
Notes
Article history
The research reported in this issue of the journal was commissioned by the HTA programme as project number 06/302/19. The contractual start date was in April 2007. The draft report began editorial review in January 2010 and was accepted for publication in October 2010. As the funder, by devising a commissioning brief, the HTA programme specified the research question and study design. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2011. This work was produced by Goodacre et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising.Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2011 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Diagnostic assessment of acute chest pain
Chest pain is responsible for around 700,000 patient attendances per year in England and Wales, and around one-quarter of hospital admissions. 1 The main reason for attendance is the possibility of acute myocardial infarction (AMI). Patients with ST-elevation myocardial infarction (STEMI) can benefit from early coronary reperfusion using primary angioplasty or intravenous thrombolysis. Patients with non-ST-elevation myocardial infarction (NSTEMI), who are much more numerous than those with STEMI, benefit from hospital admission and treatment. The potential benefits of treatment for AMI have led to public awareness campaigns encouraging people with acute chest pain to call for emergency medical help and national guidelines recommending that patients with chest pain should call for an emergency ambulance to take them to a hospital emergency department (ED) rather than contact their general practitioner (GP). 2 Thus, ED attendances with chest pain are increasing.
Standard initial assessment of patients with acute chest pain consists of a clinical history and examination, 12-lead electrocardiogram (ECG) recording and a chest radiograph. On the basis of this assessment around 34% will have a clinical diagnosis of suspected acute coronary syndrome (ACS), 11% will have an ECG diagnosis of suspected acute coronary syndrome, 19% will have a clear diagnosis of benign non-cardiac chest pain (such as muscular pain) and 12% will have suspected serious non-cardiac chest pain (such as pulmonary embolus) or comorbidity (such as heart failure). The remaining 24% have no clear diagnosis but have a significant risk (5–10%) of undiagnosed AMI. 1
Acute myocardial infarction is usually diagnosed by serial cardiac biomarker testing (‘cardiac enzymes’) typically by measurement of troponin in the blood. A patient with ischaemic symptoms (such as chest pain) would be diagnosed as having AMI according to the universal definition for acute, evolving or recent AMI if a troponin level is recorded above the 99th percentile of the values for a reference control group. 3 These patients are likely to benefit from hospital admission. 4 Patients with no troponin elevation have a low risk of adverse outcome and are unlikely to benefit from hospital admission, unless they have a serious non-coronary cause for their pain (such as pulmonary embolus) or serious comorbidity (such as heart failure or arrhythmia). Rapid and accurate identification of patients with AMI is thus a hallmark of effective emergency care for chest pain.
Current recommendations suggest that patients with chest pain due to suspected, but not proven, AMI should receive diagnostic testing with a troponin sample taken 12 hours after symptom onset,5 the delay being necessary because troponin sensitivity does not reach optimal levels until this time. This approach is inconvenient and potentially costly because it requires many patients to be unnecessarily admitted to hospital until the time delay has elapsed. Most patients presenting to the ED with suspected AMI do not actually have subsequent confirmation of AMI, so their admission will ultimately prove avoidable. Economic analysis suggests that admitting patients for cardiac marker testing is not a cost-effective use of health service resources compared with early cardiac marker testing. 6 Recent studies7,8 have suggested that new high-sensitivity troponin assays may be able to rule out AMI earlier than 12 hours, although the effect of using these assays in practice has not yet been evaluated.
Evidence also suggests that these guidelines are often not followed in a busy emergency setting where acute beds are limited. Collinson et al. 9 showed that 7% of patients discharged after ED assessment for acute chest pain had elevated troponin levels at follow-up 2 days later. Goodacre et al. 10 showed that, in the standard-care arm of a randomised trial of a chest pain unit, 14% of patients with an elevated troponin level at 2-day follow-up had been sent home from the ED. A national survey of EDs11 asked the lead consultant what proportion of patients with undifferentiated chest pain would be admitted to hospital. Estimates varied from less than 20% to over 80%. Hence, it appears that the theoretical ideal of a 12-hour troponin is not realised in practice and, as a result, patients are inadvertently discharged home with undetected AMI.
The point-of-care cardiac marker panel
Rapid point-of-care testing using a panel of markers offers an alternative approach that may be more effective and cost-effective than current practice. This technology has two elements that offer putative benefits: (1) point-of-care testing to reduce the turnaround time to results being available and (2) the use of a combination of markers, including measurement of marker gradients, to optimise early sensitivity. This is based on the idea that other markers, such as myoglobin and creatine kinase MB (mass) [CK-MB (mass)], may start to rise earlier than cardiac troponin.
Point-of-care testing involves using an analyser in the ED for marker assays. The analyser needs to be operated quickly and reliably by clinical staff. It can then allow rapid provision of marker results with short turnaround times to guide decision-making. Several randomised controlled trials (RCTs) have compared point-of-care tests with the same test performed on a laboratory analyser to determine whether use of point-of-care testing reduces turnaround times and changes practice. Collinson et al. 12 showed that the use of point-of-care troponin testing on a coronary care unit led to reduced length of coronary care unit stay and overall hospital stay. Renaud et al. 13 showed that point-of-care troponin testing in an ED reduced time to anti-ischaemic therapy and physician notification of troponin results, but did not change ED length of stay or patient outcomes. Ryan et al. 14 evaluated point-of-care troponin testing in four EDs and found that the effect varied between settings, with length of stay in the ED being increased in one hospital and decreased in another. Kendall et al. 15 evaluated a variety of point-of-care tests in an ED and found that point-of-care testing reduced turnaround times and times to decisions being made, but did not influence clinical outcomes or length of stay. Overall, therefore, there is reasonable evidence that point-of-care testing reduces turnaround times and possibly also times to decisions being made, but no consistent evidence of an effect on length of stay. One crucial component may be the need to incorporate point-of-care testing within a decision-making protocol.
Combining markers in a panel aims to overcome the limitations of individual markers. Different cardiac markers have optimal sensitivity for AMI at different times after symptom onset. 16,17 Combining markers to form a panel that is positive if any one marker is positive should optimise sensitivity and ensure that fewer cases of AMI are missed. Measuring the gradient rise of markers between baseline (when the patient arrives at hospital) and a specified time later (typically 90 minutes) has been shown to improve early sensitivity18,19 and can also improve specificity if only the gradient rise (rather than the absolute value) is considered positive.
Most studies of point-of-care cardiac marker panels have evaluated the combination of CK-MB (mass), myoglobin and troponin I measured at presentation and 90 minutes later. These have shown that the panel has high sensitivity and can accurately rule out AMI by 90 minutes after presentation. 20–27 This results in earlier identification of AMI than with laboratory testing22 and expedited decision-making with turnaround times reduced by 55%. 23 Meanwhile, comparison of patient management with the panel with previous practice showed a 40% reduction in coronary care unit admissions. 24
These studies show that the point-of-care combination of CK-MB (mass), myoglobin and troponin I measured at presentation and 90 minutes has appropriate diagnostic accuracy, but they do not reliably tell us whether the panel will alter patient care, improve outcomes or reduce health service costs. Early diagnostic accuracy and reduced turnaround times will lead to changes in practice only if clinicians act upon the additional diagnostic information. The before and after study by Ng et al. 24 may be confounded by changes in coronary care referrals over time and, originating from the USA where coronary care usage is much higher than in the UK, may not be applicable to the UK NHS. To date, there have been no randomised trials comparing marker panels with routine practice.
Existing data therefore suggest that point-of-care cardiac markers can accelerate decision-making in the ED and that a panel of markers consisting of troponin, CK-MB (mass) and myoglobin, measured at baseline and 90 minutes later, can accurately identify patients with AMI. This strategy could allow rapid and accurate diagnosis in the ED or clinical decision unit, facilitating hospital admission for those with AMI and discharge home for those without. However, such a strategy needs to be evaluated in practice and compared with current management before it can be recommended for widespread adoption throughout the NHS. Evaluation allows us to determine whether clinical decision-making is changed by using the point-of-care panel, whether hospital admissions are actually reduced in practice, whether reduced hospital admissions save sufficient health service costs to compensate for the additional costs of point-of-care testing and whether patient outcomes are improved. In addition, advances in assay methodology for troponin suggest that troponin alone may be used to rule in and rule out AMI soon after presentation, and marker panels may be unnecessary. 28 This has also not been tested in a randomised controlled manner.
Research objectives
We aimed to measure the effect of using a point-of-care cardiac marker panel upon the following outcomes in patients presenting to the ED with suspected but not proven AMI:
-
the proportion of patients successfully discharged home after ED assessment
-
health utility and satisfaction with care
-
the use of coronary care beds and cardiac treatments
-
subsequent re-attendance at and/or re-admission to hospital
-
major adverse events (death, non-fatal AMI, life-threatening arrhythmia, emergency revascularisation or hospitalisation for myocardial ischaemia)
-
health and social care costs.
In addition, we planned to undertake secondary analysis of the data of the Randomised Assessment of Treatment using Panel Assay of Cardiac markers (RATPAC) trial to evaluate the TIMI (thrombolysis in myocardial infarction)29 and GRACE (Global Registry of Acute Coronary Events)30 clinical prediction scores and store blood samples taken from participants in the intervention arm to evaluate potential new or alternative markers. The results of the evaluation of TIMI and GRACE scores are presented in this report. Funding has been sought to undertake secondary blood sample analysis.
Chapter 2 Methods
Study design
We undertook a multicentre pragmatic RCT and economic evaluation of a point-of-care cardiac marker panel in the management of patients with suspected, but not proven, AMI in six EDs in the UK.
Setting
The participating hospitals were Barnsley District General Hospital, Derriford Hospital in Plymouth, Edinburgh Royal Infirmary, Frenchay Hospital in Bristol, Leeds General Infirmary and Leicester Royal Infirmary. They were selected to provide a range of different settings that reflected the variation in current NHS practice and the variation in facilities available to manage patients with acute chest pain. Table 1 outlines the characteristics of the participating centres.
| Annual ED attendances: 1 April 2008 to 31 March 2009 | No. of acute medical bedsa | ED facilities | On-site cardiology services | |
|---|---|---|---|---|
| Barnsley | 71,678 | 462 | – | CCU, rapid access clinic |
| Derriford | 85,341 | 240 | CDU | CCU, angioplasty, cardiac surgery, rapid access clinic |
| Edinburgh | 105,378 | 843b | – | CCU, angioplasty, cardiac surgery, rapid access clinic |
| Frenchay | 62,823 | 461 | CDU | CCU, angioplasty |
| Leeds | 109,362 | 491 | CDU | CCU, angioplasty, cardiac surgery, rapid access clinic |
| Leicester | 156,053 | 290 | – | CCU, rapid access clinic |
Participants
We recruited people presenting to the ED with chest pain due to suspected but not proven AMI in whom a negative point-of-care marker test could potentially rule out AMI and allow discharge home. All patients with chest pain were considered for participation but then excluded if they met any of the following criteria:
-
Diagnostic ECG changes for AMI or high-risk acute coronary syndrome (> 1 mm ST deviation or > 3 mm inverted T waves). These patients are at high risk of adverse outcome and require inpatient care even if marker tests are negative.
-
Known coronary heart disease (CHD) presenting with prolonged (> 1 hour) or recurrent episodes of typical cardiac-type pain. These patients have unstable angina and require inpatient care for symptom control even if marker tests are negative.
-
Proven or suspected serious non-coronary pathology (e.g. pulmonary embolus) that requires inpatient care even if AMI is ruled out.
-
Comorbidity or social problems that require hospital admission even if AMI can be ruled out.
-
An obvious non-cardiac morbidity (e.g. pneumothorax or muscular pain) in a patient in whom AMI can be excluded as a possible cause without resorting to further diagnostic testing.
-
Presentation more than 12 hours after the most significant episode of pain, in which case a single troponin measurement would clearly be more appropriate than point-of-care panel assessment.
-
Previous participation in the RATPAC trial.
-
Inability to understand the trial information owing to cognitive impairment.
-
Non-English-speaking patients for whom translation facilities were not available.
For every fourth week of trial recruitment the research nurse at each hospital examined ED attendance lists to identify patients attending with chest pain and record basic demographic details and reasons for exclusion. The huge number of attendances with chest pain meant that undertaking this process throughout the whole trial would have produced an excessive workload, whereas monitoring every fourth week achieved the aim of reporting sample selection within acceptable use of resources.
Recruitment and randomisation
Research nurses and ED staff identified eligible patients, provided trial information and obtained written consent. Participants were then randomly allocated to receive either (1) diagnostic assessment using the point-of-care biochemical marker panel or (2) conventional diagnostic assessment without the panel.
The Nottingham Clinical Trials Unit (CTU) generated a simple randomisation sequence, stratified by centre, which was not revealed to any person involved in patient recruitment. Recruiting doctors and research nurses accessed a secure website provided by Nottingham CTU and entered participant details. The CTU revealed the participant’s allocated treatment group to the ED only after the participant’s details were entered, written consent was confirmed and the participant irrevocably entered into the trial.
Planned interventions
Participants were randomised to receive either:
-
diagnostic assessment using the point-of-care biochemical marker panel
or
-
conventional diagnostic assessment without the panel.
The only difference between the two arms of the trial was that patients in the intervention arm received assessment with the point-of-care panel. The use of all other tests and treatments, and decision-making in the ED, was at the discretion of the attending clinician.
The point-of-care cardiac marker panel comprised CK-MB (mass), myoglobin and troponin I, measured at presentation and 90 minutes later, using the Siemens Stratus® CS Analyser (Dade Behring, Milton Keynes, UK). Clinical staff were trained to use the test and given guidance in interpretation of the results. We provided a recommended protocol that advised a first panel test immediately after initial ED assessment and a second panel test 90 minutes later, and then advised hospital admission or discharge on the basis of point-of-care results (Appendix 1). Decisions were ultimately at the discretion of clinical staff. We did not police the use of the point-of-care protocol to ensure that it was being followed.
Other than obtaining consent, collecting data and random allocation to use of the point-of-care test, the only change to routine practice was that we asked clinical staff to take an additional sample of blood for storage (without repeating venepuncture) each time a point-of-care blood sample was required. The additional blood sample taken after point-of-care testing was transported to the hospital laboratory to be centrifuged and the serum separated and then frozen. Batches of frozen samples were then transported quarterly to St George’s Hospital for longer-term storage and future secondary analysis of new biomarker assays.
The RATPAC trial was a pragmatic trial, intended to determine whether point-of-care panel assessment should be standard practice for patients presenting to the ED with suspected AMI. It was designed to compare two pragmatic alternatives (management with and without point-of-care panel assessment) under routine conditions to determine whether use of the test changes costs or outcomes. This pragmatic design had the following implications:
-
There was no attempt to blind clinical staff, patients or carers to the allocated treatment group after randomisation.
-
The point-of-care test was provided with a recommended protocol for use but management decisions were ultimately at the discretion of the clinical staff.
-
All other diagnostic tests and the use of laboratory blood tests in the control group were at the discretion of the clinical staff. Blood samples were taken only for the purposes of clinical management. We did not take blood samples at additional time points to evaluate theoretical management strategies or to evaluate the accuracy of diagnostic assessments. The blood samples taken for storage and future analysis were taken at the same time as the point-of-care samples used for clinical care and did not require additional venepuncture.
Point-of-care assays and analyser
Cardiac troponin I, CK-MB (mass) and myoglobin were measured by point-of-care testing in the ED on the Stratus CS Analyser. The original project protocol planned to use the Biosite Triage® analyser (Biosite, San Diego, CA, USA), as this was the most widely used analyser at the time of protocol development. However, as protocol development continued it became apparent that new-generation high-sensitivity troponin assays, as well as point-of-care analysers with high-sensitivity troponin assays, were becoming available. 31 Discussions with experts in the field revealed reservations about the analytical performance of the Biosite system, particularly the analytical sensitivity of its troponin assay. In view of this, and the need to ensure the future generalisability and the development towards more sensitive troponin assays, it was deemed prudent to use a more sensitive system in the trial. Recent publications7,8 have since confirmed the potential of high-sensitivity troponin assays to improve early assessment.
The Stratus CS Analyser was selected on the basis of having most data as an instrument suitable both for the emergency laboratory and for use as a point-of-care instrument,32–34 as well as a troponin assay with performance characteristics close to current recommendations for analytical goals. It has the advantage that it can measure troponin, myoglobin and CK-MB (mass) on the same sample. 35 In addition, it has the health and safety advantages of using a closed system that does not require the operator to open the blood tube and pipette the sample on to a test strip.
Blood is drawn directly into a Vacutainer tube (a standard no-touch aseptic blood-sampling system) using lithium heparin as an anticoagulant. The tube is then inverted twice to mix the blood and anticoagulant. It is then ready for analysis. The sample is introduced directly into the machine, the sample door closed and analysis is fully automatic.
The analytical characteristics of the assays were as follows. Cardiac troponin I: detection limit 0.02 µg/l, analytical range 0.02–50 µg/l, interassay coefficient of variation (CV) 4.3–5.1% (0.03–0.22 µg/l). The 99th centile of the assay is 0.07 µg/l. Myoglobin: detection limit 1 µg/l, analytical range 1–900 µg/l, interassay CV 1.9–12.7% (56–308 µg/l); 95% reference interval, males 21–98 µg/ml, females 19–56 µg/l, combined 20–82 µg/l. CK-MB (mass): detection limit 0.3 µg/l, analytical range 0.3–150 µg/l, interassay CV 0.15–1.27% (3.7–39.3 µg/l); 95% reference interval 0.6–3.5 µg/l.
The recommended point-of-care protocol
The point-of-care protocol is outlined in Appendix 1. The sample(s) were deemed positive and hospital admission advised if any of the following were met:
-
any CK-MB (mass) exceeded 5 µg/l
-
the CK-MB (mass) gradient exceeded 1.6 µg/l
-
the myoglobin gradient increase exceeded 25% of the baseline value
-
troponin exceeded 0.02 µg/l.
The CK-MB (mass) gradient is based on data from Fesmire et al. ,18,19 suggesting that this value allows optimisation of both sensitivity and specificity. The myoglobin gradient was used alone because absolute myoglobin levels have very poor specificity, whereas using only the gradient rise maintains the value of early sensitivity without compromising specificity.
The Stratus CS Analyser is able to detect troponin levels in the range of 0.03–0.07 µg/l even though this is below the 99th centile of normal, which is conventionally used as a diagnostic threshold. Our understanding of the significance of very low troponin levels developed during the trial and is continuing to develop now (indeed we anticipate that secondary analysis of RATPAC blood samples will provide a further contribution). At the start of the trial we decided to err on the side of caution and recommended that any detectable troponin above 0.02 µg/l should be considered positive in case it represented the start of a significant rise. However, it became apparent from emerging data that levels between 0.03 and 0.07 µg/l do not typically represent an early troponin rise and where they do this is evident upon measuring the difference between baseline and 90-minute values. We therefore amended the guidance to recommend that the tests only be considered positive on the troponin assay if:
-
any troponin level exceeded 0.07 µg/l
or
-
the initial troponin is below 0.03 µg/l and the second sample is above 0.02 µg/l.
The new guidance was disseminated and implemented between December 2008 and February 2009.
Standard care
The standard-care group were managed without point-of-care panel assessment according to existing guidance for management of low-risk chest pain due to suspected ACS at each of the participating hospitals. We deliberately selected a variety of hospitals that were operating a range of different strategies and had a variety of different facilities available for chest pain management. Table 2 summarises the location of care and biomarker(s) used for low-risk patients in each hospital. Patients were referred to the cardiology team if biomarkers were positive and discharged (with or without exercise treadmill testing) if negative.
| Location | Troponin assay | Troponin threshold used (µg/l) | Laboratory analyser | Timing of troponin (hours)a | Other biomarkers | |
|---|---|---|---|---|---|---|
| Barnsley | Inpatient ward | Siemens Centaur Troponin I Ultra | < 0.20 | Siemens Centaur XP | 12 | |
| Derriford | CDU | Roche Troponin T | < 0.01 | Roche Modular E170 | 6 | |
| Edinburgh | Medical Assessment Unit | Abbott STAT Troponin I | < 0.05b | Architect i2000SR | 12 | Creatine kinase |
| Frenchay | CDU | Beckman Coulter Access Accu Troponin I | < 0.06 | Beckman Coulter Access 2 | 12 | |
| Leeds | CDU | Siemens Centaur Troponin I Ultra | < 0.05 | Siemens Centaur XP | 12 | |
| Leicester | Inpatient ward | Siemens Centaur Troponin I Ultra | < 0.06 | Siemens Centaur XP | 12 | Creatine kinase |
Outcome measures
The primary outcome was the proportion of patients successfully discharged home after ED assessment. To be considered successfully discharged the patient had to have both (1) left the hospital or be awaiting transport home with a discharge decision having been made at 4 hours after initial presentation and (2) suffered no major adverse event (as defined below) during the following 3 months.
Secondary outcomes were:
-
Health utility measured using the European Quality of Life-5 Dimensions (EQ-5D) self-complete questionnaire at 1 and 3 months after attendance.
-
Satisfaction with care measured at 1 month after attendance using a modified Group Health Association of America questionnaire that had been used successfully in previous studies of diagnostic strategies for acute chest pain. 10,36
-
The proportion of patients admitted to the coronary care unit, receiving cardiac medications (aspirin, heparin, clopidogrel or glycoprotein IIb/IIIa inhibitors) or receiving cardiac interventions [angiography, percutaneous coronary intervention (PCI) or bypass grafting].
-
Length of initial hospital stay and total inpatient days over 3 months.
-
Re-attendance at and/or re-admission to hospital and outpatient attendances over the following 3 months.
-
Adverse events (death, non-fatal AMI, life-threatening arrhythmia, emergency revascularisation or hospitalisation for myocardial ischaemia).
-
The proportion of admitted patients ultimately diagnosed as having AMI by the universal definition. 3
We selected successful discharge home as the primary outcome because the main purpose of point-of-care cardiac marker testing in this patient group is to facilitate discharge home. This outcome is beneficial for patients, who avoid the inconvenience and risks of hospital admission, and is beneficial for the health service, which avoids unnecessary admissions and pressure upon acute and emergency services. Patients who suffered an adverse event after discharge were not classified as a successful discharge home because they may have benefited from hospital admission. We also recorded the proportion of admitted patients who were ultimately diagnosed as having AMI to provide a measure of the appropriateness of admissions.
Assessment of outcomes
Recruiting staff recorded baseline data (including the variables required to calculate the TIMI or GRACE score), the results of initial assessment (including any biochemical cardiac tests) and admission or discharge from the ED. Research nurses then used ED and hospital inpatient notes to record management decisions at initial attendance and admission, extract resource use data and identify subsequent attendances/admissions and adverse events up to 3 months. Time and date of discharge for the initial admission were recorded as precisely as possible using computer records, case notes and contact with hospital staff. The total number of inpatient days over 3 months was recorded using hospital notes. An inpatient day was defined as being an overnight stay.
Research nurses checked patient status (dead or alive) at 1 and 3 months, using hospital information systems. Deceased patients were assumed to have a score of zero on EQ-5D and were excluded from other patient-based assessments. Participants who were not recorded as dead were mailed a questionnaire at 1 and 3 months from the University of Sheffield to identify adverse events and hospital attendances, health and social care resource use, and measure EQ-5D and satisfaction with care (satisfaction at 1 month only). Our previous study suggested a 70–80% response rate to this questionnaire. 10,37
A single reviewer (SG) blinded to treatment group classified all ED re-attendances, subsequent hospital admissions and outpatient reviews as either potentially chest pain related (including non-cardiac conditions that could have initially presented as chest pain) or clearly non-chest pain related (such as limb injuries).
The initial working diagnosis and final diagnosis were recorded and categorised by the research nurse, based upon the diagnosis recorded in the notes by the most senior clinician at the end of initial ED assessment and at the end of hospital admission, respectively. Patients were classified as having AMI on the basis of the presence of a rise in their troponin level above the diagnostic threshold of the relevant assay and absence of a final diagnosis of an alternative condition (such as sepsis or pulmonary embolism) that could have produced a troponin elevation. Patients with a troponin rise consistent with AMI and final diagnosis of ACS or ‘other AMI’ were classified as having AMI. Patients with no troponin rise and a final diagnosis that was not ACS or ‘other AMI’ were classified as not having AMI. A single reviewer blinded to treatment group reviewed the initial and next-day ECGs of patients with a final diagnosis of ACS and no troponin rise and categorised these patients as having AMI if an ECG showed ST elevation and coronary reperfusion was performed, otherwise they were categorised as having no AMI. Two independent reviewers blinded to treatment group reviewed case details of all patients with a troponin rise and a final diagnosis other than ACS or ‘other AMI’, and all patients with a troponin rise that was inconsistent with AMI (e.g. if a positive troponin was shortly followed by a negative troponin result). Each decided whether AMI was the most likely diagnosis. Disagreements were resolved by discussion and patients classified as having AMI or no AMI.
Proposed sample size
We planned to recruit 3130 participants to the trial. A previous randomised trial in this patient group suggested that around 50% of the control group would be successfully discharged. 10 With 1565 evaluable subjects in each arm of the trial we expected to have 80% power to detect a 5% absolute difference in the primary outcome (50% vs 55%) at the two-sided significance level of 5%. The same sample size provided 80% power to detect a 2% absolute difference (2% vs 4%) in major adverse events (death, non-fatal AMI, life-threatening arrhythmia, emergency revascularisation or hospitalisation for myocardial ischaemia), again at the two-sided 5% level of significance.
We estimated that we would require six hospitals to recruit for 12 months each to achieve the sample size of 3130, assuming that we recruited 70% of those eligible. Previous studies of this specific patient group undertaken by our team had shown that recruitment of 550 suitable patients per year is attainable at a typical hospital. 10,37–39
Previous studies had also shown a response rate of 70–80% for postal questionnaires,10,37 thus providing an effective sample size of at least 1000 in each of the two groups to evaluate health utility, satisfaction with care and health service resource use.
Statistical analysis
We planned to analyse the primary outcome through logistic regression, fitting concurrently with intervention group the effect of centre and appropriate baseline measures (including age, gender and past history of CHD), to present adjusted odds ratios (ORs) along with their corresponding 95% confidence intervals (CIs). A similar analysis was used for adverse events. Primary analysis was on an intention-to-treat basis. Secondary analysis excluded those who were not managed according to their allocated strategy.
We undertook a descriptive assessment to explore whether use of biochemical cardiac markers or admission rates changed over time in either the intervention or control group, either as a result of staff ‘learning curves’ in the intervention group or as a result of contamination of the control group.
For each patient, GRACE and TIMI scores were calculated. Two approaches were undertaken for calculating each score: one using the first available troponin sample and the second using the highest troponin level taken during the initial hospital stay. Some patients did not have all of the data items that were required to permit calculation of the score so two analyses were undertaken: one including only cases with complete data, the other including all cases with imputation to complete the data set. Imputation involved assuming that missing variables would be negative or, where appropriate, the mean value for the RATPAC population (e.g. continuous variables used in the GRACE score). We evaluated the predictive value of GRACE and TIMI by calculating the proportion with an adverse outcome at 1 and 3 months in each quintile of GRACE score and each TIMI category. We then calculated the area under the receiver-operator characteristic curve (c-statistic) for each score.
Economic evaluation
Economic evaluation was undertaken alongside the trial using recommended practice. 40 In addition, a cost-effectiveness model was developed to duplicate the trial results (as a way of validation) and extrapolate the results to longer follow-up periods. The NHS perspective was undertaken and other methods were in line with National Institute for Health and Clinical Excellence (NICE) Technology Appraisal Guidelines. 41
Resource use data were collected for all patients covering the length of time in the ED, the use of diagnostic tests, admissions, re-admissions, outpatient reviews and cardiac procedures. Cost and outcome data were collected using patient notes and self-completed questionnaires as described previously. A small microcosting study of 30–40 patients was carried out at each site, gathering data on staff times relating to the care of patients. ED cost per minute was based on a study previously undertaken by the investigators,10 and amended using the microcosting data from this study. Panel costs were based on purchase price, and the remaining costs were valued using national unit costs. 42,43 Total NHS costs up to 3 months after initial attendance were calculated. Quality-adjusted life-years (QALYs) were calculated by the trapezium rule using the EQ-5D tariff values at all follow-up points.
Economic analysis
Both cost and QALY analysis compared bootstrap estimates of the mean cost per patient of the two groups. Cost-effectiveness analysis estimated the incremental cost per QALY of using point-of-care cardiac marker testing compared with management without point-of-care panel assessment. Results were plotted on the cost-effectiveness plane and then transformed into cost-effectiveness acceptability curves with their associated frontier. 44 A sensitivity analysis was undertaken to include production losses as reported by the patient.
We anticipated that some of the resource use and QALY data would be incomplete (missing). Thus, in order to maximise the information collected from the trial, we imputed missing values using multiple imputation within Stata (StataCorp LP, College Station, TX, USA). 45 The idea of multiple imputation draws from the fact that missing values from incomplete data are unknown and the technique of multiple imputation imputes more than one likely value for the missing data; hence, providing an unbiased representation of uncertainty. 46 Thus, an additional set of results is available from the imputed cost and QALY data.
Decision-analytic model
We constructed a decision-analytic model to describe the care observed in the trial, and likely care pathways subsequent to it. This allowed us to systematically investigate the impact of subsequent costs, quality of life and survival. These values were initially based on population norms, but then replaced with literature review estimates where appropriate. Finally, they were replaced by RATPAC trial estimates where required. The decision-analytic model was probabilistic, but with conventional sensitivity analysis used to assess the impact of structural uncertainties. 47
The main purpose of the decision-analytic model was to explore the potential impact of changes in the major adverse event rate upon cost-effectiveness. We anticipated that the trial would have adequate power to detect economically important differences in resource use and EQ-5D, but would only have limited power to detect differences in major adverse events. It was therefore possible that an intervention could be apparently cost-effective (in terms of reducing costs and increasing health utility), while being associated with a statistically non-significant increase in adverse events. The decision-analytic model was intended to explore whether uncertainty around the effect of the intervention upon the major adverse event rate could influence the potential cost-effectiveness of the intervention.
The decision-analytic model used trial data to estimate costs and QALYs up to 3 months. Beyond this, we used lifetime cost and QALY estimates from a previous economic analysis of a similar population. 48 The mean lifetime cost of care for a patient with CHD was estimated to be £10,079 [standard error (SE) = £2200] and mean QALYs accrued were estimated to be 6.829 (SE = 0.34), while estimates of £0 and 20 QALYs [standard deviation (SD) = 5] were used for patients without CHD. Trial data were used to estimate the rates of death and non-fatal AMI by 3 months after management with the point-of-care panel and standard care. We then assumed that those who had died by 3 months would accrue no further costs or QALYs, while those who survived with non-fatal AMI would accrue costs and QALYs associated with CHD. Probabilistic sensitivity analysis was then used to estimate the mean lifetime costs and QALYs of patients in the two arms of the trial.
The decision-analytic model was developed and implemented in an Excel (Microsoft Corporation, Redmond, WA, USA) spreadsheet, which included a Visual Basic macro to implement the replication of outcomes from the model. The results presented were based on 10,000 outcome simulations. The pathways through the model described (1) the choice of diagnostic tool, using either point-of-care tests or standard-care approaches; (2) the disposal from the ED, either to admission or to discharge; (3) possible interventions or care once a positive diagnosis of ACS (and therefore admission) was made, including coronary artery bypass graft (CABG), PCI, thrombolysis, hospital stay with no intervention or thrombolysis treatment, and discharge without intervention in the case of a negative updated diagnosis, or death from ACS while in hospital; and (4) possible ACS events after discharge without diagnosis from the ED, which resulted in subsequent admission and treatment according to the treatments outlined in (3). Costs were accrued as simulated patients passed through the various stages in the model, and benefits were accrued at the end of each passage. Once discharged, either from ED or after admission, costs were assumed to be unaffected by the choice of diagnostic technique, and therefore costs of follow-up care or intervention were combined across both arms.
Ethical arrangements
All participants were asked to provide written, informed consent. Although participants were recruited in an emergency setting and there was only a limited amount of time available for considering trial information, the nature of the selected group (in particular the exclusion of people clearly requiring hospital treatment) ensured that eligible patients would not be incapacitated by their medical condition. We did not therefore make provision for recruitment of incapacitated patients by personal or professional legal representatives.
Ethical approval was granted by Leeds East Research Ethics Committee and review provided by the local research ethics committee (LREC) at each participating centre. The trial was conducted in accordance with Medical Research Council (MRC) Guidelines for Good Clinical Practice in Clinical Trials. 49 The University of Sheffield was the sponsor for the trial. The Trial Steering Committee (TSC) consisted of the Chief Investigator (SG), one of the co-applicants (PC), an independent chair (MF), two independent members (SH, JK) and a consumer representative (EH). We also invited a representative of the funder to join the committee but this was declined. The Data Monitoring and Ethics Committee (DMEC) consisted of an independent statistician (HT), emergency physician (WT) and cardiologist (JG), who were asked to review trial data at regular intervals and implement stopping rules in accordance with MRC guidance. The Trial Management Group (TMG) consisted of the Chief Investigator, co-applicants, Principal Investigator at each site, project manager, statistician, health economist and research nurses.
Data management
Trial data were collected on the case report form and follow-up form and then entered by the research nurses into an online database provided on a secure central server by the Sheffield CTU. The system had a full electronic audit trail. Quality control procedures were applied to validate the trial data. The project manager undertook quarterly data monitoring visits to each site, at which a random sample of data forms were checked for errors and validated against source documents. Any errors, protocol deviations or violations were reviewed with the research nurse and Principal Investigator and documented in the site files and master file. Central monitoring involved flagging discrepant or questionable data. Error reports were generated where data clarification was required. Monthly outputs were generated for the TMG and TSC summarising baseline characteristics of patients recruited, follow-up rates, data completion and adverse events by study site, but not study group. Outcome data summaries were provided for closed meetings of the DMEC. All activities were performed in accordance with Sheffield CTU standard operating procedures.
