
Notes
Article history
The research reported in this article of the journal supplement was commissioned and funded by the HTA programme on behalf of NICE as project number 06/23/01. The assessment report began editorial review in July 2007 and was accepted for publication in December 2008. See the HTA programme website (www.hta.ac.uk) for further project information. This summary of the ERG report was compiled after the Appraisal Committee’s review. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© 2009 Queen’s Printer and Controller of HMSO. This monograph may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2009 Queen’s Printer and Controller of HMSO
This paper presents a summary of the evidence review group (ERG) report into the clinical and cost-effectiveness of erlotinib for the treatment of relapsed non-small cell lung cancer (NSCLC), according to its licensed indication, based upon the evidence submission from Roche Products to the National Institute for Health and Clinical Excellence (NICE) as part of the single technology appraisal (STA) process. The submitted clinical evidence includes one randomised controlled trial (RCT) (BR21) investigating the effect of erlotinib versus placebo, which demonstrates that erlotinib significantly increases median overall survival, progression-free survival and response rate compared with placebo. The majority of patients in the trial experienced non-haematological drug-related adverse effects. Currently there are no trials that directly compare erlotinib with any other second-line chemotherapy agent. For the purposes of indirect comparison, the manufacturer’s submission provides a narrative discussion of data from 11 RCTs investigating the use of docetaxel. From these data the manufacturer concludes that erlotinib has similar clinical efficacy levels to docetaxel but results in fewer serious haematological adverse events; however, it is difficult to compare the results of BR21 with those of the docetaxel trials or with current UK clinical practice because, for example, the BR21 patient population is younger than that expected to present in UK clinical practice and almost half of the BR21 participants received erlotinib as third-line chemotherapy, with third-line chemotherapy being rare in the UK. The manufacturer’s submission included a three-state model comparing erlotinib with docetaxel, reporting an incremental cost-effectiveness ratio (ICER) of –£1764 per quality-adjusted life-year (QALY) gained for erlotinib compared with docetaxel. Rerunning the manufacturer’s economic model with varied parameters and assumptions increases the ICER to in excess of £52,000 per QALY gained. There is still a large amount of unquantifiable uncertainty in the model and it is unlikely that erlotinib could be considered to be cost-effective compared with docetaxel at a willingness to pay of £30,000 and there may even be the potential for docetaxel to dominate erlotinib. Because of the limitations of the indirect analysis undertaken by the manufacturer and the subsequent economic modelling exercise there is a need for a head-to-head trial comparing erlotinib with docetaxel. The guidance issued by NICE in February 2007 as a result of the STA states that erlotinib is not recommended for the treatment of locally advanced or metastatic NSCLC.
Introduction
The National Institute for Health and Clinical Excellence (NICE) is an independent organisation within the NHS that is responsible for providing national guidance on the treatment and care of people using the NHS in England and Wales. One of responsibilities of NICE is to provide guidance to the NHS on the use of selected new and established health technologies, based on an appraisal of those technologies.
NICE’s single technology appraisal (STA) process is specifically designed for the appraisal of a single product, device or other technology, with a single indication, for which most of the relevant evidence lies with one manufacturer or sponsor. 1 Typically, it is used for new pharmaceutical products close to launch. The principal evidence for an STA is derived from a submission by the manufacturer/sponsor of the technology. In addition, a report reviewing the evidence submission is submitted by the evidence review group (ERG), an external organisation independent of NICE. This paper presents a summary of the ERG report for the STA of erlotinib for the treatment of relapsed non-small cell lung cancer (NSCLC). 2
Description of the underlying health problem
Lung cancer is the most common cause of cancer-related death in men and the second most common cause of cancer-related death after breast cancer in women. 3 In 2002, 37,700 patients were newly diagnosed with lung cancer in the UK, accounting for one in seven new cancer cases, with an incidence of about 62–65 per 100,000 population; the incidence of NSCLC is approximately 52 per 100,000 population. 4 Lung cancer is rarely diagnosed in people under 40 years of age, but the incidence rises steeply with age thereafter, peaking in people aged 75–84 years. 4 The male–female ratio for lung cancer cases is 3:2. 4 There is a strong association between incidence and mortality rates and levels of deprivation. 4
