

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 16/61/18. The contractual start date was in February 2018. The final report began editorial review in April 2022 and was accepted for publication in August 2022. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2023 Metcalfe et al. This work was produced by Metcalfe et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2023 Metcalfe et al.
Chapter 1 Introduction
Subacromial spacer balloons
Shoulder pain is a common and disabling problem. The prevalence of shoulder pain in UK adults is approximately 16%, with rotator cuff disease accounting for 70–85% of this. 1–3 Surgery for rotator cuff disease has increased considerably; nearly 30,000 annual cases in 2009–10 in the UK, when this disease was last formally studied. 4 Individuals with a symptomatic rotator cuff tear typically present with pain, restricted movement and loss of strength and function, and the condition is associated with substantial expense to society through treatment costs and loss of work (both paid and unpaid). 5–8
The term ‘rotator cuff’ refers to the subscapularis, supraspinatus, infraspinatus and teres minor muscles and associated tendons around the shoulder, which when intact, function to keep the humerus centred on the glenoid as the shoulder moves, providing a stable fulcrum for normal glenohumeral joint motion. 9,10 A rotator cuff tear can result in loss of this stabilising function and lead to pain. The exact cause of pain is unknown but may be due to mechanical impingement between the humerus and the acromion, impingement of torn or loose tissue in the joint or biological causes such as bursitis or synovitis. 10,11
Rotator cuff repair is a widely accepted treatment for symptomatic rotator cuff tears. 12,13 However, there are multiple factors which influence whether a tear can be repaired, including the size of the tear, its chronicity, fatty infiltration of the muscle (atrophy) and the ability to bring the torn end back to its original site without excessive tension. Some tears cannot be surgically repaired (in which case they are called irreparable tears), and these can be difficult to manage.
Treatment for symptomatic irreparable rotator cuff tears includes physiotherapy, injections, arthroscopic debridement (with or without biceps tenotomy), partial repair, muscle transfers, interposition grafts and even shoulder replacements, typically reverse shoulder arthroplasty. 14–17 Arthroscopic debridement is commonly used and benefit has been demonstrated in case series studies. 18,19 However, it remains a controversial option and there are few randomised controlled trial (RCT) data on its use in the irreparable tear population. 18–20
The InSpace® (Stryker, Kalamazoo, MI, USA) subacromial balloon spacer received a Conformité Européenne (CE) mark in 2010. In 2013, the device was introduced into UK orthopaedic practice as a potential treatment option for people with irreparable rotator cuff tears. 21 At the start of our study, the cost was approximately £1250 per implant. Its introduction was underpinned by case series evidence. In May 2016, an interventional procedure guidance document was published by the UK National Institute for Health and Care Excellence (NICE), which found that there was very limited evidence for its use. Therefore, NICE recommended that the device should be restricted to use in the context of research only and a research recommendation was made to assess its effectiveness. 22 It received Food and Drug Administration (FDA) clearance in the United States in July 2021, with approximately 29,000 devices having been implanted outside the United States prior to this date. 23
The InSpace balloon is a saline-filled balloon made of biodegradable (dissolvable) synthetic material. It is inserted above the main glenohumeral joint of the shoulder at the end of an arthroscopic debridement after an irreparable tear has been identified. It is simple to deploy and typically adds less than 10 minutes to the procedure. 21,24 The balloon cushions the humerus, preventing it from pressing on the acromion above it when the deltoid is active and during abduction of the arm, potentially reducing pain. It may also assist in the biomechanics of the shoulder, resisting proximal migration of the humerus under deltoid activity. It is thought the InSpace balloon begins to deflate from 3 months, during which time it is thought to allow and improve rehabilitation of the remaining rotator cuff and deltoid, so that when it has dissolved the biomechanics of the shoulder are maintained.
The safety of the device was established in rodents. One adverse event was recorded: a fibrosarcoma thought to be unique to rodents. 25 Proof of concept was established in a series of 24 irreparable cuff tears in Slovakia in 2012, with five-year follow-up results published in 2016. 26,27
The InSpace balloon has been used in a number of centres across the UK. At the time of starting this study, data had been presented in three conference abstracts, totalling 61 cases. 28–30 These case series studies demonstrated improvements in outcomes from baseline. Complications such as balloon displacement and non-cyst forming synovitis were reported in a small number of cases (3 of 61). One retrospective non-randomised study of 23 participants (12 with the balloon) showed an improvement in outcomes compared with debridement alone. 31
Reviews in 2019–20 found between 10 and 20 studies of the InSpace balloon, all case series. 32–36 The largest of these reviews included 619 participants (513 analysed; in some studies people with complications were excluded from analysis). 32 Many case series have documented encouraging clinical results but some studies have reported poor results or cases of inflammation and pain and have concluded that comparative data are needed.
One 2021 case series study37 following 51 people who underwent balloon placement found that after a mean follow-up of 3 years, shoulder function scores improved substantially after balloon spacer insertion (measured using the Constant score). 38,39 The authors reported limited need for revision surgery, with five participants undergoing reverse total shoulder replacement, one latissimus dorsi tendon transfer and high participant satisfaction. 37 In contrast, a different case series published in 2021, followed up 22 participants for almost 3 years. 40 The balloon spacer was found to be effective in a minority of participants in the medium term, as despite an improvement in Oxford Shoulder Scores (OSS) (23.6 vs. 29.6; p < 0.02), six participants converted to reverse total shoulder replacement at a mean time of 11 months post balloon insertion, with a mean deterioration of 1.1 on the OSS, while a further six participants with the balloon still in place demonstrated either a deterioration on the OSS or an improvement less than the minimal important difference – reported as 6.0 points. 41
Prior to starting our study, we undertook a systematic review (search date September 2016) and meta-analysis of randomised trials in rotator cuff pathology. 42 We found 57 trials (n = 4542), mostly of repair, but no trials using the InSpace balloon. The review found improvements in outcome for rotator cuff tears treated surgically, with conservative care and with acromioplasty. 42 Therefore, benefits compared with baseline found in case series may not be unique to the InSpace balloon. The effectiveness of the balloon in comparison to nonoperative care or acromioplasty is not known. It could still be a substantial improvement or may be of no benefit. 20 The device is costly but there is no evidence that it is effective clinically. If the device is effective, then it would relieve pain and improve function for patients with a disabling condition that currently has few good alternative treatments and it should be recommended for widespread use. However, if the device is ineffective or harmful, alternative approaches should be sought.
One other blinded randomised trial funded by the manufacturer (previously OrthoSpace, now Stryker) has been undertaken in the United States comparing partial cuff repair with a subacromial balloon spacer (clinicaltrials.gov NCT02493660). 43 It should be noted that partial cuff repair is not a technique that is often used in the UK and is not an appropriate comparator in a UK context but is more appropriate in the United States.
A non-inferiority, prospective, single-blinded, multicentre RCT was conducted by Verma et al. 43 to compare the outcomes of arthroscopic subacromial balloon spacer implantation with partial repair in participants 40 or more years of age who had full-thickness massive rotator cuff tears. A summary of 12-month results has been posted on a trials registry but results have not, at the time of writing, been published in a peer-reviewed journal.
Participants’ baseline and follow-up data were collected to identify a non-inferiority margin of 10% difference in the proportion of responders in a composite outcome measure. This composite outcome was defined as participants who had improved from baseline on both the Western Ontario Rotator Cuff (WORC) score (≥ 275 points) and American Shoulder and Elbow Society (ASES) score (≥ 6.4 points) at 6 weeks and had maintained the improvements at 12 months without subsequent secondary surgical intervention or serious adverse device effects. Of the 184 randomised participants, 176 (88/group) completed 12-month follow-up.
At 12 months, 45 (51.1%) participants in the spacer implant group and 35 participants (39.8%) in the partial repair group had reached and maintained the primary composite end point, corresponding to an 11.43% unadjusted mean advantage for the spacer implant group. However, this composite outcome should be taken in context, as it is likely to be difficult for people who have had partial cuff repair to achieve improvements by 6 weeks as many would still be in an early postoperative recovery phase. 44,45 Both groups improved between baseline and follow-up in all WORC and ASES scores but there were no differences observed between groups in WORC and ASES data reported so far. Six participants (n = 3 balloon spacer; n = 3 partial repair) required secondary surgery; two in each group undergoing reverse shoulder arthroplasty and one participant in each group undergoing shoulder arthroscopy. 43
Trial designs in new surgical procedures
The safe introduction of new surgical procedures is essential to the delivery of high-quality surgical care. Innovative procedures may result in a step-change improvement in treatment but can also bring new risks and substantial costs. Major harm can occur when a well-meant intervention is used widely across the health service before it is formally and thoroughly evaluated. 46,47
Pharmaceuticals undergo rigorous clinical trials before being introduced but this is not always true for surgical procedures, which are often introduced on the basis of cadaveric testing or small case series data only. 48 There is a need to develop new processes and methodology to introduce surgical procedures safely, using early RCTs to determine whether a treatment is likely to be safe, clinically effective and cost-effective prior to widespread uptake. 48
To rigorously assess new surgical procedures, large multicentre RCTs which produce reliable and statistically precise evidence may be undertaken. However, these studies typically need to recruit over extended periods. 47,49 Large pragmatic surgical trials are expensive (typically £1.5–2 million or more) and can take five years or more from award to completion, as for example the Wound management of Open Lower Limb Fractures (WOLLF), Fixation of Distal Tibia Fractures (FixDT) and Ankle Injury Management trials, even disregarding the time taken over feasibility and pilot studies. 49–51 Costly, ineffective or unsafe treatments may be used for many years before they are removed from practice. There is thus a requirement for trial designs to determine efficiently and rapidly whether an intervention is ineffective or even harmful but also to demonstrate superiority if the intervention is a genuine improvement on standard care. Improvements in the efficiency of undertaking trials of surgical interventions would provide earlier answers to crucial clinical questions, providing benefits to patients and making better use of health-care resources.
Adaptive trial designs are becoming increasingly popular and their use has been encouraged by major scientific journals, the US FDA and National Institute of Health Research (NIHR) panels. 52–54 This design allows for prospectively planned modifications, such as stopping the study or discontinuing an intervention, based on emerging findings as the trial proceeds, while preserving the scientific validity and integrity of the trial. This more flexible strategy typically reduces costs and shortens timescales, without compromising the integrity, statistical power or rigour of the study. 52,55–57 Efficiency savings in terms of cost and time can be substantial (a 40% reduction in sample size in one study) without a loss in power or increase in false-positive error rate. The use of adaptive designs which are flexible in their sample size may also avoid the delay associated with prolonged pilot or feasibility studies, as they can be incorporated into the trial without delaying the main study. 58,59 Yet, despite the potential benefit of reducing the number of people exposed to a procedure that may be unnecessary or even harmful, adaptive designs remain rare in surgical trials. 52,57,60
In this trial, we applied these design principles to a multicentre clinical trial of a new surgical procedure for rotator cuff tears. We have termed this trial design REACTS (Randomised, Efficient, Adaptive Clinical Trials in Surgery). Subacromial spacer for Tears Affecting Rotator Cuff Tendons (START) is the first study using this new statistical adaptive design approach for the assessment of a new surgical procedure. The statistical principles are laid out in a 2019 methodology paper. 61 Further evaluation of the REACTS studies was also undertaken in a range of other trial settings, with the aim of establishing this trial design as the future standard for assessing new surgical procedures. In the future such an approach, if successful, could be used before a new procedure is introduced into widespread clinical practice. This would reduce costs to funders but more importantly it will ensure that high-quality evidence is delivered more rapidly to improve patient care and outcomes.
Aim
Our primary clinical aim was to assess the clinical and cost-effectiveness and safety of a subacromial InSpace balloon for patients with symptomatic irreparable rotator cuff tears. 22 Methodologically, the primary aim was to develop and implement appropriate statistical tools to allow an efficient adaptive clinical trial design. 61
Chapter 2 Main trial methods
Trial design
We conducted a participant- and assessor-blinded, adaptive, multicentre RCT with a parallel economic analysis. The setting was secondary care across the UK. The study compared arthroscopic debridement using an InSpace balloon with arthroscopic debridement alone and was designed using the REACTS framework (see Figure 1). We have published a detailed description of the protocol elsewhere. 62
FIGURE 1.
Flow diagram describing the planned trial methodology.

Patient and public involvement
Patient involvement has been central to the design, delivery and interpretation of the study and the results; this involvement will continue as we disseminate the findings. Initially, we engaged with multiple people who had previously undergone rotator cuff surgery to learn about and understand their experiences. Their insights were reassuring about the need for a trial and helped establish the design of the study, especially the choice of primary and secondary outcomes and the design of all participant facing materials. We engaged with patients during development of the study materials and trialled our study forms with a number of patients before they were used in the study.
One of the co-authors of the report has shoulder problems previously treated surgically and represents the patient view in trial management meetings, while two other patients sit on our steering committee. We will produce patient and public focused summaries of the research and disseminate this widely.
Objectives
Primary clinical objective
Our primary clinical objective was to quantify and make inferences on observed differences between arthroscopic debridement using an InSpace balloon with arthroscopic debridement alone 12 months after surgery, using the OSS as the primary outcome measure. 63,64 A 12-month time point was selected based on our meta-analysis of outcomes for randomised trials which found that shoulder scores typically reach a plateau at 12 months after any intervention for a rotator cuff tear. 42 Although we are collecting 24-month scores, they do not provide sufficient additional value to justify the increase in costs and delay in the trial result that would be required had they been used as the primary outcome.
Secondary clinical objectives
Our secondary clinical objectives were to:
-
quantify and make inferences on observed differences between the two intervention groups on the following measures; all were assessed at baseline, 3, 6 and 12 months:
-
Perform an economic analysis to assess the comparative cost-effectiveness of the two interventions (see Chapter 5).
-
Perform a magnetic resonance imaging (MRI) substudy to compare the acromiohumeral distance (AHD) on MRI scans in a sample of participants with and without the balloon at 8 weeks and at least 6 months after treatment. This was to assess the proposed mechanism of action of the InSpace balloon while still inflated (8 weeks) and to determine any persistent effects following deflation (after 6 months).
Methodological objectives
The methodological objectives of the study were to:
-
develop and implement appropriate statistical tools for an efficient adaptive clinical trial design, with the potential to stop early either for futility or efficacy, using outcome data available at 3, 6 and 12 months61
-
compare the use of frequentist and Bayesian design and analysis on the conduct and interpretation of an adaptive surgical clinical trial with reference to decision-making by data monitoring committees (DMCs) during the study and by clinicians, commissioners and other stakeholders at the conclusion of the trial
-
explore the challenges of supporting adaptive design decision-making with net benefit and expected value of information approaches to health economic analyses.
Outcome measures
Primary outcome
The Oxford Shoulder Score
The OSS is a patient-reported outcome measure with 12 questions which has been well validated to assess the degree of pain and disability caused by shoulder pathology. It is simple to complete and has proved to be valid and reliable in determining the outcome from shoulder surgery. 41,68 It is a well-known measure and has also been used in previous high-impact randomised trials of shoulder surgery. 69 A higher score (0–48) corresponds to a better outcome. 63,64
Originally, the planned primary outcome for the study was the Constant score. 38,39 This was chosen as it had been widely used in shoulder trials, is well accepted by surgeons, and has good reliability and responsiveness. 42 However, in light of the COVID-19 outbreak in March 2020, the trial management group (TMG) decided to change the primary outcome to the OSS. As the Constant score requires face-to-face contact to measure and it is usually taken in hospital clinics, it would have exposed participants to unnecessary risk during the height of the pandemic. The decision was agreed by both the trial steering committee (TSC) and DMC as well as the NIHR prior to the change. The OSS correlates well with the Constant score; both assess shoulder pain and function, are similarly responsive and have comparable effect sizes in rotator cuff pathology. 41,63,64,68,69 More details on the effect of changing the primary outcome can be found in the power calculation and sample size section below.
Secondary outcomes
The Oxford Shoulder Score at baseline, 3, 6 and 24 months
-
The Constant score at baseline, 3, 6 and 12 months: the Constant score consists of four variables that are used to assess shoulder function. The subjective variables are pain and activities of daily living (sleep, work, recreation/sport), which give a total of 35 points and the objective variables are ROM and strength, which give a total of 65 points. Each component can be reported separately or can be combined to give a score out of 100. A standardised protocol for the objective component of the score was developed based on the work of Moeller et al.,70 with training provided for all sites. 38,39
-
Range of pain-free movement in abduction and flexion of the shoulder at baseline, 3, 6 and 12 months measured using a 12.5-inch long-arm goniometer.
-
Strength of shoulder in the scapular plane at baseline, 3, 6 and 12 months measured using a supplied IsoForceControl EVO2 dynamometer (Herkules Kunststoff, Switzerland).
-
WORC index at baseline, 3, 6, 12 and 24 months. 65 The WORC is a condition-specific self-reported instrument to assess ‘quality of life’ (QoL). It comprises 21 items incorporating five domains: physical symptoms, sports/recreation, work, lifestyle and emotions. Item scores are recorded on a visual analogue scale (VAS) ranging from 0 to 100 (100 mm VAS). Individual domain scores can be summated for a total score (range 0–2100). Higher scores represent poorer QoL. 65 To present this in a more easily accessible and reproducible format, the VAS lines were modified into 11-point numerical rating scale lines (1-cm intervals labelled as 0-10). Item scores were then summed and reported as a percentage of maximum total score.
-
Health utility assessed using EQ-5D-5L at baseline, 3, 6, 12 and 24 months. 66,67 The EQ-5D-5L is a validated, generic health-related QoL measure consisting of five dimensions each with a five-level answer possibility. Each combination of answers can be converted into a health utility score using the UK crosswalk value set. It has good test–retest reliability, is simple for participants to use and gives a single preference-based index value for health status that can be used for broader cost-effectiveness comparative purposes.
-
Health-care resource use at 3, 6, 12 and 24 months. The primary analysis focused on direct intervention and health-care/personal social services (PSSs) costs, while wider impact (societal) costs were included within a sensitivity analysis. Relevant resource-use questionnaires were administered to participants at baseline and all follow-up points to collect resource-use data associated with the interventions under examination.
-
Patient Global Impression of Change (PGIC) score was collected at 3, 6, 12 and 24 months. This was collected using a simple seven-point scale assessing the participants’ perception of improvement. 71
-
Analgesia use (drug, and approximate frequency) collected at baseline and 3, 6 and 12 months.
-
MRI scans (a planned substudy of 56 patients, 6 weeks and at least 6 months postsurgery): see the ‘MRI substudy’ section.
-
Adverse events were collected from site reports sent directly to the trial office when they occurred throughout the first 12 months of each participant’s follow-up, and from participant’s 3, 6 and 12-month questionnaires.
Note that the patient-reported outcome measures (OSS, WORC, EQ-5D-5L, PGIC) collected at 24 months will be published separately so as not to delay publication of the primary 12-month outcome data.
As an exploratory analysis, baseline shoulder radiographs performed as part of standard care were collected and the shortest AHD was measured according to the method described by Kolk et al. 72 Measurements were performed independently by two blinded observers, a professor of radiology and a consultant orthopaedic surgeon, and a mean of their two scores was used as a final value.
Setting and participants
Participant identification, screening and withdrawals
Potential participants were identified by the attending clinical team in intermediate or secondary care clinics, or from surgical waiting lists. The attending clinician confirmed each patient’s eligibility for the study based on clinical assessment and standard care preoperative imaging for each site (e.g. MRI or ultrasound).
All potential participants meeting study entry criteria were checked for eligibility and their details recorded on a screening log. Potential participants suitable for inclusion and willing to be approached were given verbal and written information about the study by a suitably trained member of the research team, were given adequate time to consider the information and were invited to ask questions and discuss the study further prior to informed consent being obtained. Trial procedures including baseline assessments were not undertaken until the consent form was signed and dated by the participant.
All participants provided informed, written, signed and dated consent. As there was a delay of a number of weeks before randomisation (the waiting list for surgery), people entering the study still had the option to withdraw before treatment commenced if for any reason, they changed their mind. Any new relevant information arising during the trial was discussed with the participants and, where applicable, continuing consent was obtained using an amended consent form.
Sites agreeing to take part in both the main study and the additional MRI substudy were provided with a combined main and substudy patient information sheet and consent form.
Eligibility was also confirmed by the operating surgeon intraoperatively and participants were excluded (withdrawn) at this stage if it was discovered that the rotator cuff tear was repairable. These excluded participants were informed by letter that they were no longer taking part in the study. Baseline data were, however, retained to explore any differences between those who were deemed ineligible intraoperatively to those who were randomised. Participants randomised into the study were allowed to withdraw from follow-up at any time, without prejudice and without any effect on their current or future care.
Eligibility
Inclusion criteria
Patients were eligible to be included in the trial if they met the following inclusion criteria:
-
Rotator cuff tear deemed by the treating clinician to be technically irreparable. Multiple factors apart from size can influence whether a tear can be repaired (i.e. chronicity, retraction of the tendon ends and fat infiltration in muscle). However, patients with tears thought to be technically repairable, such as a small tear, but considered unsuitable for repair due to age or comorbidities, were not eligible for this study.
-
Experiencing intrusive symptoms (pain and loss of function) which in the opinion of the treating clinician warrants surgery.
-
Had undergone unsuccessful nonoperative management, as determined by the treating clinician.
Exclusion criteria
Patients were considered ineligible if they met any of the following criteria:
-
Advanced glenohumeral osteoarthritis on pre-operative imaging (in the opinion of the treating clinician). This was interpreted as Kellgren–Lawrence grade 3 or 4 changes on routine preoperative radiographs73 or the MRI equivalent if radiographs were not available.
-
Subscapularis deficiency,1 defined as a tear involving more than the superior 1 cm (approximately) of the subscapularis, if repaired, or any tear that is not repaired.
-
The treating clinician determines that interposition grafting or tendon transfers are indicated.
-
Pseudoparalysis (an inability to actively abduct or forward flex up to 20 degrees), as determined by the treating clinician.
-
Having any unrelated, symptomatic ipsilateral shoulder disorder that would interfere with strength measurement or ability to perform rehabilitation.
-
Other neurological or muscular conditions that would interfere with strength measurement or ability to perform rehabilitation, in the opinion of the treating clinician.
-
A previous proximal humerus fracture that could influence shoulder function, as determined by the treating clinician.
-
Previous entry into the present trial (i.e. other shoulder).
-
Unable to complete trial procedures.
-
Age under 18 years.
-
Unable to consent to the trial.
-
Unfit for surgery as defined by the treating clinician.
Randomisation
Participants were randomly allocated on a one to one basis to the two treatment groups via a central computer-based randomisation system provided by Warwick Clinical Trials Unit (WCTU), independent of the study team. A minimisation algorithm was used to determine participant allocation, using site, sex (used rather than gender as we were interested in the structural effect), age group (< 70 and ≥ 70 years) and cuff tear size (as assessed by the operating surgeon, ≥ 3 or < 3 cm, commonly used as the definition between small/medium and large/massive cuff tears) as factors, with a random element included to provide a 70% chance that the participant would receive the treatment that minimises imbalance, to ensure unpredictability.
Randomisation was performed by theatre staff, after intraoperative findings were checked (including cuff tear size) and eligibility confirmed. To maintain blinding, staff used an online system in a separate room (a 24-hour back-up automated telephone system was also available). Participants were randomised strictly sequentially at site level. Allocation concealment was maintained by the independent randomisation team who were responsible for the generation of the sequence and had no role in the randomisation of participants beyond this.
Trial interventions
Standard arthroscopic debridement (control)
The control group were randomised to arthroscopic debridement of the subacromial space with removal of inflamed tissue (bursectomy) and unstable remnants of the torn tendon, limited bone resection of the acromion, retention of the coracoacromial ligament and biceps tenotomy (if not already torn). The anaesthetic (general or local) was decided by the anaesthetist. Surgeons were permitted to use their normal surgical technique within the confines described in a trial-specific surgical guideline, available on the Warwick Research Archive Portal (http://wrap.warwick.ac.uk).
Standard arthroscopic debridement plus insertion of InSpace balloon (intervention)
Those allocated to the intervention underwent the same arthroscopic debridement as the control group plus insertion of the InSpace Balloon by a subspecialty trained shoulder surgeon. The recommended surgical technique was followed for sizing, insertion, and deployment of the balloon, as documented in the surgical manual. The content of the surgical manual was confirmed with the manufacturers (OrthoSpace at the time) prior to interventions starting in the study, to ensure the study followed recommended techniques.
For both groups, fidelity was assessed with an operative record form, arthroscopic photographs (posterior and lateral portal photographs after debridement for both groups, and a photograph from the posterior portal just before balloon inflation in the balloon group to demonstrate balloon position). In addition, the number of physiotherapy visits for each participant was also documented in both groups.
Rehabilitation
The postoperative rehabilitation programme was developed during the set-up phase of the trial. The process involved collecting current NHS rehabilitation protocols from participating sites, manufacturer protocols, scoping the literature and ratification among a panel of expert physiotherapists. As with the surgical technique, a copy was forwarded to the manufacturer, OrthoSpace, prior to starting interventions to ensure that the study did not conflict with manufacturer’s recommendations. A physiotherapy trial manual was produced and followed, to standardise rehabilitation progression and is available from: https://warwick.ac.uk/fac/sci/med/research/ctu/trials/startreacts/links/healthlinks/physiotherapy_manual_v2.2_10072018.pdf.
Postoperative rehabilitation for both groups was blind to treatment allocation and included standardised postoperative information, home exercises and the offer of a minimum of three face-to-face physiotherapy appointments. Additional physiotherapy was arranged at the discretion of the trial sites.
Blinding
Participants and assessors were blinded to treatment allocation; only the surgical teams at the time of the operation were aware of the allocation. Theatre staff were asked not to mention or discuss the balloon and to communicate the allocation in writing – that is, by using methods such as holding up a piece of paper on which the allocation was clearly written.
For those participants who were awake during surgery, drapes were used to obscure their view and arthroscopic screens were positioned in such a way that they were unable to see the procedure. Although the incisions required for the two procedures are similar one (the lateral portal) needs to be slightly larger to insert the balloon; 1.5 cm as opposed to 1 cm. Therefore, a 1.5-cm incision was used for all participants and, due to the very small change from standard care, was very unlikely to have any negative effects on any participant. Incisions were, therefore, the same for both groups and there was no external way in which the participants could detect the presence or absence of the balloon.
A standard recommended operation note template was provided to all sites and adjusted to fit local operation note systems to ensure that operation notes could be blinded and to prevent accidental unblinding (e.g. in any discharge information or during postoperative physiotherapy. The details of the operation related to the balloon were recorded in a secure online form easily accessible to the surgeon.
Although unblinding was unlikely, an unblinding plan was produced for use by NHS staff in emergency situations such as overnight admission for suspected postoperative infection. Using a predefined web-based system and two-way secure verification procedure performed using e-mail, staff used a link inserted in the operation notes to access a code to unblind the allocation group, which was only sent to an active NHS e-mail address. The trial team requested a full explanation of the clinical circumstances and the need for access to data for audit and monitoring purposes from the person performing the unblinding, and the principal investigator for the site was informed.
Participants were also asked at the 12-month time point, after collection of the primary outcome, if they were aware of their allocation.
Adverse events, adverse device effects and serious adverse device effects
Adverse events, serious adverse events (SAEs), adverse device effects and serious adverse device effects were defined using standard accepted criteria. An unanticipated serious adverse device effect was defined as an adverse effect which, by its nature, incidence, severity or outcome, was not identified in the risk analysis report. Adverse events were recorded for any participant where it was thought there may be a relationship between the event and the trial interventions or the condition being studied (in this case, any shoulder condition or related to the anaesthetic). These included device-specific deficiencies or complications such as balloon migration, which was recorded if it was identified by clinical teams during their normal practice.
All SAEs, serious adverse device effects and unanticipated serious adverse device effects occurring from the time of randomisation until 12 months post-randomisation were communicated to the sponsor within 24 hours of the research staff becoming aware of the event. All events were followed up until resolution or a final outcome was reached.
For participants lost to follow-up at or beyond the 12-month time point, their general practitioner was contacted and a short form requesting any information or health record that could be an adverse event was requested, as well as confirmation of the current contact details of the participant.
Statistical methods
All statistical analyses were carried out using R version 4.0.3 (The R Foundation for Statistical Computing, Vienna, Austria). 74
Power and sample size
Initial sample size calculations were based on the Constant score, with a target difference of 10 units, as widely used for other trials and a standard deviation (SD) of 20, giving a moderate standardised mean difference of 0.5. 20,27,75,76 In anchor-based studies the estimated target difference for the OSS is 6 with an SD of 12 being observed in multiple studies. 41,68,69,76 For an expensive invasive procedure of this nature, a smaller standardised mean difference was considered unlikely to be worthwhile. Therefore, a moderate standardised mean difference of 0.5 was considered appropriate. For a power of 90% and a (two-sided) type I error rate of 5% a study with a conventional fixed design (i.e. with no possibility of stopping early), assuming an approximate normal distribution for the score data, would require 170 participants.
The design characteristics and required sample size for the planned adaptive design were assessed and estimated in a large simulation study (see below). 61 We anticipated correlations between time points (based on Karthikeyan et al.)76 and effect sizes to be equivalent between the Constant score and OSS; thus, these simulations remained valid, despite the change of primary outcome. 68,76 The simulations showed that an adaptive design that allowed the possibility of early stopping for efficacy and/or futility, was feasible for the START:REACTS study.
Based on an assumed modest correlation between 3-, 6-, and 12-month OSS equal to 0.5, and an SD of 12 for both 3- and 6-month scores, the simulations for the selected adaptive design indicated that follow-up data from a maximum of 188 participants would be needed to detect a six-point difference in the OSS between treatment arms with 90% power and 5% (two-sided) type I error rate. Allowing for 15% lost to follow-up, while attempting to keep this below 10%, a maximum study sample size of 221 was calculated.
Primary outcome analysis
All data have been analysed and reported in accordance with the Consolidated Standards of Reporting Trials guidelines. 77,78 A detailed statistical analysis plan and a data-sharing plan were agreed with the DMC prior to any formal analyses being conducted. 79
The primary analysis investigated differences in the OSS 12 months after surgery between the two treatment groups on an intention-to-treat basis following the methods, test statistics and boundaries described by Parsons et al. 61 To preserve the integrity of the study, the exact boundaries used for testing were specified in an adaptive charter known only to the study team and independent DMC. In brief, if the study recruited to target, Parsons et al. ’s61 methods would be used to calculate the boundaries. As the study stopped early, testing proceeded using boundaries calculated by the deletion method for over-running analysis,80 with inferences following directly from widely used methods for unbiased estimates and confidence intervals (CIs) in group sequential trials. 80–82
Secondary outcome analyses
Descriptive statistics of all outcome measures data (i.e. the OSS, WORC, EQ-5D-5L, PGIC and analgesia usage) at each time point were constructed.
Adjusted estimates of treatment group differences (with 95% CIs) for the OSS at 12 months post-randomisation were calculated using a mixed-effects model including a random effect for the recruiting centre and fixed effects for variables of interest including patient age, sex and size of tear. Estimates of efficacy for the other outcome measures followed this approach to analysis. Adverse and SAEs were analysed using Fisher’s exact test. Patient-reported change in symptoms and the PGIC were analysed using adjusted proportional odds ordered regression models.
Owing to the low level of missing data in the primary outcome, no imputation was undertaken.
Exploratory analyses
To assess the impact of changing the primary outcome measure from the Constant score to the OSS, we assessed the relationship between the two scores by fitting a simple linear model between the two outcomes at each time point.
Based on discussions with clinicians at sites, proximal humeral migration on a preoperative X-ray was used by some shoulder surgeons to understand the severity of the rotator cuff tear. 83,84 Explanatory models were used to assess the relationship between acromiohumeral position and the OSS data in the trial. The routine clinical images at baseline (X-rays and MRIs) were used to measure AHD for all participants recruited (registered) into the study. To assess the effects of AHD on the primary outcome, AHD was added as an additional confounder in the adjust mixed-effects model (described above).
Subgroup analyses
Prespecified subgroup analyses were undertaken to assess whether there was evidence that the intervention effect differed with respect to:
-
the size of the rotator cuff tear as measured at the start of surgery, defined as large or massive cuff tear (≥ 3 cm) or moderate to small tear (< 3 cm)
-
sex (male or female)
-
age (≥ 70 or < 70 years).
These variables were selected as they are either important to the function of the intervention (cuff tear size) or to interpretation (sex and age). The subgroup analyses followed the methods described for the mixed-effects model for the primary analysis, with additional interaction terms incorporated into the mixed-effects regression model to assess the level of support for these hypotheses. The study was not powered to formally test these hypotheses, so they are reported as exploratory analyses only, and are secondary to the analysis reporting the main effects of the intervention in the full study population. 85
Interim analyses
Interim analyses were only undertaken following the principles laid out by Parsons et al. 61 Details of the timings and settings for the interim analyses were kept confidential so as not to prejudice the outcome of the trial based on the decision to stop the study or proceed but were recorded in the DMC meeting minutes and on date-stamped internal documents. The decision to stop the study early for efficacy or futility was not planned to be communicated outside of the DMC closed meeting. However, the TSC requested to view the interim analysis report before accepting the DMC’s recommendation to close randomisation.
Initial simulation study
A 3-month work package was undertaken at the start of study to develop appropriate methodological tools, implement a series of simulation studies using synthetic data to understand the properties of the selected design and its sensitivities to model assumptions and, in collaboration with the study DMC, agree futility and efficacy stopping boundaries. 61
Study end points for the OSS were at 3, 6 and 12 months. For the adaptive design, the 12-month primary outcome would have been too late to be used for determining futility before the study finished recruiting, and therefore, available early and late outcome data were used to determine early stopping.
A 3-month outcome was chosen, particularly as the balloon degrades after this time, with the predictive strength of the model strengthened further by the addition of 6- and 12-month outcomes. A decision to stop for futility would only be considered when there was sufficient confidence (based on this comprehensive simulation work) using all available 3-, 6- and 12-month data. A decision to stop because of clear evidence of efficacy was also considered in the modelling but especially strong evidence of efficacy (and the relationship to later outcomes) was required for early stopping.
Boundary setting
Futility stopping boundaries based on early and late observations of a single study end point with a final analysis adjusted for futility stopping have been suggested previously by Stallard. 58 Methodology developed previously by the group for two time points was extended to three time points (and also made more general) to allow the totality of observed data at planned interim analyses to be used to inform study progress (e.g. stopping).
Results from our systematic review suggested a strong association between early and late outcomes based on data from trials of interventions for rotator cuff tears. 42 A strong positive correlation between 3- and 6-month outcomes and 12-month outcomes indicated that information on the former early outcomes were strongly indicative of later outcomes, and consequently intervention efficacy or futility.
Sequential stopping boundaries were constructed that allowed stopping for futility or stopping to reject the null hypothesis (efficacy), with interim analyses determined by the results of the simulations and agreed with the TSC and DMC. 86 It was agreed that the stopping boundaries would be binding. Stopping boundaries for safety were also agreed and set as a separate criterion for early stopping.
Simulated data that replicate the metric properties of the primary outcome were then used to explore the characteristics of the design and sensitivities to likely treatment effect sizes and correlations between early and late outcomes. The aim was to understand changes in study power and rates of early stopping for a range of likely treatment effect sizes. The results of these simulation studies were presented to the TSC and DMC, and rules were agreed such that it was clearly defined as to when interim analyses should happen, what the thresholds were for early stopping, and how decisions should be communicated within the study team.
Preliminary simulations indicated that a single interim analysis using 3-month data on 53 patients per arm with the probability of early stopping under the null hypothesis set to 50% would result in only a small reduction in power to 88% for modest correlations between 3- and 12-month data. Given the findings of our systematic review of a good relationship between early and late outcomes in trials of interventions for rotator cuff tears42 and the fact that correlations were improved by considering all of the available time points, there is likely to be little loss of power.
As we expected correlations between time points (based on Karthikeyan et al.)76 and effect sizes to be equivalent between the Constant score and the OSS, these simulations remained valid, despite the change of primary outcome score. 68
As with the primary analysis, the interim analyses were performed using a frequentist approach although a Bayesian framework was explored as a substudy (see Chapter 8).
Changes to the protocol
As described above, the planned primary outcome for the study was originally the Constant score. 38,39 However, in light of the COVID-19 outbreak in March 2020, the TMG decided to change the protocol to use the OSS as the primary outcome.
Various other changes were made to the original protocol throughout the duration of the trial. These include:
-
To insert the balloon, the ‘lateral portal’ incision was slightly larger – 1.5 cm as opposed to 1 cm. To ensure blinding to study allocation, we used a 1.5-cm incision for all participants.
-
For surgeons unable to attend a training session in person we suggested that they either read the surgery manual or watch an online surgery video.
-
As eligibility was confirmed intraoperatively, patients found to be ineligible at randomisation were informed that they were no longer taking part in the study by letter.
-
The number of sites taking part increased to 16–20 from the initial proposed amount.
-
We conducted two pilot studies for the MRI substudy (MRI pilot and shoulder muscle function pilot) prior to conducting the main substudy. This was to confirm that the design of the main substudy was robust enough to achieve the objectives set out.
-
There were concerns that participants who were lost to follow-up may have had safety events that we could miss. For that reason, we decided to contact the participants’ general practitioners for those who were considered lost to follow-up at 12 months to request information in regards to adverse and SAEs.
-
We used minimisation for randomisation rather than permuted random blocks based on a simulation exercise which demonstrated a high risk of major imbalance in the study arms with random permutated blocks. The randomisation process was changed to minimisation with a random factor, with a 70% weighting towards balance across the whole study.
-
The isometer was deleted from the secondary objectives and replaced with a dynamometer, which was used for the strength measurements at 3, 6 and 12 months.
-
Images were collected at baseline from previous imaging of the shoulder to assess in a secondary exploratory analysis whether imaging findings (especially proximal migration of the humerus) predicts outcome after surgery.
-
Planned sample size increased from 212 to 221 due to recalculation to increase the power in the study after discussions with the DMC.
-
We changed the time point for the first MRI in the substudy from 6 to 8 weeks, with the window between −2 and + 4 weeks. This was to ensure that participants had longer to recover from surgery before their first MRI.
-
The time window for the 3-month follow-up decreased from −6 to −2 weeks to ensure that participants had longer to recover before they returned for the 3-month follow-up.
-
Study participants whose postoperative pain interfered with their rehabilitation programme could be referred for a steroid injection (with or without local anaesthetic in the shoulder region).
Chapter 3 Main trial results
Participants
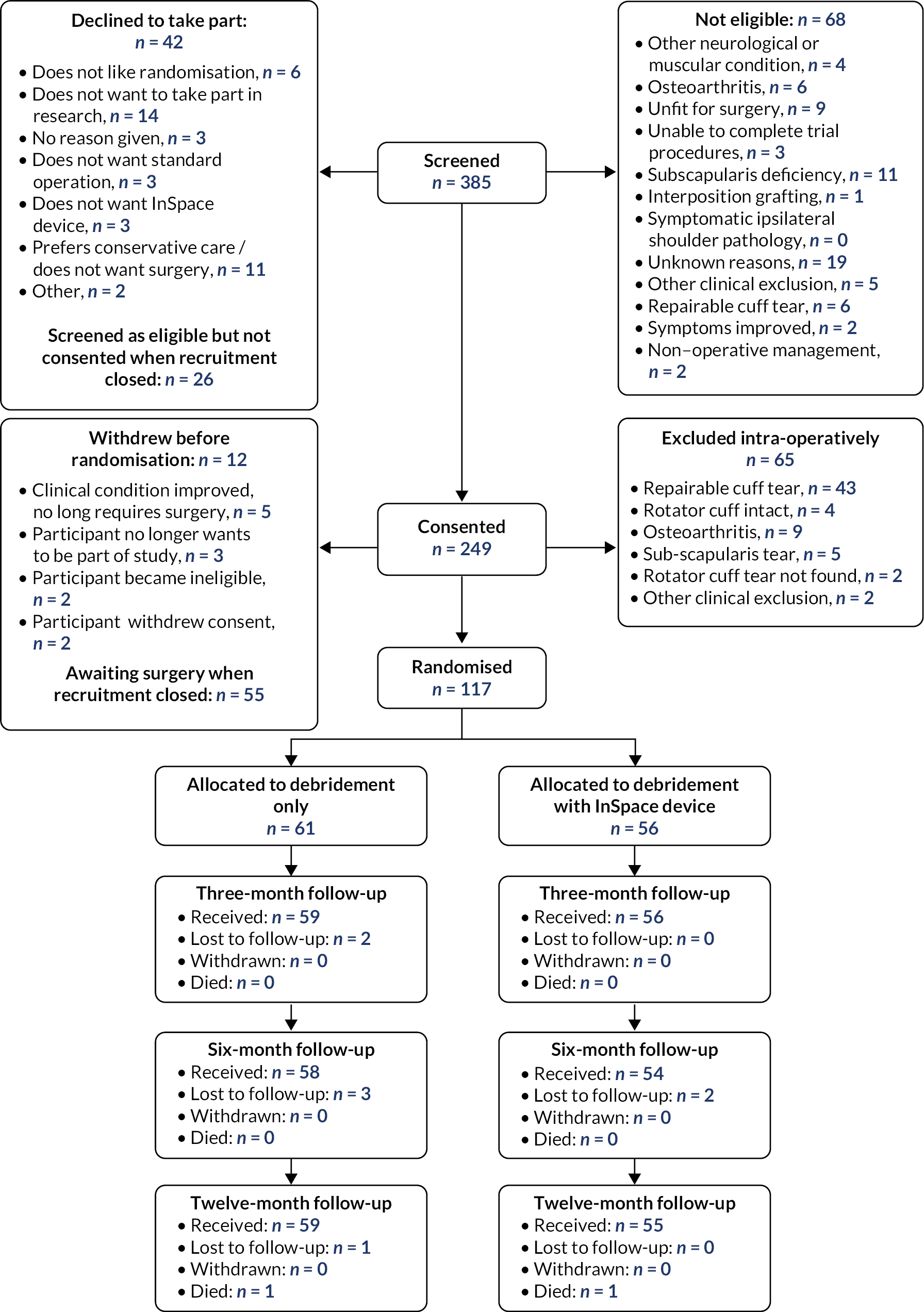
Between 1 June 2018 and 30 July 2020, we identified 317 eligible people and 249 (79%) consented to participate. A total of 117 participants were randomised into the study when recruitment was closed; 61 (52%) participants were randomised to receive arthroscopic debridement surgery alone, and 56 (48%) participants were randomised to receive arthroscopic debridement with the InSpace balloon (see Figure 2). On the 30 of July 2020, recruitment and randomisation were stopped after the futility boundary had been crossed at the first interim analysis. Stopping rules and observed data at the first interim analysis are given in Appendix 1.
FIGURE 2.
Consolidated Standards of Reporting Trials diagram.
Baseline statistics
Baseline variables were well balanced (see Table 1), although there were more male than female participants (n = 67 : 50), and slightly more under age 70 years in the InSpace balloon group. The mean age of the study participants was 67 years; 79 (68%) participants had injured their right shoulder, with 63% having their dominant shoulder affected. Mean duration of symptoms or pain was five years and 68% reported past trauma or injury may have caused the tear. Five participants had rotator cuff tears of less than 3 cm in the control group and one in the intervention group. The mean tear sizes were very similar between groups (debridement-only, mean 4.3 cm; debridement with InSpace balloon, mean 4.2 cm).
| Baseline statistics | Debridement-only (n = 61)a |
Debridement with InSpace balloon (n = 56)a |
All randomised (n = 117)a |
||
|---|---|---|---|---|---|
| Baseline details and demographics | Age (years) – mean (SD) | 67.3 (9.0) | 66.4 (7.6) | 66.9 (8.3) | |
| Age group (years), n (%) | Under 70 | 33 (54) | 36 (64) | 69 (59) | |
| Sex, n (%) | Female | 28 (46) | 22 (39) | 50 (43) | |
| Rotator cuff tear size, n (%) | Large (≥3 cm) | 56 (92) | 55 (98) | 111 (95) | |
| Medium/small (<3 cm) | 5 (8) | 1 (2) | 6 (5) | ||
| Right shoulder affected, n (%) | 42 (69) | 37 (66) | 79 (68) | ||
| Left or right-handed, n (%) | Left | 10 (16) | 5 (9) | 15 (13) | |
| Right | 51 (84) | 50 (89) | 101 (86) | ||
| Missing | 0 (0) | 1 (2) | 1 (1) | ||
| Dominant or non-dominant shoulder affected, n (%) | Dominant | 38 (62) | 36 (64) | 74 (63) | |
| Non-dominant | 22 (36) | 18 (32) | 40 (34) | ||
| Missing | 1 (2) | 2 (4) | 3 (3) | ||
| Baseline PROM mean, - (SD) | OSS | 21.7 (9.4) | 23.1 (8.5) | 22.4 (9.0) | |
| Constant score | 33.6 (13) | 29.9 (13.4) | 31.9 (13.2) | ||
| WORC | 34.4 (14.2) | 33.7 (13.1) | 34.1 (13.6) | ||
| EQ-5D-5L | 0.501 (0.258) | 0.486 (0.247) | 0.494 (0.251) | ||
| Pain-free range of motion, mean (SD) (n = 109) | Abduction angle (degrees) | 76.3 (32.8) | 63.9 (22.2) | 70.5 (28.9) | |
| Flexion angle (degrees) | 74.1 (25.1) | 67.8 (29.8) | 71.1 (27.4) | ||
| Symptom duration in years – mean (SD) | 4.3 (6.2) | 5.5 (7.1) | 4.9 (6.7) | ||
| Other medical conditions, n (%) | 53 (87) | 45 (80) | 98 (84) | ||
| Participant smokes, n (%) | 4 (7) | 5 (9) | 9 (8) | ||
| Participant has diabetes (all type II), n (%) | 9 (15) | 9 (16) | 18 (15) | ||
| Participants has unilateral or bilateral symptoms, n (%) | Unilateral | 43 (71) | 39 (70) | 82 (70) | |
| Received previous physiotherapy treatment, n (%) | 42 (69) | 44 (79) | 86 (74) | ||
| Previously received steroid injection, n (%) | 34 (56) | 36 (64) | 70 (60) | ||
| Number of steroid injections taken | Median (range) | 2 (1–6) | 2 (1–10) | 2 (1–10) | |
| Previously had surgery on shoulder, n (%) | 16 (26) | 9 (16) | 25 (21) | ||
| Surgery details | Anterior-posterior tear size (cm) | Mean (SD) | 4.3 (1.3) | 4.2 (1.3) | 4.2 (1.3) |
| Mediolateral retraction from GT attachment (cm) | Mean (SD) | 4.3 (1.0) | 4.0 (1.0) | 4.1 (1) | |
| Biceps tendon intact, n (%) | 38 (62) | 39 (70) | 77 (66) | ||
| Subscapularis torn, n (%) | 12 (20) | 14 (25) | 26 (22) | ||
| Subscapularis tear size (cm) | Mean (SD) | 0.7 (0.3) | 0.8 (0.4) | 0.8 (0.4) | |
| Subscapularis repaired, n (%) | 2 (3) | 2 (4) | 4 (3) | ||
Primary outcome: Oxford Shoulder Score
Response rates were very high across the follow-up time points. Twelve-month primary outcome data were obtained from 114 of the 117 participants (97%). Of the three instances of missing primary outcome data, two participants had died (neither trial related) and one could not be contacted. Table 2 shows the descriptive statistics of the OSS at each time point.
| Follow-up point | Statistic | Debridement-only (n = 61) |
Debridement with InSpace balloon (n = 56) |
All randomised (n = 117) |
|---|---|---|---|---|
| Baseline | Received, n (%) | 61 (100) | 56 (100) | 117 (100) |
| Missing, n (%) | 0 (0) | 0 (0) | 0 (0) | |
| Mean (SD) | 21.7 (9.4) | 23.1 (8.5) | 22.4 (9.0) | |
| 3 months | Received, n (%) | 59 (97) | 54 (96) | 113 (97) |
| Missing, n (%) | 2 (3) | 2 (4) | 4 (3) | |
| Mean (SD) | 30.4 (11.2) | 25 (10.4) | 27.8 (11.1) | |
| 6 months | Received, n (%) | 58 (95) | 54 (96) | 112 (96) |
| Missing, n (%) | 3 (5) | 2 (4) | 5 (4) | |
| Mean (SD) | 33.3 (10.4) | 28.5 (11) | 31 (10.9) | |
| 12 months | Received, n (%) | 59 (97) | 55 (98) | 114 (97) |
| Missing, n (%) | 2 (3) | 1 (2) | 3 (3) | |
| Mean (SD) | 34.3 (11.1) | 30.3 (10.9) | 32.4 (11.2) |
Figure 3 shows the mean OSS score at each time point and the two allocation groups. Scores improved compared with baseline in both groups. The debridement-only group had a relatively quicker recovery and higher scores at each of the follow-up time points.
FIGURE 3.
Oxford Shoulder Score means and 95% confidence intervals for each time point.

