

Notes
Article history
This article presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
Permissions
Copyright statement
Copyright © 2024 Ives et al. This work was produced by Ives et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Ives et al.
Chapter 1 Introduction
Clinical background
Shiga-toxin-producing Escherichia coli haemolytic uraemic syndrome (STEC HUS) is the most common single cause of paediatric acute kidney injury (AKI), and affects approximately 100 UK children each year. 1 The hallmark features of HUS are a triad of microangiopathic haemolytic anaemia, thrombocytopenia and AKI. 2 This clinical presentation occurs due to acute thrombotic microangiopathy (TMA), most commonly in the renal microvasculature. STEC HUS has a 2–3% mortality rate and considerable morbidity, with 50–60% of children requiring renal replacement therapy (RRT). 1 Approximately 20–25% of children with STEC HUS develop severe disease with extrarenal involvement, including colonic necrosis and perforation (requiring laparotomy and bowel resection), central nervous system (CNS) disturbance including seizures, focal neurological defects and coma, pancreatitis (including temporary or permanent glucose intolerance) and myocardial dysfunction (including infarction). 3 While not as common as renal involvement, neurological dysfunction represents the major cause of mortality in HUS. 4–7
Luna et al. described the phenotype of the most critically ill children with STEC HUS presenting to a single centre in Argentina over a 15-year period. 8 From a total cohort of 362 patients, the report focuses on the 44 patients (12%) with severe disease. These were patients who had required admission to the paediatric intensive care unit (PICU) within 2 days of presentation for indications including haemodynamic instability, and severe multiorgan involvement (one or more of the following conditions: major neurological involvement, serious gastrointestinal, cardiovascular, respiratory complications and/or sepsis). Most of these critically ill patients (84%) received therapeutic plasma exchange. The mortality rate was 12/362 (3.3%) in the whole cohort, and all those who died were from the critically ill group (12/44; 27%). The 32 surviving critically ill patients were followed up prospectively. Eleven of the 32 (34%) survivors had significant neurological sequelae (3% of the whole cohort), and 5 (16%) had reached end-stage kidney disease at last follow-up – no later than 15 years of age (1.4% of the whole cohort).
This and other reports reveal that long-term complications such as chronic kidney disease (CKD) or more rarely permanent brain injury occur in up to one-third of survivors of STEC HUS. 9 A meta-analysis demonstrated that 12% of patients with STEC HUS died or developed end-stage kidney disease by 4.4 years of follow-up, with long-term sequelae [hypertension, proteinuria, impaired glomerular filtration rate (GFR)] in approximately 25% of survivors. 9 Consequently, all cases require lifelong renal follow-up.
Pathophysiology of Shiga-toxin-producing Escherichia coli haemolytic uraemic syndrome
Shiga-toxin-producing E. coli infection usually occurs as a result of ingestion of contaminated food or water. 10 STEC colonise the intestinal mucosa, adhere to colonic enterocytes and produce Shiga toxin (stx). 11 The main cellular target for stx is the Globotriaosylceramide (Gb3) receptor located on the microvascular endothelium within the brain, gut and kidney. 12 Once bound to Gb3, stx enters the cell and inhibits ribosomal activity, leading to activation of apoptotic pathways, induction of inflammatory cytokines and cellular necrosis. 12 All these processes lead to the generation of a pro-inflammatory environment within the microvasculature.
The field of HUS has been transformed through the delineation of causative genes for the closely related condition, atypical haemolytic uraemic syndrome (aHUS). 13 aHUS describes patients with HUS without STEC infection, approximately 60% of whom have defects of the alternative complement pathway.
The alternative complement pathway, part of the innate immune system, is described further in Chapter 4. 14 Following activation of the pathway by cleavage of the complement protein C3, the final product is a pore-like structure, the membrane attack complex (MAC), which is a complex of complement proteins C5, C6, C7, C8 and C9. This structure forms a permeable pore in the cell membrane leading to cell lysis. Damage to host cells from over-activity of the pathway is prevented by regulator proteins. Failure of complement regulation is the key pathogenic factor in the development of aHUS.
The role of complement in Shiga-toxin-producing Escherichia coli haemolytic uraemic syndrome
While there is clear evidence that stx mediates glomerular endothelial TMA,15 there is increasingly compelling evidence that complement plays a role in the pathogenesis of STEC HUS. Complement activation was first observed in STEC HUS over 30 years ago, when it was demonstrated that children with STEC HUS had higher plasma levels of alternative complement activation products. 16,17 Patients with STEC HUS may exhibit transiently low plasma complement C3 levels during acute disease, which return to normal during convalescence, indicating complement activation and consumption. Low admission plasma C3 levels have repeatedly been shown to correlate with several measures of disease severity including dialysis requirement, neurological and other extrarenal complications, PICU admission and length of hospitalisation. 18–20 Adding to evidence for complement activation, serum complement activation products are elevated in the acute phase and correlate with disease severity. 21–23 Further evidence of complement involvement in STEC HUS is supported by the presence of circulating complement-containing microvesicles from platelets, leucocytes and erythrocytes in individuals with STEC HUS,24,25 suggesting a direct interaction between these cells and complement.
Results from animal studies are mixed. In a murine model of STEC HUS, complement blockade was protective against severe disease. 26 In contrast, no evidence of complement activation was detected in a nonhuman primate model of STEC HUS. 27 A mouse model that recapitulates STEC HUS was recently developed by targeting stx to the glomerular podocyte by exclusively expressing the Gb3 receptor on this cell type. 28 In this model, inhibition of the terminal complement pathway by C5 blockade prior to stx exposure prevented the development of STEC HUS, although no data is currently available regarding rescue C5 inhibition treatment.
Treatment of Shiga-toxin-producing Escherichia coli haemolytic uraemic syndrome
Children with confirmed or suspected STEC HUS are managed with supportive therapy (as reviewed by Walsh),2 including blood transfusion, nutritional support, RRT and PICU support if required. Despite numerous attempts, many previous studies have failed to demonstrate improved short-term or long-term outcomes in STEC HUS with interventions such as anticoagulation, plasma infusion, corticosteroids or oral therapy with a stx-binding agent. 29 Therapeutic plasma exchange has been used to treat severe STEC HUS, usually with extrarenal manifestations, based upon the rationale that it might remove proinflammatory cytokines and prothrombotic factors. 7,8,30–33 However, no definitive answers concerning its efficacy can be given within the available evidence. There is emerging evidence that early volume expansion with 0.9% saline may reduce the incidence of oligoanuria in STEC HUS and the need for RRT. 34–37
Eculizumab in Shiga-toxin-producing Escherichia coli haemolytic uraemic syndrome
Eculizumab is a humanised monoclonal antibody that binds to C5, preventing formation of the MAC. 38 Eculizumab was first approved for use in aHUS in 2011. Eculizumab is highly effective for the treatment of aHUS in both adults and children39–41 with transformational outcomes. Given the evidence for complement in the pathogenesis in STEC HUS, several authors have reported the experimental use of eculizumab in STEC HUS. In May 2011, Lapeyraque et al. described the use of eculizumab in three children with STEC HUS who had severe CNS involvement,42 all of whom showed dramatic resolution of CNS symptoms.
Including that 2011 report, a total of 30 publications have reported the use of eculizumab in STEC HUS outside of randomised controlled trials3,19,32,33,42–67 including 1 prospective study,46 10 retrospective cohort studies,19,32,43,44,48,50,58,59,62,63 2 retrospective case control studies,33,61 8 case series3,42,49,55,57,60,65,67 and 9 individual case reports. 45,47,51–54,63,64,66 Together, the 30 reports contain details of the use of eculizumab in 450 patients with a diagnosis of confirmed or suspected STEC HUS. From careful analysis of the papers, it is likely that five of these patients (all children) are doubly reported – three patients within two overlapping cohorts have the same age, STEC serotype and neurological score,19,58 and two patients were reported within a paper focusing on neurological involvement,55 who had previously been reported in an earlier paper. 48 Therefore, data are available on 445 patients.
Of these 445 patients, 307 were adults, including 268 who were among over 800 cases of STEC HUS that occurred in several European countries, mainly Germany, in 2011. 32,33,44 This outbreak was later found to be caused by a novel STEC serotype (H4:O104). Most of these adult patients (n = 198) were treated as part of a single-arm trial of the safety and efficacy of eculizumab in STEC HUS, which was rapidly convened by the drug manufacturer, with participants receiving up to six doses of eculizumab (www.clinicaltrials.gov/ct2/show/NCT01410916). The results of this trial were not published as a single report – instead data on most of the patients are contained within several reports. 32,33,44,48,49,55 These reports also contain data on patients who were treated off-label with eculizumab, which was provided for compassionate use by the manufacturer outside of the clinical trial, and it is not always possible to separate those within the industry trial from those treated off-label. The indications for treatment of patients with eculizumab were broad and varied; however, it was often given after the development of neurological symptoms or other extrarenal manifestations. Reporting of these patients focussed mainly upon survival in the acute phase.
In the largest study to emerge from the outbreak, Kielstein et al. performed a retrospective cohort analysis in 491 patients comparing the effectiveness of best supportive care (57 patients) with therapeutic plasma exchange (241 patients) and therapeutic plasma exchange with eculizumab (193 patients). 32 The authors used propensity scoring to address differences in disease severity between treatment groups. They found no significant difference in survival, neurological and renal outcomes between the three groups. In another study from the 2011 outbreak, Menne et al. performed a retrospective multicentre case control study on 298 adult patients with STEC-HUS, including 67 patients treated with eculizumab. 33 These patients were compared with a control group of 65 patients with similar disease severity who did not receive eculizumab. No statistically significant difference was noted between the groups for improvements in platelet count, lactate dehydrogenase (LDH), creatinine or haemoglobin. The rate of complications – including hypertension, chronic renal impairment, diabetes mellitus and neurological and psychiatric disease – was also similar between the two groups. These studies had several limitations – patients were not randomised and the timing of eculizumab administration was highly variable, often over a week after the onset of HUS. Long-term outcome data were also not presented. With these caveats, the data from these largest retrospective studies suggested that treatment with eculizumab did not result in significant improvement in haematological, renal or neurological outcome in adult patients with STEC HUS. In addition to the adult patients reported in these studies, 14 children who received eculizumab during the same outbreak have also been reported,44,48,49,55 most of whom were treated with eculizumab because of severe neurological involvement.
Although the data from the large studies that arose from the German outbreak did not suggest improved outcomes with eculizumab treatment, its continued use has been reported in both adults and children with severe STEC HUS. To date, the use of eculizumab has been reported in 138 children with STEC HUS in 26 papers outside of randomised controlled trials and these are summarised in Table 1.
| Author | Date | Patients < 18 years (n) | Indication for eculizumab | Eculizumab regime | STEC serotype (n) | Eculizumab firstline? | Died | CNS sequelae | Renal sequelae at last follow-upa | Cholestasis | Infections |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lapeyraque | 2011 | 3 | CNS | Not reported | Not specified | 1/2 | 0/3 | 0/3 | 2/3 | Not reported | Not reported |
| Gitiaux | 2013 | 7 | CNS | Not reported | O157 (3); O121 (2): O26 (1) | 5/7 | 2/7 | 0/5 | 3/5 | Not reported | Not reported |
| Loos, Bauer | 2012, 2014 | 13 | CNS | As per 2011 industry trial | O104 | 6/13 | 1/13 | 8/9 | Not reported | Not reported | Not reported |
| Delmas | 2014 | 1 | Main organ involvementb | As per 2011 industry trial | O104 | 1/1 | 0/1 | 0/1 | 0/1 | Not reported | Not reported |
| Ekinci | 2014 | 2 | CNS | As per 2011 industry trial | Not specified | 1/2 | 0/2 | 0/2 | 2/2 | Not reported | Not reported |
| Pape | 2015 | 11 (9)c | CNS | As per 2011 industry trial (2)d | Not specified | 11/11: 9/9e | 1/11: 1/9e | 2/10: 2/8e | Not reported | Not reported | Not reported |
| Saini | 2015 | 1 | CNS | Multiple doses | O157 | 0/1 | 1/1 | N/A | N/A | Not reported | Not reported |
| Wijnsma | 2017 | 1 | Suspected aHUS | Single dose | O80 | 1/1 | 1/1 | N/A | N/A | 1/ | Not reported |
| Rasa | 2017 | 1 | CNS | 3 doses | O157 | 1/1 | 0/1 | 0/1 | 1/1 | Not reported | Not reported |
| Matthies | 2016 | 2 | CNS (1) Cardiac (1) | Not reported | Not specified | 1/2 | 0/2 | 1/2 | 1/1 | 1/1f | Not reported |
| Percheron | 2018 | 33 | Severe STEC HUSg | Variable guided by CH50 | Not specified | 21/33 | 4/33 | 5/24 | 12/19h | Not reported | Staphylococcal septicemia (1) severe chicken pox (1) |
| Agbas | 2018 | 9 | Prolonged anuria and severe haematological or extrarenal involvement | Not reported | O104 and O157i | 9/9 | 1/9 | 8/8 | 5/8 | Not reported | Sepsis due to Gram-negative bacillusj |
| Keenswijk | 2018 | 1 | CNS | Single dose | O157 | 0/1 | 0/1 | 1/1 | 0/1 | Not reported | Not reported |
| Giordano | 2019 | 5 | CNS | 2 doses | O26 (3); O111 (2) | Not specified | 0/5 | 1/5 | 1/5 | Not reported | Not reported |
| Mauras | 2019 | 3 | CNS (2) Suspected aHUS (1) | 1, 2 or 3 doses | O26 (2); O145/O80 (1) | 3/3 | 0/3 | 0/3 | 3/3 | 3/3k | Not reported |
| Konopasek | 2022 | 4 | CNS | Single dose | O157 (1); O26 (1); nt (2) | 4/4 | 1/4 | 0/3 | 2/3 | Not reported | Not reported |
| Netti | 2020 | 10 (7)C | CNS | Not reported | O111 (2 : 1)e; O26 (5 : 3)e; O157 (2); O103 (1) | 5/10: 3/7e | 1/10: 1/7e | Not reported | Not reported | Not reported | Not reported |
| Monet-Didailler | 2020 | 18 | Severe STEC HUSl | As per 2011 industry trial up to 5 doses | Not specified | 18/18 | 0/18 | 4/10 | 13/13 | Not reported | Not reported |
| Costigan | 2022 | 8 | CNS | Not reported | Not specified | 5/8 | 1/8 | 1/7 | Not reported | Not reported | Not reported |
| Balestracci | 2021 | 1 | CNS | Single dose | O157 | 1/2 | 0/1 | 0/1 | Not reported | Not reported | Not reported |
| Yesilbas | 2021 | 1 | Cardiac | Single dose | Not specified | 0/1 | 0/1 | 0/1 | Not reported | 1/1m | Not reported |
| Umscheid | 2021 | 1 | CNS | 8 weeks | O157 | 1/1 | 0/1 | 0/1 | 1/1 | 0/1 | Not reported |
| Santangelo | 2021 | 2 | CNS | 2 doses | O111 (2) | 2/2 | 0/2 | 2/2 | Not reported | 0/2 | Not reported |
| Weber | 2021 | 1 | CNS | 2 doses | Not specified | 1/1 | 0/1 | 1/1 | Not reported | Not reported | Fever |
| Wildes | 2022 | 1 | CNS | 2 doses | O26 (2); O45 (2) | 4/4 | 0/4 | 0/4 | 0/4 | Not reported | Not reported |
In these children, the indication for eculizumab treatment was purely CNS involvement in 79/138 patients, purely cardiac involvement in 1/138 patient and multisystem TMA (not always specified and including severe haemolysis in some) in 52/138 patients. Two patients received eculizumab due to a possible diagnosis of aHUS and one patient had HUS with no specific features of severity. The indication was unknown in 3/138 patients. E. coli serotypes were: O157 in 11 patients, O104 in 14 patients; O26 in 12 patients; O111 in 5 patients; O121 in 2 patients; other in 5 patients and the serotype was unknown or not specified in 89 patients. The eculizumab dosing regimen given to the patients was highly variable. Forty-five patients received eculizumab according to the regime in the industry-sponsored trial, but some of these received a shortened course if the haematological features settled. Fourteen patients had a single dose, 13 patients had 2 doses, 2 patients had 3 doses, one patient received 8 weeks' treatment and 28 patients had a regime of multiple doses (up to 9 infusions) guided by haemolytic activity (CH50). The regime was not stated in 37 patients. Ninety-eight of the 138 patients received eculizumab as the first line treatment, while 35 patients received eculizumab following a variable length course of therapeutic plasma exchange. In five patients this information was not known.
Following eculizumab treatment for STEC HUS, 14/138 (10.1%) children are reported to have died. Of the surviving 124 patients, CNS outcome was unknown in 25 patients. For those in whom the outcome was known, 77/99 (77.8%) had a normal CNS outcome following treatment with eculizumab and 22/99 (22.2%) had an abnormal CNS outcome. It should be noted that not all of the patients who received eculizumab had CNS involvement, but the data do not permit further examination. As in the first report on the use of eculizumab,42 it is noteworthy in several reports that the improvement in neurological symptoms occurred rapidly following the first dose of eculizumab. 49,52,54,57,59
Data regarding renal outcome were reported for only 57 of the 124 surviving patients. In these 57 patients, there were no renal sequelae at last follow-up in 24 patients (42.1%) and 33 patients (57.9%) had CKD, proteinuria or hypertension, including 2/57 (3.5%) who were known to have developed end-stage kidney disease at last follow-up.
The two largest paediatric studies discussed above are presented in more detail below. 50,61 The largest paediatric cohort to date involved 33 children with STEC HUS treated with eculizumab at 15 French centres. 50 The authors divided patients based upon their outcome; favourable (n = 15) and unfavourable (n = 18) – meaning death or persistent neurological abnormalities or elevated pancreatic enzymes or estimated glomerular filtration rate (eGFR) < 60 ml/minute/1.73 m2 or proteinuria > 0.1 g/mmol at last follow‐up. Baseline characteristics were comparable. The main finding was that patients who reached complete complement blockade (assessed by CH50 activity prior to the second dose of eculizumab) were more likely to be in the favourable outcome group compared with those who did not reach complete blockade. However, this did not reach statistical significance (p = 0.12), which investigators contributed to the small size of the cohort.
Monet-Didailler reported 18 children treated with eculizumab for STEC HUS in a single centre and compared them with a historical matched control group (for age, sex, severity of renal impairment) of 36 children who did not receive eculizumab. 61 The groups were not matched with regard to neurological, cardiac and gastrointestinal complications. Indications for use of eculizumab included severe neurological involvement, cardiac injury, pulmonary oedema, severe pancreatitis and severe enterocolitis associated with persistent renal failure despite 4 days of RRT. After both short-term (1 month) and longer-term (12 months) follow-up, the evolution of haematological and renal parameters did not differ between the groups. Four of 10 children with CNS involvement who were treated with eculizumab showed long-term neurological sequelae compared to one of nine untreated patients. The authors concluded that it was not possible to determine the efficacy of eculizumab because of the retrospective observational design of the study.
In summary, the published evidence from large case-control and cohort studies in adult patients does not suggest that eculizumab improves haematological, renal or neurological outcome in STEC HUS. In paediatric patients, a number of small cohorts, case reports and case series do report spontaneous resolution of clinical symptoms, but the absence of controlled studies makes it difficult to interpret whether this improvement in clinical status is due to administration of eculizumab or due to the highly variable natural history of STEC HUS, including spontaneous resolution of clinical symptoms.
Safety of eculizumab in children with Shiga-toxin-producing Escherichia coli haemolytic uraemic syndrome
Eculizumab is licensed for children ≥ 5 kg with aHUS. The eculizumab doses administered within the Eculizumab in Shiga-toxin-producing Escherichia coli Haemolytic Uraemic Syndrome (ECUSTEC) trial were the same as those given in the Summary of Product Characteristics for aHUS. 68 As such, eculizumab for STEC HUS was a re-purposed intervention and the safety profile in the trial age group was well established. According to pharmacokinetic data, two doses of eculizumab result in complement blockade for at least 14 days, which corresponds to the timing of complement activation in STEC HUS. 22 Eculizumab is currently indicated for chronic administration in aHUS. Since STEC HUS is an acute disorder, and evidence shows that complement activation is transient,22 there is no rationale for chronic administration.
The most important side effect of treatment with eculizumab is an increase in the risk of infection with encapsulated organisms, in particular Neisseria meningitidis (meningococcus). Long-term pharmacovigilance has quantified this risk as approximately 0.25 cases of meningococcal disease per 100 patient years of eculizumab use. 69
Generally, there have been few safety concerns following the use of eculizumab in STEC HUS. However, of the 138 children who have been reported to have received eculizumab in STEC HUS outside of clinical trials, 6 have developed significant cholestasis. 3,53,60,63 Of these six patients, three have subsequently undergone liver transplant,3,60 and one died from liver failure. 53 Pathological examination of the native liver in one of these cases found TMA, but the authors could not completely rule out that eculizumab may have worsened the liver lesions. 60 Interestingly, a 2-year-old child with STEC HUS (serotype O157) was reported to have developed secondary sclerosing cholangitis and portal hypertension as a late complication, without use of eculizumab. 70 Serious infections have been reported in three patients (including one death from Gram-negative bacterial sepsis,43 one case of staphylococcal septicaemia and one case of severe varicella zoster infection). 50 No cases of meningococcal disease in patients with STEC HUS treated with eculizumab have been reported.
Rationale for the ECUSTEC trial
The use of eculizumab for the treatment of severe STEC HUS has been increasing internationally, without objective evidence of efficacy or safety, and at a significant cost to the NHS and other healthcare systems. A single dose of eculizumab costs between £3000 and £9000 (although the availability of biosimilar drugs may reduce this cost), and most publications have reported a multiple dose regimen. Until recently, there were no published prospective, controlled evaluations of eculizumab in STEC HUS. There was therefore a need to evaluate the efficacy and safety of eculizumab in children with STEC HUS in a prospective randomised controlled trial (RCT). Two recent trials have attempted to do this – the UK ECUSTEC trial (reported here) and the French Eculizumab in Shiga-toxin Related Hemolytic and Uremic Syndrome Paediatric Patients (ECULISHU) trial. The trials ran simultaneously and the results of the ECULISHU trial have recently been reported. 71 In the phase 3 RCT, 100 children were randomised to treatment with eculizumab or placebo. Patients with severe multi-organ involvement were excluded. Four patients in the placebo group were withdrawn and subsequently received eculizumab. There was no difference between treatment groups in the proportion of children who required renal replacement therapy 48 hours after randomisation. In addition, no differences between groups were seen in the secondary outcome measures of extra-renal manifestations, duration of hospitalisation and mortality. During follow-up, there was a slight difference in the proportion who exhibited renal sequelae at 12 months post-randomisation (20 patients in the eculizumab group (43.48%) and 29 patients (64.44%) in the placebo group (P = 0.04). The authors concluded that eculizumab seemed to have no impact on the course of AKI in this cohort of patients with relatively mild disease and interpreted the 12-month follow-up data with caution. No data have been published which include the role of eculizumab in patients with severe manifesations of disease in an RCT setting.
There are several important differences in the study design of the ECUSTEC and ECULISHU trials as shown in Table 2. ECULISHU was a single-blind RCT that examined whether giving three to five doses of eculizumab reduced the severity of renal disease in children with STEC HUS, but without extrarenal involvement. In contrast, ECUSTEC is a double-blind RCT comparing two doses of eculizumab (as per the first two doses of the aHUS schedule) in patients with severe extrarenal disease. In both trials, eculizumab was administered early in the disease course (corresponding with the peak of complement activation according to experimental data). This is in contrast with many of the previous retrospective studies where eculizumab was administered late in the disease process and after other therapeutic strategies. In 2013, the Clinical Studies Group of the British Association for Paediatric Nephrology, including parent representatives, identified eculizumab in STEC HUS as one of its highest research priorities.
| ECULISHU | ECUSTEC | |
|---|---|---|
| Blinding of parents/carers and clinical team | Single blinded | Double blinded |
| Extrarenal manifestations | Not eligible | Eligible |
| Number of doses of eculizumab | Three to five | Two |
| Eculizumab for patients in control group who develop severe disease | Yes | No |
| Primary outcome measure | Renal only – absence of dialysis requirement 48 hours after randomisation | Multidomain CSS |
Clinical research questions
In children aged 6 months–< 19 years:
-
Does eculizumab reduce the severity of STEC HUS compared with placebo?
-
What is the safety profile of eculizumab in STEC HUS?
-
Does eculizumab reduce the incidence of CKD following STEC HUS compared with placebo?
-
To evaluate the cost-effectiveness of administration of eculizumab in STEC HUS from the perspective of the NHS.
Mechanistic substudy research questions
-
What is the time course of systemic complement activation in STEC HUS and is it related to the severity of disease?
-
Does TMA in STEC HUS occur via a stx-mediated reduction in podocyte vascular endothelial growth factor (VEGF) production, leading to loss of complement regulation?
-
Do STEC HUS patient neutrophils deliver stx to podocytes?
-
Are there genetic variations in patients with STEC HUS that point to novel pathogenic mechanisms?
Chapter 2 Methods
Trial design
The trial was a multicentre randomised, double-blind, placebo-controlled, parallel-group trial of two doses of eculizumab in children (aged 6 months–< 19 years) with STEC HUS. The trial had an internal pilot phase and included nested mechanistic laboratory studies and a cost-effectiveness elevation, although the latter was not undertaken following the trial being stopped early. The trial had a favourable ethics opinion from the North East – Newcastle and North Tyneside 1 Research Ethics Committee (REC reference number 16/NE/0325, date of approval 23/01/2017). The full trial protocol can be accessed at www.birmingham.ac.uk/research/bctu/trials/renal/ecustec/index.aspx (last accessed 13 July 2022). A summary of protocol amendments during the course of the trial is given in Appendix 1.
Internal pilot and stopping rules
The trial included an internal pilot phase of 18 months (12 months recruitment, 6 months follow-up), the purpose of which was to determine whether the substantive trial would continue. At the end of the pilot phase, the following progression rules were used to guide the decision process as to whether the trial continued:
-
that 26 participants were recruited in 12 months
-
that 20 of the 26 recruited participants (i.e. 10 of 13 participants in each group) received the planned 2 doses of trial treatments as per the trial protocol
-
that at least 22 of the 26 recruited participants had completed 26 weeks follow-up including the completion of the primary outcome at 60 days
-
that the independent Data Monitoring Committee (DMC) had reviewed the safety and efficacy data on the first 26 participants, and did not identify any tolerability or safety concerns
-
that the DMC were satisfied there was sufficient evidence to continue the trial based on the interim data and a futility analysis of the primary outcome.
