

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 15/74/01. The contractual start date was in February 2017. The final report began editorial review in May 2022 and was accepted for publication in February 2023. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2023 Gunn et al. This work was produced by Gunn et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2023 Gunn et al.
Chapter 1 Introduction
Background and rationale
Irritable bowel syndrome (IBS), which affects around 4% of the world population,1 accounts for around 1% of the 285 million consultations per year in primary care in England and Wales,2 which is approximately 2.8 million. Around one-third of these patients meet criteria for IBS with diarrhoea (IBS-D). Symptoms include frequent, loose or watery stools with associated urgency, which can severely limit socialising, travelling and eating out. This results in a reduction in quality of life (QoL) and loss of work productivity. When patients with IBS are asked to rank symptoms in order of importance, erratic bowel habit is rated first, followed by abdominal pain and, for those with diarrhoea, urgency. 3 This can often be associated with incontinence, which is socially debilitating, but often under-reported. 4 Current over-the-counter treatments for patients with IBS-D such as loperamide reduce bowel frequency, but do not improve abdominal pain5,6 and often lead to constipation. The lack of effective treatments results in frequent referrals to secondary care, and such patients have in the past been a significant proportion of gastroenterology outpatients. A previous meta-analysis showed that the 5-hydroxytryptamine-3 receptor antagonists (5HT3RAs) alosetron and cilansetron benefitted such patients, improving stool consistency, and reducing both frequency and urgency of defaecation. 7 However, these drugs had serious side effects, including constipation in 25% of patients and, rarely, ischaemic colitis (1 in 700). Alosetron was initially withdrawn and is now available in the USA, only through a risk evaluation and mitigation strategy and is not available in Europe. Cilansetron never came to market, while ramosetron, another 5HT3RA, is only available in Japan, where it is licensed for IBS-D with several good-quality trials confirming its benefit. 8,9
Ondansetron is a potent, highly selective 5HT3RA, which blocks 5HT3 receptors in the gastrointestinal tract. Penetration across the blood–brain barrier is limited, with cerebrospinal fluid (CSF) concentration < 15% of plasma levels so central nervous system (CNS) adverse effects are few. Ondansetron was developed and is currently licenced for use in adults and children for the management of nausea and vomiting induced by cytotoxic chemotherapy and radiotherapy (mediated by local release of serotonin in the gut from enterochromaffin cells), and for the prevention and treatment of postoperative nausea and vomiting. Constipation is an unintended side effect of ondansetron, which was first shown to slow colonic transit 30 years ago. 10,11 Ondansetron is widely used and unlike alosetron has not been associated with ischaemic colitis. We previously performed a randomised double-blind, placebo-controlled, crossover pilot study of two 5-week periods of treatment recruiting 120 IBS-D patients randomised to receive either ondansetron [2 mg up to 8 mg three times a day (t.d.s)] followed by placebo, or placebo followed by ondansetron with washout period of 2–3 weeks. 12 The primary outcome measure for the study was the difference in average stool consistency in the last 2 weeks of treatment, which showed a highly significant improvement with ondansetron versus placebo. We also showed significant benefits for both urgency and stool frequency, with associated slowing of whole gut transit. 12 Despite having limitations, results of this pilot study were very encouraging, supporting our clinical experience of ondansetron’s benefits. Recently a novel formulation of ondansetron (Bekinda) comprising 3 mg immediate release and 9 mg slow-release has been shown in a 12-week randomised, placebo-controlled trial to improve stool consistency, though it was underpowered to show significant benefit for pain. Currently there is lack of understanding as to exactly how it works, nor can we predict the individual dose required for optimum effect, which varies widely. 13 One key effect we found, also seen with other 5HT3RAs,14 was a marked reduction in urgency, which may play an important role in improving QoL for patients with IBS-D. 15
Potential mechanisms of action of 5HT3 receptor antagonists
Transit
5-HT3 receptor antagonists slow transit,10,11 an effect we found particularly marked in the left colon of IBS-D patients and rectosigmoid region, but the underlying mechanism was unclear. 12
Tone and motility
An early colonic barostat study showed a reduction in the postprandial rise in colonic tone with ondansetron in both carcinoid syndrome patients16 and healthy volunteers,17 which would be predicted to slow colonic transit. Previous studies of the impact of 5HT3RAs on human colonic motility18–20 were however paradoxical in showing that the 5HT3RAs tropisetron, alosetron and cilansetron all increased periprandial frequency of colonic contractions, and mean amplitude of contractions in the left colon. We hypothesised that 5HT3RAs increase retrograde sigmoid motility, perhaps enhancing ‘brake’ function,21,22 which would be a novel mode of action.
Proteases
In our pilot study we showed the decrease in urgency associated with ondansetron use correlated directly with the reduction in faecal protease (FP),23 but whether this represents a true causal relationship, or just an epiphenomenon, is unclear. FPs have been shown to be increased in IBS-D and, at least in animal models, cause hypersensitivity to rectal distension via activation of protease-activated receptors type 2 (PAR2). 24 We have shown that most FPs are endogenous,23 representing pancreatic enzymes that have escaped degradation by colonic bacteria. We hypothesise that slowing gut transit with ondansetron reduces FP, by allowing time for bacterial degradation, and that this may contribute to the beneficial effects of ondansetron on urgency. Reducing faecal tryptase might improve the anal soreness that is commonly reported by patients with IBS-D.
Bile acids
Bile acids have also been shown to sensitise the rectum,25 and elevated faecal bile acids (FBAs) have been shown by several groups in patients with IBS-D. 26 Slowing transit may increase the time for bile acid deconjugation by colonic bacteria, and therefore enhance absorption, but how important this is in reducing rectal sensitivity, compared with the effects on FPs, is unclear.
Genetic factors altering sensitivity to 5HT3RAs
We have shown that individuals vary widely in their responsiveness to ondansetron, explaining why trials using fixed doses of 5HT3RAs result in severe constipation in around one in four patients. Meta-analysis gives a relative risk of constipation of 4.3 (3.3–5.6). 7 However, when patients were allowed to dose titrate we found that constipation was rare, occurring in only 9% of patients, most of whom responded to dose reduction and only 2% discontinuing ondansetron because of this. 12 However, the required dose of ondansetron ranged from 4 mg on alternate days to 8 mg three times a day (t.d.s. ). The reasons for this variation are unclear, but recent evidence suggests that responsiveness to 5HT3RAs might be linked to polymorphisms in the genes controlling 5HT synthesis. Serotonin availability in the rectal mucosa is thought to be determined by the activity of the rate-limiting synthetic enzyme tryptophan hydroxylase-1 (TPH-1), which produces serotonin in enterochromaffin cells. A recent small study showed TPH-1 messenger ribonucleic acid (mRNA) levels in rectal mucosa (and thus presumably serotonin synthesis rate) were approximately doubled in responders to another 5HT3RA, ramosetron, compared with non-responders, and that this was linked to the TPH-1 genotype. 27 TPH-1 rs211105 minor allele G was found in 44% of non-responders, but only 4% of responders, indicating that possessing the major allele increases responsiveness to the drug. It was also associated with worse diarrhoea, possibly because of the greater 5HT synthesis.
Objectives
The overall aim was to determine the efficacy and safety of ondansetron in patients with symptoms of IBS-D, in particular evaluating the effect on the characteristic abnormalities of stool consistency, frequency and urgency as well as abdominal pain, satisfactory relief of IBS symptoms, mood and use of rescue medication and to determine the effect of 12 weeks ondansetron over 1 month after discontinuation.
Chapter 2 Methods
Trial design
A multisite, parallel-group, randomised, double-blinded, placebo-controlled trial, with embedded mechanistic studies recruiting patients from primary and secondary care as well as the community.
Participants
Patients were screened using a standardised protocol to establish eligibility using the following criteria.
Inclusion criteria
These were as follows:
-
patients had to meet modified Rome IV criteria for IBS-D (see as previously published);
-
patients had to be aged ≥ 18 years;
-
patients should have completed standardised workup to exclude:
-
microscopic colitis (colonoscopy or flexible sigmoidoscopy with colonic biopsies);
-
bile acid diarrhoea (BDA) [Selenium homocholic acid taurine (SeHCAT) results of > 10%, C4 results of < 19 ng/ml or failed 1-week trial of a bile acid binding agent (colestyramine 4 g t.d.s. , colesevelam 625 mg t.d.s. or equivalent)];
-
lactose malabsorption (suggested but not mandated negative lactose breath hydrogen test, negative clinical challenge or failure to respond to lactose-free diet);
-
coeliac disease [tissue transglutaminase (tTG) or duodenal biopsy];
-
-
patients of childbearing potential or with partners of childbearing potential had to agree to use methods of medically acceptable forms of contraception during the trial and for 90 days after completion of trial medication;
-
for women of childbearing potential, a negative pregnancy test was performed within 72 hours of confirmation of eligibility;
-
weekly average worst pain score had to be ≥ 30 on a 0 to 100-point scale;
-
stool diary needed to show stools with a consistency of 6 or 7 on the Bristol Stool Form Scale (BSFS) for 2 or more days per week.
Modified Rome IV diagnostic criteria for irritable bowel syndrome with diarrhoea
Must fulfil the following criteria for the past 3 months:
-
recurrent abdominal pain at least weekly;
-
pain is associated with two or more of the following criteria:
-
related to defaecation;
-
associated with a change in frequency of stool;
-
associated with a change in form (appearance) of stool;
-
-
symptom onset at least 6 months prior to diagnosis;
-
> 25% of abnormal stools are loose (BSFS 6 or 7) but < 25% are hard (BSFS 1 or 2).
Reproduced under creativecommons.org/licenses/by/4.0/.
Exclusion criteria
-
Gastrectomy.
-
Intestinal resection.
-
Other known organic gastrointestinal diseases [e.g. inflammatory bowel disease (Crohn’s disease, ulcerative colitis)].
-
Unable or unwilling to stop restricted medication, including regular loperamide, antispasmodics (e.g. hyoscine butylbromide, mebeverine, peppermint oil, alverine citrate), eluxadoline, tricyclic antidepressant (TCA) doses > 30 mg/day or other drugs likely in the opinion of the investigator to alter bowel habit. These medicines should be discontinued for a 7-day washout period prior to registration.
-
Corrected QT (QTc) interval ≥ 450 milliseconds for men or ≥ 470 milliseconds for women [assessed within the last 3 months by electrocardiogram (ECG)].
-
Previous chronic use of ondansetron or contraindications to it.
-
Pulse, blood pressure (BP) and laboratory blood values outside the normal ranges according to the site’s local definition of normal [assessed within the last 3 months]. Note minor rises in alanine transaminase (ALT) (< 2 × upper limit of normal) will be acceptable, but the patient’s general practitioner (GP) will be informed if they remain elevated at the end of the trial.
-
Women who were pregnant or breastfeeding.
-
Patients currently participating in, or who have been in, a trial of an investigational medicinal product (IMP) in the previous 3 months, where the use of the IMP may cause issues with the assessment of causality in this trial.
-
Patients who had started or altered dosing of selective serotonin reuptake inhibitors or TCAs in the last 3 months, or who were planning to change the dose during the trial.
-
Patients currently taking and unwilling or unable to stop apomorphine or tramadol (which interact with ondansetron).
-
Patients with only stools of consistency 7 on the BSFS for 7 days a week.
Patients taking QT-prolonging or cardiotoxic drugs were reviewed by the local principal investigator (PI) to determine whether they were suitable for the trial.
Intervention
Participants were randomised on a 1 : 1 basis to initially receive either ondansetron 4 mg or matched placebo, one tablet daily. Both treatments were administered in oral doses which ranged from one tablet every third day to a maximum of six tablets daily for 12 weeks. The optimum dose was established by dose titration monitored closely by the research nurse using a standardised advice protocol in the first 2 weeks of the trial to avoid constipation, which at a standard dose occurs in one-quarter of patients. Patients were told to increase or decrease dosage every 2 days according to their stool consistency. If stools became hard or there was no bowel movement on day 2, they were asked to stop the drug for 1 day and recommence at a lower dose going from one tablet daily to one tablet on alternate days. If stools still remained hard or infrequent, they were asked to reduce to one tablet every third day. Continuing loose stools led to the advice to increase the daily dose by one tablet every 2 days, while constipation led to a reduction in dosage. Most patients had established a stable dose by 2 weeks. Patients were reminded to take their medication regularly on each trial visit, as well as during the phone calls within the 2-week titration period. Counts of any remaining IMP were done at visits 4 and 5.
Participant timeline
This is summarised in Appendix 1, Table 34 with details given below.
Visit 1
Potential trial candidates attended their first visit for registration and consent by the PI or delegate. If required, further tests to exclude diagnoses other than IBS-D were arranged. These included a SeHCAT scan, serum C4 level (Edinburgh centre only) or a 1-week trial of a bile acid binding agent to assess for bile acid malabsorption (unless done within the last 5 years), and a colonoscopy (unless done within 2 years, or 5 years if they also had a current normal calprotectin) to assess for microscopic colitis. Baseline serum blood tests, vital signs, demographics (date of birth, gender, ethnicity and smoking history) and an ECG were obtained. Current medications were reviewed, and those unable to discontinue drugs likely to alter bowel habit were unable to enter the trial. Patients on QT-prolonging drugs and cardiotoxic drugs were reviewed by the PI for suitability for the trial, as high-dose ondansetron may increase the risk of QT prolongation and arrhythmias. Eligible and consenting patients were registered and allocated a unique trial ID.
All patients were asked to complete a 2-week daily diary recording stool frequency, stool consistency for each stool (using the BSFS), worst abdominal pain (on a scale of 0–100), worst bowel movement urgency (on a scale 0–100) and if they had used loperamide that day. In addition, patients had the option to be sent two automated text messages each day, something most participants initially agreed to do. The first message asked the patient if they had passed a stool of consistency 6 or 7 on the BSFS. They had to reply with either a yes or no. The second text message asked what their worst abdominal pain score was that day. The patient could respond with a number from a scale of 0–100 (where 0 is no pain and 100 is the worst imaginable pain).
Visit 2
Two weeks later the patient returned to confirm eligibility. The diary was used to confirm they had stool consistency BSFS 6–7 for more than 2 days a week and did not meet the exclusion criteria of having only BSFS 7. They also had to have a weekly average worst pain score ≥ 30. For patient consenting to the whole gut transit study, Transit-Pellet capsules containing markers were dispensed and the abdominal X-ray appointment confirmed for the morning of visit 3. If the patient had consented to one or both mechanistic studies, appointments for baseline assessment were arranged prior to visit 3.
Visit 3
On visit 3, patients underwent a pregnancy test if applicable, whole gut transit assessment by abdominal X-ray (if consented), rigid sigmoidoscopy (if consented), completion of baseline questionnaire booklet [including IBS-SSS, Short-form Leeds dyspepsia questionnaire (SFLDQ), Hospital Anxiety and Depression Scale (HADS), Patient Health Questionnaire 12 (PHQ-12) and IBS-QOL questionnaires], and collection of stool, whole blood and serum samples (if consented). Patients were then randomised and given a 6-week patient diary and the trial medication in accordance with their blinded randomisation allocation. Patients were asked to record the following on a daily basis: stool frequency, consistency of each stool, worst abdominal pain experienced that day, worst bowel movement urgency, number of trial medication capsules taken and whether they had used loperamide that day. Every week the diary required them to respond to the question whether they felt that ‘they have had satisfactory relief from their IBS symptoms that week’. If they agreed, the patient continued to receive two text messages each day for the rest of the trial, asking if they had passed a stool of a consistency of six or seven that day, and what their worst abdominal pain was that day.
During the first 2 weeks, patients were contacted every 2 days by the local site team to discuss bowel habit. The dose of ondansetron or placebo was titrated as required. Additional guidance on dose titration was given to each trial site in a standard operating procedure, and to the patient in a dose titration instruction leaflet (see Report Supplementary Materials 1 and 2). A check for serious adverse events (SAEs) was performed during each telephone call. The steady dose to be taken forward for the remainder of the trial was mostly confirmed in week 2, although patients could alter this during the 12 weeks, if required, to avoid constipation.
Visit 4
At 6 weeks on the trial treatment, patients returned for their fourth visit. Diaries were collected, and the investigator asked whether any reportable adverse events (AEs) had occurred since the last visit. A pregnancy test was taken (if required) and concurrent medication reviewed to ensure these do not interfere with the trial medication. A further 6-week patient diary, trial medication and Transit-Pellet capsules (for use 6 days prior to visit 5) were dispensed. If the patient consented to mechanistic studies, appointments were confirmed and arranged as convenient between 8 and 11 weeks of treatment. Daily text messages continued for a further 6 weeks for those patients who agreed.
Visit 5
After 12 weeks on the trial medication, patients returned for visit 5. A pregnancy test was taken (if required), concurrent medication reviewed to ensure that these did not interfere with the trial medication and the investigator asked whether any reportable AEs had occurred since the last visit. Unused medication and completed patient diaries were collected. The plain abdominal X-ray to assess whole gut transit was performed for consenting patients. Serum and stool samples were collected for consenting patients, and all patients completed the 12-week questionnaire booklet, including the IBS-SSS, SFLDQ, HADS and IBS-QOL questionnaires. Patients were issued with a follow-up diary and continued to respond to text messages for a further 4 weeks.
Visit 6
Patients returned for the sixth and final visit, where the diary was collected, and the investigator asked about any reportable AEs since the last visit.
See Appendix 1, Table 34 for a breakdown of activities per visit.
Trial outcomes
Primary outcome measure
Food and Drug Administration (FDA)-defined response in relation to abnormal defaecation and abdominal pain measured over 12 weeks post randomisation by patient diary and daily text message.
Definition: A FDA responder is a patient who records both a reduction in pain intensity and an improvement in stool consistency for at least 6 weeks of the 12-week treatment period, where:
-
reduction in pain intensity is defined as ≥ 30% decrease from baseline in weekly average worst daily pain;
-
improvement in stool consistency is defined as ≥ 50% decrease in the number of days per week with at least one loose stool [BSFS (21) 6 or 7)].
The two components to response (pain intensity and stool consistency) are also reported as individual outcomes.
Secondary outcome measures
Irritable bowel syndrome with diarrhoea symptoms measured by patient diary and daily text message over 12 weeks post randomisation and calculated for weeks 1–12 and for weeks 9–12:
-
abdominal pain (mean daily pain score, scored 0 = no pain to 100 = worst possible pain);
-
urgency of defaecation (mean daily urgency score, scored 0 = no urgency to 100 = worst imaginable urgency);
-
stool consistency (mean number of days per week with at least one loose stool; mean daily BSFS);
-
stool frequency (mean number of daily stools);
-
use of rescue medication (loperamide) over 12 weeks of treatment;
-
satisfactory relief assessed through answers to ‘Overall, have you had satisfactory relief from your IBS symptoms in the past week?’ Yes or No (measured by diary at the end of each week).
Self-reported symptoms, QoL and mood at 12 weeks post randomisation:
-
IBS symptom severity [measured by the IBS Severity Scoring System (IBS-SSS)];28
-
dyspepsia (using the SFLDQ);29
-
QoL and mood (IBS-QOL);30
-
HADS questionnaires;31
-
somatic symptoms [Patient Health Questionnaire 12 Somatic Symptoms (PHQ-12SS)] questionnaire. 32
Post-treatment (weeks 13–16 post randomisation) recorded in patient diary:
-
abdominal pain;
-
urgency;
-
stool consistency;
-
stool frequency.
Safety:
-
SAEs assessed throughout 12-week treatment period;
-
AEs assessed 4 weeks after the end of treatment to determine if there are any persisting effects.
Summary of any changes to the project protocol
A number of minor amendments were made; a summary of protocol changes can be found in Appendix 1, Table 35. There were no changes to end points or drug administration.
These minor amendments included:
-
raising the permitted calprotectin level to be < 100 ng/mg in view of new national recommendations;
-
allowing a 7-day trial of colestyramine to replace SeHCAT testing to exclude BDA to overcome the problem of long delays in SeHCAT testing;
-
simplifying the information sheets and including the new data on congenital malformations which became available during the trial;
-
adding GP surgeries within the East Midlands as Participant Identification Centres (PICs), October 2018 and January 2019;
-
allowing patients who have had a colonoscopy within 10 years to exclude microscopic colitis to be enrolled in the trial. The exclusion criteria have been amended to clarify that patients would need to stop taking high-dose TCAs to 19 July 2019;
-
allowing new adverts, July 2019;
-
both the treatment patient information sheet (PIS) and the treatment + tests PIS have been updated to reflect the new safety information regarding the use of ondansetron in pregnancy, October 2019;
-
the SMS Guidance for patients has been updated. A fully tracked document has been included for review. Changes have been made to this document to make it easier to understand and to add clarity on what is involved in this aspect of the study, October 2019;
-
an SMS messages flowchart has been added. Due to an oversight the content of the SMS messages has not been previously submitted for approval, October 2019.
Mechanistic methods
The trial aimed to evaluate the possible mechanisms underlying any changes in the primary and secondary end points. The hypothesis we wished to test was that ondansetron would slow transit, particularly on the left side of the colon, possibly by stimulating retrograde sigmoid contractions. We planned to measure whole gut transit at baseline and 12 weeks (n = 400), using radio-opaque markers and an abdominal X-ray as previously described by Abrahamsson. 33 All patients were offered the opportunity to take part in high-resolution manometry (HRM) studies to be performed at baseline and after 8–11 weeks of treatment at either Nottingham or the Royal London Hospital. We provided travel expenses and where necessary overnight stay for those who needed to travel to reach one of these centres though few required this. We also hypothesised that, like alosetron, a closely related 5HT3RA, ondansetron would relax the rectum as shown by increased compliance in the rectal barostat studies which were performed in Nottingham and the Royal London Hospital. Previous studies had identified abnormalities of FPs and bile acids in IBS-D which correlated with transit, so we planned to assess their correlation with symptoms at both baseline and week 12.
We attempted to collect faecal samples at baseline and week 12 for all patients to assess faecal water %, bile acids and protease.
Transit
Aim
To assess if ondansetron altered colonic transit significantly.
Methods
Patients were asked to take the transit capsules daily for Transit-Pellets method™ supplied by Medifactia AB Limited, Stockholm, Sweden. Pellets were taken starting 6 days before their planned visit 3. Patients were instructed to take one pill at approximately 9 a.m. each day apart from day 6 when there were two pills to take, one at 9 a.m. and the second pill at around 6 p.m. This adaptation of the standard Metcalf method is designed to deal with patients with very fast transit when pellets taken at 9 a.m. may well have left the colon leaving zero pills to count. This then would raise the question of whether any pills had been taken whereas taking the second later pill makes this much less likely. On visit 3 a plain abdominal X-ray was taken and used to count the number of pellets in each of three regions designated right colon, left colon and pelvic area as defined on the plain abdominal X-ray by a vertical line to tip of the fifth lumber vertebra spinal process and then tangential to either pelvic brim as shown below. Colonic transit calculations from plain X-ray (Figure 1) were as described by Chan et al. 1 and used by Garsed et al. 2
FIGURE 1.
Colonic transit calculations from plain X-ray. Whole gut transit (WGTT) was calculated from total pellets × 2.4 hours, so in this example of a patient on ondansetron with 21 pellets, WGTT = 50.4 hours.
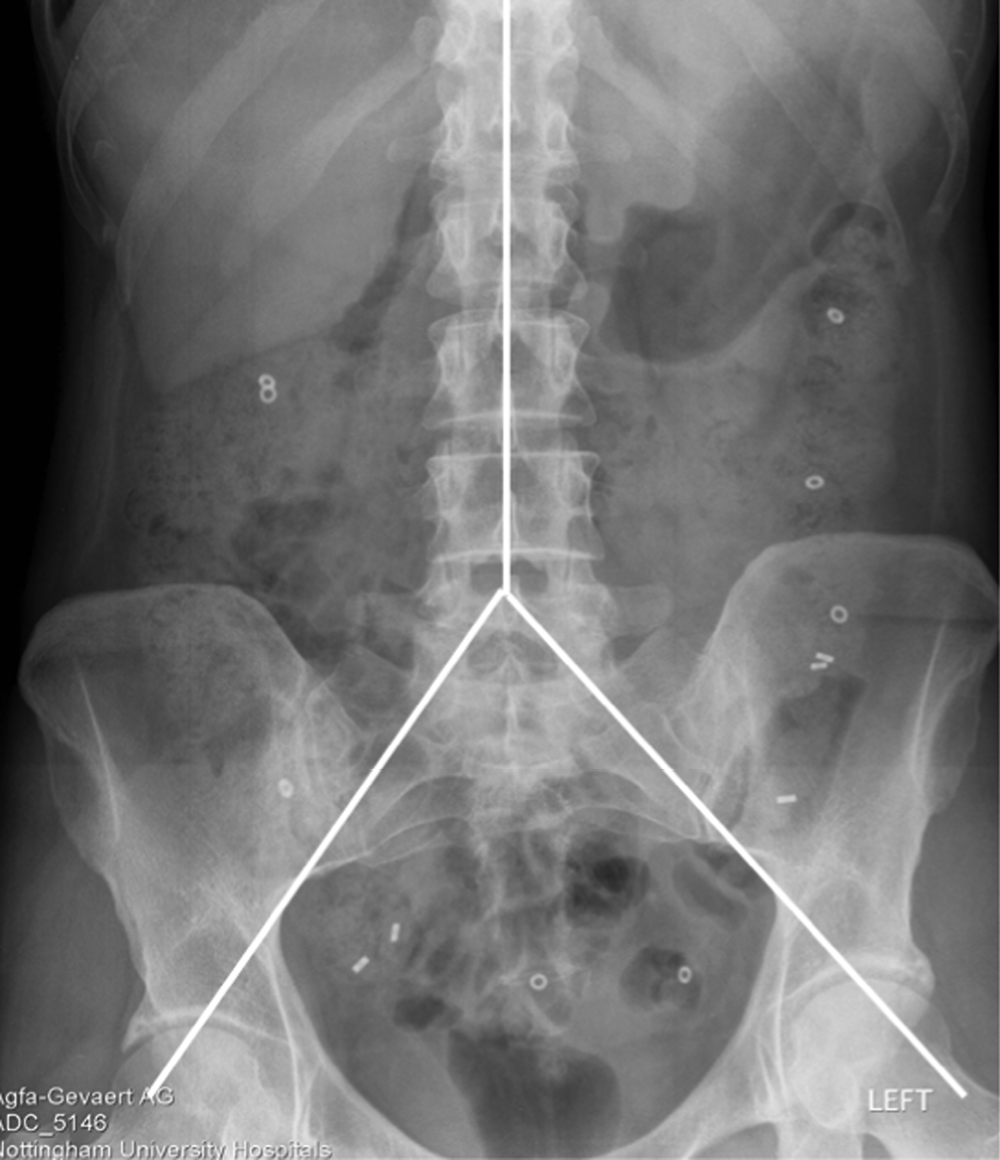
Regional transit was calculated from the number of pellets in the right, left and pelvic areas × 2.4 so in the example right transit time = 2 × 2.4 = 4.8, left = 2.4 × 3 = 7.2 and pelvic = 16 × 2.4 = 38.4 hours.
High-resolution manometry
Aim
Our aim was to characterise for the first time, the motor patterns in IBS-D using high-resolution manometry (HRM). We also planned to assess if baseline readings would relate to symptoms and whether these would alter with treatment.
This was performed at baseline and after 8–11 weeks of treatment in Nottingham and the Royal London Hospital. Patients from other sites were also offered the opportunity to travel to either site; two patients accepted this offer. The methods followed those reported by Dinning et al. 34 and subsequently used by Corsetti et al. 35,36
Methods
After a 12-hour fasting period, all patients were admitted to the Motility Unit for bowel preparation with tap water enemas. They then underwent a colonoscopy-assisted positioning of the colonic HRM catheter (UniTip High-Resolution Catheter, Unisensor AG, Attikon, Switzerland). As the colonoscope was being withdrawn after having fixed the catheter with haemoclips, four biopsies were taken. The catheter consisted of 40 sensors spaced 2.5 cm apart. After allowing 1 hour for the recording to settle following the colonoscopy, colonic pressure recording was started and continued for 2 hours before consuming a standardised meal [Tomato & Basil Dolmio PastaVita (285 kcal, 3.9 g fat, 8.7 g sugars) and Ensure® TwoCal drink (399 kcal, 17.8 g fat, 9.2 g sugars)]. During the recording, the subjects were asked to score their feeling of abdominal gas, desire to evacuate gas, desire to defaecate and urgency to defaecate every 15 minutes. Both the investigator and patient were blind as to their treatment. The manometry files were sent via secure transfer to Dr Phil Dinning at the University of Adelaide for analysis using previously published methods. 21,37
Barostat assessment
This was performed at baseline in nine subjects and after 8–11 weeks of treatment in Nottingham and the Royal London Hospital; six of whom also underwent manometry. Patients from other sites were also offered the opportunity to travel to either site but only two patients accepted this offer.
Aim
To assess if ondansetron increases rectal compliance and decreases sensitivity (manifest as increased pressure thresholds for pain and urgency) compared to placebo.
Methods
Patients were asked to fast from midnight for morning slots, or from 8 a.m. for afternoon slots prior to the assessment. They were asked to bring their usual dose of the study drug for the 8–11-week visit. After taking the dose, there was a 60-minute delay before making any measurements. On arrival patients were given instructions on how to self-administer a 500 ml body temperature tap water enema. After defaecating, patients were asked to lie down on the bed in a semi-prone left lateral decubitus position in a relaxed fashion with the foot end of the bed elevated from horizontal by 15° to decrease the effects of abdominal viscera on the bag volume. Rectal sensitivity was then assessed 60 minutes after taking the trial medication using a dual-drive barostat (Distender series II, G & J Electronics, Toronto, ON, Canada) and a polyethylene bag (600 ml) fixed on the end of a double-lumen barostat catheter (MUI Scientific, Mississauga, ON, Canada). The catheter was inserted into the rectum so that the middle of the bag was located approximately 10 cm from the anal verge and taped securely to the buttocks. The barostat bag was then unfolded by transiently inflating it with 75 ml of air and subsequently deflating it completely but leaving it in situ. Rectal pressure/volume relationships were assessed during a phasic isobaric, ascending method of limits distension protocol (4 mmHg steps to maximum toleration; 1 minute distension period, with 1 minute rest period between distensions). This was followed by a random phasic distension protocol distending to 8, 16, 24 and 36 mmHg with subjects rating sensation of gas, urgency, discomfort and pain on a 0–10 visual analogue scale (VAS). The maximum pressure used was adjusted depending on established pain threshold to avoid excessive pain and pressure was immediately released if patients reported > 80 mm of discomfort or pain on the VAS, following which higher distensions were not administered. Once the series had been completed the bag was deflated and removed. All procedures followed the written standard operating procedure which was part of the protocol.
End points
Primary: Pain pressure threshold mmHg.
Secondary: Urgency volume threshold, urgency pressure threshold, pain volume threshold.
Stool biomarkers: faecal proteases and bile acids
Aim
Previous studies had identified abnormalities of FPs and bile acids in IBS-D which correlated with transit and urgency, so we planned to assess these along with transit to see if we could confirm these findings and see if these affected symptoms.
Methods
All patients were asked to provide stool samples to assess whether ondansetron reduces total FBA and tryptase concentrations, both active mediators which might correlate with urgency.
Faecal water was measured by simple drying of weighed faecal samples in a vacuum rota-evaporator (Jouan, RC10.22, Thermo Fisher Scientific, Waltham, MA, USA) at 400 °C until constant weight was achieved.
Proteases
Using previously reported methods, samples (reproduced under creativecommons.org/licenses/by-nc/3.0/)23 (1 g) of stool stored at –70 °C were thawed and mixed in 5 ml 50 mM Tris buffer, 150 mM NaCl, pH7.2. Turbid suspensions were clarified using a sequential combination of centrifugation [10 minutes, 3000 relative centrifugal force (RCF)] and filtration (0.2 μm). Particulate-free supernatants were archived (–70 °C) until required for various assays or protein characterisation procedures.
Quantitation of total protein
Total protein quantitation was performed using the Bradford method,38 modified for low volume and high throughput. Briefly, equal volumes of sample and reagent were mixed in 96-well microplates and, following 20 minutes incubation, absorbance measurements at 595 nm were used to quantitate by reference to bovine serum albumin calibrants.
Quantitation of total protease activity
Total FP quantitation was performed using the non-specific proteolysis of azo-casein. The endoproteolysis of this liberates azo-peptides into the supernatant which are quantitated by absorbance measurement at 440 nm subsequent to protein precipitation with trichloroacetic acid. Protease activity was quantitated against bovine trypsin calibrant and expressed as trypsin units. Protein concentrations ranged from 0.08 to 1.7 mg/ml and measured protease activities were normalised against these values and expressed as units of trypsin/mg protein.
Quantification of bile acids (LC–MS)
After thawing faecal sample, 0.5 g was suspended in 2 ml of 50% (w/v) acetonitrile and extracted by vortexing and sonication for 10 minutes. The suspension was centrifuged twice at 25,000 g for 20 minutes. Supernatants were transferred to sample vials and loaded onto a liquid chromatography mass spectrometer (LC–MS) system containing online solid-phase extraction (SPE).
At the beginning of each analysis, 50 µl of the sample were transferred to SPE column at a flow of 0.1 ml/min with the loading mobile phase, aqueous 5% acetonitrile, 0.1% formic acid and 0.02% trifluoroacetic acid.
The chromatographic separation was performed using a binary system with pumps (A) and (B) (Jasco, PU-2085 Plus) connected to a degasser (Alltech, degasser, Stamford, UK). The two systems were connected using a two-position, six-port valve, used to switch automatically from loading (position 1) to injection (position 2) after 9 minutes (Valvemate®, Gilson, Dunstable. UK). Samples were analysed on a Waters Ex-bridge C18 Column (Waters, 100 × 2.1 mm; 3.5 µm particle size, Waters, Wexford, Ireland), using a gradient program. Mobile phase (A) 5 mM ammonium acetate, 0.1% ammonium hydroxide, mobile phase (B) 100% acetonitrile. Initial composition was 80% (A) was reduced to 70% (A) over 30 minutes the further reduction to 65% (A) over the next 3 minutes. The eluent composition was held at 65% for 1.5 minutes before returning to 80% (A) initial condition over the next 1.5 minutes. Flow rate 0.2 ml/min.
High-performance liquid chromatography was coupled in series with the turbo ion-spray (ESI) source of the tandem mass spectrometer (Micromass, Manchester, UK). Electrospray ionisation was performed in negative mode with nitrogen as the nebuliser gas. Detection of individual bile acids was performed using selective-ion monitoring (SIM) mode. Additional structural information was obtained via tandem MS (MS/MS) fragmentation, with collision energies ranging from 15 to 30 electron volts. Data were acquired using software MassLynX (Waters, Wexford, Ireland).
The concentration of bile acids in the samples was determined on the basis of the peak areas of individual bile acids and external standards.
End points
Primary: FP in trypsin units/mg protein, primary and secondary bile acids mmol/l stool water.
Secondary: Concentrations of individual bile acids, namely, cholic, chenodeoxycholic, deoxycholic and lithocholic acid, ratio of secondary/primary bile acids.
Mechanistic outcome measures
-