Primary efficacy analysis
In the primary intention-to-treat analysis, the mean (unadjusted) difference in the OSS at 12 months was −4.2 points (95% CI −8.2 to −0.26; p = 0.037), favouring debridement only. This estimate of the effect of the intervention is smaller than our target difference of six points, however, this difference is included in the 95% CI. It should also be noted that this was a target for a worthwhile benefit, and we had not specified what might be a meaningful harm.
Secondary efficacy analyses
The secondary prespecified efficacy analysis was a model adjusted to account for the baseline OSS, sex, tear size and age group. The results are shown in Table 3. A similar treatment effect was observed to the primary analysis, with the groups which received the InSpace balloon having worse outcomes than the group which received arthroscopic debridement alone (effect −4.2, 95% CI −7.8 to −0.6; p = 0.026). This difference was smaller than the target difference of six points. The baseline OSS score was the only other statistically significant variable, showing that those participants whose disability and pain were worse at baseline also had worse outcomes at follow-up. That is, there was an additional gain of 0.57 points (95% CI 0.4 to 0.8; p < 0.001) at follow-up for every point increase in the baseline OSS.
| Fixed effect variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 |
– |
0.026 |
| Debridement with balloon | −4.2 | (−7.8 to −0.6) | ||
| Baseline | OSS score | 0.6 | (0.4 to 0.8) | < 0.001 |
| Sex | Male | 0 | – | 0.598 |
| Female | −1.1 | (−4.9 to 3.0) | ||
| Tear size | Large | 0 |
– | 0.349 |
| Medium or small | 4.0 | (−4.4 to 12.3) | ||
| Age group (years) | < 70 | 0 | – | 0.778 |
| ≥ 70 | 0.6 | (−3.2 to 4.4) | ||
Secondary outcomes
The Constant score
Further details of the Constant scores can be found in Appendix 1.
The Constant score had high levels of missingness due to the difficulty of obtaining data during the lockdown due to COVID-19. Insufficient data were collected to allow for the planned linear regression model to be calculated. However, as earlier time points were less affected, some inferences could be made on the 3- and 6-month data. The collected data showed that the debridement-only group had better outcomes at these early time points.
Western Ontario Rotator Cuff Index
Further details of the WORC scores can be found in Appendix 1. Here it can be seen that the effect of the InSpace balloon again has a reduction on function (mean −8.4 points, 95% CI −16.7 to −0.1; p = 0.055), following the results of the OSS. Similarly, to the adjusted OSS model, smaller tear size and higher ages were associated with non-significant increases in function. Female sex was also associated with an in increase in function, but this was small (mean 1.5, 95% CI −7.3 to 11.1; p = 0.74).
EuroQol Five Dimension Five-Level score
Further details of the EQ-5D-5L scores at each time point can be found in Appendix 1. No terms were found to be statistically significantly associated with health-related quality of life (HRQoL). The InSpace balloon was associated with a small non-significant decrease on overall HRQoL (mean −0.056, 95% CI −0.15 to 0.03; p = 0.239). The older age group was associated with an increase (0.015, 95% CI −0.08 to 0.12) while female sex (−0.007, 95% CI −0.10 to 0.09) and smaller tear sizes (−0.059, 95% CI −0.28 to 0.15) were associated with reductions in HRQoL, although all these differences were non-significant.
Patient Global Impression of Change
Participants were asked to report how much better or worse their shoulder felt 12 months after their surgery as well as if there was any change in their activity limitations, symptoms, emotions, and overall QoL. Details are shown in Appendix 1. There were no statistically significant differences in either overall change (OR 0.6, 95% CI 0.3 to 1.2) or in PGIC (OR 0.5, 95% CI 0.3 to 1.1); although both questions favoured the arthroscopic debridement alone group.
Subgroup analyses
Descriptive statistics and details of the models for each of the pre-specified subgroups (age group, tear size and sex) can be found in Appendix 1. The results of each of the three subgroups are summarised visually in Figure 4.
FIGURE 4.
Forest plot of the adjusted effect of the allocation group on 12-month OSS for each pre-planned subgroup. Blue circles denote the adjusted mean difference, grey whiskers the 95% CI. Values left of zero favour the arthroscopic debridement group.