Trial progress
The project started on 1 April 2007. We planned to recruit staff and gain ethical and local governance approvals in months 1–6, recruit patients in months 7–18 and complete follow-up, data analysis, writing-up and dissemination in months 19–24. Actual trial progress was slower than anticipated: staff recruitment, ethics and regulatory approvals took up to 12 months to complete, patient recruitment was slower than expected, and the trial was terminated before the target of 3130 patients was recruited. The Gantt chart in Figure 1 outlines the planned and actual trial progress.
FIGURE 1.
Gantt chart of trial timetable and progress.
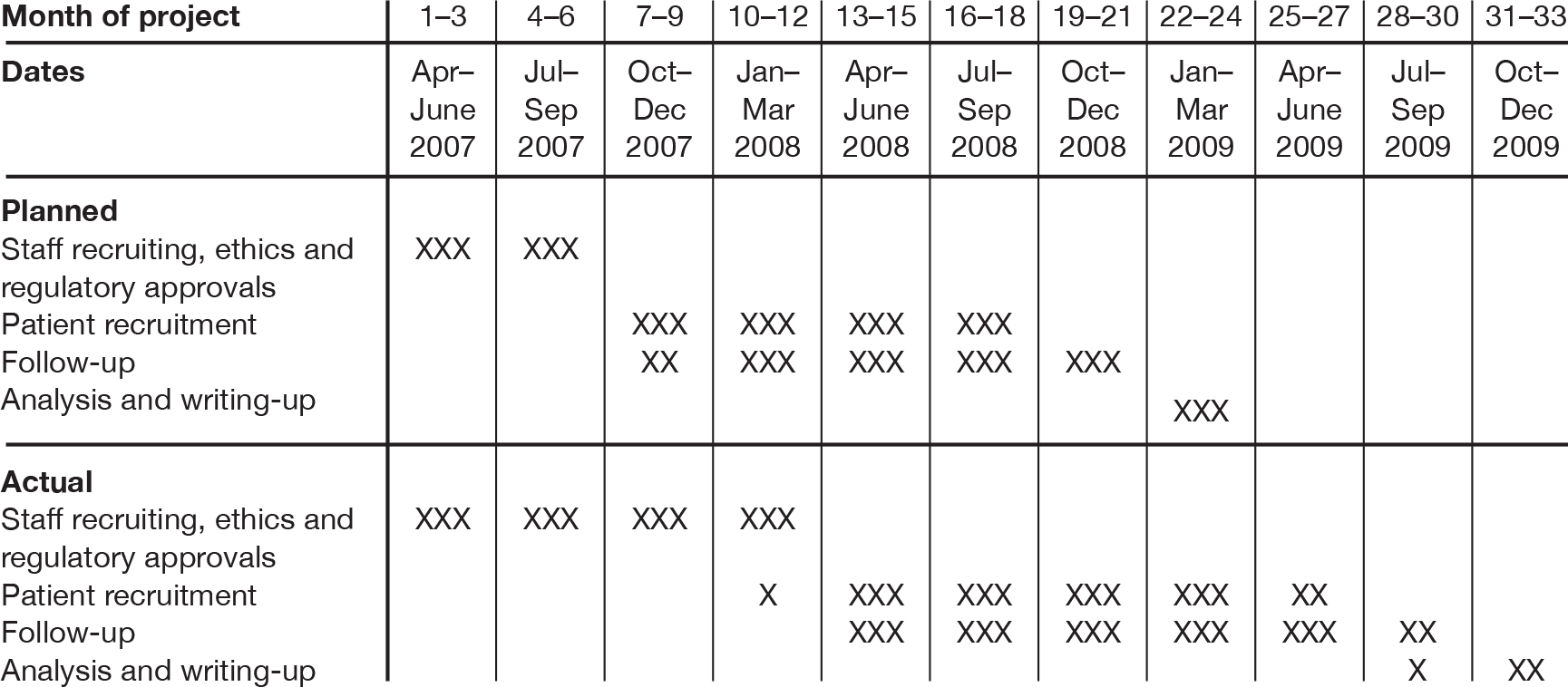
Table 3 shows the timing of processes at the six participating centres.
| Site | Contract signed with Sheffield | Memorandum of Understanding signed with Siemens | Approved by LREC | Approved by Hospital Research Office | Research nurse appointed | Recruitment started |
|---|---|---|---|---|---|---|
| Barnsley | 6 August 2007 | 14 September 2007 | 7 June 2007 | 22 May 2007 | 1 October 2007 | 30 January 2008 |
| Derriford | 22 November 2007 | 3 January 2008 | 5 July 2007 | 4 January 2008 | 26 November 2007 | 25 February 2008 |
| Edinburgh | 28 August 2007 | 4 December 2007 | 11 June 2007 | 15 June 2007 | 14 January 2008 | 2 April 2008 |
| Frenchay | 22 November 2007 | 23 October 2007 | 18 July 2007 | 28 November 2007 | 9 January 2008 | 31 March 2008 |
| Leeds | 9 September 2007 | 17 September 2007 | 5 July 2007 | 18 June 2007 | 3 March 2008a | 16 April 2008 |
| Leicester | 1 October 2007 | 5 October 2007 | 25 June 2007 | 19 September 2008 | 14 January 2008 | 25 March 2008 |
The reasons for the set-up phase being longer than expected are as follows:
-
The process of drawing up contracts between the sponsor (the University of Sheffield) and the six participating hospitals took longer than expected.
-
Local ethics and regulatory approvals could not be processed at some sites until contracting had been completed.
-
The time taken to provide local ethics and, in particular, governance approvals varied between hospitals.
-
In addition to trial training, some of the participating hospitals required all staff involved in the trial, including recruiting doctors, to have received formal training in Good Clinical Practice (GCP), even though the RATPAC trial was not a trial of an investigational medicinal product.
Patient recruitment was slower than anticipated and varied between 300 and 400 patients per centre per year of recruitment, compared with the expected 550 per centre per year. Details of patient recruitment are reported below (see Results) but it appeared that failure to achieve the expected recruitment rate was due to failure to recruit the expected proportion of eligible patients rather than any lack of eligible patients. Overall 35% (604/1719 – see Table 4) of eligible patients were recruited instead of the 70% anticipated. The following factors are likely to have accounted for this difference:
-
The requirement for GCP training resulted in doctors being unwilling to assist with trial recruitment. This varied between centres, depending upon the local regulatory requirements. In one centre the need for all doctors to receive formal GCP training resulted in most patients being recruited by research nurses. This centre recruited the lowest proportion of eligible patients (19%).
-
Service commitments, particularly the target of discharging 98% of patients from the ED within 4 hours of arrival, discouraged ED staff from recruiting patients during busy times.
-
The recall of point-of-care testpaks (described below) may have reduced the confidence of ED staff in the technology and thus their willingness to recruit patients to the trial.
| All centres | Barnsley | Derriford | Edinburgh | Frenchay | Leeds | Leicester | |
|---|---|---|---|---|---|---|---|
| Recruitment | |||||||
| No. of days recruiting | 2658 | 490 | 464 | 427 | 429 | 413 | 435 |
| No. of patients recruited | 2263 | 327 | 328 | 457 | 469 | 353 | 329 |
| No. of patients recruited/day | 0.9 | 0.7 | 0.7 | 1.1 | 1.1 | 0.9 | 0.8 |
| Screeninga | |||||||
| No. of days screening | 667 | 124 | 135 | 106 | 102 | 92 | 108 |
| No. of patients screened | 9109 | 1164 | 1632 | 1769 | 662 | 1760 | 2122 |
| No. of patients screened/day | 13.7 | 9.4 | 12.1 | 16.7 | 6.5 | 19.1 | 19.6 |
| No. (%) of patients recruited | 604 (7) | 83 (7) | 93 (6) | 123 (7) | 111 (17) | 90 (5) | 104 (5) |
| No. (%) of patients not recruited | 8505 (93) | 1081 (93) | 1539 (94) | 1646 (93) | 551 (83) | 1670 (95) | 2018 (95) |
| Reason for non-recruitment [n, (%)] | |||||||
| Diagnostic ECG changes | 1295 (14) | 74 (6) | 153 (9) | 221 (12) | 137 (21) | 401 (23) | 309 (15) |
| Known CHD with prolonged/recurrent episodes | 1378 (15) | 232 (20) | 456 (28) | 271 (15) | 58 (9) | 150 (9) | 211 (10) |
| Proven/suspected serious non-coronary pathology | 724 (8) | 54 (5) | 186 (11) | 60 (3) | 37 (6) | 219 (12) | 168 (8) |
| Comorbidity or social problems requiring admission | 414 (5) | 9 (< 1) | 70 (4) | 223 (13) | 5 (< 1) | 70 (4) | 37 (2) |
| Obvious non-cardiac | 2506 (28) | 407 (35) | 369 (23) | 488 (28) | 210 (32) | 431 (24) | 601 (28) |
| Presented > 12 hours after most significant pain | 465 (5) | 13 (1) | 45 (3) | 182 (10) | 10 (2) | 53 (3) | 162 (8) |
| Previous participant | 21 (< 1) | 1 (< 1) | 3 (< 1) | 3 (< 1) | 2 (< 1) | 8 (< 1) | 4 (< 1) |
| Unable to understand trial information | 109 (1) | 16 (1) | 23 (1) | 26 (1) | 9 (1) | 3 (< 1) | 32 (2) |
| Non-English speaking | 29 (< 1) | 2 (< 1) | 7 (< 1) | 7 (< 1) | 4 (< 1) | 0 | 9 (< 1) |
| Refused consent | 40 (< 1) | 2 (< 1) | 4 (< 1) | 8 (< 1) | 1 (< 1) | 8 (< 1) | 17 (< 1) |
| Eligible but recruitment not sought | 1115 (12) | 265 (23) | 171 (10) | 60 (3) | 37 (6) | 148 (8) | 434 (20) |
| Prisoner | 8 (< 1) | 0 | 1 (< 1) | 2 (< 1) | 0 | 4 (< 1) | 1 (< 1) |
| Other | 161 (2) | 6 (< 1) | 14 (< 1) | 29 (2) | 20 (3) | 60 (3) | 32 (2) |
| Unknown | 240 (3) | 0 | 37 (2) | 66 (4) | 21 (3) | 115 (7) | 1 (< 1) |
Changes to protocol and other unanticipated events
A number of unanticipated events occurred during the trial, two of which resulted in changes to the protocol. Most of the unanticipated events related to the intervention being evaluated. Point-of-care cardiac markers are a developing technology and clinicians are learning more about the technology as it is implemented. Even mature technologies can be subject to unanticipated problems, especially if used by a wide range of staff in a variety of settings.
The unanticipated events were:
-
change of the point-of-care analyser
-
recall of point-of-care testpaks
-
amendment of the point-of-care protocol
-
early termination of trial recruitment.
The first and third of these resulted in changes to the protocol. In addition, we became aware that we had not included life-threatening arrhythmia as a major adverse event in the trial protocol, although it was included as such in the case report form. We therefore amended the protocol to include life-threatening arrhythmia as a major adverse event.
Change of the point-of-care analyser
This occurred during protocol development and before the trial started. Details and the reasons for this are outlined above (see Point-of-care assays and analyser).
Recall of point-of-care testpaks
Siemens informed the RATPAC research team on 4 July 2008 of the need to recall point-of-care assays for troponin I because of a faulty batch that could have produced low false-positive levels. All six sites were affected, resulting in a suspension of recruitment at all sites for up to 1 week between 4 and 14 July 2008 until new assays were delivered. A meeting was held with Siemens and the biochemical expert on the Research Team (PC) to address concerns. It was concluded that quality assurance was tight, particularly with high-risk tests, so this would be a rare rather than a common occurrence. It was also discussed in length at a TMG meeting in July 2008. It was agreed that patients with a potentially false-positive point-of-care troponin result would have received appropriate follow-up based on their 12-hour troponin level and clinical condition, and therefore no additional intervention was required.
Amendment of the point-of-care protocol
Details and the reasons for this are outlined above (see The recommended point-of-care protocol).
Early termination of trial recruitment
Owing to the set-up delays and slower than anticipated recruitment, a request for additional funding was submitted to allow completion of the trial. The DMEC undertook a futility analysis (Appendix 2). This showed that the conditional power for the primary outcome recalculated using data to May 2009 was > 99.9% and the conditional power against major cardiac events was < 10%. In the light of this analysis, the trial funder declined the request for additional funding, based on clear efficacy (primary outcome) and futility (major cardiac events), and, consequently, trial recruitment was terminated on 2 June 2009, with a total of 2263 patients recruited. The findings reported here represent those as accrued at the point at which this decision to halt the trial was taken.
Chapter 3 Results
Screening, recruitment, randomisation and follow-up
Patients were recruited between 30 January 2008 and 2 June 2009. Table 4 summarises the process of screening and recruitment at each centre. All hospitals stopped recruitment on 2 June 2009 but the staggered start meant that some recruited for more days than others. Overall, 2263 patients were recruited over a total of 2658 hospital-days (0.9 patients per day). The total at each site ranged from 327 to 469, and the recruitment rate ranged from 0.7 to 1.1 per day.
Research nurses screened 9109 chest pain attendances on a total of 667 hospital-days during the trial (13.7 patients per hospital per day). Overall, 1719/9109 (19%) were eligible for recruitment (2.6 per hospital per day), of whom 604/1719 (35%) were recruited. The proportion of eligible patients recruited varied across the sites from 19% to 75%.
The proportions excluded for each exclusion criterion were as follows: 14% had ECG changes, 15% had known CHD with prolonged or recurrent pain, 8% had suspected serious non-CHD pathology, 5% had comorbidities or social problems, 28% had obvious non-cardiac pain, 5% presented > 12 hours since their worst pain, < 1% had previously participated in the trial, 2% were unable to understand the trial information owing to cognitive impairment or being non-English speaking, 2% had other exclusion criteria, 3% had an unknown reason for exclusion and < 1% declined consent. These proportions varied across the sites. We were unable to verify whether this was due to differences in coding and classification, or differences in population characteristics.
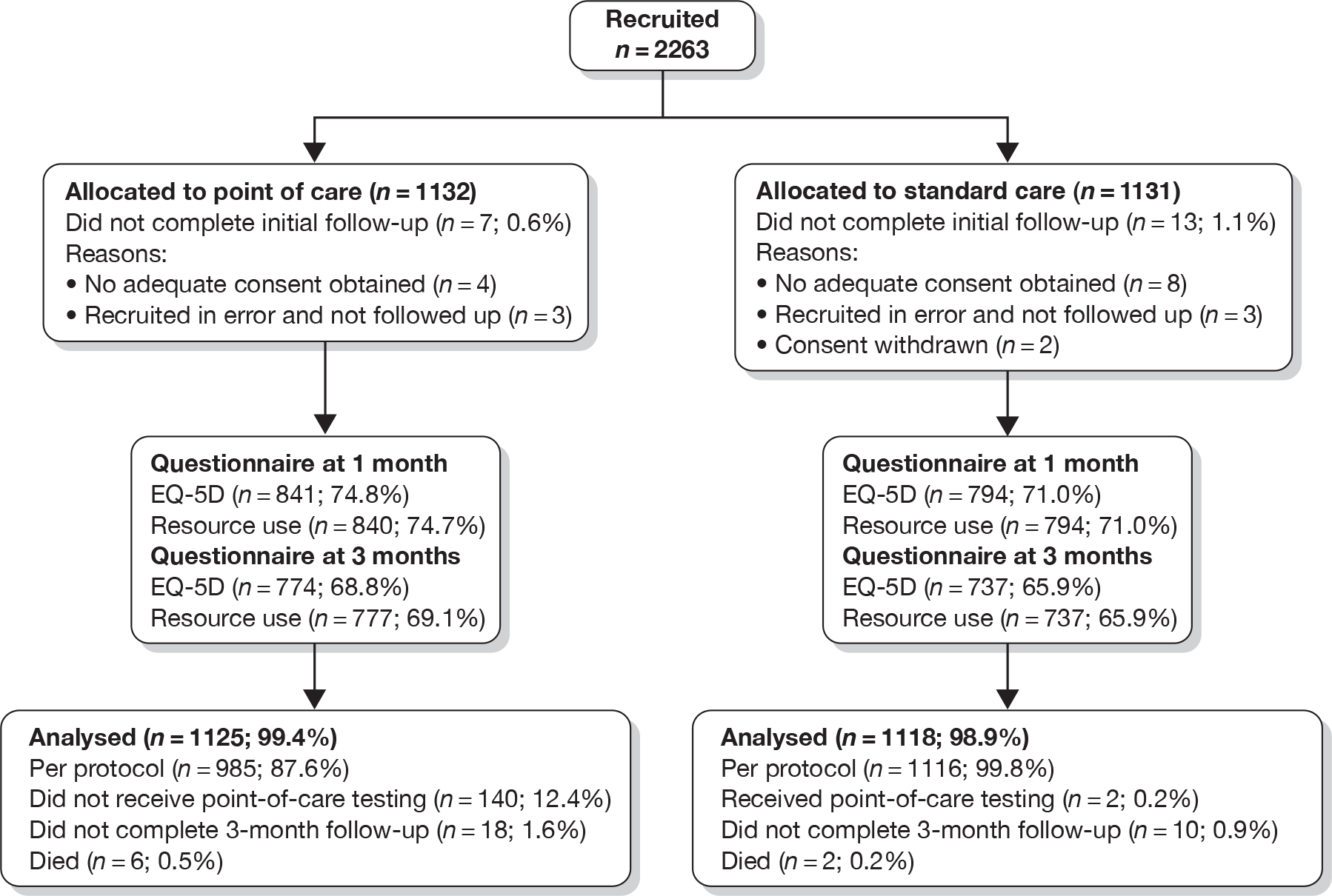
Figure 2 shows the flow of patients through the trial after recruitment and Table 5 shows withdrawals and losses to follow-up. We intended to follow up patients who were recruited in error, provided they did not withdraw their consent, so that all consented patients could be analysed as randomised. However, six patients (three in each group) were not followed up after being recruited in error for specific reasons necessitating their exclusion (prisoners, who were defined as ‘vulnerable groups’, n = 3; clinician error, n = 2; and recruitment when the trial was suspended, n = 1). In the point-of-care group seven patients withdrew or were lost to follow-up before any outcomes were recorded, and a further 18 withdrew or were lost to follow-up by the end of the trial. These numbers were 13 and 10, respectively, in the standard-care group. Questionnaire response rates were slightly higher in the point-of-care group: 75% at 30 days and 69% at 90 days, as opposed to 71% and 66%, respectively, for the standard-care group.
FIGURE 2.
CONSORT chart showing flow of patients after recruitment.
| PoC (n = 1132) | SC (n = 1131) | Total (n = 2263) | |
|---|---|---|---|
| Analysis population [n (%)] | |||
| Full analysis set | 1125 (99) | 1118 (99) | 2243 (99) |
| Failed to complete initial follow-up [n (%)] | |||
| No adequate consent obtained | 4 (< 1) | 8 (1) | 12 (1) |
| Withdrew consent before 4 hours | 0 | 2 (< 1) | 2 (< 1) |
| Recruited in error and not followed up | 3 (< 1) | 3 (< 1) | 6 (< 1) |
| Failed to complete 3-month follow-upa | 18 (2) | 10 (1) | 28 (1) |
| Incomplete questionnaire datab [n (%)] | |||
| No EQ-5D questionnaire at 1 month | 284 (25.2) | 324 (29.0) | 608 (27.1) |
| No EQ-5D questionnaire at 3 months | 351 (31.2) | 381 (34.1) | 732 (32.6) |
| No resource use questionnaire at 1 month | 285 (25.3) | 324 (29.0) | 609 (27.2) |
| No resource use questionnaire at 3 months | 348 (30.9) | 381 (34.1) | 729 (32.5) |
| No patient resource use questionnaires | 284 (25.2) | 322 (28.8) | 606 (27.0) |
Research nurses checked whether patients had been appropriately recruited and the results are shown in Table 6. In addition to the six patients who were randomised in error and not followed up (reported above), a total of 23 patients (1%) were inappropriately recruited: 12 in the point-of-care group and 11 in the standard-care group. These cases were followed up and analysed as planned on an intention-to-treat basis.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | |
|---|---|---|---|
| Number inappropriately randomised [n (%)] | 12 (1) | 11 (1) | 23 (1) |
| Reason [n (%)] | |||
| Diagnostic ECG changes | 2 (< 1) | 0 | 2 (< 1) |
| Known CHD with prolonged/recurrent episodes | 3 (< 1) | 5 (< 1) | 8 (< 1) |
| Proven/suspected serious non-coronary pathology | 1 (< 1) | 1 (< 1) | 2 (< 1) |
| Comorbidity or social problems requiring admission | 0 | 1 (< 1) | 1 (< 1) |
| Obvious non-cardiac | 2 (< 1) | 1 (< 1) | 3 (< 1) |
| Presented > 12 hours after most significant pain | 1 (< 1) | 2 (< 1) | 3 (< 1) |
| Other | 3 (< 1) | 1 (< 1) | 4 (< 1) |
Protocol violations
Some 140 patients (12.4%) in the point-of-care group did not receive testing as recommended in the protocol, i.e. two panels consisting of CK-MB (mass), myoglobin and troponin (or one panel if the first troponin was positive). Of these, 34 did not receive any point-of-care testing at all, typically because of machine failure or inability to use the machine, while the remainder had fewer tests than recommended in the protocol. Two patients (0.2%) in the standard-care group received point-of-care testing.
Baseline characteristics, emergency department findings and diagnosis
Table 7 shows the characteristics and demographics of the recruited patients. The characteristics were as anticipated, with a mean age of 54.5 years, more men than women, and 12% with a past history of CHD.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | |
|---|---|---|---|
| Age (years) | |||
| n | 1125 | 1118 | 2243 |
| Mean (SD) | 54.5 (13.8) | 54.6 (14.4) | 54.5 (14.1) |
| Median (IQR) | 53.4 (44–64) | 53.1 (44–64) | 53.2 (44–64) |
| Minimum, maximum | 22, 93 | 23, 96 | 22, 96 |
| Gender [n (%)] | |||
| Male | 683 (61) | 624 (56) | 1307 (58) |
| Female | 442 (39) | 494 (44) | 936 (42) |
| Centre [n (%)] | |||
| Barnsley | 162 (14) | 164 (15) | 326 (15) |
| Derriford | 164 (15) | 164 (15) | 328 (15) |
| Edinburgh | 228 (20) | 224 (20) | 452 (20) |
| Frenchay | 233 (21) | 231 (21) | 464 (21) |
| Leeds | 173 (15) | 171 (15) | 344 (15) |
| Leicester | 165 (15) | 164 (15) | 329 (15) |
| Past history of CHD [n (%)] | |||
| n | 1117 | 1110 | 2227 |
| No | 985 (88) | 973 (88) | 1958 (88) |
| Yes | 132 (12) | 137 (12) | 269 (12) |
| Previous MI | 60 (5) | 65 (6) | 125 (6) |
| Angina with positive diagnostic test | 46 (4) | 53 (5) | 99 (4) |
| Previous CABG | 12 (1) | 15 (1) | 27 (1) |
| Previous angioplasty | 37 (3) | 34 (3) | 71 (3) |
| Stenosis > 50% on angiography | 14 (1) | 12 (1) | 26 (1) |
| Unproven clinical label of CHD | 36 (3) | 31 (3) | 67 (3) |
| Other | 12 (1) | 10 (1) | 22 (1) |
| Risk factors [n (%)] | |||
| Diabetes | 86 (8) | 92 (8) | 178 (8) |
| Hypertension | 376 (34) | 361 (33) | 737 (33) |
| Hyperlipidaemia | 271 (26) | 282 (27) | 553 (27) |
| Present smoker | 310 (28) | 316 (29) | 626 (28) |
| Ex-smoker – last 10 years | 144 (13) | 129 (12) | 273 (13) |
| Cocaine abuse | 6 (1) | 10 (1) | 16 (1) |
| First-degree relative with angina/MI, onset age < 60 years | 344 (33) | 352 (34) | 696 (33) |
| Use of aspirin in previous 7 days | 207 (19) | 215 (20) | 422 (19) |
| > One episode rest angina in < 24 hours | 75 (7) | 74 (7) | 149 (7) |
| Source of referral [n (%)] | |||
| n | 1123 | 1115 | 2238 |
| Referred by GP | 188 (17) | 189 (17) | 377 (17) |
| Called emergency ambulance | 481 (43) | 510 (46) | 991 (44) |
| Self-referred | 419 (37) | 375 (34) | 794 (35) |
| Other | 35 (3) | 41 (4) | 76 (3) |
Table 8 shows the presenting characteristics of the patients. The median duration between the onset of worst pain and arrival at hospital was just over 2 hours, excluding the 1% who suffered their worst pain after hospital arrival. There were no marked differences between the study groups in the nature or duration of pain, or the associated features.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | |
|---|---|---|---|
| Worst pain onset [n (%)] | |||
| n | 1115 | 1103 | 2218 |
| Before attendance | 1102 (99) | 1090 (99) | 2192 (99) |
| After attendance | 13 (1) | 13 (1) | 26 (1) |
| Duration between onset of worst pain and arrival at hospital (minutes)a | |||
| n | 1102 | 1090 | 2192 |
| Mean (SD) | 241.3 (503.9) | 218.9 (325.1) | 230.2 (424.6) |
| Median (IQR) | 129.5 (79–240) | 128.5 (80–256) | 129.0 (80–246) |
| Duration of longest episode of worst pain (minutes) | |||
| n | 1083 | 1062 | 2145 |
| Mean (SD) | 97.1 (133.3) | 98.7 (127.2) | 97.9 (130.3) |
| Median (IQR) | 60.0 (20–120) | 50.0 (20–120) | 50.0 (20–120) |
| Type of pain [n (%)] | |||
| n | 1098 | 1081 | 2179 |
| Continuous | 815 (74) | 848 (78) | 1663 (76) |
| Intermittent | 283 (26) | 233 (22) | 516 (24) |
| n | 1034 | 1018 | 2052 |
| Single episode | 672 (65) | 695 (68) | 1367 (67) |
| Multiple episodes | 362 (35) | 323 (32) | 685 (33) |
| Main chest pain [n (%)] | |||
| n | 1114 | 1099 | 2213 |
| Indigestion/burning (n, %) | 81 (7) | 73 (7) | 154 (7) |
| Stabbing/sharp (n, %) | 219 (20) | 240 (22) | 459 (21) |
| Aching/dull (n, %) | 273 (25) | 294 (27) | 567 (26) |
| Gripping/crushing/heavy (n, %) | 416 (37) | 378 (34) | 794 (36) |
| Non-specific/other (n, %) | 125 (11) | 114 (10) | 239 (11) |
| Main site [n (%)] | |||
| n | 1124 | 1114 | 2238 |
| Central (n, %) | 743 (66) | 721 (65) | 1464 (65) |
| Left chest (n, %) | 285 (25) | 288 (26) | 573 (26) |
| Right chest (n, %) | 24 (2) | 26 (2) | 50 (2) |
| Upper abdomen/epigastrium (n, %) | 47 (4) | 49 (4) | 96 (4) |
| Other (n, %) | 25 (2) | 30 (3) | 55 (2) |
| Radiationb [n (%)] | |||
| None | 481 (43) | 466 (42) | 947 (43) |
| Left arm | 365 (33) | 384 (35) | 749 (34) |
| Right arm | 97 (9) | 99 (9) | 196 (9) |
| Neck | 141 (13) | 150 (14) | 291 (13) |
| Jaw | 71 (6) | 97 (9) | 168 (8) |
| Back | 179 (16) | 159 (14) | 338 (15) |
| Other | 97 (9) | 90 (8) | 187 (8) |
| Associated featuresb [n (%)] | |||
| Nausea | 362 (33) | 374 (34) | 736 (34) |
| Vomiting | 62 (6) | 67 (6) | 129 (6) |
| Dyspnoea | 492 (45) | 487 (44) | 979 (45) |
| Sweating | 503 (46) | 470 (43) | 973 (44) |
| Dyspepsia | 83 (8) | 67 (6) | 150 (7) |
| Other | 161 (15) | 181 (17) | 342 (16) |
Table 9 shows the initial ECG and examination findings. A small proportion of patients had ST-segment deviations or T-wave inversions, although these were not deemed to be characteristic of myocardial ischaemia by the research nurse or recruiting doctor.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | |
|---|---|---|---|
| First ECG in ED [n (%)] | |||
| Normal | 765 (68) | 779 (70) | 1544 (69) |
| T-wave inversion | 79 (7) | 74 (7) | 153 (7) |
| ST depression | 17 (2) | 18 (2) | 35 (2) |
| ST elevation | 8 (1) | 8 (1) | 16 (1) |
| Bundle branch block | 24 (2) | 30 (3) | 54 (2) |
| Other | 205 (18) | 179 (16) | 384 (17) |
| Unknown/missing | 27 (2) | 30 (3) | 55 (2) |
| Pulse (bpm)a | |||
| n | 1120 | 1118 | 2238 |
| Mean (SD) | 76.6 (15.2) | 76.2 (15.0) | 76.4 (15.1) |
| Median (IQR) | 75.0 (66–85) | 75.0 (65–86) | 75.0 (65–85) |
| Diastolic blood pressure (mmHg) | |||
| n | 1117 | 1116 | 2233 |
| Mean (SD) | 80.3 (14.0) | 80.1 (14.7) | 80.2 (14.3) |
| Median (IQR) | 80.0 (71–90) | 80.0 (70–90) | 80.0 (71–90) |
| Systolic blood pressure (mmHg) | |||
| n | 1117 | 1117 | 2234 |
| Mean (SD) | 140.0 (22.9) | 140.8 (22.5) | 140.4 (22.7) |
| Median (IQR) | 138.0 (124–154) | 138.0 (125–155) | 138.0 (125–154) |
| Killip class [n (%)] | |||
| Class I | 1020 (91) | 1020 (91) | 2040 (91) |
| Class II | 8 (1) | 5 (< 1) | 13 (1) |
| Class III | 0 | 0 | 0 |
| Class IV | 1 (< 1) | 0 | 1 (< 1) |
| Missing | 96 (9) | 93 (8) | 189 (8) |
Table 10 shows the events in the ED, working diagnosis after initial assessment, the next-day ECG findings and final diagnosis. The proportions in each category differed between the final and initial diagnosis in both groups. More patients had a non-specific or other final diagnosis, and fewer had a final diagnosis of angina or ACS.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | |
|---|---|---|---|
| Events in EDa [n (%)] | |||
| Cardiac arrhythmia | 8 (1) | 8 (1) | 16 (1) |
| Further pain requiring treatment | 106 (11) | 101 (10) | 207 (11) |
| New ECG changes | 8 (1) | 21 (2) | 29 (1) |
| Other symptoms requiring admission | 26 (3) | 20 (2) | 46 (2) |
| Other events | 35 (4) | 26 (3) | 61 (3) |
| Working diagnosis after initial assessment [n (%)] | |||
| Non-specific | 233 (21) | 219 (20) | 452 (20) |
| Anxiety | 51 (5) | 49 (4) | 100 (4) |
| Angina no ACS | 173 (15) | 178 (16) | 351 (16) |
| ACS | 334 (30) | 332 (30) | 666 (30) |
| Gastro-oesophageal | 117 (10) | 115 (10) | 232 (10) |
| Musculoskeletal | 108 (10) | 102 (9) | 210 (9) |
| Other | 86 (8) | 93 (8) | 179 (8) |
| Unknown/missing | 23 (2) | 30 (3) | 53 (2) |
| Next-day ECG [n (%)] | |||
| Normal | 162 (14) | 199 (18) | 361 (16) |
| T-wave inversion | 31 (3) | 23 (2) | 54 (2) |
| ST depression | 1 (< 1) | 2 (< 1) | 3 (< 1) |
| ST elevation | 5 (< 1) | 3 (< 1) | 8 (< 1) |
| Bundle branch block | 6 (1) | 8 (1) | 14 (1) |
| Other abnormality | 46 (4) | 65 (6) | 111 (5) |
| Not applicable | 633 (56) | 432 (39) | 1065 (47) |
| Unknown/missing | 35 (3) | 66 (6) | 101 (5) |
| Final diagnosis [n (%)] | |||
| Non-specific | 366 (33) | 336 (30) | 702 (31) |
| Anxiety | 36 (3) | 23 (2) | 59 (3) |
| Angina no ACS | 89 (8) | 63 (6) | 152 (7) |
| ACS | 87 (8) | 72 (6) | 159 (7) |
| Gastro-oesophageal | 126 (11) | 136 (12) | 262 (12) |
| Musculoskeletal | 143 (13) | 169 (15) | 312 (14) |
| Other | 215 (19) | 252 (23) | 467 (21) |
| Unknown/missing | 63 (6) | 67 (6) | 130 (6) |
Blood testing
Table 11 shows the number and proportion of the point-of-care group who received each test, the number and proportion with a positive test, and the mean (median) time from worst symptoms to sampling. Most positive cases arose from the first troponin sample, although about one-quarter arose from the second sample.
| Sample 1 | Sample 2 | |
|---|---|---|
| n (%) with troponin result | 1076 (95.6) | 842 (74.8) |
| n (%) with troponin > 0.02 µg/l | 272 (25.5) | 150 (17.8) |
| n (%) with troponin > 0.07 µg/l | 113 (10.5) | 28 (3.3) |
| n (%) with myoglobin result | 1078 (95.6) | 840 (74.7) |
| n (%) with myoglobin gradient > 25% | – | 67 (6.2) |
| n (%) with CK-MB (mass) result | 1076 (95.6) | 841 (74.8) |
| n (%) with both CK-MB (mass) result > 5 µg/l | – | 17 (2.0) |
| n (%) with CK-MB (mass) gradient > 1.6 µg/l | – | 2 (0.2) |
| Mean (median) time from worst symptoms to test (hours) | 6.6 (4.1) | 7.6 (5.7) |
Table 12 shows the number and proportion of patients in each study group receiving each laboratory blood test. Although most patients in the point-of-care group had negative point-of-care tests, a substantial proportion went on to have laboratory testing with troponin I or T. In the standard-care group around 90% received laboratory testing with troponin I or T. Other cardiac biomarkers were rarely used.
| PoC | SC | |
|---|---|---|
| n (%) with at least one laboratory troponin (I or T) | 661 (58.8) | 992 (88.7) |
| n (%) with at least one laboratory CK | 177 (15.7) | 155 (13.9) |
| n (%) with at least one laboratory CK-MB (mass) | 1 (< 1) | 1 (< 1) |
| n (%) with at least one LDH | 1 (< 1) | 0 |
| n (%) with at least one laboratory myoglobin | 9 (< 1) | 9 (< 1) |
Primary efficacy
Table 13 shows the results for the primary outcome, i.e. successful discharge home. This was defined as having left the hospital or as a decision to discharge made 4 hours after initial presentation, and no adverse event over the following 3 months. Some 509 patients were defined as discharged at initial presentation: 453 had left the hospital by 4 hours and 56 had a decision to discharge. However, five of these patients had a major adverse event over the following 3 months, so 504 were defined as being successfully discharged home.
| PoC [n (%)] | SC [n (%)] | Total [n (%)] | |
|---|---|---|---|
| Successfully discharged | 358 (32) | 146 (13) | 504 (22) |
| Not successfully discharged | 767 (68) | 972 (87) | 1739 (78) |
| Reason for no successful discharge | |||
| In hospital 4 hours after arrival and no decision has been made to discharge | 763 (68) | 971 (87) | 1734 (77) |
| Initially discharged but re-attended with major adverse event | 4 (< 1) | 1 (< 1) | 5 (< 1) |
| Discharge success by initial status | |||
| Initially discharged | 362 (32) | 147 (13) | 509 (23) |
| Not in hospital at 4 hours | 319 (28) | 134 (12) | 453 (20) |
| In hospital at 4 hours; decision made to discharge | 43 (4) | 13 (1) | 56 (2) |
The primary efficacy analyses were adjusted, as planned, for hospital, age, gender and known CHD. Patients in the point-of-care group were significantly more likely to be successfully discharged home (OR = 3.81, 95% CI 3.01 to 4.82, p < 0.001).
