

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 11/100/50. The contractual start date was in April 2013. The final report began editorial review in October 2021 and was accepted for publication in May 2022. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2022 Brown et al. This work was produced by Brown et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2022 Brown et al.
Chapter 1 Introduction
Background
Approximately 16,000 people die from colorectal cancer (CRC) in the UK every year, making it the second most common cause of cancer death in the UK and accounting for 10% of all cancer deaths in the country. 1 Given that the concept of one treatment for all patients with a particular type of cancer has become outdated, the major challenge for oncologists is the identification of effective treatments for patients with CRC, given the limited benefits demonstrated of bevacizumab and cetuximab and the failure of multiple other agents in recent trials. 2 To this end, the use of epidermal growth factor receptor (EGFR)-targeted therapy has highlighted the importance of BRAF, PIK3CA, KRAS and NRAS mutations for predicting a lack of response to EGFR-targeted therapy. 3 However, there is also a need for a new approach to the design of clinical trials, allowing for the evaluation of multiple treatments in patients stratified based on biomarkers that are considered to be predictive of a treatment response.
The FOCUS4 trial was designed to enable the rapid identification of patients whose tumours could be characterised based on the presence of specific mutations or validated biomarkers that characterise biological subgroups. This trial was designed to characterise the tumours and stratify eligible patients into comparison subtrials, which, by being a component of a large national study, allow efficient adaptation of the biomarker eligibility criteria within the trial and enable the rapid accrual of patients, even those with the rare subtypes, such as those with the BRAF mutation, who account for only 8% of the population of patients with metastatic CRC.
The design of the FOCUS4 trial was considered to be efficient in that it exploited the concept of a population-enriched design, with the aim of evaluating multiple treatments and multiple biomarkers and, thereby, including most patients with a given type of cancer in the trial, irrespective of their biomarker categorisation. Each treatment was evaluated first in the cohort of patients for whom the biomarker was hypothesised to be predictive of response. If appropriate, the hypothesis of the predictive ability of the biomarker was subsequently tested by evaluating the agent in the biomarker-negative patients. It was not assumed that a treatment would work only in biomarker-positive patients, although it was assumed that a treatment that did not demonstrate efficacy in the selected or enriched cohort did not require further evaluation in a biomarker-negative patient population. This more efficient sequential testing approach had been favourably compared with other existing trial designs. 4
The random assignment of each comparison with placebo within the biomarker-defined groups in the FOCUS4 trial design removed the potential bias of the prognostic effects of certain biomarker-defined groups. In addition, the FOCUS4 trial was adaptive in that random assignment to treatments that did not seem to be sufficiently active could be discontinued; new biomarkers and treatments could be introduced when warranted; and biomarkers could be refined as new information from within or outside the trial emerged.
The approach used in the FOCUS4 trial was driven by advances in the molecular understanding of CRC and the development of new targeted therapies, which demand the evaluation of these therapies in subsets of the CRC patient population that are mostly likely to derive benefit from the treatment. This need is evidenced by the failure of many classic trials to show benefit for new treatments in CRC,2 demonstrating the clear need for a new paradigm in the drive for progress.
Use of molecular classification in FOCUS4
In the FOCUS4 trial, patients were allocated to the comparison subtrials, which were initially defined by four specific molecular cohorts, with another cohort for patients whose cancer could not be classified into a specific molecular cohort. Cohorts could change as data became available during the trial.
The initial five molecular cohorts in the FOCUS4 trial, which were allocated to comparison subtrials referred to as FOCUS4-A, -B, -C, -D and -N, were as follows:
-
BRAF mutation (FOCUS4-A). BRAF mutations are more frequent in the presence of microsatellite instability (MSI) and arise more commonly in right-sided colon carcinomas and have a reasonably consistent gene expression signature. 5 In the Medical Research Council (MRC) COntinuous or INtermittent (COIN) trial,6 8% of patients had BRAF mutations. Patients with this molecular classification and with normal platelets in the intermittent arm of the COIN trial had a median overall survival (OS) of 14.8 months and a median progression-free survival (PFS) in the interval off therapy of 3.1 months.
-
PIK3CA mutation or profound loss of phosphatase and tensin homologue (PTEN) expression (FOCUS4-B). These mutations lead to activation of the protein kinase B (AKT) signalling network, and approximately half of the patients also have KRAS mutations. In addition, approximately 10% of patients have a loss of PTEN expression by various mechanisms, including mutation, methylation silencing of the promoter and microRNA inhibition. The TCGA report7 has identified increased signalling through the insulin-like growth factor (IGF) receptor owing to amplification of IGF2 as an important additional driver within this pathway. PIK3CA mutations have been identified as one of the most common mutations in cancer and were identified in 13% of patients in the COIN trial. 6 Patients with PIK3CA or PIK3R1 mutations or profound loss of PTEN expression account for approximately 20–30% of the CRC population and have activated AKT signalling. Patients with this molecular classification and with normal platelets in the intermittent arm of the COIN trial had a median OS of 16.9 months and a median PFS in the interval off therapy of 2.7 months. These figures were similar whether or not a KRAS/NRAS mutation was also present.
-
KRAS or NRAS mutation (FOCUS4-C). Expression profile analysis shows a variation in gene expression patterns in tumours with KRAS mutation, with signalling down the canonical RAS-RAF-MEK-ERK pathway dominating in about one-quarter, signalling through the PIK3-AKT-mTOR pathway in others and diverse signalling in other tumours. 8 In the COIN trial,6 44% of patients exhibited either KRAS or NRAS mutations, rising to 52% of those who also exhibited PIK3CA mutation. Patients with this molecular classification and with normal platelets in the intermittent arm of the COIN trial6 had a median OS of 18.4 months and a median PFS in the interval off therapy of 3.0 months.
-
Wild type for all the above mutations (EGFR dependent) (FOCUS4-D). Patients with these tumours are wild type for BRAF, PIK3CA, KRAS and NRAS and do not have loss of PTEN, and include the subset of patients who respond best to EGFR-targeted monoclonal antibodies. In addition, mutations or amplification in human epidermal growth factor receptor (HER) 2 occur in around 5% and overexpression of HER3 in around 50% of these patients. In the COIN trial,6 42% of patients were free from all four above mutations. Patients with this molecular classification and with normal platelets in the intermittent arm of the COIN trial6 had a median OS of 19.1 months and a median PFS in the interval off treatment of 3.3 months. In the COIN-B trial,9 the equivalent figures were 20.0 months and 4.4 months, respectively, albeit with small patient numbers, indicating that prognosis is potentially good among such patients.
-
Non-stratified group (unclassified) (FOCUS4-N). Approximately 2% of patients with CRC have tumours that cannot be classified successfully; these patients were included in the non-stratified group. In addition, this cohort included those patients eligible for comparisons that were suspended between novel therapy evaluations or patients who chose not to participate in their specific molecular comparison for reasons such as distance to an experimental therapy centre. Once a patient entered a particular comparison subtrial, they could not enter another FOCUS4 comparison at another time.
The identification of novel biomarkers and their link to the selection of patients for specific therapy is a fast-moving field. Therefore, an essential feature of FOCUS4 was the capacity to introduce novel biomarkers once they had been sufficiently validated to identify newly characterised tumour subgroups for evaluation of therapies hypothesised to be effective in the identified patient subpopulations. Therefore, the FOCUS4 trial incorporated some accepted biomarkers (e.g. KRAS mutation), some biomarkers reaching consensus (e.g. BRAF mutation) and other biomarkers for which further development and refinement was required (e.g. PTEN, mRNA for epiregulin) but could be accomplished within the trial itself.
FOCUS4 was structured to provide an overarching recruitment and biomarker identification strategy, which was linked to a series of randomised comparison subtrials between novel and control treatments for the identified subpopulations. During the trial, some of these interventions could have been shown to lack sufficient activity and would be accordingly withdrawn and replaced by new agents or by new biomarker-defined groups.
Within the individual comparison subtrials for each molecular cohort, the novel therapy comparisons were double blind and placebo controlled where possible. However, patients in FOCUS4-N were allocated to either capecitabine or active monitoring; this was, therefore, an open-label, unblinded subtrial.
Aims and objectives
The primary objectives of the FOCUS4 trial were to answer the following research questions:
-
Clinical benefit . In the interval following standard first-line chemotherapy, do the proposed interventions improve PFS and eventually OS compared with a control group in the biomarker-defined cohorts?
-
Improvement in trial design and conduct. To understand better the challenges and efficiencies of conducting a large, molecularly stratified platform trial in metastatic CRC in the UK health-care system.
Chapter 2 Methods
Trial design
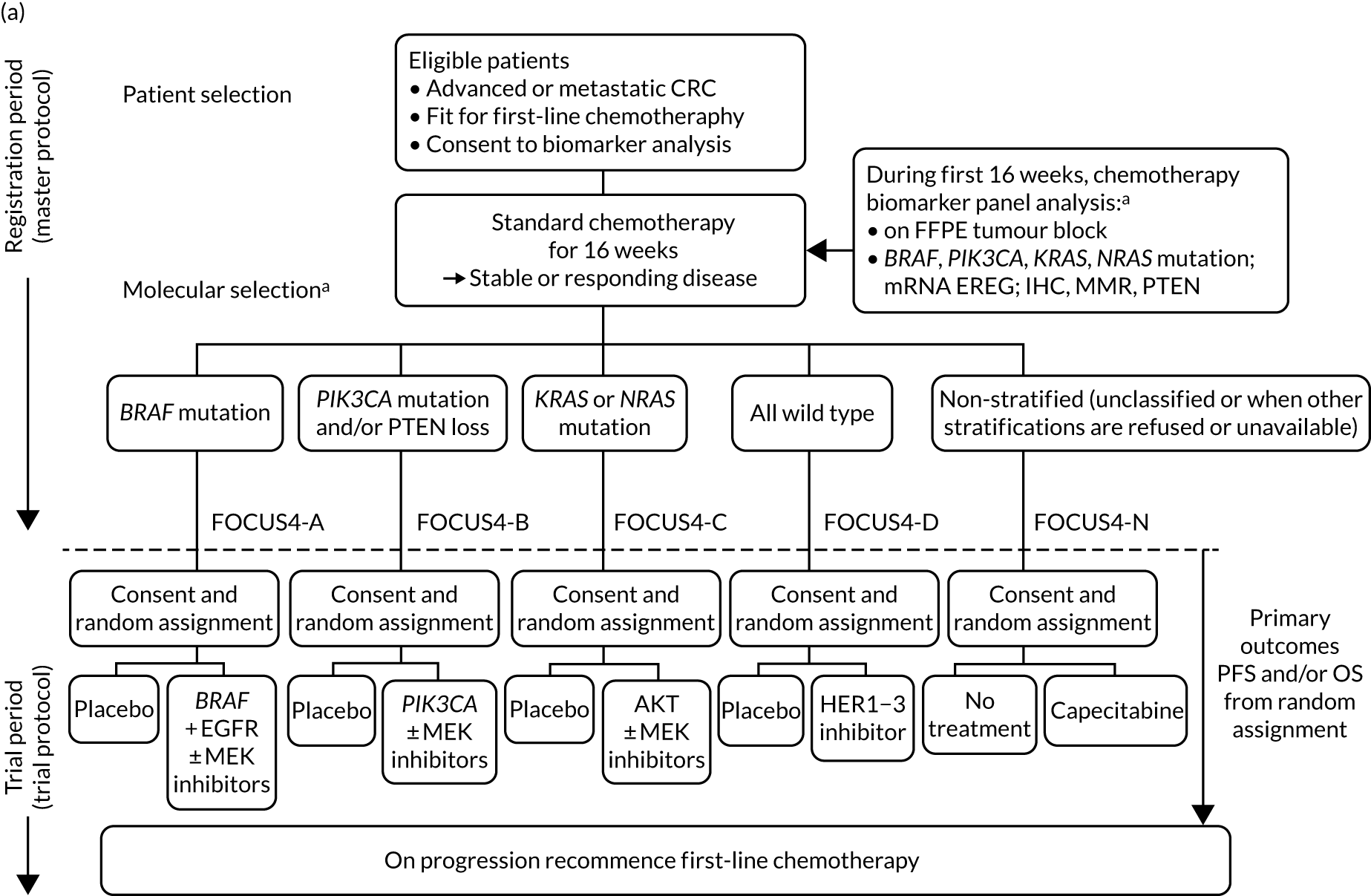
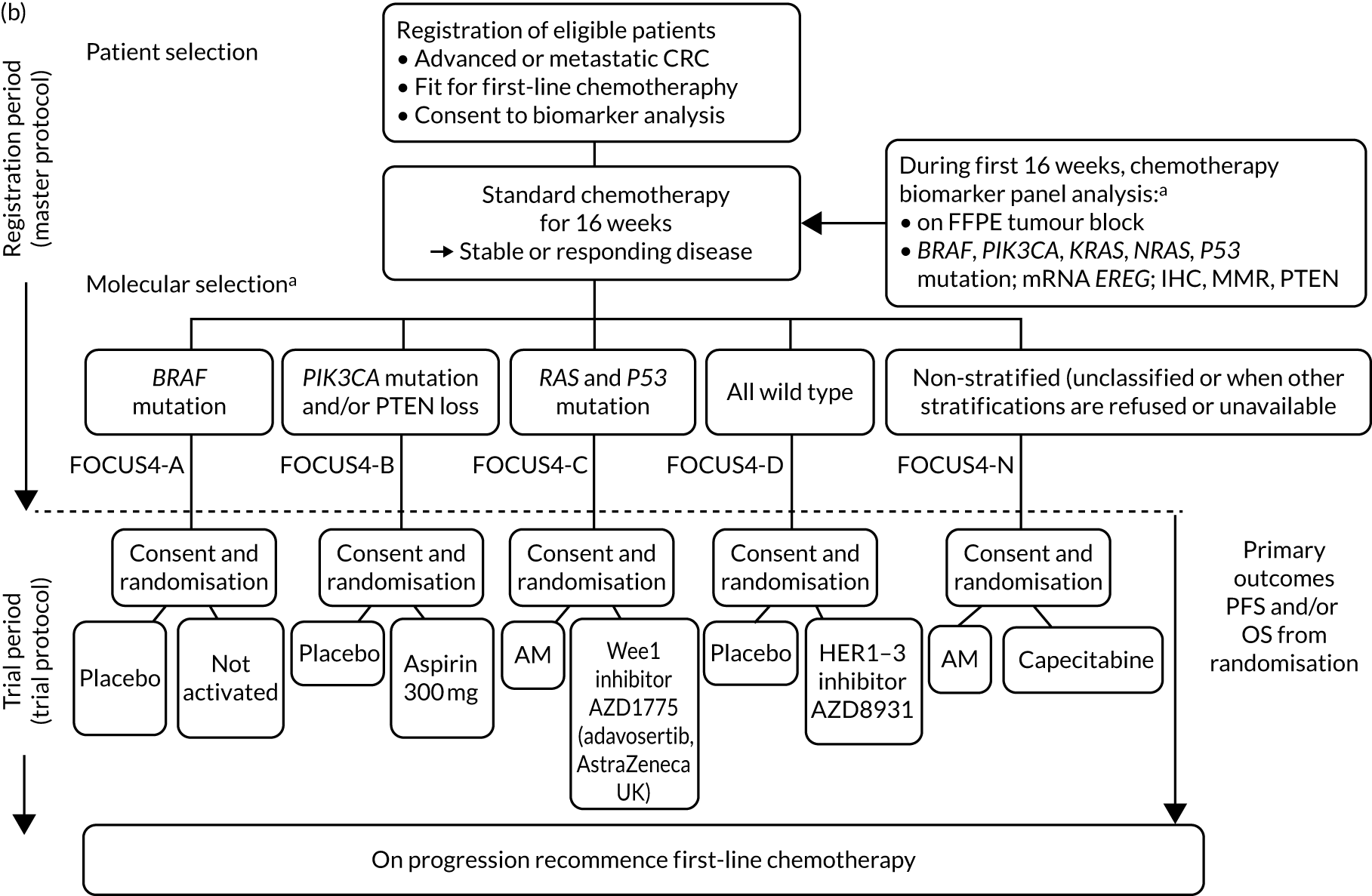
FOCUS4 used some of the methods of the multiarm, multistage (MAMS) randomised trial design. 10–12 After registration and biomarker assessment during a planned 16 weeks of standard first-line chemotherapy, patients were stratified into one of four biologically defined cohorts (A to D), as detailed in Figure 1. Figure 1a presents the original trial schema when the trial was activated in 2014, but the adaptive nature of the design allowed it to change over time, and many iterations occurred before the final schema, which is presented in Figure 1b. If stable or responding disease was confirmed at the end of 16 weeks, patients were then enrolled into the corresponding randomised trial of the novel targeted agent(s) or, for travel, logistic or technical reasons, to the one conventional chemotherapy maintenance trial (i.e. FOCUS4-N).
FIGURE 1.
FOCUS4 trial programme schema: registration, randomisation and treatment. (a) Original schema; and (b) final schema. a, The molecular cohorts are arranged in a hierarchy from left to right. For example, a patient with both a BRAF and a PIK3CA mutation are classified into the BRAF mutation cohort. AM, active monitoring; EREG, epiregulin; FFPE, formalin fixed, paraffin embedded; HER, human epidermal growth factor receptor; PTEN, phosphatase and tensin homologue.
Key principles of the FOCUS4 trial design
The design of the FOCUS4 trial was based on seven key principles, as discussed below. These principles are also described by Kaplan et al. 13
Key principle 1
Key principle 1 was to evaluate multiple treatments and biomarkers in the same protocol, including as many patients as possible with a given disease, with separate clinical questions for as many marker-defined subgroups as are supported by current evidence.
Incorporating multiple treatments across multiple population-enriched biomarker-defined trials fits into conventional clinical practice patterns, in which patients with one type of cancer are managed in a common manner with similar protocols. In CRC, this single treatment approach for all has evolved into two clinical pathways: first, for patients with KRAS wild-type tumours (for whom EGFR-targeted monoclonal antibodies may be planned) and, second, for those with KRAS-mutated tumours. As further individualisation of treatment approaches occurs, managing separately co-ordinated clinical research efforts, which often involve different collaborators and different research teams, becomes progressively more unwieldy and inefficient. The approach used in FOCUS4 filters all eligible patients into one overarching clinical trial programme; this design offers clear efficiency gains in both cost and time compared with running multiple individual trials to evaluate different treatments under separate protocols. This design also increases the likelihood that the investment leads to discovery of effective treatments and halts further development of ineffective treatments in this disease setting. Further efficiency is inherent in biomarker analysis being set up to include all diagnostic tests for the differing subgroups. So far as is scientifically feasible, an inclusive trial allows the maximum number of patients to participate and maximises the potential to recruit rare subtypes. It allows for maximum flexibility in refinement of the biomarker cohort definitions in response to developing clinical data from both within and outside the trial, and provides administrative and organisational efficiencies.
Key principle 2
For key principle 2, in the initial stages, assess each treatment in the presumptive biomarker-enriched subset (thus exploiting the putative link between biomarkers and novel treatments with corresponding mechanisms of action), but without assuming that this association would be confirmed in later stages.
In oncology, even when novel agents are found to be active, the expected biomarker selection may not apply. 14 A key strength of the FOCUS4 protocol is that it neither assumes that any encouraging outcome results are limited to the biomarker selection nor expends numbers of biomarker-negative patients until there is a positive signal from the initial staged analyses (stages 1 to 2). Therefore, entry into the earlier phases of the evaluation of a novel treatment was restricted to those patients who were thought to be most likely to respond. Once the significance level associated with the activity of the experimental treatment fell to less than a given value, there was the option to open a similar efficacy evaluation among those patients who did not show the positivity of the biomarker in their tumour (i.e. off-target effect), using the same type of lack-of-activity assessments, to refute or confirm activity in this complementary population of patients. This approach builds on the putative link between the biomarker and drug efficacy but does not presume that this is a certainity.
Key principle 3
Key principle 3 was to use randomised evidence with a control group for each biomarker/treatment cohort evaluation (eliminating confounding resulting from prognostic biomarker effects).
Constructing the protocol as a set of parallel randomised comparisons ensured that the measured or unmeasured prognostic effects of different biomarkers did not confound the assessment of treatment efficacy, meaning that any benefits were ascribed to the new treatment and not the potential prognostic effect of the marker.
Key principle 4
Key principle 4 was to ensure rapid evaluation of each new treatment, which involves (1) incorporating the flexibility of Phase II and III components into each trial and (2) targeting a reasonably large treatment effect, with discontinuation of random assignment to treatments that are unpromising or overwhelmingly effective as early and reliably as possible.
For the individual trials within the protocol, a larger effect size could be targeted than might be chosen in a more traditional trial. This could be undertaken for two reasons. First, enrichment of the population meant that a somewhat larger effect could be expected, even if enrichment excluded only a proportion of those who did not benefit. Second, there are currently a large number of potential treatments available for evaluation and, therefore, it was reasonable to seek a larger treatment effect than if only a limited number of new treatments were available for testing.
A key aspect of the FOCUS4 protocol was the flexibility to have both Phase II and Phase III components for each randomised comparison, with the potential to move seamlessly from Phase II to Phase III. The aim of the Phase III component was to determine if, in the interval after standard first-line chemotherapy, the proposed novel agents improved PFS and potentially (in some of the larger cohorts) OS, compared with placebo, within the biomarker-defined populations. Use of PFS and OS could be particularly important when agents with different mechanisms of action are being tested. This is in preference to other earlier outcomes of treatment response (e.g. disease response by response evaluation criteria in solid tumours), which may not reliably translate into longer-term outcomes of importance to the patient. For each agent-versus-placebo comparison, FOCUS4 employed a maximum of four stages:
-
stage 1 – safety and screening for sufficient activity (relevant primary end point: PFS)
-
stage 2 – screening for sufficient activity (relevant primary end point: PFS)
-
stage 3 – efficacy (relevant primary end point: PFS)
-
stage 4 – efficacy [relevant primary end point: OS (for those cohorts with sufficient patients)].
Stages 1 and 2 together can be considered analogous to a traditional Phase II trial, whereas continuing into stages 3 and 4 can be considered functionally to complete a traditional Phase III trial. Such designs can be adapted for different end points at different stages and use different decision criteria for moving from one stage to another.
For all therapies that passed activity screening in stage 2, a number of paths were possible. If no major changes were to be made to the research arm (e.g. in biomarker selection criteria or agent dosing), a seamless move to stage 3 was possible, with outcome data on all patients entered during stages 1 and 2 being used in stages 3 and 4. Alternatively, if a major change to a research arm was made, the trial could still continue to the Phase III component (stages 3 and 4), but with outcome data from only newly entered patients from that time forward contributing to the final (Phase III) analysis. However, there was considerable efficiency even in this situation. This so-called new Phase III trial did not require a new protocol and could be initiated by amendment and activated rapidly at all of the sites already participating in the single FOCUS4 protocol. A final possibility was that, for reasons external to the trial itself, the sponsor or funder could decide not to support continuation to the Phase III stages. This could also happen in the face of a positive outcome in the activity screening stages (e.g. if testing of the agent in other settings was planned). Note that such an outcome would still be appropriately viewed as useful if it served to stimulate or facilitate additional trials. In such a situation, other novel agent(s) could then be tested in the relevant cohort of FOCUS4.
For each of the trials within FOCUS4, the overall power was maintained at 80%, allowing for multiple interim data looks, with a maximum 5% two-sided overall significance level (type I error rate). To maintain 80% power overall (for each trial), the power of each trial for the primary analysis varied from 85% to 95%, depending on the number and timing of the interim analyses.
Each biomarker-defined trial was considered separately in terms of the effect size (hazard ratio) to be detected to suit issues relevant to specific biomarker cohorts or agents. An important distinction between stage 2 (lack of sufficient activity) and stage 3 (efficacy) was the difference in type I error, which was set higher for the stage 2 interim PFS analysis (one-sided 10%) than for the stage 3 main PFS efficacy analysis (one-sided 2.5%).
Data for each of the trials were reviewed by the Independent Data Monitoring Committee (IDMC) at each interim analysis. The committee could advise early closure of a trial in the event of overwhelming evidence of efficacy, using a significance level of p = 0.001 as a guideline on the Phase III efficacy outcome measure. This was considered at approximately the halfway point in terms of accrued number of events for each biomarker-defined trial.
Key principle 5
Key principle 5 was to allow the possibility of refining any biomarkers throughout the course of the trial, from either internal data or, more typically, data emerging outside the trial.
Biomarker definitions are constantly evolving. Although, for example, KRAS mutation was a multiply confirmed predictive marker for use of anti-EGFR antibody therapy, it subsequently became clear that not all KRAS mutations had identical effects, with some perhaps not even carrying the same negative predictive value (e.g. KRAS G13D). 3 Evolving data now suggest that expression of EGFR ligands, such as epiregulin and amphiregulin, may also modulate response to this class of agents. 15 Other alterations elsewhere in the RASRAF–MEK–ERK and interacting pathways [e.g. phosphoinositide 3-kinase (PI3K)–AKT–mammalian target of rapamycin (mTOR)] are almost certainly also of considerable importance. An example of biomarker refinement would be the introduction of a new platform for mutation assessment. Developments in molecular diagnostics are occurring at such a pace that older technologies are continually being outperformed by newer technologies in cost and sample requirements. Such refinements could be introduced into a continuing programme, such as FOCUS4, but with close attention to quality assurance and a pre-planned parallel evaluation using both platforms for a period of time to ensure comparability of assessment. If there proved a need to revise one or another biomarker assay during the first two (signal-seeking) analysis stages, patients assigned on the basis of the earlier assay would probably have had to be excluded from the definitive third- and fourth-stage analyses and their possible use towards registration. The third-stage sample size might need to be increased, but the overall time delay introduced in the stage 3 analysis would be minimal (months), because the trial would already be activated and recruiting from a large number of sites.
Key principle 6
Key principle 6 was to allow the possibility of introducing a new biomarker and treatment pairing into the overall trial programme when evidence warranted.
The flexibility inherent in the FOCUS4 design created opportunities to further adapt the trial to accommodate additional findings, typically from other research outside the trial. For instance, it is well known that there is a cohort of patients with MMR deficiency within CRC. In earlier disease, these patients amount to 15% of the total, but they have an improved prognosis, and series among metastatic patients reveal only approximately 4% prevalence of MMR deficiency. At the time that FOCUS4 was designed, there was no convincing case for testing a specific class of agent in this cohort. However, during this period, the rapid emergence of immune checkpoint inhibitors transformed cancer therapy and it is in the MMR-deficient subgroup of CRC that these agents have made a major impact. In principle, if such evidence emerged during the course of the study, the protocol could be amended to open up a new biomarker/treatment cohort by identifying the MMR-deficient cohort and testing the appropriate novel agent(s) compared with placebo in this group. The introduction of such a new biomarker-defined cohort during the trial would remove such patients from the other cohorts, but the introduction of a new cohort would not compromise the study design, requiring only sample size adjustments.
Key principle 7
Key principle 7 was to investigate new treatments in the earliest and most likely responsive settings that are clinically feasible. With many agents being developed and recognition that drug development is a lengthy and expensive process, it is critical to seek strong positive signals as early as possible in testing. When new agents are tested against all-comers and late in the natural history of the disease, it is usually difficult to determine whether or not observed modest improvements are likely to hold up with further testing, especially in earlier stages of disease.
In the FOCUS4 protocol, a four-stage selection process of patients for each trial was used, which improves the chance of identifying clinical benefit from novel agents. First, patients with aggressive disease, as manifested in a raised baseline platelet count, were excluded; this exclusion was later removed following full analysis of the effect of raised baseline platelet count (thrombocytosis) in other trials. Second, only those patients with stable or responding disease during 16 weeks of first-line systemic therapy were included. This, therefore, specifically selected responding patients, in comparison with most study designs, which select patients resistant to evaluate novel agents. Third, by testing the novel agents first in the molecular cohort in which theoretically they should have the greatest benefit, the likelihood of success was maximised. Finally, new agents are used on their own or in novel–novel combinations, after standard treatment, thereby avoiding unpredictable negative pharmacological or toxicity interactions with conventional chemotherapy, which has been seen repeatedly in CRC chemotherapy. Consequently, randomisation occurs in a treatment window of opportunity or treatment break, which is a clinically reasonable and safe strategy on the basis of randomised data from the COIN trial. 16 Although this strategy is somewhat unusual in CRC, there are many settings in the management of other tumours in which periods of observation of patients off treatment are standard and could be used for such window-of-opportunity trials. In FOCUS4, the setting has the advantage of allowing relatively new agents to be tested in patients before the onset of chemotherapy resistance and yet well before comprehensive data would become available with regard to combined administration along with chemotherapy.
Patient consent, registration and biomarker panel testing
Patients were approached to take part in FOCUS4 using a two-stage consent process. Initially, patient consent was obtained for registration and permission for biomarker testing of their tumour tissue; consent was also obtained when eligibility for particular comparisons had been established on the basis of the test results. Patient information sheets were provided for each stage of consent and signed consent forms were required prior to registration or randomisation.
Patients were registered via an online registration platform managed at the MRC Clinical Trials Unit (CTU) at University College London (UCL). Male or female patients aged ≥ 18 years with World Health Organization (WHO) status of 0, 1 or 2 were eligible for registration providing that they had histologically confirmed adenocarcinoma of the small bowel or colon or rectum, with an accessible diagnostic formalin-fixed paraffin-embedded (FFPE) tumour block taken prior to the commencement of standard first-line treatment. They were required to have inoperable metastatic or locoregional disease (synchronous or metachronous) that could be RECIST (Response Evaluation Criteria in Solid Tumours) reported (v1.1) with unidimensionally measurable disease identified by computed tomography (CT) no more than 6 weeks before registration.
The movement of samples was tracked by the MRC CTU from the local site pathology department to one of two mutually quality-assured laboratories in Cardiff (Department of Cellular Pathology and All Wales Molecular Genomics Laboratory, Institute of Molecular Genetics, both located at University Hospital of Wales) or one in Leeds (Division of Pathology and Data Analytics, Leeds Institute of Medical Research at St James’s, University of Leeds). Laboratory testing initially comprised pyrosequencing of the mutation hotspots, then, from August 2017, the use of whole-gene next-generation sequencing (NGS), plus immunohistochemistry (IHC) for MMR proteins and PTEN. The technical components of the biomarkers and inter-laboratory quality assurance have been described previously. 17
Initially, deoxyribonucleic acid (DNA) was extracted from FFPE tissue and analysed using pyrosequencing to obtain the tumour mutation profile across known mutation hotspots in KRAS, NRAS, PIK3CA and BRAF. Further sections were stained on a DAKO Autostainer Link 48™ (Agilent Technologies, Inc., Santa Clara, CA, USA) to determine the protein expression of four mismatch repair (MMR) markers (MLH1, MSH2, MHS6 and PMS2) and PTEN. From August 2017, when FOCUS4-C was opened, the sequencing methodology was changed to NGS to allow coverage of the full sequence of KRAS, NRAS, PIK3CA and BRAF, and the addition of TP53. The GeneRead Clinically Relevant Mutation panel (QIAGEN, Hilden, Germany) was used, adhering to the manufacturer’s instructions. Results were uploaded to the centralised trial database from each laboratory. Where FFPE tumour blocks contained insufficient tumour tissue, an alternative tumour block was requested. If an alternative was unavailable, the patient was still eligible for entry into FOCUS4-N.
