

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as award number 12/62/109. The contractual start date was in March 2014. The draft manuscript began editorial review in February 2023 and was accepted for publication in December 2023. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ manuscript and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this article.
Permissions
Copyright statement
Copyright © 2024 Sharp et al. This work was produced by Sharp et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Sharp et al.
Chapter 1 Introduction
Intrauterine growth restriction (IUGR) complicates up to 10% of pregnancies, accounting for over one-quarter of all stillbirths. 1,2 With no effective treatment available, up to 70% of pregnant women with IUGR diagnosed in the early third trimester require delivery before 32 weeks’ gestation. 3 It is well documented that these infants have substantially increased risks of neonatal death, major morbidity and prolonged neonatal admission compared with preterm infants of appropriate birthweight. 4
Early-onset IUGR most commonly occurs when the placental transfer of nutrients and oxygen is impaired due to inadequate placental implantation. The resulting fetal malnutrition and hypoxia are considered untreatable in utero. The only current option is an elective preterm delivery in order to rescue the baby from an adverse intrauterine environment. IUGR and the associated indicated preterm birth expose the fetus and neonate to significant mortality and morbidity. This diagnosis causes an important management dilemma: early delivery causes extreme prematurity with all its sequelae while delivering the baby too late risks intrauterine death or morbidity secondary to critical fetal hypoxia.
Being born too small and too early can pose significant health risks throughout the child’s life. In particular, IUGR has adverse effects on brain structure and function, which are independent of gestational age at birth5 and often compounded by poor postnatal growth, ultimately leading to an increased risk of neurological impairment, cognitive impairment, inattention and specific difficulties with executive functions and impulsivity. 6
Between 25% and 40% of surviving growth-restricted very preterm infants have developmental delay,7,8 in particular in the areas of fine and gross motor difficulties, attentional difficulties5 and language delay9 with a mean difference in intelligence quotient (IQ) of almost one standard deviation (SD) by the time they reach school age compared with preterm and term appropriate for gestational age (AGA) controls. 10,11
Intrauterine growth restriction is a key risk factor for adult diseases such as hypertension, diabetes and ischaemic heart disease due to its ability to permanently alter organ capacity and neuroendocrine regulation leading to an adverse cardiometabolic phenotype that predisposes to adult disease12 and alters reproductive health with evidence of impaired fetal growth in future generations. 13 There is strong evidence that the adverse consequences of placental insufficiency, leading to fetal growth restriction, extends beyond infancy to childhood and even adulthood. This adverse effect is above and beyond the effect of prematurity. 14 Only children who were growth restricted during their fetal life, among those born preterm, have increased arterial stiffness and evidence of metabolic dysfunction. 14 They also demonstrate greater aortic wall thickening progression, suggestive of preclinical atherosclerosis which leads to a higher risk of developing hypertension later in life. 15 To date, there has been limited progress in developing interventions to reverse the lifelong effects of IUGR.
Intrauterine growth restriction is most commonly caused by abnormal placental development and invasion of the maternal blood vessels. This process leads to placental dysfunction and poor fetal nutrition. Preclinical work16–18 and pilot studies19–21 have shown that sildenafil, a phosphodiesterase inhibitor and vasodilator, may improve uteroplacental circulation and increase fetal growth.
Sildenafil potentiates the effect of nitrous oxide (NO) and thus may cause vasodilatation of vessels responsive to NO. The incomplete remodelling of maternal spiral arteries in IUGR results in vessels with intact or partially intact muscular layers, which remain responsive to regional vascular control. Sildenafil has the potential to increase uteroplacental circulation and perfusion resulting in improved gaseous and nutrient exchange and improved fetal growth and well-being.
Use of sildenafil in an obstetric population has been limited, but several case reports and small studies now exist. Sildenafil has been used in selected cases for the treatment of maternal pulmonary arterial hypertension where there are growing data on both its safety and efficacy to improve both maternal and fetal outcomes. There are also limited data suggesting that sildenafil has the potential to increase fetal weight.
The identification of an effective therapy (such as sildenafil) could improve both the short- and long-term health outcomes for these children in addition to significantly reducing the emotional and financial burden for such individuals, their families, and the wider community.
STRIDER UK assessed the effect of sildenafil in severe early-onset fetal growth restriction where the only available treatment is early delivery. It is well documented that IUGR is often accompanied by complications such as fetal hypoxia, acidosis and inflammation, all of which are thought to have a detrimental effect on brain growth and development. Consequently, there may be a complex trade-off between the effects of longer gestational length and ongoing exposure to a suboptimal fetal environment that should be considered. 22,23 It is therefore important to evaluate long-term outcomes, regardless of short-term results, to ensure the overall balance of benefits and risks associated with sildenafil treatment are examined.
The international STRIDER collaboration included studies in the UK, Australia/New Zealand, the Netherlands and Canada with each country funding their own trial but all following a similar protocol. 24 Data from all trials will be published independently and then included in a high-quality pre-planned individual participant data (IPD) meta-analysis. 25 All trials were funded by their government funding bodies. Recruitment took place in five countries (UK, Australia, New Zealand, the Netherlands and Canada) with STRIDER UK completing recruitment first.
Chapter 2 Objectives
The STRIDER UK study was designed to answer the following objectives in two phases: phase 1 – recruitment to a randomised controlled trial of sildenafil versus placebo for the treatment of early-onset intrauterine fetal growth restriction, and phase 2 – follow-up at 2 years of age to assess how cardiovascular and neurodevelopmental outcomes effect the surviving infants.
The primary objective of the phase 1 study was to determine whether sildenafil, compared to placebo therapy, delays the need to deliver a severely growth-restricted fetus by a minimum of 1 week.
The secondary objectives were as follows:
-
To investigate the impact on fetal growth and fetal well‐being by comparing differential effect on vascular resistance in the uterine arteries, umbilical, fetal middle cerebral artery and fetal ductus venosus and differences in birthweight centiles in infants treated in utero with sildenafil and placebo.
-
To examine, through collaboration with an international consortium, the hypothesis that sildenafil therapy compared to placebo therapy increases the rate of infant survival free of major neurodisability.
-
To report frequency of adverse and serious adverse events (SAEs) associated with sildenafil use.
-
To investigate the impact on maternal cardiovascular parameters by measurements of maternal heart rate and peripheral blood pressure (BP) before and after administration of study medication.
-
To elucidate the precise mechanism and location of action of sildenafil in pregnancy by investigating the effects of sildenafil therapy on omental (representative of the wider maternal systemic vasculature), myometrial (uterine vasculature) and chorionic plate artery (placental vasculature) reactivity.
The objective of the phase 2 follow-up study was to examine neurodevelopmental and cardiovascular outcomes at 2 years of age in children born to mothers who received sildenafil compared with placebo during pregnancy.
It was hypothesised that:
-
STRIDER UK children whose mothers received sildenafil will have improved neurodevelopmental outcomes at age 2–3 years (corrected) compared with controls exposed to placebo.
-
There will be no difference in BP at 2–3 years (corrected) between STRIDER UK children whose mothers received sildenafil compared with controls exposed to placebo.
Chapter 3 Methods
Study design and participants
The STRIDER study was a Phase III clinical trial to quantify the effects of administration of sildenafil on pregnancy outcome in severe early‐onset IUGR. A total of 135 women with affected pregnancies were recruited and randomised to receive one of two treatment arms, sildenafil or placebo. The study received funding from the National Institute of Health and Care Research (NIHR) and Medical Research Council (MRC). It was co-sponsored by the University of Liverpool and Liverpool Women’s NHS Foundation Trust and co-ordinated through the Liverpool Clinical Trials Unit (LCTU, which is part of the Liverpool Trials Collaborative, UKCRC Registration 12).
All participants were recruited from one of the 19 STRIDER research sites located in the UK. All sites were leading obstetric units within the UK and successfully completed site feasibility during the green light phase of the trial set-up. Suitable collaborating sites and investigators were therefore assessed on the level of fetal medicine and neonatal service they provide and their ability to conduct the trial. Ahead of the trial starting at a site the Principal Investigators were required to agree to adhere to the good clinical practice (GCP) guidelines. In addition, all relevant regulatory and ethics approvals were required.
The study was designed as a randomised double-blind, placebo-controlled trial with sildenafil or placebo prescribed orally at a dose of 25 mg three times per day (Figure 1). This dosage regime was based on previous studies by the collaborators on the project. All participants recruited had a singleton pregnancy between 22+0 weeks’ gestation and 29+6 weeks’ gestation with a diagnosis of IUGR and had agreed to expectant management. For the purpose of study, IUGR was defined as a fetus with an estimated fetal weight or abdominal circumference below the 10th centile using local charts and absent or reversed end-diastolic flow in the umbilical artery on Doppler velocimetry.
FIGURE 1.
STRIDER trial flow chart.
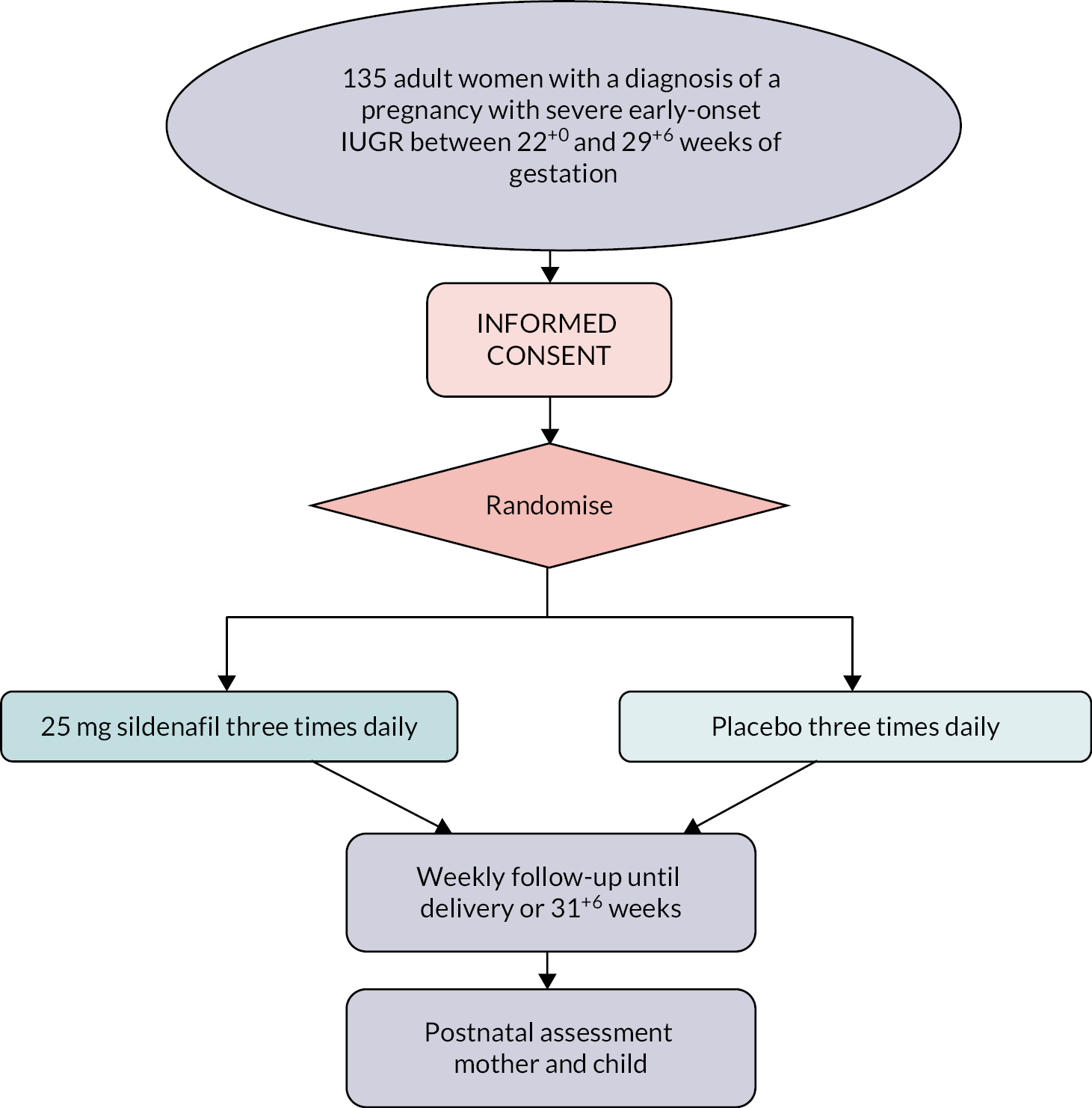
Gestational age was confirmed by first trimester ultrasound and in each case, the diagnosis of severe early-onset IUGR was confirmed by a fetal medicine expert having excluded fetal anatomical abnormalities. Following diagnosis and informed consent, a full history, measurements of maternal cardiovascular parameters (BP and pulse rate), fetal biometry and Doppler velocimetry were taken. Maternal venepuncture for angiogenic biomarkers was also performed.
All participants had further BP and pulse rate measurements and blood sampling 2 hours after receiving the first dose of the study drug. Subsequently, participants were followed up within 3–4 days and at weekly intervals thereafter, or earlier if clinically indicated. The remainder of clinical care was at the discretion of the local fetal medicine experts and included regular ultrasound assessment of growth and Doppler blood flow and antenatal cardiotocography.
Study medication was over-encapsulated (Sharp Clinical Services, Crickhowell, UK) to ensure that participants, clinicians and pharmacists were masked to the study drug. 26 Medication was dispensed in 10-day supplies with a new supply being provided weekly to ensure there was no period where medication was missed. Treatment ended at 31+6 weeks’ gestation or delivery, whichever came first. All participants were advised of the potential side effects.
Data on pregnancy outcome were collected prospectively from clinical maternity notes and entered onto a secure electronic case report form (eCRF) platform at research sites. Data quality and protocol compliance were monitored regularly by central and on-site monitoring methods.
The STRIDER UK study protocol v7.0 is available as Appendix 1
Start of study
Prior to the start of recruitment on 11 November 2014, the following approvals were obtained on the respective dates: Sponsorship – 9 October 2013, Research Ethics Committee (REC; North East – Newcastle and North Tyneside 2, Chair Dr Alasdair MacSween) – 20 March 2014, Clinical Trial Authority (CTA) – 18 July 2014 and Trial Greenlight Approval – 6 November 2014. All approvals for participating research sites were in place within 12 months of opening. The trial protocol (Appendix 1) was first registered on 31 July 2014, 4 months prior to the first participant being recruited. The first participant was recruited and randomised on 21 November 2014.
End-of-study definition
As defined in the protocol, the end of phase 1 of the STRIDER study was ‘when the last recruited woman/baby is discharged from hospital, or the baby has reached expected date of birth, whichever is later’. The last participant to be recruited to the study was on 6 July 2016 and the last infant to be discharged from hospital was on 13 February 2017. Therefore, the end of study for the STRIDER trial was reported as 13 February 2017.
Inclusion and exclusion criteria
The study inclusion criteria were as follows:
-
singleton pregnancy with severe, early‐onset IUGR between 22+0 and 29+6 weeks’ gestation AND a clinical decision to manage expectantly
-
IUGR defined as an estimated fetal weight < 10th centile OR abdominal circumference < 10th centile AND absent or reversed end-diastolic flow in the umbilical artery
-
16 years of age or older
-
consent to take part in the trial
The exclusion criteria for the study were as follows:
-
multiple pregnancy
-
known or suspected structural or chromosomal fetal abnormality
-
maternal illness (such as pre‐eclampsia) expected to require delivery for maternal reasons within 72 hours
-
maternal wish not to have active management of the pregnancy, such as a decision to have a termination of pregnancy
-
inability to give informed consent
-
cocaine use in the current pregnancy
-
contraindication to sildenafil therapy
-
known maternal cardiac disease
-
left ventricular outflow tract obstruction
-
concomitant treatment with nitrates, nitrate drugs for chest pains/heart problems including nitro-glycerine (glyceryl trinitrate, GTN), isosorbide dinitrate, isosorbide mononitrate
-
nitrates – some recreational drugs contain amyl nitrate (‘poppers’)
-
previous allergy to sildenafil, including hives, difficulty breathing, swelling of the face, lips or tongue
-
Sample size estimation
Internal audits of early‐onset IUGR cohorts revealed an average diagnosis to delivery interval of around 20 days with a SD of 11 days. In order to confirm that sildenafil could prolong pregnancy by 1 week (7 days), a total recruitment of 104 women (alpha 5%, power 90%) was required. Although loss to follow‐up was not anticipated, recruitment of 135 women was planned in order to account for any possible post‐randomisation withdrawal of consent or missing data.
The secondary hypothesis was that sildenafil will improve utero‐placental circulation and therefore delay the development of fetal cardiovascular changes (reduced short-term heart rate variability, deterioration of fetal Doppler indices) that lead to the indication for iatrogenic delivery. With a complete data set for approximately 100 participants, it was predicted that a clinically meaningful 20% difference in mean Doppler Pulsatility Index (PI) values of uterine arteries (0.86, SD 0.20), middle cerebral artery (2.21, SD 0.39) and ductus venosus (0.62, SD 0.22) would be detectable (alpha 5%, power > 80%).
Randomisation
Randomisation was performed using a web‐based randomisation service operating at the Clinical Trials Unit (CTU), British Columbia Women’s Hospital (Vancouver, Canada). Passwords and login details were provided to each STRIDER research site at the point of site ‘green light’ authorisation by the LCTU.
Treatments were allocated with equal probability by means of computer generated random permuted blocks of size two and four in equal proportions. The randomisation was stratified by two factors, the participating research sites and the gestational age at diagnosis: < 26+0 and ≥ 26+0 weeks of gestation.
As STRIDER was a double-blind placebo-controlled trial, both the participant and any clinical staff were blinded to the treatment allocation. It was a requirement that any unbinding that occurred during the running of the study was reported as a major protocol deviation. Unblinded participants would then be retained in the intention to treat (ITT) population, but removed from any per-protocol analyses.
Study end points
The primary end point for the study was the difference in length of gestation (days), defined as the time from estimated gestation until birth.
The secondary end points were divided into subgroups for fetal, infant and maternal safety and were as follows.
Fetal end points
-
Estimated fetal weight – measured in kilograms.
-
Abdominal circumference growth velocity between randomisation and discharge.
-
Measurements of gestational age adjusted Doppler PI in the umbilical artery, middle cerebral artery and ductus venosus and uterine arteries.