Scope of the ERG report
The ERG report presents the results of the assessment of the manufacturer’s (Roche Products) evidence submission regarding the use of erlotinib for the second-line treatment of patients with locally advanced or metastatic (stage III/IV) NSCLC. The report includes an assessment of both the clinical and cost-effectiveness evidence submitted by the manufacturer. Erlotinib (Tarceva®) is an orally active inhibitor of epidermal growth factor receptor/human epidermal growth factor receptor 1 (EGFR/HER1) tyrosine kinase inhibitor. In 2004, pemetrexed (Alimta®; Lilly) received a licence for use ‘as monotherapy for the treatment of patients with locally advanced or metastatic non-small cell lung cancer after prior chemotherapy’. The licensing submission for pemetrexed was supported by a phase III study comparing pemetrexed and docetaxel. 5 In 2005, erlotinib was licensed ‘for the treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of at least one prior chemotherapy regimen’. The licensing submission for erlotinib was supported by a phase III study comparing erlotinib with placebo. 6
Methods
The ERG report comprised a critical review of the evidence for the clinical effectiveness and cost-effectiveness of the technology based upon the manufacturer’s/sponsor’s submission to NICE as part of the STA process. The ERG assessed the quality of the clinical effectiveness review using a checklist and conducted a literature search. The group fitted exponential curves to the manufacturer’s Kaplan–Meier plots to calculate overall survival (OS) and also reran the manufacturer’s economic model after correcting for an inherent error and altered some of the assumptions and parameter values to recalculate the cost–utility ratios, quality-adjusted life-years (QALYs) and estimates of benefits.
Results
Summary of submitted clinical evidence
The submitted clinical evidence includes one randomised, placebo-controlled, double-blind trial (BR21)6 that investigates the effect of erlotinib within its licensed indication (treatment of relapsed NSCLC) versus placebo. The BR21 trial demonstrates that erlotinib significantly increases median OS by 42.5% compared with placebo (6.7 months versus 4.7 months respectively; p < 0.001, hazard ratio 0.70). Progression-free survival (PFS) is significantly longer in the erlotinib arm compared with the placebo arm (2.2 months versus 1.8 months respectively; p < 0.001, hazard ratio 0.61) and the overall response rate is significantly higher (8.9% versus 0.9%; p < 0.001).
The majority of patients in the BR21 trial experienced non-haematological drug-related adverse effects (AEs). The most commonly reported AEs attributed to erlotinib were rash (76%) and diarrhoea (55%), leading to a dose reduction in 12% and 5% of patients respectively. Currently there are no trials that directly compare erlotinib with any other second-line chemotherapy agent. For the purposes of indirect comparison, the manufacturer’s submission provides a narrative discussion of data from 11 randomised controlled trials (RCTs) investigating the use of docetaxel at a dose of 75 mg/m2. The manufacturer extracted detailed data from two of the 11 trials involving docetaxel: docetaxel versus best supportive care (TAX317)7 and docetaxel versus pemetrexed (JMEI)5. In these trials docetaxel showed similar efficacy levels to those of erlotinib as reported in the BR21 trial. Median OS was 7.5 months (docetaxel, TAX317), 7.9 months (docetaxel, JMEI) and 6.7 months (erlotinib, BR21). Median PFS was reported as 2.9 months (docetaxel, JMEI) and 2.2 months (erlotinib, BR21) and overall response rates were reported as 8.9% (docetaxel, JMEI) and 8.8% (erlotinib, BR21). Analyses of TAX317 and JMEI in relation to the BR21 study demonstrated the lower rates of haematological toxicities experienced by patients receiving erlotinib compared with those receiving docetaxel, particularly incidences of febrile neutropenia. The manufacturer’s submission therefore concludes that erlotinib has similar clinical efficacy levels to docetaxel but results in fewer serious haematological adverse events. When interpreting the results of BR21 a number of issues relating to the patient population must be considered. For example, the BR21 patient population is younger than that expected to present in UK clinical practice. Almost half of the trial participants received erlotinib as third-line chemotherapy, with third-line chemotherapy being rare in the UK. Furthermore, a large number of participants in the BR21 trial had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 2–3; typically patients receiving chemotherapy in UK clinical practice have a PS of 0–1. For these reasons it is difficult to compare the results of BR21 with those of TAX317 and JMEI or with current UK clinical practice.
