
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 08/42/01. The protocol was agreed in July 2009. The assessment report began editorial review in November 2009 and was accepted for publication in June 2010. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
MS has a financial interest in a consulting company that has undertaken work for Abbott, Schering-Plough and Wyeth, but not relating to psoriatic arthritis, and he has not personally participated in this consultancy work. He has personally undertaken paid consultancy for some of the comparator manufacturers, again not relating to psoriatic arthritis. IB has received speaker fees and attended Advisory Board Meetings for Abbott, Schering-Plough and Wyeth.
Permissions
Copyright statement
© 2011 Queen’s Printer and Controller of HMSO. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2011 Queen’s Printer and Controller of HMSO
Chapter 1 Background
Description of health problem
Epidemiology
Psoriatic arthritis (PsA) is defined as a unique inflammatory arthritis affecting the joints and connective tissue, and is associated with psoriasis of the skin or nails. 1 There are difficulties in estimating its prevalence due to the lack of a precise definition and diagnostic criteria for PsA. 2 The prevalence of psoriasis in the general population has been estimated at between 2% and 3%,1 and the prevalence of inflammatory arthritis in patients with psoriasis has been estimated to be up to 30%. 3 PsA affects males and females equally, with a worldwide distribution. Figures for the UK have estimated the adjusted prevalence of PsA in the primary care setting to be 0.3%, based on data from north-east England involving six general practices, covering a population of 26,348. 4 Another study reported PsA prevalence rates per 100,000 of 3.5 for males and 3.4 for females, based on data from 77 general practices in the Norwich Health Authority, with a population of 413,421. 5 Severe PsA with progressive joint lesions can be found in at least 20% of patients with psoriasis. 6
Aetiology, pathology and prognosis
Psoriatic arthritis is a hyperproliferative and inflammatory arthritis that is distinct from rheumatoid arthritis (RA). 7,8 The aetiology of PsA is not fully known; genetic susceptibility and exogenous influences might play roles in the cause of disease. 9 The expression of major histocompatibility complex antigens [e.g. human leucocyte antigen (HLA)-B27] might also predispose certain patients to develop PsA, as well as a number of environmental factors, such as trauma, repetitive motion, human immunodeficiency virus infection, and bacterial infection. 9 PsA is diagnosed when a patient with psoriasis has a distinctive pattern of peripheral and/or spinal arthropathy. 10 The rheumatic characteristics of PsA include stiffness, pain, swelling, and tenderness of the joints and surrounding ligaments and tendons. 11
Several clinical features distinguish PsA from RA. In PsA, the absolute number of affected joints is less and the pattern of joint lesion involvement tends to be asymmetric. 12 The joint distribution tends to occur in a ray pattern in PsA, with the common involvement of distal interphalangeal (DIP) joint and nail lesions. All joints of a single digit are thus more likely to be affected in PsA, whereas in RA the same joints on both sides tend to be affected. 1 Dactylitis, spondylitis and sacroiliitis are common in PsA, whereas they are not in RA. 12 In PsA the affected joints are tighter, contain less fluid, and are less tender than those in RA, with a propensity for inflammation of the entheseal sites. PsA and RA also show differences in the inflammatory reaction that accompanies each form of arthritis. 12 Extra-articular manifestations of PsA are also different from those of RA; rheumatoid nodules are particularly absent in patients with PsA. 1 Most patients with PsA develop psoriasis first, while joint involvement appears first only in 19% of patients, and concurrently with psoriasis in 16% of cases. 10 For those who develop psoriasis first, the onset time of PsA is around 10 years after the first signs of psoriasis. 1 In addition, rheumatoid factor (RF) (an antibody produced by plasma cells) may be detected in about 13% of patients with PsA, whereas it can be detected in more than 80% of patients with RA. 1
Psoriatic arthritis is a progressive disorder, ranging from mild synovitis to severe progressive erosive arthropathy. 11,13 Research has found that patients PsA presenting with oligoarticular disease progress to polyarticular disease; a large percentage of patients develop joint lesions and deformities, which progress over time. 9 Despite clinical improvement with current disease-modifying antirheumatic drug (DMARD) treatment, radiological joint damage has been shown in up to 47% of patients with PsA at a median interval of 2 years. 14 Untreated patients with PsA may have persistent inflammation and progressive joint damage. 11 The deformities resulting from PsA can lead to shortening of digits due to severe joints or bone lysis. 1 Remission can occur in PsA, especially in patients with Health Assessment Questionnaire (HAQ) levels of < 1 score. 15 Of those who can sustain clinical remission, only a small fraction of patients can discontinue medication with no evidence of damage. 16 Research has reported that the frequency of remission was 17.6% in patients with PsA, and the average duration of remission was 2.6 years, from data of 391 patients with peripheral arthritis. 16 Joint damage can occur early in the disease often prior to functional limitation. 9,17 This appears to be associated with the development of inflamed entheses close to peripheral joints, although the link still remains largely unclear. 13 It has been shown that there is an association between polyarthritis and functional disability, with higher mean HAQ scores than those in oligoarthritic patients. 18,19
A number of risk factors have been found to be predictive of the progression of PsA. A polyarticular onset (five or more swollen joints) of PsA is an important risk factor in predicting the progressive joint deformity. 20 Each actively inflamed joint in PsA is associated with a 4% risk of increased damage within 6 months. 1 HLA antigens have also been found to be predictive of the progression of joint damage. It has been shown that HLA-B27, HLA-B39 and HLA-DQW3 were associated with disease progression. 21 Other risk factors for a more progressive course of PsA include the presence of an elevated erythrocyte sedimentation rate (ESR) and being female. 1,22
A classification scheme for PsA on the basis of joint manifestations describes five patterns of disease:9,23
-
Distal interphalangeal arthritis This condition is considered as the classic form of PsA. It can occur as the sole presentation or in combination with other symptoms. It can be symmetrical or asymmetrical and can involve a few or many joints. Adjacent nails may demonstrate psoriatic changes and progressive joint erosions are common.
-
Arthritis mutilans It is a severe presentation of the disease with osteolysis of the phalanges, metatarsals and metacarpals.
-
Symmetric polyarthritis The clinical feature of symmetric polyarthritis is similar to RA, with inflammation of the metacarpals and the proximal interphalangeal joints being prominent. However, it is usually milder than RA and patients are often RF-negative.
-
Oligoarthritis This is the most common condition of PsA, which is characterised by asymmetric involvement of a small number of joints (fewer than four). Arthritis in a single knee might be the first symptom of oligoarthritis.
-
Spondylitis and/or sacroiliitis It resembles ankylosing spondylitis, but is generally less severe and less disabling. The axial skeleton tends to be involved in an atypical fashion, with the lumbar spine as the most common site of involvement.
Despite this classification, these patterns of PsA often overlap and evolve from one pattern to another as the disease progresses and diagnostic investigations become more thorough. 13 A common feature of PsA is dactylitis (or ‘sausage digit’) in which the whole digit appears swollen due to inflammation of the tendons and periosteum as well as the joints. 9,11 Radiographic features of PsA involve the distinctive asymmetric pattern of joint involvement, sacroiliitis and spondylitis, bone erosions, new bone formation, bony ankylosis, bony outgrowths in the axial skeleton, osteolysis and enthesopathy.
Significance in terms of ill health
The health burden of PsA can be considerable. PsA is a lifelong disorder and its impact on patients’ functional status and quality of life (QoL) fluctuates over time. 24 As it involves both skin and joints, PsA can result in significant impairment of QoL and psychosocial disability7,10 compared with a healthy population. Patients with PsA score significantly worse in health-related quality of life (HRQoL) assessment on physical mobility, pain, energy, sleep, social isolation and emotional reaction. 25 A comparison of HRQoL between patients with PsA and patients with RA found that both patient populations had lower physical health than healthy control patients. 26 Patients with PsA reported more role limitations due to emotional problems and more bodily pain after the adjustment of the difference in vitality and other covariates. These findings were also reflected in another comparison of disability and QoL between patients with RA and patients with PsA; this study reported that despite greater peripheral joint damage in patients with RA the function and QoL scores were similar for both groups. 27,28 These reveal that there might be unique psychological disabilities associated with the psoriasis dimension (i.e. skin lesion) of PsA. Due to the skin involvement, patients with PsA may also suffer from other psychological consequences, such as embarrassment, self-consciousness and even depression. Because of a significant reduction in a patient’s HRQoL, ideally PsA should be diagnosed early and treated aggressively in order to minimise joint damage and skin disease. 17
The severity of PsA is also reflected in increased mortality. Patients with PsA have a 60% higher risk of mortality relative to the general population. 24,29,30 The causes of premature death are similar to those noted in the general population, with cardiovascular causes being the most common. 1 The estimated reduction in life expectancy for patients with PsA is approximately 3 years. 31
The economic costs of PsA have not been well quantified. In the USA, the mean annual direct (health and social care) cost per patient with PsA is estimated as US$3638 according to data from Medstat MarketScan in 1999–2000. 32 In Germany, the mean annual direct cost per patient with PsA is estimated as €3162, with a mean indirect cost (time lost from work and normal activities) per patient of €11,075. 33 Studies of RA34–36 and psoriasis37 have shown that costs increase with the severity of both diseases, and productivity losses are significant,38,39 largely as a consequence of extensive work disability. 35 These findings are likely to be generalisable to PsA.
Studies of the economic impact of RA in the UK before the introduction of biologic therapies found that direct health-care costs represented about one-quarter of all costs, and these were dominated by inpatient and community day care,40 with DMARDs representing a minor proportion: 3%–4% of total costs and 13%–15% of direct costs. 41 Evidence from the USA suggests that expenditure on biologic therapies might represent 35% of direct cost,42 but similar data are not yet available for the UK. Increasing expenditure on biologic therapies might be at least partly offset by cost savings elsewhere,43 although, as yet, the evidence for this is only suggestive.
Assessment of treatment response in psoriatic arthritis
The assessment of effectiveness of treatments for PsA relies on there being outcome measures that accurately and sensitively measure disease activity. Overall response criteria have not yet been clearly defined; they are being developed by an international collaboration on outcome measures in rheumatology (OMERACT – Outcome Measures in Rheumatoid Arthritis Clinical Trials). There are a number of different parameters of disease activity in arthropathies, including: number of swollen joints, number of tender joints, pain, level of disability, patient’s global assessment, physician’s global assessment and biochemical markers in the blood. Selecting which to assess in clinical trials and which to appoint as the primary variable can be difficult. Different ways of combining the various outcome measures have been suggested including a simple ‘pooled index’. 44 In recent years the compound response criterion, the American College of Rheumatology 20% improvement criteria (ACR 20), has gained general acceptance for the assessment of treatments for PsA, and this has been adopted for many PsA trials. Another compound measure, Psoriatic Arthritis Response Criteria (PsARC), was developed specifically for a trial in PsA and has been adopted by the BSR. 45
American College of Rheumatology response criteria
The ACR response criteria were developed after the identification of a set of core disease activity measures. ACR 20 requires a 20% reduction in the tender joint count (TJC), a 20% reduction in the swollen joint count (SJC), and a 20% reduction in three out of five additional measures, including patient and physician global assessment, pain, disability and an acute-phase reactant. In patients with RA, ACR 20 has been confirmed as being able to discriminate between a clinically significant improvement and a clinically insignificant one. 46,47 It is unclear whether the ACR 20 has the same discriminatory validity in PsA. 48 The ACR 20 is generally accepted to be the minimal clinically important difference that indicates some response to a particular intervention. The ACR 50 reflects significant and important changes in the patient’s disease status that may be acceptable to both clinician and patient in long-term management. The ACR 70 represents a major change and approximates in most minds to a near remission. Because of the differences between PsA and RA, it is imperative that, when the ACR response criteria are used in the trials of treatment for PsA, the DIP joints are included. Rather than changes from bad to moderate synovitis in any individual joint, these criteria detect improvement from swollen to not swollen or from tender to not tender joints. Therefore, patients with oligoarthritis in a few large joints may not appear to respond as well on this outcome as patients with polyarthritis involving many smaller joints.
Psoriatic Arthritis Response Criteria
The Psoriatic Arthritis Response Criteria were developed for a trial of sulfasalazine in PsA,49 and incorporate four assessment measures (patient self-assessment, physician assessment, joint pain/tenderness score and joint swelling score). Treatment response was defined as an improvement in at least two of these four measures, one of which had to be joint pain/tenderness score or joint swelling score, with no worsening in any of these four measures. PsARC has not been validated, but responses assessed by it do parallel those identified with ACR 20. A limitation of PsARC is that although it was developed for assessment of PsA, it does not incorporate an assessment of psoriasis. The working group producing the British Society for Rheumatology (BSR) guidelines for the use of biologics in PsA50 elected to use PsARC as the primary joint response to biologic treatment, although it advocates some extra data collection, such as a patient self-assessed disability (HAQ), and a biochemical marker of disease activity, such as ESR or C-reactive protein (CRP).
Radiological assessments
In all arthropathies progression of the disease can only be truly measured by assessment of the joint damage. The radiological assessments include the Steinbrocker, Sharp and Larsen methods. A modification of the Steinbrocker method, which assigns a score for each joint has been validated for PsA. The Sharp method grades all the joints of the hand separately for erosions and joint space narrowing, each erosion being assigned a score of 0–5 and each joint space narrowing a score of 0–4. A total score (maximum 149) is calculated. The Total Sharp Score (TSS), modified to include the DIP and metatarsophalangeal joints of the feet and interphalangeal joint of the first toe, has been used in the trials of etanercept and adalimumab. 51,52 None of these methods that were developed for RA score additional radiographic changes that are specific to PsA. A new score has been tested by Wassenberg et al. ,53 but this scoring method has not yet been validated in clinical trials. Whichever method is selected it is important that trials should be stratified by baseline radiographic findings.
Health Assessment Questionnaire
The HAQ score is a well-validated tool in the assessment of patients with RA. 48 It focuses on two dimensions of health status: physical disability (eight scales) and pain, generating a score of 0 (least disability) to 3 (most severe disability). A modification of the HAQ for spondylarthropathies (HAQ-S) and for psoriasis (HAQ-SK) have been developed, but when tested against HAQ their scores were almost identical,54 suggesting either can be used in PsA. 48 The HAQ is one component of the ACR 20 (50 or 70) response criteria.
The HAQ has been tested in patients with PsA, showing a moderate-to-close correlation with disease activity as measured by the actively inflamed joint count and some measures of clinical function (including the ACR functional class). 55 Although the HAQ has been used as a disability measure and is a common outcome measure in PsA trials, it may not sufficiently incorporate all aspects of disease activity (i.e. deformity or damage resulting from disease process, especially in late PsA), therefore, clinical assessment of disease activity and both clinical and radiological assessments of joint damage remain important outcome measures in PsA. 56
Overall, the advantage of the HAQ as an instrument is that it can measure the functional and psychological impact of the disease. HAQ is conventionally used as a driver of QoL scores and costs in main economic evaluations on the use of biologics and DMARDs in RA. 57–59
Psoriasis Area and Severity Index
When evaluating the efficacy of interventions in the treatment of PsA, the outcome measures used must assess disease activity in both the joint and the skin. 48 In clinical trials of patients with psoriasis, assessment of the response to treatment is usually based on the Psoriasis Area and Severity Index (PASI). The PASI is also used in trials of PsA; given the various degrees of severity of psoriasis in these patients, not all patients are evaluable for the assessment of response – at least 3% of the body surface area (BSA) has to be affected by the skin disease in order for the PASI measure to be used. 48 Although it is widely used, the PASI measure also has a number of deficiencies: its constituent parameters have never been properly defined; it is insensitive to change in mild-to-moderate psoriasis; estimation of disease extent is notoriously inaccurate; and the complexity of the formula required to calculate the final score further increases the risk of errors. It combines an extent and a severity score for each of the four body areas (head, trunk, upper extremities and lower extremities). The extent score of 0–6 is allocated according to the percentage of skin involvement (e.g. 0 and 6 represent no psoriasis and 90%–100% involvement, respectively). The severity score of 0–12 is derived by adding scores of 0–4 for each of the qualities of erythema (redness), induration and desquamation representative of the psoriasis within the affected area. It is probable, but usually not specified in trial reports, that most investigators take induration to mean plaque thickness without adherent scale and desquamation to mean thickness of scale rather than severity of scale shedding. The severity score for each area is multiplied by the extent score and the resultant body area scores, weighted according to the percentage of total BSA that the body area represents (10% for head, 30% for trunk, 20% for upper extremities and 40% for lower extremities), are added together to give the PASI score. Although PASI can theoretically reach 72, scores in the upper half of the range (> 36) are not common even in severe psoriasis. Furthermore, it fails to capture the disability that commonly arises from involvement of functionally or psychosocially important areas (hands, feet, face, scalp and genitalia), which together represent only a small proportion of total BSA.
Although the optimum assessment outcomes for PsA trials are yet to be defined, those selected as the primary measures of efficacy in this review, namely PsARC-, ACR 20/50/70-, HAQ- and PASI-based measures, all have discriminatory capability and are generally accepted for the assessment of treatment effect. HAQ has been chosen as our primary outcome variable of arthritis in the economic evaluation because it makes it technically feasible to evaluate the impact of retarding and/or halting the progression of the disease, both in an economic sense and in terms of QoL. PASI has been chosen as the primary outcome variable of psoriasis in the economic evaluation because it is recommended to assess severity and response in the British Association of Dermatologists (BAD) guidelines and used in the majority of randomised controlled trials (RCTs).
Current service provision
The effective treatment for PsA needs to consider both skin and joint conditions, especially if both are significantly affected. In current services it is rheumatologists who manage the majority of patients with PsA. Although dermatologists focus principally on the cutaneous expression of psoriasis, they frequently use drugs, such as methotrexate (MTX) or biological agents, which may benefit both skin and joints. Patients with severe manifestations of PsA in joints and skin will tend to be managed jointly by rheumatologists and dermatologists, whereas many patients with less severe joint disease may remain under the care of dermatologists alone.
Most treatments for PsA have been borrowed from those used for RA and non-steroidal anti-inflammatory drugs (NSAIDs) are widely used. 10 There is a concern that NSAIDs may provoke a flare of the psoriasis component of the disease, but this may not be of clinical significance. 13 Local corticosteroid injections are also frequently used,10 although there is a significant risk of a serious flare in psoriasis when corticosteroids are withdrawn. Disease that is unresponsive to NSAIDs, and in particular polyarticular disease, should be treated with DMARDs in order to reduce the joint damage and prevent disability. 13 It is also suggested that aggressive treatment of early-stage progressive PsA should be used in order to improve prognosis. 13 Again, the treatments used are based on the experience in RA rather than knowledge of the pathophysiology of PsA or trial-based efficacy. Currently, MTX and sulfasalazine are considered the DMARDs of choice, despite the largely empirical evidence for MTX and the modest effects of sulfasalazine. 13 A review of the experience of 100 patients with PsA treated with DMARDs60 reported that of those treated with sulfasalazine, gold, MTX or hydroxychloroquine over 70% of patients had discontinued due to a lack of efficacy or adverse events (range 35% with MTX to 94% with hydroxychloroquine).
Another DMARD (leflunomide) has, in addition to being licensed for RA, also been licensed for use in PsA. This is the only non-biologic licensed in PsA. Leflunomide inhibits de novo pyrimidine synthesis and because activated lymphocytes require a large pyrimidine pool, it preferentially inhibits T-cell activation and proliferation. Clinical trials have demonstrated the efficacy in RA61 and PsA. 62 Evidence also suggests that clinical responses in patients with RA receiving leflunomide treatment are equivalent to those receiving MTX treatment. 63 Unlike MTX, however, leflunomide has little effect on the skin. Other drugs investigated for the treatment of PsA are auranofin, etretinate, fumaric acid, intramuscular gold, azathioprine, and Efamol Marine. 54 Ciclosporin and penicillamine are also sometimes used in clinical practice. 64
Costs of current service
Based on prices from the British National Formulary (BNF),65 weekly treatment costs with the most commonly used DMARDs in PsA, sulfasalazine and MTX are approximately £2 and < £0.50, respectively. The cost of ciclosporin is approximately £40–80 per week.
Prescriptions for DMARDs for all indications have been rising rapidly in general practice in England from 300,000 per quarter year in December 2003 to over 500,000 in December 2008, with expenditure increasing from £2M per quarter year to nearly £4.5M during this period. In addition to the cost of DMARDs, the cost of NSAIDs was almost £4M per quarter year in December 2008, although the number of prescriptions and expenditure on NSAIDS has fallen sharply in recent years. 66
Variation in service
No surveys of UK service models for PsA have been conducted. Although PsA is a disease of joints and skin it is treated mainly by rheumatologists. A study of patients with confirmed PsA in the Netherlands found considerable variations among rheumatologists in the delivery of care; 29% failed to diagnose PsA, mainly due to their failure to enquire about skin lesions. 67 Of those who did correctly diagnose PsA, only 43% referred patients to a dermatologist and 66% ordered laboratory tests. The median costs for imaging and laboratory investigations were higher for those patients who were correctly diagnosed with PsA than for the remaining patients who were incorrectly diagnosed.
Description of technology under assessment
Numerous chemokines and cytokines are believed to play an important role in triggering cell proliferation and sustaining joint inflammation in PsA. Cytokines stimulate inflammatory processes, resulting in the migration and activation of T cells, which then release tumour necrosis factor-alpha (TNF-α). TNF-α is one of several proinflammatory cytokines that have been implicated in the pathogenesis of both psoriasis and PsA. 68,69 Newer strategies for the treatment of PsA focus on modifying T cells in this disease through direct elimination of activated T cells, inhibition of T-cell activation, or inhibition of cytokine secretion or activity. 70 Etanercept, infliximab and adalimumab are among a number of these new biological agents that have been developed and investigated for the treatment of various diseases, including psoriasis and PsA. Etanercept is a human dimeric fusion protein that binds specifically to TNF and blocks its interaction with cell surface receptors. 10 Infliximab is a murine/human chimeric anti-TNF monoclonal gamma immunoglobulin that inhibits the binding of TNF to its receptor. 10 Adalimumab is a fully humanised monoclonal IgG1 antibody and TNF antagonist. 71 All three biologics are licensed in the UK for the treatment of active and progressive PsA in adults when the response to previous DMARD therapy has been inadequate.
Anticipated costs of biologic interventions
Based on the recommended dose regimen (25-mg injections administered twice weekly as a subcutaneous injection), the initial 3-month acquisition cost of etanercept is £2324, and the annual cost thereafter is £9296. The acquisition costs of adalimumab are the same, based on the recommended dose regimen (40-mg subcutaneous injections administered every other week). The recommended dose for infliximab is 5 mg/kg is given as an intravenous (i.v.) infusion over a 2-hour period followed by additional 5-mg/kg infusion doses at 2 and 6 weeks after the first infusion then every 8 weeks thereafter, each dose corresponding to three, four or five vials of infliximab depending upon the patient’s body weight. The initial 3-month acquisition cost of infliximab is estimated to be £5035, assuming four vials, and the annual cost thereafter is £10,912.
Current expenditure on biologic therapies in England is considerable. For all indications, the cost of prescribing in 2008 was £152.2M for etanercept, £102.7M for adalimumab and £77.1M for infliximab, with > 95% of these prescriptions dispensed by hospitals. 72 Expenditure for biologic drugs increased during 2008 by 15% for etanercept, 55% for adalimumab and 25% for infliximab. Among the drugs appraised by the National Institute for Health and Clinical Excellence (NICE), etanercept and adalimumab are now ranked in the top five by estimated cost of prescribing in England.
This report contains reference to confidential information provided as part of the NICE appraisal process. This information has been removed from the report and the results, discussions and conclusions of the report do not include the confidential information. These sections are clearly marked in the report.
Chapter 2 Definition of decision problem
Decision problem
The use of biologics in inflammatory disease is a rapidly evolving area. Etanercept and infliximab were previously evaluated together for their efficacy and safety in PsA in 200673 and adalimumab was separately evaluated more recently. 74 There is a need for an up-to-date evaluation of all three biological agents that are licensed for use in the treatment of PsA.
It is important to establish how well these three licensed biologics work in patients with PsA, in terms of both joint and skin response, as well as disease progression. In addition to determining the absolute efficacy of the biologics relative to placebo, it is important to determine their relative clinical effectiveness and cost-effectiveness.
Overall aims and objectives of assessment
To determine the clinical effectiveness, safety and cost-effectiveness of etanercept, infliximab and adalimumab for the treatment of active and progressive PsA in patients who have an inadequate response to standard treatment (including DMARD therapy).
Chapter 3 Assessment of clinical effectiveness
Methods for reviewing clinical effectiveness
A systematic review of the evidence for the clinical effectiveness and safety of etanercept, infliximab and adalimumab for the treatment of active and progressive PsA in patients who have an inadequate response to standard treatment (including DMARD therapy) was conducted following the general principles recommended in the guidance of the Centre for Reviews and Dissemination (CRD) guidance75 and the quality of reporting of meta-analyses (QUOROM) statement. 76
Search strategy
The following databases were searched for relevant clinical effectiveness and cost-effectiveness research:
-
MEDLINE
-
EMBASE
-
Cochrane Central Register of Controlled Trials (CENTRAL)
-
Science Citation Index (SCI)
-
Conference Proceedings Citation Index – Science (CPCI-S)
-
ClinicalTrials.gov
-
metaRegister of Current Controlled Trials (mRCT)
-
NHS Economic Evaluation Database (NHS EED)
-
Health Economic Evaluations Database (HEED)
-
EconLit.
Searches of major bibliographic databases were undertaken in three tranches – for RCTs, for economic evaluations and for studies of serious adverse effects. In the RCT and economic evaluation searches, the etanercept and infliximab search was limited by date (1 April 2004 to date), updating the searches undertaken for the 2006 Health Technology Assessment (HTA) report. 73 The search for adalimumab had no date limits. The searches for studies of adverse effects of all three drugs were not date limited. Internet resources were also searched for information on adverse effects. At the time of receiving the company submission (August 2009), update searches were conducted to ensure that the review remained up to date and covered all relevant evidence at the time of submission. No language or other restrictions were applied. In addition, reference lists of all included studies and industry submissions made to NICE were hand-searched to identify further relevant studies.
The terms for search strategies were identified through discussion between an information specialist and the research team, by scanning the background literature and browsing the MEDLINE medical subject headings (MeSH). As several databases were searched, some degree of duplication resulted. To manage this issue, the titles and abstracts of bibliographic records were imported into endnote bibliographic management software to remove duplicate records.
Inclusion and exclusion criteria
Two reviewers independently screened all titles and abstracts. Full paper manuscripts of any titles/abstracts that may be relevant were obtained where possible and the relevance of each study was assessed by two reviewers according to the criteria below. Studies were included in the review according to the inclusion criteria, described as follows. Studies that did not meet all of the criteria were excluded and their bibliographic details listed with reasons for exclusion. Any discrepancies were resolved by consensus or consulting a third reviewer if necessary.
Study design
Randomised controlled trials (including any open-label extensions of these RCTs) were included in the evaluation of efficacy. Information on the rate of serious adverse events was sought from regulatory sources [the US Food and Drug Administration (FDA), European Medicines Agency (EMEA)]. If these failed to report the necessary data to calculate event rates then non-randomised studies that provided these data for etanercept, infliximab and adalimumab were included in the review. If multiple non-randomised studies were identified, inclusion was limited to those studies reporting outcomes for a minimum of 500 patients receiving biologic therapy.
Interventions
Etanercept, infliximab and adalimumab were the interventions of interest. Comparators were placebo, another of the three listed agents, or conventional management strategies for active and progressive PsA that have responded inadequately to previous DMARD therapy, excluding TNF inhibitors.
Participants
For the evaluation of the effectiveness of etanercept, infliximab and adalimumab, included studies were of adults with active and progressive PsA with an inadequate response to previous standard therapy (including at least one DMARD). Trials of effectiveness had to specify that the patients had PsA, with the definition and/or the inclusion criteria for PsA stated. For the assessment of adverse effects, studies of patients with other conditions were eligible for inclusion in the review.
Outcomes
The eligible outcomes of effectiveness were measures of the anti-inflammatory response (PsARC, ACR 20/50/70), response of psoriatic skin lesions (PASI), functional measures (HAQ), radiological assessments of disease progression or remission, QoL assessments [e.g. Dermatology Life Quality Index (DLQI)] and overall global assessments.
In terms of the outcomes of adverse events of biologics, we provided an initial overview of previous systematic reviews of biologic safety (see Results of review of clinical effectiveness) before conducting our systematic review of adverse events of these agents. Our systematic review specifically focused on the known serious adverse events of these agents: malignancies, severe infections (i.e. those that require i.v. antibiotic therapy and/or hospitalisation or cause death) and reactivation of latent tuberculosis (TB). If additional serious adverse events have been reported to regulatory bodies then the incidence of these were also assessed. In addition, data relating to serious adverse events in indications other than PsA were also considered in our systematic review, provided it was clinically appropriate to do so.
Data extraction strategy
Data on study and participant characteristics, efficacy outcomes, adverse effects, costs to the health service and cost-effectiveness were extracted. Baseline data were extracted where reported. Data were extracted by one reviewer using a standardised data extraction form and independently checked for accuracy by a second reviewer. The results of data extraction were presented in the structured tables (see Appendix 3, Efficacy data extraction: etanercept/infliximab/adalimumab). Disagreements were resolved through consensus, or consulting a third reviewer if necessary. Attempts were made, where possible, to contact authors for missing data. Data from studies with multiple publications were extracted and reported as a single study. In the rare case of minor discrepancies for the same data between published and unpublished data, data from published sources were used.
Quality assessment strategy
The quality of RCTs and other study designs were assessed using standard checklists. 75 Regarding the additional studies reviewed for data on serious adverse events: as all observational studies are prone to confounding and bias to some extent, non-randomised studies including < 500 patients receiving biologics were excluded from the review. The assessment was performed by one reviewer and checked independently by a second. Disagreements were resolved through consensus or by consulting a third reviewer if necessary.
Data analysis
Where sufficient clinically and statistically homogeneous data were available, data were pooled using standard meta-analytic methods. The levels of clinical and methodological heterogeneity were investigated, and statistical heterogeneity was assessed using Q- and I2-statistics. Given the small number of trials available, a fixed-effects model was used to pool outcomes where pooling was appropriate. Sensitivity analyses were undertaken when permitted by sufficient data (e.g. exclusion of concomitant MTX treatment). The potential short- and long-term benefits of etanercept, infliximab and adalimumab on both the psoriasis and arthritis components of PsA were investigated. The rates of serious adverse effects of these biologic agents were synthesised narratively.
As trials conducting head-to-head comparisons of etanercept, infliximab and adalimumab were not available the possibility of conducting some form of indirect comparison was investigated. Indirect comparisons are useful analytic tools when direct evidence on comparisons of interest is absent or sparse. 77 Meta-analysis using indirect comparisons enables data from several sources to be combined, while taking into account differences between the different sources, in a similar way to, but distinct from, how a random-effects model takes into account between-trial heterogeneity. As with a mixed-treatment comparison (MTC), Bayesian indirect comparisons need a ‘network of evidence’ to be established between all of the interventions of interest. The three drugs being evaluated all have a common comparator: placebo. It is this common comparator that allows the network between etanercept, infliximab and adalimumab to be established and provide information on the benefits of these agents relative to placebo and each other.
To help inform both the clinical review and the economic modelling four separate outcomes were considered. These outcomes were: PsARC response, HAQ score conditional on PsARC response, ACR 20/50/70 responses and PASI 50/75/90 responses. All outcomes were evaluated at 12 weeks. The evidence synthesis was undertaken using winbugs (version 1.4.2). winbugs is a Bayesian analysis software tool that, through the use of Markov chain Monte Carlo, calculates posterior distributions for the parameters of interest given likelihood functions derived from data and prior probabilities. Full details of the Bayesian indirect comparison methods and the winbugs codes along for the four different analyses are presented in Appendix 5.
Results of review of clinical effectiveness
Quantity and quality of research available
A total of 1320 records were identified from both the clinical effectiveness and adverse event searches (Figure 1). Details of studies excluded at the full publication stage are provided in Appendix 4.
FIGURE 1.
Flow chart showing the number of studies identified and included.
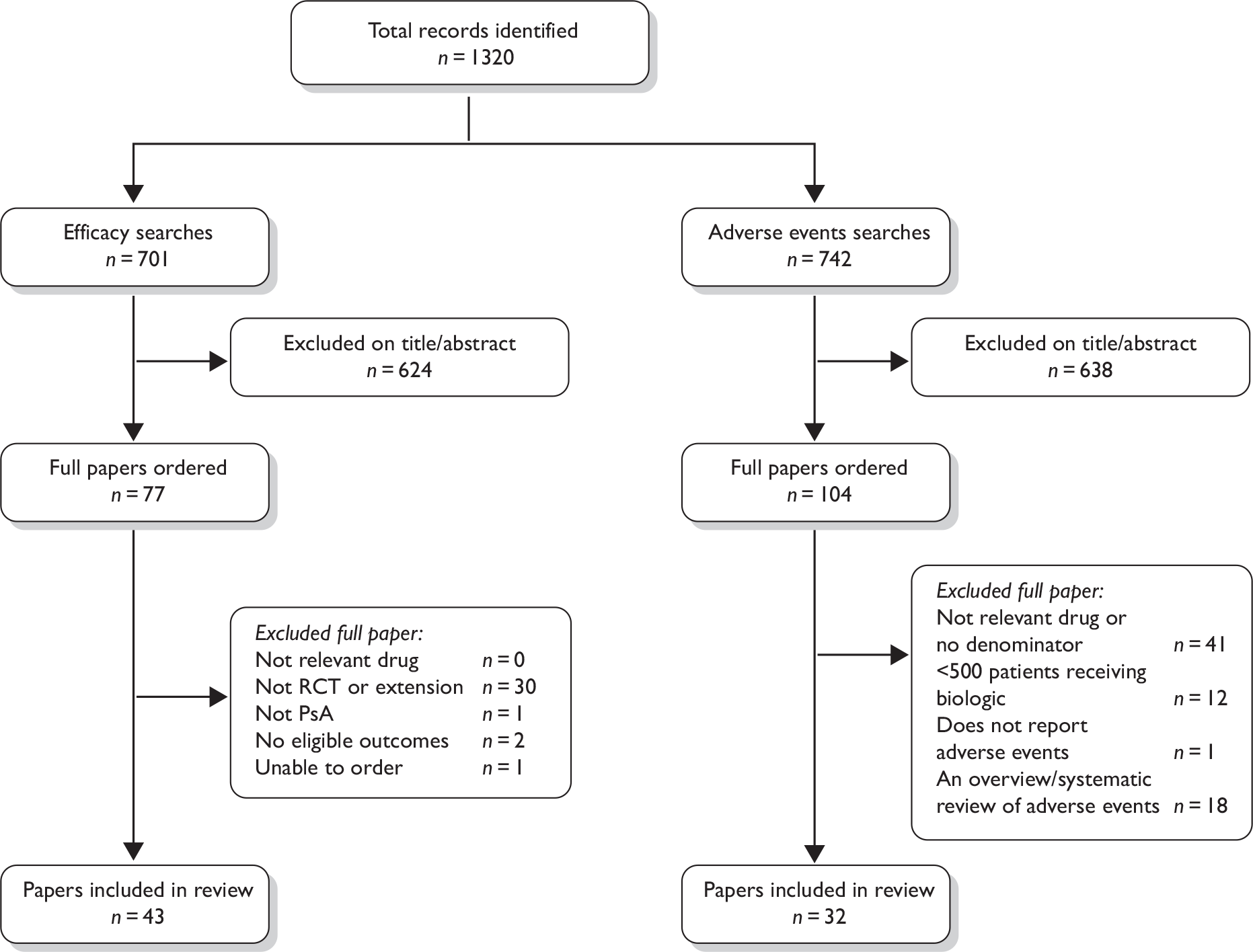
Randomised controlled trials and extensions in psoriatic arthritis
Of the 701 studies identified from the search for RCTs, a total of 43 publications, representing multiple publications of six RCTs and their extensions met the inclusion criteria for the review of efficacy. 51,52,78–118 Two placebo-controlled RCTs in patients with PsA were found for each of the three agents: etanercept,52,78,97,99,105,107,110 infliximab79–82,89–91,95,96,98,106,109,111–118 and adalimumab. 51,83,88,92,93,100–104 Baseline characteristics from all six RCTs are presented in Table 1.
| Etanercept | Infliximab | Adalimumab | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mease 200078 | Mease 200452,97,99,105,107,110 | IMPACT79–81,89,96,109,111,113–115,117,118 | IMPACT 2 82,90,91,95,98,106,112,116 | ADEPT51,88,92,93,100–104 | Genovese 200783 | |||||||
| Etanercept (n = 30) | Placebo (n = 30) | Etanercept (n = 101) | Placebo (n = 104) | Infliximab (n = 52) | Placebo (n = 52) | Infliximab (n = 100) | Placebo (n = 100) | Adalimumab (n = 151) | Placebo (n = 162) | Adalimumab (n = 51) | Placebo (n = 49) | |
| Age (years): mean (SD) | 46.0 (30.0–70.0)a | 43.5 (24.0–63.0)a | 47.6 (18–76)a | 47.3 (21–73)a | 45.7 (11.1) | 45.2 (9.7) | 47.1 (12.8) | 46.5 (11.3) | 48.6 (12.5) | 49.2 (11.1) | 50.4 (11.1) | 47.7 (11.3) |
| Male (%) | 53 | 60 | 57 | 45 | 58 | 58 | 71 | 51 | 56 | 55 | 57 | 51 |
| Duration of PsA (years): mean (SD) | 9.0 (1–31)a | 9.5 (1–30)a | 9.0 (–)a | 9.2 (–)a | 8.7 (8.0) | 8.5 (6.4) | 8.4 (7.2) | 7.5 (7.8) | 9.8 (8.3) | 9.2 (8.7) | 7.5 (7.0) | 7.2 (7.0) |
| Duration of psoriasis (years): mean (SD) | 19.0 (4–53)a | 17.5 (2–43)a | 18.3 (–)a | 19.7 (–)a | 16.9 (10.9) | 19.4 (11.6) | 16.2 (11.0) | 16.8 (12.0) | 17.2 (12.0) | 17.1 (12.6) | 18.0 (13.2) | 13.8 (10.7) |
| Number of prior DMARDs: mean (SD) | 1.5 | 2.0 | 1.6 | 1.7 | – | – | – | – | 1.5 | 1.5 | 1.7 | 2.1 |
| Proportion of patients with numbers of previous DMARDsb | – | – | 27% = 0 40% = 1 20% = 2 | 21%=0 50% =1 19% =2 | 0% = 0 52% = 1 37% = 2–3 12% = 3+ | 2% = 0 38% = 1 48% = 2–3 12% = 3+ | 71% = 1–2 12% = 2+ | 67% = 1–2 9% = 2+ | – | – | – | |
| Concomitant therapies during study (%) | ||||||||||||
| Corticosteroids | 20 | 40 | 19 | 15 | 17 | 29 | 15 | 10 | – | – | – | – |
| NSAIDs | 67 | 77 | 88 | 83 | 89 | 79 | 71 | 73 | – | – | 73 | 86 |
| MTX | 47 | 47 | 45 | 49 | 46 | 65 | 47 | 45 | 51 | 50 | 47 | 47 |
| Hydroxychloroquine | – | – | – | – | – | – | – | – | – | – | 16 | 16 |
| Sulfasalazine | – | – | – | – | – | – | – | – | – | – | 8 | 14 |
| Leflunomide | – | – | – | – | – | – | – | – | – | – | 6 | 4 |
| Other DMARDs | – | – | – | – | – | – | – | – | – | – | 2 | 6 |
| Type of PsA (%) | ||||||||||||
| DIP joints in hand and feet | – | – | 51 | 50 | – | – | – | – | – | – | – | – |
| Arthritis mutilans | – | – | 1 | 2 | – | – | – | – | 1 | 0 | 0 | 0 |
| Polyarticular arthritis | – | – | 86 | 83 | 100 | 100 | – | – | 64 | 70 | 82 | 84 |
| Asymmetric peripheral arthritis | – | – | 41 | 38 | – | – | – | – | 25 | 25 | 10 | 14 |
| Ankylosing arthritis | – | – | 3 | 4 | – | – | – | – | 1 | 0 | 2 | 2 |
| TJC: mean (SD) | 22.5 (11, 32)b | 19.0 (10, 39)b | 20.4 (–)b | 22.1 (–)b | 23.7 (13.7) | 20.4 (12.1) | 24.6 (14.1) | 25.1 (13.3) | 23.9 (17.3) | 25.8 (18.0) | 25.3 (18.3) | 29.3 (18.1) |
| SJC: mean (SD) | 14.0 (8, 23)b | 14.7 (7, 24)b | 15.9 (–)b | 15.3 (–)b | 14.6 (7.5) | 14.7 (8.2) | 13.9 (7.9) | 14.4 (8.9) | 14.3 (12.2) | 14.3 (11.1) | 18.2 (10.9) | 18.4 (12.1) |
| HAQ (0–3): mean (SD) | 1.3 (0.9, 1.6)b | 1.2 (0.8, 1.6)b | 1.1 (–)b | 1.1 (–)b | 1.2 (0.7) | 1.2 (0.7) | 1.1 (0.6) | 1.1 (0.6) | 1.0 (0.6) | 1.0 (0.7) | 0.9 (0.5) | 1.0 (0.7) |
| Patients evaluable for PASI at baseline: no. (%) | 19 (63%)c | 19 (63%)c | CiC | CiC | 22 (42%)d | 17 (33%)d | 83 (83%)c | 87 (87%)c | 70 (46%)c | 70 (43%)c | – | – |
| PASI (0–72) at baseline among patients evaluable for PASI: mean (SD) | 10.1 (2.3–30.0)a | 6.0 (1.5–17.7)a | CiC | CiC | 8.6 (6.6) | 8.1 (6.6) | 11.4 (12.7) | 10.2 (9.0) | 7.4 (6.0) | 8.3 (7.2) | – | – |
Additional adverse event studies
In total, 742 records were identified from the separate search for larger studies reporting adverse event rates for biologic agents in any indication. Of these records, 32 publications reported treatment with etanercept, infliximab or adalimumab in 500 or more patients, and reported either adverse event rates directly or provided sufficient information to calculate these rates (Figure 1). 89,97,99,119–148
Assessment of effectiveness
Efficacy of etanercept
Both trials evaluating etanercept for PsA were double-blind and placebo-controlled, and both were rated as ‘good’ on the quality assessment rating (Table 2). 52,78,97,99,105,107,110 Both trials were available as industry trial reports and journal publications.
| Quality assessment criteria | Study | |
|---|---|---|
| Mease 200078 | Mease 200452,97,99,105,107,110 | |
| Eligibility criteria specified? | Y | Y |
| Power calculation? | Y | Y |
| Adequate sample size? | Y | Y |
| Number randomised stated? | Y | Y |
| True randomisation? | Y | Y |
| Double blind? | Y | Y |
| Allocation of treatment concealed? | Y | Y |
| Treatment administered blind? | Y | Y |
| Outcome assessment blind? | Y | Y |
| Patients blind? | Y | Y |
| Blinding successful? | NR | NR |
| Adequate baseline details presented? | Y | Y |
| Baseline comparability? | Y | Y |
| Similar cointerventions? | Y | Y |
| Compliance with treatment adequate? | Y | Y |
| All randomised patients accounted for? | Y | Y |
| Valid ITT analysis? | Y | Y |
| ≥ 80% patients in follow-up assessment? | Y | Y |
| Quality rating | Good | Good |
The baseline characteristics of the trial population are summarised in Table 1. Both trials were of adults (aged 18–70 years), with active PsA (defined in both trials as at least three swollen joints and at least three tender or painful joints, although only the more recent trial52,97,99,105,107,110 specified stable plaque psoriasis). Patients in both trials had demonstrated an inadequate response to NSAIDs. Over 70% of the patients in the larger trial52,97,99,105,107,110 had previously used at least one DMARD. Over 80% of patients in the Mease et al. 52,97,99,105,107,110 trial had polyarticular disease, indicating that, overall, the disease was severe. Patients were not required to have active psoriasis at baseline, but 77% of etanercept patients and 73% of placebo patients did have. The proportion of patients with spine involvement and arthritis mutilans at baseline was reported only for the larger trial, where such patients made up only a small proportion of the trial population. These details were not available for the smaller of the two trials, so the severity of disease across that population is unknown. However, given the similarity between the trials for other measures of disease activity (TJC, SJC, HAQ at baseline, plus baseline and previous medication), significant differences between the populations in terms of overall disease severity are unlikely. Patients taking stable doses of MTX or corticosteroids were permitted to continue with that dose and randomisation was stratified for MTX use at baseline. Overall, the baseline characteristics demonstrate that the trial populations are similar and are likely to be representative of a population with PsA requiring DMARD or biologic therapy. It should be noted, however, that the populations in these trials of etanercept are not representative of the patients for whom etanercept is recommended for use: these patients, according to the BSR, would have demonstrated a lack of response to at least two DMARDs. 149
In both trials, etanercept was administered by subcutaneous (s.c.) injection twice weekly at a dose of 25 mg. Treatment with active drug or placebo was administered for 12 weeks in the smaller trial78 and for 24 weeks in the larger trial. 52,97,99,105,107,110 In both trials the controlled phase was followed by a follow-up period during which etanercept was administered in an open-label fashion to all patients.
Outcome data derived under RCT conditions are available from both trials for PsARC, ACR 20/50/70 and HAQ at week 12. The primary outcome variable in the Mease 2000 trial78 was PsARC, whereas in the Mease et al. 52,97,99,105,107,110 trial it was ACR 20. Data on PASI at week 12 are available from the small78 trial only. RCT outcome data for PsARC, ACR 20/50/70, HAQ, PASI and radiographic assessment of progression at week 24 are available from the larger trial52,97,99,105,107,110 (n = 205). In addition, a subgroup analyses by concomitant MTX use provided additional PsARC, ACR 20/50/70 data at weeks 12 and 24. As subgroup analyses in already fairly small trials, the findings generated must be interpreted with some caution. They are useful, however, to explore the influence concomitant MTX has on the main treatment effect. All outcome data are summarised in Table 3, with pooled 12-week data shown in Table 4.
| Outcomes | Etanercept: n (%) | Placebo: n (%) | RR or mean difference (95% CI) | |
|---|---|---|---|---|
| Mease 2000,78 12 weeks | ||||
| PsARCa | 26/30 (87) | 7/30 (23) | 3.71 (1.91 to 7.21) | |
| ACR 20 | 22/30 (73.0) | 4/30 (13) | 5.50 (2.15 to 14.04) | |
| ACR 50 | 15/30 (50.0) | 1/30 (3) | 15.00 (2.11 to 106.49) | |
| ACR 70 | 4/30 (13) | 0/30 (0) | 9.00 (0.51 to 160.17) | |
| HAQ% change from baseline: mean (SD) | n = 29 (64.2–38.7) | n = 30 (9.9–42.9) | CiC | |
| PASI 50 | 8/19 (42) | 4/19 (21) | 2.00 (0.72 to 5.53), p = 0.295 | |
| PASI 75 | 5/19 (26) | 0/19 (0) | 11.00 (0.65 to 186.02), p = 0.0154 | |
| Mease 2004,52,97,99,105,107,110 12 weeks | ||||
| PsARC | All pts | 73/101 (72) | 32/104 (31) | 2.35 (1.72 to 3.21), p < 0.001 |
| +MTX | 32/42 (76) | 14/43 (33) | 2.34 (1.47 to 3.72) | |
| –MTX | 41/59 (69) | 18/61 (30) | 2.35 (1.54 to 3.60) | |
| ACR 20a | All pts | 60/101 (59) | 16/104 (15) | 3.86 (2.39 to 6.23), p < 0.001 |
| +MTX | 26/42 (62) | 8/43 (19) | 3.33 (1.70 to 6.49) | |
| –MTX | 34/59 (58) | 8/61 (13) | 4.39 (2.22 to 8.7) | |
| ACR 50 | All pts | 38/101 (38) | 4/104 (4) | 9.78 (3.62 to 26.41), p < 0.001 |
| +MTX | 17/42 (40) | 1/43 (2) | 17.40 (2.42 to 124.99) | |
| –MTX | 21/59 (36) | 3/61 (5) | 7.24 (2.28 to 22.98) | |
| ACR 70 | All pts | 11/101 (11) | 0/104 (0) | 23.68 (1.41 to 396,53), p < 0.001 |
| +MTX | 4/42 (10) | 0/43 (0) | 9.21 (0.51 to 165.93) | |
| –MTX | 7/59 (12) | 0/61 (0) | 15.5 (0.91 to 265.46) | |
| HAQ% change from baseline: mean (SD) | (n = 96) 53.5, (43.4) | (n = 99) 6.3, (42.7) | CiC | |
| Mease 2004,52,97,99,105,107,110 24 weeks | ||||
| PsARC | All pts | 71/101 (70) | 24/104 (23) | 3.05 (2.10 to 4.42), p < 0.001 |
| +MTX | 31/42 (74) | 11/43 (26) | 2.89 (1.68 to 4.95) | |
| –MTX | 40/59 (68) | 13/61 (21) | 3.18 (1.90 to 5.32) | |
| ACR 20 | All pts | 50/101 (50) | 14/104 (13) | 3.68 (2.17 to 6.22), p < 0.001 |
| +MTX | 23/42 (55) | 8/43 (19) | 2.94 (1.49 to 5.83) | |
| –MTX | 27/59 (46) | 6/61 (10) | 4.73 (2.10 to 10.63) | |
| ACR 50 | All pts | 37/101 (37) | 4/104 (4) | 9.52 (3.52 to 25.75), p < 0.001 |
| +MTX | 16/42 (38) | 3/43 (7) | 5.46 (1.72 to 17.37) | |
| –MTX | 21/59 (36) | 1/61 (2) | 21.71 (3.02 to 156.30) | |
| ACR 70 | All pts | 9/101 (9) | 1/104 (1) | 9.27 (1.20 to 71.83), p = 0.009 |
| +MTX | 2/42 (5) | 0/43 (0) | 5.12 (0.25 to 103.50) | |
| –MTX | 7/59 (12) | 0/61 (0) | 15.50 (0.91 to 265.46) | |
| HAQ% change from baseline: mean (SD) | (n = 96) 53.6 (55.1) | (n = 99) 6.4 (49.6) | 47.20 (32.47 to 61.93), p < 0.001 | |
| PASI 50 | 31/66 (47) | 11/62 (18); | 2.65 (1.46 to 4.80), p < 0.001 | |
| PASI 75 | 15/66 (23) | 2/62 (3) | 7.05 (1.68 to 29.56), p = 0.001 | |
| PASI 90 | 4/66 (6) | 2/62 (3) | 1.88 (0.36 to 9.90), p = 0.681 | |
| TSS mean (SD) annualised rate of progression | All pts | (n = 101) –0.03 (0.73) | (n = 104) 0.53 (1.39) | –0.56 (–0.86 to –0.26), p = 0.0006 |
| +MTX | (n = 42) 0.06 (0.76) | (n = 43) 0.48 (1.00) | –0.42 (–0.80 to –0.04), p = 0.12345 | |
| –MTX | (n = 59) –0.09 (0.71) | (n = 61) 0.57 (1.62) | –0.66 (–1.11 to –0.21), p = 0.0014 | |
| Trial | Etanercept | Placebo | RR or mean difference (95% CI) |
|---|---|---|---|
| PsARC | |||
| Mease 200078 | 26/30 (87%) | 7/30 (23%) | 3.71 (1.91 to 7.21) |
| Mease 200452,97,99,105,107,110 | 73/101 (72%) | 32/104 (31%) | 2.35 (1.72 to 3.21), p < 0.001 |
| Pooled RR (95% CI), p-value, I 2 : 2.60 (1.96 to 3.45), p < 0.00001, I 2 = 34% | |||
| ACR 20 | |||
| Mease 200078 | 22/30 (73%) | 4/30 (13%) | 5.50 (2.15 to 14.04) |
| Mease 200452,97,99,105,107,110 | 60/101 (59%) | 16/104 (15%) | 3.86 (2.39 to 6.23), p < 0.001 |
| Pooled RR (95% CI), p-value, I 2 : 4.19 (2.74 to 6.42), p < 0.00001, I 2 = 0% | |||
| ACR 50 | |||
| Mease 200078 | 15/30 (50%) | 1/30 (3%) | 15.00 (2.11 to 106.49) |
| Mease 200452,97,99,105,107,110 | 38/101 (38%) | 4/104 (4%) | 9.78 (3.62 to 26.41), p < 0.001 |
| Pooled RR (95% CI), p-value, I 2 : 10.84 (4.47 to 26.28), p < 0.00001, I 2 = 0% | |||
| ACR 70 | |||
| Mease 200078 | 4/30 (13%) | 0/30 (0%) | 9.00 (0.51 to 160.17) |
| Mease 200452,97,99,105,107,110 | 11/101 (11%) | 0/104 (0%) | 23.68 (1.41 to 396,53), p < 0.001 |
| Pooled RR (95% CI), p-value, I 2 : 16.28 (2.20 to 120.54), p = 0.006, I 2 = 0% | |||
| HAQ% change from baseline: mean (SD) | |||
| Mease 200078 | (n = 29) –64.2 (CiC) | (n = 30) –9.9 (CiC) | –54.3 (33.47 to 75.13) |
| Mease 200452,97,99,105,107,110 | (n = 96) –53.5 (CiC) | (n = 99) –6.3 (CiC) | –47.20 (35.11 to 59.29) |
| Pooled RR (95% CI), p-value, I 2 : –48.99 (38.53 to 59.44), p < 0.00001, I 2 = 0% | |||
Uncontrolled data on all outcomes are also available at 36 weeks or 12 months (uncontrolled follow-up data). These data are summarised in Table 4.
Efficacy after 12 weeks’ treatment
The individual trial results (Table 3) and pooled estimates of effect (Table 4) demonstrate a statistically significant benefit of etanercept for all joint disease and HAQ score outcomes. There was no statistical heterogeneity for any outcome.
Across the two trials at 12 weeks almost 85% of patients treated with etanercept achieved a PsARC response, which is the only joint disease outcome measure that has been specifically defined for PsA. In addition, around 65% of patients treated with etanercept achieved an ACR 20 response, demonstrating a basic degree of efficacy in terms of arthritis-related symptoms. Around 45% of patients treated with etanercept achieved an ACR 50 response, and around 12% achieved an ACR 70 response, demonstrating a good level of efficacy. The subgroup analyses conducted on the Mease et al. 52,97,99,105,107,110 data revealed that the effect of etanercept was not dependent on patients’ concomitant use, or not, of MTX. The PASI results from Mease et al. 78 indicate some beneficial effect on psoriasis at 12 weeks; however, the data were too sparse (38 patients in total) to establish statistical significance. The statistically significant reduction in HAQ score with etanercept compared with placebo indicates a beneficial effect of etanercept on functional status.
Efficacy after 24 weeks’ treatment
At 24 weeks, the treatment effect for all joint disease outcome measures was statistically significantly greater with etanercept than with placebo, although these data were available only for one trial (see Table 3). As at 12 weeks, the subgroup analyses conducted on the Mease et al. 52,97,99,105,107,110 data revealed that the effect of etanercept was not dependent upon patients’ concomitant use, or not, of MTX. The size of treatment effect did not appear greater at 24 weeks than at 12 weeks.
At 24 weeks, the TSS annualised rate of progression was statistically significantly lower in patients treated with etanercept than in patients receiving placebo. This treatment difference did not vary with or without concomitant MTX use. However, this duration of follow-up is to be considered short and barely adequate for this outcome.
At 24 weeks, the treatment effect on psoriasis favoured etanercept with relative risks (RRs) for PASI 75 of 7.05 [95% confidence interval (CI) 1.68 to 29.56], PASI 50 of 2.65 (95% CI 1.46 to 4.80) and PASI 90 of 1.88 (95% CI 0.36 to 9.90). The result for PASI 75 and PASI 50 was statistically significant despite there being only 66 patients on etanercept evaluable for psoriasis. 52,97,99,105,107,110
Longer-term follow-up
The results for long-term follow-up are summarised in Table 5. The data are uncontrolled and therefore cannot be taken as reliable. In general, they do indicate that the improvements in patients’ joint and skin symptoms and HAQ score achieved during the controlled phase of the trials are maintained in the medium term. At 1 year, the mean annualised rate of progression TSS for all patients was –0.03 [standard deviation (SD 0.87)] indicating that on average no clinically significant progression of joint erosion had occurred. Limited 2-year data indicated little change in mean TSS, although data on patient numbers or variability were not reported.
| Trial | Type of data | Duration | Outcomes | Etanercept/placebo | |
|---|---|---|---|---|---|
| Mease 200078 | Uncontrolled | 36 weeks | PsARC | 26/30 (87%) | |
| ACR 20 | 26/30 (87%) | ||||
| ACR 50 | 19/30 (63%) | ||||
| ACR 70 | 10/30 (33%) | ||||
| HAQ% change from baseline: mean (median) | CiC | ||||
| PASI 75 | 7/19 (37%) | ||||
| PASI 50 | 11/19 (58%) | ||||
| Mease 200452,97,99,105,107,110 | Uncontrolled | 12 months | ACR results, etc. only as brief text | Maintained as at 24 weeks | |
| TSS mean (SD) annualised rate of progression | All pts | (n = 101) –0.03 (0.87) | |||
| +MTX | (n = 42) 0.01 (0.81) | ||||
| –MTX | (n = 59) –0.13 (0.91) | ||||
| 24 months | TSS mean change from baseline | Etanercept/etanercept –0.38 | |||
| Placebo/etanercept 0.50 | |||||
Summary of the efficacy of etanercept in the treatment of psoriatic arthritis
-
There is evidence from double-blind placebo-controlled trials of a good level efficacy for etanercept in the treatment of PsA. Conclusions to be drawn from these data are limited by the small sample size and the short duration of one of the trials.
-
There is evidence from two RCTs that etanercept treatment improves patients’ functional status as assessed using the HAQ score.
-
There is limited evidence from the two RCTs that etanercept treatment has a beneficial effect on the psoriasis component of the disease.
-
Uncontrolled follow-up of patients indicate that treatment benefit is maintained for at least 50 weeks; however, these data may not be reliable.
-
There are radiographic data from controlled trials for etanercept in PsA that demonstrate a beneficial effect on progression of joint disease at 24 weeks. This is a very short time over which to identify a statistically significant effect of therapy and indicates a rapid onset of action of etanercept. Data from uncontrolled follow-up indicate that, on average, disease progression may be halted for at least 1 year; however, these data may not be reliable.
Efficacy of infliximab
The literature search identified two RCTs of infliximab for the treatment of PsA. 79–82,89–91,95,96,98,106, 109,111–118 Both were rated as ‘good’ by the quality assessment (Table 6). The trials were reported in published papers and abstracts, and the industry trial report was made available.
| Quality assessment criteria | Study | |
|---|---|---|
| IMPACT79–81,89,96,109,111,113–115,117,118 | IMPACT 282,90,91,95,98,106,112,116 | |
| Eligibility criteria specified? | Y | Y |
| Power calculation? | Y | Y |
| Adequate sample size? | Y | Y |
| Number randomised stated? | Y | Y |
| True randomisation? | Y | Y |
| Double blind? | Y | Y |
| Allocation of treatment concealed? | Y | Y |
| Treatment administered blind? | Y | Y |
| Outcome assessment blind? | Y | Y |
| Patients blind? | Y | Y |
| Blinding successful? | NR | NR |
| Adequate baseline details presented? | Y | Y |
| Baseline comparability? | Y | Y |
| Similar cointerventions? | Y | Y |
| Compliance with treatment adequate? | Y | Y |
| All randomised patients accounted for? | Y | Y |
| Valid ITT analysis? | Y | Y |
| ≥ 80% patients in follow-up assessment? | Y | Y |
| Quality rating | Good | Good |
Both were double-blind, placebo-controlled trials of adult patients with active PsA, randomising a total of 304 patients. All patients had been diagnosed with PsA for at least 6 months, with a negative RF and active disease including 5+ swollen/tender joints. All patients must have had an inadequate response to at least one DMARD. 79–82,89–91,95,96,98,106,109,111–118 One trial required patients to have active plaque psoriasis with at least one qualifying target lesion (≥ 2-cm diameter). 82,90,91,95,98,106,112,116 The earlier of the two trials did not require patients to have active psoriasis at baseline, but 42% of infliximab patients and 33% of placebo patients did have (defined as PASI score of at least 2.5). 79–81,89,96,109,111,113–115,117,118 The proportion of patients with spine involvement, arthritis mutilans and erosions at baseline was not reported for either trial, so the severity of disease across the populations is unknown. The baseline characteristics of the trial populations are summarised in Table 1. These demonstrate that the trial populations are broadly similar, are likely to be representative of a population with quite severe PsA requiring further DMARD or biologic therapy, and that the treatment and placebo groups were well balanced. Relative to the patients for whom infliximab treatment is recommended in practice, these trial populations may be less severely affected, with only around one-half in the Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT)79–81,89,96,109,111,113–115,117,118 and possibly even fewer in IMPACT 2,82,90,90,95,98,106,112,116 having failed to respond to two or more DMARDs (failure to respond to DMARDs as defined by the BSR). 149
In the RCT phase of the IMPACT trial,79–81,89,96,109,111,113–115,117,118 infliximab (5 mg/kg) or placebo was infused at weeks 0, 2, 6 and 14, with follow-up at week 16. Further infusions of infliximab were administered to all patients in an open-label fashion at 8-week intervals, with further follow-up at week 50. Patients in the IMPACT 2 trial82,90,91,95,98,106,112,116 were randomised to receive infusions of placebo or infliximab, 5 mg/kg, at weeks 0, 2, 6, 14 and 22, with assessments at weeks 14 and 24. Further infusions of infliximab were administered to all patients in an open-label fashion (timing dependent upon whether they were originally randomised to infliximab, or crossed over from placebo either at week 16 or 24) with further follow-up at week 54.
The primary outcome variable in these trials was ACR 20 at 14 or 16 weeks. The two trials also reported 14-week and/or 16-week outcome data for ACR 50, ACR 70, PsARC, HAQ, PASI 50, PASI 75 and PASI 90 (RCT data). IMPACT 282,90,91,95,98,106,112,116 also maintained randomisation and reported these outcomes at week 24. Both studies reported longer-term open-label follow-up of patients after 50 and 54 weeks (IMPACT79–81,89,96,109,111,113–115,117,118 and IMPACT 2,82,90,91,95,98,105,112,115 respectively). All data are summarised in Table 7, with pooled data presented in Table 8.
| Trial | Duration (weeks) | Outcomes | Infliximab | Placebo | RR or mean difference (95% CI) | |
|---|---|---|---|---|---|---|
| IMPACT (randomised period)79–81,89,96,109,111,113–115,117,118 | 14 | PsARC | 40/52 (76.9%) | 7/52 (13.5%) | 5.71 (2.82 to 11.57) | |
| ACR 20 | All pts | 35/52 (67.3%) | 6/52 (11.5%) | 5.83 (2.68 to 12.68) | ||
| +MTX | NR | NR | – | |||
| –MTX | NR | NR | – | |||
| ACR 50 | 19/52 (36.5%) | 1/52 (1.9%) | 19.00 (2.64 to 136.76) | |||
| ACR 70 | 11/52 (21.2%) | 0/52 (0%) | 23.00 (1.39 to 380.39) | |||
| 16 | PsARC | 39/52 (75.0%) | 11/52 (21.2%) | 3.55 (2.05 to 6.13), p < 0.01 | ||
| ACR 20 | All pts | 34/52 (65.4%) | 5/52 (9.6%) | 6.80 (2.89 to 16.01) p < 0.01 | ||
| +MTX | 15/24 (62.5%) | 4/34 (11.8%) | 5.31 (2.01 to 14.03), p < 0.01 | |||
| –MTX | 19/28 (67.9%) | 1/18 (5.6%) | 12.21 (1.79 to 83.46), p < 0.01 | |||
| ACR 50 | 24/52 (46.2%) | 0/52 (0%) | 49.00 (3.06 to 785.06), p < 0.01 | |||
| ACR 70 | 15/52 (28.8%) | 0/52 (0%) | 31.00 (1.90 to 504.86), p < 0.01 | |||
| HAQ mean (SD)% change from baseline | (n = 48) –49.8 (56.8) | (n = 47) 1.6 (56.9) | –51.4 (–74.5 to –28.3), p < 0.01 | |||
| PASI 50a | 22/22 (100%) | 0/16 (0%) | 33.26 (2.17 to 510.71) | |||
| PASI 75a | 15/22 (68.2%) | 0/16 (0%) | 22.91 (1.47 to 356.81) | |||
| PASI 90a | 8/22 (36.4%) | 0/16 (0%) | 12.57 (0.78 to 203.03) | |||
| PASI mean (SD) change from baselineb | (n = 42) –4.1 (3.9) | (n = 38) 0.9 (3.7) | –5 (–6.8 to –3.3), p < 0.01 | |||
| IMPACT 2 (randomised)82,90,91,95,98,106,112,116 | 14 | PsARC | 77/100 (77%) | 27/100 (27%) | 2.85 (2.03 to 4.01) | |
| ACR 20 | All pts | 58/100 (58%) | 11/100 (11%) | 5.27 (2.95 to 9.44) | ||
| +MTX | NR | NR | – | |||
| –MTX | NR | NR | – | |||
| ACR 50 | 36/100 (36%) | 3/100 (3)% | 12.00 (3.82 to 37.70) | |||
| ACR 70 | 15/100 (15%) | 1/100 (1%) | 15.00 (2.02 to 111.41) | |||
| HAQ mean (SD)% change from baseline | (n = 100) –48.6 (43.3) | (n = 100) 18.4 (90.5) | –67.00 (–86.66 to –47.33) | |||
| PASI 50b | CiC | CiC | CiC | |||
| PASI 75b | CiC | CiC | CiC | |||
| PASI 90b | CiC | CiC | CiC | |||
| PASI mean (SD)% change from baseline | NR | NR | – | |||
| 24 | PsARC | 70/100 (70%) | 32/100 (32%) | 2.19 (1.60 to 3.00) | ||
| ACR 20 | All pts | 54/100 (54%) | 16/100 (16%) | 3.38 (2.08 to 5.48) | ||
| +MTX | NR | NR | – | |||
| –MTX | NR | NR | – | |||
| ACR 50 | 41/100 (41%) | 4/100 (4%) | 10.25 (3.81 to 27.55) | |||
| ACR 70 | 27/100 (27%) | 2/100 (2%) | 13.5 (3.30 to 55.26) | |||
| PASI 50b | CiC | CiC | CiC | |||
| PASI 75b | CiC | CiC | CiC | |||
| PASI 90b | CiC | CiC | CiC | |||
| HAQ mean (SD)% change from baseline | (n = 100) –46.0 (42.5) | (n = 100) 19.4 (102.8) | –65.40 (–87.20 to –43.60) | |||
| PASI mean (SD)% change from baseline | NR | NR | – | |||
| Total modified van der Heijde–Sharp score: mean (SD) change from baseline | –0.70 (2.53) | 0.82 (2.62) | – | |||
| Trial | Outcomes | Infliximab | Placebo | RR or mean difference (95% CI) |
|---|---|---|---|---|
| PsARC | ||||
| IMPACT79–81,89,96,109,111,113–115,117,118 | 40/52 (76.9%) | 7/52 (13.5%) | 5.71 (2.82 to 11.57) | |
| IMPACT 282,90,91,95,98,106,112,116 | 77/100 (77%) | 27/100 (27%) | 2.85 (2.03 to 4.01) | |
| Pooled RR (95% CI) to p-value | 3.44 (2.53 to 4.69), p < 0.0001 | |||
| I 2 | I2 = 68% | |||
| ACR 20 | ||||
| IMPACT79–81,89,96,109,111,113–115,117,118 | 35/52 (67.3%) | 6/52 (11.5%) | 5.83 (2.68 to 12.68) | |
| IMPACT 282,90,91,95,98,106,112,116 | 58/100 (58%) | 11/100 (11%) | 5.27 (2.95 to 9.44) | |
| Pooled RR (95% CI), p-value | 5.47 (3.43 to 8.71) | |||
| I 2 | I2 = 0% | |||
| ACR 50 | ||||
| IMPACT79–81,89,96,109,111,113–115,117,118 | 19/52 (36.5%) | 1/52 (1.9%) | 19.00 (2.64 to 136.76) | |
| IMPACT 282,90,91,95,98,106,112,116 | 36/100 (36%) | 3/100 (3%) | 12.00 (3.82 to 37.70) | |
| Pooled RR (95% CI), p-value | 13.75 (5.11 to 37.00), p < 0.0001 | |||
| I 2 | I2 = 0% | |||
| ACR 70 | ||||
| IMPACT79–81,89,96,109,111,113–115,117,118 | 11/52 (21.2%) | 0/52 (0%) | 23.00 (1.39 to 380.39) | |
| IMPACT 282,90,91,95,98,106,112,116 | 15/100 (15%) | 1/100 (1%) | 15.00 (2.02 to 111.41) | |
| Pooled RR (95% CI), p-value | 17.67 (3.46 to 90.14), p = 0.001 | |||
| I 2 | I2 = 0% | |||
| PASI 50 | ||||
| IMPACT79–81,89,96,109,111,113–115,117,118 | 22/22 (100%) | 0/16 (0%) | 33.26 (2.17 to 510.71) | |
| IMPACT 282,90,91,95,98,106,112,116 | CiC | CiC | CiC | |
| Pooled RR (95% CI), p-value | 10.58 (5.47 to 20.48), p < 0.0001a | |||
| I 2 | I2 = 0% | |||
| PASI 75 | ||||
| IMPACT79–81,89,96,109,111,113–115,117,118 | 15/22 (68.2%) | 0/16 (0%) | 22.91 (1.47 to 356.81) | |
| IMPACT 282,90,91,95,98,106,112,116 | CiC | CiC | CiC | |
| Pooled RR (95% CI), p-value | 26.68 (7.79 to 91.44), p < 0.0001a | |||
| I 2 | I2 = 0% | |||
| PASI 90 | ||||
| IMPACT79–81,89,96,109,111,113–115,117,118 | 8/22 (36.4%) | 0/16 (0%) | 12.57 (0.78 to 203.03) | |
| IMPACT 282,90,91,95,98,106,112,116 | CiC | CiC | CiC | |
| Pooled RR (95% CI), p-value | 40.01 (5.93 to 270.15), p < 0.0001a | |||
| I 2 | I2 = 0% | |||
| HAQ% change from baseline: mean (SD) | ||||
| IMPACT79–81,89,96,109,111,113–115,117,118 | (n = 48) –49.8 (56.8) | (n = 47) 1.6 (56.9) | –51.4 (–74.27 to –28.54) | |
| IMPACT 282,90,91,95,98,106,112,116 | (n = 100) –48.6 (43.3) | (n = 100) 18.4 (90.5) | –67.00 (–86.66 to –47.33) | |
| Pooled WMD (95% CI), p-value | –60.37 (–75.28 to –45.46) | |||
| I 2 | I2 = 3% | |||
Efficacy after 14–16 weeks’ treatment
At 14 weeks, both trials reported a significant improvement in the PsA-specific PsARC measure for patients receiving infliximab, relative to those receiving placebo (pooled RR 3.44, 95% CI 2.53 to 4.69 – Table 8). There was evidence of statistical heterogeneity (I2 = 68%) between the two study estimates, due to the different placebo response rates (13.5% vs 27%). PsARC response on infliximab was around 77% in both trials.
The pooled RR for ACR 20 at 14 weeks was 5.47 (95% CI 3.43 to 8.71), with an overall response of 61% in infliximab-treated patients, demonstrating a clear degree of efficacy of infliximab in terms of arthritis-related symptoms. As very few patients receiving placebo achieved an ACR 50 or ACR 70 response, the pooled RRs clearly favoured infliximab in terms of these outcomes, although the limited number of observations mean that there is considerable uncertainty around these pooled estimates, as reflected by their CIs (see Table 8). Despite the potentially large relative effects, it should also be noted that only the minority of infliximab-treated patients achieved an ACR 50 or ACR 70 response at 14 weeks (36% and 17%, respectively). Data from the IMPACT trial79–81,89,96,109,111,113–115,117,118 indicated no significant difference in ACR 20 response at 16 weeks between patients with and without concomitant MTX, although the number of patients in each of these groups was small.
As with the ACR outcomes, few patients receiving placebo demonstrated skin improvements over 14–16 weeks in terms of a PASI response; the pooled RR for PASI 50 was 10.58 (95% CI 5.47 to 20.48), demonstrating a clear degree of efficacy of infliximab in terms of skin-related symptoms. PASI 75 and PASI 90 response measures favoured infliximab even more strongly, although it should be noted that PASI outcomes were recorded only for those patients with a score of at least 2.5 at baseline. Forty-two per cent of patients receiving infliximab achieved the highest level of skin response (PASI 90), although again there is considerable uncertainty around the estimates (see Table 7).
The statistically significant pooled percentage change from baseline in HAQ score with infliximab compared with placebo [mean difference –60.37 (–75.28 to –45.46)] indicates a beneficial effect of infliximab on functional status.
Efficacy after 24 weeks
The IMPACT 2 trial82,90,91,95,98,106,112,116 maintained randomisation for 24 weeks. The data for all measures of joint disease, psoriasis and HAQ are similar to those observed at the earlier 14-week follow-up, suggesting that the benefits of infliximab are maintained up to 24 weeks of treatment (see Table 7). Change from baseline in modified van der Heijde–Sharp score significantly differed between infliximab and placebo groups, indicating that infliximab may inhibit progression of joint damage at 24 weeks (see Table 7).
Longer-term follow-up
The data for longer-term follow-up (50 or 54 weeks) from the two IMPACT trials are summarised in Table 9. These data are uncontrolled and may therefore be unreliable. Also, the duration of treatment varied between participants, as some will have crossed over from placebo treatment. However, the data broadly indicate that the levels of efficacy achieved with infliximab in terms of joint disease, psoriasis and HAQ after 14–24 weeks’ treatment might be maintained in the medium term.
| Trial | Duration (weeks) | Outcomes | Infliximab/placebo | |
|---|---|---|---|---|
| IMPACT79–81,89,96,109,111,113–115,117,118 | 50 | ACR 20 | All pts | 34/49 (69.4%) |
| +MTX | 16/22 (72.7%) | |||
| –MTX | 18/27 (66.7%) | |||
| ACR 50 | 26/49 (53.1%) | |||
| ACR 70 | 19/49 (38.8%) | |||
| PsARC | 36/49 (73.5%) | |||
| HAQ: mean (SD)% change from baseline | (n = 45) –42.5 (59.0) | |||
| PASI 50a | 19/22 (86.3%) | |||
| PASI 75a | 13/22 (59%) | |||
| PASI 90a | 9/22 (40.9%) | |||
| PASI: mean (SD) change from baselinea | (n = 35) –4.8 (5.9) | |||
| Total modified van%%der%%Heijde–Sharp score: mean (SD) change from baseline | (n = 70) –1.72 (5.82) | |||
| IMPACT 282,90,91,95,98,106,112,116 | 54 | PsARC | 67/90 (74.4%) | |
| PASI 50a | 57/82 (69.5%) | |||
| PASI 75a | 40/82 (48.8%) | |||
| PASI 90a | 32/82 (39%) | |||
| Total modified van der Heijde–Sharp score: mean (SD) change from baseline |
Infliximab/infliximab –0.94 (3.4) Placebo/infliximab 0.53 (2.6) |
|||
In terms of radiographic assessment, there was no significant change from baseline in the total modified van der Heijde–Sharp score for those infliximab-treated patients followed up at 50 or 54 weeks in the two studies, suggesting infliximab may inhibit progression of joint damage. However, as with other post-24-week outcomes, there was no placebo group for comparison.
Summary of the efficacy of infliximab in the treatment of PsA
-
There is evidence from two double-blind placebo controlled trials of a good level of efficacy for infliximab in the treatment of PsA, with beneficial effects on joint disease, psoriasis and functional status as assessed by HAQ.
-
Conclusions to be drawn from these data are limited by the short duration of the controlled trials; controlled data to evaluate long-term effects are not available.
-
Uncontrolled follow-up of patients indicate that short-term benefit is maintained for at least 50 weeks; however, these data may not be reliable.
-
Radiographic data from uncontrolled follow-up of infliximab trials suggest that the drug may delay the progression of joint disease in PsA, although these data are not of high quality.
Efficacy of adalimumab
Both trials evaluating adalimumab for PsA were double blind and placebo controlled, and both were rated as ‘good’ on the quality assessment rating (Table 10). 51,83,88,92,93,100–104
| Quality assessment criteria | Study | |
|---|---|---|
| ADEPT51,88,92,93,100–104 | Genovese 200783 | |
| Eligibility criteria specified? | Y | Y |
| Power calculation? | Y | Y |
| Adequate sample size? | Y | Y |
| Number randomised stated? | Y | Y |
| True randomisation? | Y | Y |
| Double blind? | Y | Y |
| Allocation of treatment concealed? | NR | Y |
| Treatment administered blind? | Y | Y |
| Outcome assessment blind? | Y | Y |
| Patients blind? | Y | Y |
| Blinding successful? | NR | NR |
| Adequate baseline details presented? | Y | Y |
| Baseline comparability? | Y | Y |
| Similar cointerventions? | Y | Y |
| Compliance with treatment adequate? | Y | Y |
| All randomised patients accounted for? | Y | Y |
| Valid ITT analysis? | Y | Y |
| ≥ 80% patients in follow-up assessment? | Y | Y |
| Quality rating | Good | Good |
Both trials were of adults (aged 18–70 years), with active PsA (defined in both trials as three or more swollen joints and three or more tender or painful joints, with active psoriatic skin lesions or a documented history of psoriasis). Patients in the larger trial had demonstrated an inadequate response to NSAIDs and received no concomitant DMARDs other than MTX. 51,88,92,93,100–104 All patients in the smaller trial received concomitant DMARDs or had a history of DMARD therapy with inadequate response. 83
The baseline characteristics of the trial populations are summarised in Table 1. In both trials, around one-half of the randomised patients received concomitant MTX. Other DMARDs and NSAIDs were used concomitantly by patients in the smaller trial,83 but not by those in the larger trial. 51,88,92,93,100–104 The mean number of prior DMARDs used was similar between the trials, although as seen in trials of the other biologics, the trials clearly included patients who had not yet demonstrated a lack of response to at least two DMARDs. The proportion of patients with polyarticular disease between the two trials indicated that overall the disease was moderate to severe. The proportion of patients with spine involvement, and arthritis mutilans at baseline made up only a small proportion of the trial population. The similarity of the trials on other measures of disease activity (TJC, SJC and HAQ at baseline) suggests that significant differences between the populations in terms of overall disease severity are unlikely. Overall, the baseline characteristics demonstrate that the trial populations are similar and are likely to be representative of a population with PsA requiring DMARD or biologic therapy.
In both trials adalimumab was administered by subcutaneous injection every other week at a dose of 40 mg. Treatment with active drug or placebo was administered for 12 weeks in the smaller trial (Genovese et al. )83 and for 24 weeks in the larger trial (Adalimumab Effectiveness in Psoriatic Arthritis Trial – ADEPT). 51,88,92,93,100–104 In both trials the controlled phase was followed by a follow-up period, during which adalimumab was administered in an open-label fashion to all patients.
Outcome data derived under RCT conditions are available from both trials for PsARC, ACR 20, ACR 50 and ACR 70 and HAQ at week 12. The larger of the two trials also reported these outcomes at 24 weeks. In addition, this trial reported PASI 50/70/90 outcomes at 12 and 24 weeks, as well as data on progression of joint disease at 24 weeks expressed in terms of the mean TSS. 51,88,92,93,100–104 All randomised outcome data are summarised in Table 11, with pooled data presented in Table 12.
| Trial | Duration (weeks) | Outcomes | Adalimumab | Placebo | RR or mean difference (95% CI) | |
|---|---|---|---|---|---|---|
| ADEPT51,88,92,93,100–104 | 12 | PsARC | 94/151 (62%) | 42/162 (26%) | 2.40 (1.80 to 3.20), p < 0.05 | |
| ACR 20 | All pts | 88/151 (58%) | 23/162 (14%) | 4.10 (2.75 to 6.14), p < 0.05 | ||
| +MTX | 43/77 (55%) | |||||
| –MTX | 45/74 (61%) | |||||
| ACR 50 | All pts | 54/151 (36%) | 6/162 (4%) | 9.66 (4.28 to 21.79), p < 0.05 | ||
| +MTX | 27/77 (36%) | |||||
| –MTX | 27/74 (36%) | |||||
| ACR 70 | All pts | 30/151 (20%) | 1/162 (1%) | 32.19 (4.44 to 233.11), p < 0.05 | ||
| +MTX | 13/77 (17%) | |||||
| –MTX | 17/74 (23%) | |||||
| HAQ change from baseline: mean (SD) | –0.4(0.5) | –0.1(0.5) | –0.3 (–0.41 to –0.19), p < 0.001 | |||
| PASI 50a | All pts | 50/69 (72%) | 10/69 (14%) | 5.00 (2.77 to 9.03), p < 0.05 | ||
| +MTX | 17/29 (76%) | |||||
| –MTX | 28/40 (70%) | |||||
| PASI 75a | All pts | 34/69 (49%) | 3/69 (4%) | 11.33 (3.65 to 35.17), p < 0.05 | ||
| +MTX | 17/29 (59%) | |||||
| –MTX | 17/40 (43%) | |||||
| PASI 90a | All pts | 21/69 (30%) | 0/69 (0%) | 43.00 (2.66 to 696.04), p < 0.05 | ||
| +MTX | 11/29 (38%) | |||||
| –MTX | 10/40 (25%) | |||||
| 24 | PsARC | 91/151 (60%) | 37/162 (23%) | 2.64 (1.93 to 3.60), p < 0.05 | ||
| ACR 20 | All pts | 86/151 (57%) | 24/162 (15%) | 3.84 (2.59 to 5.70), p < 0.05 | ||
| +MTX | 42/77 (55%) | |||||
| –MTX | 44/74 (59%) | |||||
| ACR 50 | All pts | 59/151 (39%) | 10/162 (6%) | 6.33 (3.34 to 12.64), p < 0.05 | ||
| +MTX | 28/77 (36%) | |||||
| –MTX | 31/74 (42%) | |||||
| ACR 70 | All pts | 35/151 (23%) | 1/162 (1%) | 37.55 (5.21 to 270.70), p < 0.05 | ||
| +MTX | 17/77 (22%) | |||||
| –MTX | 17/74 (23%) | |||||
| HAQ change from baseline: mean (SD) | –0.4(0.5) | –0.1 (0.4) | –0.3 (–0.40 to –0.20), p < 0.001 | |||
| PASI 50a | All pts | 52/69 (75%) | 8/69 (12%) | 6.50 (3.34 to 12.64), p < 0.05 | ||
| +MTX | 25/29 (86%) | |||||
| –MTX | 27/40 (68%) | |||||
| PASI 75a | All pts | 41/69 (59%) | 1/69 (1%) | 41.00 (5.80 to 289.75), p < 0.05 | ||
| +MTX | 21/29 (72%) | |||||
| –MTX | 20/40 (50%) | |||||
| PASI 90a | All pts | 29/69 (42%) | 0/69 (0%) | 59.00 (3.68 to 946.75), p < 0.05 | ||
| +MTX | 15/29 (52%) | |||||
| –MTX | 14/40 (35%) | |||||
| TSS mean change from baseline | –0.2 (n = 144) | 0.1 (n = 152) | p < 0.001 | |||
| Genovese 200783 | 12 | PsARC | 26/51 (51%) | 14/49 (24%) | 1.78 (1.06 to 3.00), p < 0.05 | |
| ACR 20 | 20/51 (39%) | 8/49 (16%) | 2.40 (1.17 to 4.94), p < 0.05 | |||
| ACR 50 | 13/51 (25%) | 1/49 (2%) | 12.49 (1.70 to 91.90), p < 0.05 | |||
| ACR 70 | 7/51 (14%) | 0/49 (0%) | 14.42 (0.85 to 5.26), p = n.s. | |||
| HAQ change from baseline: mean (SD) | –0.3 (0.5) | –0.1 (0.3) | –0.2 (–0.36 to –0.04), p = 0.015 | |||
| 24 (open-label extension) | PsARC | 38/51 (75%) | 32/46 (70%) | – | ||
| ACR 20 | 33/51 (65%) | 26/46 (57%) | – | |||
| ACR 50 | 22/51 (43%) | 17/46 (37%) | – | |||
| ACR 70 | 13/51 (27%) | 10/46 (22%) | – | |||
| HAQ change from baseline: mean (SD) | –0.3 (0.5) | –0.4 (0.4) | – | |||
| Trial | Outcomes | Adalimumab | Placebo | RR or mean difference (95% CI) |
|---|---|---|---|---|
| PsARC | ||||
| ADEPT51,88,92,93,100–104 | 94/151 (62%) | 42/162 (26%) | 2.40 (1.80 to 3.20) | |
| Genovese 200783 | 26/51 (51%) | 14/49 (24%) | 1.78 (1.06 to 3.00) | |
| Pooled RR (95% CI), p-value | 2.24 (1.74 to 2.88), p < 0.0001 | |||
| I 2 | I2 = 0% | |||
| ACR 20 | ||||
| ADEPT51,88,92,93,100–104 | 88/151 (58%) | 23/162 (14%) | 4.10 (2.75 to 6.14) | |
| Genovese 200783 | 20/51 (39%) | 8/49 (16%) | 2.40 (1.17 to 4.94) | |
| Pooled RR (95% CI), p-value | 3.65 (2.57,5.17), p < 0.0001 | |||
| I 2 | I2 = 38% | |||
| ACR 50 | ||||
| ADEPT51,88,92,93,100–104 | 54/151 (36%) | 6/162 (4%) | 9.66 (4.28 to 21.79) | |
| Genovese 200783 | 13/51 (25%) | 1/49 (2%) | 12.49 (1.70 to 91.90) | |
| Pooled RR (95% CI), p-value | 10.08 (4.74 to 21.44), p < 0.0001 | |||
| I 2 | I2 = 0% | |||
| ACR 70 | ||||
| ADEPT51,88,92,93,100–104 | 30/151 (20%) | 1/162 (1%) | 32.19 (4.44 to 233.11) | |
| Genovese 200783 | 7/51 (14%) | 0/49 (0%) | 14.42 (0.85 to 5.26) | |
| Pooled RR (95% CI), p-value | 26.05 (5.18 to 130.88), p < 0.0001 | |||
| I 2 | I2 = 0% | |||
| HAQ change from baseline [mean (SD)] | ||||
| ADEPT51,88,92,93,100–104 | –0.4 (0.5) | –0.1 (0.5) | –0.3 (–0.41 to –0.19) | |
| Genovese 200783 | –0.3 (0.5) | –0.1 (0.3) | –0.2 (–0.36 to –0.04), p = 0.015 | |
| Pooled WMD (95% CI), p-value | –0.27 (–0.36, –0.18), p < 0.0001 | |||
| I 2 | I2 = 0.6% | |||
ADEPT51,88,92,93,100–104 reported longer-term open-label follow-up of patients at 48, 104, and 144 weeks. These data are summarised in Table 13.
| Trial | Type of data | Duration (weeks) | Outcomes | Adalimumab | Adalimumab/placebo |
|---|---|---|---|---|---|
| ADEPT51,88,92,93,100–104 | Uncontrolled | 48 | ACR 20 | – | 58.7% (165/281) |
| ACR 50 | – | 42.7% (120/281) | |||
| ACR 70 | – | 27.8% (78/281) | |||
| HAQ change from baseline: mean (median) | – | (n = 298) –0.3 (0.5) | |||
| PASI 50 | 67% (46/69) | 61% (42/69) | |||
| PASI 75 | 58% (40/69) | 53% (37/69) | |||
| PASI 90 | 46% (32/69) | 44% (30/69) | |||
| Mean (SD) TSS change from baseline | (n = 115) 0.1 (1.95) | (n = 128) 0.8 (4.23) | |||
| 104 | ACR 20 | – | 57.3% (161/281) | ||
| ACR 50 | – | 45.2% (127/281) | |||
| ACR 70 | – | 29.9% (84/281) | |||
| HAQ change from baseline: mean (median) | – | (n = 271) –0.3 (0.5) | |||
| 144 | Mean (SD) TSS change from baseline | (n = 115) 0.5 (4.20) | (n = 128) 0.9 (6.36) |
Efficacy after 12 weeks’ treatment
At 12 weeks, both trials reported a significant improvement in the PsA-specific PsARC measure for adalimumab relative to placebo (pooled RR 2.24; 95% CI 1.74 to 2.88), with an overall response rate of around 59% for adalimumab. The pooled RR for ACR 20 at 12 weeks was 3.65 (95% CI 2.57 to 5.17), demonstrating a clear degree of efficacy of adalimumab in terms of arthritis-related symptoms. There was no statistically significant heterogeneity between any of the pooled outcomes. The pooled RRs for ACR 50 and ACR 70 also clearly favoured adalimumab, although as with other estimates of these outcomes their related CIs were wide (Table 12). Again, the large relative differences on these higher-response thresholds reflect some response with biologic therapy versus virtually none with placebo (e.g. 18% vs 0.5% for ACR 70). Data from the larger trial indicated little evidence of any differential ACR response at 12 weeks between patients with and without concomitant MTX. 51,88,92,93,100–104
Only one trial reported 12-week PASI response measures: in patients with psoriasis of at least 3% BSA at baseline. 51,88,92,93,100–104 Response was significantly greater for adalimumab than placebo at all three PASI thresholds (PASI 50/75/90 – see Table 11). As with the ACR outcomes, there was little evidence of any differential PASI response between patients receiving and not receiving concomitant MTX, although the number of patients in each subgroup was small.
The statistically significant pooled absolute mean change from baseline in HAQ score with adalimumab compared with placebo (mean difference –0.27; 95% CI –0.36 to –0.18) indicates a beneficial effect of adalimumab on functional status.
Efficacy after 24 weeks’ treatment
The ADEPT trial51,88,92,93,100–104 maintained randomisation for 24 weeks. The data for all measures of joint disease, psoriasis and HAQ were all similar to those observed at the earlier 14-week follow-up, suggesting that the benefits of adalimumab are maintained for up to 24 weeks of treatment (see Table 12).
In addition, this trial51,88,92,93,100–104 reported a statistically significant difference in mean change in TSS score from baseline (–0.2 vs 0.1, p < 0.001), favouring adalimumab over placebo in terms of delayed progression of joint disease. However, this duration of follow-up is to be considered short and barely adequate for this outcome.
The smaller of the two trials allowed patients to enter an open-label follow-up period from weeks 12–24. 83 The pattern of reported joint disease outcomes appears similar to those reported at the end of the 12-week randomised period; however, estimates based on these non-randomised data cannot be considered reliable.
Longer-term follow-up
The larger adalimumab trial followed patients in an open-label fashion, measuring several outcomes at 48 weeks and at 2 years (Table 13). 51,88,92,93,100–104 Both ACR response rates and mean HAQ scores at weeks 48 and 104 appeared to have remained stable relative to the randomised observations of these outcomes at weeks 12 and 24. Similarly, rates of PASI response reported at 48 weeks appeared largely consistent with the earlier randomised observations. Disease progression as measured by TSS was reported at weeks 48 and 144, with higher mean values than observed at 24 weeks, although the open-label observational nature of these open-label data makes it difficult to reliably determine any clear changes in TSS over time.
Summary of the efficacy of adalimumab in the treatment of psoriatic arthritis
-
There is evidence from two double-blind placebo-controlled trials of a good level efficacy for adalimumab in the treatment of PsA, with beneficial effects on joint disease and functional status as assessed by HAQ.
-
There is limited evidence from a single RCT that adalimumab treatment has a beneficial effect on the psoriasis component of the disease in patients with PsA, as measured by PASI.
-
Conclusions to be drawn from these data are limited by the short duration the controlled trials; large-scale controlled data to evaluate long-term effects are not available.
-
Uncontrolled follow-up of patients indicate that treatment benefits in terms of joint disease and HAQ measures may be maintained at up to 2 years; however, these data may not be reliable.
-
Radiographic data from a single controlled trial for adalimumab in PsA demonstrate a beneficial effect on progression of joint disease at 24 weeks. This is a very short time over which to identify a statistically significant effect of therapy and indicates a rapid onset of action of adalimumab. Data from uncontrolled follow-up are inadequate to determine whether any potential delay in disease progression persists at 1–2 years’ follow-up.
Efficacy of all three biologics
As described above (see Data analysis), the Bayesian indirect comparison enables a comparison to be made across all three biologics despite the lack of head-to-head trial data. The three agents were included in the analysis, with placebo being the common comparator. All of the trials identified in the systematic review were used in the analysis, although not all trials provided data for all of the outcomes analysed. Full details of the methods used are given in Appendix 5.
Psoriatic Arthritis Response Criteria response
The results of the evidence synthesis for PsARC response are in the form of probability of response (Table 14). The mean probability of a PsARC response was estimated to be 71% for etanercept, 79% for infliximab and 59% for adalimumab, compared with 25% for placebo. While the credible intervals for all three biologics overlap each other, none overlap placebo.
| Treatment | Mean | Credible intervals (%) | |
|---|---|---|---|
| 2.50 | 97.50 | ||
| Placebo | 0.249 | 0.178 | 0.317 |
| Etanercept | 0.713 | 0.567 | 0.832 |
| Infliximab | 0.795 | 0.673 | 0.886 |
| Adalimumab | 0.587 | 0.444 | 0.713 |
Changes in Health Assessment Questionnaire
The results of the evidence synthesis of HAQ conditional on response are presented as absolute changes in HAQ. These are calculated separately for the patients achieving a PsARC response (Table 15) and those who did not achieve a PsARC response (Table 16).
| Treatment | Mean | Credible intervals (%) | |
|---|---|---|---|
| 2.50 | 97.50 | ||
| Placebo | –0.244 | –0.337 | –0.151 |
| Etanercept | –0.630 | –0.805 | –0.455 |
| Infliximab | –0.657 | –0.793 | –0.523 |
| Adalimumab | –0.477 | –0.596 | –0.351 |
| Treatment | Mean | Credible intervals (%) | |
|---|---|---|---|
| 2.50 | 97.50 | ||
| Placebo | 0 | 0 | 0 |
| Etanercept | –0.190 | –0.381 | 0.000 |
| Infliximab | –0.194 | –0.333 | –0.057 |
| Adalimumab | –0.130 | –0.262 | –0.001 |
Statistically significant reductions in mean HAQ score were achieved with all four treatments compared, i.e. the credible intervals did not include zero. However, patients who responded to placebo achieved an improvement in the HAQ score of –0.244, which is below the minimum clinically significant threshold for PsA of –0.3. 150 Patients who responded to etanercept and infliximab achieved similar mean changes in HAQ (–0.630 and –0.657, respectively), whereas responders to adalimumab achieved a lower mean change in the HAQ score of –0.477, although credible intervals overlap those of the other two treatments.
For all three biologics the changes in HAQ for those patients who did not respond to treatment were below the minimum clinically significant threshold. Placebo non-responders were used as a baseline in the synthesis.
Psoriasis Area and Severity Index
The results of the evidence synthesis for a PASI response are in the form of probability of response (Table 17). The mean probability of a PASI 75 response was estimated to be 18% for etanercept, 77% for infliximab and 48% for adalimumab, compared with 4% for placebo. The credible intervals for infliximab and etanercept do not overlap each other and none for the biologics overlap placebo.
| Mean | Credible intervals (%) | ||
|---|---|---|---|
| 2.50 | 97.50 | ||
| PASI 50 | |||
| Placebo | 0.130 | 0.092 | 0.175 |
| Etanercept | 0.403 | 0.236 | 0.592 |
| Infliximab | 0.913 | 0.823 | 0.968 |
| Adalimumab | 0.738 | 0.552 | 0.881 |
| PASI 75 | |||
| Placebo | 0.044 | 0.028 | 0.065 |
| Etanercept | 0.177 | 0.085 | 0.313 |
| Infliximab | 0.769 | 0.594 | 0.901 |
| Adalimumab | 0.477 | 0.275 | 0.693 |
| PASI 90 | |||
| Placebo | 0.018 | 0.010 | 0.026 |
| Etanercept | 0.074 | 0.032 | 0.145 |
| Infliximab | 0.557 | 0.347 | 0.767 |
| Adalimumab | 0.257 | 0.120 | 0.452 |
American College of Rheumatology model
The results of the evidence synthesis for a ACR response are in the form of probability of response (Table 18). The ACR 20 is generally accepted to be the minimal clinically important difference that indicates some response to a particular intervention in terms of arthritis-related symptoms. The mean probability of an ACR 20 response was estimated to be 61% for etanercept, 68% for infliximab and 56% for adalimumab, compared with 14% for placebo. The credible intervals for all three biologics overlap each other, but none overlap those for placebo.
| Mean | Credible intervals (%) | ||
|---|---|---|---|
| 2.50 | 97.50 | ||
| ACR 20 | |||
| Placebo | 0.137 | 0.108 | 0.168 |
| Etanercept | 0.609 | 0.459 | 0.750 |
| Infliximab | 0.678 | 0.533 | 0.805 |
| Adalimumab | 0.560 | 0.429 | 0.686 |
| ACR 50 | |||
| Placebo | 0.053 | 0.040 | 0.070 |
| Etanercept | 0.362 | 0.231 | 0.516 |
| Infliximab | 0.433 | 0.288 | 0.594 |
| Adalimumab | 0.315 | 0.209 | 0.438 |
| ACR 70 | |||
| Placebo | 0.018 | 0.012 | 0.025 |
| Etanercept | 0.158 | 0.087 | 0.260 |
| Infliximab | 0.203 | 0.114 | 0.326 |
| Adalimumab | 0.131 | 0.077 | 0.205 |
Summary of evidence synthesis results
Across all outcomes – PsARC, ACR and PASI – infliximab is associated with the highest probability of response. The response in joint disease (PsARC and ACR) is greater with etanercept than with adalimumab, whereas the response in skin disease (PASI) is greater with adalimumab than with etanercept, although these differences are not statistically significant. In those patients who achieve a PsARC response to treatment the highest mean reductions in HAQ are seen with infliximab and etanercept.
Comparison of evidence synthesis results
Each of the three company submissions combined evidence derived using Bayesian evidence synthesis methods. A brief comparison of these methods and the methods used by the assessment team are presented in Table 19 and discussed below.
| Interventions: etanercept, infliximab, adalimumab | |||||
|---|---|---|---|---|---|
| Abbott151 | Schering-Plough152 | Wyeth153 | Assessment team (York) | ||
| Studies used in the analysis | Mease 2000,78 Antoni 2003,117 Mease 2004,52,97,99,105,107,110 Antoni 2005,82 Kaltwasser 2004,62 Mease 2005,51 Mease 2006,97 Genovese 2007,83 Kavanaugh 2009,154 Gottlieb 2009155 | IMPACT,79–81,89,96,109,111,113–115,117,118 IMPACT 2,82,90,91,95,98,106,112,116 Mease 2000,78 Mease 2004, 52,97,99,105,107,110 ADEPT,51,88,92,93,100–104 Genovese 2007,83 York HTA,73 GO-REVEAL156 | Mease 2004, 52,97,99,105,107,110 PRESTA,157 ADEPT,51,88,92,93,100–104 IMPACT 2,82,90,91,95,98,106,112,116 STA ADL74 | IMPACT,79–81,89,96,109,111,113–115,117,118 IMPACT 2,82,90,91,95,98,106,112,116 Mease 2000,78 Mease 2004, 52,97,99,105,107,110 ADEPT,51,88,92,93,100–104 Genovese 200783 | |
| Outcomes of interest | PsARC | 12 and 24 weeks (24-week results estimated based on the conditional 12 weeks) | 12 or 14 weeks | 12 and 24 weeks; derived from STA ADL74 | 12 weeks |
| HAQ | 12 weeks (dependent on ACR response type via multivariate regression) | Weeks 12 and 24 for adalimumab; week 14 or 16 for infliximab; week 12 for etanercept (conditional on PsARC response) | Derived from Mease 2004; changes in HAQ were predicted via PASI; assumed equal magnitude of change in HAQ for all three biologics | HAQ at 12 weeks conditional on PsARC response at 12 weeks (by biologic) | |
| PASI 25/50/75 | 12 and 24 weeks (independently modelled for both 12 and 24 weeks) | Week 24 for adalimumab; week 14 or 16 for infliximab; week 24 for etanercept | PASI 75 only (12 and 24 weeks); derived from STA ADL74 and Mease 2004 | PASI 50/70/90 at 12 weeks (by biologic) | |
| ACR 20/50/70 | 12 and 24 weeks (24 week results estimated based on the conditional 12 weeks) | Not estimated | Not estimated | ACR 50/70/90 at 12 weeks (by biologic) | |
| Model | Bivariate probit model; Bayesian fixed-effects meta-analysis of bivariate ordinal data | Two joint meta-analyses: PsARC/HAQ and PASI | Model used not reported; the results were taken from a published evidence synthesis74 | Fixed-effects meta-analysis (PsARC, HAQ, ordered logit model PASI/ACR) | |
| Results reported | PsARC, ACR and PASI responses at 12 and 24 weeks: estimated means of marginal probabilities. Joint distribution of PsARC and ACR response at 12 weeks. Joint distribution of PASI 75 at 12 and 24 weeks | Incremental HAQ change given PsARC response in treatment; incremental HAQ change given PsARC non-response in treatment; incremental HAQ change given PsARC response in placebo; incremental HAQ change given PsARC non-response in placebo | PsARC (% patients), PASI 75, HAQ change from baseline, change in PASI | Probability of response in terms of PsARC, ACR and PASI; changes in HAQ given PsARC response/non-response to treatment | |
| Comments | Results ‘borrow’ information from trials of therapies not of interest (golimumab, leflunomide, alefacept and ustekinumab) | It was not possible to fully assess the results of the evidence synthesis performed as no details were provided even in the original publication159 | |||
Two of the company submissions – Abbott151 and Schering-Plough152 – conducted evidence syntheses to derive estimates that would allow the relative efficacy of the drugs to be compared (Table 19). (Since the production of this report, Schering-Plough has merged with Merck.) Wyeth153 chose not to conduct this synthesis themselves but to use the results of a previously published single technology appraisal (STA) relating to Abbott’s adalimumab. 74
Full details of the evidence synthesis model used by Wyeth153 were not provided in the Wyeth submission. 153 Further, the methodology of the evidence synthesis from which these results were obtained was not presented in the original report. 151 The synthesis was conducted by Abbott151 on the request from the Evidence Review Group (ERG) and only the results were presented in the ERG report. For this reason no summary/critique of the methods can be presented. The following section gives a comparative overview of the evidence synthesis results obtained by Schering-Plough,152 Abbott151 and by the Assessment Group in this report.