Whole gut transit time (WGTT; in hours), assessed using radio-opaque markers and an abdominal radiograph at baseline and 12 weeks.
-
Pain pressure threshold, pain volume threshold, urgency pressure threshold and urgency volume threshold, assessed using a barostat prior to starting the trial and 8–11 weeks during the trial.
-
FBAs and proteases prior to starting the trial and at 12 weeks during the trial.
-
Number of high amplitude propagated contractions (HAPCs) fasted and postprandially, assessed by HRM at baseline and at a visit at 8–11 weeks during the trial.
-
Percentage time occupied by cyclical propagated contractions, assessed by HRM at baseline and at a visit at 8–11 weeks during the trial.
Sample size
Primary end point
Treatment of irritable bowel syndrome using titrated ondansetron trial (TRITON) planned to recruit 400 patients from up to 24 sites across England and Scotland. This would have provided 90% power at 5% significance to detect a 15% absolute difference between the randomised groups in the proportion of patients achieving the FDA-recommended end point39 of a weekly responder for pain intensity and stool consistency for at least 6 weeks of the 12-week treatment period. This difference (15%) was considered to represent the minimum clinically important difference since in practice over the last two decades new IBS drugs with a lesser margin of efficacy are rarely prescribed in the NHS. We assumed a placebo response rate of 17%, as recently reported12 using this end point and allowed for a 15% attrition rate.
Mechanistic studies
Whole gut transit
Our previous study using the same radio-opaque marker technique showed ondansetron increased WGTT by a mean [95% confidence interval (CI)] of 10 (6 to 14) hours. 12 Using 200 per group would have given us a 90% power to detect a change of 0.7 hours.
High-resolution left-sided colonic manometry
Previous studies with the closely related 5HT3RA alosetron showed an increase in motility index compared with placebo, with a mean [standard deviation (SD)] of 1.0 (1.2),19 indicating we would have a power of 80% to detect a standardised effect size of 1 with 17 patients. We aimed for 20 patients on each treatment to allow for dropouts, that is, 40 each undergoing 2 studies, a total of 80 HRM studies.
Rectal compliance and sensitivity
Previous studies with alosetron showed an increase in compliance from 5.9 (SD 1.3) to 9.8 (SD 1.2) ml/mmHg in 22 patients. 40 We aimed to study 40 patients on each treatment to calculate correlations with symptoms, which typically require much larger numbers than just showing a change in mean values.
Randomisation
Randomisation was performed on a 1 : 1 basis to receive either ondansetron or placebo, and each patient was allocated three bottles of trial medication, each with a unique IMP kit code. Minimisation was used to ensure treatment groups were well balanced with respect to the minimisation factors of registering site and whether the patient had undergone mechanistic assessments.
Blinding
The trial was double-blind, neither the patient nor those responsible for their care and evaluation (treating team and research team) knew the allocation or coding of the treatment allocation. This was achieved by identical packaging and labelling of both the over-encapsulated ondansetron and matched placebo. Each bottle of ondansetron/placebo was identified by a unique kit code. Randomisation lists containing kit allocation were generated by the safety statistician at the Clinical Trials Research Unit (CTRU) and sent to the clinical supply company that produced the kits and the code break envelopes. Management of kit codes on the kit logistics application, which was linked to the 24-hour randomisation system, was conducted by the CTRU safety statistician in addition to maintaining the backup kit-code lists for each site.
Access to the code break envelopes was restricted to the safety statistician and designated safety team. Code breaks were permitted in emergency situations, where treatment allocation knowledge was needed to optimise treatment of the patient. Unblinded interim reports provided to the Data Monitoring and Ethics Committee (DMEC) were provided by the CTRU safety statistician and the reports were securely password protected.
Statistical methods: general considerations
All hypothesis tests were two-sided and used a 5% significance level. Methods to handle missing data are described for each analysis. Analysis and reporting were in line with Consolidated Standards of Reporting Trials (CONSORT). 41 As TRITON was a double-blind trial, the trial statistician was blinded to treatment group allocation throughout the trial, until the database had been locked and downloaded for final analysis. Only the safety statistician, supervising trial statistician, backup safety statistician and authorised unblinded individuals at the CTRU had access to unblinded treatment group allocation prior to final analysis.
Frequency of analyses
Outcome data were analysed once only, at final analysis, although statistical monitoring of safety data was conducted throughout the trial and reported at agreed intervals to the DMEC. Final analysis took place 16 weeks post–last patient randomisation.
End-point analysis
All analyses were conducted on the ITT population, defined as all patients randomised, regardless of non-compliance with the intervention. A per-protocol (PP) analysis of the primary end point was carried out to indicate whether results were sensitive to the exclusion of patients who violated the protocol (e.g. those patients randomised but subsequently found to be ineligible). Primary and secondary analyses were blind to random allocation. Outcome measures were analysed by regression models appropriate to the data type. Such analyses adjusted for randomisation minimisation factors: site, completion of manometry assessment and barostat assessment, as well as baseline values where applicable, age and gender. Baseline characteristics were summarised by randomised group.
Primary analysis
The primary analysis compared the proportions of patients achieving the FDA-recommended end point between treatment groups at 12 weeks post randomisation using a logistic regression model adjusted for minimisation factors, age and gender. The plan was that any missing data were assumed missing at random (MAR) and imputed for the primary analysis. However, complete case analysis was undertaken and those without sufficient data (n = 4) to evaluate the primary end-point were assumed to be non-responders. The potential impact of missing data would be small with only 5% of missing responses. Odds ratios and corresponding 95% CIs are presented.
Sensitivity analysis was planned to assess the impact of missing data on the treatment effect was performed. This was planned to include complete case analysis and alternatives to multiple imputation (e.g. pattern mixture modelling), if missing patterns suggested data were missing not at random. However, given the small overall sample size and only four participants with missing data (5%), this was not undertaken.
Secondary analyses
The proportions of patients with satisfactory relief of IBS symptoms at 12 weeks post randomisation was compared between the treatment groups using logistic regression models, adjusting for minimisation, baseline values, age and gender. Odds ratios and corresponding 95% CIs are presented.
Differences between the treatment groups for the continuous secondary end points at 12 weeks post randomisation were compared using linear regression models, adjusted for the minimisation variables, baseline where applicable, age and gender. These end points were urgency of defaecation over the last month, stool frequency over the last month, number of days per week with at least one loose stool (BSFS > 5) over the last month, average stool consistency, number of days rescue medication used over 12 weeks, abdominal pain score, HADS depression and anxiety scores, SFLDQ score, IBS-QOL score and subscales, PHQ-12 and IBS-SSS severity scores. Treatment estimates and corresponding 95% CIs are reported.
The differences between the treatment groups post treatment over weeks 13–16 post randomisation in the following end points: stool frequency, abdominal pain and urgency of defaecation were compared using linear regression model adjusting for minimisation factors, baseline values and relevant baseline factors. Treatment estimates and corresponding 95% CIs are reported. Any missing data are assumed MAR.
Exploratory analyses on the daily measurements (worst abdominal pain, loose stools, number of stools passed, consistency of stool, worst urgency and use of loperamide) were carried out, using repeated measures models, which incorporate correlation between measurements from the same patient.
SAS software version 9.4 was used in the analyses of primary and secondary end points.
Safety analyses
All patients who receive at least one dose of trial treatment were included in the safety analysis set. The number of patients reporting a SAE (up to 28 days after the last dose of treatment), and details of all SAEs, are reported for each treatment group. The number of patients withdrawing from trial treatment is summarised by treatment arm, along with reasons for withdrawal. All safety analyses performed prior to final analysis were undertaken by the safety statistician (rather than the trial statistician), thus ensuring that the trial team remain blinded.
Subgroup analyses
No subgroup analyses were planned.
Mechanistic studies
Mechanistic studies were analysed by the site research fellow, blinded to the intervention allocation under supervision of the chief investigator and local supervising PIs. The differences between treatment groups for changes in whole gut transit times, colonic motility measures (% time of cyclical retrograde contractions and HAPC frequency), rectal compliance and thresholds for urgency and pain measured using the barostat, FBA concentrations and faecal tryptase were assessed using two-way analysis of variance (ANOVA) for analysis of treatment and time effects and t-test, Mann–Whitney U-test and Wilcoxon matched pairs test for paired comparisons between normally and non-normally distributed variables, respectively. In addition, exploratory mediator analyses were planned to explore whether treatment effects, in terms of changes in urgency or pain, are mediated through changes in FBAs or protease using Spearman/Pearson correlation as appropriate.
Patient and public involvement
Both the grant application and the design of the study were assisted by our patient participation and involvement (PPI) group, which included patients with IBS-D who supported the original application and subsequently helped with the design of patient-facing documents.
Chapter 3 Clinical trial results
Site and patient recruitment
Recruitment strategies
A number of strategies were adopted during the trial to address the recruitment difficulties that were encountered. The initial plan had been to open 11 secondary care centres for recruitment, but it soon became clear that this would not be adequate to achieve the necessary numbers required to complete the trial. The source of patients varied widely by site (see Appendix 3, Table 37). As well as increasing the number of secondary care sites to 16, we utilised numerous other strategies to attract more patients to the trial.
We opened a number of secondary PICs. The secondary care PICs would identify potential participants from their clinics and refer them on to a recruiting centre for consideration for the trial. A total of six such PICs were opened.
A key issue in recruitment of patients within secondary care seemed to be changes in referral patterns over recent years. A trial in Yorkshire showed the value of screening patients with diarrhoea with calprotectin in primary care, reducing unnecessary colonoscopy and this has strengthened existing National Institute for Health and Care Excellence (NICE) guidance42 on the use of calprotectin in primary care. Evidence from NICE suggests a substantial reduction in referral from primary care with recent onset diarrhoea (56% in Cannock Chase, Staffordshire, UK). 43 Patients are thus less likely to be referred into secondary care for investigation of diarrhoea but are being investigated and managed in primary care. We soon realised that we would need to widen our recruitment pool to include patients in primary care and also in the general population.
We set up a self-referral pathway to allow patients to refer themselves to a local recruitment centre to be considered for the trial. Advertising was used to increase awareness of the trial and interested patients could visit the TRITON trial website for further information. The website included a simple self-screening questionnaire to confirm whether the patient was broadly eligible for the trial (see Appendix 10, Figure 31). Potentially eligible patients could then contact one of the trial research fellows to obtain further information about the trial and discuss their suitability before being referred on to their local recruiting centre.
We used a number of strategies to reach patients outside the secondary care centres. We approached GP practices in the areas surrounding the recruiting centres to set them up as PICs. The practices were asked to perform a search of their patient database to identify any suitable patients for the trial. They would then mail out a letter and leaflet introducing the trial to the patient asking them to visit the trial website if they were interested. Between March 2019 and February 2020, a total of 26 GP practices were set up as PICs and mail outs were sent to a total of 1381 patients. Of all the patients screened, two were identified in this way and of these one was randomised into the trial.
Starting in October 2019, we advertised within GP practices and pharmacies using posters and leaflets. GP practices that agreed to participate as PICs were asked to display the promotional materials in their waiting rooms. We also approached the Clinical Research Networks to arrange for promotional materials (see Appendix 10, Figures 32 and 33) to be displayed in the waiting rooms of GP practices not involved as PICs, as well as pharmacies.
In addition, we instigated a publicity drive to increase awareness of the trial outside of clinical settings. To achieve this, we advertised in local press in the areas surrounding our recruiting centres. The first advertisement drive was in May 2019 and regular advertising continued until March 2020. This advert (see Appendix 10, Figure 34) consisted of a quarter page in colour in publications such as the Metro, Manchester Evening News and London Evening Standard.
In November 2019, a Communications Officer was appointed to assist with the advertising strategy for the study, being responsible for engaging with Twitter (Twitter, Inc., San Francisco, CA, USA), TV, newspaper and radio, IBS charities and other patient forums to publicise the study. The trial Twitter account was used to engage with sites and improve awareness within trial teams in the hope that it would help attract more patients to the study. We actively encouraged staff at recruiting centres and collaborators to follow and retweet as well as using appropriate hashtags and tagging any relevant organisations in our tweets. We also advertised on social media, in particular Facebook (Meta Platforms, Inc., Menlo Park, CA, USA) and Google (Google Inc., Mountain View, CA, USA).
Professor Spiller and Karen Andrews (patient and public involvement representative) did an interview for the Mail on Sunday, which was the lead article on their health page on Sunday, 23 March 2019 and produced 3000 visits to our webpage and some 430 contacts of which approximately 10 entered the trial.
ContactME-IBS is a registry run through one of the TRITON recruiting sites, County Durham and Darlington NHS Foundation Trust. ContactME-IBS holds the contact information of approximately 2766 adults interested in hearing about, and taking part in, IBS research, of which 641 were suitable for TRITON having been identified as having IBS-D and associated pain (a key eligibility criterion). A mail shot to the 641 potentially eligible registry patients was sent in November 2019. Due to the nature of the registry setup, patients are mainly centred around areas where ContactME have had a drive to recruit GP practices, with 593 of the potentially eligible registry patients located in areas affiliated with TRITON secondary care sites, and 48 in areas without a suitable TRITON site.
Summary of patient identification method and screening approach is in Table 1.
| Identification method | Screened (n = 1582)a (%) | Clinic 41 (2.6%) (%) | Telephone call 20 (1.3%) (%) | E-mail/letter 15 (0.9%) (%) | Missing 1506 (95.2%)b (%) | Randomiseda (n = 80) (%) | Clinic 17 (21.3%) | Telephone call 3 (3.8%) (%) | E-mail/letter 3 (3.8%) (%) | Missingb 57 (71.3%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Secondary care | 485 (30.7) | 41 (8.5) | 18 (3.7) | 11 (2.3) | 415 (85.6) | 58 (72.5) | 17 (29.3%) | 1 (1.7) | 1 (1.7) | 39 (67.2%) |
| Primary care and pharmacies | 10 (0.6) | 0 (0.0) | 1 (10.0) | 1 (10.0) | 8 (80.0) | 4 (5.0) | 0 (0.0%) | 1 (25.0) | 0 (0.0) | 3 (75.0%) |
| Self-referral via website | 64 (4.0) | 0 (0.0) | 1 (1.6) | 2 (3.1) | 61 (95.3) | 12 (15.0) | 0 (0.0%) | 1 (8.3) | 2 (16.7) | 9 (75.0%) |
| Self-referral – other means | 34 (2.1) | 34 (100.0) | 4 (5.0) | 0 (0.0%) | 0 (0.0) | 0 (0.0) | 4 (100.0%) | |||
| Secondary PIC | 4 (0.3) | 4 (100.0) | ||||||||
| Primary care (invitation letter) | 2 (0.1) | 2 (100.0) | 1 (1.3) | 0 (0.0%) | 0 (0.0) | 0 (0.0) | 1 (100.0%) | |||
| Patients on file | 951 (60.1) | 951 (60.1) | ||||||||
| Forwarded by CTRU | 2 (0.1) | 2 (0.1) | ||||||||
| Identified by bowel cancer charity | 1 (0.1) | 1 (0.1) | ||||||||
| Other | 1 (0.1) | 1 (0.1) | ||||||||
| Missing | 28 (1.8) | 0 (0.0) | 0 (0.0) | 1 (3.6) | 27 (96.4) | 1 (1.3) | 0 (0.0%) | 0 (0.0) | 0 (0.0) | 1 (100.0%) |
Screening and recruitment
A total of 1582 potential participants were screened for entry into TRITON, across 13 hospital sites, including those referred to local sites by the Clinical Research Fellows (section Self-referrals below). Of these, 295 (18.6%) were found to be eligible for the study, 173 (58.6% of eligible) consented to take part in the study, 149 (86.1% of consented) were registered to the study, and 80 (53.7% of registered) were randomised to the study. Eligibility varied widely between centres from 10% to 90% with some being more efficient than others in deciding who to approach (see Appendix 3, Table 36). A detailed flow of these patients in the form of a CONSORT diagram can be seen in the supplementary document. A large proportion, 55.8% (882/1582), of screened patients were ineligible due to not meeting the Rome IV criteria. Of those who passed screening and were registered, insufficient pain scores were the largest single cause of being ineligible for randomisation (31/149). Overall, patients were screened over 28 months from March 2018 until June 2020.
Self-referrals
A total of 620 people self-referred via the TRITON website (Figure 2) and were sent information sheets. Subsequently, 297 (47.9%) did not respond further. A total of 155 (25.0%) were referred onto a local site, having confirmed their interest in taking part in the TRITON study and fulfilled the initial eligibility checks.
FIGURE 2.
Self-referral flow of participants.
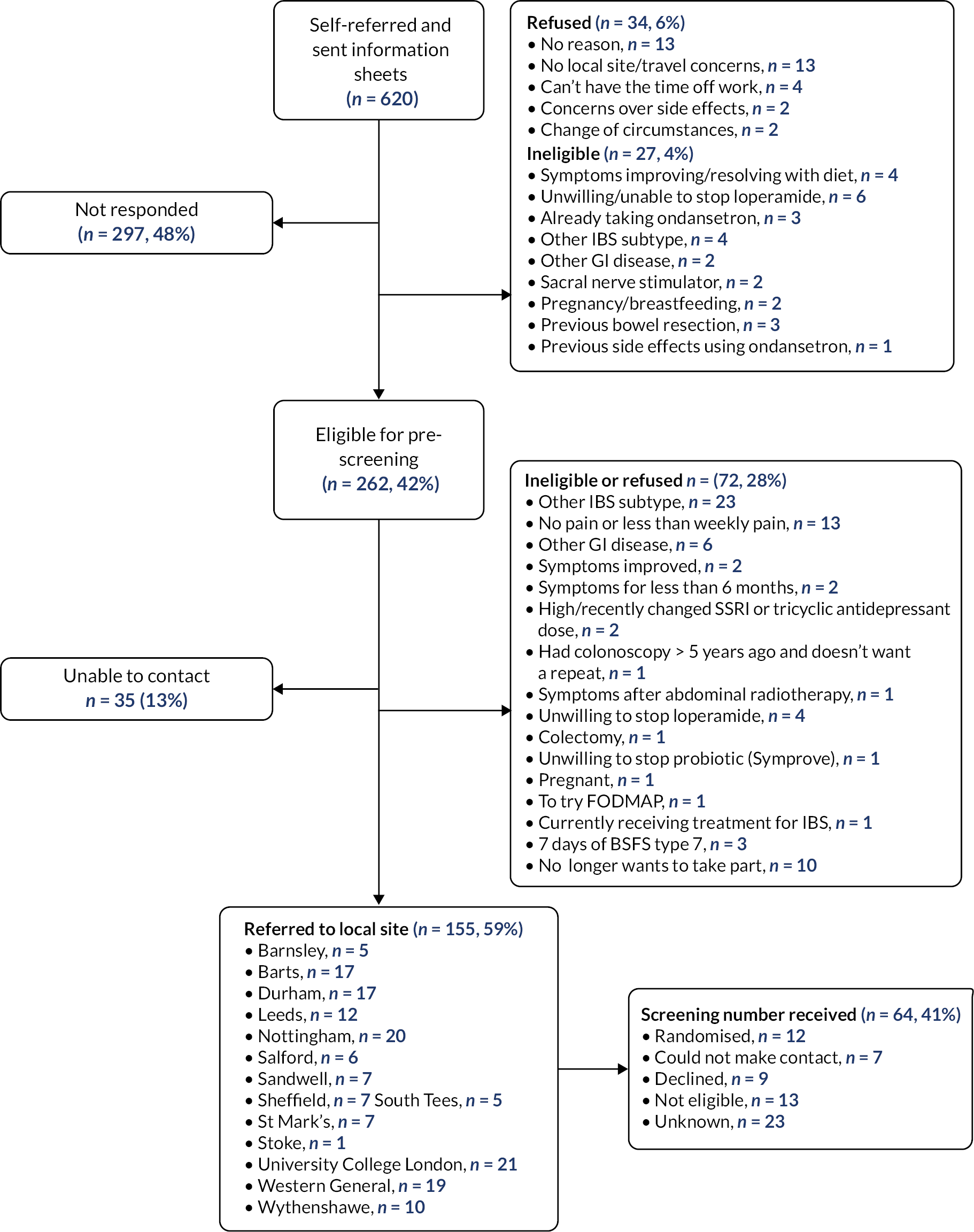
The trial Clinical Research Fellows responded to each patient’s e-mail, and 64 patients were screened through the formal screening process. Twelve patients were randomised using the self-referral route (see Table 1). Self-referrals were received and screened over 20 months from November 2018 until June 2020.
Screening characteristics
Characteristics of screened participants (age and gender) summarised by site are detailed in Table 2. One site in particular screened more potential participants than the others, 49.2% (778/1582) of the total; but most of these screening forms were missing age and gender data. The percentage who were eligible varied from 0.6% to 91.7% of those considered indicating that some sites were much more selective in who they approached. For those potential participants with screening data available, the mean age of those screened was 44.9 years old (SD 15.4), and 67.1% were female (523/780). Further screening information by site can be found in Appendix 3, Tables 36–38.
| Age (years) | Gender | |||||
|---|---|---|---|---|---|---|
| Site | Total (%) | Mean (SD) | Missing | Male (%) | Female (%) | Missing (%) |
| James Cook, South Tees | 38 (2.4) | 39.6 (12.5) | 0 | 13 (34.2%) | 25 (65.8%) | 0 (0.0%) |
| St James’s University Hospital, Leeds | 80 (5.1) | 43.5 (17.4) | 6 | 16 (20.0%) | 63 (78.8%) | 1 (1.3%) |
| Royal Hallamshire Hospital | 51 (3.2) | 48.1 (13.9) | 1 | 23 (45.1%) | 27 (52.9%) | 1 (2.0%) |
| Sandwell Hospital | 43 (2.7) | 45.2 (12.5) | 1 | 15 (34.9%) | 28 (65.1%) | 0 (0.0%) |
| University Hospital of North Durham | 23 (1.5) | 49.0 (11.8) | 4 | 6 (26.1%) | 16 (69.6%) | 1 (4.3%) |
| Wythenshawe Hospital | 12 (0.8) | 47.4 (15.7) | 0 | 5 (41.7%) | 7 (58.3%) | 0 (0.0%) |
| Royal Stoke University Hospital | 44 (2.8) | 43.6 (17.1) | 0 | 14 (31.8%) | 28 (63.6%) | 2 (4.5%) |
| Nottingham University Hospital | 58 (3.7) | 45.7 (18.2) | 4 | 25 (43.1%) | 32 (55.2%) | 1 (1.7%) |
| Barnsley District General Hospital | 216 (13.7) | 46.8 (15.5) | 5 | 54 (25.0%) | 161 (74.5%) | 1 (0.5%) |
| St Mark’s Hospital | 11 (0.7) | 56.3 (12.1) | 2 | 6 (54.5%) | 5 (45.5%) | 0 (0.0%) |
| Western General Hospital | 53 (3.4) | 43.8 (14.6) | 1 | 12 (22.6%) | 40 (75.5%) | 1 (1.9%) |
| Salford Royal Hospital | 16 (1.0) | 38.6 (15.5) | 0 | 6 (37.5%) | 10 (62.5%) | 0 (0.0%) |
| Barts and London School of Medicine and Dentistry | 159 (10.1) | 43.0 (14.3) | 67 | 43 (27.0%) | 53 (33.3%) | 63 (39.6%) |
| University College London Hospital | 778 (49.2) | 42.1 (16.1) | 740 | 19 (2.4%) | 28 (3.6%) | 731 (94.0%) |
| Total | 1582 (100) | 44.9 (15.4) | 831 |
257 (32.9% of non-missing)
(16.2% of total) |
523 (67.1% of non-missing)
(33.1% of total) |
802 (50.7%) |
Randomisation and participant flow
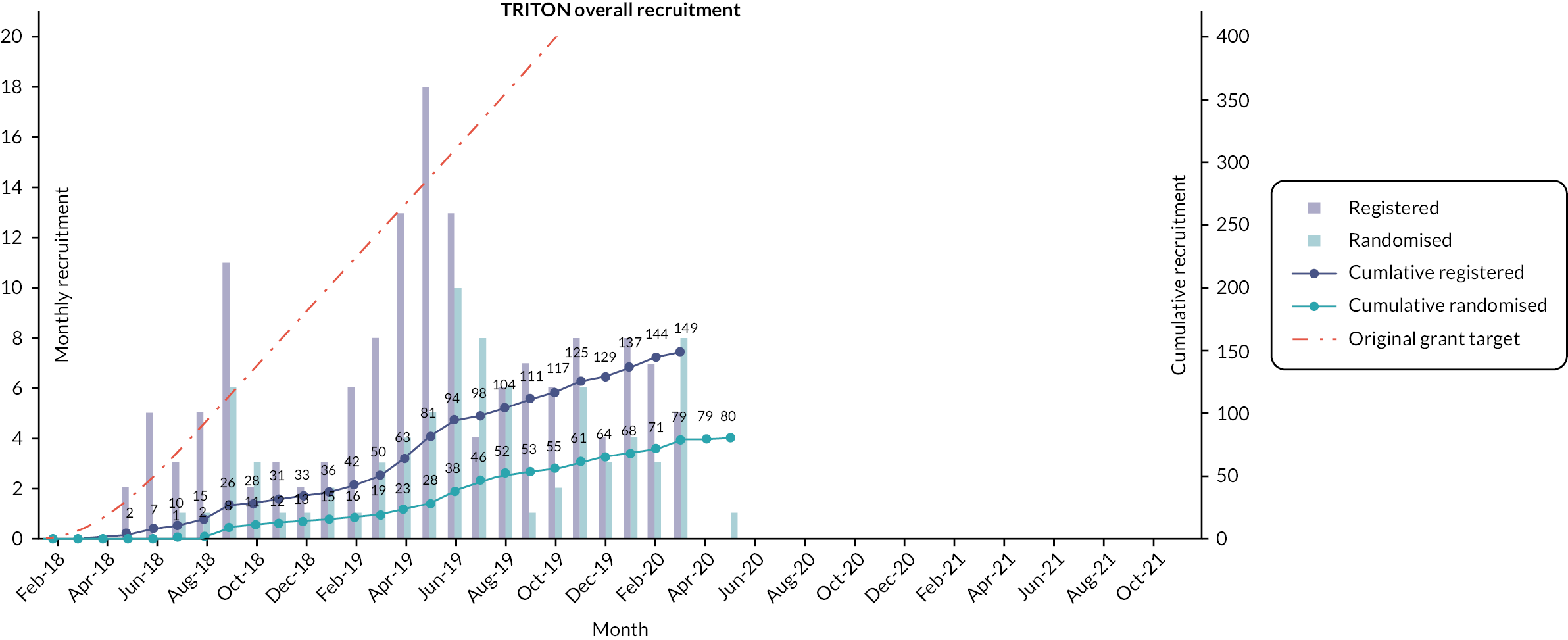
Registration data by site are provided in Appendix 3, Table 39. Participants were randomised over 23 months from July 2018 until May 2020 (Figure 3). Thirty-seven (46.3%) participants were randomised to ondansetron and 43 (53.8%) to placebo (see CONSORT diagram). By the end of treatment at 12 weeks, six participants discontinued treatment, four in the ondansetron arm (one of these patients withdrew from follow-up) and two in the placebo arm.
FIGURE 3.
Overall recruitment.
Populations
Populations are summarised in Table 3.
| Randomised to | |||
|---|---|---|---|
| Population | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
| ITT | 37 | 43 | 80 |
| PP | 36 | 40 | 76 |
| Safety | 37 | 43 | 80 |
Intention-to-treat (ITT) population. All 80 randomised participants are included in the ITT population.
Per-protocol (PP) population. Four patients of the 80 randomised were removed from the PP population due to major protocol violations. One participant was taking a restricted medication (Buscopan), two participants’ abdominal pain scores in the pre-treatment diary were too low compared with the inclusion criteria, and one participant became pregnant while in the trial.
Safety population. All 80 participants received at least 1 dose of trial medication, and there were no cases of incorrect treatment taken, so all 80 randomised participants are included in the safety population in accordance with randomised allocation.
Baseline
At baseline, randomised participants were of a similar age to all screened participants, mean age 43.9 and 44.9 years, respectively. Overall, a higher proportion of males were randomised compared with the proportion of males screened; 41.3% and 32.9%, respectively. However, a high proportion of screening records had no age and sex data making comparisons uncertain (Table 4). In both arms, males and females reported similar non-gastrointestinal somatic symptoms associated with IBS measured by PHQ-12 (Table 5).
| Randomised | Screened | ||||
|---|---|---|---|---|---|
| Demographics | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Screened (n = 1582) | Missing |
| Mean age (years) (SD) | 45.0 (15.7) | 43.0 (16.3) | 43.9 (16.0) | 44.9 (15.4) | 831a |
| Sex, N (%) | 802a | ||||
| Male | 16 (43.2%) | 17 (39.5%) | 33 (41.3%) | 257 (32.9%)b | |
| Female | 21 (56.8%) | 26 (60.5%) | 47 (58.8%) | 523 (67.1%)b | |
| Ethnicity, N (%) | |||||
| White | 34 (91.9%) | 41 (95.3%) | 75 (93.8%) | – | |
| Black | 2 (5.4%) | 1 (2.3%) | 3 (3.8%) | – | |
| Asian | 1 (2.7%) | 1 (2.3%) | 2 (2.5%) | – | |
| Current smoker, N (%) | |||||
| Missing | 4 (10.8%) 1 | 5 (11.6%) 0 | 9 (11.3%) 1 | ||
| Number of years smoked/number of cigarettes/day listing | (8/12, 10/15, 12/7, 38/9) | (5/10, 10/5, 12/7, 18/7, 37/6) | |||
| Pre-treatment diary | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
|---|---|---|---|
| Mean pain score (SD) | 61.4 (19.7) | 55.2 (16.7) | 58.0 (18.3) |
| Mean days per week with loose stool (SD) | 5.9 (1.3) | 5.4 (1.2) | 5.6 (1.3) |
| Mean urgency score (SD) | 67.5 (19.6) | 60.4 (17.8) | 63.8 (18.9) |
| Urgency score missing | 0 | 1 | 1 |
| Mechanistic test undertaken | |||
| Barostat, N (%) | 8 (21.6%) | 10 (23.3%) | 18 (22.5%) |
| Colonic manometry, N (%) | 7 (18.9%) | 6 (14.0%) | 13 (16.3%) |
| Both mechanistic tests undertaken | 6 (16.2%) | 6 (14.0%) | 12 (15.0%) |
| PHQ-12 score | |||
| Male, N | 16 | 17 | 33 |
| Mean (SD) missing | 7.5 (4.63) 0 | 7.5 (3.54) 0 | 7.5 (4.04) 0 |
| Female, N | 21 | 26 | 47 |
| Mean (SD) missing | 10.3 (4.32) 0 | 9.6 (4.63) 0 | 9.9 (4.46) 0 |
During the pre-treatment period, participants randomised subsequently to ondansetron reported, on average, higher abdominal pain scores (61.4 vs. 55.2 mean pain score), slightly more days per week with a loose stool (5.9 vs. 5.4 mean days per week) and a higher urgency to defaecate score (67.5 vs. 60.4 mean urgency score) than those randomised to placebo (see Table 5). Pre-treatment diary scores by site are provided in Appendix 3, Table 40.
Table 4 shows that the participants were well matched for demographics across randomised treatment arms. Demographic data by site are provided in Appendix 3, Table 41.
As part of the trials mechanistic substudy, participants were offered a barostat procedure and a colonic manometry. Eighteen participants underwent a barostat and 13 a colonic manometry assessment. Of these, 12 undertook both manometry assessments (see Table 5). Mechanistic test uptake by site is provided in Appendix 3, Table 42.
Further baseline summaries including summaries across time points are in Summaries and analyses of the secondary outcomes.
Losses and exclusions after randomisation
Treatment discontinuation and withdrawals
Five participants discontinued treatment, four in the ondansetron arm (two were requested by the patient and two by the clinician) and one in the placebo arm (requested by the patient) (see Table 6 and Appendix 4, Table 43). One participant in the placebo arm withdrew due to pregnancy (see Appendix 8, Table 65).
| Ondansetron (n = 37) (%) | Placebo (n = 43) (%) | Total (n = 80) (%) | |
|---|---|---|---|
| Number of discontinued treatment (% of randomised) | 4 (10.8) | 1 (2.3) | 5 (6.2) |
| Requested by patient | 2 (50.0) | 1 (100.0) | 3 (60) |
| Requested by clinician | 2 (50.0) | 0 (0.0) | 2 (40) |
| Reason for discontinuation | |||
| Patient decision | 1 (25.0) | 0 (0.0) | 1 (16.7) |
| Patient’s health compromised | 1 (25.0) | 0 (0.0) | 1 (16.7) |
| Drug intolerance | 1 (25.0) | 1 (50.0) | 2 (33.3) |
| Othera | 1 (25.0) | 0 (0.0) | 1 (16.7) |
Seventeen participants, 10 in the ondansetron and 7 in the placebo arm, withdrew from one or more parts of the study data collection processes. All 17 withdrew consent for daily text messaging, and 3 participants in the ondansetron arm also withdrew from trial follow-up (Table 7).
| Withdrawal | Ondansetron (n = 37) (%) | Placebo (n = 43) (%) | Total (n = 80) (%) |
|---|---|---|---|
| Number of withdrawn from any part of the study (% of randomised) | 10 (27.0) | 7 (16.3) | 17 (21.3) |
| Number of withdrawn consent for | |||
| Daily text messaging (% of withdrawals) | 10 (100.0) | 7 (100.0) | 17 (100.0) |
| Use of previously obtained samples (% of withdrawals) | 2 (20.0) | 0 (0.0) | 2 (11.8) |
| Optional main study assessments (% of withdrawals) | 2 (20.0) | 1 (14.3) | 3 (17.6) |
| Trial follow-up (% of withdrawals) | 3 (30.0) | 0 (0.0) | 3 (17.6) |
Protocol violations and deviations
There were four major protocol violations: one participant took a restricted medication (hyoscine), two had abdominal pain scores in the pre-treatment diary too low to meet the inclusion criteria and one became pregnant during the trial. All four were removed from the PP population.
There were 12 participants that deviated from the protocol in terms of taking more than 2 loperamide doses a week in the treatment period, 5 in the ondansetron arm and 7 in the placebo arm (Table 8). Four of those five participants in the ondansetron arm deviated from the protocol during the pre-treatment, treatment and post-treatment periods. Two of the participants in the placebo arm deviated from the protocol in the pre-treatment, treatment and post-treatment periods.
| Unique participant | Ondansetron | ||
|---|---|---|---|
| Pre-treatment | Treatment (weeks 1–12) | Follow-up (weeks 13–16) | |
| 1 | 1 | ||
| 2 | 2 | 3 | 4 |
| 3 | 3 | 1 | 1 |
| 4 | 3 | ||
| 5 | 12 | ||
| 6 | 1 | 2 | 1 |
| 7 | 2 | 3 | |
| 8 | 2 | 8 | 2 |
| Placebo | |||
| 1 | 1 | ||
| 2 | 1 | 2 | 1 |
| 3 | 1 | ||
| 4 | 1 | ||
| 5 | 5 | 3 | |
| 6 | 1 | ||
| 7 | 2 | 6 | 2 |
| 8 | 1 | ||
| 9 | 5 | ||
| 10 | 1 | ||
| 11 | 1 | ||
| 12 | 1 | ||
| 13 | 2 | 2 | |
These participants were included in the ITT analysis and additional analysis was undertaken adding the use of loperamide as an independent variable in the primary analysis model, section Supportive and sensitivity analyses.
Clinical efficacy of the intervention
Analyses of the primary outcome
Analyses were conducted on the ITT population and all 80 participants were included in the primary analysis. There were 15/37 [40.5%, 95% CI (24.7%, 56.4%)] primary end-point responders in the ondansetron arm and 12/43 [27.9%, 95% CI (14.5% to 41.3%)] in the placebo arm. Four participants (two in each arm) did not provide sufficient data to calculate the primary end point and were therefore assumed to be non-responders. Multiple imputation was not used, as there were only 5% of missing responses the potential impact of the missing data would be small44 and the overall sample size was small.
Considering the individual components of the primary end point; there were 17/37 [46.0%, 95% CI (29.9% to 62.0%)] on ondansetron who were pain responders (i.e. met FDA criteria for reduction in pain intensity) and 16/43 [37.2%, 95% CI (22.8% to 51.7%)] on placebo. As regards stool consistency, 25/37 [67.6%, 95% CI (52.5% to 82.7%)] on ondansetron and 22/43 [51.2%, 95% CI (36.2% to 66.1%)] on placebo were stool consistency responders according to FDA criteria (Tables 9 and 10). There was no statistical evidence of a difference in the FDA-defined primary end-point responder rate between arms. The odds ratio from the logistic regression model, adjusted for the treatment group, minimisation variables (undergoing manometry or barostat), age and gender was 1.93 (95% CI 0.73 to 5.11, p-value 0.1869). Site was excluded from the model due to model convergence issues (see Table 10). Additionally, when assessing individual components of the primary end point, ondansetron had greater effect on stool consistency improvement than on pain intensity reduction when comparing between arms. However, none of these differences were statistically significant.
| Primary end point | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
|---|---|---|---|
| Number of primary end-point responders, N (%) | 15 (40.5%) | 12 (27.9%) | |
| Number of non-responders, N (%) | 20 (54.1%) | 29 (67.4%) | |
| Number of participants with insufficient data to evaluate primary end point, N (%) (treated as non-responders in ITT) | 2 (5.4%) | 2 (4.7%) | 4 (5.0%) |
| Pain intensity reduction responders, N (%) | 17 (46.0%) | 16 (37.2%) | |
| Stool consistency reduction responders, N (%) | 25 (67.6%) | 22 (51.2%) | |
| Number of available data weeks | |||
| Mean (SD) | 11.2 (2.1) | 11.3 (2.0) | 11.3 (2.0) |
| Median (range) | 12.0 (4, 12) | 12.0 (0, 12) | 12.0 (0, 12) |
| Number of weeks as a responder | |||
| Mean (SD) | 4.3 (3.9) | 3.4 (3.9) | 3.8 (3.9) |
| Median (range) | 3.0 (0, 12) | 2.0 (0, 12) | 3.0 (0, 12) |
| Number of weeks as a non-responder | |||
| Mean (SD) | 7.0 (3.9) | 7.9 (4.0) | 7.5 (4.0) |
| Median (range) | 7.0 (0, 12) | 8.0 (0, 12) | 8.0 (0, 12) |
| Analysis | Responders | Model output | ||||
|---|---|---|---|---|---|---|
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
| N (%) | (95% CI) | N (%) | (95% CI) | p-value | OR (95% CI) | |
| 1. Primary end point | 15 (40.5) | (24.7 to 56.4) | 12 (27.9) | (14.5 to 41.3) | 0.1869 | 1.93 (0.73 to 5.11) |
| a. PP | 0.2493 | 1.78 (0.67 to 4.76) | ||||
| b. Sensitivity + loperamide use | 0.3076 | 1.68 (0.62 to 4.57) | ||||
| c. +interaction (treatment arm * loperamide use) | 0.3598a | 1.71 (0.54 to 5.40) | ||||
| d. +HADS anxiety score | 0.2026 | 1.96 (0.70 to 5.53) | ||||
| e. +HADS depression score | 0.0942 | 2.50 (0.86 to 7.31) | ||||
| 2. Pain intensity reduction | 17 (46.0) | (29.9 to 62.0) | 16 (37.2) | (22.8 to 51.7) | 0.3222 | 1.61 (0.63 to 4.12) |
| a. PP | 0.4229 | 1.47 (0.57 to 3.81) | ||||
| b. Sensitivity +loperamide use | 0.4976 | 1.40 (0.53 to 3.67) | ||||
| c. + interaction (treatment arm * loperamide use) | 0.3496a | 1.72 (0.55 to 5.35) | ||||
| 3. Stool consistency reduction | 25 (67.6) | (52.5 to 82.7) | 22 (51.2) | (36.2 to 66.1) | 0.0730 | 2.45 (0.92 to 6.52) |
| a. PP | 0.1042 | 2.28 (0.84 to 6.16) | ||||
| b. Sensitivity + loperamide use | 0.0647 | 2.60 (0.94 to 7.15) | ||||
| c. + interaction (treatment arma loperamide use) | 0.0595a | 3.13 (0.96 to 10.26) | ||||
Supportive and sensitivity analyses
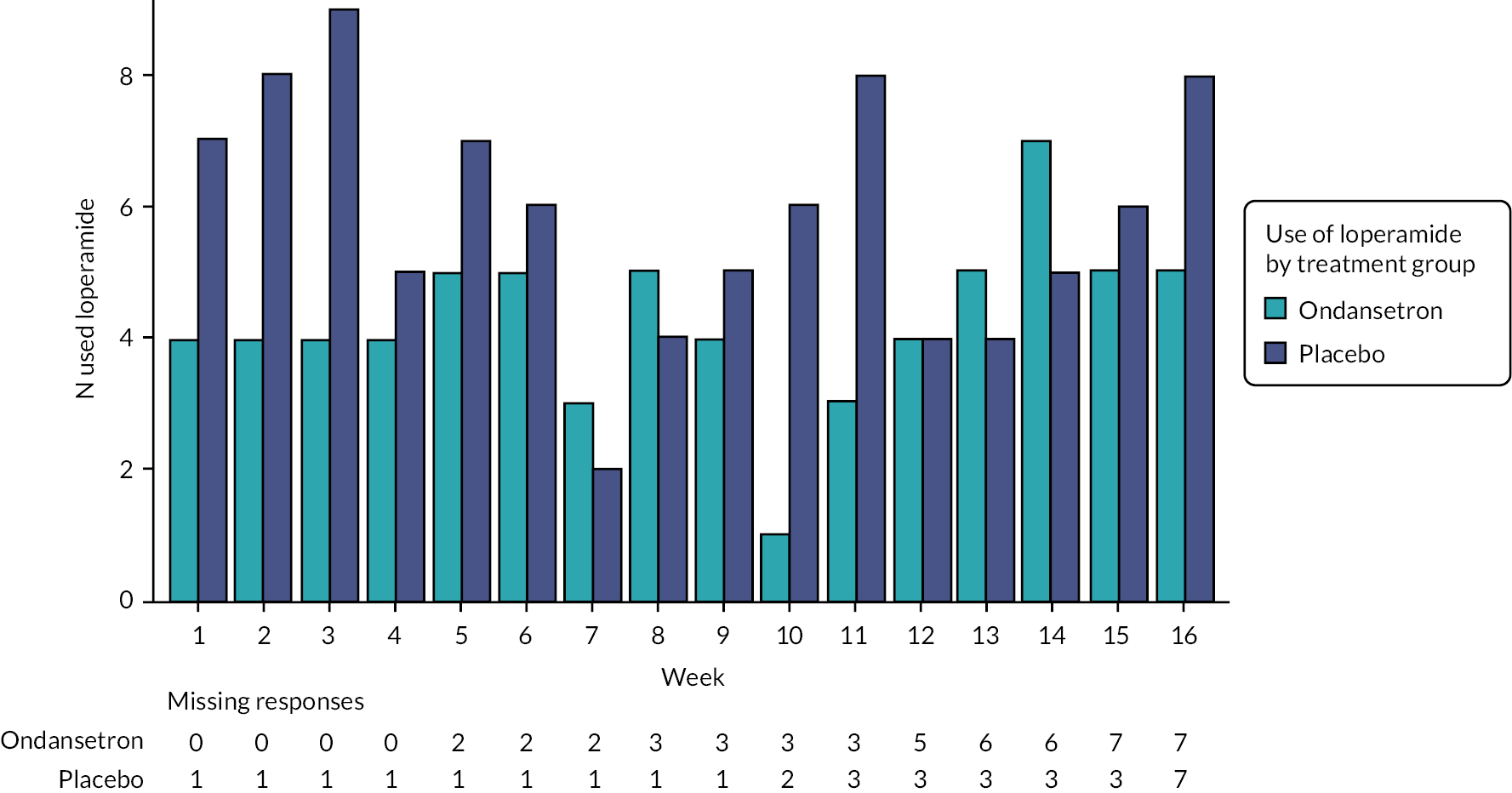
Analysis of the primary outcome on the PP population supported the primary analysis result that there is no evidence of a difference in the treatment arms (see Table 10). Throughout the study (pre-treatment, treatment and follow-up), participants were allowed to use rescue medication (loperamide) no more than twice per week. Similar proportions of participants used loperamide during pre-treatment and follow-up (off-treatment) across arms, but a higher proportion used loperamide in the placebo arm during treatment; 39.5% (n = 17) compared with 18.9% (n = 7) in ondansetron (see Appendix 6, Table 48). Therefore, as part of the sensitivity analyses, use of rescue medication was added as an independent variable and the primary analysis model was repeated. The outcome of this analysis is consistent with the primary analysis finding that there is no difference in the primary end point of FDA-defined responder rates between the treatment arms. Additionally, loperamide use was added as an interaction term with the treatment allocation and the findings were again similar. The number of responders using rescue medication is summarised in Appendix 6, Table 49; higher proportions of both responders and non-responders in the placebo arm used loperamide. Sensitivity analysis including HADS anxiety and depression scores as independent variables in the primary end-point model supports the conclusion of no evidence of a statistical difference between the treatment arms (see Table 10).
Summaries of overall responders and responders to individual components of primary outcome by week
A graphical summary of responders by week is provided in Figures 4–6. Weeks 1–12 are the treatment weeks and weeks 13–16 are the post-treatment follow-up weeks. Based on those summaries, participant response increased during the weeks in treatment in both treatment arms and decreased in the follow-up period. The largest difference between treatment arms is in stool consistency; a higher proportion of participants in the ondansetron arm were responders compared with those in the placebo arm (see Figure 6). This effect was apparent already in the first week (see Appendix 7, Table 54).
FIGURE 4.
Overall responders by week.

