
Notes
Article history
The research reported in this article of the journal supplement was commissioned and funded by the HTA programme on behalf of NICE as project number 07/66/01. The assessment report began editorial review in August 2008 and was accepted for publication in April 2009. See the HTA programme web site for further project information (www.hta.ac.uk). This summary of the ERG report was compiled after the Appraisal Committee’s review. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report. The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
Declared competing interests of authors
none
Permissions
Copyright statement
© 2009 Queen’s Printer and Controller of HMSO. This monograph may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2009 Queen’s Printer and Controller of HMSO
This paper presents a summary of the evidence review group (ERG) report into the clinical effectiveness and cost-effectiveness of telbivudine for the treatment of chronic hepatitis B (CHB) in adults based upon a review of the manufacturer’s submission to the National Institute for Health and Clinical Excellence (NICE) as part of the single technology appraisal (STA) process. The submission’s evidence came from one randomised controlled trial (RCT) (GLOBE) of reasonable methodological quality comparing telbivudine with lamivudine. One other RCT that appeared to meet the inclusion criteria was excluded from the submission. For the primary outcome of therapeutic response telbivudine was statistically superior to lamivudine at weeks 52 and 104 for hepatitis B e antigen (HBeAg)-positive patients, and at week 104 for HBeAg-negative patients. There were statistically significant differences in favour of telbivudine for some secondary outcomes at 2 years including hepatitis B virus (HBV) DNA reduction, HBV DNA non-detectability and alanine aminotransferase normalisation though not for HBeAg-positive patients. In HBeAg-positive patients there was no significant difference between treatment groups for HBeAg loss or seroconversion at any time point. The incidence of adverse events was similar between treatments. Two RCTs comparing entecavir with lamivudine were included in the indirect comparison; however, this was poorly conducted and the results should be treated with caution. The manufacturer developed two economic models to determine the cost-effectiveness of telbivudine. Evidence on the efficacy of telbivudine and lamivudine was taken from the GLOBE trial; efficacy of adefovir was based on assumption. There was a lack of critical assessment and assurance of the quality of the data used to populate the models. The manufacturer concluded that telbivudine is a cost-effective option compared with lamivudine using evidence from the viral load model [HBeAg-positive patients/HBeAg-negative patients: mean incremental cost £19,087/£49,003, mean quality-adjusted life-year (QALY) gain 1.30/4.67, incremental cost-effectiveness ratio (ICER) £14,665/£10,497 per QALY]. Resubmitted results after a request for clarification by the ERG gave less favourable ICERs (HBeAg-positive patients/HBeAg-negative patients: mean incremental cost £23,983/£41,910, mean QALY gain 1.56/2.07, ICER £15,377/£20,256 per QALY). The manufacturer concluded that telbivudine is a cost-effective option (on its own or followed by adefovir) for patients who have developed resistance to first-line telbivudine treatment; however, the presentation of the results was not ideal. In conclusion, although telbivudine was statistically superior to lamivudine for most antiviral outcomes, the difference was not clinically significant; in addition, the cost-effectiveness evidence for telbivudine presented in the manufacturer’s submission was limited. The NICE guidance issued as a result of the STA states that telbivudine is not recommended for the treatment of chronic hepatitis B and that people currently receiving telbivudine should have the option to continue therapy until they and their clinicians consider it appropriate to stop.
Introduction
The National Institute for Health and Clinical Excellence (NICE) is an independent organisation within the NHS that is responsible for providing national guidance on the treatment and care of people using the NHS in England and Wales. One of the responsibilities of NICE is to provide guidance to the NHS on the use of selected new and established health technologies, based on an appraisal of those technologies.
NICE’s single technology appraisal (STA) process is specifically designed for the appraisal of a single product, device or other technology, with a single indication, for which most of the relevant evidence lies with one manufacturer or sponsor. 1 Typically, it is used for new pharmaceutical products close to launch. The principal evidence for an STA is derived from a submission by the manufacturer/sponsor of the technology. In addition, a report reviewing the evidence submission is submitted by the evidence review group (ERG), an external organisation independent of NICE. This paper presents a summary of the ERG report for the STA of the clinical effectiveness and cost-effectiveness of telbivudine for the treatment of chronic hepatitis B (CHB) in adults.