Variation between sites
Table 14 shows the results for each individual hospital. Full details are supplied in Appendix 3. There was marked variation in the primary outcome across the six hospitals. Point-of-care panel assessment was associated with substantial increases in successful discharge rates at Barnsley and Edinburgh, modest increases at Derriford and Frenchay, and no increase at Leeds or Leicester.
| PoC [n (%)] | SC [n (%)] | ORa (95% CI) | RDb (95% CI) | p-value | |
|---|---|---|---|---|---|
| Overall | 358/1125 (32) | 146/1118 (13) | 3.81 (3.01 to 4.82) | 18.7 (15.4 to 22.1) | < 0.001 |
| Barnsley | 110/162 (68) | 43/164 (26) | 6.97 (4.18 to 11.63) | 41.3 (31.4 to 51.1) | < 0.001 |
| Derriford | 43/164 (26) | 21/164 (13) | 2.48 (1.37 to 4.49) | 13.4 (5.0 to 21.9) | 0.003 |
| Edinburgh | 104/228 (46) | 16/224 (7) | 11.07 (6.23 to 19.66) | 38.4 (31.1 to 45.6) | < 0.001 |
| Frenchay | 50/233 (21) | 9/231 (4) | 7.03 (3.35 to 14.75) | 17.4 (11.6 to 23.2) | < 0.001 |
| Leeds | 1/173 (1) | 8/171 (5) | 0.12 (0.01 to 1.03) | –4.0 (–7.2 to –0.7) | 0.054 |
| Leicester | 50/165 (30) | 49/164 (30) | 1.11 (0.66 to 1.84) | 0.4 (–9.5 to 10.3) | 0.699 |
Figure 3 shows how the proportion of patients in hospital varies with time from arrival and Figure 4 shows this for each individual hospital. Overall, point-of-care panel assessment is associated with fewer patients being in hospital between 4 and 24 hours after arrival. However, there was again variation between the hospitals. Point-of-care panel assessment was associated with markedly fewer patients being in hospital up to 24 hours at Barnsley and Edinburgh. At Derriford the difference in proportion in hospital was apparent only between 4 and 8 hours. At Frenchay the difference was marked up to 12 hours, but after 12 hours the proportion of patients in hospital was greater in the point-of-care group. At Leeds the difference between the groups did not emerge until 6 hours after attendance, but between 6 and 24 hours point-of-care panel assessment was associated with markedly fewer patients being in hospital. Finally, at Leicester there was no difference in the proportions in hospital up to 12 hours and only slightly fewer patients in hospital in the point-of-care group from 12 to 36 hours.
FIGURE 3.
Duration from arrival to discharge from hospital (all centres).
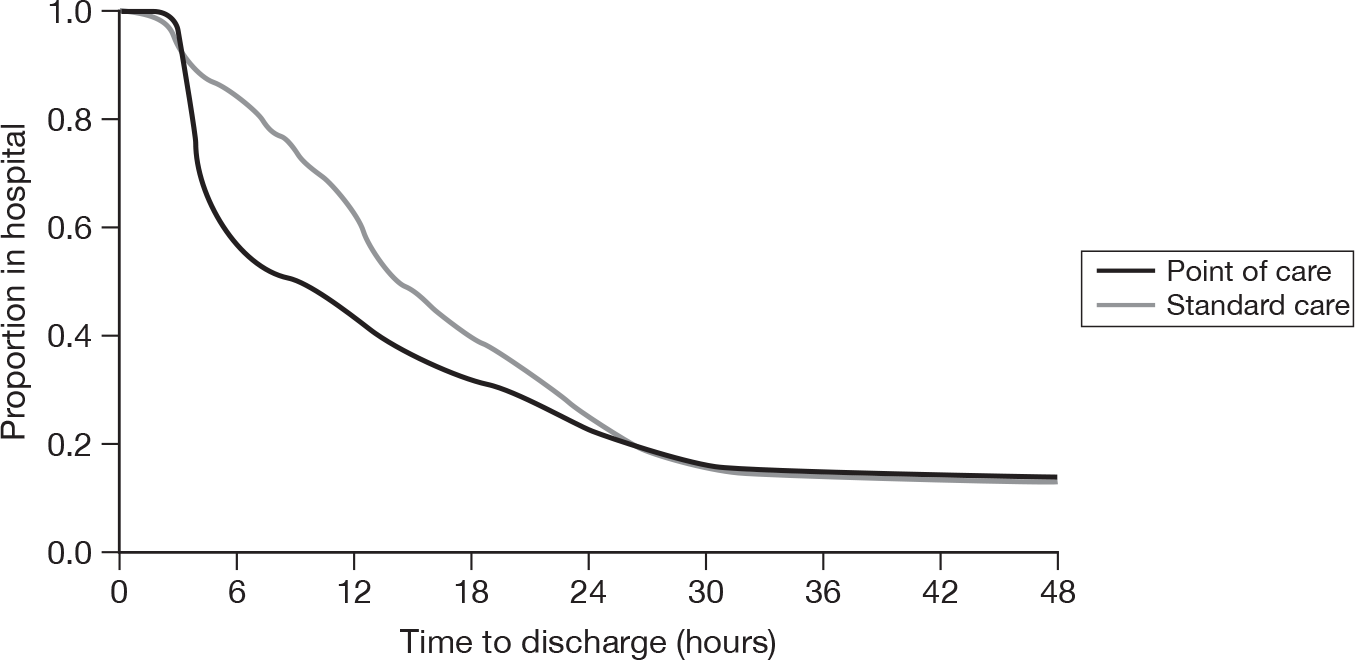
FIGURE 4.
Duration from arrival to discharge from hospital (individual centres).
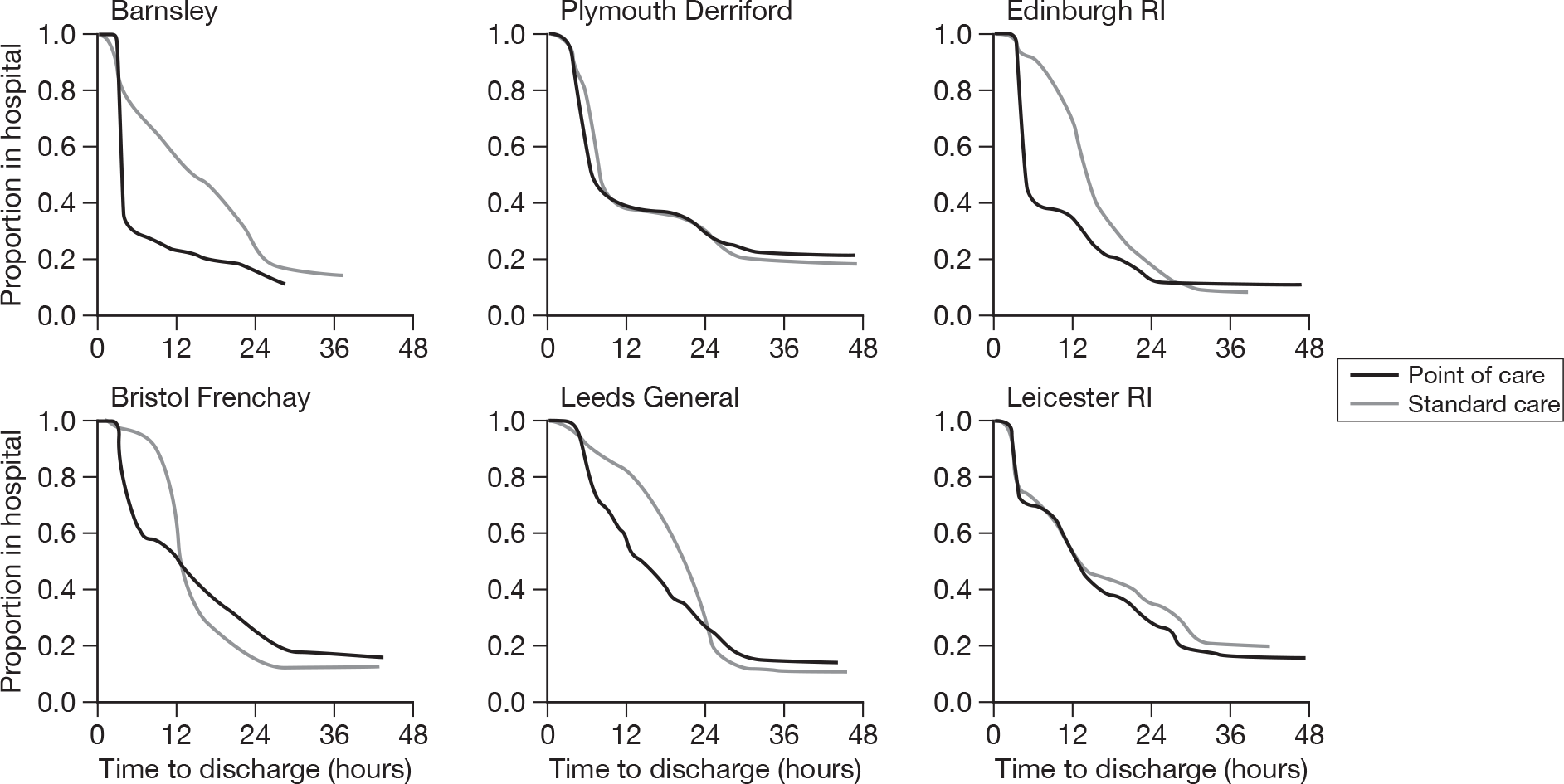
These differences probably reflect differences in standard care practice or the facilities for patients with chest pain at the hospitals. Standard care at Derriford was based on a troponin level measured 6 hours after arrival in hospital. This explains why the effect of point-of-care panel assessment at Derriford was limited to the 4- to 8-hour window. At Leeds all patients with chest pain are admitted to a clinical decision unit where diagnosis and management are undertaken without the pressure of the 4-hour target. This probably explains why the effect of point-of-care panel assessment was delayed at Leeds until 6 hours after arrival. At Leicester the point-of-care tests did not seem to alter decision-making with regards to hospital admission.
Per-protocol analysis
Table 15 shows the results of per-protocol analysis, with the 140 point-of-care patients who did not receive testing according to the protocol and the two standard-care patients who received point-of-care testing excluded. Exclusion of these patients does not markedly change the proportions in each group who are successfully discharged.
| PoC [n (%)] | SC [n (%)] | Total [n (%)] | |
|---|---|---|---|
| Successfully discharged | 326 (33) | 145 (13) | 471 (22) |
| Not successfully discharged | 659 (67) | 971 (87) | 1630 (78) |
| Reason for no successful discharge | |||
| In hospital 4 hours after arrival and no decision has been made to discharge | 655 (66) | 970 (87) | 1625 (77) |
| Initially discharged but re-attended with major adverse event | 4 (< 1) | 1 (< 1) | 5 (< 1) |
| Discharge success by initial status | |||
| Initially discharged | 330 (34) | 146 (13) | 476 (23) |
| Not in hospital at 4 hours | 292 (30) | 133 (12) | 425 (20) |
| In hospital at 4 hours, decision made to discharge | 38 (4) | 13 (1) | 51 (2) |
Variation over time
We analysed the primary outcome by each quarter-year to determine if the proportion successfully discharged varied over time due to clinical staff becoming more familiar with the intervention or changing management in the standard-care group. We also analysed the proportion of patients in the point-of-care group who did not receive testing according to the recommended protocol to determine whether increased familiarity over time led to fewer protocol deviations. The results are outlined in Table 16. The proportion successfully discharged in the point-of-care group appears to dip in the middle of the trial and then rise again at the end, whereas the proportion successfully discharged in the standard-care group appears to progressively fall throughout the trial. The proportion of patients in the point-of-care group who did not receive testing according to the recommended protocol remained relatively constant.
| Quarter | PoC protocol deviations [n (%)] | PoC successfully discharged [n (%)] | SC successfully discharged [n (%)] |
|---|---|---|---|
| January to March 2008 | 3/34 (9) | 13/34 (38) | 10/33 (30) |
| April to June 2008 | 41/286 (14) | 93/286 (33) | 42/286 (15) |
| July to September 2008 | 33/199 (17) | 57/199 (29) | 30/195 (15) |
| October to December 2008 | 24/239 (10) | 64/239 (27) | 27/235 (11) |
| January to March 2009 | 22/195 (10) | 77/195 (35) | 24/222 (11) |
| April to June 2009 | 17/150 (11) | 54/150 (36) | 13/147 (9) |
We compared the primary efficacy analysis before and after the amendment to the troponin threshold in the point-of-care protocol. Before the amendment the proportion successfully discharged in the point-of-care group was 210/729 (29%) compared with 107/721 (15%) in the standard-care group (adjusted OR = 2.79, 95% CI 2.09 to 3.72). After the amendment the proportion successfully discharged in the point-of-care group was 148/396 (37%) compared with 39/397 (10%) in the standard-care group (adjusted OR = 5.61, 95% CI 3.79 to 8.31, p < 0.001). Hence, it appeared that the effect of point-of-care panel assessment increased after amendment of the protocol, although some of this may be attributable to a lower successful discharge rate in the standard-care group.
Secondary efficacy
Length and location of hospital stay
The mean length of the initial hospital stay was 29.6 hours in the point-of-care group and 31.8 hours in the standard-care group [mean difference = 2.1 hours, 95% CI –3.7 to 8.0 hours, p = 0.462 (t-test)]. The median length of initial hospital stay was markedly shorter for both groups, reflecting the positively skewed nature of length-of-stay data: 8.8 hours for the point-of-care group and 14.2 hours for the standard-care group (p < 0.001, equality-of-medians test). The two groups had significantly different distributions of length of stay (p < 0.001, Mann–Whitney U-test). Point-of-care panel assessment was therefore associated with reduced probability of hospital admission and shorter median length of stay, but there was no significant difference in mean length of stay.
Table 17 shows the total number of days spent in hospital over the 3-month follow-up and the number of days spent in coronary care or intensive care, including the initial hospital admission. We did not record fractions of days spent in hospital for this analysis and recorded an inpatient day only if the patient stayed overnight. Hence, a proportion of patients were recorded as having no inpatient days if their initial hospital visit did not result in an overnight stay and they were not admitted on a subsequent occasion. More patients in the point-of-care group had no inpatient days recorded during follow-up (54% vs 40%, p < 0.001). This reflected the difference in the proportion of patients in Figure 5 who were no longer in hospital 12 hours after attendance, as patients who are discharged before 12 hours and are not re-admitted are likely to be recorded as having no inpatient days over follow-up.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | Percentage difference (95% CI) | p-value | |
|---|---|---|---|---|---|
| Days in hospital at any location | |||||
| n (%) with at least one day | 518 (46.0) | 670 (59.9) | 1188 (53.0) | –13.9 (–18.0 to –9.8) | < 0.001 |
| Mean days per patient | 1.8 | 1.7 | 1.7 | 0.815 | |
| Total days (all patients) | 1961 | 1908 | 3869 | ||
| Days in coronary care | |||||
| n (%) with at least one day | 50 (4.4) | 31 (2.8) | 81 (3.6) | 1.7 (0.1 to 3.2) | 0.041 |
| Mean days per patient | 0.17 | 0.09 | 0.13 | 0.033 | |
| Total days (all patients) | 190 | 105 | 295 | ||
| Days in intensive care | |||||
| n (%) with at least one day | 8 (< 1) | 3 (< 1) | 11 (< 1) | 0.4 (–0.2 to 1.0) | 0.225 |
| Mean days per patient | 0.08 | 0.01 | 0.04 | 0.259 | |
| Total days (all patients) | 76 | 14 | 90 | ||
FIGURE 5.
Histograms of number of inpatient days.
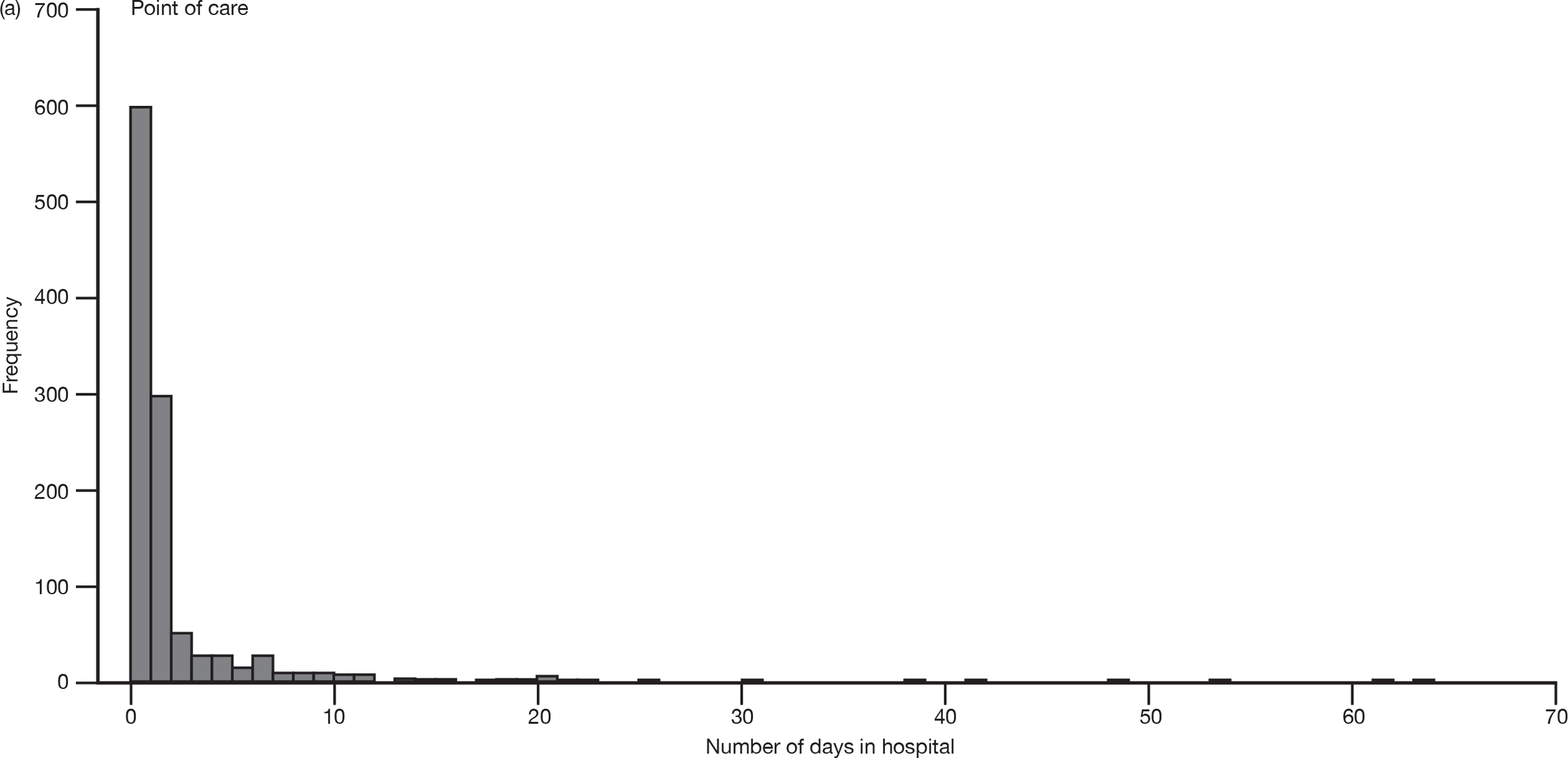
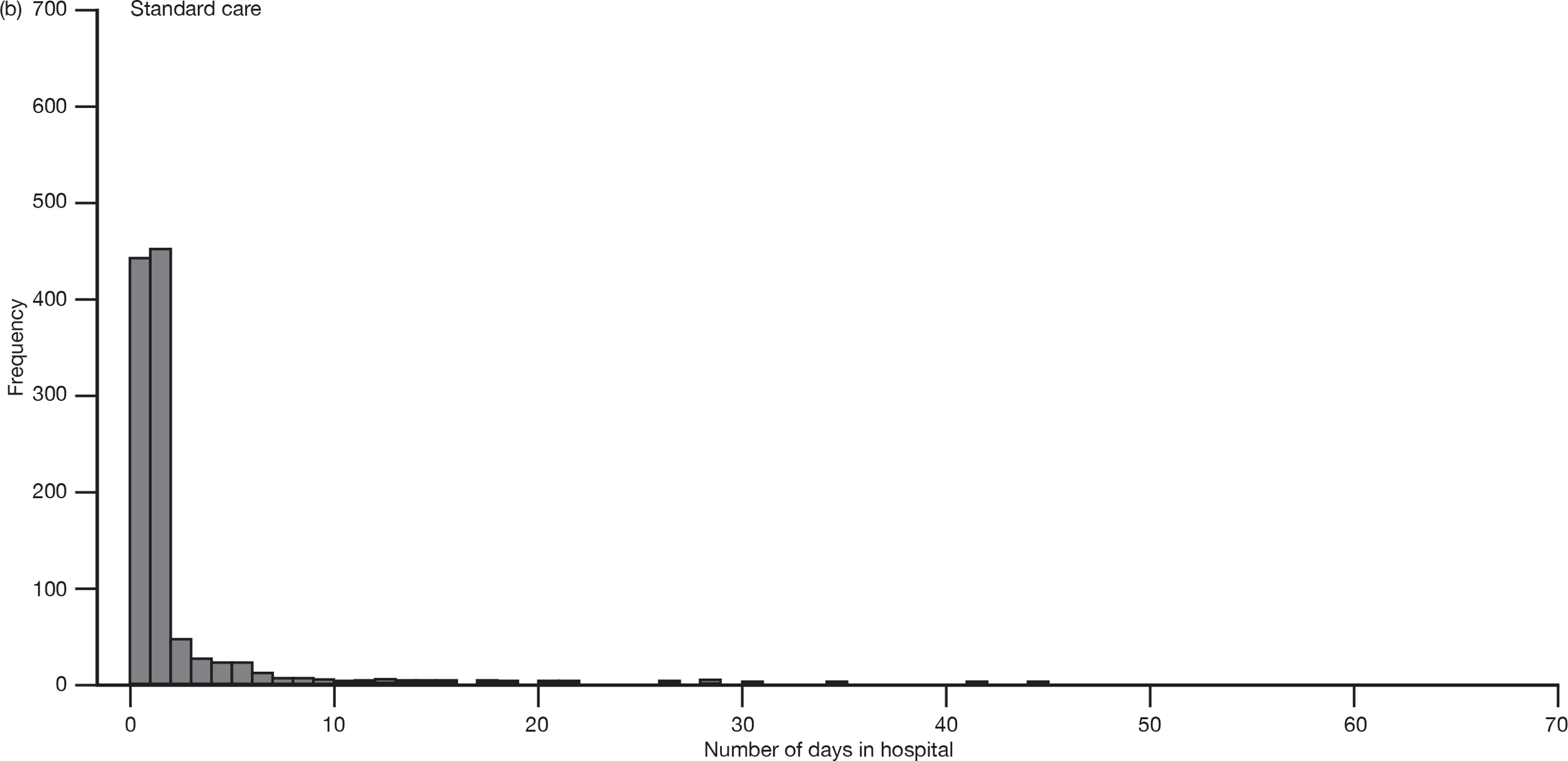
Despite this difference in the proportion with no inpatient days, there was no difference in the mean days per patient or the total inpatient days in the two groups. Figure 5 compares histograms for total days spent in hospital in the two groups to show why this was. The difference between the two groups in the proportions of patients having no inpatient stay was reflected in the difference in proportions having one inpatient day. So point-of-care panel assessment saved one inpatient day in about 14%. However, a few patients in both groups had a very high number of inpatient days (over 30). There were a few more of these in the point-of-care group, and enough to contribute disproportionately to the mean and total inpatient days for the group. Hence, point-of-care panel assessment reduced the proportion of patients completely avoiding admission but did not reduce overall inpatient days. Point-of-care panel assessment was also associated with a higher use of coronary care over follow-up (4% vs 3%, p = 0.041), although only a small minority of patients received any coronary care or intensive care.
Initial care in the first 24 hours
Table 18 shows the proportion of patients in each group receiving various elements of care in the first 24 hours after arrival. Only a small proportion in each group was admitted from the ED on to coronary care, but twice as many were admitted to coronary care in the point-of-care group (4% vs 2%, p = 0.001). There were no significant differences between point-of-care and standard care in the proportions receiving glyceryl trinitrate (GTN), heparin, glycoprotein inhibitors, antacids or beta-blockers. More patients in the point-of-care group received clopidogrel (21% vs 16%, p = 0.002), while more patients in the standard-care group received aspirin (60% vs 55%, p = 0.031). There were non-significant trends towards greater use of angiography and PCI in the point-of-care group and greater use of analgesia in the standard-care group.
| PoC (n = 1125) [n (%)] | SC (n = 1118) [n (%)] | Total (n = 2243) [n (%)] | p-value | |
|---|---|---|---|---|
| Admitted to coronary care | 50 (4) | 21 (2) | 71 (3) | 0.001 |
| Received GTN | 446 (40) | 460 (42) | 906 (41) | 0.327 |
| Received heparin | 206 (18) | 186 (17) | 392 (18) | 0.381 |
| Received glycoprotein inhibitors | 8 (1) | 7 (1) | 15 (1) | 0.822 |
| Received antacid | 90 (8) | 108 (10) | 198 (9) | 0.128 |
| Received angiography | 16 (1) | 8 (1) | 24 (1) | 0.105 |
| Received aspirin | 618 (55) | 663 (60) | 1281 (58) | 0.031 |
| Received beta-blocker | 108 (10) | 105 (10) | 213 (10) | 0.967 |
| Received clopidogrel | 237 (21) | 176 (16) | 413 (19) | 0.002 |
| Received analgesic | 373 (34) | 407 (37) | 780 (36) | 0.058 |
| Received any other drugs | 316 (29) | 340 (31) | 656 (30) | 0.150 |
| Received angioplasty/stent | 11 (1) | 4 (< 1) | 15 (1) | 0.073 |
Interventions, re-attendances, re-admissions, investigations and outpatient reviews up to 3 months
Table 19 shows the proportion of patients receiving cardiac interventions in each study group over the 3-month follow-up period, including during the initial hospital admission. Very few patients received cardiac interventions and there were no significant differences between the study groups.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | p-value | |
|---|---|---|---|---|
| n (%) needing thrombolysis | 1 (< 1) | 2 (< 1) | 3 (< 1) | 0.624 |
| n (%) needing PCI | 29 (3) | 29 (3) | 58 (3) | 1.000 |
| n (%) needing emergency PCI | 7 (1) | 12 (1) | 19 (1) | 0.260 |
| n (%) needing CABG | 11 (1) | 5 (< 1) | 16 (1) | 0.209 |
Table 20 shows the proportion of patients in each group with any ED attendance, the proportion with a potentially chest pain-related attendance, the proportion with any subsequent hospital admission (i.e. any admission other than that arising from their initial visit) and the proportion with a potentially chest pain-related hospital admission over the follow-up 3 months. There were no significant differences between the two groups in any of these outcomes.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | p-value | |
|---|---|---|---|---|
| n (%) of ED attendances | 140 (12) | 138 (12) | 278 (12) | 0.949 |
| n (%) of chest pain-related ED attendances | 107 (10) | 103 (9) | 210 (9) | 0.828 |
| n (%) of subsequent hospital admissions | 117 (10) | 122 (11) | 239 (11) | 0.732 |
| n (%) of subsequent chest pain-related admissions | 84 (7) | 99 (9) | 183 (8) | 0.247 |
Table 21 shows the proportion of patients in each group receiving each investigation over the 3-month follow-up. Most patients did not receive any of these investigations. There were no differences between the two groups in the proportions receiving any of the investigations.
| PoC (n = 1125) [n (%)] | SC (n = 1118) [n (%)] | p-value | |
|---|---|---|---|
| Exercise treadmill test | 231 (20.5) | 226 (20.2) | 0.893 |
| Echocardiography | 73 (6.5) | 60 (5.4) | 0.300 |
| Radionuclide scan | 36 (3.2) | 33 (3.0) | 0.827 |
| 24-hour tape | 12 (1.1) | 19 (1.7) | 0.270 |
| Coronary angiography | 81 (7.2) | 78 (7.0) | 0.901 |
| Abdominal ultrasound | 13 (1.2) | 20 (1.8) | 0.284 |
| Upper gastrointestinal endoscopy | 9 (0.8) | 17 (1.5) | 0.162 |
| Other test | 20 (1.8) | 17 (1.5) | 0.755 |
Table 22 shows the proportion of patients in each group who received outpatient follow-up (any or potentially chest pain related). Patients in the point-of-care group were slightly more likely to have a chest pain-related outpatient review (21% vs 18%, p = 0.05).
