
Notes
Article history
The research reported in this issue of the journal was commissioned by the HTA programme as project number 08/228/01. The contractual start date was in April 2009. The draft report began editorial review in June 2010 and was accepted for publication in November 2010. As the funder, by devising a commissioning brief, the HTA programmespecified the research question and study design. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draftdocument. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2011. This work was produced by Kaltenthaler et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2011 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Historical perspective
The National Institute for Health and Clinical Excellence (NICE) is an independent organisation that is part of the NHS and is responsible for providing guidance on the promotion of good health and the prevention and treatment of ill health to the population of England and Wales. The establishment of NICE in 1999 was the natural extension and institutionalisation of the process of using clinical effectiveness and cost-effectiveness evidence to inform clinical practice decisions. One of the key components of NICE’s work involves technology appraisals, which lead to recommendations on the use of new and existing medicines and treatments within the NHS, such as medicines and medical devices among others. NICE technology appraisal guidance is mandatory in the NHS in England and Wales, giving NICE the potential to decrease variation in the provision of care across the nations.
The processes used to establish NICE guidance on technology appraisals are based on the internationally accepted models of reviewing clinical effectiveness and cost-effectiveness evidence. These include a rigorous and systematic approach to identifying, evaluating and synthesising the available evidence (clinical and economic data) carried out by groups of academic researchers (assessment groups) aided by submissions from the involved manufacturers of the technologies. The result of this synthesis is then considered by a carefully selected group of clinicians, health economists, statisticians and patients. This group, the Appraisal Committee (AC), is responsible for weighing all of the available evidence, including submissions from manufacturers, patients and expert groups, to make recommendations regarding the clinical effectiveness and cost-effectiveness of various technologies and produce guidance to direct care in the NHS. This is known as the multiple technology appraisal (MTA) process. In addition to this process for the evaluation of evidence, opportunity is also provided for interested parties to appeal against the AC decisions.
A more rapid process became a political imperative and the newer single technology appraisal (STA) process was introduced in 2005. The STA process was specifically designed to appraise a new technology for a single indication, although there may be more than one comparator; most importantly, the STA process was designed to examine evidence in a more timely fashion than the MTA process so that guidance for new products was produced as close to their launch into the NHS as possible. The STA process differs from the MTA process in that the manufacturer’s submission (MS) to NICE forms the principal source of evidence for decision-making. The MS is expected to contain an evaluation of the clinical effectiveness and cost-effectiveness of the technology using decision-analytic approaches as outlined in the STA methods guidelines developed by NICE. 1 The timescales from time of referral of the appraisal to production of the final appraisal documentation (FAD) are intended to be much shorter for STAs, around 34 weeks compared with 51 weeks for an MTA. Initially, scoping workshops (which are an opportunity for consultees and commentators to discuss the scope and important issues related to it) did not take place for STAs; however, this has changed and scoping workshops now take place for both MTAs and STAs.
Further changes to both the MTA and the STA processes occurred in 2009, related to what has come to be known as ‘end-of-life’ criteria and technology pricing. End-of-life criteria affect the use of life-extending treatments licensed for terminal illnesses (survival < 24 months) affecting small numbers of patients, so that treatments that may have been previously ruled out as not sufficiently cost-effective for routine use in the NHS might now be recommended for use. This change, implemented within a very time-limited consultation process, is based on the assumption that the last few months of life are in fact worth more than ‘ordinary’ life and therefore there should be a willingness to pay that is higher than in other technologies. The exact amount of this extra value was not stipulated in the final policy document. 2 A second change to the technology appraisal processes took place as a result of the new Pharmaceutical Price Regulation Scheme (PPRS), launched in January 2009, which allows manufacturers to submit proposals for Patient Access Schemes (PASs) and flexible pricing schemes. 3 These schemes have the potential to lengthen the STA process. Finally, manufacturers are now invited to participate in, and contribute to, the AC meetings.
Description of the single technology appraisal process
The STA process is divided into stages. Initially, provisional topics are identified. NICE manages the topic selection process on behalf of the Department of Health (DH) and works with the DH to develop a draft scope for each topic, which defines the disease, the patients and the technology covered by the appraisal and the questions it aims to answer. Consultees and commentators are then requested to comment on the draft scope and a scoping workshop is held to discuss key issues relating to the scope. The DH formally refers topics to NICE and the STA timelines are then set. Figure 1 shows the STA process timeline from formal referral through to the issuing of NICE guidance.
FIGURE 1.
Single technology appraisal process timeline (from NICE guide to the STA process3). ACD, appraisal consultation document; CHMP, Committee for Medicinal Products for Human Use.
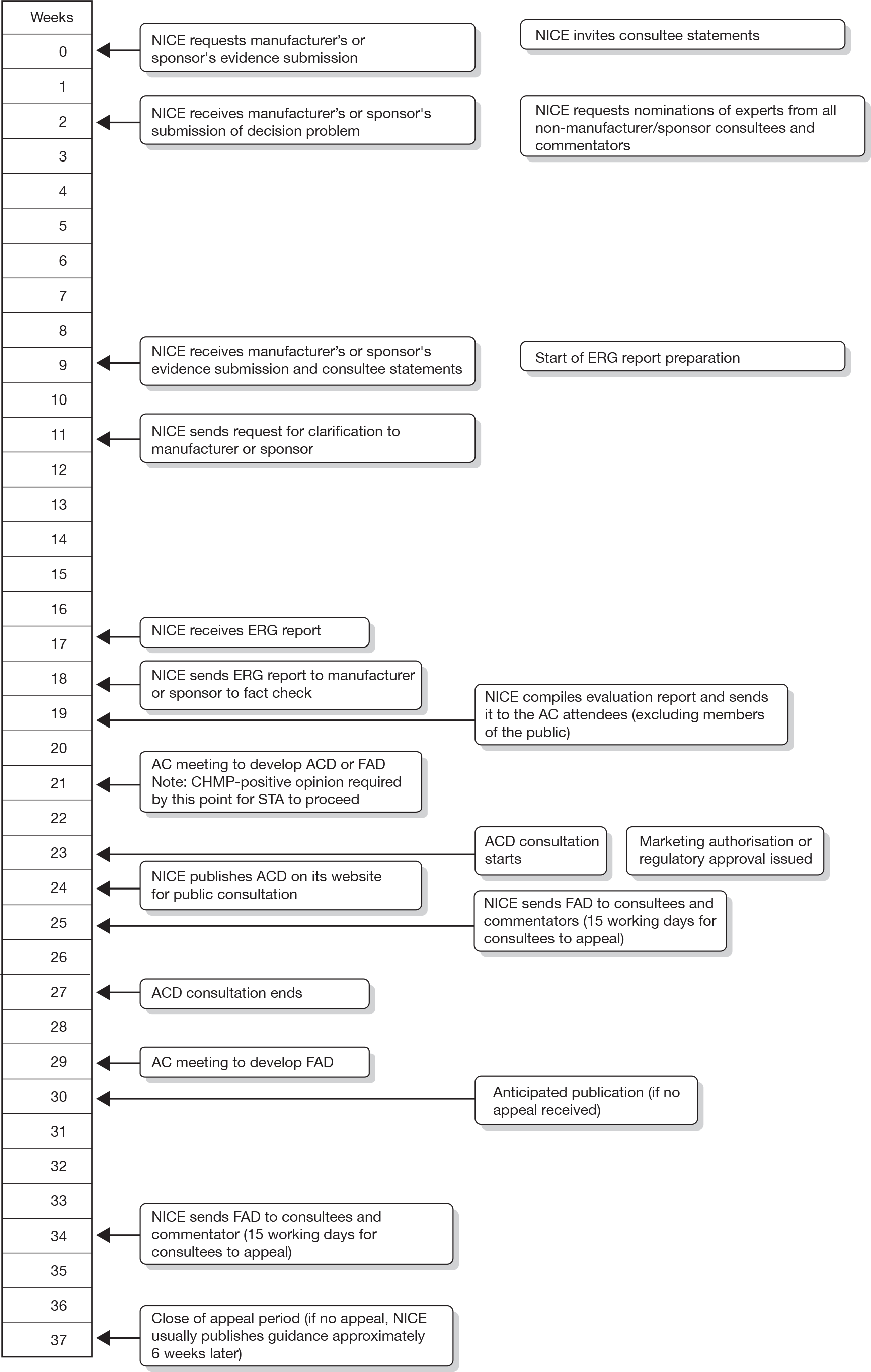
Manufacturers are invited to prepare their submission to NICE using a standard report template. Extensive guidance for manufacturers is provided in the NICE guide to the methods for technology appraisal. 1 The MS is expected to include a systematic review of the clinical effectiveness evidence for the technology under consideration, as well as a cost-effectiveness analysis. External independent Evidence Review Groups (ERGs) are based in academic centres and are charged with the task of critically appraising the MS to identify strengths, weaknesses and gaps in the evidence presented. The resultant ERG reports are then considered as a part of the evidence considered by the AC. Little guidance has been provided outlining the approaches ERG teams should take. ERG teams are expected to critically appraise the MS to determine whether the evidence presented is:
-
relevant to the issue under consideration in terms of patient groups, comparators, perspective and outcomes
-
complete (all relevant evidence must be identified)
-
inclusive of all study design information (including the type of study, the circumstances of its undertaking and the selection of outcomes and costs) and inclusive of all intention to treat patients
-
fit for purpose [contributing to an overall assessment of the clinical benefit and quality of life (QoL), preferably in such units that allow comparison of the benefits from different technologies and between different patient groups].
In addition, the ERG team must critically appraise the MS to ensure that the analyses and modelling presented by the manufacturer are methodologically sound and minimise any bias. Models must be replicable, have face validity (be plausible) and be open to external scrutiny.
Within the STA process, there are no resources for the ERG to produce an independent systematic review or cost-effectiveness model. The current ERG report template is attached as Appendix 1. Currently, NICE works with the following ERGs (three new ERGs will join these seven teams from April 2011):
-
Health Economics Research Unit and Health Services Research Unit, University of Aberdeen
-
Liverpool Reviews and Implementation Group (LRiG), University of Liverpool
-
Centre for Reviews and Dissemination (CRD) and Centre for Health Economics (CHE), University of York
-
Peninsula Technology Assessment Group (PenTAG), Peninsula Medical School, Universities of Exeter and Plymouth
-
School of Health and Related Research (ScHARR), University of Sheffield
-
Southampton Health Technology Assessment Centre (SHTAC), University of Southampton
-
West Midlands Health Technology Assessment Collaboration (WMHTAC), Department of Public Health and Epidemiology, University of Birmingham.
The process of requesting further information from the manufacturer following submission was not included in the original plan for the conduct of an STA. However, it very soon became apparent that such a mechanism was required, as the ERGs identified aspects of the submissions that were unclear or where further information was required. The process of the clarification letter was implemented and is now standard practice. The ERG is requested to submit any clarification questions for the manufacturer within 10 working days of receiving the MS. NICE contributes additional questions and sends the letter on to the manufacturer who has 10 working days to respond. The format of the letter has evolved over time and more recent letters follow a consistent format developed by NICE that separates out clinical and economic clarification issues.
The AC meets to consider the evidence presented by the manufacturer and also the critique of the evidence by the ERG and makes recommendations to NICE. There are currently four ACs. If the AC’s provisional recommendations are more restrictive than the terms of the marketing authorisation for the technology under appraisal then an appraisal consultation document (ACD) is produced and consultees and commentators are invited to comment on the ACD. The AC meets again to consider these comments and aims to produce the FAD, which forms the basis of the guidance that NICE will issue to the NHS in England and Wales. However, instead, the AC may issue another ACD inviting further comments from consultees and commentators, which are, again, considered by the AC to produce the FAD.
Both ACDs and FADs may have more than one decision within the same document; for example, for different subgroups of patients different decisions may be made. An appeal may be launched up to 15 working days after the FAD has been issued and guidance is not published until an appeal decision has been made by the appeal panel.
When the STA process was initially introduced, concerns were raised that it may represent a less robust process for producing guidance on the use of health technologies than the MTA process. 4 Concerns have also been raised regarding the need for consistency and transparency of methods used to critically appraise the MS as part of the STA process.
The NICE Decision Support Unit (DSU) is a collaboration of UK universities that is commissioned by NICE to provide research and training to support the NICE technology appraisal process. Occasionally, the DSU is requested to provide additional analysis to enable the AC to reach a decision. The NICE DSU reviewed the STA process in 20085 and looked at the first 10 STAs only. The report highlighted the potential for inconsistencies between ERGs in terms of the content of their reports. This report takes a broader perspective and provides an in-depth analysis of components of the STA process, which have changed significantly since the earlier report by the DSU.
Aims and objectives
The aims of this study were to review the methods currently used by ERG teams to critically appraise MSs within the NICE STA process and to provide recommendations on approaches that could be considered in the future. An additional aim was to map the STA process to date. There were five primary objectives:
-
to provide a map of the STA process to date
-
to identify current approaches to critical appraisal of MSs by ERGs
-
to identify recurring themes in clarification letters sent to manufacturers
-
to provide recommendations for possible alternative approaches to be used in the critical appraisal process
-
to revise the current ERG report template.
Chapter 2 describes the mapping of the STA process to date, Chapter 3 covers the documentary and thematic analysis of 30 ERG reports and Chapter 4 explores the issues identified in the 21 clarification letters. Chapter 5 includes a description of the process used to modify the ERG report template, as well as recommendations for approaches to be considered in the development of ERG reports.
The protocol for this project stated that telephone interviews would be undertaken with members of the ERGs who had worked on STAs. However, subsequently a Technology Assessment Services Collaboration (InterTASC) workshop to discuss the STA process was arranged and several issues were discussed during this meeting in April 2010. Key points debated during the workshop have been integrated into the discussion and conclusion sections of this report.
Chapter 2 Mapping exercise
Methods
Data collection
All STAs that had been identified by NICE up to, and including, March 2009 were included in the mapping exercise. The list of STAs was provided by the NIHR Evaluation, Trials and Studies Coordinating Centre (NETSCC). A mapping tool was devised using a Microsoft Excel 2003 spreadsheet (Microsoft Corporation, Redmond, WA, USA) and is attached as Appendix 2. The tool was designed to collect information on a range of topics from each STA.
The mapping tool/spreadsheet was piloted by two reviewers (EK and AB) on nine STAs. Following the pilot, amendments were made. These included standardising data entry and insertion of comment boxes where clarification of the data was required.
The majority of the data for each STA was obtained from the NICE guidance website. 6 Three reviewers (DP, AB and EK) extracted the data. After data extraction was completed a sample of nine STAs was taken and data were checked by a second reviewer (DP, EK or AB) who had not completed the initial data collection in order to verify that data extraction had been undertaken in a consistent manner by all reviewers. Where data were not available on the NICE website, for example referral date in some cases or the date of ERG report submission, the information was requested from NETSCC.
Data analysis
Data analysis was completed in Excel. The majority of analyses involved calculating frequencies for each topic considered in the mapping tool/spreadsheet. This was used, for example, to calculate the number of STA projects that were completed, in progress, suspended or terminated. The number of working days for the following time periods were calculated and then the values were divided by five to calculate the number of working weeks:
-
referral date to FAD, i.e. total length of project
-
date of commencement (date of final scope) to FAD, i.e. total length of ‘active’ project
-
referral date to final scope date
-
date of manufacturer report submission to ERG report submission
-
date of ERG report submission to first AC
-
date between first AC and second AC
-
date between second AC and FAD.
The exact date (day/month/year) was not provided on documents such as the final scope and FAD. The date was usually provided only in month/year format on the documents and an exact date of upload was provided on the NICE website. Where the month of issue differed on the scope or FAD document from the month stated on the NICE website for the document upload, the month listed on the document was used and the 28th day of the month was chosen as the ‘exact date’ in order to perform analyses between the different stages in the STA project timeline. Where the month agreed between the document and date of upload on the NICE website, the exact date listed as the upload date on the NICE website was used. For either scenario, these decisions meant that the analyses would underestimate, rather than overestimate, the time taken between stages in the STA process. Similarly, when the exact date of referral provided by NETSCC was not available, the 28th day of the month that the STA was referred was selected for each referral date. Simple descriptive statistics, such as calculating the mean and range, were applied where appropriate.
The NICE disease categories to which STA topics were allocated were obtained from the NICE website and the numbers within each category were tabulated. Information on DSU involvement and appeals was obtained from the NICE website and the information summarised in a narrative synthesis. The number of suspended or terminated appraisals was tabulated and reasons summarised. A completed STA was defined as an STA that had received a final decision (FAD) from NICE on the technology being considered. STAs that had previously received a final decision from NICE but which was subsequently withdrawn were not included in the completed STA category.
Results
Single technology appraisals by characteristics
Ninety-five STAs had been identified in the NICE appraisal process since the STA programme began in 2005 up to, and including, March 2009 as per the list provided by NETSCC.
Topic area
Single technology appraisals are categorised by topic on the NICE website. There are 21 topic categories. Table 1 shows the categorisation of topics identified in the mapping exercise. Some topics are listed under more than one category, for example ‘pemetrexed for the first-line treatment of locally advanced or metastatic non-small cell lung cancer’7 is listed under cancer and respiratory. Thirty-one STAs were listed in more than one category. The majority of topics were cancer topics. Under the cancer category, some STAs, such as those concerning satraplatin for hormone-refractory prostate cancer and capecitabine for advanced pancreatic cancer, were not listed on the NICE website and the topic category was assigned on the basis of the title of the appraisal by the authors of this report.
| STA topics in NICE disease categories | Frequency |
|---|---|
| Cancer | 46 |
| Musculoskeletal | 15 |
| Digestive system | 13 |
| Respiratory | 11 |
| Cardiovascular | 9 |
| Therapeutic procedures | 7 |
| Skin | 7 |
| Central nervous system | 5 |
| Endocrine, nutritional and metabolic | 4 |
| Infectious diseases | 3 |
| Blood and immune system | 2 |
| Gynaecology, pregnancy and birth | 2 |
| Eye | 1 |
| Injuries, accidents and wounds | 1 |
| Mental health and behavioural conditions | 1 |
| Public health | 1 |
| Urogenital | 1 |
| Diagnostic procedures | 0 |
| Ear and nose | 0 |
| Mouth and dental | 0 |
| Surgical procedures | 0 |
Single technology appraisal report and project status
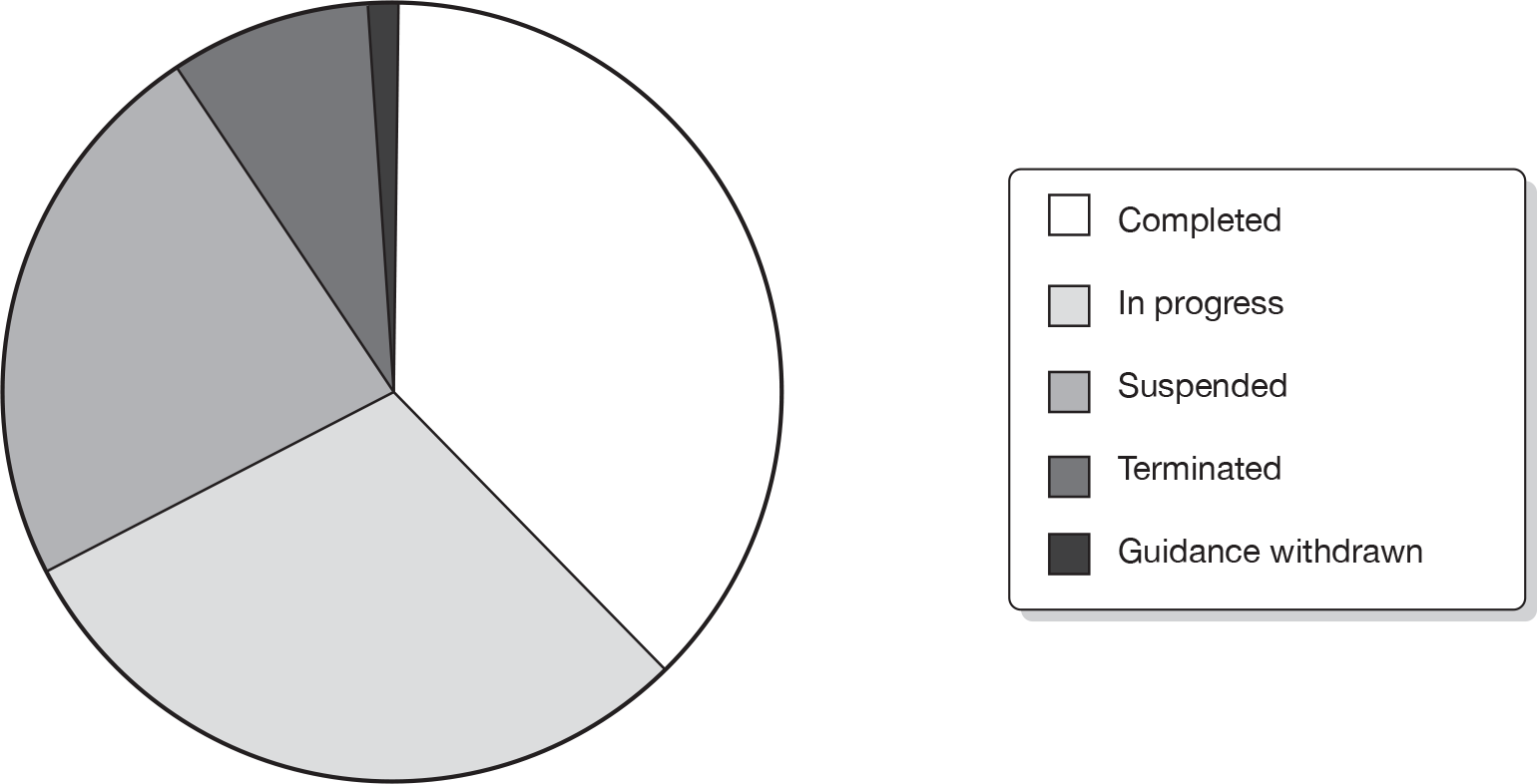
Of the 95 STAs identified, 36 were categorised as completed STAs (38%), 28 were in progress (29%), 22 (23%) had been suspended, eight terminated (8%) and one had been completed but subsequently the guidance was withdrawn (1%). Three of the 95 STAs (satraplatin for hormone-refractory prostate cancer, etanercept for moderate to severe chronic plaque psoriasis in children and adolescents and rimonabant for type 2 diabetes mellitus) had passed through the topic selection process but were not referred to NICE. See Figure 2 for STA project status.