Description of the underlying health problem
Hepatitis B is an infectious disease of the liver caused by the hepatitis B virus (HBV). The majority of people who are infected as adults recover spontaneously, but around 5% develop CHB, defined as viraemia and hepatic inflammation for more than 6 months. 2 If not successfully treated it can lead to progressive liver damage, including cirrhosis, hepatocellular carcinoma (HCC) and death. Patients with CHB may be hepatitis B e antigen (HBeAg) positive or HBeAg negative, depending on the presence or absence of the ‘e’ antigen.
The Department of Health2 and the British Liver Trust3 estimate that the prevalence of CHB in the UK is approximately 150,000–200,000, with around 7000 estimated new cases every year (mostly from immigration of established HBV carriers). However, the Hepatitis B Foundation4 recently estimated that prevalence may have increased to 325,000, and it is thought likely to increase further as a consequence of increasing rates of immigration of people from countries with a high CHB prevalence.
The main goal of antiviral therapy is to suppress the level of the virus (HBV DNA) for a prolonged period of time to reduce the risk of disease progression and HCC, and also to improve long-term outcomes. HBV DNA is one of the key markers of disease management, as well as HBeAg seroconversion, alanine aminotransferase (ALT) levels and, over the longer term, histological response.
Scope of the ERG report
The ERG critically evaluated the evidence submission from Novartis for the use of telbivudine for the treatment of CHB, in accordance with the licensed indication. Telbivudine is a synthetic thymidine nucleoside analogue that inhibits HBV DNA polymerase and thus HBV replication. It is licensed for the treatment of CHB in adult patients with compensated liver disease and evidence of viral replication, persistently elevated serum ALT levels and histological evidence of active inflammation and/or fibrosis.
The outcomes stated in the manufacturer’s definition of the decision problem were HBV DNA virological response, seroconversion rate, histological improvement, biochemical response, viral resistance, time to treatment failure, survival, health-related quality of life and adverse effects.
Methods
The ERG report comprised a critical review of the evidence for the clinical effectiveness and cost-effectiveness of the technology based upon the manufacturer’s/sponsor’s submission to NICE as part of the STA process.
The ERG checked the literature searches and applied the NICE critical appraisal checklist to the included studies and checked the quality of the manufacturer’s submission with the Centre for Reviews and Dissemination (CRD) quality assessment criteria for a systematic review. In addition, the ERG checked and provided commentary on the manufacturer’s model using standard checklists. A one-way sensitivity analysis, scenario analysis and a probabilistic sensitivity analysis (Figures 1 and 2) were undertaken by the ERG.
FIGURE 1.
CEACs from ERG probabilistic analysis using viral load model. a, CEAC for telbivudine compared with lamivudine (HBeAg-positive cohort – prior = 0); b, CEAC for telbivudine compared with lamivudine (HBeAg-negative cohort – prior = 0). CEAC, cost-effectiveness acceptability curve; ERG, evidence review group; HBeAg, hepatitis B e antigen; QALY, quality-adjusted life-year.