Psoriatic Arthritis Response Criteria response For PsARC response, all of the evidence synthesis models used a fixed-effects meta-analysis to synthesise the evidence. Both the Assessment Group and Schering-Plough152 identified and included six RCTs in their synthesis. Abbott,151 with slightly broader inclusion criteria, identified and included 10 RCTs. Abbott151 included RCTs in which the drug golimumab was administrated to the comparator arm of the RCT and, although no results were presented for this comparator, the other estimates do ‘borrow strength’ from these data. Although including the same six RCTs, both the Assessment Group and Schering-Plough152 estimated PsARC response using slightly different data. The Assessment Group used the closest follow-up outcome to 12 weeks, whereas Schering-Plough152 used the latest available end points (Table 20). This meant that, with the exception of the adalimumab data, the data inputs were principally the same. Abbott151 took a more complex bivariate approach, which enabled them to model the joint distribution of ACR/PsARC response at 12 weeks. Taking a bivariate approach allows the correlation between outcomes, if present, to be accounted for. However, if the correlation is zero then any bivariate joint modelling will arrive at the same estimates as two independent models. Given the lack of transparency of the Abbott151 evidence synthesis, it was not possible to unpick and decipher the subtleties of their model. The Assessment Group, following clinical advice, have used PsARC at 12 weeks to determine response to treatment. This follows clinical practice.
| Abbott151 | Schering-Plough152 | Assessment team (York) |
|---|---|---|
|
|
PsARC response
|
As can be seen from the results presented for the probability of response to the biologics under appraisal (and placebo) (Table 21), all of the mean estimates obtained were very similar, despite the different modelling assumptions and evidence used. There does appear to be some difference in the level of uncertainty, as presented by the confidence/credible intervals, but generally the means were close and the ranking consistent. The Abbott151 evidence synthesis model was extremely difficult to interpret; however, the analysis enabled the estimation of the joint probability of an ACR response and a PsARC response at 12 weeks. The 24-week results of the PsARC and ACR were then estimated individually, conditional on the 12-week response. Schering-Plough152 based their evidence synthesis on a previous HTA report,73 which linked two meta-analyses, one estimating PsARC the other HAQ conditional on PsARC.
| Treatment | Probability of response | |||||||
|---|---|---|---|---|---|---|---|---|
| Current assessment | Abbott151 | Schering-Plough152 | Wyeth153 | |||||
| Mean | Credible interval | Mean | Credible interval | Mean | Credible interval | Mean (%) | Credible interval (%) | |
| Placebo | 0.249 | (0.1779 to 0.3169) | 0.258 | NR | AiC | AiC | 26 | (21 to 31) |
| Etanercept | 0.713 | (0.5665 to 0.8317) | 0.743 | AiC | AiC | 76 | (46 to 96) | |
| Infliximab | 0.795 | (0.6725 to 0.8855) | 0.76 | AiC | AiC | 75 | (45 to 95) | |
| Adalimumab | 0.587 | (0.4441 to 0.713) | 0.591 | AiC | AiC | 57 | (24 to 85) | |
Health Assessment Questionnaire conditional on a Psoriatic Arthritis Response Criteria response The economic models developed by both the Schering-Plough152 and the Assessment Group required an estimate of the expected change in HAQ in the first 3 months for treatment responders and non-responders, as measured by PsARC. HAQ conditional on a PsARC response was modelled by both the Assessment Group and Schering-Plough. 152 The two modelling approaches were based on fixed-effects meta-analysis. The Schering-Plough152 approach uses two linked meta-analyses, which estimated the probability of response and then the mean reduction in HAQ score conditional on that response. The Assessment Group estimated the probability of PsARC response in one meta-analysis and then used this result to inform a second HAQ model. Both synthesis models used the same clinical trials to inform the HAQ–PsARC estimates. However, Schering-Plough152 used the latest available end points for HAQ, in contrast with the Assessment Group, who elected to use the 12- to 16-week HAQ data to reflect short-term benefits. Long-term benefits are considered explicitly in the economic model.
The results obtained (Table 22) were generally similar, with the drugs maintaining the same ranking. The differences may reflect the slightly differing modelling approaches or the difference in data used. The Assessment Group included only the five trials that reported HAQ outcomes for responders and non-responders. To enable them to include all six trials, Schering-Plough152 assumed that for the one trial where the data were not stratified by responder/non-responder78 the HAQ change for the PsARC non-responder was equivalent to the average HAQ change in the non-responders, as seen in other trials, and that the HAQ change for the PsARC responders could be inferred to match the reported mean HAQ change. The Assessment Group opted not to make this assumption, as it was not clear that it was appropriate or that it would have a significant impact on the results obtained. The Assessment Group took the decision to use only data that reported in a manner that facilitated modelling. The Schering-Plough152 report clearly states that six trials were considered; however, the detailed appendix and model code both appear to consider a seventh trial of the biologic golimumab. Although they state that this was used only to inform relationships between variables, the coding and appendix do not make this clear.
| Treatment | Current assessment | Abbott151 | Schering-Plough152 | Wyeth153 | ||
|---|---|---|---|---|---|---|
| Mean | Credible interval | Mean | Credible interval | |||
| Changes in HAQ – response | ||||||
| Etanercept | –0.630 | (–0.805 to –0.455) | NC | AiC | AiC | NC |
| Infliximab | –0.657 | (–0.793 to –0.523) | NC | AiC | AiC | NC |
| Adalimumab | –0.477 | (–0.596 to –0.351) | NC | AiC | AiC | NC |
| Changes in HAQ – no response | ||||||
| Etanercept | –0.190 | (–0.381 to 0.000) | NC | AiC | AiC | NC |
| Infliximab | –0.194 | (–0.333 to –0.057) | NC | AiC | AiC | NC |
| Adalimumab | –0.130 | (–0.1878 to 0.0652) | NC | AiC | AiC | NC |
| Placebo | NC | NC | ||||
| Changes in HAQ – response | ||||||
| All treatments | –0.244 | (–0.337 to –0.151) | NC | AiC | AiC | NC |
Abbott151 did not model HAQ conditional on response, although HAQ for the economic modelling section of their report they did state that relationships between ACR response rate and HAQ improvement, and PASI response and PASI improvement were developed in order to obtain estimates of HAQ and PASI improvement for responders and non-responders for each treatment.
This analysis estimated the expected change in HAQ in the first 3 months, conditional on treatment response. PsARC is not a baseline variable and therefore conditioning the analysis on PsARC response may be potentially biased. The analysis assumes there are no confounding factors (unrelated to treatment received) that change during the trial and affect both PsARC response and, independently, the change in HAQ.
Psoriasis Area and Severity Index 50/75/90 response The PASI outcomes were synthesised by Abbott,151 Schering-Plough152 and the Assessment Group. Schering-Plough152 elected to use absolute PASI change as their main outcome, on the basis that this was the most appropriate outcome for the economic modelling. As a result, the estimates obtained are not comparable with the Assessment Group or Abbott151 results, both of which elected to use probability of achieving each PASI outcome (50/75/90) as their main outcome. This was achieved using two different modelling approaches. The Assessment Group elected to use an ordered multivariate logit model, whereas Abbott151 chose to use a bivariate probit model. The logit and probit models are similar; both allow the different thresholds of PASI (50/75/90) to be modelled simultaneously, the ordered nature of the data to be maintained and an estimate of patients’ percentage reduction in PASI score from baseline to be obtained. The results estimated and presented in Table 23 are similar. As previously stated, the Abbott model151 was complex and (as felt by the assessment team) difficult to fully understand. As such it is not clear if data from all 10 included trials were used in the Abbott PASI model. 151 The data inputs for the Assessment Group model are reported in Appendix 5. Owing to a lack of reporting in some trials, the Assessment Group model included data from five trials, one of which provided data on only two of the outcomes (PASI 50/75).
| Treatment | Outcome | Probability of response | ||||||
|---|---|---|---|---|---|---|---|---|
| Current assessment | Abbott151 | Schering-Plough152 | Wyeth153 | |||||
| Mean | Credible interval | Mean | Credible interval | Mean (%) | Credible interval | |||
| Placebo | ||||||||
| PASI 50 | 0.1305 | (0.0917 to 0.1747) | 0.151 | NR | NC | 12 | (0.03 to 0.25) | |
| PASI 75 | 0.0445 | (0.0281 to 0.0654) | 0.049 | NR | NC | 4 | (0.01 to 0.09) | |
| PASI 90 | 0.0167 | (0.0098 to 0.0261) | 0.009 | NR | NC | |||
| Etanercept | ||||||||
| PASI 50 | 0.4026 | (0.2361 to 0.5916) | 0.393 | NR | NC | 39 | (0.03 to 0.81) | |
| PASI 75 | 0.1768 | (0.085 to 0.313) | 0.189 | NR | NC | 20 | (0.01 to 0.59) | |
| PASI 90 | 0.0737 | (0.0317 to 0.145) | 0.057 | NR | NC | |||
| Infliximab | ||||||||
| PASI 50 | 0.9128 | (0.823 to 0.968) | 0.915 | NR | NC | 82 | (0.47 to 0.97) | |
| PASI 75 | 0.7687 | (0.5943 to 0.901) | 0.774 | NR | NC | 64 | (0.2 to 0.88) | |
| PASI 90 | 0.5571 | (0.347 to 0.767) | 0.515 | NR | NC | |||
| Adalimumab | ||||||||
| PASI 50 | 0.7383 | (0.5518 to 0.881) | 0.732 | NR | NC | 65 | (0.11 to 0.92) | |
| PASI 75 | 0.4772 | (0.275 to 0.693) | 0.500 | NR | NC | 43 | (0.03 to 0.78) | |
| PASI 90 | 0.2571 | (0.119 to 0.4524) | 0.239 | NR | NC | |||
American College of Rheumatology 20/50/70 response Schering-Plough152 did not synthesise for this outcome. Both the Assessment Group and Abbott151 did, but again elected to use two differing modelling approaches, ordered logit and bivariate probit. The comparative results are presented in Table 24. The results are again similar, with the ranking of the drugs being maintained.
| Treatment | Outcome | Probability of response | |||||
|---|---|---|---|---|---|---|---|
| Current assessment | Abbott151 | Schering-Plough152 | Wyeth153 | ||||
| Mean | Credible interval | Mean | Credible interval | ||||
| Placebo | |||||||
| PASI 50 | 0.1369 | (0.108 to 0.168) | 0.132 | NR | NC | NC | |
| PASI 75 | 0.05347 | (0.04 to 0.07) | 0.048 | NR | NC | NC | |
| PASI 90 | 0.01806 | (0.013 to 0.025) | 0.012 | NR | NC | NC | |
| Etanercept | |||||||
| PASI 50 | 0.6093 | (0.459 to 0.75) | 0.578 | NR | NC | NC | |
| PASI 75 | 0.362 | (0.231 to 0.516) | 0.362 | NR | NC | NC | |
| PASI 90 | 0.1583 | (0.088 to 0.26) | 0.174 | NR | NC | NC | |
| Infliximab | |||||||
| PASI 50 | 0.6775 | (0.533 to 0.81) | 0.615 | NR | NC | NC | |
| PASI 75 | 0.4333 | (0.288 to 0.59) | 0.398 | NR | NC | NC | |
| PASI 90 | 0.2028 | (0.1138 to 0.326) | 0.199 | NR | NC | NC | |
| Adalimumab | |||||||
| PASI 50 | 0.5595 | (0.429 to 0.686) | 0.537 | NR | NC | NC | |
| PASI 75 | 0.3146 | (0.209 to 0.438) | 0.323 | NR | NC | NC | |
| PASI 90 | 0.1313 | (0.077 to 0.205) | 0.148 | NR | NC | NC | |
Abbott’s model151 produced estimates of 24-week ACR response conditional on the 12-weeks ACR response rate. The 12-week response rate was modelled as a joint distribution of 12-week PsARC and ACR response rates. The code and explanation of this modelling was not clear and therefore it was not possible to fully interpret all of the modelling conducted. As the Abbott economic model151 included both PsARC and ACR there was a need for them to estimate the correlation between these two outcomes. The correlation was estimated using the available evidence. However, it was unclear as to the number of trials informing the Abbott ACR synthesis151 and the correlation estimate. The Assessment Group have presented an ordered logit model, using data from all six trials. The estimates obtained were not used in the Assessment Group economic model, so it was not necessary to make any assumptions on the correlation between PsARC and ACR outcomes.
The annotated winbugs code, assumptions and data have been presented for all models used by the Assessment Group. Although it can be difficult to justify some of the differences in modelling assumptions taken by the various groups, the Assessment Group have tried to reflect clinical reality, minimise generalising assumptions and allow the results obtained to reflect the evidence obtained as part of the clinical review.
Review of adverse events
Overview of existing systematic reviews of adverse events
Several existing systematic reviews have investigated the safety of biologic agents. This section provides an overview of those reviews that were sufficiently rigorous to meet the Database of Abstracts of Reviews of Effects (DARE) inclusion criteria. 75 The searches (see Appendix 1) resulted in 16 potentially relevant reviews; 10 were excluded because of a failure to meet the DARE criteria or to report relevant data on adverse events of biologics. Six systematic reviews (Table 25) were therefore included in this overview.
| Study details | Intervention and patients | Searching and included studies | Analyses | Outcomes |
|---|---|---|---|---|
| Bongartz et al. 2006 158 |
Infliximab and adalimumab 5014 patients with RA |
Data sources: MEDLINE, EMBASE and the Cochrane Library were searched from inception to December 2005. The abstract databases of annual scientific meetings of EULAR and the ACR were searched from 1996 to 2005 Included studies: Nine RCTs (four RCTs of infliximab; five RCTs of adalimumab) |
Studies were combined using a fixed-effects model of Mantel–Haenszel method. Pooled ORs with 95% CIs were calculated, with a continuity correction method for sparse data. The effects for high and low doses of anti-TNFs were estimated separately. The number-needed-to-harm with 95% CI was also calculated. Statistical heterogeneity was assessed using I2-statistic. Sensitivity analyses were performed with exclusion of trials of moderate or high risk of bias, omission of malignancies diagnosed within the first 6 weeks of a trial, and omission of malignancies that were classified as NMSCs | The pooled OR for malignancy was 3.3 (95% CI 1.2 to 9.1) and for serious infection was 2.0 (95% CI 1.3 to 3.1). Malignancies were significantly more common in patients received higher doses of biologics compared with patients received lower doses of biologics. For patients with biologic treatment in included RCTs, the NNH was 154 (95% CI 91 to 500) for one additional malignancy within a treatment period of 6–12 months. For serious infections, the NNH was 59 (95% CI 39 to 125) within a treatment period of 3–12 months |
| Bongartz et al. 2009 159 |
Etanercept 3316 patients with RA |
Data sources: MEDLINE, EMBASE, the Cochrane Library and the Web of Science were searched from inception to December 2006. Pharmaceutical companies were contacted for unpublished trials Included studies: Nine RCTs |
Studies were combined using a random-effects model of DerSimonian–Laird model. Pooled HRs with 95% CIs were calculated using IPD. A survival analysis of time-to-first-event using the Cox’s proportional hazards model stratified by trial and assuming a fixed treatment effect was conducted. Sensitivity analyses were performed by omitting cancers diagnosed within 6 weeks of trial entry and omitting all NMSCs from case definition. Subgroup analyses were performed for three non-overlapping periods of follow-up time (< 6 months, 6–12 months, > 24 months). In addition, pooled ORs with 95% CIs were calculated using the Mantel–Haenszel model with a continuity correction method |
The pooled HR for malignancies based on IPD data was 1.84 (95% CI 0.79 to 4.28) in patients using etanercept compared with control patients. The random-effects model resulted in a similar estimate of an HR of 1.82 (95% CI 0.78 to 4.22). When using Mantel–Haenszel methods, the pooled OR for malignancies in patients using etanercept compared with patients receiving control treatment was 1.93 (95% CI 0.85 to 4.38). When using a random-effects DerSimonian–Laird model, the pooled HR malignancies in patients receiving etanercept compared with patients receiving control treatment was 1.71 (95% CI 0.73 to 4.01) With the exclusion of four malignancies that were diagnosed during the first 6 weeks after the first treatment dose, the HR for malignancies in patients treated with etanercept compared with the non-etanercept group was 1.87 (95% CI 0.75 to 4.62). With the exclusions of all NMSC from analyses, similar results were found (HR 1.86, 95% CI 0.62 to 5.59). When the data were stratified according to three different time points: 0–6 months; 6–12 months and > 12 months, it did not show a particular time period in which the risk of cancer was significantly increased |
| Brimhall et al. 2008 163 |
Etanercept and infliximab 7931 patients with moderate-to-severe psoriasis |
Data sources: MEDLINE, EMBASE, the Cochrane Central Register of Controlled Trials and ClinicalTrials.gov were searched from inception to June 2005, and an updating search was conducted in July 2006 to capture reports from the interim period. Industry sponsors were contacted to additional unpublished data FDA reports were reviewed Included studies: 16 RCTs |
Studies were combined in meta-analyses using the Mantel–Haenszel method, with a constant continuity correction. The synthesis results from the random-effects models were also reported. Bioequivalent or equivalent FDA-approved doses were pooled for each biologic agent. The safety of biologic agents was assessed by RR of one or more AEs and SAEs for all doses. All dosages were combined for comparison. The NNT and the NNH, with 95% CIs, were calculated. Statistical heterogeneity was measured using Q-statistic |
Etanercept: The pooled RR of one or more AEs was not significantly increased for patients receiving etanercept (RR 1.05, 95% CI 0.96 to 1.16, p = 0.28). Similar results were observed for the incidence of SAEs (RR 1.17, 95% CI 0.59 to 2.33, p = 0.66). The most common reported AEs reported were injection-site reaction, headache and URTI. The most common SAEs were malignancy (n = 10), serious infection (n = 4) and worsening psoriasis (n = 3). Both AEs and SAEs were evaluated cumulatively over 12–24 weeks of the treatment Infliximab: The pooled RR for one or more AEs was significantly associated with an increased one or more AEs compared with placebo (RR 1.18, 95% CI 1.07 to 1.29, p < 0.001), with NNH of 9 (95% CI 5.99 to 19.61). The most common reported AEs were URTI, headache, increased hepatic enzymes and infection. Infliximab was not associated with a significant increase in SAEs (RR 1.26, 95% CI 0.56 to 2.84, p = 0.58). The most common SAEs reported were malignancy (n = 12), serious infection (n = 6), serious infusion reaction (n = 4) and lupus-like syndrome (n = 4). Both AEs and SAEs were evaluated across 10–30 weeks of the treatment |
| Gartlehner et al. 2006 160 |
Etanercept, infliximab and adalimumab The review included patients with RA who have failed to respond to traditional DMARD therapy. For indirect comparison, the authors pooled data for 2354 patients receiving adalimumab (five studies), for 1151 patients receiving etanercept (five studies) and for 704 patients receiving infliximab (four studies). The total number of patients in the review was not reported |
Data sources: MEDLINE, EMBASE, The Cochrane Library, and the international pharmaceutical abstracts were searched from 1980 to 2006. Reference lists of relevant publications were searched. The Centre for Drug Evaluation and Research database was searched for unpublished research. Pharmaceutical companies were contacted for unpublished trials Included studies: 26 RCTs for efficacy and 18 studies (experimental and observational) for AEs |
Studies were combined in meta-analyses using random-effects models. Subgroup analyses were conducted for the population who had remained symptomatic despite the MTX treatment. Subgroup analyses were also performed by only including data to FDA-approved dosage ranges to achieve better equivalency across drugs Statistical heterogeneity was measured using I2-statistic and metaregression. Publication bias was assessed using funnel plots and Kendall’s tests. Where there were no direct head-to-head studies comparing an anti-TNF with another, an indirect comparison was undertaken using placebo as the common comparator. For the AE data, the evidence was summarised qualitatively |
Adalimumab: When the studies were pooled, adalimumab was associated with weighted mean incidence of diarrhoea (8.16, 95% CI 4.44 to 11.88), headache (18.23, 95%CI 6.51 to 29.95), infection site (18.98, 95% CI 9.21 to 28.76), nausea (8.84, 95% CI 5.55 to 12.13), rhinitis (14.8, 95% CI 7.26 to 22.35) and URTI (17.05, 95% CI 9.5 to 24.59) Etanercept: Etanercept was associated with weighted mean incidence of diarrhoea (18.14, 95% CI 3.45 to 32.84), headache (17.54, 95%CI 1.9 to 33.18), infection site (24.67, 95% CI 11.21 to 38.13), nausea (20.86, 95% CI 2.65 to 39.08), rhinitis (18.42, 95% CI 6.97 to 35.71) and URTI (20.89, 95% CI 6.97 to 34.82) Infliximab: Infliximab was associated with weighted mean incidence of diarrhoea (9.31, 95% CI 7.94 to 10.68), headache (17.7, 95% CI 3.03 to 33.36), rhinitis (7.77, 95% CI 0 to 18.12) and URTI (24.05, 95% CI 0 to 49.81) In addition, rare but SAEs (e.g. serious infections, lymphoma or neutropenia) were of concern in the included trials but could not be reliably assessed |
| Ravindran et al. 2008 161 |
Etanercept, infliximab and adalimumab 2039 patients with PsA, in total, receiving the treatment of anti-TNFs, sulfasalazine, gold salts, leflunomide and DMARDs (882 patients with PsA receiving anti-TNFs) |
Data sources: MEDLINE, EMBASE were searched from 1966 to June 2006. The Cochrane Clinical Trials Register and Cochrane Database for Systematic Reviews were also searched. Reference lists of relevant publications were also searched Included studies: 18 RCTs |
Studies were combined in meta-analyses using random-effects models. The pooled RRs with 95% CIs for dichotomous outcomes were calculated. The pooled Peto ORs with 95% CIs were calculated for the outcome of overall toxicity, based on withdrawals due to side-effects. Sensitivity analyses were performed. based on agents used and outcome measured. The ratio of NNT to NNH was calculated to assess the benefit vs risk of each treatment | When the studies (two RCTs of etanercept, two RCTs of infliximab and one RCT of adalimumab) were pooled, anti-TNF treatment was associated with a non-significant increase of withdrawal rate due to toxicity compared with placebos (RR 2.2, 95% CI 0.82 to 5.91, p = 0.12; five RCTs). Anti-TNFs were associated with a high ratio (0.25) of NNT) to NNH |
| Saad et al. 2008 162 |
Etanercept, infliximab and adalimumab 982 patients with PsA |
Data sources: MEDLINE, EMBASE, CINAHL, and the CCTR were searched from inception to May 2007. The US FDA and European Medicines Evaluation Agency websites were searched. Reference lists of relevant publications were also screened Included studies: Six RCTs |
Studies were combined in meta-analyses using random-effects models. The pooled RRs and RDs for dichotomous outcomes, with 95% CIs, were calculated. The WMDs for continuous outcomes, with 95%CIs were also calculated. Statistical heterogeneity was measured using chi-squared and I2-statistics. Where there were no direct head-to-head studies comparing an anti-TNF with another, an indirect comparison was undertaken using placebo as the common comparator |
There were no significant differences between biologics and placebos in the proportion of patients experiencing withdrawals for any reason (RR 0.48, 95% CI 0.20 to 1.18), withdrawal due to AEs (RR 2.14, 95% CI 0.73 to 6.27), SAEs (RR 0.98, 95% CI 0.55 to 1.77), and URTIs (RR 0.91, 95% CI 0.65 to 1.28). The pooled rate for injection site reactions were significantly higher for adalimumab and etanercept compared with placebos (RR 2.48, 95% CI 1.16 to 5.29). There was no significant difference in the proportion of patients experiencing infusion reactions with infliximab compared with placebos (RR 1.03, 95% CI 0.48 to 2.20) Significant heterogeneity was only observed in the outcome of withdrawal for any reason (I2 = 53.1%, p = 0.07). Indirect analyses did not show any significant differences between these biologics in the proportion of patients experiencing SAEs. Five RCTs (n = 922) monitored the incidence of malignancies during treatment; only one patient in the placebo group developed a basal cell carcinoma of the skin |
All of the six systematic reviews were published between 2006 and 2009. Three reviews158–160 included patients with RA and three reviews161–163 included patients with PsA or psoriasis. Almost all reviews evaluated the safety of more than two biologics. The sample size of included reviews varied from 982 to 7931. Almost all systematic reviews included RCTs to assess the safety of biologics, whereas only one review160 included both RCTs and observational studies. The search strategies were generally adequate to identify both published and unpublished studies, thereby minimising the potential of publication bias. 164,165 However, in the majority of these reviews158–161,163 it was unclear whether any language restrictions on study inclusion were made, which may have introduced the possibility of language bias. 166
There were variations in methods of pooling the adverse event data in these reviews. Five reviews158,159,161–163 used meta-analyses to synthesise the evidence of adverse event data of biologics, whereas one review used a narrative synthesis. 160 For those using meta-analyses, the included studies were combined using either a fixed-effects or random-effects model; one review by Bongartz et al. 159 also used the individual patient data (IPD) to pool the results. Where there were no direct head-to-head studies comparing one biologic with another, an indirect comparison was undertaken using placebo as the common comparator in two reviews. 158,162 Statistical heterogeneity167,168 was adequately assessed in most reviews. In addition, three reviews assessed the adverse events for more than two biologics combined,158,161,162 whereas the other reviews evaluated them for each biologic respectively. 159,160,163
A range of adverse events of biologics were evaluated in these reviews. Three reviews160,162,163 evaluated both common and serious adverse events of biologics, whereas two reviews exclusively focused on serious adverse events such as malignancy. 158,159 Two reviews161,162 used withdrawal rate due to toxicity/adverse events of biologics as the review outcome.
There were considerable variations in the effect estimations between the reviews. Brimhall et al. 163 reported that there were no significant increased incidences of one or more adverse events or serious adverse events for patients receiving etanercept; they also reported that there was no significant increase in the incidence of serious adverse events for patients receiving infliximab compared with those receiving placebo, although patients who received infliximab experienced a significant increased incidence of one or more adverse events. It should be noted that this systematic review was limited to short-term safety data of over 10–30 weeks of the biologic treatment. The review by Gartlehner et al. ,160 which principally evaluated the common adverse events of biologics, showed similar results based on the data from 18 experimental and observational studies for patients with RA. This review reported that biologics appeared to have a good tolerability profile; injection site reactions or infusion reactions were the most commonly reported adverse events for biologics of etanercept, infliximab and adalimumab. However, a lack of sound long-term safety data prevented this review from drawing a firm conclusion about the comparative safety between these three biologics for patients with RA.
Both the review by Ravindran et al. 161 and the review by Saad et al. 162 used the withdrawal rate due to toxicity/adverse events as the outcome measure to assess the safety of biologics. These are two reviews that exclusively include patients with PsA. The review by Ravindran et al. 161 reported that biologic treatment for patients with PsA was associated with a non-significant increase of withdrawal rate due to toxicity compared with placebo, when pooling the data from five RCTs of etanercept, infliximab and adalimumab. Similar results were found in the review by Saad et al. 162 on the basis of the pooled results of five RCTs (including the same four RCTs as Ravindran et al. 161), which also reported a non-significant difference between biologics and placebo in the proportion of patients with PsA experiencing withdrawals due to adverse events or serious adverse events. It should be noted that this outcome measure is associated with a methodological limitation: it is difficult to discern withdrawals due to adverse events from those due to poor efficacy, and those that result from a combination of both. In addition, the lack of long-term adverse event data in these two reviews makes it difficult to assess rare but potentially serious adverse events (e.g. malignancy or serious TB infection) of biologics for patients with PsA.
Two reviews assessed the serious adverse events of malignancy and/or serious infections due to use of biologics for patients with RA. 158,159 Bongartz et al. 158 reported that malignancies were significantly more common in patients treated with biologics compared with placebo: the pooled odds ratio (OR) for malignancy in patients receiving infliximab and adalimumab compared with placebo was 3.3 (95% CI 1.2 to 9.1) and for serious infection was 2.0 (95% CI 1.3 to 3.1). Malignancies were also significantly more common in patients receiving higher doses of biologics than in patients receiving lower doses of biologics. However, some inconsistent findings were reported in the review by Bongartz et al. ,159 which exclusively assessed the serious adverse event of malignancy for etanercept. This review reported that the pooled increased hazard ratio (HR) for malignancies based on IPD was not statistically significant (HR 1.84, 95% CI 0.79 to 4.28) in patients using etanercept compared with placebo or mixed control patients being treated with one DMARD. Similar non-significant results were also generated from the random-effects models. It is noteworthy that the pooled estimate of malignancy due to use of biologics in both of the reviews was limited to short-term follow-up; there is a necessity to evaluate the risk of malignancy of biologics on long-term follow-up durations.
Based on these reviews of adverse events of biologics, in general there is a concern that biologics may be associated with an increased risk of infection and malignancy. Due to some inconsistencies in the results and variations in methods of synthesising the data, no firm conclusions could be drawn from these reviews about the evidence of adverse events of biologics, especially for these serious adverse events. The lack of long-term adverse event data in the majority of reviews could compromise any comparative safety estimation between biologics. Furthermore, a probable exacerbation of latent TB is also considered to be potentially associated with use of biologics. 146,169–171 However, no reviews have addressed this outcome. In particular, adalimumab is a new drug for which there is only limited experience on long-term monitoring; further investigation on its safety is warranted.
In light of the outstanding uncertainties around the findings of previous reviews of biologic safety, our systematic review (see the following section) specifically focused on the serious potential adverse events of these biologics: malignancies, severe infections (i.e. those that require i.v. antibiotic therapy and/or hospitalisation or cause death) and reactivation of latent TB. Apart from RCTs, our systematic review also included observational studies in order to evaluate the long-term adverse events of biologics.
Review of primary studies
Two main sources of information on adverse events were incorporated into the review: RCTs evaluating etanercept, infliximab and adalimumab in PsA, and controlled and uncontrolled studies or registry data in which at least 500 patients with any indication received one or other of these agents.
As the identified non-randomised studies were highly heterogeneous, and because some studies using the same registry at different time points (thereby being likely to contain an overlap in patient data), the range of rates have summarised in a narrative synthesis, and no attempt has been made to pool values across studies. Reported percentage rates of adverse events are presented for randomised trials and single-arm studies. For non-randomised controlled studies in which the length of follow-up differed between groups, results are presented as the number of events per 100 patient-years where reported.
Etanercept
Randomised controlled trials in psoriatic arthritis Two placebo-controlled RCTs evaluated etanercept in patients with PsA. The first, which followed 60 patients for 12 weeks, reported more infections in the etanercept group than the placebo group for respiratory tract infection (27% vs 13%, respectively), pharyngitis (17% vs 10%), rhinitis (17% vs 13%) and sinusitis (10% vs 7%). Influenza was more commonly reported in the placebo group (0% vs 20%). 78 However, given the small numbers of patients in each group, these differences could be attributable to the play of chance. No deaths or withdrawals due to adverse events were reported for either group. Data on cancer and TB were not clearly reported.
A second, larger placebo-controlled RCT by the same authors, followed 205 patients over 24 weeks. 52,97,99,105,107,110 One patient in the placebo group died following surgical complications, and one patient from each group withdrew from the study. There were no reported cancers. Similar rates were observed between the etanercept and placebo groups for upper respiratory tract infection (URTI) (21% vs 23%), sinusitis (6% vs 8%) and urinary tract infection (6% vs 6%), although again, this efficacy study was not powered to detect a difference between groups in terms of adverse events. TB was not reported.
Non-randomised studies/large randomised controlled trials in other conditions Thirteen non-randomised studies, in which more than 500 patients received biologic agents, reported adverse event data for etanercept. The majority of treated patients had RA, although outcomes for PsA, juvenile idiopathic arthritis, ankylosing spondylitis and patients with other chronic inflammatory conditions were also reported (Table 26). Average length of follow-up ranged from 48 weeks to 7 years.
| Study | Total infections | Serious infections (%) | Cancers | TB (%) | Mortality | Withdrawals to AE |
|---|---|---|---|---|---|---|
| Brassard 2006135 | – | – | – | 1.40 | – | – |
| Carmona 2005141 | – | – | – | 0.00 | – | – |
| Dixon 2006136 | – | 5.80 | – | 0.06 | – | – |
| Dixon 2007147 | – | 11.20 | – | – | – | – |
| Favalli 2009129 | – | 4.50 | – | 0.40 | – | – |
| Feltelius 2005142 | 11 | 2.60 | 1 | – | 0.30 | 5.50 |
| Fleischmann 200699 | 54.40 | 4.90 | – | 0 | 0.90 | 6.50 |
| Gomez-Reino 2003146 | – | – | – | 0 | – | – |
| Gomez-Reino 2007132 | – | – | – | 0 | – | – |
| Horneff 2009125 | 9.60 | 4.30 | – | – | 0 | – |
| Klareskog 2006120 | 26.50 | 16.20 | 1.30 | 0 | 1.80 | 4.60 |
| Listing 2005122 | 21.30 | 6.10 | – | 0 | – | – |
| Mease 200697 | 1.80 | 0.60 | – | – | 0 | 0 |
| Moreland 2006121 | – | 13.20 | 5.70 | 0 | 3.10 | 13.60 |
The total reported rate of infections ranged from 9.6% to 54.4% (reported by five studies), with serious infections (requiring hospitalisation) ranging from 2.6% to 16.2% (nine studies). Only three studies clearly reported cancer, with rates ranging from 1% to 5.7%. Seven out of eleven studies reporting rates of TB in patients receiving etanercept found no cases. The remaining four studies reported rates ranging from 0.03% to 1.4%. Four studies reported rates of withdrawal due to adverse events, ranging from 4.6% to 13.6%. Where reported, mortality ranged from 0% to 3.1% (five studies).
Two of these studies compared adverse event rates in patients receiving etanercept against control. 99,122 One cohort study122 reported significantly more infections in patients with RA receiving etanercept than control patients (22.6 vs 6.8 infections per 100 patient-years, p < 0.01; 6.4 vs 2.3 serious infections per 100 patient-years, p < 0.01). However, a second study, an analysis of collated trial data on the use of etanercept reported no significant difference in overall infection rates between etanercept and control (placebo or MTX) across a range of conditions (54.4% vs 41.4%, p > 0.05). 99
Infliximab
Randomised controlled trials in psoriatic arthritis Two placebo-controlled RCTs evaluated infliximab in patients with PsA. 79–82,89–91,95,96,98,106,109,111–118 One RCT followed 104 patients over 16 weeks, reporting more respiratory tract infections in placebo-treated patients than in infliximab-treated patients (9.8% vs 1.9% respectively), though reported rates of bronchitis (7.8% vs 5.8%) and rhinitis (3.9% vs 5.7%) were similar between groups. 79–81,89,96,109,111,113–115,117,118 However, the very small numbers of events reported preclude any meaningful interpretation of these differences. No deaths or withdrawals were reported for either group.
The second RCT followed 200 patients over 24 weeks and reported similar rates between infliximab and placebo groups for URTI (10% vs 14%), pharyngitis (5% vs 4%) and sinusitis (5% vs 4%), although as with other RCTs, the study was not powered to detect differences in adverse events. 82,90,91,95,98,106,112,116 One patient in the placebo group developed basal cell carcinoma of the skin, although no deaths or withdrawals due to adverse events were reported.
Non-randomised studies/large randomised controlled trials in other conditions Eighteen non-randomised studies and two RCTs in indications other than PsA reported adverse event data for infliximab. Outcomes were reported for patients with PsA, juvenile idiopathic arthritis and ankylosing spondylitis, although the vast majority of patients had RA (Table 27). Average length of follow-up ranged from 22 weeks to 6 years.
| Study | Total infections (%) | Serious infections | Cancer (%) | TB (%) | Mortality | Withdrawals to AE (%) | |
|---|---|---|---|---|---|---|---|
| Antoni 200889 |
URTI 38.5 Diarrhoea 9.0 Pharyngitis 9.0 Sinusitis 5.1 UTI 5.1 |
2.6 | 5.1 | 0 | – | 6.4 | |
| Brassard 2006135 | – | – | – | 1.8 | – | – | |
| Caspersen 2008128 | – | 10.1 | 0.6 | 0.3 | 2.0 | – | |
| Carmona 2005141 | – | – | – | 4.6 | – | – | |
| Colombel 2004124 | 9.6 | 3.0 | 1.8 | – | 2.0 | – | |
| Dixon 2006136 | – | 8.9 | – | 0.2 | – | – | |
| Dixon 2007147 | – | 13.8 | – | – | – | – | |
| Favalli 2009129 | – | 8.1 | – | 0.6 | – | – | |
| Fidder 2009119 | – | 6.5 | 2.9 | 0.1 | 1.6 | – | |
| Gomez-Reino 2003146 | – | – | – | 1.1 | – | – | |
| Gomez-Reino 2007132 | – | – | – | 0.4 | – | – | |
| Listing 2005122 | 26.6 | 5.8 | 0.3 | ||||
| Oka 2006137 | – | 3.1 | – | 0.3 | 0.06 | – | |
| Schnitzler 2009127 | – | 0.8 | 0.16 | – | 1.6 | 12.8 | |
| St. Clair 2004143 |
URTI 26.7 Sinusitis 9.7 Pharyngitis 13.8 |
5.3 | 0.5 | 0.5 | 0.27 | 9.6 | |
| Takeuchi 2008130 | 8.7 |
Bacterial pneumonia 2.2 Interstitial pneumonitis 0.5 |
0.16 | 0.3 | – | – | |
| Westhovens 2006139 | 0–22 weeks |
URTI 10.8 Pharyngitis 4.7 Sinusitis 4.2 Pneumonia 0.8 TB 0.4 Cellulitis 0.3 UTI 0.3 |
Pneumonia 0.8 TB 0.4 Cellulitis 0.3 UTI 0.3 |
2.6 | 0.4 | – | 5.3 |
| 22–54 weeks | 35.4 | 3.1 | 2.6 | 0.4 | 0.4 | 8.0 | |
| Wolfe 2004144 | – | – | – | 0.06 | – | – | |
The total reported rate of infections ranged from 8.7% to 26.6% (reported by four studies). Where detailed separately, the most common infections were URTIs, ranging from 10.8% to 38.5% (three studies). Serious infections (requiring hospitalisation) ranged from 0.8% to 13.8% (12 studies). Eight studies reported total cancers, with rates ranging from 0.16% to 5.1%. Sixteen studies reported rates of TB in patients receiving infliximab, 11 of which reported rates less than 0.5%, with the overall range being 0% to 4.6%. Where reported, mortality ranged from 0.06% to 2% (seven studies). Four studies reported rates of withdrawal due to adverse events, ranging from 5.3% to 12.8%.
Four of the studies compared adverse event rates for patients receiving infliximab against some form of control group. 119,122,139,143 Two of these were RCTs of infliximab versus placebo plus MTX in RA,139,143 of which one found no difference in serious infections between groups at 22 weeks (3.3% vs 1.7%, p > 0.05),139 and one reported significantly more serious infections associated with infliximab at around 54 weeks (5.3% vs 2.1%, p < 0.05). 143 Two cohort studies compared adverse event rates between infliximab and control patients: one reported significantly higher rates of overall infections (28.3 per 100 patient-years vs 6.8 per 100 patient-years, p < 0.01) and serious infections (6.2 per 100 patient-years vs 2.3 per 100 patient-years) among patients with RA receiving infliximab,122 the second reported no significant differences in serious infections (1.6 per 100 patient-years vs 1.1 per 100 patient-years) or cancer (0.4 per 100 patient-years vs 0.5 per 100 patient-years) or mortality (0.3 per 100 patient-years vs 0.2 per 100 patient-years). 119
Adalimumab
Randomised controlled trials in psoriatic arthritis The smaller of the two RCTs evaluating adalimumab (102 patients over 12 weeks) reported more overall infections in placebo-treated than adalimumab-treated patients (32.7% vs 17.6%, respectively), with the infection classified as ‘serious’ for a single patient in each group. Reported rates of URTI were 8.2% and 13.7%, respectively. 83 As with other RCTs, small numbers of events reported limit meaningful interpretation of these differences. No deaths were reported for either group, and the small proportions of withdrawals were comparable.
The larger trial, which randomised 315 patients over 24 weeks, reported similar rates between adalimumab and placebo groups for URTI (12.6% vs 14.8%, respectively) and nasopharyngitis (9.9% vs 9.4%). 51,88,92,93,100–104 Serious infections were reported in three patients; two receiving adalimumab and one receiving placebo. No deaths were reported.
Non-randomised studies/large randomised controlled trials in other conditions Eight non-randomised studies and two RCTs in indications other than PsA reported adverse event data for adalimumab. Outcomes were reported for patients with PsA, juvenile idiopathic arthritis and ankylosing spondylitis, although, as for the other agents, most patients had RA (Table 28). Average length of study follow-up ranged from 12 weeks to 5 years.
| Study | Total infections (%) | Serious infections (%) | Cancers (%) | TB (%) | Mortality | Withdrawals to AE (%) | |
|---|---|---|---|---|---|---|---|
| Breedveld 2006140 | 9.12 | 2.20 | 1.10 | 0.18 | 0.90 | 10.70 | |
| Burmester 2007131 | – | 3.10 | 0.70 | 0.30 | 0.50 | 10.30 | |
| Carmona 2005141 | – | – | – | 0 | – | – | |
| Dixon 2006136 | – | 5.10 | – | 0.08 | – | – | |
| Dixon 2007147 | – | 7.30 | – | – | – | – | |
| Colombel 2007133 | 0–4 weeks | 15.20 | 1.20 | 0.20 | 6.30 | ||
| 4–56 weeks | 45.30 | 2.70 | 0.20 | 0.40 | 5.80 | ||
| Favalli 2009129 | – | 6.60 | – | 0.30 | – | – | |
| Gomez–Reino 2007132 | – | – | – | 0.20 | – | – | |
| Rudwaleit 2009126 | – | 0.40 | – | – | – | – | |
| Schiff 2006138 | – | 6.30 | 0.10 | 0.30 | – | – | |
The total reported rate of infections ranged from 9.1% to 45.3% (three studies), with serious infections ranging from 0.4% to 7.3% (nine studies). Four studies reported total cancer, with rates ranging from 0.1% to 1.1%. Eight studies reported rates of TB in patients receiving infliximab, ranging from 0% to 0.4%. Four studies reported rates of withdrawal due to adverse events, ranging from 5.8% to 10.7%. Where reported, mortality ranged from 0.2% to 0.9% (three studies).
Two of these studies were RCTs of adalimumab in conditions other than PsA. 133,140 One RCT of adalimumab alone or in combination with MTX against MTX alone in patients with RA, reported no difference between adalimumab monotherapy and MTX monotherapy in terms of overall infections (110 per 100 patient-years vs 119 per 100 patient-years), serious infections (0.7 per 100 patient-years vs 1.6 per 100 patient-years), or cancer (0.9 per 100 patient-years in each group). However, significantly more serious infections were observed for combined adalimumab/MTX therapy than for adalimumab monotherapy (2.9 per 100 patient-years vs 0.7 per 100 patient-years, p < 0.05). 140 The second RCT reported that, after 56 weeks of treatment in patients with Crohn’s disease, no significant differences were found between adalimumab and placebo in terms of overall (45.3% vs 36.8%) or serious infection rates (2.7% vs 3.4%). 133
Studies reporting more than one agent
No RCTs exist that provide a head-to-head comparison between any of the three agents of interest, and substantial clinical heterogeneity precludes any meaningful comparison of rates between the different uncontrolled studies summarised above. However, limited information on the relative rates of certain adverse events between agents was reported by 10 of these uncontrolled studies (Table 29).
| Study | Total infections | Serious infections (%) | Cancers | TB (%) | Mortality (%) | Withdrawals to AE |
|---|---|---|---|---|---|---|
| Brassard 2006135 | – | – | – |
Etanercept 1.4 Infliximab 1.8 |
– | – |
| Carmona 2005141 | – | – | – |
Infliximab 4.6 Etanercept 0 Adalimumab 0 |
– | – |
| Curtis 2007134 | – | 2.70 | – | – | – | – |
| Dixon 2006136 | – |
Etanercept 5.8 Infliximab 8.9 Adalimumab 5.1 |
– |
Etanercept 0.06 Infliximab 0.2 Adalimumab 0.08 |
– | – |
| Dixon 2007147 | – |
Etanercept 11.2 Infliximab 13.8 Adalimumab 7.3 |
– | – | – | – |
| Dreyer 2009148 | – | – | 0.76 | – | – | – |
| Favalli 2009129 | – |
Etanercept 4.5 Infliximab 8.1 Adalimumab 6.6 |
– |
Etanercept 0.4 Infliximab 0.6 Adalimumab 0.3 |
0.40 | – |
| Gomez-Reino 2003146 | 7.60 | 0.65 | – |
Etanercept 0 (0) Infliximab 17 (1.1) |
0.10 | – |
| Gomez–Reino 2007132 | – | – | – |
Etanercept 2 (0.1) Infliximab 5 (0.4) Adalimumab 1 (0.2) |
– | – |
| Listing 2005122 |
Etanercept 21.3 Infliximab 26.6 |
Etanercept 6.1 Infliximab 5.8 |
– |
Etanercept 0 (0) Infliximab 1 (0.3) |
0.50 | – |
Patients with RA predominated and the average length of study follow-up (where reported) ranged from 1 to 5 years. One prospective cohort study reported a total rate of infections of 21.3% (6.1% serious) and 26.6% (5.8% serious) for etanercept and infliximab, respectively. 122 Three more studies reported rates of serious infections for all three agents: etanercept (5.8%, 11.2%, 4.5%), infliximab (8.9%, 13.8%, 8.1%) and adalimumab (5.1%, 7.3%, 6.6%). 129,136,147
Rates of TB were reported in seven studies of patients receiving etanercept (0%–1.4%) and infliximab (0%–4.6%), four of which also included patients receiving adalimumab (0%–0.3%).
One large prospective cohort study of reported that 0.76% of patients treated with biologic agents developed cancer during follow-up. 148 None of the studies provided adequate data on rates of withdrawal, and no studies provided separate mortality data for each agent.
Summary of serious adverse events across all three agents
Table 30 summarises the rates of serous adverse events, where reported, among the included non-randomised studies and large RCTs. This indicates that the rates of serious adverse events cover a broadly similar range across the three different biologic agents. However, it should be noted that all of these estimates are derived from a highly heterogeneous group of studies in terms of participants (e.g. inflammatory condition, disease severity), study design (e.g. length of follow-up) and treatment regimens (e.g. dose and frequency). Consequently, reliable estimates of the relative rate of serious adverse events for each drug cannot be made.
| Drug | Serious infections (%) | Cancer (%) | TB (%) | Mortality (%) | Withdrawals due to AE (%) |
|---|---|---|---|---|---|
| Etanercept | 0.6–13.2 | 1–5.7 | 0–1.4 | 0–3.1 | 0–13.6 |
| Infliximab | 0.8–13.8 | 0.16–5.1 | 0.06–4.6 | 0.06–2.0 | 6.4–12.8 |
| Adalimumab | 0.4–5.1 | 0.1–1.1 | 0–0.4 | 0.5–0.9 | 5.8–10.7 |
Withdrawal rates due to adverse events were typically < 10% for all drugs, with the highest reported single estimate being 13.8% for one etanercept study. This would suggest that the majority of patients can tolerate biologic treatment in the medium term, although again these estimates are derived from a highly heterogeneous group of studies, therefore poorer tolerability in specific patient groups cannot be ruled out.
Discussion of clinical evaluation
Efficacy
Study design and quality
All six included studies were randomised, double-blind controlled trials. Based on the quality assessment using the pre-specified criteria, all the included trials were rated as ‘good’ quality. Concealment allocation and blinding were adequate in almost all included trials. All of the trials appeared to deal with withdrawals appropriately by using intention-to-treat (ITT) analyses. The completeness of follow-up was fairly good in all trials, with losses to follow-up of < 20%, thereby minimising attrition bias. 172 All the trials reported the use of a power calculation to determine the sample size. Five of them had an open-label extension after the randomisation period. However, it should be noted that the maximum randomised follow-up period across these trials was only 24 weeks.
Though there were some differences relating to patients’ characteristics at baseline across the trials, participants were generally similar in terms of disease activity and severity, and were likely to represent a population with moderate to severe PsA requiring further treatment. This was reflected by the lack of evidence for statistical heterogeneity in most efficacy analyses in this review. However, although the majority of patients in the trials had previously received at least one DMARD, no trial specified the failure to respond to at least two DMARDs (patients whom the current BSR guidelines consider eligible for biologic treatment) as a recruitment criterion. Therefore, trial participants were not precisely representative of patients receiving these agents in practice, and were likely to have had less severe disease, having often received biologic therapy after failing a single DMARD.
There were inconsistencies in the choice of primary outcome between included studies. Most studies used the ACR 20 as the primary outcome measure, while one trial used the PsARC as the primary outcome. However, it should be noted that ACR 20 is not frequently used in routine clinical practice to measure response to a biologic treatment.
Outcomes relating to joint disease
There were limited efficacy data from RCTs for the three biological agents. For each agent, there were two RCTs with around 200 or fewer patients receiving active treatment. However, all six trials were of good quality and provided clear indication of a response to treatment at 12–16 weeks, with continued efficacy at 24 weeks for each biologic agent.
Point estimates of effect sizes were generally moderate to large, implying that these treatment effects could be clinically significant. Moreover, although a very small number of studies were pooled for each estimate, the CIs indicate reasonable precision of these estimates. However, pooling the long-term efficacy data from trials was impossible due to lack of data.
In general, there was no significant heterogeneity in the treatment effect for almost all of the efficacy outcomes, with the PsARC in infliximab being the only exception. The radiographic data from RCTs of etanercept and adalimumab in PsA demonstrated a beneficial effect on joint disease progression at 24 weeks. Follow-up this early is often considered insufficient to detect radiological changes, although if the 24-week effect is reliable it would indicate a rapid onset of action in terms of joint disease for these agents. The open-label extensions of these RCTs also provided data on radiographic assessment at long-term follow-up, indicating that the effect on joint disease progression may persist over time. However, the reliability of these longer-term data was compromised by the lack of a control group.
Functional status (Health Assessment Questionnaire)
All three agents appeared to have beneficial effects on functional status as measured by HAQ. The estimates with relatively high precision indicated that all of the biologic therapies significantly improved the functional status of patients with PsA at around 3 months’ follow-up. The clinical significance of these effects was not entirely clear, for example, adalimumab was associated with a significant absolute mean reduction of HAQ score from baseline of –0.27 (95% CI –0.36 to –0.18). However, only changes > –0.3 have been considered as clinically meaningful improvement in PsA. 150
In this systematic review, the benefit of the biologic treatment compared with placebo on joint disease outcomes was consistent with the previous systematic review, which investigated the efficacy of etanercept and infliximab in the treatment of PsA. 73 In general, both of the systematic reviews used the same rigorous methodology and revealed similar magnitudes of the treatment effect of etanercept and infliximab. The current review also assessed effects of the recently licensed biologic agent ‘adalimumab’ and demonstrated its beneficial treatment effects compared with placebo.
Outcomes relating to skin disease (psoriasis component)
Skin outcomes (i.e. PASI response) were less commonly reported than joint response measures. Where reported, these results were generally statistically significant, although CIs were wide – possibly due to the small sample size of patients evaluable for psoriasis in the trials. Overall, biologic treatment appears to have a broadly beneficial effect on skin disease in patients with PsA. Evidence of response from trials in patients with psoriasis lay outside the scope of this evaluation. 173,174
Relative efficacy of the biologics
As data for the direct head-to-head comparison between these biologic agents were not available from trials, the relative efficacy of these biologic agents in the treatment of PsA was evaluated using Bayesian indirect comparison methodology.
The results of this evidence synthesis highlighted the superior efficacy of biologics over placebo across the outcomes evaluated. Infliximab appears to be the most effective among the three biologics. Patients treated with infliximab had a higher probability of responding to treatment regarding both the skin and arthritis aspects of disease. Additionally, we have estimated that infliximab allows improvements in the functional and psychological impact of the disease, measured by HAQ. However, patients who responded to etanercept achieved similar mean changes in HAQ (–0.6275 for infliximab and –0.6235 for etanercept) with placebo non-responders being used as a baseline in the synthesis. For all three biologics the changes in HAQ for those patients who did not respond to treatment were below the suggested minimum clinically significant threshold,150 and only those for infliximab achieved statistical significance. A comparison of the indirect comparison undertaken by the Assessment Group with those of the manufacturers shows similar mean estimates of treatment effect despite the rather different methods used.
Safety
Study design and quality
For the evaluation of adverse events of these biological agents, this review included a range of study types including RCTs, trial open-label extensions and observational studies. The quality of studies therefore varied across these different study designs; in particular, observational studies were subject to confounding, thereby threatening the internal validity of their findings. In addition, the definition of serious adverse events was also unclear in most studies.
Outcomes relating to serious adverse events
Previous systematic reviews have focused on short-term follow-up and reported conflicting findings on the risk of serious infections and cancer associated with biologic treatment. Our current systematic review contributes an evaluation of potential serious adverse events of biologic treatment in the longer term, incorporating the risk of activation of latent TB. Although the estimates of the rates of these adverse events varied widely, the findings from our review did raise a concern that treatment with etanercept, infliximab and adalimumab might be associated with an increased risk of serious infection, malignancy and activation of latent TB. The adverse event analyses demonstrated that etanercept, infliximab and adalimumab were associated with a broadly similar range of incidences of these events. However, there was considerable uncertainty around these estimates, in part due to the high degree methodological and clinical diversity between the included studies. In addition, the adverse event data were derived primarily from patients with RA or other indications, so the generalisability of these findings to patients with PsA remains unclear. Overall, the limited evidence prevents firm conclusions about the comparative safety of the three biologic agents being drawn from our systematic review.
Chapter 4 Assessment of cost-effectiveness evidence
Systematic review of existing cost-effectiveness evidence
The purpose of this section of the report is to review existing evidence on the cost-effectiveness of biologic therapy in PsA. It includes submissions made to NICE by the manufacturers of the three biologic agents included in this assessment.
Methods
A broad range of studies was considered for inclusion in the assessment of cost-effectiveness, including economic evaluations conducted alongside trials and modelling studies. Only full economic evaluations that compared two or more options, and considered both costs and consequences, were included.
The following databases were searched for relevant published literature: Cochrane Controlled Trials Register (CCTR), EMBASE, HEED, MEDLINE, National Research Register (NRR), NHS EED, PsycINFO and SCI. Full details of the main search strategy for this review are presented in Appendix 1.
Two reviewers assessed all obtained titles and abstracts for inclusion, with any discrepancies resolved by discussion. In addition, the industry submissions to NICE were included in the review.
The studies have been summarised within the text of the report. A summary of effectiveness, costs and cost-effectiveness is presented along with a critique of the studies. The quality of the cost-effectiveness studies was also assessed according to a checklist updated from that developed by Drummond et al. 175
Results
Identified studies
The systematic literature of published literature identified three studies,176–178 which met the inclusion criteria for the cost-effectiveness review (one of which is the journal publication of the previous York Assessment Report model for NICE on etanercept and infliximab73). In addition there were three industry submissions to NICE from Abbott,151 Schering-Plough152 and Wyeth. 153
Of the six cost-effectiveness studies available, described above, five of these are decision-analytic models, incorporating evidence from a variety of sources, and one is a cost-effectiveness study, using evidence from a single trial.
Available data
Table 31 summarises the data available from each of the six cost-effectiveness studies. 151–153,176–178 The studies by Olivieri et al. 178 and Bansback et al. 176 are only available as journal articles. The study by Bravo Vergel et al. 177 is available as a journal article, but also as a full assessment report with an accompanying electronic model. 73 The three industry submissions included full reports and electronic models. Where an electronic model has been made available it has been possible to provide some validation of the model by ensuring that the base-case results provided by the manufacturer in its report can be replicated. It was also possible to check parameter estimates presented in the reports against those used in the relevant models.
| Journal article | Full report | Electronic model | Additional utility regression | Clarifications | |
|---|---|---|---|---|---|
| Olivieri 2008178 | |||||
| Bansback 2006176 | ✓ | ||||
| Bravo Vergel 2007177 | ✓ | ✓ | ✓ | ||
| Abbott submission 2009151 | ✓ | ✓ | ✓ | ✓ | |
| Schering-Plough submission 2009152 | ✓ | ✓ | ✓ | ||
| Wyeth submission 2009153 | ✓ | ✓ | ✓ | ✓ |
Due to differences in the regression methods used to generate utility estimates in the industry submissions, the Assessment Group requested that each manufacturer provide new utility estimates using a common methodology (see Appendix 17) and report the results of this regression, as coefficients, a variance–covariance matrix, the number of observations, the number of clusters (if appropriate) and indicating the source of data. This information was provided by manufacturers for all three of the submissions.
In addition, a number of further clarifications on data sources and methodology were sought from the three manufacturers on data sources and methodology (full details in Appendix 6). Wyeth153 clarified that 12- and 24-week response rates were modelled independently, provided an estimation of HAQ without PASI as a predictor, and clarified how withdrawal rates were calculated (see Critique of manufacturers’ submissions and justification for current York modelling approach). Abbott151 clarified how many DMARDs were sequenced in the model, how withdrawal rates were calculated (see Chapter 4, Critique) and clarified the degree of correlation between arthritis and skin outcomes. No further clarifications were sought from Schering-Plough152 other than the additional utility regressions.
Summaries of cost-effectiveness studies
A full description of each of the six cost-effectiveness studies, along with a quality assessment checklist, is presented in Appendix 7. Table 32 summarises the key features and data sources for each of the studies.
| Olivieri178 | Bansback176 | Bravo Vergel177 | Abbott151 | Schering-Plough152 | Wyeth153 | |
|---|---|---|---|---|---|---|
| Comparators | Biologics (as a group) compared with non biologics | Etanercept, ciclosporin and leflunomide | Etanercept, infliximab and palliative care | Etanercept, infliximab, adalimumab and DMARDs (which includes different combinations of DMARDs) | Etanercept, infliximab, adalimumab and palliative care | Etanercept, infliximab, adalimumab and DMARDs |
| Model structure |
No model Economic evaluation alongside a before/after study |
Response according to PsARC determined and associated HAQ score. Changes in HAQ and further withdrawals are modelled over 10-year time horizon | Response according to PsARC determined and associated HAQ score. Changes in HAQ and further withdrawals are modelled over 40- and 10-year time horizons | Response according to the joint distribution of PsARC and ACR response rates. Associated HAQ and PASI changes by type of response. Changes in HAQ and further withdrawals are modelled over a lifetime time horizon | Response according to PsARC determined and associated HAQ score. Changes in HAQ and further withdrawals are modelled over a lifetime time horizon | Response according to PsARC determined and associated changes in HAQ and PASI. Initial change in HAQ is a function of PASI and PsARC. Longer-term changes in HAQ were modelled using observed changes in PASI score, PASI 75 response and PsARC response. Changes in HAQ and further withdrawals are modelled over a 50-year time horizon |
| Patient inputs | Single trial of 107 patients from nine tertiary referral centres in Italy | Individual sampling model using patient-level data from Mease et al.52 | Baseline HAQ is assumed to be average from the three trials (Mease et al.52,78 and Antoni et al.81) | Individual sampling model using baseline patient characteristics from the ADEPT trial88 used to determine the distribution of patients characteristics in the model | Baseline HAQ of 1.1 is assumed. Baseline PASI of 11 is assumed. The sources of these are not presented. For patients with no clinically significant psoriasis component to their disease only the change in HAQ is modelled | Individual sampling model using baseline characteristics of patients were taken from the Mease et al.52 Subgroups were: mild, moderate and severe HAQ, and mild, severe and very severe PASI |
| Sources of effectiveness evidence | Effectiveness from a single trial | Mease et al.52 used to determine response rates and HAQ | Short-term trial data (Mease et al.52,78 and IMPACT81) were used to model the PsARC response of patients | Data from 10 different sources to determine short-term efficacy | In many cases results from the York model were used as priors in the Bayesian evidence synthesis. Data from the previous York model177 along with IMPACT,81 IMPACT 2,82 Mease et al.52,78 GO-REVEAL,156 Genovese et al.83 and ADEPT51 were used in the evidence synthesis model | Data from the published MTC for adalimumab179 and the Mease et al. trial52 comparing etanercept with placebo were used to estimate effects |
| Synthesis of effectiveness evidence | Effectiveness from a single trial | Effectiveness from a single trial | A Bayesian evidence synthesis was used to generate estimates of PsARC and mean improvements in HAQ score conditional on response using the three trials via indirect comparisons methods |
A Bayesian evidence synthesis was used to determine: (1) joint distribution of 12-week PsARC and ACR response rates; (2) 24-week PsARC response conditional on the 12-week PsARC response; and (3) 24-week ACR response conditional on the 12-week ACR response Patient-level data from ADEPT88 used to estimate HAQ and PASI changes |
A Bayesian evidence synthesis was used to generate estimates of PsARC and mean improvements in HAQ and PASI score conditional on response | A published MTC for adalimumab179 and the Mease et al. trial52 was used to estimate PsARC response and improvements in HAQ and PASI |
| Sources of cost data |
Resource use collected retrospectively from patients Diagnosis-related group costs were used to cost hospitalisations. Little detail on other medical costs. Transportation costs from patients’ reports. Carers’ costs and days lost from work were costed using the human capital approach |
Drug costs were taken from MIMS and administration and monitoring costs generated using resource use recommended in the BSR guidelines The cost offsets of improving disability were also estimated using a study of patients with RA |
Drug costs were taken from the BNF. Administration and monitoring costs were estimated using industry assumptions regarding resources use and published unit costs The costs associated with PsA were estimated as a function of HAQ score using a published study in RA |
The cost of drugs was estimated using MIMS. Resource use associated with monitoring and administering drugs was estimated according to BSR guidelines The relationship between HAQ score and disease-related hospital costs was estimated using the NOAR database. A physician survey was conducted to assess the ongoing costs of psoriasis |
Resource use associated with treatment, administration and monitoring was taken from the previous York model. Health-care costs as a function of HAQ were derived from the Kobelt et al. study41 (CiC information has been removed) |
The costs of medication were taken from the BNF.65 A Administration and monitoring was costed as recommended in the BSR guidelines. Health-care costs associated with PsA were taken from an evaluation by HODaR, using data from BSRBR and THIN (reference not given). PASI are not included, as PASI is assumed to be a predictor of HAQ |
| Utilities | EQ-5D utility scores were used in the cost-effectiveness analysis. These were collected directly from patients at 6 months preceding biologics treatment, baseline, 6 months and 12 months | Leeds cohort study used to estimate utilities. The relationship between health utilities and HAQ was examined using linear regression models | Leeds cohort study used to estimate utilities. The relationship between health utilities and HAQ was examined using linear regression models | In the base-case data from the ADEPT trial88 of adalimumab was used. SF-36 was converted to EQ-5D | Two alternative methods to generate utilities were explored: the Gray et al. algorithm180 (selected as the base case) and the Brazier algorithm181 | The relationship between HAQ and EQ-5D observed in the PRESTA data set157 was used in the base case to generate utilities. The relationship between PASI and EQ-5D was not included |
| Base-case results | At 12 months there was a gain of 0.25 in utility for biologics, equating to a 0.12 gain in QALYs. Direct costs increased by €5052. This produces an ICER of €40,876 for the NHS and an ICER of €37,591 for society |
QALYs were 4.49 for etanercept, 3.67 for ciclosporin and 3.84 for leflunomide Total costs of etanercept over 10 years is estimated as £51,122, ciclosporin was £28,010 and leflunomide £26,822 This gives an ICER for etanercept of £28,000 compared with ciclosporin and £38,000 compared with leflunomide |
Infliximab is the most effective strategy in both scenarios (4.636 and 4.455 QALYs). Total mean costs were highest for infliximab in both rebound scenarios (£64,274 and £64,418, respectively) The ICERs for infliximab are unlikely to be considered reasonable. The ICER for etanercept for rebound equal to gain is £26,361 and for rebound equal to natural history is £30,628 |
Infliximab was associated with the highest QALYs (8.49) at a cost of £104,772 The ICER for infliximab is unlikely to be considered acceptable. Adalimumab has an ICER of £29,827 compared with a DMARD |
Infliximab is the most effective strategy, for all patients as a group and psoriasis patients (8.65 QALYs for all patients and 8.40 QALYs for patients with psoriasis), but is also associated with the highest cost (between £107,954 and £123,475) Infliximab is the most cost-effective strategy for a 60-kg patient, for all patients, and for psoriatic patients. For a 70-kg patient etanercept is the most cost-effective strategy for all patients and for psoriatic patients. For an 80-kg patient etanercept is the most cost-effective strategy for all patients and for psoriatic patients, with ICERs of £12,696 and £12,606 compared with adalimumab. For all patient weights, etanercept is the most cost-effective with an ICER of £12,432 compared with adalimumab for non-psoriatic patients |
Etanercept was associated with the highest gain in QALYs (6.90). Infliximab had the highest total costs (£66,867). The base-case results show that infliximab is dominated by adalimumab and adalimumab extendedly dominated by etanercept. Comparing etanercept with ciclosporin results in an ICER of £12,480 |
| Key sensitivity analysis | – | Sensitivity analysis showed that the ICER was sensitive to the baseline HAQ and annual HAQ progression | Results were sensitive to many of the changes in parameters, in particular not using a specific stopping rule for biologic therapy and instead using no response test and withdrawal rates from BSRBR and the rebound assumption | Results were sensitive to the stopping rule for BSRBR withdrawal rates and the rebound assumption | Biologics appear to be robust to the sensitivity analysis compared with palliative care, apart from changing the algorithm for estimating QoL | Results are sensitive to the rebound effect, the utility function used and annual progression on standard care |
As shown in Table 32, the six cost-effectiveness studies produce different costs and quality-adjusted life-years (QALYs), resulting in different incremental cost-effectiveness ratios (ICERs) for the various options being compared. The study by Olivieri et al. 178 is difficult to compare with the others, as all biologics were considered as a group compared with DMARDs. This produced an ICER of around €40,000 for biologics. Bansback et al. 176 produced an ICER of around £38,000 for etanercept compared with the next best strategy – leflunomide. Bravo Vergel et al. 177 produced a much lower ICER for etanercept, of between £26,361 and £30,628, depending on the rebound scenario used. The studies including all three biologics in this assessment – adalimumab, etanercept and infliximab – also show large differences in results. Abbott151 generates an ICER for adalimumab of £29,827, with etanercept dominated by adalimumab and infliximab, with an ICER over £199,000. Schering-Plough152 report results for all patients, psoriatic patients and non-psoriatic patients. For all patients, etanercept is the most cost-effective strategy, assuming a patient weight of 70 or 80 kg (ICER of £12,606 compared with adalimumab). For a 60-kg patient etanercept is the most cost-effective strategy for patients without psoriasis (ICER of £12,432 compared with adalimumab) and infliximab is the most cost-effective for psoriatic patients and all patients, dominating etanercept. Wyeth153 produces a base-case ICER for etanercept of £12,480 compared with DMARDs. All other biologics are dominated or extendedly dominated.
It is difficult to disentangle exactly why, in some cases, the six studies produce markedly different results. However, there are a number of key differences between the modelling approaches and the data sources used in the six cost-effectiveness studies that may provide some explanation.
-
Choice of comparator All biologics were grouped together in the Olivieri et al. study,178 although the majority of patients were taking etanercept. It is, therefore, not possible to estimate any differences in the cost-effectiveness between the biological agents. Bansback et al. 176 compare only etanercept with DMARDs, omitting all other biologics, whereas Bravo Vergel et al. 177 compare only infliximab and etanercept with palliative care. The models from Abbott,151 Schering-Plough152 and Wyeth153 all include the three biologics etanercept, infliximab and adalimumab. However, Abbott151 and Wyeth153 compare these with DMARDs, whereas Schering-Plough152 use palliative care as the comparator. The patient group specified by the decision problem (see Executive summary, Objectives) are those who have previously failed two DMARDs. Therefore, these patients may be unlikely to be considered for further DMARD treatment, which suggests that they would instead receive palliative care.
-
Sources and synthesis of effectiveness data Olivieri et al. 178 use a relatively small sample of patients recruited from a single site. The analysis has a limited length of follow-up (12 months) and, as PsA is a chronic disease, it is unlikely that all differences in costs and outcomes between comparators can be captured in this short time frame. This is also a before/after study, so there may be a problem of selection bias. Bansback et al. 176 similarly use data from a single phase II trial to determine effectiveness. Other relevant randomised trials are now available, and this evidence should be appropriately synthesised to inform cost-effectiveness. The models by Bravo Vergel et al. ,177 Abbott,151 Schering-Plough152 and Wyeth153 all use multiple sources to determine the short-term effectiveness of treatments, all of these synthesising data using a Bayesian methods in winbugs. However, in the Abbott151 and Schering-Plough152 models, some of these data sources relate to treatments not included as comparators in the model, such as golimumab (see Chapter 3, Results of review of clinical effectiveness). The implications of using this wider selection of treatments in the evidence synthesis are uncertain.
-
Effect of treatment on skin component of disease Although PsA is associated with psoriasis as well as an inflammation of the joints, Bansback et al. 176 and Bravo Vergel et al. 177 do not include the effect of treatments on the skin component of PsA, whereas the models by Abbott,151 Wyeth153 and Schering-Plough152 all include the effect of both conditions. In the Wyeth model,153 however, the initial change and longer-term changes in HAQ were determined, including PASI as an explanatory variable. Although PASI and HAQ are used to measure the severity of the two components of PsA, psoriasis and arthritis, there are only limited circumstances in which a patient’s psoriasis should affect their degree of functional disability or joint disease, as measured by HAQ.
-
Model structure Olivieri et al. 178 does not use a model to generate estimates of costs and QALYs and instead uses the results of an economic evaluation conducted alongside a single trial. The models by Bansback et al.,176 Bravo Vergel et al. 177 and Schering-Plough152 all determine response according to PsARC and then model the associated HAQ score. Schering-Plough152 includes PASI change from baseline to 12 weeks, but estimates this for weeks for PsARC responders/non-responders. Wyeth153 similarly determines response according to PsARC and calculates the associated change in HAQ and PASI. However, initial change in HAQ is modelled using changes in PASI and PsARC, and longer-term changes in HAQ were modelled using observed changes in PASI score, PASI 75 response and PsARC response. Abbott151 use ACR response rates in addition to PsARC to determine the joint distribution of response, and then associated HAQ and PASI changes by type of response. Schering-Plough152 assumes that changes in HAQ in the first 3 months are a function of PsARC response and the biologic used, whereas Abbott151 and Wyeth153 assume that changes in HAQ are independent of the biologic used after conditioning on other predictive clinical and demographic variables (such as ACR and age).
-
Patient characteristics Of the five model-based studies, three of these use an individual sampling approach, with baseline characteristics taken from IPD from trials. 151,153,176 Bravo Vergel et al. 177 and Schering-Plough152 both use cohort models, with common baseline HAQ/PASI scores, which are then varied in a sensitivity analysis. The individual sampling models are complex and time intensive in order to run probabilistic sensitivity analysis. They are also difficult to audit and there may be differences in methodology used in these models that the Assessment Group were not able to fully explain in the constrained timescale.
-
Sources of cost data In their trial-based evaluation, Olivieri et al. 178 collected resource use data retrospectively from patients and valued these using appropriate unit costs. The model-based studies all include the same set of costs: drug acquisition, drug administration and monitoring, and costs of disability and psoriasis (where PASI was included in the model). However, the cost estimates generated differ quite significantly between models (see Critique of manufacturers’ submissions and justification for current York modelling approach), reflecting different methodology and sources of data.
-
Sources of utility data Olivieri et al. 178 collected utilities directly from patients who were enrolled in the trial, using the European Quality of Life-5 Dimensions (EQ-5D questionnaire). These were collected for the 6 months preceding biologic treatment, baseline, and 6 months and 12 months after starting treatment. The other studies use different external data sets to generate utilities and used regression analysis to link the utility data to clinical parameters. Each of the studies assumed that utility was independent of the biologic treatment used, after conditioning on HAQ and PASI. However, each used a different function to relate utility to HAQ and PASI, and it is possible that different utility regressions result in differences in the relative impact of HAQ/PASI on utility between treatments. Bansback et al. 176 and Bravo Vergel et al. 177 both use the Leeds cohort study as a source of utility estimates. Abbott151 use the ADEPT trial88 of adalimumab, which reports Short Form questionnaire-36 items (SF-36 data), which are then converted to EQ-5D to generate utilities. Schering-Plough152 use the same approach, but use the GO-REVEAL (Golimumab-Randomized Evaluation of Safety and Efficacy in Subjects with Psoriatic Arthritis Using a Human Anti-TNF Monoclonal Antibody)156 trial data set. Wyeth153 use the relationship between HAQ and EQ-5D observed in the Psoriasis Randomized Etanercept STudy in Subjects with Psoriatic Arthritis (PRESTA) data set in the base case to generate utilities, and the relationship between PASI and EQ-5D was indirectly included only through the effect of PASI on HAQ.
Relevance of cost-effectiveness evidence for NICE decision-making
The evidence provided from the cost-effectiveness study conducted alongside a single trial178 is not considered relevant for UK decision-making because of its lack of a concurrent control group, narrow use of evidence (a single trial) and limited length of follow-up (12 months). The five modelling studies are, however, potentially relevant for UK decision-making. The current appraisal has recognised the need to assess the effect of biologics on both the arthritis and the psoriasis component of the disease. Only the three industry models include the psoriasis aspect of PsA, and therefore only these models are relevant to address the decision problem as specified by the NICE scope.
There are a number of issues with the three industry models that require further consideration. These are discussed in further detail in the section Critique of manufacturers’ submissions and justification for current York modelling approach, later in this chapter, but can be summarised as:
-
The use of DMARDs as a comparator to biologics used in the Wyeth153 and Abbott151 models. This approach can be criticised if it is considered unrealistic for patients who have previously failed two or more DMARDs, as defined in the BSR guidelines149 to receive a third DMARD.
-
In estimating the treatment effect, the Abbott151 and Schering-Plough152 models use data sources relating to comparators not included in the model, such as golimumab, and the implications of this are not clear. It is uncertain whether the relative treatment effects can be transferred from one biologic to another.
-
Also for the Wyeth submission153 data from an existing synthesis for adalimumab179 and the Mease et al. trial52 were used to estimate effects. Although data were included from a number of trials in the adalimumab MTC, new trial evidence may be available and efforts should be made to identify any new relevant data.
-
In estimating the treatment effect, it is also important to consider what treatment effect is likely to be observed in general practice. RCTs might overestimate the absolute response rates in both placebo and treatment groups. Schering-Plough152 assume that this is the case and adjust the expected effectiveness of biologics, whereas the Wyeth153 and Abbott151 models do not make any such adjustment. The models do not use sensitivity analysis to assess how much difference this adjustment makes to the results.
-
Withdrawals after 3 months due to adverse events and lack of efficacy were estimated from a single data set (BSR register) in all of the industry models. There are other potential biologic registry data sets available, which could have been synthesised.
-
The prediction of initial change in HAQ and longer-term changes in HAQ using PASI as an explanatory variable in the Wyeth model153 is questionable. There is no evidence to suggest that one component of the disease is a good predictor of the other, although there may be a correlation between joint and skin response, which has not been explored in any detail by the industry models.
-
There are some considerable differences in the sources of costs and the costing methodology used in each of the three industry models (see Critique of manufacturers’ submissions and justification for current York modelling approach). It is therefore important to understand what these differences are and to generate appropriate costs for the model.
-
The results from each of the industry models are also markedly different. There is therefore a need to develop a de novo model that considers and addresses each of these limitations. This model is presented below.
York Economic Assessment
Methods of York Economic Assessment
Introduction
The review of models detailed in published literature (including the earlier one by the York Assessment Group) and those in the company submissions to this appraisal (see Systematic review of existing cost-effectiveness evidence) indicates that a wide range of assumptions and evidence was used in model development. None of the models reviewed can be considered unequivocally superior to the others. In this section we further develop the earlier York Assessment Group model, reflecting more recent evidence about PsA and the use of biologics in its treatment. This model also provides a framework within which to compare the assumptions and evidence used in the different models and to assess their implications for the cost-effectiveness results.
Previous guidance has been issued by NICE on the use of biologics in PsA. 182,183 The main limitation of the economic assessments informing this earlier guidance was that they did not take account of the effect of the drugs on psoriasis. Therefore, a key objective of the updated York model is to assess the cost-effectiveness of etanercept, infliximab and adalimumab for PsA, taking account of the cost and health impact of the patient’s psoriasis and joint disease, and the impact of therapy.
Methods
Overview
A probabilistic decision-analytic model was developed to estimate the costs and QALYs of the three biologics over a lifetime (40 years) compared with palliative care only. The model has similarities with the earlier York Assessment Group model, but a number of changes have been implemented, necessitating a full description of the model here. The model aims to be consistent with licensed indications and current BSR149 and BAD173 guidelines for the use of biologics in PsA (Box 1).
Etanercept, infliximab and adalimumab are licensed for the treatment of active and progressive PsA in adults when the response of previous DMARD therapy has been inadequate. Infliximab should be administered in combination with MTX or alone in patients who show intolerance to MTX or for whom MTX is contraindicated
BSR guidelines for commencing biologics in PsABiologic therapy, within its licensed indications, is recommended for the treatment of adults with active PsA only when the following criteria are met:
-
The person has peripheral arthritis with three or more tender joints and three or more swollen joints on two separate occasions, at least 1 month apart, based on a 78-tender and 76-swollen-joint count
-
The PsA has not responded to adequate trials of at least two standard DMARDs, administered either individually or in combination
To be considered eligible for treatment with biologic therapy, patients must have:
-
severe disease, defined as a PASI score of 10 or more and a DLQI > 10
and
-
contraindications to (have developed, or are at risk of developing) clinically important drug-related toxicity, where phototherapy and alternative standard therapy cannot be used, or are intolerant or unresponsive to standard systemic therapy, have significant, coexistent, unrelated comorbidity that precludes use of systemic agents, such as ciclosporin or MTX, or have severe, unstable, life-threatening disease
-
Patients who have active PsA or skin disease that fulfils defined BSR or BAD guideline criteria, respectively
Patients with severe skin psoriasis and PsA who have failed, or cannot use, MTX may need to be considered for biologic treatment, given the potential benefit of such treatment on both components of psoriatic disease
The parameters of the model were obtained from published literature, manufacturers’ parameter estimates, the results of the evidence synthesis in Chapter 3, Efficacy of all three biologics and a structured elicitation of expert opinion. The model adopts the perspective of the UK NHS and Personal Social Services. The price year is 2008–9 and the annual discount rate is 3.5%. 184 The population is assumed to be 47 years old, with at least 7 years since diagnosis of PsA, based on the average characteristics of participants in the RCTs (see Table 1). The body weight is assumed to be between 60 and 80 kg, based on the mean adult weight in the UK general population (women 69.7 kg, men 83.5 kg185). Patients are assumed to have failed at least two DMARDs. In the base case, patients are assumed to fulfil BSR criteria (see Box 1). In the base case the HAQ at the start of the model is 1.05, based on the average in the RCTs (see Table 1). Although the mean HAQ when patients start biologics in the BSR register was 1.8,186 clinical opinion suggests that, in current practice, clinicians are more likely to offer biologics early in the course of the disease.
Clinical opinion suggests that about 50% of patients starting biologics have mild or minimal psoriasis (< 3% BSA or a PASI score of < 2.5), 25% have mild-to-moderate psoriasis (a baseline PASI score of between 2.5 and 10), and 25% have moderate-to-severe psoriasis (a PASI score > 10) (Ian Bruce, Arc Epidemiology Unit, University of Manchester, UK, 20 November 2009, personal communication). Approximately 50% of patients in the RCTs had < 3% BSA psoriasis or a baseline PASI < 2.5 (see Table 1), indicating the trials are broadly representative of skin involvement in general practice. We assume patients in the base case have mild-to-moderate psoriasis with a PASI score of 7.5. The effect of biologic treatments in other patient subgroups is explored in scenario analyses.
Model structure
The model is a cohort model, assuming a homogeneous baseline population. The model has a Markov structure (see Figure 2). Patients enter the model either (i) when commencing therapy with etanercept, infliximab or adalimumab or (ii) with no therapy (assumed to be palliative care only).
FIGURE 2.
Structure of the decision model, assuming patients continue beyond 3 months if they achieve a PsARC response.
Initial response at 3 months
Table 33 shows the parameters used in the base-case model. Initial response of the drug is defined in the model as PsARC for joints and PASI 75 for psoriasis, based on the BSR149 and the BAD guidelines173 (Box 2). These parameters were estimated by the evidence synthesis (see Chapter 3, Efficacy of all three biologics).
| Description | Variable name | Mean | SE | Source/appendix | |
|---|---|---|---|---|---|
| Gender male = 1, female = 0 | Male | 1 | |||
| PsA minimum duration (years) | PsA.dur | 3 | |||
| Concomitant MTX in all strategies: yes = 1, no = 0 | MTX | 1 | |||
| Baseline HAQ | HAQ0 | 1.05 | Mean of RCTs (Table 1) | ||
| Baseline PASI | PASI0 | 7.5 | Clinical opinion | ||
| Baseline age | Age | 47 | Mean of RCTs (Table 1) | ||
| Model time horizon (years) | Years | 40 | Clinical opinion | ||
| Discount rate (per year) | r | 0.035 | NICE184 | ||
| Utility function intercept | h0 | 0.897 | 0.006 | Appendix 17 | |
| Change in utility for 1 unit change in HAQ | h1 | –0.298 | 0.006 | Appendix 17 | |
| Change in utility for 1 unit change in PASI | h2 | –0.004 | 0.0003 | Appendix 17 | |
| Interaction term HAQ PASI | h3 | 0 | 10 x E-5 | Appendix 17 | |
| Cost function intercept (per 3-month period) | c0 | 233 | Appendix 15 | ||
| Change in cost for 1 unit change in HAQ | c1 | 103 | 67 | Kobelt et al.,41 Appendix 15 | |
| Three-month cost for mild-to-moderate psoriasis if uncontrolled by biologics | c2.1 | 198 | 9 | DoH Reference Costs 2007–08,187 Appendix 16 | |
| Three-month cost for psoriasis in remission | c2.2 | 16 | 1 | Hartman et al.,188 Appendix 16 | |
| Change in HAQ while on treatment per 3-month period | HAQ1.d | 0 | 0.02 | Experts, Appendix 11 | |
| Change in HAQ while not on treatment per 3-month period | HAQ1.w | 0.018 | 0.007 | NOAR, Appendix 14 | |
| Rebound in HAQ in 3 months after withdrawal (compared to HAQ at baseline) (zero means ‘rebound equal to initial gain’) | loss.w | 0 | 0.3 | Experts, Appendix 11 | |
| Intercept of regression of log-mortality vs age in men | ln.R.g.m | –10.25 | 0.046 | England and Wales life table, Appendix 19 | |
| Intercept of regression of log-mortality vs age in women | ln.R.g.f | –11.10 | 0.046 | ||
| Change in log-mortality with additional year of age in men over 40 years | a.g.m | 0.094 | 0.0006 | ||
| Change in log-mortality with additional year of age in women over 40 years | a.g.f | 0.101 | 0.0006 | ||
| Log withdrawal rate from biologics per year | ln.long.yr | –1.823 | 0.2044 | Registers, Appendix 12 | |
| Probability of PsARC response on placebo | p.psarc.plac | 0.249 | 0.0384 | Results of review of clinical effectiveness | |
| Change in HAQ given a PsARC response on placebo | HAQ.resp.plac | –0.2436 | 0.04746 | ||
| Probability of PASI 50 response on placebo | p.pasi.50.plac | 0.130 | 0.021 | Results of review of clinical effectiveness | |
| Probability of PASI 75 response on placebo | p.pasi.75.plac | 0.044 | 0.009 | ||
| Probability of PASI 90 response on placebo | p.pasi.90.plac | 0.016 | 0.004 | ||
| Standardised mortality ratio for PsA vs general population | SMRmen | 1.65 | Wong et al.,29 Appendix 19 | ||
| SMRwomen | 1.59 | ||||
| Generalisability of trial (1 = no, 2 = yes) | plac.effect | 1 | Appendix 9 | ||
| Rules on continuation (1–5) | continue | 1 | BSR and BAD | ||
| Etan (mean) | Inflix (mean) | Adal (mean) | |||
| Cost of drugs (first 3 months) | c.drug1 | £2495 | £5523 | £2495 | BSR, Appendix 13 |
| Cost of drugs for months 4–6 | c.drug2 | £2443 | £2965 | £2443 | |
| Cost of drugs, subsequent 3 months | c.drug3 | £2385 | £2965 | £2385 | |
| Probability of PsARC response on biologic | p.psarc | 0.713 | 0.795 | 0.587 | Results of review of clinical effectiveness |
| p.psarc_SE | 0.071 | 0.058 | 0.072 | ||
| Change in HAQ in first 3 months given no PsARC response of biologic | HAQ.no.resp | –0.190 | –0.194 | –0.130 | Results of review of clinical effectiveness |
| HAQ.no.resp_SE | 0.10 | 0.070 | 0.066 | ||
| Change in HAQ in first 3 months given PsARC response of biologic | HAQ.resp | –0.630 | –0.657 | –0.477 | Results of review of clinical effectiveness |
| HAQ.resp_SE | 0.090 | 0.069 | 0.062 | ||
| Probability of PASI 50 response on biologic | p.pasi.50 | 0.4026 | 0.9128 | 0.7383 | Results of review of clinical effectiveness |
| Probability of PASI 75 response on biologic | p.pasi.75 | 0.1768 | 0.7687 | 0.4772 | |
| Probability of PASI 90 response on biologic | p.pasi.90 | 0.0737 | 0.5571 | 0.2571 | |
| p.pasi.50_SE | 0.0916 | 0.0374 | 0.0853 | ||
| p.pasi.75_SE | 0.0586 | 0.0795 | 0.1085 | ||
| p.pasi.90_SE | 0.0292 | 0.1088 | 0.0863 | ||
| Correlation between PASI 75 and PsARC | Rho | 0.435 | 0.435 | 0.435 | ADEPT,51 Appendix 10 |
| rho_SE | 0.112 | 0.112 | 0.112 | ||
Primary joint response: PsARC at 12 weeks/3 months
Primary skin response: PASI 75
Treatment will be withdrawn in the event of adverse events or inefficacy, defined as patients who fail to achieve the PsARC response within 3 months of treatment
BAD guidelines for treatment responseAn adequate response to treatment is defined as either (1) a 50% or greater reduction in baseline PASI (or % BSA where the PASI is not applicable) and a ≥ 5-point improvement in DLQI or (2) a 75% reduction in PASI score compared with baseline. Initial response to therapy should be assessed at time points appropriate for the drug in question.
For patients on TNF antagonist treatment with psoriasis and PsA, treatment may be continued if there has been a sufficient response in at least one of these components (see BSR guidelines for definition of disease response in PsA).
The BAD guidelines highlight that the recommended time points for assessing the initial response vary between drugs and between guideline-making bodies. The licences for psoriasis recommend an assessment at 14 weeks for infliximab, at 12 weeks for etanercept and at 16 weeks for adalimumab. Current NICE guidelines for psoriasis recommend an assessment at 10 weeks for infliximab. In the current appraisal we do not make these distinctions and assume that an assessment is made for all drugs at ‘around 3 months’ or between 12 and 16 weeks. The assessment of effectiveness in Chapter 3 (see Assessment of effectiveness) did not find any appreciable differences in the biologics’ response rates for joint disease or psoriasis between approximately 12 weeks and 24 weeks.
In the decision model, the change in HAQ compared with baseline is conditional on whether a PsARC response was achieved. These parameters were estimated by the evidence synthesis in Chapter 3, Efficacy of all three biologics. It is uncertain whether the change in HAQ is the same for all PsARC treatment responders, or depends on the particular biologic treatment followed. In the opinion of our clinical advisor, either scenario could be plausible (Ian Bruce, personal communication). In the base-case model, we allow the change in HAQ for treatment responders to depend on PsARC response and the biologic treatment, and consider the alternative scenario as a sensitivity analysis. According to the evidence synthesis in Appendix 5, the mean change in HAQ in the first 3 months for PsARC responders, across all biologic drugs, is –0.5688 [standard error (SE) 0.0315] and the mean change in HAQ for PsARC non-responders, across all biologic drugs, is –0.1697 (SE 0.0338).
During the initial 3-month trial period the model assumes that patients on biologics have some improvement in HAQ even if they do not reach the PsARC threshold. These parameters were estimated by the evidence synthesis in Chapter 3, Efficacy of all three biologics. Patients who do not achieve the required level of response during the first 3 months and are withdrawn from therapy are assumed to return to the same HAQ score after withdrawal as patients who had palliative care only.
The model assumes that patients who achieve a PASI 75 response will gain at least a 75% improvement in psoriasis compared with baseline PASI. The calculation of the expected improvement in PASI for PASI 75 responders is described in Appendix 18. Patients who do not achieve a PASI 75 response will also have some proportionate gain in PASI while they continue taking a biologic, although this will be less than a 75% improvement (see Appendix 18).
A proportion of patients in the placebo arms of the RCTs achieved a PsARC response and an improvement in HAQ. Part of the response in both the placebo and treatment arms of RCTs may be due to non-pharmacological aspects of medical care that would be common to both arms (sometimes called a ‘placebo’ or ‘expectancy’ effect). It is uncertain whether this effect would be reproducible in general practice. 189 In the base case we assume that part of the predicted response for treatment observed in the trial is attributable to the controlled trial setting and would not be reproducible in general practice. The change in HAQ in patients using biologics is reduced by the mean change in HAQ across the placebo arms of the RCTs. A similar adjustment is made for the expected change in PASI in patients using biologic therapy. Appendix 9 gives further details of the conceptual framework and adjustments made for the possible placebo/expectancy effects. An alternative scenario assumes that the response rate to treatment in the RCTs is fully generalisable to general practice and no adjustment for placebo/expectancy effects is made.
Because there are two response variables (PsARC and PASI), there are four possible outcomes at 3 months: skin response only, joints response only, response of both and response of neither (Figure 2). The base-case model assumes that the responses to psoriasis and arthritis might be correlated. Appendix 10 reviews the evidence on the correlation between these responses and how the decision model calculates the probabilities of each of the four outcomes at 3 months. An alternative scenario assumes that the responses to psoriasis and arthritis are independent.
The BSR guidelines recommend that biologics are withdrawn if a PsARC response is not achieved at 3 months. This rule is used in the base-case analysis of the model. However, in patients who have significant skin and joint disease, some patients may achieve PsARC but not PASI 75, or achieve PASI 75 but not PsARC. In these cases, one could specify that patients should continue biologic therapy irrespective of the psoriasis response (BSR guideline), or those that respond to either can continue (BAD guidelines) or (in principle at least) only those that achieve both should continue. These alternative continuation rules are explored in sensitivity analyses.
The model assumes that no patients withdraw due to adverse events in the first 3 months. This is because the RCTs estimate responses on an ITT basis, whereby withdrawals for any reason are considered treatment failures and counted as non-response. Including withdrawals during the first 3 months in the model would, therefore, be double-counting.
Long-term outcomes and withdrawal from biologic therapy
If the decision is made to continue with the biologic therapy beyond 3 months, it is assumed that patients maintain their initial improvement in HAQ while on that therapy. This is based on evidence from an opinion elicitation exercise from clinical experts, and supported by data on HAQ and HRQoL from biologics registers. 186,190 Appendix 11 describes the opinion elicitation methods and results used to inform the model. It is assumed that patients maintain the improvement in PASI while on biologic therapy. This assumption has been made in other decision models (see Systematic review of existing cost-effectiveness evidence).
There is an ongoing risk of withdrawal from biologic therapy. Withdrawal might occur for lack of continuing efficacy (‘secondary non-response’), adverse events or other reasons. The rate of withdrawal after 3 months is assumed to be independent of the HAQ and PASI score in the model, to be independent of whether the initial response was for both psoriasis and arthritis or just arthritis, and to be constant over time. The rate is estimated from a meta-analysis of registry data from several countries to be –1.823 (SE 0.2044) on the log scale, or exp(–1.823 + 0.5 × 0.20442) = 0.165 per year (see Appendix 12). Although the registries present withdrawal rates by drug, these data are not randomised and patient cohorts starting on different biologic therapies are unlikely to be similar. 191 Therefore, the decision model assumes the same withdrawal rates for all biologics. Appendix 12 gives further details. As the withdrawal rate is constant over time after the first 3 months, patients who achieve an initial PsARC response will on average remain on biologic drugs for just over 6 years in the model (1/0.165 = 6.06 years).
Patients withdraw from biologic to palliative care only. On withdrawal, it is assumed that mean PASI returns to its initial score at baseline (rebound equal to initial gain). There is considerable uncertainty about change in HAQ associated with withdrawal (rebound). Previous modelling work assumed rebound of HAQ follows either of two alternative scenarios, with no data to inform which scenario is the more likely: rebound equal to initial gain, and rebound equal to natural history (NH). 177 These scenarios are explained in more detail in Appendix 11. The current model is informed by the expert opinion elicitation exercise conducted with five experts, described in Appendix 11. All experts suggested that not all the initial gain in HAQ is lost following late withdrawal of patients who initially responded to biologic therapy at 3 months. This scenario, that the HAQ rebound might be less than initial gain, has not been considered in any of the previous models of PsA, nor, to our knowledge, in any model of RA. Given the difficulty and limitations of eliciting expert opinion and the novelty of these findings, the current model assumes that rebound is equal to initial gain in the base case, and explores other scenarios (rebound less than initial gain and rebound equal to NH) in sensitivity analyses.
Outcomes for patients on palliative care
The PASI is assumed not to change on average compared with baseline for patients undergoing palliative care. HAQ is assumed to progressively worsen in such patients at a constant rate, estimated by an analysis requested from Deborah Symmons and colleagues at Manchester University for this appraisal using data from the Norfolk Arthritis Register (NOAR) (see details in Appendix 14).
Illustration of progression of HAQ in the model
Figure 3 illustrates the progression of HAQ over time for three different patient histories in the model. For a patient whose arthritis is controlled by biologic therapy, HAQ score is initially reduced (improves) and then maintained over time. For a patient who does not start biologic therapy, HAQ increases (deteriorates) over time to a maximum score of 3. For a patient who withdraws at 5 years, HAQ ‘rebounds’ (quickly increases) to the baseline level after withdrawal and then increases at the same rate as those who never started biologic therapy. However, in this scenario (‘rebound equal to initial gain’) the 5-year delay in progression obtained while on biologic drugs is permanently maintained after withdrawal.
FIGURE 3.
Illustration of the progression of arthritis for a patient successfully maintained on biologic, a patient without biologic and a patient who withdraws at 5 years. Note: a greater HAQ score indicates worse disability.
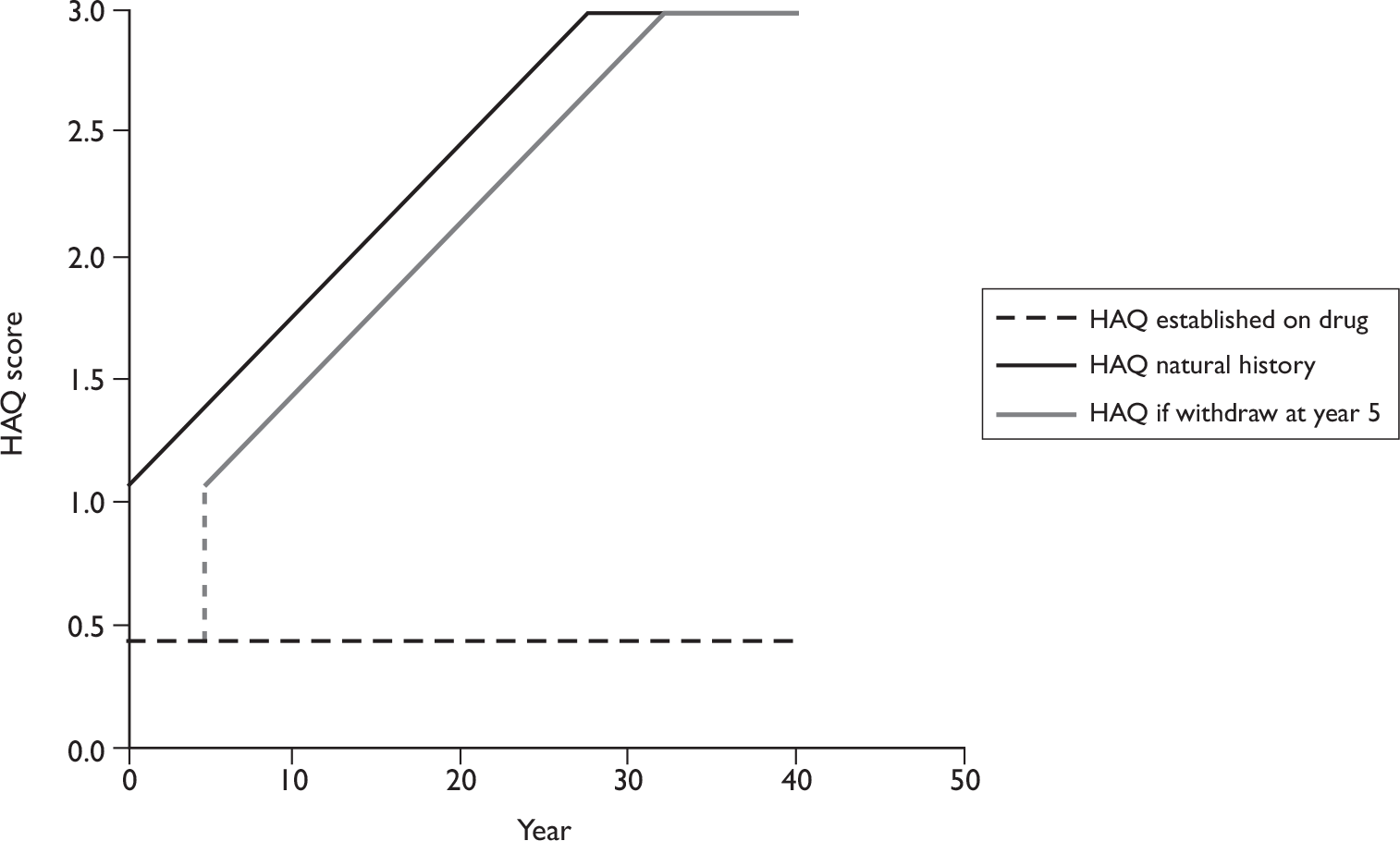
Utility
Health utility is measured as a function of HAQ and PASI. This relationship was estimated from analyses provided by the manufacturers, who carried out linear regressions of EQ-5D utility versus HAQ and PASI in participants in key RCTs (see Appendix 17). The base-case utility function is:
Other utility functions, supplied by the manufacturers, were used as sensitivity analyses.
Figure 4 illustrates the change in utility over time for different patients in the model. For a patient who is maintained on biologic therapy, utility is initially improved as a consequence of the reduction in HAQ and PASI, the latter depending on the proportion of patients who respond to psoriasis, given a response of arthritis (see Figure 2 and Appendix 10). This utility gain is assumed to be maintained over time. For a patient who did not start biologic therapy, utility deteriorates over time to a minimum value that is < 0, indicating that the general population would consider HRQoL with the severest arthritis symptoms and uncontrolled psoriasis to be worse than death. For a patient who withdraws at 5 years, utility ‘rebounds’ to the baseline level after withdrawal and then deteriorates at the same rate as those on NH. The area between these curves (area ‘A + C’ in Figure 4) represents the difference in lifetime QALYs between a patient who withdraws at 5 years and a patient who never uses biologic therapy.