FIGURE 2.
Single technology appraisal project status.
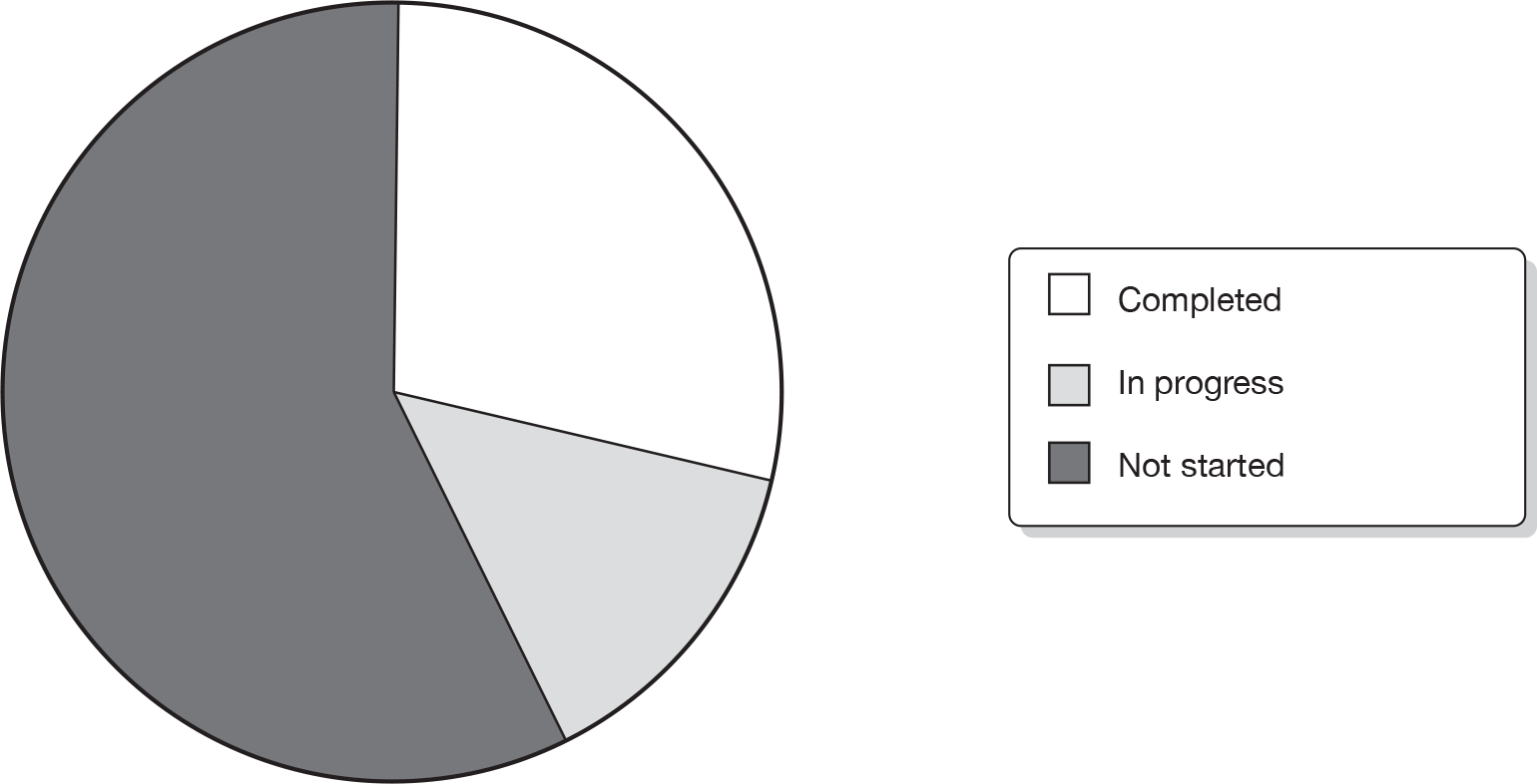
Of the 28 in-progress STAs, eight had completed ERG reports, four had ERG reports not yet completed and 16 ERG reports had not yet commenced (Figure 3).
FIGURE 3.
Evidence Review Group report status.
Of the 31 STAs that had been suspended or terminated or had their guidance withdrawn, three ERG reports had been completed. For two of these appraisals (mifamurtide for osteosarcoma and romiplostim for thrombocytopenic purpura) the appraisals were suspended due to delays in drug launch. For the third appraisal (rimonabant for type 2 diabetes mellitus) the guidance was withdrawn.
Manufacturer
Sixty-four STAs had a named manufacturer, while the remaining 31 did not have a manufacturer reported on the NICE website, usually because the STA process for those topics was still in the early stages. Twenty-seven manufacturers have been involved in the STA process to date. Table 2 lists manufacturers and the numbers of STAs they have been involved in.
| Manufacturer | No. of STAs |
|---|---|
| Roche Products Ltd | 12 |
| Schering-Plough Ltd | 6 |
| Bristol-Meyers Squibb Pharmaceuticals Ltd | 5 |
| Eli Lilly & Co | 4 |
| GlaxoSmithKline | 3 |
| Merck Serono | |
| Sanofi-aventis | |
| Abbott Laboratories Ltd | 2 |
| AstraZeneca | |
| Boehringer Ingelheim | |
| Celgene | |
| Janssen-Cilag | |
| Novartis Pharmaceuticals UK Ltd | |
| Pfizer Ltd | |
| Archimedes | 1 |
| Amgen | |
| Basilea Medical | |
| Bayer | |
| Bayer Schering Pharma | |
| Biogen | |
| Gilead Sciences | |
| IDM Pharma | |
| Ipsen Ltd | |
| PharmaMar | |
| Schering Health Care Ltd | |
| UCB | |
| Wyeth Pharmaceuticals |
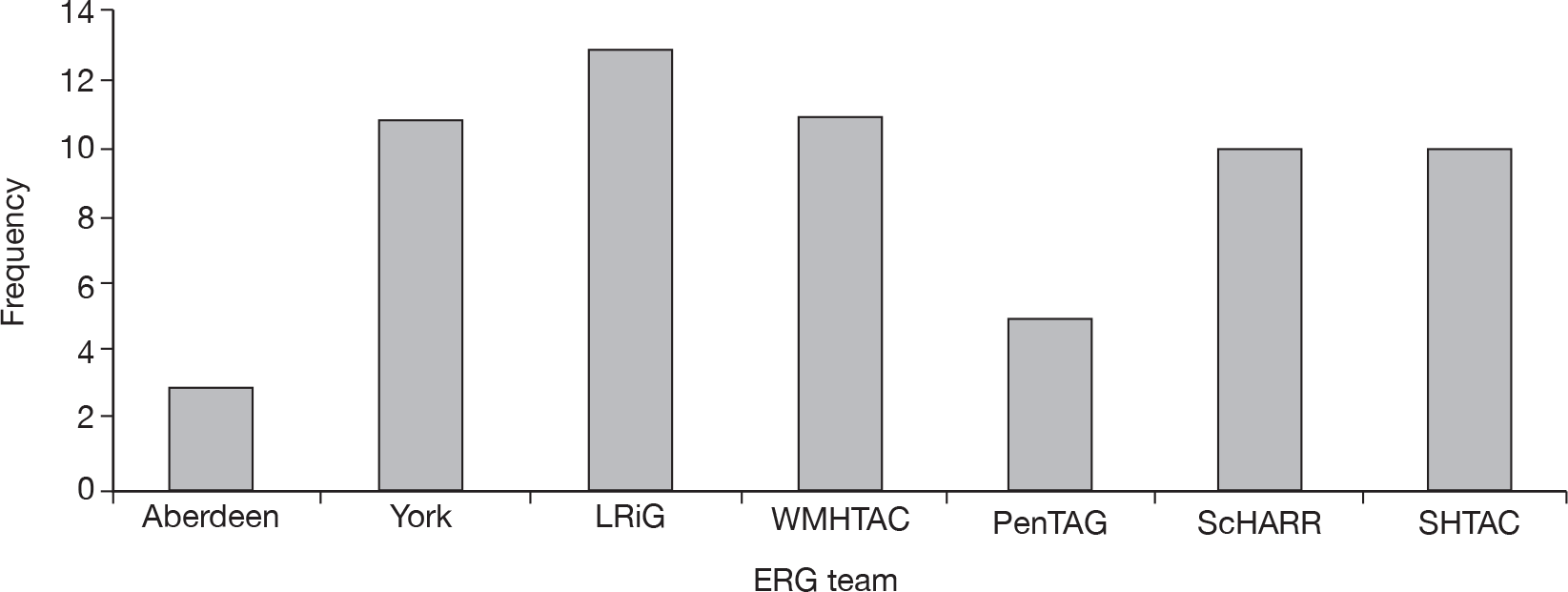
Evidence Review Group team
Sixty-three of the STAs had been assigned an ERG team. Five teams undertook 10 or more ERG reports and the remaining two teams undertook three and five reports (Figure 4).
FIGURE 4.
Evidence Review Group teams.
Decision process in completed and in-progress single technology appraisals
Incremental cost-effectiveness ratios reported
Thirty-six completed STAs and seven in-progress STAs contained figures for the base-case incremental cost-effectiveness ratios (ICERs) reported by the manufacturer within their submission. Thirty-three completed STAs and seven in-progress STAs included figures for base-case ICERS reported by the ERG within their report.
Significant differences were seen when the manufacturer and ERG base-case ICERs were compared. The manufacturer base-case ICERs were > £30,000 per quality-adjusted life-year (QALY) in 13 cases and < £30,000 per QALY in 30 cases. In contrast, the ERG base-case ICERS were reported as > £30,000 per QALY in 25 cases and < £30,000 per QALY in 15 cases (Figure 5).
FIGURE 5.
Base-case ICERs reported by ERG and manufacturer.
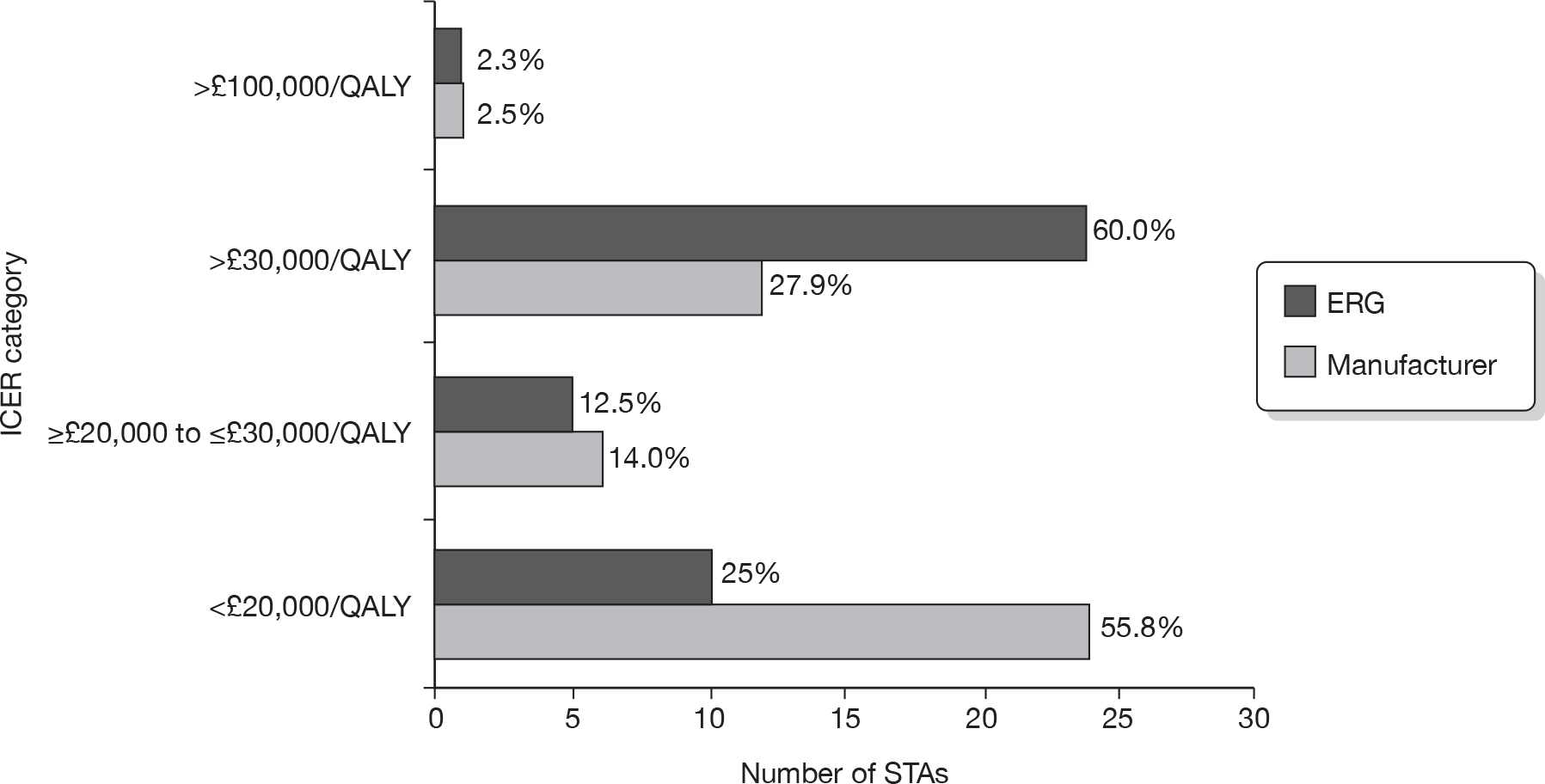
In three ERG reports, a clear figure for the base-case ICER calculated by the ERG was not reported. 8–10 One ERG report stated that a £30,000-per-QALY threshold analysis was conducted (natalizumab for the treatment of adults with highly active relapsing–remitting multiple sclerosis8); one ERG report estimated two base-case ICERs of > £30,000 and > £100,000 per QALY (pemetrexed for the treatment of relapsed non-small cell lung cancer9); and one ERG report stated that the ERG was unable to provide revised ICERs (pemetrexed for the first-line treatment of locally advanced or metastatic non-small cell lung cancer10).
Appraisal consultation document decision
The AC can make various recommendations in the ACD. Obviously there can be a clear ‘yes’, a clear ‘no’ or a ‘minded’ opinion. A ‘minded’ opinion usually means that the AC is minded to say ‘yes’ or ‘no’ but that it requires further information, usually from the manufacturer, to make its decision at the subsequent AC meeting. There were 38 STAs with ACD decisions available on the NICE website (31 completed STAs and seven in-progress STAs). Nineteen had a ‘no’ recorded as the ACD decision, nine were recorded as ‘yes’ decisions and seven had ‘minded no’ decisions within the ACD (Table 3). Two STAs had both a ‘yes’ and a ‘minded no’ decision recorded within the ACD and one STA had both a ‘no’ and a ‘minded no’ recorded in the ACD.
| ACD decisiona | Frequency (%) |
|---|---|
| Yes | 9 (23.7) |
| No | 19 (50.0) |
| ‘Minded no’ | 7 (18.4) |
| Yes and ‘minded no’ | 2 (5.3) |
| No and ‘minded no’ | 1 (2.6) |
| ACD not on website | 5 (N/A) |
Appraisal consultation documents were not made available on the NICE website for five completed STAs11–15 (trastuzumab as adjuvant therapy for early-stage breast cancer;11 varenicline for smoking cessation;12 dabigatran etexilate for the prevention of venous thromboembolism in patients undergoing elective hip and knee surgery;13 rituximab for the first-line treatment of low-grade follicular non-Hodgkin’s lymphoma;14 and rivaroxaban for the treatment of venous thromboembolism15). However, ACDs are issued only if the AC recommendations are restrictive or if the manufacturer is requested to provide further clarification, and therefore ACDs are not issued for all STAs.
Two STAs had two ACDs, of which one had a ‘no’ decision recorded in its first ACD and then a ‘yes’ decision in the second ACD,16 and the other had a ‘no’ decision recorded in its first ACD and then a ‘yes’ decision with conditions in the second ACD. 17
Final appraisal determination decision
Thirty-six completed STAs and one in-progress STA had FAD decisions. Twenty-five STAs were recorded as ‘yes’ decisions, whereas 12 were recorded as ‘no’ decisions. Thirty-two STAs had both ACD and FAD documents available on the NICE website. Table 4 shows the initial ACD decision (taken from the first ACD if more than one ACD) and subsequent FAD decision in the 32 STAs with both ACDs and FADs. All ‘yes’ decisions and 10 out of 14 ‘no’ decisions remained the same between the ACD and FAD. ‘Minded no’ and ‘yes/minded no’ decisions within the ACDs almost always became ‘yes’ decisions within the FAD. The one exception was a ‘minded no’ to ‘no’ decision between the ACD and FAD. In this case, a subsequent appeal was upheld. After additional work was undertaken by the DSU, the AC met again and the ‘no’ decision in the FAD was subsequently changed to a ‘yes with conditions’ (erlotinib for the treatment of relapsed non-small cell lung cancer18).
| ACD decision | FAD decision ‘yes’ | FAD decision ‘no’ |
|---|---|---|
| Yes | 8 | 0 |
| No | 4 | 10 |
| ‘Minded no’ | 6 | 1 |
| Yes and ‘minded no’ | 2 | 0 |
| No and ‘minded no’ | 0 | 1 |
Project timeline (dates analyses)
Table 5 shows the breakdown of project timelines for the 36 completed STAs. Some of the analyses are based on fewer than 36 STAs as some of the data were missing, for example scope dates or AC dates.
| Date analysis | Mean (range) in working weeks |
|---|---|
| Referral date to FAD | 54.9 (30.4 to 131.0) |
| Final scope to FAD | 44.0 (26.2 to 122.2) |
| Referral date to scope | 15.3 (4.4 to 83) |
| MS to ERG report submission | 10.5 (8.2 to 23.8) |
| ERG report to first AC | 5.3 (3.4 to 11.2) |
| First AC to second AC | 10.4 (8.2 to 22) |
| Second AC to FAD | 8.2 (3.0 to 29.8) |
Referral date to final appraisal determination
The NICE STA process guide3 states on p. 18, in section 3.2.1, that the STA process begins ‘after NICE has received formal referral from the Secretary of State for Health’. However, no guidance is given with regards to the length of the STA project between the referral date and the FAD. Week 0 on the STA project timeline is when manufacturers are invited to complete an evidence submission or when the final scope is issued. The time from final scope to FAD should be 34 weeks or less, as stated in the 2009 STA process guide3 (when there is no appeal). In the original STA process guide (2006)19 this was stated to be 35 weeks.
The time period between the referral date and final scope date (i.e. week 0) ranges considerably between 4.4 and 83 working weeks (see Table 5). The length of the entire STA project was taken as the number of working weeks between the referral date and the FAD date. The mean number of working weeks between the referral date and the FAD in the 36 completed STAs was 54.9. This ranged considerably between 30.4 and 131.0 working weeks. The majority of completed STAs (27/36) took between 39 and 60 weeks to complete from their referral to FAD.
Final scope to final appraisal determination
The 35-week final scope to FAD time period for a completed appraisal was used in this analysis, as some of the STAs were completed before 2006. A 2-week grace period was added to allow for holiday periods and other delays, making the cut-off point 37 weeks for a completed, on-time STA. Scopes (and scoping workshops) were not part of the initial STA process and seven STAs did not have a final scope available on the NICE website. It was therefore not possible to calculate the time period between the issue of final scope and FAD in these seven STAs. Eight of the 29 STAs where it was possible to calculate time to completion were completed within 37 working weeks, i.e. 28% of STAs were completed within a period that could be considered ‘on time’ when the final scope date is taken as the start of the process and there is no appeal process. The mean number of working weeks between the final scope and FAD in the 36 completed STAs was 44.0. This ranged considerably from 26.2 to 122.2 weeks.
The scope to FAD period for 12 STAS was delayed between 2 and 5 weeks. Possible reasons made available on the NICE website for the 2- to 5-week delay were provided for two STAs only and included appraisal suspension (omalizumab for severe persistent allergic asthma20) and transfer from the 10th to the 12th wave (natalizumab for the treatment of adults with highly active relapsing–remitting multiple sclerosis8). A third STA was recorded to have a 2- to 5-week delay between scope and FAD (docetaxel for adjuvant treatment of early breast cancer21). However, this STA had been transferred from the MTA to the STA process and thus the final scope date related to that of the original MTA (scopes were not initially provided for STAs). There did not appear to be any reasons provided on the NICE website as to why the remaining nine STAs were delayed between 2 and 5 weeks. However, it was noted that four of the STAs required extra work from the manufacturer22–25 (adalimumab for the treatment of moderate to severe psoriatic arthritis,22 rituximab for the treatment of refractory rheumatoid arthritis,23 rituximab for the treatment of recurrent or refractory stage III or IV follicular non-Hodgkin’s lymphoma,24 and entacavir for the treatment of chronic hepatitis B25) and extra work was undertaken by the ERG for one STA26 (cetuximab for the treatment of metastatic and/or recurrent squamous cell carcinoma of the head and neck26).
The scope to FAD period was delayed by > 5 weeks for two STAs,7,27 for which no apparent reasons were provided on the NICE website (ustekinumab for the treatment of moderate to severe psoriasis27 and pemetrexed for the first-line treatment of locally advanced or metastatic non-small cell cancer7). A further seven STAs had scope to FAD periods delayed by > 10 weeks. Reasons for scope to FAD periods being delayed by > 10 weeks were provided for six of these STAs and included appeal proceedings (erlotinib – non-small cell lung cancer,18 abatacept,28 febuxostat29), additional work undertaken (erlotinib18), end-of-life consideration with production of two ACDs (lenalidomide16), first AC meeting cancelled owing to clarification of European regulatory timings (febuxostat29), timings reset at the request of the manufacturer to allow for the outcome of discussions with the Committee for Medicinal Products for Human Use of the European Medicines Agency to be incorporated in the MS (abatacept28), third and fourth AC meetings (cetuximab for colorectal cancer30) and a FAD returned with comments after the second AC (sunitinib31). There were no apparent reasons stated on the NICE website to explain why the scope to FAD period for the infliximab for ulcerative colitis32 STA was delayed by 14.2 weeks.