When including an interaction term for age and allocation group, the model remained broadly consistent with the secondary effect analysis (interaction effect for InSpace balloon and 70 years and over −5.3, 95% CI −12.1 to 2.38). This was also the case for tear size (interaction effect for InSpace balloon and small tears 6.8, 95% CI −14.9 to 28.8). Note that because of the small number of small to medium tears, the marginal effects of tear size could not be estimated.
Participant sex was found to be an important confounder (interaction effect for InSpace device and female sex −9.5, 95% CI −16.5 to −2.6). Females in the InSpace balloon allocation group had a decrease in function, larger than the worthwhile difference, when compared with males. Figures depicting this effect can be found in Appendix 1.
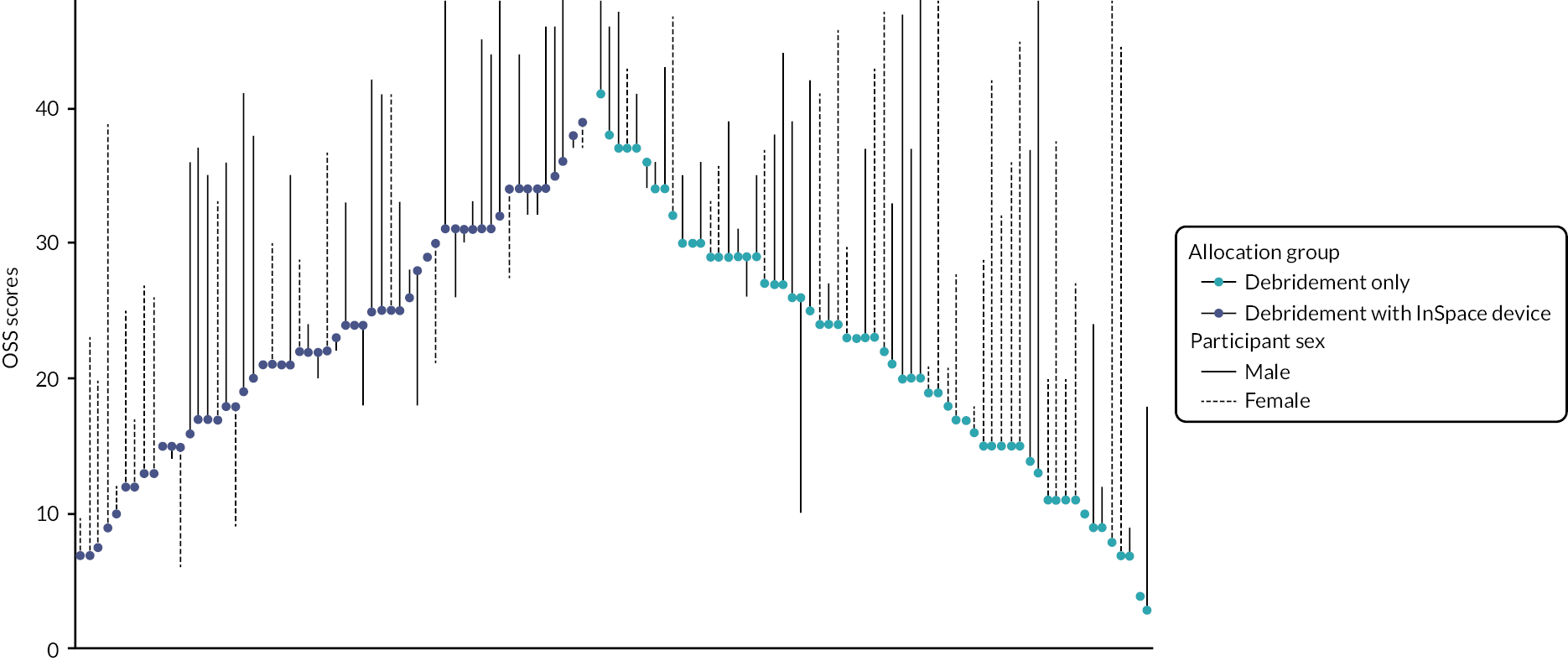
To further investigate this effect, we constructed a waterfall plot (see Figure 5). This allows us to inspect visually the effects of baseline and 12-month OSS, allocation group and participant sex. Here, it can be seen that while females tended to have lower scores at baseline, those allocated to receive debridement-only typically had larger gains than those who received the InSpace balloon. Most participants reported better function at follow-up than at baseline, although a larger number of people declined in function in the balloon group compared to baseline.
FIGURE 5.
Waterfall plot of 12-month OSS score by sex and allocation group. Scores have been ordered by baseline score (highest at centre) and allocation group (debridement-only on right-hand side). The coloured dot denotes the participant’s baseline score, which is joined to the participant’s 12-month OSS by a line (solid for males, dashed for females).
Safety data
Overall, 20 participants reported a total of 28 adverse events. Numbers were similar in the two groups (n = 9 : 11; see Table 4). There were six serious adverse events, four in the debridement with InSpace balloon group and two in the debridement-only group. Three serious adverse events were considered unrelated to the surgery and three were considered related: two (one in each group) for persistent pain or disability at 12 months (defined as requiring continuing secondary care review) and one for further surgery, a reverse shoulder replacement in the debridement with device group.
| Adverse events | Debridement-only (n = 61) | Debridement with InSpace balloon (n = 56) | Total (n = 117) |
p-values | |
|---|---|---|---|---|---|
| Participant experienced any AE (n, % of total participants in group) |
9 (15) | 11 (20) | 20 (17) | 0.624 | |
| AEs per participant, n (% of total participants with AEs) |
1 | 7 (78) | 9 (82) | 16 (80) | 0.592 |
| 2 or more | 2 (22) | 2 (18) | 4 (20) | 1 | |
| Total reported AEs (n) | 11 | 17 | 28 | 0.134 | |
| Exacerbation/persistence of shoulder pain or restrictive ROM, n (% of total AEs in group) | 5 (46) | 6 (35) | 11 (39) | 0.756 | |
| Injection into the shoulder region, n (% of total AEs in group) | 1 (9) | 3 (18) | 4 (14) | 0.348 | |
| Adhesive capsulitis, n (% of total AEs in group) | 0 | 2 (12) | 2 (7) | 0.227 | |
| Persistent muscle soreness or muscle injury, n (% of total AEs in group) | 0 | 1 (6) | 1 (4) | 0.479 | |
| Other, n (% of total AEs in group) | 5 (46) | 4 (24) | 9 (32) | 1 | |
| Related events – SAE category | |||||
| Participants experiencing ≥ 1 SAE, n (% of total participants in group) | 1 (2) | 2 (4) | 3 (5) | NA | |
| Total reported SAEs (n) | 1 | 2 | 3 | NA | |
| Hospitalisation or prolongation of existing hospitalisation, n (% of SAEs in group) | 0 | 1a (50) | 1 (33) | NA | |
| Persistent or significant disability/incapacity, n (% of SAEs in group) | 1 (100) | 1 (50) | 2 (67) | NA | |
| Was related SAE expected: Yes, n (% of related SAEs in group) | 1 (100) | 2 (100) | 3 (100) | NA | |
| Related to InSpace balloon, n (% of total in group) | NA | 2 (100) | 2 | ||
| Unrelated events – SAE category | |||||
| Participants experiencing ≥ 1 SAE, n (% of total participants in group) | 1 (2) | 2 (4 | 3 (3) | NA | |
| Total reported SAEs (n) | 1 | 2 | 3 | NA | |
| Hospitalisation or prolongation of existing hospitalisation, n (% of SAEs in group) | 0 | 1 (50) | 1 (33) | NA | |
| Persistent or significant disability/incapacity (n, % of SAEs in group) |
1 (100) | 1 (50) | 2 (67) | NA | |
Sensitivity and exploratory analyses
Effects of COVID-19
We undertook a sensitivity analysis to assess if COVID-19 could have affected the primary analysis. Data from participants who had completed the 12-month primary outcome before the first lockdown were compared with the remaining participants; 25 participants had completed their 12-month primary end point prior to the first UK lockdown (20 March 2020) and 89 participants were followed up during/after the COVID-19 lockdown period. The results of this analysis (see Appendix 1) confirm that COVID-19 did not significantly change the collected OSS or EQ-5D-5L scores at 12 months, nor did it affect the completion rates. However, since there was a major impact on collecting the 12-month Constant score (97.8% missing during or after COVID-19), this score could not be assessed.
Acromiohumeral distance and rotator cuff pathology
Since more participants than expected were excluded intraoperatively, we did additional exploratory analyses to explore whether there were differences in the pathology between the two groups at registration, prior to surgery. Hence, these analyses should be interpreted with caution and may not be generalisable.
While baseline OSS and Constant scores were both greater in the repairable cuff tear group, these differences were small and not statistically significant (OSS mean difference 2.1, 95% CI −5.0 to 0.9; Constant score mean difference 3.8, 95% CI −1.2 to 8.9). The difference in AHD was statistically significant with those which had a repairable cuff tear having a significantly larger AHD than those with a non-repairable tear (mean 1.1, 95% CI −0.11 to 2.1; p = 0.034). Full details of these analyses can be found in Appendix 1.
Acromiohumeral distance and Oxford Shoulder Score at 12-month follow-up
Including AHD into the adjusted efficacy model showed that the OSS improved by approximately 0.47 points for every 1 mm increase in the AHD; however, this was not a statistically significant result (95% CI −0.3 to 1.2; p = 0.236). The effect of the allocation group remained broadly similar to the main analyses, favouring the debridement alone group.
Relationship between the Oxford Shoulder Score and the Constant score
The original primary outcome was the Constant score (see above). As the study design was dependent upon the observed correlations at each follow-up pint, Pearson’s correlation coefficient between this outcome and the OSS were calculated to confirm that it was a suitable alternative. The OSS is scored between 0 and 48, hence we rescaled scores to a 0–100 range to allow direct comparison to the Constant score (range is 0–100). These results (see Appendix 1) suggest that the Constant score has a strong association with the OSS at each time point.
Chapter 4 Discussion of main trial results
Clinical discussion
We set out to test the effectiveness of a new surgical procedure, the InSpace subacromial balloon spacer, for people with irreparable rotator cuff tears. The study was blinded and the comparator was an otherwise identical procedure without the balloon. A futility boundary was crossed and the study stopped with 117 participants recruited as the result of a planned interim analysis. Arthroscopic debridement was found to be superior to arthroscopic debridement with the InSpace device, based on the OSS 12 months after surgery. Given that the only difference between the two interventions was the device itself, it is highly unlikely that the balloon provides any meaningful benefit, and it could be harmful.
Secondary outcomes were in agreement with the primary outcome. Although these were mostly not significant, there was a consistent pattern of change in favour of debridement-only. Significant differences were noted for some of the objective measures such as strength and ROM, although numbers for these measures at later time points were limited due to the pandemic, so these should be treated with caution. Safety data showed no differences between interventions, although as there were few adverse events, the sample size was much too small to detect any meaningful difference in this regard.
The target difference for the OSS this study was six, based on previous anchor-based studies and used in other multicentre trials. 41,68,69,76 While the point estimate was lower than this, the target difference was contained within the CI and the CI excluded zero. Minimum clinically important differences (MCIDs) are typically determined to measure benefit, they are less likely to be relevant in the setting of negative results. It may be difficult to justify using any intervention that shows statistically significant worse results on the primary outcome compared with control, no matter how small the point estimate of the difference.
In the subgroup analyses, a strong effect of sex was observed, with poor results for the device in females. We strongly caution against the use of the device in females but, even in males it is unlikely that the device provides any benefit. There was no other significant association found in the subgroup analyses, although an association with age (older people having worse results) could not be ruled out.
The lack of influence of baseline AHD on outcome should reassure surgeons that there is not a latent subgroup related to radiographic disease severity.
A blinded multicentre trial of the InSpace device compared with partial rotator cuff repair was conducted by Verma et al. 87 and was reported shortly after our study, in April 2022. This was a company-funded trial (by OrthoSpace, subsequently purchased by Stryker) and contributed to the device being awarded FDA marketing approval in the United States in 2021. The eligibility criteria were different to ours, principally as the study was focused on people with massive irreparable rotator cuff tears (over 5 cm or two or more tendons torn). However, in our study, tear size did not appear to influence the result. The study reported non-inferiority of the balloon compared to partial cuff repair, with similar outcomes across most domains at the primary outcome time point of 2 years. 43 A small improvement in the secondary outcome of ROM was seen in the balloon arm, although some differences were present at baseline, and any discrepancy in this might be related to stiffness after partial repair. Partial rotator cuff repair is rarely used in the UK in this condition and the evidence for its use is limited to case series data, with mixed results reported. 44,45,83 The trial did not directly test the effect of the balloon, as we have in the present trial.
Apart from the direct clinical impact, the findings of our study also demonstrate the critical importance of randomised trials in the early evaluation of surgical devices and techniques, before they are introduced into widespread clinical practice. The InSpace device had been used in around 29,000 cases in Europe at the time of the 2021 FDA approval, largely on the basis of case series data alone, most of which has only been published in the last few years. 23 Case series are unable to demonstrate a true benefit as a comparator is needed. We have previously shown in a meta-analysis that outcomes for people with a rotator cuff tear improve over time regardless of the treatment given, and, therefore, an improvement in patient outcome from baseline does not mean that a treatment is effective.
There was no clear pattern in the safety data and this is also an important observation. Monitoring of safety data alone cannot be used to ensure that a product is not causing harm. Such information may only be evident in a robust evaluation of clinical outcomes, such as a clinical trial reporting patient-focused outcomes.
This study identifies a deficiency in the current process for introducing new surgical devices and procedures. One solution to this would be to apply the IDEAL criteria more stringently to the introduction of new procedures, allowing registered prospective development studies and randomised trials in approved centres, but only allowing a product or technique to be more widely available when convincing proof exists that it is likely to be of benefit. 88,89 This would be a marked change to current practice but would protect patients from potential harm and prevent the health service from incurring unnecessary expense.
Adaptive design methods
In this study, we have successfully implemented a novel adaptive group sequential randomised trial, using the correlation between early end point data and the primary outcome to inform stopping decisions at two planned interim analyses. The correlations between time points were higher than expected and due to the pandemic, we continued to accrue outcome data for a 3-month period while randomisations were paused. As a result of these factors, the emerging data enabled us to stop recruitment early with robust data in what would have otherwise been a very challenging time. Assuming that we continued with the rate of recruitment that was observed prior to March 2020, the study would have taken an additional 11 months to recruit the full sample size, although it is likely that recruitment would have been further delayed by the effects of the pandemic on elective surgery. This highlights the value of the novel methodology by not only saving on research costs but also by providing much earlier impact for patients and the health service, improving patient outcomes and saving money.
Having successfully implemented this novel methodology in one study, we recognise the need for ongoing work to establish this as a technique for future use and to convince clinicians, trials methodologists and research funders that the methods could be used more widely. To address this, we undertook a large simulation study using data from multiple previous high-impact randomised trials; this work is reported in Chapter 7.
Limitations
Our study has a number of limitations which should be considered. As the study stopped early, CIs remained wide for the secondary outcomes and a larger sample size may have provided more precise estimates. The study was designed to determine if the device was effective or not, which was consistently answered by both the primary and secondary outcomes. Even if a larger sample size demonstrated further evidence of harm, the implications of the findings would remain that the device is not recommended in this population. A larger sample size would have exposed more people to risk, but it is highly unlikely that the overall findings of the study would change. The study was not designed to have adequate power for subgroup analyses, either at 117 or 221 participants, and it is unlikely that the clinical conclusions would have been changed had these analyses been formally powered. While the device could be of benefit in a different population, our eligibility was focused on the most commonly used indications for the device both in the UK and internationally. Because of the pandemic, we were not able to collect objective measures for many of the participants, especially at 12 months, although the objective outcomes we did take in the study were consistent with the primary outcome and significant differences were observed in favour of debridement-only, although with a very small amount of data.
This study was not designed to determine the effectiveness of arthroscopic debridement for people with an irreparable rotator cuff tear. It is commonly used in the UK for this indication and provided an appropriate comparator in this study. Given the results presented here, there is need for a future trial of arthroscopic debridement compared to alternative surgical procedures, placebo, or a non-surgical intervention. There is also a need for additional RCTs of other treatments, as there is a lack of evidence for all available treatment options for irreparable rotator cuff tears at present.
Conclusion
We implemented a novel group sequential, blinded, multicentre randomised trial in people with irreparable rotator cuff tears and found that arthroscopic debridement was superior to an otherwise identical procedure performed with the InSpace device. We do not recommend the InSpace balloon spacer in this population.
Chapter 5 Health economics
Overview of economic evaluation
The economic component of the study included a standard health policy-relevant economic analysis, and an exploration of how early economic data might support decision-making through the use of an adaptive design.
A prospective economic evaluation was integrated into the trial, adhering to the recommendations of the NICE reference case. 90 The evaluation was conducted with the objective of estimating the cost-effectiveness of arthroscopic debridement of the subacromial space with insertion of the InSpace balloon compared with arthroscopic debridement of the subacromial space only. A health economic analysis plan was developed and is available on request.
This economic evaluation took the form of a cost-utility analysis, expressed in terms on incremental cost per quality-adjusted life-year (QALY) gained. The primary analysis is based on an NHS and PSS perspective as recommended by NICE, while wider impact (societal) costs were included within a sensitivity analysis. 90
Mechanisms of missingness of data were explored and multiple imputation methods applied to impute missing data. Imputation sets were used in bivariate analysis of costs and QALYs to generate incremental cost per QALY estimates and confidence regions. 91–94 It was anticipated that incremental costs and benefits would be captured within the trial and that extrapolated modelling would not be required.
Relatively little research has been conducted prospectively on how interim economic analysis might inform interim decision-making. Current approaches are based mainly on net monetary benefit approaches using value of information methods. 95,96 We compared various methods using economic data (costs and QALYs) and clinical data to evaluate the practical implications and operating characteristics of stopping a trial early based on cost-effectiveness data alone, efficacy data alone or a combination of cost-effectiveness and efficacy. START:REACTS has been used to evaluate putative analytical methods, as set out within a prospectively written health economic analysis plan and parallel interim analyses, separate from the real trial analyses, exploring how interim decisions might have been influenced. Given that health economic decision rules for adaptive designs are less widely understood and used in the literature, the properties of these methods will be developed further for future trials.
Measurement of resource use and costs
We used a comprehensive strategy including estimation of intervention delivery costs and broader health and PSS costs97 to determine the incremental costs associated with the START intervention.
Costing of the intervention
The economic evaluation focused on assessing intervention implementation costs, including:
-
Health-care resource use: costed using most recently available published national reference costs (see Appendix 2, Table 60), within a common year. 22,98–100 Participants were asked about their health service contacts in connection with their shoulder condition at 3, 6 and 12 months. The numbers of contacts with community and outpatient clinics and use of hospital services were recorded within the trial case report form.
-
Additional medication/prescriptions: collected from a diary to assist participants to record this information. The costs of study medications were included using most recent English prescription cost analysis data (using British National Formulary 2020). 101 Participant-level costs were estimated as the sum of resources used weighted by their unit costs. 101
-
Direct intervention costs: including the costs of delivering arthroscopic debridement with insertion of the InSpace balloon and arthroscopic debridement only.
-
Production losses in terms of the time from work (paid or unpaid) due to receiving the intervention was also recorded (although time from work was not included as a resource from the perspective of the analysis).
Collection of broader resource-use data
Data were collected on health, PSS and broader societal resource inputs between randomisation and 24 months post-randomisation. Trial participants were required to complete resource-use questionnaires at baseline, 3, 6, 12 and 24 months post-randomisation using a modified version of the Client Service Receipt Inventory (version 1.0). Data were reported in terms of the following categories:
-
inpatient care
-
outpatient care
-
community health care
-
personal and social services
-
loss of production/work
-
pain medication.
Medication use was categorised by drug name (or active constituent), mode of administration, dosage frequency and duration.
Valuation of resource use
Resource inputs were valued using a combination of primary research and data collated from secondary national tariff sets, using standard accounting methods. Inpatient admissions during the study (overall and by type) were determined from the NHS reference costs trusts schedule (2020–21 tariffs were not available due to COVID-19). 98–100 Other hospital-based care costs were valued from 2019–20 national tariffs by applying unit costs. NHS medication prices (per milligram) were obtained from the British National Formulary. 101 Participant-level costs were estimated based on reported doses and frequencies, when available, or otherwise based on an assumed daily dose. 102
Sex-specific median earnings data were applied to occupational classifications derived from self-reported work status information to determine the costs of time taken off work by participants (or their carers). Data reported by the participants as part of the follow-up resource-use questionnaires were also used to determine other family-borne costs. The NHS Hospital and Community Health Services Pay and Prices Index Unit was used to inflate or deflate costs where necessary to 2020–21 prices (£ sterling). 103 No discounting of costs was applied because cost-effectiveness was determined over a 1-year time horizon.
Calculation of utilities and quality-adjusted life-years
The economic evaluation estimated QALY profiles for participants, based on reports of preference-based HRQoL outcomes. The HRQoL of trial participants was assessed using the EuroQol EQ-5D-5L,104 measured at baseline, 3, 6, 12 and 24 months post-randomisation. Participants completed a two-page questionnaire consisting of the EQ-5D-5L descriptive system and the EQ VAS – a self-rated vertical VAS where 100 denotes ‘best imaginable health state’ and 0 denotes ‘worst imaginable health state’. EQ-5D-5L scores were converted to health status scores using the mapping function developed by van Hout et al. 105 The analysis values each health state/level combination as a single health-related index including 0 (death) and 1 (perfect health), where negative scores are possible for some health states.
Using the trapezoidal rule, the area-under-the-curve of health status scores were calculated, providing participant-level QALY estimates. No discounting was applied to quality-adjusted survival data reflecting the follow-up period (1 year). EQ-VAS scores were entered as a separate item and reported for completeness, but not for trial-based analysis. No discounting of QALYs was applied because cost-effectiveness was determined over a 1-year time horizon.
Missing data
Multiple imputation was used to estimate effects under the missing at random assumption. Missing at random assumptions were explored using logistic regression of the missingness of costs and QALYs against baseline variables. Prognostic and stratification covariates were assessed as potential missingness predictors (including outcome measures and costs at each time point). 106
Imputation models used fully conditional methods (multiple imputation by chained equations),66,107 which are appropriate when correlation occurs between variables. Predictive mean matching drawn from the five nearest neighbours (knn = 5) was used to enhance the plausibility and robustness of imputed values, as normality could not be assumed.
A model was used to generate multiple imputed data sets for treatment groups, where missing values were estimated conditional on available covariates: these included baseline costs, baseline utilities, the size of the rotator cuff tear as measured at the start of surgery (≥ 3 or < 3 cm), sex and age (≥ 70, < 70 years). With multiple imputation, a complete data set was generated, reflecting the distributions and correlations between variables in the observed data. Each imputed data set was analysed independently using model-based approaches; estimates obtained were pooled to generate mean and variance estimates of costs and QALYs using Rubin’s rules108 to capture within and between variances for imputed samples. Information loss from finite imputation sampling was minimised using 20 data sets, resulting in minimal loss of efficiency (< 0.5%) when compared with infinite sampling. Since the fraction of information missing was reasonably low, n = 20 imputation sets were considered adequate. 95,107–109 Imputed and observed values were compared to determine the impact of imputation on estimates.
Analyses of resource use, costs and outcome data
Resource-use items were summarised by treatment group and assessment point; differences between groups were analysed using two-sample t-tests for continuous variables. Mean (SD) of each resource type, including by cost category, were estimated by trial allocation group for each time period. Costs were estimated from both an NHS and PSS perspective. This was also repeated from a broader societal perspective. Differences between groups in terms of costs, together with their respective Cis, were estimated and reported. Nonparametric bootstrap estimates using 10,000 replications90 were also calculated for differences along with their respective CIs. EQ-5D-5L utility score differences at each follow-up point between the groups were tested using two-sample t-tests assuming unequal variances.
Model-based methods (seemingly unrelated regression) were used to estimate mean incremental changes in costs and QALYs and accounted for the correlation between costs and outcomes within the data while adjusting for covariates, including baseline costs and utility scores to adjust for potential baseline imbalances. Non-parametric bootstrap methods were used to generate the joint distributions of costs and outcomes to populate the cost-effectiveness plane. Bootstrapping (using bias-corrected non-parametric bootstrapping) is a resampling method which jointly resamples costs and outcomes from the observed data while holding the sample correlation structure. From each bootstrap sample (10,000 samples), a change in costs and QALYs are estimated. Means estimates were reported with 95% CIs.
Cost-effectiveness analyses
The incremental cost-effectiveness ratio (ICER) was estimated as the difference between the trial comparators in mean total costs divided by the difference in mean total QALYs. Value for money was determined by comparing the ICER with a cost-effectiveness threshold value; typically, the NICE cost-effectiveness threshold for British studies ranges between £20,000 and £30,000 per QALY. In addition, a £15,000 cost-effectiveness threshold was used. This represents society’s willingness to pay for an additional QALY; lower ICER values than the threshold could be considered cost-effective for use in the NHS. Base-case assumptions were explored using a range of supportive sensitivity analyses.
The incremental net monetary benefit (INMB) of switching (from standard care) to the experimental intervention was also reported as a recalculation of the ICER at a range of cost-effectiveness thresholds. The INMB succinctly describes the resource gain (or loss) when investing in a new intervention when resources can be used elsewhere at the same threshold. INMB estimates were used to generate cost-effectiveness acceptability curves. The curve compares the likelihood that interventions are cost-effective as the cost-effectiveness threshold varies.
All statistical analyses and cost-effectiveness modelling were conducted in SAS® version 9.4 (SAS Institute Inc., Cary, NC, USA) on a Microsoft Windows platform.
Sensitivity and subgroup analyses
The following subgroup analyses were undertaken by the planned subgroups:
-
rotator cuff tear (≥ 3 or < 3 cm)
-
sex (male or female)
-
age (≥ 70 or < 70 years).
Treatment by subgroup interaction
A treatment by subgroup interaction term was also tested for sex and age group for each of costs and QALYs, while ensuring costs and QALYs were correlated following a bivariate regression model of the form:
by the planned subgroups: sex and age (≥ 70 years, < 70 years) in terms of QALYs and total costs. The interaction was tested at a two-sided 5% level.
Long-term cost-effectiveness model
The study protocol allowed for extrapolation of costs and consequences over a longer time horizon if the trial demonstrated statistically significant differences in medium-term outcomes. As the key primary clinical outcomes did not demonstrate clinical and statistical benefits, this was not formally undertaken. However, a sensitivity analysis for QALYs over a 2-year period was carried out.
Results
Study population
A total of 117 participants were randomised into the START trial: 56 to the debridement with InSpace balloon group and 61 to the debridement-only group. Complete baseline information was available for 117 participants, so the baseline study population for the bulk of the health economic analyses was 117 participants.
Between 96% and 100% of all health resource-use data were complete at baseline for participants randomised to debridement with InSpace balloon and between 95% and 97% for those randomised to debridement-only group. Similarly, these values ranged between 93% and 100% between 3 and 12 months, depending on the specific health resource item. A complete QALY profile was available over 12 months for 54/56 participants (96%) with debridement with InSpace balloon and 58/61 (95%) for those with debridement-only. Consequently, about 5% of QALY data and between 5% and 7% of the total cost outcomes were missing (and subsequently imputed) for the primary analysis.
Resource use and costs
Cost of intervention
The direct cost of arthroscopic debridement with InSpace balloon was £5378 and £3589 for arthroscopic debridement-only. These costs were based on healthcare resource group procedure codes HN53A (major shoulder procedures for non-trauma with CC score 4+). 22
Broader resource use
Over 12 months, for debridement with InSpace balloon and debridement-only, hospital admissions and outpatient care use were 16% versus 10% and 75% versus 67%, respectively (see Appendix 2, Table 59). The average (mean) number of physiotherapy contacts were 8.2 versus 6.4 and 20% of patients had at least one MRI in the debridement with InSpace balloon group compared with 11% in the debridement-only group.
Community health care and PSS use were 20% versus 15% and 2% versus 7% for debridement with InSpace balloon and debridement-only, respectively. Analgesic use was 10% lower in the debridement with InSpace balloon group (75% vs. 85%) as was other prescription medication usage at 29% versus 41%. The mean number of days taken off work were 79 days versus 64 days for debridement with InSpace balloon versus debridement-only. Where unit costs were not available for 2021 prices, these were inflated using the NHS Hospital and Community Health Services Pay and Prices Index. 103
Resource-use values (frequency of use) were combined with unit costs for each resource item to estimate economic costs for each resource category. Table 5 shows resource-use costs for participants with complete data by trial allocation, resource-use category and study period. The cost components are aggregated into seven components, namely: (1) physiotherapy costs; (2) hospital inpatient costs; (3) hospital outpatient costs; (4) community health-care costs; (5) travel costs, based on distances travelled by practitioners by mode of transport; (6) venue hire costs; (7) personal and social care services; (8) time of work; and (9) analgesic/medication use. These costs varied between £1 (analgesic use) and £466 (inpatient costs) depending on treatment group and cost type.
| Cost category between baseline and 12 months | Debridement with InSpace balloon (N = 56) Mean (SD) |
Debridement-only (N = 61) Mean (SD) |
Mean difference | p-valuea | Bootstrap 95% CIb | |
|---|---|---|---|---|---|---|
| Lower 85% | Upper 95% | |||||
| NHS/PPS costs | ||||||
| Inpatient costs | 466 (1126) | 353 (1181) | 113 | 0.5996 | −473 | 227 |
| Outpatient costs | 281 (375) | 269 (355) | 12 | 0.8595 | −123 | 90 |
| PSS costs | 2 (16) | 4 (19) | −2 | 0.60.32 | −7 | 4 |
| Analgesic use | 1 (0.5) | 1 (0.5) | 0 | 0.4706 | −0.14 | 0.03 |
| Other concomitant/prescription medications | 4 (22) | 1 (0.8) | 3 | 0.2884 | −1 | 9 |
| MRI | 55 (114) | 32 (81) | 23 | 0.3515 | −1 | 37 |
| Total NHS PSS costs | 809 (1337) | 659 (1284) | 150 | 0.5382 | −298 | 84 |
| Broader societal costs | ||||||
| Time off work (days) | 230 (662) | 109 (467) | 121 | 0.0502 | −1 | 223 |
| Total (societal) | 1039 (1830) | 768 (1557) | 271 | 0.1039 | −168 | 395 |
| Direct intervention costs | 5378 | 3589 | 1789 | – | – | – |
| Total PSS/NHS including intervention costs | 6187 (1337) | 4248 (1284) | 1939 | < 0.001* | 1540 | 2337 |
| Total societal including intervention costs | 6417 (1829) | 4357 (1557) | 2060 | < 0.001c | 1729 | 2811 |
Resource-use frequencies in other categories were low (see Appendix 2, Table 59), hence, meaningful comparisons could not be easily made and for the most part were not statistically significant. The cost of taking time off work was, on average, £230 versus £109 for the arthroscopic debridement with InSpace balloon group and arthroscopic debridement-only groups, respectively, and this difference was close to statistical significance (p = 0.0502).
Economic costs
There were no statistically significant differences between the trial groups in any specific component cost category, at any time point with the exception of time off work (see Table 5). Over the entire follow-up period, mean (standard error, SE) total NHS and PSS costs, inclusive of the cost of the intervention, were £6187 (£1337) in the intervention arm compared with £4248 (£1284) in the control arm, generating a mean cost difference of £1939 (bootstrap 95% CI £1540 to £2337; p < 0.001). Over the entire 12-month follow-up period, mean (SE) total societal costs, inclusive of the cost of the intervention, were £6417 (£1829) and £4357 (£1557) for debridement with InSpace balloon and debridement-only, respectively, generating a mean cost difference of £2060 (bootstrap 95% CI £1729 to £2811; p < 0.001). Hence, debridement with InSpace balloon costs were statistically higher compared with debridement-only. There was no treatment by sex interaction in terms of costs (p = 0.2153) or treatment by age group interaction (p = 0.424).
Health-related quality-of-life outcomes
There were no (statistically significant) differences in the overall EQ-5D-5L utility scores or EQ-5D-5L VAS scores between the intervention and control groups, at each of the follow-up time points. For complete cases, the mean (SE) patient-reported QALY over 12 months was estimated as 0.551 (0.0586) compared with 0.606 (0.0307) for debridement with InSpace balloon and debridement-only, respectively (p = 0.2242). In the absence of a significant effect on the EQ-5D-5L either at the primary outcome time point, or the EQ-5D-5L based QALY, usual care was dominant in health economic terms. Unless the expected costs were lower for the debridement with InSpace balloon group, usual care would almost always dominate.
There was a statistically significant treatment by subgroup interaction in terms of QALYs for sex (p = 0.0025) and age group, which is reflected in estimates of the incremental QALY in these groups (see Table 6).
| Incremental cost (95% CI)a | Incremental QALYs (95% CI) | ICERa (95% CI) | Probability of cost-effectiveness (%) | INMB | |||||
|---|---|---|---|---|---|---|---|---|---|
| pb | pc | pd | INMBa,b | INMBa,c | INMBa,d | ||||
| Base case (NHS/PSS perspective + societal) | |||||||||
| Imputed attributable costs and QALYs, covariate and baseline adjusted EQ-5D utility score | 2322 (686 to 1765) | −0.0512 (−0.0226 to 0.1259) | Dominated | 0.01 | 0.07 | 0.37 | −3074 (−4403 to −1761) | −3325 (−5010 to −1652) | −3826 (−6244 to −1427) |
| Sensitivity analyses | |||||||||
| 1. Complete cases attributable costs and QALYs, and baseline adjusted EQ-5D utility score | 2060 (1729 to 2811) | −0.0549 (−0.1439 to 0.0341) | Dominated | < 1 | < 1 | < 1 | −2884 (−3888 to −2299) | −3158 (−4607 to −2129) | −3707 (−6046 to −1788) |
| Subgroup analyses | |||||||||
| 2. Rotator cuff, imputed costs and QALYs, covariate and baseline adjusted EQ-5D utility score: | |||||||||
| Rotator cuff (≥ 3 cm) (n = 111) | 2359 (1780 to 2914) | −0.0566 (−0.209 to 0.0928) | Dominated | 1.7 | 2.93 | 7.1 | −3208 (−5551 to −912) | −3491 (−6575 to −490) | −4058 (−8661 to 405) |
| Rotator cuff (< 3 cm) (n = too small) | n/c | n/c | n/c | n/c | n/c | n/c | n/c | n/c | n/c |
| 3. Sex, imputed costs ande QALYs, covariate and baseline adjusted EQ-5D utility score: | |||||||||
| Male (n = 67) | 1953 (1332 to 2546) | 0.0529 (−0.209,0.0928) | Dominated | 12.0 | 24 | 42 | −1159 (−2757 to 453) |
−895 (−2952 to 1173) |
−366 (−3347 to 2650) |
| Female (n = 50) | 2974 (2061,3980) | −0.212 (−0.3198 to −0.10437) | Dominated | 0 | 0 | 0 | −6155 (−8026 to −4233) | −7216 (−9560 to −4811) | −9336 (−12675 to −5910) |
| 4. Age group, imputed costs and QALYs, covariate and baseline adjusted EQ-5D utility score:e | |||||||||
| ≥ 70 (n = 48) | 2307 (1880 to 2758) | −0.118 (−0.218 to −0.0215) | Dominated | 0 | <1 | <1 | −4087 (−5809 to −2388) | −4680 (−6886 to −2504) | −5867 (−9042 to −2732) |
| < 70 (n = 69) | 2180 (1350 to 3007) | 0.00537 (−0.101 to 0.111) | Dominated | 3.0 | 7.0 | 16.0 | −2099 (−3955 to −221) | −2072 (−4432 to 287) | −2019 (−5412 to 1352) |
Cost-effectiveness results
Base case analysis
The incremental cost-effectiveness of arthroscopic debridement with InSpace balloon is shown in Table 14 for the participants with costs and health outcomes data subject to multiple imputation. When a societal (NHS/PSS plus broader societal costs) perspective was adopted (i.e. that adopted for the baseline analysis) and health outcomes were measured in terms of QALYs, the mean incremental cost was £2322. The mean incremental cost-effectiveness of debridement with InSpace balloon was estimated at −£45,351 per QALY; that is, on average, the intervention was associated with a higher net cost and a lower net effect and was dominated in health economic terms.
The associated mean INMB (net loss) at cost-effectiveness thresholds of £15,000, £20,000 and £30,000 per QALY were −£3074, −£3325 and −£3826, respectively (see Table 6). The base case mean INMB was < 0, suggesting that those randomised to debridement with InSpace balloon would result in an average NHS/PSS loss of about £3074 [INMB (£) = −2158, 95% CI −3455 to −969] to [INMB (£) = −3074, 95% CI −4403 to −1761]. The cost-effectiveness plane (see Figure 6) shows that the vast majority of the ICER values lie in the north-west quadrant. These result in a probability of cost-effectiveness close to zero; that is, if decision-makers are willing to pay between £15,000 and £30,000 for an additional QALY, the probability that the debridement with InSpace balloon intervention is cost-effective is very low (< 1%).
FIGURE 6.
Cost-effectiveness plane for DB (experimental: debridement with InSpace balloon intervention) vs. D (control: debridement only): incremental cost (£) vs. incremental QALY – base case.
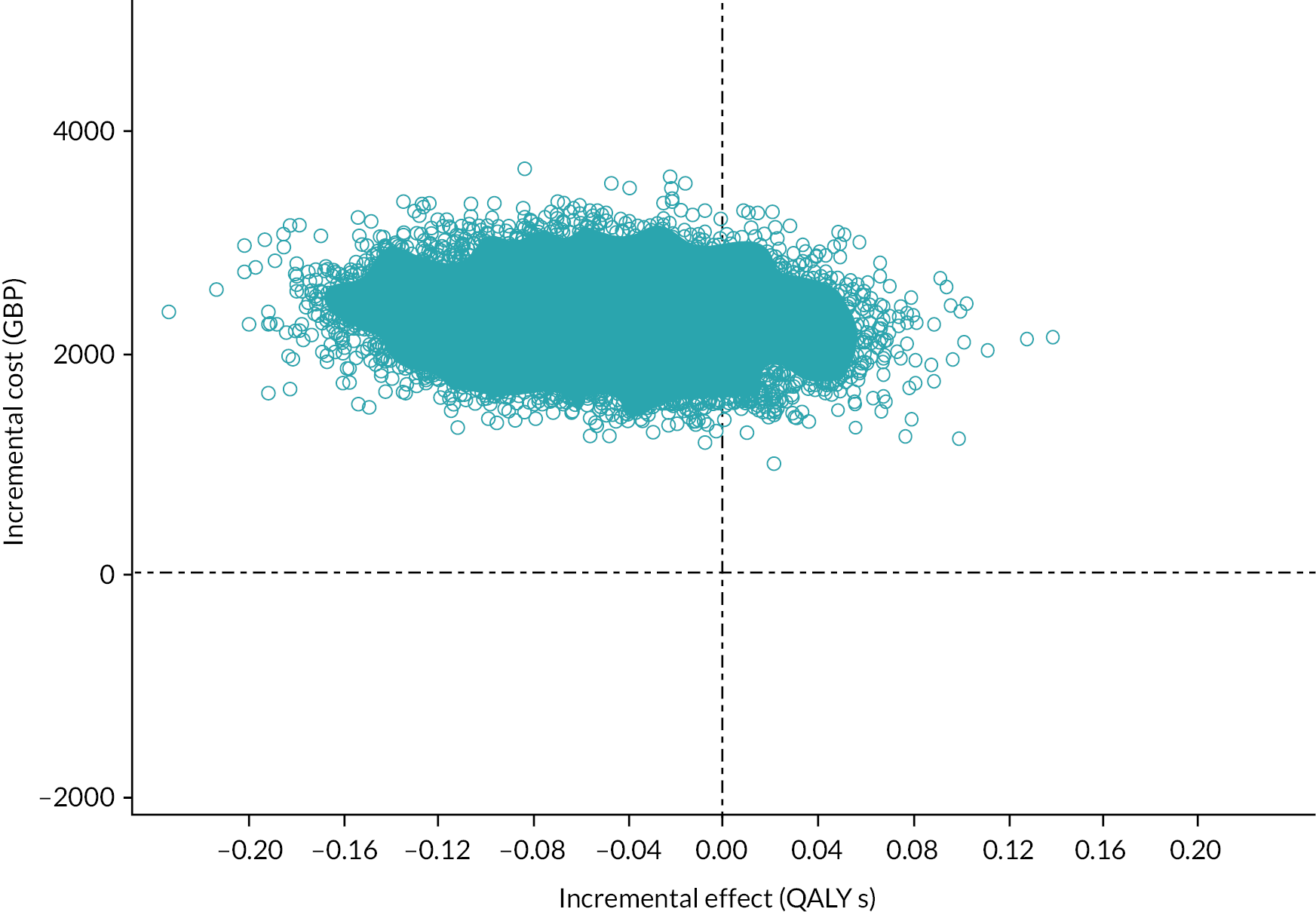
Sensitivity and subgroup analyses
Several sensitivity analyses were undertaken to assess the impact of uncertainty surrounding key parameters or methodological features on the cost-effectiveness results. The probability that the debridement with InSpace balloon intervention is cost-effective remained relatively static (< 1%) for the majority of the sensitivity analyses (see Table 6). For subgroups, only in the case of sex was the probability of cost-effectiveness higher (at 42%) for males compared with females at a CE threshold of £30,000/QALY. In all cases the average INMB was negative: all analyses showed the average INMB were unlikely to be positive as all upper limits of the 95% CI were below zero (see Table 6; Figure 7). A treatment by subgroup interaction in terms of costs and QALYs.
FIGURE 7.
Forest plot of sensitivity and subgroup analyses (impact on incremental net monetary benefit).
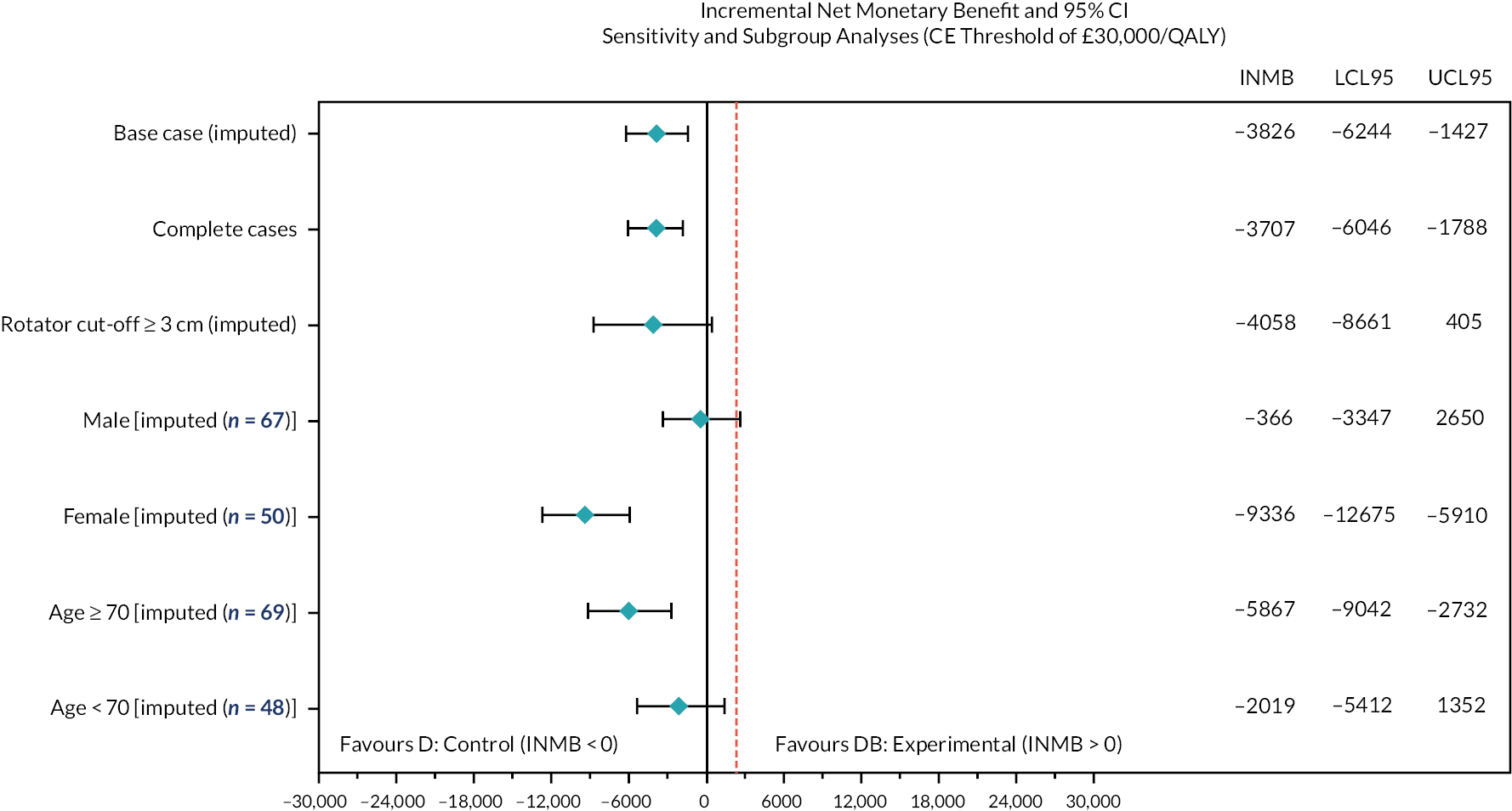
Missing data assumptions
Using a logistic regression model, the response (missing or not) was modelled against covariates age group and sex to assess whether missingness was explained by the covariates. Since the number of missing observations was small [n = 3 (5%) for debridement-only and n = 4 (7%) for the debridement with InSpace balloon group] and similar between groups, missingness was not statistically associated with the covariates (p > 0.05) (see Appendix 2, Table 61).
Conditional power for cost-effectiveness
Conditional power (CP) is a well-established method for conducting futility analyses to stop a trial early due to lack of efficacy using interim data. 110 However, such futility rules can be problematic. Trials of interventions with modest treatment effects may pass futility criteria (high CP) but have little chance of demonstrating value for money from a cost-effectiveness perspective. Conversely, trials may stop prematurely due to modest treatment effects (low CP) that might have proved cost-effective because, for example, safety improved. Futility analyses reflecting both health outcomes and costs might better inform futility decision-making. In this research, we generalise the CP expressions used for efficacy to a cost-effectiveness framework. 110,111
Expressions for the CP based on efficacy for the two-sample case (1 : 1 allocation) are extended to cost-effectiveness. The INMB, willingness to pay threshold, interim and final sample sizes, variabilities and correlations between costs and effects are examined. Data from two clinical trials are used to examine the operating characteristics of the conditional power of cost-effectiveness (CPCE).
Assuming a two (independent) group trial, the sample size formula for a two group (1 : 1 allocation) randomised trial using the standard two-sample t-test is described in equation 1:
Rewritten, this is:
where Zα, Zβ are typically the 5% and 80% (or 90%) values from the normal tables referring to the type I and type II errors respectively, σ2 is the estimated (population) pooled variance from responses across the two treatment arms and Δ is the clinically relevant difference in means (between groups) in some efficacy outcome (determined from |μ1−μ2 |, where μ1 and μ2 are the population mean responses for groups 1 and 2, respectively, and N is the total sample size).
The expressions in equations (1) and (2) can be expressed in the following form:
Where
is the observed test statistic at some fraction t where all parameters are estimated using data at the interim analysis. ΙΩ in equation (2) is the combined information fraction from, nt is the total sample size at time t. In equation (3), θINMB is the INMB under H1 (i.e. the target mean INMB, at the end of the trial, which should be > 0). Hence, expanding (Eq 3) to use the average information fraction, we get:
where INMB is the observed mean INMB at some interim period, v2c1 and v2c2 are the variances of costs for each of the two groups at some interim period; Nc1 and Nc2 are the planned sample sizes, nc1 and nc1 are the interim sample sizes, k is the cost-effectiveness threshold; s2c1 and s2c2 are the observed variances of costs at the interim which are estimates of the unknown v2c1 and v2c2; subscripts E and C refer to costs and effects (QALYs) for the respective treatment (experimental and control); The term θINMB is the expected INMB under some alternative hypothesis (usually > 0). If the value of θINMB is set to 0 (i.e. the mean INMB is non-negative), equation (4) can be simplified further. For this example, we will assume the target mean INMB is zero in the worst case, since to plan for a trial yielding a mean INMB of less than 0 would not seem to make sense.
Results of applying the above equation (4) to the START data under the assumption of zero correlation between costs and effects, with the results estimated after 50% of patients using summary statistics only at 6 months, for NHS/PSS perspective, the estimated parameters for the CP are shown in Table 62 in Appendix 2. Hence, Pt(θ) = 2.5% This calculation provides a CP for cost-effectiveness of 2.5%
This means that the chance of cost-effectiveness at the end of the trial, conditional on what was observed during the interim and assuming that future data would behave in a similar way to the interim, would be low enough to warranting terminating the trial for lack of cost-effectiveness.
Discussion
This trial-based economic evaluation revealed that the arthroscopic debridement with InSpace balloon is not cost-effective compared with arthroscopic debridement-only. The INMB estimate was negative, a finding that remained robust to several sensitivity and subgroup analyses. The main reason for lack of cost-effectiveness appears to stem from a combination of higher cost in the experimental group with no evidence of effectiveness based on EQ-5D-5L derived QALYs. The main strengths of this analysis are that the START trial was prospectively designed for a cost-effectiveness analysis using individual-level data. Costs and outcomes were carefully considered in the design of this trial with the purpose of reaching a robust conclusion with respect to cost-effectiveness in a large sample of individuals.
There were, however, several limitations to this cost-effectiveness analysis. QALYs were based on utility measurement at just four time points post-randomisation. Although the trial did not yield benefits, the assumption of linearity of HRQoL between data collection points is uncertain and more uncertain when missing data are present. Despite the longitudinal nature of the study, resource use was retrospectively recalled by trial participants, which is likely to result in recall bias. In addition, similar pilot or phase II trials may have been useful in identifying the critical costs that drive cost-effectiveness. Instead, all costs were collected which, on average, had little impact on the ICER. Many costs items did not occur (see Table 5) and a reduced form of the data collection in a similar setting may be advisable with a focus on the largest and more relevant costs. In addition, many of these costs had little impact on the results. Finally, the 95% CIs surrounding the incremental QALYs do not exclude the possibility of a small QALY benefit because the 95% CIs contain the value zero. However, the upper limit of these intervals from the sensitivity analyses, never exceeds much beyond 0.10, in which case for an observed base-case incremental cost of £2322, the ICER would be around £23,000 per QALY. However, the probability of this would be small as the upper 95% limit for the incremental QALY is a value in the tail end of the distribution.
Despite these limitations, this trial was a comparatively large prospectively designed RCT. A recent trial,87,112 used a composite end point and demonstrated non-inferiority of arthroscopic InSpace (subacromial tissue spacer system) implantation to arthroscopic partial repair of rotator cuff. Notably, a secondary outcome in this trial was a 12-month change from baseline in the EQ-5D VAS. As the results are not published in full, no conclusion can be drawn or compared with the results of this trial at this time.
Methodology summary
We explored different approaches to the use of novel methodologies to complement adaptive designs aimed at arriving earlier decisions around cost-effectiveness. Our initial approach using value of information was unlikely to be meaningful given the observed results. We therefore extended CP computations traditionally used for futility to a cost-effectiveness framework.
Conditional power is a useful monitoring tool that allows for decision-making to stop a trial unlikely to yield the desired effects based on some early interim data. The importance of futility analyses in publicly funded trials has been shown to important. 113 While CP has been used in clinical trials where the primary outcome is efficacy, its use in cost-effectiveness settings has been very limited due to a combination of complexity and absence of closed form expressions. In this research, we present a close form expression that can be used in a relatively simple way in an excel spread sheet that allows for computing the probability that a trial will be cost-effective at the end of a trial based on current (interim data). The expression also computes CPCE when costs and effects are correlated.
CPCE is a useful monitoring tool that can assist in early decision-making based on using information on costs and effectiveness. This will complement traditional CP approaches and may help in a decision to stop or continue a trial when the treatment effects in particular are modest.
Conclusion
In conclusion, the data collected in the START trial support strongly a hypothesis that arthroscopic debridement with an InSpace balloon, when compared with debridement alone is more costly and less effective.
Chapter 6 Magnetic resonance imaging substudy and development work
Introduction
In order to use MRI to enable the evaluation of the physical effect of the InSpace balloon on the shoulder, a predictable and reproducible method of activating the deltoid was sought, to determine whether humeral head migration can be observed during MRI studies. Humeral head migration towards the acromion is one of the purported mechanisms of causing pain in this cohort of people and is theorised to occur under activation of the deltoid muscle. A consistent and predictable MRI technique to study dynamic proximal migration of the humerus could be of value to a wide range of research into rotator cuff pathology and the relationship between rotator cuff tears and clinical symptoms. MRI was selected over plain radiograph to assess for humeral migration because the measurement of AHD on X-ray was thought to be too inconsistent, as it would depend on the angle the beam was taken, which may vary. MRI has been previously used for this measure and was thought to be a more sensitive measure.
The most commonly proposed action of the balloon is not as a depressor of the humeral head but to resist dynamic proximal motion of the humerus. There are two time points of interest for this study; the first is when the balloon is still inflated to see whether its proposed mechanism of action occurs in vivo. The second is when the balloon has deflated, to see whether the mechanical effect is maintained, as has been proposed by some authors. 33
Aim
The aims in the substudy had two components, the first was development and refinement of the novel MRI technique, the second was the substudy itself:
-
The aim of the development work was to refine a technique to reliably and repeatably measure humeral proximal migration on MRI when placing the deltoid muscle under load using a simple abduction task against resistance.
-
The aim of the START:REACTS MRI substudy was to scan 56 participants to assess the mechanism of action of the balloon in comparison with no balloon, when the balloons are likely to be still inflated (but when acute postoperative pain has subsided), and when they are likely to have fully deflated, to see if the proposed mechanism for continuing improvement is maintained.
Methods
Methods for developmental work
People with a rotator cuff tear listed for surgical repair were identified from waiting lists at University Hospitals Coventry and Warwickshire and invited to enter one of two studies. While the main START:REACTS study was recruiting patients with irreparable rotator cuff tears, we selected people with repairable rotator cuff tears for the development of the technique described here. The development of the technique was not impeded by selecting a similar yet distinct group of people but allowed for the retention of as many patients for potential recruitment into the main study.
Consent was taken prior to their participation. Ethical approval was obtained on 13 February 2018 (Integrated Research Application 233804). All participants completed a case report form, which included sections on demographics, the severity, duration and impact of symptoms, as well as the investigative work-up and therapy received.
The first group of 11 participants took part in an electromyography (EMG) study to assess the optimal method of activating the deltoid muscle, and then four participants underwent MRI scanning using the pilot methodology. For the EMG component of the study, participants were invited to attend research clinics where deltoid muscle activity was determined using surface EMG while undergoing a series of tasks. This allowed us to determine a technique for deltoid muscle activation that could subsequently be performed by participants in the second part of the study, who were invited for a MRI where humeral head migration was measured. All participants provided written consent after discussion with a research nurse but prior to MRI and EMG studies being done. Participants were free to stop the investigations at any time during the studies if they felt pain.
Electromyography substudy
The aim of the first study was to define an appropriate movement against resistance that would be possible in the MRI scanner and would activate the deltoid muscle. To measure activation of the deltoid muscle, surface EMG electrodes were attached to the participant while they performed the movements.
Participants were asked to perform a simple task involving abduction of the arm at the shoulder to either 5 cm or 10 cm, measured from the neutral resting position of the medial humeral epicondyle, under tension while lying down on a bed with the upper body raised to a 45-degree angle. A tape measure was placed on the couch to ensure the distance of abduction was the same between participants.
Exercise bands (TheraBandTM, Akron, OH, USA) of various tension were used to allow for reproducible resistance forces against the arm movements between participants. These bands deform predictably under normal tension to a defined distance allowing the force generated to be controlled. 19 Three bands were selected for the task, representing the lowest tension (yellow), average tension (green) and highest tension (black). The band was wrapped around the participants’ arms at the level of the elbow and hand-tied at the front while the participants were in a neutral resting position allowing them to push outwards (abduct at the shoulder) against the band. The band did not have any slack and was neither stretched nor deformed before the movements took place to prevent preconditioning of the bands. The positioning was checked independently by two researchers.
Each participant performed this task at least four times on the injured side, a baseline measure with the arm held in the neutral position (i.e. not extended), and a further three times under tension using the graded exercise bands. Each data capture trial lasted for 20 seconds, consisting of 10 seconds with the arm at rest in the neutral position, followed by the abduction of the arm, held for another 10 seconds. Total data capture duration was set to 30 seconds to allow for any delay in movement onset.
Surface EMG was undertaken at rest and during the tasks to determine deltoid activity. Two Shimmer3TM (Shimmer Sensing, Dublin, Ireland) units were used to detect EMG activity. Each device consists of a small transmitter unit which is fitted to the participant using an elasticated band; one device was attached to the arm and the other around the chest to minimise cable movement and distance to the muscles. Up to two channels of EMG can be recorded by each device. The first device on the arm was used to capture signals from electrodes placed on the posterior and lateral heads of the deltoid muscle; the device on the chest was used to capture signals from the anterior deltoid muscle. The location of the electrodes was determined using the guidelines provided in Hermens et al. 114 The devices used wireless Bluetooth connections to transmit the EMG data to a laptop.
Signal processing
Signals were captured with a sample frequency of 512 Hz and processed using MATLAB (version 2018a; MathWorks Inc., Natick, MA). Each channel was subsequently high-pass filtered with a cut-off frequency of 10 Hz using a sixth-order Butterworth filter. The resulting signals were subsequently rectified and further filtered using a moving-average filter, with a window size of 50 milliseconds.
A baseline measure of the EMG signal from each channel in the rest position was taken over the final one-second period before the arm was abducted. The mean and SD of the signal amplitude was calculated over this period. Muscle activation was then defined as any parts of the recorded signal that exceeded a threshold of three SDs above the mean (as recorded in the one-second rest period). 115 The level of activation during abduction was defined as the proportion of the 10-second abduction period that the muscle was activated (i.e. 100% activation would be recorded where the amplitude of the EMG signal for that channel exceeded the threshold for the full 10 seconds). We refer to this measure as the activation score, ranging from 0.0 to 100.0.
Four participants exclusively underwent tests using all three bands at 10 cm, however as the study progressed it became clear that a 10-cm movement may be too far to be feasible in a MRI scanner for all patients, therefore, we added 5-cm movements for subsequent participants. Three participants underwent testing at both 5 and 10 cm, and four more participants at just 5-cm abduction. This produced two groups of seven participants each, one for abduction testing at 10 cm and another group for testing at 5 cm. A mean of the activation scores for each test (e.g. all participants conducting 5-cm movements with a yellow band) was calculated. Outliers were removed as well as null values where their values exceeded two SDs from the mean. This produced a mean activation score for each test.
Magnetic resonance imaging substudy
After the EMG study, a further four participants were invited for an MRI of the shoulder using the novel protocol developed for this study. These were all participants who had been listed by the treating surgeon for a repair of the rotator cuff based on prior imaging. The participants were positioned in the MRI scanner with a custom-made plastic ‘L-shaped board’ placed under the participants back, level with the elbow.
The board was designed with two vertical slats spaced 5 cm apart. Each participant placed their arm against the first slat. A green TheraBand was applied around the participants in the same manner as for the EMG study. The first slat was then removed and the participant was asked to abduct their arm until it was touching the second slat, ensuring that each participant abducted their arm by 5 cm under the tension of the TheraBand thus allowing us to control the force provided under each deltoid activation between participants (see Figure 8).
FIGURE 8.
Images of MRI coil placement and TheraBand positioning. (a) Arm in neutral position against L-shaped device. (b) Slat removed and arm abducted by 5 cm to outer slat. Source: photograph of staff member reproduced with permission from University Hospitals Coventry and Warwickshire.
While 5- and 10-cm abduction were tested for the EMG study, only 5-cm abduction was used, as practically a 10-cm movement was not found to be consistently possible due to the 70-cm bore of the scanner and the variance in body habitus of the participants being scanned.
Imaging methods
The participants were scanned on the Optima MR 750w 3T (GE Healthcare, Chicago, IL, USA) MRI scanner using the 16-channel GEM Flex coil, allowing for easy positioning of the participants, maximum visualisation of the rotator cuff, and easier abduction of the arm away from the body. Participants were asked to lie on the magnetic resonance table with the arm abducted in a neutral position. Pads were placed under the opposite shoulder so the shoulder that was being scanned was comfortable on the coil and to reduce movement. To further minimise any movements during the scan, the coil was firmly secured. A loop of light tension (green) TheraBand was applied around the participants just above the elbow, so it was not stretched and did not have any slack as in the EMG study.
Participants were positioned using the device to achieve 5-cm abduction. A standard three-plane localiser was acquired to plan the coronal oblique proton density fat-suppressed images. The participant was then asked to release the TheraBand while keeping their arm in the same position. The coronal oblique proton density fat-suppressed images were then repeated. Each sequence had an acquisition time of 2 minutes and 22 seconds.
Measurements of humeral head migration were made using the internal measurement system of the picture archiving and communication storage system. Measurements were made in the same anatomic location for the strained and relaxed state (i.e. not against resistance but in the same position) scans. The measurement position chosen was the anterior margin of the acromion at the joint margin and perpendicular to the humeral surface.
Basic statistical analysis and production of graphs and tables were done using Microsoft Excel® (Microsoft Corporation, Redmond, WA, USA). Owing to the low numbers of participants and the exploratory nature of the work, it was not deemed necessary to determine significance scores.
Methods for magnetic resonance imaging substudy
All participants from across both treatment groups were invited to undergo two research MRI scans: one at 8 weeks post intervention and a further one after 6 months.
MRI scans were preferred to X-ray, as measures taken from X-rays would be prone to error, primarily due to variation in the angle between the shoulder joint and the beam of the X-ray. The proposed mechanism of the balloon was discussed with its inventor (Dr Assaf Dekel), who explained that it was not designed to depress the humeral head passively but to cushion the humeral head from impinging on the acromion during activity. We therefore decided that passive imaging alone was likely to be inadequate to demonstrate the function of the balloon and imaging also needed to be performed when the deltoid muscle was active, producing a proximally directed force on the humerus. 10,11
As described, we developed and piloted a novel dynamic approach to assessing the function of the rotator cuff using MRI under a mild deltoid load specifically to assess the mechanism of the balloon. We followed the methods for positioning and scanning participants described previously. We used conventional MRI with images in the oblique coronal planes as the preferred technique for imaging the rotator cuff. Evidence shows that fat-suppressed, fast spin-echo, T2-weighted images are the most accurate for the assessment of rotator cuff tears and a variation of this sequence was used and applied on the range of MRI machines across the different sites in this substudy. 116,117
Staff carrying out the scans and trial staff assessing images were blinded to participant allocation.
Outcomes
The primary outcome was the minimum AHD, on the ‘deltoid-active’ coronal sequences at 6 months, a reliable and proven measure. 118–120 Secondary measures were AHD on passive and sagittal images and the change in AHD between active and passive images. The position of the balloon was assessed on both sequences (with particular focus on the sagittal images) to check for migration and consistency of placement relative to the acromion.
Power and sample size
Based on Gumina’s120 study, the minimum AHD has SD of 1.72 mm, so to observe a minimum important difference of 1.5 mm (above the minimum detectable change of 1.3 mm established elsewhere121 with an alpha of 0.05 at 80% power, assuming a 20% loss to follow-up at 6 months, 56 participants were required for this substudy. It was estimated that five centres would be involved in recruiting participants for this substudy based on an assumption that only 50% of participants would agree to participate.
Analyses
The primary end point was between-group difference on the 6-month MRI, as that is the better indicator of long-term function determining whether the early effect of the balloon was maintained. The between-group differences on the 8-week scan were a secondary outcome. The primary analysis compared the ‘deltoid-active’ AHD on coronal images between intervention and control groups using a linear regression model adjusting for age, sex, recruitment site and tear size. The research sites standard MRI exclusion criteria was applied and a MRI safety questionnaire was administered prior to inclusion as is standard for MRI at each site.
In light of the COVID-19 pandemic and reduced safety and capacity at sites, some 6-month MRI scans could not be completed on time. It was anticipated that the shoulder condition or the underlying biomechanics would not change after the 6-month point and would be stable for a prolonged time. Therefore, the TSC decided (6 July 2020) to extend the 6-month MRI window with no end point, to allow for the delayed scans to be completed and used in the analysis.
Results
The development study
A total of 11 participants attended for EMG testing while a further 4 attended for an MRI scan (see Table 7). The median age was 64 years (range 53–73 years) for the EMG group and 63 years (range 49–72 years) for the MRI group. The median duration of symptoms was 9 years (range 6–168 years) and 24 months (range 11–36 months), respectively.
| Characteristics | EMG substudy (n = 11) |
MRI substudy (n = 4) |
||
|---|---|---|---|---|
| Demographics | ||||
| Male | Female | Male | Female | |
| Sex | 5 (45%) | 6 (56%) | 3 (75%) | 1 (25%) |
| Mean age (years) | 63.5 | 61.5 | ||
| Details of injury | ||||
| Right | Left | Right | Left | |
| Side of tear | 7 (64%) | 4 (36%) | 3 (75%) | 1 (25%) |
| Trauma | 7 (64%) | 3 (75%) | ||
| Median duration of symptoms (months) | 9 | 24 | ||
| Comorbidities | ||||
| Type 1 | Type 2 | Type 1 | Type 2 | |
| Diabetic | 0 (0%) | 1 (9%) | 0 (0%) | 0 (0%) |
| Yes | No | Yes | No | |
| Current smoker | 1 (9%) | 10 (91%) | 0 (0%) | 0 (0%) |
| Previous dislocation | 0 (0%) | 1 (25%) | ||
| Previous rotator cuff tear | 3 (27%) | 1 (25%) | ||
| Details of investigations | ||||
| MRI | 5 (46%) | 1 (25%) | ||
| Ultrasound scan | 11 (100%) | 3 (75%) | ||
| X-ray | 9 (81%) | 2 (50%) | ||
| Details of treatment | ||||
| Steroid injection | 8 (73%) | 3 (75%) | ||
| Physiotherapy | 7 (64%) | 3 (75%) | ||
In the EMG group, of 11 participants, 7 (64%) had suffered trauma to the shoulder, 8 (73%) had received a steroid injection prior to commencing the study and 7 (64%) had received physiotherapy. In the MRI group three of four participants (75%) had suffered trauma, received steroid injections and physiotherapy.
Electromyography results
Participants in this group were divided into two groups, those undertaking the tasks to 10 cm of shoulder abduction and those to 5 cm. This allowed us to compare the activation of the deltoid when moving the arm to two different distances under the same tensions as provided by the exercise bands.
Table 8 shows the mean activation scores. The highest activation score (94.3) was achieved with a black band at 5 cm of movement. While there were differences in activation between the 5- and 10-cm movements for the lower resistance (yellow) band, high levels of activation were seen for the higher resistance bands (green and black) for both movements. As a 5-cm movement was more practical within the confines of the MRI scanner, the lowest resistance required to achieve good activation was preferred to prevent participant discomfort and fatigue during the 2 minutes 22 seconds acquisition time when a scan would be performed. This was achieved with the green TheraBand.
| 10-cm abduction | 5-cm abduction | Difference | ||
|---|---|---|---|---|
| Task | 10 cm | Task | 5 cm | |
| Relax | 1.7 | Relax | 2.3 | −0.6 |
| Yellow | 86.7 | Yellow | 78.0 | 8.7 |
| Green | 90.3 | Green | 89.2 | 1.1 |
| Black | 92.1 | Black | 94.3 | −2.2 |
| Mean | 1.8 | |||
| SD | 4.8 | |||
Magnetic resonance imaging development results
All participants were able to undertake the 5-cm abduction for the requisite period. One participant reported an ‘ache’ that resolved after imaging without the need for pain relief.
These measurements consistently show a small migration of the humeral head towards the acromion on the images under load (see Table 9). A mean percentage difference of 7.8% was found between the resting position of the humeral head and under deltoid activation. The greatest humeral head position difference was 12.4% (0.73 mm/6.63 mm) from the resting position when under load, and the smallest was 5.7 (0.37 mm/6.84 mm).
| Participant | Humeral head migration measured (mm) | Difference (%) | ||
|---|---|---|---|---|
| Under strain | Relaxed | Difference | ||
| 1 | 7.47 | 7.92 | 0.45 | 6.0 |
| 2 | 6.47 | 6.84 | 0.37 | 5.7 |
| 3 | 5.91 | 6.64 | 0.73 | 12.4 |
| 4 | 4.13 | 4.43 | 0.30 | 7.3 |
Magnetic resonance imaging substudy main results
Recruitment was lower than anticipated, with a total of 24 participants recruited to the substudy before randomisation was stopped. This was partly due to the early adaptive stop from the main trial, and partly due to restrictions related to COVID-19. A summary of these participants can be found in Appendix 3. Furthermore, this substudy was greatly affected by the response to the COVID-19 pandemic, as participants could not attend scans during lockdown. Hence, only 17 (71%) had images that could be analysed. Table 10 shows the values of AHD for each scan type at the two follow-up point by allocation groups. A further analysis investigating the change on the AHD between active and passive scans can be found in Appendix 3.
| Follow-up point | Image type | Overall (n = 24) |
Debridement (n = 11) |
Debridement with InSpace balloon (n = 13) |
Mean difference and 95% CI | p-value | |||
|---|---|---|---|---|---|---|---|---|---|
| Imagesb | Mean (SD) | Imagesb | Mean (SD) | Imagesb | Mean (SD) | ||||
| Early (8 weeks) | Passive | 16 | 5.4 (3.4) | 5 | 8.1 (4.8) | 11 | 4.2 (1.5) | −3.9 (−9.8 to 2.0) | 0.141 |
| Active | 16 | 5.8 (3.8) | 5 | 8.6 (5.5) | 11 | 4.5 (1.7) | −4.2 (−11.0 to 2.6) | 0.168 | |
| Late (> 6 months) | Passive | 17 | 4.4 (2.7) | 7 | 4.9 (3.8) | 10 | 4.1 (1.7) | −0.9 (−4.4 to 2.7) | 0.592 |
| Activea | 17 | 4.6 (2.2) | 7 | 4.7 (2.9) | 10 | 4.5 (1.6) | −0.1 (−2.9 to 2.7) | 0.923 | |
AHD reduced under active loading in all study groups, in a way that was consistent with the findings from the development work. Participants who underwent debridement-only and those who underwent debridement with the InSpace device had comparable findings on both the passive and active images, and the relative differences between passive and active scans were consistent throughout. This relationship was repeated across the time points and scan types.
Discussion
The development study
The biomechanical action of the deltoid muscle causing proximal humeral migration in the absence of a functional rotator cuff is well understood in the literature. However, this action is hard to measure and define in real patients in a consistent and measurable manner. Our first aim was to produce a reproducible technique for deltoid muscle activation that could be performed within an MRI scanner and used to measure proximal humeral migration.
We found several limitations for movement within an MRI scanner, namely the size of the scanner (bore/diameter) and the ability of the participants to hold the movement under load for up to 3 minutes at a time. If the technique produced called for large movements, these would not be possible within the confined space.
Development of the technique involved us testing shoulder abduction under various tensions at two different distances while simultaneously measuring activation. Our initial thoughts were that deltoid muscle activity would be positively associated with both the distance and the force applied. While this held true for the large part, the differences in the actual measurements achieved in activating the deltoid using the same band at either 5 cm or 10 cm were minimal, except for the lowest tension band.
It became apparent when designing the task that a movement of 10 cm measured from the elbow to abduct the shoulder would not be achievable for larger participants within the confines of the MRI scanner. This combined with the minimal difference in activation scores between 5 cm and 10 cm made us implement the shorter distance for the technique in the MRI study. The scanner used in this study was a wide-bore scanner (70 cm) compared with a standard-bore scanner (55–60 cm). It was hoped that the 5-cm abduction technique would be possible on the majority of participants in standard-bore MRI scanners.
The ability of participants to hold their arm abducted under tension for approximately two and a half minutes during a MRI was also of concern. As tension increased during the EMG study the difficulty of movement became apparent, with participants fatiguing after holding the band for only a few seconds as well as moving their arm, contributing to a large number of the outliers we observed. This was particularly noticed with the black band and so this meant that we could not use this band for the final technique. This resulted in our final recommendation for using the green band at a distance of 5 cm for the activation of the deltoid muscle in a consistent and controllable manner within a MRI scanner.
One MRI participant found it difficult to perform and maintain 5 cm abduction for the allotted time, which resulted in movement artefacts on the images. The technique’s aim was to achieve a reproducible abduction distance, so if 5 cm abduction cannot be maintained, the distance of abduction should be reduced by centimetre decrements until a comfortable abduction position is achieved. The participant’s abduction distance can be noted and reproduced for subsequent visits.
Though surface EMG techniques are less accurate than conventional EMGs, it would have been inappropriate to use an invasive technique for research purposes in this setting. Other inaccuracies as a result of inconsistencies in the positioning of the probes may have contributed to some measurement error in our study. Despite using set anatomic landmarks for the positioning of the probes,19 those with a small deltoid muscle bulk may have had interference from other muscles. We did not account for repeat movements as well, and the impact that muscle fatigue, particularly in those with pre-existing shoulder injury may have on the signals obtained. We also did not control for pain and the impact this would have on the movement profile for different patients.
A small amount of humeral head migration was consistently observed while the deltoid was activated. While the true values may seem small, they are relative to a small space between the acromion and the humerus. In addition, the pilot work was performed in a group of participants who were awaiting rotator cuff repair whose rotator biomechanics may be better preserved than those with irreparable tears, who tend to have larger tears and more chronic pathology. People were recruited for these pilot studies from rotator cuff repair waiting lists so as to not impinge on recruitment to the main trial, but participants in the main study may have much greater migration under deltoid load.
To make the imaging task manageable for participants and to avoid fatigue, it was decided to restrict the number of shoulder sequences under load and use a relatively quick sequence. The use of a plastic former (constructed in house at University Hospitals Coventry and Warwickshire radiotherapy department) meant that it was easy to reproduce the position. The use of the green band gives a constant load that could be maintained by most participants for the duration of the scan. A single series of coronal oblique images was acquired while under load and then a repeat of the same sequence, while relaxed, were obtained for consistency and a consistent, reproducible measurement protocol was developed.
Abduction against and exercise band to a distance of 5 cm can be an effective way of activating the deltoid muscle, that can be applied in an MRI scanner. It can be performed safely and was successful in causing humeral head migration in our early pilot study.
Discussion of magnetic resonance imaging substudy findings
Recruitment to the MRI substudy from the main randomised trial was severely impacted by the COVID-19 pandemic, but also by the early stop in recruitment. When embedding a substudy into an adaptive randomised trial such as this, it would be best to front-load recruitment to the substudy as much as possible. We were able to achieve this in the lead site but many of our early sites were unable to participate in the substudy and it can be challenging to identify sites willing to undertake relatively time-intensive substudies. Despite this, a number of sites were open and recruiting to the substudy by the time of the pandemic, 44 participants had consented to the substudy and we were exploring imaging options at other sites that would have allowed us to resolve these issues. Unfortunately, the COVID-19 pandemic meant that people were unable attend hospitals for research visits and we were unable to increase the sample for this study.
Despite the difficulties in delivering the study during these challenging times, we found that the MRI technique did demonstrate narrowing of the acromiohumeral space under deltoid load, confirming our biomechanical theory that deltoid activation would act to draw the humerus proximally in the absence of a functioning rotator cuff. As with the development study, although these differences were small, they were consistently observed and are relative to an already narrow space.
With this small sample size, we did not observe a statistical difference between the randomised intervention groups. Given the clinical findings of the study, we might not expect a difference in MRI appearances. We did not identify any reason for the debridement performing better in this MRI study, and the reason for the clinical findings presented in Chapter 3 remains uncertain. Stopping early for futility also meant that the rationale for continuing to pursue an associated mechanistic study also changed. In this setting, we can be reassured that we did not see any clear evidence of harm on the images for either group.
Conclusion
In conclusion, we developed and refined a new dynamic MRI testing protocol and then applied it in a mechanistic substudy to test the effect of the InSpace subacromial balloon spacer on humeral head migration under deltoid load. The substudy recruitment was severely limited by both the early stop and by the coronavirus pandemic, but while the MRI technique achieved its technical aims, no evidence was seen of a between-group difference.
Chapter 7 Adaptive designs for surgical trials
Introduction
Historically, new surgical procedures have been introduced based solely on safety considerations and what a surgeon believes might benefit patients. This perceived lack of rigour and inefficiency has motivated the development of many new processes and methodologies,122–124 and a steady increase in the number of RCTs testing surgical interventions, especially over the last 10 years.
Many of the late-stage clinical trials testing surgical interventions are large, often because they use patient-reported outcome measures, typically take many (e.g. more than five) years to complete and are consequently expensive. This is particular the case with trials in the trauma and orthopaedics specialty, where interventions often require extended periods of participant follow-up to assess effectiveness. In such settings, Parsons et al. 61 suggested that novel adaptive design methods might be a means to undertake clinical trials in a much more flexible and efficient manner, while retaining trial integrity.
The approach proposed by the authors exploited the fact that it is very common in surgical trials to routinely monitor participants at a number of fixed occasions prior to collecting the definitive (final) study outcome (e.g. early outcomes might be collected at 3 and 6 months, prior to the main 12-month time point). In such settings, if an interim analysis uses information from only those participants with final outcome data, then the opportunities for early stopping are likely to be limited by time constraints, as often trial recruitment will have completed prior to sufficient final outcome data being available for stopping decisions to be made. However, if the early outcomes are correlated with the final outcome, then a group sequential analysis,125 which uses the totality of information available from both early and final outcomes to estimate the treatment effect at the final study end point, is likely to make adaptive designs feasible and lead to increases in statistical power. 58,126,127
The work described here uses a frequentist approach to the group sequential design defined by the error spent at each look with predefined information levels. 125,128,129 Bayesian methods are also widely available for adaptive group sequential designs,130 and have previously been suggested for trials in trauma and orthopaedics and emergency medicine, albeit in very different applications to those presented here. 131,132
The adaptive study design approach of Parsons et al. 61 was described in the context of START:REACTS. 62 Part of this study investigated how the adaptive design methods used in the START:REACTS study might have been implemented and whether they would have resulted in changes in trial length and decision-making in a number of recently undertaken high-profile conventional (fixed design) trials in trauma and orthopaedics. This work is reported here.
The work uses anonymised data from seven RCTs made available by Warwick Clinical Trials Unit [Warwick Medical School (WMS) https://warwick.ac.uk/fac/sci/med/research/ctu and the Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, University of Oxford; www.ndorms.ox.ac.uk]: (1) the WOLLF study;49,133 (2) the Distal Radius Acute Fracture Fixation Trial (DRAFFT);47,134 (3) the FixDT trial;50,135 (4) the UK Full RCT of Arthroscopic Surgery for Hip Impingement versus Best Conventional care study (FASHIoN);46,132 (5) the Warwick Arthroplasty Trial (WAT);46,136 (6) the Can Shoulder Arthroscopy Work (CSAW) trial;69,137 and (7) the Total or Partial Knee Arthroplasty Trial (TOPKAT). 138–140
The studies were selected because data were readily available, recruitment processes and outcomes were expected to be such that the methods used for the START:REACTS study might be applicable, and they represent a typical cross-section of recent RCTs in trauma and orthopaedics in terms of size and complexity.
Methods
This study assessed whether each of a selected number of large (pragmatic) RCTs, originally implemented using conventional fixed sample size designs, would have stopped early if an adaptive (group sequential) trial design had been used. For the purposes of this study, all the selected trials had two treatment arms (with one nominally designated as the control or standard treatment), randomised participants to treatment groups in a one to one ratio and reported a single primary outcome, with one or more assessments of the primary trial outcome measure (e.g. outcomes at 3 and 6 months or 1, 2, 3, 4 and 5 years).
To assess whether the trial would have stopped early, the temporal sequence of data accumulation was replicated in exactly the manner it was in the original study using the dates (which were expected to be available in the original trial databases) when each outcome measure was made. We call these repeated trials simulated trials hereafter, as they aimed to simulate the original fixed sample size trial with an adaptive version. However, to be clear we used the original trial data and pattern of data accrual, and not simulation models based on distributional assumptions. Using the original trial data and selected options for the number of planned interim analyses and stopping boundaries, we simulated how each study might have progressed using the methodological approach described by Parsons et al. 61 for an adaptive two-arm clinical trial using early end points to inform decision-making.
To simulate a single instance of a trial we implemented the full simulation model, shown in Figure 9. We decided on the number of interim analyses we wished to make, stopping probabilities and information levels necessary to trigger the interim analyses. These were used to determine upper and lower stopping boundaries for the test statistics. Data from the original trial were used to simulate information accumulation in the new adaptive design, using the observed ordering of data accrual from the original trial. Test statistics were compared with boundaries, with decisions on stopping following from this process. Clearly, by sticking with the original (fixed design) trial sample size our simulated adaptive trials will have lower power, relative to the original trial. In practice, if the selected trials had planned to use adaptive designs prospectively, we would have increased the sample size relative to the fixed design sample size to allow for any interim analyses.
FIGURE 9.
Overview of the process for simulating progress in an adaptive trial using data from a conventional (fixed design) trial.