Recruitment
Trial participants were recruited from paediatric nephrology units in 12 participating NHS sites across the UK, with the support of 88 Patient Identification Centres (PIC). Because the intervention needed to be given early in the disease course, screening began as soon as possible after a diagnosis of STEC HUS was suspected. If potential participants were receiving care at a PIC, their parents/guardians were provided with a brief participant information sheet prior to transfer for clinical care at a paediatric nephrology unit. At an appropriate time after transfer to the paediatric nephrology unit, if potential participants fulfilled the eligibility criteria, they were referred, with their parent/guardian’s permission, to the local research teams by their attending paediatric nephrologist. This plan of approach was made in conjunction with parents who had experience of having a child with STEC HUS.
Once eligibility was confirmed, parents/guardians/patients were approached, with permission, by researchers who were trained in Good Clinical Practice and specifically in taking consent for this trial. Parents/guardians were provided with a participant information sheet. Age-appropriate information sheets for children and young people were also available. The patient-facing documents were co-designed with parents of children who had experienced STEC HUS. Parents/guardians/patients were reassured that declining participation would not affect their child’s/their normal clinical care, and that they could withdraw from the trial at any point without this affecting their child’s/their care. Time was given to consider their involvement. If parents/guardians and, where (age) appropriate, potential participants agreed to participate in the clinical trial, written, informed consent was sought. Eligible young people aged 16–18 years provided their own consent for participation in the trial, with assent from younger children if appropriate (according to age). The mechanistic substudies were optional, all participants in the main trial were offered the opportunity to participate in the substudies, which involved providing blood and urine samples over the first 30 days of the trial.
Eligibility criteria
Participants were assessed for eligibility by an appropriately trained doctor. The participants needed to meet the following inclusion criteria:
-
Aged 6 months ≤ 19 years.
-
Weight ≥ 5 kg.
-
Diagnosis of HUS:
-
a. Micro-angiopathic haemolytic anaemia (indicated by fragmented red cells on blood film OR plasma LDH above local centre reference range).
-
-
AND
-
b. Thrombocytopenia (platelets < 150 × 109/l). (If the patient had received a platelet infusion prior to randomisation, the lowest documented platelet count prior to platelet infusion was used.)
-
-
AND
-
c. AKI: ‘injury’ or ‘failure’ category of paediatric risk/injury/failure/loss/end-stage definition ofacute kidney injury (pRIFLE) criteria [The eGFR for use in the pRIFLE criteria72 was calculated either from serum creatinine measured at the referring hospital or at the renal unit using the modified Schwartz formula: eGFR = height (cm) × 36.5/plasma creatinine (μmol/l). 73 If height could not be measured this was estimated from the corresponding centile from the child’s weight.] despite correction of hypovolaemia. [Patients who had not already received at least 10 ml/kg 0.9% saline since the diagnosis of STEC HUS were given 10 ml/kg 0.9% saline or equivalent (unless evidence of hypervolaemia) and eligibility criteria 3c was then reassessed.]
-
-
EITHER
-
Reported diarrhoea within 14 days prior to diagnosis of HUS (defined according to World Health Organization as ‘the passage of three or more loose or liquid stools per day – or more frequent passage than is normal for the individual’).
OR
-
Passage of blood per rectum within 14 days prior to diagnosis of HUS.
OR
-
Stool culture or stx polymerase chain reaction (PCR) or STEC serology result indicating STEC in the patient. (STEC positivity was not a prerequisite for eligibility since testing for STEC infection can sometimes be falsely negative.)
OR
-
Stool culture or stx PCR or STEC serology result indicating STEC in a close contact (household or institutional).
-
Patient intended to be able to receive trial drug within 48 hours of the on-call paediatric nephrologist formally taking over the care of the patient at the trial site providing diagnosis of HUS is met, or within 48 hours of meeting diagnosis of HUS if not met at the time the on-call paediatric nephrologist takes over the care of the patient. Note: since the speed with which eculizumab can be administered is believed to relate to the effectiveness, the initial aim was to keep the treatment window as short as possible. However, during the course of the trial, it became apparent that having a short treatment window was operationally unviable and therefore subsequent amendments to the protocol extended the period from 36 to 48 and then to 72 hours. The 72-hour amendment was approved, but was not be implemented due to early closure of the trial
-
Sexually active male or female patients must agree to be practising an effective, reliable and medically approved contraceptive regimen for 6 months after enrolment.
-
Sexually active female patients have provided a negative pregnancy test ≤ 48 hours prior to randomisation.
-
Patient/parent/guardian reported that vaccinations are up to date according to the routine UK (or equivalent) immunisation schedule. Note: it was required that vaccination against Haemophilus influenzae type b and pneumococcus were complete. If vaccination against other organisms e.g. MMR (measles, mumps and rubella), HPV (human papilloma virus) was incomplete, the patient remained eligible.
-
Written informed consent obtained from the patient’s parents/guardians and written assent obtained from patients (where age appropriate). Patients aged 16 years and above will provide their own written consent.
The following were exclusion criteria:
-
family history of aHUS
-
previous episode of HUS
-
known pre-existing eGFR < 90 ml/minute/1.73 m2
-
known or suspected pneumococcal infection
-
known or suspected meningococcal infection
-
prior to diagnosis, patient taking a drug known to be associated with HUS, for example calcineurin inhibitors, chemotherapy, quinine, oral contraceptive pill
-
hypersensitivity to eculizumab, murine proteins or any of the excipients listed in the Summary of Product Characteristics
-
pregnancy or lactation
-
malignancy
-
known disseminated intravascular coagulopathy. Note: Testing of coagulation was not mandatory for inclusion in trial.
-
refusal of consent, including consent for pregnancy testing, meningococcal vaccination or antibiotic prophylaxis
-
currently participating in another clinical trial of an investigational medicinal product (CTIMP).
Randomisation method and minimisation variables
Once eligibility was confirmed and informed consent obtained, the participants were commenced upon prophylactic antibiotics, and unless contraindicated [platelet count < 50 × 109/l or on systemic anticoagulation (in which case vaccination was deferred until clinically appropriate but before discharge)] or already administered, participants were also vaccinated against meningococcal infection. Participants were then randomised into the ECUSTEC trial via a secure online central randomisation system at the Birmingham Clinical Trials Unit. Participants were randomised at the level of the individual in a 1 : 1 ratio to either eculizumab or placebo, which was commenced as soon as possible after randomisation. A minimisation algorithm was used to ensure balance in the treatment allocation over the following variables:
-
recruiting centre
-
pRIFLE category (Injury or Failure category)
-
volume of 0.9% saline received in the 48 hours prior to randomisation (≤ 20 ml/kg or > 20 ml/kg).
To avoid predictability in the randomisation, a random element was included in the minimisation algorithm, so that each patient had a probability (unspecified here), of being randomised to the opposite treatment that they would have otherwise received.
Investigational medicinal product information
The investigational medicinal product (IMP) was eculizumab (Soliris®, Alexion Pharmaceuticals, Boston, MA) in the form of an intravenous (IV) infusion made up according to the manufacturer’s instructions, using 0.9% saline as diluent. 68 A total of two doses were administered by an appropriately trained professional according to the dosing regimen for aHUS in paediatric patients (Table 3). The weight of the child at randomisation was used to determine both doses. The first dose was given as soon as possible after randomisation (designated day 1), with the second dose given 7 days later (i.e. on day 8 ± 1 day).
| Patient bodyweight (kg) | Day 1 | Day 8 (± 1 day) | ||||
|---|---|---|---|---|---|---|
| Placebo arm | Active arm | Placebo arm | Active arm | |||
| Volume of 0.9% saline (ml) | Dose of eculizumab (mg) | Total infusion volume (made up with 0.9% saline) (ml) | Volume of 0.9% saline (ml) | Dose of eculizumab (mg) | Total infusion volume (made up with 0.9% saline) (ml) | |
| ≥ 40 | 180 | 900 | 180 | 180 | 900 | 180 |
| 20–< 40 | 120 | 600 | 120 | 120 | 600 | 120 |
| 10–< 20 | 120 | 600 | 120 | 60 | 300 | 60 |
| 5–< 10 | 60 | 300 | 60 | 60 | 300 | 60 |
The placebo was an equivalent volume of 0.9% saline (see Table 3) administered on day 1 and day 8. There was no detectable difference between IMP and placebo.
All participants received meningococcal vaccination (with tetravalent N. meningitidis vaccine – Nimenrix® or Menveo® – and Bexsero®), unless they had already been vaccinated as part of a routine immunisation programme, and an 8-week course of prophylactic antibiotics to reduce the risk of meningococcal infection (phenoxymethylpenicillin or erythromycin if penicillin allergy). Participants/parents/guardians were provided with an ECUSTEC Meningitis Warning Card containing information regarding the signs and symptoms of meningococcal disease, and were advised how to access medical care immediately if suspected. They were also provided with an ECUSTEC Participant Card to alert healthcare practitioners to the risk of meningococcal infection. These precautions were taken for all participants, whether randomised to receive eculizumab or placebo, in order to ensure that administration of IMP was the only difference between groups and to maintain the blinding of the trial.
In addition to the trial interventions, all participants received standard supportive care as follows:
-
RRT for refractory electrolyte imbalance, hypervolaemia, fluid restriction preventing sufficient nutrition, oligoanuria
-
red cell transfusion if haemoglobin < 70 g/l or if < 75 g/l with fall of > 20 g/l evidenced in previous 24 hours
-
a 3-month course of oral folic acid therapy was prescribed to prevent folate deficiency following acute haemolysis.
Plasma exchange was not permissible under the trial protocol and plasma infusion was only permitted when essential for correction of coagulopathy.
Blinding
All site personnel and participants/parents/guardians were blind to the randomised treatment allocation, apart from those responsible for preparing the IMP (e.g. clinical trials pharmacy). After randomisation, the pharmacy staff received the treatment allocation electronically and prepared an IV infusion bag containing either 0.9% saline with eculizumab or sodium chloride 0.9% saline (placebo) alone using aseptic technique. The prepared infusion bag was labelled, using labels approved by the Sponsor’s pharmacy and the Medicines and Healthcare products Regulatory Agency (MHRA), in an identical manner to maintain blinding. Researchers at the co-ordinating centre also remained blind to the randomised treatment allocations. The blinded trial treatment allocation was only broken for valid medical or safety reasons, such as meningococcal sepsis or pregnancy. If aHUS was suspected, it was recommended that the principal investigator (PI) or delegate contact the consultant on-call for the National aHUS Service prior to unblinding to discuss the case. In case the allocation was required immediately to assist in the medical management of a participant, clinicians were provided with a secure login and password to access the ECUSTEC online system where the allocation could be revealed. This would automatically alert the ECUSTEC Trials Office that the participant has been unblinded, but the treatment allocation would not be revealed. If it became necessary to unblind, only those who needed to know the treatment allocation would be informed, subject to clinical need. Unblinded participants remained in the trial and continued with trial follow-up assessments.
Outcome measures
The primary outcome measure was a multidomain clinical severity score (CSS) (see Appendix 2). The ECUSTEC CSS is a purpose-developed, multidomain score comprising severity of AKI and extrarenal events, developed for the trial using pilot data from 96 consecutive historic STEC HUS patients treated at 5 of the trial centres. A single score was assigned at day 60 which reflected the cumulative morbidity up until that point. The score ranges from 1 to 69; with higher scores indicating greater disease severity. Since severity of AKI is a significant prognostic factor in STEC HUS, the score was weighted for severity of the AKI. The severity score was considered an appropriate outcome measure by parents of children who had experienced STEC HUS.
Development of the ECUSTEC clinical severity score
Previous interventional trials in STEC HUS had failed to demonstrate an effective intervention. The ECUSTEC team reviewed the primary outcome measures used in these trials, including mortality and evidence of CKD at last follow-up, as candidate outcome measures. The outcomes had low event rates, and therefore would require a large number of participants to show a difference. Isolated kidney outcome measures such as number of days of dialysis or oligoanuria were also considered; however, since only 50% of children develop oligoanuria and/or the need for dialysis, many children would not reach the primary end point. In addition, number of days of dialysis does not reflect overall disease severity because it takes no account of extrarenal disease, which is strongly associated with adverse outcomes. Since no single outcome measure accurately reflected disease severity, the team developed a CSS for use as the primary outcome measure. Advice on the development of the score was provided by Dr Joanna Elson, Newcastle University.
Domains (organ systems) were selected for inclusion in the score based on the frequency of their involvement and their association with long-term sequelae, namely kidney, CNS, gastrointestinal tract, pancreas and heart. The highest score in each domain was assigned to events with permanent sequelae, thus linking higher scores with poorer long-term outcome.
Since most children with STEC HUS develop isolated kidney involvement, the kidney score was given the most weighting (maximum score 24, out of total of 69). This facilitated a wider range of scores for the kidney domain, in order to differentiate severity even at the milder end of the spectrum (e.g. a child who did not develop oligoanuria scored less than a child who did develop oligoanuria but did not need dialysis). The score increased with duration of dialysis, since duration of dialysis is linked with long-term outcome.
The CNS domain was developed in collaboration with Professor Bobby McFarland, Newcastle University. This domain was given the second highest weighting since it is linked with adverse long-term outcome – maximum score 15, out of total of 69. Children who displayed CNS features during the acute phase underwent assessment at day 60 for evidence of persistent CNS defect. Higher scores were assigned to denote persisting focal or global defects, whereas lower scores reflected transient involvement.
The pancreas and gastrointestinal domains were developed in collaboration with Dr Julian Thomas, Newcastle University. Lower pancreatic scores reflected transient, but increasingly significant involvement, whereas higher scores denoted requirement for substantial treatment and long-term sequelae (such as development of diabetes mellitus).
The cardiac domain was developed in collaboration with Dr Zdenka Reinhardt, Newcastle Hospitals and was based upon standardised echocardiogram (ECHO) and electrocardiogram (ECG) assessment of cardiac failure, ischaemia and infarction.
The key requirement of the score was to determine whether there was a meaningful clinical benefit from the IMP. We determined that a difference of five points in CSS equated to a meaningful clinical benefit, since this represented avoidance of significant morbidity (e.g. either a 5-day reduction in dialysis duration, avoiding a surgical laparotomy or avoiding development of cardiac failure).
Pilot data were collected on 94 consecutive historic STEC HUS patients treated at 5 of the planned trial centres to assess data collection and completeness. These data gave a mean CSS of 13.16 [standard deviation (SD) = 9.66; range: 2–45].
Secondary outcome measures were as follows:
-
overall survival
-
duration of RRT (days)
-
duration of thrombocytopenia (number of consecutive days until platelet count > 150 × 109/l)
-
duration of haemolysis (number of days until LDH within local centre reference range)
-
number of packed red blood cell (RBC) transfusions required and volume (ml/kg)
-
duration markers of inflammation present [number of days until neutrophil cell count and C-reactive protein (CRP) are in normal range for that centre]
-
persistent neurological defect at day 60 measured by structured expert assessment to include CNS examination, vision, hearing and neuropsychological assessment
-
CKD at 52 weeks [a composite end point of the presence of hypertension (> 95th centile for systolic blood pressure over an average of 3 readings by manual method using centile charts74 for age/sex/height), albuminuria (urine albumin-creatinine ratio > 2.5 mg/mmol on early morning urine) or eGFR < 90 ml/minute/1.73 m2 at 52 weeks]; presence of any of these will constitute CKD at 52 weeks
-
eGFR measurement using a centralised cystatin C assay at 52 weeks
-
economic evaluation of cost per CSS point, and cost per quality-adjusted life-year (QALY) gained, using Paediatric Quality of Life Inventory (PedsQL) and Child Health Utility-9D (CHU-9D) assessments to measure health-related quality of life (HRQoL). (Due to the early closure of the trial, the economic evaluation was not undertaken. The PedsQL and CHU-9D are measures of HRQoL, and since the economic evaluation was not being undertaken, it was decided to summarise and analyse the PedsQL and CHU-9D as part of the clinical data analysis.)
Scheduled trial assessments
Trial participants completed a variable number of assessments, depending upon the length of hospitalisation. After the baseline assessment (immediately prior to administration of IMP), daily assessments were carried out until either hospital discharge or day 14 (whichever was soonest). If hospital admission lasted > 14 days, then assessments continued weekly from day 14 to discharge or day 60 (whichever was soonest). The information from the in-patient assessments was collated into a single case report form (CRF). All participants underwent four further assessments at 30 and 60 days, and then at 26 and 52 weeks post randomisation. This visit schedule was designed in consultation with parents of children who had experienced STEC HUS. At assessments up to and including day 60, participants/parents/guardians were reminded of the signs and symptoms of meningococcal disease. At each visit, the participant’s and/or parent/guardian’s willingness to continue in the trial was ascertained and documented. The schedule of trial procedures and assessments is given in Box 1.
ENROLMENT
-
Eligibility assessment
-
Informed consent
-
Randomisation and allocation of study number
INTERVENTIONS
ASSESSMENTS
Day 1-
Medical history
-
Height and weight and blood pressure
-
Targeted physical exam
-
Full blood count (FBC)
-
Blood film
-
Plasma biochemistryc
-
Plasma complement C3 and C4
-
STEC investigation–stools
-
STEC investigation–serum
-
Concomitant medication check
-
Review signs and symptoms for meningococcal disease
-
Documentation of targeted eventsd
-
Completion of PedsQL and CHU-9D questionnaire
-
Blood sample obtained for DNA
-
Urine and plasma samples for exploratory studies (optional)
-
Urine and plasma samples for exploratory studies
-
Eculizumab or placebo
-
Completion of PedsQL and CHU-9D questionnaire
-
Urine and plasma samples for exploratory studies
-
FBC
-
Plasma biochemistryc
-
Concomitant medication check
-
Review signs and symptoms for meningococcal disease
-
Documentation of targeted eventsd
-
Height and weight and blood pressure
-
Targeted physical exam
-
Plasma biochemistryc
-
Concomitant medication check
-
Review signs and symptoms for meningococcal disease
-
Documentation of targeted eventsd
-
Completion of PedsQL and CHU-9D questionnaire
-
STEC investigation – stool sample
-
Urine and plasma samples for exploratory studies
-
Early morning urine sample (albumin:creatinine ratio)
-
Meningococcal vaccines if applicable
-
Height and weight and blood pressure
-
Targeted physical exam
-
Plasma biochemistryc
-
Concomitant medication check
-
Review signs and symptoms for meningococcal disease
-
Documentation of targeted eventsd
-
Optional anonymised feedback questionnaire
-
Completion of PedsQL and CHU-9D questionnaire
-
CNS examination if applicablef
-
Early morning urine sample (albumin:creatinine ratio)
-
Height and weight and blood pressure
-
Targeted physical exam
-
Plasma biochemistryc
-
Concomitant medication check
-
Documentation of targeted eventsd
-
Completion of PedsQL and CHU-9D questionnaire
-
Early morning urine sample (albumin:creatinine ratio)
-
Height and weight and blood pressure
-
Blood sample for cystatin C
-
Targeted physical exam
-
Plasma biochemistryc
-
Concomitant medication check
-
Documentation of targeted eventsd
-
Completion of PedsQL and CHU-9D questionnaire
-
Early morning urine sample (albumin:creatinine ratio)
DNA, deoxyribonucleic acid.
Antibiotic prophylaxis commenced prior to randomisation and IMP administration. Administered daily until week 8 (day 56).
Meningococcal vaccine administered prior to trial drug unless contraindicated or already received.
Plasma biochemistry comprising electrolytes, urea, creatinine, LDH, glucose, amylase, CRP, alanine transaminase.
Targeted events including RRT, urine output, administration of blood products, concomitant medication, need for abdominal surgery, occurrence of CNS symptoms, occurrence of hyperglycaemia and insulin use, need for parenteral nutrition, myocardial infarction and additional infections.
Daily assessments until hospital discharge, if admission ≥ 14 days then weekly assessments from day 14 to discharge or day 60, whichever was soonest.
CNS examination at day 60 (−3/+ 7 days) if the participant had CNS features during acute disease.
Between day 1 and day 8, a blood sample was obtained for deoxyribonucleic acid (DNA) analysis of genes previously associated with HUS, including: complement factor H (CFH), complement factor I, CD46, complement C3, complement factor B and diacylglycerol kinase (e). To determine whether eculizumab leads to prolonged STEC excretion, a stool sample was collected at day 30 to be analysed for STEC.
Health-related quality of life was measured at specific time points (see Box 1) using the parent completed CHU-9D and PedsQL questionnaires (dependent upon the participant’s age).
In order to determine the CNS component of the CSS, participants who had CNS features during acute disease underwent a comprehensive CNS assessment by a Consultant Paediatric Neurologist, a visual assessment by an optometrist and ophthalmologist, a hearing assessment by an audiologist and a neuropsychology assessment (supervised parental completion of the Adaptive Behaviour Assessment System Third Edition form) by a neuropsychologist at the day 60 assessment. If impairment was detected by any of these assessments, the assessor was asked to make a judgement about whether this impairment had occurred since the onset of STEC HUS from the information available (e.g. parental history). The results of the four assessments were collated by the paediatric neurologist and a CNS score was assigned.
At the week 52 assessment, a blood sample was obtained and sent to a central laboratory for measurement of Cystatin C to permit estimation of GFR.
In light of the COVID-19 global pandemic, a protocol amendment was submitted so that when follow-up visits were unable to be conducted face to face, staff were able to collect as much follow-up information as possible via telephone contact, providing the family concerned were happy to be contacted in this way.
Details of how and when the data for the trial outcome measures were collected is given in Table 4.
| Outcome assessed | Time point | Method | Reported by |
|---|---|---|---|
| ECUSTEC CSS | Up to day 60 | Clinical assessment of participant at follow-up visit and medical records | Research nurse/doctor |
| Survival | Up to 52 weeks | Clinical follow-up | Research nurse/doctor |
| Duration of RRT | Up to 52 weeks | Clinical assessment of participant at follow-up visit and medical records | Research nurse/doctor |
| Duration of thrombocytopenia, haemolysis and inflammation (CRP and neutrophils) | Daily until discharge from initial admission or until day 56, whichever is the soonest | Clinical assessment of participant at follow-up visit and medical records | Research nurse/doctor |
| Number of packed RBC transfusions required and volume (ml/kg) | Up to day 60 | Clinical assessment of participant at follow-up visit and medical records | Research nurse/doctor |
| Persistent neurological defect | Day 60 | Structured expert assessment to include CNS examination, vision, hearing and neuropsychological assessment | Paediatric neurologist, optometrist, audiologist and neuropsychologist |
| HRQoL | Day 1, day 8, day 30, day 60, week 26 and week 52 | PedsQL, CHU-9D | Study participant or parent/carer |
| CKD at 52 weeks | Week 52 | Clinical assessment of participant at follow-up visit and medical records | Research nurse/doctor |
| eGFR measurement | Week 52 | Centralised cystatin C assay with eGFRcys equation | Central laboratory |
| AEs | Up to week 52 | Clinical assessment of participant at follow-up visit and medical records | |
| Prolonged STEC excretion | Day 30 | Stool culture and stx PCR | National reference laboratory |
Adverse events and serious adverse events
Targeted adverse events (AEs) were collected and recorded on the trial CRFs. These comprised the development of any significant infections, infusion reactions to trial interventions and the presence of STEC in a stool sample collected at day 30. All serious adverse events (SAEs) occurring within 90 days of the first dose of meningococcal vaccination or prophylactic antibiotic (whichever occurred first) were e-mailed or faxed to the trial office within 24 hours of the research staff becoming aware of the event. SAEs that were judged to be at least possibly related to the IMP were reported irrespective of how long after IMP administration the reaction occurred. The local PI (or nominated clinician) had to assign severity, causality and expectedness (if deemed related) to the SAE before reporting. The coding of SAEs was in accordance with Common Terminology Criteria for Adverse Events v4.03. A plan was made to ensure that events categorised as suspected unexpected serious adverse reactions (SUSARs) were unblinded and reported to the chief investigator, Sponsor, Main Research Ethics Committee (REC) and MHRA within the required time frames.
Adherence monitoring
Adherence to the randomised treatment allocation was defined using the following criteria:
-
both the day 1 and day 8 doses were given and at least two-thirds of the intended dose was administered at each time point
-
no plasma infusion during period: post randomisation and up to 1 week post second dose
-
no plasma exchange during period: post randomisation and up to 1 week post second dose
-
both doses were given in the intended time window (Table 5).
| Dose | Time window for administration |
|---|---|
| Dose 1 | Within 48 hours of arriving in the renal unit (or within 48 hours of eligibility if not eligible on arrival) |
| Dose 2 | Seven days after the first dose (± 1 day) |
Adherence to the randomised allocated intervention was classified for each participant using two definitions:
-
Criteria 1–3 are met (regardless of whether criterion 4 is met).
-
Criteria 1–4 are met.
Adherence to prophylactic antibiotics and meningococcal vaccination was also monitored.
Participant withdrawal
There were no clinical situations that would mandate withdrawal from the trial. Participants were made aware that they could freely withdraw (discontinue participation) from the trial at any time. Participants who withdrew consent from the trial discontinued trial follow-up and only data collected prior to their withdrawal was used in the trial analysis. A participant who wished to cease to participate in a particular aspect of the trial was considered as having changed their status within the trial to either ‘no trial intervention’ (no further IMP but was willing to continue trial follow-up) or ‘no trial related follow-up’ (no further IMP, did not wish to attend trial visits but was willing for data collected at standard clinic visits to be used in the trial analysis).
Statistical considerations
Sample size
The planned sample size of 134 participants was based on retrospective pilot data collected on 94 consecutive historic patients with STEC HUS. These data gave a mean CSS of 13.16 (SD = 9.66; range: 2–45). A difference in CSS of five points is a moderate effect size (0.52) and equates to a meaningful clinical benefit (e.g. 5-day reduction in dialysis duration, avoiding a surgical laparotomy or avoiding development of cardiac failure). To detect a difference of 5 points in the CSS between groups using a 2-sided t-test and assuming a SD of 9.66, with 80% power and a type I error rate of 5% (α = 0.05), a total of 60 participants per group needed to be randomised – adjusting for a 10% attrition rate, 134 participants (67 per group) were planned to be recruited.