In addition, seven STAs are delayed and were still in progress at the time of data extraction (August 2009):
-
Topotecan for the treatment of recurrent and stage IVB carcinoma of the cervix (third AC meeting for end-of-life consideration). 33
-
Tocilizumab for the treatment of juvenile idiopathic arthritis (delay due to pricing issues, guidance delayed as a result). 34
-
Bevacizumab in combination with non-taxane chemotherapy within its licensed indications for the first-line treatment of metastatic breast cancer (rescheduled to align with regulatory approval). 35
-
Trabectedin for the treatment of advanced metastatic soft tissue sarcoma (extension granted for manufacturer to meet full requirements of the STA process). 36
-
The clinical effectiveness and cost-effectiveness of erlotinib monotherapy for maintenance treatment of non-small cell lung cancer after previous platinum-containing chemotherapy (rescheduled to align with regulatory expectations). 37
-
The clinical effectiveness and cost-effectiveness of erlotinib in combination with bevacizumab for maintenance treatment of non-squamous advanced or metastatic non-small cell lung cancer after previous platinum-containing chemotherapy (rescheduled to align with regulatory expectations). 38
-
Rituximab for relapsed treatment of chronic lymphocytic leukaemia (delays due to variation to marketing authorisation). 39
If these delays had been taken into account then the overall time frame for the 95 STAs would have been lengthened.
Manufacturer submission to Evidence Review Group report submission
The appendix B document in the NICE STA process guide3 (p. 56) allows 8 weeks between the manufacturer report submission and ERG report submission. The NICE guidance also stipulates that ERG teams will be given a minimum of 8 weeks to complete the ERG report. Eighteen (50%) of the completed STA ERG reports were completed within 8–10 working weeks and a further 14 (39%) STA ERG reports were submitted 10–12 working weeks after receipt of the MS. The mean number of working weeks between the manufacturer report submission and ERG report submission in the 36 completed STAs was 10.5. This ranged considerably between 8.2 and 23.8 working weeks. The majority of STAs (32/36) took 8–12 working weeks between the manufacturer report submission and ERG report submission.
Evidence Review Group report submission to first Appraisal Committee
The first AC is scheduled to take place 4 weeks after the ERG report has been submitted, and this was the case for 5 out of the 36 completed STAs. The first AC meeting was 4–5 weeks after the ERG report submission in 15 STAs and 5–7 weeks in 13 STAs. The mean number of working weeks between the ERG report submission and first AC in the 36 completed STAs was 5.3. This ranged considerably between 3.4 and 11.2 working weeks.
First Appraisal Committee to second Appraisal Committee
The second AC is scheduled to take place 8 weeks after the first AC. The mean number of working weeks between the first AC and second AC in the 36 completed STAs was 10.4. This ranged considerably between 8.2 and 22 working weeks. The time period between the first AC and second AC in two-thirds of completed STAs (24/36) was between 8 and 10 working weeks.
Second Appraisal Committee to final appraisal determination
The FAD is published (if no appeal is received) 10 weeks after the second AC meeting, although a second AC meeting is not held if an ACD is not produced at the first AC meeting. In all but four STAs this was achieved. The mean number of working weeks between the second AC and the FAD in the 36 completed STAs was 8.2 working weeks. This ranged considerably between 3 and 29.8 working weeks. The time period between the second AC and the FAD was 3–5 weeks in six STAs and 5–9 weeks in 20 STAs.
Extra work involved in the single technology appraisal process
Manufacturer
After the first AC meeting, in 11 out of 36 of the completed STAs and in one out of seven of the in-progress STAs, the manufacturer was requested to complete further work before the second AC meeting.
Evidence Review Group
Before the first AC meeting, in 5 out of 36 of the completed STAs and in three out of seven of the in-progress STAs, the ERG was requested to complete work in addition to the ERG report. After the first AC meeting, in 4 out of 36 of the completed STAs and in one out of seven of the in-progress STAs, the ERG was requested to complete further work before the second AC meeting.
Decision Support Unit involvement
Three STAs had involvement from the NICE DSU:
-
cetuximab for first-line treatment of metastatic colorectal cancer30
-
erlotinib for the treatment of relapsed non-small cell lung cancer18
-
lapatinib for breast cancer for use in women with previously treated, advanced or metastatic breast cancer. 40
In one of these STAs it appears that the decision of the AC changed from ‘no’ in the ACD to ‘yes’ with restrictions in the FAD (cetuximab30). For one of the STAs (erlotinib18) the decision changed from ‘minded no’ at ACD to ‘no’ at FAD; however, after additional work undertaken post appeal the decision was changed to ‘yes’ but with restrictions. For lapatinib,40 the decision remained ‘no’ at FAD although at the time of writing this STA is still ongoing.
End-of-life consideration
On 5 January 2009, NICE issued supplementary advice to be taken into account when appraising treatments which may be life-extending for patients with short life expectancy. 41 Four STAs16,26,33,40 have considered this supplementary advice: two completed STAs and two in-progress STAs. Lenalidomide for relapsed and/or refractory multiple myeloma (completed STA) recorded the first ACD decision as ‘no’, changing to ‘yes’ in the second ACD and FAD (requiring three ACs). 16 Cetuximab for the treatment of metastatic and/or recurrent squamous cell carcinoma of the head and neck (completed STA) recorded a ‘no’ decision within the ACD and FAD. 26 Lapatinib for breast cancer for use in women with previously treated, advanced or metastatic breast cancer (in-progress STA) recorded a ‘no’ decision within the ACD and FAD. 40 Following on from an appeal, this technology was put forward for end-of-life consideration. Topotecan for the treatment of recurrent and stage IVB carcinoma of the cervix (in-progress STA) recorded a ‘no’ in the ACD; the FAD decision is to be confirmed. 33
Appeals
In total, 8 out of the 36 completed appraisals in this analysis had an appeal. The appraisals were:
-
cetuximab in its licensed indications for refractory head and neck cancer26
-
erlotinib for the treatment of relapsed non-small cell lung cancer18
-
abatacept for the treatment of refractory rheumatoid arthritis28
-
trastuzumab as adjuvant therapy for early-stage breast cancer11
-
febuxostat for the management of hyperuricaemia in patients with gout29
-
lapatinib for breast cancer for use in women with previously treated, advanced or metastatic breast cancer40
-
pemetrexed for the treatment of relapsed non-small cell lung cancer42
-
bortezomib monotherapy for relapsed multiple myeloma. 17
Four of these appeals were dismissed (abatacept,28 trastuzumab,11 febuxostat29 and pemetrexed42). For three STAs – cetuximab,26 erlotinib18 and bortezomib17 – the appeals resulted in a change to NICE guidance. In all cases the decision became ‘yes’ with restrictions. At the time of writing, one STA (lapatinib40) was ongoing (March 2010).
Suspended/withdrawn/terminated single technology appraisals
Overall, 30 of the 95 STAs (32%) in this study were either suspended or terminated. Guidance was released and later withdrawn by NICE for one STA (rimonabant for the treatment of overweight and obese patients43).
Twenty-two STAs are listed as suspended. In two of these STAs, the ERG report had already been submitted when the appraisal was suspended. Reasons for suspension include the drug not yet launched (2), indication for the drug uncertain (1), regulatory or licensing issues (11), no MS received (1), further data awaited by manufacturer (3), referral to MTA process (1) and no reason given (3). Eight STAs are listed as terminated. Three STAs passed through topic selection but were not referred to NICE; two were terminated because no MS was received by NICE. No reasons were stated on the NICE website for the other three terminated STAs.
Record-keeping and consistency issues
Nearly all of the information used in this mapping analysis was obtained from the NICE website; therefore the information described in this report is a reflection of the record-keeping on the website and not necessarily an accurate reflection of what actually happened during the appraisal process. There are a number of issues regarding record-keeping and consistency requiring further discussion.
Dates
Full dates (day/month/year) were often not recorded within documents published on the website; often only the month and year were available and this made calculation of timings difficult. Where exact dates were not published within documents, such as the scope and FADs, we used the 28th of each month as the date of issue. Therefore, there is some inaccuracy and underestimation within these analyses.
There were also other inconsistencies with dates. Often the month of issue within the scope and FAD documents was not the same as the month stated on the website. Examples of this are romiplostim for the treatment of chronic immune or idiopathic thrombocytopenic purpura (ITP)44 and paclitaxel for the adjuvant treatment of early breast cancer. 45
Documentation
Some documents were missing from the NICE website, such as ACDs and final scopes. This could be due to the fact that initially scoping documents and ACDs were not part of the STA process and were later additions. In some appraisals there were two FAD documents and it was not always clear why.
Transparency and inconsistencies
Reasons for delays, suspensions or terminations are not always clear on the NICE website. As each appraisal does not have a unique identifying number, and there are several appraisals for the same drug within different conditions, it was sometimes difficult to find information on a specific appraisal.
Three STAs in this mapping exercise had passed through the topic selection process but were not referred to NICE: two of these are listed on the NICE website, although difficult to find, while one is not mentioned at all (etanercept for moderate to severe chronic plaque psoriasis in children and adolescents).
As described previously, there is inconsistency in the way STAs are categorised by topic. The terms ‘suspended’ and ‘terminated’ are not used consistently. NICE continues to monitor topics that are referred to it even if the appraisal has ended. STAs also occasionally have different titles within different documents relating to the same appraisal that occasionally caused confusion.
Discussion
Almost all of the necessary relevant documentation relating to each STA included in this mapping exercise was available on the NICE website. However, some information was difficult to locate and there was inconsistency in the details related to dates of topic referral. A guide for the public regarding where specific documentation is kept could be useful.
Within a relatively short time period NICE has dealt with 95 STA topics. Base-case ICERs reported by the ERGs were consistently higher than those reported by manufacturers. Extra analyses were requested of manufacturers in 28% of STAs, which translates to extra work for the ERGs. Not all of the extra work required of the ERGs was recorded on the NICE website.
Lack of consistency in recording referral dates meant it was difficult to calculate the total STA process timelines. However, it is noted that the length of time between referral date and final scope date can be substantial.
The majority (78%) of STAs in this study do not meet the established 35-week scope to FAD target. The delays are attributable not to delays in submission of ERG reports or the scheduling of AC meetings but to delays in other aspects of the process.
The first delay point relates to STAs that were suspended or terminated. We identified that 32% (30/95) of the topics were in this category, with a further seven marked as in progress but delayed. The vast majority of topics in these groups were stopped because of regulatory issues and/or non-submission by the manufacturers.
Suspended/terminated appraisals have significant resource implications. Each topic referred as part of the STA process includes a scoping workshop to which the appropriate stakeholders are invited to comment on the proposed scope for the STA. These meetings can involve as few as 10 people or as many as 30 and they take a half-day plus travel time for each participant. Delays can also occur further along in the process, resulting in substantial waste of resources.
The next delays occur after the first AC meeting. Although not always the case, requests for additional analysis from the manufacturer or the ERG have the potential to delay the second AC meeting and therefore the FAD. This is not a common occurrence but does happen. In addition, this subsequent analysis may require input from the DSU, additional AC meetings and subsequent consultation on further ACDs. These are all resource-intensive activities. Anecdotal evidence has been provided by one ERG indicating that one STA is now at its fifth AC meeting.
However, the longest delay in the process takes place when an appeal is made, usually by the manufacturer. This occurred in 22% (8/36) of completed appraisals included in this analysis, with one of these going on to judicial review. The appeal process is very time-consuming for all of those involved, although manufacturers are likely to take heart in the fact that half of the topics that went to appeal resulted in approval (although with restrictions) of their product.
The STA process is already complex and, looking to the future, is likely to become more so, and these complexities have potential to add even further delays to the release of guidance. The introduction of the consideration of end-of-life issues and PASs3 has the potential to extend the STA processes.
Chapter 3 Thematic analysis of Evidence Review Group reports
Methods
A documentary analysis of the first 30 ERG reports was undertaken. For this research we were exclusively interested in the content, rather than the context, of the reports. Attention was focused neither on the context within which the documents were produced nor on their subsequent impact on external decision-making processes, but rather on the content of the reports, making this a content analysis approach. Content analysis is a strategy ‘to identify consistencies and meanings’ in the text, with the aim of identifying patterns, themes and categories; the document is viewed as ‘a container of static and unchanging information’. 46 The 30 ERG reports were anonymised and are referred to in this report as numbers 1–30.
Data extraction was conducted by three team members (AB, CC, PF), using forms developed for this project and piloted on two ERG reports (see Appendix 4). The aim of the extraction was to retrieve data on key elements of the MSs, the processes undertaken by ERGs, and the strengths and weaknesses of the MSs. To address these first two issues, data were extracted on key elements, such as the number of trials included, whether or not a meta-analysis or sensitivity analysis (SA) was performed, and whether or not ERGs replicated submissions searches. These findings are presented as descriptive statistics (see Descriptive statistics from the 30 Evidence Review Group reports, below).
Second, data were extracted on the ERGs’ assessment of the strengths and weaknesses of MSs (see Thematic analysis results, below). These data consisted of text, i.e. statements or summaries by the ERG, so an appropriate form of analysis was applied. Thematic analysis was chosen as this method is grounded in the data and permitted the generation of a novel theoretical framework reflecting the ERGs’ assessments of the strengths and weaknesses of MSs to the STA process. 47 This method of synthesis is interpretive and the first stage is data reduction, i.e. to reduce statements, comments, quotations or findings to a single theme, which captures or reflects those data. This process was initially performed by one reviewer (CC) on the extractions of the strengths, weaknesses, key issues and uncertainties sections in the executive summaries of a random sample of 10 ERG reports. This section of the ERG report was chosen for the primary analysis because it should reflect most if not all of an ERG’s comments on the submission that appear elsewhere in the ERG report. If themes identified in this way were considered to be related then they were placed under a broader theme that captured them all; for example, ‘inappropriate analysis’ and ‘issues with data in the analysis’ were both placed within the broader theme of ‘processes’, and ‘population issues’ and ‘comparator issues’ within the broader theme of ‘satisfying objectives’. Definitions were developed for each theme in order to produce greater reliability in the coding of data, especially when this was carried out by more than one reviewer. Two members of the project team (RD and EK) then independently assessed whether these interpretations of the data were both credible and appropriate, and whether the themes identified reflected the data. This led to a small number of revisions: the relabelling of one theme, the reassignment of some data to different themes, and some further clarification of the themes’ definitions.
The remaining textual data in the data extraction forms, i.e. from the strengths, weaknesses, key issues and uncertainties sections of the other 20 ERG reports, as well as all other textual data extracted from all 30 ERG reports relating to the MSs, were then coded using these agreed themes following a process akin to that outlined in framework analysis. 48 This was performed by two reviewers (CC and EK), each working on the data extracted from 15 of the ERG reports.
Results
Descriptive statistics from the 30 Evidence Review Group reports
Description of manufacturers’ submissions
The numbers of pivotal trials included in the MSs are shown in Figure 6. Less than half of MSs included only one pivotal trial.
FIGURE 6.
Number of pivotal trials included by manufacturers.
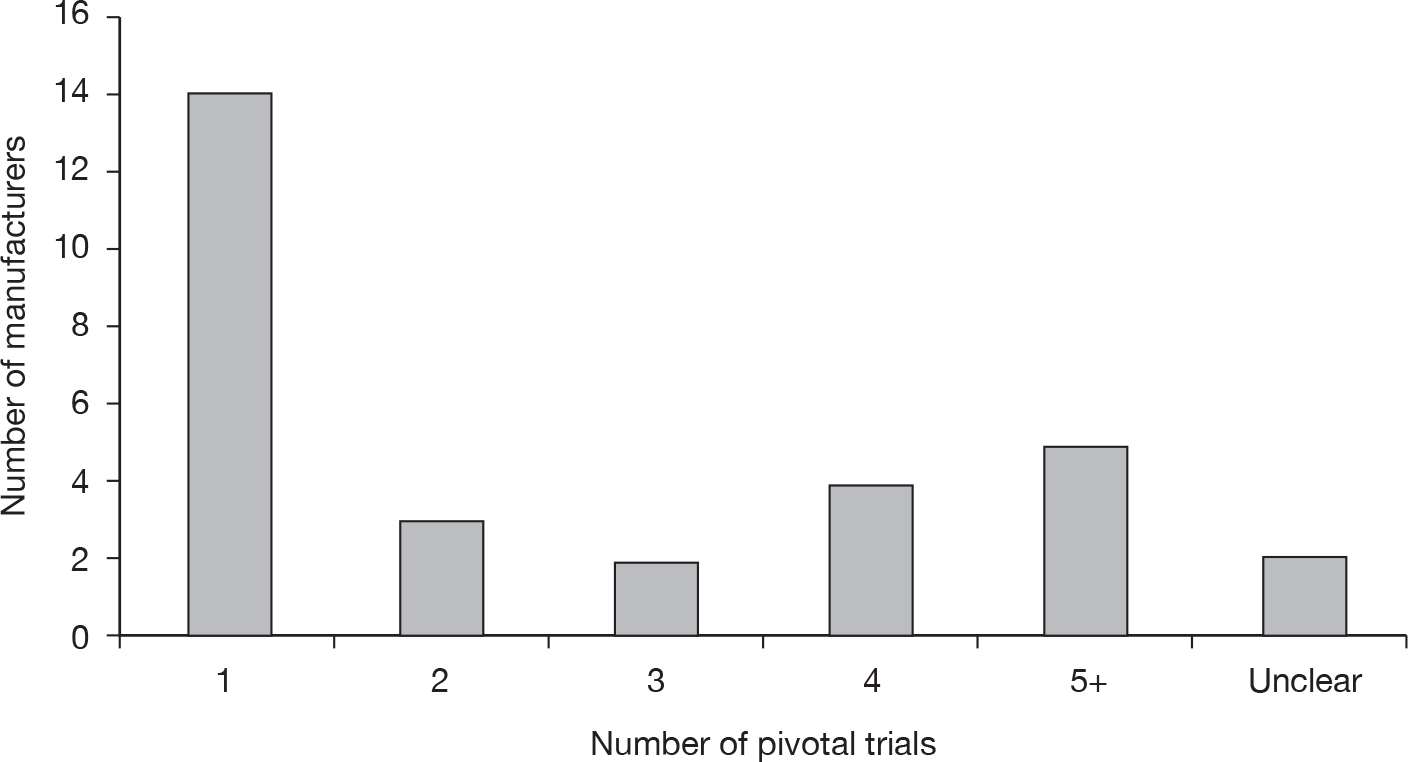
Eight manufacturers (27%) included a meta-analysis in their submission, although as 14 out of 30 submissions included only one pivotal trial then a meta-analysis would not have been appropriate in these submissions. Nineteen manufacturers (63%) undertook an indirect comparison, 29 (97%) undertook SAs and 28 (93%) carried out probabilistic sensitivity analyses (PSAs).
Description of Evidence Review Group approaches to critical appraisal of clinical evidence
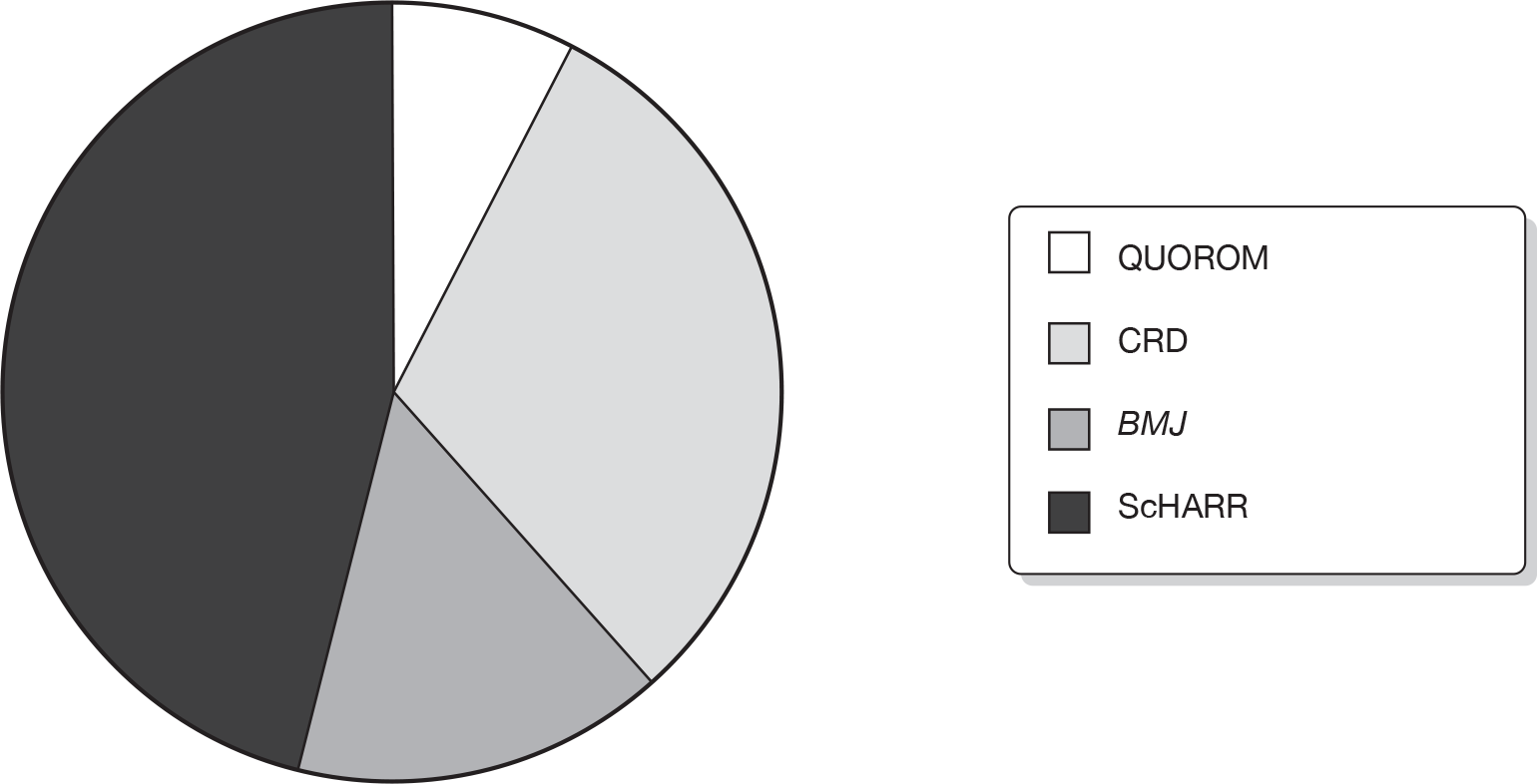
In 26 (87%) ERG reports the team commented on the searches in the MS and 14 (47%) replicated the searches. Methods used to critically appraise the manufacturer’s systematic review were not always clear. Thirteen (43%) ERG reports stated that a published critical appraisal tool was used to assess the quality of the review. These included the QUOROM (quality of reporting of meta-analyses),49 CRD,50 BMJ (British Medical Journal)51 and ScHARR52 tools. Figure 7 shows the tools used in the 13 ERG reports reporting that published tools were used.
