
Notes
Article history
The research reported in this issue of the journal was funded by the HS&DR programme or one of its preceding programmes as project number 11/2003/27. The contractual start date was in October 2012. The final report began editorial review in July 2015 and was accepted for publication in November 2015. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HS&DR editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Bruce Guthrie has been a member of the National Institute for Health Research (NIHR) Health Services and Delivery Research researcher-led panel since April 2014, and is the chairperson of the guideline development group of the National Institute for Health and Care Excellence (NICE) multimorbidity clinical guideline. Phil Alderson is employed by NICE, which produces clinical guidelines for the NHS in England and Wales, and is a member of the NIHR Systematic Reviews Programme Advisory Group and Cochrane panel. Moray Nairn is employed by the Scottish Intercollegiate Guidelines Network, which produces clinical guidelines for the NHS in Scotland.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2017. This work was produced by Guthrie et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Background to the project
In the last 20 years, clinical guidelines (CGs) have become a key method for disseminating evidence of effective practice. With success in improving quality of care and reducing variation in practice1 has come increasing criticism of the single-disease focus of most guidelines because of concern that guidelines do not adequately account for the large number of people who have multiple conditions. 2–7 In part, this reflects the fact that the evidence on which guidelines are based is largely focused on single diseases. Although it will of course never be possible to have good evidence for every possible combination of conditions, the starting point for this project (known as the Better Guidelines project) was a belief that single-disease guidelines could better account for multimorbidity. The overall aim was therefore to work collaboratively with the National Institute for Health and Care Excellence (NICE) and the Scottish Intercollegiate Guidelines Network (SIGN) to explore how this could be achieved within the context of existing guideline development.
Defining multimorbidity and comorbidity
Multimorbidity is usually defined as the presence of two or more long-term conditions in an individual, where no condition is given primacy. Comorbidity is the other term commonly used in this field. Comorbidity is defined as the presence of one or more other condition in someone with a particular condition of interest. Although this project is framed in terms of multimorbidity (the general problem), it is worth noting that, from the perspective of a single-disease guideline development group (GDG), the actual problem is comorbidity (the extent to which people with the single disease that is the focus of the guideline have other conditions). We therefore use both terms in this report, using ‘multimorbidity’ when describing the general problem, but ‘comorbidity’ when appropriate.
Valderas et al. 8 helpfully provide a framework for conceptualising the definitions of multimorbidity and comorbidity, distinguishing between:
-
multimorbidity and comorbidity defined in terms of the presence of multiple diseases
-
morbidity burden, which is additionally influenced by non-disease characteristics such as age, sex, frailty and other health-related individual attributes
-
complexity, which is additionally influenced by non-health-related individual attributes. 8
However, although this is a very useful conceptual framework, it is not straightforward to use in practice. This is because diseases are often social constructions rather than natural categories, and the distinction between disease and health-related individual attributes is therefore not fixed. As an example, it is arguable whether obesity, mild cognitive impairment without clear dementia, and some elements of frailty are diseases or health-related individual attributes. In addition, because diseases often share causes or are caused by each other,8 it is often unclear where one disease ends and another begins. A person with diabetes who is blind from diabetic retinopathy and with end-stage kidney disease from diabetic nephropathy can be considered to have one condition (complicated diabetes) or three (diabetes, blindness and renal failure). The focus of this study is on multimorbidity in the wider context of morbidity burden as defined above. This is because the implications of multimorbidity for guideline developers are often framed by how multimorbidity interacts with the wider context of an individual.
Epidemiology of multimorbidity
There have been many studies of the epidemiology of multimorbidity. Most of them examine prevalence in relation to patient demography and a smaller number examine patterns of commonly co-occurring conditions. 9,10 The estimated prevalence of multimorbidity in the most recent systematic review varied from 13% to 95% depending on the population studied and the way in which morbidity data are collected and recorded. 9 Prevalence also varies with how many conditions are counted; the estimated prevalence of multimorbidity based on morbidities recorded in general practice was 16% when conditions included in the UK Quality and Outcomes Framework were counted and 58% when a larger number of conditions were counted. 11 This makes it difficult to compare different studies or to generalise from studies in one country or context to the UK or any other specific country,12 although consistent findings across studies are that multimorbidity increases with age and with lower socioeconomic status and is somewhat more common in women than in men. 9
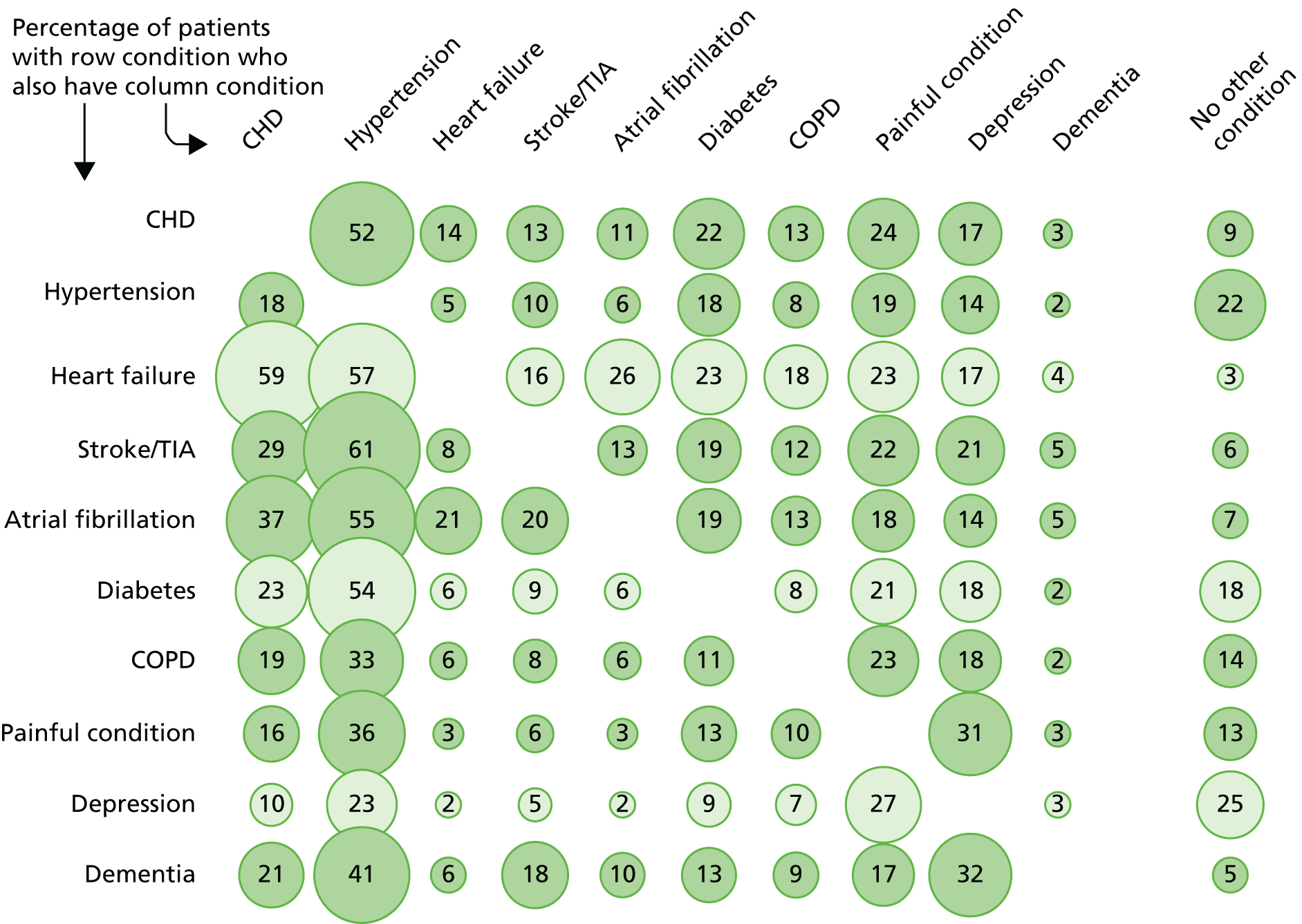
Since the focus of the study is UK CGs, the remainder of this section therefore describes the epidemiology using data from our previous study of the prevalence of multimorbidity. This used data extracted from general practice clinical systems for 1.75 million people in Scotland (approximately one-third of the Scottish population, and representative in terms of age, sex and deprivation). 13 Based on counting 40 common, long-term conditions, 42% of people have one or more conditions and 23% are multimorbid, meaning that a small majority of those with any of the 40 chronic conditions included have more than one. The prevalence of multimorbidity rises steeply with age, with the majority of those aged > 65 years having two or more conditions, and the majority of those aged > 75 years having three or more (Figure 1). However, because there are more middle-aged than older people, the absolute number of people aged < 65 years with multimorbidity is slightly greater than the absolute number aged ≥ 65 years with multimorbidity. 13 People living in more deprived areas have approximately twice the prevalence of multimorbidity in middle age as those living in the most affluent areas. Put another way, on average they become multimorbid 10–15 years earlier than those living in the most affluent areas. 13 The type of multimorbidity experienced varies with age, with a higher proportion being physical–mental health multimorbidity in younger people, particularly in deprived areas, where a common combination is comorbid physical disease and depression. 14
FIGURE 1.
Prevalence of comorbidity for selected common conditions. Source: based on data from the study reported by Barnett et al. 13 CHD, coronary heart disease; COPD, chronic obstructive pulmonary disease; TIA, transient ischaemic attack.
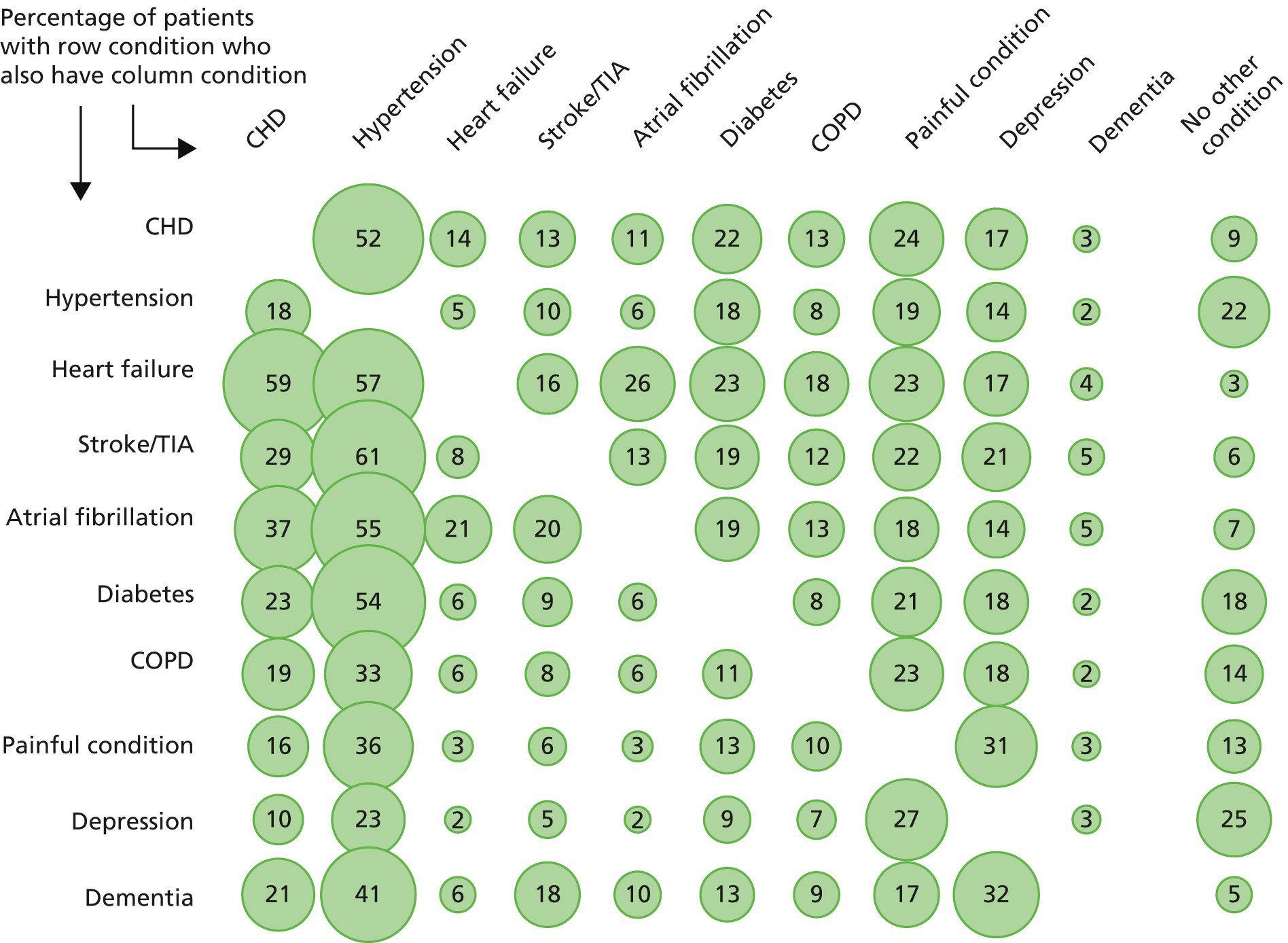
Figure 1 shows patterns of comorbidity for selected common conditions. Of note is that, for the conditions shown, a maximum of 25% of people have only that condition. The figure can be read both horizontally and vertically, and the implications of reading in each direction can differ. For example, people with dementia have many comorbidities (reading horizontally) but dementia is rarely a comorbidity of any one of the other conditions (reading vertically). Hypertension is the reverse, in that a relatively small proportion of people with hypertension have any one of the other conditions (reading horizontally), but hypertension is very commonly a comorbidity of the other conditions (reading vertically).
The implications of having multimorbidity depend on which combination of conditions an individual has. One framework for considering this is in terms of whether conditions are concordant or discordant, or whether any one condition currently dominates (e.g. potentially curative cancer treatment will take priority over almost all other conditions in the short term). 15 Piette and Kerr15 originally defined these terms in relation to diabetes as follows:
-
Clinically dominant conditions are ‘comorbid chronic conditions that are so complex and serious that they eclipse the management of other health problems’.
-
Concordant conditions ‘represent parts of the same overall pathophysiologic risk profile and are more likely to be the focus of the same disease and self-management plan’.
-
Discordant conditions are ‘not directly related in either their pathogenesis or management’.
Much like conceptual frameworks for multimorbidity and comorbidity, deciding whether two conditions are concordant or discordant in practice requires judgement, since concordance is often a spectrum rather than a clear dichotomy, and partly depends on context and purpose, since, for example, pathogenesis might be concordant but management discordant. For example, diabetes and hypertension are commonly comorbid but we judged them to be concordant in that there are few implications for an individual in terms of disease management or care organisation. This is because most of the management of hypertension is accounted for by the management of diabetes. In contrast, coronary heart disease (CHD) and depression are also commonly comorbid but we judged them to be discordant. This is because the presence of each makes outcomes for the other worse; physical and mental health care usually operate in distinct silos, which may complicate care co-ordination; and there are important treatment interactions [e.g. between antiplatelet drugs used in CHD and selective serotonin reuptake inhibitor (SSRI) antidepressants, both of which cause gastrointestinal (GI) bleeding16]. Of note is that almost one in five people with any of the specified conditions have depression, which is one reason that NICE has developed a guideline for people with depression who also have a chronic physical health problem. 17
Why multimorbidity matters
Multimorbidity matters because people with multimorbidity have increased mortality, reduced quality of life, higher use of health services, higher treatment burden and worse experience of care partly because of greater fragmentation. 13,18–26 However, the exact impact of multimorbidity on an individual is mediated by other factors. For example, as might be expected, having any chronic condition is associated with a decrement in health-related quality of life (HRQoL), and having more than one chronic condition is associated with a greater decrement than having only one. 26 However, the decrement in quality of life from multimorbidity is greater in people living in deprived areas than those living in affluent areas, and is greatest in younger people living in deprived areas. 26 Similarly, as might be expected, rates of unscheduled admission to acute hospitals (almost entirely for physical health admissions) increase progressively with the number of physical conditions that an individual has. 22,27 However, admission rates also increase with socioeconomic deprivation and with having a mental health condition. 22 The impact of multimorbidity is therefore likely to be mediated by the particular combination of conditions an individual has (with, for example, the presence of a mental health condition influencing physical health care or outcomes) and by the social context in which multimorbidity is experienced. 24–26
Guidelines and multimorbidity
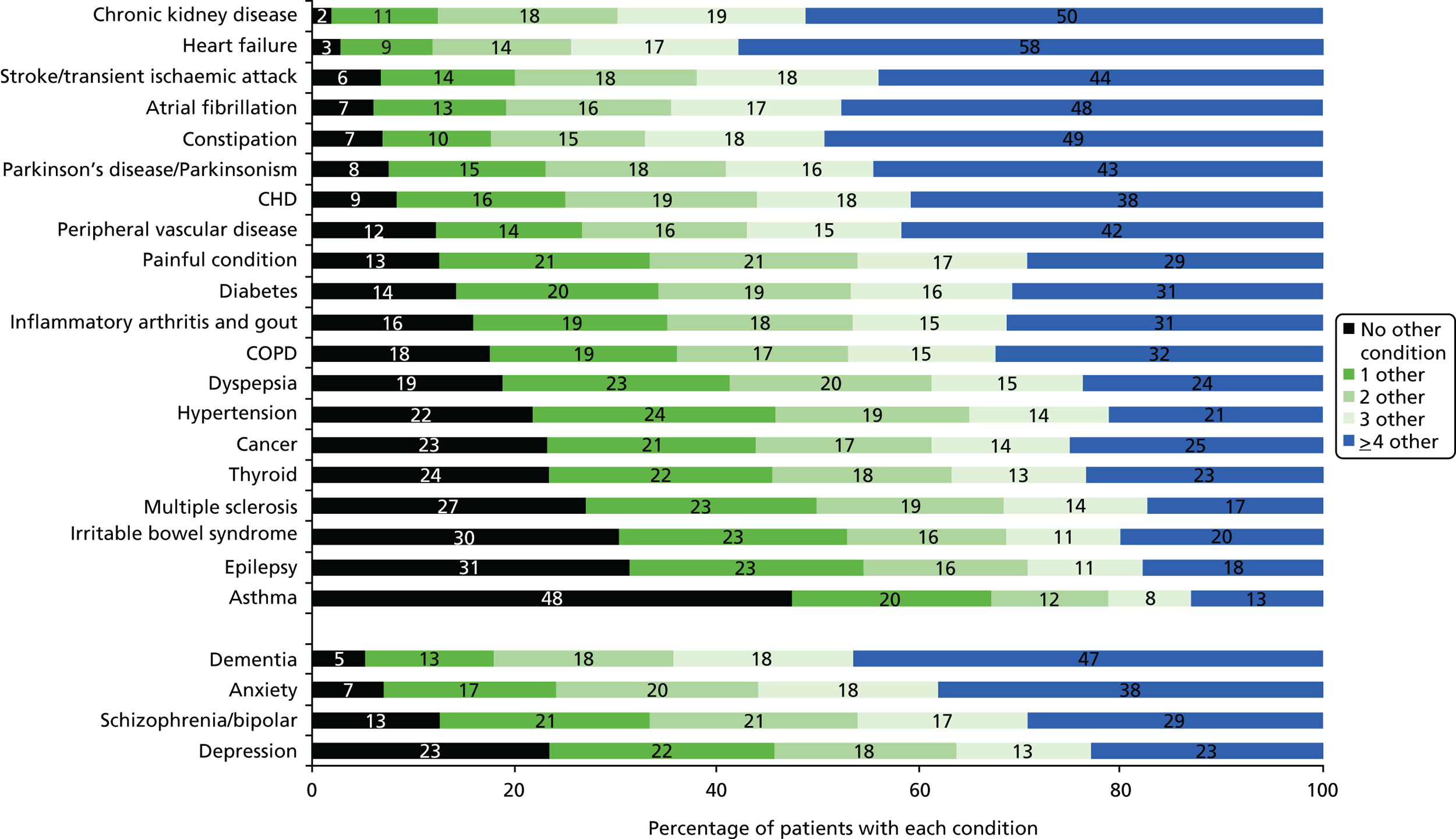
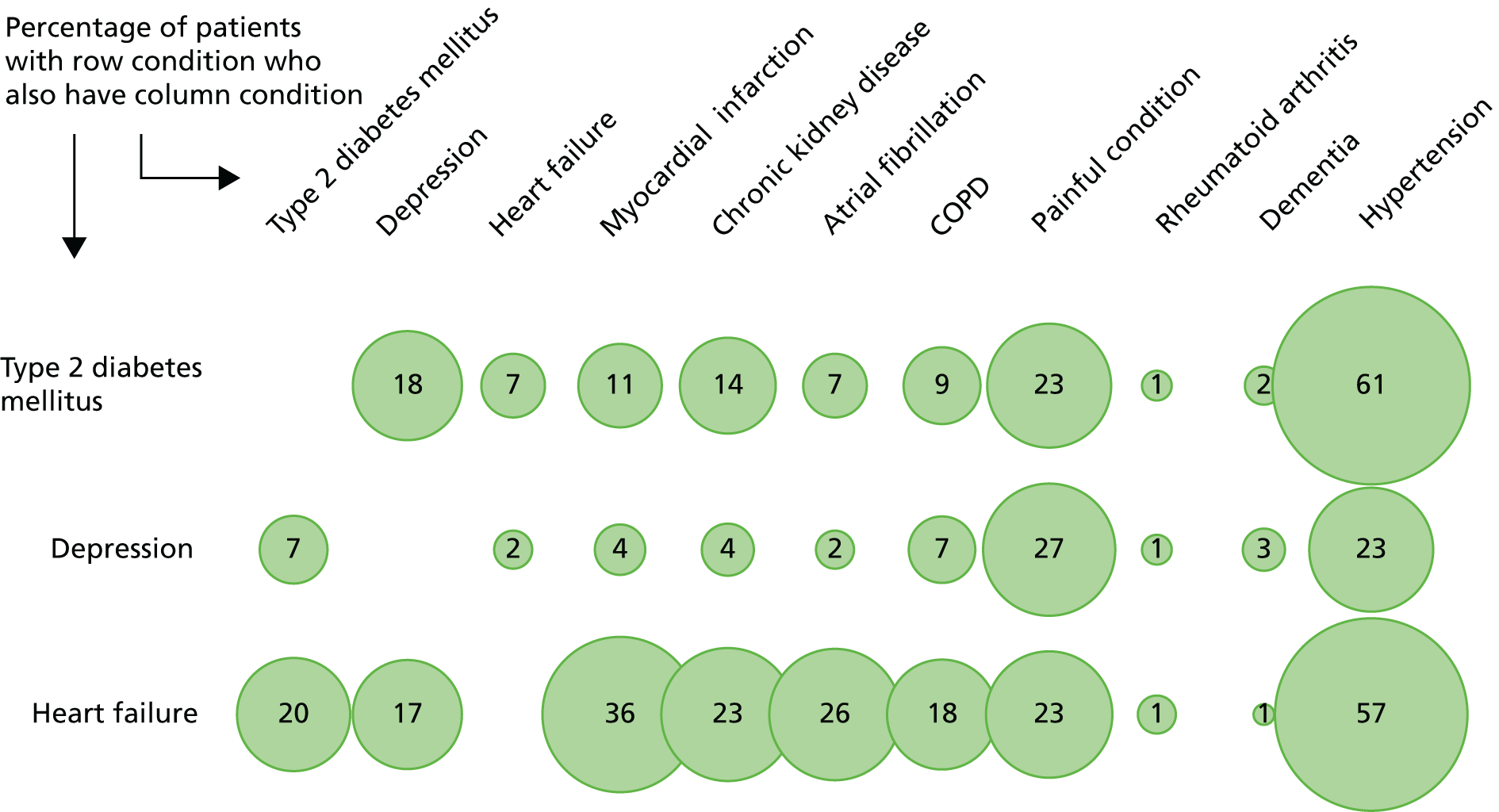
From a guideline perspective, multimorbidity matters because people with multimorbidity have the highest health needs and are the highest users of health care. Ensuring appropriate treatment for them is therefore a priority. More specifically, it matters because the majority of people who are the target of most adult single-disease guidelines will have comorbidity (Figure 2). A challenge for guideline developers is, therefore, appropriately accounting for relevant comorbidity (e.g. because one condition worsens outcomes or because treatments for one condition are contraindicated by or synergistic with treatments for another condition) without creating an unusable guideline that attempts to account for all possible comorbidities.
FIGURE 2.
Prevalence of comorbidity for common chronic conditions. Source: based on data from the study reported by Barnett et al. 13
In the face of multimorbidity, clinicians and patients may struggle to balance the benefits and risks of multiple recommended treatments,24 partly because of the complexity caused by drug–drug and drug–disease interactions and by reduced life expectancy or frailty, and partly because the application of clinical and economic evidence is appropriately influenced by personal preference. 28 Meta-ethnography of qualitative research with clinicians about managing patients with multimorbidity found that the ‘inadequacy of guidelines and evidence-based medicine’29 was one of four common themes across studies, particularly in relation to extrapolation of evidence from trials to people with multimorbidity, and the relevance of disease-specific outcomes in relation to the use of preventative treatments in this population. In response, general practitioners (GPs) often sought to find satisfactory and sufficient treatments in people with multimorbidity, which often involved deviation from guideline recommendations. 29,30
Such variation from guideline-recommended best practice reflects the fact that, even if every guideline recommendation is rational because it is based on robust synthesis of clinical and economic evidence, the cumulative impact of following multiple guideline recommendations can easily be harmful or lead to burdensome overall treatment regimens. 2,20 Boyd et al. 2 demonstrated this for US guidelines by examining recommendations for an older person with chronic obstructive pulmonary disease (COPD), type 2 diabetes, osteoporosis, hypertension and osteoarthritis. Four out of the five guidelines examined did not explicitly acknowledge the potential for the patient to have comorbidity, and the recommendations made were sometimes contradictory and implied a burdensome drug and self-care treatment regimen that would be unfeasible for many patients. 2
Similar issues apply to NICE guidelines in the UK. 3 For example, consider a 78-year-old woman with five conditions – previous myocardial infarction (MI), type 2 diabetes, osteoarthritis, COPD and depression – who smokes and has a body mass index (BMI) of 29 kg/m2. NICE guidelines recommend offering 11 medications as a minimum, with up to 10 other drugs if risk factor control or symptom control is inadequate. Guidelines also advise her to routinely engage in nine lifestyle alterations or self-care programmes, attend four to six GP appointments for routine planned follow-up and attend 8–30 smoking cessation, psychosocial intervention and pulmonary rehabilitation appointments if she accepted referral. 3 Not unexpectedly, therefore, polypharmacy is one important potential consequence of guideline recommendations in people with multimorbidity. 2,3
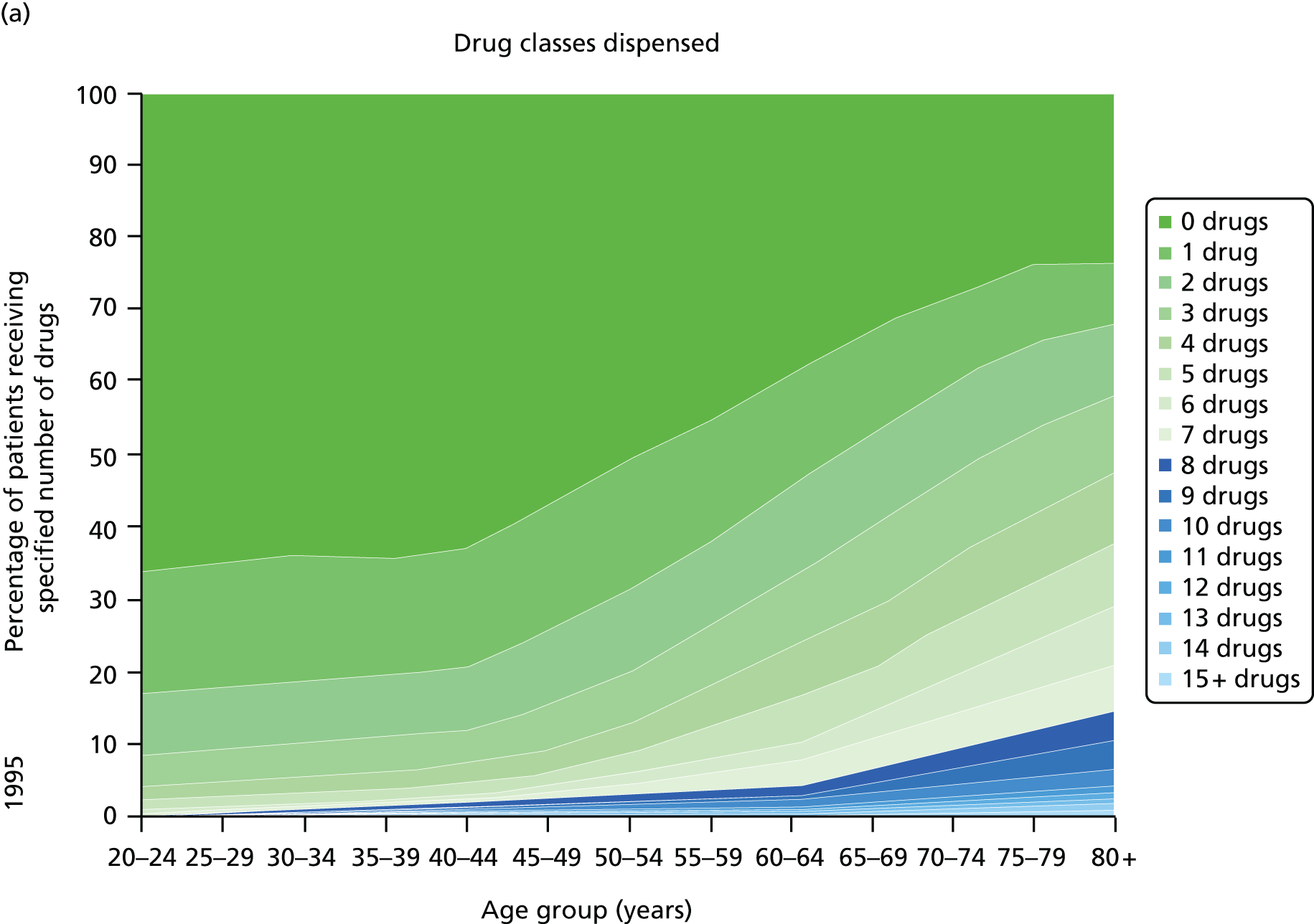
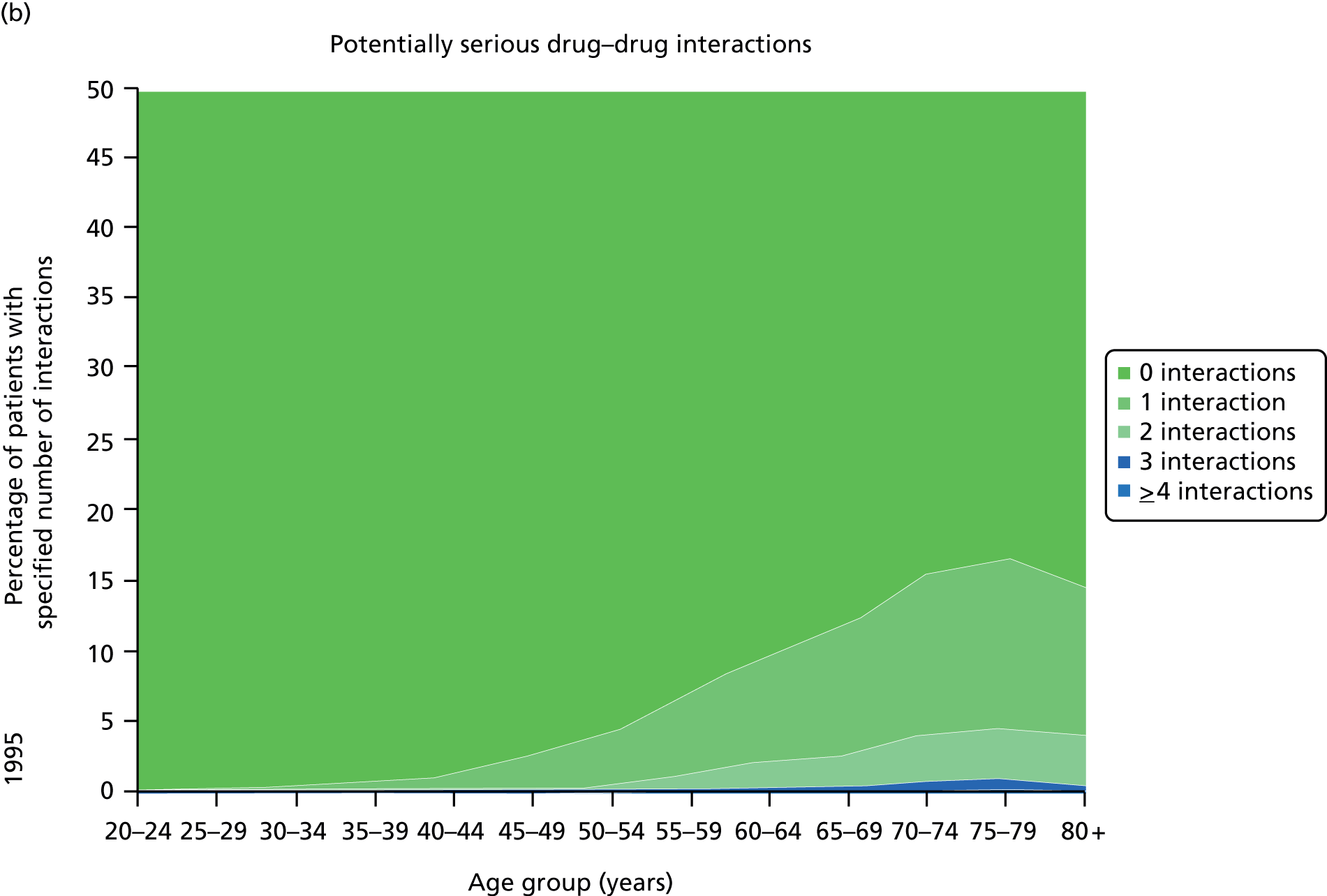
Figure 3 shows the number of pharmacologically active medications dispensed to all ≈ 310,000 adult residents of the NHS Tayside region of Scotland in 1995 and 2010 stratified by age, and the proportion of each age group prescribed pairs of drugs with a ‘potentially serious’31 (p. 852) drug–drug interaction as defined by the British National Formulary (BNF). 31,32 The proportion of adults dispensed ≥ 5 drugs (the most common definition of polypharmacy) rose from 11.5% to 20.8% between 1995 and 2010, and the proportion dispensed ≥ 10 tripled to 5.8%. The number of drugs dispensed rose markedly with age. The proportion of adults with potentially serious drug–drug interactions rose from 5.8% in 1995 to 13% in 2010, and the number of drugs dispensed was the characteristic most strongly associated with this (10.9% if dispensed two to four drugs vs. 80.8% if dispensed ≥ 15 drugs). 32
FIGURE 3.
Changes in polypharmacy and ‘potentially serious’ drug–drug interactions between 1995 and 2010 in a stable geographic population. (a) Drug classes dispensed 1995; (b) potentially serious drug–drug interactions; (c) drug classes dispensed 2010; and (d) potentially serious drug–drug interactions. Reproduced from © 2015 Guthrie et al. 32; licensee BioMed Central. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited.
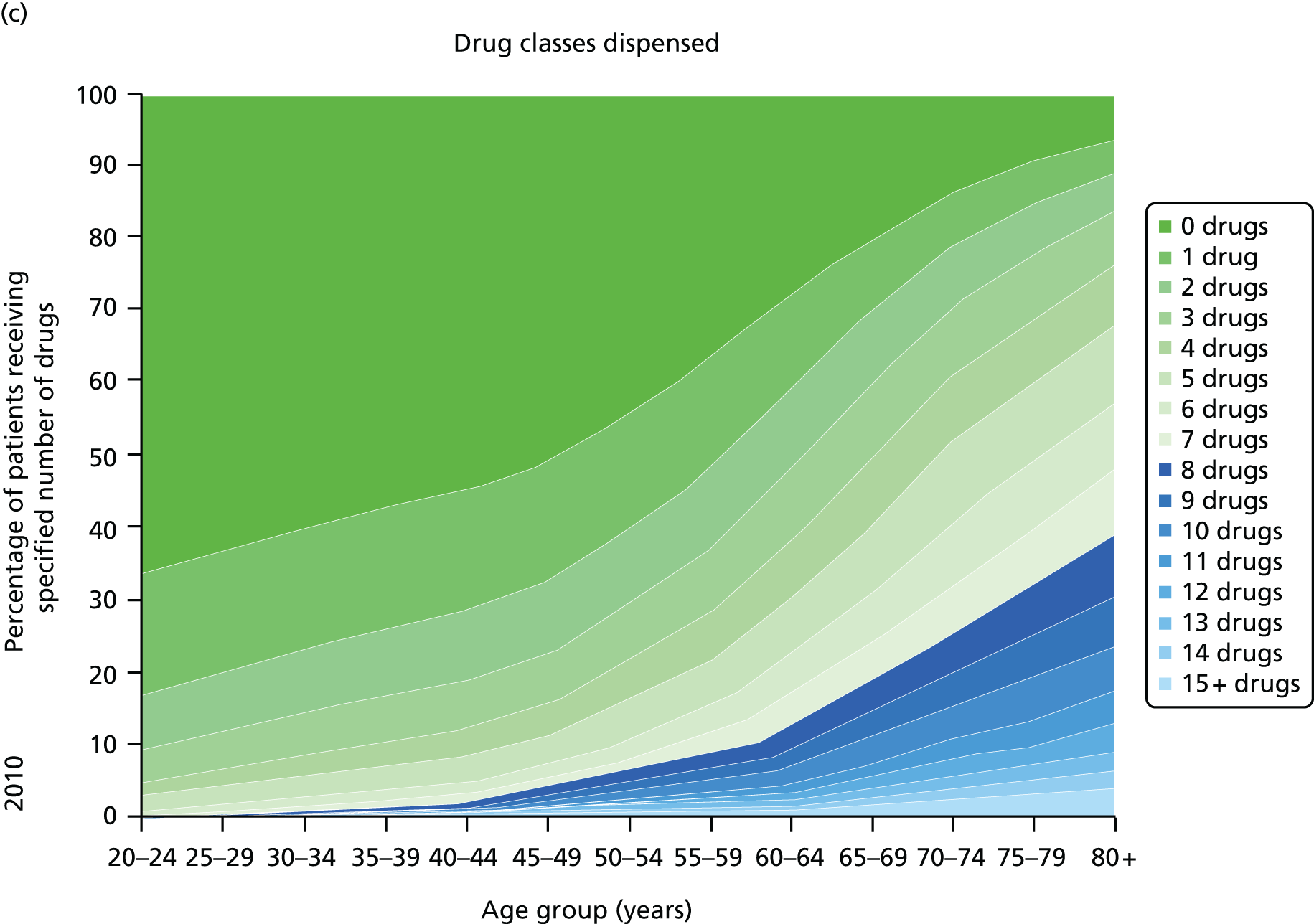
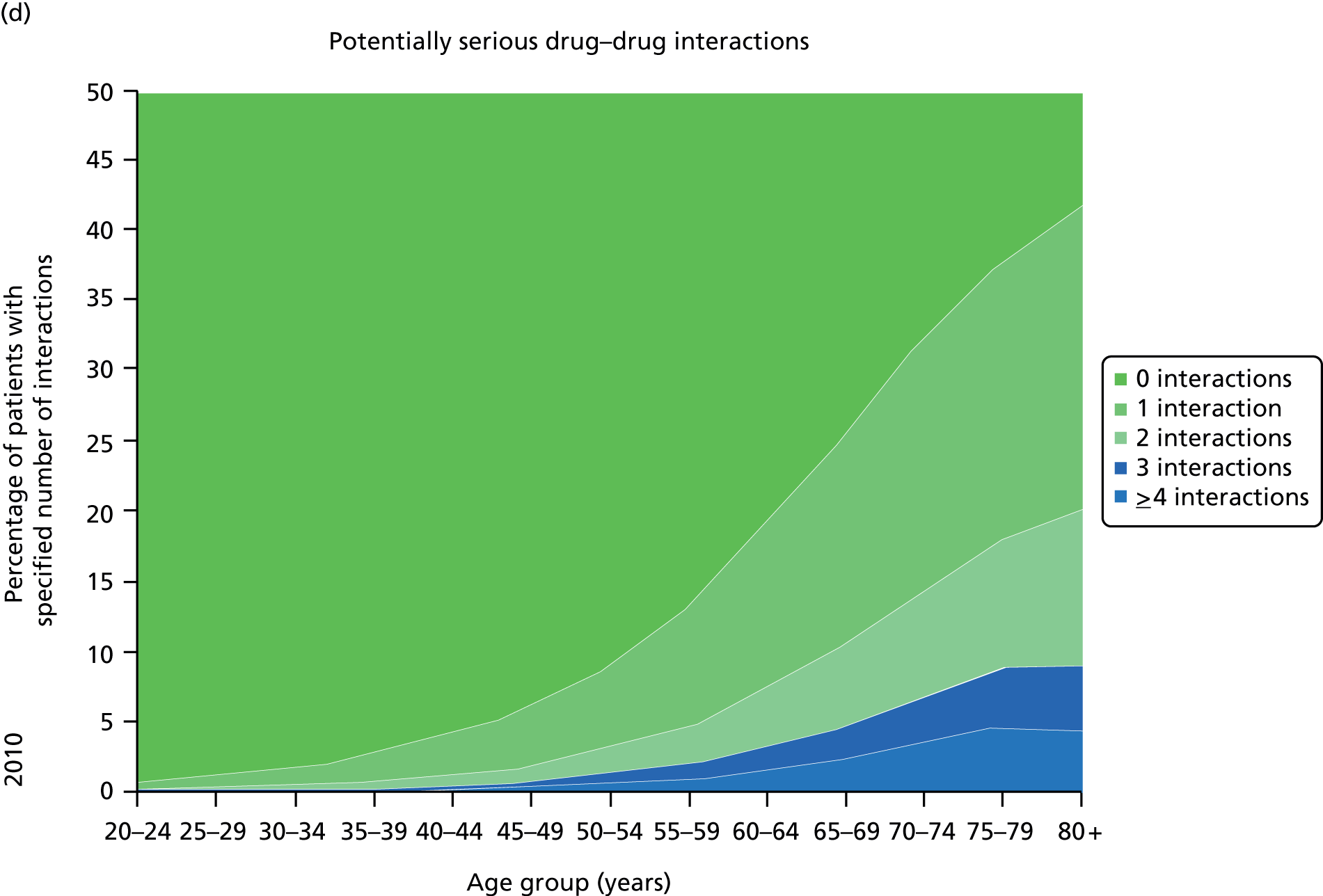
Guidelines are only one driver of increasing polypharmacy, and it is important to recognise that polypharmacy in itself is not inappropriate. However, polypharmacy is often a problem because it is consistently associated with high-risk or potentially inappropriate prescribing33–35 and increased treatment burden. 20 To the extent that guideline recommendations contribute to increasing polypharmacy and its associated risks, it is noteworthy that single-disease guidelines rarely systematically consider the applicability of trial evidence to individuals with multimorbidity and limited life expectancy who may have little likelihood of benefit from long-term preventative treatment. 28,36
Systematic examination of how guidelines account for multimorbidity
The Boyd et al. 2 and Hughes et al. 3 studies referred to above are based on carefully selected vignettes, chosen to emphasise the more extreme implications of following guideline recommendations in people with multimorbidity. However, more systematic examinations of published CGs also show they at best only partly account for multimorbidity. Lugtenberg et al. 37 examined 20 published guidelines for four common conditions (COPD, depressive disorder, type 2 diabetes and osteoarthritis). The study found that, although 85% made some mention of comorbidity and there was a mean of three treatment recommendations with a specific variation in the presence of a comorbidity, most such variations were in relation to single concordant15 comorbidities (e.g. a glycaemic control treatment recommendation in people with acute MI). Vitry and Zhang6 examined 17 Australian CGs and found that 53% made at least one specific recommendation for people with one comorbid condition, but only 12% did the same for people with several. 6 The time required to benefit from treatment in the context of limited life expectancy was discussed in only 18%, and treatment burden in 29%. A study of 16 Canadian guidelines using the same methods had similar findings, although more guidelines (94%) made at least one specific recommendation for people with one comorbid condition. 7 Cox et al. 38 examined 14 Canadian guidelines with similar results in terms of the proportion of guidelines making at least one ‘comorbidity’ recommendation. Twelve of the 14 guidelines also provided one or more age-varied recommendations for those aged 65–79 years, and five for those aged ≥ 80 years. However, of the total 1189 recommendations made, only 33 (2.8%) and 7 (0.6%) were actually age varied for these two age groups, consistent with a rather more limited accounting for multimorbidity than implied by counting any mention of comorbidity in an entire guideline.
Single-disease guidelines explicitly designed to account for multimorbidity
A small number of guidelines have been specifically created to account for multimorbidity, with two broad types. The first type provides general guidance on how to manage people with multimorbidity. The American Geriatrics Society (AGS) guiding principles for the care of older people with multimorbidity is the most comprehensive published example,39–41 and the NICE multimorbidity CG that is in development also takes this approach. 42 However, the focus of this project is accounting for multimorbidity in single-disease guidelines, so these are not directly relevant. The second type is single-disease guidelines that explicitly aim to account for comorbidity, notably the California HealthCare Foundation and International Diabetes Federation guidelines on diabetes care in older people,43,44 and the NICE guideline on the management of depression in adults with a chronic physical health problem. 17
The California HealthCare Foundation guideline for diabetes care in older people has several features that distinguish it from whole-population diabetes guidelines. It more explicitly considers the applicability of trial evidence to older people, and clearly states that many recommendations should be applied to all or most older people. However, it specifically varies some recommendations in people with significant comorbidity, frailty or reduced life expectancy because of consensus in the GDG that benefits are less likely to be realised and harms are more likely to happen. Finally, it also includes specific reference to geriatric syndromes such as falls and urinary incontinence because these are common in older people and may be precipitated or exacerbated by diabetes treatment. 43 For example, based on consensus, the recommendation in relation to the treatment target for glycated haemoglobin is:
For older persons, target hemoglobin A1c (A1C) should be individualized . . . For frail older adults, persons with life expectancy of less than 5 years, and others in whom the risks of intensive glycaemic control appear to outweigh the benefits, a less stringent target such as 8% is appropriate.
California HealthCare Foundation43
The International Diabetes Federation guideline takes this stratification of recommendations one step further, by making distinct recommendations for older people who are functionally independent, who are functionally dependent (with impairments in activities of daily living, with further variation for two subgroups of the functionally dependent: people who are frail and people with dementia) and who are at the end of life. Using the example of glycated haemoglobin again, the recommended target levels are 7.0–7.5% for the functionally independent, 7.0–8.0% for the functionally dependent without frailty or dementia, ≤ 8.5% for people with frailty or dementia, and treating only if symptomatic at end of life. In addition, recommended treatments are varied, with increasing emphasis on avoiding hypoglycaemia as frailty increases and life expectancy decreases. 44
In contrast, the NICE guideline on depression in adults with a chronic physical health problem17 largely copies recommendations from the single-disease depression guideline, which was developed in parallel, although with several important modifications. 45 Recommendations are varied where the evidence in people with a chronic physical problem is different (e.g. some psychological interventions are not recommended) and physical condition treatment recommendations are added where there is some evidence of impact on depression (e.g. peer support focused on physical condition). The guideline also provides much more detailed advice on drug–drug interactions, reflecting the fact that coprescribing of physical health drugs will be much more common, with risks of adverse drug events (ADEs).
A feature of all three of the modified single-disease guidelines is that general-population recommendations are varied for important subgroups (older people, people with physical comorbidities). This is either because there was an available evidence base in the subgroup, which informed variation in recommendations, or more commonly because it was the considered consensus judgement of the GDG that a variation was appropriate. However, although these guidelines provide models for how recommendations might be varied, they do not clearly state a more general process by which a GDG could decide when and how it might be appropriate to vary or qualify recommendations.
Published guidance on how to account for multimorbidity in guidelines
Since the original proposal was written, two papers have been published addressing how single-disease guidelines could account for multimorbidity. In the context of respiratory disease, Fabbri et al. 46 suggested that guideline developers needed to:
-
explicitly consider whether or not the trials underpinning treatment recommendations included people with the most common comorbidities, and if there is usable information from subgroup analyses
-
use expected absolute risk reduction (ARR) to inform treatment decisions, or at a minimum consider the range of variation in absolute benefit based on variation in baseline risk and competing risk of death from other causes
-
specify the actual outcomes that each therapy improves, since individual patients may vary in their preferences for which outcome matters most to them
-
present data on the average and extremes of length of therapy required to achieve expected absolute benefit, because ‘time to benefit from therapy’46 is essential in decision-making in patients with competing risks, who may have reduced life expectancy
-
address interactions that are common or important given the prevalence of specific comorbidities. 46
More recently, Uhlig et al. 47 proposed a more comprehensive framework for guideline development to account for multimorbidity, which specifically identified when during guideline development these issues should be addressed and how the strength of recommendations might vary as a result. 47
Of interest is the considerable overlap between both of these frameworks and the work that was proposed for this study, including in relation to applicability of evidence, interactions, the importance of absolute benefit to compare across guidelines, and the temporal dimension of benefit in the context of short life expectancy. A key difference is that both papers take the form of general guidance and do not examine how these elements could be feasibly integrated into guideline development. For example, time to benefit (TTB) is a very appealing idea, but is difficult to operationalise meaningfully. The focus of the Better Guidelines project was therefore on how existing guideline development could be practically altered to address the problems identified by previous research.
Aims and objectives
The aim of this project was to test the methodological feasibility of new approaches to summarising and creating evidence for guidelines for the management of people with multimorbidity.
There were two specific objectives, although the intention was to be guided by the project reference group (PRG) in terms of focusing on what they perceived to be the main problems requiring addressing and in response to emerging findings:
-
to systematically collate and summarise the evidence of benefit, harm and cost-effectiveness for guideline recommendations for three common conditions, including where recommendations are mutually reinforcing or contradictory, in order to examine the value and feasibility of making existing evidence and guideline recommendations more useful for people with multimorbidity
-
to develop and evaluate exploratory modelling methods to estimate expected benefit, TTB, risk and health-care costs for people with selected multiple conditions, in order to examine the value and feasibility of new approaches to evidence creation for guidelines for people with multimorbidity.
Project management and public involvement
The project was designed to be conducted collaboratively with coapplicants from NICE and collaborators from SIGN, with the intention of being focused on work that could plausibly be incorporated into existing guideline development. As well as having a project team and study steering committee (SSC), we therefore also convened an expert PRG. The PRG comprised professional and public members with experience of guideline development who worked with us to interpret findings and to ensure the project focused on issues important to the practicalities of guideline development. In the original proposal, the PRG was described as a GDG but NICE requested a name change to make clear that NICE was not bound by its decisions in the way that it is by GDG decisions. The roles of these three groups were:
-
A project team consisting of the coapplicants plus a collaborator from SIGN. The project team had responsibility for delivery of the project.
-
A PRG, which provided expert advice to help select key recommendations from the three exemplar guidelines studied for more detailed examination in regard to both objectives, and informed the economic modelling. The PRG was particularly concerned with prioritising work that would most contribute to dealing with what the group perceived to be the main problems posed by multimorbidity in guideline development, and therefore that was most likely to improve how guideline development is done. The independent chairperson of the PRG additionally contributed to a number of project team meetings, ensuring that PRG perspectives were represented in project delivery. PRG members are listed in the acknowledgments at the end of the main text.