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | p-value | |
|---|---|---|---|---|
| n (%) with any outpatient attendance | 334 (30) | 322 (29) | 656 (29) | 0.676 |
| n (%) with a chest pain-related outpatient attendance | 241 (21) | 202 (18) | 443 (20) | 0.050 |
Patient-reported use of health services
Tables 23 and 24 show the number and proportion of patients in each group who reported having used various health services over the previous month in their 1- and 3-month resource use questionnaires. The only significant difference was in the proportion admitted to hospital. There may have been some confusion in responses to this question, with some patients reporting the initial hospital admission while others reported only subsequent admissions.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | p-value | |
|---|---|---|---|---|
| Questionnaires returned [n (%)] | 841 (74.8) | 795 (71.1) | 1636 (72.9) | |
| n (%) using telephone health advice | 232/760 (31) | 221/711 (31) | 453/1471 (31) | 0.821 |
| n (%) using GP surgery consultations | 555/799 (69) | 537/758 (71) | 1092/1557 (70) | 0.580 |
| n (%) using GP home visits | 45/749 (6) | 43/702 (6) | 88/1451 (6) | 1.000 |
| n (%) using nurse home visits | 44/756 (6) | 46/698 (7) | 90/1454 (6) | 0.587 |
| n (%) using social worker visits | 14/751 (2) | 18/698 (3) | 32/1449 (2) | 0.376 |
| n (%) attending ED | 282/765 (37) | 280/728 (38) | 562/1493 (38) | 0.557 |
| n (%) attending outpatients | 334/778 (43) | 277/726 (38) | 611/1504 (41) | 0.066 |
| n (%) using other health services | 83/733 (11) | 76/660 (12) | 159/1393 (11) | 0.933 |
| n (%) admitted to hospital | 182/832 (22) | 208/786 (26) | 390/1618 (24) | 0.032 |
| n (%) operated on | 33/829 (4) | 32/784 (4) | 65/1613 (4) | 1.000 |
| n (%) having ECG | 224/813 (28) | 228/765 (30) | 452/1578 (29) | 0.344 |
| n (%) having exercise stress test | 184/818 (22) | 157/769 (20) | 341/1587 (21) | 0.328 |
| n (%) having heart monitor | 43/806 (5) | 58/757 (8) | 101/1563 (6) | 0.064 |
| n (%) having abdominal ultrasound | 59/806 (7) | 54/746 (7) | 113/1552 (7) | 1.000 |
| n (%) having coronary angiography | 91/813 (11) | 79/756 (10) | 170/1569 (11) | 0.685 |
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | p-value | |
|---|---|---|---|---|
| Questionnaires returned [n (%)] | 787 (70.0) | 745 (66.6) | 1532 (68.3) | |
| n (%) using telephone health advice | 151/661 (23) | 145/633 (23) | 296/1294 (23) | 1.000 |
| n (%) using GP surgery consultations | 512/746 (69) | 457/701 (65) | 969/1447 (67) | 0.180 |
| n (%) using GP home visits | 27/646 (4) | 40/626 (6) | 67/1272 (5) | 0.080 |
| n (%) using nurse home visits | 32/645 (5) | 39/622 (6) | 71/1267 (6) | 0.330 |
| n (%) using social worker visits | 12/642 (2) | 21/621 (3) | 33/1263 (3) | 0.112 |
| n (%) attending ED | 106/659 (16) | 112/635 (18) | 218/1294 (17) | 0.459 |
| n (%) attending outpatients | 281/693 (41) | 265/661 (40) | 546/1354 (40) | 0.868 |
| n (%) using other health services | 84/625 (13) | 80/598 (13) | 164/1223 (13) | 1.000 |
| n (%) admitted to hospitals | 76/779 (10) | 79/742 (11) | 155/1521 (10) | 0.611 |
| n (%) operated on | 27/772 (3) | 21/734 (3) | 48/1506 (3) | 0.558 |
| n (%) having ECG | 97/762 (13) | 74/720 (10) | 171/1482 (12) | 0.144 |
| n (%) having exercise stress test | 83/761 (11) | 71/724 (10) | 154/1485 (10) | 0.497 |
| n (%) having heart monitor | 32/760 (4) | 28/712 (4) | 60/1472 (4) | 0.794 |
| n (%) having abdominal ultrasound | 45/758 (6) | 42/716 (6) | 87/1474 (6) | 1.000 |
| n (%) having coronary angiography | 39/755 (5) | 31/717 (4) | 70/1472 (5) | 0.465 |
Patient-reported time taken off work
Tables 25 and 26 show the number and proportion of patients in each group who reported at 1 and 3 months that they had taken time off work during the last month, had not taken time off, were retired and were not in paid employment. They also show the median (IQR) number of days taken off. Around one-half of those in paid employment had taken time off during the first month and around one-third had taken time off during the third month. There were no differences in time taken off work between the two groups.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | p-value | |
|---|---|---|---|---|
| Questionnaires returned [n (%)] | 841 (74.8) | 795 (71.1) | 1636 (72.9) | |
| n (%) taken time off | 226 (27) | 215 (28) | 441 (27) | 0.841 |
| n (%) not taken time off | 227 (27) | 222 (28) | 449 (28) | |
| n (%) retired | 107 (13) | 65 (8) | 172 (11) | |
| n (%) not in paid employment | 267 (32) | 279 (36) | 546 (34) | |
| Median (IQR) days off | 8.0 (3–20) | 10.0 (4–20) | 10.0 (3–20) | 0.29 |
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | p-value | |
|---|---|---|---|---|
| Questionnaires returned [n (%)] | 787 (70.0) | 745 (66.6) | 1532 (68.3) | |
| n (%) taken time off | 136 (17) | 123 (17) | 259 (17) | 0.498 |
| n (%) not taken time off | 275 (35) | 278 (38) | 553 (37) | |
| n (%) retired | 89 (11) | 61 (8) | 150 (10) | |
| n (%) not in paid employment | 279 (36) | 271 (37) | 550 (36) | |
| Median (IQR) of days off | 11.0 (4–40) | 9.0 (3–35) | 10.0 (3–40) | 0.399 |
Health utility
Table 27 compares mean EQ-5D scores at 1 and 3 months between the point-of-care and standard-care groups. There were no significant differences in mean EQ-5D scores at either time point.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | Difference between means (95% CI) | p-value | |
|---|---|---|---|---|---|
| 1 month | |||||
| n (%) of responses | 825 (73.3) | 770 (68.9) | 1595 (71.1) | ||
| Mean (SD) | 0.742 (0.289) | 0.759 (0.267) | 0.750 (0.279) | –0.017 (–0.04 to 0.01) | 0.614 |
| Median (IQR) | 0.796 (0.69–1.00) | 0.796 (0.69–1.00) | 0.796 (0.69–1.00) | ||
| 3 months | |||||
| n (%) of responses | 756 (67.2) | 717 (64.1) | 1473 (65.7) | ||
| Mean (SD) | 0.752 (0.291) | 0.759 (0.279) | 0.755 (0.285) | –0.007 (–0.03 to 0.02) | 0.638 |
| Median (IQR) | 0.796 (0.69–1.00) | 0.796 (0.69–1.00) | 0.796 (0.69–1.00) | ||
Patient satisfaction
Table 28 shows the proportion of responses in each category for each question of the patient satisfaction questionnaire. Most patients were satisfied with most aspects of their care, with only a small proportion rating their care as poor. The exception was the question on advice about ways to avoid illness and stay healthy, where 19% rated their care as poor and only 14% rated it as excellent.
| PoC (n = 1125) | SC (n = 1118) | Total (n = 2243) | |
|---|---|---|---|
| n (%) of questionnaires returned | 841 (74.8) | 796 (71.2) | 1637 (73.0) |
| The urgency with which you were assessed | |||
| n | 838 | 791 | 1629 |
| Poor | 10 (1) | 16 (2) | 26 (2) |
| Fair | 40 (5) | 51 (6) | 91 (6) |
| Good | 132 (16) | 160 (20) | 292 (18) |
| Very good | 317 (38) | 257 (32) | 574 (35) |
| Excellent | 339 (40) | 307 (39) | 646 (40) |
| p-value | 0.038 | ||
| The thoroughness of your assessment | |||
| n | 837 | 791 | 1628 |
| Poor | 9 (1) | 10 (1) | 19 (1) |
| Fair | 41 (5) | 37 (5) | 78 (5) |
| Good | 140 (17) | 162 (20) | 302 (19) |
| Very good | 325 (39) | 295 (37) | 620 (38) |
| Excellent | 322 (38) | 287 (36) | 609 (37) |
| p-value | 0.163 | ||
| Explanations given to you about medical procedures and tests | |||
| n | 837 | 789 | 1626 |
| Poor | 19 (2) | 20 (3) | 39 (2) |
| Fair | 55 (7) | 57 (7) | 112 (7) |
| Good | 167 (20) | 177 (22) | 344 (21) |
| Very good | 315 (38) | 301 (38) | 616 (38) |
| Excellent | 281 (34) | 234 (30) | 515 (32) |
| p-value | 0.066 | ||
| Attention given to what you have to say | |||
| n | 837 | 792 | 1629 |
| Poor | 24 (3) | 21 (3) | 45 (3) |
| Fair | 62 (7) | 67 (8) | 129 (8) |
| Good | 207 (25) | 193 (24) | 400 (25) |
| Very good | 346 (41) | 317 (40) | 663 (41) |
| Excellent | 198 (24) | 194 (24) | 392 (24) |
| p-value | 0.996 | ||
| Advice you got about ways to avoid illness and stay healthy | |||
| n | 831 | 776 | 1607 |
| Poor | 151 (18) | 161 (21) | 312 (19) |
| Fair | 142 (17) | 121 (16) | 263 (16) |
| Good | 210 (25) | 192 (25) | 402 (25) |
| Very good | 209 (25) | 197 (25) | 406 (25) |
| Excellent | 119 (14) | 105 (14) | 224 (14) |
| p-value | 0.487 | ||
| Friendliness and courtesy shown to you by hospital staff | |||
| n | 839 | 790 | 1629 |
| Poor | 7 (1) | 8 (1) | 15 (1) |
| Fair | 39 (5) | 28 (4) | 67 (4) |
| Good | 121 (14) | 141 (18) | 262 (16) |
| Very good | 307 (37) | 272 (34) | 579 (36) |
| Excellent | 365 (44) | 341 (43) | 706 (43) |
| p-value | 0.605 | ||
| Personal interest in you and your medical problems | |||
| n | 837 | 788 | 1625 |
| Poor | 24 (3) | 26 (3) | 50 (3) |
| Fair | 77 (9) | 89 (11) | 166 (10) |
| Good | 204 (24) | 206 (26) | 410 (25) |
| Very good | 308 (37) | 281 (36) | 589 (36) |
| Excellent | 224 (27) | 186 (24) | 410 (25) |
| p-value | 0.044 | ||
| Respect shown to you, and attention to your privacy | |||
| n | 837 | 788 | 1625 |
| Poor | 20 (2) | 21 (3) | 41 (3) |
| Fair | 65 (8) | 67 (9) | 132 (8) |
| Good | 174 (21) | 179 (23) | 353 (22) |
| Very good | 338 (40) | 276 (35) | 614 (38) |
| Excellent | 240 (29) | 245 (31) | 485 (30) |
| p-value | 0.870 | ||
| Reassurance and support offered to you by hospital staff | |||
| n | 836 | 787 | 1623 |
| Poor | 31 (4) | 33 (4) | 64 (4) |
| Fair | 79 (9) | 81 (10) | 160 (10) |
| Good | 207 (25) | 182 (23) | 389 (24) |
| Very good | 288 (34) | 275 (35) | 563 (35) |
| Excellent | 231 (28) | 216 (27) | 447 (28) |
| p-value | 0.870 | ||
| Amount of time the hospital staff gave you | |||
| n | 839 | 789 | 1628 |
| Poor | 32 (4) | 28 (4) | 60 (4) |
| Fair | 108 (13) | 127 (16) | 235 (14) |
| Good | 246 (29) | 230 (29) | 476 (29) |
| Very good | 278 (33) | 247 (31) | 525 (32) |
| Excellent | 175 (21) | 157 (20) | 332 (20) |
| p-value | 0.210 | ||
| Overall, how satisfied are you with the service you received? | |||
| n | 839 | 788 | 1627 |
| Poor | 20 (2) | 22 (3) | 42 (3) |
| Fair | 51 (6) | 68 (9) | 119 (7) |
| Good | 153 (18) | 172 (22) | 325 (20) |
| Very good | 326 (39) | 284 (36) | 610 (37) |
| Excellent | 289 (34) | 242 (31) | 531 (33) |
| p-value | 0.008 | ||
Patients receiving point-of-care panel assessment were generally more likely to rate their care as excellent or very good, while those receiving standard care were more likely to rate their care as good, fair or poor. However, this trend was clearly significant only on the final question of overall satisfaction with the service received, whilst being borderline significant for questions relating to the urgency of assessment, explanations given and personal interest shown in the patient and his or her medical problems.
Table 29 shows the characteristics of responders and non-responders in the two groups. At both 1 and 3 months the responders were older and more likely to be female.
| PoC (n = 1125) | SC (n = 1118) | |||
|---|---|---|---|---|
| Responders | Non-responders | Responders | Non-responders | |
| (n = 825) | (n = 300) | (n = 771) | (n = 347) | |
| 1 month | ||||
| Age (years) [mean (SD)] | 56.7 (13.1) | 48.5 (14.0) | 57.1 (13.9) | 48.9 (13.8) |
| Gender | ||||
| Male [n (%)] | 468 (56.7) | 215 (71.7) | 410 (53.2) | 214 (61.7) |
| Female [n (%)] | 357 (43.3) | 85 (28.3) | 361 (46.8) | 133 (38.3) |
| Previous CHD | ||||
| Yes [n (%)] | 50 (6.1) | 10 (3.4) | 45 (5.9) | 20 (5.8) |
| No [n (%)] | 767 (93.9) | 288 (96.6) | 719 (94.1) | 324 (94.9) |
| 3 months | ||||
| Age (years) [mean (SD)] | 57.1 (13.3) | 49.0 (13.2) | 58.1 (13.7) | 48.1 (13.3) |
| Gender | ||||
| Male [n (%)] | 439 (57.2) | 244 (68.2) | 386 (53.3) | 238 (60.4) |
| Female [n (%)] | 328 (42.8) | 114 (31.8) | 338 (46.7) | 156 (39.6) |
| Previous CHD | ||||
| Yes [n (%)] | 49 (6.5) | 11 (3.1) | 43 (6.0) | 22 (5.6) |
| No [n (%)] | 710 (93.5) | 345 (96.9) | 675 (94.0) | 368 (94.4) |
Adverse events
Table 30 shows the number of major adverse events in each study group up to 3 months. The causes of death were MI (two cases), cancer (two cases), multiorgan failure, pneumonia and unknown (two cases). Adverse event rates were very low, with an overall death rate of 3.6 per thousand, non-fatal MI rate of 4.5 per thousand and emergency revascularisation rate of 10.7 per thousand. The analyses were adjusted as planned for age, gender and known CHD. There were no significant differences in adverse event rates between the two groups.
| PoC (n = 1125) [n (%)] | SC (n = 1118) [n (%)] | Total (n = 2243) [n (%)] | OR (95% CI)a | p-valuea | |
|---|---|---|---|---|---|
| Any event | 36 (3) | 26 (2) | 62 (3) | 1.31 (0.78 to 2.20) | 0.313 |
| Death | 6 (1) | 2 (< 1) | 8 (< 1) | 3.4 (0.7 to 17.3) | 0.142 |
| Non-fatal AMI | 5 (< 1) | 5 (< 1) | 10 (< 1) | 0.9 (0.3 to 3.2) | 0.903 |
| Hospitalisation for ACS without AMI | 18 (2) | 9 (1) | 27 (1) | 1.8 (0.8 to 4.1) | 0.149 |
| Life-threatening arrhythmia | 6 (1) | 2 (< 1) | 8 (< 1) | 3.2 (0.6 to 15.9) | 0.160 |
| Emergency revascularisation | 10 (1) | 14 (1) | 24 (1) | 0.7 (0.3 to 1.5) | 0.324 |
As shown in Table 13 (primary efficacy), five of the adverse events occurred in patients who were discharged after initial assessment. As these could potentially represent episodes of suboptimal care, these are described in Table 31.
| Age and gender | Study group | Testing at initial presentation | Working diagnosis | Adverse event |
|---|---|---|---|---|
| 43, female | Point of care | PoC tests negative, taken 505 and 600 minutes after worst pain | Musculoskeletal pain | Hospitalisation for ACS 50 days later |
| 63, male | Point of care | PoC tests negative, taken 721 and 824 minutes after worst pain | Musculoskeletal pain | Hospitalisation for ACS 1 day later |
| 64, female | Standard care | Troponin negative 1606 minutes after worst pain | Angina, no ACS | Non-fatal AMI 32 days later |
| 78, female | Point of care | PoC tests negative, taken 800 and 900 minutes after worst pain | Gastro-oesophageal pain | Died from metastatic pancreatic carcinoma 75 days later |
| 78, male | Point of care | CK-MB and myoglobin negative, troponin 0.06 μg/l and 0.07 μg/l at 909 and 1005 minutes after worst pain | Angina, no ACS | Life-threatening arrhythmia (supraventricular tachycardia) 48 days later |
The proportion of admitted patients ultimately diagnosed as having myocardial infarction
Overall 158/2243 (7%) patients were diagnosed as having AMI during their initial presentation: 157 were diagnosed on the basis of a troponin rise and one developed ST elevation and received immediate reperfusion.
The proportion diagnosed with AMI was slightly higher in the point-of-care group [82/1125 (7.3%) vs 76/1118 (6.8%)] but this was not statistically significant (p = 0.650).
The proportion of admitted patients diagnosed with AMI was 82/763 in the point-of-care group and 75/971 in the standard-care group (10.7% vs 7.7%, p = 0.029), suggesting that point-of-care panel assessment was associated with a higher proportion of admitted patients having AMI. This reflects a difference in the denominator between these two proportions, rather than the numerator, i.e. point-of-care was associated with fewer admissions rather than more diagnosis of AMI.
Economic analysis
Tables 32 and 33 show the unit costs used in the main analysis and the microcosting, respectively.
| Trial costs | Cost (£)/event | Sourcea | |
|---|---|---|---|
| PoC panel assessment | See Table 34 | ||
| Cardiac interventions (adjusted for cost of mean length of stay – see below) | |||
| CABG | 6806.86 | HRG code | Average of EA14Z–EA16Z |
| PCI for stenting | 2012.97 | HRG code | Average of EA31Z–EA32Z |
| Thrombolysis | 780.00 | BNF 50 | Alteplase: accelerated regimen, 15 mg IV injection + 50 mg IV infusion (30 minutes) + 35 mg IV infusion (1 hour) |
| Diagnostic procedures | |||
| Angiogram | 129.96 | HRG code | Average of RA16Z–RA18Z – contrast fluoroscopy procedures |
| Radionuclide scan | 221.32 | HRG code | Average of RA35Z–RA39Z – nuclear medicine |
| Exercise treadmill test | 116.04 | HRG code | EA47Z – ECG monitoring and stress testing |
| ECG | 116.04 | HRG code | EA47Z – ECG monitoring and stress testing |
| 24-hour monitoring test | 116.04 | HRG code | EA47Z – ECG monitoring and stress testing |
| Echocardiogram | 176.39 | HRG code | EA46Z – simple echocardiogram |
| Transoesophageal echocardiogram | 104.69 | HRG code | EA45Z – complex echocardiogram |
| Abdominal ultrasound | 57.42 | HRG code | AVERAGE of RA23Z–RA24Z, across both outpatient and admitted care |
| Upper gastrointestinal endoscopy | 258.62 | HRG code | FZ03A – diagnostic and intermediate procedures on the upper gastrointestinal tract 19 years and over |
| MRI scan | 198.53 | HRG code | Average of RA01Z–RA03Z |
| Outpatient clinic/centre visits | |||
| Cardiology – first attendance | 160.08 | HRG code | 320 – cardiology clinic (assume consultant led) |
| Cardiology – follow-up visit | 70.83 | HRG code | 320 – cardiology clinic (assume non-consultant led) |
| Cardiac surgery – first attendance | 270.45 | HRG code | 172 – cardiac surgery (assume consultant led) |
| Cardiac surgery – follow-up attendance | 272.85 | HRG code | 172 – cardiac surgery (assume non-consultant led) |
| Cardiothoracic surgery – first attendance | 202.27 | HRG code | 170 – cardiothoracic surgery (assume consultant led) |
| Cardiothoracic surgery – follow-up attendance | 215.03 | HRG code | 170 – cardiothoracic surgery (assume non-consultant led) |
| Cardiac rehabilitation | 195 | HRG code | average of VB38Z – rehabilitation for AMI and other cardiac disorders (across levels 1 and 2) |
| Rapid-access chest pain clinic | 569.78 | £458 (from Goodacre et al.51) inflated according to Hospital & Community Health Services (pay and prices index)52 | |
| Hospital cost per day | |||
| Coronary care | 1115.78 | HRG code | Average of XC01Z–XC07Z – adult critical care (cardiac ICU) |
| Intensive care | 1410.54 | HRG code | Average of XC01Z–XC07Z – adult critical care (ITU) |
| Medical assessment unit | 276.21 | HRG code | VEB10Z Actual or suspected MI – observation ward – inflated according to Hospital & Community Health Services (pay and prices index)52 |
| Clinical ward | 276.21 | Assumed same as observation ward | |
| Community health-care costs | |||
| GP – surgery (17.2 minutes’ contact) | 52.00 | 8.8b GP52 (p. 109) | |
| GP – home visit (23.4 minutes) | 58.00 | 8.8b GP52 (p. 109) | |
| Nurse – home visit (quarter hour) | 21.65 | 8.4 nurse specialist (community)52 | |
| Social worker visit (1 hour) | 28.46 | 9.2 social worker (adult)52 | |
| Resource | Cost (£) | Source52 |
|---|---|---|
| Consultant | 163 | 13.4 Consultant: medical (all duties/patient contact) |
| Trainee specialist doctor, year 4 | 52 | 13.3 Specialty doctor (40-hour week) |
| Trainee specialist doctor, year 3 | 51 | 13.3 Specialty doctor (40-hour week) |
| Trainee specialist doctor, year 2 | 49 | 13.3 Specialty doctor (40-hour week) |
| Trainee specialist doctor, year 1 | 47 | 13.3 Specialty doctor (40-hour week) |
| Staff grade | 60 | Staff grade (Table 3, p, 169) |
| Senior house officer | 32 | 13.2 Foundation house officer 2 (56-hour week) |
| House officer | 27 | 13.1 Foundation house officer 1 (56-hour week) |
| Nursing director (band 8/9) | 85 | Table 2, p, 168 (band 8b) (patient contact) |
| Nurse manager (band 7) | 74 | 12.1 Nurse team manager (patient contact) |
| Nurse team leader (band 6) | 65 | 12.2 Nurse team leader (patient contact) |
| Staff nurse (band 5) | 47 | 12.3 Nurse, 24-hour ward/RN (patient contact) |
| Clinical/health-care assistant | 23 | 12.5 Clinical support worker (patient contact) |
Table 34 shows the unit costs used to estimate the cost per patient of providing point-of-care panel assessment. The total cost per patient of point-of-care panel assessment was estimated as the sum of a number of components: the cost of the machine and of periodic maintenance, the cost of calibration and quality control during testing, and the annual testing rate for the machine. Information was provided by Siemens Healthcare Diagnostics. Machine and maintenance costs were annualised to the index year (2009). The cost applied to the analyses reported here is that for an annual rate of 1500 full panels (£21.33 per panel). It is worth noting that the relationship between cost and annual rate of testing is known with certainty only for the rates reported in Table 34.
| Cost component | Cost (£) |
|---|---|
| Panel costs (includes reagent, machine and maintenance) | |
| Based on 1500 full panelsa | 20.54 |
| Based on 3000 full panelsa | 15.70 |
| Based on 3000 troponin-only panelsa | 5.70 |
| Calibration and quality control for full panel (per annum)b | 1426 |
| Calibration and quality control for troponin only panel (per annum)b | 1397 |
| Total cost per panel | |
| Based on 1500 full panels | 21.33 |
| Based on 3000 full panels | 16.18 |
| Based on 3000 troponin-only panels | 6.17 |
Data were collected from 246 patients for the microcosting study. The average age of this sample was 54 years, 52% were male (slightly lower than the overall percentage: 58% – Table 7) and 12% had a history of CHD. The results are shown in Table 35. The point-of-care panel assessment added £38.13 per patient managed in this arm of the trial. Staff costs were £11.55 higher in the point-of-care group, reflecting the increased level of staff involvement required to deliver the intervention. Overall, the microcosting study showed that point-of-care panel assessment added £53.16 to the costs of ED management.
| Item cost | PoC (£) (n = 122) | SC (£) (n = 124) |
|---|---|---|
| Staff costsa | 60.76 | 49.21 |
| PoC tests (£21.33/panel) | 38.13b | 0 |
| ED overheads (£5.38/hour) | 20.81 | 17.32 |
| Total costs | ||
| Mean | 119.70 | 66.54 |
| Minimum | 52.04 | 26.41 |
| Maximum | 268.46 | 151.81 |
Table 36 shows the number of patients in each group receiving other interventions during their initial assessment, the number of interventions received and the cost per patient of providing these interventions. The small differences noted in the use of aspirin and clopidogrel between the two groups did not produce a significant difference in the cost per patient of medications. The standard-care group received more laboratory blood tests, leading to an excess cost of £9.42 per patient compared with the point-of-care group. The standard-care group also received more ECGs than the point-of-care group, although this resulted in only a small (£1.95) additional cost per patient. More patients in the point-of-care group received angiography or PCI, but the differences in mean cost per patient attributable to these two interventions were not statistically significant. A cost for ED stay for discharged patients was estimated on a pro rata basis using the ward cost for a day (£276.21) for patients who were discharged from the ED without any subsequent hospital admission. This cost was higher by £5.83 for patients discharged from the point-of-care group, who spent about a half-hour longer in the ED.
| Resource item | No. of patients | Frequencies | Cost (£) | p-valueb | |||
|---|---|---|---|---|---|---|---|
| PoC | SC | PoCa | SCa | PoCa | SCa | ||
| Medications | 827 | 891 | 2086 | 2112 | 5.56 | 5.21 | 0.703 |
| Laboratory tests | 1071 | 1075 | 2079 | 2546 | 28.58 | 38.00 | < 0.001 |
| ECGs | 1125 | 1118 | 1411 | 1484 | 33.44 | 35.39 | < 0.001 |
| Angiograms | 16 | 8 | 16 | 8 | 1.85 | 0.92 | 0.104 |
| ED stayc | 598 | 446 | – | – | 40.36 | 34.53 | < 0.001 |
Table 37 shows resource use after the initial assessment. Items of resource use were costed only if they were chest pain related and could be matched with an appropriate unit cost. This explains why the numbers in this table for some items are slightly lower than corresponding data in the main outcomes analysis (Tables 16–21). Patients in the point-of-care group received more coronary care and intensive care, while patients in the standard-care group received more general inpatient care. None of these resulted in significant differences in mean cost, although the differences in the point estimates for costs relating to coronary care and intensive care were marked, with point-of-care panel assessment being associated with an additional £70.77 and £62.58 per patient, respectively. Patients in the point-of-care group received more outpatient follow-up. Standard care is associated with more nurse home visits and social work visits, but with relatively small differences in mean cost per patient.
| Itema | Resource use | Cost (£) | p-valueb | ||||
|---|---|---|---|---|---|---|---|
| PoC | SC | PoC | SC | ||||
| Patients | Frequency | Patients | Frequency | ||||
| Coronary care days | 47 | 176 | 31 | 104 | 174.56 | 103.79 | 0.064 |
| Intensive care days | 7 | 64 | 3 | 14 | 80.24 | 17.66 | 0.352 |
| Other inpatient days | 429 | 1353 | 564 | 1467 | 330.96 | 362.43 | 0.420 |
| Outpatient attendances | 191 | 222 | 155 | 183 | 35.18 | 27.67 | 0.045 |
| Diagnostic tests (non-laboratory) | 344 | 450 | 327 | 430 | 51.41 | 49.28 | 0.573 |
| Interventions | 47 | 54 | 38 | 40 | 149.83 | 91.25 | 0.061 |
| Post-discharge events (ED attendances) | 100 | 140 | 104 | 155 | 13.30 | 14.82 | 0.507 |
| Community health supportc | |||||||
| GP surgery visits | 562/626 | 1977 | 519/666 | 1816 | 154.36 | 150.85 | 0.646 |
| GP home visits | 44/553 | 98 | 47/535 | 109 | 10.28 | 11.82 | 0.602 |
| Nurse home visits | 44/554 | 128 | 46/529 | 262 | 5.00 | 10.72 | 0.046 |
| Social worker visits | 14/549 | 45 | 26/529 | 164 | 2.33 | 8.82 | 0.091 |
Table 38 shows resource use and costs for community health service support use when missing values for each item are imputed by the mean cost for the corresponding treatment arm. Note that this approach usually underestimates the variability in the data so that p-values are slightly underestimated.
| Item | Resource use | Cost (£) | p-valuea | ||||
|---|---|---|---|---|---|---|---|
| PoC | SC | PoC | SC | ||||
| Patients | Frequency | Patients | Frequency | ||||
| GP surgery visits | 1125 | 2668 | 1118 | 2440 | 123.32 | 113.48 | 0.065 |
| GP home visits | 1125 | 166 | 1118 | 186 | 8.56 | 9.64 | 0.497 |
| Nurse home visits | 1125 | 206 | 1118 | 400 | 3.96 | 7.75 | 0.008 |
| Social worker visits | 1125 | 77 | 1118 | 226 | 1.95 | 5.75 | 0.040 |
| Work time lost (days) | 1125 | 1118 | 7.58 | 8.28 | 0.3776b | ||
Table 39 shows the mean cost per patient by treatment group at each site and across all sites. Inferential analyses were conducted on imputed data; totals based on complete case means are included for comparison. Across all sites the mean cost per patient was higher in the point-of-care group by £211.22 in the imputed analysis (p = 0.056). Mean differences at individual sites ranged from £214.49 less in the point-of-care group at Leeds to £646.57 more in the point-of-care group at Edinburgh, with only the difference at Edinburgh being statistically significant (p = 0.025). An ANOVA test across centres (based on permuted test summed across the imputed data sets) yielded a p-value of 0.0803.
| PoC (£) | SC (£) | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| n | Mean | n | Mean | |||
| Barnsley | 162 | 1058.33 | 164 | 923.13 |
135.20 (–306.97 to 598.44) |
0.538 |
| Derriford | 164 | 1466.81 | 164 | 1307.95 |
158.86 (–326.84 to 679.23) |
0.529 |
| Edinburgh | 228 | 1356.35 | 224 | 709.78 |
646.57 (73.12 to 1612.71) |
0.025 |
| Frenchay | 233 | 1162.53 | 231 | 1058.33 |
104.20 (–288.11 to 511.34) |
0.625 |
| Leeds | 173 | 785.00 | 171 | 999.49 |
–214.49 (–657.10 to 132.56) |
0.345 |
| Leicester | 165 | 1495.54 | 164 | 1115.41 |
380.13 (–181.53 to 914.82) |
0.148 |
| Total (imputed data) | 1125 | 1217.14 | 1,118 | 1005.91 |
211.23 (–16.53 to 442.90) |
0.056 |
| Total (complete case means) | 1216.18 | 1008.94 |
207.24 (2.98 to 431.62) |
0.047 | ||
Tables 40 and 41 show the mean EQ-5D scores at 30 and 90 days using complete cases only (i.e. only those with data at both time points) and with imputation of missing values. There was no significant difference at either time point. Imputation did not markedly change the mean scores in either group at either time point.
| PoC | SC | p-valuea | |||
|---|---|---|---|---|---|
| n | Mean | n | Mean | ||
| Score at 1 month | 697 | 0.747 | 650 | 0.761 | 0.369 |
| Score at 3 months | 697 | 0.753 | 650 | 0.759 | 0.710 |
| Item | PoC | SC | p-valuea | ||
|---|---|---|---|---|---|
| n | Mean | n | Mean | ||
| Score at 1 month | 1125 | 0.753 | 1118 | 0.769 | 0.158 |
| Score at 3 months | 1125 | 0.764 | 1118 | 0.772 | 0.433 |
Table 42 shows the total QALYs accrued (the difference) over 3 months at each site for both treatment groups. Data are reported assuming that EQ-5D was zero at baseline, although means for any baseline score between 0 and 1 can be estimated by adding a constant k/24, where k is the baseline EQ-5D score of interest. As 3 months is approximately one-quarter of a year, the maximum possible number of QALYs accrued is 0.25 (assuming EQ-5D was 1 at baseline). In practice, there is very little difference between treatment arms evident (a difference of 0.00273 is equivalent to 1 day), and there were no significant differences in QALYs accrued at any site or across all sites.
| PoC | SC | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| n | Mean | n | Mean | |||
| Barnsley | 162 | 0.153 | 164 | 0.157 |
–0.004 (–0.016 to 0.010) |
0.621 |
| Derriford | 164 | 0.158 | 164 | 0.156 |
0.002 (–0.011 to 0.013) |
0.743 |
| Edinburgh | 228 | 0.158 | 224 | 0.162 |
–0.004 (–0.014 to 0.006) |
0.529 |
| Frenchay | 233 | 0.157 | 234 | 0.163 |
–0.006 (–0.016 to 0.004) |
0.267 |
| Leeds | 173 | 0.162 | 171 | 0.162 |
0.000 (–0.010 to 0.011) |
0.687 |
| Leicester | 165 | 0.158 | 164 | 0.163 |
–0.005 (–0.015 to 0.006) |
0.401 |
| Total | 1125 | 0.158 | 1118 | 0.161 |
–0.003 (–0.007 to 0.002) |
0.250 |
The cost-effectiveness plane based on the resampled cost-effectiveness data points for the point-of-care test strategy (Figure 6, with mean values shown by grey target) shows that, owing to the negative sign of the incremental QALYs, there is a high probability that the strategy is dominated (empirical probability 0.888). Conversely, the probability of the point-of-care test strategy being cost-effective at £20,000 per QALY is (understandably) very low (0.004).
FIGURE 6.
Cost-effectiveness plane for point-of-care treatment strategy.
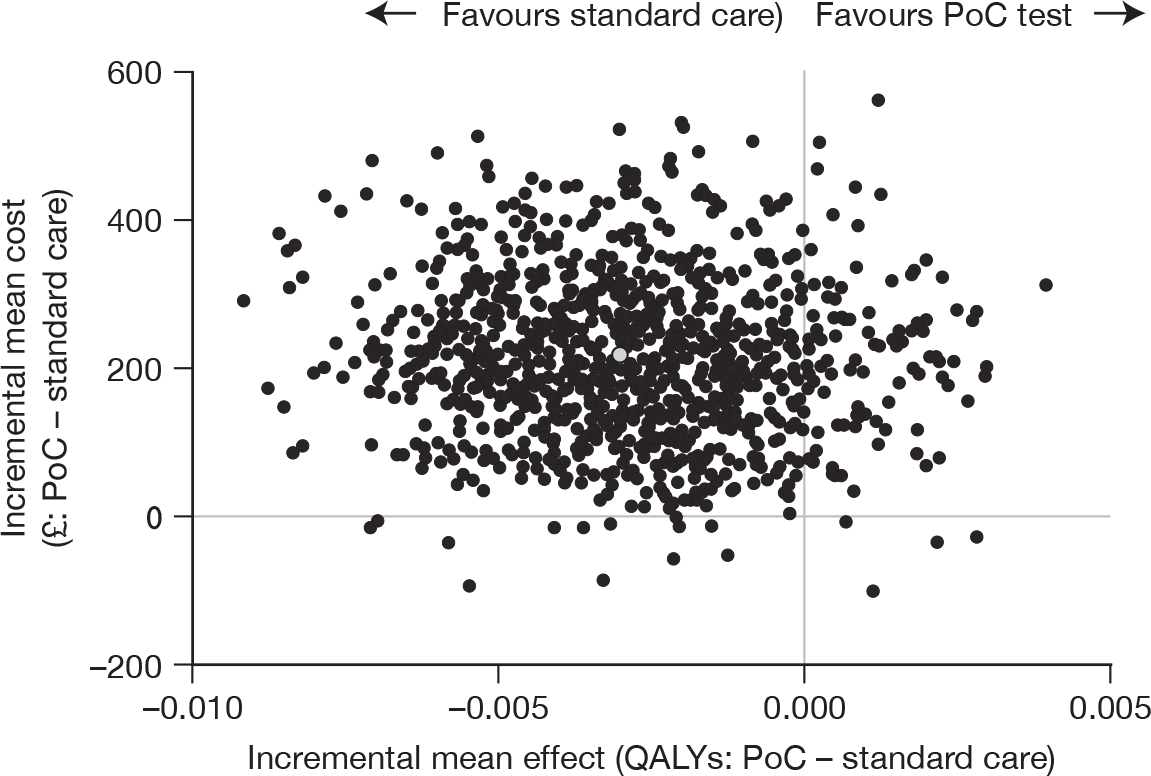
Cost-effectiveness results are presented in Table 43. Empirical probabilities of a policy being dominated [i.e. has higher cost and is less effective than the index (standard care) policy], and of being cost-effective (i.e. having an incremental cost-effectiveness ratio no larger than £20,000 when the policy is more effective), are also presented.
| Policy | Incremental means | Probability of | ||
|---|---|---|---|---|
| Cost (£) | QALYs | Being dominated | Cost-effectiveness at £20,000 per QALY | |
| SC | 0 | 0 | – | – |
| PoC | 211.22 | –0.0028154 | 0.888 | 0.004 |
| PoC test using a single troponin test | 179.26 | –0.0028154 | 0.869 | 0.006 |
| PoC costs excluding intensive care | 153.59 | –0.0028154 | 0.876 | 0.006 |
Table 43 also shows the results of two analyses used to explore whether the findings were robust to two important assumptions. First, we explored whether a cheaper point-of-care test could be cost-effective by assuming that the test performance for a single troponin test, which at £6.17 incurs the lowest feasible point-of-care test cost, is the same as that of the trial strategy. In this scenario only costs would be affected, and test costs are reduced from £38.13 to £6.17, so that the mean cost difference would reduce to £179.26. Despite this, the probability of the point-of-care strategy being cost-effective remained low. Second, we explored whether excluding intensive care costs would alter the findings. Intensive care costs are only weakly related to the intervention and one patient in the point-of-care group had spent 52 days on intensive care, incurring substantial costs and increasing the variance of cost estimates. The results show that excluding intensive care costs reduced the mean cost per patient of point-of-care but did not markedly alter the probability that this strategy would be cost-effective.