FIGURE 4.
Illustration of utility (HRQoL) of a patient successfully maintained on biologic, a patient without biologic and a patient who withdraws at 5 years. Note: EQ-5D utility takes a maximum value of 1, indicating full health; values of < 0 correspond to health states that are considered worse than death by the general population.
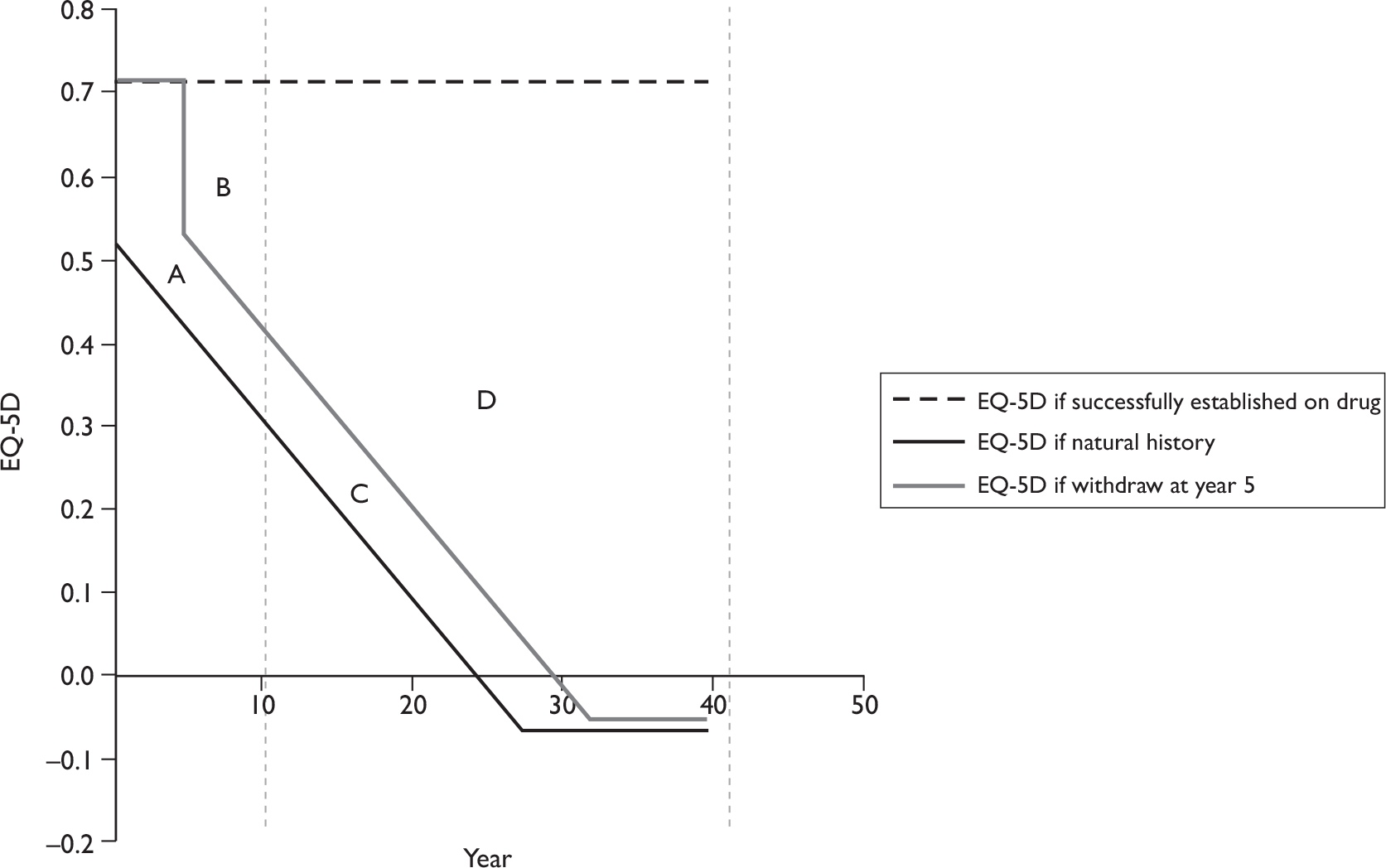
Time horizon for maintaining treatment effects
It is uncertain whether the effectiveness of biologic therapy is maintained in the very long term. Previous models considered a scenario where it is assumed that all patients withdraw from biologic therapy at 10 years, and all gains in HAQ with respect to NH are lost at this point. 177 Figure 4 illustrates the effect on utility of this ‘10-year time horizon for treatment effects’ scenario compared with the base case that assumes that treatment effects are maintained over the lifetime.
The difference in lifetime QALYs for a patient who is maintained successfully on a biologic, compared with NH, is area A + B + C + D. However, if is assumed that treatment effects last for only 10 years, the difference in QALYs over 10 years between being on a biologic and NH is only area A + B. For a patient who withdraws from a biologic at 5 years, the difference in lifetime QALYs compared with NH is area A + C. The difference in QALYs between assuming a 10-year time horizon and assuming a 40-year time horizon for a patient who withdraws from therapy at 5 years is area ‘C’. Biologic therapy appears much more effective if it is assumed that treatment effects in those who withdraw and those who do not withdraw are maintained over the long term. The base-case model assumes that the benefits of biologic therapy are maintained for a lifetime. Time horizons for treatment remaining effective for up to 10 years and up to 20 years are considered in sensitivity analyses.
Health service costs
The acquisition costs of the drugs and of their administration and monitoring were obtained from BSR recommendations and pharmaceutical list prices65 (see Appendix 13). The base case assumes that four vials of infliximab are administered and that vial sharing is not permitted.
Health-care costs increase with severity of both arthritis36 and psoriasis. 37 The health service costs of treating arthritis were measured from a UK-based study that estimated the effect of HAQ on costs in patients with RA41,59 (see Appendix 15). The NHS costs used for treating mild-to-moderate psoriasis in patients who do not use biologics or who do not respond to biologics were obtained from NHS unit costs of phototherapy187 and a UK RCT. 192 No UK studies based on prospective IPD were identified to estimate the health service costs of treating moderate or severe psoriasis in patients who do not use biologics or who do not respond to biologics. In the model these costs were obtained from a Dutch RCT and adjusted to UK price levels188 (see Appendix 16).
All-cause mortality
All-cause mortality was estimated from UK life tables. A Gompertz function was fitted to these data (see Appendix 19). The base case uses a published estimate of the additional mortality risk in PsA. 29 The effect of biologics on mortality in PsA is uncertain. The US VA study of MTX in psoriasis and patients with RA found that MTX was associated with significantly reduced incidence of vascular disease. 193 Long-term control of chronic inflammation may reduce mortality. However, long-term use of biologics might increase other mortality risks. The decision model assumes that there is no difference in mortality rates between treatments, or between biologic treatments and no treatment.
Subgroup analyses
The base-case model assumes a cohort of patients with PsA with baseline HAQ of 1.05, the mean of HAQ across the RCTs (see Table 1), and mild-to-moderate psoriasis (baseline PASI of 7.5). The model considered other cohorts in subgroup analyses:
-
A more severe baseline HAQ of 1.8, which is the mean HAQ of patients entering the British Society for British Society for Rheumatology Biologics Register (BSRBR). 186
-
No skin involvement, with PASI of 0. Clinical opinion suggests 50% of patients with PsA starting biologics in clinical practice would have mild or no skin involvement (Ian Bruce, personal communication).
-
A baseline PASI of 12.5, corresponding to moderate-to-severe psoriasis. 194,195 Clinical opinion suggests that 25% of patients with PsA starting biologics in clinical practice would have a baseline PASI > 10 (Ian Bruce, personal communication).
The review described in Chapter 3 did not find any evidence with which to assess whether treatment effects might differ by baseline severity, and, consequently, these analyses assume no change in relative treatment effects and focus just on variation between subgroups in baseline severity.
The base-case model assumes patients have failed at least two DMARDs, but are naive to biologics at baseline. The model was also used to estimate the cost-effectiveness of biologics used as a second course of therapy, if the first biologic is withdrawn. For example, if etanercept has been tried and failed, then the next alternative in sequence is adalimumab, infliximab or no biologic therapy. The reason why the patient failed the first course of therapy is potentially important information in deciding on the second course. Therefore, we consider two subgroups: one who failed the first biologic because of adverse events, and another who failed because of lack of efficacy. No RCTs have evaluated outcomes in these subgroups, and we estimate treatment response and withdrawal rates for these subgroups from observational data from the BSR register, which showed that if a patient failed first-line therapy for lack of efficacy, then the risk of failing the second-line therapy for lack of efficacy increased by 2.7 (95% CI 2.1 to 3.4). If a patient failed first-line therapy because of an adverse event then the risk of failing the second-line therapy for adverse events increased by 2.3 (95% CI 1.9 to 2.9). 196 Appendix 20 describes how these data were used to estimate the probability of initial response and later withdrawal for biologic therapies used as second line.
Analytic methods
The uncertainty in each parameter was represented using a probability distribution. The probabilities in Table 33 were assigned beta distributions. If p∼Beta(α,β) then α = E(p) × E(p) × (1 – E(p))/Var(p) and β = E(p) × (1 – E(p)) × (1 – E(p))/Var(p). The rate of change of HAQ while not on treatment was assigned a gamma distribution to ensure that values are strictly positive. If x∼Gamma(a,s) then a = E(x) × E(x)/Var(x) and s = Var(x)/E(x). All other uncertain parameters were assigned normal distributions with the mean and SE shown in Table 33. Probabilistic sensitivity analysis was carried out using Monte Carlo simulation.
The results of the model are presented in two ways. First, mean lifetime costs and QALYs for the three strategies are reported and their cost-effectiveness compared, estimating ICERs using standard decision rules. 197 Briefly, the alternative strategies are ranked by mean cost. Strategies that are more costly than another, but offer no greater expected benefit are known as ‘dominated’, and excluded. Strategies that are dominated by a linear combination of other strategies are considered subject to ‘extended domination’ and are also excluded. ICERs are then calculated for each of the remaining strategies, compared with the next best alternative. Although NICE does not specify a particular cost-effectiveness threshold, a strategy is more likely to be considered cost-effective if the ICER were < £20,000 per QALY, and less likely to be considered cost-effective if the ICER were > £30,000 per QALY. 184 Second, the decision uncertainty is shown as the probability that each intervention is the most cost-effective for a given cost-effectiveness threshold.
A series of alternative scenarios is also presented to explore the effect of changing one or more parameters/assumptions in the model.
Results of York Economic Assessment
Estimated probabilities of response at 3 months in the base case
Based on the results of the evidence synthesis in Chapter 3 (see Results of review of clinical effectiveness), and an estimate of the correlation between PsARC and PASI 75 outcomes in biologic therapy from an RCT,51 the model estimated the probability that a patient would respond for psoriasis only, joints only, both outcomes or neither outcome with each biologic therapy. These outcomes are shown under two assumptions: positive correlation (base case) and independence (Table 34).
| Response | Etanercept | Infliximab | Adalimumab |
|---|---|---|---|
| Positive correlation ρ = 0.435 | |||
| Skin only | 0.000 | 0.083 | 0.090 |
| Joints only | 0.536 | 0.110 | 0.200 |
| Both | 0.177 | 0.685 | 0.387 |
| Neither | 0.287 | 0.122 | 0.323 |
| No correlation (independence) | |||
| Skin only | 0.051 | 0.157 | 0.197 |
| Joints only | 0.587 | 0.184 | 0.307 |
| Both | 0.126 | 0.611 | 0.280 |
| Neither | 0.236 | 0.047 | 0.216 |
Results of the base-case cost-effectiveness analysis
The results of the base-case cost-effectiveness analysis are shown in Table 35, and univariate sensitivity analyses in Table 36. The base-case analysis suggests that infliximab is the most effective treatment (in terms of expected QALYs), followed by etanercept then adalimumab. Infliximab is also the most costly treatment, followed by etanercept then adalimumab. The ICER of etanercept compared with palliative care is about £18,000, and the ICER of infliximab compared with etanercept is about £44,000 per QALY. Of the three biologic therapies, etanercept has the highest probability of being cost-effective at a threshold of between £20,000 and £30,000 per QALY. Etanercept is the most cost-effective strategy in 44% of simulations of the base-case model, at a threshold ICER of £20,000 and in 48% of simulations at a threshold of £30,000 per QALY.
| Strategy | QALY | Cost (£) | Incremental QALY | Incremental cost (£) | ICER | PCE | |
|---|---|---|---|---|---|---|---|
| 20K | 30K | ||||||
| N | 5.171 | 42,168 | NA | 0.472 | 0.309 | ||
| A | 6.580 | 68,638 | 1.409 | 26,470 | Ex dom | 0.046 | 0.032 |
| E | 7.001 | 74,841 | 0.422 | 6203 | 17853 | 0.436 | 0.475 |
| I | 7.308 | 88,442 | 0.307 | 13,601 | 44326 | 0.046 | 0.184 |
| Scenario | Description | Trt | QALY | Cost (£) | ICER | PCE | |
|---|---|---|---|---|---|---|---|
| 20K | 30K | ||||||
| 1 | Base case | N | 5.171 | 42,168 | NA | 0.472 | 0.309 |
| A | 6.580 | 68,638 | Ex dom | 0.046 | 0.032 | ||
| E | 7.001 | 74,841 | 17,853 | 0.436 | 0.475 | ||
| I | 7.308 | 88,442 | 44,326 | 0.046 | 0.184 | ||
| 2 | Rebound in HAQ is small after withdrawal (base case = initial gain) | N | 5.171 | 42,168 | NA | 0.214 | 0.114 |
| A | 7.225 | 67,710 | Ex dom | 0.051 | 0.029 | ||
| E | 7.792 | 73,706 | 12,035 | 0.609 | 0.521 | ||
| I | 8.188 | 87,174 | 34,006 | 0.126 | 0.336 | ||
| 3 | Rapid worsening in HAQ with no treatment (upper 95% of CI) | N | 3.309 | 44,434 | NA | 0.358 | 0.187 |
| A | 4.967 | 70,829 | Ex dom | 0.047 | 0.029 | ||
| E | 5.447 | 76,985 | 15,221 | 0.528 | 0.544 | ||
| I | 5.786 | 90,609 | 40,248 | 0.067 | 0.240 | ||
| 4 | Log-PASI utility function (Abbott151) (base case linear) | N | 4.558 | 42,168 | NA | 0.459 | 0.308 |
| A | 6.001 | 68,638 | Ex dom | 0.069 | 0.040 | ||
| E | 6.390 | 74,841 | 17,835 | 0.400 | 0.390 | ||
| I | 6.769 | 88,442 | 35,898 | 0.072 | 0.262 | ||
| 5 | No correlation between PASI 75 and PsARC (base case = 0.4) | N | 5.171 | 42,168 | NA | 0.479 | 0.311 |
| A | 6.571 | 68,968 | Ex dom | 0.040 | 0.032 | ||
| E | 6.997 | 74,990 | 17,979 | 0.434 | 0.476 | ||
| I | 7.303 | 88,641 | 44,558 | 0.047 | 0.181 | ||
| 6 | RCT results fully generalisable to clinical practice (no adjustment for placebo effect) | N | 5.171 | 42,168 | NA | 0.451 | 0.282 |
| A | 6.637 | 68,561 | Ex dom | 0.053 | 0.037 | ||
| E | 7.068 | 74,752 | 17,178 | 0.446 | 0.482 | ||
| I | 7.381 | 88,344 | 43,371 | 0.050 | 0.199 | ||
| 9 | Exponential HAQ-cost function (Abbott151) (base case linear) | N | 5.171 | 63,052 | NA | 0.375 | 0.266 |
| A | 6.580 | 82,129 | Ex dom | 0.048 | 0.032 | ||
| E | 7.001 | 86,502 | 12,813 | 0.477 | 0.457 | ||
| I | 7.308 | 99,045 | 40,878 | 0.100 | 0.245 | ||
| 12 | Inpatient treatment for uncontrolled psoriasis | N | 5.171 | 151,496 | NA | 0.255 | 0.151 |
| A | 6.580 | 165,282 | 9787 | 0.114 | 0.055 | ||
| I | 7.308 | 175,157 | 13,557 | 0.621 | 0.769 | ||
| E | 7.001 | 178,530 | Dom | 0.010 | 0.025 | ||
| 13 | Cost per 3 months per 1-unit change in HAQ is £183 (US data)42 (base case £103) | N | 5.171 | 52,548 | NA | 0.444 | 0.303 |
| A | 6.580 | 77,518 | Ex dom | 0.047 | 0.032 | ||
| E | 7.001 | 83,224 | 16,761 | 0.453 | 0.467 | ||
| I | 7.308 | 96,562 | 43,468 | 0.056 | 0.198 | ||
| 14 | Change in utility per 1-unit change in HAQ is –0.45 (Wyeth153) (base case = 0.29) | N | 0.846 | 42,168 | NA | 0.312 | 0.203 |
| A | 2.905 | 68,638 | Ex dom | 0.024 | 0.011 | ||
| E | 3.589 | 74,841 | 11,913 | 0.522 | 0.474 | ||
| I | 3.954 | 88,442 | 37,280 | 0.142 | 0.312 | ||
| 15 | HAQ improves while on drug (lower 95% of CI) (base case no change) | N | 5.171 | 42,168 | NA | 0.029 | 0.007 |
| A | 7.845 | 66,823 | Ex dom | 0.075 | 0.023 | ||
| E | 8.492 | 72,704 | 9194 | 0.712 | 0.516 | ||
| I | 8.959 | 86,065 | 28,635 | 0.184 | 0.454 | ||
| 16 | High rate of withdrawal (upper 95% of CI) | N | 5.171 | 42,168 | NA | 0.464 | 0.316 |
| A | 6.302 | 62,085 | Ex dom | 0.041 | 0.029 | ||
| E | 6.635 | 66,604 | 16,690 | 0.436 | 0.460 | ||
| I | 6.876 | 77,323 | 44,451 | 0.059 | 0.195 | ||
| 17 | Low rate of withdrawal (lower 95% of CI) | N | 5.171 | 42,168 | NA | 0.485 | 0.322 |
| A | 6.891 | 76,566 | Ex dom | 0.060 | 0.035 | ||
| E | 7.411 | 84,811 | 19,038 | 0.427 | 0.462 | ||
| I | 7.793 | 101,890 | 44,731 | 0.028 | 0.181 | ||
| 18 | All treatments have the same probability of PsARC response at 3 months | N | 5.197 | 41,416 | NA | 0.472 | 0.312 |
| A | 7.104 | 77,174 | Ex dom | 0.176 | 0.193 | ||
| E | 7.236 | 78,115 | 17,999 | 0.351 | 0.467 | ||
| I | 7.316 | 87,889 | 122,073 | 0.001 | 0.028 | ||
| 19 | All treatments have the same probability of psoriasis responses (PASI 50/75/90) at 3 months | N | 5.273 | 41,746 | NA | 0.418 | 0.275 |
| A | 6.722 | 67,892 | Ex dom | 0.016 | 0.016 | ||
| E | 7.186 | 72,834 | 16,254 | 0.554 | 0.602 | ||
| I | 7.414 | 87,951 | 66,219 | 0.012 | 0.107 | ||
| 20 | Cost of drugs as in Wyeth submission153 | N | 5.171 | 42,168 | NA | 0.425 | 0.273 |
| A | 6.580 | 65,847 | Ex dom | 0.067 | 0.057 | ||
| E | 7.001 | 71,478 | 16,015 | 0.505 | 0.614 | ||
| I | 7.308 | 92,632 | 68,944 | 0.003 | 0.056 | ||
| 22 | All biologics have the same change in HAQ at 3 months for a PsARC responder | N | 5.171 | 42,168 | NA | 0.470 | 0.314 |
| A | 6.659 | 68,526 | 17,717 | 0.165 | 0.174 | ||
| E | 6.949 | 74,920 | 22,056 | 0.341 | 0.395 | ||
| I | 7.217 | 88,573 | 50,806 | 0.024 | 0.117 | ||
| 23 | Three vials of infliximab (base case: four vials) | N | 5.171 | 42,168 | NA | 0.423 | 0.259 |
| A | 6.580 | 68,638 | Ex dom | 0.000 | 0.000 | ||
| E | 7.001 | 74,841 | Ex dom | 0.034 | 0.061 | ||
| I | 7.308 | 76,550 | 16,809 | 0.543 | 0.680 | ||
| 26 | Rebound to NH after withdrawal (base case: rebound to initial gain) | N | 5.171 | 42,168 | NA | 0.983 | 0.687 |
| A | 5.846 | 69,701 | Ex dom | 0.004 | 0.038 | ||
| E | 6.104 | 76,145 | 36,408 | 0.013 | 0.273 | ||
| I | 6.307 | 89,900 | 67,759 | 0.000 | 0.002 | ||
| 31 | No costs of psoriasis (base case: UK data187,192) | N | 5.171 | 28,908 | NA | 0.485 | 0.317 |
| A | 6.580 | 56,792 | Ex dom | 0.037 | 0.022 | ||
| E | 7.001 | 62,209 | 18,196 | 0.459 | 0.513 | ||
| I | 7.308 | 77,704 | 50,499 | 0.019 | 0.148 | ||
| 32 | Schering-Plough estimates152 of cost per PASI point excluding phototherapy152 | N | 5.171 | 55,479 | NA | 0.456 | 0.298 |
| A | 6.580 | 80,496 | Ex dom | 0.065 | 0.042 | ||
| E | 7.001 | 87,252 | 17,361 | 0.414 | 0.423 | ||
| I | 7.308 | 99,438 | 39,715 | 0.065 | 0.237 | ||
| 33 | Schering-Plough estimates152 of cost per PASI point including phototherapy152 | N | 5.171 | 112,633 | NA | 0.370 | 0.237 |
| A | 6.580 | 131,482 | 13,381 | 0.146 | 0.057 | ||
| E | 7.001 | 141,118 | Ex dom | 0.145 | 0.161 | ||
| I | 7.308 | 146,187 | 20,188 | 0.339 | 0.545 | ||
| 99 | The effectiveness of biologic therapy lasts no longer than 10 years compared with palliative care | N | 5.171 | 42,168 | NA | 0.861 | 0.534 |
| A | 5.875 | 66,044 | Ex dom | 0.017 | 0.038 | ||
| E | 6.130 | 71,556 | 30,645 | 0.122 | 0.408 | ||
| I | 6.325 | 83,779 | 62,746 | 0.000 | 0.020 | ||
| 35 | Continue on biologic after 3 months if respond to either PsARC or PASI 75 (base case: PsARC only) | N | 5.171 | 42,168 | NA | 0.475 | 0.312 |
| A | 6.763 | 72,421 | Ex dom | 0.078 | 0.040 | ||
| E | 7.006 | 74,934 | 17,859 | 0.376 | 0.382 | ||
| I | 7.476 | 92,890 | 38,194 | 0.071 | 0.266 | ||
| 38 | Assume that adalimumab and etanercept are equally effective for PsARC response, HAQ change and PASI response | N | 5.231 | 41,524 | NA | 0.509 | 0.335 |
| E or A | 7.033 | 74,489 | 18,296 | 0.441 | 0.611 | ||
| I | 7.338 | 88,405 | 45,557 | 0.050 | 0.054 | ||
Adalimumab is extendedly dominated by palliative care and etanercept. This means that if NICE were considering adalimumab, the ICER relative to palliative care would be £26,470/1.409 = £18,786. However, the expected QALY per patient achieved with etanercept is greater than for adalimumab (7.00 vs 6.58), while the ICER of etanercept versus palliative care is £17,853. Therefore, it would not, on average, be cost-effective to recommend adalimumab because a greater QALY gain can be achieved from etanercept within the threshold of £20,000 per QALY.
Expected QALYs are low in this model. The total lifetime discounted health associated with palliative care is about 5.17 QALYs. This is because the base-case scenario assumes that utility declines fairly rapidly in patients with uncontrolled arthritis, and may be < 0 in later years (see Figure 4). For comparison, if HAQ and PASI could be reduced to 0 for the complete time horizon of the model (40 years), the model predicts that this cohort would expect 15 QALYs, given the rate of mortality, the intercept of the utility function and the discount rate. Figure 5 partitions the lifetime discounted QALYs gained by biologic therapies into those associated with improving arthritis and those associated with improving psoriasis, relative to palliative care. In the base case, utility gains as a result of improvement in arthritis are predicted to be much greater than utility gains as a result of improvement in the psoriasis component of PsA.
FIGURE 5.
Gains in lifetime discounted QALYs associated with treating arthritis and psoriasis in PsA with biologic therapies relative to palliative care.
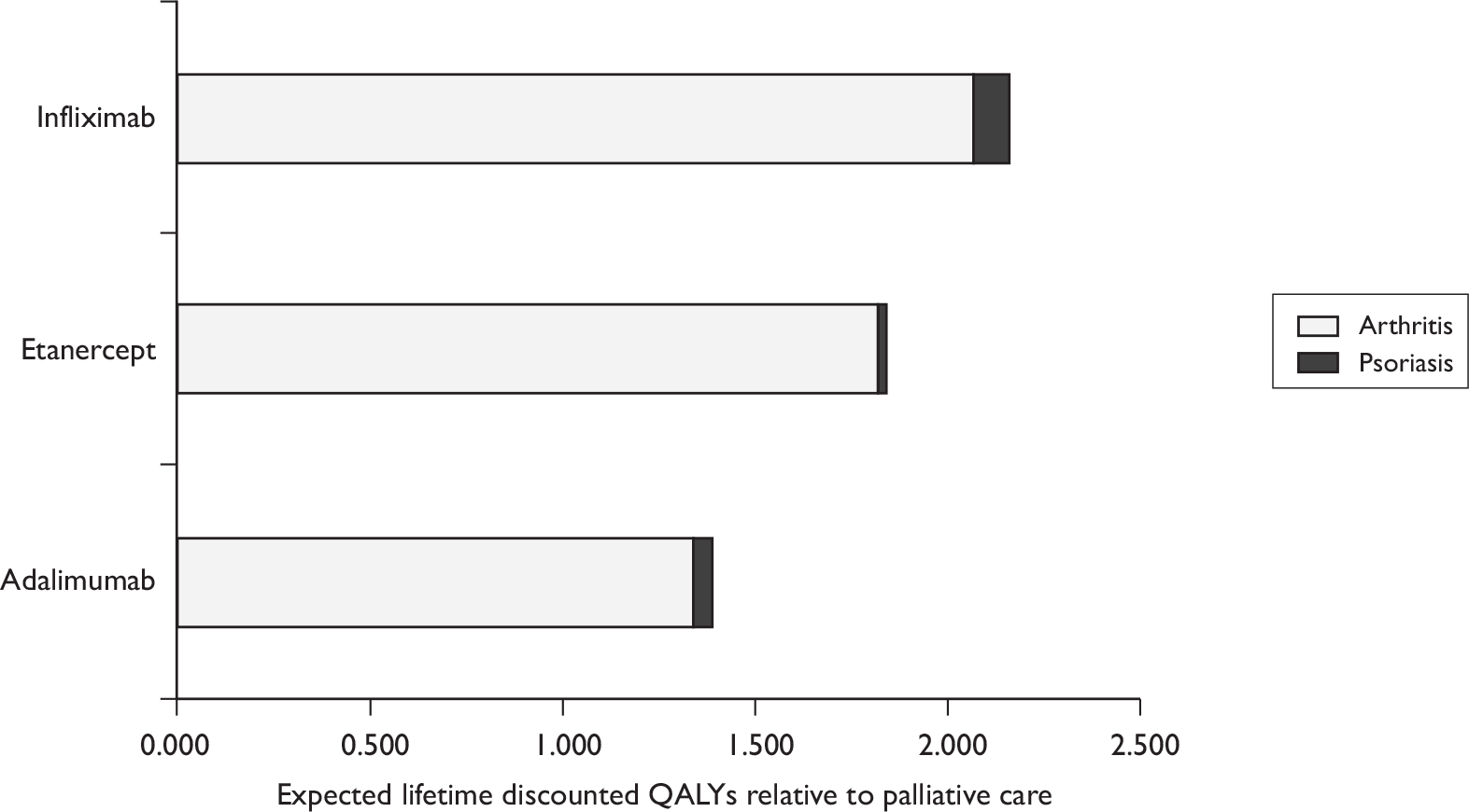
The expected lifetime (40-year) discounted costs without biologics (palliative care only) are about £42,000 in the base case for a patient with PsA and mild-to-moderate psoriasis. This can be partitioned into £29,000 for the treatment of arthritis and £13,000 for the treatment of psoriasis. Figure 6 partitions the total lifetime discounted health-care costs of the strategies into costs associated with the acquisition, monitoring and administration cost of the biologic drugs, the cost savings associated with treating arthritis (i.e. the reduction in HAQ score) and the cost savings associated with treating psoriasis (i.e. the reduction in PASI score). All costs are shown relative to the costs of palliative care.
FIGURE 6.
Lifetime discounted costs of biologic drugs, and cost savings for arthritis and psoriasis relative to non-biologic treatments for PsA.
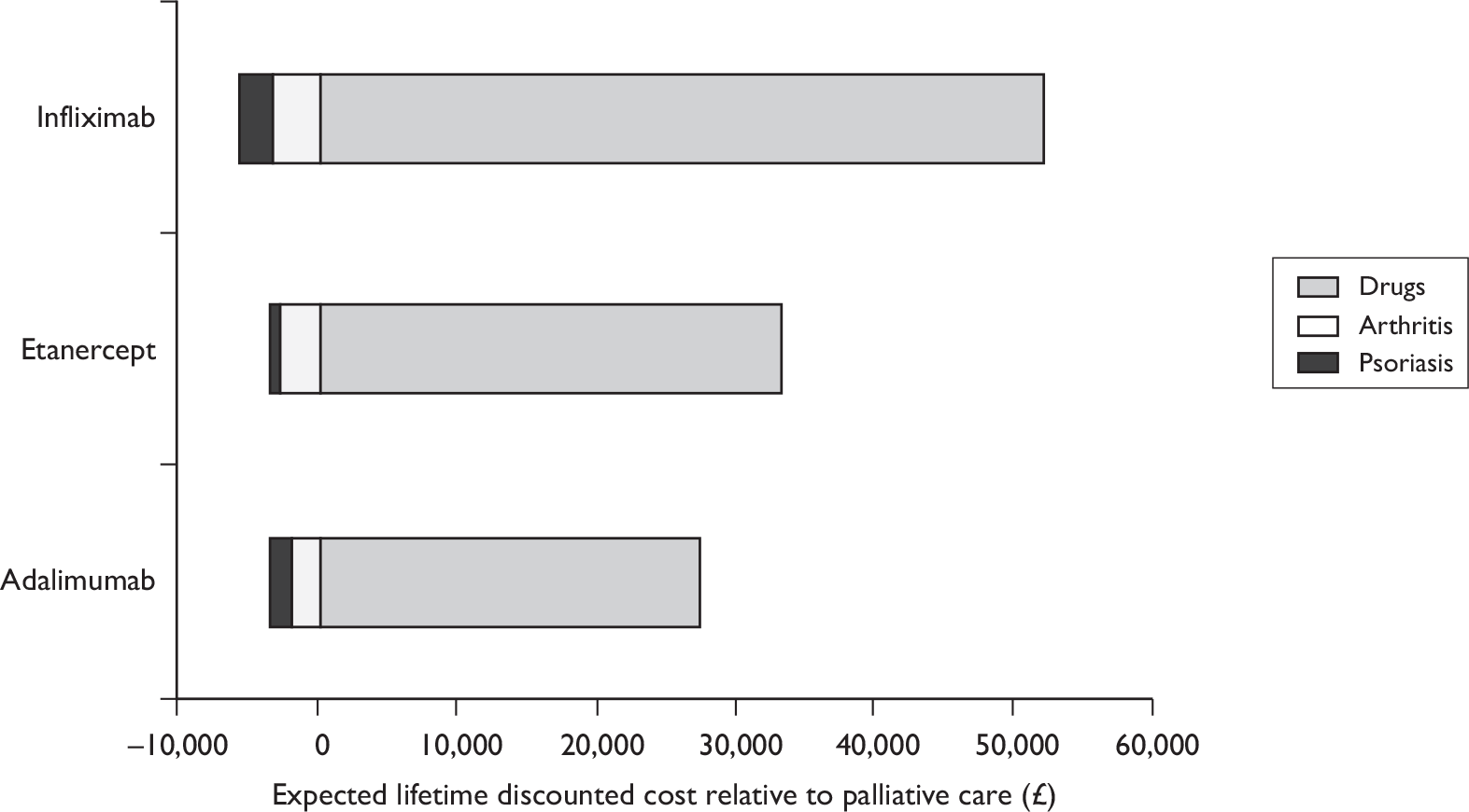
The lifetime discounted acquisition, administration and monitoring cost of infliximab is about £52,000; etanercept is about £33,000 and adalimumab is about £27,000. These prescribing costs are much greater than any offset health-care cost savings elsewhere. Infliximab is associated with the greatest gains in PASI and HAQ, and the greatest cost savings. Adalimumab has the second greatest gains in PASI and associated cost savings, and etanercept has the second greatest gains in HAQ and associated cost savings.
Results of sensitivity analyses
Table 36 shows the results of the univariate sensitivity analyses. Table 37 shows the cost-effectiveness of the alternatives in each of the scenarios, assuming that an ICER of £20,000 or less is likely to be cost-effective and a strategy with an ICER of ≥ £30,000 is unlikely to be accepted.
| Scenario | Description | Adalimumab | Etanercept | Infliximab |
|---|---|---|---|---|
| 1 | Base case | Ex dom | < 20K | > 30K |
| 2 | Rebound in HAQ is small after withdrawal (base case = initial gain) | Ex dom | < 20K | > 30K |
| 3 | Rapid worsening in HAQ with no treatment (upper 95% of CI) | Ex dom | < 20K | > 30K |
| 4 | Log-PASI utility function (Abbott151) (base case linear) | Ex dom | < 20K | > 30K |
| 5 | No correlation between PASI 75 and PsARC (base case = 0.4) | Ex dom | < 20K | > 30K |
| 6 | RCT results fully generalisable to clinical practice (no adjustment for placebo effect) | Ex dom | < 20K | > 30K |
| 9 | Exponential HAQ-cost function (Abbott151) (base case linear) | Ex dom | < 20K | > 30K |
| 12 | Inpatient treatment for uncontrolled psoriasis | < 20K | Dom | < 20K |
| 13 | Cost per 3 month per 1 unit change in HAQ is £183 (US data)42 (base case £103) | Ex dom | < 20K | > 30K |
| 14 | Change in utility per 1 unit change in HAQ is –0.45 (Wyeth153) (base case –0.29) | Ex dom | < 20K | > 30K |
| 15 | HAQ improves while on drug (lower 95% of CI) (base case no change) | Ex dom | < 20K | 20–30K |
| 16 | High rate of withdrawal (upper 95% of CI) | Ex dom | < 20K | > 30K |
| 17 | Low rate of withdrawal (lower 95% of CI) | Ex dom | < 20K | > 30K |
| 18 | All treatments have the same probability of PsARC response at 3 months | Ex dom | < 20K | > 30K |
| 19 | All treatments have the same probability of psoriasis responses (PASI 50, 75 and 90) at 3 months | Ex dom | < 20K | > 30K |
| 20 | Cost of drugs as in Wyeth submission153 | Ex dom | < 20K | > 30K |
| 22 | All biologics have the same change in HAQ at 3 months for a PsARC responder | < 20K | < 20K | > 30K |
| 23 | Three vials of infliximab (base case: four vials) | Ex dom | Ex dom | < 20K |
| 26 | Rebound to NH after withdrawal (base case: rebound to initial gain) | Ex dom | > 30K | > 30K |
| 31 | No costs of psoriasis (base case: UK data) | Ex dom | < 20K | > 30K |
| 32 | Schering-Plough estimates152 of cost per PASI point without phototherapy152 | Ex dom | < 20K | > 30K |
| 33 | Schering-Plough estimates152 of cost per PASI point with phototherapy152 | < 20K | Ex dom | < 20K |
| 99 | The effectiveness of biologic therapy lasts no longer than 10 years, compared with palliative care | Ex dom | 20–30K | > 30K |
| 35 | Continue on biologic after 3 months if respond to either PsARC or PASI 75 (base case: PsARC only) | Ex dom | 20–30K | > 30K |
| 38 | Assume that adalimumab and etanercept are equally effective for PsARC response, HAQ change and PASI response | < 20K | < 20K | > 30K |
The ICER of adalimumab falls below £20,000 per QALY and is no longer dominated by other strategies in any of the following univariate sensitivity analyses, assuming all other variables take mean values as in the base case:
-
All responders to PsARC have the same change in HAQ at 3 months, regardless of biologic therapy used.
-
If etanercept and adalimumab are considered equally effective for PsARC response, HAQ change and PASI response.
-
A patient who does not respond for psoriasis, or does not use biologic therapy, undergoes annual inpatient psoriasis treatment rather than annual ultraviolet light, type B (UVB), treatment.
The higher cost per PASI point (including phototherapy) from the Schering-Plough model152 are used.
The ICER of etanercept increases above £20,000 per QALY or is dominated by other strategies in any of the following univariate sensitivity analyses, assuming that all other variables take mean values, as in the base case:
-
A patient who does not achieve a PASI 75 response is offered one course of therapy as a hospital inpatient per year to treat psoriasis. The base case assumed that these patients are offered UV therapy.
-
HAQ rebounds after withdrawal from biologic to NH rather than to initial gain.
-
Biologic treatment becomes ineffective (relative to no treatment) after 10 years.
-
If the Schering-Plough152 estimates of the cost of treating psoriasis with phototherapy are used in the York Assessment Group model.
The ICER of infliximab falls below £30,000 per QALY in any of the following univariate sensitivity analyses, assuming that all other variables take mean values as in the base case:
-
A patient who does not respond for psoriasis, or does not use biologic therapy, undergoes annual inpatient psoriasis treatment rather than annual UVB treatment.
-
Infliximab requires three vials rather than four vials per administration.
-
The higher cost per PASI point (including phototherapy) from the Schering-Plough152 model are used.
-
HAQ improves while on a biologic drug.
No biologic appears cost-effective at a threshold of £30,000 per QALY if rebound of HAQ is to NH, rather than initial gain. In the scenario where treatment remains effective for only up to 10 years, the ICER for etanercept versus palliative care is £31,000 per QALY and is therefore is likely to be on the boundary of what would be considered cost-effective. If treatment remains effective for up to 20 years then the ICER of etanercept versus palliative care is £19,000 per QALY and the ICER for infliximab versus etanercept is £60,000 per QALY.
It should be noted that these are univariate analyses, where one variable in the base case is changed, holding others constant. Changes in combinations of variables might generate different results.
Results of subgroup analyses
Table 38 shows the results of the subgroup analyses.
| Scenario | Description | Trt | QALY | Cost (£) | ICER | PCE | |
|---|---|---|---|---|---|---|---|
| 20K | 30K | ||||||
| 10 | Baseline HAQ 1.8 (BSRBR186) (base case 1.05) | N | 2.090 | 46,594 | NA | 0.528 | 0.350 |
| 10 | A | 3.397 | 73,207 | Ex dom | 0.044 | 0.029 | |
| 10 | E | 3.804 | 79,431 | 19,156 | 0.389 | 0.447 | |
| 10 | I | 4.101 | 93,046 | 45,898 | 0.039 | 0.174 | |
| 11 | Baseline PASI 12.5 (base case 7.5) | N | 4.810 | 66,811 | NA | 0.431 | 0.274 |
| 11 | A | 6.257 | 90,422 | 16,310 | 0.115 | 0.057 | |
| 11 | E | 6.661 | 98,214 | 19,319 | 0.294 | 0.269 | |
| 11 | I | 7.012 | 107,988 | 27,778 | 0.160 | 0.400 | |
| 7 | Baseline PASI 12.5, and continue after 3 months only if respond to both PsARC and PASI 75 (base-case PsARC only) | N | 4.810 | 66,811 | NA | 0.399 | 0.246 |
| 7 | E | 5.315 | 74,865 | Ex dom | 0.030 | 0.039 | |
| 7 | A | 5.790 | 81,637 | 15,125 | 0.174 | 0.073 | |
| 7 | I | 6.717 | 101,796 | 21,739 | 0.397 | 0.642 | |
| 8 | Baseline PASI 12.5, and continue after 3 months if respond to either PsARC or PASI 75 | N | 4.810 | 66,811 | NA | 0.435 | 0.278 |
| 8 | A | 6.448 | 93,601 | 16,349 | 0.170 | 0.076 | |
| 8 | E | 6.665 | 98,293 | 21,609 | 0.208 | 0.177 | |
| 8 | I | 7.187 | 111,940 | 26,177 | 0.187 | 0.469 | |
| 21 | Baseline PASI 12.5, and annual inpatient treatment for uncontrolled psoriasis (base-case UVB) | N | 4.810 | 171,746 | NA | 0.185 | 0.079 |
| 21 | A | 6.257 | 183,184 | 7,901 | 0.101 | 0.053 | |
| 21 | I | 7.012 | 191,216 | 10,636 | 0.710 | 0.855 | |
| 21 | E | 6.661 | 197,741 | Dom | 0.004 | 0.013 | |
| 30 | Baseline PASI zero (base case 7.5) | N | 5.713 | 28,908 | NA | 0.498 | 0.330 |
| 30 | A | 7.064 | 56,792 | Ex dom | 0.018 | 0.019 | |
| 30 | E | 7.512 | 62,209 | 18,512 | 0.471 | 0.549 | |
| 30 | I | 7.752 | 77,704 | 64,744 | 0.013 | 0.102 | |
Biologics are slightly less cost-effective if the baseline HAQ is 1.8; however, etanercept still has an ICER below £20,000 per QALY. In this model, the size of the absolute gain in HAQ for responders is assumed to be independent of baseline HAQ, although there is a ceiling effect as the maximum HAQ score is 3. There is less scope for biologics to alter the course of the disease if they are started when patients already have a high degree of disability.
Etanercept is the most cost-effective strategy in patients with negligible baseline psoriasis. The ICER of infliximab versus etanercept increases to £65,000 per QALY. If baseline PASI were moderate-to-severe (12.5 instead of 7.5) the ICER of adalimumab versus palliative care would be < £16,000 per QALY, the ICER of etanercept versus adalimumab would be around £19,000 per QALY and the ICER of infliximab versus etanercept would be about £28,000 per QALY. If patients with uncontrolled moderate-to-severe psoriasis receive annual inpatient treatment instead of annual UVB the ICER for infliximab is below £20,000 per QALY and it is likely to be the most cost-effective strategy.
If the patient is indicated for biologics because of both severe skin disease and severe joint disease, we can consider alternative rules for continuing therapy. The base case follows the BSR guidelines, i.e. treatment is withdrawn from patients who fail to achieve the PsARC response within 3 months of treatment. Alternative decision rules (see Box 2) can change the conclusions. If patients with PsA and moderate-to-severe psoriasis are allowed to continue beyond 3 months if they respond to either PsARC or PASI 75 then all biologics have a similar probability of being cost-effective at a threshold of £20,000 per QALY, and infliximab has the highest probability of being cost-effective at a threshold of £30,000 per QALY. If patients with PsA and moderate-to-severe psoriasis are allowed to continue beyond 3 months only if they respond to both PsARC and PASI 75 then infliximab has the highest probability of being cost-effective at thresholds of £20,000 and £30,000 per QALY.
Table 39 shows the outcomes for each strategy if the biologic drugs are used as a second course of therapy after a first biologic has failed for patients with PsA with mild-to-moderate skin disease. The ICERs depend on which drug was used as first-line therapy, and which is therefore ineligible for use as second-line.
-
For patients who failed adalimumab as first line for inefficacy, etanercept has an ICER of < £20,000, and the ICER for infliximab is above £40,000 per QALY.
-
For patients who failed etanercept as first line for inefficacy, adalimumab has an ICER of < £20,000 and infliximab is around £25,000 per QALY.
-
For patients who failed infliximab as first line for inefficacy, etanercept has an ICER of < £20,000 per QALY and adalimumab is extendedly dominated compared with palliative care and etanercept.
-
The ICERs are broadly similar for patients who failed first-line therapy for adverse effects compared with results for those who failed first-line therapy for inefficacy.
| Scenario | Description | Trt | QALY | Cost (£) | ICER assuming: | ||
|---|---|---|---|---|---|---|---|
| I was used first line | E was used first line | A was used first line | |||||
| 24 | Second-line biologic if first failed for inefficacy | N | 5.171 | 42,168 | |||
| 24 | A | 5.827 | 54,394 | 18,652 | |||
| 24 | E | 6.142 | 58,783 | 17,114 | 17,114 | ||
| 24 | I | 6.410 | 68,630 | 24,406 | 36,746 | ||
| 25 | Second-line biologic if first failed for AEs | N | 5.171 | 42,168 | |||
| 25 | A | 6.273 | 61,430 | 17,486 | |||
| 25 | E | 6.597 | 65,780 | 16,554 | 16,554 | ||
| 25 | I | 6.831 | 76,205 | 26,445 | 44,569 | ||
Comparison of the York Economic Assessment with the manufacturers’ models
The following sections compare the assumptions and data sources used in each of the industry models with the current York model (see York Economic Assessment). A full description of the three industry models is provided in Appendix 7 and a critique is detailed in Appendix 8.
Summary of the models’ results
The three industry models, along with the current York model, are all potentially relevant to address the decision problem as specified by the NICE scope. However, each generates a different set of results. Abbott’s base case151 is for a 40-year time horizon, baseline HAQ = 1.3, baseline PASI = 6.9, proportion with psoriasis = 40%, and rebound of HAQ after withdrawal from biologic therapy equal to initial gain. Only results averaged across all patients are presented in the base case. The results show that infliximab was associated with the highest QALYs (8.49), followed by etanercept and adalimumab (both 8.33) and then DMARDs (7.47). Infliximab is the most costly strategy (£104,772). The ICER for adalimumab compared with DMARDs is £29,827. Etanercept is dominated by adalimumab and infliximab has an ICER of £199,596 compared with adalimumab.
Schering-Plough’s base case152 is for a 40-year time horizon, baseline HAQ = 1.14, baseline PASI = 11, proportion with psoriasis = 66% and rebound equal to gain. Results are reported for all patients, psoriatic patients and non-psoriatic patients. The results show that palliative care is the strategy associated with the lowest QALYs in all base-case scenarios (5.79–6.68, depending on the group of patients). Infliximab is the most effective strategy for all patients with PsA and those with a psoriasis component (8.65 QALYs for all patients and 8.40 QALYs for patients with psoriasis). For patients without psoriasis, etanercept is the most effective (9.14 QALYs). For all patients the model estimates a total cost of £64,704 for palliative care, £99,278 for adalimumab, £108,481 for etanercept and between £107,954 and £123,475 for infliximab, depending on the weight of patients. Similar estimates were generated for minimal psoriasis and psoriasis patients separately. Therefore, for all patients, etanercept has an ICER of £12,606 (compared with adalimumab) assuming a patient weight of 70 or 80 kg. For a 60-kg patient etanercept has an ICER of £12,432, compared with adalimumab, for patients without psoriasis. Infliximab dominates etanercept for psoriatic patients and all patients.
Wyeth’s base case153 is for a 40-year time horizon, baseline HAQ = 0.69, baseline PASI = 3.39, proportion with psoriasis = 62.4% and rebound equal to gain. Only results for all patients are presented in the base case. The results show that etanercept was associated with the highest gain in QALYs (6.90) followed by adalimumab (6.54), infliximab (6.39) and then ciclosporin (5.96). Ciclosporin was associated with the lowest cost (£53,860). Infliximab had the highest total costs (£66,867). Etanercept is the most cost-effective strategy with an ICER of £12,480.
The base-case analysis in the York model assumes a lifetime time (40-year) horizon for costs and QALYs a baseline HAQ = 1.05, baseline PASI = 7.5, rebound equal to gain and incorporates the correlation between PsARC and PASI 75 outcomes. The results for the base case for patients with PsA and mild-to-moderate psoriasis show that infliximab is the most effective treatment followed by etanercept then adalimumab. Infliximab is also the most costly treatment, followed by etanercept then adalimumab. The ICER of etanercept compared with palliative care is £18,000. Adalimumab is extendedly dominated. The ICER for infliximab compared with etanercept is £44,000 per QALY. Results are also presented for other baseline subgroups: HAQ = 1.8, PASI = 0 and PASI = 12.5.
Critique of manufacturers’ submissions and justification for current York modelling approach
There are large differences in the results generated by each of the four models. In order to determine which model provides the most appropriate estimates of the cost-effectiveness of biologics for the treatment of PsA, the key features of the models are compared and contrasted in more detail in the sections below. Justification for the approach taken in the current York model is also presented. A full critique of the industry models is also presented in Appendix 8. Table 40 shows the key features of each of the models. A full description of the three industry models is provided in Appendix 7.
| Wyeth153 | Schering-Plough152 | Abbott151 | Current York model | |
|---|---|---|---|---|
| Comparators | Ciclosporin (a DMARD) | Palliative care | Unspecified DMARD | Palliative care |
| Model structure | Initial response determined. HAQ and PASI tracked over time, accounting for withdrawals | Initial response determined. HAQ and PASI tracked over time, accounting for withdrawals | Initial response determined. HAQ and PASI tracked over time, accounting for withdrawals | Initial response determined. HAQ and PASI tracked over time, accounting for withdrawals |
| Patient characteristics | Heterogeneous cohort (first-order simulation) | Homogeneous cohort | Heterogeneous cohort (first-order simulation) | Homogeneous cohort |
| Baseline HAQ = 0.69 | Baseline HAQ = 1.14 | Baseline HAQ = 1.3 | Baseline HAQ = 1.05 | |
| Baseline PASI = 3.39 | Baseline PASI = 11 | Baseline PASI = 6.9 | Baseline PASI = 7.5 | |
| Proportion with psoriasis = 60.4% | Proportion with psoriasis = 66% | Proportion with psoriasis = 40% | ||
| Adjustment for placebo effect | No adjustment. Assumes comparator group represents effect of ciclosporin | Average HAQ gain in placebo arm is subtracted from HAQ gain in responders and non-responders on treatment | No adjustment. Assumes comparator group represents effect of DMARD | Average HAQ gain in placebo arm is subtracted from HAQ gain in responders and non-responders on treatment in the base case |
| Sequencing after failure of first drug | Patients withdraw from biologic drug to no treatment | Patients withdraw from biologic drug to no treatment | Sequence of unspecified DMARDs. There is a 24% reduction in response (i.e. increased probability of withdrawal) for each successive treatment in sequence compared with the previous | Patients withdraw from biologic drug to no treatment |
| Outcomes of evidence synthesis |
PsARC and PASI 75 at 12 and 24 weeks (from previous adalimumab MTC) Regression to predict 4 week PsARC from 12-week PsARC |
PsARC at 12 weeks: In subgroup with > 3% body skin area: PASI change from baseline at 12 weeks, by PsARC response/no response HAQ change from baseline at 12 weeks by PsARC response/no response and treatment drug (CiC information has been removed) |
Four regressions specified:
|
PsARC at 12 weeks: HAQ by PsARC response/no response and specific biologic treatment PASI 50/75/90 at 12 weeks |
| Decision to withdraw depending on initial response(s) | Withdrawal will be made if patient is a PsARC non-responder at either 12 weeks or 24 weeks | Withdrawal will be made if patient is a PsARC non-responder at 12 weeks | Withdrawal will be made if patient is a PsARC non-responder at 12 weeks |
Base case: withdrawal will be made if patient is a PsARC non-responder at 12 weeks Model considers other stopping decisions, e.g. PsARC or PASI responder |
| Initial change in HAQ for responders and non responders | HAQ at 4, 12 and 24 weeks predicted from PASI, PsARC, any biologic and baseline HAQ. HAQ does not differ by biologic drug after conditioning on other predictive variables |
HAQ by PsARC response and treatment from evidence synthesis. HAQ differs by the biologic used, after conditioning on PsARC For responders: maintain HAQ gain for 24 weeks from week 0 to 24 For non-responders (on biologics): maintain HAQ from week 0 to 12 |
HAQ at 12 and 24 weeks predicted from ACR response (20, 50, etc.), baseline HAQ, age, gender, baseline PsA duration, whether on MTX and whether on any biologic (from ADEPT51,88,92,93,100–104 data). HAQ does not differ by biologic drug after conditioning on the other predictive variables |
HAQ by PsARC response and treatment from evidence synthesis HAQ differs by the biologic used, after conditioning on PsARC |
| HAQ progression while on biologic and responder | 0 | Assumes a HAQ improvement for first year while on biologics, then 0 | Worsening by 0.0005 per year (Bath data set) | 0 |
| HAQ progression when on DMARD | 0.028 per year (Sokoll) | Not applicable | 0.024 per year (Leeds) | Not applicable |
| HAQ progression while on therapy and ACR < 20 | Not applicable | Not applicable | 0.066 per year (Leeds) | Not applicable |
| HAQ progression while not on therapy | 0.069 per year (Leeds) | 0.071 per year (Leeds) | 0.066 per year (Leeds) | 0.072 per year (NOAR) |
| Initial change in psoriasis on biologic | Initial improvement in PASI (weeks 4, 12 and 24) was estimated using multivariate regression models |
PASI change from baseline to 12 weeks for PsARC Responder/non-responder from evidence synthesis |
PASI at 12 and 24 weeks predicted from baseline PASI, age, gender, baseline PsA duration, MTX, whether PASI 50/75/90 response | Predicted from baseline PASI and proportion who are PASI 50/75/90 response |
| Correlation between PASI and PsARC responses | Assumes PASI is a predictor of HAQ | Predicts PASI by PsARC response, generating a different PASI change for PsARC responders and non responders The change in PASI is dependent on the biologic used, after conditioning on PsARC | Assumed independent | Correlation of PsARC and PASI 75 estimated from ‘ADEPT’ trial to estimate the joint pdf |
| Psoriasis progression on biologic | 0 | 0 | 0 | 0 |
| Psoriasis progression not on biologic | 0 | 0 | 0 | 0 |
| HAQ rebound when stopping therapy | To initial gain OR to NH | To initial gain OR to NH | To initial gain | To initial gain and using elicited values in sensitivity analysis |
| Psoriasis rebound when stopping therapy | To initial gain | To initial gain | To initial gain | To initial gain |
| Withdrawal rate: biologics | Different withdrawal rates for each biologic (Saad et al.191). Weibull estimated using data from three time points | 11% per year (Geborek et al.198) per year | Average withdrawal rate across all biologics (Saad et al. 191). Weibull estimated using data from three time points |
Average withdrawal rate across all biologics (meta-analysis of observational studies). 16% per year |
| Withdrawal rate- DMARD | 0.34 per year | Not applicable | Weibull distribution used. Unclear how this was specified as only one data point reported (Malesci et al.199) | Not applicable |
| Utility (HRQoL) | Predicted from HAQ, age and gender (PRESTA) | Predicted from HAQ and PASI, HAQ-squared and PASI-squared, using regression, (no interaction term) (GO-REVEAL data) | Predicted from HAQ and PASI (no interaction term) (ADEPT) | Wyeth:153 additional utility regression as the base case and other functions as sensitivity analyses |
| Mortality | Same rate for all treatments and no treatment (Wong et al.29) | Same rate for all treatments and no treatment (Wong et al.29) | Same rate for all treatments and no treatment (Wong et al.29) | Same rate for all treatments and no treatment (Wong et al.29) |
| Costs of treatments | Assumes no wastage of Infliximab | Results shown assuming three vials of infliximab (60 kg), three and a half vials (and four vials (80 kg) | Assumes no wastage of Infliximab (four vials, 80-kg weight) | Assumes no vial sharing (four vials, 70–80 kg weight) in base case. Three vials for a 60-kg patients considered in sensitivity analysis |
| Costs of start-up, administration and monitoring | BSR recommendations | From York model | BSR recommendations | BSR recommendations validated by clinical opinion |
| Cost depending on HAQ | THIN data set. HAQ was not recorded in this data, and was predicted based on relationship between HAQ, age, number of prior DMARDs in BSR data set | RA data set (Kobelt et al.59) | NOAR | RA data set (Kobelt et al.59) in base case |
| Cost of psoriasis | Not included (other than through HAQ which is in part predicted by PASI) | Physician survey | Physician survey | For mild-moderate psoriasis: Poyner et al.192 |
| Patient subgroups | Mild, moderate, severe HAQ and mild, moderate/severe and very severe PASI |
With psoriasis Without psoriasis |
Varying severity of HAQ and PASI at baseline | Varying severity of HAQ and PASI at baseline |
Choice of comparator
The choice of comparator is crucial in determining the relative cost-effectiveness of biologics. In comparing biologics to DMARDs while using the effectiveness estimates of placebo from randomised trials (i.e. assume DMARD cost and placebo effectiveness), Wyeth153 and Abbott151 may artificially inflate the cost-effectiveness of biologics, as DMARDs are liable to be more effective than palliative care in practice. It is also unlikely that patients who have failed two previous DMARDs would be considered for further DMARD treatment, and such patients are likely to receive palliative care (as assumed in the York and Schering-Plough152 models).
Heterogeneity
Although patients included in the model will be similar in terms of their exposure to DMARDs and the fact that they will be biologic naive, they may be a heterogeneous group in many other respects. The Abbott151 and Wyeth153 models use an individual sampling approach, where observed heterogeneity in the group of patients is modelled by sampling over a set of patient characteristics, taken from Mease et al. 200 This approach effectively averages over the heterogeneity between patients. In contrast, the Schering-Plough152 and current York models use a cohort approach which assumes a homogeneous group of patients. To account for any heterogeneity in a cohort model, the models can be ran separately for each homogeneous group to generate estimates of cost-effectiveness, conditional on each set of observed characteristics. In principle, separate NICE decisions can then made for each group of patients. This difference in how heterogeneity is reflected in the different models may partly explain the variation in their results.
Baseline characteristics differ quite markedly between models. In the Wyeth model153 the baseline HAQ and PASI are both low, at 0.69 and 3.39, respectively. These are higher in the Abbott151 model at 1.3 for HAQ and 6.9 for PASI. In the Schering-Plough152 model baseline HAQ is about the average for the RCTs at 1.14; however, a baseline PASI score of 11 suggests that patients have relatively severe psoriasis. The Schering-Plough152 model also includes the highest proportion of patients with psoriasis at 66%; however, these are run as a separate subgroup to those without any significant psoriasis rather than as a model input. The current York model also distinguishes between those with little or no psoriasis (PASI scores < 5) and moderate or severe psoriasis (PASI scores > 5) with 7.5 as the base case. Baseline HAQ in the York model is 1.05, based on the average observed in the RCTs (see Table 1). The current York model also run a series of scenarios to vary base-case HAQ and PASI scores (see Tables 36 and 38). For patients with a high baseline PASI (12.5) adalimumab is no longer extendedly dominated (ICER is £16,000 compared with palliative care). The ICER for etanercept is similar to the base case at £19,000 compared to adalimumab and the ICER for infliximab falls to £28,000 compared to etanercept. These changes in ICERs are because of the differences in PASI response rates between the drugs. For more severe psoriasis (high baseline PASI), treatments with a better effect on PASI will be more cost-effective. For patients without any significant psoriasis aspect to their disease, the ICER for etanercept increases slightly to £19,000 compared with palliative care. For patients with a higher baseline HAQ (1.8) the ICER for etanercept also increases to £19,000 compared with palliative care.
Model structure
The basic structure in each of the four models is similar. Each determines initial response to treatment and then tracks HAQ and PASI scores over a lifetime, taking account of any withdrawals from treatment.
Measurement of initial response for joints
All models use PsARC to measure the initial response for joints. All models used a Bayesian evidence synthesis to estimate PsARC. However, the results differ, partly because different RCTs are included in the analyses (see Table 19). Schering-Plough152 and the York model predict that infliximab is the most effective drug for PsARC response, then etanercept, then adalimumab. Wyeth153 predict etanercept is the most effective, then infliximab, then adalimumab (see Table 21). Using a bivariate meta-analysis to inform the economic model, Abbott151 predicts that infliximab is most effective for PsARC and ACR responses, then adalimumab, then etanercept. These differences have a substantial effect on the results of the economic analysis. The sensitivity analysis shown in Table 36 shows that by assuming that all treatments have the same probability of psoriasis responses (PASI 50/75/90) at 3 months, the ICER for etanercept falls to £16,000, adalimumab remains extendedly dominated and the ICER for infliximab increases to over £66,000. This is because infliximab had a much higher probability of skin response in the base case. Applying the same PsARC response at 3 months to all treatments also has a minimal effect on the ICERs of adalimumab and etanercept, but increases the ICER for infliximab compared with etanercept to over £100,000. This is because infliximab was associated with a much higher PsARC response in the base case (see Table 34).
Continuation on biologic treatment after initial assessment
All of the industry models assume that patients are withdrawn from treatment if they are PsARC non-responders at 12 weeks (and 24 weeks for Wyeth153), irrespective of PASI response. The current York model also uses this assumption in the base case, but additionally explores alternative scenarios for discontinuation for patients who are indicated for both moderate-to-severe psoriasis and arthritis. The BAD guidelines recommend that patients continue if they achieve PsARC or PASI 75, although this continuation rule does not change the conclusions for patients with PsA and mild-to-moderate psoriasis.
Correlation between skin and join response
If patients have both joints and skin involvement at baseline then in determining the initial response to treatment it is important to incorporate any correlation between the joint and skin responses, measured by PsARC and PASI respectively. The current York model incorporates the correlation between PsARC and PASI 75 using data from the ADEPT trial88 and the results of the evidence synthesis in Chapter 3 (see Results of review of clinical effectiveness) to estimate the probability of a response to both psoriasis and joints, the probability of a response to neither, and the probability of a response to one but not the other. The industry models, in contrast, do not afford this issue as much attention. Abbott151 assumes that PsARC and PASI responses are independent (see Appendix 7 for further detail). The Schering-Plough model152 predicts PASI by PsARC response, thus generating a different PASI change for PsARC responders and non-responders by drug. This implicitly incorporates a correlation between PsARC and PASI responses, but is difficult to vary in sensitivity analysis. The Wyeth model153 assumes PASI is a linear predictor of HAQ (see Appendix 7 for further detail). This is a strong assumption that is difficult to vary in sensitivity analysis, and Wyeth153 did not support this by a clinical justification. The York model also considers a scenario where there is no correlation between PASI 75 and PsARC (see Table 36). The impact on the ICER for etanercept is minimal, however, with the ICER for etanercept increasing to £16,106.
Effect on joints and skin for responders and non-responders
The models differ in the variables used to predict the change in HAQ for responders. Wyeth153 estimate HAQ from PsARC response and PASI. Abbott151 estimate HAQ from ACR response (assumed correlated with PsARC) and other clinical and demographic variables. Abbott151 assumes the ACR response varies by biologic drug, after conditioning on PsARC. Schering-Plough152 and the York model estimate HAQ from PsARC response, and assume that HAQ varies by biologic received, after conditioning on PsARC response.
Given the initial response (or lack of response) to treatment, all models then determine an associated HAQ and PASI score. The current York model uses the same approach as Schering-Plough,152 and predicts HAQ by PsARC response and treatment, and this is estimated by the evidence synthesis model. Abbott151 predicts HAQ from the ACR response as an explanatory variable, and other clinical and demographic explanatory variables. The same HAQ gain is assumed for all treatments, after conditioning on ACR. Despite this, the Abbott model151 allows a different HAQ gain for PsARC responders for each treatment. This is because ACR response is assumed to differ by biologic drug, and ACR was correlated with PsARC in the Abbott151 evidence synthesis (see Table 19). Although this seems an attractive method of predicting changes in HAQ, it was decided not to use this approach in the York model as the Abbott151 evidence synthesis was very complex. Furthermore, it is not clear what data used in the Abbott151 evidence synthesis inform their economic model. In the clinical section of the Abbott report,151 Table 2.7.2.2 shows the marginal probabilities of PsARC and ACR responses were estimated to be higher for etanercept than adalimumab (these results are reproduced in Tables 5.21 and 5.24 of the York Assessment Group report and are similar to those of the York Assessment Group). However, Table 3.4.3.1.1 of the economic section of the Abbott report151 shows the contingent or joint probabilities of ACR and PsARC, and appears to contradict the results of their clinical section; in this table, adalimumab is shown as more effective for PsARC and ACR response than etanercept. It appears that the Abbott meta-analysis151 that informed their economic section made use of different data to the clinical section, including data from biologics that are not relevant to this appraisal.
Wyeth153 estimates the initial change in HAQ, including changes in PASI in the regression. The same HAQ gain is used for all treatments. The use of the skin component of PsA to predict the arthritis component of the disease is considered of doubtful clinical validity. There is no evidence to suggest that one component of the disease is a good predicator of the other: patients can have differing degrees of both components and those with severe arthritis will not necessary have severe psoriasis and vice versa.
The fact that two of the models use treatment specific HAQ gains (Schering-Plough152 and York) and two use the same HAQ gain for all treatments may explain some of the variability in results. The results of a sensitivity analysis on the York model (see Table 36) show that by assuming all biologics have the same change in HAQ at 3 months for a PsARC responder, the results differ quite significantly from those in the base case. Adalimumab is no longer extendedly dominated and the ICER for etanercept increases to £22,000 compared with adalimumab. This is because in the base case etanercept was associated with a much higher HAQ gain for a PsARC responder (–0.63) than adalimumab (–0.48).
To determine the initial change in PASI, the current York model and the Abbott151 model predict the initial 12-week (and 24-week for Abbott151) change in PASI, using baseline PASI and the proportion of patients who are PASI 50/75/90 responders, thereby using all information on PASI response. Wyeth153 uses only PASI-75 to generate the initial improvement in PASI, thereby ignoring all of the other PASI information. Schering-Plough152 also estimates PASI change (not specifying which proportion) in the initial period, but do this for PsARC responders/non-responders in their evidence synthesis model. It is not clear why PASI change was estimated for PsARC responders and non-responders and not for PASI responders. Determining the initial differences in PASI response between treatments is likely to be a key driver of the cost-effectiveness results. All of the evidence syntheses predicted that infliximab is most effective for psoriasis response, then adalimumab, then etanercept (see Table 23). However, Wyeth153 predicted that infliximab was less effective in absolute terms than the York and Abbott151 models. The sensitivity analysis shown in Table 36 shows that by assuming that all treatments have the same probability of psoriasis responses (PASI 50/75/90) at 3 months, etanercept appears more cost-effective and the other biologic drugs less cost-effective than the base case.
Placebo effects
In determining this initial impact of treatment, it is important to account for any overestimate of the absolute response rates in both placebo and treatment groups in RCTs, compared with what would be expected in routine practice. This is termed the placebo adjustment. However, the York sensitivity analysis found that this adjustment had a minor effect on results.
Health Assessment Questionnaire progression when not on a biologic
For those patients who do not respond to treatment, or who are assigned palliative care, HAQ and PASI progression must be tracked over the model. To determine HAQ progression off treatment, all of the industry models use data from the Leeds cohort study201 data. As detailed in Appendix 14, however, the Leeds data set does have some limitations. The current York model therefore uses data from patients enrolled in the NOAR (see Appendix 14) data set to estimate HAQ change in patients who have uncontrolled PsA. The 3-month progression rates are similar to those generated using the Leeds data (0.018 in NOAR compared with 0.016 in Leeds data set) and is unlikely to lead to major differences in the results.
Withdrawal from biologics
For those patients who do initially respond to biologic treatment, each of the models considers the possibility that they may withdraw from treatment beyond the initial period due to either loss of efficacy or adverse events. Each of the industry models makes use of a single data set to estimate withdrawals. Schering-Plough152 uses the same rates as used in the previous York model (0.11 per year from Geborek et al. 198). Wyeth153 and Abbott151 use evidence from a recent paper by Saad et al.,191 which used data from the BSRBR registry to estimate parameters of a Weibull distribution to quantify the rate of withdrawal over time. All models assumed that withdrawal rates did not vary by treatment. However, the sensitivity analysis reported in Table 36 shows that there is very little impact of changes withdrawal rates within the current York model.
Sequential biologic therapies
Once patients withdraw from biologic treatment they are assumed to move to either palliative care or DMARDs. None of the four models consider the use of sequential biologics in the base-case scenario. The sequential use of biologics is common in clinical practice; however, a lack of data on the effectiveness of biologics beyond first line use limits the scope to consider such an analysis. The current York model conducts an exploratory sensitivity analysis on the issue of sequencing biologics (see Appendix 20), utilising available registry data on response rates for subsequent lines of biologics.
Utility and cost estimates
The utilities and costs assigned to treatments are of paramount importance in determining the cost-effectiveness of the included treatments. It is, therefore, important to note that each of the models uses different methodology and data sources to link HAQ and PASI to utilities and to determine the associated costs of treatments. In generating utilities each of the industry models uses both different data sources and different models to predict utilities from HAQ and PASI. To disentangle these two effects the current York model explores various scenarios using regression results provided on request from each of the manufacturers (see Appendix 17), which are estimated using a common methodology. In addition, the York model explores the use of alternative assumptions regarding the calculation of utilities in sensitivity analysis (see Table 36). Only the scenario where a higher estimate of the effect of a unit change in HAQ on utility is taken from the Wyeth153 submission (–0.45) has a discernible impact on the results. Etanercept is more cost-effective (ICER is £12,000 compared with palliative care) and the ICER for infliximab falls to £37,000.
Resource use assumed in establishing drug, administration and monitoring costs differs between the industry models. In particular, there were varying assumptions regarding the number of doses given for each of the drugs (see Appendix 8) and the number of laboratory tests required for monitoring patients. The costs attached to hospital visits also differed between models. In the Abbott model,151 it was not possible to validate the resource use and costs used, and the total costs given in the report could not be replicated in terms of the resource use items and unit costs presented. That is, using the resource use multiplied by the respective unit costs gave different total costs to those presented in the model report. These also differed from those used in the model.
The current York model therefore sought to generate costs for each of the treatments using resource use specified by the BSR guidelines and validated by clinical collaborators (see Appendix 13). These differences in costing methodology produce quite different estimates of total costs. For example, in the initial 3-month period the cost of infliximab in the base-case analysis is £5459 in the Abbott model,151 £4386, in the Schering-Plough model,152 £6286 in the Wyeth model153 and £5522 in the current York model. The sensitivity analysis in Table 36 shows the impact of varying drug costs in the current York model. Using the costs presented in the Wyeth submission153 in the York model (which are higher for infliximab (see Appendix 8) but lower for adalimumab and etanercept than the York estimates, increases the cost-effectiveness of etanercept and increases the ICER for infliximab (£69,000). Reducing the number of vials used for each infliximab infusion from four to three greatly increases the cost-effectiveness of infliximab and reduces the relative cost-effectiveness of the other biologics.
In addition to the costs of drugs, administration and monitoring each of the models considers the ongoing health-service costs of PsA as a function of a patient’s HAQ score. Abbott151 and Schering-Plough152 also include health-service costs according to PASI scores. The costs associated with PASI score, in particular differ quite markedly (see Appendix 8). Abbott151 and Schering-Plough152 rely on surveys of clinicians’ opinions, based on vignettes of ‘typical cases’ to estimate the costs associated with treating psoriasis. The York model estimates the costs of treating mild-to-moderate psoriasis that is uncontrolled by biologic drugs from a UK RCT and the costs of treating moderate-to-severe psoriasis that is uncontrolled by biologic drugs from a Dutch RCT. The sensitivity analysis in Table 36 shows the impact of varying ongoing costs of PsA as a function of a patient’s HAQ and PASI score in the current York model. Using the exponential HAQ cost function from the Abbott model151 reduces the ICER for etanercept to £13,000. Adding in a high inpatient cost of uncontrolled psoriasis had a much more dramatic impact on model results: etanercept is dominated by infliximab, which is itself associated with an ICER of £13,000 compared with adalimumab. This reflects the beneficial effect of infliximab in terms of reducing PASI score compared with other biologics. Infliximab is associated with an ICER likely to be below the threshold when the cost estimates per PASI point (including phototherapy) from Schering-Plough152 are used. In this situation the ICER for infliximab is £20,000. Other sensitivity analysis on costs dependent on HAQ and PASI had little impact on the model results.
Summary
The key differences between the three industry models and the current York model have been discussed. This has highlighted a number of potentially important limitations with the three industry models, in particular: the choice of comparator, averaging across patient heterogeneity; failure to consider alternative correlations between response types; how initial PsARC response is determined; how the change in HAQ is determined; no consideration of alternative decision rules about continuing beyond the initial 3-month period; generating withdrawals rates from a single observational study; the costs of drugs; drug administration and monitoring; and the health-care costs associated with treating arthritis and psoriasis if these are uncontrolled by biologics.
Discussion of York Economic Assessment
The economic model has evaluated the cost-effectiveness of three alternative biologic therapies and palliative care only. Under base-case assumptions, for patients with PsA and mild-to-moderate skin disease, the ICER of etanercept versus palliative care is about £18,000 per QALY and the ICER of infliximab versus etanercept is about £44,000 per QALY. Adalimumab is extendedly dominated. On average, given the base-case assumptions in the York model, etanercept would be considered the most cost-effective strategy if the threshold for cost-effectiveness were £20,000 per QALY or £30,000 per QALY. The probability etanercept is the most cost-effective treatment is 0.44 at a threshold of £20,000 per QALY and 0.48 at a threshold of £30,000 per QALY. The expected lifetime prescription costs of biologic therapies is considerably greater than the offset cost savings elsewhere in the NHS.
These results are sensitive to several of the scenarios tested in univariate sensitivity analyses:
-
All biologics appear less cost-effective if they are assumed to remain effective for a maximum of 10 years rather than 40 years, or if HAQ rebounds to NH after withdrawal.
-
Results are sensitive to assumptions about the prescription cost. If three vials of infliximab are required rather than four, infliximab is much more cost-effective and the other biologics are not cost-effective.
-
Results are sensitive to assumptions about the cost of treating patients who do not achieve a response to biologics for the psoriasis component of PsA. If these costs are high, etanercept appears less cost-effective as it is considerably less effective in treating psoriasis than the other biologics.
-
Results are sensitive to assumptions about the progression of HAQ on and off treatment. If the prognosis for patients without biologics is worse than the base case, or HAQ improves while on biologic drugs, all biologics appear more cost-effective.
-
Results are sensitive to assumptions about whether clinical effectiveness differs between the therapies.
Cost-effectiveness also varies between different subgroups of patients:
-
For patients with PsA and moderate-to-severe skin disease, and who continue with biologic therapy if they achieve a response for either psoriasis or joint disease, the ICER of adalimumab versus palliative care is about £16,000 per QALY, the ICER of etanercept versus adalimumab is about £21,000 per QALY and the ICER for infliximab versus etanercept is about £26,000 per QALY.
-
For patients with PsA and negligible skin involvement, the ICER of etanercept versus palliative care is about £19,000 per QALY, and the ICER of infliximab versus etanercept is about £65,000 per QALY. Adalimumab is extendedly dominated in this group.
-
For patients who have failed adalimumab or infliximab as first-line therapy for either adverse events or inefficacy, etanercept is cost-effective at a threshold of £20,000 per QALY. For patients who have failed etanercept as first-line therapy for either adverse events or inefficacy, adalimumab is cost-effective at a threshold of £20,000 per QALY, though infliximab is more likely to be cost-effective if the threshold is £30,000 per QALY.
These are univariate sensitivity and subgroup analyses. Multivariate sensitivity analyses may lead to different conclusions.
The decision model and data sources have several limitations and uncertainties. BAD guidelines recommend that both PASI and DLQI are used to measure the psoriasis component of PsA. Few RCTs measured DLQI and so this criterion could not be used in the decision model. PASI may be less well correlated with HRQoL than DLQI. 194 The decision model assumes that mean changes in HAQ are a function of PsARC response and the biologic therapy used. This approach has been used in other decision models of PsA (see Systematic review of existing cost-effectiveness evidence). 152, 177 Changes in HAQ may be more accurately predicted by other clinical and demographic variables, such as ACR and age. The Abbott model151 estimated a joint distribution of ACR and PsARC, and predicted HAQ from ACR responses. Although this is an attractive method, we considered the evidence synthesis required to undertake this modelling to be very complex and appeared to use data relating to biologics that are not currently licensed for PsA (see Systematic review of existing cost-effectiveness evidence).
There is some debate about whether the RCTs have recruited similar cohorts of patients and the effect on the results. Our review of the trial patients’ characteristics (see Table 1) indicated that the trials were similar enough to conduct a meta-analysis, and any resultant treatment differences were included in the model. However, other experts suggest that biologic therapies are very similar in their effectiveness and suggest that the RCTs show superior effects for infliximab only because these trials recruited a greater proportion of patients with polyarticular disease who may have better response rates than other types of PsA (Phillip Halliwell, London, UK, 16 February 2010, evidence to NICE committee).
The base-case model assumes that patients who fail therapy will be placed on palliative care. In practice many patients are tried with a second or third biologic. The use of biologics as the second line in a sequence is explored in a secondary, subgroup analysis. This analysis relies on non-randomised comparisons and therefore should be considered with caution.
Some of the patients included in RCTs did not use at least two DMARDs before trialling a biologic, as recommended by the BSR. In contrast, data on the NH of PsA without biologic therapy are from an observational study of rheumatoid factor-negative inflammatory polyarthritis patients with at least three tender joints and three swollen joints, who have failed at least two DMARDs.
Data on withdrawal rates after 3 months are from a meta-analysis of observational studies. In this model, withdrawal rates are assumed to be exogenous, i.e. independent of other variables in the model. In practice, withdrawal may depend on other factors, such as the biologic therapy used, obtaining a continuing response of both arthritis and psoriasis, and the options for switching to other biologics. Adverse events are not included in the model other than through their influence on withdrawal rates in the biologics register. In practice, there may be longer-term consequences and costs of adverse events, such as cancers and infections.
There is little good-quality data on the effect of arthritis and psoriasis on health-service costs in the UK. The base-case model uses UK data176 on the effect of HAQ on costs, but is rather dated, the methods used to analyse the data are not clearly reported and are likely to underestimate the impact of very severe HAQ on health and Personal Social Services costs. The base-case model uses data from a UK study of 272 patients with mild-to-moderate psoriasis to estimate the health service costs if biologics are not used or patients do not respond to biologics. 192 The model uses data from the Netherlands to estimate the health service costs of treating moderate-to-severe psoriasis if biologics are not used or patients do not respond to biologics. 188
It is assumed that there is no progression of HAQ for patients using biologics, based on elicitation of opinion from experts. There is considerable uncertainty about the ‘rebound’ of HAQ after withdrawal. The results of the expert elicitation seemed to indicate that experts believed that HAQ would rebound by less than the initial gain. This scenario increased the cost-effectiveness of all biologics, but did not materially change the conclusions of the model compared with the base case.
There is uncertainty about how the results of RCTs should be generalised to clinical practice. The base-case model assumed that the results in the placebo arm of the trials represented ‘non-pharmacological’ aspects of medical care that might not be reproduced outside the trial setting. The results of the trials were adjusted to take out this ‘placebo effect’. An alternative scenario that assumed these non-pharmacological aspects of medical care would be generalisable to general practice slightly increased the cost-effectiveness of all biologics, but did not materially alter the conclusions of the base-case analysis.
We compared the results from the current York model with those of other models and, in particular, the industry submissions to this appraisal. The current York model is essentially very similar in methods (and results) to the earlier York Assessment Group model reported by Bravo Vergel et al. 177 if there is no skin involvement, the time horizon is 40 years and the HAQ rebound after withdrawal from biologic is equal to initial gain. Adalimumab was not included in Bravo Vergel et al. model,177 but the current York model finds that adalimumab would not be cost-effective in this subgroup.
Abbott151 (manufacturers of adalimumab) found that adalimumab has an ICER of just below £30,000 per QALY compared with palliative care and other biologics are not cost-effective. The Abbott model151 calculated the ‘average’ cost-effectiveness of the biologics over all patients with PsA, 40% of whom were assumed not to have psoriasis, and assumed a mean PASI of 6.9 in the 60% of the population with psoriasis. The York model found that for patients with PsA and mild-to-moderate psoriasis adalimumab is extendedly dominated and is therefore unlikely to be the most cost-effective treatment. The reasons for the differences between the York and the Abbott model151 are difficult to pinpoint, not least because the sources of data for the Abbott151 bivariate evidence synthesis are unclear. Abbott151 estimate a higher response rate for PsARC and ACR with adalimumab than etanercept.
Schering-Plough152 (the manufacturers of infliximab) found that infliximab was cost-effective for patients of 60 kg weight if vial sharing is allowed, or if patients use three vials per administration. If vial sharing is not allowed or patients require four vials per administration then Schering-Plough152 concluded that etanercept was the most cost-effective strategy at a threshold of £20,000 per QALY in patients without psoriasis and with psoriasis. These conclusions are broadly consistent with those of the York model.
Wyeth153 (the manufacturer of etanercept) found that etanercept was the most effective and cost-effective biologic, and dominated or extendedly dominated infliximab and adalimumab. This is not consistent with the results of the York model, which found infliximab to be the most effective and most costly biologic. The main differences between the models are likely to be:
-
The estimates of PsARC response Wyeth153 found that etanercept had the highest probability of PsARC response, whereas the York evidence synthesis (and those of the other manufacturers) found infliximab to be the most effective for PsARC.
-
The assumption made by Wyeth153 that changes in HAQ are proportional to changes in PASI This is a strong assumption and Wyeth153 did not provide any clinical justification to support it.
Despite the differences in data and model structure outlined above (see Comparison of the York Economic Assessment with the manufacturers’ models), the results of the York model are broadly consistent with those of Schering-Plough,152 taking account of assumptions about vial sharing. The Abbott model151 appears to have overestimated the effectiveness of adalimumab in terms of PsARC and ACR responses. The Wyeth model153 appears to have overestimated the effectiveness of etanercept, in terms of PsARC response, and makes strong and arguably unjustified assumptions about the relationship between HAQ and PASI.
Chapter 5 Assessment of factors relevant to the NHS and other parties
The results of this technology assessment have some implications for clinical practice. At present, most patients with PsA who receive biologic therapy are managed by a rheumatologist. However, patients with PsA primarily concerned with improvements in their skin may benefit from being managed by a dermatologist who can tailor any ongoing topical therapy appropriately. Some patients with severe skin and joint disease may need dual management of both specialties, although it has implications in terms of additional administration, costs and communication between the specialties and primary care.
For patients with joint disease who respond to biologic treatment, potential cost savings might include reduced need for contact with services (e.g. physiotherapy) and monitoring costs for certain DMARDs. For patients responding in terms of skin disease, there may be the potential for avoiding inpatient admissions resulting from severe psoriasis.
There is a choice of measures available for assessing joint response (ACR or PsARC). BSR guidelines currently recommend PsARC, but also suggest this is supplemented with measures of HAQ, ESR and CRP. The choice of outcome measure will therefore have resource use as well as methodological implications.
The mode of delivery varies among the biologics included in this evaluation. Provision of infliximab requires the treatment centre to have the appropriate capacity in terms of staff and facilities to delivered scheduled i.v. infusions of the agent. In contrast, etanercept and adalimumab are delivered by self-administered injection. This may have short-term implications for initial training of patients, although with potential cost savings in the longer term.
As the rate of serious adverse events for these biologic agents has yet to be well established, all patients should be monitored by a specialist. In addition, relevant data for the BSRBR should be collected and appropriate measures for infection screening should be used.
The potential benefits of these agents on physical function and QoL might result in reduced demand on social services and carers, and the potential (although not yet fully demonstrated) for slowing disease progression could potentially reduce the demand for joint replacement surgery and associated services.
Chapter 6 Discussion
Statement of principal findings
The systematic review of clinical efficacy found a limited amount of high-quality data suggesting that etanercept, infliximab and adalimumab all produce significant improvements in joint response measures relative to placebo. Some evidence suggesting beneficial effects for these agents in terms of skin response, although data on this outcome are sparse. Although short-term data on joint progression are promising, longer-term controlled data on this outcome are lacking. The range of incidences of serious adverse events did not appear to differ remarkably between agents.
An indirect comparison of the three drugs indicated that infliximab is associated with the highest probability of response on joint and skin outcomes. The response in joint disease appeared greater with etanercept than with adalimumab, whereas the skin response appeared greater with adalimumab than with etanercept, though these differences are not statistically significant. In those patients who achieve a PsARC response to treatment the highest mean reductions in HAQ are seen with infliximab and etanercept.
Under base-case assumptions the York economic model found that for patients with mild-to-moderate skin disease, the ICER of etanercept versus palliative care is about £18,000 per QALY, the ICER of infliximab versus etanercept is about £44,000 per QALY and adalimumab is extendedly dominated. On average, given these base-case assumptions, etanercept would be considered the most cost-effective strategy if the threshold for cost-effectiveness were £20,000 or £30,000 per QALY. The probability etanercept is the most cost-effective treatment is 0.44 at a threshold of £20,000 per QALY and 0.48 at a threshold of £30,000 per QALY. The expected lifetime prescription costs of biologic therapies is considerably greater than the offset cost savings elsewhere in the NHS. These results were sensitive to several of the scenarios tested in univariate sensitivity analyses
Strengths and limitations of the assessment
Strengths
We conducted a rigorous systematic review that addressed clear research questions using predefined inclusion criteria. Comprehensive literature searches were performed to locate all relevant published and unpublished studies without any language restrictions, thereby minimising both publication and language biases. 164,166 Efforts were also made to identify additional studies by hand-searching company submissions, clinical trial reports and reference lists of relevant publications. Compared with the previous review,174 the current updated review has included a larger body of evidence (e.g. additional inclusion of two RCTs of adalimumab). In addition, data on serious adverse events of biologic treatment were also systematically reviewed. We are therefore confident that we have been able to include all the relevant studies in the evaluation of efficacy and adverse events of etanercept, infliximab and adalimumab.
Our review included RCTs to assess the efficacy of biologic agents in the treatment of PsA. That uncontrolled trials would be particularly unreliable for the purpose of evaluating treatments for PsA was demonstrated by the trials of treatment interventions for PsA in which the uniform improvement of symptoms was consistently observed in the placebo group. 54 It is important to note that all the included trials were rated as ‘good’ quality using the prespecified criteria, which ensured the internal validity of their research findings.
In the review process, sufficient attempts have been taken to reduce the potential for reviewer errors and biases. The study selection, data extraction and quality assessment were performed in duplicate. In particular, statistical heterogeneity was assessed and appropriate meta-analyses methods were adopted in the evaluation of efficacy. In terms of the evaluation of adverse events, the level of clinical heterogeneity between studies has been fully investigated. Owing to the high degree of clinical heterogeneity identified between included studies, a narrative synthesis was therefore appropriately adopted.
In the absence of head-to-head comparison evidence on the efficacy between the alternative biologic therapies, an indirect comparison was undertaken using Bayesian approaches to estimate the relative efficacy of these biologic agents in terms of both skin and joint symptom improvement. These estimates, together with other parameters were subsequently used to inform the independent economic model as an overall framework for the cost-effectiveness evaluation of biologic treatment.
This review has addressed many of the limitations of the previous economic assessment of biologic therapies for PsA. It is based on an updated evidence synthesis that includes infliximab, etanercept and adalimumab and includes responses of both psoriasis and arthritis. The model assesses the cost-effectiveness of biologic therapies for patients with different degrees of severity of psoriasis and arthritis at baseline. The model takes account of potential correlations between responses of arthritis and skin disease to biologic, and considers alternative rules about continuation on therapy beyond the initial 3 months. Withdrawal rates are estimated from a synthesis of data from several registers. The model takes account of the health-care costs associated with treating psoriasis if this is uncontrolled by biologics. The appraisal undertook an elicitation of expert clinical opinion to inform the estimate of the change in HAQ following withdrawal from biologic drugs. The economic analysis explores the potential for sequencing biologic drugs.
Limitations
The main limitation of this systematic review was that there were limited efficacy data available. Although all the included trials were judged as good quality, the analyses for each efficacy outcome were limited to only two RCTs. Some trials also recruited a small number of participants. Most trials had short follow-up period of either 12/14 or 24 weeks, which were often considered inadequate to assess radiographic changes in response to the treatment. There was a lack of controlled data on long-term outcomes, such as radiographic assessments. Given the fact that the treatment effect on the joint disease is more accurately reflected by the more objective radiographic measure, radiographic long-term data could provide more generalisable estimates of the biologic treatment effect. In addition, a lack of direct comparison evidence between biologic agents also made it difficult to draw firm conclusions on the relative effectiveness of these biological agents.
Another limitation of this systematic review resulted from the difficulties in assessing PsA activity and its response to the biologic therapy. Although a number of outcome measures were used in estimating the treatment effects, no outcome measure has been clearly identified as optimal for PsA. In this review we have attempted to use the best available outcome measures. In the clinical evaluation, we used a number of efficacy outcome measures as reported in the various clinical trials including PsARC, ACR 20/50 /70, HAQ and PASI. These measures are not ideal but are the best available, especially when data for joint and skin are both used. We also used the outcome of radiological assessment to address the long-term joint disease progression despite the data being sparse in included trials.
Despite the fact that we have incorporated both joint and skin aspects of treatment effects in the clinical effectiveness and cost-effectiveness evaluation, the data of biologic efficacy on the skin condition were very sparse.
Limitations of the adverse event evaluation in this review reflected on the non-randomised design of the majority of included studies and its reliance on uncontrolled data. Although we also included the data from RCTs, the adverse event data from these RCTs were often limited by a very short-term follow-up. The majority of data in the evaluation of adverse events for the treatment with etanercept, infliximab and adalimumab were derived from the observational studies and open-label extension of RCTs; however, the reliability of these data was questionable due to lack of a control group.
The new York cost-effectiveness model measures the severity of skin disease using the PASI score. PASI may not be well correlated with HRQoL. BAD recommends that both DLQI and PASI are used to assess the severity of arthritis. DLQI was not recorded by many trials, and so could not be measured in the evidence synthesis or model. The model measures the severity of joint disease using the HAQ score and assumes initial changes in HAQ are a function of PsARC response and treatment. Changes in HAQ may be more accurately predicted by a richer set of clinical and demographic variables such as ACR response and age. ACR responses from the RCTs were synthesised in Chapter 3 (see Results of review of clinical effectiveness), but incorporating PASI 75, PsARC and ACR responses in the model was considered to be very complex.
The cost-effectiveness model relied on observational data to estimate withdrawal rates and changes in HAQ for patients not using biologic therapy. However, it is unlikely that long-term randomised data would ever be available. The model uses observational data to estimate how the effectiveness of second-line therapy differs from first-line therapy. However, a randomised study comparing second-line use of biologics, depending on the reason for failing the first-line therapy, might be difficult to design. The model assumes patients withdraw to palliative care. If sequential use of biologics were included in the model this might change the estimate of the cost-effectiveness of first-line biologic therapy. The elicitation of expert opinion included only five experts and the results should be considered exploratory.
The model only includes adverse events to the extent that they influence the assessment of initial response and long-term withdrawal rates. Serious adverse events such as cancers and infections are rare, but may have long-term consequences. Biologics may have an effect on mortality, either for better (through reduced coronary events) or worse (through serious adverse events). Data on mortality attributable to the use of biologics in PsA is sparse and these effects therefore have been excluded.
There are few good quality data on the effect of arthritis and psoriasis on health-service costs in the UK. The model excludes productivity losses and private health-care expenditure in accordance with the NICE reference case, but these costs to society from PsA are likely to be substantial.
Uncertainties
-
The treatment effect of each biologic agent for the joint and skin conditions in this systematic review is based on only two RCTs with limited sample size. In particular, few patients provided data on the psoriasis response to biologics.
-
Bayesian indirect comparison analyses provide evidence of the relative effectiveness of these biological agents; however, those findings may be considered more uncertain than would be provided in head-to-head RCTs.
-
The patients recruited in most trials are not precisely representative of the populations recommended for biologic therapy in current guidelines. It is unclear whether the observed beneficial effects are similar in those populations.
-
The evidence of risk of serious adverse events (serious infection, malignancy and activation of latent TB) for treatment with these biologic agents remains uncertain because there are large uncertainties associated with these estimates, as well as the unreliable nature of the majority of the data.
-
The adverse event data for etanercept, infliximab and adalimumab are derived primarily from patients with RA or other indications. The generalisability of these findings to patients with PsA remains unclear.
-
The results of the York economic model are sensitive to several of the scenarios tested in univariate sensitivity analyses.
-
– The model assumes that biologics are effective in treating joint disease in two ways: (1) for patients successfully maintained on treatment, biologics reduce symptoms and prevent the progression of arthritis; and (2) biologics are assumed to permanently delay the progress of joint disease in patients, even if they withdraw from treatment, relative to a patient who had never used biologics. Results are sensitive to these assumptions about the progression of HAQ on and off treatment and the length of time over which biologics are assumed to be effective.
-
– The elicitation of expert opinion found that clinicians believed the change in HAQ following withdrawal from biologic drugs would be less than the initial gain on starting biologic therapy. This is an important parameter in the model and should be investigated further.
-
– The estimate of the prescription cost of the therapies relies on BSR guidelines and expert opinion about the number of vials required. This should be supported with empirical evidence on actual resource use. Results are sensitive to alternative data about the costs of treating psoriasis of different levels of severity. Results are sensitive to alternative assumptions about the relationship between utility and the severity of arthritis and psoriasis.
-
Chapter 7 Conclusions
Implications for service provision
-
The limited data available indicate that etanercept, infliximab or adalimumab are efficacious in the treatment of PsA compared with placebo, with beneficial effects on both joint and skin symptoms and on functional status. Short-term data suggest that these three biologic agents can delay joint disease progression.
-
Despite the limited data in the evaluation of clinical effectiveness of etanercept, infliximab and adalimumab, the evidence to support their efficacy in the treatment of PsA is convincing given the size of treatment effect and quality of data.
-
An indirect comparison of the three drugs indicated that infliximab is associated with the highest probability of response on PsARC, ACR and PASI outcomes. In those patients who achieve a PsARC response to treatment the highest mean reduction in HAQ are seen with infliximab and etanercept.
-
This review cannot rule out concerns about increased risk of rare serious adverse events (serious infection, malignancy and activation of latent TB) of the biologic agents investigated. Until further data are available, appropriate measures for screening and monitoring of patients should be used.
-
Under base-case assumptions, the York model indicated that etanercept would be considered the most cost-effective strategy if the threshold for cost-effectiveness were £20,000 per QALY or £30,000 per QALY. The expected lifetime prescription costs of biologic therapies are considerably greater than offset cost savings elsewhere in the NHS.
-
For patients with PsA and mild-to-moderate psoriasis who have failed adalimumab or infliximab as first-line therapy for either adverse events or inefficacy, etanercept is cost-effective at a threshold of £20,000 per QALY. For patients who have failed etanercept as first-line therapy for either adverse events or inefficacy, adalimumab is cost-effective at a threshold of £20,000 per QALY, although infliximab is more likely to be cost-effective if the threshold is £30,000 per QALY.
-
The present value prescription costs per person of biologic therapy over 40 years are estimated to be around £52,000 for infliximab, £33,000 for etanercept and £27,000 for adalimumab (at a discount rate of 3.5% per year). Most of these liabilities will accrue to NHS hospital trusts. Offset cost savings elsewhere in the NHS from less need for arthritis and psoriasis treatments are likely to be relatively modest. For patients with PsA with minimal psoriasis or mild-to-moderate psoriasis, who are thought to make up about 75% of the population, the present value of lifetime offset cost savings are expected to be no greater than about £5000.
Suggested research priorities
-
Long-term observational studies with large sample sizes of patients with PsA are required to demonstrate that beneficial effects for joint and skin disease and improvement of function are maintained. In particular data on the effects of joint disease progression (e.g. radiographic assessment), long-term HAQ progression while responding to biologic agents and HRQoL are required. Withdrawal rates due to lack of efficacy and adverse events should also be reported.
-
Further monitoring of the safety profiles of the biologic agents (e.g. through the BSBR) is required. Future research should also establish whether long-term patterns of adverse events of these biologic agents in PsA are similar to those in RA.
-
Further investigation is required to reduce uncertainties around the following parameters identified in the economic model:
-
– The length of time over which biologics are assumed to be effective.
-
– The change in HAQ following withdrawal from biologic drugs.
-
– Evidence from general practice about the prescribing, administration and monitoring costs of biologic therapy.
-
– The NHS costs of treating psoriasis of different levels of severity.
-
– The progression of HAQ on and off biologic treatment.
-
– The effectiveness and withdrawal rates of biologics used as second-line therapy.
-
-
Future studies should assess how the biologic treatment of both arthritis and psoriasis affects patients’ QoL using generic preference-based utility instruments.
-
The cost effectiveness of sequential use of biologic therapies should be evaluated further.
-
Although indirect analysis is useful, future trials comparing one biologic agent with another in the treatment of PsA are warranted.
The effectiveness and cost-effectiveness of biologics in patients who might not quite reach the current BSR/BAD criteria for either psoriasis or arthritis, but might nevertheless benefit from biologic therapy, should also be examined.
Acknowledgements
We would like to thank the following for their assistance in providing additional data or advice: Suzanne Verstappen and Deborah Symmons at the ARC Epidemiology Unit, University of Manchester, for analyses of the NH of PsA; Carolyn Davies for advice on the costs of biologics; Darren Ashcroft at the University of Manchester for an advance draft of an article submitted for publication and additional analyses; Neil Hawkins at Oxford Outcomes for advice on accounting for placebo effects; and the manufacturers who provided additional data where requested.
Contribution of authors
Mark Rodgers was responsible for study selection, data extraction, validity assessment, data analysis and writing the report. David Epstein and Laura Bojke were responsible for the review of cost-effectiveness evidence and the overall development of the economic model. Huiqin Yang contributed to study selection, data extraction, validity assessment, data analysis and writing the report. Dawn Craig and Tiago Fonseca were responsible for the evidence synthesis section. Lindsey Myers devised the search strategy, carried out the literature searches and wrote the search methodology sections of the report. Ian Bruce and Robert Chalmers provided clinical advice and commented on drafts of the report. Sylwia Bujkiewicz was responsible for developing Transparent Interactive Decision Interrogator (TIDI) interface and wrote the chapter on TIDI development. Monica Lai contributed to programming of the TIDI interface and liaised between York and Leicester teams. Alex Sutton, Nicola Cooper, Keith Abrams and David Spiegelhalter contributed original ideas and oversaw the development of TIDI, commented on drafts of the chapter on TIDI and provided statistical advice on the decision model and evidence synthesis. Mark Sculpher contributed to all aspects of the economic sections, and Nerys Woolacott contributed to all aspects of the clinical effectiveness sections. All authors contributed to and commented on the final draft of the report.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis 2005;64:14-7.
- Helliwell PS, Taylor WJ. Classification and diagnostic criteria for psoriatic arthritis. Ann Rheum Dis 2005;64:3-8.
- Zachariae H. Prevalence of joint disease in patients with psoriasis: implications for therapy. Am J Clin Dermatol 2003;4:441-7.
- Kay LJ, Parry-James JE, Walker DJ. The prevalence and impact of psoriasis and psoriatic arthritis in the primary care population in North East England. Arthritis Rheum 1999;42.
- Harrison BJ, Silman AJ, Barrett EM, Scott DG, Symmons DP. Presence of psoriasis does not influence the presentation or short-term outcome of patients with early inflammatory polyarthritis. J Rheumatol 1997;24:1744-9.
- Feuchtenberger M, Kleinert S, Tony HP, Kneitz C. Psoriatic arthritis: therapeutic principles. Clin Dermatol 2008;26:460-3.
- Ruderman E. Evaluation and management of psoriatic arthritis: the role of biologic therapy. J Am Acad Dermatol 2003;49:125-32.
- Patel S, Veale D, FitzGerald V, McHugh N. Psoriatic arthritis: emerging concepts. Rheumatology 2001;40:243-6.
- Krueger G. Clinical features of psoriatic arthritis. Am J Manag Care 2002;8:160-70.
- Galadari H, Fuchs B, Lebwohl M. Newly available treatments for psoriatic arthritis and their impact on skin psoriasis. Int J Dermatol 2003;42:231-7.
- Gottlieb A, Korman NJ, Gordon KB, Feldman SR, Lebwohl M, Koo JYM, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 2. Psoriatic arthritis: overview and guidelines of care for treatment with an emphasis on the biologics. J Am Acad Dermatol 2008;58:851-64.
- Gladman D. Effectiveness of psoriatic arthritis therapies. Semin Arthritis Rheum 2003;33:29-37.
- Pipitone N, Kingsley G, Manzo A, Scott D, Pitzalis C. Current concepts and new developments in the treatment of psoriatic arthritis. Rheumatology 2003;42:1138-48.