-
Measurements of short-term variability of the fetal heart rate recorded by transabdominal cardiotocography.
Infant end points
-
Gestational age at birth.
-
Survival to discharge.
-
Birthweight centile (adjusted for gestational age and gender).
-
Length of admission on the Neonatal Intensive Care Unit.
-
Oxygen dependency at day 28 and 36 weeks corrected age.
-
Necrotising enterocolitis.
-
Retinopathy of prematurity.
-
Significant (grade III/IV) cerebral haemorrhage detected by cerebral ultrasound.
-
Number of doses of surfactant.
-
Ventilator days.
-
Supplemental oxygen days.
-
Number of days to full feeds.
Maternal safety
-
Mode of delivery.
-
Standardised BP and pulse monitoring during treatment.
-
Postpartum haemorrhage.
-
Recording of the side effects, for example headache, facial flushing.
-
Inpatient postnatal stay.
Statistical analysis
Participants’ groups for analysis were defined on an ITT basis. Unadjusted estimates with Kaplan–Meier estimates were presented and analysed with linear regression techniques, including the stratification factor as a main effect. The treatment effect was reported as the mean difference between groups. Statistical significance was determined as p = 0.05 or less and participants randomised before 26+0 weeks and at 26+0 weeks or later were included in the subgroup analyses.
For continuous data, the analysis of secondary end points matched the analysis for the primary end point. Binary data were compared across treatment groups using a chi-squared (χ2) test or Fisher’s exact test as appropriate and reported using RR with 95% confidence intervals (95% CI). All analyses were performed using the statistical software package, R (version 3.3.3).
A Statistical Analysis Plan (SAP) was not produced prior to the analysis being undertaken.
Quality control and data validation
The STRIDER study was subject to regular data checks and reviews as set out in the trial-specific Data Management Plan. The study was also subject to both central and on-site monitoring as set out in the trial-specific Monitoring Plan. Regular Central Monitoring Reports were produced and reviewed by the Trial Management Team. In addition, on-site monitoring visits were carried out for each research site following the hospital discharge of the first participant and surviving infant. All visits were completed and any outstanding issues identified were actioned and closed accordingly.
An Independent Trial Steering Committee (TSC – Chair Professor Alan Cameron) and Safety and Data Monitoring Committee (ISDMC – Chair Professor Ed Juszczak) were established to provide oversight for the study. These committees met prior to the study opening, twice yearly while it was running and then one final time at the end of the study to review and approve the results. No significant issues relating to the management of the study or the safety of the participants were escalated.
The STRIDER randomisation list was reviewed to ensure provision of the correct number of strata, adequate randomisation numbers per stratum, appropriate block sizes and treatment allocations, and balanced allocation of treatments for various cumulative totals. This was found to be accurate. In line with the regular safety and efficacy review of the data by the ISDMC, checks were carried out for omitted, or, out of sequence allocations and balanced in treatment allocations.
At the end of the trial, a multiple logistic regression model with treatment arm as response and baseline variables as explanatory variables was used to confirm whether the best-fitting (minimum AIC) model was the one with no explanatory variables – that is, the baseline variable was uninformative as to treatment allocation.
All statistical coding relating to the analysis of the STRIDER data deriving the primary outcome variable was reviewed by an independent statistician who performed checks to ensure that the number of participants from the database matched the number in the analyses. A random check of at least 10% of participants was also performed to ensure that the derived gestational time and birth date was correct.
Adverse events and compliance
Adverse events (AEs) and treatment adherence were assessed and recorded at weekly clinical visits from recruitment to delivery. Participants were encouraged to record any side effects or AEs, which were then reviewed and documented during each clinical visit. Adherence was assessed weekly during clinical review, with any temporary discontinuation in treatment being recorded. Treatment adherence was considered to be good if the reported intake of tablets was 90% or more of the total expected to have been taken between randomisation and the visit date.
Phase 2 – 2-year follow-up
All surviving children of mothers recruited to the STRIDER UK study were eligible and invited for follow-up. Nominated members of the core research team were responsible for accessing the original confidential trial data in order to determine the contact details of all potential participants. As part of the original STRIDER UK trial, an audit of all consent forms took place to ensure that all participants had consented to further contact in relation to future research. In addition, a check was also made to ensure that the infants of all such participants survived. This was carried out via a number of methods including a thorough audit of all SAEs (which detail all fetal and neonatal deaths) and a final check at the local research site on the child’s health status. All participants received newsletters which provided information on the trial and also give the option of opting out of any further contact. Any participant who contacted the trial management team and requested to opt out of future correspondence and participation in further research was removed from the list of potential participants for the follow-up study.
A study invitation pack was sent to all parents/carers of surviving children. This included an invitation letter, participant information sheet and informed consent form. Participants were asked to read the information carefully and discuss their child’s participation in the study with either close friends, family and/or a relevant health professional. A named contact, telephone number and e-mail address were included on all correspondence so that parents/carers were able to contact the research team and discuss their child’s participation further, if required. If they were interested in taking part, they were asked to contact the research team in order to give verbal consent to take part in the study and to arrange a convenient date and time for their child’s assessment. Those participants who did not contact the research team within 2 weeks of the invitation pack being sent were contacted by a member of the core research team. For those participants who wanted to take part, an assessment date and time was arranged. Once an assessment date and time had been confirmed, an assessment pack was sent to the participant, as detailed below who have given expressed verbal consent to take part in the study.
The assessment pack included confirmation of the (already agreed) assessment date, time and location, a map (if necessary), details of what will take place during the assessment, who will carry out the assessment and a study questionnaire pack.
Assessments took place at either a local outpatient facility or in the home. A preference was made to carry out all assessments in a controlled setting (i.e. clinical research setting), however, where necessary assessments also took place in the child’s home after the researcher had assessed the home environment for suitability. Where assessments were planned to take place in the child’s home, further information was provided to parents on how to prepare their home (e.g. clearing an open space on the floor, providing a small table/work surface and turning off any distractions such as the television and or radio). Every effort was made to ensure that assessment dates were booked to suit the needs of the participant; however, it was preferred that assessments took place in the morning as this is a time that children are often well rested, fed and able to concentrate.
Participants were given contact details should they need to cancel or rearrange their assessment. Furthermore, they received a reminder telephone call 3 days prior to the assessment date and a text reminder the day before.
All assessments were performed by a suitably trained senior research psychologist with expertise in developmental assessment techniques in young infants. This researcher was also suitably trained in the specific cardiovascular assessment techniques used within this study and blinded to treatment allocation for the main STRIDER UK study. Additional oversight and supervision were provided by co-investigator, Professor Brigitte Vollmer for neurodevelopmental assessment techniques and Dr Andrew Sharp for cardiovascular assessment procedures.
Initially, the researcher facilitated introductions between all present, following on from this the study was explained and the child and parent/carer were given the opportunity to ask any questions relating to their child’s participation. Once this was complete and informed written consent had been received, the research assessment began. The researcher explained to the parent/carer the importance of allowing the child to complete all activities independently. The parent/carer was allowed to remain in the room while the assessment took place; however, they were asked not to interrupt or assist the child with these tasks unless otherwise invited to by the researcher.
The questionnaires sent to parents/carers in the assessment pack were reviewed by the researcher and any missing items were highlighted and discussed. If the parent/carer had any questions relating to the completion of the questionnaires, this was addressed so that the parent/carer could complete them during the assessment. Other factors were determined from the core STRIDER UK data set and where necessary neonatal records including gestational age, birthweight, APGAR (Activity, Pulse, Grimace, Appearance, Respiration) scores, duration of ventilation, chronic lung disease, persistent arterial ductus, neonatal sepsis, retinopathy of prematurity, necrotising enterocolitis and focal brain injury.
Once this was complete, the researcher played with the child to build a rapport so the child was relaxed and at ease during the assessment activities. Once the researcher deemed that the child was ready, the formal assessments were introduced to the child in the order set out in Table 1. These included the Cognitive, Language and Motor Subscales of the Bayley Scales of Infant and Toddler Development – III (BSID-III);27 Hempel’s Neurological Examination for Toddler Age28 which was used to identify major neurological impairment (cerebral palsy; CP) and to detect subtle deviations from typical neurological and neuromotor function and a cardiovascular assessment which included brachial systolic BP and diastolic BP and arterial stiffness, assessed as aortic (central) augmentation index (AIx).
| Assessment/procedure | Completed by | Time to complete |
|---|---|---|
| Introduction/consent | Parent/researcher | 15–20 minutes |
| Questionnaire review | Parent/researcher | 5–10 minutes |
| General health and well-being | Parent/researcher | 5–10 minutes |
| Break | ||
| Introductory play session | Child/researcher | 10–15 minutes |
| BSID: mental/cognitive assessment | Child/researcher | 10–15 minutes |
| BSID: fine motor assessment | Child/researcher | 10–15 minutes |
| Break | ||
| BSID: gross motor assessment | Child/researcher | 10–15 minutes |
| Hempel neuromotor assessment | Child/researcher | 20–30 minutes |
| Break | ||
| Cardiovascular assessment | Child/researcher | 15 minutes |
| Thank you and assessment summary | Parent/child/researcher | 5 minutes |
The assessments took approximately 2.5 hours, excluding rest breaks (Table 1). Additional data were collected by the parental questionnaire and completed prior to the follow-up assessment. The assessment included well-established, valid and reliable standardised measures selected to provide a comprehensive neurodevelopmental and cardiovascular assessment together with the collection of core information on the child’s general health and well-being.
Once the assessments were complete, the researcher thanked the child and their parent/carer for their participation and parents/carers and children were given the opportunity to ask any further questions. They were advised that once the results of the assessment had been scored and interpreted, they would receive a report which would summarise the neurodevelopmental findings. Where specifically requested, a more detailed report was sent to the child’s GP and/or paediatrician who they would be able to contact if they should require any further information regarding these outcomes. The children were given a pack for participating in the study which included a certificate and a small gesture of thanks (e.g. age appropriate book) and parents/carers were given a voucher to the value of £10 to reimburse them for their time and any travel expenses.
Where potential participants cancelled or failed to attend follow-up appointments on more than three occasions, they were invited to participate remotely. All such participants received a follow-up questionnaire pack which included all questionnaires detailed as part of the main study in addition to the Ages and Stages Questionnaire-3 (in place of the BSID-III, neurodevelopmental assessment). A separate participant information sheet and consent form was also included in this pack, which had been modified to reflect the changes for remote participation in the study. This pack was also sent to potential participants who were hard-to-reach in the initial contact phase. Those participants invited to take part remotely who did not respond within 4 weeks of the remote participation pack being sent were contacted by telephone by a member of the core research team. This was to check whether the participant had received their pack and if they would like to discuss their potential participation further. During this telephone contact, participants interested in taking part were given the option to provide consent and complete the follow-up questionnaires via the paper-based method or verbally over the telephone.
Assessment methods
The Bayley Scales of Infant and Toddler Development – III
Bayley Scales27 were used as a standard measure of cognitive, language and motor development. This is an individually administered instrument designed to measure the developmental functioning of infants and toddlers. Specific purposes of the BSID-III are to identify possible developmental delay, inform professionals about specific areas of strength or weakness when planning a comprehensive intervention, and provide a method of monitoring a child’s developmental progress. The BSID-III is appropriate for administration to children between the ages of 1 and 42 months (although norms extend downward to age 16 days). 29
Hempel’s neurological examination for toddler age
The Hempel neurological examination for toddlers28 was used to identify major neurological impairment/CP and to detect subtle deviations from typical neurological and neuromotor function. The Hempel examination is a video-recorded assessment that has been developed to evaluate minor neurological dysfunction (MND) at preschool age. The Hempel examination assesses MND in five domains of function: fine motor, gross motor, posture and muscle tone, reflexes and visuo-motor function. 30 Each domain is scored as typical or deviant. All findings are then classified as being major neurological dysfunction, complex MND, simple MND, or neurologically normal. Major neurological dysfunction implies the presence of a distinct neurological syndrome, such as CP. In order to be categorised as complex MND, the presence of two or more deviant domains is required; simple MND implies the presence of just one deviant domain. Neurologically normal must have no deviant domains or the presence of only deviant reflexes. 30 Simple MND has limited clinical significance and reflects the presence of a normal, but non-optimally wired brain. On the other hand, complex MND represents the clinically relevant form of MND and is associated with behavioural and learning disorders. 31 The reliability of the Hempel examination is satisfactory (κ scores for various items: 0.62–1.00).
Where it was not feasible to administer the Hempel assessment scores, owing to difficulties/lack of consent in recording the session parental report of, a CP diagnosis was used.
Health status classification system – preschool version
The health status classification system – preschool version (HSCS-PS) is a parental (or clinician) proxy measurement of the health status of a child. The instrument includes 10 mutually exclusive domains, that is, ‘Vision’, ‘Hearing’, ‘Speech’, ‘Mobility’, ‘Dexterity’, ‘Self-care’, ‘Emotion’, ‘Learning and remembering’, ‘Thinking and problem solving’, ‘Pain and discomfort’ as well as two additional parent-reported single-item measures: ‘General health’ and ‘Behaviour’. There are 10 domains each with 3–5 levels, and the two additional items. The overall health status is described as a 10-element vector consisting of one level for each of the domains. In this study, to facilitate comparisons between groups, a total ‘disability score’ for the overall health state of a child was calculated as the sum of the level codes for the original domains. Therefore, the range of the disability score varied from 10 (no disability on any domain) to 41 (maximum disability on all 10 domains). 32
Child behaviour checklist 1.5–5 parent form
The child behaviour checklist (CBCL)33 was used to assess emotional and behavioural difficulties. The CBCL includes 100 items that address emotional and behavioural problems, which are scored by parents on a three-point scale: not true, somewhat or sometimes true, and very true or very often true. The sum of all questions results in the total problem score (TPS), an internalising problem score (IPS: emotionally reactive, anxious or depressed, somatic complaints, and withdrawn) and an externalising problem score (EPS: attention problems and aggressive behaviour). Raw scores are normalised into T-scores (mean: 50, SD: 10). Higher T-scores represent more problematic behaviour. T-scores below 60 are in the normal range, T-scores of 60–63 (84th–90th percentile) are in the borderline range, and T-scores above 63 (above 90th percentile) are in the clinical range. The T-scores are dichotomised into typical (scores in the normal range) and atypical (scores in the borderline and clinical range). The reliability and validity of the CBCL are good. 33
Behaviour rating inventory of executive function – preschool version
Behaviour rating inventory of executive function – preschool version (BRIEF-P)34 is a parent questionnaire for early assessment of executive function. The BRIEF is a standardised questionnaire completed by the primary caregiver or parent and has extensive evidence from research and clinical settings to assess severity of executive dysfunction in day-to-day situations. 34 It comprises three broad indices (General Executive Composite, the Metacognitive Index and the Behavior Regulation Index) and eight subscales. The Metacognitive Index has a further five subscales: initiate (how well an individual independently initiates tasks), working memory (holding information in mind, manipulating information in mind), planning/organisation (using systematic, well planned approaches to tasks), organisation of materials and monitor (monitoring behaviour, or task approach). The Behavior Regulation Index has three subscales: inhibit (an index of impulsive behaviour or acting before thinking), shift (the ability to maintain a flexible approach to problem solving or behaviour) and emotional control (the ability to manage and regulate emotional responses). Age-based T-scores are computed for each subscale and index, and a score of 65 or higher is considered a clinically significant problem.
Cardiovascular
Cardiovascular assessments were carried out using standard BP equipment. Prior to the assessments the researcher ensured that the following was controlled: (1) room temperature – environment kept at 22°C ± 1°C; (2) participants were asked to be in a recumbent, supine position; and (3) the researcher was aware of the effect of cardiac arrhythmia and white coat hypertension on measurements. Where children were fearful or distressed, a note was made in the assessment notes, and if necessary the assessment was stopped and the most recent information from the child’s medical notes where used (where applicable).
Chapter 4 Results
Trial population
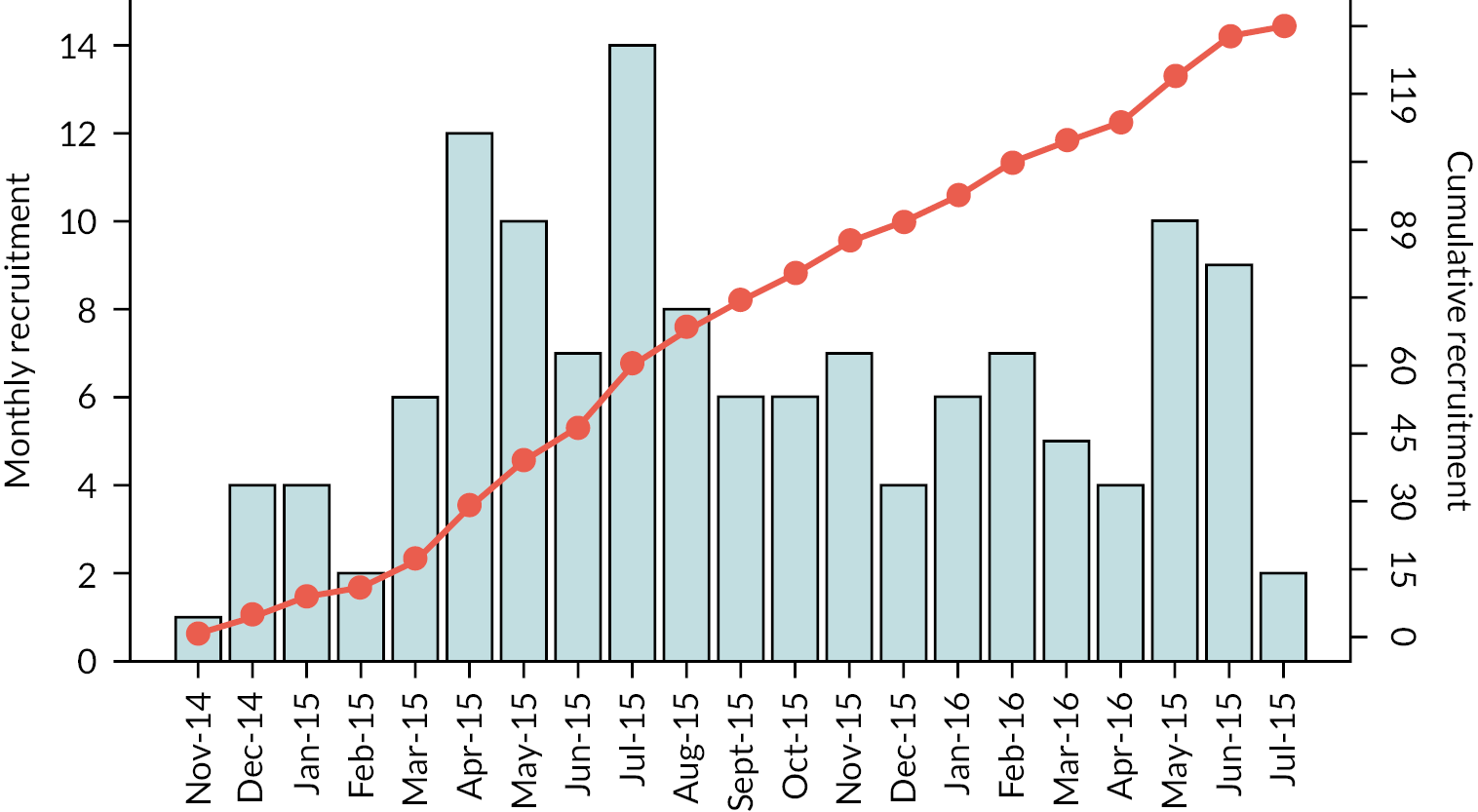
One hundred and thirty-five participants were recruited to the STRIDER trial between 21 November 2014 and 6 July 2016 from 19 fetal medicine units within the UK (Figure 2). A number of 75 participants were recruited before 26+0 weeks’ gestation and 60 between 26+0 and 29+6 weeks’ gestation. A number of 70 participants were randomly assigned to receive sildenafil and 65 to receive the placebo. None of the participants withdrew their consent nor were lost to follow-up; therefore, additional ‘per-protocol’ analysis was not performed.