These sensitivity analyses do not vary the results of the base case (i.e. trial) cost-effectiveness analysis, which might be expected as the alternative scenarios affect only costs.
Figure 7 shows estimated 95% confidence ellipses, assuming bivariate normality, of incremental costs and QALYs, for each of the hospital sites. Note that the plots are based on smaller sample sizes than the overall result and this is reflected in the higher variability. The plots generally reflect the overall result, and highlight, for example, the greater incremental cost variability of Edinburgh (where the patient with the long intensive care stay was recruited) and the possibility that, for Derriford or Leeds, the strategy may have a reasonable probability of being cost-effective.
FIGURE 7.
Cost-effectiveness plane for point-of-care treatment strategy, by site.
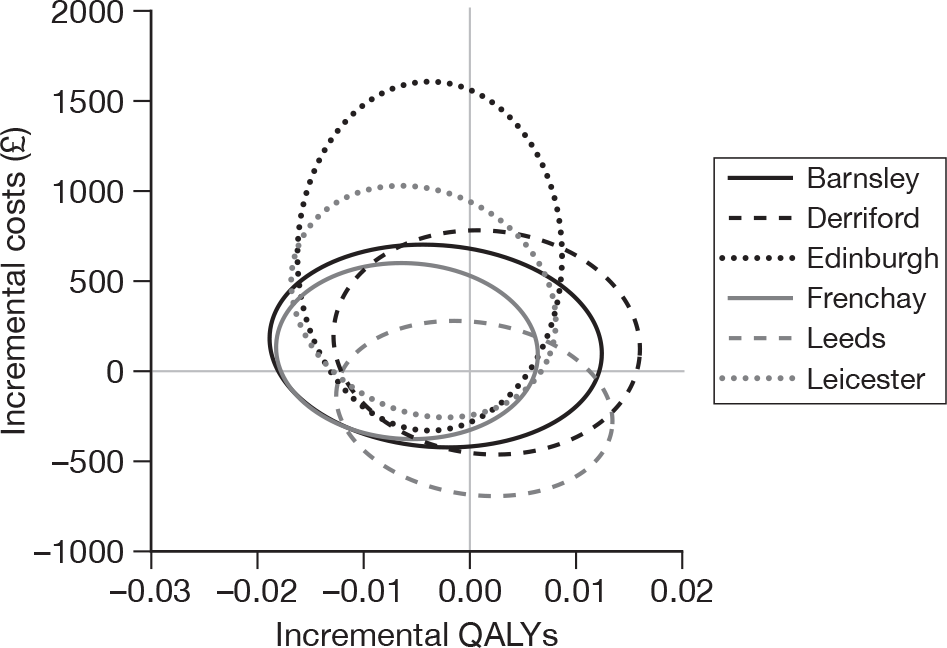
Decision-analytic modelling
The parameters used in the model are shown in Appendix 4. The probabilistic analyses based on the decision-analytic model yielded a mean difference in costs (point of care minus standard care) of £169 (empirical 95% CI –£1229 to £1658) and in QALYs of –0.002 (95% CI –0.108 to 0.104), which correspond reasonably with the trial-based results, particularly in the light of the simplifications outlined above (see Methods). The estimated probability that point-of-care panel assessment is cost-effective for a willingness to pay of £20,000 per QALY under these circumstances was 0.223. The model extrapolated to lifetime survival resulted in a mean cost difference of £329 (95% CI –£1312 to £2028) and corresponding value for QALYs of –0.087 (95% CI –12.3 to 12.2) accrued over a lifetime. The probability of cost-effectiveness of point-of-care panel assessment based on lifetime survival was estimated at 0.336.
Evaluation of GRACE and TIMI scores
Table 44 shows the number and proportion of patients with and without a major adverse event up to 1 month in each quintile of the GRACE score and each category of the TIMI score. Table 45 shows the same data for major adverse events up to 3 months. The proportion with a major adverse event at either 1 month or 3 months increases progressively with increasing quintiles of GRACE score regardless of the method used to incorporate troponin or the method used to analyse the data. The relationship between TIMI score and proportion with a major adverse event is not as clear, especially at 1 month. Patients with a TIMI score of 2 had the highest rate of major adverse events by 1 month.
| Score | No. of patients | Major adverse event [n (%)] | No major adverse event [n (%)] |
|---|---|---|---|
| GRACE score | |||
| Complete case analysis | |||
| Any troponin positive | |||
| < 60 | 280 | 0 | 280 (100.0) |
| 60–79 | 402 | 5 (1.2) | 397 (98.8) |
| 80–89 | 251 | 3 (1.2) | 248 (98.8) |
| 90–109 | 384 | 11 (2.9) | 373 (97.1) |
| > 109 | 422 | 19 (4.5) | 403 (95.5) |
| First troponin positive | |||
| < 60 | 304 | 1 (0.3) | 303 (99.7) |
| 60–79 | 429 | 4 (0.9) | 425 (99.1) |
| 80–89 | 260 | 5 (1.9) | 255 (98.1) |
| 90–109 | 363 | 11 (3.0) | 352 (97.0) |
| > 109 | 392 | 17 (4.3) | 375 (95.7) |
| Imputed analysis | |||
| Any troponin positive | |||
| < 60 | 389 | 0 | 389 (100.0) |
| 60–79 | 520 | 5 (1.0) | 515 (99.0) |
| 80–89 | 327 | 5 (1.5) | 322 (98.5) |
| 90–109 | 477 | 13 (2.7) | 464 (97.3) |
| > 109 | 530 | 20 (3.8) | 510 (96.2) |
| First troponin positive | |||
| < 60 | 414 | 1 (0.2) | 413 (99.8) |
| 60–79 | 547 | 4 (0.7) | 543 (99.3) |
| 80–89 | 338 | 7 (2.1) | 331 (97.9) |
| 90–109 | 459 | 13 (2.8) | 446 (97.2) |
| > 109 | 485 | 18 (3.7) | 467 (96.3) |
| TIMI score | |||
| Complete case analysis | |||
| Any troponin positive | |||
| 0 | 764 | 5 (0.7) | 759 (99.3) |
| 1 | 565 | 14 (2.5) | 551 (97.5) |
| 2 | 314 | 13 (4.1) | 301 (95.9) |
| 3 | 106 | 4 (3.8) | 102 (96.2) |
| 4 or 5 | 33 | 1 (3.0) | 32 (97.0) |
| First troponin positive | |||
| 0 | 839 | 6 (0.7) | 833 (99.3) |
| 1 | 531 | 15 (2.8) | 516 (97.2) |
| 2 | 305 | 13 (4.3) | 292 (95.7) |
| 3 | 89 | 2 (2.2) | 87 (97.8) |
| 4 or 5 | 31 | 1 (3.2) | 30 (96.8) |
| Imputed analysis | |||
| Any troponin positive | |||
| 0 | 969 | 6 (0.6) | 963 (99.4) |
| 1 | 695 | 15 (2.2) | 680 (97.8) |
| 2 | 403 | 16 (4.0) | 387 (96.0) |
| 3 | 138 | 5 (3.6) | 133 (96.4) |
| 4 or 5 | 38 | 1 (2.6) | 37 (97.4) |
| First troponin positive | |||
| 0 | 1046 | 8 (0.8) | 1038 (99.2) |
| 1 | 656 | 15 (2.3) | 641 (97.7) |
| 2 | 386 | 16 (4.1) | 370 (95.9) |
| 3 | 120 | 3 (2.5) | 117 (97.5) |
| 4 or 5 | 35 | 1 (2.9) | 34 (97.1) |
| Score | No. of patients | Major adverse event [n (%)] | No major adverse event [n (%)] |
|---|---|---|---|
| GRACE score | |||
| Complete case analysis | |||
| Any troponin positive | |||
| < 60 | 280 | 0 | 280 (100.0) |
| 60–79 | 402 | 7 (1.7) | 395 (98.3) |
| 80–89 | 251 | 7 (2.8) | 244 (97.2) |
| 90–109 | 384 | 12 (3.1) | 372 (96.9) |
| > 109 | 422 | 29 (6.9) | 393 (93.1) |
| First troponin positive | |||
| < 60 | 304 | 1 (0.3) | 303 (99.7) |
| 60–79 | 429 | 6 (1.4) | 423 (98.6) |
| 80–89 | 260 | 9 (3.5) | 251 (96.5) |
| 90–109 | 363 | 12 (3.3) | 351 (96.7) |
| > 109 | 392 | 27 (6.9) | 365 (93.1) |
| Imputed analysis | |||
| Any troponin positive | |||
| < 60 | 389 | 0 | 389 (100.0) |
| 60–79 | 520 | 7 (1.3) | 513 (98.7) |
| 80–89 | 327 | 10 (3.1) | 317 (96.9) |
| 90–109 | 477 | 14 (2.9) | 463 (97.1) |
| > 109 | 530 | 31 (5.8) | 499 (94.2) |
| First troponin positive | |||
| < 60 | 414 | 1 (0.2) | 413 (99.8) |
| 60–79 | 547 | 6 (1.1) | 541 (98.9) |
| 80–89 | 338 | 12 (3.6) | 326 (96.4) |
| 90–109 | 459 | 15 (3.3) | 444 (96.7) |
| > 109 | 485 | 28 (5.8) | 457 (94.2) |
| TIMI score | |||
| Complete case analysis | |||
| Any troponin positive | |||
| 0 | 764 | 8 (1.0) | 756 (99.0) |
| 1 | 565 | 17 (3.0) | 548 (97.0) |
| 2 | 314 | 18 (5.7) | 296 (94.3) |
| 3 | 106 | 6 (5.7) | 100 (94.3) |
| 4 or 5 | 33 | 2 (6.1) | 31 (93.9) |
| First troponin positive | |||
| 0 | 839 | 9 (1.1) | 830 (98.9) |
| 1 | 531 | 20 (3.8) | 511 (96.2) |
| 2 | 305 | 16 (5.2) | 289 (94.8) |
| 3 | 89 | 4 (4.5) | 85 (95.5) |
| 4 or 5 | 31 | 2 (6.5) | 29 (93.5) |
| Imputed analysis | |||
| Any troponin positive | |||
| 0 | 969 | 9 (0.9) | 960 (99.1) |
| 1 | 695 | 19 (2.7) | 676 (97.3) |
| 2 | 403 | 24 (6.0) | 379 (94.0) |
| 3 | 138 | 8 (5.8) | 130 (94.2) |
| 4 or 5 | 38 | 2 (5.3) | 36 (94.7) |
| Troponin positive | |||
| 0 | 1046 | 11 (1.1) | 1035 (98.9) |
| 1 | 656 | 22 (3.4) | 634 (96.6) |
| 2 | 386 | 21 (5.4) | 365 (94.6) |
| 3 | 120 | 6 (5.0) | 114 (95.0) |
| 4 or 5 | 35 | 2 (5.7) | 33 (94.3) |
Table 46 reports the area under the receiver-operator characteristic curve for GRACE and TIMI scores for predicting major adverse events up to 1 month and 3 months, and Figure 8 shows the curves. As before, these are presented for both methods of incorporating the troponin measurement and both methods of analysing the data. The method of analysis made little difference but the use of any troponin measurement resulted in consistently higher discriminant value than the use of the first troponin only. Both scores tended to predict events slightly better over the longer follow-up period. Overall, GRACE had better discriminant value for predicting adverse events than TIMI, although neither score predicted events with the kind of accuracy required to be clinically useful (i.e. with an area under the receiver-operator characteristic curve greater than 0.8). The area under the receiver-operator characteristic curve for age alone as a predictor was 0.66 for major adverse events up to 1 month and 0.69 for events up to 3 months. Hence, knowing the TIMI score appears to be no better than knowing the patient’s age for predicting subsequent major adverse events in this cohort.
| Area under the ROC curve | 95% CI | ||||
|---|---|---|---|---|---|
| Lower | Upper | ||||
| 1 month | |||||
| GRACE score | Complete case analysis | Any troponin positive | 0.722 | 0.700 | 0.743 |
| First troponin positive | 0.706 | 0.683 | 0.727 | ||
| Imputed analysis | Any troponin positive | 0.717 | 0.698 | 0.735 | |
| First troponin positive | 0.705 | 0.686 | 0.724 | ||
| TIMI score | Complete case analysis | Any troponin positive | 0.678 | 0.656 | 0.700 |
| First troponin positive | 0.668 | 0.645 | 0.689 | ||
| Imputed analysis | Any troponin positive | 0.682 | 0.662 | 0.701 | |
| First troponin positive | 0.665 | 0.645 | 0.684 | ||
| 3 months | |||||
| GRACE score | Complete case analysis | Any troponin positive | 0.731 | 0.709 | 0.752 |
| First troponin positive | 0.721 | 0.699 | 0.742 | ||
| Imputed analysis | Any troponin positive | 0.726 | 0.707 | 0.745 | |
| First troponin positive | 0.715 | 0.696 | 0.734 | ||
| TIMI score | Complete case analysis | Any troponin positive | 0.680 | 0.657 | 0.701 |
| First troponin positive | 0.668 | 0.645 | 0.689 | ||
| Imputed analysis | Any troponin positive | 0.693 | 0.674 | 0.712 | |
| First troponin positive | 0.675 | 0.655 | 0.694 | ||
FIGURE 8.
Receiver-operator characteristic curves for GRACE and TIMI scores. (a) TIMI score and 30-day major adverse events (MAEs). (b) GRACE score and 30-day MAEs. (c) TIMI score and 90-day MAEs. (d) GRACE score and 90-day MAEs.
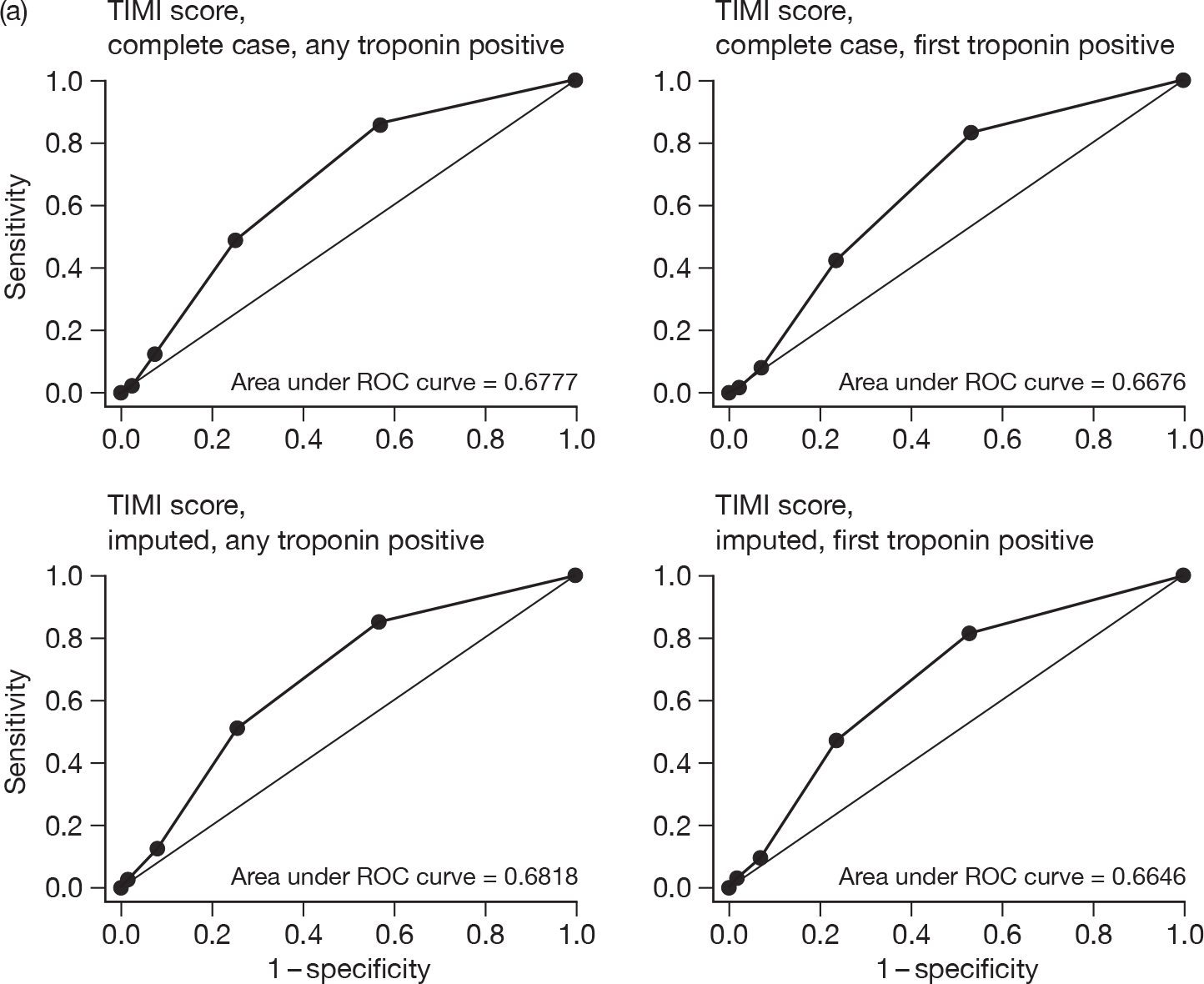
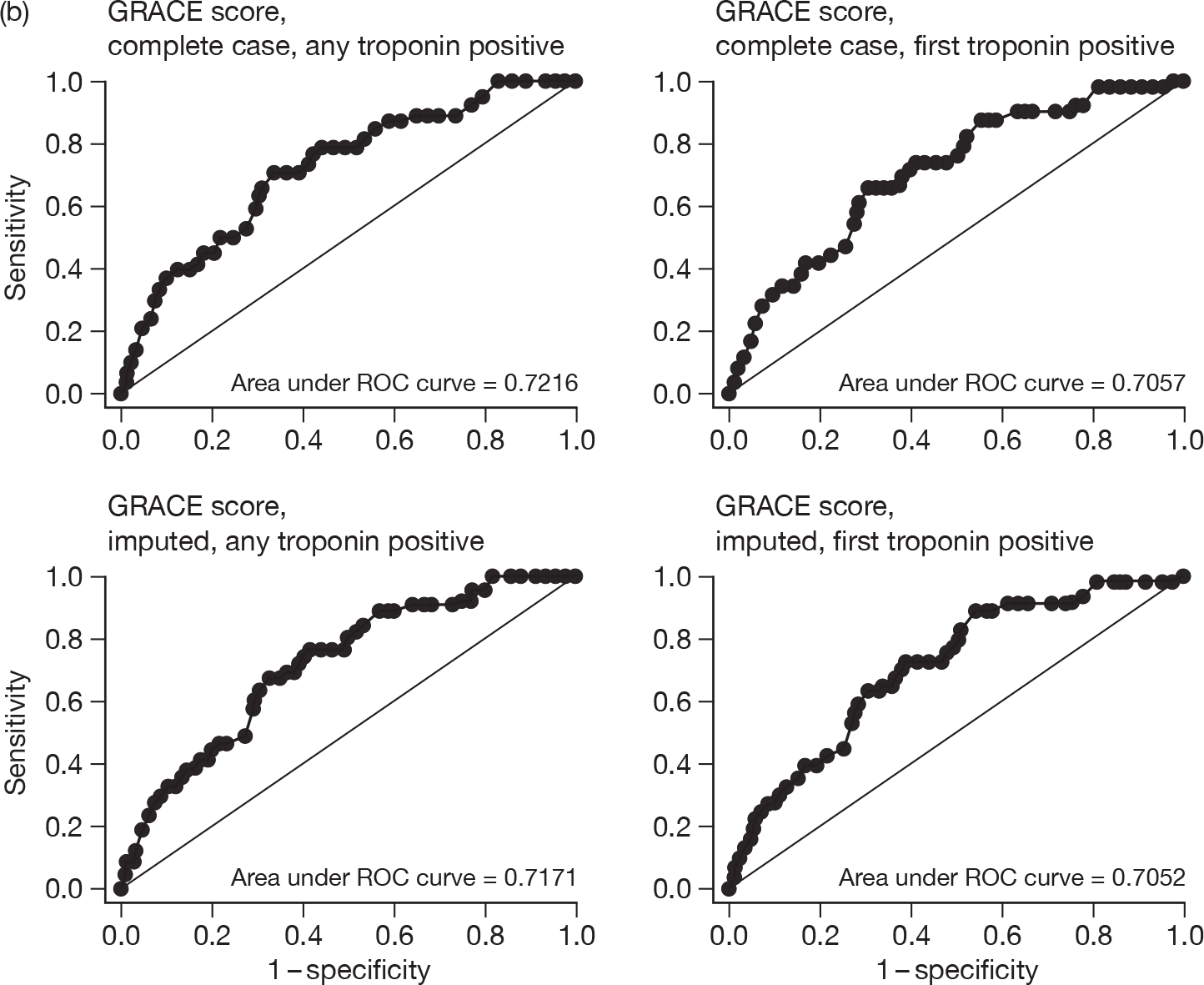
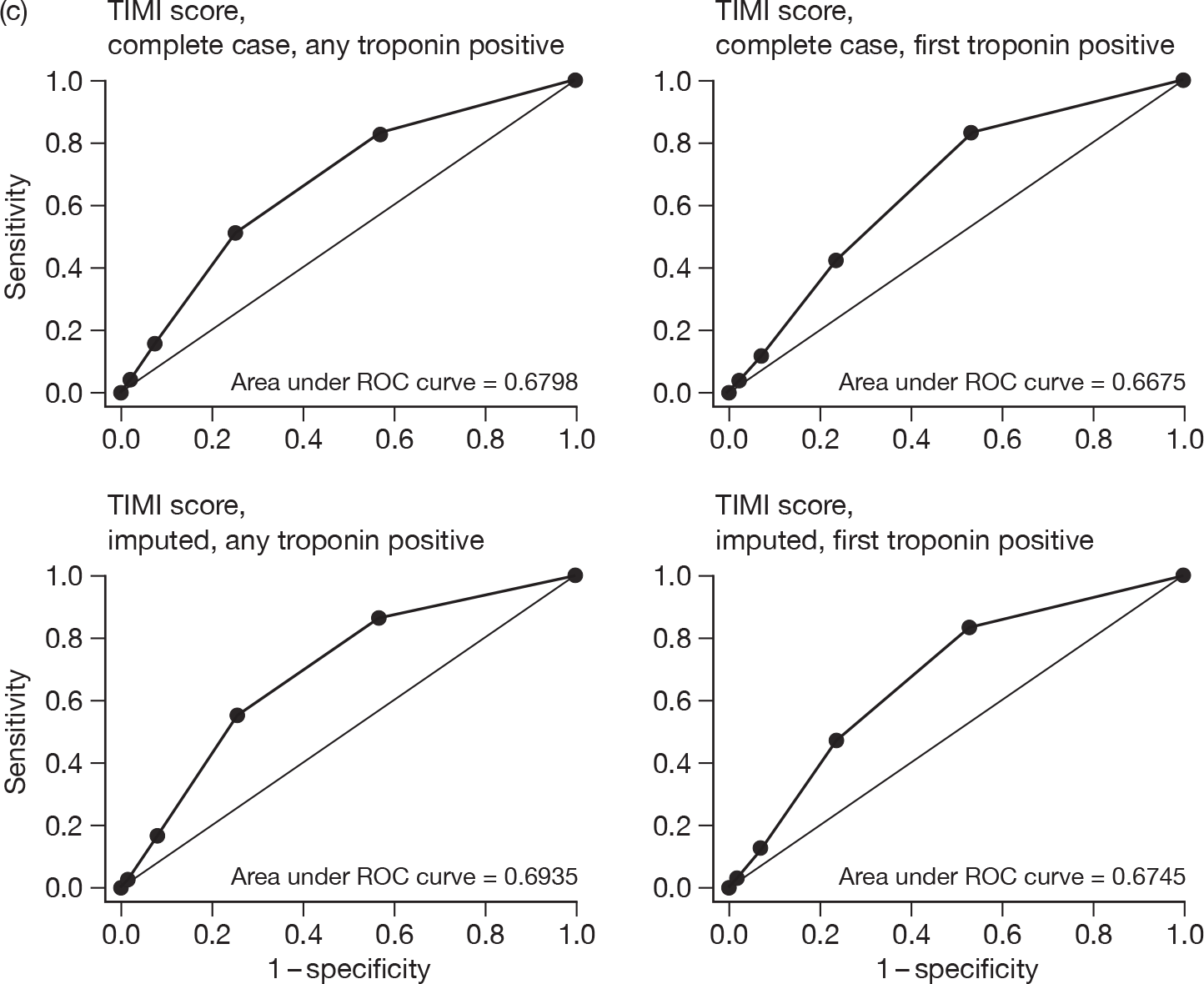
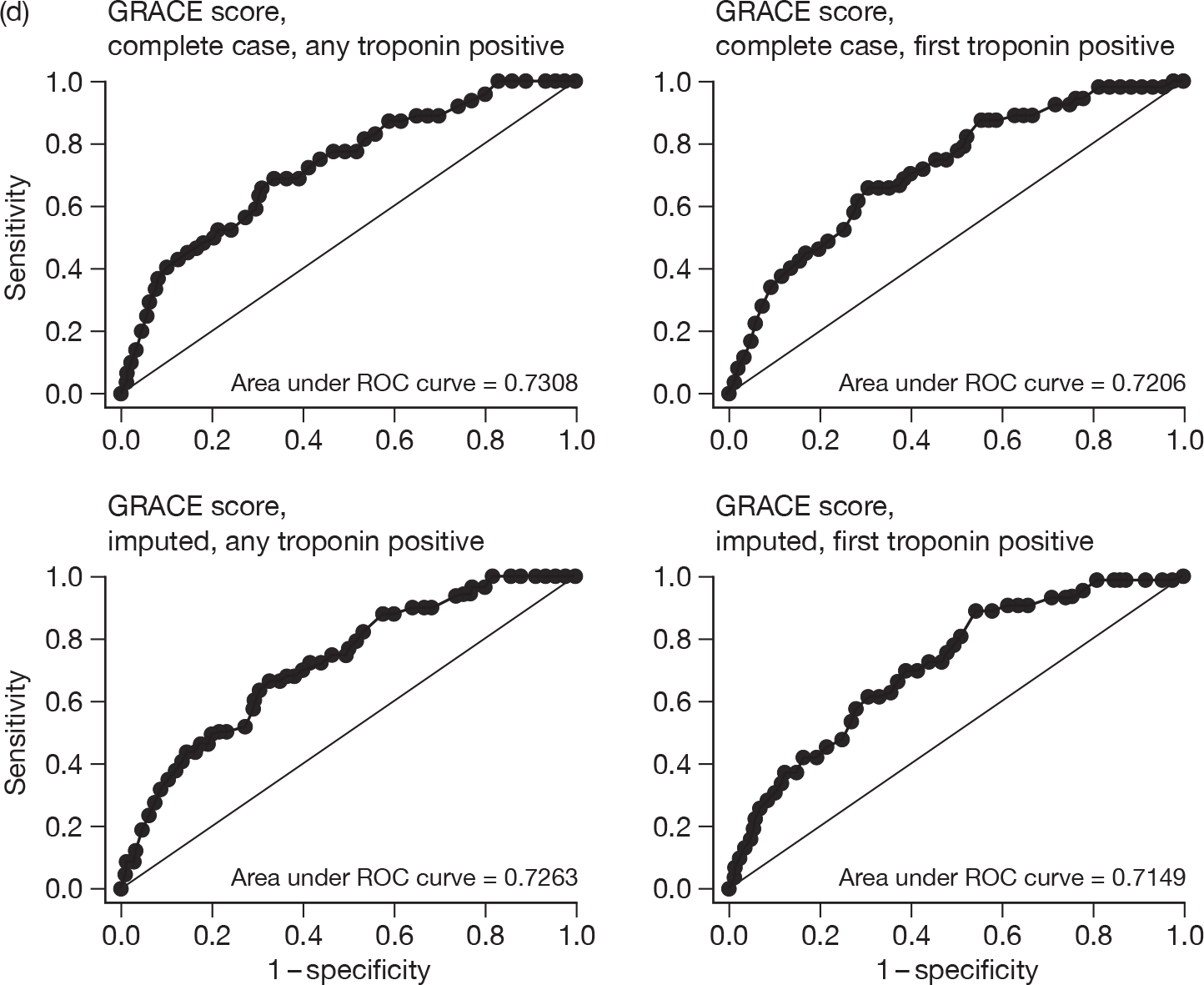
Chapter 4 Discussion
Effectiveness of point-of-care panel assessment
Point-of-care panel assessment with CK-MB (mass), myoglobin and troponin at baseline and 90 minutes increased the proportion of patients with acute undifferentiated chest pain successfully discharged home after ED assessment (defined as having left the hospital or with a decision to discharge by 4 hours after attendance and no major adverse event over the following 3 months). The difference in the proportion of patients in hospital persisted until 24 hours after attendance and resulted in a shorter median length of initial hospital stay for patients in the point-of-care group and a greater proportion of the point-of care group having no inpatient days over the 3 months of the trial. However, it did not lead to any difference in the mean length of initial hospital stay or any difference in the mean number of inpatient days over the 3-month follow-up. This appeared to be due to a small number of patients with very long hospital stays and/or large number of inpatient-days that increased the variance for these variables and which were over-represented in the point-of-care group.
These findings suggest that the benefit of point-of-care panel assessment may depend upon one’s perspective. The proportion successfully discharged and the median length of stay describe the experience of the typical patient. Patients and clinicians may perceive benefit from point-of-care panel assessment, reducing the proportion of patients admitted and the duration of hospital stay for a typical patient. Hospital bed use is best estimated from the mean length of stay and mean number of inpatient days. When these are multiplied by the number of patients they produce an estimate of the total bed-days used by the patient cohort. Point-of-care panel assessment may reduce patient turnover (i.e. the number of patients being admitted and using hospital beds), but our data suggest that it will not significantly reduce hospital bed use.
Point-of-care panel assessment was associated with increased use of coronary care and administration of clopidogrel, but reduced use of aspirin. The last two findings appear to match each other, with 5% more patients in the point-of-care group receiving clopidogrel and 5% fewer receiving aspirin. These findings suggest a potential benefit from point-of-care panel assessment but one that is difficult to extrapolate into measures that are meaningful to the patient, such as improved survival or quality of life. They do, however, incur additional costs and are considered in the economic analysis.
Point-of-care panel assessment was not associated with any significant differences in investigations, cardiac interventions, ED attendances and subsequent hospital admissions over the following 3 months, although the non-significant differences in these areas of resource use contributed to the economic analysis. Point-of-care panel assessment was associated with a borderline significant increase in chest pain-related outpatient attendances, but this was one of many hypothesis tests and could be a chance finding.
From the patient’s perspective, point-of-care panel assessment was associated with no significant differences in health utility at 1 or 3 months, or time taken off work. It was associated with higher overall satisfaction with care, with more patients in the point-of-care group assessing their care as being excellent or very good, and fewer assessing it as good, fair or poor. Point-of-care panel assessment received a more favourable rating on 2 of the 10 dimensions of satisfaction measured: the urgency of assessment and the personal interest shown in the patient and his or her medical problems. There were no significant differences between the study groups on the other dimensions.
These findings suggest a modest patient preference for point-of-care panel assessment. However, patients were not blinded and would have been aware of whether they were receiving the experimental intervention or standard care. Their preference for point-of-care panel assessment may therefore reflect a positive response to receiving the ‘new’ intervention in a trial rather than a specific preference for the intervention.
Major adverse event rates were low, with only 62 out of 2243 patients (3%) having any major adverse event over 3 months, including only eight deaths and 10 non-fatal MIs. There were no significant differences in overall major adverse event rates or the rates of any individual event. CIs for adverse event rates were wide and the study was only originally powered to detect a relatively high 2% absolute increase in major adverse event rates.
We looked in detail at the major adverse events occurring in patients who were discharged after ED assessment, as these are the patients in whom a causal link between initial diagnostic assessment and major adverse event might be strongest. For example, if a patient was discharged after ED assessment had failed to identify MI then this could increase the risk of adverse outcome. Four patients in the point-of-care arm and one in the standard-care arm suffered adverse events after ED discharge. In all but one case the adverse event occurred more than 1 month after initial assessment. This relatively long time between intervention and outcome makes it difficult to assert a causal link. Furthermore, all five patients presented relatively late after their pain and received biomarker testing after a relatively long delay from worst symptoms. It is therefore very unlikely that any of these patients had a missed diagnosis of MI at their initial presentation.
Cost-effectiveness of point-of-care panel assessment
Overall, the economic analysis showed that point-of-care panel assessment was unlikely to be cost-effective compared with standard care. The main analysis produced a 0.888 probability that standard care dominated point-of-care panel assessment (i.e. was cheaper and more effective). The cost-effectiveness analysis was planned as an analysis of the incremental cost per QALY of point-of-care panel assessment. However, the main difference between the strategies and the main driver of cost-effectiveness was cost. The difference in QALYs between the two groups was small (0.003 QALYs) and non-significant (p = 0.250). The difference in costs between the two groups was £211 and bordered on being significant (p = 0.056). Hence, it appears that point-of-care panel assessment may increase costs despite reducing hospital admissions and inpatient days.
