
Notes
Article history
The research reported in this issue of the journal was commissioned by the HTA programme as project number 08/83/01. The contractual start date was in August 2009. The draft report began editorial review in August 2010 and was accepted for publication in December 2010. As the funder, by devising a commissioning brief, the HTA programme specified the research question and study design. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Susan Howell has received funding for symposium attendance and travel, and also one advisory board fee from Novartis for discussing the role of letrozole in early breast cancer. William Newman has received research funding support from Roche Molecular Systems. Professor Tom Walley is Editor-in-Chief of Health Technology Assessment, although he was not involved in the editorial processes for this report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2011. This work was produced by Fleeman et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2011 Queen’s Printer and Controller of HMSO 2011
Chapter 1 Introduction to CYP2D6 and CYP2D6 testing
Pharmacogenetic testing and the use of testing in clinical practice is a relatively new, evolving and complex topic. This short summary provides an introduction to the basic concepts that need to be considered in relation to cytochrome P450 2D6 (CYP2D6) and CYP2D6 testing.
Enzymes, genes and pharmacogenetics
Differences in the response of individuals to the same drug at the same dose may occur as a result of interindividual differences in enzymes (e.g. CYP2D6) responsible for metabolising the drug. These differences may be inherited and occur as a result of differences in the genes (e.g. CYP2D6) that encode the enzyme.
In humans, each gene is composed of two alleles, one inherited from each parent, and a person may have two copies of the same allele (homozygous) or one copy of two different alleles (heterozygous). Alleles that differ from the normal or common form are known as polymorphisms [variant (vt)], while a normal allele is referred to as wild type (wt). It is from these differences that an individual’s genotype is derived, for example the homozygous wt (i.e. wt/wt) genotype.
A phenotype is the observable physical trait of an organism, which, in pharmacogenetics, relates to an individual’s reaction to a drug, usually as a result of the way in which the drug is metabolised. The phenotype is largely determined by the overall genetic make-up of a person, although it may also be influenced by environmental factors (e.g. diet and smoking).
The cytochrome P450 (CYP450) enzyme system, to which CYP2D6 belongs, has been identified as a major metabolic pathway for many drugs and a source of interindividual variability in patient response. It is believed to play a prominent role in the way in which tamoxifen (TAM) is metabolised and thus may explain differences in responses in individual patients to the same dose as it is known that TAM is metabolised to its active metabolites (which are thought to affect patient response, rather than TAM itself) by a number of CYP450 enzymes (including CYP2D6).
Based on studies that have examined the urinary metabolic ratios of drugs such as debrisoquine and/or dextromethorphan to their metabolites (4-hydroxydebrisoquine and dextrorphan, respectively), an association between CYP2D6 genotypes (genetic make-up) and phenotypes (response to treatment) is believed to exist. It is thus also believed that patients experiencing a normal response at a normal dose of TAM would be CYP2D6 extensive metabolisers (EMs). These individuals are thought to be homozygous for the wt allele. Patients experiencing reduced clinical effects owing to deficient alleles are referred to as poor metabolisers (PMs) and are thought to be homozygous (and possibly heterozygous) for the vt allele.
However, there are a number of different vt alleles, some which result in decreased enzyme activity and others that result in a complete lack of enzyme activity (i.e. the differing extent to which the drug is metabolised). PMs must possess at least one of these complete lack of function alleles (e.g. *4; see Table 1).
| CYP2D6 variant | Predicted enzymatic function via enzymes encoded by the gene |
|---|---|
| wt alleles | |
| *1, *2, *35 | Normal (associated with EMs) |
| vt alleles | |
| *3, *4, *5, *6, *9 | Loss of function, i.e. a complete lack of enzyme activity (associated with PMs) |
| *10, *17, *41 | Decreased activity (associated with IMs) |
| Multiple alleles | |
| e.g. *1 × N, *2 × N | Increased activity (usually associated with UMs, although this is not always the case) |
Patients are sometimes also considered to be intermediate metabolisers (IMs) if clinical effects lie somewhere between EMs and PMs. Generally, IMs are thought to possess at least one decreased activity allele (e.g. *10). Patients are also sometimes considered to be ultrarapid metabolisers (UMs) when there are multiple copies of an allele (e.g. *2 × 2, *2 × 3, etc.). However, multiple copies of an allele do not necessarily result in increased activity. Furthermore, for CYP2D6 there is no uniformly agreed way in which to relate genotype to phenotype. While it is acknowledged by all that a patient with a wt/wt genotype would be an EM and a patient with the *4/*4 genotype would be a PM, some would also classify patients heterozygous for these alleles differently, for example a patient with the wt/*4 genotype could be considered an EM, IM or PM.
Genotyping for CYP2D6
There is growing anticipation that genotyping for CYP2D6 may be used to assist in treatment decision-making. A number of these tests have been developed and are described in the literature, and have been used for a wide range of drugs and diseases, not just TAM and breast cancer. However, not all tests will be the same.