- Kane D, Stafford L, Bresnihan B, FitzGerald O. A prospective, clinical and radiological study of early psoriatic arthritis: an early synovitis clinic experience. Rheumatology 2003;42:1460-8.
- Husted J, Gladman D, Long J, Farewell V. A modified version of the Health Assessment Questionnaire (HAQ) for psoriatic arthritis. Clin Exp Rheumatol 1995;13:439-43.
- Gladman D, Hing E, Schentag C, Cook R. Remission in psoriatic arthritis. J Rheumatol 2001;28:1045-8.
- Gottlieb A. Psoriatic arthritis: a guide for dermatology nurses. Dermatol Nurs 2003;15:107-19.
- McHugh N, Balachrishnan C, Jones S. Progression of peripheral joint disease in psoriatic arthritis: a 5-year prospective study. Rheumatology 2003;42:778-83.
- Gladman D, Stafford-Brady F, Chang C, Lewandowski K, Russell M. Longitudinal study of clinical and radiological progression in psoriatic arthritis. J Rheumatol 1990;17:809-12.
- Queiro-Silva R, Torre-Alonso J, Tinture-Eguren T, Lopez-Lagunas I. A polyarticular onset predicts erosive and deforming disease in psoriatic arthritis. Ann Rheum Dis 2003;62:68-70.
- Gladman D, Farewell V. The role of HLA antigens as indicators of disease progression in psoriatic arthritis Multivariate relative risk model. Arthritis Rheum 1995;38:845-50.
- Lindqvist URC, Alenius G-M, Husmark T, Theander E, Holmstrom G, Larsson PT, et al. The Swedish early psoriatic arthritis register: 2-year follow-up: a comparison with early rheumatoid arthritis. J Rheumatol 2008;35:668-73.
- Moll J, Wright V. Psoriatic arthritis. Semin Arthritis Rheum 1973;3:55-78.
- Mease P, Goffe B. Diagnosis and treatment of psoriatic arthritis. J Am Acad Dermatol 2005;52:1-19.
- Borman P, Toy GG, Babaoğlu S, Bodur H, Ciliz DAN. A comparative evaluation of quality of life and life satisfaction in patients with psoriatic and rheumatoid arthritis. Clin Rheumatol 2007;26:330-4.
- Husted J, Gladman D, Farewell V, Cook R. Health-related quality of life of patients with psoriatic arthritis: a comparison with patients with rheumatoid arthritis. Arthrit Care Res 2001;45:151-8.
- Curran S, Winchester R, Costello P, Peterson K, Bresnihan B, FitzGerald O. Methotrexate therapy reduces polyclonal T cell infiltration in the psoriaitc arthritis synovium, revealing expanded CD4 and CD8 T-cell clones. Arthritis Rheum 1999;42.
- Haake H, Koneke J, Amann K, vom Dahl J, Janssen U. [Development of systemic lupus erythematosus with focal proliferative lupus nephritis during anti-TNF-alpha therapy for psoriatic arthritis]. Med Klin 2007;102:852-7.
- Wong K, Gladman DD, Husted J, Long J, Farewell VT. Mortality studies in psoriatic arthritis: results from a single outpatient clinic. Causes and risk of death. Arthritis Rheum 1997;40:1868-72.
- Gladman D, Farewell V, Wong K, Husted J. Mortality studies in psoriatic arthritis: results from a single outpatient center. II Prognostic indicators for death. Arthritis Rheum 1998;41:1103-10.
- Ali Y, Tom BD, Schentag CT, Farewell VT, Gladman DD. Improved survival in psoriatic arthritis with calendar time. Arthritis Rheum 2007;56:2708-14.
- Williams JP, Meyers JA. Immune-mediated inflammatory disorders (I.M.I.D.s): the economic and clinical costs. Am J Manag Care 2002;8:S664-81.
- Huscher D, Merkesdal S, Thiele K, Ziedler H, Schneider M, Zink A. Cost of illness in rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis and systemic lupus erythematous in Germany. Ann Rheum Dis 2006;65:1175-83.
- Jonsson B, Rehnberg C, Borgquist L, Larsson S. Locomotion status and costs in destructive rheumatoid arthritis. A comprehensive study of 82 patients from a population of 13,000. Acta Orthop Scand 1992;63:207-12.
- Kvien T. Epidemiology and burden of illness of rheumatoid arthritis. Pharmacoeconomics 2004;22:1-12.
- Pugner K, Scott D, Holmes J, Hieke K. The costs of rheumatoid arthritis: an international long-term view. Semin Arthritis Rheum 2000;129:305-20.
- Feldman S, Fleischer A, Reboussin D, Rapp S, Bradham D, Clark A. The economic impact of psoriasis increases with psoriasis severity. J Am Acad Dermatol 1997;37:564-9.
- Jonsson B, Kaarela K, Koblet G. Economic consequences of the progression of rheumatoid arthritis: a Markov model. Stockholm: Stockholm School of Economics; 1997.
- Kobelt G, Eberhardt K, Jonsson L, Jonsson B. Economic consequences of the progression of rheumatoid arthritis in Sweden. Arthritis Rheum 1999;42:347-56.
- McIntosh E. Clinical audit: the cost of rheumatoid arthritis. Br J Rheumatol 1996;35:781-90.
- Kobelt G, Jonsson L, Lindgren P, Young A, K. E. Modelling the progression of rheumatoid arthritis:a two-country model to estimate costs to estimate costs and consequences of rheumatoid arthritis. Arthritis Rheum 2002;46:2310-19.
- Michaud K, Messer J, Choi H, Wolfe F. Direct medical costs and their predictors in patients with rheumatoid arthritis. Arthritis Rheum 2003;48:2750-62.
- Armstrong D, McCausland E, Wright G. The impact of anti-TNF-alpha therapy on the nature of service provision. Rheumatology 2006;45.
- Goldsmith C, Smythe H, Helewa A. Interpretation and power of a pooled index. J Rheumatol 1993;20:575-8.
- Strangfeld A, Listing J, Herzer P, Liebhaber A, Rockwitz K, Richter C, et al. Risk of herpes zoster in patients with rheumatoid arthritis treated with anti-TNF-alpha agents. JAMA 2009;301:737-44.
- Felson D, Anderson J, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727-35.
- Felson D, Anderson J, Lange M, Wells G, LaValley M. Should improvement in rheumatoid arthritis clinical trials be defined as fifty percent or seventy percent improvement in core set measures, rather than twenty percent?. Arthritis Rheum 1998;41:1564-70.
- Gladman DD, Helliwell P, Mease PJ, Nash P, Ritchlin C, Taylor W. Assessment of patients with psoriatic arthritis: a review of currently available measures. Arthritis Rheum 2004;50:24-35.
- Clegg D, Reda D, Mejias E, Cannon G, Weisman M, Taylor T, et al. Comparison of sulfasalazine and placebo in the treatment of psoriatic arthritis. A Department of Veterans Affairs Cooperative Study. Arthritis Rheum 1996;39:2013-20.
- McHugh N, Chandler D, Griffiths C, Helliwell P, Lewis J, McInnes I, et al. BSR guideline for anti-TNFa therapy in psoriatic arthritis. London: British Society for Rheumatology; 2004.
- Mease PJ, Gladman DD, Ritchlin CT, Ruderman EM, Steinfeld SD, Choy EH, et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum 2005;52:3279-89.
- Mease PJ, Kivitz AJ, Burch FX, Siegel EL, Cohen SB, Ory P, et al. Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Arthritis Rheum 2004;50:2264-72.
- Wassenberg S, Fischer-Kahle V, Herborn G, Rau R. A method to score radiographic change in psoriatic arthritis. Z Rheumatol 2001;60:156-66.
- Jones G, Crotty M, Brooks P. Interventions for treating psoriatic arthritis. Cochrane Database Syst Rev 2000;3. 10.1002/14651858.CD000212.
- Taccari E, Spadaro A, Rinaldi T, Riccieri V, Sensi F. Comparison of the Health Assessment Questionnaire and Arthritis Impact Measurement Scale in patients with psoriatic arthritis. Rev Rhum 1998;65:751-8.
- Blackmore M, Gladman D, Husted J, Long J, Farewell V. Measuring health status in psoriatic arthritis: the Health Assessment Questionnaire and its modification. J Rheumatol 1995;22:886-93.
- Wong J, Singh G, Kavanaugh A. Estimating the cost-effectiveness of 54 weeks of infliximab for rheumatoid arthritis. Am J Med 2002;113:400-8.
- Haroon M, Bond U, Phelan M. Sinusitis: a possible link with adalimumab. Clin Rheumatol 2008;27:1189-90.
- Kobelt G, Jonsson L, Young A, Eberhardt K. The cost-effectiveness of infliximab (Remicade) in the treatment of rheumatoid arthritis in Sweden and the United Kingdom based on the ATTRACT study. Rheumatology 2003;42:326-35.
- Marguerie L, Flipo R, Grardel B, Beaurain D, Duquesnoy B, Delcambre B. Use of disease-modifying antirheumatic drugs in patients with psoriatic arthritis. Joint Bone Spine 2002;69:275-81.
- Alldred A, Emery P. Leflunomide: A novel DMARD for the treatment of rheumatoid arthritis. Expert Opin Pharmacother 2001;2:125-37.
- Kaltwasser J, Nash P, Gladman D. Efficacy and safety of leflunomide in the treatment of psoriatic arthritis and psorisasis. Arthritis Rheum 2004;50:1939-50.
- Strand V, Cohen S, Schiff M, Weaver A, Fleischmann R, Cannon G, et al. Treatment of active rheumatoid arthritis with leflunomide compared with placebo and methotrexate. Arch Intern Med 1999;159:2542-50.
- Nash P, Clegg D. Psoriatic arthritis and psoriasis: treatment. Ann Rheum Dis 2005;64:74-7.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary. 2009.
- NHS Business Services Authority . Prescribing Analysis Charts 2009. www.nhsbsa.nhs.uk/PrescriptionServices/2585.aspx (accessed 13 November 2009).
- Gorter S, van der Heijde DMFM, van der Linden S, Houben H, Rethans J-J, Scherpbier AJJA, et al. Psoriatic arthritis: performance of rheumatologists in daily practice. Ann Rheum Dis 2002;61:219-24.
- Pariser D. Management of moderate to severe plaque psoriasis with biologic therapy. Manag Care 2003;12:36-44.
- Gniadecki R, Zachariae C, Calverley M. Trends and developments in the pharmacological treatment of psoriasis. Acta Derm Venereol 2002;82:401-10.
- Prinz J. The role of T cells in psoriasis. J Eur Acad Derm Venereol 2003;17:257-70.
- Cvetkovic RS, Scott LJ. Adalimumab: a review of its use in adult patients with rheumatoid arthritis. Biodrugs 2006;20:293-311.
- The NHS. Information Centre. Hospital Prescribing, 2008: England n.d. www.ic.nhs.uk/ (accessed 13 November 2009).
- Woolacott N, Bravo Vergel Y, Hawkins N, Kainth A, Khadjesari Z, Misso K, et al. Etanercept and infliximab for the treatment of psoriatic arthritis: a systematic review and economic evaluation. Health Technol Assess 2006;10.
- NICE Evidence Review Group . Adalimumab for the Treatment of Moderate to Severe Psoriatic Arthritis 2009.
- Centre for Reviews and Dissemination (CRD) . Manual for Selecting Reviews and Writing Abstracts for the Database of Abstracts of Reviews of Effects (DARE) 2008. www.crd.york.ac.uk/crdweb/html/help.htm.
- Moher D, Cook D, Eastwood S, Olkin I, Rennie D, Stroup D. Improving the quality of reports of meta-analyses of randomised controlled trials: the QUOROM statement. Lancet 1999;354:1896-900.
- Higgins J, Whitehead J. Borrowing strength from external trials in meta-analysis. Stat Med 1996;15:2733-49.
- Mease PJ, Goffe BS, Metz J, VanderStoep A, Finck B, Burge DJ. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet 2000;356:385-90.
- Centocor . A Multicenter Placebo-Controlled, Double-Blind, Randomised Study of Anti-TNF Chimeric Monoclonal Antibody (cA2, Infliximab) in Patients With Active Psoriatic Arthritis (IMPACT): Protocol No P02114 [industry Submission] 2003.
- Schering-Plough Ltd . Remicade in the Treatment of Psoriatic Arthritis in the United Kingdom: A Submission to the National Institute for Clinical Excellence [industry Submission] 2004.
- Antoni CE, Kavanaugh A, Kirkham B, Tutuncu Z, Burmester GR, Schneider U, et al. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthritis Rheum 2005;52:1227-36.
- Antoni C, Krueger GG, de Vlam K, Birbara C, Beutler A, Guzzo C, et al. Infliximab improves signs and symptoms of psoriatic arthritis: results of the IMPACT 2 trial. Ann Rheum Dis 2005;64:1150-7.
- Genovese MC, Mease PJ, Thomson GT, Kivitz AJ, Perdok RJ, Weinberg MA, et al. Safety and efficacy of adalimumab in treatment of patients with psoriatic arthritis who had failed disease modifying antirheumatic drug therapy. J Rheumatol 2007;34:1040-50.
- Genovese MC, Mease PJ, Thomson GTD, Kivitz AJ, Perdok RJ, Weinberg MA. Adalimumab efficacy in patients with psoriatic arthritis who failed prior DMARD therapy [abstract FRI0187]. Ann Rheum Dis 2005;64.
- Mease PJ, Gladman DD, Ritchlin CT, Ruderman EM, Choy EHS, Steinfeld SD, et al. Clinical efficacy and safety of adalimumab for psoriatic arthritis: 48-week results of ADEPT [abstract 500]. Arthritis Rheum 2005;52.
- Mease PJ, Sharp JT, Ory PA, Gladman DD, Richlin CT, Choy EH. Adalimumab treatment effects on radiographic progression of joint disease in patients with psoriatic arthritis: results from ADEPT (abstract FRI0212). Ann Rheum Dis 2005;64.
- Van den Bosch F, Reece R, Manger B, Goupille P, Roedevand E, Holck P, et al. Adalimumab (HUMIRA®) Is Effective and Safe in Treating Psoriatic Arthritis (PsA) in Real-Life Clinical Practice: Preliminary Results of the STEREO Trial n.d.
- Mease PJ, Ory P, Sharp JT, Ritchlin CT, Van den Bosch F, Wellborne F, et al. Adalimumab for long-term treatment of psoriatic arthritis: 2-year data from the Adalimumab Effectiveness in Psoriatic Arthritis Trial (ADEPT). Ann Rheum Dis 2009;68:702-9.
- Antoni CE, Kavanaugh A, van der Heijde D, Beutler A, Keenan G, Zhou B, et al. Two-year efficacy and safety of infliximab treatment in patients with active psoriatic arthritis: findings of the Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT). J Rheumatol 2008;35:869-76.
- van der Heijde D, Kavanaugh A, Gladman DD, Antoni C, Krueger GG, Guzzo C, et al. Infliximab inhibits progression of radiographic damage in patients with active psoriatic arthritis through one year of treatment: results from the induction and maintenance psoriatic arthritis clinical trial 2. Arthritis Rheum 2007;56:2698-707.
- Kavanaugh A, Krueger GG, Beutler A, Guzzo C, Zhou B, Dooley LT, et al. Infliximab maintains a high degree of clinical response in patients with active psoriatic arthritis through 1 year of treatment: results from the IMPACT 2 trial. Ann Rheum Dis 2007;66:498-505.
- Gladman DD, Mease PJ, Ritchlin CT, Choy EHS, Sharp JT, Ory PA, et al. Adalimumab for long-term treatment of psoriatic arthritis: forty-eight week data from the adalimumab effectiveness in psoriatic arthritis trial. Arthritis Rheum 2007;56:476-88.
- Gladman DD, Mease PJ, Cifaldi MA, Perdok RJ, Sasso E, Medich J. Adalimumab improves joint-related and skin-related functional impairment in patients with psoriatic arthritis: patient-reported outcomes of the Adalimumab Effectiveness in Psoriatic Arthritis Trial. Ann Rheum Dis 2007;66:163-8.
- Vander Cruyssen B, De Keyser F, Kruithof E, Mielants H, Van den Bosch F. Comparison of different outcome measures for psoriatic arthritis in patients treated with infliximab or placebo. Ann Rheum Dis 2007;66:138-40.
- Kavanaugh A, Antoni C, Mease P, Gladman D, Yan S, Bala M, et al. Effect of infliximab therapy on employment, time lost from work, and productivity in patients with psoriatic arthritis. J Rheumatol 2006;33:2254-9.
- Kavanaugh A, Antoni CE, Gladman D, Wassenberg S, Zhou B, Beutler A, et al. The Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT): results of radiographic analyses after 1 year. Ann Rheum Dis 2006;65:1038-43.
- Mease PJ, Kivitz AJ, Burch FX, Siegel EL, Cohen SB, Ory P, et al. Continued inhibition of radiographic progression in patients with psoriatic arthritis following 2 years of treatment with etanercept. J Rheumatol 2006;33:712-21.
- Kavanaugh A, Antoni C, Krueger GG, Yan S, Bala M, Dooley LT, et al. Infliximab improves health related quality of life and physical function in patients with psoriatic arthritis. Ann Rheum Dis 2006;65:471-7.
- Fleischmann R, Baumgartner SW, Weisman MH, Liu T, White B, Peloso P. Long term safety of etanercept in elderly subjects with rheumatic diseases. Ann Rheum Dis 2006;65:379-84.
- Choy E, Gladman D, Sasso E. Efficacy of adalimumab by disease duration in psoriatic arthritis: subanalysis of ADEPT (abstract P2748). J Am Acad Dermatol 2007;56.
- Gladman D, Mease P, Ritchlin C, Okun M. PASI-100 is associated with better dermatology-specific patient-reported outcomes compared with PASI-75e99 in adalimumab-treated patients with psoriatic arthritis: subanalysis of ADEPT (abstract P2746). J Am Acad Dermatol 2007;56.
- Gladman D, Mease P, Kavanaugh A, Weinberg M. Adalimumab is efficacious in treating skin disease in psoriatic arthritis: subanalysis of moderate versus severe psoriasis in the ADEPT trial (abstract P40). J Am Acad Dermatol 2006;54:10-1.
- Mease P, Gladman D, Ritchlin C, Weinberg M. Clinical efficacy, safety and inhibition of joint destruction of adalimumab in the treatment of moderate to severe psoriatic arthritis: 48-week results of the ADEPT trial (abstract P2865). J Am Acad Dermatol 2006;54.
- Mease P, Sharp J, Ory P, Gladman D, Ritchlin C, Choy E, et al. Adalimumab treatment effects on radiographic progression of joint disease in patients with psoriatic arthritis: results from ADEPT (abstract P06.12). J Eur Acad Dermatol Venereol 2005;19.
- Lebwohl M, Gottlieb A, Goffe BS, Jahreis A. Etanercept in psoriatic arthritis: sustained improvement in skin and joint disease and inhibition of radiographic progression at 2 years [abstract 155–09]. J Am Acad Dermatol 2005;52.
- Papp K, Menter A, Antoni C, Kavanaugh A, Gottlieb AB, Poulin Y, et al. Rapid and persistent improvement of psoriasis in patients with active psoriatic arthritis treated with infliximab: 24-week results of the impact 2 trial (abstract P028). J Eur Acad Dermatol Venereol 2004;18.
- Wanke LA, Mease PJ, Gottlieb AB. Sustained improvement in functional status and vitality of psoriatic arthritis patients treated with etanercept (abstract 382). J Invest Dermatol 2004;122.
- Sterry W, Ortonne JP, Kirkham B, Robertson D, Molta C, Pedersen R, et al. Results of a randomized, double-blind study to evaluate the efficacy and safety of etanercept in patients with psoriasis and psoriatic arthritis: the PRESTA trial (abstract P29). Br J Dermatol 2008;159:1410-11.
- Kavanaugh A, Krueger GG, Birbara C, Halter D, Geusens P, de Vlam K, et al. Treatment with infliximab is associated with ‘major clinical response’ in psoriatic arthritis patients treated with infliximab: analysis of two double-blind placebo controlled trials (abstract OP0104). Ann Rheum Dis 2006;65.
- Mease PJ, Woolley M, Singh A, Tsuji W, Dunn M, Chiou CF. Patient-reported outcomes in a randomized comparison of etanercept and placebo for treatment of psoriatic arthritis (abstract SAT0279). Ann Rheum Dis 2006;65.
- Kavanaugh A, Krueger GG, Birbara C, Halter D, Geusens P, de Vlam K, et al. Treatment with infliximab is associated with ‘major clinical response’ in psoriatic arthritis patients treated with infliximab: analysis of two double-blind placebo controlled trials (abstract 121). J Invest Dermatol 2007;127.
- Kavanaugh A, Krueger GG, Birbara C, Halter D, Geusens P, de Vlam K, et al. Treatment with infliximab is associated with ‘major clinical response’ in psoriatic arthritis patients treated with infliximab: analysis of two double-blind placebo controlled trials (abstract L18). Arthritis Rheum 2005;52.
- Antoni CE, Kavanaugh A, Kirkham B, Tutuncu ZN, Burmester G, Schneider U, et al. Two-year data: infliximab maintains clinical response in psoriatic arthritis patients: data from the Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT) (abstract 485). Arthritis Rheum 2005;52.
- Antoni CE, Kavanaugh A, Kirkham B, Tutuncu Z, Burmester GR, Schneider U, et al. Two-year data: Infliximab maintains clinical response in psoriatic arthritis patients – an analysis of the year 2 data from the Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT) (abstract OP0080). Ann Rheum Dis 2005;64.
- Antoni CE, Kavanaugh A, Gladman D, Wassenberg S, Zhou B, Beutler A, et al. The infliximab multinational psoriatic arthritis controlled trial (IMPACT): results of radiographic analyses after 1 year [abstract OP0156]. Ann Rheum Dis 2005;64.
- Kavanaugh A, Krueger GG, de Vlam K, Birbara C, Beutler A, Guzzo C, et al. Infliximab significantly improves joint and skin involvement in psoriatic arthritis to a substantial extent and irrespective of baseline joint involvement or MTX use: analysis of clinical response from the IMPACT2 trial (abstract 1637). Arthritis Rheum 2004;50.
- Antoni CE, Kavanaugh A, Kirkham B, Burmester G, Manger B, Tutuncu Z, et al. The infliximab multinational psoriatic arthritis controlled trial (IMPACT): substantial efficacy on synovitis and psoriatic lesions with or without concomitant DMARD therapy (abstract OP0082). Ann Rheum Dis 2003;62.
- Antoni CE, Kavanaugh A, Kirkham B, Burmester G, Manger B, Schneider U, et al. The infliximab multinational psoriatic arthritis controlled trial (IMPACT1): the concomitant use of DMARDs does not influence the efficacy and safety of infliximab over a one year period (abstract SAT0083). Ann Rheum Dis 2004;63:411-12.
- Fidder H, Schnitzler F, Ferrante M, Noman M, Katsanos K, Segaert S, et al. Long-term safety of infliximab for the treatment of inflammatory bowel disease: a single-centre cohort study. Gut 2009;58:501-8.
- Klareskog L, Gaubitz M, Rodriguez-Valverde V, Malaise M, Dougados M, Wajdula J. A long-term, open-label trial of the safety and efficacy of etanercept (Enbrel) in patients with rheumatoid arthritis not treated with other disease-modifying antirheumatic drugs. Ann Rheum Dis 2006;65:1578-84.
- Moreland LW, Weinblatt ME, Keystone EC, Kremer JM, Martin RW, Schiff MH, et al. Etanercept treatment in adults with established rheumatoid arthritis: 7 years of clinical experience. J Rheumatol 2006;33:854-61.
- Listing J, Strangfeld A, Kary S, Rau R, von Hinueber U, Stoyanova-Scholz M, et al. Infections in patients with rheumatoid arthritis treated with biologic agents. Arthritis Rheum 2005;52:3403-12.
- Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J Dermatolog Treat 2004;15:280-94.
- Colombel J-F, Loftus EV, Tremaine WJ, Egan LJ, Harmsen WS, Schleck CD, et al. The safety profile of infliximab in patients with Crohn’s disease: the Mayo clinic experience in 500 patients. Gastroenterology 2004;126:19-31.
- Horneff G, De Bock F, Foeldvari I, Girschick HJ, Michels H, Moebius D, et al. Safety and efficacy of combination of etanercept and methotrexate compared to treatment with etanercept only in patients with juvenile idiopathic arthritis (JIA): preliminary data from the German JIA Registry. Ann Rheum Dis 2009;68:519-25.
- Rudwaleit M, Olivieri I, Boki KA, Griep EN, Jarvinen P, Wong RL, et al. Adalimumab is effective and well tolerated in treating patients with ankylosing spondylitis who have advanced spinal fusion. Rheumatology 2009;48:551-7.
- Schnitzler F, Fidder H, Ferrante M, Noman M, Arijs I, Van Assche G, et al. Long-term outcome of treatment with infliximab in 614 patients with Crohn’s disease: results from a single-centre cohort. Gut 2009;58:492-500.
- Caspersen S, Elkjaer M, Riis L, Pedersen N, Mortensen C, Jess T, et al. Infliximab for inflammatory bowel disease in Denmark 1999–2005: clinical outcome and follow-up evaluation of malignancy and mortality. Clin Gastroenterol Hepatol 2008;6:1212-17.
- Favalli EG, Desiati F, Atzeni F, Sarzi-Puttini P, Caporali R, Pallavicini FB, et al. Serious infections during anti-TNF alpha treatment in rheumatoid arthritis patients. Autoimmun Rev 2009;8:266-73.
- Takeuchi T, Tatsuki Y, Nogami Y, Ishiguro N, Tanaka Y, Yamanaka H, et al. Postmarketing surveillance of the safety profile of infliximab in 5000 Japanese patients with rheumatoid arthritis. Ann Rheum Dis 2008;67:189-94.
- Burmester GR, Mariette X, Montecucco C, Monteagudo-Saez I, Malaise M, Tzioufas AG, et al. Adalimumab alone and in combination with disease-modifying antirheumatic drugs for the treatment of rheumatoid arthritis in clinical practice: the Research in Active Rheumatoid Arthritis (ReAct) trial. Ann Rheum Dis 2007;66:732-9.
- Gomez-Reino JJ, Carmona L, Descalzo MA. Risk of tuberculosis in patients treated with tumor necrosis factor antagonists due to incomplete prevention of reactivation of latent infection. Arthritis Rheum 2007;57:756-61.
- Colombel J, Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Panaccione R, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s disease: the CHARM trial. Gastroenterology 2007;132:52-65.
- Curtis JR, Patkar N, Xie A, Martin C, Allison JJ, Saag M, et al. Risk of serious bacterial infections among rheumatoid arthritis patients exposed to tumor necrosis factor alpha antagonists. Arthritis Rheum 2007;56:1125-33.
- Brassard P, Kezouh A, Suissa S. Antirheumatic drugs and the risk of tuberculosis. Clin Infect Dis 2006;43:717-22.
- Dixon WG, Watson K, Lunt M, Hyrich KL, Silman AJ, Symmons DPM. Rates of serious infection, including site-specific and bacterial intracellular infection, in rheumatoid arthritis patients receiving anti-tumor necrosis factor therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum 2006;54:2368-76.
- Oka H, Nishioka K, Togo M, Ochi T. The efficacy of infliximab for patients with rheumatoid arthritis in Japan: results of 5000 cases by post-marketing surveillance data. APLAR J Rheum 2006;9:142-5.
- Schiff MH, Burmester GR, Kent JD, Pangan AL, Kupper H, Fitzpatrick SB, et al. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis 2006;65:889-94.
- Westhovens R, Yocum D, Han J, Berman A, Strusberg I, Geusens P, et al. The safety of infliximab, combined with background treatments, among patients with rheumatoid arthritis and various comorbidities: a large, randomized, placebo-controlled trial. Arthritis Rheum 2006;54:1075-86.
- Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, Van Vollenhoven R, et al. The PREMIER study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 2006;54:26-37.
- Carmona L, Gomez-Reino JJ, Rodriguez-Valverde V, Montero D, Pascual-Gomez E, Mola EM, et al. Effectiveness of recommendations to prevent reactivation of latent tuberculosis infection in patients treated with tumor necrosis factor antagonists. Arthritis Rheum 2005;52:1766-72.
- Feltelius N, Fored CM, Blomqvist P, Bertilsson L, Geborek P, Jacobsson LT, et al. Results from a nationwide postmarketing cohort study of patients in Sweden treated with etanercept. Ann Rheum Dis 2005;64:246-52.
- St. Clair EW, Van Der Heijde DMFM, Smolen JS, Maini RN, Bathon JM, Emery P, et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis Rheum 2004;50:3432-43.
- Wolfe F, Michaud K, Anderson J, Urbansky K. Tuberculosis infection in patients with rheumatoid arthritis and the effect of infliximab therapy. Arthritis Rheum 2004;50:372-9.
- Nahar IK, Shojania K, Marra CA, Alamgir AH, Anis AH. Infliximab treatment of rheumatoid arthritis and Crohn’s disease. Ann Pharmacother 2003;37.
- Gomez-Reino JJ, Carmona L, Rodriguez Valverde V, Mola EM, Montero MD. Treatment of rheumatoid arthritis with tumor necrosis factor inhibitors may predispose to significant increase in tuberculosis risk: a multicenter active-surveillance report. Arthritis Rheum 2003;48:2122-7.
- Dixon WG, Symmons DPM, Lunt M, Watson KD, Hyrich KL. British Society for Rheumatology Biologics Register Control Centre Consortium, et al. Serious infection following anti-tumor necrosis factor alpha therapy in patients with rheumatoid arthritis: lessons from interpreting data from observational studies. Arthritis Rheum 2007;56:2896-904.
- Dreyer L, Mellemkjaer L, Hetland ML. Cancer in arthritis patients after anti-tumour necrosis factor therapy. Ugeskr Laeger 2009;171:506-11.
- Kyle S, Chandler D, Griffiths CEM, Helliwell P, Lewis J, McInnes I, et al. Guideline for anti-TNF-alpha therapy in psoriatic arthritis. Rheumatology 2005;44:390-7.
- Mease P, Ganguly R, Wanke L, Yu E, Singh A. How much improvement in functional status is considered important by patients with active psoriatic arthritis: applying the outcome measures in rheumatoid arthritis clinical trials (OMERACT) group guidelines. Ann Rheum Dis 2004;63.
- Abbott Laboratories Ltd . Adalimumab (HUMIRA): Multiple Technology Appraisal of Adalimumab, Etanercept and Infliximab for Psoriatic Arthritis National Institute for Health and Clinical Excellence (NICE) Health Technology Appraisal 2009.
- Schering-Plough . REMICADE (infliximab): Remicade in the Treatment of Psoriatic Arthritis (PsA) in the United Kingdom. A Submission to the National Institute of Clinical Excellence 2009.
- Wyeth Pharmaceuticals . Etanercept (ENBREL): Appraisal of the Clinical and Cost-Effectiveness of Etanercept, Infliximab and Adalimumab for the Treatment of Psoriatic Arthritis. An Appraisal Submission for the National Institute of Health and Clinical Excellence 2009.
- Kavanaugh A, McInnes I, Mease P, Krueger DG, Gladman D, Gomez-Reino J. Golimumab, a new human tumour necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: Twenty-four-week efficacy and safety results of a randomized, placebo-controlled study. Arthritis Rheum 2009;60:976-86.
- Gottlieb A, Menter A, Mendelsohn A, Shen YK, Li S, Guzzo C, et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet 2009;373:633-40.
- REVEAL (M03–656) . Clincial Study Report n.d.
- Sterry WEA. Results of a Randomised, Double-Blind Study to Evaluate the Efficacy and Safety of Etanercept in Patients With Psoriasis psoriatic Arthritis: The PRESTA Trial. Gene to Clinic Conference 2009.
- Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006;295:2275-85.
- Bongartz T, Warren FC, Mines D, Matteson EL, Abrams KR, Sutton AJ. Etanercept therapy in rheumatoid arthritis and the risk of malignancies: a systematic review and individual patient data meta-analysis of randomised controlled trials. Ann Rheum Dis 2009;68:1177-83.
- Gartlehner G, Hansen RA, Jonas BL, Thieda P, Lohr KN. The comparative efficacy and safety of biologics for the treatment of rheumatoid arthritis: a systematic review and metaanalysis. J Rheumatol 2006;33:2398-408.
- Ravindran V, Scott DL, Choy EH. A systematic review and meta-analysis of efficacy and toxicity of disease modifying anti-rheumatic drugs and biological agents for psoriatic arthritis. Ann Rheum Dis 2008;67:855-59.
- Saad AA, Symmons DPM, Noyce PR, Ashcroft DM. Risks and benefits of tumor necrosis factor-alpha inhibitors in the management of psoriatic arthritis: systematic review and metaanalysis of randomized controlled trials. J Rheumatol 2008;35:883-90.
- Brimhall AK, King LN, Licciardone JC, Jacobe H, Menter A. Safety and efficacy of alefacept, efalizumab, etanercept and infliximab in treating moderate to severe plaque psoriasis: a meta-analysis of randomized controlled trials. Br J Dermatol 2008;159:274-85.
- Sutton AJ, Duval SJ, Tweedie RL, Abrams KR, Jones DR. Empirical assessment of effect of publication bias on meta-analyses. BMJ 2000;320:1574-7.
- Easterbrook PJ, Berlin JA, Gopalan R, Matthews DR. Publication bias in clinical research. Lancet 1991;337:867-72.
- Egger M, Zellweger-Zähner T, Schneider M, Junker C, Lengeler C, Antes G. Language bias in randomised controlled trials published in English and German. Lancet 1997:350-9.
- Patsopoulos NA, Evangelou E, Ioannidis JP. Sensitivity of between-study heterogeneity in meta-analysis: proposed metrics and empirical evaluation. Int J Epidemiol 2008;37:1148-57.
- Thompson SG. Systematic Review: Why sources of heterogeneity in meta-analysis should be investigated. BMJ 1994;309:1351-5.
- Mohan AK, Cote TR, Block JA, Manadan AM, Siegel JN, Braun MM. Tuberculosis following the use of etanercept, a tumor necrosis factor inhibitor. Clin Infect Dis 2004;39:295-9.
- Iliopoulos A, Psathakis K, Aslanidis S, Skagias L, Sfikakis PP. Tuberculosis and granuloma formation in patients receiving anti-TNF therapy. Int J Tuberc Lung Dis 2006;10:588-90.
- Toussirot E, Streit G, Wendling D. Infectious complications with anti-TNFalpha therapy in rheumatic diseases: a review. Recent Pat Inflamm Allergy Drug Discov 2007;1:39-47.
- Dumville J, Torgerson D, Hewitt C. Reporting attrition in randomised controlled trials. BMJ 2006;332:969-71.
- Smith C, Anstey A, Barker J, Burden A, Chalmers R, Chandler D, et al. British Association of Dermatologists’ Guidelines for Biologic Interventions for Psoriasis. Br J Dermatol 2009;161:987-1019.
- Woolacott N, Hawkins N, Mason A, Kainth A, Khadjesari Z, Vergel YB, et al. Etanercept and efalizumab for the treatment of psoriasis: a systematic review. Health Technol Assess 2006;10.
- Drummond MF, O’Brien BJ, Stoddart GL, Torrance GW. Methods for the economic evaluation of health care programmes. New York, NY: Oxford University Press; 1997.
- Bansback NJ, Ara R, Barkham N, Brennan A, Fraser AD, Conway P, et al. Estimating the cost and health status consequences of treatment with TNF antagonists in patients with psoriatic arthritis. Rheumatology 2006;45:1029-38.
- Bravo Vergel Y, Hawkins NS, Claxton K, Asseburg C, Palmer S, Woolacott N, et al. The cost-effectiveness of etanercept and infliximab for the treatment of patients with psoriatic arthritis. Rheumatology 2007;46:1729-35.
- Olivieri I, de Portu S, Salvarani C, Cauli A, Lubrano E, Spadaro A, et al. The psoriatic arthritis cost evaluation study: a cost-of-illness study on tumour necrosis factor inhibitors in psoriatic arthritis patients with inadequate response to conventional therapy. Rheumatology 2008;47:1664-70.
- Turner D, Picot J, Cooper K, Loveman E. Adalimumab for the treatment of psoriasis. Health Technol Assess 2009;13:49-54.
- Gray A, Rivero-Arias O, Clarke P. Estimating the association between SF-12 responses and EQ-5D utility values by response mapping. Med Decis Making 2006;26:18-29.
- Brazier JE, J. R. The estimation of a preference-based measure of health from the SF-12. Med Care 2004;42:851-9.
- NICE . TA104 Psoriatic Arthritis, Etanercept and Infliximab – Guidance 2006.
- NICE . TA125 Psoriatic Arthritis (moderate to Severe) – Adalimumab: Guidance. 2007.
- NICE . Updated Guide to the Methods of Technology Appraisal 2008.
- The NHS Information Centre . Health Survey for England 2007: Adult Trends Tables 2008.
- Saad A, Ashcroft D, Watson K, Symmonds D, Noyce P, Hyrich K. Improvements in quality of life and functional status in patients with psoriatic arthritis receiving anti-TNF therapies. Arthritis Care Res 2010;62:345-53.
- Department of Health (DoH) . Reference Costs 2007–08 2009.
- Hartman M, Prins M, Swinkels OQJ, Severens JL, De Boo T, Van Der Wilt GJ, et al. Cost-effectiveness analysis of a psoriasis care instruction programme with dithranol compared with UVB phototherapy and inpatient dithranol treatment. Br J Dermatol 2003;147:538-44.
- Spiegel D. Placebos in practice. BMJ 2004;329:927-8.
- Gulfe A, Kristensen L, Saxne T, Jacobsson L, Petersson I, Geborek P. Rapid and sustained health utility gain in anti-TNF treated inflammatory arthritis. Observational data during seven years in southern Sweden. Ann Rheum Dis 2010;69:352-7.
- Saad A, Ashcroft D, Watson K, Hyrch K, Noyce P, Symmons D. Persitence with anti-tumour necrosis factor therapies in patients with psoriatic arthritis: observation study from the British Society for Rheumatology Biologics register. Arthritis Res Ther 2009;11.
- Poyner T, Wall A, Adnitt P, Menday A. Economic impact of psoriasis treatment on the patient and on the National Health Service. J Dermatolog Treat 1999;10:25-9.
- Prodanowich S, Fangchao M, Taylor J, Pezon C, Fasihi T, Kirsner R. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. Am Acad Dermatol 2005;52.
- Finlay A. Current severe plaque psoriasis and the rule of tens. Br J Dermatol 2005;152:861-7.
- Reich K, Mrowietz U. Treatment goals in psoriasis. JDDG 2007;5:556-74.
- Hyrich K, Lunt M, Watson K, Symmons D, Silman A. Outcomes after switching from one anti tumor necrosis factor alpha agent in patients with rheumatoid arthritis. Results from a large UK national cohort study. Arthritis Rheum 2007;56:13-20.
- Johannesson M, Weinstein S. On the decision rules of cost-effectiveness analysis. J Health Econ 1993;12:459-67.
- Geborek P, Bladstrom A, Turesson C, Gulfe A, Petersson IF, Saxne T, et al. Tumour necrosis factor blockers do not increase overall tumour risk in patients with rheumatoid arthritis, but may be associated with an increased risk of lymphomas. Ann Rheum Dis 2005;64:699-703.
- Malesci D, Tirri R, Buono R, G LM. Leflunomide in psoriatic arthritis: a retrospective study of discontinuation rate in daily clinical practice compared with methotrexate. Rheumatol 2007;25:881-4.
- Mease P. TNF-alpha therapy in psoriatic arthritis and psoriasis. Ann Rheum Dis 2004;63:755-8.
- Scott DL, Pugner K, Kaarela K, Doyle DV, Woolf A, Holmes J, et al. The links between joint damage and disability in rheumatoid arthritis. Rheumatol Int 2000;39:122-32.
- Lefebvre C, Manheimer E, Glanville J, Higgins JPT, Green S. Cochrane handbook for systematic reviews of interventions, version 5.0.1 (updated September 2008). Oxford: The Cochrane Collaboration; 2008.
- Wong S, Wilczynski N, Haynes R. Developing optimal search strategies for detecting clinically sound treatment studies in EMBASE. J Med Libr Assoc 2006;94:41-7.
- Centre for Reviews and Dissemination (CRD) . Identifying Studies for Inclusion in NHS EED 2009. www.crd.york.ac.uk/crdweb/html/helpdoc.htm#item17 (accessed 11 June 2009).
- Sutton A, Ades A, Cooper N, Abrams KR. Use of indirect and mixed treatment comparisons for technology assessment. Pharmacoeconomics 2008;26:753-67.
- Ades A, Welton N, Caldwell D, Price M, Goubar A, Lu G. Multiparameter evidence synthesis in epidemiology and medical decision-making. J Health Serv Res Policy 2008;13:12-2.
- Lunn DJ, Thomas A, Best N, Spiegelhalter D. Winbugs: a Bayesian modelling framework: concepts, structure, and extensibility. Stat Comput 2000;10:325-37.
- Jansen J, Crawford B, Bergman G, Stam W. Bayesian meta-analysis of multiple treatment comparisons: an introduction to mixed treatment comparisons. Value Health 2008;11:956-64.
- Ades A, Lu G, Higgins J. The interpretation of random-effects meta-analysis in decision models. Med Decis Making 2005;25:646-54.
- Fahrmeir L, Tutz G. Multivariate statistical modelling based on generalized linear models. New York, NY: Springer-Verlag; 2001.
- Wailoo AJ, Bansback N, Brennan A, Michaud K, Nixon RM. F W . Biologic drugs for rheumatoid arthritis in the Medicare Program. Arthritis Rheum 2008;58:939-46.
- Kind P, Spilker B. Quality of life and pharmacoeconomics in clinical trials. Philadelphia: Lippincott-Raven Publishers; 1996.
- Fraser AD, van Kuijk AW, Westhovens R, Karim Z, Wakefield R, Gerards AH, et al. A randomised, double blind, placebo controlled, multicentre trial of combination therapy with methotrexate plus ciclosporin in patients with active psoriatic arthritis. Ann Rheum Dis 2005;64:859-64.
- Aletaha D, Stamm T, Kapral T, Eberl G, Grisar J, Machold KP, et al. Survival and effectiveness of leflunomide compared with methotrexate and sulfasalazine in rheumatoid arthritis: a matched observational study. Ann Rheum Dis 2003;62:944-51.
- Kristensen LE, Gulfe A, Saxne T, Geborek P. Efficacy and tolerability of anti-tumour necrosis factor therapy in psoriatic arthritis patients: results from the South Swedish Arthritis Treatment Group register. Ann Rheum Dis 2008;67:364-9.
- Monthly Index of Medical Specialties (MIMS) 2009. www.mims.co.uk/home/.
- Curtis L. Unit costs of health and social care 2008. London: PSSRU; 2008.
- Hrobjartsson A, Gotzsche P. Placebo interventions for all clinical conditions. Cochrane Database Syst Rev 2004;3.
- Bedford T, Cooke R. Probabilistic risk analysis: foundations and methods. Cambridge: Cambridge University Press; 2004.
- Garthwaite PH, Kadane JB, O’Hagan A. Statistical methods for eliciting probability distributions. J Am Stat Assoc 2005;100:680-70.
- O’Hagan A, Buck CE, Daneshkhah A, Eiser JR, Garthwaite PH, Jenkinson DJ, et al. Uncertain judgements: eliciting experts’ probabilities. Chichester: Wiley; 2006.
- van Noortwijk JM, Dekker R, Cooke RM, Mazzuchi TA. Expert judgement in maintenance optimization. IEEE T Reliab 1992;41:427-32.
- Cooke RM. Experts in uncertainty: opinion and subjective probability in science. New York, NY: Oxford University Press; 1991.
- Genest C, Zidek JV. Combining probability distributions. Stat Sci 1986;1:114-47.
- Pocock SJ, Assmann SE, Enos LE, Kasten LE. Subgroup analysis, covariate adjustment and baseline comparisons in clinical trial reporting. Stat Med 2000;21:2917-30.
- Heiberg MS, Koldingsnes W, Mikkelsen K, Rodevand E, Kaufmann C, Mowinckel P, et al. The comparative one-year performance of anti-tumor necrosis factor alpha drugs in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: results from a longitudinal, observational, multicenter study. Arthritis Rheum 2008;59:234-40.
- Heiberg MS, Kaufmann C, Rødevand E, Mikkelsen K, Koldingsnes W, Mowinckel P, et al. The comparative effectiveness of anti-TNF therapy and methotrexate in patients with psoriatic arthritis: 6 month results from a longitudinal, observational, multicentre study. Ann Rheum Dis 2007;66:1038-42.
- Gomez-Reino JJ, Carmona L, Group B. Switching TNF antagonists in patients with chronic arthritis: an observational study of 488 patients over a four-year period. Arthritis Res Ther 2006;8.
- Lu G, Ades A, Sutton A, Cooper N, Briggs A, Caldwell D. Meta-analysis of mixed treatment comparisons at multiple follow up times. Stat Med 2007;26:3681-99.
- Department of Health (DoH) . Reference Costs 2008–09 2010.
- Morgan C, Lunt M, Bunn D, Scott DGI, Symmons DPM. Five-year outcome of a primary-care-based inception cohort of patients with inflammatory polyarthritis plus psoriasis. Rheumatology 2007;46:1819-23.
- OECD . Purchasing Power Parities 2005–2008 n.d. www.oecd.org/dataoecd/61/54/18598754.pdf.
- Wiles N, Cooper N, Symmons D. Resource Use Within the Norfolk Arthritis Register (NOAR) Cohort During the First Five Years of Disease: Report for Roche (NICE Data on File) 2005.
- Sizto S, Bansback N, Feldman S, Willian M, Anis A. Economic evaluation of systemic therapies for moderate to severe psoriasis. Br J Dermatol 2009;160:1264-72.
- Lloyd A, Reeves P, Conway P, Reynolds A, Baxter G. Economic evaluation of etanercept in the management of chronic plaque psoriasis. Br J Dermatol 2008;160:380-6.
- Colombo G, Altomare G, Peris K, Martini P, Quarta G, Congedo M, et al. Moderate and severe plaque psoriasis: cost-of-illness study in Italy. Ther Clin Risk Manag 2008;4:559-68.
- Marchetti A, LaPensee K, An P. A pharmacoeconomic analysis of topical therapies for patients with mild-to-moderate stable plaque psoriasis: a US study. Clin Ther 1998;20:851-69.
- National Psoriasis Foundation n.d. www.psoriasis.org (accessed 22 November 2009).
- Gefland J, Troxel A, Lewis J, Kohli Kurd S, Shin D, Wang X, et al. The risk of mortality in patients with psoriasis: results from a population-based study. Arch Dermatol 2007;143:1493-9.
- Gagnon Y, Levy A, Spencer M, Hurley J, Frost F, Mapel D, et al. Economic evaluation of treating chronic obstructive pulmonary disease with inhaled corticosteroids and long-acting [beta]2-agonists in a health maintenance organization. Respir Med 2005;99:1534-45.
- Coates LC, Cawkwell LS, Ng NWF, Bennett AN, Bryer DJ, Fraser AD, et al. Sustained response to long-term biologics and switching in psoriatic arthritis: results from real life experience. Ann Rheum Dis 2008;67:717-19.
- Carmona L, Gomez-Reino JJ. Survival of TNF antagonists in spondylarthritis is better than in rheumatoid arthritis. Data from the Spanish registry BIOBADASER Group. Arthritis Res Ther 2006;8.
- Walkenbach J. Excel 2007 Bible. Indianapolis, IN: Wiley Publishing; 2007.
- Walkenbach J. Excel 2007 Power programming with VBA. Indianapolis, IN: Wiley Publishing; 2007.
- Baier T, Neuwirth E. Excel: COM:: R. Computational Statistics 2007;22:91-108.
- Heiberger R, Neuwirth E. R through Excel: a spreadsheet interface for statistics, data analysis, and graphics. Hamden, CT: Springer; 2009.
- Sturtz S, Ligges U, . R2WinBUGS: a package for running WinBUGS from R. J Stat Softw 2005;12:1-16.
Appendix 1 Literature search strategies
Full details of all databases searched and search strategies are provided below. Numbers in brackets reflect the number of hits retrieved.
The search strategy was designed for searching MEDLINE through the OvidSP interface and was adapted as appropriate for all other databases searched, taking into account differences in indexing terms and search syntax for each database.
Clinical effectiveness: search for RCTS
MEDLINE: OvidSP
The MEDLINE search covered the date range 1950 to week 5 May 2009 for adalimumab and 1 April 2004 to week 5 May 2009, using the search field ‘ed: Entry Date’, for etanercept and infliximab. The search was carried out on 9 June 2009 and identified 399 records.
The strategy uses the Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE, sensitivity-maximising version (lines 1–11). 202
-
randomized controlled trial.pt. (272,711)
-
controlled clinical trial.pt. (79,394)
-
randomized.ab. (182,345)
-
placebo.ab. (112,659)
-
drug therapy.fs. (1,317,603)
-
randomly.ab. (132,262)
-
trial.ab. (189,408)
-
groups.ab. (909,284)
-
or/1-8 (2,406,033)
-
(animals not (humans and animals)).sh. (3,290,537)
-
9 not 10 (2,040,011)
-
Arthritis, Psoriatic/ (2223)
-
(psoria$adj2 (arthrit$or arthropath$)).ti,ab. (3596)
-
12 or 13 (4138)
-
(etanercept or enbrel).ti,ab,rn. (2085)
-
(infliximab or remicade).ti,ab,rn. (4715)
-
15 or 16 (5890)
-
11 and 14 and 17 (450)
-
(200404$or 200405$or 200406$or 200407$or 200408$or 200409$ or 200410$or 200411$or 200412$or 2005$or 2006$or 2007$or 2008$ or 2009$).ed. (3,555,234)
-
18 and 19 (356)
-
(adalimumab or humira or D2E7 or (D2 adj E7)).ti,ab,rn. (1161)
-
11 and 14 and 21 (143)
-
20 or 22 (399)
MEDLINE In-Process & Other Non-Indexed Citations: OvidSP
The MEDLINE In-Process & Other Non-Indexed Citations search, database dated 8 June 2009, was carried out on 9 June 2009 and identified five records.
The strategy sess the Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE, sensitivity-maximising version (lines 1–11).
-
randomized controlled trial.pt. (387)
-
controlled clinical trial.pt. (40)
-
randomized.ab. (7406)
-
placebo.ab. (3160)
-
drug therapy.fs. (20)
-
randomly.ab. (8231)
-
trial.ab. (7527)
-
groups.ab. (42,954)
-
or/1-8 (56,348)
-
(animals not (humans and animals)).sh. (8)
-
9 not 10 (56,346)
-
Arthritis, Psoriatic/ (1)
-
(psoria$adj2 (arthrit$or arthropath$)).ti,ab. (125)
-
12 or 13 (125)
-
(etanercept or enbrel).ti,ab,rn. (164)
-
(infliximab or remicade).ti,ab,rn. (287)
-
(adalimumab or humira or D2E7 or (D2 adj E7)).ti,ab,rn. (110)
-
or/15-17 (438)
-
11 and 14 and 18 (5)
EMBASE: OvidSP
The EMBASE search covered the date range 1980–2009 week 23 for adalimumab and 1 January 2004 to week 23 2009, using the search field ‘em: Entry Week’, for etanercept and infliximab. The search was carried out on 9 June 2009 and identified 369 records.
The strategy uses the Hedges Team best-sensitivity strategy for detecting clinically sound treatment studies in EMBASE (lines 17–20). 203
Note: A pragmatic approach was taken to reduce the number of irrelevant records retrieved and to negate the over indexing of records in EMBASE; EMTREE drug terms were focused in this strategy.
-
Psoriatic Arthritis/ (4225)
-
(psoria$adj2 (arthrit$or arthropath$)).ti,ab. (3339)
-
1 or 2 (5024)
-
*Etanercept/ (1973)
-
(etanercept or enbrel).ti,ab. (2192)
-
*Infliximab/ (3482)
-
(infliximab or remicade).ti,ab. (3991)
-
or/4-7 (6134)
-
(2004$or 2005$or 2006$or 2007$or 2008$or 2009$).em. (3,193,493)
-
8 and 9 (4694)
-
*Adalimumab/ (881)
-
(adalimumab or humira or D2E7 or (D2 adj E7)).ti,ab. (958)
-
11 or 12 (1236)
-
3 and 10 (500)
-
3 and 13 (219)
-
14 or 15 (561)
-
random$.tw. (399,406)
-
clinical trial$.mp. (608,378)
-
exp Health Care Quality/ (802,714)
-
or/17-19 (1,446,048)
-
16 and 20 (369)
CENTRAL: The Cochrane Library
Issue 2, 2009, of The Cochrane Library was searched to identify trials on CENTRAL. The etanercept and infliximab search covered the date range 2004–2009. The search for adalimumab had no date limits. The search was carried out on 9 June 2009 and identified 37 records.
#1 MeSH descriptor Arthritis, Psoriatic, this term only (99)
#2 (psoria* NEAR/2 arthrit*) in Clinical Trials (132)
#3 (psoria* NEAR/2 arthropath*) in Clinical Trials (6)
#4 (#1 OR #2 OR #3) (199)
#5 (etanercept or enbrel):ti,ab,kw, from 2004 to 2009 in Clinical Trials (184)
#6 (infliximab or remicade):ti,ab,kw, from 2004 to 2009 in Clinical Trials (224)
#7 (adalimumab or humira or D2E7 or (D2 adj E7)):ti,ab,kw in Clinical Trials (91)
#8 (#5 OR #6 OR #7) (579)
#9 (#4 AND #8) (37)
SCI: ISI Web of Knowledge
The SCI search covered the date range 1990–2009 for adalimumab and 2004–9 for etanercept and infliximab. The search was carried out on 9 June 2009 and identified 302 records.
The strategy uses the terms used in the 2006 HTA report73 to identify RCTs in the SCI (lines #1–7).
# 13 302 #10 or #12
Databases=SCI-EXPANDED Timespan=All Years
# 12 108 #7 and #8 and #11
Databases=SCI-EXPANDED Timespan=All Years
# 11 1,676 TS=(adalimumab or humira or D2E7 or “D2 E7”)
Databases=SCI-EXPANDED Timespan=All Years
# 10 275 #7 and #8 and #9
Databases=SCI-EXPANDED Timespan=2004–2009
# 9 9,327 TS=(etanercept or enbrel or infliximab or remicade)
Databases=SCI-EXPANDED Timespan=All Years
# 8 4,706 TS=((psoria* same arthrit*) or (psoria* same arthropath*))
Databases=SCI-EXPANDED Timespan=All Years
# 7 >100,000 #5 not #6
Databases=SCI-EXPANDED Timespan=All Years
# 6 >100,000 TS=(animal or animals or dog or dogs or hamster* or mice or mouse or rat or rats
or bovine or sheep or guinea*)
Databases=SCI-EXPANDED Timespan=All Years
# 5 >100,000 #1 or #2 or #3 or #4
Databases=SCI-EXPANDED Timespan=All Years
# 4 >100,000 TS=(placebo* or random* or control* or prospectiv* or volunteer*)
Databases=SCI-EXPANDED Timespan=All Years
# 3 >100,000 TS=(clinic* same trial*)
Databases=SCI-EXPANDED Timespan=All Years
# 2 >100,000 TS=((singl* or doubl* or trebl* or tripl*) SAME (blind* or mask*))
Databases=SCI-EXPANDED Timespan=All Years
# 1 >100,000 TS=((study or studies) SAME design*)
Databases=SCI-EXPANDED Timespan=All Years
CPCI-S: ISI Web of Knowledge
The CPCI-S search covered the date range 1990–2009 for adalimumab and 2004–9 for etanercept and infliximab. The search was carried out on 9 June 2009 and identified 37 records.
The strategy uses the terms used in the 2006 HTA report73 to identify RCTs in the CPCI-S (previously ISI Science and Technology Proceedings) (lines #1–7).
# 13 37 #10 or #12
Databases=CPCI-S Timespan=1990–2009
# 12 12 #7 and #8 and #11
Databases=CPCI-S Timespan=1990–2009
# 11 635 TS=(adalimumab or humira or D2E7 or “D2 E7”)
Databases=CPCI-S Timespan=1990–2009
# 10 29 #7 and #8 and #9
Databases=CPCI-S Timespan=2004–2009
# 9 2,588 TS=(etanercept or enbrel or infliximab or remicade)
Databases=CPCI-S Timespan=1990–2009
# 8 797 TS=((psoria* same arthrit*) or (psoria* same arthropath*))
Databases=CPCI-S Timespan=1990–2009
# 7 >100,000 #5 not #6
Databases=CPCI-S Timespan=1990–2009
# 6 >100,000 TS=(animal or animals or dog or dogs or hamster* or mice or mouse or rat or rats
or bovine or sheep or guinea*)
Databases=CPCI-S Timespan=1990–2009
# 5 >100,000 #1 or #2 or #3 or #4
Databases=CPCI-S Timespan=1990–2009
# 4 >100,000 TS=(placebo* or random* or control* or prospectiv* or volunteer*)
Databases=CPCI-S Timespan=1990–2009
# 3 22,210 TS=(clinic* same trial*)
Databases=CPCI-S Timespan=1990–2009
# 2 15,096 TS=((singl* or doubl* or trebl* or tripl*) SAME (blind* or mask*))
Databases=CPCI-S Timespan=1990–2009
# 1 >100,000 TS=((study or studies) SAME design*)
Databases=CPCI-S Timespan=1990–2009
ClinicalTrials.gov
The ClinicalTrials.gov registry was searched for ongoing trials information. The search was carried out on 9 June 2009 and identified 27 studies.
Basic Search: ((psoriatic arthritis OR psoriatic arthropathy) AND (etanercept OR enbrel OR infliximab OR remicade OR adalimumab or humira or D2E7 or ‘D2 E7’))
mRCT
The mRCT was searched for ongoing trials information. The search was carried out on 10 June 2009 and identified 41 studies.
SEARCH FOR [all registers]: ((“psoriatic arthritis” OR “psoriatic arthropathy”) AND (etanercept OR enbrel OR infliximab OR remicade OR adalimumab or humira or D2E7 or “D2 E7”))
Cost-effectiveness search
MEDLINE: OvidSP
The MEDLINE search covered the date range 1950 to week 1 June 2009 for adalimumab and 1 April 2004 to week 1 June 2009, using the search field ‘ed: Entry Date’, for etanercept and infliximab. The search was carried out on 11 June 2009 and identified 24 records.
The strategy uses the CRD NHS EED strategy for identifying economic evaluations in MEDLINE (lines 13–39). 204
-
Arthritis, Psoriatic/ (2225)
-
(psoria$adj2 (arthrit$or arthropath$)).ti,ab. (3601)
-
1 or 2 (4143)
-
(etanercept or enbrel).ti,ab,rn. (2086)
-
(infliximab or remicade).ti,ab,rn. (4731)
-
4 or 5 (5906)
-
3 and 6 (488)
-
(200404$or 200405$or 200406$or 200407$or 200408$or 200409$ or 200410$or 200411$or 200412$or 2005$or 2006$or 2007$ or 2008$or 2009$).ed. (3,568,700)
-
7 and 8 (387)
-
(adalimumab or humira or D2E7 or (D2 adj E7)).ti,ab,rn. (1164)
-
3 and 10 (152)
-
9 or 11 (432)
-
economics/ (25,433)
-
exp “Costs and Cost Analysis”/ (143,147)
-
VALUE OF LIFE/ (5039)
-
economics, dental/ (1776)
-
exp economics, hospital/ (15,981)
-
economics, medical/ (7044)
-
economics, nursing/ (3784)
-
economics, pharmaceutical/ (2048)
-
(econom$or cost or costs or costly or costing or price or prices or pricing or pharmacoeconom$).ti,ab. (300,152)
-
(expenditure$not energy).ti,ab. (12,542)
-
(value adj1 money).ti,ab. (12)
-
budget$.ti,ab. (12,911)
-
or/13-24 (407,009)
-
((energy or oxygen) adj cost).ti,ab. (2082)
-
(metabolic adj cost).ti,ab. (512)
-
((energy or oxygen) adj expenditure).ti,ab. (11,540)
-
or/26-28 (13,584)
-
25 not 29 (403,828)
-
letter.pt. (654,164)
-
editorial.pt. (239,274)
-
historical article.pt. (272,822)
-
or/31-33 (1,155,003)
-
30 not 34 (381,317)
-
Animals/ (4,399,394)
-
Humans/ (10,777,302)
-
36 not (36 and 37) (3,292,558)
-
35 not 38 (361,076)
-
12 and 39 (24)
MEDLINE In-Process & Other Non-Indexed Citations: OvidSP
The MEDLINE In-Process & Other Non-Indexed Citations search, database dated 11 June 2009, was carried out on 12 June 2009 and identified one record.
The strategy uses the CRD NHS EED strategy for identifying economic evaluations in MEDLINE (lines 9–35).
-
Arthritis, Psoriatic/ (1)
-
(psoria$adj2 (arthrit$or arthropath$)).ti,ab. (130)
-
1 or 2 (130)
-
(etanercept or enbrel).ti,ab,rn. (174)
-
(infliximab or remicade).ti,ab,rn. (298)
-
(adalimumab or humira or D2E7 or (D2 adj E7)).ti,ab,rn. (113)
-
or/4-6 (457)
-
3 and 7 (21)
-
economics/ (1)
-
exp “Costs and Cost Analysis”/ (7)
-
VALUE OF LIFE/ (0)
-
economics, dental/ (0)
-
exp economics, hospital/ (11)
-
economics, medical/ (0)
-
economics, nursing/ (0)
-
economics, pharmaceutical/ (0)
-
(econom$or cost or costs or costly or costing or price or prices or pricing or pharmacoeconom$).ti,ab. (15,266)
-
(expenditure$not energy).ti,ab. (422)
-
(value adj1 money).ti,ab. (2)
-
budget$.ti,ab. (620)
-
or/9-20 (15,898)
-
((energy or oxygen) adj cost).ti,ab. (103)
-
(metabolic adj cost).ti,ab. (14)
-
((energy or oxygen) adj expenditure).ti,ab. (435)
-
or/22-24 (536)
-
21 not 25 (15,762)
-
letter.pt. (14,507)
-
editorial.pt. (8936)
-
historical article.pt. (2)
-
or/27-29 (23,445)
-
26 not 30 (15,515)
-
Animals/ (12)
-
Humans/ (105)
-
32 not (32 and 33) (8)
-
31 not 34 (15,515)
-
8 and 35 (1)
EMBASE: OvidSP
The EMBASE search covered the date range 1980–2009 week 23 for adalimumab and 1 January 2004–9 week 23, using the search field “em: Entry Week”, for etanercept and infliximab. The search was carried out on 12 June 2009 and identified 80 records.
The strategy uses the CRD NHS EED strategy for identifying economic evaluations in EMBASE (lines 17–43).
Note: A pragmatic approach was taken to reduce the number of irrelevant records retrieved and to negate the over indexing of records in EMBASE; EMTREE drug terms were focused in this strategy.
-
Psoriatic Arthritis/ (4225)
-
(psoria$adj2 (arthrit$or arthropath$)).ti,ab. (3339)
-
1 or 2 (5024)
-
*Etanercept/ (1973)
-
(etanercept or enbrel).ti,ab. (2192)
-
*Infliximab/ (3482)
-
(infliximab or remicade).ti,ab. (3991)
-
or/4-7 (6134)
-
(2004$or 2005$or 2006$or 2007$or 2008$or 2009$).em. (3,193,493)
-
8 and 9 (4694)
-
*Adalimumab/ (881)
-
(adalimumab or humira or D2E7 or (D2 adj E7)).ti,ab. (958)
-
11 or 12 (1236)
-
3 and 10 (500)
-
3 and 13 (219)
-
14 or 15 (561)
-
Health Economics/ (10,611)
-
exp Economic Evaluation/ (104,472)
-
exp “Health Care Cost”/ (107,017)
-
exp PHARMACOECONOMICS/ (56,975)
-
(econom$or cost or costs or costly or costing or price or prices or pricing or pharmacoeconomic$).tw. (234,263)
-
(expenditure$not energy).ti,ab. (9859)
-
(value adj2 money).ti,ab. (462)
-
budget$.ti,ab. (8863)
-
or/17-24 (347,643)
-
(metabolic adj cost).ti,ab. (388)
-
((energy or oxygen) adj cost).ti,ab. (1707)
-
((energy or oxygen) adj expenditure).ti,ab. (10,088)
-
or/26-28 (11,689)
-
25 not 29 (345,077)
-
(letter or note or editorial).pt. (925,192)
-
30 not 31 (298,277)
-
exp Animal/ (18,276)
-
exp Animal Experiment/ (1,298,147)
-
Nonhuman/ (3,232,877)
-
(rat or rats or mouse or mice or hamster or hamsters or animal or animals or dog or dogs or cat or cats or bovine or sheep).ti,ab. (1,737,766)
-
or/33-36 (3,643,672)
-
exp human/ (6,568,828)
-
exp Human Experiment/ (257,542)
-
38 or 39 (6,569,696)
-
37 not (37 and 40) (2,983,952)
-
32 not 41 (274,297)
-
16 and 42 (80)
CENTRAL: The Cochrane Library
A search of CENTRAL was not repeated for cost-effectiveness evidence. The search carried out on 9 June 2009 (shown in Clinical effectiveness: search for RCTs) was not limited by study design and would also have identified economic evaluations.
SCI: ISI Web of Knowledge
The SCI search covered the date range 1900–2009 for adalimumab and 2004–9 for etanercept and infliximab. The search was carried out on 12 June 2009 and identified 31 records.
The strategy uses the terms used in the 2006 HTA report73 to identify economic evaluations in the SCI (lines #7–10).
# 10 31 #8 not #9
Databases=SCI-EXPANDED Timespan=1900–2009
# 9 >100,000 TS=(animal or animals or dog or dogs or hamster* or mice or mouse or rat or rats or bovine or sheep or guinea*)
Databases=SCI-EXPANDED Timespan=1900–2009
# 8 33 #6 and #7
Databases=SCI-EXPANDED Timespan=1900–2009
# 7 >100,000 TS=(econom* or cost or costs or costly or costing or price or prices or pricing or pharmacoeconom* or budget*)
Databases=SCI-EXPANDED Timespan=1900–2009
# 6 666 #3 or #5
Databases=SCI-EXPANDED Timespan=1900–2009
# 5 211 #1 and #4
Databases=SCI-EXPANDED Timespan=1900–2009
# 4 1,699 TS=(adalimumab or humira or D2E7 or “D2 E7”)
Databases=SCI-EXPANDED Timespan=1900–2009
# 3 570 #1 and #2
Databases=SCI-EXPANDED Timespan=1900–2009
# 2 7,383 TS=(etanercept or enbrel or infliximab or remicade)
Databases=SCI-EXPANDED Timespan=2004–2009
# 1 4,736 TS=((psoria* same arthrit*) or (psoria* same arthropath*))
Databases=SCI-EXPANDED Timespan=1900–2009
CPCI-S: ISI Web of Knowledge
The CPCI-S search covered the date range 1990–2009 for adalimumab and 2004–9 for etanercept and infliximab. The search was carried out on 12 June 2009 and identified three records.
The strategy uses the terms used in the 2006 HTA report to identify economic evaluations in the CPCI-S (previously ISI Science and Technology Proceedings) (lines #7–10).
# 10 3 #8 not #9
Databases=CPCI-S Timespan=1990–2009
# 9 >100,000 TS=(animal or animals or dog or dogs or hamster* or mice or mouse or rat or rats or bovine or sheep or guinea*)
Databases=CPCI-S Timespan=1990–2009
# 8 3 #6 and #7
Databases=CPCI-S Timespan=1990–2009
# 7 >100,000 TS=(econom* or cost or costs or costly or costing or price or prices or pricing or pharmacoeconom* or budget*)
Databases=CPCI-S Timespan=1990–2009
# 6 196 #3 or #5
Databases=CPCI-S Timespan=1990–2009
# 5 62 #1 and #4
Databases=CPCI-S Timespan=1990–2009
# 4 651 TS=(adalimumab or humira or D2E7 or “D2 E7”)
Databases=CPCI-S Timespan=1990–2009
# 3 140 #1 and #2
Databases=CPCI-S Timespan=1990–2009
# 2 2,192 TS=(etanercept or enbrel or infliximab or remicade)
Databases=CPCI-S Timespan=2004–2009
# 1 814 TS=((psoria* same arthrit*) or (psoria* same arthropath*))
Databases=CPCI-S Timespan=1990–2009
NHS EED
The NHS EED was searched for economic evaluations. As no records were identified in the 2006 HTA review, no date limits were set. The search was carried out on 12 June 2009 and identified seven records.
Note: The strategy was run across the entire CRD databases and the final results shown here, 20 records, relate to the total number of records found.
-
# 1 MeSH Arthritis, Psoriatic (22)
-
# 2 (psoria* NEAR arthrit*) (43)
-
# 3 (psoria* NEAR arthropath*) (1)
-
# 4 #1 or #2 or #3 (44)
-
# 5 etanercept OR enbrel OR infliximab OR remicade (165)
-
# 6 adalimumab OR humira OR D2E7 OR “D2 AND E7” (48)
-
# 7 #5 or #6 (182)
-
# 8 #4 and #7 (20)
HEED
The HEED was searched for economic evaluations. As no records were identified in the 2006 HTA review, no date limits were set. The search was carried out on 12 June 2009 and identified eight records.
Compound Search
All Data: ((psoria* AND arthrit*) OR (psoria* AND arthropath*))
AND
All Data:etanercept OR enbrel OR infliximab OR remicade OR adalimumab OR humira OR D2E7 OR ‘D2 E7’
EconLit: OvidSP
The American Economic Association's electronic bibliography, EconLit, database was searched for economic evaluations. The search carried out on 12 June 2009, covering the date range 1969–May 2009, identified no records.
-
(psoria$adj2 (arthrit$or arthropath$)).ti,ab. (0)
-
(etanercept or enbrel or infliximab or remicade or adalimumab or humira or D2E7 or “D2 E7”).ti,ab. (3)
-
#1 and #2 (0)
Additional searches
Side-effects/adverse effects search
The following resources were searched for information on side-effects:
-
Center for Drug Evaluation and Research. Drugs@FDA. Silver Spring, MD: US Food and Drug Administration [cited 2009 Jun 08]. URL: www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm
-
EPARs for authorised medicinal products for human use. London: European Medicines Agency [cited 8 June 2009]. URL: www.emea.europa.eu/htms/human/epar/a.htm
Additional information on side-effects was gathered by supplementary searches. The following searches were designed to capture the major side-effects that had been identified as arising from the use of etanercept, infliximab or adalimumab: urinary tract infections, lower respiratory tract infections, skin infections, bone infections, joint infections, malignancy, and the reactivation of latent TB.
A pragmatic approach to searching was adopted for the supplementary side-effects search. This can be seen in the reliance of indexed terms to search for the side-effects and the use of subheadings linked to specific side-effects, such as the MeSH subheading ‘Chemically Induced’ and the EMTREE subheading ‘Side Effect’. This search approach enhances the precision of a search but has an unknown effect on its sensitivity.
MEDLINE: OvidSP
The MEDLINE search covered the date range 1950 to week 1 June 2009. The search was carried out on 16 June 2009 and identified 60 records.
-
(etanercept or enbrel).ti,ab. (2086)
-
(infliximab or remicade).ti,ab. (3743)
-
(adalimumab or humira or D2E7 or (D2 adj E7)).ti,ab. (878)
-
or/1-3 (5297)
-
Safety/ (26,929)
-
(safe or safety).ti,ab. (271,847)
-
(side effect or side effects).ti,ab. (130,142)
-
treatment emergent.ti,ab. (867)
-
undesirable effect$.ti,ab. (1448)
-
tolerability.ti,ab. (19,551)
-
Drug Toxicity/ (2820)
-
toxicity.ti,ab. (173,622)
-
Adverse Drug Reaction Reporting Systems/ (3900)
-
adrs.ti,ab. (975)
-
(adverse adj3 (effect or effects or reaction or reactions or event or events or outcome or outcomes)).ti,ab. (147,732)
-
(undesir$adj2 (effect or effects or reaction or reactions or event or events or outcome or outcomes)).ti,ab. (4632)
-
Drug Hypersensitivity/ (17,725)
-
(hypersensit$or hyper sensit$).ti,ab. (45,094)
-
harm$.ti,ab. (54,739)
-
or/5-19 (750,762)
-
4 and 20 (1654)
-
exp Infection/ci [Chemically Induced] (2859)
-
exp Urinary Tract Infections/ci [Chemically Induced] (61)
-
exp Respiratory Tract Infections/ci [Chemically Induced] (3678)
-
exp Skin Diseases, Infectious/ci [Chemically Induced] (451)
-
exp Bone Diseases, Infectious/ (27,676)
-
exp Arthritis, Infectious/ci [Chemically Induced] (55)
-
exp Neoplasms/ci [Chemically Induced] (50,219)
-
exp Tuberculosis/ci [Chemically Induced] (315)
-
or/22-29 (84,100)
-
21 and 30 (60)
-
(animals not (humans and animals)).sh. (3,292,558)
-
31 not 32 (60)
EMBASE: OvidSP
The EMBASE search covered the date range 1980–2009 week 24. The search was carried out on 17 June 2009 and identified 648 records.
Note: A pragmatic approach was taken to reduce the number of irrelevant records retrieved and to negate the over indexing of records in EMBASE; EMTREE drug terms were focused in this strategy.
-
(etanercept or enbrel).ti,ab. (2202)
-
(infliximab or remicade).ti,ab. (3999)
-
(adalimumab or humira or D2E7 or (D2 adj E7)).ti,ab. (960)
-
or/1-3 (5648)
-
*Etanercept/ (1979)
-
*Infliximab/ (3486)
-
*Adalimumab/ (882)
-
or/5-7 (5086)
-
4 or 8 (6595)
-
(safe or safety).ti,ab. (246,785)
-
side effect$.ti,ab. (123,415)
-
treatment emergent.ti,ab. (963)
-
undesirable effect$.ti,ab. (1421)
-
tolerability.ti,ab. (22,410)
-
toxicity.ti,ab. (164,169)
-
adrs.ti,ab. (1214)
-
(adverse adj3 (effect or effects or reaction or reactions or event or events or outcome or outcomes)).ti,ab. (144,000)
-
Safety/or Drug Safety/ (183,510)
-
Side Effect/ (94,185)
-
Adverse Drug Reaction/ (95,592)
-
Drug Tolerability/ (54,359)
-
Toxicity/or Drug Toxicity/ (47,998)
-
Drug Surveillance Program/ (7235)
-
Adverse Outcome/ (1414)
-
hypersensit$.ti,ab. (35,011)
-
harm$.ti,ab. (46,014)
-
Drug Hypersensitivity/ (25,074)
-
or/10-27 (892,235)
-
9 and 28 (2822)
-
*Etanercept/ae, to [Adverse Drug Reaction, Drug Toxicity] (917)
-
*Infliximab/ae, to [Adverse Drug Reaction, Drug Toxicity] (1636)
-
*Adalimumab/ae, to [Adverse Drug Reaction, Drug Toxicity] (442)
-
or/30-32 (2470)
-
29 or 33 (3651)
-
Urinary Tract Infection/si [Side Effect] (2059)
-
Lower Respiratory Tract Infection/si [Side Effect] (144)
-
Skin Infection/si [Side Effect] (488)
-
Bone Infection/si [Side Effect] (26)
-
Infectious Arthritis/si [Side Effect] (55)
-
Neoplasm/si [Side Effect] (452)
-
Tuberculosis/si [Side Effect] (1297)
-
or/35-41 (4150)
-
34 and 42 (648)
Appendix 2 Quality assessment tool
All of the criteria listed below should be scored with one of the following responses:
-
yes (Y)
-
no (N)
-
partial (P)
-
not stated (NS)
-
not applicable (NA)
-
unclear (U).
Study
| 1 | Were the eligibility criteria for the study adequately specified? |
| Adequate study population clearly defined | |
| 2 | Was an a priori power calculation for adequate sample size performed? |
| 3 | Was the sample size adequate for the analysis of the primary outcome variable? |
| 4 | Was the number of participants who were randomised stated? |
| 5 | Was the method used to assign participants to treatment groups truly random? |
| Adequate computer-generated random numbers, random number tables | |
| Inadequate alternation, case record numbers, birth dates, days of the week | |
| 6 | Was the trial described as double blind? |
| 7 | Was allocation of treatment concealed? |
| Adequate centralised or pharmacy controlled assignment, serially numbered containers, serially numbered opaque envelopes, on-site computer-based systems where assignment is unreadable until after allocation, other robust measures to prevent revelation of a participant’s treatment | |
| Inadequate alternation, case record numbers, days of the week, open random number lists | |
| 8 | Were the individuals administering the treatment blinded to the treatment allocation? |
| 9 | Were the outcome assessors blinded to the treatment allocation? |
| 10 | Were the participants blinded to the treatment allocation? |
| 11 | Was the blinding procedure successful? |
| 12 | Were adequate details of the treatment groups at baseline presented? |
| Adequate information on age, nature and severity of psoriasis, previous treatments | |
| 13 | Were the treatment groups comparable at baseline? |
| Answer ‘yes’ if no important differences or if appropriate adjustments had been made for any differences in the baseline characteristics of the treatment groups | |
| 14 | Were the treatment groups similar in terms of co-interventions that could influence the results? |
| 15 | Was participant compliance with the assigned treatment adequate? |
| 16 | Were all participants who were randomised accounted for at the end of the trial? |
| 17 | Was a valid ITT analysis performed? |
| Adequate all participants randomised included in efficacy analysis, all randomised participants who took at least one dose of trial medication included in efficacy analysis | |
| 18 | Were at least 80% of those randomised included in the follow-up assessment? |
| Answer ‘yes’ if at least 80% of those randomised provided complete data with regard to the primary outcome(s) |
Quality rating:
-
Excellent The answer is ‘Yes’ to all of the criteria.
-
Good The answer is ‘Yes’ to all of the following criteria: 1, 3, 4, 6, 10, 12–14, 16–18.
-
Satisfactory The answer is ‘Yes’ to all of the following criteria: 1, 3, 6, 13, 17.
-
Poor The answer is not ‘Yes’ to one or more of the criteria listed for ‘Satisfactory’.