FIGURE 2.
Cumulative monthly recruitment to the STRIDER trial.
There were no clinically important differences found between the sildenafil arm and the placebo arm for ethnicity, age, body mass index (BMI), parity and pre-existing pre-eclampsia, but more participants self-reported smoking in pregnancy in the sildenafil arm (17% compared 3%; Table 2).
| Sildenafil N = 70 |
Placebo N = 65 |
|
|---|---|---|
| Age (years) | 29 (26–34) | 33 (28–36) |
| Height (cm) | 164 (158–167) | 163 (158–166) |
| Weight (kg) | 68 (59–82) | 70 (60–82) |
| BMI | 25 (23–32) | 26 (23–31) |
| Ethnicity | ||
| White | 48 (69%) | 43 (66%) |
| Asian | 6 (9%) | 8 (12%) |
| African | 6 (9%) | 7 (11%) |
| Other | 10 (14%) | 7 (11%) |
| Current smoker | 12 (17%) | 2 (3%) |
| Non-smoker | 58 (83%) | 63 (97%) |
| Nulliparous | 35 (50%) | 25 (38%) |
| Pre-eclampsia | 13 (19%) | 11 (17%) |
| Gestational hypertension | 12 (17%) | 23 (35%) |
| Current antihypertensive treatment | 25 (36%) | 27 (42%) |
| Gestational diabetes | 2 (3%) | 3 (5%) |
| Antepartum haemorrhage | 1 (1%) | 0 |
| Preterm prelabour rupture of membranes | 0 | 1 (2%) |
| Gestation at randomisation (weeks) | 25.1 (24.0–27.5) | 25.6 (24.1–27.4) |
| Gestation < 26+0 weeks | 40 (57%) | 35 (54%) |
| Umbilical artery Doppler | ||
| End-diastolic flow absent | 46 (66%) | 45 (69%) |
| End-diastolic flow Reversed | 24 (34%) | 20 (31%) |
| Absent ductus venosus a-wave | 4 (6%) | 4 (6%) |
| Uterine artery doppler abnormala | 50/63 (79%) | 45/63 (78%) |
| Estimated fetal weight (g) | 451 (352–613) | 436 (326–594) |
| Estimated fetal weight < 500 g | 33 (47%) | 36 (55%) |
| Systolic blood pressure (mmHg) | 135.5 (125.5–147.5) | 134.0 (120.5–144.5) |
| Diastolic blood pressure (mmHg) | 88.5 (80.5–95.5) | 86.5 (78.0–94.5) |
| Mean arterial pressure (mmHg) | 103 (12) | 109 (38) |
| Creatinine (μmol/L) | 57.4 (1.9) | 62.4 (2.7) |
| Urea (mmol/L) | 4.0 (0.2) | 4.4 (0.5) |
| Uric acid (mmol/L) | 300.6 (13.4) | 288.6 (14.7) |
| Aspartate transaminase (U/L) | 26.0 (3.3) | 32.4 (5.7) |
| Albumin (g/L) | 31.8 (0.7) | 32.4 (0.7) |
| Platelets (×109/L) | 277.1 (10.2) | 233.5 (9.5) |
The median gestation at randomisation was 24.4 weeks [interquartile range (IQR) 24.0–27.5; see Table 2]. At randomisation, a reversed Doppler umbilical artery end-diastolic flow was detected in 44 (33%) participants (see Table 2). An absent umbilical artery end-diastolic flow was detected in all of the remaining participants. The fetal ductus venosus a-wave was found to be absent or reversed in 8 (6%) participants (see Table 2). The estimated fetal weight at randomisation was 445 g (IQR 344–608; Table 2). A total of 69 (51%) fetuses had an estimated fetal weight below 500 g (see Table 2). Two babies were postnatally diagnosed with Down syndrome (one allocated to the sildenafil arm and the other to the placebo arm) and two had confirmed cytomegalovirus infection (one allocated to the sildenafil arm and the other to placebo arm); all four babies were included in the ITT analysis.
The phase 2 follow-up study of surviving infants (aged 2–3 years corrected), born to mothers who took part in STRIDER UK, included the 75 babies discharged alive from the neonatal unit. One baby died after discharge and before follow-up (treatment arm – placebo). After extensive attempts to contact participants, 3 declined further involvement and 10 were uncontactable; therefore, 61 babies were included in the follow-up phase. Core descriptive statistics of the study sample can be found in Table 4. The mothers of 32 of the babies assessed had received sildenafil when pregnant and 29 had received placebo. There was no difference in the sex, birthweight, gestation at delivery (median 29.214 weeks vs. 29.857 weeks), mode of delivery or oxygen usage between babies whose mothers had received sildenafil or placebo.
Primary end point
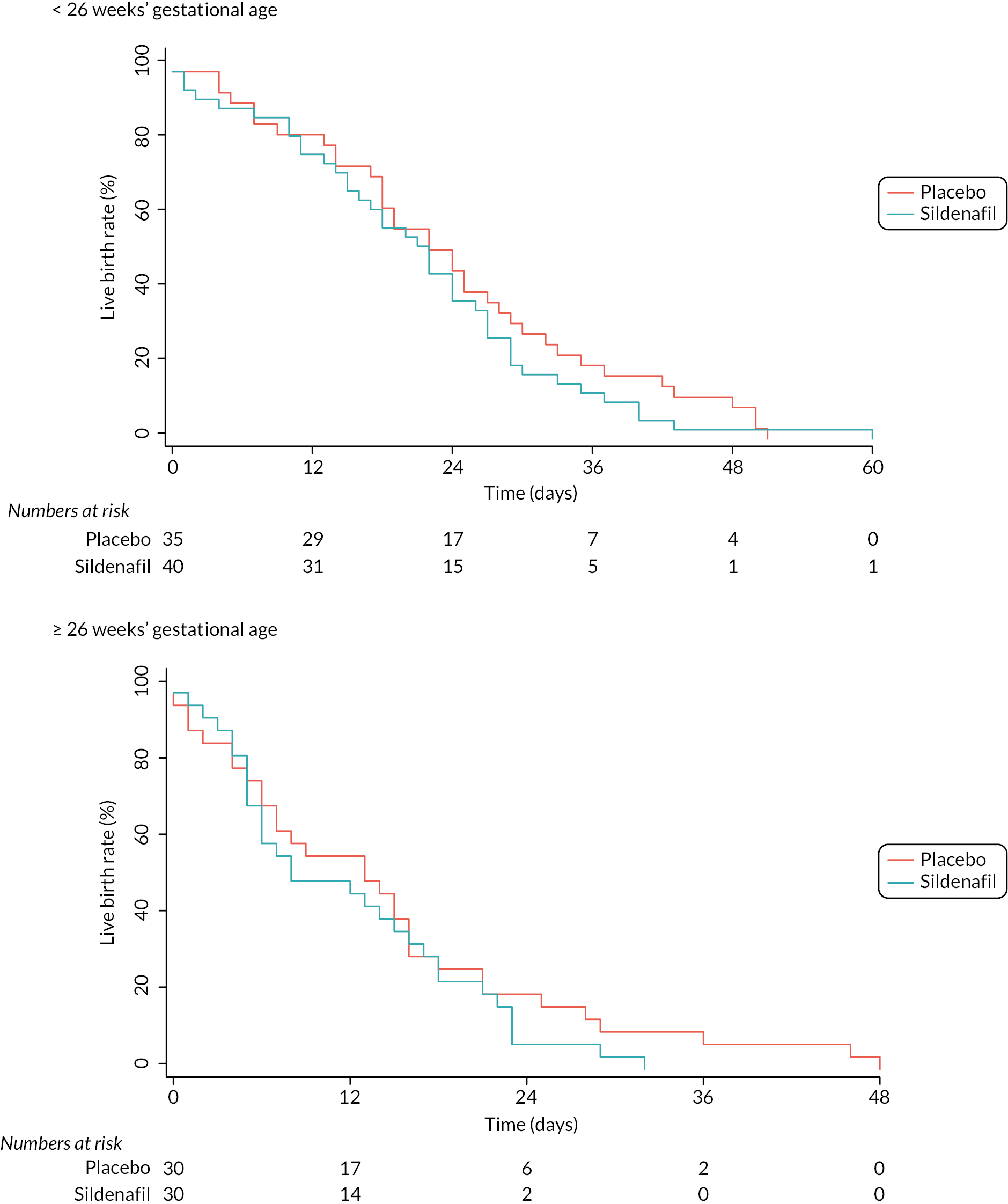
The median time between randomisation and delivery was 18 days (IQR 8–27); 17 days (7–24) in the sildenafil arm and 18 days (8–28) in the placebo arm (p = 0.23; Table 3 and Figure 3). Linear regression showed that time to delivery did not differ between the two treatment arms for all participants (2.7 days, 95% CI −1.3 to 6.8; p = 0.19; Figure 3).
| Sildenafil N = 70 |
Placebo N = 65 |
p-value | |
|---|---|---|---|
| Randomisation to delivery interval (days) | 17 (7–24) | 18 (8–24) | 0.23 |
| < 26+0 weeks’ gestation | 22 (14–29) | 24 (16–33) | 0.36 |
| ≥ 26+0 weeks’ gestation | 10 (5–8) | 14 (6–20) | 0.34 |
| Gestation (weeks) | 28.1 (26.7–29.7) | 28.4 (27.3–30.1) | 0.28 |
| < 26+0 weeks’ gestation | 26.9 (26.1–28.3) | 27.6 (26.3–28.5) | 0.55 |
| ≥ 26+0 weeks’ gestation | 29.7 (28.3–30.7) | 29.6 (28.4–30.9) | 0.41 |
FIGURE 3.
Kaplan–Meier plot of interval from randomisation to birth. (a) < 26 weeks’ gestational age. (b) ≥ 26 weeks’ gestational age.
Secondary end points
Of the 135 participants recruited to the STRIDER trial, 98 (73%) had at least two separate umbilical artery Doppler measurements with a minimum 48 hours apart, 93 (69%) for ductus venosus, 90 (67%) for middle cerebral artery and 87 (64%) for uterine arteries (see Table 3). Ductus venosus a-wave deteriorated over time in more participants treated with sildenafil than with placebo (Table 5). Eighteen (95%) of the 19 babies in whom the ductus venosus deteriorated were randomly assigned before 26+0 weeks’ gestation (see Table 4). Between-group differences were not observed in the pattern of Doppler changes for any of the other fetal vessels examined (middle cerebral artery, umbilical artery and uterine arteries; see Table 4).
| Sildenafil | Placebo | RR (95% CI) | |
|---|---|---|---|
| Umbilical artery Doppler (all) | N = 51 | N = 47 | |
| Improvement | 5 (10%) | 5 (11%) | 0.92 (0.28 to 2.98) |
| No change | 25 (49%) | 25 (53%) | 0.92 (0.63 to 1.36) |
| Deterioration | 21 (41%) | 17 (36%) | 1.14 (0.69 to 1.88) |
| Umbilical artery Doppler (< 26+0 weeks) | N = 35 | N = 28 | |
| Improvement | 4 (11%) | 3 (11%) | 1.07 (0.26 to 4.38) |
| No change | 16 (46%) | 14 (50%) | 0.91 (0.55 to 1.53) |
| Deterioration | 15 (43%) | 11 (39%) | 1.09 (0.60 to 1.99) |
| Ductus venosus a-wave (all) | N = 51 | N = 42 | |
| Improvement | 0 | 0 | – |
| No change | 36 (71%) | 38 (90%) | 0.78 (0.64 to 0.96) |
| Deterioration | 15 (29%) | 4 (10%) | 3.09 (1.11 to 8.60) |
| Ductus venosus a-wave (< 26+0 weeks) | N = 35 | N = 24 | |
| Improvement | 0 | 0 | – |
| No change | 21 (60%) | 20 (83%) | 0.72 (0.52 to 1.00) |
| Deterioration | 14 (40%) | 4 (17%) | 2.40 (0.90 to 6.41) |
| Middle cerebral artery (all) | N = 50 | N = 40 | |
| Improvement | 4 (8%) | 2 (5%) | 1.60 (0.31 to 8.30) |
| No change | 33 (66%) | 24 (60%) | 1.10 (0.80 to 1.52) |
| Deterioration | 13 (26%) | 14 (35%) | 0.74 (0.40 to 1.39) |
| Middle cerebral artery (< 26+0 weeks) | N = 34 | N = 25 | |
| Improvement | 1 (3%) | 1 (4%) | 0.74 (0.05 to 11.2) |
| No change | 23 (68%) | 16 (64%) | 1.06 (0.73 to 1.54) |
| Deterioration | 10 (29%) | 8 (32%) | 0.92 (0.42 to 1.99) |
| Uterine artery Doppler (all) | N = 45 | N = 42 | |
| Improvement | 41 (91%) | 36 (86%) | 1.06 (0.91 to 1.24) |
| No change | 1 (2%) | 3 (7%) | 0.31 (0.03 to 2.88) |
| Deterioration | 3 (7%) | 3 (7%) | 0.93 (0.19 to 4.38) |
| Abdominal circumference change (mm) | N = 46 | N = 41 | |
| All participants | 14 (6–20) | 18 (8–25) | −4.5 (−9.5 to 4.5) |
| < 26+0 weeks | 14 (8–21) | 15 (7–29) | 1.0 (−10.1 to 6.5) |
| ≥ 26+0 weeks | 12 (4–17) | 19 (14–21) | −7.0 (−17.6 to 8.5) |
| Pre-eclampsia | |||
| All participants | 15/70 (21%) | 12/65 (18%) | 1.16 (0.59 to 2.29) |
| < 26+0 weeks | 8/40 (20%) | 6/35 (17%) | 1.17 (0.45 to 3.04) |
| Antenatal corticosteroids | |||
| All participants | 41/70 (59%) | 37/65 (57%) | 1.03 (0.77 to 1.37) |
| < 26+0 weeks | 21/40 (53%) | 17/35 (49%) | 1.08 (0.69 to 1.70) |
| MgSO4 for neuroprotection | |||
| All participants | 40/70 (57%) | 25/65 (38%) | 1.49 (1.03 to 2.14) |
| < 26+0 weeks | 20/40 (50%) | 12/35 (34%) | 1.46 (0.84 to 2.54) |
| Caesarean section | |||
| All participants | 47/70 (67%) | 43/65 (66%) | 1.01 (0.80 to 1.29) |
| < 26+0 weeks | 20/40 (50%) | 15/35 (43%) | 1.17 (0.71 to 1.91) |
The exposure to antenatal corticosteroids and magnesium sulphate, given for neuroprotection, was similar in both treatment arms (see Table 4). There was also no difference in the caesarean section rate between the arms (see Table 4), with 98% (90 of 92) of all livebirths being delivered by caesarean section (Table 5).
