributed to the overall trial design, interpretation of the findings, the writing of the mechanistic sections of the report, critical review of the report and final approval of the report.
Professor Philip C Calder (https://orcid.org/0000-0002-6038-710X) (Professor) was a co-investigator and contributed to the overall trial design, interpretation of the findings, the writing of the mechanistic sections of the report, critical review of the report and final approval of the report.
Helen Stanton (https://orcid.org/0000-0003-0197-3667) (Research Assistant) was the data manager and led the design and implementation of the qualitative substudy and contributed to the acquisition and interpretation of data, the writing of Chapters 2 and 5 of the report, critical review of the report and final approval of the report.
Dr Mandy Wootton (https://orcid.org/0000-0002-2227-3355) (Consultant Scientist/Operational Manager, Specialist Antimicrobial Chemotherapy Unit) was a co-investigator and lead microbiologist, and contributed to the overall trial design, acquisition of data, interpretation of the findings, the writing of the microbiology sections of the report, critical review of the report and final approval of the report.
Vivian Castro Herrera (https://orcid.org/0000-0003-0422-5496) (PhD Student) was a co-investigator and contributed to the laboratory work, interpretation of the findings, the writing of the mechanistic sections of the report, critical review of the report and final approval of the report.
Professor Antony Bayer (https://orcid.org/0000-0002-7514-248X) (Professor of Geriatric Medicine) was a co-investigator and contributed to the overall trial design, interpretation of the findings, critical review of the report and final approval of the report.
Dr Jane Davies (https://orcid.org/0000-0002-9409-8605) (Research Associate) was a RN and contributed towards acquisition of data and interpretation of the findings, critical review of the report and final approval of the report.
Alison Edwards (https://orcid.org/0000-0002-9603-2618) (Research Associate) was a RN and contributed towards acquisition of data and interpretation of the findings, critical review of the report and final approval of the report.
Mina Davoudianfar (https://orcid.org/0000-0002-2930-9437) (Clinical Trial Manager) was the trial manager and contributed to the trial design and implementation, acquisition of data and final approval of the report.
Heather Rutter (https://orcid.org/0000-0002-7297-2234) (Senior RN) was a RN and contributed to acquisition of data and interpretation of the findings, critical review of the report and final approval of the report.
Professor Kerenza Hood (https://orcid.org/0000-0002-5268-8631) (Professor of Trials and Director of Centre for Trials Research) was a co-investigator and contributed to the conception of the study and the overall trial design, supervised the statistical analysis and contributed to the critical review of the report and final approval of the report.
Professor Michael Moore (https://orcid.org/0000-0002-5127-4509) (Professor of Primary Care Research) was a co-investigator and contributed to the overall trial design, interpretation of the findings, critical review of the report and final approval of the report.
Professor Paul Little (https://orcid.org/0000-0003-3664-1873) (Professor of Primary Care Research) was a co-investigator and contributed to the overall trial design, interpretation of the findings, critical review of the report and final approval of the report.
Dr Victoria Shepherd (https://orcid.org/0000-0002-7687-0817) (Research Associate) was a co-investigator and contributed to the overall trial design, interpretation of the findings, critical review of the report and final approval of the report. She also co-ordinated the compilation, formatting, proofreading and final approval of the report.
Dr Rachel Lowe (https://orcid.org/0000-0002-7306-0085) (Research Fellow) was the Senior Trial Manager and contributed to the trial design and implementation, acquisition of data, critical review of the report and final approval of the report.
Elizabeth A Miles (https://orcid.org/0000-0002-8643-0655) (Lecturer) was a co-investigator and contributed to the overall trial design, interpretation of the findings, the writing of the mechanistic sections of the report, critical review of the report and final approval of the report.
Julia Townson (https://orcid.org/0000-0001-8679-3619) (Research Fellow) was the Senior Trial Manager and contributed to the trial design and implementation, acquisition of data, critical review of the report and final approval of the report.
FD Richard Hobbs (https://orcid.org/0000-0001-7976-7172) (Professor and Head of Primary Care Health Sciences) was a co-investigator and contributed to interpretation of the findings, critical review of the report and final approval of the report.
Professor Nick A Francis (https://orcid.org/0000-0001-8939-7312) (GP and Professor of Primary Care Research) was a co-investigator and contributed to obtaining funding, the overall trial design, interpretation of the findings, critical review of the report and final approval of the report submission.
Publication
Owen-Jones E, Lowe R, Lown M, Gillespie D, Addison K, Bayer T, et al. Protocol for a double-blind placebo-controlled trial to evaluate the efficacy of probiotics in reducing antibiotics for infection in care home residents: the Probiotics to Reduce Infections iN CarE home reSidentS (PRINCESS) trial. BMJ Open 2019;9:e027513.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Owen-Jones E, Lowe R, Lown M, Gillespie D, Addison K, Bayer AJ, et al. Protocol for a double-blind placebo-controlled trial to evaluate the efficacy of probiotics in reducing antibiotics for infection in care home residents: the Probiotics to Reduce Infections iN CarE home reSidentS (PRINCESS) trial. BMJ Open 2019;9. https://doi.org/10.1136/bmjopen-2018-027513.
- University of Southampton, National Institute for Health Research (NIHR) Dissemination Centre . Advancing Care: Research With Care Homes 2017. https://evidence.nihr.ac.uk/wp-content/uploads/2020/03/Advancing-Care-Final.pdf (accessed 20 January 2021).
- World Health Organization (WHO), Food and Agriculture Organization of the United Nations . Health and Nutritional Properties and Guidelines for Evaluation: Report of a Joint FAO WHO Expert Consultation on Evaluation of Health and Nutritional Properties of Probiotics in Food Including Powder Milk With Live Lactic Acid Bacteria 2006. http://fao.org/3/a-a0512e.pdf (accessed 20 January 2021).
- Boge T, Rémigy M, Vaudaine S, Tanguy J, Bourdet-Sicard R, van der Werf S. A probiotic fermented dairy drink improves antibody response to influenza vaccination in the elderly in two randomised controlled trials. Vaccine 2009;27:5677-84. https://doi.org/10.1016/j.vaccine.2009.06.094.
- Yamamoto S, Tsukamoto T, Terai A, Kurazono H, Takeda Y, Yoshida O. Genetic evidence supporting the fecal-perineal-urethral hypothesis in cystitis caused by Escherichia coli. J Urol 1997;157:1127-9. https://doi.org/10.1016/S0022-5347(01)65154-1.
- Graham JE, Christian LM, Kiecolt-Glaser JK. Stress, age, and immune function: toward a lifespan approach. J Behav Med 2006;29:389-400. https://doi.org/10.1007/s10865-006-9057-4.
- McFarland LV. Evidence-based review of probiotics for antibiotic-associated diarrhoea and Clostridium difficile infections. Anaerobe 2009;15:274-80. https://doi.org/10.1016/j.anaerobe.2009.09.002.
- Hempel S, Newberry S, Ruelaz A, Wang Z, Miles JN, Suttorp MJ, et al. Safety of probiotics used to reduce risk and prevent or treat disease. Evid Rep Technol Assess 2011;200:1-645.
- Butler CC, Duncan D, Hood K. Does taking probiotics routinely with antibiotics prevent antibiotic associated diarrhoea?. BMJ 2012;344. https://doi.org/10.1136/bmj.e682.
- Castro-González JM, Castro P, Sandoval H, Castro-Sandoval D. Probiotic lactobacilli precautions. Front Microbiol 2019;10. https://doi.org/10.3389/fmicb.2019.00375.
- Hempel S, Newberry SJ, Maher AR, Wang Z, Miles JN, Shanman R, et al. Probiotics for the prevention and treatment of antibiotic-associated diarrhea: a systematic review and meta-analysis. JAMA 2012;307:1959-69. https://doi.org/10.1001/jama.2012.3507.
- Bafeta A, Koh M, Riveros C, Ravaud P. Harms reporting in randomized controlled trials of interventions aimed at modifying microbiota: a systematic review. Ann Intern Med 2018;169:240-7. https://doi.org/10.7326/M18-0343.
- Carvour ML, Wilder SL, Ryan KL, Walraven C, Qeadan F, Brett M, et al. Predictors of Clostridium difficile infection and predictive impact of probiotic use in a diverse hospital-wide cohort. Am J Infect Control 2019;47:2-8. https://doi.org/10.1016/j.ajic.2018.07.014.
- Carlsson M, Gustafson Y, Haglin L, Eriksson S. The feasibility of serving liquid yoghurt supplemented with probiotic bacteria, Lactobacillus rhamnosus LB 21, and Lactococcus lactis L1A – a pilot study among old people with dementia in a residential care facility. J Nutr Health Aging 2009;13:813-19. https://doi.org/10.1007/s12603-009-0218-3.
- Beerepoot MA, ter Riet G, Nys S, van der Wal WM, de Borgie CA, de Reijke TM, et al. Lactobacilli vs antibiotics to prevent urinary tract infections: a randomized, double-blind, noninferiority trial in postmenopausal women. Arch Intern Med 2012;172:704-12. https://doi.org/10.1001/archinternmed.2012.777.
- Zaky A. Interventions to Prevent the Acquisition of Resistant Gram-Negative Bacteria in Critically Ill Patients – A Systematic Review and Meta-Analysis 2012.
- Manley KJ, Fraenkel MB, Mayall BC, Power DA. Probiotic treatment of vancomycin-resistant enterococci: a randomised controlled trial. Med J Aust 2007;186:454-7. https://doi.org/10.5694/j.1326-5377.2007.tb00995.x.
- Hao Q, Dong BR, Wu T. Probiotics for preventing acute upper respiratory tract infections. Cochrane Database Syst Rev 2015;2. https://doi.org/10.1002/14651858.CD006895.pub3.
- King S, Tancredi D, Lenoir-Wijnkoop I, Gould K, Vann H, Connors G, et al. Does probiotic consumption reduce antibiotic utilization for common acute infections? A systematic review and meta-analysis. Eur J Public Health 2019;29:494-9. https://doi.org/10.1093/eurpub/cky185.
- Turchet P, Laurenzano M, Auboiron S, Antoine JM. Effect of fermented milk containing the probiotic Lactobacillus casei DN-114001 on winter infections in free-living elderly subjects: a randomised, controlled pilot study. J Nutr Health Aging 2003;7:75-7.
- Guillemard E, Tanguy J, Flavigny A, de la Motte S, Schrezenmeir J. Effects of consumption of a fermented dairy product containing the probiotic Lactobacillus casei DN-114 001 on common respiratory and gastrointestinal infections in shift workers in a randomized controlled trial. J Am Coll Nutr 2010;29:455-68. https://doi.org/10.1080/07315724.2010.10719882.
- Makino S, Ikegami S, Kume A, Horiuchi H, Sasaki H, Orii N. Reducing the risk of infection in the elderly by dietary intake of yoghurt fermented with Lactobacillus delbrueckii ssp. bulgaricus OLL1073R-1. Br J Nutr 2010;104:998-1006. https://doi.org/10.1017/S000711451000173X.
- Van Puyenbroeck K, Hens N, Coenen S, Michiels B, Beunckens C, Molenberghs G, et al. Efficacy of daily intake of Lactobacillus casei Shirota on respiratory symptoms and influenza vaccination immune response: a randomized, double-blind, placebo-controlled trial in healthy elderly nursing home residents. Am J Clin Nutr 2012;95:1165-71. https://doi.org/10.3945/ajcn.111.026831.
- Office for National Statistics (ONS) . Changes in the Older Resident Care Home Population Between 2001 and 2011 2014.
- Hood K, Nuttall J, Gillespie D, Shepherd V, Wood F, Duncan D, et al. Probiotics for Antibiotic-Associated Diarrhoea (PAAD): a prospective observational study of antibiotic-associated diarrhoea (including Clostridium difficile-associated diarrhoea) in care homes. Health Technol Assess 2014;18. https://doi.org/10.3310/hta18630.
- Lingard L. Health Needs Assessment of the Residents of Nursing Homes in South Tyneside Executive Summary 2011. https://southtyneside.gov.uk/media/12231/Nursing-Homes/pdf/Nursing_Homes.pdf (accessed 20 January 2021).
- Costelloe C, Metcalfe C, Lovering A, Mant D, Hay AD. Effect of antibiotic prescribing in primary care on antimicrobial resistance in individual patients: systematic review and meta-analysis. BMJ 2010;340. https://doi.org/10.1136/bmj.c2096.
- Butler CC, Hillier S, Roberts Z, Dunstan F, Howard A, Palmer S. Antibiotic-resistant infections in primary care are symptomatic for longer and increase workload: outcomes for patients with E. coli UTIs. Br J Gen Pract 2006;56:686-92.
- Van Hecke O, Wang K, Lee JJ, Roberts NW, Butler CC. The implications of antibiotic resistance for patients’ recovery from common infections in the community: a systematic review and meta-analysis. Clin Infect 2017;65:371-82. https://doi.org/10.1093/cid/cix233.