Participating sites
A total of 104 hospitals were activated in FOCUS4 across all four devolved UK nations. All sites were able to register patients into FOCUS4; however, given that the drugs being tested in the randomised comparisons varied widely, and included novel unlicensed drugs and generic therapies, sites were assessed for relevant capacity and expertise for participation in each of the comparisons. Sites were classified into three levels:
-
Level 1 sites (n = 51). Hospitals with clinical trial experience but without the required expertise for testing unlicensed therapies. Level 1 sites could register patients and recruit into FOCUS4-B (testing aspirin) and FOCUS4-N (testing capecitabine).
-
Level 2 sites (n = 29). Hospitals with experience of testing both licensed and unlicensed drugs but without extensive early phase experience. Level 2 sites could register patients and randomise into FOCUS4-B, FOCUS4-D, FOCUS4-N and eventually into FOCUS4-C when safety and tolerability of the wee-1 inhibitor had been assessed by the IDMC.
-
Level 3 sites (n = 23). Hospitals with early phase experience and extensive clinical trials experience of licensed and unlicensed drugs. Level 3 sites could register and randomise into all comparisons.
Statistical design and methods
FOCUS4 was designed to allow comparisons to be added into the platform as new agents became available or drop agents if prespecified interim analyses indicated a lack of sufficient drug activity. Decisions on the stopping of particular comparisons were based on MAMS statistical methodology such that the IDMC were provided with prespecified stopping guidelines for each comparison and asked to review the data in confidence at interim analyses and make recommendations on whether to continue or stop the comparison; the statistical analysis plan (SAP) is presented at www.fundingawards.nihr.ac.uk/award/11/100/50. These recommendations were considered by the Trial Steering Committee (TSC) and Trial Management Group (TMG) without sight of any data before a stop/go decision was made for that comparison.
Molecular assays
Details of the molecular assays used for molecular characterisation of the tumour samples are presented in the following sections. Parts of this section are reproduced by Richman et al. 17 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: https://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
In addition, a copy of the Biomarker Laboratory Manual is provided in Report Supplementary Material 1.
Sample processing
A series of 5-mm-thick sections were taken from each block, the first of which was used for haematoxylin and eosin staining, to identify the area of greatest tumour density, and the rest made available for DNA extraction and whole-section IHC. From the residual blocks, tissue microarrays (TMAs) were created, comprising four 0.6-mm tumour tissue cores and one core, if available, of ‘tumour-associated’ normal tissue. To reduce tissue use, the TMAs were prepared only once, in Cardiff, and then shipped to Leeds, where sections were cut and used for IHC.
DNA macrodissection and extraction
The spare sections from the resection blocks were marked out for the richest areas of neoplastic cell content, using the corresponding haematoxylin- and eosin-stained section as a guide, and were macrodissected. DNA was extracted in Leeds using the QIAGEN QIAamp DNA Extraction Kit and in Cardiff using the QIAGEN EZ1 following the manufacturer’s standard protocol.
Mutation detection
The analysis of mutation hotspots was carried out by pyrosequencing within KRAS codons 12, 13, 61 and 146 (exons 2, 3 and 4); BRAF codon 600 (exon 15); NRAS codons 12, 13 and 61 (exons 2 and 3); and PIK3CA codons 542, 545, 546 and 1047 (exons 9 and 20). Pyrosequencing was carried out in each laboratory using a PyroMark Q96 (QIAGEN, Hilden, Germany). A negative water control and a positive control for each assay were included in every sample run. Raw data files were used to generate pyrograms for interpretation by qualified personnel.
Next-generation sequencing
Next-generation sequencing was used for the detection of mutations in the KRAS, NRAS, BRAF, PIK3CA and TP53 genes. All samples were analysed for mutations at specified codons/exons within the following genes: BRAF codons 599, 600 and 601 (exon 15); KRAS codons 12 and 13 (exon 2), 61 (exon 3), 117 and 146 (exon 4); NRAS codons 12 and 13 (exon 2), 61 (exon 3), 117 and 146 (exon 4); PIK3CA codons 542, 545, 546 (exon 10*), 1047 and 1049 (exon 21*); and TP53. *Note: PIK3CA numbering corrected from previous standard operating procedures versions in line with ref sequence NM_006218.2 (previously referred to as exons 9 and 20).
Any changes in these specific codons were reported, as long as they were not considered to be polymorphisms. Full details of the NGS methods can be found in the Laboratory Manual.
Mismatch repair status determination
All four immunohistochemical analyses were carried out on a DAKO Autostainer Link 48 (Ely, UK) using DAKO pre-programmed protocols, which were available with the Autostainer. Antigen retrieval was performed in the accompanying PT-Link chamber with high-pH DAKO target retrieval solution, in accordance with manufacturer’s instructions. Slides were rinsed with DAKO wash buffer prior to loading into the Autostainer. DAKO ready-to-use antibodies were used for MLH1 (IR079), MSH2 (IR085) and MSH6 (IR086). DAKO PMS2 (M3674) was used at a dilution of 1 : 40. Sections from the two validation TMAs were stained with each antibody, then corresponding whole sections were also stained in cases where the cores appeared negative or equivocal or for cases where all cores had been lost from the TMA section. Tumours were deemed positive if any proportion of the tumour nuclei was positively stained, or negative where all discernible tumour nuclei were negative in the local presence of positively staining stromal and infiltrating lymphocytic cells. Any samples appearing wholly negative with respect to both tumour and stromal components were deemed to be of indeterminate status.
Phosphatase and tensin homologue protein expression
Immunohistochemical staining was carried out using the DAKO Autostainer Link 48. Antigen retrieval was carried out in the accompanying PT-Link chamber with high-pH DAKO target retrieval solution, in accordance with manufacturer’s instructions. Slides were rinsed with DAKO wash buffer prior to loading into the Autostainer. DAKO PTEN antibody (M3627) was used at a predetermined dilution of 1 : 100. Both validation TMAs were stained, along with all corresponding whole sections. The presence and intensity grade of cytoplasmic staining in the tumour component was noted (0 = negative; 1 = weak cytoplasmic staining, less intense than the surrounding stroma; 2 = moderate cytoplasmic staining, where staining is equal in intensity to the adjacent stromal staining; and 3 = strong cytoplasmic staining, where staining is stronger in intensity to the adjacent stromal staining). For the purposes of randomised stratification of patients, any positive result was reported as ‘no loss’ of PTEN, whereas the negative result was reported as ‘absence’ of PTEN. Three FFPE cell lines (LNCaP, PTEN negative; ZR-75–1, a weak expresser of PTEN; and MCF7, which overexpresses PTEN) were used to create a mini control TMA, which was stained along with each section. A suspension was generated from each cell line, which was subsequently spun down, fixed in 10% neutral-buffered formalin, added to 12% Noble agar at a 1 : 1 ratio, processed and paraffin embedded. Three cores were taken from each and embedded into a new paraffin block to create the mini ‘control TMA’. A section of this was cut onto the same slide as each of the 97 validation samples.
Data validation
Each laboratory sent the results of all analyses directly to the MRC CTU for independent cross-referencing. Any discrepant results were discussed between the biomarker teams from both laboratories until a final unanimous result was agreed on.
A copy of the master protocol and of the protocols for the comparison subtrials can be found on the FOCUS4 trial website (www.focus4trial.org/; accessed 28 October 2022).
Chapter 3 Results from the registration period
Details of the methods used for the registration period can be found in Chapter 2.
Activation and recruitment
FOCUS4 was approved by the UK National Ethics Committee Oxford – Panel C (reference 13/SC/0111) and by the relevant regulatory body, the Medicines and Healthcare products Regulatory Agency (MHRA) (CTA 20363/0400/001 and EudraCT 2012-005111-12), in May 2013. FOCUS4 opened to recruitment in January 2014, with two subtrials activated (FOCUS4-D and FOCUS4-N). Over the following 6 years, 103 UK sites were opened and subsequent substantial amendments approved the inclusion of two further subtrials (FOCUS4-B and FOCUS4-C). The trial centres, investigators and co-investigators are listed in Report Supplementary Material 2. Considerable challenges were encountered in engaging pharmaceutical companies to test therapies in the platform, with 20 drug combinations explored for inclusion. Of the five subtrials that had been planned to open at the design stage (FOCUS4-A, -B, -C, -D and -N), only four were activated (FOCUS4-B, -C, -D and -N) and only three reported results (FOCUS4-C, -D and -N), with FOCUS4-B stopping early on grounds of futility (see Figure 1).
Results from registration
Between January 2014 and October 2020, 1434 patients were registered from 88 UK hospitals, and 316 were randomised into a comparison subtrial (25%). A flow chart of patients during the registration period is presented according to the Consolidated Standards of Reporting Trials (CONSORT) criteria in Figure 2.
FIGURE 2.
The CONSORT flow diagram of patients during registration period of FOCUS4.

Biomarker analysis was undertaken on 1382 samples and the median time from registration to the availability of the biomarker results was 6 weeks [interquartile range (IQR) 4.3–8.3 weeks]. Molecular stratification was successful in 1291 out of 1382 (93%) samples received and was processed with mutations identified in the following genes: BRAF (10%), PIK3CA (14%), KRAS (53%), NRAS (6%), TP53 (69%), RAS + TP53 (32%), BRAF-PIK3CA-RAS wild type (26%) and deficient MMR (3%), as presented in Table 1.
| Biomarker | Total with successful test result (n)a | Total with mutations, PTEN loss or MSI (n) | Observed (%) | Expected (%)b |
|---|---|---|---|---|
| BRAF mutation | 1260 | 125 | 10 | 8 |
| PIK3CA mutation | 1245 | 179 | 14 | 13 |
| KRAS mutation | 1264 | 666 | 53 | 42 |
| NRAS mutation | 1264 | 72 | 6 | 4 |
| PTEN loss | 1260 | 91 | 7 | 10 |
| Wild type (i.e. none of the above, c.f. FOCUS4-D) | 1243 | 328 | 26 | 42 |
| TP53 | 805 | 556 | 69 | NR |
| TP53 + RAS mutation (cf. FOCUS4-C) | 805 | 332 | 41 | 23 |
| Presence of deficient MMR | 1255 | 33 | 3 | 4 |
Three molecularly targeted subtrials were activated:
-
FOCUS4-D –
-
February 2014 to March 2016
-
evaluated ErbB kinase inhibitor sapitinib (AZD8931, AstraZeneca UK, Cambridge, UK) in the BRAF-PIK3CA-RAS wild-type subgroup.
-
-
FOCUS4-B –
-
February 2016 to July 2018
-
evaluated aspirin in the PIK3CA-mutant subgroup.
-
-
FOCUS4-C –
-
June 2017 to October 2020
-
evaluated adavosertib (AstraZeneca Ltd, Cambridge, UK) in the RAS + TP53 double-mutant subgroup.
-
-
FOCUS4-A for the BRAF-mutant subgroup was not activated, despite a full protocol being prepared, because the data emerging from studies of the planned triplet combination (dabrafenib, panitumumab and trametinib) were determined not to warrant further investigation by the company involved.
In addition, FOCUS4-N was active throughout and evaluated capecitabine monotherapy compared with a treatment break.
Of the 1315 patients completing 16 weeks of first-line therapy, 908 (68%) had disease stabilisation or response and were potentially eligible for randomisation. Of these, 361 were randomly allocated to one of the comparison subtrials: FOCUS4-B (n = 6), FOCUS4-C (n = 69), FOCUS4-D (n = 32) and FOCUS4-N (n = 254).
The choice of first-line therapies used during the registration period is summarised in Table 2.
| Chemotherapy regimen (ordered by frequency) | All registered patients, n (%) |
|---|---|
| FOLFIRI | 422 (29) |
| CAPOX | 301 (22) |
| FOLFIRI + CET/PAN | 218 (15) |
| FOLFOX | 215 (15) |
| CAP alone | 47 (3) |
| FOLFOX + CET/PAN | 46 (3) |
| FOLFOXIRI | 28 (2) |
| CAPIRI | 20 (1) |
| FOLFOX + BEV | 16 (1) |
| CAPOX + CET/PAN | 15 (1) |
| FOLFIRI + BEV | 10 (1) |
| Other | 46 (3) |
| Data not available | 50 (4) |
Treatment with EGFR-targeted monoclonal antibodies was administered according to NHS access or Cancer Drug Fund restrictions.
Details on biomarker prevalence and rates of disease progression during the registration period are presented in Table 3 (see also Table 1).
| Molecular cohort | Randomisation status | Disease assessment, n (%) | |||
|---|---|---|---|---|---|
| CR/PR/stable disease | PD | Missinga | Total | ||
| BRAF mutant | Not randomised | 43 (48) | 40 (45) | 6 | 89 |
| FOCUS4-N | 30 | 0 | 0 | 30 | |
| Subtotal | 73 (61) | 40 (34) | 6 | 119 | |
| PIK3CA mutantb | Not randomised | 60 (60) | 38 (38) | 2 | 100 |
| FOCUS4-B | 6 | 0 | 0 | 6 | |
| FOCUS4-N | 26 | 0 | 0 | 26 | |
| Subtotal | 92 (70) | 38 (29) | 2 | 132 | |
| RAS mutantc | Not randomised | 142 (59) | 82 (34) | 18 | 242 |
| FOCUS4-N | 103 | 0 | 0 | 103 | |
| Subtotal | 245 (71) | 82 (24) | 18 | 345 | |
| RAS + TP53 mutantd | Not randomised | 85 (47) | 87 (48) | 10 | 182 |
| FOCUS4-C | 69 | 0 | 0 | 69 | |
| FOCUS4-N | 13 | 0 | 0 | 13 | |
| Subtotal | 167 (63) | 87 (33) | 10 | 264 | |
| All wild type | Not randomised | 159 (65) | 74 (30) | 12 | 245 |
| FOCUS4-D | 32 | 0 | 0 | 32 | |
| FOCUS4-N | 57 | 0 | 0 | 57 | |
| Subtotal | 248 (74) | 74 (22) | 12 | 334 | |
| Non-stratified | Not randomised | 58 (60) | 35 (36) | 3 | 96 |
| FOCUS4-N | 25 | 0 | 0 | 25 | |
| Subtotal | 83 (69) | 35 (29) | 3 | 121 | |
| Overall | Not randomised | 547 (57) | 356 (37) | 51 | 954 |
| FOCUS4-B | 6 | 0 | 0 | 6 | |
| FOCUS4-C | 69 | 0 | 0 | 69 | |
| FOCUS4-D | 32 | 0 | 0 | 32 | |
| FOCUS4-N | 254 | 0 | 0 | 254 | |
| Total | 908 (69) | 356 (27) | 51 | 1315 | |
Failed endeavours
The continuous pursuit of new agents for testing in the FOCUS4 platform accounted for a lot of the investigator’s time. During the course of the trial, a total of 20 therapeutic interventions were explored and presented to the joint National Institute for Health and Care Research (NIHR) Efficacy and Mechanism Evaluation (EME)/Cancer Reserch UK funding subboard for peer-review approval, but only four culminated in an activated comparison (Table 4). Reasons for non-activation were predominantly failure of drugs at early clinical testing in this advanced metastatic disease setting; however, protocol development and contract negotiations had often progressed a long way and considerable resource had been used up for ultimately futile collaborations. Other endeavours failed bceause of strategic shifts
| Cohort | Biomarker | Biomarker incidence | Intervention | Outcome |
|---|---|---|---|---|
| A1 | BRAF V600E mutation | 10% | BRAF inhibitor and MEK inhibitor | Science evolved |
| A2 | BRAF V600E mutation | 10% | Dabrafenib (tafinlar, Novartis Pharmaceuticals UK Ltd, London, UK), trametinib (mekinist, Novartis Pharmaceuticals UK Ltd) and panitumumab (vectibix, Amgen, Thousand Oaks, CA, USA) | GlaxoSmithKline plc (Brentford, UK) sold oncology portfolio to Novartis (Basel, Switzerland). Novartis found activity insufficient to support trial |
| B1 | PIK3CA mutant or PTEN loss on IHC | 22% | Dual PI3K inhibitor/mTOR inhibitor | Insufficient evidence of benefit with evolving data to obtain pharmaceutical engagement |
| B2 | PIK3CA mutation | 12% | Aspirin | FOCUS4-B trial |
| C1 | KRAS/NRAS mutation | 45% | MEK inhibitor and PI3K inhibitor | Found to be too toxic in early studies |
| C2 | RAS mutation + HLA A-2 | 20% | IMA 190 peptide vaccine (Immatics, Tuebingen, Germany) | Company did not commit |
| C3A | H3K36me3 loss | < 2% | Wee1 inhibitor, AZD1775 (adavosertib, AstraZeneca UK) | Biomarker validation showed very low incidence of loss in CRC |
| C3B | RAS + TP53 double mutation | 30% | Wee1 inhibitor, AZD1775 | FOCUS4-C trial |
| C3C | ATM loss | 6% | ATM inhibitor, AZD6738 | Company did not support concept |
| D1 | KRAS, NRAS and BRAF wild type | 40% | pan-HER inhibitor, AZD7941 | FOCUS4-D trial |
| D2 | KRAS, NRAS and BRAF wild type | 40% | MM151 | Company sold asset prior to contract |
| D3 | Triple wild type, HER2 negative | 25% | Cetuximab and CDK4/6 inhibitor | Data from SCCHN19 |
| D4 | Triple wild type, HER2 overexpressed | 2% | Trastuzumab (Herceptin, F. Hoffman-La Roche Ltd, Basel, Switzerland) and CDK4/6 inhibitor | Biomarker incidence too low |
| E1 | MMR deficient and POLE mutant | 4% | Avelumab (Bavencio, Merck KGaA, Darmstadt, Germany and Pfizer Inc., New York, NY, USA) | Company did not support concept |
| E2 | MMR deficient or TGFb activated | 30% | Bintrafusp alpha | EME programme did not extend grant |
| F | Wnt ligand dependant, axin 2 overexpressed | 9% | RXC004 porcupine inhibitor | EME programme did not extend grant |
| G | ALK/ROS rearrangements | 2% | Crizotinib (Zalkori, Pfizer Inc.) | Biomarker incidence too low |
| N1 | Non-stratified group | N/A | Capecitabine | FOCUS4-N trial reported and met primary end point |
| N2 | Non-stratified group | N/A | Add TAS-102 randomisation | Global company did not support concept |
| N3 | Non-stratified group | N/A | Metronomic cyclophosphamide | EME programme did not extend grant |
within companies, including the selling of assets suddenly and unexpectedly. Markers with incidence below 5% were also generally deemed too infrequent to pursue, although we have seen the very successful evaluation and licensing of immune checkpoint inhibitors in the MSI-high subgroup, which occurs in only 4% of metatastic CRCs.
Discussion and conclusions
Parts of this section have been reproduced with permission from Brown et al. 20 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Adaptive trials not only provide significant advantages in the evaluation of multiple novel therapies in a disease setting, but also provide major challenges in their design, funding and delivery. FOCUS4 was jointly funded by the MRC/NIHR EME programme and Cancer Research UK (CRUK), with a combined budget (£3.6M) that was large in 2012 but small compared with current major molecularly stratified platform studies. Joint funding added a delay to trial initiation and review of amendments (performed via a sub-board representing both funders). For these trials to fulfil their aim of flexibility and nimbleness, it is important to learn and implement the lessons of undertaking complex trials in the era of the pandemic, particularly in terms of protocol and amendment approval, as recently espoused in the UK. 21
The key features of the FOCUS4 design have all proved to be robust in the application. The use of PFS in the maintenance setting as the primary end point has been shown to be an effective indicator of agents with activity, which may not have been revealed through a more conventional assessment of response in end-stage patients. This is of particular relevance for trials in the maintenance therapy setting. It was not possible to progress a therapy as planned through to evaluate whether or not the activity was specific to the molecular subgroup initially identified. Nor did it prove possible to move from Phase II to Phase III testing. These limitations were due to timing and funding availability rather than design failure.
The setting of the interval after 16 weeks of induction chemotherapy for patients with metastatic disease is a novel one. The registration of olapraib (Lynparza, Astrazeneca) in this setting of olaparib in platinum-sensitive ovarian cancer has made this a more recognisable route to registration. It builds on a pattern of practice of allowing patients a complete break from therapy following several months of induction chemotherapy, which is evidence based. Although this method is not unique to the UK, it is perhaps more widely utilised in the UK than in many other countries. This may change as a result of the COVID-19 pandemic, as patients now choose to spend more time away from hospitals and many have preferred a move to remote monitoring.
Adding the complexity of molecular stratification to allocate patients into the optimal biomarker-defined subgroup adds an extra layer of challenges, including the developmental status of the biomarker, the predictive power of the biomarker and selectivity for the therapy being evaluated (which is usually underdeveloped for any novel agent). Timely and reproducible delivery of the biomarker-testing panel, understanding the prognostic impact of the biomarker selection and the need to accommodate biomarker-negative patients remains critical to success. During FOCUS4, the simple mutation-based stratification process for many of the biomarkers that we tested has become part of standard care. In the future, despite the now more widespread routine availability of NGS-based tumour profiling, trial-based stratification testing will continue to need to provide transcriptomic and other more sophisticated analyses. It may be that the revolution in digital pathology and artificial intelligence will enable some biological stratification to be achieved directly from routine images, as shown recently with consensus molecular subgroup subtyping. 22
Rates of allocation into biomarker-selected groups are often very low in precision medicine studies, as exemplified by the Lung-Matrix Trial23 and the Lung-Map Trial. 24 In the largest study of this kind, National Cancer Institute (NCI)-Match,25 5954 patients were enrolled with refractory malignancies, of whom 17.8% were assigned to a targeted therapy. Among these, 848 patients with CRC were registered, 13.7% of whom were assigned and 10% enrolled into a specific therapy trial. 26 In FOCUS4, rather than assigning patients in non-randomised cohorts to therapies with hypothesised efficacy, 361 out of 1434 (25%) registered patients were randomised into a specified subtrial, and 107 (7.5%) of the registered patients were randomised into molecularly stratified subtrials. This attrition related to three main factors: (1) inadequacy of sample submission or analysis (10%), (2) death or progressive disease during induction therapy (27%) or (3) lack of availability of a suitable molecular subtrial or lack of patient consent when the patient was eligible for randomisation (60% of those potentially eligible). However, as shown in the evaluation of adavosertib in FOCUS4-C,27 randomisation against a control arm was critical to demonstrating efficacy, which would probably have been overlooked in a non-randomised design.
It is notable from the RECOVERY trial28 that negative results are more likely but are just as important as positive results in tackling diseases of high unmet clinical need. To date, the RECOVERY trial28 has identified four positive results [dexamethasone, tocilizumab, baricitinib (Olumiant®, Eli Lilly and Company, Indianapolis, IND, USA) and the combination of Casirivimab and imdevimab (Ronapreve, Roche, Basel, Switzerland)] and seven negative results since its set-up in March 2020. NCI-Match has 39 cohorts: 12 have published final results to date,29 of which six have shown positive results. In FOCUS4, two positive results (capecitabine and adavosertib) were reported, one clear negative result and one feasibility failure.
Although non-commercial organisations may be best placed to set up and run such studies enabling collaboration with different pharmaceutical companies for different agents, engagement with the pharmaceutical and biotechnology industries is critical to success. A large amount of investigator time was spent negotiating with companies to test their agents in the platform, but only 4 out of 20 drug combinations explored came to fruition. To obtain approval from companies to include their agents, a very robust approach to the development of preclinical data packages to support the selection of particular drug/biomarker combinations is essential. This requires a well-funded, preclinical testing collaboration, ideally with disease subtype-specific models using genetically engineered mouse models, patient-derived xenograft and patient-derived organoids linked to detailed disease stratification information. From this basis, strong applications can be made to pharma to include agents in such precision medicine studies with a higher likelihood of success than was observed in FOCUS4. Only in the last 2 years has this been available through separate funding streams (the MRC stratified medicine consortium S:CORT30 and the ACRCelerate collaboration31 jointly funded by CRUK, the Italian Association for Cancer Research and the Spanish Association Against Cancer) and the fruit of this is yet to be seen to feed into an updated precision medicine study in CRC.
The enterprise of precision medicine adaptive platform trials is a massive exercise in team science. It is a tribute to the large body of investigators, research nurses and data managers at sites, laboratory scientists and trial unit staff, with the support from the institutions, funders and pharmaceutical companies, and, central to all of this, our patients, who have enabled us to complete this FOCUS4 trial.
Chapter 4 Results from FOCUS4-B
Parts of this chapter have been reproduced with permission from Brown et al. 20 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Scientific rationale for FOCUS4-B
PIK3CA is one of the most commonly altered genes in CRC, with mutations found in 12–20% of cases depending on whether the sequencing is limited to hotspot exons or covers the entire gene (i.e. broader genetic testing of the entire gene identifies a higher number of mutations than testing limited to the most common sites of mutation). 32 PIK3CA encodes the catalytic p110α subunit of PI3K, a key node in the PI3K–AKT pathway, which regulates cellular proliferation, apoptosis and metabolism. 33 Approximately two-thirds of PIK3CA mutations cluster to codons 542 and 545 in the exon 9 helical domain or to codon 1047 in the exon 20 kinase domain. Although they act through different mechanisms, both helical and kinase domain substitutions cause constitutive activation of the PI3K–AKT pathway and are strongly oncogenic in preclinical models. 34 PIK3CA mutations in CRC are often associated with KRAS-activating mutations35 (and also with NRAS- and BRAF-activating mutations) and are reported to correlate with other clinicopathological features, such as right-sided cancers and tumoural MMR deficiency. 36,37 Importantly, in contrast to other molecular aberrations, such as MMR deficiency, the frequency of PIK3CA mutation in metastatic CRC is broadly similar (12.7%) to that observed in early-stage disease,18 consistent with data indicating that neither exon 9 nor exon 20 mutations are prognostic in isolation in early CRC. 38 Similarly, PIK3CA mutation does not appear to be prognostic in metastatic disease. 18 Although it has been suggested that CRCs with mutation of both PIK3CA exon 9 and PIK3CA exon 20 display poor outcome,35 the low frequency (approximately 0.6%) of such double mutants means that validation is required. Emerging data suggest that, similar to KRAS, BRAF and NRAS mutations, PIK3CA mutation may predict resistance to cetuximab in metastatic CRC; however, the current evidence is conflicting (perhaps because of the high frequency of concurrent activating mutations in KRAS and NRAS) and there are insufficient analyses to inform practice. 39,40
Unsurprisingly, given the high frequency of PIK3CA mutation in CRC and other solid tumours, much attention has focused on the development of PI3K inhibitors. Although the high degree of conservation of PI3K with other kinases has made the design of specific agents challenging, compounds with nanomolar affinity have recently entered clinical practice. 41 Disappointingly, emerging data from current early phase clinical trials42 indicate that these drugs lack significant activity against CRC (either alone or in combination with MEK inhibitors) and, therefore, are poor candidates for further development in this particular disease. Our initial efforts to obtain these agents were, therefore, unfruitful. Therefore, at present, no specific therapeutic strategies exist for the subgroup of patients with PIK3CA-mutant CRC.
Aspirin as an anti-cancer agent in colorectal cancer
Over the past two decades, multiple retrospective and prospective studies have unequivocally demonstrated that regular use of aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs) protects against the development of colorectal adenoma and CRC. 43–46 More recently, aspirin has also been shown to reduce the incidence of metastatic disease following CRC resection. 47,48 In both cases, the antineoplastic activity of aspirin is not only seen with high, multiple-daily dosing (300–600 mg four times per day) required for its systemic anti-inflammatory effect owing to inhibition of cyclo-oxygenase 2 (COX-2), but also seen with low, once-daily dosing (70–100 mg once daily) at which the action of aspirin is thought to be limited to an antiplatelet effect from irreversible inhibition of cyclo-oxygenase 1. 48,49 Platelets promote tumour growth and metastasis through multiple mechanisms, including protection of circulating tumour cells from immune destruction by microthrombi formation, facilitation of tumour cell extravasation at distant sites and secretion of tumourigenic growth factors, including platelet-derived growth factor and vascular endothelial growth factor. 50 Thrombocytosis predicts poor outcome in patients with metastatic CRC,51 and platelet depletion inhibits tumour growth in animal models. 52–54 These data, together with the recent demonstration that tumour self-seeding by circulating tumour cells is an important promoter of cancer growth in mice,55 suggest that aspirin may demonstrate anti-tumour activity in the context of advanced disease. Such an evaluation would complement the investigation of aspirin as adjuvant therapy in CRC, which is to be determined by large randomised trials that are currently recruiting (ASCOLT56) or in set-up (Add-Aspirin57).
PIK3CA mutation as an aspirin biomarker
Two large, retrospective studies35,58 have suggested that mutations in exons 9 and 20 of PIK3CA strongly predict benefit of aspirin in CRC. The first, reported by Liao et al. ,35 retrospectively analysed 964 patients with a mixture of disease stages (stages 1–4). The subgroup of patients with stage 4 CRC in this study was small (64 patients, 7% of the total of 964 patients). The study demonstrated that, among patients with PIK3CA-mutant tumours, reduction in CRC-specific mortality was significantly greater in those who were regular users of aspirin after diagnosis than in non-users of aspirin [multivariate adjusted hazard ratio (HR) 0.18, 95% confidence interval (CI) 0.06 to 0.61; p = 0.0001]. By contrast, patients with tumours lacking PIK3CA mutations did not appear to benefit from aspirin use (HR 0.96, 95% CI 0.69 to 1.32; p = 0.76). Statistical analysis demonstrated a significant interaction between PIK3CA mutation and aspirin use (p = 0.02).
These data were supported by analysis of the VICTOR study: a large, randomised controlled trial (RCT) that compared the cyclo-oxygenase-2-specific inhibitor rofecoxib with placebo following completion of adjuvant therapy for stage 2/3 CRC. 58 It should be noted that there were no patients with stage 4 CRC in this study. Although the study protocol did not permit concomitant NSAID or high-dose aspirin use, low-dose (≤ 100 mg daily) aspirin was allowed and was recorded both at randomisation and during follow-up. As in the study by Liao et al. ,35 the benefit of aspirin appeared to be restricted to patients with PIK3CA-mutant cancers, with no recurrences in the subgroup of regular aspirin users whose tumours harboured PIK3CA exon 9 or 20 mutations (multivariate adjusted HR 0.11, 95% CI 0.001 to 0.832; p = 0.027; p-interaction = 0.024), compared with similar outcome of aspirin users and non-users in those lacking tumour PIK3CA mutation (HR 0.92, 95% CI 0.60 to 1.42; p = 0.71).