Group sequential design
The primary interest of all the RCTs discussed in this substudy was to estimate the effect of the test treatment, compared to the control treatment, on the study outcome at the definitive (final) end point at time t (the primary study end point), which we call βt hereafter. For all studies, we used the same primary or final end point as was originally used.
To implement the approach suggested by Parsons et al. ,61 we needed a single early outcome or a series of early outcomes in addition to the final outcome for each participant in the trial (e.g. primary outcome at 6 months with early outcome at 3 months or primary outcome at 9 months with early outcomes at 3 and 6 months). To assess whether a trial should be stopped at a particular time point during the course of follow-up, we used data not only from those participants with final outcome data but also from those participants with early outcome data.
The early outcomes provide information on the final outcome due to the correlation between the early and the final outcomes for each participant. A strong correlation between, for instance, 3- and 6-month outcomes suggests that a good (or poor) outcome at three months will be indicative of a good (or poor) outcome at 6 months. Independent of these correlations between successive outcomes for study participants, treatment effects for the early outcomes per se do not provide information on treatment effects for the final trial outcome βt. This approach has been described previously by a number of authors for one, two and more generally any number of early outcomes. 58,126,127,141 The notation used here for the effect size estimate (βt) reflects the fact that estimation follows from fitting a longitudinal linear model to the totality of outcome data (i.e. data from all trial time points). Methods for estimating (βt) and var(βt), using all the data available at any time point during follow-up, are described by Parsons et al. 61
The test statistic Z = βt/sd(βt) was used to make stopping decisions at the interim analyses using estimates of the covariance parameters (i.e. the correlations between outcomes and SDs of the outcomes) without constraints (i.e. the covariances parameters can take any value). In addition, as a means to assess the importance of the early outcome data in modifying estimates of the βt and var(βt), an analysis that forces all the correlations to be zero was also undertaken. We designate these parameters, which show the evidence for treatment effects using final outcome data only, as β0t and var (β0t), with Z = β0t/sd(β0t).
Stopping rules
Timing of interim analysis
The number of feasible interim analyses was determined, in large part, by the patterns of recruitment and data accrual for each RCT. Interim analyses needed to occur during the window of opportunity bookended at the start by the earliest time sufficient data were available for a sensible analysis to occur and at the end by the time when recruitment was complete. After the latter time point, there is no advantage to stopping a study, as there is a requirement to complete follow-up for all participants recruited into the trial. The number of possible interim analyses for each RCT was determined, before simulating data accumulation for the adaptive design, by a consideration of the likely width of the window of opportunity, which is itself determined by the likely pattern of recruitment and follow-up.
At all times, we endeavoured to use only the information that would have been available to those designing the trials at the initial stages when decisions about the likely number of analyses would need to have had to be made. The lead statistician from all the selected trials was consulted on these issues and the knowledge gained from them and from the published protocols for all the trials was used to inform the designs described in the following sections.
The timing of the interim analyses could, in principle, be triggered in a number of ways (e.g. when data were available from a preset number of participants who had completed follow-up in each arm of the RCT). Here, we adopted the most widely used approach, that is to monitor information as the trial progresses and implement the interim analyses when preset information thresholds (I1,I2, … Iw) were reached. 125 The information was determined by the number of outcomes available for both the primary and early end points, and the variances and correlations between end points. 61 In general, the larger the number of data points available for analysis, the smaller the variances, and the stronger the correlations between early end points and the primary end point, the greater the information. Although, in practice as information is monitored during the course of recruitment, it can decrease as well as increase as the amount of data increases, as estimates of variances and correlations will also change as data are accumulated.
Using the approach adopted by Parsons et al. ,61 we set information thresholds to trigger the interim analyses, at the start of recruitment, at planned occasions based on expected rates of recruitment and using a priori estimates of the covariances of the outcomes. Clearly, in a real trial, we would not know the true values of the covariances, as we do in this setting. However, we would need to have sensible (reasonably precise) estimates of the covariances otherwise it is unrealistic to believe that an adaptive trial would ever have been planned. Information is monitored constantly during simulated recruitment in the adaptive design. Therefore, if correlations are lower than expected, interim analyses will be later than planned (or abandoned) and if higher than expected, interim analyses will be earlier than planned.
Boundaries
Given the practical constraints imposed by the need for interim analyses to take place during the window of opportunity (after primary outcome data become available and before recruitment finishes), we restricted this study to a maximum of three interim analyses within any trial. The primary focus of this study was to assess whether and under what circumstances an adaptive design may have resulted in the selected trials stopping early. The selected trials are all pragmatic in outlook, testing interventions that are often already being widely used and, as such, the boundaries we choose needed to reflect the fact that stopping is most likely to be for treatment futility rather than efficacy. In many such pragmatic trials, one might argue that settings for the upper (efficacy) boundaries should favour the strategy of collecting as much information as possible if there is emerging evidence of efficacy; that is, not stopping early for efficacy unless there is very strong evidence.
Parsons et al. 61 suggested a range of possible futility boundaries defined by stopping probabilities in the setting of up to three interim analyses, that represented a sequence of four increasingly aggressive options, from a low probability of stopping for futility, labelled as (a), to a high probability, labelled as (d), with (b) and (c) intermediate to these (see Table 8).
In Table 11, at overall one-sided test level α and under the null hypothesis, α*u are the probabilities of stopping and rejecting the null hypothesis (H0) in favour of alternative (efficacy), and α*l are the probabilities of stopping without rejecting H0 (futility). All the futility scenarios (a) to (d) in Table 11 were tested for every trial, but for any trial stopping boundaries were defined by one or more of α*l and α*u.
| Interim analyses | α*l | α*u | |||
|---|---|---|---|---|---|
| (a) | (b) | (c) | (d) | ||
| (i) One | |||||
| 1 | 0.160 | 0.320 | 0.480 | 0.640 | 0.005 |
| End | 0.975 | 0.975 | 0.975 | 0.975 | 0.025 |
| (ii) Two | |||||
| 1 | 0.080 | 0.160 | 0.240 | 0.320 | 0.001 |
| 2 | 0.160 | 0.320 | 0.480 | 0.640 | 0.010 |
| End | 0.975 | 0.975 | 0.975 | 0.975 | 0.025 |
| (iii) Three | |||||
| 1 | 0.080 | 0.160 | 0.240 | 0.320 | 0.001 |
| 2 | 0.160 | 0.320 | 0.480 | 0.640 | 0.005 |
| 3 | 0.240 | 0.480 | 0.720 | 0.960 | 0.010 |
| End | 0.975 | 0.975 | 0.975 | 0.975 | 0.025 |
The stopping probabilities from Table 11 were used to construct appropriate boundaries for standardised test statistics at each of the planned interim analyses for each trial. This required us to make some assumptions, based on what we believed the trial team may have thought prior to the commencement of recruitment, about (1) the number of possible interim analyses, (2) the covariances between the primary end points and (3) the number of data points that may have been available at each of the interim analyses. Using these data, we calculated the information necessary to trigger each interim analysis, which allowed us to define stopping boundaries for the observed test statistics.
Information monitoring
Data were generated using the observed trial outcome data and the observed recruitment and follow-up patterns, such that they represented the order that data would have accumulated in real time (i.e. the outcome data in the order they accumulated in the original trial). Information monitoring begins, after sufficient data are available to estimate accumulated information, and continues on a regular basis (every 2 weeks) to reflect what would likely have happened in the trial, if an adaptive design had been implemented. Once the required information level is reached, the test statistic is calculated and compared to the stopping boundaries, and decisions about whether the trial would have stopped or continued to the next interim analysis are recorded together with summary information on the current recruitment, time point, and outcome data summaries. This process is continued for subsequent interim analyses if necessary.
In summary, the simulated group sequential design for a trial proceeds as follows:
-
Trial data are accumulated, in the order they were in the original study, and the observed information is estimated as I* = 1/var(βt).
-
When I* reaches the first preset threshold I1, parameters βt1 and var(βt1) are estimated to give the test statistic Z1 = βt1/sd(βt1).
-
Data accumulation continues until I* reaches the next and subsequent information thresholds and test statistics Zw calculated.
-
At the study end the observed information I* is noted and data recorded.
Decisions about whether the simulated trial would have stopped, either for efficacy or futility, were made by comparing the estimated test statistics at each interim analysis to the stopping boundaries for the four scenarios (a) to (d). Also, at each interim analysis data on all those study participants recruited up to that point were used to estimated model parameters for the over-running analysis. 80,142 This analysis comprised all the data that would eventually have accumulated on those participants already recruited and is the definitive analysis that would have been reported, had the trial stopped recruiting at the interim analysis. In the over-running analysis, boundaries were recalculated based on the observed information at the final analysis and used to draw interference on the observed treatments effects.
Trial data
Each of the selected seven RCTs are briefly described in Appendix 4 With full details available from the published protocols and papers describing the main clinical results.
Group sequential designs
WOLLF
The study recorded Disability Rating Index (DRI) at 3, 6, 9 and 12 months, with the primary outcome at 12 months. From the WOLLF study protocol,49 the planned sample size was n = 412, based on a SD σ12m = 25 and MCID = 8 for DRI; allowing 10% loss to follow-up, gave n = 460.
If recruitment had proceeded to target as originally planned, and assuming that n012m = n112m = 206, then the information at the study end would be given by IEnd = 103/252 = 0.165. Inspection of the planned recruitment schedules for the trial suggested that three interim analyses would have been feasible for WOLLF. For the purposes of planning the interim analyses, it was assumed that the numbers in the intervention arms are always equal (i.e. n0t = n1t = nt, where the total number of participants providing data at time t is Nt = 2nt), and that interim analyses could have feasibly occurred when n12m = 25, n12m = 50 and n12m = 75. The estimated numbers of early outcomes at these three interim analyses could have been as follows, based on the patterns of follow-up that might have been expected before recruitment started: n3m = 60, 120, 180 and n6m = 50, 100, 150 and n9m = 35, 70, 105 and n12m = 25, 50, 75.
Using the approach of Parsons et al. ,61 we further assumed that the SDs were the same for the early outcomes as for the final outcome σ3m = σ6m = σ9m = σ3m = 25, and that the correlations between outcomes were all equal such that ρ3m,6m = ρ3m,9m = ρ3m,12m = ρ6m,9m = ρ6m,12m = ρ9m,12m = 0.5. In this instance, the information required at the three interim analyses were as follows, I1 = 0.025, I2 = 0.05 and I3 = 0.075. Using the four options for futility (cumulative) stopping probabilities from Table 15, (a) α*l = (0.080, 0.160, 0.240, 0.975), (b) α*l = (0.160, 0.320, 0.480, 0.975), (c) α*l = (0.240, 0.480, 0.720 0.975) and (d) α*l = (0.320, 0.640, 0.960, 0.975) and efficacy stopping probabilities (for all options) α*u = (0.001, 0.005, 0.010, 0.025), gave the lower and upper stopping boundaries for the test statistic (Z) at each interim analysis, which are shown in Appendix 4. These stopping boundaries were used to simulate the progress of the trial and assess whether the study would have stopped at each of the interim analyses.
DRAFFT
The study recorded early patient-rated wrist evaluation (PRWE) outcome at 3 and 6 months, with the primary outcome at 12 months. From the DRAFFT study,134 planned sample size n = 350, based on SD σ12m = 20 and MCID = 6 for PRWE. Allowing 10% loss to follow-up, this gave n = 390. As a result of faster than expected recruitment to the trial, and with the permission of the review board, the sample size was increased resulting in n = 461 participants. Recruitment to the trial was expected to be rapid, as this is a common injury (particularly in the winter months), therefore the window of opportunity for interim analyses would have been narrow, as recruitment could feasibly have been completed before any 12 months outcome data were available. For this reason, it seems likely that one interim analysis would have been the most likely option to select at planning.
If recruitment had proceeded to target as originally planned, and we assume that n012m = n112m = 175 then the information at the study end would be given by IEnd= 87.5/202 = 0.219.
Inspection of the planned recruitment schedules for the trial suggested that only one interim analysis would have been feasible for DRAFFT. Again, it was presumed, for the purposes of planning the interim analysis, that numbers in the intervention arms are always equal, and that the interim analysis could have feasibly occurred when n12m = 50. The estimated numbers of early outcomes at the interim analysis could have been as follows, based on the patterns of follow-up that might have been expected before recruitment started: n3m = 100 and n6m = 70 and n12m = 50.
Assuming that the SDs were the same for the early outcomes as for the final outcome σ3m = σ6m = 20, and that the correlations between outcomes were such that ρ3m,6m = ρ3m,12m = ρ6m,12m = 0.5, then the information required at the interim analysis was as follows, I1 = 0.073. Using the following four options for futility (cumulative) stopping probabilities, (a) α*l = (0.160, 0.975), (b) α*l = (0.320, 0.975), (c) α*l = (0.480, 0.975) and (d) α*l = (0.640, 0.975) and efficacy stopping probabilities (for all options) α*u = (0.005, 0.025), gave the lower and upper stopping boundaries for the test statistic (Z) at each interim analysis and the study end, which are shown in Appendix 4. These stopping boundaries were used to simulate the progress of the trial and assess whether the study would have stopped at each of the interim analyses.
FixDT
The study recorded an early DRI outcome at 3 months, with the primary outcome at 6 months. From the FixDT trial protocol,50 the planned sample size was n = 264, based on SD σ6m = 20 and MCID = 8 for DRI (DRI; 0–100). Allowing 20% loss to follow-up, gave n = 320.
If recruitment had proceeded to target as originally planned, and we assume that n06m = n16m = 132, then the information at the study end would be given by IEnd= 66/202 = 0.165. Inspection of the planned recruitment schedules for the trial suggested that two interim analyses would have been feasible for FixDT. For the purposes of planning the interim analyses, it was assumed that numbers in the intervention arms are always equal, and that analyses could have feasibly occurred when n6m = 25 and n6m = 50. The estimated numbers of early outcomes at the interim analyses could have been as follows, based on the patterns of follow-up that might have been expected before recruitment started: n3m = 50, 100 and n6m = 25, 50.
Assuming that the SDs were the same for the early outcomes as for the final outcome σ3m = σ6m = 20, and that the correlations between outcomes were such that ρ3m,6m = 0.5, then the information required at the two interim analyses were as follows, I1 = 0.036 and I2 = 0.071. Using the following four options for futility (cumulative) stopping probabilities, (a) α*l = (0.080, 0.160, 0.975), (b) α*l = (0.160, 0.320, 0.975), (c) α*l = (0.240, 0.480 0.975) and (d) α*l = (0.320, 0.640, 0.975) and efficacy stopping probabilities (for all options) α*u = (0.001, 0.010, 0.025), gave the lower and upper stopping boundaries for the test statistic (Z) at each interim analysis and the study end, which are shown in Appendix 4. These stopping boundaries were used to simulate the progress of the trial and assess whether the study would have stopped at each of the interim analyses.
FASHIoN
The FASHIoN trial recorded an early International Hip Outcome Tool (iHOT-33) outcome at 6 months, with the primary outcome at 12 months. From the FASHIoN trial protocol,143 the planned sample size was n = 292, based on SD σ12m = 16 and MCID = 6 for iHOT-33. Allowing 15% loss to follow-up gave n = 344. However, this plan was based on the very limited information that was available when the study started. iHOT-33 was a newly developed outcome measure, with little data available on its natural variation in the target population of the study, other than from pilot work and baseline data. 144 Therefore, at the design stage of the original study it was evident that the selected value for σ12m was likely to be an unreliable (imprecise) estimate. It quickly became clear as the study proceeded that σ12m = 16 was far too low, with emerging data suggesting that σ12m = 24 was more realistic. However, the study sample was not changed after recruitment started for FASHIoN, as the new working sample size model based on σ12m = 24 and MCID = 8, gave the same effect size estimate (0.38) and overall sample size as the original model. Given that the timing of the interim analyses in the adaptive design is driven in large part by σ12m, via accumulated information, we chose this latter formulation to motivate our design.
If recruitment had proceeded to target as originally planned, and we assume that n012m = n112m = 146, then the information at the study end would be given by IEnd= 73/242 = 0.127. Inspection of the planned recruitment schedules for the study suggested that two interim analyses would have been feasible for FASHIoN. It was assumed, for the purposes of planning the interim analyses, that numbers in the intervention arms are always equal, and that analyses could have feasibly occurred when n12m = 25 and n12m = 50. The estimated numbers of early outcomes at the interim analyses could have been as follows, based on the patterns of follow-up that might have been expected before recruitment started: n6m = 50, 100 and n12m = 25, 50.
Assuming that the SDs were the same for the early outcomes as for the final outcome σ6m = σ12m = 24, and that the correlations between outcomes were such that ρ6m,12m = 0.5, then the information required at the two interim analyses were as follows, I1 = 0.025 and I2 = 0.050. Using the following four options for futility (cumulative) stopping probabilities, (a) α*l = (0.080, 0.160, 0.975), (b) α*l = (0.160, 0.320, 0.975), (c) α*l = (0.240, 0.480 0.975) and (d) α*l = (0.320, 0.640, 0.975) and efficacy stopping probabilities (for all options) α*u = (0.001, 0.010, 0.025), gave the lower and upper stopping boundaries for the test statistic (Z) at each interim analysis and the study end, which are shown in Appendix 4. These stopping boundaries were used to simulate the progress of the trial and assess whether the study would have stopped at each of the interim analyses.
WAT
The trial reported early outcomes for Oxford Hip Score (OHS) at 6 weeks, 3 months and 6 months, with the primary outcome at 12 months. From the WAT study protocol, the planned sample size was n = 104 for 80% power, based on SD σ12m = 9 and MCID = 5 for OHS. 136 Allowing 10% loss to follow-up gave n = 120. For the selected study interventions, there is often a long wait from recruitment until the operation takes place. Therefore, outcomes during follow-up were also expected to often be later than would have been anticipated from the recruitment date alone. For instance, if the operation took place 2 months after recruitment, the 6-week, 3-month, 6-month and 12-month follow-ups, which were based on the operation date, were consequently delayed by 2 months. This results in a relatively narrow window of opportunity for interim analyses and, therefore, it seemed likely that one interim analysis would have been the most likely option to select at planning.
If recruitment had proceeded to target as originally planned, and we assume that n012m = n112m = 52, then the information at the trial end would be given by IEnd= 26/92 = 0.32. Inspection of the planned recruitment schedules for the study suggested that one interim analysis would have been feasible for WAT. Assume now, for the purposes of planning the interim analysis, that numbers in the intervention arms are always equal, and that the interim analysis could have feasibly occurred when n12m = 20. The estimated numbers of early outcomes at the interim analysis could have been as follows, based on the patterns of follow-up that might have been expected before recruitment started: n6w = 40 and n3m = 35 and n6m = 30 and n12m = 20.
Assuming that the SDs were the same for the early outcomes as for the final outcome σ6w = σ3m = σ6m = 9, and that the correlations between outcomes were such that ρ6w,3m = ρ6w,6m = ρ6w,12m = ρ3m,6m = ρ3m,12m = ρ6m,12m = 0.5, then the information required at the interim analysis was as follows, I1 = 0.150. Using the following four options for futility (cumulative) stopping probabilities, (a) α*l = (0.160, 0.975), (b) α*l = (0.320, 0.975), (c) α*l = (0.480, 0.975) and (d) α*l = (0.640, 0.975) and efficacy stopping probabilities (for all options) α*u = (0.005, 0.025), gave the lower and upper stopping boundaries for the test statistic (Z) at each interim analysis and the study end, which are shown in Appendix 4. These stopping boundaries were used to simulate the progress of the trial and assess whether the study would have stopped at each of the interim analyses.
CSAW
The study recorded OSS at 6 and 12 months, with the primary outcome at 6 months. From the CSAW study,137 planned sample size n = 85, based on SD σ6m = 9 and MCID = 4.5 for OSS, based on 90% power at the 5% level. Allowing for 15% loss to follow-up, the final study sample size was n = 100 in each of the three study intervention arms.
Unusually among the trauma and orthopaedics trials discussed here, the CSAW trial did not collect an early outcome prior to the definitive (primary) outcome at 6 months. For this reason, it was not possible to use the approach of Parsons et al. ,61 which required early outcome data to fully implement the methodological approach. However, in the case of no early outcome data the method of Parsons et al. 61 reduces to the conventional group sequential setting with stopping decisions based on the 6-month primary outcome data only. In this setting, the treatment effect estimate (βt) is given simply by the difference in 6-month OSS treatment group means and SD(βt) by the usual expression for the SE of the difference in means. Also, of note in this simpler setting is that test statistics Z and Z0 gave exactly equivalent results, as there is no early outcome data to inform decision-making.
If recruitment had proceeded to target as originally planned, and we assume that n06m = n16m = 85, then the information at the study trial end would be given by IEnd= 42.5/92 = 0.525. Inspection of the actual and planned recruitment schedules for the study suggested that two interim analyses would have been feasible for CSAW. Assume now, for the purposes of planning the interim analyses, that numbers in the intervention arms are always equal, and that the interim analyses could have feasibly occurred when n6m = 20 and n6m = 40.
Assuming a SD σ6m = 9, then the information required at the interim analyses were as follows, I1 = 0.123 and I2 = 0.247. Using the following four options for futility (cumulative) stopping probabilities, (a) α*l = (0.080, 0.160, 0.975), (b) α*l = (0.160, 0.320, 0.975), (c) α*l = (0.240, 0.480 0.975) and (d) α*l = (0.320, 0.640, 0.975) and efficacy stopping probabilities (for all options) α*u = (0.001, 0.010, 0.025), gave the lower and upper stopping boundaries for the test statistic (Z) at each interim analysis and the study end, which are shown in Appendix 4. These stopping boundaries were used to simulate the progress of the trial and assess whether the study would have stopped at each of the interim analyses.
TOPKAT
The study recorded early Oxford Knee Score (OKS) outcome at 1, 2, 3 and 4 years, with the primary outcome at 5 years. From the TOPKAT study,138 planned sample size n = 500, based on SD σ5y = 8 and MCID = 2 for OKS.
Recruitment to the trial was rapid (just over 3.5 years), relative to the definitive study end point at five years of follow-up. Therefore, the window of opportunity, between some five-year final outcome data being available and recruitment completion was non-existent. That is, no five-year outcome data were available, prior to recruitment completing and as such the methodology we are investigating here, assessing possible early stopping of the trial, could not have been used.
Results
WOLLF: simulated group sequential trial
Figure 10 shows the observed number of participants recruited and followed up at 3, 6, 9 and 12 months for WOLLF, the window of opportunity available for interim analyses and the times when the interim analyses would have occurred in the simulated adaptive design.
FIGURE 10.
Recruitment (―), follow-up (―) and numbered interim analyses (+) for WOLLF; window of opportunity for interim analyses is shaded.
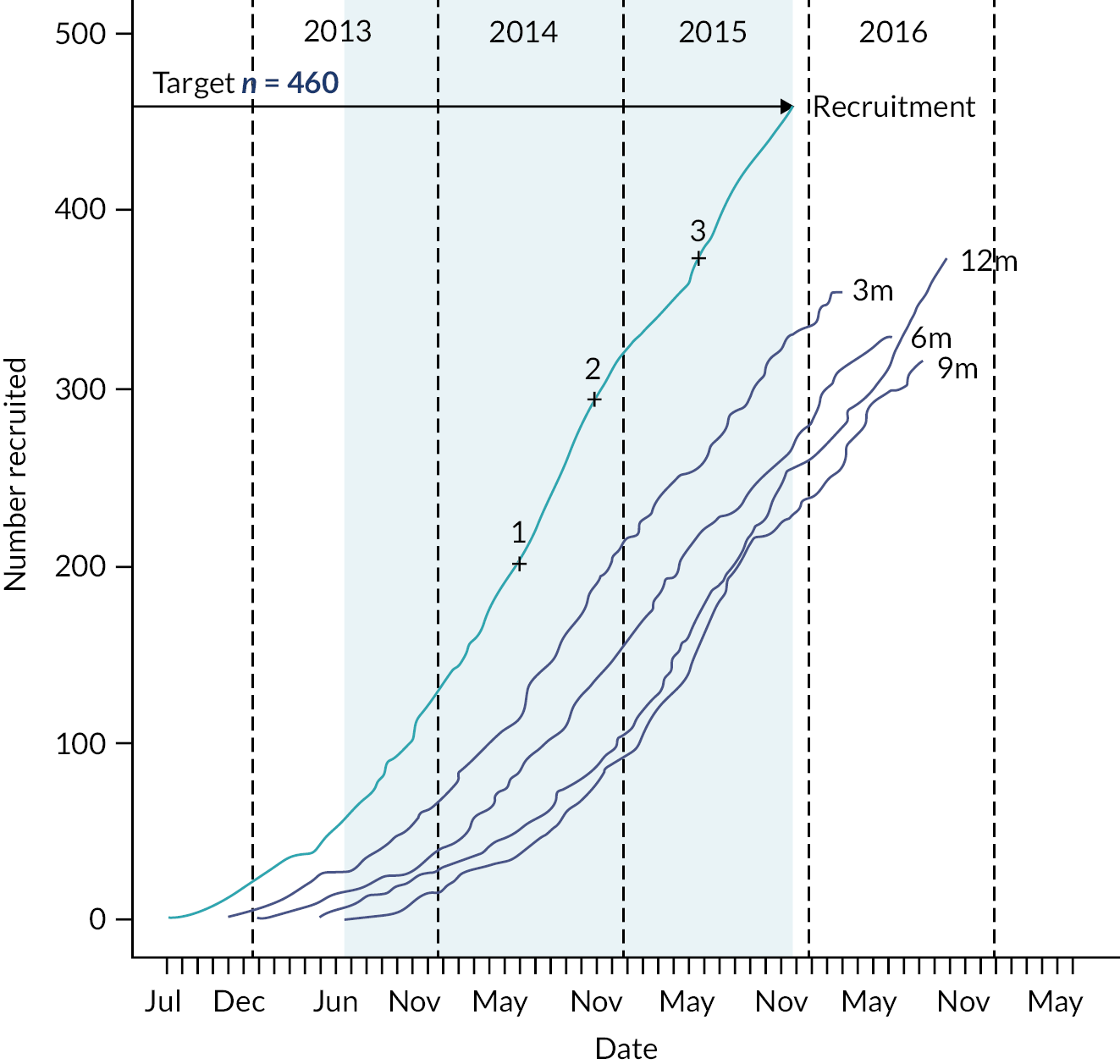
The trial hit the recruitment target of n = 460, and all the three planned interim analyses could feasibly have occurred. The numbers of observed trial participants providing data at each interim analysis is shown in Table 12. Estimated model parameters and test statistics are shown in Table 13.
| Analysis | Observed | Expected | ||||||
|---|---|---|---|---|---|---|---|---|
| N 3m | N 6m | N 9m | N 12m | N 3m | N 6m | N 9m | N 12m | |
| 1 | 115 | 84 | 51 | 37 | 120 | 100 | 70 | 50 |
| 2 | 188 | 136 | 85 | 74 | 240 | 200 | 140 | 100 |
| 3 | 255 | 217 | 173 | 156 | 360 | 300 | 210 | 150 |
| End | 354 | 329 | 314 | 374 | 412 | 412 | 412 | 412 |
| Analysis | m112m | m012m | Δ | βt | var(βt) | Z | I* | I | β0t | Var(β0t) | Z0 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 44.67 | 42.88 | −1.79 | −0.36 | 39.72 | −0.06 | 0.025 | 0.025 | −1.79 | 63.67 | −0.22 |
| 2 | 40.06 | 44.17 | 4.11 | −2.76 | 19.59 | −0.62 | 0.051 | 0.050 | 4.11 | 31.40 | 0.73 |
| 3 | 41.98 | 44.13 | 2.16 | 0.19 | 13.25 | 0.05 | 0.075 | 0.075 | 2.16 | 17.82 | 0.51 |
| End | 45.51 | 42.36 | −3.14 | −3.65 | 6.97 | −1.38 | 0.143 | 0.165 | −3.14 | 7.29 | −1.16 |
The estimated correlations and SDs at each interim analysis are shown in Appendix 4. The estimated correlations were all generally larger than those used in the model to determine the timings of the interim analyses (i.e. ρ = 0.5 for all pairs of times) and SDs were much as expected (i.e. σ = 25 for all times), at all interim analyses.
Figure 11a shows the stopping boundaries and Z and Z0 from Table 13, at each interim analysis and study end. Test statistic Z is in the continuation region (between upper and lower boundaries) at interim analysis 1, for all boundary settings a to d. However, at interim analysis 2, Z falls below the lower boundaries for settings b to d, indicating that the study would have been stopped for futility. Z does not fall below the lower boundary or above the upper boundary for setting a, so the study would not have stopped for this setting.
FIGURE 11.
Stopping boundaries and Z and Z0 for decision-making. (a) For stopping decisions using data available at each interim analysis. (b) Using over-running data.

The over-running analysis (see Figure 11b) confirms the results of the stopping decisions based on the interim data and indicate stopping for futility at settings b to d (see Appendix 4 for full results of the over-running analysis).
Estimates of the treatment effects βt, from the longitudinal model, are much closer to raw differences in means (Δ) for the over-running analysis as there is less information available from the early outcomes at this analysis. If there were complete follow-up data available for all participants, then (βt) would be equal Δ. However, in practice it is almost always the case that some participants who did not provide 12-month outcomes had early outcomes (at one or more than one of 3, 6 and 9 months), which provide some information on the 12-month outcomes. Estimates of treatment effects β0t, from the models that force correlations (between all outcomes) to be zero, are always equal to Δ and have larger variances than (βt).
WOLLF: summary
The results of the simulated group sequential design for WOLLF can be summarised as follows:
-
For three of the four boundary settings tested, the WOLLF study would have stopped at the second interim analysis, when data were available from n = 74 participants with 12-month outcomes, n = 85 with 9 months, n = 136 with 6 months and n = 188 with 3-month outcomes.
-
At this interim analysis, N = 293 participants had been recruited into the study and follow-up would have been completed in 27 months; this compares with N = 460 and 50 months for the original study.
-
The estimated treatment effect, for 12-month DRI, after completing follow-up for the stopped study (over-running analysis) was −3.5 favouring the control treatment; the estimate of the treatment effect in the original (fixed design) study was −3.1 (95% CI −8.5 to 2.2).
-
At the second interim analysis when the study was stopped, the estimated treatment effect from the model was −2.8 (favouring the control), and the raw difference between groups using 12-month data only was 4.1 [favouring negative pressure wound therapy (NPWT)].
-
There were extremely strong correlations between the early outcomes (3, 6 and 9 months) and the primary outcome (12 months) for DRI, meaning that at interim analysis inferences based on the modelling approach, that used all the data, gave much better estimates of the true end of trial treatment effect, than simple differences in primary outcome (12 months) group means.
-
The stronger than expected correlations also meant that the interim analyses generally occurred early than expected. That is, the observed number of participants providing 3-, 6-, 9- and 12-month outcome data at each interim analysis was less than the expected number.
DRAFFT: simulated group sequential trial
Figure 12 shows the observed number of participants recruited and followed up at 3, 6 and 12 months for DRAFFT, the window of opportunity available for interim analyses and the times when the interim analyses would have occurred in the simulated adaptive design.
FIGURE 12.
Recruitment (―), follow-up (―) and numbered interim analyses (+) for DRAFFT; window of opportunity for interim analyses is shaded.

The study surpassed the recruitment target of n = 390, and the planned interim analysis could feasibly have occurred. The numbers of study observed trial participants providing data at the interim analysis is shown in Table 14. Estimated model parameters and test statistics are shown in Table 15.
| Analysis | Observed | Expected | ||||
|---|---|---|---|---|---|---|
| N 3m | N 6m | N 12m | N 3m | N 6m | N 12m | |
| 1 | 205 | 135 | 26 | 200 | 140 | 100 |
| End | 423 | 414 | 415 | 350 | 350 | 350 |
| Analysis | m112m | m012m | Δ | βt | var(βt) | Z | I* | I | β0t | Var(β0t) | Z0 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 12.29 | 16.68 | 4.39 | 1.41 | 12.54 | 0.40 | 0.080 | 0.073 | 4.39 | 24.42 | 0.89 |
| End | 13.93 | 15.30 | 1.37 | 1.51 | 2.59 | 0.94 | 0.387 | 0.219 | 1.37 | 2.60 | 0.85 |
The estimated correlations and SDs at each interim analysis are shown in Appendix 4. The estimated SDs for the 12-month outcome were much smaller than expected when planning the study; σ12m was expected to around 20 but was actually nearer to 15. This explains why the numbers of participants at the interim analysis was much smaller than planned, as it took fewer participants to accumulate the required information, due to the smaller than expected value for σ12m. Also, the fast rate of recruitment and small number of 12-month outcomes meant that it was problematic to hit the required information level exactly; the value for I*1 is larger than I1.
Figure 13a shows the stopping boundaries and Z and Z0 from Table 15, at each interim analysis and study end. Test statistic Z is in the continuation region (between upper and lower boundaries) at interim analysis 1 for all boundary settings a to d, indicating that the study would not have stopped.
FIGURE 13.
Stopping boundaries and Z and Z0 for decision-making. (a) For stopping decisions using data available at each interim analysis and (b) using over-running data.

The results of the over-running analysis (see Figure 13b) confirm the stopping decisions based on the interim data, that the study would not have stopped at the interim analysis (see Appendix 4 for full results of the over-running analysis).
DRAFFT: summary
The results of the simulated group sequential design for DRAFFT can be summarised as follows:
-
For all four boundary settings tested, the DRAFFT study would not have stopped at the interim analysis, when data when available from n = 26 participants with 12-month outcomes, and n = 135 with 6-month outcomes and n = 205 with 3-month outcomes.
-
At this interim analysis N = 294 participants had been recruited into the study and follow-up would have been completed in 15 months; this compares to N = 461 and 34 months for the original study.
-
The estimate of the SD of the primary outcome (σ12m), used in the original sample size calculation, and used to build the group sequential design, was smaller than the true value (15 vs. 20). This caused the interim analysis to take place at a much earlier time than planned (i.e. with fewer participants with 12-month outcomes than expected; N12m = 26 vs. N12m = 100).
-
In reality, the original DRAFFT study recruited at an even faster rate than expected, and the sample size was increased from n = 390 to n = 461. This gave greater precision in the estimate of the treatment effect, which was important for inferences and the health economics analysis particularly. 145
FixDT: simulated group sequential trial
Figure 14 shows the observed number of participants recruited and followed up at 3, 6, and 12 months for FixDT, the window of opportunity available for interim analyses and the times when the interim analyses would have occurred in the simulated adaptive design.
FIGURE 14.
Recruitment (―), follow-up (―) and numbered interim analyses (+) for FixDT; window of opportunity for interim analyses is shaded.

The trial hit the recruitment target of n = 320, and the planned interim analyses could both feasibly have occurred. The numbers of observed trial participants providing data at each interim analysis is shown in Table 16. Estimated model parameters and test statistics are shown in Table 17.
| Analysis | Observed | Expected | ||
|---|---|---|---|---|
| N 3m | N 6m | N 3m | N 6m | |
| 1 | 105 | 79 | 100 | 50 |
| 2 | 178 | 146 | 200 | 100 |
| End | 273 | 282 | 264 | 264 |
| Analysis | m16m | m06m | Δ | βt | var(βt) | Z | I* | I | β0t | var(β0t) | Z0 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 30.02 | 32.32 | 2.29 | 1.14 | 27.06 | 0.22 | 0.037 | 0.036 | 2.29 | 30.77 | 0.41 |
| 2 | 32.44 | 31.92 | −0.52 | −2.85 | 13.86 | −0.76 | 0.072 | 0.071 | −0.52 | 15.14 | −0.13 |
| End | 33.80 | 29.84 | −3.96 | −4.27 | 8.03 | −1.51 | 0.125 | 0.165 | −3.96 | 8.08 | −1.39 |
The estimated correlations and SDs at each interim analysis are shown in Appendix 4. The estimated SDs for the 6-month outcome were much larger than expected when planning the study; σ6m was expected to around 20 but was actually nearer to 24. This explains why the numbers of participants at the interim analysis were larger than planned, as it took more participants to accumulate the required information, due to the larger than expected value for σ6m.
Figure 15a shows the stopping boundaries and Z and Z0 from Table 17, at each interim analysis and study end. Test statistic Z is in the continuation region (between upper and lower boundaries) at interim analysis 1, for all boundary settings a to d. However, at interim analysis 2, Z falls below the lower boundaries for settings b to d, indicating that the study would have been stopped for futility.
FIGURE 15.
Stopping boundaries and Z and Z0 for decision-making. (a) For stopping decisions using data available at each interim analysis and (b) using over-running data.

The over-running analyses (see Figure 15b) confirms the results of the stopping decisions based on the interim data, and indicate stopping for futility at settings b to d. In fact, the over-running analyses provided stronger evidence in favour of stopping at all interim analyses and settings a to d (see Appendix 4 for full results of the over-running analysis).
FixDT: summary
The results of the simulated group sequential design for FixDT can be summarised as follows:
-
For three of the four boundary settings tested, the FixDT study would have stopped at the second interim analysis, when data when available from n = 146 participants with 6-month outcomes, and n = 178 with 3-month outcomes.
-
At this interim analysis, N = 243 participants had been recruited into the study and follow-up would have been completed in 27 months; this compares with N = 321 and 42 months for the original study.
-
The estimated treatment effect after completing follow-up for the stopped study (over-running analysis) was −6.2 favouring the control treatment; the estimate of the treatment effect in the original (fixed design) study was −4.0 (95% CI −9.6 to 1.6).
-
At the second interim analysis when the study was stopped, the estimated treatment effect was −2.9 (favouring the nail), and the raw difference between groups was −0.5 (favouring the nail).
-
There was a strong correlation between the early outcome (3 months) and the primary outcome (6 months) for DRI.
-
The estimate of the SD of the primary outcome (σ6m) used in the original sample size calculation, and used to build the group sequential design, was a marked underestimate of the true value (20 vs. 24). This caused the study to be underpowered and the interim analyses to be at later times than planned (i.e. with more participants than expected).
FASHIoN: simulated group sequential trial
Figure 16 shows the observed number of participants recruited and followed up at 3, 6 and 12 months for FASHIoN, the window of opportunity available for interim analyses and the times when the interim analyses would have occurred in the simulated adaptive design.
FIGURE 16.
Recruitment (―), follow-up (―) and numbered interim analyses (+) for FASHIoN; window of opportunity for interim analyses is shaded.
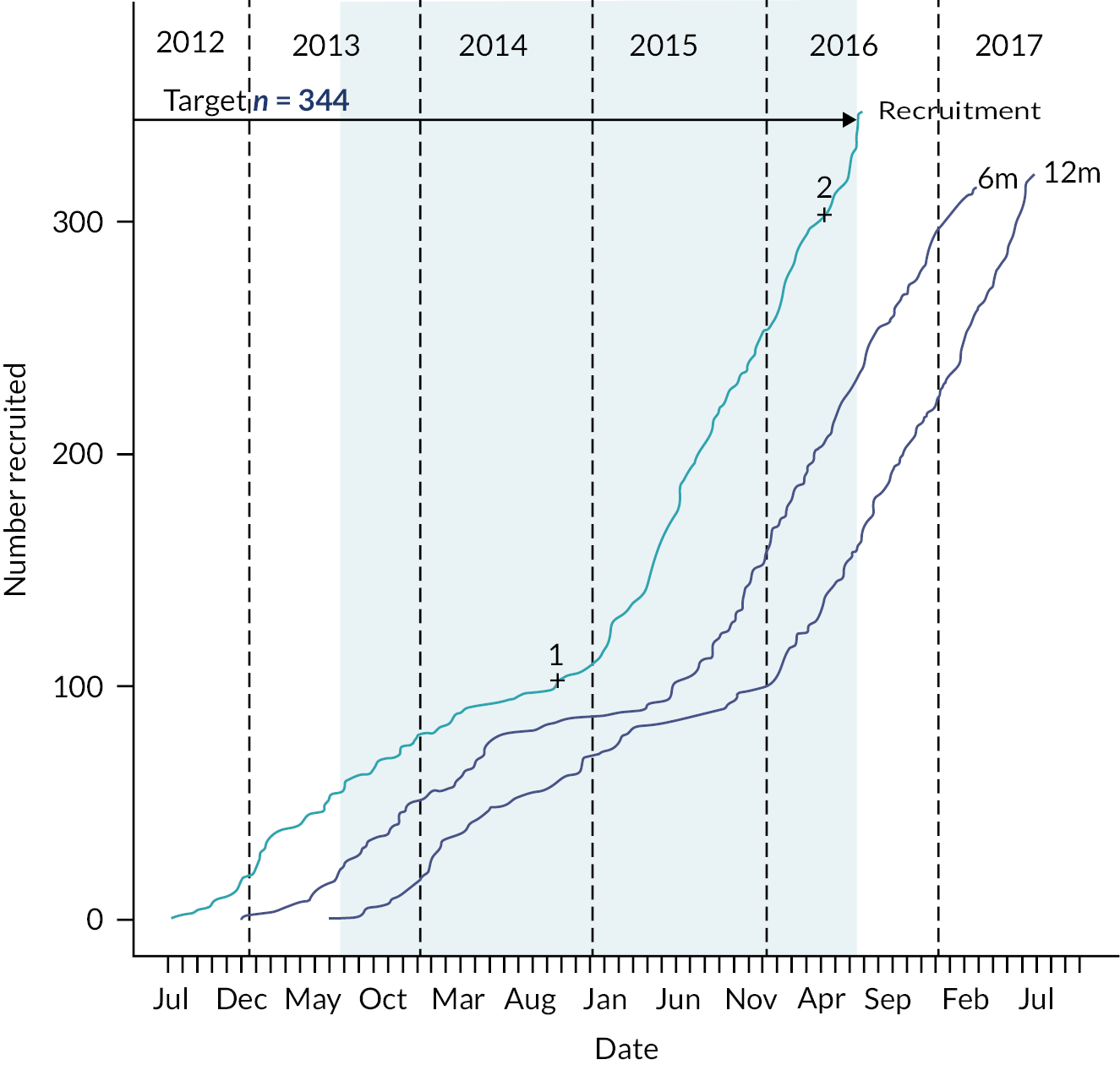
The trial hit the recruitment target of n = 344, and the planned interim analyses could both feasibly have occurred. The numbers of observed trial participants providing data at each interim analysis is shown in Table 18. Estimated model parameters and test statistics are shown in Table 19.
| Analysis | Observed | Expected | ||
|---|---|---|---|---|
| N 6m | N 12m | N 6m | N 12m | |
| 1 | 86 | 62 | 100 | 50 |
| 2 | 208 | 141 | 200 | 100 |
| End | 315 | 321 | 292 | 292 |
| Analysis | m112m | m012m | Δ | βt | var(βt) | Z | I* | I | β0t | var(β0t) | Z0 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 56.09 | 49.02 | 7.08 | 3.60 | 39.08 | 0.58 | 0.026 | 0.025 | 7.08 | 42.64 | 1.08 |
| 2 | 55.90 | 49.13 | 6.77 | 6.50 | 18.86 | 1.50 | 0.053 | 0.050 | 6.77 | 20.97 | 1.48 |
| End | 58.76 | 49.68 | 9.08 | 8.74 | 8.53 | 2.99 | 0.117 | 0.127 | 9.08 | 8.64 | 3.09 |
The estimated correlations and SDs at each interim analysis are shown in Appendix 4. The estimated SDs for the 12-month outcome are slightly larger than expected when planning the study; σ12m was expected to around 24 but was actually nearer to 26. This explains why the numbers of participants at the interim analysis were slightly larger than planned, as it took more participants to accumulate the required information, due to the larger than expected value for σ12m.
Figure 17a shows the stopping boundaries and Z and Z0 from Table 19, at each interim analysis and study end. Test statistic Z is in the continuation region (between upper and lower boundaries) at interim analysis 1 and 2, for all boundary settings a to d. Figure 17b shows that the interim analyses based on the over-running data indicate stopping for efficacy for all settings a to d, at the second interim analysis. This is consistent with the final data analysis of the original fixed design study (see Appendix 4 for full results of the over-running analysis).
FIGURE 17.
Stopping boundaries and Z and Z0 for decision-making. (a) For stopping decisions using data available at each interim analysis. (b) Using over-running data.
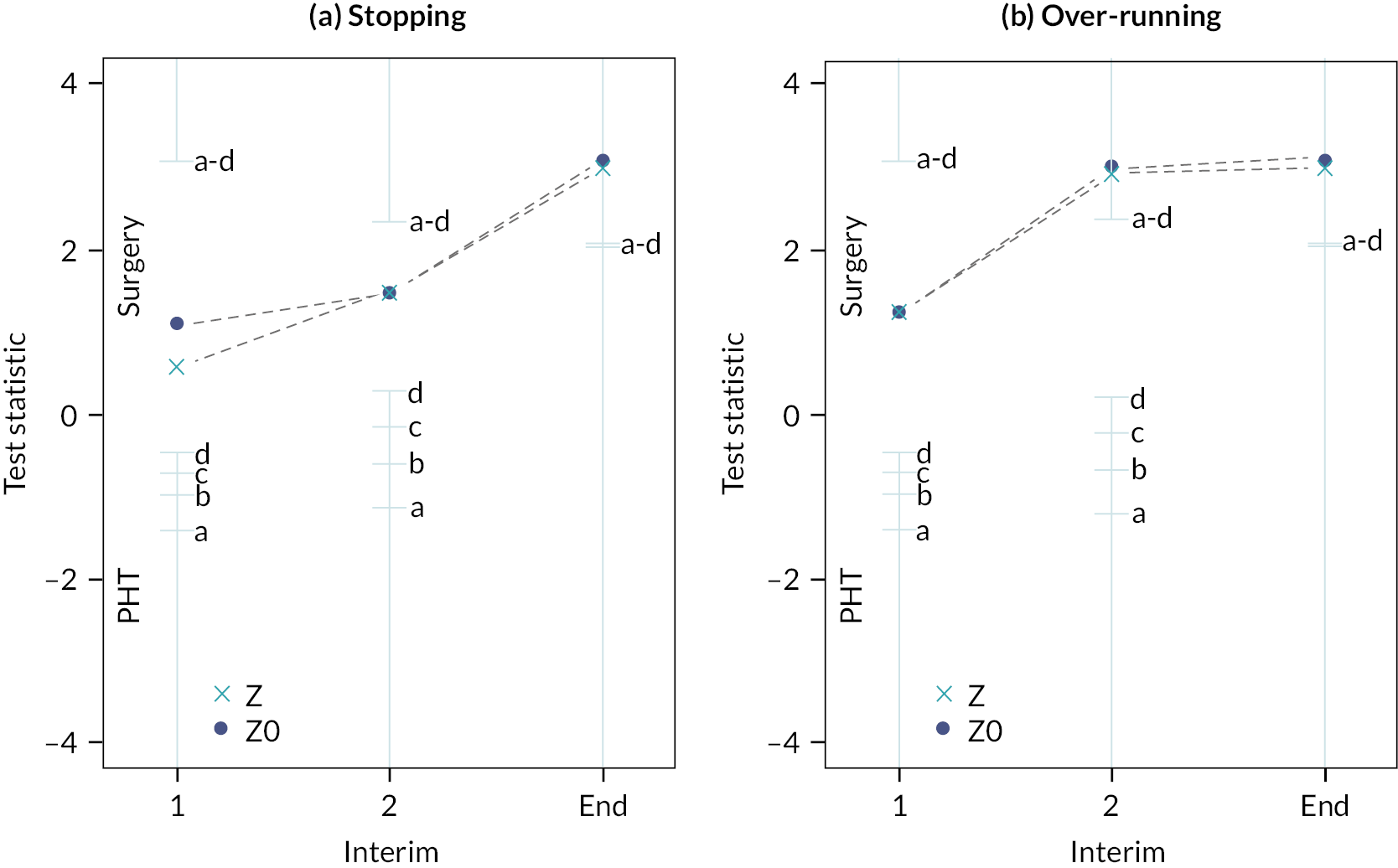
FASHIoN: summary
The results of the simulated group sequential design for FASHIoN can be summarised as follows:
-
The recruitment and follow-up profiles (see Figure 16) for FASHIoN were unusual and reflected the phased approach to the study with an initial feasibility/pilot stage at a small number of sites, followed be more rapid recruitment as many more sites were opened.
-
This resulted in relatively little benefit available from the early outcomes at the interim analyses – for example, at the first interim analysis 12-month data were available from 62 participants and 6-month data from 86 participants.
-
Of the four boundary settings tested, the FASHIoN study would not have stopped at either interim analysis for futility or efficacy.
-
The estimated treatment effect after completing follow-up for the second interim analysis (over-running analysis) was consistent with result of the original study.
-
The estimate of the treatment effect in the original (fixed design) study was 9.1 (95% CI 3.3 to 14.9), which is consistent with the over-running analysis from the second interim analysis (9.1) suggesting that the study could legitimately have stopped at the second interim analysis.
WAT: simulated group sequential trial
Figure 18 shows the observed number of participants recruited and followed up at 6 weeks, 3, 6, and 12 months for WAT, the window of opportunity available for interim analyses and the times when the interim analyses would have occurred in the simulated adaptive design.
FIGURE 18.
Recruitment (―), follow-up (―) and numbered interim analyses (+) for WAT; window of opportunity for interim analyses is shaded.

The trial hit the recruitment target of n = 120, and the planned interim analysis could feasibly have occurred. The numbers of observed trial participants providing data at each interim analysis is shown in Table 20. Estimated model parameters and test statistics are shown in Table 21.
| Analysis | Observed | Expected | ||||||
|---|---|---|---|---|---|---|---|---|
| N 6w | N 3m | N 6m | N 12m | N 6w | N 3m | N 6m | N 12m | |
| 1 | 49 | 43 | 29 | 10 | 80 | 70 | 60 | 40 |
| End | 119 | 119 | 122 | 120 | 104 | 104 | 104 | 104 |
| Analysis | m112m | m012m | Δ | βt | var(βt) | Z | I* | I | β0t | var(β0t) | Z0 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 45.00 | 38.20 | 6.80 | 4.30 | 6.58 | 1.68 | 0.152 | 0.150 | 6.80 | 14.64 | 1.78 |
| End | 40.40 | 38.17 | 2.23 | 2.18 | 3.54 | 1.16 | 0.283 | 0.321 | 2.23 | 3.59 | 1.18 |
The estimated correlations and SDs at each interim analysis are shown in Appendix 4. The estimated SDs for the 12-month outcome at the interim analysis was much smaller than expected; σ12m was expected to around 9 but was actually nearer to 5. Also, the correlations were generally larger than expected. These factors explain why the numbers of participants at the interim analysis was much smaller than planned, as it took many fewer participants to accumulate the required information, due to the larger than expected correlations and smaller than expected value for σ12m.
Figure 19a shows the stopping boundaries and Z and Z0 from Table 21, at each interim analysis and study end. Test statistic Z is in the continuation region (between upper and lower boundaries) at the interim analysis, for all boundary settings a to d.
FIGURE 19.
Stopping boundaries and Z and Z0 for decision-making. (a) For stopping decisions using data available at each interim analysis. (b) Using over-running data.