Appendix 3 Data extraction tables
Efficacy data extraction: etanercept
| Study details and design | Participant details | Intervention/outcome/analyses details | Results |
|---|---|---|---|
|
Mease, 2000, USA 78 Type of publication: Full publication Funding: Immunex Corporation Study design: Stage 1: Double-blind RCT, parallel group monotherapy; Stage 2: Open-label follow-up Setting: Outpatient Duration of follow-up: Stage 1: 12 weeks, stage 2: 24 weeks Frequency of follow-up: Stage 1: Baseline, 4, 8 and 12 weeks Stage 2: 16 and 36 weeks Extracted by: HY Checked by: MR |
Inclusion/exclusion criteria: Adults between 18 and 70 years of age with active PsA (defined as three or more swollen joints and three or more tender or painful joints) and an inadequate response to NSAIDs, and were thought candidates for immunomodulatory therapy. Patients taking a stable dose of MTX (≤ 25 mg/week) were permitted to continue with that dose. Other DMARDs were discontinued at least 2 weeks prior to the trial. Corticosteroids were allowed during the study at a dose of ≤ 10 mg/day of prednisone if it was stable for at least 2 weeks prior to the trial and maintained during the trial. For patients with skin involvement psoriasis, therapies had to be discontinued (phototherapy 4 weeks before and topical therapies and oral retinoids 2 weeks before) No. randomised: 60 Age (median age, range) Etanercept: 46.0 years (30.0–70.0 years) Placebo: 43.5 years (24.0–63.0) Gender Etanercept, male 16/30 (53%) Placebo, male 18/30 (60%) |
Intervention: etanercept Dose regimen: 25 mg etanercept twice per week Length of treatment: 12 weeks No. randomised: 30 No. completed: 30 Comparator: placebo Dose regimen: placebo twice per week Length of treatment: 12 weeks No. randomised: 30 No. completed: 26 Primary outcome The proportion of patients meeting the PsARC at 12 weeks Sample size calculation Assuming that a response rate of 30% on placebo and 75% on etanercept, the sample size of 30 patients per group gives 80% power to detect a significant difference between treatments in the primary outcome, with α = 0.05 (two-sided) |
EFFICACY OUTCOMES (STAGE 1, RANDOMISED) ACR 20 Etanercept 25 mg, 12 weeks: 22/30 (73%); placebo 12 weeks: 4/30 (13%); p < 0.0001 ACR 50 Etanercept 25 mg, 12 weeks: 15/30 (50%); placebo 12 weeks: 1/30 (3%); p = 0.0001 ACR 70 Etanercept 25 mg, 12 weeks: 4/30 (13%); placebo 12 weeks: 0/30 (0%); p = 0.0403 PsARC Etanercept 25 mg, 4 weeks: 23/30 (77%); placebo 4 weeks: 4/30 (14%); p < 0.0001 Etanercept 25 mg, 8 weeks: 25/30 (83%); placebo 4 weeks: 8/30 (27%); p < 0.0001 Etanercept 25 mg, 12 weeks: 26/30 (87%); placebo 12 weeks: 7/30 (23%); p < 0.0001 HAQ Median (25th and 75th percentiles): Etanercept 25 mg, baseline 1.3 (CiC information has been removed),12 weeks 0.1 (CiC information has been removed) Placebo baseline 1.2 (CiC information has been removed), 12 weeks 1.1 (CiC information has been removed); p < 0.001 (at 12 weeks) Mean (SD): Etanercept 25 mg, baseline 1.2 (CiC information has been removed), 12 weeks 0.5 (CiC information has been removed) Placebo baseline 1.2 (CiC information has been removed), 12 weeks 1.1 (CiC information has been removed) Percentage improvement at 12 weeks (mean, SD): Etanercept 25 mg (n = 29) 64.2 (CiC information has been removed) Placebo (n = 30) 9.9 (CiC information has been removed) Median (range) PASI at baseline Etanercept 25 mg = 10.1 (2.3–30.0) Placebo = 6.0 (1.5–17.7) PASI 50 Etanercept 25 mg, 12 weeks: 8/19 (42%) Placebo 12 weeks: 4/19 (21%) Treatment difference p = 0.295 |
|
PsA history Duration of PsA (median, range): Etanercept 9.0 years (1.0–31.0 years) Placebo: 9.5 years (1.0–30.0 years) Psoriasis history Duration of psoriasis (median, range): Etanercept 19.0 years (4.0–53.0 years) Placebo: 17.5 years (2.0–43.0 years) Psoriasis evaluation Patients with ≥ 3% BSA affected with psoriasis: Etanercept: 19/30 (63%) Placebo: 19/30 (63%) Concurrent therapies Patients taking a stable dose of MTX (≤ 25 mg/week) were permitted to continue with that dose if it had been stable for 4 weeks prior to study entry and remained constant during the study. Corticosteroids were allowed during the study at a dose of ≤ 10 mg/day prednisolone if the dose had been stable at study entry and if it was maintained during the trial Concomitant therapy during trial Corticosteroids: Etanercept group 6/30 (20%) Placebo group 12/30 (40%) NSAIDS: Etanercept group 20/30 (67%) Placebo group 23/30 (77%) MTX: Etanercept group 14/30 (47%) Placebo group 14/30 (47%) |
Statistical analyses Proportions of patients’ responding were compared using the Mantel–Haenszel chi-squared test adjusted for MTX use. Continuous variables were ranked and analysed by a general linear model with factors of treatment, MTX use and their interaction. The Breslow–Day test was used to test for heterogeneity of relative response between MTX use strata. The LOCF approach was used for imputing missing data ITT analysis All randomised patients were included in the analysis |
PASI 75 Etanercept 25 mg, 12 weeks: 5/19 (26%) Placebo 12 weeks: 0/19 (0%); p = 0.0154 100% improvement in physician global assessment Etanercept 25 mg, 12 weeks: 6/30 (20%) Placebo 12 weeks: 0/30 (0%) 100% improvement in patient global assessment Etanercept 25 mg, 12 weeks: 5/30 (17%) Placebo 12 weeks: 0/30 (0%) ADVERSE EVENTS (STAGE 1, RANDOMISED) Infectious adverse events (n, %) [Placebo (P), n = 30; etanercept (E), n = 30] Respiratory tract infection: P, 4 (13%); E, 8 (27%) Pharyngitis: P, 3 (10%), E, 5 (17%) Rhinitis: P, 4 (13%); E, 6 (20%) Sinusitis: P, 2 (7%); E, 3(10%) Influenza syndrome: P, 6 (20%); E, 0 Infections that required hospitalisation or i.v. antibiotics Etanercept: 0 Placebo: 0 Cancer: Not reported Reactivation of latent TB: Not reported Deaths: None Withdrawals due to adverse events: None EFFICACY OUTCOMES (STAGE 2, OPEN LABEL) PsARC Etanercept 25 mg, 16 weeks: 26/30 (87%); placebo/etanercept 16 weeks: 19/28 (68%) Etanercept 25 mg, 36 weeks: 26/30 (87%); placebo/etanercept 36 weeks: 21/28 (75%) ACR 20 Etanercept 25 mg, 16 weeks: 22/30 (73%); placebo/etanercept 16 weeks: 12/28 (43%) Etanercept 25 mg, 36 weeks: 26/30 (87%); placebo/etanercept 36 weeks: 17/28 (61%) ACR 50 Etanercept 25 mg, 16 weeks: 13/30 (43%); placebo/etanercept 16 weeks: 8/28 (29%) Etanercept 25 mg, 36 weeks: 19/30 (63%); placebo/etanercept 36 weeks: 13/28 (46%) ACR 70 Etanercept 25 mg, 16 weeks: 7/30 (23%); placebo/etanercept 16 weeks: 0/28 Etanercept 25 mg, 36 weeks: 10/30 (33%); placebo/etanercept 36 weeks: 7/28 (25%) HAQ (CiC information has been removed) PASI (patients evaluable for psoriasis only) PASI 50: Etanercept 25 mg, 36 weeks: 11/19 (58%); placebo/etanercept 36 weeks: 10/18 (56%) PASI 75: Etanercept 25 mg, 36 weeks: 7/19 (37%); placebo/etanercept 36 weeks: 5/18 (28%). ADVERSE EVENT OUTCOMES (STAGE 2, OPEN LABEL, 24 WEEKS) [Placebo (P), n = 28; etanercept (E), n = 30] Infectious adverse events, including any serious infections occurring in > 5% of patients by treatment: Respiratory tract infection: P, 9 (32%); E, 7 (23%) Pharyngitis: P, 2 (7%); E, 1 (3%) Influenza syndrome: P, 4 (14%); E, 3 (10%) Urinary tract infection: P, 2 (7%); E, 0 Infection (not specified) : P, 0; E, 2 (7%) Cancer: None Other non-infectious serious adverse events: (CiC information has been removed) Deaths: (CiC information has been removed) Withdrawals due to adverse events: (CiC information has been removed) Comments: All efficacy data in Stage 2 relates to non-randomised patients. All patients in Stage 2 had received etanercept |
|
|
Mease, 2004, USA 52 , 97 , 99 , 105 , 107 , 110 Type of publication: Full publication Funding: Immunex Corporation Study design: Stage 1: Double-blind placebo-controlled RCT Stage 2: Maintenance period Stage 3: Open-label follow-up Duration of follow-up: Stage 1: 24 weeks Stage 2: < 24 weeks Stage 3: 48 weeks |
Inclusion criteria: Patients between 18 and 70 years of age with active PsA and stable plaque psoriasis (target lesion > 2-cm diameter) with more than three swollen joints and more than tender joints. Patients had at least one of the following subtypes of PsA: DIP joint involvement, polyarticular arthritis, arthritis mutilans, asymmetric peripheral arthritis, or ankylosing spondylitis-like arthritis. Patients taking a stable dose of MTX (≤ 25 mg/week) for 2 months were permitted to continue with that dose. Other DMARDs were discontinued at least 4 weeks prior to the trial |
Intervention: etanercept Stage 1: Dose regimen: 25 mg s.c. twice per week Duration/frequency of treatment: 24 weeks No. of participants: 101 Stage 2: After completing stage 1, patients could chose to continue on their blinded study treatment in this maintenance period until all patients had completed 24 weeks of study treatment and the database was locked Dose regimen: 25 mg s.c. twice per week Duration/frequency of treatment: < 24 weeks (CiC information has been removed) |
STAGE 1: EFFICACY OUTCOMES PsARC Etanercept 25 mg 4 weeks: 57 (56%); placebo 4 weeks: 25 (24%); p < 0.001 Etanercept 25 mg 12 weeks: 73 (72%); placebo 12 weeks: 32 (31%); p < 0.001 Etanercept 25 mg 24 weeks: 71 (70%); placebo 24 weeks: 24 (23%); p < 0.001 Subgroup analysis (with and without MTX): Etanercept + MTX 12 weeks: 32/42 (76%); placebo 12 weeks: 14/43 (33%) Etanercept – MTX 12 weeks: 41/59 (69%); placebo 12 weeks: 18/61 (30%) Etanercept + MTX 24 weeks: 31/42 (74%); placebo 24 weeks: 11/43 (26%) Etanercept – MTX 24 weeks: 40/59 (68%); placebo 24 weeks: 13/61 (21%) ACR 20 Etanercept 25 mg 4 weeks: 38 (38%); placebo 4 weeks: 11 (11%); p < 0.001 Etanercept 25 mg 12 weeks: 60 (59%); placebo 12 weeks: 16 (15%); p < 0.001 Etanercept 25 mg 24 weeks: 50 (50%); placebo 24 weeks: 14 (13%); p < 0.001 Subgroup analysis (with and without MTX): Etanercept + MTX 12 weeks: 26/42 (62%); placebo 12 weeks: 8/43 (19%) Etanercept – MTX 12 weeks: 34/59 (58%); placebo 12 weeks: 8/61 (13%) Etanercept + MTX 24 weeks: 23/42 (55%); placebo 24 weeks: 8/43 (19%) Etanercept – MTX 24 weeks: 27/59 (46%); placebo 24 weeks: 6/61 (10%) |
|
Frequency of follow-up: Stage 1: Baseline, 4, 12 and 24 weeks Stage 2: 12-week intervals thereafter Stage 3: 48 weeks Extracted by: HY Checked by: MR |
Corticosteroids were allowed during the study at a dose of ≤ 10 mg/day of prednisone if it was stable for at least 4 weeks prior to the trial. For patients with skin involvement psoriasis, phototherapy therapies had to be discontinued prior to the trial Oral retinoids, tropical vitamin A or D-analogue preparations, and dithranol were not allowed. Tropical therapies were only permitted on the scalp, axillae and groin No. randomised and treated Stage 1: 205 (CiC information has been removed) Stage 3: 168 Age (mean) Etanercept: 47.6 years Placebo: 47.3 years Gender Etanercept: male 58/101(57%) Placebo: male 47/104 (45%) PsA history Duration of PsA, mean: Etanercept: 9.0 years Placebo: 9.2 years Psoriasis history Duration of psoriasis, mean: Etanercept: 18.3 years Placebo: 19.7 years Psoriasis evaluation Patients with ≥ 3% BSA affected with psoriasis: Etanercept: 66/101 (65%) Placebo: 62/104 (60%) |
Stage 3: After the database was locked all patients (CiC information has been removed) were eligible to enter a 48-week open-label extension Dose regimen: (CiC information has been removed) Duration/frequency of treatment: 48 weeks No. of participants: 168 (87 previously on etanercept; 81 stage 1 previously on placebo) (CiC information has been removed) Comparator: placebo Stage 1: Placebo (n = 104): equivalent Stage 2: Placebo (n = 59): equivalent Primary outcome The proportion of patients meeting the ACR 20 at 24 weeks Sample size calculation Assuming that an ACR 20 rate of 60% on etanercept and 30% on placebo, a sample size of 100 patients per group gives a power of 90% power to detect a significant difference between treatments in the primary outcome, with α = 0.05 (two-sided) Statistical analyses Binary response rates were compared using the Cochran–Mantel–Haenszel test or Fisher’s exact test. Continuous variables were analysed by Wilcoxon rank-sum test, using LOCF for missing data or early termination ITT analysis All randomised patients who received at least one dose of blinded study drug were included in the analysis |
ACR 50 Etanercept 25 mg 4 weeks: 11 (11%); placebo 4 weeks: 2 (2%); p = 0.009 Etanercept 25 mg 12 weeks: 38 (38%); placebo 12 weeks: 4 (4%); p < 0.001 Etanercept 25 mg 24 weeks: 37 (37%); placebo 24 weeks: 4 (4%); p < 0.001 Subgroup analysis (with and without MTX): Etanercept + MTX 12 weeks: 17/42 (40%); placebo 12 weeks: 1/43 (2%) Etanercept – MTX 12 weeks: 21/59 (36%); placebo 12 weeks: 3/61 (5%) Etanercept + MTX 24 weeks: 16/42 (38%); placebo 24 weeks: 3/43 (7%) Etanercept – MTX 24 weeks: 21/59 (36%); placebo 24 weeks: 1/61 (2%) ACR 70 Etanercept 25 mg 4 weeks: 1 (1%); placebo 4 weeks: 0; p = 0.493 Etanercept 25 mg 12 weeks: 11 (11%); placebo 12 weeks: 0; p < 0.001 Etanercept 25 mg 24 weeks: 9 (9%); placebo 24 weeks: 1 (1%); p = 0.009 Subgroup analysis (with and without MTX) Etanercept + MTX 12 weeks: 4/42 (10%); placebo 12 weeks: 0/43 (0%) Etanercept – MTX 12 weeks: 7/59 (12%); placebo 12 weeks: 0/61 (0%) Etanercept + MTX 24 weeks: 2/42 (5%); placebo 24 weeks: 0/43 (0%) Etanercept – MTX 24 weeks: 7/59 (12%); placebo 24 weeks: 0/61 (0%) HAQ Mean (SD) absolute values : Etanercept 25 mg, baseline (n = 101) 1.1 (CiC information has been removed); placebo baseline (n = 104) 1.1 (CiC information has been removed) Etanercept 25 mg, 4 weeks (n = 101) 0.7 (CiC information has been removed); placebo 4weeks (n = 104) 1.0 (CiC information has been removed) Etanercept 25 mg, 12 weeks (n = 101) 0.6 (CiC information has been removed); placebo 12 weeks (n = 104) 1.0 (CiC information has been removed) Etanercept 25 mg, 24 weeks (n = 101) 0.5 (CiC information has been removed); placebo 24 weeks (n = 104) 1.0 (CiC information has been removed) Mean (SD) % changes from baseline: Etanercept 25 mg, 4 weeks (n = 96) 35.1 (CiC information has been removed); placebo 4 weeks (n = 99) 8.0 (CiC information has been removed); p < 0.001 Etanercept 25 mg, 12 weeks (n = 96) 53.5 (CiC information has been removed); placebo 12 weeks (n = 99) 6.3 (CiC information has been removed); p < 0.001 Etanercept 25 mg, 24 weeks (n = 96) 53.6 (CiC information has been removed); placebo 24 weeks (n = 99) 6.4 (CiC information has been removed); p < 0.001 |
|
Concurrent therapies Concomitant therapy at baseline : MTX: etanercept 42/101 (42%); placebo 43/104 (41%) Corticosteroids: etanercept 19/101 (19%); placebo 16/104 (15%) NSAIDS: etanercept 89/101(88%); placebo 86/104(83%) |
Comments Patients receiving MTX were randomised separately |
TOTAL SHARP SCORE Mean (SD) annualised rate of progression at 6 months: Etanercept (n = 101) –0.03 (0.73); placebo (n = 104) 0.53 (1.39); p = 0.0006 Subgroup analysis (with and without MTX) (mean, SD): Etanercept + MTX (n = 42) (CiC information has been removed); placebo (n = 43) (CiC information has been removed) Etanercept – MTX (n = 59) (CiC information has been removed); placebo (n = 61) (CiC information has been removed) Mean PASI score at baseline: (CiC information has been removed) PASI 50 No. (%) improvement in PASI 50: Etanercept 25 mg, 24 weeks (n = 66): 31 (47%); placebo 24 weeks (n = 62): 11 (18%); p < 0.001 PASI 75 No. (%) improvement in PASI 75: Etanercept 25 mg 24 weeks (n = 66): 15 (23%); placebo 24 weeks (n = 62): 2 (3%); p = 0.001 PASI 90 No. (%) improvement in PASI 90: Etanercept 25 mg, 24 weeks (n = 66): 4 (6%); placebo 24 weeks (n = 62): 2 (3%); p = 0.681 Target lesion score No. (%) with 50% improvement from baseline: Etanercept 25 mg, 24 weeks (n = 101): 43 (43%); placebo 24 weeks (n = 104): 18 (17%); p < 0.001 No. (%) with 75% improvement from baseline: Etanercept 25 mg, 24 weeks (n = 101): 22 (22%); placebo 24 weeks (n = 104): 10 (10%); p = 0.017 Physician global assessment Mean (median) % improvement from baseline: Etanercept 25 mg, 4 weeks 36.0 (50.0); placebo 4 weeks 2.9 (0); p < 0.001 Etanercept 25 mg, 12 weeks 44.9 (50); placebo 12 weeks 0.3 (0); p < 0.001 Etanercept 25 mg, 24 weeks 47.2 (50); placebo 24 weeks 2.3 (0); p < 0.001 Patient global assessment Mean (median) % improvement from baseline: Etanercept 25 mg, 4 weeks 21.6 (25.0); placebo 4 weeks 1.3 (0); p < 0.001 Etanercept 25 mg, 12 weeks 36.1 (33.3); placebo 12 weeks –0.3 (0); p < 0.001 Etanercept 25 mg, 24 weeks 40.4 (50.0); placebo 24 weeks –3.9 (0); p < 0.001 SF-36 – physical component score Mean (median) % changes from baseline: Etanercept 25 mg, 4 weeks 5.8 (5.1); placebo 4 weeks 0.5 (0.7); p < 0.001 Etanercept 25 mg, 12 weeks 8.9 (6.8); placebo 12 weeks 1.2 (1.6); p < 0.001 Etanercept 25 mg, 24 weeks 9.3 (7.7); placebo 24 weeks 0.7 (0.5); p < 0.001 STAGE 1: ADVERSE EVENTS Infectious adverse events [n, (%) – after 24 weeks] [Etanercept (E), n = 101; placebo (P), n = 104] Any infection: P, 40 (40%); E, 45 (43%) Upper respiratory infection: P, 21 (21%): E, 24 (23%) Sinusitis: P, 6 (6%); E, 8 (8%) Urinary tract infection: P, 6 (6%); E, 6 (6%) Infections that required hospitalisation or use of i.v. antibiotics Etanercept: 0/101 Placebo: 1/104 (1 gastroenteritis) Cancer: None Reactivation of latent TB: Not reported Deaths (no. of patients) Etanercept: 0 Placebo: one – surgery complications for perforated bowel Withdrawals due to adverse events (no. of patients) Etanercept: one – elevated liver enzymes Placebo: one – increased psoriasis STAGE 2: EFFICACY OUTCOMES Not reported STAGE 3: EFFICACY OUTCOMES ACR 20/50/70 responses were maintained or improved over the open follow-up stage of the trial in those patients who had taken etanercept from baseline. Data reported in graphical form only (not extractable) Radiographic results Total Sharp Score Mean (SD) annualised rate of progression at 12 months: Etanercept (n = 101) –0.03 (CiC information has been removed); placebo (n = 104) 1.00 (CiC information has been removed); p = 0.0001 Subgroup analysis (with and without MTX) (mean, SD): (CiC information has been removed) Total Sharp Score excluding DIP joints Mean (SE) annualised rate of progression at 12 months (CiC information has been removed) Erosion score: mean rate of change (units/year): Etanercept (n = 101) –0.08; placebo (n = 104) 0.69; p = 0.0001 Joint space narrowing: mean rate of change (units/year): Etanercept (n = 101) 0.06; placebo (n = 104) 0.35; p = 0.04 PsA-specific radiographic features: (CiC information has been removed) STAGE 2: ADVERSE EVENTS (CiC information has been removed) STAGE 3: ADVERSE EVENTS (CiC information has been removed) Serious infection n = 1 (pneumonia) STAGE 2 AND STAGE 3 COMBINED: ADVERSE EVENTS (CiC information has been removed) |
Efficacy data extraction: infliximab
| Study details and design | Participant details | Intervention/outcome/analyses details | Results |
|---|---|---|---|
|
IMPACT, 2005, USA 79 – 81 , 89 , 96 , 109 , 111 , 112 – 115 , 117 , 118 Type of publication: Full publication Funding: Centocor and Schering-Plough152 Study design: Double-blind RCT with open uncontrolled extension Setting: Outpatient, multicentre Duration of follow-up: Stage 1: 16 weeks Stage 2: > 34 weeks Frequency of follow-up: Stage 1: Baseline, 2, 6, 14 and 16 weeks Stage 2: 18, 22, 30, 46 and 50 weeks Extracted by: HY Checked by: MR |
Inclusion/exclusion criteria Adults aged 18 years or above, diagnosed with PsA for at least 6 months, with negative results of the serum tests for RF. Patients must have active peripheral polyarticular arthritis (defined as five or more swollen and tender joints), with at least one of the following criteria: ESR ≥ 28 mm/hour, CRP level ≥ 15 mg/l, and/or morning stiffness lasting 45 minutes or longer. Patients must have failed to the treatment of at least one DMARD No. randomised: 104 Mean age (SD): Infliximab: 45.7 years (11.1) Placebo: 45.2 years (9.7) Gender (% male): Infliximab: 30/52 (57.7%) Placebo: 30/52 (57.7%) PsA history: Mean (SD) duration: Infliximab: 11.7 years (9.8) Placebo: 11.0 years (6.6) Psoriasis history Mean (SD) duration: Infliximab: 16.9 years (10.9) Placebo: 19.4 years (11.6) Psoriasis evaluation Patients with baseline PASI ≥ 2.5: Infliximab: 22/52 Placebo: 17/52 |
Intervention: infliximab Dose regimen: 5 mg/kg at weeks 0, 2, 6 and 14 Length of treatment: 16 weeks No. randomised: 52 No. completed: 49 Comparator: Placebo Dose regimen: Equivalent Length of treatment: 16 weeks No. randomised: 52 No. completed: 50 Patients in the placebo group in Stage 1 received 5 mg/kg of infliximab at weeks 16, 18, 22, 30, 38 and 46. Patients that were in the infliximab group in Stage 1 received placebo at weeks 16 and 18, and 5 mg/kg of infliximab at weeks 22, 30, 38 and 46 Primary outcome ACR 20 at week16 Sample size calculation Assuming an ACR 20 rate of 50% on infliximab and 20% on placebo, a sample size of 45 patients per group gave 80% power to detect a significant difference between treatments on the primary outcome, with α = 0.05 (two-sided) Statistical analyses Categorical outcomes (including ACR 20) were compared using the chi-squared test The Mantel–Haenszel test was conducted to estimate the ORs of the two treatment groups. Continuous outcomes were analysed using one-way ANOVA |
STAGE 1: EFFICACY OUTCOMES ACR 20 Infliximab 14 weeks 67.3% (35/52); placebo 14 weeks 11.5% (6/52); p < 0.01 Infliximab 16 weeks 65.4% (34/52); placebo 16 weeks 9.6% (5/52); p < 0.001 ACR 50 Infliximab 14 weeks 36.5% (19/52); placebo 14 weeks 1.9% (1/52); p < 0.01 Infliximab 16 weeks 46.2% (24/52); placebo 16 weeks 0% (0/52); p < 0.001 ACR 70 Infliximab 14 weeks 21.2% (11/52); placebo 14 weeks 0% (0/52); p < 0.01 Infliximab 16 weeks 28.8% (15/52); placebo 16 weeks 0% (0/52); p < 0.001 PsARC Infliximab 14 weeks 76.9% (40/52); placebo 14 weeks 13.5% (7/52); p < 0.01 Infliximab 16 weeks 75% (39/52); placebo 16 weeks 21% (11/52); p < 0.001 HAQ (mean, SD) Infliximab baseline: 1.2 (0.7); placebo baseline = 1.2 (0.7) (CiC information has been removed) HAQ mean (SE) % change from baseline Infliximab 16 weeks (n = 48) –49.8 (8.2); placebo 16 weeks (n = 47) 1.6 (8.3) Mean (SD) PASI at baseline for all patients measured Infliximab (n = 52): 5.1 (5.9); placebo (n = 52) = 4.2 (5.8) (CiC information has been removed) PASI 50 Infliximab 16 weeks 100% (22/22); placebo 16 weeks 0% (0/16) PASI 75 Infliximab 16 weeks 68.2% (15/22); placebo 16 weeks 0% (0/16) PASI 90 Infliximab 16 weeks 36.4% (8/22); placebo 16 weeks 0% (0/16) Patient global assessment of disease mean (SE) Infliximab 16 weeks –47.5 (7.4); placebo 16 weeks 13.9 (7.5); p < 0.001 Physician global assessment of disease mean (SE) Infliximab 16 weeks –58.4 (6.0); placebo 16 weeks 4.7 (6.0); p < 0.001 STAGE 1: ADVERSE EVENTS Infectious adverse events including any serious infections (Placebo, P; infliximab, I) Bronchitis: P, 4/51 (7.8%); I, 3/52 (5.8%) Rhinitis: P, 2/51 (3.9%); I, 3/52 (5.7%) Upper respiratory tract infection: P, 5/51 (9.8%); I, 1/52 (1.9%) |
|
Concurrent therapies: Patients receiving one on the following DMARDs were eligible; MTX, leflunomide, sulfasalazine, hydroxychloroquine, intramuscular gold, penicillamine, and azathioprine. Patients receiving a DMARD must have received a stable dosage for at least 4 weeks prior to the trial and throughout the investigation. Dosages of corticosteroids and NSAIDs were permitted to remain stable throughout the study if the dosages had been stable for at least 2 weeks prior to screening. Stable dose of topical treatment for psoriatic lesions (e.g. topical steroids) were also permitted. Therapy with PUVA was not permitted. Patients could not receive any investigational drug within 3 months of screening or any previous treatment with a monoclonal antibody or fusion protein Concomitant therapy at baseline Concomitant DMARD at baseline: Placebo 41/52 (79%) Infliximab 33/52 (63%) Note: the most commonly used DMARD was MTX |
ITT analysis The analyses were performed on an ITT basis |
Infections that required hospitalisation or use of i.v. antibiotics: Not reported Non-infectious adverse events Infliximab: one – synovitis (culture negative) Placebo: one – rectal bleeding due to diverticulitis Cancer: None Reactivation of latent TB: None Deaths: Not reported Withdrawals due to adverse events (no. of patients): Not reported STAGE 2: EFFICACY OUTCOMES ACR 20 response Infliximab 18 weeks 77.6% (38/49); placebo/infliximab 18 weeks 52.0% (26/50) Infliximab 22 weeks 71.4% (35/49); placebo/infliximab 22 weeks 62.0% (31/50) Infliximab 30 weeks 65.3% (32/49); placebo/infliximab 30 weeks 66.0% (33/50) Infliximab 38 weeks 57.1% (28/49); placebo/infliximab 38 weeks 62.0% (31/50) Infliximab 46 weeks 57.1% (28/49); placebo/infliximab 46 weeks 66.0% (33/50) Infliximab 50 weeks 69.4% (34/49); placebo/infliximab 50 weeks 68.0% (34/50) Subgroup results (baseline MTX or no baseline MTX) at 50 weeks: (CiC information has been removed) ACR 50 response Infliximab 18 weeks 49.0% (24/49); placebo/infliximab 18 weeks 26.0% (13/50) Infliximab 22 weeks 38.8% (19/49); placebo/infliximab 22 weeks 36.0% (18/50) Infliximab 30 weeks 42.9% (21/49); placebo/infliximab 30 weeks 44.0% (22/50) Infliximab 38 weeks 40.8% (20/49); placebo/infliximab 38 weeks 48.0% (24/50) Infliximab 46 weeks 49.0% (24/49); placebo/infliximab 46 weeks 46.0% (23/50) Infliximab 50 weeks 53.1% (26/49); placebo/infliximab 50 weeks 42.0% (21/50) ACR 70 response Infliximab 18 weeks 28.6% (14/49); placebo/infliximab 18 weeks 8.0% (4/50) Infliximab 22 weeks 22.4% (11/49); placebo/infliximab 22 weeks 20.0% (10/50) Infliximab 30 weeks 26.5% (13/49); placebo/infliximab 30 weeks 22.0% (11/50) Infliximab 38 weeks 26.5% (13/49); placebo/infliximab 38 weeks 28.0% (14/50) Infliximab 46 weeks 32.7% (16/49); placebo/infliximab 46 weeks 24.0% (12/50) Infliximab 50 weeks 38.8% (19/49); placebo/infliximab 50 weeks 34.0% (17/50) Mean (SD) % ACR improvement (CiC information has been removed) PsARC Infliximab 18 weeks 81.6% (40/49); placebo/infliximab 18 weeks 70.0% (35/50) Infliximab 22 weeks 77.6% (38/49); placebo/infliximab 22 weeks 74.0% (37/50) Infliximab 30 weeks 73.5% (36/49); placebo/infliximab 30 weeks 78.0% (39/50) Infliximab 38 weeks 71.4% (35/49); placebo/infliximab 38 weeks 82.0% (41/50) Infliximab 46 weeks 69.4% (34/49); placebo/infliximab 46 weeks 74.0% (37/50) Infliximab 50 weeks 73.5% (36/49); placebo/infliximab 50 weeks 76.0% (38/50) HAQ (0–3) (CiC information has been removed) HAQ (0–3) mean (SE) % change from baseline Infliximab 50 weeks (n = 45) –42.5 (8.8); (CiC information has been removed) Change in PASI mean (SE) change from baseline Infliximab 50 weeks (n = 35) –4.8 (1.0); placebo/infliximab 50 weeks (n = 37) –2.7 (1.0) PASI 50 Infliximab 86.3% (19/22); placebo/infliximab 68.8% (11/16) PASI 75 Infliximab 59.1% (13/22); placebo/infliximab 50% (8/16) PASI 90 Infliximab 40.9% (9/22); placebo/infliximab 37.5% (6/16) Mean (SD) total modified van der Heijde–Sharp score Baseline: Infliximab (n = 37), 69.2 (94.9); placebo/infliximab (n = 35), 32.3 (39.7) Week 50 change from baseline: Infliximab (n = 37), –1.52 (NR); placebo/infliximab (n = 33), –1.95 (NR); combined (n = 70) –1.72 (5.82) STAGE 2: ADVERSE EVENTS (CiC information has been removed) Serious infection: one patient on infliximab/placebo – Salmonella infection |
|
|
IMPACT 2, 2005, USA 82 , 90 , 91 , 95 , 98 , 106 , 112 , 116 Type of publication: Full publication Funding: Centocor and Schering-Plough152 Study design: Double-blind RCT and open-label extension Setting: Outpatient, multicentre Duration of follow-up: Stage 1: 24 weeks RCT Stage 2: Open-label follow-up to 54 weeks Frequency of follow-up: Baseline, 2, 6, 14, 24 and 54 weeks Extracted by: HY Checked by: MR |
Inclusion/exclusion criteria Adult patients diagnosed with active PsA at least 6 months before the first infusion of infliximab, with five or more swollen and tender joints and either CRP of ≥ 15 mg/l and/or morning stiffness lasting 45 minutes or longer. Patient must have had an inadequate response to current or previous DMARDs or NSAIDs. Patient had a negative RF and active plaque psoriasis with at least one qualifying target lesion (≥ 2-cm diameter) No. randomised: 200 Mean age (SD) Infliximab: 47.1 years (12.8) Placebo: 46.5 years (11.3) Gender (% male) Infliximab: 71% Placebo: 51% PsA history Mean (SD) duration: Infliximab: 8.4 years (7.2) Placebo: 7.5 years (7.8) Psoriasis history Mean (SD) duration: Infliximab: 16.8 years (12.0) Placebo: 16.2 years (11.0) Psoriasis evaluation Patients with ≥ 3% BSA affected with psoriasis: Infliximab: 83/100 (83%) Placebo: 87/100 (87%) |
Intervention: infliximab Dose regimen: 5 mg/kg at weeks 0, 2, 6, 14, and 22 Length of treatment: 24 weeks No. randomised: 100 No. completed: 93 Comparator: placebo Dose regimen: equivalent Length of treatment: 24 weeks No. randomised: 100 No. completed: 92 Further infusions of infliximab were administered to all patients in an open-label fashion (timing dependent upon whether they were originally randomised to infliximab, or crossed over from placebo at either week 16 or 24) with further follow-up at week 54 Primary outcome ACR 20 at week 14 Sample size calculation: Assuming that an ACR 20 rate of 42% on infliximab and 20% on placebo, a sample size of 100 patients per group gives 90% power to detect a significant difference between treatments on the primary outcome, with α = 0.05 (two-sided). Statistical analyses Cochran–Mantel–Haenszel chi-squared test stratified by baseline MTX use was used to analyse categorical outcomes. A two-sided F-test using ANOVA with baseline MTX as a factor was used to analyse continuous data. The LOCF approach was used for imputing missing data ITT analysis The analyses were performed on an ITT basis |
STAGE 1: EFFICACY OUTCOMES ACR 20 Infliximab 14 weeks: 58% (58/100); placebo 14 weeks: 11% (11/100); p < 0.001 Infliximab 24 weeks: 54% (54/100); placebo 24 weeks = 16% (16/100); p < 0.001 ACR 50 Infliximab 14 weeks: 36% (36/100); placebo 14 weeks = 3% (3/100); p < 0.001 Infliximab 24 weeks: 41% (41/100); placebo 24 weeks: 4% (4/100); p < 0.001 ACR 70 Infliximab 14 weeks: 15% (15/100); placebo 14 weeks = 1% (1/100); p < 0.001 Infliximab 24 weeks: 27% (27/100); placebo 24 weeks: 2% (2/100); p < 0.001 PsARC Infliximab 14 weeks: 77% (77/100); placebo 14 weeks: 27% (27/100); p < 0.001 Infliximab: 24 weeks = 70% (70/100); placebo 24 weeks: 32% (32/100); p < 0.001 Mean (SD) HAQ at baseline Infliximab = 1.1 (0.6); placebo = 1.1 (0.6) HAQ % change from baseline (SD) Infliximab 14 weeks: 48.6 (43.3); placebo 14 weeks: –18.4 (90.5); p < 0.001 Infliximab 24 weeks: 46.0 (42.5); placebo 24 weeks: –19.4 (102.8); p < 0.001 HAQ improvement (≥ 0.3 decrease) Infliximab 14 weeks: 59%; placebo 14 weeks: 19%; p < 0.001 Infliximab 24 weeks: 52%; placebo 24 weeks: 20%; p < 0.001 PASI 50 (in patients with ≥ 3% BSA psoriasis) Infliximab 14 weeks: 82% (68/83); placebo 14 weeks: 9% (8/87); p < 0.001 Infliximab 24 weeks: 75% (62/83); placebo 24 weeks: 8% (7/87); p < 0.001 PASI 75 (in patients with ≥ 3% BSA psoriasis) Infliximab 14 weeks: 64% (53/83); placebo 14 weeks: 2% (2/87); p < 0.001 Infliximab 24 weeks: 60% (50/83); placebo 24 weeks: 1% (1/87); p < 0.001 PASI 90 (in patients with ≥ 3% BSA psoriasis) Infliximab 14 weeks: 41% (34/83); placebo 14 weeks: 0% (0/87); p < 0.001 Infliximab 24 weeks: 39% (32/83); placebo 24 weeks: 0% (0/87); p < 0.001 PASI 50 (in patients with PASI ≥ 2.5 at baseline) (CiC information has been removed) PASI 75 (in patients with PASI ≥ 2.5 at baseline) (CiC information has been removed) PASI 90 (in patients with PASI ≥ 2.5 at baseline) (CiC information has been removed) |
| Concurrent therapies: Concomitant MTX (up to 25 mg/week) was permitted at least 3 months prior to the first infusion and was maintained at a stable dose for at least 4 weeks prior to first infusion. A stable dose ( 10 mg) of oral prednisone was permitted. DMARDs or intra-articular corticosteroids were prohibited within 4 weeks before the first infusion. DMARDs other than MTX were not permitted during the trial. Systematic or topical treatment for psoriasis was not permitted (except for low potency topical corticosteroids on face or groin) |
Mean (SD) SF-36 at baseline Physical component: Infliximab = 33.0 (9.4); placebo = 31.0 (9.0) Mental component: Infliximab = 45.5 (11.9); placebo = 47.0 (11.9) SF-36 mean change from baseline (SD) Physical component: Infliximab 14 weeks: 9.1 (9.3); placebo 14 weeks = 1.1 (8.4); p < 0.001 Infliximab 24 weeks: 7.7 (9.8); placebo 24 weeks: 1.3 (8.2); p = 0.001 Mental component: Infliximab 14 weeks: 3.8 (11.1); placebo 14 weeks: –1.2 (9.3); p < 0.001 Infliximab 24 weeks: 3.9 (11.9); placebo 24 weeks: 0.4 (11.6); p = 0.05 Mean (SD) total modified van der Heijde–Sharp score Week 24 change from baseline: Infliximab –0.70 (2.53); placebo 0.82 (2.62) STAGE 1: ADVERSE EVENTS Infectious adverse events, including any serious infections (up to week 24) [Placebo (P), n = 97; infliximab (I), all patients who received an infliximab dose, n = 150] Upper respiratory tract infection: P, 14 (14%); I, 15 (10%) Pharyngitis: P, 4 (4%); I, 8 (5%) Sinusitis: P, 4 (4%); I, 8 (5%) Infections that required hospitalisation or use of i.v. antibiotics: Not reported Malignancy Placebo: one – basal cell carcinoma of skin Infliximab: 0 Reactivation of latent TB: None Deaths: None Total serious adverse events Placebo: 6 (6%) Infliximab: 13 (9%) Withdrawals due to adverse events (no. of patients) Infliximab: 6 Placebo: 1 STAGE 2: EFFICACY OUTCOMES PsARC Infliximab 54 weeks: 74.4% (67/90); placebo/infliximab 54 weeks: 81.9% (68/83) PASI 50 (in patients with ≥ 3% BSA psoriasis) Infliximab 54 weeks: 69.5% (57/82); placebo/infliximab 54 weeks: 80% (64/80) PASI 75 (in patients with ≥ 3% BSA psoriasis) Infliximab 54 weeks: 48.8% (40/82); placebo/infliximab 54 weeks: 58.8% (47/80) PASI 90 (in patients with ≥ 3% BSA psoriasis) Infliximab 54 weeks: 39% (32/82); placebo/infliximab 54 weeks: 81.9% (68/80) Mean (SD) total modified van der Heijde–Sharp score Baseline: Infliximab 30.3 (61.4); placebo/infliximab 39.1 (82.8) Week 54 change from baseline: Infliximab –0.94 (3.4); placebo/infliximab 0.53 (2.6) STAGE 2: ADVERSE EVENTS Infectious adverse events including any serious infections (through week 54) Combined infliximab/placebo (all who received an infliximab dose, n ≥ 173) (CiC information has been removed) Infections that required hospitalisation or use of i.v. antibiotics: Not reported Malignancy: Two (one basal cell carcinoma, one Hodgkin’s lymphoma) Reactivation of latent TB: None Deaths: None Total serious adverse events: 22 (11.5%) Withdrawals due to adverse events (no. of patients): 16 (8.4%) |
Efficacy data extraction: adalimumab
| Study details and design | Participant details | Intervention/outcome/analyses details | Results |
|---|---|---|---|
|
ADEPT 2005, USA 51 , 88 , 92 , 93 , 100 – 104 Type of publication: Full publication Funding: Abbott Study design: Stage 1: Double-blind RCT Stage 2: Open-label extension Setting: Outpatient Duration of follow-up: Stage 1: 24 weeks Stage 2: 24–144 weeks Frequency of follow-up: Baseline, 2, 4, 8, 12, 16, 20 and 24 weeks Extracted by: HY Checked by: MR |
Inclusion/exclusion criteria: Adults aged 18 years or above diagnosed with moderately or severely PsA (defined as ≥ 3 swollen and tender or painful joints). Patients must have either active psoriatic skin lesions or a documented history of psoriasis, with an adequate response or intolerance to NSAIDs. Patients were excluded if they had the following treatment: (1) within 4 weeks of the baseline visit with ciclosporin, tacrolimus, DMARDs other than MTX, or oral retinoids; (2) topical therapy for psoriasis within 2 weeks of baseline, other than medicated shampoos or low-potency topical steroids; (3) concurrent therapy with MTX at dosage > 30 mg/week and/or corticosteroids in a prednisone-equivalent dosage of > 10 mg/day; and (4) biologic therapy at any time No. randomised: 315 Mean age (SD) Adalimumab: 48.6 years (12.5) Placebo: 49.2 years (11.1) Gender (% male) Adalimumab: 85/151(56.3%) Placebo: 89/162 (54.9%) PsA history Mean (SD) duration: Adalimumab: 9.8 years (8.3) Placebo: 9.2 years (8.7) |
Intervention: adalimumab Dose regimen: 40 mg every other week Length of treatment: 24 weeks No. randomised: 153 No. completed: 140 Comparator: placebo Dose regimen: Equivalent Length of treatment: 24 weeks No. randomised: 162 No. completed: 149 Primary outcome ACR 20 at week 12 and the change in TSS of structural damage on radiographs of the hands and feet at week 24 Sample size calculation Assuming that the effect size of anticipated change in the modified TSS is 0.325, the sample size of 150 per treatment group gave 80% power to detect a significant difference between treatments on this primary outcome, with α = 0.05 (two-sided) Statistical analyses Proportions of patients responding were compared using the Cochran–Mantel–Haenszel mean score test adjusted for the MTX use. Continuous data were analysed by ANOVA with factors of treatment, baseline, MTX use and extent of psoriasis. Non-responder imputation was used, in which participants who discontinued or had missing data were counted as non-responders. Patients who received rescue therapy were considered to be non-responders at the time that rescue therapy was initiated |
STAGE 1: EFFICACY OUTCOMES ACR 20 Adalimumab 12 weeks: 58% (88/151); placebo 12 weeks:14% (23/162); p < 0.001 Adalimumab 24 weeks: 57% (86/151); placebo 24 weeks: 15% (24/162); p < 0.001 Adalimumab + MTX 12 weeks: 55% (42/77); adalimumab alone 12 weeks: 61% (45/74); p = 0.511 Adalimumab + MTX 24 weeks: 55% (42/77); adalimumab alone 24 weeks: 59% (44/74), p = 0.622 ACR 50 Adalimumab 12 weeks: 36% (54/151); placebo 12 weeks: 4% (6/162); p < 0.001 Adalimumab 24 weeks: 39% (59/151); placebo 24 weeks: 6% (10/162); p < 0.001 Adalimumab + MTX 12 weeks: 36% (28/77); Adalimumab alone 12 weeks: 36% (27/74), p > 0.999 Adalimumab + MTX 24 weeks: 36% (28/77); Adalimumab alone 24 weeks: 42% (31/74), p = 0.509 ACR 70 Adalimumab 12 weeks: 20% (30/151); placebo 12 weeks: 1% (1/162); p < 0.001 Adalimumab 24 weeks: 23% (35/151); placebo 24 weeks: 1% (1/162); p < 0.001 Adalimumab + MTX 12 weeks: 17% (13/77); adalimumab alone 12 weeks: 23% (17/74); p = 0.416 Adalimumab + MTX 24 weeks: 22% (17/77); adalimumab alone 24 weeks: 23% (17/74); p > 0.999 PsARC Adalimumab 12 weeks: 62% (94/151); placebo 12 weeks: 26% (42/162) Adalimumab 24 weeks: 60% (91/151); placebo 24 weeks: 23% (37/162) Mean HAQ at baseline (SD) Adalimumab: 1.0 (0.6); placebo: 1.0 (0.7) HAQ mean change from baseline (SD) Adalimumab 12 weeks: –0.4(0.5); placebo 12 weeks: –0.1(0.5); p < 0.001 Adalimumab 24 weeks: –0.4(0.5); placebo 24 weeks: –0.1(0.4); p < 0.001 Adalimumab + MTX 12 weeks: –0.3 (0.4); adalimumab alone 12 weeks: –0.4 (0.5); p = 0.188 Adalimumab+ MTX 24 weeks: –0.4 (0.5); adalimumab alone 24 weeks: –0.4 (0.5); p = 0.690 |
|
Psoriasis history Mean (SD) duration: Adalimumab: 17.2 years (12) Placebo: 17.1 years (12.6) Psoriasis evaluation Patients with > 3% BSA affected with psoriasis: Adalimumab: 70/151 (46.4%) Placebo: 70/162 (43.2%) Concurrent therapies MTX use was permitted if it had been taken for ≥ 3 months previously, with a stable dose for ≥ 4 weeks prior to the trial Concomitant therapy at baseline Concomitant MTX at baseline: Adalimumab 77/151 (51%) Placebo 81/162 (50%) |
ITT analysis The analyses were performed on an ITT basis |
12-week HAQ mean change conditional on PsARC response at 12 weeks PsARC responders: Adalimumab (n = 93): –0.5 (0.4); placebo (n = 42): –0.3 (0.5) PsARC non-responders: Adalimumab (n = 58): –0.1 (0.4); placebo (n = 120): –0.0 (0.4) 24 week HAQ mean change conditional on PsARC response at 12 weeks PsARC responders: Adalimumab (n = 90): –0.5 (0.49); placebo (n = 37): –0.3 (0.49) PsARC non-responders: Adalimumab (n = 61): –0.1 (0.39); placebo (n = 125): –0.1 (0.39) Mean PASI at baseline (SD) Adalimumab: 7.4 (6.0); placebo: 8.3 (7.2) PASI 50 Adalimumab 12 weeks: 72% (50/69); placebo 12 weeks: 15% (10/69); p < 0.001 Adalimumab 24 weeks: 75% (52/69); placebo 24 weeks: 12% (8/69); p < 0.001 Adalimumab + MTX 12 weeks: 76% (17/29); adalimumab alone 12 weeks: 70% (28/40); p = 0.785 Adalimumab + MTX 24 weeks: 86% (25/29); adalimumab alone 24 weeks: 68% (27/40); p = 0.094 PASI 75 Adalimumab 12 weeks: 49% (34/69); placebo 12 weeks: 4% (3/69); p < 0.001 Adalimumab 24 weeks: 59% (41/69); placebo 24 weeks: 1% (1/69); p < 0.001 Adalimumab + MTX 12 weeks: 59% (17/29); adalimumab alone 12 weeks: 43% (17/40); p = 0.227 Adalimumab + MTX 24 weeks: 72% (21/29); adalimumab alone 24 weeks: 50% (20/40); p = 0.083 PASI 90 Adalimumab 12 weeks: 30% (21/69); placebo 12 weeks: 0% (0/69); p < 0.001 Adalimumab 24 weeks: 42% (29/69); placebo 24 weeks: 0% (0/69); p < 0.001 Adalimumab + MTX 12 weeks: 38% (11/29); adalimumab alone 12 weeks: 25% (10/40); p = 0.295 Adalimumab + MTX 24 weeks: 52% (/1529); adalimumab alone 24 weeks: 35% (14/40); p = 0 Concurrent joint and skin response (PsARC and PASI 75) Adalimumab 12 weeks: 42% (29/69); placebo 12 weeks: 1% (1/69); p < 0.001 Adalimumab 24 weeks: 42% (29/69); placebo 24 weeks: 0% (0/69); p < 0.001 TSS change from baseline Adalimumab 24 weeks: –0.2 (n = 144); placebo 24 weeks: 0.1 (n = 152); p < 0.001 SF-36 mean change from baseline (SD) Physical component summary: Adalimumab baseline: 33.2 (9.9); placebo baseline: 33.3 (9.8); p < 0.001 Change, adalimumab 12 weeks: 9.3 (10.0); placebo 12 weeks: 1.4 (8.7); p < 0.001 Change, adalimumab 24 weeks: 9.3 (10.1); placebo 24 weeks: 1.4 (9.6); p < 0.001 Mental component summary: Adalimumab baseline: 48.1 (10.2); placebo baseline: 46.6 (12.2); p < 0.001 Change, adalimumab 12 weeks: 1.6 (10.1); placebo 12 weeks: 1.2 (10.2); p = 0.71 Change, adalimumab 24 weeks: 1.8 (9.3); placebo 24 weeks: 0.6 (10.4); p = 0.29 STAGE 1: ADVERSE EVENTS Infectious adverse events including any serious infections (Placebo, P; adalimumab, A) Upper respiratory tract infection: P, 24/162 (14.8%); A 19/151 (12.6%) Nasopharyngitis: P, 15/162 (9.3%); A 15/151 (9.9%) Diarrhoea: P, 9/162 (5.6%); A, 3/151 (2.0%) Infections that required hospitalisation or use of i.v. antibiotics Adalimumab: 1/151 (one – viral meningitis) Placebo: 2/162 (one – pericarditis, one – cellulitis) Malignancy: None Reactivation of latent TB: Not reported Deaths: None Withdrawals due to adverse events (no. of patients) Adalimumab: 3 Placebo: 1 STAGE 2: EFFICACY OUTCOMES (24–144 WEEKS) ACR 20 Adalimumab 48 weeks: 58.7% (165/281) Adalimumab 104 weeks: 57.3% (161/281) ACR 50 Adalimumab 48 weeks: 42.7% (120/281) Adalimumab 104 weeks: 45.2% (127/281) ACR 70 Adalimumab 48 weeks: 27.8% (78/281) Adalimumab 104 weeks: 29.9% (84/281) HAQ mean change from baseline (SD) Adalimumab (n = 298) 48 weeks: –0.3 (0.5) Adalimumab (n = 271) 104 weeks: –0.3 (0.5) HAQ percentage change from baseline (SD) Adalimumab 48 weeks: –41.9% (114/271) Adalimumab 104 weeks: –42.7% (116/271) Mean changes in modified TSS Adalimumab (n = 115) 48 weeks: 0.1 (1.95); adalimumab/placebo (n = 128) 48 weeks: 0.8 (4.23) Adalimumab (n = 115) 144 weeks: 0.5 (4.20); adalimumab/placebo (n = 128) 144 weeks: 0.9 (6.36) Percentage changes (increase) in modified TSS Adalimumab 48 weeks: 26.6% (34/115); adalimumab/placebo 48 weeks: 11.3% (13/128) Adalimumab 144 weeks: 20.9% (24/115); adalimumab/placebo 144 weeks: 31.3% (40/128) PASI 50 Adalimumab 48 weeks: 67% (46/69); adalimumab/placebo 48 weeks: 61% (42/69) PASI 75 Adalimumab 48 weeks: 58% (40/69); adalimumab/placebo 48 weeks: 53% (37/69) PASI 90 Adalimumab 48 weeks: 46% (32/69); adalimumab/placebo 48 weeks: 44% (30/69) STAGE 2: ADVERSE EVENTS (24–144 WEEKS) Any serious adverse events Adalimumab exposure: 16.8% (50/298) Infections that required hospitalisation or use of i.v. antibiotics Adalimumab exposure: 5% (15/298) Cancer Any malignancies: 1.3% (4/298) Lymphoma: 0.3% (1/298) Non-melanoma skin cancers: 0.7% (2/298) Other malignancies: 0.3% (1/298) Reactivation of latent TB Adalimumab exposure: 0.3% (1/298) Deaths Adalimumab exposure: 1.0% (3/298) Withdrawals due to AEs (no. of patients) Adalimumab exposure: 6.7% (20/298) |
|
|
Genovese, 2007, USA 83 Type of publication: Full publication Funding: Abbott Study design Stage 1: Double-blind RCT Stage 2: Open-label extension Setting: Outpatient Duration of follow-up Stage 1: 0–12 weeks Stage 2: 12–24 weeks Frequency of follow-up Baseline, 2, 4, 8, 12, 14, 18 and 24 weeks Extracted by: HY Checked by: MR |
Inclusion/exclusion criteria Adults aged 18 years or above had generally good health based on medical history, physical examination, laboratory profile, chest radiograph, and 12-lead electrocardiogram. Patient must have three or more swollen and tender or painful joints, and either an active cutaneous lesion of chronic plague psoriasis or a documented history of chronic plague psoriasis. All patients received concomitant DMARD therapy or had a history of DMARD therapy with an inadequate response Patients were excluded if they had the following treatment: (1) previous biologic therapy; (2) i.v. infusion or intra-articular injections of corticosteroids within 4 weeks of baseline; (3) topical psoriasis therapies within 2 weeks of baseline; (4) UVA phototherapy or use of tanning booth within 2 weeks of baseline; (5) oral retinoids within 4 weeks of the baseline visit, alefacept or siplizumab within 12 weeks, or any other biologic or investigational therapy within 6 weeks of the baseline visit; and (6) antiretroviral therapy at any time No. randomised: 102 Mean age (SD) Adalimumab: 50.4 years (11.0); Placebo: 47.7 years (11.3) Gender Adalimumab: Male 29/51 (56.9%) Placebo: Male 25/49 (51%) PsA history Mean (SD) duration: Adalimumab: 7.5 years (7.0) Placebo: 7.2 years (7.0) Psoriasis history Mean (SD) duration: Adalimumab: 18.0 years (13.2) Placebo: 13.8 years (10.7) Psoriasis evaluation (CiC information has been removed) Concurrent therapies: All patients were permitted to use concomitant DMARD therapy or had a history of DMARD therapy with an inadequate response. Oral corticosteroids were permitted to use if the dosage did not exceed the equivalent of prednisone 10 mg/day and had been stable during the 4 weeks prior to the trial. Concomitant treatments with MTX or other DMARD, with the exception of ciclosporin and tacrolimus received within 4 weeks of the baseline visit, were permitted if the patient had received a minimum of 3 months of therapy and the dosage was stable during the 4 weeks prior to the trial. The maximum allowable MTX dosage was 30 mg/week Concomitant therapy at baseline Concomitant MTX at baseline: Adalimumab 24/51 (47.1%) Placebo 23/49 (46.9%) |
Intervention: adalimumab Dose regimen: 40 mg every other week Length of treatment: 12 weeks No. randomised: 51 No. completed: 50 Comparator: Placebo Dose regimen: Equivalent Length of treatment: 12 weeks No. randomised: 51 No. completed: 46 Primary outcome ACR 20% criteria for improvement in RA (ACR 20) at week 12 Sample size calculation: Assuming that a response rate of 25% on placebo and 60% on adalimumab, the sample size of 50 patients per groups gave 90% power to detect a significant difference between treatments on the primary outcome, with α = 0.05 (two-sided) Statistical analyses: Proportions of patients responding were compared using the Cochran–Mantel–Haenszel test, with baseline DMARD use as the stratification factor. ACR 20 at response rates at time points except for week 12, and ACR 50 and ACR 70 rates at all time points were analysed using Fisher’s exact test, combining baseline DMARD use categories. Continuous data were analysed using ANOVA with factors of baseline DMARD use and treatment. Non-responder imputation for missing data was used for analyses of ACR and PsARC responses, and LOCF was used for all other efficacy measures ITT analysis The analyses were performed on an ITT basis |
STAGE 1: EFFICACY OUTCOMES ACR 20 Adalimumab 12 weeks: 39% (20/51); placebo 12 weeks: 16% (8/49); p < 0.05 ACR 50 Adalimumab 12 weeks: 25% (13/51); placebo 12 weeks: 2% (1/49); p < 0.001 ACR 70 Adalimumab 12 weeks: 14% (7/51); placebo 12 weeks: 0% (0/49); p < 0.05 PsARC Adalimumab 12 weeks: 51% (26/51); placebo 12 weeks: 24% (12/49); p = 0.007 Mean HAQ at baseline (SD) Adalimumab: 0.9(0.5); placebo: 1.0(0.7) HAQ mean change from baseline (SD) Adalimumab 12 weeks: –0.3(0.5); placebo 12 weeks: –0.1(0.3); p < 0.01 12-week HAQ mean change conditional on PsARC response at 12 weeks PsARC responders: Adalimumab (n = 26): –0.4 (0.4); placebo (n = 12): –0.2 (0.3) PsARC non-responders: Adalimumab (n = 26): –0.1 (0.4); placebo (n = 12): –0.1 (0.3) Patient global assessment of disease activity (improvement from baseline) Adalimumab 12 weeks: –14.8 (24.5); placebo 12 weeks: –0.4 (24.9); p < 0.004 Physician global assessment of disease activity (improvement from baseline) Adalimumab 12 weeks: –21.4 (22.4); placebo 12 weeks: –9.7 (18.2); p < 0.005 Physician global assessment for psoriasis (‘clear’ or ‘minimal’) Adalimumab 12 weeks: 40.6% (13/32); placebo 12 weeks: 6.7% (2/30); p < 0.002 Target lesion score mean change from baseline (SD) Adalimumab 12 weeks: –3.7 (3.3); placebo 12 weeks: –0.3 (3.1); p < 0.001 Mean (SD) SF-36 at baseline Physical component summary: Adalimumab: 34.9 (9.2); placebo: 32.7 (11.3) Mental component summary: Adalimumab: 48.1 (10.2); placebo: 46.6 (10.2) SF-36 mean change from baseline (SD) Physical component summary: Adalimumab 12 weeks: 5.7 (8.5); placebo 12 weeks: 2.8 (7.1); p = 0.08. Mental component summary: Adalimumab 12 weeks: 1.1 (7.4); placebo 12 weeks: –0.6 (7.8); p = 0.24 DLQI mean change from baseline (SD) Adalimumab 12 weeks: –3.4 (4.5); placebo 12 weeks: –1.7 (5.3); p = 0.171 STAGE 1: ADVERSE EVENTS Infectious adverse events including any serious infections (Placebo, P; adalimumab, A) Any infectious adverse events: P, 16/49 (32.7%); A, 9/51 (17.6%) Upper respiratory tract infection: P, 4/49(8.2%); A, 7/51(13.7%) Diarrhoea: P, 3/49 (6.1%); A, 1/51 (2.0%) Infections that required hospitalisation or use of i.v. antibiotics Adalimumab: 1/51 Placebo: 1/49 Non-infectious serious adverse events Adalimumab: 1/51 (diverticulitis) Placebo: 2/49 (one sublingual abscess, one benign paraganglioma neoplasm) Cancer: None Reactivation of latent TB: None Deaths: None Withdrawals due to adverse events (no. of patients) Adalimumab: 1 Placebo: 2 STAGE 2: EFFICACY OUTCOMES ACR 20 Adalimumab 24 weeks: 65% (33/51); adalimumab/placebo 24 weeks: 57% (26/46) ACR 50 Adalimumab 24 weeks: 43% (22/51); adalimumab/placebo 24 weeks: 37% (17/46) ACR 70 Adalimumab 24 weeks: 27% (13/51); adalimumab/placebo 24 weeks: 22% (10/46) PsARC Adalimumab 24 weeks: 75% (38/51); adalimumab/placebo 24 weeks: 70% (32/46) HAQ mean change from baseline (SD) Adalimumab 24 weeks: –0.3(0.5); adalimumab/placebo 24 weeks: –0.4(0.4) Physician global assessment for psoriasis (‘clear’ or ‘minimal’) Adalimumab 24 weeks: 56.3% (18/32); adalimumab/placebo 24 weeks: 50% (13/26) SF-36 mean change from baseline (SD) Physical component summary: Adalimumab 24 weeks: 8.6 (7.4); adalimumab/placebo 24 weeks: 11.7(9.1) Mental component summary: Adalimumab 24 weeks: 1.9 (8.2); adalimumab/placebo 24 weeks: 0.3 (9.7) DLQI mean change from baseline (SD) Adalimumab 24 weeks: –3.5 (5.1); adalimumab/placebo 24 weeks: –3.9 (6.4) STAGE 2: ADVERSE EVENTS (WEEKS 12–24) Infectious adverse events including any serious infections (Adalimumab/placebo) Any infectious adverse events: 29/97 (29.9%) Upper respiratory tract infection: 6/97 (6.2%) Diarrhoea: 2/97 (2.1%) Infections that required hospitalisation or use of i.v. antibiotics Adalimumab/placebo: 0% (0/97) Malignancy: Three cases (one non-Hodgkin’s lymphoma, one squamous cell carcinoma of the skin and one adenocarcinoma of the prostate) Reactivation of latent TB: None Deaths: None Withdrawals due to adverse events (no. of patients): Not reported |
Adverse events data extraction
| Study details and design | Intervention and duration of follow-up | No. of patients receiving biologics | No. of patients with any infection | Infections that required hospitalisation or use of i.v. antibiotics (no. of patients) | Malignancy (no. of patients) | TB (no. of patients) | Deaths (no. of patients) | Withdrawals due to adverse events (no. of patients) |
|---|---|---|---|---|---|---|---|---|
| Multiple biologics | ||||||||
|
Brassard 2006 135 Weeks control study |
Etanercept and infliximab 373.9 days (mean) |
Etanercept: 2349 patients with RA Infliximab: 1074 patients with RA |
NR | NR | NR |
Etanercept: 32 (1.4%) Infliximab: 19 (1.8%) |
NR | NR |
|
Carmona 2005 141 Multicenter surveillance study |
Etanercept, infliximab and adalimumab 5 years |
Total: 4092 patients of RA, AS, PsA, juvenile idiopathic arthritis and other chronic inflammatory rheumatic conditions It includes 2833 (69%) patients with RA Etanercept: 2227 Infliximab: 739 Adalimumab: 154 |
NR | NR | NR |
Infliximab: 34 (4.6%), of whom 28 had RA Etanercept: None (0%) Adalimumab: None (0%) |
One patient with TB died of liver failure | NR |
|
Curtis 2007 134 Retrospective cohort study |
Etanercept, infliximab and adalimumab 20 months (mean) |
Etanercept: 1201 Infliximab: 792 Adalimumab: 118 More than one biologic: 282 Total: 2393 patients with RA |
NR | 65 (2.7%) | NR | NR | NR | NR |
|
Dixon 2006 136 Prospective cohort study |
Etanercept, infliximab and adalimumab 1.26 years (median) |
Etanercept: 3596 Infliximab: 2878 Adalimumab: 1190 Total: 7664 patients with RA |
NR |
Etanercept: 209 (5.8%) Infliximab: 255 (8.9%) Adalimumab: 61 (5.1%) |
NR |
Etanercept: 2 (0.06%) Infliximab: 7 (0.2%) Adalimumab: 1 (0.08%) |
NR | NR |
|
Dixon 2007 147 Prospective cohort study |
Etanercept, infliximab and adalimumab 24 months |
Etanercept: 3844 Infliximab: 2944 Adalimumab: 1871 Total: 8659 patients with RA |
NR |
Etanercept: 432 (11.2%) Infliximab: 405 (13.8%) Adalimumab: 138 (7.3%) |
NR | NR | NR | NR |
|
Dreyer 2009 148 Prospective cohort study |
Etanercept, infliximab and adalimumab 6092 patient-years |
Total: 3688 | NR | NR | NR | 30 cancers in 28 patients (0.76%) | NR | NR |
|
Favalli 2009 129 Cohort study |
Etanercept, infliximab and adalimumab 24.21 months |
Etanercept: 242 Infliximab: 519 Adalimumab: 303 Total: 1064 patients with RA |
NR |
Etanercept: 11 (4.5%) Infliximab: 42 (8.1%) Adalimumab: 20 (6.6%) |
NR |
Etanercept: 1 (0.4%) Infliximab: 3 (0.6%) Adalimumab: 1 (0.3%) |
Total (all serious infection): 4 (0.4%) | NR |
|
Gomez-Reino 2003 146 Multicenter surveillance study |
Etanercept Infliximab 1.1 years (mean) |
1540 patients of RA, PsA and AS | 118 (7.6%) | 10 sepsis (0.65%) | NR |
Etanercept: 0 (0%) Infliximab: 17 (1.1%) |
Serious infection: 2 (0.1%) | NR |
|
Gomez-Reino 2007 132 Multicenter surveillance study132 |
Etanercept, infliximab and adalimumab NR |
Etanercept: 1336 Infliximab: 1137 Adalimumab: 615 Total: 3088 patients with rheumatic diseases |
NR | NR | NR |
Etanercept: 2 (0.1%) Infliximab: 5 (0.4%) Adalimumab: 1 (0.2%) |
NR | NR |
|
Listing 2005 122 Prospective cohort study |
Etanercept and infliximab 12 months |
Etanercept: 512 patients with RA Infliximab: 346 patients with RA |
Etanercept: 109 (21.3%) Infliximab: 92 (26.6%) |
Etanercept: 31 (6.1%) Infliximab: 20 (5.8%) |
NR |
Etanercept: 0 (0%) Infliximab: 1 (0.3%) |
Serious infection: 4 (0.5%) | NR |
| Etanercept | ||||||||
|
Fleischmann 2006 99 Integrated data of trials |
Etanercept NR |
Etanercept: 3132 patients with RA, PsA and AS Control: 1190 patients receiving placebo or MTX |
Etanercept: 1704 (54.4%) Control (placebo or MTX): 493 (41.4%) |
Etanercept: 155 (4.9%) Control (placebo or MTX): 25 (2.1%) |
NR | None | Etanercept and control: 41 (0.9%) |
Etanercept: 204 (6.5%) Control (placebo or MTX): 57 (4.8%) |
|
Horneff 2009 125 Open, non-randomised study |
Etanercept 12 months |
604 patients of juvenile idiopathic arthritis | 58 (9.6%) | 26 (4.3%) | NR | NR | None (0%) | NR |
|
Klareskog 2006 120 Open-label extension |
Etanercept 5 years |
549 patients with RA | 146 (26.5%) | 89 (16.2%) |
Total: 7 (1.3%) Lung cancer: 2 (0.4%) Breast cancer: 3 (0.5%) Lymphoma: 1 (0.2%) Basocellular skin cancer: 2 (0.4%) |
None |
Total: 10 (1.8%) Serious infection: 7 (1.3%) |
25 (4.6%) |
|
Mease 2006 97 Open-label extension |
Etanercept 48 weeks |
169 patients with PsA | 3 (1.8%) | 1 (0.6%) | NR | NR | None (0%) | None (0%) |
|
Moreland 2006 121 Data from RCTs or open-label extension |
Etanercept 7 years |
714 patients with RA | NR | 94 (13.2%) |
Total: 41 (5.7%) Squamous cell carcinoma of larynx: 1 Lymphoma: 7 Lung cancer: 5 Ovarian cancer: 4 Breast cancer: 3 Leukaemia: 2 Prostate cancer: 2 Malignant melanoma: 2 Squamous cell skin carcinomas: 4 Basal cell skin carcinomas: 11 |
None |
Total: 22 (3.1%) Serious infection: 2 Malignancy: 3 |
97 (13.6%; due to adverse events and deaths) |
|
Feltelius 2005 142 Nationwide postmarketing cohort study |
Etanercept 24 months |
1073 patients with RA | 120 (11%) |
Total: 28 (2.6%) Sepsis: 8 Pneumonia: 8 Osteitis: 3 Infectious arthritis: 2 Soft tissue abscess: 2 Gastroenteritis: 2 Recurrent fever: 1 Skin inflammation: 1 Encephalitis: 1 |
Total: 11 (1%) Lymphoma: 3 Benign respiratory tract neoplasm: 2 Unspecified liver cancer: 1 Primary liver cancer: 1 Benign gastrointestinal neoplasm: 1 Ovarian cancer: 1 Cervical cancer: 1 Rectal cancer: 1 |
NR |
Total: 3 (0.3%) Serious infection: 1 Malignancy: 1 |
59 (5.5%) |
| Infliximab | ||||||||
|
Antoni 2008 89 Open-label extension |
Infliximab 98 weeks |
78 patients with PsA |
URTI: 30 (38.5%) Diarrhoea: 7 (9.0%) Pharyngitis: 7 (9.0%) Sinusitis: 4 (5.1%) Urinary tract infection: 4 (5.1%) |
2 (2.6%; one knee wound, one bowel) |
Total: Four neoplasms (5.1%) Benign abdominal mucinous systoma: 1 Non-resectable pancreatic ductal adenocarcinoma: 1 Mild haemangioma: 1 Leucocythaemia: 1 |
None (0%) | NR | 5 (6.4%) |
|
Caspersen 2008 128 Cohort study |
Infliximab 6 years |
651 patients with Crohn’s disease | NR |
Total: 66 (10.1%) Abscesses: 34 Pneumonia: 16 Sepsis: 8 Pleuritis: 2 Aspergillus pneumonia: 2 Keratoconjunctivitis: 2 Bone infection in jaw: 1 Exacerbation of osteomyelitis: 1 |
Total: 4 (0.6%) Relapse of breast cancer: 1 |
2 (0.3%) |
Total: 13 (2.0%) Serious infection: 4 Malignancy: 1 |
NR |
|
Colombel 2004 124 Retrospective cohort study |
Infliximab 17 months (median) |
500 patients with Crohn’s disease | 48 (9.6%) |
Total: 15 (3.0%) Sepsis: 2 Pneumonia: 8 Histoplasmosis: 1 Viral infections: 1 Abscesses: 2 Cutaneous infections: 1 |
Total: 9 (1.8%) Cancer: 7 (two lung cancer, one abdominal carcinomatosis, two squamous cell carcinoma, two basal cell carcinoma) Non-Hodgkin’s lymphoma: 1 Hodgkin’s lymphoma: 1 |
NR |
Total: 10 (2%) Serious infection: 4 Malignancy: 2 |
NR |
|
Fidder 2009 119 Retrospective cohort study |
Infliximab 58 months (median) |
734 patients with IBD | NR | 48 (6.5%) | 21 (2.9%) | 1 (0.1%) |
Total: 12 (1.6%) Serious infection: 1 Malignancy: 3 |
NR |
|
Oka 2006 137 Postmarketing surveillance data |
Infliximab22 weeks | 5000 patients with RA | NR | Lung infections: 155 (3.1%) | NR | 14 (0.3%) |
Total: 3 (0.06%) Serious infection: 3 |
NR |
|
Schnitzler 2009 127 Retrospective cohort study |
Infliximab 55 months (median) |
614 Crohn’s disease patients | NR |
5 serious infections (0.8%):1 fatal Aspergillus, 1 abdominal TB |
1 pancreatic carcinoma (0.16%) | NR | Total: 10 (1.6%) 1 fatal Aspergillus infection | 70 (12.8%) |
|
St. Clair 2004 143 RCT |
Infliximab + MTX ~54 weeks |
749 early patients with RA |
URTI: 200 (26.7%) Sinusitis: 73 (9.7%) Pharyngitis: 103 (13.8%) |
At least 1 serious infection: 40 (5.3%) Pneumonia: 15 (2.0%) TB: 4 (0.5%) Sepsis: 3 (0.4%) Bronchitis: 2 (0.27%) Septic bursitis: 2 (0.27%) |
Total: 4 (0.5%) 1 endometrial cancer 1 pancreatic cancer 1 colon adenocarcinoma 1 acute myeloid leukaemia |
4 (0.5%) |
Total: 2 (0.27%) 1 pancreatic cancer |
69/722 (9.6%) |
|
Takeuchi 2008 130 Prospective cohort study |
Infliximab 6 months |
5000 patients with RA | Total: 433 (8.7%) |
Bacterial pneumonia: 108 (2.2%) (Suspected Pneumocytitis jirovecii pneumonia: 22 (0.4%) Interstitial pneumonitis: 25 (0.5%) |
All neoplasms: 8 (0.16%) | 14 (0.3%) | NR | NR |
|
Westhovens 2006 139 RCT |
Infliximab + MTX 22 weeks 54 weeks |
721 patients with RA at 22 weeks 1001 patients with RA at 54 weeks |
0–22 weeks URTI: 78 (10.8%) Pharyngitis: 34 (4.7%) Sinusitis: 30 (4.2%) Pneumonia: 6 (0.8%) TB: 3 (0.4%) Cellulitis: 2 (0.3%) UTI: 2 (0.3%) |
0–22 weeks Pneumonia: 6 (0.8%) TB: 3 (0.4%) Cellulitis: 2 (0.3%) UTI: 2 (0.3%) |
Total: 26 (2.6%) Details reported |
0–22 weeks 3 (0.4%) |
Total: 4 (0.4%) 1 TB |
0–22 weeks 38/721 (5.3%) |
|
22–54 weeks Total: 354 (35.4%) |
22–54 weeks Total: 31 (3.1%) Pneumonia: 12 (1.2%) TB: 4 (0.4%) Abscess: 6 (0.6%) Pyelonephritis: 3 (0.3%) |
22–54 weeks 4 (0.4%) |
22–54 weeks 87/1084 (8.0%) |
|||||
|
Wolfe 2004 144 Prospective cohort study |
Infliximab 2.5 years |
6460 patients with RA | NR | NR | NR | 4 (0.06%) | NR | NR |
| Adalimumab | ||||||||
|
Breedveld 2006 140 RCT |
Adalimumab ± MTX 2 years |
542 patients with RA | Total: 9.12% (estimated) |
Total: 12 (2.2%) Pulmonary infection: 4 (0.74%) Sinus infection: 1 (0.18%) Wound infection: 1 (0.18%) Septic arthritis: 2 (0.37%) Infected hygroma: 1 (0.18%) Cellulitis: 2 (0.37%) UTI: 1 (0.18%) |
Total: 6 (1.1%) | 1 (0.18%) |
Total: 5 (0.9%) Cancer: 3 (0.55%) |
58/542 (10.7%) |
|
Burmester 2007 131 Uncontrolled open-label study |
Adalimumab ± DMARD Median: 211 days |
6610 patients with RA | NR | 202 (3.1%) | 43 (0.7%) | 21 (0.3%) |
Total: 35 (0.5%) TB: 1 |
682/6610 (10.3%) |
|
Colombel 2007 133 RCT |
Adalimumab 56 weeks |
CD patients: 0–4 weeks, n = 854; 4–56 weeks, n = 517 |
0–4 weeks 130 (15.2%) |
0–4 weeks 10 (1.2%) |
0–4 weeks 54/854 (6.3%) |
|||
|
4–56 weeks 234 (45.3%) |
4–56 weeks 14 (2.7%) |
4–56 weeks 1 breast cancer (0.2%) |
4–56 weeks 2 (0.4%) |
4–56 weeks 1 (0.2%) |
4–56 weeks 30/517 (5.8%) |
|||
|
Rudwaleit 2009 126 Uncontrolled open-label study |
Adalimumab Median: 12 weeks |
969 AS patients with advanced spinal fusion | NR | 4 (0.4%) | NR | NR | NR | NR |
|
Schiff 2006 138 Analysis of clinical trial safety database |
Adalimumab NR |
10,050 patients with RA | NR | 638 (6.3%) | 15 lymphomas (0.1%) | 34 (0.3%) | NR | NR |
Appendix 4 Table of excluded studies with rationale
Studies excluded from efficacy search
| Study | Reason for exclusiona |
|---|---|
| Anandarajah AP, Ritchlin CT. Etanercept in psoriatic arthritis. Expert Opin Biol Ther 2003;3(1):169–77. | 2 |
| Antoni CE. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT) (errata). Arthritis Rheum 2005;52(9):2951. | 2 |
| Bathon J, Fleischmann R, Peloso P, Chon Y, Hooper M, Lin SL. Rates of cardiovascular events in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis treated with etanercept or placebo in clinical trials. Arthritis Rheum 2006;54(Suppl. 9):188. | 2 |
| Bongiorno MR, Pistone G, Doukaki S, Arico M. Adalimumab for treatment of moderate to severe psoriasis and psoriatic arthritis. Dermatol Ther 2008;21(Suppl. 2):15–20. | 2 |
| Brodszky V, Pentek M, Gulacsi L. Efficacy of adalimumab, etanercept, and infliximab in psoriatic arthritis based on ACR 50 response after 24 weeks of treatment. Scand J Rheumatol 2008;37(5):399–400. | 2 |
| Colombel JF. Efficacy and safety of adalimumab for the treatment of Crohn’s disease in adults. Expert Rev Gastroenterol Hepatol 2008;2(2):163–76. | 2 |
| Cruyssen BV, De Keyser F, Kruithof E, Mielants H, Van den Bosch F. Comparison of different outcome measures for psoriatic arthritis in patients treated with infliximab or placebo. Ann Rheum Dis 2006;65(Suppl. 2):546–7. | 2 |
| Frankel EH, Strober BE, Crowley JJ, Fivenson DP, Woolley JM, Yu EB, et al. Etanercept improves psoriatic arthritis patient-reported outcomes: results from EDUCATE. Cutis 2007;79(4):322–6. | 2 |
| Gottlieb AB, Kircik L, Eisen D, Jackson JM, Boh EE, Strober BE, et al. Use of etanercept for psoriatic arthritis in the dermatology clinic: the Experience Diagnosing, Understanding Care, and Treatment with Etanercept (EDUCATE) study. J Dermatolog Treat 2006;17(6):343–52. | 2 |
| Hamza S, Chon Y, Hooper M, MacPeek D, Lin S. Rates of serious infectious events and opportunistic infections in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis treated with etanercept or placebo in clinical trials. Ann Rheum Dis 2007;66(Suppl. 2):171–2. | 2 |
| Kimball AB, Jackson JM, Sobell JM, Boh EE, Grekin S, Pharmd EBY, et al. Reductions in healthcare resource utilization in psoriatic arthritis patients receiving etanercept therapy: results from the educate trial. J Drugs Dermatol 2007;6(3):299–306. | 2 |
| Kristensen LE, Gulfe A, Saxne T, Geborek P. Efficacy and tolerability of anti-tumour necrosis factor therapy in psoriatic arthritis patients: results from the South Swedish Arthritis Treatment Group register. Ann Rheum Dis 2008;67(3):364–9. | 2 |
| Kvien TK, Heiberg MS, Lie E, Kaufmann C, Mikkelsen K, Nordvag BY, et al. A Norwegian DMARD register: prescriptions of DMARDs and biological agents to patients with inflammatory rheumatic diseases. Clin Exp Rheumatol 2005;23(Suppl. 39):188–94. | 2 |
| McHugh N, van den Bosch F, Manger B, Goupille P, Cooper R, Kron M, et al. Adalimumab treatment is effective in patients with psoriatic arthritis (PSA) in day-to-day clinical practice: results from the stereo trial. Rheumatology 2008;47(Suppl. 2):ii, 76. | 2 |
| Mease P. Infliximab (Remicade) in the treatment of psoriatic arthritis. Ther Clin Risk Manag 2006;2(4):389–400. | 2 |
| Mease PJ, Choy EHS, Atkins CJ, Sasso EH. Effectiveness of adalimumab in psoriatic arthritis patients with oligoarticular arthritis: subanalysis of ADEPT. Fourth European Academy of Dermatology and Venereology (EADV) Spring Symposium Saariselka, Lapland, Finland, 9–12 February, 2006: P-022. | 5 |
| Ravindran V, Scott DL, Choy EH. A systematic review and meta-analysis of efficacy and toxicity of disease modifying anti-rheumatic drugs and biological agents for psoriatic arthritis. Ann Rheum Dis 2008;67(6):855–9. | 2 |
| Revicki D, Willian MK, Saurat JH, Papp KA, Ortonne JP, Sexton C, et al. Impact of adalimumab treatment on health-related quality of life and other patient-reported outcomes: results from a 16-week randomized controlled trial in patients with moderate to severe plaque psoriasis. Br J Dermatol 2008;158(3):549–57. | 3 |
| Rinaldi F, Provenzano G, Termini A, Spinello M, La Seta F. Long term infliximab treatment for severe psoriatic arthritis: evidence of sustained clinical and radiographic response. Ann Rheum Dis 2005;64(9):1375–6. | 2 |
| Ritchlin C. Efficacy and safety of infliximab for the treatment of psoriatic arthritis. Nat Clin Pract Rheumatol 2006;2(6):300–1. | 2 |
| Romero-Mate A, Garcia-Donoso C, Cordoba-Guijarro S. Efficacy and safety of etanercept in psoriasis/psoriatic arthritis: an updated review. Am J Clin Dermatol 2007;8(3):143–55. | 2 |
| Saad AA, Symmons DPM, Noyce PR, Ashcroft DM. Risks and benefits of tumor necrosis factor-alpha inhibitors in the management of psoriatic arthritis: systematic review and metaanalysis of randomized controlled trials. J Rheumatol 2008;35(5):883–90. | 2 |
| Scheinfeld N. Adalimumab: a review of side effects. Expert Opin Drug Saf 2005;4(4):637–41. | 2 |
| Simpson D, Scott LJ. Adalimumab: in psoriatic arthritis. Drugs 2006;66(11):1487–96. | 2 |
| Spadaro A, Ceccarelli F, Scrivo R, Valesini G. Life-table analysis of etanercept with or without methotrexate in patients with psoriatic arthritis. Ann Rheum Dis2008;67(11):1650–1. | 2 |
| Strober B, Teller C, Yamauchi P, Miller JL, Hooper M, Yang YC, et al. Effects of etanercept on C-reactive protein levels in psoriasis and psoriatic arthritis. Br J Dermatol 2008;159(2):322–30. | 4 |
| Toussirot E, Streit G, Wendling D. Infectious complications with anti-TNFalpha therapy in rheumatic diseases: a review. Recent Pat Inflamm Allergy Drug Discov 2007;1(1):39–47. | 2 |
| Van den Bosch F, Reece R, Manger B, Goupille P, Roedevand E, Holck P, et al. Adalimumab (HUMIRA (R)) is effective and safe in treating psoriatic arthritis (PsA) in real-life clinical practice: preliminary results of the STEREO trial. Arthritis Rheum 2006;54(Suppl. 9):S719–20. | 2 |
| Van den Bosch F, McHugh NJ, Reece R, Cooper R, Manger B, Goupille P, et al. Treatment with adalimumab (Humira (R)) is safe and effective in psoriatic arthritis (PsA) patients in real-life clinical practice: preliminary results of the stereo trial. Rheumatology 2007;46(Suppl. 1):i,52–3. | 2 |
| Van den Bosch F, Manger B, Goupille P, McHugh N, Roedevand E, Holck P, et al. Adalimumab (Humira (R)) is effective in treating patients with psoriatic arthritis (PSA) in real-life clinical practice: results of the STEREO trial [abstract OP0147]. Ann Rheum Dis 2007;66(Suppl. 2):98. | 2 |
| Van den Bosch F, Manger B, Goupille P, Kron M, Kary S, Kupper H. Clinical remission and good clinical responses in patients with psoriatic arthritis (PsA) treated with adalimumab (HUMIRA (R)): results of the STEREO trial. Arthritis Rheum 2008;58(Suppl. 9):576. | 2 |
| Van Kuijk AWR, Gerlag DM, Vos K, Wolbink G, Zwinderman AH, Dijkmans BAC, et al. A randomized, placebo-controlled study to identify biomarkers associated with active treatment in psoriatic arthritis: effects of adalimumab treatment on synovial biomarkers. Arthritis Rheum 2008;58(Suppl. 9):415. | 4 |
| Winterfield LS, Menter A. Infliximab. Dermatol Ther 2004;17(5):409–26. | 2 |
| Winthrop KL, Siegel JN, Jereb J, Taylor Z, Lademarco MF. Tuberculosis associated with therapy against tumor necrosis factor alpha. Arthritis Rheum 2005;52(10):2968–74. | 2 |
Studies excluded from adverse event searches
| Study | Reason for exclusiona |
|---|---|
| Anandarajah AP, Ritchlin CT. Etanercept in psoriatic arthritis. Expert Opin Biol Ther 2003;3(1):169–77. | 2 |
| Author not found. [Active tuberculosis after use of infliximab (Remicade).] Geneesmiddelenbulletin 2001;35(3):33. | 2 |
| Author not found. Infection risk with infliximab. Pharm J 2001;266(7129):7. | 2 |
| Baldin B, Dozol A, Spreux A, Chichmanian RM. [Tuberculosis and infliximab treatment: national surveillance from January 1, 2000, through June 30, 2003.] Presse Med 2005;34(5):353–7. | 2 |
| Boehncke WH, Prinz J, Gottlieb AB. Biologic therapies for psoriasis. A systematic review. J Rheumatol 2006;33(7):1447–51. | 4 |
| Brimhall AK, King LN, Licciardone JC, Jacobe H, Menter A. Safety and efficacy of alefacept, efalizumab, etanercept and infliximab in treating moderate to severe plaque psoriasis: a meta-analysis of randomized controlled trials. Br J Dermatol 2008;159(2):274–85. | 4 |
| Brown SL, Greene MH, Gershon SK, Edwards ET, Braun MM. Tumor necrosis factor antagonist therapy and lymphoma development: twenty-six cases reported to the Food and Drug Administration. Arthritis Rheum 2002;46(12):3151–8. | 2 |
| Caviglia R, Boskoski I, CiCala M. Long-term treatment with infliximab in inflammatory bowel disease: safety and tolerability issues. Expert Opin Drug Saf 2008;7(5):617–32. | 4 |
| Colombel JF. The CHARM trial of adalimumab in Crohn’s disease. Gastroenterol Hepatol (N Y) 2006;2(7):486–8. | 4 |
| Colombel JF. Efficacy and safety of adalimumab for the treatment of Crohn’s disease in adults. Expert Rev Gastroenterol Hepatol 2008;2(2):163–76. | 4 |
| Drosou A, Kirsner RS, Welsh E, Sullivan TP, Kerdel FA. Use of infliximab, an anti-tumor necrosis alpha antibody, for inflammatory dermatoses. J Cutan Med Surg 2003;7(5):382–6. | 2 |
| Dunlop H. Infliximab (Remicade) and etanercept (Enbrel): serious infections and tuberculosis. Can Med Assoc J 2004;171(8):992–3. | 1 |
| Emery P, Breedveld FC, Hall S, Durez P, Chang DJ, Robertson D, et al. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double-blind, parallel treatment trial. Lancet 2008;372(9636):375–82. | 1 |
| European Medicines Agency. Assessment report for Remicade [Internet]: London: EMEA; 2007. | 1 |
| European Medicines Agency. Assessment report for Enbrel [Internet]: London: EMEA; 2008. | 1 |
| European Medicines Agency. Assessment report for Humira [Internet]: London: EMEA; 2008. | 1 |
| European Medicines Agency. Humira EMEA/H/C/481/II/06: scientific discussion [Internet]: London: EMEA; 2004. | 1 |
| European Medicines Agency. Humira EMEA/H/C/481/II/21: scientific discussion [Internet]: London: EMEA; 2005. | 1 |
| European Medicines Agency. Humira EMEA/H/C/481/II/22: scientific discussion [Internet]: London: EMEA; 2005. | 1 |
| European Medicines Agency. Humira EMEA/H/C/481/II/38: scientific discussion [Internet]: London: EMEA; 2007. | 1 |
| European Medicines Agency. Humira EMEA/H/C/481/II/43: scientific discussion [Internet]: London: EMEA; 2007. | 1 |
| European Medicines Agency. Humira/Trudexa EMEA/H/C/481–482/II/33: scientific discussion [Internet]: London: EMEA; 2007. | 1 |
| European Medicines Agency. Product information: Enbrel [Internet]: London: EMEA; 2009. | 1 |
| European Medicines Agency. Product information: Humira [Internet]: London: EMEA; 2009. | 2 |
| European Medicines Agency. Product information: Remicade [Internet]: London: EMEA; 2009. | 2 |
| European Medicines Agency. Remicade EMEA/H/C/240/II/65: scientific discussion [Internet]: London: EMEA; 2006. | 1 |
| European Medicines Agency. Remicade EMEA/H/C/240/II/73: scientific discussion [Internet]: London: EMEA; 2006. | 1 |
| European Medicines Agency. Remicade EMEA/H/C/240/II/100: scientific discussion [Internet]: London: EMEA; 2007. | 1 |
| European Medicines Agency. Scientific discussion [Internet]. London: EMEA; 2004. | 1 |
| European Medicines Agency. Scientific discussion [Internet]. London: EMEA; 2004. | 1 |
| European Medicines Agency. Scientific discussion [Internet]. London: EMEA; 2005. | 1 |
| Food and Drug Administration. Approval package for: application number: BL 103772/1007 [Internet]. Rockville, MD: US Food and Drug Administration, Center for Drug Evaluation and Research; 2000. | 1 |
| Food and Drug Administration. Approval package for: application number: 103795/S-5097 [Internet]: Rockville, MD: US Food and Drug Administration, Center for Drug Evaluation and Research; 2003. | 1 |
| Food and Drug Administration. Approval package for: application number: 103795/S-5109 [Internet]. Rockville, MD: US Food and Drug Administration, Center for Drug Evaluation and Research; 2003. | 1 |
| Food and Drug Administration. Medical review(s). Approval package for: application number 103795/5123 [Internet]. Rockville, MD: US Food and Drug Administration, Center for Drug Evaluation and Research; 2003. | 1 |
| Food and Drug Administration. Medical review(s). Application number: sBLA 125057/110 [Internet]. Rockville, MD: US Food and Drug Administration, Center for Drug Evaluation and Research; 2008. | 1 |
| Food and Drug Administration. Medical/statistical review(s). Approval package for application number STN 103795/5102 [Internet]. Rockville, MD: US Food and Drug Administration, Center for Drug Evaluation and Research; 2003. | 1 |
| Food and Drug Administration. Review of BLA submission 98–0012 [Internet]: Rockville, MD: US Food and Drug Administration, Center for Biologic Evaluation and Research; 1998. | 1 |
| Food and Drug Administration. Review of BLA submission 99-O 128. Infliximab (REMICADE) for signs and symptoms of rheumatoid arthritis [Internet]. Rockville, MD: US Food and Drug Administration, Center for Biologic Evaluation and Research; 1999. | 1 |
| Food and Drug Administration. Risk assessment and risk mitigation review(s). Application number: sBLA 125057/110 [Internet]. Rockville, MD: US Food and Drug Administration, Center for Drug Evaluation and Research; 2008. | |
| Food and Drug Administration. Statistical review. Application number: sBLA 125057/110 [Internet]. Rockville, MD: US Food and Drug Administration, Center for Drug Evaluation and Research; 2008. | 1 |
| Furst DE, Schiff MH, Fleischmann RM, Strand V, Birbara CA, Compagnone D, et al. Adalimumab, a fully human anti-tumor necrosis factor-alpha monoclonal antibody, and concomitant standard antirheumatic therapy for the treatment of rheumatoid arthritis: results of STAR (Safety Trial of Adalimumab in Rheumatoid Arthritis). J Rheumatol 2003;30(12):2563–71. | 1 |
| Gartlehner G, Hansen RA, Jonas BL, Thieda P, Lohr KN. The comparative efficacy and safety of biologics for the treatment of rheumatoid arthritis: a systematic review and metaanalysis. J Rheumatol 2006;33(12):2398–408. | 4 |
| Goekoop-Ruiterman YPM, De Vries-Bouwstra JK, Allaart CF, Van Zeben D, Kerstens PJSM, Hazes JMW, et al. Comparison of treatment strategies in early rheumatoid arthritis: a randomized trial. Ann Intern Med 2007;146(6):406–15. | 1 |
| Gordon KB, Gottlieb AB, Leonardi CL, Elewski BE, Wang A, Jahreis A, et al. Clinical response in psoriasis patients discontinued from and then reinitiated on etanercept therapy. J Dermatolog Treat 2006;17(1):9–17. | 1 |
| Kamm MA. Safety issues relating to biological therapies, with special reference to infliximab therapy. Research and Clinical Forums 2002;24(1):79–86. | 4 |
| Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001;345(15):1098–104. | 1 |
| Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum 2004;50(5):1400–11. | 1 |
| Klareskog L, Van Der Heijde D, De Jager JP, Gough A, Kalden J, Malaise M, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet 2004;363(9410):675–81. | 1 |
| Langley RG, Gupta AK, Cherman AM, Inniss KA. Biologic therapeutics in the treatment of psoriasis. Part 1: review. J Cutan Med Surg 2007;11(3):99–122. | 4 |
| McLeod C, Bagust A, Boland A, Dagenais P, Dickson R, Dundar Y, et al. Adalimumab, etanercept and infliximab for the treatment of ankylosing spondylitis: a systematic review and economic evaluation. Health Technol Assess 2007;11(28). | 4 |
| Mikuls TR, Weaver AL. Lessons learned in the use of tumor necrosis factor-alpha inhibitors in the treatment of rheumatoid arthritis. Curr Rheumatol Rep 2003;5(4):270–7. | 4 |
| Montilla Salas J, Munoz Gomariz E, Collantes E. [Meta-analysis of efficacy of anti-TNF alpha therapy in ankylosing spondylitis patients]. Reumatología Clínica 2007;3(5):204–12. | 4 |
| Moss AC, Farrell RJ. Infliximab for induction and maintenance therapy for ulcerative colitis. Gastroenterology 2006;131(5):1649–51. | 4 |
| Neven N, Vis M, Voskuyl AE, Wolbink GJ, Nurmohamed MT, Dijkmans BAC, et al. Adverse events in patients with rheumatoid arthritis treated with infliximab in daily clinical practice. Ann Rheum Dis 2005;64(4):645–6. | 2 |
| Orlando A, Mocciaro F, Civitavecchia G, Scimeca D, Cottone M. Minimizing infliximab toxicity in the treatment of inflammatory bowel disease. Dig Liver Dis 2008;40(Suppl. 2):S236–46. | 4 |
| Panes J, Gomollon F, Taxonera C, Hinojosa J, Clofent J, Nos P. Crohn’s disease: a review of current treatment with a focus on biologics. Drugs 2007;67(17):2511–37. | 4 |
| Papoutsaki M, Costanzo A, Mazzotta A, Gramiccia T, Soda R, Chimenti S. Etanercept for the treatment of severe childhood psoriasis. Br J Dermatol 2006;154(1):181–3. | 2 |
| Papp KA. The long-term efficacy and safety of new biological therapies for psoriasis. Arch Dermatol Res 2006;298(1):7–15. | 4 |
| Pariente A, Gregoire F, Fourrier-Reglat A, Haramburu F, Moore N. Impact of safety alerts on measures of disproportionality in spontaneous reporting databases: the notoriety bias. Drug Saf 2007;30(10):891–8. | 2 |
| Romero-Mate A, Garcia-Donoso C, Cordoba-Guijarro S. Efficacy and safety of etanercept in psoriasis/psoriatic arthritis: an updated review. Am J Clin Dermatol 2007;8(3):143–55. | 4 |
| Scheinfeld N. Adalimumab: a review of side effects. Expert Opin Drug Saf 2005;4(4):637–41. | 4 |
| Subramanian V, Pollok RCG, Kang JY, Kumar D. Systematic review of postoperative complications in patients with inflammatory bowel disease treated with immunomodulators. Br J Surg 2006;93(7):793–9. | 1 |
| Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 2006;367(9504):29–35. | 1 |
| Tyring S, Gordon KB, Poulin Y, Langley RG, Gottlieb AB, Dunn M, et al. Long-term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch Dermatol 2007;143(6):719–26. | 3 |
| Van Der Heijde D, Klareskog L, Rodriguez-Valverde V, Codreanu C, Bolosiu H, Melo-Gomes J, et al. Comparison of etanercept and methotrexate, alone and combined, in the treatment of rheumatoid arthritis: two-year clinical and radiographic results from the TEMPO study, a double-blind, randomized trial. Arthritis Rheum 2006;54(4):1063–74. | 1 |
| Van Der Heijde D, Klareskog L, Landewe R, Bruyn GAW, Cantagrel A, Durez P, et al. Disease remission and sustained halting of radiographic progression with combination etanercept and methotrexate in patients with rheumatoid arthritis. Arthritis Rheum 2007;56(12):3928–39. | 1 |
| Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis 2004;38(9):1261–5. | 1 |
| Weisman MH, Paulus HE, Burch FX, Kivitz AJ, Fierer J, Dunn M, et al. A placebo-controlled, randomized, double-blinded study evaluating the safety of etanercept in patients with rheumatoid arthritis and concomitant comorbid diseases. Rheumatology 2007;46(7):1122–5. | 1 |
| Winterfield LS, Menter A. Infliximab. Dermatol Ther 2004;17(5):409–26. | 2 |
| Winthrop KL, Siegel JN, Jereb J, Taylor Z, Iademarco MF. Tuberculosis associated with therapy against tumor necrosis factor alpha. Arthritis Rheum 2005;52(10):2968–74. | 4 |
| Wong A, Fonseca MCM, Sandron CA. [Descriptive analyses of safety data for anti-TNF therapies using related outcomes from Uppsala Monitoring Centre (UMC) of World Health Organization (WHO).] Rev Bras Med 2007;64(7):323–33. | 1 |
Appendix 5 Evidence synthesis overview
Background
A Bayesian MTC (indirect comparison) is an extension of a meta-analysis, but where a meta-analysis includes only direct evidence an MTC analysis draws on both direct and indirect evidence. 205 As in a meta-analysis, it is the summary treatment effect from each study that is utilised in the MTC analysis; hence the benefit of randomisation in each study is retained.
A standard meta-analysis combines the results from two or more studies that have comparable populations, interventions, comparators and outcomes. Study quality and other study characteristics are also assumed to be similar. Similarly, to make indirect comparisons, it is assumed that the study characteristics are comparable. This is known as exchangeability, which can be investigated through the consistency of the direct and indirect evidence. 206
These types of evidence syntheses require a ‘network of evidence’ between all the treatments of interest. In the context of the present review this would mean that the network is required to comprise trials of adalimumab, etanercept, infliximab and placebo, where each treatment has been compared either directly or indirectly with every other. For example, although adalimumab and etanercept may not have been directly compared within a single trial, they can be compared indirectly if both have been assessed against a common comparator, placebo. The common comparator need not be placebo and, within a MTC, there can be more than one common comparator. Within a MTC all of the available trials’ data on a treatment for the specified indication should be included.
In the present analysis all six trials compared one of the three biologics with placebo. Several outcomes were deemed clinically relevant to determining the effectiveness of the biologics and a Bayesian indirect comparison was conducted for each of these outcomes. All included trials were assessed as part of the clinical review and it was determined that the population, intervention protocols, outcomes and other study characteristics were sufficiently exchangeable for synthesis to be conducted. The analysis was undertaken using winbugs version 1.4.2. 207 winbugs is a Bayesian analysis software that, through the use of Monte Carlo Markov chains, calculates posterior distributions for the parameters of interest, given likelihood functions derived from data and prior probabilities. The Monte Carlo Markov chain simulation begins with an approximate distribution and, if the model is a good fit to the data, the distribution converges to the true distribution. For all models used in the present analysis the first 10,000 iterations were considered to be ‘burn in’ and excluded, and a further 100,000 iterations were performed in order to calculate the results. The winbugs codes for the different analyses are presented in winbugs code, below. All of the data used in the evidence synthesis are presented in Tables 41–44.
| Study | Treatment | Response | n |
|---|---|---|---|
| Mease 200078 | Placebo | 7 | 30 |
| Etanercept | 26 | 30 | |
| Mease 200452,97,99,105,107,110 | Placebo | 32 | 104 |
| Etanercept | 73 | 101 | |
| IMPACT79 | Placebo | 7 | 52 |
| Infliximab | 40 | 52 | |
| IMPACT 282 | Placebo | 27 | 100 |
| Infliximab | 77 | 100 | |
| ADEPT51 | Placebo | 42 | 162 |
| Adalimumab | 94 | 151 | |
| Genovese 200783 | Placebo | 12 | 49 |
| Adalimumab | 26 | 51 |
| HAQ given PsARC response | SE | HAQ given no PsARC response | SE | ||
|---|---|---|---|---|---|
| Placebo | –0.258 | 0.006 | Placebo | –0.002 | 0.042 |
| Etanercept | –0.635 | 0.062 | Etanercept | –0.196 | 0.072 |
| Placebo | –0.27 | 0.14 | Placebo | 0.02 | 0.05 |
| Infliximab | –0.65 | 0.09 | Infliximab | –0.2 | 0.09 |
| Placebo | –0.16 | 0.096 | Placebo | 0.07 | 0.042 |
| Infliximab | –0.58 | 0.057 | Infliximab | –0.11 | 0.06 |
| Placebo | –0.3134 | 0.0761 | Placebo | 0.0260 | 0.0366 |
| Adalimumab | –0.5 | 0.0445 | Adalimumab | –0.1198 | 0.0525 |
| Placebo | –0.1771 | 0.0624 | Placebo | –0.0574 | 0.0530 |
| Adalimumab | –0.4231 | 0.0809 | Adalimumab | –0.1500 | 0.0904 |
| Trial | Treatment | Outcome (% change in PASI) | n |
|---|---|---|---|
| Mease 200078 | Placebo | < 50 | 15 |
| 50–75 | 4 | ||
| 75 | 0 | ||
| > 90 | No data | ||
| Etanercept | < 50 | 11 | |
| 50–75 | 3 | ||
| 75 | 5 | ||
| > 90 | No data | ||
| Mease 200452 | Placebo | < 50 | 51 |
| 50–75 | 9 | ||
| 75–90 | 0 | ||
| > 90 | 2 | ||
| Etanercept | < 50 | 35 | |
| 50–75 | 16 | ||
| 75–90 | 11 | ||
| > 90 | 4 | ||
| IMPACT79–81,89,96,109,111,113–115,117,118 | Placebo | < 50 | 16 |
| 50–75 | 0 | ||
| 75–90 | 0 | ||
| > 90 | 0 | ||
| Infliximab | < 50 | 0 | |
| 50–75 | 7 | ||
| 75–90 | 7 | ||
| > 90 | 8 | ||
| IMPACT 282,90,91,95,98,106,112,116 | Placebo | < 50 | 79 |
| 50–75 | 6 | ||
| 75–90 | 2 | ||
| > 90 | 0 | ||
| Infliximab | < 50 | 15 | |
| 50–75 | 15 | ||
| 75–90 | 19 | ||
| > 90 | 34 | ||
| ADEPT51 | Placebo | <50 | 59 |
| 50–75 | 7 | ||
| 75–90 | 3 | ||
| > 90 | 0 | ||
| Adalimumab | < 50 | 19 | |
| 50–75 | 16 | ||
| 75–90 | 13 | ||
| > 90 | 21 |
| Trial | Treatment | Outcome (% change in ACR data) | n |
|---|---|---|---|
| Mease 200078 | Placebo | < 20 | 26 |
| 20–50 | 3 | ||
| 50–75 | 1 | ||
| > 75 | 0 | ||
| Etanercept | < 20 | 8 | |
| 20–50 | 7 | ||
| 50–75 | 11 | ||
| > 75 | 4 | ||
| Mease 200452,97,99,105,107,110 | Placebo | < 20 | 88 |
| 20–50 | 12 | ||
| 50–75 | 4 | ||
| > 75 | 0 | ||
| Etanercept | < 20 | 41 | |
| 20–50 | 22 | ||
| 50–75 | 27 | ||
| > 75 | 11 | ||
| IMPACT79–81,89,96,109,111,113–115,117,118 | Placebo | < 20 | 46 |
| 20–50 | 5 | ||
| 50–75 | 1 | ||
| > 75 | 0 | ||
| Infliximab | < 20 | 17 | |
| 20–50 | 16 | ||
| 50–75 | 8 | ||
| > 75 | 11 | ||
| IMPACT 282,90,91,95,98,106,112,116 | Placebo | < 20 | 89 |
| 20–50 | 8 | ||
| 50–75 | 2 | ||
| > 75 | 1 | ||
| Infliximab | < 20 | 42 | |
| 20–50 | 22 | ||
| 50–75 | 21 | ||
| > 75 | 15 | ||
| ADEPT51,88,92,93,100–104 | Placebo | < 20 | 139 |
| 20–50 | 17 | ||
| 50–75 | 5 | ||
| > 75 | 1 | ||
| Adalimumab | < 20 | 63 | |
| 20–50 | 34 | ||
| 50–75 | 24 | ||
| > 75 | 30 | ||
| Genovese 200783 | Placebo | < 20 | 41 |
| 20–50 | 7 | ||
| 50–75 | 1 | ||
| > 75 | 0 | ||
| Adalimumab | < 20 | 31 | |
| 20–50 | 7 | ||
| 50–75 | 6 | ||
| > 75 | 7 |
An evidence synthesis was conducted for each of the four main outcomes. The primary outcome of this analysis was the probability of response to treatment in terms of PsARC (PsARC response) at 12 weeks following the BSR guidelines. The changes in HAQ score are conditional on a PsARC response to treatment, the probability of achieving the PASI 50/75/90 response, and the probability of achieving the ACR 20/50/70 response were also calculated. Three different models were produced to allow the separate outcomes to be synthesised. An overview of each model, along with the formal model is presented in the following section.
Psoriatic Arthritis Response Criteria response
The probability of initial response to each treatment, as determined by the PsARC outcome at 12 weeks, was modelled using a common-effects meta-analysis. Outcomes at 14 weeks were included in the analysis and assumed equivalent to outcomes at 12 weeks. Data were available from all six trials (two for each active treatment) for this outcome measure (see Table 41). Each trial reported the number of events in the control group (rCi) and the number of events under active treatment (rTi), where i represents a trial (i = Fleischmann et al. ,99 Kavanaugh et al. ,109 Lebowhl et al. ,105 Mease,52,78,97 Wanke et al. ,107 IMPACT,79 IMPACT 2,82 ADEPT,51 Genovese et al. 83). It was assumed that both rCi and rTi are binomially distributed.
The common baseline for each treatment effect was the probability of response to placebo. In order to achieve this, a meta-analysis on the placebo arms of the six RCTs was conducted. Each of the individual studies estimate the same true treatment effect δi (i.e. the underlying effect), and that differences between studies are solely due to chance. The observed effect of each study equals a fixed effect that is common to all studies plus sampling error;208 In the Bayesian evidence synthesis, δi was assigned a non-informative normal prior distribution. Formally:
Treatment effects on probability of response were additive to the placebo probability of response on the log-odds scale. The probability of response to the intervention is given by:
with Tk = μ + δk being the treatment effect on the intervention k (k = placebo, etanercept, infliximab, adalimumab) and being the true treatment effect of the intervention k (on a log-odds scale).
The common effects model was compared with a random-effects model for both fit, as measured by the deviation information criterion (DIC), convergence and correlation. The data for these models are presented in Table 41. The DIC statistic combines model deviance and the effective number of parameters. The DIC statistics were very similar: 128.288 for the common-effects model versus 128.274 for the random-effects model. Convergence and autocorrelation were assessed using graphical tools available within winbugs. The common-effects model was a good fit, converged well and did not display any issues with autocorrelation. The random-effects model did not converge well and displayed issues with autocorrelation. For these reasons the common-effects model was used.
Changes in Health Assessment Questionnaire
Trials that reported the absolute changes in HAQ from baseline, conditional on whether the patient responds to therapy at 12 weeks were modelled using a random-effects meta-analysis. Data were available from five of the six trials for this outcome measure: etanercept data were not available from the Mease et al. 78 trial.
Let ‘TR’ be the treatment responders, ‘TNR’ be the treatment non-responders, ‘PR’ be the placebo responders and ‘PNR’ be the placebo non-responders. Also, let i represent the trial and j the alternative treatments. We have assumed changes in HAQ given placebo non-responders as common baseline (μPNR) – a non-informative normal distribution was assign to this parameter. The effects of treatment response (δ.diffTRij) and non-response (δ.diffTNRij) on HAQ change are assumed to be treatment specific and additive to the placebo probability of non-response on the log-odds scale as illustrated below:
For each of the different trials the true effect may be study specific and vary across studies although remain common across biologics. These true effects are described by a normal distribution. Hence, the variation in observed individual study results is caused not only by sampling error (as with the common-effects approach), but also by the variation in the true (underlying) effects of each study. 209
When estimating HAQ separately for those who responded to PsARC we investigated a number of alternative modelling scenarios including:
-
a fixed-effects model, assuming that all biologics have the same effectiveness after conditioning on PsARC response
-
a random-effects model, assuming that all biologics have the same effectiveness after conditioning on PsARC response, and that heterogeneity in effects is the same for responders and non-responders
-
a random-effects model with all biologics having different (non-related) effectiveness after conditioning on PsARC response, assuming heterogeneity in effects is the same for responders and non-responders
-
a random-effects model assuming that all biologics have the same effectiveness after conditioning on PsARC response, including a response effect as a fixed effect and an interaction term to allow treatment/response interaction.
Due to the volume of data informing the synthesis, and the need to derive clinically relevant estimates for the economic model, the decision was made to limit the choice to a fixed/common-effects model, assuming all biologics have the same effectiveness (after conditioning on PsARC response) and a random effects model, with all biologics having different (non-related) effectiveness (after conditioning on PsARC response), while assuming heterogeneity in effects is the same for responders and non-responders. Finally, two alternative modelling scenarios were tested in an attempt to identifying the most appropriate model. The data for these two alternatives are presented in Table 42. The DIC statistic, convergence and autocorrelation were all assessed and informed model selection. The DIC statistics were –42.925 for the random-effects model and –55.095 for the fixed/common-effects model. As there was no issues with convergence or autocorrelation, the random-effects model was selected for use in the base weeks of the economic decision model, and the common treatment effect evidence synthesis estimate was used in a sensitivity analysis of the economic decision model. The results of the common effect model have been presented in Table 45 at the end of this appendix, not in the main clinical chapter.
Psoriatic Arthritis Response Criteria 50/75/90
Data were available from five of the six trials for this outcome measure: adalimumab data were not available from the Genovese et al. trial. 83 All responses are measured at 12–16 weeks apart from the IMPACT 2 trial,82,90,91,95,98,106,112,116 which reported only PASI responses at 24 weeks. A coefficient was included in the linear predictor to estimate whether the difference in follow-up time for this trial was significant. The probability of response in terms of the PASI 50/75/90 scores was modelled using an ordered multinomial logit model. In the ordered logit model the probability of an outcome is calculated by estimating a latent variable as a linear function of the independent variable plus a set of thresholds/cut-off points. In this analysis these thresholds represent the different outcomes of PASI 50/75/90. The probability of observing the latent variable equals the probability that the estimated linear function is within the cut-off points estimated for the outcome. This type of model allows the ordered nature of the outcomes to be maintained. Outcomes estimated are the probability of achieving each of the three PASI levels. A number of assumptions were made to facilitate modelling:
-
A common-effects model was used to estimate baseline; this was estimated using data from placebo non-responders (i.e. those receiving placebo and not achieving PASI 50).
-
Common effects were assumed for each treatment class (etanercept, infliximab and adalimumab).
-
Thresholds were assumed to be fixed across trials.
-
The baseline latent variable was assumed fixed.
The response of a patient to treatment for psoriasis is measured using the PASI scoring system. The RCTs typically measure the change in psoriasis in each participant by comparing the percentage change in PASI with the score at baseline, and report the number of patients who achieved the following responses, in trial i and treatment j, where j = 0 is placebo, and j = 1, 2, 3 are the three biologic therapies:
-
PASI 50ij is at least a 50% change.
-
PASI 75ij is at least a 75% change.
-
PASI 90ij is at least a 90% change.
The statistical analysis used a multicategorical response model to analyse these data. The multivariate response variable rij is a vector of the number of participants in arm j of study i reporting one of the four possible values:
In a trial arm of size Nij, rij is multinomially distributed:
where
We estimate the probability that patients have a PASI 50, 75 or 90 response by a cumulative logistic model. We define Zij to be a latent variable representing the mean improvement in psoriasis in arm j of trial i. The latent variable is determined by the explanatory variables in a linear form:
Where ai represents the mean improvement in the placebo arm of trial i and coefficient bj represents the mean improvement that can be attributed to treatment j, for j = 1, 2, 3, and Tij is a dummy variable for the biologic that was trialled in RCT i. Coefficient ai is a fixed-effects for trial i and coefficient bj is assumed to be common across all trials for treatment j. As this is an ordered logit model, coefficient bj can be interpreted as the log-treatment effect of drug j relative to placebo.
R and Z are connected by:
for r = 2, 3, 4 where
The parameters θr represent thresholds for observing a particular psoriasis response, rather than a less strong response. The error term eij was assumed to take a logistic distribution function Pr(eij ≤ e) = F(e) = 1/[1 + exp(–e)].
We define variable Yijr to be the cumulative probability of achieving a response r or greater, so that Yij1 is the probability of a patient achieving a PASI 50 response in trial i and treatment j, Yij2 is the probability of achieving a PASI 75 response, and Yij3 the probability of achieving a PASI 90 response.
Therefore,
Parameter θ2 is not estimated as it is co-linear with the intercept term.
It follows that:
To avoid problems with estimation that may occur if the thresholds are very similar, the thresholds θ3 and θ4 were reparameterised by210 θ3 = ω3 and θ4 = ω3 + exp(ω4).
In the Bayesian evidence synthesis, all parameters of the model (ai, bj, and ωr) were assigned non-informative normal prior distributions.
One of the aims of the model was to provide predictions of PASI 50/75/90 response rates for each treatment. This requires an estimate of parameter a, the intercept of the linear latent variable function. This was made by assuming it is equivalent to the pooled (mean) log-odds of a PASI 50 response across all the placebo arms of the RCTs.
As with the other evidence synthesis models, different modelling scenarios were assessed using criteria such as the DIC statistic, convergence and autocorrelation graphs. These models included an ordered probit model and random-effects versions of both the ordered logit and probit. The model selected was the best fit and presented good convergence and no sign of autocorrelation. The data for these models are presented in Table 43. The ordered logit models both had lower DIC statistics than the ordered probit models: 146.301 for the common effects versus 147.421 for the random effects. As with other models, issues with convergence and autocorrelation made the common effects a better choice. The ordered probit models, although behaving quite well in terms of convergence did show signs of autocorrelation. Additionally, both the common- and random-effects models produced DIC statistics in excess of 1800.
American College of Rheumatology 20/50/70
Data were available from all of the six trials for this outcome, across all three thresholds. As with the PASI data, the ACR data were modelled using an ordered multinomial logit model.
The same set of modelling assumptions that were applied to the PASI model was used for the ACR model. As stated previously, different modelling scenarios were assessed using criteria such as the DIC statistic, convergence and autocorrelation graphs. These models included an ordered probit model and random-effects versions of both the ordered logit and probit. The model selected was the best fit, and presented good convergence and no sign of autocorrelation. The data for these models are presented in Table 44. Like the PASI models, the ACR ordered probit models behaving well in terms of convergence although they also showed signs of autocorrelation. They again produced DIC statistics in excess of 1800. Both the ordered logit models both had lower DIC statistics: 200.88 for the common effects and 202.069 for the random effects. Again, the random-effects model having some issues with autocorrelation, hence making the common effects model a better choice.
The formal model for the ACR data is extremely similar to the PASI model outlined above.
Results for Health Assessment Questionnaire/Psoriatic Arthritis Response Criteria common effect
Table 45 shows the results for the evidence synthesis of HAQ conditional on PsARC response assuming that all three biologics have the same underlying treatment effect. The results are presented here as they were used in a sensitivity analysis scenario in the economic decision model.
| HAQ | response: common treatment effects (common baseline) | Mean | Credible interval (%) | |
|---|---|---|---|
| 2.50 | 97.50 | ||
| Treatment changes in HAQ | response | –0.5688 | –0.6305 | –0.5073 |
| Treatment changes in HAQ | no response | –0.1697 | –0.2362 | –0.1038 |
| Placebo changes in HAQ | response | –0.2606 | –0.3149 | –0.2062 |
winbugs code
Evidence synthesis models winbugs code
Model one: probability of PsARC response to each treatment (and placebo)
Model two: Health Assessment Questionnaire conditional on Psoriatic Arthritis Response Criteria response
Model three: probability of achieving Psoriasis Area and Severity Index response
Model four: probability of achieving American College of Rheumatology response
Appendix 6 Clarifications from manufacturers
Wyeth153
Decision to withdraw depending on initial response
The model requires patients to withdraw from biologic therapy if no response is achieved at either 12 or 24 weeks. How are responses at 12 and 24 weeks correlated? Is there a regression model to link response at 12 weeks with response at 24 weeks?
No, it was not possible to include any correlation between the response rates at 12 weeks and 24 weeks, given the evidence available (MTC – STA). Data from a previous published MTC (STA – Adalimumab) was used to model the response rate at either 12 or 24 weeks independently. It is believed that data presented in the MTC for the response rate at 24 weeks is independent to the response at 12 weeks when looking at the sample size of patients included in the MTC. For instance, all of the patients randomised in the etanercept arm in the Mease 2004 trial52,97,99,105,107,110 or in the infliximab arm in the IMPACT 2 trial82,90,91,95,98,106,112,116 were included at 24 weeks in the MTC, whether or not they responded at 12 weeks. Consequently, this suggests that response rates reported in the MTC at 12 and 24 weeks were not conditional of each other. The response rates at 12 and 24 weeks were therefore sampled independently of each other. It was not possible to sample the response rate jointly (taking into account the correlation) in the absence of patient data for other treatments.
Health Assessment Questionnaire for responders and non-responders
Wyeth153 estimates a regression of HAQ given PsARC and PASI (Tables 9 and 10). The Assessment Group would like to request that Wyeth153 rerun this regression without PASI. This is for two reasons. First, each of the manufacturers has submitted a different model and we would like to compare estimates of parameters from different sources. Wyeth’s model153 is the only one that uses PASI to predict HAQ. Second, this will enable the York Assessment Group to use Wyeth’s data153 to inform HAQ in the York economic model.
Our model included PASI to predict HAQ, given the possible correlation between HAQ and PASI. A full regression model, including different covariates, was estimated initially. Non-significant covariates were then excluded (significance level of 0.05). PASI was found to be a significant predictor of HAQ in addition to PsARC. PASI thus explain part of the variance in HAQ in addition to PsARC. Removing PASI would remove part of the explained variance in HAQ. Our method was also justified by the absence of relationship between Cost, HAQ and PASI.
However, as requested by the Assessment Group, regression models for HAQ without PASI were rerun.
The Assessment Group would also like to use the data on mean HAQ conditional on response from the Mease 2004 trial,52,97,99,105,107,110 which was commercial-in-confidence (CiC) in the previous NICE appraisal. Please could you consider releasing this data from the CiC restriction?
We are in contact with our Global Medical Affairs department to clarify whether this data can be released from the CiC restriction.
Long-term withdrawal rate from biologics
Wyeth153 has estimated Weibull models for the rate of withdrawal from biologics, from data published from the BSR register. The York Assessment Group is not clear what calculations were made to estimate these parameters. Please clarify how these parameters were worked out from the data?
The BSR paper191 reported the proportion of patients on etanercept at 1 year (86%), 2 years (79%) and 3 years (65%). A Weibull curve was fitted to these three values by calibrating the two parameters of the Weibull function (scale and shape) in order to minimise the error between the observed and predicted proportion of patients still treated with etanercept. The observed and predicted proportions of patients treated with etanercept at 1, 2 and 3 years are reported below. The root mean square error between the observed and predicted proportion was 0.01961.
| Year | Observed | Predicted |
|---|---|---|
| – | 1.00 | 1.00 |
| 1 | 0.86 | 0.88 |
| 2 | 0.79 | 0.76 |
| 3 | 0.65 | 0.66 |
The Weibull function was assumed to follow the following equation (as defined in stata):
Utility conditional on Psoriatic Area and Severity Index and Health Assessment Questionnaire
Wyeth153 has presented regression models to predict utility from HAQ and PASI. However, the Assessment Group is unable to easily compare this with the other models because each has used a different source of data and different covariates in the regression. To enable us to compare the submissions, and include estimates from different sources in the York model, we would like to request that you rerun this regression in a comparable way. We suggest the following set of untransformed covariates is included in the regression: Constant, HAQ, PASI and HAQ × PASI (interaction term). We would like to request the results of this regression as coefficients, variance–covariance matrix, number of observations and number of clusters (if appropriate), indicating the source of data.
The regression model to predict utility from PRESTA was rerun to include HAQ, PASI and the interaction between HAQ and PASI as requested by ERG. A second model was also generated without the interaction between HAQ and PASI given the non-significance of the coefficient for the interaction.
Abbott151
Sequencing
The Abbott model151 allows a sequence of DMARDs after failure of biologic therapy. Is there always 10 DMARDs in this sequence? What treatment (or no treatment) is given after failure of the last DMARD in the sequence?
The model151 is structured to allow for a maximum of 10 different DMARD treatments (which includes different combinations of DMARDs). The model151 assumes that patients will continue to try different combinations of DMARDs rather than receive no active treatment. Consequently, no response test is used for DMARD therapies, and patients withdraw from these treatments based on the long-term withdrawal rate. Once the patient reaches the last DMARD combination in the sequence, they have effectively run out of options and so will continue on that treatment until they die.
Long-term withdrawal rate from biologics
Abbott151 has estimated Weibull models for the rate of withdrawal from biologics, from data published from the BSRBR register. The York Assessment Group is not clear what calculations were made to estimate these parameters. Please can you clarify how these parameters were worked out from the data?
A crude survival analysis is made using the reported figures in Table 46 of Saad et al. 191 As can be seen in Figure 7, the analysis used survival rates reported by Saad et al. 191 for all biologics in year 1 (0.82), in year 2 (0.70) and in year 3 (0.59). Survival rates beyond the initial 3-year period were modelled assuming a Weibull distribution following the shape of survival curves observed for other rheumatic diseases. 211
| Parameter | Estimate | SE | 95% confidence limits | Z | Probability > |Z | | |
|---|---|---|---|---|---|---|
| Intercept | 0.8862 | 0.0182 | 0.8506 | 0.9217 | 48.82 | <.0001 |
| HAQ | –0.2317 | 0.0248 | –0.2803 | –0.1831 | –9.35 | <.0001 |
| PASI | –0.0025 | 0.0015 | –0.0054 | 0.0004 | –1.69 | 0.0906 |
| HAQ × PASI | –0.0039 | 0.002 | –0.0079 | 0 | –1.94 | 0.0523 |
| No. observations used: 386 | ||||||
| No. clusters: 138 | ||||||
FIGURE 7.
Observed versus predicted survival for all biologics. Utility conditional on the PASI and HAQ.