FIGURE 7.
Published tool used to appraise quality of manufacturer’s systematic review.
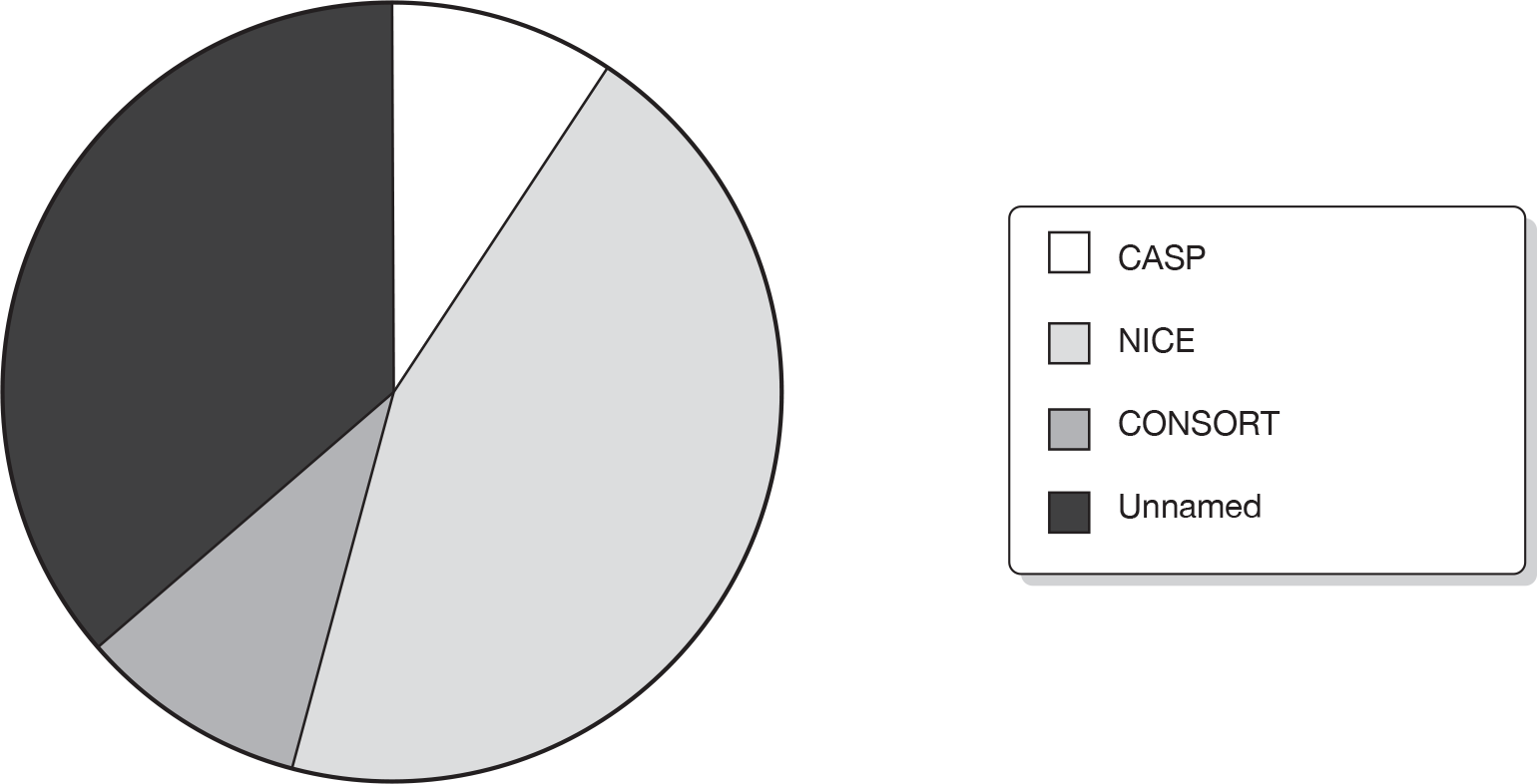
Only 11 (37%) used a published critical appraisal tool to assess the quality of the manufacturer’s included trials. In 12 (40%) methods for assessing the quality of the trials were unclear. Figure 8 shows the tools used in the 11 ERG reports reporting the use of a published tool, which included the CASP (Critical Appraisal Skills Programme),53 NICE1 and CONSORT (consolidated standards of reporting trials)54 checklists.
FIGURE 8.
Tools used in 11 ERG reports to assess trial quality.
In 11 (37%) ERG reports the team undertook additional analysis of the clinical data. Table 6 shows the reported number of clinicians used by the ERGs, either listed as an author or mentioned in the acknowledgement section in the 30 ERG reports.
| No. of clinicians | ERG reports |
|---|---|
| 0 | 1 (3%) |
| 1 | 10 (33%) |
| 2 | 11 (37%) |
| 3 | 1 (3%) |
| 4 or more | 4 (13%) |
| Unclear | 3 (10%) |
Description of Evidence Review Group approaches to critical appraisal of cost-effectiveness evidence
Sixteen (53%) of the ERG reports commented on the manufacturers’ searches for economic evidence and five (17%) replicated the searches. Only two (7%) of the ERG reports stated that a published tool was used to critically appraise the quality of included cost-effectiveness studies. In both cases the tool was the Drummond checklist. 55 Fourteen (47%) reports stated that the MS followed the NICE reference case1 (see Appendix 3). Table 7 shows the reported critique of the MSs in the ERG reports from the point of the view of items in the NICE reference case. Table 8 provides an overview of the elements used in the critique of the submitted model(s) by the ERGs. In the 30 ERG reports included in this study, 23 (77%) reported that extra cost-effectiveness analyses were undertaken. Table 9 shows the frequency of various approaches reported by the ERG teams to assess the models presented in the 30 MSs.
| n | % | |
|---|---|---|
| Perspective | ||
| Yes | 5 | 17 |
| No | 21 | 70 |
| Uncertain | 4 | 13 |
| Time horizon | ||
| Yes | 6 | 20 |
| No | 22 | 73 |
| Uncertain | 2 | 7 |
| Patient population | ||
| Yes | 13 | 43 |
| No | 14 | 47 |
| Uncertain | 3 | 10 |
| Choice of comparators | ||
| Yes | 12 | 40 |
| No | 15 | 50 |
| Uncertain | 3 | 10 |
| Choice of outcomes | ||
| Yes | 5 | 17 |
| No | 24 | 80 |
| uncertain | 1 | 3 |
| Costs | ||
| Yes | 19 | 63 |
| No | 11 | 37 |
| Uncertain | ||
| Alternative consideration of costs | ||
| Yes | 9 | 30 |
| No | 16 | 53 |
| Uncertain | 5 | 17 |
| Benefits | ||
| Yes | 16 | 53 |
| No | 13 | 43 |
| Uncertain | 1 | 3 |
| Alternative consideration of benefits | ||
| Yes | 9 | 30 |
| No | 15 | 50 |
| Uncertain | 6 | 20 |
| n | % | |
|---|---|---|
| Choice of model structure | ||
| Yes | 14 | 47 |
| No | 15 | 50 |
| Uncertain | 1 | 3 |
| Health states | ||
| Yes | 9 | 30 |
| No | 20 | 67 |
| Uncertain | 1 | 3 |
| Sequencing of treatments | ||
| Yes | 4 | 13 |
| No | 9 | 30 |
| Uncertain | 16 | 53 |
| Incorporation of efficacy data | ||
| yes | 20 | 67 |
| no | 6 | 20 |
| uncertain | 4 | 13 |
| Incorporation of adverse events | ||
| Yes | 17 | 57 |
| No | 11 | 37 |
| Uncertain | 2 | 7 |
| Incorporation of survival data | ||
| Yes | 15 | 50 |
| No | 3 | 10 |
| Uncertain | 12 | 40 |
| Incorporation of QoL data | ||
| Yes | 18 | 60 |
| No | 11 | 37 |
| Uncertain | 1 | 3 |
| Mapping of health states | ||
| Yes | 6 | 20 |
| No | 4 | 13 |
| Uncertain | 20 | 67 |
| Replication of trial results | ||
| Yes | 10 | 33 |
| No | 4 | 13 |
| Uncertain | 16 | 53 |
| Type of continuity correction | ||
| Yes | 7 | 23 |
| No | 9 | 30 |
| Uncertain | 14 | 47 |
| Approach to discounting | ||
| Yes | 4 | 13 |
| No | 26 | 87 |
| Uncertain | ||
| Question | Response | Results | |
|---|---|---|---|
| n | % | ||
| Did the ERG comment on the methods used by the manufacturer to determine internal validity? | Yes | 17 | 57 |
| No | 9 | 30 | |
| Unclear/not stated | 4 | 13 | |
| Did the ERG comment on the transparency of the model? | Yes | 16 | 53 |
| No | 8 | 27 | |
| Unclear/not stated | 6 | 20 | |
| Did the ERG comment on the level of detail in the annotation to the model provided by the manufacturer? | Yes | 7 | 23 |
| No | 10 | 33 | |
| Unclear/not stated | 13 | 43 | |
| Did the ERG undertake additional economic analysis? | Yes | 23 | 77 |
| No | 7 | 23 | |
| Did the ERG run the manufacturer’s model with different parameter values? | Yes | 21 | 70 |
| No | 4 | 13 | |
| Unclear/not stated | 5 | 17 | |
| Did the ERG run the manufacturer’s model after making corrections to the model? | Yes | 12 | 40 |
| No | 8 | 27 | |
| Unclear/not stated | 10 | 33 | |
| Did the ERG carry out separate analysis of evidence to augment/replace aspects of the manufacturer’s submitted analysis? | Yes | 10 | 33 |
| No | 14 | 47 | |
| Unclear/not stated | 6 | 20 | |
| Did the ERG estimate revised ICERs as part of their additional analysis? | Yes | 20 | 67 |
| No | 2 | 7 | |
| Unclear/not stated | 8 | 27 | |
| How many economic advisors/economists did the ERG use? | 0 advisors | 20 | 67 |
| 1 advisor | 8 | 27 | |
| 2 advisors | 2 | 7 | |
Thematic analysis results
The thematic analysis generated a large number of themes. However, five broader themes emerged, under which themes related to one another could be meaningfully grouped. These five themes related to the ‘processes’ applied in the MSs; the ‘reporting’ of the submissions review and analysis processes (sometimes strong, sometimes poor); the ‘satisfaction of objectives’ by the submissions; the ‘reliability’ and ‘validity of the submissions’ findings; and the ‘content’ of the submissions (e.g. the amount and quality of evidence contained in the submissions). Numbers in parentheses refer to unique, anonymised identifiers relating to the ERG reports containing data that helped to generate that particular theme. The result was the creation of a framework of a priori themes with associated definitions (Table 10).
| Theme | Definition | Subthemes |
|---|---|---|
| Process | How various methodologies have been applied, e.g. in the performance of the review – searching, screening, extraction, appraisal; in the modelling – in the meta-analysis, in the performance of the review |
Conduct of review Conduct of modelling |
| Was an analysis performed? May include failure to include or perform all necessary analyses (e.g. in a model); inadequate conduct of the review or analysis | No or inadequate analysis | |
| Was the analysis method applied appropriate? May include inappropriate combining of data; inappropriate data being used to populate the model | Inappropriate analysis | |
| Issues concerning data used, especially in the modelling, including costs, parameters and assumptions | Issues with data used in analysis | |
| Reporting | Provision of sufficient or insufficient details about searching, selection, extraction, criteria, analyses performed and their rationale; descriptions and definitions provided in the Background section |
Adequate reporting Inadequate reporting |
| Satisfying objectives | Success or failure to answer question(s) set or for the submission to reflect the decision problem and its scope in terms of the target population, the intervention and its dose, relevant comparators and outcomes, and the NICE base case for the model |
Population issues Intervention issues Comparator issues Outcome issues NICE base case |
| Reliability and validity of findings | Excessive uncertainty surrounding results or model due to lack of evidence, bias within or across included trials or potential exaggerated effect of intervention; explicit concerns regarding validity |
Uncertainty due to absence of evidence Uncertainty due to possible bias or exaggerated effect |
| Findings reported as being reliable (as opposed to uncertain as above) and not uncertain | Reliability of findings | |
| Issues affecting external validity, e.g. specified differences between the trials and data and what exists in the UK, including population, dose, comparator, licensing, real-world/current practice; future developments that may change key parameters | External validity | |
| Content | Weaknesses or strengths inherent in the trial evidence. Amount – concerns issues with number of trials included (e.g. often only one); quality – concerns how good the included evidence is, e.g. very good or very poor? | Amount and quality of evidence |
Processes
The ‘processes’ theme emerged from five subthemes, which were interpretations of ERG comments on the conduct or performance of the reviewing and modelling in the submissions and, in particular, the conduct of the analyses. How well or how badly the manufacturer performed these key processes for the submission was a frequent source of criticism or comment within ERG reports. The five subthemes identified from the data were the conduct of both the review and the modelling processes; the inadequacy or inappropriateness of the analyses performed; and reported concerns with the data that were used in the analyses.
More than half of the ERG reports (17/30) explicitly criticised the conduct of the review, for example the quality of the searching, screening or quality assessment processes (1, 2, 5, 11, 13–17, 20, 27); the definition or application of inclusion criteria, especially for indirect comparisons (5, 7, 13, 15–17, 19, 20); the presence of errors in, or a failure to perform, meta-analyses (5, 16, 19); failure to control for confounders (6, 28) or perform a key subgroup analysis (28); or even the complete absence of a formal systematic review (9, 30). On some occasions, elements of the review process, such as searching and data extraction, prompted positive comments from an ERG (8, 10, 19, 20, 22, 25) or a statement that the review generally was reasonable or good (21, 26).
Evidence Review Group reports rarely commented explicitly positively on the approach taken in the submission to reviewing, modelling or analysis. However, half of all ERG reports (15/30) did pass positive comments on the appropriateness of the structure of the economic model presented or the reasonableness of the modelling approach taken (8, 9, 12, 14, 16–19, 21–23, 25–27, 30), albeit often with some criticism of certain elements of the model. Such weaknesses, highlighted in 20 ERG reports, included a failure to incorporate or capture adequate levels of uncertainty (1, 10, 17); calculation errors in the model requiring revision or correction (2, 3, 25); other technical, structural or design errors in the model or SA (4, 5, 7, 10, 11, 15, 17, 21, 22, 28, 30); and the failure to control for confounders (6) or queries over a key assumption (13), missing data (13, 17, 24, 29) or the absence of a SA (16).
The analyses performed by the manufacturers and reported in the submissions constitute another element of the effectiveness review process that was frequently critiqued by the ERGs. Criticisms of the inadequacies of the analyses presented consisted of the failure to address heterogeneity of trials (5); the failure to perform a relevant analysis, for example a meta-analysis or SA or PSA (6, 7, 16, 18, 19, 21, 23, 27); no validation of the model (6, 13); or the inadequate analysis of safety outcomes (15). However, it was not simply the failure to perform a necessary analysis, but rather the performance of an inappropriate analysis also, that generated criticism in 17 ERG reports, i.e. that the combination, pooling or comparison of effectiveness data was highly questionable (2, 7, 11, 17, 20, 21, 23–25) or that the methods used for the reported analysis (6, 8, 11, 13, 15, 20, 21, 27) or modelling (1, 5, 7, 29) were deemed to be inappropriate.
The other major criticism relating to analyses contained within submissions focused on issues with the data being used, especially the data used in the cost-effectiveness model. Two-thirds of ERG reports (20/30) contained such criticisms. These criticisms related to the efficacy data being used from both direct and indirect comparisons (8, 9, 14, 17, 18, 20, 23, 24); the cost data used (1, 5, 7, 18–20, 30); the utility or QoL data (1, 25, 27, 29, 30); the data on population, comparators, outcomes or various model parameters (5, 6, 8–10, 12, 18, 19, 23, 28); the use of unpublished data (23, 25, 29); and the data being used in the SAs or PSAs (7, 21). By contrast, the data used in the direct and indirect comparisons performed for the effectiveness review generated explicit criticism in only four ERG reports (5, 24, 25, 27).
Reporting
The ‘reporting’ theme was derived from explicit comments made in ERG reports on the quality of the manufacturers’ description of the conduct of both the reviewing and the modelling. Only in a small number of cases did ERGs make explicit, positive comments on the description of the technology or processes within MSs. Nine ERGs commented on the accuracy and adequacy of the background section provided in the submission (6–8, 10, 11, 13, 20, 27, 29), albeit in some cases stating that further information would still have been useful (11, 13, 20, 27). The reporting of the searches for the effectiveness review was praised in four ERG reports (3, 21–23) and the description of the model and its data sources in five ERG reports (21–23, 26, 29). One ERG specifically described good reporting of statistics (1), and another a well-reported multiple treatment comparison process (18).
It was far more common, however, for ERGs to perceive as inadequate the reporting or description of the processes being undertaken or the sources of data being used. A total of 26 out of 30 ERG reports described some form of inadequacy in the reporting of a submission. These included poor descriptions of the searches undertaken, prohibiting replication, for both clinical effectiveness and cost-effectiveness reviews (2, 4–6, 8, 20, 26, 27); an explicit criticism in seven ERG reports regarding the lack of transparency of the clinical review processes generally, for both direct and indirect comparisons (e.g. screening, extraction and quality appraisal) (2, 5, 7, 11, 12, 20, 23); and, quite frequently, in eight ERG reports, inadequate reporting concerning the number of included studies and their characteristics for both clinical effectiveness and cost-effectiveness reviews (2, 4, 18, 19, 22–25). Five ERGs also considered the background section to be inadequate and lacking key information (14–16, 29, 30). The description of the analyses in submissions was also a major source of criticism, i.e. the failure to report data and data sources in full (1, 29, 30); the failure to describe all of the methods being used in direct (5, 20, 24, 27) or indirect comparisons (12, 17–19); the failure to provide adequate descriptions of the analyses more generally (6, 11, 18), and the poor reporting of included studies and data in tables (4, 5, 16). However, the submissions’ reporting of the cost-effectiveness model was the most common criticism by ERGs. Eleven submissions were deemed to be affected by such inadequacies as a failure to describe adequately the parameters or assumptions behind the model, the generation or source of various values, or the impact of bias from missing data (4, 11, 17–22, 27, 28, 30).
Satisfaction of objectives
The ‘satisfaction of objectives’ theme emerged from issues surrounding the relationship between the decision problem and whether, or how far, the elements of the decision problem were satisfactorily addressed in the MSs. The five subthemes identified under this theme relate to the objectives determined by the decision problem, i.e. the population, intervention, comparator, outcomes and the NICE base case.
Ten submissions were not criticised for the relevance of their included trial populations to the populations required by the scope, but 20 out of the 30 ERG reports did raise issues with the trial populations being presented and considered in the submission (1–3, 6, 7, 9–11, 15, 16, 18–22, 25–28, 30). These concerned differences between the population defined in the decision problem and the population being evaluated in the submissions’ trials (3, 9–11, 19, 20, 22, 25–27, 30); differences between the UK population and the trial population based on age, treatment or current practice (1, 2, 6, 7, 15, 27); differences between the licensed population and the trial population (9, 28); a failure to consider the effect of the treatment on different, relevant subgroups of patients (1, 7, 18, 19, 21, 30), including in the model (18); and, finally, problems with the definition of the population in the submission (16, 20, 26, 30), again including in the model (26).
The second major disparity between submissions and the requirements of the decision problem concerned the comparators. Twelve ERG reports found nothing to criticise in this regard, but 18 did raise various issues with the comparators considered in submissions. The principal issue involved a submission’s failure to consider one or more of the comparators designated in the decision problem, or the submission’s use of a combination of comparators not admitted in the decision problem: this was raised in 13 reports (1, 3–5, 7–9, 11, 13, 15, 17, 22, 30). In two cases, this issue was raised for the cost-effectiveness model too (8, 11). Three reports were concerned that the comparator being evaluated was not in use in the UK (6, 15, 26) or that a non-optimal dose of the comparator was being assessed (26), and two reports also identified problems with a lack of definition for the chosen comparator (25, 30). One submission reported a non-optimal treatment duration for the comparator in the model (28) and one conducted a mixed-treatment comparison (MTC) using comparators other than the principal comparator named in the scope (18).
By contrast, few ERGs reported a submission’s failure to satisfy the intervention or outcome elements of the decision problem. Twenty-one ERG reports found no problem with the intervention being evaluated in the submission. In nine instances the actual definition of the intervention was an issue, i.e. the inclusion or failure to include the intervention as combination therapy or monotherapy (1, 16, 18, 29, 30); differences between the trial interventions and UK practice (22, 26); differences between the licensed intervention and the trial intervention (3); and a specific instance where the dose being evaluated was an issue (11). Twenty-one reports (1–7, 9, 10, 12, 13, 18, 19, 21, 23–28, 30) also did not have any criticism to make of the outcomes presented in the submissions. The principal issue in the remaining reports concerned uncertainties regarding the appropriateness of the outcome being measured (8, 11, 14, 20, 22, 29). Otherwise, comments were limited to a lack of clarity on how QoL was being measured (14, 16), the inadequacy of the safety measures presented (15), a failure to report outcomes (17) or concerns about a trial’s lack of power to detect the secondary outcomes being presented (17).
The vast majority of the issues relating to the satisfaction of objectives were raised in the appraisal of the clinical effectiveness evidence. Very few, for example only those relating to the measurement of QoL as an outcome (14, 16), were issues raised principally or exclusively in the cost-effectiveness sections of the reports. In relation to satisfying objectives, the principal focus of the cost-effectiveness section concerned the submission’s failure or otherwise to satisfy the requirements of the NICE base case when developing the model. The majority of submissions appear to have adhered to the NICE base-case scenario and prompted no criticism: only eight ERGs reported either multiple deviations from the base case (23, 29) or specific deviations in relation to either the calculation of utilities (3, 5, 13) or the comparator (17, 20, 27).