The test statistic from the over-running analysis (see Figure 19b) confirms the results of the stopping decisions based on the interim data and indicates that the study would not have stopped for any of the settings a to d (see Appendix 4 for full results of the over-running analysis).
Estimates of the treatment effects βt, from the longitudinal model, are close to raw differences in means (Δ) for the over-running analysis as there is less information available from the early outcomes at this analysis. If there were complete follow-up data available for all participants, then βt would be equal Δ. However, in practice it is almost always the case that some participants who did not provide 12-months outcomes had early outcomes (at one or more than one of 6 weeks, 3 months and 6 months), which provide some information on the 12-month outcomes. Estimates of treatment effects β0t, from the models that force correlations (between all outcomes) to be zero, are always equal to Δ, and have larger variances than βt).
WAT: summary
The results of the simulated group sequential design for WAT can be summarised as follows:
-
For all four boundary settings tested, the WAT study would not have stopped at the interim analysis, when data were available from n = 10 participants with 12-month outcomes, n = 29 with 6-month outcomes, n = 43 with 3-month outcomes and n = 49 with 6-week outcomes.
-
At this interim analysis N = 75 participants had been recruited into the study and follow-up would have been completed in 17 months; this compares with N = 126 and 48 months for the original study.
-
The estimate of the SD of the primary outcome (σ12m = 9), used in the original sample size calculation, and used to build the group sequential design, was much larger than the observed value at the interim analysis. Also, many of the correlations were significantly larger (e.g. ρ3m,12m = 0.71, ρ3m,6m = 0.90, ρ6w,3m = 0.86) than anticipated (e.g. ρ3m,12m = ρ3m,6m = ρ6w,3m = 0.5).
-
This caused the interim analysis to take place at a much earlier time than planned (i.e. with fewer participants with 12-month outcomes than expected; n = 10 vs. n = 40).
-
Given the small, but not clinically significant, result observed in the original study, it seems unlikely that any sensible stopping rule would have caused the WAT study to stop early.
CSAW: simulated group sequential trial
Figure 20 shows the observed number of participants recruited and followed up at 6 months for CSAW, the window of opportunity available for interim analyses and the times when the interim analyses would have occurred in the simulated adaptive design.
FIGURE 20.
Recruitment (―), follow-up (―) and numbered interim analyses (+) for CSAW; window of opportunity for interim analyses is shaded.
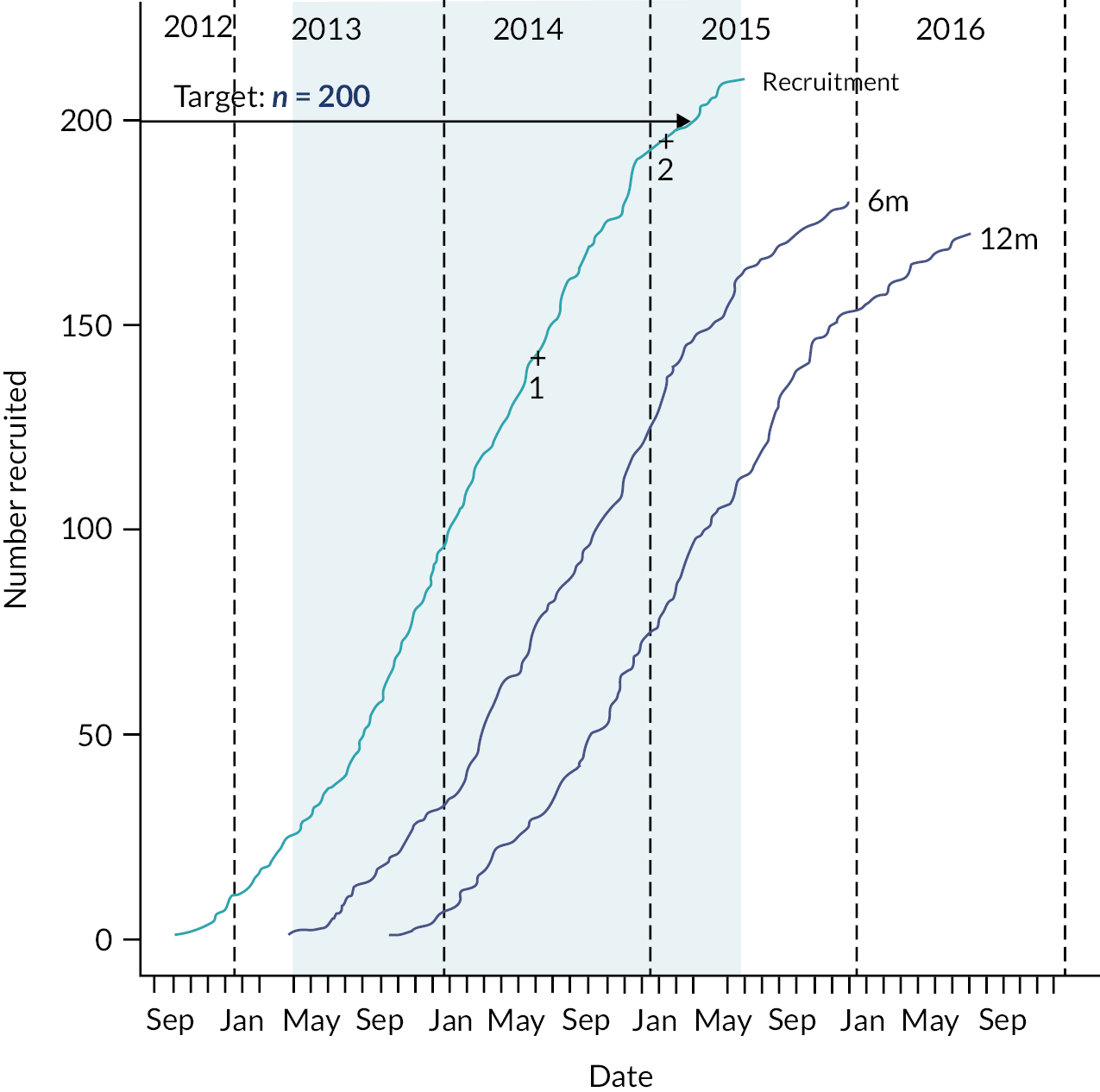
The trial hit the recruitment target of n = 200, and the planned interim analyses could feasibly have occurred. The numbers of observed trial participants providing data at each interim analysis is shown in Table 22. Estimated model parameters and test statistics are shown in Table 23.
| Analysis | Observed | Expected |
|---|---|---|
| N 6m | N 6m | |
| 1 | 79 | 40 |
| 2 | 137 | 80 |
| End | 180 | 170 |
| Analysis | m16m | m06m | Δ | βt | var(βt) | Z | I* | I | β0t | var(β0t) | Z0 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 30.05 | 31.47 | 1.42 | 1.42 | 7.49 | 0.52 | 0.134 | 0.123 | 1.42 | 7.49 | 0.52 |
| 2 | 29.56 | 31.72 | 2.17 | 2.17 | 4.01 | 1.08 | 0.249 | 0.247 | 2.17 | 4.01 | 1.08 |
| End | 29.37 | 32.68 | 3.31 | 3.31 | 3.08 | 1.89 | 0.325 | 0.525 | 3.31 | 3.08 | 1.89 |
The estimated SDs at each interim analysis were as follows: σ6m = 12.2, 11.7, 11.8. The estimated SDs for the 6-month outcome at the interim analysis was larger than expected; σ6m was expected to around 9 but was actually nearer to 12. This explains why the numbers of participants at the interim analysis were larger than planned, as it took more participants than expected to accumulate the required information.
Figure 21a shows the stopping boundaries and Z and Z0 from Table 23, at each interim analysis and study end. Test statistic Z is in the continuation region (between upper and lower boundaries) at the interim analysis, for all boundary settings a to d. Note that because we have no early outcome data for CSAW, Z is equal to Z0 at all analyses.
FIGURE 21.
Stopping boundaries and Z and Z0 for decision-making. (a) For stopping decisions using data available at each interim analysis. (b) Using over-running data.
The test statistics (Z) from the over-running analysis (see Figure 21b) confirm the results of the stopping decisions based on the interim data and indicate that the study would not have stopped for any of the settings a to d.
There is no evidence in the simulated study to believe that an adaptive design would have caused CSAW to stop early. Indeed, there is some reason to believe that an adaptive design may have complicated the inferences as in the simulated designs described here, in contrast to the original result of the study,69 the final (over-running) analysis does not reject the null hypothesis (although Figure 21b shows that the test statistic is close to the threshold). This is because by spending some of the overall error testing at interim analyses, we make it to harder to reject at the end of the study.
CSAW: summary
The results of the simulated group sequential design for CSAW can be summarised as follows:
-
For all four boundary settings tested, the CSAW study would not have stopped at the interim analyses, when data were available from n = 79 and n = 137 participants with 6-month outcomes.
-
At these interim analysis N = 142 and N = 195 participants had been recruited into the study and follow-up would have been completed in 21 and 28 months, respectively; this compares with N = 210 and 39 months for the original study.
-
The estimate of the SD of the primary outcome (σ6m = 9), used in the original sample size calculation and used to build the group sequential design, was smaller than the observed value at the interim analyses.
-
This caused the interim analysis to take place at a much later than planned (i.e. with more participants with 6-month outcomes than expected; n = 79 vs. n = 40 and n = 137 vs. n = 80).
-
Given the small but not clinically significant result observed in the original study, it seems unlikely that any sensible stopping rule would have caused the CSAW study to stop early for efficacy, unless some early outcome data (e.g. 3 months) had been available.
TOPKAT: simulated group sequential trial
Figure 22 shows the observed number of participants recruited and followed up at 2 months, 1, 2, 3, 4 and 5 years for TOPKAT. The recruitment for TOPKAT completed in June 2013 and the first five-year (primary) outcome data were not available until March 2015. Therefore, the window of opportunity, between some five-year final outcome data being available and recruitment completion was non-existent and, as such, the methodology we are investigating here, assessing possible early stopping of the trial, could not have been used.
FIGURE 22.
Recruitment (―), follow-up (―) for TOPKAT; there is no window of opportunity for interim analyses.