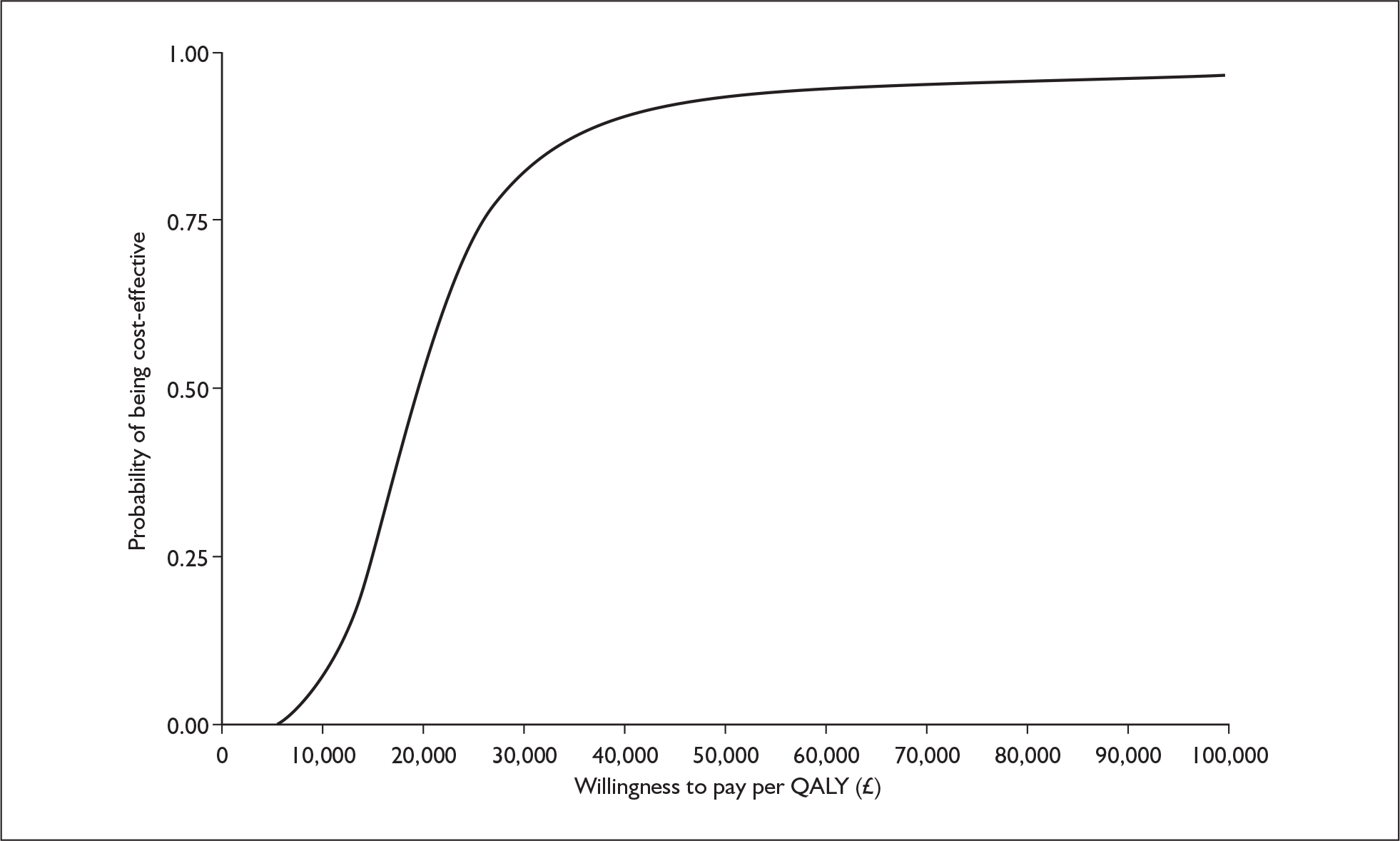
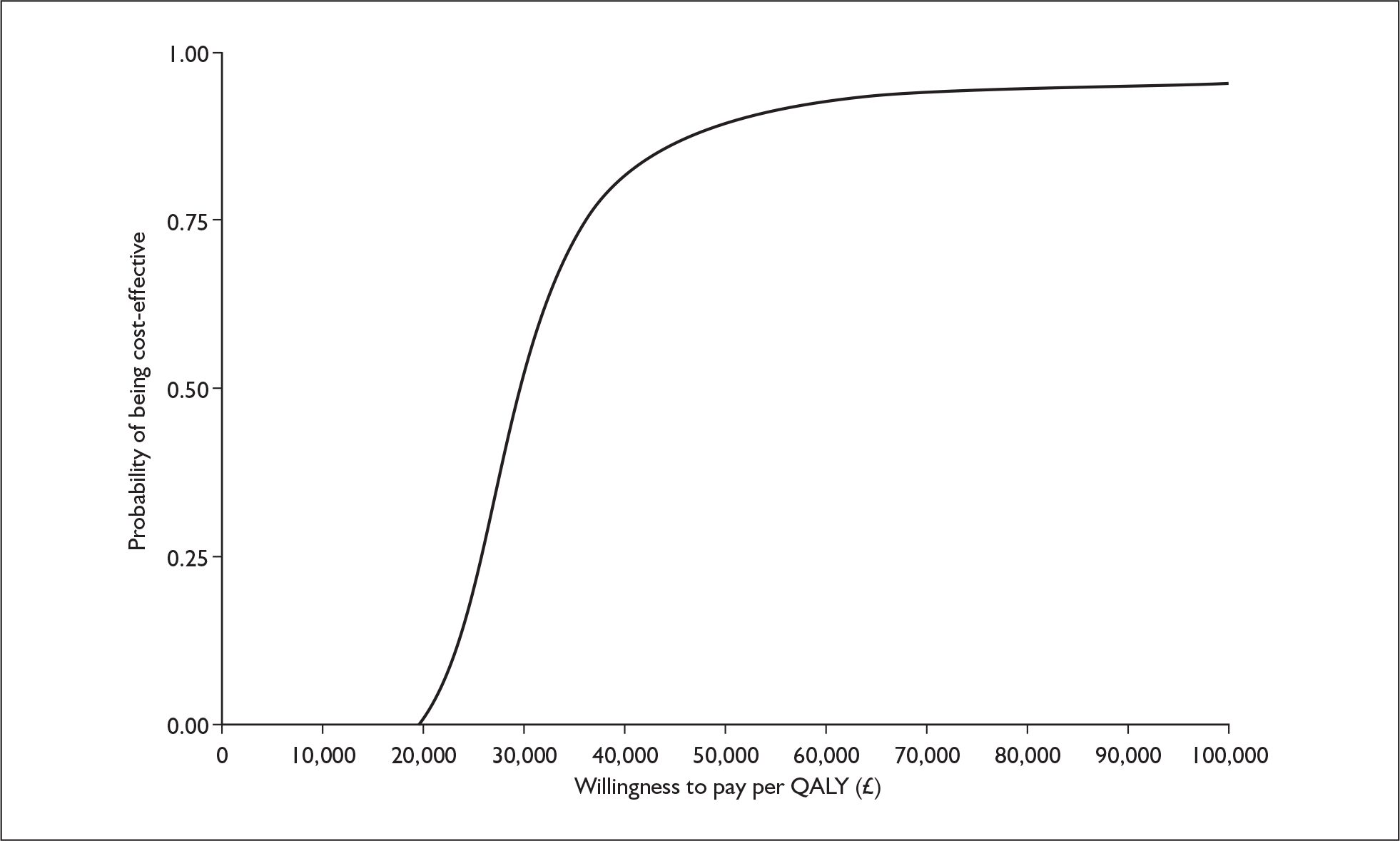
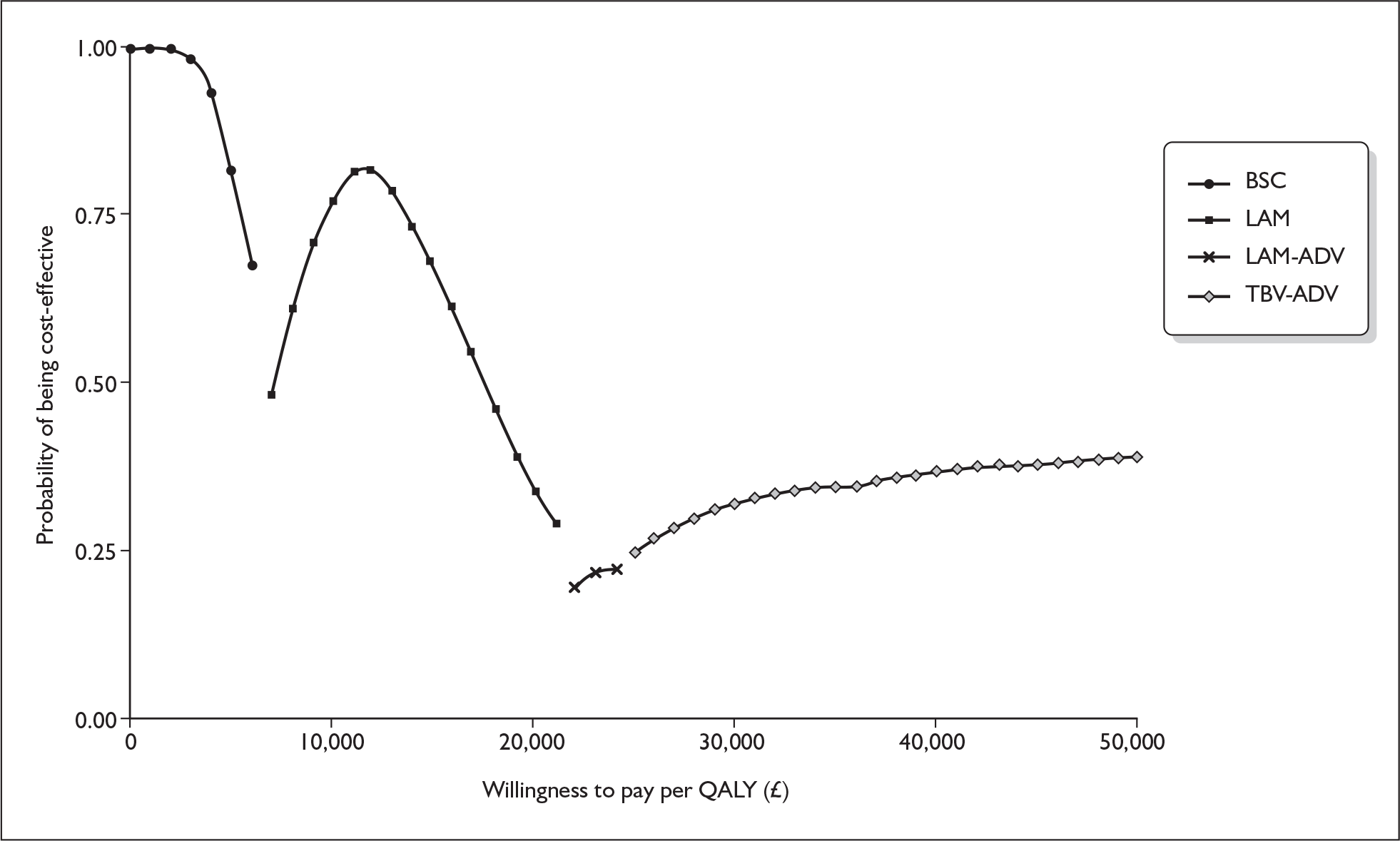
FIGURE 2.
Cost-effectiveness frontier from seroconversion model (ERG’s probabilistic sensitivity analysis). BSC, best supportive care; ERG, evidence review group; LAM, lamivudine; LAM→ADV; lamivudine followed by adefovir; QALY, quality-adjusted life-year;TBV→ADV, telbivudine followed by adefovir.
Results
Summary of submitted clinical evidence
The manufacturer’s submission presented clinical evidence for telbivudine in patients with compensated CHB based on one multicentre, international, double-blind randomised controlled trial (RCT) (the GLOBE trial). 5 This was the pivotal registration trial for telbivudine. The trial compared telbivudine with lamivudine in patients with HBeAg-positive and HBeAg-negative CHB for 104 weeks. The 2-year data presented throughout the manufacturer’s submission are unpublished, although publications of earlier results from the GLOBE trial are available.
For the primary outcome of therapeutic response (suppression of HBV DNA < 5 log copies/ml plus either clearance of detectable HBeAg or ALT normalisation) telbivudine was statistically superior to lamivudine at weeks 52 and 104 for HBeAg-positive patients, and at week 104 for HBeAg-negative patients.
In terms of secondary outcomes there were statistically significant differences in favour of telbivudine for HBV DNA reduction, HBV DNA non-detectability, ALT normalisation (although not for HBeAg-negative patients), virological breakthrough and HBV resistance at 2 years. In HBeAg-positive patients there was no significant difference between treatment groups for HBeAg loss or seroconversion at any time point. There were no significant differences in histological response or change in fibrosis score at 1 year, with the exception of histological improvement in HBeAg-positive patients, which was greater in telbivudine patients than in lamivudine patients. In terms of adverse events there appeared to be no difference between treatments.