Although these data are provocative, it is important to note that other retrospective analyses have not confirmed improved outcome with aspirin use in PIK3CA-mutant CRC and have indeed suggested other biomarkers that may predict benefit from aspirin (Table 5). 60 Furthermore, these studies were all limited by their retrospective nature, variable aspirin dose used, differing end points (e.g. relapse-free survival, CRC-specific mortality, OS) and small numbers of patients with advanced CRC. Therefore, prospective evaluation in a randomised, controlled trial is essential to determine the efficacy of aspirin as an anti-cancer therapy in PIK3CA-mutant stage 4 CRC.
| Study | Stage | End point | PIK3CA wild type | PIK3CA mutant | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No aspirin (n) | Aspirin (n) | HR (95% CI) | p-value | No aspirin (n) | Aspirin (n) | HR (95% CI) | p-value | ||||
| Liao X et al.35 | I–IV | CRC-specific survival | 466 | 337 | 0.93 (0.68 to 1.28) | 0.76 | 95 | 66 | 0.18 (0.05 to 0.6) | < 0.001 | 34 |
| OS | 0.94 (0.75 to 1.17) | 0.96 | 0.54 (0.31 to 0.94) | 0.01 | |||||||
| Domingo E et al.58 | II, III | RFS | 681 | 111 | 0.92 (0.6 to 1.42) | 0.71 | 90 | 14 | 0.11 (0.001 to 0.81) | 0.027 | 57 |
| OS | 1.05 (0.62 to 1.77) | 0.85 | 0.29 (0.04 to 2.33) | 0.245 | |||||||
| Murphy C et al.59 | II, III | RFS | 235 | 66 | 0.77 (0.34 to 1.73) | 0.53 | 40 | 16 | 0.45 (0.06 to 3.70) | 0.48 | 63 |
| OS | NR | NR | 2.50 (1.46 to 4.28) | 0.0008 | 1.76 (0.51 to 6.04) | 0.37 | |||||
| Reimers MS et al.60 | I–IV | OS | 384 | 147 | 0.55 (0.40 to 0.75) | 0.001 | 73 | 27 | 0.73 (0.33 to 1.63) | 0.4 | 58 |
Potential toxicities of aspirin
Although concerns regarding the potential toxicities of aspirin, particularly that of gastrointestinal haemorrhage, have limited the use of aspirin as a chemopreventative agent,61 the absolute increase in risk of significant toxicities from low-dose therapy for patients without established risk factors for such toxicity is modest. A meta-analysis62 of RCTs that investigated the benefits of low-dose aspirin (75–500 mg daily) for primary cardiovascular prevention, including over 95,000 partipants, demonstrated that aspirin increased the incidence of serious extracranial bleeding (usually defined as requiring transfusion or resulting in death) from 0.07% per year to 0.1% per year (HR 1.54, 95% CI 1.30 to 1.82). Furthermore, although participants in the recent CAPP2 study63 were perhaps younger than the anticipated population in FOCUS4-B, in the CAPP2 study aspirin at 600 mg daily for 25 months was not associated with an excess of adverse events compared with placebo.
To minimise potential harm to trial participants, several measures proven to reduce the risk of serious adverse events from aspirin were recommended,64 including exclusion of patients with a high risk of complications (e.g. previous peptic/duodenal ulcer or gastrointestinal bleed); careful management and treatment of symptoms, such as dyspepsia; blood pressure monitoring; and avoidance of concomitant NSAID use. Testing and eradication of Helicobacter pylori was also recommended and proton pump inhibitors were used where appropriate.
Aspirin dosing
Although evidence indicates that aspirin at doses of 75–100 mg daily exerts an antineoplastic effect, at present the mechanism of action of aspirin is unclear and potentially multifactorial. Taking into account the possibility that some or all of these mechanisms could be dose dependent, and the data suggesting that the impact on CRC mortality is greater for patients taking the highest aspirin doses (> 6× 325 mg tablets per week vs. 2–5 × 325 mg per week),65 while recognising that gastrointestinal toxicity is likely to increase with doses higher than 325 mg daily, the recommended aspirin dose for patients in FOCUS4-B is 300 mg daily. This dose permits the evaluation of both the antiplatelet effect of aspirin and possible additional mechanisms underlying its antineoplastic effect.
FOCUS4-B research objectives
FOCUS4-B aims to determine whether or not regular, intermediate-dose aspirin improves PFS when used as maintenance therapy following initial disease stabilisation or response from first-line treatment in patients with PIK3CA exon 9/exon 20 mutant CRC.
Translational studies, including analysis of tumour blocks for other putative aspirin biomarkers and analysis of cell-free DNA, were planned. The purpose of the former was to increase our understanding of determinants of response to antiplatelet therapy in malignant disease, whereas the latter could generate insights into the dynamics of tumour clones under therapy if efficacy was demonstrated.
Study methods
Trial approvals, patient eligibility and recruitment
The trial and subsequent amendments were approved by the UK National Ethics Committee Oxford, panel C, and by the MHRA. Patients with newly diagnosed metastatic CRC were registered into the FOCUS4 trial programme from 88 UK hospitals (Figure 3). Patients were randomised into the FOCUS4-B trial in all participating hospitals, starting in February 2016. Patients aged ≥ 18 years with newly diagnosed locally advanced or metastatic CRC were assessed for eligibility for FOCUS4-B if their tumour was confirmed to have a PIK3CA exon 9 or exon 20 mutation (using a NGS platform), and if they had remained stable or were responding after 16 weeks of first-line treatment. Patients were required to undergo baseline CT within 4 weeks prior to randomisation; a minimum 3-week washout period after the last dose of chemotherapy or biological therapy and before the first dose of aspirin or matched placebo; adequate renal (creatinine clearance of > 50 ml/minute) and liver function; and a WHO performance status of 0–2.
FIGURE 3.
FOCUS4-B schema.

The aspirin 300-mg tablets and matched placebo were supplied by Bayer AG (Leverkusen, Germany). The packaging, labelling and distribution of aspirin were undertaken by Alcura UK Ltd (Northampton, UK).
Patients randomised to receive aspirin or placebo continued to take the drug until disease progression, death or intolerable toxicity. The patients received 300 mg of aspirin or matched placebo once daily. Patients underwent clinical evaluation after the first 4 weeks from day 1 of randomised trial treatment by a research nurse or doctor to determine if there were any toxicity or tolerability issues. Patients were assessed by CT every 8 weeks from randomisation, together with full reporting of safety outcomes. PFS was the primary outcome measure for FOCUS4-B and, therefore, clinicians were required to use a consistent approach to treatment duration. Trial treatment was planned to be continued until progressive disease was identified on radiological grounds (RECIST v1.1), the development of cumulative toxicity or the patient chose to stop treatment. Patients were considered to be off trial treatment (but still in the trial) if there was a continuous break in their trial treatment of more than 28 days. If this occurred, trial medication was permanently discontinued and this was appropriately documented.
However, patients who discontinued the trial drug for reasons other than objective disease progression were to be followed up with tumour assessments every 8 weeks until objective disease progression as assessed by RECIST v1.1, even if they had started subsequent anti-cancer therapies. Toxicity assessments were to be carried out until the patient stopped trial treatment. On disease progression, patients were to restart their first-line treatment or move onto a standard second-line therapy at the discretion of the treating clinician. After progression, a progress electronic case report form was to be completed at 3 months and then every 6 months until patient death or the end of the trial.
Statistical methods
Treatment allocation
Once patients had been consented and deemed eligible for FOCUS4-B, they were randomly assigned to one of the following treatment arms (in a 2 : 1 ratio for aspirin vs. placebo):
-
arm B1 – placebo once daily
-
arm B2 – aspirin 300 mg once daily.
Both patients and treating clinicians were blinded to patient allocation to 300 mg aspirin once daily or placebo.
Outcome measures
The primary outcome for FOCUS4-B was PFS, defined as the time from randomisation to either disease progression (according to RECIST criteria) or death from any cause. OS was a secondary outcome, defined as the time from randomisation to death from any cause. Other secondary outcomes included safety, toxicity and tumour response.
Sample size calculation
For FOCUS4-B, a randomisation ratio of 2 : 1 was used in favour of the active arm. A summary of the operating characteristics for FOCUS4-B, with predicted timelines for the staged interim analyses, is presented in Table 6. It was anticipated that all data were to be released at the end of stage 3 to allow an open decision on whether or not to proceed to an assessment of OS.
| Characteristic | Stage 1: safety | Stage 2: LSA | Stage 3: efficacy for PFS |
|---|---|---|---|
| Outcome | PFS | PFS | PFS |
| One-sided alpha | 0.5 | 0.25 | 0.025 |
| Power (overall power maintained at 80%) | 0.95 | 0.95 | 0.82 |
| Target HR | 0.65 | 0.65 | 0.65 |
| Critical HR | 1.00 | 0.88 | 0.75 |
| Time required (months) | 24.5 | 15.9 | 17.3 |
| Cumulative time (months) | 24.5 | 40.4 | 57.6 |
| Cumulative events required in control arm (total) | 24 (65) | 45 (126) | 68 (195) |
| Total expected cumulative randomisations | 98 | 162 | 231 |
Statistical analysis
All analyses were performed in accordance with a predefined SAP that was agreed before database lock and were undertaken using Stata statistical software, version 16.1 (Stata Corporation, TX, USA). Given that this was a Phase II efficacy signal-seeking study, the primary analysis was prespecified as the per-protocol analysis, which was defined by patients who had completed at least one cycle of trial treatment. Analyses were also performed according to intention to treat. Patients were censored according to the following criteria:
-
For survival status, patients were censored on the date that they were last known to be alive because they collected a prescription from their hospital pharmacy or attended a follow-up visit or a CT appointment.
-
For PFS, patients were censored without progression on the date of the last CT scan, showing no progression.
-
For patients who died before any follow-up visit or CT appointment, the date of death was used as the date of the event and assumed death without progression providing the death occurred within 3 months of randomisation or any previous CT scan confirming no progression.
Kaplan–Meier curves were used to present survival data and Cox regression modelling to estimate HRs between randomised groups. Unadjusted HRs were estimated, as well as HRs adjusted for the stratification factors that were used to minimise patients into allocated groups (primary analysis). A further analysis also adjusted for resection status, timing of metastatic disease, alkaline phosphatase, white blood cell count, age of tumour sample and use of aspirin at baseline. Deviation from non-proportional hazards was assessed using regression of scaled Schoenfeld residuals against the log of time.
Study results
Recruitment and patient characteristics
By December 2017, a total of 47 PIK3CA-mutated patients had been identified. Of these patients, six entered FOCUS4-B, four entered FOCUS4-N, 12 progressed, nine refused owing to patient or clinician decision, two were not randomised owing to toxicity concerns, five were ineligible for various reasons, two died during their first-line therapy and seven were still in the registration phase. Between June 2016 and September 2017, six patients were randomised into FOCUS4-B using a 2 : 1 ratio: four pateints to aspirin and two patients to placebo. An overview of the baseline and demographic characteristics for the six patients randomised into FOCUS4-B is presented in Table 7.
| Characteristic | Placebo (N = 2) | Aspirin (N = 4) |
|---|---|---|
| Age (years), mean (SD) | 67.0 (7.1) | 49.6 (13.5) |
| Sex, n (%) | ||
| Male | 1 (50) | 2 (50) |
| Female | 1 (50) | 2 (50) |
| Current WHO performance status, n (%) | ||
| 0 | 2 (100) | 2 (50) |
| 1 | 0 (0) | 2 (50) |
| Site of primary of tumour, n (%) | ||
| Right colon | 1 (50) | 1 (25) |
| Left colon | 0 (0) | 2 (50) |
| Rectum | 1 (50) | 1 (25) |
| Current state of primary tumour, n (%) | ||
| Resected primary | 2 (100) | 3 (75) |
| Unresected primary | 0 (0) | 1 (25) |
| Timing of metastases, n (%) | ||
| Metachronous | 1 (50) | 2 (50) |
| Synchronous | 1 (50) | 2 (50) |
| Number of metastatic sites, n (%) | ||
| One | 1 (50) | 2 (50) |
| Two or more | 1 (50) | 2 (50) |
| Disease assessment at end of first-line treatment, n (%) | ||
| Partial response | 0 (0) | 3 (75) |
| Stable disease | 2 (100) | 1 (25) |
| First-line treatment regimen, n (%) | ||
| FOLFIRI | 2 (100) | 1 (25) |
| FOLFIRI + CET | 0 (0) | 1 (25) |
| CAPOX | 0 (0) | 2 (50) |
| RAS mutation status, n (%) | ||
| Mutation | 2 (100) | 2 (50) |
| Wild type | 0 (0) | 2 (50) |
| Total | 2 (100) | 4 (100) |
Outcome data
All four patients allocated to aspirin were recorded as having commenced trial treatment within 1 week of randomisation. At the time of database lock (4 November 2020), three of the four patients were recorded as having stopped trial treatment (at 28 days, 56 days and 61 days after randomisation).
At the time of database lock, all six patients had progressed. Five patients progressed at between 53 and 56 days (approximately 8 weeks) after randomisation, and the sixth patient (allocated to aspirin) progressed at 84 days (approximately 12 weeks) after randomisation.
In addition, three of the six patients were recorded as having died: two patients allocated to aspirin (with death occurring at 6 months and 22 months after randomisation) and one patient allocated to placebo (with death occurring at 8 months after randomisation). Only one moderate toxicity symptom was recorded: a grade 3 hypertension occurring in a patient allocated to aspirin. No adverse events were recorded.
Early closure of FOCUS4-B
In view of the very poor recruitment for FOCUS4-B, discussions took place between the FOCUS4 TMG and the oversight committees (FOCUS4 TSC and IDMC) on whether or not to close FOCUS4-B on the grounds of recruitment futility. Both committees recommended in July 2018 that this comparison should be closed and that data cleaning and analysis of the data for these six patients should be completed.
Discussion and conclusions
This cohort in FOCUS4 investigated the use of aspirin in a molecular-defined cohort of patients in whom previous laboratory and considerable epidemiological research had suggested potential for considerable benefit. Unfortunately, our enthusiasm for this subtrial was not matched by the successful recruitment of patients; therefore, early closure was necessary after we had enrolled only six patients. We made repeated efforts to boost recruitment, but all of these were ultimately unsuccessful. We explored the reasons for this and they were very varied, but more common findings were:
-
Patients felt that they could buy aspirin themselves over the counter if they strongly believed our hypothesis and that it may benefit them.
-
Conversely, some patients and doctors felt that a common drug being repurposed like aspirin was highly unlikely to be beneficial despite all the supporting laboratory and epidemiological data.
Although hugely disappointing, the results are important in showing the challenges involved in using repurposed and freely available drugs in this experimental setting. Aspirin continues to be of considerable interest to the cancer research community and is being investigated in many other cancer trials, including in the adjuvant Add-Aspirin trial57 in earlier stage colorectal and other cancers.
Chapter 5 Results from FOCUS4-C
Scientific rationale for FOCUS4-C
The full report of the FOCUS4-C trial has been published. 27 Parts of this chapter have been reproduced by Seligmann et al. 27 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: https://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
The cellular DNA damage response (DDR) is mediated through complex pathways, which trigger the mobilisation of cell cycle checkpoints and DNA repair proteins to halt cell cycle progression. This allows the repair of damaged DNA prior to critical phases of replication or entry into mitosis, maintaining genomic stability. However, genome instability is considered an ‘enabling characteristic’ of cancer66 and a wide range of DDR genes are mutated in cancer cells. 67
Targeting the DDR has been an effective therapeutic strategy in several tumour sites, including ovarian and pancreatic cancer. 68,69 These agents can be used as monotherapy in cancers with defective DDR, where a ‘synthetic lethality’ interaction might be expected: two pathways together perform an essential function and the loss of one pathway (e.g. owing to mutation) is tolerated, but loss of both pathways leads to cell death. 70
Wee1 is a nuclear tyrosine kinase that has a central role in cell cycle regulation, including being the key regulator of the G2/M checkpoint through actions on CDK1, optimising DNA–histone stoichiometry prior to mitotic entry71 and modulating CDK1/2 during the intra-S-phase to block replication initiation. 72 Inhibition of Wee1 causes unscheduled entry into mitosis, aberrant firing of replication origins leading to deoxyribonucleotide triphosphate shortage and replication stress, and accumulation of DNA damage during S-phase, which all lead to increased reliance on the G1/S checkpoint. 71 Adavosertib is the first small molecule inhibitor of Wee1 kinase and has been tested in combination with chemotherapy and radiotherapy,73,74 but more recently as monotherapy to generate synthetic lethality in tumours with DDR defects.
In FOCUS4-C, adavosertib was tested in RAS- and TP53-mutant (RAS-/TP53-mutant) metastatic CRC, with the hypothesis that it would be sensitive to Wee1 inhibition. TP53 is a key regulator of the G1/S checkpoint;75 loss of function leads to dependence on the intra-S and G2/M checkpoints to detect DNA damage and initiate repair. 76 In preclinical studies, adavosertib demonstrated preferential killing of TP53-deficient compared with TP53 wild-type tumours. 77 Mutant RAS, as well as having recognised actions through downstream MAPK-AKT pathway signalling, also drives cell cycle progression, leading to replication stress during S-phase. 78 In preclinical studies, mutant RAS was demonstrated to drive cells into S-phase through regulation of the CDK4 or CDK6 complex, and provides sustained mitogenic signals through sustained CDK2 activity. These effects activate the replication stress response, including checkpoint activation. 79 Adavosertib plus mTOR inhibition in KRAS-mutant CRC was associated with inhibition of cell growth in in vitro models. 80
Theoretically, a tumour with both TP53 loss and RAS mutation will be highly vulnerable to adavosertib: such a tumour will have G1 checkpoint failure and evidence of replication stress and will be reliant on the intra-S-phase and G2/M checkpoints. In a post-adavosertib treatment biopsy of a RAS-/TP53-mutant patient, the tumour had invoked both mechanisms of response to Wee1 inhibition, with evidence of DDR with Cdk2 activation and mitogenic signalling, compared with baseline. 81 Therefore, it was hypothesised that the combination of these two mutations was likely to produce a more potent synthetically lethal interaction with adavosertib than either alone.
FOCUS4-C was designed to assess the safety and efficacy of adavosertib compared with active monitoring in patients with metastatic CRC whose tumours were RAS-mutant and TP53 mutant and have achieved disease stability following induction chemotherapy.
Study methods
Trial approvals, patient eligibility and recruitment
The trial and subsequent amendments were approved by the UK National Ethics Committee Oxford – panel C – and by the MHRA. Patients with newly diagnosed metastatic CRC were registered into the FOCUS4 trial programme from 88 UK hospitals. Patients were randomised into the FOCUS4-C trial in a subset of 25 hospitals between June 2017 and March 2020. Patients aged ≥ 18 years with newly diagnosed locally advanced or metastatic CRC were assessed for eligibility for FOCUS4-C if their tumour was confirmed to have both RAS and TP53 mutations (using NGS platform) and if they remained stable or responding after 16 weeks of first-line treatment. Patients were required to have a baseline randomisation CT scan performed within 4 weeks prior to randomisation; a minimum 3-week washout period after the last dose of chemotherapy or biological therapy and before the first dose of adavosertib; adequate renal (creatinine clearance greater than 50 ml/minute) and liver function; a WHO performance status of 0–2; and no evidence of prolonged QT interval on electrocardiogram.
Trial procedures
Adavosertib was supplied by AstraZeneca Ltd (Cambridge, UK). The packaging, labelling and distribution of adavosertib were undertaken by Fisher Services (Horsham, UK). Patients randomised to adavosertib continued to receive the drug until disease progression, death or intolerable toxicity. The first 21 patients received 250 mg of adavosertib once daily on days 1–5 and 8–12 of a 3-week cycle. The next 23 patients received 300 mg of adavosertib once daily on the same schedule. Patients took an oral 5-HT3 antagonist with each dose, and 4 mg of oral dexamethasone was given on days 1 and 8 of each cycle unless clinically contraindicated. Patients were asked to complete a diary card to record the day and number of pills taken.
Owing to the mandatory supportive medication for nausea and vomiting, for which a placebo was not available, blinding was not possible and active monitoring was used as the control arm. Patients randomised to active monitoring followed the same follow-up schedule and remained off any other anti-cancer treatment until clinical or radiological evidence of disease progression.
Patient tumour status was assessed every 8 weeks by CT scan, which was reviewed at the treating hospital site according to RECIST, version 1.1. Toxicities and symptoms were assessed locally every 4 weeks from randomisation or the start of treatment using the NCI common terminology criteria for adverse events (version 3.0). Patients were followed until progressive disease occurred, at which point the patient was recommended to restart first-line chemotherapy.
Treatment was stopped in the event of grade 3 or worse toxic effects or persistent toxicities judged to be medically significant or not tolerated by the patient, until the toxicity resolved to grade 1 or better.
Statistical methods
Treatment allocation
Patients were allocated to adavosertib or active monitoring using a 2 : 1 allocation ratio by minimisation with a random element of 20% by a centrally managed telephone service at the MRC CTU at UCL. Minimisation factors were as follows: treating hospital site, site of primary tumour [right colon (defined as proximal to and including splenic flexure), left colon or rectum], WHO performance status (0 or 1), 16-week CT scan result (stable disease, partial response or complete response), number of metastatic sites (none, one, or two or more) and first-line chemotherapy regimen.
Outcome measures
The primary outcome for FOCUS4-C was PFS, defined as the time from randomisation to either disease progression (according to RECIST criteria) or death from any cause. OS was a secondary outcome, defined as the time from randomisation to death from any cause. Other secondary outcomes included safety, toxicity and tumour response.
Sample size calculation
FOCUS4 used a MAMS design that allowed pre-planned signal-seeking analyses to inform a decision on whether or not to continue the trial. The nstage package under Stata version 14.1 was used to calculate the operating characteristics for each molecular trial. A median PFS of 3.6 months in the active monitoring arm was assumed based on a similar group of patients being followed up in the COIN trial. 16 A target HR of 0.5 was sought for PFS, with a randomisation ratio of 2 : 1. Overall power was maintained at 80% at the one-sided 0.025 significance level, corrected for one closed interim and one final analysis. The recruitment target of 113 patients was based on the projected number of patients that we anticipated would be recruited when a target of 26 PFS events had occurred in the active monitoring arm. In addition, a prespecified interim analysis for futility was triggered when at least 13 PFS events had occurred in the active monitoring arm.
Statistical analysis
All analyses were carried out in accordance with a predefined SAP that was agreed before database lock and were undertaken using Stata version 16.1. Given that this was a Phase II efficacy signal-seeking study, the primary analysis was prespecified as the per-protocol analysis, which was defined as patients who had completed at least one cycle of trial treatment. Analyses were also carried out according to intention to treat (ITT). Patients were censored using the following criteria:
-
For survival status, patients were censored on the date that they were last known to be alive, which was recorded as the latest of either the date of collection of a prescription from their hospital pharmacy or the date of a follow-up visit or CT scan.
-
For PFS, patients were censored without progression on the date of the last CT scan showing no progression.
-
For patients who died before a follow-up visit or CT scan, the date of death was used as the date of the event and assumed death without progression providing that the death occurred within 3 months of randomisation or any previous CT scan confirming no progression.
Kaplan–Meier curves were used to present survival data and Cox regression modelling was used to estimate HRs between randomised arms. Unadjusted HRs were estimated, as well as HRs adjusted for the stratification factors that were used to minimise patients into allocated groups (primary analysis). A further analysis also adjusted for resection status, timing of metastatic disease, alkaline phosphatase, white blood cell count, age of tumour sample and use of aspirin at baseline. Deviation from non-proportional hazards was assessed using regression of scaled Schoenfeld residuals against the log of time.
Tolerability, toxicity and safety reviews
Two tolerability assessments were undertaken by the IDMC, which made recommendations on (1) which sites were permitted to recruit patients into FOCUS4-C on the basis of their experience of unlicensed drugs and (2) whether or not a higher dose of medication may be tested. The trial commenced with a starting dose of 250 mg but with the possibility of this dose increasing to 300 mg if the lower dose was well-tolerated.
Exploratory analyses based on molecular profiles
The RAS cases were divided into two molecular subtypes: KRAS versus NRAS mutants, and KRAS codons 12 and 13 versus NRAS/KRAS mutants outside codons 12 and 13. TP53 mutations were divided based on the presence of missense or truncating mutations (including nonsense, frameshift and splice site mutations).
To understand the prognostic implications of mutation subtypes, mutation data from 438 patients from the S:CORT programme30 from the FOCUS trial82 were retrieved, which represented a similar clinical setting as FOCUS4-C. Tumours had been successfully sequenced (Illumina; San Diego, CA, USA) with a targeted panel (SureSelect; Agilent, Santa Clara, CA, USA). Among other genes, the panel spanned all coding exons of KRAS, NRAS, BRAF and TP53. 83 Variant calling was performed with CaVEMan (Genome Research Ltd, Wellcome Trust Genome Campus, Saffron Walden, UK) for single-point mutations and Pindel (Genome Research Ltd) for insertions/deletions. Cases showing either MSI or BRAF mutation without MSI were grouped separately. The remaining cases were classified into four groups according to their combination of mutational status by RAS genes (KRAS/NRAS) and TP53. Kaplan–Meier plots and univariable survival analysis by Cox proportional hazard model were carried out according to the final six molecular subgroups.
Study results
Recruitment and patient characteristics
Between April 2017 and March 2020, 817 patients were registered into the FOCUS4 trial with successful biomarker profiling (Figure 4). A total of 247 patients (34%) had tumours confirmed to have both RAS and TP53 mutations (RAS/TP53 mutant). Of these patients, 151 had stable or responding disease after 16 weeks of first-line treatment and 69 were randomised using a 2 : 1 ratio: 44 to adavosertib and 25 to active monitoring.
FIGURE 4.
Flow of patients through the trial. CR, complete response; PR, partial response; RECIST, response evaluation criteria in solid tumours.
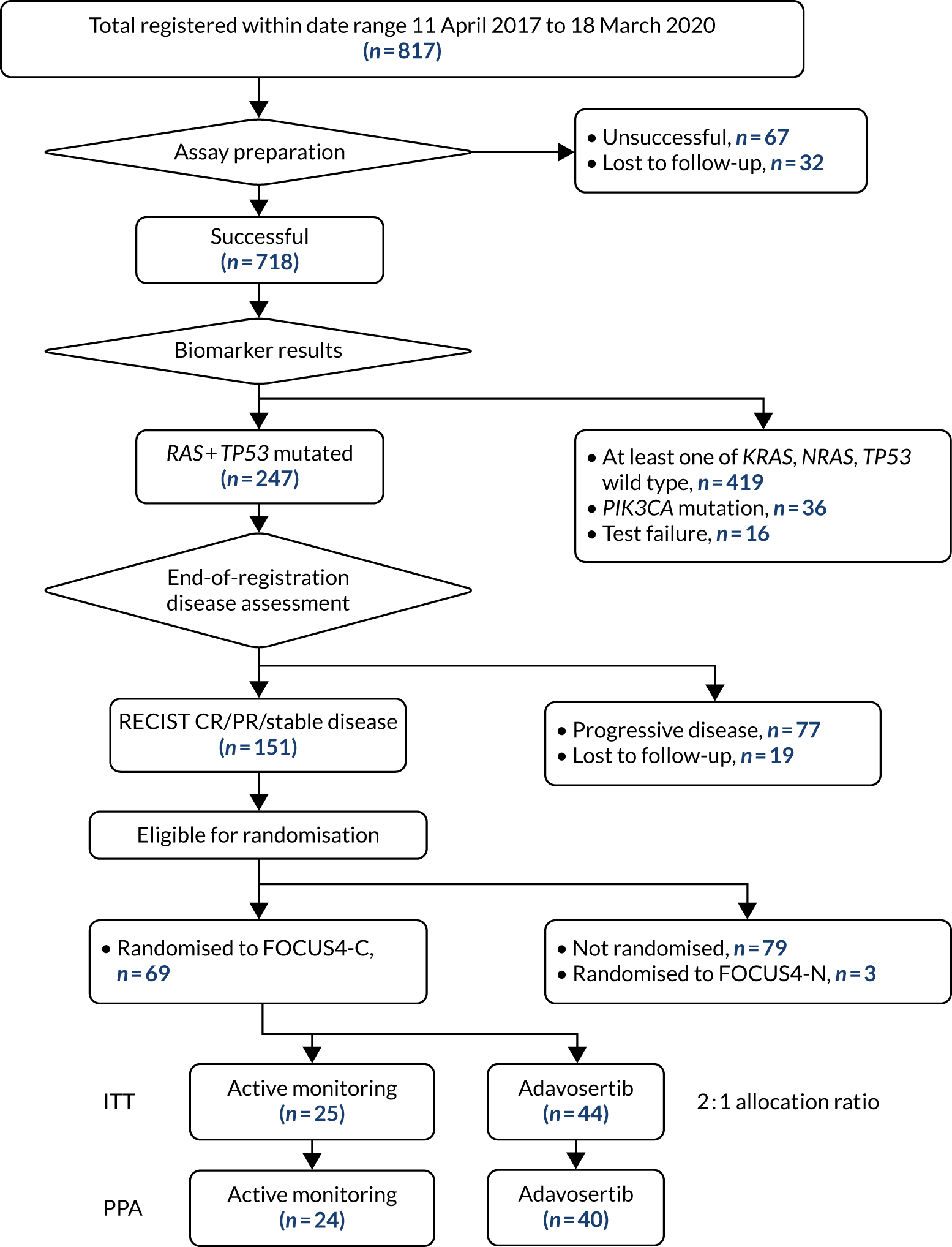
Owing to the COVID-19 pandemic, the trial was suspended in March 2020 just before reaching the required number of control arm PFS events (n = 26) to trigger final analysis. Given that recruitment had been planned to close in May 2020, the FOCUS4 IDMC were asked to review the efficacy data in a closed meeting to determine whether recruitment should re-open or whether the trial could close on the grounds of adequate data for determining any signal of drug activity. The IDMC recommended the latter and, following endorsement by the TSC, recruitment was formally closed. Follow-up and data cleaning continued to October 2020, when database lock took place.
A summary of the patients’ baseline characteristics is presented in Table 8. There were several minor imbalances favouring the adavosertib arm, which were corrected for in the adjusted analysis (primary model). More patients in the active monitoring arm than in the adavosertib arm had two or more metastatic sites and an unresected primary tumour. There were no differences in the frequency of other molecular alterations between the arms.
| Characteristic | Active monitoring (N = 25) | Adavosertib (N = 44) |
|---|---|---|
| Age (years), mean (SD) | 61.9 (12.2) | 59.2 (12.8) |
| Sex, n (%) | ||
| Male | 15 (60) | 31 (70) |
| Female | 10 (40) | 13 (30) |
| Current WHO performance status, n (%) | ||
| 0 | 17 (68) | 35 (80) |
| 1 | 8 (32) | 9 (20) |
| Site of primary of tumour, n (%) | ||
| Right colon | 9 (36) | 13 (30) |
| Left colon | 6 (24) | 13 (30) |
| Rectum | 10 (40) | 18 (41) |
| Current state of primary tumour, n (%) | ||
| Resected primary | 9 (36) | 23 (52) |
| Unresected primary | 16 (64) | 19 (43) |
| Unresected local recurrence | 0 (0) | 2 (5) |
| Timing of metastases, n (%) | ||
| Metachronous | 4 (16) | 13 (30) |
| Synchronous | 21 (84) | 31 (70) |
| Number of metastatic sites, n (%) | ||
| One | 6 (24) | 16 (36) |
| Two or more | 19 (76) | 28 (64) |
| Disease assessment at end of first-line treatment, n (%) | ||
| Complete response | 0 (0) | 1 (2) |
| Partial response | 13 (52) | 26 (59) |
| Stable disease | 12 (48) | 17 (39) |
| First-line treatment regimen, n (%) | ||
| FOLFOX | 7 (28) | 15 (34) |
| FOLFIRI | 8 (32) | 14 (32) |
| CAPOX | 6 (24) | 11 (25) |
| FOLFOXIRI | 3 (12) | 3 (7) |
| Other | 1 (4) | 1 (2) |
| PIK3CA mutation status, n (%) | ||
| Mutation | 1 (4) | 1 (2) |
| Wild type | 24 (96) | 43 (98) |
| Total | 25 (100) | 44 (100) |
There were no significant differences in the registration period chemotherapy regimens used between the adavosertib arm and the active monitoring arm. Of the 69 randomised patients, 22 (32%) received leucovorin, fluorouracil and oxaliplatin (FOLFOX), 22 (32%) received leucovorin, fluorouracil and irinotecan (FOLFIRI), 17 (25%) received CAPOX, and 6 (9%) received leucovorin, fluorouracil, oxaliplatin and irinotecan (FOLFOXIRI) (see Table 8). Slightly more patients in the active monitoring arm than in the adavosertib arm had stable disease at the end of first-line chemotherapy (48% vs. 39%, respectively).