-
A SSC with an independent chairperson and a public member: the composition of the SSC was agreed with the National Institute for Health Research (NIHR), and was smaller than is usual given that the study did not involve any intervention and because of the existence of the PRG. The SSC provided oversight of study progress and adherence to the original proposal, although it was sensitive to the guidance on focus from the PRG. SSC members are listed in the acknowledgements at the end of the main text.
The PRG was recruited from professionals and members of the public who had experience of guideline development through serving on one or more NICE or SIGN GDGs or having other involvement in NICE or SIGN guideline development. We initially recruited the chairperson and the 10 members through an advert circulated to previous NICE GDG members, and interviewed applicants using NICE procedures for recruitment to GDGs including defining the role and number of public members. All the professional members were recruited this way, but no public members applied. We therefore recruited two public members through an advert circulated to previous SIGN GDG or SIGN Patient Network members. The PRG met for four 1-day meetings during the project, and provided guidance on the direction and focus of the project, as well as advice on the interpretation of findings. One or both of the two public members attended every PRG meeting. They played an active role in PRG discussions about interpretation of findings and decisions about the overall direction and focus of the project. They particularly contributed to discussion about the evidence base for non-pharmacological compared with pharmacological interventions, the value of absolute benefit and how to communicate it to patients, and understanding the implications of applicability.
The methods used for each of these are described in subsequent chapters, but, in brief, although all coapplicants and collaborators contributed to all aspects of the project:
-
objective 1 was predominantly the responsibility of the University of Dundee, and the method used was examination of guidelines, the relevant research literature and epidemiological data relating to the three exemplar conditions. The PRG was particularly interested in absolute benefit, which was one of the key features of the original proposal, and the applicability of evidence, which was present but less prominent in the proposal. The latter is specifically addressed in Chapter 3
-
objective 2 was predominantly the responsibility of the University of Manchester, and the methods used were examining modifications of existing economic models, and de novo economic modelling informed by formal elicitation methods. The PRG was particularly interested in the temporal dimension of benefit and this is specifically addressed in Chapter 5.
Almost all elements of the study either were literature based or involved modelling, and therefore did not require ethical review. The exception was the elicitation study, which was reviewed by the University of Manchester Research Ethics Committee 3, which raised no objection to its conduct (reference AJ/Ethics/1809/13). Chapter 2 reports the initial scoping of guidelines for three exemplar conditions to examine the extent to which they accounted for multimorbidity, the extent to which recommendations were underpinned by economic evidence and synergy, contradiction and interaction between their recommendations. Chapter 3 examines the applicability of randomised controlled trial (RCT) evidence to the population that has the condition actually treated with recommended drugs. Chapter 4 explores how the absolute benefit of treatments with conditions could be compared, both for clinical outcomes and for absolute quality-adjusted life-year (QALY) gain. Chapter 5 discusses the temporal dimension of benefit, and examines how existing economic models could be used to make this more explicit to GDGs through ideas such as the pay-off time and QALY profiles. Chapter 6 details a model-based discrete event simulation (DES) cost-effectiveness analysis (CEA) which accounts for both depression and CHD including potential harms from drug–drug interactions. Finally, Chapter 7 summarises the findings and their implications.
Chapter 2 Initial work examining how three exemplar guidelines accounted for multimorbidity
Background
The work reported in this chapter particularly relates to objective 2, and specifically to:
-
cross-referencing recommendations in the guidelines for the three exemplar conditions to identify where there is synergy (recommendations are consistent or reinforce each other), inconsistency (recommendations do not agree or there are potentially serious interactions) or contradiction (recommendations contradict each other)
-
examining the extent to which economic evidence informs recommendations in the guidelines for the three exemplar conditions, to improve understanding of how new uses for existing economic models or new types of economic model would fit with existing guideline development.
The National Institute for Health and Care Excellence guideline development process
Although GDGs have considerable discretion in how they interpret evidence and what recommendations they write, NICE requires them to work according to a well-defined process specified in the Guidelines Manual:48
-
Topics are referred to NICE by stakeholders such as the English Department of Health, or may be identified internally in terms of existing guidelines requiring updating.
-
A draft guideline scope is created and finalised after stakeholder consultation. For guideline updates, the scope may cover the whole guideline or only selected aspects of it. The populations and settings that will be covered are defined by the scope, which also identifies the key issues and questions that will be considered.
-
A suitably representative GDG is convened, which agrees the clinical research questions (CRQs) required to cover the scope and which CRQs will be prioritised for new economic modelling to examine cost-effectiveness. Relevant evidence for these questions is systematically searched for and synthesised, and additional evidence from stakeholders is sought if required. Evidence syntheses are discussed by the GDG, which uses a deliberative and consensus process to formulate recommendations that are structured and worded based on a common framework. Recommendations can be both for care (diagnosis, treatment, follow-up, service organisation) and for future research to inform future guidelines.
-
A draft guideline is published for stakeholder consultation and amended as required before final publication. There are a number of versions of guidelines produced, and the format of these has changed over time. Currently, NICE publishes both a NICE guideline and a full guideline. The NICE guideline essentially lists the recommendations and priorities for implementation. The full guideline is a much larger document and details the evidence and the GDG discussion that connects evidence to the recommendations made. Historically, a quick reference guide was also usually published, but this has since been replaced with a web-based NICE pathway. The NICE guideline and the quick reference guide or NICE pathway are the documents that clinicians and patients are most likely to use, with the full guideline serving as a reference document detailing how the guideline was created.
Three exemplar guidelines
Analysis focuses on exemplar NICE guidelines for type 2 diabetes (CG6649 and its partial update CG8750), depression in adults (CG9045 and the accompanying guideline for depression in adults with a chronic physical health problem CG9117) and chronic heart failure (CG10851). Where necessary, other guidelines are also examined, as for the interactions work below, which required a broader perspective, and for some of the economic modelling, where we were constrained by model availability. We chose these three conditions because they:
-
are individually important because they are common and are associated with a large burden of population morbidity and health service resource use
-
are commonly comorbid with each other, and people with each condition commonly have a range of other comorbidities in addition (Figure 4)
-
had a recent NICE guideline with economic modelling carried out for at least some of the individual treatment recommendations
-
included both physical and mental health conditions where co-occurrence is known to worsen outcomes of both conditions,52 and where there are some published trials of the effectiveness of treatment of physical and mental health conditions in the presence of the other53,54
-
included a physical condition where treatment is primarily long-term and preventative (type 2 diabetes) and one where life expectancy is much reduced but treatment has a major benefit over short periods [heart failure, particularly moderate/severe left ventricular systolic dysfunction (LVSD)].
FIGURE 4.
Common comorbidities of the three index conditions compared with other conditions. Source: based on data from the study reported by Barnett et al. 13 TIA, transient ischaemic attack.
Structure of this chapter
This chapter describes initial work examining the NICE guidelines for the three exemplar conditions, in terms of:
-
examining and cross-referencing CRQs and treatment recommendations
-
quantifying the extent to which guideline development is informed by any economic evidence, and specifically by new economic analysis
-
quantifying the potential for drug–drug and drug–disease interactions in people with multimorbidity.
The background, methods and findings for each of these is presented in turn with a short summary, and the implications of all the findings are discussed at the end of the chapter.
Examining and cross-referencing treatment recommendations in the three exemplar guidelines
Background
As described in Chapter 1, a number of commentators have identified that multimorbidity poses challenges to guideline developers,4,5 and several studies have described situations where cumulative guideline recommendations might be irrational or troublesome. 2,3 More systematic examination of guidelines from a range of international sources has shown that, although most guidelines do qualify at least some recommendations for people with comorbidity, this is relatively infrequent and usually relates to concordant comorbidity, although some guidelines account at least partly for comorbidity by making recommendations for older people, in whom comorbidity will be more common. 6,7,37,38 However, guideline developers may be reluctant to routinely make age-qualified recommendations in order to minimise unconsidered non-treatment of older people, particularly given that age is now a protected characteristic in the UK under the Equality Act 2010. 55
We therefore systematically examined treatment recommendations made by guidelines for the three exemplar conditions, specifically in relation to:
-
the extent to which comorbidity or specific age groups were accounted for in the CRQs
-
the extent to which statements about the recommended treatments accounted for comorbidity or specific age groups
-
the extent to which the research recommendations that were made accounted for comorbidity or specific age groups.
Methods
We examined relevant guideline documents for our three exemplar conditions as previously described, including those publicly available on the NICE website and (in the case of the CRQs) internal NICE documents listing these, since they are not explicit in all published guideline documents. Although in principle it is straightforward to count how often CRQs or recommendations accounted for comorbidity, several problems were encountered in practice. First, guidelines are evolving documents, which are often partially rather than fully updated. This means that at any one moment a guideline may contain unchanged recommendations copied in from a previous version and recommendations amended or created in the light of new evidence. Mapping which CRQs are relevant to any one guideline is therefore not always straightforward. For example, the CG87 type 2 diabetes guideline was a partial update of CG66, meaning that many recommendations in it are copied in from CG66. The CRQs for six guidelines were therefore examined (CG66 and CG87 for type 2 diabetes; CG90 and CG91 for depression; and CG05 and CG108 for heart failure). Of note is that the two depression guidelines were unusual in that they were commissioned at the same time and developed in parallel. It therefore makes no real sense to consider them separately because CG90 (depression in adults) effectively accounts for comorbid physical health problems even if it never explicitly mentions them because it cross-references CG91 (Depression in Adults with a Chronic Physical Health Problem,17 which to our knowledge is the only published NICE guideline specifically addressing commonly occurring comorbidity). Second, counting how often guidelines account for comorbidity is complicated because it is not always clear what constitutes a recommendation. NICE ‘recommendations’ are numbered, but any single numbered piece of text may contain a number of separate statements recommending different aspects of care, and recommendations for the use of any single treatment (e.g. metformin in type 2 diabetes) may be defined across multiple numbered recommendations. For the purposes of this analysis, we therefore examined recommended treatments defined as pharmacological and non-pharmacological interventions of various kinds, and considered whether or not comorbidity was accounted for in these treatments across all relevant numbered recommendations.
Similarly to previous studies,6,7,37,38 three reviewers first examined the three most recent guidelines for the exemplar conditions (CG87, CG108, CG90) to classify text into discrete recommendations, and classified these recommendations in terms of whether or not they related to treatment. For each recommendation, the same reviewers then made a judgement about:
-
whether or not comorbidity was addressed in the way the recommendation was worded:
-
without cross-reference to other NICE guidance and unlinked to a recommended treatment
-
by cross-reference to other NICE guidance and unlinked to a recommended treatment
-
in relation to a recommended treatment
-
-
when comorbidity was addressed in the way the recommendation was worded, the proportion of times that this was in relation to a concordant comorbidity as defined by Piette and Kerr15
-
whether or not use of a recommended treatment in older people was addressed. Although age and comorbidity are not the same constructs, comorbidity is much commoner in older people, so we believed it relevant to consider qualifications by age as at least partial proxies for comorbidity38
-
whether or not use of a recommended treatment was addressed in relation to life expectancy. Although life expectancy can be shortened by single conditions, multimorbidity is commonly perceived to be important partly because it is associated with reduced life expectancy independent of single-condition severity46,47
-
explicitly contradictory treatment recommendations, which we defined as the same treatment stated to be indicated in one guideline and contraindicated in another, and explicitly synergistic treatment recommendations, which we defined as the same treatment stated to be indicated in more than one of the three guidelines. This is examined across more guidelines in the interactions analysis later in this chapter
-
synergistic treatment recommendations.
Findings
Clinical research questions
The CRQs examined rarely explicitly included any consideration of comorbidity or relevant subgroups, such as older people, in whom comorbidity is very likely. None of the 60 CRQs for the diabetes guidelines (40 in CG66 and 20 in CG87) made any mention of comorbidity or relevant subgroups. Two of the 19 CRQs in CG90 (depression in adults) were specifically framed in terms of older adults:
CRQ 090-12: What is the clinical effectiveness of pharmacological/physical interventions in the treatment of depression in older adults?
CRQ 090-13: What is the clinical effectiveness of other pharmacological and physical management of depression for people (older adults) who have not adequately responded to treatment, and relapse prevention?
CG9045
However, all 11 of the CRQs in CG91 (depression in adults with a chronic physical condition) were by definition framed in terms of comorbidity. Three of the 32 CRQs in CG05 or CG108 (heart failure) accounted for comorbidity either explicitly (cross-referencing to other NICE guidance for the management of comorbid anxiety and comorbid depression) or implicitly (consideration of when to start end-of-life care; consideration of drugs for other conditions to avoid in heart failure). A further CRQ related to whether or not there were subgroups of patients with heart failure who should be treated differently, although it did not indicate which subgroups were of interest. None of the 12 CRQs in CG108 (heart failure) explicitly considered comorbidity.
Recommended treatments
All three guidelines made at least one general statement that one or more comorbidities should be considered as part of care for the index condition, although without any specific advice about how this should be achieved (Table 1). All made at least one cross-reference to specific NICE guidance. The diabetes guideline referenced only the depression guideline, whereas the heart failure guideline referenced guidelines for depression and three concordant physical conditions (hypertension, MI, type 2 diabetes). The depression guideline referenced NICE anxiety guidance.
| If and how comorbidity was addressed | Type 2 diabetes (CG8750) | Heart failure (CG10856) | Depression (CG9045)a |
|---|---|---|---|
| Number of recommended treatments | 18 | 13 | 8 |
| Comorbidity addressed without cross-reference to other NICE guidance and unlinked to a recommended treatment | Two statements (one relating to specialist care for complications, which is not covered, the other to care for people with physical, sensory or learning disabilities) | Two statements (one saying that comorbidity should be managed according to relevant NICE guidance, the other referring to assessment of cognitive ability when sharing information) | One statement about assessing depression in the context of comorbid physical and mental disorders |
| Comorbidity addressed by cross-reference to other NICE guidance and unlinked to a recommended treatment | Depression | Depression, hypertension, MI, type 2 diabetes | Anxiety |
| Comorbidity addressed in relation to a recommended treatment | Eight specific examples | Seven specific examples | All recommendations effectively qualified by the presence of a chronic physical health problem (cross-referenced to CG9117)a Seven other specific examples |
| Proportion for which these were for concordant conditions | 50% (four of the specific examples) | 71% (five of the specific examples) | 14% (one of the specific examples) |
| Use of a recommended treatment in older people addressed | Three specific examples all related to choice of hypoglycaemic therapy | Two specific examples, one providing general advice about prescribing in older people, the other related to use of beta-blockers | Two specific examples related to using age-appropriate dosing, accounting for general physical health and coprescribing, and being cautious in the use of electroconvulsive therapy in older people |
| Use of a recommended treatment in relation to life expectancy addressed | No | No (although palliative care recommended where appropriate) | No |
| Explicitly contradictory treatment recommendations | None | None | None |
| Synergistic treatment recommendations | Two synergistic with heart failure relating to ACE inhibitor and aspirin use | Two synergistic with type 2 diabetes relating to ACE inhibitor and aspirin use | None |
Summary
All three guidelines had a number of specific qualifications in treatment recommendations for people with comorbidity. There were eight such examples in the type 2 diabetes guideline, of which four were for concordant conditions, and seven in the heart failure guideline, of which five were for concordant conditions. All of the treatment recommendations in the main depression guideline (CG90) were effectively qualified by the presence of a chronic physical health problem by cross-reference to CG91, and there were seven other specific examples, of which only one was for a concordant condition (see Table 1).
The diabetes guideline had three treatments where the recommendation to use was qualified for older people (all in relation to the risk of hypoglycaemia with some drugs). The heart failure guideline had two qualified treatment recommendations (one providing general advice about dosing and adverse effects of recommended drugs in older people, the other emphasising that age was not a restriction to beta-blocker use). The depression guideline had two qualified treatment recommendations (one providing general advice about dosing of recommended drugs and interactions related to physical health conditions in older people, the other relating to risks of electroconvulsive therapy and associated anaesthesia). None of the recommended treatments was qualified in relation to life expectancy (although the heart failure guideline stated that palliative care was central to the care of many people with heart failure). There were no explicitly contradictory recommendations across the three guidelines. There were two recommended treatments where the diabetes and heart failure guidelines were considered to be synergistic in that both recommended the same drugs, with closely related beneficial outcomes in at least a subset of people with each condition.
Research recommendations
None of the 14 research recommendations in the two type 2 diabetes guidelines related to comorbidity or age. None of the 10 research recommendations in the standard depression guideline, CG90, related to comorbidity or age, whereas all seven in CG91 (depression in people with a chronic physical health problem) were framed in terms of comorbidity. Specifically, two were identical research recommendations to those made in CG90 but qualified for the subgroup of people with a chronic physical health problem, four were specific to people with a chronic physical health problem and one was specific to people with COPD. None of the five research recommendations in the heart failure guideline related to comorbidity or age.
Our analysis is broadly similar to previous studies in this area. 6,7,37,38 Comorbidity and older age were not part of the routine framing of NICE CRQs (although this in itself does not prevent GDGs from asking for subgroup analysis or considering subgroups). Relatively few recommendations were qualified in terms of comorbidity or older age (although the number of such qualifications was higher than found in previous research, which could mean either that NICE guidelines better account for comorbidity or that there has been a broader secular trend in such accounting). However, the qualification was usually relatively narrow. For example, the type 2 diabetes guideline qualified some hypoglycaemic drug recommendations in relation to the risk of symptomatic hypoglycaemia caused by some drugs, but did not specifically qualify the general recommendation to achieve tight glycaemic control. In other words, although this recommendation does say that less tight glycaemic control targets may be appropriate in some patients, it gives no guidance about which patients. In practice then, the qualification made was about drug choice, not overall treatment goals, and it is the latter that are likely to drive intensity of treatment and therefore treatment burden.
The potential implications of short life expectancy was not addressed in any guideline, although in practice this would be most relevant in conditions where recommended treatments accrue benefit over a long time, such as type 2 diabetes and to a lesser extent heart failure. Across these three guidelines, there was little clear synergy and no clear contradiction between recommended treatments. Relative contraindication in relation to drug–drug and drug–disease interactions is examined later in the chapter.
Finally, none of the research recommendations in the single-condition guidelines explicitly called for research in population subgroups such as people with comorbidity or older people, although CG91, on depression in adults with a chronic physical health problem, did make a number of such research recommendations.
Use of economic data in guidelines for the three exemplar conditions
Background
What is economic evidence?
Economic evaluation is increasingly seen as important in the health-care setting, facilitating the allocation of scarce resources in the most efficient manner by taking account of opportunity cost. 57 In the UK, model-based CEA has become the preferred vehicle and method of economic evaluation to generate evidence on incremental costs and QALYs. 58–61 Model-based CEA is now routinely used in a diverse range of countries in the evaluation of technologies,62 such as pharmaceuticals and diagnostics, and also public health interventions. 59,63,64 In contrast, the use of information on resource use and cost data to inform CG creation is controversial and remains fairly limited. 65 A notable exception is the production of CGs by NICE, in the UK, where guideline developers are required to take account of the available evidence on the relative cost-effectiveness of interventions before recommendations can be made. 66
Economic evidence in the National Institute for Health and Care Excellence guideline development process
The National Institute for Health and Care Excellence requires that economic evidence be incorporated into GDG decision-making where possible, typically in the form of model-based CEA. 63 For CGs, this explicit social value judgement requires that decision-makers must take into account evidence on the incremental costs and benefits of an intervention as well as its possible effectiveness or harms. 67 The level of influence of CEA on the CG production process at NICE is unique among large guideline-producing bodies and, combining this with its reputation for rigour and transparency, NICE is recognised as a world leader in guideline methodology and development. 65 The NICE Guidelines Manual defines how economic evidence is used in guideline development:48
-
The requirement to produce economic evidence related to one or more CRQs is considered by NICE early in the scoping process. The importance of prioritisation of effort is recognised within the Guidelines Manual, as evidence on cost-effectiveness from the literature will rarely be of sufficient quality and relevance for decision-making for the NHS, and in an ideal world many of the CRQs would be subject to further analysis. 48 NICE takes the view that a new economic analysis to inform CGs is most useful when the topic (1) is important, which is a function of the population and the expected incremental costs and benefits, and (2) has a high level of uncertainty in the current evidence base, with the likelihood that a new analysis will reduce this uncertainty. If a CRQ is deemed to be important by the GDG and further analysis is likely to reduce current uncertainty, then de novo modelling is likely to be prioritised. However, no formal analysis to understand the potential value of generating new evidence is conducted as part of the guideline development process, such as estimating the expected value of perfect information, which is becoming embedded in research prioritisation decisions made by NIHR as part of the Health Technology Assessment (HTA) programme. 68
-
During initial GDG meetings, CRQs are agreed that describe key decision points in the patient care pathway that will be covered by the CG. These are usually framed in the PICO (population, intervention, comparator, outcome) format, for example as ‘in patient group X in the treatment of condition Y, what is the clinical effectiveness of drug A compared with drug B in relation to total mortality?’. In parallel, an economic plan is produced that lists CRQs prioritised for economic evaluation, using either a systematic review of published economic analyses or de novo analysis using a model-based CEA. Model-based CEAs tend to use decision trees, Markov models or DES. 69 The specific methods used to produce the de novo analysis are described in the NICE ‘reference case’, which specifies how economic analysis should usually be done, with departures from the reference case requiring justification. 48,59 NICE uses a common reference case to ensure continuity between the evidence considered by committees of decision-makers for technology appraisal, diagnostic assessment and guideline development. Consequently CEA is the preferred form of economic evidence, in which costs are quantified in keeping with the perspective of the NHS and the QALY is used as the preferred measure for patient benefit. 59
-
Ultimately, decisions on recommendations included in the CG are made as a collective view of the GDG reached during a deliberative process. The economic evidence (either from systematic reviews of published studies or from a de novo analysis) is presented within GDG meetings alongside evidence on effectiveness, harms and risks. After discussion of the evidence and clarification of where there is uncertainty in the evidence base, recommendations are drafted and their wording fine-tuned. When economic evidence for a particular CRQ is not available and the CRQ has not been prioritised for synthesis of economic evidence by the health economist, the GDG will make a qualitative judgement of the cost-effectiveness of a particular recommendation.
The intention, and feasibility, for NICE to use evidence of the relative cost-effectiveness of interventions within CGs is limited by two factors: (1) the availability, quality and applicability of cost-effectiveness evidence within the existing literature; and (2) the time and resources required to produce de novo evidence of relative cost-effectiveness. Whereas health technology assessments with a tight clinical focus usually have de novo CEA to support decision making, CGs do not normally have such evidence for all CRQs. However, the type and extent of evidence of relative cost-effectiveness evidence used to inform the clinical questions within a CG has not been examined before.
A key overall aim of the Better Guidelines project was to assess economic evidence within exemplar CGs produced by NICE. The aim of this specific study was to assess the extent to which economic evidence was currently available for consideration by NICE GDGs for the three exemplar conditions where comorbidity is common.
Methods
The use of economic evidence within guideline development was evaluated by one researcher (AT), who searched systematically through the published full guideline versions of CG66 (type 2 diabetes),49 CG90 and CG91 (depression)17,45 and CG108 (heart failure)51 to identify CRQs and match them to relevant sections of the full guidelines and economic plans. Three strategies were used to establish the extent of the economic evidence used. This was necessary because, although CRQs are central to guideline development, the published full guidelines are not necessarily explicitly structured in the same way. The full guideline layout has also changed over time and varies between the national collaborating centres that produce them:
-
For each CRQ, key terms contained in the CRQ were used to search within the full document. Once the key term had highlighted the relevant section within the guideline discussing the relevant CRQ, the associated section within the guideline was then screened manually and the nature of the economic evidence available was recorded and categorised.
-
Each guideline was examined to identify use of variants of the term ‘cost-effectiveness’ or ‘health economics’ until all the findings within the guideline had been exhaustively identified. Classifications generated from this approach were based around guideline chapters.
-
The referenced list of economic evidence was retrospectively manually mapped to the CRQs.
The lack of consistency between the CGs and the need for three strategies to extract the relevant data means that the findings are only indicative in terms of how quantifiable they are, since it was not possible to fully recreate the process by which economic evidence was used.
Findings
The number of CRQs ranged from 11 in CG91 to 40 in CG66 (Table 2). In the two depression guidelines, 26% and 27% of CRQs were supported by at least some economic evidence, compared with 53% and 92% of CRQs in the type 2 diabetes and chronic heart failure guidelines. A much smaller proportion of CRQs were supported by producing a de novo economic model. This at least partly reflects the resources available to NICE in terms of health economics capacity and expertise, in the context of having to generate answers to the substantial number of CRQs underlying most CGs. De novo modelling was available for 9% and 11% of depression CRQs, compared with 20% of diabetes CRQs and 8% of heart failure CRQs.
| Criterion | CG66: type 2 diabetes | CG90: depression | CG91: depression and chronic physical health problems | CG108: chronic heart failure |
|---|---|---|---|---|
| Number of CRQs | 40 | 19 | 11 | 12 |
| Number of CRQs with economic evidence from existing literature (number of studies referred to) | 13 (25) | 3 (10) | 2 (4) | 11 (13) |
| Number (%) of CRQs using evidence from a de novo model | 8 (20) | 2 (11) | 1 (9) | 1 (8) |
| Number (%) of CRQs informed by some form of economic evidence | 21 (53) | 5 (26) | 3 (27) | 11 (92) |
| Number (%) of CRQs with no economic evidence | 19 (47) | 14 (74) | 8 (73) | 1 (8) |
Summary
The extent and type of economic evidence used were hard to quantify systematically because of the variability between how a CRQ was specified and how it was reported or made explicit in the full guideline reporting economic evidence. However, systematically identifying when and how economic evidence was used was helpful for generating a qualitative understanding of the process. Half (51%) of all the CRQs examined were not informed by any economic evidence, although this varied considerably across guidelines (range 8–74%), and only a minority (14.6%) of CRQs were informed by de novo economic modelling, again with variation between guidelines (range 8–20%). This will partly reflect the quality and scope of the clinical evidence, and partly the resources available to the GDG to create de novo CEA models.
With regard to multimorbidity, the development of CGs follows the way that health care and health-care research are currently organised in relatively discrete disease-specific silos. 13 Economic evaluation, and methods of model-based CEA, as subdisciplines of health economics, have also evolved within the biomedical model of disease and thus share preferences for probabilistic evidence sourced from systematic reviews, meta-analyses and RCTs as well as a focus on populations with a single index condition. Therefore, model-based CEAs commonly address one particular physiological condition rather than a decision problem that involves a patient with multiple morbidities. This review has confirmed that current uses of economic evidence are unlikely to routinely support guideline development that accounts for multimorbidity.
Drug–drug and drug–disease interactions
Background
As described in Chapter 1, the prevalence of polypharmacy and potentially serious drug–drug interactions has risen considerably since 1995,32 with evidence that ADEs have also become more common. 34 The harm from ADEs is considerable, with 6.5% of emergency hospital admissions in the UK being attributable to ADEs, of which about half are judged preventable. 70 A study of older people in Italy found that 31.5% of reported ADEs in older people were judged to be caused by drug interactions, with 35.9% of ADEs being severe enough to require hospitalisation. 71 The drugs associated with ADEs causing hospital admission are typically those that guidelines recommend for common conditions such as hypertension, diabetes and CHD, reflecting the fact that most harm is caused by commonly prescribed drugs with relatively low risk rather than rarely prescribed drugs with high risk. 72 However, the observed increases in interactions and ADEs are not necessarily inappropriate, since not all ADEs are predictable (e.g. an anaphylactic reaction in a patient not known to be allergic to a drug), and increases in drug-related harm could in principle be more than balanced by the benefit of prescribing that drug. 32 The appropriate balancing of benefits and harms including consideration of drug–disease and drug–drug interactions has been identified as a key element of optimal care for older adults with multimorbidity. 40
We consider that it would be difficult for CGs to list all possible drug–drug interactions in a helpful way, and, in the UK at least, such a list would also duplicate information available elsewhere or embedded in GP electronic prescribing systems. NICE guidelines usually make this clear; for example, the heart failure guideline says: ‘It is not possible in the development of a CG to complete extensive systematic literature reviews of all pharmacological toxicity. NICE expect the guidelines to be read alongside the Summaries of Product Characteristics’ (p. 24). 51 However, most guidelines do contain comment about some interactions, although it is not always clear why these have been selected.
The study reported in this section therefore aimed to quantify how often the drugs recommended by NICE guidelines for the three exemplar conditions have drug–disease interactions in the presence of other commonly comorbid conditions, or have potentially serious drug–drug interactions with drugs recommended by guidelines for these conditions. The findings of the study were published in the British Medical Journal in 2015, and the findings and figures are reported here under the terms of the Creative Commons Attribution (CC-BY) open access licence that applies, which allows free sharing and adaptation of material for any purpose. 73
Methods
Our starting point was single-disease guidelines for heart failure,56 type 2 diabetes74 and depression. 75 We then selected nine other NICE guidelines to examine, choosing common and chronic conditions that were commonly comorbid with the exemplar conditions, and with recently published NICE guidelines that included recommendations for the initiation of a drug treatment (Figure 5). After discussion with the PRG, the nine selected other guidelines were for atrial fibrillation,76 osteoarthritis,77 COPD,78 hypertension,79 secondary prevention following MI,80 dementia,81 rheumatoid arthritis,82 chronic kidney disease (CKD)83 and neuropathic pain. 84 These were selected on the basis of being common and important problems, for which there was recent NICE guidance available, and with varying prevalence of comorbidity for the three exemplar conditions (see Figure 5).
FIGURE 5.
Percentage of people with the three index conditions who have each of the other conditions. Morbidity data were not available for osteoarthritis or neuropathic pain; the ‘painful condition’ data shown are defined by receipt of four or more prescriptions for non-over-the-counter analgesics in the previous 12 months. Reproduced from Dumbreck et al. 73 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.
A panel of three clinicians reviewed all 12 guidelines to identify and classify recommendations about initiation of chronic drug treatments. Drugs were defined as ‘first-line’ if they were recommended as a treatment for all or nearly all people with the condition [e.g. angiotensin-converting enzyme (ACE) inhibitors for people with heart failure]. Drugs that were recommended for only some patients with the condition under some circumstances were defined as ‘second-line’ (e.g. spironolactone for people with heart failure and high levels of symptoms despite first-line treatment).
Drug–disease and drug–drug interactions were then identified and classified, and the published guidelines examined to identify if these were explicitly discussed. For each of the three exemplar index guidelines, the BNF was systematically searched to identify drug–disease warnings for guideline-recommended drugs, taking account of the pre-defined 11 conditions (the other two index conditions and the nine others). 85 Drug–disease interactions were defined as being significant if a disease was stated to be a contraindication in relation to all or most people with the condition, or if the BNF stated that drugs should be used only with caution accompanied by a clear statement to avoid in all or most people with the condition. For CKD but not for other conditions, BNF warnings frequently recommended dose adjustment, and this was additionally counted for CKD.
For drug–drug interactions, we counted those that the BNF categorised as ‘potentially serious’, which is when ‘concomitant administration of the drugs involved should be avoided (or only undertaken with caution and appropriate monitoring)’ (p. 852). 85 These interactions are defined as potentially serious based on the severity of the potential harm, not the likelihood of the interaction. Potentially serious interactions were identified between drugs recommended by each of the three index guidelines and drugs recommended by any of the 12 guidelines (since two drugs recommended in the same guideline can interact), and the nature of the harm caused was classified, with any disagreement between expert panel members resolved by discussion. The nature of harm was categorised as bleeding risk, central nervous system toxicity, cardiovascular adverse effect (including change in blood pressure, or effect on heart rate or rhythm), effect on renal function or serum potassium, or other (which included harms associated with changes in level of narrow therapeutic index drugs such as lithium carbonate, digoxin and theophylline). 86 These classification categories were chosen to reflect the types of ADEs associated with emergency hospital admission. 70
Interaction findings
Table 3 shows the number of drugs recommended as first or second line for each of the 12 guidelines, which varied from 1 to 9 and from 1 to 19, respectively. Table 4 shows how often drugs recommended for each of the three index conditions would be contraindicated by, or should be avoided in the presence of, any of the other 11 conditions. Drug–disease interactions were not common, with the exception of those related to CKD, which affected type 2 diabetes in particular. CKD was involved in 27 of the identified 32 drug–disease interactions for drugs recommended in the type 2 diabetes CG and all of the 6 and 10 drug–disease interactions for the depression and heart failure guidelines, respectively. The guidelines for type 2 diabetes and heart failure each specifically discussed just one of these identified drug–disease interactions. For type 2 diabetes, this recommendation was regarding the need to avoid thiazolidinedione treatment in people with comorbid heart failure. For heart failure, it was identified that amlodipine should be considered for the treatment of comorbid hypertension and/or angina in patients with heart failure, but verapamil, diltiazem or short-acting dihydropyridine agents should be avoided. The depression guideline did not discuss any of the identified drug–disease interactions.
| Guideline | Guideline number | Year published | Drugs recommended first line (n)a | Drugs recommended second line (n)b |
|---|---|---|---|---|
| Type 2 diabetes | CG87 | 2009 | 4 | 19 |
| Depression | CG90 | 2009 | 1 | 12 |
| Heart failure | CG108 | 2010 | 2 | 9 |
| Atrial fibrillation | CG36 | 2006 | 4 | 7 |
| Dementia | CG42 | 2006 | 3 | 1 |
| Secondary prevention post MI | CG48 | 2007 | 4 | 13 |
| Osteoarthritis | CG59 | 2008 | 2 | 5 |
| CKD | CG73 | 2008 | 1 | 6 |
| Rheumatoid arthritis | CG79 | 2009 | 9 | 9 |
| Neuropathic pain | CG96 | 2010 | 2 | 5 |
| COPD | CG101 | 2010 | 2 | 8 |
| Hypertension | CG127 | 2011 | 4 | 3 |
| Other condition | Type 2 diabetes | Depression | Heart failure | |||
|---|---|---|---|---|---|---|
| First linea | Second lineb | First line | Second line | First line | Second line | |
| CKD (dose change) | 3 | 11 | 1 | 2 | 2 | 3 |
| CKD (avoid) | 2 | 11 | 0 | 3 | 1 | 4 |
| Heart failure | 0 | 5 | 0 | 0 | N/A | 0 |
| Depression | 0 | 0 | N/A | 0 | 0 | 0 |
| Type 2 diabetes | N/A | 0 | 0 | 0 | 0 | 0 |
| Atrial fibrillation | 0 | 0 | 0 | 0 | 0 | 0 |
| Osteoarthritis | 0 | 0 | 0 | 0 | 0 | 0 |
| COPD | 0 | 0 | 0 | 0 | 0 | 0 |
| Hypertension | 0 | 0 | 0 | 0 | 0 | 0 |
| Post MI | 0 | 0 | 0 | 0 | 0 | 0 |
| Dementia | 0 | 0 | 0 | 0 | 0 | 0 |
| Rheumatoid arthritis | 0 | 0 | 0 | 0 | 0 | 0 |
| Neuropathic pain | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 5 | 27 | 1 | 5 | 3 | 7 |
Potentially serious drug–drug interactions were common (Figure 6). There were 133 potentially serious drug–drug interaction pairs identified for the type 2 diabetes guideline, of which 25 (19%) involved one of the four drugs recommended as first-line treatments (the diabetes guideline had a total of four drugs, or classes of drug, recommended as first-line treatments and 19 second-line treatments; see Appendix 1). Nine of the recommended drugs for diabetes did not have any potentially serious drug–drug interactions. For the depression guideline, 89 potentially serious drug–drug interaction pairs were identified, of which 19 (21%) involved the one drug class recommended as first line (SSRI antidepressants) rather than the 12 drugs or drug classes recommended as second-line treatments for depression. For heart failure, 111 potentially serious drug–drug interaction pairs were identified, of which 21 (19%) involved the two drug classes recommended as first line rather than the nine drugs (or drug classes) recommended as second line for heart failure.
FIGURE 6.
Number of potentially serious drug–drug interactions for each of the three exemplar guidelines. This figure has been reproduced from Dumbreck et al. 73 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.
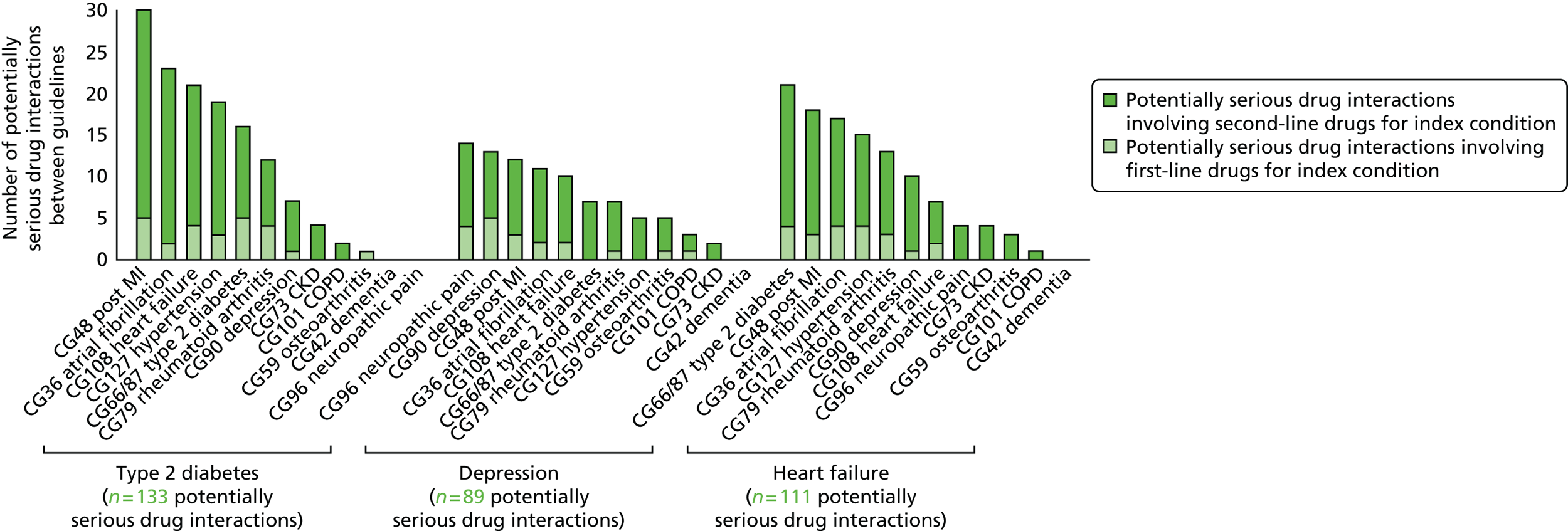
Figure 7 summarises the types of harm expected from potentially serious drug–drug interactions by index condition (see Appendix 2 for further details). For type 2 diabetes, cardiovascular-related harm such as significant hypotension or bradycardia was the most frequent category, followed by ‘other’ (which includes increased lithium or digoxin levels causing risk of toxicity, and myopathy with statin therapy), and harms associated with renal or serum potassium. For depression, bleeding risks were the most commonly identified harm, particularly involving SSRI antidepressants recommended as first line, followed by ‘other’ harms (most commonly relating to lithium toxicity), and cardiovascular and central nervous system toxicity. The majority of cardiovascular adverse effects in the depression guideline were related to increased risk of ventricular arrhythmias. The most common potentially serious drug interactions for the heart failure guideline were for bleeding events, but also drug interactions causing severe hypotension or related to increased digoxin or lithium levels causing risk of toxicity.
FIGURE 7.
Type of harm of potentially serious drug–drug interactions for each of the three exemplar guidelines. This figure has been reproduced from Dumbreck et al. 73 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.

A very limited number of the identified drug–drug interactions were highlighted in the index guidelines. In the guideline for type 2 diabetes, only two interactions were mentioned: potassium-sparing diuretics with ACE inhibitors, and potassium-sparing diuretics with angiotensin receptor blockers (ARBs). The depression guideline highlighted only the increased risk of bleeding with SSRIs plus non-steroidal anti-inflammatory drugs (NSAIDs) or aspirin. None of the recommendations in the heart failure guideline contained an explicit discussion of the 111 potentially serious drug–drug interactions identified.
Summary
Potentially serious drug–drug interactions were found to be relatively common among guideline recommendations for each of the three index conditions and 11 other common conditions. In contrast, drug–disease interactions were found to be relatively uncommon, with the exception of interactions when an individual has comorbid CKD. The types of harm potentially introduced by coprescription of drugs varied by CG and was most commonly cardiovascular and ‘other’ for diabetes; bleeding and ‘other’ for depression; and bleeding and cardiovascular for heart failure.
Previous studies of the implications of following single-disease guidelines in people with multimorbidity have usually considered single, hypothetical patients with carefully selected multiple conditions, an approach that is likely to overstate the scale of the problem. 2,3 Using US population survey data, Lorgunpai et al. 87 estimated a much higher rate of drug–disease interactions (which they termed ‘therapeutic competition’), with one-fifth of older American adults being prescribed drugs for one condition that have the potential to worsen another. 87 However, their study included interactions that did not reach our threshold of being recommended to avoid in all or most patients. For example, the use of beta-blockers for CHD in people with COPD was common in their study, but, although it carries some risk, it is not stated as a contraindication or recommendation to avoid in the BNF, because the benefits outweigh the harms in most patients.
One key potential limitation is the use of a selection of CGs as exemplar case studies, and some other guidelines do discuss interactions in more detail. For example, NICE has produced a guideline for depression in people with a chronic physical health problem,17 which includes extensive discussion about drug interactions (although in a full guideline appendix, which will not be commonly read by clinicians), and a guideline on management of bipolar disorder, which includes detailed recommendations about safe use of lithium. However, we would not expect the pattern of findings to be substantially different for other guidelines that include a reasonable number of recommendations for chronic drug treatment. Any recommendations for commencing drugs for acute conditions were excluded from this analysis, but it should be noted that interactions with drugs such as antibiotics and NSAIDs used for short-term intercurrent illness are common and important. 33 The inclusion of additional guidelines would have further increased the number of potential interactions identified. Both of these exclusions imply that our findings are likely to be conservative.
This study systematically examined recent national guidelines produced by NICE for important and common clinical conditions, using data on interactions drawn from a single, authoritative UK source. Defining contraindications and potentially serious interactions was not straightforward, reflecting the facts that the risk of such events is often poorly quantified and information sources vary in what they rate to be significant. 88 This study used the BNF because it is the reference source used by most UK-based clinicians. The BNF draws on data from manufacturer summaries of product characteristics, NICE guidance, the medical literature and expert opinion, but other reference sources might not be consistent with this, and a databases of listed potentially serious drug interactions might have yielded different results. For example, a summary of product characteristics for amitriptyline from the online electronic medicines compendium of up-to-date, approved and regulated prescribing information for licensed medicines89 includes cardiac arrhythmias and history of MI as contraindications but these are not listed in the BNF. Of note is that the focus of this study has been negative interactions, and there will of course be situations where drug–condition interactions are positive in that one drug has benefits for more than one condition. In addition, the way in which we defined both drug–drug and drug–condition interactions prioritises interactions more strongly associated with harm. Less severe interactions will sometimes cause harm but are also much more common. 87 Our judgement was that focusing on interactions more strongly associated with harm was appropriate to help ensure feasibility in a guidelines context.
Discussion
Summary of the three studies
Accounting for comorbidity in National Institute for Health and Care Excellence guidelines for the three exemplar conditions
Clinical research questions for the guidelines examined were only rarely framed in terms of comorbidity or older age (as a proxy for comorbidity), and relatively few treatment recommendations were qualified in terms of comorbidity or older age. None of the guidelines examined included any qualification in relation to people with short life expectancy. There was little clear synergy and no clear contradiction between recommended treatments across the guidelines examined. None of the research recommendations in the single-condition guidelines explicitly called for research in population subgroups such as people with comorbidity or older people.
Use of economic evidence in National Institute for Health and Care Excellence guidelines for the three exemplar conditions
Half (51%) of all the CRQs examined were not informed by any economic evidence, although this varied considerably across guidelines (range 8–74%), and only a minority (14.6%) of CRQs were informed by de novo economic modelling, again with variation between guidelines (range 8–20%). This will partly reflect the quality and scope of the clinical evidence, and partly the resources available to the GDG to create de novo CEA models. Like the CRQs on which they were based, economic analyses were focused on single diseases, although it is routine to carry out sensitivity analyses examining cost-effectiveness in different age groups.
Drug–drug and drug–disease interactions in National Institute for Health and Care Excellence guidelines for the three exemplar conditions
Potentially serious drug–drug interactions were common between drugs recommended in guidelines for the three exemplar conditions and drugs recommended in guidelines for 11 other conditions. In contrast, drug–disease interactions were relatively uncommon, with the exception of interactions when an individual has comorbid CKD. The types of harm potentially introduced by coprescription of drugs varied by CG and were most commonly cardiovascular and ‘other’ for diabetes; bleeding and ‘other’ for depression; and bleeding and cardiovascular for heart failure.
Interpretation and implications of the three studies
Accounting for comorbidity in National Institute for Health and Care Excellence guidelines for the three exemplar conditions
The purpose of this analysis was largely to define current practice to make sure that we understood the context within which suggested changes to guideline development would have to happen. Our interpretation is that GDGs do take account of comorbidity to at least some extent, but that this is predominantly for concordant conditions or for conditions where there is broad acceptance that comorbidity matters. This is most explicit in the pairing of chronic physical problems and depression, where both the type 2 diabetes and heart failure guidelines refer to the depression guidelines, and NICE has produced a specific depression guideline for people with a chronic physical condition. However, the way in which treatment recommendations were qualified was usually quite narrow, and there was little or no specific guidance on how clinicians or patients should respond in the presence of other morbidities (including age or frailty as a proxy for, or consequence of, this) or reduced life expectancy. With the exception of the guideline for depression in adults with a chronic physical health problem (CG91), no research recommendations were made about evidence generation in people with a comorbidity or older people. In contrast, the majority of research recommendations in CG91 were specific to people with comorbid depression and chronic physical health problems, implying that such research recommendations are feasible to make. Chapter 3 further addresses this issue by examining the applicability of evidence used in guideline development.
Use of economic evidence in National Institute for Health and Care Excellence guidelines for the three exemplar conditions
Economic evaluation has a central place in GDG decision-making, but is deployed sparingly because of resource and data constraints. In the guidelines studied, half of CRQs were not informed by economic evidence and only 8–20% were informed by de novo model-based CEA. Like the underlying CRQs, such analysis was essentially focused on single diseases. When interpreting the results of a model-based CEA, significant comorbidity or multimorbidity within the patient population is likely to undermine the validity of statements on the relative cost-effectiveness of the interventions being compared. On the resource use side of the evaluation there could be economies of scale or scope when treating diseases that are concordant (similar pathogenesis), thus permitting overlaps in management or treatment. Evidence for this hypothesis has been found on a macro scale for primary care costs in England; for example, a recent observational study found that hypertension associated with other cardiovascular diseases (CVDs) was cost limiting, in that the combined costs in people with particular patterns of comorbidity was lower than the sum of the costs of the conditions individually. 90 In contrast, discordant conditions requiring different management or treatments could lead to increased costs, with the same study finding depression associated with other conditions to be cost increasing. In relation to patient benefits, patients with increasing numbers of diseases are known to have generally lower HRQoL, but this relationship is far from simple, with the impact varying depending on the nature and combination of diseases.
Model-based CEAs that overlook these properties are unlikely to give representative estimates of the costs and benefits of interventions for patient populations with multiple morbidities. Therefore, even if analysis of relative cost-effectiveness for an intervention is correct on average for the whole population, if large subgroups with multiple diseases have different cost-effectiveness estimates then there is the potential for resources to be used more efficiently by stratifying recommendations within CGs accordingly. 85,91 In principle, subgroup analysis that varies baseline risk or treatment effects for patients with multiple conditions could play an important role in integrating the impact of comorbidities on cost-effectiveness results. Given resource constraints in current guideline development, there are several approaches that could improve the contribution of economic evidence to guideline development that accounts for multimorbidity.
-
Existing model-based CEAs can be used in novel ways that are potentially useful for decision-making regarding patients with multimorbidity, and we have examined two such uses. In Chapter 4 we show how the absolute QALY could theoretically be used to compare and prioritise treatments for patients with multimorbidity where multiple treatments are recommended. In Chapter 5 we show how the ‘pay-off time’ concept can be applied to current economic evidence to provide a new temporal dimension on the value of interventions for populations who are likely to have multimorbidities.
-
More ambitiously, and with greater resource implications, economic analysis for populations with multiple morbidities could go beyond the adjustment of input parameters in existing single-disease models. Instead, the structure of the analysis itself can be changed to incorporate at least some of the characteristics of the conditions that are of consequence. As an exemplar of this approach, the de novo model described in Chapter 6 attempts to incorporate the dual characteristics of both depression and CHD into a single model structure.
Drug–drug and drug–disease interactions in National Institute for Health and Care Excellence guidelines for the three exemplar conditions
For the conditions examined in this study, major drug–disease interactions were relatively rare, with the exception of CKD, where they were more common. We believe that it would be useful for guideline developers to use epidemiological data to decide explicitly whether or not CKD (and other comorbidities if relevant) is common enough in people with the condition that is the target of the guideline to require comment or modification of recommendations. For the three index conditions examined here, CKD comorbidity prevalence was 4.1% in depression, 13.5% in type 2 diabetes and 23.0% in heart failure (see Figure 5). The implication might be that guideline developers should consider CKD with heart failure, possibly consider it with type 2 diabetes and possibly not consider it with depression. The actual decision would depend on GDG judgement, but should be informed by epidemiological evidence rather than the necessarily unpredictable knowledge and expertise that individual members bring to the GDG.
Potentially serious drug–drug interactions were much more common, but there are too many for all of them to be specifically mentioned by guidelines. This means that there will have to be some selection process. From this perspective, CGs produced and disseminated on paper will only ever be able to account adequately for a minority of potential drug–drug interactions. Electronic versions of CGs could in principle account for more, but some selection based on importance would still be important. The implication is that it would be useful for guideline developers to use epidemiological data to estimate the likely frequency and severity of drug–drug interactions to inform a judgement about whether or not a treatment recommendation should be qualified. Frequency will be determined by whether or not the drug being recommended is first line (intended for all or nearly all people with the condition), by how commonly interacting drugs are used (which will depend on rates of comorbidity) and by how commonly the ADE in question occurs.
Of note is the requirement for detailed epidemiological information about the real-world population for which the guideline is making recommendations. This is currently much less commonly used in guideline development than trial data from narrowly selected populations. With the growth of large electronic primary care data sets, it is increasingly straightforward to define the population for which recommendations are being made, and describe its demography, comorbidity and current prescribing. As an example, there is a potentially serious interaction between statins (recommended as first line for patients with type 2 diabetes) and ciclosporin (recommended as second line for patients with rheumatoid arthritis), due to risk of myopathy and rhabdomyolysis. Given that only 1.4% of people with type 2 diabetes also have rheumatoid arthritis, and ciclosporin is recommended only as second line for rheumatoid arthritis, this will only ever be a very rare drug–drug interaction and so is very unlikely to reach the threshold for explicit consideration by a GDG. In contrast, coprescription of SSRI antidepressants (recommended as first line for depression) and tramadol (recommended as second line for painful conditions) is likely to be common because tramadol is commonly used for pain in the UK and 27.1% of people with depression also have painful conditions. 13,92 However, the risk of serious or fatal serotonin syndrome appears to be low (although this is poorly quantified93) and a GDG would have to make a judgement about whether or not the interaction requires specific mention to inform clinicians and patients to be aware of the signs and symptoms of serotonin syndrome should they occur. The key implication is that we believe that interactions and risks should be systematically assessed and explicit decisions made about whether or not they require discussion, like the requirement for treatment benefits to be systematically and explicitly assessed. The systematic assessment of the likelihood of interactions in relation to particular guideline recommendations is further examined in Chapter 3.