- Cai J, Zhao C, Du Y, Zhang Y, Zhao M, Zhao Q. Comparative efficacy and tolerability of probiotics for antibiotic-associated diarrhea: systematic review with network meta-analysis. United Eur Gastroenterol J 2018;6:169-80. https://doi.org/10.1177/2050640617736987.
- Allen SJ, Wareham K, Wang D, Bradley C, Hutchings H, Harris W, et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2013;382:1249-57. https://doi.org/10.1016/S0140-6736(13)61218-0.
- Koch AM, Eriksen HM, Elstrøm P, Aavitsland P, Harthug S. Severe consequences of healthcare-associated infections among residents of nursing homes: a cohort study. J Hosp Infect 2009;71:269-74. https://doi.org/10.1016/j.jhin.2008.10.032.
- Vihta KD, Stoesser N, Llewelyn MJ, Quan TP, Davies T, Fawcett NJ, et al. Trends over time in Escherichia coli bloodstream infections, urinary tract infections, and antibiotic susceptibilities in Oxfordshire, UK, 1998–2016: a study of electronic health records. Lancet Infect Dis 2018;18:1138-49. https://doi.org/10.1016/S1473-3099(18)30353-0.
- Heginbothom MH, Howe R. Antibacterial Resistance in Wales 2005–2013. Cardiff: Public Health Wales; 2014.
- Lasseter G, Charlett A, Lewis D, Donald I, Howell-Jones R, McNulty CA. Staphylococcus aureus carriage in care homes: identification of risk factors, including the role of dementia. Epidemiol Infect 2010;138:686-96. https://doi.org/10.1017/S0950268810000233.
- Rooney PJ, O’Leary MC, Loughrey AC, McCalmont M, Smyth B, Donaghy P, et al. Nursing homes as a reservoir of extended-spectrum beta-lactamase (ESBL)-producing ciprofloxacin-resistant Escherichia coli. J Antimicrob Chemother 2009;64:635-41. https://doi.org/10.1093/jac/dkp220.
- Suez J, Zmora N, Segal E, Elinav E. The pros, cons, and many unknowns of probiotics. Nat Med 2019;25:716-29. https://doi.org/10.1038/s41591-019-0439-x.
- Snydman DR. The safety of probiotics. Clin Infect Dis 2008;46:104-11. https://doi.org/10.1086/523331.
- Liu S, Hu P, Du X, Zhou T, Pei X. Lactobacillus rhamnosus GG supplementation for preventing respiratory infections in children: a meta-analysis of randomized, placebo-controlled trials. Indian Pediatr 2013;50:377-81. https://doi.org/10.1007/s13312-013-0123-z.
- Hojsak I, Abdović S, Szajewska H, Milosević M, Krznarić Z, Kolacek S. Lactobacillus GG in the prevention of nosocomial gastrointestinal and respiratory tract infections. Pediatrics 2010;125:e1171-7. https://doi.org/10.1542/peds.2009-2568.
- Hojsak I, Snovak N, Abdović S, Szajewska H, Misak Z, Kolacek S. Lactobacillus GG in the prevention of gastrointestinal and respiratory tract infections in children who attend day care centers: a randomized, double-blind, placebo-controlled trial. Clin Nutr 2010;29:312-16. https://doi.org/10.1016/j.clnu.2009.09.008.
- Davidson LE, Fiorino AM, Snydman DR, Hibberd PL. Lactobacillus GG as an immune adjuvant for live-attenuated influenza vaccine in healthy adults: a randomized double-blind placebo-controlled trial. Eur J Clin Nutr 2011;65:501-7. https://doi.org/10.1038/ejcn.2010.289.
- Toiviainen A, Jalasvuori H, Lahti E, Gursoy U, Salminen S, Fontana M, et al. Impact of orally administered lozenges with Lactobacillus rhamnosus GG and Bifidobacterium animalis subsp. lactis BB-12 on the number of salivary mutans streptococci, amount of plaque, gingival inflammation and the oral microbiome in healthy adults. Clin Oral Investig 2015;19:77-83. https://doi.org/10.1007/s00784-014-1221-6.
- Liu PT, Krutzik SR, Modlin RL. Therapeutic implications of the TLR and VDR partnership. Trends Mol Med 2007;13:117-24. https://doi.org/10.1016/j.molmed.2007.01.006.
- Ly NP, Litonjua A, Gold DR, Celedon JC. Gut microbiota, probiotics, and vitamin D: interrelated exposures influencing allergy, asthma, and obesity?. J Allergy Clin Immunol 2011;127:1087-94. https://doi.org/10.1016/j.jaci.2011.02.015.
- Smith TJ, Rigassio-Radler D, Denmark R, Haley T, Touger-Decker R. Effect of Lactobacillus rhamnosus LGG® and Bifidobacterium animalis ssp. lactis BB-12® on health-related quality of life in college students affected by upper respiratory infections. Br J Nutr 2013;109:1999-2007. https://doi.org/10.1017/S0007114512004138.
- Savard P, Lamarche B, Paradis ME, Thiboutot H, Laurin É, Roy D. Impact of Bifidobacterium animalis subsp. lactis BB-12 and, Lactobacillus acidophilus LA-5-containing yoghurt, on fecal bacterial counts of healthy adults. Int J Food Microbiol 2011;149:50-7. https://doi.org/10.1016/j.ijfoodmicro.2010.12.026.
- Taipale T, Pienihäkkinen K, Isolauri E, Larsen C, Brockmann E, Alanen P, et al. Bifidobacterium animalis subsp. lactis BB-12 in reducing the risk of infections in infancy. Br J Nutr 2011;105:409-16. https://doi.org/10.1017/S0007114510003685.
- Rautava S, Salminen S, Isolauri E. Specific probiotics in reducing the risk of acute infections in infancy – a randomised, double-blind, placebo-controlled study. Br J Nutr 2009;101:1722-6. https://doi.org/10.1017/S0007114508116282.
- Kajander K, Myllyluoma E, Rajilić-Stojanović M, Kyrönpalo S, Rasmussen M, Järvenpää S, et al. Clinical trial: multispecies probiotic supplementation alleviates the symptoms of irritable bowel syndrome and stabilizes intestinal microbiota. Aliment Pharmacol Ther 2008;27:48-57. https://doi.org/10.1111/j.1365-2036.2007.03542.x.
- Pitkala KH, Strandberg TE, Finne Soveri UH, Ouwehand AC, Poussa T, Salminen S. Fermented cereal with specific bifidobacteria normalizes bowel movements in elderly nursing home residents. A randomized, controlled trial. J Nutr Health Aging 2007;11:305-11.
- Murakami T, Miyahara H, Yukisato S, Nakamura R, Kanno H, Kotakemori M, et al. Safety and effect of yoghurt containing Bifidobacterium lactis BB-12® on improvement of defecation and faecal microflora in healthy volunteers. J Nutr Food 2006;9:15-26.
- Council for International Organizations of Medical Sciences (CIOMS) in collaboration with the World Health Organization (WHO) . International Ethical Guidelines for Health-Related Research Involving Humans 2016.
- Great Britain . Medicines for Human Use (Clinical Trials) Regulations 2004 2004.
- Shepherd V, Hood K, Sheehan M, Griffith R, Jordan A, Wood F. Ethical understandings of proxy decision making for research involving adults lacking capacity: a systematic review (framework synthesis) of empirical research. AJOB Empir Bioeth 2018;9:267-86. https://doi.org/10.1080/23294515.2018.1513097.
- Wood F, Prout H, Bayer A, Duncan D, Nuttall J, Hood K, et al. PAAD Study Team . Consent, including advanced consent, of older adults to research in care homes: a qualitative study of stakeholders’ views in South Wales. Trials 2013;14. https://doi.org/10.1186/1745-6215-14-247.
- Shepherd V, Nuttall J, Hood K, Butler CC. Setting up a clinical trial in care homes: challenges encountered and recommendations for future research practice. BMC Res Notes 2015;8. https://doi.org/10.1186/s13104-015-1276-8.
- Great Britain . Mental Capacity Act 2005 2005.
- Department of Health, Scientific Development and Bioethics Division . Guidance on Nominating a Consultee for Research Involving Adults Who Lack Capacity to Consent 2008. www.manchester.gov.uk/download/downloads/id/12218/guidance_on_nominating_a_consultee_for_research_involving_adults_who_lack_capacity_to_consent.pdf (accessed 20 January 2021).
- Great Britain . Data Protection Act 2018 2018.
- European Parliament and Council of European Union . Regulation (EU) 2016 679 of the European Parliament and of the Council n.d. http://legislation.gov.uk/ukpga/2018/12/contents/enacted (accessed 2 February 2021).
- Janssen MF, Pickard AS, Golicki D, Gudex C, Niewada M, Scalone L, et al. Measurement properties of the EQ-5D-5L compared to the EQ-5D-3L across eight patient groups: a multi-country study. Qual Life Res 2013;22:1717-27. https://doi.org/10.1007/s11136-012-0322-4.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- Grewal I, Lewis J, Flynn T, Brown J, Bond J, Coast J. Developing attributes for a generic quality of life measure for older people: preferences or capabilities?. Soc Sci Med 2006;62:1891-901. https://doi.org/10.1016/j.socscimed.2005.08.023.
- Coast J, Flynn TN, Natarajan L, Sproston K, Lewis J, Louviere JJ, et al. Valuing the ICECAP capability index for older people. Soc Sci Med 2008;67:874-82. https://doi.org/10.1016/j.socscimed.2008.05.015.
- National Institute for Health and Care Excellence (NICE) . Diarrhoea – Adult'S Assessment. 2018 n.d. https://cks.nice.org.uk/topics/diarrhoea-adults-assessment/ (accessed 20 January 2021).
- World Health Organization (WHO) . WHO Guidelines on Drawing Blood: Best Practices in Phlebotomy 2010.
- Public Health England . Seasonal Influenza Vaccination Annual Report 2017 18: South West 2018.
- The European Committee on Antimicrobial Susceptibility Testing . Breakpoint Tables for Interpretation of MICs and Zone Diameters, Version 9.0 2019. https://eucast.org/ast_of_bacteria/previous_versions_of_documents/ (accessed 19 April 2019).
- Doron S, Snydman DR. Risk and safety of probiotics. Clin Infect Dis 2015;60:129-34. https://doi.org/10.1093/cid/civ085.
- Hatakka K, Ahola AJ, Yli-Knuuttila H, Richardson M, Poussa T, Meurman JH, et al. Probiotics reduce the prevalence of oral candida in the elderly – a randomized controlled trial. J Dent Res 2007;86:125-30. https://doi.org/10.1177/154405910708600204.
- Aunan JR, Watson MM, Hagland HR, Soreide K. Molecular and biological hallmarks of ageing. BJS 2016;103:e29-46. https://doi.org/10.1002/bjs.10053.
- Sikora E, Arendt T, Bennett M, Narita M. Impact of cellular senescence signature on ageing research. Ageing Res Rev 2011;10:146-52. https://doi.org/10.1016/j.arr.2010.10.002.
- Tiihonen K, Ouwehand AC, Rautonen N. Human intestinal microbiota and healthy ageing. Ageing Res Rev 2010;9:107-16. https://doi.org/10.1016/j.arr.2009.10.004.
- Dong H, Rowland I, Thomas LV, Yaqoob P. Immunomodulatory effects of a probiotic drink containing Lactobacillus casei Shirota in healthy older volunteers. Eur J Nutr 2013;52:1853-63. https://doi.org/10.1007/s00394-012-0487-1.
- Wang H, Gao K, Wen K, Allen IC, Li G, Zhang W, et al. Lactobacillus rhamnosus GG modulates innate signaling pathway and cytokine responses to rotavirus vaccine in intestinal mononuclear cells of gnotobiotic pigs transplanted with human gut microbiota. BMC Microbiol 2016;16. https://doi.org/10.1186/s12866-016-0727-2.
- O’Toole PW, Jeffery IB. Gut microbiota and aging. Science 2015;350:1214-15. https://doi.org/10.1126/science.aac8469.
- Vitetta L, Saltzman ET, Thomsen M, Nikov T, Hall S. Adjuvant probiotics and the intestinal microbiome: enhancing vaccines and immunotherapy outcomes. Vaccines 2017;5. https://doi.org/10.3390/vaccines5040050.
- Castro-Herrera V, Lown M, Lewith G, Miles EA, Calder PC. Influence of delayed sample processing on blood immune cell phenotypes, immune cell responses and serum anti-influenza vaccine antibody titres. J Immunol Methods 2018;458:8-14. https://doi.org/10.1016/j.jim.2018.03.012.
- Sampson EL, Feast A, Blighe A, Froggatt K, Hunter R, Marston L, et al. Evidence-based intervention to reduce avoidable hospital admissions in care home residents [the Better Health in Residents in Care Homes (BHiRCH) study]: protocol for a pilot cluster randomised trial. BMJ Open 2019;9. https://doi.org/10.1136/bmjopen-2018-026510.
- Biernacki P, Waldorf D. Snowball sampling: problems and techniques of chain referral sampling. Sociol Meth Res 1981;10:141-63. https://doi.org/10.1177/004912418101000205.