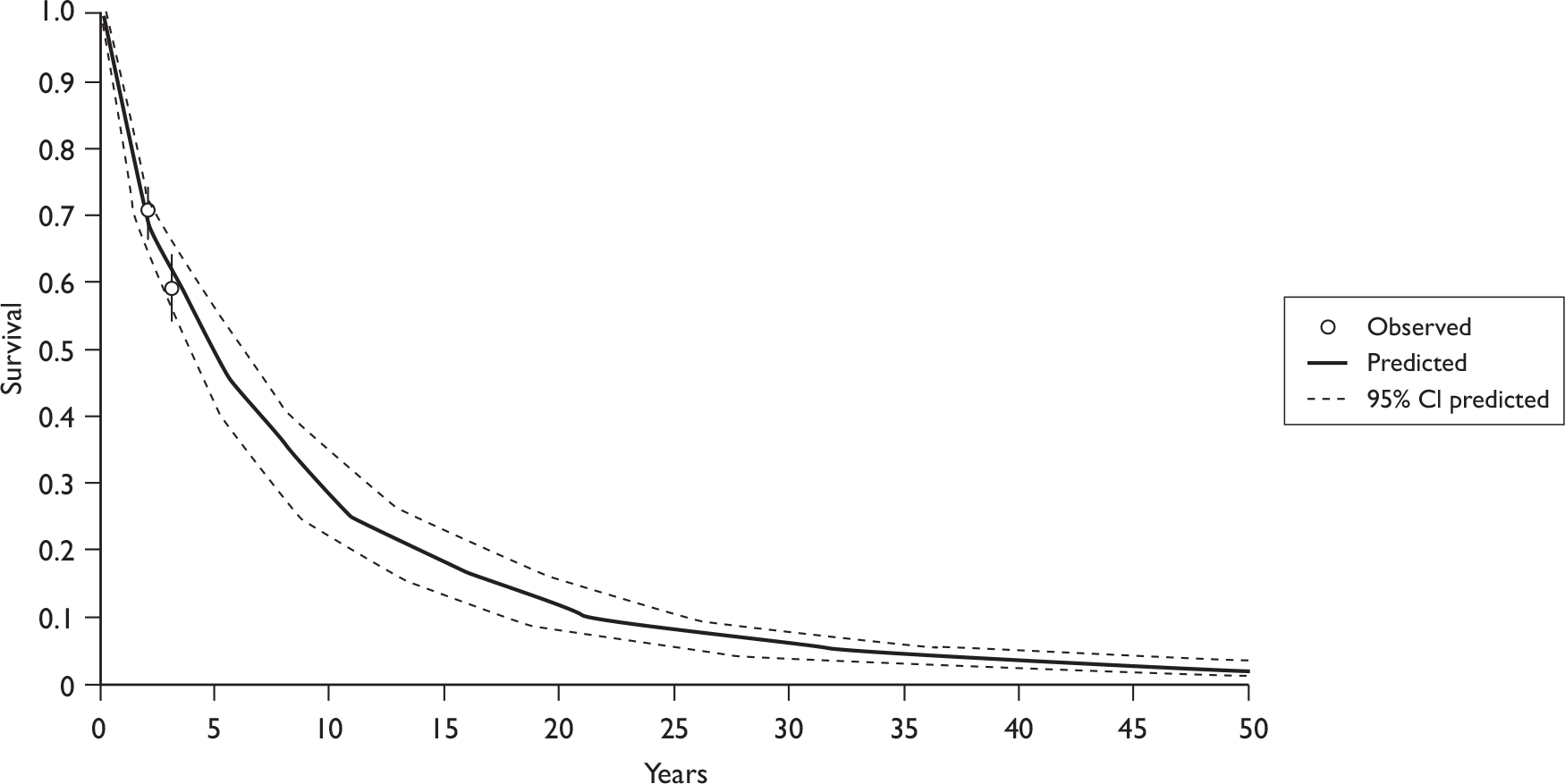
Abbott151 has presented a regression model to predict utility from HAQ and PASI. However, the Assessment Group is unable to easily compare your model with the others because each model has used a different source of data and different covariates in the regression. To enable us to compare the submissions, and include estimates from different sources in the York model, we would like to request that you rerun this regression in a comparable way. We suggest the following set of untransformed covariates is included in the regression: Constant, HAQ, PASI and HAQ × PASI (interaction term). We would also like to request the results of this regression as coefficients, variance–covariance matrix, number of observations, number of clusters (if appropriate), indicating the source of data.
The utility regression estimates are shown in Table 46, and the covariance matrix is shown in Table 47. It should be noted that in the ADEPT trial88 a proportion of patients had a HAQ score of 0. It was therefore impossible for these patients to experience an improvement in their HAQ score. In order to ensure the utility regressions truly capture the impact a change in HAQ has on a patient’s utility score, these patients have been excluded from the analysis.
| Intercept | HAQ | PASI | HAQ × PASI | |
|---|---|---|---|---|
| Intercept | 0.0003295 | –0.000292 | –0.000014 | 0.0000126 |
| HAQ | –0.000292 | 0.0006146 | 0.0000129 | –0.000033 |
| PASI | –0.000014 | 0.0000129 | 2.1946E-06 | –0.000001607 |
| HAQ × PASI | 0.0000126 | –0.000033 | –0.000001607 | 4.0944E-06 |
Correlation between outcomes
There is no evidence presented to support the correlation across outcomes. How large are the correlations? What were the data restrictions that meant a trivariate analysis could not be completed? Can the data be presented?
Spearman correlations have been calculated using patient-level data from the ADEPT clinical trial. 88 There is a positive correlation between the two measures of the arthritis component of the disease (PsARC and ACR), indicating that a PsARC responder is also likely to be an ACR responder, although this correlation is not as strong as would be expected if these two measures were truly interchangeable (Table 48). As can be seen in Table 49, approximately 80% of PsARC responders were ACR 20 responders at week 12 in the treatment group in the ADEPT trial,88 with a κ-coefficient of 0.56 (moderate agreement).
| PsARC | ACRb | Treatmenta | |
|---|---|---|---|
| Adalimumab (n = 151) | Placebo (n = 162) | ||
| PsARC (week 12) | ACR 20 (week 12) | 0.57 (p < 0.0001) | 0.57 (p < 0.0001) |
| PsARC (week 24) | ACR 20 (week 24) | 0.64 (p < 0.0001) | 0.69 (p < 0.0001) |
| Week 12 PsARC | Week 12 ACR 20 | |
|---|---|---|
| Non-responders: n (%) | Responders: n (%) | |
| Non-responder | 45 (77.5) | 13 (22.4) |
| Responders | 19 (20.4) | 74 (79.5) |
| κ-coefficient | 0.56 (moderate agreement) | |
As can be seen in Table 50 there is a significant and positive correlation between all three outcomes observed between week 12 and week 24. This is particularly high for ACR 20 response rates and is stronger in the adalimumab arm than in the placebo arm of the trial. It is anticipated that the lower correlation in the placebo arm is due to the fact that these patients may be classed as responders by chance rather than because they are actually responding to treatment. The probability that patients in the placebo arm who respond to treatment at week 12 are still responding to treatment at week 24 is therefore lower than for those patients in the adalimumab arm. Correlations are higher between ACR responses at week 12 and week 24 compared with PsARC response rates indicating that the ACR is a more robust measure of response than the PsARC.
| Weeks | Treatmenta | ||
|---|---|---|---|
| 12 | 24 | Adalimumab (n = 151) | Placebo (n = 162) |
| PsARC | PsARC | 0.61 (p < 0.0001) | 0.37 (p < 0.0001) |
| ACRb | ACRb | 0.79 (p < 0.0001) | 0.33 (p < 0.001) |
| (n = 69) | (n = 69) | ||
| PASIc | PASIc | 0.64 (p < 0.0001) | 0.39 (p < 0.0001) |
The correlations presented in Table 51 indicate that there is a weak correlation between skin response and arthritis response. This suggests that patients who observe improvements in their skin symptoms may not observe similar improvements in their arthritis symptoms. Table 52 indicates that approximately 62% of ACR 20 responders were also PASI 75 responders at week 12 in the ADEPT trial,88 with a κ-coefficient of 0.31 (fair agreement). When interpreting these data it is important to remember that only a subset of patients in the ADEPT trial88 were eligible for PASI assessment, thus reducing the statistical power of the analysis.
| Response measure | Treatmenta | ||
|---|---|---|---|
| Arthritis | Skin | Adalimumab (n = 69) | Placebo (n = 69) |
| PsARC (week 12) | PASIc (week 12) | 0.49 (p < 0.0001) | 0.13 (p = 0.2969) |
| PsARC (week 24) | PASIc (week 24) | 0.36 (p = 0.0023) | 0.26 (p = 0.304) |
| ACRb (week 12) | PASIc (week 12) | 0.42 (p = 0.0004) | 0.23 (p = 0.0614) |
| ACRb (week 24) | PASIc (week 24) | 0.38 (p = 0.0014) | 0.23 (p = 0.0612) |
| Week 12 ACR 20 response | Week 12 PASI 75 response | |
|---|---|---|
| Non-responders, n (%) | Responders, n (%) | |
| Non-responder | 19 (70.3) | 8 (29.6) |
| Responders | 16 (38) | 26 (61.9) |
| κ-coefficient | 0.31 (fair agreement) | |
A trivariate analysis could not be completed for several reasons. First, in the ADEPT trial,88 PASI was measured only in patients with a BSA ≥ 3%, meaning that PASI, PsARC and ACR response data were available only for 43.2% of patients (n = 69). Excluding those patients with no PASI scores would have meant discarding most of the data on arthritis response, thus significantly reducing the power of the analysis. Including these patients would result in an error and the model would not be able to run due to the absence of PASI scores.
A further barrier to conducting a trivariate analysis was the computational burden required for such a complex analysis. For example, the model examining the relationship between ACR 20 at 12 weeks and at 24 weeks took approximately 5 hours to compile; for the fixed-effects model it took a total of 50 hours to run three chains, while for random-effects models it took 500 hours. Expanding to a trivariate analysis would require many times this. It is therefore not possible to present the results of a trivariate analysis.
Schering-Plough152
Regression of Quality of Life on Health Assessment Questionnaire and Psoriasis Area and Severity Index
-
NICE request – 29 September 2009.
NICE requested a linear regression of QoL on the following covariates:
-
Intercept
-
HAQ
-
PASI
-
HAQ × PASI interaction term.
Two options are available for estimating the QoL data:
-
SF-36 to EQ-5D via Gray algorithm
-
EQ-5D.
The data source used here is the IMPACT 2 study82,90,91,95,98,106,112,116 (excel files from Ewen Cummins’ e-mails, 21 March 2009, Schering-Plough). EQ-5D was converted to a QoL index score using the published UK tariffs212 (Brazier algorithm).
Results
Patients with missing values for baseline EQ-5D, HAQ or PASI have been removed from both analyses. Multiple observations in the same patient were treated as independent observations, no cluster-based analysis was used. Sample size in both cases: n = 740 observations.
Using the Short Form questionnaire-36 items data via Gray algorithm
| Covariate | Mean | Variance–covariance matrix | |||
|---|---|---|---|---|---|
| Intercept | HAQ | PASI | HAQ × PASI | ||
| Intercept | 8.712e-01 | 5.978e-07 | –4.215e-07 | –3.698e-08 | 2.632e-08 |
| HAQ | –2.490e-01 | –4.215e-07 | 5.107e-07 | 2.679e-08 | –3.024e-08 |
| PASI | –2.485e-03 | –3.698e-08 | 2.679e-08 | 9.536e-09 | –6.684e-09 |
| HAQ × PASI | 5.928e-05 | 2.632e-08 | –3.024e-08 | –6.684e-09 | 6.405e-09 |
Using the European Quality of Life-5 Dimensions data
| Covariate | Mean | Variance–covariance matrix | |||
|---|---|---|---|---|---|
| Intercept | HAQ | PASI | HAQ × PASI | ||
| Intercept | 7.862e-01 | 9.233e-08 | –6.510e-08 | –5.712e-09 | 4.065e-09 |
| HAQ | –1.437e-01 | –6.510e-08 | 7.888e-08 | 4.139e-09 | –4.670e-09 |
| PASI | –2.648e-03 | –5.712e-09 | 4.139e-09 | 1.437e-09 | –1.032e-09 |
| HAQ × PASI | 9.927e-04 | 4.065e-09 | –4.670e-09 | –1.032e-09 | 9.893e-10 |
Appendix 7 Reviews of cost-effectiveness studies and checklists
Review of Olivieri et al.178
-
The PsA cost evaluation study: a cost-of-illness study on TNF inhibitors in patients with PsA with inadequate response to conventional therapy. 178
Overview
This is a before/after study that evaluated the costs and benefits of biologics (as a group) compared with no biologics. The study was undertaken in Italy and included 107 patients from nine tertiary referral centres. Both NHS and societal costs were included and HRQoL was measured using the EQ-5D. Results were expressed using a third-party payer and a societal perspective.
Summary of effectiveness data
The following outcomes were collected before and after biologics treatment: laboratory parameters, TJC/SJC, numbers of digits with dactylitis, Maastricht Ankylosing Spondylitis Enthesitis Score, Bath Ankylosing Spondylitis Disease Activity Index, Bath Ankylosing Spondylitis Functional Index, occiput to wall distance, chest expansion, modified Schober’s test, visual analogue scale (VAS), duration of morning stiffness, PASI, HAQ, EQ-5D, SF-36, demographic characteristics, clinical characteristics, surgical procedures, use of health-care resources, days off work due to illness and caregiver time. Patients were interviewed using a structured electronic weeks report form. This was administered and completed by a physician. Resource use and HRQoL were collected for the 6 months preceding biologics treatment, at baseline, 6 months and 12 months following initiation of treatment.
Both the EQ-5D (VAS and utility) and the SF-36 were used to evaluate HRQoL. Only the EQ-5D utility scores were used in the cost-effectiveness analysis. The EQ-5D utilities were converted to QALYs by computing the difference between average per patient utility at enrolment (before biologics) and average utility after initiation of treatment. This difference was then multiplied by 0.5 (6 months).
At the end of the 12-month observation period there was a gain of 0.25 in utility, equating to a 0.12 gain in QALYs.
Summary of resource utilisation and cost data
As described above, resource use was retrospectively collected from patients, for the 6 months preceding biologics and for the 12-months after initiation of treatment. Resource use data collected were from surgical procedures, hospitalisations, visits to the physician, medications and other non-health-care items, including days off work, caregivers’ time and transport to/from hospital visits. Case record forms were designed to collect all of this information from patients. This was administered and completed by physicians.
Medical costs were calculated by multiplying the items of resource use by the associated unit costs. The diagnosis-related group costs were used to represent the unit costs of hospitalisations. The authors did not state the sources for other medical costs. The costs of transportation were taken directly from patients’ reports. Carers’ costs and days lost from work were costed using the human capital approach.
At the end of the 12-month follow-up, direct costs increased by €5052. There were some decreases in hospitalisation costs (€142) and indirect costs (costs to the patient and carers – €413).
Summary of cost-effectiveness
Incremental cost-effectiveness ratios were appropriately calculated using the differences in costs and QALYs described above.
The increase in costs is somewhat offset by the 0.12 increase in QALYs to produce an ICER of €40,876 for the NHS and an ICER of €37,591 for society.
The uncertainty regarding the estimates of costs and QALYs were expressed using cost-effectiveness acceptability curves (CEACs), showing the probability that biologic were cost-effective at various thresholds for a QALY gained. If a decision-makers’ willingness-to-pay threshold was €45,000 then the probability that biologics is cost-effective is 0.82.
Comments
All TNFs were grouped together, although the majority of patients were taking etanercept. It is therefore not possible to estimate any differences in cost-effectiveness between the biologic drugs.
The analysis has a limited length of follow-up (6 months). PsA is a chronic disease and it is therefore likely that all differences in costs and outcomes between comparators can be captured in this short time frame.
Internal validity
This is a before/after study, so there may be a problem of confounding. It is possible that patients will get better over time as a result of increased monitoring as part of the study. It is not possible to disentangle these effects.
External validity
This is a relatively small sample of patients recruited from a single site. Patients, however, seem fairly typical of the PsA population in terms of disease markers.
Checklist for Olivieri et al.178
| ✓ or ✗ | ||
|---|---|---|
| Study question | Grade | Comments |
| 1. Costs and effects examined | ✓ | |
| 2. Alternatives compared | ✗ | |
| 3. The viewpoint(s)/perspective of the analysis is clearly stated (e.g. NHS, society) | ✗ | Two perspectives chosen; confusing statements about which is used for costing |
| Selection of alternatives | ||
| 4. All relevant alternatives are compared (including ‘do nothing’ if applicable) | ✗ | |
| 5. The alternatives being compared are clearly described (who did what, to whom, where and how often) | ✓ | |
| 6. The rationale for choosing the alternative programmes or interventions compared is stated | ||
| Form of evaluation | ||
| 7. The choice of form of economic evaluation is justified in relation to the questions addressed | ✗ | |
| 8. If a cost-minimisation design is chosen, have equivalent outcomes been adequately demonstrated? | NA | |
| Effectiveness data | ||
|
9. The source(s) of effectiveness estimates used are stated (e.g. single study, selection of studies, systematic review, expert opinion) |
✓ | |
| 10. Effectiveness data from RCT or review of RCTs | ✗ | |
| 11. Potential biases identified (especially if data not from RCTs) | ✗ | |
| 12. Details of the method of synthesis or meta-analysis of estimates are given (if based on an overview of a number of effectiveness studies) | NA | |
| Costs | ||
| 13. All of the important and relevant resource use included | ✓ | |
| 14. All of the important and relevant resource use measured accurately (with methodology) | ✓ | |
| 15. Appropriate unit costs estimated (with methodology) | ✗ | |
| 16. Unit costs reported separately from resource use data | ✗ | |
| 17. Productivity costs treated separately from other costs | ✓ | |
| 18. The year and country to which unit costs apply is stated with appropriate adjustments for inflation and/or currency conversion | ✗ | |
| Benefit measurement and valuation | ||
| 19. The primary outcome measure(s) for the economic evaluation are clearly stated | ✓ | |
| 20. Methods to value health states and other benefits are stated | ✓ | |
| 21. Details of the individuals from whom valuations were obtained are given | ✓ | |
| Decision modelling | ||
| 22. Details of any decision model used are given (e.g. decision tree, Markov model) | NA | |
| 23. The choice of model used and the key input parameters on which it is based are adequately detailed and justified | NA | |
| 24. All model outputs described adequately | NA | |
| Discounting | ||
| 25. Discount rate used for both costs and benefits | NA | |
| 26. Do discount rates accord with NHS guidance? | NA | |
| Allowance for uncertainty | ||
| Stochastic analysis of patient-level data | ||
| 27. Details of statistical tests and CIs are given for stochastic data | ✓ | |
| 28. Uncertainty around cost-effectiveness expressed (e.g. CI around ICER, CEACs) | ✓ | |
| 29. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | ✓ | |
| Stochastic analysis of decision models | ||
| 30. Are all appropriate input parameters included with uncertainty? | NA | |
| 31. Is second-order uncertainty (uncertainty in means) included rather than first order (uncertainty between patients)? | NA | |
| 32. Are the probability distributions adequately detailed and appropriate? | NA | |
| 33. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | NA | |
| Deterministic analysis | ||
| 34. The approach to sensitivity analysis is given (e.g. univariate, threshold analysis, etc.) | No deterministic sensitivity analysis performed | |
| 35. The choice of variables for sensitivity analysis is justified | ||
| 36. The ranges over which the variables are varied are stated | ||
| Presentation of results | ||
| 37. Incremental analysis is reported using appropriate decision rules | ✓ | |
| 38. Major outcomes are presented in a disaggregated as well as aggregated form | ✓ | |
| 39. Applicable to the NHS setting | ✗ | Biologics not evaluated separately; problems with internal validity |
Review of Bansback et al.176
-
Estimating the cost and health status consequences of treatment with TNF antagonists in patients with PsA. 176
Overview
This paper aimed to generate estimates of the long-term benefits (in terms of HRQoL) of biologics (etanercept) in PsA. In addition, they assessed the cost-effectiveness of biologics compared with conventional therapies. The model is based on that used in the Wyeth submission153 to the previous NICE appraisal of biologic drugs. 73 The Health Assessment Questionnaire Disability Index (HAQ-DI) was used to measure benefit and linked to utilities to generate QALYs. A third-party payer perspective was used for the analysis.
An individual sampling model was used to simulate costs and benefits over a 10-year time horizon, using data from a variety of sources, including RCTs, open-label and observational data. The authors do not state which software was used to programme the model.
Following failure on conventional DMARDs, sequencing of three comparators was evaluated. Etanercept was compared with combination therapy on MTX and ciclosporin or leflunomide.
Summary of effectiveness data
To estimate the initial (3-month) effect of etanercept, patient-level data from a phase III randomised trial was obtained (Mease et al. 52). HAQ was measured at 4, 12 and 24 weeks, after which patients were invited to join an open-label extension of the trial and be treated with etanercept. The randomised data was used within a multivariate regression model to predict 3-month HAQ change. The open-label extension data was used to estimate HAQ progression beyond 3 months.
A cohort study containing moderate-to-severe patients with PsA from the Academic Unit of Musculoskeletal Disease at the University of Leeds201 was used to estimate health–state utilities. The relationship between health utilities and HAQ was examined by fitting linear regression models that were estimated by generalised estimating equation algorithms.
The data set was also used to estimate long-term progression on best standard care and to explore the effect of adding the skin component (PASI) to the prediction of health utilities. The effect of PASI was found to be very small and not statistically significant. This may have been due to the relatively homogeneous PASI scores in the Leeds data set. 201
Withdrawal from etanercept was taken from the literature and assigned values of 34%213 and 42%214 for psoriatic arthritis and rheumatoid arthritis respectively. Patients that withdrew from treatment were assumed to worsen instantaneously by the same magnitude as they initially improved. This assumption is based on the ‘rebound’ effect observed in a previous economic evaluation of etanercept in RA.
Discounted 10-year QALYs were 4.49 for etanercept, 3.67 for ciclosporin and 3.84 for leflunomide.
Summary of resource utilisation and cost data
Costs included all direct costs attributable to patients with PsA, including drug costs, monitoring, administration and hospitalisation costs. The cost offsets of improving disability were also estimated using a study of patients with RA.
Total costs of etanercept over 10 years is estimated as £51,122, ciclosporin £28,010 and leflunomide £26,822.
Summary of cost-effectiveness
An individual sampling model was used to estimate costs and benefits over 10 years. Baseline characteristics were sampled from the demographics from the Mease 2004 trial. 200 The model tracks the decision to continue treatment at 3-monthly intervals. At each interval a decision about whether to continue treatment was randomly sampled. Biologics were assumed to halt the progression of disease while treatment is continued.
One-way and probabilistic sensitivity analysis were used to explore uncertainties in the data and the model structure.
The results show that at 6 months etanercept gives an additional 0.4 QALYs at an additional cost of £3000, which gives an ICER of around £70,000. At 10 years, the QALY benefit increased giving and ICER of £28,000 compared with ciclosporin and £38,000 compared with leflunomide.
Sensitivity analysis showed that the ICER was sensitive to the baseline HAQ and annual HAQ progression. The probabilistic sensitivity analysis showed the decision to recommend etanercept as the optimum treatment was uncertain at 10 years, with a probability that is it cost-effective of 0.58 (at a threshold of £30,000 per QALY).
Comments
This is a good-quality evaluation of biologics for PsA. However, only the biologic etanercept was evaluated and therefore the study cannot inform the question as to which biologic is most cost-effective (adalimumab, infliximab and etanercept). It only addresses the question of if biologics are cost-effective compared with ciclosporin and leflunomide. In addition, only data from a single phase II trial was used to determine effectiveness. More trials are now available and this evidence should be appropriately synthesised.
The skin component of PsA was not included. The effect of PASI was explored using the Leeds data set201 and found not to be statistically significant. However, this may have been due to the relatively homogeneous PASI scores in the Leeds data set. 201 Alternative data sets to explore the effect on PASI should have been explored.
Only a single scenario (rebound to gain) was used to represent the uncertainty regarding the effect of withdrawal from treatment on HAQ. Other scenarios, such as rebound to NH were not explored.
Internal validity
There are no major issues with internal validity.
External validity
The use of a single trial to estimate the initial response to treatment may be expected to produce less robust estimates and limit generalisability. In addition, the study is of little use in determining the relative cost-effective of alternative biologics, as the use of biologics was limited to etanercept. This is a major limitation to the study’s generalisability.
Checklist for Bansback et al.176
| ✓ or ✗ | ||
|---|---|---|
| Study question | Grade | Comments |
| 1. Costs and effects examined | ✓ | |
| 2. Alternatives compared | ✗ | Only looks at the biologic etanercept |
| 3. The viewpoint(s)/perspective of the analysis is clearly stated (e.g. NHS, society) | ✓ | |
| Selection of alternatives | ||
| 4. All relevant alternatives are compared (including ‘do nothing’ if applicable) | ✗ | A ‘do-nothing’ (palliative care) option is not considered |
| 5. The alternatives being compared are clearly described (who did what, to whom, where and how often) | ✓ | |
| 6. The rationale for choosing the alternative programmes or interventions compared is stated | ✓ | |
| Form of evaluation | ||
| 7. The choice of form of economic evaluation is justified in relation to the questions addressed | ✓ | |
| 8. If a cost-minimisation design is chosen, have equivalent outcomes been adequately demonstrated? | NA | |
| Effectiveness data | ||
|
9. The source(s) of effectiveness estimates used are stated (e.g. single study, selection of studies, systematic review, expert opinion) |
✓ | But limited to a single study |
| 10. Effectiveness data from RCT or review of RCTs | ✓ | |
| 11. Potential biases identified (especially if data not from RCTs) | ✗ | Fact that the skin component not considered is not discussed |
| 12. Details of the method of synthesis or meta-analysis of estimates are given (if based on an overview of a number of effectiveness studies) | NA | |
| Costs | ||
| 13. All of the important and relevant resource use included | ✓ | |
| 14. All of the important and relevant resource use measured accurately (with methodology) | ✓ | |
| 15. Appropriate unit costs estimated (with methodology) | ✓ | |
| 16. Unit costs reported separately from resource use data | ✗ | |
| 17. Productivity costs treated separately from other costs | ✗ | |
| 18. The year and country to which unit costs apply is stated with appropriate adjustments for inflation and/or currency conversion | ✓ | |
| Benefit measurement and valuation | ||
| 19. The primary outcome measure(s) for the economic evaluation are clearly stated | ✓ | |
| 20. Methods to value health states and other benefits are stated | ✓ | |
| 21. Details of the individuals from whom valuations were obtained are given | ✓ | But only limited information presented |
| Decision modelling | ||
| 22. Details of any decision model used are given (e.g. decision tree, Markov model) | ✓ | |
| 23. The choice of model used and the key input parameters on which it is based are adequately detailed and justified | ✗ | Not clear why it was appropriate to use an individual sampling model |
| 24. All model outputs described adequately. | ✓ | |
| Discounting | ||
| 25. Discount rate used for both costs and benefits | ✓ | Also explored in the sensitivity analysis |
| 26. Do discount rates accord with NHS guidance? | ✓ | |
| Allowance for uncertainty | ||
| Stochastic analysis of patient-level data | ||
| 27. Details of statistical tests and CIs are given for stochastic data | NA | |
| 28. Uncertainty around cost-effectiveness expressed (e.g. CI around incremental cost-effectiveness ratio (ICER), CEACs) | NA | |
| 29. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | NA | |
| Stochastic analysis of decision models | ||
| 30. Are all appropriate input parameters included with uncertainty? | ✗ | Costs presented as fixed |
| 31. Is second-order uncertainty (uncertainty in means) included rather than first order (uncertainty between patients)? | Both are presented | |
| 32. Are the probability distributions adequately detailed and appropriate? | ✓ | |
| 33. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | ✓ | |
| Deterministic analysis | ||
| 34. The approach to sensitivity analysis is given (e.g. univariate, threshold analysis, etc.) | ✓ | |
| 35. The choice of variables for sensitivity analysis is justified | ✓ | |
| 36. The ranges over which the variables are varied are stated | ✓ | |
| Presentation of results | ||
| 37. Incremental analysis is reported using appropriate decision rules | ✗ | Compares etanercept with all other comparators not just against next-best strategy |
| 38. Major outcomes are presented in a disaggregated as well as aggregated form | ✓ | |
| 39. Applicable to the NHS setting | ✗ | Use of a single trail to determine effectiveness potentially limits generalisability |
Review of Bravo Vergel et al.177
-
The cost-effectiveness of etanercept and infliximab for the treatment of patients with PsA. 177
Overview
The aim of the study was to estimate the cost-effectiveness of etanercept and infliximab for the treatment of active and progressive PsA in patients who have inadequate response to standard treatment (palliative care), including DMARD therapy. The analysis is based on the York Assessment Group model developed as part of the previous NICE appraisal of biologic therapies for PsA. 73 A probabilistic cohort model was developed in excel and used over 10- and 40-year time horizons. A third-party payer perspective was used for the analysis.
Summary of effectiveness data
Short-term trial data57,78,81 was used to model the response of patients (measured by PsARC criteria) to biologics. A Bayesian evidence synthesis was used to link the trials via indirect comparisons methods. A winbugs synthesis model was also used to estimate the mean improvements in HAQ score conditional on response. The placebo effect was deducted from the estimates of effect as the comparison strategy was palliative care (‘do nothing’). The mean HAQ change for non-responders was also estimated by the synthesis model and incorporated into the decision model for the initial 3-month period.
The absolute change in HAQ conditional on response from the Mease et al,52,78 and IMPACT trials81 was obtained from the pharmaceutical companies. HAQ progression for palliative-care patients was taken from the Leeds cohort study. 201
The posterior distributions estimated by the synthesis model were used to populate the decision model. In addition the probability of withdrawals from treatment was taken from Geborek et al. 198 Standard UK mortality rates were used and no excess mortality risk for patients with PsA was assumed.
Utility data was taken from a previous cost-effectiveness analysis for biologics in PsA176 in which the relationship between health–state utility and the HAQ-DI was examined by fitting a regression model to the Leeds data set. 201
The results show that infliximab is the most effective strategy in both scenarios (4.636 and 4.455 QALYs for rebound to gain and rebound to NH, respectively) and etanercept the next most effective (4.514 and 4.356 for both scenarios). Palliative care is the least effective strategy.
Summary of resource utilisation and cost data
Drug costs (including acquisition, administration and monitoring) were inputted into the model as fixed costs. Drug costs were taken from the BNF. 65 The issue of vial sharing for infliximab was explored as a sensitivity analysis. Administration and monitoring costs were estimated using industry assumptions regarding resources use and published unit costs.
The costs associated with PsA were estimated as a function of HAQ score using a published study in RA. These costs were assumed to include the costs of palliative care.
The results show that total mean costs were highest for infliximab in both rebound scenarios (£64,274 and £64,418 for rebound to gain and rebound to NH respectively). Etanercept is the next most costly (£44,111 and £44,169 for both scenarios) and palliative care the least costly (£10,718 and £10,679 for both scenarios).
Summary of cost-effectiveness
A modified decision tree was used to model the cohort of patients with PsA over time. The model was run separately for males and females.
Patients have a probability of responding the biologics in an initial 3-month period. This response is measured using the PsARC criteria. The associated HAQ change for responders is then estimated, this accounts for the progressive nature of the disease. For responders there is an annual risk of withdrawal (for any reason) from treatment. Once patients have withdrawn from treatment they experience a worsening in HAQ.
Uncertainty regarding parameters was characterised using the posterior distributions from the evidence synthesis and by assigning probability to other parameters. Monte Carlo simulation was used to generate lifetime costs and QALYs for the three strategies. Scenario analysis was used to explore some of the other uncertainties in the model, such as the rebound for patients withdrawing from treatment (rebound equal to gain and rebound equal to NH), time horizon, discount rate and number of vials of infliximab.
The ICERs for infliximab are unlikely to be considered reasonable at £165,363 and £205,345 compared to etanercept for rebound to gain and rebound to NH, respectively. The ICER for etanercept may or may not be acceptable depending on the threshold for cost-effectiveness and the scenario for rebound believed to be correct. The ICER for rebound equal to gain is £26,361 and the ICER for rebound equal to NH is £30,628. Both of these ICERs are compared to palliative care.
Etanercept has the highest probability of being cost effective in the rebound equal to gain scenario (0.693 at a £30,000 threshold), whereas palliative care has the highest probability of being cost-effective in the rebound equal to NH scenario (0.554 at a £30,000 threshold).
Comments
This is a good quality evaluation of biologics for PsA. Its limitations are not considering the use of the biologics adalimumab, simply presenting the uncertainty about the rebound effect as scenarios and exclusion of the skin component.
Internal validity
There are no major issues with internal validity.
External validity
The psoriasis component (measured using PASI) was not included in the model. HRQoL for patients with PsA is influenced by both the arthritis component and the psoriasis component. Failure to capture the effect of treatments on the psoriasis component of disease represents a major limitation of the study.
In addition, the uncertainty regarding the effect of withdrawal from treatment on HAQ was only presented as two alternative scenarios. It is therefore difficult to determine the value of further research to reduce this uncertainty.
Checklist for Bravo Vergel177
| ✓ or ✗ | ||
|---|---|---|
| Study question | Grade | Comments |
| 1. Costs and effects examined | ✓ | |
| 2. Alternatives compared | ✓ | |
| 3. The viewpoint(s)/perspective of the analysis is clearly stated (e.g. NHS, society) | ✓ | |
| Selection of alternatives | ||
| 4. All relevant alternatives are compared (including ‘do nothing’ if applicable) | ✓ | |
| 5. The alternatives being compared are clearly described (who did what, to whom, where and how often) | ✓ | |
| 6. The rationale for choosing the alternative programmes or interventions compared is stated | ✗ | Does not justify why a ‘do-nothing’ strategy is more appropriate than an active comparator such as other DMARDs |
| Form of evaluation | ||
| 7. The choice of form of economic evaluation is justified in relation to the questions addressed | ✓ | |
| 8. If a cost-minimisation design is chosen, have equivalent outcomes been adequately demonstrated? | NA | |
| Effectiveness data | ||
|
9. The source(s) of effectiveness estimates used are stated (e.g. single study, selection of studies, systematic review, expert opinion) |
✓ | |
| 10. Effectiveness data from RCT or review of RCTs | ✓ | |
| 11. Potential biases identified (especially if data not from RCTs) | ✗ | Comparability of studies not discussed; fact that the skin component not considered is not discussed |
| 12. Details of the method of synthesis or meta-analysis of estimates are given (if based on an overview of a number of effectiveness studies) | ✓ | |
| Costs | ||
| 13. All of the important and relevant resource use included | ✓ | |
| 14. All of the important and relevant resource use measured accurately (with methodology) | ✓ | |
| 15. Appropriate unit costs estimated (with methodology) | ✓ | |
| 16. Unit costs reported separately from resource use data | ✗ | Although further details available in HTA report |
| 17. Productivity costs treated separately from other costs | ✗ | Not considered |
| 18. The year and country to which unit costs apply is stated with appropriate adjustments for inflation and/or currency conversion | ✓ | |
| Benefit measurement and valuation | ||
| 19. The primary outcome measure(s) for the economic evaluation are clearly stated | ✓ | QALYs |
| 20. Methods to value health states and other benefits are stated | ✓ | Fact that the skin component not considered is not discussed |
| 21. Details of the individuals from whom valuations were obtained are given | ✗ | Does reference a separate publication |
| Decision modelling | ||
| 22. Details of any decision model used are given (e.g. decision tree, Markov model) | ✓ | |
| 23. The choice of model used and the key input parameters on which it is based are adequately detailed and justified | ✓ | |
| 24. All model outputs described adequately | ✓ | |
| Discounting | ||
| 25. Discount rate used for both costs and benefits | ✓ | |
| 26. Do discount rates accord with NHS guidance? | ✓ | |
| Allowance for uncertainty | ||
| Stochastic analysis of patient-level data | ||
| 27. Details of statistical tests and CIs are given for stochastic data | NA | |
| 28. Uncertainty around cost-effectiveness expressed (e.g. CI around ICER, CEACs) | NA | |
| 29. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | NA | |
| Stochastic analysis of decision models | ||
| 30. Are all appropriate input parameters included with uncertainty? | ✗ | Costs presented as fixed |
| 31. Is second-order uncertainty (uncertainty in means) included rather than first order (uncertainty between patients)? | ✓ | |
| 32. Are the probability distributions adequately detailed and appropriate? | ✓ | |
| 33. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | ✓ | |
| Deterministic analysis | ||
| 34. The approach to sensitivity analysis is given (e.g. univariate, threshold analysis, etc.) | ✓ | |
| 35. The choice of variables for sensitivity analysis is justified | ✓ | |
| 36. The ranges over which the variables are varied are stated | ✓ | |
| Presentation of results | ||
| 37. Incremental analysis is reported using appropriate decision rules | ✓ | |
| 38. Major outcomes are presented in a disaggregated as well as aggregated form | ✓ | |
| 39. Applicable to the NHS setting | ✓ | |
Review of Abbott submission151
An individual sampling model is used to assess the cost-effectiveness of adalimumab compared with etanercept, infliximab and conventional DMARDs. Third-, fourth- and fifth-line treatments are modelled with fourth- and fifth-line treatments always comprising DMARDs. The patients included in the model were assumed to have not responded to at least two DMARDs, individually or in combination. A third-party payer perspective was used for the analysis. The model is programmed in ‘R’ and a lifetime time horizon is assumed.
Summary of effectiveness data
Baseline patient characteristics from the ADEPT trial88 were used determine the baseline distribution of patients characteristics in the model.
Long-term outcomes were expressed as QALYs. To generate QALYs, short- and long-term outcomes were estimated. These longer-term outcomes were then regressed on to utilities. Short-term efficacy was determined using PsARC, ACR and PASI responses. Longer-term outcomes were HAQ and PASI.
In the base-case model, 12-week PsARC response rates were used to determine continuation of therapy beyond the trial period. A mixed-treatment fixed-effects meta-analysis was used to determine response rates. The evidence synthesis was undertaken using winbugs, and utilised data from 10 different source studies,51,52,62,78,81,82,88,83,154,155 each of which compares different treatment, some of which that are not included in this appraisal. Three Bayesian bivariate analyses were conducted to determine: (1) joint distribution of 12-week PsARC and ACR response rates; (2) 24-week PsARC response conditional on the 12-week PsARC response; and (3) 24-week ACR response conditional on the 12-week ACR response. The joint distribution of 12- and 24-week PASI response rate is modelled independently. The associated winbugs code was presented. In a sensitivity analysis, continuation beyond 12 weeks was estimated directly from the BSRBR and so PsARC response rates were not used to determine continuation.
Patient-level data from the ADEPT88 study were then used to estimate HAQ and PASI changes dependent on the magnitude of response. Patients who had previously failed two or more DMARDs and had a baseline HAQ > 0 were included in the analysis. A forward stepwise regression analysis was used to select significant variables in predicting HAQ and PASI improvement, including ACR response type, HAQ at baseline, demographics, disease duration and treatment. In order to estimate the PASI, the data were transformed by log(PASI + 0.5). The authors state that this was done ‘to obtain normality’. It is important to note that this log-transformation assumes that a 1% improvement in PASI will lead to a constant change in utility, regardless of the absolute change in PASI. For example, this regression assumes that a reduction in PASI score from 16 to 0 leads to the same change in HRQoL as a reduction in PASI score from 8 to 0. A linear regression on the other hand assumes that a reduction in PASI by 16 points gives twice the HRQoL benefit of a reduction in PASI by 8 points, regardless of the baseline. A similar regression was specified for HAQ at 24 weeks.
Placebo response rates from trials were used to represent the DMARD efficacy data. A common efficacy was used for all DMARDs. A reduction multiplier was applied to response rates for subsequent DMARDs (24% reduction in receiving response). Alternative reduction multipliers were examined in sensitivity analysis.
Long-term progression of HAQ while on biologics was assumed to be 0.0005 per year. This was taken from a longitudinal analysis of the Bath Psoriatic Arthritis Database (reference not given). Progression on DMARDs was 0.024 per year. Progression of patients who do not respond (defined as ACR 20) is assumed to be 0.06 per year. These were both estimated using the Leeds data set. 201 PASI is assumed to halt for responders.
The model assumes that patients withdrawn from therapy at 12 months due to inefficacy reflect the PsARC response rates in practice. Rates of withdrawal from therapy between 1 and 3 years, due to either adverse events or loss of efficacy, were estimated using data from the BSRBR registry162 and specified using a Weibull distribution. No differences between drugs were assumed due to selection bias. Sensitivity analysis explored differential biologics withdrawal and the use of data from Kristensen et al. 215 Withdrawal rates for conventional DMARDs were taken from a smaller study by Malesci et al. 199 and were again specified using a Weibull distribution. It is unclear how the parameters for either of these Weibull distributions were derived from the referenced data. Following withdrawal from treatment patients HAQ is assumed to rebound equivalent to the initial gain and PASI rebound to the starting level. The rate of HAQ progression following stopping biologics therapy was assumed to be the same as for patients who do not respond to therapy (0.066).
Two sources of data were used to estimate the improvement in health utility through a direct linear relationship with HAQ and PASI. Base case uses the ADEPT trial88 of adalimumab. SF-36 was converted to EQ-5D. In a sensitivity analysis, data from the Bath Psoriatic Arthritis Database was used. Functions for health utilities reported with and without skin effect. Any interaction between HAQ and PASI was not explored.
The model used PsA specific mortality inflators29 along with UK life tables.
Infliximab was associated with the highest QALYs (8.49), followed by etanercept and adalimumab (8.33) and then DMARDs (7.47).
Summary of resource utilisation and cost data
The costs of all drugs were estimated using MIMS (online and print prescribing database for health professionals)216 as opposed to the BNF. 65 Infliximab costs were calculated assuming that four vials were used per infusion based on an average patient weight of 80 kg.
Resource use associated with monitoring and administering drugs was estimated according to BSR guidelines. Assumes infliximab requires a half-day hospital visit for each infusion. A single outpatient visit is required for adalimumab and etanercept. Gives references for each unit cost used to cost these items of resource use.
The relationship between HAQ score and disease-related hospital costs was estimated using the NOAR database. A physician survey was conducted to assess the ongoing costs of psoriasis, therefore estimating the relationship between PASI. This was done for four hypothetical patients with differing PASI scores. The median responses on resource utilisation were to generate costs. A logarithmic regression was then fitted to the data points to estimate cost based on a continuous PASI scale.
The base-case results show that infliximab is the most costly strategy (£104,772).
Summary of cost-effectiveness
An individual sampling model is used to simulate the disease progression of a cohort of patients with PsA over a lifetime horizon. The model is written in ‘R’ with an accompanying evidence synthesis model written in winbugs.
Initial response to treatment is determined according to the PsARC criteria at the end of the initial 3-month period. Patients who do not respond according to PsARC take the next available treatment in the sequence. Patients who respond according to PsARC criteria remain on treatment unless they withdraw due to either loss of efficacy or toxicity. Three-monthly cycles are used.
It is assumed that patients who do not receive an biologics agent after failure of two conventional DMARDs would continue treatment with an alternative conventional DMARD.
The ICER for infliximab is unlikely to be considered acceptable given current levels for the threshold (ICER = £199,596 compared with adalimumab). Etanercept is dominated by adalimumab. Adalimumab has an ICER of £29,827 compared with a DMARD.
Probabilistic sensitivity analysis was conducted and shows that there is considerable uncertainty regarding the optimum strategy. Adalimumab had a probability of < 0.5 of being cost-effective at thresholds up to £30,000. This rose to around 0.7 at thresholds of > £60,000.
Multiple univariate sensitivity analysis were conducted to assess the models sensitivity to effectiveness parameters, withdrawal rates, disease progression estimates, utilities, costs, rebound effect, characteristics of patients and discounting. Results were sensitive to many of the changes in parameters, in particular the stopping rule for BSRBR withdrawal rates and the rebound assumption. The impact on decision uncertainty using alternative parameter assumptions was not presented.
Comments
This is a comprehensive evaluation of biologics for the treatment of PsA. There are, however, a number of limitations. In particular, the model assumes that after failing biologics, patients will receive another DMARD, or combinations of DMARDs. This is un-realistic as patients have previously failed two or more DMARDs. Placebo response rates from trials were also used to represent the DMARD efficacy data. This means that DMARDs will have no effect but will incur costs, biasing against DMARDs. The authors do not give a clear rationale for not choosing palliative care as the comparator to biologics.
Withdrawals were calculated using data from a single data set. There are other potential registry data sets available, which could have been synthesised with the data by Saad et al. 162 In addition, parameters for a Weibull distribution were derived using longitudinal data from three time points and the data were assumed to be independent. This assumption is incorrect, because the same patients contribute data to the probability of survival at 2 years as 1 year. Only one scenario was used to determine HAQ following rebound – that patients will rebound equivalent to the initial gain.
Internal validity
There are no major issues with internal validity.
The model results have been checked and verified by the assessment team. There are some issues with the cost estimates used in the model. These cannot be ratified with the costs presented in the report. In particular the drug, monitoring and administration costs in the model differ from those presented in the report.
External validity
The use of DMARDs as a comparator to biologics is a major limitation. As discussed, DMARDs are unlikely to be considered for patients withdrawing from biologic treatment, as this cohort of patients will have previously failed two or more DMARDs.
In addition, the evidence synthesis uses all available evidence to generate estimates of effect, using data from 10 different sources. However, some of these data sources relate to treatments not included as comparators in the model, such as golimumab. It is not clear if the relative treatment effects can be transferred from one biologic to another.
Checklist for Abbott submission151
| ✓ or ✗ | ||
|---|---|---|
| Study question | Grade | Comments |
| 1. Costs and effects examined | ✓ | |
| 2. Alternatives compared | ||
| 3. The viewpoint(s)/perspective of the analysis is clearly stated (e.g. NHS, society) | ✓ | |
| Selection of alternatives | ||
| 4. All relevant alternatives are compared (including ‘do nothing’ if applicable) | ✗ | Biologics compared with DMARDs and no palliative care |
| 5. The alternatives being compared are clearly described (who did what, to whom, where and how often) | ✗ | Does not describe what the series of DMARDs are |
| 6. The rationale for choosing the alternative programmes or interventions compared is stated | ✓ | |
| Form of evaluation | ||
| 7. The choice of form of economic evaluation is justified in relation to the questions addressed | ✓ | |
| 8. If a cost-minimisation design is chosen, have equivalent outcomes been adequately demonstrated? | NA | |
| Effectiveness data | ||
|
9. The source(s) of effectiveness estimates used are stated (e.g. single study, selection of studies, systematic review, expert opinion) |
✓ | |
| 10. Effectiveness data from RCT or review of RCTs | ✓ | |
| 11. Potential biases identified (especially if data not from RCTs) | ✗ | Limitations of using registry data not discussed |
| 12. Details of the method of synthesis or meta-analysis of estimates are given (if based on an overview of a number of effectiveness studies) | ✓ | Evidence synthesis model is not well annotated and thus is difficult to interpret |
| Costs | ||
| 13. All of the important and relevant resource use included | ✓ | |
| 14. All of the important and relevant resource use measured accurately (with methodology) | ✓ | |
| 15. Appropriate unit costs estimated (with methodology) | ✓ | |
| 16. Unit costs reported separately from resource use data | ✓ | |
| 17. Productivity costs treated separately from other costs | NA | |
| 18. The year and country to which unit costs apply is stated with appropriate adjustments for inflation and/or currency conversion | ✗ | |
| Benefit measurement and valuation | ||
| 19. The primary outcome measure(s) for the economic evaluation are clearly stated | ✓ | |
| 20. Methods to value health states and other benefits are stated | ✓ | |
| 21. Details of the individuals from whom valuations were obtained are given | ✓ | |
| Decision modelling | ||
| 22. Details of any decision model used are given (e.g. decision tree, Markov model) | ✓ | |
| 23. The choice of model used and the key input parameters on which it is based are adequately detailed and justified | ✓ | Do not give adequate justification for why an individual sampling model is used |
| 24. All model outputs described adequately | ✗ | Calculation of withdrawal rates is not clear |
| Discounting | ||
| 25. Discount rate used for both costs and benefits | ✓ | |
| 26. Do discount rates accord with NHS guidance? | ✓ | |
| Allowance for uncertainty | ||
| Stochastic analysis of patient-level data | ||
| 27. Details of statistical tests and CIs are given for stochastic data | ✓ | |
| 28. Uncertainty around cost-effectiveness expressed (e.g. CI around ICER, CEACs) | ✓ | |
| 29. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | ✓ | |
| Stochastic analysis of decision models | ||
| 30. Are all appropriate input parameters included with uncertainty? | ✓ | Costs are fixed |
| 31. Is second-order uncertainty (uncertainty in means) included rather than first order (uncertainty between patients)? | ✓ | Both |
| 32. Are the probability distributions adequately detailed and appropriate? | ✓ | |
| 33. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | ✓ | |
| Deterministic analysis | ||
| 34. The approach to sensitivity analysis is given (e.g. univariate, threshold analysis, etc.) | ✓ | |
| 35. The choice of variables for sensitivity analysis is justified | ✓ | |
| 36. The ranges over which the variables are varied are stated | ✓ | |
| Presentation of results | ||
| 37. Incremental analysis is reported using appropriate decision rules | ✓ | |
| 38. Major outcomes are presented in a disaggregated as well as aggregated form | ✓ | |
| 39. Applicable to the NHS setting | ✓ | |
Review of Schering-Plough submission152
A cohort model was developed to assess the cost-effectiveness of four treatment alternatives: adalimumab, etanercept, infliximab and DMARDs (assumed to represent palliative care) for patients with PsA. Sequential use of biologics was not considered. The report states that a sequence of DMARDs was considered.
The model was programmed in excel with evidence synthesis undertaken in winbugs. A third-party payer perspective was used for the analysis.
Summary of effectiveness data
The primary outcome was QALYs, estimated using both HAQ and PASI. An evidence synthesis model was used to determine the response to biologics and the associated HAQ and PASI change for responders. The evidence synthesis model used to generate initial HAQ and PASI changes and the data used are presented. In many cases results from the York model were used as priors. Data from the previous York model177 along with IMPACT,81 IMPACT 2,82 Mease et al. ,52,78 GO-REVEAL,156 Genovese et al. 83 and ADEPT51 were used in the evidence synthesis model. As change in absolute PASI was modelled, absolute changes in PASI were inferred form relative changes reported in trials. It is also assumed that the average HAQ change in non-responders can be used when data are not reported by responders/non-responders. From this HAQ for responders can be inferred from the aggregate data.
At the end of the first cycle (12 weeks), patients were categorised as responders or not responders according to their PsARC response. Responders continued with treatment, whereas non-responders discontinued treatment and instead received palliative care. The results of the evidence synthesis showed that PASI was not different in individuals with and without a PsARC response. This was concluded using data for golimumab, but assumed for all drugs. All patients start with the same PASI score. PASI change is not assumed to be correlated with baseline score.
The same HAQ and PASI change is assumed for the two 12-week cycles for responders. In addition, a HAQ reduction is also assumed for the third cycle (CiC information has been removed). The HAQ reductions for the second and third cycles are taken from the GO-REVEAL trial156 (this is a trial of golimumab that is not included in the appraisal; however, relationships observed in this trial were assumed across all biologics). For non-responders the HAQ and PASI change is only applied for the first cycle. The placebo effect is then subtracted from the treatment effect (on HAQ) estimated by the evidence synthesis model; however, palliative care in this model is DMARDs (active treatment). This will not bias the comparison between biologic, but may affect the comparison with palliative care.
HAQ is not assumed to progress for patients responding to treatment and is not correlated with initial HAQ change. A sensitivity analysis is conducted assuming that progression for responders is the same as NH. Patients on palliative care (in this case actually DMARDs) will progress in line with NH (0.0719 annual). This is estimated from the Leeds study. 201 The distribution placed on this assumes that the value can only be non-negative. The NH of PASI was assumed to be flat, based on expert opinion (source for this is not stated). Following rebound patients rebounding are assumed to return to their original PASI score.
Two alternative methods to generate utilities were explored: the Gray algorithm180 (selected as the base case) and the Brazier algorithm. 181 The Gray algorithm180 converts SF-36 to EQ-5D then EQ-5D to utilities, whereas the Brazier algorithm181 estimates utilities directly from SF-36. Explanatory variables used in the model were: HAQ, PASI, HAQ2 and PASI2. Interaction between PASI and HAQ was not explored. The GO-REVEAL data was used to estimate the regression.
Annual withdrawals from treatment were taken from the Geborek et al. study198 and are 11.4% per annum. The same withdrawal rate was applied to all strategies. After withdrawal patients will go onto palliative care. Patients also have an annual risk of death. PsA specific mortality multipliers are also included. 29
The results show that palliative care is the strategy associated with the lowest QALYs in all base-case scenarios (5.79 to 6.68 depending on the group of patents). Infliximab is the most effective strategy for all base-case scenarios, for all patients as a group and psoriasis patients (8.65 QALYs for all patients and 8.40 QALYs for patients with psoriasis). For patients without psoriasis etanercept is the most effective (9.14 QALYs).
Summary of resource utilisation and cost data
Resource use associated with treatment, administration and monitoring was taken from the previous York model. Costs associated with adalimumab were assumed to be the same as etanercept. The BNF65 was used to cost medications. Costs for infliximab were calculated using 60-, 70- and 80-kg weights for patients, in addition to the use of four and three and a half vials.
Ongoing costs as a function of HAQ were derived from the Kobelt et al. study. 41 Patients on treatment incur only 85% of these costs, whereas those withdrawing from treatment incur 100%. (CiC information has been removed.)
The base-case results for all patients produce a total cost of £64,704 for palliative care, £99,278 for adalimumab, £108,481 for etanercept, and between £107,954 and £123,475 for infliximab, depending on the weight of patients. Similar patterns were observed separately for patients with minimal psoriasis and patients with psoriasis.
Summary of cost-effectiveness
An initial two cycles of 12 weeks were modelled followed by annual cycles. Half-cycle correction is applied. In the first cycle, patient’s response to PsARC is assessed and his/her associated HAQ and PASI change is determined. PsARC responders on continue with current treatment, whereas those do not respond will move on to palliative care. PsARC responders will then experience an annual risk of withdrawal from treatment with an associated HAQ loss. Two scenarios were modelled for the rebound: rebound equal to gain (followed by NH after 3 months) and rebound equal to NH.
For approximately one-third of patients with no clinically significant psoriasis component to their disease (estimated from the IMPACT79–81,89,96,109,111,113–115,117,118 and IMPACT 282,90,91,95,98,106,112,116 trials) only the change in HAQ is modelled. The PASI impact on QoL is not included for these patients. Costs and QALYs are reported separately for psoriatic and non-psoriatic patients as well as the group as a whole.
The base-case results are presented for 60-, 70- and 80-kg patients and for patients with psoriasis, minimal psoriasis and all patients. For a 60-kg patient, infliximab is the most cost-effective strategy for all patients, and for psoriatic patients, dominating etanercept and extendedly dominating adalimumab. For a 70-kg patient, etanercept is the most cost-effective strategy for all patients and for psoriatic patients, with an ICER of £12,696 compared with adalimumab (however, this is extendedly dominated so should be compared with palliative care, which gives an ICER over £16K) for psoriatic patients and £12,606 for all patients. For an 80-kg patient, etanercept is again the most cost-effective strategy for all patients and for psoriatic patients, with ICERs of £12,696 and £12,606, respectively, compared with adalimumab. For all patient weights, etanercept is the most cost-effective with an ICER of £12,432 compared with adalimumab for non-psoriatic patients.
A number of univariate sensitivity analyses were conducted: reduction in the baseline HAQ, HAQ reduction beyond week 12, non-zero HAQ progression for responders after week 12, reduction in the baseline PASI score, 20-year time horizon as opposed to lifetime, exclusion of phototherapy costs, reduction in annual withdrawals from 11.4% to 5.7%, reduction of NH progression to 0.036 annually and using the Brazier algorithm to calculate utilities. Vial optimisation is not considered in the sensitivity analysis.
Results for the sensitivity analysis are presented as ICERs versus palliative care and ICERs versus other biologics. It is not clear from the results if these results are for psoriatic, non-psoriatic or all patients. The results of the sensitivity analysis appear sensible given the changes in parameter assumptions made, for example, increasing the lifetime of the model makes all biologics more cost-effective.
Biologics appear to be robust to the sensitivity analysis compared with palliative care, apart from changing the algorithm for estimating QoL. This generated ICERs of > £36,000 for all biologics compared with palliative care. For patients with a body weight of < 70 kg, infliximab remained the most cost-effective strategy compared with other biologics, apart from when the baseline HAQ is reduced from 1.14 to 0.90, no HAQ change beyond first cycle is assumed, and HAQ of responders to etanercept, infliximab and adalimumab progress at the same rate as NH after initial HAQ improvement.
Probabilistic sensitivity analysis is also conducted. This shows a great deal of decision uncertainty for the optimum strategies given each of the base-case assumptions.
Comments
This is a good quality evaluation of the relevant biologics for the treatment of PsA. There are, however, a number of issues that are of concern. In particular, the use of data from a trial of golimumab to inform a number of model parameters, the use of DMARDs to represent the comparator, the addition of HAQ gains beyond the initial cycle, and the use of a single data source to estimate withdrawals.
Internal validity
There are no major issues with internal validity.
We were able to replicate the deterministic results. The probabilistic results could not be replicated; however, differences were small and the interpretation of results was the same in terms of ordering of strategies.
External validity
Data from a number of sources were used to estimate benefits of treatments. However, data (CiC information has been removed) from a trial of golimumab was also used to inform a number of parameters, in particular HAQ and PASI changes. This biologic was not included in the model and it is unclear if the relationships observed in this trial can be assumed to transfer across to other biologics. In addition, the estimated placebo effect has been subtracted from the treatment effect (on HAQ); however, palliative care in this model is actually DMARDs (active treatment). This will not bias the comparison between biologics, but may affect the comparison with palliative care.
Withdrawals were also estimated from a single data source, and it was unclear if this is a representative data source. It is of concern that identification of studies to generate withdrawal rates was not more systematic.
Checklist for Schering-Plough submission152
| ✓ or ✗ | ||
|---|---|---|
| Study question | Grade | Comments |
| 1. Costs and effects examined | ✓ | |
| 2. Alternatives compared | ✓ | |
| 3. The viewpoint(s)/perspective of the analysis is clearly stated (e.g. NHS, society) | ✓ | |
| Selection of alternatives | ||
| 4. All relevant alternatives are compared (including ‘do nothing’ if applicable) | ✓ | |
| 5. The alternatives being compared are clearly described (who did what, to whom, where and how often) | ✓ | |
| 6. The rationale for choosing the alternative programmes or interventions compared is stated | ✓ | |
| Form of evaluation | ||
| 7. The choice of form of economic evaluation is justified in relation to the questions addressed | ✓ | |
| 8. If a cost-minimisation design is chosen, have equivalent outcomes been adequately demonstrated? | NA | |
| Effectiveness data | ||
|
9. The source(s) of effectiveness estimates used are stated (e.g. single study, selection of studies, systematic review, expert opinion) |
✓ | |
| 10. Effectiveness data from RCT or review of RCTs | ✓ | |
| 11. Potential biases identified (especially if data not from RCTs) | ✗ | Potential biases of using registry/survey data not discussed |
| 12. Details of the method of synthesis or meta-analysis of estimates are given (if based on an overview of a number of effectiveness studies) | ✓ | winbugs code presented |
| Costs | ||
| 13. All of the important and relevant resource use included | ✓ | |
| 14. All of the important and relevant resource use measured accurately (with methodology) | ✓ | |
| 15. Appropriate unit costs estimated (with methodology) | ✓ | |
| 16. Unit costs reported separately from resource use data | ✓ | |
| 17. Productivity costs treated separately from other costs | ✓ | |
| 18. The year and country to which unit costs apply is stated with appropriate adjustments for inflation and/or currency conversion | ✓ | |
| Benefit measurement and valuation | ||
| 19. The primary outcome measure(s) for the economic evaluation are clearly stated | ✓ | |
| 20. Methods to value health states and other benefits are stated | ✓ | |
| 21. Details of the individuals from whom valuations were obtained are given | ✓ | |
| Decision modelling | ||
| 22. Details of any decision model used are given (e.g. decision tree, Markov model) | ✓ | |
| 23. The choice of model used and the key input parameters on which it is based are adequately detailed and justified | ✓ | |
| 24. All model outputs described adequately | ✓ | Not clear why PASI was predicted for PsARC responders and non-responders |
| Discounting | ||
| 25. Discount rate used for both costs and benefits | ✓ | |
| 26. Do discount rates accord with NHS guidance? | ✓ | |
| Allowance for uncertainty | ||
| Stochastic analysis of patient-level data | ||
| 27. Details of statistical tests and CIs are given for stochastic data | NA | |
| 28. Uncertainty around cost-effectiveness expressed (e.g. CI around ICER, CEACs) | NA | |
| 29. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | NA | |
| Stochastic analysis of decision models | ||
| 30. Are all appropriate input parameters included with uncertainty? | ✓ | |
| 31. Is second-order uncertainty (uncertainty in means) included rather than first order (uncertainty between patients)? | ✓ | |
| 32. Are the probability distributions adequately detailed and appropriate? | ✓ | |
| 33. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | ✓ | |
| Deterministic analysis | ||
| 34. The approach to sensitivity analysis is given (e.g. univariate, threshold analysis, etc.) | ✓ | |
| 35. The choice of variables for sensitivity analysis is justified | ✓ | |
| 36. The ranges over which the variables are varied are stated | ✓ | |
| Presentation of results | ||
| 37. Incremental analysis is reported using appropriate decision rules | ✓ | |
| 38. Major outcomes are presented in a disaggregated as well as aggregated form | ✓ | |
| 39. Applicable to the NHS setting | ✓ | |
Review of Wyeth submission153
An individual patient-based model (discrete event simulation) was developed to assess the cost-effectiveness of etanercept in comparison with infliximab, adalimumab, ciclosporin and best supportive care (BSC) for the treatment of chronic patients with PsA in the UK. Sequences were not considered; instead, patients are given BSC after treatment failure.
In addition to the primary analysis using the patient-level data, subgroups were also defined in the sensitivity analysis. These were mild, moderate and severe HAQ, and mild, severe and very severe PASI.
The model was programmed in excel and the evidence synthesis in winbugs. The model used a 50-year time horizon and a third-party payer perspective. Subgroups at baseline were defined in terms of mild, moderate and severe HAQ, and mild, moderate and severe PASI.
Summary of effectiveness data
Baseline characteristics of patients were taken from the Mease et al. trial. 52 Characteristics at baseline were age, gender, disease duration, HAQ, eligibility for PASI assessment, PASI score, polyarthritis, and concurrent use of MTX. In total, 37.6% of patients in the trial were not eligible for PASI assessment, and were assigned a PASI score of 0.
The benefit of treatments was measured using QALYs. These were estimated using PsARC response and changes in HAQ and PASI. Data from the published MTC for adalimumab179 and the Mease et al. trial52 comparing etanercept with placebo were used to estimate effects. The results from the MTC excluding the data from the open-label study were used as the base case. The inclusion of this study in the MTC was examined in sensitivity analysis. The benefits of ciclosporin are assumed to be equivalent to that of placebo and the data taken from the MTC. PsARC response used to model initial withdrawal from treatment at 12 and 24 weeks. Non-responders according to PASI are assumed not to withdraw.
Response rate at 4 weeks (from Mease et al. 52) applied together with the 12- and 24-week rates from the MTC for adalimunab. 179 Regressions were used to find the relationship between response rates at 12 and 4 weeks (results presented). The initial improvement in PASI 75 (week 4, 12 and 24) was estimated using multivariate regression models and the relationship between patient characteristics.
Response rates by subgroup population were not available from the MTC. Instead response rates, subgrouped according to baseline severity of HAQ or PASI, for etanercept were obtained from the Mease et al. trial. 52 The ratio of etanercept response rates from the MTC179 compared with the etanercept subgroup response rates from Mease et al. 52 were then used in conjunction with the treatment specific response rates from the MTC to estimate subgroup response rates for each of the treatments modelled.
Initial change in HAQ (4, 12 and 24 weeks) was modelled using changes in PASI and PsARC (again from Mease et al. 52 and adalimumab STA179). The same magnitude of change is assumed for all three biologics agents.
Longer-term changes in HAQ were modelled using observed changes in PASI score, PASI 75 response and PsARC response. Changes in PASI are predicted and the results used together with PsARC response to predict changes in HAQ. Results from the regressions are presented.
It is assumed that patients who remain and respond to biologics experience a lack of progression on HAQ. Annual HAQ progression of 0.028 is used for ciclosporin (Sokoll, no reference given). The annual HAQ progression rate (mean = 0.07) for patients on BSC was obtained from the Leeds data set. 201
Longer-term withdrawals (made up on adverse events and loss of efficacy) according to HAQ, were estimated using data from Saad et al. 162 (using the BSRBR registry). A Weibull function was fitted to etanercept data at 1, 2 and 3 years. HRs between infliximab and ETN, and adalimumab and ETN were used to derive survivor functions for infliximab and adalimumab. Ciclosporin is given an annual withdrawal of 34% and assumes patients withdraw exponentially. The effect of withdrawing from treatment is assumed to be either equal to gain or back up to NH.
The relationship between HAQ and EQ-5D observed in the PRESTA data set was used in the base case to generate utilities. The relationship between PASI and EQ-5D was not included, as PASI is already included as a predictor of HAQ. PRESTA is a 24-week clinical study comparing two forms of etanercept. A linear mixed-effect model was used to explore the relationship. Regression results are reported. Other data sets are used in the sensitivity analysis (including the Leeds study used in the original York model201).
Patients have an annual risk of death, taken from UK life tables. PsA specific mortality multipliers are also included. 29
The base-case results show that etanercept was associated with the highest gain in QALYs (6.90) followed by adalimumab (6.54), infliximab (6.39) and then ciclosporin (5.96).
Summary of resource utilisation and cost data
The costs of medication were taken from the BNF. 65 A weight of 70 kg was assumed for infliximab and vial sharing was used. Administration and monitoring was costed as recommended in the BSR guidelines. Etanercept and adalimumab were assumed to be self-administered and thus received zero cost for baseline apart from one outpatient visit at baseline. Infliximab had a half-day care hospital cost assigned for each infusion.
Health-care costs associated with PsA were taken from an evaluation by the Health Outcomes Data Repository (HODaR) using data from BSRBR and The Health Improvement Network (THIN). The THIN database does not include HAQ, thus variables in the BSRBR data set, which were also available in the THIN data, were used to predict HAQ values for the THIN data. Regression results from THIN are reported. Ongoing costs associated with PASI are not included as PASI is assumed to be a predictor of HAQ.
The costs of BSC are assumed to be included in the health-care costs associated the PsA. A sensitivity analysis was conducted to test this assumption.
The base-case results show that ciclosporin was associated with the lowest cost (£53,860). Infliximab had the highest total costs (£66,867).
Summary of cost-effectiveness
An initial two cycles of 12 weeks were modelled followed by annual cycles. Half-cycle correction is applied. Costs and QALYs were discounted by 3.5%.
The base-case results show that infliximab is dominated by adalimumab, and adalimumab is extendedly dominated by etanercept. Comparing etanercept to ciclosporin results in an ICER of £12,480.
A number of univariate sensitivity analyses were conducted: HAQ progression rates, rebound of HAQ on withdrawal from treatment, utility functions, discount rates, monitoring cost for BSC, using results from the MTC, including an open-label study of adalimumab at 24 weeks, withdrawal rates from treatment and subgroups by baseline severity of PsA and PASI. Results are sensitive to the rebound effect, the utility function used and the annual progression on standard care. The results appear to make sense in terms of the changes made to parameters assumptions. For example, increasing the rate of HAQ progressing while receiving biologics increases costs slightly and decreases QALYs for adalimumab, etanercept and infliximab.
Probabilistic sensitivity analysis is also conducted (using 2000 iterations) to generate distributions of total costs and QALYs. This shows a great deal of decision uncertainty for the optimum strategies given each of the base-case assumptions. Probabilistic sensitivity analysis shows there is a 0.65 probability that etanercept will be cost-effective at a threshold of £20,000.
Comments
This is a good-quality evaluation of biologics for the treatment of PsA. There are, however, a number of issues that may cause concern. In particular, the initial change in HAQ and longer-term changes in HAQ were determined including PASI as an explanatory variable. Although PASI and HAQ are used to measure the severity of the two components of PsA, psoriasis and arthritis, there is no clear clinical rationale to suggest that a patient’s psoriasis should affect their degree of functional disability or joint disease, as measured by HAQ. In addition, the same magnitude of initial HAQ change is assumed for all three biologic agents.
Another limitation of the model is the use of ciclosporin as a comparator to biologics as opposed to palliative care; however, the benefits of are assumed to be equivalent to that of placebo. Thus, although the drugs cost are incurred for ciclosporin, no additional benefit beyond that of palliative care is used. This could be expected to bias against ciclosporin.
In addition, withdrawals were calculated using data from a single data set162 and assuming that data from three time points were independent and could be used to derive parameters for a Weibull distribution. The assumption of independence is unlikely to be valid (see Appendix 12). Withdrawal rates could potentially have a large impact on the results, as patients are essentially either in the on treatment or off treatment states, and so it is of concern that identification of studies to generate withdrawal rates was not more systematic.
Internal validity
There are no major issues with internal validity.
It was not possible to replicate the deterministic model results as there was a runtime error in the visual basic macro. Given this, and the anticipated 24 hour + simulation time, we did not attempt to replicate the results of the probabilistic model.
External validity
Data from an existing MTC for adalimumab179 and the Mease et al. trial52 were used to estimate effects. Although data were included from a number of trials in the adalimumab MTC, the original review used to identify trials to populate this MTC was restricted to a review of clinical trials including adalimumab as an intervention.
As discussed above, the use of ciclosporin as a comparator to biologics as opposed to palliative care is unlikely to be appropriate, given that the patients relevant for treatment with biologics will have failed at least two previous DMARDs.
Checklist for Wyeth submission153
| ✓ or ✗ | ||
|---|---|---|
| Study question | Grade | Comments |
| 1. Costs and effects examined | ✓ | |
| 2. Alternatives compared | ✓ | |
| 3. The viewpoint(s)/perspective of the analysis is clearly stated (e.g. NHS, society) | ✓ | |
| Selection of alternatives | ||
| 4. All relevant alternatives are compared (including ‘do nothing’ if applicable) | ✗ | Ciclosporin used as comparator not palliative care |
| 5. The alternatives being compared are clearly described (who did what, to whom, where and how often) | ✓ | |
| 6. The rationale for choosing the alternative programmes or interventions compared is stated | ✓ | |
| Form of evaluation | ||
| 7. The choice of form of economic evaluation is justified in relation to the questions addressed | ✓ | |
| 8. If a cost-minimisation design is chosen, have equivalent outcomes been adequately demonstrated? | NA | |
| Effectiveness data | ||
|
9. The source(s) of effectiveness estimates used are stated (e.g. single study, selection of studies, systematic review, expert opinion) |
✓ | |
| 10. Effectiveness data from RCT or review of RCTs | ✓ | |
| 11. Potential biases identified (especially if data not from RCTs) | ✗ | Does not discuss the bias associated with using registry and survey data. |
| 12. Details of the method of synthesis or meta-analysis of estimates are given (if based on an overview of a number of effectiveness studies) | ✓ | |
| Costs | ||
| 13. All of the important and relevant resource use included | ✗ | Does not include the costs of PASI, as these are used to predict HAQ |
| 14. All of the important and relevant resource use measured accurately (with methodology) | ✓ | |
| 15. Appropriate unit costs estimated (with methodology) | ✓ | Unclear how the costs of HAQ have been used in the model |
| 16. Unit costs reported separately from resource use data | ✓ | |
| 17. Productivity costs treated separately from other costs | NA | |
| 18. The year and country to which unit costs apply is stated with appropriate adjustments for inflation and/or currency conversion | ✓ | |
| Benefit measurement and valuation | ||
| 19. The primary outcome measure(s) for the economic evaluation are clearly stated | ✓ | PASI incorrectly used to predict HAQ |
| 20. Methods to value health states and other benefits are stated | ✓ | |
| 21. Details of the individuals from whom valuations were obtained are given | ✓ | |
| Decision modelling | ||
| 22. Details of any decision model used are given (e.g. decision tree, Markov model) | ✗ | The need to use an individual sampling model was not justified sufficiently |
| 23. The choice of model used and the key input parameters on which it is based are adequately detailed and justified | ✓ | |
| 24. All model outputs described adequately | ✓ | |
| Discounting | ||
| 25. Discount rate used for both costs and benefits | ✓ | |
| 26. Do discount rates accord with NHS guidance? | ✓ | |
| Allowance for uncertainty | ||
| Stochastic analysis of patient-level data | ||
| 27. Details of statistical tests and CIs are given for stochastic data | ✓ | |
| 28. Uncertainty around cost-effectiveness expressed (e.g. CI around ICER, CEACs | ✓ | |
| 29. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | ✓ | |
| Stochastic analysis of decision models | ||
| 30. Are all appropriate input parameters included with uncertainty? | ✓ | |
| 31. Is second-order uncertainty (uncertainty in means) included rather than first order (uncertainty between patients)? | ✓ | |
| 32. Are the probability distributions adequately detailed and appropriate? | ✗ | Not clear how the uncertainty in HAQ costs is propagated |
| 33. Sensitivity analysis used to assess uncertainty in non-stochastic variables (e.g. unit costs, discount rates) and analytic decisions (e.g. methods to handle missing data) | ✓ | |
| Deterministic analysis | ||
| 34. The approach to sensitivity analysis is given (e.g. univariate, threshold analysis, etc.) | ✓ | |
| 35. The choice of variables for sensitivity analysis is justified | ✓ | |
| 36. The ranges over which the variables are varied are stated | ✓ | |
| Presentation of results | ||
| 37. Incremental analysis is reported using appropriate decision rules | ✓ | |
| 38. Major outcomes are presented in a disaggregated as well as aggregated form | ✓ | |
| 39. Applicable to the NHS setting | ✓ | |
Appendix 8 Critique of the manufacturers’ models
Choice of comparator(s)
The submission by Schering-Plough152 compares etanercept, infliximab and adalimumab with palliative care. Wyeth153 and Abbott151 use DMARDs as the comparator to the biologics. Wyeth153 specifies ciclosporin as the DMARD. Patients who fail on biologics or ciclosporin then receive BSC, presumed the same as palliative care. Abbott151 uses a series of unspecified DMARDs as comparators with fourth- and fifth-line treatments always being DMARDs. Although Wyeth153 and Abbott151 compare biologics to DMARDs, they assign effectiveness estimates from the placebo arms of trials. Therefore, the effectiveness of biologics is likely to be artificially inflated.
Patient characteristics
The Schering-Plough model152 uses a homogeneous cohort of patients that was considered to be representative of the groups of patients eligible for biologic therapies to treat PsA, i.e. patients who have failed two or more conventional DMARDs.
Wyeth153 and Abbott,151 however, model heterogeneous cohorts using individual patient simulation. Both of the individual sampling models are difficult to critique and require a significant time to run probabilistic sensitivity analysis. In the Wyeth model153 patients’ characteristics are taken from the Mease et al. 52 trial comparing etanercept and placebo. Characteristics at baseline were age, gender, disease duration, HAQ, eligibility for PASI assessment, PASI score, polyarthritis and concurrent use of MTX. As 37.6% of patients in the trial were not eligible for PASI assessment, these patients were assigned a PASI score of 0. In the Abbott submission, baseline patient characteristics from the ADEPT trial88 were used to determine the baseline distribution of patients characteristics in the model. The ADEPT trial88 compared adalimumab with placebo. Only patients who had failed at lease two DMARDs were included in the analysis. Patients’ characteristics that were included were age, disease duration, gender, presence of psoriasis, percentage on MTX, PASI and HAQ score.
Adjustment for placebo effect
A placebo adjustment accounts for any overestimate of the absolute response rates in both placebo and treatment groups, compared with what would be expected in general practice.
There may be a need to adjust for the placebo effect observed in the clinical trials if the placebo effects in the trials are assumed not to occur in usual practice (see Appendix 9).
The Wyeth153 and Abbott151 models do not make an adjustment for placebo response. Both assume the comparator group represents the effect of DMARD. However, for both of these models the effects observed in the placebo arms of trials are used to represent the effectiveness of DMARDs. In other words, these models assume that DMARDs are no more effective than placebo in these patients.
In the Schering-Plough model,152 the placebo effect is subtracted from the treatment effect (on HAQ) for responders and non-responders on biologics, estimated by the evidence synthesis model. However, palliative care in this model is DMARDs (active treatment). As an inactive treatment is not actually included in any of these three models, the use of a placebo adjustment should have little impact on the results or their interpretation. It will also not bias the comparison between biologics, but may overstate the effectiveness of biologics.
Sequencing
None of the four models considers the use of sequential biologics in the base-case scenario. The Abbott model151 uses a series of unspecified DMARDs, following failure of treatment with any biologic (up to fifth line), but the use of subsequent DMARDs for patients who have previously failed two or more DMARDs is unlikely in practice. A reduction multiplier is applied to response rates for subsequent DMARDs (24% reduction in receiving response in the base case). This reduction is justified using estimates from the BSRBR of the percentage of patients that withdraw on their second biologic at year 1 compared with the first course. A reference for these figures is not given.
The sequential use of biologics is likely to be feasible in practice; however, a lack of data on the effectiveness of biologics beyond first line limits the possibilities to consider such an analysis.
Outcomes of the evidence synthesis
Each of the three industry models uses an evidence synthesis component (implemented in winbugs) to generate estimates of treatment effect (see Chapter 3, Assessment of effectiveness). The Wyeth study153 uses the evidence synthesis from a previous STA of adalimumab179 and does not develop a de novo synthesis for this appraisal. The need for an evidence synthesis component is primarily because of the lack of head-to-head data from trials for the three biologics, thus there is a need to use a MTC model. Each model, however, generates different parameters using different data.
The model by Wyeth153 generates estimates of PsARC and PASI 75 at 12 and 24 weeks using data from the published MTC for adalimumab179 and the Mease et al. trial. 52 A regression was undertaken to predict 4-week PsARC (from Mease et al. 52) from 12-week PsARC. Response rate at 4 weeks is applied, together with the 12- and 24-week rates, from the MTC for adalimunab. 179 The initial improvement in PASI 75 (weeks 4, 12 and 24) was estimated using multivariate regression models and the relationship between patient characteristics.
Schering-Plough152 estimates PsARC at 12 weeks for responders and non-responders. In the subgroup with > 3% body skin area PASI change from baseline at 12 weeks by PsARC response/no response was estimated. The prediction of PASI change by PsARC response is somewhat questionable. Schering-Plough152 also determine HAQ change at 12 weeks by PsARC response/no response and treatment drug was also estimated. In many cases the results from the previous York model were used as priors. The Abbott study151 used a mixed-treatment fixed-effects meta-analysis to determine: (1) joint distribution of 12-week PsARC and ACR response rates; (2) 24-week PsARC response conditional on the 12-week PsARC response; and (3) 24-week ACR response conditional on the 12-week ACR response. The joint distribution of 12- and 24-week PASI response rate is modelled independently. The results of the bivariate meta-analysis to determine the joint distribution of PsARC and ACR responses appears to differ from the estimates of the marginal probabilities of these two outcomes, shown in Tables 22 and 24. In these tables, infliximab is most effective, followed by etanercept, then adalimumab. In the bivariate meta-analysis (Table 3.4.3.1.1 of the Abbott submission151), Abbott151 find that adalimumab is more effective than etanercept for PsARC and ACR responses. The reason for this discrepancy is not clear.
Decision to withdraw depending on initial response(s)
All of the industry models assume that patients are withdrawn from treatment if they are PsARC non-responders at 12 weeks, irrespective of PASI response. In addition, the Wyeth model153 also allows patients to be withdrawn from treatment if they are non-responders at 24 weeks. Abbott151 conduct a sensitivity analysis in which continuation beyond 12 weeks is estimated directly from the BSRBR,162 and so PsARC response rates are not used to determine continuation. None of the industry models considers the possibility of different scenarios for discontinuation, for example, the possibility that there may be a response on either PsARC or PASI or both.
Initial change in Health Assessment Questionnaire for responders and non-responders
Schering-Plough152 predicts HAQ by PsARC response and treatment from the evidence synthesis. The latest available end points for HAQ were used to reflect short-term benefits. The same HAQ change is assumed for the two initial 12-week cycles for responders. In addition, a HAQ reduction is also assumed for the third cycle (–0.0313). The HAQ reductions for the second and third cycles are taken from (CiC information has been removed). For non-responders, the HAQ change is only applied for the first cycle, after which a NH progression is assumed.
The Abbott study151 predicts HAQ at 12 and 24 weeks as a function of ACR response (20, 50, etc.), baseline HAQ, age, gender, baseline PsA duration, concomitant MTX and if receiving biologic drugs (ADEPT88). HAQ does not differ by biologic drug.
The Wyeth study153 estimates the initial change in HAQ (4, 12 and 24 weeks) using changes in PASI, baseline HAQ and PsARC (from Mease et al. 52 and adalimumab STA179). The same magnitude of change is assumed for all three biologic agents. Despite the justification given in the report for using PASI to predict HAQ, the use of the skin component of PsA to predict the arthritis component of the disease is of doubtful validity. There is no evidence to suggest that one component of the disease is a good predicator of the other: patients can have differing degrees of both components and those with severe arthritis will not necessary have severe psoriasis and vice versa.
Health Assessment Questionnaire progression while responding on a biologic therapy
As in the earlier York Assessment Group model, Wyeth153 and Schering-Plough152 assume that HAQ does not progress for patients who are responding to a biologic therapy. The Schering-Plough model152 incorporates a slight improvement in HAQ over the first year. The Abbott model151 assumes that HAQ will worsen by 0.0005 per year. This figure was taken form a longitudinal analysis of the Bath Psoriatic Arthritis Database (reference not given).
The Abbott model151 also models a subgroup of patients where ACR < 20 separately and uses a HAQ progression rate of 0.066 per year from the Leeds cohort. 201
Health Assessment Questionnaire progression when on disease-modifying antirheumatic drugs
In the Schering-Plough model152 the comparator is palliative care, and thus progression is assumed to be that of NH (0.066 per year). 201 For the Abbott151 and Wyeth153 models, DMARDs are used as comparators. Abbott151 uses an annual rate of progression of 0.024 from the Leeds cohort study. 201 Wyeth153 uses a similar rate of 0.028 from Sokoll (reference not given).
Health Assessment Questionnaire progression while not on biologic therapy
All of the industry models use the Leeds cohort study201 data to estimate HAQ progression while not on biologic therapy (also called NH progression). The Abbott study151 estimates this as a 0.066 increase in HAQ per year, Wyeth153 an 0.069 increase and Schering-Plough152 an 0.071 increase per year. It is not clear why the same data source appears to generate three slightly different estimates, but these differences are unlikely to have major impacts on the cost-effectiveness results.
The Leeds data set is small, including only 24 patients. In addition, patients surveyed do not meet the requirements for this analysis in that many have not failed at least two previous DMARDs. It is also not clear if patients met the current guideline criteria for initiating biologics for PsA (three tender and three swollen joints).
Initial change in psoriasis severity while on biologic therapy
Each of the models uses a different approach to estimate the initial change in psoriasis severity after treatment with a biologic. The Wyeth study153 generates the initial improvement in PASI 75 (weeks 4, 12 and 24) using multiple regression models and the relationship between patient characteristics. Schering-Plough152 estimates the PASI change from baseline to 12 weeks for PsARC responders/non-responders in their evidence synthesis model. As change in absolute PASI was modelled, absolute changes in PASI were inferred form relative changes reported in trials. It is not clear why PASI change was estimated for PsARC responders and non-responders, and not for PASI responders. Abbott151 predict the initial (12-week) change in PASI, using baseline PASI and proportion who are PASI 50/75/90 responders. Abbott151 also predicts this at 24 weeks.
Correlation between Psoriasis Area and Severity Index and Psoriatic Arthritis Response Criteria responses
Biologics are intended to treat both joint disease and psoriasis. Clinical response at 3 months is measured using the PsARC for joints and PASI 75 for skin conditions for these two aspects, respectively. The PsARC and PASI 75 responses are not necessarily independent (see Appendix 10).
Each of the industry models uses a different approach to account for any correlation between PASI and PsARC responses. The Wyeth model153 assumes that PASI is a predictor of HAQ (see Appendix 8 for further detail), which is unlikely. Abbott151 assumes that they are independent and thus models them separately (see Appendix 8 for further detail). The Schering-Plough model152 predicts PASI by PsARC response, thus generating a different PASI change for PsARC responders and non-responders, by drug.
Psoriasis progression on and off biologic therapy
Each of the models assumes that psoriasis will not progress on or off treatment, i.e. psoriasis will not worsen over time. This assumption is justified quoting clinical opinion, although this is not referenced.
Health Assessment Questionnaire rebound after discontinuation of biologic therapy
Following withdrawal from treatment, either due to adverse events or loss of efficacy, it can be expected that there will be some change in patients’ HAQ scores. The previous York model177 looked at two possible scenarios for this: rebound by the same amount as initial gain and rebound back to NH progression (see Appendix 11). The models from Wyeth153 and Schering-Plough152 also explore these two scenarios. The ICERs for all biologics increase significantly. The Abbott model151 uses only the rebound to initial gain scenario, as it states that rebound to NH is unlikely to be possible as halting joint destruction does have an impact on long-term disability.
Psoriasis rebound when stopping therapy
Each of the industry models assume that following withdrawal from treatment, patients PASI score will rebound by the original gain. As PASI is not assumed to progress while receiving treatment, the rebound will be to the original PASI score. Clinical opinion is cited as the source of this evidence, but no reference is given.
Withdrawal rates
To estimate the probability of withdrawal while receiving biologics, due to either loss of efficacy or adverse events, Schering-Plough152 uses the same rates as used in the previous York model (0.11 per year from Geborek et al. 198 beyond the initial 12-week period) for biologics. As the comparator is palliative care (in active treatment) no withdrawals were seen in the comparator arm.
Wyeth153 and Abbott151 use evidence from a recent paper by Saad et al. ,191 which used data from the BSBDR registry to estimate parameters of a Weibull distribution in order to quantify the rate of withdrawal over time. This is used to represent a common withdrawal probability for all biologics. On seeking clarification from Wyeth,153 they confirmed that a Weibull curve was fitted to the proportion of patients on etanercept at 1, 2 and 3 years. Calibrating the two parameters of the Weibull function was undertaken in order to minimise the error between the observed and predicted proportion of patients still treated with etanercept. The root mean square error between the observed and predicted proportion was 0.01961. On seeking clarification from Abbott151 they confirmed that the reported figures in Table 2 of Saad et al. 191 These are slightly lower than the values fitted in the Wyeth analysis. 153 A diagram showing observed versus predicted survival was presented. (CiC information has been removed.) No further details of this study were presented.
There are a number of issues with the Wyeth153 and Abbott151 approach. First, no justification was given for the choice of Weibull distributions rather than other parametric distributions. It may be that other distributions offered a better fit. Second, the 1-year rates from the BSRBR are likely to include non-responders to biologics in addition to those who withdraw due to loss of efficacy or adverse events after the initial 3-month period. As these initial withdrawals are already counted as non-responders, there is a degree of double counting. Third, this approach assumes that the data points are independent, which is unlikely.
Utility estimates
Each of the industry models uses different methodologies and data sets to link changes in HAQ and PASI to utilities, in order to generate QALYs (Table 53).
| Regression estimates | |
|---|---|
| aWyeth153 | HAQ = –0.45586 (SE = 0.027047) |
| Age = –0.00096 (SE = 0.000511) | |
| Gender = 0.020057 (SE = 0.012448) | |
| Age: HAQ = 0.003089 (SE = 0.000516) | |
| Male: HAQ = –0.03876 (SE = 0.011613) | |
| Intercept = 0.899592 (SE = 0.025597) | |
| bSchering-Plough152 | Intercept = 0.6442260 (SE = 0.0115177) |
| sHAQ = –0.1610008 (SE = 0.0087963) | |
| sPASI = –0.0375632 (SE = 0.0132345) | |
| sHAQ2 = –0.0050072 (SE = 0.0067073) | |
| sPASI2 = 0.0051515 (SE = 0.0030365) | |
| cAbbott151 | Intercept = 0.9144 (SE = 0.0186) |
| HAQ = –0.2512 (SE = 0.0189) | |
| PASI_t = –0.0355 (SE = 0.0096) |
The Wyeth model153 uses the relationship between HAQ and EQ-5D, observed in the PRESTA data set (a clinical of etanercept including 752 patients),157 to generate utilities. The relationship between PASI and EQ-5D was not included, as PASI is already included as a predictor of HAQ in the Wyeth model. 153 PRESTA is a 24-week clinical study comparing two forms of etanercept. A linear mixed-effect model was used to explore the relationship. The use of other data sets is explored in sensitivity analysis, including the Leeds study and the Mease et al. data. 52 The ICER of etanercept compared with ciclosporin was £12,666 (using the function from Leeds), and £15,795 (using the function from patients receiving adalimumab) compared with £31,828 when using the function from Mease et al.
The Schering-Plough152 model explores two alternative methods to generate utilities: the Gray algorithm180 and the Brazier algorithm. 181 The Gray algorithm180 converts SF-36 profiles to EQ-5D profiles, and then EQ-5D profiles to utilities. The Brazier algorithm181 estimates utilities directly from SF-36. The Gray algorithm180 was used in the base-case analysis. The GO-REVEAL156 trial data were used in a multiple regression model using HAQ, PASI, HAQ2 and PASI2, with no interaction terms, as explanatory variables. The Abbott model151 uses the ADEPT trial88 of adalimumab versus placebo to estimate utility through a direct linear relationship with HAQ and PASI collected in the trial. The base case uses the SF-36, collected in the trial, converted to EQ-5D. In a sensitivity analysis, data from the Bath Psoriatic Arthritis Database was used (no reference given). Again any interaction between HAQ and PASI was not explored.
There is some uncertainty regarding which of the industry regression models is appropriate to generate utilities.
Mortality
All of the industry models use UK life tables along with PsA specific mortality multipliers29 to estimate mortality. Each also uses the same mortality rate for all treatments and no treatment (i.e. there is not differential impact of the alternative therapies on mortality). This assumption is reasonable, although there may be a beneficial effect of biologics on mortality; however, data to quantify this are not available.
Costs of treatment, start-up, administration and monitoring
Each industry model presents information, to a differing degree, on the resource use and unit costs used to cost drug treatment, administration of drugs and monitoring of patients. Of concern is the fact that in the Abbott model151 the total costs given in the report could not be replicated in terms of the resource use items and unit costs presented. These also appear to differ from the costs used in the model, where drug costs are split into direct and indirect costs with no accompanying definition provided in the report.
The BNF65 was used to cost medications in the Wyeth153 and Schering-Plough152 submissions. MIMS216 was used in the Abbott submission. 151 However, unit costs are consistent across the industry models: £419.62 per vial of infliximab, £89.38 per vial of etanercept and £357.50 per vial of adalimumab. Despite the consistency in unit costs, there are some differences in the medication costs for the industry models (Table 54). A number of differences in costing methodology explain this. First, different assumptions were made regarding the use of vials and patient weight for infliximab. The Abbott study151 assumes that four vials were used per infusion, based on an average patient weight of 80 kg. The Wyeth study153 assumes a patient weight of 70 kg and allows vial sharing. The Schering-Plough study152 explores various scenarios to cost infliximab, using 60-, 70- and 80-kg weights for patients, in addition to the use of four and three and a half vials. All models assume that 5-mg infliximab is given per kg. Second, there are some differences in the number of vials used for the biologics in the different time periods. Schering-Plough152 and Abbott151 assume that three doses of infliximab are given in the initial 3-month period (at 0, 2 and 6 weeks). This is followed by doses every 8 weeks. Wyeth153 gives infliximab at 0, 2 and 6 weeks and then every 6–8 weeks. Thus, four doses are given in the initial 3-month period, as opposed to three in the Schering-Plough152 and Abbott151 models. All three industry models assume that six vials of adalimumab are given in the first period. Abbott151 then assumes that seven vials are given in months 3–6, followed by six and a half vials in subsequent 3-month periods. Wyeth153 assumes that six vials are given in all subsequent cycles. Schering-Plough152 assumes that six vials for the 3- to 6-month period, followed by six and a half vials for subsequent 3-month periods. All three models assume that 24 vials of etanercept are given in the initial 3-month period. Wyeth153 continues to give 24 vials for all subsequent 3-month periods. Schering-Plough152 gives 24 vials for months 3–6, followed by 26 for subsequent 3-month periods. Abbott151 gives 28 vials in the 3- to 6-month period, followed by 26 vials in all subsequent periods.
| Manufacturer and time frame | Strategy | Costs (£) | ||||
|---|---|---|---|---|---|---|
| Drug | Administration | Monitoring | Total | |||
| aAbbott151 | ||||||
| From report | ||||||
| 0–12 weeksb | Etanercept | 2324 | 194.5 | 2518.5 | ||
| Adalimumab | 2324 | 194.5 | 2518.5 | |||
| Infliximab | 4196 | 1263 | 5459 | |||
| DMARD | 70.5 | 363.5 | 434c | |||
| 12–24 weeks | Etanercept | 2324 | 194.5 | 2518.5 | ||
| Adalimumab | 2324 | 194.5 | 2518.5 | |||
| Infliximab | 4196 | 1263 | 5459 | |||
| DMARD | 70.5 | 363.5 | 434 | |||
| 24 weeks + (3-month costs) | Etanercept | 2324 | 152 | 2476 | ||
| Adalimumab | 2324 | 152 | 2476 | |||
| Infliximab | 2727.5 | 1018.5 | 3746 | |||
| DMARD | 70.5 | 328 | 398.5 | |||
| From model code | ||||||
| 0–12 weeks | Etanercept |
2145.12 (direct), 2239.64 (indirect) |
236.73 | |||
| Adalimumab |
2145 (direct), 2239.52 (indirect) |
236.73 | ||||
| Infliximab |
5035.44 (direct), 5319 (indirect) |
1507.73 | ||||
| DMARD |
65.15 (direct), 85.49 (indirect) |
399.07 | ||||
| 12–24 weeks | Etanercept |
2502.64 (direct), 2597.16 (indirect) |
151.98 | |||
| Adalimumab |
2502.5 (indirect), 2597.02 (indirect) |
151.98 | ||||
| Infliximab |
3356.96 (direct), 3546 (indirect) |
1018.48 | ||||
| DMARD |
76.01 (direct), 93.96 (indirect) |
328.04 | ||||
| 24 weeks + (3-month costs) | Etanercept |
2323.88 (direct), 2418.40 (indirect) |
151.98 | |||
| Adalimumab |
2323.75 (direct), 2418.27 (indirect) |
151.98 | ||||
| Infliximab |
2727.53 (direct), 2881.13 (indirect) |
1018.48 | ||||
| DMARD |
70.58 (direct), 87.60 (indirect) |
328.04 | ||||
| dSchering-Plough152 | ||||||
| 0–12 weeks | Infliximab |
4 vials 3.5 vials 3 vials |
5035 4406 3776 |
372 | 225.78 | 4374.36e |
| Etanercept | 2145 | 394.09 | 225.78 | 2764.99 | ||
| Adalimumab | 2145 | 394.09 | 225.78 | 2764.87 | ||
| 12–24 weeks | Infliximab |
4 vials 3.5 vials 3 vials |
3356 2937 2517 |
248 | 50.39 | 2816.11f |
| Etanercept | 2145 | 0 | 90.40 | 2235.52 | ||
| Adalimumab | 2145 | 0 | 90.40 | 2235.40 | ||
| 24 week + (3-month costs) | Infliximab |
4 vials 3.5 vials 3 vials |
2727.53 2386.58 2045.65 |
201.5 | 54.59 | 2301.74f |
| Etanercept | 2323.88 | 0 | 97.93 | 2421.81 | ||
| Adalimumab | 2323.75 | 0 | 97.93 | 2421.68 | ||
| gWyeth153 | ||||||
| First 3 months | Etanercept | 2145.12 | 71 | 66 | 2282.12 | |
| Adalimumab | 2145 | 71 | 66 | 2282.00 | ||
| Infliximab | 5874.68 | 345.69 | 65.98 | 6286.35 | ||
| MTX | 9.11 | 0 | 144.64 | 224.75 | ||
| Ciclosporin | 498.23 | 71 | 139.95 | 709.17 | ||
| Between 3 and 6 months | Etanercept | 2145.12 | 0 | 33 | 2178.12 | |
| Adalimumab | 2145 | 0 | 33 | 2178.00 | ||
| Infliximab | 2937.34 | 230.46 | 32.99 | 3200.79 | ||
| MTX | 9.11 | 0 | 58.32 | 67.43 | ||
| Ciclosporin | 498.23 | 0 | 33.96 | 532.18 | ||
| 6 months + (3-month costs) | Etanercept | 2145.12 | 0 | 16.50 | 2161.62 | |
| Adalimumab | 2145 | 0 | 16.50 | 2161.50 | ||
| Infliximab | 2937.34 | 230.46 | 16.49 | 3184.29 | ||
| MTX | 9.11 | 0 | 58.32 | 67.43 | ||
| Ciclosporin | 498.23 | 0 | 33.96 | 532.18 | ||
All of the three submissions state that they use the BSR guidelines to determine the resource use associated with administering drugs and monitoring patients; however, there are differences in the estimates of administration and monitoring costs in the various time periods.
The Abbott model151 assumes that etanercept and adalimumab were self-administered and incur the cost of a single outpatient visit (£115) in the initial 3-month period. This assumption was also made in the Wyeth153 and the Schering-Plough152 models; however, an outpatient visit is assigned a cost of £222.71 in the Schering-Plough model152 and a cost of £71 in the Wyeth model. 153 The Schering-Plough model152 also assumes an additional 4 hours of staff nursing time for follow-up (£150.58).
In the Abbott model,151 infliximab has a half-day care hospital cost assigned for each infusion (£462 multiplied by three infusions). This cost is taken from NHS Reference Costs2007–08 for a day case for inflammatory spine, joint or connective tissue disorders without complications. The Wyeth model153 also assumes a hospital cost for each infusion of infliximab; however, this is much lower, at £115.23 per half day for each infusion, taken from published hospital costs. 217 The Schering-Plough model152 uses a cost of £124 per half day, citing results of a multiple technology appraisal (MTA).
In terms of monitoring costs, for the initial 3-month period the Schering-Plough model152 assumes a second outpatient visit for all biologics at £135.71 per visit. In addition, there is £90.07 of laboratory costs. This includes the cost of a full blood count (FBC), ESR, liver function test (LFT), urea and electrolytes (U&E) test, chest radiograph, TB Heaf test, antinuclear antibodies (ANAs) and DNA binding [double-stranded (dsDNA)]. Outpatients visits are then reduced to 0.23 of a visit for infliximab and 0.46 for etanercept and adalimumab in the 3- to 6-month period. Laboratory costs are also reduced to £19.07 for all biologics. In periods beyond 6 months patients receiving infliximab are assumed to require 0.25 of an outpatient visit, and patients being treated with etanercept and adalimumab are assumed to require 0.5 of a visit. Laboratory costs are £20.66 for all biologics.
The Wyeth model153 assumes that all biologics patients will require one FBC at £5.50, one ESR at £3.86, one LFT at £12 and one U&E test at £11.64 in the first 3 months. For subsequent 3-month periods they will incur only 50% of these costs. The Abbott151 model assumes that all biologics patients will receive two FBCs at £15.19 each, two ESRs at zero cost, two LFTs at £8.43 each, two comprehensive metabolic panel (CMP) tests at £8.43 each and one chest radiograph at £27.25 in the first 3 months. In the subsequent 3-month periods, patients will receive tests at the same intensity, but will not require a chest radiograph.