Table 2 presents examples of three possible CYP2D6 tests that could be used. As can be seen, in test A, patients are simply tested for *4. Those who are found not to possess *4 are considered to be wt. Even with this simple test, it is possible to classify a patient with the wt/*4 genotype in three different ways: EM, IM or PM. As the number of alleles tested for increases (test B), the chances of detecting IMs and/or PMs are increased and the classification is complicated somewhat by the inclusion of the decreased activity allele (*10) in test C.
| Test | Alleles tested | Possible genotypes and phenotypes | |||
|---|---|---|---|---|---|
| Genotypes | Classification | ||||
| 1 | 2 | 3 | |||
| A | *4 | wt/wt | EM | EM | EM |
| wt/*4 | IM | PM | EM | ||
| *4/*4 | PM | PM | PM | ||
| B | *3, *4, *5 | wt/wt | EM | EM | EM |
| wt/*3 | IM | PM | EM | ||
| wt/*4 | IM | PM | EM | ||
| wt/*5 | IM | PM | EM | ||
| *3/*3 | PM | PM | PM | ||
| *3/*4 | PM | PM | PM | ||
| *3/*5 | PM | PM | PM | ||
| *4/*4 | PM | PM | PM | ||
| *4/*5 | PM | PM | PM | ||
| *5/*5 | PM | PM | PM | ||
| C | *3, *4, *10 | wt/wt | EM | EM | EM |
| wt/*3 | IM | PM | EM | ||
| wt/*4 | IM | PM | EM | ||
| wt/*10 | IM | IM | EM | ||
| *3/*3 | PM | PM | PM | ||
| *3/*4 | PM | PM | PM | ||
| *3/*10 | IM | PM | IM | ||
| *4/*4 | PM | PM | PM | ||
| *4/*10 | IM | PM | IM | ||
| *10/*10 | IM | IM | IM | ||
These examples can also be used to show that the construction of phenotypes can change the way in which results are interpreted. Thus, if we had 30 patients and found from test A that 15 had the wt/*4 genotype and that 10 of these patients had a side effect from taking TAM that was not detected in the other 20 patients then, depending on which classification we used, we would describe these patients as being IM, PM or EM. Consequently, we would assume from this sample of patients that there was an association between the phenotype and the side effects.
In addition, these examples also show that as a larger number of alleles are tested for, the chances of detecting IMs and PMs are increased. For example, a patient with the *3/*5 genotype identified by test B and labelled a PM would not have been detected as a PM by test A, which did not test for these two alleles, and so he or she would have been classified as wt/wt, i.e. EM. Test C may also be unable to identify this patient as a PM, not because of the number of alleles tested but because of the types of alleles tested, here this patient being identified as *3/wt. Thus, the types of alleles tested for are just as crucial as the number tested.
To date, the majority of these tests are designed bespoke, ‘in house’, for specific research projects, often using commercially available technologies such as TaqMan® (Roche Molecular Systems). The only commercially available complete test that is available and used in clinical practice, albeit rarely, is the AmpliChip® (Roche Molecular Systems), which tests for 33 different alleles.
Chapter 2 Background
Description of health problem
Incidence/prevalence and health impact
Breast cancer is the most common cancer affecting women in the UK. In England and Wales, in 2007, around 45,000 new cases of breast cancer were diagnosed1 and there were nearly 11,000 deaths due to breast cancer. 2 Breast cancer incidence rates increase with age; around 80% of breast cancers occur in women aged > 50 years, and women have a one in nine lifetime risk of developing breast cancer. 3 Breast cancer prevalence is around 172,000 women in the UK according to the most recently published data. 4 This relatively high prevalence rate has been attributed to high incidence rates combined with 5-year survival rates of > 75%. 5
Aetiology
Breast cancer is the uncontrolled, abnormal growth of malignant breast tissue affecting predominantly women. The strongest risk factor for breast cancer (after gender) is age – the older the woman, the higher her risk – but other genetic and hormonal risk factors have also been identified in the aetiology of breast cancer. 5,6
Carriers of the breast cancer 1 (BRCA1) or 2 (BRCA2) gene mutations7,8 and women with a family history of breast cancer9 both have an increased risk of developing breast cancer. Higher concentrations of some endogenous hormones appear to increase breast cancer risk. 10 Risk factors associated with endogenous oestrogen – including early age at menarche, late natural menopause, later age at first full-term pregnancy and never breastfeeding – are all associated with an increased risk of breast cancer,11 while childbearing and a higher number of full-term pregnancies increase the protection. 11 Risk factors associated with the use of exogenous hormones, such as oral contraception, oestrogen replacement therapy and combined anti-oestrogen therapy, increase the risk of breast cancer, as do other factors such as breast density (a risk factor independent of endogenous hormones), a body mass index of > 25 kg/m2 in postmenopausal women, moderate to heavy alcohol intake and a sedentary lifestyle. 11 Patients with a history of breast cancer12 and radiation exposure13 are also at increased risk.