Livebirth rates and neonatal deaths did not differ between the treatment arms (see Table 5). Forty-three (72%) of the 60 deaths reported occurred in utero and 48 (80%) deaths occurred in the subgroup randomly assigned before 26+0 weeks’ gestation (see Table 5). No clinically significant differences were observed between the two treatment arms for any of the other pre-specified secondary end points (see Table 5).
| Sildenafil | Placebo | RR | p-value | |
|---|---|---|---|---|
| (N = 70) | (N = 65) | (95% CI) | ||
| Live births | 49 (70%) | 43 (66%) | 1.06 (0.84 to 1.33) | 0.62 |
| < 26+0 weeks’ gestation | 22 (31%) | 15 (23%) | 1.28 (0.8 to 2.06) | 0.31 |
| ≥ 26+0 weeks’ gestation | 27 (39%) | 28 (43%) | 0.96 (0.83 to 1.12) | 0.59 |
| Fetal death | 21 (30%) | 22 (34%) | 0.89 (0.54 to 1.45) | 0.64 |
| < 26+0 weeks’ gestation | 18 (26%) | 20 (31%) | 0.79 (0.5 to 1.23) | 0.31 |
| ≥ 26+0 weeks’ gestation | 3 (4%) | 2 (3%) | 1.50 (0.27 to 8.34) | 0.64 |
| Neonatal death | 10 (14%) | 7 (11%) | 1.33 (0.54 to 3.28) | 0.53 |
| < 26+0 weeks’ gestation | 6 (9%) | 4 (6%) | 1.31 (0.40 to 4.28) | 0.65 |
| ≥ 26+0 weeks’ gestation | 4 (6%) | 3 (5%) | 1.33 (0.33 to 5.45) | 0.69 |
| Neonatal morbidity | 37/49 (76%) | 28/43 (65%) | 1.23 (0.86 to 1.75) | 0.25 |
| < 26+0 weeks’ gestation | 20/22 (91%) | 13/15 (87%) | 1.35 (0.79 to 2.29) | 0.27 |
| ≥ 26+0 weeks’ gestation | 17/27 (63%) | 15/28 (54%) | 1.13 (0.70 to 1.82) | 0.62 |
| Infants with composite perinatal adverse outcome (perinatal death or neonatal morbidity) | 58 (83%) | 50 (77%) | 1.08 (0.91 to 1.28) | 0.38 |
| < 26+0 weeks’ gestation | 37/40 (93%) | 33/35 (94%) | 0.98 (0.87 to 1.11) | 0.74 |
| ≥ 26+0 weeks’ gestation | 21/30 (70%) | 17/30 (57%) | 1.24 (0.84 to 1.83) | 0.28 |
| Birthweight (g) | 604 (496–766) | 590 (430–842) | −14 (−100 to 126) | 0.81 |
| < 26+0 weeks’ gestation | 520 (355–602) | 450 (356–579) | −70 (−123 to 40) | 0.32 |
| ≥ 26+0 weeks’ gestation | 750 (663–1073) | 856 (611–1015) | 106 (−129 to 236) | 0.25 |
| Infants admitted to NICU | 47/49 (96%) | 38/43 (88%) | 1.09 (0.96 to 1.23) | 0.17 |
| < 26+0 weeks’ gestation | 21/22 (95%) | 14/15 (93%) | 1.02 (0.87 to 1.20) | 0.81 |
| ≥ 26+0 weeks’ gestation | 26/27 (96%) | 24/28 (86%) | 1.12 (0.95 to 1.33) | 0.19 |
| Days on NICU | 25 (10–50) | 16 (8–55) | −9 (−18 to 2) | 0.24 |
| < 26+0 weeks’ gestation | 25 (11–58) | 39 (12–57) | 15 (−17 to 31) | 0.24 |
| ≥ 26+0 weeks’ gestation | 25 (10–46) | 15 (7–35) | −11 (−28 to 11) | 0.29 |
| Age at NICU discharge (days) | 79 (50–106) | 73 (51–100) | −6 (−24 to 11) | 0.50 |
| < 26+0 weeks’ gestation | 97 (73–109) | 87 (74–112) | 10 (−32 to 27) | 0.48 |
| ≥ 26+0 weeks’ gestation | 59 (46–84) | 63 (46–94) | 4 (−24 to 16) | 0.73 |
| Oxygen dependency at 28 days | 23/49 (47%) | 14/43 (33%) | 1.44 (0.85 to 2.43) | 0.17 |
| < 26+0 weeks’ gestation | 12/22 (55%) | 6/15 (40%) | 1.36 (0.66 to 2.82) | 0.41 |
| ≥ 26+0 weeks’ gestation | 11/27 (41%) | 8/28 (29%) | 1.43 (0.68 to 2.99) | 0.34 |
| Oxygen dependency at 36 weeks | 10/49 (20%) | 7/43 (16%) | 1.25 (0.52 to 3.01) | 0.62 |
| < 26+0 weeks’ gestation | 6/22 (27%) | 2/15 (13%) | 2.05 (0.48 to 8.80) | 0.33 |
| ≥ 26+0 weeks’ gestation | 4/27 (15%) | 5/28 (18%) | 0.83 (0.25 to 2.77) | 0.76 |
| Necrotising enterocolitis | 8/49 (16%) | 12/43 (28%) | 0.59 (0.26 to 1.30) | 0.20 |
| < 26+0 weeks’ gestation | 5/22 (23%) | 5/15 (33%) | 0.68 (0.24 to 1.95) | 0.47 |
| ≥ 26+0 weeks’ gestation | 3/27 (11%) | 7/28 (25%) | 0.44 (0.13 to 1.54) | 0.19 |
| Retinopathy of prematurity | 6/49 (12%) | 10/43 (23%) | 0.54 (0.21 to 1.36) | 0.20 |
| < 26+0 weeks’ gestation | 3/22 (14%) | 4/15 (27%) | 0.54 (0.14 to 2.05) | 0.37 |
| ≥ 26+0 weeks’ gestation | 3/27 (11%) | 6/28 (21%) | 0.52 (0.14 to 1.87) | 0.32 |
| Any intracranial haemorrhage | 13/39 (33%) | 8/33 (24%) | 1.37 (0.65 to 2.91) | 0.41 |
| < 26+0 weeks’ gestation | 6/19 (32%) | 4/12 (33%) | 0.95 (0.34 to 2.68) | 0.92 |
| ≥ 26+0 weeks’ gestation | 7/20 (35%) | 4/21 (19%) | 1.84 (0.63 to 5.33) | 0.26 |
| Surfactant used | 37/49 (76%) | 25/43 (58%) | 1.30 (0.96 to 1.75) | 0.078 |
| < 26+0 weeks’ gestation | 19/22 (86%) | 9/15 (60%) | 1.44 (0.92 to 2.25) | 0.11 |
| ≥ 26+0 weeks’ gestation | 18/27 (67%) | 16/28 (57%) | 1.17 (0.77 to 1.77) | 0.46 |
| Ventilator dependency | 40/49 (82%) | 28/43 (65%) | 1.25 (0.97 to 1.62) | 0.073 |
| < 26+0 weeks’ gestation | 19/22 (86%) | 12/15 (80%) | 1.08 (0.80 to 1.46) | 0.62 |
| ≥ 26+0 weeks’ gestation | 21/27 (78%) | 16/28 (57%) | 1.36 (0.93 to 1.99) | 0.11 |
| Ventilator days | 7 (2–21) | 10 (3–27) | −3 (−12 to 7) | 0.60 |
| < 26+0 weeks’ gestation | 12 (5–23) | 12 (7–24) | 0 (−17 to 11) | 1.00 |
| ≥ 26+0 weeks’ gestation | 3 (1–17) | 6 (3–29) | −3 (−16 to 11) | 0.66 |
Serious adverse events and adherence
Adverse events (AEs) were recorded as part of routine medical data review. AEs were not graded.
A total of eight SAEs, graded as per medDRA, were reported during the course of the study; none of these were attributed to sildenafil. Three (38%) were maternal hospital admissions in the placebo arm; one antepartum haemorrhage, one general malaise (unwell, dizzy and light-headed), and one hospital admission following a stillbirth with drowsiness. There were two reported neonatal SAEs in the sildenafil arm; a baby with Down syndrome with an atrioventricular septal defect and a fetal intracranial haemorrhage grade 1, which was detected on an antenatal MRI performed in the context of a separate research study. In the placebo arm, three neonatal SAEs were reported; a baby with Down syndrome, a fetal intracranial haemorrhage grade 1 and a baby with bone fractures that were postnatally attributed to osteopenia/metabolic bone disease.
Overall, 42 women reported 94 side effects: 24 (34%) in the sildenafil arm and 18 (28%) in the placebo arm (RR 1.24, 95% CI 0.74 to 2.06; p = 0.41). The majority of the side effects reported were for facial flushing [45 of 94 (48%)]. Other reported side effects included nasal congestion, a dry mouth and headaches.
Good treatment adherence was reported; of the 265 recorded cycles of therapy, 257 (97%) reported that drug adherence was at least 90%. At a participant level, 130 (96%) of the 135 participants had study drug compliance of at least 90% for all cycles of therapy.
Phase 2 end points
Child neurodevelopmental assessments were performed on all eligible infants in their home setting or a local hospital at the parents request as described above. Demographic data are presented in Table 6.
| Covariate | Sildenafil N = 32 |
Placebo N = 29 |
Relative risk (95% CI) |
p-value | |
|---|---|---|---|---|---|
| Gestation at birth (weeks) | Median (IQR) | 29.21 (28.07, 30.28) |
29.85 (28.42, 31) |
– | 0.28 |
| Mode of delivery | Emergency caesarean section | 12 (38%) | 8 (28%) | 1.36 (0.65 to 2.85) |
|
| Pre-labour caesarean section | 18 (56%) | 21 (72%) | 0.78 (0.53 to 1.14) |
||
| Vaginal | 2 (6%) | 0 (0%) | – | – | |
| Sex of child | Female | 12 (38%) | 11 (38%) | 0.99 (0.60 to 1.62) |
0.97 |
| Birthweight | Median (IQR) | 750 (597.5, 945.75) |
800 (610, 1000) |
– | 0.81 |
| Oxygen dependency at 28 days | Yes | 16 (50%) | 11 (38%) | 1.32 (0.74 to 2.36) |
0.44 |
| Oxygen dependency at 36 weeks | Yes | 9 (28%) | 4 (13%) | 2.04 (0.70 to 5.92) |
0.41 |
| Surfactant use | Yes | 24 (75%) | 16 (55%) | 1.36 (0.93 to 1.99) |
0.11 |
| Ventilator dependency | Yes | 25 | 18 | 1.26 (0.89 to 1.77) |
0.26 |
The Bayley assessment of child neurodevelopment at 2 years of age (corrected) is shown in Table 7. No significant differences in cognitive, language (including receptive and expressive language) or motor (including fine and gross motor) subscales were found between children of sildenafil- and placebo-treated mothers. Total scores reveal a trend of being lower than expected across all three domains compared with standard population norms (i.e. 100, SD = 15); however, this was not found to be statistically significant. No difference was found between the sildenafil and placebo groups for presence of CP at 2 years, as reported by the parent.
| Covariate | Sildenafil N = 32 |
Placebo N = 29 |
p-value | |
|---|---|---|---|---|
| Survivors assessed for neurodevelopment | 26 (80%) | 24 (83%) | ||
| BSID COGNITIVE Composite score | Median (IQR) | 92.5 (90, 103.75) | 90 (80, 100) | 0.139a |
| BSID LANGUAGE Composite score | Median (IQR) | 89 (86, 91) | 86 (78.5, 91) | 0.352a |
| BSID MOTOR Composite score | Median (IQR) | 88 (82, 94) | 91 (84.25, 100) | 0.507a |
| Cerebral palsy | Yes | 4 | 5 |
Table 8 shows the physical characteristics of the population. There was no difference in height or weight. Head circumference was slightly larger in those treated with sildenafil (49.25, 46.4–50.262) versus placebo (47.175, 44.713–48.95). There was no difference between systolic and diastolic BP between those children treated with sildenafil or placebo. Median values were appropriate for children aged 2 years.
| Covariate | Sildenafil N = 32 |
Placebo N = 29 |
p-value | |
|---|---|---|---|---|
| Survivors assessed | 26 (80%) | 24 (83%) | ||
| Head circumference (cm) | Median (IQR) | 49.25 (46.43, 50.26) |
47.18 (44.71, 48.95) |
0.02 |
| Height (cm) | Median (IQR) | 86.35 (83.08, 90.19) |
85.23 (80.95, 87.65) |
0.30 |
| Weight (kg) | Median (IQR) | 10.53 (9.80, 11.77) |
10.08 (9.30, 11.61) |
0.37 |
| Systolic blood pressure (mmHg) | Median (IQR) | 95.25 (90.75, 104.63) |
100.25 (91.38, 107.38) |
0.38 |
| Diastolic blood pressure (mmHg) | Median (IQR) | 60 (58.50, 62.88) |
62.25 (58.13, 65.13) |
0.43 |
The BRIEF-P (Table 9) demonstrated no difference in adjusted T-scores between sildenafil and placebo for any of the assessed domains. The median total CBCL scores and adjusted T-scores (Table 10) showed no difference between infants whose mothers were treated with sildenafil versus placebo for any of the assessed domains.
| Sildenafil N = 32 |
Placebo N = 29 |
p-value | ||
|---|---|---|---|---|
| Inhibit | Sum | 1 (0, 2.75) [0, 5] | 1 (0, 3) [0, 14] | |
| T-score | 49 (48, 52) [24, 54] | 51 (50, 53) [50, 60] | 0.826 | |
| Shift | Sum | 4 (2, 6) [0, 14] | 4 (2.75, 9) [0, 19] | |
| T-score | 53 (51, 56) [48, 66] | 53 (52, 59) [48,73] | 0.561 | |
| Emotional control | Sum | 3 (0.5, 7) [0, 16] | 3 (2, 10.25) [0, 17] | |
| T-score | 53 (50, 58) [50, 70] | 53 (52, 62) [50, 71] | 0.391 | |
| Working memory | Sum | 8 (4, 15) [1, 26] | 10.5 (3, 14) [0, 27] | |
| T-score | 55 (52, 59) [51, 65] | 56 (52, 58) [50, 65] | 0.600 | |
| Plan/organize | Sum | 3 (1, 6.5) [0, 15] | 4 (2, 6) [0, 16] | |
| T-score | 53 (51, 57) [50, 66] | 54 (51, 56) [50, 67] | 0.813 |
| Sildenafil | Placebo | p-value | ||
|---|---|---|---|---|
| Emotionally reactive | Sum | 1 (0, 2.75) [0, 5] | 1 (0, 3) [0, 14] | |
| T-score | 50 (50, 54) [50, 62] | 50 (50, 55) [50, 87] | 0.700 | |
| Anxiety/depression | Sum | 1.5 (0, 3) [0, 7] | 2 (0.75, 2.25) [0, 12] | |
| T-score | 50 (50, 51) [50, 66] | 50 (50, 50.25) [50, 74] | 0.594 | |
| Somatic complaints | Sum | 1.5 (1, 3.75) [0, 9] | 2 (1, 3) [0, 8] | |
| T-score | 51.5 (50, 61) [50, 74] | 53 (50, 58) [50, 72] | 0.941 | |
| Withdrawn | Sum | 1 (0, 2.75) [0, 7] | 1 (1, 2.5) [0, 14] | |
| T-score | 51 (50, 59) [50, 73] | 51 (51, 57.75) [50, 94] | 0.472 | |
| Sleep problems | Sum | 1 (0, 2.75) [0, 10] | 2 (0, 3.25) [0, 10] | |
| T-score | 50 (50, 52.5) [50, 76] | 51 (50, 53.75) [50, 76] | 0.405 | |
| Attention problems | Sum | 3 (1, 5) [0, 7] | 2 (1, 4) [0, 10] | |
| T-score | 53 (50, 62) [50, 70] | 51 (50, 57) [50, 80] | 0.384 | |
| Aggressive behaviour | Sum | 7.5 (5.25, 12) [0, 23] | 6 (3, 11.5) [0, 19] | |
| T-score | 51 (51, 53) [50, 68] | 51 (50, 53) [50, 63] | 0.333 | |
| Internalising | Sum | 5 (3, 11.5) [0, 27] | 6 (4.5, 9.5) [0, 41] | |
| T-score | 52 (50.312, 55.94) [50, 68] | 51.8 (51, 54.8) [50, 76.2] | 0.6024 | |
| Externalising | Sum | 10 (6.25, 16.75) [0, 28] | 7 (4, 16) [0, 26] | |
| T-score | 52 (50.5, 56.875) [50, 65] | 51 (50, 55.25) [50, 69.5] | 0.2873 |
The HSCS-PS scores are shown as a total score by domain and as individual components (Table 11). There was no difference between infants who had received sildenafil and those who had received placebo for any of the domains assessed.
| Question | Treatment | Answer | ||||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | ||
| Vision_a | Placebo (n = 28) | 3 (11%) | 23 (82%) | 2 (7%) | 0 (0%) | 0 (0%) |
| Sildenafil (n = 30) | 6 (20%) | 22 (73%) | 2 (7%) | 0 (0%) | 0 (0%) | |
| Total (n = 58) | 9 (16%) | 45 (78%) | 4 (7%) | 0 (0%) | 0 (0%) | |
| Vision_b | Placebo (n = 28) | 2 (7%) | 24 (86%) | 1 (4%) | 0 (0%) | 1 (4%) |
| Sildenafil (n = 30) | 3 (10%) | 25 (83%) | 2 (7%) | 0 (0%) | 0 (0%) | |
| Total (n = 58) | 5 (9%) | 49 (84%) | 3 (5%) | 0 (0%) | 1 (2%) | |
| Hearing | Placebo (n = 28) | 0 (0%) | 26 (93%) | 1 (4%) | 1 (4%) | 0 (0%) |
| Sildenafil (n = 29) | 1 (3%) | 27 (93%) | 1 (3%) | 0 (0%) | 0 (0%) | |
| Total (n = 57) | 1 (2%) | 53 (93%) | 2 (4%) | 1 (2%) | 0 (0%) | |
| Speaking | Placebo (n = 28) | 0 (0%) | 8 (29%) | 10 (36%) | 9 (32%) | 1 (4%) |
| Sildenafil (n = 30) | 0 (0%) | 7 (23%) | 15 (50%) | 6 (20%) | 2 (7%) | |
| Total (n = 58) | 0 (0%) | 15 (26%) | 25 (43%) | 15 (26%) | 3 (5%) | |
| Mobility | Placebo (n = 28) | 0 (0%) | 21 (75%) | 6 (21%) | 1 (4%) | 0 (0%) |
| Sildenafil (n = 30) | 1 (3%) | 19 (66%) | 8 (28%) | 1 (3%) | 0 (0%) | |
| Total (n = 58) | 1 (2%) | 40 (70%) | 14 (25%) | 2 (4%) | 0 (0%) | |
| Dexterity | Placebo (n = 28) | 0 (0%) | 24 (86%) | 4 (14%) | 0 (0%) | 0 (0%) |
| Sildenafil (n = 30) | 1 (3%) | 19 (63%) | 8 (27%) | 1 (3%) | 1 (3%) | |
| Total (n = 58) | 1 (2%) | 43 (74%) | 12 (21%) | 1 (2%) | 1 (2%) | |
| Self-care | Placebo (n = 28) | 0 (0%) | 15 (54%) | 6 (21%) | 5 (18%) | 2 (7%) |
| Sildenafil (n = 30) | 1 (3%) | 15 (50%) | 11 (37%) | 3 (10%) | 0 (0%) | |
| Total (n = 58) | 1 (2%) | 30 (52%) | 17 (29%) | 8 (14%) | 2 (3%) | |
| Emotion | Placebo (n = 28) | 0 (0%) | 20 (71%) | 6 (21%) | 2 (7%) | 0 (0%) |
| Sildenafil (n = 30) | 1 (3%) | 22 (73%) | 7 (23%) | 0 (0%) | 0 (0%) | |
| Total (n = 58) | 1 (2%) | 42 (72%) | 13 (22%) | 2 (3%) | 0 (0%) | |
| Learning and remembering | Placebo (n = 28) | 0 (0%) | 17 (61%) | 8 (29%) | 3 (11%) | 0 (0%) |
| Sildenafil (n = 30) | 1 (3%) | 20 (67%) | 8 (27%) | 1 (3%) | 0 (0%) | |
| Total (n = 58) | 1 (2%) | 37 (64%) | 16 (28%) | 4 (7%) | 0 (0%) | |
| Thinking and problem solving | Placebo (n = 28) | 0 (0%) | 11 (39%) | 12 (43%) | 2 (7%) | 3 (11%) |
| Sildenafil (n = 30) | 1 (3%) | 16 (53%) | 10 (33%) | 3 (10%) | 0 (0%) | |
| Total (n = 58) | 1 (2%) | 27 (47%) | 22 (38%) | 5 (9%) | 3 (5%) | |
| Pain and discomfort | Placebo (n = 28) | 0 (0%) | 19 (68%) | 8 (29%) | 1 (4%) | 0 (0%) |
| Sildenafil (n = 30) | 0 (0%) | 26 (87%) | 4 (13%) | 0 (0%) | 0 (0%) | |
| Total (n = 58) | 0 (0%) | 45 (78%) | 12 (21%) | 1 (2%) | 0 (0%) | |
| General health | Placebo (n = 28) | 0 (0%) | 15 (54%) | 11 (39%) | 2 (7%) | 0 (0%) |
| Sildenafil (n = 30) | 4 (13%) | 15 (50%) | 11 (37%) | 0 (0%) | 0 (0%) | |
| Total (n = 58) | 4 (7%) | 30 (52%) | 22 (38%) | 2 (3%) | 0 (0%) | |
| Behaviour | Placebo (n = 28) | 0 (0%) | 20 (71%) | 5 (18%) | 2 (7%) | 1 (4%) |
| Sildenafil (n = 30) | 1 (3%) | 18 (60%) | 10 (33%) | 1 (3%) | 0 (0%) | |
| Total (n = 58) | 1 (2%) | 38 (66%) | 15 (26%) | 3 (5%) | 1 (2%) | |
Methodological notes
There were a number of methodological issues that affected the collection of secondary end points. First, owing to either (1) lack of parental consent to film the Hempel assessment or (2) impracticality of filming assessments reliably in the home setting. It was not possible to record the assessment and therefore fully undertake the Hempel assessments that require scoring to be carried out and validated by a trained assessor using the recording provided. Second, it was noted that the NICOM cardiovascular assessment was found to cause a significant amount of undue stress to children taking part in the study. A number of mitigating actions were put in place to try and calm children prior to, and during the assessment, e.g. performing the assessment on a doll or teddy bear and inviting the child to join in with the task before attempting to assess the child, and carrying out the assessment towards the end of the assessment when the child would be most familiar with the assessor. Unfortunately, for the majority of children neither action allowed children to tolerate the administration of the NICOM cardiovascular test, leaving BP as the sole assessment of infant cardiovascular status.