Progression-free survival (per protocol)
Five patients were excluded from the per-protocol analysis: four patients did not start treatment (adavosertib arm) and one patient was found to have had progressive disease at the point of randomisation and was, therefore, not eligible (active monitoring arm). One patient was censored early when they received fluorouracil as an anti-cancer treatment prior to progression (active monitoring arm).
Within the primary per-protocol analysis (n = 64), there were 40 PFS events in the adavosertib arm (n = 40 patients) and 22 PFS events in the active monitoring arm (n = 24 patients). Patients treated with adavosertib had a longer PFS than those on active monitoring (3.61 vs. 1.87 months, respectively). Both the unadjusted (HR 0.52, 95% CI 0.30 to 0.89; p = 0.022) and the adjusted (HR 0.35, 95% CI 0.18 to 0.68; p = 0.0022) analysis were statistically significant. Kaplan-Meier curves are presented in Figure 5.
FIGURE 5.
Kaplan–Meier curves for PFS in the per-protocol analysis.

Progression-free survival (intention to treat)
All patients were included in the ITT analysis but four patients were censored the day after randomisation: three patients in the adavosertib arm (two because of patient withdrawal and one without any post-randomisation CT scan assessments) and one in the active monitoring arm without any post-randomisation CT scan assessments.
There were 41 PFS events in the adavosertib arm (n = 44 patients) and 23 PFS events in the active monitoring arm (n = 25 patients). Consistent with the per-protocol analysis, the ITT PFS analysis showed a PFS advantage of adavosertib compared with active monitoring in both the unadjusted (HR 0.55, 95% CI 0.32 to 0.94; p = 0.032) and the adjusted (HR 0.40, 95% CI 0.21 to 0.75; p = 0.0051) analysis.
Overall survival (intention to treat)
There were 27 deaths in the adavosertib arm (n = 44 patients) and 16 deaths in the active monitoring arm (n = 25 patients). There was no significant OS benefit of adavosertib compared with active monitoring (median survival 14.0 vs. 12.8 months, respectively) (unadjusted HR 0.79, 95% CI 0.42 to 1.48; p = 0.47) (adjusted HR 0.92, 95% CI 0.44 to 1.94; p = 0.93).
Tumour control
Adavosertib was associated with a higher proportion of patients with disease control than active monitoring (47% vs. 28%, respectively, at any time during the trial), including one patient with a documented partial response to adavosertib.
Subgroup analyses
The impact of adavosertib compared with active monitoring on PFS was explored in prespecified subgroups (Figure 6). The most marked difference in adavosertib effect was for the site of the primary tumour location (PTL); patients with a right PTL had no PFS advantage with adavosertib compared with active monitoring (1.87 vs. 1.91 months, respectively; HR 1.02, 95% CI 0.41 to 2.56), whereas those with a left PTL did have a PFS advantage (3.61 vs. 1.87 months, respectively; HR 0.24, 95% CI 0.11 to 0.51) (interaction p = 0.043) (Figures 7 and 8).
FIGURE 6.
Subgroup analyses for PFS by ITT. WHO, World Health Organization.
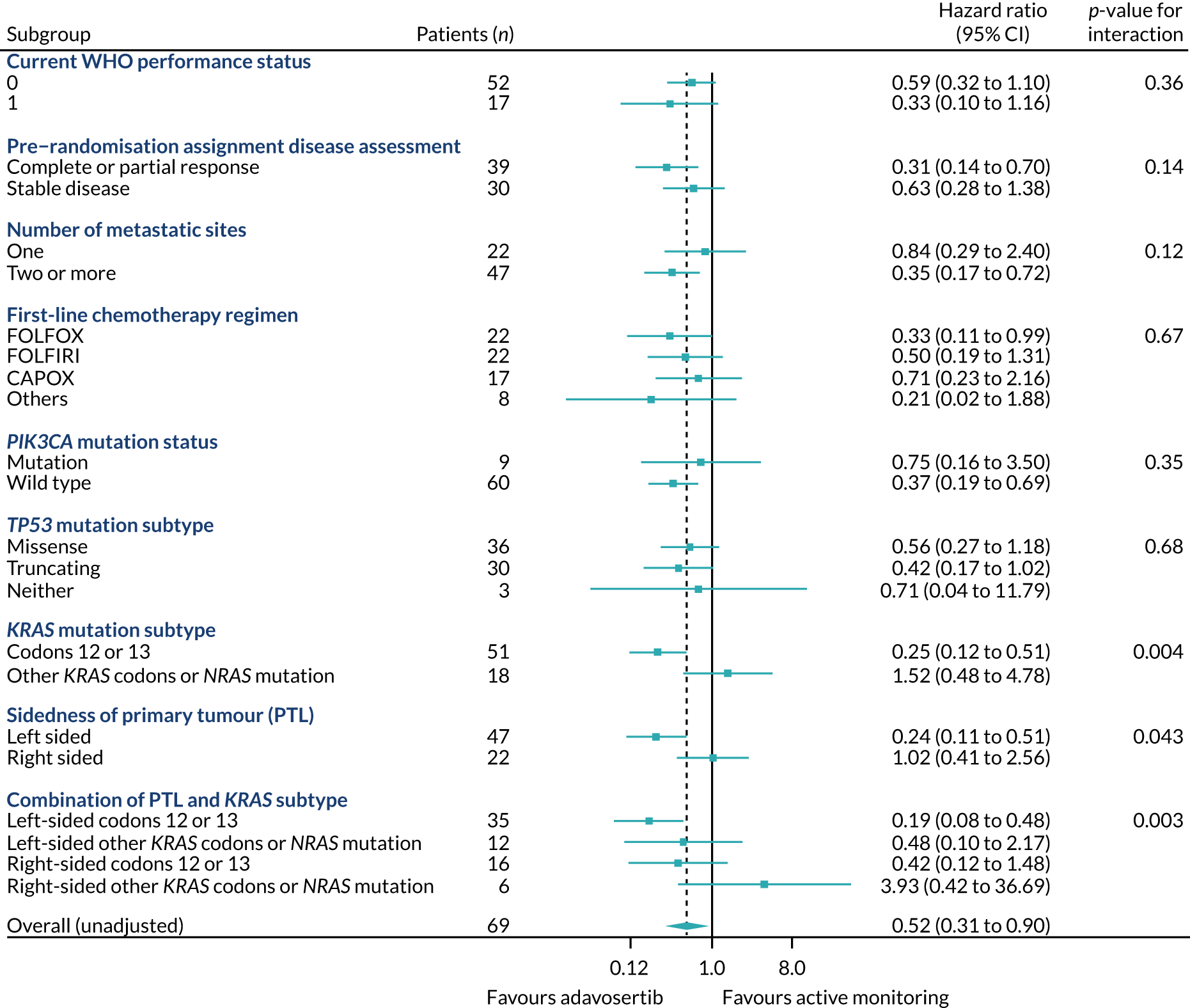
FIGURE 7.
Kaplan–Meier curves for PFS (ITT) by PTL: (a) left sided (n = 47); and (b) right sided (n = 22).


FIGURE 8.
Swimmer plot by randomised group and location of primary tumour. Light-blue horizontal strip, right sided; navy strip, left sided (including rectum).

This prompted an unplanned subgroup analysis of PTL on OS and, despite that the number of events was small, the interaction was even more marked. The median OS was 14.1 versus 11.3 months for adavosertib versus active monitoring, respectively, for left PTL (adjusted HR 0.40, 95% CI 0.17 to 0.97), but was 6.5 versus 15.5 months, respectively, for right PTL (HR 6.46, 95% CI 1.34 to 33.6) (interaction p = 0.0032). In terms of response, 38% of right-sided adavosertib patients compared with 42% of right-sided active monitoring patients reported disease stability or response at least once while on the trial, whereas for left-sided tumours the figures were 53% compared with 19%, respectively.
Patients who had responded to induction chemotherapy (vs. stable disease) and who had two or more metastatic sites appeared to benefit more from adavosertib, albeit to a lesser degree than those with only one metastatic site (interaction p = 0.14 for response to induction; p = 0.12 for number of metastatic sites) (see Figure 6).
External analyses to further characterise the RAS/TP53-mutant biomarker population
To the best of our knowledge, the RAS/TP53-mutant population has not been previously described. To understand the prognostic implication of this alteration, we analysed the outcomes of a subset (n = 438) of the FOCUS trial in whom the S:CORT consortium30 had analysed a wider panel of CRC genes, including KRAS, NRAS, BRAF, MSI and TP53. The RAS/RAF wild-type group was the reference population (median OS 21.6 months). The RAS/TP53-mutant population was distinct from and had a worse prognosis than the population with either mutation alone (RAS or TP53), with a median OS of 14.9 months (HR 2.06, 95% CI 1.08 to 3.93; p = 0.028). This suggests that the RAS/TP53-mutant population constitutes a poor prognosis subgroup, but the prognosis is not as poor as it is for patients with a BRAF-mutant or MSI-high tumour.
Effect of RAS and TP53 mutation subtypes on adavosertib activity
We observed that patients with KRAS codon 12/13 mutations (n = 51) experienced a significant benefit from adavosertib (interaction p = 0.004), whereas no detectable benefit was observed in those with KRAS mutations at other codons or with NRAS mutation (n = 18). Furthermore, the interaction effects of KRAS subtype and of PTL on PFS may be additive, as there is a significant benefit from adavosertib within the subgroup of left PTL KRAS codon 12/13 subtypes (HR 0.19, 95% CI 0.08 to 0.48) and a potential disbenefit within the subgroup of right PTL non-codon 12/13 subtypes (HR 3.93, 95% CI 0.42 to 36.69) (Figure 9). The subtype of TP53 mutation or the co-occurrence of PIK3CA mutation did not affect the outcome.
FIGURE 9.
Forest plot for PFS (ITT) by PTL and KRAS codon 12/13 status.
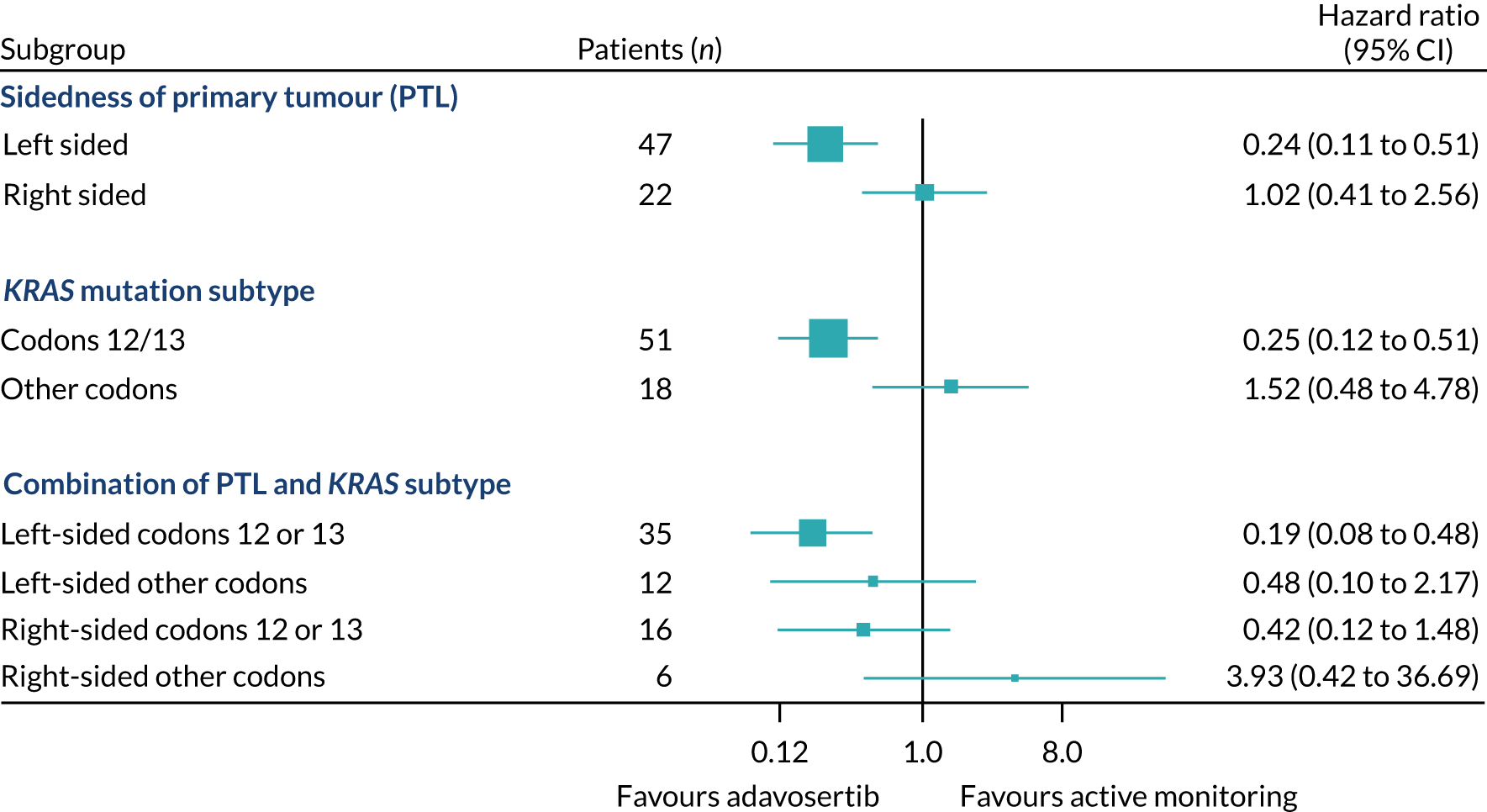
Toxicity and compliance
There was good compliance with the randomised allocation, and adavosertib was well tolerated but with some increased toxicity. Adavosertib was associated with an increase in reported toxicity (grade 1 or worse) compared with active monitoring, most notably increased frequency of diarrhoea (61% vs. 28%, respectively), fatigue (75% vs. 56%), nausea (68% vs. 32%) and vomiting (41% vs. 4%). However, the majority of such toxicity was of low grade; in the adavosertib arm, 9% of patients reported diarrhoea, 11% reported fatigue, 5% reported nausea and 2% reported vomiting of grade 3 or worse, compared with no patients in the active monitoring arm.
Impact of adavosertib dosing
There was an increase in the dose of adavosertib from 250 mg to 300 mg during the trial. PFS was 2.2 months (HR 0.58, 95% CI 0.31 to 1.06) for the 250-mg dose and 3.7 months (HR 0.47, 95% CI 0.25 to 0.89) for the 300-mg dose; this difference was non-significant (p = 0.48). The increase in dose from 250 mg to 300 mg was associated with an increased frequency of grade 3 diarrhoea (4% vs. 14%, respectively), but otherwise the toxicity profile was similar. Rates of dose modifications were similar at doses of 250 mg and 300 mg: dose delays 16% vs. 7%, respectively; dose reductions 4% vs. 5%; and dose omissions 19% vs. 17%.
Discussion and conclusions
FOCUS4-C met its primary end point: patients with RAS/TP53-mutant metastatic CRC had a PFS advantage with adavosertib compared with active monitoring following induction chemotherapy. This activity was clearly limited to patients with left PTL. An assessment of the impact on longer-term outcomes showed an OS benefit with adavosertib compared with active monitoring in RAS/TP53-mutant patients with left PTL. These results are particularly encouraging because patients with RAS/TP53-mutant metastatic CRC are a poor prognostic population and have limited treatment options. Adavosertib was well tolerated at both of the doses evaluated, and it is possible that nearly half of the adavosertib patients were dosed suboptimally (250 mg once daily instead of 300 mg once daily). It is encouraging that a clear efficacy signal was reached for both doses despite the forced early closure as a result of the COVID-19 pandemic.
The overarching aim of the FOCUS4 trial programme was to test novel agents efficiently with specified biomarker subgroups in metastatic CRC using a ‘window of opportunity’ following 16 weeks of induction chemotherapy, with the MAMs design allowing for an early signal of drug inactivity. 13 FOCUS4-C represents the success of this approach, utilising a PFS end point and control arm. The intermittent treatment strategy employed in FOCUS4 follows the demonstration of no detriment in OS in the MRC COIN trial, which has been further substantiated by an individual patient data (IPD) meta-analysis. 35 Therefore, we designed FOCUS4 to specifically use this window following first-line induction chemotherapy to test novel agents in biomarker-specified groups, prior to the evolution of multiple resistance mechanisms. 13
Wee1 inhibition induces failure of the G2 and intra-S-phase checkpoints, which increases the reliance on the G1/S checkpoint, and deoxyribonucleotide triphosphate shortage due to unscheduled replication fork firing, which further exacerbates replication stress during S-phase. Tumours containing RAS mutations, which drive replication stress and loss of G1 checkpoint control subsequent to loss of TP53 function, were hypothesised to benefit from this treatment.
Although the clinical implications of the RAS/TP53 mutation in metastatic CRC are not well studied, each alteration is individually well characterised. FOCUS4-C has shown that the double-mutant subgroup carries a moderately poor prognosis, which is largely driven by the KRAS mutation. Other approaches to targeting KRAS-mutant tumours include inhibition of the G18C mutation specifically; we show here that patients in FOCUS4-C have a similar benefit with adavosertib as those treated in a Phase I trial with sotorasib (Lumakras, Amgen), a promising small molecule targeting KRAS p.G12C (PFS 3.6 vs. 4 months). 84
There was marked and significant heterogeneity in the benefit of adavosertib by PTL and KRAS mutation subtype. Adavosertib activity was limited to left colon and rectal PTL, with little activity observed in right PTL. Although the analysis by PTL was prespecified, the analysis by KRAS mutation subtype was not prespecified. Having observed the significant subgroup effects on PFS, the impact on OS was assessed. It is interesting to see that OS was significantly improved in patients with left-sided tumours, with an increase in median OS from randomisation from 11.3 months to 14.1 months (HR 0.40, 95% CI 0.17 to 0.97). There is also a possibility that adavosertib may adversely affect outcome in patients with right PTL. However, the numbers of patients and events were limited and, therefore, any conclusions in relation to this observed effect on OS.need to be drawn with caution. Differences in CRC by PTL are well documented in terms of biology, prognosis and treatment response, and ongoing translational studies may provide a molecular basis for this observation. In addition, adavosertib had the most PFS effect in patients with KRAS codon 12,13-/TP53-mutant tumours, with lesser effect in those patients with extended KRAS or NRAS mutations; functional differences between RAS isoforms are documented. 85
Adavosertib has demonstrated an acceptable safety profile; the main toxicity was diarrhoea. There was only minimal additional toxicity when the dose was increased from 250 mg to 300 mg, and a suggestion of additional activity with the higher dose. The 300-mg dosing is, therefore, recommended to progress to further clinical studies.
Alterations in the DDR are emerging as novel targets for cancer therapeutics, with notable successes with poly (ADP -ribose) polymerase inhibitors in pancreatic and ovarian cancers that harbour breast cancer gene mutations68,69 or with defects in other genes belonging to the human hairless gene family, as seen in prostate cancer. 86 This strategy is less developed in CRC, but the prevalence of alterations in DDR genes are estimated to be between 10% and 30%87 and more frequent in the MSI-high subgroup. One limitation has been the lack of standard assays to identify CRC tumours harbouring clinically relevant DDR defects. The studies of poly (ADP -ribose) polymerase inhibitors in CRC have been limited by lack of patient selection and toxicity with chemotherapy combinations; promising preclinical work may serve the basis for further therapeutic development. 14
Adavosertib has been tested in multiple tumour sites as monotherapy or in combination with radiotherapy or chemotherapy. 73,74 Disappointing results have been observed in unselected populations. However, clinical benefit was recently shown from the combination of adavosertib plus gemcitabine compared with gemcitabine alone in high-grade serous ovarian cancer, a TP53-mutated tumour type with high replication stress, similar to our selected population. 73 FOCUS4-C has highlighted the importance of optimisation of patient selection for therapeutics targeting the DDR.
There are limitations to this study. FOCUS4-C was not placebo controlled because of the requirement of premedication with dexamethasone and a 5HT3 inhibitor owing to concerns regarding adavosertib toxicity. It is possible, therefore, that the PFS effect observed was influenced by investigator and patient preference to restart first-line chemotherapy sooner in the active monitoring arm. However, a marked difference in effect was observed between the right and the left PTL groups treated with adavosertib, suggesting a lesser effect on the primary analysis owing to this potential bias.
Although these studies were not designed or powered for cross-study comparison, comparison of the effect of maintenance capecitabine (in FOCUS4-N) compared with adavosertib within a RAS/TP53-mutant population shows similar effect (PFS HR 0.44, 95% CI 0.30 to 0.63). However, the purpose of FOCUS4-C was to demonstrate activity of adavosertib within a biomarker-selected population, which has been achieved. Adavosertib has shown similar efficacy to capecitabine, the most active agent in CRC, within this poor prognosis subgroup, and this is promising for its further use in this disease.
Further clinical development is warranted given the efficacy and safety profile demonstrated, particularly within a population of unmet need. Translational work to understand the mechanism of this response and resistance is ongoing by the S:CORT30 and ACRCelerate31 programmes. Suggestions for development include other stages of the clinical pathway (treatment resistant setting) as monotherapy, in combination with other agents, or in combination with radiotherapy.
Chapter 6 Results from FOCUS4-D
The full report of the FOCUS4-D trial has been published. 88 Parts of this chapter have been reproduced from Adams et al. 88 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: https://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Scientific rationale for FOCUS4-D
Signalling through HER receptors (e.g. EGFR, HER2, HER3 and HER4) and their downstream pathways is a key mechanism that promotes proliferation and the malignant phenotype in cancer. Epidermal growth factor has been recognised as a key pathogenic surface receptor in CRC for many years. A greater understanding of CRC biology followed by use of EGFR-targeted treatments has led to the discovery of the importance of BRAF, PIK3CA, KRAS and NRAS mutations in predicting a lack of response to EGFR-targeted therapy. 3 The monoclonal antibodies cetuximab and panitumumab were developed to target EGFR on the surface of cancer cells. After licensing, cetuximab and panitumumab were reported to be ineffective in patients whose cancer expressed a somatic mutation in the NRAS or KRAS genes. 14 Simple EGFR inhibition, therefore, has limitations because both de novo and acquired resistance can arise and result in a lack of benefit for most of these biomarker-selected patients, thus driving interest in novel approaches to inhibit this pathway. However, EGFR remains a valid target for CRC within the population of patients who are wild type for KRAS, NRAS and BRAF, with proven clinical benefit.
The proteins HER2 and HER3 (also known as ERBB2 and ERBB3, respectively) heterodimerise with EGFR and are mechanisms of resistance to EGFR inhibition. Human epidermal growth factor-3 is a membrane-bound receptor protein that has extracellular heregulin- and neuregulin-binding domains but does not have an intracellular kinase domain, relying on heterodimerisation to other family members for downstream effects. Human epidermal growth factor-3 expression is associated with poor prognosis in CRC. 89 The protein has a central role in driving oncogenic signals in tumours,90 and preclinical and clinical data have led to the hypothesis that HER3 is an escape pathway to EGFR blockade through a compensatory shift to HER3 signalling, predominantly through the PI3K/AKT pathway. 91–93 Moreover, clinical data indicate that HER3 overexpression predicts the lack of efficacy of panitumumab. 94 Human epidermal growth factor-2 is another member of the EGFR family of membrane-bound receptors. It is an orphan receptor, having no known associated ligands, but is activated through homodimerism and heterodimerisation with other EGFR family members, resulting in intracellular phosphorylation and cascaded downstream signalling. Growing in vitro and in vivo evidence suggests that HER2 might be overexpressed more frequently in patients with EGFR-dependent (all wild type) tumours. 95 Furthermore, this protein might be upregulated in acquired EGFR inhibitor resistance, and concomitant blockade of HER2 could increase the efficacy and prolong activity in a synergistic fashion. Bertotti et al. 96 developed a range of CRC xenograft models from genetically well-characterised, metastatic CRC samples. A cohort of these so-called xenopatients showed amplification of HER2, particularly in RAS wild-type tumours, suggesting enrichment in this cohort, which showed high and sustained sensitivity to combination EGFR and HER2 inhibition. Inhibition of EGFR, HER2 and HER3 signalling is postulated to reduce de novo resistance, thereby increasing the proportion of patients showing benefit when compared with inhibition of EGFR only. Furthermore, this inhibition might slow the development of acquired resistance in patients with initially EGFR-dependent tumours, thereby providing greater clinical benefit than EGFR inhibition alone.
FOCUS4-D was designed to assess the efficacy of AZD8931 in patients with CRC whose tumours were wild type for BRAF, PIK3CA, KRAS and NRAS mutations, and reached its first interim analysis trigger point in March 2016.
AZD8931 is an orally active, equipotent tyrosine kinase inhibitor of EGFR, HER2 and HER3 signalling. In vitro analysis of AZD8931 in ligand-driven cell assays shows potency exceeding that of gefitinib (Iressa, AstraZeneca) and lapatinib (Tykerb, Novartis) in this system for EGFR, HER2 and HER3 inhibition. 97 In pharmacokinetic and dynamic analyses, a direct relation was recorded between the total concentration of AZD8931 in plasma and the inhibition of EGFR phosphorylation. 97 Patients with PTEN loss or somatic BRAF, PIK3CA, KRAS and NRAS mutations were excluded from FOCUS4-D. The rationale for the exclusion of patients with BRAF mutations relates to two specific sets of data. First, BRAF-mutant CRC has significantly worse prognosis than disease with wild-type KRAS and NRAS. 98 Second, despite some conflicting evidence, BRAF-mutant tumours gain no additional benefit from targeted EGFR inhibition. 99 Patients harbouring tumours with a PIK3CA mutation or PTEN loss were excluded specifically because data suggest that the PI3K/AKT pathway is a resistance mechanism to EGFR inhibition. 100
In the Phase II MRC COIN-B trial,9 patients who had metastatic KRAS wild-type tumours were treated with an intermittent strategy of oxaliplatin, intravenous fluorouracil and folinic acid and were randomised to either intermittent cetuximab (with intermittent chemotherapy) or continuous cetuximab (including single-agent maintenance through the interval between chemotherapy). These trial data showed that the use of maintenance cetuximab in the interval between chemotherapy was associated with an improvement in the duration of the progression-free interval from 3 months to 6 months. 9 FOCUS4-D builds on these data to test if, in a more specific molecular cohort, AZD8931 could improve PFS in the interval off chemotherapy.
Study methods
Patients’ registration and biomarker assessment
The FOCUS4-D trial was undertaken at 18 hospital sites in the UK. Patients aged ≥ 18 years with newly diagnosed locally advanced or metastatic CRC were eligible for registration in the FOCUS4 trial programme, either at the start of a 16-week regimen of first-line chemotherapy or up to 12 weeks into the regimen. Patients could not be registered into FOCUS4 unless a CT scan had been carried out within 4 weeks of starting chemotherapy (pre-registration CT scan). CT scans were obtained 8 weeks and 16 weeks after chemotherapy was started. A CT scan performed at the end of the 16-week regimen was compared with the preregistration CT scan by local radiologists, using RECIST, version 1.1. Patients whose tumours had progressed were ineligible for randomisation and any further involvement with FOCUS4 ended at this time.
The 16-week duration of first-line chemotherapy was determined by reviewing data from three key trials: COIN,16 CAIRO3101 and AIO0207. 102 This period accounted for factors including maximum tumour response and tolerability by patients.
During the 16-week period of first-line chemotherapy, a sample of the patient’s tumour was sent to one of two dedicated FOCUS4 biomarker laboratories in Leeds and Cardiff (UK), respectively, for assessment and stratification of the patient into one of four molecular groups. The molecular stratification hierarchy for the FOCUS4 trial programme is presented in Chapter 2 (see Figure 1). The technical components of the biomarkers and interlaboratory quality assurance are also described in Chapter 2. 17
Patients whose tumours had remained stable or responded to treatment, as shown by the CT scan, were assessed for eligibility for FOCUS4-D. The biomarker assessment had to show wild-type status for BRAF, PIK3CA, KRAS and NRAS, as well as PTEN expression on immunohistochemistry. The FOCUS4-D trial design with eligibility criteria is presented in Figure 10. Full inclusion and exclusion criteria are available from the trial website (www.focus4trial.org; accessed 13 May 2022).
FIGURE 10.
FOCUS4-D trial schema. a, Criteria were aged older than 17 years, no brain metastases, adequate organ function, WHO performance status of 0–2, not pregnant and CT scan within 4 weeks before randomisation. PTEN, phosphatase and tensin homologue; RECIST, Response Evaluation Criteria in Solid Tumours.

In addition to the eligibility criteria, patients had to have had a minimum of a 3-week gap after the last dose of chemotherapy or biological therapy and before the first dose of trial drug; a WHO performance status of 0–2; and adequate organ function, which was ascertained by an estimated creatinine clearance > 50 ml/minute (according to local estimation method), serum bilirubin less than 1.5 times the upper limit of normal (ULN), alanine aminotransferase, aspartate aminotransferase and alkaline phosphatase less than 2.5 × ULN in the absence of liver metastases and less than 3.0 × ULN in presence of liver metastases, and a left ventricular ejection fraction greater than 50% by multigated acquisition scan or echocardiography. Ethics approval was granted by the National Research Ethics Service South Central Oxford—Panel C ethics committee. Regulatory approval was granted by the UK MHRA. All trial procedures and processes complied with the International Conference On Harmonisation’s Good Clinical Practice guidelines. 103 Patients were asked to sign a consent form for registration and for analysis of the biomarker panel; additional written informed consent was obtained before randomisation into FOCUS4-D.
Randomisation and masking
Randomisation was undertaken by telephone to a centrally managed service at the MRC CTU at UCL. Patients were allocated to either AZD8931 or placebo by minimisation with a random element of 20% (both patients and clinicians were masked to treatment allocation). Minimisation factors were treating hospital site, site of primary tumour (right colon, left colon or rectum), WHO performance status (0–2), 16-week CT scan result (stable disease, partial response or complete response), number of metastatic sites (none, one, or two or more) and first-line chemotherapy regimen (fluorouracil, capecitabine or neither; both oxaliplatin and irinotecan, oxaliplatin only, irinotecan only, or neither; and cetuximab or panitumumab, bevacizumab, or no monoclonal antibody). AZD8931 and placebo were identical in appearance and were supplied by AstraZeneca Ltd. The packaging, labelling and distribution of AZD8931 and placebo were all undertaken by Fisher Services. Treatment was administered double-blind using an interactive web-based drug delivery system provided by Cenduit (Nottingham, UK).
Procedures
Patients were asked to take oral AZD8931 (40 mg twice daily) or placebo until disease progression, death or toxic effects. Patients were also requested to complete a diary card on which the number of pills taken every day was recorded. Patients were assessed every 8 weeks by CT scan, which was reviewed at the treating hospital site according to RECIST, version 1.1. Measurements were collated centrally to ensure appropriate delineation of response and progression. Toxic effects and symptoms were assessed locally every 4 weeks from the start of trial treatment, using the NCI Common Terminology Criteria for Adverse Events (version 3.0). 104 Patients were followed up until progressive disease was identified on a CT scan, at which point the patient was recommended to restart first-line chemotherapy. Treatment was stopped in the event of grade 3 or worse toxic effects or persistent toxicities judged medically significant or not tolerated by the patient, until the toxicity resolved to grade 1 or better. After stopping treatment (for a maximum of 28 days), trial therapy could be re-initiated at a reduced dose. For the first event, treatment was restarted at 40 mg in the morning and 20 mg at night; for the second event, treatment was restarted at 20 mg twice per day. A third event resulted in treatment discontinuation. Any stoppage for ≥ 28 days was not permitted and the trial therapy was discontinued from.