Pathology, clinical staging and diagnosis
Breast cancer is classified into clinical stages according to tumour size, spread of cancer to lymph nodes and distant metastases. A number of different classification systems exist, including the tumour/nodes/metastasis (TNM) staging system developed and maintained by the American Joint Committee on Cancer (AJCC)14 and the Union Internationale Contre le Cancer (UICC). 15 In this system, ‘T’ refers to the size of the tumour and its spread, ‘N’ to the number of lymph nodes involved and ‘M’ to the presence of metastases (Table 3). The TNM system can be categorised further into disease stages (Table 4).
The stage of disease is an indication of prognosis. Data reported by Cancer Research UK in 2004 and cited by Ward et al. 6 suggested that the 5-year survival rate was around 90% for those with stage I disease, dropping to 75% for stage II, 42% for stage III and 14% for stage IV.
| Tumour stage (T) | |
|---|---|
| Tx | Cannot be assessed |
| Tis | Carcinoma in situ |
| T0 | No evidence of primary tumour |
| T1 | Tumour < 2 cm in greatest dimension |
| T2 | Tumour 2–5 cm |
| T3 | Tumour > 5 cm |
| T4 | Tumour of any size with direct extension to skin or chest wall |
| Lymph node stage (N) | |
| Nx | Cannot be assessed |
| N0 | No nodal metastases |
| N1 | Metastases to ipsilateral nodes |
| N2 | Metastases to ipsilateral nodes that are fixed to one another or other structures |
| N3 | Metastasis to ipsilateral supraclavicular or infraclavicular nodes |
| Metastasis stage (M) | |
| Mx | Cannot be assessed |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| Stage | Description of disease | T | N | M |
|---|---|---|---|---|
| 0 | Ductal carcinoma in situ – cancer cells are located within a duct and have not invaded the surrounding fatty breast tissue | Tis | N0 | M0 |
| I | The tumour is ≤ 2 cm in diameter and has not spread to lymph nodes or distant sites | T1 | N0 | M0 |
| IIA | No tumour is found in the breast but it is in one to three axillary lymph nodes, or the tumour is < 2 cm and has spread to one to three axillary lymph nodes or has been found by sentinel node biopsy as microscopic disease in internal mammary nodes but not on imaging studies or by clinical examination, or the tumour is > 2 cm in diameter and < 5 cm but has not spread to axillary nodes | T0 | N1 | M0 |
| T1 | N1 | M0 | ||
| T2 | N0 | M0 | ||
| IIB | The tumour is > 2 cm in diameter and < 5 cm and has spread to one to three axillary lymph nodes or has been found by sentinel node biopsy as microscopic disease in internal mammary nodes, or the tumour is > 5 cm and does not grow into the chest wall and has not spread to lymph nodes | T2 | N1 | M0 |
| T3 | N0 | M0 | ||
| IIIA | The tumour is < 5 cm in diameter and has spread to four to nine axillary lymph nodes or has been found by imaging studies or clinical examination to have spread to internal mammary nodes, or the tumour is > 5 cm and has spread to one to nine axillary nodes or to internal mammary nodes | T0 | N2 | M0 |
| T1 | N2 | M0 | ||
| T2 | N2 | M0 | ||
| T3 | N1 | M0 | ||
| T3 | N2 | M0 | ||
| IIIB | The tumour has grown into the chest wall or skin and may have spread to no lymph nodes or as many as nine axillary nodes | T4 | N(any) | M0 |
| T(any) | N3 | M0 | ||
| IV | The cancer has spread from the breast to another part of the body (metastasis) | T(any) | N(any) | M(any) |
Alternatively, many clinicians in the UK use prognostic tools, such as the Nottingham Prognostic Index (NPI)16 or the web-based tool ‘Adjuvant! Online’. 17 The NPI takes into account three of the major prognostic factors, namely tumour size, lymph nodal status and grade according to the following formula:
The formula gives scores, which fall into the following categories:
-
excellent-prognosis group ≤ 2.4
-
good-prognosis group > 2.4 and ≤ 3.4
-
moderate-prognosis group > 3.4 and ≤ 5.4
-
poor-prognosis group > 5.4.
The 10-year predictive survival rates are as follows:18
-
excellent-prognosis group = 96%
-
good-prognosis group = 93%
-
moderate-prognosis group = 53%
-
poor-prognosis group = 39%.
‘Adjuvant! Online’ also incorporates tumour oestrogen receptor (ER) status and patient comorbidity, and provides an estimate of the potential benefit of treatment, derived from clinical trial data. This programme also has the feature of a modifiable prognostic calculator to factor in other known poor prognostic features, such as lymphovascular invasion and human epidermal growth factor receptor 2 (HER2) expression.
Current service provision
Treatment for breast cancer can be divided into surgical treatment to control the disease locally (within the breast and axillary lymph nodes) and adjuvant treatment after surgical removal of the primary cancer. The aim of adjuvant treatment is to prevent recurrence and may involve radiotherapy, chemotherapy, biological therapy or anti-oestrogen therapy.