Both issues were raised as part of ongoing central monitoring and oversight of the study with several mitigations put in place to try and resolve the issues; however, as detailed above, these did not prove to be successful. It was therefore agreed by the CI and study management team that parental report of CP (Does your child currently have a diagnosis of CP?) would be used to confirm the CP diagnostic status. These responses were also confirmed by a review of the child’s medical notes following the assessment. Where possible, all children were encouraged to complete a BP assessment which would be used as a measure of cardiovascular function. Although still noted in the researcher assessment notes as stressful for some children, this was not to the same extent as the NICOM assessment which often resulted in refusal to continue with the assessment by children and their parent. Long-term functional assessments are detailed in Tables 9 and 10.
Chapter 5 Discussion
The STRIDER RCT (randomised controlled trial) of sildenafil versus placebo, for the treatment of women carrying a singleton pregnancy affected by severe early-onset IUGR, was a pragmatic design to ensure that clinically relevant findings were assessed. The study was based on good laboratory and animal evidence of benefits of treatment with sildenafil to recover placental function. In addition, there were emerging human data showing a beneficial effect on both placental function and fetal growth as demonstrated by improvements in some assessments of fetal weight and fetal Dopplers. Sildenafil was also known to be safe in women and while not advised for use in pregnancy, there was no evidence of harm.
Of greater concern was the emerging anecdotal evidence that clinicians across the world were already beginning to prescribe sildenafil for the treatment of growth restriction with no RCT evidence of benefit. The pragmatic design to use a mixed population from across the UK and to have a primary outcome of prolongation of pregnancy by 1 week was chosen to ensure that any positive findings would be immediately translatable to clinical practice.
The study was completed on time and within budget for the RCT with good engagement from clinicians and women. Recruitment was excellent with > 80% of screened women randomised, which is particularly noticeable as this was a CTIMP (Clinical Trial of an Investigational Medicinal Product) study in pregnancy. The subsequent extended funding for the follow-up phase allowed us to ensure that any benefits or harms from treatment with sildenafil would be identified in the perinatal period and in infancy.
The STRIDER UK study showed no beneficial effect for any perinatal outcome for mother or baby from treatment with 25 mg sildenafil TDS for severe early-onset IUGR. In fact, the interval between randomisation and delivery was on average 2.7 days shorter in the sildenafil arm, although this difference did not reach conventional statistical significance in the gestational age adjusted logistic regression analysis (p = 0.19). The study also showed no clinically important differences in mortality or short-term neonatal morbidity, although the trial was not adequately powered for these secondary end points.
It was anticipated that, if sildenafil was effective, there may be a beneficial effect on placental function, as assessed by uteroplacental and fetal Doppler studies, even in the absence of a clear benefit on substantive clinical outcomes. The observed higher proportion of babies in whom Doppler findings in ductus venosus deteriorated with sildenafil treatment may have been a chance finding, but is also potentially worrying, particularly if linked to the somewhat shorter randomisation to delivery interval in this treatment arm. Interestingly, no such adverse effect from sildenafil on the blood flow in uterine arteries, umbilical artery or middle cerebral artery was found. It was not possible to obtain two separate measurements for all babies, but in this placebo-controlled study, it is very unlikely that Doppler measurements were somehow systematically biased. At present, a plausible pathophysiological explanation cannot be offered for the possible adverse effect of sildenafil on the fetal blood flow in the STRIDER cohort.
The findings of the study are in contrast with animal and several previously reported clinical studies. 16–19,21,35–40 The sildenafil dose used was based on the consensus from researchers with most experience in clinical evaluation of sildenafil in pregnancy at the time17,20 and a higher dose could possibly have been more effective. A recent systematic review identified 16 studies of sildenafil in human pregnancies, of which only four exceeded our daily dose of 75 mg in three divided doses. Three reports of improved uteroplacental perfusion in IUGR pregnancies used a 50-mg dose once daily and recruited participants at later gestations with umbilical end-diastolic flow present in most cases. 21,35,36 As pharmacokinetic studies of sildenafil in pregnancy are currently not available, it would be difficult to determine an ideal dosing schedule for future studies although other dosing regimens of sildenafil have been proposed. More importantly, a possibility that the current dose of 25 mg three times daily may have a deleterious effect on blood flow in the ductus venosus and would require extreme caution in any future studies with a higher dose, particularly in fetuses with absent or reversed end-diastolic flow in the umbilical artery.
Another possibility is that the study’s definition of growth restriction included fetuses with such advanced disease that it was not possible to improve or reverse the process. The STRIDER study recruited more than half of the IUGR babies before 26 weeks’ gestation and all fetuses had severely compromised umbilical circulation with absent or reversed end-diastolic flow; overall mortality was around 45%. In comparison, the average gestational age at randomisation in the study by Dastjerdi et al. was 35 weeks. The authors did not report the proportion of babies with absent or reversed umbilical artery blood flow, but given the reported gestation, it is likely that these babies would have been delivered rather than recruited. 21 El-Sayed et al. 35 reported that only 11 (20%) of 54 babies developed absent or reversed end-diastolic umbilical artery blood flow at some point after randomisation, whereas in the study by Trapani et al. ,36 reversed umbilical artery blood flow was, in fact, an exclusion criterion. None of the studies reported any perinatal deaths or long-term follow-up data and it is, therefore, far too early to speculate that the reported improvements in uteroplacental perfusion in less severe IUGR at later gestation would lead to improved survival and better long-term outcomes.
Although there was no firmly agreed fetal monitoring protocol, or uniform triggers for the delivery of compromised babies in this study, all participating units had access to fetal medicine experts, detailed Doppler assessment of fetal and uteroplacental circulation and antenatal cardiotocography. It is therefore not surprising that the overall survival observed is broadly in agreement with other recent studies that included severe early-onset IUGR with abnormal umbilical artery Doppler. 3,41
The phase 2 follow-up STRIDER study had good retention and engagement in a very high-risk population with complex needs with > 80% of liveborn babies having neurodevelopmental and behavioural assessment at 2 years of age.
However, the follow-up phase encountered delays due to the significant impact of the COVID-19 pandemic on the availability of research staff and the challenges in accessing patients, which may have contributed to some women declining participation in this phase of the study.
Although the study was not specifically designed to assess neurodevelopmental or behavioural outcomes between 2 and 3 years of age in the surviving infants, the findings revealed that maternal treatment with sildenafil did not yield any statistically significant beneficial effects in the parameters studied, compared to the placebo. Importantly, no harmful effects were observed either. There was a statistically significant increase in head circumference in those babies treated with sildenafil. Given that there were no other clinically important differences in clinical outcomes between the two groups, and that due to follow-up issues and perinatal deaths the children assessed can no longer be deemed to be randomised this increase of 2.1 cm on average is likely to be a chance finding. There was also no effect on infant BP from treatment with sildenafil. Given these results, further investigation is warranted to explore the potential long-term effects and outcomes of maternal treatment with sildenafil. Future research could delve into assessing subtle neurodevelopmental and behavioural phenotypes as the children progress into school age and beyond. This could involve evaluating aspects such as attention, emotional regulation, and various cognitive functions. Additionally, it is crucial to emphasise the value of continuing to follow this well-characterised cohort into the school years, with access to linked data encompassing health and school records. Such comprehensive follow-up would provide invaluable insights into the potential benefits or risks associated with maternal sildenafil treatment, contributing to a deeper understanding of its implications.
After the conclusion of the UK and the New Zealand/Australia STRIDER trials, the Dutch STRIDER trial was halted early due to evidence from a planned interim review of increased perinatal mortality in the Sildenafil-treated group. 42 Further assessment deemed this to be predominantly due to persistent pulmonary hypertension of the neonate (PPHN), which has been proposed to be a pathophysiological mechanism of ‘rebound’ vasoconstriction after cessation of sildenafil. 43
Both the UK and the New Zealand/Australia STRIDER Phase I trials reviewed their data using the same criteria for PPHN as the Dutch STRIDER trial and did not find an increased mortality. 44 This will be the subject of a planned IPD of all the international STRIDER studies. 24
The STRIDER RCT, and its international collaborating studies, has shown no beneficial effect for mothers or babies affected by severe early-onset IUGR from treatment with 25 mg sildenafil 8 hourly. On current evidence, the researchers do not believe that there is likely to be any beneficial effect on fetal growth or perinatal outcomes in this patient group and would advise that further use of sildenafil in this population should be stopped. Prior to any further studies using PDE5 inhibitors to treat IUGR being performed, pharmacokinetic and pharmacodynamic experiments specific to pregnancy should be performed to establish an efficacious therapeutic dose.
Therefore, as sildenafil was neither beneficial nor harmful to these infants, the cohort could now be interpreted as a study of a population of severe early-onset IUGR which would be beneficial to clinicians and families for counselling. Furthermore, when combined with less severe cohorts from the TRUFFLE3,22 and POPS45 studies, it would give a counselling picture for fetuses affected with growth restriction from 22 weeks to term.
In conclusion, when sildenafil was administered to pregnant women carrying a severely growth-restricted fetus, it did not prolong pregnancy, improve survival or reduce short-term neonatal morbidity.
Implications for decision-makers
This study has demonstrated that there is no benefit from the use of oral sildenafil given to mothers carrying a fetus affected by severe early-onset IUGR. Therefore, the study recommend that future guidance on the management of the IUGR fetus should declare that sildenafil should not be used for this condition outside of a research study.
Recommendations for future research
The study recommend that future research should focus on the use of differing doses of sildenafil and the use of sildenafil in less severe cohorts of IUGR. Further assessment of the long-term implications of severe early-onset IUGR on neurodisability and behaviour could also be gained from studying the surviving children from the STRIDER study.
Study registration
ISRCTN Ref: ISRCTN39133303
EudraCT Ref: 2013-005398-32
MHRA CTA Ref: 04196/0032/001-0001
REC Ref: 14/NE/0011 (phase 1)
REC Ref: 16/LO/2225 (phase 2)
Patient and public involvement
The study incorporated patient and public involvement (PPI) through various key contributors. The Stillbirth and Neonatal Death Society (SANDS) played a pivotal role in shaping and informing the study, as they were actively involved in its design and served as members of the study steering committee. Additionally, the antenatal results and choice (ARC) charity played a crucial role in facilitating PPI efforts throughout the study’s duration.
Antenatal results and choice, in particular, was instrumental in the development of patient information and played a significant part in disseminating the study findings. Their involvement in the STRIDER study began from its initial design stages and continued all the way through to the study’s execution and the subsequent dissemination of its results.
Equality, diversity and inclusion
Promoting equality, diversity and inclusion (EDI) was a fundamental principle embedded in the STRIDER UK study. Although the rarity of severe early-onset IUGR posed challenges in conducting specific diversity characterisations, the researchers made concerted efforts to ensure that the selection of recruiting sites encompassed a comprehensive geographical representation across the UK. By embracing this approach, our recruitment was able to encompass diverse populations from both rural and urban areas, which inherently exhibit distinct demographic compositions across recruiting sites.
Chapter 6 Summary of key findings
-
Sildenafil did not prolong pregnancy in severe early-onset IUGR compared to placebo.
-
Sildenafil did not improve perinatal outcomes in severe early-onset IUGR.
-
Sildenafil did not improve maternal cardiovascular parameters in severe early-onset IUGR.
-
Sildenafil did not affect infant neurodevelopmental function at age 2 years.
-
Sildenafil did not affect infant emotional or behavioural status at age 2 years.
Additional information
Acknowledgements
The researchers would like to thank all the women who participated in this study during such a distressing time for them and their families. They would also like to thank the members of the Trial Steering Committee (Professor Alan Cameron – Chair, Professor Elizabeth Draper, Professor Paul Clarke, Dr Laura Price, Dr Laura Bonnett, Mr Alex Astor, Ms Louise Hardman, and Miss Karen Wilding), Independent Safety and Data Monitoring Committee (Professor Ed Juszczak – Chair, Professor Christoph Lees and Professor Ben Stenson) and all the individuals who helped with the management and conduct of the STRIDER UK study. The researchers are also grateful to Sharp Clinical Services and The University of British Columbia (UBC), Canada for supporting the provision of blinded drug to research sites and to staff of pharmacy and research and development departments in all of the participating hospitals. The researchers would like to thank UBC for the development and support of the STRIDER randomisation and electronic data capture systems.
The researchers would like to thank the NIHR who funded this programme of work.
Contributions of authors
Andrew Sharp (https://orcid.org/0000-0003-3396-7464) (Senior lecturer in Obstetrics, Obstetrician) wrote the initial submission for funding, wrote the submission for phase 2 funding for the study, supervised the conduct of the RCT and wrote the manuscript.
Christine Cornforth (https://orcid.org/0000-0002-6047-8634) (Director of Partnerships & Programmes, Senior Trial Manager) wrote the submission for Phase 2 funding for the study, supervised the conduct of the RCT, performed neurodevelopmental assessments, reviewed the neurodevelopmental data and wrote the manuscript.
Richard Jackson (https://orcid.org/0000-0002-7814-5088) (Senior Lecturer in Statistics, Statistician) performed statistical analysis.
Jane Harrold (https://orcid.org/0000-0002-9281-1446) (Doctor, Trial Manager) collated trial data.
Mark A Turner (https://orcid.org/0000-0002-5299-8656) (Professor of Neonatology, Neonatologist) supervised neonatal outcomes.
Louise Kenny (https://orcid.org/0000-0002-9011-759X) (Professor of Obstetrics and Executive Pro-Vice-Chancellor, Obstetrician) developed the STRIDER study consortium.
Philip N Baker (https://orcid.org/0000-0002-4592-6427) (Professor of Obstetrics and Pro Vice-Chancellor, Research and Enterprise, Obstetrician) conceived the idea for the study and developed the STRIDER study consortium.
Edward D Johnstone (https://orcid.org/0000-0001-9732-8544) (Professor of Obstetrics, Obstetrician) reviewed trial data.
Asma Khalil (https://orcid.org/0000-0003-2802-7670) (Professor of Fetal Medicine, Obstetrician) assessed the cardiovascular results.
Peter von Dadelszen (https://orcid.org/0000-0003-4136-3070) (Professor of Global Women’s Health, Obstetrician) developed the STRIDER study consortium.
Aris T Papageorghiou (https://orcid.org/0000-0001-8143-2232) (Professor of Fetal Medicine, Obstetrician) developed the STRIDER study consortium.
Brigitte Vollmer (https://orcid.org/0000-0003-4088-5336) (Professor of Perinatal Neurology) provided oversight relating to the neurodevelopmental outcomes assessment and interpretation, reviewed the neurodevelopmental data and wrote the manuscript.
Zarko Alfirevic (https://orcid.org/0000-0001-9276-518X) (Emeritus Professor of Fetal Medicine, Obstetrician) developed the STRIDER study consortium, wrote the initial submission for funding, wrote the submission for phase 2 funding for the study, supervised the conduct of the RCT and wrote the manuscript.
All authors reviewed the final manuscript and prepared the results for publication.
Disclosure of interests
Full disclosure of interests: Completed ICMJE forms for all authors, including all related interests, are available in the toolkit on the NIHR Journals Library report publication page at https://doi.org/10.3310/WAKV3677.
Primary conflicts of interest: Andrew Sharp, Asma Khalil, Richard Jackson, Edward D Johnstone, Jane Harrold, Louise Kenny, Peter von Dadelszen, Zarko Alfirevic, Christine Cornforth, Mark A Turner, Brigitte Vollmer declare no conflicts of interest.
Philip N Baker is a minority shareholder of Metabolomic Diagnostics, a spin out company which seeks to develop screening tests for pregnancy complications.