FIGURE 5.
Weekly abdominal pain responders.
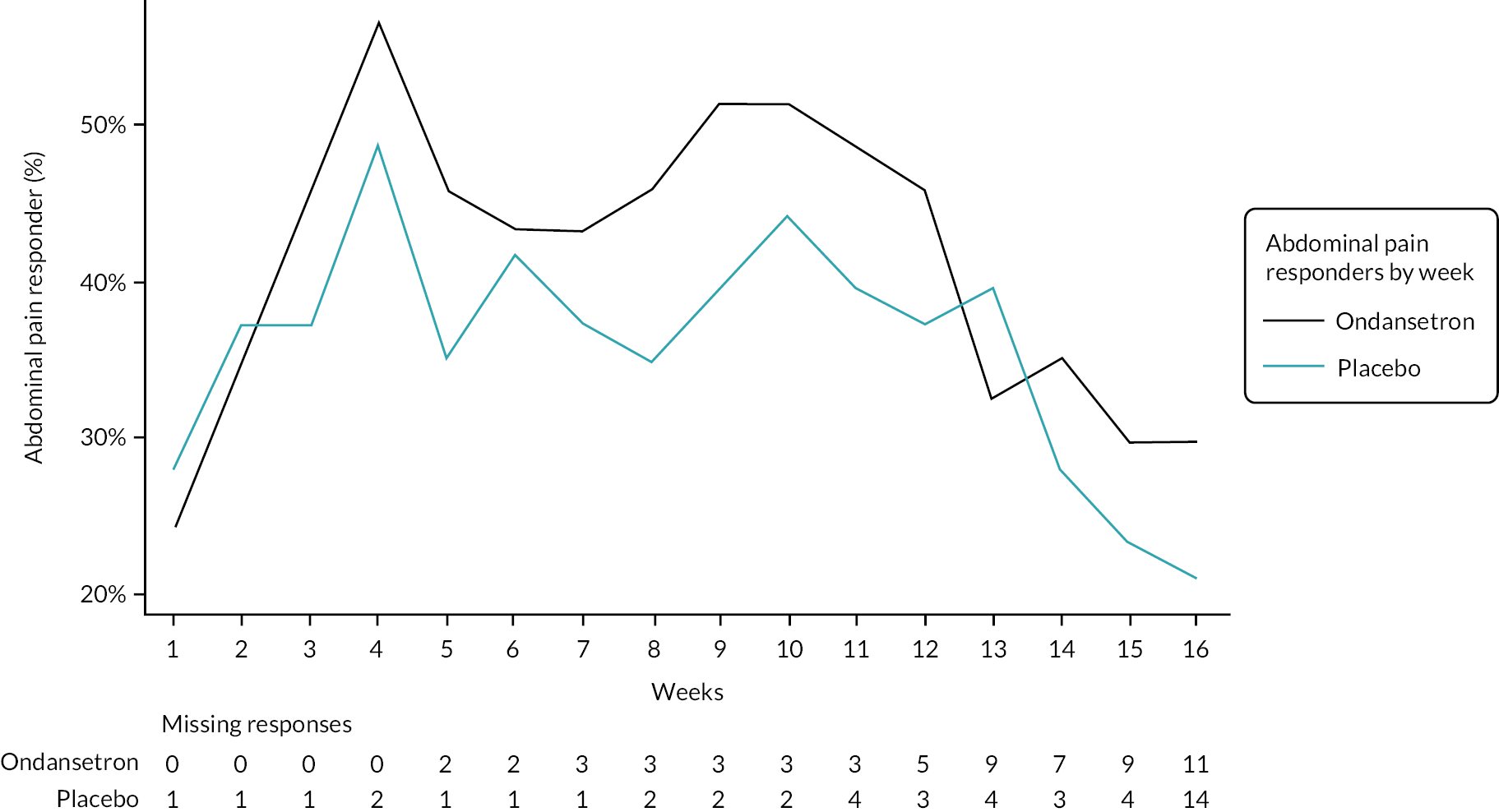
FIGURE 6.
Weekly stool consistency responders.
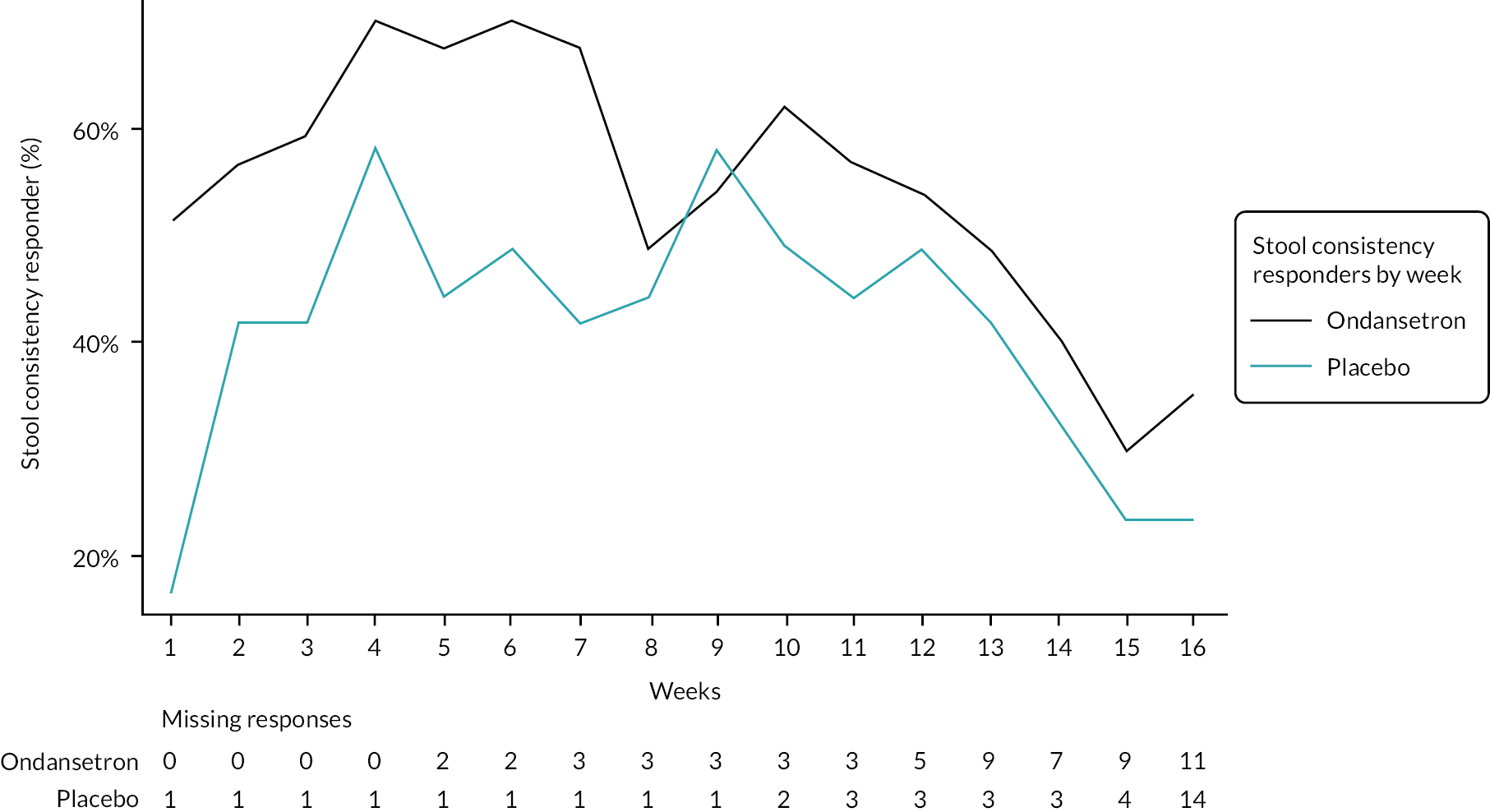
The number and proportion of participants using loperamide as a rescue medication by week are summarised in Figures 7 and 8. Additionally, the proportion of participants using loperamide was added to the figures to complement the sensitivity analyses. Tables summarising number and proportion of responders by week are in Appendix 7, Tables 52–56.
FIGURE 7.
Loperamide use (proportion of participants).
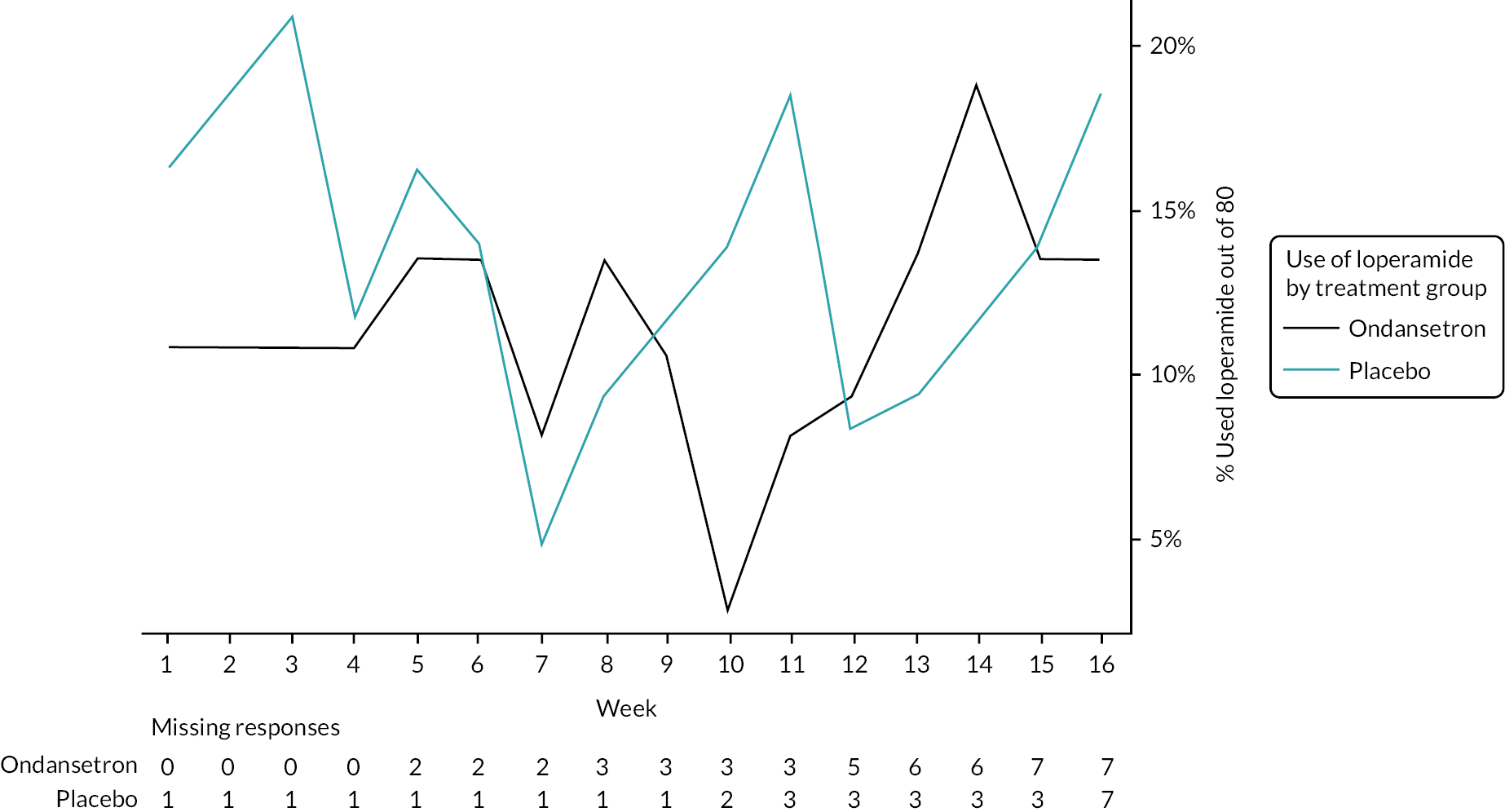
FIGURE 8.
Loperamide use (number of participants).
Summaries and analyses of the secondary outcomes
Abdominal pain
Mean abdominal pain scores were higher in the ondansetron arm prior to randomisation compared with placebo (see Table 5), but were similar across the treatment arms during the treatment and showed a similar increase in the follow-up period (Table 11), indicating a greater reduction in pain scores in the ondansetron arm during treatment (see Appendix 6, Tables 44 and 53). However, there was no evidence of a statistically significant difference in abdominal pain scores between the treatment arms during or after the treatment periods (Tables 12–14).
| Weeks 1–12 | Weeks 9–12 | Off-treatment weeks 13–16 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | |
| Abdominal pain score (0–100) | |||||||||
| Mean (SD) missing | 44.6 (23.13) 2 | 42.6 (21.25) 1 | 43.5 (22.00) 3 | 42.7 (25.28) 3 | 41.0 (25.02) 4 | 41.8 (24.98) 7 | 47.4 (27.08) 10 | 46.2 (24.06) 4 | 46.7 (25.14) 14 |
| Median (range) | 44.2 (5.5, 100.0) | 47.3 (6.0, 81.9) | 46.4 (5.5, 100.0) | 44.0 (0.4, 100.0) | 44.6 (1.4, 81.5) | 44.5 (0.4, 100.0) | 47.3 (3.2, 100.0) | 48.0 (0.0, 91.5) | 47.7 (0.0, 100.0) |
| Urgency score (0–100) | |||||||||
| Mean (SD) missing | 44.2 (27.54) 0 | 45.1 (22.35) 3 | 44.7 (24.82) 3 | 40.7 (29.09) 5 | 43.4 (24.01) 4 | 42.2 (26.26) 9 | 48.0 (29.04) 10 | 50.9 (23.95) 6 | 49.7 (26.04) 16 |
| Median (range) | 38.9 (3.1, 100.0) | 47.3 (7.1, 81.6) | 44.9 (3.1, 100.0) | 37.2 (0.7, 100.0) | 46.2 (0.0, 78.2) | 44.3 (0.0, 100.0) | 45.7 (0.8, 100.0) | 53.8 (0.4, 85.5) | 51.5 (0.4, 100.0) |
| Analysis (linear models) – weeks 9–12 | Adjusted mean ondansetron | Adjusted mean placebo | Difference in adjusted means | SE | p-value | 95% CI Lower | 95% CI Upper | N analysed |
|---|---|---|---|---|---|---|---|---|
| Abdominal pain score (0–100) | 40.0 | 41.6 | –1.6 | 4.67 | 0.741 | –10.9 | 7.8 | 73 |
| Stool urgency (0–100) | 35.6 | 42.5 | –6.8 | 5.7 | 0.236 | –18.2 | 4.6 | 70 |
| Days/week with loose stool | 2.4 | 3.0 | –0.6 | 0.51 | 0.2652 | –1.6 | 0.4 | 76 |
| Stool consistency (BSFS) (1–7) | 4.4 | 5.0 | –0.5 | 0.25 | 0.0415 | –1.0 | 0.02 | 71 |
| Stool frequency (n stools/day) | 2.5 | 2.5 | 0.1 | 0.28 | 0.8166 | –0.5 | 0.6 | 73 |
| Analysis (linear models) follow-up weeks 13–16 | Adjusted mean ondansetron | Adjusted mean placebo | Difference in adjusted means | SE | p-value | 95% CI lower | 95% CI upper | N analysed |
|---|---|---|---|---|---|---|---|---|
| Abdominal pain score (0–100) | 44.4 | 46.0 | –1.5 | 4.55 | 0.7389 | –10.6 | 7.6 | 66 |
| Stool urgency (0–100) | 43.9 | 53.3 | –9.3 | 4.94 | 0.0639 | –19.2 | 0.6 | 63 |
| Days week with loose stool | 3.1 | 3.8 | –0.7 | 0.54 | 0.2076 | –1.8 | 0.4 | 70 |
| Stool consistency (BSFS) (1–7) | 5.1 | 5.2 | –0.1 | 0.23 | 0.6166 | –0.6 | 0.3 | 67 |
| Stool frequency (n stools/day) | 2.6 | 2.9 | –0.2 | 0.3 | 0.4254 | –0.9 | 0.4 | 70 |
| Questionnaire | Adjusted mean ondansetron | Adjusted mean placebo | Difference in adjusted means | SE | p-value | 95% CI lower | 95% CI upper | N analysed |
|---|---|---|---|---|---|---|---|---|
| HADS anxiety score (0–21) | 8.4 | 9.4 | –1 | 0.84 | 0.2464 | –2.7 | 0.7 | 72 |
| HADS depression score (0–21) | 5.3 | 6.1 | –0.8 | 0.85 | 0.3659 | –2.5 | 0.9 | 72 |
| IBS-SSS questionnaire score (0–500) | 228 | 254.5 | –26.5 | 32.51 | 0.4183 | –91.5 | 38.5 | 69 |
| SFLDQ (0–32) | 6.1 | 9.3 | –3.2 | 1.43 | 0.0275 | –6.1 | –0.4 | 66 |
| IBS-QOL questionnaire score (0–100) | 61.9 | 53.1 | 8.8 | 4.96 | 0.0817 | –1.1 | 18.7 | 72 |
Urgency
Mean urgency of defaecation at baseline was higher in the ondansetron arm compared with placebo (see Table 5). During the treatment period and in the follow-up, unadjusted urgency scores were slightly higher in the placebo arm compared with ondansetron, having reduced more in the ondansetron arm from baseline (see Table 11). Urgency scores increased in both arms in the follow-up period compared with the treatment period. There was no evidence of a difference in urgency of defaecation scores between the treatment arms during or after the treatment periods (see Tables 12, 13 and 15).
| Analysis (linear models) – weeks 1–12 | Adjusted mean ondansetron | Adjusted mean placebo | Difference in adjusted means | SE | p-value | 95% CI lower | 95% CI upper | N analysed |
|---|---|---|---|---|---|---|---|---|
| Abdominal pain score (0–100) | 41.6 | 43.5 | –1.8 | 3.88 | 0.6391 | –9.6 | 5.9 | 77 |
| Stool urgency (0–100) | 38.4 | 44.9 | –6.5 | 4.8 | 0.1788 | –16.1 | 3.1 | 76 |
| Days/week with loose stool | 2.3 | 3.2 | –1 | 0.45 | 0.0362 | –1.9 | –0.1 | 79 |
| Stool consistency (BSFS) (1–7) | 4.4 | 5.0 | –0.7 | 0.19 | 0.0013 | –1.0 | –0.3 | 75 |
| Stool frequency (number of stools/day) | 2.5 | 2.8 | –0.3 | 0.25 | 0.2036 | –0.8 | 0.2 | 76 |
Number of days with loose stool, stool consistency and frequency
In all time periods reported, participants in the placebo arm had a higher mean number of days per week with loose stool and a higher mean stool consistency compared with those in the ondansetron arm. Mean daily number of stools reported by participants was similar during the treatment period and in the follow-up period (Table 16). There was a significant difference in stool consistency in favour of ondansetron between the treatment arms both in the final month of treatment, adjusted mean difference –0.5 [standard error (SE) 0.25, 95% CI (–1.0 to –0.02), p = 0.0415] (see Table 12) and during overall treatment period, adjusted mean difference –0.7 (SE 0.19, 95% CI (–1.0 to –0.3), p = 0.0013) (see Table 15). Weekly values are given in Table 13. There was no statistically significant difference in mean stool consistency in the follow-up period (see Table 13).
| Weeks 1–12 | Weeks 9–12 | Off-treatment weeks 13–16 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | |
| Number of days/week with loose stool | |||||||||
| Mean (SD) missing | 2.4 (2.13) 0 | 3.1 (2.06) 1 | 2.8 (2.11) 1 | 2.5 (2.38) 3 | 2.9 (2.18) 1 | 2.7 (2.26) 4 | 3.1 (2.52) 7 | 3.6 (2.17) 3 | 3.4 (2.32) 10 |
| Median (range) | 2.0 (0.0, 7.0) | 2.7 (0.2, 7.0) | 2.3 (0.0, 7.0) | 1.9 (0.0, 7.0) | 2.6 (0.0, 7.0) | 2.4 (0.0, 7.0) | 2.5 (0.0, 7.0) | 3.6 (0.0, 7.0) | 3.1 (0.0, 7.0) |
| Mean stool consistency | |||||||||
| Mean (SD) missing | 4.4 (1.01) 3 | 5.1 (0.77) 1 | 4.8 (0.94) 4 | 4.4 (1.32) 4 | 5.0 (0.85) 4 | 4.7 (1.12) 8 | 5.0 (1.36) 8 | 5.2 (0.82) 4 | 5.1 (1.09) 12 |
| Median (range) | 4.2 (2.1, 6.5) | 5.3 (3.9, 6.7) | 4.9 (2.1, 6.7) | 4.5 (1.6, 6.5) | 5.2 (3.2, 6.8) | 5.0 (1.6, 6.8) | 5.1 (0.0, 6.8) | 5.4 (3.4, 6.8) | 5.2 (0.0, 6.8) |
| Mean daily number of stools | |||||||||
| Mean (SD) missing | 2.6 (1.45) 2 | 2.7 (1.74) 1 | 2.7 (1.60) 3 | 2.7 (1.57) 3 | 2.5 (1.43) 3 | 2.6 (1.49) 6 | 2.6 (1.69) 6 | 2.8 (1.80) 3 | 2.7 (1.74) 9 |
| Median (range) | 2.5 (0.0, 7.0) | 2.2 (0.1, 9.4) | 2.3 (0.0, 9.4) | 2.4 (0.0, 7.3) | 2.0 (0.0, 8.1) | 2.2 (0.0, 8.1) | 2.5 (0.0, 7.6) | 2.3 (0.0, 10.0) | 2.4 (0.0, 10.0) |
There was no evidence of a statistically significant difference in mean number of days per week with loose stool or mean daily stool frequency between treatment arms in the final month of treatment (see Table 12), in the follow-up period (see Table 13), or during the entire duration of treatment (see Table 15).
Hospital anxiety and depression scale
At baseline, participants in both treatment arms had similar HADS anxiety scores, and those in the ondansetron arms had higher depression scores. At 12 weeks, both anxiety and depression scores decreased in both arms, but were generally lower in the ondansetron arm (see Appendix 6, Tables 45 and 46). A higher proportion of participants on ondansetron reported reduction in anxiety and/or depression at 12 weeks compared with those on placebo. Anxiety and depression categories are summarised in Appendix 6, Table 45. There was no evidence of statistically significant differences between the arms at the end of the treatment period in either HADS anxiety or depression scores (see Table 14).
Irritable bowel syndrome severity scoring system
At baseline, participants on ondansetron reported higher IBS symptom severity scores compared with those on placebo. At the end of the treatment period, symptom severity reduced more in the ondansetron arm and a higher proportion of participants in the ondansetron arm reported symptoms that were in remission or mild compared with placebo (Table 17). A higher proportion of participants in the ondansetron arm 19/31 (61.2%) than in the control arm 18/43 (41.8%) reported a reduction of 50 or more points in IBS-SSS at 12 weeks compared to baseline, but this was not significant (p = 0.15), Fisher Exact test (see Appendix 6, Table 47). There was no evidence of statistically significant differences between ondansetron and placebo in IBS-SSS scores at the end of treatment period (see Table 14).
| Baseline | 12 weeks | |||||
|---|---|---|---|---|---|---|
| IBS-SSS | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
| Overall score | ||||||
| Mean (SD) missing | 387.6 (89.06) 0 | 336.5 (82.21) 1 | 360.4 (88.73) 1 | 258.6 (137.80) 6 | 263.6 (113.85) 4 | 261.4 (124.09) 10 |
| Median (range) | 385.0 (155.0, 700.0) | 335.0 (190.0, 500.0) | 350.0 (155.0, 700.0) | 270.0 (0.0, 500.0) | 260.0 (30.0, 475.0) | 265.0 (0.0, 500.0) |
| Categorical | ||||||
| Remission (0–74) | 3 (8.1%) | 3 (7.0%) | 6 (7.5%) | |||
| Mild (75–174) | 1 (2.7%) | 0 (0.0%) | 1 (1.3%) | 8 (21.6%) | 5 (11.6%) | 13 (16.3%) |
| Moderate (175–299) | 4 (10.8%) | 11 (25.6%) | 15 (18.8%) | 6 (16.2%) | 15 (34.9%) | 21 (26.3%) |
| Severe (300–500) | 31 (83.8%) | 31 (72.1%) | 62 (77.5%) | 14 (37.8%) | 16 (37.2%) | 30 (37.5%) |
| Missing | 1 (2.7%) | 1 (2.3%) | 2 (2.5%) | 6 (16.2%) | 4 (9.3%) | 10 (12.5%) |
Short-form Leeds dyspepsia questionnaire
Participants in the ondansetron arm at baseline reported higher severity of dyspeptic symptoms compared with placebo. At the end of 12 weeks, both groups reported reduction of severity of symptoms, although the reduction was greater in the ondansetron arm. The largest reduction was in symptoms of indigestion and nausea (see Appendix 6, Table 50). There was evidence of a statistically significant difference between ondansetron and placebo in the total SFLDQ score, with an adjusted mean difference in scores, –3.2 points [SE 1.43, 95% CI (–6.1 to –0.4), p = 0.0275], suggesting that participants in the ondansetron arm experienced lower severity of dyspeptic symptoms (see Table 14). However, the result needs to be considered with caution due to the small sample size and missing data.
Irritable bowel syndrome quality of life
At baseline, participants in the placebo arm reported better IBS-specific QoL compared with those in the ondansetron arm. At 12 weeks, participants in both arms reported improvements in QoL, those in the ondansetron arm reported a greater improvement; there was improvement reported in all subscales of the IBS-QOL (see Appendix 6, Table 51). There was no statistically significant difference in IBS-specific QoL between the treatment arms (see Table 14).
Satisfactory relief of IBS symptoms (weeks 1–12)
There was no evidence of a statistically significant difference between treatment arms in terms of satisfactory relief of IBS symptoms (Table 18).
| Secondary outcome | Number of participants (proportions) | Model output | |||||
|---|---|---|---|---|---|---|---|
| Ondansetron (n = 37) | Placebo (n = 43) | ||||||
| N (%) missing | (95% CI) | N (%) missing | (95% CI) | p-value | OR (95% CI) | N | |
| Satisfactory relief of IBS symptoms (Yes) weeks 1–12 | 15 (40.5%) 12 | (24.7% to 56.4%) | 17 (39.5%) 9 | (24.9% to 54.1%) | 0.5332 | 1.43 (0.46 to 4.41) | 58 |
| Urgency responders (Yes) weeks 1–12 | 18 (48.6%) 0 | (32.5% to 64.8%) | 16 (37.2%) 4 | (22.8% to 51.7%) | 0.6041 | 1.3 (0.49 to 3.46) | 76 |
| Loperamide use (0 days of loperamide use) | |||||||
| Weeks 1–12 treatment | 23 (62.2%) 7 | (46.5% to 77.8%) | 22 (51.2%) 8 | (36.2% to 66.1%) | 0.1962 | 0.43 (0.12 to 1.55) | 61 |
| Weeks 9–12 only | 23 (62.2%) 8 | (46.5% to 77.8%) | 23 (56.5%) 13 | (38.6% to 68.4%) | 0.3619 | 0.48 (0.10 to 2.31) | 56 |
| Weeks 13–16 post treatment | 21 (56.8%) 8 | (40.1% to 72.7%) | 23 (56.5%) 13 | (38.6% to 68.4%) | 0.6277 | 1.39 (0.36 to 5.35) | 56 |
Use of rescue medication – number of days taken loperamide over weeks 1–12
Loperamide was used as a rescue medication by only a small per cent of subjects, mostly those that were not treatment responders (see Appendix 5, Table 55 and Figures 28 and 29). The number of days participants took loperamide over weeks 1–12 was planned to be evaluated. However, data on the number of days were right-skewed with considerable proportion of participants not using loperamide at all (see Appendix 7, Table 55). We attempted to fit two-stage model; however due to low frequencies, we were unable to fit it. Logistic regression was used instead (participant used loperamide 0 days or used it at least for 1 day during the time period). There was no evidence of statistically significant differences between the treatment groups in using loperamide during treatment or post-treatment period (see Table 18). Abdominal pain response and stool response according to loperamide use are detailed in Appendix 5, Figures 30 and 31).
Exploratory analyses on end points measured weekly, repeated measures
Weekly summaries
Figures 9–14 provide graphical summaries of abdominal pain, stool frequency, stool consistency, number of days with loose stool and satisfactory relief by week. The largest observed difference between the treatment arms was in mean stool consistency during the treatment period. Participants in the ondansetron arm had improved stool consistency measured by the BSFS compared with those in the placebo arm; these differences diminished in the post-treatment period in weeks 13–16 (see Figure 12).
FIGURE 9.
Abdominal pain by week.
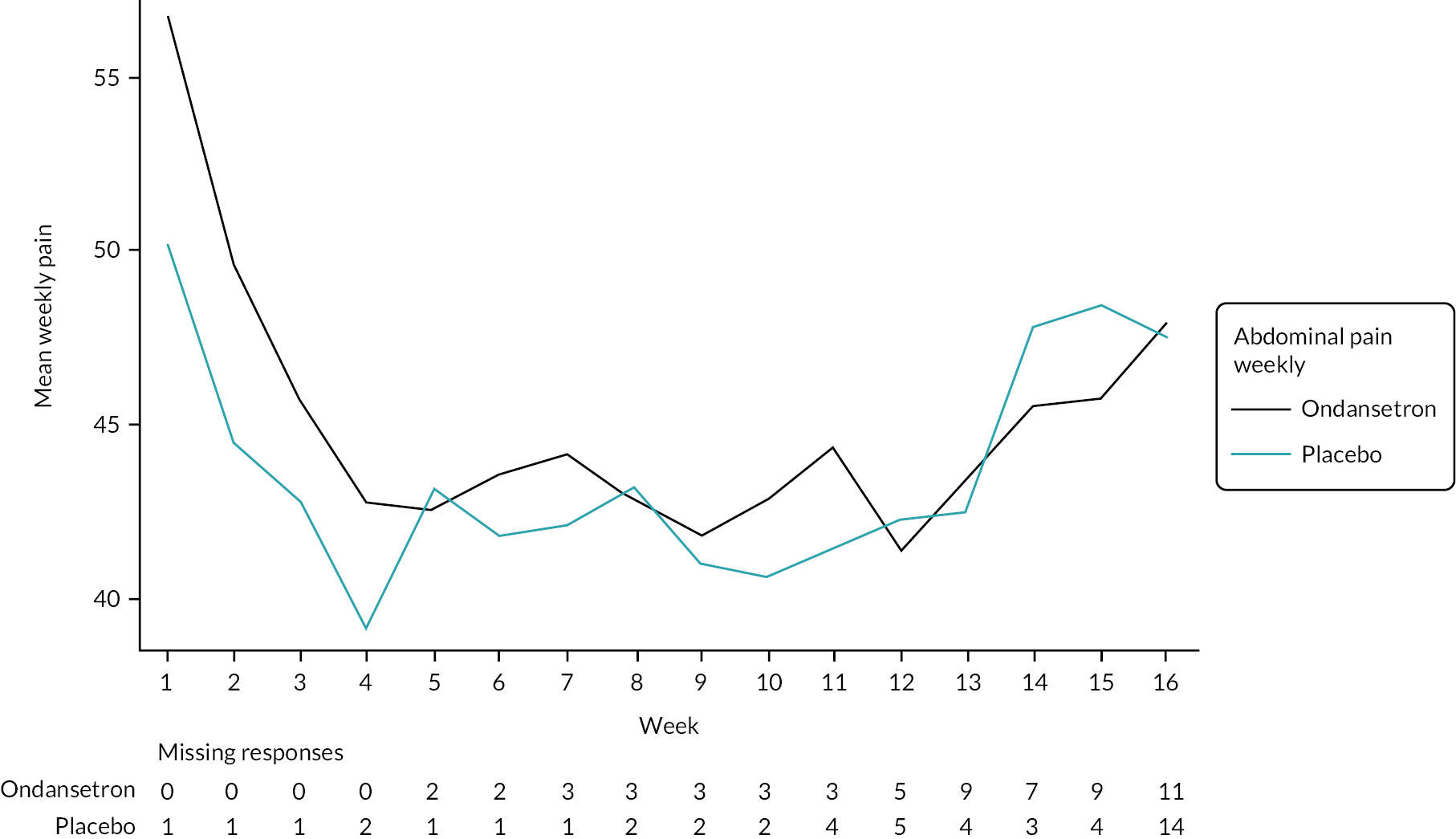
FIGURE 10.
Stool urgency by week.