Statistical analysis
A comprehensive statistical analysis plan (SAP) was produced. In light of the trial closing early to recruitment, and the small sample size, statistical analysis is only presented for the primary outcome. The analysis methods in the SAP for the secondary outcomes were followed in order to summarise the data, but no statistical analysis of this data is presented.
Categorical baseline data were summarised using frequencies and percentages. Normally distributed continuous variables were summarised using means and SDs; otherwise, medians and interquartile ranges (IQRs) were presented. No formal statistical tests were performed on the baseline data.
The primary comparison groups are composed of those randomised to eculizumab versus those randomised to placebo. In the first instance, participants were analysed in the treatment group to which they were randomised (intention to treat), irrespective of adherence with the treatment protocol. For primary and secondary outcomes, summary statistics were reported for each treatment group. Estimates of differences between groups for the primary outcome are presented with two-sided confidence intervals (CIs) and p-values. The placebo group is the reference group. All analyses were undertaken in SAS (version 9.4).
For the primary outcome, the mean and SD of the CSS for each group were reported alongside an adjusted mean difference (with a 95% CI), which was estimated using a linear regression model adjusting for the minimisation parameters (volume of 0.9% saline received in the 48 hours prior to randomisation, pRIFLE category and recruiting centre; all included as fixed effects). Statistical significance of the treatment group parameter was determined from the p-value generated by the model. In the first instance, the primary analysis was only performed on participants with complete CSS data. For participants who did not have a CSS due to death, a maximum score of 69 was assigned and a secondary analysis was performed which included such participants.
Continuous secondary outcome measures [eGFR at 52 weeks, PedsQL, volume of RBC transfusions (ml/kg)] were summarised using means and SDs. Binary outcomes (CKD at 52 weeks, overall survival and persistent neurological defect at day 60) were summarised using frequencies and percentages. For secondary outcome measures which measure counts (number of RBC transfusions, number of days on RRT), data were summarised using medians with IQRs. Time-to-event outcomes (duration of thrombocytopenia, duration of haemolysis and duration markers of inflammation) were summarised using medians and IQRs. A Kaplan–Meier plot was produced to assess the data visually where appropriate. Time to event analyses for the duration of haemolysis and duration markers of inflammation outcomes only included participants who had elevated values at baseline.
Sensitivity analyses were performed on the primary outcome. These included:
-
A per-protocol analysis (using definition A and definition B as outlined in the adherence monitoring section above).
-
An analysis to assess the impact of missing outcome data. For participants who did not have a CSS due to death a maximum score of 69 was assigned to these participants in all scenarios. For participants who did not have a CSS due to missing scores in specific domains of the CSS, a maximum or minimum score was applied to that domain (and subdomain where relevant). Four different scenarios were considered:
-
Maximum score applied (in each missing component/domain) in both the eculizumab and placebo groups.
-
Minimum score applied (in each missing component/domain) in both the eculizumab and placebo groups.
-
Maximum score applied (in each missing component/domain) in the eculizumab group, minimum score applied (in each missing component/domain) in the placebo group.
-
Minimum score applied (in each missing component/domain) in the eculizumab group, maximum score applied (in each missing component/domain) in the placebo group.
-
-
An analysis to assess the impact of timing of completion of follow-up assessments by excluding those with assessments filled in outside of the mandated time window for completion.
A sensitivity analysis was also included for the secondary outcome measure of duration of RRT, where days of RRT prior to randomisation (where the participant started RRT pre randomisation and remained on RRT at the point of randomisation) were included in the count of the number of days on RRT.
Pre-planned subgroup analyses (limited to the primary outcome measure only) were completed for the following: (1) pRIFLE category (injury/failure) and (2) volume of 0.9% saline received in the 48 hours prior to randomisation (≤ 20 ml/kg/> 20 ml/kg). The effects of these subgroups were examined by adding a subgroup by treatment-group interaction parameter to the linear regression model. The p-value from the interaction terms were presented alongside the effect estimate and 95% CI within subgroups.
Trial oversight
Study oversight was provided by a Trial Steering Committee (TSC) that was chaired by Professor David Jayne (Cambridge University) and a DMC that was chaired by Professor David Wheeler (University College London). The TSC provided independent oversight of the trial, and provided advice to the chief investigator and co-investigators on all aspects of the trial throughout the study. Parents of children who had experienced STEC HUS were among the lay membership. The DMC adopted the DAMOCLES charter to define its terms of reference and operation in relation to oversight of the ECUSTEC trial.
Interim analyses of effectiveness and safety outcomes were provided to the DMC during the trial at approximately 6-month intervals, one of which occurred at the end of the internal pilot phase. Formal stopping rules were not adopted, instead a difference of at least p < 0.001 (similar to Haybittle-Peto stopping boundary) in an interim analysis of a major end point would have been needed to justify halting, or modifying, the study prematurely.
Chapter 3 Results of the clinical trial
Recruitment
Recruitment took place over 31 months in 10 UK NHS hospitals from July 2017 to July 2020. The contribution from each site is shown in Table 6. Two sites were open to recruitment but did not recruit any participants.
| Centre | Number of patients recruited |
|---|---|
| Bristol Royal Hospital for Children | 6 |
| Evelina Children’s Hospital | 1 |
| Great North Children’s Hospital | 5 |
| Great Ormond Street Hospital | 5 |
| Leeds General Infirmary | 4 |
| Nottingham University Hospital | 4 |
| Royal Hospital for Sick Children (Glasgow) | 4 |
| Royal Manchester Children’s Hospital | 4 |
| Southampton General Hospital | 1 |
| University Hospital of Wales | 2a |
In agreement with the funder, the internal pilot phase was extended, and a revised review date of July 2019 was agreed. At this point:
-
Twenty-four participants had been recruited in 24 months (July 2017–June 2019; rather than the 12 months originally planned).
-
Twenty-two of 23 participants who had returned treatment forms at the point of data review had received the planned two doses of trial treatments as per the trial protocol.
-
Twenty-one of 24 participants had completed 26 weeks follow-up including the completion of the primary outcome at 60 days. (Note: Two participants had not yet reached 26 weeks follow-up assessment point, and another participant had withdrawn before this point.)
-
The independent DMC reviewed the safety and efficacy data, and did not identify any tolerability or safety concerns. They also reviewed the primary outcome data and a futility analysis of this outcome, and were supportive of the trial continuing.
Although recruitment during the internal pilot phase was slower than anticipated, the DMC and TSC were supportive of the trial continuing, and this was agreed with the funder. Unfortunately however, recruitment remained a challenge throughout the trial (see below). Figure 1 shows that recruitment was behind target prior to the start of the COVID-19 global pandemic. Recruitment was paused in March 2020 at the onset of the pandemic, and was then closed early following a review by the funder, due to low recruitment and the impact of the pandemic. The last participant was therefore randomised in February 2020, and the trial closed to recruitment with 36 participants randomised. The last follow-up assessment visit of the final participant to be recruited took place in February 2021.
FIGURE 1.
Number of participants randomised by month. Pts participants; Shaded areas correspond to STEC HUS peak season.
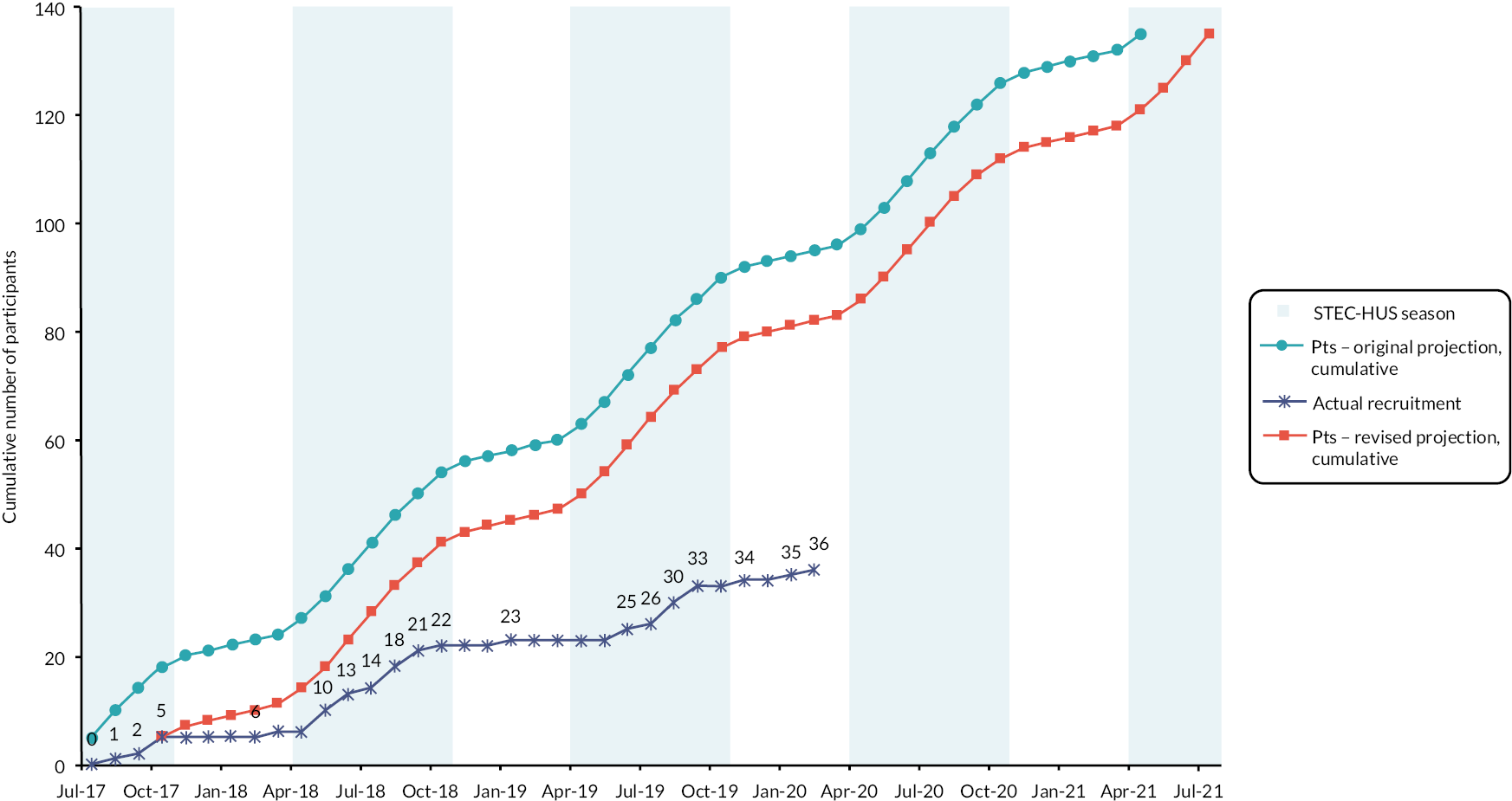
Reasons for slow recruitment were explored and addressed throughout the trial. Our collaborations with national public health bodies in participating nations enabled us to establish that the incidence of STEC HUS fell during the trial. Based on the previous incidence, we would have anticipated approximately 230 cases of STEC HUS in children during the recruitment period. However, we were only aware of 145 cases occurring during that time. We are confident that our surveillance captured the majority of cases of STEC HUS, and therefore this represents an approximate 37% reduction in incidence.
Another key reason for slow recruitment was a lack of out-of-hours infrastructure for undertaking acute CTIMPs in children. Early administration of trial treatment was an essential part of the trial. Children with STEC HUS typically present out of normal working hours and therefore this meant that the treatment window often fell out of hours. Only 2 of 12 trial centres were able to facilitate out of hours delivery of IMP. As a direct result of this, 22/108 (20%) of potentially eligible participants could not be approached or recruited. We addressed this during the trial, by making a protocol amendment that increased the treatment window from 36 to 48 hours, which was approved and implemented.
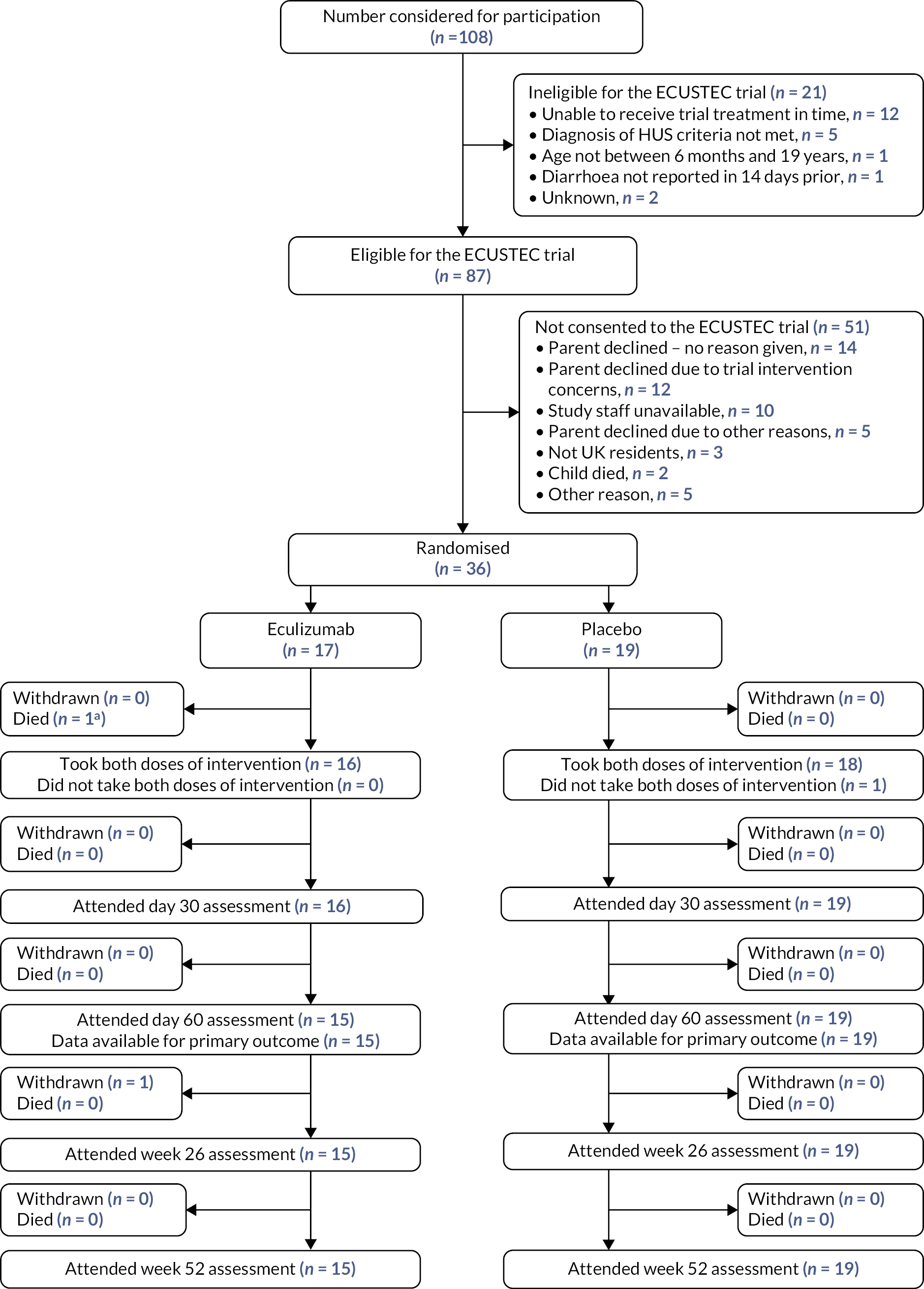
The flow of participants through the trial is shown in the Consolidated Standards of Reporting Trials (CONSORT) diagram in Figure 2. 75 At the point the trial was stopped, 108 individuals had been screened for participation, of which 87 were initially considered eligible based on clinical criteria. Of these 87 individuals, 36 were randomised, 31 did not consent to participation, 10 were not able to receive treatment in the required time frame, 2 patients died before being approached for the trial and 8 were not randomised for other reasons. Of the 36 children consented and randomised (27% of the 134 target sample size), 17 participants were randomised to the eculizumab group and 19 to the placebo group. One participant withdrew from the trial (withdrew consent) and one participant died.
FIGURE 2.
The CONSORT diagram of the flow of participants through the ECUSTEC trial. aParticipant had taken day 1 dose of intervention.
Participant characteristics
The baseline characteristics of the 36 randomised participants are shown in Tables 7 and 8. The randomisation minimisation algorithm ensured balance between groups in terms of the proportion with a pRIFLE category of Injury or Failure, the proportion of participants who had received > 20 ml/kg 0.9% saline prior to randomisation and the treatment centre. The groups were well balanced and comparable in all other baseline characteristics with the exception of age and weight – the eculizumab group had a mean age of 4.8 years (SD 3.2) compared with 6.4 years (SD 4.5) in the placebo group. There was a corresponding difference in weight with mean weight of 18.9 kg (SD 10.6) in the eculizumab group compared with 26.0 kg (SD 16.6) in the placebo group. There was also a difference in the proportion of patients who were anuric for > 12 hours (9/15, 60% of the eculizumab group and 2/18, 11% of the placebo group).
| Eculizumab N = 17 (%) |
Placebo N = 19 (%) |
|
|---|---|---|
| Minimisation variables | ||
| pRIFLE categorya | ||
| Injury | 2 (12) | 1 (5) |
| Failure | 15 (88) | 18 (95) |
| Volume of 0.9% saline (ml/kg)a | ||
| ≤ 20 | 13 (76) | 13 (68) |
| > 20 | 4 (24) | 6 (32) |
| Mean (SD, N) | 21.1 (22.9, 17) | 25.7 (26.2, 19) |
| Demographic and other clinical baseline variables | ||
| Age at randomisation (years) | ||
| Mean (SD, N) | 4.8 (3.2, 17) | 6.4 (4.5, 19) |
| Minimum–maximum | 1.5–13.4 | 0.7–14.7 |
| Sex | ||
| Female | 10 (59) | 10 (53) |
| Male | 7 (41) | 9 (47) |
| Weight (kg) | ||
| Mean (SD, N) | 18.9 (10.6, 17) | 26.0 (16.6, 19) |
| Minimum–maximum | 8.8–53.9 | 7.8–67.6 |
| Height (cm) | ||
| Mean (SD, N) | 107.5 (22.3, 17) | 118.2 (30.4, 19) |
| Minimum–maximum | 75.0–159.0 | 68.0–170.2 |
| Systolic blood pressure (mmHg) | ||
| Mean (SD, N) | 106.8 (13.6, 17) | 108.1 (14.0, 19) |
| Minimum–maximum | 89–144 | 82–140 |
| eGFR at randomisation (ml/minute/1.73 m2) | ||
| Mean (SD, N) | 13.2 (12.7, 14) | 12.9 (11.1, 19) |
| Minimum–maximum | 0–38 | 0–38 |
| Urine output < 0.5 ml/kg/hour for 16 hoursb | N = 2 | N = 1 |
| Yes | 0 (0) | 0 (0) |
| No | 2 (100) | 1 (100) |
| Missing | 0 (0) | 0 (0) |
| Urine output < 0.3 ml/kg/hour for 24 hoursc | N = 15 | N = 18 |
| Yes | 5 (33) | 6 (33) |
| No | 6 (40) | 6 (33) |
| Missing | 4 (27) | 6 (33) |
| Urine output (ml/kg/hour)d | ||
| Mean (SD, N) | 0.05 (0.05, 5) | 0.15 (0.10, 6) |
| Minimum–maximum | 0.00–0.10 | 0.01–0.27 |
| Anuria for 12 hoursc | N = 15 | N = 18 |
| Yes | 9 (60) | 2 (11) |
| No | 3 (20) | 11 (61) |
| Missing | 3 (20) | 5 (28) |
| CNS symptoms (in 48 hours pre randomisation) | 3 (18) | 2 (11) |
| Altered consciousnesse | 3/3 (100) | 2/2 (100) |
| Single seizure | 1/3 (33) | 1/2 (50) |
| Two or more seizures 24 hours apart | 1/3 (33) | 0/2 (0) |
| RRT (pre randomisation) | 8 (47) | 13 (68) |
| STEC HUS diagnosis | ||
| Diarrhoea | 17 (100) | 19 (100) |
| Bloody diarrhoea | 17/17 (100) | 12/19 (63) |
| STEC positive | ||
| Yes | 11 (65) | 13 (68) |
| No | 6 (35) | 6 (32) |
| Household/institutional contact STEC positive | ||
| Yes | 2 (12) | 2 (11) |
| No | 11 (65) | 17 (89) |
| Missing | 4 (23) | 0 (0) |
| Medical therapy (in 7 days pre randomisation) | ||
| Paracetamol | 16/17 (94) | 16/19 (84) |
| Missing | 0 | 0 |
| Ibuprofen | 2/16 (13) | 4/19 (21) |
| Missing | 1 | 0 |
| Codeine | 1/16 (6) | 2/19 (11) |
| Missing | 1 | 0 |
| Loperamide | 0/16 (0) | 1/18 (6) |
| Missing | 1 | 1 |
| Other anti-motility agent | 1/15 (7) | 0/18 (0) |
| Missing | 2 | 1 |
| Other antibiotics | 8/17 (47) | 3/19 (16) |
| Missing | 0 | 0 |
| Treatment | Eculizumab N = 17 (%) |
Placebo N = 19 (%) |
|---|---|---|
| Bloods and biochemistry at baseline (day 1) | ||
| Platelet count (× 109/l) at randomisation | ||
| Mean (SD, N) | 43.9 (20.5, 17) | 64.1 (36.8, 19) |
| Minimum–maximum | 13–94 | 19–146 |
| Neutrophils (109/l) | ||
| Mean (SD, N) | 11.3 (8.9, 17) | 8.7 (4.8, 18) |
| Minimum–maximum | 3.8–32.8 | 3.1–20.3 |
| Not done | 0 (0) | 1 (5) |
| Within local normal rangea | 11/17 (65) | 9/18 (50) |
| White blood cell count (109/l) | ||
| Mean (SD, N) | 16.5 (10.1, 17) | 15.2 (7.0, 19) |
| Minimum–maximum | 7.2–44.8 | 6.2–31.4 |
| Not done | 0 (0) | 0 (0) |
| Within local normal rangea | 11/17 (65) | 10/19 (53) |
| CRP (mg/l) | ||
| Mean (SD, N) | 69.1 (74.7, 12) | 33.8 (27.4, 13) |
| Minimum–maximum | 5–241 | 8–99 |
| Not done | 5 (29) | 6 (32) |
| Within local normal rangea | 2/12 (17) | 0/13 (0) |
| LDH (U/l) | ||
| Mean (SD, N) | 5218 (2621, 15) | 5221 (2681, 17) |
| Minimum–maximum | 1254–9158 | 2126–10,374 |
| Not done | 2 (12) | 2 (11) |
| Within local normal rangea | 0/15 (0) | 0/17 (0) |
| Creatinine (umol/l) | ||
| Mean (SD, N) | 346.2 (161.9, 17) | 428.6 (218.6, 19) |
| Minimum–maximum | 83–690 | 106–898 |
| Not done | 0 (0) | 0 (0) |
| Within local normal rangea | 1/17 (6) | 0/19 (0) |
| Glucose (mmol/l) | ||
| Mean (SD, N) | 5.1 (1.2, 10) | 5.6 (1.8, 10) |
| Minimum–maximum | 4.1–7.8 | 3.6–8.7 |
| Not done | 7 (41) | 9 (47) |
| Within local normal rangea | 10/10 (100) | 8/10 (80) |
| Amylase (U/l) | ||
| Mean (SD, N) | 95.3 (68.9, 8) | 156.5 (107.0, 12) |
| Minimum–maximum | 41–229 | 35–434 |
| Not done | 9 (53) | 7 (37) |
| Within local normal rangea | 5/8 (63) | 4/12 (33) |
| Alanine transaminase (U/l) | ||
| Mean (SD, N) | 151.5 (99.7, 14) | 137.4 (129.3, 17) |
| Minimum–maximum | 20–303 | 12–537 |
| Not done | 3 (18) | 2 (11) |
| Within local normal rangea | 3/14 (21) | 4/17 (24) |
| Urea (mmol/l) | ||
| Mean (SD, N) | 29.5 (9.2, 17) | 30.6 (12.7, 18) |
| Minimum–maximum | 8.1–47.2 | 7.4–49.8 |
| Not done | 0 (0) | 1 (5) |
| Within local normal rangea | 1/17 (6) | 1/18 (6) |
| Sodium (mmol/l) | ||
| Mean (SD, N) | 136.3 (5.8, 17) | 136.7 (4.7, 19) |
| Minimum–maximum | 118–143 | 127–148 |
| Not done | 0 (0) | 0 (0) |
| Within local normal rangea | 15/17 (88) | 13/18b (72) |
| Plasma C3 concentration (g/l) | ||
| Mean (SD, N) | 1.06 (0.17, 11) | 0.99 (0.29, 13) |
| Minimum–maximum | 0.82–1.29 | 0.44–1.51 |
| Not done | 6 (35) | 5 (26) |
| Within local normal rangea | 11/11 (100) | 11/13 (85) |
| Plasma C4 concentration (g/l) | ||
| Mean (SD, N) | 0.17 (0.05, 11) | 0.23 (0.19, 13) |
| Minimum–maximum | 0.10–0.26 | 0.06–0.84 |
| Not done | 6 (35) | 5 (26) |
| Within local normal rangea | 7/11 (64) | 8/13 (62) |
Adherence to trial treatment
Adherence to treatment allocation is shown in Table 9. According to definition A (detailed above), 16 out of the 16 (100%) participants in the eculizumab group who could receive both doses were considered adherent, compared with 18 out of 19 (95%) participants in the placebo group. One participant in the eculizumab group received dose 1 but died prior to administration of dose 2 and therefore was not assessed for adherence. One participant in the placebo group showed clinical improvement between randomisation and planned administration of trial treatment, and a clinical decision was made not to administer trial treatment. Trial follow-up was completed in this patient. According to definition B, 15 out of 16 (94%) participants in the eculizumab group were considered adherent, compared with 18 out of 19 (95%) participants in the placebo group. One participant in the eculizumab group received the second dose 9 days after the first dose which was outside the intended window (7 days ± 1 day after the first dose).