Reliability and validity
The ‘reliability and validity’ theme emerged from four subthemes that were interpretations of ERG comments on the robustness or limitations of the submissions’ findings. The four subthemes are uncertainty due to the possibility of bias or an exaggerated estimate of effect; uncertainty due to the absence of evidence; the reliability (rather than uncertainty) of the findings; and external validity, i.e. how far the ERG considered the submission to be externally valid for the intended population and service.
The level of uncertainty surrounding the findings presented by the submissions was a frequent cause of criticism in ERG reports. A total of 27 out of 30 ERG reports (1–13, 15–18, 20–24, 26–30) stressed the presence of bias within the analyses, thus highlighting the lack of certainty surrounding the results presented in the submissions. The principal causes of uncertainty identified by ERGs concerned the possible exaggerated effect of the technology from the analysis (13, 15, 21, 27, 29), especially the relative efficacy of the technology versus relevant comparators (1, 5, 9, 11, 12, 23, 24); the safety of the technology (11); and uncertain levels of risk for different populations (30). These uncertainties within the clinical effectiveness analyses impacted on the models, which were also subject to other biases, such as uncertainties due to issues with the parameters or values in the model (3, 4, 6, 7, 15, 17, 20, 24, 28, 29); an exaggeration or overestimate of benefits or costs (1, 2, 8, 9, 18, 22, 23); excessive extrapolation from the data (2, 9, 16, 26); errors in the model itself (10, 13, 16); and uncertainty due to the model’s high degree of sensitivity to values or assumptions within the model (2, 5, 8, 12, 20, 21).
By contrast, few ERG reports stated that it was the lack of evidence rather than the quality of the evidence or its analysis that generated uncertainty in the submissions’ findings. Only seven ERG reports stated that the available trial evidence was insufficient to generate robust findings on follow-up or treatment duration (3, 4, 14, 16, 28), including survival in the model (1, 14). There was also uncertainty regarding relative efficacy of technologies as no head-to-head trial had been performed (3, 23, 30) or because the findings were based on only a single trial (21). ERG reports did sometimes also explicitly comment that the findings of the effectiveness or cost-effectiveness analyses were strong and reliable, but this was relatively infrequent. In seven reports, ERGs stated that the submission offered a convincing case for the technology (3, 5), an unbiased estimate of treatment effect (17–19) or a fair interpretation of the trial data (26, 28). In terms of the cost-effectiveness analysis, only three ERG reports considered that the submission presented a reasonable estimation of the cost-effectiveness of the technology versus relevant comparators (10), that the modelling of the costs of the technology appeared sound (15), or that the model was superior to previous published models (28).
Finally, the external validity of the submission’s findings was not criticised in 13 ERG reports, but was certainly queried explicitly by 17 ERG teams. These issues principally related to differences between the trial and UK populations (1, 6, 12, 15, 26, 27), including subgroups of UK patients likely to receive the treatment (7); differences between the treatment practices being evaluated in trials and clinical practice in the UK (2, 8, 14, 15, 22, 26) and those in Europe (5); or, simply, differences between the trials and ‘real-world’ clinical practice generally (10). One model was also criticised for not reflecting ‘real-world’ decisions (11), two others for using non-UK sources of data for the model (13, 16) and one ERG reported that the efficacy findings could not be extrapolated to all of the PSA populations (28).
Content
The ERGs often commented on the amount and quality of the trial evidence that formed the basis for the submission, as this affected both the internal and external validity of the submission and its reviews. However, unlike the other themes, this element is not readily addressed by the improved conduct of reviews, analyses, modelling or reporting of the processes used. Nevertheless, it is an element of the submission that attracted comment from ERGs and so is represented in this analysis. The limited amount of relevant trial evidence, for example the fact that the submission may be based on the evaluation of a single randomised controlled trial (RCT), or perhaps two such trials only, was explicitly raised as a point by 10 ERGs (1, 3, 7, 17–19, 21, 22, 26, 28). The absence of any head-to-head trials of relevant technologies was another issue that was sometimes specifically mentioned by ERGs (3, 9, 22). The quality of the included trials was also often commented on by ERGs, as this had implications for the validity of the review, model and submission: included trials, even if there was only one, were more often explicitly reported as being of good or reasonable quality (1, 8–12, 17, 18, 22) than low quality (14). Small sample sizes in trials (22) and limited follow-up (20, 26) were other factors affecting the amount and quality of the evidence that drew comments from ERGs.
Discussion
It is important to note that this analysis is based on what was reported in the ERG reports and may not accurately reflect the work actually undertaken by the teams. If teams did not report undertaking certain critical appraisal methods, then even if they were carried out they would not be reflected in this analysis. The role of the ERG team is to critically appraise the MS, so emphasis is on identifying aspects where there are concerns rather than all the occasions where something has been undertaken appropriately.
Many MSs are based on a single RCT, which means that indirect comparisons are inevitable. This often entails a different review with different criteria, although this was rarely carried out by manufacturers, which usually only attempt a standard systematic review based on direct trial evidence only. The ERGs do not always clearly report what processes they are using or applying, and these can differ between STAs. They do not use the same tools for critical appraisal and do not always address clearly whether or not the MS has satisfied the decision problem and addressed the licensing issues. It may be unavoidable for manufacturers to include trials with data that do not satisfy the decision problem, but this should be acknowledged.
More than half of the ERG reports explicitly criticised the conduct of the systematic review within the MS. However, half of all ERG reports did pass positive comments on the appropriateness of the structure of the economic model presented or the reasonableness of the modelling. Criticisms covered both failure to perform a necessary analysis and the performance of an inappropriate analysis. The issues identified are similar to those identified by Hill et al. 56 a decade ago when they reviewed the economic analysis submitted as part of the appraisal process in Australia. Two-thirds of ERG reports contained criticisms related to the data being used, especially the data used in the cost-effectiveness model. There were major issues regarding poor reporting of processes used in MSs. These included poor descriptions of searching and lack of transparency in the description of the processes used for both the clinical effectiveness and the cost-effectiveness sections of the MSs, with reporting of the cost-effectiveness model being the most common criticism by ERGs. The population and comparator represented the key items in the decision problem that the ERGs assessed as being either poorly or completely inadequately addressed by manufacturers. Some of these themes relate either directly or indirectly to one another. An ERG’s assessment of the deficiencies in the processes performed in a submission, for example the failure to perform an analysis, the presence of bias within analyses, or issues with the data used, directly impacted also on its assessment of the reliability and validity of the findings of the submission, another key theme. The failure of submissions to address or satisfy the objectives as outlined in the decision problem directly informed ERGs’ assessments of the external validity of the findings as they related to clinical practice in the UK. This suggests that if a submission addresses issues relating to the processes being used, and presents clear rationale for the choices being made and unavoidable limitations of evidence or analysis then ERG assessments of the reliability and validity of the submission’s findings will be positively affected also.
Chapter 4 Clarification letters
Methods
Clarification letters were, in the first instance, identified from the NICE website. Letters from a total of 17 of the 30 STAs used in the documentary analysis exercise were identified. A request was made through NETSCC to NICE for copies of the remaining letters and a further five were received. Three were provided by NETSCC in the form of ERG reports, where the letters and responses had been embedded in the document. In one of these reports the data were not easily extractable. Two other clarification letters were provided by NICE. A decision was taken to analyse the available 21 letters.
Letters were downloaded into ATLASti® version 6 (Scientific Software Development GmbH), a software package used for qualitative data analysis. This software allowed for the sorting of specific sections of the clarification letters into prespecified coded categories. A set of open codes was established for the categorisation of the data. These are presented in Table 11 with brief definitions. The establishment of these codes evolved from a primary list drawn up and revised by the reviewer during pilot data extraction, using three random clarification letters. Data coding was conducted by one person (RD).
| Initial coding | ||
|---|---|---|
| Clinical | General | Any general query |
| Explanation | Any request for clarification of clinical data | |
| Additional data | Requests for additional clinical data | |
| Additional analyses | Request for additional clinical data analysis | |
| Economic | General | General economic topic issues |
| Explanation | Any request for clarification of economic data | |
| Additional data | Requests for additional data | |
| Additional analyses | Request for additional economic data analysis | |
| Indirect comparisons/MTC | Any clarification requested regarding these analyses | |
| Licensing | Any issues related to the license or change in license | |
| Systematic review methods | Any issues related to the conduct of the systematic review requiring clarification | |
| Logistics/report quality | Any issues related to the STA process or the quality of the MSs | |
There were almost equal numbers of clarification points in the clinical and economic sections. One reviewer (RD) examined the clinical points as well as issues related to report quality. These were then grouped into three categories: report quality, systematic review methods and clinical data. After initial coding, the economic clarification points were examined and categorised by two health economists (PF and AB). One health economist (PF) went on to regroup and categorise the clarification points into a matrix of three new categories: clarification/explanation, data/method and further analysis. Examples of all of these categories are presented here.
Results
The data are presented here in relation to four categories (report quality, systematic review methods, clinical and economic) and include > 400 clarification points. The purpose of this analysis is to provide an overview of the key issues raised by the ERGs and not to provide quantitative analysis. Samples of actual requests are included in each section. A substantive number of examples are provided as it is felt that they represent the range of issues raised by the ERGs.
Report quality
General issues related to the overall quality of the MSs as well as issues related to errors and general inconsistencies within the submissions are included within this category. The first point indicates that the standard of reporting required improvement, while the second indicates that submissions were at times lacking basic information:
The text of the document contains an over abundance of errors and does not appear to have been copy edited. Formatting of references is inconsistent in the text and in the reference list with some incomplete references.
Please provide a simple list of all the studies used in the submission, a list of outcomes evaluated for each study. Also, please provide the characteristics and quality assessment of the following trials …
Linked to report quality but somewhat different were issues that were raised related to the presentation of data in the submissions. Clarifications included examples of specific copy-editing details as well as internal consistencies:
Table 5.9 the numbers for the patients in the *** and *** groups should be 42 and 45 respectively as per Table 5.3.2, and not 141 and 145 respectively – we presume this is a typographical error carried over from Table 5.8, please clarify.
Not all numbers are consistent with the published study, please check and revise flowchart or explain.
Table 7 on page 49 of the submission appears to be incorrect. The assessment group were particularly interested in the numbers for *** involvement that do not sum up across the rows. In addition many of the totals in individual rows do not sum up to the number at the top of the column? Is there a reason for this? Please provide us with the correct figure.
Please explain why secondary endpoints reported here do not correspond with the secondary endpoints as defined in Tables 15 and 16 previously.
Systematic review methods
Comments related to review methods were categorised into issues related to searching and trial flow, decisions regarding inclusion criteria and general methods issues. Examples of each are presented here. The comments indicate that the submissions frequently failed to report the use of robust methods for the conduct of their systematic reviews of the literature:
Please provide a QUOROM flow diagram or diagrams for the various searches (i.e. those for RCTs, observational studies, systematic reviews and evaluations). [Note: a variation of this request was seen in a further five letters. ]
Please perform a search for trials involving the submission’s stated comparators (*** and ***) and report the results of that search.
The methods and results of the economic searches are unclear. Please can you provide further details on the economic searches undertaken (e.g. summary table of number of identified studies by database; summary of the inclusion and exclusion terms used). Please can you confirm that all of the papers identified (n = 73) could have been identified by searching NHS EED and HEED only.
Please provide a description of the processes undertaken in applying the inclusion and exclusion criteria, the data extraction and the quality assessment of the trials.
Please explain the processes by which this trial was identified, and how any other studies were identified and excluded.
Please provide, a simple list of all the studies used in the submission, a list of outcomes evaluated for each study. Also, please provide the characteristics and quality assessment of the following trials …
Provide further detail of trial flow and baseline characteristics of groups in the included studies. Identify and discuss any differences within studies and respond to all critical appraisal items included in the submission template.
Clinical data analysis
Clarification points relating to specific clinical issues are discussed in relation to general or explanatory queries (including specific clinical questions), requests for additional clinical data or additional data analysis and, importantly, requests for the provision of rationale for the analysis that had been undertaken.
General queries
As would be expected, there were instances when there was a need to clarify the specific data presented in the submission:
Table 5.7.1 – provide a definition of (any) severe adverse event.
Please be explicit that the **** trial is currently published as an abstract only. This is important since a great deal of the data reported to be from this study are marked confidential in the submission and cannot be verified with reference to a published paper.
Please explain the differences, if any, between the populations receiving the different doses of *** (Table 12).
Please clarify how the **** risk reduction values used in the model were derived from the *** trial. It does not seem as though they were drawn from the published paper.
Please provide a list of the 18 RCTs (section 5.2.1, page 17) which were excluded because the population was not aligned with the EU/UK licence; with the specific reason (s) for exclusion for each one.
The values of n change for the different analyses due to withdrawals, crossovers and pooling of different studies (as highlighted in Table 1). Not only are the reasons for these differences complex there are occasions where it is not possible to determine why certain patients were included and excluded e.g. the long term efficacy following repeated treatments.
As can be seen from these examples, the lack of information or inconsistency of the provided data would limit the ability of the ERG to move forward with assessment of the MS.
In addition to these general queries, there were three STA clarification letters that included specific questions requiring clinical expertise and related to issues of patient pathway/response and drug dosage:
Please clarify if a switch from *** to *** is expected to happen in clinical practice and provide a rationale for why a sequential approach of prescribing *** to all patients and then *** to patients who do not respond was not considered.
The diagnostic criteria for *** have not been made clear in the submission. Clear diagnostic criteria are required to identify the patient population and give an indication of incidence – is this available from the manufacturer?
Based on the figures presented in the ‘validation’ sheet of the submitted model it can be seen that the patients in the comparator arm in the USA trials have a better prognosis than the patients in the comparator arm from the *** study (84.8% in the joint US analysis compared with 77.4% from the *** trial). A more detailed discussion of this issue and why these differences exist would be most useful to the Committee in its discussions.
Please clarify whether there is evidence for no dose adjustment in the elderly? Reference 9 appears to be based on clinical opinion rather than trial evidence.
Please clarify in a clinical context why within the model the dosing of *** is allowed to be altered (letting patients move from 80 mg to 120 mg) but the dose of *** remains constant.
Additional clinical data
In addition to requests for additional data, there were indications from the clarification points that there were data that the ERG might have expected to see in the submission but which were omitted from the MS (e.g. selective reporting):
In the *** trial, the most common adverse event leading to withdrawal was abnormal liver-function test results which accounted for withdrawal of 5 patients … In addition, more patients receiving *** (80 or 120 mg/d) discontinued the study because of rashes. It is not clear, why this has not been reported in the STA submission.
Only data for the last 4 weeks of the *** and *** trials have been reported (Figures 5–6 do not report n/N at each time point). It is noteworthy, that in the *** trial, nearly 70% of patients in the *** groups experienced *** flares after discontinuing prophylaxis i.e. between week 9 and 52 (no data reported for *** trial from week 1 to 24).
Please provide further detail of discontinuation rates for the *** trial? It is unclear why this was not reported in the submission. Please provide a table with baseline characteristics for the trials other than ***. Section 2.3.2 only provides baseline characteristics for ***. Although it is appreciated that only limited information may be available for ***, NEJM [New England Journal of Medicine] articles are available for ***, *** and ***.
Present response results (as in Table 5.6.3 and including change in *** scores) for the 12 week trials excluded from the cost-effectiveness study reported in Table 5.6.2.
Please provide the response data that are entered into the model, including the adjustment for the differing skin involvement in the patient populations of the trials.
Please provide appropriate data in table format; section 5.4.5 (secondary endpoints: proportion of patients requiring treatment for ****) has been poorly reported with important omissions. It appears that limited/selective data have been reported, particularly on the number of patients that experience **** from day 1 to end of study.
Selective reporting has been recognised as an important issue in relation to clinical trial reports and the MSs demonstrated that the ERGs had identified instances of such reporting. 57
Additional clinical analysis
These clarification points represent examples of requests for additional clinical analysis. As can be seen, a number of these requests would require a considerable amount of additional analysis to be carried out by the manufacturer:
Please provide the additional analyses of clinical trial data relating to the table of assumptions (on page 109) about scheduling of response rates.
Please provide working Excel spreadsheet which describes how investigator-assessed response rates were pooled for use in the SA reported in table. In particular, please provide a copy of time-to-event analyses for overall survival and time-to-disease progression in trials.
Please provide a full indirect comparison, including methodology, between *** and ***/***. If this is not available, please provide pairwise comparisons (including associated Confidence Intervals) for: weight loss; waist circumference; change from baseline in BMI; and the proportion with 5% and 10% weight loss.
Could you provide a meta-analysis of the safety and efficacy evidence (as noted in guidance section 5.5) using individual patient level data or aggregate data for all outcomes of interest and not just significant results?
Both fixed effect and random effects model based results are required for all analyses. [Requests of this kind were seen in five letters.]
Please undertake a full synthesis of PsARC response and other outcome parameters at 12 weeks for ***, *** and *** using data from all available studies (including both 12 and 24 week studies).
Please provide a subgroup analysis of the co-primary results.
Please provide statistical evaluations of heterogeneity for the studies from which absolute efficacy estimates were pooled.
Please provide either forest plots, or the results of the tests for heterogeneity, for all the pooled results in Table 20.
Analysis decisions/rationale
The majority of issues in this category related to requests for information explaining how analyses were carried out or why various decisions were taken in relation to the analysis that was presented in the submission:
Please provide additional clarification on the approach used to estimate separate treatment effects for each subgroup (main subgroups and those considered in the sensitivity analyses). In particular, clarify whether the data was from the trials was simply split into the different subgroups or whether formal statistical interactions were estimated and employed.
Please provide a rationale for presenting the pooled analysis of individual patient data from the *** and *** trial and discuss the methodological limitations of doing so.
Expand description of the mixed treatment comparison (MTC). Specify which studies have been included and explain process for estimation of adjusted indirect comparison.
Please explain the process of ‘estimation by adjusted indirect comparison’ used to generate RRs for *** and extended *** versus nil in the single intervention meta-analyses, and why no adjustment was possible for ***.
Please provide reasons why you have not considered using a ‘multiple treatment comparison’ approach to answering all the comparisons presented in your decision problem.
Please provide a more descriptive explanation of the method of indirect comparison used to compare *** with *** and provide a critique of the pros and cons of this approach.
Please provide more detail about how and why the adverse event data for *** was pooled (page 89).
Please provide further rationale for the use of last observation carried forward to account for missing data and provide an alternative analysis of the clinical data using a best case/worst case scenario.
Please provide a rationale for not undertaking any subgroup analyses when in this disease’s context specific subgroups may imply different disease management (e.g. patients with renal impairment) and eventually require different clinical endpoints (e.g. patients with **** requiring a quicker reduction in sUA levels) from the overall population.
Provide pooled analyses for primary endpoints and safety data from studies, or an explanation for not doing so. Include analysis of bleeding outcomes. Full description of methodology should be provided.
Currently, pooled analyses have only been performed on the secondary efficacy endpoint and no explanation has been given for not performing this analysis on the primary efficacy endpoint (or safety endpoints).
Please clarify which analyses used IPD data, whether data relating to subgroups was obtained from different trials, and which trials contributed to which outcomes.
As can be seen the requests range across a number of issues including, subgroup analysis, methods for indirect and MT comparisons, endpoints and outcomes.
Economic data
Clarification/explanations
As would be expected, this is the largest category and spans all aspects of the submissions. In terms of clarification of calculations, the queries ranged from requests for simple explanations of the sources of data to more complex explanations that were related to estimates used in the model:
Please provide a step by step explanation of how you got from the initial figure (–0.15) to a treatment effect of –7.32% and why you did not consider the adjusted figure in the same document (–0.21) appropriate for use.
Please clarify the methodology for obtaining the CEACs. Traditionally this would not have more than 3 dp as only 1,000 simulations were provided, whereas the figure reported for being less than £20k at 24 months is 0.6234.
Please clarify whether cause-elimination approaches were used to estimate these mortality estimates.
Please explain whether the following analysis does not constitute double counting? Cell J54 on the ‘Outputs sheet’ already contains the first 5 years of utility. In cell L31 lifetime and 0–5 years are added together.
Other queries highlighted inconsistencies between the clinical and economic sections of the submission as well as inconsistencies within the economic sections:
Why are the inclusion criteria, namely the duration of study, different between the clinical (no restriction) and cost (minimum 12 weeks) section?
The odds ratio for death at 6 months following *** event is taken from a meta analysis that includes the *** 1993 study. This study was excluded in the clinical effectiveness section. Please explain why this study has been included in the economics.
Please provide clarification on Table 6.4 in the context of the model. The mean data for *** is stated in the table as 0.2016, but is in the model as 0.2108. The distribution from the raw data is also unlikely to be normal. Clarify that the *** data is also correct within the model.
Clarification issues also included queries related to the sources of data and their use within the economic analysis:
Please provide clarification on the data and sources for these assumptions and how these compare with epidemiological studies of *** levels in patients treated for ***.
Please confirm that survival analyses for 2-arm model use only data from time of second randomisation. Was the same truncation point (1500 days from first randomisation) used for these analyses as for the 4-arm model?
Please explain further how you used the above information to ‘identify the most likely distribution of responses’ for the rest of treatment options, and how you sample the type of response for the next 5 timepoint from the joint distribution of ***, *** and ***.
Justify combining of minor bleed and clinically relevant bleed in the economic evaluation.
Please justify the selection of an initial 6-month cycle when the majority of trial outcomes data relates to 90 days results.
In addition, and similar to the clinical section, there were queries related to why explicit clinical and modelling decisions were taken:
Please provide further rationale for the use of your choice of comparator considering this argument.
Please provide justification for excluding strategies in which the drug or comparator drug is discontinued if a specific target is not achieved.
Please explain why your indirect comparison data is not used in the model.
Please provide a rationale for the inclusion or exclusion of variables used in the PSA.
Please clarify the clinical justifications of transition probabilities from the ‘Flare d/t resistance’ health state to other states (Response, CHB Resist Salvage Tx, CHB no TX, etc.).
Data/methods
Data queries were broad and, as can be seen from the examples, the majority of the requests were critical to the ERG as they attempted to appraise the submission:
Please provide the absolute event numbers on which these transition probabilities are based and the details of the methodology used to obtain the probabilities.
Please make clear where in the model (which cells) were used to adjust the utility for surgery in the first year.
Please define what counts as an event (e.g. first or subsequent? Local, regional, distant recurrence, contralateral?) for each clinical outcome used in the model, including whether death without breast cancer recurrence, or the diagnosis of a non-related cancer counts as an event.
Please provide further clarification of the meaning of the adverse event rates used in the model (for example does the adverse event data refer to the number of events, or the number of patients for whom any event occurred at any time).