Conclusion
Comorbidity is partly considered in existing NICE guidelines but there is no documented process for such consideration in the NICE Guidelines Manual. It is also not always clear from guideline documentation why some comorbidity or interaction issues are chosen to be highlighted, and with some exceptions those that are identified are for closely related conditions. Drug–drug and drug–disease interactions are one important aspect of the problems posed by comorbidity, and the analysis quantifies the frequency of potentially serious interactions and ways in which GDGs could more systematically identify and judge which of them require accounting for. Systematic use of appropriate epidemiological evidence could allow GDGs to make transparent decisions relating to the development of specific recommendations for population subgroups with particular comorbidities or other important characteristics such as reduced life expectancy.
Chapter 3 The applicability of evidence used to inform clinical guideline treatment recommendations
Background
Randomised controlled trials are usually carried out in selected populations. 94–96 This creates a potential problem for guideline developers, because their treatment recommendations may be based on an extrapolation from evidence in one group of people with a condition to another group of people with the same condition. This issue is described using a number of terms including ‘the external validity or generalisability of evidence’, ‘the directness of evidence’ and ‘the applicability of evidence’, which is the term we shall use. This chapter initially discusses literature relevant to applicability. It then uses a combination of information about the participants in trials that provide evidence for CG recommendations and epidemiological data about the real-world treated population to examine how GDGs could more systematically consider applicability when making treatment recommendations.
How commonly is applicability of evidence a potential problem?
Several studies have shown that many or most people with a condition would not be eligible for the RCTs providing evidence of treatment effectiveness. Of a sample of 283 trials published in high-impact general medical journals, 81% excluded people with common comorbidities, 54% excluded people taking other commonly prescribed drugs and 72% excluded people on the basis of age. 94 When considering single diseases, fewer than half of people in Scotland with newly diagnosed type 2 diabetes would have been eligible for the landmark UK Prospective Diabetes Study (UKPDS) 33 and UKPDS34 studies of initial hypoglycaemic treatment. 97 Only 13–25% of older people discharged from hospital with heart failure in the USA would have been eligible for inclusion in three seminal trials of heart failure treatment. 98 A maximum of 20% of people with COPD in the community would have been eligible for the 18 key trials underlying the Global Initiative for Chronic Obstructive Lung Disease guidelines for COPD (median 5%). 99 A maximum of 43% of people with asthma would have been eligible for the Global Initiative for Asthma guidelines (median 6%). 100 Depending on the trial, only 17–71% of women with breast cancer would have been eligible for 12 major breast cancer trials that changed clinical practice. 101 Evidence of treatment benefit from RCTs is therefore often based on highly selected subgroups of people with the target condition.
Of note is that, in most such studies, what is being evaluated is exclusion due to easily measured factors such as age, and that there will be additional exclusions from less well specified criteria such as ‘any life-threatening disease (other than heart failure)’102 or ‘any factors likely to limit adherence to interventions’. 103 Such criteria are open to the recruiting researcher’s judgement and would be expected to exclude people of all ages who are sicker or less likely to adhere to treatment, or perceived to be at increased risk of harm. For example, patients in carotid endarterectomy trials who were judged unfit for surgery (i.e. were believed to have very high risk from surgery) had much worse outcomes in terms of stroke risk and death even though their baseline characteristics as measured by trial variables were not different from those judged fit for surgery. 96 Excluded patients will therefore plausibly have different baseline risks of the outcomes targeted by the trial, different risks of harm and potentially different likelihoods of benefit (although the standard assumption is that relative risk is constant across populations). However, although older and sicker people, who are often excluded from trials, are more likely to be harmed by treatment (e.g. the Action to Control Cardiovascular Risk in Diabetes study excluded those aged ≥ 80 years because the pilot study showed higher risk of serious hypoglycaemia in older people103), it is often difficult to predict the extent to which net benefit is likely to vary.
Extrapolation of evidence into recommendations in guideline development
Guideline recommendations for whole populations of people with a particular condition will therefore often require extrapolation from the available evidence, and GDGs use their judgement to make such extrapolations. The 2009 NICE Guidelines Manual identifies a number of challenges when writing recommendations, one of which relates to applicability and extrapolation (Box 1).
The clinical evidence is not directly applicable to the population covered by the guideline, for example because of a different age group.
Possible solutionsThe GDG may wish to extrapolate to the recommendations from the evidence – for example, from high-quality evidence in a largely similar patient group. The GDG will need to make its approach explicit, stating the basis it has used for extrapolating from the data and the assumptions that have been made.
The 2014 update to the NICE Guidelines Manual also discusses this issue, but using the example of patients with different conditions in the same care setting.
. . . a review of systems for managing medicines in care homes for people with dementia may identify good practice that is relevant in other care home settings . . . extrapolation must be considered carefully by the Committee, with explicit consideration of the features of the condition or interventions that allow extrapolation.
p. 16748
Of note is the assumption in the 2014 guidelines manual that the use of GRADE working group profiles will account for the problem of applicability.
How does Grading of Recommendations Assessment, Development and Evaluation deal with applicability?
The GRADE working group has produced detailed recommendations for guideline development, and current NICE guideline development draws on these with some modifications. 105 Applicability is dealt with as part of whether evidence is direct or indirect, and judgement of directness is used to assess the strength of the evidence underlying recommendations. 106 In GRADE, indirectness can arise from one or more of four mechanisms:
-
differences between trial populations and the population specified as being the focus of the guideline (as defined in the PICO question), which GRADE considers a problem of applicability
-
differences between the intervention used in the trial and the intervention that is the focus of the guideline, which GRADE considers a problem of applicability
-
differences between the outcomes used in trials and outcomes that matter to patients, which GRADE discusses in terms of surrogate outcomes
-
the use of indirect comparisons, where choices are being made about two treatments for the same condition that have been evaluated in trials against placebo, but not against each other.
The presence of serious indirectness leads to the quality of the evidence in the GRADE profile being downgraded, but this in itself may not alter the actual recommendation made. Where trial populations are completely or substantially different from the population for which recommendations are being made, then it is fairly easy to say that the evidence is of low quality for applying to the latter (e.g. evidence from adults with one type of lymphoma applied to children with another). Much more common, however, is that trial populations are a subset of the population for which recommendations are being made. In that sense, the trial evidence is directly applicable to some of the real-world population, but is indirect for another part of the population. In this situation, downgrading the quality of the evidence is unreasonable for people who would have been eligible for the trial, but not downgrading for people who were not eligible may be problematic. For this reason, we believe that the GRADE application of directness does not adequately deal with the problem of applicability, since it is applied only at the evidence synthesis stage, whereas it also applies when GDGs create recommendations based on the evidence. Whether or not applicability is a problem will depend on the extent to which trial populations are very different in their expected response to the intervention from the populations for which GDGs make recommendations, or are very different from a substantial subset of the guideline population.
The aim of this element of the project was therefore to examine the applicability of trial evidence by comparing the populations included in trials underlying selected treatment recommendations for the three exemplar conditions with the actual population for which recommendations were being made.
Methods
With the advice of the PRG, we first selected key first-line drug treatment recommendations for each condition. As described in the interactions study in Chapter 2, we defined a drug as first line if it was recommended as a treatment for all or nearly all people with the condition, whereas drugs that were recommended for only some patients with the condition under some circumstances were defined as second line. With the advice of the PRG, for type 2 diabetes, we selected initial drug treatment with metformin and sulphonylureas for persistent hyperglycaemia after lifestyle change. For heart failure, we selected ACE inhibitor and beta-blocker treatment for people with heart failure due to LVSD. For depression, we selected SSRI antidepressants for people with moderate to severe depression.
We then identified the trials cited by guidelines as the evidence underlying these recommendations. We defined the populations included in these trials by examining trial published inclusion and exclusion criteria as reported in the main trial publication or any cited published protocol or in the full guideline, and by examining the actual patients included as defined by their reported baseline characteristics. For type 2 diabetes and heart failure, we selected trials measuring patient-centred outcomes such as mortality and major morbidity (as opposed to surrogate outcomes such as glycaemic control), and larger trials with long follow-up (> 26 weeks). Almost all the cited trials for depression were relatively small and with relatively short follow-up (reflecting the fact that they were usually examining initial treatment response), and we therefore included all trials cited in the two relevant guidelines, CG90 and CG91. Of note is that inclusion and exclusion criteria in the depression trials were less well reported and that trials varied in whether they reported only the age range within which patients were eligible, only the mean or median age of those actually participating, both or (sometimes) neither.
We defined the characteristics of the population for which the recommendations are assumed to be made, using epidemiological data from two sources:
-
A study of multimorbidity using data recorded in GP clinical information technology systems, which had data on the presence of 40 conditions for 1.76 million people in Scotland in 2007. 13 Type 2 diabetes and heart failure were two of the conditions included, as defined by the presence of relevant read codes. Depression is poorly coded and was defined as the presence of a read code for depression in the last year or receipt of four or more prescriptions for an antidepressant [excluding low-dose tricyclic antidepressants (TCAs), which are predominantly used for pain, night sedation and anxiety] in the previous year.
We used this data set to measure rates of common, important comorbidities in people with each of the three exemplar conditions. For each condition, we examined the distribution of patients by age to compare with populations included/excluded by relevant trials, and the distribution of comorbidity by age. Using the same methodology as the interactions work reported in Chapter 2, we additionally used this data set to identify drug–disease interactions for recommended drugs. The BNF was used to identify whether the disease was stated to be a contraindication in relation to all or most people with the condition, or the BNF stated that drugs should be used only with caution, accompanied by a clear statement to avoid in all or most people with the condition, or requiring dose adjustment in all people with the condition.
-
A study of polypharmacy using data for all 318,000 adult residents of the NHS Tayside region of Scotland in 2010, including all drugs dispensed by community pharmacies. In this data set, type 2 diabetes was defined using the regional diabetes register, which is > 99% complete. Heart failure was defined as ever having been admitted to hospital with heart failure (and therefore covered a selected population of people with more severe heart failure). Depression was defined as current treatment with a SSRI, a monoamine oxidase inhibitor (MAOI) or an ‘other’ antidepressant (reflecting the fact that depression is a less clearly defined condition than diabetes or heart failure and there are no central registers to draw on). 32 Tricyclic antidepressants were not included in the definition of depression, since in the UK these are used almost exclusively for conditions other than depression, such as chronic pain. However, a weakness in the depression data is therefore that some patients taking these drugs will be being treated for conditions other than depression.
For the guideline-recommended drugs being examined, we used the BNF to identify rates of prescription of all drugs stated to have a potentially serious interaction, which the BNF defines as ones where ‘concomitant administration of the drugs involved should be avoided (or only undertaken with caution and appropriate monitoring)’. 85 In the paper BNF such interactions are marked with a black dot and in the online BNF they are marked in red.
Findings for initial management of hypoglycaemia in type 2 diabetes with metformin or sulphonylurea
Characteristics of the trial populations
The UKPDS was the only trial that assessed long-term patient-centred outcomes for initial treatment with sulphonylureas or insulin107 and, in overweight people, initial treatment with metformin. 108 It was carried out in the UK for people with newly diagnosed type 2 diabetes aged 25–65 years. Exclusion criteria included ketonuria, creatinine > 175 µmol/l (at least moderate CKD), recent or multiple vascular events, and ‘severe concurrent illness that would limit life or require extensive systemic treatment’. 107
One-third of patients screened were excluded,108 and patients were randomised to active treatment intended to maintain fasting plasma glucose < 6 mmol/l or to diet with drugs, only if symptomatic or fasting plasma glucose > 15 mmol/l (i.e. what would now be considered poor or very poor control). Patients who were overweight (> 120% ideal bodyweight) were randomly assigned to either metformin or diet. Patients who were not overweight were randomly assigned to sulphonylurea, insulin or diet [although analysis was of ‘intensive’ treatment (i.e. sulphonylurea or insulin) vs. diet]. The mean age of patients in all arms was 53 years.
Comparison of trial population and the population for which recommendations are being made
Using data for all newly diagnosed people in Scotland in 2008, Saunders et al. 97 found that 49.3% of patients would have been excluded from UKPDS33 and 68.2% from UKPDS34. Using our data, 44% of 6647 people newly diagnosed with type 2 diabetes in 2006/7 (approximately one-third of the Scottish population) would not have been eligible for UKPDS based on age alone, with some younger people excluded by other criteria. The remainder of the analysis focuses on age, since this is the main reason for exclusion.
Table 5 shows rates of comorbidity by age group (the UKPDS population were all aged < 65 years). Not surprisingly, older people newly diagnosed with diabetes who were not eligible for the trial were substantially more likely to have other comorbidities, including vascular and non-vascular conditions, notably CKD (2.8% of those < 65 years compared with 25.5% of those > 75 years), stroke/transient ischaemic attack (TIA) (3.2% vs. 15.5%), atrial fibrillation (1.4% vs. 14.9%), dementia (0.2% vs. 5.7%), recent cancer (3.3% vs. 10.6%), CHD (10.6% vs. 33.1%), heart failure (1.7% vs. 10.7%) and COPD (5.2% vs. 11.4%). The main exceptions to this were depression and pain, which were present in between one-sixth and one-fifth of patients at all ages, with depression a little more common in those aged < 65 years.
| Condition | Eligible for trials, aged < 65 years (n = 3744) | Ineligible for trials, aged 65–74 years (n = 1718) | Ineligible for trials, aged ≥ 75 years (n = 1185) | Overall (n = 6647) |
|---|---|---|---|---|
| Hypertension | 40.9 | 62.0 | 64.8 | 50.6 |
| Painful condition | 17.5 | 21.7 | 22.1 | 19.4 |
| CHD | 10.6 | 26.0 | 33.1 | 18.6 |
| Depression | 17.6 | 15.4 | 14.3 | 16.4 |
| Thyroid disorders | 7.3 | 11.7 | 12.6 | 9.4 |
| CKD | 2.8 | 11.5 | 25.5 | 9.1 |
| COPD | 5.2 | 11.2 | 11.4 | 7.9 |
| Anxiety or insomnia | 5.8 | 8.1 | 10.3 | 7.2 |
| Stroke/TIA | 3.2 | 9.8 | 15.5 | 7.1 |
| Cancer in last 5 years | 3.3 | 7.8 | 10.6 | 5.8 |
| Atrial fibrillation | 1.4 | 7.8 | 14.9 | 5.4 |
| Heart failure | 1.7 | 5.8 | 10.7 | 4.4 |
| Peripheral vascular disease | 1.9 | 5.0 | 7.3 | 3.7 |
| Dementia | 0.2 | 0.8 | 5.7 | 1.4 |
| Number of other conditions | ||||
| 0 | 22.6 | 7.6 | 4.5 | 15.5 |
| 1 | 29.8 | 20.6 | 12.2 | 24.3 |
| 2 | 19.2 | 22.5 | 18.0 | 19.8 |
| 3 | 12.1 | 19.0 | 20.0 | 15.3 |
| ≥ 4 | 16.3 | 30.3 | 45.3 | 25.1 |
Drug–disease and drug–drug interactions
For common comorbidities (> 5% prevalence) the only stated drug–disease interaction in the BNF was renal impairment for metformin (dose reduction recommended if estimated glomerular filtration rate < 45 ml/min and avoidance if estimated glomerular filtration rate < 30 ml/min) and sulphonylureas (use ‘with care in mild to moderate renal impairment’ and avoid in more severe renal impairment).
Table 6 lists drugs with potentially severe interactions with sulphonylureas (metformin does not have any such interactions), and the percentage of the population with type 2 diabetes who are dispensed them. There are relatively few interactions and, with the exception of coumarin anticoagulants and NSAIDs, most are with drugs that are infrequently used.
| Recommended drug for type 2 diabetes with inadequate glycaemic control and potentially seriously interacting drugs | Eligible for trials, aged < 65 years (n = 7495) | Ineligible for trials, aged 65–74 years (n = 5217) | Ineligible for trials, aged ≥ 75 years (n = 5167) |
|---|---|---|---|
| Chloramphenicol (oral) | 0 | 0 | 0 |
| Coumarins | 5.2 | 11.7 | 15.7 |
| Fluconazole | 1.4 | 0.6 | 0.9 |
| Miconazole | < 0.01 | 0 | 0 |
| NSAIDs | 9.4 | 7.3 | 3.9 |
| Rifamycins | < 0.01 | < 0.01 | < 0.01 |
| Sulfinpyrazone | < 0.01 | < 0.01 | < 0.01 |
| Bosentan | 0 | 0 | 0 |
Are there applicability issues with metformin and sulphonylureas for the initial management of hyperglycaemia in people with newly diagnosed type 2 diabetes?
Applicability of evidence
Making recommendations for all people newly diagnosed with type 2 diabetes requires that evidence derived from younger people be extrapolated to older people, and older people are a large proportion of the newly diagnosed population. Older people have many more comorbidities and are prescribed more drugs.
Drug–disease interactions
Chronic kidney disease is common in people with type 2 diabetes at diagnosis (9.1% overall, 25.5% in those aged > 75 years), and is relevant to metformin and sulphonylurea prescribing.
Drug–drug interactions
There were two common potentially serious drug–drug interactions. The first, between sulphonylureas and coumarin anticoagulants, is likely to cause problems in practice only when sulphonylureas are started or stopped in people taking coumarins. The second is between sulphonylureas and NSAIDs, where NSAIDs may increase the risk of hypoglycaemia.
Findings for heart failure due to left ventricular systolic dysfunction: treatment with angiotensin-converting enzyme inhibitors and beta-blockers
Characteristics of the trial populations
Four of the five large ACE inhibitor trials were carried out in people with significant LVSD [ejection fraction (EF) ≤ 40% and often lower] although with varying levels of symptoms, and one in people with normal EF (Table 7). In principle, the Survival and Ventricular Enlargement study (SAVE) and the two Studies of Left Ventricular Dysfunction (SOLVD) trials recruited patients aged up to 80 years, but the mean age of those actually participating in these three trials was 59–61 years. The Trandolapril Cardiac Evaluation (TRACE) study recruited only adults, with a mean age of 67 years. All trials excluded people with varying degrees of CKD (although typically at least moderately severe with creatinine more than ≈ 170 mmol/l), and several stated that they excluded people with limited life expectancy from causes other than heart failure.
| Treatment | Trial | Selected inclusion and exclusion criteria | Mean age of those randomised (years) |
|---|---|---|---|
| ACE inhibitor | SAVE109 (n = 2231) | Aged 21–80 years with LVSD EF ≤ 40% in the 3 days after acute MI. Exclusions included significant CKD and ‘other conditions believed to limit survival’ | 59 |
| SOLVD-treatment (n = 2569)110 | Aged 21–80 years with LVSD EF ≤ 35%, NYHA class III or IV (i.e. significant symptoms). Exclusions included various comorbidities including significant CKD, and ‘any other disease that might substantially shorten survival or impede participation in a long-term trial’ | 61 | |
| SOLVD-prevention111 (n = 4228) | Aged 21–80 years with LVSD EF ≤ 35%, NYHA class I or II (i.e. few symptoms). Exclusions included various comorbidities including ‘renal failure’, ‘any other disease that might shorten life or impede participation’, ‘likelihood of no adherence’ and ‘other life-threatening disease’ | 59 | |
| TRACE112 (n = 1749) | Aged ≥ 18 years with LVSD in the week after acute MI. Multiple disease exclusions including significant CKD | 67 | |
| Beta-blocker | Beta-Blocker Evaluation Survival Trial (BEST)113 (n = 2708) | Aged ≥ 18 years with LVSD EF ≤ 35%, and NYHA class III–IV (i.e. moderate to high level of symptoms). Exclusion criteria included various comorbidities including significant CKD, ‘if they had a life expectancy of less than three years’ or conditions ‘that could adversely affect the safety or efficacy of the study drug’ | 60 |
| Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalisation in Seniors with Heart Failure (SENIORS)114 (n = 2128) | Aged ≥ 70 years with hospital admission with heart failure in previous year or known EF ≤ 35%. Exclusions included various comorbidities including significant CKD and current prescription of various agents including calcium-channel blockers, tricyclics and beta-agonists, and ‘other major medical conditions that may have reduced survival during the period of the study’ | 76 | |
| Australia–New Zealand (ANZ) heart failure collaborative group115 (n = 415) | People with LVSD EF ≤ 45% due to ischaemic heart disease, and NYHA class II or III (i.e. low to moderate symptoms). Exclusions included various comorbidities including significant CKD and ‘any life threatening non-cardiac disease’ | 61 | |
| Cardiac Insufficiency Bisoprolol Study (CIBIS)-2116 (n = 2647) | Aged 18–80 years with LVSD EF ≤ 35%, and NYHA class III–IV (i.e. moderate to high level of symptoms). Exclusions included various comorbidities including significant CKD and coprescription of calcium-channel antagonists | 61 | |
| Metoprolol CR/XL Randomised Intervention Trial in congestive Heart Failure (MERIT-HF)117 (n = 3991) | Aged 40–80 years with LVSD EF ≤ 40%, and NYHA class II–IV (i.e. low to high level of symptoms). Exclusions included various vascular comorbidities and ‘poor compliance’ during the run-in phase | 64 | |
| US carvedilol heart failure study group118 (n = 1904) | Symptomatic heart failure with LVSD EF ≤ 35% despite treatment with diuretic and ACE inhibitor. Exclusions included various cardiac comorbidities and current prescription of calcium-channel blockers and beta-agonists | 58 | |
| Carvedilol Prospective Randomized Cumulative Survival Study (COPERNICUS)119 (n = 2289) | Severe chronic heart failure defined as LVSD EF ≤ 25% despite optimal diuretic and ACE inhibitor/ARB treatment and high level of symptoms. Exclusions included various comorbidities including ‘severe primary pulmonary, renal or hepatic disease’ and current prescription of calcium-channel blockers and beta-agonists | 63 |
The beta-blocker trials were also largely carried out in people with significant LVSD and significant symptoms. Six of the seven trials recruited patients with mean ages of 58–64 years, and one [Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalisation in Seniors with Heart Failure (SENIORS)] recruited patients aged ≥ 70 years with a mean age of 76 years, although not all participants in SENIORS had LVSD. All trials excluded people with a range of physical comorbidities (most commonly CKD, although again usually moderate to severe rather than mild) and coprescribing (most commonly calcium-channel blockers and beta-agonists), and several explicitly excluded people with limited life expectancy.
Comparison of trial population and the population for which recommendations are being made
The majority (52.4%) of people with heart failure recorded on GP registers are aged ≥ 75 years, and it is unlikely that trials that were not focused on those aged > 70 years recruited many patients in this age range. Trials largely recruited patients with more severe heart failure assessed by left ventricular EF and/or symptoms. Trials additionally excluded people with a range of comorbid conditions, notably moderate to severe CKD and life-limiting conditions (the latter was explicitly stated by only a minority of trials, although conversely no study explicitly includes such people).
Table 8 shows rates of comorbidity by age group. Comorbidity is common at all ages in people with heart failure, but substantially more common in older people, where a majority of those aged > 65 years with heart failure had at least four other conditions. CKD was much more common in those aged > 75 years than < 65 years (30.6% vs. 7.5%), as were peripheral vascular disease (11.5% vs. 5.4%), recent cancer (11.2% vs. 3.9%), dementia (6.7% vs. 0.5%), stroke/TIA (20.0% vs. 7.4%), atrial fibrillation (32.4% vs. 11.7%) and COPD (19.3% vs. 10.6%). As with diabetes, pain affected approximately one in five people and depression affected approximately one in six people with heart failure, irrespective of age.
| Comorbid conditions | Eligible for trials, aged < 65 years (n = 4186) | Eligible for trials but under-represented, aged 65–74 years (n = 4903) | Eligible for a few trials, seriously under-represented, aged ≥ 75 years (n = 9865) | Overall (n = 18,954) |
|---|---|---|---|---|
| CHD | 44.1 | 65.3 | 62.6 | 59.2 |
| Hypertension | 49.6 | 59.1 | 58.7 | 56.8 |
| Atrial fibrillation | 11.7 | 24.8 | 32.4 | 25.9 |
| Diabetes | 20.1 | 29.5 | 21.5 | 23.2 |
| CKD | 7.5 | 21.0 | 30.6 | 23.0 |
| Painful condition | 19.9 | 26.6 | 22.2 | 22.8 |
| COPD | 10.6 | 20.4 | 19.3 | 17.7 |
| Depression | 17.3 | 16.6 | 16.7 | 16.8 |
| Stroke/TIA | 7.4 | 14.8 | 20.0 | 15.9 |
| Thyroid disorders | 7.9 | 13.0 | 17.0 | 14.0 |
| Anxiety or insomnia | 7.7 | 10.8 | 13.8 | 11.7 |
| Peripheral vascular disease | 5.4 | 11.4 | 11.5 | 10.1 |
| Cancer in last 5 years | 3.9 | 8.4 | 11.2 | 8.9 |
| Asthma and no COPD | 5.9 | 5.6 | 3.9 | 4.8 |
| Dementia | 0.5 | 1.3 | 6.7 | 3.9 |
| Number of other conditions | ||||
| 0 | 9.3 | 1.1 | 0.9 | 2.8 |
| 1 | 21.0 | 8.4 | 4.7 | 9.3 |
| 2 | 20.9 | 14.1 | 10.3 | 13.6 |
| 3 | 16.8 | 18.3 | 15.7 | 16.6 |
| ≥ 4 | 32.9 | 58.1 | 68.4 | 57.7 |
Drug–disease and drug–drug interactions
The BNF states that ACE inhibitors should be used with caution in people who may have undiagnosed renovascular disease (those with peripheral vascular disease and widespread atherosclerotic disease) and those with renal impairment. Older people, who were usually excluded from trials, were much more likely to have peripheral vascular disease or other atherosclerotic disease (although these were all common in younger people as well) and CKD.]
The BNF states that beta-blockers are relatively contraindicated in asthma and COPD, as well as in heart block, unstable heart failure and diabetes. COPD was twice as common in those aged > 75 years with heart failure as in those < 65 years, although there is good evidence that beta-blockers improve outcomes in this population. Asthma was less common in the older age group (3.9% vs. 5.9%), and diabetes had a similar prevalence.
Table 9 lists drugs with potentially severe interactions with ACE inhibitors and beta-blockers, and the percentage of the population with hospital-admitted heart failure who are currently dispensed them. The most commonly prescribed drugs that potentially interacted with ACE inhibitors were all ones that are commonly used to treat heart failure – ARBs (which are usually used in people intolerant of ACE inhibitors), diuretics and aldosterone antagonists in particular – although coprescription of these may increase the risk of renal adverse effects. Drugs that interacted with beta-blockers were rarely used and most have effective alternatives.
| Recommended drug for heart failure due to LVSD | Potentially seriously interacting drugs | Eligible for trials, aged < 65 years (n = 826) | Eligible for trials but under-represented, aged 65–74 years (n = 941) | Eligible for a few trials, seriously under-represented, aged ≥ 75 years (n = 2085) |
|---|---|---|---|---|
| ACE inhibitor | Aliskiren | 0 | 0 | 0.05 |
| ARB | 13.3 | 19.4 | 14.1 | |
| Ciclosporin | 1.2 | 0.7 | 0.05 | |
| Diuretics | 44.2 | 57.4 | 72.6 | |
| Potassium-sparing diuretics | 1.0 | 1.5 | 1.2 | |
| Aldosterone antagonists | 20.0 | 17.4 | 16.7 | |
| Everolimus | 0 | 0 | 0 | |
| Lithium | 0.2 | 0.2 | 0.1 | |
| Potassium | 0.5 | 0.2 | 0.2 | |
| Gold | 0.1 | 0 | 0 | |
| Beta-blocker | Alpha-blocker | 3.5 | 5.6 | 4.0 |
| Antiarrhythmics | 2.7 | 2.9 | 1.9 | |
| Clonidine | 0.04 | 0 | 0 | |
| Diltiazem | 3.9 | 5.6 | 2.6 | |
| Dobutamine | 0 | 0 | 0 | |
| Fingolimod | 0 | 0 | 0 | |
| Moxisylyte | 0 | 0 | 0 | |
| Nifedipine | 0.01 | 1.7 | 1.4 | |
| Verapamil | 0.01 | 0.01 | 0.01 |
Are there applicability issues with angiotensin-converting enzyme inhibitors and beta-blockers for the management of heart failure due to left ventricular systolic dysfunction?
Applicability of evidence
Making recommendations for all people with heart failure requires an extrapolation to older people of evidence derived from younger people. Although there have been large trials of both ACE inhibitors and beta-blockers in older people (aged ≥ 70 years) with heart failure, those aged > 75 years are a very large proportion of the population with heart failure and so are significantly under-represented.
Drug–disease interactions
Chronic kidney disease and conditions associated with significant undiagnosed renovascular disease are common in people with heart failure, and much more common in older people with heart failure.
Drug–drug interactions
Coprescribing of drugs that have renal adverse effects in addition to ACE inhibitors is common.
Findings for depression treatment with selective serotonin reuptake inhibitor antidepressant
Characteristics of the trial populations
There were a large number of relatively small trials of SSRI antidepressants in people with depression (CG9045) and in people with depression with a chronic physical health problem (CG9117). Not all trials reported detailed inclusion or exclusion criteria, or the age range within which patients were eligible, or the mean age of those included. We therefore focus on age in this section. Trials included in CG90 usually had lower age limits of 18–21 years, and the majority (23 of 39 trials that reported age range for eligibility) had upper age limits between 56 and 65 years. However, as Figure 8 shows, the mean age of recruited patients was well below this, being between 36 and 45 years for 28 of the 31 trials reporting mean age. Among trials included in CG91, the majority selected patients based on them having one condition, most commonly stroke (seven trials), cancer (four trials), diabetes (four trials) and Parkinson’s disease (three trials). The mean age of included patients was higher than for the depression alone trials, but patients were more middle aged than older (e.g. the four depression with cancer trials had mean participant ages of 53, 54, 56 and 60 years).
FIGURE 8.
Mean age of patients included in studies of SSRI antidepressants for the management of depression.
Comparison of trial population and the population for which recommendations are being made
Using epidemiological data to make this comparison is more problematic than for type 2 diabetes and heart failure because depression is not well coded in clinical records, so the epidemiological data are for people treated with antidepressants. We therefore measured depression in a large data set derived from GP clinical records, defining it as ‘depression Read Code recorded in the last year OR receipt of four or more antidepressants (excluding low dose tricyclic antidepressants) in the last year’. 13 The population being compared with the trial population is therefore people with ‘recently recorded or currently treated depression’. 13 Of note is that clinically diagnosed or drug-treated depression is likely to include people with persistent mild to moderate symptoms of depression, who would not have been eligible for trials. Using this definition, 75.0% of people with depression were aged < 65 years, and 12.6% aged ≥ 75 years (Table 10).
| Condition | Eligible for trials, aged < 65 years (n = 108,264) | Eligible for some trials but seriously under-represented, aged 65–74 years (n = 17,827) | Eligible for a few trials, seriously under-represented, aged ≥ 75 years (n = 18,212) | Overall (n = 144,303) |
|---|---|---|---|---|
| Painful condition | 23.9 | 40.8 | 32.7 | 27.1 |
| Hypertension | 14.2 | 46.3 | 52.7 | 23.0 |
| Anxiety or insomnia | 18.0 | 25.8 | 32.9 | 21.2 |
| CHD | 4.2 | 22.8 | 28.5 | 9.6 |
| Diabetes | 6.5 | 18.7 | 16.1 | 9.3 |
| Thyroid disorders | 7.2 | 14.5 | 16.8 | 9.3 |
| COPD | 4.5 | 14.8 | 13.9 | 6.9 |
| Stroke/TIA | 2.0 | 11.5 | 18.4 | 5.2 |
| Cancer in last 5 years | 2.7 | 8.3 | 10.2 | 4.4 |
| CKD | 1.1 | 9.2 | 17.0 | 4.1 |
| Peripheral vascular disease | 1.5 | 6.4 | 7.6 | 2.9 |
| Dementia | 0.3 | 3.1 | 15.8 | 2.6 |
| Heart failure | 0.7 | 4.6 | 9.1 | 2.2 |
| Atrial fibrillation | 0.5 | 4.5 | 10.5 | 2.2 |
| Number of other conditions | ||||
| 0 | 29.9 | 4.9 | 2.3 | 23.3 |
| 1 | 26.4 | 12.5 | 7.6 | 22.3 |
| 2 | 18.6 | 18.8 | 13.7 | 18.0 |
| 3 | 11.7 | 19.0 | 17.0 | 13.3 |
| ≥ 4 | 43.3 | 44.8 | 59.4 | 23.1 |
| Any physical conditiona | ||||
| 0 | 40.3 | 8.0 | 4.7 | 31.8 |
| 1 | 26.5 | 16.5 | 11.9 | 23.4 |
| 2 | 16.0 | 21.0 | 17.4 | 16.8 |
| 3 | 9.0 | 19.3 | 19.2 | 11.5 |
| ≥ 4 | 8.2 | 35.2 | 46.8 | 16.4 |
Comorbidity was common at all ages, including physical comorbidity, but was much commoner in older people. Of people with depression aged < 65 years, 59.7% had at least one of the 33 physical conditions counted, with almost one-quarter having a painful condition and 14.2% hypertension. More than 90% of those aged > 65 years had a physical condition. In those aged > 75 years, 84.4% had two or more comorbid physical conditions; 28.5% had CHD and 16.1% diabetes, which are the two conditions in which comorbid physical disease and depression have been most studied.
Drug–disease and drug–drug interactions
The BNF states only acute mania as a disease contraindication for a SSRI, but this could not be reliably examined using GP clinical data. To examine drug–drug interactions, we used dispensing data for all patients resident in the NHS Tayside region of Scotland to define a population of people with treated depression in terms of ‘current dispensing’ of SSRIs (drugs in BNF chapter 4.3.3) or MAOIs (BNF chapter 4.3.2) or ‘other’ antidepressants (BNF chapter 4.3.4). 85 Current dispensing was defined as the dispensing of one or more of these drugs in the 84 days before 31 March 2010.
Table 11 lists drugs with potentially severe interactions with SSRI antidepressants, and the percentage of the population with depression who are currently dispensed them. Strikingly, more than one-third of older people with depression treatment were prescribed aspirin, with significant percentages prescribed oral anticoagulants, NSAIDs and clopidogrel. SSRIs increase bleeding risk, with cumulative increases in risk when coprescribed with other drugs causing bleeding, and further increases in risk with increasing age. 16 Other commonly coprescribed drugs were tramadol, TCAs and antiepileptics (although in practice the interaction – lowering of seizure threshold – does not apply to most people taking such drugs because they are prescribed them for pain modification).
| Recommended drug for depression and potentially seriously interacting drugs | Eligible for trials, aged < 65 years (n = 19,430) | Eligible for some trials but seriously under-represented, aged 65–74 years (n = 2826) | Eligible for a few trials, seriously under-represented, aged ≥ 75 years (n = 2998) |
|---|---|---|---|
| Aspirin | 6.4 | 32.1 | 42.8 |
| Oral anticoagulants | 0.7 | 4.6 | 6.9 |
| Clopidogrela,b | 0.7 | 2.9 | 4.1 |
| NSAIDs | 11.3 | 9.9 | 4.6 |
| Lithium | 1.2 | 1.1 | 1.3 |
| TCAs | 6.3 | 8.7 | 5.9 |
| MAOIs | 0.5 | 0.7 | 0.2 |
| Tramadol | 6.1 | 8.4 | 7.1 |
| Tamoxifena,c | 0.03 | 0.7 | 0.5 |
| Antiepileptics | 8.9 | 9.8 | 6.6 |
| Carbamazepinea,b | 1.5 | 1.9 | 1.2 |
| Phenytoina,b | 0.2 | 0.7 | 0.4 |
| Pimozide | 0 | 0 | 0 |
| Haloperidola,d,e | 0.2 | 0.4 | 1.1 |
| Aripiprazolea,c | 0.5 | < 0.01 | < 0.01 |
| Clozapinea,b,c,f | 0.1 | < 0.01 | < 0.01 |
| Droperidola,b,f | 0 | 0 | 0 |
| Rasagiline or selegelinea,b,c,f | < 0.01 | 0.5 | 0.7 |
| Ritonavir | 0 | 0 | 0 |
| Antiarrhythmicsd,e | 0.1 | 0.6 | 0.7 |
| Mizolastined,e | < 0.01 | < 0.01 | 0 |
| Mobeclomidea,b,d,e,f | 0 | 0 | 0 |
| Moxifloxacind,e | < 0.01 | 0 | < 0.01 |
| Telithromycine | 0 | 0 | 0 |
| 5HT1 antagonistse | 1.7 | 0.4 | < 0.01 |
| Sumatriptana,b,c,d | 0.7 | 0.1 | < 0.01 |
| Antimalarials (includes quinine)e | 1.0 | 5.4 | 5.4 |
| Aminophylline/theophyllineb | 0.4 | 1.6 | 0.9 |
| Metoprololc | < 0.01 | 0.4 | 0.4 |
| Sotalold,e | < 0.01 | 0.01 | 0.4 |
Are there applicability issues with selective serotonin reuptake inhibitor treatment for depression?
Applicability of evidence
Current evidence is derived from the main population of people with treated depression: those aged < 65 years without and with chronic physical disease. Although older people are only a relatively small proportion of people with depression (in contrast to heart failure and to type 2 diabetes), they are a population who almost all have chronic physical disease and who have very high rates of coprescribing of drugs with potentially serious interactions.
Drug–disease interactions
There are no significant drug–disease interactions that need considering.
Drug–drug interactions
There a high prevalence of significant drug–drug interactions, particularly those associated with GI bleeding.
Discussion
Summary of findings
The analysis shows that it is feasible to use a combination of trial inclusion and exclusion criteria and epidemiological data to examine more systematically whether or not applicability of evidence is likely to be a problem, and whether or not there are important drug–disease or drug–drug interactions. An important limitation is that the study was not funded to carry out de novo data extraction and analysis. Our ability to completely match guideline populations was therefore incomplete. For example, for the examination of drug–drug interactions, heart failure was defined as hospital-admitted heart failure, in which category people are likely to have more severe symptoms, and depression was defined as drug treatment with selected antidepressants. In addition, it is likely that epidemiological data will be of higher quality for some conditions (e.g. type 2 diabetes, where diagnostic criteria are clear) than others (e.g. depression, where diagnosis and recording in routine records are more variable). Strikingly, the three exemplar conditions have distinct patterns of applicability and interaction problems.
Making recommendations for the initial treatment of hyperglycaemia in all people newly diagnosed with type 2 diabetes requires an extrapolation to older people of evidence derived from much younger people. Older people have many more comorbidities and are prescribed more drugs. They are at a higher baseline risk of CVD, meaning that in principle they would benefit more from effective treatment, but there is little evidence that tight glycaemic control per se improves cardiovascular outcomes in the first 10 years of treatment. Metformin treatment does improve morbidity and mortality, but the effect sizes are relatively small over a relatively long time. In this situation, where benefit accrues over a long time and where adverse events from treatment are probably common, our interpretation is that consideration of the potential importance of reduced life expectancy due to comorbidity/competing risks is indicated. Drug–disease interactions are common for CKD, which is common in people with type 2 diabetes at diagnosis (9.1% overall, 25.5% in those aged > 75 years), and is relevant to metformin and sulphonylurea prescribing. Our interpretation is that consideration of whether or not this requires noting in recommendations is indicated. There are two fairly common potentially serious drug–drug interactions to consider between sulphonylureas and coumarins, and between sulphonylureas and NSAIDs.
Making recommendations for the use of ACE inhibitors and beta-blockers for all people with heart failure requires an extrapolation to older people of evidence derived in younger people. Although there have been large trials of both ACE inhibitors and beta-blockers in older people (aged ≥ 70 years) with heart failure, those aged > 75 years are a very large proportion of the population with heart failure and so are significantly under-represented overall in the body of evidence informing guideline recommendations. In addition, trials are largely in people with significant (and often severe) LVSD and at least moderate symptoms despite best treatment, and exclude people with significant comorbidities. However, given evidence of substantial benefit in trial populations, it is unlikely that older populations would not benefit, although the risks of treatment will be greater, at a minimum because of greater comorbidity and coprescribing. Drug–disease interactions for ACE inhibitors are common for comorbid CKD and conditions associated with significant undiagnosed renovascular disease, both of which are common in people with heart failure, and much more common in older people with heart failure. Our interpretation is that consideration of whether or not this requires noting in recommendations or inclusion of recommendations to mitigate risk is indicated. Coprescribing of drugs that have renal adverse effects in addition to ACE inhibitors is common. Our interpretation is that consideration of whether or not explicit accounting for renal function requires noting in relevant recommendations or inclusion of recommendations to mitigate risk is indicated.
Making recommendations for SSRI treatment of depression requires less extrapolation in the sense that current evidence is derived from the main population of people with treated depression: those aged < 65 years without and with chronic physical disease. However, although older people are only a relatively small proportion of people with depression, they are a population who almost all have chronic physical disease and who have very high rates of coprescribing of drugs with potentially serious interactions. Extrapolation to this group is therefore potentially problematic because harms are likely to be much more common than in trial populations, and our interpretation is that this should be explicitly considered in recommendations. There are no significant drug–disease interactions that need considering. There is a high prevalence of significant drug–drug interactions, particularly those associated with GI bleeding, and our interpretation is that these require consideration when writing recommendations.
Of note is that the heart failure and depression (including depression with a physical condition) guidelines did address some of the identified issues, although the type 2 diabetes guideline did not (beyond a general statement to agree individualised glycaemic control targets where appropriate). However, a formal consideration of the epidemiology of the population for which recommendations are being made at scoping would be potentially useful to ensure that applicability is explicitly considered by GDGs and problems of applicability are responded to appropriately. Such formal consideration will always be limited by the data available, in terms of both the detail that trial reports provide on participants (which was poorer for depression trials than for the much larger trials considered for the two other conditions) and the quality of the epidemiological data on the characteristics of the treated population. In terms of the latter, guideline producers such as NICE or SIGN would ideally create a single data set describing patterns of morbidity and prescribing, which could then be used in the development of multiple guidelines.
Implications of findings
Given access to large and representative epidemiological data sets that characterise the population for which guideline recommendations are being made, it is feasible to examine more systematically the extent of the extrapolations being made in making treatment recommendations to inform GDG decisions about whether or not and how these recommendations should be qualified. It is worth noting that GDGs do already do this, but, as we understand it, this is largely driven by the knowledge and expertise of the individual members who happen to have been recruited. However, we believe that applicability and interactions are sufficiently important that they should be addressed more systematically, in the same way that evidence synthesis is, rather than being left to being judged in the context of the informal knowledge and expertise of GDG members.
Of note is that we do not believe that applicability is sufficiently dealt with if GRADE methods are used. GRADE consideration of applicability takes place during evidence synthesis both for individual trials and for the body of evidence in any meta-analysis. Applicability is one of four criteria used to judge whether evidence is direct or indirect. As defined by GRADE:
We are more confident in the results when we have direct evidence. By direct evidence, we mean research that directly compares the interventions in which we are interested delivered to the populations in which we are interested and measures the outcomes important to patients.
p. 1304106
However, the consequence of indirectness in GRADE is that the quality of the evidence that may support a recommendation is downgraded, which influences the strength with which recommendations are made. Our conclusion is that applicability either needs explicitly accounting for by defining pre-specified subgroups to examine during evidence synthesis, if there is enough prior evidence to support this approach, or needs explicitly re-examining after evidence synthesis and drawing on the findings of all included studies, in order to inform the writing of recommendations (Figure 9). This is because there may be direct evidence for some people in the guideline population but not for others, and therefore it may be appropriate to make different recommendations for subgroups based on different quality of evidence in those subgroups.
FIGURE 9.
Directness and applicability at different stages of guideline development.
Then GDGs will have to consider whether they wish to make a single recommendation for all people with a condition, or stratified or otherwise qualified recommendations for different subgroups. Such judgements already happen but we believe that systematic use of epidemiological data to inform them is required. Based on discussion with the PRG, factors which GDGs might consider when making such judgements include:
-
the nature of the treatment, in terms of whether or not its mechanism of effect is likely to apply across all patients, and its potential for harm
-
the duration over which the treatment will be used, which is relevant when extrapolating to populations with limited life expectancy due to other conditions, age or general frailty
-
the absolute size of the observed benefit in the trials, which is relevant, since large benefits are less likely to be sensitive to small variations in benefit or harm when used in people not eligible for trials
-
the nature of the differences between trial and non-trial populations, including age, comorbidity, coprescribing and likely life expectancy, in terms of whether or not these are large enough to matter in the context of the previous factors.
Such considerations are particularly likely to apply when the outcomes being improved by treatment are not observable by clinicians or patients. For example, clinicians and patients can observe change in pain during treatment with an analgesic. In contrast, most preventative treatments require clinicians and patients to take it on trust that meaningful outcomes are better, because a prevented heart attack or other prevented future event is not observable in an individual.
Conclusion
Problems of applicability of evidence are the norm in guideline development, because there is usually extrapolation of evidence from trial populations to groups of the population who were excluded from trials but who are nevertheless potential candidates for the treatment. The extent of this extrapolation varied across the treatment recommendations examined, as did the potential implications depending on exactly how trial and non-trial populations differed. More systematic use of epidemiological information has the potential to usefully inform how GDGs account for applicability and interactions.
Chapter 4 Comparing treatments in terms of absolute benefit
Background
People with multimorbidity often experience significant treatment burden, because of the number of treatments they are required to use (medicines, rehabilitation of various kinds, self-care interventions) and the number of health-care providers that they are asked to attend in community, primary and hospital care. 20 The single most commonly used treatment is medication, and one important aspect of treatment burden is polypharmacy. Polypharmacy is conventionally defined as taking either five or more, or sometimes 10 or more, different drugs. 32,120,121 Polypharmacy is associated with higher rates of high-risk prescribing, potential drug–drug interactions and adverse drug effects,32–34 presenting clinicians and patients with complex decisions about optimising treatment to maximise benefit and minimise adverse effects. An important problem that clinicians and patients often face in this situation is making decisions about which drugs are most likely to be of benefit, including whether or not and when it is rational to stop drugs that are recommended by guidelines.
Current NICE guidelines and NICE pathways (the two short versions of guidelines) do not give any indication of the actual benefit of drugs being recommended, although the full versions of more recent guidelines do usually have some statement about the absolute benefits of treatment. This makes it difficult for clinicians and patients to make rational decisions when optimising complex medication regimens. One aim of the overall project was therefore to examine how the benefit of drugs for different conditions could be compared. This topic has also been prioritised for the NICE ‘Multimorbidity: clinical assessment and management’ guideline. 42 The final scope of the multimorbidity guideline notes that a key issue to be covered is ‘Ranking absolute risks and benefits of interventions for prevention or improving prognosis of common morbidity (for example, treatments to improve glucose and blood pressure control, statins, angiotensin-converting enzyme [ACE] inhibitors, drugs for osteoporosis)’ (p. 4). 42
How best to express the comparative benefits or harms of treatment is uncertain, although the most frequently proposed method is to compare treatments in terms of their absolute benefit across one or more outcomes.
Using absolute benefit expressed in terms of trial outcomes
Historically, binary outcome treatment effects measured in RCTs were usually only reported in terms of relative risks or risk ratios (RRs), odds ratios, relative risk reductions (RRRs) or hazard ratios. Not all patient-centred outcomes are binary, but such outcomes are common, including mortality, serious events such as stroke or heart attack, and admission to hospital or a care home. The evidence-based medicine movement highlighted that reporting binary outcomes in terms of relative benefit was often misleading, since the absolute benefit to which this translates often varies widely depending on the baseline risk of the outcome. 122 For example, a treatment that reduces death by 60% (RR 0.4, RRR 0.6) sounds very impressive, but does not actually prevent many deaths if only 10 in 1000 people not treated die compared with 4 in 1000 people who are treated. Instead the recommendation was that clinicians focus on absolute benefit, which in the example above would be six avoided deaths per 1000 people treated (ARR of 0.006 deaths per patient treated). However, since ARR is not always easy to grasp, it was proposed that this could be usefully expressed as a ‘number needed to treat’ (NNT), which is defined as 1/ARR and conventionally rounded up to the nearest whole number. In the hypothetical example given, the NNT would be 1/0.006 = 167 (i.e. 167 patients would need to receive the treatment to avoid one death).
One important rationale of the NNT was to create a measure of treatment effect that was more meaningful to clinicians and patients, and therefore better able to support decision-making. There is some evidence that clinicians feel that NNTs are meaningful123 and that they vary their treatment recommendations in response to vignettes more appropriately when presented with NNTs than RRRs. 124 However, although there is evidence that both clinicians and patients are both less likely to choose a theoretical treatment if benefit is presented as a NNT or ARR than when it is presented as a RR or RRR, it is unclear whether or not the use of NNTs actually influences practice, and NNT does not appear to be as helpful to patients. 125,126 Stovring et al. 127 suggest that all numbers may be confusing to patients, but that ARR is marginally easier for them to interpret.
Of note, however, is that, despite the appealing face validity of the example above, such simple statements of NNTs are in practice misleading. For example, ‘NNT of 167 to avoid one death’ leaves much unsaid that has to be explicitly considered, including the nature of the treatment and what it is being compared with, the duration of treatment and the length of follow-up. The interpretation of that NNT would be very different if the treatment were a tablet with no side effects, taken once, with the death avoided in the next 24 hours, from if it were lifelong treatment with a drug that caused persistent low-level nausea in everyone who took it, with the death avoided after 30 years of treatment. 128,129
Overall, there is general approval of the idea of reporting treatment effects in terms of absolute risk; for example, the Consolidated Standards of Reporting Trials statement for trial reporting states ‘For binary outcomes, presentation of both absolute and relative effect sizes is recommended’. 130 However, a number of important problems have been identified in using absolute benefit and NNT. In practice, the NNT in particular is not routinely calculated or reported in trials, systematic reviews or guidelines (although the increasing use of the GRADE approach means that guidelines increasingly report net benefit of treatments in a standardised way105,131,132).
Critiques of the number needed to treat
A major problem with estimates of absolute benefit and the NNT is that they are very dependent on baseline risk. This means that simply estimating a NNT from a single trial or from a meta-analysis will be helpful only if the population or individual to whom that NNT is applied has the same or a similar baseline risk. However, trial populations often have different baseline risks from each other and from real-world populations, and secular trends in baseline risk further complicate interpreting a NNT derived from meta-analysis. 133 Several studies of trials including in large numbers of meta-analyses have shown that, in contrast to NNT estimates, which vary between trials depending on baseline risk of outcomes, estimates of relative treatment effect are usually fairly stable irrespective of the population being studied. 134,135 In other words, if a treatment reduces risk of an outcome by 10% in one population, it usually – but not always – reduces it by a similar percentage in a different population. One specific example is the reduction in CVD from cholesterol-lowering treatments, which appears to be reasonably constant across different levels of baseline risk. 136 However, even in this context other studies have found some evidence of heterogeneity, with lower RRRs in people with hypertension than those without (although, since people with hypertension have higher baseline risk of CVD, they still obtained similar absolute benefit despite a lower relative reduction from treatment). 137
Although the majority of treatments do appear to have stable RRRs across different levels of baseline risk, it is also clear that this is not universal,138 with Schmid et al. 139 finding that relative risk or odds ratio varied depending on baseline risk in approximately one in seven of 115 meta-analyses of different treatments. To some extent, this may depend on the nature of the treatment. It is particularly likely for surgical treatments and others where there is a large initial risk from treatment that may not vary much by baseline risk of the outcome, and then a variable absolute benefit that depends on baseline risk, meaning that relative risk is not stable across populations. 128,138,140 It may also depend on the nature of the outcome, particularly for total mortality where mortality in trial populations may be dominated by outcomes that the treatment affects, whereas mortality in non-trial populations will sometimes or often have other, more important, determinants. In this situation, where there are competing risks of mortality, trial estimates of RRR are likely to be too high. 128,138 In practice, most systematic reviews report absolute benefit calculated by applying a pooled relative risk from a meta-analysis to a pooled baseline risk from the control arms of the trials in the meta-analysis (e.g. the median baseline risk across all included trials), and this is the approach generally recommended in GRADE and used by NICE.