| Eculizumab N = 17 (%) |
Placebo N = 19 (%) |
|
|---|---|---|
| Adherent (definition A) | ||
| Yes | 16 (100) | 18 (95) |
| No | 0 (0) | 1a (5) |
| Unable to define | 1b | 0 |
| Adherent (definition B) | ||
| Yes | 15 (94) | 18 (95) |
| No | 1 (6) | 1a (5) |
| Unable to define | 1b | 0 |
| Dose 1 | ||
| Received dose 1 | 17 (100) | 18a (95) |
| Received full intended dose 1 | 16/17 (94) | 17/18 (94) |
| Proportion of dose 1 receivedc | ||
| Median (IQR, N) | 0.74 (-, 1) | 0.92 (-, 1) |
| Minimum–maximum | - | - |
| Received dose 1 in intended time windowd | 17/17 (100) | 18/18 (100) |
| Did not receive dose 1 | 0 (0) | 1a (5) |
| Dose 2 | ||
| Received dose 2 | 16 (94) | 18 (95) |
| Received full intended dose 2 | 14/16 (88) | 18/18 (100) |
| Proportion of dose 2 receivedb | ||
| Median (IQR, N) | 0.83 (0.78–0.88, 2) | - |
| Minimum–maximum | 0.78–0.88 | - |
| Received dose 2 in intended time windowe | 15/16f (94) | 18/18 (100) |
| Did not receive dose 2 | 0 (0) | 1 (5) |
| Plasma infusion (during period: post randomisation and up to 1 week post second dose) | ||
| Received plasma infusion | 0 (0) | 0 (0) |
| Plasma exchange (during period: post randomisation and up to 1 week post second dose) | ||
| Received plasma exchange | 0 (0) | 0 (0) |
Adherence to vaccination and prophylactic antibiotics is summarised in Table 10. One participant in the placebo group did not receive trial treatment, and therefore vaccinations and prophylactic antibiotics were not administered. One participant in the eculizumab group received the first dose of prophylactic antibiotics, but died before ACWY vaccination was able to be administered. The remainder of participants in both groups (34 of 36) received prophylactic antibiotics for 8 weeks and ACWY vaccination as part of the trial. All participants who received trial treatment received Bexsero vaccination within the trial (21 of 35) or had previously received it as part of the UK immunisation programme (14 of 35).
| Eculizumab N = 17 (%) |
Placebo N = 19 (%) |
|
|---|---|---|
| Prophylactic antibiotics | ||
| Received first dose | 17 (100) | 18 (95)a |
| Trial-mandated antibiotic cover for 2 weeks post-discharge prescribed | 16 (94)b | 18 (95)a |
| Antibiotic cover by the GP confirmed, in line with the ECUSTEC Initial GP Letter | 15 (88)c | 17 (89)d |
| ACWY vaccine | ||
| Received vaccination | 16 (94) | 18 (95) |
| Did not receive vaccination | 1 (6)b | 1 (5)a |
| Bexsero vaccine | ||
| Received vaccination as part of the UK immunisation programme | 8 (47) | 6 (32) |
| Received vaccination as part of the ECUSTEC trial | 9 (53) | 12 (63) |
| Did not receive vaccination | 0 (0) | 1 (5)a |
Primary outcome
The mean CSS at day 60 for participants randomised to eculizumab was 11.5 (SD 8.4) compared to 14.6 (SD 7.7) for participants randomised to placebo. The adjusted mean difference between the two groups was −2.5 points (95% CI −7.8 to 2.8; p = 0.3) shown in Table 11. When we included the participant in the eculizumab group who died (assigned the highest possible severity score of 69), the adjusted mean difference between groups was 3.4 points (95% CI −5.5 to 12.3). The breakdown of CSS components in each group is shown in Table 11.
| Eculizumab, N = 17 (%) | Placebo, N = 19 (%) | Mean differencea (95% CI) | p-value | |
|---|---|---|---|---|
| CSS (excluding any participants who have died)b | ||||
| Mean (SD, N) | 11.5 (8.4, 15) | 14.6 (7.7, 19) | −2.5 (−7.8 to 2.8) | 0.3 |
| Minimum–maximum | 1–28 | 2–29 | ||
| CSS (including any participants who have died)b | ||||
| Mean (SD, N) | 15.1 (16.5, 16) | 14.6 (7.7, 19) | 3.4 (−5.5 to 12.3) | 0.4 |
| Minimum–maximum | 1–69 | 2–29 | ||
| Domain scores | ||||
| Renal domain | ||||
| Lowest eGFR > 50 | 1 (7) | 0 (0) | - | - |
| Lowest eGFR 26–50, no oligoanuria | 1 (7) | 2 (11) | ||
| Lowest eGFR ≤ 25, no oligoanuria | 2 (13) | 1 (5) | ||
| Oligoanuria (no RTT) | 0 (0) | 0 (0) | ||
| Dialysis/RRT < 48 hours | 1 (7) | 0 (0) | ||
| Dialysis/RRT 2 days | 0 (0) | 0 (0) | ||
| Dialysis/RRT 3 days | 2 (13) | 0 (0) | ||
| Dialysis/RRT 4 days | 0 (0) | 0 (0) | ||
| Dialysis/RRT 5 days | 0 (0) | 3 (16) | ||
| Dialysis/RRT 6 days | 0 (0) | 1 (5) | ||
| Dialysis/RRT 7 days | 0 (0) | 0 (0) | ||
| Dialysis/RRT 8 days | 1 (7) | 1 (5) | ||
| Dialysis/RRT 9 days | 0 (0) | 2 (11) | ||
| Dialysis/RRT 10 days | 2 (13) | 1 (5) | ||
| Dialysis/RRT 11 days | 0 (0) | 0 (0) | ||
| Dialysis/RRT 12–13 days | 1 (7) | 2 (11) | ||
| Dialysis/RRT 14–17 days | 3 (20) | 4 (21) | ||
| Dialysis/RRT 18–20 days | 0 (0) | 1 (5) | ||
| Dialysis/RRT 21–27 days | 1 (7) | 0 (0) | ||
| Dialysis/RRT 28–34 days | 0 (0) | 1 (5) | ||
| Dialysis/RRT 35–41 days | 0 (0) | 0 (0) | ||
| Dialysis/RRT 42–48 days | 0 (0) | 0 (0) | ||
| Dialysis/RRT 49–55 days | 0 (0) | 0 (0) | ||
| Dialysis/RRT > 55 days | 0 (0) | 0 (0) | ||
| CNS domain | ||||
| No obvious CNS involvement | 13 (86) | 15 (80) | - | - |
| Altered consciousness | 0 (0) | 1 (5) | ||
| Single seizure | 0 (0) | 1 (5) | ||
| Two or more seizures 24 hours apart | 0 (0) | 0 (0) | ||
| Transient focal neurological defect | 1 (7) | 1 (5) | ||
| Persistent focal neurological defect | 1 (7) | 1 (5) | ||
| Persistent global | 0 (0) | 0 (0) | ||
| Pancreas domain | ||||
| No clinical/biochemical evidence pancreatitis | 14 (93) | 12 (63) | - | - |
| Raised amylase and/or lipase without clinical symptoms/signs | 1 (7) | 6 (32) | ||
| Hyperglycaemia without insulin requirement | 0 (0) | 0 (0) | ||
| Pancreatitis with sequelae | 0 (0) | 1 (5) | ||
| Chronic sequelae of pancreatitis | 0 (0) | 0 (0) | ||
| Gastrointestinal domain | ||||
| No abdominal surgery required | 15 (100) | 19 (100) | - | - |
| Laparoscopy/laparotomy required for abdominal symptoms | 0 (0) | 0 (0) | ||
| Intestinal perforation AND/OR bowel resection required | 0 (0) | 0 (0) | ||
| Stoma formation | 0 (0) | 0 (0) | ||
| Cardiac domain | ||||
| No cardiac involvement | 15 (100) | 19 (100) | - | - |
| Cardiac failure confirmed by ECHO | 0 (0) | 0 (0) | ||
| Cardiac failure confirmed by ECHO with dilated cardiomyopathy | 0 (0) | 0 (0) | ||
| Myocardial infarction | 0 (0) | 0 (0) | ||
Sensitivity analyses for the primary outcome are shown in Table 12. In the per-protocol analyses, including only the participants defined as adherent to trial treatment (using both definition A and definition B) and the analysis excluding assessments completed outside the time window, the mean differences for the CSS changed only marginally from the intention to treat analysis. Not surprisingly, the point estimates were more sensitive and changed more, when analyses were conducted to assess the impact of missing data based on applying minimum and maximum scores to participants with missing data.
| Eculizumab N = 17 |
Placebo N = 19 |
Mean differencea (95% CI) |
|
|---|---|---|---|
| Per-protocol analyses | |||
| CSS (definition A)b | |||
| Mean (SD, N) | 11.5 (8.4, 15) | 15.3 (7.3, 18) | −2.2 (−8.2 to 3.7) |
| Minimum–maximum | 1–28 | 2–29 | |
| CSS (definition B)b | |||
| Mean (SD, N) | 11.9 (8.6, 14) | 15.3 (7.3, 18) | −2.2 (−8.2 to 3.7) |
| Minimum–maximum | 1–28 | 2–29 | |
| Missing responses analyses | |||
| CSS (scenario A)c | |||
| Mean (SD, N) | 18.1 (20.2, 17) | 14.6 (7.7, 19) | 6.6 (−3.5 to 16.8) |
| Minimum–maximum | 1–69 | 2–29 | |
| CSS (scenario A)c excluding participant who died | |||
| Mean (SD, N) | 14.9 (15.8, 16) | 14.6 (7.7, 19) | 1.4 (−6.9 to 9.8) |
| Minimum–maximum | 1–66 | 2–29 | |
| CSS (scenario B)c | |||
| Mean (SD, N) | 14.9 (16.0, 17) | 14.6 (7.7, 19) | 2.8 (−5.7 to 11.3) |
| Minimum–maximum | 1–69 | 2–29 | |
| CSS (scenario B)c excluding participant who died | |||
| Mean (SD, N) | 11.6 (8.1, 16) | 14.6 (7.7, 19) | −2.9 (−7.9 to 2.2) |
| Minimum–maximum | 1–28 | 2–29 | |
| Time of completion analysis | |||
| CSSd | |||
| Mean (SD, N) | 13.5 (8.5, 11) | 14.8 (8.2, 13) | −3.1 (−10.9 to 4.7) |
| Minimum–maximum | 1–28 | 2–29 | |
Two subgroup analyses (based on pRIFLE category and volume of saline prior to randomisation) were carried out for the primary outcome. There was no evidence that the treatment effect differed across the subgroup categories for either subgroup analysis (Table 13).
| Eculizumab | Placebo | Interaction p-value |
Mean differencea (95% CI) |
|
|---|---|---|---|---|
| CSSb | ||||
| pRIFLE category | ||||
| Injury | 1.5 (0.7, 2) | 2.0 (-, 1) | 0.83 | −4.6 (−25.3 to 16.1) |
| Failure | 13.1 (7.9, 13) | 15.3 (7.3, 18) | −2.2 (−8.2 to 3.7) | |
| Volume of 0.9% saline (ml/kg) | ||||
| ≤ 20 | 11.4 (9.1, 12) | 14.5 (7.5, 13) | 0.65 | −2.1 (−8.4 to 4.2) |
| > 20 | 12.0 (6.2, 3) | 14.7 (8.9, 6) | −5.2 (−17.8 to 7.4) | |
Secondary outcome results
There was one death (on day 2) reported during the trial. This participant received one dose of eculizumab, and the death was considered disease related.
Participants in the eculizumab group received a median of 8 days of RRT (IQR 1–12) and those in the placebo group received a median of 9 days of RRT (IQR 5–14). This analysis only included RRT following randomisation. A secondary analysis including days of RRT prior to randomisation (where the participant started RRT pre randomisation and remained on RRT at the point of randomisation) gave similar results [median of 8 (IQR 1–12)] and 9 days (IQR 5–16) for the eculizumab and placebo groups, respectively (Table 14).
| Eculizumab N = 17 |
Placebo N = 19 |
|
|---|---|---|
| Number of days on RRT | ||
| Median (IQR, N) | 8 (1–12, 17) | 9 (5–14, 19) |
| Minimum–maximum | 0–26 | 0–31 |
| Time to resolution of thrombocytopenia (days) | ||
| Number who resolveda | 13/17 | 18/19 |
| Median (IQR, N) | 6 (4–7, 13) | 7.5 (6–8, 18) |
| Minimum–maximum | 1–13 | 1–12 |
| Time to resolution of haemolysis (days) b | ||
| Number who resolveda | 3/15 | 2/17 |
| Median (IQR, N) | 6 (3–17, 3) | 11 (2–19, 2) |
| Minimum–maximum | 3–17 | 2–19 |
| Time to resolution of inflammation of neutrophil cell count (days) c | ||
| Number who resolveda | 5/6 | 9/9 |
| Median (IQR, N) | 4 (2–6, 5) | 5 (2–7, 9) |
| Minimum–maximum | 1–20 | 2–11 |
| Time to resolution of inflammation of CRP (days)d | ||
| Number who resolveda | 6/10 | 10/13 |
| Median (IQR, N) | 10.5 (4–13, 6) | 9.0 (4–12, 10) |
| Minimum–maximum | 3–17 | 1–17 |
| Number of packed RBC transfusions | ||
| Median (IQR, N) | 2 (1–3, 17) | 2 (0–2, 19) |
| Minimum–maximum | 0–4 | 0–8 |
| Total volume of RBC transfusion (ml/kg) | ||
| Mean (SD, N) | 25.0 (17.3, 17) | 23.6 (28.6, 18) |
| Minimum–maximum | 0–62 | 0–99 |
Several secondary outcomes assessed the time to resolution of abnormal parameters commonly seen in STEC HUS – thrombocytopenia, raised LDH (marker of haemolysis) and raised neutrophil count and CRP (markers of inflammation) (see Table 14). In those who resolved, the median time to resolution of thrombocytopenia was 6 days (IQR 4–7) in the eculizumab group compared with 7.5 days (IQR 6–8) in the placebo group. Figure 3 shows a Kaplan–Meier plot of the time to resolution of thrombocytopenia. Ten participants in the eculizumab group and 13 in the placebo group had raised CRP at baseline. This resolved in 6/10 participants after a median time of 10.5 days (IQR 4–13) in the eculizumab group compared with 10/13 participants after 9.0 days (IQR 4–12) in the placebo group. Of the 32 participants with abnormal LDH levels at baseline, only 5 resolved prior to discharge (3/15 in the eculizumab group and 2/17 in the placebo group). Baseline neutrophil count was only raised in 15/36 (41.7%) participants, with 14 resolving before discharge (5/6 in the eculizumab group and 9/9 in the placebo group). The number and volume of packed RBC transfusions were similar across the two groups.
FIGURE 3.
Kaplan–Meier plot of time to resolution of thrombocytopenia (days).
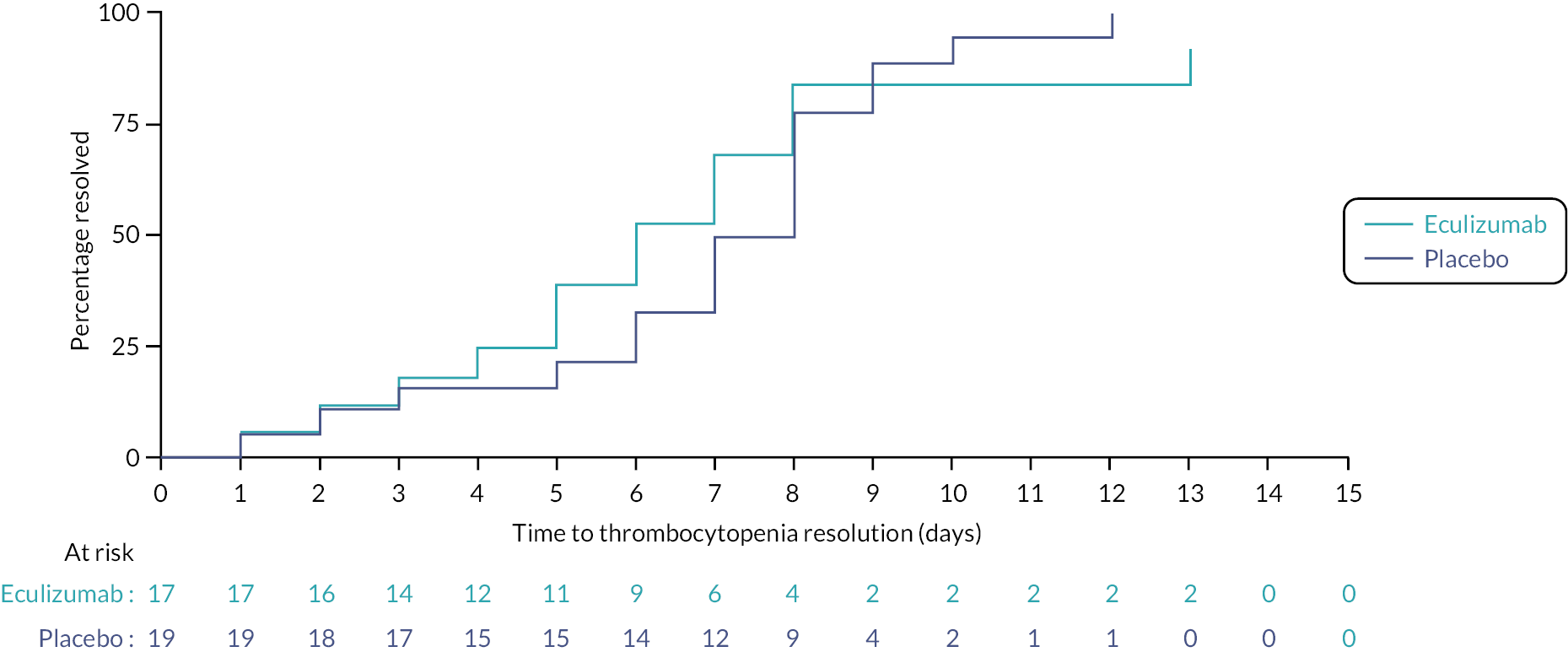
Two participants (5.6% of the cohort) had a persistent neurological defect at day 60 – one in each group. Both were persistent focal defects. At 52 weeks, the incidence of CKD was similar between the 2 groups: 5/15 (33%) in the eculizumab group compared with 6/19 (32%) in the placebo group. The eGFR measured by centralised Cystatin C assay at 52 weeks was 84.9 ml/minute/1.73 m2 in the eculizumab group compared with 84.0 ml/minute/1.73 m2 in the placebo group.
Quality of life
Since the health economic evaluation was not performed, it was decided to analyse the HRQoL PedQL and CHU-9D data as part of the clinical outcomes. The PedsQL data are shown in Figures 4–6 and the CHU-9D data are shown in. PedsQL is based upon recall of the past one month. For all PedsQL scores, perhaps not surprisingly, there was a decrease in score (worsening in HRQoL) between baseline and day 8, and then an increase in score up to day 60, followed by a plateau. It was interesting that by 52 weeks, the scores were not that dissimilar to those observed at baseline. CHU-9D score covers recall of today/last night and scores were lowest at baseline (Figure 7).
FIGURE 4.
Longitudinal plot of PedsQL Psychosocial Health Summary Score by treatment group (with standard error bars).
FIGURE 5.
Longitudinal plot of PedsQL Physical Health Summary Score by treatment group (with standard error bars).
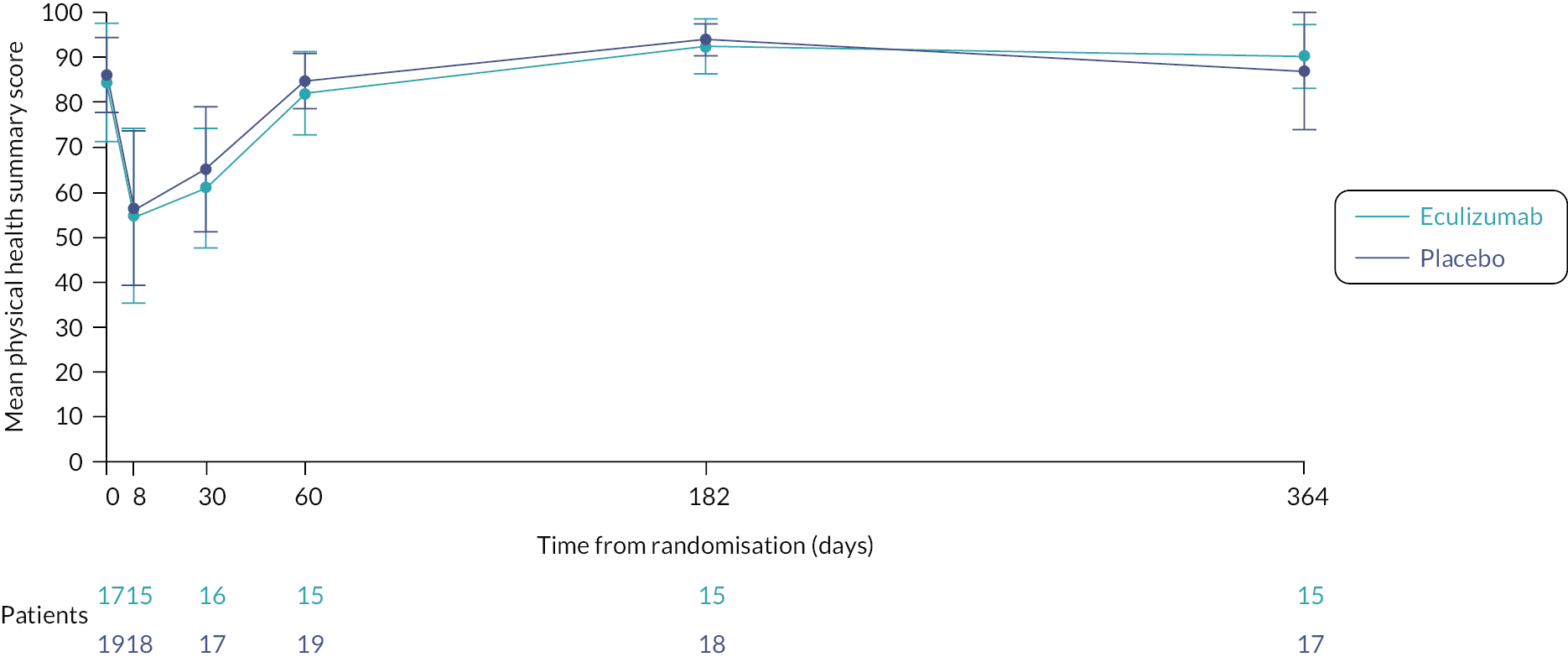
FIGURE 6.
Longitudinal plot of PedsQL Total Summary Score by treatment group (with standard error bars).
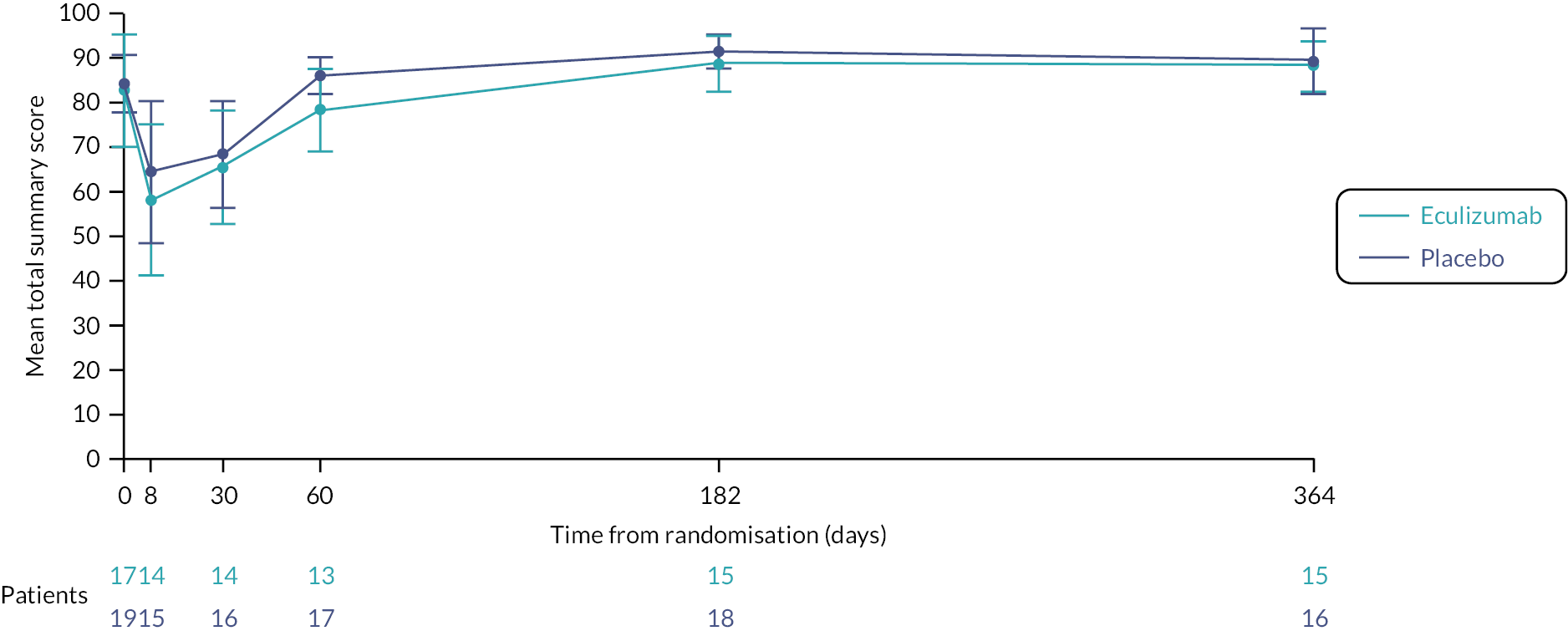
FIGURE 7.
Longitudinal plot of CHU-9D Score by treatment group (with standard error bars).