Summary of submitted cost-effectiveness evidence
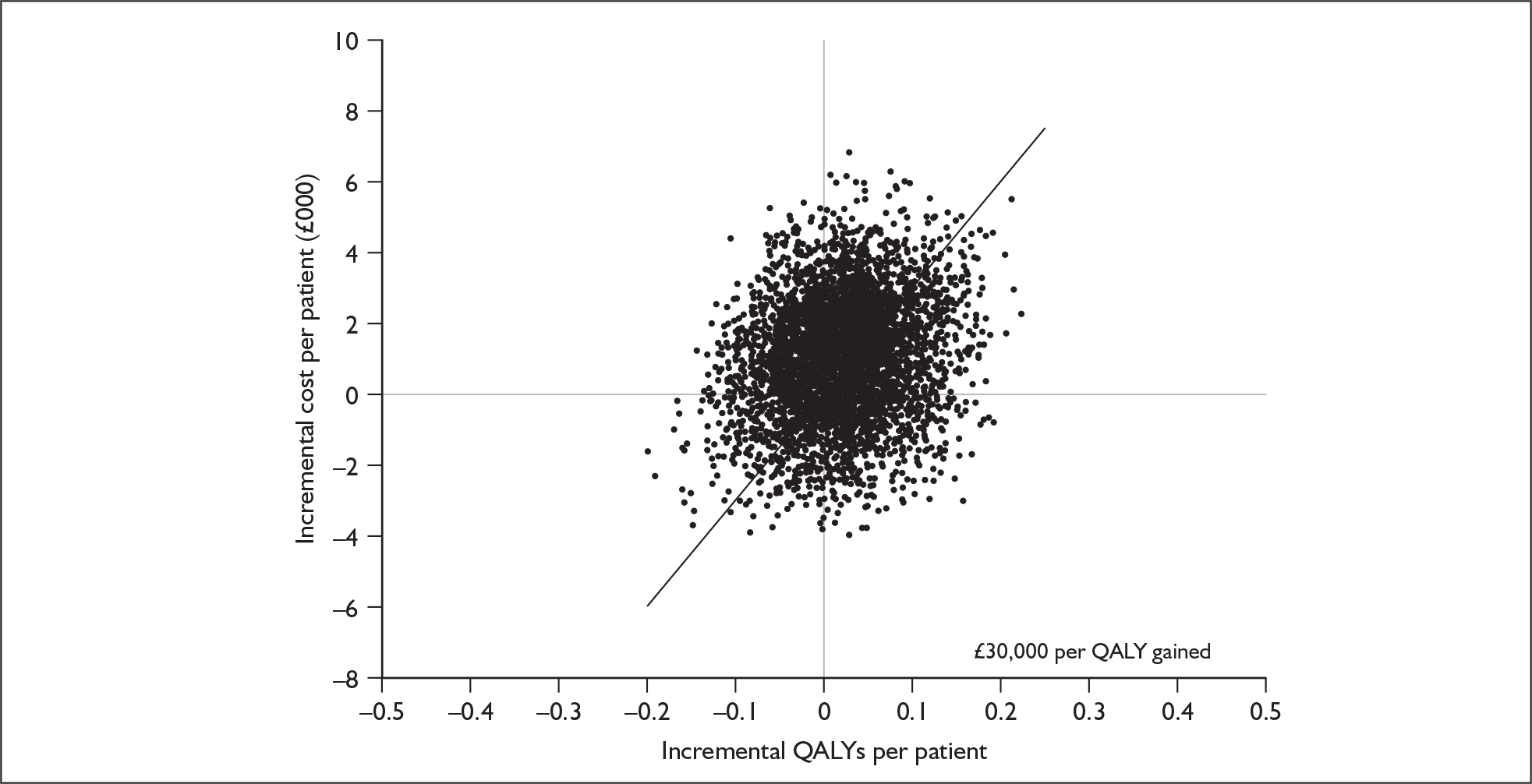
The economic model submitted in support of the manufacturer’s submission is a basic three-state model comparing erlotinib with docetaxel, furnished with clinical data from the TAX317 and BR21 trials. The manufacturer reports an incremental cost-effectiveness ratio (ICER) of –£2941 per QALY for erlotinib compared with docetaxel, with a 68% probability that erlotinib is cost-effective at a willingness to pay (WTP) of £30,000 per QALY gained. After adjustment for the double counting of half-cycle correction, the manufacturer’s model yields a corrected ICER of –£1764. However, a number of key assumptions and parameters in the model do not seem to be clinically and/or economically justified, particularly in terms of costs. For example, the manufacturer underestimates the acquisition cost of erlotinib and overestimates the acquisition cost of docetaxel. Once these assumptions are adjusted to reflect more realistic estimates, the ICER increases to £52,098 per QALY as shown in Table 1, with a 44% probability that erlotinib is cost effective at a WTP of £30,000. A modified cost-acceptability curve using manufacturer probabilistic sensitivity analysis (PSA) results adjusted for average incremental cost and outcome alterations and a modified cost-effectiveness uncertainty scatter plot using manufacturer PSA results adjusted for average incremental cost and outcome alterations are shown in Figures 1 and 2 respectively.