An alternative is to apply the pooled relative risk to a population cohort estimate of baseline risk or to an estimated baseline risk for an individual. 129,141 This requires an assumption that the relative risk of treatment is stable across populations. Although this assumption will sometimes be wrong,142 any attempt to generalise from results in trial populations to other populations for which there is no direct evidence typically has to assume that RRR is stable, although this assumption is not always made explicit. This highlights the importance of being clear about what assumptions are made when creating measures of absolute benefit.
In a review 20 years after the NNT was first proposed, McAlister128 summarises much of the relevant literature, highlighting that NNT depends on a comparison between two therapies in a particular context, and is influenced by three variables beyond the treatment effect as expressed by the relative risk, namely:128
-
Baseline risk. This varies between populations and over time in the same population as a result of long-term trends and the impact of other treatments for the same condition becoming widely used.
-
The time frame over which outcomes are measured. For preventative treatments, ARR will typically increase over time (although, in the long run, we will of course all die) meaning that the NNT over a long period would be expected to be smaller than the NNT over a short period.
-
The outcome being measured. Trials typically measure multiple outcomes, each of which can have a NNT calculated for it that will need interpreting and weighing against other outcomes.
Of note is that all these critiques apply equally to relative risk and RRR. Although relative risk is (usually) stable across populations and, therefore, a more attractive summary measure of treatment effect in meta-analysis, in practice relative risk is uninterpretable without knowledge of baseline risk. Relative risk also has the same problem of meaning different things over different time periods (because baseline risk over a long period will usually be higher than baseline risk over a short period), and relative risk estimates from trials are also reported for a range of different outcomes that may be difficult to compare directly. In practice, therefore, the key issue is whether NICE, SIGN or other guideline organisations are willing and able (in a resource-constrained environment) to address these three problems. Potential ways of addressing them include acquiring and publishing data to inform implementation (e.g. population baseline risk data), making the many required assumptions in generating comparative absolute risk estimates (e.g. by recalculating absolute risk estimates so that they are all over the same time period to facilitate comparability) and/or investing in developing effective ways of presenting large amounts of comparative data to clinicians and patients.
In what contexts might measures of absolute benefit such as numbers needed to treat be used?
In the original evidence-based medicine context, NNTs were intended to be used to inform specific treatment decisions for individuals: should this patient take this treatment for this condition? Even those who are cautious about using NNTs generally conclude that they have a place in this context, notably when choosing between two treatments for the same condition tested in people with similar baseline risk and with benefit measured over the same time frame. As McAlister says, ‘If it is to be used to compare treatments, the therapies must have been tested in similar populations with the same condition at the same stage, using the same comparator, time period and outcomes’. 128 This use of the NNT in decision-making is particularly suited to comparing relatively short-duration treatments, such as choosing which antidepressant to use in someone with new-onset depression.
This is very different from the context of use envisaged in this project, which is to provide clinicians and patients with information about the relative benefits of treatments for different conditions, for example when treatment is burdensome to the patient or when treatments of different conditions are incompatible with each other. In such circumstances, decisions may be made about treatment optimisation where there is little direct evidence because trials typically exclude people with complex comorbidity, and information about which treatments are likely to provide greater benefit may be useful. We consider this to be particularly important for preventative therapies, where the benefit is unobservable in an individual, in contrast to treatments for symptoms, where both clinician and patient can judge effectiveness more directly.
Using estimated lifetime quality-adjusted life-year gain as a single metric for comparison
The QALY presents an attractive solution to at least one of the three problems highlighted by McAlister128 in his critique of the NNT: that of disparate outcome measures. In principle, the QALY provides a standardised outcome by which treatments can be benchmarked and prioritised for those with multimorbidity based on absolute health gains.
The QALY has become the cornerstone of CEA used to inform resource allocation decisions because it provides a generic measure of patient benefit that, when combined with costs, allows mutually exclusive alternative interventions to be compared and prioritised. The QALY calculation has two dimensions that are combined: survival multiplied by HRQoL. Generating values for the survival dimension can be argued to involve few value judgements. In contrast, generating values for the HRQoL dimension requires a fundamental judgement regarding what constitutes ‘quality’. The NICE methods guide for technology appraisal recommends using the European Quality of Life-5 Dimensions (EQ-5D) as the generic HRQoL measure. 59 The EQ-5D is a multiattribute measure of HRQoL comprising five domains with three levels (a newer five-level version is now available), which generates a total of 243 health states. 143 To use it in the context of economic evaluations, it is necessary to understand the preference weight attached to each health state. The commonly agreed scale used is anchored at 1 for full health and 0 for health states equivalent to dead. NICE recommends that that the values attached to the HRQoL dimension should be informed by societal preferences. The preference weights currently used in the UK were collected as part of a study called the Social Tariff survey. 143 The preference weights were estimated using a time trade-off exercise with a representative set of the UK population.
Traditionally the QALY has been limited to the domain of economic evaluation. In this domain, the benefits of the QALY are clear, as it provides a common currency by which scarce health-care resources can be allocated in the most efficient manner. The characteristics of the QALY, calculated as a combination of morbidity and mortality into a single measure of absolute benefit, make it attractive outside this domain. 144
If communicated using clear and explicit language suitable for the audience, the use of the absolute QALY could potentially provide a useful measure of clinical benefit to stakeholders, such as patients and clinicians, as an aid to their treatment decisions. In addition, guideline developers focusing on multimorbidity could use estimates of the absolute QALY to complement current cost-effectiveness evidence as an additional source of evidence. The absolute QALY, once a set of interventions have been found to be individually cost-effective, could be used to prioritise treatments within bundles of interventions.
For the purposes of this work, the absolute QALY is defined as the number of QALYs gained from receiving intervention A minus the number of QALYs gained without receiving intervention A. The ‘absolute’ part of the absolute QALY is reliant on the comparator being defined as the ‘do-nothing’ option.
Figure 10 is adapted from a NICE Decision Support Unit (DSU) document that was used as supportive evidence to inform discussions on whether or not to preferentially value interventions that treat conditions in which the ‘burden of illness’ is large. 145 It can be used to illustrate the absolute QALY concept. As part of this evidence base, the absolute QALY shortfall was put forward as a potential proxy measure of ‘burden of illness’. When showing the importance of burden of illness, the starting point is X in Figure 10, where patients are already getting treatment for their condition. The burden of illness is then defined as the difference in the area that lies between O1 and O2 and between O1 and X (labelled areas D, E and F in Figure 10). Each of these areas, with its own interpretation, represents the absolute QALYs a patient would lose on an intervention compared with the total possible number of QALYs if in, age-adjusted, full health. The QALY shortfall has two dimensions: a HRQoL impact (measured by D) and a survival impact (measured by F). The area E represents the consequential impact of both HRQoL and survival impact together. In contrast, the starting point for the absolute QALY calculation starts at the value of QALYs assuming patients are not on treatment (point Y on Figure 10). At point Y the baseline QALYs for no treatment are represented by the area Z. The absolute QALY gain is calculated as the difference between X and Y (areas A, B and C). Survival gain is measured by the change in S and HRQoL gain is measured by the change in H. The combined impact of the improvement in survival (area F) and improvement in HRQoL (C and A) is represented by area B.
FIGURE 10.
The use of absolute QALY shortfall as a proxy for burden of illness. Source: adapted from the NICE DSU document. 145
Theoretical application of the absolute quality-adjusted life-year concept
In this section, a theoretical application of the absolute QALY concept is described. Consider two interventions, A and B. Intervention A improves both the HRQoL and survival domains of the QALY but the survival gain is relatively short. Intervention B improves only the survival gain but does so with a longer duration of survival.
Table 12, when used in conjunction with Figure 10, illustrates how, in this scenario, intervention B would result in a larger absolute QALY gain of 1.25 than intervention A with an absolute QALY gain of 1.17.
| Intervention | HRQoL without treatment (utility) | Life expectancy without treatment (years) | Life expectancy with treatment (years) | HRQoL gain with treatment (utility) | QALYs without treatment | QALYs with treatment | Absolute QALY gain |
|---|---|---|---|---|---|---|---|
| A | 0.5 | 5.5 | 5.6 | 0.7 | 2.75 | 3.92 | 1.17 |
| B | 0.5 | 5.5 | 8.0 | 0.5 | 2.75 | 4.00 | 1.25 |
| Corresponding area on Figure 10 | |||||||
| Area F | Area G | N/A | N/A | Area Z | Areas A + B + C + Z | Areas A + B + C | |
Comparing interventions using the absolute quality-adjusted life-year concept
To compare interventions using the absolute QALY concept it is necessary to use a broad set of principles:
-
interventions that allow comparison with a null option
-
standard length of time for analysis
-
comparable baseline risk
-
standardised reference case
-
understanding of uncertainty.
The general aim of these principles is to be clear about, and when feasible limit, some of the inherent uncertainty when comparing interventions using the absolute QALY concept. These five principles are now described.
Principle 1: interventions that allow comparison with a null option. The use of the absolute QALY to prioritise interventions is most meaningful when the comparison of a potential new intervention is with a do-nothing comparator as a realistic and feasible scenario. This implies that the absolute QALY is potentially useful in two areas: (1) for preventative interventions, either primary or secondary, which could include ongoing medical management such as statin therapy or screening programmes, compared with not having these interventions; and (2) for improving the prognosis of an ongoing chronic morbidity, such as pharmacotherapy for osteoporosis.
Principle 2: standard length of time for analysis. The size of the absolute QALY gain is dependent on the length of time over which the analysis has been conducted (known as the ‘time horizon’ for the analysis). Analyses that are based on shorter time horizons will have lower absolute QALY gains than those analyses that take a lifetime horizon, because patients will have had less chance to accrue benefit. Equally important is the start point for the analysis in terms of the age of the patient population. Patients who are older will have less opportunity to accrue absolute QALYs than patients who are younger. Therefore, direct comparisons between treatments using absolute QALY gains will need to be standardised by two criteria: (1) starting age of the patient population and (2) use of a consistent (ideally) lifetime horizon.
Principle 3: comparable baseline risk. Ideally the baseline risks used within the analysis to generate the absolute QALY should be similar across the various options under consideration. In practice, this might not always be feasible where interventions are targeting different underlying biological mechanisms in different disease areas. A minimum requirement to be met should be an equivalent risk of all-cause mortality between the alternatives being compared.
Principle 4: standardised reference case. Absolute QALYs should be compared only where they have been produced using a common overarching framework within the same jurisdiction. This framework should standardise, among others, the discount rate, the outcome measures used (e.g. the EQ-5D) and the study perspective. The NICE Methods Guide for Technology Appraisal59 (hereafter called the NICE reference case) provides a good example of a standardised approach. By applying a reference case and using results from a single jurisdiction, many of the context-specific variation in methods can be limited. It is also important to take account of the fact that even with a standardised reference case there is potential for methods to be non-compliant with the suggested methods (because of either time constraints or limitations in the data for other reasons).
Principle 5: understanding of uncertainty. Understanding the uncertainty associated with the absolute QALY is likely to be complex. Philips et al. 146 suggest four potential sources of uncertainty:
-
Stochastic uncertainty is the random variation that may exist between any two identical patients due to random fluctuations in an outcome unrelated to their characteristics.
-
Parameter uncertainty is our uncertainty in relation to a parameter of interest, such as the risk reduction associated with any particular treatment.
-
Heterogeneity relates to the variability between patients explained by their known underlying differences in characteristics.
-
Structural uncertainty relates to the differences in outcomes caused by the assumptions made within any particular model.
Ideally, as with any CEA, the sources of uncertainty should be identified and reported explicitly. The NICE reference case recommends that a probabilistic sensitivity analysis (PSA) be included in the submitted evidence for all technology appraisals, to provide a measure of parameter uncertainty and variability about mean incremental costs and QALYs. 59 There is no similar recommendation for economic evidence used to inform NICE CGs. Anecdotal evidence suggests there is variability in the type of sensitivity analysis used in model-based CEA to inform NICE CGs. This lack of consistency poses a challenge for producing measures of uncertainty around estimates of absolute QALYs, because the basis of the analysis will be existing model-based CEA used to produce NICE CGs. If there is no submitted PSA then it will not be possible to generate a measure of uncertainty around the absolute QALY.
How to generate evidence to provide absolute quality-adjusted life-years: model- or trial-based analysis?
Cost-effectiveness analysis can be conducted using one of two ‘vehicles’ for the evaluation: (1) an analysis based entirely on evidence from a single clinical trial; or (2) an analysis based on evidence from multiple sources as inputs into a decision-analytic model-based CEA. The two vehicles of evaluation each have their relative advantages and disadvantages. 58,147
One advantage of a trial-based analysis is that it is likely to have the best properties for robust internal validity. 147 The measure of absolute QALYs generated within a clinical trial is likely to be the most robust estimate for the stated trial population for the time period studied. One limitation is linked with principle 2, which outlined the need for a standardised time horizon, ideally lifetime. This principle is unlikely to be upheld within a trial-based analysis, and consequently the benefits of using model-based economic analysis to generate absolute QALYs are clear. Model-based economic analyses allow the results from a clinical trial, such as the estimated hazard ratio between treatment and comparator, to be extrapolated beyond the time frame found within the trial and be converted into QALYs. Therefore, the problem of short-term trials or trials of differing lengths is removed. Model-based analyses also have the potential to generate QALY estimates using multiple data sources and allow flexibility in the type of relevant comparator used. It is relatively straightforward to introduce a do-nothing comparator in a model-based analysis because meta-analysis or network meta-analysis can be used to provide the relevant data for the do-nothing comparator. A further advantage of a model-based approach is that model parameters can be adjusted to standardise the populations under comparison, for example the baseline risk. This makes the model the more amenable vehicle to produce statistics that allow absolute QALY comparisons to be made.
A key drawback of model-based analyses, however, is that they have the potential to be viewed as black boxes with the structural assumptions hidden from decision-makers. It is important, therefore, that key assumptions be reported in a transparent manner and that a reference case be followed, such as the NICE reference case, which details the methods-based approach to be used for the analysis. 59
Aim
The aim of this study was to implement two methods of estimating the absolute benefit of treatment using clinical outcomes as measured in trials and absolute QALY as a composite outcome, and to make explicit the assumptions required in creating such estimates.
Methods
For clinical outcomes, after discussion with the PRG, we focused on two of the three exemplar conditions (heart failure and type 2 diabetes) because the treatments for these conditions are relatively long-term, the aim of treatment is often (although not always) preventative, and preventative benefits cannot be observed in individuals. For each guideline, we focused on first-line drug treatments as already described in Chapter 2 and defined in Appendix 1, and examined trials and systematic reviews referenced by the relevant guidelines for heart failure,51 type 2 diabetes50 and lipid modification148 as key evidence informing recommendations. For each selected treatment, and drawing on GRADE recommendations for the initial steps:149
-
We estimated the relative risk associated with a treatment versus comparator using published data in guidelines or meta-analyses.
-
We estimated the ARR and NNT, by applying that relative risk to the baseline risk of the outcome in the comparator arm of the trial population or to the mean or median trial duration (depending on what the guideline reported).
-
We calculated annualised NNT by assuming that the observed benefit of an intervention in trials conducted over a longer period would be accrued evenly over time, and that the observed benefit of an intervention in trials conducted over a shorter period would continue to accrue after the trial completed.
-
Where available, we estimated ARR and NNT for real-world populations using population data on baseline risk where available, using a range of estimates of baseline risk to give a sense of the range of likely absolute benefit depending on patient characteristics.
To examine the use of absolute QALY gain as an outcome, we chose two case studies to provide examples of a potential application of how to generate and use absolute QALYs. The case studies were selected to (1) minimise the potential problems in comparability between interventions as described in the five principles and (2) include only interventions that have been recommended as being most cost-effective, within existing NICE CGs. The case studies were identified from two guidelines produced by the National Clinical Guideline Centre on behalf of NICE:
-
pharmacological treatment for patients with hypertension from the 2011 NICE hypertension guideline (CG127)150
-
pharmacological treatment for the prevention of CVD from the 2014 NICE guideline on lipid modification (CG181). 148
Both models used a Markov structure, using 1-yearly cycles, and assumed a lifetime horizon. Table 13 summarises the key model attributes.
| Model attribute | Pharmacological treatment in hypertension150 | Pharmacological treatment for lipid management148 |
|---|---|---|
| Published date | August 2011 | July 2014 |
| Collaborating centre | National Clinical Guideline Centre | National Clinical Guideline Centre |
| Guideline methods manual | 2009 | 2012 |
| Evaluation type | Model-based | Model-based |
| Model type | Markov (cycle = 1 year) | Markov (cycle = 1 year) |
| Horizon taken | Lifetime, to 100 years old | Lifetime, to 100 years old |
| Starting age | 65 years old | 60 years old |
| Model period | 35 years | 40 years |
| Comparators |
|
|
| Do-nothing included? | Yes | Yes |
| Intervention type | Primary preventative | Primary preventative |
| Population | Essential hypertension seen in primary care, excluding those with pre-existing CVD (annual 1% risk), heart failure (annual 2% risk) or diabetes (annual 1.1% risk) | Primary prevention for people without existing CVD and without diabetes, using the primary model, calibrated to relate to CVD risk as predicted by the QRisk2 tool (version 2015-0, ClinRisk Ltd, Nottingham) |
| Cost-effective option | Calcium-channel blockers | High-intensity statin treatment |
Calculating absolute quality-adjusted life-years
Quality-adjusted life-years were calculated using the same data inputs as in the original model used to inform the NICE CGs. All QALYs were discounted at a rate of 3.5%. To make the models comparable for the purpose of estimating absolute QALYs, some of the baseline assumptions that were used to inform the original guideline recommendations were adapted. The base-case results in the antihypertensives model were for a 65-year-old with a 2% annual risk of CVD. This risk was lowered to a 1% annual risk to bring it in line with the lipid modification guideline, which used a 10% 10-year risk of CVD. Subgroup analyses for different starting ages were also conducted.
Absolute QALYs were calculated by subtracting the expected QALYs for no treatment from the expected QALYs of treatment with the cost-effective intervention. It was not feasible to produce a measure of the uncertainty around the mean absolute QALYs because the model-based CEA used as exemplars did not include the necessary type of PSA as part of the original evidence submitted to inform the NICE CGs. The PSA used included only a selection of parameter values.
Results
Comparing absolute benefit using trial outcomes
Tables 14 and 15 show a version of the GRADE profile used by NICE modified for our purposes to focus on relative and absolute benefits and omitting assessment of quality of evidence for reasons of space. Stang et al. 129 recommend that a NNT should never be quoted without clarity about which treatments are being compared, the period of treatment and follow-up, and the direction of the effect towards benefit or harm.
| Outcome and condition | Intervention, n (%) of patients with outcome | Comparator, n (%) of patients with outcome | Relative risk (95% CI) | Absolute benefit (95% CI) | NNT (95% CI) | Duration of treatment |
|---|---|---|---|---|---|---|
| Total mortality in heart failure due to LVSD151 | ACE inhibitors, 1467 of 6391 (23.0) | Placebo, 1710 of 6372 (26.8) | 0.86 (0.81 to 0.91) | 39 more people benefit per 1000 (from 24 more to 54 more) | 27 (20 to 42) | Mean trial duration 35 months (15–42 months) |
| Readmission to hospital with heart failure151 | ACE inhibitors, 876 of 6391 (13.7) | Placebo, 1202 of 6372 (18.9) | 0.73 (0.67 to 0.79 | 52 more people benefit per 1000 (from 39 more to 65 more) | 20 (16 to 26) | Mean trial duration 35 months (15–42 months) |
| Stroke with heart failure151 | ACE inhibitors, 239 of 6391 (3.7) | Placebo, 249 of 6372 (3.9) | 0.96 (0.80 to 1.14) | 2 more people benefit per 1000 (from 5 fewer to 9 more) | No significant difference | Mean trial duration 35 months (15–42 months) |
| Total mortality in heart failure due to LVSD152 | Beta-blockers, 440 of 5378 (8.2) | Placebo (majority are on ACE inhibitors or ARBs), 622 of 4642 (13.4) | 0.61 (0.54 to 0.69) | 53 more people benefit per 1000 (from 40 more to 65 more) | 20 (17 to 25) | Mean trial duration 11 months |
| Hospitalisation with heart failure152 | Beta-blockers, 613 of 5301 (11.6) | Placebo (majority are on ACE inhibitors or ARBs), 833 of 4827 (17.3) | 0.67 (0.61 to 0.74) | 57 more people benefit per 1000 (from 44 more to 71 more) | 18 (15 to 24) | Mean trial duration 11 months |
| Total mortality in people with newly diagnosed type 2 diabetes with BMI > 30 kg/m2108 | Metformin, 50 of 342 (14.6) | Diet (plus drugs only if very poor control), 89 of 411 (21.7) | 0.68 (0.49 to 0.93) | 71 more people benefit per 1000 (from 16 more to 125 more) | 15 (10 to 66) | Median 10.7 years |
| Diabetes-related death in people with newly diagnosed type 2 diabetes with BMI > 30 kg/m2108 | Metformin, 28 of 342 (8.2) | Diet (plus drugs only if very poor control), 55 of 411 (13.4) | 0.61 (0.40 to 0.94) | 52 more people benefit per 1000 (from 8 more to 96 more) | 20 (11 to 125) | Median 10.7 years |
| MI in people with newly diagnosed type 2 diabetes with BMI > 30 kg/m2108 | Metformin, 39 of 342 (11.4) | Diet (plus drugs only if very poor control), 73 of 411 (17.8) | 0.64 (0.45 to 0.92) | 64 more people benefit per 1000 (from 14 more to 114 more) | 16 (9 to 74) | Median 10.7 years |
| Stroke in people with newly diagnosed type 2 diabetes with BMI > 30 kg/m2108 | Metformin, 12 of 342 (3.5) | Diet (plus drugs only if very poor control), 23 of 411 (5.6) | 0.63 (0.32 to 1.24) | 21 more people benefit per 1000 (from 9 fewer to 51 more) | No significant difference | Median 10.7 years |
| Microvascular outcomes in people with newly diagnosed type 2 diabetes with BMI > 30 kg/m2108 | Metformin, 24 of 342 (7.0) | Diet (plus drugs only if very poor control), 38 of 411 (9.2) | 0.76 (0.46 to 1.24) | 23 more people benefit per 1000 (from 17 fewer to 62 more) | No significant difference | Median 10.7 years |
| Total mortality in people with newly diagnosed type 2 diabetes107 | Sulphonylurea or insulin, 489 of 2729 (17.9) | Diet (plus drugs only if very poor control), 213 of 1138 (18.7) | 0.94 (0.80 to 1.10) | 8 more people benefit per 1000 (from 19 fewer to 35 more) | No significant difference | Median 10.7 years |
| Diabetes-related death in people with newly diagnosed type 2 diabetes107 | Sulphonylurea or insulin, 285 of 2729 (10.4) | Diet (plus drugs only if very poor control), 129 of 1138 (11.3) | 0.90 (0.73 to 1.11) | 9 more people benefit per 1000 (from 13 fewer to 31 more) | No significant difference | Median 10.7 years |
| MI in people with newly diagnosed type 2 diabetes107 | Sulphonylurea or insulin, 387 of 2729 (14.2) | Diet (plus drugs only if very poor control), 186 of 1138 (16.3) | 0.84 (0.71 to 1.00) | 22 more people benefit per 1000 (from 4 fewer to 47 more) | No significant difference | Median 10.7 years |
| Stroke in people with newly diagnosed type 2 diabetes107 | Sulphonylurea or insulin, 148 of 2729 (5.4) | Diet (plus drugs only if very poor control), 55 of 1138 (4.8) | 1.11 (0.81 to 1.51) | 6 fewer people benefit per 1000 (from 10 fewer to 21 more) | No significant difference | Median 10.7 years |
| Microvascular outcomes in people with newly diagnosed type 2 diabetes107 | Sulphonylurea or insulin, 225 of 2729 (8.2) | Diet (plus drugs only if very poor control), 121 of 1138 (10.6) | 0.75 (0.60 to 0.93) | 24 more people benefit per 1000 (from 4 more to 45 more) | 42 (23 to 312) | Median 10.7 years |
| Total mortality in people with newly diagnosed type 2 diabetes148 | Statin, 149 of 2869 (5.2) | Placebo, 178 of 2831 (6.3) | 0.83 (0.67 to 1.02) | 11 more people benefit per 1000 (from 2 fewer to 23 more) | No significant difference | 2–5.4 years |
| Cardiovascular mortality in people with newly diagnosed type 2 diabetes148 | Statin, 95 of 3026 (3.1) | Placebo, 111 of 3010 (3.7) | 0.85 (0.65 to 1.11) | 6 more people benefit per 1000 (from 4 fewer to 15 more) | No significant difference | 2–5.4 years |
| Non-fatal MI in people with newly diagnosed type 2 diabetes148 | Statin, 60 of 1940 (3.1) | Placebo, 106 of 1936 (5.5) | 0.56 (0.41 to 0.77) | 24 more people benefit per 1000 (from 12 more to 37 more) | 42 (28 to 91) | 2–5.4 years |
| Stroke in people with newly diagnosed type 2 diabetes148 | Statin, 223 of 5230 (4.3) | Placebo, 309 of 5234 (5.9) | 0.72 (0.61 to 0.85) | 17 more people benefit per 1000 (from 8 more to 25 more) | 61 (41 to 126) | 2–5.4 years |
| Outcome and condition | Intervention, n (%) of patients with outcome | Comparator, n (%) of patients with outcome | Relative risk (95% CI) | NNT (95% CI) | Duration of treatment |
|---|---|---|---|---|---|
| Total mortality in heart failure due to LVSD151 | ACE inhibitors, 1467 of 6391 (23.0) | Placebo, 1710 of 6372 (26.8) | 0.86 (0.81 to 0.91) | 27 (20 to 42) | Mean trial duration 35 months (15–42 months) |
| Total mortality in heart failure due to LVSD152 | Beta-blockers, 440 of 5378 (8.2) | Placebo (majority are on ACE inhibitors or ARBs), 622 of 4642 (13.4) | 0.61 (0.54 to 0.69) | 20 (17 to 25) | Mean trial duration 11 months |
| Total mortality in people with newly diagnosed type 2 diabetes with BMI > 30 kg/m2108 | Metformin for glycaemic control, 50 of 342 (14.6) | Diet (plus drugs only if very poor control), 89 of 411 (21.7) | 0.68 (0.49 to 0.93) | 15 (10 to 66) | 10.7 years |
| Total mortality in people with newly diagnosed type 2 diabetes107 | Sulphonylurea or insulin for glycaemic control, 489 of 2729 (17.9) | Diet (plus drugs only if very poor control), 213 of 1138 (18.7) | 0.94 (0.80 to 1.10) | No significant difference | 10.7 years |
| Total mortality in people with type 2 diabetes148 | Statin, 149 of 2869 (5.2) | Placebo, 178 of 2831 (6.3) | 0.83 (0.67 to 1.02) | No significant difference | 2–5.4 years |
Table 14 shows the relative risk and ARR for multiple outcomes for five different treatment/condition combinations: ACE inhibitors and beta-blockers in heart failure due to left systolic dysfunction, metformin and sulphonylureas/insulin in newly diagnosed type 2 diabetes, and statins in people with type 2 diabetes. The size of the table highlights that providing clinicians and patients with comparative data for treatments for different conditions is likely to fairly rapidly require decisions about which outcomes to compare, and paper-based ways of delivering this information such as NICE already produce153 are unlikely to be feasible. Of note is that, as McAlister128 points out, comparing across multiple outcomes is challenging. Table 15 shows the same data but only for the total mortality outcome, which makes direct comparisons more feasible. The NNTs are estimated from the trial data (i.e. by applying the estimated trial RRR to the baseline risk in the control group), and where the relative risk is not significantly different from 1 then NNT has not been calculated. Of note is that all NNTs are of a similar order (most are 15–30, the largest is ≈ 60), but this cannot be easily interpreted because trial duration varies more than 10-fold, and the NNT in itself is not meaningfully interpretable without simultaneous consideration of the duration of treatment. In addition, extrapolating these NNTs to other populations is straightforward only if the baseline risk of the outcome is the same as or very similar to that observed in the trial population. 133
Estimating annualised absolute risk reduction and number needed to treat to improve comparability
Table 16 reports the same total mortality outcomes as Table 15 and is based on applying pooled relative risk to the estimate of baseline risk for the trial populations, but also includes an estimate of the annualised ARR and annualised NNT. This is intended to make it easier to compare trials with very different lengths of treatment and follow-up, and requires two important assumptions.
| Outcome and condition | Intervention, n (%) of patients with outcome | Comparator, n (%) of patients with outcome | Relative risk (95% CI) | NNT (95% CI) | Duration of treatment | Annualised NNT (95% CI) |
|---|---|---|---|---|---|---|
| Total mortality in heart failure due to LVSD151 | ACE inhibitors, 1467 of 6391 (23.0) | Placebo, 1710 of 6372 (26.8) | 0.86 (0.81 to 0.91) | 27 (20 to 42) | Average trial duration 35 months (15–42 months) | 79 (59 to 123) |
| Total mortality in heart failure due to LVSD152 | Beta-blockers, 440 of 5378 (8.2) | Placebo (majority are on ACE inhibitors or ARB), 622 of 4642 (13.4) | 0.61 (0.54 to 0.69) | 20 (17 to 25) | Average trial duration 11 months | 19 (16 to 23) |
| Total mortality in people with newly diagnosed type 2 diabetes with BMI > 30 kg/m2108 | Metformin, 50 of 342 (14.6) | Diet (plus drugs only if very poor control), 89 of 411 (21.7) | 0.68 (0.49 to 0.93) | 15 (10 to 66) | 10.7 years | 160 (107 to 704) |
| Total mortality in people with newly diagnosed type 2 diabetes with BMI > 30 kg/m2107 | Sulphonylurea or insulin, 489 of 2729 (17.9) | Diet (plus drugs only if very poor control), 213 of 1138 (18.7) | 0.94 (0.80 to 1.10) | No significant difference | 10.7 years | No significant difference |
| Total mortality in people with type 2 diabetes (NICE lipid guideline)148 | Statin, 149 of 2869 (5.2) | Placebo, 178 of 2831 (6.3) | 0.83 (0.67 to 1.02) | No significant difference | 2–5.4 years | No significant difference |
The first assumption is that the observed benefit of an intervention in trials conducted over a longer period is accrued evenly over time. There is some evidence that this is not always the case. For example, the RRR from ACE inhibitors in LVSD is greater in the first 90 days than subsequently. 128,138 RRR from statins in people at high risk of cardiovascular events in the Anglo-Scandinavian Cardiac Outcomes Trial was also initially larger,128 although, conversely, individual meta-analysis of major statin trials found that the RRR was lower in the first year of treatment than subsequently. 154 Further caution is required if treatments have large initial harms relative to annually accruing benefits, which applies particularly to surgical and some screening treatments. Annualisation from a longer follow-up in this situation may be very misleading and overestimate initial benefit. 128
The second assumption is that the observed benefit of an intervention in trials conducted over a shorter period would continue to accrue after the trial completed. Since the longer-term outcome of a short-term trial is by definition unobservable, this is a stronger assumption. If initial RRR is lower than later RRR, then this assumption would underestimate annual absolute benefit, and vice versa if initial RRR is higher. More broadly, the longer a short trial benefit is extrapolated, the less plausible the extrapolation, since competing risks of death are likely to become more common. 128
As expected given the varying trial durations, annualised NNT was more variable than trial-estimated NNT, with a range from 59 to 150 for the three treatments with a significant effect on total mortality, compared with 15 to 27 in the original estimates over different durations of treatment. The key advantage of annualising is that it simplifies interpretation because the reader has to read only one number rather than simultaneously interpret two (NNT and duration of treatment). The assumptions underlying this simplification are non-trivial. However, they are already implicit in normal practice, in that clinicians simultaneously considering a NNT value and a trial duration are likely to make them, and presentations of absolute benefit in the NICE evidence tables do not state duration, implying that absolute benefit is interpretable independent of this. The assumptions are also less problematic if comparisons are being made between drugs taken for prevention of future events with trial follow-up over relatively long periods (1–5 years), than if comparing a drug treatment taken every day with a surgical intervention.
Using more realistic estimates of baseline risk
An important limitation of absolute risk estimates calculated using the baseline rate of the outcome in trial control arms is that these may not be very representative, and therefore may over- or underestimate likely absolute benefits in the treated population or in individuals. Depending on how the trial population has been selected, control-arm baseline risk may be higher or lower than baseline risk in the real-world population, and baseline risks may vary considerably between different groups of the real-world population.
From a guideline development perspective, for conditions where there are published data on the distribution of baseline risk across the population, it would be feasible to estimate the likely range of absolute benefit across the actual range of baseline risk in the population for which recommendations are being written. This would potentially allow a GDG to define if there are subgroups for whom recommendations could or should be varied. For individual patient decision-making, baseline risk can be estimated by a suitable prediction tool (if available). Such tools would ideally have been developed using a representative population cohort and validated in an independent cohort. Commonly used examples of such calculators include the Framingham and QRisk scores for risk of CVD, and the FRAX™ (Sheffield, UK version, World Health Organization Collaborating Centre for Metabolic Bone Diseases) and QFracture (version 2012-1, ClinRisk Ltd, Nottingham) scores for risk of fracture. For heart failure, there are two risk prediction tools available for estimating total mortality risk. The Seattle Heart Failure model is derived from a single trial population and validated in five other trial populations. 155 The Meta-Analysis Global Group in Chronic heart failure (MAGGIC) model156 is derived from six trial and 24 observational populations and has been externally validated using the Swedish national heart failure registry. 157 For type 2 diabetes, the NICE lipid modification guideline recommended using QRisk2 to estimate cardiovascular risk in people with type 2 diabetes, on the basis that it was derived from a much larger and more recent cohort than the UKPDS Risk Engine or the Framingham study. 148 In practice, baseline risk estimates in real-world populations are often not available, and, for the illustration here, we chose to use MAGGIC estimates of 1-year survival in heart failure, and QRisk2 estimates of cardiovascular risk in type 2 diabetes.
Variation in baseline risk and absolute benefit within a population
Table 17 illustrates the range of MAGGIC-estimated baseline risk of death at 3 years observed in the Swedish national heart failure registry (Ulrik Sartipy, Karolinska Institute, Sweden, 2014, personal communication). Baseline risk of mortality at 3 years varied from 13.5% in the lowest-risk group to 70.5% in the highest-risk group. Assuming that the RRR from ACE inhibitor treatment versus placebo is constant, there is therefore almost fivefold variation in estimated absolute benefit. Of note here is that the assumption of constant RRR across the whole population is relatively strong for total mortality, since ACE inhibitor treatment would be expected to influence only heart failure mortality. Although heart failure mortality is a very large proportion of total mortality in people with heart failure, the proportion varies somewhat with age (with a higher proportion of non-heart failure mortality in older people with heart failure). 158 However, although we have not annualised the estimate of absolute benefit, the mean duration of treatment in the trials from which the RR estimate is derived was 35 months, meaning that the assumptions underlying annualisation are more likely to be reasonable. This emphasises that the plausibility of the assumptions required to use absolute benefit in practice are at least partly context dependent.
| MAGGIC risk groupa | Patients (n)b | Predicted 3-year mortality (%) | RR (95% CI) | ARR (range) | NNT (95% CI) |
|---|---|---|---|---|---|
| 1 | 7674 | 13.5 | 0.86 (0.81 to 0.91) | 18 fewer deaths per 1000 (from 12 fewer to 25 fewer) | 53 (39 to 83) |
| 2 | 8043 | 22.3 | 0.86 (0.81 to 0.91) | 31 fewer deaths per 1000 (from 20 fewer to 42 fewer) | 33 (24 to 50) |
| 3 | 11,445 | 30.9 | 0.86 (0.81 to 0.91) | 43 fewer deaths per 1000 (from 27 fewer to 78 fewer) | 24 (18 to 36) |
| 4 | 11,279 | 41.2 | 0.86 (0.81 to 0.91) | 57 fewer deaths per 1000 (from 37 fewer to 78 fewer) | 18 (13 to 27) |
| 5 | 7849 | 53.5 | 0.86 (0.81 to 0.91) | 74 fewer deaths per 1000 (from 48 fewer to 101 fewer) | 14 (10 to 21) |
| 6 | 4753 | 70.5 | 0.86 (0.81 to 0.91) | 98 fewer deaths per 1000 (from 63 fewer to 134 fewer) | 11 (8 to 16) |
Comparing treatments using annualised number needed to treat across plausible ranges of baseline risk
For heart failure, we used published data on the distribution of predicted risk score in the Swedish national heart failure register (which have a near normal distribution) to estimate average, lower and higher baseline risks for mortality at 1 year defined as MAGGIC scores of 25, 17 and 32 (approximately encompassing the interquartile range of risk). 157 Using these baseline risk estimates and estimates of relative risk from meta-analyses, we estimated 1-year ARR and NNT for ACE inhibitors versus placebo and for beta-blockers versus placebo.
For type 2 diabetes, we are not aware of any published data on the individual distribution of cardiovascular risk. We therefore chose to model 10-year baseline risks of CVD at 10%, 15% and 20%, representing the range between current (10%) and previously recommended (20%) thresholds for use of statins for primary prevention of CVD. The published guidelines do not provide a relative risk for reduction in total CVD events, and we therefore applied the estimated RR for preventing MI using metformin versus diet (plus drugs, only if glycaemic control were very poor) from UKPDS34108 and using statins versus placebo. 148 Since statins are less effective at preventing stroke, this is likely to overestimate the benefits of statins on total CVD events. We then annualised the NNT to account for the 10-year follow-up implied by the QRisk baseline risk estimate.
Table 18 shows the estimated ARR and annualised NNT. Annualised NNTs across the range of estimated baseline risk examined were 9–34 for beta-blockers versus placebo and 25–93 for ACE inhibitors versus placebo to prevent one death in people with LVSD. NNTs ranged from 139 to 278 for metformin versus diet and from 114 to 228 for statins versus placebo to prevent one cardiovascular event in people with type 2 diabetes.
| Outcome and condition (source reference) | Treatment and comparator | Relative risk (95% CI) | Baseline risk (%) | Absolute benefit | NNT (95% CI) | Assumed duration of treatmenta | Annualised NNT (95% CI) |
|---|---|---|---|---|---|---|---|
| Total mortality in heart failure due to LVSD151 | ACE inhibitor vs. placebo | 0.86 (0.81 to 0.91) | 7.7 (MAGGIC risk score 17b) | 10 fewer per 1000 (from 6 fewer to 14 fewer) | 93 (69 to 145) | 1 year | 93 (69 to 145) |
| Total mortality in heart failure due to LVSD151 | ACE inhibitor vs. placebo | 0.86 (0.81 to 0.91) | 16.1 (MAGGIC risk score 25b) | 22 fewer per 1000 (from 14 fewer to 30 fewer) | 45 (33 to 70) | 1 year | 45 (33 to 70) |
| Total mortality in heart failure due to LVSD151 | ACE inhibitor vs. placebo | 0.86 (0.81 to 0.91) | 29.3 (MAGGIC risk score 32b) | 41 fewer per 1000 (from 26 fewer to 55 fewer) | 25 (18 to 38) | 1 year | 25 (18 to 38) |
| Total mortality in heart failure due to LVSD152 | Beta-blockers vs. placebo | 0.61 (0.54 to 0.69) | 7.7 (MAGGIC risk score 17b) | 30 fewer per 1000 (from 23 fewer to 35 fewer) | 34 (29 to 42) | 1 year | 34 (29 to 42) |
| Total mortality in heart failure due to LVSD152 | Beta-blockers vs. placebo | 0.61 (0.54 to 0.69) | 16.1 (MAGGIC risk score 25b) | 62 fewer per 1000 (from 49 fewer to 74 fewer) | 16 (14 to 21) | 1 year | 16 (14 to 21) |
| Total mortality in heart failure due to LVSD152 | Beta-blockers vs. placebo | 0.61 (0.54 to 0.69) | 29.3 (MAGGIC risk score 32b) | 11 fewer per 1000 (from 9 fewer to 13 fewer) | 9 (8 to 11) | 1 year | 9 (8 to 11) |
| MI or stroke in people with type 2 diabetes108 | Metformin vs. diet (+ drug treatment if very poor glycaemic control) | 0.64 (0.45 to 0.92)c | 10d | 36 fewer per 1000 (from 8 fewer to 55 fewer) | 28 (19 to 125) | 10 years | 278 (182 to 1250) |
| MI or stroke in people with type 2 diabetes108 | Metformin vs. diet (+ drug treatment if very poor glycaemic control) | 0.64 (0.45 to 0.92)c | 15d | 54 fewer per 1000 (from 12 fewer to 82 fewer) | 19 (13 to 84) | 10 years | 186 (122 to 834) |
| MI or stroke in people with type 2 diabetes108 | Metformin vs. diet (+ drug treatment if very poor glycaemic control) | 0.64 (0.45 to 0.92)c | 20d | 72 fewer per 1000 (from 16 fewer to 110 fewer) | 14 (10 to 63) | 10 years | 139 (91 to 625) |
| MI or stroke in people with type 2 diabetes (NICE lipid guideline)148 | Statin vs. placebo | 0.56 (0.41 to 0.77)c | 10d | 44 fewer per 1000 (from 23 fewer to 59 fewer) | 23 (17 to 44) | 10 years | 228 (170 to 435) |
| MI or stroke in people with type 2 diabetes (NICE lipid guideline)148 | Statin vs. placebo | 0.56 (0.41 to 0.77)c | 15d | 66 fewer per 1000 (from 34 fewer to 88 fewer) | 16 (12 to 29) | 10 years | 152 (113 to 290) |
| MI or stroke in people with type 2 diabetes (NICE lipid guideline)148 | Statin vs. placebo | 0.56 (0.41 to 0.77)c | 20d | 88 fewer per 1000 (from 46 fewer to 118 fewer) | 12 (9 to 22) | 10 years | 114 (85 to 218) |
Comparing treatments using absolute quality-adjusted life-year gain
Table 19 presents the estimated absolute QALY gains from pharmacological treatment of hypertension and lipid management based on the model-based CEAs used to inform the relevant NICE guidelines. The results show that the lifetime absolute QALY gain from the use of calcium-channel blockers to treat hypertension is much larger than the lifetime gain for high-intensity statin treatment to manage a similar level of cardiovascular risk.
| Patients | Calcium-channel blockers to treat hypertension (age 65 years at start of model) | High-intensity statins to treat cardiovascular risk (age 60 years at start of model) |
|---|---|---|
| Men | 0.85 | 0.20 |
| Women | 0.91 | 0.25 |
Discussion
Comparing the absolute benefit of treatments using trial clinical outcomes
There are no major technical barriers to using a consistent method to produce different estimates of the absolute benefit of treatments for different conditions, but all such estimates rely on making a number of significant assumptions, namely:
-
Relative risk is assumed to be constant across all populations. This is a standard assumption and it is difficult to extrapolate any trial evidence without making it. There is evidence that it is usually but not always true. 138
-
Competing risks of death are assumed to be insignificant. This is also a standard assumption that is rarely examined, and is unlikely to be true in the presence of other conditions with a high risk of death. For example, mortality in heart failure trial populations is likely to be more driven by heart failure than by other conditions, whereas mortality in real-world populations with higher rates of comorbidity and greater age-related frailty than trial populations is likely to have a larger component that is not amenable to heart failure treatment. As an example, a young patient without comorbidity who is in the highest risk group of MAGGIC scores is likely to have severe, symptomatic LVSD and this will drive their mortality risk. An older patient with comorbidity may be in the same risk group despite having asymptomatic mild LVSD, and a larger proportion of their mortality will be from causes that will not be affected by heart failure treatment. In populations with significant competing risks of death, benefits are therefore likely to be overestimated by the method applied.
-
Baseline risk is assumed to be measured without error, at both population level (e.g. median baseline risk in a trial) and individual level if a risk calculator is used in individual decision-making. 159 The confidence intervals (CIs) around ARR and NNT estimates are therefore likely to be falsely narrow. Although there are methods available to account for this, baseline risk calculators do not normally provide CIs around baseline risk estimates. 160 A broader concern is that all baseline risk estimates are at risk of a number of forms of bias. For example, observational data in principle provide better estimates of baseline risk in the population who will actually receive the treatment than trial estimates. However, observational data are more likely to underascertain outcomes because they usually rely on routine clinical recording of incident conditions. In addition, both trial and observational data may not reflect changes due to changes over time in baseline risk, for example because treatment with other effective interventions has become routine. 159
-
Estimates of relative risk and estimates of baseline risk may not be available for the same outcome. Here, they were not both available for metformin and statins; the relative risk used is for the outcome of MI, whereas the baseline risk estimate is for total cardiovascular risk, which includes fatal and non-fatal stroke. Since the estimated relative risk for stroke reduction is somewhat smaller in both cases, the actual absolute benefit is also likely to be smaller and so the NNT somewhat larger.
-
Harm is assumed to be constant across populations, which is unlikely given that older people and those taking multiple drugs are known to be more likely to experience ADEs. 34 For metformin, there is evidence that age, coprescribing and genetic factors all contribute to metformin discontinuation in the absence of treatment failure (as a proxy for adverse effects), with age dominating. 161 For people on ACE inhibitors, risk of acute kidney injury in the face of coprescribing of other nephrotoxic drugs is strongly associated with increasing age. 162 Net benefit may therefore be reduced in some populations because of increased harm.
Are measures of absolute benefit and number needed to treat helpful?
The idea of using absolute benefit and NNT estimates to inform clinical decision-making is very appealing and is feasible in at least some cases, but is troublesome because it requires significant assumptions. However, most of the problems also apply to any attempt to use a relative-risk or RRR estimate to inform clinical decision-making, since this requires the same set of assumptions, although these are not usually made explicit.
In principle, the ideal approach in decision-making for an individual would be to use a validated risk prediction tool to estimate their baseline risk of the outcomes that they prioritise, and then apply trial-derived relative risks to estimate the benefit that that individual can expect from treatment. A key limitation is that baseline risk prediction tools may not exist, be validated or estimate the right outcomes (either those an individual prioritises or those where there are trial estimates of relative risk). Assuming that suitable baseline risk calculators exist, then annualising absolute benefits simplifies comparison, albeit at the cost of more assumptions, although again these are commonly implicitly made anyway. However, this approach usually ignores the problems of competing risks.
Failing this, then using annualised, wholly trial-derived absolute risk estimates (see Table 20, for example) gives an indication of treatments of which likely absolute benefits are different by an order of magnitude. This in itself may meaningfully inform individual clinical decision-making when there is a wish to prioritise treatments, for example when life expectancy is short or when treatment burden is intolerable. In principle, such tables could be created as part of single-disease guideline development, since the steps required need only minimal adaptation of what is already done. However, making such data feasible to use in practice is likely to require an electronic method of delivery whereby the user selects which conditions and which treatments to see data for.
Comparing absolute benefit using quality-adjusted life-years
Absolute QALYs gained are a potentially attractive concept that could potentially inform prioritisation between two treatments. In the example given, where key principles were satisfied, then antihypertensive treatment on average leads to three- to four-fold QALY gain compared with statin treatment. For example, there is a QALY gain of 0.85 in men aged 60 years with antihypertensive treatment compared with 0.20 with statin treatment (although, even in this case, the models do not entirely match, as the starting age is not identical). This approach specifically addresses one of McAlister’s128 three critiques relating to the problem of comparing the benefit of treatments when they affect different outcomes. However, making such comparisons is not as straightforward as it might first appear, and we propose that it should be underpinned by five guiding principles that help make assumptions explicit, and that can be reported and provided to decision-makers wishing to use the information to guide prioritisation.
There are a number of important considerations to take account of when using absolute QALY gains to compare between interventions. The decision-analytic models used in this analysis were similar in terms of a number of key attributes, but differed in the time horizon for the analysis, as the antihypertensive model ran for 35 years compared with 40 years for the lipid modification model. The length of time in which absolute QALYs have had chance to accrue is a key variable that may drive the results of an analysis estimating absolute QALYs. Economic evaluations are disparate in terms of the assumed time horizon for the analysis. 163 The relevant time horizon will be guided by the relevant patient population and the need to capture the key differences between intervention and comparators during the time period for the analysis. For the selected case studies, there was a relatively small difference of 5 years. This difference in time horizon is unlikely to change the findings substantially but analysts will have to be transparent and inform decision-makers when the difference in time horizon between models is likely to bias the ability to compare between interventions using absolute QALYs. They also differed in the characteristics of the starting cohort. Two model-based CEAs could have the same time horizon (lifetime) but, if the starting age of the original cohort is different, then the length of time in which the QALYs have to accrue will clearly be different. Similarly, even for treatments affecting the same outcome (in our example, CVD), then the way in which each model defines the population studied in terms of baseline risk may have large effects on the conclusions drawn.
Following the approach recommended in the NICE reference case, a de novo model-based CEA created to inform guideline development usually reports the incremental QALY comparing the intervention with the next best alternative. This approach is underpinned by the logic that NICE is assessing the cost-effectiveness of mutually exclusive, non-interacting interventions. However, incremental comparisons against the next best alternative are not the same as absolute gains against a do-nothing approach. In the analysis estimating absolute QALYs gained, we assumed a do-nothing alternative to allow direct comparison between interventions. This approach is not new. The recognition that this standard method of generating incremental QALYs from CEA may not be not appropriate when interactions between interventions are important has been recognised by the World Health Organization in its generalised cost-effectiveness framework. 164
The generalisability of findings from model-based CEA has been the topic of ongoing debates between health economists and users of economic evidence. 163,165 Some health economists argue that the limited generalisability of economic evaluations means that producing an assimilated evidence base is not useful. 166 A review of cost-effectiveness studies found some 26 factors that influenced how results from studies would vary from location to location and from time to time. 163 In the analysis presented here, the issue of poor generalisability, when generating absolute QALYs for comparison between interventions, was taken into account by a number of key assumptions. Costs were not included and, therefore, many of the socioeconomic and organisational factors that vary between organisations affecting resource use and cost were eliminated. Most of the jurisdiction-specific factors were also limited by adopting a standardised approach, such as the NICE reference case, and comparing only outcomes that have been produced in agreement with this approach.
The QALY has become a key input into model-based CEA, and the current NICE reference case recommends using published public preference weights for health states generated using the EQ-5D. 59 This approach has become accepted practice when using model-based CEA to inform guidance produced by NICE as part of its technology appraisal or guideline development processes. The estimate of an absolute QALY within either a trial- or model-based framework does not imply that all patients can be expected to receive such an outcome, in the same way that estimates of life expectancy do not mean all individuals will live for the same length. The absolute QALY is an aggregate measure calculated either within a hypothetical cohort of patients or within the trial sample. Some patients would probably gain fewer QALYs and some would gain more, but the absolute QALY measure is what would be expected over the cohort or sample. This variability in absolute QALYs will have no impact when using the values to inform decisions affecting populations of patients but may limit the interpretation of the relevance of absolute QALY values at the individual patient and clinician level.