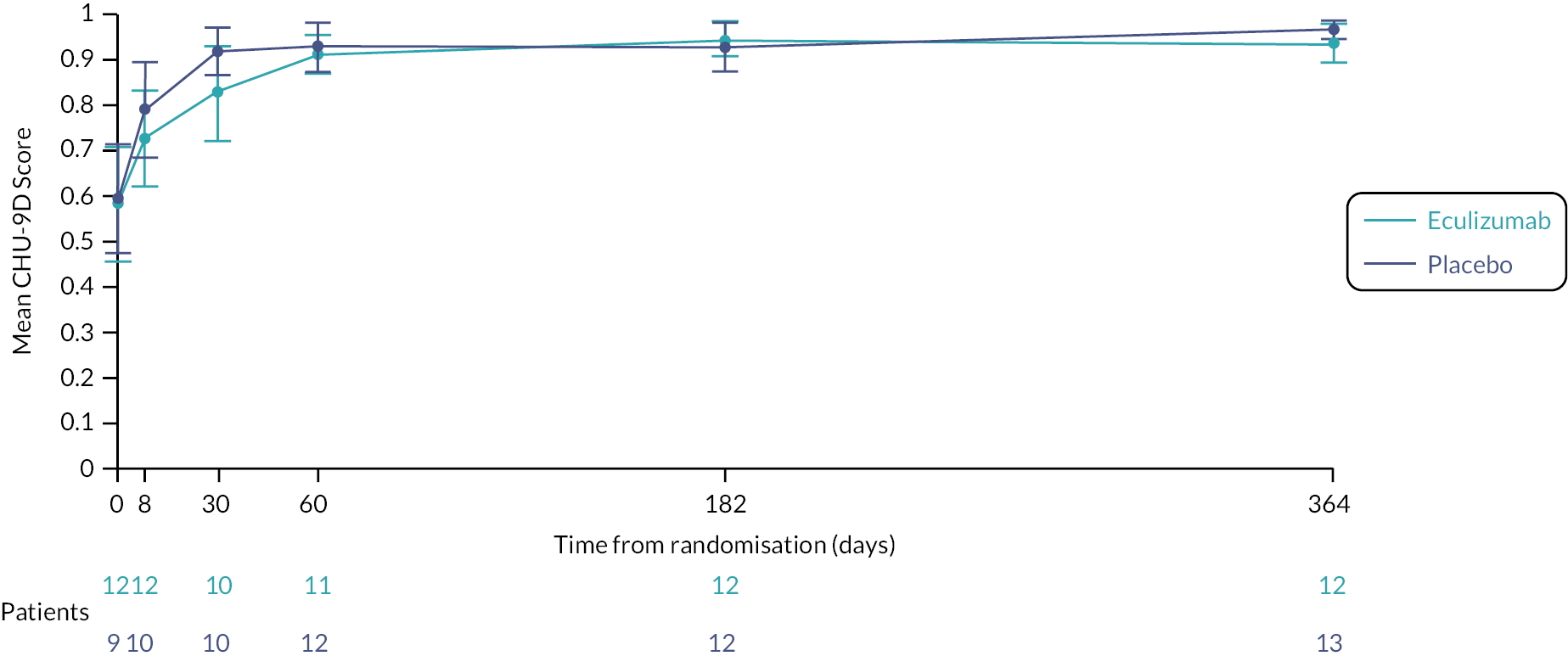
Health resource utilisation
A full health economic evaluation was planned; however, due to the early closure of the trial this was not completed. A summary of the health resource utilisation data collected for the evaluation is given in Table 15.
| Eculizumab N = 17 | Placebo N = 19 | ||
|---|---|---|---|
| Primary care visits | |||
| N (%) | 12 (71) | 9 (47) | |
| All visits | N | 31 | 23 |
| Mean (SD, n) | 2.6 (2.4, 12) | 2.6 (2.0, 9) | |
| Range | 1–9 | 1–6 | |
| Visits related to HUS | N | 15 | 5 |
| Mean (SD, n) | 1.3 (2.0, 12) | 0.6 (0.9, 9) | |
| Range | 0–5 | 0–2 | |
| GP visits | Mean (SD, n) | 2.2 (2.4, 10) | 2.7 (1.7, 7) |
| Range | 1–9 | 1–5 | |
| Nurse visits | Mean (SD, n) | 1.0 (0.0, 2) | 1.0 (-, 1) |
| Range | 1.0–1.0 | 1.0 | |
| Other visits | Mean (SD, n) | 3.5 (0.7, 2) | 1.0 (0.0, 3) |
| Range | 3.0–4.0 | 1.0–1.0 | |
| Outpatient visits | |||
| N (%) | 8 (47) | 9 (47) | |
| All visits | N | 49 | 29 |
| Mean (SD, n) | 6.1 (9.5, 8) | 3.2 (1.6, 9) | |
| Range | 1–29 | 1–6 | |
| Visits related to HUS | N | 41 | 29 |
| Mean (SD, n) | 5.1 (7.5, 8) | 3.2 (1.6, 9) | |
| Range | 1–23 | 1–6 | |
| Doctor visits | Mean (SD, n) | 5.8 (5.8, 5) | 3.0 (1.0, 7) |
| Range | 1–15 | 2–4 | |
| Nurse visits | Mean (SD, n) | 5.3 (7.5, 3) | 1.4 (0.5, 5) |
| Range | 1–14 | 1–2 | |
| Other visits | Mean (SD, n) | 2.0 (0.0, 2) | 1.0 (-, 1) |
| Range | 2–2 | 1 | |
| A and E visits | |||
| Number participants (%) | 6 (35) | 5 (26) | |
| All visits | N | 9 | 10 |
| Mean (SD, n) | 1.5 (0.8, 6) | 2.0 (1.7, 5) | |
| Range | 1–3 | 1–5 | |
| Visits related to HUS | N | 6 | 9 |
| Mean (SD, n) | 1.0 (0.6, 6) | 1.8 (1.9, 5) | |
| Range | 0–2 | 0–5 | |
| Hospital admissions | |||
| N (%) | 6 (35) | 5 (26) | |
| N | 7 | 7 | |
| Elective | 1 | 4a | |
| Emergency | 6 | 3 | |
| Mean (SD, n) | 1.2 (0.4, 6) | 1.4 (0.9, 5) | |
| Range | 1–2 | 1–3 | |
| Emergency length of stay | Mean (days) (SD, n) | 1.6 (1.3, 5) | 3.0 (-, 1) |
| Range | 1–4 | 3 | |
Genetic studies
Results of the genetic analyses were not complete at the time of publication – these will be published within a subsequent manuscript.
Adverse events
Five participants (three eculizumab, two placebo) experienced an AE. The detection of STEC in a stool sample was a targeted AE collected at day 30. It was not possible to obtain data regarding STEC in the day 30 stool sample for all participants. Of 19 forms returned, there were four participants with STEC detected in their day 30 stool sample. Three of these were in the eculizumab group (3/12, 25%) and one was in the placebo group (1/7, 14%). One participant in the placebo group reported experiencing a significant infection. There were seven SAEs in six participants. Five participants in the eculizumab group experienced six SAEs (anaemia, rash, gastroenteritis, Horner syndrome, prolonged nasogastric feeding, death due to severe brain injury) compared with one patient in the placebo group (serum amylase increased, general anaesthetic for dialysis central line). All SAEs were assessed as either unrelated, or unlikely to be unrelated, to the trial intervention. No participants experienced a SUSAR.
Chapter 4 Results of the mechanistic substudies
Introduction
Pathophysiology of Shiga-toxin-producing Escherichia coli haemolytic uraemic syndrome
Shiga-toxin-producing E. coli infection usually occurs as a result of ingestion of contaminated food or water. 10 STEC colonise the intestinal mucosa and have a number of virulence factors that result in adhesion to colonic enterocytes and subsequent production of stx. 11 Once secreted, stx traverses the intestinal wall and enters the bloodstream,12 where it binds to circulating polymorphonuclear leucocytes and is transported to distal sites. 76 The main cellular target for stx is the Gb3 receptor located on the microvascular endothelium within the brain, gut and kidney. 12 Within the kidney, in addition to the endothelium, Gb3 is expressed on the surface of tubular cells, mesangial cells and, in primates, podocytes. 77 Once bound to Gb3, stx enters the cell via endocytosis and is trafficked through the Golgi apparatus and endoplasmic reticulum, before being released into the cytosol. 37 Once in the cytosol, stx exerts its effect via inhibition of the ribosomal activity and subsequent blockage of protein transcription. These events lead to activation of apoptotic pathways, induction of inflammatory cytokines and cellular necrosis. 12 All these processes lead to the generation of a pro-inflammatory environment within the microvasculature.
The field of HUS has been transformed through the delineation of causative genes for the closely related condition, aHUS. 13 aHUS describes patients with HUS without STEC infection, approximately 60% of whom have defects of the alternative complement pathway.
The complement system
The complement system is a complex cascade of over 30 proteins that form part of the innate immune system. 14 It is comprised of three pathways, namely the classical, alternative and lectin-binding pathways. The alternative pathway is constitutively active at a low level via the spontaneous hydrolysis of circulating C3 molecules, which interact with factor B and factor D to produce a C3 convertase which forms the basis of a C3 amplification loop. The convertase cleaves further C3 into C3a and C3b, and the C3b generated by this process binds to the C3 convertase, forming the C5 convertase (C3BbC3b). 78 The C5 convertase cleaves circulating C5–C5a (anaphylatoxin) and C5b. Finally, C5b complexes with C6, C7, C8 and C9, forming the MAC. This structure forms a permeable pore in the cell membrane leading to cell lysis. To prevent overactivity of the pathway and to protect host cells from damage by complement, a number of fluid phase {CFH, I [CFI (complement factor I)]} and membrane-bound [CD46, DAF(decay accelerating factor) and CD59] regulators exist.
The role of complement in Shiga-toxin-producing Escherichia coli haemolytic uraemic syndrome
While there is clear evidence that stx mediates glomerular endothelial TMA,15 in STEC HUS there is increasingly compelling evidence that complement plays a role in pathogenesis. Complement activation was first observed in STEC HUS over 30 years ago, when it was demonstrated that children with STEC HUS had higher plasma levels of the alternative complement activation products, C3b, C3c, C3d and factor B. 16,17 Patients with STEC HUS may exhibit transiently low plasma complement C3 levels during acute disease which return to normal during convalescence, indicating complement activation and consumption. Low admission plasma C3 levels have repeatedly been shown to correlate with several measures of disease severity including dialysis requirement, neurological and other extrarenal complications, PICU admission and length of hospitalisation. 18–20 Adding to evidence for complement activation, serum complement activation products are elevated in the acute phase and correlate with disease severity, including increased levels of C5 convertase and the common end point of complement activation soluble C5b-C9 [or terminal complement complex (TCC)], a fluid phase form of MAC. 21–23 Further evidence of complement involvement in STEC HUS is supported by the presence of circulating complement-containing microvesicles from platelets, leucocytes and erythrocytes in individuals with STEC HUS,24,25 suggesting a direct interaction between these cells and complement.
Experimental studies have been undertaken to understand the possible mechanism of this activation. Activation of the alternative complement pathway by stx2 has been demonstrated. 79 In the same study, stx2 was shown to bind to and inhibit the function of CFH. In a separate study, stx exposure reduced the expression of CD59, a cell surface complement regulator. 80 stx-treated microvascular endothelial cells demonstrate C3 surface deposition via activation of the alternate complement pathway when treated with human serum. Taken together, these results indicate that stx activates the alternative complement pathway and may also result in increased susceptibility of microvascular endothelial cells to complement-mediated damage through a reduction in complement regulation by CFH and CD59.
Prior to undertaking mechanistic studies within the ECUSTEC trial, results from animal studies were mixed. In a murine model of STEC HUS, complement blockade was protective against severe disease. 26 In contrast, no evidence of complement activation was detected in a nonhuman primate model of STEC HUS. 27
Most work on STEC HUS views the glomerular endothelial cell as the target of stx. However, our previous work, both in vivo and in vitro, supports the hypothesis that the podocyte is a central target of stx damage, which disrupts endothelial complement regulation via a reduction in podocyte VEGF secretion, resulting in TMA. 81 Podocytes normally produce VEGF, which maintains the healthy glomerular endothelial phenotype. 82 A concept changing study by Eremina et al. 83 demonstrated that reduced podocyte production of VEGF leads to glomerular endothelial TMA (the hallmark of HUS). We have generated a considerable body of preliminary work showing that stx directly targets human podocytes to reduce podocyte VEGF secretion. Alongside this we have shown that VEGF upregulates protective complement factors on glomerular endothelial cells, and in vivo that podocyte specific VEGF knock out results in decreased glomerular endothelial Factor H expression and increased complement deposition in the glomerular endothelium. 84
Objectives
-
To investigate the time course of systemic complement activation in STEC HUS and its relation to the severity of disease.
-
To determine whether TMA in STEC HUS occurs via a stx-mediated reduction in podocyte VEGF production, leading to loss of complement regulation.
-
To test whether neutrophils derived from patients with acute STEC HUS deliver stx to podocytes.
-
To assess whether any genetic variations in patients with STEC HUS point to novel pathogenic mechanisms.
Methods
Urine samples were collected from patients at specified time points – some patients could not provide urine samples at all time points because of anuria. Urine CFH levels were measured in urine samples by enzyme-linked immunosorbent assay (ELISA) (Abcam) at days 1, 2, 4, 6, 8 and 30. Assays were performed according to the manufacturer’s instructions.
Urine VEGF levels were measured by ELISA (R + D systems) at days 1, 2, 4, 6, 8 and 30. Assays were performed according to the manufacturer’s instructions.
Urine podocytes from two patients were spun and quantified by Western blot analysis of podocyte specific proteins. Urinary markers of podocyte damage [nephrin and Wilms tumour-1 (WT-1)] were measured by ELISA of urine cell pellets according to methods previously described. 85
Plasma CFH levels were measured by ELISA (Abcam) at days 1 and 30 according to the manufacturer’s instructions. Plasma VEGF levels were measured by ELISA (R + D systems) at days 1 and 30 according to manufacturer’s instructions.
Measurement of serum complement activation products (Bb, C3a, C4a and sC5b9) by ELISA was outsourced to Exsera BioLabs, Aurora, USA.
Complement activation was also assessed by a novel technique, termed ‘degradomics’, in collaboration with Professor Markus Rinschen (University of Koln). This technique detects substrates degraded by proteases (e.g. components of the complement activation pathway). This employs a new field of positional proteomics or terminomics aimed at identifying protein N- or C-terminal modifications of protease substrates using mass spectrometry. Degradomics was performed based on the techniques described by the Rinschen group. 86 The method was modified in order to use ethylenediaminetetraacetic acid plasma samples from patients.
Exploration of the mechanism of stx on surface complement factor expression was performed using co-culture experiments. Co-culture models of human conditionally immortalised podocytes and glomerular endothelial cells were set up, to explore the hypothesis that stx acts via podocytes to tune down complement regulators on the surface of endothelial cells, as shown in Figure 8. 87,88 Cells were cultured in endothelial media without VEGF. They were serum starved for one hour before stx experiments. Co-cultures were performed in transwells with endothelial cells at the bottom, podocytes at the top and stx added to podocytes. The final stx concentration was 0.1 ng/ul. Glomerular cell surface CFH and C3d were assessed by confocal microscopy and semiquantified.
FIGURE 8.
Schematic showing co-culture experiments using podocytes and glomerular endothelial cells.
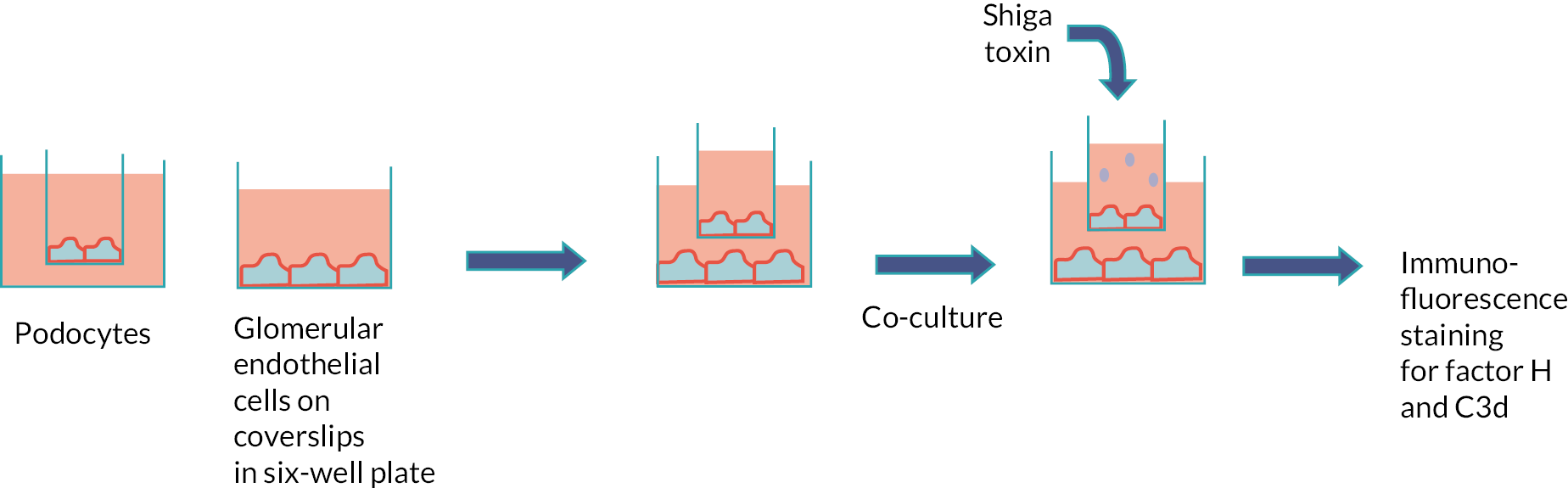
Insufficient patient samples were obtained to allow assessment of the delivery of stx to podocytes from patient-derived neutrophils.
Whole exome sequencing for 30× coverage and variant calling was provided by Eurofins Genomics, Wolverhampton, UK.
Serum anti-CFH antibodies were measured by ELISA as previously described. 89
Outcome measures
-
Plasma and urine VEGF and CFH levels.
-
Urinary markers of podocyte damage (nephrin and WT-1).
-
Plasma complement activation products.
-
Immunofluorescence staining and quantitative ELISA for endothelial cell factor H and C3d following co-culture experiments.
-
Whole exome sequencing, plasma anti-CFH antibody levels.
Results
Of the 36 participants recruited to the main trial, 32 consented to take part in the mechanistic studies and provided blood and/or urine samples. In anuric participants, only blood samples were collected.
Urine complement factor H and vascular endothelial growth factor levels in serial samples
We explored the hypothesis that stx-mediated damage to podocytes will lead to shedding of CFH and VEGF in the urine during the acute phase of the disease. The highest urine CFH levels were at day 1 (150 ng/ml), diminishing by day 4 (30 ng/ml) and completely normalising by day 30 (undetectable) as shown in Figure 9.
FIGURE 9.
Concentration of factor H in serial urine samples of participants in the ECUSTEC trial.
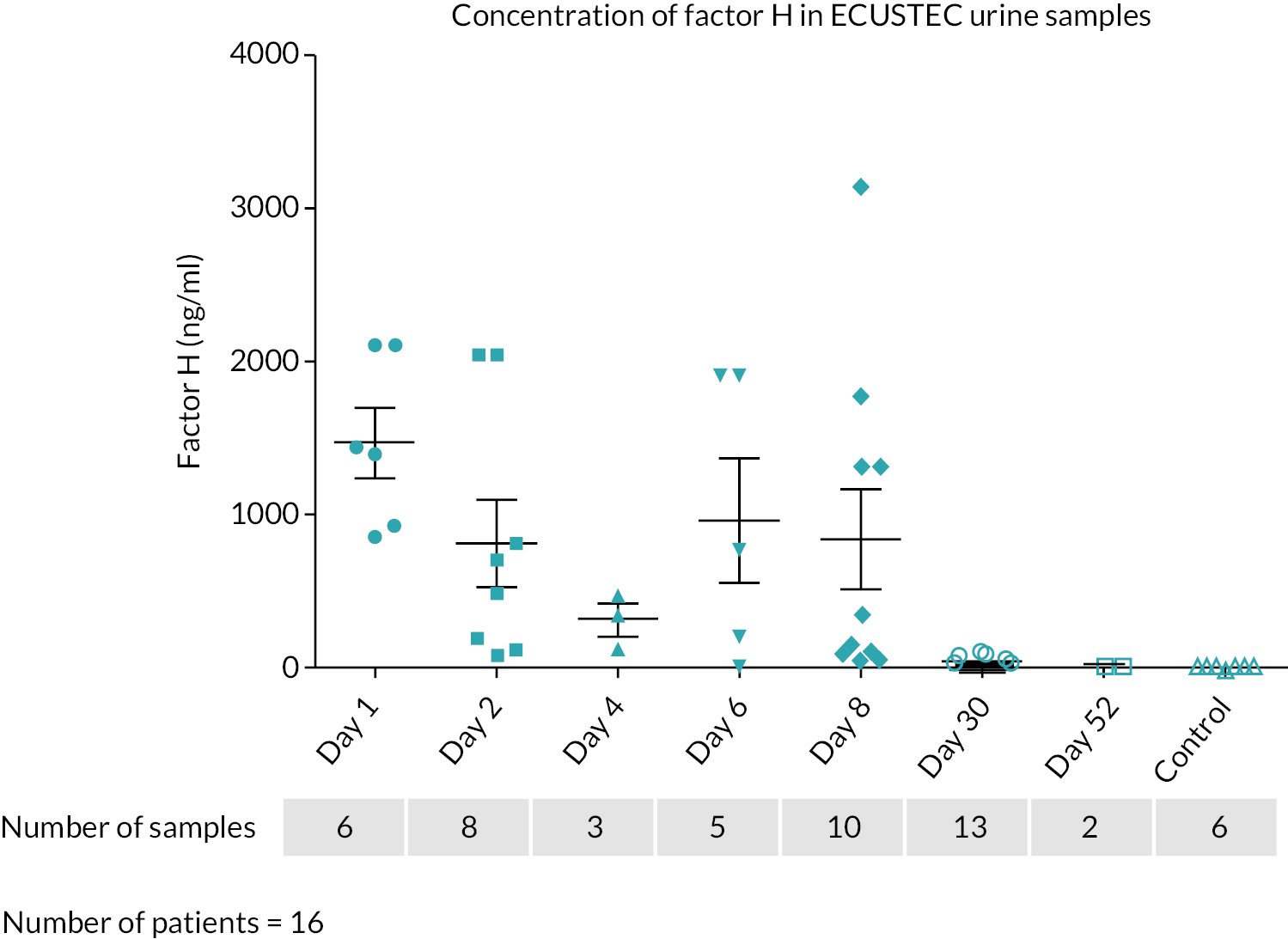
The highest urine VEGF levels were at day 1 (average 1300 ng/ml) and by day 30 the levels were below 20 ng/ml, as shown in Figure 10.
FIGURE 10.
Concentration of VEGF in serial urine samples of participants in the ECUSTEC trial.
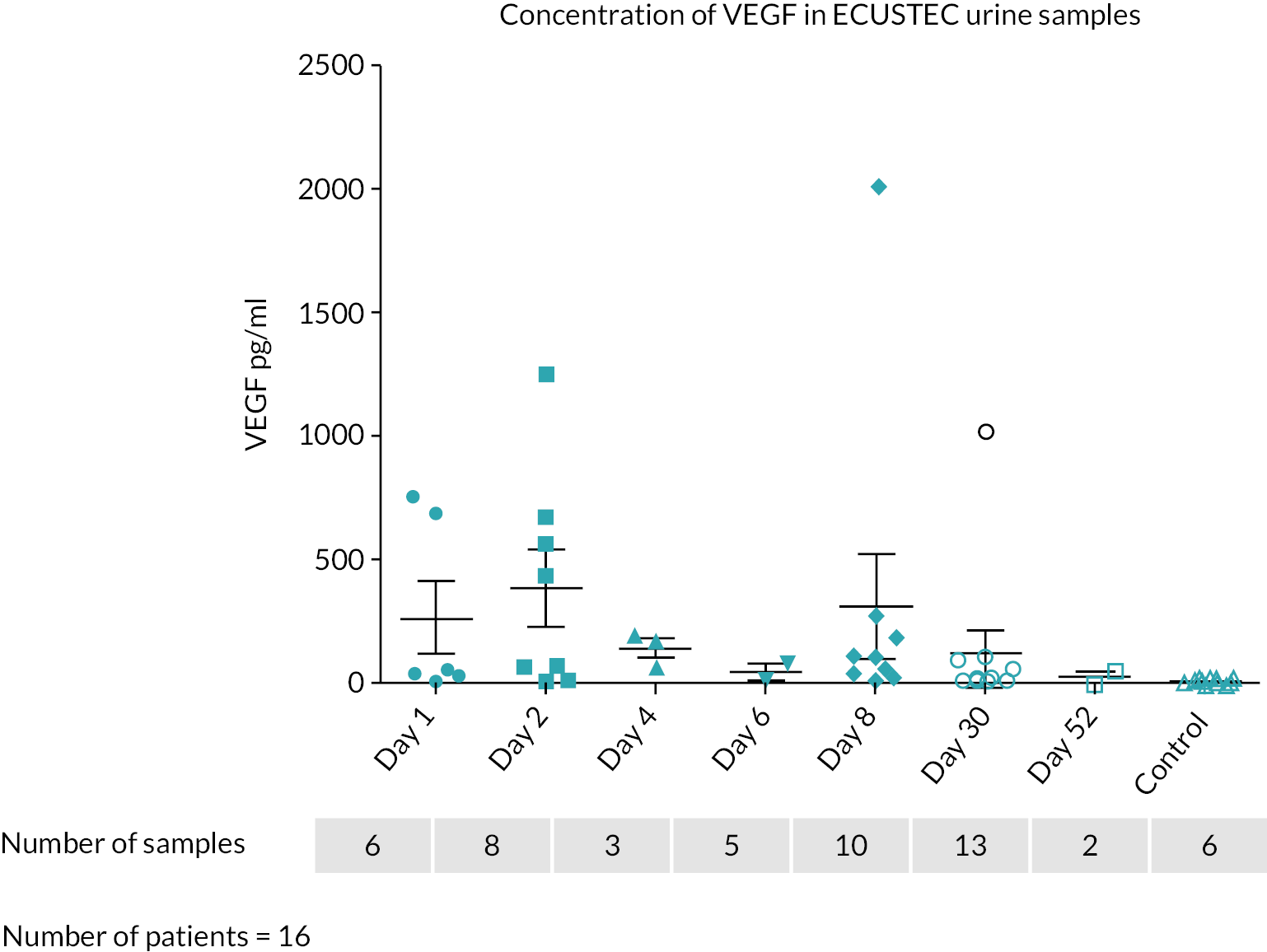
Markers of podocyte damage
In both participants studied, nephrin and WT-1 were clearly evident in the urine at day 1 as shown in Figure 11. WT-1 remained detectable in the urine until day 8 but was undetectable by day 30 in both patients. This is compatible with acute podocyte loss during active disease.