| Erlotinib | Docetaxel | Increment | |
|---|---|---|---|
| Costs per patient | |||
| Drug acquisition | £7164 | £5022 | £2142 |
| Drug administration and monitoring | £473 | £839 | –£365 |
| Adverse event treatment | £113 | £374 | –£261 |
| Other preprogression care | £1034 | £859 | £175 |
| Postprogression care | £4699 | £5444 | –£745 |
| Total cost | £13,482 | £12,536 | £946 |
| Outcomes per patient | |||
| Overall mean survival (months) | 9.03 | 9.03 | 0.00 |
| PFS (months) | 4.11 | 3.33 | 0.78 |
| PPS (months) | 4.92 | 5.70 | –0.78 |
| PFS QALYs | 0.1591 | 0.1139 | 0.0452 |
| PPS QALYs | 0.0953 | 0.1224 | –0.0271 |
| Total QALYs | 0.2544 | 0.2362 | 0.0182 |
| Incremental cost per QALY | £52,098 | ||
FIGURE 1.
Modified cost-effectiveness curve using manufacturer probabilistic sensitivity analyses results adjusted for average incremental cost and outcome alterations. WTP, willingness to pay.
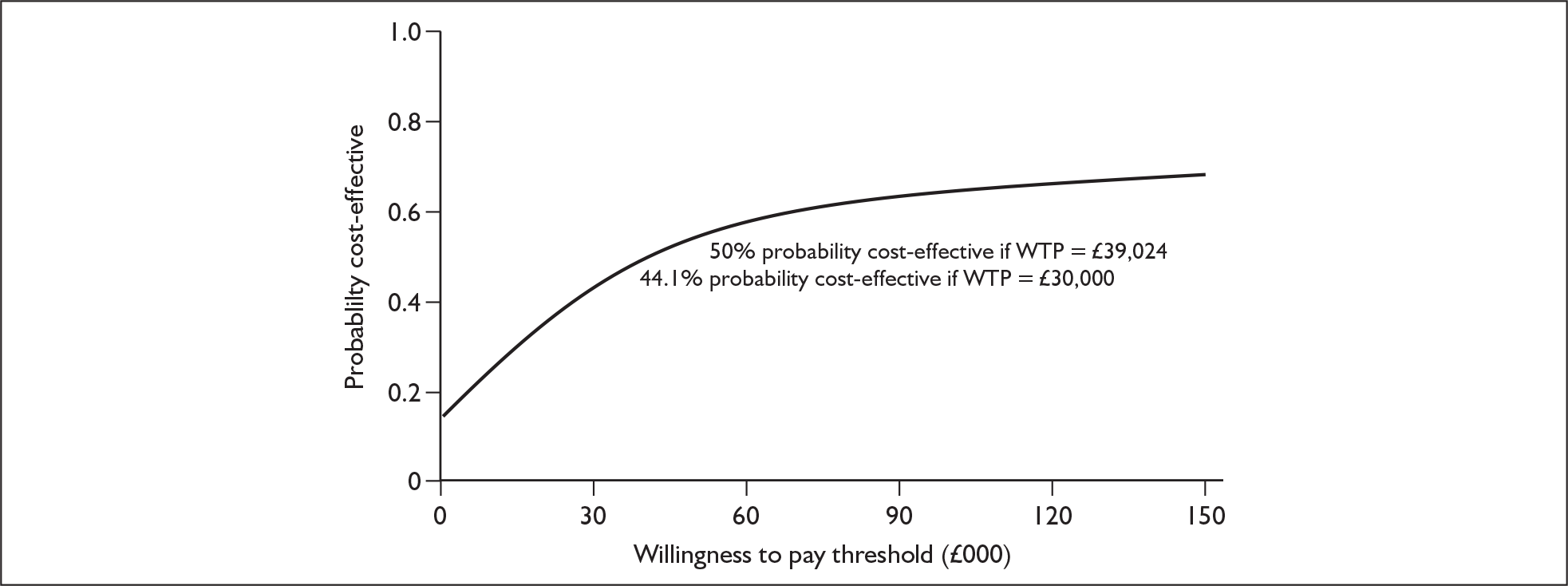
FIGURE 2.