The absolute QALY gain is suggested as another input for decision-makers developing CGs for populations of patients. There is potential, however, for decision-making based on the absolute QALY to diverge from other decision-making criteria, such as recommendations based on cost-effectiveness. For example, an intervention that had a large absolute QALY gain but was expensive might not be a cost-effective option, with the incremental benefit not justifying the incremental cost for a given cost-effectiveness threshold. Likewise, the text-book approach when making decisions between mutually exclusive options based on cost-effectiveness is to compare interventions with the next best alternative. Comparisons with a do-nothing approach (e.g. mean cost-effectiveness) may produce misleading estimates of the true displaced activities when compared with the next best alternative. The suggested solution to this challenge is to generate estimates of absolute QALYs only once the relative cost-effectiveness and most cost-effective option for an intervention in a defined patient population have been determined.
Finally, from a guideline development perspective, a key limitation of the absolute QALY gain approach is that it relies on having access to a suitable economic model. As shown in Chapter 2, fewer than one-fifth of CRQs have an associated de novo economic model, and about half have no economic evidence associated with them at all. From this perspective, comparing absolute benefits in terms of trial clinical outcomes is more straightforward, since these may be more easily available.
Conclusion
It is feasible to compare the absolute benefit of treatments for different conditions, provided that guideline developers and users of the information are willing to make the required assumptions (although many existing treatment decisions involve the implicit acceptance of these assumptions anyway). Guideline developers will also have to be willing to invest the resource required. From the latter perspective, the current NICE guideline development process uses the GRADE framework and therefore already produces estimates of the absolute benefit of treatment on outcomes that the GDG has chosen as critical and important. These estimates are based on the relative risk of single trials or the pooled relative risk from meta-analyses and some measure of the baseline risk of the outcome in the trial population, but are not readily comparable because current evidence tables do not make the duration of treatment explicit. Using estimates of absolute benefit based on a plausible range of population baseline risks and accounting for duration of treatment by annualising estimates are two relatively simple (although assumption-laden) methods for making it easier to compare absolute benefit. However, it seems inevitable that delivering such data to clinicians and patients will require an electronic rather than a paper platform, to allow users to select which conditions, treatments and outcomes to compare. In addition, previous research suggests that there is no single way of presenting absolute benefit (ARR or NNT expressed as numbers or as text, or graphical displays of various kinds) that is preferred by clinicians and patients, so allowing users to choose how to view absolute benefit estimates would also seem valuable (although beyond the scope of this project). Absolute QALY gain is an alternative metric that is feasible to use when appropriate economic models exist, and where the principles outlined in this chapter apply. However, the QALY is a metric that may have greater value for GDGs than in individual decision-making.
Chapter 5 Including a temporal dimension in model-based cost-effectiveness analysis: an application to the use of statins in primary prevention of cardiovascular disease
Introduction
Clinicians involved with selecting treatments for patients with multimorbidity are increasingly encouraged to consider the evidence on benefits and harms alongside a temporal dimension, for example that decisions should take account of life expectancy. The application of this temporal dimension has been referred to using a number of related but distinct terms including TTB, time horizon to benefit or, alternatively, pay-off time. In 2012, the AGS outlined some key principles for the care of patients with multimorbidity. 40 One key principle recommended by the AGS was that the time it takes for benefits of treatments to be realised, together with the absolute size of the treatment benefit, should be used as an integral part of the clinical decision-making process. 40 The rationale underpinning this suggestion is clear. Patients with limited life expectancy resulting from comorbid conditions, age or frailty may never accrue the benefits from interventions with deferred effects. Where there are also upfront harms associated with an intervention there is also the potential for overall net harm.
Current CGs generally do not provide explicit information on the temporal dimensions of benefit. In some instances, the temporal dimension of benefit is implicitly alluded to in the evidence provided to inform the CG, where trial duration is presented in the reference tables. 167 The lack of explicit information on the potential impact of a temporal dimension of benefit will make it difficult for clinicians, patients and guideline developers to prioritise among multiple potentially relevant interventions. The AGS recommended that interventions forming part of complex regimens should ideally be selected and prioritised based on evidence that indicates whether or not net benefit occurs within a patient’s expected lifespan.
The overall aim of this study was to investigate whether or not interventions can be compared by making explicit reference to a temporal dimension of benefit. The estimation of the pay-off time in the context of generating economic evidence from a model-based CEA is suggested as a readily applicable concept to quantify the potential impact of including a temporal dimension of benefit to inform the development of CGs. In this chapter, the key underlying concepts are first defined and described. An empirical example of the application of generating the pay-off time is then presented. The chapter concludes with a discussion on the potential use of the pay-off time when developing CGs for patients with multimorbidity.
Key underlying concepts
The key underlying concepts that provide the context for the empirical study are TTB and time horizon to benefit; pay-off time; cumulative QALYs and QALY profiles; and direct treatment disutility (DTD).
Time to benefit
Time to benefit and time horizon to benefit are analogous terms used in the literature (hereafter we will use TTB). The literature on TTB has focused on the need to identify the time point at which the benefits of an intervention (exclusive of its harms) occurred. TTB can be demarcated, and interpreted, as either a biological or an epidemiological concept. 168 The biological interpretation means that it is feasible to calculate a precise TTB from an intervention for an individual. Practically, this interpretation involves generating a measure of the time it takes for an intervention, generally a pharmacological therapy, to reach its intended therapeutic target and reduce a measurable outcome by a certain pre-set threshold. An example of this biological interpretation would be the time for plasma levels of LDL cholesterol to reach the pre-defined target after starting a statin therapy. The biological interpretation allows an individualised TTB to be calculated based on observed responses. However, individualised times would often be suitable only for surrogate clinical outcomes, such as the reduction in LDL cholesterol, rather than the patient-relevant final end point, such as a cardiovascular event. This limits the practical relevance of using the biological interpretation of TTB in informing clinical decision-making. To expand TTB so that it takes account of outcomes that clinicians and patients value, such as a reduction in the risk of mortality, TTB needs to be interpreted as an epidemiological construct. TTB as an epidemiological construct requires assessment of reduction in risk at the population level, and evidence generated from appropriately designed clinical trials. 168
To date, however, there has been relatively little work examining TTB, reflecting the fact that clinical trials are typically designed to run for, and examine effectiveness after, a defined period of follow-up. 168 This observation is not surprising given the lack of formal statistical methods developed to estimate TTB in an unbiased manner, although some statistical methods have been proposed. Lee et al. suggest an inference about TTB can be made by visual inspection of the published Kaplan–Meier curves and identifying the point of separation. 169 In the absence of any formal way of assessing when divergence happens, this seems likely to overestimate the presence of significant risk reduction when one does not exist, while also underestimating the actual TTB. 170
Another potential statistical approach to estimate TTB is to use published evidence post hoc, with the associated caveats for inference associated with post-hoc analysis. Ray and Cannon171 provided an example of this post-hoc approach when they re-examined a published clinical trial to assess whether or not the benefits of high-intensity statin treatment (40 mg of atorvastatin daily) occurred more quickly than low-intensity treatment (40 mg of pravastatin daily) for those patients with a recent diagnosis of acute coronary syndrome. The original clinical trial, published by Cannon et al. 172 in 2004, identified a significant statistical difference between treatments at mean 2-year follow-up in favour of the high-intensity approach (RRR 0.16; p = 0.005) when using a composite outcome of mortality or a serious cardiovascular event. The authors of the original clinical trial also made reference to an observed divergence of the Kaplan–Meier curves, suggesting that the clinical beneficial effects may occur sooner than the full follow-up time at which the formal statistical test was applied in the pre-specified main trial analysis. Ray and Cannon171 identified that a statistically significant benefit occurred at 4 months’ follow-up, implying a much shorter TTB than trial duration. Lee et al. 169 also retrospectively fitted statistical models to published meta-analysed data to identify the TTB for colorectal and breast cancer screening programmes. This analysis made use of a large sample of pooled meta-analysis data and found that it took 10.7 years (95% CI 4.4 to 21.6) before one death from breast cancer screening was prevented per 1000 patients screened. Similar results for colorectal cancer were also found, with a TTB of 10.3 years for 1000 patients screened (95% CI 6.0 to 16.4). 169 However, all such essentially observational post-hoc analysis risks finding spurious results in a similar way to post-hoc subgroup analysis. 173
In practice, therefore, it is more usual for TTB to be defined simply as the median or mean follow-up in clinical trials showing that treatment is effective. This is likely to be conservative but is consistent with the original, pre-specified trial design and power. Theoretically, therefore, interventions in different disease areas could be compared where TTB for a standardised outcome is assumed to be trial duration. For a common outcome such as a reduction in all-cause mortality, then, all things being equal, the intervention with the shortest trial duration, and therefore the shortest TTB, would be preferred. However, a range of important caveats must be taken into account even when using the concept of trial length as TTB. For example, as Holmes et al. 170 describe, the TTB estimated in this way will be inherently linked to the trial design. Accordingly, all things being equal, larger trials will indicate shorter TTB than smaller trials because trial duration is likely to be shorter, as outcomes will become statistically significantly different earlier. 170
Consequently, developments in methods are required to allow quantification of statistical estimates of TTB ex ante to trial design, which are likely to parallel developments in the analysis of trials with early stopping rules. Stopping rules are built into trial protocol and involve setting interim time periods to test for differences between experiment and control, often to protect patients from clearly harmful drugs. 174 However, the use of stopping rules is associated with controversy, most notably when stopping trials early for a perceived benefit of the experiment versus the control. As trials are typically powered to find clinically meaningful differences at trial follow-up, it has been found that truncated trials, which stop early because of an apparent treatment effect, can provide misleading and biased estimates of treatment effect compared with those studies that are not truncated. 175 Similar problems will be relevant to using trials to estimate TTB. Repeated statistical tests increase the likelihood of a false positive, while large random fluctuations early in the trial, where the sample size is small, could wrongly suggest statistical TTB and overestimate treatment effect.
In summary, the highest-quality evidence on statistical TTB is likely to come from trials that have been designed from the outset to identify interim time points at which benefit occurs within the trial, but methods to validate such an approach are still required. 168 A further, less robust approach, which would generate a conservative view of TTB, is to assume that it occurred at trial completion or at median trial follow-up. 171 Alternatively, TTB could be estimated from ex-hoc analysis of already published evidence or using qualitative visual inspection of where published survival curves appeared to separate. All of these methods of analysis are still in their infancy and in need of further development.
Given the current state of play of the literature, the incorporation of a statistical TTB as the means by which a temporal dimension could be incorporated within CGs was judged not to be feasible. Therefore, the use of a temporal dimension in already existing model-based CEAs supplemented by the pay-off time framework was explored as an alternative way of informing the development of CGs.
Pay-off time
The following text has been adapted from Thompson A, Guthrie B, Payne K. Using the ‘pay-off time’ in decision-analytic models: a case study for statins in primary prevention. Med Decis Making 2017. Published online 25 April 2017. 182 This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 3.0 License (http://www.creativecommons.org/licenses/by-nc/3.0/) which permits non-commercial use, reproduction and distribution of the work without further permission provided the original work is attributed as specified on the SAGE and Open Access page (https://us.sagepub.com/en-us/nam/open-access-at-sage).
The pay-off time is defined as the minimum elapsed time in which the expected cumulative net benefits of an intervention exceeds its expected cumulative harms. 176 The pay-off time concept, first proposed by Braithwaite et al. 176 in 2009, more readily captures a temporal dimension that can be readily incorporated into a model-based CEA to inform CGs. The rationale for the pay-off time approach is similar to statistical TTB in that short-term harms must be balanced against longer-term benefits, and stakeholders can use this evidence to prioritise care. When using the pay-off time approach, less attention is placed on the explicit timing of benefit within trial and more focus is on the relative timing of benefit compared with harms from the intervention. Braithwaite et al. 176 argued that CG recommendations should not be implemented in individuals if the overall ratio of expected benefits to harms is likely to be negative within their life expectancy. 176 Equivalently, this can be expressed as that an intervention should be used if the life expectancy of a patient is greater than the pay-off time from the intervention. 36,177,178
When applying the pay-off time concept, benefits are classified as outcomes that can improve mortality, morbidity or both in a patient. Harms are outcomes that can worsen mortality, morbidity or both. Initially the pay-off time concept was applied using disparate outcomes such as reductions in mortality (for benefit) as well as adverse events (for harms). 177 In a later application, benefits and harms were standardised using a common metric, the QALY, to measure both benefits and harms in a single measure. 36 In this context, Braithwaite used the QALY in the pay-off time approach to assess whether or not to (1) screen chronically ill 50-year-old women for colon cancer and (2) use intensive glucose control for chronically ill diabetic patients. In 2013, Yuo et al. ,178 including Braithwaite, applied the pay-off time framework to assess whether or not the potential upfront harms of revascularisation for patients with asymptomatic carotid artery stenosis (where the operation itself causes stroke in 1–3% of patients) are worth the deferred benefits (a 5% absolute stroke risk reduction over 5 years). 178 These published applications of the pay-off time concept all examined interventions with upfront, one-off harms, and the pay-off time was readily estimated using algebraic mathematical calculations. It was clear from these applications that the QALY metric together with the pay-off time concept is a potentially useful measure to capture harms and benefits in a way that allows comparison across interventions and an understanding of the impact of a temporal dimension.
The published examples of using the pay-off time approach show a move away from simple algebraic calculations to using a decision-analytic approach in which outcomes and probabilities are combined to provide a systematic assimilation of multiple evidence sources and calculation of the probability-weighted outcomes (QALYs). 36,176–179 The decision-analytic approach proposed is similar to the use of model-based analysis to generate estimates of incremental costs and benefits to inform CEA. McCabe et al. have demonstrated how a pay-off based approach can be integrated within a decision-analytic model to generate information for reimbursement agencies. 180,181 Such an approach can produce an estimate on the time it takes for a new innovative technology to ‘pay off’ given an original cost. In a similar way it is, therefore, straightforward to combine the use of the pay-off time concept, QALYs and model-based CEA to incorporate a temporal dimension into existing model-based CEAs that follow patients, or cohorts of patients, over their lifetime and estimate when the benefits of an intervention should outweigh the harms or when the intervention pays off.
Cumulative quality-adjusted life-years and quality-adjusted life-year profiles
To calculate a pay-off time, it is necessary to generate estimates of cumulative QALYs. Cumulative QALYs are the total sum of QALYs gained (or lost) overall at pre-defined time intervals, such as yearly. Figure 11 illustrates how cumulative QALYs can be plotted to represent a QALY profile that shows the cumulative QALYs gained over time.
FIGURE 11.
Quality-adjusted life-year profile plot showing cumulative QALYs over time. This figure has been adapted from figure 1B in Thompson A, Guthrie B, Payne K. Using the ‘pay-off time’ in decision-analytic models: a case study for statins in primary prevention. Med Decis Making 2017. Published online 25 April 2017. 182 This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 3.0 License (http://www.creativecommons.org/licenses/by-nc/3.0/) which permits non-commercial use, reproduction and distribution of the work without further permission provided the original work is attributed as specified on the SAGE and Open Access page (https://us.sagepub.com/en-us/nam/open-access-at-sage).
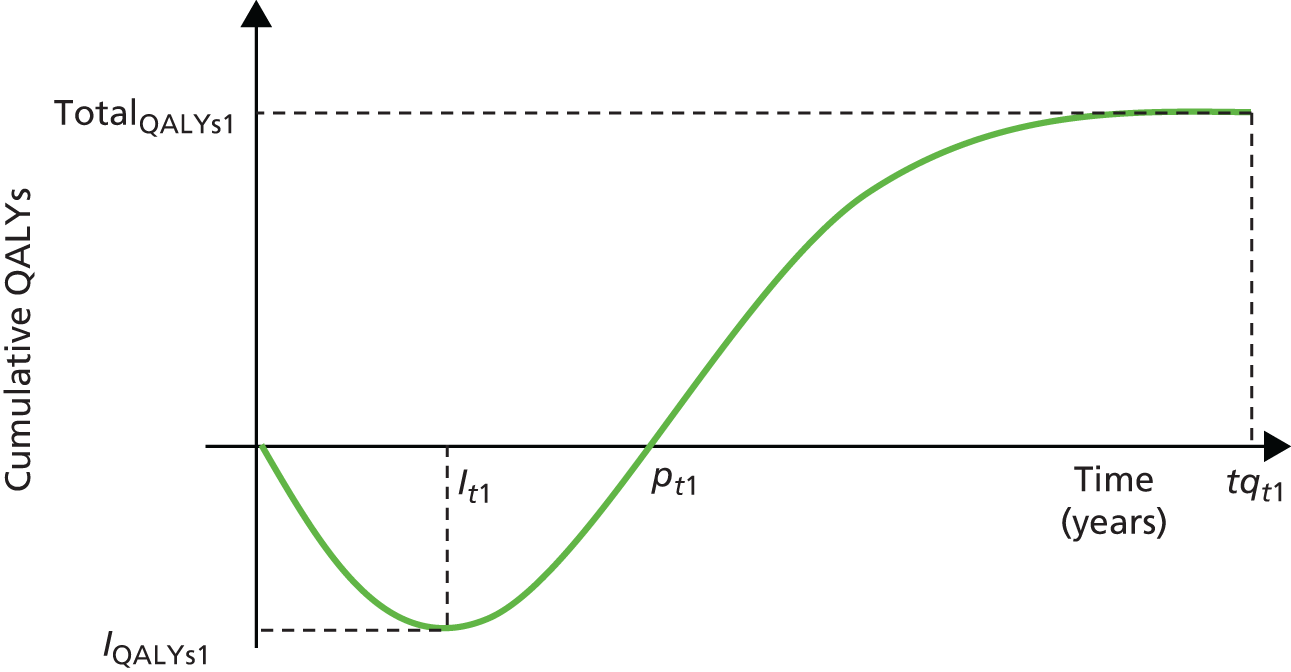
The pay-off time is the point shown on Figure 11 where the overall net benefit or cumulative QALYs become positive (crossing the y-axis at pt1). Total QALYs gained are reached at time tqt1. From Figure 11 it is also possible to calculate the peak investment, which has both an associated time period (It1) and an associated size measured in QALYs (IQALYs1). The peak investment represents the time when the intervention has the most negative cumulative QALYs (or alternatively maximum harm). It is the period at which patients and clinicians will need to have the most trust that eventually, if taken for a sufficiently long time, the intervention will recoup the built-up harm to produce an overall balance in positive QALYs. Value judgements have to be made by decision-makers, who may prefer to measure peak investment in terms of a shorter time or, alternatively, by minimising the size of the peak investment in QALYs.
The relevant comparator is a key criterion when defining a model that is fit for the purpose of addressing a specific decision problem. Generally, when used to inform the development of CGs or in technology appraisals, the relevant comparators should be (1) established clinical practice, (2) cost-effective practice relative to another available treatment and the (3) natural history of the condition without suitable treatment. 59 To allow comparisons between disparate interventions and between conditions, it is necessary to make use of this last comparator as a standard benchmark in the pay-off time approach. We define this comparator as a do-nothing approach (see Chapter 4 for a more detailed description of this issue). Figure 11 shows the plot of cumulative QALYs over time compared with a do-nothing comparator for a hypothetical intervention with persistent and constant harms over time and benefits that accrue over time. If there were no difference between comparators, the curve would run along the x-axis in perpetuity. At the end of a patient’s lifetime, the cumulative QALY equals the total or absolute QALYs gained from the intervention over the comparator. Again, if no difference between comparators existed, the absolute benefit would be zero.
The application of cumulative QALY in practice requires only a minor extension over the way QALYs are currently calculated and presented in model-based CEA used to inform CGs. Most model-based CEAs use Markov models to capture the relevant health states affected by the intervention and the associated costs and benefits that will accrue for a patient population over time. 69 A typical Markov model would begin with a cohort of patients, for example n = 1000, in an initial health state (e.g. a healthy state). In each cycle of the model, a proportion of the patient cohort would move (according to a defined transition probability) into the next defined health state (or remain in the initial healthy state). The cycle length needs to be specified and relevant to the condition and intervention, which is typically 1 year for long-term models looking at interventions such as statins and antihypertensives. Transition probabilities can be identified from published sources and ideally from meta-analyses of clinical trials. 183 Models with a lifetime horizon will typically be run until the point at which all patients will have died.
Each health state also has an associated utility score and cost, again identified from published sources. 183 When the utility score is combined with the time the cohort spends in that health state, this is used to calculate QALYs. The pooled total number of QALYs per cycle (i.e. per year) is then estimated to be the weighted proportion of the cohort within each health state and by utility for the health state. The model would then be run for the various comparators, with the treatment probabilities being adapted according to the relative risk as identified from the relevant published data sources. The intervention and comparator will each have their own assigned transition probabilities, which will affect the progress of the starting patient cohort through the defined health states in the Markov model. The total costs and QALYs are then estimated for the intervention and relevant comparator. It is here that the simple extension to measure cumulative QALYs over time (per model cycle) can be used to examine the pay-off time. A further simple extension is to include the impact on costs and cumulative QALYs by the use of net benefit. Net benefit is the balance between costs and QALYs,184 transformed into a monetary metric, and is estimated by making reference to a threshold of willingness to pay for an additional QALY, for example £20,000 per QALY. 60
The majority of published model-based CEAs for primary preventative interventions with medications would show no pay-off time (in purely QALY terms), as a benefit (assuming the intervention is clinically effective) would typically be realised within the first cycle (first year) of the model, as no immediate harm from the intervention is generally taken into account. Note here a common assumption in CEA models that the statistical TTB, which at its conservative value would be taken to occur at the median trial length, actually occurs within the first cycle of the model. This approach is generally viewed as sufficient for decision-making, as model-based CEAs are currently used to identify whether or not the incremental benefits outweigh the incremental costs of an intervention compared with current practice. 60 However, the pay-off time specifically aims to capture harms compared with benefits and in this scenario a known harm must be specified and measured in the model. Yuo et al. 178 used a one-off surgical intervention to illustrate immediate harm. For preventative treatments, which are taken in the long term, such a one-off harm is not generally clinically meaningful. This is where the concept of DTD becomes relevant.
Direct treatment disutility
There is a small but growing evidence base that suggests that treatments cause inconvenience or disutility to a patient beyond the intrinsic unwanted harms, adverse outcomes or effects that an intervention can potentially induce. 185 There are many theoretical reasons for a disutility associated with treatment. A drug per day for life may come with a small psychological harm, as well as the physical inconvenience of taking the drug. The drug is likely to come at a financial cost to the patient, require ongoing maintenance (in terms of visiting GPs and other health-care professionals) and require regularly ordering prescriptions and collecting drugs, which includes both inconvenience and cost to the patient. All of this can add up to disutility associated with the intervention that is not related to harm caused by any particular drug’s specific adverse effects. Collectively this concept can be termed a DTD to distinguish the concept from the more conventional unwanted ADEs. Currently, DTD is assumed to be trivial in comparison with either the costs of an intervention or the potential health benefits and so is generally not included in model-based CEA (e.g. see Heather et al. 186).
Some model-based CEAs, however, have started to consider the impact of DTD for specific interventions taken for chronic conditions. To identify some examples, a rapid review of published model-based CEAs for a selected intervention (statins) for a chronic condition (CVD) was conducted. The rapid review was conducted on 26 January 2015 and used the search strategy shown in Appendix 3. It was run in OVID for four databases (MEDLINE, EMBASE, PsycINFO and the American Economic Association’s electronic bibliography, EconLit).
This rapid review identified six studies that had included some DTD. 187–193 DTD was typically included as a 1-year disutility value. From the review of the literature, low values of DTDs were found to be increasingly used in economic models associated with statins for primary and secondary prevention of cardiovascular events. 185,189–191,193 Low values of utility decrements were applied periodically with values of disutility ranging from 0.00384 (equivalent to 2 weeks of full health traded to avoid 10 years on statins) to 0.02 (10 weeks of full health traded to avoid 10 years on statins) in sensitivity analyses. The results of the presented CEA were extremely sensitive to inclusion of DTDs, which changed the relative cost-effectiveness of the interventions being evaluated.
There is an emerging literature that has elicited values for DTD. Four published empirical studies have been identified anecdotally that relate to the concept of DTD. Table 20 summarises the identified studies. The elicited values for DTD were in the order of a disutility size of 0.01. A 0.01 decrement in QALYs is equivalent to a loss of ≈ 3.6 days of perfect health over 1 year. These values should be treated with caution because of some methodological limitations of the methods used to elicit the values. However, they do serve as a useful indicator of the potential impact of DTD for the purpose of estimating pay-off time associated with a patient population taking a treatment for the remainder of their lives. The impact of including DTD in combination with pay-off time needs to be assessed in an empirical study.
| Study | Clinical scenario | Elicitation method | Study sample | Direct treatment disutility |
|---|---|---|---|---|
| Gage et al. (1996), USA194 | Taking aspirin or warfarin for stroke prevention | Time trade-off | 70 patients with atrial fibrillation | For warfarin = 0.997 |
| Standard gamble | For aspirin = 1.000 | |||
| Hutchins et al. (2015), USA195 | One pill a day for cardiovascular prevention | Time trade-off | 1000 US residents aged ≥ 30 years | Time trade-off 0.990 (95% CI 0.988 to 0.992) |
| Standard gamble | Standard gamble 0.991 (95% CI 0.989 to 0.993) | |||
| Willingness to pay | Willingness to pay US$1445, ≈ 0.994 (95% CI 0.940 to 0.997) | |||
| Fontana et al. (2014), UK185 | Idealised preventative pill | Gain of an expected x days of life | 360 members of London public | 1 day to > 10 years of life (median 6 months, interquartile range 1–36 months) |
| Hutchins et al. (2015), USA196 | One pill a day for cardiovascular prevention | Time trade-off | 708 health-care employees aged ≥ 18 years | Time trade-off 0.9972 (95% CI 0.9962 to 0.9980) |
| Standard gamble | Standard gamble 0.9967 (95% CI 0.9954 to 0.9979) | |||
| Willingness to pay | Willingness to pay 0.9989 (95% CI 0.9986 to 0.9991) |
Empirical study aim and objectives
This empirical study aimed to identify the impact of including a temporal dimension in a model-based CEA by using DTD and the pay-off time concept. There were four objectives:
-
to identify an existing model-based CEA to use as a case study to quantify the impact of including DTD and pay-off time
-
to present cumulative QALY profiles, for example values for DTD and specific patient population characteristics
-
to present cumulative net-benefit for example values for DTD and specific patient population characteristics
-
to quantify the pay-off time, for example values for DTD and specific patient population characteristics.
Methods
A published model-based CEA, developed by the National Clinical Guideline Centre, which had been used to inform an existing NICE CG, was used as the framework for the methods used in this study. 197
Case study selection
The feasibility of using the pay-off time in a model-based CEA within guidelines for the three pre-selected exemplar conditions (type 2 diabetes, depression and chronic heart failure) was explored. These exemplar CGs did not use model-based CEA based on Markov-type models, which was needed to implement the pay-off time approach. Therefore, a manual search of published CGs relevant to the management of long-term conditions was conducted, and identified the 2014 lipid modification guidance (CG181) as suitable. 197 The decision-analysts named in published full NICE guidance were then contacted using e-mail. We were then granted access to a full executable version of the de novo model developed by the National Clinical Guidelines Centre under the standard NICE licence.
The model
Table 21 summarises the key attributes for the de novo model used in this analysis.
| Model attribute | Model-based cost-effectiveness analysis of statin therapy for primary and secondary prevention of cardiovascular disease |
|---|---|
| Published date | 2014 |
| Collaborating centre | National Clinical Guideline Centre |
| Methods manual | NICE (2009) |
| Model type | Markov (cycle = 1 year) |
| Study perspective | NHS |
| Horizon taken | Lifetime, to 100 years old |
| Valuation of benefits | QALYs |
| Discount rate | 3.5% for costs and benefits |
| Starting age | Varied |
| Comparators | Low-intensity statins (21–29% reduction in LDL cholesterol): 20 mg of fluvastatin per day; 40 mg of fluvastatin per day; 10 mg of pravastatin per day; 20 mg of pravastatin per day; 40 mg of pravastatin per day; 10 mg of simvastatin per day |
| Medium-intensity statins (32–38% reduction in LDL cholesterol): 80 mg of fluvastatin per day; 20 mg of simvastatin per day; 40 mg of simvastatin per day; 10 mg of atorvastatin per day; 5 mg of rosuvastatin per day | |
| High-intensity statins (42–55% reduction in LDL cholesterol): 80 mg of simvastatin per day; 20 mg of atorvastatin per day; 40 mg of atorvastatin per day; 80 mg of atorvastatin per day; 10 mg of rosuvastatin per day; 20 mg of rosuvastatin per day; 40 mg of rosuvastatin per day | |
| No treatment | |
| Do-nothing included? | Yes |
| Intervention type | Primary prevention |
| Population | Adults in England and Wales without CVD |
| Cost-effective option | High-intensity statins for patients with QRisk-estimated 10-year CVD risk ≥ 10% |
The model included 15 health states, and is fully documented in the published NICE guideline CG181. 198 The model was populated with the same data as used for the production of CG181. The model was designed to support the use of the QRisk2 tool for predicting risk in people without diabetes being considered to receive statins for primary prevention. QRisk2 (10 years) is a cardiovascular risk tool developed by Hippisley-Cox et al. 199 based on the QResearch UK primary care cohort. It estimates an individual’s risk of experiencing any of fatal or non-fatal angina, MI, TIA or stroke over the following 10 years, and can be found at www.qrisk.org/index.php. The model allows subgroup analysis based upon sex, age (40, 50, 60 and 70 years) and baseline QRisk2 (10 years).
In 2005, NICE conducted a technology appraisal of the use of statins for the primary prevention of CVD (published in 2006). 200 In 2007, the NIHR funded a HTA,201 with a subsequent report published by the same authors as the original 2005 work. Neither the NICE technology appraisal nor the NIHR HTA-funded study included quantification of the impact of DTDs in the base-case analysis, the sensitivity analysis or a scenario analysis. 200,201 The justification for this omission was given as:
. . . as adverse events and side-effects are rare, patients receiving statin treatment in the model do not receive a penalty utility due to their medication. As statins are prescribed for life, there may be a disutility associated with this, but it is assumed that this is small in comparison to the benefits received and as such is not modelled.
Ward et al. 201
The potential for disutility losses associated with adverse events and side effects following treatment was considered but thought to be inconsequential in comparison with the potential benefits. In 2014, a subsequent economic model was developed for the NICE CG on lipid modifications (CG181) appraising the use of statins for both primary and secondary prevention. This model had a similar structure to the two existing models and did not include DTD either in the base-case or in scenario analysis. Instead scenario analysis was used to explore the impact of assumptions about patients’ desire to continue with the treatment on 6-month adherence to capture some negative impact associated with taking the medicine. The justification given for the lack of inclusion of DTDs in the sensitivity analysis was that the analysis should be consistent with the previous NICE technology appraisal (2005)200 and published NIHR-funded HTA report. 201
For the purpose of this study, additional harms were then built into the model to reflect DTD. All patients in the model cohort were assumed to suffer from an annual disutility associated with treatment. The values for the DTD were informed from the rapid review of the literature described previously.
Patient vignettes
Patient vignettes were used to provide clinically relevant scenarios to quantify the impact of pay-off time. Three patient vignettes were created with input from clinicians on the project team (n = 3) and the PRG (n = 4). The patient vignettes were built around scenarios defined by three 10-year QRisk2-estimated CVD risks of 10%, 15% and 20% (Table 22), which span the range between the currently recommended treatment threshold of 10% 10-year CVD risk and the previously recommended threshold of 20%.
| Patient vignette | QRisk2-estimated 10-year risk of CVD (%) |
|---|---|
| 1: 60-year-old white man, no relevant comorbidity, non-smoker, systolic blood pressure 150 mmHg, height 178 cm, weight 75 kg, BMI 23.7 kg/m2. His fasting lipid test shows a total/high-density lipoprotein cholesterol ratio of 3.3 | 10 |
| 2: 60-year-old white man, no relevant comorbidity, light smoker, systolic blood pressure of 140 mmHg, height 178 cm, weight 80 kg, BMI 25.25 kg/m2. His fasting lipid test shows a total/high-density lipoprotein cholesterol ratio of 4.2 | 15 |
| 3: 70-year-old white man, no relevant comorbidity, non-smoker, systolic blood pressure 140 mmHg, height 178 cm, weight 85 kg, BMI of 26.8 kg/m2. His fasting lipid test shows total/high-density lipoprotein cholesterol ratio of 4.1 | 20 |
Analysis
Quality-adjusted life-year profiles were generated for each of the three patient vignettes, representing three different levels of baseline QRisk2, and also stratified according to different DTDs for treatment with high-intensity statins compared with do-nothing. The following were calculated: pay-off time in QALYs; pay-off time in net benefit assuming a threshold of £20,000 per QALY gained; absolute QALYs; time for the peak investment in QALYs gained (years); and size of the peak investment (net benefit, £). DTDs were applied by ‘adding’ in the defined (negative utility) value to the total utilities for each cycle and assuming that the whole cohort had the DTD. This approach means that a constant utility decrement is applied to each cycle in the model for the lifetime of each patient in the cohort who is taking the treatment. This is consistent with the current methods of eliciting DTDs, which often use time–trade-off exercises (with questions spanning different hypothetical time periods, typically 10 years) to estimate the utility decrement to use for each 1-year time period. 185,194–196
Exploratory analysis
In a supplementary exploratory analysis, the pay-off time was calculated for a sample of hypothetical patients with multimorbidity. Values for all-cause mortality used in model-based CEA are commonly taken from life tables from the Office for National Statistics (ONS). 202 From these life tables, age- and sex-adjusted mortality rates can be calculated. However, these rates represent population average mortality for men and women at different ages. A reasonable hypothesis is that those with multimorbidity are likely to have a higher risk of all-cause mortality. Menotti et al. 203 found evidence to support an increasing relative risk of all-cause death for patients with one condition, two conditions and three or more conditions, for populations of patients from Finland, the Netherlands and Italy, respectively. In this exploratory analysis, it was assumed that patients’ risk of all-cause mortality would increase in proportion to the number of conditions. A range of pay-off times were calculated by varying the value for relative risk, assumed to increase with increasing numbers of conditions associated with multimorbidity. Five examples of values for the relative risk (1, 0.5, 2.0, 3.0 and 4.0) were used. Two values of DTD (0.005 and 0.010) were used as examples of harm from the treatment.
Results
This section presents the analysis of pay-off time for three patient vignettes and also the supplementary exploratory analysis for a patient population with a hypothetical number of more than one condition, representing multimorbidity. For different assumed values of DTD and the equivalent number of full health days traded, Table 23 summarises the calculated values for pay-off time in QALYs; pay-off time in net benefit assuming a threshold of £20,000 per QALY gained; absolute QALYs; peak investment in QALYs (years); investment size in net benefit (£); and peak investment size (QALYs).
| DTD (harm) | Equivalent number of full health days tradeda | QALYs pay-off time (years) | Net-benefit pay-off time (years)b | Absolute QALY gain | Peak investment (years) | Net benefit (£)b | Peak investment size (QALYs) |
|---|---|---|---|---|---|---|---|
| Patient vignette 1 | |||||||
| 0 | 0 | Immediatec | 7.5 | 0.20 | N/A | 2865 | 0 |
| 0.00274 | 30 | 4.6 | 11.9 | 0.16 | 4.5 | 2011 | –0.003 |
| 0.00500 | 55 | 8.5 | 16.0 | 0.12 | 6.5 | 1307 | –0.010 |
| 0.00800 | 88 | 14.2 | 24.6 | 0.08 | 8.5 | 373.41 | –0.024 |
| 0.01000 | 110 | 18.9 | Neverc | 0.05 | 9.5 | –249 | –0.036 |
| 0.01500 | 164 | Neverc | Neverc | –0.03 | 13.5 | –1806 | –0.077 |
| 0.02000 | 219 | Neverc | Neverc | –0.11 | 18.5 | –3364 | –0.128 |
| Patient vignette 2 | |||||||
| 0 | 0 | Immediated | 4.4 | 0.29 | N/A | 4936 | 0 |
| 0.00274 | 30 | 3.0 | 7.1 | 0.25 | 3.5 | 4095 | –0.002 |
| 0.00500 | 55 | 5.5 | 9.5 | 0.21 | 4.5 | 3400 | –0.006 |
| 0.00800 | 88 | 9.1 | 13.1 | 0.17 | 5.5 | 2479 | –0.016 |
| 0.01000 | 110 | 11.7 | 15.7 | 0.14 | 6.5 | 1864 | –0.025 |
| 0.01500 | 164 | 19.7 | 27.3 | 0.06 | 9.5 | 328 | –0.054 |
| 0.02000 | 219 | Neverc | Neverc | –0.02 | 11.5 | –1207 | –0.092 |
| Patient vignette 3 | |||||||
| 0 | 0 | Immediated | 3.3 | 0.22 | 0.5 | 3847 | 0.000 |
| 0.00274 | 30 | 2.2 | 5.3 | 0.19 | 1.5 | 3212 | –0.001 |
| 0.00500 | 55 | 4.2 | 7.2 | 0.17 | 2.5 | 2687 | –0.005 |
| 0.00800 | 88 | 6.9 | 10.0 | 0.13 | 3.5 | 1991 | –0.012 |
| 0.01000 | 110 | 8.9 | 12.2 | 0.11 | 4.5 | 1527 | –0.019 |
| 0.01500 | 164 | 15.2 | 20.6 | 0.05 | 6.5 | 367 | –0.041 |
| 0.02000 | 219 | Neverc | 0.0 | –0.01 | 8.5 | –793 | –0.070 |
Patient vignette 1
The following text has been adapted from Thompson A, Guthrie B, Payne K. Using the ‘pay-off time’ in decision-analytic models: a case study for statins in primary prevention. Med Decis Making 2017. Published online 25 April 2017. 182 This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 3.0 License (http://www.creativecommons.org/licenses/by-nc/3.0/) which permits non-commercial use, reproduction and distribution of the work without further permission provided the original work is attributed as specified on the SAGE and Open Access page (https://us.sagepub.com/en-us/nam/open-access-at-sage).
This patient is a 60-year-old man whose cardiovascular risk is largely driven by age and sex. Table 23 shows that when there is no DTD the high-intensity statin treatment is a cost-effective option, with a positive net benefit of £2865 for a cost-effectiveness threshold of £20,000. Likewise, the absolute QALY gain was estimated to be 0.20 QALYs per patient, equal to approximately 73 days at full health. Figure 12 displays much of the same information as QALY profiles for this vignette. Of note is that even low DTD is associated with pay-off times measured in years, and the expected absolute QALY gain is sensitive to the presence of DTD. When the size of the DTD is set at ≥ 0.015 then statin treatment never pays off and the intervention is no longer cost-effective at a decision-maker’s threshold of £20,000 per QALY.
FIGURE 12.
Quality-adjusted life-year profiles for patient vignettes 1–3 with four levels of DTD (harm) from treatment (0–0.015 QALYs annually). (a) Patient vignette 1: 60-year-old man with 10% 10-year cardiovascular risk; (b) Patient vignette 2: 60-year-old man with 15% 10-year cardiovascular risk; and (c) Patient vignette 3: 70-year-old man with 20% 10-year cardiovascular risk. This figure has been adapted from figure 3 in Thompson A, Guthrie B, Payne K. Using the ‘pay-off time’ in decision-analytic models: a case study for statins in primary prevention. Med Decis Making 2017. Published online 25 April 2017. 182 This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 3.0 License (http://www.creativecommons.org/licenses/by-nc/3.0/) which permits non-commercial use, reproduction and distribution of the work without further permission provided the original work is attributed as specified on the SAGE and Open Access page (https://us.sagepub.com/en-us/nam/open-access-at-sage).
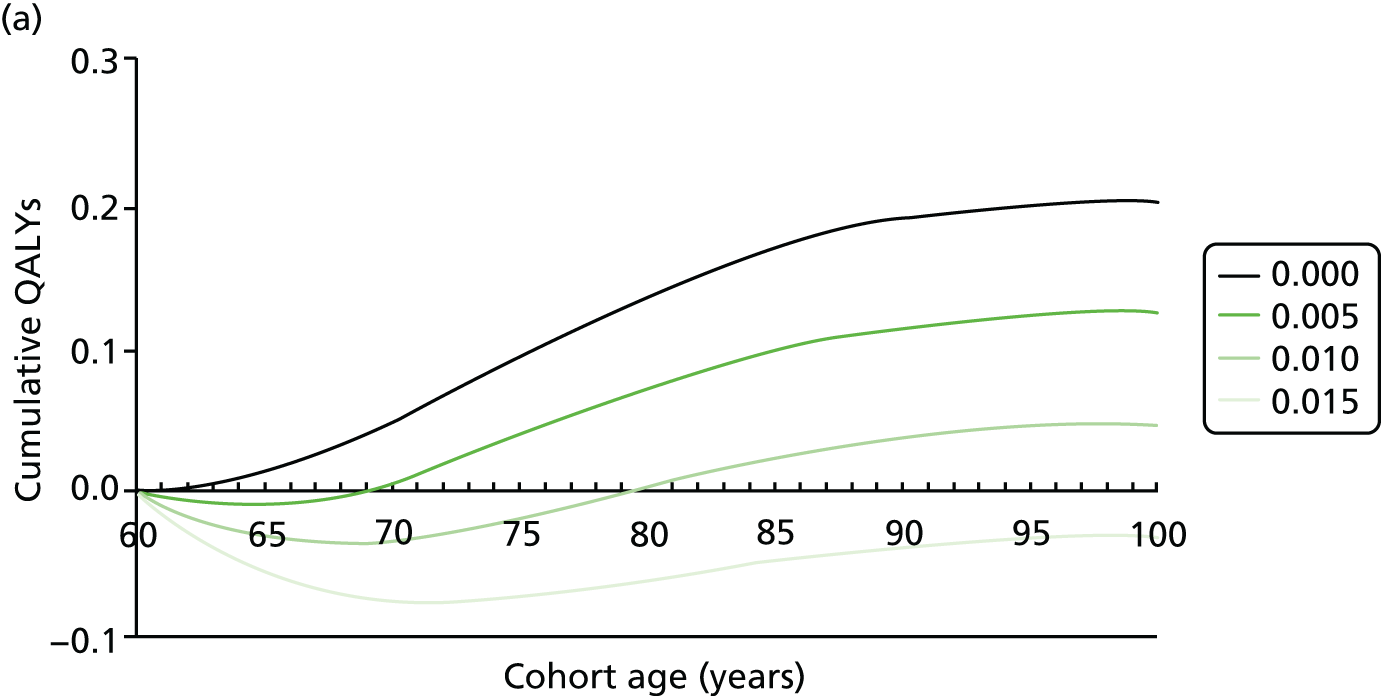
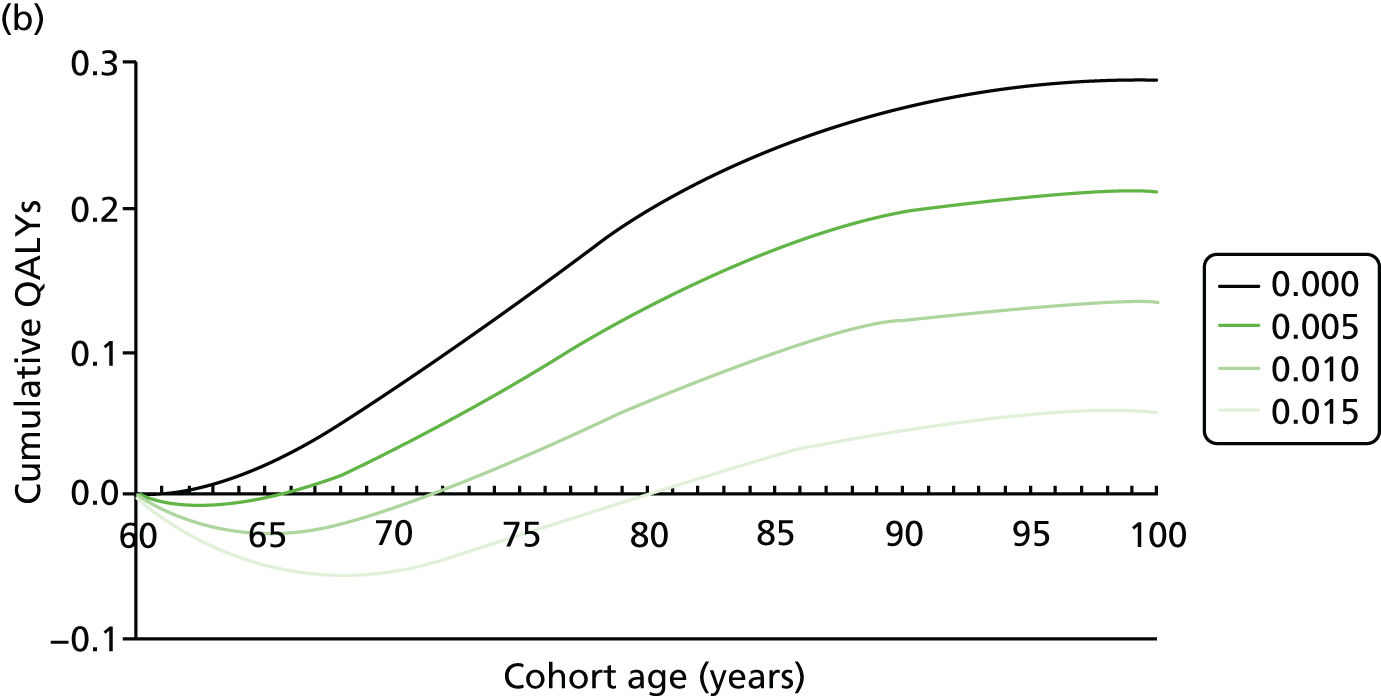
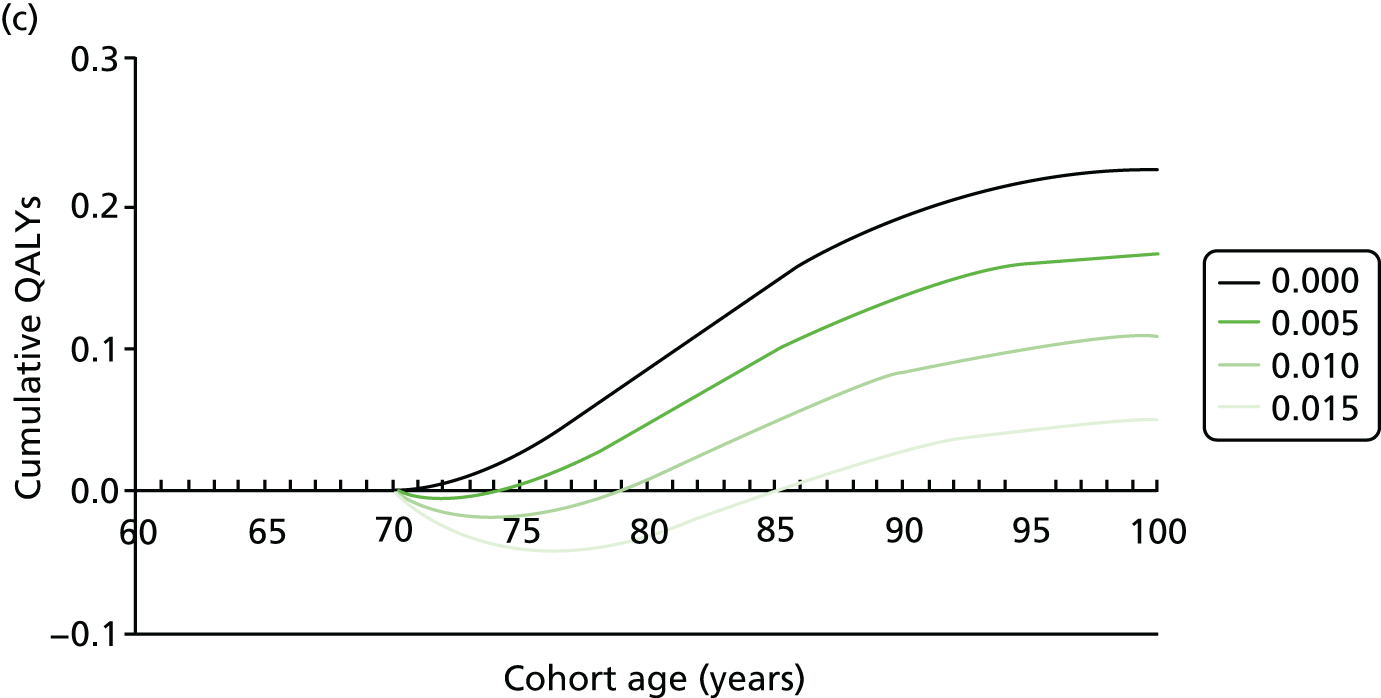
Patient vignette 2
This patient is a 60-year-old man whose 10-year cardiovascular risk of 15% is largely driven by age, sex, smoking and lipid profile. Table 23 shows that when there is no DTD the high-intensity statin treatment is a cost-effective option, with a positive net benefit of £4936 for a cost-effectiveness threshold of £20,000. Likewise, the absolute QALY gain was estimated to be 0.29 QALYs per patient, equal to approximately 106 days at full health. Figure 12 shows the QALY profile for this vignette. Of note is that even low DTD is associated with pay-off times measured in years (although, as expected, pay-off times are lower and absolute QALY gains are greater when baseline risk of CVD increases), and the expected absolute QALY gain is sensitive to the presence of DTD. When the size of the DTD is set at ≥ 0.020 then statin treatment never pays off and the intervention is no longer cost-effective at a decision-maker’s threshold of £20,000 per QALY.
Patient vignette 3
This patient is a 70-year-old man whose 10-year cardiovascular risk of 10% is driven by age, sex and lipid profile. Table 23 shows that when there is no DTD the high-intensity statin treatment is a cost-effective option, with a positive net benefit of £3847 for a cost-effectiveness threshold of £20,000. Likewise, the absolute QALY gain was estimated to be 0.22 QALYs per patient, equal to approximately 79 days at full health. As there is no harm included in the model the pay-off time is within the first cycle (year) and not relevant. Figure 12 shows the QALY profile for this vignette. Of note is that even low DTD is associated with pay-off times measured in years (although again, as expected, pay-off times are lower and absolute QALY gains are greater when baseline risk of CVD increases), and the expected absolute QALY gain is sensitive to the presence of DTD. When the size of the DTD is set at ≥ 0.020 then statin treatment never pays off and the intervention is no longer cost-effective at a decision-maker’s threshold of £20,000 per QALY.
Competing risks of death in patient populations with multimorbidity
Table 24 shows the results for a hypothetical patient population with an assumed baseline QRisk2 score of 10%, with different levels of multimorbidity represented by increasing levels of relative risk of death from all-cause mortality. Two analyses are presented for two assumed levels of DTD. Figure 13 shows the equivalent QALY profiles for each assumed level of DTD, 0.005 and 0.010.
| Relative risk total mortalitya | Pay-off time (QALYs) | Pay-off time (net benefit)b | Absolute QALY gain | Peak investment (years) | Net benefit (£)b | Peak investment size (from net benefit, £) |
|---|---|---|---|---|---|---|
| Scenario 1 with DTD = 0.005 | ||||||
| 0.5 | 8.5 | 15.7 | 0.16 | 6.5 | 1820 | –666 |
| 1.0b | 8.5 | 16.0 | 0.12 | 6.5 | 1308 | –659 |
| 2.0 | 8.5 | 16.5 | 0.09 | 6.5 | 746 | –646 |
| 3.0 | 8.5 | 17.3 | 0.07 | 6.5 | 429 | –633 |
| 4.0 | 8.5 | 18.4 | 0.05 | 6.5 | 223 | –620 |
| Scenario 2 with DTD = 0.010 | ||||||
| 0.5 | 18.5 | 37.0 | 0.07 | 9.5 | 47 | –1402 |
| 1.0b | 18.9 | 0.0 | 0.05 | 9.5 | –250 | –1380 |
| 2.0 | 19.9 | 0.0 | 0.02 | 9.5 | –565 | –1338 |
| 3.0 | 21.5 | 0.0 | 0.01 | 9.5 | –725 | –1297 |
| 4.0 | 26.6 | 0.0 | 0.00 | 9.5 | –816 | –1259 |
FIGURE 13.