There is no information in the submission concerning the timing of, and reason for, withdrawals in the two arms of the trial. The ERG request a detailed table of withdrawals by week and by reason (serious AE, reaction, clinical advice, patient request, lack of efficacy, etc.).
Further analysis
The queries raised in this section highlight some of the additional analyses that were requested by the ERGs. The most commonly requested additional analysis related to SA around various parameters. One example requested four such analyses, considering cost, outcomes, time horizon and confidence intervals, and demonstrates that important analyses were missing from the original submission:
Please provide additional analysis including:
increased drug cost including cost of administration to £25,000 and £30,000
improving/reducing the survival estimates in the comparator arm
sensitivity analysis around extrapolation of benefits of *** beyond the trial period, by setting the relative risk of *** equal to 1 after (a) 10 years (b) 5 years
widening the confidence intervals around RFS, DDFS and RFM curves over time to reflect uncertainty in these measures.
Other examples include:
Re-run the base case analysis based on a complete synthesis of all 12-week *** data for all comparators (based on all trials of at least 12 weeks duration).
Re-run the base case analysis without the ‘conventional ***s’ drug cost.
Could you provide SA on the extension of the first phase of the model up to 6 months, noting that *** Guidelines state that initial prophylactic treatment can go up to 6 months.
Please provide a SA on the effect of vial sharing on the cost-effectiveness.
Specific requests for cost-effectiveness analysis indicate that there may have been cases when basic information/analysis was missing from the original submission, as indicated by the requested information in the following examples from a single clarification letter:
Please present summary cost-effectiveness results for the base-case populations and the populations considered in the SA.
We request that the cost-effectiveness results are also presented based on this re-run synthesis. We would be grateful if ***would replicate Tables B1.2, B1.3, B1.4, B1.5 and C4.4 for this analysis.
There are examples where the ERG requested a different modelling/analysis approach:
Please provide a simultaneous comparison of *** (lifetime treatment duration), *** (lifetime treatment duration), *** (1-year treatment duration) and diet/exercise as opposed to separate pairwise comparisons.
Please assess the use of relative risks compared to odds ratios on the central estimates and SA results.
Provide incremental cost-effectiveness ratios (ICERs) using both random effects and fixed effect results.
A mean of 307 days is quoted in the submission for the time between doses of ***. This is used to estimate the cost per treatment. The ERG request a detailed table AND Kaplan Meier analysis of the time to second treatment in the intervention arm of the trial, censoring all treatment terminations. We also request the provision of total numbers of first and second treatments given in the intervention arm of the trial.
Please perform a full PSA across a wider range of parameters, including as a minimum all of those which are varied in the one- and multi-way sensitivity analyses. Please state the number of iterations performed.
In addition, present the associated cost-effectiveness acceptability curves and report the probabilities that each intervention is cost-effective at a threshold of £20,000, £30,000 and £40,000 per QALY for the base-case populations.
Other clarification letters requested analyses to be carried out related to different subgroups of patients:
Please present summary cost-effectiveness results for the base-case populations and the populations considered in the SA.
Please consider subgroup analyses for patients in different OTT intervals (e.g. 0–90 and 91–180 minutes).
Please provide separate incremental cost-effectiveness ratios for patients with recurrent cancer and for those patients with metastatic cancer
There are other interesting queries that raise the question of what data were used in the original submission to populate the economic model:
Please provide additional cost-effectiveness results showing SA with utilities derived from the clinical trial data.
Discussion
Although access to data for the mapping exercise was relatively straightforward, this did not prove to be the case when attempts were made to locate the clarification letters. Some sort of signposting to where various documents are filed on the NICE website and consistency in placement would have made the task simpler.
It is unlikely that any MS could ever include all of the information required by the ERG and the NICE appraisal process, and we can therefore expect that the clarification process will remain in place. However, receipt of comprehensive, well-structured and clearly written submissions has the potential to decrease the number of clarification questions that arise.
Analysis of the clinical and economic clarification points was conducted in somewhat different ways but identified very similar issues. After examining 21 clarification letters it is easy to understand why manufacturers involved in the STA process have been known to claim that they are ‘dying the death of a thousand pinpricks’ when they receive the clarification letter (personal communication from a manufacturer representative who wished to remain anonymous). The current system does not formally include an opportunity to provide positive feedback on MSs so it is difficult to see how this can be resolved. Although there has been standardisation of the format of the clarification letters, there is variation in the comments that are included and, as would be expected, the amount of work that is required of the manufacturer to address them.
It is not unreasonable for NICE and the ERGs to expect to receive MSs that are comprehensive, clearly written, internally consistent and appropriately referenced. Analysis of the clarification letters demonstrates that this is not always the case. In addition, internal inconsistencies in the document are a particular problem as, until they are clarified, the ERG may be unable to move forward with the analysis of the conflicting information.
There are internationally accepted standards that include recommendations for the conduct and reporting of systematic reviews of clinical effectiveness and cost-effectiveness evidence, and NICE has a methods guide. 1 In cases where there are multiple trials, then the systematic review should adhere to accepted research standards for its conduct and reporting. The clarification issues raised in the sampled letters demonstrate that this frequently is not the case and, in the extreme, requests such as those asking the manufacturer to conduct a search using the names of the treatments being assessed demonstrate that submission authors are not always using these standards.
A further issue is related to when it might be appropriate to conduct and report additional searches of the literature. This is not just an issue for STAs but a methodological question regarding all reviews of clinical effectiveness and cost-effectiveness evidence. As noted above, the current STA template requires a systematic review of the literature for the primary research question when there is more than one study. However, a number of points in the clarification letters reflect the need to apply these robust methods when identifying studies used to inform indirect comparisons and utilities. The new MS template does provide some guidance to manufacturers as to the preferred presentation of methods and results when reporting indirect comparisons.
Analysis of the clarification letters revealed that requests for further clinical and economic analyses varied. Such requests could indicate that insufficient data were provided in the submission to allow the ERG to conduct the analysis, or that the ERG believed that such analyses were the responsibility of the manufacturer. It is not possible to tell which from the clarification letters. The majority of requests were for additional meta-analysis (using fixed or random effects) or inclusion of different subgroups or asked for provision of rationale related to the analysis that had been conducted. It is clear from the clarification points that often the ERGs considered that alternative strategies might have been more appropriately used by manufacturers and that sufficient rationale for the methods used was not provided.
There is no mechanism in place to provide positive feedback to manufacturers, which makes the ERG comments appear to be very critical. The clarification points identify that key information is missing from the submissions examined and although not directly examined it is clear that to address the clarification points would require a significant amount of time.
It needs to be acknowledged that the MS template for submission does little to assist in the production of a smooth-flowing report, and in fact the template itself calls for significant repetition of data. The new (January 2010) MS template is even longer (75 pages in total) and includes headings for all possible data that might need to be considered by the ERG and the AC. A number of recent submissions have included all the headings and guidance from the template to indicate that all of the data requests have been considered, or perhaps to make a point about the template.
The NICE recommended report length limit of 100 pages could be considered an impediment to the production of a comprehensive submission. In fact, some have said that being able to produce a 100-page document when the template is 75 pages is inconsistent. However, the fact is that no submission has ever been rejected on the basis of length, although recently a manufacturer was asked to shorten a report that was > 400 pages plus appendices.
It would be worth considering renaming the current MS template and considering it more as a checklist for manufacturers to cross-check that completed reports contain all the information required in the submission. This has the potential to shorten reports without the loss of critical information.
Chapter 5 Discussion
Statement of principal findings
Ninety-five STAs were included in the mapping exercise. Nearly all necessary information for this mapping exercise was available on the NICE website, but access to clarification letters was somewhat more problematic. The majority of STAs were not completed within the agreed STA process recommended timelines. ICERs reported by manufacturers were consistently lower than those estimated by the ERGs. Overall, 32% of referred topics for appraisal were either suspended or terminated, the majority due to issues related to regulations or manufacturers’ decisions to decline the invitation to submit. An appeal was undertaken in 22% of the completed STAs against a ‘no’ decision. The introduction of PAS has the potential to extend the length of the STA process further.
Thematic analysis was performed on data extracted from the first 30 completed ERG reports. This approach offered a means of clarifying and describing the strengths and weaknesses of MSs and generated the five themes of ‘process’, ‘reporting’, ‘satisfaction of objectives’, ‘reliability and validity of findings’ and ‘content’. ‘Process’ concerned how the various relevant methodologies had been applied in the performance of the review, analyses or modelling; ‘reporting’ how well these processes had been described or justified in the submissions; ‘satisfaction of objectives’ how far the submission complied with or addressed the scope or decision problem; and ‘reliability and validity’ how far the findings of the submission were affected by uncertainty or bias.
These themes were inter-related. The adequacy of reporting in the submission influenced the assessment of the processes being conducted, and both the content and the conduct of the review or modelling directly affected the reliability and validity of a submission’s findings. In the same way, a submission’s success or failure to address the objectives set by the scope or decision problem affected the external validity of the submission.
The STA process may be improved if manufacturers more carefully address the following issues in their submissions: transparency in the reporting, conduct and justification of review and modelling processes and analyses, as well as choice of data, and either adhering to or addressing differences from the scope or decision problem. These changes might substantially reduce ERGs’ criticisms in the assessment of submissions. The analysis conducted here did not consider whether differences in approach by different ERGs actually adversely affect outcomes, i.e. to what extent discrepancies between the processes conducted by different ERGs have an impact on the decisions or decision-making processes of ACs. It may be that such discrepancies are of no consequence and autonomy and flexibility within the basic requirements should be permitted to ERGs. It may not even be an appropriate consideration as the wide variation in MSs necessitates some variation in approach on the part of the ERGs. However, the existence of a more strictly defined appraisal process for ERGs with established criteria, appraisal tools, etc. might act as a clear guide and benchmark for manufacturers and enhance the quality of submissions, and thus facilitate the STA process as a whole.
In terms of this project it was assumed that examination of clarification letters would assist in identifying the context of the clarifications that had been requested and thus allow us to determine if there were any consistencies across the 30 included STAs being examined in detail. The response to these letters is not part of this analysis. Analysis of the clarification letters has identified a number of consistent areas where MSs could be improved and in so doing may lead to a decrease in the number of clarifications and additional analyses requested by ERGs. Recommendations for the improvement of MSs are provided below (see Recommendations for manufacturers) and manufacturers may choose to cross-check these prior to forwarding their submissions to NICE.
Strengths and limitations of the assessment
This report presents the first independent mapping exercise of the NICE STA process to date. Nearly all data were obtained from the NICE website; therefore, any errors in the data on the website will be reflected in the mapping analysis presented in this report. Missing data for the mapping exercise do limit the generalisability of the findings. As many of the STAs included in the mapping exercise were still ongoing at the time of data extraction (August 2009) and even ‘completed’ STAs have been changed since then, this report presents a ‘snapshot’ of data available at that time. The mapping exercise covered the first 95 STAs only and may therefore not be an accurate reflection of what currently happens with STAs.
The thematic analysis of the ERG reports used validated methods and multiple reviewers to check and verify analysis. Only 30 ERG reports were included in the thematic framework analysis due to time constraints. These were the first 30 STAs and critical appraisal methods used by the ERG teams may have developed over time and may not be accounted for in these analyses. There may be some misinterpretations in these analyses, as only documentary analysis was used to collect data. As ERG teams gain more experience in developing their reports and build up their expertise, they may use different methods. Analysis is limited to what is reported in the ERG report. Much of the ERG work for critiquing the MS is not presented in the report itself and therefore to establish current approaches would need a much deeper investigation than was possible for this report. We were therefore not able to provide an in-depth analysis of the critical appraisal approaches used by the ERG teams and identify those most useful to the ACs. This issue is a key recommendation for further research.
Only 21 clarification letters were examined in this analysis. The analysis here has been pragmatic; however, the approach provides an overview of the commonalities of the queries put forward in the clarification letters and, as such, provides direction for the ERGs, NICE and the manufacturers. Scoping workshops are now part of the STA process and may have a bearing on how manufacturers produce their submissions. Clarification letters are currently developed using a more structured approach. Recently, there have been changes to the MS template. All of these changes may impact on the issues presented in current clarification letters, which will not be reflected in this analysis.
Uncertainties
The whole STA process is very time constrained and some variation in the critical appraisal approaches undertaken by the ERGs is inevitable when time is limited and the remit of the ERG is not well defined. The approach taken by the manufacturer in its submission will have important implications for the approaches taken by the ERG team. The role of the ERG has extended over time, in that attendance at scoping workshops is now compulsory and greater time is allocated to work post ACD. There is still little guidance provided to the ERGs regarding expectations for their work. This lack of clarity as to the objectives of the ERG critical appraisal and tasks explains some of the variation in approaches. More clarity as to requirements and processes is needed, which will, in turn, help to standardise ERG team approaches to the critical appraisal of the MS. It is also uncertain whether or not the issues dealt with in this report are the same issues that are of concern to the ACs, an issue highlighted for further research.
The manufacturers’ STA template was revised in January 2010 and may have some impact on the quality of future submissions. Scoping workshops have been ongoing for several years now and may have an impact on the clarity of the scope that the MS needs to address. Finally, methodological work undertaken by NICE may assist manufacturers and ERG teams with issues such as approaches used for indirect comparisons.
Evidence Review Group report template
The current ERG report template is attached as Appendix 1. Modifications were made to the template based on recommendations from reviewers and modellers from the LRiG and ScHARR. These included removal of some sections to avoid repetition and the inclusion of a section to critique any indirect comparisons undertaken by the manufacturer. The other five InterTASC teams were invited to comment. All comments were incorporated into a revised template (see Appendix 5). This will be trialled for a period of 6 months (January–June 2011), when the template will be used to develop all ERG reports written during this time period. Consultations will then be undertaken with the seven teams along with representatives from NICE at an InterTASC meeting to determine what if any further modifications are necessary. As currently happens, it would be expected that teams modify the template as appropriate for each STA and that the template will serve as more of a checklist rather than be strictly adhered to by the ERGs.
Recommendations
Single technology appraisals vary considerably due to differences in technologies and approaches used by manufacturers in their submission. Below are suggestions for ERG teams, manufacturers and NICE to ensure that the process is as transparent and effective as possible. These recommendations are based on the findings from Chapters 2–4 of this report, as well as discussions held at the InterTASC workshop held in April 2010. At this workshop experienced researchers who have been involved in the STA process for several years made recommendations for undertaking STAs.
Recommendations for Evidence Review Group teams
-
The reporting in ERG reports should be clear and transparent.
-
The ERG needs to anticipate the needs of the AC prior to undertaking their critical appraisal and development of the ERG report.
-
The focus of the ERG report should be on the key issues in the systematic review and model.
-
The role of the ERG is to point out what is missing and what the problems are with the MS.
-
Prioritisation of tasks for critical appraisal is crucial. ERGs should focus on identifying the key parameters of the economic model that drive the decision regarding cost-effectiveness, and then focus their critical appraisal on these aspects.
-
Clinicians may be helpful with identifying any key trials that may be missing from the MS.
-
There is a need to be flexible and pragmatic and there are time constraints on what it is practical to accomplish within the 8 weeks for development of the ERG report.
Searching
-
Re-running the manufacturers’ searches may be considered but as a minimum it is important to look at the numbers of identified studies in the MS. Depending on circumstances there may be time to critique and correct the searches if necessary. Comments should be made on the adequacy of the search strategy and database selection.
-
Searches for ongoing studies and background information may be considered.
-
There may be specific tasks that arise, for example finding a study hinted at in the MS, finding utility information, key parameter values or other economic evaluations and checking consistency in different sections of the MS related to the searching.
Critical appraisal tools
-
The use of critical appraisal tools for assessing the quality of the manufacturer’s systematic review and model is considered to be important, as this increases transparency and provides a framework or starting point for appraising the MS. However, the use of a critical appraisal tool alone is not sufficient and teams should not be constrained by them.
-
Critical appraisal tools are needed to assess the manufacturers’ included studies.
-
There is a need to focus on those studies that are driving the results. Levels of critical appraisal may depend on their importance in the decision. This is not always clear and can change.
-
The use of critical appraisal tools is potentially of more use to more inexperienced ERG members.
Recommendations for manufacturers
-
The submission should be comprehensive, clearly written, appropriately copy-edited and internally consistent.
-
Definitions for all key terms and abbreviations in the MS should be provided.
-
There should be transparency in the reporting, conduct and justification of review and modelling processes and analyses (especially model parameters), as well as choice of data, and the submission should either adhere to or address differences from the scope or decision problem.
-
In cases in which reviews of the clinical effectiveness and cost-effectiveness literature are conducted, the submission should adhere to internationally accepted standards for conducting and reporting these reviews. Standardised tools such as PRISMA58 and Drummond55 could be considered for use in preparing submissions.
-
In cases in which only one trial provides data on the intervention being reviewed then the submission should state this and substantiate not conducting a full search for clinical effectiveness evidence. In such a case there may still be a need to conduct appropriate searches for data related to any indirect comparisons that are described in the submission and to report the conduct of such searches.
-
In cases of STAs with a single clinical study conducted by the manufacturer, the MS should include the submission of the clinical study report and trial protocol in an addendum to the submission.
-
There should be clear reporting of methods and results used for indirect comparisons.
-
The MS should provide relevant and sufficiently detailed data related to clinical progression, outcomes and adverse events.
-
Clear and concise rationale for the synthesis of clinical data and, if appropriate, rationale for not using alternative methods that might have been considered need to be provided.
-
Clear rationale should be provided for the types of analyses chosen for use in the MS and explanations for any analyses not carried out that would normally be expected.
-
Clear and concise rationale for the development of economic models and the assumptions used to develop models need to be provided. If appropriate, rationale for not using alternative methods that might have been considered should also be included.
-
A systematic review of utility values should be included, where appropriate, in the MS.
-
A systematic review may not be appropriate for all parameter values but reviews of parameter values should be comprehensive and transparent – how comprehensive depends on the criticality of the parameter.
Recommendations to the National Institute for Health and Clinical Excellence
For the website
-
When categorising topics it may be appropriate to double list them in more than one category but this needs to be consistent to avoid confusion.
-
Documents should be recorded with exact dates (day/month/year).
-
The current search engine on the NICE website does not provide enough detail to find all relevant information. There needs to be a mapping tool for site visitors to tell them where to find documents for ongoing and completed projects. It is easy to find guidance but there are limited markers for the process documents (scopes, ACDs, ERG reports, etc.).
-
As noted in other parts of this report, documentation for the STAs should be consistently placed on the NICE website.
-
Unique identifiers for each topic on the website would make differentiation between topics easier, especially when a single intervention may be appraised for a number of different conditions.
-
Standardising the referral date to final scope time period could be considered or making this process more transparent on the NICE website so that it is clear what activity is taking place during this time period.
General points
-
There needs to be consideration of a requirement for a comprehensive review to identify studies to inform the conduct of indirect comparisons or MTCs and other additional aspects of the modelling exercise (e.g. utility data, cost data, etc.).
-
Procedures to reduce the number of suspended, delayed and terminated STAs need to be considered.
Chapter 6 Conclusions
The NICE website is transparent and nearly all relevant documentation for the mapping analysis in this report was available there. However, this is not true for all information and the current search engine is of limited use in the identification of specific documentation beyond actual guidance.
The STA process is time and resource intensive for all of those involved. A significant proportion (> 30%) of referred topics do not go on to be appraised, meaning that the resource invested in them is lost capacity. Once an appraisal begins, the primary points of delay are related to the need for additional consideration of data (either from the manufacturer or the ERG) and the appeal process. Further delays to the STA process are expected to result from the introduction of PAS.
The ERG reports highlighted the strengths and weaknesses of MSs to the STA process, and the thematic analysis of these data offered a means of clarifying and describing these aspects of the submissions. ERGs raised particular concerns and criticisms around a number of related themes: the conduct of reviews, analyses and modelling, and submissions’ reporting of these elements; submissions’ frequent failure to satisfy the requirements of the scope or decision problem regarding populations and comparators; and the resulting impact of all of these issues, directly and indirectly, on the reliability and validity of the findings being presented in the submissions.
Points from clarification letters were analysed and presented in four main categories: report quality, systematic review methods, clinical data analysis and economic data analysis. The majority of clarification points related to the clinical data analysis and economic data analysis and covered issues such as inconsistencies between the clinical and economic sections of the submission, queries regarding sources of data and their use in the economic analysis and queries about modelling decisions and data queries, as well as requests for additional analyses. The examination of the letters also identified that the majority of reports failed to use internationally accepted standards for the conduct of systematic reviewing of clinical effectiveness and cost-effectiveness literature.
Currently, there are significant shortfalls in MSs and there is no mechanism for giving positive feedback. There is no ‘one-size-fits-all’ approach to the critical appraisal of MSs by ERGs, and flexibility in approach should be maintained.
Implications for service provision
Several recommendations have been presented in this report. It is hoped that suggested changes to the NICE website would enable easier access to relevant information and ensure that the process for accessing information is facilitated. Recommendations provided for ERG teams will help to guide teams in the future and train new members. Guidance for manufacturers will help to ensure that more appropriate submissions are received in the future. Finally, changes to the ERG report template will ensure that the reports have less repetition and that it is easier to find relevant information.
Suggested research priorities
Several research priorities have been identified and include the following:
-
Research involving AC members is needed to determine what they deem to be important information in ERG reports. This would include identifying what types of information and what level of information are most useful for making decisions. This may include comments on the layout of the ERG report template, which may also affect how easy it is for AC members to access relevant information.
-
The MS report template needs to be analysed to determine whether or not it is ‘fit for purpose’ and ensures that all information relevant to the AC is collected. Data also need to be collected on how manufacturers apply the template in their submissions.
-
This report focused on approaches reported in the ERG reports. A deeper analysis of critical appraisal approaches should be undertaken, potentially involving in-depth interviews and focus groups with ERG teams to develop a better understanding of how critical appraisal takes place. Observational studies of ERG report development could also be undertaken. Specific questions could be asked of teams, such as whether the team signs up to the concept of the ERG ‘base-case ICER’? More in-depth analysis is needed to determine approaches used to appraise electronic models presented in the MSs, as well as methods used by the ERG teams to determine internal validity.
-
More research is needed to identify appropriate methods for the systematic reviewing of utility data as well as other parameter values.
-
More research is needed into the use of appropriate critical appraisal tools for appraising systematic reviews, trials and economic evaluations within MSs and whether or not there would be benefit in standardising the use of these tools.