- Moore GF, Audrey S, Barker M, Bond L, Bonell C, Hardeman W, et al. Process evaluation of complex interventions: Medical Research Council guidance. BMJ 2015;350. https://doi.org/10.1136/bmj.h1258.
- Ritchie J, Spencer L, Bryman A, Burgess RG. Analyzing Qualitative Data. London: Routledge; 1994.
- Spencer L, Ritchie J, Ormston R, O’Conner W, Barnard M, Ritchie J, et al. Qualitative Research Practice: A Guide For Social Science Students and Researchers. London: SAGE Publications Ltd; 2003.
- Gale NK, Heath G, Cameron E, Rashid S, Redwood S. Using the framework method for the analysis of qualitative data in multi-disciplinary health research. BMC Med Res Methodol 2013;13. https://doi.org/10.1186/1471-2288-13-117.
- Gobat NH, Gal M, Butler CC, Webb SAR, Francis NA, Stanton H, et al. Talking to the people that really matter about their participation in pandemic clinical research: a qualitative study in four European countries. Health Expect 2018;21:387-95. https://doi.org/10.1111/hex.12634.
- Gordon AL, Franklin M, Bradshaw L, Logan P, Elliott R, Gladman JRF. Health status of UK care home residents: a cohort study. Age Ageing 2014;43:97-103. https://doi.org/10.1093/ageing/aft077.
- Jokanovic N, Tan EC, Dooley MJ, Kirkpatrick CM, Bell JS. Prevalence and factors associated with polypharmacy in long-term care facilities: a systematic review. J Am Med Dir Assoc 2015;16:535.e1-12. https://doi.org/10.1016/j.jamda.2015.03.003.
- Resnick B, Galik E, Boltz M, Holmes S, Fix S, Vigne E, et al. Polypharmacy in assisted living and impact on clinical outcomes. Consult Pharm 2018;33:321-30. https://doi.org/10.4140/TCP.n.2018.321.
- Bond Sutton L, Erlen JA, Glad JM, Siminoff LA. Recruiting vulnerable populations for research: revisiting the ethical issues. J Prof Nurs 2003;19:106-12. https://doi.org/10.1053/jpnu.2003.16.
- Ewing G, Rogers M, Barclay S, McCabe J, Martin A, Todd C. Recruiting patients into a primary care based study of palliative care: why is it so difficult?. Palliat Med 2004;18:452-9. https://doi.org/10.1191/0269216304pm905oa.
- Kars MC, van Thiel GJ, van der Graaf R, Moors M, de Graeff A, van Delden JJ. A systematic review of reasons for gatekeeping in palliative care research. Palliat Med 2016;30:533-48. https://doi.org/10.1177/0269216315616759.
- Mody L, Miller DK, McGloin JM, Freeman M, Marcantonio ER, Magaziner J, et al. Recruitment and retention of older adults in aging research. J Am Geriatr Soc 2008;56:2340-8. https://doi.org/10.1111/j.1532-5415.2008.02015.x.
- Shepherd V. Research involving adults lacking capacity to consent: the impact of research regulation on ‘evidence biased’ medicine. BMC Med Ethics 2016;17. https://doi.org/10.1186/s12910-016-0138-9.
- Bruhn H, Treweek S, Duncan A, Shearer K, Cameron S, Campbell K, et al. Estimating Site Performance (ESP): can trial managers predict recruitment success at trial sites? An exploratory study. Trials 2019;20. https://doi.org/10.1186/s13063-019-3287-6.
- Cox LE, Mainiero F. Perceptions of communication and respect among coworkers: a comparison of employees with and without experience in long-term care environments. J Hum Rights Soc Work 2017;2:134-42. https://doi.org/10.1007/s41134-017-0041-4.
- Jansen BW, Brazil K, Passmore P, Buchanan H, Maxwell D, McIlfatrick SJ, et al. Exploring healthcare assistants’ role and experience in pain assessment and management for people with advanced dementia towards the end of life: a qualitative study. BMC Palliat Care 2017;16. https://doi.org/10.1186/s12904-017-0184-1.
- Edvardsson D, Baxter R, Corneliusson L, Anderson RA, Beeber A, Boas PV, et al. Advancing long-term care science through using common data elements: candidate measures for care outcomes of personhood, well-being, and quality of life. Gerontol Geriatr Med 2019;5. https://doi.org/10.1177/2333721419842672.
- European Parliament, Council of the European Union . Clinical Trials Regulation (EU) No. 536 2014 n.d. https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-1/reg_2014_536/reg_2014_536_en.pdf (accessed 20 January 2021).
- Couzner L, Crotty M, Norman R, Ratcliffe J. A comparison of the EQ-5D-3L and ICECAP-O in an older post-acute patient population relative to the general population. Appl Health Econ Health Policy 2013;11:415-25. https://doi.org/10.1007/s40258-013-0039-8.
- Global Market Insights Inc . Probiotics Market Size to Exceed USD 64 Billion by 2023: Global Market Insights Inc. 2016. https://prnewswire.co.uk/news-releases/probiotics-market-size-to-exceed-usd-64-billion-by-2023-global-market-insights-inc-578769891.html (accessed 20 January 2021).
- Laxminarayan R, Duse A, Wattal C, Zaidi AK, Wertheim HF, Sumpradit N, et al. Antibiotic resistance-the need for global solutions. Lancet Infect Dis 2013;13:1057-98. https://doi.org/10.1016/S1473-3099(13)70318-9.
- King S, Glanville J, Sanders ME, Fitzgerald A, Varley D. Effectiveness of probiotics on the duration of illness in healthy children and adults who develop common acute respiratory infectious conditions: a systematic review and meta-analysis. Br J Nutr 2014;112:41-54. https://doi.org/10.1017/S0007114514000075.
- Wachholz PA, Nunes VDS, Polachini do Valle A, Jacinto AF, Villas-Boas PJF. Effectiveness of probiotics on the occurrence of infections in older people: systematic review and meta-analysis. Age Ageing 2018;47:527-36. https://doi.org/10.1093/ageing/afy006.
- Suez J, Zmora N, Zilberman-Schapira G, Mor U, Dori-Bachash M, Bashiardes S, et al. Post-antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell 2018;174:1406-23.e16. https://doi.org/10.1016/j.cell.2018.08.047.
- Lomax AR, Calder PC. Probiotics, immune function, infection and inflammation: a review of the evidence from studies conducted in humans. Curr Pharm Des 2009;15:1428-518. https://doi.org/10.2174/138161209788168155.
Appendix 1 Protocol amendments
The following substantial amendments were made to the trial protocol and were communicated to all trial sites.
| Amendment number | Amendment date | Version number | Details of changes made |
|---|---|---|---|
| 1 | 19 July 2016 | PRINCESS protocol v2.0 15 July 2016 |
Trial team details updated Details of randomisation website added Secondary outcome of GP visits, A&E visits and prophylactic antibiotic use removed The word ‘serum’ added to clarify that this refers to vitamin D levels in blood rather than in faecal specimens Section 3.1 Trial Schema and 3.2 Participant Flow Diagram updated Section 4.1 Background modified to further clarify the objectives of the study Reference to ‘hospital records’ changed to ‘discharge summaries’ Reference to ‘microbiome analysis’ removed Reference to ‘record increased level of care required’ via analysis of MAR sheets removed Section 8.3 Exclusion criteria amended to include CHRs currently involved in another CTIMP or who have participated in a CTIMP during the last 30 days Section 10.4 updated to change Unblinding SOP to unblinding procedure, and individual carrying out unblinding from Quality Assurance Manager to suitable delegated individual, and addition of two trial packs at initial allocation Clinical Rating Scale removed from the table in section 23, and text Section 13 Adverse Events (Safety) updated to include General Definitions, Expectedness, updated flowchart, and removal of ‘not assessable’ outcome, Reporting Procedures. Section 14 Adverse Events (Human Tissue) added |
| 2 | 1 September 2016 | PRINCESS poster for care homes version 1.0 dated 1 September 2016 | PRINCESS poster for care homes version 1.0 dated 1 September 2016 |
| 3 | 2 February 2017 | PRINCESS protocol version 2.2 dated 2 February 2017 |
Richard Fuller removed as co-applicant; Victoria Shepherd replaced by Alison Edwards Additional secondary outcome added (incidence and duration of diarrhoea) |
| 4 | 10 April 2017 |
PRINCESS template letter to consultee if capacity lost v1.0 10 April 2017 PRINCESS letter to consultee if capacity lost (anon) v1.0 10 April 2017 |
Letter to consultee of a trial participant who initially had capacity, but who lost capacity: PRINCESS template letter to consultee if capacity lost v1.0 10.04.2017; and PRINCESS letter to consultee if capacity lost (anon) v1.0 10.04.2017 |
| 5 | 20 June 2017 |
PRINCESS Informed Consent Procedure v1.1 13 June 2017 PRINCESS care home letter to potential consultee v1.0 13 June 2017 PRINCESS care home reminder letter to potential consultee v1.0 13 June 2017 PRINCESS protocol version 2.3 dated 20 June 2017 |
Professor Robling’s details updated Consent Procedure for nominated consultee: text added to the following sentence – ‘All consultees who have not responded will be contacted by phone or sent a follow-up letter at least once by the individual delegated to undertake the declaration process; the three month follow-up window changed to –2/+4 weeks’ |
| 6 | 25 April 2019 |
PRINCESS protocol version 3.0 dated 25 April 2018 PRINCESS participant information booklet update v1.0 25 April 2018 PRINCESS consultee information booklet update v1.0 25 April 2018 PRINCESS thank-you letter to participant on completion of trial dated 25 April 2018 PRINCESS thank-you letter to consultee on completion of trial dated 25 April 2018 |
Change of data manager and RN; change of surname of senior trial manager; changes to trial schema and flow diagram Shortened follow-up and clarification of objective and outcome terminologies: changes to terminology and items in synopsis table; clarification and update to secondary end points/outcomes; Table 5.1 changes to terminology and items in table; change in wording to sample size to ‘Between 258 and 270 participants, with an upper limit of 330’; ‘Description of outcome’ amended under Secondary – mechanistic: immune parameters heading; change to timing and name of 12-month interview; timing of SP; detail of follow-ups amended; change to trial closure definition |
| 7 | 30 April 2019 |
PRINCESS protocol version 4.0 dated 15 April 2019 PRINCESS interview postcard v3.0 dated 4 February 2019 PRINCESS nterview poster v2.0 dated 29 January 2019 PRINCESS interview consent form v1.0 5 November 2018 PRINCESS interview cover letter care home staff v1.0 dated 5 November 2018 PRINCESS interview cover letter participant v1.0 dated 5 November 2018 PRINCESS interview cover letter consultee v1.0 dated 5 November 2018 PRINCESS interview cover email care home manager v1.0 dated 28 January 2019 PRINCESS interview cover email research nurse v1.0 dated 22 January 2019 PRINCESS interview participant information sheet care home staff v1.0 dated 6 November 2018 PRINCESS interview participant information sheet participant v1.0 dated 6 November 2018 PRINCESS interview participant information sheet consultee v1.0 dated 6 November 2018 PRINCESS interview participant information sheet care home manager v1.0 dated 6 November 2018 PRINCESS interview participant information sheet research nurse v1.0 dated 6 November 2018 |
Section 16 and Appendix 1 addition of qualitative substudy |
Appendix 2 The PRINCESS trial interview topic guide for care home management and staff members
Introduction
(Completed written consent should have been sent to Helen Stanton prior to conducting the interview.)
-
I would like to ask you about your experiences of carrying out the PRINCESS trial in the care home you work in, and to ask you what it has been like to be a part of the PRINCESS trial.
-
This interview will be audio-recorded. The recording will be treated with the strictest confidentiality and may be listened to by the research team but by no-one else. The recording will not be labelled with your name, and any written record or report derived from it will be fully anonymised.
-
Are there any questions you would like to ask me before we start?
Carrying out the PRINCESS trial
In the PRINCESS trial, we asked [care home management]/[care home team members] such as yourself to carry out various activities for the trial with residents, consultees and the PRINCESS researchers who came in to your care home.
-
To get us started, could you talk me through how your care home became involved in the PRINCESS trial?
[Prompts: Management only: What did you think when the researchers at Cardiff or Oxford invited your care home to take part? What made you feel this trial was right for your care home? Was there any aspect of the trial/how you were approached that confirmed that you were willing for the home to participate?]
-
What were your roles and responsibilities on the trial?
[Prompts: Were you involved more heavily at some stages of the trial (e.g. set-up, recruitment, follow-up)? Did you take charge of any particular tasks associated with the trial (e.g. paperwork, study product, recruitment, management and oversight)?]
-
What were your experiences of your initial contact with the researchers/nurses on the trial?
[Prompts: Did you meet with the PRINCESS researchers/nurses before or during trial set-up? How did you (and the care home team) adapt to carrying out PRINCESS trial tasks in the care home? How did you (and the care home team) and the residents adapt to the PRINCESS researchers/nurses coming in to the care home to carry out PRINCESS trial tasks? How did you build a rapport with the PRINCESS researchers/nurses?]
-
If not stated previously in the interview: Were you involved in the recruitment process itself?
-
If yes, proceed to next question.