In the elevated ALT subset analysis of the HBeAg-positive subgroup, telbivudine was statistically superior to lamivudine for most outcomes. In the ethnicity subgroup analysis, telbivudine was significantly more favourable than lamivudine in Asian patients, but there were no statistically significant differences between treatments for HBeAg-positive Caucasian patients and few differences for HBeAg-negative Caucasian patients.
Two RCTs comparing entecavir with lamivudine were included in the indirect comparison, one in HBeAg-positive patients6 and one in HBeAg-negative patients. 7 In the indirect comparison of telbivudine and entecavir, the manufacturer’s submission reported that there were no statistically significant differences for any efficacy outcome.
Summary of submitted cost-effectiveness evidence
The manufacturer’s submission presented evidence on the cost-effectiveness of telbivudine using two economic models, referred to as the viral load and seroconversion models. Evidence on the efficacy of telbivudine and lamivudine, in terms of reducing viral load, the probability of normalising ALT and HBeAg seroconversion, was taken from the GLOBE trial for a subgroup of patients with ALT levels ≥ two times the upper limit of normal (ULN). The benefit of these outcomes is that they are associated with reduced probability of progression to advanced liver disease. Efficacy of adefovir was based on assumption.
The viral load model, the manufacturer’s preferred approach, stratified response to treatment and the development of resistance by five viral load levels and regarded reducing viral load as a key determinant of disease progression. This model is relevant both to patients with HBeAg-positive CHB and to those with HBeAg-negative CHB. The viral load model incorporated a multivariate risk model to derive transition probabilities for the development of progressive liver disease based on viral load levels, the probability of ALT normalisation and HBeAg serological status (for HBeAg-positive patients). Two versions of the viral load model were submitted. The first used the observed proportion of patients moving between states to estimate transition probabilities (referred to as ‘zero prior’). In the second model an arbitrary value of 0.5 was added to all numerators and denominators (referred to as ‘0.5 prior’).
The seroconversion model was an attempt to replicate the model used in a recent NICE assessment2 and was structured with HBeAg seroconversion as the key determinant of disease progression. By definition this model is relevant only to patients with HBeAg-positive CHB.
Both models adopted a lifetime horizon and extrapolated lifetime costs and quality-adjusted life-years (QALYs) for patients treated with telbivudine and each of the included comparators. Incremental cost-effectiveness ratios (ICERs) were estimated against different comparators (depending on the model used) in the manufacturer’s submission. The comparator in the viral load model was lamivudine, whereas in the seroconversion model there were multiple competing interventions (lamivudine, telbivudine and adefovir alone or in sequence as well as best supportive care). All ICERs in the seroconversion model were calculated relative to best supportive care.
The manufacturer’s submission concluded that telbivudine is a cost-effective option compared with lamivudine using evidence from the viral load model (mean incremental cost of £19,087, mean QALY gain of 1.30 with an ICER of £14,665 per QALY gained for HBeAg-positive patients and mean incremental cost of £49,003, mean QALY gain of 4.67 with an ICER of £10,497 per QALY gained for HBeAg-negative patients). In response to a request for clarification from the ERG the manufacturer noted that there were errors in the models originally submitted and therefore in the results reported in the submission. Resubmitted results gave less favourable ICERs, particularly for HBeAg-negative patients (mean incremental cost of £23,983, mean QALY gain of 1.56 with an ICER of £15,377 per QALY gained for HBeAg-positive patients and mean incremental cost of £41,910, mean QALY gain of 2.07 with an ICER of £20,256 per QALY gained for HBeAg-negative patients).