FIGURE 11.
Urinary cells from two participants in ECUSTEC trial probed with antibodies against podocyte specific proteins, nephrin and WT-1.
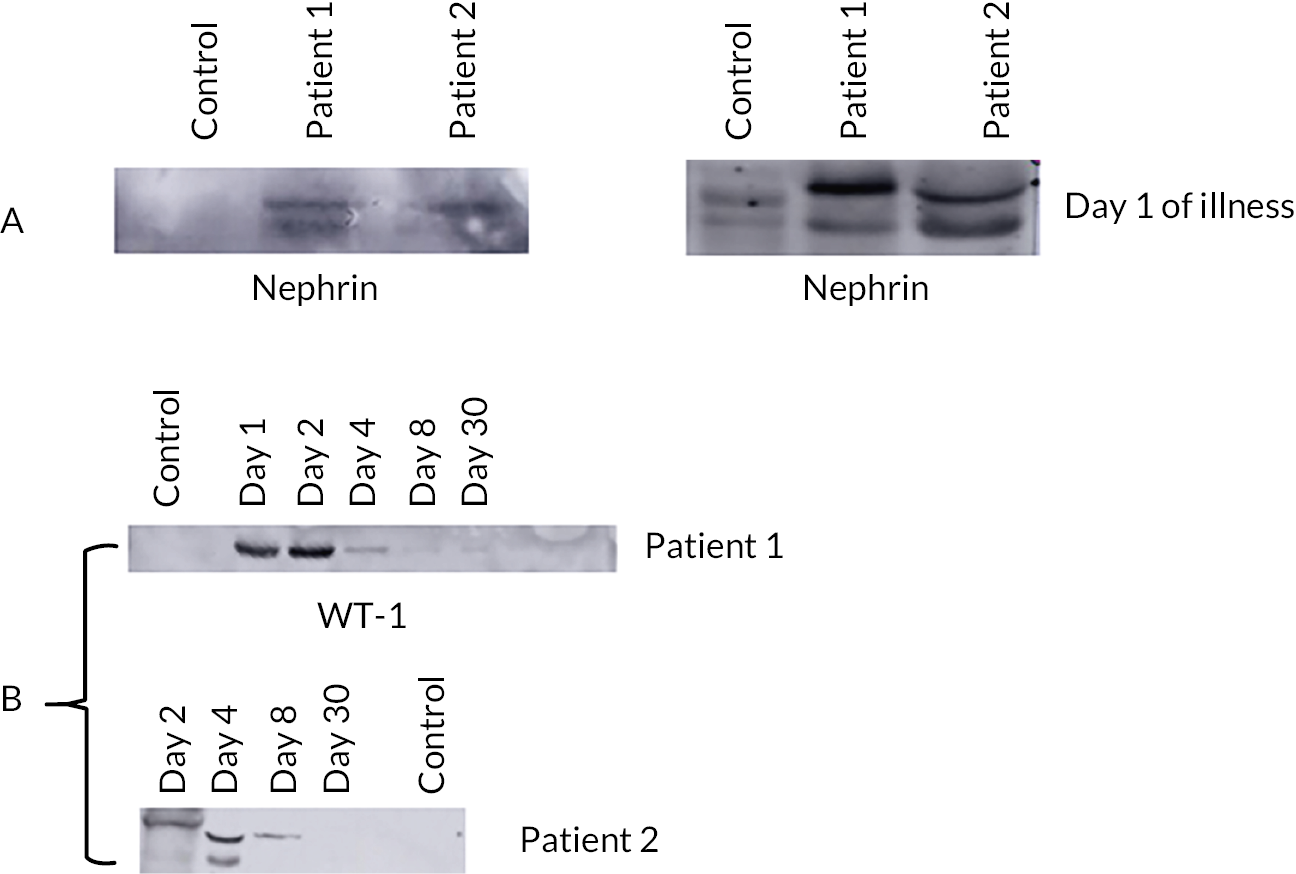
Plasma complement factor H and vascular endothelial growth factor levels
No difference was seen in the plasma levels of CFH (as shown in Figure 12) and VEGF (data not shown) between day 1 and day 30.
FIGURE 12.
Concentration of factor H in plasma samples of participants in the ECUSTEC trial.
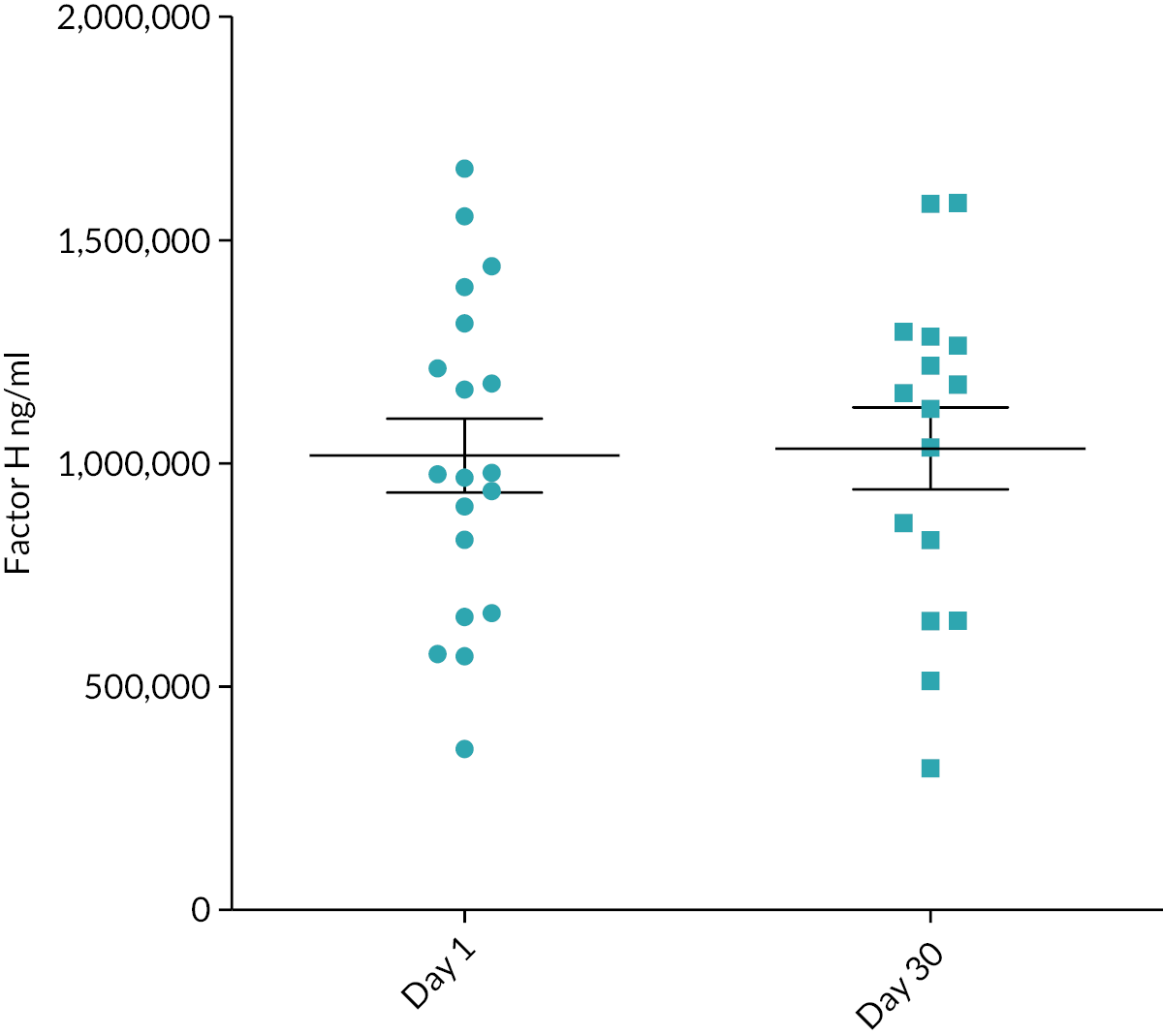
Plasma degradomics analysis
We tested samples (at days 1 and 3) from five participants in a pilot experiment. The N-termini consistent with C3 and C4 activation were much more abundant at day 1 (with overall estimated protein abundance as the control) than at day 30, as shown in Figure 13.
FIGURE 13.
C3 and C4 degradomics data for five participants in the ECUSTEC trial.
Plasma complement activation products
Table 16 shows the results of ELISA assays for plasma levels of complement activation products at serial time points.
| Eculizumab | Placebo | |||
|---|---|---|---|---|
| Analyte | ||||
| Bb | Day 1 | Mean (SD, n) | 4.38 (2.06, 9) | 10.38 (16.77, 9) |
| Range | 0.32–7.50 | 0.06–54.41 | ||
| Day 2 | Mean (SD, n) | 5.91 (7.41, 9) | 4.09 (3.47, 8) | |
| Range | 0.35–25.03 | 0.35–10.92 | ||
| Day 4 | Mean (SD, n) | 4.90 (5.53, 7) | 3.16 (2.17, 6) | |
| Range | 1.31–16.88 | 0.29–5.92 | ||
| Day 6 | Mean (SD, n) | 1.37 (0.81, 5) | 6.21 (5.79, 6) | |
| Range | 0.33–2.25 | 1.01–14.41 | ||
| Day 8 | Mean (SD, n) | 1.42 (0.62, 11) | 3.47 (6.14, 9) | |
| Range | 0.32–2.34 | 0.89–19.81 | ||
| Day 30 | Mean (SD, n) | 1.05 (0.46, 8) | 0.80 (0.38, 6) | |
| Range | 0.62–1.96 | 0.33–1.23 | ||
| C3a | Day 1 | Mean (SD, n) | 175.55 (64.24, 9) | 195.95 (71.10, 9) |
| Range | 84.88–259.61 | 134.41–371.61 | ||
| Day 2 | Mean (SD, n) | 168.35 (82.12, 9) | 163.53 (92.44, 8) | |
| Range | 82.30–282.51 | 59.71–320.43 | ||
| Day 4 | Mean (SD, n) | 134.53 (87.20, 7) | 203.44 (69.09, 6) | |
| Range | 33.62–256.84 | 102.19–292.20 | ||
| Day 6 | Mean (SD, n) | 100.64 (48.82, 5) | 253.35 (163.03, 6) | |
| Range | 47.90–178.93 | 76.84–444.85 | ||
| Day 8 | Mean (SD, n) | 173.94 (188.68, 11) | 212.91 (101.89, 9) | |
| Range | 51.22–721.40 | 92.69–377.40 | ||
| Day 30 | Mean (SD, n) | 44.76 (10.02, 8) | 68.39 (57.95, 6) | |
| Range | 35.79–61.88 | 33.77–181.05 | ||
| C4a | Day 1 | Mean (SD, n) | 3852.01 (1138.20, 9) | 3969.93 (1547.68, 9) |
| Range | 2559.29–6471.08 | 2260.00–7745.64 | ||
| Day 2 | Mean (SD, n) | 3025.55 (791.09, 9) | 3572.99 (2545.37, 8) | |
| Range | 2007.41–3945.87 | 1747.86–9447.02 | ||
| Day 4 | Mean (SD, n) | 3422.65 (2292.23, 7) | 2672.90 (792.35, 6) | |
| Range | 977.13–8089.71 | 1445.92–3503.02 | ||
| Day 6 | Mean (SD, n) | 3067.15 (715.18, 5) | 4347.56 (2865.62, 6) | |
| Range | 2125.91–3674.70 | 2254.52–9966.68 | ||
| Day 8 | Mean (SD, n) | 3424.80 (2203.63, 11) | 2843.56 (974.85, 9) | |
| Range | 1584.54–9614.02 | 705.28–3879.71 | ||
| Day 30 | Mean (SD, n) | 1622.61 (453.32, 8) | 1992.23 (936.04, 6) | |
| Range | 856.26–2347.47 | 1161.74–3511.49 | ||
| sC5b9 | Day 1 | Mean (SD, n) | 349.72 (198.12, 9) | 467.62 (192.20, 9) |
| Range | 29.77–721.67 | 225.66–806.90 | ||
| Day 2 | Mean (SD, n) | 255.62 (127.24, 9) | 356.03 (241.36, 8) | |
| Range | 34.58–463.50 | 36.99–790.98 | ||
| Day 4 | Mean (SD, n) | 354.88 (124.05, 7) | 487.14 (363.27, 6) | |
| Range | 203.43–569.20 | 41.19–1100.19 | ||
| Day 6 | Mean (SD, n) | 442.52 (323.09, 5) | 514.44 (318.86, 6) | |
| Range | 127.99–794.18 | 163.85–1093.58 | ||
| Day 8 | Mean (SD, n) | 282.74 (258.57, 11) | 367.11 (193.06, 9) | |
| Range | 41.79–1017.96 | 183.63–838.15 | ||
| Day 30 | Mean (SD, n) | 222.86 (136.62, 8) | 117.57 (69.01, 6) | |
| Range | 116.92–541.47 | 30.37–179.32 | ||
Mean plasma levels of Bb were elevated in both groups at baseline, day 2 and day 4. At day 6 and day 8, Bb levels remained elevated in the placebo group but were normal in the eculizumab group. In both groups, mean Bb levels were in the normal range at day 30 (Figure 14).
FIGURE 14.
Plasma levels of Bb in ECUSTEC participants.

Mean plasma levels of C3a were elevated in both groups at baseline and at days 2 and 4. At days 6 and 8, mean levels remained elevated in the placebo group while mean levels in the eculizumab group were in the normal range. Levels were in the normal range for both groups by day 30 (Figure 15). Mean plasma levels of C4a were elevated at all time points in both groups but fell at day 30 (Figure 16). Mean plasma levels of sC5b9 were high throughout baseline to day 8 in both groups and fell by day 30, when mean levels in the placebo group were normal and in the eculizumab group were marginally elevated (Figure 17).
FIGURE 15.
Plasma levels of C3a in ECUSTEC participants.
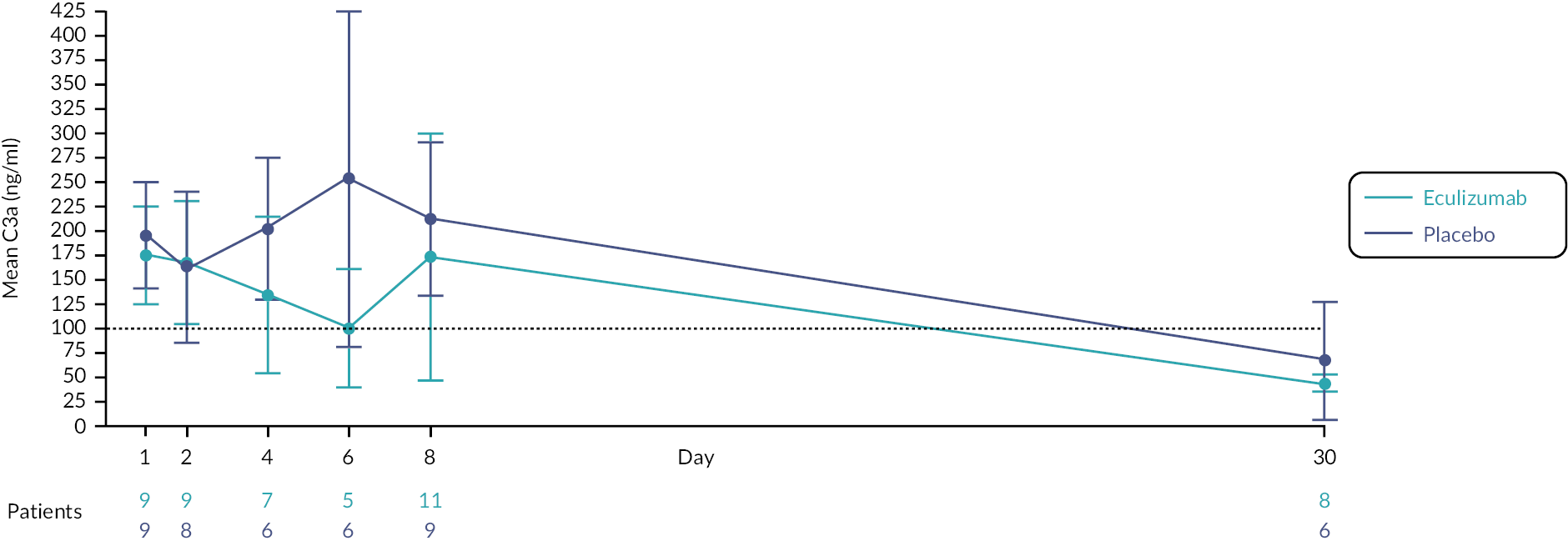
FIGURE 16.
Plasma levels of C4a in ECUSTEC participants.
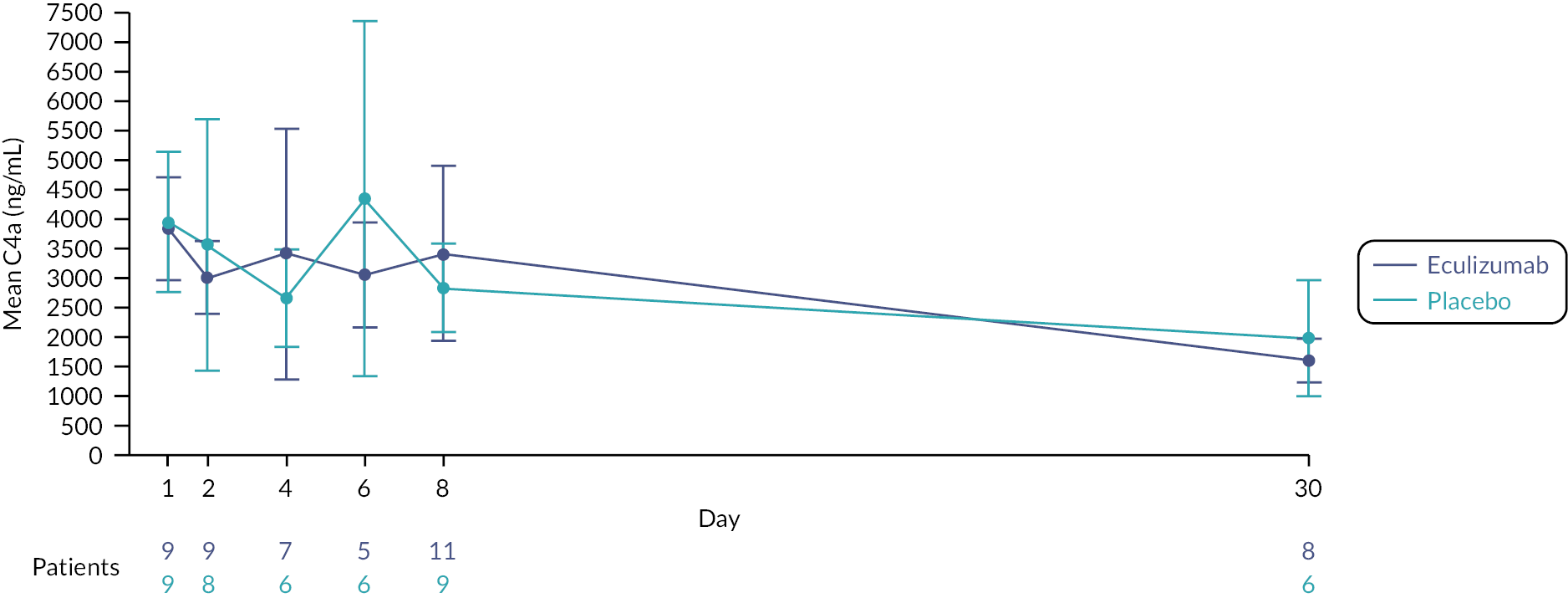
FIGURE 17.
Plasma levels of sC5b9 in ECUSTEC participants.
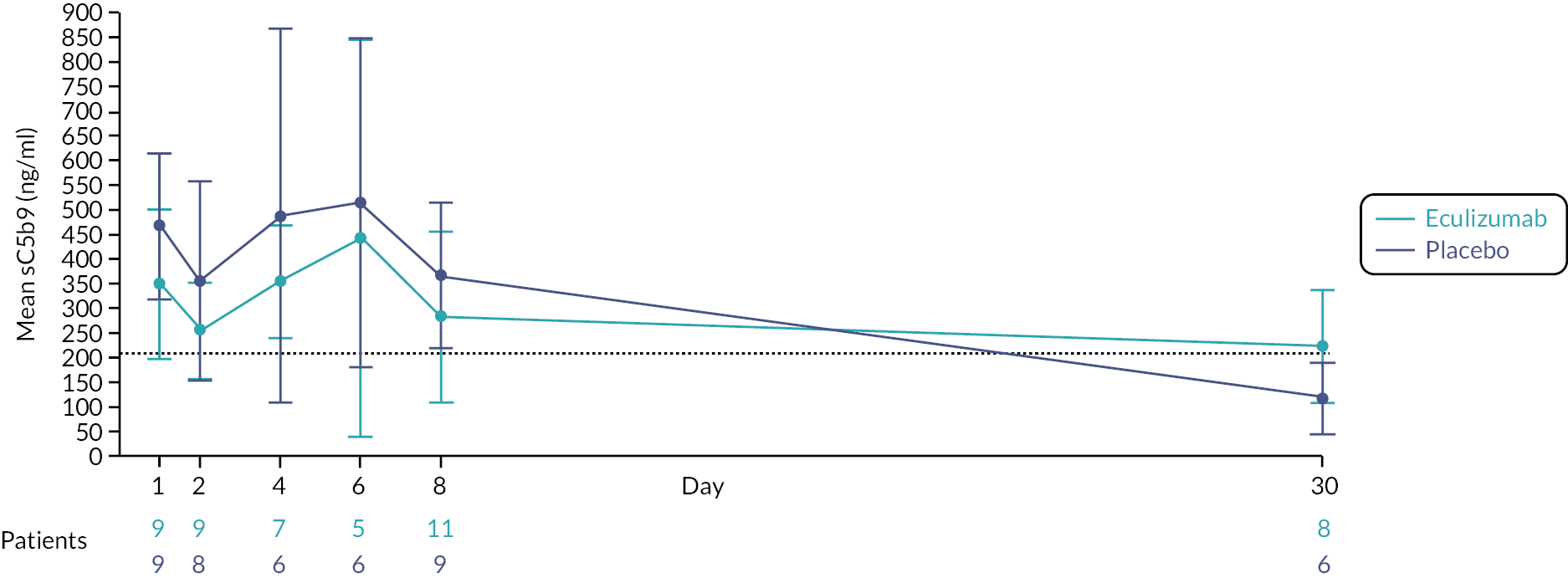
In the placebo group, a linear relationship was not established between CSS and baseline Bb (r = 0.43, p = 0.2); C3a (r = −0.16, p = 0.7); C4a (r = 0.15, p = 0.7) or sC5b9 (r = −0.17, p = 0.7) as demonstrated in Figure 18. Similarly, a linear relationship was not established between CSS and the maximum value of Bb (r = 0.45, p = 0.1); C3a (r = 0.15, p = 0.6); C4a (r = 0.23, p = 0.4); or sC5b9 (r = −0.22, p = 0.5) also shown in Figure 19.
FIGURE 18.
Relationship between baseline plasma complement activation products and CSS (placebo group only).
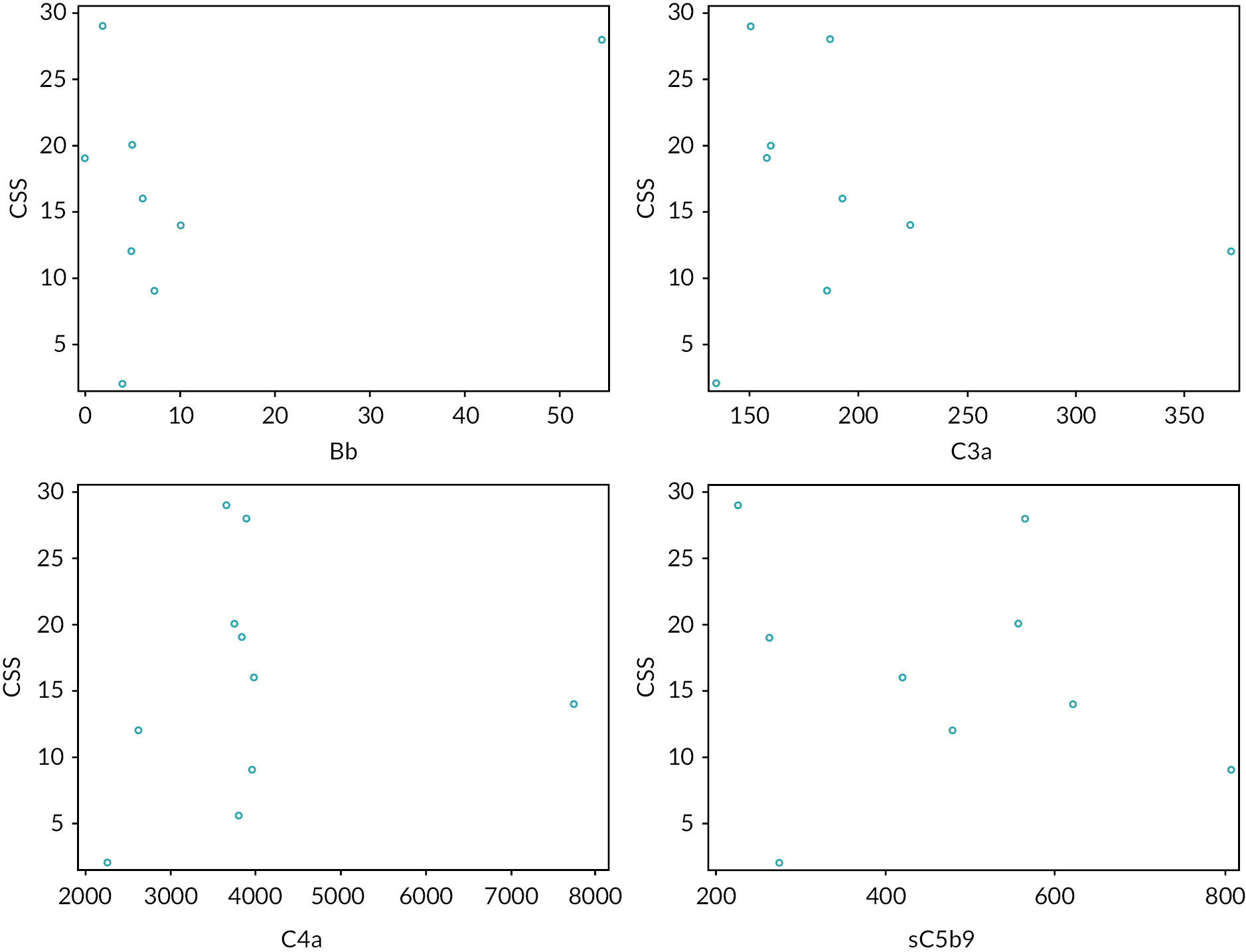
FIGURE 19.