-
If no, skip to question 7.
-
-
What was your role in the recruitment process as a whole?
[Prompts: How did you acquire information about the resident that would affect their suitability for the trial? Were there any participants who were eligible on paper, but you did not recommend approaching or recruiting? If so, why? How did you work with relatives (i.e. consultees) to recruit residents? How did you work with PRINCESS researchers/nurses to recruit residents? Did you undertake assessments of mental capacity for the purposes of recruitment? How did you find this? What was the most challenging aspect of recruiting residents? Did you have concerns about any part of the process?]
-
How did you feel about the consultee process?
[Prompts: Were you involved in identifying consultees? How did you feel about older, perhaps more vulnerable individuals being recruited to a randomised controlled trial such as PRINCESS? Did you (or any of the care home team) act as nominated consultees for any residents? Did you contact/utilise any other resources/agencies for advice or information around decision-making where residents lacked capacity?]
The PRINCESS trial was a placebo-controlled trial, where the study product given to participating residents was either an inactive capsule or a capsule containing a probiotic. If the resident was initially allocated the probiotic their capsules would contain probiotic throughout the trial, and if they were initially allocated the inactive capsule (the placebo) they would continue taking placebo throughout.
-
If not stated previously in the interview: Were you involved in the handling or distribution of the study product?
-
If yes, proceed to next question.
-
If no, skip to question 11.
-
-
The study product could be taken as a capsule, in liquid or sprinkled on food. How did this go?
[Prompts: Were there any issues with residents taking the study product? Did you have any concerns about the methods of ingestion?]
-
Were there any factors which you think may have affected the residents taking the study product daily?
[Prompts: Were there any barriers to ensuring that residents received their study product? Do you think the routines at your care home lend themselves to giving out study product?]
-
How did you know whether the resident had taken the study product?
[Prompts: Did you record ingestion on MAR charts? Did you ever have to recall from memory about study product consumption, dose and method of ingestion? To what extent were you confident that residents received the correct study product that they were allocated?]
Working within the care home environment
We asked the researchers/nurses employed to work on the PRINCESS trial to record study product adherence, as well as incidences of infections, antibiotics, diarrhoea and hospitalisations on weekly record forms.
-
If not stated previously in the interview: Were you also involved in the data collection for the trial?
-
If yes, proceed to next question.
-
If no, skip to question 13.
-
-
How did you go about acquiring the information you needed to complete the data collection forms?
[Prompts: Please explain how you collected information about the residents? Did you find care home records easy to locate? How easy was it to derive information from written care home notes and MAR sheets? To what extent did you speak with residents or consultees about data collection?]
-
To what extent did you work with the PRINCESS researchers/nurses to collect information with/about the resident?
[Prompts: What was your working relationship like with researchers/nurses in the care home? Were you able to help researchers/nurses collect information that they needed about the resident? Did you feel you had time to discuss participating residents with them? Were care home staff knowledgeable about the residents taking part? Did researchers/nurses ask you to take responsibility for any particular trial activities (e.g. taking samples)? How did you communicate about tasks?]
-
More generally, what was it like hosting trial activities in your care home?
[Prompts: Were there any barriers/challenges to carrying out research activities (or helping the PRINCESS researchers/nurses to carry these out) in the care home environment? How did you attempt to overcome these? Going forward what do you think would make carrying out research in care homes easier?]
Working with vulnerable older people and consultees
The PRINCESS trial participants were older, perhaps very vulnerable residents. Those who lacked the mental capacity to provide consent to be part of the trial could take part if consultee advice/agreement was provided.
-
How did you feel about carrying out PRINCESS trial activities with vulnerable older adults in the care home environment?
[Prompts: Did you have concerns about any aspect of the trial/carrying out research activities? Did you experience any challenges/barriers to assisting with the research activities?]
-
How do you think the residents and consultees felt about taking part in the trial?
[Prompts: Did residents/consultees seem engaged/interested in the research? Did anything about the trial or trial activities cause any particular worry to the residents, consultees/relatives? (or anyone else?)]
At baseline, 3-month and 12-month time points researchers were also asked to collect responses to questionnaires (ICECAP and EQ-5D-5L) that measure health and capability outcomes.
-
If not stated previously in the interview: Did you carry out the ICECAP and EQ-5D-5L questionnaires with the resident? Did you ever act as a proxy on behalf of the resident and complete the questionnaires as you felt the resident would’ve answered if they were able?
-
If yes, proceed to next question.
-
If no, skip to question 20.
-
-
What was your experience of asking residents to answer these questions?
[Prompts: Did you feel comfortable asking these types of questions? How relevant did you feel these questions were to older people or to residents lacking capacity?]
-
How did you feel putting yourself in the resident’s shoes and responding to the questions as you think they might have done?
Practical aspects of the research and training
I’d like to explore some of practical, hands-on aspects of the trial and the training provided.
-
Did you have enough room to store trial materials (e.g. trial site files, sample containers) and study product at the care home?
-
(If not already mentioned) How did you find taking and sending samples at the three follow-up time points (baseline, 3 months and 12 months)?
[Prompts: What type(s) of samples were you involved in collecting? What was your typical procedure for taking, storing and sending samples? Were there any barriers/challenges to collecting samples? Did you complete the forms to go with the samples? How did you find this?]
-
What training (if any) did you receive for the PRINCESS trial?
[Prompts: How was the training delivered? Was it useful? Did you have enough information? Is there any other information you would have liked? How much time did it take to complete? Was this OK? What would you change about the training?]
Trial intervention, probiotics and infections
So far, we’ve been talking specifically about the PRINCESS trial. I’d like to ask you a few more general questions about your views on probiotics and managing infections in older people in care homes.
-
What was your overall opinion of the intervention that was being trialled (i.e. probiotics)?
[Prompts: Did you have an opinion on probiotics before your home became involved in the trial? Did you have any safety (or other) concerns about use of probiotics in the older population? Has it changed the way you view probiotic supplements?]
-
What do you think are the main challenges with managing infections in older people in care homes?
-
In terms of health-care services for older people in care homes (including those lacking capacity to make decisions for themselves), what do you think would be the most important thing to improve?
Conducting research in care homes/research process
Almost at the end of the interview; we’re interested in your overall experience of being involved in the PRINCESS trial and your thoughts on conducting research in care homes.
-
Could you tell me what it has been like to take part in the PRINCESS trial?
[Prompts: Were there any problems? What were the best things about taking part? What were the worst things about taking part?]
-
Before you were involved in the PRINCESS trial did you have any prior experience hosting or carrying out research in your care home or in other care homes you have worked in?
-
Lastly, what are your views about care home research in general?
-
Do you have any other comments?
Thank you very much for taking part in this interview.
Appendix 3 The PRINCESS trial interview topic guide for research professionals
Introduction
(Completed written consent should have been sent to Helen Stanton prior to conducting the interview.)
-
I would like to ask you about your experiences of carrying out the PRINCESS trial in care homes, and to ask you what it has been like to be a part of the PRINCESS trial.
-
This interview will be audio-recorded. The recording will be treated with the strictest confidentiality and may be listened to by the research team but by no-one else. The recording will not be labelled with your name, and any written record or report derived from it will be fully anonymised.
-
Are there any questions you would like to ask me before we start?
Carrying out the PRINCESS trial
In the PRINCESS trial, we asked research professionals such as yourself to carry out various research activities with residents, consultees and the care home staff in the care home.
-
To get us started, could you talk me through your involvement in the PRINCESS trial, and what your roles and responsibilities were on the trial?
[Prompts: Did you have a particular interest in the study, or were you allocated the role as part of your overall work responsibilities? Did you work on the trial at any particular stage of the trial (e.g. set-up, recruitment, follow-up)? Did you take charge of any particular tasks associated with the trial (e.g. collecting samples, follow-ups, study product, recruitment, management and oversight)?]
-
What were your experiences of your initial contact with the care home?
[Prompts: Were you involved in visiting the care homes before or during trial set-up? What reception did you get from management? How did you adapt to carrying out PRINCESS trial tasks in the care home? How do you think the care home staff and residents adapted to you being in the care home to carry out PRINCESS trial tasks? How did you build a rapport with the care home staff and residents?]
-
If not stated previously in the interview: Were you involved in the recruitment process itself?
-
If yes, proceed to next question.
-
If no, skip to question 7.
-
-
How did you assess resident eligibility for the trial?
[Prompts: How easy was it to acquire information about the resident that would affect their suitability for the trial? Were there any participants who were eligible on paper, but you did not approach or recruit? If so, why?]
-
How did you approach the task of recruiting residents in to the trial?
[Prompts: Talk me through the stages that you went through to recruit residents with or without capacity? How did you work with relatives (i.e. consultees) to recruit residents? How did you work with care home staff to recruit residents? Did you undertake assessments of mental capacity for the purposes of recruitment? How did you find this? What was the most challenging aspect of recruiting residents? Did you have concerns about any part of the process?]
-
How did you feel about the consultee process?
[Prompts: How did you feel about recruiting older, perhaps more vulnerable individuals in to a randomised controlled trial such as PRINCESS? How did you identify consultees? Did you have to approach care home staff to act as nominated consultees for any residents? Did you contact/utilise any other resources/agencies for advice or information around decision making where residents lack capacity?]
The PRINCESS trial was a placebo-controlled trial, where the study product given to participating residents was either an inactive capsule or a capsule containing a probiotic.
-
If not stated previously in the interview: Were you involved in the handling or distribution of the study product?
-
If yes, proceed to next question.
-
If no, skip to question 11.
-
-
The study product could be taken as a capsule, in liquid, or sprinkled on food. How did this go?
[Prompts: Were there any issues with residents taking the study product? Did you have any concerns about the methods of ingestion?]
-
Were there any factors which you think may have affected study product adherence?
[Prompts: Were there any barriers to ensuring that residents received their study product? Did the care home routines lend themselves to giving out study product?]
-
How did you monitor adherence to study product?
[Prompts: Did you look at MAR charts? How well were these completed? Did you ever have to use verbal reports about study product consumption, dose and method of ingestion? To what extent were you confident that residents received the correct study product that they were allocated?]
Working within the care home environment
We asked researchers to record study product adherence, as well as incidences of infections, antibiotics, diarrhoea and hospitalisations on weekly record forms.
-
If not stated previously in the interview: Were you involved in the data collection for the trial?
-
If yes, proceed to next question.
-
If no, skip to question 14.
-
-
How did you go about acquiring the information you needed to complete the data collection forms?
[Prompts: please explain how you sourced information about the residents? Did you find care home records easy to locate? How easy was it to derive information from written care home notes and MAR sheets? Were care home staff helpful/knowledgeable about the residents taking part? To what extent did you speak with residents about their data?]
-
To what extent did you work with the care home staff to collect information with/about the resident?
[Prompts: What was your working relationship like with staff in the care home? Were care home staff helpful in assisting you to collect information about the resident? Did you feel care home staff had time to discuss participating residents with you? Were care home staff knowledgeable about the residents taking part? Did you delegate any particular trial activities to care home staff (e.g. taking samples)? How did you communicate about tasks?]
-
More generally, what was working in the care home like?
[Prompts: Were there any barriers/challenges to carrying out research activities in the care home environment? How did you attempt to overcome these? Going forward what do you think would make carrying out research in care homes easier?]
Working with vulnerable older people and consultees
The PRINCESS trial participants were older, perhaps very vulnerable residents. Those who lacked the mental capacity to provide consent to be part of the trial could take part if consultee advice/agreement was provided.
-
How did you feel about carrying out PRINCESS trial activities with vulnerable older adults in the care home environment?
[Prompts: Did you have concerns about any aspect of the trial/carrying out research activities? Did you experience any challenges/barriers to carrying out the research activities?]
-
How do you think the residents and consultees felt about taking part in the trial?
[Prompts: Did residents/consultees seem engaged/interested in the research? Did anything about the trial or trial activities cause any particular worry to the residents, consultees/relatives? (or anyone else?)]
At baseline, 3-month and 12-month time points researchers were also asked to collect responses to questionnaires (ICECAP and EQ-5D-5L) that measure health and capability outcomes.
-
If not stated previously in the interview: Did you carry out the ICECAP and EQ-5D-5L questionnaires with the resident or a proxy acting on behalf of the resident?
-
If yes, proceed to next question.
-
If no, skip to question 19.
-
-
What was your experience of asking residents and proxies to answer these questions?
[Prompts: Did you feel comfortable asking these types of questions? How relevant did you feel these questions were to older people or to residents lacking capacity?]
Practical aspects of the research and training
I’d like to explore some of practical, hands-on aspects of the trial and the training provided.
-
Did you have enough room to store study materials (e.g. trial site files, sample containers) and study product at the care home?
-
How did you find the data collection materials and using the trial database?
[Prompts: Did you find any of the information difficult/awkward to collect? What would you change about the data collection? How did you find using an online database, versus completing paper forms?]
-
How did you find taking and sending samples at the three follow-up time points?
-
What did you think of the training provided for the PRINCESS trial?
[Prompts: How was the training delivered? Was it useful? Did you have enough information? Is there any other information you would have liked? How much time did it take to complete? Was this OK? What would you would change about the training?]