FIGURE 11.
Daily number of stools by week.
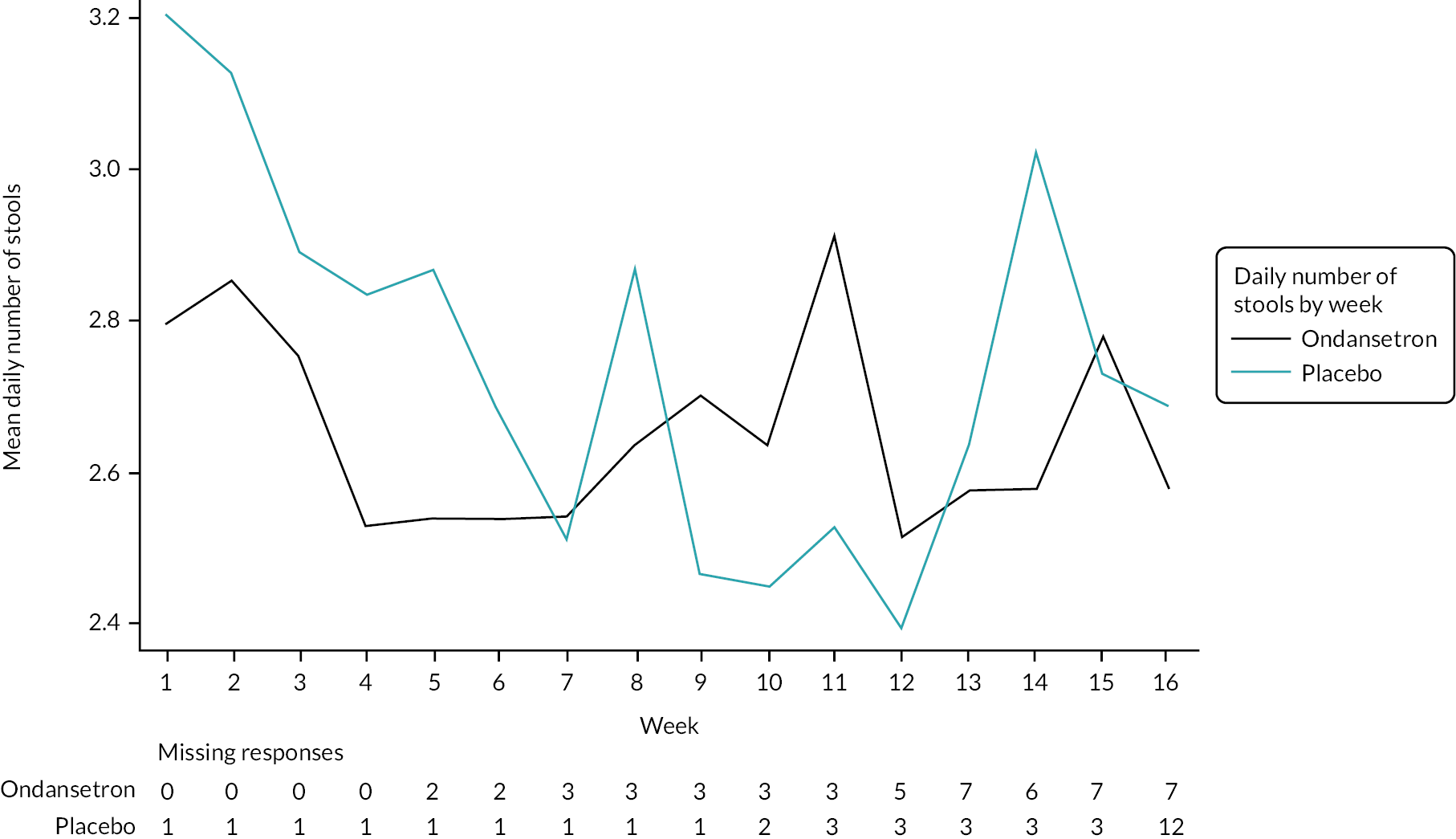
FIGURE 12.
Mean stool consistency by week.
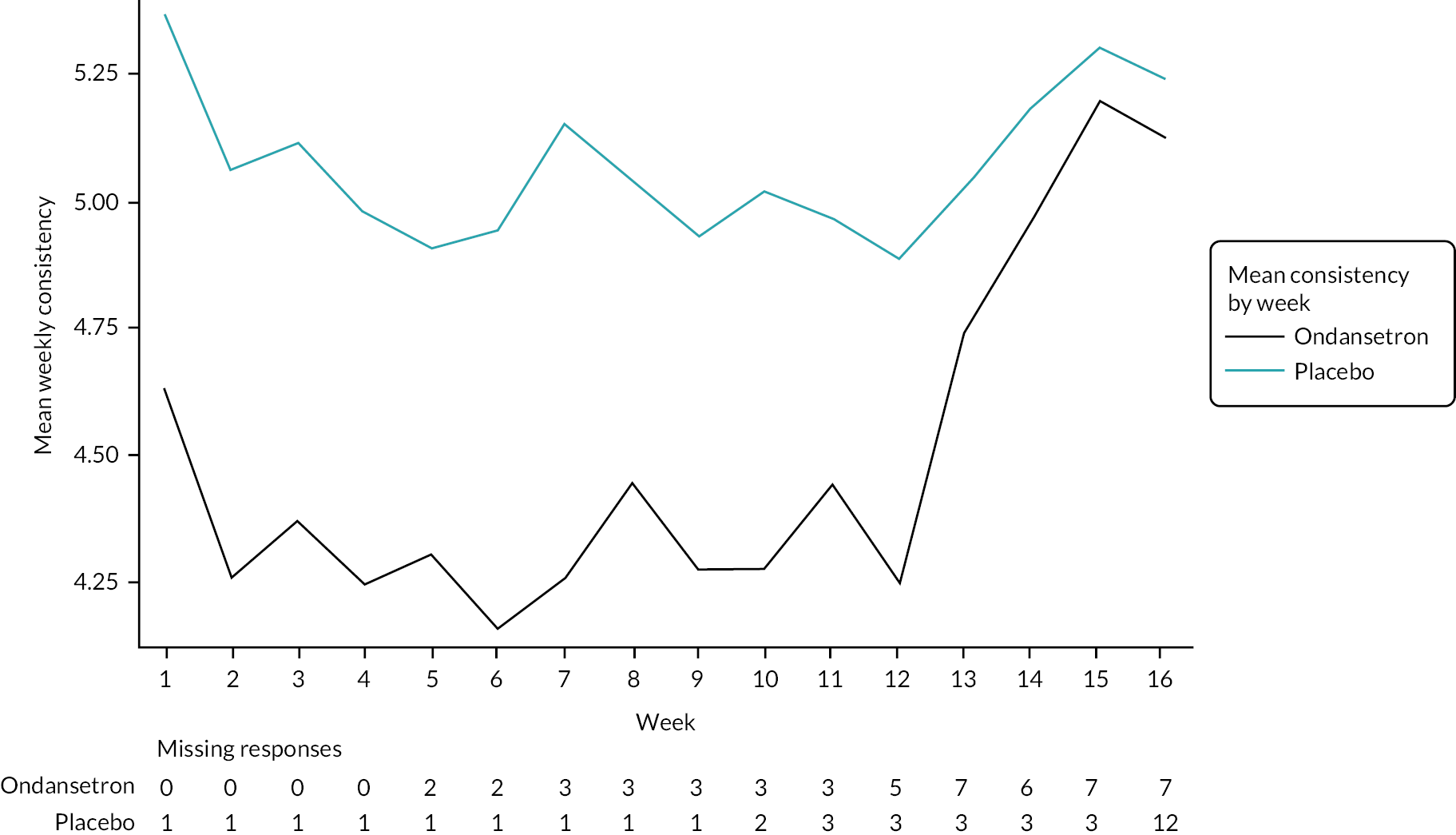
FIGURE 13.
Number of days/week with loose stools by week.
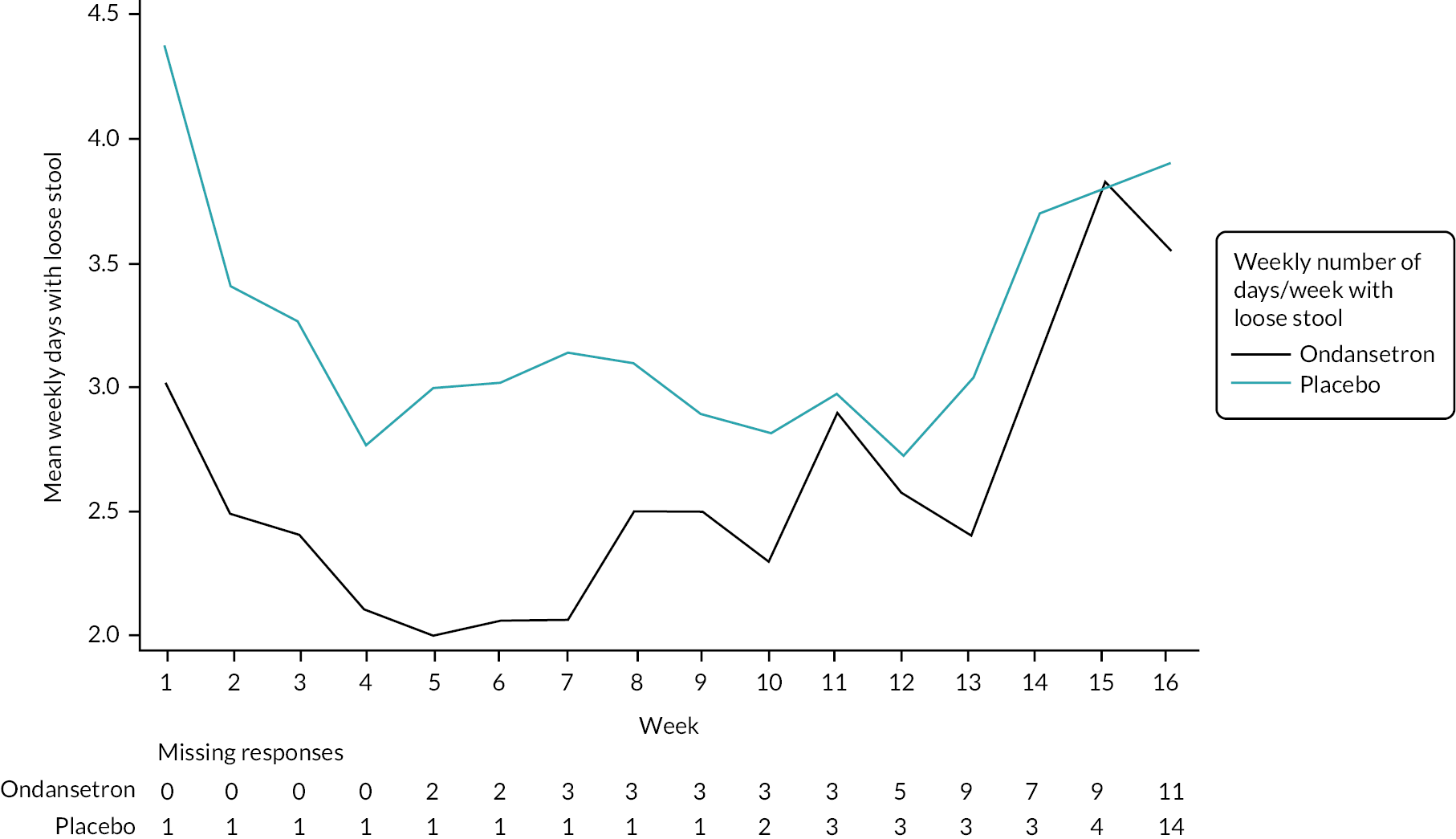
FIGURE 14.
Satisfactory relief by week.

Tabulated summaries of numbers, proportions, means and SDs of the end points described above are in Appendix 7, Tables 57–61.
Repeated measures models
In all repeated measures models, a week was fitted as a time variable and interaction between time and treatment arm was included in the model. Unstructured covariance structure provided the best fit for models. Models were run for all 12 weeks of treatment, with numerical results presented for the last 4 weeks of treatment due to space issues. The small number of participants needs to be considered in interpretation of repeated measures models described below.
Abdominal pain
There were no statistically significant differences between the treatment arms in weekly abdominal pain scores (Table 19 and Figure 15).
| Ondansetron, adjusted abdominal pain score | Placebo, adjusted abdominal pain score | Difference | |||||
|---|---|---|---|---|---|---|---|
| Week | Est (SE) | (95% CI) | Est (SE) | (95% CI) | Est (SE) | (95% CI) | p-value |
| 9 | 39.7 (3.51) | (32.7 to 46.7) | 42.3 (3.37) | (35.6 to 49.0) | –2.6 (4.40) | (–11.4 to 6.2) | 0.5607 |
| 10 | 40.6 (3.59) | (33.5 to 47.8) | 42.6 (3.45) | (35.7 to 49.4) | –1.9 (4.53) | (–11.0 to 7.1) | 0.6680 |
| 11 | 42.9 (4.03) | (34.9 to 50.9) | 42.8 (3.83) | (35.2 to 50.4) | 0.1 (5.16) | (–10.2 to 10.4) | 0.9849 |
| 12 | 39.4 (4.06) | (31.4 to 47.5) | 44.7 (3.85) | (37.1 to 52.4) | –5.3 (5.19) | (–15.6 to 5.0) | 0.3105 |
FIGURE 15.
Abdominal pain, output from repeated measures model, adjusted scores and 95% CI by week.
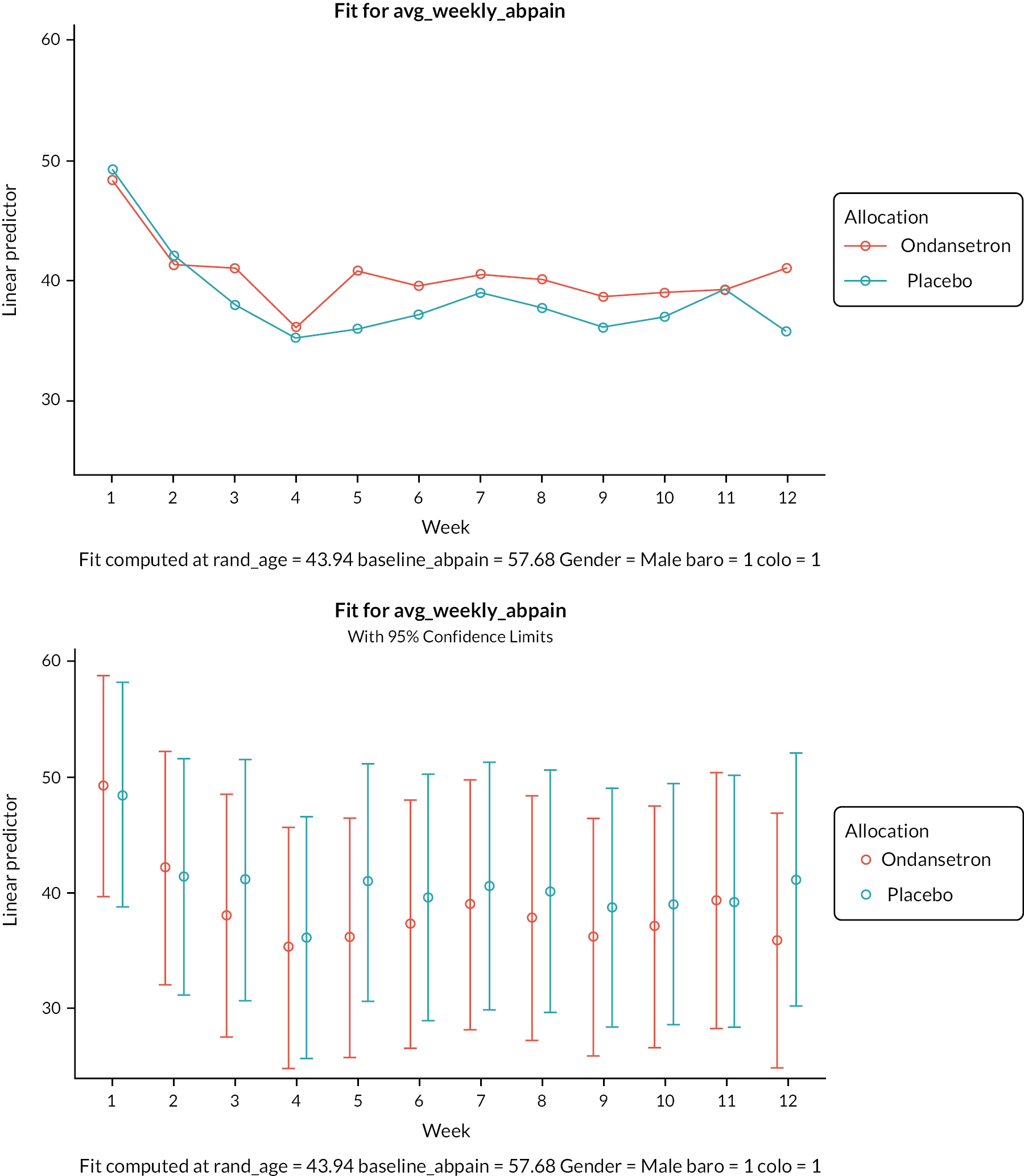
Stool urgency
There were no statistically significant differences between the treatment arms in stool urgency scores (see Table 20 and Figures 10 and 16).
| Ondansetron, adjusted stool urgency score | Placebo, adjusted stool urgency score | Difference | |||||
|---|---|---|---|---|---|---|---|
| Week | Est (SE) | (95% CI) | Est (SE) | (95% CI) | Est (SE) | (95% CI) | p-value |
| 9 | 36.1 (4.35) | (27.5 to 44.8) | 44.5 (4.42) | (35.7 to 53.3) | –8.4 (5.51) | (–19.4 to 2.6) | 0.1326 |
| 10 | 37.7 (4.51) | (28.7 to 46.7) | 41.8 (4.54) | (32.7 to 50.8) | –4.1 (5.74) | (–15.5 to 7.4) | 0.4806 |
| 11 | 40.4 (5.02) | (30.4 to 50.4) | 44.9 (4.99) | (34.9 to 54.8) | –4.5 (6.48) | (–17.4 to 8.4) | 0.4915 |
| 12 | 35.3 (4.74) | (25.9 to 44.7) | 45.6 (4.72) | (36.2 to 55.0) | –10.3 (6.06) | (–22.4 to 1.8) | 0.0930 |
FIGURE 16.
Stool urgency, output from repeated measures model, adjusted scores and 95% CI by week.

Urgency
Urgency responders were defined as participants that reported reduction in urgency score by at least 30% in at least 6 weeks of 12 weeks treatment. There was no evidence of a difference in proportions of responders between the treatment arms (see Appendix 6, Table 45 and Figure 17).
FIGURE 17.
Urgency responders by treatment arm.