The manufacturer’s submission concluded that telbivudine is a cost-effective option – on its own or followed by adefovir – for patients who have developed resistance to first-line telbivudine treatment. The manufacturer’s submission reported ICERs for seven treatment strategies relative to best supportive care. This is not an ideal presentation of the results of competing treatment strategies. The ERG derived appropriate comparisons, based on the manufacturer’s results, using the cost-effectiveness frontier, estimating ICERs of £7887, £19,680 and £24,277 per QALY gained for lamivudine, telbivudine and telbivudine followed by adefovir respectively. The sequence of treatment options implied is problematic as the strategy of using telbivudine followed by adefovir (for patients who develop resistance to telbivudine) is not accessible to patients who have lamivudine as their first-line treatment. To provide the treatment strategy of telbivudine followed by adefovir (which yields the greatest QALY gain of all of the strategies in the seroconversion model and which is optimal at a willingness to pay greater than £25,000 per QALY) telbivudine must be available as a first-line treatment.
Commentary on the robustness of submitted evidence
Strengths
The manufacturer conducted a systematic search for clinical effectiveness studies of telbivudine. It appears unlikely that any additional trials would have met the inclusion criteria had the search been widened to include other databases.
The GLOBE trial appears to be of reasonable methodological quality (with some limitations) and measured a range of outcomes that are as appropriate and clinically relevant as possible, although health-related quality of life was not reported. On the whole, the manufacturer’s submission appears to represent an unbiased estimate of the antiviral treatment effect of telbivudine based on the results of one trial.
The methods adopted for the economic evaluation of telbivudine were broadly consistent with those adopted for previous evaluations of antiviral treatment of CHB, including the recent NICE assessment of adefovir and pegylated interferon. 2
Weaknesses
The manufacturer’s submission did not include all of the comparators specified in the scope.
Despite a systematic search and screen of the literature, only one RCT was included. The manufacturer’s submission is therefore largely dependent upon this one trial. Further high-quality RCT evidence for the effectiveness of telbivudine in the patient group meeting the licensed indication would be beneficial.
Literature searches were poorly documented, lacking clarity and transparency throughout. Search filters were extremely precise at the expense of sensitivity. The processes undertaken by the manufacturer for data extraction and applying quality criteria to the GLOBE trial were not detailed and no formal quality assessment was undertaken on the comparator trials. These factors limit the robustness of the systematic review. In addition, one RCT8 that appeared to meet the inclusion criteria was excluded from the submission.
The indirect comparison with entecavir was poorly conducted and should be treated with caution. It was reported as a visual comparison and then as a statistical comparison, which the manufacturer deemed invalid. An inadequate description of the methodology was provided and the conclusions are based largely on a visual comparison of efficacy outcomes.
The economic models used data from a subgroup of patients in the GLOBE study and these data were not presented in detail in the clinical evidence section of the manufacturer’s submission. No information was given on the baseline characteristics of the subgroup of patients with ALT levels ≥ two times the ULN.
In the cost-effectiveness section of the submission the manufacturer paid insufficient attention to appraising the data used to populate the economic models. Denominators used for calculation of some transition probabilities appear inconsistent and some input values (e.g. resistance rates calculated using data reported in appendices) are substantially lower than those reported for all patients in the GLOBE study. These discrepancies were not discussed in the submission.
The electronic models submitted are complex and highly reliant on Visual Basic programming to produce any analyses. There is a large amount of reprocessing of data within the models that was not clearly documented or readily apparent to the user.
There was little discussion in the manufacturer’s submission of uncertainty around the mean estimates reported as the base case for both the viral load and seroconversion models. The NICE guide to methods of technology appraisal describes confidence ellipses and scatter plots on the cost-effectiveness plane and cost-effectiveness acceptability curves as the most appropriate ways of presenting uncertainty in probabilistic sensitivity analysis. These were not presented for all comparisons and were submitted in appendices, without commentary, rather than in the main body of the report.
Conclusions
Areas of uncertainty
The results of the key efficacy outcomes were broken down by HBeAg status, study treatment and (1) race/ethnicity or (2) ALT levels. It is not clear whether the GLOBE study was powered to detect differences in these subgroups. Without confidence intervals and standard deviations in the reporting of the results it is not possible to ascertain how much variance there was among the subgroups/patients.
The rates of viral resistance to entecavir were not reported in the manufacturer’s submission and therefore do not allow for a comparison with the resistance rates for telbivudine.