Relationship between maximum plasma complement activation products and CSS (placebo group only).
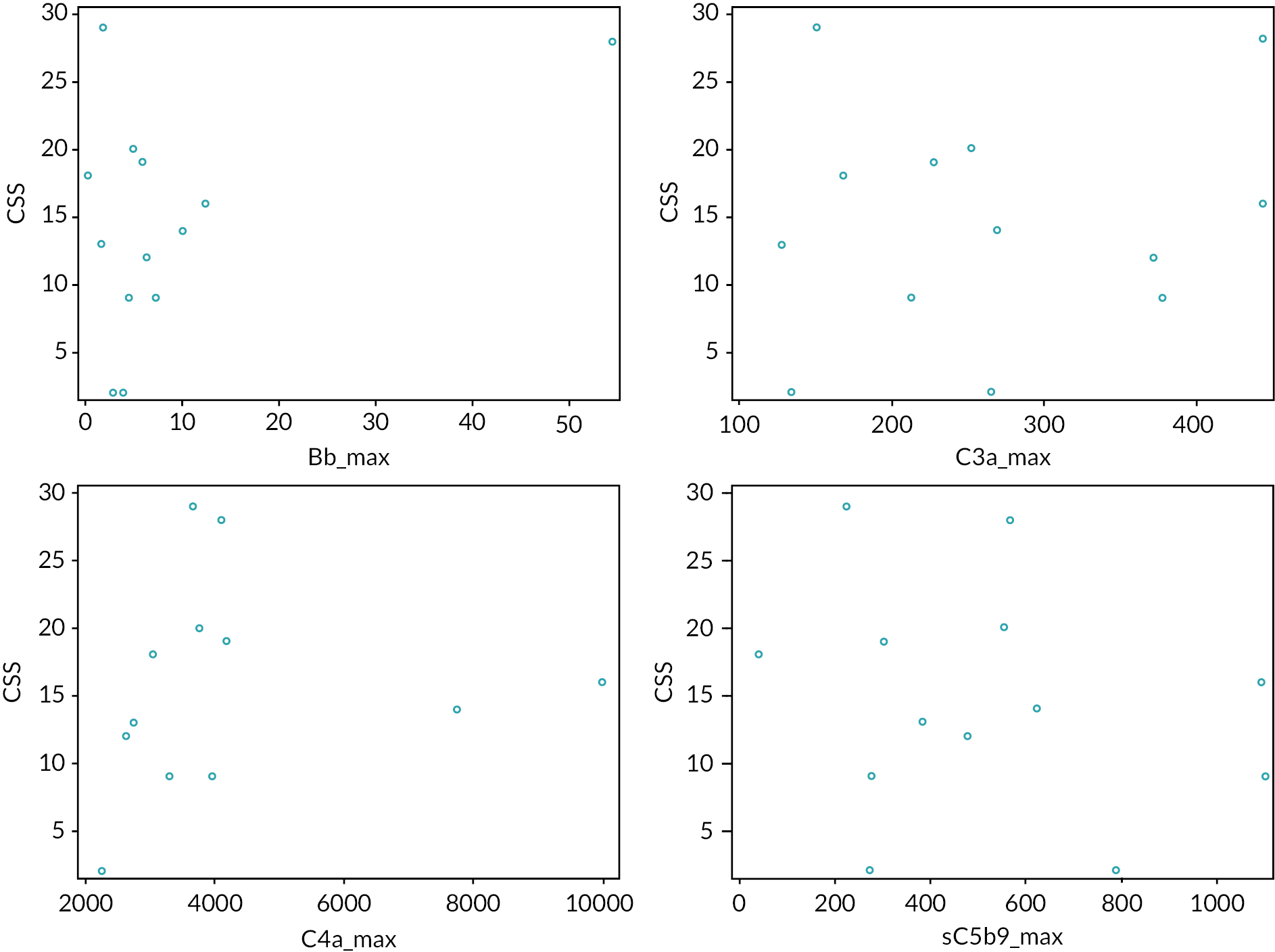
In vitro cell co-culture work to explore the mechanism of stx on surface complement factor expression
In co-culture models of human conditionally immortalised podocytes and glomerular endothelial cells, stx caused a decrease in CFH and an increase in C3d (a complement activation product) on glomerular endothelial cells (as shown in Figure 20). The presence of podocytes was critical for this change since stx had no effect on these factors when added to endothelial cells alone. We also showed that supernatant from podocytes exposed to stx had the same effect on glomerular endothelial cells (Figure 21).
FIGURE 20.
Endothelial cell surface CFH and C3d levels after exposure to stx in co-culture with podocytes.
FIGURE 21.
Surface endothelial factor H in response to supernatant from stx-treated podocytes.
Genetic variations in patients with Shiga-toxin-producing Escherichia coli haemolytic uraemic syndrome
Data from whole exome sequencing have been obtained and analysis is ongoing.
Discussion
Overall, we have explored the mechanistic hypothesis that complement activity occurs early in the glomerulus in STEC HUS, and is initiated by stx targeting the podocyte, leading to cross-talk to glomerular endothelial cells, potentially via the soluble mediator VEGF. We hypothesised that this cross-talk leads to downregulation of the protective CFH on the surface of endothelial cells and concurrent activation of complement on the endothelial cell surface. All of the lines of evidence we have gathered strongly support this hypothesis.
Firstly, we tested the hypothesis that stx-mediated damage to podocytes leads to shedding of CFH and VEGF in the urine during the acute phase of the disease. This led us to demonstrate that CFH and VEGF levels in the urine can be explored as biomarkers of the earliest phase of disease. As a control experiment, there was no change in plasma levels of CFH or of VEGF.
Next, we tested whether we could detect complement activation in the plasma, again at the earliest stage of disease. Using a novel proteomics technology that enables proteome-wide identification, mapping and quantification of protein N-termini to comprehensively characterise cleaved plasma proteins, we could demonstrate upregulation of activated fragments of C3 and C4 complement components in a pilot study of five patient samples at days 1 and 3, with no change in overall abundance of C3 and C4. In addition, we assessed a range of complement activation products using ELISAs. These results showed evidence of both alternative and classical complement pathway activation in the acute phase of disease.
Finally, we tested whether podocytes are responsible for initiating changes in glomerular endothelial cell complement activation, by employing a co-culture model of human podocytes and glomerular endothelial cells. The results clearly demonstrate that stx downregulates CFH levels on glomerular endothelial cells and leads to loss of protection from complement activation only in the presence of podocytes. We also showed that supernatant from podocytes exposed to stx had the same effect. Therefore, we propose that there is a soluble mediator or mediators, released by the podocyte, that downregulates glomerular endothelial CFH levels, thus rendering them susceptible to complement activation (demonstrated in our model by C3d deposition). Further work has started to use proteomics to analyse the podocyte supernatant in order to discover the soluble factor(s) responsible.
Conclusions
We have established that urine CFH and VEGF levels are sensitive measures of disease activity in STEC HUS and could therefore be explored as new biomarkers of acute disease. We have demonstrated complement activation early in disease in plasma from patients with STEC HUS using sophisticated proteomics technology and ELISAs. Co-culture cell work demonstrated that podocyte cross-talk is responsible for reducing glomerular endothelial cell CFH expression and that this results in complement activation on the glomerular endothelial cell surface. Collectively, this work strongly supports the mechanistic hypothesis of a STEC HUS as a complement-mediated disease, driven via the podocyte as the target cell for stx.
Future work
We plan to undertake proteomics analysis of podocyte supernatant after treatment with stx in order to discover the relevant soluble cross-talk factors influencing endothelial cell complement regulators. We also plan to further develop early plasma and urine biomarkers for clinical detection and diagnosis of STEC HUS, including the degradomics-based complement assay and urine biomarkers such as CFH and VEGF. One specific aim would be to test whether urine VEGF can be used as a biomarker of STEC HUS versus aHUS, in order to make a definitive early diagnosis in cases where there is clinical doubt.
We will explore testing of patient plasma and neutrophils for the presence of outer membrane vesicles containing stx.
Subsequent to this work, Professor Saleem and colleagues have developed a mouse model that recapitulates STEC HUS by targeting stx to the glomerular podocyte by exclusively expressing the Gb3 receptor on this cell type. 28 In this model, inhibition of the terminal complement pathway by C5 blockade prior to stx exposure prevented the development of STEC HUS. They have also undertaken further co-culture work to confirm that stx binds to Gb3 on human podocytes, leading to reduced cell surface heparan sulphate expression and CFH binding on human glomerular endothelial cells, and evidence of endothelial cell surface complement activation (C3b and C5b-9 deposition).
Chapter 5 Discussion
Main findings
In this multicentre, randomised, double-blind, placebo-controlled trial in children with STEC HUS, we found that two doses of eculizumab did not reduce disease severity. The mean CSSs at day 60 were similar between those randomised to eculizumab and those randomised to placebo. However, the trial was stopped early due to low recruitment, and only recruited 36 of the 134 target sample size, meaning the trial was under powered, and so this cannot be interpreted as evidence of no effect of the intervention.
Although a reduction in disease incidence contributed to the low recruitment to the trial, the lack of out-of-hours infrastructure to deliver a CTIMP that required urgent intervention in children is important to consider when developing future trials for children with this and other acute conditions. STEC HUS is a rare disease and small outbreaks can have a moderate impact on annual incidence. A reduction in incidence during the trial period could have been due to a minor difference in the number of small outbreaks and does not necessarily mean that there is a permanent reduction in incidence.
In the mechanistic studies, we have established that urine CFH and VEGF levels are sensitive measures of disease activity in STEC HUS. We have demonstrated complement activation in plasma from patients with STEC HUS using sophisticated proteomics technology. Co-culture cell work demonstrated that podocyte cross-talk is responsible for reducing glomerular endothelial cell CFH expression and that this results in complement activation on the glomerular endothelial cell surface. Collectively, this work strongly supports the mechanistic hypothesis of STEC HUS as a complement-mediated disease, driven via the podocyte as the target cell for stx.
Strengths
The robust study design included blinding of the treatment allocation to participants and investigators. In addition, all participants received vaccination and prophylactic antibiotics regardless of treatment allocation in order to maintain blinding. All investigators demonstrated true equipoise regarding the intervention, in that no patients in the control group were unblinded and given eculizumab.
The primary outcome measure, the ECUSTEC CSS, captured overall disease severity at day 60. Previous trials in STEC HUS have used purely renal outcome measures, which only represents part of the disease course. This CSS gives a global assessment of disease severity. We believe that this CSS is a promising tool to test future treatments for STEC HUS, although further validation of the score is required.
The involvement of patients and families in all stages of our trial design and implementation was a key strength. Approximately 50% of families who were approached for the trial consented to their child’s participation. In any trial setting, this would be an excellent acceptance rate, but given the acute circumstances, this demonstrates the success of patient and family involvement in the trial design, and the skill and sensitivity of the clinical teams across the country.
The trial protocol was developed with and approved by representatives from all trial centres (which comprised 12 of the 13 UK centres), including an agreement to standardise supportive care, which demonstrates the commitment of clinical teams to testing a potential treatment in this condition. Adherence to the protocol was very high, with only one patient who did not receive treatment according to protocol. The retention of participants and the completeness of their follow-up following randomisation was also very high. This further demonstrates the commitment of clinical teams to the trial.
The inclusion of mechanistic substudies is also a strength of this study. These have furthered the evidence for complement activation in STEC HUS and show a clear mechanism for this activation. They have also provided novel biomarkers of STEC HUS for further exploration, which may lead to clinically useful predictors of severe disease.
Limitations
The main limitation of our study was that, due to the trial being closed early to recruitment, we had limited data and the study was under-powered to conclude whether there was a difference between groups. We calculated the sample size based upon achieving a clinically meaningful reduction in CSS score of 5 points. The original sample size of 134 participants would have provided 80% power to detect such a reduction. Since the trial recruited only 36 participants, there are insufficient data and power to make any conclusions on the use of eculizumab in this clinical setting.
A number of strategies were employed to increase recruitment. The administration window for the treatment was increased. Consideration was also given to international collaboration, but this was not considered feasible. The onset of the COVID-19 pandemic meant that any mitigation strategies were unlikely to make up for previous poor recruitment, and a decision was made to close the trial early by the funder.
It is possible that some differences between treatment groups at baseline may have affected outcome measures (see Table 7). The groups were well balanced and comparable in all baseline characteristics except for age, weight and proportion with anuria – the eculizumab group had a lower mean age than the placebo group (4.8 years compared with 6.4 years) and there was a corresponding difference in mean weight (18.9 kg in the eculizumab group compared with 26.0 kg in the placebo group). A higher proportion of patients in the eculizumab group were anuric for > 12 hours (9/15, 60% of the eculizumab group and 2/18, 11% of the placebo group). These are likely to be chance imbalances, since patients were randomised into the trial, and is likely to have occurred due to the small number of participants. There is conflicting evidence about whether age at onset impacts outcome of STEC HUS – some studies indicate that younger age is associated with worse outcome, while other studies do not. The trial did not show a difference in severity score between groups, and it is unlikely that the imbalance in age, weight and proportion with anuria > 12 hours between groups is of significance in the interpretation of these data.
Interpretation of findings
Due to the ECUSTEC trial being closed early to recruitment, we are unable to answer the research question of whether treatment with eculizumab reduces the severity of STEC HUS in children aged 6 months–< 19 years. Following our experience with this trial, we recommend that consideration is given to reinforcing children’s research infrastructure, so that CTIMPs can be delivered out of standard working hours, so that treatments for acutely unwell children can be assessed in clinical trials in this setting, and thus improve the future treatment of these children.
Public and patient involvement
We have been supported before and during the trial by the charity HUSH (HUSH haemolytic uraemic syndrome) E. coli and in particular its founder. We were also supported by parent members of the paediatric nephrology clinical studies group and parents whose children had experienced STEC HUS. Patient and parent involvement (PPI) was crucial for designing a trial that was acceptable to the families of acutely unwell children. Our patient and parent information material was reviewed and modified by our patient and parent partners, both at the outset of the trial and when an amendment was required after feedback from local teams. Our patient and parent partners helped us to gather participant experience from the trial in order to review acceptability as the trial progressed. We particularly appreciate the support of our partners when liaising with the funder regarding continuation of the trial. We will engage with our partners regarding the reporting and dissemination of this study. We are sorry that we have been unable to answer the primary research question for our patient and parent partners. A summary of the PPI for the ECUSTEC trial is presented in Table 17.
| Section and topic | Item |
|---|---|
| 1: Aim | The aims of PPI in the ECUSTEC trial were to:
|
| 2: Methods | Parent members of the paediatric nephrology clinical studies group and families of children who had experienced STEC HUS were approached. The patient support group HUSH E. coli was also approached. Interested individuals formed a focus group that worked with the chief investigator, supported by PPI co-ordinator from the Sponsor organisation. The focus group was convened at appropriate times during the course of the study. The PPI partners devised a questionnaire to obtain feedback from participants in case any aspects of the trial were distressing to families. |
| 3: Study results | PPI was a very positive factor in the trial. Our trial design was acceptable to families, and the patient facing information contributed to a high participation rate in those approached for the trial. PPI was also crucial in us continuing the trial, despite recruitment difficulties, by helping highlight the importance of the trial to the funder. |
| 4: Discussion and conclusions | We think the high acceptability of the trial and excellent retention were largely down to the involvement of our PPI partners. |
| 5: Reflections/critical perspective | It is important to start PPI as early as possible in the process, before even considering a research question or trial protocol. While it can take time to build the right group and ensure good representation, it is an essential step in developing and delivering a high-quality trial. |
Equality, diversity and inclusion
Our trial centred upon acutely unwell children and their families – children are an under-represented group in research. In order to maximise participation, we included parents of children who had experienced STEC HUS in the design of our protocol. We also set up participant identification centres in referring units so that information could be given to families prior to their arrival at the renal unit.
The age profile of our participants reflects the typical age range of patients with STEC HUS, so we do not think that particular age groups were under-represented in our trial. Local research teams were permitted to utilise translators to recruit participants without English as a first language; however, we could have improved representation by providing information sheets in a number of languages.
Gaps in prior evidence were identified by literature review and by informal consultation with international colleagues with STEC HUS expertise and experience.
Our patient information was reviewed by a panel of parents and young people to make sure it was inclusive and accessible to different age groups. The panel was geographically representative of the patient population. During the trial, some of the information was simplified into an infographic following feedback. In order to increase accessibility and inclusivity in future studies, it would be important to ensure diversity in the patient panel.
Our research team was diverse in terms of age, gender, ethnicity and disability. Junior members of the team were given opportunities to contribute with skilled supervision from senior members.
Chapter 6 Conclusions
Implications for practice
No conclusions can be drawn about the efficacy of eculizumab in STEC HUS from the ECUSTEC trial, and so unfortunately, the data are unable to inform clinical practice. However, it does add to the knowledge base of the use of eculizumab in STEC HUS, and may contribute to future meta-analyses.
From our mechanistic studies, we have established that urine CFH and VEGF levels are sensitive measures of disease activity in STEC HUS and could therefore be explored as new biomarkers of acute disease. We have demonstrated complement activation early in disease in plasma from patients with STEC HUS using sophisticated proteomics technology and ELISAs. Co-culture cell work demonstrated that podocyte cross-talk is responsible for reducing glomerular endothelial cell CFH expression and that this results in complement activation on the glomerular endothelial cell surface. Collectively, this work strongly supports the mechanistic hypothesis of STEC HUS as a complement-mediated disease, driven via the podocyte as the target cell for stx.
Recommendations for future research
There remains a significant unmet need for children with STEC HUS which has no effective treatment. We await the results of a trial of administration of a polyclonal antibody in children with STEC infection prior to the development of HUS, which began subsequent to commencement of the ECUSTEC trial (NCT04132375, Immunova S.A.), and of a trial of azithromycin in children with STEC HUS (ZITHROSHU) (NCT02336516). However, as potential agents to treat STEC HUS arise, it is vital that the infrastructure to deliver an intervention to acutely unwell children is in place. For future trials, we think that the ECUSTEC CSS is a useful tool for future studies. We also think that overall survival, duration of thrombocytopenia, evidence of CKD at 52 weeks, eGFR measurement using a centralised cystatin C assay at 52 weeks, and persistent neurological defect at day 60 measured by structured expert assessment are feasible end points in this clinical group.
This trial attempted to evaluate an urgent intervention in acutely unwell children. One of the main factors leading to low recruitment was insufficient out-of-hours children’s research infrastructure. Specifically, IMP could not be administered outside of normal working hours due to a lack of suitably trained research staff on duty. We recommend that this provision is reviewed in order to successfully deliver similar trials in the future.
With regard to mechanistic studies, work is planned to undertake proteomics analysis of podocyte supernatant after treatment with stx in order to discover the relevant soluble cross-talk factors influencing endothelial cell complement regulators. We also plan to further develop early plasma and urine biomarkers for detection and diagnosis of STEC HUS, including the degradomics-based complement assay and urine biomarkers such as CFH and VEGF.
Additional information
Contributions of authors
Natalie Ives (https://orcid.org/0000-0002-1664-7541) (Senior Trial Statistician) contributed to the design and analysis of the trial, the interpretation of all components and drafting and editing of the final report.
Rebecca Woolley (https://orcid.org/0000-0001-5119-1431) (Trial Statistician) performed the interim and final analyses for the trial, contributed to the interpretation of the trial and the drafting and editing of the final report.
Moin A Saleem (https://orcid.org/0000-0002-9808-4518) (Professor of Paediatric Renal Medicine and Honorary Consultant Paediatric Nephrologist) contributed to the design, delivery and interpretation of the trial, the design, analysis and interpretation of the mechanistic studies and contributed to the drafting and editing of the final report.
Catherine A Moakes (https://orcid.org/0000-0002-0473-0532) (Trial Statistician) performed the final data cleaning and the final analyses for the trial, contributed to the interpretation of the trial and the drafting and editing of the final report.
Aoife Waters (https://orcid.org/0000-0003-4520-5635) (Consultant Paediatric Nephrologist) contributed to the design, delivery and interpretation of the trial and the drafting and editing of the final report.
Rodney D Gilbert (https://orcid.org/0000-0001-7426-0188) (Consultant Paediatric Nephrologist) contributed to the design, delivery and interpretation of the trial and the drafting and editing of the final report.
Hugh Jarrett (https://orcid.org/0000-0001-7374-3700) (Trials Management Team Leader) was responsible for the day-to-day management and delivery of the trial, contributed to drafting the report and editing of the final report.
Elizabeth Brettell (https://orcid.org/0000-0002-0669-3085) (Trials Management Team Leader) contributed to the design and delivery of the trial and editing of the final report.
Steve Nash (Campaigner on Escherichia Coli O157) contributed to the design and delivery of the trial and to the review of the final report.
Louise K Farmer (https://orcid.org/0000-0002-9462-2606) (Research Associate) contributed to the design, analysis and interpretation of the mechanistic studies.
Khadija Ourradi (https://orcid.org/0000-0002-9031-1931) (Research Associate) contributed to the design, analysis and interpretation of the mechanistic studies and the drafting of the report.
Sally A Johnson (https://orcid.org/0000-0001-9221-1347) (Consultant Paediatric Nephrologist and Chief Investigator) contributed to the design, delivery, analysis and interpretation of the trial, the drafting of the report and overall editing of the final report.
Acknowledgements
The ECUSTEC trial was made possible by the collaborative work of the following people:
Co-investigators
Sally Johnson, Newcastle Upon Tyne Hospitals NHS Foundation Trust, Newcastle, UK.
Moin Saleem, Bristol University, Bristol, UK.
Nicholas Webb, Manchester University NHS Foundation Trust, Manchester, UK (until May 2018).
Munir Ahmed, Worcester Acute Hospitals NHS Trust, Worcester, UK.
Rodney Gilbert, University Hospital Southampton NHS Trust, Southampton, UK.
Aoife Waters, Great Ormond Street Hospital for Children NHS Foundation Trust, London, UK.
Steve Nash, Consumer representative
Julia Newton, Newcastle University, Newcastle, UK (From June 2018).
Natalie Ives, University of Birmingham, Birmingham, UK.
Elizabeth Brettell, University of Birmingham, Birmingham, UK.
Trial Management Group
Sally Johnson, Newcastle Upon Tyne Hospitals NHS Foundation Trust, Newcastle, UK.
Moin Saleem, Bristol University, Bristol, UK.
Rebecca Woolley, University of Birmingham, Birmingham, UK.
Natalie Ives, University of Birmingham, Birmingham, UK.
Hugh Jarrett, University of Birmingham, Birmingham, UK.
Kelsey Thomas, Newcastle Upon Tyne Hospitals NHS Foundation Trust, Newcastle, UK.
Christopher Price, Newcastle Upon Tyne Hospitals NHS Foundation Trust, Newcastle, UK.
Birmingham Clinical Trials Unit staff
Emma Barsoum
Elizabeth Brettell
Raquel Fernández del Río
Matt Hill
Terry Hughes
Natalie Ives
Hugh Jarrett
Catherine Moakes
Rebecca Woolley
Collaborators
Claire Jenkins, Gastrointestinal Bacteria Reference Unit, UK Health Security Agency (formerly Public Health England), Colindale, London, UK.
Principal investigators and recruiting sites
Dr Caroline Jones, Alder Hey Children’s Hospital, Alder Hey Children’s NHS Foundation Trust, Liverpool, UK.
Dr Larissa Kerecuk, Birmingham Children’s Hospital, Birmingham Women’s and Children’s NHS Foundation Trust, Birmingham, UK.
Professor Moin Saleem, Bristol Royal Hospital for Children, University Hospitals Bristol NHS Foundation Trust, Bristol, UK.
Dr Manish Sinha, Evelina Children’s Hospital London, Guy’s and St Thomas’ NHS Foundation Trust, London, UK.
Dr Aoife Waters, Great Ormond Street Hospital, Great Ormond Street Hospital for Children NHS Foundation Trust, London, UK.
Dr Eric Finlay, Leeds General Infirmary, The Leeds Teaching Hospitals NHS Trust, Leeds, UK.
Dr Andrew Lunn, Nottingham Children’s Hospital, Nottingham University Hospitals NHS Trust, Nottingham, UK.
Dr Ben Reynolds, Royal Hospital for Children Glasgow, NHS Greater Glasgow and Clyde, Glasgow, UK.
Dr Amrit Kaur, Royal Manchester Children’s Hospital, Central Manchester University Hospitals NHS Foundation Trust, Manchester, UK.
Dr Rodney Gilbert, Southampton Children’s Hospital, University Hospital Southampton NHS Foundation Trust, Southampton, UK.
Dr Sally Johnson, The Great North Children’s Hospital, The Newcastle Upon Tyne Hospitals NHS Foundation Trust, Newcastle, UK.
Dr Shivaram Hegde, University Hospital of Wales, Cardiff and Vale University Health Board, Cardiff, UK.
Trial Steering Committee
Professor David Jayne, (Chairperson) University of Cambridge School of Clinical Medicine, Cambridge, UK.
Dr Nicole van de Kar, (Paediatric nephrologist) Radboud umc, Nijmegen, The Netherlands.