Stool frequency
We were unable to fit the Poisson model due to dispersion, and a negative binomial model did not converge, so stool frequency was fitted using a linear regression model instead as stool frequency was fitted as continuous data. There were no statistically significant differences in daily stool frequency between participants in the ondansetron and placebo arms (see Table 21, Appendix 7, Table 58, Figures 11 and 18).
| Ondansetron, adjusted stool frequency | Placebo, adjusted stool frequency | Difference | |||||
|---|---|---|---|---|---|---|---|
| Week | Est (SE) | (95% CI) | Est (SE) | (95% CI) | Est (SE) | (95% CI) | p-value |
| 9 | 2.72 (0.238) | (2.25 to 3.20) | 2.96 (0.240) | (2.49 to 3.44) | –0.24 (0.294) | (–0.83 to 0.35) | 0.4155 |
| 10 | 2.62 (0.259) | (2.10 to 3.13) | 2.70 (0.257) | (2.19 to 3.21) | –0.08 (0.325) | (–0.73 to 0.57) | 0.8066 |
| 11 | 2.93 (0.278) | (2.38 to 3.48) | 2.76 (0.272) | (2.22 to 3.30) | 0.17 (0.352) | (–0.54 to 0.87) | 0.6364 |
| 12 | 2.60 (0.268) | (2.06 to 3.13) | 2.74 (0.264) | (2.22 to 3.27) | –0.14 (0.337) | (–0.82 to 0.53) | 0.6693 |
FIGURE 18.
Stool frequency, output from repeated measures model, adjusted scores and 95% CI by week.
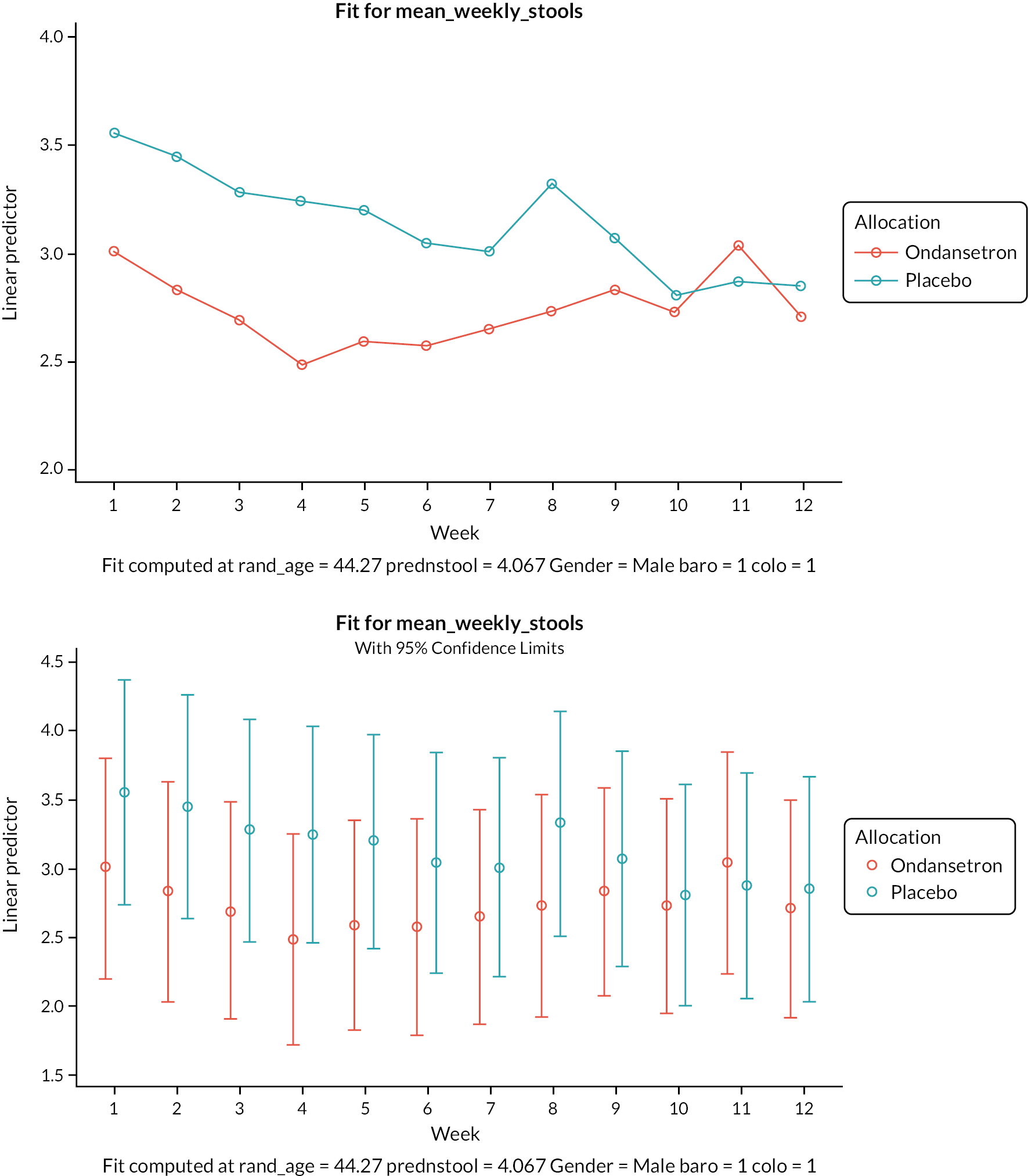
Stool consistency
Due to the small number of participants, we were not able to fit a proportional odds model for stool consistency measured by BSFS. A linear model was fitted instead. Values from weeks 9–12 are shown in Table 22 and Figure 19. There was no evidence of statistically significant differences in mean daily stool consistency between participants in the ondansetron and placebo arms, Allocation*week interaction, p-value 0.3988.
| Ondansetron, adjusted mean stool consistency | Placebo, adjusted mean stool consistency | Difference | |||||
|---|---|---|---|---|---|---|---|
| Week | Est (SE) | (95% CI) | Est (SE) | (95% CI) | Est (SE) | (95% CI) | p-value |
| 9 | 4.09 (0.257) | (3.57 to 4.60) | 4.64 (0.254) | (4.14 to 5.15) | –0.56 (0.323) | (–1.20 to 0.09) | 0.0891 |
| 10 | 3.95 (0.264) | (3.42 to 4.47) | 4.45 (0.259) | (3.94 to 4.97) | –0.51 (0.332) | (–1.17 to 0.16) | 0.1319 |
| 11 | 4.14 (0.285) | (3.57 to 4.70) | 4.49 (0.277) | (3.94 to 5.04) | –0.36 (0.362) | (–1.08 to 0.37) | 0.3292 |
| 12 | 4.01 (0.302) | (3.41 to 4.61) | 4.31 (0.292) | (3.73 to 4.89) | –0.30 (0.387) | (–1.07 to 0.47) | 0.4409 |
FIGURE 19.
Stool consistency, output from repeated measures model, adjusted scores and 95% CI by week.
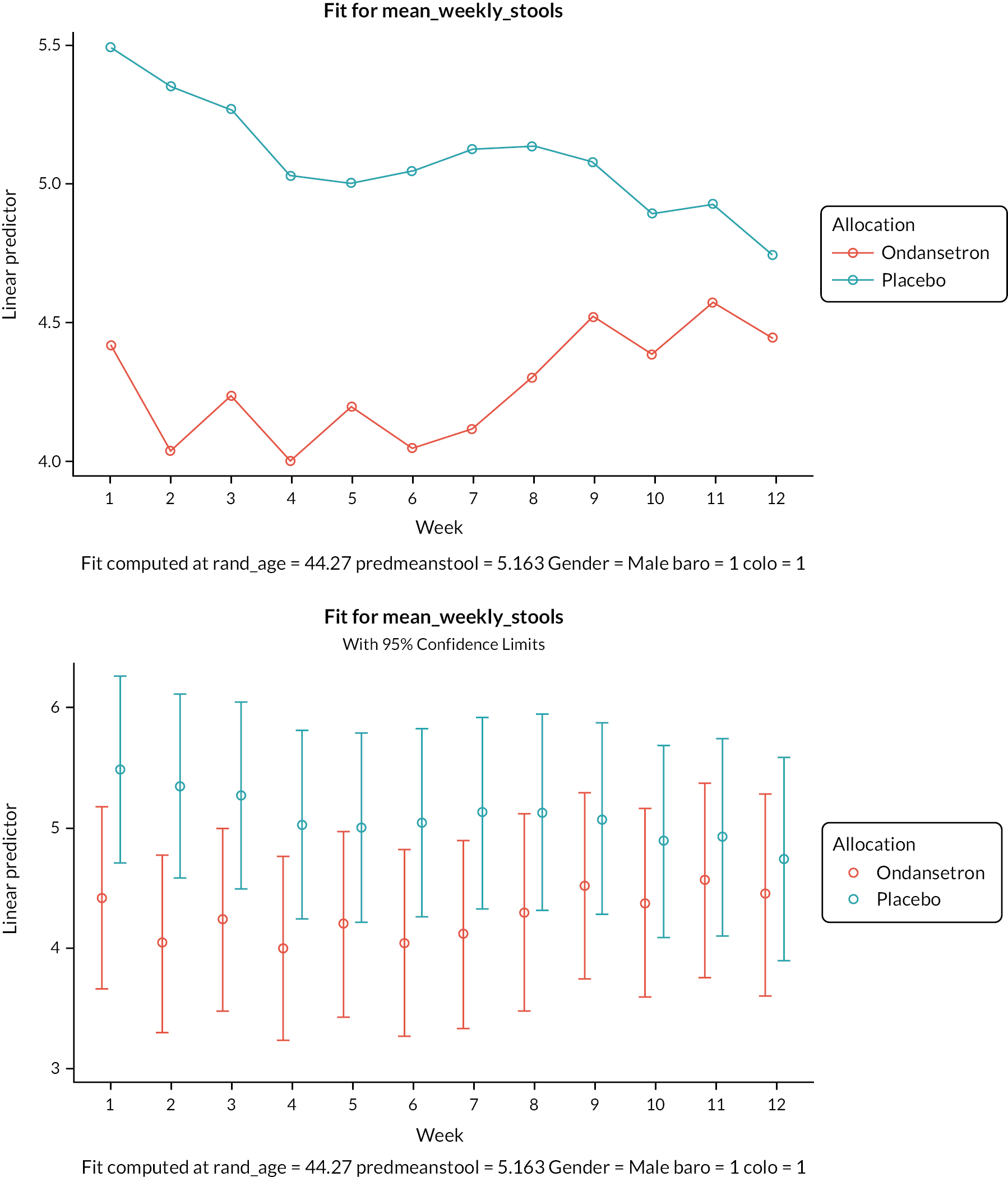
Number of days with loose stools
Number of days/week with loose stool was fitted as a model outcome. A Poisson regression model did not converge and so a linear regression model was used instead. There was no evidence of a statistically significant difference in the number of days with loose stools between the treatment arms (see Table 23, Appendix 7, Table 60, Figures 13 and 20).
| Ondansetron adjusted n of days/week with loose stool | Placebo adjusted n of days/week with loose stool | Difference | |||||
|---|---|---|---|---|---|---|---|
| Week | Est (SE) | (95% CI) | Est (SE) | (95% CI) | Est (SE) | (95% CI) | p-value |
| 9 | 6.97 (0.133) | (6.71 to 7.24) | 6.83 (0.122) | (6.59 to 7.07) | 0.15 (0.177) | (–0.20 to 0.49) | 0.4048 |
| 10 | 7.01 (0.133) | (6.75 to 7.27) | 6.85 (0.122) | (6.61 to 7.09) | 0.16 (0.177) | (–0.18 to 0.51) | 0.3566 |
| 11 | 6.98 (0.133) | (6.72 to 7.24) | 6.87 (0.122) | (6.63 to 7.11) | 0.11 (0.177) | (–0.24 to 0.45) | 0.5521 |
| 12 | 6.42 (0.135) | (6.16 to 6.69) | 6.68 (0.123) | (6.44 to 6.93) | –0.26 (0.179) | (–0.61 to 0.09) | 0.1478 |
FIGURE 20.
Number of days with loose stool/week, output from repeated measures model, adjusted scores and 95% CI.

Relief model
A logistic regression model was fitted; however odds ratios between the treatment arms need to be interpreted with caution due to missing data. Estimated treatment odds ratio varied by week (i.e. below and above 1) and there was no evidence of a statistically significant difference between arms.
Loperamide use model
A logistic regression model was fitted to the data and similarly to the relief model, odds ratios need to be interpreted with caution due to missing data and the small number of participants. Estimated treatment odds ratio varied by week and there was no evidence of a statistically significant difference between treatment arms.
Safety
There were no SAEs, serious adverse reactions or suspected unexpected serious adverse reactions reported. Comparison between treatment arms needs to be considered with caution as there were small numbers of participants in both arms (see Appendix 8, Table 62). During treatment at both 6 and 12 weeks, a higher proportion of participants in the ondansetron arm reported constipation compared with placebo. In the majority of cases, constipation was mild. Severe constipation was rare, 5% on ondansetron and 0% on placebo (see Appendix 7, Table 61).
At all time points (including follow-up), a higher proportion of participants in the placebo arm reported abdominal pain or bloating. However, the severity of abdominal pain/bloating was mild or moderate in the majority of participants (> 80%) at both 6 and 12 weeks. During follow-up, 17 (27.9%) participants overall reported severe abdominal pain/bloating.
At 6 weeks, a higher proportion of participants in the ondansetron arm reported headache, but at 12 weeks and during follow-up, proportions of participants with headache between arms were similar. The majority of headaches were mild or moderate with few rated severe.
A higher proportion of participants in the placebo arm reported nausea at 6 and 12 weeks. The majority of cases of nausea were mild or moderate. Less than 10% of participants reported vomiting during the treatment period and only one participant on ondansetron reported severe vomiting at that time.
A slightly higher proportion of participants in the placebo arm reported rectal bleeding at all time points. The proportion of participants with rectal bleeding was small, 12.5% overall. One participant in the placebo arm had severe rectal bleeding; the remaining participants had only minor rectal bleeding. At 6 weeks, all but 1 of the 10 participants with rectal bleeding had a clinical review; none were thought to need colonoscopy or flexible sigmoidoscopy performed to exclude ischaemic colitis (see Appendix 8, Table 63).
At 12 weeks, 60 (75%) participants overall had pulse and BP reported. All those with data had pulse within the normal range and two participants (one in each arm) recorded BP outside the normal range (see Appendix 8, Table 64).
There was one pregnancy reported in the placebo arm, with a positive pregnancy test confirmed after the treatment period, 96 days post randomisation; the outcome was a healthy live birth (see Appendix 8, Table 65).
At 6 weeks, five participants had commenced a new medication (two in the ondansetron and three in the placebo arm), all of which were considered by the PI as not restricted or prohibited and the participants were considered suitable for continuation in the trial (see Appendix 8, Table 66).
Additional summaries
Dose titration
Participants were allowed to self-titrate during the treatment period. Based on clinical experience, it was considered unlikely that participants would need to alter their dose after 6 weeks. The recommended titrated dose at 2 weeks, the requested dose at 6 weeks and the number and percentage of patients taking a constant dose from 6 weeks onwards are reported in Appendix 8, Table 67. A higher proportion of participants in the placebo arm required higher titrated doses of IMP after 2 weeks of treatment. Consequently, a higher number of bottles was requested for placebo participants at 6 weeks of replenishment.
In the majority of cases 54/80 (72%) overall, the number of capsules returned at 12 weeks was not consistent with the dose at 6 weeks (see Appendix 8, Table 62).
Unplanned trial drug usage
No incorrect bottles were provided to participants and there was one replacement of two damaged bottles.
Blinding and exit poll
There were no unblindings reported.
An exit poll completed by the study nurse is summarised in Table 24. There are differences in blinding index between the treatment arms. The blinding index45 ranges from –1 to +1 and a blinding index of 0 indicates compete blinding. The blinding index from the study nurse perspective was 0.40 [95% CI (0.17 to 0.63)] in the ondansetron arm compared with 0.10 [95% CI (–0.15 to 0.35)] in the placebo arm. As the lower limit of the 95% CI for the ondansetron arm is above zero; this suggests failure in masking in the ondansetron arm.
| Randomised allocation | Exit poll response | Missing response | Total | Blinding index | (95% CI) | ||
|---|---|---|---|---|---|---|---|
| Ondansetron | Placebo | Don’t know | |||||
| Ondansetron | 22 | 8 | 5 | 2 | 37 | 0.40 | (0.17 to 0.63) |
| Placebo | 17 | 21 | 2 | 3 | 43 | 0.10 | (–0.15 to 0.35) |
An exit poll completed by participants is in Table 25. There are differences in blinding index in participants’ responses. The participant blinding index in ondansetron was 0.42 [95% CI (0.18 to 0.67)] and in placebo 0.05 [95% CI (–0.18 to 0.28)], suggesting unblinding in the ondansetron arm.
| Randomised allocation | Exit poll response | Missing response | Total | Blinding index | (95% CI) | ||
|---|---|---|---|---|---|---|---|
| Ondansetron | Placebo | Don’t know | |||||
| Ondansetron | 22 | 8 | 3 | 4 | 37 | 0.42 | (0.18 to 0.67) |
| Placebo | 16 | 18 | 7 | 2 | 43 | 0.05 | (–0.18 to 0.28) |
Possible explanations for this occurrence in terms of response could be that participants in the ondansetron arm were more likely to experience (and/or report to the study nurse) some therapeutic effects. However, results of the exit poll should be treated with caution due to the small number of participants.
Certainty of choice on the scale 1–10 and reasons for choice which were completed by the site study nurse are summarised in Appendix 9, Tables 68 and 69.
Chapter 4 Mechanistic studies results
Transit
A total of 65 patients successfully completed whole gut transit time (WGTT) measurements at baseline, 28 on ondansetron and 37 on placebo. Their results are shown in Table 26. There were no significant differences between treatments at baseline; however when differences in WGTT between baseline and week 12 were compared, ondansetron tended to show a bigger increase (Figure 21), mean (SD) being 3.8 (9.1) on ondansetron versus a decrease –2.2 (10.4) on placebo, p = 0.011. Comparing the changes in regional transit times (right, left and rectosigmoid colon) between ondansetron and placebo (Table 27), only the increase in rectosigmoid transit was statistically significant.
| Baseline WGTT | Week 12 WGTT: change from baseline | |||
|---|---|---|---|---|
| Treatment | n | Mean (SD) | ||
| Ondansetron | 28 | 6.8 (6.3) | 27 | 3.8 (9.1)* |
| Placebo | 37 | 10.4 (11.7) | 37 | –2.2 (10.4) |
FIGURE 21.
Change in WGTT in hours. This shows the change in WGTT in hours, which increased significantly more on ondansetron compared to placebo, p = 0.011.
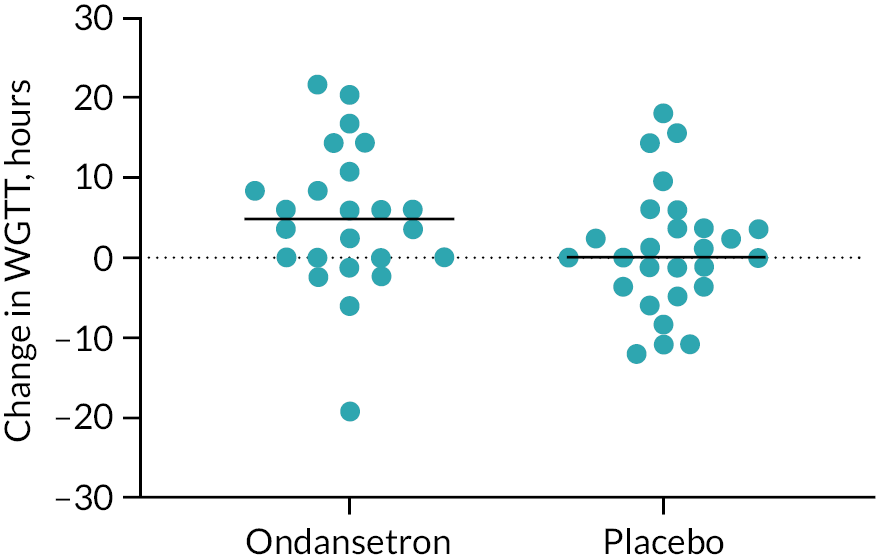
| Baseline | Week 12: change from baseline | |||||||
|---|---|---|---|---|---|---|---|---|
| WGTT | Right | Left | RS | WGTT | Right | Left | RS | |
| Ondansetron | 4.8 (2.7, 9.3) |
2.4 (0.0, 6.0) |
1.2 (0.0,3.3) |
0.0 (0.0, 1.2) |
3.6** (–2.4, 8.4) |
0.0 (–2.7, 3.6) |
0.0 (–1.2, 2.7) |
1.2* (0.0, 5.1) |
| Placebo | 7.2 (0.0, 4.8) |
3.6 (0.6, 5.4) |
1.2 (0.0, 4.2) |
1.2 (0.0, 4.8) |
–1.2 (–7.8, 3.0) |
–1.2 (–2.4, 0.0) |
0.0 (–1.2, 2.4) |
0.0 (–1.2, 1.2) |
Barostat
Data are available on just eight patients allocated to ondansetron and 10 on placebo of whom only seven and six, respectively, also underwent a second study on treatment, meaning we were seriously underpowered to assess any effect. See Tables 28 and 29 below for further details at baseline.
| Ondansetron, n = 8 | Placebo, n = 10 | p-value | |
|---|---|---|---|
| Age, median (min–max) | 50 (23–75) | 57 (22–72) | 0.76 |
| Gender, M/F | 3/4 | 1/5 | 0.6 |
| Baseline anxiety (mean ± SD) | 7 ± 3 | 11 ± 5 | 0.15 |
| Baseline urgency volume threshold, ml (mean ± SD) | 204 ± 76 | 196 ± 89 | 0.86 |
| Baseline pain threshold, mmHg (mean ± SD) | 29 ± 8 | 29 ± 12 | 0.98 |
| Ondansetron | Placebo | p-value | |
|---|---|---|---|
| n | 7 | 6 | |
| Pr1/2 (mmHg, mean ± SD) | 3.5 ± 5.2 | 2 ± 5.6 | 0.64 |
| Change in urgency threshold pressure (mmHg, mean ± SD) | 9 ± 8 | 5 ± 6 | 0.27 |
| Change in urgency threshold volume (ml, mean ± SD) | 84 ± 61 | 38 ± 48 | 0.16 |
| Change in pain threshold pressure (mmHg, mean ± SD) | 5 ± 9 | –1 ± 15 | 0.43 |
| Change in pain threshold volume (ml, mean ± SD) | 57 ± 70 | 24 ± 65 | 0.39 |
Thus, despite showing a substantial change in the mean volume to reach urgency threshold (Figure 22), which on ondansetron rose by 84 (SD 61) ml while on placebo rose from 38 to 76, mean difference 38 (SD 48) ml, there was wide variability in this effect, which was non-significant, p = 0.26.
FIGURE 22.
Change in urgency threshold volume by treatment. The change from baseline on treatment was assessed by barostat and showed a non-significant increase in volume on ondansetron.

Thus, using these figures, we calculate that to detect the difference in volume threshold urgency with 90% power would require 16 subjects per group showing as suspected, we were significantly underpowered. However, using a crossover design and paired test in the same individual we would need to study only 10 individuals, which could easily be done separate from any long-term trial.
The difference in the change in pain threshold pressure (Figure 23) was –3.500 ± 7.0 mmHg, which would however have required 85 per group to achieve 90% power to detect such a difference emphasising the small effect size on pain. Unlike alosetron there was an even smaller effect size for compliance, which increased just –1.5 ± 3.1, to detect such a difference would require 97 in each group suggesting that ondansetron does not affect compliance. Given the very minimal effect on pressure pain threshold, it seems likely that it acts on the local reflexes to reduce urgency rather than a purely sensory effect.
FIGURE 23.
Pain threshold in mmHg. This was assessed from the barostat using the ascending method of limits at baseline and after 12 weeks treatment showing no significant effect of either time, treatment or interaction, p = 0.89, 0.4 and 0.4, respectively.

High-resolution colonic manometry
Regrettably very few subjects completed the two manometry visits before and after treatment. Nine completed baseline assessment (5 allocated to ondansetron, 4 to placebo) and there was 1 dropout on ondansetron at week 12 assessment, leaving 4 and 4 to compare. Another complication was the wide variability in the depth of insertion of the manometry catheter. Some were only recording rectal activity [Figure 24, Panel (a)], while others reached the ascending colon [see Figure 24, Panel (b)].
FIGURE 24.
Plain X-ray at end of recording showing a very variable positioning achieved. Panel (a) shows recording ports mostly in the rectosigmoid region, while panel (b) shows recording ports in transverse colon missing the rectosigmoid altogether.

Obviously, these data can only be used to develop hypotheses to be tested by further studies but nevertheless there were some interesting results.
We did look at the frequency of HAPCs in the 4 hours of recording, which at baseline (n = 8) were (median, range) 1 (0–3) and on ondansetron (n = 4) were 0, 1, 11, 6 and on placebo, (n = 4) 0, 0, 2, 0. With such small numbers there were no significant differences between treatments, p = 0.25, Mann–Whitney.
Subject 78 showed marked clinical improvement on ondansetron with large falls in urgency (80.7 falling to 10.0), stool frequency (4.9 falling to 2.1) and BSFS (6.5 falling to 3.0). The results are displayed in Figures 25 and 26. There is a striking change in patterns of motility with the baseline showing many rapid moving, almost simultaneous contractions which would be predicted to cause urgent defaecation, which fits perfectly with the patient’s symptoms that were extreme.
FIGURE 25.
Manometry: example of response to a meal. Left-hand image shows position of manometry catheter, and the upper tracing shows pressure waves before ondansetron during a meal when contractions appear to rapidly traverse the sigmoid colon and rectum. In contrast, the lower trace made at the same time during a meal while taking ondansetron shows a very different, more normal pattern, with retrograde movements in both sigmoid and rectum. The right-hand panels depict the power spectrum of the pressure waves showing most are around 2–3 contractions per minute with greater power at baseline compared to after treatment.

FIGURE 26.
Manometry: example of effect of ondansetron. Raw data shown in the upper panel demonstrate increased activity in sigmoid colon on ondansetron. The bottom left panel shows an increase in the power in the retrograde contraction (negative phase difference), which is predominantly in the 2–4 cycles per minute frequency spectrum.