Quality-adjusted life-year profile for patients with different levels of total mortality due to varying multimorbidity (expressed as relative risk of total mortality) assuming a DTD of 0.005 and 0.01. (a) Absolute QALY gain for a 60-year-old man with 10% 10-year CVD risk and DTD 0.005; and (b) Absolute QALY gain for a 60-year-old man with 10% 10-year CVD risk and DTD 0.01.
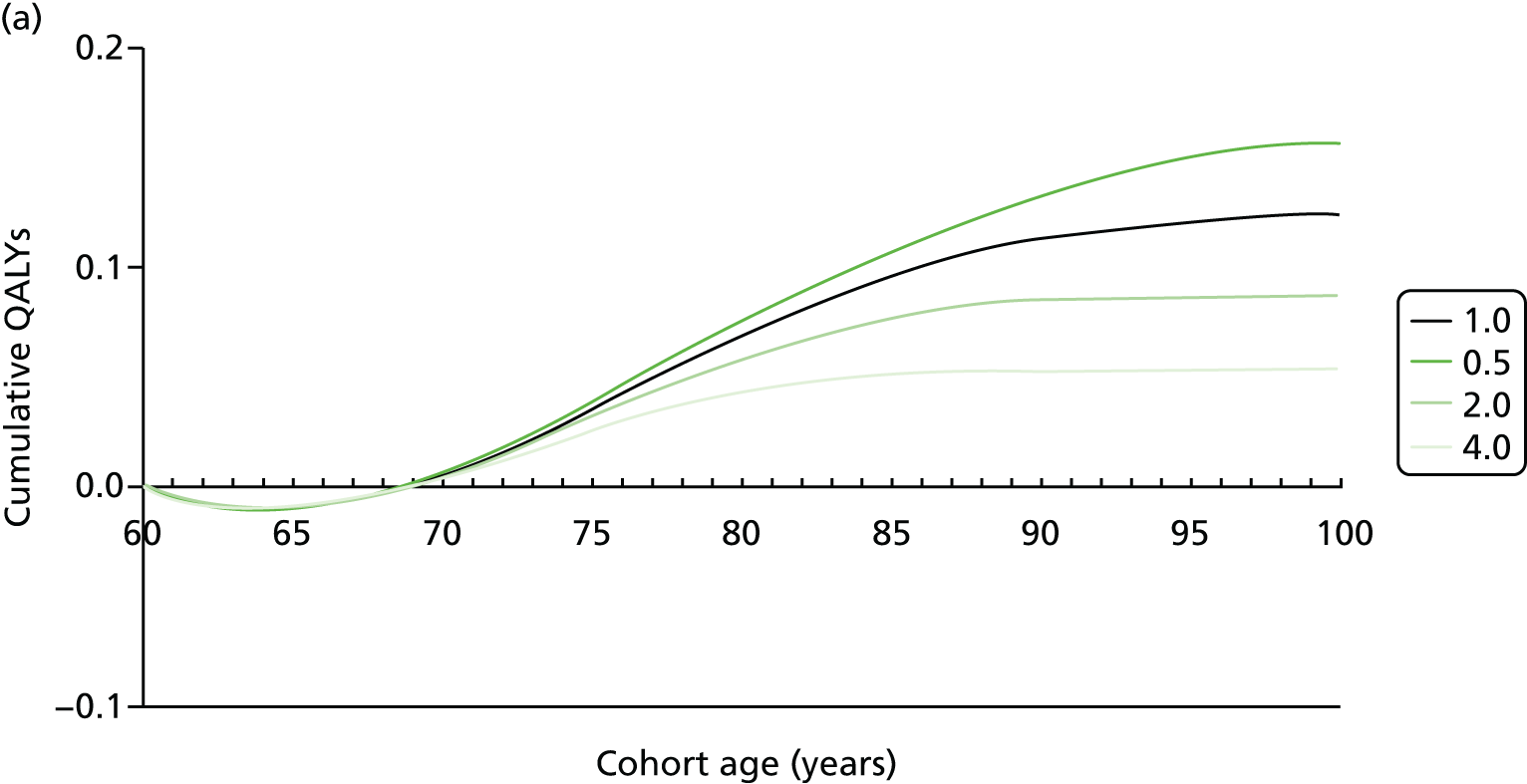
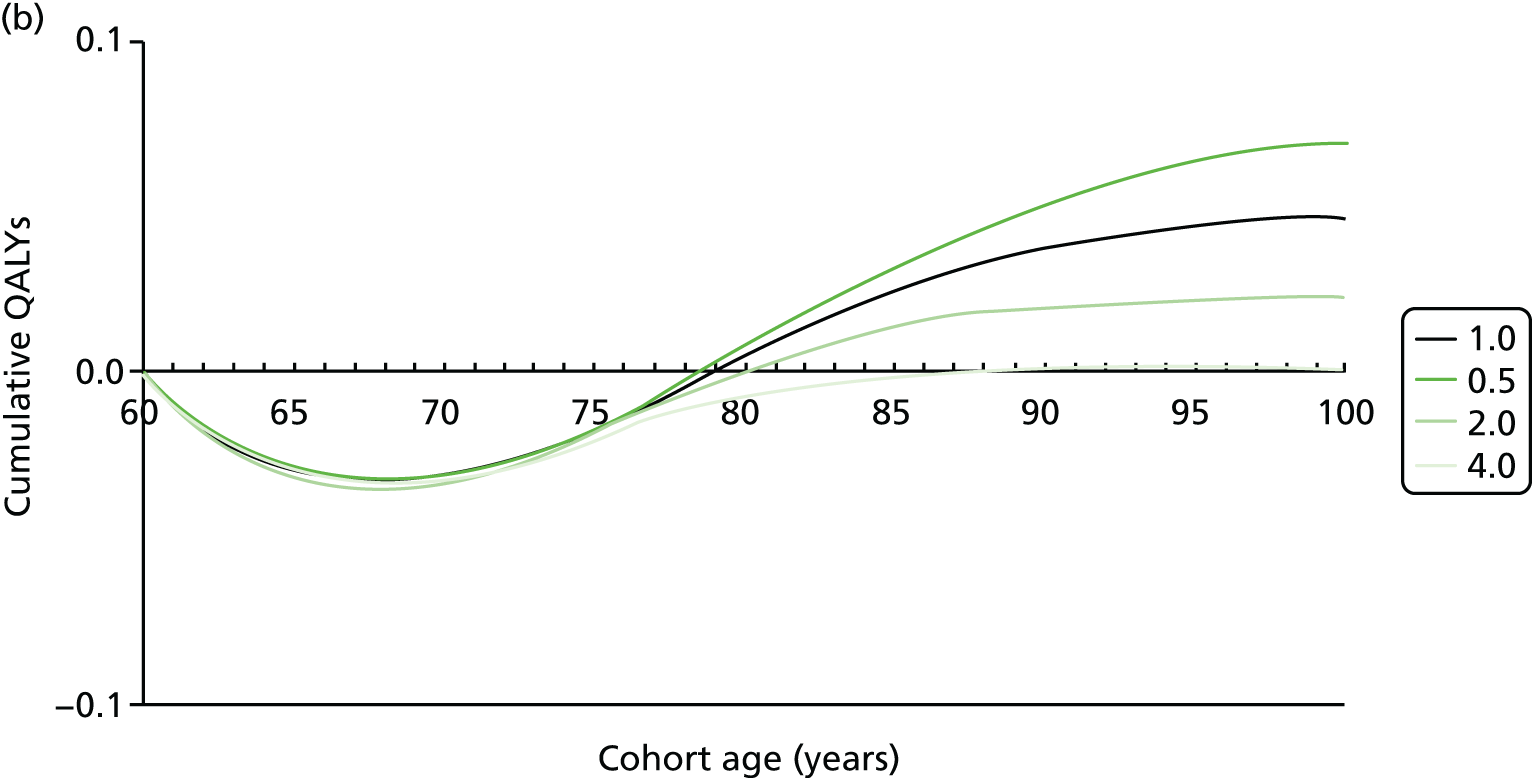
Making an adjustment to the assumed relative risk for all-cause mortality has a large impact on the absolute QALY gain. In scenario 1, when the DTD is set at 0.005, for individuals with a fourfold increased risk of all-cause mortality, the absolute QALY gain is estimated to be fewer than half of individuals with unadjusted all-cause mortality (0.12 vs. 0.05). In scenario 2, when the DTD is set at 0.010, the absolute QALY gain for the base-case analysis, with no adjustment to the risk of all-cause mortality, is now much lower, at 0.05. In scenario 2, the absolute QALY gain is estimated to be zero in individuals with a fourfold increased risk of all-cause mortality. In scenario 2, individuals never acrue net benefit from treatment unless there is a reduced all-cause mortality of half that in the base-case analysis. In both scenarios, the adjustment on relative risk has no effect on pay-off time, which is the time to reach peak investment (in years).
Discussion
This empirical study applied the concepts of pay-off time, DTD and graphical QALY profiles from an existing model-based CEA used for the production of CGs for the primary prevention of CVD with statins. These three concepts have the potential to be an informative approach to include a temporal dimension in model-based CEA. The calculation of pay-off time together with using visual presentation of the information (QALY profiles) provides a potentially useful tool to complement existing cost-effectiveness results and potentially aid guideline decision-making when choosing between interventions with different pay-off times.
The original CG, CG181, recommended that high-intensity statin treatment should be the preferred method of primary prevention for individuals with a 10-year cardiovascular risk of 10%. This guideline was informed by a Markov model-based CEA that used a lifetime horizon, which made it amenable to the generation of estimates of pay-off time by including assumed values of DTD. Cardiovascular risk is heavily driven by the age of the patient and to a lesser extent by sex. If DTD is ignored, using a threshold of 10% 10-year estimated risk, the model-based CEA supported recommending that virtually all men aged > 60 years and women aged > 65 years be offered a statin to prevent CVD, irrespective of levels of formal risk factors such as blood pressure or cholesterol. Therefore, CG181 effectively recommended offering statin therapy to a whole population. This recommendation was made without taking into account the potential for some members of that patient population to have negative views about the action of taking a medicine every day for the rest of their lives. There is emerging evidence to suggest that disutility from taking medicines every day can occur irrespective of whether or not individuals experience drug-specific adverse effects. 185 This empirical study illustrated how the expected pay-off time for the average person within a patient population will increase as the value of the DTD increases.
The pay-off concept is versatile and, when combined with graphical QALY profiles to aid interpretation by GDG members, could potentially be used to help decision-makers clarify the impacts over time of interventions that have deferred benefits but quantifiable harms. The method can be easily and rapidly applied using current model-based CEA. The pay-off time can be stratified according to various baseline characteristics that are pre-built into economic models and it can be calculated as a pay-off time in purely QALY terms or it can also be inclusive of financial costs associated with an intervention by using net benefit. Another potential extension, which would make the pay-off time informative in the context of patients with multimorbidity, is to explore the impact of adjusting the relative risk of all-cause mortality, which is generally a parameter included in Markov model-based CEAs. In this scenario, the adjustment in the relative risk of all-cause mortality is used as a proxy for a patient population with competing risk of death from other morbidities. One approach would be to use a plausible range of values for all-cause mortality, for example the interquartile range of population mortality rather than just the median. An alternative would be to use expert judgement, elicited from the members of a GDG or from individual clinicians, as the basis for deciding what the appropriate adjustment factor should be for different types and levels of morbidity within a patient population.
There are some potential limitations with using the proposed pay-off time approach in the context of using model-based CEA to inform the development of NICE CGs. The obvious challenge is identifying a robust source of DTD. This is a potential topic for future research using well-designed stated preference studies in appropriate samples of the patient population and members of the public. In the interim, the use of pre-defined DTD could enable some exploration in sensitivity analysis of the potential impact if patients do attach some level of harm to taking a long-term treatment. Related to this challenge is the analytical issue of how DTDs are incorporated into the utility values for the patient cohort being run through a Markov model. In this study a constant harm (DTD value) was used and the utility decrement was taken from the total utility for the relevant model cycle. This simple additive assumption may not be appropriate and research is needed to explore these assumptions around how DTDs are incorporated into the calculation of utility values used in the model-based analysis. This need for more research is related to the issue raised by Ara and Wailoo,204 who have analysed the impact of different analytical methods to include utility values associated with comorbidities into model-based CEA.
An important analytical problem with this empirical study was that it was not feasible to generate estimates of uncertainty around the pay-off time. This was because the analysis focused on providing an empirical application of the concept using an existing model-based CEA, which is how the approach could be used in practice. However, the ability to generate measures of uncertainty around the point estimates of pay-off time for different levels of DTD is reliant on the original model-based CEA presenting a PSA. Further research is required to inform how best to present the impact of joint uncertainty on point estimates of pay-off time for use in guideline development decision-making.
A key conceptual problem relates to when it is appropriate to apply population-level estimates to an individual when translating the findings from the model to a clinician–patient interaction. The pay-off time as applied in this empirical study was an aggregation of both HRQoL and mortality improvements. Fundamentally this means it involves a utilitarian calculation with the associated value-based judgements of equivalence in the value of saving and improving life. In the calculations of a QALY it is assumed that the value of saving one life is equal to the value of improving HRQoL in 10 patients by a value of 0.1. The analogy with the pay-off time concept is that a value of 5 years represents the average of different patient types: (1) some patients who are healthy but die quickly; (2) some patients who are very ill but live a long time; (3) some patients who are very healthy and live a long time; and (4) some patients who are sick and die young. Consequently, the pay-off time is best considered, and used, in the context of population-level decision-making, which does make it useful in the context of developing NICE CGs. Within the CG context, the pay-off time can be used as a tool to give a quality-adjusted benefit–harm calculation that takes account of a temporal dimension. If the absolute benefit of treatments were being compared using absolute QALYs as discussed in Chapter 4, then further accounting for TTB using the pay-off time would provide additional potentially useful information for decision-makers. Caution is advised if the pay-off time concept is applied as an individualised decision-making aid, between a clinician and patient, which was how it was first conceptualised. 176
Conclusion
Generating estimates of the pay-off time, using the QALY or QALY inclusive of costs (net benefit) form, can potentially be used as a complement to the outputs from existing model-based CEA already used to inform NICE CGs. A series of one-way sensitivity analyses can be used to present the impact of using different values for DTD and also the potential impact of comorbidity by using different adjustment factors for the included values of the relative risk for all-cause mortality. Ideally, if the original model-based CEA included PSA, then a joint measure of uncertainty should be presented as evidence to inform guideline development. QALY profiles are an attractive and comprehensible way for GDGs to visualise the potential impact of DTD on pay-off over time for interventions with deferred benefits. Practically, pay-off time should be explored only in the context of interventions that have already been shown to be a cost-effective use of resources in the main model-based CEA. Further research is required to generate better empirical estimates of DTD and inform how best to present joint uncertainty around point estimates for pay-off time in an informative way for use in NICE CG development.
Chapter 6 Quantifying the impact of multimorbidity in a model-based cost-effectiveness analysis
This chapter reports the background and rationale for the need to extend existing model-based CEA to take account of the impact of interventions for populations with multimorbidity. It presents the aim, objectives, methods and results of a model-based CEA that focuses on a relevant case study identified by the PRG: depression and CHD.
Background
Current NICE CGs make use of cost-effectiveness evidence when making recommendations. The ultimate goal for model-based CEA is to compare the costs and benefits in order to promote (and recommend) interventions that provide the best value for money for the health service. 205 The NICE DSU describes a mathematical model as being a
representation of the real world . . . characterised by the use of mathematics to represent the parts of the real world that are of interest and the relationships between those parts.
p. 14183
Current methods used in NICE CG development take a predominantly single-disease perspective. This approach, in theory, entails assessing mutually exclusive interventions for a single disease against one another and funding being allocated (or recommendations made) on an ‘all or nothing’ basis. Based on the selected disease, and the CRQ, a range of relevant indicated interventions are identified. The economic analysis assesses the impact of the interventions on the natural history of the disease and on the resulting patient outcomes and resource use. This approach to economic analysis is congruent with the way evidence is currently generated in clinical trials. An alternative to model-based CEA is to piggyback an economic evaluation onto a prospective RCT. However, there are problems with basing an economic analysis solely on one RCT. 58 Model-based CEAs are useful in that they can extrapolate beyond the time horizon of any individual clinical trial. Models can also link intermediate clinical end points to final health outcomes, for example depression scores to QALYs. Models also allow different forms of evidence, as well as those from RCTs, to be integrated. This structured assimilation of evidence in a model-based CEA can then provide decision-makers with more relevant and generalisable information on whether or not health interventions represent a good use of public resources. 206
To date, few published examples of model-based CEA consider patients with comorbidities of the main condition of interest. A review conducted by this research team suggested that there were also limited recommendations within health economics guidelines on how to incorporate comorbidities within economic models. Model-based CEAs that fail to account for the particular characteristics of a multimorbid population have the potential to lack validity in much the same way as clinical evidence from single-disease populations (as discussed in terms of applicability in Chapter 3). However, there are additional implications for the face validity of CEA results when patients with multimorbidity undergo numerous treatments. One example, where estimates of the economic benefit could be wrong or misleading, is the addition of a new treatment to a patient already undergoing treatment for comorbidities. In this scenario, all things being equal, the size of the additional absolute benefit (in health measured by QALYs) of the medicine is likely to be smaller than if the patient were receiving no treatment at all for the comorbidity. This follows from at least two potential mechanisms. The first comes from the quality-of-life weighting system, which is anchored between 0 and 1. For patients with multiple possible interventions for multiple diseases the initial treatment improves quality of life but at some point the gains to be made by increasing treatments diminish because of the anchoring system on the quality-of-life scale. Second, the mechanism by which any absolute benefit occurs (however measured) is generated typically from applying a relative risk adjustment to a baseline risk. However, if the baseline risk is lowered by a concordant treatment, the absolute gains for any new treatment will be reduced compared with when no treatment was given.
Another example of where CEA results may be unreliable is where the impact of a new intervention negatively impacts on the existing treatments for an existing condition, increasing the risks of harm associated with a new intervention. The harm, such as an ADE, can have an impact on both the benefit to patients (through the QALY) and the costs (e.g. a visit to accident and emergency department or an additional attendance at primary care). The inclusion of ADEs is generally considered to be important in economic models but it is not clear how they should be incorporated and tackled within an economic analysis. 207 Moreover, harm is typically less well quantified than benefit because RCTs are primarily designed to quantify benefit in populations selected to be at lower risk of harm (by being younger and having fewer potentially interacting comorbidities and coprescriptions), whereas harms are only sometimes quantified in observational studies. However, it is increasingly being recognised that incorporating ADEs within economic models is important and should be formalised if CEA results are to be valid and useful for decision-making. 186
The inclusion of more than one condition of interest for the relevant patient population poses a number of challenges to the normal conduct of economic analysis, generally, and to model-based CEA conducted for a CG, specifically. The most obvious challenge for a model, which is a purposeful simplification of reality in order to provide useful information, is how to (1) identify the important conditions to model simultaneously and then (2) capture the interactions between the various entities (e.g. the progress of the diseases simultaneously) mathematically into a structured model. The inclusion of more than one condition makes the decision problem (what is the most cost-effective treatment/intervention?) much more complex. With comorbidity there is the potential for interactions between the intervention and the other conditions patients have (which may be either harmful or beneficial), as well as between interventions for different conditions. There are long-standing debates on how to tackle the trade-off between simplicity and complexity in models, and there are significant time constraints on producing timely and robust evidence useful for national-level resource allocation decisions. 208 A fundamental challenge is how to populate new, more complex, models with robust evidence.
Aim
This study aimed to structure a model-based CEA of treatment strategies to inform a CG for a patient population with more than one condition.
Study objectives
This study had seven objectives:
-
to select a relevant case study to model the impact of including more than one condition in a model-based CEA suitable for informing a CG
-
to conceptualise and structure a decision-analytic model to identify the relative costs and benefit of interventions relevant to the selected case study
-
to populate the decision-analytic model to identify the relative costs and benefit of interventions relevant to the selected case study
-
to use structured expert elicitation methods to quantify parameter values that cannot be identified in the published literature
-
to identify the incremental costs and benefits of interventions relevant to the selected case study
-
to characterise the uncertainty in the incremental costs and benefits
-
to estimate absolute measures of benefit that could be compared with other treatment options for patients with multimorbidity.
Selection of the case study
The PRG was involved in the selection of the case study to model the impact of including more than one condition in a model-based CEA suitable for informing a CG. The PRG was guided by three pre-defined criteria to select a relevant case study. First, there must be an accessible existing model-based CEA that has been used to inform a CG produced by NICE. Second, to make the modelling approach feasible, two conditions that are commonly comorbid should be selected. Third, the conditions to be considered should occur together with sufficient frequency to provide a clinically relevant scenario of multimorbidity. On this basis, and after extensive discussion at the first meeting of the PRG, the case study selected was pharmacological interventions in patients with depression and CHD. In CG91, patients who had not benefited from low-intensity psychosocial interventions were recommended an antidepressant (usually a SSRI) or high-intensity psychological treatment. 17 The discussion within the PRG acknowledged that much of the specific guidance provided in the CG for patients with depression and chronic physical conditions (CG91) on the selection of antidepressants was contingent on the side-effect profile of the antidepressants and the potential interacting effects the antidepressants had on the patient’s chronic physical health condition. CG91 also recommended not using SSRIs where patients were also likely to be using medications for CHD, such as warfarin and other antiplatelets or anticoagulants, because of potential antiplatelet effects. Moreover, given the growing literature investigating the impacts of depression on CHD outcomes and vice versa, there was thought to be a sufficient pool of evidence on which to generate exploratory economic analysis and cost-effectiveness results investigating some of the interactions between these conditions.
Scope of the model-based cost-effectiveness analysis
Box 2 summarises the research question and scope for the selected base-case analysis for the model-based CEA.
Decision problem: To estimate the relative cost-effectiveness of SSRIs, duloxetine, venlafaxine extended release and mirtazapine for the treatment of major depressive disorder in primary care for patients who are also likely to go on and receive treatment for CHD. Secondary analysis to conduct exploratory analysis investigating interaction effects and the calculation of the absolute QALY.
Comparators: Citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline, duloxetine, mirtazapine, reboxetine, venlafaxine.
Model type: A DES.
Population: Male patients with moderate to severe depression (major depressive disorder) at risk of CHD, starting treatment for a new episode of depression in primary care, aged ≥ 60 years.
Perspective: NHS and personal social services.
Time horizon: 1 year, 5 years, 10 years and lifetime (to 100 years of age).
Effectiveness data: Existing network meta-analysis of placebo-controlled and head-to-head RCTs.
Harms data: Probability of ADE taken from the same study as for the effectiveness data. Proxies ADEs with dropout rate. For patients who go on to develop CHD, risk of ADE is taken from an expert elicitation exercise.
Costs (£, 2014): Direct medical costs (drugs, GP visits, costs associated with drug-to-drug reactions, ADEs, CHD states).
Benefits: QALYs and absolute QALYs.
Discounting: 3.5% for both costs and benefits.
Analysis of uncertainty: PSAs to quantify uncertainty and scenario sensitivity analysis to assess the impact of model structures and to conduct more exploratory analysis, such as drug–disease interactions.
Cost-effectiveness threshold: NICE recommended threshold of £20,000 per QALY gained.
Modelling approach
This study used a DES model with a lifetime horizon to capture the costs and benefits of antidepressant treatment for patients with depression at risk of CHD (some of whom will go on to develop CHD). Caro209 and Caro et al. 210 provide useful discussions of the advantages of DES models.
Economic models used in the context of model-based CEA can be categorised in many ways, but one useful way is the aggregation level of the object of interest (the patient). Generally there are two classifications. The first is the cohort approach (used in the NICE CG90 model assessing the cost-effectiveness of 12 new-generation antidepressants), whereby the patient population as a whole is modelled at an aggregate level. 45 The patient population is described as having certain characteristics that affect the transitions within the models between health states. By making the population homogeneous (example characteristics: male, aged 65 years, smoker), transition probabilities will be that of the mean profile conditional on the various characteristics. The vast majority of models take a cohort approach. Decision trees and Markov models are usually cohort based. A problem with these cohort-type models is that they lack the flexibility to readily incorporate parameters such as risk when the inputs are not usually normally correlated (they are skewed), or to incorporate patient characteristics that may interact or correlate.
A key problem pertinent to cohort models is the way that time is captured. In decision trees the time element associated with events and treatments is not adequately captured. In Markov models a time element can be captured but this is done by setting arbitrary cycle lengths within the model structure. Another key problem is the ability to incorporate memory within the model. When a patient cohort moves from one state to another within a Markov model, the process is blind to the previous disease history of the cohort. Therefore, a patient who has had three recurrent depressive episodes and is now in remission is bundled within the same health state as a patient who has had only one previous episode. The only way to avoid this omission, and capture the impact of memory within a Markov model, is to use tunnel states, which massively increase the complexity of the model yet still do not adequately capture the time element.
Rather than adopt a cohort approach, it is possible to model the individuals in the patient population. Using this approach means that each individual’s characteristics then determines his or her progress. Patient attributes can be tracked as individuals progress, dynamically changing the risk of moving between health states. Moreover, time is explicitly captured in the model, allowing patients to age within the model and again dynamically altering the risk structure between the various competing risks. There are some published DES models relevant to depression, but none has included another condition. 211–215 The characteristics of depression alone, and particularly the characteristics of depression alongside another condition, make DES the ideal modelling approach to address the stated research question.
Model conceptualisation
Model conceptualisation is a key stage in developing a model structure. This study followed recommendations published by the International Society for Pharmacoeconomics and Outcomes Research/Society for Medical Decision Making and also referred to in a NICE DSU document on model conceptualisation. 183,216 The process of conceptualisation followed a problem-orientated approach. The problem-orientated approach is intended to elicit current clinical knowledge of the relevant characteristics of the disease as well as the important events associated with the disease. Once the important aspects have been captured, the model conceptualisation moves on to how best to capture these aspects in a mathematical model structure. This process generally includes a review of existing modelling structures relevant to the research question.
Systematic review studies were identified that investigated the use of economic models for depression. 212,215,217 The most recent review, by Ali Afzali et al. ,218 investigates 14 depression models from the literature and finds evidence of substantial heterogeneity in model structures and types, techniques employed, time horizons and data sources used. The authors identified two key potential problems arising in some of the models reviewed: first, a short time horizon for analysis, particularly where decision trees were employed, which limited the ability of the analysis to capture long-term effects of the interventions (e.g. on suicide); second, the inability of some models to fully capture the natural course of depression in the health states (e.g. relapse and remission). Consequently Ali Afzali et al. 218 proposed to use a DES model with a lifetime horizon, which is in keeping with a growing body of model developers using discrete event models for cost-effectiveness modelling in depression. 212–215
Key attributes that were identified as being attractive for using a DES model for depression and CHD were the ability to:
-
model long-term disease effects beyond the initial acute stage
-
adjust the risk of relapse dependent on the number previous episodes (or CHD status) so that individuals within the model have memory
-
incorporate competing risk more simply by modelling time to event
-
capture adverse drug reactions more completely.
Following a review of the available DES models, the model by Vataire et al. 214 was considered to be the most relevant DES model for depression and was used as the basis for informing the structure and events within the proposed new model.
Cost-effectiveness models obtained through NICE were also used to help conceptualise both the depression and CHD aspects of the economic model. In particular, the NICE cost-effectiveness model investigating the use of statins for the prevention of CHD was considered in detail.
The key attributes of the depression and CHD model were then conceptualised to be:
-
the natural course of the two diseases and how this could be translated into associated health states
-
the impact that the health states have on HRQoL and costs
-
the impact that the comparator has on the movement between the health states
-
the potential interactions between the two diseases.
An initial meeting was conducted with the PRG to decide upon the research area for the CEA. Follow-up meetings were used in order to provide peer-review to the model structure, to ensure relevant attributes were included and to identify relevant sources of data to populate the model. The conceptualisation of the model underwent numerous iterations to reflect the feedback from the clinical experts as well the limitations of the evidence base. Prior to each meeting, the model was explained using non-technical, non-mathematical language as well as through the use of graphical methods. In total, four meetings were held, and the final one concluded with a presentation of some preliminary results from the model-based CEA.
Model overview
The cost-effectiveness research question was answered by structuring a DES model developed using Simul8 software (version 2014; Simul8 Corporation, Boston, MA, USA). The model tracks patients with depression who go on to develop CHD and so includes events linked to both disease types. Current non-DES economic models for depression typically use a time frame ranging from 6 to 15 months. The time horizon used in this analysis was selected to capture all relevant benefits and costs for depression and CHD (and varied from 1 year through 5 and 10 years to lifetime). The perspective adopted was that of the NHS and personal social services, in keeping with the NICE reference case. 59 Life expectancy statistics were sourced from ONS interim life tables. 202 Patients were followed up from initial treatment until death, which has multiple potential causes (all-cause, suicide, CHD mortality), or until the time horizon is reached. Patients with depression were assumed to go through periods of response and remission and to be at risk of relapse, and were also assumed to be at risk of developing CHD with its associated costs and its impact on ADE probability. Patients treated for CHD were assumed to have a higher risk of an ADE from the antidepressant.
Model structure
Figure 14 shows the general model structure. Patients (male and aged 60 years) enter the model when they experience a new episode of major depression for which they are going to be offered treatment with an antidepressant following a GP consultation.
FIGURE 14.
Schematic of the overall model structure.

Treatment effectiveness and adverse drug reactions
Patients were assumed to either respond, or fail to respond, to the first selected antidepressant (first-line intervention) within 8 weeks of starting treatment. Response probabilities for first-line intervention were taken from a published network meta-analysis conducted by Cipriani et al. ,219 which was also used in the economic analysis conducted for NICE CG90. These data were selected because (1) they were deemed to be of high quality, (2) they were consistent with the NICE guideline and (3) they provided a set of parameters for all the comparators. In the original network meta-analysis, the population was not described explicitly as including patients with both depression and CHD, but for this analysis it was assumed that the response rates to antidepressants would not be affected by the existence of the second condition (CHD) in the population. Uncertainty around the point estimates for response was represented by using normal probability distributions making use of the upper and lower CI for the treatment probabilities in the PSA (see Table 25).
| Comparator | Probability of response in 8 weeks (95% CI)a | Probability of ADE: patients have no CHD (95% CI)a | Probability of ADE: patients have CHDb |
|---|---|---|---|
| Citalopram | 0.57 (0.53 to 0.62) | 0.26 (0.22 to 0.30) | 0.30 |
| Escitalopram | 0.62 (0.58 to 0.62) | 0.24 (0.21 to 0.28) | 0.30 |
| Fluoxetine | 0.55 (0.55 to 0.55) | 0.28 (0.20 to 0.32) | 0.30 |
| Fluvoxamine | 0.54 (0.48 to 0.60) | 0.32 (0.26 to 0.38) | 0.30 |
| Paroxetine | 0.55 (0.52 to 0.59) | 0.30 (0.27 to 0.33) | 0.30 |
| Sertraline | 0.60 (0.57 to 0.64) | 0.25 (0.22 to 0.29) | 0.30 |
| Duloxetine | 0.55 (0.49 to 0.60) | 0.31 (0.26 to 0.38) | 0.35 |
| Mirtazapine | 0.63 (0.58 to 0.67) | 0.28 (0.24 to 0.33) | 0.37 |
| Reboxetine | 0.45 (0.39 to 0.51) | 0.35 (0.29 to 0.42) | 0.36 |
| Venlafaxine | 0.61 (0.58 to 0.64) | 0.29 (0.26 to 0.32) | 0.38 |
A rapid review was conducted to find the most suitable data on utilities and combined with the evidence synthesised from the systematic review by Ali Afzali et al. 218 Patients who had responded to their treatment were assumed to experience an increase in their health status that was reflected by a higher utility value. Table 26 summarises the utility values used in the model.
| Depression state | CHD state | ||||||
|---|---|---|---|---|---|---|---|
| Stable angina | Unstable angina | MI | TIA | Stroke | Heart failure | No CHD symptoms | |
| Depression | 0.406a | 0.368a | 0.358a | 0.406a | 0.226a | 0.281a | 0.424b |
| Response | 0.742a | 0.544a | 0.694a | 0.742a | 0.562a | 0.617a | 0.760c |
| None | 0.808d | 0.770d | 0.760d | 0.808d | 0.628d | 0.683d | 0.826e |
Figure 15 illustrates that the model assumed patients could experience an ADE following either non-response or response to a treatment. ADEs that did occur were assumed to be experienced within the first 4 weeks of treatment, prompting a repeat GP consultation at this time. The probability of any ADE was estimated using evidence on dropout rates for the different types of antidepressants from Cipriani et al. 219 as a proxy measure. A priori an assumption made was that patients who have had a CHD event were more likely to be on concomitant treatment and, therefore, the chance of any ADE was likely to be higher. For patients who had undergone a CHD event, the probability of any ADE (see Table 25) was sourced from an expert elicitation exercise, which used the Sheffield Elicitation Framework technique223 to elicit values from members of the PRG. Appendix 4 summarises the design and analysis of the expert elicitation exercise.
FIGURE 15.
Schematic of the ‘response’ section of the model. ADR, Adverse Drug Reaction.
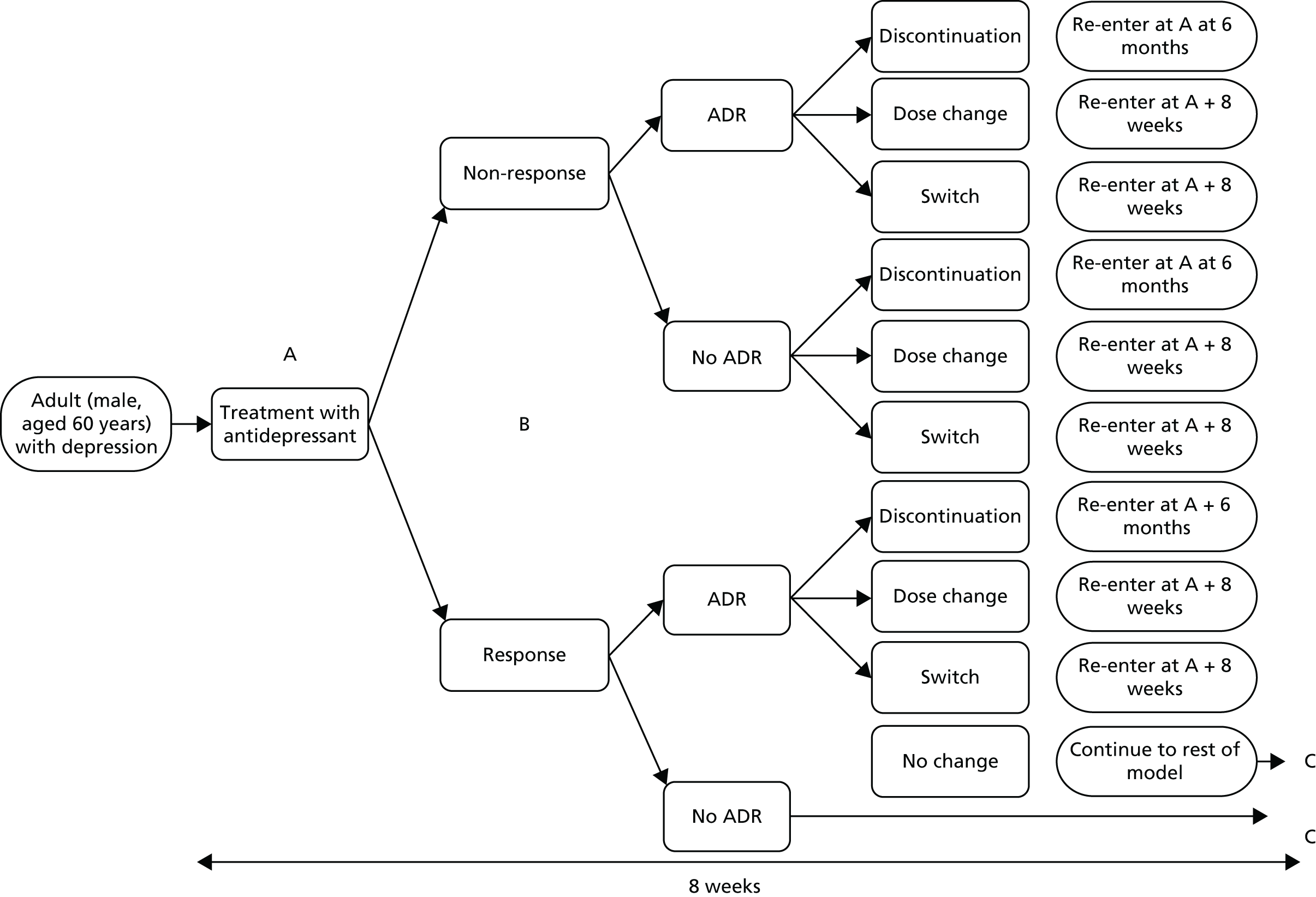
For patients who respond, and who have an ADE, the model assumed that patients would require an additional GP consultation in which the GP had the choice of (1) switching, (2) changing the dose or (3) keeping things as they are. Appendix 5 summarises the assumed probabilities for these events.
The model also accounted for the probability of patients discontinuing treatment following an ADE (see Appendix 5). Patients who discontinued treatment were assumed to have a period of down-time (6 months) in which they did not interact with the health-care system. For those who did not respond to treatment, an assumption was made that clinicians could not keep things as they were but must change treatment (to start a second-line treatment). It was assumed that the second-line antidepressant has the same clinical effectiveness as first-line treatment.
If a patient experienced an ADE, a temporary disutility was used to reflect the decrease in health status associated with that adverse event (Table 27). The disutility was assumed to last for a 4-week period. The ADE disutilities were applied additively.
Subsequent response and recovery
Patients who responded following either a first or a second line of treatment were assumed to have an increase in their HRQoL. Patients were also assumed to be at risk of a relapse of their depression. Time to relapse was modelled as being dependent on the number of previous episodes of depression the patients had experienced. Weibull curves were fitted to evidence taken from a 10-year multicentre naturalistic study to simulate this effect. 225 Figure 16 shows the diminishing expected time it was assumed to take for relapse to occur with each increasing number of episodes of depression. Patients who did not relapse within 6 months were said to be in the state of ‘recovery’. The utility value for the recovery was set as the same value as response.
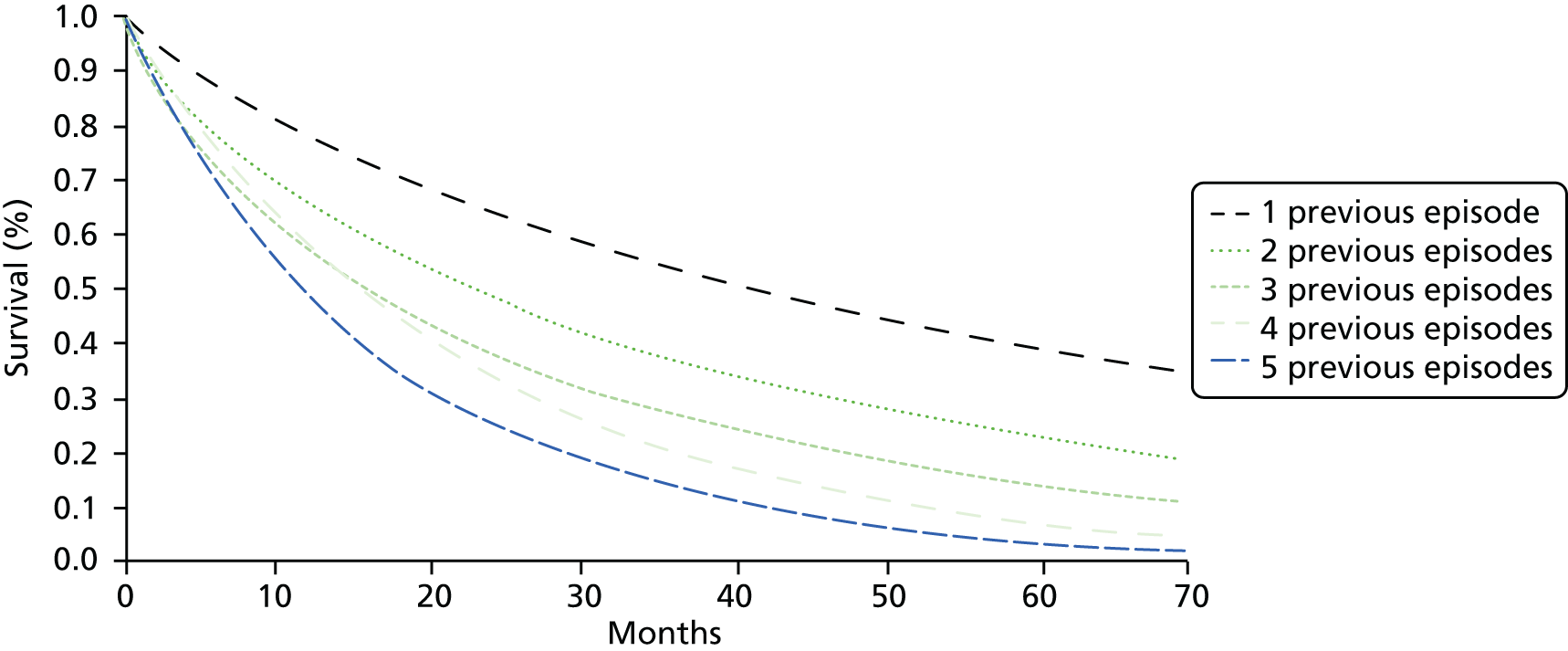
Long-term events
Patients were assumed to be at risk of a long-term event. This risk was pre-specified at the beginning of the model and took account of (1) all-cause mortality (excluding suicide and CHD death), (2) suicide and (3) a CHD event. The likelihoods of all-cause mortality and suicide were taken from ONS interim life tables and causes of death statistics. 202 Suicide risk for depressed patients was assumed to be twice that of the general population.
The baseline risk and the type of first CHD event were taken from Ward et al. 201 (Table 28). The potential CHD health states were:
-
stable angina
-
unstable angina
-
MI
-
TIA
-
stroke
-
heart failure.
| Age (years) | Stable angina (%) | Unstable angina (%) | MI (%) | TIA (%) | Stroke (%) | CHD death (%) | Total cardiovascular event rate per 1000 per annum |
|---|---|---|---|---|---|---|---|
| 55–64 | 32.8 | 7.1 | 17.2 | 8.9 | 20.6 | 8.6 | 13.7 |
| 65–74 | 21.4 | 8.3 | 17.3 | 10.0 | 27.0 | 9.7 | 24.3 |
| 75–84 | 19.1 | 8.1 | 16.1 | 8.0 | 34.3 | 6.3 | 37.5 |
| ≥ 85 | 21.4 | 9.6 | 18.6 | 1.6 | 35.1 | 5.5 | 42.6 |
Following a first CHD event, the time to the next secondary CHD event was sampled from a transition matrix of time-to-event data. The type of secondary event CHD probabilities and time-to-event data were adjusted using 5-year age bands as reported by Ward et al. 201 and the shortest time to event was selected (see Appendix 6 for the transition matrix).
The model accounted for the impact on the patient’s health status measured in utility values from experiencing a CHD event. Where patients experienced both a CHD event and a depression state they were allocated a joint health state utility value. To calculate the joint health state utility value, the utility value for the depressed health state (depression, response) was combined with the CHD health state utility score using the additive method as detailed by Ara and Wailoo. 226 Currently there is no best-practice recommended method for combining joint health state utility values204 so the additive method was chosen for simplicity.
Resource use and costs
The model assumed a NHS and personal social services perspective. Costs used were from the price year 2013/14. The key items of resource use and costs in the model are described in Table 29 for depression and Table 30 for CHD. The costs associated with time spent within CHD health states were sourced from the assumptions made within NICE CG181. 148 Costs associated with depression were taken from the resources used by patients in attending GP appointments (first and follow-ups) as well as the ongoing antidepressant cost.
| Item | Unit cost (£) | Per annum cost (£) | Source |
|---|---|---|---|
| GP appointment | 46 | N/A | Curtis (2014)227 |
| Citalopram | 1.09a | 14.21b | BNF 69228 |
| Escitalopram | 25.20a | 657.00b | BNF 69228 |
| Fluoxetine | 1.16a | 14.11b | BNF 69228 |
| Fluvoxamine | 17.01a | 103.48b | BNF 69228 |
| Paroxetine | 2.29a | 27.86b | BNF 69228 |
| Sertraline | 1.46a | 19.03b | BNF 69228 |
| Duloxetine | 27.72a | 361.35b | BNF 69228 |
| Mirtazapine | 1.60a | 20.86b | BNF 69228 |
| Reboxetine | 18.91a | 57.52b | BNF 69228 |
| Venlafaxine | 2.65a | 34.54b | BNF 69228 |
| GI bleed | 2850 | N/A | Campbell et al. (2015)229 |
| Event state | First year (£) | Follow-up year (£) |
|---|---|---|
| No CHD | 0 | 0 |
| Stable angina | 7736 | 240 |
| MI | 3313 | 385 |
| TIA | 3337 | 788 |
| Stroke | 578 | 1234 |
| Heart failure | 4092 | 155 |
| CHD death | 2297 | 0 |
The model assumed that there was no cost burden associated with ADEs apart from the additional GP consultation, with the exception of patients who have a GI bleed, who were assumed to cost an additional £2850 because of the need for in-hospital and post-discharge care associated with bleeds. 229
The cost for each CHD event was calculated by multiplying the time spent in the health state with the per-day unit cost of the health state. These costs were adjusted according to whether the CHD health state was in its first year or subsequent follow-up years (see Table 30).
Model logic
Patients were assumed to move through the events in the model, and experience the response and recovery periods as specified. The associated QALYs and costs were tracked for each event throughout the model. Patients were also assumed to be at risk of long-term CHD events, all-cause mortality and suicide. If the cumulative time a patient spent within the model exceeded the shortest of the long-term events, then a patient was assumed to move to these states. Patients were assumed to leave the model if their time in the model exceeded the shortest of the long-term events (all-cause mortality, suicide, CHD death) or if the time horizon was exceeded.
Calculation of quality-adjusted life-years, costs and net benefit
A total of 5000 patients were simulated moving through the model. This number of patients was used to allow stability in the model outputs. Each patient’s QALYs and costs were calculated. Costs and QALYs were discounted at 3.5% equally. Incremental net benefit was calculated using the formula for a cost-effectiveness threshold (λ) of £20,000:
Model verification, structural stability and sensitivity analyses
The recommendations contained within the NICE DSU document on DES model was used to inform model verification processes. 230 In particular, the model was verified by:
-
stepping through the experience of individual patients within the model
-
recording interim outputs
-
comparing the results with ex-ante expectations for face validity
-
comparing deterministic and probabilistic results
-
internal peer-review by the analyst responsible for building the model
-
discussions with clinical experts within the PRG and research team on the model structure.
The model was run separately for each comparator. For the deterministic analysis, the mean value for each parameter was calculated to capture mean QALYs, costs and net benefit. For the PSA, pre-defined parameters within the model had an associated distribution from which a value was sampled in order to capture uncertainty. For the PSA, 1000 samples (bootstrap replicates) were completed, as convergence in the results was assumed to occur at this point.
Results
Table 31 shows the results from the deterministic analysis. The final column in this table shows the results from the fully incremental analysis, in which the antidepressants are first ordered in terms of costs and then the incremental cost-effectiveness ratios are calculated as appropriate. The two most cost-effective treatment options were identified to be sertraline and citalopram.
| Antidepressant | 1 year | 5 years | 10 years | Lifetime | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mean cost (£) | Mean QALYs | Mean cost (£) | Mean QALYs | Mean cost (£) | Mean QALYs | Mean cost (£) | Mean QALYs | Lifetime ICERa | |
| Mirtazapine | 176 | 0.586 | 413 | 3.115 | 1035 | 5.638 | 3411 | 9.943 | – |
| Fluoxetine | 189 | 0.581 | 431 | 3.105 | 1062 | 5.640 | 3467 | 9.972 | Extendedly dominated by mirtazapine and citalopram |
| Citalopram | 179 | 0.589 | 394 | 3.127 | 1037 | 5.662 | 3484 | 9.984 | £1743 per QALY |
| Sertraline | 167 | 0.600 | 407 | 3.149 | 1010 | 5.700 | 3532 | 9.997 | £3807 per QALY |
| Paroxetine | 189 | 0.583 | 478 | 3.110 | 1066 | 5.651 | 3692 | 9.975 | Dominated |
| Venlafaxine | 187 | 0.578 | 466 | 3.094 | 1195 | 5.621 | 3732 | 9.967 | Dominated |
| Reboxetine | 271 | 0.512 | 684 | 2.868 | 1433 | 5.337 | 4005 | 9.589 | Dominated |
| Fluvoxamine | 221 | 0.578 | 639 | 3.100 | 1422 | 5.638 | 4522 | 9.936 | Dominated |
| Duloxetine | 312 | 0.564 | 1056 | 3.054 | 2655 | 5.579 | 7327 | 9.917 | Dominated |
| Escitalopram | 342 | 0.604 | 1456 | 3.158 | 3935 | 5.703 | 10,924 | 10.006 | £868,101 per QALY |
Figure 17 shows the cost-effectiveness plane that plots the mean expected QALYs against the mean expected costs for each antidepressant, assuming a time horizon of a lifetime.
FIGURE 17.
Cost-effectiveness plane for the selected 10 antidepressants for a lifetime horizon.