Aris T Papageorghiou is a co-founder of and shareholder of Intelligent Ultrasound, a University spin out company.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Ethics statement
Ethical approval for phase 1 of this study was obtained on 20 March 2014, Research Ethics Committee (REC; North East – Newcastle and North Tyneside 2, REC Ref: 14/NE/0011) and for phase 2 was obtained on 21 December 2016 (REC; London – Brent Research Ethics Committee, REC Ref: 16/LO/2225).
Information governance statement
The University of Liverpool and Liverpool Women’s Hospital have a steadfast commitment to adhering to the UK Data Protection Act (2018) and the General Data Protection Regulation (EU GDPR) 2016/679 when it comes to managing personal information. As per the Data Protection legislation, the University of Liverpool serves as the Data Controller. For detailed information on how personal data is handled, including guidance on exercising individual rights and contacting the Data Protection Officer, please refer to this link (legal@liverpool.ac.uk).
Disclaimers
This article presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
References
- Gardosi J, Kady SM, McGeown P, Francis A, Tonks A. Classification of stillbirth by relevant condition at death (ReCoDe): population based cohort study. BMJ 2005;331:1113-7.
- Flenady V, Koopmans L, Middleton P, Frøen JF, Smith GC, Gibbons K, et al. Major risk factors for stillbirth in high-income countries: a systematic review and meta-analysis. Lancet 2011;377:1331-40.
- Lees C, Marlow N, Arabin B, Bilardo CM, Brezinka C, Derks JB, et al. TRUFFLE Group . Perinatal morbidity and mortality in early-onset fetal growth restriction: cohort outcomes of the trial of randomized umbilical and fetal flow in Europe (TRUFFLE). Ultrasound Obstet Gynecol 2013;42:400-8.
- Engineer N, Kumar S. Perinatal variables and neonatal outcomes in severely growth restricted preterm fetuses. Acta Obstet Gynecol Scand 2010;89:1174-81.
- Tolsa CB, Zimine S, Warfield SK, Freschi M, Sancho Rossignol A, Lazeyras F, et al. Early alteration of structural and functional brain development in premature infants born with intrauterine growth restriction. Pediatr Res 2004;56:132-8.
- Baschat AA. Neurodevelopment following fetal growth restriction and its relationship with antepartum parameters of placental dysfunction. Ultrasound Obstet Gynecol 2011;37:501-14.
- Torrance HL, Bloemen MCT, Mulder EJH, Nikkels PGJ, Derks JB, de Vries LS, et al. Predictors of outcome at 2 years of age after early intrauterine growth restriction. Ultrasound Obstet Gynecol 2010;36:171-7.
- Shand AW, Hornbuckle J, Nathan E, Dickinson JE, French NP. Small for gestational age preterm infants and relationship of abnormal umbilical artery Doppler blood flow to perinatal mortality and neurodevelopmental outcomes. Aust N Z J Obstet Gynaecol 2009;49:52-8.
- Levine TA, Grunau RE, McAuliffe FM, Pinnamaneni RM, Foran A, Alderdice FA. Early childhood neurodevelopment after intrauterine growth restriction: a systematic review. Pediatrics 2015;135:126-41.
- Morsing E, Asard M, Ley D, Stjernqvist K, Marsál K. Cognitive function after intrauterine growth restriction and very preterm birth. Pediatrics 2011;127:e874-82.
- McCarton CM, Wallace IF, Divon M, Vaughan HG. Cognitive and neurologic development of the premature, small for gestational age infant through age 6: comparison by birth weight and gestational age. Pediatrics 1996;98:1167-78.
- Barker DJ. Developmental origins of adult health and disease. J Epidemiol Community Health 2004;58:114-5.
- Gallo LA, Tran M, Moritz KM, Mazzuca MQ, Parry LJ, Westcott KT, et al. Cardio-renal and metabolic adaptations during pregnancy in female rats born small: implications for maternal health and second generation fetal growth. J Physiol 2012;590:617-30.
- Chan PY, Morris JM, Leslie GI, Kelly PJ, Gallery EDM. The long-term effects of prematurity and intrauterine growth restriction on cardiovascular, renal, and metabolic function. Int J Pediatr 2010;2010.
- Zanardo V, Visentin S, Trevisanuto D, Bertin M, Cavallin F, Cosmi E. Fetal aortic wall thickness: a marker of hypertension in IUGR children?. Hypertens Res 2013;36:440-3.
- Wareing M, Myers JE, O’Hara M, Baker PN. Sildenafil citrate (Viagra) enhances vasodilatation in fetal growth restriction. J Clin Endocrinol Metab 2005;90:2550-5.
- Wareing M, Myers JE, O’Hara M, Kenny LC, Taggart MJ, Skillern L, et al. Phosphodiesterase-5 inhibitors and omental and placental small artery function in normal pregnancy and pre-eclampsia. Eur J Obstet Gynecol Reprod Biol 2006;127:41-9.
- Stanley JL, Andersson IJ, Poudel R, Rueda-Clausen CF, Sibley CP, Davidge ST, et al. Sildenafil citrate rescues fetal growth in the catechol-O-methyl transferase knockout mouse model. Hypertension 2012;59:1021-8.
- von Dadelszen P, Dwinnell S, Magee LA, Carleton BC, Gruslin A, Lee B, et al. Research into Advanced Fetal Diagnosis and Therapy (RAFT) Group . Sildenafil citrate therapy for severe early-onset intrauterine growth restriction. BJOG 2011;118:624-8.
- Samangaya RA, Mires G, Shennan A, Skillern L, Howe D, McLeod A, et al. A randomised, double-blinded, placebo-controlled study of the phosphodiesterase type 5 inhibitor sildenafil for the treatment of preeclampsia. Hypertens Pregnancy 2009;28:369-82.
- Dastjerdi MV, Hosseini S, Bayani L. Sildenafil citrate and uteroplacental perfusion in fetal growth restriction. J Res Med Sci 2012;17:632-6.
- Lees CC, Marlow N, van Wassenaer-Leemhuis A, Arabin B, Bilardo CM, Brezinka C, et al. TRUFFLE Study Group . 2 year neurodevelopmental and intermediate perinatal outcomes in infants with very preterm fetal growth restriction (TRUFFLE): a randomised trial. Lancet 2015;385:2162-72.
- Thornton JG, Hornbuckle J, Vail A, Spiegelhalter DJ, Levene M. GRIT Study Group . Infant wellbeing at 2 years of age in the Growth Restriction Intervention Trial (GRIT): multicentred randomised controlled trial. Lancet 2004;364:513-20.
- Liauw J, Groom K, Ganzevoort W, Gluud C, McKinlay CJD, Sharp A, et al. STRIDER Consortium . Short-term outcomes of phosphodiesterase type 5 inhibitors for fetal growth restriction: a study protocol for a systematic review with individual participant data meta-analysis, aggregate meta-analysis, and trial sequential analysis. Syst Rev 2021;10.
- Ganzevoort W, Alfirevic Z, von Dadelszen P, Kenny L, Papageorghiou A, van Wassenaer-Leemhuis A, et al. STRIDER: Sildenafil Therapy In Dismal prognosis Early-onset intrauterine growth Restriction – a protocol for a systematic review with individual participant data and aggregate data meta-analysis and trial sequential analysis. Syst Rev 2014;3.
- Sharp A, Cornforth C, Jackson R, Harrold J, Turner MA, Kenny LC, et al. STRIDER Group . Maternal sildenafil for severe fetal growth restriction (STRIDER): a multicentre, randomised, placebo-controlled, double-blind trial. Lancet Child Adolesc Health 2018;2:93-102.
- Bayley N. Bayley Scales of Infant and Toddler Development. San Antonio, TX: Psychcorp; 2006.
- Hempel MS. Neurological development during toddling age in normal children and children at risk of developmental disorders. Early Hum Dev 1993;34:47-5.
- Albers CA, Grieve AJ. Test review: Bayley, N. (2006). Bayley Scales of Infant and Toddler Development – Third Edition San Antonio, TX: Harcourt Assessment. J Psychoedu Assess 2007;25:180-90.
- Hadders-Algra M. The neuromotor examination of the preschool child and its prognostic significance. Ment Retard Dev Disabil Res Rev 2005;11:180-8.
- Hadders-Algra M. Two distinct forms of minor neurological dysfunction: perspectives emerging from a review of data of the Groningen Perinatal Project. Dev Med Child Neurol 2002;44:561-71.
- Fang X, Bai G, Windhorst DA, Feeny D, Saigal S, Duijts L, et al. Feasibility and validity of the Health Status Classification System – Preschool (HSCS-PS) in a large community sample: the Generation R study. BMJ Open 2018;8.
- Achenbach TM, Rescorla L. Manual for the ASEBA Preschool Forms and Profiles. Burlington, VT: University of Vermont, Research Center for Children, Youth and Families; 2000.
- Isquith P, Gioia G. Behavior Rating Inventory of Executive Function® – Preschool Version (BRIEF®-P). Odessa, FL: Psychological Assessment Resources, Inc.; 2008.
- El-Sayed MA, Saleh SA, Maher MA, Khidre AM. Utero-placental perfusion Doppler indices in growth restricted fetuses: effect of sildenafil citrate. J Matern Fetal Neonatal Med 2018;31:1045-50.
- Trapani A, Gonçalves LF, Trapani TF, Franco MJ, Galluzzo RN, Pires MMS. Comparison between transdermal nitroglycerin and sildenafil citrate in intrauterine growth restriction: effects on uterine, umbilical and fetal middle cerebral artery pulsatility indices. Ultrasound Obstet Gynecol 2016;48:61-5.
- Refuerzo JS, Sokol RJ, Aranda JV, Hallak M, Hotra JW, Kruger M, et al. Sildenafil citrate and fetal outcome in pregnant rats. Fetal Diagn Ther 2006;21:259-63.
- Sanchez-Aparicio P, Mota-Rojas D, Nava-Ocampo AA, Trujillo-Ortega ME, Alfaro-Rodríguez A, Arch E, et al. Effects of sildenafil on the fetal growth of guinea pigs and their ability to survive induced intrapartum asphyxia. Am J Obstet Gynecol 2008;198:127.e1-6.
- Oyston C, Stanley JL, Oliver MH, Bloomfield FH, Baker PN. Maternal administration of sildenafil citrate alters fetal and placental growth and fetal – placental vascular resistance in the growth-restricted ovine fetus. Hypertension 2016;68:760-7.
- Dilworth MR, Andersson I, Renshall LJ, Cowley E, Baker P, Greenwood S, et al. Sildenafil citrate increases fetal weight in a mouse model of fetal growth restriction with a normal vascular phenotype. PLOS ONE 2013;8.
- Lawin-O’Brien AR, Dall’Asta A, Knight C, Sankaran S, Scala C, Khalil A, et al. Short-term outcome of periviable small-for-gestational-age babies: is our counseling up to date?. Ultrasound Obstet Gynecol 2016;48:636-41.
- Pels A, Derks J, Elvan-Taspinar A, van Drongelen J, de Boer M, Duvekot H, et al. Dutch STRIDER Trial Group . Maternal sildenafil vs placebo in pregnant women with severe early-onset fetal growth restriction: a randomized clinical trial. JAMA Netw Open 2020;3.
- Pels A, Onland W, Berger RMF, van Heijst AFJ, Lopriore E, Reiss IKM, et al. Neonatal pulmonary hypertension after severe early-onset fetal growth restriction: post hoc reflections on the Dutch STRIDER study. Eur J Pediatr 2022;181:1709-18.
- Sharp A, Cornforth C, Jackson R, Harrold J, Turner MA, Kenny L, et al. Mortality in the UK STRIDER trial of sildenafil therapy for the treatment of severe early-onset fetal growth restriction. Lancet Child Adolesc Health 2019;3:e2-3.
- Sovio U, White IR, Dacey A, Pasupathy D, Smith GCS. Screening for fetal growth restriction with universal third trimester ultrasonography in nulliparous women in the Pregnancy Outcome Prediction (POP) study: a prospective cohort study. Lancet 2015;386:2089-97.
Appendix 1
STRIDER UK: phase 2 follow-up
Version 7.0, 11 October 2022
SPONSORS: The University of Liverpool
SPONSOR REFERENCE: UoL001286
FUNDERS: National Institute for Health and Care Research (NIHR) Efficacy and Mechanism Evaluation (EME)
FUNDING REFERENCE: 12/62/109
RESEARCH ETHICS COMMITTEE: London – Brent Research Ethics Committee
REC REFERENCE: 16/LO/2225
Research study team
Chief Investigator: Dr Andrew Sharp, The University of Liverpool
Co-investigators: Dr Christine Cornforth, The University of Liverpool
Dr Brigitte Vollmer, The University of Southampton
Professor Neil Marlow, University College London
Dr Samantha Johnson, The University of Leicester
Professor Aris Papageorghiou, St George’s Hospital London
Professor Asma Kahlil, St George’s Hospital London
Professor Philip Baker, The University of Leicester
Professor Louise Kenny, The University of Liverpool
Professor Mark Turner, The University of Liverpool
Dr Edward Johnstone, The University of Manchester
Statistician: Dr Richard Jackson, The University of Liverpool
Research Manager: Dr Jane Harrold, The University of Liverpool
Protocol approval
I, the undersigned, hereby approve this clinical study protocol.
| Signature: | Date: |
| Dr Andrew Sharp | |
| Chief Investigator | |
| Department of Women’s and Children’s Health The University of Liverpool |
|
| Signature: | Date: |
| Miss Karen Wilding | |
| Senior Clinical Research Governance Manager Clinical Directorate Faculty of Health and Life Sciences |
|
| The University of Liverpool, Sponsor |
Clinical queries
Clinical queries should be directed to Professor Zarko Alfirevic who will direct the query to the appropriate person.
Sponsors
This study is sponsored by The University of Liverpool. For further information regarding the sponsorship conditions, please contact
Miss Karen Wilding
Senior Clinical Research Governance Manager
Clinical Directorate
The University of Liverpool
Research Support Office
2nd Floor Block C Waterhouse Building
3 Brownlow Street
Liverpool L69 3GL
Tel.: 0151 794 8373
Funder
This study is funded by the National Institute for Health and Care Research (NIHR) Efficacy and Mechanism Evaluation (EME) funding body. This funding has been awarded as a costed extension to the STRIDER trial.
Study summary
This protocol describes the STRIDER UK: phase 2 follow-up study and provides information about procedures for participants. Every care was taken in its drafting, but corrections or amendments may be necessary. These will be circulated to investigators in the study. Problems relating to this study should be referred, in the first instance, to the Chief Investigator.
This study will adhere to the principles outlined in the NHS Research Governance Framework for Health and Social Care (2nd edition). It will be conducted in compliance with the protocol, the Data Protection Act and other regulatory requirements as appropriate.
Executive summary
Severe early-onset IUGR has no effective treatment and often results in stillbirth or iatrogenic preterm birth. Being born too small and too early can cause both immediate and long-term health problems. STRIDER UK is the MRC EME randomised placebo-controlled trial recruiting women with pregnancies complicated by severe IUGR to investigate whether sildenafil improves fetal growth. The results of this study will also contribute to the international STRIDER collaboration assessing the effects of sildenafil on neonatal survival free of major morbidity.
This is a follow-up of the STRIDER UK trial to examine the balance of longer-term benefits and risks associated with this novel therapy. All eligible surviving children from the trial will be invited for assessment between the ages of 2 and 3 years corrected. This will allow us to determine the effect of sildenafil on neurodevelopmental and cardiovascular outcomes in infancy in comparison with a placebo-controlled group of IUGR infants. Importantly, this follow-up will facilitate the maintenance of contact with this well-characterised unique cohort as they develop throughout childhood.
List of abbreviations
| HRA | Health Research Authority |
| REC | Research Ethics Committee |
| NIHR | National Institute for Health and Care Research |
| NREC | National Research Ethics Committee |
| IUGR | intrauterine growth restriction |
| BP | blood pressure |
| PP | pulse pressure |
| BRIEF | behaviour rating index for executive function |
| BDAI | Bayley Developmental Assessment Index |
| CBCL | child behaviour checklist |
| MND | minor neurological dysfunction |
Keywords Neonate, preterm, intrauterine growth restriction, neurodevelopment, cardiovascular, sildenafil
| TITLE: | STRIDER UK: phase 2 follow-up |
| STUDY DESIGN: | Follow-up study of surviving infants (aged between 2 and 3 years corrected) born to mothers who took part in STRIDER UK, a multicentre double-blind randomised placebo-controlled trial (please refer to the full STRIDER protocol for further details). |
| STUDY OBJECTIVES: | Primary outcomes |
|
|
| Secondary outcomes | |
|
|
| STUDY POPULATION: | All surviving children of mothers recruited to the STRIDER UK study will be eligible and invited for follow-up. This approach will maximise the scientific value of the STRIDER study data that will be available when the children are aged 2–3 years corrected. The inclusion of a gestational age- and sex-matched control group has been considered; however, this was not considered feasible, therefore standardised tests will be used to classify outcomes with reference to a norm for comparing rates of impairment between the two groups. |
| DURATION: | 70 months |
Table of contents
| INTRODUCTION | 43 |
| BACKGROUND | 43–44 |
| RESEARCH OUTCOMES | 44–45 |
| STUDY DESIGN/ASSESSMENT PROCEDURE | 45 |
| STUDY PROCEDURE | 45–49 |
| PARTICIPANT ENTRY | 49 |
| INCLUSION CRITERIA | 49 |
| EXCLUSION CRITERIA | 49 |
| WITHDRAWAL CRITERIA | 49 |
| ASSESSMENT METHODS | 50–51 |
| STATISTICAL ANALYSES | 51–53 |
| REGULATORY ISSUES | 53 |
| ETHICS APPROVAL | 53 |
| CONSENT | 53–54 |
| CONFIDENTIALITY | 54 |
| INDEMNITY | 54 |
| SPONSOR | 54 |
| FUNDING | 54 |
| AUDITS | 54 |
| PROJECT MANAGEMENT | 55 |
| END OF STUDY | 55 |
| SUBSTUDIES | 55 |
| VASCULAR PROFILING | 55 |
| PLACENTAL BIOBANKING | 55 |
| INDIVIDUAL PATIENT DATA META-ANALYSIS | 56 |
| ARCHIVING | 56 |
| PUBLICATION | 56 |
| PROTOCOL AMENDMENTS | 56–57 |
| REFERENCES | 58–59 |
Introduction
Background
Intrauterine growth restriction complicates up to 10% of pregnancies, accounting for over one-quarter of all stillbirths. 1,2 With no effective treatment available, up to 70% of pregnant women with IUGR diagnosed in the early third trimester require delivery before 32 weeks’ gestation. 3 It is well documented that these infants have substantially increased risks of neonatal death, major morbidity and prolonged neonatal admission compared with preterm infants of appropriate birthweight. 4
Being born too small and too early can pose significant health risks throughout the child’s life. In particular, IUGR has adverse effects on brain structure and function, which are independent of gestational age at birth5 and often compounded by poor postnatal growth, ultimately leading to an increased risk of neurological impairment, cognitive impairment, inattention, and specific difficulties with executive functions and impulsivity. 6
Between 25% and 40% of surviving growth-restricted very preterm infants have developmental delay,7,8 in particular in the areas of fine and gross motor difficulties, attentional difficulties5 and language delay9 with a mean difference in IQ of almost one standard deviation by the time they reach school age compared with preterm and term AGA controls. 10,11
Intrauterine growth restriction is well recognised as a key risk factor for adult diseases such as hypertension, diabetes and ischaemic heart disease. 12
It is also known to permanently alter organ capacity and neuroendocrine regulation leading to an adverse cardiometabolic phenotype that predisposes to adult disease12 and alters reproductive health with evidence of impaired fetal growth in future generations. 13 To date, there has been limited progress in developing interventions to reverse the lifelong effects of IUGR. The identification of an effective therapy (such as sildenafil) could improve both the short- and long-term health outcomes for these children in addition to significantly reducing the emotional and financial burden for such individuals, their families, and the wider community. There is strong evidence that the adverse consequences of placental insufficiency, leading to fetal growth restriction, extends beyond infancy to childhood and even adulthood. This adverse effect is above and beyond the effect of prematurity, as demonstrated by Chan et al. 14 Only children who were growth restricted during their fetal life, among those born preterm, have increased arterial stiffness and evidence of metabolic dysfunction. 14 They also demonstrate greater aortic wall thickening progression, suggestive of preclinical atherosclerosis which leads to a higher risk of developing hypertension later in life. 15
Intrauterine growth restriction is most commonly caused by abnormal placental development and invasion of the maternal blood vessels. This process leads to placental dysfunction and poor fetal nutrition. Preclinical work16–18 and pilot studies19–21 have shown that sildenafil, a phosphodiesterase inhibitor and vasodilator, may improve uteroplacental circulation and increase fetal growth. STRIDER UK will assess the effect of sildenafil in severe early-onset fetal growth restriction where the only available treatment is early delivery. It is well documented that IUGR is often accompanied by complications such as fetal hypoxia, acidosis and inflammation, all of which are thought to have a detrimental effect on brain growth and development. Consequently, there may be a complex trade-off between the effects of longer gestational length and ongoing exposure to a suboptimal fetal environment that should be considered. 22,23 It is therefore important to evaluate long-term outcomes, regardless of short-term results, to ensure the overall balance of benefits and risks, associated with sildenafil treatment, are examined.