Subject 58 also had good traces at both baseline and on ondansetron. Figure 27 shows an increase in postprandial contraction of ondansetron and a shift in the phase spectrum indicating an increase in retrograde propagated contractions. These would push material from the rectum into the sigmoid, which would reduce urgency induced by liquid reaching the sensitive anorectal margin.
FIGURE 27.
Change in WGTT in hours.
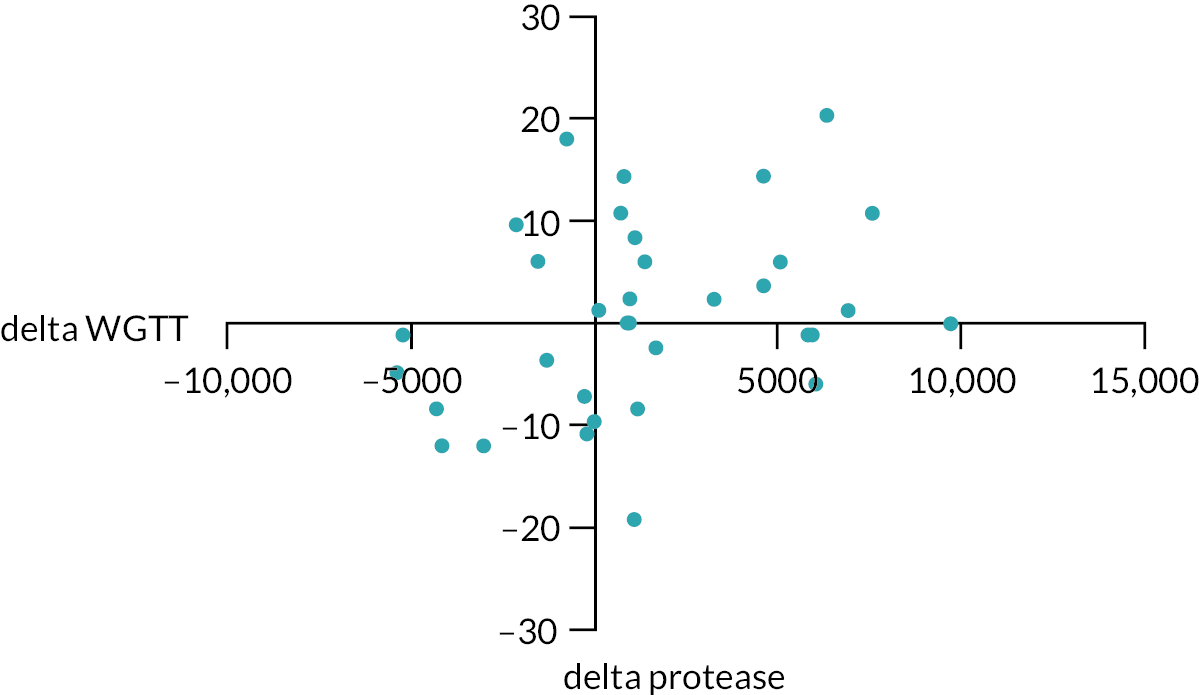
Thus, manometry is inconclusive. However, it does provide pilot data and a plausible hypothesis, namely that ondansetron stimulates retrograde activity in the rectosigmoid region, and this could be tested in future studies.
Stool % water
Combining all participants’ stools water content at baseline was mean (SD) 72.4 (6.5) %, n = 48. There were no significant differences between ondansetron or placebo groups, either at baseline or week 12 (Table 30). There was no significant correlation with transit time, r = 0.02, p = –0.88, n = 48.
| Baseline | Week 12 | |||
|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |
| Ondansetron | 20 | 72.0 (6.5) | 16 | 72.83 (5.7) |
| Placebo | 27 | 72.9 (6.6) | 16 | 74.20 (6.4) |
Loose stool responders to ondansetron (defined by < 1 day reduction in days per week with loose stools) tended to have less watery stool % water = 71.3 (6.3), n = 17 versus non-responders, stool % water = 75.5 (3.3), n = 4, but the difference was not significant, p = 0.16.
At baseline, there was no correlation between stool % water and urgency score, Pearson’s r = 0.11, n = 52, p = 0.42, nor between stool % water and days with loose stool, stool consistency nor number of stools per day, p = 0.8, 0.8 and 0.2, respectively.
Faecal proteases
These were assessed at baseline and week 12 and the results are shown in Table 31. There was wide variability but no significant change by visit or treatment, p = 0.8, two-way ANOVA.
| Baseline | Week 12 | |||||
|---|---|---|---|---|---|---|
| Mean | SD | n | Mean | SD | n | |
| Placebo | 2849 | 2620 | 23 | 2867 | 2600 | 17 |
| Ondansetron | 4017 | 2685 | 16 | 3753 | 2867 | 13 |
Contrary to our previous findings,12 we found no correlation between baseline protease and WGTT, Spearman’s r = –0.0037, p = 0.98, n = 39. However, when looking at the change from baseline in FP and WGTT at 12 weeks, Spearman’s r = mean (95% CI) 0.41 (0.03 to 0.69), there is a statistically significant correlation p = 0.03 (Figure 27).
Change in WGTT in hours [delta WGTT plotted against associated change in FP (delta protease) in trypsin units/mg protein for all subjects]. Spearman’s r = mean (95% CI) 0.32 (0.02 to 0.60), n = 34, p = 0.03.
Faecal bile salts
Faecal bile acids were assessed at baseline as shown in Table 32. Stools with more watery stool had lower total bile acid concentration, correlation with stool % water, Spearman’s r = 0.56, N = 48.
| Bile acids | CA (mM) |
CDCA (mM) |
DCA (mM) |
LA (mM) |
Total bile acids (mM) |
|---|---|---|---|---|---|
| Baseline | 0.22 (0.13) | 0.14 (0.17) | 1.55 (1.05) | 2.06 (1.5) | 3.9 (2.1) |
As Table 33 shows, at baseline there was wide variability. After 11–12 weeks of treatment, there was a numerical fall in primary bile acids on both ondansetron and placebo with a non-significant rise in the secondary/primary ratio.
| Baseline | Week 11–12 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P | S | S/P ratio | P | S | S/P ratio | p * | |||||
| N = 23 | N = 17 | ||||||||||
| Ondansetron | 0.35 (0.24) | 3.4 (1.77) | 9.72 (7.08) | 0.18 (0.09) | 3.44 (1.88) | 21.4 (32.9) | 0.85 | ||||
| N = 30 | N = 18 | ||||||||||
| Placebo | 0.36 (0.26) | 3.40 (2.29) | 22.84 (58.23) | 0.25 (0.18) | 3.435 (1.80) | 28.61 (31.42) | 0.56 | ||||
Using the total sample of 51 at baseline, there was no correlation between the secondary/primary bile acids ratio and WGTT, Spearman’s r = –0.08 (0.39–0.75), p = 0.9.
Chapter 5 Discussion
Recruitment failure and lessons learnt
This study was ended prematurely because of failure to recruit the required number of patients, despite strenuous efforts by the research team as documented in Chapter 6 Conclusions. We had previously recruited well in secondary care,12,46 so the current failure is most likely due to a substantial shift in referral practice, which markedly reduces the numbers of IBS-D patients referred to secondary care. Previously many patients suffering from pain and diarrhoea in primary care would have been referred to secondary care to exclude inflammatory bowel disease (IBD), particularly Crohn’s disease. However, this leads to extensive negative investigations and high healthcare costs, particularly those associated with colonoscopy. Current evidence-based advice is for primary care practitioners to screen such patients with a faecal calprotectin and not to refer to secondary care for those that have normal values. This care pathway is associated with a substantial reduction in costs while still ensuring that inflammatory bowel disease (IBD) cases are appropriately referred. 42
Although we responded to this reduction in referral by spreading our recruitment net wider, the alternative routes we used via advertising in media and screening via primary care patient lists proved very much less efficient. Of 620 self-referred patients who approached the trial staff for information, only 12 (2%) were ultimately randomised compared to 58/485 (12%) of referrals from secondary care. This emphasises that patients being referred to secondary care are already highly selected and well-motivated to take part in trials as their symptoms are severe enough that they want treatment. By contrast, given the fluctuating nature of IBS-D severity, our impression was that many patients approached using GP lists are often either not motivated or fail to meet severity criteria currently.
Given our small numbers, our ability to generalise is weak and the CIs for estimates are large. As such, we lacked power to demonstrate the efficacy of ondansetron in the treatment of IBS-D. Like many other trials, we found a strong placebo effect with substantial improvements in pain and stool consistency in the first 4 weeks such that, overall, in the present study around 30% met FDA criteria for response. The size predicted for this placebo effect led to our power calculation indicating we would need to recruit 400 patients to prove superiority over placebo.
We found many with a diagnosis of IBS-D did not suffer abdominal pain ≥ 1 days per week and, therefore, using Rome IV criteria to define IBS, they were not eligible. Although this change in criteria was intended to raise the bar for diagnosis so that patients meeting criteria would warrant treatment, it has changed the nature of the population and made recruitment more difficult.
Impact on primary end point
We were unable to demonstrate a significant increased response rate in the ondansetron arm, as defined by the FDA composite end point compared with placebo. As already mentioned, our data support our initial power calculations suggesting we needed > 400 patients to achieve adequate power (80%). As in our previous pilot study,12 and the recently reported Bekinda trial using modified release ondansetron,47 the effect on responder rate for stool consistency was higher than for pain. Although the current trial result did not confirm the efficacy of ondansetron, the size and direction of the effect were very similar, suggesting there is a genuine effect.
Impact on stool consistency, days with loose stool and stool urgency
Despite our lack of statistical power, we did show statistically significant differences between treatment arms with improvement in stool consistency as assessed by the BSFS and reduction in days with loose stool in the ondansetron arm.
Ondansetron treatment in our trial was associated with a lower average BSFS over weeks 1–12 with a mean difference from placebo of 0.7 (0.19), p = 0.0013. It was also associated with fewer days per week with loose stool, a mean difference of –1.0 day (0.45), p = 0.03, compared with placebo. Although we showed a reduction in stool urgency in the ondansetron arm, there was no evidence of a statistically significant difference between the arms.
Dose titration
Our trial differed from most IBS trials in mimicking clinical practice and using dose titration, which prior clinical experience had suggested as optimum. We had previously reported that when patients were allowed to adjust the dose, they choose a wide range of optimum doses. A subgroup who needed very small doses were more sensitive to the drug as shown by a larger increase in transit time despite the lower dose. 12 The same study had a much-reduced incidence of constipation, just 9% compared with 33% in a recent study of alosetron, where fixed dosing was used. 48 Evidence from our study supports this with only 1/37 (2%) of our patients on ondansetron discontinuing treatment because of constipation, no different from placebo in whom one patient also discontinued because of constipation. We suggest that future trials should also use the titration method as it avoids early dropout due to unacceptable constipation.
Time course of effect
Analysis of the weekly scores showed that the effect of ondansetron on stool form was rapid, being seen in the first week in keeping with long clinical experience from prescribing this drug. Thus, 51.4% were stool consistency responders on ondansetron at week 1 compared with 16.3% on placebo. This difference was maximal in week 1 but over time differences lessened; there were no statistically significant differences between the arms over weeks 1–12. Interpretation of this was complicated by loperamide use, which was greater in the placebo group, a not unexpected feature that would however have tended to minimise treatment differences. The option of not allowing loperamide use was considered, but rejected, as it was felt that this would prove a severe barrier to recruitment and would lead to extensive dropouts from the placebo arm. Furthermore, when we performed a sensitivity analysis including loperamide use in the model, we found it made very little difference. Nearly 80% of subjects did not use loperamide and its use does complicate interpretation. A simpler alternative would be to prohibit loperamide and treat dropouts with uncontrolled diarrhoea as treatment failures.
Mode of action
Our transit studies confirmed earlier reports that ondansetron significantly slowed transit, an effect particularly noticeable in the rectosigmoid region. This was reflected clinically in the significant increase in the proportion of stool consistency responders. Unexpectedly, the water content of stool did not correlate with either the objective measure of WGTT or any of the expected symptoms, such as urgency and days with loose stool. This was despite quite reasonable numbers, especially at baseline. One problem is the erratic nature of the IBS bowel habit, so perhaps it would have been necessary to average several stools to get a more meaningful value, as has been suggested with analysis of the microbiota. The other conclusion could be that other factors such as sensitisation of pelvic nerves or central sensitisation are more important than stool % water in determining symptoms. Regrettably for most of the remaining studies, we had far too few patients to be able to reliably define the mode of action.
The barostat study was underpowered and there was a large placebo effect but there was a tendency for ondansetron to increase the volume eliciting a sense of urgency. We calculated that we would only need 16 per group to show this difference significant at 90% power or even 10 using a crossover design, so this should probably be tested further.
Protease results showed no treatment effect, nor did we confirm our original finding that baseline protease correlated negatively with transit, although we did show that changes in transit correlated with changes in protease, supporting the concept that fast transit increases FP levels by reducing the time for protease degradation, as we had previously shown using an osmotic laxative to accelerate transit. 23 We performed an individual patient data meta-analysis of the change in transit and change in FP combining our data with the first arm of the Garsed crossover trial, which comes to a similar conclusion (Spearman’s r = 0.25, n = 60, p = 0.048).
Sensitivity as assessed by baseline barostat pain threshold, however, did not correlate significantly with baseline FPs [Pearson’s r = 0.23 (–0.7–0.8), p = 0.65], which gives no support for the idea that proteases are critical targets for future therapies.
Recognising the high prevalence of BAD in patients thought to have IBS-D, we deliberately excluded those meeting conventional criteria for BAD. Studies at the Mayo Clinic have previously reported on 74 IBS-D patients in whom BAD was excluded and showed a weak correlation with transit measured as geometric centre after 24 hours. We found a similar tendency, but this was not significant, likely due to our smaller numbers. Our values for total bile acids of 3.9 (2.1) mM were in the lower range of those reported by the Mayo group of 9.6 (7.1) mM for normal subjects. 49 None of our patients’ values for concentration of total bile acids were above the mean + 2SD reported by Peleman and colleagues,49 suggesting that we had effectively excluded BAD.
Equality, diversity and inclusion
We attempted to include all patients presenting with IBS-D using a wide range of approaches. However, the distribution of patients with 93.8% white people and only 3.8% black people and 2.5% Asian suggests failure to recruit from minorities. Future studies should explore the use of alternative ways of reaching these groups.
Recommendations for future trials
Our experience suggests that the change in referral practice over the last decade has substantially reduced referrals of IBS-D patients to secondary care. This means that, to improve recruitment, future trials should recruit mainly from primary care. We have recently had good experience using enthusiastic primary care doctors to successfully recruit 463 IBS patients to a the HTA-funded ATLANTIS trial of amitriptyline, despite the ongoing COVID pandemic. 50 This was achieved using 56 general practices across 3 regions of England. The trial was adapted to the pandemic, with as many remote processes (including data entry) as possible, other than screening bloods and informed consent, which required face-to-face visits. The barriers to recruitment could be reduced by adopting national guidelines using recommended simple screening to exclude IBD and coeliac disease [calprotectin, full blood count (FBC) and coeliac serology]. The threshold for entry could be the Rome IV clinical practice modifications with loose stools ‘usually’ or > 25% of the abnormal stools passed. 51 The symptom threshold would be ≥ 1 day per week with pain but not requiring an average pain > 3 out of 10, since this is not part of the Rome criteria and may exclude the moderate cases who might do best. A 1-week trial of colestyramine should be used to exclude BAD and the trial should use titrated ondansetron starting with 4 mg daily. Titration undoubtedly accounts for the low rate of constipation and the fact that dropouts due to constipation were very low (2%) compared to other studies on 5HT3RAs. The primary end point should be the responder rate, defined clinically. We followed recent commercial trials and used the FDA composite end point responder with stool consistency, frequency, urgency and abdominal pain scores as secondary end points. Using our own data, we can calculate that using the FDA responder rate, we would need 222 per arm to achieve 80% power with type I error of 5% and allowing 15% attrition would require 522 to be recruited in total. However, there are alternatives to the FDA end points. Several recent trials, particularly for treatments which do not directly affect bowel habit such as diets, have used the IBS-SSS responder rate, defining responders as those with a > 5052 or 7553-point fall in IBS-SSS score, which is a more global assessment of IBS impact on QoL. Using IBS-SSS score as an end point and a responder definition of a fall > 50 points in IBS-SSS [which the originators of the IBS-SSS give as the minimal clinically important difference (MCID)], we could achieve a 90% power to detect the difference we observed (61.8% vs. 41.8%) using 306. Allowing for 15% dropout, the study would need to recruit a total of 360 patients, which might be considered more feasible for a non-commercial study. Loperamide use complicated interpretation of results with more taken on placebo likely to have reduced differences observed in stool consistency and frequency. However, at any one time only 10–15% took loperamide and if we had excluded loperamide use and taken its use as a failure of treatment, it would have made the study more powerful to detect such differences, accepting that some patients might have refused to take part.
Using Rome III criteria, and so including those with less pain or abdominal discomfort alone, would facilitate recruitment. From registration to randomisation, we lost 31/149 (20.1%) of those willing to take part because of this requirement. The logic of including those with lesser or absent abdominal pain is that the drug appears most effective at controlling loose stools and urgency. We found many patients actually had only minor pain and for them control of diarrhoea would be ample gain to justify the use of ondansetron, now that it is available in an inexpensive generic form throughout the world.
Chapter 6 Conclusions
This trial closed prematurely owing to failure to recruit. However, it did show that titrated ondansetron is well tolerated in IBS-D and does slow whole gut transit and improve stool consistency. We were underpowered to prove that ondansetron improves IBS symptoms using the FDA-recommended composite end point, but our results were consistent with other trials of ondansetron which did so. We believe the treatment should be more widely available but ensuring that this is accepted by regulatory authorities will require a further larger trial in primary care.
Acknowledgements
We would like to thank all the members of the various committees listed below who gave their time and effort to support the trial.
Contribution of authors
David Gunn (https://orcid.org/0000-0003-1436-7754) was a clinical research fellow and contributed to the trial implementation including recruitment, data acquisition and delivery of mechanistic aspects of the trial as well as data analysis.
Ron Fried (https://orcid.org/0000-0002-7327-055X) was a clinical research fellow and contributed to the trial implementation including recruitment, data acquisition and delivery of mechanistic aspects of the trial as well as data analysis.
Rabia Topan (https://orcid.org/0000-0001-8722-9024) was a clinical research fellow and contributed to the trial implementation including recruitment, data acquisition and delivery of mechanistic aspects of the trial as well as data analysis.
Ivana Holloway (https://orcid.org/0000-0002-9542-883X) (Senior Medical Statistician) provided statistical input into the trial design, implementation and statistical analysis plan, under the supervision of Amanda Farrin.
Richard Brindle (https://orcid.org/0000-0002-1848-6219) (Medical Statistician) provided statistical input into the trial design, implementation and statistical analysis plan, under the supervision of Amanda Farrin.
Suzanne Hartley (https://orcid.org/0000-0003-2346-9461) (Head of Trial Management) was responsible for operational delivery of the trial.
Lorna Barnard (https://orcid.org/0000-0001-5436-6882) (Senior Trial Manager) contributed to the protocol development and implementation and co-ordination of the data acquisition and trial reporting.
Maura Corsetti (https://orcid.org/0000-0003-2957-4684) (Clinical Associate Professor in Gastroenterology) patient enrolment and data acquisition.
S Mark Scott (https://orcid.org/0000-0002-7997-1533) (Director of GI Physiology Unit, Barts Health NHS Trust) designed the TRITON trial.
Adam Farmer (https://orcid.org/0000-0003-1902-2640) (Consultant Gastroenterologist and Senior Lecturer) designed the TRITON trial, patient enrolment and data acquisition.
Ayesha Akbar (https://orcid.org/0000-0002-3924-6505) (Consultant Gastroenterologist) designed the TRITON trial, patient enrolment and data acquisition.
Maria Eugenicos (https://orcid.org/0000-0002-2630-6396) (Senior Lecturer and Specialist in Gastroenterology) designed the TRITON trial, patient enrolment and data acquisition.
Nigel Trudgill (https://orcid.org/0000-0002-8040-8158) (Consultant Gastroenterologist) designed the TRITON trial, patient enrolment and data acquisition.
Kapil Kapur (https://orcid.org/0000-0003-0269-6681) (Consultant Gastroenterologist) designed the TRITON trial, patient enrolment and data acquisition.
John McLaughlin (https://orcid.org/0000-0001-6158-5135) (Professor of Gastroenterologist and Senior Lecturer) designed the TRITON trial, patient enrolment and data acquisition.
David S Sanders (https://orcid.org/0000-0002-3739-0124) (Consultant Gastroenterologist) designed the TRITON trial, patient enrolment and data acquisition.
Arvind Ramadas (https://orcid.org/0000-0001-7353-2516) (Consultant Gastroenterologist) patient enrolment and data acquisition.
Peter Whorwell (https://orcid.org/0000-0002-5220-8474) (Professor of Medicine and Gastroenterology) designed the TRITON trial, patient enrolment and data acquisition.
Lesley Houghton (https://orcid.org/0000-0002-5351-0229) (Professor of Neurogastroenterology) designed the TRITON trial and data acquisition.
Phil G Dinning (https://orcid.org/0000-0001-8910-2801) (Senior Clinical Scientist) designed the TRITON trial and performed blinded review of the manometry examinations.
Qasim Aziz (https://orcid.org/0000-0002-2718-2065) (Professor of Neurogastroenterology) designed the TRITON trial patient enrolment and data acquisition.
Alexander C Ford (https://orcid.org/0000-0001-6371-4359) (Honorary Consultant Gastroenterologist) designed the TRITON trial, shared responsibility in his role as co-chief investigator, patient enrolment and data acquisition.
Amanda Farrin (https://orcid.org/0000-0002-2876-0584) (Professor of Clinical Trials and Evaluation of Complex Interventions and Director of Complex Interventions Division) designed the TRITON trial and took overall responsibility for statistical design, implementation and analysis.
Robin Spiller (https://orcid.org/0000-0001-6371-4500) (Professor of Gastroenterology) conceived and designed the TRITON trial and had overall responsibility in his role as chief investigator.
All authors contributed to the writing of the report and had the opportunity to revise it prior to submission.
Trial Steering Committee
Dr Duncan Loft, TSC Chair (University Hospitals Coventry & Warwickshire), Dr David Elphick (Chesterfields Royal Hospitals NHS Foundation Trust), Prof Graeme MacLennan (University of Aberdeen) and Karen Andrews (PPI representative), DMEC.
Dr Charlie Murray, DMEC Chair (Royal Free London NHS Foundation Trust), Dr James Turvill (York Teaching Hospitals NHS Foundation Trust) and Natalie Rowland (Birmingham Clinical Trials Unit).
Leeds Institute of Clinical Trials Research
Sadie Reed, Catherine Olivier, Rachel Ellison, Thomas Smith, Melise Harland and Gina Bianco.
Patient and public involvement
Karen Andrews and Peter Rutherford.
Participating sites
With thanks to all the research staff at the participating centres who provided and cared for trial participants and collected trial data:
Barnsley Hospital NHS Foundation Trust (Dr Kapil Kapur), Barts and The London School of Medicine and Dentistry (Prof Qasim Aziz), County Durham and Darlington NHS Foundation Trust (Prof Yan Yiannakou), Doncaster and Bassetlaw Teaching Hospitals NHS Foundation Trust (Dr Anurag Agrawal), Leeds Teaching Hospitals NHS Trust (Prof Alex Ford), London North West Healthcare Trust (Dr Ayesha Akbar), Manchester University NHS Foundation Trust (Prof Peter Whorwell), NHS Lothian (Dr Maria Eugenicos), Nottingham University Hospitals NHS Trust (Dr Maura Corsetti), Salford Royal NHS Foundation Trust (Prof John McLaughlin), Sandwell and West Birmingham NHS Trust (Dr Nigel Trudgill), Sheffield Teaching Hospitals NHS Foundation Trust (Prof David Sanders), South Tees NHS Trust (Dr Arvind Ramadas), St Georges University Hospital NHS Trust (Dr Jamal Hayat), University College London Hospitals NHS Foundation Trust (Prof Anton Emmanuel) and University Hospitals of North Midlands NHS Trust (Dr Adam Farmer).
Participants
We are grateful to all the trial participants for their essential contribution to the trial.
All data requests should be submitted to the corresponding author for consideration and would be subject to review by a subgroup of the trial team, which will include the data guarantor, Professor Farrin. Access to anonymised data may be granted following this review. All data-sharing activities would require a data-sharing agreement.
Ethical approval
Ethical approval for the study was given by Leeds West Research Ethics Committee in November 2017 (reference number 17/YH/0262). Confirmation of capacity and capability was obtained from the recruiting centres as well as the participant identification centres in primary and secondary care trusts. The trial was registered with the International Standard Randomised Controlled Trial Register (ISRCTN) under the reference number 17508514. A summary of the changes made to the original protocol is provided in Table 35.
Funding
This project was funded by the National Institute for Health and Care Research (NIHR) Efficacy and Mechanism Evaluation programme (project number 15/74/01) and will be published in full in Efficacy and Mechanism Evaluation; Vol. 10, No. 9. See the NIHR Journals Library website for further project information.
Nottingham University Hospitals NHS Trust agreed to act as sponsor for the research and the study was adopted by the CRN.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
References
- Sperber AD, Bangdiwala SI, Drossman DA, Ghoshal UC, Simren M, Tack J, et al. Worldwide prevalence and burden of functional gastrointestinal disorders, results of Rome foundation global study. Gastroenterology 2020;160:99-114.e3. https://doi.org/10.1053/j.gastro.2020.04.014.
- Hobbs FDR, Bankhead C, Mukhtar T, Stevens S, Perera-Salazar R, Holt T, et al. National Institute for Health Research School for Primary Care Research . Clinical workload in UK primary care: a retrospective analysis of 100 million consultations in England, 2007–14. Lancet 2016;387:2323-30. https://doi.org/10.1016/S0140-6736(16)00620-6.
- Tillisch K, Labus JS, Naliboff BD, Bolus R, Shetzline M, Mayer EA, et al. Characterization of the alternating bowel habit subtype in patients with irritable bowel syndrome. Am J Gastroenterol 2005;100:896-904.
- Atarodi S, Rafieian S, Whorwell PJ. Faecal incontinence-the hidden scourge of irritable bowel syndrome: a cross-sectional study. BMJ Open Gastroenterol 2014;1. https://doi.org/10.1136/bmjgast-2014-000002.
- Lavo B, Stenstam M, Nielsen AL. Loperamide in treatment of irritable bowel syndrome: a double-blind placebo controlled study. Scand J Gastroenterol 1987;22:77-80.
- Hovdenak N. Loperamide treatment of the irritable bowel syndrome. Scand J Gastroenterol 1987;22:81-4.
- Andresen V, Montori VM, Keller J, West CP, Layer P, Camilleri M. Effects of 5-hydroxytryptamine (serotonin) type 3 antagonists on symptom relief and constipation in nonconstipated irritable bowel syndrome: a systematic review and meta-analysis of randomized controlled trials. Clin Gastroenterol Hepatol 2008;6:545-55.
- Fukudo S, Ida M, Akiho H, Nakashima Y, Matsueda K. Effect of ramosetron on stool consistency in male patients with irritable bowel syndrome with diarrhea. Clin Gastroenterol Hepatol 2014;12:953-9. https://doi.org/10.1016/j.cgh.2013.11.024.
- Fukudo S, Kinoshita Y, Okumura T, Ida M, Akiho H, Nakashima Y, et al. Ramosetron reduces symptoms of irritable bowel syndrome with diarrhea and improves quality of life in women. Gastroenterology 2016;150:358-66. https://doi.org/10.1053/j.gastro.2015.10.047.
- Gore S, Gilmore IT, Haigh CG, Brownless SM, Stockdale H, Morris AI. Colonic transit in man is slowed by ondansetron (GR38032F), a selective 5-hydroxytryptamine receptor (type 3) antagonist. Aliment Pharmacol Ther 1990;4:139-44.
- Talley NJ, Phillips SF, Haddad A, Miller LJ, Twomey C, Zinsmeister AR, et al. GR 38032F (ondansetron), a selective 5HT3 receptor antagonist, slows colonic transit in healthy man. Dig Dis Sci 1990;35:477-80.
- Garsed K, Chernova J, Hastings M, Lam C, Marciani L, Singh G, et al. A randomised trial of ondansetron for the treatment of irritable bowel syndrome with diarrhoea. Gut 2014;63:1617-25. https://doi.org/10.1136/gutjnl-2013-305989.
- Gunn D, Garsed K, Lam C, Singh G, Lingaya M, Wahl V, et al. Abnormalities of mucosal serotonin metabolism and 5-HT3 receptor subunit 3C polymorphism in irritable bowel syndrome with diarrhoea predict responsiveness to ondansetron. Aliment Pharmacol Ther 2019;50:538-46. https://doi.org/10.1111/apt.15420.
- Lembo AJ, Olden KW, Ameen VZ, Gordon SL, Heath AT, Carter EG. Effect of alosetron on bowel urgency and global symptoms in women with severe, diarrhea-predominant irritable bowel syndrome: analysis of two controlled trials. Clin Gastroenterol Hepatol 2004;2:675-82.
- Spiegel B, Strickland A, Naliboff BD, Mayer EA, Chang L. Predictors of patient-assessed illness severity in irritable bowel syndrome. Am J Gastroenterol 2008;103:2536-43.
- von der Ohe MR, Camilleri M, Kvols LK. A 5HT3 antagonist corrects the postprandial colonic hypertonic response in carcinoid diarrhea. Gastroenterology 1994;106:1184-9.
- von der Ohe MR, Hanson RB, Camilleri M. Serotonergic mediation of postprandial colonic tonic and phasic responses in humans. Gut 1994;35:536-41.
- Stacher G, Gaupmann G, Schneider C, Stacher-Janotta G, Steiner-Mittelbach G, Abatzi TA, et al. Effects of a 5-hydroxytryptamine3 receptor antagonist (ICS 205-930) on colonic motor activity in healthy men. Br J Clin Pharmacol 1989;28:315-22. https://doi.org/10.1111/j.1365-2125.1989.tb05432.x.
- Clemens CH, Samsom M, Van Berge Henegouwen GP, Fabri M, Smout AJPM. Effect of alosetron on left colonic motility in non-constipated patients with irritable bowel syndrome and healthy volunteers. Aliment Pharmacol Ther 2002;16:993-1002. https://doi.org/10.1046/j.1365-2036.2002.01252.x.
- Stacher G, Weber U, Stacher-Janotta G, Bauer P, Huber K, Holzäpfel A, et al. Effects of the 5-HT3 antagonist cilansetron vs placebo on phasic sigmoid colonic motility in healthy man: a double-blind crossover trial. Br J Clin Pharmacol 2000;49:429-36.
- Lin AY, Du P, Dinning PG, Arkwright JW, Kamp JP, Cheng LK, et al. High-resolution anatomic correlation of cyclic motor patterns in the human colon: evidence of a rectosigmoid brake. Am J Physiol Gastrointest Liver Physiol 2017;312:G508-15. https://doi.org/10.1152/ajpgi.00021.2017.
- Lin AY, Dinning PG, Milne T, Bissett IP, O’Grady G. The ‘rectosigmoid brake’: review of an emerging neuromodulation target for colorectal functional disorders. Clin Exp Pharmacol Physiol 2017;44:719-28. https://doi.org/10.1111/1440-1681.12760.
- Tooth D, Garsed K, Singh G, Marciani L, Lam C, Fordham I, et al. Characterisation of faecal protease activity in irritable bowel syndrome with diarrhoea: origin and effect of gut transit. Gut 2014;63:753-60. https://doi.org/10.1136/gutjnl-2012-304042.
- Gecse K, Roka R, Ferrier L, Leveque M, Eutamene H, Cartier C, et al. Increased faecal serine protease activity in diarrhoeic IBS patients: a colonic lumenal factor impairing colonic permeability and sensitivity. Gut 2008;57:591-9.
- Edwards CA, Brown S, Baxter AJ, Bannister JJ, Read NW. Effect of bile acid on anorectal function in man. Gut 1989;30:383-6.
- Bajor A, Tornblom H, Rudling M, Ung K-A, Simrén M. Increased colonic bile acid exposure: a relevant factor for symptoms and treatment in IBS. Gut 2015;64:84-92. https://doi.org/10.1136/gutjnl-2013-305965.
- Shiotani A, Kusunoki H, Ishii M, Imamura H, Manabe N, Kamada T, et al. Pilot study of Biomarkers for predicting effectiveness of ramosetron in diarrhea-predominant irritable bowel syndrome: expression of S100A10 and polymorphisms of TPH1. Neurogastroenterol Motil 2015;27:82-91. https://doi.org/10.1111/nmo.12473.
- Francis CY, Morris J, Whorwell PJ. The irritable bowel severity scoring system: a simple method of monitoring irritable bowel syndrome and its progress. Aliment Pharmacol Ther 1997;11:395-402.
- Fraser A, Delaney BC, Ford AC, Qume M, Moayyedi P. The Short-Form Leeds Dyspepsia Questionnaire validation study. Aliment Pharmacol Ther 2007;25:477-86. https://doi.org/10.1111/j.1365-2036.2006.03233.x.
- Hahn BA, Kirchdoerfer LJ, Fullerton S, Mayer E. Patient-perceived severity of irritable bowel syndrome in relation to symptoms, health resource utilization and quality of life. Aliment Pharmacol Ther 1997;11:553-9.
- Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983;67:361-70.
- Spiller RC, Humes DJ, Campbell E, Hastings M, Neal KR, Dukes GE, et al. The Patient Health Questionnaire 12 Somatic Symptom scale as a predictor of symptom severity and consulting behaviour in patients with irritable bowel syndrome and symptomatic diverticular disease. Aliment Pharmacol Ther 2010;32:811-20.
- Törnblom H, Van OL, Sadik R, Abrahamsson H, Tack J, Simrén M. Colonic transit time and IBS symptoms: what’s the link?. Am J Gastroenterol 2012;107:754-60. https://doi.org/10.1038/ajg.2012.5.
- Dinning PG, Wiklendt L, Maslen L, Gibbins I, Patton V, Arkwright JW, et al. Quantification of in vivo colonic motor patterns in healthy humans before and after a meal revealed by high-resolution fiber-optic manometry. Neurogastroenterol Motil 2014;26:1443-57. https://doi.org/10.1111/nmo.12408.
- Corsetti M, Thys A, Harris A, Pagliaro G, Deloose E, Demedts I, et al. High-resolution manometry reveals different effect of polyethylene glycol, bisacodyl, and prucalopride on colonic motility in healthy subjects: an acute, open label, randomized, crossover, reader-blinded study with potential clinical implications. Neurogastroenterol Motil 2020;33. https://doi.org/10.1111/nmo.14040.
- Corsetti M, Pagliaro G, Demedts I, Deloose E, Gevers A, Scheerens C, et al. Pan-colonic pressurizations associated with relaxation of the anal sphincter in health and disease: a new colonic motor pattern identified using high-resolution manometry. Am J Gastroenterol 2017;112:479-89. https://doi.org/10.1038/ajg.2016.341.
- Wiklendt L, Costa M, Scott MS, Brookes SJH, Dinning PG. Automated analysis using a Bayesian functional mixed-effects model with Gaussian process responses for wavelet spectra of spatiotemporal colonic manometry signals. Front Physiol 2020;11. https://doi.org/10.3389/fphys.2020.605066.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248-54.
- FDA . Irritable Bowel Syndrome – Clinical Evaluation of Products for Treatment 2012.
- Delvaux M, Louvel D, Mamet JP, Campos-Oriola R, Frexinos J. Effect of alosetron on responses to colonic distension in patients with irritable bowel syndrome. Aliment Pharmacol Ther 1998;12:849-55.
- Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. PLOS Med 2010;7. https://doi.org/10.1371/journal.pmed.1000251.
- Turvill J, O’Connell S, Brooks A, Bradley-Wood K, Laing J, Thiagarajan S, et al. Evaluation of a faecal calprotectin care pathway for use in primary care. Prim Health Care Res Dev 2016;17:428-36. https://doi.org/10.1017/S1463423616000049.
- National Institute for Health and Care Excellence . Implementing Faecal Calprotectin Testing in Primary Care 2015 2022. https://www.nice.org.uk/sharedlearning/implementing-faecal-calprotectin-testing-in-primary-care (accessed 3 August 2022).
- Jakobsen JC, Gluud C, Wetterslev J, Winkel P. When and how should multiple imputation be used for handling missing data in randomised clinical trials – a practical guide with flowcharts. BMC Med Res Methodol 2017;17. https://doi.org/10.1186/s12874-017-0442-1.
- Bang H, Park JJ. Blinding in clinical trials: a practical approach. J Altern Complement Med 2013;19:367-9. https://doi.org/10.1089/acm.2012.0210.
- Lam C, Tan W, Leighton M, Hastings M, Lingaya M, Falcone Y, et al. A mechanistic multicentre, parallel group, randomised placebo-controlled trial of mesalazine for the treatment of IBS with diarrhoea (IBS-D). Gut 2016;65:91-9. https://doi.org/10.1136/gutjnl-2015-309122.
- Plasse TF, Barton G, Davidson E, . Bimodal release ondansetron improves stool consistency and symptomatology in diarrhea-predominant irritable bowel syndrome: a randomized, double-blind, trial. Am J Gastroenterol 2020;11:1466-73. https://doi.org/10.14309/ajg.0000000000000727.
- Olden KW, Chey WD, Shringarpure R, Nicandro JP, Chuang E, Earnest DL. Alosetron versus traditional pharmacotherapy in clinical practice: effects on resource use, health-related quality of life, safety and symptom improvement in women with severe diarrhea-predominant irritable bowel syndrome. Curr Med Res Opin 2019;35:461-72. https://doi.org/10.1080/03007995.2018.1533456.
- Peleman C, Camilleri M, Busciglio I, Burton D, Donato L, Zinsmeister AR. Colonic transit and bile acid synthesis or excretion in patients with irritable bowel syndrome-diarrhea without bile acid malabsorption. Clin Gastroenterol Hepatol 2017;15:720-7. https://doi.org/10.1016/j.cgh.2016.11.012.
- Alderson SL, Wright-Hughes A, Ford AC, Farrin A, Hartley S, Fernandez C, et al. Amitriptyline at low-dose and titrated for irritable bowel syndrome as second-line treatment (The ATLANTIS trial): protocol for a randomised double-blind placebo-controlled trial in primary care. Trials 2022;23. https://doi.org/10.1186/s13063-022-06492-6.
- Drossman DA, Tack J. Rome foundation clinical diagnostic criteria for disorders of gut-brain interaction. Gastroenterology 2022;162:675-9. https://doi.org/10.1053/j.gastro.2021.11.019.
- Goyal O, Batta S, Nohria S, Kishore H, Goyal P, Sehgal R, et al. Low fermentable oligosaccharide, disaccharide, monosaccharide, and polyol diet in patients with diarrhea-predominant irritable bowel syndrome: a prospective, randomized trial. J Gastroenterol Hepatol 2021;36:2107-15. https://doi.org/10.1111/jgh.15410.
- Serrano-Falcon B, Delgado-Aros S, Mearin F, Ciriza de Los Ríos C, Serra J, Mínguez M, et al. Clinical response to linaclotide at week 4 predicts sustained response in irritable bowel syndrome with constipation and improvements in digestive and extra-digestive symptoms. Therap Adv Gastroenterol 2019;12. https://doi.org/10.1177/1756284819857358.
Appendix 1 Activities per visit
| Time point | Week 12 | Week 10 | Weeks 8–11 | Week 0 | Week 6 | Weeks 8–11 | Week 12 | Week 16 | |
|---|---|---|---|---|---|---|---|---|---|
| Activity/assessment | Visit 1: Registration | Visit 2: Eligibility confirmation | Mechanistic assessments | Visit 3: Randomisation | Visit 4: 6 week visit | Mechanistic assessments | Visit 5: 12 week visit | Visit 6: Follow-up visit | |
| Enrolment | |||||||||
| Informed consent | X | ||||||||
| Demographicsa | X | ||||||||
| Vital signs | X | X | |||||||
| Haematology and biochemistryb | X | X | |||||||
| 12 Lead ECG | X | ||||||||
| Pregnancy test | X | X | X | X | X | ||||
| Confirmation of eligibility | X | ||||||||
| Randomisation | X | ||||||||
| Interventions | |||||||||
| Daily ondansetron or placeboc | |||||||||
| Assessments | |||||||||
| Symptom diary startd | X | ||||||||
| SMS messages starte | X | ||||||||
| Questionnairesf | X | X | |||||||
| Colonic transit assessment | X | X | |||||||
| Research biopsiesg | X | ||||||||
| Research bloodh | X | X | |||||||
| Stool samplesi | X | X | |||||||
| High-resolution colonic manometryj | X | X | |||||||
| Barostat assessmentk | X | X | |||||||
| Exit poll | X | ||||||||
| Date | |
|---|---|
| To allow all participants to consent to optional biopsy collection not just those consenting to mechanistic studies | 22 February 2018 |
| Including patients with a prolonged QT interval or taking drugs known to cause this with caution | |
| Change of eligibility criteria regarding QTc interval from > 420 milliseconds to > 450 milliseconds for men and > 470 milliseconds for women | 12 June 2018 |
| Change of maximum allowed time period between consent and the eligibility confirmation visit from 4 to 8 weeks | |
| Change from requirement to use double contraceptives to conventional contraception | 6 November 2018 |
| Addition of a trial of bile acid binding agent as an alternative to a SeHCAT scan to exclude BDA as an alternative diagnosis | |
| Allow patients with minor rises in ALT (< 2 × upper limit of normal) to be included | |
| Allow patients assessed as ineligible to be rescreened at a later date | |
| Increase the time since colonoscopy required for eligibility from 5 to 10 years with stable symptoms and a normal faecal calprotectin | 19 July 2019 |
| Patients taking high-dose TCAs need to stop taking them to participate in the trial | |
| Extension of the recruitment period from 18 to 20 months | |
| Change to allow patients with pulse, BP or laboratory blood values that are out of normal range but deemed not clinically significant to enter the trial | 25 September 2019 |
| Addition of information about new safety information on the use of ondansetron in pregnancy and the risk of orofacial malformations | October 2019 |
Appendix 2 Quantification of bile acids (LC–MS)
After thawing, 0.5 g of the faecal sample was suspended in 2 ml of 50% (w/v) acetonitrile and extracted by vortexing and sonication for 10 minutes. The suspension was centrifuged twice at 25,000g for 20 minutes. Supernatants were transferred to sample vials and loaded onto a LC–MS system containing online SPE. At the beginning of each analysis 50 µl of the sample were transferred to SPE column at a flow of 0.1 ml/min with the loading mobile phase, aqueous 5% acetonitrile, 0.1% formic acid and 0.02% trifluoroacetic acid. The chromatographic separation was performed using a binary system with pumps (A) and (B) (Jasco, PU-2085 Plus, Cremella, Italy) connected to a degasser (Alltech, degasser, Stamford, UK). The two systems were connected using a two-position, six-port valve, used to switch automatically from loading (position 1) to injection (position 2) after 9 minutes (Valvemate, Gilson, Dunstable, UK). Samples were analysed on a Waters Ex-bridge C18 column (Waters, 100 × 2.1 mm; 3.5 µm particle size), using a gradient program. Mobile Phase (A) 5 mM ammonium acetate, 0.1% ammonium hydroxide, mobile phase (B) 100% acetonitrile. Initial composition was 80% (A), which was reduced to 70% (A) over 30 minutes with further reduction to 65% (A) over the next 3 minutes. The eluent composition was held at 65% for 1.5 minutes before returning to 80% (A) initial condition over the next 1.5 minutes. Flow rate was 0.2 ml/min.
High-performance liquid chromatography was coupled in series with the turbo ion-spray (ESI) source of the tandem mass spectrometer (Micromass, Manchester, UK). Electrospray ionisation was performed in negative mode with nitrogen as the nebuliser gas. Detection of individual bile acids was performed using SIM mode. Additional structural information was obtained via tandem MS (MS/MS) fragmentation, with collision energies ranging from 15 to 30 electron volts. Data were acquired using software MassLynX (Waters, Wexford, Ireland).
The concentration of bile acids in the samples was determined on the basis of the peak areas of individual bile acids and external standards.
Appendix 3 Screening summary
| Identification method | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Site | Total N (%) | Secondary care (%) | Primary care and pharmacy (%) | Self-referral via website (%) | Self-referral other (%) | Secondary PIC (%) | Primary care invitation (%) | Other identification method (%) | Missing (%) |
| James Cook, South Tees | 38 (2.4) | 26 (68.4) | 0 (0.0) | 3 (7.9) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 9 (23.7) | 0 (0.0) |
| St James’s University Hospital, Leeds | 80 (5.1) | 19 (23.8) | 0 (0.0) | 1 (1.3) | 8 (10.0) | 0 (0.0) | 0 (0.0) | 51 (63.8) | 1 (1.3) |
| Royal Hallamshire Hospital | 51 (3.2) | 20 (39.2) | 0 (0.0) | 1 (2.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 29 (56.9) | 1 (2.0) |
| Sandwell Hospital | 43 (2.7) | 18 (41.9) | 0 (0.0) | 4 (9.3) | 3 (7.0) | 0 (0.0) | 0 (0.0) | 16 (37.2) | 2 (4.7) |
| University Hospital of North Durham | 23 (1.5) | 9 (39.1) | 0 (0.0) | 12 (52.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (8.7) | 0 (0.0) |
| Wythenshawe Hospital | 12 (0.8) | 3 (25.0) | 3 (25.0) | 4 (33.3) | 2 (16.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Royal Stoke University Hospital | 44 (2.8) | 29 (65.9) | 0 (0.0) | 0 (0.0) | 0 (0.0%) | 0 (0.0) | 0 (0.0) | 14 (31.8) | 1 (2.3) |
| Nottingham University Hospital | 58 (3.7) | 23 (39.7) | 5 (8.6) | 10 (17.2) | 6 (10.3) | 0 (0.0) | 2 (3.4) | 11 (19.0) | 1 (1.7) |
| Barnsley District General Hospital | 216 (13.7) | 195 (90.3) | 0 (0.0) | 0 (0.0) | 7 (3.2) | 0 (0.0) | 0 (0.0) | 1 (0.5) | 13 (6.0) |
| St Mark’s Hospital | 11 (0.7) | 5 (45.5) | 0 (0.0) | 5 (45.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (9.1) | 0 (0.0) |
| Western General Hospital | 53 (3.4) | 42 (79.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 9 (17.0) | 2 (3.8) |
| Salford Royal Hospital | 16 (1.0) | 2 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 14 (87.5) | 0 (0.0) |
| Barts and London School of Medicine and Dentistry | 159 (10.1) | 67 (42.1) | 2 (1.3) | 17 (10.7) | 1 (0.6) | 4 (2.5) | 0 (0.0) | 67 (42.1) | 1 (0.6) |
| University College London Hospital | 778 (49.2) | 28 (3.6) | 0 (0.0) | 7 (0.9) | 6 (0.8) | 0 (0.0) | 0 (0.0) | 731 (94.0) | 6 (0.8) |
| Total | 1582 (100) | 486 (30.7) | 10 (0.6) | 64 (4.0) | 33 (2.1) | 4 (0.3) | 2 (0.1) | 955 (60.4) | 28 (1.8) |
| Eligibility status | |||||
|---|---|---|---|---|---|
| Site | Total N | Eligible (%) | Not eligible (%) | Further investigation required (%) | Missing (%) |
| James Cook, South Tees | 38 | 10 (26.3) | 12 (31.6) | 16 (42.1) | 0 (0.0) |
| St James’s University Hospital, Leeds | 80 | 15 (18.8) | 56 (70.0) | 9 (11.3) | 0 (0.0) |
| Royal Hallamshire Hospital | 51 | 12 (23.5) | 35 (68.6) | 1 (2.0) | 3 (5.9) |
| Sandwell Hospital | 43 | 7 (16.3) | 24 (55.8) | 9 (20.9) | 3 (7.0) |
| University Hospital of North Durham | 23 | 4 (17.4) | 3 (13.0) | 16 (69.6) | 0 (0.0) |
| Wythenshawe Hospital | 12 | 11 (91.7) | 0 (0.0) | 1 (8.3) | 0 (0.0) |
| Royal Stoke University Hospital | 44 | 20 (45.5) | 24 (54.5) | 0 (0.0) | 0 (0.0) |
| Nottingham University Hospital | 58 | 41 (70.7) | 15 (25.9) | 1 (1.7) | 1 (1.7) |
| Barnsley District General Hospital | 216 | 29 (13.4) | 176 (81.5) | 0 (0.0) | 11 (5.1) |
| St Mark’s Hospital | 11 | 4 (36.4) | 3 (27.3) | 3 (27.3) | 1 (9.1) |
| Western General Hospital | 53 | 5 (9.4) | 20 (37.7) | 28 (52.8) | 0 (0.0) |
| Salford Royal Hospital | 16 | 1 (6.3) | 15 (93.8) | 0 (0.0) | 0 (0.0) |
| Barts and London School of Medicine and Dentistry | 159 | 38 (23.9) | 105 (66.0) | 14 (8.8) | 2 (1.3) |
| University College London Hospital | 778 | 5 (0.6) | 761 (97.8) | 0 (0.0) | 12 (1.5) |
| Total | 1582 | 202 (12.8) | 1249 (79.0) | 98 (6.2) | 33 (2.1) |
| Consented to study | If not consented, reasons for non-consent | |||||||
|---|---|---|---|---|---|---|---|---|
| Site | Total N | Yes (%) | No (%) | Status missing (%) | Refused consent (%) | Cannot get hold of patient (%) | Other reason (%) | Missing (%) |
| James Cook, South Tees | 38 | 10 (26.3) | 16 (42.1) | 12 (31.6) | 9 (56.3) | 1 (6.3) | 5 (31.3) | 1 (6.3) |
| St James’s University Hospital, Leeds | 80 | 15 (18.8) | 9 (11.3) | 56 (70.0) | 1 (11.1) | 5 (55.6) | 3 (33.3) | 0 (0.0) |
| Royal Hallamshire Hospital | 51 | 12 (23.5) | 1 (2.0) | 38 (74.5) | 0 (0.0) | 0 (0.0) | 1 (100.0) | 0 (0.0) |
| Sandwell Hospital | 43 | 9 (20.9) | 7 (16.3) | 27 (62.8) | 5 (71.4) | 1 (14.3) | 1 (14.3) | 0 (0.0) |
| University Hospital of North Durham | 23 | 11 (47.8) | 9 (39.1) | 3 (13.0) | 4 (44.4) | 4 (44.4) | 1 (11.1) | 0 (0.0) |
| Wythenshawe Hospital | 12 | 12 (100.0%) | 0 (0.0) | 0 (0.0) | ||||
| Royal Stoke University Hospital | 44 | 18 (40.9) | 2 (4.5) | 24 (54.5) | 0 (0.0) | 0 (0.0) | 2 (100.0) | 0 (0.0) |
| Nottingham University Hospital | 58 | 42 (72.4) | 0 (0.0) | 16 (27.6) | ||||
| Barnsley District General Hospital | 216 | 8 (3.7) | 21 (9.7) | 187 (86.6) | 8 (38.1) | 9 (42.9) | 4 (19.0) | 0 (0.0) |
| St Mark’s Hospital | 11 | 1 (9.1) | 6 (54.5) | 4 (36.4) | 5 (83.3) | 0 (0.0) | 1 (16.7) | 0 (0.0) |
| Western General Hospital | 53 | 7 (13.2) | 26 (49.1) | 20 (37.7) | 7 (26.9) | 2 (7.7) | 17 (65.4) | 0 (0.0) |
| Salford Royal Hospital | 16 | 1 (6.3) | 0 (0.0) | 15 (93.8) | ||||
| Barts and London School of Medicine and Dentistry | 159 | 23 (14.5) | 21 (13.2) | 115 (72.3) | 13 (61.9) | 2 (9.5) | 6 (28.6) | 0 (0.0) |
| University College London Hospital | 778 | 4 (0.5) | 1 (0.1) | 773 (99.4) | 1 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Total | 1582 | 173 (10.9) | 119 (7.5) | 1290 (81.5) | 53 (44.5) | 24 (20.2) | 41 (34.5) | 1 (0.8) |
| Registered to study | Reasons for non-registration | |||||
|---|---|---|---|---|---|---|
| Site | Total N | Yes (%) | No (%) | Status missing (%) | No longer eligible (%) | Other reason (%) |
| James Cook, South Tees | 38 | 7 (15.8) | 4 (10.5) | 28 (73.7) | 1 (25.0) | 3 (75.0) |
| St James’s University Hospital, Leeds | 80 | 13 (16.3) | 2 (2.5) | 65 (81.3) | 2 (100.0) | 0 (0.0) |
| Royal Hallamshire Hospital | 51 | 12 (23.5) | 0 (0.0) | 39 (76.5) | ||
| Sandwell Hospital | 43 | 6 (14.0) | 3 (7.0) | 34 (79.1) | 2 (66.7) | 1 (33.3) |
| University Hospital of North Durham | 23 | 4 (17.4) | 7 (30.4) | 12 (52.2) | 5 (71.4) | 2 (28.6) |
| Wythenshawe Hospital | 12 | 12 (100.0) | 0 (0.0) | 0 (0.0) | ||
| Royal Stoke University Hospital | 44 | 18 (40.9) | 0 (0.0) | 26 (59.1) | ||
| Nottingham University Hospital | 58 | 39 (67.2) | 3 (5.2) | 16 (27.6) | 0 (0.0) | 3 (100.0) |
| Barnsley District General Hospital | 216 | 7 (3.2) | 1 (0.5) | 208 (96.3) | 1 (100.0) | 0 (0.0) |
| St Mark’s Hospital | 11 | 1 (9.1) | 0 (0.0) | 10 (90.9) | ||
| Western General Hospital | 53 | 6 (11.3) | 1 (1.9) | 46 (86.8) | 0 (0.0) | 1 (100.0) |
| Salford Royal Hospital | 16 | 1 (6.3) | 0 (0.0) | 15 (93.8) | ||
| Barts and London School of Medicine and Dentistry | 159 | 19 (11.9) | 2 (1.3) | 138 (86.8) | 1 (50.0) | 0 (0.0) |
| University College London Hospital | 778 | 4 (0.5) | 0 (0.0) | 774 (99.5) | ||
| Total | 1582 | 149 (9.4) | 23 (1.5) | 1411 (89.2) | 12 (52.2) | 10 (43.5) |
| Abdominal pain score | Days with loose stool | Urgency score | |||
|---|---|---|---|---|---|
| Site | Total N (%) | Mean score (0–100) (SD) | Mean days (per week) (SD) | Mean urgency score (0–100) (SD) | Urgency score missing |
| James Cook, South Tees | 4 (5.0) | 66.2 (23.4) | 6.1 (1.0) | 69.9 (18.8) | 0 |
| St James’s University Hospital, Leeds | 10 (12.5) | 66.3 (19.5) | 5.8 (1.3) | 76.0 (17.5) | 0 |
| Royal Hallamshire Hospital | 4 (5.0) | 48.4 (11.9) | 4.9 (1.1) | 55.5 (11.6) | 0 |
| Sandwell Hospital | 3 (3.8) | 61.6 (4.6) | 5.1 (1.6) | 49.3 (12.9) | 0 |
| University Hospital of North Durham | 2 (2.5) | 71.4 (7.1) | 6.3 (1.1) | 76.4 (17.2) | 0 |
| Wythenshawe Hospital | 6 (7.5) | 43.5 (5.8) | 6.2 (0.6) | 60.7 (13.9) | 0 |
| Royal Stoke University Hospital | 11 (13.8) | 59.6 (19.1) | 6.2 (0.8) | 63.4 (18.9) | 1 |
| Nottingham University Hospital | 21 (26.3) | 54.4 (10.2) | 5.2 (1.4) | 58.7 (15.5) | 0 |
| Barnsley District General Hospital | 2 (2.5) | 55.4 (20.7) | 6.3 (1.1) | 65.9 (6.4) | 0 |
| St Mark’s Hospital | 1 (1.3) | 98.6 (.) | 7.0 (.) | 96.1 (.) | 0 |
| Western General Hospital | 6 (7.5) | 56.3 (29.4) | 6.0 (1.4) | 60.0 (22.3) | 0 |
| Barts and London School of Medicine and Dentistry | 9 (11.3) | 63.0 (21.4) | 5.1 (1.6) | 68.6 (27.6) | 0 |
| University College London Hospital | 1 (1.3) | 21.7 (.) | 5.0 (.) | 39.2 (.) | 0 |
| Total | 80 (100) | 58.0 (18.3) | 5.6 (1.3) | 63.8 (18.9) | 1 |
| Age | Gender | Ethnicity | |||||
|---|---|---|---|---|---|---|---|
| Site | Total N (%) | Mean age (years) (SD) | Male (%) | Female (%) | White (%) | Black (%) | Asian (%) |
| James Cook, South Tees | 4 (5.0) | 38.3 (12.1) | 1 (25.0) | 3 (75.0) | 4 (100.0) | 0 (0.0) | 0 (0.0) |
| St James’s University Hospital, Leeds | 10 (12.5) | 37.3 (12.6) | 3 (30.0) | 7 (70.0) | 10 (100.0) | 0 (0.0) | 0 (0.0) |
| Royal Hallamshire Hospital | 4 (5.0) | 36.0 (15.7) | 2 (50.0) | 2 (50.0) | 4 (100.0) | 0 (0.0) | 0 (0.0) |
| Sandwell Hospital | 3 (3.8) | 44.3 (6.5) | 2 (66.7) | 1 (33.3) | 2 (66.7) | 0 (0.0) | 1 (33.3) |
| University Hospital of North Durham | 2 (2.5) | 53.0 (4.2) | 0 (0.0) | 2 (100.0) | 2 (100.0) | 0 (0.0) | 0 (0.0) |
| Wythenshawe Hospital | 6 (7.5) | 43.5 (15.4) | 3 (50.0) | 3 (50.0) | 6 (100.0) | 0 (0.0) | 0 (0.0) |
| Royal Stoke University Hospital | 11 (13.8) | 42.9 (19.4) | 3 (27.3) | 8 (72.7) | 11 (100.0) | 0 (0.0) | 0 (0.0) |
| Nottingham University Hospital | 21 (26.3) | 45.0 (19.4) | 10 (47.6) | 11 (52.4) | 20 (95.2) | 1 (4.8) | 0 (0.0) |
| Barnsley District General Hospital | 2 (2.5) | 50.0 (12.7) | 1 (50.0) | 1 (50.0) | 2 (100.0) | 0 (0.0) | 0 (0.0) |
| St Mark’s Hospital | 1 (1.3) | 55.0 (.) | 0 (0.0) | 1 (100.0) | 1 (100.0) | 0 (0.0) | 0 (0.0) |
| Western General Hospital | 6 (7.5) | 39.5 (13.8) | 3 (50.0) | 3 (50.0) | 5 (83.3) | 1 (16.7) | 0 (0.0) |
| Barts and London School of Medicine and Dentistry | 9 (11.3) | 52.9 (13.6) | 5 (55.6) | 4 (44.4) | 7 (77.8) | 1 (11.1) | 1 (11.1) |
| University College London Hospital | 1 (1.3) | 61.0 (.) | 0 (0.0) | 1 (100.0) | 1 (100.0) | 0 (0.0) | 0 (0.0) |
| Total | 80 (100) | 43.9 (16.0) | 33 (41.3) | 47 (58.8) | 75 (93.8) | 3 (3.8) | 2 (2.5) |
| Site | Total N (%) | Barostat undertaken (%) | Colonic manometry (%) |
|---|---|---|---|
| James Cook, South Tees | 4 (5.0) | 0 (0.0) | 0 (0.0) |
| St James’s University Hospital, Leeds | 10 (12.5) | 0 (0.0) | 0 (0.0) |
| Royal Hallamshire Hospital | 4 (5.0) | 0 (0.0) | 0 (0.0) |
| Sandwell Hospital | 3 (3.8) | 1 (33.3) | 1 (33.3) |
| University Hospital of North Durham | 2 (2.5) | 0 (0.0) | 0 (0.0) |
| Wythenshawe Hospital | 6 (7.5) | 0 (0.0) | 0 (0.0) |
| Royal Stoke University Hospital | 11 (13.8) | 3 (27.3) | 3 (27.3) |
| Nottingham University Hospital | 21 (26.3) | 9 (42.9) | 5 (23.8) |
| Barnsley District General Hospital | 2 (2.5) | 0 (0.0) | 0 (0.0) |
| St Mark’s Hospital | 1 (1.3) | 0 (0.0) | 0 (0.0) |
| Western General Hospital | 6 (7.5) | 0 (0.0) | 0 (0.0) |
| Barts and London School of Medicine and Dentistry | 9 (11.3) | 4 (44.4) | 3 (33.3) |
| University College London Hospital | 1 (1.3) | 1 (100.0) | 1 (100.0) |
| Total | 80 (100) | 18 (22.5) | 13 (16.3) |
Appendix 4 Treatment discontinuation
| Patient | Allocation | Who requested discontinuation | Last dose (days post randomisation) | Reason for discontinuation | Further information | Withdrawn from trial follow-up |
|---|---|---|---|---|---|---|
| 1 | Ondansetron | Clinician | 18 | Other (patient not eligible) | Patient not eligible as per section 8.5 of protocol as unable to discontinue Buscopan | Yes |
| 2 | Ondansetron | Patient | 26 | Drug intolerance | Became very constipated and found it intolerable to continue | Yes |
| 3 | Ondansetron | Patient | 27 | Patient decision | Side effects | Yes |
| 4 | Ondansetron | Clinician | 28 | Patient’s health compromised | No | |
| 5 | Placebo | Patient | 12 | Drug intolerance | Severe constipation. The participant discontinued treatment but did not withdraw from the trial | No |
Appendix 5 Responders versus loperamide use by week
FIGURE 28.
Overall responders vs. loperamide use by week.
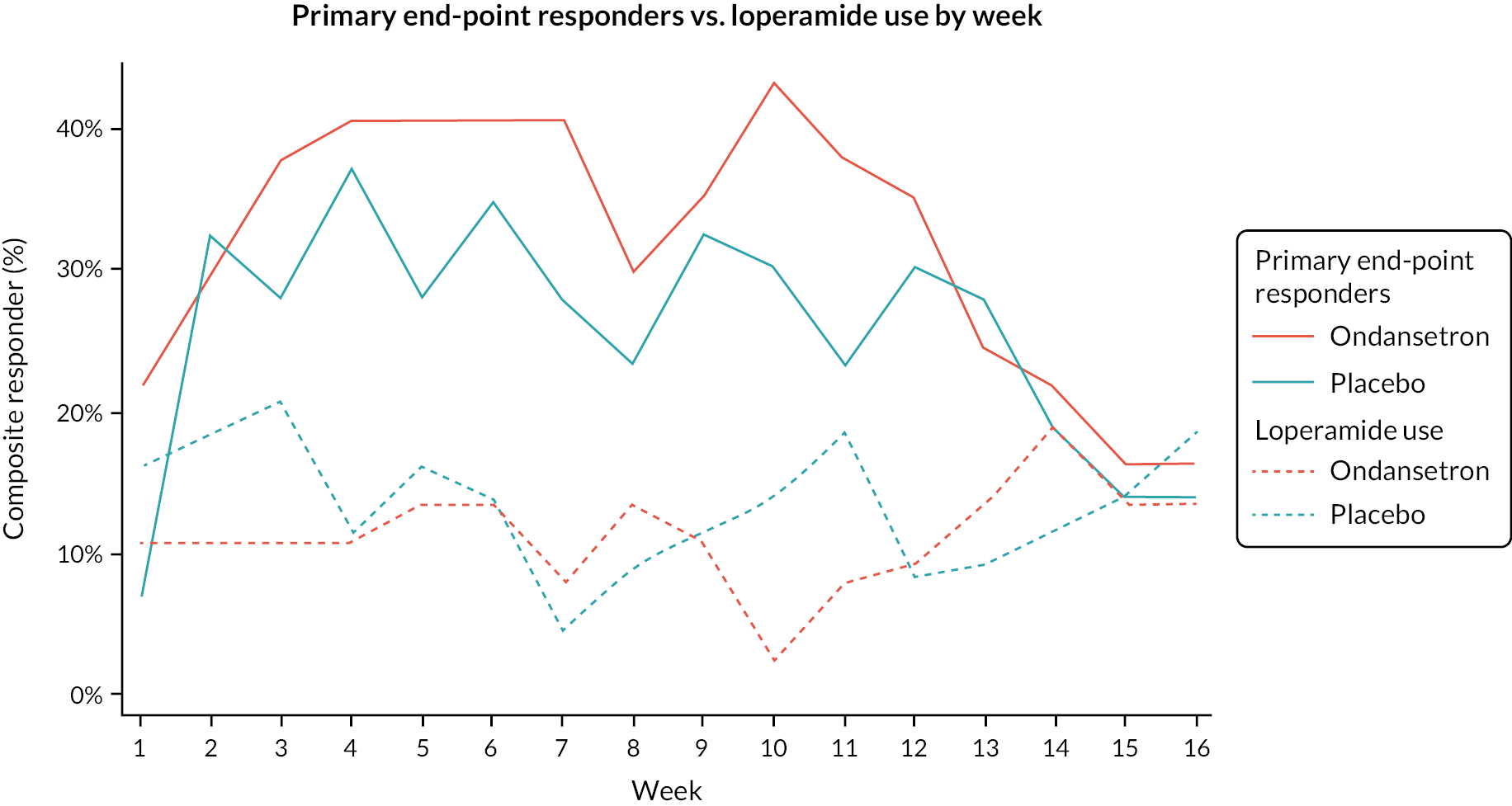
FIGURE 29.
Proportion of responders vs. loperamide use by week.