Trial intervention, probiotics and infections
So far, we’ve been talking specifically about the PRINCESS trial. I’d like to ask you a few more general questions about your views on probiotics and managing infections in older people in care homes.
-
What was your overall opinion of the intervention that was being trialled (i.e. probiotics)?
[Prompts: Did you have an opinion on probiotics before you became part of the study? Did you have any safety (or other) concerns about use of probiotics in the older population? Has it changed the way you view probiotic supplements?]
-
What do you think are the main challenges with managing infections in older people in care homes?
-
In terms of health-care services for older people in care homes (including those lacking capacity to make decisions for themselves), what do you think would be the most important thing to improve?
Conducting research in care homes/research process
Almost at the end of the interview; we’re interested in your overall experience of taking part in the PRINCESS trial and your thoughts on conducting research in care homes.
-
Could you tell me what it has been like to take part in the PRINCESS trial?
[Prompts: Were there any problems? What were the best things about taking part? What were the worst things about taking part?]
-
Before you took part in the PRINCESS trial did you have any prior experience with conducting research in care homes?
-
Lastly, what are your views about care home research in general?
-
Do you have any other comments?
Thank you very much for taking part in this interview.
Appendix 4 The PRINCESS trial interview topic guide for care home residents and consultees
Introduction
(Completed written consent should have been sent to Helen Stanton prior to conducting the interview)
-
I would like to ask you about your experiences of [taking part in]/[providing advice on behalf of your relative for] the PRINCESS trial.
-
This interview will be audio-recorded. The recording will be treated with the strictest confidentiality and may be listened to by the research team but by no-one else. The recording will not be labelled with your name and any written record or report derived from it will be fully anonymised.
-
Are there any questions you would like to ask me before we start?
Carrying out the PRINCESS trial
In the PRINCESS trial, we asked [people such as yourself]/[older people living in care homes] to take part in the research that would help us determine if a probiotic supplement can help reduce infections and, ultimately, reduce the consumption of antibiotics.
-
To get us started, could you talk me through how [you]/[your relative] became involved in the PRINCESS trial?
{Prompts: What did you think when [you]/[your relative] were invited to take part? What made you feel this trial was right for [you]/[your relative] to take part in?}
-
What happened when [you]/[your relative] were approached to take part in the trial?
{Prompts: What were your experiences of your initial contact with the researchers/nurses who worked on the trial? What was it like having other people you weren’t familiar with coming in to the home and discussing the trial with [you]/[you on behalf of your relative]?}
[For consultees only] How did you feel about providing advice about the trial on behalf of your relative?
[Prompts: How did you feel about your relative, whom you might consider vulnerable being recruited in to a randomised controlled trial such as PRINCESS? Were there any aspects of this process you found particularly challenging? Did you speak to anyone else about the study, or consult anyone else for advice, about whether it was a good idea for your relative to take part in PRINCESS?]
-
How were channels of communication about the trial?
[Prompts: Were updates and information about aspects of the trial forthcoming? Did you feel like you were included in plans (e.g. visits from researchers) and decisions about the trial?]
PRINCESS was a placebo-controlled trial, where the study product given to participating residents was either an inactive capsule, or a capsule containing a probiotic. If the resident was initially allocated the probiotic their capsules would contain probiotic throughout the trial, and if they were initially allocated the inactive capsule (the placebo) they would continue taking placebo throughout.
-
How did you feel about [your relative] taking the study product?
{Prompts: Did [you]/[they] experience any issues taking the study product? Did you feel you had enough information about the study product and the potential effects it could have?}
-
The study product could be taken as a capsule, in liquid, or sprinkled on food. Did you have any concerns about the method in which it was taken?
-
Were there any factors which you think may have affected [you]/[your relative] taking the study product daily?
{Prompts: Were there any barriers to ensuring that residents received their study product? Do you think the routines at [your]/[your relative’s] home lend themselves to giving out study product?}
-
Did you feel [you]/[your relative] were always receiving the correct study product for [you]/[them]?
Working with the care home team and PRINCESS researchers/nurses
We asked the researchers/nurses employed to work on the PRINCESS trial to collect data about [you]/[your relative]. This included whether [you]/[your relative] had taken study product each day, and whether [you]/[your relative] had shown signs of infection, taken antibiotics, had diarrhoea, or had been admitted to hospital.
-
To what extent did the PRINCESS researchers/nurses speak with you to collect the information needed for the trial?
-
What was your impression of the way the PRINCESS researchers/nurses carried out the trial in [your]/[your relative’s] home?
{Prompts: Did you see the PRINCESS researchers/nurses often at [your]/[the] home? Do you think they enjoyed coming in to [your]/[the] home? Did you get an opportunity to chat with the researchers/nurses when they visited? What kind of relationship did you have with the PRINCESS researchers/nurses?}
-
How do you think the staff at [your]/[the] home found hosting and carrying out the PRINCESS trial with [you]/[your relative] and others at [your]/[the] home?
{Prompts: What impact do you think having the trial at [your]/[the] home had on the staff and residents at [your]/[the] home? Do you think doing the trial at [your]/[the] home changed anything about the way routines usually run? If yes, what and why do you think this was?}
Practical aspects of the research
We asked for permission to collect samples from [you]/[your relative] at the beginning of the trial, and at 3 months and 12 months after initially joining the study.
-
(If not already mentioned) Did [you]/[your relative] have samples taken at the three follow-up time points (baseline, 3 months and 12 months)?
{Prompts: What type(s) of samples did [you]/[your relative] have taken? Did you have any concerns about any aspect of the sample-taking? Is there anything that could have been improved regarding this part of the trial?}
Trial intervention, probiotics and infections
So far, we’ve been talking specifically about PRINCESS trial. I’d like to ask you a few more general questions about your views on probiotics and managing infections.
-
What was your overall opinion of the intervention that was being trialled (i.e. probiotics)?
[Prompts: Did you have an opinion on probiotics before your home became involved in the trial? Did you have any safety (or other) concerns about use of probiotics in the older population? Has it changed the way you view probiotic supplements?]
-
[For consultees only] What do you think are the main challenges with managing infections in older people in care homes?
-
[For consultees only] In terms of health-care services for older people in care homes (including those lacking capacity to make decisions for themselves), what do you think would be the most important thing to improve?
Conducting research in care homes/research process
Almost at the end of the interview; we’re interested in your overall experience of being involved in the PRINCESS trial as a [participant]/[consultee on behalf of your relative].
-
[As a participant] Could you tell me what it has been like to take part in the PRINCESS trial? [As a consultee] How do you think your relative has found taking part in the PRINCESS trial?
[Prompts: Were there any problems? What were the best things about taking part? What were the worst things about taking part?]
-
Before you were involved in PRINCESS [as a participant]/[in an advisory capacity on behalf of your relative] had you taken part in research before?
-
Lastly, what are your thoughts about doing research in this environment?
-
Do you have any other comments?
Thank you very much for taking part in this interview.
Appendix 5 Additional tables and figures
| Care home identifier | Characteristic | |||||
|---|---|---|---|---|---|---|
| Age (years), median (IQR) | Capacity, n (%) | Sex, n (%) | Duration of residence (years), median (IQR) | |||
| Has | Lacks | Female | Male | |||
| 1 | 87 (82.5–88) | 1 (6.7) | 14 (93.3) | 10 (66.7) | 5 (33.3) | 2 (1–3) |
| 2 | 86.5 (79–91) | 10 (33.3) | 20 (66.7) | 19 (63.3) | 11 (36.7) | 0 (0–2) |
| 3 | 88.5 (84–93) | 8 (44.4) | 10 (55.6) | 11 (61.1) | 7 (38.9) | 0.5 (0–3) |
| 4 | 86 (83–89) | 13 (50.0) | 13 (50.0) | 15 (57.7) | 11 (42.3) | 1 (0–3) |
| 5 | 83 (79–88) | 2 (9.5) | 19 (90.5) | 16 (76.2) | 5 (23.8) | 2 (1–3) |
| 6 | 88 (79.5–92.5) | 8 (53.3) | 7 (46.7) | 9 (60.0) | 6 (40.0) | 1 (0–2) |
| 7 | 86 (82.5–88) | 7 (43.8) | 9 (56.3) | 10 (62.5) | 6 (37.5) | 0 (0–1) |
| 8 | 85 (79–91) | 7 (41.2) | 10 (58.8) | 9 (52.9) | 8 (47.1) | 0 (0–1) |
| 9 | 86 (83–87) | 5 (38.5) | 8 (61.5) | 9 (69.2) | 4 (30.8) | 1 (0–3) |
| 10 | 82.5 (79–91) | 3 (50.0) | 3 (50.0) | 2 (33.3) | 4 (66.7) | 2 (0–6) |
| 11 | 87 (80–93) | 6 (46.2) | 7 (53.8) | 9 (69.2) | 4 (30.8) | 2 (0–4) |
| 12 | 80 (74–88) | 4 (30.8) | 9 (69.2) | 9 (69.2) | 4 (30.8) | 1 (0–2) |
| 13 | 91.5 (88–93) | 3 (21.4) | 11 (78.6) | 9 (64.3) | 5 (35.7) | 0.5 (0–1) |
| 14 | 88 (82.5–91) | 8 (53.3) | 7 (46.7) | 12 (80.0) | 3 (20.0) | 1 (0–1.5) |
| 15 | 90 (86–94) | 1 (50.0) | 1 (50.0) | 2 (100.0) | 0 (0.0) | N/A |
| 16 | 88 (82–88) | 2 (22.2) | 7 (77.8) | 9 (100.0) | 0 (0.0) | 2 (0–2) |
| 17 | 88 (84–90) | 2 (40.0) | 3 (60.0) | 3 (60.0) | 2 (40.0) | 3 (2–4) |
| 18 | 86 (80–91) | 7 (46.7) | 8 (53.3) | 10 (66.7) | 5 (33.3) | 1 (0–1.5) |
| 19 | 86 (80–90) | 0 (0.0) | 14 (100.0) | 12 (85.7) | 2 (14.3) | 2.5 (1–6) |
| 20 | 90 (86–95) | 3 (30.0) | 7 (70.0) | 5 (50.0) | 5 (50.0) | 0 (0–3) |
| 21 | 86.5 (84–90) | 0 (0.0) | 10 (100.0) | 6 (60.0) | 4 (40.0) | 0 (0–0) |
| 22 | 83 (70–86) | 4 (40.0) | 6 (60.0) | 9 (90.0) | 1 (10.0) | 0.5 (0–2) |
| 23 | 84 (83.5–90) | 2 (66.7) | 1 (33.3) | 2 (6.7) | 1 (33.3) | 3 (2.5–3.5) |
FIGURE 15.
Median age of CHR (years) by care home.
FIGURE 16.
Percentage of recruited CHRs with lack of capacity by care home.

FIGURE 17.
Clinical Frailty Scale score by care home.
FIGURE 18.
Yeasts present in CHRs’ saliva at baseline.

| Outcome measure | Trial arm | Adjusted IRRa | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | ||||||||
| n | Mean | SE | n | Mean | SE | ||||
| Incidence of AAD | 153 | 0.6 | 0.15 | 152 | 0.8 | 0.16 | 1.39 | 0.79 to 2.46 | 0.254 |
| Cumulative days of AAD | 153 | 4.4 | 1.30 | 152 | 6.8 | 1.81 | 1.83 | 0.95 to 3.54 | 0.072 |
| Incidence of all-cause diarrhoea | 153 | 1.6 | 0.28 | 152 | 1.8 | 0.31 | 1.05 | 0.71 to 1.57 | 0.799 |
| Cumulative days of all-cause diarrhoea | 153 | 4.4 | 0.88 | 152 | 4.4 | 0.82 | 1.23 | 0.77 to 1.97 | 0.388 |
| Mean duration of diarrhoea episodesb | |||||||||
| At least one all-cause diarrhoea | 61 | 39.9% | 64 | 42.1% | 1.04c | 0.61 to 1.77 | 0.890 | ||
| Non-zero duration | 61 | 1.4 | 0.08 | 64 | 1.4 | 0.07 | 0.06 | –0.05 to 0.18 | 0.266 |
FIGURE 19.
Cumulative number of infections by trial arm. This is the absolute number of infection events, without adjusting for death, participant withdrawal, or truncated follow-up time.
FIGURE 20.
Forest plot for all infection outcomes (values are adjusted IRRs and 95% CIs).

FIGURE 21.
Forest plot for all diarrhoea outcomes (values are adjusted IRRs and 95% CIs).

FIGURE 22.
Yeasts detected in CHRs’ saliva samples at the second follow-up time point.