Discussion
Overview
For five of the selected trauma and orthopaedic trials discussed here (WOLLF, FixDT, DRAFFT, FASHIoN and WAT), the methodology of Parsons et al. ,61 that used early outcome data in addition to final outcome data to inform stopping decisions at interim analyses, proved to be feasible. All of the putative group sequential designs described for these five studies used only information that was known (or thought to be known) or could have reasonably been speculated on (e.g. the numbers and patterns of patient data), at the study design and planning stage. The designs do not knowingly use any information from the observed trial publications or data. For this reason, we believe that the results of the simulated trials that used the observed data and the known dates when data were collected for each trial are a true test of whether the design would have been possible, whether the trial would have stopped early and if so whether the result would have been consistent with that obtained from the original (fixed) design.
The CSAW and TOPKAT studies were different from the other trials discussed here in two key respects that made an adaptive design of the type under discussion here impossible, the lack of early outcome data for CSAW and the lack of a window of opportunity for TOPKAT. For these reasons, these two trials are discussed after the other five studies.
WOLLF, FixDT, DRAFFT, FASHIoN and WAT
Looking at each of these five trials in turn, the WOLLF study would have stopped early at the second interim analysis for three of the four boundary settings tested, when 293 participants had been recruited into the study (c.f. 460 in the original trial) and follow-up would have been completed in 27 months (c.f. 50 months for the original study).
Inferences for the stopped studies would have been very similar to the original study; the over-running analysis gave an estimate of the treatment effect equal to −3.5 (favouring the control), whereas the treatment effect estimate in the original (fixed design) study was −3.1 (95% CI −8.5 to 2.2). Of particular note in WOLLF were the extremely strong correlations between the early outcomes (3, 6 and 9 months) and the primary outcome (12 months) for DRI. The strong correlations are important for two reasons. First, they allowed the modelling approach, that used all the data, to give better (more precise) estimates of the true end of trial treatment effect, than simple between-group mean differences for the primary 12-month outcome. Second, the stronger than expected correlations also meant that information accrued rapidly causing the interim analyses to occur earlier than might have been expected based on the number of participants with 12-month outcomes alone.
At the second interim analysis when the study was stopped, the estimated treatment effect from the model was −2.8 (favouring the control). In marked contrast, the raw difference between groups using 12-month data only was 4.1 (favouring NPWT). As we note above, the strong correlations meant that the model estimate was a much better estimate of the true treatment effect than simple between-group difference in 12-month outcomes. However, it is worth considering how this might have played out if this had been the situation in the real trial. Would the trial data monitoring and safety committee have had the confidence in the model estimate to stop the study for futility when the difference in the means for 12-month data alone was strongly favouring NPWT?
The FixDT trial was of a similar overall design to WOLLF, as it ran concurrently with and was designed by the same research team. As for WOLLF, for three of the four boundary settings tested, the FixDT study would have stopped at the second interim analysis, when 243 (c.f. 321 in the original study) participants had been recruited into the study and follow-up would have been completed in 27 months (c.f. 42 months for the original study). Therefore, as for WOLLF, there would have been a considerable saving in time and cost, if an adaptive design had been used.
The treatment estimate for FixDT after completing follow-up for the stopped study was −6.2 favouring the control (nail) treatment and the estimate of the treatment effect in the original (fixed design) study was −4.0 (95% CI −9.6 to 1.6). The estimate of the SD of the primary outcome (σ6m) used in the original sample size calculation, and used to build the group sequential design, for FIXDT was a marked underestimate of the true value (20 vs. 24). This caused the original study to be underpowered and the interim analyses to be at later times than planned. That is, when there were more participants than expected (146 vs. 100) with 6-month outcome data.
WOLLF and FixDT both reported results in the original studies favouring the control treatments; intramedullary nail fixation in FixDT and standard dressing in WOLLF. The boundaries for our designs reflected the wish to stop for futility if there were a lack of emerging evidence to support better outcomes for the comparator test treatments (locking-plate fixation and NPWT, respectively). Locking-plate fixation and NPWT both proved unlikely to be a cost-effective compared with intramedullary nail fixation and a standard dressing respectively in reported health economic analyses. 146,147
In contrast to WOLLF and FixDT, the DRAFFT trial provided treatment effects throughout the study that tended to marginally favour the test locking-plate treatment over wire fixation control treatment. Therefore, it is perhaps not surprising given the asymmetry of the boundaries that the DRAFFT study would not have stopped at the interim analysis for any of the four boundary settings tested in the simulated studies.
The estimate of the SD of the primary outcome for DRAFFT (σ12m), used in the original sample size calculation and to build the group sequential design, was much smaller than expected (15 vs. 20). This caused the interim analysis to take place much earlier than planned when there were many fewer participants with 12-month outcomes than expected; 26 rather than the expected 100. The stronger than expected correlations also in part contributed to the early interim analysis. As did the extremely rapid recruitment to DRAFFT caused by a surge in recruitment due to the harsh winter weather causing a surge in distal radius fracture as a consequence of falls. However, given the consistent positive treatment effects in favour of the plate intervention it seems unlikely that better estimates of the covariance parameters or change of boundaries (with reason) would have caused the study to stop early for futility.
The FASHIoN study had a quite different recruitment profile from the other trials (see Figure 19) due to the phased approach with an initial feasibility/pilot stage at a small number of sites, followed be more rapid recruitment as sites were opened. This caused there to be relatively little benefit available from early outcomes (6 months) in addition to that provided by the final 12-month outcomes.
Of the four boundary settings tested, the FASHIoN study would not have stopped at either interim analysis for futility or efficacy. The two interim analyses both provided evidence in favour of the surgical intervention, but test statistics were not of sufficient magnitude to cause the trial to stop. Although, the estimated treatment effect after completing follow-up for the second interim analysis (over-running analysis) was consistent with result of the original study, which reported a positive result in favour of the surgery intervention. The lack of stopping (for efficacy) for FASHIoN is in large part due to the asymmetric selection of boundaries that made it relatively harder to stop for efficacy. The boundaries reflected the widespread view within trauma and orthopaedic medicine that much stronger evidence is required to cause a trial to stop for efficacy than futility, with many clinicians believing that if there is emerging evidence for efficacy then a trial should complete recruitment to target in order to provide a precise estimate of the treatment effect and capture as much safety information as possible (e.g. adverse events). The trials reported here tested interventions that typically were available within the NHS, so there was no ethical imperative to end trials as soon as possible and offer all patients the superior intervention, as for instance might be case when testing novel pharmaceutical drugs.
Given the relatively small sample size and the small (clinically insignificant) result observed in the original WAT trial, it seems unlikely that any sensible stopping rule would have caused the study to stop early. For all four boundary settings tested, the WAT study would not have stopped at the interim analysis. As for some of the other trials discussed here, the estimate of the SD of the primary outcome, used in the original sample size calculation, and used to build the group sequential design, was much larger than the observed value at the interim analysis. Also, for WAT, many of the estimated correlations were significantly larger than anticipated. These together caused the interim analysis to take place at a much earlier time than planned, when there were many fewer participants with 12-month outcomes than expected (n = 10 vs. n = 40).
CSAW and TOPKAT
For the CSAW and TOPKAT trials it was not possible to use the methodology of Parsons et al. 61 directly as for the former study there was no early outcome data available and for the latter no final outcome data were available prior to recruitment completing. Therefore, for TOPKAT we cannot proceed to simulate an adaptive study based on early assessment of the treatment effect on the final outcome.
For CSAW, although no early outcome data were available, we can simulate how the study would have proceeded via a group sequential design based simply on the final outcome data alone. This is amounts to using the test statistic Z0, rather than Z, to make stopping decisions; Z0 forces all the correlations between the final and early outcomes to be zero, and by so doing in practice uses only final outcome data for decision-making. Using this methodological approach, there was no evidence in the simulated study to believe that an adaptive design would have caused CSAW to stop early. Indeed, there is some reason to believe that an adaptive design may have complicated the inferences as in the simulated designs described here, in contrast to the original result of the study,69 the final (over-running) analysis does not reject the null hypothesis (although Figure 20b shows that the test statistic is close to the threshold). This is because by spending some of the overall error testing at interim analyses, we make it to harder to reject at the end of the study. However, there is reason to think that if an early outcome had been collected (e.g. at 3 months) then given that it correlated reasonably stronger with the final outcome, this may have caused us to stop for efficacy at the second interim analysis. This is not an unreasonable suggestion, as the over-running analysis at this occasion very nearly crossed the upper stopping boundary.
Summary
The results for five of the studies reported here (WOLLF, FixDT, DRAFFT, FASHIoN and WAT) showed that adaptive design using early outcome data would have been feasible and likely to provide designs that were at least as efficient, and possibly more efficient, than the original fixed sample size designs. For WOLLF and FixDT the simulations showed that it was highly likely these studies would have (correctly) stopped early for futility, saving potential considerable effort and resources. WOLLF particularly showed the important part that early outcome data particularly can play, as analyses based purely on the final outcome data alone would have meant that stopping (for any reason) would have been unlikely. The boundaries selected here favoured stopping for futility, at the cost of making stopping for efficacy unlikely, unless there were very strong evidence available. For this reason, the two studies that showed modest effect estimates at interim analyses in favour of the test treatment (WAT and DRAFFT), did not stop early. This was consistent with the final results of these studies. The FASHIoN trial showed good evidence in favour of the test surgical intervention in the final analysis but fell short of stopping at the interim analyses. For this study it would have been possible to select different, but sensible, boundaries that would have resulted in early stopping for efficacy.
For all the studies it was clear that the feasibility and practicality of using the methods proposed by Parsons et al. 61 was determined in large part by: (1) the width of the window of opportunity for stopping; (2) the available of early outcome data and their correlations with final outcomes; (3) recruitment and outcome data follow-up accrual profiles; and (4) the veracity of the estimates of the covariance parameters available at the design planning stage. We assumed that the authors had a high degree of confidence in the primary outcomes, which would be a requisite for the method presented, although it may be argued this is true for conventional sample size calculations too. The timing of the first interim analysis could be chosen to ensure sufficient data are also available on other outcomes (e.g. sufficient economic data in the case of an efficacy stopping rule).
The first of the three issues we highlight here were evident for all the trials. If there were little or no final outcome data available at interim analyses, and little or no early outcome data were available or uncorrelated with final outcomes then decision-making for early stopping were simply not possible. The pattern of data accrual and follow-up were important determinants of the feasibility of the methods used. However, more work is needed to fully understand the impact of different approaches to recruitment and follow-up on the widespread applicability of the methods. For instance, whether limiting or increasing recruitment at certain stages of a trial may be beneficial in certain circumstances. It was also clear for a number of the trials that the times when interim analyses occurred were either much earlier or later than expected. This was largely due to estimates of the covariance estimates used in the initial planning being markedly different from the observed values. For instance, if correlations between outcomes were stronger than expected and variances smaller, then interim analyses would occur sooner than might have been expected. This in itself is not necessarily problematic, as we deliberately motivated stopping based on information rather than purely sample size considerations. However, in instances where interim analyses occurred particularly early (e.g. in the DRAFFT study an interim analysis occurred when there were final outcome data from 26 participants rather than the expected 100), it is likely that in practice it would have been difficult for the trial DMC, TMG and TSC to make and confirm stopping decisions and justify these to the funding body based on so few data. In practice, either minimum sample sizes might have to be prespecified or interim analyses plans be modified as the study proceeds (e.g. by using blinded re-estimation of the covariance parameters as data accumulates to update the trial plans).
In many of the trials, correlations between outcomes were much stronger than expected (e.g. > 0.7). If there are such strong correlations between early and final outcomes, then it may also be that there are strong correlations between treatment effect estimates (e.g. treatment effects for an early outcome at 6 months were much the same as those for the primary outcome at 12 months). If this is the case, then arguably we might want to consider using the 6 months outcome as the primary. If this is the case, then this would be a simpler strategy to shorten the trial and save costs. With an earlier primary outcome, the adaptive designs presented here would also become even more efficient, with more early primary outcome data available at each interim analysis and a better likelihood of a strong correlation between (for example) 3- and 6-month outcomes.
There are a number of limitations to this study. We have chosen to use the same sample size in the adaptive designs as was used in the original fixed sample size designs. In practice to maintain power, the sample size for the adaptive designs would have needed to be increased relative to the fixed designs, to allow for the interim analyses and possibility of early stopping. However, by increasing the sample size it would have not been possible to use just the original data and observed recruitment and follow-up patterns, as we would have been forced to, for instance, simulate some or all the data using a model or resample data. This would have diminished some of the clinical impact of the work, as the simulated trials would have deviated considerably from the original trials. Therefore, given that we were predominantly interested in assessing whether the simulated adaptive trials would have stopped, we decided to not to modify the original sample sizes.
We have tried, as strictly as possible, to avoid using information that was only known after trial data were available when planning the adaptive designs. For instance, by using estimates of variances from the published protocols and, where possible, details of recruitment and follow-up strategies from the trial teams. However, it may be that the results or knowledge of the selected trials may have unconsciously influenced the adaptive designs (e.g. the timing and number of interim analyses). We have used the date when outcome data were ‘collected’ as a proxy for when it would have been available to make stopping decisions. However, in reality in a trial it often takes some time to enter the data on the study database and extract data (e.g. freeze and check data ready for analysis). These data would then need to be sent to the trial statistician to undertake an analysis, circulate to colleagues on a data and safety monitoring committee to meet and discuss the results and make a recommendation to the TMG/TSC to finalise the decision. This would typically take some time – a number of weeks at least. We have not accounted for these delays in the simulation study, so it maybe that our assessments of the savings an adaptive design might have made (in terms of time and cost) may be somewhat optimistic. In reality, however, many of these tasks could be better planned, streamlined and automated to a large extent, if an adaptive design were being used.
In summary, adaptive clinical trials may be applied in a number of settings. The applicability and efficiency of these methods are dependent on trial design, especially in terms of the timing of the relevant outcomes and the correlation between early and late outcomes, as well as the expected speed of recruitment. In two of the trials evaluated here, major time savings could have been made in terms of completing the study and reporting early. This aligns with our experience from the START:REACTS trial presented earlier in this monograph. Clearly not all studies will stop early if the methods are applied correctly, but across a body of trials, major efficiency savings could be made. Such savings could reduce research costs and reduce the number of people exposed to potential harm in trials. However, they may also deliver clinical trial results quicker, resulting in earlier changes in practice that also result in major cost savings for the health service. In the IDEAL classification, it may allow more efficient progress between stages 2b and 3. This may be particularly important in the introduction of new surgical procedures, where risks of harm are more likely than in established procedures, and these may be identified earlier and prevented across the community.
The application of more complicated statistical techniques such as this inevitably adds some cost, although this would be expected to reduce substantially as statistical code become readily available and experience is gained across the statistical community. These development costs would likely be offset not only by reduced overall research costs, but also by major savings made across the health-care community by the earlier delivery of important trial results, although this would be a topic for future research.
Chapter 8 Interim analysis interpretation study
Background
It is not clear when designing adaptive trials whether they are better designed, monitored and analysed using a frequentis or Bayesian approach. Decision-making in the monitoring of phase II/III adaptive trials can be difficult,148 and some researchers suggest that frequentist analyses of trials may be misinterpreted by clinicians and other stakeholders. 149 It has also been argued that Bayesian analyses are more intuitive for stakeholders to interpret at the end of a trial. 150 An additional substudy was therefore conceived to produce, using Bayesian adaptive methodology, alternative designs for trials in progress and to evaluate the effects that this would have on decision-making during the trial. To achieve this, we carried out parallel interim analyses on simulated data separate from the main trial analyses, to determine whether different interim decisions would have been made using Bayesian or frequentist designs and analyses.
Aims
We aimed to investigate whether the type of information presented to a DMC (Bayesian vs. frequentist) influenced the decision process at an interim analysis point in a group sequential design clinical trial. Specifically, we aimed to investigate whether there was a concordance of stopping decisions or differing conclusions between the information modes.
Methods
This study received separate ethical approval to the main study on 14 December 2021, reference: BSREC.45/21-22.
We used three different scenarios of treatment effectiveness, chosen to present different challenges for the DMC decision-making process. They were as follows:
-
a recommendation to stop recruitment due to strong evidence of futility
-
a recommendation to stop recruitment due to strong evidence of efficacy
-
a recommendation to continue, with weak evidence of efficacy.
We did not use real trial data so as not to compromise the integrity of the main trial. All data were simulated using R (version 4.0.3).
The main results for frequentist and the Bayesian analogue were simulated to achieve the target scenarios. The frequentist results were simulated using the methods of Parsons et al. 61 and scenarios were chosen from those used to plan the main trial (as explained above). To create analogous scenarios for the Bayesian method, we explored the properties of the probability boundaries which provided similar characteristics to the frequentist rules, as well as simulating and exploring which data sets achieved the target scenarios. The mock DMC reports focused on the primary outcome measures with information relating to the follow-up presented the stopping rules, test statistics and summaries of the simulated data, with limited information on the recruitment. The frequentist report followed the layout of the report used for the main trial. The key components of the Bayesian report had boundary rules, statistical properties of the data and boundaries and prior and posterior probability distributions. Both reports contained visual charts and graphs to aid interpretation of key data. Examples of the reports can be found in Appendix 5.
We initially planned to hold these mock DMCs in person but because of the COVID-19 measures, they were held virtually (via Microsoft Teams). We requested volunteers take the role of members of the mock DMCs and to review two statistical reports (one Bayesian, one frequentist, each randomly chosen from the three scenarios), then decide whether the hypothetical trial should continue or stop recruitment. We then asked the mock DMC members to answer a questionnaire to establish their thoughts and which report style they preferred, if any.
Participants were recruited through local staff and postgraduate student networks (WMS statistics book and journal club; WCTU staff mailing list and the WMS staff newsletter); and presented our substudy at the WCTU statisticians group meeting. Members of the START:REACTS main study were also invited to join the mock DMCs, with the caveat that if they opted into taking part as a participant, they would be unable to take part in the analysis. Participants were asked to contact the substudy team directly if they were interested in taking part. All digitally signed and returned consent forms via e-mail before being sent a meeting invite and the mock DMC reports.
Each mock DMC was allocated a Bayesian and frequentist scenario at random, with each scenario and mode appearing twice in the six meetings. Reports were then randomly labelled as ‘Report 1’ and ‘Report 2’. Participants were allocated to the DMC based on availability.
At the start of each meeting, committee members were reminded that there were ‘no correct’ answers, and all opinions were important. After the first report was presented, participants were given a short interval to complete the questionnaire before discussing the second report.
Results
Six mock DMCs were held in January 2022, hosted on Microsoft Teams® (Microsoft Corporation, Redmond, WA, USA) by AH and HP. Each meeting was attended by between two and four ‘independent members’, one of whom was a trained WCTU trial statistician. As shown in Table 24, over half of the participants identified themselves as statisticians, and approximately half reported prior experience of being on an oversight committee (9/19 participants reported either independent or non-independent oversight experience). Four (40%) of the statisticians reported no previous experience of sitting on an oversight committee. The six who reported non-clinical or statistical roles included: trial administration staff (e.g. trial manager), WCTU quality assurance managers and medical school administration staff.
| Participants | |||
|---|---|---|---|
| N (total = 19) | (%) | ||
| Role | Statistician | 10 | 53 |
| Clinical or allied health professional | 3 | 16 | |
| Administration/trial management | 6 | 32 | |
| Experience in oversight committeea | As independent member | 4 | 21 |
| As non-independent member | 7 | 37 | |
| No oversight experience | 10 | 53 | |
| Confidence in statistical methods | Bayesian | 4 | 21 |
| Frequentist | 11 | 58 | |
| None | 5 | 26 | |
The DMC recommendations for each of the 12 reports reviewed are shown in Table 25. Here, it can be seen that 92% (n = 11) resulted in an agreed recommendation from the committee. For the remaining scenario (Bayesian, weak efficacy), DMC members could not reach agreement on what action to recommend. On inspection of the table, it appears that while the DMCs were relatively comfortable stopping for futility, they were more cautious when stopping for efficacy, wanting to continue to see more convincing evidence.
| Report recommendation | DMC decision (N = 12; % of each scenario) | ||
|---|---|---|---|
| Continue n (%) |
Stop n (%) |
Other decision n (%) |
|
| Continue (weak efficacy) | 3 (75) | 0 | 1 (25) |
| Stop for efficacy | 2 (50) | 2 (50) | 0 |
| Stop for futility | 1 (25) | 3 (75) | 0 |
Table 26 shows the self-reported understanding of the DMC reports; 11 (60%) participants understood the Bayesian report and 14 (73%) understood the frequentist report. Participants reported that the Bayesian reports were more difficult to interpret how the rules apply for stopping. A few participants (n = 4, 10%) reported not understanding the explanation, only one of who was a statistician. Again, the majority of participants who did not understand the interpretation of the rules for either report (n = 8 participants, 24%) were non-statisticians (seven of eight participants who responded).
| Understanding the reporta | Bayesian reportsa (n = 18) |
Frequentist reports (n = 19) |
Total (n = 37a) |
|---|---|---|---|
| n (%) | n (%) | n (%) | |
| Understood all | 11 (60) | 14 (73) | 25 (66) |
| Did not understand explanation | 2 (10) | 2 (10) | 4 (10) |
| Did not understand interpretation | 6 (32) | 3 (16) | 9 (24) |
| Did not understand another part of report | 2 (11) | 1 (5) | 3 (8) |
Participants were also asked, on a five-point Likert scale (strongly disagree = 1; disagree = 2; undecided = 3; agree = 4; strongly agree = 5), for their personal agreement and confidence with the DMC’s recommendation. Most participants agreed (gave a score > 3) with the recommendation of the report (n = 28/36, 71%) and were confident with their recommendation (scored > 3; n = 24/36, 63%). Agreement was lower for the Bayesian reports (Bayesian: n = 12/19, 63%, frequentist: 16/19, 84%); but confidence in the recommendation was the same (n = 12, 63% for both report types). Only a third of those who did not agree (gave a score ≤ 3) with the recommendation of the Bayesian reports (n = 3/9, 33%) were confident in the DMC’s decision to stop or continue the trial. There were no participants who strongly disagreed (gave a score of 1) with the report recommendations.
When splitting respondents by previous participation in an oversight committee; participants with no oversight experience reported that the Bayesian report was more helpful (see Table 27).
| Which report is more helpful? | Experience in oversight committee (independent or non-independent including TSC or DMC) | ||
|---|---|---|---|
| No experience (n = 10) |
Any experience (n = 9) |
Total (n = 19) |
|
| Bayesian report | 4 (40) | 1 (11) | 5 (26) |
| Frequentist report | 3 (30) | 3 (33) | 6 (32) |
| Both reports | 3 (30) | 4 (44) | 7 (37) |
| Neither report | 0 | 1 (11) | 1 (5) |
Focusing on those who identified as statisticians; more of those reporting no previous oversight experience preferred the Bayesian reports compared to the number preferring frequentist reports (see Table 28). For the non-statisticians (see Table 28), the frequentist report was preferred more.
| Which report was more helpful? | Statistician (n = 10) |
Non-statistician (n = 9) |
Total (n = 19) |
||
|---|---|---|---|---|---|
| Experience | No experience | Experience | No experience | ||
| Bayesian report | 1 | 3 | 0 | 1 | 5 |
| Frequentist report | 2 | 0 | 2 | 3 | 7 |
| Both reports | 2 | 1 | 1 | 2 | 6 |
| Neither report | 1 | 0 | 0 | 0 | 1 |
Discussion
The substudy recruited 19 participants, consisting of medical statisticians, clinical academics and university administration staff. The participants confidence in their knowledge of statistics was surveyed and found a large difference between confidence in Bayesian statistics and frequentist statistics (21% confident in Bayesian to 58% frequentist). This was quite evident in the results as participants were reporting more often that they understood the frequentist scenarios more than the Bayesian reports. The non-statisticians often reported finding it difficult to apply the stopping rules of the Bayesian adaptive method. It was found that participants with no oversight experience were more likely to find the Bayesian report more helpful compared to someone with oversight experience. This pattern was primarily for statisticians but the opposite for non-statisticians where over half the participants exclusively preferred the frequentist report.
One limitation from the study was the difference in the way the Bayesian and frequentist were presented. A key difference in the frequentist reports was that the stopping rules and test statistics were graphically presented in such way that the Bayesian was not; that is, a chart visualising the thresholds and with the test statistic. We received feedback that the charts were helpful in decision-making. This could be an important item as non-statisticians did not know how to apply the rules for Bayesian. Another key difference was an extra part in the report explaining statistical properties to the Bayesian stopping thresholds, whereas the frequentist report did not contain these details.
A qualitative observation from the substudy was how DMCs decided to continue the study in a few scenarios while seeing sufficient evidence, from a statistical viewpoint, that one intervention was better than the other. We found that DMC members wanted further information to support their decision. As the reports emphasised some of the adaptive approaches of the two methods and focused on the primary outcome measure to ‘simplify’ the interim analyses, this could have made it more difficult for DMCs members to decide. It was found that the DMCs were more cautious to stop early for efficacy, and experienced members felt more comfortable to continue the trial. The mock DMC reports were made very similar to the real report shown to the START:REACTS DMC members; a key difference was that the rules were binding in the real trial, therefore, the DMC agreed in advance to decide strictly based on observing the test statistics; this may have made an easier decision for the DMC as the members relied on the expert opinion of the statistician interpreting the test results which suggested to stop. However, the TSC questioned the DMC decision and requested an explanation as to why the study should stop early.
These patterns may indicate that the research community may not be entirely comfortable with adaptive designs given how cautious they are of stopping early, the requests for additional information did not comply with how trials are usually designed on a single primary outcome. This implies that adaptive designs rely on there being a high degree of confidence in the primary outcome.
A study by Snowden et al.,151 looking at qualitative accounts from the BRACELET study, found that while statistical guidance for stopping was helpful, the decision about stopping/continuing a trial was informed by a wider set of considerations and discussions. This was also the experience for the START:REACTS trial and concurs with the results and experience of this substudy.
In conclusion, regarding the preference of reports it appears that it is not entirely clear which report is preferred as the numbers show only small difference. Experienced members were more cautious about stopping for efficacy and inexperienced were more likely to stop. Given the trial experience and observing other studies it suggests that with either method of reporting, an important consideration is the supplementary data that will aid the DMCs in understanding the impact of stopping or continuing the study, clear explanations to aid interpretation of the data, and clear figures to visually demonstrate both the data and the decision rules.
Chapter 9 Conclusion
In this monograph, we present a programme of work designed to fully address the NIHR EME programme’s 2016 commissioned call for research into the evaluation of new surgical procedures through the use of novel study designs.
We evaluated a new surgical procedure in a randomised trial, arthroscopic debridement with the InSpace subacromial spacer balloon for people with irreparable rotator cuff tears. To do this, we used a novel group sequential adaptive design, utilising the correlation between early and late outcomes to improve the efficiency of a set of early stopping rules. We applied this design to a multicentre blinded RCT to test the clinical and cost-effectiveness of arthroscopic debridement and biceps tenotomy with the InSpace subacromial balloon spacer against an otherwise identical procedure without the device. We undertook a parallel within-trial health economic analysis.
We developed and refined a technique for dynamic MRI of a shoulder under deltoid load, using EMG to establish an effective method for activating the deltoid muscle and a rapid scanning protocol to collect the images. We used this technique in a mechanistic substudy.
To understand the novel adaptive design methodology in more detail, we undertook a large simulation study, applying the design principles that were developed for the main study to a number of previous high-impact randomised trials in trauma and orthopaedic surgery. Finally, to understand the influence of different approaches to adaptive trail design on the conduct of DMC decisions about interim analyses, we performed an exploratory study of the interpretation of Bayesian and frequentist interim analysis reports by potential committee members.
We found the following main conclusions:
-
In the pre-specified primary analysis of OSS at 12 months, arthroscopic debridement alone was superior to arthroscopic debridement with the InSpace device.
-
The use of the device is both more costly and less effective and was dominated by debridement-only. The InSpace device is highly unlikely to be cost-effective.
-
The adaptive design allowed the study to stop early, meaning that we were able to deliver a robust trial result despite the coronavirus pandemic and are likely to achieve clinical impact much earlier than we would have done with a traditional fixed sample size.
-
We developed an effective technique for dynamic MRI of the shoulder. Although the sample size was greatly reduced both by the early stop and the pandemic, we observed no difference between people in the two intervention arms of the study.
-
In studies where early time point data are captured and there is a sufficient window to conduct interim analyses, the novel adaptive methodology is likely to provide designs that are at least as efficient, and in some cases much more efficient, than the original fixed sample size designs without compromising the main findings of the trial.
-
DMC members do not always choose to follow the stopping rules that are presented and would benefit from more ancillary information to support their decision-making both in terms of wider trial information as well as information to support their understanding of the analysis, regardless of the statistical framework used.
Overall, this monograph presents a body of work that has the potential to substantially change the evaluation of new surgical procedures and technologies in the future. Randomised trials are needed early in the evaluation of new technologies to prevent harm to patients and cost to society, but also to allow effective treatments to be offered widely. Such trials could be delivered more efficiently and provide more rapid answers using adaptive designs, as has been presented here. By delivering efficient, effective trial designs early in the introduction of new procedures and technologies, we will make major cost savings for the health service and deliver better patient outcomes both now and into the future.
Research recommendations
Based on the findings in this monograph, we present the following recommendations for future research:
-
There is an urgent need for high-quality research into interventions for people with irreparable rotator cuff tears as there is a lack of good evidence for all available treatment options at present.
-
Further research into the balloon itself may focus either on a different patient population or if the balloon itself is changed in terms of either design or size. Evidence of effectiveness would need to be demonstrated if a new indication or design was to be recommended. Trials should include a control arm that allows the effect of the balloon to be isolated, as was used in this trial.
-
Further studies are recommended to validate the MRI technique presented here before it is used as an objective outcome in shoulder trials.
-
Further research into the utility of group sequential adaptive designs is needed to explore a wider range of operating parameters, and to examine their use across a broader range of clinical fields, trial designs and outcome measures.
-
The cost-effectiveness of using adaptive trials more widely is yet to be defined, such analyses should include both research costs and the impact of early decisions on the health-care service and wider society.
-
The potential to use CP for cost-effectiveness analyses needs further exploration to determine its utility in supporting interim analysis decision-making in adaptive clinical trials.
-
Future research is recommended into improving the information given to DMCs, especially to assist in making complex decisions such as in interim analyses.
Acknowledgements
This project (funders reference 16/61/18) is funded by the Efficacy and Mechanism Evaluation (EME) Programme, a Medical Research Council and National Institute for Health Research partnership. The views expressed in this publication are those of the authors and not necessarily those of the Medical Research Council, NIHR or the Department of Health and Social Care.
To reduce treatment costs to trial sites, 23 InSpace balloons were provided for free under an agreement with the manufacturers of the balloon (initially OrthoSpace, Israel, and later Stryker, USA). OrthoSpace also funded some of the training centre costs of a study-related cadaveric course to train participating surgeons in the use of the balloon at the start of the study. The full independence of the trial team is protected by legal agreements.
The trial was co-sponsored by the University Hospital of Coventry and Warwickshire NHS Trust and the University of Warwick.
In accordance with the trial protocol, and with the help of their teams, the START:REACTS principal investigators oversaw the participation of their site research team in the study, recruitment of participants and their involvement throughout the study.
Investigators at sites (site principal investigator listed first).
Bristol: Iain Packham, Elizabeth Barnett, Rian Witham, Mark Crowther, Richard Murphy, Katherine Coates, Josephine Morley, Stephen Barnfield, Sukhdeep Gill, Alistair Jones, Ruth Halliday, Sarah Dunn, James Fagg, Peter Dacombe.
North Tees: Rajesh Nanda, Deborah Wilson, Lesley Boulton, Raymond Liow, Richard Jeavons, Andrea Meddes.
Cambridge: Niel Kang, Leila Dehghani, Aileen Nacorda, Anuj Punnoose.
London North-West: Nicholas Ferran, Gbadebo Adewetan, Temi Adedoyin, Arun Pall, Matthew Sala, Tariq Zaman.
Bournemouth: Richard Hartley, Charif-a-Sayyad, Luke Vamplew, Elizabeth Howe, Norbert Boker.
Guys & St Thomas’s, London: Steve Corbett, Robert Moverley, Elise Cox.
Yeovil: Oliver Donaldson, Michael Jones, Diane Wood, Jess Perry, Alison Lewis, Linda Howard, Kate Beesley, Luke Harries.
Salisbury: Ahmed Elmorsy, Sridhar Sampalli, Katherine Wilcocks, Kate Shean, Sarah Diment, Ben Wilson, Helen Pidgeon, Victoria King.
West Suffolk: Soren Sjolin, Angharad Williams, Joanne Kellett, Lora Young, Michael Dunne, Tom Lockwood.
Kingston: Mark Curtis, Nashat Siddiqui, India Mckenley, Sarah Morrison, Charlotte Quamina, Tracey O’Brien, Isabel Bradley, Kenneth Lambatan.
Robert Jones and Agnes Hunt, Oswestry: Cormac Kelly, Charlotte Perkins, Teresa Jones, Tessa Rowlands, Dawn Collins, Claire Nicholas, Claire Birch, Julie Lloyd-Evans, Pouya Akhbari, Jefin Jose Edakalathur.
Southampton: Campbell Hand, Andy Cole, Debbie Prince, Kerry Thorpe, Louise Rooke, Maria Baggot, Matt Morris, Dima Ivanova, David Baker.
Cardiff: Tim Matthews, Jessica Falatoori, Heather Jarvis, Debbie Jones, Matthew Williams, Richard Evans.
Royal Gwent Hospital, Newport: Huw Pullen, Gemma Hodkinson, Nicola Vannet, Alison Davey, Emma Poyser, Angela Hall, Hemang Mehta, Devi Prakash Tokola, Clare Connor, Caroline Jordan.
Prince Philip Hospital, Llanelli: Owain Ennis, Zohra Omar, Tracy Lewis, Angharad Lisa Owen, Andrew Morgan, Ravi Ponnada, Waheeb Al-Azzani, Carolyn Williams, Liam Knox.
Leicester: Harvinder Singh, Tracy Lee, Kathryn Robinson, Dileep Kumar, Alison Armstrong.
Royal Orthopaedic Hospital, Stanmore: Addie Majed, Mark Falworth, David Butt, Deborah Higgs, Will Rudge, Ben Hughes, Esther Hanison, Deirdre Brooking, Amit Patel, Andrew Symonds, Jenifer Gibson, Rodney Santiago.
Wrexham: David Barlow, Joanne Lennon.
Royal Devon and Exeter: Christopher Smith, Jane Hall, Emily Griffin, Rebecca Lear, William Thomas.
Maidstone and Tunbridge Wells: David Rose, Janet Edkins, Helen Samuel, Hagen Jahnich.
Nottingham: John Geoghegan, Ben Gooding, Siobhan Hudson, Jess Nightingale.
Doncaster: Madhavan Papanna, Tom Briggs, Rebecca Pugh, Amy Neal, Lisa Warrem, Veronica Maxwell, Robert Chadwick.
Study oversight committees
Data monitoring committee members
Professor Thomas Jaki (Chair), Professor Stephen Gwilym, Ms Loretta Davies.
Trial steering committee members
Professor Rod Taylor (Chair), Professor Angus Wallace, Dr Christopher Littlewood, Dr Anthony Howard, Mr Geoffrey Abel, Mr John Graham.
Magnetic resonance imaging substudy
We acknowledge University Hospitals Coventry and Warwickshire Radiotherapy Workshop for constructing the L-shaped board for the MRI substudy.
We also acknowledge Andrew Weedall for his contribution to the MRI development work.
Adaptive design data
We acknowledge the following chief investigators who kindly provided the anonymised trial data that were used in the analyses presented in Chapter 7.
WOLLF: Matthew L Costa, Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences (ox.ac.uk)
FixDT: Matthew L Costa
DRAFFT: Matthew L Costa
FASHIoN: Damian R Griffin, Professor of Trauma and Orthopaedic Surgery (warwick.ac.uk)
WAT: Matthew L Costa
CSAW: David J Beard, Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences (ox.ac.uk)
TOPKAT: David J Beard
Contributions of authors
Andrew Metcalfe (https://orcid.org/0000-0002-4515-8202) (Professor of Orthopaedics) was chief investigator, led on the drafting and co-ordination of the completion of the report, and oversaw the analysis.
Susanne Arnold (https://orcid.org/0000-0001-8152-7610) (Research Fellow) contributed to data acquisition and interpretation, created the first draft, and contributed to the complete report.
Helen Parsons (https://orcid.org/0000-0002-2765-3728) (Associate Professor) was co-applicant, contributed and supervised the statistical analysis of the main study, contributed to study design, data acquisition and interpretation, wrote sections of the report, designed, and lead the Bayesian substudy, and was responsible for the statistical management of the main trial and MRI substudy.
Nicholas Parsons (https://orcid.org/0000-0001-9975-888X) (Professor) was co-applicant, was responsible for the simulation study for the trial design, contributed to the analyses and interpretation, wrote sections of the report and designed and lead the adaptive designs for surgical trials substudy.
Gev Bhabra (https://orcid.org/0000-0002-3352-1852) (Consultant Shoulder and Elbow Surgeon) was a shoulder surgeon and provided expertise in the MRI substudy development and design, and contributed to the final report.
Jaclyn Brown (https://orcid.org/0000-0003-3110-8594) was the senior project manager and was involved on the management, interpretation and contributed to the completed report.
Howard Bush (https://orcid.org/0000-0001-9360-0504) was an expert shoulder physiotherapist and helped develop the rehabilitation plan and assisted with the study design and conduct and contributed to the final report.
Michael Diokno (https://orcid.org/0000-0002-6282-4270) (MRI Research Radiographer) took part in the MRI substudy development work, helped to develop the image acquisition protocol, co-ordinated the MRI substudy and contributed to sections of the report.
Mark Elliott (https://orcid.org/0000-0003-4000-0198) (Associate Professor) was an EMG expert, performed the EMG measurements and analysis for the MRI development work, and contributed to the report.
Josephine Fox (https://orcid.org/0000-0003-0740-1096) was the patient lead, a co-applicant and was involved in the management and interpretation, and contributed to the completed report.
Simon Gates (https://orcid.org/0000-0003-3193-0975) (Professor of Clinical Trials) was co-applicant and was involved in the design of the study.
Elke Gemperlé Mannion (https://orcid.org/0000-0002-6116-2420) was trial manager for the START:REACTS trial and contributed to the completed report.
Aminul Haque (https://orcid.org/0000-0003-3589-6751) (Associate Statistician) conducted the statistical analyses for the main trial and MRI substudy, managed the Bayesian substudy and wrote sections of the report.
Charles Hutchinson (https://orcid.org/0000-0003-3387-9229) (Professor of Imaging) was co-applicant and led the radiological interpretation of the main trial, supervised the MRI substudy and contributed to the completed report.
Rebecca Kearney (https://orcid.org/0000-0002-8010-164X) (Professor) was co-applicant and the physiotherapy lead and was involved in conception, design, management and analysis, and contributed to the completed report.
Iftekhar Khan (https://orcid.org/0000-0001-6041-8837) (Senior Research Fellow) performed the health economic analysis of the main trial and wrote sections of the report.
Tom Lawrence (https://orcid.org/0000-0002-1296-1891) (Consultant Shoulder and Elbow Surgeon) was co-applicant, a shoulder surgeon for the trial, led fidelity assessments, was involved in conception, design, management, analysis and edited the final draft of the report.
James Mason (https://orcid.org/0000-0001-9210-4082) (Professor of Health Economics) was co-applicant, supervised the health economic analysis of the report and contributed to the completed report.
Usama Rahman (https://orcid.org/0000-0002-7558-689X) took part in the MRI substudy development work and wrote sections of the report
Nigel Stallard (https://orcid.org/0000-0001-7781-1512) (Professor of Medical Statistics and Epidemiology) was co-applicant, was involved in conception, design, management, analysis and contributed to the completed report.
Sumayyah Ul-Rahman (https://orcid.org/0000-0002-1188-0264) was the data entry clerk for the main trial and contributed to the completed report.
Aparna Viswanath (https://orcid.org/0000-0001-5018-6761) (Consultant Orthopaedic Surgeon) was a shoulder surgeon and led the acromiohumeral distance measurement analysis with Professor Hutchinson and contributed to the report.
Sarah Wayte (https://orcid.org/0000-0002-1565-6171) took part in the MRI substudy development work, led the development of the image acquisition protocol, contributed to the MRI substudy and contributed to sections of the report.
Stephen Drew (https://orcid.org/0000-0002-9523-682X) (Consultant Trauma and Orthopaedic Surgeon) was co-applicant, a senior shoulder surgeon for the trial, contributed to the fidelity assessments, was involved in conception, design, management and analysis, and edited the final draft of the report. He was joint senior author with Professor Underwood.
Martin Underwood (https://orcid.org/0000-0002-0309-1708) (Professor of Primary Care Research) was co-applicant, contributed to data acquisition and interpretation, and edited formal report for key intellectual content. He was joint senior author with Mr Drew.
Ethics, registration and oversight
The trial was conducted in accordance with the principles of the Declaration of Helsinki and to Medical Research Council good clinical practice guidelines as well as all applicable UK legislation and University of Warwick standard operating procedures. A DMC and TSC provided trial oversight, both made up of mostly independent members and conducted according to Warwick standard operating procedures. The sponsors undertook monitoring and audit according to a monitoring plan. The study was registered on the International Standard Randomised Controlled Trials Number Registry on 6 April 2018. The trial received full research ethical approval (REC Reference 18/WM/0025) on 13 February 2018, prior to commencing recruitment.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
References
- Ostor AJ, Richards CA, Prevost AT, Speed CA, Hazleman BL. Diagnosis and relation to general health of shoulder disorders presenting to primary care. Rheumatology 2005;44:800-5.
- Teunis T, Lubberts B, Reilly BT, Ring D. A systematic review and pooled analysis of the prevalence of rotator cuff disease with increasing age. J Shoulder Elbow Surg 2014;23:1913-21.
- Urwin M, Symmons D, Allison T, Brammah T, Busby H, Roxby M, et al. Estimating the burden of musculoskeletal disorders in the community: the comparative prevalence of symptoms at different anatomical sites, and the relation to social deprivation. Ann Rheum Dis 1998;57:694-55.
- Judge A, Murphy RJ, Maxwell R, Arden NK, Carr AJ. Temporal trends and geographical variation in the use of subacromial decompression and rotator cuff repair of the shoulder in England. Bone Joint J 2014;96-B:70-4.
- Largacha M, Parsons IMt, Campbell B, Titelman RM, Smith KL, Matsen F. Deficits in shoulder function and general health associated with sixteen common shoulder diagnoses: a study of 2674 patients. J Shoulder Elbow Surg 2006;15:30-9.
- Minns Lowe CJ, Moser J, Barker K. Living with a symptomatic rotator cuff tear ‘bad days, bad nights’: a qualitative study. BMC Musculoskelet Disord 2014;15.
- Virta L, Joranger P, Brox JI, Eriksson R. Costs of shoulder pain and resource use in primary health care: a cost-of-illness study in Sweden. BMC Musculoskelet Disord 2012;13.
- Whittle S, Buchbinder R. In the clinic. Rotator cuff disease. Ann Int Med 2015;162:ITC1-16.
- Funk L. 2005. https://www.shoulderdoc.co.uk/article/384 (accessed 14 July 2022).
- Huegel J, Williams AA, Soslowsky LJ. Rotator cuff biology and biomechanics: a review of normal and pathological conditions. Curr Rheumatol Rep 2015;17.
- Burkhart SS. Reconciling the paradox of rotator cuff repair versus debridement: a unified biomechanical rationale for the treatment of rotator cuff tears. Arthroscopy 1994;10:4-19.
- Carr AJ, Cooper CD, Campbell MK, Rees JL, Moser J, Beard DJ, et al. Clinical effectiveness and cost-effectiveness of open and arthroscopic rotator cuff repair [the UK Rotator Cuff Surgery (UKUFF) randomised trial]. Health Technol Assess 2015;19:1-218.
- Acevedo DC, Paxton ES, Williams GR, Abboud JA. A survey of expert opinion regarding rotator cuff repair. J Bone Joint Surg 2014;96.
- Anley CM, Chan SK, Snow M. Arthroscopic treatment options for irreparable rotator cuff tears of the shoulder. World J Orthop 2014;5:557-65.
- Leung B, Horodyski M, Struk AM, Wright TW. Functional outcome of hemiarthroplasty compared with reverse total shoulder arthroplasty in the treatment of rotator cuff tear arthropathy. J Shoulder Elbow Surg 2012;21:319-23.
- Harreld KL, Puskas BL, Frankle MA. Massive rotator cuff tears without arthropathy: when to consider reverse shoulder arthroplasty. J Bone Joint Surg 2011;93:973-84.
- Mulieri P, Dunning P, Klein S, Pupello D, Frankle M. Reverse shoulder arthroplasty for the treatment of irreparable rotator cuff tear without glenohumeral arthritis. J Bone Joint Surg 2010;95:2544-56.
- Rockwood CA, Williams GR, Burkhead WZ. Debridement of degenerative, irreparable lesions of the rotator cuff. J Bone Joint Surg 1995;77:857-66.
- Liem D, Lengers N, Dedy N, Poetzl W, Steinbeck J, Marquardt B. Arthroscopic debridement of massive irreparable rotator cuff tears. Arthroscopy 2008;24:743-8.
- Kukkonen J, Joukainen A, Lehtinen J, Mattila KT, Tuominen EK, Kauko T, et al. Treatment of non-traumatic rotator cuff tears: A randomised controlled trial with one-year clinical results. Bone Joint J 2014;96-B:75-81.
- Savarese E, Romeo R. New solution for massive, irreparable rotator cuff tears: the subacromial ‘biodegradable spacer’. Arthrosc Tech 2012;1:e69-74.
- National Institute for Health and Care Excellence . Biodegradable Acromial Spacer Insertion for Rotator Cuff Tears. Interventional Procedures Guidance IPG558 2016. www.nice.org.uk/guidance/ipg558 (accessed 14 July 2022).
- Stryker . Stryker announces the FDA clearance of the first biodegradable subacromial balloon spacer, filling a gap in the shoulder continuum of care 2021. https://www.stryker.com/us/en/about/news/2021/stryker-announces-the-fda-clearance-of-the-first-biodegradable-s.html (accessed 6 May 2023).
- Szollosy G, Rosso C, Fogerty S, Petkin K, Lafosse L. Subacromial spacer placement for protection of rotator cuff repair. Arthrosc Tech 2014;3:e605-9.
- Ramot Y, Nyska A, Markovitz E, Dekel A, Klaiman G, Zada MH, et al. Long-term local and systemic safety of poly(L-lactide-co-epsilon-caprolactone) after subcutaneous and intra-articular implantation in rats. Toxicol Pathol 2015;43:1127-40.
- Senekovic V, Poberaj B, Kovacic L, Mikek M, Adar E, Dekel A. Prospective clinical study of a novel biodegradable sub-acromial spacer in treatment of massive irreparable rotator cuff tears. Eur J Orthop Surg Traumatol 2013;23:311-6.
- Senekovic V, Poberaj B, Kovacic L, Mikek M, Adar E, Markovitz E, et al. The biodegradable spacer as a novel treatment modality for massive rotator cuff tears: a prospective study with 5-year follow-up. Arch Orthop Trauma Surg 2017;131:95-103.
- Bakti N, Bhat M, Gulihar A, Prasad V, Singh B. Subacromial balloon interpositional arthroplasty for the management of irreparable rotator cuff tears: five-year results. Open Ortho J 2019;13:89-96.
- Henderson DJ, Simeson K, Venkateswaran B. Sub-acromial spacer vs partial rotator cuff repair; a single surgeon cohort study. Should Elb 2016;8.
- Prasad VR, Fung M, Borowsky KA, Tolat AR, Singh B. Outcomes of InSpace balloon arthroplasty for irreparable cuff tear at two years: a longitudinal study. Ortho Proceed 2018;98-B.
- Holschen M, Brand F, Agneskirchner JD. Subacromial spacer implantation for massive rotator cuff tears: clinical outcome of arthroscopically treated patients. Obere Extremitat 2017;12:38-45.
- Viswanath A, Drew S. Subacromial balloon spacer – where are we now?. J Clin Orthop Trauma 2021;17:223-32.
- Johns WL, Ailaney N, Lacy K, Golladay GJ, Vanderbeck J, Kalore NV. Implantable subacromial balloon spacers in patients with massive irreparable rotator cuff tears: a systematic review of clinical, biomechanical, and financial implications. Arthrosc Sports Med Rehabil 2020;2:e855-e72.
- Liu F, Dong J, Kang Q, Zhou D, Xiong F. Subacromial balloon spacer implantation for patients with massive irreparable rotator cuff tears achieves satisfactory clinical outcomes in the short and middle of follow-up period: a meta-analysis. Knee Surg Sports Traumatol Arthrosc 2021;29:143-53.
- Stewart RK, Kaplin L, Parada SA, Graves BR, Verma NN, Waterman BR. Outcomes of Subacromial Balloon Spacer Implantation for Massive and Irreparable Rotator Cuff Tears: A Systematic Review. Orthop J Sports Med 2019;7.
- Osti L, Milani L, Ferrari S, Maffulli N. Subacromial spacer implantation: an alternative to arthroscopic superior capsular reconstruction. A systematic review. Br Med Bull 2021;139:59-72.
- Familiari F, Nayar SK, Russo R, De Gori M, Ranuccio F, Mastroianni V, et al. Subacromial balloon spacer for massive, irreparable rotator cuff tears is associated with improved shoulder function and high patient satisfaction. Arthroscopy 2021;37:480-6.
- Constant CR, Murley AH. A clinical method of functional assessment of the shoulder. Clin Ortho Relat Res 1987;214:160-4.
- Constant CR, Gerber C, Emery RJ, Sojbjerg JO, Gohlke F, Boileau P. A review of the Constant score: modifications and guidelines for its use. J Shoulder Elbow Surg 2008;17:355-61.
- Oderuth ENH, Morris DLJ, Manning PA, Geoghegan JM, Gooding BW, Wijeratna MD. The balloon spacer improves outcomes in only a minority of patients with an irreparable rotator cuff tear. J Arthr Joint Surg 2021;8:64-70.
- van Kampen DA, Willems WJ, van Beers LW, Castelein RM, Scholtes VA, Terwee CB. Determination and comparison of the smallest detectable change (SDC) and the minimal important change (MIC) of four-shoulder patient-reported outcome measures (PROMs). JOrthop Surg 2013;8.
- Khatri C, Ahmed I, Parsons H, Smith NA, Lawrence TM, Modi CS, et al. The natural history of full-thickness rotator cuff tears in randomized controlled trials: a systematic review and meta-analysis. Am J Sports Med 2019;47:1734-43.
- Verma NN, Srikumaran U, Roden C, Rogusky E, Lapner P, Abboud J, et al. Shoulder innovation research award paper: balloon subacromial spacer vs. partial repair for massive rotator cuff tears: a randomized trial. Arthroscopy 2021;37:e19-e20.
- Shon MS, Koh KH, Lim TK, Kim WJ, Kim KC, Yoo JC. Arthroscopic partial repair of irreparable rotator cuff tears: preoperative factors associated with outcome deterioration over 2 years. Am J Sports Med 2015;43:1965-75.
- Cuff DJ, Pupello DR, Santoni BG. Partial rotator cuff repair and biceps tenotomy for the treatment of patients with massive cuff tears and retained overhead elevation: midterm outcomes with a minimum 5 years of follow-up. Elbow Surg 2016;25:1803-9.
- Costa ML, Achten J, Parsons NR, Edlin RP, Foguet P, Prakash U, et al. Total hip arthroplasty versus resurfacing arthroplasty in the treatment of patients with arthritis of the hip joint: single centre, parallel group, assessor blinded, randomised controlled trial. BMJ 2012;344.
- Costa ML, Achten J, Parsons NR, Rangan A, Griffin D, Tubeuf S, et al. Percutaneous fixation with Kirschner wires versus volar locking plate fixation in adults with dorsally displaced fracture of distal radius: randomised controlled trial. BMJ 2014;349.
- McCulloch P. Developing appropriate methodology for the study of surgical techniques. J R Soc Med 2009;102:51-5.
- Achten J, Parsons NR, Bruce J, Petrou S, Tutton E, Willett K, et al. Protocol for a randomised controlled trial of standard wound management versus negative pressure wound therapy in the treatment of adult patients with an open fracture of the lower limb: UK Wound management of Lower Limb Fractures (UK WOLLF). BMJ Open 2015;5.
- Achten J, Parsons NR, McGuinness KR, Petrou S, Lamb SE, Costa ML. UK Fixation of Distal Tibia Fractures (UK FixDT): protocol for a randomised controlled trial of ‘locking’ plate fixation versus intramedullary nail fixation in the treatment of adult patients with a displaced fracture of the distal tibia. BMJ Open 2015;5.
- Keene DJ, Mistry D, Nam J, Tutton E, Handley R, Morgan L, et al. The Ankle Injury Management (AIM) trial: a pragmatic, multicentre, equivalence randomised controlled trial and economic evaluation comparing close contact casting with open surgical reduction and internal fixation in the treatment of unstable ankle fractures in patients aged over 60 years. Health Technol Assess 2016;20:1-158.
- Bhatt DL, Mehta C. Adaptive designs for clinical trials. N Engl J Med 2016;375:65-74.
- US Food and Drug Administration . Adaptive Designs for Medical Device Clinical Studies Final Guidance 2016. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/adaptive-designs-medical-device-clinical-studies (accessed 11 January 2022).
- Dimairo M, Boote J, Julious SA, Nicholl JP, Todd S. Missing steps in a staircase: a qualitative study of the perspectives of key stakeholders on the use of adaptive designs in confirmatory trials. Trials 2015;16.
- Campbell G, Yue LQ. Statistical innovations in the medical device world sparked by the FDA. J Biopharm Stat 2016;26:3-16.
- Elsaser A, Regnstrom J, Vetter T, Koenig F, Hemmings RJ, Greco M, et al. Adaptive clinical trial designs for European marketing authorization: a survey of scientific advice letters from the European Medicines Agency. Trials 2014;15.
- Kairalla JA, Coffey CS, Thomann MA, Muller KE. Adaptive trial designs: a review of barriers and opportunities. Trials 2012;13.
- Stallard N. A confirmatory seamless phase II/III clinical trial design incorporating short-term endpoint information. Stat Med 2010;29:959-71.
- Stallard N, Todd S. Seamless phase II/III designs. Stat Methods Med Res 2011;20:623-34.
- Hatfield I, Allison A, Flight L, Julious SA, Dimairo M. Adaptive designs undertaken in clinical research: a review of registered clinical trials. Trials 2016;17.
- Parsons N, Stallard N, Parsons H, Wells P, Underwood M, Mason J, et al. An adaptive two-arm clinical trial using early endpoints to inform decision making: design for a study of sub-acromial spacers for repair of rotator cuff tendon tears. Trials 2019;20.
- Metcalfe A, Gemperlé Mannion E, Parsons H, Brown J, Parsons N, Fox J, et al. Protocol for a randomised controlled trial of Subacromial spacer for Tears Affecting Rotator cuff Tendons: a Randomised, Efficient, Adaptive Clinical Trial in Surgery (START:REACTS). BMJ Open 2020;10.
- Dawson J, Fitzpatrick R, Carr A. Questionnaire on the perceptions of patients about shoulder surgery. J Bone Joint Surg Br 1996;78:593-600.
- Dawson J, Rogers K, Fitzpatrick R, Carr A. The Oxford shoulder score revisited. Arch Orthop Trauma Surg 2009;129:119-23.
- Holtby R, Razmjou H. Measurement properties of the Western Ontario rotator cuff outcome measure: a preliminary report. J Shoulder Elbow Surg 2005;14:506-10.
- Brooks R. EuroQol: the current state of play. Health Policy 1996;37:53-72.
- Janssen MF, Pickard AS, Golicki D, Gudex C, Niewada M, Scalone L, et al. Measurement properties of the EQ-5D-5L compared to the EQ-5D-3L across eight patient groups: a multi-country study. Qual Life Res 2013;22:1717-27.
- Christiansen DH, Frost P, Falla D, Haahr JP, Frich LH, Svendsen SW. Responsiveness and minimal clinically important change: a comparison between 2 shoulder outcome measures. J Orthop Sports Phys Ther 2015;45:620-5.
- Beard DJ, Rees JL, Cook JA, Rombach I, Cooper C, Merritt N, et al. Arthroscopic subacromial decompression for subacromial shoulder pain (CSAW): a multicentre, pragmatic, parallel group, placebo-controlled, three-group, randomised surgical trial. Lancet 2018;391:329-38.
- Moeller AD, Thorsen RR, Torabi TP, Bjoerkman AS, Christensen EH, Maribo T, et al. The Danish version of the modified Constant-Murley shoulder score: reliability, agreement, and construct validity. J Orthop Sports Phys Ther 2014;44:336-40.
- Kamper S, Maher C, Mackay G. Global rating of change scales: a review of strengths and weaknesses and considerations for design. J Man Manip Ther 2009;17:163-70.
- Kolk A, Overbeek CL, de Groot JH, Nelissen RGHH, Nagels J. Reliability and discriminative accuracy of 5 measures for craniocaudal humeral position: an assessment on conventional radiographs. JSES International 2020;4:189-96.
- Kellgren JH, Lawrence JS. Radiological assessment of osteo-arthrosis. Ann Rheum Dis 1957;16:494-502.
- R Foundation n.d. www.r-project.org (accessed 9 October 2021).
- Haahr JP, Ostergaard S, Dalsgaard J, Norup K, Frost P, Lausen S, et al. Exercises versus arthroscopic decompression in patients with subacromial impingement: a randomised, controlled study in 90 cases with a one year follow up. Ann Rheum Dis 2005;64:760-4.
- Karthikeyan S, Kwong HT, Upadhyay PK, Parsons N, Drew SJ, Griffin D. A double-blind randomised controlled study comparing subacromial injection of tenoxicam or methylprednisolone in patients with subacromial impingement. J Bone Joint Surg 2010;92:77-82.
- Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomised trials. Lancet 2001;357:1191-4.
- Dimairo M, Coates E, Pallmann P, Todd S, Julious SA, Jaki T, et al. Development process of a consensus-driven CONSORT extension for randomised trials using an adaptive design. BMC Med 2018;16.
- Parsons H, Haque A. Sub-acromial spacer for Tears Affecting Rotator cuff Tendons: A Randomised, Efficient, Adaptive Clinical Trial in Surgery (START:REACTS): Statistical Analysis Plan. Warwick: University of Warwick; 2021.
- Whitehead J. Overrunning and underrunning in sequential clinical trials. Control Clin Trials 1992;13:106-21.
- Liu A, Hall WJ. Unbiased estimation following a group sequential test. Biometrika 1999;86:71-8.
- Todd S, Whitehead J, Facey KM. Point and interval estimation following a sequential clinical trial. Biometrika 1996;83:453-61.
- Cvetanovich GL, Waterman BR, Verma NN, Romeo AA. Management of the Irreparable Rotator Cuff Tear. J Am Acad Orthop Surg 2019;27:909-17.
- Hamada K, Yamanaka K, Uchiyama Y, Mikasa T, Mikasa M. A radiographic classification of massive rotator cuff tear arthritis. Clin Orthop Relat Res 2011;469:2452-60.
- Pincus T, Miles C, Froud R, Underwood M, Carnes D, Taylor SJ. Methodological criteria for the assessment of moderators in systematic reviews of randomised controlled trials: a consensus study. BMC Med Res Methodol 2011;11.
- Stallard N, Todd S. Sequential designs for phase III clinical trials incorporating treatment selection. Stat Med 2003;22:689-703.
- Verma N, Srikumaran U, Roden CM, Rogusky EJ, Lapner P, Neill H, et al. InSpace Implant Compared with Partial Repair for the Treatment of Full-Thickness Massive Rotator Cuff Tears. J Bone Joint Surg 2022;104:1250-62.
- McCulloch P, Altman DG, Campbell WB, Flum DR, Glasziou P, Marshall JC, et al. No surgical innovation without evaluation: the IDEAL recommendations. Lancet 2009;374:1105-12.
- Sedrakyan A, Campbell B, Merino JG, Kuntz R, Hirst A, McCulloch P. IDEAL-D: a rational framework for evaluating and regulating the use of medical devices. BMJ 2016;353.
- National Institute for Health and Care Excellence . Guide to the Methods of Technology appraisal 2013. Process and Methods Guides PMG9 2013. https://www.ncbi.nlm.nih.gov/books/NBK395867 (accessed 14 July 2022).
- Manca A, Hawkins N, Sculpher MJ. Estimating mean QALYs in trial-based cost-effectiveness analysis: the importance of controlling for baseline utility. Health Econ 2005;14:487-96.
- Sterne JA, White IR, Carlin JB, Spratt M, Royston P, Kenward MG, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ 2009;338.
- White IR, Horton NJ, Carpenter J, Pocock SJ. Strategy for intention to treat analysis in randomised trials with missing outcome data. BMJ 2011;342.
- White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 2011;30:377-99.
- Chen MH, Willan AR. Determining optimal sample sizes for multistage adaptive randomized clinical trials from an industry perspective using value of information methods. Clin Trials 2013;10:54-62.
- Willan A, Kowgier M. Determining optimal sample sizes for multi-stage randomized clinical trials using value of information methods. Clin Trials 2008;5:289-300.
- Lamb SE, Mistry D, Alleyne S, Atherton N, Brown D, Copsey B, et al. Aerobic and strength training exercise programme for cognitive impairment in people with mild to moderate dementia: the DAPA RCT. Health Technol Assess 2018;22.
- NHS England n.d. www.england.nhs.uk/national-cost-collection (accessed 10 January 2022).
- Curtis L, Burns A. Unit Costs of Health and Social Care 2020. Canterbury: Personal Social Services Research Unit, University of Kent; 2020.
- NHS England . 2019/20/National/Cost/Collection/Data/Publication 2022. www.england.nhs.uk/publication/2019-20-national-cost-collection-data-publication (accessed 10 January 2022).
- National Institute for Health and Care Excellence n.d. https://bnf.nice.org.uk (accessed 10 January 2022).
- Office for National Statistics . Annual Survey of Hours and Earnings: 2015 Provisional Results 2021. www.ons.gov.uk/employmentandlabourmarket/peopleinwork/earningsandworkinghours/bulletins/annualsurveyofhoursandearnings/2015provisionalresults (accessed 10 January 2022).
- NHS . 2020/21/National/Tariff/Payment/System 2020. www.england.nhs.uk/wp-content/uploads/2021/02/20-21_National-Tariff-Payment-System.pdf (accessed 10 January 2022).
- Dolan P. Modeling valuations for EuroQol health states. Med Care 1997;35:1095-108.
- van Hout B, Janssen MF, Feng YS, Kohlmann T, Busschbach J, Golicki D, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health 2012;15:708-15.
- Knowles CH, Booth L, Brown SR, Cross S, Eldridge S, Emmett C, et al. Non-drug therapies for the management of chronic constipation in adults: the CapaCiTY research programme including three RCTs. Programme Grants Appl Res 2021;9. https://doi.org/10.3310/pgfar09140.
- Rubin DB. Multiple Imputation for Nonresponse in Surveys. New York, NY: Wiley; 1987.
- Graham JW, Olchowski AE, Gilreath TD. How many imputations are really needed? Some practical clarifications of multiple imputation theory. Prev Sci 2007;8.
- Willan AR, Briggs AH, Hoch JS. Regression methods for covariate adjustment and subgroup analysis for non-censored cost-effectiveness data. Health Econ 2004;13:461-75.
- Davis BR, Hardy RJ. Data monitoring in clinical trials: the case for stochastic curtailment. J Clin Epidemiol 1994;47:1033-42.
- Lan KKG, Simon R, Halperin M. Stochastically curtailed tests in long-term clinical trials. Commun Stat C Sequen Analysis 1982;1:207-19.
- OrthoSpace Ltd . A Pivotal Study to Assess the InSpace™ Device for Treatment of Full Thickness Massive Rotator Cuff Tears 2022. https://clinicaltrials.gov/ct2/show/NCT02493660 (accessed 10 January 2022).
- Sully BGO, Julious SA, Nicholl J. An investigation of the impact of futility analysis in publicly funded trials. Trials 2014;15.
- Hermens HJ, Freriks B, Merletti R, Stegeman D, Blok J, Rau G, et al. European recommendations for surface electromyography. Roessingh Res Dev 1999;8:13-54.
- Hodges PW, Bui BH. A comparison of computer-based methods for the determination of onset of muscle contraction using electromyography. Electroencephalogr Clin Neurophysiol 1996;101:511-9.
- Ardic F, Kahraman Y, Kacar M, Kahraman MC, Findikoglu G, Yorgancioglu ZR. Shoulder impingement syndrome: relationships between clinical, functional, and radiologic findings. Am J Phys Med Rehabil 2006;85:53-60.
- Burks RT, Crim J, Brown N, Fink B, Greis PE. A prospective randomized clinical trial comparing arthroscopic single- and double-row rotator cuff repair: magnetic resonance imaging and early clinical evaluation. Am J Sports Med 2009;37:674-82.
- McCreesh K, Crotty J, Lewis J. Acromiohumeral distance measurement in rotator cuff tendinopathy: is there a reliable, clinically applicable method? A systematic review. Br J Sports Med 2015;49:298-305.
- Tempelaere C, Pierrart J, Lefevre-Colau M-M, Vuillemin V, Cuenod C-A, Hansen U, et al. Dynamic three-dimensional shoulder Mri during active motion for investigation of rotator cuff diseases. PLOS ONE 2016;11.
- Gumina S, Arceri V, Fagnani C, Venditto T, Catalano C, Candela V, et al. Subacromial space width: does overuse or genetics play a greater role in determining it? An MRI study on elderly twins. J Bone Joint Surg Br 2015;97:1647-52.
- Kalra N, Seitz AL, Boardman ND, Michener LA. Effect of posture on acromiohumeral distance with arm elevation in subjects with and without rotator cuff disease using ultrasonography. J Orthop Sports Phys Ther 2010;40:633-40.
- Cook JA, McCulloch P, Blazeby JM, Beard DJ, Marinac-Dabic D, Sedrakyan A, et al. IDEAL framework for surgical innovation 3: randomised controlled trials in the assessment stage and evaluations in the long-term study stage. BMJ 2013;346.
- Ergina PL, Barkun JS, McCulloch P, Cook JA, Altman DG. IDEAL Group . IDEAL framework for surgical innovation 2: observational studies in the exploration and assessment stages. BMJ 2013;346.
- McCulloch P, Cook JA, Altman DG, Heneghan C, Diener MK. IDEAL Group . IDEAL framework for surgical innovation 1: the idea and development stages. BMJ 2013;346.
- Jennison C, Turnbull BW. Group Sequential Methods with Applications to Clinical Trials. Boca Raton: Chapman & Hall/CRC; 2000.
- Galbraith S, Marschner IC. Interim analysis of continuous long-term endpoints in clinical trials with longitudinal outcomes. Stat Med 2003;22:1787-805.
- Engel B, Walstra P. Increasing precision or reducing expense in regression experiments by using information from a concomitant variable. Biometrics 1991;47:13-20.
- Slud E, Wei LJ. Two-sample repeated significance tests based on the modified Wilcoxon statistic. J Am Statist Assoc 1982;77:862-8.
- Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika 1983;70:659-63.
- Stallard N, Todd S, Ryan EG, Gates S. Comparison of Bayesian and frequentist group-sequential clinical trial designs. BMC Med Res Methodol 2020;20.
- Ryan EG, Lamb SE, Williamson E, Gates S. Bayesian adaptive designs for multi-arm trials: an orthopaedic case study. Trials 2020;21.
- Ryan EG, Stallard N, Lall R, Ji C, Perkins GD, Gates S. Bayesian group sequential designs for phase III emergency medicine trials: a case study using the PARAMEDIC2 trial. Trials 2020;21.
- Costa ML, Achten J, Bruce J, Tutton E, Petrou S, Lamb SE, et al. Effect of negative pressure wound therapy vs standard wound management on 12-month disability among adults with severe open fracture of the lower limb: The WOLLF Randomized Clinical Trial. JAMA 2018;319:2280-8.
- Costa ML, Achten J, Parsons NR, Rangan A, Edlin RP, Brown J, et al. UK DRAFFT – a randomised controlled trial of percutaneous fixation with kirschner wires versus volar locking-plate fixation in the treatment of adult patients with a dorsally displaced fracture of the distal radius. BMC Musculoskelet Disord 2011;12.
- Costa ML, Achten J, Griffin J, Petrou S, Pallister I, Lamb SE, et al. Effect of locking plate fixation vs intramedullary nail fixation on 6-month disability among adults with displaced fracture of the distal tibia: the UK FixDT Randomized Clinical Trial. JAMA 2017;318:1767-76.
- Achten J, Parsons NR, Edlin RP, Griffin DR, Costa ML. A randomised controlled trial of total hip arthroplasty versus resurfacing arthroplasty in the treatment of young patients with arthritis of the hip joint. BMC Musculoskelet Disord 2010;11.
- Beard D, Rees J, Rombach I, Cooper C, Cook J, Merritt N, et al. The CSAW Study (Can Shoulder Arthroscopy Work?) – a placebo-controlled surgical intervention trial assessing the clinical and cost effectiveness of arthroscopic subacromial decompression for shoulder pain: study protocol for a randomised controlled trial. Trials 2015;16.
- Beard D, Price A, Cook J, Fitzpatrick R, Carr A, Campbell M, et al. Total or Partial Knee Arthroplasty Trial – TOPKAT: study protocol for a randomised controlled trial. Trials 2013;14.
- Beard DJ, Davies LJ, Cook JA, MacLennan G, Price A, Kent S, et al. Total versus partial knee replacement in patients with medial compartment knee osteoarthritis: the TOPKAT RCT. Health Technol Assess 2020;24:1-98.
- Beard DJ, Davies LJ, Cook JA, MacLennan G, Price A, Kent S, et al. The clinical and cost-effectiveness of total versus partial knee replacement in patients with medial compartment osteoarthritis (TOPKAT): 5-year outcomes of a randomised controlled trial. Lancet 2019;394:746-56.
- Van Lancker K, Vandebosch A, Vansteelandt S. Improving interim decisions in randomized trials by exploiting information on short-term endpoints and prognostic baseline co-variates. Pharm Stat 2020;19:583-601.
- Baldi I, Azzolina D, Soriani N, Barbetta B, Vaghi P, Giacovelli G, et al. Overrunning in clinical trials: some thoughts from a methodological review. Trials 2020;21.
- Griffin DR, Dickenson EJ, Wall PD, Donovan JL, Foster NE, Hutchinson CE, et al. Protocol for a multicentre, parallel-arm, 12-month, randomised, controlled trial of arthroscopic surgery versus conservative care for femoroacetabular impingement syndrome (FASHIoN). BMJ Open 2016;6.
- Griffin DR, Dickenson EJ, Wall PDH, Realpe A, Adams A, Parsons N, et al. The feasibility of conducting a randomised controlled trial comparing arthroscopic hip surgery to conservative care for patients with femoroacetabular impingement syndrome: the FASHIoN feasibility study. J Hip Preserv Surg 2016;3:304-11.
- Tubeuf S, Yu G, Achten J, Parsons NR, Rangan A, Lamb SE, et al. Cost effectiveness of treatment with percutaneous Kirschner wires versus volar locking plate for adult patients with a dorsally displaced fracture of the distal radius: analysis from the DRAFFT trial. Bone Joint J 2015;97-B:1082-9.
- Petrou S, Parker B, Masters J, Achten J, Bruce J, Lamb SE, et al. Cost-effectiveness of negative-pressure wound therapy in adults with severe open fractures of the lower limb: evidence from the WOLLF randomized controlled trial. Bone Joint J 2019;101-B:1392-401.
- Maredza M, Petrou S, Dritsaki M, Achten J, Griffin J, Lamb SE, et al. A comparison of the cost-effectiveness of intramedullary nail fixation and locking plate fixation in the treatment of adult patients with an extra-articular fracture of the distal tibia: economic evaluation based on the FixDT trial. Bone Joint J 2018;100-B:624-33.
- Grant A. Stopping clinical trials early. BMJ 2004;329:525-6.
- Freedman LS, Spiegelhalter DJ. Comparison of Bayesian with group sequential methods for monitoring clinical trials. Control Clin Trials 1989;10:357-67.
- Dmitrienko A, Wang MD. Bayesian predictive approach to interim monitoring in clinical trials. Stat Med 2006;25:2178-95.
- Snowdon C, Brocklehurst P, Tasker R, Ward Platt M, Elbourne D. ‘You have to keep your nerve on a DMC.’ Challenges for Data Monitoring Committees in neonatal intensive care trials: Qualitative accounts from the BRACELET Study. PLOS ONE 2018;13.
- Griffin DR, Dickenson EJ, Wall PDH, Achana F, Donovan JL, Griffin J, et al. Hip arthroscopy versus best conservative care for the treatment of femoroacetabular impingement syndrome (UK FASHIoN): a multicentre randomised controlled trial. Lancet 2018;391:2225-35.
- Jeffreys H. Theory of Probability. Oxford: Oxford University Press; 1961.
Appendix 1 Adaptive design parameters and process for early stopping
The information required to trigger interim analyses and stopping boundaries for the test statistic for the group-sequential design are shown in Table 29; details of the calculation of the test statistics are given in Parsons et al. 61 The information accrued in the study was monitored monthly as 12-month outcome score data accumulated. The information depended on both the correlation between time points and the variance of the outcomes.
| Interim analysis | Information boundary required to trigger analysis (estimated no. of 12-month follow-up data points needed) | Lower boundary (li) | Upper boundary (ui) |
|---|---|---|---|
| 1 | 0.102 (50) | l1 = −0.706 | u1 = ∞ |
| 2 | 0.139 (70) | l2 = 0.581 | u2 = 3.090 |
When the information had reached that required to trigger the first interim analysis, data were extracted from the study database and the treatment effect, variance and test statistic were estimated by the study statistician and made available for the consideration of the DMC only. The test statistic was found to have crossed the lower (futility) boundary. After seeing these findings and summaries of study data, the DMC stated that ‘We recommend that recruitment is stopped with immediate effect, but follow-up continued for patients already enrolled in the study’. The TSC was informed and agreed with the DMC recommendation, and trial sites were asked to stop recruitment with immediate effect.
| Balloon | Control | ||
|---|---|---|---|
| No. of study participants providing OSS data | N12m | 23 | 24 |
| N6m | 41 | 45 | |
| N3m | 53 | 59 | |
| Correlations between OSS data | ρ 3m,6m | 0.728 | |
| ρ 3m,12m | 0.760 | ||
| ρ 6m,12m | 0.765 | ||
| Mean of OSS data | μ 12m | 24.72 | 30.44 |
| μ 6m | 28.42 | 31.87 | |
| μ 3m | 29.96 | 34.58 | |
| SD of OSS data | σ 12m | 11.66 | |
| σ 6m | 10.57 | ||
| σ 3m | 10.82 | ||
| Mean group difference of OSS data | ∇μ12m | −5.72 | |
| ∇μ6m | −3.45 | ||
| ∇μ3m | −4.62 | ||
| Information | I1 | 0.110 | |
| Test statistic (observed) | S1 | −0.881 | |
Recruitment
A total of 117 participants were recruited into the study when recruitment was closed; 61 (52%) participants were randomised to receive arthroscopic surgery alone and 56 (48%) participants were randomised to receive arthroscopy with the InSpace device. Table 31 shows the number of participants randomised at each site by allocation group.
| Site | Debridement-only (n = 61) | Debridement with InSpace balloon (n = 56) | All randomised (n = 117) |
|---|---|---|---|
| A | 7 (11.5) | 10 (17.9) | 17 (14.5) |
| B | 7 (11.5) | 3 (5.4) | 10 (8.5) |
| C | 2 (3.3) | 2 (3.6) | 4 (3.4) |
| D | 6 (9.8) | 3 (5.4) | 9 (7.7) |
| E | 0 (0) | 1 (1.8) | 1 (0.9) |
| F | 3 (4.9) | 3 (5.4) | 6 (5.1) |
| G | 4 (6.6) | 2 (3.6) | 6 (5.1) |
| H | 6 (9.8) | 5 (8.9) | 11 (9.4) |
| I | 6 (9.8) | 7 (12.5) | 13 (11.1) |
| J | 3 (4.9) | 0 (0) | 3 (2.6) |
| K | 1 (1.6) | 2 (3.6) | 3 (2.6) |
| L | 0 (0) | 1 (1.8) | 1 (0.9) |
| M | 0 (0) | 1 (1.8) | 1 (0.9) |
| N | 2 (3.3) | 4 (7.1) | 6 (5.1) |
| O | 1 (1.6) | 0 (0) | 1 (0.9) |
| P | 6 (9.8) | 6 (10.7) | 12 (10.3) |
| Q | 2 (3.3) | 0 (0) | 2 (1.7) |
| R | 3 (4.9) | 4 (7.1) | 7 (6) |
| S | 0 (0) | 0 (0) | 0 (0) |
| T | 0 (0) | 1 (1.8) | 1 (0.9) |
| U | 0 (0) | 0 (0) | 0 (0) |
| V | 2 (3.3) | 1 (1.8) | 3 (2.6) |
| W | 0 (0) | 0 (0) | 0 (0) |
| X | 0 (0) | 0 (0) | 0 (0) |
| Y | 0 (0) | 0 (0) | 0 (0) |
Primary outcome: Oxford Shoulder Score
Table 32 shows the study primary outcome, the OSS at each time point. Box plots illustrating the participants scores over time are shown in Figure 23.
| Follow-up point (months) | Statistic | Debridement-only (n = 61) | Debridement with InSpace balloon (n = 56) | All randomised (n = 117) |
|---|---|---|---|---|
| Baseline | n | 61 (100) | 56 (100) | 117 (100) |
| Missing | 0 (0) | 0 (0) | 0 (0) | |
| Mean (SD) | 21.7 (9.4) | 23.1 (8.5) | 22.4 (9.0) | |
| 3 | n | 59 (96.7) | 54 (96.4) | 113 (96.6) |
| Missing | 2 (3.3) | 2 (3.6) | 4 (3.4) | |
| Mean (SD) | 30.4 (11.2) | 25 (10.4) | 27.8 (11.1) | |
| 6 | n | 58 (95.1) | 54 (96.4) | 112 (95.7) |
| Missing | 3 (4.9) | 2 (3.6) | 5 (4.3) | |
| Mean (SD) | 33.3 (10.4) | 28.5 (11) | 31 (10.9) | |
| 12 | n | 59 (96.7) | 55 (98.2) | 114 (97.4) |
| Missing | 2 (3.3) | 1 (1.8) | 3 (2.6) | |
| Mean (SD) | 34.3 (11.1) | 30.3 (10.9) | 32.4 (11.2) |
FIGURE 23.
Box plot of the OSS at each allocation group and time point.
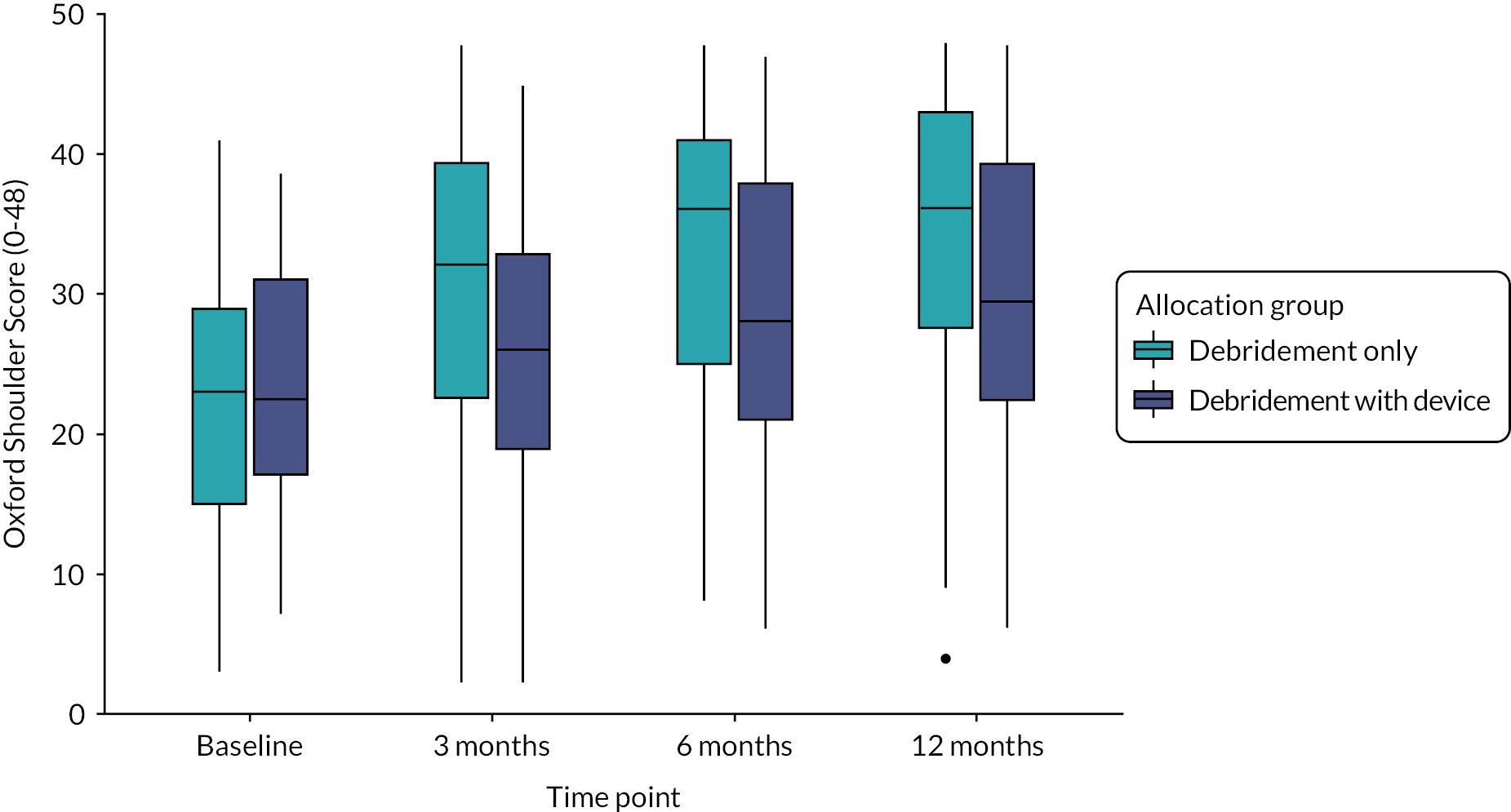
Subgroup analyses
Here, we report additional analyses for the pre-specified subgroups of tear size, age group and sex. Table 33 shows the descriptive statistics of each of the pre-planned subgroups.
| Subgroup | Debridement-only (N = 61) | Debridement with InSpace balloon (N = 56) | Overall (N = 117) | ||||
|---|---|---|---|---|---|---|---|
| n | OSS, mean (SD) | n | OSS, mean (SD) | n | OSS, mean (SD) | ||
| Sex | Male | 33 | 33.8 (12.1) | 34 | 34.5 (9.5) | 67 | 34.2 (10.8) |
| Female | 28 | 35 (10.1) | 22 | 24 (9.9) | 50 | 30.1 (11.3) | |
| Age group (years) | Under 70 | 33 | 31.6 (11.9) | 36 | 30.6 (11.6) | 69 | 31 (11.7) |
| 70 and over | 28 | 37.4 (9.6) | 20 | 29.9 (9.7) | 48 | 34.3 (10.2) | |
| Tear size | Large | 56 | 34.2 (10.9) | 55 | 30 (10.8) | 111 | 32.1 (11) |
| Medium or small | a | a | a | a | 6 | 37.5 (14.1) | |
Sex
Figure 24 show box plots of the OSS at 12 months postrandomisation by participant sex and allocation group. Table 34 shows the full details of the adjusted model of the OSS at 12 months with the allocation group and sex interaction term.
FIGURE 24.
Box plot of OSS at 12 months by sex and allocation group.
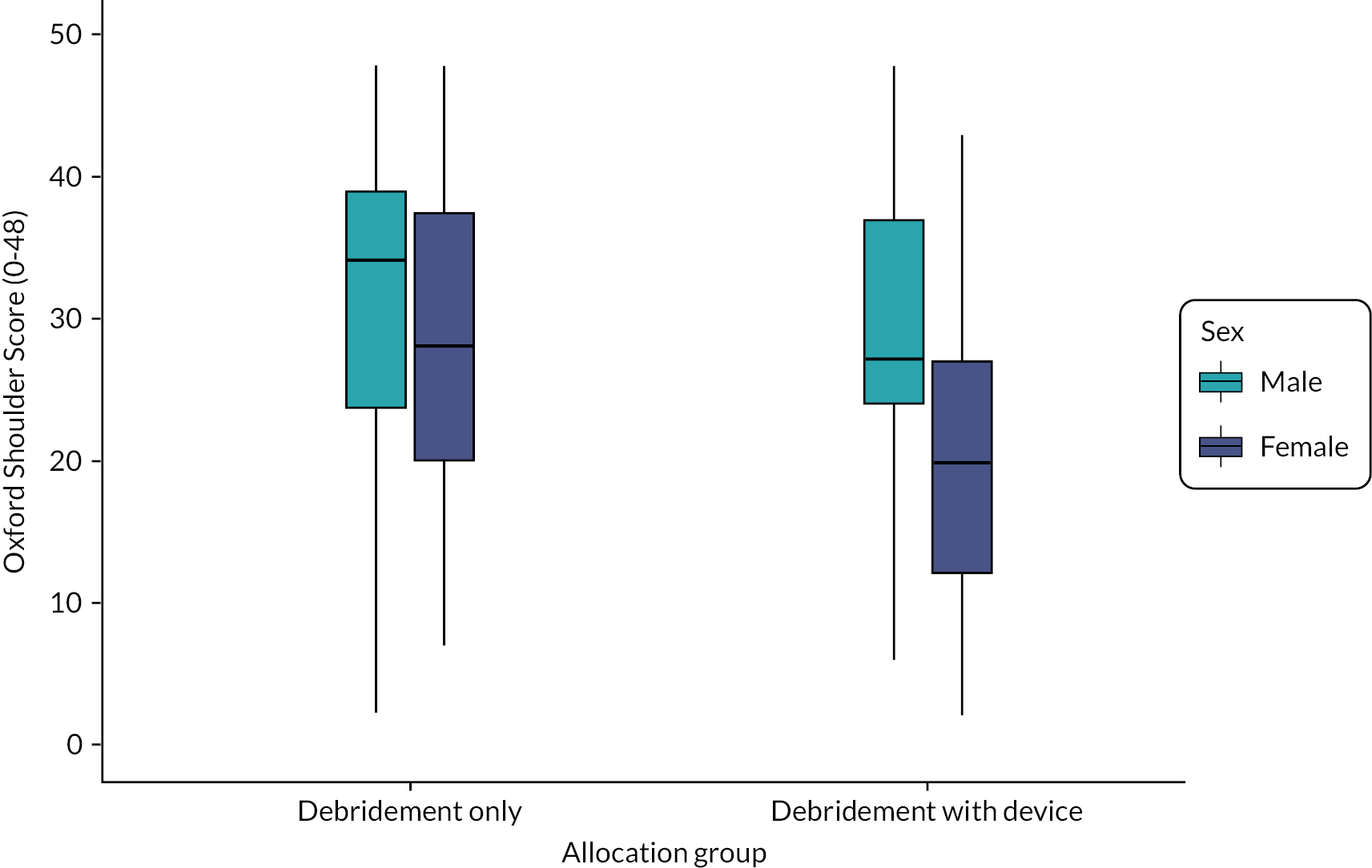
| Model coefficienta | 95% CI | p-value | ||
|---|---|---|---|---|
| Intervention group | Arthroscopy | 0 | − | 0.913 |
| Arthroscopy with device | −0.25 | (−4.8 to 4.2) | ||
| Baseline | OSS score | 0.6 | (0.3 to 0.8) | p < 0.001 |
| Sex | Male | 0 | – | 0.187 |
| Female | 3.5 | (−1.5 to 8.6) | ||
| Interaction term | Female*arthroscopy with device | −9.5 | (−16.5 to −2.6) | 0.01 |
| Tear size | Large | 0 | – | 0.607 |
| Medium or small | 2.2 | (−6.0 to 10.3) | ||
| Age group (years) | Under 70 | 0 | – | 0.788 |
| 70 and over | 0.5 | (−3.1 to 4.2) | ||
Here, the effects of age and tear size remain in line with the main efficacy model, and while the effect of the InSpace device is much reduced, it still favours the arthroscopy only group. Effects of sex are reversed with females now performing slightly better than males. However, the interaction term suggests that females have poorer outcome when in the device allocation group. This can be seen visually in Figure 25, which shows the group means with their 95% CIs for allocation group and participant sex.
FIGURE 25.
Means and 95% CIs of OSS at 12 months by sex and intervention.