Radiotherapy is routinely given to women after breast-conserving surgery. After mastectomy, it is given to those who are considered to be at high risk of breast cancer recurrence. Owing to its side effects, adjuvant chemotherapy is usually given only to women at significant risk of recurrence, or if their cancers are ER negative (ER–). Biological therapy is given to women whose cancers overexpress the HER2 receptor. The majority of women who have been diagnosed with ER positive (ER+) breast cancers receive anti-oestrogen therapy, which typically comprises TAM and/or aromatase inhibitors. Anti-oestrogen therapy is not used for women with ER– breast cancers.
Because aromatase inhibitors are ineffective in women whose ovaries are functional and produce oestrogen,19 TAM is considered the standard of care for premenopausal women with ER+ breast cancer. TAM is a selective ER modulator, i.e. it is a compound that competes with oestrogen for binding to the ER.
For postmenopausal women with ER+ early breast cancer, the most recent National Institute for Health and Clinical Excellence (NICE) guidelines18 state that in the UK ‘Current practice is to give low-risk patients TAM for five years’. Risk is based on the NPI, and low-risk patients are those in the excellent- or good-prognosis groups. NICE recommends that women who are considered to be at higher risk of disease recurrence should be offered an aromatase inhibitor [anastrozole (ANA) or letrozole] as their adjuvant treatment. 18 Aromatase inhibitors (exemestane or ANA) are recommended for patients who have already received 2–3 years of adjuvant therapy with TAM but are not considered low risk for disease recurrence, who are intolerant of TAM or for whom TAM is contraindicated. After 5 years of treatment with TAM, aromatase inhibitor treatment (letrozole) is also recommended by NICE for 2–3 years for women with lymph node positive (LN+) ER+ early invasive breast cancer.
The National Institute for Health and Clinical Excellence also recommends the use of TAM and aromatase inhibitors for some women with ER+ advanced breast cancer. 20 TAM is the recommended first-line treatment for premenopausal and perimenopausal women not previously treated with TAM. In postmenopausal women, aromatase inhibitors are recommended for women with no prior history of anti-oestrogen therapy or for those who have been previously treated with TAM.
The NICE guidelines regarding the use of TAM and aromatase inhibitors in early breast cancer are based on randomised controlled trial (RCT) evidence. Two RCTs [ATAC21 (Arimidex, Tamoxifen, Alone or in Combination) and BIG (Breast International Group) 1-9822] report 5 years of aromatase inhibitors to have modestly improved outcomes over 5 years of TAM use in terms of disease-free survival (DFS). Other RCTs also report a switch to an aromatase inhibitor after 2–3 years of TAM to be more efficacious than TAM alone for 5 years [ABCSG-6a,23 ABSCG-8 (Austrian Breast and Colorectal Cancer Study Group)/ARNO-95 (Arimidex/Nolvadex),24 IES25 (Intergroup Exemestane Study), ITA26 (Italian tamoxifen anastrozole)]. In addition, the MA.17 trial27 has reported improved outcomes in patients who were given letrozole after 5 years of TAM. All of these findings have also been summarised in three systematic reviews,28–30 and in an additional earlier review31 that included three of the switching strategy trials.
As can be seen from Figure 1, significant differences between TAM and aromatase inhibitors are not evident in overall survival (OS). However, significantly modest improvements in DFS after 5 years of aromatase inhibitor (ANA or letrozole) or switching to an aromatase inhibitor (exemestane) 2–3 years after TAM treatment have been reported. Disease recurrence has also been reported to be significantly improved by 5 years’ treatment with an aromatase inhibitor (ANA or letrozole) and switching to ANA after 2–3 years of TAM. The most recent systematic review28 pooled findings for mortality and recurrence in meta-analyses. For 5 years of treatment with an aromatase inhibitor or TAM, the absolute difference in breast cancer mortality was 1.1% at 5 years (4.8% for aromatase inhibitor vs 5.9% for TAM; p = 0.1) and there was an absolute 2.9% decrease in recurrence (9.6% for aromatase inhibitor vs 12.6% for TAM; p < 0.001). The switching strategy resulted in an absolute difference of 0.7% at the same time point, which was also approximately 3 years since divergence from TAM (1.7% for aromatase inhibitor vs 2.4% for TAM since divergence; p = 0.02) and an absolute 3.1% decrease in recurrence (5.0% for aromatase inhibitor vs 8.1% for TAM since divergence; p < 0.001).
FIGURE 1.
Differences in outcomes in patients receiving (a) either an aromatase inhibitor or TAM for 5 years and (b) an aromatase inhibitor after 2–3 years of TAM therapy compared with similar duration of TAM monotherapy. Data taken from the review by Eisen et al. 29 CI, confidence interval.