Outcomes
The primary outcome of FOCUS4-D was PFS, defined as the time from randomisation to either disease progression (according to RECIST criteria) or death from any cause. The trial could be extended to include OS as a secondary outcome if adequate drug activity was seen for PFS in the early interim analyses. Additional secondary end points included safety and toxicity.
Statistical methods
FOCUS4 used a MAMS design, which allows pre-planned analyses to be carried out to inform a decision on whether or not to continue the trial. 105 The nstage MAMS function in Stata, version 14.1, was used to calculate the operating characteristics for each molecular trial. 106 For FOCUS4-D, a recruitment rate of nine patients per month was anticipated if 100 sites were open. For the placebo group, a median PFS of 4.6 months was assumed, based on a similar group of patients being followed up in the COIN trial. 16 A target HR of 0.5 was sought for PFS, with a randomisation ratio of 1 : 1. A summary of the operating characteristics for FOCUS4-D, with predicted timelines for the staged interim analyses, is presented in Table 9. Power was maintained at 85% after correction for two interim analyses and one final analysis. The target recruitment was 174 patients, with the first interim analysis timed to occur when nine PFS events had occurred in the control group. At every interim analysis, the observed HR was compared with the critical HR for that interim analysis (see Table 9). If the HR was above the critical value, trial closure could be considered on the grounds of an absence of sufficient drug activity. When an interim analysis was triggered by the prespecified number of PFS events in the control group, data were analysed and the results presented at a closed and confidential meeting of the IDMC, who were not involved with the conduct of the study. Recommendations to continue or close the study were made to the TSC, who would recommend their decision. If a decision was made to close the study, the TSC would allow the TMG to see the data to ensure that there were no objections to study closure. When all committees agreed to trial closure, study sites, the pharmaceutical company (AstraZeneca Ltd) and the trial funders were to be informed, and no further patients were to be permitted entry into the study.
| Characteristic | Stage 1: safety and LSA | Stage 2: LSA | Stage 3: efficacy |
|---|---|---|---|
| Outcome | PFS | PFS | PFS |
| One-sided alpha | 0.5 | 0.2 | 0.025 |
| Power (overall power maintained at 85%) | 0.91 | 0.95 | 0.95 |
| Target HR | 0.5 | 0.5 | 0.5 |
| Critical HR | 1.00 | 0.81 | 0.70 |
| Time required (months) | 5.9 | 6.3 | 7.1 |
| Cumulative time (months) | 5.9 | 12.3 | 19.4 |
| Cumulative events required in control arm (total) | 9 (15) | 30 (50) | 59 (101) |
| Total expected cumulative randomisations | 54 | 110 | 174 |
All analyses were undertaken according to a predefined SAP, which was agreed before any data inspection. The primary outcome was prespecified to be analysed by ITT (final analysis). An interim analysis was undertaken in the per-protocol sample, which was defined as patients who completed at least one cycle of trial treatment (≥ 28 days). Per-protocol analyses were carried out for sensitivity. Data were analysed with Stata, version 14.1.
Patients were censored according to the following criteria. For survival status, patients were censored on the date that they were last known to be alive, which was recorded as the latest of either the date of collection of a prescription from their hospital pharmacy or the date of a follow-up visit or CT scan. For patients who died before any follow-up visit or CT scan, the date of death was used as the date of the event and assumed death without progression. For PFS, patients were censored without progression on the date of the last CT scan showing no progression.
Kaplan–Meier curves were used to present survival data and Cox regression modelling to estimate HRs, which were adjusted for the stratification factors that were used to minimise patients into allocated groups. Because the numbers of patients and events were small, adjustments were undertaken using the method of inverse probability weighting107 and using the bootstrap method to estimate CIs. The proportional hazards assumption was tested by regressing scaled Schoenfeld residuals against the log of time. 108 If evidence showed significant violation, a sensitivity analysis using restricted mean survival analysis was performed. 105
Study results
Between 7 July 2014 and 7 March 2016, 32 patients were randomised (16 into each treatment group) across 18 hospitals in the UK (Figure 11). The groups were well balanced in terms of baseline characteristics (Table 10). The first interim analysis was triggered in March 2016, when nine PFS events had occurred in the placebo group. The IDMC reviewed the data as part of a closed confidential meeting and made a recommendation to close the trial based on insufficient drug activity; the observed HR did not fall below the critical HR threshold of 1.0, which was predefined as part of the multistage sample size calculations (see Table 9). This decision was subsequently endorsed by both the TSC and the TMG. Trial recruitment was closed, and during subsequent follow-up of patients already entered into the study further PFS events occurred, such that 31 out of 32 patients had a PFS event by the time of the final analysis on 1 August 2016. No patients were lost to follow-up. The median PFS was 3.48 months (95% CI 1.51 to 5.09 months) with placebo and 2.96 months (95% CI 1.94 to 5.62 months) with AZD8931 (adjusted HR 1.10, 95% CI 0.47 to 3.57; p = 0.95) (Figure 12).
FIGURE 11.
FOCUS4-D trial profile. a, Reasons for non-randomisation not recorded. PTEN, phosphatase and tensin homologue. CR, complete response; PR, partial response.
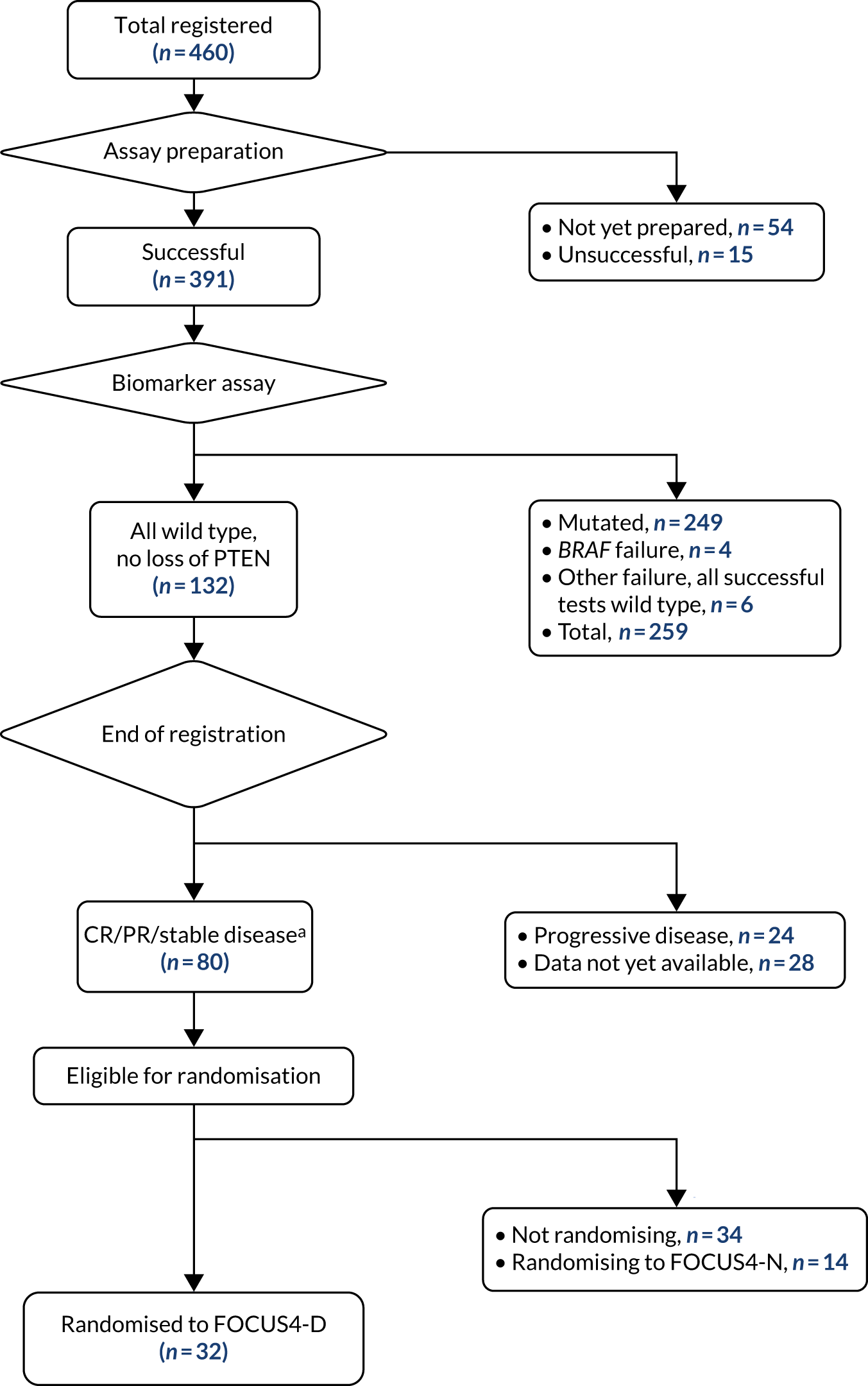
| Characteristic | Placebo | AZD8931 |
|---|---|---|
| Age (years), mean (SD) | 65 (11) | 64 (11) |
| Sex, n (%) | ||
| Male | 13 (81) | 13 (81) |
| Female | 3 (19) | 3 (19) |
| Current state of primary tumour, n (%) | ||
| Resected primary | 11 (69) | 8 (50) |
| Unresected primary | 5 (31) | 7 (44) |
| Unresected local recurrence | 0 (0) | 1 (6) |
| Timing of metastases, n (%) | ||
| Metachronous | 4 (25) | 4 (25) |
| Synchronous | 12 (75) | 11 (75) |
| Site of primary of tumour, n (%) | ||
| Right colon | 5 (31) | 5 (31) |
| Left colon | 6 (38) | 6 (38) |
| Rectum | 5 (31) | 5 (31) |
| Current WHO performance status, n (%) | ||
| 0 | 12 (75) | 11 (69) |
| ≥ 1 | 4 (25) | 5 (31) |
| Disease assessment at end of first-line treatment, n (%) | ||
| Partial response | 8 (50) | 7 (44) |
| Stable disease | 8 (50) | 9 (56) |
| Number of metastatic sites, n (%) | ||
| One | 6 (38) | 9 (56) |
| Two or more | 10 (63) | 7 (44) |
| Fluoropyrimidine drug used during first-line treatment, n (%) | ||
| Fluorouracil | 11 (69) | 10 (63) |
| Capecitabine | 5 (33) | 6 (38) |
| Oxaliplatin or Irinotecan used during first-line treatment, n (%) | ||
| Oxaliplatin only | 6 (38) | 6 (38) |
| Irinotecan only | 9 (56) | 9 (56) |
| Neither | 1 (6) | 1 (6) |
| Monoclonal antibody used during first-line treatment, n (%) | ||
| Cetux/panitumumab | 4 (25) | 3 (19) |
| Bevacizumab | 1 (6) | 1 (6) |
| No antibody | 11 (69) | 12 (75) |
| Total, n (%) | 16 (100) | 16 (100) |
FIGURE 12.
Progression-free survival (ITT analysis) FOCUS4-D.

Two patients did not receive any treatment (one patient in each group) and one further patient in the placebo group did not complete their first cycle of treatment. Therefore, 29 patients were included in the per-protocol sample (14 in the placebo group and 15 in the AZD8931 group). The Cox regression analysis within the per-protocol sample (for sensitivity) generated an adjusted HR of 1.15 (95% CI 0.49 to 4.75; p = 0.92) in favour of placebo. No statistical evidence was found that the proportional hazards assumption had been violated (Grambsch–Therneau test on log-time scale: p = 0.25 for the ITT analysis, and p = 0.35 for the per-protocol analysis); however, as a result of the small sample size, a sensitivity analysis using restricted mean survival time analysis at 4 months was carried out. However, this test did not alter the results for either the ITT analysis [adjusted restricted mean 2.90 months (95% CI 2.34 to 3.46 months) in the placebo group vs. 2.95 months (95% CI 2.37 to 3.53 months) in the AZD8931 group; p = 0.90 for difference] or the per-protocol analysis [adjusted restricted mean 3.08 months (95% CI 2.61 to 3.56 months) vs. 3.05 months (95% CI 2.48 to 3.63 months); p = 0.94 for difference].
Of the 32 randomised patients, 11 (34%) received first-line irinotecan and fluorouracil. After disease progression, only four (36%) of these patients returned to their first-line therapy. Reasons for not returning to their first-line therapy were the clinician’s decision because of toxicity or disease progression. A further eight (25%) patients received first-line oxaliplatin plus capecitabine, and four (50%) of these patients returned to this regimen after disease progression. Reasons for not returning to first-line therapy were the clinician’s decision because of toxicity (n = 1), disease progression (n = 1) and no symptoms being reported (n = 1) and the patient entering another trial (n = 1). A further seven (22%) patients received irinotecan and fluorouracil plus cetuximab or panitumumab as first-line therapy, and only one (14%) patient returned to their first-line regimen after disease progression. The reasons for not returning to first-line therapy were patient’s choice (n = 4) and the patient was considered for resection (n = 1), and there was also missing data on the reasons provided (n = 1). Other first-line therapies included oxaliplatin and capecitabine plus bevacizumab (n = 2), oxaliplatin and fluorouracil (n = 2), capecitabine alone (n = 1) and fluorouracil alone (n = 1). In total, 11 out of 32 (34%) patients restarted their first-line therapy at the time of data collection.
No patterns were noted in the reasons given for not restarting therapy. As at November 2017, not all data for post-progression treatments were available. Five of the 32 randomised patients needed dose reductions: one in the placebo group and four in the AZD8931 group. The patient in the placebo group had their evening dose reduced from 40 mg to 20 mg on their second cycle because of diarrhoea (grade 3). The dose reductions in the AZD8931 group were all evening dose reductions from 40 mg to 20 mg on the second or third cycles and were because of skin rash (grade 2, n = 1; grade 3, n = 3).
One patient in the AZD8931 group subsequently had a morning dose reduction from 40 mg to 20 mg on their fourth cycle, which was also because of skin rash (grade 3), and another patient subsequently had a morning dose reduction from 40 mg to 20 mg on their sixth cycle, which was for dry eyes (grade 3). All dose reductions were maintained within the currently available data. Only one patient explicitly gave the reason for stopping trial treatment as toxicity. This patient was assigned AZD8931 and reported grade 3 dry eyes, grade 2 skin ulceration and grade 2 skin rash. No serious adverse events were reported for this patient. Overall, few toxic effects were reported. Skin rash was the most frequent grade 3 adverse event, recorded in 3 out of 15 patients (20%) in the AZD8931 group compared with no patients in the placebo group. Diarrhoea was the most frequent grade 3 adverse event in the placebo group, recorded in one patient (6%) compared with one patient (7%) in the AZD8931 group (Table 11). No grade 4 adverse events were reported. Five serious adverse events required admission to hospital: four in the AZD8931 group [epigastric pain (grade 3); back pain (grade 3), hyperbilirubinaemia (grade 4) and device-related infection (grade 2); duodenal ulcer (grade 3); and dehydration (grade 3, but subsequently fatal)] and one in the placebo group [chest infection (grade 3)]. No treatment-related deaths were reported. Patients’ compliance was 85% in the placebo group and 75% in the AZD8931 group.
| Toxicity | Placebo, n (%) | AZD8931, n (%) |
|---|---|---|
| Nausea | 0 (0) | 0 (0) |
| Vomiting | 0 (0) | 0 (0) |
| Diarrhoea | 1 (6) | 1 (7) |
| Stomatitis | 0 (0) | 0 (0) |
| Dry skin | 0 (0) | 0 (0) |
| Skin rash | 0 (0) | 3 (20) |
| Acne | 0 (0) | 0 (0) |
| PPE | 0 (0) | 0 (0) |
| Anaemia | 0 (0) | 1 (7) |
| Neutropenia | 0 (0) | 0 (0) |
| Thrombocytopenia | 0 (0) | 0 (0) |
| Hyperbilirubinaemia | 0 (0) | 1 (7) |
| Transaminitis | 0 (0) | 0 (0) |
| Hypomagnesaemia | 0 (0) | 0 (0) |
| Cardiac toxicity | 0 (0) | 0 (0) |
| Pneumonitis | 0 (0) | 0 (0) |
| Infection | 0 (0) | 1 (7) |
| Dry eyes | 0 (0) | 1 (7) |
| Photophobia | 0 (0) | 0 (0) |
| Blurred vision | 0 (0) | 0 (0) |
| Conjunctivitis | 0 (0) | 0 (0) |
| Corneal ulcer | 0 (0) | 0 (0) |
| Fatigue | 0 (0) | 0 (0) |
| Paronychia | 0 (0) | 0 (0) |
| Epistaxis | 0 (0) | 0 (0) |
| Cystitis | 0 (0) | 0 (0) |
| Total | 16 (100) | 15 (100) |
Discussion and conclusions
In FOCUS4-D, AZD8931 failed to pass the first stage of assessment within the MAMS trial design, that is the predefined critical HR of 1.0 at stage 1 (lack of sufficient activity) was not reached. However, this trial has set a new paradigm in molecularly stratified trials in CRC.
Two Phase II, randomised, combination trials of AZD8931 have been carried out previously, both in patients with breast cancer. The MINT study109 was stopped because of a lack of efficacy (NCT01151215). In this trial, AZD8931 (40 mg or 20 mg twice daily) was compared with placebo in combination with anastrozole in postmenopausal women with hormone receptor-positive locally advanced or metastatic breast cancer who had never received endocrine treatment. The second randomised trial (THYME)110 did not meet its primary objective of prolonged PFS when AZD8931 was added to weekly paclitaxel in patients with advanced breast cancer and low expression of HER2 (NCT00900627). The primary hypothesis in the THYME study was that low HER2 expression was a driver for heterodimerisation, which would be inhibited by AZD8931.
In FOCUS4-D, it was not possible to investigate subgroups effectively because of the small sample size. Specific areas of interest include the effect of anti-EGFR agents in the chemotherapy induction phase and pre randomisation (six patients in total) and the role of PTL as a marker of response to treatment. The findings of these three trials indicate a need for greater understanding of EGFR family heterodimers and their dynamic interaction with targeted agents. To date, heterodimers have been poorly assessed in vitro and in vivo because of a paucity of effective methods, which might have hampered more complete scrutiny of therapeutic agents, such as FOCUS4-D, which was the first of the FOCUS4 molecular cohorts to reach its first MAMS-defined interim analysis. The observed HR at the interim analysis was above the critical threshold for continuation of the trial, and the decision to close the trial was clear cut and unanimous across all oversight committees. This outcome was considered to be a success of the trial design, in that a decision to continue might have wasted clinical trial resources on recruiting and following up a further 142 patients, and closing the trial prevented other patients from being given an ineffective but potentially harmful treatment. FOCUS4-D was designed to detect a target HR of 0.5. Therefore, the probability of a final analysis of 174 patients showing a HR of 0.5 when the noted HR at this interim analysis was 1.10 was exceptionally small.
A limitation of the FOCUS4-D study was that there was some deviation from the assumptions that we made in the sample size calculations. The final sample size was small, with recruitment of only 32 of the maximum expected target of 174 patients. The median PFS in the placebo group was 3.5 months, which is lower than that anticipated in the sample size (4.6 months), and the higher event rate brought forward the trigger point for analysis (nine events in the placebo group). Owing to delays in site opening of this complex trial, recruitment was also slower than anticipated; therefore, this trigger occurred after randomisation of fewer patients than had been expected (32 instead of 54 patients). However, since all but one patient had a PFS event by the time of the final analysis, it was reassuring that further follow-up would not alter the conclusions of the study, and there is little reason to suspect that a further 22 patients (which would take the total sample size to the expected 54 patients at this analysis) would have responded any differently to the 32 patients reported here. In addition, the smaller number of patients in the stage 1 analysis of lack of sufficient activity might increase any effect on outcome between the two treatment groups posed by trial imbalances in prognostic factors and choice of first-line treatment. For instance, the trial size makes it impossible to assess with any meaningful interpretation the effects of previous first-line treatment by EGFR inhibition with cetuximab or panitumumab (four patients in the placebo group and three patients in the AZD8931 group).
FOCUS4-D has shown no evidence of efficacy of single-agent EGFR, HER and HER3 inhibition with AZD8931 in patients with advanced CRC whose tumours are wild type for BRAF, PIK3CA, KRAS and NRAS after first-line induction therapy. Toxic effects were low, apart from skin rash in about 20% of patients. Early planned interim analyses with prespecified efficacy thresholds for lack of sufficient drug activity might be helpful in closing trials that are very unlikely to show benefit, which could facilitate more efficient use of clinical trial resources. New agents are currently being investigated for testing in this cohort of patients with all wild-type tumours.
Chapter 7 Results from FOCUS4-N
The full report of the FOCUS4-N trial has been published. 111 Parts of this chapter have been reproduced by Adams et al. 111 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: https://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Scientific rationale for FOCUS4-N
Treatment breaks in patients receiving palliative chemotherapy for metastatic CRC reduce toxicity burden and improve quality of life. 16 However, current standards either mandate or recommend a strategy of continuing therapy until disease progression or excess toxicity. Standard maintenance strategies in high-income countries favour a combination of capecitabine orally with intravenous bevacizumab thrice weekly,112,113 based on the Phase III CAIRO3114 and AIO-0207102 studies. Health economic evaluation of this approach has previously indicated a lack of cost-effectiveness driven by the non-significant improvement in OS and relatively high cost of intravenous bevacizumab (drug plus administration); a 93% cost reduction would be required for this strategy to become cost-effective. 115 Prior studies have evaluated a range of strategies to either completely stop therapy as a ‘treatment holiday’, reducing toxicities and visits to hospital, or attenuate the therapy, removing certain drugs as a maintenance therapy in comparison with the historic standard of care of continuing maximum tolerated dose of treatment. Meta-analysis of these approaches overall shows no difference in OS. 116 Notably, maintenance strategies, almost uniformly, demonstrate an improvement in PFS but at the expense of ongoing, yet attenuated, toxicity and unending, multiple visits to hospital for intravenous therapy. To the best of our knowledge, the FOCUS4-N trial is the first trial to explore the oral strategy of capecitabine only versus active monitoring to assess the potential impact on PFS, toxicity and quality of life, and enable patients and clinicians to choose an optimum approach tailored to the individual.
FOCUS4-N was the non-stratified comparison in the FOCUS4 trial programme and was designed for patients in whom a molecularly stratified comparison was unavailable or the biomarker tests failed for the patient’s tumour tissue. FOCUS4-N investigated the efficacy of capecitabine as a maintenance therapy compared with active monitoring in patients with metastatic CRC. The methods used for patient registration and biomarker testing in FOCUS4-N have been described in Chapter 2.
Study methods
Trial approvals, patient eligibility and recruitment
The trial and subsequent amendments were approved by the UK National Ethics Committee Oxford – Panel C and by the relevant regulatory body MHRA (CTA# 20363/0400/001 and EudraCT# 2012-005111-12).
Patients aged ≥ 18 years with newly diagnosed locally advanced or metastatic CRC were eligible for registration in the FOCUS4 trial programme. 20 Patients whose tumours had remained stable or responded to treatment according to their 16-week CT were assessed for eligibility for the FOCUS4-N comparison. In addition to the registration eligibility criteria, patients were required to have a baseline randomisation CT scan performed within 4 weeks prior to randomisation; a minimum 3-week washout period after the last dose of chemotherapy or biological therapy and before the first dose of capecitabine; adequate renal (creatinine clearance > 50 ml/minute) and liver function; and a WHO performance status of 0–2. Patients who were eligible for either FOCUS4-N or a molecularly stratified trial were offered entry into either trial and given the option of which study to participate in.
In the first phase of the FOCUS4 trial, between January 2014 and June 2017, patients with a raised baseline platelet count (thrombocytosis) were considered ineligible based on previous data from the COIN trial, which indicated a significant detriment in survival in this group of patients receiving an intermittent strategy. 16 A subsequent individual-patient data meta-analysis of Phase III intermittent strategy trials did not confirm this observation in COIN. 117 Thus, between June 2017 and March 2020, eligibility criteria were adapted to allow this group of patients with thrombocytosis to be included.
Trial procedures
Patients randomised to receive capecitabine were asked to continue taking the drug until disease progression, death or intolerable toxicity. Capecitabine was sourced from local hospital stock and dosed in accordance with standard cycle guidelines: oral capecitabine twice per day for 14 days, followed by a 7-day rest period of no capecitabine tablets.
Patient tumour status was assessed every 8 weeks by CT scan, which was reviewed at the treating hospital site in accordance with RECIST, version 1.1. 118 Toxicities and symptoms were assessed locally every 4 weeks from randomisation or the start of treatment, using the NCI common terminology criteria for adverse events (version 3.0). Patients were followed until progressive disease occurred, at which point the patient was recommended to restart first-line chemotherapy.
Treatment was stopped in the event of grade 3 or worse toxic effects or persistent toxicities judged medically significant or not tolerated by the patient, until the toxicity resolved to grade 1 or better. After stopping treatment (for a maximum of 28 days), capecitabine could be re-initiated at a reduced dose. Any stoppage for ≥ 28 days was not permitted, and the patient was discontinued from trial therapy but remained under follow-up.
Quality-of-life data were collected using EuroQol-5 Dimensions (EQ-5D) at multiple time points: at randomisation, every 8 weeks until progression, 4 weeks after end of trial treatment (progression, toxicity or patient choice), 3 months after progression and 6-monthly.
Statistical methods
Treatment allocation
Patients were allocated to capecitabine or active monitoring by a centrally managed telephone service at the MRC CTU at UCL, using a 1 : 1 allocation ratio by minimisation with a random element of 20%. Minimisation factors were treating hospital site, site of primary tumour (right colon, left colon or rectum), WHO performance status (0–2), 16-week CT scan result (stable disease, partial response or complete response), number of metastatic sites (none, one, or two or more) and first-line chemotherapy regimen (fluorouracil, capecitabine or neither; both oxaliplatin and irinotecan, oxaliplatin only, irinotecan only or neither; and cetuximab or panitumumab, bevacizumab or no monoclonal antibody).
Outcome measures
The primary outcome for FOCUS4-N was PFS, defined as the time from randomisation to either disease progression (in accordance with RECIST criteria) or death from any cause. Patients without a PFS event were censored at the time of their last recorded CT scan. OS was a secondary outcome, defined as time from randomisation to death from any cause, with patients censored at last recorded disease assessment, blood measurement or anti-cancer treatment. Other secondary outcomes included safety, toxicity, quality of life and tumour response. Quality of life was analysed using mixed-effects linear modelling with patient-level random intercepts and time slopes, with differences by treatment arm tested by evaluating the area under the curve from the model.
Sample size calculation
The target sample size for FOCUS4-N was calculated using the analysis of resources for trials programme implemented in Stata software. Given that the rate of recruitment into FOCUS4-N was dependent on the availability of other molecular comparisons, failure of biomarker testing or patient choice, exact recruitment figures were not known at the start of the trial. Various scenarios were used to estimate the recruitment rate over 5 years and a median PFS of 4 months was assumed in the active monitoring arm (based on data in the COIN trial). A total of 644 patients (635 events) would provide 80% power of detecting a HR of 0.8 at the two-sided 5% significance level.
In March 2020, the COVID-19 pandemic resulted in temporary closure of FOCUS4 to new recruitment and, following IDMC review and recommendation, a decision was taken to close recruitment permanently in April 2020 because trial funding was nearing its end. A previous review of the implications of reduced recruitment on the statistical power of FOCUS4-N had been reviewed by the funders, who recommended that, despite reduced power, the trial should close in 2020 and report the data accrued up to that point.
Statistical analysis
All analyses were performed in accordance with a predefined SAP that was agreed before database lock. Analyses were performed using Stata statistical software, version 16.1. The primary analysis was performed in accordance with ITT, with a secondary per-protocol analysis defined by patients who completed at least one cycle of trial treatment (≥ 28 days). Patients were censored in accordance with the following criteria. For survival status, patients were censored on the date that they were last known to be alive, either via collection of prescription from their hospital pharmacy or attendance at a follow-up visit or CT scan. For PFS, patients were censored without progression on the date of the last CT scan showing no progression. For patients who died before any follow-up visit or CT scan, the date of death was used as the date of the event and assumed death without progression providing that the death occurred within 3 months of randomisation or any previous CT scan confirming no progression.
Kaplan–Meier curves were used to present survival data and Cox regression modelling was used to estimate the HR between randomised groups. Unadjusted HRs were estimated, as well as HRs adjusted for the stratification factors that were used to minimise patients into allocated groups (primary analysis). A further analysis also adjusted for resection status, timing of metastatic disease, alkaline phosphatase, white blood cell count, age of tumour sample and use of aspirin at baseline. Deviation from non-proportional hazards was assessed using regression of scaled Schoenfeld residuals against the log of time.
Study results
Recruitment and compliance
Across 88 UK hospitals, between January 2014 and March 2020, a total of 1434 patients were registered into the FOCUS4 trial, of whom 924 underwent successful biomarker assessment and completed 16 weeks of first-line therapy with stable disease, partial response or complete response. Of these patients, 254 were randomised into FOCUS4-N (Figure 13), 127 patients to active monitoring and 127 patients to maintenance capecitabine monotherapy.
FIGURE 13.
Patient flow through the FOCUS4-N trial. CR, complete response; PR, partial response.