There appear to be a number of reasons for this. First, point-of-care panel assessment is associated with an additional £53 per patient for initial ED assessment. Most of this (£38) is related to the point-of-care machine, but additional staff time to undertake the tests also adds to the costs. It is possible that using the machine for other ED patients (thus reducing the amount charged for equipment) or using a simple protocol (such as a single troponin measurement) could reduce the additional costs associated with point-of-care panel assessment. We undertook a sensitivity analysis assuming that the machine costs were only £6.17 per patient instead of £38.13, but this did not markedly change the overall result, with there still being a 0.869 probability that standard care was dominant.
Point-of-care panel assessment was also associated with markedly higher mean costs for cardiac interventions, coronary care and intensive care. These failed to compensate for a £31 per patient saving from reduced inpatient days. The cost differences were not statistically significant but still added to the overall cost difference between point-of-care panel assessment and standard care. It could be argued that intensive care admissions are unlikely to be related to the initial diagnostic process. Indeed, one patient who suffered serious complications after CABG accounted for most of the intensive care days. We therefore undertook a sensitivity analysis with intensive care costs excluded. Although the difference in costs was reduced from £211 to £154, the probability of standard care being dominant remained high at 0.876.
The finding that there is a very low probability of point-of-care panel assessment being cost-effective is perhaps surprising given that the trial showed it was associated with a statistically significant improvement in its primary outcome measure, the proportion of patients successfully discharged home. However, it appears that earlier diagnosis may have an impact on other clinical decisions in addition to discharge home from the ED. In particular, admissions to coronary care and cardiac interventions were more frequent in the point-of-care group. One possible explanation for the increase in coronary care costs, for example, is that early recognition of troponin elevation may prompt admission to the coronary care unit, whereas an elevated 12-hour troponin after admission to a general ward is unlikely to prompt transfer to coronary care.
Variation between the participating sites
The analysis of the primary outcome and cost-effectiveness analysis showed marked differences between the participating sites. As described in the primary efficacy results section (see Chapter 3), these differences are probably due to differences in the facilities available for managing patients with chest pain and the standard care processes normally undertaken at the hospital. All hospitals, except Leicester, showed an effect from point-of-care panel assessment upon the primary outcome. The most marked effect occurred at hospitals in which chest pain patients were subject to a target of spending no more than 4 hours in the ED and standard care involved a 12-hour troponin. In these hospitals there was a significant difference in the primary outcome, which persisted until about 24 hours after attendance. At Leeds, where all patients with chest pain were cared for on a clinical decision unit that was not subject to a 4-hour target, the effect of point-of-care panel assessment was delayed until around 6 hours after attendance. At Derriford, where the standard care guidelines recommended a 6-hour troponin, the effect of point-of-care panel assessment was short-lived and not noticeable after 8 hours from attendance. The exception of Leicester is probably explained by trial factors. The Leicester Research Office stipulated that only doctors who had received full training in GCP could undertake trial recruitment. This meant that the research nurses were overwhelmingly responsible for recruiting patients and producing point-of-care test results. This probably meant that the clinical decision-makers (doctors) were not engaged in the trial and did not act on the test results.
The differences between the sites in the primary outcome are not reflected in differences in mean costs per patient. This is perhaps unsurprising as the overall analysis showed that the increase in the proportion successfully discharged with point-of-care panel assessment did not translate into cost savings. Mean costs were higher in the point-of-care arm at five hospitals and lower at one. It is possible that the overall conclusion that point-of-care panel assessment is unlikely to be cost-effective does not apply at some of the sites, such as Leeds and Derriford. However, we should be careful about drawing conclusions from individual site cost data because cost variances are large and most of the cost differences at individual sites were not statistically significant. The apparently dramatically higher costs associated with point-of-care panel assessment at Edinburgh, in particular, can be at least in part accounted for by two patients in the point-of-care arm with very high costs due to prolonged hospital stays.
However, in general terms it is possible that inpatient treatment patterns differ between hospitals and that in some settings point-of-care panel assessment in the ED does not lead to higher costs of inpatient care. If this were the case then point-of-care would have the potential to be cost-effective, as is suggested with the confidence ellipses in Figure 7. Previous work on chest pain units has tried to identify the combination of costs and admission rates that would make such initiatives cost-effective. 53 Similar studies could be worthwhile here.
Variation over time
The effect of point-of-care panel assessment could change over time as a result of clinicians becoming familiar with the technology and changing their decision-making in response to the point-of-care results. In particular, the change in the recommended decision-making threshold for point-of-care troponin could have changed the proportion of patients successfully discharged after point-of-care assessment. Conversely, standard care could change over time and start to mimic point-of-care panel assessment, thus leading to contamination of the control group and a bias towards no effect.
We analysed the primary outcome and deviations from the recommended point-of-care protocol at quarterly intervals to explore whether the effect of point-of-care panel assessment changed over time. Only two patients in the control group received the combination of tests used in the point-of-care machine (CK-MB, myoglobin and troponin) either from the machine itself or from the hospital laboratory, so we did not examine deviations from standard care over time. Deviations from the recommended point-of-care protocol remained relatively constant over time, so there was no evidence that developing experience with the technology eliminated these problems. However, the proportion successfully discharged in the point-of-care arm decreased during the middle third of the trial and increased in the last third, while the proportion successfully discharged in the standard-care arm steadily fell over the trial. The first finding may be explained by the amendment to the troponin threshold in the recommended point-of-care protocol that advised discharge instead of admission for patients with a non-rising troponin in the range 0.03–0.07 µg/l. The second finding may be due to clinical staff becoming progressively more cautious during the trial. However, we must be careful not to overinterpret these findings as they are based on relatively small numbers.
Other studies of point-of-care cardiac markers
The RATPAC trial is the first randomised trial to evaluate whether a panel of point-of-care cardiac markers designed to rule out MI in ED patients with acute chest pain can change clinical decision-making and lead to changes in patient care. Previous studies22,23 have evaluated the point-of-care panel and laboratory testing in the same cohort to compare the times taken to receive a test result and have shown that turnaround times are shorter with point-of-care testing, but few studies have compared practice in cohorts with or without point-of-care testing. Ng et al. 24 compared management with the point-of-care panel with previous practice without the test and found a 40% reduction in coronary care unit admissions. This contrasts with RATPAC, in which we found that point-of-care panel assessment was associated with increased coronary care use. There are two potential explanations for this discrepancy. First, the study by Ng et al. 24 may have been confounded by changes in practice occurring over time that would have been controlled for in RATPAC by the randomised design. Second, the study by Ng et al. 24 was undertaken in a health-care system and patient cohort with much higher coronary care use, so there was much greater potential to reduce admissions. This illustrates the difficulties in extrapolating findings relating to health service use between diverse settings.
Other studies, including randomised trials, have evaluated point-of-care testing by directly comparing use of a point-of-care troponin assay with use of a laboratory assay, rather than evaluating a point-of-care protocol as we did in RATPAC. As outlined in Chapter 1, such studies have produced mixed results, with Collinson et al. 12 finding reduced length of coronary care unit stay and overall hospital stay, Renaud et al. 13 reporting reduced time to anti-ischaemic therapy and physician notification of results, but no change in ED length of stay or patient outcomes, and Ryan et al. 14 finding that the effect of point-of-care testing varied between settings, with length of stay in the ED being increased in one hospital and decreased in another. These studies are not directly comparable with RATPAC because they compare only the process of using a point-of-care versus a laboratory assay, whereas RATPAC evaluated a point-of-care protocol involving a specific panel of tests designed to rule out MI in patients with acute chest pain. However, the inconsistencies noted between the findings of these studies and the variability between the sites involved in RATPAC suggest that the effects of point-of-care testing are inconsistent, whether one is simply examining the effect of point-of-care testing itself or point-of-care testing is being used to deliver a specific protocol.
Strengths and weaknesses of the study
As a randomised trial, RATPAC has the strength of providing an unbiased comparison between the point-of-care protocol and standard care. Allocation of treatment group was concealed until the patient was irreversibly entered into the trial and the low rate of withdrawals before the primary outcome, coupled with analysis being as randomised, meant that the benefits of randomisation were preserved. The trial was pragmatic, in that the only difference between study groups was the availability of point-of-care panel assessment, and involved six diverse hospitals, so findings from the study can be generalised across the NHS. We measured a range of important outcomes and undertook cost-effectiveness analysis using data from real patients, thus avoiding having to make assumptions about how patients would be managed. The economic analysis used standard methods recommended by NICE41 to allow comparison of the cost-effectiveness of point-of-care panel assessment with other competing demands for NHS resources.
The study also had a number of limitations that should be taken into account when interpreting the findings. We obviously could not blind patients or carers to whether they had been randomised to intervention or control. In some respects this does not matter for a pragmatic trial where we are simply interested in whether an intervention works rather than how or why it works, but it may be important in interpreting patient-reported outcomes. Patients who are randomised to the standard-care group may feel that they have had a potentially valuable intervention withheld and may respond negatively when asked for their opinion. Response rates for the patient questionnaire were, as anticipated, around 70%, so there is the potential for responder bias. It is notable in this respect that response rates were lower in the standard-care group, perhaps reflecting a reduced level of engagement in those who did not receive the study intervention.
Point-of-care testing, and troponin assays in particular, is a rapidly developing area of technology. This created inevitable difficulties in selecting the appropriate machine and assay to use, and in providing guidance for the interpretation of results. We believe that our decision to change from the Biosite troponin assay to the more sensitive Stratus CS Analyser has been vindicated by recent publications7,8 that show the value of high-sensitivity troponin assays and suggest that these will become standard. However, working with a relatively new assay created problems with interpreting low positive results and led to our having to change the recommended protocol during the trial. These issues are not limited to the RATPAC trial and the introduction of highly sensitive troponin assays into practice is likely to be complicated by difficulties deciding how to manage patients with levels close to, or just below, the diagnostic threshold. Thus, it can be argued that the difficulties encountered in the trial in terms of machine malfunction and assay interpretation merely reflect the difficulties encountered in normal practice. However, an inevitable side effect of undertaking evaluation of a developing technology may be a degree of bias against the new technology, in that clinicians may take time to develop familiarity and the technology may ultimately perform better when it is in widespread use.
The trial was terminated before the target of 3130 patients was reached so it did not have the originally planned power. For the primary outcome this was compensated by the actual control admission rate being higher than the original estimate of the baseline admission rate. The trial was originally powered to detect a doubling of the major adverse event rate from 2% to 4%. In the event, at the time it was terminated it only had conditional power of < 10% to detect such a difference. It is therefore possible that point-of-care panel assessment may achieve increased successful discharges at the expense of an increase in major adverse events that the trial was not powered to detect. However, detailed examination of the adverse events suggests that there is unlikely to be a strong association between point-of-care panel assessment and adverse events. Most of the events occurred in patients who had been admitted after ED assessment. Of the five cases that occurred after discharge, only one occurred within the time span of a typical hospital admission. In general, the low adverse event rate in this study, especially deaths and non-fatal MI, should provide some reassurance that in this selected low-risk population the risk of adverse outcome shortly after attendance is very low.
The trial was deliberately designed as a pragmatic evaluation of a specific point-of-care protocol. The intervention was therefore a composite of the point-of-care technology and the cardiac marker panel. This type of pragmatic evaluation carries the disadvantage that we cannot clearly isolate which element of the intervention is effective (or ineffective). It is thus possible that the effects recorded in this study were due to the biomarker panel, rather than point-of-care technology, and could have been achieved using laboratory assays if turnaround times were sufficiently rapid.
The trial was undertaken at six diverse NHS hospitals and all participating centres contributed substantially to recruitment. The variation in the participating hospitals is likely to reflect variation in practice across the NHS. 11 This variation means that although we may conclude that out findings apply in general across the NHS it is possible that point-of-care panel assessment may be more or less effective or cost-effective in specific hospitals. The methods used for cost-effectiveness in particular are appropriate for NHS-wide decision-making but may not reflect the costs, effects and priorities of specific individual hospitals.
Imputation methods were used to estimate missing values on the basis of observed values. Recent methods favour the approach of generating multiple imputed data sets (multiple imputation). The main potential limitation of multiple imputation arises from the implicit assumption that the observed data are representative of those that are missing. In other words, these data must be missing at random. With regard to the imputation of staff costs using the microcosting study data, it is likely that this assumption holds, as the sample on which this study was conducted was chosen at random. In the case of the imputation of community health service costs, it may be that the assumption is more tenuous. For example, it may be that patients who are well are less likely to respond to the follow-up survey. Imputation may overestimate costs in these cases. On the other hand, severely ill patients may also have higher rates of non-response. In this case, imputation would probably underestimate costs. However, given the follow-up procedures of the RATPAC study, it is less likely that data from such patients were not recorded.
Other limitations of imputation methods include small numbers and a proportion of missing values higher than some percentage, typically set at 30%. In the case of the staff costs imputed from the microcosting data, this assumption is violated, as the missing data rate is about 90%. However, the method of imputation used in this case was one of choosing the value from the patient with complete data which matches closest that of the incomplete case, which is less prone to result in a biased estimate, although it may result in underestimation of the variability.
Implications for practice
The RATPAC trial has shown that point-of-care panel assessment with CK-MB (mass), myoglobin and troponin leads to increased successful discharge after ED assessment, but probably costs more than standard care, and is unlikely to be considered a cost-effective use of NHS resources. This means that from an NHS perspective it would not be appropriate to recommend widespread adoption of the point-of-care panel unless specific reasons can be identified as to why it might be worth paying more to reduce the proportion of patients requiring hospital admission. From the perspective of an individual hospital, the decision of whether or not to use the point-of-care panel will depend upon the specific needs of the ED and acute specialties, and whether it may be considered worthwhile to pay the potential additional costs of point-of-care panel assessment to achieve a specific service outcome, such as discharging more patients from the ED within the 4-hour time frame.
It is important to recognise that RATPAC evaluated a specific point-of-care combination for a specific purpose and that care should be taken not to extrapolate findings beyond this specific role. The RATPAC trial results suggest that a rule-out diagnostic testing protocol, which would be expected to lead to a substantial reduction in admissions, may, in practice, have only a modest effect and that a reduction in hospital admissions may not lead to health service cost savings. Although these findings may provide some useful insights when considering the wider role of point-of-care testing, they should not be extrapolated to all point-of-care tests in all settings.
Other findings from the RATPAC trial may have implications for practice and future developments in chest pain care. In particular, the very low rate of adverse events and the observation that most occur after the typical time span of hospital admission suggest that the value of hospital admission for these selected low-risk patients should be brought into question. Most of the investigations and interventions that may benefit patients can be delivered without hospital admission. If the purpose of hospital admission is simply to monitor patients thought to be at risk of adverse outcome then the data from this study suggest that admission for this purpose is unlikely to be worthwhile.
Implications for future research
The very low adverse event rates suggest that further research is required to evaluate the benefit of hospital admission and explore ways of managing acute chest pain that do not require hospital admission. RATPAC has shown that rapid diagnostic testing can make a modest impact upon hospital admissions. It may be that a substantial reduction in admissions can be achieved only through a fundamental restructuring of the way in which acute chest pain is assessed. This would involve moving from a ‘rule-out’ approach, where the aim is to identify low-risk patients who can be discharged home (with the rest being admitted), to a ‘rule-in’ approach, where the aim is to identify high-risk patients for hospital admission and treatment (with the rest being discharged home).
The RATPAC proposal recognised that this is a rapidly developing field and made provisions for future studies accordingly. We have stored blood samples from all participants in the point-of-care group and will be using these samples to test new and emerging assays. This should be coupled with economic evaluation and modelling to explore the potential impact of new early biomarkers with different levels of sensitivity and specificity. Development of biomarkers has tended to accept loss of specificity in the attempt to optimise sensitivity. However, poor specificity has potentially important economic consequences that need to be explored and better understood.
Analysis of the individual sites involved in the RATPAC trial showed marked variation in standard practice and in the effect of the intervention upon the primary outcome. This suggests that point-of-care panel assessment could be more effective or cost-effective in specific settings. Further research should be undertaken to assess in what circumstances point-of-care panel assessment could be effective and cost-effective.
Analysis of GRACE and TIMI scores
We undertook this analysis as a secondary objective of the study unrelated to the primary objectives of evaluating point-of-care panel assessment. The GRACE and TIMI scores have been developed and validated on patients with diagnosed ACS to predict adverse events, usually up to 1 month. Our analysis suggests that they have limited prognostic value in low-risk patients with undifferentiated chest pain, with TIMI performing no better than patient age as a predictor and GRACE performing only slightly better (and also being more complex to calculate). This is perhaps not surprising since both were developed in populations with diagnosed ACS and a high risk of major adverse events. Developing a new clinical risk score in the low-risk population with undifferentiated chest pain may produce an instrument with greater prognostic value. Alternatively, it could be argued that the risk of adverse outcome in this population is so low that attempts to discriminate between those at very low risk and those at moderately low risk are unlikely to be successful.
Chapter 5 Conclusions
Point-of-care panel assessment with CK-MB (mass), myoglobin and troponin increases the proportion of patients with acute undifferentiated chest pain who are successfully discharged home after ED assessment. This leads to a reduced proportion of patients being in hospital at any time over the next 3 months and a reduced median length of initial hospital stay, but no change in mean hospital stay or inpatient days. Point-of-care panel assessment is also associated with increased use of coronary care and may be associated with increased use of other interventions. It is unlikely to be considered cost-effective in the NHS, with a 0.888 probability that standard care is dominant. Cost-effectiveness is mainly driven by differences in mean cost, with point estimates suggesting that point-of-care panel assessment is £211 per patient more expensive than standard care.
Acknowledgements
We would like to thank Margaret Jane for her invaluable clerical assistance; David Gaze at St George’s Hospital, London, for his helpful advice and assistance with all things blood tube related; staff involved at the NHS sites for all their hard work; Francis Morris for advice on ED data; and Siemens for providing the Stratus CS Analyser machines and technical support and assistance.
Contributions of authors
Steve Goodacre (Professor of Emergency Medicine, Health Services Research, University of Sheffield) Chief Investigator, conception and design of the study, analysis and interpretation of the data, writing the report and final approval of the version to be published.
Liz Cross (Research Associate, Health Services Research, University of Sheffield) Project management, analysis and interpretation of the data, drafting and revising the report and final approval of the version to be published.
Mike Bradburn (Senior Medical Statistician, Clinical Trials Research Unit, University of Sheffield) Trial statistician, analysis and interpretation of the data, writing the report and final approval of the version to be published.
Patrick Fitzgerald (Research Fellow, Health Economics and Decision Science, University of Sheffield) Health economics, analysis and interpretation of the data, writing the report and final approval of the version to be published.
Alasdair Gray (Consultant in Emergency Medicine, Royal Infirmary of Edinburgh) Conception and design of the study, analysis and interpretation of the data, revising the report and final approval of the version to be published.
Alistair Hall (Professor of Clinical Cardiology, University of Leeds) Conception and design of the study, analysis and interpretation of the data, revising the report and final approval of the version to be published.
Paul Collinson (Consultant in Chemical Pathology, St George’s Hospital, London) Conception and design of the study, analysis and interpretation of the data, drafting and revising the report and final approval of the version to be published.
List of contributors
Project Management Group
Charlotte Arrowsmith (RATPAC Research Nurse, Derriford Hospital, Plymouth); Julian Barth (Consultant in Chemical Pathology, Leeds General Infirmary/Co-applicant); Jonathan Benger (Professor of Emergency Care, University of the West of England/Co-applicant); Mike Bradburn (Senior Medical Statistician, Clinical Trials Research Unit, University of Sheffield); Simon Capewell (Professor of Epidemiology, University of Liverpool/Co-applicant); Tim Chater (Database Manager, Clinical Trials Research Unit, University of Sheffield); Tim Coats (Professor of Emergency Medicine/Co-applicant); Paul Collinson (Consultant in Chemical Pathology, St George’s Hospital, London/Co-applicant); Cindy Cooper (Director, Clinical Trials Research Unit, University of Sheffield); Mandy Cooper (RATPAC Research Nurse, Leicester Royal Infirmary); Judy Coyle (RATPAC Research Nurse, Edinburgh Royal Infirmary); Liz Cross (Trial Manager, Health Services Research, University of Sheffield); Simon Dixon (Professor of Health Economics, Health Economics & Decision Science/Co-applicant); Patrick Fitzgerald (Research Fellow, Health Economics & Decision Science, University of Sheffield); Emma Gendall (RATPAC Research Nurse, Frenchay Hospital, Bristol); Steve Goodacre (Professor of Emergency Medicine, Health Services Research, University of Sheffield/Chief Investigator); Emma Goodwin (RATPAC Research Nurse, Barnsley Hospital); Alasdair Gray (Consultant in Emergency Medicine, Royal Infirmary of Edinburgh/Co-applicant); Alistair Hall (Professor of Clinical Cardiology, University of Leeds/Co-applicant); Kevin Hall (RATPAC Research Nurse, Barnsley Hospital); Taj Hassan (Consultant in Emergency Medicine, Leeds General Infirmary/Co-applicant); Julian Humphrey (Consultant in Emergency Medicine, Barnsley Hospital); Steven Julious (Senior Lecturer in Medical Statistics, Medical Statistics Group, Health Services Research, University of Sheffield/Co-applicant); Jason Kendall (Consultant in Emergency Medicine, Frenchay Hospital, Bristol); Vanessa Lawlor (RATPAC Research Nurse, Frenchay Hospital, Bristol); Sue Mackness (RATPAC Research Nurse, Leicester Royal Infirmary); Yvonne Meades (RATPAC Research Nurse, Leeds General Infirmary); David Newby (Professor of Cardiology, University of Edinburgh/Co-applicant); Dawn Newell (RATPAC Research Nurse, Leicester Royal Infirmary); Doris Quartey (RATPAC Research Nurse, Leeds General Infirmary); Karen Robinson (RATPAC Research Nurse, Leicester Royal Infirmary); Glen Sibbick (RATPAC Research Nurse, Leicester Royal Infirmary); Jason Smith (Consultant in Emergency Medicine, Derriford Hospital, Plymouth); and Roz Squire (RATPAC Research Nurse, Derriford Hospital, Plymouth).
Trial Steering Committee
Simon Carley, Consultant in Emergency Medicine, Manchester Royal Infirmary (Independent Member); Paul Collinson, Consultant in Chemical Pathology, St George’s Hospital, London (Co-applicant); Liz Cross, Research Associate, Health Services Research, University of Sheffield (Trial Manager); Sue Dodd, CHD Emergency & Acute Care Manager (Vascular Programme), Department of Health (Independent Member); Marcus Flather, Director Clinical Trials & Evaluation Unit, Royal Brompton Hospital, London (Chair); Steve Goodacre, Professor of Emergency Medicine, Health Services Research, University of Sheffield (Chief Investigator); Sara Hilditch, Statistician, Director of Statistical Services Unit, Sheffield University (Independent Member); Enid Hirst (Lay Representative); Richard Hudson, Quality Assurance Manager, Research Office, Academic Division, University of Sheffield (Sponsor Representative); Jason Kendall, Consultant in Emergency Medicine, Frenchay Hospital, Bristol (Investigator Representative); and Rebecca Whitlock-Moss, Programme Manager, NIHR HTA (Funder Representative).
Data Monitoring Committee
Mike Bradburn, Senior Medical Statistician, Clinical Trials Research Unit, University of Sheffield (Trial Statistician); Liz Cross, Research Associate, Health Services Research, University of Sheffield (Trial Manager); Steve Goodacre, Professor of Emergency Medicine, Health Services Research, University of Sheffield (Chief Investigator); John Greenwood, Senior Lecturer/Consultant Cardiologist, Academic Unit of Cardiovascular Medicine, Leeds General Infirmary (Independent Clinician); Jon Nicholl, Professor of Health Services Research, University of Sheffield (Acting Chair); Helen Thorpe, Principal Statistician, Clinical Trials Research Unit, University of Leeds (Chair and Independent Statistician); and William Townend, Consultant in Emergency Medicine, Hull Royal Infirmary (Independent Clinician).
Publications
Goodacre SW, Bradburn M, Cross E, Collinson PO, Gray A, Hall AS on behalf of the RATPAC research team. The RATPAC trial (Randomised Assessment of Treatment using Panel Assay of Cardiac markers): a randomised controlled trial of point-of-care cardiac markers in the emergency department. Heart 2011;97:190–6.
Bradburn M, Goodacre S, Fitzgerald P, Coats TJ, Gray AJ, Hassan TB, et al. Inter-hospital variation in the RATPAC trial (Randomised Assessment of Treatment using Panel Assay of Cardiac markers). Emerg Med J 2011. In press.
Fitzgerald P, Goodacre SW, Cross E, Dixon S. Cost-effectiveness of point-of-care biomarker assessment for suspected myocardial infarction: the RATPAC trial (Randomised Assessment of Treatment using Panel Assay of Cardiac markers). Acad Emerg Med 2011;18:488–95.
Goodacre SW, Bradburn M, Mohamed A, Gray A. Evaluation of Global Registry of Acute Cardiac Events and Thrombolysis in Myocardial Infarction scores in patients with suspected acute coronary syndrome. Am J Emerg Med 2011. In press. doi: 10.1016/j.ajem.2010.09.013.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Goodacre S, Cross E, Arnold J, Angelini K, Capewell S, Nicholl J. The health care burden of acute chest pain. Heart 2005;91:229-30.
- Department of Health (DoH) . The National Service Framework for Coronary Heart Disease 2000.
- Thygesen K, Alpert JS, White HD. Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2007;50:2173-95.
- Ebell MH, White LL, Weismantel D. A systematic review of troponin T and I values as a prognostic tool for patients with chest pain. J Fam Pract 2000;49:746-53.
- Bertrand ME, Simoons ML, Fox KAA, Wallentin LC, Hamm CW, McFadden E, et al. Task Force on the Management of Acute Coronary Syndromes of the European Society for Cardiology. Management of acute coronary syndromes in patients presenting without ST elevation. Eur Heart J 2002;23:1809-40.
- Goodacre S, Calvert N. Cost effectiveness of diagnostic strategies for patients with acute, undifferentiated chest pain. Emerg Med J 2003;20:429-33.
- Reichlin T, Hochholzer W, Bassetti S, Steuer S, Stelzig C, Hartwiger S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med 2009;361:858-67.
- Keller T, Zeller T, Peetz D, Tziwas S, Roth A, Czyz E, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med 2009;361:868-72.
- Collinson PO, Premachandram S, Hashemi K. Prospective audit of incidence of prognostically important myocardial damage in patients discharged from the emergency department. BMJ 2000;320:1702-5.
- Goodacre S, Nicholl J, Dixon S, Cross E, Angelini K, Arnold J, et al. Randomised controlled trial and economic evaluation of a chest pain observation unit compared with routine care. BMJ 2004;328:254-7.
- Goodacre S, Nicholl J, Beahan J, Quinney D, Capewell S. National survey of emergency department management of patients with acute, undifferentiated chest pain. Br J Cardiol 2003;10:50-4.
- Collinson PO, John C, Lynch S, Rao A, Canepa-Anson R, Carson E, et al. A prospective randomised controlled trial of point-of-care testing on the coronary care unit. Ann Clin Biochem 2004;41:397-404.
- Renaud B, Maison P, Ngako A, Cunin P, Santin A, Hervé J, et al. Impact of point-of-care testing in the emergency department evaluation and treatment of patients with suspected acute coronary syndromes. Acad Emerg Med 2008;15:216-24.
- Ryan RJ, Lindsell CJ, Hollander JE, O’Neil B, Jackson R, Schreiber D, et al. A multicenter randomized controlled trial comparing central laboratory and point-of-care cardiac marker testing strategies: the Disposition Impacted by Serial Point of Care Markers in Acute Coronary Syndromes (DISPO-ACS) trial. Ann Emerg Med 2009;53:321-8.
- Kendall J, Reeves B, Clancy M. Point of care testing: randomised controlled trial of clinical outcomes. BMJ 1998;316:1052-7.
- Ebell MH, Flewelling D, Flynn CA. A systematic review of troponin T and I for diagnosing acute myocardial infarction. J Fam Pract 2000;49:550-6.
- Balk EM, Ioannidis JPA, Salem D, Chew PW, Lau J. Accuracy of cardiac biomarkers to diagnose acute cardiac ischaemia in the emergency department: a meta-analysis. Ann Emerg Med 2001;37:478-94.
- Fesmire FM, Percy RF, Bardoner JB, Wharton DR, Calhoun FB. Serial creatinine kinase (CK) MB testing during the emergency department evaluation of chest pain: utility of a 2-hour deltaCK-MB of +1.6 ng/ml. Am Heart J 1998;136:237-44.
- Fesmire FM, Christensen RH, Focy EP, Feintuch TA. Delta creatinine kinase MB outperforms myoglobin at two hours during the emergency department identification and exclusion of troponin positive non-ST elevation acute coronary syndromes. Ann Emerg Med 2004;44:12-9.
- McCord J, Nowak RM, McCullough PA, Foreback C, Borzak S, Tokarski G, et al. Ninety-minute exclusion of acute myocardial infarction by use of quantitative point-of-care testing of myoglobin and troponin I. Circulation 2001;104:1483-8.
- Apple FS, Christensen RH, Valdes R, Andriak Aj, Berg A, Duh SH, . Simultaneous rapid measurement of whole blood myoglobin, creatinine kinase MB and cardiac troponin I by the triage cardiac panel for detection of myocardial infarction. Clin Chem 1999;45:199-205.
- Newby LK, Storrow AB, Gibler WB, Garvey JL, Tucker JF, Kaplan AL, et al. Bedside multimarker testing for risk stratification in chest pain units: the CHECKMATE study. Circulation 2001;103:1832-7.
- Caragher TE, Fernandez BB, Jacobs FL, Barr LA. Evaluation of quantitative cardiac biomarker point of care testing in the emergency department. J Emerg Med 2002;22:1-7.
- Ng SM, Krishnaswamy P, Morissey R, Clopton P, Fitzgerald R, Maisel AS, et al. Ninety-minute accelerated critical pathway for chest pain evaluation. Am J Cardiol 2001;88:611-17.
- Hamilton AJ, Swales LA, Neill J, Murphy JC, Darragh KM, Rocke LG, et al. Risk stratification of chest pain patients in the emergency department by a nurse utilizing a point of care protocol. Eur J Emerg Med 2008;15:9-15.
- Rathore S, Knowles P, Mann APS, Dodds PA. Is it safe to discharge patients from accident and emergency using a rapid point of care triple cardiac marker test to rule out acute coronary syndrome in low to intermediate risk patients presenting with chest pain?. Eur J Int Med 2008;19:537-40.
- Straface AL, Myers JH, Kirchick HJ, Blick KE. A rapid point-of-care cardiac marker testing strategy facilitates the rapid diagnosis and management of chest pain patients in the emergency department. Am J Clin Pathol 2008;129:788-95.
- Eggers KM, Oldgren J, Nordenskjold A, Lindahl B. Diagnostic value of serial measurement of cardiac markers in patients with chest pain: limited value of adding myoglobin to troponin I for exclusion of myocardial infarction. Am Heart J 2004;148:574-81.
- Antman EM, Cohen M, Berninck PJ, Horacek T, Papuchis G, Mautner B, et al. The TIMI risk score for unstable angina/non-ST elevation MI: a method for prognostication and therapeutic decision making. JAMA 2000;284:835-42.
- Eagle KA, Lim MJ, Dabbous OH, Pieper KS, Goldberg RJ, Van de Werf F, et al. A validated prediction model for all forms of acute coronary syndrome. JAMA 2004;291:2727-33.
- Burtonwood C, Halloran S. Report 06020: Three Point of Care Devices for Troponin Measurement 2006.
- Panteghini M, Pagani F, Yeo KT, Apple FS, Christenson RH, Dati F, et al. Evaluation of imprecision for cardiac troponin assays at low-range concentrations. Clin Chem 2004;50:327-32.
- Heeschen C, Goldmann BU, Moeller RH, Hamm CW. Analytical performance and clinical application of a new rapid bedside assay for the detection of serum cardiac troponin I. Clin Chem 1998;44:1925-30.
- Heeschen C, Goldmann BU, Langenbrink L, Matschuck G, Hamm CW. Evaluation of a rapid whole blood ELISA for quantification of troponin I in patients with acute chest pain. Clin Chem 1999;45:1789-96.
- Altinier S, Mion M, Cappelletti A, Zaninotto M, Plebani M. Rapid measurement of cardiac markers on Stratus CS. Clin Chem 2000;46:991-3.
- Goodacre S, Cross E, Lewis C, Nicholl J, Capewell S. on behalf of the ESCAPE Research Team . The ESCAPE cluster randomised trial: effectiveness and safety of chest pain assessment to prevent emergency admissions. BMJ 2007;335:659-62.
- Goodacre S, Mason S, Arnold J, Angelini K. Psychological morbidity and health-related quality of life of patients assessed on a chest pain observation unit. Ann Emerg Med 2001;38:369-76.
- Conway Morris A, Caesar D, Gray S, Gray A. The TIMI risk score accurately risk stratifies patients with undifferentiated chest pain presenting to an emergency department. Heart 2006;92:1333-4.
- Keating L, Benger J, Beetham R, Bateman S, Veysey S, Kendall J, et al. The PRIMA study: presentation IMA in the emergency department. Emerg Med J 2006;23:764-8.
- Ramsey S, Willke R, Briggs A, Brown R, Buxton M, Chawla A, et al. Good research practices for cost-effectiveness analysis alongside clinical trials: The ISPOR RCT-CEA Task Force Report. Value Health 2001;8:521-33.
- National Institute for Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2004.
- Netten A, Curtis L. Unit costs of health and social care 2004. Canterbury: University of Kent, Personal Social Services Research Unit; 2004.
- Department of Health (DoH) . NHS Reference Costs 2005 n.d. www.dh.gov.uk/PolicyAndGuidance/OrganisationPolicy/FinanceAndPlanning/NHSReferenceCosts/fs/en.
- Fenwick E, Claxton K, Sculpher M. Representing uncertainty: the role of cost effectiveness acceptability curves. Health Econ Lett 2001;10:779-87.
- Schafer J. Analysis of incomplete multivariate data. London: Chapman & Hall/CRC; 2000.