| Variable | Primary outcome data | Total | ||||
|---|---|---|---|---|---|---|
| Not complete (N = 109) | Complete (N = 201) | |||||
| Number of CHRs | % | Number of CHRs | % | Number of CHRs | % | |
| Trial arm | ||||||
| Probiotic | 57 | 36.8 | 98 | 63.2 | 155 | 100.0 |
| Placebo | 52 | 33.5 | 103 | 66.5 | 155 | 100.0 |
| Sex | ||||||
| Female | 70 | 34.0 | 136 | 66.0 | 206 | 100.0 |
| Male | 39 | 37.5 | 65 | 62.5 | 104 | 100.0 |
| Age (years) at consent | ||||||
| Minimum to maximum | 67 to 97 | 65 to 102 | 65 to 102 | |||
| Mean (SD) | 86.5 (6.5) | 84.7 (7.75) | 85.3 (7.39) | |||
| Median (IQR) | 87 (82–91) | 86 (80–90) | 86 (81–91) | |||
| Duration of residence in care home (years) (consent: date)a | ||||||
| Minimum to maximum | 0 to 10 | 0 to 15 | 0 to 15 | |||
| Mean (SD) | 1.6 (2.19) | 1.7 (2.45) | 1.7 (2.36) | |||
| Median (IQR) | 1 (0–2) | 1 (0–2) | 1 (0–2) | |||
| Capacity to consent | ||||||
| Lacks | 79 | 38.7 | 125 | 61.3 | 204 | 100.0 |
| Has | 30 | 28.3 | 76 | 71.7 | 106 | 100.0 |
| Clinical Frailty Scale | ||||||
| Very fit to managing well | 6 | 19.4 | 25 | 80.6 | 31 | 100.0 |
| Vulnerable to moderately frail | 35 | 30.4 | 80 | 69.6 | 115 | 100.0 |
| Severely frail to terminally ill | 68 | 41.5 | 96 | 58.5 | 164 | 100.0 |
| Domain | Self-reported | Proxy-reported | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | Total | Placebo | Probiotic | Total | |||||||
| n | % | n | % | n | % | n | % | n | % | n | % | |
| Mobility | ||||||||||||
| No problems | 5 | 9.4 | 16 | 24.6 | 21 | 17.8 | 24 | 15.6 | 28 | 18.3 | 52 | 16.9 |
| Slight problems | 13 | 24.5 | 8 | 12.3 | 21 | 17.8 | 27 | 17.5 | 31 | 20.3 | 58 | 18.9 |
| Moderate problems | 12 | 22.6 | 13 | 20.0 | 25 | 21.2 | 23 | 14.9 | 30 | 19.6 | 53 | 17.3 |
| Severe problems | 9 | 17.0 | 10 | 15.4 | 19 | 16.1 | 15 | 9.7 | 16 | 10.5 | 31 | 10.1 |
| Extreme problems | 14 | 26.4 | 18 | 27.7 | 32 | 27.1 | 65 | 42.2 | 48 | 31.4 | 113 | 36.8 |
| Self-care | ||||||||||||
| No problems | 16 | 30.2 | 25 | 38.5 | 41 | 34.7 | 9 | 5.8 | 12 | 7.8 | 21 | 6.8 |
| Slight problems | 11 | 20.8 | 7 | 10.8 | 18 | 15.3 | 25 | 16.2 | 27 | 17.6 | 52 | 16.9 |
| Moderate problems | 9 | 17.0 | 11 | 16.9 | 20 | 16.9 | 24 | 15.6 | 27 | 17.6 | 51 | 16.6 |
| Severe problems | 7 | 13.2 | 12 | 18.5 | 19 | 16.1 | 12 | 7.8 | 18 | 11.8 | 30 | 9.8 |
| Extreme problems | 10 | 18.9 | 10 | 15.4 | 20 | 16.9 | 84 | 54.5 | 68 | 45.1 | 153 | 49.8 |
| Usual activities | ||||||||||||
| No problems | 14 | 26.4 | 18 | 27.7 | 20 | 27.1 | 28 | 18.2 | 27 | 17.6 | 55 | 17.9 |
| Slight problems | 6 | 11.3 | 12 | 18.5 | 18 | 15.3 | 23 | 14.9 | 27 | 17.6 | 50 | 16.3 |
| Moderate problems | 14 | 26.4 | 14 | 21.5 | 28 | 23.7 | 41 | 26.6 | 32 | 20.9 | 73 | 23.8 |
| Severe problems | 10 | 18.9 | 9 | 13.8 | 19 | 16.1 | 24 | 15.6 | 27 | 17.6 | 51 | 16.6 |
| Extreme problems | 9 | 17.0 | 12 | 18.5 | 21 | 17.8 | 38 | 24.7 | 40 | 26.1 | 78 | 25.4 |
| Pain/discomfort | ||||||||||||
| No problems | 24 | 45.3 | 31 | 47.7 | 55 | 46.6 | 64 | 42.1 | 58 | 38.2 | 122 | 40.1 |
| Slight problems | 13 | 24.5 | 17 | 26.2 | 30 | 25.4 | 55 | 36.2 | 46 | 30.3 | 101 | 33.2 |
| Moderate problems | 15 | 28.3 | 13 | 20.0 | 28 | 23.7 | 27 | 17.8 | 41 | 27.0 | 68 | 22.4 |
| Severe problems | 1 | 1.9 | 3 | 4.6 | 4 | 3.4 | 5 | 3.3 | 5 | 3.3 | 10 | 3.3 |
| Extreme problems | 0 | 0.0 | 1 | 1.5 | 1 | 0.8 | 1 | 0.7 | 2 | 1.3 | 3 | 1.0 |
| Anxiety/depression | ||||||||||||
| No problems | 23 | 43.4 | 33 | 51.6 | 55 | 47.9 | 63 | 40.9 | 70 | 45.8 | 133 | 43.3 |
| Slight problems | 16 | 30.2 | 14 | 21.9 | 30 | 25.6 | 45 | 29.2 | 51 | 33.3 | 96 | 31.3 |
| Moderate problems | 13 | 24.5 | 15 | 23.4 | 28 | 23.9 | 39 | 25.3 | 22 | 14.4 | 61 | 19.9 |
| Severe problems | 1 | 1.9 | 1 | 1.6 | 2 | 1.7 | 7 | 4.5 | 8 | 5.2 | 15 | 4.9 |
| Extreme problems | 0 | 0.0 | 1 | 1.6 | 1 | 0.9 | 0 | 0.0 | 2 | 1.3 | 2 | 0.7 |
| Domain | Self-reported | Proxy-reported | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | Total | Placebo | Probiotic | Total | |||||||
| n | % | n | % | n | % | n | % | n | % | n | % | |
| Mobility | ||||||||||||
| No problems | 7 | 16.3 | 14 | 26.9 | 21 | 22.1 | 23 | 17.7 | 26 | 19.8 | 49 | 18.8 |
| Slight problems | 5 | 11.6 | 9 | 17.3 | 14 | 14.7 | 25 | 19.2 | 27 | 20.6 | 52 | 19.9 |
| Moderate problems | 13 | 30.2 | 6 | 11.5 | 19 | 20.0 | 16 | 12.3 | 22 | 16.8 | 38 | 14.6 |
| Severe problems | 5 | 11.6 | 8 | 15.4 | 13 | 13.7 | 7 | 5.4 | 9 | 6.9 | 16 | 6.1 |
| Extreme problems | 13 | 30.2 | 15 | 28.8 | 28 | 29.5 | 59 | 45.4 | 47 | 35.9 | 106 | 40.6 |
| Self-care | ||||||||||||
| No problems | 15 | 34.9 | 19 | 36.5 | 34 | 35.8 | 13 | 10.0 | 12 | 9.2 | 25 | 9.6 |
| Slight problems | 7 | 16.3 | 8 | 15.4 | 15 | 15.8 | 12 | 9.2 | 13 | 9.9 | 25 | 9.6 |
| Moderate problems | 9 | 20.9 | 6 | 11.5 | 15 | 15.8 | 22 | 16.9 | 29 | 22.1 | 51 | 19.5 |
| Severe problems | 2 | 4.7 | 8 | 15.4 | 10 | 10.5 | 16 | 12.3 | 16 | 12.2 | 32 | 12.3 |
| Extreme problems | 10 | 23.3 | 11 | 21.2 | 21 | 22.1 | 67 | 51.5 | 61 | 46.6 | 128 | 49.0 |
| Usual activities | ||||||||||||
| No problems | 13 | 30.2 | 15 | 28.8 | 28 | 29.5 | 29 | 22.3 | 31 | 23.7 | 60 | 23.0 |
| Slight problems | 6 | 14.0 | 10 | 19.2 | 16 | 16.8 | 29 | 22.3 | 24 | 18.3 | 53 | 20.3 |
| Moderate problems | 15 | 34.9 | 10 | 19.2 | 25 | 26.3 | 29 | 22.3 | 29 | 22.1 | 58 | 22.2 |
| Severe problems | 5 | 11.6 | 7 | 13.5 | 12 | 12.6 | 21 | 16.2 | 17 | 13.0 | 38 | 14.6 |
| Extreme problems | 4 | 9.3 | 10 | 19.2 | 14 | 14.7 | 22 | 16.9 | 30 | 22.9 | 52 | 19.9 |
| Pain/discomfort | ||||||||||||
| No problems | 25 | 58.1 | 21 | 40.4 | 46 | 48.4 | 64 | 49.2 | 56 | 42.7 | 120 | 46.0 |
| Slight problems | 8 | 18.6 | 15 | 28.8 | 23 | 24.2 | 47 | 36.2 | 48 | 36.6 | 95 | 36.4 |
| Moderate problems | 10 | 23.3 | 10 | 19.2 | 20 | 21.1 | 16 | 12.3 | 22 | 16.8 | 38 | 14.6 |
| Severe problems | 0 | 0.0 | 5 | 9.6 | 5 | 5.3 | 3 | 2.3 | 5 | 3.8 | 8 | 3.1 |
| Extreme problems | 0 | 0.0 | 1 | 1.9 | 1 | 1.1 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| Anxiety/depression | ||||||||||||
| No problems | 23 | 53.5 | 24 | 46.2 | 47 | 49.5 | 58 | 44.6 | 55 | 42.0 | 113 | 43.3 |
| Slight problems | 7 | 16.3 | 14 | 26.9 | 21 | 22.1 | 40 | 30.8 | 43 | 32.8 | 83 | 31.8 |
| Moderate problems | 10 | 23.3 | 13 | 25.0 | 23 | 24.2 | 26 | 20 | 25 | 19.1 | 51 | 19.5 |
| Severe problems | 2 | 4.7 | 0 | 0.0 | 2 | 2.1 | 6 | 4.6 | 6 | 4.6 | 12 | 4.6 |
| Extreme problems | 1 | 2.3 | 1 | 1.9 | 2 | 2.1 | 0 | 0.0 | 2 | 1.5 | 2 | 0.8 |
| Domain | Self-reported | Proxy-reported | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Probiotic | Total | Placebo | Probiotic | Total | |||||||
| n | % | n | % | n | % | n | % | n | % | n | % | |
| Mobility | ||||||||||||
| No problems | 5 | 15.6 | 6 | 15.0 | 11 | 15.3 | 12 | 12.4 | 21 | 21.2 | 33 | 16.8 |
| Slight problems | 5 | 15.6 | 10 | 25.0 | 15 | 20.8 | 20 | 20.6 | 23 | 23.2 | 43 | 21.9 |
| Moderate problems | 8 | 25.0 | 9 | 22.5 | 17 | 23.6 | 15 | 15.5 | 14 | 14.1 | 29 | 14.8 |
| Severe problems | 3 | 9.4 | 3 | 7.5 | 6 | 8.3 | 6 | 6.2 | 7 | 7.1 | 13 | 6.6 |
| Extreme problems | 11 | 34.4 | 12 | 30.0 | 23 | 31.9 | 44 | 45.4 | 34 | 34.3 | 78 | 39.8 |
| Self-care | ||||||||||||
| No problems | 12 | 37.5 | 17 | 42.5 | 29 | 40.3 | 6 | 6.2 | 6 | 6.1 | 12 | 6.1 |
| Slight problems | 1 | 3.1 | 6 | 15.0 | 7 | 9.7 | 11 | 11.3 | 10 | 10.1 | 21 | 10.7 |
| Moderate problems | 6 | 18.8 | 6 | 15.0 | 12 | 16.7 | 19 | 19.6 | 24 | 24.2 | 43 | 21.9 |
| Severe problems | 7 | 21.9 | 6 | 15.0 | 13 | 18.1 | 12 | 12.4 | 16 | 16.2 | 28 | 14.3 |
| Extreme problems | 6 | 18.8 | 5 | 12.5 | 11 | 15.3 | 49 | 50.5 | 43 | 43.4 | 92 | 46.9 |
| Usual activities | ||||||||||||
| No problems | 7 | 21.9 | 13 | 32.5 | 20 | 27.8 | 14 | 14.4 | 21 | 21.2 | 35 | 17.9 |
| Slight problems | 7 | 21.9 | 7 | 17.5 | 14 | 19.4 | 17 | 17.5 | 14 | 14.1 | 31 | 15.8 |
| Moderate problems | 5 | 15.6 | 10 | 25.0 | 15 | 20.8 | 27 | 27.8 | 33 | 33.3 | 60 | 30.6 |
| Severe problems | 9 | 28.1 | 3 | 7.5 | 12 | 16.7 | 15 | 15.5 | 12 | 12.1 | 27 | 13.8 |
| Extreme problems | 4 | 12.5 | 7 | 17.5 | 11 | 15.3 | 24 | 24.7 | 19 | 19.2 | 43 | 21.9 |
| Pain/discomfort | ||||||||||||
| No problems | 14 | 43.8 | 23 | 57.5 | 37 | 51.4 | 39 | 40.2 | 37 | 37.4 | 76 | 38.8 |
| Slight problems | 12 | 37.5 | 5 | 12.5 | 17 | 23.6 | 34 | 35.1 | 33 | 33.3 | 67 | 34.2 |
| Moderate problems | 6 | 18.8 | 7 | 17.5 | 13 | 18.1 | 23 | 23.7 | 25 | 25.3 | 48 | 24.5 |
| Severe problems | 0 | 0.0 | 4 | 10.0 | 4 | 5.6 | 1 | 1.0 | 4 | 4.0 | 5 | 2.6 |
| Extreme problems | 0 | 0.0 | 1 | 2.5 | 1 | 1.4 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| Anxiety/depression | ||||||||||||
| No problems | 17 | 53.1 | 20 | 52.6 | 37 | 51.4 | 28 | 28.9 | 33 | 33.3 | 61 | 31.1 |
| Slight problems | 8 | 25.0 | 5 | 13.2 | 17 | 23.6 | 38 | 39.2 | 35 | 35.4 | 73 | 37.2 |
| Moderate problems | 7 | 21.9 | 9 | 23.7 | 14 | 19.4 | 28 | 28.9 | 25 | 25.3 | 53 | 27.0 |
| Severe problems | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 2 | 2.1 | 5 | 5.1 | 7 | 3.6 |
| Extreme problems | 0 | 0.0 | 4 | 10.5 | 4 | 5.6 | 1 | 1.0 | 1 | 1.0 | 2 | 1.0 |
FIGURE 23.