Tear size
Table 35 shows the adjusted model results for the OSS at 12 months for the tear size subgroup and Figure 26 shows these results graphically. These results suggest that participants with small and medium tears have better OSS values at 12 months. However, the number of participants with small or medium tears is very low (n = 6, 5% of randomised participants), leading to an imprecise estimate of effect size.
| Variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | – | 0.023 |
| Debridement with InSpace balloon | −4.3 | (−8.1 to −0.8) | ||
| Baseline | OSS score | 0.6 | (0.34 to 0.8) | p < 0.001 |
| Sex | Male | 0 | – | 0.634 |
| Female | −1.0 | (−4.8 to 3.1) | ||
| Tear size | Large | 0 | – | 0.578 |
| Medium or small | 2.7 | (−6.8 to 11.9) | ||
| Interaction Term: tear size*intervention | Tear size: medium; intervention: debridement with InSpace balloon | 6.8 | (−14.9 to 28.8) | 0.553 |
| Age group (years) | Under 70 70 and over |
0.75 | (−3.0 to 4.7) | 0.707 |
FIGURE 26.
Interaction effects of the allocation group and tear size on the OSS at 12 months. Owing to the small number of medium or small tears in the device group, this CI could not be computed.

To further explore the effects of tear size; instead of using the tear size category, the two measurements of tear taken during surgery were used as continuous measures. The adjusted model results are shown in Tables 36 and 37 shows the summary statistics of the two tear measurements, which is broadly similar to the model using the categories of tear size.
| Model variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | – | 0.025 |
| Debridement with InSpace balloon | −4.2 | (−7.7 to −0.7) | ||
| Baseline | OSS score | 0.6 | (0.37 to 0.8) | <0.001 |
| Sex | Male | 0 | – | 0.709 |
| Female | −0.8 | (−7.7 to −0.7) | ||
| Age group (years) | Under 70 | 0 | – | 0.735 |
| 70 and over | 0.7 | (−3.0 to 4.5) | ||
| Tear size | Anteroposterior | −0.9 | (2.9 to 1.1) | 0.374 |
| Mediolateral | 1.9 | (−0.3 to 4.0) | 0.097 | |
| Tear measurement (cm) | Summary statistic | Debridement only (n = 61) | Debridement with InSpace balloon (n = 56) | Total (n = 117) |
|---|---|---|---|---|
| Anteroposterior | Minimum | 2.0 | 2.5 | 2.0 |
| Lower quartile | 3.5 | 3.1 | 3.4 | |
| Mean | 4.3 | 4.2 | 3.4 | |
| Upper quartile | 5.0 | 5.0 | 5.0 | |
| Maximum | 9.0 | 8.0 | 9.0 | |
| Mediolateral | Minimum | 1.6 | 1.0 | 1.0 |
| Lower quartile | 3.5 | 3.5 | 3.5 | |
| Mean | 4.3 | 4 | 4.1 | |
| Upper quartile | 5.0 | 4.5 | 5.0 | |
| Maximum | 7.0 | 8.5 | 8.5 |
The two tear sizes were modelled separately as interaction effects; the anteroposterior measurement is shown in Table 38 and the mediolateral in Table 39. Both interaction effect models show large changes to the observed effect of the allocation group, however, the overall effect of the InSpace device remains below that of the debridement only group.
| Model variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | – | 0.148 |
| Debridement with InSpace balloon | −9.3 | (−21.5 to −3.6) | ||
| Baseline | OSS score | 0.6 | (0.4 to 0.8) | < 0.001 |
| Sex | Male | 0 | – | 0.620 |
| Female | −1.0 | (−4.9 to −3.1) | ||
| Age group (years) | Under 70 | 0 | – | 0.8089 |
| 70 and over | 0.5 | (−3.2 to 4.3) | ||
| Tear size (cm) | Anteroposterior | −1.6 | (−4.1 to 1.1) | 0.213 |
| Mediolateral | 2.0 | (−0.2 to 4.2) | 0.077 | |
| Interaction term | Debridement with InSpace balloon*anteroposterior | 1.2 | (−1.7 to 4.0) | 0.401 |
| Model variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | – | 0.021 |
| Debridement with InSpace balloon | −16.7 | (−30.2 to −2.6) | ||
| Baseline | OSS score | 0.6 | (0.4 to 0.8) | < 0.001 |
| Sex | Male | 0 | – | 0.597 |
| Female | −1.1 | (−4.9 to 2.9) | ||
| Age group (years) | Under 70 | 0 | – | 0.921 |
| 70 and over | 0.2 | (−3.5 to 3.9) | ||
| Tear size (cm) | Anteroposterior | −1.0 | (−3.0 to 1.0) | 0.312 |
| Mediolateral | 0.6 | (−1.9 to 3.2) | 0.631 | |
| Interaction term | Debridement with InSpace balloon*mediolateral | 3.0 | (−0.3 to 6.2) | 0.073 |
Age group
Table 40 shows the results of the model including the interaction term of age group and allocation group, which is shown graphically in Figure 27. These results remain broadly consistent with the secondary efficacy model.
| Fixed effect variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | (−6.7 to 2.4) | 0.38 |
| Debridement with InSpace balloon | −2.1 | |||
| Baseline | OSS score | 0.6 | (0.3 to 0.8) | < 0.001 |
| Sex | Male | 0 | − | 0.511 |
| Female | −1.3 | (−5.2 to 2.7) | ||
| Tear size | Large | 2.6 | (−6.0 to 11.0) | 0.566 |
| Medium or small | ||||
| Age group (years) | Under 70 | 3.1 | (−2.1 to 8.4) | 0.255 |
| 70 and over | ||||
| Interaction term | Over 70*debridement with InSpace balloon | −5.3 | (−12.8 to 2.3) | 0.18 |
FIGURE 27.
Interaction effects of the allocation and age groups on the OSS at 12 months.

Secondary outcomes
Constant score
Descriptive statistics for the Constant score at each follow-up point are shown in Table 41 and Figure 28. The high level of missing data is due to the lack of in-person follow-up during the COVID-19 pandemic. Note that while there appears to be a large difference between allocation groups at 12-month follow-up, approximately 80% of the data at that time point are missing. Due to the high level of missing, the Constant score has not been modelled.
| Follow-up point | Statistic | Debridement only (n = 61) | Debridement with InSpace balloon (n = 56) |
All randomised (n = 117) |
|---|---|---|---|---|
| (1) Baseline | n (%) | 60 (99) | 54 (96) | 114 (97) |
| Missing (%) | 1 (2) | 2 (4) | 3 (3) | |
| Mean (SD) | 33.6 (13) | 29.9 (13.4) | 31.9 (13.2) | |
| (2) 3 months | n (%) | 45 (76) | 41 (73) | 86 (75) |
| Missing (%) | 14 (24) | 15 (27) | 29 (25) | |
| Mean (SD) | 46 (15.7) | 36.7 (21) | 41.6 (18.9) | |
| (3) 6 months | n (%) | 29 (50) | 26 (48) | 55 (49) |
| Missing (%) | 29 (50) | 28 (52) | 57 (51) | |
| Mean (SD) | 49 (18.6) | 45.2 (19.9) | 47.2 (19.1) | |
| (4) 12 months | n (%) | 11 (19) | 11 (20) | 22 (20) |
| Missing (%) | 48 (81) | 43 (80) | 91 (81) | |
| Mean (SD) | 63.6 (11.2) | 47.5 (13.2) | 55.5 (14.5) |
FIGURE 28.
Box plot of the Constant score at each follow-up point by allocation group.