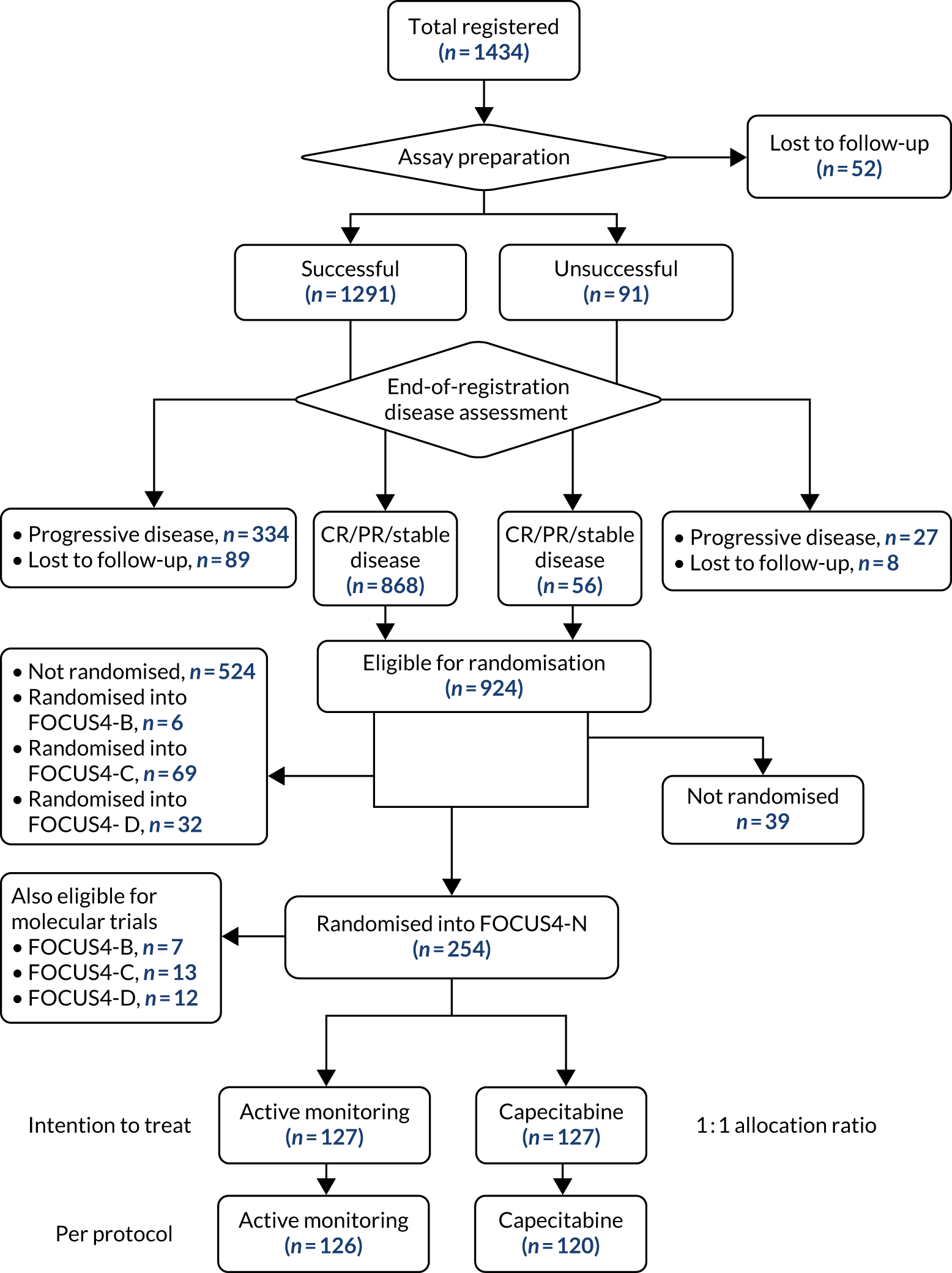
Baseline demographic and clinical characteristics were well balanced between the study arms (Table 12). The majority of patients had widespread synchronous metastatic disease, with approximately half of the patients having no resection of their primary tumour. A right-sided PTL was present in approximately one-third of patients. The majority of patients were treated with doublet chemotherapy (irinotecan based, 57%) without a monoclonal antibody because bevacizumab is not reimbursed in the UK. The molecular characteristics are shown in Table 12, which shows that only 37% had a RAS wild-type tumour, reflecting the NHS England policy not to allow treatment breaks for patients on EGFR monoclonal antibodies.
| Characteristic | Active monitoring | Capecitabine |
|---|---|---|
| Age (years), mean (SD) | 63.7 (10.9) | 64.7 (9.6) |
| Sex | ||
| Male | 76 (60) | 86 (68) |
| Female | 51 (40) | 41 (32) |
| Baseline WHO performance status, n (%) | ||
| 0 | 76 (60) | 80 (63) |
| 1 | 49 (39) | 45 (35) |
| 2 | 2 (2) | 2 (2) |
| Site of primary of tumour, n (%) | ||
| Right colon | 45 (35) | 47 (37) |
| Left colon | 32 (25) | 33 (26) |
| Rectum | 50 (39) | 47 (37) |
| Current state of primary tumour, n (%) | ||
| Resected primary | 62 (49) | 54 (43) |
| Unresected primary | 61 (48) | 68 (54) |
| Unresected local recurrence | 4 (3) | 5 (4) |
| Number of metastatic sites, n (%) | ||
| No metastases | 2 (2) | 4 (3) |
| One site | 41 (32) | 40 (31) |
| Two or more sites | 84 (66) | 83 (65) |
| Timing of metastases, n (%) | ||
| Metachronous | 40 (31) | 21 (17) |
| Synchronous | 85 (67) | 101 (80) |
| No metastases | 2 (2) | 4 (3) |
| Missing data | 0 (0) | 1 (1) |
| Disease assessment at end of first-line treatment, n (%) | ||
| Complete response | 3 (2) | 5 (4) |
| Partial response | 75 (59) | 71 (56) |
| Stable disease | 49 (39) | 51 (40) |
| Fluoropyrimidine drug used during first-line treatment, n (%) | ||
| Fluorouracil | 95 (75) | 97 (76) |
| Capecitabine | 32 (25) | 30 (24) |
| Oxaliplatin or irinotecan used during first-line treatment, n (%) | ||
| Both oxaliplatin and irinotecan | 2 (2) | 2 (2) |
| Oxaliplatin only | 50 (39) | 50 (39) |
| Irinotecan only | 73 (57) | 71 (56) |
| Neither | 2 (2) | 4 (3) |
| Monoclonal antibody used during first-line treatment, n (%) | ||
| Cetux/panitumumab | 25 (20) | 20 (16) |
| Bevacizumab | 6 (5) | 7 (6) |
| No antibody | 96 (76) | 100 (79) |
| PIK3CA mutation status, n (%) | ||
| Mutation | 15 (12) | 14 (11) |
| Wild type | 96 (76) | 100 (79) |
| Fail | 7 (6) | 5 (4) |
| Insufficient tumour | 9 (7) | 8 (6) |
| BRAF mutation status, n (%) | ||
| Mutation | 13 (10) | 17 (13) |
| Wild type | 103 (81) | 98 (77) |
| Fail | 2 (2) | 4 (3) |
| Insufficient tumour | 9 (7) | 8 (6) |
| RAS mutation status, n (%) | ||
| Mutation | 68 (54) | 68 (54) |
| Wild type | 47 (37) | 48 (38) |
| Fail | 3 (2) | 3 (2) |
| Insufficient tumour | 9 (7) | 8 (6) |
| p53 mutation status, n (%) | ||
| Mutation | 61 (48) | 62 (49) |
| Wild type | 33 (26) | 28 (22) |
| Fail | 3 (2) | 2 (2) |
| Could not be tested | 18 (14) | 24 (19) |
| Insufficient tumour | 12 (9) | 11 (9) |
| MSI status, n (%) | ||
| MSS | 108 (85) | 104 (82) |
| MSI | 2 (2) | 3 (2) |
| Fail | 2 (2) | 4 (3) |
| Could not be tested | 6 (5) | 8 (6) |
| Insufficient tumour | 9 (7) | 8 (6) |
Compliance with randomised allocation was good for only one patient in the active monitoring arm receiving capecitabine approximately 6 months prior to progression. Patients in the capecitabine arm received a median of four cycles (IQR 2–8 cycles).
Primary outcome: progression-free survival
There were 122 PFS events in the active monitoring arm (n = 127 patients) and 117 in the capecitabine arm (n = 127 patients). The median PFS in the capecitabine arm was 3.88 months (IQR 2.10–7.39 months) and 1.87 months (IQR 1.64–3.65 months) in the active monitoring arm. The unadjusted and adjusted HRs were 0.44 (95% CI 0.33 to 0.57; p = 2.8 × 10–10) and 0.40 (95% CI 0.21 to 0.75; p = 3.9 × 10–10), respectively; the Kaplan–Meier curves are presented in Figure 14. Per-protocol analyses demonstrated very similar findings: the unadjusted and adjusted HRs were 0.42 (95% CI 0.32 to 0.55; p = 6.9 × 10–10) and 0.38 (95% CI 0.28 to 0.51; p = 9.5 × 10–11), respectively. There was no evidence to suggest that there was deviation from the proportional hazards assumption (p = 0.084).
FIGURE 14.
Kaplan–Meier curve for PFS in FOCUS4-N. Minimisation factors: location of primary tumour (left, right, rectum), baseline WHO performance status, baseline disease assessment, number of metastases, first-line therapy (fluoropyrimidine, oxaliplatin or irinotecan, monoclonal antibody), Biomarker cohort and stratified for FOCUS4 trial time points.
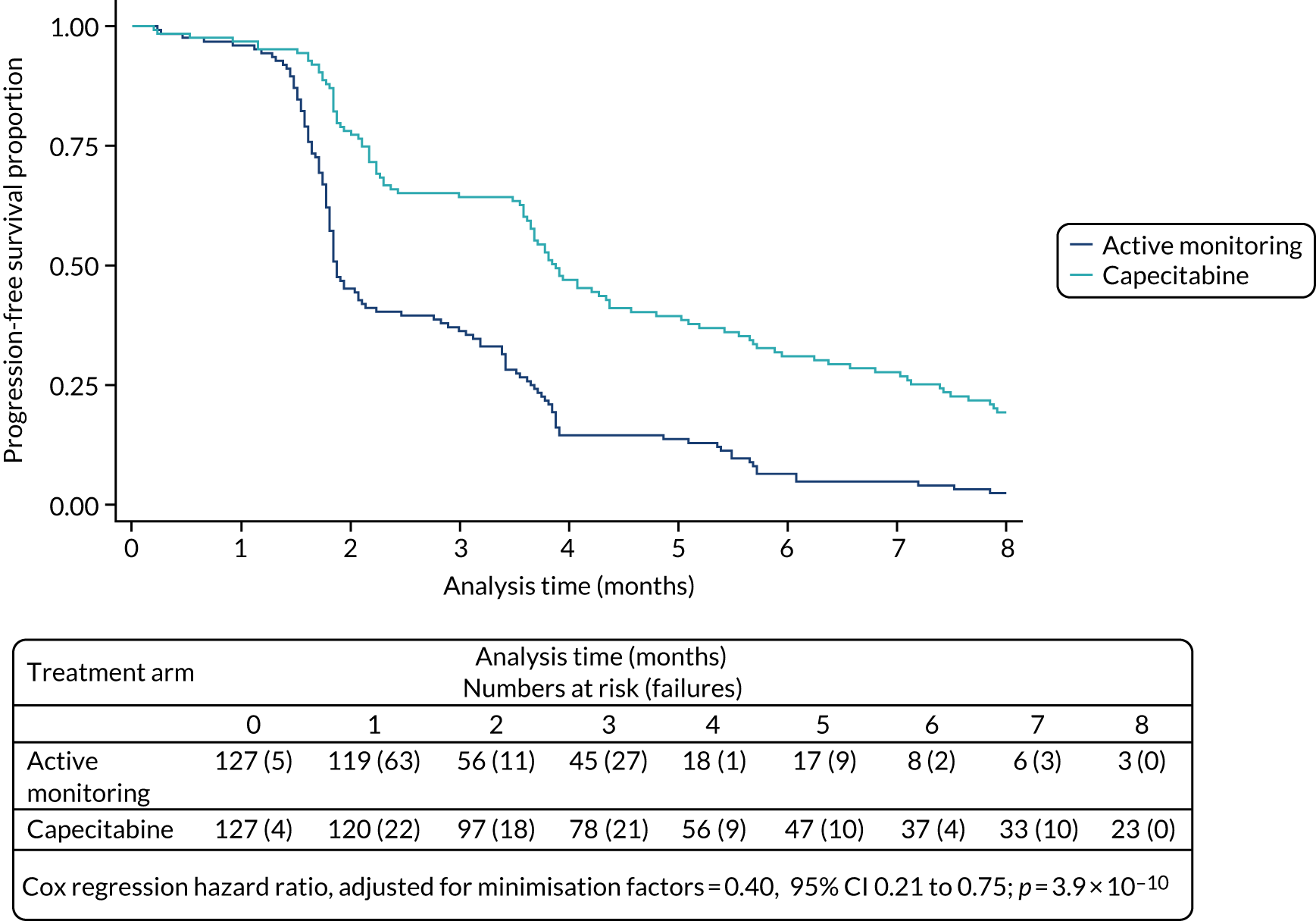
Overall survival
There were 90 deaths in the active monitoring arm (n = 127 patients) and 99 deaths in the capecitabine arm (n = 127 patients). The median time to death was 15.2 months (IQR 8.8–24.0 months) in the active monitoring arm, compared with 14.8 months (IQR 10.2–21.8 months) in the capecitabine arm, with a non-significant difference in survival between the two arms. The unadjusted and adjusted HRs were 1.00 (95% CI 0.75 to 1.33; p = 0.98) and 0.93 (95% CI 0.69 to 1.27; p = 0.66), respectively; the Kaplan–Meier curves are presented in Figure 15. There was no evidence to suggest that there was deviation from the proportional hazards assumption (p = 0.58).
FIGURE 15.
Kaplan–Meier curve for OS in FOCUS4-N. Minimisation factors: location of primary tumour (left, right, rectum), baseline WHO performance status, baseline disease assessment, number of metastases, first-line therapy (fluoropyrimidine, oxaliplatin or irinotecan, monoclonal antibody), biomarker cohort and stratified for FOCUS4 trial time points.

Subgroup analyses
The pre-planned subgroup analysis for PFS (Figure 16) demonstrated a non-significant trend towards better PFS with a maintenance strategy in left-sided tumours (HR 0.38 vs. 0.56 for right sided; interaction p = 0.13); a similar trend was seen with OS (HR 0.82 for left sided vs. 1.37 for right sided; interaction p = 0.076; Figure 17). There was a suggestion that patients with loss of PTEN and PIK3CA-mutant tumours may show less benefit than other molecular subgroups of maintenance capecitabine (PFS HR 0.74, OS HR 1.47), although this was not statistically significant. Patients with the double mutation in RAS and TP53 – that is, the same molecular characteristics as selected for the FOCUS4-C trial27 – showed the same beneficial effect of maintenance capecitabine as for the whole trial (PFS HR 0.44, 95% CI 0.30 to 0.63). For OS, the only other notable subgroup effect was that those with stable disease at randomisation appeared to benefit from maintenance capecitabine, whereas those with partial response did not (OS HR 0.63 and 1.42, respectively; interaction p = 0.005; see Figure 17). Swimmer plots showed the distribution of individual patient PFS duration and timing of CT scans by left-sided versus right-sided disease (Figure 18).
FIGURE 16.
Forest plot of subgroup analyses for PFS (unadjusted HRs) in FOCUS4-N. CET, cetuximab; PAN, panitumumab; WHO, World Health Organization.
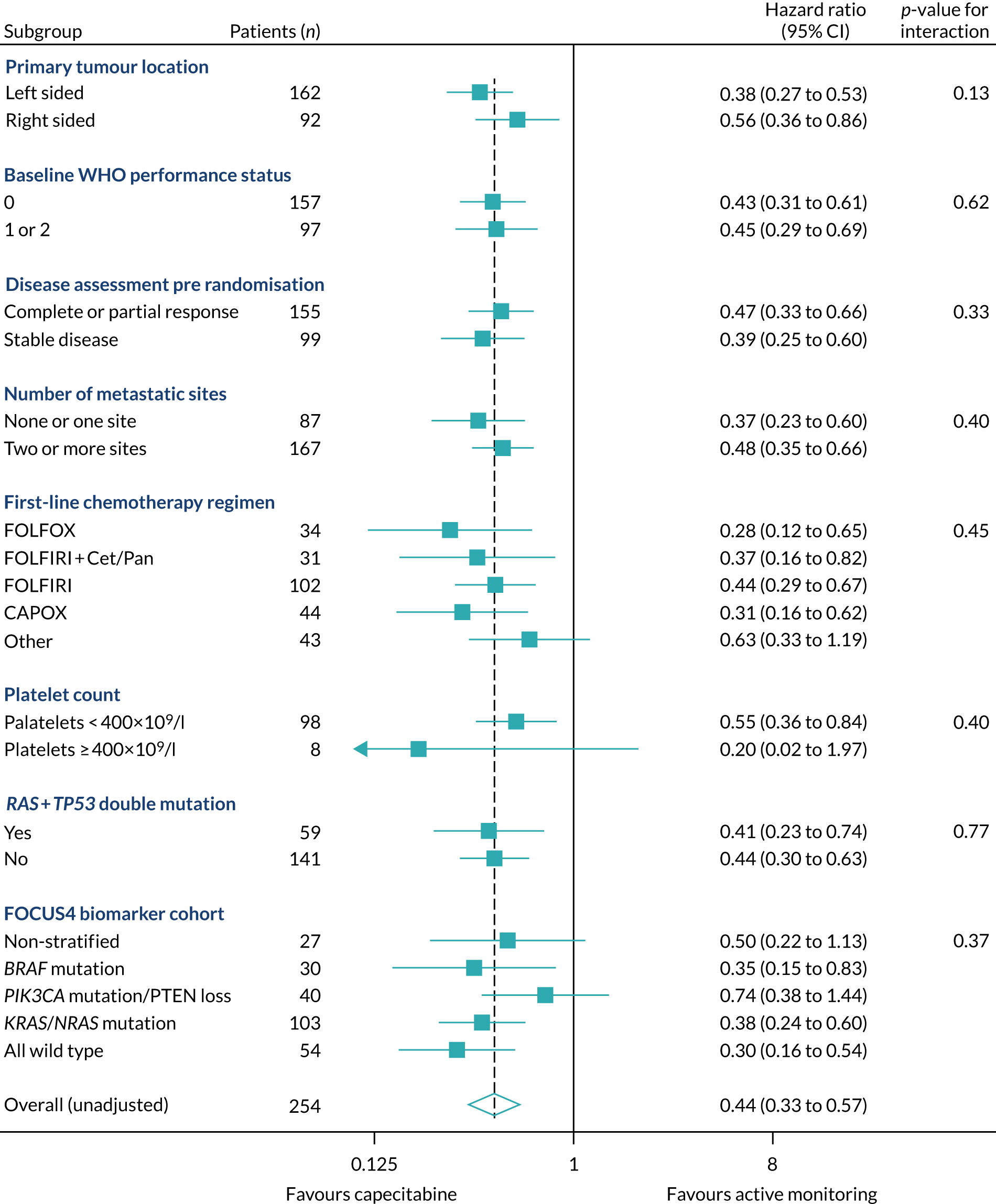
FIGURE 17.
Forest plot of subgroup analyses for OS (unadjusted HRs) in FOCUS4-N. CET, cetuximab; PAN, panitumumab; PTEN, phosphatase and tensin homologue; WHO, World Health Organization.
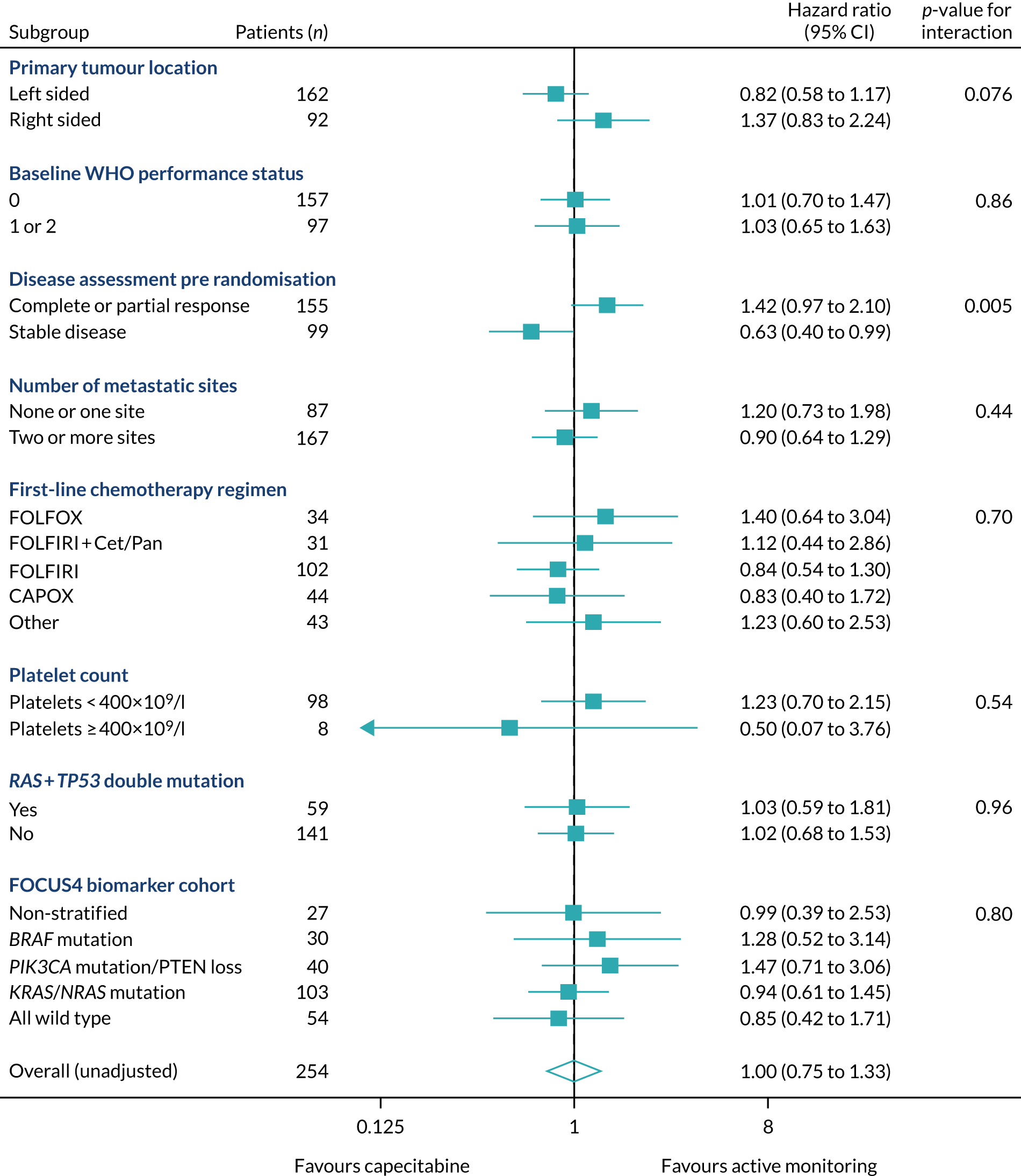
FIGURE 18.
Swimmer plot for FOCUS-N, by location of primary tumour in FOCUS4. Light blue horizontal strip, right-sided; navy strip, left-sided (including rectum).

Toxicity
Cumulative toxicities were significantly less in the active monitoring arm, with increased toxicities associated with capecitabine maintenance, including diarrhoea, dry skin, fatigue, nausea and palmar–plantar erythema, as presented in Figure 19. Ideally, a maintenance therapy should result in no toxicity. The incidences of grade zero toxicity as the worst toxicity reported per patient are, therefore, instructive and were as follows for active monitoring and capecitabine maintenance, respectively: nausea 74% versus 67%; diarrhoea 72% versus 46%; stomatitis 90% versus 77%; dry skin 83% versus 77%; palmar–plantar erythema 87% versus 44%; and anaemia 69% versus 54%. There were no notable differences in the incidence of grade zero toxicities for neutropenia, skin rash, nail dystrophy or vomiting.
FIGURE 19.
Cumulative reported toxicity by randomised group in FOCUS4-N. (a) Active monitoring; and (b) capecitabine. PPE, pruritic papular eruption.
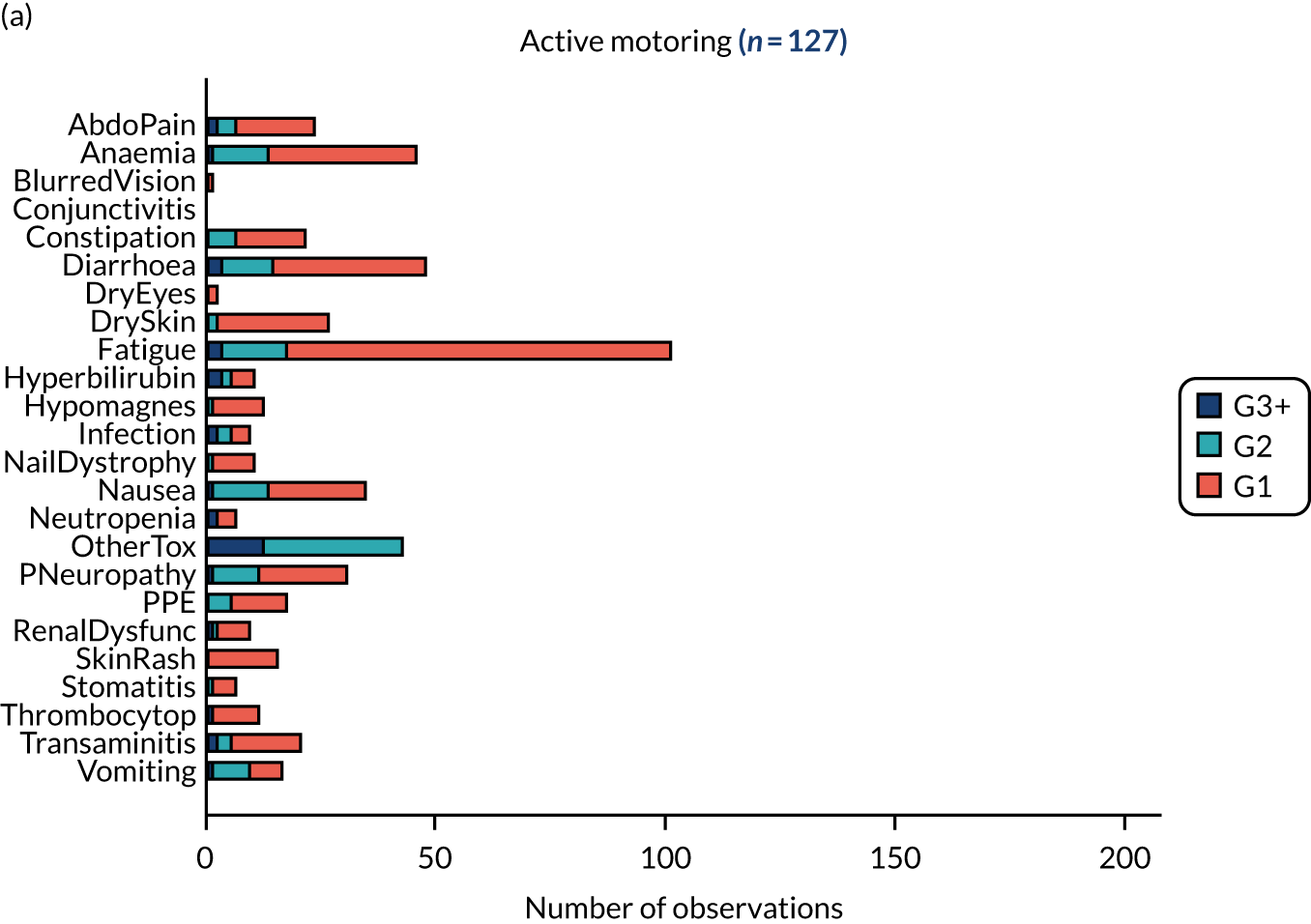

During the trial, 51% of capecitabine patients had at least one cycle delayed, 37% had a dose reduction and 34% had at least one missed dose (within a cycle). A total of 50% of capecitabine patients commenced at least four cycles and 25% commenced at least eight cycles.
Quality of life
The EQ-5D quality-of-life forms were completed for 93% (active monitoring) and 90% (capecitabine) of patients at baseline (pre randomisation but post induction chemotherapy). The protocol mandated 8-weekly completion until progression and 6-monthly thereafter; for analysis purposes, all available forms were forced into an 8-weekly schedule. On this basis, 63%, 45% and 33% of randomised patients had data available at 8, 16 and 24 weeks, respectively, with continued decline thereafter. Modelling was applied to data up to 48 weeks, given that data became too sparse beyond that point. No notable differences were seen in mobility, self-care, usual activities, anxiety and depression. A non-significant difference (p = 0.11) was seen in pain and discomfort in favour of the capecitabine maintenance strategy (Figure 20). This may be a result of symptoms associated with increased rates of progression in the active monitoring arm.
FIGURE 20.
Quality of life measured by EQ-5D by randomised group in FOCUS4-N. (a) Mobility; (b) self-care; (c) usual activities; (d) pain and discomfort; and (e) anxiety and depression.