Costs depending on Health Assessment Questionnaire and costs of psoriasis
Each of the models estimates the ongoing costs of PsA in relation to HAQ and PASI scores (Table 55). The Abbott model151 estimates the relationship between HAQ score and disease-related hospital costs using data on resource use by HAQ from the NOAR database. It is difficult to assess the validity of this approach, as the NOAR report used in the Abbott submission151 was not made available to the Assessment Group on request. As the NOAR data did not include any measure of uncertainty in the mean estimates of resource use, the estimates of the SEs of mean costs in the Abbott submission151 cannot be valid. The Schering-Plough model152 derives these estimates from the UK data of a study by Kobelt et al. ,41 which was used in the previous York Assessment Group model. The Kobelt et al. data41 include the costs of RA drugs, primarily DMARDs. As per the previous York model, patients on biologic treatment incur only 85% of these costs, whereas those withdrawing from biologic treatment incur 100%. The Wyeth model153 uses an evaluation by HODaR, utilising data from BSRBR and THIN to estimate the costs associated with HAQ. The THIN database does not include HAQ, thus variables in the BSRBR data set that were also available in the THIN data were used to predict HAQ values for the THIN data. A general linear modelling approach was taken and regression results from THIN were reported. However, prediction errors from the BSRBR/THIN regression were not included in the first regression of predicted HAQ values on to the observed costs. As such, the goodness of fit and uncertainty estimates do not reflect all of the uncertainty in the prediction. The costs used in the Wyeth submission153 are difficult to interpret and the costs by HAQ score are not presented. It is also not clear how estimates of uncertainty were derived.
| Costs (£) | |
|---|---|
| HAQ | PASI |
| Abbott151 | |
| By HAQ score:a | PASI state 1: score = 1.5 (1.5 to 2.7) = 153.68b |
| 0.0 < 0.5 = 121 (59–173) | PASI state 2: score = 9 (7 to 11.2) = 933.62 |
| 0.5 < 1.0 = 77 (43–109) | PASI state 3: score = 15 (12.6 to 16.8) = 859.35 |
| 1.0 < 1.5 = 269 (141–382) | PASI state 4: score = 40 (32.4 to 43.2) = 1002.83 |
| 1.5 < 2.0 = 388 (206–550) | |
| 2.0 < 2.5 = 909 (459–1295) | |
| 2.5, 3.0 = 1945 (958–2778) | |
| Schering-Plough152 | |
| Constant: mean = 1325, SE = 466 | (CiC information has been removed) |
| Slope: mean = 401, SE = 259 | |
| Wyeth153 | |
| Does not present HAQ by score. Uses £2.05 per 3 months from sum of regression coefficients (also does this for SE). Cannot determine how this has been used in the model | – |
The Abbott151 and Schering-Plough152 models both conduct separate physician surveys to assess the ongoing costs of psoriasis in relation to PASI. Abbott151 uses four hypothetical patients with differing PASI scores to generate costs. A logarithmic regression was then fitted to the median responses to estimate 6-month costs, based on a continuous PASI scale. It is not clear how many physicians were surveyed. Schering-Plough152 sample from 20 dermatologists to determine NHS costs associated with various PASI scores. The report does not say how the responses were synthesised. Wyeth153 does not generate costs associated with PASI, as PASI was assumed to be a predictor of HAQ in their model. Each of the industry models relies on survey data to estimate the costs associated with psoriasis. This could be associated with a number of biases.
Patient subgroups
The Schering-Plough model152 reports results separately for psoriatic and non-psoriatic patients. For approximately one-third of patients with no clinically significant psoriasis (estimated from the IMPACT81 and IMPACT 282 trials) only the change in HAQ is modelled. The PASI impact on HRQoL is not included for these patients. They do not consider variation in baseline HAQ.
The Wyeth153 and Abbott151 models use the variation in baseline disease severity (measured using both HAQ and PASI) to explore the cost-effectiveness of treatments for subgroups. This is preferred to the approach used by the Schering-Plough model,152 as it allows the comparison of a greater number of subgroups, defined not only by the presence or absence of psoriasis, but also by their severity of disease according to PASI and HAQ.
Appendix 9 Generalising the results of randomised controlled trials to general practice
Introduction
Chapter 3, Results of review of clinical effectiveness, showed that biologic drugs are much more effective than placebo controls in the experimental setting. The RCT is generally accepted as the best method to estimate an unbiased measure of the relative effectiveness of the treatment, in this case versus a placebo control, whether that relative effect is measured on a proportionate scale, such as an OR, or as a difference in means between groups. However, RCTs are not necessarily predictive of the absolute effectiveness of the intervention in general practice.
Any medical intervention can be thought of as a complex set of factors, of which the active pharmaceutical ingredients are only one component, albeit usually an important one. Other components of the intervention might include the relationship between the doctor and patient, interventions by other health professionals, and the patient’s expectations, all of which to a greater or lesser extent, and for better or worse, contribute towards the overall outcome. Selection effects, or ‘regression to the mean’, may also play a part. These ‘non-pharmacological’ components of the intervention can be thought of as acting equally in the intervention and placebo arms of clinical trials, assuming that both doctors and patients are blinded as to the treatment arm. In these circumstances, the effect observed in the placebo arm of the trial measures the effectiveness of these non-pharmacological components, while the ‘treatment difference’ measures the independent effectiveness of the pharmacological component of the intervention.
Predicting the absolute effectiveness of the intervention in general practice requires some assumption to be made about whether the protocols, procedures and general ‘quality of care’ of the RCT are similar to general practice. A Cochrane Review218 found little evidence that using a placebo improved symptoms, with the exception of pain relief. However, the key question is not whether the ‘placebo effect’ is operating in every case, but whether outcomes associated with non-pharmacological components of the treatment are generalisable from RCTs to clinical practice. In other words, it matters less how the treatment works than whether it works. 189
This generalisability would not matter too much if the decision model were comparing ‘placebo’ with ‘biologic therapy’, as both groups would experience the same non-pharmacological components of therapy. However, NICE will not compare an active therapy with a placebo, even if it were shown to be effective: it compares active therapies with ‘standard practice’ which in this case is assumed to be palliative care only. Adding the doctor’s caring to the medical care component of biologic therapy might affect the patient’s experience of treatment and may, for example, reduce pain and affect outcome. The ‘no-treatment’ group might or might not receive equivalent non-pharmacological care.
We can represent these possibilities as two scenarios:
-
Scenario 1 The ‘no-treatment group’ receives similar care (with similar mean outcomes) to the placebo arm in an RCT.
-
Scenario 2 The ‘no-treatment group’ receives less care than the placebo arm in an RCT, and does not achieve the response rate of the placebo arm in an RCT.
Conceptual framework
Figure 8 shows the mean change in HAQ ΔYjr from 0 to 12 weeks in the RCTs in the treatment group j = 1 and placebo group j = 0, depending on response, r = 1,0. These parameters were estimated in the evidence synthesis in Chapter 3. Variable α represents the change in HAQ over 3 months if there is no response for patients with placebo. Variable™ represents the mean difference in the change in HAQ between placebo non-responders and placebo responders. Variable ®j represents the mean difference in the change in HAQ between placebo non-responders and non-responders with treatment, j. Variable ©j represents the mean difference in the change in HAQ between placebo non-responders and responders with treatment, j.
FIGURE 8.
Change in HAQ from 0 to 12 weeks in treatment groups estimated by RCTs.
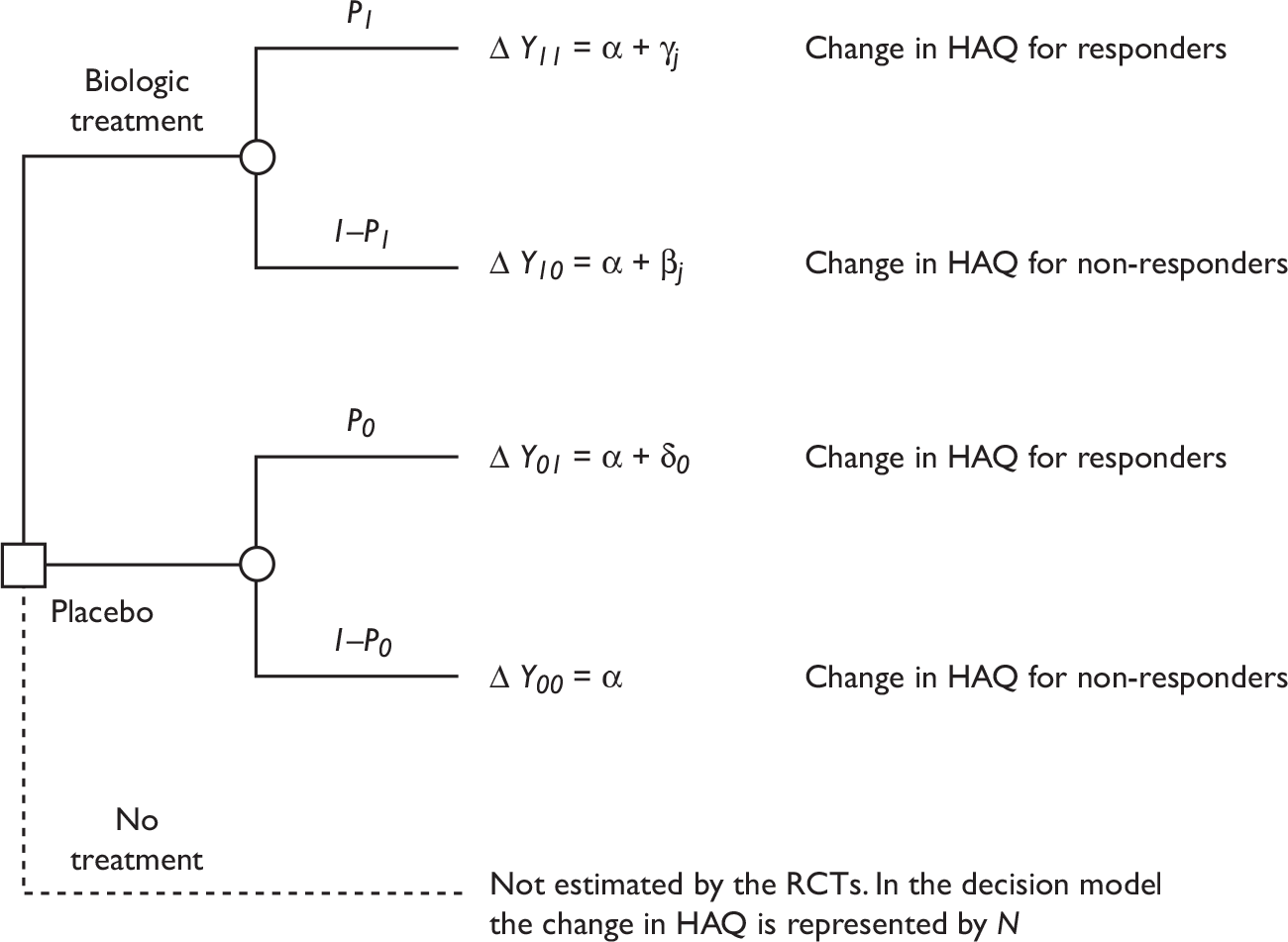
The average change in HAQ (over responders and non-responders) in the placebo arm is:
We can represent these scenarios by our beliefs about the relationship between the NH (i.e. the change in HAQ N in 3 months observed in general practice with no treatment) and the change in HAQ for non-responders in a placebo group (α), if both ‘placebo’ and ‘no treatment’ were compared in general practice.
Scenario 1: Results with ‘no treatment’ in practice are similar to placebo arms of randomised controlled trials
If N is approximately equal to α + p0δ (the average change in HAQ in the placebo group), this represents a scenario where we think the results obtained in a group given placebo, averaged across responders and non-responders, would be the same as what would have been observed if no treatment had been given.
In scenario 1, the absolute difference in the change in HAQ between treatment in practice and no treatment (difference-in-difference) can be estimated by substituting N = α + p0δ into the parameters shown in Figure 8 and so the difference-in-difference for responders is estimated to be (α + γj) − N + α + γj − (α + p0δ) = γj − p0δ and for non-responders βj − p0δ.
Scenario 2: The ‘no-treatment group’, in practice, gets worse outcomes than the placebo arm in an randomised controlled trial
In this scenario, patients with no treatment would not achieve the response rates observed in the placebo arms of RCTs. It is assumed that they would have the same outcomes as patients with ‘no response’ in the placebo group of an RCT. This implies that N is approximately equal to α. In this scenario, if placebo were to be given in practice, there would be some lasting average benefit over and above NH equal to: (α + p0δ) − N = α + p0δ − α = p0δ.
This might imply a lasting psychological benefit of the act of taking medication or could be due to beneficial interactions between the doctor and patient that occur both in trials and in the regular clinical setting. By extension, this ‘placebo effect’ would also partly explain the results in the treatment group, and would be expected equally in the trials and in general clinical practice. Therefore, we would expect that if biologic therapy and no treatment were compared in general practice, the absolute difference in the change in HAQ between treatment and no treatment (difference-in-difference) would be α + γj − N = γj for responders and βj for non-responders.
It is difficult to test these alternative hypotheses, because the scenarios represent our hypothetical beliefs about a counterfactual argument: what would happen if ‘no treatment’, ‘placebo’ and ‘treatment’ were compared in general practice.
Conclusion
We conclude by setting out the implications for predicting the HAQ score in the decision model under each scenario.
In the decision model, variable N (the long-term NH in the untreated patients) is informed by observational evidence independent of the RCTs and is assumed to be constant over time. Therefore, in either scenario the HAQ score in the untreated group at time t after the start of the model is calculated as N × t.
If responders on treatment are assumed not to progress (worsen) over time, then the HAQ(t,j) score at time t for responders while still on treatment j is:
-
Scenario 1 Results with ‘no treatment’ are similar to average in placebo arms of RCTs (N = α + p0™). HAQ(t,j)=α+γj=N−p0δ+γj
-
Scenario 2 The ‘no-treatment group’ achieves worse outcomes than the average in placebo arms of RCTs (N = α). HAQ(t,j)=α+γj=N+γj
We assume that scenario 1 is the base case, consistent with the assumptions made in the previous Assessment Group model,177 and that scenario 2 is a sensitivity analysis.
Appendix 10 Estimation of probability of achieving both Psoriatic Arthritis Response Criteria and Psoriasis Area and Severity Index 75 response
Introduction
Biologic therapy may be indicated to treat both joints disease and psoriasis. Clinical response at 3 months is measured using the PsARC for joints and PASI 75 for skin conditions.
Because there are two response variables, there are four possible outcomes at 3 months: skin response only, joints response only, response of both and response of neither. Furthermore, the PsARC and PASI 75 responses are not necessarily independent.
The meta-analysis in Chapter 3 estimated the marginal probability of each type of response. However, this analysis did not estimate the bivariate probability, that is, the probability of observing both a response on arthritis and skin disease together.
This appendix shows how the bivariate probability density function (pdf) of PASI 75 and PsARC was estimated from the clinical trial evidence, to be used in the decision model for patients who have both skin and arthritis involvement at baseline, and assessed for PASI and PsARC responses at 3 months.
Estimate of correlation between Psoriatic Arthritis Response Criteria and Psoriasis Area and Severity Index 75 outcomes in the ADEPT trial51
No published papers reported the correlation between PsARC and PASI 75. The Assessment Group requested this from the manufacturers. One manufacturer (Abbott151) provided this data, based on the ADEPT trial,51 comparing adalimumab with placebo. In this appendix, we use the estimate of the correlation coefficient derived from the ADEPT trial51 and the estimates of the marginal pdfs of each type of response from the meta-analysis to estimate the bivariate pdf.
Table 56 shows the outcomes of the ADEPT trial,51 in the 66 patients who were assessed for both outcomes at 12 weeks. We refer to PsARC as variable x and PASI 75 as variable y. The responses are dichotomous, where 0 represents no response and 1 represents a response. To distinguish between the results of the meta-analysis and the results of the ADEPT trial,51 we label the pdfs from the ADEPT trial51 as f(x) and f(y) and the corresponding pdfs for the population estimated from the meta-analysis as Pr(x = 1) and Pr(y = 1). Similarly, the joint pdf from the ADEPT trial is f(x,y) and the (predicted) joint pdf for the population as Pr(x = 1,y = 1).
| PsARC (x) | PASI 75 (y) | n | f(x,y) |
|---|---|---|---|
| 0 | 0 | 18 | 0.27 |
| 0 | 1 | 5 | 0.08 |
| 1 | 0 | 14 | 0.21 |
| 1 | 1 | 29 | 0.45 |
where the trial estimate of covx,y = E(XY) –E(X)E(Y) = f(x = 1,y = 1) – f(x = 1)f(y = 1) and the trial estimate of sx = SD(X) = √{f(x = 1)[1 – f(x = 1)]}.
From the ADEPT trial, covx,y = [29/66 – (34/66)(43/66)] = 0.103
This value of ρ is significant at the 5% level [t = 3.31 with 65 degrees of freedom (df), p = 0.0015].
The SE is SE(ρ) = √[(1– ρ2)/(N – 2)] = 0.112, and t is distributed according to a Student’s t-distribution with N – 2 df.
The ADEPT trial found that responses were uncorrelated for the placebo group, with an estimated correlation coefficient of 0.02 (Table 57) (t = 0.16, 67 df, p = 0.87).
| PsARC (x) | PASI 75 (y) | n | f(x,y) |
|---|---|---|---|
| 0 | 0 | 49 | 0.72 |
| 0 | 1 | 2 | 0.03 |
| 1 | 0 | 17 | 0.24 |
| 1 | 1 | 1 | 0.01 |
Estimate of joint pdf of Psoriatic Arthritis Response Criteria and Psoriasis Area and Severity Index 75 in the population
We can use these relationships to estimate the bivariate probability of PASI 75 and PsARC in the population Pr(x = 1, y = 1).
We assume the correlation coefficient ρ between response types from the ADEPT trial is an unbiased estimate for all biologics in the population. This represents the correlation between outcomes in the population, and is a measure of variability not uncertainty.
where sx and sy are estimates of variability of X and Y in the population, and not the uncertainty σx and σy in the mean E(X) = Pr(x = 1) and E(Y) = Pr(y = 1). An estimate of sx in the population is SD(X) = √{Pr(x = 1)[1 – Pr(x = 1)]}
From the definition of the covariance [E(XY) = Pr(x = 1, y = 1) × 1 × 1 + Pr(x = 0, y = 1) × 0 × 1 + Pr(x = 1, y = 0) × 1 × 0 + Pr(x = 0, y = 0) × 0 × 0 = Pr(x = 1, y = 1)]:
Rearranging Equation 1 and substituting in Equation 2 gives:
The contingent probabilities of the joint outcomes are:
There are constraints on Pr(x = 1, y = 1) and Pr(x = 0, y = 0):
Substituting Equation 3 in these constraints, and rearranging, implies that:
where
Implications for the decision model
We show an example of the implications of these assumptions for the decision model. For illustrative purposes, assume that the probability of PsARC for treatment j is estimated to be Pr(x = 1) = 0.80, and the probability of PASI 75 is Pr(y = 1) = 0.5.
In this example, odds(x = 1) = 0.8/0.2 = 4 and odds (y = 1) = 0.5/0.5 = 1. Given Pr(x = 1) and Pr(y = 1), the constraints on ρ are: –0.5 ≤ ρ ≤ 0.5
If we assume there is no correlation between these outcomes ρ = 0, then:
If we estimate that the correlation between X and Y is ρ = 0.5, then:
Appendix 11 Elicitation exercise
A number of parameters within the model either did not have adequate evidence, or did not have any evidence at all, with which to populate them. This latter issue, in particular, poses a potential problem. One option would be to assign uninformative priors to these. However, this uninformative prior may not truly represent the current level of knowledge regarding these parameters. As an alternative to uninformative priors, elicitation techniques can be used to generate subjective priors for the unknown parameters in the absence of actual data. 219 An elicitation method is used to link an expert’s underlying beliefs to an expression of these in a statistical form.
An elicitation exercise was designed to generate subjective prior estimates of the unknown parameters in the model, the effect of withdrawal from biologics, along with two other parameters for which evidence may be poor.
The following sections first describe the uncertainties and then go onto describe the elicitation exercise used to generate prior information to characterise these uncertainties. Finally, the results of the elicitation exercise are presented.
Uncertainties in the psoriatic arthritis model
The rate of disease progression beyond the initial Health Assessment Questionnaire change
The rate of progression following a response to etanercept or infliximab is uncertain. In the original York model, an assumption was made that beyond the initial HAQ gain, disease progression will stop (rate of progression = 0 in Figure 9) following response to biologics. There is some uncertainty, however, about the extent to which this truly reflects the longer-term efficacy of biologics. Colloquial evidence suggests that patients may either improve their disease following a response to biologics or may experience some disease progression at a slower rate than the NH of the disease. Recent observational evidence from national biologics registers suggests that HAQ and health utility remain stable for patients with PsA while on biologics. Gulfe et al. 190 analysed data from 574 patients in south Sweden between May 2002 and December 2008, and found health utilities remained largely unchanged for PsA over 7 years. (CiC information has been removed.) The limitation of these registry data for the purposes of the decision model is that the data do not distinguish between outcomes for patients who persisted with their initial biologic and those who withdrew completely or switched to another drug.
FIGURE 9.
Natural history of PsA measured using the HAQ.
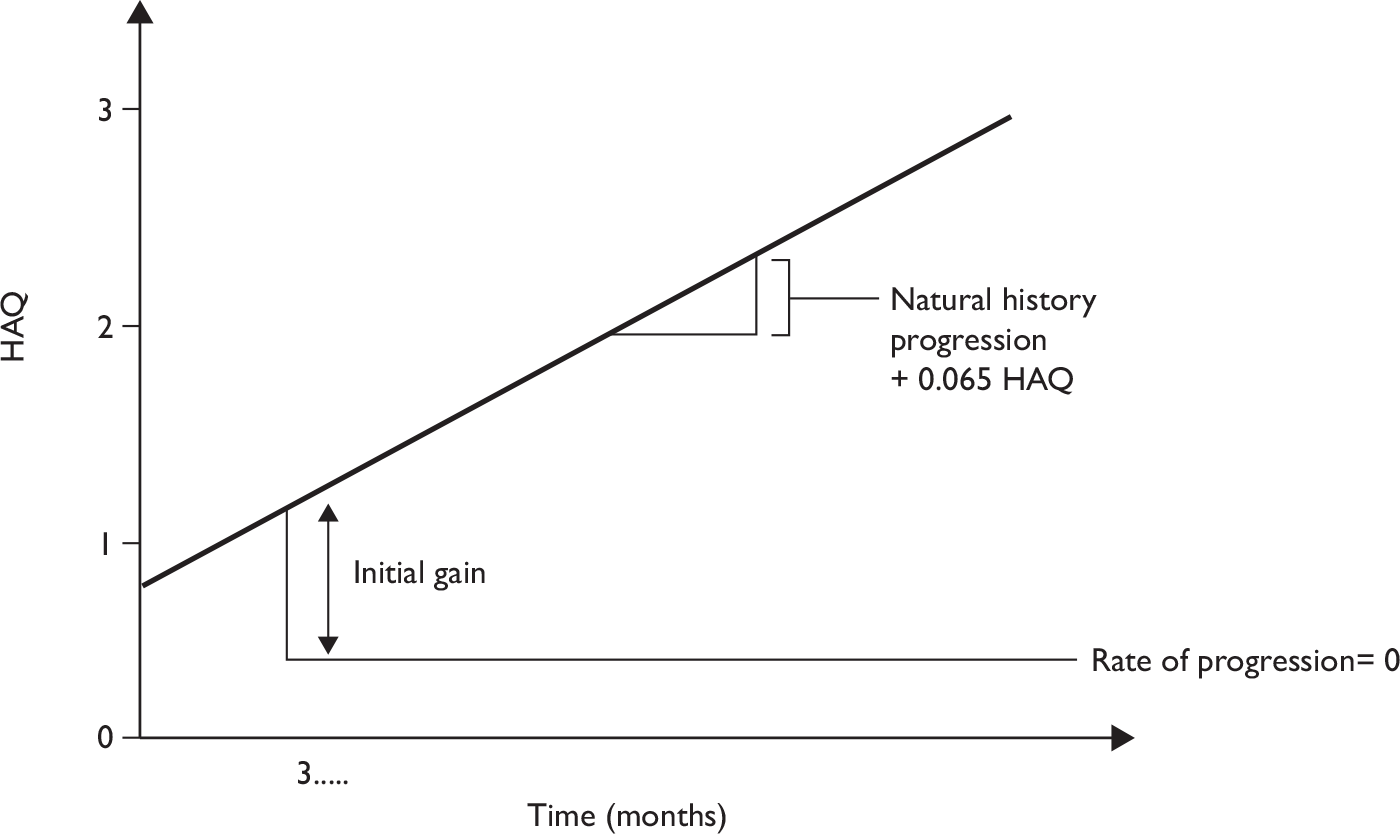
In the original York model, progression following a response was simply assigned a fixed value of 0 and no scenarios were specified for this assumption. It is therefore not possible to determine the sensitivity of the model to this assumption.
The rebound effect
Patients who withdraw from biologic treatment, due to either adverse events or loss of efficacy, will then have some worsening in HAQ score (the ‘rebound’). There are no data on the rate of disease progression for the 3-month period immediately following withdrawal from treatment (given an initial response on the PsARC criteria). Clinical opinion suggests that there will be some kind of rebound (back up to NH progression), but the degree of rebound is unknown. In the original York model, therefore, two rebound scenarios were considered (Figure 10):
-
When patients fail therapy (after initially responding), their HAQ score deteriorates by the same amount by which it improved when patients initially responded to therapy (rebound equal to gain in Figure 10).
-
When patients fail therapy, their HAQ score returns to the level and subsequent trajectory it would have been had they not initially responded to therapy (rebound to NH in Figure 10).
FIGURE 10.
Disease progression following treatment failure.
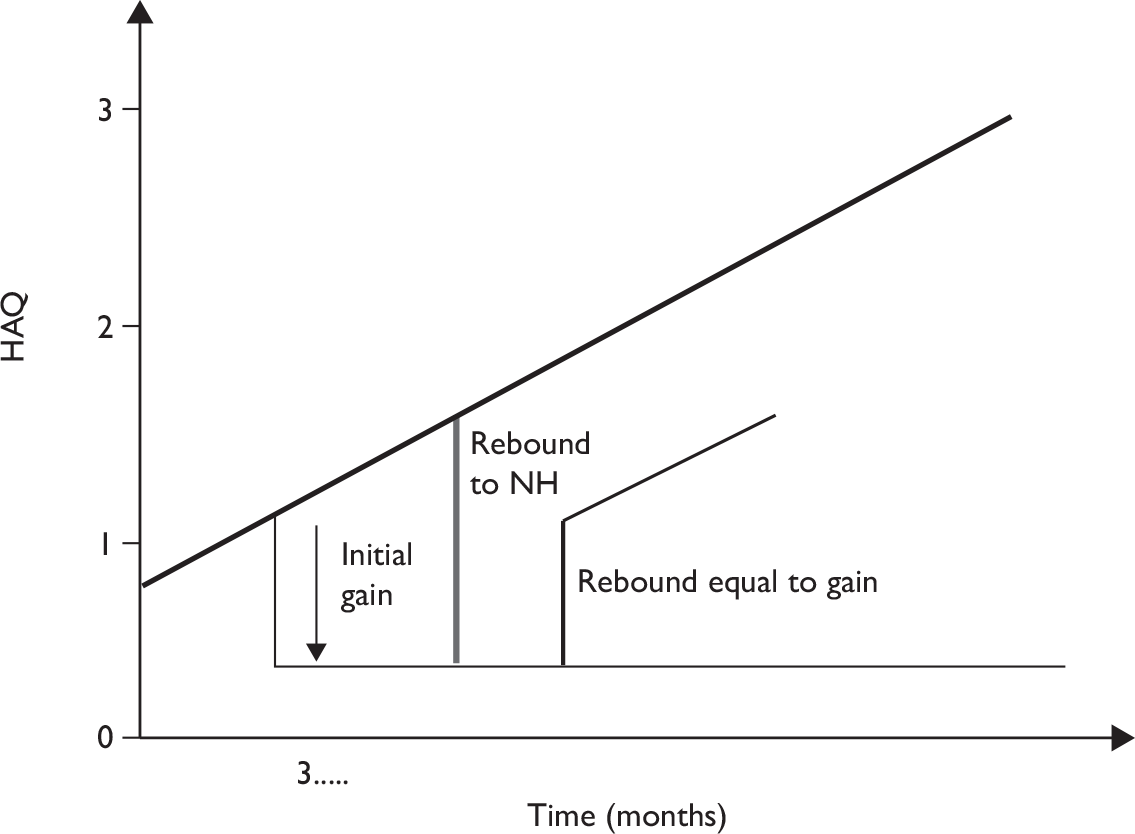
The two rebound scenarios for progression following relapse produced two different estimates of the cost-effectiveness of etanercept and infliximab. By specifying the rebound as equal to NH progression, the ICER for etanercept increases from £26,361 to £30,628 in the 10-year model compared with the rebound equal to initial gain. This increase in the ICER may be sufficient to change the adoption decision if the threshold is > £26,361, but < £30,628.
The rate of disease progression beyond the rebound effect
The original York model assumed that following a change in HAQ after withdrawing from biologics (the rebound effect) patients would immediately return to the NH progression rate. Clinical opinion suggests that this might not be the case. That is when withdrawing from treatment, having received, and responded to, biologics alters the course of the disease for a given period of time after withdrawal. This issue was not explored in the previous York model.
Methods of the elicitation
The parameters described above were elicited from multiple experts individually, followed by appropriate synthesis. Clinical opinion suggests that the first two uncertain parameters may be correlated, i.e. the degree of rebound following relapse is conditional upon the extent of gain when responding. In addition, clinical opinion also suggested that extent of gain when responding may be conditional upon the extent of initial HAQ change following a PsARC response. The exercise, therefore, incorporates these relationships when eliciting data from experts.
To enable experts to express the extent of gain when responding conditional upon the extent of initial HAQ change following a PsARC response, this HAQ change was also elicited from experts during the exercise. These data are not used directly in the decision model, which takes estimates of initial HAQ gain from the evidence synthesis in Chapter 3.
Format and content of elicitation
A spreadsheet (excel)-based, interactive elicitation exercise was designed to generate estimates of initial HAQ change, disease progression while responding to treatment, disease progression for the 3 months following a relapse and longer-term disease progression following withdrawal. An interactive format was used as the elicitation exercise was also designed to incorporate any correlation between the first three parameters. To build in the correlation between parameters, responses for some questions were conditional upon responses to previous questions. This method is an appropriate way to incorporate conditional dependence suggested by Garthwaite et al. 220
In accordance with good elicitation practice, background to the elicitation was presented at the start of the exercise along with a guide to completion. 221 The background information presented can be seen below. Experts were told the rationale for the elicitation exercise, to obtain data on unknown parameters to inform a decision-analytic model, and reminded of the HAQ scoring method and expected NH progression (progression without treatment). Experts were presented with an illustration of the trajectory of disease progression without treatment and change in HAQ score. Experts were given examples of the question format and invited to complete practice questions.
The histogram approach222 is used in this elicitation. For each question, a discretised numerical scale was predefined and experts were asked to place 20 crosses on a frequency chart, representing their beliefs about the distribution of a particular quantity. Each cross represents 5% of the distribution.
Once the expert had read through the supporting material and completed example questions, they were asked to start the elicitation questions. Experts were then taken to a separate worksheet where the four questions were arranged into sections, which they were asked to complete sequentially.
Initial Health Assessment Questionnaire gain following treatment with etanercept, infliximab or adalimumab
Experts were asked to provide an estimate of the known parameter (HAQ gain) following treatment with infliximab, etanercept or adalimumab. Experts could choose to group all three biologics together or complete separate histograms for each biologic.
Experts were asked for their estimates of HAQ score following treatment (3-month response) and were asked to place 20 crosses on a grid running from 0 to +3.
Rate of progression while still responding to treatment
Experts were asked to provide an estimate of disease progression for patients who have responded to treatment on etanercept, infliximab or adalimumab. Again experts could choose to group all three biologics together or complete separate histograms for each biologic. In addition, experts were asked if they believed that the rate of progression while responding was related to the initial HAQ gain (separately for each biologic if appropriate). If experts responded ‘yes’ they were requested to complete grids for each of the 0–25, 25–50, 50–75 and 75–100th percentiles from the winbugs output of HAQ score for infliximab, adalimumab and etanercept (see Chapter 3, Assessment of effectiveness). If experts responded no, they completed a single grid, assuming no relationship between the two parameters.
Again experts were asked to place 20 sets of crosses on each grid. Experts were reminded prior to answering these questions that we estimated the NH rate of progression of HAQ (progression without treatment) to be +0.016 per 3 months. 176
Rate of progression in the 3-month period after withdrawal from treatment
Experts were asked to provide an estimate of disease progression for the 3 months following a treatment failure (after an initial response); this was termed the ‘rebound’. Again experts could choose to group all three biologics together or complete separate histograms for each biologic. In addition experts were asked if they believed that the rate of progression after withdrawal from treatment was related to the rate of progression while responding (separately for each biologic if appropriate). If experts responded yes they were requested to complete grids for each of the 0–25, 25–50, 50–75 and 75–100th percentiles. These ranges were generated by sampling from the responses to question 2, given the likelihood of observing a particular conditional HAQ gain (question 1). The likelihood of observing particular ranges for HAQ gain was again taken from the winbugs output of the current York model. If experts responded ‘no’, they completed a single grid, assuming no relationship between the two parameters.
Rate of progression following the 3-month rebound
Experts were asked to provide an estimate of disease progression for period following the 3-month rebound. Again, experts were reminded that this was for patients who had previously responded to biologics using the PsARC criteria but who had now withdrawn from treatment either due to adverse effects or loss of efficacy.
Experts were asked, for each of the three biologics, if they believed that the rate of progression would return to NH. If they answered ‘yes’ then the questionnaire was complete. If they answered ‘no’ then they were asked to complete a grid (for each biologic separately if appropriate) expressing their belief about the progression rate following the rebound period. They were then asked for the number of months they would expert to observe this progression rate before patients retuned to NH.
Study sample
Sixteen experts were sent the questionnaire. These experts were chosen to represent a range of clinical opinion nationally. Experts were chosen on the basis of the clinical advice from a ‘lead expert’.
Questionnaires were sent by e-mail, along with a covering letter. This format was chosen because of the wide national distribution of experts in the original sample of 16. Experts were then sent a reminder e-mail inviting them to complete the questionnaire. A number of experts expressed a desire to be guided through the questionnaire by telephone. The remainder completed the questionnaire independently and returned it via e-mail.
Questionnaire responses were received from five experts. A large number of the remaining 11 experts expressed a conflict of interest that prevented them from taking part in the exercise. The remainder stated that due to other commitments they were unable to participate. Experts were anonymised here and are referred to as experts 1–5.
Synthesis of experts’ histograms
Linear opinion pooling is the synthesis method most commonly applied in expert elicitation. 223 In linear pooling, experts’ probabilities or weights are aggregated using simple linear combinations. If p(θ) is the probability distribution for unknown parameter θ in linear pooling, experts’ probabilities or weights are aggregated using simple linear combination, p(θ) = Σiwi × pi(θ) where wi is expert i’s weight.
This method is akin to generating a ‘super’ distribution by pooling the five experts’ assessments. From this we can generate an arithmetic mean and associated uncertainty. 224 This method assumes that by gathering more priors (eliciting from more experts) we do not necessarily become any more certain about the rate of progression during response or relapse. The linear pooling method considers each expert’s distributions as separate priors with no relationship between experts’ distributions assumed. Here linear pooling was carried out using equal weights for experts.
Results
Questionnaire responses
Responses to the elicitation questions varied, reflecting different clinical opinion regarding treatment. The histograms for each of the, questions, for each of the five experts are presented below. Table 58 also shows the means and SEs of the means for each of the elicited parameters.
| Expert | HAQ gain | Progression while responding | Progression in 3 months after withdrawal | LT progression after withdrawal | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E | I | A | E | I | A | E | I | A | E | I | A | |
| 1 | –1 (0.18) | –1 (0.18) | –1 (0.18) | –0.0035 (0.007) | –0.0035 (0.007) | –0.0035 (0.007) | 0.08 (0.016) | 0.08 (0.016) | 0.08 (0.016) | 0.016 | 0.016 | 0.016 |
| 2 | –0.805 (0.135) | –0.805 (0.135) | –0.805 (0.135) | –0.009 (0.007) | –0.009 (0.007) | –0.009 (0.007) | 0.393 (0.006) | 0.393 (0.006) | 0.393 (0.006) | 0.016 | 0.016 | 0.016 |
| 3 | –0.72 (0.16) | –0.72 (0.16) | –0.72 (0.16) | –0.017 (0.01) | –0.017 (0.01) | –0.017 (0.01) | 0.013 (0.007) | 0.013 (0.007) | 0.013 (0.007) | 0.02 (0.01) | 0.019 (0.008) | 0.02a (0.008) |
| 4 | –0.82 (0.24) | –0.82 (0.24) | –0.82 (0.24) | –0.0017 (0.014) | –0.0017 (0.014) | –0.0017 (0.014) | 0.037 (0.034) | 0.037 (0.034) | 0.037 (0.034) | 0.016 | 0.016 | 0.016 |
| 5 | –0.39 (0.17) | –0.39 (0.17) | –0.39 (0.17) | 0.04 (0.011) | 0.04 (0.011) | 0.04 (0.011) | 0.12 (0.007) | 0.12 (0.007) | 0.12 (0.007) | 0.016 | 0.016 | 0.016 |
None of the experts expressed any difference between the initial HAQ changes for the three biologics. Elicited means ranged from 0.39 to 1, with a mean of 0.747. This figure is not dissimilar to the initial HAQ changes generated by the evidence synthesis model (see Chapter 3, Assessment of effectiveness). Many of the experts believed that HAQ progression for responders would be negative, i.e. patients would continue to improve over time while receiving biologics. The elicited ‘rebound’ effect is neither similar to the original ‘rebound to initial HAQ gain’ nor the ‘rebound back to NH’ scenarios. Experts believed that there was a continued effect of biologics even for patients discontinuing treatment due to either adverse events of loss of efficacy. Four out of five of the experts believed that long-term progression would be equivalent to NH.
Synthesised beliefs
Two if the experts that stated that there was a correlation between initial HAQ gain and progression while responding to treatment and/or progression while responding to treatment and progression for the 3 months after withdrawal from treatment. These correlations, however, were very small. Given the complexity involved in building this correlation into the decision model, it was therefore decided to assume that there was in fact no correlation between elicited parameters (as expressed by the majority of experts). Table 59 shows the results from the synthesis of elicited parameters [mean (SE)] assuming no correlation between parameters.
| HAQ gain | Progression while responding | Progression after relapse | LT progression | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| E | I | A | E | I | A | E | I | A | E | I | A |
| –0.747 (0.268) | –0.747 (0.268) | –0.747 (0.268) | 0.002 (0.022) | 0.002 (0.022) | 0.002 (0.022) | 0.13 (0.14) | 0.13 (0.14) | 0.13 (0.14) | 0.0168 (0.004) | 0.0166 (0.003) | 0.0168 (0.004) |
The ‘synthesised progression while responding’ rate is very close to 0 at 0.002 (SE = 0.022). The rebound progression is 0.13 (SE = 0.14) increase in HAQ for 3 months. Again, this is somewhat different to the initial HAQ gain, contradicting the ‘rebound to initial gain’ assumption. It is further still from the ‘rebound to NH’ assumption.
Using the elicited data in the decision model
The elicitation was designed to inform the following three parameters in the decision model:
-
The rate of change of HAQ for patients on biologic therapies (HAQ1.d).
-
The change or rebound in HAQ in the 3-month period immediately after withdrawing from biologic therapy (loss.w).
-
The rate of change in HAQ in the long term after withdrawing from biologic therapy (HAQ1.w).
The base-case decision model will assume that the mean value of HAQ1.d is 0 (SE 0.02), consistent with the elicitation and the limited observational evidence from biologics registers.
For convenience, the decision model expresses the value of parameter loss.w relative to baseline HAQ. Its magnitude can be estimated as the difference between the absolute initial gain and the rebound. A value of 0 means that the rebound is equal in absolute terms to the initial gain on starting biologics, a positive value means the rebound is between the initial gain and ‘NH’, and a negative value means the rebound is less in absolute terms than the initial gain (see Figure 9). The results of the elicitation (see Table 59) suggest that loss.w is negative. Mean (initial HAQ gain) + mean (progression in 3 months after withdrawal) = –0.75 + 0.13 = –0.62 (SE = 0.29). Given the limitations of the exercise and some uncertainty about whether this accurately represents the views of the experts, we assume that the base-case mean value of loss.w is zero, with a normal distribution with a wide SE of 0.5 to indicate the considerable uncertainty. We use the mean value of loss.w = –0.62 as a sensitivity analysis.
The experts were almost unanimous that the long-term rate of change of HAQ after withdrawal would be equal to the rate of change of HAQ of patients who never used biologics (the NH). We therefore set these parameters to be equal in the decision model.
Discussion
There are a number of issues with the elicitation exercise that are worth noting. First it is likely that there is a degree of heterogeneity between experts. Possible reasons are clinical knowledge, clinical experience (types of patients seen and/or drugs used), interpretation and understanding of elicitation questions, and true underlying heterogeneity about the treatment effect. Unfortunately, it is not possible, with five experts, to incorporate these factors, as covariates, into a model. To do this would require many more experts to have any power to detect any difference. 225
Second, the selection of experts for the elicitation questionnaire was undertaken by a single lead expert and the number of experts that completed the questionnaire was very limited. While the problem with gathering sufficient experts is common in elicitation exercises conducted to inform HTA decision models, we cannot be sure that the sample of experts included is truly representative of the current level of knowledge.
Perhaps the most striking conclusion from the elicitation exercise is that the ‘rebound’ effect is neither similar to the original ‘rebound to initial HAQ gain’ nor the ‘rebound back to NH’ scenarios. Experts believed that there was a continued effect of biologics even for patients discontinuing treatment due to either adverse events of loss of efficacy. The majority of experts then believed that patients would return to a NH rate of progression beyond this rebound period. It is possible that the longer term implications of this were not clear in the exercise. In particular, the fact that by assuming that patients only return to NH rate of progression after this period meant that the progression of patients no longer on treatment would never return to the NH line of progression (see Figure 9). It is possible that the complexity of the exercise posed a significant cognitive burden on the experts. This may have been eased by including a visual expression of the resulting line of progression. Therefore, there may well be a trade-off between obtaining information on specific model parameters, the complexity of the exercise and cognitive burden on experts.
Background information presented to experts
Elicited histograms
Expert 1: Health Assessment Questionnaire gain (all drugs)
Expert 2: Health Assessment Questionnaire gain (all drugs)
Expert 3: Health Assessment Questionnaire gain (all drugs)
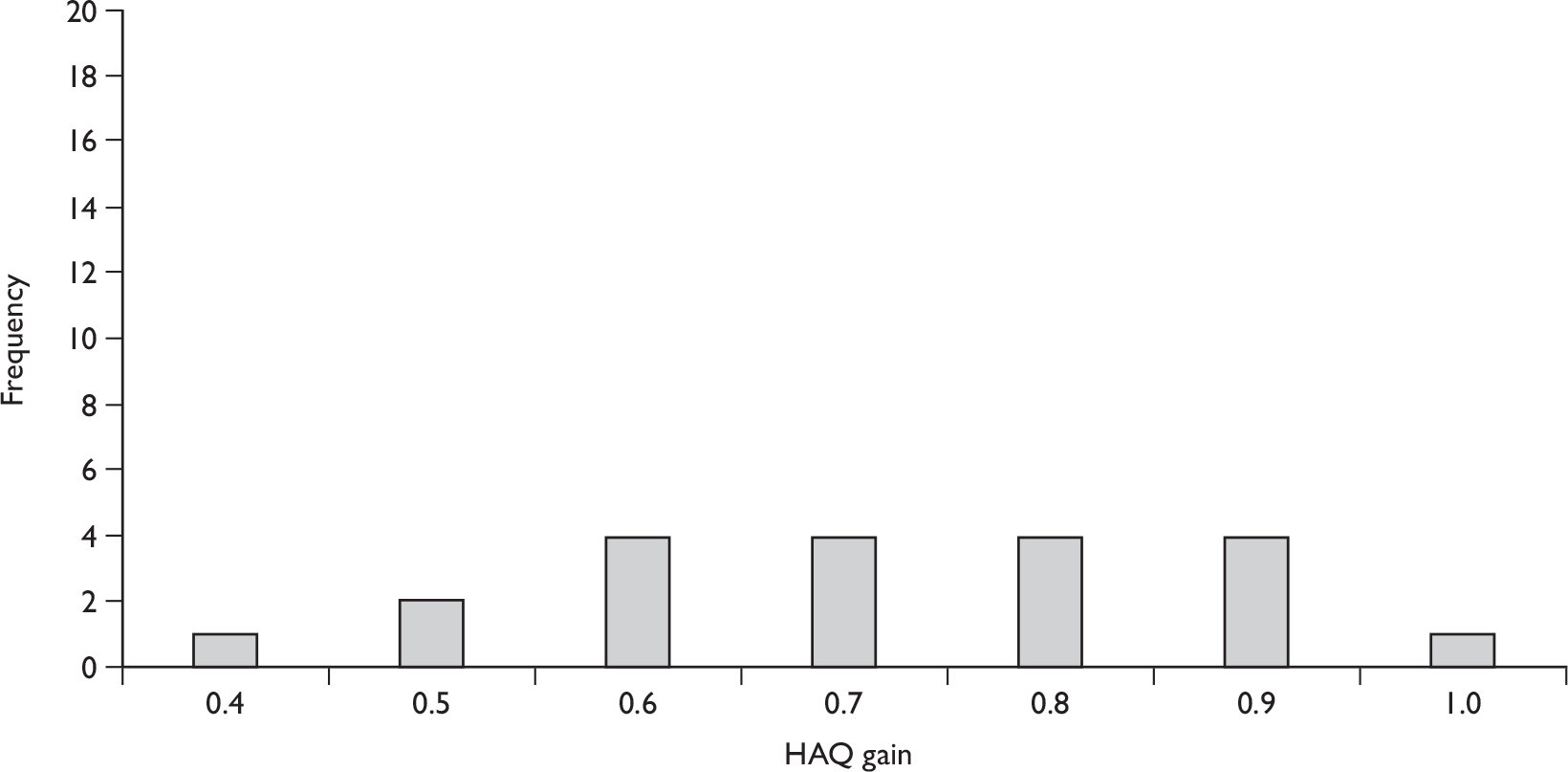
Expert 4: Health Assessment Questionnaire gain (all drugs)
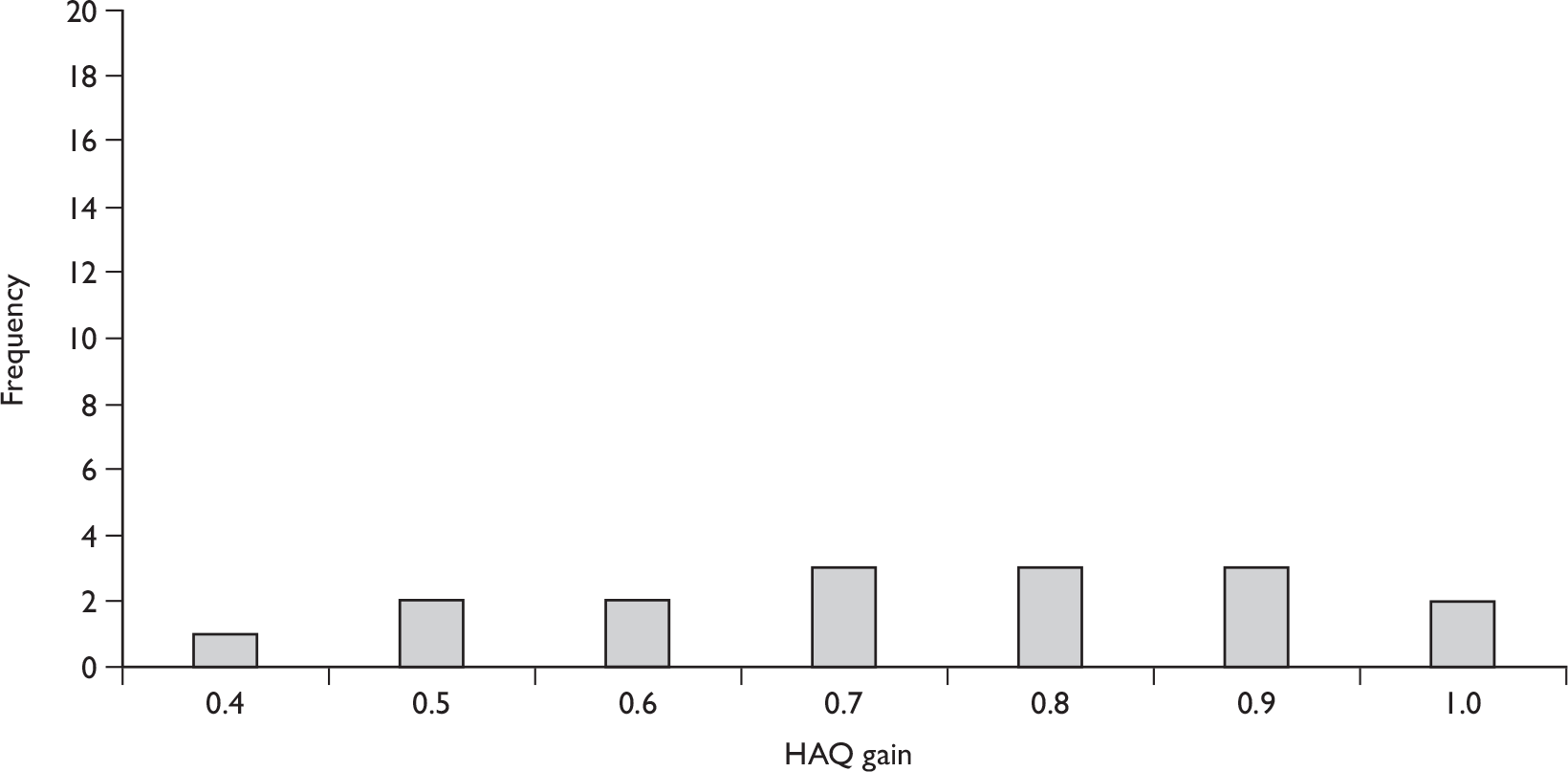
Expert 5: Health Assessment Questionnaire gain (all drugs)
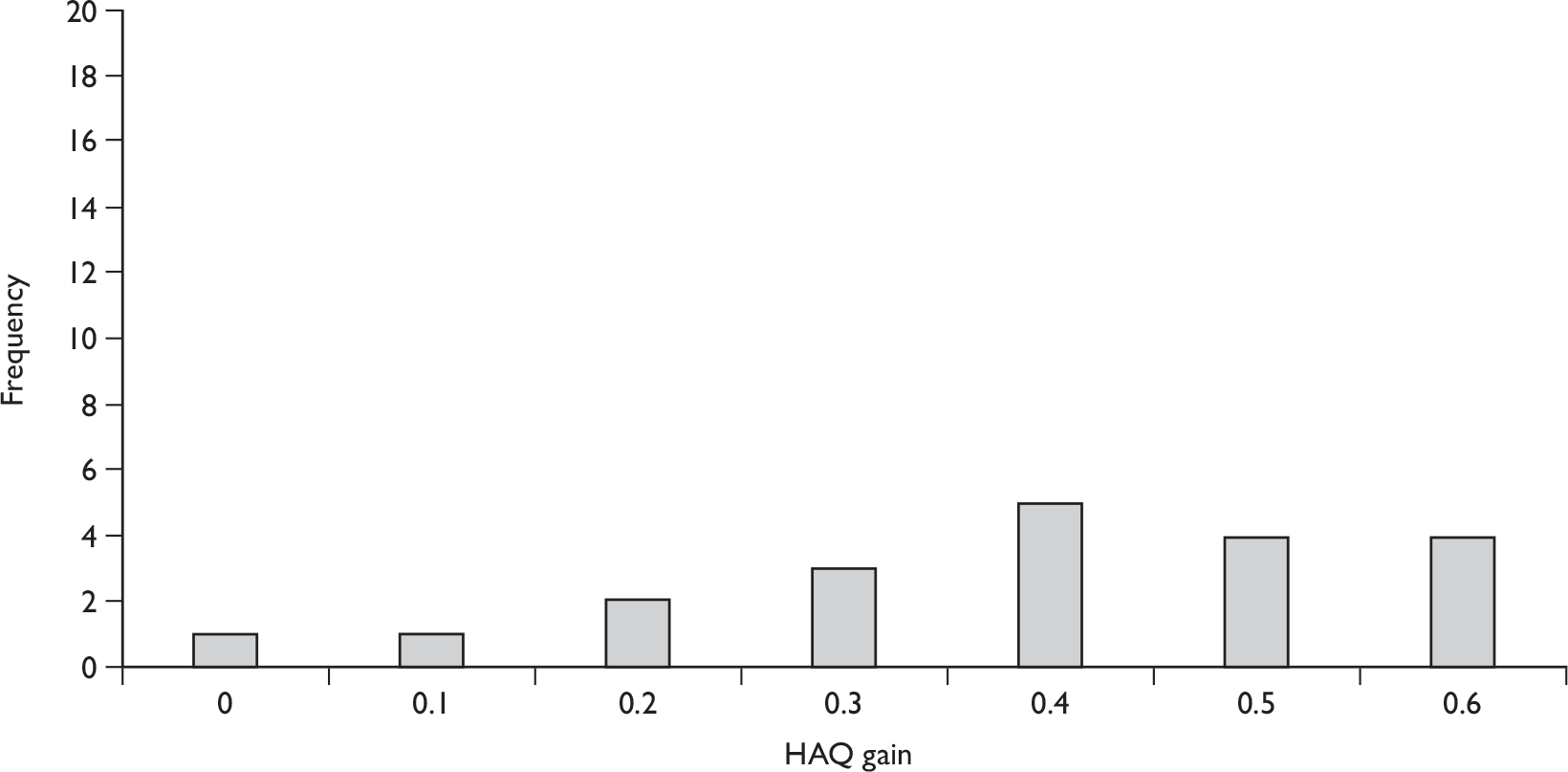
Expert 1: Progression while responding
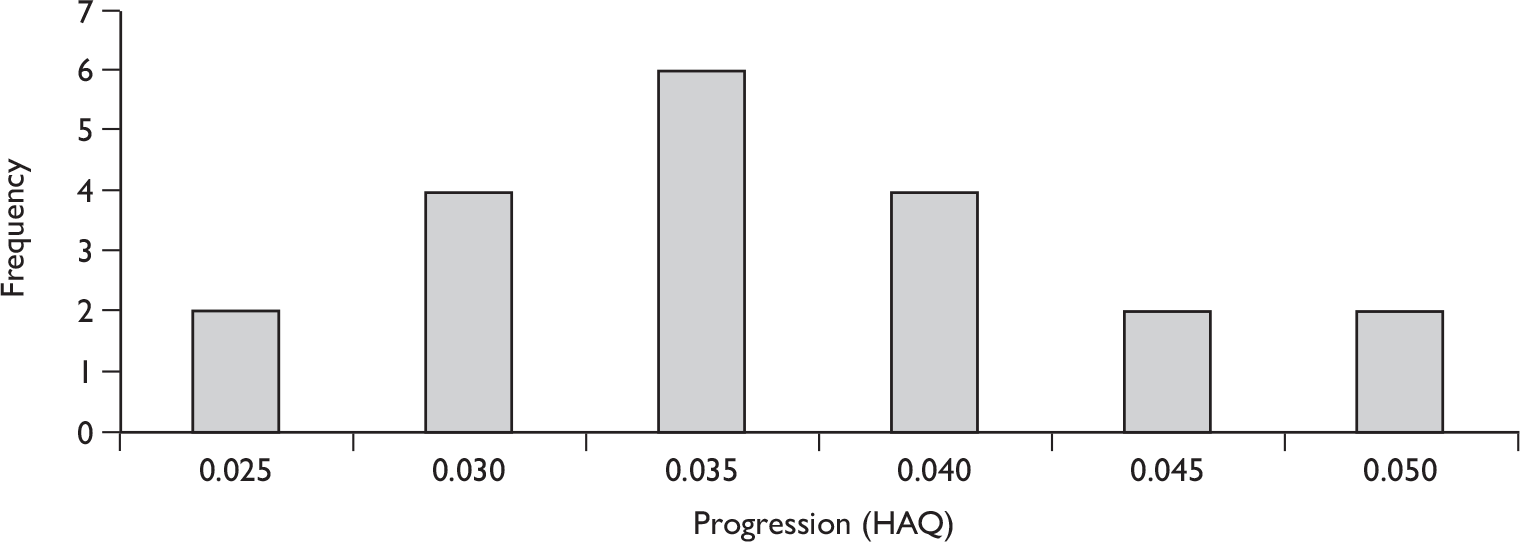
Expert 2: Progression while responding
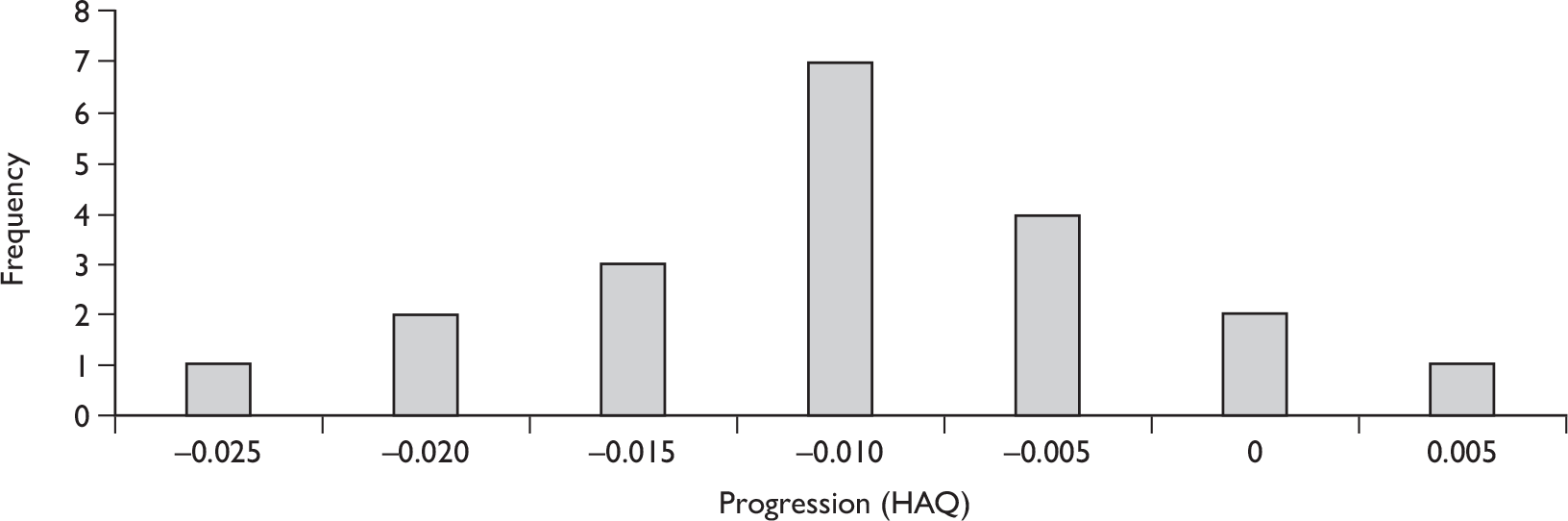
Expert 3: Progression while responding
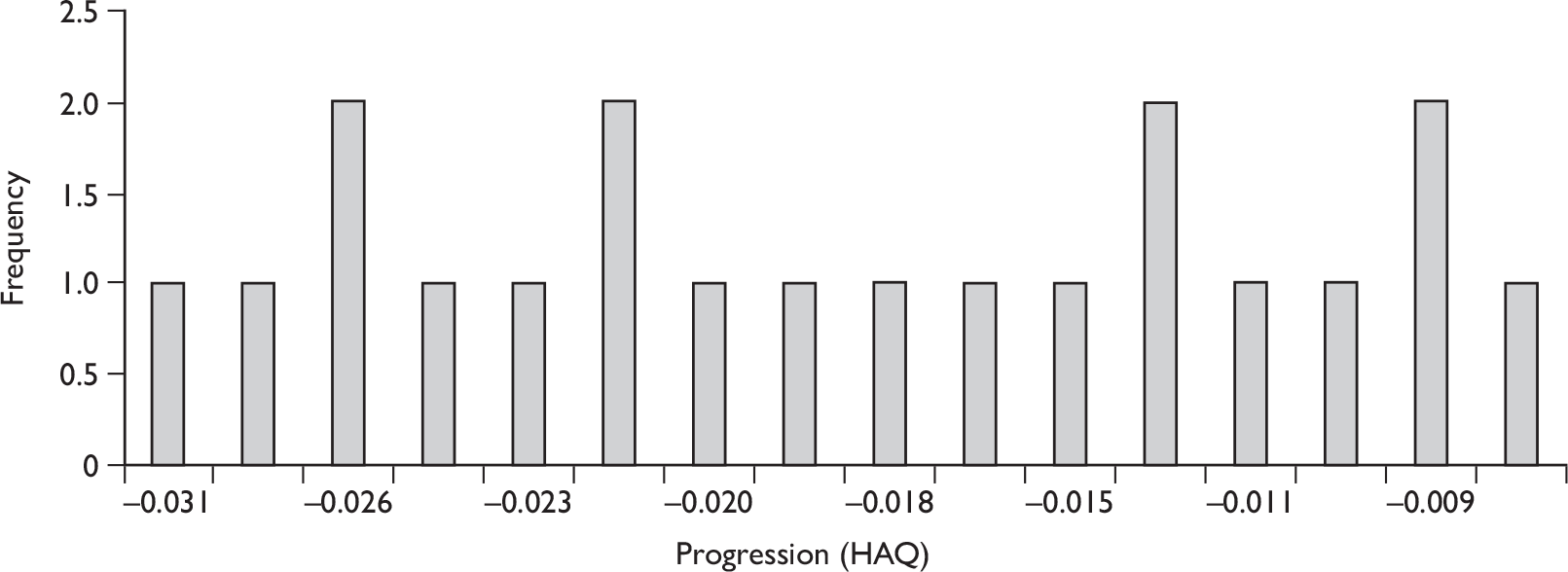
Expert 4: Progression while responding
Expert 5: Progression while responding
Expert 1: Progression during rebound period
Expert 2: Progression during rebound period
Expert 3: Progression during rebound period
Expert 4: Progression during rebound period
Expert 5: Progression during rebound period
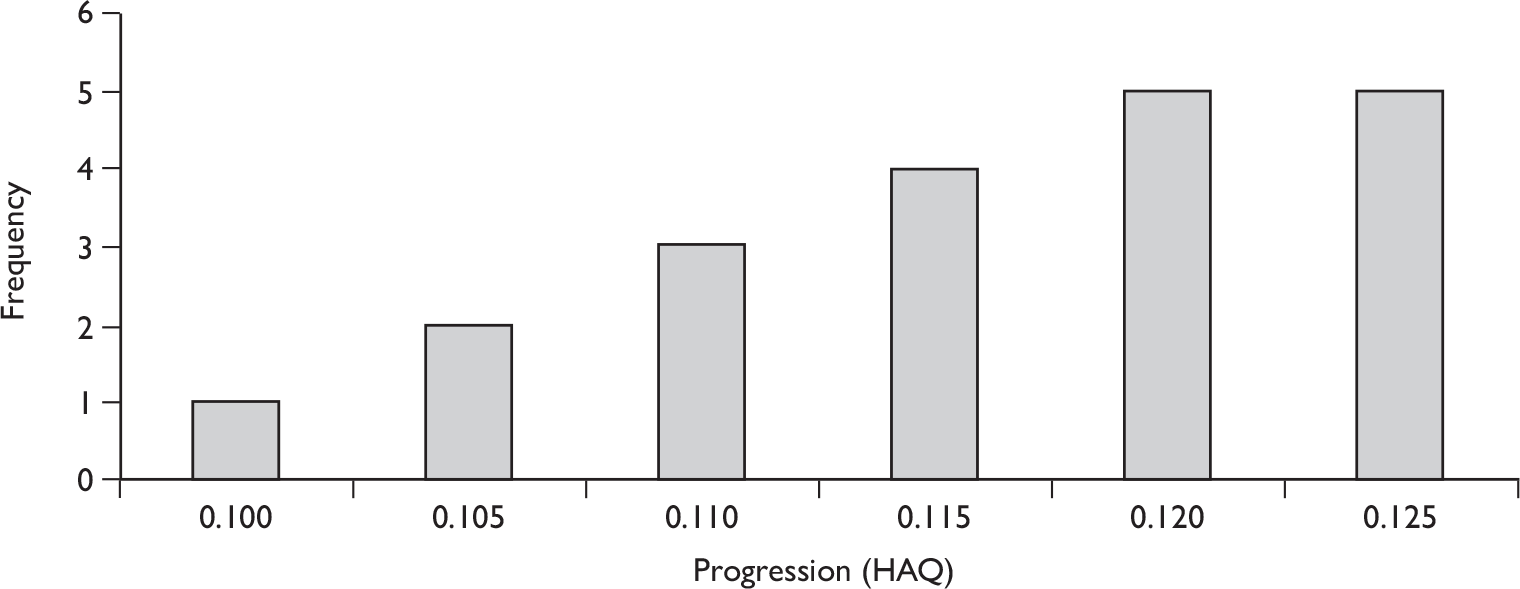
Appendix 12 Withdrawal rates from biologic therapies in patients with psoriatic arthritis
Introduction
This paper estimates persistence with initial biologics in patients with PsA. There are now registers in several countries that follow the progress of patients using biologic therapies and record the time to discontinuation. This paper undertakes a review of relevant registries to identify papers reporting drug discontinuation rates (or related data). A synthesis of relevant evidence is then undertaken in order to estimate the rate of withdrawal from initial biologic therapy. The paper considers whether this rate may vary over time, and whether there may be differences in withdrawal rates between etanercept, infliximab and adalimumab. All evidence is drawn from national biologic registers and is based on published summary data only. As withdrawal rates of patients with PsA are different from other types of chronic arthritis, all patients in this analysis have a diagnosis of PsA.
The estimates from the evidence synthesis will be used in a decision model, and extrapolated beyond the horizon of the studies to predict withdrawal over the patient’s lifetime.
Methods
Literature search
A literature search was carried out to identify published papers from biologics registers of patients with PsA who reported survival probabilities of remaining on first biologic therapy at 3 months or more, and number of patients at risk or CIs to estimate the uncertainty in the parameters. The search strategies can be seen in the annex at the end of this section.
This search identified 154 publications of registry data that were potentially relevant. In total, 130 of these were excluded based on the abstract as they were found not to be relevant, therefore leaving 24 publications that were considered in full. Of these 24 publications the information available can be summarised as:
-
reports rate of drug withdrawals, n = 8
-
reports second-line success given reason for first-line failure, n = 4
-
reports HAQ progression, n = 14
-
reports PASI progression, n = 1.
Of the eight publications reporting rates of drug withdrawals, just six of these reported rates for patients with PsA separately, and in a format that could be used in the analysis. Data from patients registered between 2000 and 2006 in NOR-DMARD (Norwegian DMARD register) were published by Heiberg et al. (2008)226 and Heiberg et al. (2007). 227 The latter was excluded as a majority of patients are likely to be included in both publications. Thus five publications were included in the analysis. These were Kristensen et al. ,215 Gulfe et al. ,190 Gomez-Reino et al. ,228 Saad et al. 162 and Heiberg et al. 226
Included studies
In the five papers included in the analysis, the majority report the average unadjusted Kaplan–Meier probabilities of survival, apart from Kristensen et al. ,215 who reported results stratified by use of concomitant MTX. Only one of the publication includes UK patients;162 Kristensen et al. 215 and Gulfe et al. 190 include Swedish patients, Gomez-Reino et al. 228 include Spanish patients and Heiberg et al. 226 include Norwegian patients. A brief summary of the papers is given in Table 60.
| Author | Year | Register | Condition | No. patients at baseline | Biologic treatment? | Parameter(s) |
|---|---|---|---|---|---|---|
| Gomez-Reino 2006228 | 2006 | BIOBADASER | PsA | 289 | Yes |
1-year drug survival, first and second line Reasons for withdrawal |
| Kristensen 2008215 | 2008 | SSATG | PsA | 261 | Yes |
~5-year drug survival for etanercept Risk of withdrawal relative to infliximab |
| Heiberg 2008226 | 2008 | NOR-DMARD | PsA | 172 | Yes | 1-year drug survival |
| Saad 2008162 | 2008 | BSRBR | PsA | 566 | Yes |
1-, 2- and 3-year drug survival, reason for withdrawal Reported by individual drug |
| Gulfe 2010190 | 2009 | SSATG | PsA | 344 | Yes |
~5-year drug survival for etanercept Risk of withdrawal relative to infliximab |
Kristensen et al. 215 (study 1) included 161 patients starting first biologic between April 1999 and September 2006 in the SSATG registry. Gulfe et al. 190 (study 2) included 344 patients, starting first biologic between May 2002 and December 2008 from the Southern Swedish Antirheumatic Therapy Group registry. We included data from both these publications in the evidence synthesis on the assumption that a minority of the patients would be included twice.
Table 61 shows the number at risk at the start of each follow-up and the probability of surviving on first biologic therapy until at least the end of the period.
| Study | Observational period | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||||||||||||||
| Start | End | N | S | St|St-1 | Start | End | N | S | St|St-1 | Start | End | N | St | St|St-1 | Start | End | N | St | St|St-1 | |
| 1a | 1 | 12 | 161 | 0.82 | 0.820 | 13 | 24 | 103 | 0.72 | 0.878 | 25 | 36 | 54 | 0.6 | 0.833 | 37 | 48 | 17 | 0.5 | 0.833 |
| 2b | 1 | 3 | 344 | 0.902 | 0.902 | 4 | 6 | 216 | 0.81 | 0.898 | 7 | 12 | 144 | 0.699 | 0.863 | 13 | 24 | 136 | 0.598 | 0.856 |
| 3c | 1 | 12 | 289 | 0.87 | 0.870 | |||||||||||||||
| 4d | 1 | 12 | 566 | 0.82 | 0.820 | 13 | 24 | 422 | 0.7 | 0.854 | ||||||||||
| 5e | 1 | 12 | 172 | 0.773 | 0.773 | |||||||||||||||
Synthesis of registry data
The evidence synthesis is carried out using Monte Carlo Markov chain estimation. The model is based on a method for meta-analysis at multiple follow-up times by Lu et al. (2007). 229
We define an ‘event’ as withdrawal from initial biologic therapy. The literature tends to report survival probabilities at a series of follow-up times, Pr(Tj > tu’) = S(tu’), and the number observed at the start of each period Nju’ (see Table 61). Unconditional survival probabilities are difficult to synthesise, as probabilities reported at successive time points in the same data set are correlated.
We therefore define the conditional probability of an event occurring between time u´ and u in trial j for those who do not have an event up to time u as Fju’u. If Tj is the withdrawal time of patients in study j then:
where tu’ is the beginning of segment u’ and tu is the end point of segment u. The data Fju’u are conditionally independent. We index the time segments 1–3 months, 3–6 months, 6–12 months, 12–24 months, 24–36 months and 36–48 months by u = 1, 2, 3, 4, 5, 6. The observation periods are, therefore, made up of adjacent time segments, of unequal length. Not all studies report the same observation periods. For example, Saad et al. 2009191 reports survival probabilities at 12 and 24 months, while Gulfe et al. 190 2009 reports survival probabilities at 3, 6, 12 and 24 months.
We assume that Fju’u is drawn from a normal distribution with mean pju’u and variance Fju’u × (1 – Fju’u)/Nju’. Other versions of the model might consider other distributions, such as the beta.
The hazard hju represents the failure rate of patients in trial j during segment u. The rate of withdrawal may vary over time. This might be represented in the model in various ways, such as a piece-wise constant hazard, or as a fully parametric function such as a Weibull distribution. The guidelines for the use of biologic therapies in PsA state that an assessment should be made at 3 months of whether the patient has responded on the PsARC and PASI 75 scales, and that drugs should be withdrawn or switched if there is no initial response. 149 Discontinuation after 3 months is likely to be a function of adverse events and/or continued response. It is therefore likely that the rate of withdrawal in the first 3 months is different from later time periods. Given we only have a few studies there is probably insufficient data to model changes in the hazard after the first 3 months. We therefore specify a piece-wise hazard that is constant after the first 3 months.
If an observation period spans segments u’ to u, for a piecewise constant hazard:
The meta-analysis is undertaken on the log-hazard scale.
Parameter μj takes random effects, and v is a constant in the base-case model. I(u = 1) is an indicator function that takes value 1 if u = 1 and 0 otherwise. Parameter v represents the additive effect of the first 3 months on the log-hazard scale. The prior of v is a non-informative normal, but in principle might be informed by non-response rates at 3 months estimated by the evidence synthesis in Chapter 3 (see Results of review of clinical effectiveness).
Differences in withdrawal between biologics
We conducted a meta-analysis of HRs for differences in withdrawal rates between biologics, assuming fixed treatment effects. Data were included from studies identified in the literature search that reported HRs for withdrawal for one biologic compared with another and its SE or CI. This analysis was conducted in stata 10 using the ‘metan’ command.
Results
Results from the winbugs model are shown in Table 62.
| Description | Mean | SE |
|---|---|---|
| Mean annual hazard in month 1 exp(MU + v) | 0.320 | 0.071 |
| Mean annual hazard in month m ≥ 2 [exp(MU)] | 0.165 | 0.031 |
| Between-study SE (log scale) (SE) | 0.332 | 0.229 |
The model predicts the pooled mean hazard is 0.17 per year across all studies and all drugs. The hazard is double in the first 3 months, and the predicted probability of withdrawal in the first 3 months is 1 – exp(–0.32 × 3/12) = 0.077.
Two studies identified in the literature review162,215 reported HRs between therapies for discontinuation from first biologic for any reason for patients with PsA. Both studies adjusted for other factors using multiple regression in a Cox proportional hazards model. The data and results of the meta-analysis are shown in Table 63. Data from Kristensen et al. 215 have been read from a graph. The authors declined our request to provide the precise HRs and CIs (Pierre Geborek, Department of Clinical Sciences, Lund University, Lund, Sweden, 22 September 2009, personal communication).
| Studya | Mean HRb | Lower 2.5% | Upper 97.5% |
|---|---|---|---|
| Etanercept vs adalimumab | |||
| Kristensen 2008215 | 1.00 | 0.30 | 3.00 |
| Saad 2009191 | 1.00 | 0.66 | 1.43 |
| Pooled | 1.00 | 0.68 | 1.46 |
| Etanercept vs infliximab | |||
| Kristensen 2008215 | 0.50 | 0.30 | 0.90 |
| Saad 2009191 | 0.36 | 0.27 | 0.47 |
| Pooled | 0.38 | 0.30 | 0.49 |
Conclusions
-
This study synthesises data on time to withdrawal from first biologic in patients with a diagnosis of PsA from national registries.
-
The estimated rate of withdrawal after the first 3 months is 0.17 per year. This value will be used as the long-term withdrawal rate in the base case of the decision model.
-
This rate is rather higher than the rate estimated in the previous appraisal of these drugs (0.11 per year), which was obtained from a longitudinal study of patients with RA in south Sweden, enrolled between March 1999 and November 2000.
-
This analysis finds that, according to this observational data, on average 7.7% of patients withdraw in the first 3 months.
-
This is much lower that the non-response rate on the PsARC scale recorded in the RCTs (about 16%). This might suggest that, in clinical practice, some patients remain on the drug even though they might not have achieved PsARC response at 12 weeks.
-
This might be because of improvement in the skin condition (not captured by PsARC) and/or the clinician’s belief that response might be achieved later than 12 weeks.
-
There does not appear to be any difference in withdrawal rates between etanercept and adalimumab. Infliximab appears to have a significantly higher withdrawal rate than etanercept.
-
However, these HRs between drugs may not be reliable.
-
The HRs were estimated over the whole follow-up time, and do not distinguish between the first 3 months and later periods. Early withdrawal is a function of initial response, while later withdrawal is a function of continuing response and adverse effects
-
Estimates of differences between drugs may be biased because infliximab was the first biologic to be marketed and may have been used on severe patients with low expectation of maintaining drug therapy.
Limitations
-
As with all observational data, results may be subject to selection bias and confounding.
-
Observed withdrawal rates are likely to depend on the options available to the clinician for switching patients to other biologics.
-
The two studies from the south Sweden register may include some of the same patients.
-
We assumed a normal distribution for probabilities. This should not be a problem if probabilities are not close to 0 or 1 and n is large.
-
Withdrawal rates may be lower in patients receiving concomitant MTX. In this synthesis, one study215 did not report average survival probabilities, but reported only results stratified by use of concomitant MTX or not. Excluding data from Kristensen et al. 215 increased the estimate of the withdrawal rate after 3 months from 0.17 (SE 0.03) to 0.20 (SE 0.72) per year, but the parameters failed to converge correctly.
Annex
Search strategy
Information was identified during a number of stages:
-
The endnote library psoriaticarthritic2009-MASTER.enl containing all the records identified by the searches was, in itself, searched for records containing the words ‘register’ or ‘registry’. This identified 25 records.
-
A search of MEDLINE OvidSP (1950 to week 2, July, 2009) was carried out on 16 July 2009. The search strategy consisted of: Arthritis, Psoriatic/OR (psoria$adj2 (arthrit$or arthropath$)).ti,ab. AND (register$or registr$).ti,ab. The results were scanned for relevance and 16 potentially relevant records were identified.
-
A search for named registries was carried out on 17 July 2009 on MEDLINE OvidSP (1950 to week 2, July, 2009), the named registries identified by the previous stages. This approach identified 112 additional records.
winbugs code
Appendix 13 Costs used in the York model
Each of the industry models presents different resource use assumptions and unit costs, which are used to cost drug treatment and administration/monitoring of patients. Different assumptions are used regarding the dosing of drugs and resource use for administration and monitoring (see Chapter 4, Comparison of the York Economic Assessment with the manufacturers’ models). The current York model sought to generate appropriate costs for each of the treatment options using clinical advice and BSR guidelines to determine the resource use associated with administering drugs and monitoring patients. These items are valued using recently published unit costs and prices. The following sections describe the assumptions made in costing, the associated resource use assumptions, unit costs and cost inputs for the decision model.
Resource use
The current York model assumes that infliximab vials cannot be shared and adopts separate scenarios regarding the use of three or four vials per patient. Infliximab is given at 0, 2 and 6 weeks, followed by every 8 weeks (1.625 every 3 months). Six and a half vials of adalimumab are given in every 3-month cycle. Twenty-six vials of etanercept are given in every 3-month cycle. These assumptions were made in consultation with an expert pharmacist (Carolyn Davies, Central Manchester University Hospitals NHS Foundation Trust, 2009, personal communication).
The York model also assumes a half-day inpatient hospital cost for each infusion of infliximab. A single outpatient visit is assumed for etanercept and adalimumab in the initial 3-month period, followed by a review visit between 3 and 6 months and then every 6 months thereafter.
In the York model it is assumed that, at baseline (in the initial 3-month period), patients will require a FBC, ESR, LFT, U&E, chest radiograph, TB Heaf test, ANA and a dsDNA test. All of these resource use assumptions are taken from the previous York model following the BSR guidelines for the use of biologics.
The resource use assumed as part of drug use, administration and monitoring for the various treatment options are shown in Table 64. All resource use was validated by clinical input.
| Drugs | Administration | Monitoring | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vials per visit | Doses | Outpatient visit | Infusion cost | FBC | ESR | LFT | U&E | Chest radiograph | TB Heaf test | ANA | dsDNA | |
| 0–3 months | ||||||||||||
| Etanercept | 1 | 26 | 1 | 0 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 |
| Adalimumab | 1 | 6.5 | 1 | 0 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 |
| Infliximab (four vials)a | 4 | 3 | 0 | 3 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 |
| Infliximab (three vials)b | 3 | 3 | 0 | 3 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 |
| 3–6 months | ||||||||||||
| Etanercept | 1 | 26 | 1 | 0 | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 | 0 |
| Adalimumab | 1 | 6.5 | 1 | 0 | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 | 0 |
| Infliximab (four vials)a | 4 | 1.625 | 0 | 1.625 | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 | 0 |
| Infliximab (three vials)b | 3 | 1.625 | 0 | 1.625 | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 | 0 |
| 6 months + (3-monthly) | ||||||||||||
| Etanercept | 1 | 26 | 0.5 | 0 | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 | 0 |
| Adalimumab | 1 | 6.5 | 0.5 | 0 | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 | 0 |
| Infliximab (four vials)a | 4 | 1.625 | 0 | 1.625 | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 | 0 |
| Infliximab (three vials)b | 3 | 1.625 | 0 | 1.625 | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 | 0 |
Unit costs
All drug costs were taken from the recent version of the BNF. 65 The costs of inpatient hospital visits were taken from NHS Reference Costs 2008–09230 and is for an elective excess bed-day for inflammatory spine, joint or connective tissue disorders without complications. An inpatient day is assigned a cost of £144 per half day. The cost of an outpatient visit is also taken from NHS Reference Costs 2008–09 and is for a follow-up visit in rheumatology. Each outpatient visit costs £116. Costs associated with laboratory tests relating to the monitoring of patents, were taken from the previous York model,177 updated to reflect 2009 prices. All unit costs used in the current York model are shown below in Table 65.
| £ (2009) | Source | |
|---|---|---|
| Drugs | ||
| Infliximab (100-mg vial) | 419.62 | BNF 5865 |
| Etanercept (25-mg syringe) | 89.38 | BNF 5865 |
| Adalimumab (40-mg syringe) | 357.5 | BNF 5865 |
| Hospital costs | ||
| Half inpatient-day | 144 | NHS Reference Costs 2008–09230 – elective inpatient excess bed-day for inflammatory spine, joint or connective tissue disorders without complications |
| Outpatient rheumatology, first attendance | 205 | NHS Reference Costs 2008–09230 – rheumatology outpatient first attendance |
| Outpatient rheumatology, follow-up attendance | 116 | NHS Reference Costs 2008–09230 – rheumatology outpatient follow up |
| Laboratory tests | ||
| FBC | 2.74 | York NHS Trust – 2005 costs updated to 2009 |
| ESR | 2.71 | |
| LFT | 0.69 | |
| U&E | 1.27 | |
| Chest radiograph | 24.04 | |
| TB Heaf test | 8.01 | NHS Reference Costs 2003 updated to 2009 |
| ANAs | 4.27 | York NHS Trust – 2005 costs updated to 2009 |
| DNA binding (dsDNA) | 4.27 | |
Costs used in the current York model
The resource use items presented in Table 64 were multiplied by the unit costs in Table 65 to generate cost inputs for the decision model. Costs were calculated for the initial 3-month period, 3- to 6-month period, and all subsequent 3-month periods. These costs are presented in Table 66.
| Drugs (£) | Administration (£) | Monitoring (£) | Total (£) | |
|---|---|---|---|---|
| 0–3 months | ||||
| Etanercept | 2323.88 | 116.00 | 55.43 | 2495.31 |
| Adalimumab | 2323.75 | 116.00 | 55.43 | 2495.18 |
| Infliximab (four vials) | 5035.44 | 432.00 | 55.43 | 5522.87 |
| Infliximab (three vials) | 3776.58 | 432.00 | 55.43 | 4264.01 |
| 3–6 months | ||||
| Etanercept | 2323.88 | 116.00 | 3.71 | 2443.59 |
| Adalimumab | 2323.75 | 116.00 | 3.71 | 2443.46 |
| Infliximab (four vials) | 2727.53 | 234.00 | 3.71 | 2965.24 |
| Infliximab (three vials) | 2045.65 | 234.00 | 3.71 | 2283.36 |
| 6 months plus | ||||
| Etanercept | 2323.88 | 58.00 | 3.71 | 2385.59 |
| Adalimumab | 2323.75 | 58.00 | 3.71 | 2385.46 |
| Infliximab (four vials) | 2727.53 | 234.00 | 3.71 | 2965.24 |
| Infliximab (three vials) | 2045.65 | 234.00 | 3.71 | 2283.36 |
Appendix 14 Natural history of patients with psoriatic arthritis eligible for biologic therapy
Introduction
The decision model estimates long-term outcomes in terms of HAQ and PASI for patients with and without biologic therapy. As NICE would not recommend a placebo, the comparator is ‘NH’, a counterfactual state where no biologic therapy is available.
Previous decision models of PsA have estimated what the change in HAQ would have been if no biologic therapy had been offered. Bansback et al. 177 used data from a long-term, open-label follow-up of 35 patients who had originally been entered in a clinical trial comparing MTX with and without ciclosporin in the Leeds Musculoskeletal Unit. These patients had previously not been controlled on MTX alone. In total, 24 responses were received to a postal questionnaire. At the end of the trial, their mean HAQ was 1.13. After ‘some 4.2 years’ follow-up’ (it is not stated if this is the maximum, minimum, mean or median), mean HAQ was 1.4, a mean annual change of 0.07 (SD 0.03).
Possible limitations of this analysis for the purposes of the current decision modelling are:
-
Small sample size.
-
Possibility of selection bias among responders to the postal questionnaire.
-
Patients have failed one DMARD (MTX), rather than two as required by NICE guidelines.
-
It is not stated in the paper if patients met the current guideline criteria for initiating biologics in PsA (three tender and three swollen joints).
No other published estimates were found of long-term outcomes in patients who had been uncontrolled on DMARDs. Morgan et al. 231 investigated outcomes in patients enrolled in NOAR between 1990 and 1994, with and without psoriasis. The median HAQ score for n = 79 patients with inflammatory polyarthritis plus psoriasis at baseline was 0.625 [interquartile range (IQR) 0.25 to 1.375] and was 0.75 (IQR 0.125 to 1.75) at 5 years, indicating a very small annual change in HAQ (0.025 per year). However, these data are not in patients who are necessarily uncontrolled with DMARD.
The NOAR data was reanalysed by the ARC Epidemiology Unit at the University of Manchester to estimate HAQ change in patients who are uncontrolled (with three tender joints three swollen joints) and have previously tried two or more DMARDs. This paper describes how HAQ progression was estimated and used in the decision model.
Methods
The NOAR database is a primary care-based cohort of patients with inflammatory polyarthritis. NOAR has been recruiting patients since 1990. Not all variables were assessed and recorded at follow-ups for the cohort registered between 1995 and 2000 and so this cohort was excluded from the analyses. HAQ and other outcomes are recorded at annual follow-ups. Baseline is the visit when the patient was first seen by the research nurse to be included into the NOAR register. NOAR did not record a diagnosis of PsA. As patients with inflammatory polyarthritis plus psoriasis are thought to have similar prognosis to those who are seronegative without psoriasis, patients who were RF-negative at baseline were selected from the NOAR register. At each time point (baseline, year 1, year 2, year 3 and year 5) we evaluated whether patients fulfilled the following criteria:
-
three tender joints (TJC) and three swollen joints (SJC) using the 51-joint count
-
previous use of two or more DMARDs, implemented as all patients who had used two DMARDs or were still using two DMARDs for at least 30 days.
These criteria are intended to select patients who would be eligible for use of biologics. The BSR recommend that the 78 TJC and 76 SJC is used,149 but this was not available in NOAR. The annual change in HAQ over the following 2 years was estimated from the time when a patient first fulfilled the criteria. The total score is based on the inclusion of all patients who fulfilled the criteria at different time points and their change in HAQ score since that time point. For example, from the data in Table 67, there were 216 patients in total: 24 patients at baseline, + 50 patients at year 1, + 46 patients at year 2, and + 52 patients at year 3 and + 44 patients at year 5. It is therefore possible that some patients are accounted for multiple times in the total score.
| Years from baseline until patient first fulfils criteria | Median symptom duration at baseline | Mean (SD) HAQ score at baseline | No. of patients fulfilling criteria with 1-year follow-up HAQ score data available | Mean (SD) annual change in HAQ score measured over subsequent year | No. of patients fulfilling criteria with 2- year follow-up HAQ score data available | Mean (SD) annual change in HAQ score measured over subsequent 2 years |
|---|---|---|---|---|---|---|
| Baseline | 2.72 | 1.55 (0.84) | 27 | –0.046 (0.513) | 24 | –0.060 (0.279) |
| 1 | 0.99 | 1.52 (0.72) | 53 | –0.104 (0.427) | 50 | –0.019 (0.236) |
| 2 | 0.69 | 1.41 (0.73) | 68 | 0.029 (0.352) | 46 | –0.053 (0.214) |
| 3 | 0.90 | 1.52 (0.73) | 56 | 0.045 (0.389) | 52 | 0.077 (0.228) |
| 5 | 0.91 | 1.51 (0.74) | NAa | 44 | 0.018 (0.180) | |
| Total score | 204 | –0.011 (0.408) | 216 | 0.000 (0.228) |
Results
The results are shown in Table 67. For all patients regardless of when they first became eligible for biologics, the data suggests that there was little change in HAQ over 2 years (mean annual change 0.00, SD 0.228) (n = 216).
For patients who met the eligibility criteria at baseline, their mean HAQ score at baseline was 1.55 (SD 0.84), and the mean change in HAQ over 2 years was –0.060 per year (SD 0.279) (n = 24). These patients had a median of 2.72 years from first onset of symptoms of disease until entry to NOAR. As a higher HAQ score represents worse disability, a negative change is an improvement.
For patients who met the eligibility criteria 3 years after entry to NOAR, the mean change in HAQ over 2 years was 0.077 per year (0.228) (n = 52), i.e. a worsening of disability. These patients had a median of 3.9 years from first onset of symptoms of disease until meeting the eligibility criteria for biologics.
The following sensitivity analyses were carried out:
-
Patients who (had) used a DMARD/DMARDs for > 90 days at time of assessment were included in the analyses. In addition, patients who had used two or more DMARDs for at least 30 days were also included in the analyses.
-
All patients who had used a DMARD/DMARDs or were still using a DMARD/DMARDs, irrespective of duration and number of DMARDs, were eligible at that time point.
-
Tender and swollen joints assessed using the 28-joint count (DAS28).
-
Patients with a nurse assessment of psoriasis as baseline.
The same trends observed in the primary analysis were also found in the sensitivity analyses.
Discussion
This paper has estimated the change in HAQ from the time at which RF-negative patients with inflammatory polyarthritis would have been eligible for biologics under current BSR guidelines. It finds that overall there is little or no change in HAQ over 1 or 2 years.
-
For patients with symptoms for less than about 3 years before they became eligible for biologics, the data suggest that HAQ tends to improve over the following 1 or 2 years.
-
For patients who have had symptoms of inflammatory polyarthritis for more than about 3 years before they became eligible for biologics, the data suggest that HAQ tends to worsen over the following 1 or 2 years.
These analyses have several limitations:
-
The data set cannot identify patients with a consultant diagnosis of PsA.
-
Biologics were licensed around the year 2000. Patients whose arthritis was not considered adequately controlled after this date would probably have been assessed against the criteria for biologics. In this study, we excluded patients who used a biologic agent at any time. Therefore, the patients who did not use biologics are likely to be those whose disability was less severe or progressed more slowly.
-
The criteria for commencement of biologics require patients to satisfy three tender and three swollen joints twice at least 1 month apart, and in these data we only have a single measure.
-
The criteria of three TJCs and three SJCs in some cases will be only moderate disease, and the patient and clinician might not consider that a failure. Patients in NOAR who satisfy the three TJC and three SJC criteria might go on to try other options such as increasing the dose of DMARDs, combination therapy or steroid injections.
-
Patients in NOAR seem to satisfy the three TJC and three SJC criteria having been treated with two or more DMARDs for starting biologic therapy much earlier than patients in RCTs. This may be because RCTs tended to recruit patients who may have worse disease than the minimum entry criteria in the licence.
Conclusion
The York decision model will use as its base case the mean progression of HAQ for patients not using biologics estimated in the NOAR data in patients with long-standing disease (about 3 years since onset of symptoms), i.e. 0.077 per year (SE = 0.228/sqrt(52) = 0.032). This value is very similar to that estimated by Bansback et al. 176 (mean change per year 0.07). Sensitivity analyses will estimate model results at the upper and lower CIs of this parameter.
Appendix 15 Impact of Health Assessment Questionnaire on health service costs
Introduction
This appendix reviews the published literature to estimate the impact of changes in functional status and disability, as measured by the HAQ, on health service and Personal Social Services costs. These estimates will be used in the decision model to predict health service costs over the patients’ lifetimes.
Methods
This is a very broad literature and an exhaustive review was beyond the time constraints of this project. Instead, a rapid review was undertaken of the following sources:
-
evidence presented to previous NICE appraisals of PsA treatments
-
the manufacturers’ submissions to the current appraisal
-
PubMed in October 2009 with the search string: ‘costs health assessment questionnaire arthritis’.
Relevant cost data for the economic model must satisfy the following criteria:
-
The data should be relevant to patients with PsA. There are few cost data specifically measured in this disease, but many studies have analysed the relationship between HAQ and costs in other forms of chronic arthritis. It is assumed here that these data are generalisable to PsA. The cohort should include patients across the full spectrum of HAQ scores, from mild to severe disability.
-
The data must show a causal relationship from HAQ to subsequent health-service utilisation and costs. Ideally, the analysis should exclude potential bias from confounding (the effect of other factors on both HAQ and costs) and endogeneity (the use of health services on subsequent disability). A retrospective or cross-sectional analysis, where patients are asked about their current disability and previous use of health services, might not capture the correct causal relationship. For example, surgery may improve function and so reduce HAQ. A prospective study design is therefore preferred, where HAQ is measured first and the costs are those accrued over the following period.
-
The data should report mean costs conditional on HAQ and measures of sampling uncertainty. If the data are longitudinal, and individuals HAQ and subsequent cost are measured more than once during the study, then the analysis should properly account for the autocorrelation between repeated measures.
-
The data should measure costs not charges or prices.
-
Preferably data would be taken from the UK. Where this is not possible, it is important to assess whether studies from other countries are likely to be generalisable to the UK, particularly countries with mixed public/private financing such as the USA.
-
The data should measure all direct health-care costs in the hospital, outpatient and community. Productivity losses should be reported separately. The base-case model excludes productivity losses in accordance with the NICE reference case.
-
The data should estimate the costs of DMARDs and biologic separately from those of other health services. The economic model includes these costs separately from the effect of HAQ on costs.
-
The study should have collected both HAQ and subsequent resource use as primary data and not use a proxy, such as expected HAQ predicted from other variables.
-
The data should state the price year, the currency and other data to allow adjustment to the UK in 2009.
Papers were excluded if a rapid review of their title or abstract showed they did not meet one or more of the above criteria. The remainder were examined in more detail.
Results
The PubMed search identified 149 papers. There were three submissions by manufacturers to the current appraisal, and three submissions from the same manufacturers to previous NICE appraisals of biologics for PsA. Excluding duplicates, five papers were reviewed in more detail and their results are described below.
The estimates of costs used in the Wyeth submission153 to the current appraisal was excluded because the IPD did not include HAQ, and the analysis used ‘predicted HAQ’ as a proxy. In Chapter 4, the section Systematic review of existing cost-effectiveness evidence gives more details of this study.
Kobelt et al.41
The Wyeth economic model153 for the previous NICE appraisal of PsA182 estimated the direct costs as a function of HAQ based on data in Kobelt et al. 41 The same source was used by the York Assessment Group to populate the economic model for the previous NICE appraisal182 and by Schering-Plough152 in their submission to the current NICE appraisal. The data published by Kobelt et al. 41 are shown in Table 68.
| HAQ score range | Proportion of patientsa | Direct (1999 US$) | Indirect (1999 US$) | Total (1999 US$) | Direct (2008 GBP) |
|---|---|---|---|---|---|
| 0–0.6 | 0.35 | 1228 | 148 | 1,376 | 1094 |
| 0.6–1.1 | 0.16 | 3,152 | 2524 | 5676 | 2809 |
| 1.1–1.6 | 0.15 | 2091 | 3474 | 5565 | 1864 |
| 1.6–2.1 | 0.14 | 3087 | 5300 | 8387 | 2751 |
| 2.1–2.6 | 0.11 | 3401 | 8070 | 11,471 | 3031 |
| 2.6–3 | 0.08 | 2697 | 8407 | 11,104 | 2404 |
The UK study began in 1987 and the cost component included 916 patients with RA with between 5 and 9 years of follow-up. Direct health-care resources were collected prospectively for all patients for hospitalisations, surgical interventions and RA medications. Details of outpatient visits and community services were collected retrospectively in a subsample of 107 patients. All observations for patients in a given state, at any year in the follow-up, were used to calculate the mean annual cost for each state. The paper states that few patients were in the worst HAQ state and no surgery was undertaken in these patients. The authors warn that results for this group may not represent general practice and should be treated with caution.
The analysis has several limitations. The paper does not explain the method of analysis used to estimate the costs in Table 58 in much detail. It is not clear if repeated measures on the same patients were included in the analysis (as their HAQ evolved). As outpatient costs were only collected for a subsample of patients, it is not clear if imputation was used to estimate these costs in the other patients in the study. No indication is given of uncertainty in the primary data such as SEs or CIs. The price year used in the analysis is not stated, although is likely to be 1999 or 2000. Table 68 shows the mean annual direct costs in 1999 US dollars (US$) and 2008 UK pounds sterling (GBP) assuming purchasing power parity index of US$ = 0.6542 GBP,232 and the UK health sector pay and prices inflation factor from 1999 to 2008 is 1.36. 217
Bansback et al.176
Based on the data in Table 68, Bansback et al. 176 carried out a linear regression and reported the coefficients as:
The study does not give much detail of the regression method used, but it is likely that this is an ordinary least-squares regression using the mid-point of the HAQ score as the independent variable and direct cost as the dependent variable, with six data points. If so then the SEs estimated in the regression do not correctly reflect the uncertainty in the mean of costs in the population, as each of these six data points is a sample mean conditional on HAQ score and has been measured with sampling error.
The assumption by Bansback et al. 176 that mean costs are a linear function of HAQ across all HAQ ranges does not appear to be supported by the data shown in Table 68. In particular, it appears that mean direct costs increase rapidly between the first and second HAQ band, but after this subsequent increases in HAQ do not seem to be associated with increasing direct cost, although the association seems stronger for indirect costs. However, there were few patients with severe HAQ states.
It is not clear if the regression estimates relate to the study price year 1999–2000 or have been adjusted for inflation to the price year used by Bansback et al. 176 (not stated by probably 2004 or 2005).
Kobelt et al. 41 estimated that RA drugs, such as DMARDs, represent, on average, 13%–15% of direct costs. The previous York Assessment group model182 reduced the means and SEs of the regression estimates by 15% to populate the decision model. This adjustment assumes that DMARD use is a constant proportion of overall direct costs for all HAQ scores. If costs are reduced by 15% to reflect expenditure on DMARDs then mean direct health-care costs per 3 months in 2008 GBP are estimated as:
Abbott submission,151 Wiles et al.233
The Abbott submission151 to the current appraisal is based on an analysis of resource use in the NOAR register. This is a UK primary care-based cohort established in 1989. The data from the Abbott submission151 are shown in Table 69.
| HAQ band | Inpatient daysa | Joint replacement proceduresa | Total cost (£), (IQR) |
|---|---|---|---|
| 0.0–0.5 | 0.26 | 0.00 | 121, (59–173) |
| 0.5–1.0 | 0.13 | 0.01 | 77, (43–109) |
| 1.0–1.5 | 0.51 | 0.02 | 269, (141–382) |
| 1.5–2.0 | 0.72 | 0.03 | 388, (206–550) |
| 2.0–2.5 | 1.86 | 0.04 | 909, (459–1295) |
| 2.5–3.0 | 4.16 | 0.05 | 1945, (958–2778) |
The reporting of these data has several limitations:
-
The Abbott submission151 states that the data are taken from Wiles et al. ,233 a report commissioned by Roche as part of a previous NICE appraisal (rituximab). However, the Assessment Group has not been granted access to the original report by Wiles et al. 233 Therefore, we cannot establish key details of how the data were collected or analysed.
-
It is not stated if the cost data are prospective or retrospective, relative to when the HAQ assessment was made.
-
It is not stated how many patients were included in the analysis in each HAQ range.
-
It is not stated if HAQ is measured at baseline or longitudinally. If the latter, it is not clear if patients were included in the analysis more than once.
-
It is not stated when the data were collected.
-
It is not clear over what time period the data reported in Table 69 were accrued. As the cycle length of the Abbott model151 is 3 months, we assume that the data in Table 69 also represent resource use and costs over 3 months.
-
No SEs or other measure of uncertainty are shown.
Based on these resource use data and published unit costs, Abbott151 calculated mean costs for each HAQ band. The ‘IQR’ estimates are based on the variability of mean unit costs between NHS hospitals in the NHS Reference Cost database.
Abbott151 fitted an exponential curve through the mean costs of the six HAQ bands.
The submission states that using the IQR, estimates of the values of α and β were calculated to be α = 54.1 (SE 15.31) and β = 1.237 (SE 0.051). The β-coefficient can be interpreted as a unit change in HAQ on average leads to a 24% increase in expenditure.
These SEs for α and β are based on the variability of unit costs between providers, and do not properly reflect the uncertainty in mean costs conditional on HAQ. This should include uncertainty in the mean number of inpatient days and joint replacement procedures conditional on HAQ, which is not given in the data on which this regression is based.
Pugner et al.36
Pugner et al. 36 reviewed cost studies undertaken between 1978 and 1998 in patients with RA in eight countries (Europe, USA and Canada). They found that costs tended to increase more than proportionately to changes in HAQ, consistent with the exponential cost function used by Abbott. 151 However, the data they present appear to be charges rather than costs and so are not suitable to use unadjusted in the UK setting.