Objective shoulder function
While performing the assessment for the Constant score, the exact measurements of participant shoulder function were also recorded. These data are summarised below in Table 42, Figure 29 and Figure 30. Owing to the high level of missing, these objective assessments have not been modelled.
| Domain | Time point (months) | Statistic | Debridement only (n = 61) | Debridement with InSpace balloon (n = 56) | All randomised (n = 117) |
|---|---|---|---|---|---|
| Pain-free abduction angle (degrees) | Baseline | n (%) | 58 | 51 | 109 |
| Mean (SD) | 76.3 (32.8) | 63.9 (22.2) | 70.5 (28.9) | ||
| 3 | n (%) | 45 | 41 | 86 | |
| Mean (SD) | 88.8 (36.6) | 69.7 (39.7) | 79.7 (39.1) | ||
| 6 | n (%) | 28 | 26 | 54 | |
| Mean (SD) | 97.5 (34.5) | 87.8 (41.9) | 92.9 (38.2) | ||
| 12 | n (%) | 12 | 11 | 23 | |
| Mean (SD) | 124.1 (37) | 87.1 (32.1) | 106.4 (38.9) | ||
| Pain-free flexion angle (degrees) | Baseline | n (%) | 58 | 51 | 109 |
| Mean (SD) | 74.1 (25.1) | 67.8 (29.8) | 71.1 (27.4) | ||
| 3 | n (%) | 45 | 41 | 86 | |
| Mean (SD) | 96.6 (36.1) | 84.2 (44.7) | 90.7 (40.7) | ||
| 6 | n (%) | 28 | 26 | 54 | |
| Mean (SD) | 103.9 (30.4) | 100.3 (46.4) | 102.2 (38.6) | ||
| 12 | n (%) | 12 | 11 | 23 | |
| Mean (SD) | 139.1 (26.4) | 98.8 (40.1) | 119.8 (38.8) |
FIGURE 29.
Box plot of shoulder flexion angle by allocation group and time point.
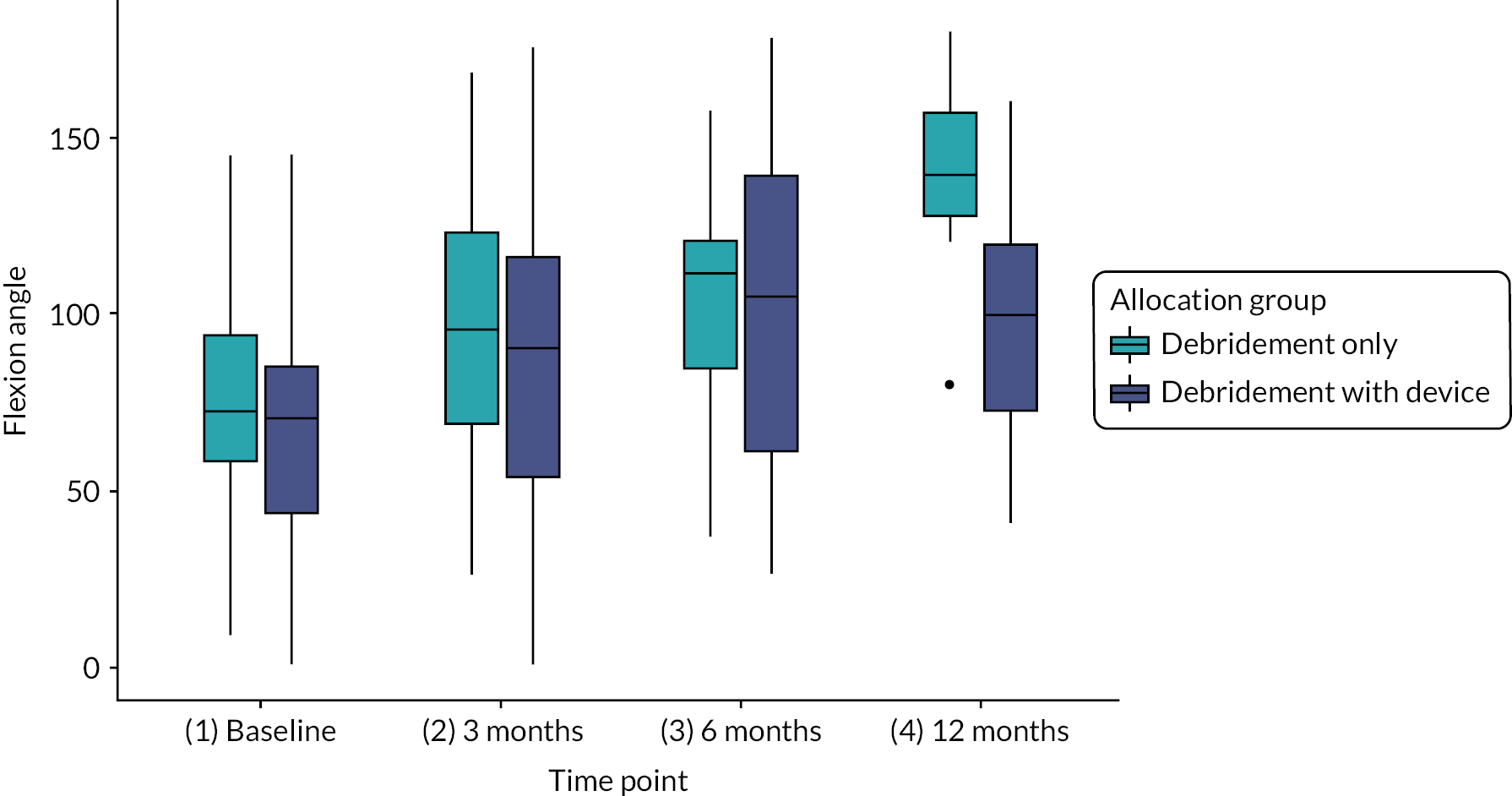
FIGURE 30.
Box plot of shoulder abduction angle by allocation group and time point.
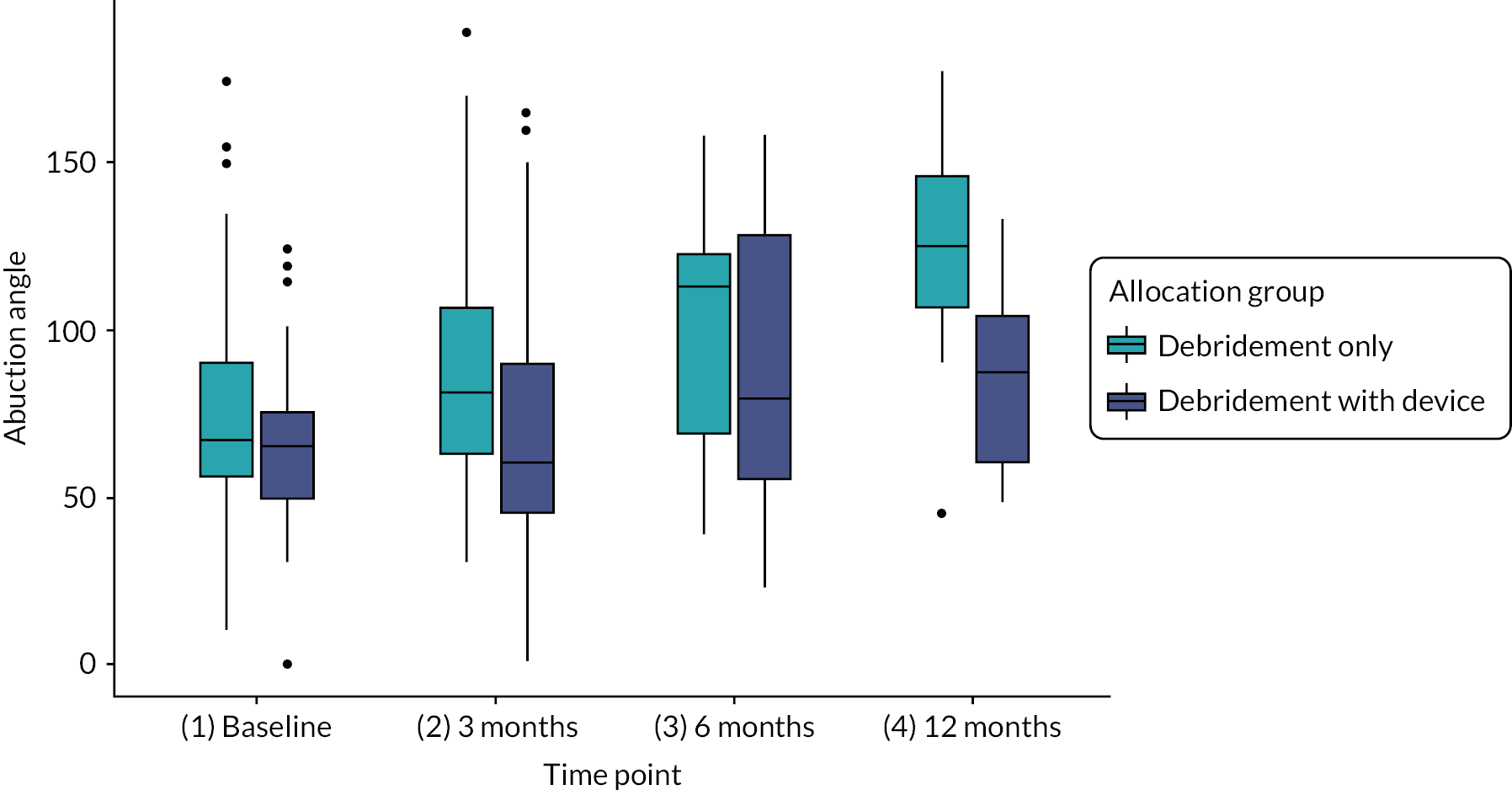
Western Ontario Rotator Cuff Index
The WORC index is a patient-reported quality-of-life outcome measure designed for patients with rotator cuff disease. Scores were calculated on a 0–100 scale such that higher score mean better function. The summaries of the data are shown in Table 43 and Figure 31.
| Follow-up point (months) | Statistic | Debridement only (n = 61) | Debridement with InSpace balloon (n = 56) | All randomised (n = 117) |
|---|---|---|---|---|
| Baseline | n (%) | 61 (100) | 55 (98) | 116 (99) |
| Missing (%) | 0 (0) | 1 (2) | 1 (1) | |
| Mean (SD) | 34.4 (14.2) | 33.7 (13.1) | 34.1 (13.6) | |
| 3 | n (%) | 56 (92) | 52 (93) | 108 (92) |
| Missing (%) | 5 (8) | 4 (7) | 9 (8) | |
| Mean (SD | 54.8 (24.5) | 40.2 (19.2) | 47.8 (23.2) | |
| 6 | n (%) | 53 (87) | 52 (93) | 105 (90) |
| Missing (%) | 8 (13) | 4 (7) | 12 (10) | |
| Mean (SD) | 60.2 (25.7) | 49.1 (22.6) | 54.7 (24.7) | |
| 12 | n (%) | 56 (92) | 51 (91) | 107 (92) |
| Missing (%) | 5 (8) | 5 (9) | 10 (9) | |
| Mean (SD) | 61.6 (25.7) | 51.7 (23.5) | 56.9 (25.1) |
FIGURE 31.
Box plot of WORC score by allocation group and time point.
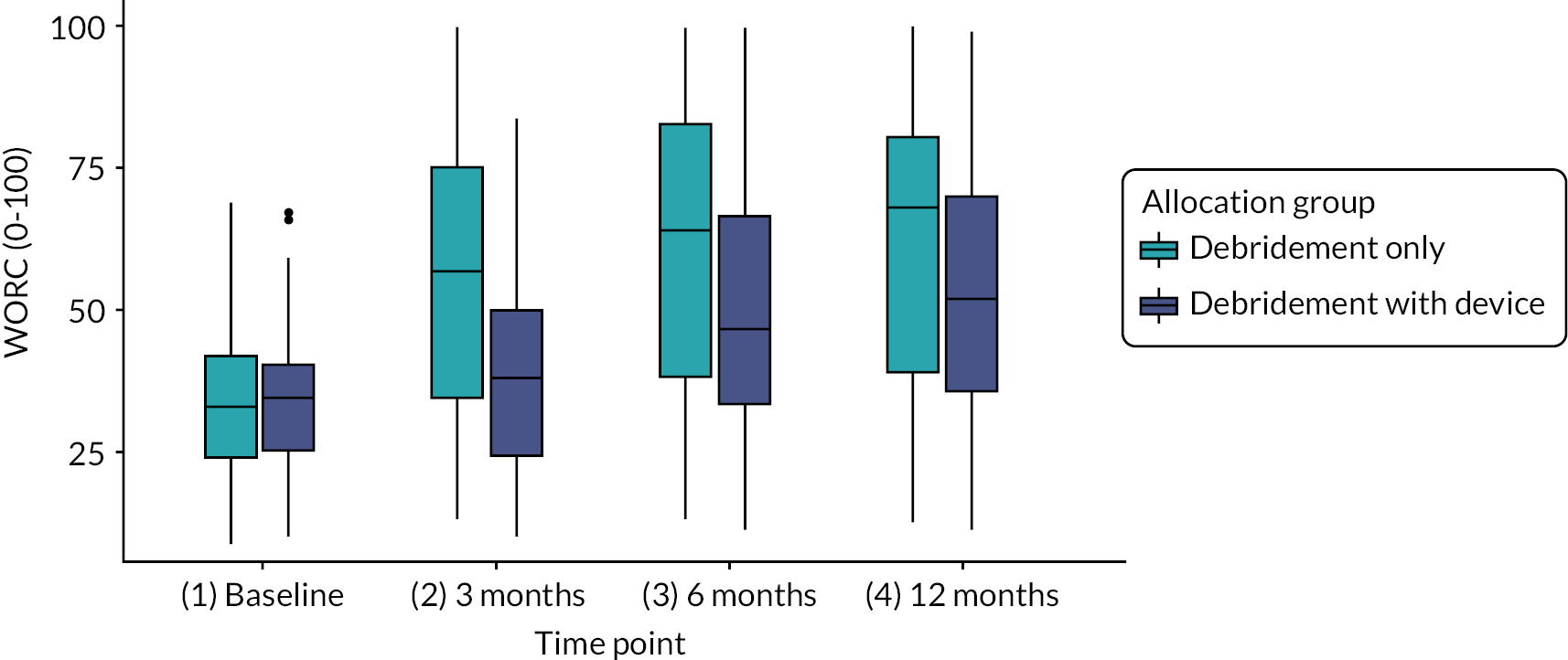
Table 44 shows the results of the adjusted model of the WORC score at 12 months postrandomisation. The model is consistent with the OSS models, with the effect of the InSpace device reducing shoulder function at the 12-month follow-up point.
| Model variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | – | 0.055 |
| Debridement with InSpace balloon | −8.4 | (−16.7 to −0.1) | ||
| Baseline | WORC score | 0.75 | (0.4 to 1.1) | < 0.001 |
| Sex | Male | 0 | – | 0.74 |
| Female | 1.5 | (−7.3 to 11.1) | ||
| Tear size | Large | 0 | – | 0.147 |
| Medium or small | 14.4 | (−4.94 to 33.1) | ||
| Age group (years) | Under 70 | 0 | – | 0.482 |
| 70 and over | 3.3 | (−5.7 to 12.5) | ||
EQ-5D-5L
The EQ-5D-5L is a validated quality-of-life utility score, calibrated such that 1 represents perfect health and 0 represents death. Descriptive statistics of the EQ-5D-5L by allocation group and time point are shown in Table 45 and Figure 32. The adjusted model results for the EQ-5D-5L can be found in Table 46.
| Follow-up point (months) | Statistic | Debridement only (n = 61) | Debridement with InSpace balloon (n = 56) | All randomised (n = 117) |
|---|---|---|---|---|
| Baseline | n (%) | 61 (100) | 56 (100) | 117 (100) |
| Missing (%) | 0 (0) | 0 (0) | 0 (0) | |
| Mean (SD) | 0.501 (0.258) | 0.486 (0.247) | 0.494 (0.251) | |
| 3 | n (%) | 59 (97) | 55 (98) | 114 (97) |
| Missing (%) | 2 (3) | 1 (2) | 3 (3) | |
| Mean (SD) | 0.632 (0.237) | 0.556 (0.275) | 0.596 (0.257) | |
| 6 | n (%) | 58 (95) | 54 (96) | 112 (96) |
| Missing (%) | 3 (5) | 2 (4) | 5 (4) | |
| Mean (SD) | 0.666 (0.253) | 0.592 (0.254) | 0.63 (0.255) | |
| 12 | n (%) | 58 (95) | 55 (98) | 113 (97) |
| Missing (%) | 3 (5) | 1 (2) | 4 (3) | |
| Mean (SD) | 0.667 (0.287) | 0.590 (0.286) | 0.63 (0.288) |
FIGURE 32.
Box plot of EQ-5D-5L score by allocation group and time point.
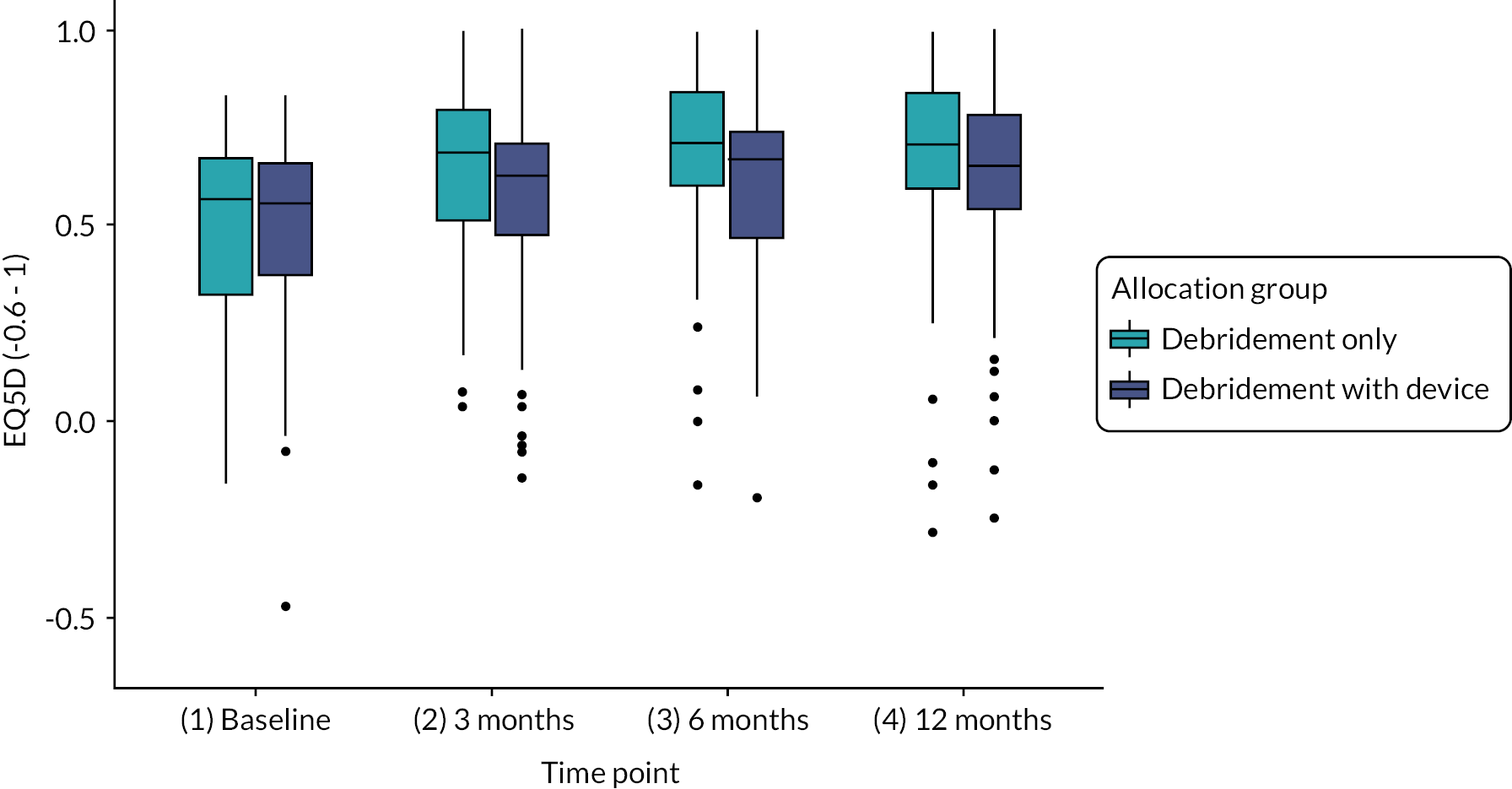
| Fixed effect variables | Coefficient | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | – | 0.239 |
| Debridement with InSpace balloon | −0.056 | (−0.150 to 0.035) | ||
| Baseline | EQ-5D-5L score | 0.579 | (0.380 to 0.777) | < 0.001 |
| Sex | Male | 0 | – | 0.896 |
| Female | −0.007 | (−0.105 to 0.095) | ||
| Tear size | Large | 0 | – | 0.593 |
| Medium or Small | −0.059 | (−0.274 to 0.152) | ||
| Age group (years) | Under 70 | 0 | – | 0.765 |
| 70 and over | 0.015 | (−0.082 to 0.119) | ||
Patient reported change
In addition to answering the patient-reported outcome measures, participants were also asked to report about their general shoulder function as compared to before their operation (PGIC). Participants were asked to report their assessment of their change in shoulder function and the more specific assessment of their ‘activity limitations, symptoms, emotions and overall quality of life’ since their operation. Table 47 shows the summary of responses of the change in shoulder function. Table 48 shows the response to the more specific prompt.
| Responses | Debridement only n (%) | Debridement with InSpace balloon n (%) | Total n (%) |
|---|---|---|---|
| Substantially better | 24 (39) | 16 (29) | 40 (34) |
| Moderately better | 17 (28) | 19 (34) | 36 (31) |
| No difference | 12 (20) | 7 (13) | 19 (16) |
| Moderately worse | 2 (3) | 6 (11) | 8 (7) |
| Substantially worse | 4 (7) | 7 (13) | 11 (9) |
| Missing | 2 (3) | 1 (2) | 3 (3) |
| Responses | Debridement only n (%) | Debridement with InSpace balloon n (%) | Total n (%) |
|---|---|---|---|
| No change or worse | 7 (12) | 14 (25) | 21 (18) |
| Almost the same | 5 (8) | 6 (11) | 11 (9) |
| A little better, no noticeable change | 6 (10) | 3 (5) | 9 (8) |
| Somewhat better, change has not made a difference | 4 (7) | 6 (11) | 10 (9) |
| Moderately better, slight but noticeable change | 7 (12) | 6 (11) | 13 (11) |
| Better, definite improvement with a difference | 17 (28) | 13 (23) | 30 (26) |
| Considerable improvement making a huge difference | 13 (21) | 7 (13) | 20 (17) |
| Missing | 2 (3) | 1 (2) | 3 (3) |
Exploratory and sensitivity analyses
Relationship between the OSS and the Constant score
The original primary outcome was the Constant score (see main text). To confirm that the OSS was a suitable alternative, the correlations between the Constant score and the OSS were calculated.
The OSS is scored between 0 and 48, hence we rescaled scores to a 0–100 range to allow direct comparison to the Constant score (range is 0–100). These results suggest that there is a moderate to strong correlation between the two outcomes at each time point (see Table 49).
| Months of follow-up | Participants analysed (n, % of randomised) | Pearson’s correlation | 95% CI |
|---|---|---|---|
| Baseline | 114 (97.4) | 0.65 | 0.53 to 0.75 |
| 3 | 85 (73.9) | 0.80 | 0.71 to 0.87 |
| 6 | 56 (50.0) | 0.78 | 0.65 to 0.86 |
| 12 | 22 (19.3) | 0.74 | 0.47 to 0.89 |
Participants with repairable tears
This analysis was added (prospectively, before recruitment closed) to investigate if there were any observable differences between participants who were randomised into the study and those who were excluded because their cuff tears were repairable.
Table 50 shows the participants baseline information for the randomised group and the excluded intraoperative group due to having a repairable tear.
| Baseline data | Randomised (n = 117) | Repairable tear (n = 43) | Mean difference with 95% CI | p-valuea | |
|---|---|---|---|---|---|
| Age group (years) | Under 70 70 or older Mean age |
69 (59.0) | 27 (62.8) | – | – |
| 48 (41.0) | 16 (37.2) | – | – | ||
| 66.9 (8.3) | 66.2 (7.3) | – | – | ||
| Sex | Female Male |
50 (42.7) | 11 (25.6) | – | – |
| 67 (57.3) | 32 (74.4) | – | – | ||
| Forward flexion without pain angle | 71.1 (27.4) | 80.1 (33.0) | – | – | |
| Abduction without pain angle | 70.5 (28.9) | 77.9 (30.2) | – | – | |
| Constant score | n (%) | 114 (97.4) | 43 (100) | – | – |
| Mean (SD) | 31.9 (13.2) | 35.7 (14.5) | 3.8 (−1.2 to 8.9) | 0.139 | |
| OSS | n (%) | 117 (100) | 43 (100) | – | – |
| Mean (SD) | 22.4 (9.0) | 24.4 (7.9) | 2.1 (−5.0 to 0.9) | 0.165 | |
A t-test was performed between the randomised group and the participants with repairable rotator cuff tears that were excluded intraoperatively. The result showed that the repairable tear group had higher Constant score by 3.8 points, and a higher score for the OSS by 2.1 points. Neither test results showed statistical difference.
Tables 51 and 52 show the linear regression model results for the OSS and the Constant score, respectively.
| Fixed effect variables | Coefficient | 95% CI | p-value | |
|---|---|---|---|---|
| Group | Randomised | 0 | – | 0.456 |
| Repairable tear | 1.1 | (−1.8 to 4.0) | ||
| Sex | Male | 0 | – | < 0.001 |
| Female | −6.3 | (−9.0 to −3.7) | ||
| Age group (years) | Under 70 | 0 | – | 0.002 |
| 70 and over | 3.2 | (0.6 to 5.8) | ||
| Fixed effect variables | Coefficient | 95% CI | p-value | |
|---|---|---|---|---|
| Group | Randomised | 0 | – | 0.252 |
| Repairable tears | 2.7 | (2.0 to 7.4) | ||
| Sex | Male | 0 | – | 0.001 |
| Female | −7.3 | (−11.6 to −3.0) | ||
| Age group (years) | Under 70 | 0 | – | 0.027 |
| 70 and over | 4.8 | (0.6 to 8.9) | ||
Large tears
At the request of the oversight committees, a sensitivity analyses restricted to only those participants who had large tears was conducted. However, this reduction in the data set resulted in the mixed-effects model to fail (singular boundary). Hence, a linear regression model was then fit without the site term and the results are shown in Table 53.
| Fixed effect variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | − | 0.0139 |
| Debridement with InSpace balloon | −4.6 | (−8.2 to −0.9) | ||
| Baseline | OSS score | 0.6 | (0.3. 0.8) | < 0.001 |
| Sex | Male | 0 | − | 0.400 |
| Female | −1.7 | (−5.7 to 2.3) | ||
| Age group (years) | Under 70 | 0 | − | 0.568 |
| 70 and over | 1.1 | (−2.7 to 4.9) | ||
Follow-up collected out of visit window
There was a total of 13 participants who had follow-up data recorded outside the target window for data collection. There were four participants outside of the 3-month window; eight outside of the 6-month window, and one outside of the 12-months window. A sensitivity analyses was carried out to observe if this affected the model results. The results obtained are shown in Table 54 and can be seen to be consistent for the adjusted efficacy model.
| Fixed effect variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | – | 0.034 |
| Debridement with InSpace balloon | −4.0 | (−7.6 to −0.4) | ||
| Baseline | OSS score | 0.6 | (0.4 to 0.8) | < 0.001 |
| Sex | Male | 0 | – | 0.554 |
| Female | −1.2 | (−5.1 to 2.9) | ||
| Tear size | Large | 0 | − | 0.335 |
| Medium or Small | 4.2 | (−4.4 to 12.3) | ||
| Age group (years) | Under 70 | 0 | – | 0.836 |
| 70 and over | 0.4 | (−3.3 to 4.3) | ||
Acromiohumeral distance analysis
Acromiohumeral distance and reparable tears
As a larger than anticipated number of registered patients were excluded during surgery because their rotator cuff tears were found to be reparable, we investigated if there were other radiological features to help identify if tears were reparable. The AHD was measured for each participant registered into the trial, so was investigated as a potential feature.
Baseline scans for each participant were used to measure the AHD. Each measurement was extracted by two separate readers and the intraobserver agreement rating (intracluster correlation coefficient) was calculated as 0.59 with 95% CI 0.34 to 0.73. This indicates moderate reliability for the AHD measures.
Table 55 shows the AHD for registered participants who were found to have a reparable tear intraoperatively (i.e. were excluded from randomisation) and the AHD for participants who were found to have a repairable tear (i.e. were randomised into the trial). Here, it can be seen that the reparable tears have statistically significantly larger AHDs.
| AHD (mm) | Repairable tear (excluded) N = 43 | Irreparable tear (randomised) N = 117 | Mean difference (95% CI) | p-value |
|---|---|---|---|---|
| n valid (% of group) | 28 (65) | 93 (80) | ‐ | ‐ |
| Mean (SD) | 7.9 (2.0) | 6.8 (2.4) | 1.1 (0.1 to 2.1) | 0.03 |
Acromiohumeral distance and shoulder function
An exploratory analysis to investigate the relationship between AHD at baseline and OSS at 12-months was conducted. A simple linear regression model showed a positive relationship, with OSS scores increasing 0.7 points for every mm increase in AHD (95% CI −0.087 to 1.420; p = 0.0823). As shown in Table 56, adding AHD into the adjusted model did not change the effect of allocation group.
| Fixed effect variables | Coefficienta | 95% CI | p-value | |
|---|---|---|---|---|
| Intervention group | Debridement-only | 0 | – | 0.019 |
| Debridement with InSpace balloon | −5.2 | (−9.5 to −0.9) | ||
| Baseline OSS | 0.4 | (0.2 to 0.7) | ||
| Sex | Male | 0 | – | 0.738 |
| Female | −0.8 | (−5.6 to 4.0) | ||
| Tear size | Large | 0 | – | 0.094 |
| Medium or small | 0.03 | (−9.8 to 9.84) | ||
| Age group (years) | Under 70 | 0 | – | 0.618 |
| 70 and over | 1.1 | (3.4 to 5.7) | ||
| AHD (mm) | 0.40 | (−0.5 to 1.3) | ||
COVID-19 sensitivity analysis
This sensitivity analysis was to assess whether the COVID-19 pandemic had any impact on the primary analysis results. To check this question, data from participants who had completed the 12-month primary outcome before the first UK national lockdown on the 23 of March 2020 were compared with the remaining participants with follow-ups during the COVID-19 lockdown (see Table 57).
| Outcome | Statistic | Follow-up in/after COVID-19 (n = 91) |
Follow-up complete before COVID-19 (n = 26) |
All follow-up data (n = 117) |
|---|---|---|---|---|
| Constant score | n (%) | 2 (2.2) | 20 (76.9) | 22 (18.8) |
| Mean (SD) | 62 (21.2) | 54.9 (14.3) | 55.5 (14.5) | |
| Missing (%) | 89 (97.8) | 6 (23.1) | 95 (81.2) | |
| OSS | n (%) | 89 (97.8) | 25 (96.2) | 114 (97.4) |
| Mean (SD) | 31.7 (11.5) | 34.7 (9.6) | 32.4 (11.2) | |
| Missing (%) | 2 (2.2) | 1 (3.8) | 3 (2.6) | |
| EQ-5D-5L | n (%) | 88 (96.7) | 25 (96.2) | 113 (96.5) |
| Mean (SD) | 0.609 (0.311) | 0.704 (0.167) | 0.63 (0.288) | |
| Missing (%) | 3 (3.3) | 1 (3.8) | 4 (3.4) |
Table 58 shows the results of the sensitivity analyses, which suggest that COVID-19 did not have a significant impact on the participants primary outcome at 12-months for either the OSS or the EQ-5D-5L. The Constant score could not be collected during lockdown, so could not be assessed for change.
| Outcome | Coefficient | 95% CI | p-value | |
|---|---|---|---|---|
| OSS | During COVID-19 | 0 | – | 0.233 |
| Pre-COVID-19 | 3.0 | −2.0 to 8.0 | ||
| EQ-5D-5L | During COVID-19 | 0 | – | 0.143 |
| Pre-COVID-19 | 0.095 | −0.032 to 0.224 | ||
Appendix 2 Additional health economics information
Table 59 reports the frequency and average amounts of resource use by cost category. The data were not suitable for reporting at granularity other than what has been provided as the type of health resource use varied and also contained free text. However, the unit costs and sources for these are provided in Table 60 which informs the type of health resource used. Where unit costs were not available for 2021 prices, these were inflated using the NHS Hospital and Community Health Services Pay and Prices Index. 103
Table 61 is a summary of the numbers of missing items for each questionnaire.
| Resource category (unit) | Debridement with InSpace balloon (n = 56) | Debridement only (n = 61) |
|---|---|---|
| Inpatient care – hospital admission, n (%) post baseline to 12 months | 9 (16) | 6 (10) |
| Specialty codea | ||
| 1 | 2 (4) | 1 (2) |
| 2 | 2 (4) | 4 (7) |
| 4 | 8 (14) | 2 (3) |
| 97 | 4 (7) | 6 (10) |
| Inpatient care – hospital admission, n (%) | ||
| Baseline | Not collected/NA | Not collected/NA |
| 3 months | 7 (13) | 3 (5) |
| 6 months | 9 (16) | 4 (7) |
| 12 months | 8 (14) | 6 (10) |
| Outpatient care – at least one visit/contact, n (%): post baseline to 12 months | 42 (75) | 41 (67) |
| Specialty codeb | ||
| 1 | 15 (27) | 13 (21) |
| 2 | 1 (2) | 0 (0) |
| 3 | 3 (5) | 1 (2) |
| 4 | 3 (5) | 4 (7) |
| 6 | 1 (2) | 0 (0) |
| 97 | 19 (34) | 23 (38) |
| Outpatient care – at least one visit/contact, n (%): post baseline to 12 months | 33 (59) | 29 (48) |
| Baseline | Not collected/NA | Not collected/NA |
| 3 months | 33 (59) | 29 (48) |
| 6 months | 17 (30) | 20 (33) |
| 12 months | 22 (39) | 19 (31) |
| Physiotherapy, n (%): post baseline to 12 months; mean number of contacts (SD) | 55 [8.2 (4.8)] | 56 [6.4 (4.8)] |
| Baseline | Not collected/NA | Not collected/NA |
| 3 months | 55 (98) | 56 (92) |
| 6 months | 39 (70) | 37 (61) |
| 12 months | 20 (36) | 17 (28) |
| MRI at least one visit/contact, n (%): post baseline to 12 months | 11 (20) | 7 (11) |
| Baseline | 11 (20) | 6 (10) |
| 3 months | 10 (18) | 7 (11) |
| 6 months | 0 | 0 |
| 12 months | 0 | 0 |
| Community health services (at least one contact), n (%) post baseline to 12 months | 11 (20) | 9 (15) |
| Baseline | Not collected/NA | Not collected/NA |
| 3 months | 10 (18) | 9 (15) |
| 6 months | 11 (20) | 4 (7) |
| 12 months | 10 (18) | 7 (11) |
| PSS (at least one visit/contact), n (%): post baseline to 12 months | 1 (2) | 4 (7) |
| Baseline | Not collected/NA | Not collected/NA |
| 3 months | 0 | 4 (7) |
| 6 months | 1 (2) | 1 (2) |
| 12 months | 0 | 1 (2) |
| Analgesic use (at least one use), n (%): post baseline to 12 months | 42 (75) | 52 (85) |
| Baseline | 41 (73) | 51 (84) |
| 3 months | 37 (66) | 38 (62) |
| 6 months | 34 (61) | 38 (62) |
| 12 months | 30 (54) | 30 (49) |
| Other medications (at least 1 type/use), n (%): post baseline to 12 months | 16 (29) | 25 (41) |
| Time off work – no. of patients with at least 1 day off, n (%): post baseline to 12 months (mean number of contacts, SD) | 79 (76) | 64 (75) |
| Baseline | Not collected/NA | Not collected/NA |
| 3 months | 20 (36) [77 (65)] | 15 (25) [53 (33] |
| 6 months | 11 (20) [103 (102)] | 9 (15) [107 (46)] |
| 12 months | 5 (9) [39 (43)] | 1 (2) [30 (n/c)] |
| Cost category | Resource item | Unit cost (£) | Unit of analysis | Source |
|---|---|---|---|---|
| Intervention arm | The balloon procedure codes to an HRG grouper of HN52A | 5378 | Per patient procedure | www.nice.org.uk/guidance/ipg558 (2021) |
| The control (standard care) procedure codes to a HRG grouper of HN53A | 3589 | Per patient procedure | www.nice.org.uk/guidance/ipg558 (2021) | |
| Inpatient admission | Hospital stay | 587 | Day visit | NHS Digital National schedule of reference costs 2016–17. Available: https://improvement.nhs.uk/resources/reference-costs/ |
| Outpatient | General medical ward | 378 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trusta |
| MRI | 147.5 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Gastrointestinal – colonoscopy | 116 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Cancer | 147 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Physiotherapy | 65 | Contact/consultancy | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Surgery | 140 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Ophthalmology | 95 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Accident and emergency | 117 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Rheumatology | 147 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Neurology | 142 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Cardiology | 122 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Gynaecology | 147 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Orthopaedics | 140 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Dental | 177 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Ear, nose and throat | 107 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Respiratory | 129 | Day visit | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Community health service | Physiotherapist – Band 5 | 36 | Per hour | Unit costs of Health and Social care (2020)a |
| Sports therapy massage | 36 | Per hour | Unit costs of Health and Social care (2020) | |
| Osteopath | 45 | Per hour | Unit costs of Health and Social care (2020) | |
| Orthopaedic nurse – Band 6 | 49 | Per hour | Unit costs of Health and Social care (2020) | |
| Personal and social care services | Support worker mental health CPN/CMHN – office | 48 | Per hour | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trusta |
| Equipment and adaptations various aidsb | Per item | Range: 4.8–930.0 | Unit costs of health and social care (2014–15) Page 199, 12.1a | |
| Care support worker – home | 24 | Per hour | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Occupational therapist – office – hospital | 45 | Per hour | National Cost Collection: Schedule of NHS costs – 2018–19; NHS trust and NHS Foundation Trust | |
| Medications | Paracetamol | 0.86 | Per pack of 12 | https://bnf.nice.org.uk/medicinal-forms (2021) |
| Co-codamol | 2.52 | Per pack of 32 | https://bnf.nice.org.uk/medicinal-forms (2021) | |
| Codeine | 1.15 | Per pack 28 | https://bnf.nice.org.uk/medicinal-forms (2021) | |
| Tramadol | 9.68 | Per pack 60 | https://bnf.nice.org.uk/medicinal-forms (2021) | |
| Other prescription medications | Various | 0.12–82.00 | https://bnf.nice.org.uk/medicinal-forms (2021) | |
| Time taken off work | National average | 530.0 | Per 37.5-hour week | www.ons.gov.uk/employmentandlabourmarket/peopleinwork/earningsandworkinghours/bulletins/annualsurveyofhoursandearnings/2015provisionalresults |
| Resource-use item | Time point (months) | Debridement only (n = 61) | Debridement with InSpace balloon (n = 56) |
||
|---|---|---|---|---|---|
| Completed, n (%) | Missing, n (%) | Completed,a n (%) | Missing,b n (%) | ||
| Physiotherapy sessions | 3 | 59 (97) | 2 (3) | 56 (100) | 0 (0) |
| 6 | 58 (95) | 3 (5) | 54 (96) | 2 (4) | |
| 12 | 59 (97) | 2 (3) | 55 (98) | 1 (2) | |
| 24 | 20 (33) | 41 (67) | 17 (30) | 39 (70) | |
| Hospital admissions | 3 | 59 (97) | 2 (3) | 56 (100) | 0 (0) |
| 6 | 58 (95) | 3 (5) | 54 (96) | 2 (4) | |
| 12 | 59 (97) | 2 (3) | 55 (98) | 1 (2) | |
| 24 | 20 (33) | 41 (67) | 17 (30) | 39 (70) | |
| Outpatient visits | 3 | 59 (97) | 2 (3) | 56 (100) | 0 (0) |
| 6 | 58 (95) | 3 (5) | 54 (96) | 2 (4) | |
| 12 | 59 (97) | 2 (3) | 55 (98) | 1 (2) | |
| 24 | 20 (33) | 41 (67) | 17 (30) | 39 (70) | |
| General community health services | 3 | 59 (97) | 2 (3) | 56 (100) | 0 (0) |
| 6 | 58 (95) | 3 (5) | 54 (96) | 2 (4) | |
| 12 | 59 (97) | 2 (3) | 55 (98) | 1 (2) | |
| 24 | 20 (33) | 41 (67) | 17 (30) | 39 (70) | |
| PSS | 3 | 59 (97) | 2 (3) | 56 (100) | 0 (0) |
| 6 | 58 (95) | 3 (5) | 54 (96) | 2 (4) | |
| 12 | 59 (97) | 2 (3) | 55 (98) | 1 (2) | |
| 24 | 20 (33) | 41 (67) | 17 (30) | 39 (70) | |
| Time off work | 3 | 59 (97) | 2 (3) | 56 (100) | 0 (0) |
| 6 | 58 (95) | 3 (5) | 54 (96) | 2 (4) | |
| 12 | 59 (97) | 2 (3) | 55 (98) | 1 (2) | |
| 24 | 20 (33) | 41 (67) | 17 (30) | 39 (70) | |
| Medications: analgesic use | Baseline | 59 (97) | 2 (3) | 53 (95) | 3 (5) |
| 3 | 58 (95) | 3 (5) | 56 (100) | 0 (0) | |
| 6 | 57 (93) | 4 (7) | 54 (96) | 2 (4) | |
| 12 | 59 (97) | 2 (3) | 55 (98) | 1 (2) | |
| 24 | 20 (33) | 41 (67) | 17 (30) | 39 (70) | |
| EQ-5D-5L | Baseline | 61 (100) | 0 (0) | 56 (100) | 0 (0) |
| 3 | 59 (97) | 2 (3) | 56 (100) | 0 (0) | |
| 6 | 58 (95) | 3 (5) | 54 (96) | 2 (4) | |
| 12 | 59 (97) | 2 (3) | 55 (98) | 1 (2) | |
| 24 | 20 (33) | 41 (67) | 20 (36) | 36 (64) | |
| Time point (months) | Parameter | Statistic | Debridement with InSpace balloon | Debridement only |
|---|---|---|---|---|
| 6 | Costs | n | 33 | 30 |
| Mean costs | 5980 | 3920 | ||
| SD costs | 2234 | 1968 | ||
| 6 | Effects (QALY) | n | 32 | 30 |
| Mean QALY | 0.501 | 0.582 | ||
| SD | 0.361 | 0.224 | ||
| INMB (interim) | (20000 × −0.081) − 2060 = −3680 | |||
| 12 | Costs | n | 56 | 61 |
| Mean costs | 6187 | 4248 | ||
| SD costs | 1337 | 1284 | ||
| 12 | Effects (QALY) | n | 56 | 61 |
| Mean QALY | 0.551 | 0.606 | ||
| SD | 0.336 | 0.168 | ||
Appendix 3 Magnetic resonance imaging substudy
Demographic details of those who took part in the MRI substudy are shown in Table 63. Table 64 shows the exploratory analysis of comparing the change in AHD between the active and passive scans.
| Debridement only (n = 11) | Debridement with balloon (n = 13) | Overall (n = 24) |
||
|---|---|---|---|---|
| Sex | Male | 6 (55) | 8 (62) | 14 (58) |
| Female | 5 (45) | 5 (38) | 10 (42) | |
| Age group (years) | Under 70 | 5 (45) | 6 (46) | 11 (46) |
| Over 70 | 6 (55) | 7 (54) | 13 (54) | |
| Tear size | Large | 8 (73) | 13 (100) | 21 (88) |
| Small | 3 (23) | 0 | 3 (12) |
Appendix 4 Descriptions of the selected trauma and orthopaedic randomised controlled trials
WOLLF
The WOLLF study was a multicentre randomised trial performed in the UK Major Trauma Network, recruiting 460 patients aged 16 years or older with a severe open fracture of the lower limb from July 2012 to December 2015. 49,133 The main objective of the study was to assess the disability of patients with a severe open fracture of the lower limb treated with either NPWT or standard wound management after the first surgical debridement of the wound. The primary outcome was the DRI score (range, 0 = no disability to 100 = completely disabled) at 12 months (12m) with early outcomes measured at 3, 6 and 9 months. Participants were recruited into the study and randomised in a one to one ratio to either NPWT (n1 = 226) or Standard (n0 = 234) wound treatment. There was no statistically significant difference in DRI score at 12 months; the mean score in the NPWT group was 45.5 (n112m = 179) and in the standard dressing group 42.4 (n012m = 195), a difference of −3.1 (95% CI −8.5 to 2.2; see Table 2 in main clinical paper). 133 The study took 50 months (recruitment plus follow-up) to complete.
DRAFFT
The DRAFFT, study compared Kirschner wire fixation (Wire) with volar locking- plate fixation (Plate) for participants with a dorsally displaced fracture of the distal radius. 47,134 The trial used the PRWE score at 12 months (12m) after surgery to assess participant outcomes; PRWE measures a patient’s experience of pain and disability to give a score out of 100 (with 100 being the worst score). The study recruited 461 participants from July 2010 to July 2012, randomising n0 = 230 to the Wire group (control) and n1 = 231 to the Plate group, on a one to one basis. Early PRWE outcomes were assessed at three and 6 months. There was no statistically significant difference in PRWE score at 12 months; the mean score in the Wire group was 15.3 (n012m = 211) and in the Plate group 13.9 (n112m = 204), a difference of 1.4 (95% CI −1.8 to 4.5; see Table 3 in main clinical paper). 47 The study took 34 months (recruitment plus follow-up) to complete.
FixDT
The FixDT trial compared intramedullary nail fixation with locking-plate fixation for adult patients with a displaced fracture of the distal tibia. 50,135 FixDT recruited 321 patients after a displaced fracture of the distal tibia between April 2013 and April 2016. The main objective of the study was to assess the disability of patients after surgical repair with either intramedullary nail fixation (Nail, control) or locking-plate fixation (Plate). The primary outcome of the study was the DRI score (range, 0 = no disability to 100 = completely disabled) at 6 months (6m), with early outcome measured at 3 months, with long-term outcomes also measured at 12 months. Participants were recruited into the study and randomised in a one to one ratio to either Nail (n0 = 161) or Plate (n1 = 160) fixation. The primary result of the study was that there was no statistically significant difference in DRI score at 6 months; the mean score in the Nail group was 29.8 (n16m = 142) and in the Plate group 33.8 (n06m = 140), a difference of −4.0 (95% CI −9.6 to 1.6; see Table 2 in main paper). 135 The study took 42 months (recruitment plus follow-up) to complete (excluding the time taken for the completion of the 12 months of long-term follow-up).
FASHIoN
The FASHIoN study was a pragmatic, multicentre, assessor-blinded RCT, undertaken at 23 NHS hospitals in the UK. 143,144,152 FASHIoN recruited 348 adult patients with femoroacetabular impingement syndrome who presented at these hospitals and randomly allocated them (1 : 1) to receive either hip arthroscopic surgery (n1 = 171) or personalised hip therapy (PHT; n0 = 177), between July 2012 and July 2016. PHT (control) is an individualised, supervised and progressive physiotherapist-led programme of conservative care. The primary outcome was hip-related QoL, as measured by the patient-reported International Hip Outcome Tool (iHOT-33) at 12 months (12m) after randomisation, with early outcome assessed at 6 months. iHOT-33 provides a 100-point score, with 100 representing no pain and perfect function, and lower scores indicating pain and poor function. The primary result of the study was that there was a statistically significant difference in iHOT-33 score at 12 months; the mean score in the surgery group was 58.8 (n112m = 158) and in the PHT group 49.7 (n012m = 163), a difference of 9.1 (95% CI 3.3 to 14.9) (see Table 2 in main clinical paper). 152 The study took 60 months (recruitment plus follow-up) to complete.
WAT
WAT was a single-centre, two arm, parallel-group, assessor-blinded, RCT with one to one treatment allocation conducted in the UK,136 recruiting 126 patients aged 18 years and over who were medically fit for an operation, and suitable for a resurfacing arthroplasty (RSA) of the hip. Patients were recruited between May 2007 and February 2010 from hip replacement clinics at the University Hospitals Coventry and Warwickshire NHS Trust (Coventry, UK) and randomly assigned on a one to one basis to receive either a total hip arthroplasty (THA) or a RSA. The primary outcome was hip function, as measured by the patient-reported OHS at 12 months (12m) after operation, with early outcome assessed at 6 weeks, 3 months and 6 months. The OHS provides a score on a scale from 0 to 48, with 48 representing no pain and perfect function, with lower scores indicating pain and poor function. There was no statistically significant difference in OHS at 12 months; the mean score in the RSA group was 40.4 (n112m = 57) and in the THA group 38.2 (n012m = 63), a difference of 2.23 (95% CI −0.51 to 12.58; see Table 2 in main clinical paper). 46
CSAW
The CSAW trial was a three-group, pragmatic, randomised (1 : 1 : 1) controlled study that compared arthroscopic subacromial decompression (ASAD), arthroscopy only and active monitoring with specialist reassessment (AMSR; no surgical treatment). 69,137 The trial used the OSS at 6 months (6m) after randomisation to assess participant outcomes. OSS was also assessed at 12 months after randomisation but no early assessment of OSS was made before the 6-month primary end point. The primary objective of the study was to compare ASAD against the non-surgery ASMR arm to assess efficacy. The study recruited n = 210 participants (to the ASAD and AMSR arms) from September 2012 to June 2015, randomising n1 = 106 to ASAD and n0 = 104 to AMSR. The primary result of the study was that there was a statistically significant difference in OSS at 6 months between ASAD and AMSR; the mean score in the ASAD group was 32.7 (n16m = 90) and in the AMSR group 29.4 (n06m = 90), a difference of 3.3 (95% CI −0.2 to 6.8) (see Table 2 in main clinical paper for adjusted difference). 69 Although statistically significant, the difference was not considered to be of clinical importance. The study took 48 months (recruitment plus 12 months of follow-up) to complete.
TOPKAT
The TOPKAT study compared total knee replacement (TKR) with partial knee replacement (PKR) for patients with medial compartment osteoarthritis of the knee. 138–140 The trial used the OKS at five years (5y) after randomisation to assess participant outcomes; OKS measures a patient’s experience of knee pain and function using 12 items scored from 0 to 4 and summed to give a score from 0 to 48 (with 0 being the worst score). The study recruited n = 528 participants from January 2010 to September 2013, randomising n0 = 264 to TKR and n1 = 264 to PKR, on a one to one basis. Early outcomes were assessed on a yearly basis at one, two, three and four years. There was no statistically significant difference in OKS at five years; the mean score in the TKR group was 37.0 (n05y = 231) and in the PKR group 38.0 (n05y = 233), a difference of 1.0 (95% CI −0.4 to 2.5; see Table 2 in main clinical paper). 140 The study took 96 months (recruitment plus follow-up) to complete.
Lower and upper stopping boundaries for the selected trauma and orthopaedics randomised controlled trials
Tables 65 to 70 describe the lower and upper stopping boundaries for each RCT.
Estimates of correlations and standard deviations for the selected trauma and orthopaedics randomised controlled trials
Tables 71 to 75 show the estimates of correlations and SDs for each RCT included in the adaptive designs for surgical trials substudy.
| Interim | 1 | 2 | 3 | End | 1 | 2 | 3 | End | ||
|---|---|---|---|---|---|---|---|---|---|---|
| a | la | −1.41 | −1.14 | −0.91 | 2.09 | ua | 3.09 | 2.62 | 2.45 | 2.09 |
| b | lb | −0.99 | −0.59 | −0.20 | 2.08 | ub | 3.09 | 2.62 | 2.45 | 2.08 |
| c | lc | −0.71 | −0.15 | 0.49 | 2.04 | uc | 3.09 | 2.62 | 2.45 | 2.04 |
| d | ld | −0.47 | 0.29 | 1.75 | 1.36 | ud | 3.09 | 2.62 | 2.45 | 1.36 |
| Interim | 1 | End | 1 | End | ||
|---|---|---|---|---|---|---|
| a | la | −0.99 | 2.02 | ua | 2.58 | 2.02 |
| b | lb | −0.47 | 2.01 | ub | 2.58 | 2.01 |
| c | lc | −0.05 | 2.00 | uc | 2.58 | 2.00 |
| d | ld | 0.36 | 1.97 | ud | 2.58 | 1.97 |
| Interim | 1 | 2 | End | 1 | 2 | End | ||
|---|---|---|---|---|---|---|---|---|
| a | la | −1.41 | −1.14 | 2.09 | ua | 3.09 | 2.34 | 2.09 |
| b | lb | −0.99 | −0.59 | 2.09 | ub | 3.09 | 2.34 | 2.09 |
| c | lc | −0.71 | −0.15 | 2.08 | uc | 3.09 | 2.34 | 2.08 |
| d | ld | −0.47 | 0.29 | 2.06 | ud | 3.09 | 2.34 | 2.06 |
| Interim | 1 | 2 | End | 1 | 2 | End | ||
|---|---|---|---|---|---|---|---|---|
| a | la | −1.41 | −1.14 | 2.10 | ua | 3.09 | 2.34 | 2.10 |
| b | lb | −0.99 | −0.59 | 2.09 | ub | 3.09 | 2.34 | 2.09 |
| c | lc | −0.71 | −0.15 | 2.08 | uc | 3.09 | 2.34 | 2.08 |
| d | ld | −0.47 | 0.29 | 2.06 | ud | 3.09 | 2.34 | 2.06 |
| Interim | 1 | End | 1 | End | ||
|---|---|---|---|---|---|---|
| a | la | −0.99 | 2.01 | ua | 2.58 | 2.01 |
| b | lb | −0.47 | 2.01 | ub | 2.58 | 2.01 |
| c | lc | −0.05 | 2.00 | uc | 2.58 | 2.00 |
| d | ld | 0.36 | 1.98 | ud | 2.58 | 1.98 |
| Interim | 1 | 2 | End | 1 | 2 | End | ||
|---|---|---|---|---|---|---|---|---|
| a | la | −1.41 | −1.14 | 2.08 | ua | 3.09 | 2.34 | 2.08 |
| b | lb | −0.99 | −0.59 | 2.08 | ub | 3.09 | 2.34 | 2.08 |
| c | lc | −0.71 | −0.15 | 2.07 | uc | 3.09 | 2.34 | 2.07 |
| d | ld | −0.47 | 0.29 | 2.06 | ud | 3.09 | 2.34 | 2.06 |
| Analysis | ρ3m,6m | ρ3m,9m | ρ3m,12m | ρ6m,9m | ρ6m,12m | ρ9m,12m | σ3m | σ6m | σ9m | σ12m |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.66 | 0.61 | 0.59 | 0.83 | 0.77 | 0.88 | 22.65 | 23.50 | 26.67 | 25.71 |
| 2 | 0.62 | 0.61 | 0.58 | 0.78 | 0.78 | 0.89 | 22.08 | 23.94 | 24.75 | 24.72 |
| 3 | 0.59 | 0.52 | 0.48 | 0.72 | 0.71 | 0.82 | 21.15 | 23.59 | 25.16 | 26.45 |
| End | 0.65 | 0.55 | 0.58 | 0.67 | 0.73 | 0.74 | 21.25 | 23.92 | 25.59 | 26.22 |
| Analysis | ρ3m,6m | ρ3m,12m | ρ6m,12m | σ3m | σ6m | σ12m |
|---|---|---|---|---|---|---|
| 1 | 0.81 | 0.78 | 0.72 | 22.34 | 17.45 | 13.51 |
| End | 0.75 | 0.61 | 0.73 | 22.57 | 18.21 | 16.63 |
| Analysis | ρ3m,6m | σ3m | σ6m |
|---|---|---|---|
| 1 | 0.61 | 20.13 | 24.56 |
| 2 | 0.65 | 20.06 | 23.70 |
| End | 0.65 | 20.01 | 24.07 |
| Analysis | ρ6m,12m | σ6m | σ12m |
|---|---|---|---|
| 1 | 0.56 | 22.90 | 25.81 |
| 2 | 0.56 | 23.88 | 27.27 |
| End | 0.57 | 24.14 | 26.32 |
| Analysis | ρ6w,3m | ρ6w,6m | ρ6w,12m | ρ3m,6m | ρ3m,12m | ρ6m,12m | σ6w | σ3m | σ6m | σ12m |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.86 | 0.80 | 0.60 | 0.90 | 0.71 | 0.50 | 9.93 | 10.43 | 9.36 | 5.58 |
| End | 0.80 | 0.69 | 0.59 | 0.83 | 0.72 | 0.80 | 10.06 | 9.84 | 8.95 | 10.37 |
Appendix 5 Bayesian interim analysis study: Bayesian sample report
Overview of Bayesian adaptive trial
The reason for the Bayesian substudy is to understand whether adaptive trials are better designed, monitored and analysed using a Bayesian approach or a frequentist one. The aim of this report is to present a Bayesian analysis and understand the how it affects decision-making during the trial compared to a frequentist report. The key information about the Bayesian set-up are described below:
-
Overall study sample size: n = 240, with n = 120 in each group.
-
The treatment intervention is debridement with InSpace balloon and the control is debridement-only.
-
Primary end point is the OSS at 12 months after randomisation. The MCID has been set to 6 points.
-
There are two planned interim analyses. The first upon observing 50 participants for the first interim analysis and 90 participants for the second.
-
Trial will stop early upon examining the probabilities of the mean difference and if they meet the boundary conditions.
-
The parameter (θ) denotes the difference between the intervention groups, where the treatment effect is positive in this report (θ > 0) then the evidence is in favour of the treatment group (debridement with InSpace balloon).
-
The following boundary/stopping rules are set out below:
-
Stop for futility if the probability of θ ≤ 0 is high; p(θ ≤ 0|data) > 0.8. (F1).
-
Stop for efficacy if the probability of the treatment is better than the control, based on the MCID of 6 OSS units. For this Bayesian approach, we set two criteria that needs to be met to match a typical frequentist set-up.
-
The probability of seeing a MCID difference in favour of the treatment is strictly high:
p(θ>6|data) > 0.8 (E1)
-
The probability that there is a difference between the intervention is very likely:
p(θ>0|data) > 0.8 (E2)
-
-
-
Bayes factor (BF) is calculated as a supplementary test, equivalent to a frequentist t-test, and is interpreted by the following rules:153
-
BF = 1 – no evidence
-
1 < BF ≤ 3 – anecdotal
-
3 < BF ≤ 10 – moderate
-
10 < BF ≤ 30 – strong
-
30 < BF ≤ 100 – very strong
-
BF > 100 – extreme.
-
Reminder of the operating characteristics
At the beginning of the trial, we derived the probabilities of stopping the trial for different observed values of the mean difference (θ). The following section serves as a reminder of the operating characteristics and an interpretation of the simulation results.
Efficacy:
-
The probability of incorrectly stopping early for efficacy when there is no treatment effect (θ = 0) is 0% and rises to 18% when θ ≤ 5.
-
The overall probability of stopping early due to efficacy, when θ ≥ 6, is at least 30%, with the probabilities increasing as the difference (θ) increases.
-
When θ ≥ 6, the total probability of declaring efficacy by the end of the final analyses is 38% and rises as the treatment difference (θ) increases.
Futility:
-
The probability of incorrectly declaring futility when the true difference is equal to or greater than the MCID of six units is 0%.
-
The overall probability of correctly stopping early for when there is no difference in treatment effect (θ = 0) is 30% and increases as the effect size is more negative (in favour of control); p(θ ≤ 0)> 0.3.
-
When θ ≤ 0, the total probability of declaring futility by the end of the final analyses is 38% and rises as the treatment difference (θ) becomes more negative (in favour of the control), p(θ ≤ 0) > 0.38.
Interim analysis results
The results show that InSpace balloon group has much lower scores by a large difference and the boundary conditions for futility had been met. We note the key results in Table 76.
| Statistic | Interim 1 |
|---|---|
| Participants (N) | 50 |
| Difference in means | −6.7 |
| 95% credible interval | −20.5 to 8.0 |
| Standard effect size | −0.6 |
| Posterior mean | −6.3 |
| Posterior SD | 8.7 |
| p(θ< 0| data) | 0.86 |
| p(θ> 0| data) | 0.24 |
| p(θ< 6| data) | 0.08 |
-
P(θ<0|data1) = 0.86 ≥ 0.80. This means the probability that the difference between the treatments is less than zero had met the stopping threshold of 0.8.
-
The 95% credible interval is (−21.2 to 8.2), therefore there is a 95% chance the mean treatment difference lies within this region.
-
Bayes factor provides a test of for the probabilities by comparing p(θ) between the initial prior and the posterior distribution.
-
The Bayes factor is 4.3 when testing for p(θ< 0), therefore, the probability of a negative treatment effect is more likely with the new posterior distribution then the prior with mean difference of zero.
The results showed that the probability bounds have been met and additional testing showed that it is unlikely for the treatment effect to be positive, therefore the trial should stop due to futility.
Summary data of both arms have been presented in Table 77 and Figure 33, the posterior distribution of the data at this time is shown in Figure 34.
| Statistic | Treatment | Control |
|---|---|---|
| Participants (N) | 28 | 22 |
| Mean | 35.9 | 42.6 |
| SD | 10.7 | 10.6 |
FIGURE 33.
Box plot summary of interim data.
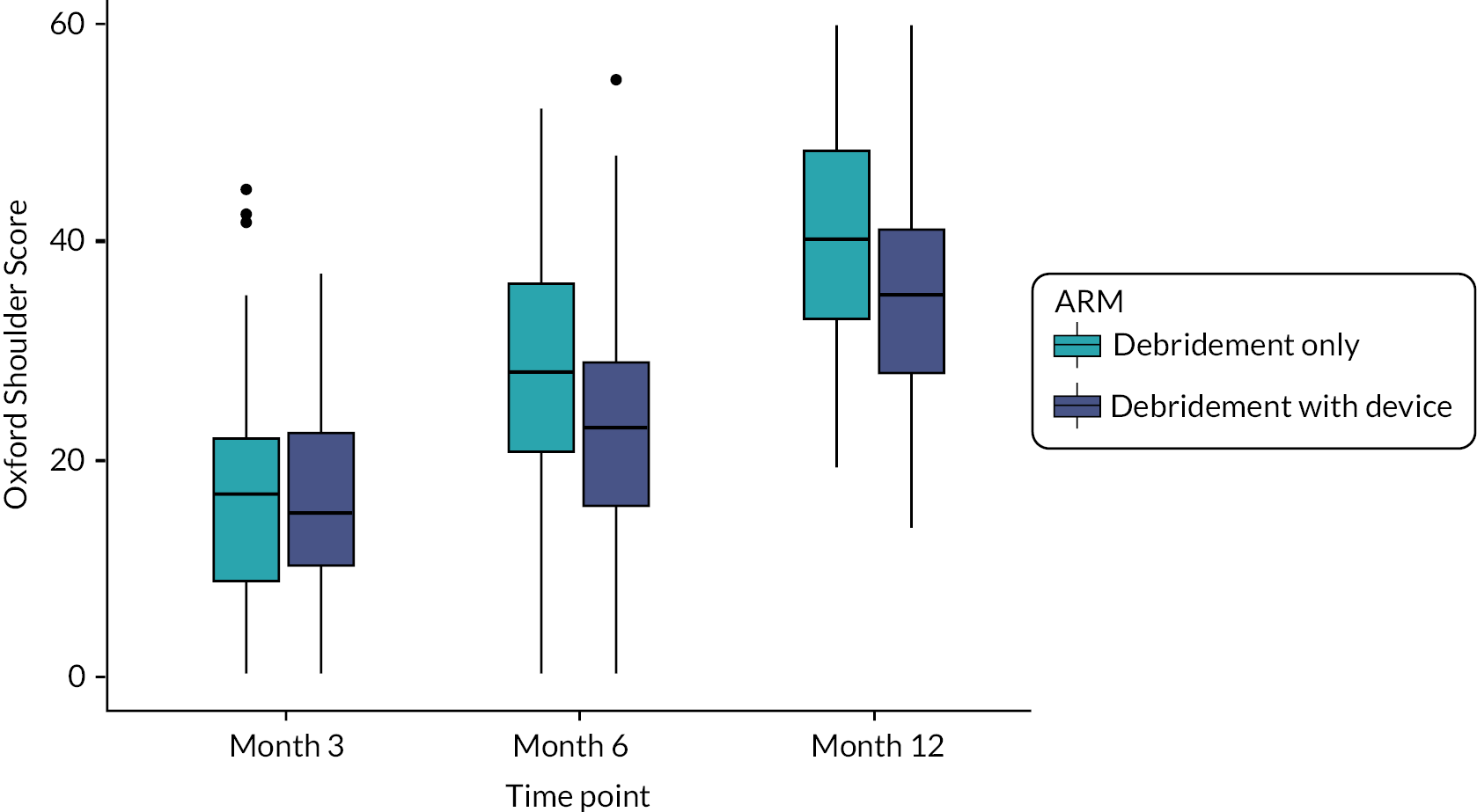
FIGURE 34.
Prior and posterior distribution at interim analysis.
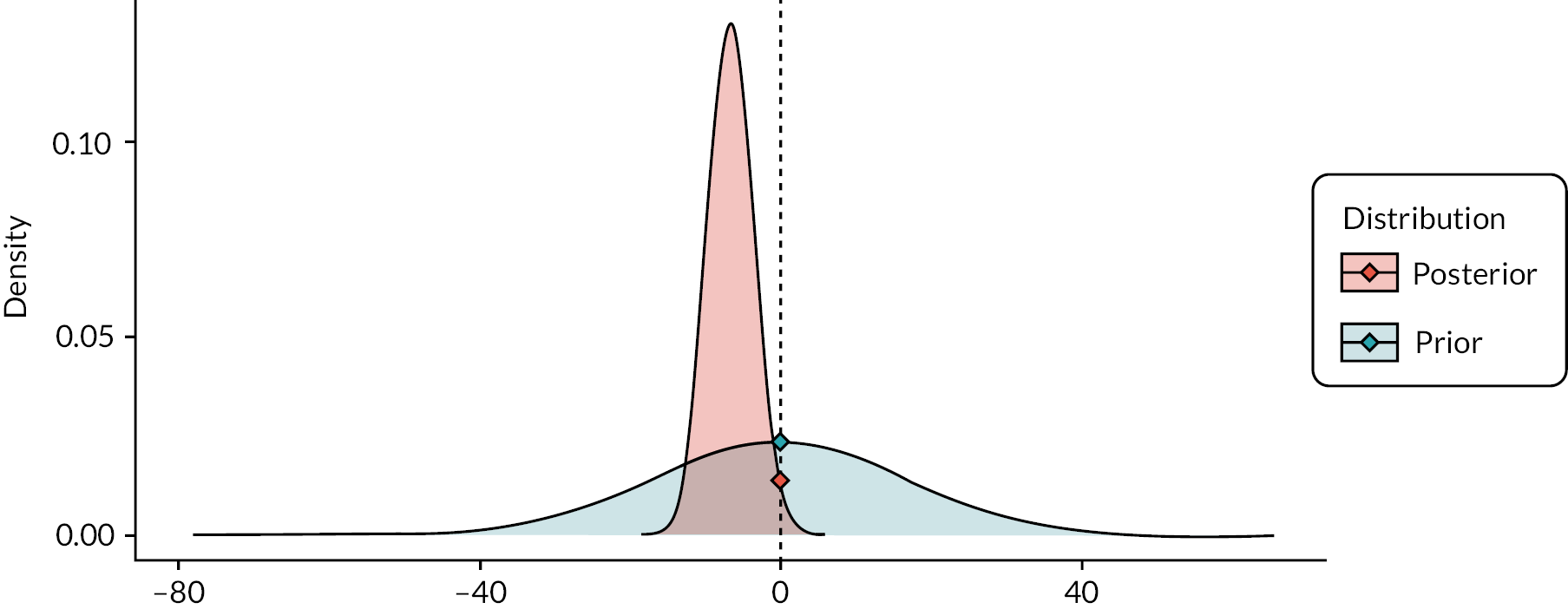
Bayesian interim analysis study: frequentist sample report
Overview of frequentist adaptive trial
The reason for the Bayesian substudy is to understand if adaptive trials are better designed, monitored and analysed using a Bayesian approach instead the frequentist method. This report is based on frequentist adaptive trial where the study can be stopped when sufficient evidence is observed at preset interim analysis points. We note that the boundaries for the interim analysis have been determined at the beginning of the study. The following summarises the features of the study:
-
Overall study sample size: n = 240, with n = 120 in each group.
-
The treatment intervention is debridement with InSpace balloon, and the control is debridement-only.
-
Primary end point is the Constant score at 12 months after randomisation. The MCID has been set to 6 points.
-
There are two planned interim analyses. The first interim observing approximately 50 participants and 90 participants for the second interim analysis. These are based on some model assumptions that gives us Information statistic Ik, which we need to meet to perform the interim analysis.
-
Trial will stop early upon examining the test statistics Sk and if they meet the boundary conditions (lk,uk) for interim analysis k = 1, 2.
-
The test statistic Sk=Bksd(Bk), where Bk is based on treatment μ12m–control μ12m
-
The treatment effects are only based on the 12-month outcomes, and not the early 3- and 6-month outcomes.
-
The early outcomes are only used to measure the correlation with 12 months outcomes and effects the Information statistic (Ik) which informs of the timing for the interim analyses.
-
lk denotes the lower futility bound, if Sk<lk, then the study should stop for futility.
-
uk denotes the upper efficacy bound, if Sk<uk, then the study should stop for efficacy.
-
The trial will recruit to full sample size if the boundary conditions aren’t met in either Interim analysis.
Statistical commentary
It is estimated that the first interim analysis will occur when obtaining data from approximately 50 participants at 12-month follow-up, and data from 60 and 45 participants for 3-month and 6-month follow-up. The estimated accrued information for the first interim analysis I1 = 0.103.
As we have now met the information boundary after careful monitoring, I1 = 0.109 > 0.103, it is now time to perform the first interim analysis.
Interim analysis results
In summary, the results show a negative value favouring the control group (debridement only). It is significantly low enough to stop the study for efficacy. The statistical results are shown in Table 78, Figure 35 and Figure 36 and summarised below:
FIGURE 35.
Estimated mean differences (•) in CS (12 months) between intervention arms (left), with 95% CIs, and test statistic S1, with stopping boundaries, at the second interim analysis (right).
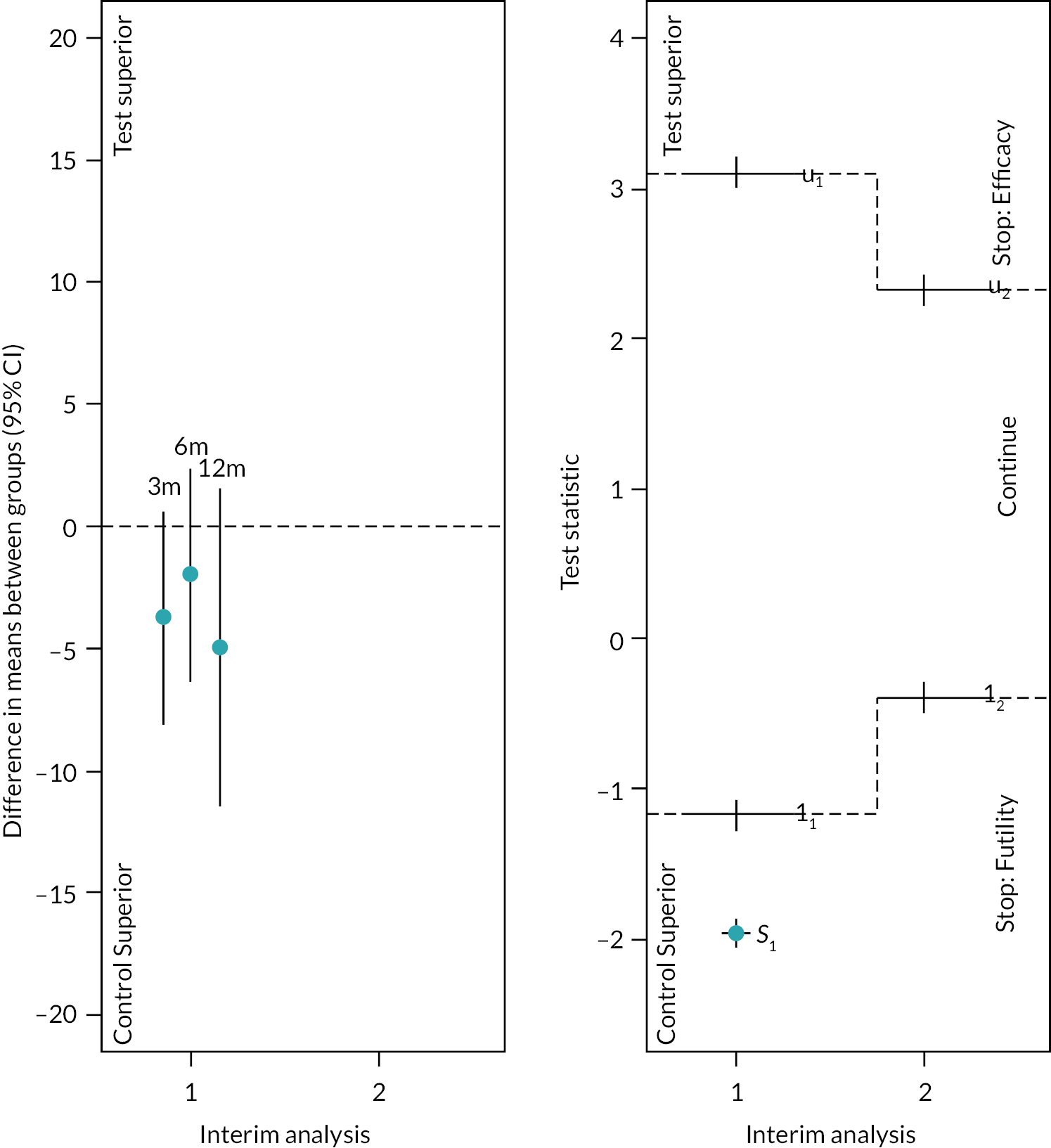
FIGURE 36.
Box plot of Constant scores for each time point.

| Expected | Observed | ||||
|---|---|---|---|---|---|
| Balloon | Control | Balloon | Control | ||
| No. of study participants providing OSS data | N 12m | 25 | 25 | 25 | 22 |
| N 6m | 45 | 45 | 47 | 40 | |
| N 3m | 60 | 60 | 70 | 59 | |
| Correlations between CS data | ρ 3m,6m | 0.5 | 0.51 | ||
| ρ3m,12m | 0.5 | 0.45 | |||
| ρ6m,12m | 0.5 | 0.52 | |||
| Mean of CS data | μ 12m | – | – | 57.5 | 62.5 |
| μ 6m | – | – | 49.0 | 51.0 | |
| μ 3m | – | – | 38.1 | 41.9 | |
| SD of CS data | σ12m | 12 | 11.2 | ||
| σ6m | 12 | 10.2 | |||
| σ3m | 12 | 12.4 | |||
| Information | I 1 | 0.103 | 0.109 | ||
| Treatment difference | B 1 | – | −5.97 | ||
| sd(B1) | – | 3.03 | |||
| Test statistic | S 1 | – | −1.97 | ||
| Lower boundary | l 1 | −1.17 | – | ||
| Upper boundary | u 1 | 3.09 | – | ||
-
If the value of test statistic is positive then results are in favour of the treatment group and a negative value favours the control group.
-
The test statistic is S1=B1sd(B1)=−1.97.
-
The lower boundary for the first interim is l1 = –1.17.
-
If the value of S1 is lower than the lower bound, then we have evidence in favour of the study being stopped for futility.
-
We have S1 = –1.97 < l1 = –1.17.
-
As the value of test statistic exceeds the lower bound, we recommend that the study stops for futility as there is sufficient evidence against treatment group.
List of abbreviations
- AHD
- acromiohumeral distance
- AMSR
- active monitoring with specialist reassessment
- ASAD
- arthroscopic subacromial decompression
- ASES
- American Shoulder and Elbow Society
- BF
- Bayes factor
- CE
- Conformité Européenne
- CI
- confidence interval
- CP
- conditional power
- CPCE
- conditional power of cost-effectiveness
- DMC
- Data monitoring committee
- DRI
- Disability Rating Index
- EMG
- electromyography
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- FDA
- Food and Drug Administration
- FixDT
- Fixation of Distal Tibia Fractures
- HRQoL
- health-related quality of life
- ICER
- incremental cost-effectiveness ratio
- iHOT-33
- International Hip Outcome Tool
- INMB
- incremental net monetary benefit
- MCID
- minimum clinically important difference
- MRI
- magnetic resonance imaging
- NICE
- National Institute for Health and Care Excellence
- NPWT
- negative pressure wound therapy
- OHS
- Oxford Hip Score
- OKS
- Oxford Knee Score
- OSS
- Oxford Shoulder Score
- PGIC
- Patient Global Impression of Change
- PHT
- personalised hip therapy
- PKR
- partial knee replacement
- PRWE
- patient-rated wrist evaluation
- PSSs
- personal social services
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- REACTS
- Randomised, Efficient, Adaptive Clinical Trial in Surgery
- ROM
- range of motion
- RSA
- resurfacing arthroplasty
- SAE
- serious adverse event
- SD
- standard deviation
- START
- Subacromial spacer for Tears Affecting Rotator cuff Tendons
- TSC
- Trial steering committee
- VAS
- visual analogue scale
- WCTU
- Warwick Clinical Trials Unit
- WMS
- Warwick Medical School
- WOLLF
- Wound Management of Open Lower Limb Fractures
- WORC
- Western Ontario Rotator Cuff