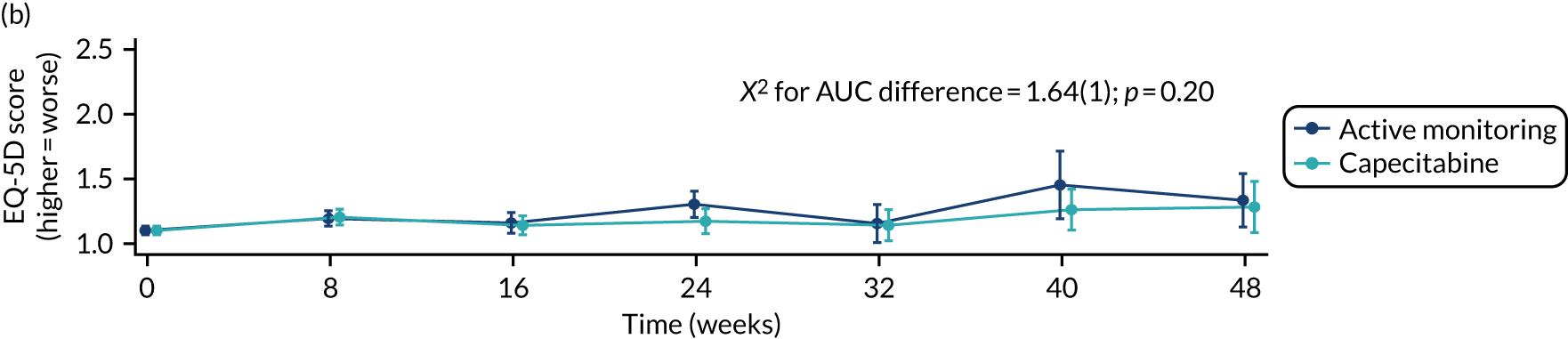
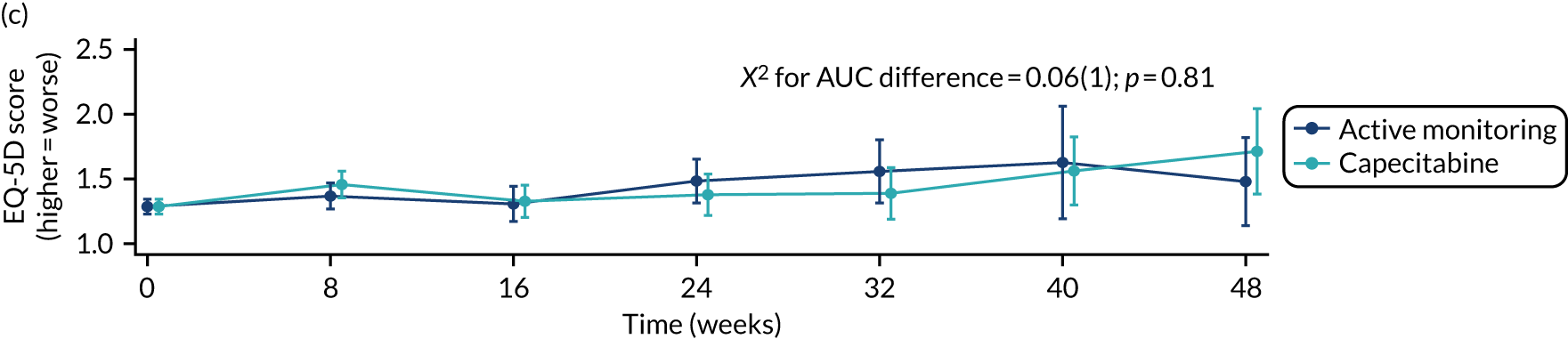

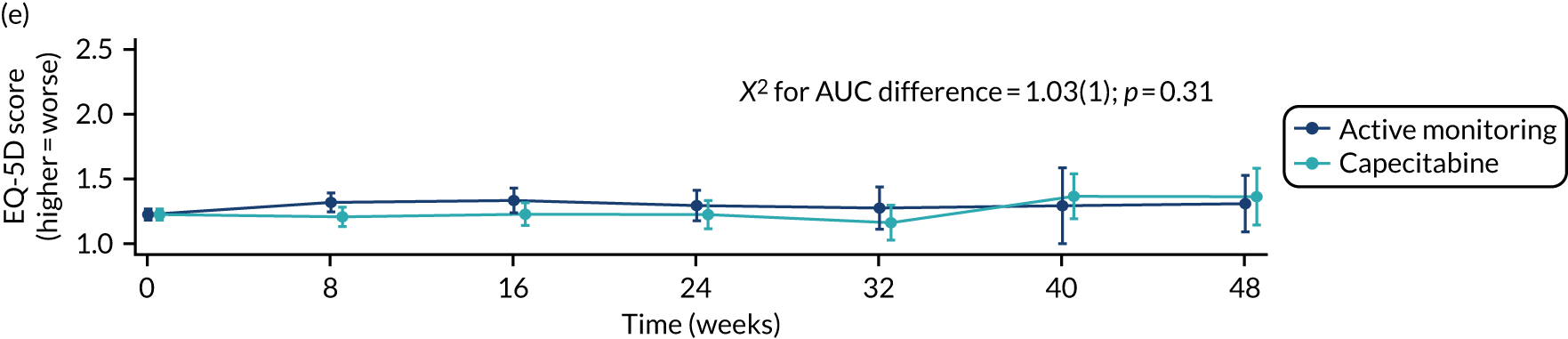
Discussion and conclusions
Choices on how to proceed with palliative treatment in the majority of patients with metastatic, incurable CRC, with responding or stable disease after 16 weeks of first-line chemotherapy, need careful consideration with the patient at the core. Discussions must be informed by the impact of receiving systemic anti-cancer therapy over the preceding period. This should include an evaluation of the burden of toxicity and quality of life, as well as the response to treatment. Pooled data from key Phase II and III trials suggest minimal impact on OS from a maintenance or continuation strategy but do show the ability to delay a return to ‘full’ combination therapy by implementation of a maintenance therapy. Notably, the FOCUS4-N data support the use of an oral-only therapy, ‘capecitabine’, to extend PFS and delay a return to combination therapy by an average of 2 months. There is a clear cost to the patient for this improvement in PFS seen with maintenance oral capecitabine, which includes worse toxicity in terms of diarrhoea, fatigue, nausea, skin rash and plantar–palmar erythema, albeit most frequently at grade 1 and 2 levels; these factors should be used to further inform decision-making processes. There was no difference in quality-of-life scores between the two arms. It is notable that the swimmer plots suggested that approximately one-third of patients gain a significant increment in PFS with maintenance capecitabine, suggesting significant fluoropyrimidine sensitivity, while one-third of patients demonstrate relative insensitivity to a fluoropyrimidine monotherapy and may indicate a further need to explore predictive biomarkers of efficacy for this strategy. Pre-planned subgroup analysis suggests that patients with stable disease at the end of the 16-week induction period may gain a significant survival benefit from maintenance capecitabine, but this was not borne out in other studies in which the same phenomenon was assessed. 117
Although this study was underpowered to evaluate OS, it demonstrated very similar median OS values of 14.8 versus 15.2 months in the capecitabine arm and active monitoring arm, respectively, with a HR of 0.93 (p = 0.66) when adjusted for minimisation factors. It is informative to compare these data with those of CAIRO3, which is a very similar study that compared an active monitoring strategy with capecitabine plus bevacizumab maintenance with comparable effects on PFS (HR 0.38; p < 0.0001), akin to the adjusted HR (0.40, p < 0.0001) in FOCUS4-N and OS (HR 0.86, p = 0.1). 114 Although cross-trial comparisons carry significant caveats and need to be undertaken with caution, this does suggest that the main driver of PFS improvement when using capecitabine plus bevacizumab is from the capecitabine. IPD meta-analysis has also shown no OS benefit from current maintenance therapy strategies. 117
Based on a subgroup analysis from the much larger Phase III COIN study,16 which demonstrated a survival detriment in those patients with a baseline thrombocytosis receiving a complete treatment break (HR 1.55; p = 0.0018), it was decided not to recruit patients to the FOCUS4 trial programme from January 2014 to June 2017. Wishing to validate or refute this finding, an IPD meta-analysis was undertaken to assess thrombocytosis as a predictive marker of the benefits or otherwise of an intermittent or continuous therapy strategy. 117 This evaluation did not validate the COIN finding on thrombocytosis and, thus, the trial eligibility was adapted to allow these patients to enter the study. Within FOCUS4-N overall, 3% (n = 8) of patients had a baseline thrombocytosis and, therefore, this study was underpowered to explore this predictive phenomenon further. Because of the conservative approach, FOCUS4-N under-represents the approximately 25% of patients with metastatic CRC who typically have thrombocytosis at baseline and form a poorer prognosis group. However, given the findings in the IPD meta-analysis, this is not considered to undermine the more general conclusions, which are independent of baseline platelet count.
Owing to funding restrictions in the UK NHS, bevacizumab is not routinely available for patients with metastatic CRC, and in patients with RAS wild-type tumours EGFR monoclonal antibodies are available only in the first-line setting, with restrictions in England also preventing treatment interruption of cetuximab or panitumumab for longer than 6 weeks. In addition, during the FOCUS4-D trial recruitment period,88 patients with RAS wild-type and BRAF wild-type tumours were eligible for randomisation and were preferentially recruited to that trial. These factors make for a slightly selective group of patients recruited to FOCUS4-N during that time. From a molecular perspective, 59% of patients randomised in the FOCUS4-N trial had a RAS mutation and 15% had a BRAF mutation. Reassuringly, the forest plots (see Figures 16 and 17) do not show any significant differences in PFS based on these molecular criteria.
The molecular stratification of patients in the FOCUS4 trial programme has allowed us to explore the impact of a capecitabine maintenance therapy from FOCUS4-N in the same molecular subgroup of patients who would otherwise have been able to receive novel maintenance therapy as a parallel cohort. Thus, patients recruited to FOCUS4-N who had a RAS and TP53 mutation within their tumour were compared with patients recruited into FOCUS4-C who had the same double-mutant status but were randomised to receive the novel Wee1 inhibitor AZD1775 (adavosertib, AstraZeneca UK) versus active monitoring. 27 There was no evidence to suggest a treatment benefit difference between these trials; however, subgroup analyses have demonstrated interesting differences between the trials, which are explored more fully in the FOCUS4-C paper. 27
Despite strong evidence of disease control with maintenance therapy, OS remains unaffected and FOCUS4-N provides additional evidence to support the use of treatment breaks as a safe management alternative for patients entering treatment de-escalation after 16 weeks of induction therapy for metastatic CRC. If maintenance therapy is selected following consideration of the advantages and disadvantages in consultation with a particular patient, capecitabine without bevacizumab may be used to extend PFS in the interval after doublet or triplet therapy, essentially doubling the period prior to recommencing full-dose induction therapy. Notably, these data also provide the tools to best inform the dialogue between patients and clinicians on the pros and cons of the different approaches and their trade-offs.
Chapter 8 Challenges and lessons learned
Parts of this chapter have been reproduced from Brown et al. 20 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: https://creativecommons.org/licenses/by/4.0/. The text below includes minor additions and formatting changes to the original text.
Reflections from stakeholder groups
Complex, adaptive stratified medicine studies are feasible but delivery can present numerous challenges. We have already published a series of papers on the practical aspects of running these sorts of complex platform trials. 119–122 With FOCUS4 now closed and learn more from the 10-year experience and provide insight for those in the research community hoping to undertake similar studies, we sought feedback from a number of stakeholders who were involved in the delivery of the trial. Each of the following eight stakeholder groups were asked to provide learning points from their experience: (1) co-chief investigators, (2) subtrial chief investigators on the TMG, (3) clinical research fellow trainees, (4) MRC CTU, (5) biomarker-testing laboratories, (6) patient/carer representatives, (7) oversight committees and (8) funders.
In addition, a participating site survey was distributed via site principal investigators to understand the experiences of site staff. Principal investigators at each of the 88 sites that registered at least one patient were sent a link to an anonymised survey, asked a series of questions about their practice and encouraged to provide additional comments on aspects of the trial that had worked well and not so well.
Feedback from stakeholder groups
Feedback was received from 19 out of 38 individuals, including at least one representative from each of the eight stakeholder groups. Experiences were very positive despite an acknowledgement of the substantial challenges encountered. There was considerable overlap in a number of the points raised, and the comments naturally grouped into three main areas: (1) resource and infrastructure, (2) biomarker-testing process and (3) trial design. A summary of the 20 learning points synthesised from the feedback is presented in Table 13. The most consistent learning points related to the following four key areas:
-
Understand resource capacity and ensure that adequate funding is secured for staff. These designs probably save time and speed up getting answers but still require similar amounts of resources per research question. The challenge for funders is to find a mechanism for funding and review of trial adaptations that facilitates delivery and minimises burden, while also managing the risks involved.
-
The biomarker-testing process should be kept as simple as possible and, for the UK, as much as possible should be kept within the NHS infrastructure.
-
Position the trial within the optimal phases of drug development and try to ensure that there are parallel research initiatives to support the trial in relation to ever-shifting biology and preclinical workup. FOCUS4 may have worked better as a Phase I/IIb platform trial.
-
Platform trials need to be nimble and able to adapt quickly with emerging new biological discoveries. This is difficult in a sometimes turgid clinical trial regulatory framework.
| Learning point | Resource and infrastructure |
|---|---|
| 1 | Secure adequate funding |
| 2 | Delivering all desired outcomes for a platform trial is clearly challenging. The challenge for funders is to find a mechanism for funding and review of adaptations that facilitates delivery and minimises burden while also managing the risks involved |
| 3 | Ideally, these trials should be conducted only in CTUs that have good core funding resources and a ballast of trained in-house trial and data managers who can be drawn on temporarily at times of intense activity |
| 4 | Activate fewer sites and stagger opening |
| 5 | Leadership: the chief investigator role is paramount and must not be underestimated, with far more pressure than being a CI on a more standard trial. An engaged and enthusiastic core TMG is vital |
| 6 | The CTU staff must feel comfortable and encouraged to escalate any site issues to senior TMG members quickly |
| 7 | A great training experience for CTU staff and clinical research fellows. Provide basic clinical trial training for research fellows to aid learning |
| 8 | Site enthusiasm was inconsistent between registration and randomisation. Understand local motivations or obstacles to recruitment |
| 9 | Trial longevity can lead to poor continuity of CTU and site staff, which is disruptive in a complex trial in which the design keeps adapting |
| 10 | For trials that last many years, trial participants need better opt-in/opt-out arrangements on how they can be kept informed on trial progress |
| Biomarker-testing process | |
| 11 | Regular quality assurance and review of sample-testing processes to identify any glitches that require modification |
| 12 | Important to spend adequate time on biomarker work-up and optimisation as well as understanding prevalence early on before taking further |
| 13 | Keep biomarker testing within the NHS infrastructure as much as possible with as few middle men as possible to avoid data privacy obstacles |
| 14 | Important to have an engaged and dedicated biomarker team who can manually step in and overcome any delays to prevent patient distress |
| Trial design | |
| 15 | The MAMS adaptive design worked well at cutting losses on poorly performing drugs early |
| 16 | The requirement for a control arm in each comparison was important in determining any prognostic biomarker effects |
| 17 | The need for a catch-all non-stratified trial (FOCUS4-N) proved to be successful at maximising trial opportunities for patients |
| 18 | Important to get the protocol structure right and consult with regulatory bodies on advice for what is acceptable within the design |
| 19 | The main issues were mostly related to pharma engagement and drug-target identification in the specific disease setting of the study. Earlier engagement in the developmental pathway for new therapies is required so that when the therapy is ready to drop into a trial, all parties have been engaged and involved with the biomarker optimisation and early drug activity assessments |
| 20 | Funding of complementary feeder collaborations, such as S:CORT30 (which focused on understanding the biology) and ACRCelerate31 (which focused on preclinical novel agent development), might have been beneficial if run in parallel with FOCUS4 |
Results from the site survey
Feedback was received from 52 out of 88 sites (59%), with representation from all level 1, level 2 and level 3 sites. Sites were asked about standard treatment for patients with unresectable stage 4 metastatic CRC; the majority used either a block of chemotherapy followed by a complete break or intermittent chemotherapy with a complete break. In some settings, treatment to progression was considered, as well as maintenance chemotherapy, but both were rare. Most sites were supportive of using the maintenance setting to test new therapies.
The remaining results of the survey have been summarised in Table 14 and show that, although sites experienced a number of challenges in delivering FOCUS4, sites were positive about their involvement and supportive of the use of these trials in the future, if resourced adequately.
| Question and response | Take home message |
|---|---|
Did inability to restart an EGFR inhibitor impact on patient selection?
|
NHS rulings on the use of EGFR inhibitors restricted recruitment and may have been a barrier to finding alternative or better therapies relevant in the RAS wild-type group |
Was having an unselected FOCUS4-N trial important?
|
An important aspect of the design that was strongly supported by sites and patients |
| What were the advantages and disadvantages of conducting this trial in the maintenance setting? | Advantages:
|
| Did you experience any particular study challenges? | Staff and infrastructure:
|
| What went well? |
|
| Are platform trials the future? |
|
Chapter 9 Discussion and conclusions
The place of FOCUS4 in the evolution of trial methodology and delivery in the UK
FOCUS4 has successfully demonstrated that the development of Clinical Research Networks enables the delivery of these large-scale, complex trials. The development of the adaptive trial methodology and MAMS by the MRC CTU, which was first implemented at scale in prostate cancer, has now been successfully used in both FOCUS4 and the RECOVERY trial. 28
FOCUS4 was the first platform to test at scale the additional complexity of molecular stratification, which was initially piloted in FOCUS3. 123 This built on initial work in the earlier haematological studies in acute myeloid leukaemia, but bridged this into solid tumour oncology. The place for FOCUS4 in the evolution of trial methodology is demonstrated by the observation that it has been rapidly mimicked by a major pharmaceutical company (Roche MODUL trial) and is also being used in other solid tumour oncology studies in the UK and elsewhere.
Was FOCUS4 in fact initiated too early before the evidence was really present for stratification in CRC? Colleagues in the USA devised a similar study, but it was not funded by the NCI because the NCI considered that there were insufficient therapeutics ready for testing in such a study. Perhaps the NCI’s view is correct. However, the attempt to undertake this study has moved the field in solid tumour oncology to think more deeply about the utility of umbrella trials – of which this is a prime example – and of basket studies. On review, the evidence shows that basket studies – testing the same intervention across multiple indications – are easier to perform and have been generally more successful in identifying positive indications for novel therapies. The great advantage of basket studies is that the triallists are dealing with one intervention, one biomarker and one company. They are nearly always initiated and sponsored by the pharmaceutical company and, therefore, overcome the key difficulty of engaging different pharma to collaborate on a generic platform.
Did FOCUS4 fulfil its ambitions?
FOCUS4 almost fulfilled its original ambitions in that four subtrials were activated (i.e. B, C, D and N), although five subtrials had been planned (i.e. A, B, C, D and N); three subtrials reported results (C, D and N),27,88,111 with FOCUS4-B stopping early on grounds of futility.
However, it is also worth noting that the group attempted to open a total of 20 trials, but the others were not activated for multiple reasons (see Table 4). During the course of the trial, a total of 20 drugs or drug combinations were explored and presented to the joint NIHR EME/CRUK funding subboard for peer-review approval but only four culminated in an activated comparison. Reasons for non-activation were predominantly because of the failure of drugs at early clinical testing in this advanced metastatic disease setting. Other endeavours failed owing to strategic shifts within companies, including the selling of assets suddenly and unexpectedly.
Recruitment to the trial was not as fast as originally planned. The proposed acrual number was 2400, which is comparable in size to the predecessor COIN trial. There were several drivers of slower recruitment, notably the lack of ‘exciting’ comparisons to energise investigators and the challenge of the NHS England rules regarding the use of EGFR inhibitors in all wild-type patients. We were able to negotiate with the National Cancer Research Network that registrations, as well as randomisations, attracted activity-based funding. We managed to obtain high-quality follow-up for FOCUS4, but this was based on the CTU commiting to funding per-patient payments at risk pending subsequent commercial support.
In terms of methodology, the following key features of the FOCUS4 design, as discussed Chapter 2, Key principles of the FOCUS4 trial design, all proved to be robust in the application:
-
using molecularly enriched cohorts to maximise the possibility of detecting promising new treatments
-
using multistage statistical design for early detection of insufficient activity
-
providing a trial opportunity for all patients regardless of biomarker status
-
adapting biomarker structure to include new biomarkers and exclude current biomarkers, as guided by evolving research findings
-
testing multiple treatments at the same time, each against its own control.
In addition, the use of PFS in the maintenance setting as the primary end point was shown to be an effective indicator of agents with activity, which may not have been revealed through a more conventional assessment of response in end-stage patients.
How could FOCUS4 have carried out better?
A key area of potential improvement is the understanding of the molecular stratification of CRC. Over this period, it has been shown that gene expression signatures (consensus molecular subtypes)124 are an important window of CRC that is not reflected directly from genetic testing. The funding of the MRC stratification in CRC (S:CORT) consortium30 was provided in 2015 and with time has enabled deeper understanding of the subtypes within CRC and how they may be targeted. 83
Timely and reproducible delivery of the biomarker-testing panel, understanding the prognostic impact of biomarker selection and the need to accommodate biomarker-negative patients was critical to success. During FOCUS4, the simple mutation-based stratification process for many of the biomarkers that were tested in FOCUS4 has become part of standard care. In the future, despite the now more widespread routine availability of NGS-based tumour profiling, trial-based stratification testing will continue to need to provide transcriptomic and other more sophisticated analyses. It may be that the revolution in digital pathology and artificial intelligence will enable some biological stratification to be achieved directly from routine images, as shown recently with consensus molecular subgroup subtyping. 23
Better preclinical testing of novel agent combinations may have improved the success of FOCUS4. As noted in Chapter 3, Failed endeavours, a key drain on investigator time was the continual pursuit of new agents for testing in the FOCUS4 platform. During the course of the trial, a total of 20 drug combinations were explored but only four culminated in an activated comparison. Reasons for non-activation were predominantly because of the failure of drugs at early clinical testing in this advanced metastatic disease setting, but often protocol development and contract negotiations had progressed a long way and considerable resource had been used up for ultimately futile collaborations.The disease positioning of genetically engineered mouse models has increased exponentially in the last few years and this provides a strong platform on which to test novel therapeutic concepts in defined biological settings more representative of human cancer. The CRUK-funded ACRCelerate programme,31 which focused on preclinical novel agent development, might have been beneficial if run in parallel with or before FOCUS4 because of its ability to provide greater depth of scientific rationale for the specific therpautics within the biomarker subsets.
Collaboration with pharmaceutical companies could have been more efficient from the onset of the FOCUS4 trials. How this can be accomplished is one of the ongoing challenges in academic-led clinical trials. High-level engagment with companies with a broad portfolio of agents is one potential approach that is more likely to be successful, as in the National Lung Matrix trial,23 which was mediated through high-level and effective support from the funder CRUK. Major governmental involvement through the NCI in the USA is also a very powerful mechansim, as shown in the IMPACT study. In the DISCOVERY trial, a committee at the Department of Health and Social Care level was also able to enable access to medicines within the context of a pandemic in a much more effective way that a research group can. A further reason for the failed endeavours to engage company support was the change in strategies within the collaborating pharmaceutical companies, including the sudden and unexpected selling of assets. One strategy, which is more within the grasp of academic triallists, is to have the strongest preclinical rationale for the specific intervention in the proposed subset. Obtaining that data set is challenging and requires timely funding of both stratification efforts and preclinical platforms to bring together data to make the case more strongly to pharma.
The alternatives to using a platform design of this umbrella style are to undertake individual molecularly stratified trials that each have their own design, entry criteria and setting. This is frequently undertaken and benefits from flexibilty – fitting the design to the specific intervention – but suffers from requiring every trial to be set up individually and a plethora of biomarker labs using differing techniques. Basket studies are very efficient when testing one intervention across a number of differing cohorts. These are very widely used by pharmaceutical companies looking to establish clinical efficacy of their drug/combination in whichever patient cohort may be sensitive. We did not pursue this because our focus was on CRC patients and we wanted to investigate treatments of potential benefit for our patient group.
Perhaps a better way ahead is for an academic funder to support a biomarker/diagnostic platform, complementary to what is available in the routine health-care setting. To this would be linked a number of discrete trials, all using the shared biomarker infrastructure, but allowing the flexibility and simplicity of individual interventions. This would leverage the benefits of national genomic diagnostics, but would enable the use of more advanced or emerging diagnostic methods, such as transcriptomic-, proteomic- or digital pathology-based allocation, supported by the biomarker research-funded infrastructure.
Summary of clinical findings from FOCUS4
In addition to the aforementioned trial methodology findings, the clinical findings from FOCUS4 are summarised below.
Data from FOCUS4-C showed that this subtrial met its primary end point; patients with RAS/TP53-mutant metastatic CRC had PFS advantage with the Wee1 inhibitor adavosertib compared with active monitoring following induction chemotherapy. This activity was clearly limited to the two-thirds of patients with primary tumours arising in the left colon and rectum (left PTL). Assessment of longer-term outcomes also showed an OS benefit with adavosertib compared with active monitoring in RAS/TP53-mutant patients with left PTL. These results are particularly encouraging because RAS/TP53-mutant metastatic CRC patients are a poor prognostic population who have limited treatment options and warrant further testing, which is now in discussion. Adavosertib was well tolerated and patients found it preferable to conventional chemotherapy.
Data from FOCUS4-D showed that the pan-HER inhibitor AZD8931 does not improve outcome in all wild-type CRC, as it failed to pass the first stage of assessment within the MAMS trial design, that is the predefined critical HR of 1.0 at stage 1 (lack of sufficient activity) was not reached.
FOCUS4-N was the non-stratified arm of FOCUS4 and shows the importance of including at least one non-stratified component within trials of this type. This trial built on previous UK trials exploring treatment holidays and confirmed that capecitabine is as effective as a maintenance therapy and probably as effective as capecitabine plus bevacizumab, the current international standard in this setting. Capecitabine alone, however, is much more cost-effective and also completely avoids the need for intravenous administration. However, none of these conventional agents improve OS (as we showed in the IPD meta-analysis117) so there is a balance to be struck for individual patient management about use of maintenance therapy. In future studies, capecitabine monotherapy would be a suitable control for testing of novel maintenance therapies.
Leadership of complex studies
The experience from FOCUS4 has shown that leadership of these complex studies is challenging, with strong input required from a well-funded CTU. In addition, multiple skill sets are essential to the successful leadership of such trials, including expert contribution from the clinical, pathology, biomarker, trial delivery, governance and statistical analysis aspects.
Furthermore, a strong TMG is critical to success. The FOCUS4 TMG met monthly over the decade of activity for 1 hour by teleconference, with detailed minutes and review of action points at the start of each meeting; this was an effective mechanism. Face-to-face meetings were interspersed especially to discuss broader trial design or novel interventions. Feedback from FOCUS4 stakeholders showed that, with respect to leadership, the role of the chief investigator was paramount and more critical to trial success that that of the co-ordinating investigator in a standard trial.
Funder perspectives
FOCUS4 was jointly funded, with an equal split in costs between the MRC–NIHR-funded EME programme and CRUK. Extensive work was required by the research team, the NIHR evaluation, trials and studies co-ordinating centre EME team and the CRUK team to carefully divide and calculate costs in accordance with the different NIHR and CRUK funding rules. Although this resulted in agreement of costs between the funders, it was clearly a complex and time-consuming process. Learning from this project helped to develop the simpler and more streamlined approaches for future use.
Patient and public involvement perspectives
Throughout FOCUS4, there was a strong patient voice on the TMG, provided by Malcolm and Janet Pope, to whom we are deeply indebted. This was supported by strong patient and public involvement representation on the Trial Steering Committee. These studies are strongly attractive to patients who appreciate the efforts to tailor treatments to their own tumour characteristic. In contrast to the triallists perspective, patients with a particular diagnosis – in this case CRC – value the umbrella trial methodology because there is one study for the whole group of patients with the same disease, and each may find their particular space under that umbrella. This can lead to a sense of collaboration, partnership and collegiality: ‘this is our study and we all have our place within it’. By contrast, biomarker-selective studies can appear exclusive: you get in only if you have the ‘right’ tumour. In a fruitful collaboration with Joshua Hordern (Faculty of Theology and Religion, University of Oxford, UK), we explored the moral aspects of the promissory nature of precision medicine and the danger of hype, wherein targeted or personalised therapies can appear to be of much greater worth than appears to be the case. 125 Furthermore, we explored the superiority that this inflated value may imply for the selected group and what effects that might have on the other people who end up in ‘the molecularly unstratified group’. 126 We made an application to the Wellcome Trust (London, UK) for a multidisciplinary analysis of this area, which unfortunately was not supported. This interest in the well-being of patients extended to a concern for those staff running studies, which revealed an interesting mixture of excitement, sense of privilege and unending stress for the personnel working in the trials unit on major platform studies of this nature. 121
The COVID-19 pandemic
FOCUS4 came to an end as the COVID-19 pandemic developed. Thankfully, for this study, sufficient patients had already participated in the study and we had accrued sufficient data to be able to present interim findings to the IDMC. The interim analyses for both FOCUS4-C and FOCUS4-N were so resoundingly positive that the IDMC were very ready to advise that we could close recruitment, follow-up the remaining patients for a few more months and then report. We are hugely grateful to the unseen work of both our IDMC and our Trial Steering Committee members in their oversight of the study and enabling us to draw it to its conclusion. We recognise that, as a result, the FOCUS4 studies under-accrued compared with the original statistical estimates. Despite this, FOCUS4-C, FOCUS4-D and FOCUS4-N all have reported clear outcomes.
Beyond the COVID-19 pandemic, our oversight committees consisted of highly experienced oncologists, statisticians and patient representatives. The members provided constant support and guidance at some particularly difficult times during the study. In particular, the chairpersons navigated the issues with clear and prompt recommendations despite the relative newness of this approach to trial design. We thank them.
Conclusions
FOCUS4 was an experiment at multiple levels. It was an experiment in clinical trial design that largely succeeded; certainly, the key points from the methodology paper were largely shown to be feasible and correct. Each individual subtrial was a defined experiment, two of which were successful in terms of reaching their primary end point; another was shown not to be feasible and one was negative. Sixteen others were developed but failed to be implemented for various reasons.
The prior knowledge that is required before setting out on such an enterprise needs to include in-depth understanding of the therapeutic vulnerabilities of different subgroups of the disease in question. That, in turn, requires detailed multi-omic stratification of the disease linked to clinical outcome to define those subgroups with different biology and preclinical evidence of efficacy of the selected therapeutic class or agent.
Such studies are a manifestation of team science: the clinicians, biomarker scientists, pathologists, statisticians and clinical trial team at the CTU; the research delivery personnel in the network and in multiple hospitals; the patients and their carers; the physicians and nurses who enrol, care for and monitor those patients; and the various companies who collaborate through provision of their novel agents. Without this whole team working together, the enterprise cannot hope to accomplish its goals.
Acknowledgements
A full list of those involved in the study is available in Report Supplementary Material 2.
We acknowledge the expert contribution of Justina Orleans-Lindsay, specialist medical writer, for her assistance in drafting this final report.
An AstraZeneca Ltd educational grant in 2017 funded the translational aspects of FOCUS4-C. Supply and distribution of AZD8931 for FOCUS-D and AZD1775 for FOCUS4-C was provided by AstraZeneca Ltd. Supply and distribution of aspirin and placebo for FOCUS4-B was provided by Bayer AG (Leverkusen, Germany). The MRC Clinical Trials Unit at University College London receives core funding from the Medical Research Council within UKRI (budget: MC_UU_12023/20).
Trial Management Group
Maughan TS (chairperson), Wilson RH, Adams R, Seymour M, Seligmann JF, Graham J, Wasan H, Pope M, Pope J, Samuel L, Shiu K, Church D, Middleton G, Steward W, Twelves C, Wellman S, Hodgkinson E, Stoner N, Beety J, Duggleby K, Dutton G, MRC CTU team (see below) and laboratory staff (see below).
MRC CTU trial co-ordinating centre
Brown LC, Kaplan R, Parmar M, Fisher D, Campos M, Yates E, Santana S, Fiddament A, Harper L, Bara A, Pugh C, Bathia R, Letchemanan K, Przybył B, Bhogal S, Gopalakrishnan G, Purvis C, Diaz-Montana C, Mohamed F, Townsend S, Cragg W, Masters L, Van Looy N and Rauchenberger M.
Laboratories
Quirke P, Richman SD, Hemmings G, Davis J, Gallop N, Wilkinson L, Butler R, Roberts H, Jasani B, White R, Dodds R, James M and Morgan M.
Independent Data Monitoring Committee
Cameron D (chairperson), Souhami R, Peeters M, Billingham C, Griffiths G and Brown J.
Trial Steering Committee
Johnson P (chairperson), Rudd R, Whelan J and Russell A.
Ethics approval
FOCUS4 was approved by the UK National Ethics Committee Oxford – Panel C (reference 13/SC/0111) and by the relevant regulatory body, the Medicines and Healthcare products Regulatory Agency (MHRA) (CTA# 20363/0400/001 and EudraCT# 2012–005111–12) in May 2013. Subsequent amendments were also reviewed and approved by the UK National Ethics Committee Oxford – Panel C.
Contributions of authors
Louise C Brown (https://orcid.org/0000-0003-2827-6634) (Senior Statistician, MRC Trials Unit) was the project lead for FOCUS4 at the MRC CTU, designed the trial statistics and oversaw statistical analyses.
David Fisher (https://orcid.org/0000-0002-2512-2296) (Statistician, MRC Trials Unit) performed all statistical analyses.
Richard Adams (https://orcid.org/0000-0003-3915-7243) (Professor of Clinical Trials and Consultant Clinical Oncologist) was a core member of the TMG, and chief investigator of the FOCUS4-D and FOCUS4-N subtrials.
Jenny Seligmann (https://orcid.org/0000-0003-4379-6005) (Clinical Trial fellow and then Senior Lecturer in Medical Oncology) was a trial fellow and co-chief investigator of FOCUS4-C.
Matthew Seymour (https://orcid.org/0000-0002-2441-9629) (Professor of Gastrointestinal Cancer Medicine, National Cluster Lead for Cancer for NIHR CRN) was co-chief investigator for FOCUS4-C.
Richard Kaplan (https://orcid.org/0000-0002-0189-8348) (Professor of Medical Oncology) was programme lead at MRC Clinical Trials Unit and provided experienced triallist advice throughout.
Susan D Richman (https://orcid.org/0000-0003-3993-5041) (Senior Laboratory Scientist) oversaw the biomarker-testing laboratory in Leeds.
Philip Quirke (https://orcid.org/0000-0002-3597-5444) (Professor of Pathology) was the lead consultant pathologist with expertise in colorectal cancer and provided expert input for molecular diagnostics.
Rachel Butler (Head of Molecular Genetics Laboratory) provided expert leadership in genetic diagnostics.