FIGURE 30.
Weekly abdominal pain responders and loperamide use.
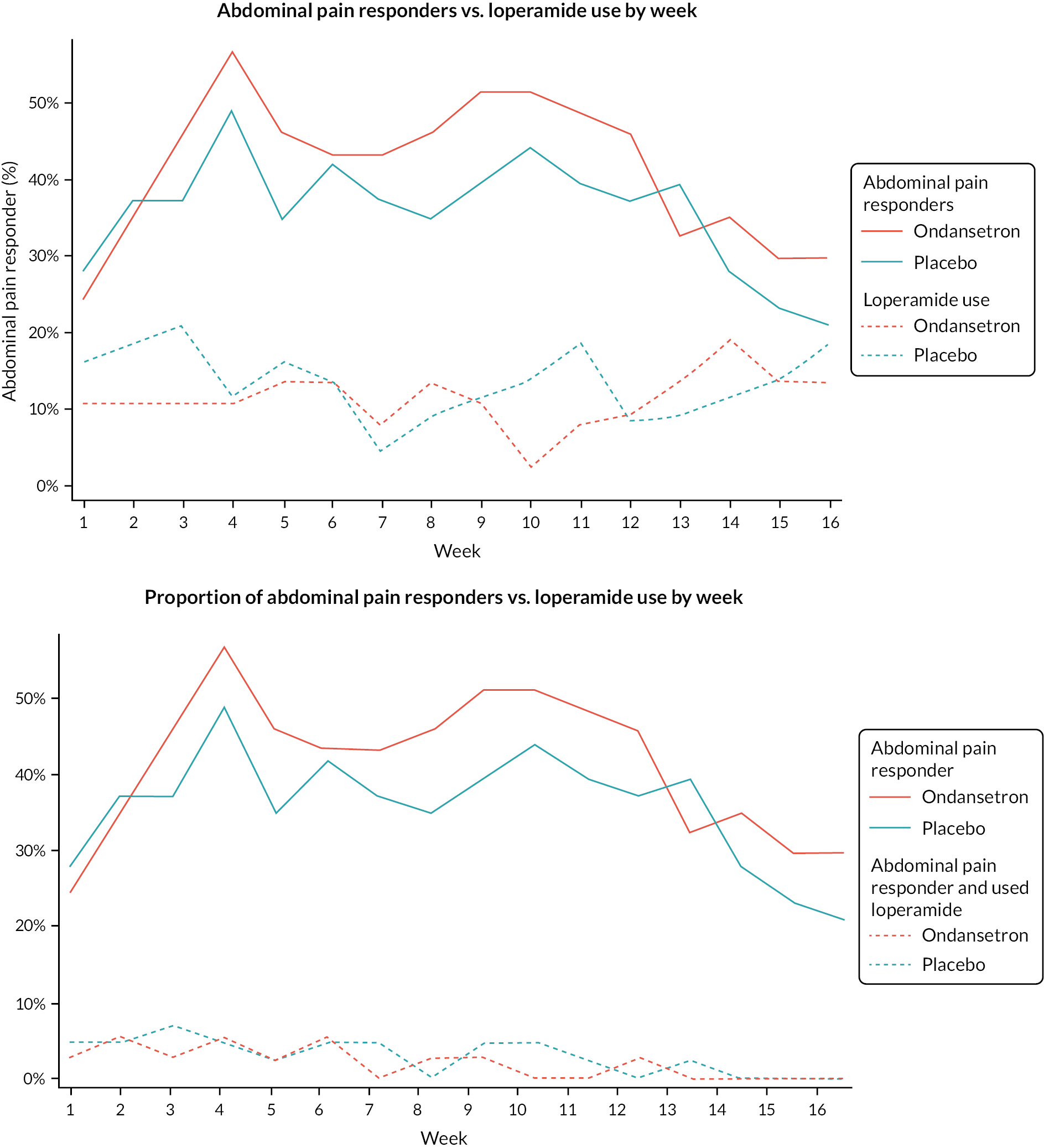
FIGURE 31.
Weekly stool consistency responders vs. loperamide use.