One hundred stacked bar charts for baseline EQ-5D-5L by trial arm: (a) proxy-reported; and (b) self-reported.


FIGURE 24.
One hundred stacked bar charts for the 3-month follow-up EQ-5D-5L by trial arm: (a) proxy-reported; and (b) self-reported.

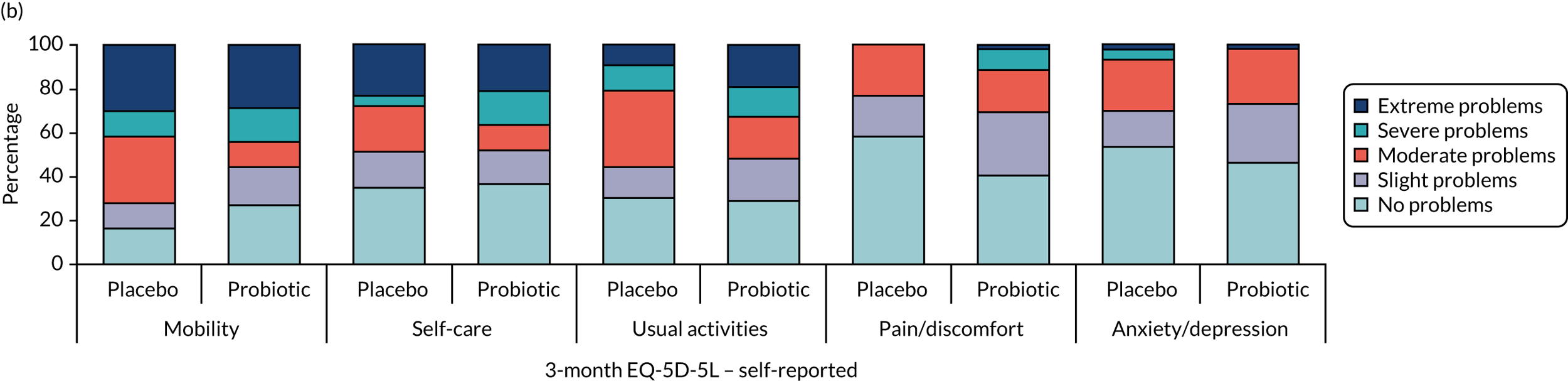
FIGURE 25.
One hundred stacked bar charts for the second follow-up EQ-5D-5L by trial arm: (a) proxy-reported; and (b) self-reported.

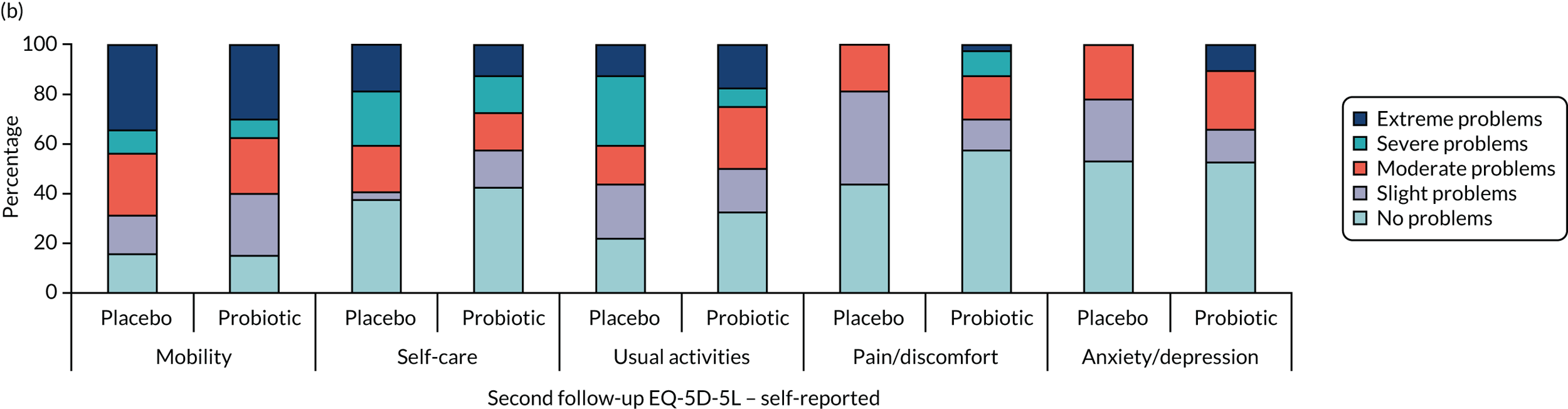
| Immune mediator | Trial arm | Adjustment | Adjusted mean difference | 95% CI | p-value | |||
|---|---|---|---|---|---|---|---|---|
| Placebo (n = 18) | Probiotic (n = 18) | |||||||
| Mean | SE | Mean | SE | |||||
| ICAM-1 (ng/ml) | ||||||||
| Baseline | 459.84 | 41.83 | 424.71 | 33.11 | Trial arm | 0.042 | –0.032 to 0.117 | 0.252 |
| Post intervention | 424.81 | 64.45 | 440.07 | 53.15 | Sex | 0.066 | –0.01 to 0.142 | 0.087 |
| Baseline | 0.916 | 0.74 to 1.092 | < 0.0001 | |||||
| IL-1Ra (ng/ml) | ||||||||
| Baseline | 2.24 | 0.34 | 2.23 | 0.27 | Trial arm | 0.032 | –0.148 to 0.212 | 0.718 |
| Post intervention | 2.49 | 0.68 | 2.52 | 0.44 | Sex | 0.12 | –0.062 to 0.303 | 0.189 |
| Baseline | 0.761 | 0.496 to 1.026 | < 0.0001 | |||||
| E-selectin (ng/ml) | ||||||||
| Baseline | 24.96 | 2.25 | 25.11 | 1.87 | Trial arm | 0.011 | –0.108 to 0.129 | 0.853 |
| Post intervention | 24.20 | 2.70 | 25.43 | 3.78 | Sex | –0.032 | –0.15 to 0.087 | 0.591 |
| Baseline | 0.757 | 0.523 to 0.991 | < 0.0001 | |||||
| VCAM-1 (ng/ml) | ||||||||
| Baseline | 1040.44 | 74.50 | 803.78 | 52.65 | Trial arm | –0.003 | –0.12 to 0.115 | 0.964 |
| Post intervention | 1151.47 | 199.48 | 913.90 | 107.99 | Sex | 0.025 | –0.092 to 0.141 | 0.671 |
| Baseline | 1.013 | 0.693 to 1.334 | < 0.0001 | |||||
| MCP-1 (ng/ml) | ||||||||
| Baseline | 0.44 | 0.04 | 0.39 | 0.03 | Trial arm | 0.073 | –0.047 to 0.193 | 0.224 |
| Post intervention | 0.41 | 0.06 | 0.43 | 0.05 | Sex | 0.049 | –0.074 to 0.172 | 0.423 |
| Baseline | 0.67 | 0.348 to 0.991 | < 0.0001 | |||||
| IP-10 (ng/ml) | ||||||||
| Baseline | 0.16 | 0.01 | 0.17 | 0.01 | Trial arm | –0.002 | –0.162 to 0.158 | 0.981 |
| Post intervention | 0.14 | 0.02 | 0.13 | 0.02 | Sex | –0.008 | –0.176 to 0.16 | 0.923 |
| Baseline | 0.409 | –0.016 to 0.834 | 0.059 | |||||
| IL-17A (pg/ml) | ||||||||
| Baseline | 2.28 | 0.51 | 2.52 | 0.42 | Trial arm | 0.076 | –0.191 to 0.342 | 0.567 |
| Post intervention | 2.99 | 0.85 | 3.09 | 0.73 | Sex | –0.183 | –0.445 to 0.08 | 0.166 |
| Baseline | 0.353 | 0.041 to 0.664 | 0.028 | |||||
| TNFR-2 (ng/ml) | ||||||||
| Baseline | 4.04 | 0.32 | 4.98 | 0.42 | Trial arm | 0.016 | –0.075 to 0.106 | 0.728 |
| Post intervention | 3.25 | 0.35 | 4.03 | 0.52 | Sex | 0.07 | –0.019 to 0.159 | 0.12 |
| Baseline | 0.685 | 0.473 to 0.898 | < 0.0001 | |||||
| IL-6 (pg/ml) | ||||||||
| Baseline | 7.89 | 1.52 | 12.50 | 4.21 | Trial arm | –0.006 | –0.299 to 0.287 | 0.966 |
| Post intervention | 9.82 | 3.41 | 6.56 | 1.58 | Sex | 0.049 | –0.246 to 0.344 | 0.738 |
| Baseline | 0.199 | –0.221 to 0.62 | 0.34 | |||||
| IL-10 (pg/ml) | ||||||||
| Baseline | 0.94 | 0.20 | 0.87 | 0.11 | Trial arm | –0.098 | –0.369 to 0.172 | 0.463 |
| Post intervention | 0.65 | 0.12 | 0.71 | 0.22 | Sex | 0.342 | 0.065 to 0.62 | 0.017 |
| Baseline | 0.639 | 0.361 to 0.917 | < 0.0001 | |||||
| IL-12p70 (pg/ml) | ||||||||
| Baseline | 1.51 | 0.29 | 1.60 | 0.20 | Trial arm | 0.154 | –0.285 to 0.593 | 0.448 |
| Post intervention | 1.22 | 0.36 | 1.64 | 0.36 | Sex | –0.087 | –0.554 to 0.381 | 0.685 |
| Baseline | 0.268 | –0.607 to 1.143 | 0.506 | |||||
| TNF-α (pg/ml) | ||||||||
| Baseline | 16.97 | 1.09 | 19.01 | 1.56 | Trial arm | 0 | –0.076 to 0.077 | 0.991 |
| Post intervention | 17.50 | 1.15 | 17.45 | 1.20 | Sex | 0.052 | –0.03 to 0.135 | 0.205 |
| Baseline | 0.626 | 0.325 to 0.927 | < 0.0001 | |||||
| Descriptive statistics categorised according to trial arm | ANCOVA | |||||||
|---|---|---|---|---|---|---|---|---|
| Immune mediator | Trial arm | Adjustment | Adjusted mean difference | 95% CI | p-value | |||
| Placebo (n = 18) | Probiotic (n = 18) | |||||||
| Mean | SE | Mean | SE | |||||
| IL-10 (pg/ml) | ||||||||
| Baseline | 345.6 | 57.6 | 583.3 | 132.5 | Trial arm | –0.103 | –0.349 to 0.142 | 0.396 |
| Post intervention | 577.9 | 93.4 | 504.0 | 98.9 | Sex | –0.272 | –0.524 to –0.02 | 0.035 |
| Baseline | –0.354 | –0.614 to –0.094 | 0.009 | |||||
| TNF-α (pg/ml) | ||||||||
| Baseline | 2376.6 | 726.4 | 3765.8 | 1002.9 | Trial arm | –0.123 | –0.407 to 0.162 | 0.386 |
| Post intervention | 2675 | 494.9 | 2129.7 | 407.4 | Sex | –0.114 | –0.393 to 0.166 | 0.413 |
| Baseline | –0.061 | –0.352 to 0.23 | 0.672 | |||||
| IL-6 (ng/ml) | ||||||||
| Baseline | 36.2 | 7.2 | 70.9 | 17.7 | Trial arm | –0.12 | –0.401 to 0.16 | 0.388 |
| Post intervention | 54.9 | 7.3 | 54.0 | 10.3 | Sex | –0.274 | –0.556 to 0.009 | 0.057 |
| Baseline | –0.148 | –0.457 to 0.162 | 0.339 | |||||
| IL-1B (pg/ml) | ||||||||
| Baseline | 165.7 | 45.1 | 321.4 | 123.0 | Trial arm | –0.244 | –0.639 to 0.15 | 0.216 |
| Post intervention | 339.7 | 93.8 | 214.2 | 60.7 | Sex | –0.137 | –0.54 to 0.266 | 0.492 |