An international IPD collaboration has been agreed between the five STRIDER collaborating sites in the UK, New Zealand, the Netherlands, Ireland and Canada. Data from all trials will be published independently and then included in a high-quality pre-planned IPD meta-analysis. 24 All trials have been funded by their government funding bodies. Recruitment is ongoing in three countries (UK, New Zealand and the Netherlands) with STRIDER UK projected to complete recruitment first.
Medical Research Council EME funded STRIDER UK (EudraCT Ref: 2013-005398-32) is currently recruiting across 19 sites. The primary aim of the study is to elucidate the underlying mechanisms that may lead to the prolongation of gestation. The study therefore hypothesise that this will lead to improved fetal growth and reduced neonatal morbidity and mortality. If the study findings support this hypothesis, sildenafil therapy will become a treatment that could have a significant impact on health and well-being later in life. The study will therefore apply for funding to follow this unique cohort of children to assess the longer-term effects of sildenafil therapy on neurodevelopment, cardiovascular and general health at 2–3 years (corrected), which is a fundamental step in the evaluation of this potential first treatment for IUGR. An age 2–3 years (corrected) follow-up has been chosen for this study as there is evidence of the cost-effectiveness of early interventions25–27 based on directly modifying risks at this age, including cardiovascular health,28 behavioural/emotional difficulties and cognitive delays. 29,30
The overall aim of this study is to examine neurodevelopmental and cardiovascular outcomes in children born to mothers who received sildenafil compared with placebo during pregnancy.
It is hypothesised that:
-
STRIDER UK children whose mothers received sildenafil will have improved neurodevelopmental outcomes at age 2–3 years (corrected) compared with controls exposed to placebo and,
-
there will be no difference in BP at 2–3 years (corrected) between STRIDER UK children whose mothers received sildenafil compared with controls exposed to placebo.
Research outcomes
The primary research outcomes for this study are:
-
survival free of neurodevelopmental delay at 2–3 years corrected age, defined by absence of cerebral palsy, deafness, blindness, developmental delay (BDAI-III cognitive, language or motor score < 85), and
-
survival free of cardiovascular impairment as defined by normal (similar to that in controls) systolic, diastolic BP or AIx (arterial stiffness) at 2–3 years corrected age.
The secondary research outcomes for this study are:
-
brachial pulse pressure (PP), brachial mean arterial pressure, heart rate, central systolic BP, central PP, aortic augmentation pressure, peripheral Alx and pulse wave velocity,
-
presence of MND, categorised as MND1 and MND2,
-
delay defined as a CBCL, internalising, externalising and overall subscale score > 2 SD, and
-
difference in mean BRIEF-P scores overall subscale score > 2 SD.
Study design/assessment procedure
This is a follow-up study of surviving infants (aged 2–3 years corrected) born to mothers who took part in STRIDER UK, a multicentre double-blind randomised placebo-controlled trial. Infants were born between December 2014 and July 2016. Core descriptive statistics of the study sample will be available upon completion of the main trial analyses.
Study procedure
Participants
All surviving children of mothers recruited to the STRIDER UK study will be eligible and invited for follow-up. Prior to contacting participants and inviting them to take part in the study, a thorough review of the child’s medical records will be carried out by designated members of the core research team to ensure the infant has survived. Contact information will be available for all mothers who took part in the main STRIDER UK trial (Figure 1).
Nominated members of the core research team will request access to the original confidential trial data in order to determine the contact details of all potential participants. As part of the original STRIDER UK trial, an audit of all consent forms will take place to ensure that all participants have consented to further contact in relation to future research. In addition, a check will also be made to ensure that the infants of all such participants survived. This will be carried out via a number of methods including a thorough audit of all serious adverse events (SAEs; which detail all fetal and neonatal deaths) and a final check at the local research site on the child’s health status. All participants receive newsletters which provide information on the trial and also give the option of opting out of any further contact regarding the trial. Any participant who has made contact with the trial management team and requested to opt out of future correspondence and participation in further research will also be removed from the list of potential participants for the follow-up study.
FIGURE 1.
Data access process for the identification of eligible participants for the follow-up study.
Pre-contact
STRIDER UK participants receive regular newsletters which provide an update on the STRIDER UK trial together with information on the follow-up. This will give participants the opportunity to contact the research team and get more information about the follow-up, if required. Potential participants will then be contacted again prior to their child reaching the age of 2 years corrected. This contact will be made by sending the parent/carer an invitation pack, detailed below. Additional efforts will be made by the research team to contact participants using a wide range of methods, including a letter, telephone call, e-mail and a newsletter. This will be carried out by experienced research staff who have a proven track record of working on longitudinal follow-up within this study population.
Invitation pack
A study invitation pack will be sent to all parents/carers of surviving children. This will include an invitation letter, participant information sheet and informed consent form. Participants will be asked to read the information carefully and discuss their child’s participation in the study with either close friends, family and/or a relevant health professional. There will also be a named contact, telephone number and e-mail address included on all correspondence so that parents/carers are able to contact the research team and discuss their child’s participation further, if required. If the parents are interested in taking part they will be asked to contact the research team in order to give verbal consent to take part in the study and to arrange a convenient date and time for their child’s assessment. Those participants who do not make contact with the research team within 2 weeks of the invitation pack being sent, will be contacted by a member of the core research team. This will be to check whether the participant has received their invitation pack and if they would like to discuss their potential participation further. For participants who do wish to take part an assessment date and time will be made (between the age of 2 and 3 years corrected ±3 months), additional time will be given to participants who would like to seek further advice about their child’s participation and a note will be made for those participants who are not contactable or who do not wish to take part in this phase of the study. Once an assessment date and time has been confirmed an assessment pack will be sent to the participant, as detailed below.
Assessment pack
Once an assessment date and time has been agreed, an assessment pack will be sent to participants who have given expressed verbal consent to take part in the follow-up. This pack will include confirmation of the (already agreed) assessment date, time and location, a map (if necessary), details of what will take place during the assessment, who will carry out the assessment, and a study questionnaire pack.
Location of assessments
Assessments will take place at either a local outpatient facility or in the home. A preference will be made to carry out all assessments in a controlled setting (i.e. clinical research setting), however, where necessary assessments will take place in the child’s home. However, under such circumstances, the researcher will need to assess the home environment for suitability beforehand. Where assessments are planned to take place in the child’s home, further information will be provided to parents on how to prepare their home (e.g. clearing an open space on the floor, providing a small table/work surface and turning off any distractions such as the television and or radio). Every effort will be made to ensure that assessment dates are booked to suit the needs of the participant; however, it is preferred that assessments take place in the morning as this is a time that children are often well rested, fed and able to concentrate.
Participants will be provided with contact details should they need to cancel or rearrange their assessment. Furthermore, they will receive a reminder telephone call 3 days prior to the assessment date and a text reminder the day before. Where the participant does not have access to a mobile phone number and the research team have been unable to make contact by telephone 3 days prior to the assessment date, further telephone calls by landline will be made to confirm the appointment is going ahead.
The follow-up assessment
The assessment will be performed by a suitably trained senior research psychologist with expertise in developmental assessment techniques in young infants. They will also be suitably trained in the specific cardiovascular assessment techniques used within this study and blinded to treatment allocation for the main STRIDER UK study.
Initially, the researcher will facilitate introductions between all present, following on from this the study will be explained and the child and parent/carer will be given the opportunity to ask any questions relating to their/their child’s participation. Once this is complete and informed written consent has been received, the research assessment will begin. The researcher will explain to the parent/carer the importance of allowing the child to complete all activities independently. The parent/carer is allowed to remain in the room while the assessment is taking place; however, they are asked not to interrupt or assist the child with these tasks unless otherwise invited to by the researcher.
The questionnaires sent to parents/carers in the assessment pack will be reviewed by the researcher and any missing items will be highlighted and discussed. If the parent/carer has any questions relating to the completion of the questionnaires, this will be addressed so that the parent/carer can complete them during the assessment. The researcher will also complete a general questionnaire with the parent/carer to determine information on a core set of variables that may affect research outcomes including, general health, since discharge from the neonatal unit and socio economic status. Other factors will be determined from the core STRIDER UK data set and where necessary neonatal records including gestational age, birthweight, APGAR scores, duration of ventilation, chronic lung disease, persistent arterial ductus, neonatal sepsis, retinopathy of prematurity, necrotising enterocolitis and focal brain injury. For the cardiovascular parameters, the study will collect information on systolic BP, diastolic BP and AIx adjusted for sex, weight and height. The study will also correct AIx for heart rate in view of the inverse linear relationship between AIx and heart rate.
Once this is complete, the researcher will play with the child to build a rapport so the child is relaxed and at ease during the assessment activities. Once the researcher deems that the child is ready, the formal assessments will be introduced to the child in the order set out in Table 1. These include the Cognitive, Language and Motor Subscales of the Bayley Scales of Infant and Toddler Development – III (BDAI-III);31 Hempel’s Neurological Examination for Toddler Age32 which will be used to identify major neurological impairment (cerebral palsy; CP) and to detect subtle deviations from typical neurological and neuromotor function and a cardiovascular assessment which includes brachial systolic BP and diastolic BP and arterial stiffness, assessed as aortic (central) AIx.
| Assessment/procedure | Completed by | Time to complete |
|---|---|---|
| Introduction/consent | Parent/researcher | 15–20 minutes |
| Questionnaire review | Parent/researcher | 5–10 minutes |
| General health and well-being | Parent/researcher | 5–10 minutes |
| Break | ||
| Introductory play session | Child/researcher | 10–15 minutes |
| BDAI: mental/cognitive assessment | Child/researcher | 10–15 minutes |
| BDAI: fine motor assessment | Child/researcher | 10–15 minutes |
| Break | ||
| BDAI: gross motor assessment | Child/researcher | 10–15 minutes |
| Hempel neuromotor assessment | Child/researcher | 20–30 minutes |
| Break | ||
| Cardiovascular assessment | Child/researcher | 15 minutes |
| Thank you and assessment summary | Parent/child/researcher | 5 minutes |
All participants will be asked to consent to the assessment being recorded for the purpose of assessing rater-reliability. For those consenting participants, all recordings will be labelled by the participant ID, date and time of assessment. These recordings will be stored electronically on a secure, password protected drive within the University of Liverpool. This drive will only be accessed by members of the core research team.
The assessments will take a total of 2.5 hours, excluding rest breaks (see Table 1). Additional data will be collected by parental questionnaire and completed prior to the follow-up assessment. The assessment includes well-established valid and reliable standardised measures selected to provide a comprehensive neurodevelopmental and cardiovascular assessment together with the collection of core information on the child’s general health and well-being. It is planned that all assessments will be completed by one trained and experienced senior researcher; however other researchers will also be trained if the situation arises where additional support is required. All assessors will be blinded to treatment allocation, formally trained in assessment techniques prior to commencing data collection and rater-reliability will be assessed throughout the study.
Once the assessments are complete, the researcher will thank the child and their parent/carer for their participation and parents/carers and children will be given the opportunity to ask any further questions. They will be advised that once the results of the assessment have been scored and interpreted, they will receive a report which will summarise the neurodevelopmental findings. With their consent, a more detailed report will be sent to the child’s GP and/or paediatrician who they will be able to contact if they should require any further information regarding these outcomes. The children will be given a pack for participating in the study which will include a certificate and a small gesture of thanks (e.g. age appropriate book) and parents/carers will be given a voucher to the value of £10 to reimburse them for their time and travel expenses.
Thanks and feedback pack
Approximately 2 weeks after the assessment date, participants will receive a Thank you/Feedback pack which will include a general letter and the participant’s version of the assessment feedback report. As outlined previously, a more detailed version of this report will be sent to the child’s GP and/or paediatrician.
As this is a research activity, it will be in addition to standard neonatal follow-up which should normally take place between the ages of 2 and 3 years (corrected for prematurity), though we will endeavour to co-ordinate appointments to both maximise recruitment and make attendance easier for parents. Consent will be requested for data from this assessment to be made available to clinicians carrying out the routine standard neonatal, aged 2–3 years, follow-up and the child’s GP and/or paediatrician.
Where potential participants cancel or fail to attend follow-up appointments, they will be invited to participate remotely. All such participants will receive a follow-up questionnaire pack which will include all questionnaires detailed as part of the main study in addition to the Ages and Stages Questionnaire (in place of the BDAI-III, neurodevelopmental assessment). A separate participant information sheet and consent form will also be included in this pack, which have been modified to reflect the changes for remote participation in the study. This pack will also be sent to potential participants who have been hard-to-reach in the initial contact phase. Those participants invited to take part remotely who do not respond within 4 weeks of the remote participation pack being sent will be contacted by telephone by a member of the core research team. This will be to check whether the participant has received their pack and if they would like to discuss their potential participation further. During this telephone contact, participants interested in taking part will be given the option to provide consent and complete the follow-up questionnaires via the paper-based method or verbally over the telephone.
Participant entry
Participants in this study are children of women who took part in the STRIDER UK trial.
Inclusion criteria
All children included in this study must be surviving children of mothers recruited to the STRIDER UK study born between December 2014 and July 2016.
Exclusion criteria
There are no exclusion criteria for this study.
Withdrawal criteria
Parents may decide to withdraw their child from this study at any time. They will be advised that this will not affect the subsequent care they or their child receives.
Assessment methods
The assessment includes well-established valid and reliable standardised measures selected to provide a comprehensive neurodevelopmental, cardiovascular and general health assessment at age 2–3 years corrected. Assessment measures are detailed overleaf.
The Bayley Scales of Infant and Toddler Development – III (BDAI-III)27 will be used as a standard measure of neurological Cognitive, Language and Motor Scales. This is an individually administered instrument designed to measure the developmental functioning of infants and toddlers. Other specific purposes of the BDAI-III are to identify possible developmental delay, inform professionals about specific areas of strength or weakness when planning a comprehensive intervention, and provide a method of monitoring a child’s developmental progress. The BDAI-III is appropriate for administration to children between the ages of 1 month and 42 months (although norms extend downward to age 16 days).
Hempel’s Neurological Examination for Toddler Age28 will be used to identify major neurological impairment (CP) and to detect subtle deviations from typical neurological and neuromotor function. The Hempel examination has been developed to evaluate MND at preschool age. It assesses MND in five domains of functions: fine motor function, gross motor function, posture and muscle tone, reflexes, and visuo-motor function. 30 Each of the domains can be scored as typical or deviant. Findings are classified as major neurological dysfunction, complex MND, simple MND, or neurologically normal. Major neurological dysfunction implies the presence of a distinct neurological syndrome, such as CP. Complex MND implies the presence of two or more deviant domains; simple MND implies the presence of one deviant domain. Neurologically normal implies the absence of deviant domains or the presence of only deviant reflexes. 30 Simple MND has limited clinical significance and reflects the presence of a normal, but non-optimally wired brain. On the other hand, complex MND represents the clinically relevant form of MND and is associated with behavioural and learning disorders. 31 The reliability of the Hempel examination is satisfactory (κ scores for various items: 0.62–1.00).
Where it is not feasible to calculate the Hempel scores owing to difficulties/lack of consent in recording the session, the assessment will continue to be completed with the following core variables collected: limb tone and reflexes, ankle and foot (e.g. clonus right and left ankle, Babinski reflex), presence and severity of cerebral palsy. These variables form part of the 2 to 3-years neurological outcomes for the STRIDER IPD analysis.