Michaud et al.42
This is a longitudinal study of 7527 patients completing a total of 25,000 semiannual (6-monthly) questionnaires from January 1999 to December 2001 in the USA. The study design and analysis have several features that suggest a high internal validity, although it is difficult to establish the degree of generalisability to the UK.
-
Patients were recruited from the practices of US rheumatologists. Patients enrolled in the database as part of pharmaceutical company-sponsored registers were excluded from this study.
-
The study is prospective, that is, HAQ was measured first and, subsequently, health service use.
-
The data were collected during the era when biologics were licensed and entering clinical practice. About 25% of patients used biologic drugs.
-
Direct costs are given in three categories: ‘outpatient’, including health-worker visits, medications, diagnostic tests and procedures, ‘hospital costs’ and ‘drugs’ including DMARDs, biologics, NSAIDS, gastrointestinal medications and non-RA drugs.
-
The price year is given (2001).
-
All direct medical costs are included, regardless of the payer. This is important because almost all medical expenditures are covered by the NHS in the UK. The paper presents data stratified by health insurer and for uninsured patients to allow the effect of financing on expenditures to be assessed.
-
The study reports costs not charges.
-
The analysis is based on primary data, allowing accurate estimation of uncertainty of the mean coefficients.
-
The analysis uses generalised estimating equations, which accounts for the panel structure of the data and repeated measurements on the same individuals.
-
The analysis uses multiple regression allowing control for other factors.
-
Both log-linear and linear models of the effect of HAQ on costs were undertaken.
The results are shown in Table 70 for the mean direct costs and the effect of HAQ on direct costs estimated in the multiple regression.
| Drug costs | Hospital costs | Outpatient costs | ||||
|---|---|---|---|---|---|---|
| Mean | SE | Mean | SE | Mean | SE | |
| Beta-coefficient from multivariable analysisa | ||||||
| HAQ | 434 | 43 | 325 | 46 | 112 | 14 |
| 2001 direct medical costs for 7527 patients with RA, by cost type (per 6 months) | ||||||
| 6-month cost | 3162 | 38 | 786 | 31 | 770 | 10 |
The currency conversion index (purchasing power parity, 2008) is US$ = 0.6542 GBP,232 and the UK health sector pay and prices inflation factor from 2001 to 2008 is 1.31. 217 Given these conversion indices, hospital and outpatient costs as a function of HAQ are:
-
change in 3-month hospital cost for a 1-unit change in HAQ = £139 (SE £20)
-
change in 3-month outpatient cost for a 1-unit change in HAQ = £48 (SE £6).
There are limitations to the generalisability of these data to the UK.
Resource use is influenced by the type of insurance held by the patient and it is thought to be greater in fully insured individuals in the USA than the average in the UK. Michaud et al. 42 found that for a given HAQ score, semiannual costs were US$590 lower for drugs, US$328 lower for hospital services and US$235 lower for outpatient services for those having no insurance compared with similar individuals with private insurance, independently of HAQ. Income also influenced expenditure on outpatient procedures in the USA independently of HAQ.
Michaud et al. 42 found that health indicators, such as fatigue and depression, and other clinical indicators, such as the Rheumatoid Arthritis Disease Activity score, influenced expenditure on outpatient procedures independently of HAQ. These are not measured in the current decision model. Relative unit costs may differ in the USA from the UK. If so, deflating or inflating by a constant conversion rate might not reflect expenditure patterns in the UK. Michaud et al. 42 lists the unit costs in 2001 as US$49.50 for a physician visit, US$688 for a gall bladder procedure and US$4083 for hospitalisation for conditions involving major joints of the lower extremity. In the UK, a specialist visit costs £253 (TCLFUSFF 313), a gall bladder day-case procedure costs £1389 (TDC GA10B) and major foot procedures £2963 (TEI HB31Z). Although it is difficult to match US DRGs with UK Healthcare Resource Groups, these data suggest that unit costs of outpatient and day-case procedures may be more expensive relative to inpatient procedures in the UK than in the USA.
The data do not include use of community nursing and nursing home services, which could be relevant to those with very severe disability.
Conclusion
This paper has reviewed published literature on the relationship between HAQ and costs of non-drug health-care services. Table 71 compares the studies and their key strengths and weaknesses with respect to the decision model in the current appraisal.
| Study, years undertaken | Country, sample size, patient group | Resources covered | Strengths | Weaknesses |
|---|---|---|---|---|
| Kobelt 2002,41 years 1987–96 | UK, 917?, RA | Inpatient, outpatient (?), community (?) | UK data | Dated, few patients in severe HAQ state, includes RA drug costs, analysis poorly reported, no SE |
| Abbott 2005,151 years unknown | UK, sample size unknown, IP | Inpatient | UK data | Analysis poorly reported, incorrectly calculated SE |
| Bansback 2006,176 years unknown | UK, 917?, RA | Inpatient, outpatient (?), community (?) | UK data | As Kobelt et al.,41 incorrectly calculated SE |
| Michaud 2002,42 years 1999–2001 | US, 7527, RA | Inpatient, outpatient, diagnostic tests | Analysis based on IPD and clearly described, drugs separately reported | US data |
The study by Michaud et al. 42 has the highest internal validity, and appears to be the only study to correctly estimate SEs from the primary data, taking account of repeated measures on the same individuals. Michaud et al. 42 estimated (in 2008 UK currency):
-
mean change in 3-month hospital cost for a 1-unit change in HAQ = £139 (SE £20)
-
mean change in 3-month outpatient cost for a 1-unit change in HAQ = £48 (SE 6)
-
mean change in 3-month total cost for a 1-unit change in HAQ = £187 (SE 21).
The main limitation of these data for the decision model is that differences between the US and UK health-care systems limit the generalisability of these data to the UK.
The UK studies are poorly reported, and therefore it is difficult to be assured of their validity and precision. Based on the data in the Kobelt et al. study,41 Bansback et al. 176 estimated (in 2008 UK currency):
-
mean change in 3-month total cost for a 1-unit change in HAQ = £103 (SE 67).
The mean costs per unit change in HAQ estimated by Michaud et al. 42 are greater than those estimated by Bansback et al. ,176 and the SEs considerably smaller. However, given the limitations of the Bansback et al. analysis,176 these data are not easily comparable. It is unclear whether the Kobelt et al. data41 include outpatient costs or not, whether the adjustment to the Kobelt et al. data41 for DMARD costs is correct, whether the Kobelt et al. data41 includes costs for the most severe patients, the price year of the Bansback et al. regression176 is not stated and the SEs have not been calculated from the IPD in the Bansback et al. regression. 176
Despite these limitations, the mean coefficient represents a useful approximate linear relationship between HAQ and health service costs that is generalisable to the current decision model. The base-case decision model will use a linear relationship between HAQ and direct hospital and outpatient costs estimated by Bansback et al. 176 Drug costs will be estimated separately in the decision model. The intercept is not important to the decision model because it applies to all health states and all treatments in all cycles of the model. The Michaud et al. estimate42 and the Abbott estimate151 will be used in a sensitivity analysis.
Appendix 16 Impact of psoriasis on costs
Introduction
This paper describes the impact of psoriasis on health service and social care costs. These estimates will be used in the decision model to predict health service costs over the patients’ lifetimes.
Psoriasis is a chronic skin disease that can seriously impair patients’ QoL. Treatment often leads to a period of remission, after which further treatment is necessary. Therefore, the costs of psoriasis treatments can be substantial. A wide range of treatments are available including topical treatments, systemic drugs and photo(chemo)therapy.
Methods of literature search
A rapid literature search was carried out of the following sources:
-
evidence presented to previous NICE appraisals of PsA and psoriasis treatments
-
the manufacturers’ submissions to the current appraisal
-
PubMed in October 2009 with the search string: ‘costs psoriasis’.
To be used in the decision model, estimates were needed of NHS health and/or social care costs according to the severity of psoriasis, for example, by PASI score, or expected costs of controlled and uncontrolled psoriasis according to some response criterion such as PASI 75. Ideally, the estimates of costs would be based on prospectively collected data on resource use in individual patients, rather than expert opinion. Data should be from the UK or a country with a similar universal, publicly-financed health-care system.
Results of literature search
Most estimates of costs or resource use in the literature were based on expert opinion. A previous model of psoriasis treatments174 assumed one inpatient stay per year for patients with non-response of biologic therapy, based on expert opinion. The manufacturers’ submissions from Abbott151 and Schering-Plough152 in the current appraisal of biologic therapies for PsA also estimated the costs of managing psoriasis, based on expert opinion. Abbott151 estimated that costs of managing psoriasis varied from £153 per 6 months for a PASI score of about 1.5, £934 for a PASI score of 9, £859 for a PASI score of 15 and £1003 per 6 months for a PASI score of 40. Schering-Plough152 estimated 3-monthly costs of managing psoriasis as £167 per PASI point if phototherapy was used and £53 per PASI point if phototherapy was not used (see Chapter 4, Systematic review of existing cost-effectiveness evidence). Two other economic evaluations of psoriasis treatments234,235 made similar assumptions to Woolacott et al. 174 based on expert opinion. Colombo et al. 236 found the mean cost for patients with moderate plaque psoriasis (PASI ≤ 20) was €5226.04, while the mean cost for patients with more severe disease (PASI > 20) was €11,434.40 per year in Italy in 2004. Marchetti et al. 237 estimated a year of fluocinonide therapy for mild-to-moderate plaque psoriasis (< 20% of BSA) would cost an average of US$3394 in the USA at 1998 prices, corresponding to £788 per 3 months at 2008 UK prices.
Two studies were found that estimated costs in controlled and uncontrolled patients with moderate-to-severe psoriasis based on prospectively collected IPD. Hartman et al. 188 conducted a RCT in the Netherlands comparing day-case dithranol treatment, UVB therapy and inpatient dithranol treatment for 219 patients with a mean PASI at baseline of 15.3 (SD 6.9) and a mean BSA of 21% (SD 13.8%). Patients did not receive biologic therapy in the RCT. Resource-use data were collected on drugs, UVB sessions, consultations, nursing time, inpatient days, outpatient visits, primary health care, and time lost from normal activity. Hartman et al. 188 defined ‘treatment success’ as a reduction of the baseline area of at least 90% during the treatment period and ‘relapse’ as a return of 50% or more of the baseline area of psoriasis. Hartman et al. 188 report the numbers of patients who fail initial treatment, the number with initial success but relapse during the year and the number who have 1-year remission.
The results of Hartman et al. 188 are shown in Tables 72 and 73.
| Initial treatment: mean, median (IQR) € | Per month without relapse: mean (€) | Per month after relapse: mean (€) | |
|---|---|---|---|
| Day case | 765, 723 (554–988) | 19 | 264 |
| UVB | 600, 585 (458–744) | 5 | 219 |
| Inpatient | 6823, 6380 (5200–8519) | 25 | 220 |
| n | Pr (treatment fails) | Pr (initial success then relapse within 1 year) | Pr (1-year remission) | |
|---|---|---|---|---|
| Day case | 94 | 0.37 | 0.24 | 0.39 |
| UVB | 70 | 0.41 | 0.35 | 0.25 |
| Inpatient | 52 | 0.09 | 0.65 | 0.26 |
Poyner et al. 192 recorded private expenditures and NHS costs (general practitioner consultations and treatments) for 272 patients with mild-to-moderate psoriasis after a 12-week course of either calcipotriol or dithranol. Mean health-care expenditure by the NHS over 6 months was £55.61 at 1999 prices (£79 at 2008 prices). The cost of treating psoriasis (excluding the initial course of treatment) was greater to the patient than to the NHS.
The mean NHS cost of an outpatient session of phototherapy is £116. 187 Guidelines suggest that patients typically undergo 4–10 sessions. 238 Six sessions would cost £696.
Estimate of costs of psoriasis in the decision model
The decision model requires the health service costs of patients who do not use biologic therapies, or those whose psoriasis does not respond to biologic therapy, according to severity of psoriasis at baseline. Many of the studies in the literature review concluded that costs vary by baseline severity, although there does not appear to be a uniform classification of mild, moderate and severe psoriasis across the different studies, with some using PASI, some DLQI and others the percentage of BSA. Reich and Mrowietz195 define PASI > 10 or BSA > 10% as ‘at least moderate’, and PASI ≤ 10 as ‘mild to moderate’. 195
For ‘moderate-to-severe’ patients, we assume that ‘treatment responders’ to biologic therapy, as measured by PASI 75, incur the monthly costs of patients in remission estimated by Hartman et al. 188 The initial treatment cost of UVB therapy estimated by Hartman et al. 188 is very similar to NHS Reference Costs for England, indicating that these data are likely to be generalisable to the UK. Patients who are not using biologic therapy, or not responding to biologic therapy, will undergo one course of UVB treatment per year. Of these, those that fail UVB treatment incur subsequent monthly costs estimated by Hartman et al. 188 for patients after relapse. Those that initially succeed but relapse during the year are assumed to be in remission for 6 months.
We choose UVB because it is a widely used therapy for moderate-to-severe psoriasis in the UK. Evaluating the most effective and cost-effective psoriasis treatment for patients who are not using biologic therapy or in whom biologic therapy is ineffective is beyond the scope of this study. We use the costs of inpatient dithranol as a sensitivity analysis.
The currency conversion rate in purchasing power parity is US$ = €0.883 and US$ = £0.654,230 and the inflation index from 1998 to 2008 is 1.42. 215
The mean cost of UVB in 2008 UK prices is:
-
initial treatment = 600 × 1.42/0.883 × 0.654 = £631.04
-
per month without relapse = 5 × 1.42/0.883 × 0.654 = £5.26
-
per month after relapse = 219 × 1.42/0.883 × 0.654 = £230.33.
Given these data, we estimate the annual cost for each health state following UVB as follows:
-
annual cost if treatment succeeds = 631.04 + 12 × 5.26 = £694
-
annual cost if treatment relapse at 6 months = 631.04 + 6 × 5.26 + 6 × 230.33 = £2045
-
annual cost if treatment fails = 631.04 + 12 × 230.33 = £3394.
The weighted mean annual cost if UVB treatment is given is therefore:
-
mean annual cost = 3394 × 0.41 + 2045 × 0.34 + 694 × 0.25 = £2262.
The annual cost if the psoriasis were controlled by biologic drugs and no UVB treatment were given would be 12 × 5.26 = £63.
The mean costs of moderate-to-severe psoriasis used in the decision model per 3-month period are:
-
for patients using biologics and achieving PASI 75 response: £63/4 = £16(SE 1)
-
for patients not achieving PASI 75 response from using biologics:£2262/4 = £566 (SE 25)
-
for patients not using biologic therapy: £2262/4 = £566 (SE 25).
The SEs are calculated from the IQRs given in Hartman et al. 188 assuming normal distributions for costs. The costs of biologic therapies and the costs of treating disability are estimated separately in the decision model. If it is assumed that patients without biologics or without response of biologics will undergo one course of inpatient therapy per year instead of UVB, the cost increases to £8532 per year or £2133(SE 93) per 3-month period.
For ‘mild-to-moderate’ patients, the treatment cost estimated by Marchetti et al. 237 (£788 per 3 months) is US data and likely to overestimate the cost in the UK. We assume that patients with mild-to-moderate psoriasis who are not using biologic therapy or are uncontrolled by biologic therapy undergo one course of UVB therapy per year, costing £636. 187 The mean cost after treatment (averaged over responders and non-responders) is estimated from Poyner et al. 192 The total cost over the year is 636 + 2 × 79 = £794.
The mean costs of mild-to-moderate psoriasis used in the decision model per 3-month period are:
-
for patients using biologics and achieving PASI 75 response: £16 (SE 1)
-
for patients not achieving PASI 75 response from using biologics: £794/4 = £198(SE 9)
-
for patients not using biologic therapy: £198 (SE 9).
Conclusions
This paper describes the impact of psoriasis on health-service costs for patients using biologic therapy and not using biologic therapy. The estimates used in the base-case decision model for mild-to-moderate patients are based on UK resource use and cost data. Costs are based on the results of a Dutch RCT for moderate-to-severe patients. The health system in the Netherlands is a social insurance system, but results are likely to be generalisable to the UK. This analysis does not account for adverse effects of repeated psoriasis treatments, such as skin cancers.
Appendix 17 Estimation of the effect of Health Assessment Questionnaire and Psoriasis Area and Severity Index on utility in the decision model
Introduction
Clinical benefit is captured in the decision model by estimating expected HAQ and PASI at each time point at each state in the model (on and off biologic drugs). This appendix describes the relationship between HAQ, PASI and utility (a preference-based measure of HRQoL).
Methods
Chapter 4, Systematic review of existing cost-effectiveness evidence describes the Assessment Group’s critical review of the manufacturers’ submissions to the current appraisal. Each company analysed the relationship between HAQ, PASI and utility in a different way. It was difficult to assess whether differences in these results arose from differences in the primary data or from the chosen method of analysis. Consequently, the Assessment Group requested that each company estimate a similar regression analysis on their data, to assess whether results were comparable (see Appendix 6). The Assessment Group requested that the analysis should be an ordinary least-squares regression of utility versus HAQ, PASI and an interaction term HAQ × PASI.
Results
All three manufacturers reanalysed their data and the results are shown in Table 74.
| Coefficients | Variance–covariance matrices | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | SE | z | p > z | Intercept | HAQ | PASI | HAQ × PASI | ||
| Wyeth153 | |||||||||
| Intercept | 0.895 | 0.007 | 128.652 | 0.000 | Intercept | 0.000048430 | |||
| HAQ | –0.295 | 0.008 | –37.157 | 0.000 | HAQ | –0.000030080 | 0.000062880 | ||
| PASI | –0.004 | 0.000 | –9.039 | 0.000 | PASI | –0.000001640 | 0.000000947 | 0.000000207 | |
| HAQ × PASI | 0.000 | 0.000 | –0.669 | 0.504 | HAQ × PASI | 0.000001311 | –0.000002207 | –0.000000136 | 0.000000183 |
| Schering-Plough152 | |||||||||
| Intercept | 0.871 | 0.001 | 1126.782 | 0.000 | Intercept | 0.000000598 | |||
| HAQ | –0.249 | 0.001 | –348.431 | 0.000 | HAQ | –0.000000422 | 0.000000511 | ||
| PASI | –0.002 | 0.000 | –25.447 | 0.000 | PASI | –0.000000037 | 0.000000027 | 0.000000010 | |
| HAQ × PASI | 0.000 | 0.000 | 0.741 | 0.459 | HAQ × PASI | 0.000000026 | –0.000000030 | –0.000000007 | 0.000000006 |
| Abbott151 | |||||||||
| Intercept | 0.886 | 0.018 | 48.692 | 0.000 | Intercept | 0.000329500 | |||
| HAQ | –0.232 | 0.025 | –9.343 | 0.000 | HAQ | –0.000292000 | 0.000614600 | ||
| PASI | –0.003 | 0.002 | –1.667 | 0.096 | PASI | –0.000014000 | 0.000012900 | 0.000002195 | |
| HAQ × PASI | –0.004 | 0.002 | –1.950 | 0.051 | HAQ × PASI | 0.000012600 | –0.000033000 | –0.000001607 | 0.000004094 |
Conclusions
The results of these regressions are similar in all data sets. This indicates that the relationship between HAQ, PASI and utility is stable across independent clinical trials, and gives us confidence that the results are generalisable to the general population.
The interaction between HAQ and PASI does not reach statistical significance at the 5% level in any data set, but is very close to the 5% level in the Abbott data. 151
The results of the regressions in Table 74 are very similar and the decision about which data set we use in the York model is not likely to change the conclusions. We use the Wyeth results153 without the interaction term as the base case and other functions as sensitivity analyses.
Appendix 18 Estimation of Psoriasis Area and Severity Index score for treatment responders in the decision model
Introduction
The PASI is a scoring system to evaluate baseline and response of therapy in psoriasis. The BAD173 recommend PASI 75 for measuring primary response of psoriasis in patients with PsA. PASI 75 is a binary outcome that indicates a 75% or greater improvement in PASI from baseline. RCTs commonly report this and other measures of response, such as PASI 50 and PASI 90. In Chapter 3, the section Results of review of clinical effectiveness estimates the mean probability across all trials of achieving PASI 50, PASI 75 and PASI 90 for each biologic therapy and placebo using summary data from the RCTs.
These multivariate response indicators (PASI 50/75/90) indicate the probability of achieving a minimum percentage improvement in PASI compared with baseline. However, the decision model requires the mean absolute or percentage change in PASI as an input parameter, given each type of biologic therapy and no therapy.
This appendix describes how the mean absolute change in PASI is calculated in the decision model.
Methods
We calculate the marginal probabilities of each mutually exclusive outcome:
Figure 11 shows a segment of the decision tree for the psoriasis response and non-response for a given drug. Pr(< PASI 50| < PASI 75) indicates the probability of a change in PASI of between 0% and 49%, given improvement of less than PASI 75, and is calculated as:
FIGURE 11.
Segment of decision tree showing the mean change in PASI for psoriasis response and psoriasis non-response.
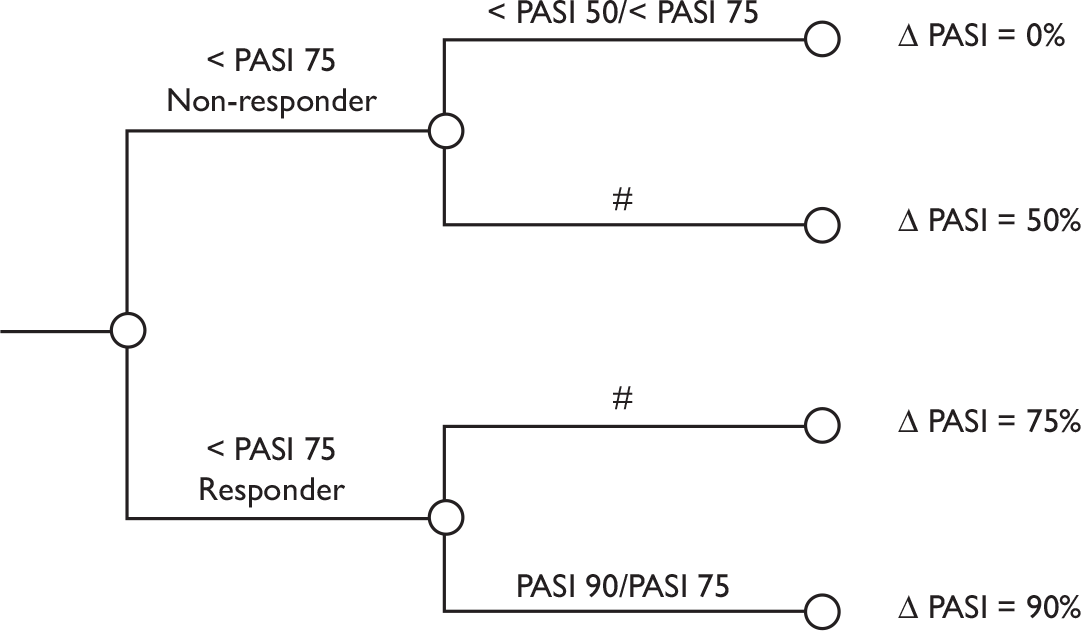
We know that the improvement for this group is within the range 0%–50%, and in the base case we (conservatively) assume that the relative improvement in PASI for this group is 0. For change in PASI between 50% and 74%, we assume the change is 50%. For a change between 75% and 89%, we assume the change is 75%, and between 90% and 100%, we assume the change is 90%.
Consequently, if baseline absolute PASI is P0, the mean absolute change in PASI for those achieving a PASI 75 response (while on therapy) is:
The mean absolute change in PASI for those not achieving a PASI 75 response (while on biologic therapy) is:
Conditioning the change in PASI on PASI 75 allows the consequences to be explored of using different decision rules about whether to withdraw biologic therapy or not if a PASI 75 response is not achieved, or to withdraw if a PASI 75 response is achieved, but a PsARC response is not.
Sensitivity analysis
Simple sensitivity analyses will assume different values of the thresholds for the change in PASI, such as using the upper end of the range, or the mid-point. For example, for PASI response between 50% and 74%, we could assume that the change is 74% or 57% (the mid-point). Note that, a priori, we have no reason to expect the distribution of percentage changes in PASI within a given range to be uniformly distributed within that range, and so we have no reason to expect the mid-point to better estimate the mean change than other values.
An alternative sensitivity analysis is suggested by data from the Abbott submission. 151 Abbott151 used regression to estimate the relationship between PASI response and the mean absolute change in PASI. Their results are reproduced in Table 75.
| Description | Covariate | Parameter estimate | SE | t-value | Pr > |t| |
|---|---|---|---|---|---|
| Intercept | α | 0.36879 | 0.28977 | 1.27 | 0.212 |
| Baseline PASI_t | β1 | 1.01496 | 0.08344 | 12.16 | < 0.0001 |
| Baseline age | β2 | –0.00461 | 0.00541 | –0.85 | 0.3997 |
| Gender (1 = male) | β3 | 0.08901 | 0.10511 | 0.85 | 0.4032 |
| Baseline PsA duration | β4 | 0.00075643 | 0.00666 | 0.11 | 0.9103 |
| Whether on MTX (1 = yes) | β5 | 0.00433 | 0.10234 | 0.04 | 0.9665 |
| Whether a PASI 50–75 responder | β6 | –0.85124 | 0.16655 | –5.11 | < 0.0001 |
| Whether a PASI 75–90 responder | β7 | –1.13011 | 0.15625 | –7.23 | < 0.0001 |
| Whether a PASI 90+ responder | β8 | –1.89522 | 0.18899 | –10.03 | < 0.0001 |
| Treatment = biologic | β9 | –0.50235 | 0.12880 | –3.90 | 0.0004 |
Results
Table 75 illustrates the calculation of the change in PASI for responders and non-responders using the probabilities of psoriasis response given in Chapter 3 (see Results of review of clinical effectiveness) and the assumptions in the methods section above. For convenience, these probabilities are shown again in Table 76.
| Etanercept | Infliximab | Adalimumab | Placebo | |
|---|---|---|---|---|
| Pr(PASI 50) | 0.40 | 0.91 | 0.74 | 0.13 |
| Pr(PASI 75) | 0.18 | 0.77 | 0.48 | 0.04 |
| Pr(PASI 90) | 0.07 | 0.56 | 0.26 | 0.02 |
| Percentage change in PASI for PASI 75 non-responders | 13.70 | 31.10 | 24.00 | NAa |
| Percentage change in PASI for PASI 75 responders | 81.20 | 85.90 | 83.10 | 80.60 |
Conclusion
On average, infliximab is predicted to give the greatest probability of a psoriasis response and the greatest change in PASI in both responders and non-responders. Adalimumab is the second-most effective and etanercept is predicted to be the least effective in terms of psoriasis.
Appendix 19 All-cause mortality
Introduction
All-cause mortality rates for a given age are higher in people with PsA than the general population. Wong et al. 29 found that men attending a PsA clinic have a 65% greater mortality rate than the general population in Canada and women 59% greater mortality. A UK population-based study using the General Practice Research Database found 50% greater mortality in patients with severe psoriasis than the general population and no change in this standardised mortality ratio after excluding patients with PsA, indicating that patients with PsA have similar mortality risk to those with severe psoriasis. 239 However, there is no clear evidence that biologic therapies change these mortality risks.
Published life tables give mortality risks in the general population for a given age and gender. However, it has been shown that in developed countries, all-cause mortality hazards increase at an exponential rate after the age of 40 years, and a Gompertz function closely approximates these hazards. 240 Using a parametric function instead of looking up the hazards directly from life tables requires fewer parameters in the decision model and arguably saves computation time. Furthermore, a parametric hazard function might allow more accurate interpolation of the hazards between years if the cycle length of the model is < 1 year.
This paper describes the estimation of the Gompertz function to predict all-cause mortality hazards.
Methods
In the Gompertz function, mortality hazards h(x) at age x (where x ≥ 40) are:
Taking log:
This linear relationship is straightforward to estimate from life-table hazards using ordinary least-squares regression of log-hazards versus age. These hazards can be adjusted for the PsA population by multiplying by the standardised mortality ratio for the disease.
Results
The results of the regression of log(life-table hazards) versus age in years are shown in Table 77 for the general population in men and women for the years 2006–8.
| Mean coefficient | SE | 95% CI | |
|---|---|---|---|
| Men | |||
| Age | 0.0946 | 0.00067 | 0.0932 to 0.0959 |
| Constant (log R) | –10.2570 | 0.04600 | –10.3490 to –10.1650 |
| Adj R-squared | 0.9965 | ||
| Women | |||
| Age | 0.1010 | 0.00067 | 0.0999 to 0.1027 |
| Constant (log R) | –11.1090 | 0.04600 | –11.2030 to –11.0170 |
| Adj R-squared | 0.9969 | ||
Conclusion
The Gompertz function can estimate general population life table all-cause hazards with a high degree of precision.
Appendix 20 Sequential use of biologic therapy
Introduction
The base-case decision model assumes that patients who enter the model are ‘biologic naive’, and that those who fail therapy have no further options, and, consequently, receive palliative care only. In practice, it many patients who withdraw from their first biologic agent will switch to another. 241 It is potentially important that the decision model takes account of this option. Hence the model was extended to consider, as far as available evidence allows, the cost-effectiveness of sequential use biologics in patients who have failed on earlier biologic therapy.
This appendix describes the literature search and methods used to obtain the response and withdrawal parameters to undertake this modelling. The results of the cost-effectiveness analysis in the subgroup of patients who switch to another biologic drug are presented in Chapter 4 (see York Economic Assessment).
Methods
The approach taken here is to consider the effectiveness and cost-effectiveness of alternative strategies for a subgroup of patients who have failed a first course of biologic therapy. For example, if etanercept had been tried and failed, the choice would be between a second trial with adalimumab or infliximab, or no further biologic therapy.
The reason why the patient failed the first course of therapy is potentially important information in deciding on the second course. Therefore, we consider two subgroups: (1) patients who has failed etanercept because of adverse events; and (2) those who failed because of lack of efficacy. We do not make a distinction here between those who had complete lack of response (measured by PsARC at 3 months) and those who had secondary loss of treatment efficacy.
We search the clinical literature and publications from UK and other registers to find response and/or withdrawal rates from a second drug for patients in PsA or RA who failed a first drug because of lack of efficacy or adverse events.
The base-case decision model has two measures of initial response (PASI 75 for psoriasis and PsARC for arthritis) and an estimated rate of withdrawal after the first 3 months. Some of the clinical literature report RRs (such as HRs) of failing a second biologic drug, compared to failing a first drug. We assume the odds of PsARC for a drug used as second-line therapy are equal to the odds as first therapy (estimated by the evidence synthesis in Chapter 3), multiplied by the RR for failing second therapy versus first therapy. We make a similar assumption to estimate the hazards of withdrawal after 3 months for a second course of biologic therapy. Given that in the base-case model patients are not withdrawn for failing to obtain PASI 75, we assume that the probabilities of PASI 75 in the second course of therapy are the same as in the first course. All of the other parameters of the model are the same as in the base case.
Results of the literature search
A review the literature did not find any RCTs that had studied these subgroups. However, the review of publications from biologics registers found four papers that included some relevant information about second-course biologic therapies.
Table 78 shows the results of three papers that estimated the probabilities of remaining on therapy (‘persistence’) in patients with PsA for first and second courses of biologic drugs. In all of the studies the probability of persistence up to 1 year is lower for second biologic than first biologic. These papers did not report withdrawal for second biologic conditional on the reason for withdrawal from the first biologic. Gomez-Reino et al. 228 also estimated the rates of withdrawal for adverse events and inefficacy for each biologic. These data show that in all of the biologic therapies at first course, patients tended to be more likely to withdraw for adverse events than inefficacy. Rates of withdrawal from infliximab when used as second-line therapy tend to be higher than other drugs used as second-line therapy. However, SEs are not reported so this may be due to chance. Perhaps more importantly, these are not randomised data and patients cohorts are unlikely to be similar between the drugs.
| Course of treatment | No. starting | No. failed | Percentage failed | Reason failed | Pr survival 1-year |
|---|---|---|---|---|---|
| Coates 2008,241 UK, patients with PsA | |||||
| First | 60 | 14 | 23 | All reasons | NA |
| Second | 12 | 7 | 58 | All reasons | NA |
| Saad 2009,191 UK, patients with PsA | |||||
| First | 566 | NA | All reasons | 0.82 (0.79 to 0.85) | |
| Second | 178 | NA | All reasons | 0.74 (0.71 to 0.78) | |
| First | 566 | NA | Inefficacy | 0.92 (0.89 to 0.94) | |
| Second | 178 | NA | Inefficacy | 0.70 (0.63 to 0.75) | |
| First | 566 | NA | Adverse events | 0.96 (0.94 to 0.97) | |
| Second | 178 | NA | Adverse events | 0.76 (0.69 to 0.81) | |
| Gomez-Reino 2006,228 Spain, patients with PsA | |||||
| First | 289 | 55 | 19 | All reasons | 0.87 (0.83 to 0.9) |
| Second | 15 | 8 | 53 | All reasons | 0.81 (0.65 to 0.9) |
| Gomez-Reino 2006,228 Spain, all chronic arthritis patients | |||||
| Course of treatment | Reason failed | Rate of failure per 100 patient-years treated | |||
| First, infliximab | Adverse events | 6.5 | |||
| First, infliximab | Inefficacy | 4.7 | |||
| Second, infliximab | Adverse events | 32.7 | |||
| Second, infliximab | Inefficacy | 38.5 | |||
| First, etanercept | Adverse events | 3.8 | |||
| First, etanercept | Inefficacy | 3.6 | |||
| Second, etanercept | Adverse events | 6.1 | |||
| Second, etanercept | Inefficacy | 9.3 | |||
| First, adalimumab | Adverse events | 7.2 | |||
| First, adalimumab | Inefficacy | 3.2 | |||
| Second, adalimumab | Adverse events | 12.5 | |||
| Second, adalimumab | Inefficacy | 12.5 | |||
Table 79 shows the result of one paper that reported HRs for withdrawal from second course of therapy compared with the first course of therapy. 196 The paper distinguishes between outcomes for patients who start a second course of biologics after adverse events in the first course, and patients who start a second course of biologics following lack of efficacy in the first course. The data are for patients with RA, rather than PsA, and are from patients in the UK BSR register who had at least 6 months’ follow-up by the end of April 2005.
| Course of treatment | n starting | No. failed | Percentage failed | Reason failed | HR for discontinuation of second therapy, compared with rate for first therapyb |
|---|---|---|---|---|---|
| First | 6739 | 2360 | 35 | All reasons | |
| First | 6739 | 841 | 12 | Inefficacy | |
| First | 6739 | 1023 | 15 | Adverse events | |
| First | 6739 | 496 | 7 | Other reason | |
| Second | inefficacy in first | 503 | 78 | 16 | Inefficacy | 2.7 (2.1 to 3.4) |
| Second | adverse event in first | 353 | 33 | 9 | Inefficacy | 1.2 (0.8 to 1.6) |
| Second | inefficacy in first | 503 | 50 | 10 | Adverse events | 1.1 (0.9 to 1.5) |
| Second | adverse event in first | 353 | 71 | 20 | Adverse events | 2.3 (1.9 to 2.9) |
Parameters in the decision model
There are four sets of parameters to estimate to implement the model for switching biologic therapy (Table 80). We assume the HRs for failing a second biologic compared with failing the first biologic are the same for all biologics.
| Reason for discontinuation of first course of biologic therapy | |||
|---|---|---|---|
| Inefficacy | Adverse event | ||
| Reason for discontinuation of second course of biologic therapy | Initial PsARC response (at 3 months), by drug j | p.psarcj2 (first inefficacy) | p.psarcj2 (first adverse event) |
| Rate of secondary non-response or adverse event after 3 months | p.long2 (first inefficacy) | p.long2 (first adverse event) | |
Initial Psoriatic Arthritis Response Criteria response given patient discontinued first course because of a lack of efficacy
Based on the data in Table 79, we assume that if the first biologic agent was discontinued due to inefficacy, the odds of achieving a PsARC response in the first 3 months on the second agent was reduced on average 2.7-fold (95% CI 2.1 to 3.4). Therefore, if the odds of a PsARC response at 3 months in drug j used as first biologic are o.psarcj1 = p.psarcj1/(1 – p.psarcj1) then the odds of a PsARC response at 3 months in drug j used as second biologic given the first was discontinued for lack of efficacy are:
Initial Psoriatic Arthritis Response Criteria response given patient discontinued first course because of an adverse event
The probability of an initial PsARC response for the second agent, given the first was discontinued for an adverse event is unchanged, so:
Withdrawal after first 3-month trial period given patient discontinued first course because of a lack of efficacy
Based on the data in Table 79, we assume that if the first biologic agent was discontinued due to inefficacy, the risk of withdrawal after 3 months due to inefficacy was increased 2.7-fold. However, the odds of withdrawal due to adverse events was unchanged, given the 95% CI includes 1.
In Table 79, 6739 patients started a first biologic. Of these, 2360 patients withdrew – 841 (36%) for inefficacy and 1023 (43%) for adverse events. If the rate of withdrawal after 3 months from the first biologic agent for any reason is ‘p.long1’ then the rate of withdrawal from the first biologic agent for inefficacy is: p.long1 × 0.36. We assume that the rate of withdrawal after 3 months for the second agent, given the first was discontinued for lack of efficacy, is:
Withdrawal after first 3-month trial period given patient discontinued first course because of an adverse event
Given the data in Table 79, we assume that if the first biologic agent was discontinued due to adverse events, the risk of withdrawal from the second biologic due to adverse events was increased by 2.3 (95% CI 1.9 to 2.9). The overall expected rate of withdrawal after 3 months for the second agent, given the first was discontinued for an adverse event is:
The HRs in Table 79 will be entered into the model as probability distributions. The HR on a log-scale for continuing lack of efficacy has a mean of 0.993 (SE 0.120), and the HR on a log-scale for continuing adverse events has mean of 0.832 (SE 0.106).
Conclusions
This subgroup analysis is necessarily exploratory, given the limitations of the data for outcomes after switching biologic therapies. These limitations include:
-
The data on outcomes after switching comes from patients with RA not PsA. Data of withdrawal by type of disease suggest that there may be differences in withdrawal rates between RA and PsA. 226,242 However, the data on outcomes after switching from patients with PsA were not reported in sufficient detail for the decision model. We assume in the decision model that even if there are differences in absolute withdrawal rates between RA and PsA, the HRs comparing withdrawal from first-line therapy with second-line therapy do not differ by disease.
-
The data are from observational studies. Therefore, there is the possibility of selection bias and other confounding factors. However, Hyrich et al. 196 cautions that designing a randomised experiment for patients to receive a second agent on the basis of their outcome (inefficacy or toxicity) would present considerable practical and ethical difficulties. Therefore, observational studies may be the best data that can be obtained.
The data cannot differentiate between those who had complete lack of response (such as PsARC at 3 months) and those who had secondary loss of treatment efficacy. The decision model has therefore assumed the HRs apply equally to both types of response.
Appendix 21 R programme for the York economic analysis
Appendix 22 Sensitivity analysis comparing results from the stochastic and deterministic models
| Sa | Description | TrT | Stochastic | Deterministic | Difference (absolute value) | Difference (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY | Cost (£) | ICER | QALY | Cost (£) | ICER | QALY | Cost (£) | ICER | QALY | Cost (£) | ICER | |||
| Average (SD) | –0.12 (0.31) | 35 (2148) | 806 (4340) | –2.16 (5.74) | 0.02 (2.84) | 4.75 (9.95) | ||||||||
| 1 | Base case | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 6.58 | 68,638 | Ex dom | 6.370075 | 68,465.66 | Ex dom | –0.21 | –172 | –3.19 | –0.25 | ||||
| E | 7.001 | 74,841 | 17,853 | 6.775944 | 74,519.33 | 19,023.27 | –0.23 | –322 | 1170 | –3.21 | –0.43 | 6.56 | ||
| I | 7.308 | 88,442 | 44,326 | 7.067269 | 88,103.69 | 46,629.52 | –0.24 | –338 | 2304 | –3.29 | –0.38 | 5.20 | ||
| 2 | Rebound in HAQ is small after withdrawal (base case = initial gain) | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 7.225 | 67,710 | Ex dom | 7.314953 | 67,159.74 | Ex dom | 0.09 | –550 | 1.25 | –0.81 | ||||
| E | 7.792 | 73,706 | 12,035 | 7.923641 | 72,933.09 | 10,853.98 | 0.13 | –773 | –1181 | 1.69 | –1.05 | –9.81 | ||
| I | 8.188 | 87,174 | 34,006 | 8.346960 | 86,335.03 | 31,659.22 | 0.16 | –839 | –2347 | 1.94 | –0.96 | –6.90 | ||
| 3 | Rapid worsening in HAQ with no treatment (upper 95% of CI) | N | 3.309 | 44,434 | NA | 3.225453 | 44,348.90 | –0.08 | –85 | –2.52 | –0.19 | |||
| A | 4.967 | 70,829 | Ex dom | 4.842184 | 70,577.36 | Ex dom | –0.12 | –252 | –2.51 | –0.36 | ||||
| E | 5.447 | 76,985 | 15,221 | 5.313432 | 76,540.66 | 15,417.67 | –0.13 | –444 | 197 | –2.45 | –0.58 | 1.29 | ||
| I | 5.786 | 90,609 | 40,248 | 5.647306 | 90,066.22 | 40,510.98 | –0.14 | –543 | 263 | –2.40 | –0.60 | 0.65 | ||
| 4 | Log-PASI utility function (Abbott151) (base case linear) | N | 4.558 | 42,168 | NA | 5.843696 | 41,817.64 | 1.29 | –350 | 28.21 | –0.83 | |||
| A | 6.001 | 68,638 | Ex dom | 7.005181 | 68,465.66 | Ex dom | 1.00 | –172 | 16.73 | –0.25 | ||||
| E | 6.39 | 74,841 | 17,835 | 7.316303 | 74,519.33 | 22,206.66 | 0.93 | –322 | 4372 | 14.50 | –0.43 | 24.51 | ||
| I | 6.769 | 88,442 | 35,898 | 7.643365 | 88,103.69 | 41,534.5 | 0.87 | –338 | 5637 | 12.92 | –0.38 | 15.70 | ||
| 5 | No correlation between PASI 75 and PsARC (base case = 0.4) | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 6.571 | 68,968 | Ex dom | 6.361208 | 68,808.52 | Ex dom | –0.21 | –159 | –3.19 | –0.23 | ||||
| E | 6.997 | 74,990 | 17,979 | 6.771297 | 74,673.95 | 19,165.02 | –0.23 | –316 | 1186 | –3.23 | –0.42 | 6.60 | ||
| I | 7.303 | 88,641 | 44,558 | 7.062126 | 88,314.90 | 46,903.65 | –0.24 | –326 | 2346 | –3.30 | –0.37 | 5.26 | ||
| 6 | RCT results fully generalisable to clinical practice (no adjustment for placebo effect) | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 6.637 | 68,561 | Ex dom | 6.429905 | 68,387.96 | Ex dom | –0.21 | –173 | –3.12 | –0.25 | ||||
| E | 7.068 | 74,752 | 17,178 | 6.847586 | 74,426.29 | 18,210.23 | –0.22 | –326 | 1032 | –3.12 | –0.44 | 6.01 | ||
| I | 7.381 | 88,344 | 43,371 | 7.146598 | 88,000.67 | 45,397.44 | –0.23 | –343 | 2026 | –3.18 | –0.39 | 4.67 | ||
| 9 | Exponential HAQ cost function (Abbott151) (base case linear) | N | 5.171 | 63,052 | NA | 5.056908 | 63,146.27 | –0.11 | 94 | –2.21 | 0.15 | |||
| A | 6.580 | 82,129 | Ex dom | 6.370075 | 81,718.09 | Ex dom | –0.21 | –411 | –3.19 | –0.50 | ||||
| E | 7.001 | 86,502 | 12,813 | 6.775944 | 85,961.47 | 13,272.09 | –0.23 | –541 | 459 | –3.21 | –0.62 | 3.58 | ||
| I | 7.308 | 99,045 | 40,878 | 7.067269 | 98,402.16 | 42,703.76 | –0.24 | –643 | 1826 | –3.29 | –0.65 | 4.47 | ||
| 12 | Inpatient treatment for uncontrolled psoriasis | N | 5.171 | 151,496 | NA | 5.056908 | 15,1186.30 | –0.11 | –310 | –2.21 | –0.20 | |||
| A | 6.580 | 165,282 | 9787 | 6.370075 | 165,046.80 | 10,555.06 | –0.21 | –235 | 768 | –3.19 | –0.14 | 7.85 | ||
| I | 7.308 | 175,157 | 13,557 | 7.067269 | 174,843.20 | 14,051.12 | –0.24 | –314 | 494 | –3.29 | –0.18 | 3.64 | ||
| E | 7.001 | 178,530 | Dom | 6.775944 | 178,192.40 | Dom | –0.23 | –338 | –3.21 | –0.19 | ||||
| 13 | Cost per 3 months per 1-unit change in HAQ is £183 (US data)42 (base case £103) | N | 5.171 | 52,548 | NA | 5.056908 | 52,668.62 | –0.11 | 121 | –2.21 | 0.23 | |||
| A | 6.580 | 77,518 | Ex dom | 6.370075 | 77,868.59 | Ex dom | –0.21 | 351 | –3.19 | 0.45 | ||||
| E | 7.001 | 83,224 | 16,761 | 6.775944 | 83,422.65 | 17,890.27 | –0.23 | 199 | 1129 | –3.21 | 0.24 | 6.74 | ||
| I | 7.308 | 96,562 | 43,468 | 7.067269 | 96,747.99 | 45,740.41 | –0.24 | 186 | 2272 | –3.29 | 0.19 | 5.23 | ||
| 14 | Change in utility per 1-unit change in HAQ is –0.45 (Wyeth153) (base case –0.29) | N | 0.846 | 42,168 | NA | 0.649240 | 41,817.64 | –0.20 | –350 | –23.26 | –0.83 | |||
| A | 2.905 | 68,638 | Ex dom | 2.564543 | 68,465.66 | Ex dom | –0.34 | –172 | –11.72 | –0.25 | ||||
| E | 3.589 | 74,841 | 11,913 | 3.225364 | 74,519.33 | 12,694.14 | –0.36 | –322 | 781 | –10.13 | –0.43 | 6.56 | ||
| I | 3.954 | 88,442 | 37,280 | 3.567963 | 88,103.69 | 39,650.89 | –0.39 | –338 | 2371 | –9.76 | –0.38 | 6.36 | ||
| 15 | HAQ improves while on drug (lower 95% of CI) (base case no change) | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 7.845 | 66,823 | Ex dom | 7.903268 | 66,346.63 | Ex dom | 0.06 | –476 | 0.74 | –0.71 | ||||
| E | 8.492 | 72,704 | 9194 | 8.564890 | 72,046.82 | 8617.258 | 0.07 | –657 | –577 | 0.86 | –0.90 | –6.27 | ||
| I | 8.959 | 86,065 | 28,635 | 9.046183 | 85,368.63 | 27,679.20 | 0.09 | –696 | –956 | 0.97 | –0.81 | –3.34 | ||
| 16 | High rate of withdrawal (upper 95% of CI) | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 6.302 | 62,085 | Ex dom | 6.046479 | 61,904.14 | Ex dom | –0.26 | –181 | –4.05 | –0.29 | ||||
| E | 6.635 | 66,604 | 16,690 | 6.349696 | 66,326.52 | 18,958.16 | –0.29 | –277 | 2268 | –4.30 | –0.42 | 13.59 | ||
| I | 6.876 | 77,323 | 44,451 | 6.567394 | 77,024.66 | 49,141.99 | –0.31 | –298 | 4691 | –4.49 | –0.39 | 10.55 | ||
| 17 | Low rate of withdrawal (lower 95% of CI) | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 6.891 | 76,566 | Ex dom | 6.746643 | 76,437.41 | Ex dom | –0.14 | –129 | –2.09 | –0.17 | ||||
| E | 7.411 | 84,811 | 19,038 | 7.273595 | 84,472.37 | 19,242.56 | –0.14 | –339 | 205 | –1.85 | –0.40 | 1.07 | ||
| I | 7.793 | 101,890 | 44,731 | 7.651982 | 101,556.7 | 45,150.50 | –0.14 | –333 | 420 | –1.81 | –0.33 | 0.94 | ||
| 18 | All treatments have the same probability of PsARC response at 3 months | N | 5.197 | 41,416 | NA | 5.056908 | 41,817.64 | –0.14 | 402 | –2.70 | 0.97 | |||
| A | 7.104 | 77,174 | Ex dom | 6.827082 | 77,272.01 | Ex dom | –0.28 | 98 | –3.90 | 0.13 | ||||
| E | 7.236 | 78,115 | 17,999 | 6.970410 | 78,067.87 | 18,944.44 | –0.27 | –47 | 945 | –3.67 | –0.06 | 5.25 | ||
| I | 7.316 | 87,889 | 122,073 | 7.067269 | 88,103.69 | 103,611.90 | –0.25 | 215 | –18,461 | –3.40 | 0.24 | –15.12 | ||
| 19 | All treatments have the same probability of psoriasis responses (PASI 50/75/90) at 3 months | N | 5.273 | 41,746 | NA | 5.056908 | 41,817.64 | –0.22 | 72 | –4.10 | 0.17 | |||
| A | 6.722 | 67,892 | Ex dom | 6.388919 | 67,859.27 | Ex dom | –0.33 | –33 | –4.96 | –0.05 | ||||
| E | 7.186 | 72,834 | 16,254 | 6.834826 | 72,816.43 | 17,435.44 | –0.35 | –18 | 1181 | –4.89 | –0.02 | 7.27 | ||
| I | 7.414 | 87,951 | 66,219 | 7.067269 | 88,103.69 | 65,767.68 | –0.35 | 153 | –451 | –4.68 | 0.17 | –0.68 | ||
| 20 | Cost of drugs as in Wyeth submission153 | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 6.580 | 65,847 | Ex dom | 6.370075 | 65,677.86 | Ex dom | –0.21 | –169 | –3.19 | –0.26 | ||||
| E | 7.001 | 71,478 | 16,015 | 6.775944 | 71,178.79 | 17,080.00 | –0.23 | –299 | 1065 | –3.21 | –0.42 | 6.65 | ||
| I | 7.308 | 92,632 | 68,944 | 7.067269 | 92,271.65 | 72,403.13 | –0.24 | –360 | 3459 | –3.29 | –0.39 | 5.02 | ||
| 22 | All biologics have the same change in HAQ at 3 months for a PsARC responder | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 6.659 | 68,526 | 17,717 | 6.448051 | 68,357.89 | 19,078.01 | –0.21 | –168 | 1361 | –3.17 | –0.25 | 7.68 | ||
| E | 6.949 | 74,920 | 22,056 | 6.705676 | 74,616.44 | 24,293.30 | –0.24 | –304 | 2237 | –3.50 | –0.41 | 10.14 | ||
| I | 7.217 | 88,573 | 50,806 | 6.957824 | 88,254.95 | 54,089.25 | –0.26 | –318 | 3283 | –3.59 | –0.36 | 6.46 | ||
| 23 | Three vials of infliximab (base case four vials) | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 6.580 | 68,638 | Ex dom | 6.370075 | 68,465.66 | Ex dom | –0.21 | –172 | –3.19 | –0.25 | ||||
| E | 7.001 | 74,841 | Ex dom | 6.775944 | 74,519.33 | Ex dom | –0.23 | –322 | –3.21 | –0.43 | ||||
| I | 7.308 | 76,550 | 16,809 | 7.067269 | 76,286.94 | 17,145.82 | –0.24 | –263 | 337 | –3.29 | –0.34 | 2.00 | ||
| 26 | Rebound to NH after withdrawal (base case: rebound to initial gain) | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 5.846 | 69,701 | Ex dom | 5.756078 | 69,314.27 | Ex dom | –0.09 | –387 | –1.54 | –0.55 | ||||
| E | 6.104 | 76,145 | 36,408 | 6.030152 | 75,550.09 | 34659.80 | –0.07 | –595 | –1748 | –1.21 | –0.78 | –4.80 | ||
| I | 6.307 | 89,900 | 67,759 | 6.235706 | 89,252.99 | 66,663.29 | –0.07 | –647 | –1096 | –1.13 | –0.72 | –1.62 | ||
| 31 | No costs of psoriasis (base case: UK data187,192) | N | 5.171 | 28,908 | NA | 5.056908 | 28,577.05 | –0.11 | –331 | –2.21 | –1.14 | |||
| A | 6.580 | 56,792 | Ex dom | 6.370075 | 56,648.07 | Ex dom | –0.21 | –144 | –3.19 | –0.25 | ||||
| E | 7.001 | 62,209 | 18,196 | 6.775944 | 61,912.54 | 19,391.97 | –0.23 | –296 | 1196 | –3.21 | –0.48 | 6.57 | ||
| I | 7.308 | 77,704 | 50,499 | 7.067269 | 77,381.28 | 53,097.84 | –0.24 | –323 | 2599 | –3.29 | –0.42 | 5.15 | ||
| 32 | Schering-Plough estimates of cost per PASI point excluding phototherapy152 | N | 5.171 | 55,479 | NA | 5.056908 | 55,158.54 | –0.11 | –320 | –2.21 | –0.58 | |||
| A | 6.580 | 80,496 | Ex dom | 6.370075 | 80,366.29 | Ex dom | –0.21 | –130 | –3.19 | –0.16 | ||||
| E | 7.001 | 87,252 | 17361 | 6.775944 | 86,959.47 | 18,499.28 | –0.23 | –293 | 1138 | –3.21 | –0.34 | 6.56 | ||
| I | 7.308 | 99,438 | 39,715 | 7.067269 | 99,146.39 | 41,832.70 | –0.24 | –292 | 2118 | –3.29 | –0.29 | 5.33 | ||
| 33 | Schering-Plough estimates of cost per PASI point including phototherapy152 | N | 5.171 | 112,633 | NA | 5.056908 | 112,333.80 | –0.11 | –299 | –2.21 | –0.27 | |||
| A | 6.580 | 131,482 | 13,381 | 6.370075 | 131,382.80 | 14,506.15 | –0.21 | –99 | 1125 | –3.19 | –0.08 | 8.41 | ||
| E | 7.001 | 141,118 | Ex dom | 6.775944 | 140,834 | Ex dom | –0.23 | –284 | –3.21 | –0.20 | ||||
| I | 7.308 | 146,187 | 20,188 | 7.067269 | 145,961.90 | 20,911.05 | –0.24 | –225 | 723 | –3.29 | –0.15 | 3.58 | ||
| 34 | The effectiveness of biologic therapy lasts no longer than 10 years, compared with palliative care | N | 5.171 | 42,168 | NA | 5.056908 | 41,817.64 | –0.11 | –350 | –2.21 | –0.83 | |||
| A | 5.875 | 66,044 | Ex dom | 6.730591 | 76,090.30 | Ex dom | 0.86 | 10,046 | 14.56 | 15.21 | ||||
| E | 6.130 | 71,556 | 30,645 | 7.252346 | 84,039.00 | 19,231.40 | 1.12 | 12,483 | –11414 | 18.31 | 17.45 | –37.24 | ||
| I | 6.325 | 83,779 | 62,746 | 7.626992 | 100,971.10 | 45,194.96 | 1.30 | 17,192 | –17551 | 20.58 | 20.52 | –27.97 | ||
| Sa | Description | Trt | Stochastic | Deterministic | Difference (absolute value) | Difference (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY | Cost (£) | ICER | QALY | Cost (£) | ICER | QALY | Cost (£) | ICER | QALY | Cost (£) | ICER | |||
| Average (SD) | –0.12 (0.31) | 35 (2148) | 806 (4340) | –2.16 (5.74) | 0.02 (2.84) | 4.75 (9.95) | ||||||||
| 10 | Baseline HAQ 1.8 (BSR register186) (base case 1.05) | N | 2.090 | 46,594 | NA | 1.943630 | 46,120.50 | –0.15 | –473.50 | –7.00 | –1.02 | |||
| A | 3.397 | 73,207 | Ex dom | 3.078577 | 73,014.84 | Ex dom | –0.32 | –192.16 | –9.37 | –0.26 | ||||
| E | 3.804 | 79,431 | 19,156 | 3.446191 | 79,121.38 | 21,963.09 | –0.36 | –309.62 | 2807.090 | –9.41 | –0.39 | 14.65 | ||
| I | 4.101 | 93,046 | 45,898 | 3.712620 | 92,740.15 | 51,115.92 | –0.39 | –305.85 | 5217.920 | –9.47 | –0.33 | 11.37 | ||
| 11 | Baseline PASI 12.5 (base case 7.5) | N | 4.810 | 66,811 | NA | 4.695834 | 66,426.42 | –0.11 | –384.58 | –2.37 | –0.58 | |||
| A | 6.257 | 90,422 | 16,310 | 6.047895 | 90,197.16 | 17,581.12 | –0.21 | –224.84 | 1271.120 | –3.34 | –0.25 | 7.79 | ||
| E | 6.661 | 98,214 | 19,319 | 6.435715 | 97,846.56 | 19724.06 | –0.23 | –367.44 | 405.060 | –3.38 | –0.37 | 2.10 | ||
| I | 7.012 | 107,988 | 27,778 | 6.771620 | 107,620.70 | 29,098.09 | –0.24 | –367.30 | 1320.090 | –3.43 | –0.34 | 4.75 | ||
| 7 | Baseline PASI 12.5, and continue after 3 months only if respond to both PsARC and PASI 75 | N | 4.810 | 66,811 | NA | 4.695834 | 66,426.42 | –0.11 | –384.58 | –2.37 | –0.58 | |||
| E | 5.315 | 74,865 | Ex dom | 5.178952 | 74,614.15 | Ex dom | –0.14 | –250.85 | –2.56 | –0.34 | ||||
| A | 5.790 | 81,637 | 15,125 | 5.618303 | 81,512.32 | 16,353.82 | –0.17 | –124.68 | 1228.820 | –2.97 | –0.15 | 8.12 | ||
| I | 6.717 | 101,796 | 21,739 | 6.510411 | 101,692.60 | 22,620.91 | –0.21 | –103.40 | 881.910 | –3.08 | –0.10 | 4.06 | ||
| 8 | Baseline PASI 12.5, and continue after 3 months if respond to either PsARC or PASI 75 | N | 4.810 | 66,811 | NA | 4.695834 | 66,426.42 | –0.11 | –384.58 | –2.37 | –0.58 | |||
| A | 6.448 | 93,601 | 16,349 | 6.211228 | 93,310.24 | 17,740.49 | –0.24 | –290.76 | 1391.490 | –3.67 | –0.31 | 8.51 | ||
| E | 6.665 | 98,293 | 21,609 | 6.435715 | 97,846.56 | 20,207.45 | –0.23 | –446.44 | –1401.550 | –3.44 | –0.45 | –6.49 | ||
| I | 7.187 | 111,940 | 26,177 | 6.930332 | 111,524.30 | 27,653.19 | –0.26 | –415.70 | 1476.190 | –3.57 | –0.37 | 5.64 | ||
| 21 | Baseline PASI 12.5, and annual inpatient treatment for uncontrolled psoriasis (base case UVB) | N | 4.810 | 171,746 | NA | 4.695834 | 171,214.40 | –0.11 | –531.60 | –2.37 | –0.31 | |||
| A | 6.257 | 183,184 | 7901 | 6.047895 | 182,733.20 | 8519.481 | –0.21 | –450.80 | 618.481 | –3.34 | –0.25 | 7.83 | ||
| I | 7.012 | 191,216 | 10,636 | 6.77162 | 190,727.30 | 11,045.79 | –0.24 | –488.70 | 409.790 | –3.43 | –0.26 | 3.85 | ||
| E | 6.661 | 197,741 | Dom | 6.435715 | 197,177.50 | Dom | –0.23 | –563.50 | –3.38 | –0.28 | ||||
| 30 | Baseline PASI zero (base case 7.5) | N | 5.713 | 28,908 | NA | 5.598518 | 28,577.05 | –0.11 | –330.95 | –2.00 | –1.14 | |||
| A | 7.064 | 56,792 | Ex dom | 6.853345 | 56,648.07 | Ex dom | –0.21 | –143.93 | –2.98 | –0.25 | ||||
| E | 7.512 | 62,209 | 18,512 | 7.286287 | 61,912.54 | 19,751.22 | –0.23 | –296.46 | 1239.220 | –3.00 | –0.48 | 6.69 | ||
| I | 7.752 | 77,704 | 64,744 | 7.510744 | 77,381.28 | 68,916.40 | –0.24 | –322.72 | 4172.40 | –3.11 | –0.42 | 6.44 | ||
| Sa | Description | Trt | Stochastic | Difference (absolute value) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY | Cost | ICER assuming I was used first line | ICER assuming E was used first line | ICER assuming A was used first line | QALY | Cost | ICER assuming I was used first line | ICER assuming E was used first line | ICER assuming A was used first line | |||
| 24 | Second-line biologic if first failed for inefficacy | N | 5.171 | 42,168 | –0.11 | –350.36 | ||||||
| A | 5.827 | 54,394 | 18,652 | NA | –0.21 | –484.84 | 3064.93 | NA | ||||
| E | 6.142 | 58,783 | 17,114 | NA | 17,114 | –0.26 | –663.09 | 2530.46 | NA | 2530.46 | ||
| I | 6.410 | 68,630 | NA | 24,406 | 36,746 | –0.29 | –695.10 | NA | 3084.47 | 4647.19 | ||
| 25 | Second-line biologic if first failed for AEs | N | 5.171 | 42,168 | –0.11 | –350.36 | ||||||
| A | 6.273 | 61,430 | 17,486 | NA | –0.26 | –176.32 | 2828.18 | NA | ||||
| E | 6.597 | 65,780 | 16,554 | NA | 16,554 | –0.29 | –265.68 | 2408.59 | NA | 2408.59 | ||
| I | 6.831 | 76,205 | NA | 26,445 | 44,569 | –0.31 | –278.95 | NA | 2714.41 | 4940.98 | ||
| Sa | Description | Trt | Deterministic | Difference (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY | Cost | ICER assuming I was used first line | ICER assuming E was used first line | ICER assuming A was used first line | QALY | Cost | ICER assuming I was used first line | ICER assuming E was used first line | ICER assuming A was used first line | |||
| 24 | Second-line biologic if first failed for inefficacy | N | 5.056908 | 41,817.64 | –2.21 | –0.83 | ||||||
| A | 5.613686 | 53,909.16 | Ex dom | 21,716.93 | NA | –3.66 | –0.89 | 16.43 | NA | |||
| E | 5.886774 | 58,119.91 | 19,644.46 | NA | 19,644.46 | –4.16 | –1.13 | 14.79 | NA | 14.79 | ||
| I | 6.123890 | 67,934.90 | NA | 27,490.47 | 41,393.19 | –4.46 | –1.01 | NA | 12.64 | 12.65 | ||
| 25 | Second-line biologic if first failed for AEs | N | 5.056908 | 41,817.64 | –2.21 | –0.83 | ||||||
| A | 6.013680 | 61,253.68 | Ex dom | 20,314.18 | NA | –4.13 | –0.29 | 16.17 | NA | |||
| E | 6.306562 | 65,514.32 | 18,962.59 | NA | 18,962.59 | –4.40 | –0.40 | 14.55 | NA | 14.55 | ||
| I | 6.516858 | 75,926.05 | NA | 29,159.41 | 49,509.98 | –4.60 | –0.37 | NA | 10.26 | 11.09 | ||
Appendix 23 Development of a Transparent Interactive Decision Interrogator to facilitate the decision-making process
Introduction
The aim of this report was to determine the clinical effectiveness, safety and cost-effectiveness of etanercept, infliximab and adalimumab for the treatment of active and progressive PsA in patients who have had an inadequate response to standard treatment. To allow readers to make fully informed decisions based on the available evidence, a range of different scenario analyses were presented in Chapter 4 (see Results of York Economic Assessment) of this report. However, as the authors cannot anticipate every potential scenario that might be of interest to decision makers, NICE agreed to allow the development and use of interfaces in their appraisal process to be undertaken alongside this report.
Transparent Interactive Decision Interrogator has been developed to aid the decisions made by NICE committees during their consultations. The aim of the development of TIDI was to make the NICE decision process more transparent, by making the decision models more accessible to critique by a wider range of decision makers and also by using flexible and clear model components developed in R and winbugs. TIDI also aims to make the decision-making process more efficient by allowing the (re-)running of models potentially in real time during the NICE appraisal meetings.
This user friendly, excel-based interface allows decision makers to have access to all components of a decision model developed in R and winbugs, without a need of knowledge of these specialised software programmes. It makes it possible not only to change model parameters and rerun models under different scenarios in real time, but also provides a control over the model assumptions. By allowing this, TIDI can make the process of evaluating uncertainty faster and more efficient, while avoiding arbitrary limitations of preprepared analysis of a restricted number of scenarios. TIDI also provides interactive access to supplementary analyses, such as meta-analyses and influence analysis, which can help to establish which parameters have most impact on the cost-effectiveness estimates. This interface has been developed for the York decision model, presented in Chapter 4.
Software
Transparent Interactive Decision Interrogator is an excel-based interface, programmed in Visual Basic for Applications (VBA). 243,244 It allows all of the model parameters and options to be changed by using controls set out on the excel spreadsheet. excel,245,246 which is an add-in to excel, provides communication between excel and R. All data used by the model components, stored in the excel spreadsheets, can be transferred to R workspace and various actions, for example execution of the model or its components, can be activated also using controls located on the excel spreadsheet. Having this possibility of running programs developed in R from excel allows the user also to execute (from excel) additional model components, for example evidence syntheses, developed in winbugs (using r2winbugs). 247
Interface capabilities: model building
The controls for the model parameters and actions are set out on the front page of TIDI, called ‘SetupAndRun’. These controls allow for change of any parameters that inform the decision model. Figure 12 shows the layout of these controls and scrolling down the spreadsheet will reveal more parameters and options of this complex model as shown in Figure 13. These controls allow for the parameter change, as well as switching between alternative options of modelling the cost-effectiveness and then further for the change of the parameters corresponding to these alternative options. This gives an in-depth access to the model parameters and assumptions leading to building new model scenarios.
FIGURE 12.
Layout of the parameter and action controls on the front page (‘SetupAndRun’) of TIDI.
FIGURE 13.
Further controls of model parameters and assumptions on the front page (‘SetupAndRun’) of TIDI.
For example, the radio buttons on top of Figure 13 allow the user to choose how the utility and additional cost over time are modelled. The parameters that were used to fit utility and cost in the base-case model or those used by the Abbot submission to NICE (scenarios 4 and 9, results of which were shown in Chapter 4, Results of York Economic Assessment) can be further changed here, allowing for much wider range of scenarios to be considered in real time.
Further options are available to choose the stopping rule or whether or not to include placebo effect (scenarios 6–8 and base case). The possibility to switch between these options combined with any change of the model parameters give a lot more flexibility in considering the most appropriate scenarios.
The TIDI also allows for a change of the parameters used in the scenarios that consider second-line treatment. The controls allow the user to choose, for example between reasons for switching (inefficacy or adverse event as in scenarios 24 and 25) and then to adjust the parameters specific to these scenarios.
All the above options make TIDI a flexible tool that can run not only all the scenarios listed in Chapter 4 (see Results of York Economic Assessment) of this report, but also, in addition, infinite numbers of new ones.
Model execution and results
Once a new model scenario is built, all model parameters are loaded to R workspace where the R code is executed and cost, QALYs and ICERs are being calculated. Note that to be able to execute the York economic model in real time, the model was run deterministically. The time required to run the full stochastic model was too long to allow running it during a committee meeting. However, the deterministic version gives a good approximation of the cost-effectiveness point estimates. The full analysis of agreement between deterministic and stochastic model for each scenario is presented in Appendix 22. The average differences between the stochastic and deterministic models were 2.16% (SD = 5.74%) for QALY, 0.02% (2.84%) for cost and 4.75% (9.95%) difference in ICER.
The results of the model are then displayed in the ‘Output’ spreadsheet. As shown in the example in Figure 14, the ‘Output’ spreadsheet lists QALYs, cost, ICERs relative to placebo, and, finally, ICERs resulting from the final selection of the most effective strategy (following exclusion of strategies being dominated or extendedly dominated). The results are shown along with the final ICERs from the base-case model listed below the main set of results for comparison. The final results of the decision model are also represented graphically. In the plot included in Figure 14, the circles denoted N, A, E and I represent each strategy on the cost-effectiveness plane (‘Cost’ vs ‘QALY’), and the slopes of the lines linking these circles correspond to the ICERS. Dashed line marks the strategy (in this case adalimumab) that has been excluded as being extendedly dominated by another strategy (in this case etanercept).
FIGURE 14.
Results of the decision model in the ‘Output’ spreadsheet (results correspond to scenario 2).
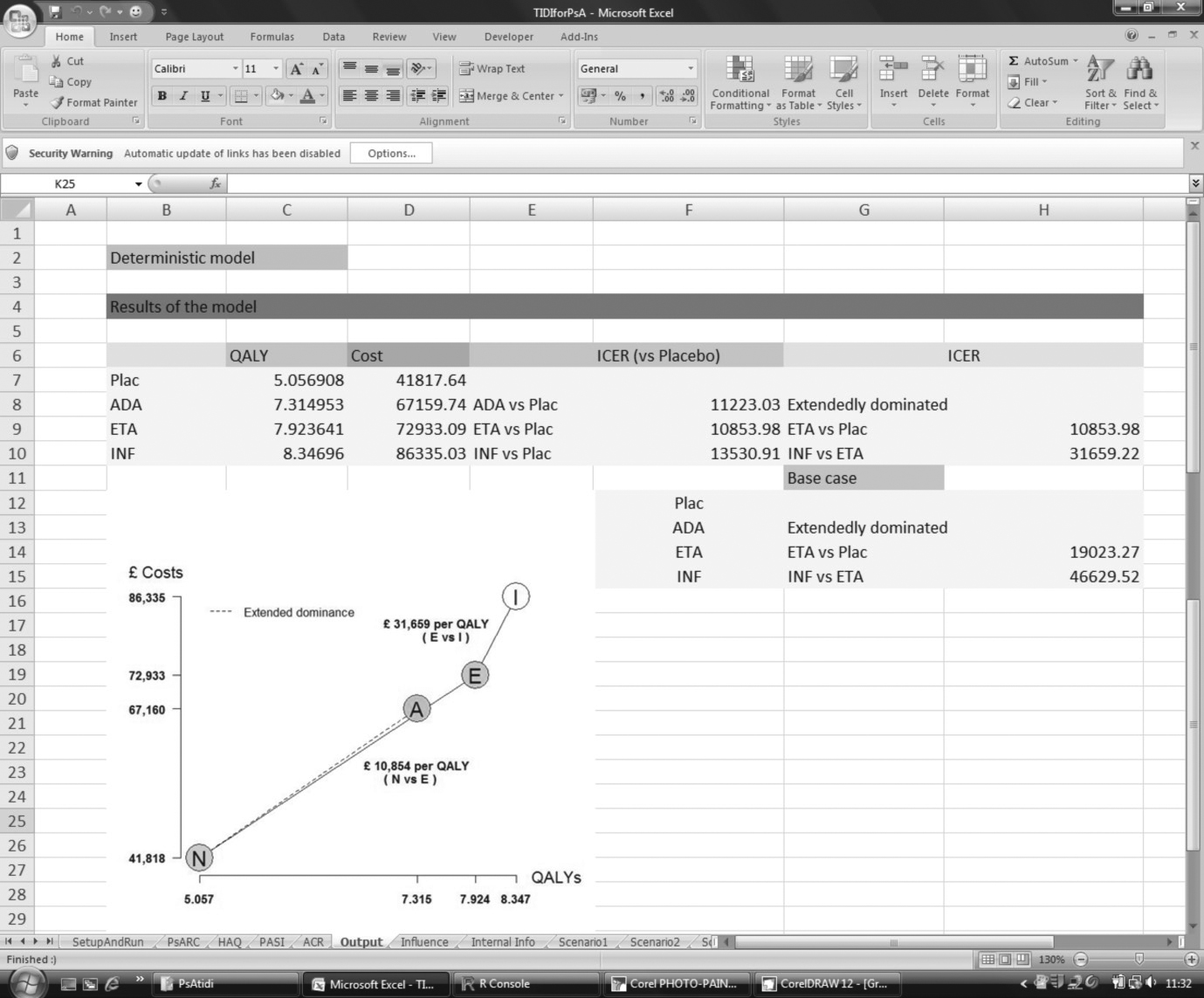
The set of the scenario parameters and corresponding results can be saved in a separate spreadsheet ‘ScenarioN’, creating a library of scenarios considered by decision makers for later viewing. The whole set of parameters and settings of any saved scenario can also be uploaded back to the ‘SetupAndRun’ front page of the TIDI workbook for further considerations and amendments, after which the model can be rerun.
Further interface applications: meta-analysis
Effectiveness estimates that inform the decision model are obtained from the meta-analyses presented in Chapter 3 of this report and these can also be accessed and adjusted from TIDI. The interface of meta-analysis is designed in such a way to allow selection of any subset of available studies for which pooled outcomes can be recalculated. The resulting estimates are then displayed in tabular form as well as using interactive forest plots. The outcome of the new meta-analysis can then be uploaded into the front spreadsheet containing the model parameters and the model can be rerun with the amended effectiveness estimates. This sensitivity analysis can be vital in situations when some of the available studies are, for example, of questionable quality or do not represent the considered population of patients.
An example of the PsARC meta-analysis is shown in Figure 15. This example shows how this tool can be used to carry out a sensitivity analysis adjusting for relevance. Here to demonstrate this, the Genovese et al. 83 study has been removed. By pressing the button ‘Run’ the user reruns the meta-analysis and new estimates are presented in tables below the ‘Run’ button, as well as in the forest plots. Now the estimates for Genovese et al. study have been greyed out as the study no longer contributes to the meta-analysis and the pooled result has now changed which is plotted alongside with the greyed out original pooled result of the meta-analysis (that includes all the available studies) for comparison. The first two forest plots show ORs of comparisons of each anti-TNF with placebo, and pooled probability of response in the placebo group. The resulting probability of response in treatment groups is showed in the final forest plot providing estimates for the decision model.
FIGURE 15.
Interactive meta-analysis.
Apart from the option of uploading the new estimates to the decision model, the interface provides the pooled common effect estimates, which can also be uploaded and used to inform the decision model. Here the common effect was precalculated (using all six studies available) and saved on the spreadsheet holding the data. In the future applications this can be extended by adding an interactive option allowing for the common treatment to be calculated in real time for chosen subsets of studies.
Influence analysis: tornado plots
Health economic models, such as the York model described in this report, are informed by a large number of parameters related to cost and effectiveness of treatments. It is infeasible to carry out the sensitivity analysis that explores all possible scenarios, considering different values of all parameters. Influence analysis can help to identify those parameters that have highest impact on the cost-effectiveness estimates, such as ICERs or incremental net benefit. The influence analysis presented here was carried out by means of tornado plots. Each parameter of the decision model has a defined range of plausible values. This range can be simply a 95% CI if we know the SE of the parameter. The minimum and the maximum value of each parameter were used in the model one at a time, leaving the remaining parameters set to the same values as in the base-case scenario. This gives the ICERs corresponding to the minimum and maximum values of each parameter, and if those ICERs are far apart for a given parameter, it suggests that a change in this parameter will have an impact on the final result of the decision model.
Figure 16 shows the ‘Influence’ spreadsheet containing values and ranges of all the model parameters. Clicking on the ‘Tornado Plot’ button shows a user form that allows the choice of a tornado plot from six pair-wise comparisons of ICERs and additional differences in ICERs that can help in understanding the influence of all the parameters better. Figure 17 shows an example of the tornado plot for ICERs of adalimumab compared with Infliximab. Ranges of ICERs for each parameter are sorted and plotted from the widest on top to the narrowest on the bottom (only the top 34 have been plotted for clarity) forming the tornado plot. The parameters most influential for this ICER are the cost of infliximab in period 2, the amount by which the patients progress in terms of HAQ while on treatment, and cost of adalimumab in period 3.
FIGURE 16.
Influence analysis spreadsheet.
FIGURE 17.
Influence analysis: tornado plot.
This analysis is limited because of more than two treatments being compared and in such cases tornado plots cannot provide definitive answers. Further development of adequate methodology is required to face these limitations. However, even these limited tools can shed some light on what is important in the decision model.
Summary and discussion
The TIDI has been created as a concept aiming to help the NICE decision process to be more transparent and efficient. It provides a tool to critique and explore further the decision models by a wider community of decision makers, not only those familiar with specialised software such as R and winbugs. It allows the user to have an in-depth access to, and control over, all model parameters and assumptions, and, at the same time, uses flexible, clear and hence transparent model components developed in R and winbugs. By making it possible to run models in real time, it makes the decision process to be more efficient. Being able to rerun a model under new scenarios in real time not only allows sensitivity analysis that potentially can change the final decision, but also can simply provide reassurance that, for example uncertainty of a parameter does not have much effect on the cost-effectiveness estimates. Any required additional model scenarios can be considered during the committee meetings without the need for the committee to delay a decision and having to reconvene. This interface, developed for the model presented in this HTA report, was used by NICE in the committee meeting to support their decision process.
The TIDI, as with every new concept, has some limitations and could be developed further. One of the disadvantages for this appraisal was that it was not possible to use it to run the full stochastic model in real time because it takes too long a time to run. However, it was possible to use deterministic model as an approximation. If any NICE committee decision was going to be influenced by the results of this simplified model, the decision could be later confirmed by the full stochastic analysis. Further work can be carried out to optimise complex models, such as the York model used here, so that it is possible to run them in real time during the committee meetings. Further considerations should also aim to develop tools equivalent to the tornado plots that will allow quantifying the influence of parameters on the model estimates that are based on comparisons of more than two strategies.
Glossary
- Acitretin
- A synthetic derivative of vitamin A, which is taken orally. It is indicated for severe psoriasis.
- Adverse effect
- An abnormal or harmful effect caused by, and attributable to, exposure to a chemical (e.g. a drug), which is indicated by some result such as death or a physical symptom or visible illness. An effect may be classed as adverse if it causes functional or anatomical damage, causes irreversible change in the homeostasis of the organism or increases the susceptibility of the organism to other chemical or biological stress.
- American College of Rheumatology 20% improvement criteria (ACR 20)
- ACR 20 is a response measure that requires a 20% reduction in the tender joint count, a 20% reduction in the swollen joint count, and a 20% reduction in at least three out of five additional measures, including patient and physician global assessment, pain, disability and an acute-phase reactant.
- American College of Rheumatology 50% improvement criteria (ACR 50)
- ACR 50 is a response measure that requires a 50% reduction in the tender joint count, a 50% reduction in the swollen joint count, and a 50% reduction in at least three out of five additional measures, including patient and physician global assessment, pain, disability and an acute-phase reactant.
- American College of Rheumatology 70% improvement criteria (ACR 70)
- ACR 70 is a response measure that requires a 70% reduction in the tender joint count, a 70% reduction in the swollen joint count, and a 70% reduction in at least three out of five additional measures, including patient and physician global assessment, pain, disability and an acute-phase reactant.
- Ankylosing spondylitis
- A rheumatic disease that affects the spine and may lead to some degree of stiffness in the back. As the inflammation goes and healing takes place, bone grows out from both sides of the vertebrae and may join the two together; this stiffening is called ankylosis.
- Arthritis
- A term meaning inflammation of the joint(s), but which is often used to include all joint disorders. Sometimes joints are damaged through the disease process of arthritis.
- Articular
- Of, or relating to, the joints.
- Autoimmune disease
- A disorder of the body’s defence mechanism (immune system), in which antibodies and other components of the immune system attack the body’s own tissue, for example, lupus (systemic lupus erythematosus).
- Biologic (biological) therapies
- Medical preparations derived from living organisms. Includes biologic drug and other new drugs that target the pathologically active T cells involved in psoriasis and psoriatic arthritis.
- Ciclosporin
- A medication originally developed to prevent the immune system from rejecting transplanted organs, but which has also proved helpful in treating psoriasis.
- Confidence interval (CI)
- The typical (‘classical’ or ‘frequentist’) definition is the range within which the ‘true’ value (e.g. size of effect of an intervention) would be expected to lie if sampling could be repeated a large number of times (e.g. 95% or 99%).
- Corticosteroid
- A synthetic hormone similar to that produced naturally by the adrenal glands, which is available in pill, topical and injectable forms.
- Cost–benefit analysis
- An economic analysis that converts the effects or consequences of interventions into the same monetary terms as the costs and compares them using a measure of net benefit or a cost–benefit ratio.
- Cost-effectiveness analysis
- An economic analysis that expresses the effects or consequences of interventions on a single dimension. This would normally be expressed in ‘natural’ units (e.g. cases cured, life-years gained, additional strokes prevented). The difference between interventions in terms of costs and effects is typically expressed as an incremental cost-effectiveness ratio (e.g. the incremental cost per life-year gained).
- Cost–utility analysis
- The same as a cost-effectiveness analysis, but the effects or consequences of interventions are expressed in generic units of health gain, usually quality-adjusted life-years (QALYs).
- Credible interval
- In Bayesian statistics, a credible interval is a posterior probability interval estimation that incorporates problem-specific contextual information from the prior distribution. Credible intervals are used for the purposes similar to those of confidence intervals in frequentist statistics.
- C-reactive protein (CRP)
- Concentrations of this protein in the blood can be measured as a test of inflammation or disease activity, for example in rheumatoid arthritis.
- Crohn’s disease
- An inflammatory condition of the digestive tract; rheumatic diseases are often associated with it and ulcerative colitis is related to it.
- Disease-modifying antirheumatic drugs (DMARDs)
- DMARDs are drugs capable of modifying the progression of rheumatic disease. The term is, however, applied to what are now considered to be traditional disease-modifying drugs, in particular sulfasalazine, methotrexate and ciclosporin, as well as azathioprine, cyclophosphamide, antimalarial drugs, penicillamine and gold. The newer agent, leflunomide, may be included as a DMARD. The biologics, such as etanercept and infliximab, are not generally referred to as DMARDs.
- Effect size
- A generic term for the estimate of effect for a study.
- Emollient
- An agent that holds moisture in the skin and by doing so softens or soothes it.
- Erythrocyte sedimentation rate (ESR)
- One of the tests designed to measure the degree of inflammation.
- Fixed-effects model
- A statistical model that stipulates that the units under analysis (e.g. people in a trial or study in a meta-analysis) are the ones of interest, and thus constitute the entire population of units. Only within-study variation is taken to influence the uncertainty of results (as reflected in the confidence interval) of a meta-analysis using a fixed-effects model.
- Health Assessment Questionnaire (HAQ)
- HAQ is a validated, self-administered questionnaire that measures two dimensions of health status, including physical disability and pain. The physical disability comprises eight subscales: dressing, grooming, arising, hygiene, reach, eating, walking, and grip and activities. HAQ is scored from 0 (able to function without difficulty) to 3 (unable to function).
- Heterogeneity
- In systematic reviews, heterogeneity refers to variability or differences between studies in the estimates of effects. A distinction is sometimes made between ‘statistical heterogeneity’ (differences in the reported effects), ‘methodological heterogeneity’ (differences in study design) and ‘clinical heterogeneity’ (differences between studies in key characteristics of the participants, interventions or outcome measures).
- Immunomodulator
- A substance that alters the body’s immune response.
- Intention to treat (ITT)
- An ITT analysis is one in which all of the participants in a trial are analysed according to the intervention to which they were allocated, whether they received it or not.
- Methotrexate (MTX)
- One of the oldest chemotherapy drugs that is used to treat cancer. Used in the treatment of psoriasis.
- Mixed-treatment comparison (MTC)
- Mixed-treatment comparison is a form of meta-analysis used to strengthen inference concerning the relative efficacy of two treatments. It uses data based on direct comparisons (A vs B and B vs C trials) and indirect comparisons (A vs C trials). Also, it facilitates simultaneous inference regarding all treatments in order to select the best treatments.
- Monoclonal antibody
- An antibody produced in a laboratory from a single clone, which recognises only one antigen.
- Non-steroidal anti-inflammatory drugs (NSAIDs)
- Consist of a large range of drugs of the aspirin family, prescribed for different kinds of arthritis, which reduce inflammation and control pain, swelling and stiffness.
- Placebo
- An inactive substance or procedure administered to a patient, usually to compare its effects with those of a real drug or other intervention, but sometimes for the psychological benefit to the patient through a belief that s/he is receiving treatment.
- Plaque psoriasis
- The most common form of psoriasis, also known as psoriasis vulgaris, recognised by red, raised lesions covered by silvery scales. About 80% of patients with psoriasis have this type.
- Psoriasis
- A chronic skin disease characterised by inflammation and scaling. Scaling occurs when cells in the outer layer of skin reproduce faster than normal and pile up on the skin’s surface. It is understood to be a disorder of the immune system.
- Psoriasis Area and Severity Index (PASI) score
- A number representing the size, redness, thickness and scaliness of a person’s psoriasis.
- Psoriatic arthritis (PsA)
- This disease is characterised by stiffness, pain, and swelling in the joints, especially of the hands and feet. It affects about 23% of people with psoriasis. Early diagnosis and treatment can help inhibit the progression of joint deterioration.
- Psoriatic Arthritis Response Criteria (PsARC)
- PsARC is a composite response measure that incorporates patient global self-assessment, physician global assessment, and tender and swollen joint scores.
- Quality-adjusted life-year (QALY)
- An index of health gain where survival duration is weighted or adjusted by the patient’s quality of life during the survival period. QALYs have the advantage of incorporating changes in both quantity (mortality) and quality (morbidity) of life.
- Quality of life (QoL)
- A concept incorporating all the factors that might impact on an individual’s life, including factors such as the absence of disease or infirmity, as well as other factors that might affect their physical, mental and social well-being.
- Random-effects model
- A statistical model sometimes used in meta-analysis in which both within-study sampling error (variance) and between-studies variation are included in the assessment of the uncertainty (confidence interval) of the results of a meta-analysis.
- Randomised controlled trial (RCT) (synonym: randomised clinical trial)
- An experiment in which investigators randomly allocate eligible people into intervention groups to receive or not to receive one or more interventions that are being compared.
- Relative risk (RR) (synonym: risk ratio)
- The ratio of risk in the intervention group to the risk in the control group. The risk (proportion, probability or rate) is the ratio of people with an event in a group to the total in the group. A RR of one indicates no difference between comparison groups. For undesirable outcomes an RR that is less than one indicates that the intervention was effective in reducing the risk of that outcome.
- Remission
- A lessening or abatement of the symptoms of a disease.
- Rheumatoid arthritis (RA)
- A chronic autoimmune disease characterised by pain, stiffness, inflammation, swelling, and, sometimes, destruction of joints.
- Sensitivity analysis
- An analysis used to determine how sensitive the results of a study or systematic review are to changes in how it was done. Sensitivity analyses are used to assess how robust the results are to uncertain decisions or assumptions about the data and the methods that were used.
- Squamous cell carcinoma
- A form of skin cancer that is more aggressive than basal cell carcinoma. People who have received psoralen combined with ultraviolet A may be at risk of this type of skin cancer.
- Statistical significance
- An estimate of the probability of an association (effect) as large or larger than what is observed in a study occurring by chance, usually expressed as a p-value.
- T cell
- A type of white blood cell that is part of the immune system that normally helps protect the body against infection and disease.
- Thrombocytopenia
- A disorder, sometimes associated with abnormal bleeding, in which the number of platelets (cells that help blood to clot) is abnormally low.
- Topical agent
- A treatment such as a cream, salve or ointment that is applied to the surface of the skin.
- Tumor necrosis factor (TNF)
- One of the cytokines, or messengers, known to be fundamental to the disease process that underlies psoriasis. It often plays a key role in the onset and the continuation of skin inflammation.
- Variance
- A measure of the variation shown by a set of observations, defined by the sum of the squares of deviations from the mean, divided by the number of degrees of freedom in the set of observations.
- Visual analogue scale (VAS)
- Direct rating where raters are asked to place a mark at a point between two anchor states appearing at either end of the line. It is used as a method of valuing health states.
- Weighted mean difference (WMD) (in meta-analysis)
- A method of meta-analysis used to combine measures on continuous scales, where the mean, standard deviation and sample size in each group are known. The weight given to each study is determined by the precision of its estimate of effect and, is equal to the inverse of the variance. This method assumes that all of the trials have measured the outcome on the same scale.
List of abbreviations
- ACR
- American College of Rheumatology
- ADEPT
- Adalimumab Effectiveness in Psoriatic Arthritis Trial
- ANA
- antinuclear antibody
- ANOVA
- analysis of variance
- BAD
- British Association of Dermatologists
- BNF
- British National Formulary
- BSA
- body surface area
- BSC
- best supportive care
- BSR
- British Society for Rheumatology
- BSRBR
- British Society for Rheumatology Biologics Register
- CEAC
- cost-effectiveness acceptability curve
- CENTRAL
- Cochrane Central Register of Controlled Trials
- CI
- confidence interval
- CiC
- commercial-in-confidence
- CPCI-S
- Conference Proceedings Citation Index – Science
- CRD
- Centre for Reviews and Dissemination
- CRP
- C-reactive protein
- DARE
- Database of Abstracts of Reviews of Effects
- df
- degrees of freedom
- DIC
- deviation information criterion
- DIP
- distal interphalangeal
- DLQI
- Dermatology Life Quality Index
- DMARD
- disease-modifying antirheumatic drug
- dsDNA
- double-stranded DNA
- EQ-5D
- European Quality of Life-5 Dimensions
- ERG
- Evidence Review Group
- ESR
- erythrocyte sedimentation rate
- FBC
- full blood count
- HAQ
- Health Assessment Questionnaire
- HAQ-DI
- Health Assessment Questionnaire-Disability Index
- HEED
- Health Economic Evaluations Databases
- HLA
- human leucocyte antigen
- HODaR
- Health Outcomes Data Repository
- HR
- hazard ratio
- HRQoL
- health-related quality of life
- HTA
- Health Technology Assessment
- IQR
- interquartile range
- i.v.
- intravenous
- ICER
- incremental cost-effectiveness ratio (e.g. incremental cost per QALY gained)
- IMPACT
- Infliximab Multinational Psoriatic Arthritis Controlled Trial
- IPD
- individual patient data
- ITT
- intention to treat
- LFT
- liver function test
- LOCF
- last observation carried forward
- MeSH
- medical subject heading
- MIMS
- online and print prescribing database for health professionals
- mRCT
- metaRegister of Current Controlled Trials
- MTC
- mixed-treatment comparison
- MTX
- methotrexate
- NH
- natural history
- NHS EED
- NHS Economic Evaluation Database
- NICE
- National Institute for Health and Clinical Excellence
- NOAR
- Norfolk Arthritis Register
- NRR
- National Research Register
- NSAID
- non-steroidal anti-inflammatory drug
- OMERACT
- Outcome Measures in Rheumatoid Arthritis (Rheumatology) Clinical Trials
- OR
- odds ratio
- PASI
- Psoriasis Area and Severity Index
- probability density function
- PRESTA
- Psoriasis Randomized Etanercept STudy in Subjects with Psoriatic Arthritis
- PsA
- psoriatic arthritis
- PsARC
- Psoriatic Arthritis Response Criteria
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RA
- rheumatoid arthritis
- RCT
- randomised controlled trial
- RF
- rheumatoid factor
- RR
- relative risk
- SCI
- Science Citation Index
- SD
- standard deviation
- SE
- standard error
- SF-36
- Short Form questionnaire-36 items
- SJC
- swollen joint count
- STA
- single technology appraisal
- TB
- tuberculosis (infection)
- THIN
- The Health Improvement Network
- TIDI
- Transparent Interactive Decision Interrogator
- TNF
- tumour necrosis factor
- TJC
- tender joint count
- TSS
- Total Sharp Score
- U&E
- urea and electrolytes
- URTI
- upper respiratory tract infection
- UVB
- ultraviolet light, type B
- VAS
- visual analogue scale
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
NoteThis monograph is based on the Technology Assessment Report produced for NICE. The full report contained a considerable amount of data that was deemed commercial-in-confidence and academic-in-confidence. The full report was used by the Appraisal Committee at NICE in their deliberations. The full report with each piece of commercial-in-confidence and academic-in-confidence data removed and replaced by the statement ‘commercial-in-confidence and academic-in-confidence information (or data) removed’ is available on the NICE website: www.nice.org.uk.
The present monograph presents as full a version of the report as is possible while retaining readability, but some sections, sentences, tables and figures have been removed. Readers should bear in mind that the discussion, conclusions and implications for practice and research are based on all the data considered in the original full NICE report.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Professor of General Practice, Department of Primary Health Care, University of Oxford Programme Director,
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Deborah Ashby, Professor of Medical Statistics and Clinical Trials, Queen Mary, Department of Epidemiology and Public Health, Imperial College London
-
Professor Peter Brocklehurst, Director, National Perinatal Epidemiology Unit, University of Oxford
-
Professor John Cairns, Professor of Health Economics, London School of Hygiene and Tropical Medicine
-
Professor Peter Croft, Director of Primary Care Sciences Research Centre, Keele University
-
Professor Jenny Donovan, Professor of Social Medicine, University of Bristol
-
Professor Jonathan Green, Professor and Acting Head of Department, Child and Adolescent Psychiatry, University of Manchester Medical School
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Freddie Hamdy, Professor of Urology, Head of Nuffield Department of Surgery, University of Oxford
-
Professor Allan House, Professor of Liaison Psychiatry, University of Leeds
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norris, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor James Raftery, Chair of NETSCC and Director of the Wessex Institute, University of Southampton
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norris Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor James Raftery, Chair of NETSCC and Director of the Wessex Institute, University of Southampton
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Catherine Moody, Programme Manager, Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire Country Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Professor Carol Tannahill, Glasgow Centre for Population Health
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Sarah Tyson, Senior Research Fellow & Associate Head of School, University of Salford
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr David P Britt, Public contributor
-
Mr Sankaran ChandraSekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seamus Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor David Taggart, Consultant Cardiothoracic Surgeon, John Radcliffe Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Professor Nicholas James, Professor of Clinical Oncology, School of Cancer Sciences, University of Birmingham
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/ Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Mr John Chapman, Public contributor
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Ms Kylie Gyertson, Oncology and Haematology Clinical Trials Manager, Guy’s and St Thomas’ NHS Foundation Trust London
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Martin Shelly, General Practitioner, Silver Lane Surgery, Leeds
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Expert Advisory Network
-
Professor Douglas Altman, Professor of Statistics in Medicine, Centre for Statistics in Medicine, University of Oxford
-
Professor John Bond, Professor of Social Gerontology & Health Services Research, University of Newcastle upon Tyne
-
Professor Andrew Bradbury, Professor of Vascular Surgery, Solihull Hospital, Birmingham
-
Mr Shaun Brogan, Chief Executive, Ridgeway Primary Care Group, Aylesbury
-
Mrs Stella Burnside OBE, Chief Executive, Regulation and Improvement Authority, Belfast
-
Ms Tracy Bury, Project Manager, World Confederation of Physical Therapy, London
-
Professor Iain T Cameron, Professor of Obstetrics and Gynaecology and Head of the School of Medicine, University of Southampton
-
Professor Bruce Campbell, Consultant Vascular & General Surgeon, Royal Devon & Exeter Hospital, Wonford
-
Dr Christine Clark, Medical Writer and Consultant Pharmacist, Rossendale
-
Professor Collette Clifford, Professor of Nursing and Head of Research, The Medical School, University of Birmingham
-
Professor Barry Cookson, Director, Laboratory of Hospital Infection, Public Health Laboratory Service, London
-
Dr Carl Counsell, Clinical Senior Lecturer in Neurology, University of Aberdeen
-
Professor Howard Cuckle, Professor of Reproductive Epidemiology, Department of Paediatrics, Obstetrics & Gynaecology, University of Leeds
-
Professor Carol Dezateux, Professor of Paediatric Epidemiology, Institute of Child Health, London
-
Mr John Dunning, Consultant Cardiothoracic Surgeon, Papworth Hospital NHS Trust, Cambridge
-
Mr Jonothan Earnshaw, Consultant Vascular Surgeon, Gloucestershire Royal Hospital, Gloucester
-
Professor Martin Eccles, Professor of Clinical Effectiveness, Centre for Health Services Research, University of Newcastle upon Tyne
-
Professor Pam Enderby, Dean of Faculty of Medicine, Institute of General Practice and Primary Care, University of Sheffield
-
Professor Gene Feder, Professor of Primary Care Research & Development, Centre for Health Sciences, Barts and The London School of Medicine and Dentistry
-
Mr Leonard R Fenwick, Chief Executive, Freeman Hospital, Newcastle upon Tyne
-
Mrs Gillian Fletcher, Antenatal Teacher and Tutor and President, National Childbirth Trust, Henfield
-
Professor Jayne Franklyn, Professor of Medicine, University of Birmingham
-
Mr Tam Fry, Honorary Chairman, Child Growth Foundation, London
-
Professor Fiona Gilbert, Consultant Radiologist and NCRN Member, University of Aberdeen
-
Professor Paul Gregg, Professor of Orthopaedic Surgical Science, South Tees Hospital NHS Trust
-
Bec Hanley, Co-director, TwoCan Associates, West Sussex
-
Dr Maryann L Hardy, Senior Lecturer, University of Bradford
-
Mrs Sharon Hart, Healthcare Management Consultant, Reading
-
Professor Robert E Hawkins, CRC Professor and Director of Medical Oncology, Christie CRC Research Centre, Christie Hospital NHS Trust, Manchester
-
Professor Richard Hobbs, Head of Department of Primary Care & General Practice, University of Birmingham
-
Professor Alan Horwich, Dean and Section Chairman, The Institute of Cancer Research, London
-
Professor Allen Hutchinson, Director of Public Health and Deputy Dean of ScHARR, University of Sheffield
-
Professor Peter Jones, Professor of Psychiatry, University of Cambridge, Cambridge
-
Professor Stan Kaye, Cancer Research UK Professor of Medical Oncology, Royal Marsden Hospital and Institute of Cancer Research, Surrey
-
Dr Duncan Keeley, General Practitioner (Dr Burch & Ptnrs), The Health Centre, Thame
-
Dr Donna Lamping, Research Degrees Programme Director and Reader in Psychology, Health Services Research Unit, London School of Hygiene and Tropical Medicine, London
-
Professor James Lindesay, Professor of Psychiatry for the Elderly, University of Leicester
-
Professor Julian Little, Professor of Human Genome Epidemiology, University of Ottawa
-
Professor Alistaire McGuire, Professor of Health Economics, London School of Economics
-
Professor Neill McIntosh, Edward Clark Professor of Child Life and Health, University of Edinburgh
-
Professor Rajan Madhok, Consultant in Public Health, South Manchester Primary Care Trust
-
Professor Sir Alexander Markham, Director, Molecular Medicine Unit, St James’s University Hospital, Leeds
-
Dr Peter Moore, Freelance Science Writer, Ashtead
-
Dr Andrew Mortimore, Public Health Director, Southampton City Primary Care Trust
-
Dr Sue Moss, Associate Director, Cancer Screening Evaluation Unit, Institute of Cancer Research, Sutton
-
Professor Miranda Mugford, Professor of Health Economics and Group Co-ordinator, University of East Anglia
-
Professor Jim Neilson, Head of School of Reproductive & Developmental Medicine and Professor of Obstetrics and Gynaecology, University of Liverpool
-
Mrs Julietta Patnick, Director, NHS Cancer Screening Programmes, Sheffield
-
Professor Robert Peveler, Professor of Liaison Psychiatry, Royal South Hants Hospital, Southampton
-
Professor Chris Price, Director of Clinical Research, Bayer Diagnostics Europe, Stoke Poges
-
Professor William Rosenberg, Professor of Hepatology and Consultant Physician, University of Southampton
-
Professor Peter Sandercock, Professor of Medical Neurology, Department of Clinical Neurosciences, University of Edinburgh
-
Dr Philip Shackley, Senior Lecturer in Health Economics, Sheffield Vascular Institute, University of Sheffield
-
Dr Eamonn Sheridan, Consultant in Clinical Genetics, St James’s University Hospital, Leeds
-
Dr Margaret Somerville, Director of Public Health Learning, Peninsula Medical School, University of Plymouth
-
Professor Sarah Stewart-Brown, Professor of Public Health, Division of Health in the Community, University of Warwick, Coventry
-
Dr Nick Summerton, GP Appraiser and Codirector, Research Network, Yorkshire Clinical Consultant, Primary Care and Public Health, University of Oxford
-
Professor Ala Szczepura, Professor of Health Service Research, Centre for Health Services Studies, University of Warwick, Coventry
-
Dr Ross Taylor, Senior Lecturer, University of Aberdeen
-
Dr Richard Tiner, Medical Director, Medical Department, Association of the British Pharmaceutical Industry
-
Mrs Joan Webster, Consumer Member, Southern Derbyshire Community Health Council
-
Professor Martin Whittle, Clinical Co-director, National Co-ordinating Centre for Women’s and Children’s Health, Lymington