Modified cost-effectiveness uncertainty scatter plot using manufacturer probabilistic sensitivity analyses results adjusted for average incremental cost and outcome alterations. QALY(s), quality-adjusted life-year(s).
In terms of health outcomes a further issue is the use of visual analogue scale (VAS) scores from the Oxford Outcomes study; the scores were not adjusted to zero for death and conflict with the tariff values calculated using responses from the same sample of healthy volunteers. As presented in Table 2, reanalysis of the model rescaling the VAS PFS utility scores to ensure that death has zero utility further increased the ICER (£68,673 per QALY gained). Similarly, reanalysis using tariff PFS utility values led to an ICER slightly above the WTP threshold of £30,000 (£31,261 per QALY gained). Joint exploration of uncertainty in the cost of docetaxel and the degree of variation in dosing introduced by clinical judgement yields a range of ICER estimates between £41,943 and £70,418 per QALY gained.
| Erlotinib | Docetaxel | Increment | |
|---|---|---|---|
| Using rescaled VAS values in PFS | |||
| PFS QALYs (rescaled VAS) | 0.1292 | 0.0883 | 0.0409 |
| PPS QALYs (the ERG estimate) | 0.0953 | 0.1224 | -0.0271 |
| Total QALYs | 0.2245 | 0.2107 | 0.0138 |
| Incremental cost per QALY | £68,673 | ||
| Using tariff values in PFS | |||
| PFS QALYs (tariff) | 0.1337 | 0.0763 | 0.0573 |
| PPS QALYs (the ERG estimate) | 0.0953 | 0.1224 | -0.0271 |
| Total QALYs | 0.2289 | 0.1987 | 0.0303 |
| Incremental cost per QALY | £31,261 | ||
There is also a large amount of unquantifiable uncertainty in the model relating to AEs, postprogression survival and PFS health state costs, and the length of PFS. These areas of ambiguity could potentially further increase the ICER and may even result in docetaxel dominating erlotinib.
Commentary on the robustness of submitted evidence
A major limitation in the manufacturer’s submission is the reliance on the BR21 trial (currently the only available erlotinib study) which compares erlotinib with placebo, rather than an accepted chemotherapy regimen. As a consequence, the manufacturer’s submission is forced to compare erlotinib and docetaxel indirectly; such comparisons have inherent difficulties and are subject to biases.
Further to this, there are a number of differences between the patient population in the BR21 trial and the TAX317 study, of which the most important are the number of prior chemotherapy regimens and the performance status of patients.
In addition, the best supportive care component of treatment may not be comparable between the trials, which could potentially inflate a treatment response in one of the trials unjustifiably. This confounding issue was not discussed in the manufacturer’s submission, but should have been considered when the indirect comparison was undertaken.
A number of unquantifiable areas of uncertainty were found and relate to AEs, pre- and postprogression health state costs and progression-free survival. There is a note in the manufacturer’s table of event probabilities for AEs, which seems to imply that the model does not allow patients to suffer multiple adverse events. If this is so it is a severe and unrealistic constraint, as individual patients frequently suffer multiple events either concurrently (e.g. rash with diarrhoea) or serially. In addition, the resources assumed to be incurred each month for patients before and after disease progression were exclusively determined by five clinical experts without use of any observational data. The main elements contributing to the increase in such costs postprogression are shown in Table 3. Clearly hospital episodes constitute the dominant component in these estimates. It seems disappointing that no attempt has been made to sample routine hospital records and statistics to validate the expert opinion in this respect. The ERG raised issues about the validity of the claims of equivalence in overall survival and of improved PFS with erlotinib. These are of profound importance to the economic evaluation of erlotinib as if either of these assertions proves to be untenable then most of the modest outcome gains claimed for erlotinib will disappear, other than the very small short-term quality of life benefits associated with oral administration and reduced AEs. In the context of important increases in drug acquisition costs this would mean that erlotinib could not be considered cost-effective and might in fact be dominated by docetaxel (more expensive and less effective).