Acknowledgements
The authors would like to thank Andrew Booth, Myfanwy Lloyd Jones, Matt Stevenson, Roberta Ara and Janette Greenhalgh who contributed to the design of this project. The authors would also like to thank members of the InterTASC teams who acted as peer reviewers for an earlier draft of the report and participants at the InterTASC workshop held in April 2010 who contributed to the recommendations outlined in this report. Thanks also to Andrea Shippam for formatting the report.
Contributions of authors
Eva Kaltenthaler acted as lead, and contributed to the mapping section and the thematic analysis of the ERG reports.
Angela Boland contributed to the mapping data extraction and documentary analysis data extraction.
Christopher Carroll designed the thematic framework analysis and contributed to the documentary analysis data extraction.
Rumona Dickson undertook the analysis of the clarification letters.
Diana Papaioannou contributed to the mapping section.
Patrick Fitzgerald contributed to the documentary analysis data extraction and the analysis of the clarification letters.
All authors contributed to the design and writing of the report.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- NICE . Updated Guide to the Methods of Technology Appraisal – June 2008 2010. www.nice.org.uk/media/B52/A7/TAMethodsGuideUpdatedJune2008.pdf (accessed 17 March 2010).
- NICE . Appraising Life-Extending, End of Life Treatments. Supplementary Advice to the Appraisal Committees 2009. www.nice.org.uk/media/E4A/79/SupplementaryAdviceTACEoL.pdf (accessed 17 March 2010).
- NICE . Guide to the Single Technology Appraisal Process 2009. www.nice.org.uk/media/913/06/Guide_to_the_STA-proof_6–26–10–09.pdf (accessed 17 March 2010).
- Buxton MJ, Akehurst R. How NICE is the UK’s fast-track system?. Scriptmag 2006:24-5.
- Wailoo AJ, Pickstone CA. Review of the NICE single technology appraisal process. Report by the Decision Support Unit. London: NICE; 2010.
- NICE . National Institute for Health and Clinical Excellence 2010. www.nice.org.uk/.
- NICE . Pemetrexed for the First Line Treatment of Non Small Cell Lung Cancer 2009. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11715%26history=t (accessed 17 March 2010).
- NICE . Natalizumab for the Treatment of Multiple Sclerosis 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11701%26history=t (accessed 17 March 2010).
- NICE . Lung Cancer (non-Small Cell) – Pemetrexed: Final Appraisal Determination 2010. www.nice.org.uk/guidance/index.jsp?action=download%26o=35192 (accessed 17 March 2010).
- NICE . Lung Cancer (non-Small-Cell, First Line Treatment) – Pemetrexed 2010. www.nice.org.uk/guidance/index.jsp?action=download%26o=43758 (accessed 17 March 2010).
- NICE . Breast Cancer (early) – Trastuzumab 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11585%26history=t (accessed 17 March 2010).
- NICE . Varenicline for Smoking Cessation 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11718%26history=t (accessed 17 March 2010).
- NICE . Dabigatran Etexilate for the Prevention of Venous Thromboembolism After Hip or Knee Replacement Surgery in Adults 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11939%26history=t (accessed 17 March 2010).
- NICE . Rituximab As First Line Treatment for Follicular Lymphoma 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11591%26history=t (accessed 17 March 2010).
- NICE . Rivaroxaban for the Prevention of Venous Thromboembolism 2010. www.nice.org.uk/guidance/index.jsp?action=folder%26o=43416 (accessed 17 March 2010).
- NICE . Lenalidomide for Multiple Myeloma in People Who Have Received at Least One Prior Therapy 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11937%26history=t (accessed 17 March 2010).
- NICE . Bortezomib Monotherapy for Relapsed Multiple Myeloma 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11713%26history=t (accessed 17 March 2010).
- NICE . Erlotinib for the Treatment of Non-Small Cell Lung Cancer 2010. www.nice.org.uk/guidance/index.jsp?action=byID%26o=11714#keydocs (accessed 17 March 2010).
- NICE . Guide to the Single Technology Appraisal (STA) Process 2006. www.nice.org.uk/media/8DE/74/STA_Process_Guide.pdf (accessed 17 March 2010).
- NICE . Omalizumab for Severe Persistent Allergic Asthma 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11686%26history=t (accessed 17 March 2010).
- NICE . Docetaxel for Adjuvant Treatment of Early Breast Cancer 2006. http://guidance.nice.org.uk/index.jsp?action=byId%26o=11593%26history=t (accessed 17 March 2010).
- NICE . Adalimumab for the Treatment of Moderate to Severe Psoriatic Arthritis 2010. http://guidance.nice.org.uk/TA/Wave12/31 (accessed 17 March 2010).
- NICE . Rituximab for the Treatment of Refractory Rheumatoid Arthritis 2010. http://guidance.nice.org.uk/TA/Wave12/32 (accessed 17 March 2010).
- NICE . Rituximab for the Treatment of Recurrent or Refractory Stage III or IV Follicular Non-Hodgkin’s Lymphoma 2008. http://guidance.nice.org.uk/index.jsp?action=byId%26o=11730%26history=t (accessed 17 March 2010).
- NICE . Entecavir for the Treatment of Chronic Hepatitis B 2008. http://guidance.nice.org.uk/TA/Wave14/19 (accessed 17 March 2010).
- NICE . Cetuximab for the Treatment of Head and Neck Cancer 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11697%26history=t (accessed 17 March 2010).
- NICE . Ustekinumab for the Treatment of Moderate to Severe Psoriasis 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=12038%26history=t (accessed 17 March 2010).
- NICE . Abatacept for the Treatment of Rheumatoid Arthritis 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11720%26history=t (accessed 17 March 2010).
- NICE . Febuxostat for the Management of Hyperuricaemia in Patients With Gout 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11830%26history=t (accessed 17 March 2010).
- NICE . Cetuximab for the First Line Treatment of Metastatic Colorectal Cancer 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11918%26history=t (accessed 17 March 2010).
- NICE . Sunitinib for the Treatment of Gastrointestinal Stromal Tumours 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=12040%26history=t (accessed 17 March 2010).
- NICE . Infliximab for the Sub-Acute Manifestations of Ulcerative Colitis 2009. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11732%26history=t (accessed 17 March 2010).
- NICE . Topotecan for the Treatment of Recurrent Carcinoma of the Cervix 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=12034%26history=t (accessed 17 March 2010).
- NICE . Tocilizumab for the Treatment of Rheumatoid Arthritis 2010. http://guidance.nice.org.uk/TA/Wave18/5 (accessed 17 March 2010).
- NICE . Bevacizumab for the Treatment of Advanced and Metastatic Breast Cancer 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11818%26history=t (accessed 17 March 2010).
- NICE . Trabectedin for the Treatment of Advanced Soft Tissue Sarcoma 2010. http://guidance.nice.org.uk/TA/Wave18/26 (accessed 17 March 2010).
- NICE . Erlotinib Monotherapy for the Maintenance Treatment of Non-Small-Cell Lung Cancer After Previous Platinum Containing Chemotherapy 2010. http://guidance.nice.org.uk/TA/Wave19/55 (accessed 17 March 2010).
- NICE . Erlotinib, in Combination With Bevacizumab for the Maintenance Treatment of Non-Squamous Advanced or Metastatic Non-Small-Cell Lung Cancer After Previous Platinum-Containing Chemotherapy 2010. http://guidance.nice.org.uk/TA/Wave19/8 (accessed 17 March 2010).
- NICE . Rituximab for the Treatment of Relapsed Chronic Lymphocytic Leukaemia 2010. http://guidance.nice.org.uk/TA/Wave18/23 (accessed 17 March 2010).
- NICE . Lapatinib for Breast Cancer (for Use in Women With Previously Treated Advanced or Metastatic Breast Cancer) 2010. www.nice.org.uk/guidance/index.jsp?action=folder%26o=42288 (accessed 17 March 2010).
- NICE . Appraising Life-Extending, End of Life Treatments 2009. www.nice.org.uk/media/88A/F2/SupplementaryAdviceTACEoL.pdf (accessed 17 March 2010).
- NICE . Pemetrexed for the Treatment of Non-Small Cell Lung Cancer 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11715%26history=t (accessed 17 March 2010).
- NICE . Rimonabant for the Treatment of Overweight and Obese Patients 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11738%26history=t (accessed 17 March 2010).
- NICE . Romiplostim for the Treatment of Chronic Immune or Idiopathic Thrombocytopenic Purpura 2010. http://guidance.nice.org.uk/TA/Wave17/16 (accessed 17 March 2010).
- NICE . Paclitaxel for the Treatment of Early Breast Cancer 2010. www.nice.org.uk/guidance/index.jsp?action=byId%26o=11595%26history=t (accessed 17 March 2010).
- Miller FA, Alvarado K. Incorporating documents into qualitative nursing research. J Nurs Scholarship 2005;37:348-53.
- Miles M, Huberman A. Qualitative data analysis. Thousand Oaks, CA: Sage; 1994.
- Ritchie J, Spencer L, Bryman A, Burgess R. Analysing qualitative data. London: Routledge; 1993.
- Moher D, Cook DJ, Eastwood S, Olkin I, Rennie D, Stroup DF. Improving the quality of reports of meta-analyses of randomised controlled trials: the QUOROM statement. Lancet 1999;354:1896-900.
- CRD . Undertaking Systematic Reviews of Research on Effectiveness 2001.
- Oxman AD, Chalmers I, Altman DG. Systematic reviews. London: BMJ Publishing; 1995.
- ScHARR . ScHARR Critical Appraisal for Reviews Checklist n.d. www.shef.ac.uk/scharr/ir/units/critapp/apprev.htm (accessed 17 March 2010).
- CASP . 10 Questions to Help You Make Sense of Randomised Controlled Trials 2008. https://www.phru.nhs.uk/Doc_Links/rct%20appraisal%20tool.pdf (accessed 17 March 2010).
- CONSORT . CONSORT Checklist n.d. www.consort-statement.org (accessed 17 March 2010).
- Drummond M, Jefferson T. Guidelines for authors and peer reviewers of economic submissions to the BMJ. BMJ 1996;313:275-83.
- Hill SR, Mitchell AS, Henry DA. Pharmaceutical Benefits Scheme. Analyses: a review of submissions to the Australian Pharmaceutical Benefits Scheme. JAMA 2000;283:2116-21.
- Dwan K. Within Study Selective Reporting Bias in Meta-Analysis 2010.
- Moher D, Liberati A, Tetzlaff J, Altman DG. The PRISMA Group (2009) preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 2009;6.
Appendix 1 Current Evidence Review Group report template
Evidence Review Group’s report template
This template should be completed with reference to NICEs ‘Guide to the Methods of Single Technology Appraisal’ (June 2008).
Title: The title should reflect that used in the NICE scoping document.
Produced by: Name of TAR Centre
Authors: Name/s, job title, department/institution
Correspondence to: Name and address
Date completed: Date completed
Source of funding: This report was commissioned by the NIHR HTA Programme as project number **/**/**.
Declared competing interests of the authors
Description of any pecuniary relationship with sponsors, both personal and of the TAR Centre. If there are none, please state ‘none’.
Acknowledgements
Names and job titles of individuals who provided advice.
Rider on responsibility for report
The views expressed in this report are those of the authors and not necessarily those of the NIHR HTA Programme. Any errors are the responsibility of the authors.
This report should be referenced as follows:
Contributions of authors
Please refer to the International Committee of Medical Journal Editors (ICMJE) Uniform Requirements for Manuscripts Submitted to Biomedical Journals; see www.icmje.org/
1. Summary
1.1 Scope of the submission
1.2 Summary of submitted clinical effectiveness evidence
1.3 Summary of submitted cost-effectiveness evidence
1.4 Commentary on the robustness of submitted evidence
1.4.1 Strengths
1.4.2 Weaknesses
1.4.3 Areas of uncertainty
1.5 Key issues
For example, comment if there are major concerns with the comparators which have been identified in the submission.
2. Background
2.1 Critique of manufacturer’s description of underlying health problem
For example, if the appraisal is about metastatic hormone-refractory prostate cancer, and the submission gives details predominantly about prostate cancer in general, are those details relevant to the appraisal?
2.2 Critique of manufacturer’s overview of current service provision
For example, does the submission concord with opinions of clinical and patient experts? Has sufficient backing evidence been given about how often the comparators and intervention are used? Are the constraints of UK market authorisations considered?
3. Critique of manufacturer’s definition of decision problem
3.1 Population
Description of relevant patient population(s) and comment on whether they are appropriately defined in the submission.
3.2 Intervention
What is the technology and what is its relevant or proposed marketing authorisation/CE mark?
3.3 Comparators
Relevant comparators in an NHS context, justification for choice of comparators. For example, where hard evidence is not available, has the manufacturer asked an unbiased clinical panel, or done its own survey, and does it agree with what the clinical experts for the appraisal say?
3.4 Outcomes
Including clinical effectiveness, adverse events, QoL and health economic outcomes and a discussion of appropriate mechanisms for measuring these outcomes? Critique of whether focus of submission is on the appropriate outcomes. Comment on whether the analysis has been limited to non-ideal outcomes.
3.5 Time frame
3.6 Other relevant factors
4. Clinical effectiveness
4.1 Critique of manufacturer’s approach
4.1.1 Description of manufacturer’s search strategy and comment on whether the search strategy was appropriate
Databases and other sources including unpublished sources, any restrictions.
4.1.2 Statement of the inclusion/exclusion criteria used in the study selection and comment on whether they were appropriate
4.1.3 Table of identified studies. What studies were included in the submission and what were excluded?
4.1.4 Details of any relevant studies that were not included in the submission?
4.1.5 Description and critique of manufacturer’s approach to validity assessment
4.1.6 Description and critique of manufacturer’s outcome selection
4.1.7 Describe and critique the statistical approach used
4.1.8 Summary statement
Describe the completeness of the submission with regard to relevant studies and relevant data within those studies. Reference should also me made concerning the extent to which the submitted evidence reflects the decision problem defined in the submission.
4.2 Summary of submitted evidence
4.2.1 Summary of results
4.2.2 Critique of submitted evidence syntheses
4.2.3 Summary
Does the submission contain an unbiased estimate of the technology’s (relative and absolute) treatment effect in relation to relevant outcomes and the comparators of interest?
5. Economic evaluation
5.1 Overview of manufacturer’s economic evaluation
Including one-page summary of structure, assumptions and sources, with signposting to tables with numerical inputs and their distributions where appropriate.
5.1.1 Natural history
5.1.2 Treatment effectiveness within the submission
5.1.3 Health-related QoL
5.1.4 Resources and costs
5.1.5 Discounting
5.1.6 Sensitivity analyses
5.1.7 Model validation
5.2 Critique of approach used
5.3 Results included in manufacturer’s submission
Including one-page summary of sensitivity analyses and resulting range of ICERs: one-way, threshold and probabilistic.
5.4 Comment on validity of results presented with reference to methodology used
5.5 Summary of uncertainties and issues
6. Additional ‘exploratory’ or other work undertaken by the ERG
Including a full explanation of any technical changes made to the company model and programming code, to allow consultees to replicate the ERG’s analyses.
7. Discussion
7.1 Summary of clinical effectiveness issues
7.2 Summary of cost-effectiveness issues
7.3 Implications for research
Appendix 1
List of those involved with developing the ERG scope.
Appendix 2
Quality assessment using ScHARR-TAG economic modelling checklist:
-
title
-
a statement of the problem
-
a discussion of the need for modelling
-
a description of the relevant factors and outcomes
-
a description of the model including type of model, time frame, perspective and setting
-
a description of data sources, with description of respective strengths and weaknesses
-
key assumptions relating to model structure and data stated
-
disease-specific factors included within modelling (items to be specified in conjunction with expert clinical input)
-
validation
-
results
-
sensitivity analysis results.
Appendix 2 Mapping template
| STA title |
|---|
| Project reference |
| Wave of work |
| STA report status |
| Project status |
| Team |
| Manufacturer |
| Technology name |
| Technology indication |
| Referral date |
| Date of commencement/date of final scope |
| Closing date for invited submissions |
| Date of ERG report submission |
| ICERs reported by manufacturer |
| ICERs reported by ERG |
| Extra work required from ERG before first AC? |
| Date of first AC |
| AC decision |
| Extra work required/volunteered from manufacturer? |
| Extra work required from ERG? |
| Date of ACD |
| Date of second AC |
| Date of FAD issue |
| Decision stated in FAD |
| Did the DSU get involved? |
| Details of DSU involvement |
| End of life considered |
| Appeal date |
| Result of appeal |
| Date of appeal result |
| Changes to timeline and/or deferral – details |
| Comments |
Appendix 3 Summary of the NICE reference case
| Element of health technology assessment | Reference case |
|---|---|
| Defining the decision problem | The scope developed by the Institute |
| Comparator | Therapies routinely used in the NHS, including technologies regarded as current best practice |
| Perspective on costs | NHS and PSS |
| Perspective on outcomes | All health effects on individuals |
| Type of economic evaluation | Cost-effectiveness analysis |
| Synthesis of evidence on outcomes | Based on a systematic review |
| Measure of health effects | QALYs |
| Source of data for measurement of health-related QoL | Reported directly by patients and/or carers |
| Source of preference data for valuation of changes in health-related QoL | Representative sample of the public |
| Discount rate | An annual rate of 3.5% on both costs and health effects |
| Equity weighting | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit |
Appendix 4 Evidence Review Group documentary analysis data extraction template
| Title of STA |
|---|
| ERG |
| Identifier |
| Executive summary |
| Key strengths of the MS identified by the ERG (i) |
| Key strengths of the MS identified by the ERG (ii) |
| Key weaknesses of the MS identified by the ERG (i) |
| Key weaknesses of the MS identified by the ERG (ii) |
| Areas of uncertainty in the MS identified by the ERG (i) |
| Areas of uncertainty in the MS identified by the ERG (ii) |
| Key issues raised by the ERG (i) |
| Key issues raised by the ERG (ii) |
| Background |
| Did the ERG critique the background chapter in the MS? |
| Context/statement of the decision problem |
| Did the ERG identify any areas where the manufacturer’s statement of the decision problem did not match the scope? |
| Population |
| Intervention |
| Comparator |
| Outcome |
| Subgroup |
| Did the manufacturer seek any special consideration? |
| For what reason did the manufacturer seek special consideration? |
| Literature review |
| Did the ERG comment on the manufacturer’s searches? |
| Did the ERG replicate the manufacturer’s search strategies? |
| Did the ERG agree that the manufacturer’s stated inclusion/exclusion criteria were appropriate? |
| Did the MS include a QUOROM diagram? |
| How many pivotal trials were identified for inclusion in the review? |
| Was other trial evidence considered? |
| Was the ERG satisfied no other studies were available for inclusion? |
| Did the ERG consider that the manufacturer used an appropriate method of quality assessment for critique of the main clinical trial evidence included in the review? (Please state tool used) |
| Appraisal of effectiveness review |
| Did the ERG use a published tool to appraise the quality of the review? e.g. QUOROM (Please state tool used) |
| Main source(s) of clinical effectiveness data |
| How many trials were used to demonstrate the clinical effectiveness of the technology under consideration? |
| Did the ERG quality assess the included trial(s)? (Please state tool used) |
| Did the ERG conclude that the quality of the trial(s) was good? |
| Did the ERG state that the nature of the clinical trial evidence cited was sufficient to answer the question posed in the decision problem? |
| Was the study design appropriate? |
| Was the population appropriate? |
| Was the comparator appropriate? |
| Was the primary outcome appropriate? |
| Generalisability of main randomised controlled trial |
| Did the ERG comment on whether or not relevance to UK is considered in the MS? |
| Licensed indication |
| Clinical practice |
| Population |
| Comparator |
| Outcome |
| Other strengths/weaknesses of review |
| Strengths highlighted by ERG (i) |
| Strengths highlighted by ERG (ii) |
| Weaknesses highlighted by ERG (i) |
| Weaknesses highlighted by ERG (ii) |
| Evidence synthesis |
| Did the manufacturer conduct a meta-analysis? |
| Did the ERG comment on the approach to meta-analysis? |
| Details of critique (i) |
| Details of critique (ii) |
| Did the manufacturer use a different method (i.e. not meta-analysis) for evidence synthesis? |
| Did the ERG comment on the approach used? |
| Details of critique (i) |
| Details of critique (ii) |
| Indirect analysis |
| Did the manufacturer conduct an indirect comparison? |
| What method of indirect analysis was undertaken by the manufacturer? |
| Did the ERG comment on the approach to indirect analysis? |
| Details of critique (i) |
| Details of critique (ii) |
| Did the manufacturer provide search strategies and results related to the indirect comparison exercise? |
| Did the ERG have confidence in the results of the indirect comparison? |
| Subgroup analysis |
| Did the manufacturer undertake subgroup analysis? |
| Did the ERG consider that the manufacturer conducted appropriate subgroup analysis? |
| Critique of subgroup analysis by ERG (i) |
| Critique of subgroup analysis by ERG (ii) |
| Did the ERG have confidence in the results of the subgroup analysis? |
| Additional information |
| Did the ERG undertake additional analysis related to clinical evidence? |
| What methods did the ERG use in their additional analysis? |
| Other strengths/weaknesses of clinical effectiveness evidence presented |
| Strengths highlighted by ERG (i) |
| Strengths highlighted by ERG (ii) |
| Weaknesses highlighted by ERG (i) |
| Weaknesses highlighted by ERG (ii) |
| Miscellaneous |
| Number of clinical advisors (not ERG staff) listed in the acknowledgements of the ERG report? |
| Number of clinicians (not ERG staff) listed as authors in the ERG report? |
| Literature review |
| Did the ERG comment on the manufacturer’s searches? |
| Did the ERG replicate the manufacturer’s search strategies? |
| Did the ERG agree that all the manufacturer’s stated inclusion/exclusion criteria were appropriate? |
| Did the MS include a flow chart of economic studies? |
| How many economic studies were identified for inclusion in the review? |
| Did the MS identify economic studies for modelling purposes that were not included in the review? |
| Was the ERG satisfied no other relevant economic studies were available for inclusion in the review? |
| Did the ERG consider that the manufacturer used an appropriate method of quality assessment for critique of the main economic studies included in the review? (Please state tool used) |
| Did the ERG use an evaluation tool to appraise the quality of the cost-effectiveness studies included in the review? e.g. Drummond checklist (Please state tool used) |
| Other strengths/weaknesses of cost-effectiveness review |
| Strengths highlighted by ERG (i) |
| Strengths highlighted by ERG (ii) |
| Weaknesses highlighted by ERG (i) |
| Weaknesses highlighted by ERG (ii) |
| Reference case |
| Did the ERG state if the manufacturer followed the NICE reference case? |
| Did the ERG comment on any areas where the manufacturer deviated from the reference case? |
| Quality of the economic evaluation/model |
| Did the ERG consider that the manufacturer offered an appropriate critique of its submitted economic evaluation/model? (If yes, please state tool) |
| Did the ERG use a critical appraisal tool to critique the manufacturer’s economic evaluation/model? (If yes, please state tool) |
| Did the ERG conclude that the quality of the economic evaluation/model was good? |
| Did the ERG state that the evaluation/model was reliable? |
| Did the ERG state that the evaluation/model was flawed? |
| Did the ERG state that the evaluation/model structure was appropriate for decision-making? |
| Did the ERG critique the following in the manufacturer’s economic evaluation/model? |
| Perspective |
| Time horizon |
| Patient population/subgroups |
| Choice of comparators |
| Choice of outcomes |
| Costs |
| Did the ERG offer any alternative regarding consideration of costs? |
| Benefits |
| Did the ERG offer any alternatives regarding consideration of benefits? |
| Did the ERG critique the methods described in the economic model in relation to the …? |
| Choice of model structure |
| Health states described in the model |
| Sequencing of treatments (if any) |
| Incorporation of efficacy data |
| Incorporation of adverse events |
| Incorporation of survival data |
| Incorporation of QoL data |
| Mapping of health states to European Quality of Life-5 Dimensions (EQ-5D) scores or other utility measure |
| Replication of the original clinical trial results |
| Type of continuity correction used |
| Approach to discounting employed |
| SA/PSA |
| Did the manufacturer undertake a SA? |
| Which type of SA was conducted in the MS? |
| Did the ERG comment on the SA in the MS? |
| Critique of SA by the ERG (i) |
| Critique of SA by the ERG (ii) |
| Did the manufacturer conduct PSA? |
| Did the ERG comment on the PSA in the MS? |
| Critique of PSA by the ERG (i) |
| Critique of PSA by the ERG (ii) |
| Internal validity of the economic model |
| Did the ERG comment on the methods used by the manufacturer to determine internal validity? |
| Did the ERG describe their attempts to determine internal validity? |
| Did the ERG state that the correct formulae are used in the model? |
| Did the ERG state that the results are consistent with the input data? |
| Did the ERG state that the results are meaningful/realistic? |
| Transparency |
| Did the ERG comment on the transparency of the model, i.e. were all clinical and cost data sources appropriately referenced? |
| Did the ERG comment on the manufacturer’s attempts to explain the methods used to generate model parameters? |
| Did the ERG comment on the level of detail in the annotation to the model provided by the manufacturer? |
| Additional analysis |
| Did the ERG undertake additional economic analysis? |
| What methods were used by the ERG in its additional economic analysis? |
| Did the ERG run the manufacturer’s model with different parameter values? |
| Did the ERG run the manufacturer’s model after making corrections to the model? |
| Did the ERG carry out separate analysis of evidence (either from published sources or from the trial) to augment/replace aspects of the manufacturer’s submitted analysis? |
| Did the ERG estimate revised ICERs as part of their additional analysis? |
| Did the ERG state whether additional modelling would have been desirable/helpful in assisting decision-making? |
| Errors |
| Did the ERG identify any errors in the model? |
| Type of error (i) |
| Type of error (ii) |
| Type of error (iii) |
| Clarification letters/response |
| Was the clarification letter to the manufacturer mentioned in the ERG report? |
| Was the clarification response from the manufacturer mentioned in the ERG report? |
| Did the ERG comment on whether the response from the manufacturer addressed the issues/requests in the ERG’s letter? |
| Miscellaneous |
| Number of economic advisors (not ERG staff) listed in the acknowledgements of the ERG report? |
| Number of economists (not ERG staff) listed as authors in the ERG report? |
| Other strengths/weaknesses of cost-effectiveness evidence presented |
| Strengths highlighted by ERG (i) |
| Strengths highlighted by ERG (ii) |
| Weaknesses highlighted by ERG (i) |
| Weaknesses highlighted by ERG (ii) |
Appendix 5 Revised Evidence Review Group report template*
*This template serves as a checklist only; it may be necessary to modify, add, omit or move sections as appropriate.