Helen Roberts (https://orcid.org/0000-0001-5664-7151) (Head of Solid Tumour Services, Molecular Genetics Unit, Cardiff) provided expert analysis of NGS sequencing and TMG membership.
Janet Graham (https://orcid.org/0000-0002-2048-2698) (Consultant Medical Oncologist) was a member of the Trial Management Group.
Richard H Wilson (https://orcid.org/0000-0001-8018-7730) (Professor of Gastrointestinal Oncology) was co-chief investigator of FOCUS4 and CI of FOCUS4-B.
Timothy S Maughan (https://orcid.org/0000-0002-0580-5065) (Professor of Clinical Oncology) was co-chief investigator of FOCUS4, FOCUS4-C and FOCUS4-N, and was the chairperson of the TMG.
Publications
Adams RA, Fisher DJ, Graham J, Seligmann JF, Seymour M, Kaplan R, et al. Capecitabine versus active monitoring in stable or responding metastatic colorectal cancer after 16 weeks of first-line therapy: results of the randomised FOCUS4-N trial. J Clin Oncol 2021;39:3693–704. https://doi.org/10.1200/JCO.21.01436
Hague D, Townsend S, Masters L, Rauchenberger M, Van Looy N, Diaz-Montana C, et al. Changing platforms without stopping the train: experiences of data management and data management systems when adapting platform protocols by adding and closing comparisons. Trials 2019;20:294. https://doi.org/10.1186/s13063-019-3322-7
Kaplan R, Maughan T, Crook A, Fisher D, Wilson R, Brown L, Parmar M. Evaluating many treatments and biomarkers in oncology: a new design. J Clin Oncol 2013;31:4562–8. https://doi.org/10.1200/JCO.2013.50.7905
Brown LC, Graham J, Fisher D, Adams R, Seligmann J, Seymour M, et al. Experiences of running a stratified medicine adaptive platform trial: challenges and lessons learned from 10 years of the FOCUS4 trial in metastatic colorectal cancer. Clin Trials 2022;19:146–57. https://doi.org/10.1177/17407745211069879
Morrell L, Hordern J, Brown L, Sydes MR, Amos CL, Kaplan RS, et al. Mind the gap? The platform trial as a working environment. Trials 2019;20:297. https://doi.org/10.1186/s13063-019-3377-5
Seligmann JF, Fisher DJ, Brown LC, Adams RA, Graham J, Quirke P, et al. Inhibition of WEE1 is effective in TP53- and RAS-mutant metastatic colorectal cancer: a randomised trial (FOCUS4-C) comparing adavosertib (AZD1775) with active monitoring. J Clin Oncol 2021;39:3705–15. https://doi.org/10.1200/JCO.21.01435
Adams R, Wilson R, Brown L, Maughan T. Reply to A. Kurreck et al and M.S. Copur et al. J Clin Oncol 2022;40:1263–4. https://doi.org/10.1200/JCO.21.02806
Richman SD, Hemmings G, Roberts H, Gallop N, Dodds R, Wilkinson L, et al. FOCUS4 biomarker laboratories: from the benefits to the practical and logistical issues faced during 6 years of centralised testing [published online ahead of print 7 March 2022]. J Clin Pathol 2022. https://doi.org/10.1136/jclinpath-2022-208233
Schiavone F, Bathia R, Letchemanan K, Masters L, Amos C, Bara A, et al. This is a platform alteration: a trial management perspective on the operational aspects of adaptive and platform and umbrella protocols. Trials 2019;20:264. https://doi.org/10.1186/s13063-019-3216-8
Data-sharing statement
FOCUS4 adopts a controlled access data sharing policy. Applications for use of the data can be made via the corresponding author for review by the MRC CTU data access committee.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
References
- Cancer Research UK . Cancer Statistics for the UK n.d. www.cancerresearchuk.org/health-professional/cancer-statistics-for-the-uk (accessed 15 November 2022).
- Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther 2020;5. https://doi.org/10.1038/s41392-020-0116-z.
- De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010;11:753-62. https://doi.org/10.1016/S1470-2045(10)70130-3.
- Freidlin B, Sun Z, Gray R, Korn EL. Phase III clinical trials that integrate treatment and biomarker evaluation. J Clin Oncol 2013;31:3158-61. https://doi.org/10.1200/JCO.2012.48.3826.
- Popovici V, Budinska E, Tejpar S, Weinrich S, Estrella H, Hodgson G, et al. Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J Clin Oncol 2012;30:1288-95. https://doi.org/10.1200/JCO.2011.39.5814.
- Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet 2011;377:2103-14. https://doi.org/10.1016/S0140-6736(11)60613-2.
- The Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330-7. https://doi.org/10.1038/nature11252.
- Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466-74. https://doi.org/10.1200/JCO.2009.23.3452.
- Wasan H, Meade AM, Adams R, Wilson R, Pugh C, Fisher D, et al. Intermittent chemotherapy plus either intermittent or continuous cetuximab for first-line treatment of patients with KRAS wild-type advanced colorectal cancer (COIN-B): a randomised phase 2 trial. Lancet Oncol 2014;15:631-9. https://doi.org/10.1016/S1470-2045(14)70106-8.
- Parmar MK, Barthel FM, Sydes M, Langley R, Kaplan R, Eisenhauer E, et al. Speeding up the evaluation of new agents in cancer. J Natl Cancer Inst 2008;100:1204-14. https://doi.org/10.1093/jnci/djn267.
- Royston P, Barthel FM, Parmar MK, Choodari-Oskooei B, Isham V. Designs for clinical trials with time-to-event outcomes based on stopping guidelines for lack of benefit. Trials 2011;12. https://doi.org/10.1186/1745-6215-12-81.
- Royston P, Parmar MK, Qian W. Novel designs for multi-arm clinical trials with survival outcomes with an application in ovarian cancer. Stat Med 2003;22:2239-56. https://doi.org/10.1002/sim.1430.
- Kaplan R, Maughan T, Crook A, Fisher D, Wilson R, Brown L, et al. Evaluating many treatments and biomarkers in oncology: a new design. J Clin Oncol 2013;31:4562-8. https://doi.org/10.1200/JCO.2013.50.7905.
- Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66:3992-5. https://doi.org/10.1158/0008-5472.CAN-06-0191.
- Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 2007;25:3230-7. https://doi.org/10.1200/JCO.2006.10.5437.
- Adams RA, Meade AM, Seymour MT, Wilson RH, Madi A, Fisher D, et al. Intermittent versus continuous oxaliplatin and fluoropyrimidine combination chemotherapy for first-line treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet Oncol 2011;12:642-53. https://doi.org/10.1016/S1470-2045(11)70102-4.
- Richman SD, Adams R, Quirke P, Butler R, Hemmings G, Chambers P, et al. Pre-trial inter-laboratory analytical validation of the FOCUS4 personalised therapy trial. J Clin Pathol 2016;69:35-41. https://doi.org/10.1136/jclinpath-2015-203097.
- Smith CG, Fisher D, Claes B, Maughan TS, Idziaszczyk S, Peuteman G, et al. Somatic profiling of the epidermal growth factor receptor pathway in tumors from patients with advanced colorectal cancer treated with chemotherapy ± cetuximab. Clin Cancer Res 2013;19:4104-13. https://doi.org/10.1158/1078-0432.CCR-12-2581.
- Adkins D, Ley J, Worden F, Sacco AG, Palka K, Grilley-Olson JE, et al. Palbociclib and cetuximab in platinum-resistant and in cetuximab-resistant human papilomavirus-unrelated head and neck cancer: a multicentre, multigroup, phase 2 trial. Lancet Oncol 2019;20:1295-30. https://doi.org/10.1016/S1470-2045(19)30405-X.
- Brown LC, Graham J, Fisher D, Adams R, Seligmann J, Seymour M, et al. Experiences of running a stratified medicine adaptive platform trial: challenges and lessons learned from 10 years of the FOCUS4 trial in metastatic colorectal cancer. Clin Trials 2022;19:146-57. https://doi.org/10.1177/17407745211069879.
- National Institute for Health and Care Research . NIHR Welcomes New Vision for the Future of UK Clinical Research Delivery 2021. www.nihr.ac.uk/news/nihr-welcomes-new-vision-for-the-future-of-uk-clinical-research-delivery/27308 (accessed 23 March 2021).
- Sirinukunwattana K, Domingo E, Richman SD, Redmond KL, Blake A, Verrill C, et al. Image-based consensus molecular subtype (imCMS) classification of colorectal cancer using deep learning. Gut 2021;70:544-54. https://doi.org/10.1136/gutjnl-2019-319866.
- Middleton G, Fletcher P, Popat S, Savage J, Summers Y, Greystoke A, et al. The National Lung Matrix Trial of personalized therapy in lung cancer. Nature 2020;583:807-12. https://doi.org/10.1038/s41586-020-2481-8.
- Clinicaltrials.gov . Lung-MAP: Biomarker-Targeted Second-Line Therapy in Treating Patients With Recurrent Stage IV Squamous Cell Lung Cancer n.d. www.clinicaltrials.gov/ct2/show/NCT02154490 (accessed 9 September 2022).
- Flaherty KT, Gray R, Chen A, Li S, Patton D, Hamilton SR, et al. The Molecular Analysis for Therapy Choice (NCI-MATCH) trial: lessons for genomic trial design. J Natl Cancer Inst 2020;112:1021-9. https://doi.org/10.1093/jnci/djz245.
- Flaherty KT, Gray RJ, Chen AP, Li S, McShane LM, Patton D, et al. Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J Clin Oncol 2020;38:3883-94. https://doi.org/10.1200/JCO.19.03010.
- Seligmann J, Fisher DJ, Brown LC, Adams RA, Graham J, Quirke P, et al. Inhibition of WEE1 is effective in TP53 and RAS mutant metastatic colorectal cancer (mCRC): a randomised phase II trial (FOCUS4-C) comparing adavosertib (AZD1775) with active monitoring. J Clin Oncol 2021;39. https://doi.org/10.1200/JCO.21.01435.
- RECOVERY . Results n.d. https://recoverytrial.net/results (accessed 14 November 2022).
- ECOG-ACRIN Cancer Research Group . NCI-MATCH/EAY131/Findings/and/Publications n.d. https://ecog-acrin.org/nci-match-eay131-findings (accessed 10 September 2022).
- S:CORT . S-CORT Stratification in Colorectal Cancer n.d. https://www.s-cort.org (accessed 16 November 2022).
- The Beatson Institute . ACRCelerate n.d. https://www.beatson.gla.ac.uk/ACRCelerate/acrcelerate.html (accessed 16 November 2022).
- Catalogue of Somatic Mutations in Cancer . COSMIC v96, Released 31-May-22 n.d. http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/ (accessed 15 November 2022).
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009;9:550-62. https://doi.org/10.1038/nrc2664.
- Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A 2008;105:2652-7. https://doi.org/10.1073/pnas.0712169105.
- Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 2012;367:1596-606. https://doi.org/10.1056/NEJMoa1207756.
- Velho S, Oliveira C, Ferreira A, Ferreira AC, Suriano G, Schwartz S, et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer 2005;41:1649-54. https://doi.org/10.1016/j.ejca.2005.04.022.
- Day FL, Jorissen RN, Lipton L, Mouradov D, Sakthianandeswaren A, Christie M, et al. PIK3CA and PTEN gene and exon mutation-specific clinicopathologic and molecular associations in colorectal cancer. Clin Cancer Res 2013;19:3285-96. https://doi.org/10.1158/1078-0432.CCR-12-3614.
- Ogino S, Liao X, Imamura Y, Yamauchi M, McCleary NJ, Ng K, et al. Predictive and prognostic analysis of PIK3CA mutation in stage III colon cancer intergroup trial. J Natl Cancer Inst 2013;105:1789-98. https://doi.org/10.1093/jnci/djt298.
- Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 2010;28:1254-61. https://doi.org/10.1200/JCO.2009.24.6116.
- Karapetis CS, Jonker D, Daneshmand M, Hanson JE, O’Callaghan CJ, Marginean C, et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer – results from NCIC CTG/AGITG CO.17. Clin Cancer Res 2014;20:744-53. https://doi.org/10.1158/1078-0432.CCR-13-0606.
- Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 2013;10:143-53. https://doi.org/10.1038/nrclinonc.2013.10.
- Mishra R, Patel H, Alanazi S, Kilroy MK, Garrett JT. PI3K inhibitors in cancer: clinical implications and adverse effects. Int J Mol Sci 2021;22. https://doi.org/10.3390/ijms22073464.
- Giovannucci E, Rimm EB, Stampfer MJ, Colditz GA, Ascherio A, Willett WC. Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Ann Intern Med 1994;121:241-6. https://doi.org/10.7326/0003-4819-121-4-199408150-00001.
- Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010;376:1741-50. https://doi.org/10.1016/S0140-6736(10)61543-7.
- Sandler RS, Halabi S, Baron JA, Budinger S, Paskett E, Keresztes R, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med 2003;348:883-90. https://doi.org/10.1056/NEJMoa021633.
- Cole BF, Logan RF, Halabi S, Benamouzig R, Sandler RS, Grainge MJ, et al. Aspirin for the chemoprevention of colorectal adenomas: meta-analysis of the randomized trials. J Natl Cancer Inst 2009;101:256-66. https://doi.org/10.1093/jnci/djn485.
- Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol 2012;13:518-27. https://doi.org/10.1016/S1470-2045(12)70112-2.
- Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 2012;379:1591-601. https://doi.org/10.1016/S0140-6736(12)60209-8.
- Thun MJ, Jacobs EJ, Patrono C. The role of aspirin in cancer prevention. Nat Rev Clin Oncol 2012;9:259-67. https://doi.org/10.1038/nrclinonc.2011.199.
- Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer 2011;11:123-34. https://doi.org/10.1038/nrc3004.
- Köhne CH, Cunningham D, Di Costanzo F, Glimelius B, Blijham G, Aranda E, et al. Clinical determinants of survival in patients with 5-fluorouracil-based treatment for metastatic colorectal cancer: results of a multivariate analysis of 3825 patients. Ann Oncol 2002;13:308-17. https://doi.org/10.1093/annonc/mdf034.
- Karpatkin S, Pearlstein E, Ambrogio C, Coller BS. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J Clin Invest 1988;81:1012-19. https://doi.org/10.1172/JCI113411.
- Gasic GJ, Gasic TB, Stewart CC. Antimetastatic effects associated with platelet reduction. Proc Natl Acad Sci U S A 1968;61:46-52. https://doi.org/10.1073/pnas.61.1.46.
- Pearlstein E, Ambrogio C, Karpatkin S. Effect of antiplatelet antibody on the development of pulmonary metastases following injection of CT26 colon adenocarcinoma, Lewis lung carcinoma, and B16 amelanotic melanoma tumor cells into mice. Cancer Res 1984;44:3884-7.
- Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, et al. Tumor self-seeding by circulating cancer cells. Cell 2009;139:1315-26. https://doi.org/10.1016/j.cell.2009.11.025.
- ClinicalTrials.gov . Aspirin for Dukes C and High Risk Dukes B Colorectal Cancers (ASCOLT) n.d. https://clinicaltrials.gov/ct2/show/NCT00565708?term=NCT00565708%26draw=2%26rank=1 (accessed 16 November 2022).
- ClinicalTrials.gov . Add-Aspirin: A Trial Assessing the Effects of Aspirin on Disease Recurrence and Survival After Primary Therapy in Common Non Metastatic Solid Tumours n.d. https://clinicaltrials.gov/ct2/show/NCT02804815?term=NCT02804815%26draw=2%26rank=1 (accessed 16 November 2022).
- Domingo E, Church DN, Sieber O, Ramamoorthy R, Yanagisawa Y, Johnstone E, et al. Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. J Clin Oncol 2013;31:4297-305. https://doi.org/10.1200/JCO.2013.50.0322.
- Murphy C, Turner N, Wong H-L, Sinnathamby M, Tie J, Lee B, et al. Examining the impact of regular aspirin use and PIK3CA mutations on survival in stage 2 colon cancer. Intern Med J 2017;47:88-9. https://doi.org/10.1111/imj.13312.
- Reimers MS, Bastiaannet E, Langley RE, van Eijk R, van Vlierberghe RL, Lemmens VE, et al. Expression of HLA class I antigen, aspirin use, and survival after a diagnosis of colon cancer. JAMA Intern Med 2014;174:732-9. https://doi.org/10.1001/jamainternmed.2014.511.
- Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol 2009;10:501-7. https://doi.org/10.1016/S1470-2045(09)70035-X.
- Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009;373:1849-60. https://doi.org/10.1016/S0140-6736(09)60503-1.
- Burn J, Gerdes AM, Macrae F, Mecklin JP, Moeslein G, Olschwang S, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011;378:2081-7. https://doi.org/10.1016/S0140-6736(11)61049-0.
- Bhatt DL, Scheiman J, Abraham NS, Antman EM, Chan FK, Furberg CD, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol 2008;52:1502-17. https://doi.org/10.1016/j.jacc.2008.08.002.
- Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA 2009;302:649-58. https://doi.org/10.1001/jama.2009.1112.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74. https://doi.org/10.1016/j.cell.2011.02.013.
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010;40:179-204. https://doi.org/10.1016/j.molcel.2010.09.019.
- Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med 2019;381:317-27. https://doi.org/10.1056/NEJMoa1903387.
- Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2018;379:2495-505. https://doi.org/10.1056/NEJMoa1810858.
- Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005;5:689-98. https://doi.org/10.1038/nrc1691.
- Beck H, Nähse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol 2012;32:4226-36. https://doi.org/10.1128/MCB.00412-12.
- Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov 2012;2:524-39. https://doi.org/10.1158/2159-8290.CD-11-0320.
- Lheureux S, Cristea MC, Bruce JP, Garg S, Cabanero M, Mantia-Smaldone G, et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021;397:281-92. https://doi.org/10.1016/S0140-6736(20)32554-X.
- Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, et al. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol 2019;37:2643-50. https://doi.org/10.1200/JCO.19.00730.
- Molinari M. Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif 2000;33:261-74. https://doi.org/10.1046/j.1365-2184.2000.00191.x.
- Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 2004;3:513-19. https://doi.org/10.1158/1535-7163.513.3.4.
- Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res 2011;17:2799-806. https://doi.org/10.1158/1078-0432.CCR-10-2580.
- Murcia L, Clemente-Ruiz M, Pierre-Elies P, Royou A, Milán M. Selective killing of RAS-malignant tissues by exploiting oncogene-induced DNA damage. Cell Rep 2019;28:119-31.e4. https://doi.org/10.1016/j.celrep.2019.06.004.
- Fikaris AJ, Lewis AE, Abulaiti A, Tsygankova OM, Meinkoth JL. Ras triggers ataxia-telangiectasia-mutated and Rad-3-related activation and apoptosis through sustained mitogenic signaling. J Biol Chem 2006;281:34759-67. https://doi.org/10.1074/jbc.M606737200.
- Weisberg E, Nonami A, Chen Z, Liu F, Zhang J, Sattler M, et al. Identification of Wee1 as a novel therapeutic target for mutant RAS-driven acute leukemia and other malignancies. Leukemia 2015;29:27-3. https://doi.org/10.1038/leu.2014.149.
- Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol 2015;33:3409-15. https://doi.org/10.1200/JCO.2014.60.4009.
- Seymour MT, Maughan TS, Ledermann JA, Topham C, James R, Gwyther SJ, et al. Different strategies of sequential and combination chemotherapy for patients with poor prognosis advanced colorectal cancer (MRC FOCUS): a randomised controlled trial. Lancet 2007;370:143-52. https://doi.org/10.1016/S0140-6736(07)61087-3.
- Malla SB, Fisher DJ, Domingo E, Blake A, Hassanieh S, Redmond KL, et al. In-depth clinical and biological exploration of DNA damage immune response as a biomarker for oxaliplatin use in colorectal cancer. Clin Cancer Res 2021;27:288-300. https://doi.org/10.1158/1078-0432.CCR-20-3237.
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med 2020;383:1207-17. https://doi.org/10.1056/NEJMoa1917239.
- Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci 2016;129:1287-92. https://doi.org/10.1242/jcs.182873.
- de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 2020;382:2091-102. https://doi.org/10.1056/NEJMoa1911440.
- Mauri G, Arena S, Siena S, Bardelli A, Sartore-Bianchi A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann Oncol 2020;31:1135-47. https://doi.org/10.1016/j.annonc.2020.05.027.
- Adams R, Brown E, Brown L, Butler R, Falk S, Fisher D, et al. Inhibition of EGFR, HER2, and HER3 signalling in patients with colorectal cancer wild-type for BRAF, PIK3CA, KRAS, and NRAS (FOCUS4-D): a phase 2–3 randomised trial. Lancet Gastroenterol Hepatol 2018;3:162-71. https://doi.org/10.1016/S2468-1253(17)30394-1.
- Kapitanović S, Radosević S, Slade N, Kapitanović M, Andelinović S, Ferencić Z, et al. Expression of erbB-3 protein in colorectal adenocarcinoma: correlation with poor survival. J Cancer Res Clin Oncol 2000;126:205-11. https://doi.org/10.1007/s004320050034.
- Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 2009;9:463-75. https://doi.org/10.1038/nrc2656.
- Scartozzi M, Mandolesi A, Giampieri R, Bittoni A, Pierantoni C, Zaniboni A, et al. The role of HER-3 expression in the prediction of clinical outcome for advanced colorectal cancer patients receiving irinotecan and cetuximab. Oncologist 2011;16:53-60. https://doi.org/10.1634/theoncologist.2010-0119.
- Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008;27:3944-56. https://doi.org/10.1038/onc.2008.19.
- Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007;445:437-41. https://doi.org/10.1038/nature05474.
- Seligmann JF, Hatch AJ, Richman SD, Elliott F, Jacobs B, Brown S, et al. Association of tumor HER3 messenger RNA expression with panitumumab efficacy in advanced colorectal cancer. JAMA Oncol 2018;4:564-8. https://doi.org/10.1001/jamaoncol.2017.3168.
- Richman SD, Southward K, Chambers P, Cross D, Barrett J, Hemmings G, et al. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J Pathol 2016;238:562-70. https://doi.org/10.1002/path.4679.
- Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts (‘xenopatients’) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 2011;1:508-23. https://doi.org/10.1158/2159-8290.CD-11-0109.
- Hickinson DM, Klinowska T, Speake G, Vincent J, Trigwell C, Anderton J, et al. AZD8931, an equipotent, reversible inhibitor of signaling by epidermal growth factor receptor, ERBB2 (HER2), and ERBB3: a unique agent for simultaneous ERBB receptor blockade in cancer. Clin Cancer Res 2010;16:1159-69. https://doi.org/10.1158/1078-0432.CCR-09-2353.
- Seligmann JF, Fisher D, Smith CG, Richman SD, Elliott F, Brown S, et al. Investigating the poor outcomes of BRAF-mutant advanced colorectal cancer: analysis from 2530 patients in randomised clinical trials. Ann Oncol 2017;28:562-8. https://doi.org/10.1093/annonc/mdw645.
- Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, et al. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer 2015;112:1888-94. https://doi.org/10.1038/bjc.2015.173.
- Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 2007;97:1139-45. https://doi.org/10.1038/sj.bjc.6604009.
- Koopman M, Antonini NF, Douma J, Wals J, Honkoop AH, Erdkamp FL, et al. Sequential versus combination chemotherapy with capecitabine, irinotecan, and oxaliplatin in advanced colorectal cancer (CAIRO): a phase III randomised controlled trial. Lancet 2007;370:135-42. https://doi.org/10.1016/S0140-6736(07)61086-1.
- Hegewisch-Becker S, Graeven U, Lerchenmüller CA, Killing B, Depenbusch R, Steffens CC, et al. Maintenance strategies after first-line oxaliplatin plus fluoropyrimidine plus bevacizumab for patients with metastatic colorectal cancer (AIO 0207): a randomised, non-inferiority, open-label, phase 3 trial. Lancet Oncol 2015;16:1355-69. https://doi.org/10.1016/S1470-2045(15)00042-X.
- Good Clinical Practice Network . ICH Harmonised Guideline Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice ICH E6(R2) n.d. https://ichgcp.net (accessed 16 November 2022).
- National Cancer Institute . Common Terminology Criteria for Adverse Events v3.0 (CTCAE) n.d. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf (accessed 16 November 2022).
- Royston P, Parmar MK. Restricted mean survival time: an alternative to the hazard ratio for the design and analysis of randomized trials with a time-to-event outcome. BMC Med Res Methodol 2013;13. https://doi.org/10.1186/1471-2288-13-152.
- Barthel FMS RP, Parmar MKB. A menu-driven facility for sample-size calculation in novel multi-arm, multi-stage randomized controlled trials with a time-to-event outcome. Stata J 2009;9:505-23. https://doi.org/10.1177/1536867X0900900401.
- Austin PC. Variance estimation when using inverse probability of treatment weighting (IPTW) with survival analysis. Stat Med 2016;35:5642-55. https://doi.org/10.1002/sim.7084.
- Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika 1994;81:515-26. https://doi.org/10.1093/biomet/81.3.515.
- Johnston S, Basik M, Hegg R, Lausoontornsiri W, Grzeda L, Clemons M, et al. Inhibition of EGFR, HER2, and HER3 signaling with AZD8931 in combination with anastrozole as an anticancer approach: Phase II randomized study in women with endocrine-therapy-naïve advanced breast cancer. Breast Cancer Res Treat 2016;160:91-9. https://doi.org/10.1007/s10549-016-3979-5.
- Baselga J, Hegg R, Vidal Losad M, Vidaurre T, Lluch A, Petrakova K, et al. Abstract LB-146: a phase II randomized placebo-controlled study of AZD8931, an inhibitor of EGFR, HER2, and HER3 signaling, plus paclitaxel (P) vs P alone in patients (pts) with low HER2-expressing advanced breast cancer (BC) (THYME). Cancer Res 2013;73.
- Adams R, Fisher DJ, Graham J, Seligmann JF, Seymour M, Kaplan R, et al. Capecitabine versus active monitoring in stable or responding metastatic colorectal cancer after 16 weeks of first-line therapy: results of the randomized FOCUS4-N trial. J Clin Oncol 2021;39:3693-704. https://doi.org/10.1200/JCO.21.01436.
- NCCN . NCCN Guidelines for Patients: Colon Cancer. n.d. www2.tri-kobe.org/nccn/guideline/colorectal/english/colon.pdf (accessed 31 January 2022).
- Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27:1386-422. https://doi.org/10.1093/annonc/mdw235.
- Simkens LH, van Tinteren H, May A, ten Tije AJ, Creemers GJ, Loosveld OJ, et al. Maintenance treatment with capecitabine and bevacizumab in metastatic colorectal cancer (CAIRO3): a phase 3 randomised controlled trial of the Dutch Colorectal Cancer Group. Lancet 2015;385:1843-52. https://doi.org/10.1016/S0140-6736(14)62004-3.
- Sherman SK, Lange JJ, Dahdaleh FS, Rajeev R, Gamblin TC, Polite BN, et al. Cost-effectiveness of maintenance capecitabine and bevacizumab for metastatic colorectal cancer. JAMA Oncol 2019;5:236-42. https://doi.org/10.1001/jamaoncol.2018.5070.
- Sonbol MB, Mountjoy LJ, Firwana B, Liu AJ, Almader-Douglas D, Mody K, et al. The role of maintenance strategies in metastatic colorectal cancer: a systematic review and network meta-analysis of randomized clinical trials. JAMA Oncol 2020;6. https://doi.org/10.1001/jamaoncol.2019.4489.
- Adams R, Goey K, Chibaudel B, Koopman M, Punt C, Arnold D, et al. Treatment breaks in first line treatment of advanced colorectal cancer: an individual patient data meta-analysis. Cancer Treat Rev 2021;99. https://doi.org/10.1016/j.ctrv.2021.102226.
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228-47. https://doi.org/10.1016/j.ejca.2008.10.026.
- Hague D, Townsend S, Masters L, Rauchenberger M, Van Looy N, Diaz-Montana C, et al. Changing platforms without stopping the train: experiences of data management and data management systems when adapting platform protocols by adding and closing comparisons. Trials 2019;20. https://doi.org/10.1186/s13063-019-3322-7.
- Schiavone F, Bathia R, Letchemanan K, Masters L, Amos C, Bara A, et al. This is a platform alteration: a trial management perspective on the operational aspects of adaptive and platform and umbrella protocols. Trials 2019;20. https://doi.org/10.1186/s13063-019-3216-8.
- Morrell L, Hordern J, Brown L, Sydes MR, Amos CL, Kaplan RS, et al. Mind the gap? The platform trial as a working environment. Trials 2019;20. https://doi.org/10.1186/s13063-019-3377-5.
- Antoniou M, Kolamunnage-Dona R, Wason J, Bathia R, Billingham C, Bliss JM, et al. Biomarker-guided trials: challenges in practice. Contemp Clin Trials Commun 2019;16. https://doi.org/10.1016/j.conctc.2019.100493.
- Maughan TS, Meade AM, Adams RA, Richman SD, Butler R, Fisher D, et al. A feasibility study testing four hypotheses with phase II outcomes in advanced colorectal cancer (MRC FOCUS3): a model for randomised controlled trials in the era of personalised medicine?. Br J Cancer 2014;110:2178-86. https://doi.org/10.1038/bjc.2014.182.
- Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350-6. https://doi.org/10.1038/nm.3967.
- Feiler T, Gaitskell K, Maughan T, Hordern J. Personalised medicine: the promise, the hype and the pitfalls. New Bioeth 2017;23:1-12. https://doi.org/10.1080/20502877.2017.1314895.
- Hordern J, Maughan T, Feiler T, Morrell L, Horne R, Sullivan R. The ‘molecularly unstratified’ patient: a focus for moral, psycho-social and societal research. Biomed Hub 2017;2:146-53. https://doi.org/10.1159/000480422.
List of abbreviations
- AKT
- protein kinase B
- CI
- confidence interval
- COIN
- COntinuous or Intermittent
- CONSORT
- Consolidated Standards of Reporting Trials
- CRC
- colorectal cancer
- CRUK
- Cancer Research UK
- CT
- computed tomography
- CTU
- clinical trials unit
- DDR
- DNA damage response
- DNA
- deoxyribonucleic acid
- EGFR
- epidermal growth factor receptor
- EME
- Efficacy and Mechanism Evaluation
- EQ-5D
- EuroQol-5 Dimensions
- FFPE
- formalin-fixed paraffin embedded
- HER
- human epidermal growth factor receptor
- HR
- hazard ratio
- IDMC
- Independent Data Monitoring Committee
- IHC
- immunohistochemistry
- IPD
- individual patient data
- IQR
- interquartile range
- ITT
- intention to treat
- MAMS
- multiarm, multistage
- mCRC
- metastatic colorectal cancer
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MMR
- mismatch repair
- MRC
- Medical Research Council
- MSI
- microsatellite instability
- mTOR
- mammalian target of rapamycin
- NCI
- National Cancer Institute
- NGS
- next-generation sequencing
- NIHR
- National Institute for Health and Care Research
- NSAID
- non-steroidal anti-inflammatory drug
- OS
- overall survival
- PFS
- progression-free survival
- PTEN
- phosphatase and tensin homologue
- PTL
- primary tumour location
- RCT
- randomised controlled trial
- RECIST
- response evaluation criteria in solid tumours
- SAP
- statistical analysis plan
- TMA
- tissue microarray
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
- UCL
- University College London
- ULN
- upper limit of normal
- WHO
- World Health Organization
Notes
-
Participating hospitals in descending order of number of patients registered with all staff listed ( n = 2076)
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/HTNB6908).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.