| Baseline | –0.049 | –0.316 to 0.218 | 0.711 | |||||
| Descriptive statistics categorised according to trial arm | ANCOVA | |||||||
|---|---|---|---|---|---|---|---|---|
| Immune mediator | Trial arm | Adjustment | Adjusted mean difference | 95% CI | p-value | |||
| Placebo (n = 18) | Probiotic (n = 18) | |||||||
| Mean | SE | Mean | SE | |||||
| IL-10 (ng/ml) | ||||||||
| Baseline | 4.3 | 1.7 | 4.2 | 1.4 | Trial arm | –0.044 | –0.301 to 0.213 | 0.73 |
| Post intervention | 3.7 | 0.7 | 3.9 | 0.8 | Sex | –0.127 | –0.388 to 0.134 | 0.328 |
| Baseline | 0.376 | 0.128 to 0.625 | 0.004 | |||||
| TNF-α (ng/ml) | ||||||||
| Baseline | 10.9 | 2.3 | 13.7 | 2.6 | Trial arm | –0.051 | –0.28 to 0.179 | 0.655 |
| Post intervention | 11.4 | 1.9 | 12.1 | 2.4 | Sex | –0.099 | –0.326 to 0.129 | 0.384 |
| Baseline | 0.249 | –0.053 to 0.551 | 0.103 | |||||
| IL-6 (ng/ml) | ||||||||
| Baseline | 40.2 | 5.3 | 47.6 | 6.9 | Trial arm | –0.037 | –0.209 to 0.135 | 0.664 |
| Post intervention | 43.8 | 4.6 | 45.7 | 6.5 | Sex | –0.081 | –0.256 to 0.094 | 0.351 |
| Baseline | 0.261 | –0.036 to 0.557 | 0.083 | |||||
| IL-1B (ng/ml) | ||||||||
| Baseline | 3.8 | 0.8 | 6.4 | 2.8 | Trial arm | 0.095 | –0.205 to 0.395 | 0.522 |
| Post intervention | 4.3 | 0.7 | 15.1 | 8.8 | Sex | –0.274 | –0.576 to 0.029 | 0.075 |
| Baseline | 0.171 | –0.238 to 0.581 | 0.399 | |||||
| Descriptive statistics categorised according to trial arm | ANCOVA | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Immune mediator | Trial arm | Adjustment | Adjusted mean difference | 95% CI | p-value | ||||
| Placebo (n = 18) | Probiotic (n = 18) | ||||||||
| Mean | SE | Mean | SE | ||||||
| IFN-γ (pg/ml) | |||||||||
| Baseline | 23.2 | 17.1 | 21.3 | 5.3 | Trial arm | 0.045 | –0.777 | 0.867 | 0.912 |
| Post intervention | 18.5 | 9.4 | 16.5 | 6.5 | Sex | 0.079 | –0.713 | 0.87 | 0.841 |
| Baseline | 0.383 | –0.019 | 0.785 | 0.061 | |||||
| TNF-α (pg/ml) | |||||||||
| Baseline | 1342.5 | 155.8 | 2013.2 | 275.2 | Trial arm | –0.024 | –0.197 | 0.148 | 0.776 |
| Post intervention | 1804.2 | 223.5 | 2094.9 | 306.0 | Sex | 0.015 | –0.15 | 0.179 | 0.854 |
| Baseline | 0.298 | –0.015 | 0.61 | 0.061 | |||||
FIGURE 26.
Anti-influenza titres for the strain A/Michigan/2015 according to allocation: (a) placebo; and (b) probiotic.
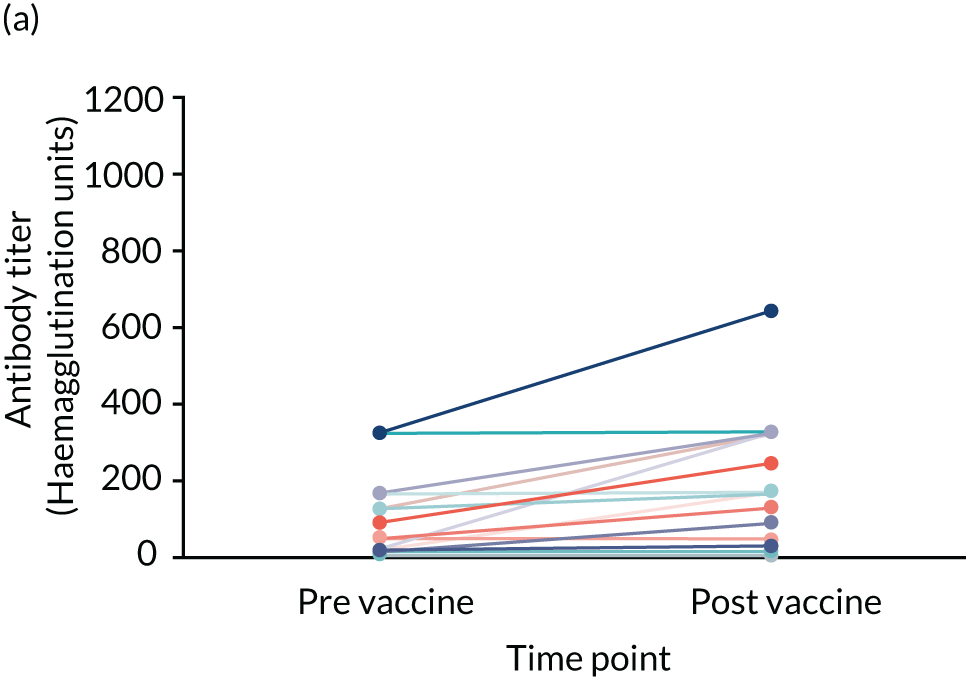

FIGURE 27.
Anti-influenza titres for the strain A/Hong Kong/2014 according to allocation: (a) placebo; and (b) probiotic.


FIGURE 28.
Anti-influenza titres for the strain B/Brisbane/2008 according to allocation: (a) placebo; and (b) probiotic.

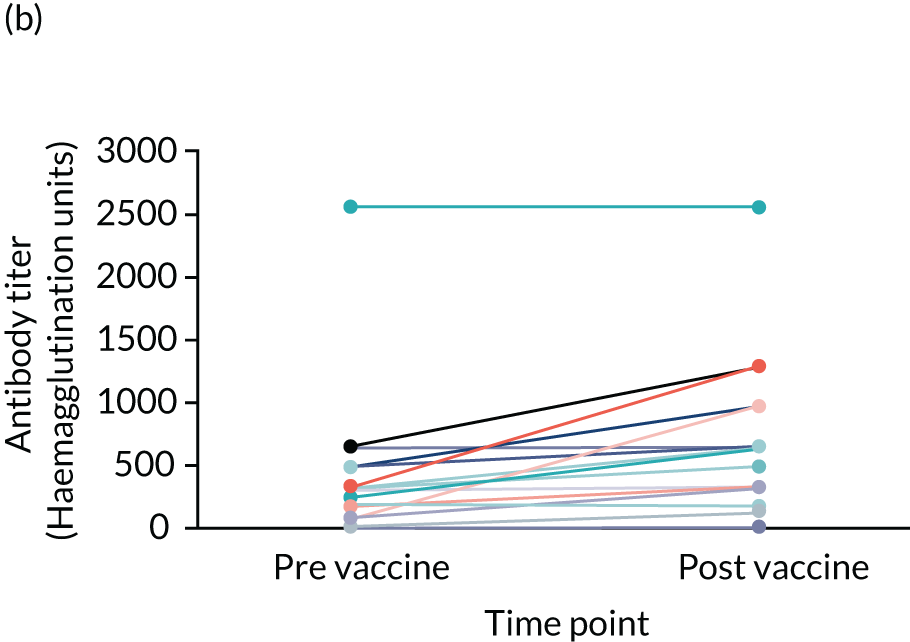
FIGURE 29.
Anti-influenza titres for the strain B/Phuket/2013 according to allocation: (a) placebo; and (b) probiotic.
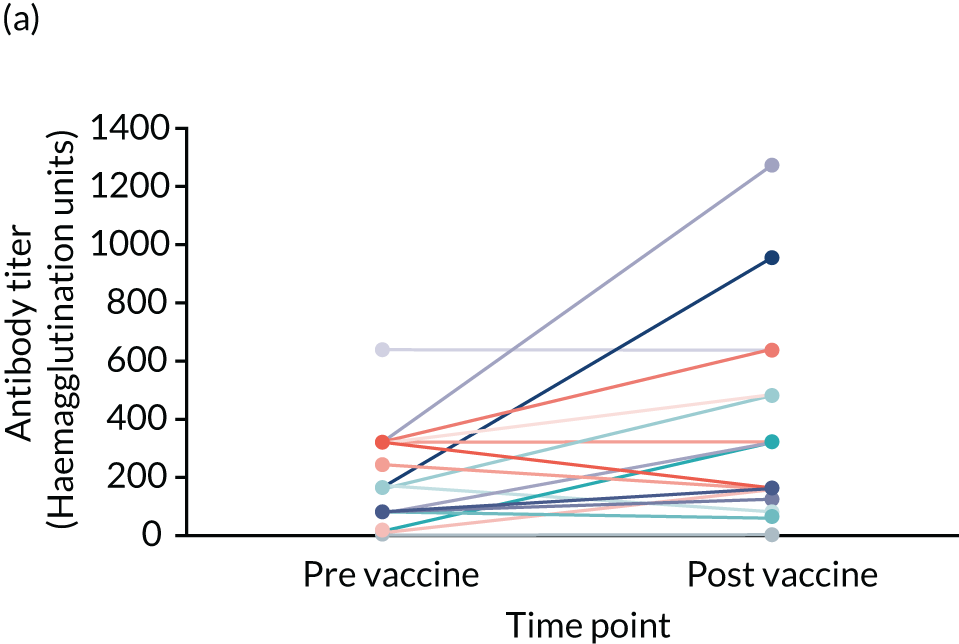

List of abbreviations
- AAD
- antibiotic-associated diarrhoea
- AE
- adverse event
- AMR
- antimicrobial resistance
- ANCOVA
- analysis of covariance
- CAAD
- cumulative antibiotic administration days
- CBA
- Columbia blood agar
- CD
- cluster of differentiation
- CFU
- colony-forming unit
- CHM
- care home manager
- CHR
- care home resident
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CRE
- carbapenem-resistant Enterobacterales
- CRF
- case report form
- CRN
- Clinical Research Network
- CTIMP
- clinical trial of an investigational medicinal product
- CTR
- Centre for Trials Research, Cardiff University
- EDTA
- ethylenediaminetetraacetic acid
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- ESBL
- extended-spectrum beta-lactamase
- FAA
- fastidious anaerobe agar
- FBC
- full blood count
- GI
- gastrointestinal infection
- GP
- general practitioner
- HAI
- haemagglutination inhibition assay
- ICAM-1
- intracellular adhesion molecule 1
- ICECAP-O
- Investigating Choice Experiments for the Preferences of Older People CAPability measure for Older people
- IFN-γ
- interferon gamma
- IL-6
- interleukin 6
- IL-10
- interleukin 10
- IL-1Ra
- interleukin 1 receptor antagonist
- IP-10
- inducible protein 10
- IQR
- interquartile range
- IRR
- incidence rate ratio
- LPS
- lipopolysaccharide
- LRTI
- lower respiratory tract infection
- MALDI-TOF
- matrix-assisted laser desorption/ionisation time of flight
- MAR
- medication administration record
- MCP-1
- monocyte chemoattractant protein 1
- NICE
- National Institute for Health and Care Excellence
- NK
- natural killer
- OR
- odds ratio
- PGN
- peptidoglycan
- PHA
- phytohaemagglutinin
- PI
- principal investigator
- PRINCESS
- Probiotics to Reduce Infections iN CarE home reSidentS
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- RN
- research nurse
- RR
- risk ratio
- RTI
- respiratory tract infection
- SAE
- serious adverse event
- SAR
- serious adverse reaction
- SD
- standard deviation
- SE
- standard error
- SP
- study product
- TLR
- toll-like receptor
- TNF-α
- tumour necrosis factor alpha
- TNFR-2
- tumour necrosis factor receptor 2
- URTI
- upper respiratory tract infection
- UTI
- urinary tract infection
- VCAM-1
- vascular cell adhesion molecule 1
- VRE
- vancomycin-resistant enterococci
- WBC
- white blood cell