- Rubin D. Multiple imputation for nonresponse in surveys. New York, NY: J Wiley & Sons; 1987.
- Briggs A. Probabilistic analysis of cost-effectiveness models: statistical representation of parameter uncertainty. Value Health 2005;8:1-2.
- Oluboyede Y, Goodacre S, Wailoo AJ. on behalf of the ESCAPE Research Team . Cost-effectiveness of chest pain unit care in the NHS. BMC Health Serv Res 2008;8.
- Medical Research Council . Guidelines for Good Clinical Practice in Clinical Trials 1998. www.mrc.ac.uk/Utilities/Documentrecord/index.htm?d=MRC002416.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary 2008.
- Goodacre S, Morris F, Arnold J, Angelini K. Is a chest pain observation unit likely to be cost saving in a British hospital?. Emerg Med J 2001;18:11-4.
- Curtis L. PSSRU unit costs of health and social care. Canterbury: PSSU; 2008.
- Goodacre S, Dixon S. Is a chest pain observation unit likely to be cost effective at my hospital? Extrapolation of data from a randomised controlled trial. Emerg Med J 2005;22:418-22.
- Department of Health . NHS Reference Costs 2007–8 n.d. www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/documents/digitalasset/dh_098949.xls.
Appendix 1 Point-of-care protocol

Step 1
-
Collect an Investigation Pack from near RATPAC machine (Stratus CS Analyser).
-
Contains two green top tubes, two yellow top tubes, four small clear top tubes, two rotors, two cannulae, one Lab Form.
Note: the small tubes are for the labs, please do not remove from bag.
Step 2 – sample 1
-
Take point-of-care (PoC) blood samples (one yellow top and one green top) and routine bloods (normal tubes).
-
Label the yellow top sample with bar code and attach corresponding bar code label to Lab Form in pack.
-
Label green top sample with same bar code.
-
Scan bar coded green top tube in Stratus.
-
Enter patient study number (given by randomisation system) into Stratus and also add to Lab Form.
-
Please process green top sample in Stratus within 20 minutes.
-
Return yellow top sample and Lab Form to pack and store in fridge.
Step 3
-
Collect three reagent strips from fridge: troponin I (green), myoglobin (red), creatine kinase MB (CK-MB) (blue).
-
Invert green top tube gently several times (do not shake).
-
Process green top tube in Stratus as per instructions.
-
Results produced in 20 minutes.
-
After processing remove cannula unit and tube from the Stratus and discard appropriately.
Step 4 – interpretation of sample 1 results
-
Fix results slip in patient notes (important):
-
Troponin I ≥ 0.03 µg/l = positive result. Admit to hospital.
-
If no detectable troponin or < 0.03 µg/l = negative result. Finish assessing patient for 90 minutes then take sample 2.
-
Step 5 – sample 2
-
At least 90 minutes from first sample, take second blood samples (one yellow top and one green top).
-
Label as before with corresponding bar codes on tubes and Lab Form.
-
Return yellow top to pack and store in fridge.
-
Process green top as before in Stratus.
-
Results produced in 20 minutes – keep printout with notes again.
Step 6
-
Remove cannula unit and tube from the Stratus and discard appropriately.
-
Add corresponding small labels (without bar code) to two small tubes for each yellow top sample.
-
Send tubes in pack with Lab Form to Clinical Biochemistry Lab (within 12 hours).
Note: Ignore remaining labels.
Step 7 – interpretation of sample results
-
Calculate CK-MB (mass) gradient = second CK-MB (mass) – first CK-MB (mass), for example 4.52 – 3.11 = 1.41 (negative).
-
Calculate percentage increase in myoglobin (m) = 100 × [(second m – first m)/first m], for example 100 × [(40 – 25)/25] = 60% (positive).
-
If any of the following, admit to hospital (positive cardiac markers):
-
– troponin I ≥ 0.03 µg/l
-
– CK-MB (mass) > 5 µg/l on both samples
-
– CK-MB (mass) gradient on second sample 1.6 µg/l higher than first sample
-
– myoglobin on second sample 25% higher than first sample.
-
-
If cardiac markers negative, consider for discharge home.
-
Appendix 2 Futility analysis
-
Following the problems surrounding the inadvertent release of some RATPAC data to the HTA, Professor Tom Walley, Director of the HTA programme, asked me to act as an interim chair of RATPAC’s Data Monitoring and Ethics Committee (DMEC) in order to advise on whether the data supported the extension request.
-
I have met the RATPAC statisticians and reviewed the method that they used to calculate the conditional power of RATPAC. They used a method favoured by HTA, by calculating the power of achieving a statistically significant result given the current data and accruing new data up to the proposed sample size in accordance with the effect size specified in the alternative hypothesis in the protocol.
-
The current conditional power for the primary outcome (discharge home from the ED) recalculated using data to May 2009 is > 99.9%. The conditional power against the one secondary outcome that was originally powered in the protocol, major cardiac events, is < 10%. The current conditional power cannot be calculated for other secondary outcomes as no effect sizes were specified in alternative hypotheses in the protocol.
-
The conditional power calculation suggests that there could be a case for stopping for efficacy. There are no specific efficacy stopping criteria approved by the RATPAC DMEC in their charter, and this was appropriate because the primary outcome is not related to any direct clinical end point. Nevertheless, the absence of such criteria cannot be taken to mean that the trial should continue irrespective of any possible outcome, and the charter makes it clear that the DMEC can recommend stopping if the results are ‘convincing’ or futile.
-
In effect, the futility analysis suggests that the data must already be highly significant and that there is no chance that future data could change the result. This is not a situation that I had ever envisaged, but is, in fact, simply another type of futility.*
-
There are some other important considerations. First, the trial is being operated in several centres and the statisticians tell me that the results are not uniform across the centres, so that there may be important information on the variation in effect. Second, there are the other secondary outcomes, such as EQ-5D scores, satisfaction with care, re-attendance and readmission, which we know nothing about. Most importantly, the question of the cost-effectiveness of the intervention may not have been resolved, and this is the critical outcome for the NHS.
-
In summary, there are grounds for stopping the trial for futility. There is no chance that future data can change the result for the primary end point. However, as with all trials, RATPAC is also providing a great deal of other information on variability between centres, subgroups, and important secondary end points, whose current status we do not know about. The value of this information has to be weighed against the cost of the extension request.
Jon Nicholl
Acting as the interim chair of the RATPAC DMEC
12 May 2009
*Futility is usually understood to mean that there is little or no chance that future data could lead to a significant result. However, what we should mean by futility is that there is little or no chance future data could change the result. Thus, continuing a trial with a significant result that cannot be changed is as futile as continuing a trial with a non-significant result that cannot be changed. This is not the same as stopping for efficacy. Stopping for efficacy is based on a test of the results under H0, not on the conditional power of achieving a significant result under H1. It is possible to stop for efficacy, even though it would not be technically futile to continue (i.e. the result could change). It is also possible to set the stopping rules for efficacy so tight that the reverse could happen, i.e. it is technically futile to continue though the efficacy stopping rule has not been reached.
Appendix 3 Primary outcome details at each hospital
Supplemental tables
| PoC [n (%)] | SC [n (%)] | Total [n (%)] | |
|---|---|---|---|
| Successfully discharged | 110 (68) | 43 (26) | 153 (47) |
| Not successfully discharged | 52 (32) | 121 (74) | 173 (53) |
| Reason for no successful discharge | |||
| In hospital 4 hours after arrival and no decision has been made to discharge | 50 (31) | 121 (74) | 171 (52) |
| Initially discharged but re-attended with major adverse event | 2 (1) | 0 | 2 (1) |
| Discharge success by initial status | |||
| Initially discharged | 112 (69) | 43 (26) | 155 (48) |
| Not in hospital at 4 hours | 105 (65) | 43 (26) | 148 (45) |
| In hospital at 4 hours, decision made to discharge | 7 (4) | 0 | 7 (2) |
| PoC [n (%)] | SC [n (%)] | Total [n (%)] | |
|---|---|---|---|
| Successfully discharged | 43 (26) | 21 (13) | 64 (20) |
| No successfully discharged | 121 (74) | 143 (87) | 264 (80) |
| Reason for no successful discharge | |||
| In hospital 4 hours after arrival and no decision has been made to discharge | 121 (74) | 142 (87) | 263 (80) |
| Initially discharged but re-attended with major adverse event | 0 | 1 (1) | 1 (< 1) |
| Discharge success by initial status | |||
| Initially discharged | 43 (26) | 22 (13) | 65 (20) |
| Not in hospital at 4 hours | 29 (18) | 18 (11) | 47 (14) |
| In hospital at 4 hours, decision made to discharge | 14 (9) | 4 (2) | 18 (5) |
| PoC [n (%)] | SC [n (%)] | Total [n (%)] | |
|---|---|---|---|
| Successfully discharged | 104 (46) | 16 (7) | 120 (27) |
| Not successfully discharged | 124 (54) | 208 (93) | 332 (73) |
| Reason for no successful discharge | |||
| In hospital 4 hours after arrival and no decision has been made to discharge | 123 (54) | 208 (93) | 331 (73) |
| Initially discharged but re-attended with major adverse event | 1 (< 1) | 0 | 1 (< 1) |
| Discharge success by initial status | |||
| Initially discharged | 105 (46) | 16 (7) | 121 (27) |
| Not in hospital at 4 hours | 90 (39) | 16 (7) | 106 (23) |
| In hospital at 4 hours, decision made to discharge | 15 (7) | 0 | 15 (3) |
| PoC [n (%)] | SC [n (%)] | Total [n (%)] | |
|---|---|---|---|
| Successfully discharged | 50 (21) | 9 (4) | 59 (13) |
| Not successfully discharged | 183 (79) | 222 (96) | 405 (87) |
| Reason for no successful discharge | |||
| In hospital 4 hours after arrival and no decision has been made to discharge | 182 (78) | 222 (96) | 404 (87) |
| Initially discharged but re-attended with major adverse event | 1 (< 1) | 0 | 1 (< 1) |
| Discharge success by initial status | |||
| Initially discharged | 51 (22) | 9 (4) | 60 (13) |
| Not in hospital at 4 hours | 44 (19) | 5 (2) | 49 (11) |
| In hospital at 4 hours, decision made to discharge | 7 (3) | 4 (2) | 11 (2) |
| PoC [n (%)] | SC [n (%)] | Total [n (%)] | |
|---|---|---|---|
| Successfully discharged | 1 (1) | 8 (5) | 9 (3) |
| Not successfully discharged | 172 (99) | 163 (95) | 335 (97) |
| Reason for no successful discharge | |||
| In hospital 4 hours after arrival and no decision has been made to discharge | 172 (99) | 163 (95) | 335 (97) |
| Initially discharged but re-attended with major adverse event | 0 | 0 | 0 |
| Discharge success by initial status | |||
| Initially discharged | 1 (1) | 8 (5) | 9 (3) |
| Not in hospital at 4 hours | 1 (1) | 8 (5) | 9 (3) |
| In hospital at 4 hours, decision made to discharge | 0 | 0 | 0 |
| PoC [n (%)] | SC [n (%)] | Total [n (%)] | |
|---|---|---|---|
| Successfully discharged | 50 (30) | 49 (30) | 99 (30) |
| Not successfully discharged | 115 (70) | 115 (70) | 230 (70) |
| Reason for no successful discharge | |||
| In hospital 4 hours after arrival and no decision has been made to discharge | 115 (70) | 115 (70) | 230 (70) |
| Initially discharged but re-attended with major adverse event | 0 | 0 | 0 |
| Discharge success by initial status | |||
| Initially discharged | 50 (30) | 49 (30) | 99 (30) |
| Not in hospital at 4 hours | 50 (30) | 44 (27) | 94 (29) |
| In hospital at 4 hours, decision made to discharge | 0 | 5 (3) | 5 (2) |
Appendix 4 Health economic parameter values used directly in model
| Item | Deterministic value | Distributions for probabilistic sensitivity analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Initial type | Parameter 1 | Parameter 2 | Transformed (if required) | Parameter 1 | Parameter 2 | No. In RATPAC data | ||
| Alpha | Beta | |||||||
| Overall prevalence of ACS | 11.8% | Beta | 264 | 1979 | 2243 | |||
| Probabilities | ||||||||
| Point-of-care arm | ||||||||
| 12.3% | Beta | 138 | 987 | 1125 | ||||
| Frequency of PoC two-panel test | 78.8% | Beta | 886 | 239 | 1125 | |||
| Test outcome positive given ACS (sensitivity) | 74.6% | Beta | 103 | 35 | 138 | |||
| Test outcome negative given no ACS (specificity) | 98.7% | Beta | 974 | 13 | 987 | |||
| Admission positive test | 100.0% | Fixed | 116 | |||||
| Admission negative test | 0.0% | Fixed | 1009 | |||||
| Intervention (given admission) | Alphas | |||||||
| Thrombolysis | 0.9% | Dirichlet | 1 | 116 | ||||
| PCI | 16.4% | 19 | 116 | |||||
| CABG | 8.6% | 10 | 116 | |||||
| 2 × PCI | 2.6% | 3 | 116 | |||||
| PCI + CABG | 0.9% | 1 | 116 | |||||
| Thrombolysis + PCI + CABG | 0.9% | 1 | 116 | |||||
| No intervention | 69.8% | 81 | 116 | |||||
| Death after admission (assumes no intervention) | 0.9% | Beta | 1 | 115 | 116 | |||
| Discharge | 99.1% | Complement | 116 | |||||
| Subsequent diagnosis given negative test | 3.5% | Beta | 35 | 974 | 1009 | |||
| Subsequent intervention (admitted) after discharge | ||||||||
| PCI | 31.4% | Dirichlet | 11 | 35 | ||||
| PCI + CABG | 2.9% | 1 | 35 | |||||
| No intervention | 65.7% | 23 | 35 | |||||
| Standard care arm | ||||||||
| Prevalence of ACS | 11.3% | Beta | 126 | 992 | 1118 | |||
| Test outcome positive given ACS (sensitivity) | 82.5% | Beta | 104 | 22 | 126 | |||
| Test outcome negative given no ACS (specificity) | 99.6% | Beta | 988 | 4 | 992 | |||
| Admission given positive test | 100% | Fixed | 108 | |||||
| Admission given negative test | 0.0% | Fixed | 1010 | |||||
| Intervention (given admission) | ||||||||
| Thrombolysis | 0.9% | Dirichlet | 1 | 108 | ||||
| PCI | 22.2% | 24 | 108 | |||||
| CABG | 3.7% | 4 | 108 | |||||
| 2 × PCI | 0.9% | 1 | 108 | |||||
| PCI + CABG | 0.9% | 1 | 108 | |||||
| No intervention | 71.3% | 77 | 108 | |||||
| Death after admission (assumes no intervention) | 0.9% | Beta | 1 | 107 | 108 | |||
| Discharge | 99.1% | Complement | 108 | |||||
| Subsequent diagnosis given negative test | 2.2% | Beta | 22 | 987 | 1009 | |||
| Subsequent intervention (admitted) after discharge | ||||||||
| PCI | 22.7% | Dirichlet | 5 | 17 | 22 | |||
| CABG | 4.5% | 1 | 21 | 22 | ||||
| No intervention | 72.7% | 16 | 6 | 22 | ||||
| Costs | ||||||||
| Point-of-care arma | ||||||||
| PoC test per panel costb | £21.33 | Fixed | ||||||
| ED costsa | ||||||||
| Mean | SD | Mean | Variance | |||||
| Staffc | £60.76 | Log normal | 60.76 | 34.15 | Normal | 3.970 | 0.275 | 122 |
| Overheads | £19.32 | Log normal | 19.32 | 6.06 | Normal | 2.914 | 0.094 | 1125 |
| Cardiac marker tests | £28.58 | Log normal | 28.58 | 16.35 | Normal | 3.211 | 0.283 | 1125 |
| Medications | £5.56 | Log normal | 5.56 | 22.70 | Normal | 0.280 | 2.872 | 1125 |
| Angiograms | £1.85 | Log normal | 1.85 | 15.39 | Normal | –1.513 | 4.254 | 1125 |
| ECGs | £33.44 | Log normal | 33.44 | 11.61 | Normal | 3.453 | 0.114 | 1125 |
| Stay (if not admitted) | £21.62 | Log normal | 21.62 | 21.81 | Normal | 2.723 | 0.702 | 1125 |
| Hospital stay (admitted from first ED presentation) | ||||||||
| PCI | £3058.46 | Log normal | 3058.46 | 2216.03 | Normal | 7.815 | 0.422 | 19 |
| CABG | £7155.76 | Log normal | 7155.76 | 4003.15 | Normal | 8.740 | 0.272 | 10 |
| 2 × PCI | £2960.83 | Log normal | 2960.83 | 1587.27 | Normal | 7.867 | 0.253 | 3 |
| PCI + CABG | £5247.99 | Fixed | 5247.99 | 1 | ||||
| Thrombolysis + PCI + CABG | £4452.18 | Fixed | 4452.18 | 1 | ||||
| No intervention | £2036.35 | Log normal | 2036.35 | 3013.93 | Normal | 7.039 | 1.160 | 81 |
| Hospital stay (after discharge from ED) | ||||||||
| Thrombolysis | £0.00 | Fixed | ||||||
| PCI | £1386.02 | Log normal | 1386.02 | 1831.44 | Normal | 6.729 | 1.010 | 11 |
| PCI + CABG | £2762.10 | Fixed | 2762.10 | 1 | ||||
| No intervention | £1648.11 | Log normal | 1648.11 | 1595.36 | Normal | 7.077 | 0.661 | 23 |
| Hospital stay if no ACS diagnosed | £201.76 | Log normal | 201.76 | 728.86 | Normal | 3.986 | 2.643 | 974 |
| Standard care arm | ||||||||
| ED costs | ||||||||
| Staffc | £49.21 | Log normal | 49.21 | 22.54 | Normal | 3.801 | 0.190 | 124 |
| Overhead costs | £17.09 | Log normal | 17.09 | 6.82 | Normal | 2.764 | 0.148 | 1118 |
| Cardiac marker tests | £38.00 | Log normal | 38.00 | 14.98 | Normal | 3.565 | 0.144 | 1118 |
| Medications | £5.21 | Log normal | 5.21 | 20.91 | Normal | 0.230 | 2.840 | 1118 |
| Angiograms | £0.93 | Log normal | 0.93 | 10.96 | Normal | –2.543 | 4.941 | 1118 |
| ECGs | £35.39 | Log normal | 35.39 | 12.52 | Normal | 3.507 | 0.118 | 1118 |
| Stay (if not admitted) | £13.86 | Log normal | 13.86 | 18.58 | Normal | 2.115 | 1.028 | 1118 |
| Hospital stay (admitted from first ED presentation) | ||||||||
| Thrombolysis | £1381.05 | Fixed | 1381.05 | 1 | ||||
| PCI | £2320.10 | Log normal | 2320.10 | 1973.05 | Normal | 7.477 | 0.544 | 24 |
| CABG | £6449.23 | Log normal | 6449.23 | 2233.36 | Normal | 8.715 | 0.113 | 4 |
| 2 × PCI | £8573.45 | Fixed | 8573.45 | 1 | ||||
| Thrombolysis + PCI | £4728.39 | Fixed | 4728.39 | 1 | ||||
| No intervention | £1875.44 | Log normal | 1875.44 | 2311.45 | Normal | 7.075 | 0.924 | 77 |
| Hospital stay (after discharge from ED) | ||||||||
| PCI | £939.11 | Log normal | 939.11 | 693.28 | Normal | 6.627 | 0.435 | 5 |
| CABG | £6683.74 | Fixed | 6683.74 | 1 | ||||
| No intervention | £1398.31 | Log normal | 1398.31 | 1373.55 | Normal | 6.905 | 0.675 | 16 |
| Hospital stay if no ACS diagnosed | £229.95 | Log normal | 229.95 | 661.58 | Normal | 4.324 | 2.228 | 988 |
| Either arm | ||||||||
| Hospital diagnostic test costs | £50.35 | Log normal | 50.35 | 89.34 | Normal | 3.208 | 1.423 | 2243 |
| Further ED attendances with no admission | £14.06 | Log normal | 14.06 | 54.19 | Normal | 1.261 | 2.764 | 2243 |
| Outpatient clinic visits | £31.44 | Log normal | 31.44 | 88.76 | Normal | 2.351 | 2.194 | 2243 |
| Planned follow-up care | ||||||||
| GP surgery visits | £152.66 | Log normal | 152.66 | 137.14 | Normal | 4.732 | 0.592 | 1292 |
| GP home visits | £11.03 | Log normal | 11.03 | 48.61 | Normal | 0.893 | 3.016 | 1088 |
| Nurse home visits | £7.80 | Log normal | 7.80 | 47.18 | Normal | 0.240 | 3.627 | 1083 |
| Social worker home visits | £5.52 | Log normal | 5.52 | 62.97 | Normal | –0.731 | 4.877 | 1078 |
| Intervention | ||||||||
| CABG54 | £6806.86 | Fixed | ||||||
| PCI54 | £2012.97 | Fixed | ||||||
| Thrombolysis (medication only)50 | £780.00 | Fixed | ||||||
| Mean | SD | |||||||
| Long-term care cost of treatmente | £10,748 | 2005–6 cost | £10,079 | £2200 | Normal | £10,748 | £2346 | |
| (Inflated to 2007–8 values)52 | ||||||||
| Health-related quality of life | ||||||||
| Point-of-care arm | ||||||||
| QALYs (from admission after first ED presentation) | ||||||||
| Thrombolysis | 0.107 | Fixed | 0.107 | 1 | ||||
| PCI | 0.154 | Normal | 0.154 | 0.038 | 19 | |||
| CABG | 0.157 | Normal | 0.157 | 0.047 | 10 | |||
| 2 × PCI | 0.169 | Normal | 0.169 | 0.034 | 3 | |||
| PCI + CABG | 0.096 | Fixed | 0.096 | 1 | ||||
| Thrombolysis + PCI + CABG | 0.208 | Fixed | 0.208 | 1 | ||||
| No intervention | 0.154 | Normal | 0.154 | 0.044 | 81 | |||
| QALYs (after discharge from ED) | ||||||||
| PCI | 0.153 | Normal | 0.153 | 0.014 | 11 | |||
| PCI + CABG | 0.169 | Fixed | 0.169 | 1 | ||||
| No intervention (if admitted) | 0.139 | Normal | 0.139 | 0.053 | 23 | |||
| If no ACS diagnosed | 0.156 | Normal | 0.156 | 0.047 | 974 | |||
| Standard-care arm | ||||||||
| QALYs (from admission after first ED presentation) | ||||||||
| Thrombolysis | 0.208 | Fixed | 0.208 | 1 | ||||
| PCI | 0.160 | Normal | 0.160 | 0.040 | 24 | |||
| CABG | 0.139 | Normal | 0.139 | 0.029 | 4 | |||
| 2 × PCI | 0.073 | Fixed | 0.073 | 1 | ||||
| Thrombolysis + PCI | 0.208 | Fixed | 0.208 | 1 | ||||
| No intervention | 0.155 | Normal | 0.155 | 0.039 | 77 | |||
| QALYs (after discharge from ED) | ||||||||
| PCI | 0.160 | Normal | 0.160 | 0.026 | 5 | |||
| PCI + CABG | 0.158 | Fixed | 0.158 | 1 | ||||
| No intervention (if admitted) | 0.148 | Normal | 0.148 | 0.044 | 16 | |||
| If no ACS diagnosed | 0.158 | Normal | 0.158 | 0.042 | 988 | |||
| Other | ||||||||
| Utility – death | 0 | |||||||
| QALYs for long-term care with CHDd | 6.829 | Normal | 6.829 | 0.340 | ||||
| Lifetime accumulated QALYs for a UK person 55 years old in 2008 (30.3 years for males, 33.3 years for females)e | 20 | Normal | 20 | 5 | ||||
Appendix 5 The RATPAC trial protocol
Project title: The RATPAC trial (HTA 06/302/19) (Randomised Assessment of Treatment using Panel Assay of Cardiac markers)
A randomised controlled trial (RCT) of point-of-care cardiac markers in the emergency department.
Planned investigation
Research objectives
We will evaluate the clinical effectiveness and cost-effectiveness of the most promising point-of-care cardiac marker panel currently used in the emergency department (ED).
In patients presenting to the ED with suspected but not proven acute myocardial infarction (AMI)*, we will measure the effect of using a point-of-care cardiac marker panel upon:
-
the proportion of patients successfully discharged home after ED assessment
-
health utility and satisfaction with care
-
the use of coronary care beds and cardiac treatments
-
subsequent re-attendance at and/or re-admission to hospital
-
major adverse events (death, non-fatal AMI, life-threatening arrhythmia, emergency revascularisation or hospitalisation for myocardial ischaemia)
-
health and social care costs.
We also plan to use trial data and blood samples to evaluate:
-
clinical prediction rules, such as the TIMI (thrombolysis in myocardial infarction) and GRACE (Global Registry of Acute Coronary Events) scores
-
potential new or alternative markers, such as ischaemia-modified albumin, ultrasensitive cardiac troponin, B-type natriuretic peptide, myeloperoxidase and fatty acid binding protein.
*Throughout this proposal, we define AMI according to European Society of Cardiology/American College of Cardiology (ESC/ACC) criteria for acute, evolving or recent AMI. 1 According to this definition, a troponin level above the 99th percentile of the values for a reference control group is considered positive, and in the context of a patient with ischaemic symptoms (i.e. chest pain) would satisfy the diagnosis for AMI. This definition identifies patients who are most likely to benefit from treatments that usually require hospital admission. Hence, it provides a pragmatic definition of a patient group whose suspected condition requires hospital admission. Setting a higher threshold for positivity would risk excluding from the definition patients who might benefit from hospital admission, whereas broadening the definition to include cases of troponin-negative acute coronary syndrome (ACS) would involve relying upon subjective (clinician determined) definitions of ACS and would define many patients who would not benefit from admission to hospital as having ACS.
Existing research
Chest pain due to suspected but not proven AMI is responsible for a substantial number of ED attendances and emergency hospital admissions in the UK NHS. 2 Current recommendations suggest that these patients should receive diagnostic testing, with a troponin sample taken 12 hours after their symptom onset,3,4 the delay being necessary because troponin sensitivity does not reach optimal levels until this time. This approach is inconvenient and potentially costly because it requires many patients to be unnecessarily admitted to hospital until the time delay has elapsed. Most patients with suspected AMI do not actually have AMI, so their admission will ultimately prove avoidable. Cost-effectiveness analysis suggests that admitting patients for cardiac marker testing is not a cost-effective use of health service resources. 5
Evidence also suggests that these guidelines are often not followed in a busy emergency setting in which acute beds are limited. Collinson et al. 7 showed that 7% of patients discharged after ED assessment for acute chest pain had elevated troponin levels at follow-up 2 days later. Goodacre et al. 8 showed that, in the routine care arm of a randomised trial of a chest pain unit, 14% of patients with an elevated troponin level at 2-day follow-up had been sent home from the ED. Our recent national survey of EDs9 asked the lead consultant what proportion of patients with undifferentiated chest pain would be admitted to hospital. Estimates varied from < 20% to > 80%. Hence, it appears that the theoretical ideal of a 12-hour troponin is not realised in practice and, as a result, patients are inadvertently discharged home with AMI.
Rapid point-of-care testing using a panel of markers offers an alternative approach that may be more effective and cost-effective than current practice. 10 A combination of markers is measured on arrival and a short time later (usually 90 minutes). The gradient of these markers (the difference between the presentation and 90-minute levels) has been shown to provide improved early sensitivity (95%) without unacceptably compromising specificity. 11,12 A typical panel will use a combination of early markers, such as myoglobin or creatinine kinase (CK-MB) (mass), and a more definitive marker, such as troponin I or T. Because the point-of-care tests can be used quickly in the ED, they can potentially rule out AMI during ED assessment, thus avoiding hospital admission and the pressure to select only high-risk patients for further diagnostic assessment.
Meta-analyses have estimated the diagnostic accuracy of individual cardiac markers,13,14 but there have been no systematic reviews of point-of-care cardiac panels. 15 Our literature review found that studies of the diagnostic accuracy of point-of-care markers have focussed upon a panel using CK-MB (mass), myoglobin and troponin I measured at presentation and 90 minutes later. These studies have shown that the panel has high sensitivity (over 95%) and can accurately rule out AMI by 90 minutes after presentation. 16–20 This results in earlier identification of AMI than laboratory testing18 and expedited decision-making with turnaround times reduced by 55%. 19 Meanwhile, comparison of patient management with the panel with previous practice showed a 40% reduction in coronary care unit admissions. 20
These studies show that the point–of-care combination of CK-MB (mass), myoglobin and troponin I measured at presentation and 90 minutes has appropriate diagnostic accuracy, but they do not reliably tell us whether the panel will alter patient care, improve outcomes or reduce health service costs. Early diagnostic accuracy and reduced turnaround times will only lead to changes in practice if clinicians act upon the additional diagnostic information. Although interesting, the before and after study by Ng et al. 20 may be confounded by changes in coronary care referrals over time and, originating from the USA, where coronary care usage is much higher than in the UK, may not be applicable to the UK NHS. Audit data from the UK suggest that point-of-care cardiac testing can reduce hospital admissions, but this finding is based on before/after audit that has yet to be published in a peer-reviewed journal. 21
Randomised trials of point-of-care testing are few in number and report conflicting results. The only randomised trial specifically of cardiac tests, by Collinson et al. ,22 showed that point-of-care measurement of troponin T in patients admitted to a coronary care unit reduced overall length of hospital stay. By comparison, Kendall et al. 23 showed that use of a variety of point-of-care tests for a heterogeneous group of patients in the ED produced shorter decision times, but did not reduce overall length of stay in the department. There are no published randomised trials evaluating the clinical impact of point-of-care cardiac markers in diagnostic assessment of acute chest pain.
We have searched the National Research Register and ClinicalTrials.gov for research in progress into point-of-care cardiac markers and have identified one relevant study. 24 This is a randomised trial being undertaken in the USA to compare point-of-care troponin I testing with laboratory testing in acute coronary syndrome to determine whether bedside use leads to shorter decision times in emergency care. It will therefore provide useful data for North American decision-makers to determine whether replacing laboratory with point-of-care troponin testing leads to more efficient patient processing. However, it will not determine whether a rapid rule-out point-of-care strategy is more effective or cost-effective than routine care, particularly in the UK.
Research methods
We will undertake a pragmatic randomised controlled trial and economic evaluation of a point-of-care cardiac marker panel in the management of patients with suspected, but not proven, AMI in six EDs in the UK.
Emergency department staff will identify eligible patients, provide trial information and obtain written consent. Participants will then be randomly allocated to receive either (1) diagnostic assessment using the point-of-care biochemical marker panel or (2) conventional diagnostic assessment without the panel.
The Sheffield Clinical Trials Research Unit (CTRU) will generate a simple randomisation sequence, stratified by centre, which will not be revealed to any person involved in patient recruitment. ED staff will telephone the CTRU randomisation service when they recruit a participant and will provide full participant details to the CTRU. The CTRU will reveal the participant’s allocated treatment group to the ED only after the participant’s details have been recorded, written consent has been confirmed and the participant irrevocably entered into the trial.
This is a pragmatic trial that is intended to determine whether point-of-care testing should be standard practice for patients presenting to the ED with suspected AMI. It is designed to compare two pragmatic alternatives (management with and without point-of-care testing) under routine conditions to determine whether use of the test changes costs or outcomes. This pragmatic design has the following implications:
-
After randomisation we will not attempt to blind clinical staff, patients or carers to the allocated treatment group.
-
Although the point-of-care test will be provided with a recommended protocol for use, management decisions will ultimately be at the discretion of the clinical staff.
-
All other diagnostic tests and the use of laboratory blood tests in the control group will be at the discretion of the clinical staff.
-
Blood samples will only be taken for the purposes of clinical management. We will not take additional blood samples to evaluate theoretical management strategies or to evaluate the accuracy of diagnostic assessments. We will not take additional samples to evaluate new markers (as set out in the secondary objectives) but will use residual blood from point-of-care tests.
Justification for choice of research methods
During the development of this proposal, we considered two other alternative methods of evaluation:
-
systematic review and modelling
-
cluster randomised trial.
We propose a pragmatic trial, as opposed to systematic review and modelling, because key pieces of information that are central to the estimation of cost-effectiveness are not yet available. First, as outlined above, while there are abundant data available to estimate the diagnostic accuracy of the constituent point-of-care panel tests, only limited data are available to estimate the diagnostic performance of the overall panel, with no studies based in the UK. This is potentially important as differences in patient characteristics and presentation patterns are likely to have an impact on sensitivity and specificity.
Second, even if these data were used, the behavioural consequences of the test results are unknown: which patients will and will not be admitted, how long will they be admitted for? Likewise, as identified by our previous work in this area,5,8 it is very difficult to determine how patients receiving point-of-care testing would have been managed if the point-of-care test were not available. Assuming that all patients would have been admitted to hospital for laboratory troponin testing at 12 hours after symptom onset is inappropriate and would overestimate the comparative cost-effectiveness of point-of-care testing.
Finally, if we were to model admission rates as a function of sensitivity/specificity using our previous work, and then interpolate the sensitivity/specificity estimates for point-of-care panels, we would have to make cavalier assumptions about the form that the relationship takes due to the paucity of data points.
Taken together, we firmly believe that there would currently be excessive uncertainty around key parameters in any cost-effectiveness model. However, based on the results of this study and others that may be published in the meantime, we feel that sufficient evidence will be available for different panels to be evaluated using the model developed as part of this proposal. The need for any further research will also be evaluated using a value of information analysis based around this model. 25
We propose to randomise individual patients, rather than using cluster randomised methods, because the advantages of cluster randomised methods, of reducing the risk of contamination or non-compliance in the control group, are outweighed by the disadvantages of selection bias due to loss of allocation concealment and loss of statistical power.