Child Behaviour Checklist 1.5–5 Parent Form (CBCL)33 will be used to assess emotional and behavioural difficulties. The CBCL includes 100 items that address emotional and behavioural problems, which are scored by parents on a three-point scale: not true, somewhat or sometimes true, and very true or very often true. The sum of all questions results in the TPS, an internalising problem score (IPS: emotionally reactive, anxious or depressed, somatic complaints and withdrawn), and an externalising problem score (EPS: attention problems and aggressive behaviour). Raw scores are normalised into T-scores (mean: 50, SD: 10). Higher T-scores represent more problematic behaviour. T-scores below 60 are in the normal range, T-scores of 60–63 (84th to 90th percentile) are in the borderline range, and T-scores above 63 (above 90th percentile) are in the clinical range. The T-scores are dichotomised into typical (scores in the normal range) and atypical (scores in the borderline and clinical range). The reliability and validity of the CBCL are good. 33 Where a child is attending nursery or daycare, the Caregiver–Teacher Report Form for Ages 1.5–5 will also be requested by post.
Behaviour Rating Inventory of Executive Function – Preschool version (BRIEF-P)34 is a parent questionnaire for early assessment of executive function. The BRIEF is a standardised questionnaire completed by the primary caregiver or parent that has been widely used in research and clinical settings to assess the presence and severity of executive dysfunction in day-to-day situations. 34 It is composed of three broad indices (General Executive Composite, the Metacognitive Index and the Behaviour Regulation Index) and eight subscales. The Metacognitive Index is comprised of five subscales: initiate (how well an individual independently initiates tasks), working memory (holding information in mind, manipulating information in mind), planning/organisation (using systematic, well planned approaches to tasks), organisation of materials and monitor (monitoring one’s behaviour or task approach). The Behaviour Regulation Index is comprised of three subscales, including inhibit (an index of impulsive behaviour or acting before thinking), shift (the ability to maintain a flexible approach to problem solving or behaviour) and emotional control (the ability to manage and regulate emotional responses). Age-based T-scores are computed for each subscale and index, and a score of 65 or higher is considered a clinically significant problem.
Cardiovascular assessments will be carried out using standard BP equipment. Prior to the assessments being carried out, the researcher will ensure that the following is controlled: (1) room temperature – environment kept at 22°C ± 1°C; (2) participants will be asked to be in a recumbent, supine position; and (3) the researcher must be aware of the effect of cardiac arrhythmia, white coat hypertension on measurements. Where children are fearful or distressed, a note will be made in the assessment notes and if necessary the assessment will be stopped and the most recent information will be used from the child’s medical notes (where applicable).
Statistical analyses
The main STRIDER UK trial recruited a total of 135 participants. It is estimated that approximately 86 participants (43 per group: 80% response rate) will take part in this follow-up phase. This could vary depending upon the final number of neonatal deaths reported for the study.
Neurodevelopmental outcomes
Based on published literature of preterm births, it is anticipated that approximately 70% of infants whose mothers receive placebo will be free of any neurodevelopmental delay. We can assume that neonatal responses should follow a binomial distribution. The key efficacy parameter here will be the comparison between the sildenafil to the placebo group which will be calculated using odds ratios and estimates based on the 95% confidence interval of the log odds ratio (Table 2). As there is some uncertainty over both the rate of the response in the placebo group as well as the allocation between the two groups, a simulation approach is taken. The results are shown in Table 2 and show the average (log) odds ratio obtained from a study along with size of half a 95% confidence interval (1.96*st.err) obtained for differing assumptions of the baseline rate and the differing observed allocation between the two groups. For equal response rates for placebo and sildenafil (e.g. both 80%), we would have a log (odds ratio) of zero theoretically. In practice, we would obtain a value close to zero and a 95% CI of length two. So, from the table for a pair of response rates, for example 70% versus 85%, we look to see if the value quoted for the log (odds ratio) minus the 1.96*st.err. is greater than zero or less than zero (−0.89 + 1.1 = 0.21). If less than zero, the study is well powered to detect this particular difference in response ratios. Therefore, looking at all the pairs of response ratios, we need a difference of about 20% points for the power to be 80% or more for example 70% versus 90%, 65% versus 85% or 60% versus 80%.
| Survival free from disability – age 2 years | Sildenafil | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (%) | 70% | 75% | 80% | 85% | 90% | ||||||
| Placebo | 60 | −0.44 | 0.93 | −0.69 | 0.96 | −0.98 | 1 | −1.33 | 1.07 | −1.79 | 1.21 |
| 65 | −0.23 | 0.94 | −0.48 | 0.97 | −0.77 | 1.01 | −1.12 | 1.08 | −1.58 | 1.22 | |
| 70 | 0 | 0.96 | −0.25 | 0.98 | −0.54 | 0.89 | −0.89 | 1.1 | −1.35 | 1.23 | |
| 75 | 0 | 1.01 | −0.29 | 1.05 | −0.64 | 1.12 | −1.1 | 1.26 | |||
| 80 | 0 | 1.1 | −0.35 | 1.16 | −0.81 | 1.29 | |||||
Cardiovascular outcomes
Power calculations for cardiovascular outcomes are based on diastolic and systolic BP. Based on a continuous outcome, previous data14,15 suggest an IQR of 20 mmHg which translates into a standard deviation of 15 mmHg. Based on this, the study will have sufficient power to detect a difference of 10 mmHg in either diastolic or systolic BP when using a two-sided alpha level of 0.05 (Table 3).
| Difference in BP (diastolic/systolic) | 5 mmHg | 10 mmHg | 15 mmHg |
|---|---|---|---|
| Power | 30% | 83% | 99% |
Analyses
All continuous data will be summarised as median (interquartile ranges) with categorical data summarised as frequencies of counts and associated percentages. Analysis of neonatal outcomes will be carried out assuming a binomial distribution. The key efficacy parameter of interest is the odds ratio comparing sildenafil against placebo. The primary analysis shall be carried out using a chi-square test unless low cell frequencies (< 6) are obtained in which case a Fisher’s exact test shall be used. Logistic regression analyses shall only be performed if > 20 events (instances of neurodevelopmental impairment or cardiovascular pathology at age 2–3 years corrected) have been observed based on the rule of thumb of at least 10 events being required for each covariate and the treatment groups being forced into the model. Comparisons of cardiovascular outcomes will be compared across groups using a t-test or a Wilcoxon test if data are sparse. Multivariable analyses will be carried out using standard linear regression techniques. Further exploratory analysis using multivariate techniques (hierarchical clustering, principal component analyses) shall be performed in an attempt to identify the number and identity of factors that may influence participant response. The results of the multivariate analyses shall be combined with the primary end point should the data allow. These analyses will be considered hypothesis generating.
As this is a follow-up study of randomly assigned treatments, the primary analysis will be carried out on an intention-to-treat (ITT) principle retaining participants in their initially randomised groups irrespective of any protocol violators and not depending on participants’ adherence to treatment. Secondary analyses shall be carried out on a per-protocol basis with further exploratory analyses adjusting for the amount of therapy received.
The reliability of neurological assessment will be tested by the review of video recordings of the on-site examinations by another assessor (Research Fellow, University of Southampton) blinded to the results of the original on-site assessment. Cases where clinically significant differences are found will be adjudicated by a third assessor (Co-applicant, Dr Brigitte Vollmer) and only agreed results will be used for the final analysis.
While every effort will be made to reduce non-compliance, analyses will be carried out initially on an ITT principle (retaining non-compliant participants in their initially randomised groups) and using a complete case data set (only analysing participants with complete sets of data). Secondary sensitivity analyses shall be carried out using per-protocol analyses with/without multiple imputations to account for any missing data. Analysing initially on an ITT principle will protect against any attrition bias that may arise due to lack of compliance.
Comparing the results of the initial and sensitivity analyses will give some indication of the effect that non-compliance has on assessing the overall treatment effect. It is expected that primary outcome data will be collected on 80% of STRIDER UK infants.
Regulatory issues
Ethics approval
As part of the IPD analysis, the main STRIDER UK trial has existing ethical approval to contact participants in order to take part in a follow-up of surviving children born to mothers who have taken part in the study.
The Chief Investigator will obtain approval from the London – Brent Research Ethics Committee and Health Research Authority (HRA). The study will be submitted to each proposed research site for Confirmation of Capacity and Capability. The study will be conducted in accordance with the recommendations for physicians involved in research on human subjects adopted by the 18th World Medical Assembly, Helsinki 1964 and later revisions.
This phase of the STRIDER UK study does NOT involve the testing of any Investigational Medicinal Products (IMPs); therefore approval from the Medicines and Healthcare products Regulatory Agency (MHRA) is not required.
This study has been reviewed by an independent patient representative and independent members of ARC and SANDS who were also involved in the main STRIDER UK trial.
Consent
Stage 1: In the first instance, ‘informed verbal consent’ will be sought over the telephone from a parent or nominated carer of the child. The parents/carers of each child will receive information regarding the follow-up phase of the study in newsletters sent by the trial management team. They will also receive a personalised Invitation Pack (detailed in Study procedure of this protocol) approximately 3 months prior to their child reaching the age of 2 years (corrected). The Invitation Pack will request that parents/carers review the information carefully and if they are interested in taking part in the study, to contact the research team in order to arrange a time and date for the follow-up assessment to take place. If the parent/carer does not contact the research team within 2 weeks of the invitation pack being sent they will receive a telephone call from a member of the team to determine (1) whether the pack has been received and (2) whether they would like to discuss their child’s participation in the follow-up study. Additional time will be given to parents/carers who would like to consider further their child’s participation. For parents/carers who give expressed verbal consent for their child to take part in the study, a date and time will be arranged for the follow-up assessment to take place (between the age of 2 and 3 years corrected ± 3 months). A note will be made in the study confidential research files for those parents/carers who decline to take part. For parents/carers who are hard-to-reach, up to three attempts will be made to contact by telephone e-mail, or post to arrange the follow-up assessment and following this a note will be included in the confidential research file that the research team were unable to contact the participant. After the third attempt, a remote study participation pack containing an information sheet, a consent form and the follow-up questionnaires will be sent to these participants, as detailed in Study procedure of this protocol. Parents/carers who do not respond within 4 weeks of the remote participation pack being sent will receive one final telephone call from a member of the research team. Parents/carers interested in taking part will be given the option to participate remotely via the paper-based method or verbally over the telephone. Where verbal consent and questionnaire completion is requested, this will be completed by a trained member of the research team and recorded. An additional consent item will be included for parents/carers who opt to participate over the telephone, this will be to consent for the research team to hold a copy of the recorded consent until the end of the study.
Stage 2: On the day of the follow-up assessment, prior to the assessment beginning, the researcher will review the trial information with the parent/carer of the child together with giving them the opportunity to ask any further questions. Following this, the parent/carer will be asked to provide written consent for their child to take part in the study. This will be initialled and signed by the parent/carer and the researcher at the time of the assessment. The original will be given to the parent/carer and a copy will be stored in the participant’s confidential research folder. In addition, the researcher will talk to the child and explain what they will be doing at an age appropriate level. The researcher will only complete the research assessment if the child is willing and able to take part. Where children become unwell or experience difficulty with concentration or unfamiliar surroundings the researcher will discuss with the parent/carer splitting the assessment over one or two days. Every effort will be made by the researcher to ensure that the child is relaxed and comfortable during the research assessment and that their rights as participants are fully respected.
The right of the participant to refuse to participate without giving reasons will be respected. All participants are free to withdraw at any time from the study without giving reasons and without prejudicing further treatment or support services.
Confidentiality
The Chief Investigator will preserve the confidentiality of participants taking part in the study and will abide by the Data Protection Act. They will ensure that participant’s anonymity will be maintained and that their identities are protected from unauthorised parties. Participants will not be identified by their names on any study documentation, but by an identification code. The investigator will keep a separate participant enrolment log showing codes, names and addresses. A confidential research folder for each participant will be kept at the lead site (Liverpool Women’s NHS Foundation Trust) containing any documentation that will include the participants details (e.g. consent form, participant contact letters and the assessment feedback report). These will also be stored electronically in a separate database which will be password protected and only accessed by designated members of the core research team.
Indemnity
The University of Liverpool holds indemnity and insurance cover with Marsh UK Ltd, which apply to this study. Further details can be obtained from the Chief Investigator.
Sponsor
The University of Liverpool will act as sponsor for this study. It is recognised that as an employee of the University and an honorary employee of the Liverpool Women’s NHS Foundation Trust, the Chief Investigator has been delegated specific duties as detailed in the Sponsorship Approval letter.
Funding
This study is funded as a costed extension to the STRIDER UK trial by the NIHR EME funding body. Further details of funding arrangements can be provided upon request to the Chief Investigator.
Audits
The study may be subject to inspection and audit by the University of Liverpool under their remit as sponsor. In addition, other regulatory bodies may audit this study to ensure adherence to GCP and the NHS Research Governance Framework for Health and Social Care (2nd edition).
Project management
The Project Management Group will be responsible for overseeing progress of the project on a day-to-day basis; this will consist of the Chief Investigator, Principal Investigators and core research staff.
A Project Steering Committee will be set up consisting of members of the main STRIDER UK trial oversight committees. This committee will oversee the progress of the project and will meet when required unless otherwise requested by the Project Management Group. This committee will formally meet once all data has been collected to review the study findings.
End of study
The study will be complete when all participating infants have had their notes retrospectively audited for the IPD data outcomes and completed their age 2–3-year (corrected) assessment.
Substudies
The main STRIDER UK trial protocol (EudraCT Ref: 2013-005398-32) included four ancillary studies linked to additional analyses. These were: (1) vascular profiling, (2) placental biobanking, (3) IPD meta-analysis and (4) cardiovascular profiling. Detailed below are the substudies that will be linked to this follow-up study and possible subsequent analyses.
Vascular profiling
The effect of sildenafil upon maternal angiogenic biomarkers was assessed by maternal blood sampling. Blood was drawn at randomisation: 2 hours post first treatment and every 3–4 days up to 2 weeks post randomisation (maximum of six collections per participant). Each sample obtained was 30 ml, with a total of up to 180 ml drawn over the length of the study. Blood samples were taken by a clinician trained in venepuncture and processed and stored in accordance with the specific SOP. Samples were sent to the Centre for Women’s Health Research, University of Liverpool and the Royal Liverpool University Hospital for analyses co-ordinated by Professor Zarko Alfirevic. All sample transfers to the Centre for Women’s Health Research were covered by an MTA embedded within the individual research site agreements and to the Royal Liverpool University Hospital by a specific MTA. This was a standard part of the main STRIDER trial research protocol and all sites were expected to participate; exceptions were given if the trial participant declined venepuncture, that is needle phobia.
All remaining samples are currently stored at the Centre for Women’s Health Research, University of Liverpool and will be used for further biomarker identification and validation analyses. The study have recently produced a predictive model that includes the biomarkers analysed as part of the main STRIDER UK trial and anticipate that the neurodevelopmental outcomes from this follow-up study will be of value in refining the model to provide a prognostic test for clinicians and parents to make informed decisions about their care.
Placental biobanking
Trial participants at some research sites donated their placenta after delivery. Placental samples and fetal blood were taken from the umbilical cord after it was separated from the fetus and was prepared and transferred to the Centre for Women’s Health Research, University of Liverpool for storage and analysis. Future funding will be sought for functional placental studies linking to overall study outcomes. It is anticipated that we will be able to use these placental samples to link the antenatal clinical features, angiogenic features with placental histology and neonatal and neurodevelopmental outcomes.
Individual patient data meta-analysis
The researchers are planning to conduct an IPD meta-analysis in collaboration with the STRIDER consortium across the world. This prospective analysis will address the issues of sildenafil effectiveness and safety focusing on substantive short and long-term clinical outcomes. A separate protocol will be developed for this work. The data capture and analysis will be co-ordinated by the CTU British Columbia Women’s Hospital (Vancouver, Canada).
Archiving
Data and all appropriate documentation should be stored for a minimum of 10 years after the completion of the study, including the follow‐up period, unless otherwise directed by the funder/sponsor/regulatory bodies.
Publication
STRIDER UK and this follow-up study will be published in peer-reviewed scientific journals and presented at clinical and academic conferences. In addition, the STRIDER IPD collaboration is in place to ensure that longer-term outcomes are assessed in the most efficient way so that if sildenafil is safe and effective, timely change in clinical practice and subsequent improvements in health will be achieved. Dissemination of results will be aided by the IPD collaboration (part of the Global Obstetric Network). Full details on publication will be defined following the first Project Oversight Meeting.
Protocol amendments
| Original version | Original date | New version | New date | Submitted to | Summary of changes |
|---|---|---|---|---|---|
| Version 2.0 | 5 December 2016 | n/a | n/a | Sponsor REC |
None |
| Version 2.0 | 5 December 2016 | Version 3.0 | 20 October 2018 | Sponsor REC HRA |
Amendment 01:
|
|
|||||
| Version 3.0 | 20 October 2018 | Version 4.0 | 15 February 2019 | Sponsor REC HRA |
Amendment 02:
|
| Version 4.0 | 15 February 2019 | Version 5.0 | 5 July 2019 | Sponsor REC HRA |
Amendment 03:
|
| Version 5.0 | 5 July 2019 | Version 6.0 | 19 October 2020 | Sponsor REC HRA |
Amendment 04:
|
| Version 6.0 | 19 October 2020 | Version 7.0 | 11 October 2022 | Sponsor REC HRA |
Amendment 05:
|
List of abbreviations
- AE
- adverse event
- AGA
- appropriate for gestational age
- AIx
- augmentation index
- BMI
- body mass index
- BP
- blood pressure
- BRIEF-P
- behaviour rating inventory of executive function – preschool version
- BSID
- Bayley Scales of Infant and Toddler Development
- CBCL
- child behaviour checklist
- CP
- cerebral palsy/major neurological dysfunction
- CTU
- Clinical Trials Unit
- eCRF
- electronic case report form
- EME
- Efficacy and Mechanism Evaluation
- GCP
- good clinical practice
- HSCS-PS
- health status classification system – preschool
- IPD
- individual participant data
- IQR
- interquartile range
- ISDMC
- Independent Safety and Data Monitoring Committee
- ITT
- intention to treat
- IUGR
- intrauterine growth restriction
- LCTU
- Liverpool Clinical Trials Unit
- MND
- minor neurological dysfunction
- NIHR
- National Institute of Health and Care Research
- NO
- nitrous oxide
- PPHN
- persistent pulmonary hypertension of the neonate
- PPI
- patient and public involvement
- REC
- Research Ethics Committee
- RR
- relative risk
- SAE
- serious adverse event
- SANDS
- Stillbirth and Neonatal Death Society
- SD
- standard deviation
- TPS
- total problem score