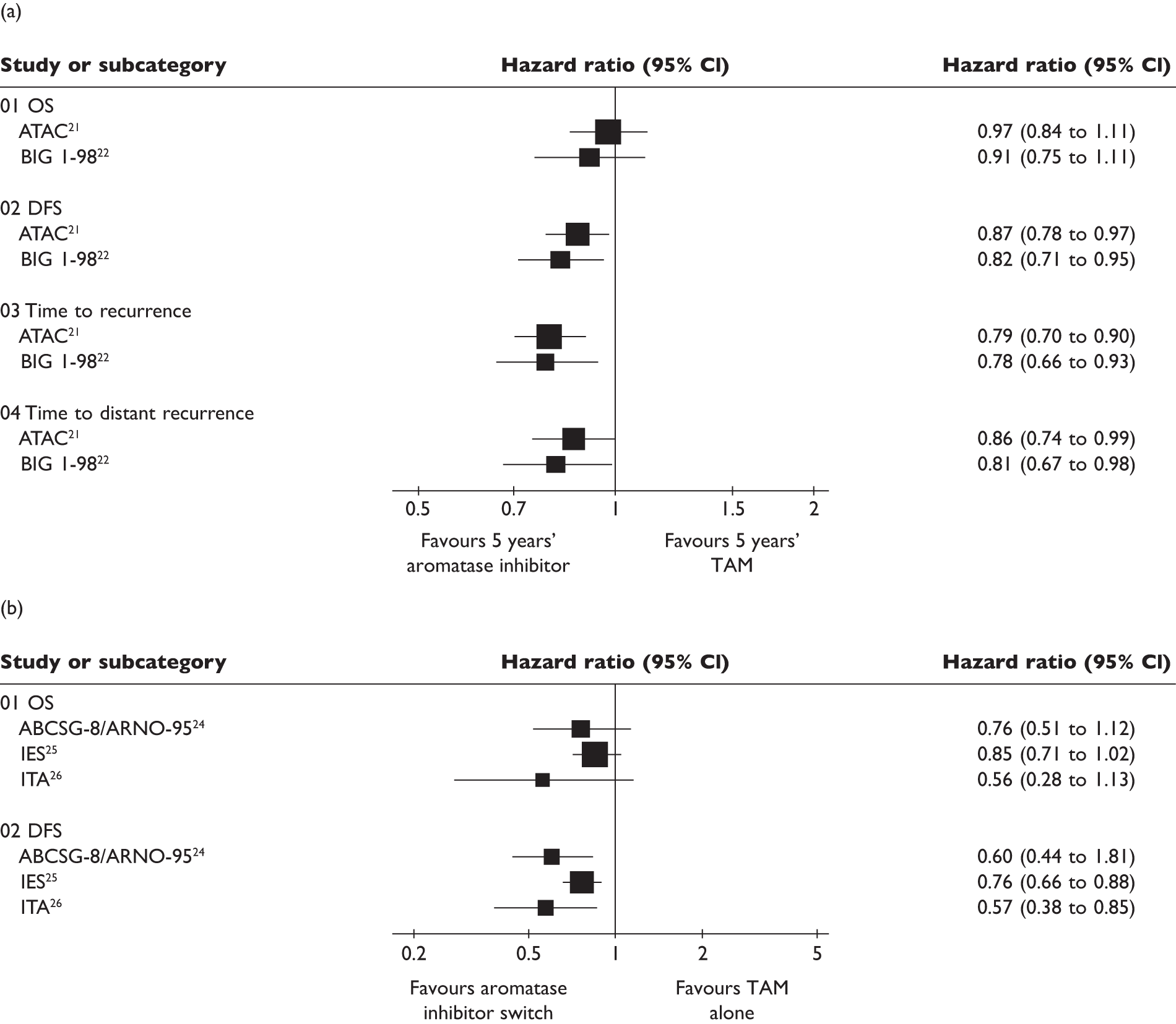
Side effect profiles differ between TAM and aromatase inhibitors. The long-term use of TAM may be associated with vaginal bleeding, endometrial thickening and increased risk of endometrial cancer and thromboembolic events. 30 Aromatase inhibitors have been reported to result in fewer hot flushes but are also associated with increased joint pain and bone fractures, and may also be associated with increased cardiovascular risk. 30 This cardiovascular risk has also been reported in a subsequent meta-analysis,32 although it was noted that the absolute difference was relatively low, and between 160 and 180 patients had to be treated to produce one event.
Assuming that these proportional benefits over TAM are maintained over 10 years, the cost per quality-adjusted life-year (QALY) gained for 5 years of ANA or letrozole compared with TAM has been reported to be between £10,000 and £12,000. 3,30 For the switch to exemestane or ANA after 2–3 years of TAM compared with TAM for 5 years, the estimated incremental cost per QALY gained was approximately £5000, and unplanned switching to letrozole compared with placebo after 5 years of TAM resulted in an incremental cost per QALY gained that was estimated to be £3000. 3,30
There are limited data available on the use of adjuvant therapy in breast cancer. 18 However, it has been reported that aromatase inhibitor use has increased at the expense of TAM, with a US study finding an increase from 4.1% in 2000 to 40% in 2003 in postmenopausal women with ER+ breast cancer. 33 This increase has been attributed to the evidence base28–31 suggesting aromatase inhibitors to be more efficacious.