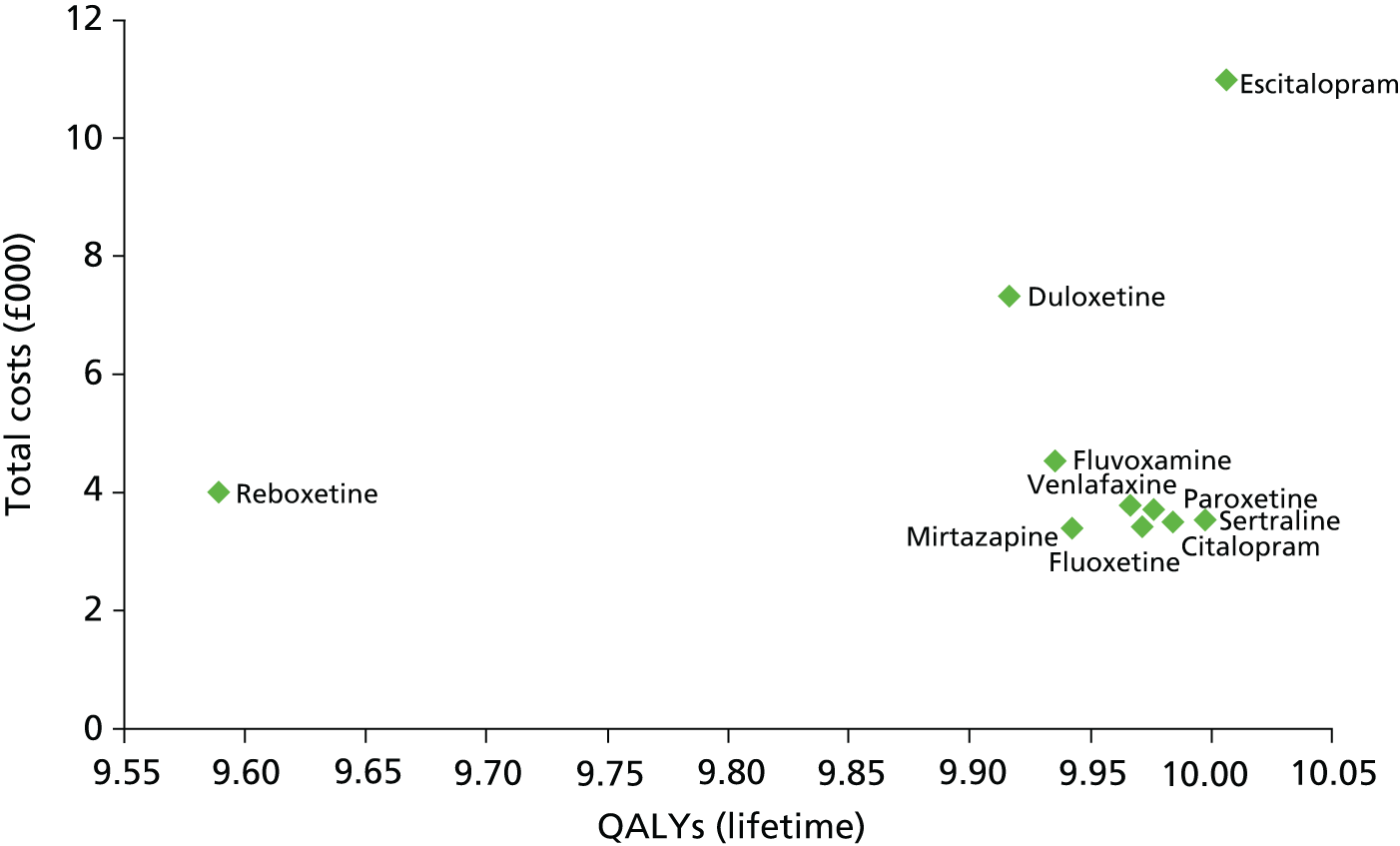
Table 32 summarises the results from the PSA and shows the uncertainty around the mean estimates of costs and QALYs for a lifetime horizon.
| Comparator | Costs, £ (95% CI) | QALYs (95% CI) | Net benefit, £ (95% CI) |
|---|---|---|---|
| Citalopram | 3412 (3256.95 to 3587.26) | 9.93 (8.13 to 10.57) | 196,206 (159,229 to 208,001) |
| Escitalopram | 10,749 (9954.26 to 11,008.73) | 9.99 (8.29 to 10.58) | 189,189 (155,761 to 200,738) |
| Fluoxetine | 3427 (3265.35 to 3610.1) | 9.90 (8.05 to 10.59) | 195,980 (157,515 to 208,346) |
| Fluvoxamine | 4447 (4281.53 to 4623.02) | 9.92 (8.02 to 10.53) | 194,191 (155,868 to 206,068) |
| Paroxetine | 3577 (3413.88 to 3759.25) | 9.94 (8.11 to 10.54) | 195,806 (158,513 to 207,281) |
| Sertraline | 3452 (3288.44 to 3608.62) | 9.99 (8.20 to 10.57) | 196,411 (160,577 to 207,849) |
| Duloxetine | 7324 (6854.82 to 7558.22) | 9.84 (7.87 to 10.54) | 191,003 (150,572 to 203,321) |
| Mirtazapine | 3464 (3312.12 to 3612.93) | 9.89 (8.11 to 10.59) | 195,444 (158,587 to 208,355) |
| Reboxetine | 4059 (3872.45 to 4272.36) | 9.54 (8.01 to 10.22) | 187,777 (155,991 to 200,348) |
| Venlafaxine | 3622 (3469.65 to 3778.49) | 9.84 (8.03 to 10.59) | 195,616 (155,991 to 200,348) |
Figure 18 shows the associated cost-effectiveness acceptability curve for each of the 10 antidepressants. Sertraline was identified as the most cost-effective option when using a threshold willingness to pay for a gain of 1 QALY within the range of £20,000 to £30,000 per QALY.
FIGURE 18.
Cost-effectiveness acceptability curve for the 10 antidepressants for a lifetime horizon.
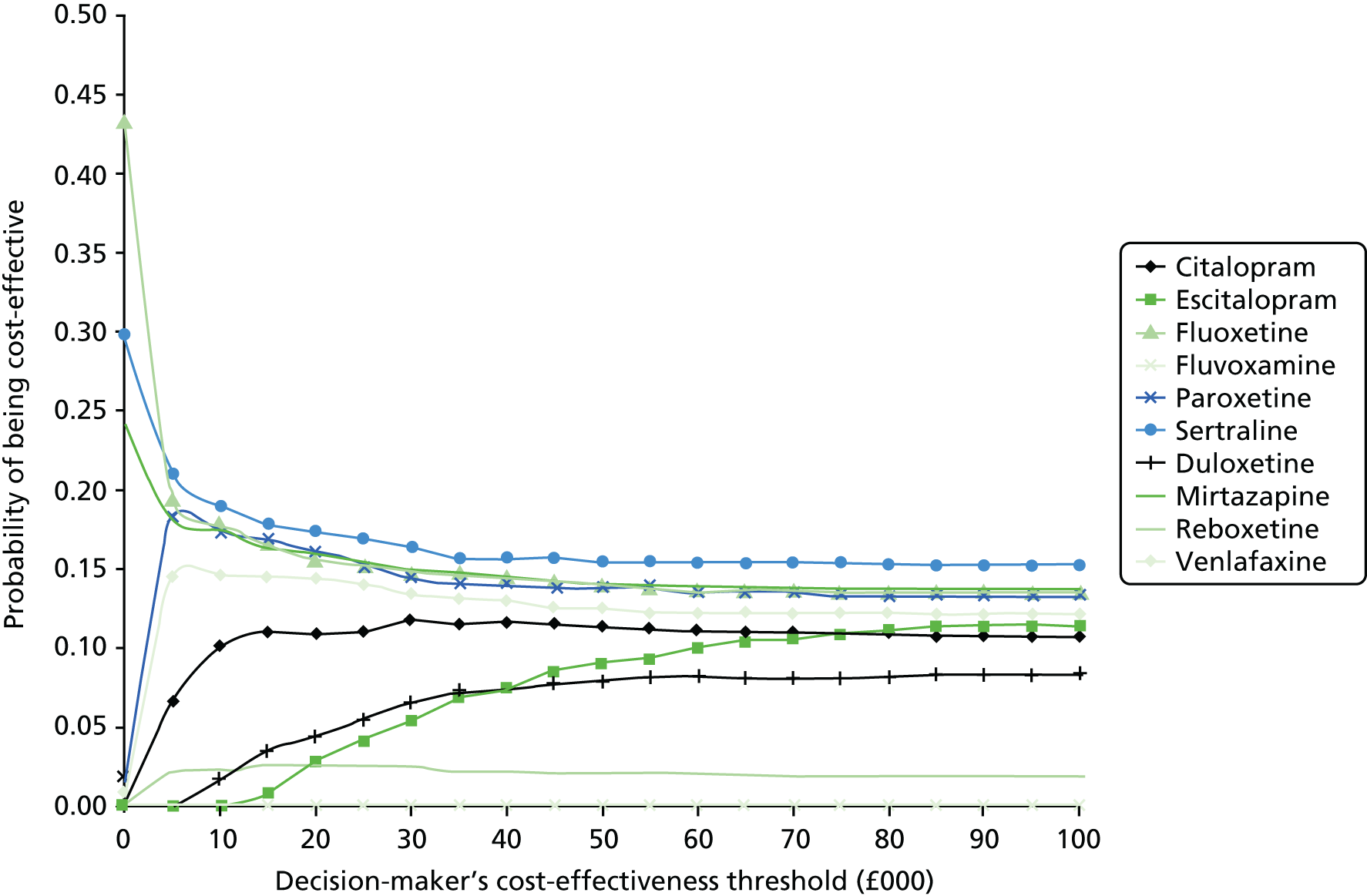
Exploratory scenario analyses: impact of disease–disease interaction
Two exploratory scenario analyses were conducted to account for an interaction between CHD and depression. Conducting these scenario analyses demonstrates the potential flexibility of the discrete event model to allow a range of scenario analyses to be performed.
Several prospective studies have shown evidence of the predictive role of depression to determine the onset of CHD. This evidence base suggests that depression may be an independent risk factor for CHD. 231,232 There is also evidence of depression having an unfavourable impact on patients with new-onset CHD in terms of their risk of mortality and future CHD events. In theory, therefore, the effective treatment of depression, using for example a SSRI, could potentially improve CHD outcomes including mortality risk through at least two potential mechanisms:233
-
behavioural – for example, an improvement in depression could improve smoking habits, medication adherence, alcohol consumption or physical activity, which could improve CHD outcomes
-
physiological – SSRIs are thought to attenuate platelet function, which therefore leads to cardiac benefits.
The first scenario analysis assumed no risk of CHD in the population (scenario 1) and the second scenario analysis assumed that treatment with a SSRI antidepressant lowered the risk of developing CHD by 5% (see scenario 2). Table 33 presents the results for two most cost-effective treatment options identified in the base-case analysis: citalopram and sertraline.
| Scenario | Treatment | 1 year | 5 years | 10 years | Lifetime | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean cost (£) | Mean QALY | Mean cost (£) | Mean QALY | Mean cost (£) | Mean QALY | Mean cost (£) | Mean QALY | ||
| Base case | Citalopram | 176 | 0.586 | 413 | 3.115 | 1035 | 5.638 | 3411 | 9.943 |
| Sertraline | 187 | 0.578 | 466 | 3.094 | 1195 | 5.621 | 3732 | 9.967 | |
| 1: no risk of CHD | Citalopram | 157 | 0.603 | 241 | 3.200 | 336 | 5.896 | 538 | 11.685 |
| Sertraline | 150 | 0.613 | 231 | 3.221 | 338 | 5.924 | 579 | 11.711 | |
| 2: CHD benefit | Citalopram | 175 | 0.589 | 396 | 3.137 | 1035 | 5.680 | 3276 | 10.062 |
| Sertraline | 185 | 0.580 | 418 | 3.116 | 1041 | 5.653 | 3289 | 10.057 | |
Calculation of absolute quality-adjusted life-years: an exploratory analysis
A third exploratory analysis was conducted to identify the potential gains in absolute QALYs from the two treatment options identified as being the most effective options in the base-case analysis. This analysis involved redefining the relevant comparator to be ‘do-nothing’. To calculate the absolute QALYs for the comparators it was necessary to make an assumption on some of the key parameters for the do-nothing treatment option. For simplicity the model structure was kept the same but the values for the probability of response to treatment and probability of an ADE were changed. The new values were assumed to be 0.26 for the probability of response within 8 weeks and 0.15 for the probability of an ADE. Absolute QALYs were calculated by subtracting the QALYs that would have been received without any treatment from those gained from treatment with either citalopram or sertraline. Table 34 shows the results from the deterministic analysis and also incorporates two scenario analyses (scenario analysis 1 and scenario analysis 2).
| Scenario | Treatment | Absolute QALYs | |||
|---|---|---|---|---|---|
| 1 year | 5 years | 10 years | Lifetime | ||
| Base case | Citalopram | 0.116 | 0.375 | 0.434 | 0.458 |
| Sertraline | 0.127 | 0.397 | 0.472 | 0.471 | |
| 1: no risk of CHD | Citalopram | 0.128 | 0.411 | 0.486 | 0.538 |
| Sertraline | 0.138 | 0.432 | 0.513 | 0.564 | |
| 2: CHD benefit | Citalopram | 0.115 | 0.386 | 0.451 | 0.536 |
| Sertraline | 0.127 | 0.408 | 0.476 | 0.550 | |
Figure 19 shows the absolute QALYs gained for sertraline compared with a do-nothing approach for different assumptions regarding the model time horizon and for the base case and two scenario analyses (scenario analysis 1 and scenario analysis 2). For the base-case analysis, it can be seen that most of the absolute QALYs gain occurred within the first 5 years of treatment. The gain in absolute QALY then falls as the time horizon increases, for two likely reasons: (1) the smaller population size as patients begin to leave the model because of the competing risk of the CHD and (2) the diminishing effect that discounting has on the quality-of-life gains.
FIGURE 19.
Absolute QALYs gain for sertraline for the base case and two scenario analyses with different model time horizons.
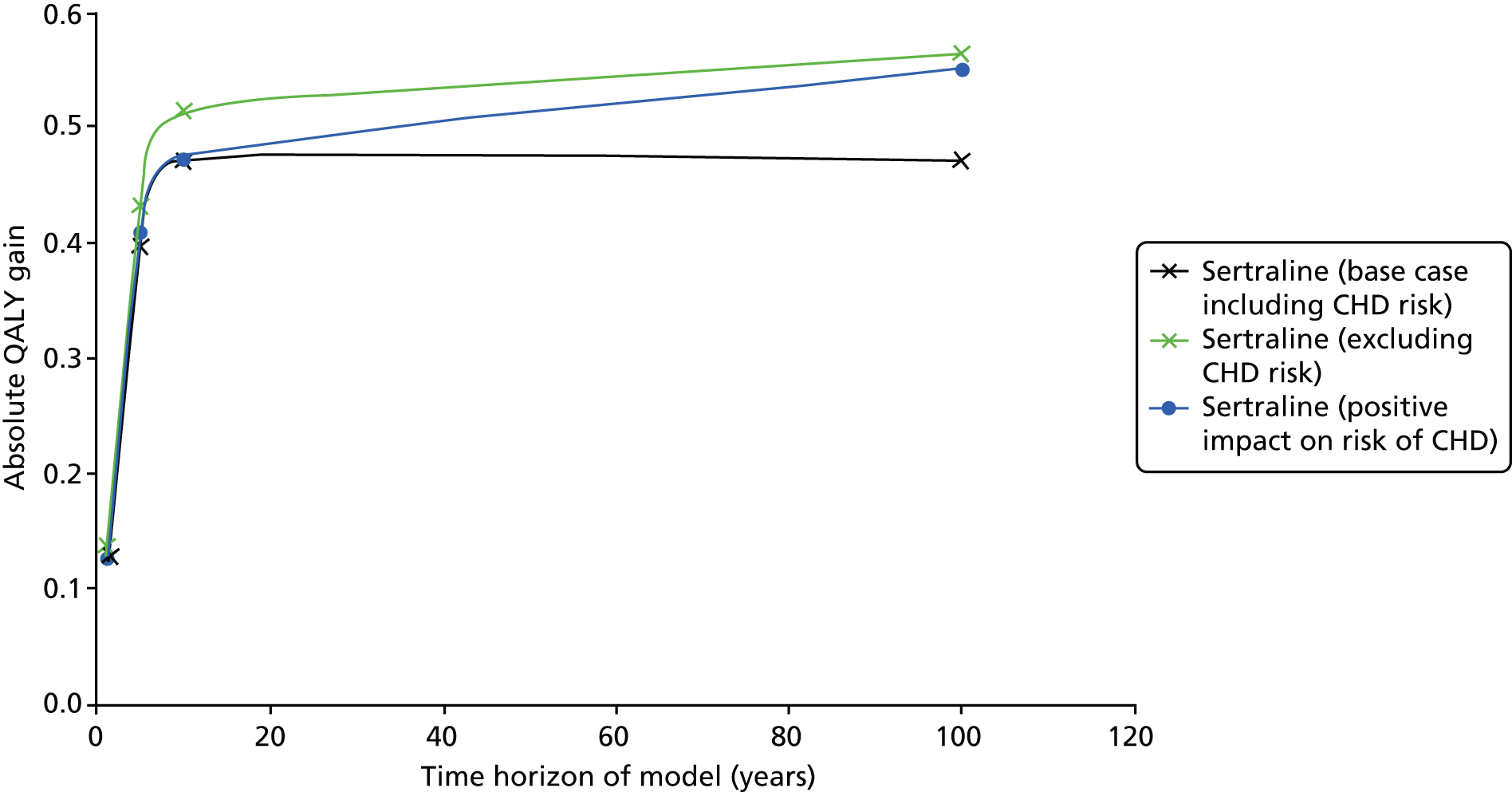
In scenario 1, it was assumed that there was no competing risk of CHD included in the model. The results of this analysis give the largest absolute QALY gains, indicating the negative impact that the competing risk of CHD has on the potential for patients to accrue QALY gains. This persists from a 1-year time horizon up until the lifetime horizon.
In scenario 2, the absolute QALY gains were calculated assuming that there is a risk of CHD as in the base-case analysis but the antidepressant reduces the risk of developing CHD. There was very little difference between the results in the base case and for this scenario until the model time horizon reaches lifetime, in which case the reduction in risk starts to lower the impact of having CHD on the absolute QALY gained.
Discussion
This study has presented a model-based CEA to estimate the relative cost-effectiveness of SSRI antidepressants, duloxetine, venlafaxine and mirtazapine for the treatment of major depressive disorder in primary care for patients who are also likely to go on and receive treatment for CHD. This case study was selected by the PRG as a useful exemplar because it included two conditions that are distinct but commonly comorbid, and because there was the potential for significant interactions between treatments for the two conditions and between the conditions themselves. The model-based CEA suggested that sertraline was likely to be the most cost-effective option for patients with these two conditions but there was great uncertainty around the mean incremental costs and benefits. By comparison, the analysis conducted for CG90, which had a short-term time horizon and did not account for CHD, found mirtazapine to be the most cost-effective option.
To deal with the potential complexity associated with capturing the impact of treating patients with two conditions, a DES model was used. Using a DES model had the advantage of being able to capture the impact of complex events and care pathways. Using this more complex modelling approach, rather than a decision tree or Markov model, was limited by a lack of data to populate all aspects of the DES model. The DES model allowed the analysis to capture the key attributes of depression and CHD and has the flexibility to accommodate greater complexity as and when more data become available. However, building and populating the model was a time-consuming exercise and required substantial analytical skills.
The flexibility of using a DES model-based CEA allowed an exploration of the impact of different time horizons on the sensitivity of the results. On balance, using a longer time horizon means that the analysis is more likely to take account of all the relevant costs and benefits, and particularly so when the interventions have impacts across the diseases. However, there is the associated challenge that as model data inputs and assumptions are extrapolated further there is more likely to be increased uncertainty around the point estimates for expected costs and QALYs.
The model was populated with existing data taken from published NICE CGs and supplemented with a published network meta-analysis219 and an expert elicitation exercise. There were a number of necessary key assumptions that limited the quality and direct relevance of some of the data inputs for the selected patient population with two conditions. The treatment response data for each antidepressant were assumed to be transferable from the published network analysis to a patient population with depression and CHD. The key mechanism used within the model to capture the impact of CHD comorbidity was changing the probability of an ADE, which increased when patients had both depression and CHD, as the result of an assumed interaction effect between the medicines used to treat these two conditions. The assumed impact on the probability of an ADE was informed by an expert elicitation exercise because of the lack of published data. The expert elicitation exercise resulted in wide variation around the point estimate for all of the antidepressants, which was not accounted for in the deterministic analysis but was in the PSA.
The results of the model-based CEA provided useful information to guide recommendations for CGs. Additional insights can be gained by using a measure of absolute QALYs gained, to prioritise treatments for patients with multimorbidity. An exploratory analysis was used to generate absolute QALYs for two selected antidepressants that showed they were the most likely to be cost-effective options in the base-case analysis: citalopram and sertraline. The analysis showed, as expected, that absolute QALYs gained will be dependent on the assumed time horizon for the analysis. The largest absolute QALY gained was generated from sertraline. The estimated absolute QALY gains were in the range of 0.48 to 0.57 QALYs, which are larger than those estimated for treatment with statins (≈ 0.2 QALYs) but smaller than those estimated for treating hypertension (≈ 1.0 QALYs) (see Chapter 4).
Conclusion
This study has shown that it is feasible to develop a model-based CEA for an intervention aimed at a patient population with more than one condition. However, this analysis was limited by the lack of available data for the relevant population and only included two conditions, albeit two that are commonly comorbid. The modelling exercise was time-consuming, which, when combined with the lack of time and number of health economists to produce models, may prohibit the general application of such analyses when informing NICE CGs. One potential solution to the problem would be to set up a national repository of models for selected conditions that can be sourced for guideline development. This would reduce the need for de novo models for some aspects of informing NICE CGs, and hence free up health economists to tackle more challenging model-based analyses such as those that take account of important interacting conditions.
Chapter 7 Summary and conclusions
Summary of findings
The aim of this project was to test the methodological feasibility of new approaches to summarising and creating evidence for guidelines for the management of people with multimorbidity. This was primarily in relation to how single-disease guidelines are created, and the specific focus was significantly determined by the advice of the multidisciplinary PRG, consisting of members with experience of working on NICE or SIGN GDGs.
The initial exploratory work reported in Chapter 2 focused on guidelines for three exemplar conditions (type 2 diabetes, depression and chronic heart failure). It found that the CRQs that determine the guideline focus were only rarely framed in terms of comorbidity or older age (as a proxy for comorbidity). Relatively few treatment recommendations were qualified in terms of comorbidity or older age, and none of the guidelines examined included any qualification in relation to people with short life expectancy. There was little clear synergy and no clear contraindication between recommended treatments across the guidelines examined. None of the research recommendations in the single-disease guidelines explicitly called for research in population subgroups such as people with comorbidity or older people. With some variation across the guidelines examined, economic evidence was available for half of CRQs, but only one in seven CRQs had an associated model-based CEA carried out for it de novo, and all of these were single disease focused.
Potentially serious drug–drug interactions were common between drugs recommended in guidelines for the three exemplar conditions and drugs recommended in guidelines for 11 other conditions. Drug–disease interactions were relatively uncommon, with the exception of interactions when an individual has comorbid CKD.
The analysis of the applicability of evidence in Chapter 3 showed that it is feasible to use a combination of trial inclusion and exclusion criteria and epidemiological data to examine more systematically whether or not applicability of evidence is likely to be a problem, and whether or not there are important drug–disease or drug–drug interactions. Of note is that the three exemplar conditions had distinct patterns of applicability and interaction problems, indicating that, although there is a general problem of applicability when moving from evidence to recommendations, the implications are likely to be condition specific and so need expert judgement to inform extrapolation. However, unlike current guideline development practice, that expert judgement could itself be systematically informed by use of epidemiological data about the population for which the guideline is making recommendations, including data on comorbidity, coprescription and ideally life expectancy.
Chapter 4 showed that it is feasible to compare the absolute benefit of treatments for different conditions, although it requires making a set of important assumptions, which will not always hold and should be made explicit (although many existing treatment decisions already require making these assumptions implicitly). Absolute benefit can be compared in terms of the clinical outcomes reported in trials or absolute QALY gain estimated from model-based CEA repurposed to examine absolute benefit. The latter has the advantage that it combines many different outcomes into a single metric, but, as identified in Chapter 2, the required models are available only for a minority of CRQs and QALYs are not a natural metric for clinicians and patients.
The impact of considering the temporal dimension of benefit was examined in Chapter 5. Previous literature has generally framed this either in terms of benefit being assumed to accrue over the duration of the clinical trials evaluating treatments, or less commonly in terms of making some assessment when survival curves comparing different treatments in trials diverge. Neither is without problems, and we chose to focus on the concept of time, using existing models for CEA to examine change in QALYs over time, and examining how sensitive the findings of such models were to DTD and to an increased competing risk of mortality. Using the model created for the NICE guideline on lipid modification, we showed that even very low levels of DTD were associated with pay-off times measured in years and with reduced lifetime QALY gain, and that increased mortality risk had large effects on lifetime QALY gain. Displaying these findings graphically using a QALY profile over time is an attractive way of summarising these results to inform GDG discussion.
Finally, Chapter 6 showed that it is feasible but challenging and time-consuming to create model-based CEAs for interventions in multimorbid patient populations. The challenges arose from the lack of available data for the relevant population, and such modelling is likely to be feasible only for conditions that very commonly co-occur rather than accounting for multiple combinations of conditions. It is unlikely that the existing NICE guideline development process could routinely accommodate such modelling within existing resources, although wider sharing of UK model-based CEAs might reduce the need for some of the modelling that NICE already does, freeing up resources for more complex modelling in situations where it is judged to be particularly important.
Of note is that, although the project was not deliberately structured to do this, it has contributed to examining feasible methods for implementing four of the five ways that single-disease guidelines could better account for multimorbidity proposed by Fabbri et al. 46 (applicability, use of ARR, accounting for TTB and accounting for interactions).
Implications of findings
We believe that there are three main implications of the findings for single-disease guideline development, which guideline developers could consider piloting or implementing within their existing processes.
The use of epidemiological data to characterise the guideline population
In contrast to the very systematic identification and synthesis of evidence relating to treatment benefit or diagnostic accuracy in guideline development, characterisation of the guideline population is more haphazard. GDGs do already sometimes qualify recommendations in terms of comorbidity and highlight some interactions, but as we understand it this is largely driven by the knowledge and expertise of the individual GDG members who happen to have been recruited. There is now reasonably straightforward access to large and representative UK epidemiological data sets. These can be used to characterise the population for which guideline recommendations are being made. It is therefore feasible to examine more systematically the extent of the extrapolations being made and the interactions that are likely to happen when making treatment recommendations to inform GDG decisions about if and how these recommendations should be qualified.
Guideline development groups would then have to consider whether they wish to make a single recommendation for all people with a condition, or stratified or otherwise qualified recommendations for different subgroups. Such judgements already happen but we believe that systematic use of epidemiological data to inform them is required. Based on discussion with the PRG, factors that GDGs might consider when making such judgements about drug–drug and drug–disease interactions include (Figure 20):
-
how common coprescription is likely to be in the guideline population (based on whether drugs are recommended for all patients with the condition or a subset, and on how commonly interacting drugs are used in the guideline population), or how common interacting conditions are in the guideline population
-
the severity of any interaction in terms of the harm that the interaction causes
-
how common the interaction is in people coprescribed two drugs or prescribed a drug in the presence of an interacting condition.
FIGURE 20.
Using epidemiological data to inform single-disease guideline development.

Based on discussion with the PRG, for issues of applicability and extrapolation from trial populations to important groups of the guideline population who would have been excluded from trials, factors that GDGs might consider when making such judgements include:
-
the nature of the treatment, in terms of whether or not its mechanism of effect is likely to apply across all patients, and its potential for harm
-
the duration over which the treatment will be used, which is relevant when extrapolating to populations with limited life expectancy from other conditions, age or general frailty
-
the absolute size of the observed benefit in the trials, which is relevant because large benefits are less likely to be sensitive to small variations in benefit or harm in people not eligible for trials
-
the nature of the differences between trial and non-trial populations, including age, comorbidity, coprescribing and likely life expectancy, in terms of whether or not these are large enough to matter in the context of the previous factors.
Such considerations are particularly likely to apply when the outcomes being improved by treatment are not observable by clinicians or patients. For example, clinicians and patients can observe change in pain during treatment with an analgesic. In contrast, most preventative treatments require clinicians and patients to take it on trust that meaningful outcomes are better because a prevented heart attack or other prevented future event is not observable in an individual.
The creation of measures of absolute benefit to allow comparison of net benefit across treatments for different conditions
There are no major technical barriers to using a consistent method to produce estimates of the absolute benefit of treatments for different conditions across a range of clinical outcomes. However, all such estimates rely on making a number of significant assumptions, including that relative risk of benefit is constant across populations, that competing risks of death are not significant, that baseline risk has been accurately measured in the guideline population or its important subgroups and that the relative risk of harm is constant across populations. All of these assumptions will be untrue at least sometimes, but it is important to note that any clinician who prescribes a treatment to a patient who would not have been eligible for the original trial is making some or all of these assumptions implicitly. Similarly, there are no technical barriers to using absolute QALYs to compare the benefit of treatments, although again only provided that guiding principles are followed to maximise comparability. In practice, an absolute QALY approach will be restricted in the short term by the relative lack of suitable economic models for many clinical interventions. In both cases, we believe that absolute benefits should be estimated only for interventions with demonstrated clinical benefit at a minimum, and ideally cost-effectiveness.
The NICE multimorbidity guideline due to be published in September 2016 will develop a method for extracting absolute benefit information from existing NICE guidelines,42 which is likely to form the core of a resource to compare the absolute benefit of treatments for different conditions. In practice, we believe that this will be of most value when the benefit of treatment is not observable by clinicians and patients. For treatments of symptoms, irrespective of the average benefit in the population, both clinician and patient can at least partly judge if treatment is successful. In contrast, for treatments that reduce the risk of future events, treatment decisions are largely informed by trial evidence, and it is in this context that we believe that comparisons of the absolute benefit of different treatment would be most useful, to inform decision-making shared with people with limited life expectancy and/or significant treatment burden.
However, current ways of delivering guidelines using downloadable versions, electronic pathways online and paper-based decision support tools will not make the large amounts of information about absolute benefit easily usable, and any absolute benefit comparison resource will require regular refreshing as guidelines are updated. We therefore believe that effective delivery will require the creation of a suitable electronic resource where clinicians and patients can choose which conditions and which treatments to compare, can calculate baseline risk or choose plausible values for it, and can choose how to view absolute benefit (in words, in numbers of various kinds or graphically). Creating and evaluating such a resource was beyond the scope of this project, but will probably require investment from either a research funder or a national decision-making body such as NICE.
The use of alternative output of model-based cost-effectiveness analysis to help guideline development groups examine the implications of time to benefit and competing risks
Our empirical study applied the concepts of pay-off time, DTD and graphical QALY profiles from an existing model-based CEA used for the production of CGs for the primary prevention of CVD with statins. These three concepts have the potential to be an informative approach to include a temporal dimension in model-based CEA. The calculation of pay-off time together with using visual presentation of the information (QALY profiles) provides a potentially useful tool to complement existing cost-effectiveness results and potentially aid guideline decision-making when choosing between interventions with different pay-off times, or considering the implications of estimated pay-off times in people with limited life expectancy.
For surgical or screening interventions with clear upfront harms, pay-off time is clearly relevant, and we believe it should be actively considered by GDGs by examination of QALY profiles. For long-term drug treatments, pay-off time is more driven by the extent to which DTD applies. However, in this context, even small harms may mean that pay-off times exceed the life expectancy of important subgroups of the population given the presence of life-limiting comorbidity.
The method can be easily and rapidly applied using current model-based CEA. The pay-off time can be stratified according to various baseline characteristics that are pre-built into economic models, and it can be calculated as a pay-off time in purely QALY terms or it can also be inclusive of financial costs associated with an intervention by using net benefit. Another potential extension, which would make the pay-off time informative in the context of patients with comorbidity, is to explore the impact of adjusting the relative risk of all-cause mortality, which is generally a parameter included in Markov model-based CEA. In this scenario, the adjustment in the relative risk of all-cause mortality is used as a proxy for a patient population with competing risk of death from comorbidities.
Generating estimates of the pay-off time, using the QALY or QALY inclusive of costs (net benefit) form, could therefore be used by GDGs as a complement to the outputs from existing model-based CEA already used to inform NICE CGs. Although there have been some empirical studies of it, more research is needed to better quantify DTD using well-designed stated preference studies in appropriate samples of the patient population and members of the public. In the interim, the use of pre-defined DTD could enable exploration in sensitivity analysis of the potential impact if patients do associate some level of harm with taking a long-term treatment. A series of one-way sensitivity analyses could therefore be used to present the impact of plausible levels of DTD, and also the potential impact of comorbidity on mortality risk. Practically, we believe that pay-off time should be explored only in the context of interventions that have already been shown to be a cost-effective use of resources in the main model-based CEA.
Recommendations for research
We believe that all of these recommendations are important and therefore list them with no particular priority.
-
Research is needed to optimise the design and effectiveness of different ways of presenting comparative absolute benefit to clinicians and patients, with subsequent research to understand if and how such information is used by clinicians and patients, either alone or in shared decision-making.
-
Evaluation of the impact of the use of epidemiological data and TTB data on GDG deliberations and decision-making is needed. GDGs apply expert judgement to create guideline recommendations that are based on but not solely determined by evidence, not least because the evidence is often incomplete or not wholly applicable. It is therefore difficult to be certain that recommendations from one process are better than recommendations from another, but it is possible to examine whether or not the process is better in the sense of being more transparent or more systematic. Such research is therefore likely to be observational and probably ethnographic, aiming to understand how the process by which GDGs reach conclusions is different when epidemiological and TTB data are used from when they are not, and whether or not any differences are judged to be improvements in process.
-
Further research is required to generate robust empirical estimates of DTD using well-designed stated preference studies in appropriate samples of the patient population and members of the public. In addition, there is a need to better define how DTD should be best incorporated into the utility values for the patient cohort being run through a Markov model, which is related to the broader issue of how best to include utility values associated with comorbidities in a model-based CEA. 204
-
Properly accounting for multimorbidity in CGs requires synthesising data from multiple sources, including evidence of benefit from trials, and evidence from observational studies about harm, baseline risk, competing risks and life expectancy. For selected conditions and treatments, it would be appropriate to evaluate interventions in more comorbid and older patients, who are commonly excluded from trials. A common problem is that observational data are not always available. In general, harm is poorly quantified, baseline risk is well quantified for only some conditions (although almost all baseline risk calculators ignore competing risks of death, which is a potential major limitation of their use in older people and those with life-limiting comorbidities) and there are few good tools to predict life expectancy. For all of these reasons, further research to reduce uncertainty would be helpful, focusing initially on high-prevalence conditions and very commonly used treatments.
Acknowledgements
Project team
University of Dundee: Bruce Guthrie, Siobhan Dumbreck and Angela Flynn.
University of Manchester: Katherine Payne, Alex Thompson and Matt Sutton.
University of Aberdeen: Shaun Treweek.
University of Glasgow: Stewart Mercer.
NICE: Phil Alderson, Tim Stokes and Bhash Naidoo.
SIGN: Moray Nairn.
Project reference group
Ian Lewin (chairperson), Alison Allen (public member), Graham Bell (public member), Claudette Allerdyce, Carolyn Chew-Graham, Mark Davis, Sarah Davis, Roger Gadsby, John Hindle, Suzanne Lucas and Hugh McIntyre.
We would like to thank the PRG for its invaluable advice and contribution to the project.
Study steering committee
Brian McKinstry (University of Edinburgh, chairperson), Sue Kinsey (public member).
We would like to thank the SSC for its invaluable advice and support for the project.
Contributions of authors
Bruce Guthrie (Professor of Primary Care Medicine, University of Dundee) was the overall chief investigator, contributed to the conceptualisation, conduct and interpretation of the study, co-ordinated the writing of the report, led the writing of Chapters 1 and 7, and wrote elements of Chapters 2, 3 and 4.
Alexander Thompson (Health Economist, University of Manchester) was the employed researcher in Manchester, contributed to the conduct and interpretation of the study, provided comment on and editing of the report, and cowrote Chapters 5 and 6 and elements of Chapters 2 and 4 with Katherine Payne.
Siobhan Dumbreck (Research Pharmacist, University of Dundee) was an employed researcher in Dundee, contributed to the conduct and interpretation of the study, provided comment on and editing of the report, and wrote elements of Chapters 2, 3 and 4.
Angela Flynn (Research Pharmacist, University of Dundee) was an employed researcher in Dundee, contributed to the conduct and interpretation of the study, provided comment on and editing of the report, and wrote elements of Chapters 2, 3 and 4.
Phil Alderson (Associate Director, Centre for Clinical Practice, NICE) contributed to the conceptualisation, conduct and interpretation of the study, and provided comment on and editing of the report.
Moray Nairn (Programme Manager, SIGN) contributed to the conduct and interpretation of the study, and provided comment on and editing of the report.
Shaun Treweek (Chairperson in Health Services Research, University of Aberdeen) contributed to the conceptualisation, conduct and interpretation of the study, and provided comment on and editing of the report.
Katherine Payne (Professor of Health Economics, University of Manchester) led the health economics work, contributed to the conceptualisation, conduct and interpretation of the study, provided comment and editing of the report, and cowrote Chapters 5 and 6 and elements of Chapters 2 and 4 with Alexander Thompson.
Publications
Peer-reviewed papers
Guthrie B, Payne K, Alderson P, McMurdo MET, Mercer SW. Adapting clinical guidelines to take account of multimorbidity. BMJ 2012;345:e6341.
Dumbreck S, Flynn A, Nairn M, Wilson M, Treweek S, Mercer SW, et al. Drug–disease and drug–drug interactions: systematic examination of recommendations in 12 UK national clinical guidelines. BMJ 2015;350:h949.
Thompson A, Guthrie B, Payne K. Do pills have no ills? Capturing the impact of direct treatment disutility. Pharmacoeconomics 2016;34:333–36.
Thompson A, Guthrie B, Payne K. Using the ‘pay-off time’ in decision-analytic models: a case study for statins in primary prevention. Med Decis Making 2017. Published online 25 April 2017.
Presentations
Alderson P. Addressing Multimorbidity in Clinical Guidelines: Experience From NICE and Future Plans. Ariadne International Symposium, Frankfurt, Germany, October 2012.
Guthrie B. Polypharmacy and Changing Emphasis Towards the End of Life. British Geriatrics Society autumn meeting, Dunfermline, UK, October 2013.
Guthrie B. New Approaches for Clinical Trials and Guidelines. REPOSI International Seminar, Aging, Multimorbidity and Polypharmacy: Strategies for the Third Millennium, Milan, Spain, September 2013.
Dumbreck S, Flynn A, Guthrie B. Better Guidelines for Better Care: Accounting for Multimorbidity in Clinical Guidelines. Poster, SIGN 20th Anniversary Event, Glasgow, UK, December 2013.
Guthrie B. How Useful are Evidence-Based Guidelines to Primary Care? DECIDE international Conference, Edinburgh, UK, June 2014.
Thompson A, Payne K, Nairn M, Alderson P, Sutton M, Guthrie B. Accounting for Multimorbidity in Economic Models: Implications for Guideline Development. Guidelines International Network Conference, Melbourne, Australia, August 2014.
Thompson A, Payne K. Economics and Multimorbidity. NICE Evidence Synthesis Network, Economics Event, Manchester, UK, June 2014.
Thompson A, Payne K. Economics and Multimorbidity: The Context. NICE Evidence Synthesis Network, Economics Event, Manchester, UK, June 2014.
Guthrie B, Thompson A, Payne K. Multimorbidity in Guidelines. NICE Technical Workshop, Manchester, UK, May 2015.
Guthrie B. Multimorbidity in Guidelines. Briefing document and oral presentation, SIGN Strategy Group, Edinburgh, UK, June 2015.
Guthrie B. Multimorbidity in Guidelines. Briefing document and oral presentation, NICE Board Strategy day, London, UK, June 2015.
Guthrie B. The Challenges of Making Guidelines in a World of Multimorbidity. SIGN/Health Improvement Scotland, Edinburgh, UK, June 2015.
Guthrie B. Multimorbidity: New Paradigm of the Emperor’s New Clothes? Society for Academic Primary Care Conference, Oxford, UK, July 2015.
Dumbreck S, Flynn A. Comparing Treatment Effectiveness Across Guidelines to Support Decision Making for People with Multimorbidity. Guidelines International Network Conference, Amsterdam, the Netherlands, October 2015.
Thompson A, Payne K. Including a Temporal Dimension in Model-Based CEA: An Application to Clinical Guidelines for the Use of Statins. Guidelines International Network Conference, Amsterdam, the Netherlands, October 2015.
Guthrie B. Multimorbidity in Guidelines. Guidelines International Network Conference, Amsterdam, the Netherlands, October 2015.
Data sharing statement
There are no specific data to archive beyond what are reported here. Data may be available to share; please contact the corresponding author to discuss.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HS&DR programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HS&DR programme or the Department of Health.
References
- Grimshaw J, Thomas R, MacLennan G, Fraser C, Ramsay CR, Vale L. Effectiveness and efficiency of guideline dissemination and implementation strategies. Health Technol Assess 2004;8. http://dx.doi.org/10.3310/hta8060.
- Boyd CM, Darer J, Boult C, Fried LP, Boult L, Wu AW. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases. JAMA 2005;294:716-24. http://dx.doi.org/10.1001/jama.294.6.716.
- Hughes L, McMurdo MET, Guthrie B. Guidelines for people not for diseases: the challenges of applying UK clinical guidelines to people with multimorbidity. Age Ageing 2013;42:62-9. http://dx.doi.org/10.1093/ageing/afs100.
- Dawes M. Co-morbidity: we need a guideline for each patient not a guideline for each disease. Family Practice 2010;27:1-2. http://dx.doi.org/10.1093/fampra/cmp106.
- van Weel C, Schellevis FG. Comorbidity and guidelines: conflicting interests. Lancet 2006;367:550-1. http://dx.doi.org/10.1016/S0140-6736(06)68198-1.
- Vitry A, Zhang Y. Quality of Australian clinical guidelines and relevance to the care of older people with multiple comorbid conditions. Med J Aust 2008;189:360-5.
- Fortin M, Contant E, Savard C, Hudon C, Poitras M-E, Almirall J. Canadian guidelines for clinical practice: an analysis of their quality and relevance to the care of adults with comorbidity. BMC Fam Pract 2011;12. http://dx.doi.org/10.1186/1471-2296-12-74.
- Valderas JM, Starfield B, Sibbald B, Salisbury C, Roland M. Defining comorbidity: implications for understanding health and health services. Ann Fam Med 2009;7:357-63. http://dx.doi.org/10.1370/afm.983.
- Violan C, Foguet-Boreu Q, Flores-Mateo G, Salisbury C, Blom J, Freitag M, et al. Prevalence, determinants and patterns of multimorbidity in primary care: a systematic review of observational studies. PLOS ONE 2014;9. http://dx.doi.org/10.1371/journal.pone.0102149.
- Prados-Torres A, Calderón-Larrañaga A, Hancco-Saavedra J, Poblador-Plou B, van den Akker M. Multimorbidity patterns: a systematic review. J Clin Epidemiol 2014;67:254-66. http://dx.doi.org/10.1016/j.jclinepi.2013.09.021.
- Salisbury C, Johnson C, Purdy S, Valderas JM, Montgomery A. Epidemiology and impact of multimorbidity in primary care: a retrospective cohort study. Br J Gen Pract 2011;582:e12-21. http://dx.doi.org/10.3399/bjgp11X548929.
- van den Akker M, Buntinx F, Roos S, Knottnerus JA. Problems in determining occurrence rates of multimorbidity. J Clin Epidemiol 2001;54:675-9. http://dx.doi.org/10.1016/S0895-4356(00)00358-9.
- Barnett K, Mercer SW, Norbury M, Watt G, Wyke S, Guthrie B. Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet 2012;380:37-43. http://dx.doi.org/10.1016/S0140-6736(12)60240-2.
- McLean G, Gunn J, Wyke S, Guthrie B, Watt GCM, Blane DN. The influence of socioeconomic deprivation on multimorbidity at different ages: a cross-sectional study. Br J Gen Pract 2014;64:e440-7. http://dx.doi.org/10.3399/bjgp14X680545.
- Piette JD, Kerr EA. The impact of comorbid chronic conditions on diabetes care. Diabetes Care 2006;29:725-31. http://dx.doi.org/10.2337/diacare.29.03.06.dc05-2078.
- Labos C, Dasgupta K, Nedjar H, Turecki G, Rahme E. Risk of bleeding associated with combined use of selective serotonin reuptake inhibitors and antiplatelet therapy following acute myocardial infarction. CMAJ 2011;183:1835-43. http://dx.doi.org/10.1503/cmaj.100912.
- Depression in Adults with a Chronic Physical Health Problem: Treatment and Management. London: National Institute for Health and Care Excellence; 2009.
- Fortin M, Lapointe L, Hudon C, Vanasse A, Ntetu A, Maltais D. Multimorbidity and quality of life in primary care: a systematic review. Health Qual Life Outcomes 2004;2. http://dx.doi.org/10.1186/1477-7525-2-51.
- Fortin M, Hudon C, Dubois M, Almirall J, Lapointe F, Soubhi H. Comparative assessment of three different indices of multimorbidity for studies on health-related quality of life. Health Qual Life Outcomes 2005;3. http://dx.doi.org/10.1186/1477-7525-3-74.
- May C, Montori VM, Mair FS. We need minimally disruptive medicine. BMJ 2010;339. http://dx.doi.org/10.1136/bmj.b2803.
- Gallacher K, Batty GD, McLean G, Mercer S, Guthrie B, Langhorne P. Stroke, comorbidity and polypharmacy in a nationally representative sample of 1,424,378 patients in Scotland: implications for treatment burden. BMC Med 2014;12. http://dx.doi.org/10.1186/s12916-014-0151-0.
- Payne RA, Abel GA, Guthrie B, Mercer SW. The effect of physical multimorbidity, mental health conditions and socioeconomic deprivation on unplanned admissions to hospital: a retrospective cohort study. CMAJ 2013;185:E221-8. http://dx.doi.org/10.1503/cmaj.121349.
- Duerden M, Avery A, Payne R. Polypharmacy and Medicines Optimisation: Making It Safe and Sound. London: The King’s Fund; 2013.
- O’Brien R, Wyke S, Guthrie B, Watt G, Mercer S. An ‘endless struggle’: a qualitative study of general practitioners’ and practice nurses’ experiences of managing multimorbidity in socio-economically deprived areas of Scotland. Chronic Illn 2011;7:45-59. http://dx.doi.org/10.1177/1742395310382461.
- O’Brien R, Wyke S, Guthrie B, Watt G, Mercer SW. The ‘everyday work’ of living with multimorbidity in socioeconomically deprived areas of Scotland. J Comorbidity 2014;4:1-10. http://dx.doi.org/10.15256/joc.2014.4.32.
- Lawson K, Mercer S, Wyke S, Grieve E, Guthrie B, Watt G. Double trouble: the impact of multimorbidity and deprivation on preference-weighted health related quality of life a cross sectional analysis of the Scottish Health Survey. Int J Equity Health 2013;12. http://dx.doi.org/10.1186/1475-9276-12-67.
- Wolff J, Starfield B, Anderson G. Prevalence, expenditures, and complications of multiple chronic conditions in the elderly. Arch Intern Med 2002;162:2269-76. http://dx.doi.org/10.1001/archinte.162.20.2269.
- Steinman MA, Hanlon JT. Managing medications in clinically complex elders. JAMA 2010;304:1592-601. http://dx.doi.org/10.1001/jama.2010.1482.
- Sinnott C, Mc Hugh S, Browne J, Bradley C. GPs’ perspectives on the management of patients with multimorbidity: systematic review and synthesis of qualitative research. BMJ Open 2013;3. http://dx.doi.org/10.1136/bmjopen-2013-003610.
- Sinnott C, Hugh SM, Boyce MB, Bradley CP. What to give the patient who has everything? A qualitative study of prescribing for multimorbidity in primary care. Br J Gen Pract 2015;65:e184-91.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2010.
- Guthrie B, Makubate B, Hernandez-Santiago V, Dreischulte T. The rising tide of polypharmacy and drug–drug interactions: population database analysis 1995–2010. BMC Med 2015;13. http://dx.doi.org/10.1186/s12916-015-0322-7.
- Guthrie B, McCowan C, Davey P, Simpson CR, Dreischulte T, Barnett K. High risk prescribing in primary care patients particularly vulnerable to adverse drug events: cross sectional population database analysis in Scottish general practice. BMJ 2011;342. http://dx.doi.org/10.1136/bmj.d3514.
- Bourgeois FT, Shannon MW, Valim C, Mandl KD. Adverse drug events in the outpatient setting: an 11-year national analysis. Pharmacoepidemiol Drug Saf 2010;19:901-10. http://dx.doi.org/10.1002/pds.1984.
- Barnett K, McCowan C, Evans JMM, Gillespie ND, Davey PG, Fahey T. Prevalence and outcomes of use of potentially inappropriate medicines in older people: cohort study stratified by residence in nursing home or in the community. BMJ Qual Saf 2011;20:275-81. http://dx.doi.org/10.1136/bmjqs.2009.039818.
- Braithwaite RS. Can life expectancy and QALYs be improved by a framework for deciding whether to apply clinical guidelines to patients with severe comorbid disease?. Med Decis Making 2011;4:582-95. http://dx.doi.org/10.1177/0272989X10386117.
- Lugtenberg M, Burgers JS, Clancy C, Westert GP, Schneider EC. Current guidelines have limited applicability to patients with comorbid conditions: a systematic analysis of evidence-based guidelines. PLOS ONE 2011;6. http://dx.doi.org/10.1371/journal.pone.0025987.
- Cox L, Kloseck M, Crilly R, McWilliam C, Diachun L. Underrepresentation of individuals 80 years of age and older in chronic disease clinical practice guidelines. Can Fam Physician 2011;57:e263-9.
- Guiding Principles for the Care of Older Adults with Multimorbidity Pocket Card. New York, NY: American Geriatrics Society; 2012.
- American Geriatrics Society Expert Panel on the Care of Older Adults with Multimorbidity . Guiding principles for the care of older adults with multimorbidity: an approach for clinicians. J Am Geriatr Soc 2012;60:E1-25. http://dx.doi.org/10.1111/j.1532-5415.2012.04188.x.
- American Geriatrics Society Expert Panel on the Care of Older Adults with Multimorbidity . Patient-centered care for older adults with multiple chronic conditions: a stepwise approach from the American Geriatrics Society. J Am Geriatr Soc 2012;60:1957-68. http://dx.doi.org/10.1111/j.1532-5415.2012.04187.x.
- Final Scope: Multimorbidity: The Assessment, Prioritisation and Management of Care for People with Commonly Occurring Multimorbidity. London: National Institute for Health and Care Excellence; 2014.
- California HealthCare Foundation/American Geriatrics Society Panel in Improving Care for Elders with Diabetes . Guidelines for improving the care of the older person with diabetes mellitus. J Am Geriatr Soc 2003;51:265-80. http://dx.doi.org/10.1046/j.1532-5415.51.5s.1.x.
- Sinclair A, Dunning T, Colagiuri S. Managing Older People with Type 2 Diabetes: Global Guideline. Brussels: International Diabetes Federation; 2013.
- Depression: The Treatment and Management of Depression in Adults. London: National Institute for Health and Care Excellence; 2009.
- Fabbri LM, Boyd C, Boschetto P, Rabe KF, Buist AS, Yawn B, et al. How to integrate multiple comorbidities in guideline development. Proc Am Thorac Soc 2012;9:274-81. http://dx.doi.org/10.1513/pats.201208-063ST.
- Uhlig K, Leff B, Kent D, Dy S, Brunnhuber K, Burgers JS, et al. A framework for crafting clinical practice guidelines that are relevant to the care and management of people with multimorbidity. J Gen Intern Med 2014;29:670-9. http://dx.doi.org/10.1007/s11606-013-2659-y.
- Developing NICE Guidelines: The Manual. London: National Institute for Health and Care Excellence; 2014.
- Type 2 Diabetes: National Clinical Guideline for Management in Primary and Secondary Care. London: Royal College of Physicians; 2008.
- Type 2 Diabetes: Newer Agents for Blood Glucose Control in Type 2 Diabetes. London: National Institute for Health and Care Excellence; 2008.
- Chronic Heart Failure: National Clinical Guideline for Diagnosis and Management in Primary and Secondary Care. London: Royal College of Physicians; 2010.
- Naylor C, Parsonage M, McDaid D, Knapp M, Fossey M, Galea A. Long-Term Conditions and Mental Health: The Cost of Co-morbidities. London: The King’s Fund and Centre for Mental Health; 2012.
- Katon WJ, Rutter CM, Lin E, Simon G, Von Korff M, Bush T, et al. Collaborative care for patients with depression and chronic illnesses. New Engl J Med 2010;363:2611-20. http://dx.doi.org/10.1056/NEJMoa1003955.
- Coventry P, Lovell K, Dickens C, Bower P, Chew-Graham C, McElvenny D, et al. Integrated primary care for patients with mental and physical multimorbidity: cluster randomised controlled trial of collaborative care for patients with depression comorbid with diabetes or cardiovascular disease. BMJ 2015;350. http://dx.doi.org/10.1136/bmj.h638.