The adjustments to the Cox proportional hazards models used to estimate the probability of developing compensated cirrhosis and HCC were inadequately reported as was the process of recalibration. These values enter the viral model deterministically – there is no assessment of parameter uncertainty for the risk models used in the viral model, nor of the methodological uncertainty around the adjustment or recalibration.
The lack of quality assurance of input data for both models introduces uncertainty – the impact of the prior value (zero or 0.5) on the model outcomes suggests that sparsity of data may be a problem, particularly for the model of HBeAg-negative patients. This is not surprising, given that data on around 250 patients were stratified across viral load levels, ALT and serological status. The manufacturer’s submission contained no discussion of alternative modelling strategies that might reduce the impact of sparsity of data nor did it clearly indicate which input variables were most affected by differences in prior values.
Key issues
Although telbivudine was statistically superior to lamivudine for most antiviral outcomes, the difference was not clinically significant, having an effectiveness advantage of only about 2% in patients treated between the two drugs. Viral breakthrough (> 1 log increase over nadir) for telbivudine was 28.6% at 2 years; although this is significantly lower than that for lamivudine (45.5%), the ERG’s clinical advisor asserts that it is still high in clinical terms.
The conclusions from the indirect comparison were based largely on a visual comparison of efficacy outcomes and a statistical indirect comparison, which the manufacturer’s submission states was not considered valid in the absence of any meta-analyses. Telbivudine seems to have approximately the same efficacy as entecavir for viral suppression, but markedly higher rates of viral resistance (as per the rates for entecavir reported in the published trials).
The exclusion of entecavir from all of the economic models and the restricted comparison included in the viral load model – telbivudine versus lamivudine, with no follow-up antiviral treatments – means that the cost-effectiveness evidence for telbivudine presented in the manufacturer’s submission is limited. Lack of critical assessment and assurance of the quality of the data used to populate the model (apparent inconsistencies and incomplete data for lamivudine and telbivudine from the GLOBE trial along with the absence of systematic searches for evidence on the comparative effectiveness of adefovir) further limits the evidence reported in the manufacturer’s submission.
Summary of NICE guidance issued as a result of the STA
NICE guidance, published August 2008,9 states that:
1.1 Telbivudine is not recommended for the treatment of chronic hepatitis B.
1.2 People currently receiving telbivudine should have the option to continue therapy until they and their clinicians consider it appropriate to stop.
Disclaimers
The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Department of Health.
Key references
- National Institute for Health and Clinical Excellence . Guide to the Single Technology (STA) Process 2006. www.nice.org.uk/page.aspx?o=STAprocessguide.
- National Institute for Health and Clinical Excellence . Adefovir Dipivoxil and Peginterferon Alfa-2a for the Treatment of Chronic Hepatitis B 2006.
- British Liver Trust . How Prevalent Is HBV? n.d. www.britishlivertrust.org.uk/home/healthprofessionals/literature-for-professionals/aprofessionals-guide-to-hepatitis-b/how-prevalent-ishbv.aspx (accessed 17 December 2007).
- The Hepatitis B Foundation . Rising Curve: Chronic Hepatitis B Infection in the UK n.d. www.hepb.org.uk/news/archive/spring_2008_03_03/rising_curve_chronic_hepatitis_b_infection_in_the_uk (accessed 29 January 2008).
- GLOBE study NV-02B-007. Week 104 Clinical Study Report 2007.
- Chang TT, Gish RG, de Man R, Gadano A, Sollano J, Chao YC, et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med 2006;354:1001-10.
- Lai CL, Shouval D, Lok AS, Chang TT, Cheinquer H, Goodman Z, et al. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N Engl J Med 2006;354:1011-20.
- Hou J, Yin Y-K, Xu D, Tan D, Niu J, Zhou X, et al. Telbivudine versus lamivudine in Chinese patients with chronic hepatitis B: results at 1 year of a randomized, double-blind trial. Hepatology 2007;47:447-54.
- National Institute for Heath and Clinical Excellence . Telbivudine for the Treatment of Chronic Hepatitis B 2008.