Tamoxifen metabolism and pharmacogenetics
Wide variability in the response of individuals to drugs of the same dose may occur as a result of interindividual differences that may be inherited (pharmacogenetics). The CYP450 enzyme system has been identified as a major metabolic pathway for many drugs and a source of interindividual variability in patient response. 34 TAM is metabolised to its active metabolites N-desmethyl TAM and 4-hydroxytamoxifen by a number of CYP450 enzymes, including CYP2D6, CYP3A4, CYP2C9, CYP2C19 and CYP2B6. 35 N-desmethyl TAM is further metabolised to endoxifen by CYP2D6. 36 Endoxifen is 30- to 100-fold more potent than TAM in suppressing oestrogen-dependent cell proliferation, and is considered an entity that is responsible for significant pharmacological effects of TAM. 35
Genes are made up of alleles that determine an individual’s genotype and control the instructions that produce enzymes. The CYP2D6 enzyme is highly polymorphic (i.e. it can exist in many variant forms); there are > 70 different alleles of the CYP2D6 gene. These polymorphisms may be deficient or overactive in enzyme activity.
Based on studies which have examined the urinary metabolic ratios of debrisoquine and/or dextromethorphan to their metabolites, 4-hydroxydebrisoquine and dextrorphan, respectively, there is also believed to be an association between genotype and phenotype (i.e. expected drug effects). Sachse et al. 37 reported significant differences in metabolic ratio between carriers of one or two functional alleles. Thus, for patients with normal enzyme activity (commonly referred to as EMs) who are given TAM, usual doses should result in expected drug concentrations and normal therapeutic response. Patients with deficient alleles (commonly recognised as PMs) would be expected to have compromised clinical effects in terms of efficacy and possibly also adverse events (AEs). 35
This study classified patients as only EMs or PMs, despite identifying the presence of slightly or moderately reduced activity alleles (e.g. *2 and *10, respectively) and patients with multiple alleles (e.g. *2 × 2), who were all classified as EMs. However, other studies have considered individuals with these alleles to be separate to, or subsets of, EMs. Thus, the literature also discusses both IMs (patients with decreased activity resulting from decreased activity alleles) and UMs (patients with increased enzymatic activity resulting from multiple alleles). Patients classified as IMs would be expected to experience effects from a drug somewhere between EMs and PMs, whereas UMs would be expected to have reduced efficacy and/or increased risk of AEs as a result of the faster metabolism of the drug.
CYP2D6 enzyme activity may also be affected by co-administration of drugs that inhibit the metabolic activity of enzyme. In particular, it has been reported that the selective serotonin reuptake inhibitors (SSRIs) fluoxetine and paroxetine effectively alter the EM phenotype to PM in some individuals. 38 Patients treated with TAM are commonly prescribed SSRIs for depression or to alleviate AEs such as hot flushes, and co-administration of such substances therefore needs to be taken into consideration. The most recent NICE guidelines state that paroxetine and fluoxetine should be offered only to breast cancer patients who are not taking TAM. 18
Prevalence of CYP450 gene polymorphisms vary across populations. Table 5 presents a summary of frequencies of CYP2D6 alleles in various populations, and also describes the predicted enzymatic function arising from genotypes derived from common alleles. Given that the four most common loss-of-function alleles – *3, *4, *5 and *6 – are associated with up to 98% of the PM phenotypes, and given that the prevalence of these differs substantially by ethnicity, it is no surprise to find that there are ethnic differences in metaboliser status. For example, following a review of many studies examining CYP2D6 allelic variation and frequency in various populations published in 2002,39 it is commonly cited that around 7% of Caucasians are PMs compared with 1% of Asians. However, fewer Asians metabolise CYP2D6 substrates normally, and so there are fewer EMs in the Asian population. This is largely because of high frequencies of the *10 allele, which is thought to result in a higher prevalence of IMs in this population. It has been estimated that up to 51% of Asian populations may consist of IMs. 40 UMs are typically as uncommon as PMs, being around 4–5% in American Caucasians and African Americans, although it has been estimated that they may account for 29% of Ethiopians. 40
| CYP2D6 variant | Predicted enzymatic function | Associated phenotype(s) | Caucasian (%) | African American (%) | Asian (%) |
|---|---|---|---|---|---|
| *1 | Normal | EM | 30–40 | 28–50 | 20–40 |
| *2 | Normal | EM | 20–35 | 10–80 | 9–20 |
| *3 | Loss of function | PM, where the other variant is also loss of function, or IMa | 1–4 | < 1 | ≤ 1 |
| *4 | Loss of function | PM, where the other variant is also loss of function, or IMa | 12–23 | 2–9 | ≤ 3 |
| *5 | Loss of function | PM, where the other variant is also loss of function, or IMa | < 2–7 | ≤ 7 | 4–6 |
| *6 | Loss of function | PM, where the other variant is also loss of function, or IMa | ≤ 1 | < 1 | – |