- Equality Act 2010. London: The Stationery Office; 2010.
- Chronic Heart Failure: Management of Chronic Heart Failure in Adults in Primary and Secondary Care. London: National Institute for Health and Care Excellence; 2010.
- Drummond M, Sculpher M, Torrance G, O’Brien B, Stoddart G. Methods for the Economic Evaluation of Health Care Programmes Paperback. Oxford: Oxford University Press; 2005.
- Sculpher M, Claxton K, Drummond M, McCabe C. Whither trial-based economic evaluation for health care decision making?. Health Econ 2006;15:677-87. http://dx.doi.org/10.1002/hec.1093.
- Methods Guide for Technology Appraisal. London: National Institute for Health and Care Excellence; 2013.
- Dakin H, Devlin N, Feng Y, Rice N, O’Neill P, Parkin D. The influence of cost-effectiveness and other factors on NICE decisions. Health Econ 2014;24:1256-71. http://dx.doi.org/10.1002/hec.3086.
- Working with SMC: A Guide for Manufacturers. Edinburgh: Scottish Medicines Consortium; 2014.
- International Society for Pharmacoeconomics and Outcomes Research . Pharmacoeconomic Guidelines Around the World 2015. www.ispor.org/peguidelines/index.asp (accessed 6 June 2015).
- Methods for the Development of NICE Public Health Guidance. London: National Institute for Health and Care Excellence; 2012.
- Diagnostic Assessment Programme Manual. London: National Institute for Health and Care Excellence; 2011.
- Eccles M, Mason J. How to develop cost-conscious guidelines. Health Technol Assess 2001;5. http://dx.doi.org/10.3310/hta5160.
- Lord J, Willis S, Eatock J, Tappenden P, Trapero-Bertran M, Miners A, et al. Economic modelling of diagnostic and treatment pathways in National Institute for Health and Care Excellence clinical guidelines: the Modelling Algorithm Pathways in Guidelines (MAPGuide) project. Health Technol Assess 2013;17. http://dx.doi.org/10.3310/hta17580.
- Social Value Judgements: Principles for the Development of NICE Guidance. London: National Institute for Health and Care Excellence; 2008.
- Mohiuddin S, Payne K, Fenwick E. The use of value of information methods in Health Technology Assessments in the UK. Int J Technol Assess Health Care 2015;30:553-70. http://dx.doi.org/10.1017/S0266462314000701.
- Brennan A, Chick SE, Davies R. A taxonomy of model structures for economic evaluation of health technologies. Health Econ 2006;15:1295-310. http://dx.doi.org/10.1002/hec.1148.
- Pirmohamed M, James S, Meakin S, Green C, Scott AK, Walley TJ, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ 2004;329:15-9. http://dx.doi.org/10.1136/bmj.329.7456.15.
- Marengoni A, Pasina L, Concoreggi C, Martini G, Brognoli F, Nobili A. Understanding adverse drug reactions in older adults through drug–drug interactions. Eur J Intern Med 2014;25:843-6. http://dx.doi.org/10.1016/j.ejim.2014.10.001.
- Howard R, Avery A, Slavenburg S, Royal S, Pipe G, Lucassen P, et al. Which drugs cause preventable admissions to hospital? A systematic review. Br J Clin Pharmacol 2007;63:136-47. http://dx.doi.org/10.1111/j.1365-2125.2006.02698.x.
- Dumbreck S, Flynn A, Nairn M, Wilson M, Treweek S, Mercer SW, et al. Drug–disease and drug–drug interactions: systematic examination of recommendations in 12 UK national clinical guidelines. BMJ 2015;350. http://dx.doi.org/10.1136/bmj.h949.
- The Management of Type 2 Diabetes. London: National Institute for Health and Care Excellence; 2010.
- The Treatment and Management of Depression in Adults. London: National Institute for Health and Care Excellence; 2009.
- The Management of Atrial Fibrillation. London: National Institute for Health and Care Excellence; 2006.
- The Care and Management of Osteoarthritis in Adults. London: National Institute for Health and Care Excellence; 2008.
- Management of Chronic Obstructive Pulmonary Disease in Adults in Primary and Secondary Care (Partial Update). London: National Institute for Health and Care Excellence; 2010.
- Clinical Management of Primary Hypertension in Adults. London: National Institute for Health and Care Excellence; 2011.
- Secondary Prevention in Primary and Secondary Care for Patients Following a Myocardial Infarction. London: National Institute for Health and Care Excellence; 2007.
- Supporting People with Dementia and Their Carers in Health and Social Care. London: National Institute for Health and Care Excellence; 2006.
- The Management of Rheumatoid Arthritis in Adults. London: National Institute for Health and Care Excellence; 2009.
- Early Identification and Management of Chronic Kidney Disease in Adults in Primary and Secondary Care. London: National Institute for Health and Care Excellence; 2008.
- The Pharmacological Management of Neuropathic Pain in Adults in Non-specialist Settings. London: National Institute for Health and Care Excellence; 2010.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2013.
- Kesselheim AS, Misono AS, Lee JL, Stedman MR, Brookhart A, Choudhry NK, et al. Clinical equivalence of generic and brand-name drugs used in cardiovascular disease: a systematic review and meta-analysis. JAMA 2008;300:2514-26. http://dx.doi.org/10.1001/jama.2008.758.
- Lorgunpai SJ, Grammas M, Lee DSH, McAvay G, Charpentier P, Tinetti ME. Potential therapeutic competition in community-living older adults in the U.S.: use of medications that may adversely affect a coexisting condition. PLOS ONE 2014;9. http://dx.doi.org/10.1371/journal.pone.0089447.
- Tan K, Petrie KJ, Faasse K, Bolland MJ, Grey A. Unhelpful information about adverse drug reactions. BMJ 2014;349. http://dx.doi.org/10.1136/bmj.g5019.
- Electronic medicines compendium . Amitriptyline Tablets BP 10 Mg (Actaris UK Ltd) n.d. www.medicines.org.uk/emc/medicine/23736 (accessed 15 July 2016).
- Brilleman SL, Purdy S, Salisbury C, Windmeijer F, Gravelle H, Hollinghurst S. Implications of comorbidity for primary care costs in the UK: a retrospective observational study. Br J Gen Pract 2013;63:e274-82. http://dx.doi.org/10.3399/bjgp13X665242.
- Sculpher M. Subgroups and heterogeneity in cost-effectiveness analysis. Pharmacoeconomics 2008;26:799-806. http://dx.doi.org/10.2165/00019053-200826090-00009.
- Ruscitto A, Smith BH, Guthrie B. Changes in opioid and other analgesic use 1995–2010: repeated cross-sectional analysis of dispensed prescribing for a large geographical population in Scotland. Eur J Pain 2015;19:59-66. http://dx.doi.org/10.1002/ejp.520.
- Boyer EW, Shannon M. The serotonin syndrome. New Engl J Med 2005;352:1112-20. http://dx.doi.org/10.1056/NEJMra041867.
- Van Spall HGC, Toren A, Kiss A, Fowler RA. Eligibility criteria of randomized controlled trials published in high-impact general medical journals: a systematic sampling review. JAMA 2007;297:1233-40. http://dx.doi.org/10.1001/jama.297.11.1233.
- Rothwell P. Can overall results of clinical trials be applied to all patients?. Lancet 1995;345:1616-19. http://dx.doi.org/10.1016/S0140-6736(95)90120-5.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply?. Lancet 2005;365:82-93. http://dx.doi.org/10.1016/S0140-6736(04)17670-8.
- Saunders C, Byrne CD, Guthrie B, Lindsay RS, McKnight JA, Philip S, et al. External validity of randomized controlled trials of glycaemic control and vascular disease: how representative are participants?. Diabet Med 2013;30:300-8. http://dx.doi.org/10.1111/dme.12047.
- Masoudi FA, Havranek EP, Wolfe P, Gross CP, Rathore SS, Steiner JF, et al. Most hospitalized older persons do not meet the enrollment criteria for clinical trials in heart failure. Am Heart J 2003;146:250-7. http://dx.doi.org/10.1016/S0002-8703(03)00189-3.
- Travers J, Marsh S, Caldwell B, Williams M, Aldington S, Weatherall M, et al. External validity of randomized controlled trials in COPD. Respir Med 2007;101:1313-20. http://dx.doi.org/10.1016/j.rmed.2006.10.011.
- Travers J. External validity of randomised controlled trials in asthma: to whom do the results of the trials apply?. Thorax 2007;62:219-23. http://dx.doi.org/10.1136/thx.2006.066837.
- Treweek S, Dryden R, McCowan C, Harrow A, Thompson A. Do participants in major, practice-changing breast cancer trials reflect the breast cancer patient population?. Trials 2013;14. http://dx.doi.org/10.1186/1745-6215-14-S1-P33.
- Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. New Engl J Med 1999;341:709-17. http://dx.doi.org/10.1056/NEJM199909023411001.
- ACCORD Study Group . Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: design and methods. Am J Cardiol 2007;99:S21-33. http://dx.doi.org/10.1016/j.amjcard.2007.03.003.
- The Guidelines Manual. London: National Institute for Health and Care Excellence; 2009.
- Thornton J, Alderson P, Tan T, Turner C, Latchem S, Shaw E, et al. Introducing GRADE across the NICE clinical guideline program. J Clin Epidemiol 2013;66:124-31. http://dx.doi.org/10.1016/j.jclinepi.2011.12.007.
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 8. Rating the quality of evidence – indirectness. J Clin Epidemiol 2011;64:1303-10. http://dx.doi.org/10.1016/j.jclinepi.2011.04.014.
- UK Prospective Diabetes Study Group . Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837-53. http://dx.doi.org/10.1016/S0140-6736(98)07019-6.
- UK Prospective Diabetes Study Group . Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998;352:854-65. http://dx.doi.org/10.1016/S0140-6736(98)07037-8.
- Pfeffer MA, Braunwald E, Moyé LA, Basta L, Brown EJ, Cuddy TE, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. New Engl J Med 1992;327:669-77. http://dx.doi.org/10.1056/NEJM199209033271001.
- SOLVD Investigators . Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. New Engl J Med 1991;325:293-302. http://dx.doi.org/10.1056/NEJM199108013250501.
- SOLVD Investigators . Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. New Engl J Med 1992;327:685-91. http://dx.doi.org/10.1056/NEJM199209033271003.
- Køber L, Torp-Pedersen C, Carlsen JE, Bagger H, Eliasen P, Lyngborg K, et al. A clinical trial of the angiotensin-converting–enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. New Engl J Med 1995;333:1670-6. http://dx.doi.org/10.1056/NEJM199512213332503.
- The BEST Steering Committee . Design of the Beta-Blocker Evaluation Survival Trial (BEST). Am J Cardiol 1995;75:1220-3. http://dx.doi.org/10.1016/S0002-9149(99)80766-8.
- Flather MD, Shibata MC, Coats AJS, Van Veldhuisen DJ, Parkhomenko A, Borbola J, et al. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005;26:215-25. http://dx.doi.org/10.1093/eurheartj/ehi115.
- Australia/New Zealand Heart Failure Research Collaborative Group . Randomised, placebo-controlled trial of carvedilol in patients with congestive heart failure due to ischaemic heart disease. Lancet 1997;349:375-80. http://dx.doi.org/10.1016/S0140-6736(97)80008-6.
- CIBIS-II Investigators and Committees . The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999;353:9-13. http://dx.doi.org/10.1016/S0140-6736(98)11181-9.
- MERIT-HF Study Group . Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999;353:2001-7. http://dx.doi.org/10.1016/S0140-6736(99)04440-2.
- Packer M, Bristow M, Cohn J, Colucci W, Fowler M, Gilbert E, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. New Engl J Med 1996;334:1349-55. http://dx.doi.org/10.1056/NEJM199605233342101.
- Packer M, Coats AJS, Fowler MB, Katus HA, Krum H, Mohacsi P, et al. Effect of carvedilol on survival in severe chronic heart failure. New Engl J Med 2001;344:1651-8. http://dx.doi.org/10.1056/NEJM200105313442201.
- Bjerrum L, Rosholm J, Hallas J, Kragstrup J. Methods for estimating the occurrence of polypharmacy by means of a prescription database. Eur J Clin Pharmacol 1997;53:7-11. http://dx.doi.org/10.1007/s002280050329.
- Hovstadius B, Hovstadius K, Astrand B, Petersson G. Increasing polypharmacy: an individual-based study of the Swedish population 2005–2008. BMC Clin Pharmacol 2010;10. http://dx.doi.org/10.1186/1472-6904-10-16.
- Sackett DL, Haynes RB, Guyatt GH, Tugwell P. Clinical Epidemiology: A Basic Science for Clinical Medicine. London: Little, Brown and Company; 1991.
- McColl A, Smith H, White P, Field J. General practitioners’ perceptions of the route to evidence based medicine: a questionnaire survey. BMJ 1998;316. http://dx.doi.org/10.1136/bmj.316.7128.361.
- Halvorsen PA, Kristiansen IS, Aasland OG, Førde OH. Medical doctors’ perception of the ‘number needed to treat’ (NNT). Scand J Prim Health Care 2003;21:162-6. http://dx.doi.org/10.1080/02813430310001158.
- Kristiansen IS, Gyrd-Hansen D, Nexøe J, Nielsen JB. Number needed to treat: easily understood and intuitively meaningful? Theoretical considerations and a randomized trial. J Clin Epidemiol 2002;55:888-92. http://dx.doi.org/10.1016/S0895-4356(02)00432-8.
- Halvorsen PA, Kristiansen IS. Decisions on drug therapies by numbers needed to treat: a randomized trial. Arch Intern Med 2005;165:1140-6. http://dx.doi.org/10.1001/archinte.165.10.1140.
- Stovring H, Gyrd-Hansen D, Kristiansen I, Nexoe J, Nielsen J. Communicating effectiveness of intervention for chronic diseases: what single format can replace comprehensive information?. BMC Med Inform Decis Mak 2008;8. http://dx.doi.org/10.1186/1472-6947-8-25.
- McAlister FA. The ‘number needed to treat’ turns 20 – and continues to be used and misused. CMAJ 2008;179:549-53. http://dx.doi.org/10.1503/cmaj.080484.
- Stang A, Poole C, Bender R. Common problems related to the use of number needed to treat. J Clin Epidemiol 2010;63:820-5. http://dx.doi.org/10.1016/j.jclinepi.2009.08.006.
- Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340. http://dx.doi.org/10.1136/bmj.c869.
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336:924-6. http://dx.doi.org/10.1136/bmj.39489.470347.AD.
- Pottie K, Connor Gorber S, Singh H, Joffres M, Lindsay P, Brauer P, et al. Estimating benefits and harms of screening across subgroups: the Canadian Task Force on Preventive Health Care integrates the GRADE approach and overcomes minor challenges. J Clin Epidemiol 2012;65:1245-8. http://dx.doi.org/10.1016/j.jclinepi.2012.06.018.
- Smeeth L, Haines A, Ebrahim S. Numbers needed to treat derived from meta-analyses: sometimes informative, usually misleading. BMJ 1999;318:1548-51. http://dx.doi.org/10.1136/bmj.318.7197.1548.
- Deeks JJ. Issues in the selection of a summary statistic for meta-analysis of clinical trials with binary outcomes. Stat Med 2002;21:1575-600. http://dx.doi.org/10.1002/sim.1188.
- Furukawa TA, Guyatt GH, Griffith LE. Can we individualize the ‘number needed to treat’? An empirical study of summary effect measures in meta-analyses. Int J Epidemiol 2002;31:72-6. http://dx.doi.org/10.1093/ije/31.1.72.
- Cholesterol Treatment Trialists’ (CTT) Collaborators . The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012;380:581-90. http://dx.doi.org/10.1016/S0140-6736(12)60367-5.
- Sacks FM, Tonkin AM, Shepherd J, Braunwald E, Cobbe S, Hawkins CM, et al. Effect of pravastatin on coronary disease events in subgroups defined by coronary risk factors: the prospective pravastatin pooling project. Circulation 2000;102:1893-900. http://dx.doi.org/10.1161/01.CIR.102.16.1893.
- McAlister FA. Commentary: relative treatment effects are consistent across the spectrum of underlying risks . . . usually. Int J Epidemiol 2002;31:76-7. http://dx.doi.org/10.1093/ije/31.1.76.
- Schmid CH, Lau J, McIntosh MW, Cappelleri JC. An empirical study of the effect of the control rate as a predictor of treatment efficacy in meta-analysis of clinical trials. Stat Med 1998;17:1923-42. http://dx.doi.org/10.1002/(SICI)1097-0258(19980915)17:17<1923::AID-SIM874>3.0.CO;2-6.
- Yusuf S, Zucker D, Passamani E, Peduzzi P, Takaro T, Fisher L, et al. Effect of coronary artery bypass graft surgery on survival: overview of 10-year results from randomised trials by the Coronary Artery Bypass Graft Surgery Trialists Collaboration. Lancet 1995;344:563-70. http://dx.doi.org/10.1016/S0140-6736(94)91963-1.
- Marx A, Bucher HC. Numbers needed to treat derived from meta-analysis: a word of caution. Evid Based Med 2003;8:36-7. http://dx.doi.org/10.1136/ebm.8.2.36.
- Rothwell PM. Subgroup analysis in randomised controlled trials: importance, indications, and interpretation. Lancet 2005;365:176-86. http://dx.doi.org/10.1016/S0140-6736(05)17709-5.
- Donal P, Kind P, Williams A. A Social Tariff for EuroQol: Results from a UK General Population Survey. York: Centre for Health Economics, University of York; 1995.
- Kind P, Lafata J, Matuszewski K, Raisch D. The use of QALYs in clinical and patient decision-making: issues and prospects. Value Health 2009;12:27-30. http://dx.doi.org/10.1111/j.1524-4733.2009.00519.x.
- Miners A, Cairnes J, Wailoo A. Burden of Illness into Value Based Pricing: A Description and Critique. London: National Institute for Health and Care Excellence; 2013.
- Philips Z, Bojke L, Sculpher M, Claxton K, Golder S. Good practice guidelines for decision-analytic modelling in health technology assessment: a review and consolidation of quality assessment. Pharmacoeconomics 2006;24:355-71. http://dx.doi.org/10.2165/00019053-200624040-00006.
- Petrou S. Rationale and methodology for trial-based economic evaluation. Clin Invest 2012;2:1191-200. http://dx.doi.org/10.4155/cli.12.121.
- Clinical Guideline 181: Lipid Modification: Cardiovascular Risk Assessment and the Modification of Blood Lipids for the Primary and Secondary Prevention of Cardiovascular Disease. London: National Institute for Health and Care Excellence; 2014.
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction: GRADE evidence profiles and summary of findings tables. J Clin Epidemiol 2011;64:383-94. http://dx.doi.org/10.1016/j.jclinepi.2010.04.026.
- Hypertension: The Clinical Management of Primary Hypertension in Adults. London: National Institute for Health and Care Excellence; 2011.
- Flather M, Yusuf S, Køber L, Pfeffer M, Hall A, Murray G, et al. Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. Lancet 2000;355:1575-81. http://dx.doi.org/10.1016/S0140-6736(00)02212-1.
- Shibata M, Flather M, Wang D. Systematic review of the impact of beta blockers on mortality and hospital admissions in heart failure. Eur J Heart Fail 2001;3:351-7. http://dx.doi.org/10.1016/S1388-9842(01)00144-1.
- Patient Decision Aid: Taking a Statin to Reduce the Risk of Coronary Heart Disease and Stroke. London: National Institute for Health and Care Excellence; 2015.
- Cholesterol Treatment Trialists’ (CTT) Collaborators . Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005;366:1267-78. http://dx.doi.org/10.1016/S0140-6736(05)67394-1.
- Levy WC, Mozaffarian D, Linker DT, Sutradhar SC, Anker SD, Cropp AB, et al. The Seattle Heart Failure Model: prediction of survival in heart failure. Circulation 2006;113:1424-33. http://dx.doi.org/10.1161/CIRCULATIONAHA.105.584102.
- Pocock SJ, Ariti CA, McMurray JJV, Maggioni A, Køber L, Squire IB, et al. Predicting survival in heart failure: a risk score based on 39 372 patients from 30 studies. Eur Heart J 2013;34:1404-13. http://dx.doi.org/10.1093/eurheartj/ehs337.
- Sartipy U, Dahlström U, Edner M, Lund LH. Predicting survival in heart failure: validation of the MAGGIC heart failure risk score in 51 043 patients from the Swedish Heart Failure Registry. Eur J Heart Fail 2014;16:173-9. http://dx.doi.org/10.1111/ejhf.32.
- Rickenbacher P, Pfisterer M, Burkard T, Kiowski W, Follath F, Burckhardt D, et al. Why and how do elderly patients with heart failure die? Insights from the TIME-CHF study. Eur J Heart Fail 2012;14:1218-29. http://dx.doi.org/10.1093/eurjhf/hfs113.
- Spencer FA, Iorio A, You J, Murad MH, Schünemann HJ, Vandvik PO, et al. Uncertainties in baseline risk estimates and confidence in treatment effects. BMJ 2012;345. http://dx.doi.org/10.1136/bmj.e7401.
- Newcombe RG, Bender R. Implementing GRADE: calculating the risk difference from the baseline risk and the relative risk. Evid Based Med 2013;19:6-8. http://dx.doi.org/10.1136/eb-2013-101340.
- Dujic T, Zhou K, Donnelly LA, Tavendale R, Palmer CN, Pearson ER. Association of organic cation transporter 1 with intolerance to metformin in type 2 diabetes: a GoDARTS study. Diabetes Care 2015;64:1786-93. http://dx.doi.org/10.2337/db14-1388.
- Dreischulte T, Morales DR, Bell S, Guthrie B. Combined use of nonsteroidal anti-inflammatory drugs with diuretics and/or renin-angiotensin system inhibitors in the community increases the risk of acute kidney injury. Kidney Int 2015;88:396-403. http://dx.doi.org/10.1038/ki.2015.101.
- Sculpher M, Pang F, Manca A, Drummond M, Golder S, Urdahl H. Generalisability in economic evaluation studies in healthcare: a review and case studies. Health Technol Assess 2004;8. http://dx.doi.org/10.3310/hta8490.
- Murray C, Evans D, Acharya A, Baltussen R. Development of WHO guidelines on generalized cost-effectiveness analysis. Health Econ 2000;9:235-51. http://dx.doi.org/10.1002/(SICI)1099-1050(200004)9:3<235::AID-HEC502>3.0.CO;2-O.
- Drummond M, Barbieri M, Cook J, Glick HA, Lis J, Malik F, et al. Transferability of economic evaluations across jurisdictions: ISPOR Good Research Practices Task Force report. Value Health 2009;12:409-18. http://dx.doi.org/10.1111/j.1524-4733.2008.00489.x.
- Anderson R. Systematic reviews of economic evaluations: utility or futility?. Health Econ 2010;364:350-64. http://dx.doi.org/10.1002/hec.1486.
- Guthrie B, Payne K, Alderson P, McMurdo MET, Mercer SW. Adapting clinical guidelines to take account of multimorbidity. BMJ 2012;345. http://dx.doi.org/10.1136/bmj.e6341.
- Holmes H, Min L, Yee M, Varadhan R, Basran J, Dale W, et al. Rationalizing prescribing for older patients with multimorbidity: considering time to benefit. Drugs Aging 2013;30:655-66. http://dx.doi.org/10.1007/s40266-013-0095-7.
- Lee SJ, Boscardin WJ, Stijacic-Cenzer I, Conell-Price J, O’Brien S, Walter LC. Time lag to benefit after screening for breast and colorectal cancer: meta-analysis of survival data from the United States, Sweden, United Kingdom, and Denmark. BMJ 2013;346. http://dx.doi.org/10.1136/bmj.e8441.
- Holmes HM, Min L, Boyd C. Lag time to benefit for preventive therapies. JAMA 2014;311. http://dx.doi.org/10.1001/jama.2014.2320.
- Ray KK, Cannon CP. Early time to benefit with intensive statin treatment: could it be the pleiotropic effects?. Am J Cardiol 2005;96:54-60. http://dx.doi.org/10.1016/j.amjcard.2005.06.027.
- Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, et al. Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004;350:1495-504. http://dx.doi.org/10.1056/NEJMoa040583.
- Oxman AD, Guyatt GH. A consumer’s guide to subgroup analyses. Ann Intern Med 1992;116:78-84. http://dx.doi.org/10.7326/0003-4819-116-1-78.
- Whitehead J. Stopping clinical trials by design. Nat Rev Drug Discov 2004;3:973-7. http://dx.doi.org/10.1038/nrd1553.
- Bassler D, Briel M, Montori VM, Lane M, Glasziou P, Zhou Q, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA 2010;303:1180-7. http://dx.doi.org/10.1001/jama.2010.310.
- Braithwaite RS, Fiellin D, Justice AC. The payoff time: a flexible framework to help clinicians decide when patients with comorbid disease are not likely to benefit from practice guidelines. Med Care 2009;47:610-17. http://dx.doi.org/10.1097/MLR.0b013e31819748d5.
- Braithwaite R, Concato J, Chang C, Roberts M, Justice A. A framework for tailoring clinical guidelines to comorbidity at the point of care. Arch Intern Med 2007;167:2361-5. http://dx.doi.org/10.1001/archinte.167.21.2361.
- Yuo TH, Roberts MS, Braithwaite RS, Chang C-CH, Kraemer KL. Applying the payoff time framework to carotid artery disease management. Med Decis Making 2013;33:1039-50. http://dx.doi.org/10.1177/0272989X13491462.
- Braithwaite RS, Meltzer DO, King JTJ, Leslie D, Roberts MS. What does the value of modern medicine say about the $50,000 per quality-adjusted life-year decision rule?. Med Care 2008;46:349-56. http://dx.doi.org/10.1097/MLR.0b013e31815c31a7.
- McCabe C, Edlin R, Hall P. Navigating time and uncertainty in health technology appraisal: would a map help?. Pharmacoeconomics 2013;31:731-7. http://dx.doi.org/10.1007/s40273-013-0077-y.
- Edlin R, Hall P, Wallner K, McCabe C. Sharing risk between payer and provider by leasing health technologies: An affordable and effective reimbursement strategy for innovative technologies?. Value Heal 2014;17:438-44. http://dx.doi.org/10.1016/j.jval.2014.01.010.
- Thompson A, Guthrie B, Payne K. Using the ‘pay-off time’ in decision-analytic models: a case study for statins in primary prevention. Med Decis Making 2017.
- Kaltenthaler E, Tappenden P, Paisley S, Squires H. NICE Decision Support Unit Technical Support Document 13: Identifying and Reviewing Evidence to Inform the Conceptualisation and Population of Cost-Effectiveness Models. London: National Institute for Health and Care Excellence; 2011.
- Fenwick E, Claxton K, Schulper M. Representing uncertainty: the role of cost-effectiveness acceptability curves. Health Econ 2001;10:779-87. http://dx.doi.org/10.1002/hec.635.
- Fontana M, Asaria P, Moraldo M, Finegold J, Hassanally K, Manisty CH, et al. Patient-accessible tool for shared decision making in cardiovascular primary prevention: balancing longevity benefits against medication disutility. Circulation 2014;129:2539-46. http://dx.doi.org/10.1161/CIRCULATIONAHA.113.007595.
- Heather E, Payne K, Harrison M, Symmons DM. Including adverse drug events in economic evaluations of anti-tumour necrosis factor-α drugs for adult rheumatoid arthritis: a systematic review of economic decision analytic models. Pharmacoeconomics 2014;32:109-34. http://dx.doi.org/10.1007/s40273-013-0120-z.
- Augustovski F, Cantor S, Thach C, Spann S. Aspirin for primary prevention of cardiovascular events. J Gen Intern Med 1998;13:824-35. http://dx.doi.org/10.1046/j.1525-1497.1998.00246.x.
- Greving J, Visseren F, de Wit G, Algra A. Statin treatment for primary prevention of vascular disease: whom to treat? Cost-effectiveness analysis. BMJ 2011;342.
- Lazar LD, Pletcher MJ, Coxson PG, Bibbins-Domingo K, Goldman L. Cost-effectiveness of statin therapy for primary prevention in a low-cost statin era. Circulation 2011;124:146-53. http://dx.doi.org/10.1161/CIRCULATIONAHA.110.986349.
- Pletcher MJ, Lazar L, Bibbins-Domingo K, Moran A, Rodondi N, Coxson P, et al. Comparing impact and cost-effectiveness of primary prevention strategies for lipid-lowering. Ann Intern Med 2009;150:243-54. http://dx.doi.org/10.7326/0003-4819-150-4-200902170-00005.
- Pletcher MJ, Pignone M, Earnshaw S, McDade C, Phillips KA, Auer R, et al. Using the coronary artery calcium score to guide statin therapy: a cost-effectiveness analysis. Circ Cardiovasc Qual Outcomes 2014;7:276-84. http://dx.doi.org/10.1161/CIRCOUTCOMES.113.000799.
- Timbie JW, Hayward RA, Vijan S. Variation in the net benefit of aggressive cardiovascular risk factor control across the US population of patients with diabetes mellitus. Arch Intern Med 2010;170:1037-44. http://dx.doi.org/10.1001/archinternmed.2010.150.
- Pignone M, Earnshaw S, Tice JA, Pletcher MJ. Aspirin, statins, or both drugs for the primary prevention of coronary heart disease events in men: a cost–utility analysis. Ann Intern Med 2006;144:326-36. http://dx.doi.org/10.7326/0003-4819-144-5-200603070-00007.
- Gage BF, Cardinalli AB, Owens DK. The effect of stroke and stroke prophylaxis with aspirin or warfarin on quality of life. Arch Intern Med 1996;156:1829-36. http://dx.doi.org/10.1001/archinte.1996.00440150083009.
- Hutchins R, Viera AJ, Sheridan SL, Pignone MP. Quantifying the utility of taking pills for cardiovascular prevention. Circ Cardiovasc Qual Outcomes 2015;8:155-63. http://dx.doi.org/10.1161/CIRCOUTCOMES.114.001240.
- Hutchins R, Pignone MP, Sheridan SL, Viera AJ. Quantifying the utility of taking pills for preventing adverse health outcomes: a cross-sectional survey. BMJ Open 2015;5. http://dx.doi.org/10.1136/bmjopen-2014-006505.
- Lipid Modification: Cardiovascular Risk Assessment and the Modification of Blood Lipids for the Primary and Secondary Prevention of Cardiovascular Disease (CG181). London: National Institute for Health and Care Excellence; 2014.
- Lipid Modification: Cardiovascular Risk Assessment and the Modification of Blood Lipids for the Primary and Secondary Prevention of Cardiovascular Disease. London: National Clinical Guideline Centre; 2014.
- Hippisley-Cox J, Coupland C, Vinogradova Y, Robson J, Minhas R, Sheikh A, et al. Predicting cardiovascular risk in England and Wales: prospective derivation and validation of QRISK2. BMJ 2008;336:1475-82. http://dx.doi.org/10.1136/bmj.39609.449676.25.
- Statins for the Prevention of Cardiovascular Events. London: National Institute for Health and Care Excellence; 2006.
- Ward S, Lloyd Jones M, Pandor A, Holmes M, Ara R, Ryan A, et al. A systematic review and economic evaluation of statins for the prevention of coronary events. Health Technol Assess 2007;11. http://dx.doi.org/10.3310/hta11140.
- Office for National Statistics . Interim Life Tables, England &Amp; Wales: Period Expectation of Life for the Years 2010–2012 2013. www.ons.gov.uk/ons/taxonomy/index.html?nscl=Interim+Life+Tables#tab-data-tables (accessed 5 June 2015).
- Menotti A, Mulder I, Nissinen A, Giampaoli S, Feskens E, Kromhout D. Prevalence of morbidity and multimorbidity in elderly male populations and their impact on 10-year all-cause mortality: the FINE study (Finland, Italy, Netherlands, Elderly). J Clin Epidemiol 2001;54:680-6. http://dx.doi.org/10.1016/S0895-4356(00)00368-1.
- Ara R, Wailoo A. NICE Decision Support Unit Technical Support Document 12: The Use of Health State Utility Values in Decision Models. London: National Institute for Health and Care Excellence; 2011.
- Wonderling D, Sawyer L, Fenu E, Lovibond K, Laramée P. National Clinical Guideline Centre cost-effectiveness assessment for the National Institute for Health and Clinical Excellence. Ann Intern Med 2011;154:758-65. http://dx.doi.org/10.7326/0003-4819-154-11-201106070-00008.
- Barton P, Bryan S, Robinson S. Modelling in the economic evaluation of health care: selecting the appropriate approach. J Health Serv Res Policy 2004;9:110-18. http://dx.doi.org/10.1258/135581904322987535.
- Craig D, McDaid C, Fonseca T, Stock C, Duffy S, Woolacott N. Are adverse effects incorporated in economic models? An initial review of current practice. Health Technol Assess 2009;13. http://dx.doi.org/10.3310/hta13620.
- Christie M, Cliffe A, Dawid P, Senn S. Simplicity, Complexity and Modelling. London: John Wiley & Sons; 2011.
- Caro JJ. Pharmacoeconomic analyses using discrete event simulation. Pharmacoeconomics 2005;23:323-32. http://dx.doi.org/10.2165/00019053-200523040-00003.
- Caro JJ, Möller J, Getsios D. Discrete event simulation: the preferred technique for health economic evaluations?. Value Health 2010;13:1056-60. http://dx.doi.org/10.1111/j.1524-4733.2010.00775.x.
- Le Lay A, Despiegel N, Francois C, Duru G. Can discrete event simulation be of use in modelling major depression?. Cost Effectiveness Resource Allocation 2006;4. http://dx.doi.org/10.1186/1478-7547-4-19.
- Ali Afzali HH, Karnon J, Gray J. A proposed model for economic evaluations of major depressive disorder. Eur J Health Econ 2012;13:501-10. http://dx.doi.org/10.1007/s10198-011-0321-3.
- Saylan M, Treur MJ, Postema R, Dilbaz N, Savas H, Heeg BM, et al. Cost-effectiveness analysis of aripiprazole augmentation treatment of patients with major depressive disorder compared to olanzapine and quetiapine augmentation in Turkey: a microsimulation approach. Value Health Regional Issues 2013;2:171-80. http://dx.doi.org/10.1016/j.vhri.2013.06.004.
- Vataire A-L, Aballéa S, Antonanzas F, Roijen LH, Lam RW, McCrone P, et al. Core discrete event simulation model for the evaluation of health care technologies in major depressive disorder. Value Health 2014;17:183-95. http://dx.doi.org/10.1016/j.jval.2013.11.012.
- Zimovetz E, Wolowacz S, Classi P, Birt J. Methodologies used in cost-effectiveness models for evaluating treatments in major depressive disorder: a systematic review. Cost Effectiveness Resource Allocation 2012;10. http://dx.doi.org/10.1186/1478-7547-10-1.
- Roberts M, Russell LB, Paltiel AD, Chambers M, McEwan P, Krahn M. Conceptualizing a model: a report of the ISPOR-SMDM Modeling Good Research Practices Task Force-2. Med Decis Making 2012;32:678-89. http://dx.doi.org/10.1177/0272989X12454941.
- Jones MT, Cockrum PC. A critical review of published economic modelling studies in depression. Pharmacoeconomics 2000;17:555-83. http://dx.doi.org/10.2165/00019053-200017060-00003.
- Ali Afzali H, Karnon J, Gray J. A critical review of model-based economic studies of depression. Pharmacoeconomics 2012;30:461-82. http://dx.doi.org/10.2165/11590500-000000000-00000.
- Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JPT, Churchill R, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet 2009;373:746-58. http://dx.doi.org/10.1016/S0140-6736(09)60046-5.
- Ward S, Lloyd Jones M, Pandor A, Holt J, Ara R, Ryan A. Statins for the Prevention of Coronary Events: Technology Assessment Report Commissioned by the HTA Programme on Behalf of the National Institute for Clinical Excellence. London: HTA Programme; 2005.
- Sapin C, Fantino B, Nowicki M-L, Kind P. Usefulness of EQ-5D in assessing health status in primary care patients with major depressive disorder. Health Qual Life Outcomes 2004;2. http://dx.doi.org/10.1186/1477-7525-2-20.
- Coventry P, Lovell K, Dickens C, Bower P, Chew-Graham C, Cherrington A, et al. Collaborative Interventions for Circulation and Depression (COINCIDE): study protocol for a cluster randomized controlled trial of collaborative care for depression in people with diabetes and/or coronary heart disease. Trials 2012;13. http://dx.doi.org/10.1186/1745-6215-13-139.
- Oakley J, O’Hagan A. SHELF: The Sheffield Elicitation Framework (v2.0). Sheffield: School of Mathematics and Statistics, University of Sheffield; 2010.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004;18:911-32. http://dx.doi.org/10.2165/00023210-200418130-00006.
- Solomon D, Keller M, Leon A, Mueller TI, Lavori PW, Shea MT, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry 2000;157:229-33. http://dx.doi.org/10.1176/appi.ajp.157.2.229.
- Ara R, Wailoo AJ. Estimating health state utility values for joint health conditions: a conceptual review and critique of the current evidence. Med Decis Making 2013;33:139-53. http://dx.doi.org/10.1177/0272989X12455461.
- Curtis L. Unit Costs of Health and Social Care 2014. Canterbury: University of Kent, Personal Social Services Research Unit; 2014.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2015.
- Campbell HE, Stokes EA, Bargo D, Logan RF, Mora A, Hodge R, et al. Costs and quality of life associated with acute upper gastrointestinal bleeding in the UK: cohort analysis of patients in a cluster randomised trial. BMJ Open 2015;5. http://dx.doi.org/10.1136/bmjopen-2014-007230.
- Davis S, Stevenson M, Tappenden P, Wailoo A. NICE Decision Support Unit Technical Support Document 15: Cost-Effectiveness Modelling Using Patient-Level Simulation. London: National Institute for Health and Care Excellence; 2014.
- Rugulies R. Depression as a predictor for coronary heart disease. Am J Prev Med 2002;23:51-6. http://dx.doi.org/10.1016/S0749-3797(02)00439-7.
- Wulsin LR, Singal BM. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom Med 2003;65:201-10. http://dx.doi.org/10.1097/01.PSY.0000058371.50240.E3.
- Lett HS, Blumenthal JA, Babyak MA, Sherwood A, Strauman T, Robins C, et al. Depression as a risk factor for coronary artery disease: evidence, mechanisms, and treatment. Psychosom Med 2004;66:305-15.
- O’Hagan A, Buck C, Daneshkhah A, Eiser JR, Garthwaite PH, Jenkinson DJ, et al. Uncertain Judgements: Eliciting Experts’ Probabilities. London: John Wiley & Sons; 2006.
- Morris DE, Oakley JE, Crowe JA. A web-based tool for eliciting probability distributions from experts. Environmental Modelling &Amp; Software 2014;52:1-4. http://dx.doi.org/10.1016/j.envsoft.2013.10.010.
Appendix 1 First- and second-line drugs recommended in the 12 guidelines examined in Chapter 2
Reproduced from Dumbreck et al. 73 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.
© BMJ Publishing Group Ltd 2015.
Appendix 2 Details of expected harm from the identified potentially serious drug–drug interactions for each of the three index conditions
Reproduced from Dumbreck et al. 73 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.
© BMJ Publishing Group Ltd 2015.
Appendix 3 Rapid review search strategy for direct treatment disutility in Chapter 5
-
(statins or statin).ti,ab.
-
(lipids or lipid).ti,ab.
-
1 or 2
-
Quality-Adjusted Life Years/
-
quality adjusted life.tw.
-
(qaly$ or qald$ or qale$ or qtime$).tw.
-
(euroqol or euro qol or eq5d or eq 5d).tw.
-
(hql or hqol or h qol or hrqol or hr qol).tw.
-
health utilit$.tw.
-
utilit$.tw.
-
or/4-10
-
(disutilit$ or dis-utilit$).tw.
-
decrement.tw.
-
12 or 13
-
3 and 11 and 14
Appendix 4 The expert elicitation exercise
Introduction
Key parameters in cost-effectiveness models are typically populated with evidence sourced from a review of the literature. However, robust evidence on patients with multimorbidity is known to be poor, as typically such patients are excluded from clinical trials. 94,97–100
Aim
An expert elicitation process was designed to help to inform and supplement the current available evidence for populating the model-based CEA reported in Chapter 6.
Method
The elicitation was conducted following the SHELF, based on elicitation practice recommended by Oakley and O’Hagan. 223,234,235 The experts (n = 6) used for the elicitation exercise consisted of generalist practitioners and geriatricians from the PRG. Informed consent was supplied from each participant.
The elicitation exercise informed the parameter incorporating the risk of ADE when CHD is present and the proportion of patients with depression and CHD who have an ADE when receiving an antidepressant within 28 days (4 weeks). This is a key parameter within the economic model, as it drives the potential cost-effectiveness of the comparators between the scenarios where patients have just depression or depression and CHD.
Experts were asked to consider each of the following antidepressants:
-
SSRIs as a class
-
duloxetine
-
mirtazapine
-
venlafaxine.
To elicit a probability distribution for the parameters of interest, the experts were asked what proportion of patients they thought would have an ADE when prescribed the antidepressant, knowing they also have CHD. The quartile method was used to derive a probability distribution on their beliefs for this proportion. Here the experts were encouraged to provide estimates of (1) the plausible range; (2) the interquartile range; and (3) the median from which a best-fitting probability distribution is derived. The elicitation of probability distributions is preferable to an individual parameter estimate, as uncertainty in the experts’ beliefs regarding the parameter can be captured and then used to inform the analysis.
A web-based portal called MATCH (Multidisciplinary Assessment of Technology Centre for Healthcare) was used to facilitate the elicitation process,235 whereby the exercise would be conducted remotely; experts would use their own computers in conjunction with a teleconference with the facilitator.
To derive a pooled estimate from all the experts, the individual responses were combined using linear aggregation methods whereby each expert’s estimation was given an equal weight. For the deterministic analysis, the mean proportion from the combined estimate was used (see Appendix 5, Tables 35 and 36); for the probabilistic analysis, samples were drawn from the empirical combined distribution (Figure 21).
FIGURE 21.
Proportion of patients who have an ADE within 28 days who had CHD: the empirical pooled distribution from a selection of experts (n = 6).
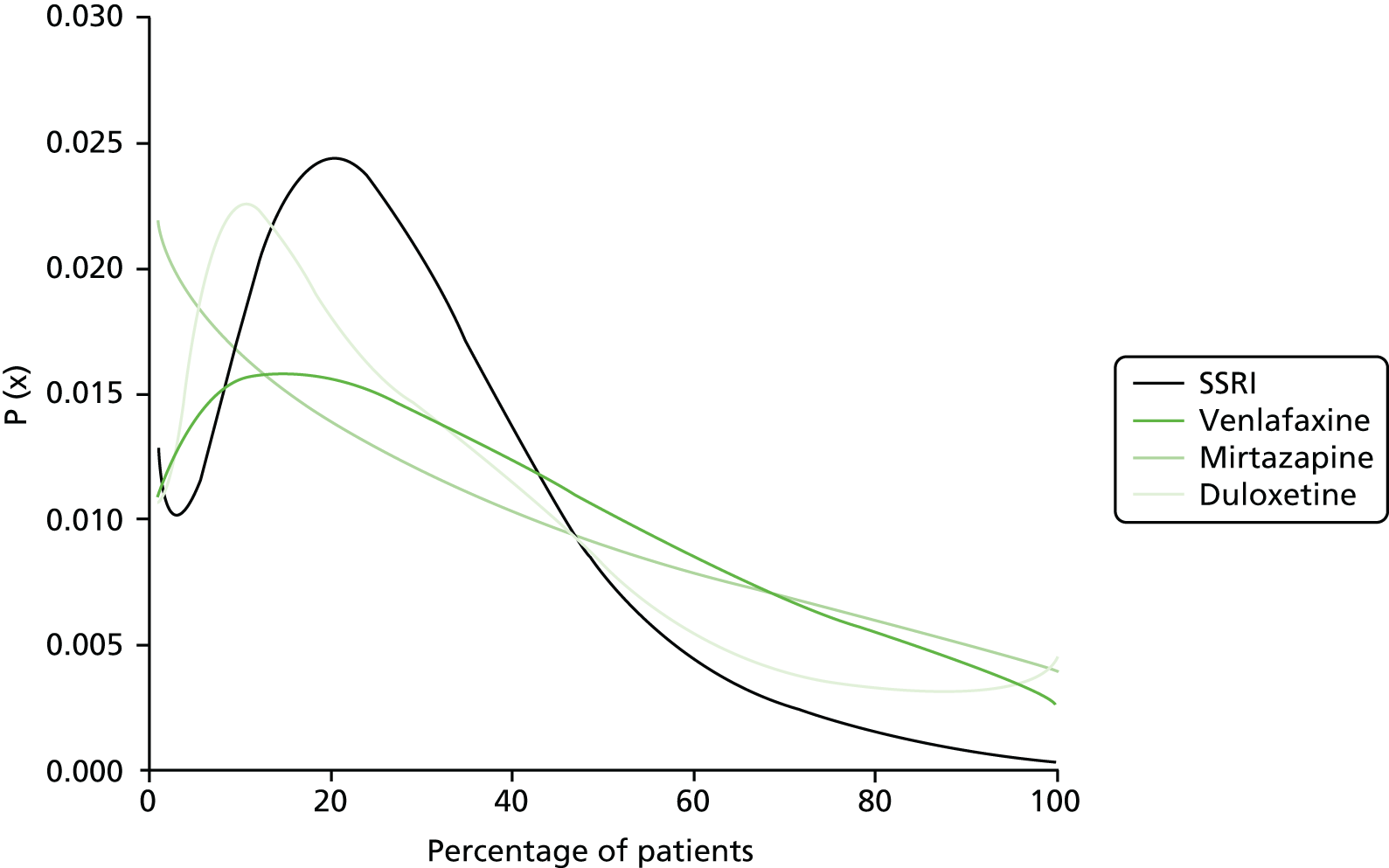
Appendix 5 Probability of adverse drug event assumptions
| Adverse events | Probabilitya | Scaled to 100 | Probability of occurring | Probability of discontinuationb | Probability of dose changeb | Probability of switchb | No changeb | Probability of discontinuation and event | Probability of dose change and event | Probability of switch and event | Probability of no change and event |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nausea | 19 | 22 | 8 | 25 | 25 | 25 | 25 | 1.9 | 1.9 | 1.9 | 1.9 |
| Headache | 16 | 18 | 6 | 25 | 25 | 25 | 25 | 1.6 | 1.6 | 1.6 | 1.6 |
| Diarrhoea | 12 | 14 | 5 | 25 | 25 | 25 | 25 | 1.2 | 1.2 | 1.2 | 1.2 |
| Insomnia | 11 | 12 | 4 | 25 | 25 | 25 | 25 | 1.1 | 1.1 | 1.1 | 1.1 |
| GI bleed | 2 | 2 | 1 | 50 | 0 | 50 | 0 | 0.4 | 0.0 | 0.4 | 0.0 |
| Other AEs | 29 | 33 | 12 | 25 | 25 | 25 | 25 | 3.0 | 3.0 | 3.0 | 3.0 |
| Summed probability of ADEc | 88 | 100 | 36 | ||||||||
| Adverse events | Probabilitya | Scaled to 100 | Probability of occurring | Probability of discontinuationb | Probability of dose changeb | Probability of switchb | No changeb | Probability of discontinuation and event | Probability of dose change and event | Probability of switch and event | Probability of no change and event |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nausea | 19 | 22 | 8 | 33 | 33 | 33 | 2.6 | 2.6 | 2.6 | 0.0 | |
| Headache | 16 | 18 | 6 | 33 | 33 | 33 | 2.1 | 2.1 | 2.1 | 0.0 | |
| Diarrhoea | 12 | 14 | 5 | 33 | 33 | 33 | 1.6 | 1.6 | 1.6 | 0.0 | |
| Insomnia | 11 | 12 | 4 | 33 | 33 | 33 | 1.4 | 1.4 | 1.4 | 0.0 | |
| GI bleed | 2 | 2 | 1 | 50 | 0 | 50 | 0.4 | 0.0 | 0.4 | 0.0 | |
| Other AEs | 29 | 33 | 12 | 33 | 33 | 33 | 4.0 | 4.0 | 4.0 | 0.0 | |
| Summed probability of ADEc | 88 | 100 | 36 | ||||||||
Appendix 6 Mean time (years) to transition between coronary heart disease states
| First health state | Second health state | Age band (years) | |||
|---|---|---|---|---|---|
| 55–64 | 65–74 | 75–84 | ≥ 85 | ||
| SA | UA | 344 | 166 | 109 | 81 |
| SA | MI | 161 | 90 | 63 | 48 |
| SA | CHD death | 285 | 142 | 142 | 142 |
| UA | MI | 20 | 20 | 21 | 23 |
| UA | ST | 71 | 71 | 71 | 71 |
| UA | HF | 22 | 22 | 22 | 22 |
| UA | CHD death | 15 | 9 | 5 | 3 |
| MI | UA | 133 | 133 | 133 | 133 |
| MI | ST | 312 | 147 | 70 | 35 |
| MI | HF | 39 | 39 | 39 | 39 |
| MI | CHD death | 30 | 15 | 8 | 5 |
| TIA | MI | 322 | 181 | 124 | 96 |
| TIA | ST | 55 | 23 | 12 | 10 |
| TIA | CVM | 61 | 28 | 19 | 18 |
| ST | UA | 322 | 181 | 124 | 96 |
| ST | MI | 322 | 181 | 124 | 96 |
| ST | HF | 138 | 78 | 53 | 41 |
| ST | CHD death | 45 | 19 | 8 | 4 |
| HF | UA | 123 | 123 | 123 | 123 |
| HF | MI | 123 | 123 | 123 | 123 |
| HF | ST | 516 | 516 | 516 | 516 |
| HF | CHD death | 21 | 21 | 21 | 21 |
List of abbreviations
- ACE
- angiotensin-converting enzyme
- ADE
- adverse drug event
- AGS
- American Geriatrics Society
- ARB
- angiotensin receptor blocker
- ARR
- absolute risk reduction
- BMI
- body mass index
- BNF
- British National Formulary
- CEA
- cost-effectiveness analysis
- CG
- clinical guideline
- CHD
- coronary heart disease
- CI
- confidence interval
- CKD
- chronic kidney disease
- COPD
- chronic obstructive pulmonary disease
- CRQ
- clinical research question
- CVD
- cardiovascular disease
- DES
- discrete event simulation
- DSU
- Decision Support Unit
- DTD
- direct treatment disutility
- EF
- ejection fraction
- EQ-5D
- European Quality of Life-5 Dimensions
- GDG
- guideline development group
- GI
- gastrointestinal
- GP
- general practitioner
- GRADE
- Grading of Recommendations Assessment, Development and Evaluation
- HRQoL
- health-related quality of life
- HTA
- Health Technology Assessment
- LVSD
- left ventricular systolic dysfunction
- MAGGIC
- Meta-Analysis Global Group in Chronic heart failure
- MAOI
- monoamine oxidase inhibitor
- MI
- myocardial infarction
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- NNT
- number needed to treat
- NSAID
- non-steroidal anti-inflammatory drug
- ONS
- Office for National Statistics
- PICO
- population, intervention, comparator, outcome
- PRG
- project reference group
- PSA
- probabilistic sensitivity analysis
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- RR
- risk ratio
- RRR
- relative risk reduction
- SENIORS
- Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalisation in Seniors with Heart Failure
- SIGN
- Scottish Intercollegiate Guidelines Network
- SSC
- study steering committee
- SSRI
- selective serotonin reuptake inhibitor
- TCA
- tricyclic antidepressant
- TIA
- transient ischaemic attack
- TTB
- time to benefit
- UKPDS
- UK Prospective Diabetes Study