Cluster methods based upon randomising periods of time, such as days of the week, would not significantly reduce the risks of contamination, so we would have to randomise large clusters, such as members of staff or whole hospitals. This would involve substantial loss of statistical power. More importantly, there would be a substantial risk of selection bias because recruiting staff would be aware of whether patients would be allocated to point-of-care testing or not and might apply exclusion criteria in a differential manner, depending upon whether they wanted to use point-of-care testing or not. This could result in patients being recruited to the point-of-care arm of the trial if they were considered appropriate for point-of-care testing, and recruited to the control arm if they were considered appropriate for routine care. This would represent a substantial flaw.
Individual patient randomisation allows us to achieve allocation concealment and avoid the risk of selection bias. Although it carries the risk of contamination and non-compliance in the control group we can explore for evidence of contamination by examining changes in control group practice and admission rates over time. We will minimise the risk of non-compliance in the control group by limiting the availability of point-of-care testing to consecutively numbered test ‘strips’ that are used only in recruited intervention group patients, and are recorded and accounted for at the end of the trial.
Planned interventions
Participants will be randomised to receive either:
-
diagnostic assessment using the point-of-care biochemical marker panel
or
-
conventional diagnostic assessment without the panel.
The only difference between the two arms of the trial will be that patients in the intervention arm will receive testing with the point-of-care panel. The use of all other tests and treatments, and decision-making in the ED, will be at the discretion of the attending clinician.
The point-of-care cardiac marker panel will comprise CK-MB (mass), myoglobin and troponin I, measured at presentation and 90 minutes later, using the Stratus CS point-of-care analyser. As outlined above, this combination has been widely evaluated in practice. 16–20 Of the systems currently available, or soon to be available, the latest version of the Dade Behring Stratus CS Analyser has the most data as an instrument suitable both for the emergency laboratory and for use as a point-of-care testing (POCT) instrument. 26
Clinical staff will be trained to use the test and give guidance in interpretation of the results. We will provide a recommended protocol that will advise a first panel test immediately after initial ED assessment and a second panel test 90 minutes later. Other than obtaining consent, collecting data, and random allocation to use of the point-of-care test, the only change to routine practice will be that we will ask clinical staff to take an additional quantity of blood for storage (without repeating venepuncture) each time a blood sample is required.
The additional blood remaining after POCT has been performed will be transported to the hospital laboratory, where it will be centrifuged and refrigerated. Batches of samples will be transported quarterly to St George’s Hospital for analysis to address the secondary objectives of the study.
Planned inclusion/exclusion criteria
We will recruit people presenting to the ED with chest pain due to suspected but not proven AMI in whom a negative point-of-care marker test could potentially rule out AMI and allow discharge home.
We will exclude the following
-
Patients with diagnostic ECG changes for AMI or high-risk acute coronary syndrome (> 1 mm ST deviation or > 3 mm inverted T waves). These patients are at high risk of adverse outcome and require inpatient care, even if marker tests are negative.
-
Patients with known coronary heart disease (CHD) presenting with prolonged (> 1 hour) or recurrent episodes of typical cardiac-type pain. These patients have unstable angina and require inpatient care for symptom control even if marker tests are negative.
-
Patients with proven or suspected serious non-coronary pathology (e.g. pulmonary embolus) that requires inpatient care, even if AMI is ruled out.
-
Patients with comorbidity or social problems that require hospital admission even if AMI can be ruled out.
-
Patients with an obvious non-cardiac cause (e.g. pneumothorax or muscular pain), in whom AMI can be excluded as a possible cause without resorting to further diagnostic testing.
-
Patients presenting more than 12 hours after their most significant episode of pain, for whom a single troponin measurement would clearly be more appropriate than point-of-care panel testing.
-
Previous participants in the RATPAC trial.
-
Patients who are unable to understand the trial information due to cognitive impairment.
-
Non-English speaking patients for whom translation facilities are not available.
The research nurse at each hospital will regularly check ED attendance lists to identify patients attending with chest pain and record basic demographic details and reason for exclusion, thus allowing completion of a CONSORT flow chart.
Proposed outcome measures
The primary outcome will be the proportion of patients successfully discharged home after ED assessment, defined as discharge with no adverse event (as defined below) during the following 3 months.
Secondary outcomes will include:
-
Health utility measured using the European Quality of Life-5 Dimensions (EQ-5D) self-complete questionnaire at 1 and 3 months after attendance.
-
Satisfaction with care measured at 1 month after attendance using a modified Group Health Association of America questionnaire that has been used successfully in previous studies of diagnostic strategies for acute chest pain.
-
The proportion of patients managed on the coronary care unit, receiving cardiac medications (such as heparin, clopidogrel or glycoprotein IIb/IIIa inhibitors) or receiving cardiac interventions (such as angiography, percutaneous intervention or bypass grafting).
-
Re-attendance at and/or re-admission to hospital over the following 3 months.
-
Adverse events (death, non-fatal AMI, life-threatening arrhthymia, emergency revascularisation or hospitalisation for myocardial ischaemia).
-
The proportion of admitted patients ultimately diagnosed as having AMI by ESC/ACC criteria. 1
We have selected successful discharge home as the primary outcome because the main purpose of point-of-care cardiac marker testing in this patient group is to facilitate discharge home. This outcome is beneficial for patients, who avoid the inconvenience and risks of hospital admission, and is beneficial for the health service, which avoids unnecessary admissions and pressure upon acute and emergency services. Patients who suffer an adverse event after discharge will not be classified as a successful discharge home because it is possible that they would have benefited from hospital admission. We will also record the proportion of admitted patients who are ultimately diagnosed as having AMI to provide a measure of the appropriateness of admissions.
Assessment of outcomes
Recruiting staff will record baseline data, the results of initial assessment (including any biochemical cardiac tests), data required for TIMI27 and GRACE28 scoring and admission/discharge decision from the ED. Research nurses will use ED and hospital inpatient notes to record management decisions at initial attendance and admission, extract resource use data and identify subsequent attendances/admissions and adverse events up to 3 months.
Research nurses will check patient status (dead or alive) at 1 and 3 months, using hospital information systems. Deceased patients will be assumed to have a score of zero on EQ-5D and will be excluded from other patient-based assessments. Participants who are not recorded as dead will be mailed a questionnaire at 1 and 3 months from the University of Sheffield to identify adverse events and hospital attendances, health and social care resource use, and to measure EQ-5D and satisfaction with care (satisfaction at 1 month only). Our previous study suggests a 70–80% response rate to this questionnaire. 8 We will therefore contact the general practitioner of all participants who do not respond at 3 months after attendance to identify any serious adverse events or deaths that have not been recorded by hospital information systems or case notes. Classification of cases of AMI and adverse events will be done by blind independent review of the relevant data.
Proposed sample size (N = 3130)
We anticipate that 50% of subjects will be successfully discharged in the group managed without the marker panel. 8 With 1565 evaluable subjects in each arm of the trial we will have 80% power to detect a 5% improvement (to 55% of patients successfully discharged) at the two-sided significance level of 5%. The same sample size will provide 80% power to detect a reduction from 4% to 2% in major cardiac events (death, non-fatal AMI, emergency revascularisation or hospitalisation for myocardial ischaemia), again at the two-sided 5% level of significance.
Based on previous studies by members of our research team, we estimate that we will require six hospitals to recruit for 12 months each to achieve the sample size of 3130, assuming that we recruit 70% of those eligible. 8,29–31 We have undertaken a number of studies of this specific patient group and have shown that recruitment of 550 suitable patients per year is attainable at a typical hospital.
Previous studies have also shown that we can anticipate a response rate of 70–80% for postal questionnaires,8,29 thus providing an effective sample size of at least 1000 in each of the two groups to evaluate health utility, satisfaction with care and health service resource use.
Statistical analysis
The primary outcome will be analysed through logistic regression, fitting concurrently with intervention group the effect of centre and appropriate baseline measures (including age, gender and past history of CHD). The results will be presented as adjusted odds ratios along with their corresponding 95% CIs. A similar analysis will be undertaken on major cardiac events. The primary analysis will be undertaken on an intention-to-treat basis. A secondary analysis will exclude those who were not managed according to their allocated strategy.
We will undertake a descriptive assessment to explore whether use of biochemical cardiac markers or admission rates change over time in either the intervention or control group, either as a result of staff ‘learning curves’ in the intervention group or as a result of contamination of the control group.
Analysis of secondary objectives
The secondary objectives of evaluating clinical prediction rules, such as the GRACE and TIMI scores, will be addressed by analysing the proportion of participants in each risk stratum of the score who suffer an adverse event over 30-day follow-up. Receiver-operator characteristic (ROC) curves will be constructed to estimate the discriminant power of the scores for adverse events.
Blood stored at St George’s Hospital will be used to analyse potential alternative cardiac markers and any new cardiac markers that are developed. We will analyse the association between marker levels and adverse events within 30 days. The data will then be split into derivation and validation data sets. ROC curves will be constructed using the derivation data set to estimate the discriminant power of the markers and to identify, alongside economic modelling, optimal thresholds for decision-making. The validation set will then be used to estimate sensitivity and specificity at the optimal threshold.
Economic evaluation
An economic evaluation will be undertaken alongside the trial using recommended practice. 32 To supplement this analysis, a cost-effectiveness model will be developed to duplicate the trial results (as a way of validation), extrapolate the results to longer follow-up periods, and incorporate a value of information analysis. The NHS perspective undertaken and other methods will be in line with NICE Technology Appraisal Guidelines,33 although data on production losses will be collected for a supplementary analysis.
Resource-use data will be collected for all patients covering the length of time in the ED, the use of diagnostic tests, admissions, re-admissions, outpatient reviews, cardiac procedures, and time off work. Cost and outcome data will be collected using patient notes and self-completed questionnaires as described previously. A small microcosting study will also be carried out at each site for a fortnight (to include around 30 patients), gathering data on staff times relating to the care of patients. ED cost per minute will be based on a study previously undertaken by the investigators,8 and amended using the microcosting data from this study. Panel costs will be based on purchase price, and the remaining costs will be valued using national unit costs. 34,35 Total NHS cost up to 3 months after initial attendance will then be calculated. Quality-adjusted life-years (QALYs) will be calculated by the trapezium rule using the EQ-5D tariff values at all follow-up points.
Economic analysis
Both cost and QALY analysis will compare bootstrap estimates of the mean cost per patient of the two groups. Cost-effectiveness analysis will estimate the incremental cost per QALY of using point-of-care cardiac marker testing compared with management without point-of-care testing. Results will be plotted on the cost-effectiveness plane and then transformed into cost-effectiveness acceptability curves with their associated frontier. 36 A sensitivity analysis will be undertaken that will include production losses as reported by the patient.
We anticipate that some of the resource use and QALY data will be incomplete (missing). Thus, in order to maximise the information that is collected from the trial we will impute missing values using multiple imputation. 37 The idea of multiple imputation draws from the fact that missing values from incomplete data are unknown and the technique of multiple imputation imputes more than one likely value for the missing data; hence, providing a representation of uncertainty. 38 Thus an additional set of results will be produced including the imputed cost and QALY data.
Decision-analytic model
We will also construct a decision-analytic model to describe the care observed in the trial, and likely care pathways subsequent to it. This will allow us to systematically investigate the impact of subsequent costs, quality of life and survival. These values will initially be based on population norms, but replaced with literature review estimates where appropriate. The decision-analytic model will be probabilistic, but with conventional sensitivity analysis used to assess the impact of structural uncertainties. 39
One important aspect of the model will be to investigate the relationship between sensitivity/specificity and admission rates. This will be modelled using all available studies of diagnostic testing in EDs. This part of the model is important, as it will help us to estimate cost-effectiveness of different panels in the future.
The decision-analytic model will be used to produce a value of information analysis. In particular, the analysis will generate partial expected value of perfect information estimates for each parameter in order to help prioritise future research.
Ethical arrangements
All participants will be asked to provide written, informed consent. This will include consent to allow research staff to examine their hospital records and contact their GP. Although participants will be recruited in an emergency setting and there will only be a limited amount of time available for considering trial information, the nature of the selected group (in particular the exclusion of people clearly requiring hospital treatment) ensures that eligible patients should not be incapacitated by their medical condition. We do not therefore plan to recruit incapacitated patients, and do not need to make provision for recruitment by personal or professional legal representatives.
Risks to participants are small, but include the following:
-
Inappropriate recruitment of high-risk patients or those with other serious non-cardiac pathology leading to risk of inappropriate discharge home. We will minimise this risk using regular review by the research nurses to identify inappropriately recruited participants. Inappropriate discharge of high-risk patients is, of course, a risk outside the confines of this trial. Indeed, a potential benefit to participants is that inclusion in a carefully audited trial should reduce their risk of mismanagement.
-
Failure of point-of-care testing to identify AMI leading to inappropriate discharge home. We have minimised this risk by choosing a widely used point-of-care test that has been shown to have high sensitivity for AMI in previous studies. 16–20 Furthermore, our own previous studies7,8 show that routine care (i.e. without point-of-care testing) is associated with a significant risk of inappropriate discharge home that appears to be reduced when rapid diagnostic testing protocols are available.
-
Distress to participants or their relatives if the postal questionnaire is sent to someone who is seriously ill or recently deceased. We will minimise this risk by ensuring that the research nurses check patient status on hospital information systems at 1 and 3 months, before questionnaires are mailed.
Submission to a Multicentre Research Ethics Committee is currently under way. We will complete Local Research Ethics Committee reviews during the first 6 months of the timetable.
Research governance
This trial will be conducted in accordance with MRC Guidelines for GCP in Clinical Trials. It does not involve a medicinal product and is not covered by the Medicine for Human Use (Clinical Trials) Regulations 2004. The University of Sheffield will act as the sponsor for the trial.
Three committees will be established to govern the conduct of this study:
-
Trial Steering Committee
-
Independent Data Monitoring and Ethics Committee (DMEC)
-
Trial Management Group.
The Trial Steering Committee will consist of the Principal Investigator, one of the co-applicants, an independent chair, two independent members and a consumer representative (Enid Hirst). We will also invite a representative of the HTA board to join the committee. The DMEC will consist of a minimum of an independent statistician, emergency physician and cardiologist, who will be asked to review trial data at regular intervals and implement stopping rules in accordance with MRC guidance. The Trial Management Group will consist of the Principal Investigator, co-applicants, project manager, statistician, health economists and research nurses.
Data management
Trial data will be entered into a validated database system, built to a specification agreed between Sheffield CTRU and the Principal Investigator. The system will be accessible remotely via a web browser, with the data stored securely on a central server. Access will be controlled by the use of assigned logins and encrypted passwords. The system will have a full electronic audit trail and will be regularly backed up. Quality control procedures will be applied to validate the trial data. Error reports will be generated where data clarification is required. Output for analysis will be generated in a format and at intervals to be agreed between Sheffield CTRU and the Principal Investigator. All activities will be performed in accordance with Sheffield CTRU Standard Operating Procedures.
Project timetable and milestones
The project will start on 1 April 2007. Months 1–6 will involve staff recruitment and local ethics and research governance; months 7–18 will involve patient recruitment; and months 19–24 will involve completion of follow-up, data analysis, writing up and dissemination. The project will be completed by 31 March 2009. The GANTT below outlines the key milestones and shows when project staff will be employed.
| Month of project | ||||||||
|---|---|---|---|---|---|---|---|---|
| 1–3 | 4–6 | 7–9 | 10–12 | 13–15 | 16–18 | 19–21 | 22–24 | |
| Trial manager | XXX | XXX | XXX | XXX | XXX | XXX | XXX | XXX |
| Trial researcher | XXX | XXX | XXX | XXX | ||||
| Clerical assistant | XXX | XXX | XXX | XXX | XXX | XXX | XXX | XXX |
| Research nurses × 6 | XX | XXX | XXX | XXX | XXX | XXX | X | |
| Staff recruiting, ethics, R&D | XXX | XXX | ||||||
| Patient recruitment | XXX | XXX | XXX | XXX | ||||
| Follow-up | XX | XXX | XXX | XXX | XXX | |||
| Analysis | XXX | |||||||
| Writing up | XXX | |||||||
We will submit 6-monthly progress reports corresponding with the following milestones:
-
completion of ethics and governance procedures and commencement of recruitment in all six sites
-
mid-point of recruitment, with a target of 1200 participants recruited (allowing for initial lag phase in recruitment)
-
end of recruitment, with 3130 participants recruited
-
completion of analysis and final report.
Expertise
The trial will be coordinated by a trial manager in the Sheffield CTRU, working with full statistical, clinical trials and health economic support. The applicants are a multidisciplinary team with expertise in health service research, emergency medicine, cardiology, chemical pathology, epidemiology, health economics and statistics. The researchers are leading experts in the management of acute chest pain and have undertaken previous landmark investigations in this field, including the ESCAPE trial of chest pain units8,40 (SG, SC, SD), randomised evaluation of point-of-care cardiac markers in coronary care22 (PC), evaluation of ischaemia-modified albumin in emergency care31,41 (JB, PC, SG), and evaluation of cardiac biomarkers (PC). We have also collaborated to successfully undertake a previous HTA-funded multicentre trial in emergency care, the 3CPO Trial42 (AG, SG, DN, JB). The co-applicants from Leeds (AH, JB, TH) have recently undertaken studies of biomarkers in patients with acute chest pain and studies in AMI. 43,44
Service users
Enid Hirst, a health service user representative, has provided valuable input into previous projects undertaken by our team. She has agreed to provide user involvement in the development of the proposal and be user representative on the Trial Steering Group. She has also created a CHD user group consisting of five people with CHD and their main carer/relative. This group has provided guidance to previous projects, notably our evaluation of the National Infarct Angioplasty Pilots. We are using this group to develop our proposal: specifically to identify relevant outcome measures, and ensure appropriate procedures are used for consent and follow-up.
References
- Joint European Society of Cardiology/American College of Cardiology Committee . Myocardial infarction redefined: a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. Eur Heart J 2000;21:1502-13.
- Goodacre S, Cross E, Arnold J, Angelini K, Capewell S, Nicholl J. The health care burden of acute chest pain. Heart 2005;91:229-30.
- Task Force on the Management of Acute Coronary Syndromes of the European Society for Cardiology . Management of acute coronary syndromes in patients presenting without ST elevation. Eur Heart J 2002;23:1809-40.
- Department of Health (DoH) . The National Service Framework for Coronary Heart Disease 2000.
- Goodacre S, Calvert N. Cost effectiveness of diagnostic strategies for patients with acute, undifferentiated chest pain. Emerg Med J 2003;20:429-33.
- Collinson PO, Premachandram S, Hashemi K. Prospective audit of incidence of prognostically important myocardial damage in patients discharged from the emergency department. BMJ 2000;320:1702-5.
- Goodacre S, Nicholl J, Dixon S, Cross E, Angelini K, Revill C, et al. Randomised controlled trial and economic evaluation of a chest pain observation unit compared with routine care. BMJ 2004;328:254-7.
- Goodacre S, Nicholl J, Beahan J, Quinney D, Capewell S. National survey of emergency department management of patients with acute, undifferentiated chest pain. Br J Cardiol 2003;10:50-4.
- Price CP. Point of care testing. BMJ 2001;322:1285-8.
- Fesmire FM, Percy RF, Bardoner JB, Wharton DR, Calhoun FB. Serial creatinine kinase (CK) MB testing during the emergency department evaluation of chest pain: utility of a 2-hour deltaCK-MB of +1.6 ng/ml. Am Heart J 1998;136:237-44.
- Fesmire FM, Christensen RH, Focy EP, Feintuch TA. Delta creatinine kinase MB outperforms myoglobin at two hours during the emergency department identification and exclusion of troponin positive non-ST elevation acute coronary syndromes. Ann Emerg Med 2004;44:12-9.
- Ebell MH, Flewelling D, Flynn CA. A systematic review of troponin T and I for diagnosing acute myocardial infarction. J Fam Pract 2000;49:550-6.
- Balk EM, Ioannidis JPA, Salem D, Chew PW, Lau J. Accuracy of cardiac biomarkers to diagnose acute cardiac ischaemia in the emergency department: a meta-analysis. Ann Emerg Med 2001;37:478-94.
- Craig J, Bradbury I, Collinson P, Emslie C, Findlay I, Hunt K, et al. Health Technology Assessment Report Number 4: The Organisation of Troponin Testing Services in Acute Coronary Syndromes. 2003.
- McCord J, Nowak RM, McCullough PA, Foreback C, Borzak S, Tokarski G, et al. Ninety-minute exclusion of acute myocardial infarction by use of quantitative point-of-care testing of myoglobin and troponin I. Circulation 2001;104:1483-8.
- Apple FS, Christensen RH, Valdes R, Andriak Aj, Berg A, Duh SH, et al. Simultaneous rapid measurement of whole blood myoglobin, creatinine kinase MB and cardiac troponin I by the triage cardiac panel for detection of myocardial infarction. Clin Chem 1999;45:199-205.
- Newby LK, Storrow AB, Gibler WB, Garvey JL, Tucker JF, Kaplan AL, et al. Bedside multimarker testing for risk stratification in chest pain units: the CHECKMATE Study. Circulation 2001;103:1832-7.
- Caragher TE, Fernandez BB, Jacobs F, Barr LA. Evaluation of quantitative cardiac biomarker point of care testing in the emergency department. J Emerg Med 2002;22:1-7.
- Ng SM, Krishnaswamy P, Morissey R, Clopton P, Fitzgerald R, Maisel AS, et al. Ninety-minute accelerated critical pathway for chest pain evaluation. Am J Cardiol 2001;88:611-17.
- Rocke LGR, McNicholl BP, Hughes D, Dunn F. Chest Pain Observation Units: Are They Really Necessary? n.d. URL: http://bmj.bmjjournals.com/cgi/eletters/328/7434/254#49899.
- Collinson PO, John C, Lynch S, Rao A, Canepa-Anson R, Carson E, et al. A prospective randomised controlled trial of point-of-care testing on the coronary care unit. Ann Clin 2004;41:397-404.
- Kendall J, Reeves B, Clancy M. Point of care testing: randomised controlled trial of clinical outcomes. BMJ 1998;316:1052-7.
- Griffs JC. Diagnosis and Treatment of ACS in the ED: The Impact of Rapid Bedside CTnI Testing on Outcomes n.d. URL: http://clinicaltrials.gov/ct/show/NCT00222352.
- Claxton K, Sculpher M, Drummond M. A rational framework for decision making by the National Institute for Clinical Excellence (NICE). Lancet 2002;360:711-15.
- Panteghini M, Pagani F, Yeo KT, Apple FS, Christenson RH, Dati F, et al. Evaluation of imprecision for cardiac troponin assays at low-range concentrations. Clin Chem 2004;50:327-32.
- Antman EM, Cohen M, Berninck PJ, Horacek T, Papuchis G, Mautner B, et al. The TIMI risk score for unstable angina/non-ST elevation MI: a method for prognostication and therapeutic decision making. JAMA 2000;284:835-42.
- Eagle KA, Lim MJ, Dabbous OH, Pieper KS, Goldberg RJ, Van de Werf F, et al. A validated prediction model for all forms of acute coronary syndrome. JAMA 2004;291:2727-33.
- Goodacre S, Mason S, Arnold J, Angelini K. Psychological morbidity and health-related quality of life of patients assessed on a chest pain observation unit. Ann Emerg Med 2001;38:369-76.
- Conway Morris A, Caesar D, Gray S, Gray A. The TIMI risk score accurately risk stratifies patients with undifferentiated chest pain presenting to an emergency department. Heart 2006;92:1333-4.
- Keating L, Benger J, Beetham R, Bateman S, Veysey S, Kendall J, et al. The PRIMA study: presentation IMA in the emergency department. Emerg Med J 2006;23:764-8.
- Ramsey S, Willke R, Briggs A, Brown R, Buxton M, Chawla A, et al. Good research practices for cost-effectiveness analysis alongside clinical trials: the ISPOR RCT-CEA Task Force Report. Value Health 2005;8:521-33.
- National Institute for Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2004.
- Netten A, Curtis L. Unit costs of health and social care 2004. University of Kent: Personal Social Services Research Unit; 2004.
- Department of Health (DoH) . NHS Reference Costs 2005 n.d. URL: www.dh.gov.uk/PolicyAndGuidance/OrganisationPolicy/FinanceAndPlanning/NHSReferenceCosts/fs/en.
- Fenwick E, Claxton K, Sculpher M. Representing uncertainty: the role of cost effectiveness acceptability curves. Health Econ Lett 2001;10:779-87.
- Schafer J. Analysis of incomplete multivariate data. London: Chapman & Hall/CRC; 2000.
- Rubin D. Multiple imputation for nonresponse in surveys. New York: J Wiley & Sons; 1987.
- Briggs A. Probabilistic analysis of cost-effectiveness models: statistical representation of parameter uncertainty. Value Health 2005;8:1-2.
- NHS Service Delivery and Organisation (NHSSDO) . The ESCAPE Multicentre Trial of the Role of Chest Pain Units in the NHS (NHS Service Delivery and Organisation Programme) n.d. URL: www.shef.ac.uk/∼scharr/escape.
- Collinson PO, Gaze DC, Bainbridge K, Morris F, Morris B, Price A, et al. Utility of admission cardiac troponin and ischemia modified albumin (IMA) measurements for rapid evaluation and rule out of suspected acute myocardial infarction in the emergency department. Emerg Med J 2006;23:256-61.
- Gray A, Goodacre S, Newby D, Masson M, Sampson F, Nicholl J. on behalf of the 3CPO trialists. Effectiveness of non-invasive ventilation in patients with acute severe cardiogenic pulmonary oedema. New Engl J Med 2008;359:142-51.
- Hall A, Barth J, Hassan T, Farrin A. H-FABP (heart-type fatty acid binding protein) and other markers in early diagnosis and risk stratification in suspected acute coronary syndrome: the FAB study; 2006 (unpublished) n.d.
- Tsang DM, Owen AM, Collinson PO, Barth JH. Surveys on the use of cardiac markers in the United Kingdom. Ann Clin Biochem 2003;40:138-42.
List of abbreviations
- ACS
- acute coronary syndrome
- AMI
- acute myocardial infarction
- CABG
- coronary artery bypass graft
- CHD
- coronary heart disease
- CI
- confidence interval
- CK-MB <br/>(mass)
- creatine kinase MB (mass)
- CTU
- Clinical Trials Unit
- CV
- coefficient of variation
- DMEC
- Data Monitoring and Ethics Committee
- ECG
- electrocardiogram
- ED
- emergency department
- EQ-5D
- European Quality of Life-5 Dimensions
- GCP
- Good Clinical Practice
- GP
- general practitioner
- GRACE
- Global Registry of Acute Coronary Events (risk score)
- GTN
- glyceryl trinitrate
- LREC
- local research ethics committee
- MI
- myocardial infarction
- MRC
- Medical Research Council
- NICE
- National Institute for Health and Clinical Excellence
- NSTEMI
- non-ST-elevation myocardial infarction
- OR
- odds ratio
- PCI
- percutaneous coronary intervention
- QALY
- quality-adjusted life-year
- RATPAC
- Randomised Assessment of Treatment using Panel Assay of Cardiac markers
- RCT
- randomised controlled trial
- SD
- standard deviation
- SE
- standard error
- STEMI
- ST-elevation myocardial infarction
- TIMI
- thrombolysis in myocardial infarction (risk score)
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Peter Brocklehurst, Professor of Women’s Health, Institute for Women’s Health, University College London
-
Professor Jenny Donovan, Professor of Social Medicine, University of Bristol
-
Professor Jonathan Green, Professor and Acting Head of Department, Child and Adolescent Psychiatry, University of Manchester Medical School
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Freddie Hamdy, Professor of Urology, Head of Nuffield Department of Surgery, University of Oxford
-
Professor Allan House, Professor of Liaison Psychiatry, University of Leeds
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Expert Advisory Network
-
Professor Douglas Altman, Professor of Statistics in Medicine, Centre for Statistics in Medicine, University of Oxford
-
Professor John Bond, Professor of Social Gerontology & Health Services Research, University of Newcastle upon Tyne
-
Professor Andrew Bradbury, Professor of Vascular Surgery, Solihull Hospital, Birmingham
-
Mr Shaun Brogan, Chief Executive, Ridgeway Primary Care Group, Aylesbury
-
Mrs Stella Burnside OBE, Chief Executive, Regulation and Improvement Authority, Belfast
-
Ms Tracy Bury, Project Manager, World Confederation of Physical Therapy, London
-
Professor Iain T Cameron, Professor of Obstetrics and Gynaecology and Head of the School of Medicine, University of Southampton
-
Professor Bruce Campbell, Consultant Vascular & General Surgeon, Royal Devon & Exeter Hospital, Wonford
-
Dr Christine Clark, Medical Writer and Consultant Pharmacist, Rossendale
-
Professor Collette Clifford, Professor of Nursing and Head of Research, The Medical School, University of Birmingham
-
Professor Barry Cookson, Director, Laboratory of Hospital Infection, Public Health Laboratory Service, London
-
Dr Carl Counsell, Clinical Senior Lecturer in Neurology, University of Aberdeen
-
Professor Howard Cuckle, Professor of Reproductive Epidemiology, Department of Paediatrics, Obstetrics & Gynaecology, University of Leeds
-
Professor Carol Dezateux, Professor of Paediatric Epidemiology, Institute of Child Health, London
-
Mr John Dunning, Consultant Cardiothoracic Surgeon, Papworth Hospital NHS Trust, Cambridge
-
Mr Jonothan Earnshaw, Consultant Vascular Surgeon, Gloucestershire Royal Hospital, Gloucester
-
Professor Martin Eccles, Professor of Clinical Effectiveness, Centre for Health Services Research, University of Newcastle upon Tyne
-
Professor Pam Enderby, Dean of Faculty of Medicine, Institute of General Practice and Primary Care, University of Sheffield
-
Professor Gene Feder, Professor of Primary Care Research & Development, Centre for Health Sciences, Barts and The London School of Medicine and Dentistry
-
Mr Leonard R Fenwick, Chief Executive, Freeman Hospital, Newcastle upon Tyne
-
Mrs Gillian Fletcher, Antenatal Teacher and Tutor and President, National Childbirth Trust, Henfield
-
Professor Jayne Franklyn, Professor of Medicine, University of Birmingham
-
Mr Tam Fry, Honorary Chairman, Child Growth Foundation, London
-
Professor Fiona Gilbert, Consultant Radiologist and NCRN Member, University of Aberdeen
-
Professor Paul Gregg, Professor of Orthopaedic Surgical Science, South Tees Hospital NHS Trust
-
Bec Hanley, Co-director, TwoCan Associates, West Sussex
-
Dr Maryann L Hardy, Senior Lecturer, University of Bradford
-
Mrs Sharon Hart, Healthcare Management Consultant, Reading
-
Professor Robert E Hawkins, CRC Professor and Director of Medical Oncology, Christie CRC Research Centre, Christie Hospital NHS Trust, Manchester
-
Professor Richard Hobbs, Head of Department of Primary Care & General Practice, University of Birmingham
-
Professor Alan Horwich, Dean and Section Chairman, The Institute of Cancer Research, London
-
Professor Allen Hutchinson, Director of Public Health and Deputy Dean of ScHARR, University of Sheffield
-
Professor Peter Jones, Professor of Psychiatry, University of Cambridge, Cambridge
-
Professor Stan Kaye, Cancer Research UK Professor of Medical Oncology, Royal Marsden Hospital and Institute of Cancer Research, Surrey
-
Dr Duncan Keeley, General Practitioner (Dr Burch & Ptnrs), The Health Centre, Thame
-
Dr Donna Lamping, Research Degrees Programme Director and Reader in Psychology, Health Services Research Unit, London School of Hygiene and Tropical Medicine, London
-
Professor James Lindesay, Professor of Psychiatry for the Elderly, University of Leicester
-
Professor Julian Little, Professor of Human Genome Epidemiology, University of Ottawa
-
Professor Alistaire McGuire, Professor of Health Economics, London School of Economics
-
Professor Neill McIntosh, Edward Clark Professor of Child Life and Health, University of Edinburgh
-
Professor Rajan Madhok, Consultant in Public Health, South Manchester Primary Care Trust
-
Professor Sir Alexander Markham, Director, Molecular Medicine Unit, St James’s University Hospital, Leeds
-
Dr Peter Moore, Freelance Science Writer, Ashtead
-
Dr Andrew Mortimore, Public Health Director, Southampton City Primary Care Trust
-
Dr Sue Moss, Associate Director, Cancer Screening Evaluation Unit, Institute of Cancer Research, Sutton
-
Professor Miranda Mugford, Professor of Health Economics and Group Co-ordinator, University of East Anglia
-
Professor Jim Neilson, Head of School of Reproductive & Developmental Medicine and Professor of Obstetrics and Gynaecology, University of Liverpool
-
Mrs Julietta Patnick, Director, NHS Cancer Screening Programmes, Sheffield
-
Professor Robert Peveler, Professor of Liaison Psychiatry, Royal South Hants Hospital, Southampton
-
Professor Chris Price, Director of Clinical Research, Bayer Diagnostics Europe, Stoke Poges
-
Professor William Rosenberg, Professor of Hepatology and Consultant Physician, University of Southampton
-
Professor Peter Sandercock, Professor of Medical Neurology, Department of Clinical Neurosciences, University of Edinburgh
-
Dr Philip Shackley, Senior Lecturer in Health Economics, Sheffield Vascular Institute, University of Sheffield
-
Dr Eamonn Sheridan, Consultant in Clinical Genetics, St James’s University Hospital, Leeds
-
Dr Margaret Somerville, Director of Public Health Learning, Peninsula Medical School, University of Plymouth
-
Professor Sarah Stewart-Brown, Professor of Public Health, Division of Health in the Community, University of Warwick, Coventry
-
Dr Nick Summerton, GP Appraiser and Codirector, Research Network, Yorkshire Clinical Consultant, Primary Care and Public Health, University of Oxford
-
Professor Ala Szczepura, Professor of Health Service Research, Centre for Health Services Studies, University of Warwick, Coventry
-
Dr Ross Taylor, Senior Lecturer, University of Aberdeen
-
Dr Richard Tiner, Medical Director, Medical Department, Association of the British Pharmaceutical Industry
-
Mrs Joan Webster, Consumer Member, Southern Derbyshire Community Health Council
-
Professor Martin Whittle, Clinical Co-director, National Co-ordinating Centre for Women’s and Children’s Health, Lymington