Dr Ly-Mee Yu (Statistician), University of Oxford, Oxford, UK.
Mrs Sarah Bryan (Patient advisor).
Mrs Sandra Cope (Patient advisor).
Mrs Wendy Cook (Patient advisor).
Mr Richard Patterson (Patient advisor).
Data and Safety Monitoring Committee
Professor David Wheeler, (Chairperson) University College London, London, UK.
Dr Mark Taylor, retired Consultant Paediatric Nephrologist.
Dr Matthew Sydes, University College London, London, UK.
Mechanistic study team
Professor Moin Saleem, Bristol University, Bristol, UK.
Dr Louise Farmer, Bristol University, Bristol, UK.
Dr Khadija Ourrada, Bristol University, Bristol, UK.
Dr Gavin Welsh, Bristol University, Bristol, UK.
Sponsor
The Sponsor was Newcastle Upon Tyne Hospitals NHS Foundation Trust.
Patient data statement
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it is important that there are safeguards to make sure that they are stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Ethics statement
The trial had a favourable ethics opinion from the North East – Newcastle and North Tyneside 1 Research Ethics Committee (REC reference number 16/NE/0325, date of approval 23/01/2017).
Information governance statement
Newcastle Upon Tyne Hospitals is committed to handling all personal information in line with the UK Data Protection Act (2018) and the General Data Protection Regulation (EU GDPR) 2016/679. Under the Data Protection legislation, Newcastle Upon Tyne Hospitals is the Data Controller, and you can find out more about how we handle personal data, including how to exercise your individual rights and the contact details for our Data Protection Officer here: https://www.newcastle-hospitals.nhs.uk/help/privacy/privacy-notice-for-patients/.
Disclosure of interests
Full disclosure of interests: Completed ICMJE forms for all authors, including all related interests, are available in the toolkit on the NIHR Journals Library report publication page at https://doi.org/10.3310/RFTY4766.
Primary conflicts of interest: Sally A Johnson’s institution has received funding from Alexion Pharmaceuticals Inc. and Novartis for being a Scientific Advisory Board member and for giving talks. Rodney D Gilbert has received honoraria for giving talks and attending advisory boards from Alexion Pharmaceuticals. He has also received fees for acting as a national co-ordinator for an Alexion global registry. Moin A Saleem has patents filed for adeno-associated virus renal gene therapy indications. He has received fees from Retrophin Inc., as an advisory board member and from Purespring Therapeutics for consultancy services. He has stock options for Purespring Therapeutics. Kidney Research UK awarded £24,000 for specific genomic and proteomic aspects of the trial.
Publications
Walsh PR, Johnson S. Eculizumab in the treatment of Shiga toxin Haemolytic Uraemic Syndrome. Pediatr Nephrol 2019;34(9):1485–92. 90
Disclaimers
This article presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
References
- Lynn RM, O’Brien SJ, Taylor CM, Adak GK, Chart H, Cheasty T, et al. Childhood hemolytic uremic syndrome, United Kingdom and Ireland. Emerg Infect Dis 2005;11:590-6.
- Walsh PR, Johnson S. Treatment and management of children with Haemolytic Uraemic Syndrome. Arch Dis Child 2018;103:285-91.
- Matthies J, Hunseler C, Ehren R, Volland R, Korber F, Hoppe B, et al. Extrarenal manifestations in Shigatoxin-associated haemolytic uremic syndrome. Klin Padiatr 2016;228:181-8.
- Oakes RS, Siegler RL, McReynolds MA, Pysher T, Pavia AT. Predictors of fatality in postdiarrheal hemolytic uremic syndrome. Pediatrics 2006;117:1656-62.
- Sheth KJ, Swick HM, Haworth N. Neurological involvement in hemolytic-uremic syndrome. Ann Neurol 1986;19:90-3.
- Siegler RL. The hemolytic uremic syndrome. Pediatr Clin North Am 1995;42:1505-29.
- Nathanson S, Kwon T, Elmaleh M, Charbit M, Launay EA, Harambat J, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol 2010;5:1218-28.
- Luna M, Kamariski M, Principi I, Bocanegra V, Valles PG. Severely ill pediatric patients with Shiga toxin-associated hemolytic uremic syndrome (STEC HUS) who suffered from multiple organ involvement in the early stage. Pediatr Nephrol 2021;36:1499-509.
- Garg AX, Suri RS, Barrowman N, Rehman F, Matsell D, Rosas-Arellano MP, et al. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: a systematic review, meta-analysis, and meta-regression. JAMA 2003;290:1360-70.
- Rangel JM, Sparling PH, Crowe C, Griffin PM, Swerdlow DL. Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982–2002. Emerg Infect Dis 2005;11:603-9.
- Chan YS, Ng TB. Shiga toxins: from structure and mechanism to applications. Appl Microbiol Biotechnol 2016;100:1597-610.
- Melton-Celsa AR. Shiga toxin (Stx) classification, structure, and function. Microbiol Spectr 2014;2:2-4.
- Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol 2013;33:508-30.
- Walport MJ. Complement. First of two parts. N Engl J Med 2001;344:1058-66.
- Cheung V, Trachtman H. Hemolytic uremic syndrome: toxins, vessels, and inflammation. Front Med (Lausanne) 2014;1.
- Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med 1977;89:845-50.
- Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol 1980;13:168-71.
- Balestracci A, Meni Bataglia L, Toledo I, Beaudoin L, Alvarado C. C3 levels and acute outcomes in Shiga toxin-related hemolytic uremic syndrome. Pediatr Nephrol 2020;35:331-9.
- Netti GS, Santangelo L, Paulucci L, Piscopo G, Torres DD, Carbone V, et al. Low C3 serum levels predict severe forms of STEC HUS with neurologic involvement. Front Med (Lausanne) 2020;7.
- Robson WL, Leung AK, Fick GH, McKenna AI. Hypocomplementemia and leukocytosis in diarrhea-associated hemolytic uremic syndrome. Nephron 1992;62:296-9.
- Ferraris JR, Ferraris V, Acquier AB, Sorroche PB, Saez MS, Ginaca A, et al. Activation of the alternative pathway of complement during the acute phase of typical Haemolytic Uraemic Syndrome. Clin Exp Immunol 2015;181:118-25.
- Thurman JM, Marians R, Emlen W, Wood S, Smith C, Akana H, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol 2009;4:1920-4.
- Westra D, Volokhina EB, van der Molen RG, van der Velden TJ, Jeronimus-Klaasen A, Goertz J, et al. Serological and genetic complement alterations in infection-induced and complement-mediated hemolytic uremic syndrome. Pediatr Nephrol 2017;32:297-309.
- Arvidsson I, Stahl AL, Hedstrom MM, Kristoffersson AC, Rylander C, Westman JS, et al. Shiga toxin-induced complement-mediated hemolysis and release of complement-coated red blood cell-derived microvesicles in hemolytic uremic syndrome. J Immunol 2015;194:2309-18.
- Stahl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011;117:5503-13.
- Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol 2011;187:172-80.
- Lee BC, Mayer CL, Leibowitz CS, Stearns-Kurosawa DJ, Kurosawa S. Quiescent complement in nonhuman primates during E coli Shiga toxin-induced hemolytic uremic syndrome and thrombotic microangiopathy. Blood 2013;122:803-6.
- Bowen EE, Hurcombe JA, Barrington F, Keir LS, Farmer LK, Wherlock MD, et al. Shiga toxin targets the podocyte causing hemolytic uremic syndrome through endothelial complement activation. Med 2023;4:761-77.
- Michael M, Elliott EJ, Ridley GF, Hodson EM, Craig JC. Interventions for Haemolytic Uraemic Syndrome and thrombotic thrombocytopenic purpura. Cochrane Database Syst Rev 2009;2009.
- Colic E, Dieperink H, Titlestad K, Tepel M. Management of an acute outbreak of diarrhoea-associated Haemolytic Uraemic Syndrome with early plasma exchange in adults from southern Denmark: an observational study. Lancet 2011;378:1089-93.
- Keenswijk W, Raes A, De Clerck M, Vande Walle J. Is plasma exchange efficacious in Shiga toxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Ther Apher Dial 2019;23:118-25.
- Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, Hafer C, et al. Collaborators of the DGfN STEC HUS registry . Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC HUS registry. Nephrol Dial Transplant 2012;27:3807-15.
- Menne J, Nitschke M, Stingele R, Abu-Tair M, Beneke J, Bramstedt J, et al. EHEC-HUS consortium . Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced Haemolytic Uraemic Syndrome: case-control study. BMJ 2012;345.
- Ake JA, Jelacic S, Ciol MA, Watkins SL, Murray KF, Christie DL, et al. Relative nephroprotection during Escherichia coli O157:H7 infections: association with intravenous volume expansion. Pediatrics 2005;115:e673-80.
- Ardissino G, Tel F, Possenti I, Testa S, Consonni D, Paglialonga F, et al. Early volume expansion and outcomes of hemolytic uremic syndrome. Pediatrics 2016;137.
- Bilkis MD, Bonany P. Hydration in hemolytic uremic syndrome. Arch Argent Pediatr 2021;119:62-6.
- Hickey CA, Beattie TJ, Cowieson J, Miyashita Y, Strife CF, Frem JC, et al. Early volume expansion during diarrhea and relative nephroprotection during subsequent hemolytic uremic syndrome. Arch Pediatr Adolesc Med 2011;165:884-9.
- Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 2007;25:1256-64.
- Greenbaum LA, Fila M, Ardissino G, Al-Akash SI, Evans J, Henning P, et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int 2016;89:701-11.
- Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013;368:2169-81.
- Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int 2015;87:1061-73.
- Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med 2011;364:2561-3.
- Agbas A, Goknar N, Akinci N, Yildirim ZY, Tasdemir M, Benzer M, et al. Outbreak of Shiga toxin-producing Escherichia-coli-associated hemolytic uremic syndrome in Istanbul in 2015: outcome and experience with eculizumab. Pediatr Nephrol 2018;33:2371-81.
- Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, Lacraz A, et al. Outbreak of Escherichia coli O104:H4 Haemolytic Uraemic Syndrome in France: outcome with eculizumab. Nephrol Dial Transplant 2014;29:565-72.
- Dinh A, Anathasayanan A, Rubin LM. Safe and effective use of eculizumab in the treatment of severe Shiga toxin Escherichia coli: associated hemolytic uremic syndrome. Am J Health Syst Pharm 2015;72:117-20.
- Gitiaux C, Krug P, Grevent D, Kossorotoff M, Poncet S, Eisermann M, et al. Brain magnetic resonance imaging pattern and outcome in children with haemolytic-uraemic syndrome and neurological impairment treated with eculizumab. Dev Med Child Neurol 2013;55:758-65.
- Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr 2018;177:311-8.
- Loos S, Ahlenstiel T, Kranz B, Staude H, Pape L, Hartel C, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis 2012;55:753-9.
- Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical Hemolytic Uremic Syndrome (HUS) with neurological involvement. Medicine (Baltim) 2015;94.
- Percheron L, Gramada R, Tellier S, Salomon R, Harambat J, Llanas B, et al. Eculizumab treatment in severe pediatric STEC HUS: a multicenter retrospective study. Pediatr Nephrol 2018;33:1385-94.
- Rasa M, Musgrave J, Abe K, Tanaka L, Xoinis K, Shiramizu B, et al. A case of Escherichia coli hemolytic uremic syndrome in a 10-year-old male with severe neurologic involvement successfully treated with eculizumab. J Investig Med High Impact Case Rep 2017;5.
- Saini A, Emke AR, Silva MC, Perlman SJ. Response to Eculizumab in Escherichia coli O157: H7-induced hemolytic uremic syndrome with severe neurological manifestations. Clin Pediatr (Phila) 2015;54:387-9.
- Wijnsma KL, Schijvens AM, Rossen JWA, Kooistra-Smid A, Schreuder MF, van de Kar N. Unusual severe case of hemolytic uremic syndrome due to Shiga toxin 2d-producing E. coli O80:H2. Pediatr Nephrol 2017;32:1263-8.
- Balestracci A, Meni Battaglia L, Martin SM, Toledo I, Puyol I, Beaudoin L, et al. Hemolytic uremic syndrome due to Shiga toxin-producing Escherichia coli and hypocomplementemia with favorable response to eculizumab: a case report. Rev Fac Cien Med Univ Nac Cordoba 2021;78:188-92.
- Bauer A, Loos S, Wehrmann C, Horstmann D, Donnerstag F, Lemke J, et al. Neurological involvement in children with E. coli O104:H4-induced hemolytic uremic syndrome. Pediatr Nephrol 2014;29:1607-15.
- Costigan C, Raftery T, Carroll AG, Wildes D, Reynolds C, Cunney R, et al. Neurological involvement in children with hemolytic uremic syndrome. Eur J Pediatr 2022;181:501-12.
- Ekinci ZB, Bek K, Aytac MB, Karadenizli A, Sabri Hancer V. Renal outcome with eculizumab in two diarrhoea-associated hemolytic-uremic syndrome cases with severe neurologic involvement. Hong Kong J Nephrol 2014;16:46-9.
- Giordano P, Netti GS, Santangelo L, Castellano G, Carbone V, Torres DD, et al. A pediatric neurologic assessment score may drive the eculizumab-based treatment of Escherichia coli-related hemolytic uremic syndrome with neurological involvement. Pediatr Nephrol 2019;34:517-27.
- Konopasek P, David J, Marejkova M, Simankova N, Vondrak K, Zieg J. The Czech experience with eculizumab in severe paediatric Shiga toxin-producing Escherichia coli-associated haemolytic uremic syndrome patients. Klin Padiatr 2022;234:48-51.
- Mauras M, Bacchetta J, Duncan A, Lavocat MP, Rohmer B, Javouhey E, et al. Escherichia coli-associated hemolytic uremic syndrome and severe chronic hepatocellular cholestasis: complication or side effect of eculizumab?. Pediatr Nephrol 2019;34:1289-93.
- Monet-Didailler C, Chevallier A, Godron-Dubrasquet A, Allard L, Delmas Y, Contin-Bordes C, et al. Outcome of children with Shiga toxin-associated Haemolytic Uraemic Syndrome treated with eculizumab: a matched cohort study. Nephrol Dial Transplant 2020;35:2147-53.
- Travert B, Dossier A, Jamme M, Cointe A, Delmas Y, Malot S, et al. Centre de Référence des Microangiopathies Thrombotiques2 . Shiga toxin-associated hemolytic uremic syndrome in adults, France, 2009–2017. Emerg Infect Dis 2021;27:1876-85.
- Yesilbas O, Yozgat CY, Akinci N, Sonmez S, Tekin E, Talebazadeh F, et al. Acute myocarditis and eculizumab caused severe cholestasis in a 17-month-old child who has hemolytic uremic syndrome associated with Shiga toxin-producing Escherichia coli. J Pediatr Intensive Care 2021;10:216-20.
- Umscheid JH, Nevil C, Vasudeva R, Farhan Ali M, Agasthya N. Treatment of Shiga-Toxin Hus With Severe Neurologic Features With Eculizumab. Case Rep Pediatr 2021;2021.
- Santangelo L, Netti GS, Torres DD, Piscopo G, Carbone V, Losito L, et al. Peripheral nervous system manifestations of Shiga toxin-producing E. coli-induced haemolytic uremic syndrome in children. Ital J Pediatr 2021;47.
- Weber B, Chan D, Hammer S. Eculizumab use in a temporarily dialysis-dependent patient with Shiga toxin-producing Escherichia coli hemolytic uremic syndrome with neurological complications. J Pediatr Pharmacol Ther 2022;27:90-5.
- Wildes DM, Harvey S, Costigan CS, Sweeney C, Twomey É, Awan A, et al. Eculizumab in STEC-HUS: a paradigm shift in the management of pediatric patients with neurological involvement. Pediatr Nephrol 2024;39:315-24.
- Soliris 300mg Concentrate for Solution for Infusion. SmPC 2021. medicines.org.uk/emc/product/362/smpc.
- Socie G, Caby-Tosi MP, Marantz JL, Cole A, Bedrosian CL, Gasteyger C, et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical Haemolytic Uraemic Syndrome: 10-year pharmacovigilance analysis. Br J Haematol 2019;185:297-310.
- Urushihara N, Ariki N, Oyama T, Chouda Y, Yagi T, Inoue T, et al. Secondary sclerosing cholangitis and portal hypertension after O157 enterocolitis: extremely rare complications of hemolytic uremic syndrome. J Pediatr Surg 2001;36:1838-40.
- Garnier A, Brochard K, Kwon T, Sellier-Leclerc A-L, Lahoche A, Launay EA, et al. Efficacy and safety of Eculizumab in pediatric patients affected by Shiga Toxin-Related Hemolytic and Uremic Syndrome: a randomized, placebo-controlled trial. J Am Soc Nephrol 2023;34:1561-73.
- Akcan-Arikan A, Zappitelli M, Loftis LL, Washburn KK, Jefferson LS, Goldstein SL. Modified RIFLE criteria in critically ill children with acute kidney injury. Kidney Int 2007;71:1028-35.
- Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol 2009;20:629-37.
- National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents . The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics 2004;114:555-76.
- Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomised trials. Lancet 2001;357:1191-4.
- te Loo DM, Monnens LA, van Der Velden TJ, Vermeer MA, Preyers F, Demacker PN, et al. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000;95:3396-402.
- Obrig TG, Karpman D. Shiga toxin pathogenesis: kidney complications and renal failure. Curr Top Microbiol Immunol 2012;357:105-36.
- Pangburn MK, Rawal N. Structure and function of complement C5 convertase enzymes. Biochem Soc Trans 2002;30:1006-10.
- Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, et al. Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol 2009;182:6394-400.
- Ehrlenbach S, Rosales A, Posch W, Wilflingseder D, Hermann M, Brockmeyer J, et al. Shiga toxin 2 reduces complement inhibitor CD59 expression on human renal tubular epithelial and glomerular endothelial cells. Infect Immun 2013;81:2678-85.
- Keir LSSR, Welsh GI, Coward RJ, Satchell S, Richards A, Saleem MA. European Society of Paediatric Nephrology. Krakow: Springer; 2012.
- Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, et al. Glomerular-specific alterations of VEGF – a expression lead to distinct congenital and acquired renal diseases. J Clin Invest 2003;111:707-16.
- Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 2008;358:1129-36.
- Keir LS, Firth R, Aponik L, Feitelberg D, Sakimoto S, Aguilar E, et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. J Clin Invest 2017;127:199-214.
- Lennon R, Welsh GI, Singh A, Satchell SC, Coward RJ, Tavare JM, et al. Rosiglitazone enhances glucose uptake in glomerular podocytes using the glucose transporter GLUT1. Diabetologia 2009;52:1944-52.
- Rinschen MM, Hoppe AK, Grahammer F, Kann M, Volker LA, Schurek EM, et al. N-degradomic analysis reveals a proteolytic network processing the podocyte cytoskeleton. J Am Soc Nephrol 2017;28:2867-78.
- Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 2002;13:630-8.
- Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, von Ruhland CJ, et al. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int 2006;69:1633-40.
- Watson R, Lindner S, Bordereau P, Hunze EM, Tak F, Ngo S, et al. Standardisation of the factor H autoantibody assay. Immunobiology 2014;219:9-16.
- Walsh PR, Johnson S. Eculizumab in the treatment of Shiga toxin Haemolytic Uraemic Syndrome. Pediatr Nephrol 2019;34:1485-92.
- Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, et al. Chamber Quantification Writing Group . Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 2005;18:1440-63.
- Zoghbi WA, Enriquez-Sarano M, Foster E, Grayburn PA, Kraft CD, Levine RA, et al. American Society of Echocardiography . Recommendations for evaluation of the severity of native valvular regurgitation with two-dimensional and Doppler echocardiography. J Am Soc Echocardiogr 2003;16:777-802.
- Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, et al. Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction . Third universal definition of myocardial infarction. Circulation 2012;126:2020-35.
Appendix 1 Summary of ECUSTEC trial protocol amendments
| Amendment number | Date of amendment | Protocol version number | Type of amendment | Summary of amendment |
|---|---|---|---|---|
| 1 | 12 December 2016 | Version 2.0 | Substantial | Changes to the initial protocol requested by the MHRA including information about contraception, pregnancy testing, more frequent CNS examinations and SUSAR reporting. |
| 2 | 1 April 2017 | Version 3.0 | Substantial | Changes to incorporate those requested by REC for Version 1.0 7 September 2016 and MHRA requested changes for Version 2.0 12 December 2 reviewed the updated stool SOP 016. Additional inclusion criteria and wording of an exclusion criteria. Further detail added regarding confirmation of vaccinations. Amendments to the assessments schedule, data collection, samples guidance and AE reporting sections. Other minor changes. |
| 3. | 18 January 2018 | Version 4.0 | Substantial | The treatment window has been extended by 12 hours due to the operational difficulty of treating patients. Other minor changes. |
| 4. | 24 June 2019 | Version 5.0 | Substantial | The wording for inclusion criteria 4 has been amended to include ‘OR Passage of blood per rectum within 14 days prior to diagnosis of HUS’. Also refined household contact to: Stool culture or Shiga toxin PCR or STEC serology result indicating STEC in a close contact (household or institutional). Other changes include an update to the UK Data Protection Act 2018, re-wording of events that do not require expedited reporting and other minor changes. |
| 5. | 20 May 2020 | Version 6.0a | Substantial | The treatment window has been extended by 24 hours due to the operational difficulty of treating patients within the current window. Other minor changes. |
Appendix 2 ECUSTEC clinical severity score
| Renal | Lowest eGFR > 50 | 1 |
| Lowest eGFR 26–50, no oligoanuriaa | 2 | |
| Lowest eGFR ≤ 25, no oligoanuriaa | 3 | |
| Oligoanuriaa but no dialysis (or RRT) required | 4 | |
| Dialysis/RRT < 48 hours | 5 | |
| Dialysis/RRT 2 days | 6 | |
| Dialysis/RRT 3 days | 7 | |
| Dialysis/RRT 4 days | 8 | |
| Dialysis/RRT 5 days | 9 | |
| Dialysis/RRT 6 days | 10 | |
| Dialysis/RRT 7 days | 11 | |
| Dialysis/RRT 8 days | 12 | |
| Dialysis/RRT 9 days | 13 | |
| Dialysis/RRT 10 days | 14 | |
| Dialysis/RRT 11 days | 15 | |
| Dialysis/RRT 12–13 days | 16 | |
| Dialysis/RRT 14–17 days | 17 | |
| Dialysis/RRT 18–20 days | 18 | |
| Dialysis/RRT 21–27 days | 19 | |
| Dialysis/RRT 28–34 days | 20 | |
| Dialysis/RRT 35–41 days | 21 | |
| Dialysis/RRT 42–48 days | 22 | |
| Dialysis/RRT 49–55 days | 23 | |
| Dialysis/RRT > 55 days | 24 | |
| CNS | No obvious CNS involvement | 0 |
| Altered consciousness (agitation, irritability, hallucinations, confusion, excessive drowsiness) | 2 | |
| Single seizure | 4 | |
| Two or more seizures 24 hours apartb | 6 | |
| Transient focal neurological defect (> 24 hoursc but < 1 week) | 7 | |
| Persistent focal neurological defect (present at day 60 and persistent for more than 1 week) | 10 | |
| Persistent global (≥ 2 brain functions – vision/hearing/cognitive/motor/sensory/memory) neurological defect at day 60 | 15 | |
| Pancreas | No clinical or biochemical evidence pancreatitis | 0 |
| Raised amylase and/or lipased without clinical symptoms/signs | 2 | |
| Hyperglycaemia without insulin requirement | 6 | |
| Pancreatitis with sequelae (laparotomy, parenteral nutrition,e insulin required) | 8 | |
| Chronic sequelae of pancreatitis at day 60 (parenteral nutrition,e insulin, other) | 10 | |
| Gastrointestinal | No abdominal surgery required (except related to peritoneal dialysis catheter) | 0 |
| Laparoscopy/laparotomy required for abdominal symptoms | 5 | |
| Intestinal perforation AND/OR bowel resection required | 8 | |
| Stoma formation | 10 | |
| Cardiac | No cardiac involvement (normal CVS examination – except hypertension/volume overload) | 0 |
| Cardiac failure confirmed by ECHOf (impaired systolic ventricularg function or chamber enlargementh or valve regurgitationi) | 4 | |
| Cardiac failure confirmed by ECHO with dilated cardiomyopathy | 6 | |
| Myocardial infarction (on standard ECG ± troponin ± ECHO evidence)j | 10 |
List of abbreviations
- AE
- adverse event
- aHUS
- atypical Haemolytic Uraemic Syndrome
- AKI
- acute kidney injury
- CFH
- complement factor H
- CHU-9D
- Child Health Utility 9D instrument
- CKD
- chronic kidney disease
- CNS
- central nervous system
- CONSORT
- Consolidated Standards of Reporting Trials
- COVID-19
- coronavirus disease of 2019
- CRF
- case report form
- CRP
- C reactive protein
- CSS
- clinical severity score
- CTIMP
- Clinical Trial of an Investigational Medicinal Product
- DMC
- Data Monitoring Committee
- DNA
- deoxyribonucleic acid
- ECHO
- echocardiogram
- eGFR
- estimated glomerular filtration rate
- ELISA
- enzyme-linked immunosorbent assay
- Gb3
- globotriaosylceramide
- HRQoL
- health-related quality of life
- HUS
- haemolytic uraemic syndrome
- IMP
- investigational medicinal product
- IV
- intravenous
- LDH
- lactate dehydrogenase
- MAC
- membrane attack complex
- MHRA
- Medicines and Healthcare products Regulatory Agency
- PCR
- polymerase chain reaction
- PedsQL
- Paediatric Quality of Life Inventory
- PIC
- Patient Identification Centres
- PICU
- paediatric intensive care unit
- PPI
- patient and parent involvement
- pRIFLE
- paediatric risk/injury/failure/loss/end-stage definition of acute kidney injury
- RBC
- red blood cell
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- RRT
- renal replacement therapy
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- STEC
- Shiga-toxin-producing Escherichia coli
- stx
- Shiga toxin
- SUSAR
- suspected unexpected serious adverse reactions
- TMA
- thrombotic microangiopathy
- TSC
- Trial Steering Committee
- VEGF
- vascular endothelial growth factor
- WT1
- Wilms tumour-1 gene