| *9 | Decreased activity | IM | ≤ 3 | < 1 | – |
| *10 | Decreased activity | IM | ≤ 8 | 3–8 | 40–70 |
| *17 | Decreased activity | IM | < 1 | 10–30 | < 1 |
| *35 | Normal | EM | 4–6 | – | – |
| *41 | Decreased activity | IM | 8–20 | – | – |
| *1 × N | Increased activity, where N ≥ 2 | UM | ≤ 1 | < 5 | < 1 |
| *2 × N | Increased activity, where N = 2, 3, 4, 5 or 13 | UM | < 2 | < 2 | 0–1 |
| *4 × N | Loss of function, where N ≥ 2 | PM, where the other variant is also loss of function, or IMa | < 1 | 2–3 | – |
| *10 × N | Loss of function, where N ≥ 2 | PM, where the other variant is also loss of function, or IMa | – | – | – |
| *17 × 2 | Normal | EM | – | – | – |
| *35 × 2 | Increased activity | UM | – | – | – |
| *41 × 2 | Normal | EM | – | – | – |
Tests currently available for genotyping for CYP2D6
There is evidence suggesting that the AmpliChip is a highly accurate test (analytic validity),42 and this test is the first pharmacogenetic test to be granted market approval in the USA and European Union, based on evidence demonstrating that the test had high analytical (but not clinical) validity,43 increasing the possibilities that this may be one of the first licensed pharmacogenetic tests to be routinely used in clinical practice. Indeed, this is the only known commercially available CYP2D6 test currently available, although it is known that other laboratories are producing their own tests ‘in house’, which focus on fewer alleles than in the AmpliChip. Such tests are often developed using commercially available technologies, such as TaqMan, mainly for research rather than clinical application purposes. The AmpliChip has been cited as costing between US$600 and US$1300 in the USA in June 200744 and £300 in the UK in April 2008. 45 These costs include administration fees and platform costs, and the actual cost of the AmpliChip is dependent on the laboratory purchasing the test. Eight laboratories were known to be using the AmpliChip in the USA as of June 2007, and a recent survey (March 2010) of breast oncologists in the UK found that 97% of the 69 clinicians who responded did not offer CYP2D6 testing before commencing TAM treatment. Reasons cited were a lack of test availability (52%), insufficient evidence to recommend use (29%), cost (8%) or a combination of these reasons.
Rationale for the current review
There is growing anticipation among scientists, health-care providers and the general public that tests will soon be widely available to identify genetic differences and direct the prescribing of therapeutic agents and thus improve our ability to personalise therapies and subsequently improve clinical outcomes. 46
Tests that are used for genotyping should have both analytical and clinical validity. Analytical validity relates to the accuracy and reliability of assays and commercial tests to appropriately identify the genotype, whereas clinical validity relates to whether or not the test is an accurate measure of a biomarker that reflects the effect of the specific gene on the development of the disease and/or metabolism of the drug in question, i.e. can relevant outcomes be predicted by genotype? However, pragmatically of greatest importance is whether or not a test has clinical utility, i.e. can the information from analytical and clinical validity be used in clinical practice, to change drugs and/or dose, and have an impact on health outcomes as a result? Finally, tests that are used for genotyping in clinical practice will also need to show they are cost-effective compared with a treatment strategy in which no genotyping is conducted.
Despite a US Food and Drug Administration (FDA) expert advisory panel announcing that the CYP2D6 gene was considered to be a predictor of TAM efficacy, no consensus on whether testing should be recommended or considered an option has yet been reached. 47 In 2008, a review published by the Blue Cross and Blue Shield Association47 reported that there was a lack of clinical evidence (clinical validity and clinical utility) to support the routine use of CYP2D6 genotyping for patients being treated with TAM; this review did not consider cost-effectiveness.
It is important to note that in determining the cost-effectiveness of a pharmacogenetic test, it is not simply the additional cost and benefit of the test itself which need to be considered but also the impact of the test on subsequent choice of therapies and on patient care pathways and associated resource use. For example, it is likely that the number of women who are currently prescribed TAM and aromatase inhibitors would differ should a CYP2D6 test be offered routinely, and there would thus be implications for future pathways of care.
Thus, the aim of this current review is to consider the evidence for the clinical effectiveness and cost-effectiveness of CYP2D6 testing in relation to the use of TAM in women with ER+ breast cancer. The objectives of this review are listed in Box 1.
In patients treated with TAM:
-
Do women with breast cancer identified as EMs for CYP2D6 have similar or different clinical outcomes to those identified as PMs, IMs or UMs?
-
Is there a relationship between CYP2D6 status and endoxifen concentrations?
-
Are endoxifen concentrations related to clinical outcomes?