Evidence Review Group’s report template
This template should be completed with reference to the NICE ‘Guide to the Methods of Single Technology Appraisal’.
Title: The title should reflect that used in the NICE scoping document.
Produced by: Name of TAR Centre
Authors: Name/s, job title, department/institution
Correspondence to: Name and address
Date completed: Date completed (dd/mm/yyyy)
Source of funding: This report was commissioned by the NIHR HTA Programme as project number **/**/**.
Declared competing interests of the authors
Description of any pecuniary relationship with sponsors, both personal and of the TAR Centre. If there are none, please state ‘none’.
Acknowledgements
Names and job titles of individuals who provided advice.
Rider on responsibility for report
The views expressed in this report are those of the authors and not necessarily those of the NIHR HTA Programme. Any errors are the responsibility of the authors.
This report should be referenced as follows:
Order of Authors. Title of the ERG report followed by: A Single Technology Appraisal. TAR team name, Year.
Contributions of authors
Please refer to the International Committee of Medical Journal Editors (ICMJE) Uniform Requirements for Manuscripts Submitted to Biomedical Journals – see www.icmje.org/
1. Summary
1.1 Scope of the manufacturer submission
1.2 Summary of clinical effectiveness evidence submitted by the manufacturer
1.3 Summary of the ERG’s critique of clinical effectiveness evidence submitted
1.4 Summary of cost-effectiveness evidence submitted by the manufacturer
1.5 Summary of the ERG’s critique of cost-effectiveness evidence submitted
1.6 ERG commentary on the robustness of evidence submitted by the manufacturer
1.6.1 Strengths
1.6.2 Weaknesses and areas of uncertainty
1.7 Summary of additional work undertaken by the ERG
2. Background
2.1 Critique of manufacturer’s description of underlying health problem
Does the ERG believe that the manufacturer’s description of the underlying health problem is appropriate and relevant to the decision problem under consideration?
2.2 Critique of manufacturer’s overview of current service provision
Does the ERG believe that the manufacturer’s overview of current service provision is appropriate and relevant to the decision problem under consideration?
3. Critique of manufacturer’s definition of decision problem
3.1 Population
To what extent does the clinical evidence submitted by the manufacturer match the patient population described in the final scope? Where there is a mismatch, provide further details. Does the clinical evidence submitted by the manufacturer reflect the characteristics of the patient population in England and Wales eligible for treatment? If not, provide further comment.
3.2 Intervention
Does the intervention described in the MS match the intervention described in the final scope? What is the technology and what is its relevant or proposed marketing authorisation/CE mark?
3.3 Comparators
Do the comparators described in the MS match the comparators described in the final scope? If not, provide further details. Where evidence is limited or not available for relevant comparators has the manufacturer asked an unbiased clinical panel, or carried out its own survey, and do the views elicited agree with what the clinical advisors to the ERG advocate?
3.4 Outcomes
Do the outcomes in the MS match the outcomes described in the final scope? If not, provide further details. Consider clinical effectiveness, adverse events, QoL and health economic outcomes and a discussion of appropriate mechanisms for measuring these outcomes. Is the focus of the submission on appropriate outcomes or has it been limited to non-ideal outcomes?
3.5 Other relevant factors
For example: Does the MS include a section on equity considerations? Is there an ongoing PAS application?
4. Clinical effectiveness
4.1 Critique of the methods used by the manufacturer to systematically review clinical effectiveness evidence
4.1.1 State objective of systematic review. Provide description of manufacturer’s search strategy and comment on whether the search strategy was appropriate. If the manufacturer did not perform a systematic review, was this appropriate?
List databases and other sources of information including unpublished sources, describe any restrictions.
4.1.2 State the inclusion/exclusion criteria used in the study selection and comment on whether they were appropriate
4.1.3 What studies were included in the clinical effectiveness review and what were excluded? Provide a table of identified studies. Please identify the most important clinical effectiveness studies
4.1.4 Provide details of any relevant studies not discussed in the submission. Why were these studies excluded and how were these studies identified by the ERG?
4.2 Summary and critique of submitted clinical effectiveness evidence
If there is more than one RCT described in the MS, it may be appropriate to discuss each trial individually using the headings described.
4.2.1 Summary of submitted clinical evidence for each relevant trial
4.2.2 Describe and critique the manufacturer’s approach to validity assessment for each relevant trial
4.2.3 Describe and critique the statistical approach used within each relevant trial
4.2.4 Describe and critique the manufacturer’s approach to outcome selection within each relevant trial
4.2.5 To what extent does each relevant trial include the patient population(s), intervention(s), comparator(s) and outcomes as defined in the final scope?
4.2.6 Where appropriate, describe and critique any meta-analysis, indirect comparisons and/or mixed treatment analysis carried out by the manufacturer
This section should include a summary of the manufacturer’s methods and results as described in the MS. The ERG should critique the methods used and interpret the results in light of the methods used by the manufacturer and generalisability to patients in England and Wales.
4.2.7 Additional clinical work conducted by the ERG
Provide details of any additional work conducted by the ERG in relation to clinical effectiveness. If the results of any of the additional work affect the size of the ICER, refer the reader to the summary table in Section 6.
4.3 Conclusions
Describe the completeness of the MS with regard to relevant clinical studies and relevant data within those studies. Does the submission contain an unbiased estimate of the technology’s (relative and absolute) treatment effects in relation to relevant populations, interventions, comparators and outcomes? Are there any remaining uncertainties about the reliability of the clinical effectiveness evidence? Reference should also be made concerning the extent to which the submitted evidence reflects the decision problem defined in the final scope.
5. Cost-effectiveness
5.1 ERG comment on manufacturer’s review of cost-effectiveness evidence
5.1.1 State objective of cost-effectiveness review. Provide description of manufacturer’s search strategy and comment on whether the search strategy was appropriate. If the manufacturer did not perform a systematic review, was this appropriate?
5.1.2 State the inclusion/exclusion criteria used in the study selection and comment on whether they were appropriate
5.1.3 What studies were included in the cost-effectiveness review and what were excluded? Where appropriate, provide a table of identified studies. Please identify the most important cost-effectiveness studies
5.1.4 What does the review conclude from the data available? Does the ERG agree with the conclusions of the cost-effectiveness review? If not, provide details
5.2 Summary and critique of manufacturer’s submitted economic evaluation by the ERG
Summarise and critique the cost-effectiveness evidence submitted by the manufacturer (headings 5.2.1–5.2.11 are suggested headings). It is noted that the ERGs may prefer not to combine the summary and critique of the submitted economic evidence and instead report summary and critique sections separately.
5.2.1 NICE reference case checklist (table only)
5.2.2 Model structure
5.2.3 Population
5.2.4 Interventions and comparators
5.2.5 Perspective, time horizon and discounting
5.2.6 Treatment effectiveness
5.2.7 Health-related QoL
5.2.8 Resources and costs
5.2.9 Cost-effectiveness results
5.2.10 Sensitivity analyses
5.2.11 Model validation
5.3 Additional work undertaken by the ERG
Provide details of any additional work conducted by the ERG in relation to cost-effectiveness. If the results of any of the additional work affect the size of the ICER, refer the reader to the summary table in Section 6.
5.4 Conclusions
Describe the completeness of the MS with regard to relevant cost-effectiveness studies and data described in any de novo economic evaluations. Does the submission contain an unbiased estimate of the technology’s ICERs in relation to relevant populations, interventions comparators and outcomes? Are there any remaining uncertainties about the reliability of the cost-effectiveness evidence? Reference should also be made concerning the extent to which the submitted evidence reflects the decision problem defined in the final scope.
6. Impact on the ICER of additional clinical and economic analyses undertaken by the ERG
Where appropriate, this section should include a table that shows (1) the effect of any major clinical or cost parameter change or structural change on the size of the base-case ICER and (2) the effect of making all changes simultaneously on the size of the base-case ICER.
7. End of life
Where appropriate, this section should summarise the manufacturer’s case for using the NICE end-of-life treatment criteria and discuss to what extent the manufacturer’s argument is valid.
8. Conclusions
The section should focus on any difference(s) of opinion between the manufacturer and the ERG that might influence the size of the ICER. Priority should be focused on discussing information that will be useful to the AC, including strengths, weaknesses and remaining uncertainties. Further summary of evidence is not required in this section.
8.1 Implications for research
Appendix 1
Quality assessment using ScHARR-TAG economic modelling checklist:
-
title
-
a statement of the problem
-
a discussion of the need for modelling
-
a description of the relevant factors and outcomes
-
a description of the model including type of model; time frame; perspective; and setting
-
a description of data sources, with description of respective strengths and weaknesses
-
key assumptions relating to model structure and data stated
-
disease-specific factors included within modelling (items to be specified in conjunction with expert clinical input)
-
validation
-
results
-
sensitivity analysis results.
List of abbreviations
- AC
- Appraisal Committee
- ACD
- appraisal consultation document
- CASP
- Critical Appraisal Skills Programme
- CRD
- Centre for Reviews and Dissemination
- DH
- Department of Health
- DSU
- Decision Support Unit
- ERG
- Evidence Review Group
- FAD
- final appraisal determination
- ICER
- incremental cost-effectiveness ratio
- InterTASC
- Technology Assessment Services Collaboration
- LRiG
- Liverpool Reviews and Implementation Group
- MS
- manufacturer’s submission
- MTA
- multiple technology appraisal
- MTC
- mixed-treatment comparison
- NETSCC
- NIHR Evaluation, Trials and Studies Coordinating Centre
- NICE
- National Institute for Health and Clinical Excellence
- PAS
- Patient Access Scheme
- PenTAG
- Peninsula Technology Assessment Group
- PPRS
- Pharmaceutical Price Regulation Scheme
- PSA
- probabilistic sensitivity analysis
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- SA
- sensitivity analysis
- ScHARR
- School of Health and Related Research
- SHTAC
- Southampton Health Technology Assessment Centre
- STA
- single technology appraisal
- WMHTAC
- West Midlands Health Technology Assessment Collaboration
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Professor of General Practice, Department of Primary Health Care, University of Oxford Programme Director,
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Deborah Ashby, Professor of Medical Statistics and Clinical Trials, Queen Mary, Department of Epidemiology and Public Health, Imperial College London
-
Professor Peter Brocklehurst, Director, National Perinatal Epidemiology Unit, University of Oxford
-
Professor John Cairns, Professor of Health Economics, London School of Hygiene and Tropical Medicine
-
Professor Peter Croft, Director of Primary Care Sciences Research Centre, Keele University
-
Professor Jenny Donovan, Professor of Social Medicine, University of Bristol
-
Professor Jonathan Green, Professor and Acting Head of Department, Child and Adolescent Psychiatry, University of Manchester Medical School
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Freddie Hamdy, Professor of Urology, Head of Nuffield Department of Surgery, University of Oxford
-
Professor Allan House, Professor of Liaison Psychiatry, University of Leeds
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norris, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor James Raftery, Chair of NETSCC and Director of the Wessex Institute, University of Southampton
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Catherine Moody, Programme Manager, Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire Country Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Professor Carol Tannahill, Glasgow Centre for Population Health
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Sarah Tyson, Senior Research Fellow & Associate Head of School, University of Salford
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr David P Britt, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Professor Nicholas James, Professor of Clinical Oncology, School of Cancer Sciences, University of Birmingham
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Mr John Chapman, Public contributor
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Ms Kylie Gyertson, Oncology and Haematology Clinical Trials Manager, Guy’s and St Thomas’ NHS Foundation Trust London
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Martin Shelly, General Practitioner, Silver Lane Surgery, Leeds
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Expert Advisory Network
-
Professor Douglas Altman, Professor of Statistics in Medicine, Centre for Statistics in Medicine, University of Oxford
-
Professor John Bond, Professor of Social Gerontology & Health Services Research, University of Newcastle upon Tyne
-
Professor Andrew Bradbury, Professor of Vascular Surgery, Solihull Hospital, Birmingham
-
Mr Shaun Brogan, Chief Executive, Ridgeway Primary Care Group, Aylesbury
-
Mrs Stella Burnside OBE, Chief Executive, Regulation and Improvement Authority, Belfast
-
Ms Tracy Bury, Project Manager, World Confederation of Physical Therapy, London
-
Professor Iain T Cameron, Professor of Obstetrics and Gynaecology and Head of the School of Medicine, University of Southampton
-
Professor Bruce Campbell, Consultant Vascular & General Surgeon, Royal Devon & Exeter Hospital, Wonford
-
Dr Christine Clark, Medical Writer and Consultant Pharmacist, Rossendale
-
Professor Collette Clifford, Professor of Nursing and Head of Research, The Medical School, University of Birmingham
-
Professor Barry Cookson, Director, Laboratory of Hospital Infection, Public Health Laboratory Service, London
-
Dr Carl Counsell, Clinical Senior Lecturer in Neurology, University of Aberdeen
-
Professor Howard Cuckle, Professor of Reproductive Epidemiology, Department of Paediatrics, Obstetrics & Gynaecology, University of Leeds
-
Professor Carol Dezateux, Professor of Paediatric Epidemiology, Institute of Child Health, London
-
Mr John Dunning, Consultant Cardiothoracic Surgeon, Papworth Hospital NHS Trust, Cambridge
-
Mr Jonothan Earnshaw, Consultant Vascular Surgeon, Gloucestershire Royal Hospital, Gloucester
-
Professor Martin Eccles, Professor of Clinical Effectiveness, Centre for Health Services Research, University of Newcastle upon Tyne
-
Professor Pam Enderby, Dean of Faculty of Medicine, Institute of General Practice and Primary Care, University of Sheffield
-
Professor Gene Feder, Professor of Primary Care Research & Development, Centre for Health Sciences, Barts and The London School of Medicine and Dentistry
-
Mr Leonard R Fenwick, Chief Executive, Freeman Hospital, Newcastle upon Tyne
-
Mrs Gillian Fletcher, Antenatal Teacher and Tutor and President, National Childbirth Trust, Henfield
-
Professor Jayne Franklyn, Professor of Medicine, University of Birmingham
-
Mr Tam Fry, Honorary Chairman, Child Growth Foundation, London
-
Professor Fiona Gilbert, Consultant Radiologist and NCRN Member, University of Aberdeen
-
Professor Paul Gregg, Professor of Orthopaedic Surgical Science, South Tees Hospital NHS Trust
-
Bec Hanley, Co-director, TwoCan Associates, West Sussex
-
Dr Maryann L Hardy, Senior Lecturer, University of Bradford
-
Mrs Sharon Hart, Healthcare Management Consultant, Reading
-
Professor Robert E Hawkins, CRC Professor and Director of Medical Oncology, Christie CRC Research Centre, Christie Hospital NHS Trust, Manchester
-
Professor Richard Hobbs, Head of Department of Primary Care & General Practice, University of Birmingham
-
Professor Alan Horwich, Dean and Section Chairman, The Institute of Cancer Research, London
-
Professor Allen Hutchinson, Director of Public Health and Deputy Dean of ScHARR, University of Sheffield
-
Professor Peter Jones, Professor of Psychiatry, University of Cambridge, Cambridge
-
Professor Stan Kaye, Cancer Research UK Professor of Medical Oncology, Royal Marsden Hospital and Institute of Cancer Research, Surrey
-
Dr Duncan Keeley, General Practitioner (Dr Burch & Ptnrs), The Health Centre, Thame
-
Dr Donna Lamping, Research Degrees Programme Director and Reader in Psychology, Health Services Research Unit, London School of Hygiene and Tropical Medicine, London
-
Professor James Lindesay, Professor of Psychiatry for the Elderly, University of Leicester
-
Professor Julian Little, Professor of Human Genome Epidemiology, University of Ottawa
-
Professor Alistaire McGuire, Professor of Health Economics, London School of Economics
-
Professor Neill McIntosh, Edward Clark Professor of Child Life and Health, University of Edinburgh
-
Professor Rajan Madhok, Consultant in Public Health, South Manchester Primary Care Trust
-
Professor Sir Alexander Markham, Director, Molecular Medicine Unit, St James’s University Hospital, Leeds
-
Dr Peter Moore, Freelance Science Writer, Ashtead
-
Dr Andrew Mortimore, Public Health Director, Southampton City Primary Care Trust
-
Dr Sue Moss, Associate Director, Cancer Screening Evaluation Unit, Institute of Cancer Research, Sutton
-
Professor Miranda Mugford, Professor of Health Economics and Group Co-ordinator, University of East Anglia
-
Professor Jim Neilson, Head of School of Reproductive & Developmental Medicine and Professor of Obstetrics and Gynaecology, University of Liverpool
-
Mrs Julietta Patnick, Director, NHS Cancer Screening Programmes, Sheffield
-
Professor Robert Peveler, Professor of Liaison Psychiatry, Royal South Hants Hospital, Southampton
-
Professor Chris Price, Director of Clinical Research, Bayer Diagnostics Europe, Stoke Poges
-
Professor William Rosenberg, Professor of Hepatology and Consultant Physician, University of Southampton
-
Professor Peter Sandercock, Professor of Medical Neurology, Department of Clinical Neurosciences, University of Edinburgh
-
Dr Philip Shackley, Senior Lecturer in Health Economics, Sheffield Vascular Institute, University of Sheffield
-
Dr Eamonn Sheridan, Consultant in Clinical Genetics, St James’s University Hospital, Leeds
-
Dr Margaret Somerville, Director of Public Health Learning, Peninsula Medical School, University of Plymouth
-
Professor Sarah Stewart-Brown, Professor of Public Health, Division of Health in the Community, University of Warwick, Coventry
-
Dr Nick Summerton, GP Appraiser and Codirector, Research Network, Yorkshire Clinical Consultant, Primary Care and Public Health, University of Oxford
-
Professor Ala Szczepura, Professor of Health Service Research, Centre for Health Services Studies, University of Warwick, Coventry
-
Dr Ross Taylor, Senior Lecturer, University of Aberdeen
-
Dr Richard Tiner, Medical Director, Medical Department, Association of the British Pharmaceutical Industry
-
Mrs Joan Webster, Consumer Member, Southern Derbyshire Community Health Council
-
Professor Martin Whittle, Clinical Co-director, National Co-ordinating Centre for Women’s and Children’s Health, Lymington