| Component | Cost per month | Proportion |
|---|---|---|
| Hospital episodes | £547.97 | 55.4% |
| Health professionals | £331.54 | 33.5% |
| Medications | £39.46 | 4.0% |
| Tests | £69.83 | 7.1% |
| Total | £988.80 | 100.0% |
Conclusions
The manufacturer’s submission presents a case for the replacement of docetaxel by erlotinib as second-line chemotherapy for NSCLC patients with advanced or metastatic disease. However, there is a proportion of NSCLC patients whose poor health status precludes them from receiving docetaxel; for these patients best supportive care is currently the only treatment option available. It may be argued that some of these patients could be considered for erlotinib instead of docetaxel as it is a less demanding oral regimen.
The ERG attempted to rectify several of the limitations in the clinical and cost-effectiveness evidence submitted, generating much higher incremental cost-effectiveness ratios than those generated in the manufacturer’s submission (in excess of £52,000). This extreme sensitivity is due to the very small value of incremental benefit, which renders the ICER highly unstable to small changes. There is still a large amount of unquantifiable uncertainty, however at the current price it is unlikely that erlotinib could be considered to be cost effective compared with docetaxel at a WTP of £30,000. There may even be the potential for docetaxel to dominate erlotinib (i.e. be more effective yet less expensive). This means that adoption of erlotinib would need to be justified on grounds out with the factors included in the model (for example, patient preference for oral self-medication and service pressures to limit or reduce demand for hospital administered chemotherapy).
Given the limitations of the indirect analysis undertaken by the manufacturer and the subsequent economic modelling exercise there is a need for a head-to-head trial comparing erlotinib with docetaxel.
Summary of NICE guidance issued as a result of the STA
The guidance issued by NICE in February 2007 states that:
-
1.1 Erlotinib is recommended, within its licensed indication, as an alternative to docetaxel as a second-line treatment option for patients with non-small-cell lung cancer (NSCLC) only on the basis that it is provided by the manufacturer at an overall treatment cost (including administration, adverse events and monitoring costs) equal to that of docetaxel.
-
1.2 The decision to use erlotinib or docetaxel (as outlined in section 1.1) should be made after a discussion between the responsible clinician and the individual about the potential benefits and adverse effects of each treatment.
-
1.3 Erlotinib is not recommended for the second-line treatment of locally advanced or metastatic NSCLC in patients for whom docetaxel is unsuitable (that is, where there is intolerance of or contraindications to docetaxel) or for third-line treatment after docetaxel therapy.
-
1.4 People currently receiving treatment with erlotinib, but for whom treatment would not be recommended according to section 1.3, should have the option to continue treatment until they and their clinicians consider it appropriate to stop.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
Key references
- National Institute for Health and Clinical Excellence . Guide to the Single Technology (STA) Process 2006. www.nice.org.uk/page.aspx?o=STAprocessguide (accessed June 2007).
- McLeod C, Bagust A, Boland A, Hockenhull J, Dundar Y, Proudlove C, et al. Erlotinib for the Treatment of Relapsed Non-Small Cell Lung Cancer 2006.
- National Collaborating Centre for Acute Care . The Diagnosis and Treatment of Lung Cancer. Methods, Evidence and Guidance 2005. www.rcseng.ac.uk (accessed February 2005).
- Cancer Research UK . Cancer Facts and Figures 2006. www.cancerresearchuk.org/.
- Hanna N, Shepherd FA, Fossella FV, Pereira JR, Demarinis F, Von Pawel J, et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol 2004;22:1589-97.
- Shepherd FA, Pereira JR, Ciuleanu T, Eng HT, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005;353:123-32.
- Shepherd FA, Dancey J, Ramlau R, Mattson K, Gralla R, O’Rourke M, et al. Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy [see comment]. J Clin Oncol 2000;18:2095-103.
- Australian Department of Health . Public Summary Document for Erlotinib 2006. www.health.gov.au/internet/wcms/publishing.nsf/Content/pbac-psderlotinib-mar06 (accessed 2006).