Appendix 6 Additional outcome summaries
| Weeks 1–12 | Weeks 9–12 | Off-treatment weeks 13–16 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Change in pain score | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
| Mean (SD) missing | –15.6 (17.30) 2 | –12.7 (16.44) 1 | –14.0 (16.78) 3 | –17.3 (19.56) 3 | –14.8 (19.16) 4 | –16.0 (19.26) 7 | –12.1 (18.55) 10 | –9.7 (17.88) 4 | –10.7 (18.06) 14 |
| Mean (95% CI) | –15.6 (–21.3, –9.8) | –12.7 (–17.6, –7.7) | –14.0 (–17.7, –10.2) | –17.3 (–23.9, –10.8) | –14.8 (–20.8, –8.8) | –16.0 (–20.4, –11.5) | –12.1 (–19.1, –5.1) | –9.7 (–15.3, –4.1) | –10.7 (–15.0, –6.3) |
| Baseline | 12 weeks | |||||
|---|---|---|---|---|---|---|
| HADS scores | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
| Anxiety score (continuous) | ||||||
| Mean (SD) missing | 9.9 (5.01) 0 | 10.2 (4.54) 0 | 10.1 (4.74) 0 | 8.4 (5.40) 5 | 9.3 (4.83) 3 | 8.9 (5.08) 8 |
| Median (range) | 9.0 (0.0–19.0) | 10.0 (2.0–19.0) | 9.5 (0.0–19.0) | 8.0 (1.0–19.0) | 9.0 (1.0–18.0) | 8.5 (1.0–19.0) |
| Depression score (continuous) | ||||||
| Mean (SD) missing | 8.0 (4.16) 0 | 6.9 (4.00) 0 | 7.4 (4.09) 0 | 6.0 (5.43) 5 | 5.7 (4.19) 3 | 5.8 (4.74) 8 |
| Median (range) | 9.0 (1.0–17.0) | 6.0 (1.0–15.0) | 8.0 (1.0–17.0) | 5.0 (0.0–18.0) | 5.0 (0.0–14.0) | 5.0 (0.0–18.0) |
| Anxiety category | ||||||
| Normal | 12 (32.4%) | 15 (34.9%) | 27 (33.8%) | 15 (40.5%) | 17 (39.5%) | 32 (40.0%) |
| Borderline abnormal | 11 (29.7%) | 7 (16.3%) | 18 (22.5%) | 5 (13.5%) | 6 (14.0%) | 11 (13.8%) |
| Abnormal | 14 (37.8%) | 21 (48.8%) | 35 (43.8%) | 12 (32.4%) | 17 (39.5%) | 29 (36.3%) |
| Missing | 0 | 0 | 0 | 5 (13.5%) | 3 (7.0%) | 8 (10.0%) |
| Depression category | ||||||
| Normal | 15 (40.5%) | 23 (53.5%) | 38 (47.5%) | 22 (59.5%) | 25 (58.1%) | 47 (58.8%) |
| Borderline abnormal | 13 (35.1%) | 11 (25.6%) | 24 (30.0%) | 4 (10.8%) | 9 (20.9%) | 13 (16.3%) |
| Abnormal | 9 (24.3%) | 9 (20.9%) | 18 (22.5%) | 6 (16.2%) | 6 (14.0%) | 12 (15.0%) |
| Missing | 0 | 0 | 0 | 5 (13.5%) | 3 (7.0%) | 8 (10.0%) |
| Ondansetron (n = 37) (%) | Placebo (n = 43) (%) | Total (n = 80) (%) | |
|---|---|---|---|
| Anxiety | |||
| Reduction/better by two categories | 1 (2.7) | 2 (4.7) | 3 (3.8) |
| Reduction/better by one category | 7 (18.9) | 5 (11.6) | 12 (15.0) |
| No change | 22 (59.5) | 30 (69.8) | 52 (65.0) |
| Increase/worse by one category | 1 (2.7) | 2 (4.7) | 3 (3.8) |
| Increase/worse by two categories | 1 (2.7) | 1 (2.3) | 2 (2.5) |
| Missing | 5 (13.5) | 3 (7.0) | 8 (10.0) |
| Depression | |||
| Reduction/better by two categories | 1 (2.7) | 1 (2.3) | 2 (2.5) |
| Reduction/better by one category | 12 (32.4) | 6 (14.0) | 18 (22.5) |
| No change | 17 (45.9) | 30 (69.8) | 47 (58.8) |
| Increase/worse by one category | 2 (5.4) | 3 (7.0) | 5 (6.3) |
| Missing | 5 (13.5) | 3 (7.0) | 8 (10.0) |
| Reduction by | Ondansetron (n = 37) (%) | Placebo (n = 43) (%) | Total (n = 80) (%) |
|---|---|---|---|
| < 50 points | 12 (32.4) | 20 (46.5) | 32 (40.0) |
| ≥ 50 points | 19 (51.4) | 18 (41.9) | 37 (46.3) |
| Missing | 6 (16.2) | 5 (11.6) | 11 (13.8) |
| Used loperamide N (%) missing | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
|---|---|---|---|
| Pre-treatment | 8 (21.6%) 0 | 9 (20.9%) 2 | 17 (21.3%) 2 |
| Treatment (weeks 1–12) | 7 (18.9%) 0 | 17 (39.5%) 1 | 24 (30.0%) 1 |
| Weeks 9–12 only | 5 (13.5%) 3 | 11 (25.6%) 1 | 16 (20.0%) 4 |
| Follow-up | 8 (21.6%) 6 | 9 (20.9%) 3 | 17 (21.3%) 9 |
| Primary end-point responder | Abdominal pain responder | Stool consistency responder | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Responder? | Used loperamide? | Ondansetron (n = 37) (%) | Placebo (n = 43) (%) | Total (n = 80) (%) | Ondansetron (n = 37) (%) | Placebo (n = 43) (%) | Total (n = 80) (%) | Ondansetron (n = 37) (%) | Placebo (n = 43) (%) | Total (n = 80) (%) | |
| Yes | AND | Yes | 2 (5.4) | 3 (7.0) | 5 (6.3) | 2 (5.4) | 5 (11.6) | 7 (8.8) | 5 (13.5) | 10 (23.3) | 15 (18.8) |
| Yes | AND | No | 13 (35.1) | 9 (20.9) | 22 (27.5) | 15 (40.5) | 11 (25.6) | 26 (32.5) | 20 (54.1) | 12 (27.9) | 32 (40.0) |
| No | AND | Yes | 5 (13.5) | 13 (30.2) | 18 (22.5) | 5 (13.5) | 11 (25.6) | 16 (20.0) | 2 (5.4) | 7 (16.3) | 9 (11.3) |
| No | AND | No | 15 (40.5) | 16 (37.2) | 31 (38.8) | 13 (35.1) | 14 (32.6) | 27 (33.8) | 8 (21.6) | 13 (30.2) | 21 (26.3) |
| Missing | 2 (5.4) | 2 (4.7) | 4 (5.0) | 2 (5.4) | 2 (4.7) | 4 (5.0) | 2 (5.4) | 1 (2.3) | 3 (3.8) | ||
| Baseline | 12 weeks | |||||
|---|---|---|---|---|---|---|
| SFLDQ | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
| SFLDQ score (0–32) | ||||||
| Mean (SD) missing | 14.9 (7.75) 1 | 11.7 (6.91) 1 | 13.2 (7.43) 2 | 7.5 (8.27) 8 | 9.4 (6.84) 4 | 8.6 (7.49) 12 |
| Median (range) | 15.0 (0.0–31.0) | 13.0 (0.0–24.0) | 14.0 (0.0–31.0) | 4.0 (0.0–28.0) | 8.0 (0.0–21.0) | 6.0 (0.0–28.0) |
| SFLDQ missing items | ||||||
| 0 | 36 (97.3%) | 42 (97.7%) | 78 (97.5%) | 29 (78.4%) | 39 (90.7%) | 68 (85.0%) |
| 1 | 1 (2.7%) | 1 (2.3%) | 2 (2.5%) | 2 (5.4%) | 0 (0.0%) | 2 (2.5%) |
| All questionnaire missing | 6 (16.2%) | 4 (9.3%) | 10 (12.5%) | |||
| Most troublesome in last 2 months | ||||||
| Heartburn | 2 (5.4%) | 5 (11.6%) | 7 (8.8%) | 3 (8.1%) | 3 (7.0%) | 6 (7.5%) |
| Regurgitation | 2 (5.4%) | 4 (9.3%) | 6 (7.5%) | 1 (2.7%) | 2 (4.7%) | 3 (3.8%) |
| Indigestion | 9 (24.3%) | 10 (23.3%) | 19 (23.8%) | 6 (16.2%) | 6 (14.0%) | 12 (15.0%) |
| Nausea | 9 (24.3%) | 15 (34.9%) | 24 (30.0%) | 7 (18.9%) | 9 (20.9%) | 16 (20.0%) |
| None of these have troubled me | 5 (13.5%) | 3 (7.0%) | 8 (10.0%) | 10 (27.0%) | 12 (27.9%) | 22 (27.5%) |
| Missing | 10 (27.0%) | 6 (14.0%) | 16 (20.0%) | 10 (27.0%) | 11 (25.6%) | 21 (26.3%) |
| Baseline | 12 weeks | |||||
|---|---|---|---|---|---|---|
| IBS-QOL | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
| Overall score (0–100) | ||||||
| Mean (SD) missing | 39.7 (19.20) 0 | 43.6 (20.69) 0 | 41.8 (19.98) 0 | 57.7 (28.61) 5 | 53.3 (25.35) 3 | 55.2 (26.74) 8 |
| Median (range) | 41.9 (5.1–77.2) | 39.7 (6.6–89.7) | 40.8 (5.1–89.7) | 58.1 (4.4–97.8) | 50.0 (14.0–98.5) | 52.2 (4.4–98.5) |
| Subscale: Mean score (SD) | ||||||
| Dysphoria | 38.9 (25.28) | 39.2 (24.56) | 39.1 (24.74) | 61.5 (32.65) | 52.0 (30.03) | 56.2 (31.37) |
| Inference with activity | 29.2 (19.06) | 34.9 (22.63) | 32.2 (21.12) | 50.9 (33.81) | 50.2 (28.07) | 50.5 (30.53) |
| Body image | 37.0 (22.60) | 43.2 (23.62) | 40.3 (23.21) | 51.6 (32.18) | 51.3 (28.03) | 51.4 (29.73) |
| Health worry | 57.9 (22.99) | 60.1 (21.86) | 59.1 (22.27) | 67.4 (22.24) | 67.7 (19.08) | 67.6 (20.39) |
| Food avoidance | 23.4 (21.86) | 33.3 (29.10) | 28.8 (26.32) | 45.6 (34.26) | 39.6 (31.51) | 42.2 (32.66) |
| Social reaction | 41.4 (24.75) | 43.6 (26.92) | 42.6 (25.80) | 59.4 (30.66) | 51.1 (30.48) | 54.8 (30.63) |
| Sexual | 49.0 (35.89) | 63.1 (33.40) | 56.6 (35.07) | 58.6 (35.70) | 59.7 (35.41) | 59.2 (35.29) |
| Relationship | 60.1 (30.12) | 56.4 (25.51) | 58.1 (27.62) | 70.8 (25.84) | 64.8 (28.27) | 67.5 (27.20) |
Appendix 7 Additional end-point summaries by week
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Composite responder | Composite responder | |||||
| Week | Yes | % | Missing | Yes | % | Missing |
| 1 | 8 | 21.6 | 0 | 3 | 7.0 | 1 |
| 2 | 11 | 29.7 | 0 | 14 | 32.6 | 1 |
| 3 | 14 | 37.8 | 0 | 12 | 27.9 | 1 |
| 4 | 15 | 40.5 | 0 | 16 | 37.2 | 2 |
| 5 | 15 | 40.5 | 2 | 12 | 27.9 | 1 |
| 6 | 15 | 40.5 | 2 | 15 | 34.9 | 1 |
| 7 | 15 | 40.5 | 3 | 12 | 27.9 | 1 |
| 8 | 11 | 29.7 | 3 | 10 | 23.3 | 2 |
| 9 | 13 | 35.1 | 3 | 14 | 32.6 | 2 |
| 10 | 16 | 43.2 | 3 | 13 | 30.2 | 2 |
| 11 | 14 | 37.8 | 3 | 10 | 23.3 | 4 |
| 12 | 13 | 35.1 | 5 | 13 | 30.2 | 5 |
| 13 | 9 | 24.3 | 9 | 12 | 27.9 | 4 |
| 14 | 8 | 21.6 | 7 | 8 | 18.6 | 3 |
| 15 | 6 | 16.2 | 9 | 6 | 14.0 | 4 |
| 16 | 6 | 16.2 | 11 | 6 | 14.0 | 14 |
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Abdominal pain responder | Abdominal pain responder | |||||
| Week | Yes | % | Missing | Yes | % | Missing |
| 1 | 9 | 24.3 | 0 | 12 | 27.9 | 1 |
| 2 | 13 | 35.1 | 0 | 16 | 37.2 | 1 |
| 3 | 17 | 45.9 | 0 | 16 | 37.2 | 1 |
| 4 | 21 | 56.8 | 0 | 21 | 48.8 | 2 |
| 5 | 17 | 45.9 | 2 | 15 | 34.9 | 1 |
| 6 | 16 | 43.2 | 2 | 18 | 41.9 | 1 |
| 7 | 16 | 43.2 | 3 | 16 | 37.2 | 1 |
| 8 | 17 | 45.9 | 3 | 15 | 34.9 | 2 |
| 9 | 19 | 51.4 | 3 | 17 | 39.5 | 2 |
| 10 | 19 | 51.4 | 3 | 19 | 44.2 | 2 |
| 11 | 18 | 48.6 | 3 | 17 | 39.5 | 4 |
| 12 | 17 | 45.9 | 5 | 16 | 37.2 | 5 |
| 13 | 12 | 32.4 | 9 | 17 | 39.5 | 4 |
| 14 | 13 | 35.1 | 7 | 12 | 27.9 | 3 |
| 15 | 11 | 29.7 | 9 | 10 | 23.3 | 4 |
| 16 | 11 | 29.7 | 11 | 9 | 20.9 | 14 |
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Stool consistency responder | Stool consistency responder | |||||
| Week | Yes | % | Missing | Yes | % | Missing |
| 1 | 19 | 51.4 | 0 | 7 | 16.3 | 1 |
| 2 | 21 | 56.8 | 0 | 18 | 41.9 | 1 |
| 3 | 22 | 59.5 | 0 | 18 | 41.9 | 1 |
| 4 | 26 | 70.3 | 0 | 25 | 58.1 | 1 |
| 5 | 25 | 67.6 | 2 | 19 | 44.2 | 1 |
| 6 | 26 | 70.3 | 2 | 21 | 48.8 | 1 |
| 7 | 25 | 67.6 | 3 | 18 | 41.9 | 1 |
| 8 | 18 | 48.6 | 3 | 19 | 44.2 | 1 |
| 9 | 20 | 54.1 | 3 | 25 | 58.1 | 1 |
| 10 | 23 | 62.2 | 3 | 21 | 48.8 | 2 |
| 11 | 21 | 56.8 | 3 | 19 | 44.2 | 3 |
| 12 | 20 | 54.1 | 5 | 21 | 48.8 | 3 |
| 13 | 18 | 48.6 | 9 | 18 | 41.9 | 3 |
| 14 | 15 | 40.5 | 7 | 14 | 32.6 | 3 |
| 15 | 11 | 29.7 | 9 | 10 | 23.3 | 4 |
| 16 | 13 | 35.1 | 11 | 10 | 23.3 | 14 |
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Week | N | % | Missing response | N | % | Missing response |
| 1 | 4 | 10.8 | 0 | 7 | 16.3 | 1 |
| 2 | 4 | 10.8 | 0 | 8 | 18.6 | 1 |
| 3 | 4 | 10.8 | 0 | 9 | 20.9 | 1 |
| 4 | 4 | 10.8 | 0 | 5 | 11.6 | 1 |
| 5 | 5 | 13.5 | 2 | 7 | 16.3 | 1 |
| 6 | 5 | 13.5 | 2 | 6 | 14.0 | 1 |
| 7 | 3 | 8.1 | 2 | 2 | 4.7 | 1 |
| 8 | 5 | 13.5 | 3 | 4 | 9.3 | 1 |
| 9 | 4 | 10.8 | 3 | 5 | 11.6 | 1 |
| 10 | 1 | 2.7 | 3 | 6 | 14.0 | 2 |
| 11 | 3 | 8.1 | 3 | 8 | 18.6 | 3 |
| 12 | 4 | 9.3 | 5 | 4 | 8.3 | 3 |
| 13 | 5 | 13.5 | 6 | 4 | 9.3 | 3 |
| 14 | 7 | 18.9 | 6 | 5 | 11.6 | 3 |
| 15 | 5 | 13.5 | 7 | 6 | 14.0 | 3 |
| 16 | 5 | 13.5 | 7 | 8 | 18.6 | 7 |
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Urgency responders | Urgency responders | |||||
| Week | Yes | % | Missing | Yes | % | Missing |
| 1 | 14 | 37.8 | 2 | 7 | 16.3 | 5 |
| 2 | 16 | 43.2 | 3 | 11 | 25.6 | 7 |
| 3 | 18 | 48.6 | 4 | 14 | 32.6 | 7 |
| 4 | 23 | 62.2 | 3 | 19 | 44.2 | 8 |
| 5 | 21 | 56.8 | 4 | 16 | 37.2 | 6 |
| 6 | 19 | 51.4 | 4 | 18 | 41.9 | 7 |
| 7 | 19 | 51.4 | 5 | 16 | 37.2 | 8 |
| 8 | 18 | 48.6 | 5 | 16 | 37.2 | 8 |
| 9 | 19 | 51.4 | 5 | 15 | 34.9 | 8 |
| 10 | 19 | 51.4 | 6 | 17 | 39.5 | 8 |
| 11 | 19 | 51.4 | 6 | 13 | 30.2 | 8 |
| 12 | 17 | 45.9 | 11 | 14 | 32.6 | 10 |
| 13 | 14 | 37.8 | 12 | 18 | 41.9 | 8 |
| 14 | 14 | 37.8 | 10 | 10 | 23.3 | 8 |
| 15 | 15 | 40.5 | 11 | 12 | 27.9 | 7 |
| 16 | 14 | 37.8 | 11 | 9 | 20.9 | 16 |
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Pain score (0–100) | Pain score (0–100) | |||||
| Week | Mean | SD | Missing | Mean | SD | Missing |
| 1 | 56.8 | 23.0 | 0 | 50.3 | 17.8 | 1 |
| 2 | 49.5 | 25.5 | 0 | 44.5 | 20.8 | 1 |
| 3 | 45.7 | 26.7 | 0 | 42.8 | 24.5 | 1 |
| 4 | 42.7 | 25.1 | 0 | 39.1 | 20.7 | 2 |
| 5 | 42.6 | 26.4 | 2 | 43.2 | 22.2 | 1 |
| 6 | 43.6 | 26.8 | 2 | 41.8 | 23.8 | 1 |
| 7 | 44.1 | 26.9 | 3 | 42.1 | 25.0 | 1 |
| 8 | 42.9 | 26.5 | 3 | 43.2 | 25.3 | 2 |
| 9 | 41.8 | 25.5 | 3 | 41.0 | 23.4 | 2 |
| 10 | 42.9 | 25.4 | 3 | 40.6 | 24.4 | 2 |
| 11 | 44.4 | 27.5 | 3 | 41.5 | 26.6 | 4 |
| 12 | 41.3 | 27.9 | 5 | 42.3 | 28.3 | 5 |
| 13 | 43.4 | 28.2 | 9 | 42.5 | 26.0 | 4 |
| 14 | 45.5 | 28.0 | 7 | 47.8 | 25.2 | 3 |
| 15 | 45.7 | 28.1 | 9 | 48.5 | 24.7 | 4 |
| 16 | 47.9 | 29.7 | 11 | 47.4 | 27.4 | 14 |
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Number of stools | Number of stools | |||||
| Week | Mean | SD | Missing | Mean | SD | Missing |
| 1 | 2.8 | 1.6 | 0 | 3.2 | 2.3 | 1 |
| 2 | 2.9 | 1.8 | 0 | 3.1 | 2.1 | 1 |
| 3 | 2.8 | 2.0 | 0 | 2.9 | 2.2 | 1 |
| 4 | 2.5 | 1.9 | 0 | 2.8 | 2.1 | 1 |
| 5 | 2.5 | 1.6 | 2 | 2.9 | 2.1 | 1 |
| 6 | 2.5 | 1.8 | 2 | 2.7 | 2.0 | 1 |
| 7 | 2.5 | 1.8 | 3 | 2.5 | 2.1 | 1 |
| 8 | 2.6 | 1.7 | 3 | 2.9 | 2.3 | 1 |
| 9 | 2.7 | 1.6 | 3 | 2.5 | 1.7 | 1 |
| 10 | 2.6 | 1.6 | 3 | 2.4 | 1.7 | 2 |
| 11 | 2.9 | 1.9 | 3 | 2.5 | 1.7 | 3 |
| 12 | 2.5 | 1.5 | 5 | 2.4 | 1.8 | 3 |
| 13 | 2.6 | 1.6 | 7 | 2.6 | 1.8 | 3 |
| 14 | 2.6 | 1.7 | 6 | 3.0 | 2.1 | 3 |
| 15 | 2.8 | 1.8 | 7 | 2.7 | 1.8 | 3 |
| 16 | 2.6 | 1.8 | 7 | 2.7 | 1.6 | 12 |
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Mean stool consistency | Mean stool consistency | |||||
| Week | Mean | SD | Missing | Mean | SD | Missing |
| 1 | 4.6 | 1.2 | 0 | 5.4 | 0.7 | 1 |
| 2 | 4.3 | 1.3 | 0 | 5.1 | 1.0 | 1 |
| 3 | 4.4 | 1.3 | 0 | 5.1 | 1.0 | 1 |
| 4 | 4.2 | 1.3 | 0 | 5.0 | 0.9 | 1 |
| 5 | 4.3 | 1.2 | 2 | 4.9 | 1.0 | 1 |
| 6 | 4.2 | 1.3 | 2 | 4.9 | 1.1 | 1 |
| 7 | 4.3 | 1.2 | 3 | 5.2 | 0.8 | 1 |
| 8 | 4.4 | 1.2 | 3 | 5.0 | 0.9 | 1 |
| 9 | 4.3 | 1.4 | 3 | 4.9 | 1.0 | 1 |
| 10 | 4.3 | 1.4 | 3 | 5.0 | 0.8 | 2 |
| 11 | 4.4 | 1.5 | 3 | 5.0 | 0.9 | 3 |
| 12 | 4.2 | 1.4 | 5 | 4.9 | 1.1 | 3 |
| 13 | 4.7 | 1.4 | 7 | 5.0 | 1.0 | 3 |
| 14 | 5.0 | 1.4 | 6 | 5.2 | 0.8 | 3 |
| 15 | 5.2 | 1.2 | 7 | 5.3 | 0.8 | 3 |
| 16 | 5.1 | 1.0 | 7 | 5.2 | 0.9 | 12 |
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Number of days with loose stool | Number of days with loose stool | |||||
| Week | Mean | SD | Missing | Mean | SD | Missing |
| 1 | 3.0 | 2.6 | 0 | 4.4 | 2.0 | 1 |
| 2 | 2.5 | 2.4 | 0 | 3.4 | 2.6 | 1 |
| 3 | 2.4 | 2.5 | 0 | 3.3 | 2.4 | 1 |
| 4 | 2.1 | 2.2 | 0 | 2.8 | 2.4 | 1 |
| 5 | 2.0 | 2.4 | 2 | 3.0 | 2.4 | 1 |
| 6 | 2.1 | 2.6 | 2 | 3.0 | 2.6 | 1 |
| 7 | 2.1 | 2.5 | 3 | 3.1 | 2.5 | 1 |
| 8 | 2.5 | 2.3 | 3 | 3.1 | 2.5 | 1 |
| 9 | 2.5 | 2.8 | 3 | 2.9 | 2.6 | 1 |
| 10 | 2.3 | 2.5 | 3 | 2.8 | 2.3 | 2 |
| 11 | 2.9 | 2.6 | 3 | 3.0 | 2.4 | 3 |
| 12 | 2.6 | 2.8 | 5 | 2.7 | 2.5 | 3 |
| 13 | 2.4 | 2.6 | 9 | 3.0 | 2.5 | 3 |
| 14 | 3.1 | 2.8 | 7 | 3.7 | 2.5 | 3 |
| 15 | 3.8 | 2.7 | 9 | 3.8 | 2.3 | 4 |
| 16 | 3.5 | 2.8 | 11 | 3.9 | 2.4 | 14 |
| Ondansetron (n = 37) | Placebo (n = 43) | |||||
|---|---|---|---|---|---|---|
| Satisfactory relief | Satisfactory relief | |||||
| Week | Yes | % | Missing | Yes | % | Missing |
| 1 | 8 | 21.6 | 8 | 10 | 23.3 | 12 |
| 2 | 10 | 27.0 | 12 | 15 | 34.9 | 11 |
| 3 | 15 | 40.5 | 12 | 17 | 39.5 | 11 |
| 4 | 15 | 40.5 | 10 | 14 | 32.6 | 14 |
| 5 | 12 | 32.4 | 14 | 11 | 25.6 | 17 |
| 6 | 13 | 35.1 | 15 | 14 | 32.6 | 19 |
| 7 | 12 | 32.4 | 17 | 13 | 30.2 | 16 |
| 8 | 16 | 43.2 | 14 | 12 | 27.9 | 15 |
| 9 | 14 | 37.8 | 17 | 14 | 32.6 | 13 |
| 10 | 13 | 35.1 | 16 | 14 | 32.6 | 12 |
| 11 | 14 | 37.8 | 15 | 11 | 25.6 | 14 |
| 12 | 14 | 37.8 | 17 | 12 | 27.9 | 18 |
| 13 | 7 | 18.9 | 18 | 8 | 18.6 | 18 |
| 14 | 5 | 13.5 | 18 | 6 | 14.0 | 19 |
| 15 | 5 | 13.5 | 19 | 4 | 9.3 | 18 |
| 16 | 4 | 10.8 | 19 | 2 | 4.7 | 27 |
Appendix 8 Safety
| 6 weeks | 12 weeks | Follow-up | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Experienced N (%) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) |
| Constipation | |||||||||
| Yes | 17 (45.9%) | 11 (25.6%) | 28 (35.0%) | 12 (32.4%) | 10 (23.3%) | 22 (27.5%) | 5 (13.5%) | 8 (18.6%) | 13 (16.3%) |
| Missing | 2 (5.4%) | 3 (7.0%) | 5 (6.3%) | 3 (8.1%) | 3 (7.0%) | 6 (7.5%) | 3 (8.1%) | 2 (4.7%) | 5 (6.3%) |
| If yes: Severity | |||||||||
| Mild | 11 (64.7%) | 6 (54.5%) | 17 (60.7%) | 9 (75.0%) | 8 (80.0%) | 17 (77.3%) | 4 (80.0%) | 3 (37.5%) | 7 (53.8%) |
| Moderate | 5 (29.4%) | 5 (45.5%) | 10 (35.7%) | 2 (16.7%) | 2 (20.0%) | 4 (18.2%) | 0 (0.0%) | 5 (62.5%) | 5 (38.5%) |
| Severe | 1 (5.9%) | 0 (0.0%) | 1 (3.6%) | 1 (8.3%) | 0 (0.0%) | 1 (4.5%) | 1 (20.0%) | 0 (0.0%) | 1 (7.7%) |
| Abdominal pain/bloating | |||||||||
| Yes | 26 (70.3%) | 36 (83.7%) | 62 (77.5%) | 23 (62.2%) | 33 (76.7%) | 56 (70.0%) | 25 (67.6%) | 36 (83.7%) | 61 (76.3%) |
| Missing | 2 (5.4%) | 2 (4.7%) | 4 (5.0%) | 3 (8.1%) | 3 (7.0%) | 6 (7.5%) | 3 (8.1%) | 2 (4.7%) | 5 (6.3%) |
| If yes: Severity | |||||||||
| Mild | 10 (38.5%) | 20 (55.6%) | 30 (48.4%) | 12 (52.2%) | 14 (42.4%) | 26 (46.4%) | 7 (28.0%) | 9 (25.0%) | 16 (26.2%) |
| Moderate | 10 (38.5%) | 11 (30.6%) | 21 (33.9%) | 7 (30.4%) | 15 (45.5%) | 22 (39.3%) | 11 (44.0%) | 17 (47.2%) | 28 (45.9%) |
| Severe | 6 (23.1%) | 5 (13.9%) | 11 (17.7%) | 4 (17.4%) | 4 (12.1%) | 8 (14.3%) | 7 (28.0%) | 10 (27.8%) | 17 (27.9%) |
| Headache | |||||||||
| Yes | 17 (45.9%) | 8 (18.6%) | 25 (31.3%) | 11 (29.7%) | 14 (32.6%) | 25 (31.3%) | 10 (27.0%) | 12 (27.9%) | 22 (27.5%) |
| Missing | 2 (5.4%) | 4 (9.3%) | 6 (7.5%) | 3 (8.1%) | 3 (7.0%) | 6 (7.5%) | 3 (8.1%) | 2 (4.7%) | 5 (6.3%) |
| If yes: Severity | |||||||||
| Mild | 11 (64.7%) | 7 (87.5%) | 18 (72.0%) | 3 (27.3%) | 8 (57.1%) | 11 (44.0%) | 6 (60.0%) | 8 (66.7%) | 14 (63.6%) |
| Moderate | 5 (29.4%) | 1 (12.5%) | 6 (24.0%) | 5 (45.5%) | 5 (35.7%) | 10 (40.0%) | 3 (30.0%) | 4 (33.3%) | 7 (31.8%) |
| Severe | 1 (5.9%) | 0 (0.0%) | 1 (4.0%) | 3 (27.3%) | 1 (7.1%) | 4 (16.0%) | 1 (10.0%) | 0 (0.0%) | 1 (4.5%) |
| Nausea | |||||||||
| Yes | 6 (16.2%) | 12 (27.9%) | 18 (22.5%) | 10 (27.0%) | 18 (41.9%) | 28 (35.0%) | 9 (24.3%) | 12 (27.9%) | 21 (26.3%) |
| Missing | 2 (5.4%) | 4 (9.3%) | 6 (7.5%) | 3 (8.1%) | 2 (4.7%) | 5 (6.3%) | 3 (8.1%) | 2 (4.7%) | 5 (6.3%) |
| If yes: Severity | |||||||||
| Mild | 4 (66.7%) | 9 (75.0%) | 13 (72.2%) | 7 (70.0%) | 10 (55.6%) | 17 (60.7%) | 3 (33.3%) | 6 (50.0%) | 9 (42.9%) |
| Moderate | 2 (33.3%) | 3 (25.0%) | 5 (27.8%) | 1 (10.0%) | 5 (27.8%) | 6 (21.4%) | 4 (44.4%) | 3 (25.0%) | 7 (33.3%) |
| Severe | 2 (20.0%) | 3 (16.7%) | 5 (17.9%) | 2 (22.2%) | 3 (25.0%) | 5 (23.8%) | |||
| Vomiting | |||||||||
| Yes | 3 (8.1%) | 3 (7.0%) | 6 (7.5%) | 1 (2.7%) | 4 (9.3%) | 5 (6.3%) | 4 (10.8%) | 6 (14.0%) | 10 (12.5%) |
| Missing | 2 (5.4%) | 4 (9.3%) | 6 (7.5%) | 3 (8.1%) | 3 (7.0%) | 6 (7.5%) | 3 (8.1%) | 2 (4.7%) | 5 (6.3%) |
| If yes: Severity | |||||||||
| Mild | 2 (66.7%) | 2 (66.7%) | 4 (66.7%) | 0 (0.0%) | 1 (16.7%) | 1 (10.0%) | |||
| Moderate | 1 (33.3%) | 1 (33.3%) | 2 (33.3%) | 0 (0.0%) | 4 (100.0%) | 4 (80.0%) | 3 (75.0%) | 2 (33.3%) | 5 (50.0%) |
| Severe | 1 (100.0%) | 0 (0.0%) | 1 (20.0%) | 1 (25.0%) | 3 (50.0%) | 4 (40.0%) | |||
| Rectal bleeding | |||||||||
| Yes | 3 (8.1%) | 7 (16.3%) | 10 (12.5%) | 4 (10.8%) | 6 (14.0%) | 10 (12.5%) | 4 (10.8%) | 6 (14.0%) | 10 (12.5%) |
| Missing | 2 (5.4%) | 2 (4.7%) | 4 (5.0%) | 3 (8.1%) | 3 (7.0%) | 6 (7.5%) | 3 (8.1%) | 2 (4.7%) | 5 (6.3%) |
| If yes: Severity | |||||||||
| Mild | 3 (100.0%) | 6 (85.7%) | 9 (90.0%) | 4 (100.0%) | 6 (100.0%) | 10 (100.0%) | 4 (100.0%) | 6 (100.0%) | 10 (100.0%) |
| Moderate | |||||||||
| Severe | 0 (0.0%) | 1 (14.3%) | 1 (10.0%) | ||||||
| Patient | Clinical opinion obtaineda | Colonoscopy/flexi sigmoidoscopy performed | Allocation |
|---|---|---|---|
| 1 | Yes | No | Ondansetron |
| 2 | Yes | No | Ondansetron |
| 3 | Yes | No | Ondansetron |
| 4 | Yes | No | Placebo |
| 5 | Yes | No | Placebo |
| 6 | Yes | No | Placebo |
| 7 | Yes | No | Placebo |
| 8 | Yes | No | Placebo |
| 9 | Yes | No | Placebo |
| 10 | No | No | Placebo |
| Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | |
|---|---|---|---|
| Pulse within normal range | |||
| Yes | 29 (78.4%) | 31 (72.1%) | 60 (75.0%) |
| Missing | 8 (21.6%) | 12 (27.9%) | 20 (25.0%) |
| BP within normal range | |||
| Yes | 28 (75.7%) | 30 (69.8%) | 58 (72.5%) |
| No | 1 (2.7%) | 1 (2.3%) | 2 (2.5%) |
| Missing | 8 (21.6%) | 12 (27.9%) | 20 (25.0%) |
| Treatment allocation | Details | Pregnancy outcome | Number of days between randomisation and date of positive pregnancy test |
| Placebo | Trial participant is pregnant | Live birth | 96 |
| Patient | New medications since previous visit | PI confirmation of medications not being restricted or prohibited | Allocation |
|---|---|---|---|
| 1 | Yes | Yes | Ondansetron |
| 2 | Yes | Yes | Placebo |
| 3 | Yes | Yes | Placebo |
| 4 | Yes | Yes | Placebo |
| 5 | Yes | Yes | Ondansetron |
| Ondansetron (n = 37) | Placebo (n = 43) | Total (n = 80) | |
|---|---|---|---|
| Recommended titrated dose after 2 weeks of treatment | |||
| One capsule (4 mg) every 3 days | 1 (2.7%) | 2 (4.7%) | 3 (3.8%) |
| One capsule (4 mg) every 2 days | 7 (18.9%) | 3 (7.0%) | 10 (12.5%) |
| One capsule (4 mg) daily | 6 (16.2%) | 3 (7.0%) | 9 (11.3%) |
| Two capsules (8 mg) daily | 8 (21.6%) | 10 (23.3%) | 18 (22.5%) |
| Three capsules (12 mg) daily | 2 (5.4%) | 5 (11.6%) | 7 (8.8%) |
| Four capsules (16 mg) daily | 2 (5.4%) | 3 (7.0%) | 5 (6.3%) |
| Five capsules (20 mg) daily | 5 (13.5%) | 3 (7.0%) | 8 (10.0%) |
| Six capsules (24 mg) daily | 3 (8.1%) | 12 (27.9%) | 15 (18.8%) |
| Missing | 3 (8.1%) | 2 (4.7%) | 5 (6.3%) |
| Number of bottles requested 6 weeks post randomisation | |||
| One bottle (one capsule or less per day) | 7 (18.9%) | 6 (14.0%) | 13 (16.3%) |
| Two bottles (two capsules per day) | 8 (21.6%) | 9 (20.9%) | 17 (21.3%) |
| Three bottles (three capsules per day) | 3 (8.1%) | 5 (11.6%) | 8 (10.0%) |
| Four bottles (four capsules per day) | 3 (8.1%) | 5 (11.6%) | 8 (10.0%) |
| Five bottles (five capsules per day) | 3 (8.1%) | 2 (4.7%) | 5 (6.3%) |
| Six bottles (six capsules per day) | 5 (13.5%) | 10 (23.3%) | 15 (18.8%) |
| Missing | 8 (21.6%) | 6 (14.0%) | 14 (17.5%) |
| Number of bottles requested 6 weeks post randomisation consistent with the recommended titrated dose after 2 weeks? | |||
| Yes | 19 (51.4%) | 29 (67.4%) | 48 (60.0%) |
| No | 8 (21.6%) | 6 (14.0%) | 14 (17.5%) |
| Missing | 10 (27.0%) | 8 (18.6%) | 18 (22.5%) |
| Dose at 2 weeks compared with 6 weeks N participants | 10 | 7 | 17 |
| Participant on a lower dose at 6 weeks compared with 2 weeks | 2 (20.0%) | 2 (28.6%) | 4 (23.5%) |
| Participant on a higher dose at 6 weeks compared with 2 weeks | 8 (80.0%) | 5 (71.4%) | 13 (76.5%) |
| Number of capsules returned at 12 weeks consistent with dose at 6 weeks N participants | 33 | 42 | 75 |
| Yes | 7 (21.2%) | 7 (16.7%) | 14 (18.7%) |
| No | 22 (66.7%) | 32 (76.2%) | 54 (72.0%) |
| Missing | 4 (12.1%) | 3 (7.1%) | 7 (9.3%) |
| Dose at 12 weeks compared with 6 weeks N | 24 | 32 | 56 |
| Participant on a higher dose at 12 weeks compared with 6 weeks | 9 (37.5%) | 20 (62.5%) | 29 (51.8%) |
| Participant on a lower dose at 12 weeks compared with 6 weeks | 15 (62.5%) | 12 (37.5%) | 27 (48.2%) |
Appendix 9 Exit poll
| Certainty of choice | Reasons for choice of those that guessed ondansetron | ||||||
|---|---|---|---|---|---|---|---|
| Ondansetron (n = 22) | Treatment benefited the patient (n = 21) | AE(s) consistent with ondansetron (n = 1) | Placebo (n = 17) | Treatment benefited the patient (n = 16) | Othera (n = 1) | Total (n = 39) | |
| 4 | 5 (29.4%) | 5 (31.3%) | 5 (12.8%) | ||||
| 5 | 1 (4.5%) | 1 (4.8%) | 2 (11.8%) | 1 (6.3%) | 1 (100.0%) | 3 (7.7%) | |
| 6 | 2 (9.1%) | 2 (9.5%) | 2 (11.8%) | 2 (12.5%) | 4 (10.3%) | ||
| 7 | 5 (22.7%) | 5 (23.8%) | 3 (17.6%) | 3 (18.8%) | 8 (20.5%) | ||
| 8 | 6 (27.3%) | 6 (28.6%) | 3 (17.6%) | 3 (18.8%) | 9 (23.1%) | ||
| 9 | 2 (9.1%) | 2 (9.5%) | 1 (5.9%) | 1 (6.3%) | 3 (7.7%) | ||
| 10 | 6 (27.3%) | 5 (23.8%) | 1 (100.0%) | 1 (5.9%) | 1 (6.3%) | 7 (17.9%) | |
| Reasons for choice of those that guessed placebo | |||||||
| Ondansetron ( n = 8) | Treatment had no benefit ( n = 8) | Placebo ( n = 21) | Treatment had no benefit ( n = 21) | Total ( n = 29) | |||
| 4 | 1 (12.5%) | 1 (12.5%) | 2 (9.5%) | 2 (9.5%) | 3 (10.3%) | ||
| 5 | 1 (4.8%) | 1 (4.8%) | 1 (3.4%) | ||||
| 6 | 1 (12.5%) | 1 (12.5%) | 2 (9.5%) | 2 (9.5%) | 3 (10.3%) | ||
| 7 | 2 (25.0%) | 2 (25.0%) | 3 (14.3%) | 3 (14.3%) | 5 (17.2%) | ||
| 8 | 1 (12.5%) | 1 (12.5%) | 3 (14.3%) | 3 (14.3%) | 4 (13.8%) | ||
| 9 | 2 (25.0%) | 2 (25.0%) | 7 (33.3%) | 7 (33.3%) | 9 (31.0%) | ||
| 10 | 1 (12.5%) | 1 (12.5%) | 3 (14.3%) | 3 (14.3%) | 4 (13.8%) | ||
| Reasons for choice of those that responded ‘Don’t know’ b | |||||||
| Ondansetron ( n = 5) | Placebo ( n = 2) | Total ( n = 7) | |||||
| 0 | 1 (50.0%) | 1 (14.3%) | |||||
| 5 | 2 (40.0%) | 2 (28.6%) | |||||
| 6 | 1 (50.0%) | 1 (14.3%) | |||||
| 7 | 1 (20.0%) | 1 (14.3%) | |||||
| 9 | 2 (40.0%) | 2 (28.6%) | |||||
| Certainty of choice | Reasons for choice of those that guessed ondansetron | ||||||
|---|---|---|---|---|---|---|---|
| Ondansetron (n = 22) | Treatment worked (n = 18) | I had a side effect (n = 3) | Other (n = 1)a | Placebo (n = 16) | Treatment worked (n = 16) | Total (n = 38) | |
| 2 | 1 (4.5%) | 1 (33.3%) | 1 (2.6%) | ||||
| 5 | 0 (0.0%) | 1 (6.3%) | 1 (6.3%) | 1 (2.6%) | |||
| 6 | 4 (18.2%) | 3 (16.7%) | 0 (0.0%) | 1 (100.0%) | 1 (6.3%) | 1 (6.3%) | 5 (13.2%) |
| 7 | 2 (9.1%) | 2 (11.1%) | 0 (0.0%) | 3 (18.8%) | 3 (18.8%) | 5 (13.2%) | |
| 8 | 4 (18.2%) | 3 (16.7%) | 1 (33.3%) | 2 (12.5%) | 2 (12.5%) | 6 (15.8%) | |
| 9 | 1 (4.5%) | 1 (5.6%) | 0 (0.0%) | 3 (18.8%) | 3 (18.8%) | 4 (10.5%) | |
| 10 | 10 (45.5%) | 9 (50.0%) | 1 (33.3%) | 6 (37.5%) | 6 (37.5%) | 16 (42.1%) | |
| Reasons for choice of those that guessed placebo | |||||||
| Ondansetron ( n = 8) | Treatment didn’t work ( n = 6) | Treatment worked ( n = 1) | Other ( n = 1) b | Placebo ( n = 18) | Treatment didn’t work ( n = 18) | Total ( n = 26) | |
| 4 | 2 (25.0%) | 2 (33.3%) | 2 (7.7%) | ||||
| 5 | 1 (12.5%) | 1 (100.0%) | 1 (5.6%) | 1 (5.6%) | 2 (7.7%) | ||
| 6 | 1 (12.5%) | 1 (100.0%) | 1 (5.6%) | 1 (5.6%) | 2 (7.7%) | ||
| 7 | 2 (11.1%) | 2 (11.1%) | 2 (7.7%) | ||||
| 8 | 6 (33.3%) | 6 (33.3%) | 6 (23.1%) | ||||
| 9 | 5 (27.8%) | 5 (27.8%) | 5 (19.2%) | ||||
| 10 | 4 (50.0%) | 4 (66.7%) | 3 (16.7%) | 3 (16.7%) | 7 (26.9%) | ||
| Reasons for choice of those that responded ‘Don’t know’ c | |||||||
| Ondansetron ( n = 3) | Placebo ( n = 7) | Total ( n = 10) | |||||
| 2 | 1 (33.3%) | 1 (10.0%) | |||||
| 4 | 1 (14.3%) | 1 (10.0%) | |||||
| 5 | 3 (42.9%) | 3 (30.0%) | |||||
| 6 | 1 (14.3%) | 1 (10.0%) | |||||
| 7 | 1 (33.3%) | 2 (28.6%) | 3 (30.0%) | ||||
| 10 | 1 (33.3%) | 1 (10.0%) | |||||
Appendix 10 Advertisement
FIGURE 32.
Website self-assessment summary flowchart.

FIGURE 33.
Clinic poster.

FIGURE 34.
Clinic leaflet page 1; clinic leaflet page 2.


FIGURE 35.
Press advertisement.

List of abbreviations
- 5HT
- 5-hydroxytryptamine
- 5HT3RA
- 5-HT3 receptor antagonist
- AE
- adverse event
- ALT
- alanine transaminase
- ANOVA
- analysis of variance
- BAD
- bile acid diarrhoea
- BP
- blood pressure
- BSFS
- Bristol Stool Form Scale/Score
- CI
- confidence interval
- CNS
- central nervous system
- CONSORT
- Consolidated Standards of Reporting Trials
- CTRU
- clinical trials research unit
- DMEC
- Data Monitoring and Ethics Committee
- ECG
- electrocardiogram
- FBA
- faecal bile acid
- FBC
- full blood count
- FDA
- Food and Drug Administration
- FP
- faecal protease
- FW
- faecal water
- GP
- general practitioner
- HADS
- Hospital Anxiety and Depression Scale
- HAPC
- high-amplitude propagated contraction
- HRM
- high-resolution manometry
- IBD
- inflammatory bowel disease
- IBS
- irritable bowel syndrome
- IBS-D
- irritable bowel syndrome with diarrhoea
- IBS-QOL
- irritable bowel syndrome–quality of life
- IBS-SSS
- Irritable Bowel Syndrome Severity Scoring System
- IMP
- investigational medicinal product
- ISRCTN
- International Standard Randomised Controlled Trial Register
- ITT
- intention to treat
- MAR
- missing at random
- NICE
- National Institute for Health and Care Excellence
- PHQ-12
- Patient Health Questionnaire 12
- PI
- principal investigator
- PIC
- Participant Identification Centre
- PIS
- patient information sheet
- PP
- per protocol
- PPI
- patient participation and involvement
- QoL
- quality of life
- QTc
- corrected QT interval
- SAE
- serious adverse events
- SD
- standard deviation
- SE
- standard error
- SeHCAT
- selenium homocholic acid taurine
- SFLDQ
- short-form Leeds dyspepsia questionnaire
- SPE
- solid-phase extraction
- TCA
- tricyclic antidepressant
- t.d.s.
- three times a day
- TPH-1
- tryptophan hydroxylase-1
- VAS
- visual analogue scale
- WGTT
- whole gut transit time
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/YTFW7874).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.