-
Do women with breast cancer who are identified as EMs for CYP2D6 have similar or different clinical outcomes with TAM compared with aromatase inhibitors?
-
What is the relative cost-effectiveness of CYP2D6 testing as a management option for women with breast cancer?
Chapter 3 Assessment of clinical effectiveness
Methods for reviewing effectiveness
Evidence for the clinical effectiveness of genotyping for CYP2D6 for the management of women with breast cancer was assessed by conducting a systematic review of published research evidence. The review was undertaken following the general principles published in the Centre for Reviews and Dissemination’s guidance for undertaking reviews in health care. 48
In order to ensure that adequate clinical input was obtained, an advisory panel comprising clinicians and experts in the field was established. The role of this panel was to comment on the draft report and answer specific clinical questions as the review progressed.
Identification of studies
The search aimed to identify all studies relating to the genotyping of CYP2D6 in the management of breast cancer, specifically related to TAM treatment. The following databases were searched on 19 June 2009: MEDLINE, EMBASE, The Cochrane Library (Cochrane Database of Systematic Reviews and Cochrane Controlled Trials Register), Web of Science (for the Science Citation Index and Conference Proceedings Citation Index) and the Centre for Reviews and Dissemination databases (Database of Abstracts of Reviews of Effects, NHS Economic Evaluation Database and Health Technology Assessment). Searches were not restricted by publication type. Because CYP2D6 genotyping is a relatively new area, and because the earliest study49 identified in the previous review of pharmacogenetics of TAM treatment was from 2003,47 searches were limited to the year 2000 and onwards. To assess the link between endoxifen plasma concentrations and clinical outcomes, a further search of MEDLINE was conducted on 21 July 2009, in which the inclusion criteria were extended to include studies considering the link between endoxifen concentrations and clinical outcomes, regardless of whether or not subjects had been genotyped for CYP2D6. The search strategies are listed in Appendix 1.
There was additional searching of the Human Genome Epidemiology Network Published Literature database, Proceedings of the American Society of Clinical Oncology, the San Antonio Breast Cancer Symposium and the European Society for Medical Oncology. Current research was identified from database citations through searching the National Research Register), the Current Controlled Trials register, the Medical Research Council Clinical Trials Register and the US National Institutes of Health website (ClinicalTrials.gov). Relevant reviews were hand searched in order to identify any further studies. Further studies that became known to the authors via relevant conferences or e-mail alerts from an automatically updated search of the Scopus database were also included as they became available, up to and including 17 March 2010.
Two reviewers (NF and RD) independently screened all titles and abstracts. Full-paper manuscripts of any titles/abstracts that were considered relevant by either reviewer were obtained. The relevance of each study was assessed (NF and RD) according to the inclusion and exclusion criteria listed in Box 2. Studies that did not meet the criteria were excluded and their bibliographic details were listed alongside reasons for their exclusion. Any discrepancies were resolved by consensus and, where necessary, a third reviewer was consulted.
Women with ER+ breast cancer treated with TAM and genotyped for CYP2D6
Any study design other than single case reports
One or more of the following relevant clinical outcomes:
-
OS, defined as hazard of death from any cause after any follow-up or the time to death from any cause expressed in months
-
DFS, however defined
-
local and distant recurrence, however defined
-
AEs, however defined
-
health-related quality of life, however defined
-
plasma concentrations of endoxifen
Studies of men with breast cancer
Editorials, opinions and reviews
Data extraction strategy
Data were extracted by one reviewer (NF) using a standardised data extraction form in Microsoft Word 2007 (Microsoft Corporation, Redmond, WA, USA) and checked independently by a second (JH). Disagreements were resolved by discussion.
Quality assessment strategy
As no universally accepted quality assessment criteria exist for assessing studies of pharmacogenetic testing, a tool based on elements of checklists developed to assess the methodological quality of prognostic factor studies50 and pharmacogenetic studies51 was used to assess specific issues considered important in terms of the reliability of such studies. Quality was independently assessed by two reviewers (NF and YD) and disagreements were resolved by discussion.
Methods of data synthesis
The results of the data extraction and quality assessment are summarised in structured tables and as a narrative description. Prespecified outcomes were tabulated and discussed within a descriptive synthesis. Additional relevant outcomes [breast cancer mortality and recurrence-free survival (RFS)] were also included.
Meta-analyses were planned in which binary outcomes were to be compared in terms of odds ratios, using either a fixed-effects or random-effects approach, depending on the degree of heterogeneity (to be assessed by visually inspecting the forest plots and by calculating the I2-statistic,52 which measures the proportion of variation across studies that is due to genuine differences rather than due to random error). In view of the controversy surrounding possible confounding from population stratification, and in keeping with the approach suggested in The HuGENet HuGE review handbook,53 in which studies differed in terms of the ethnicity of included patients, separate effect estimates were planned for each ethnic group. When studies differed in terms of their study design, sensitivity analyses were planned including only studies of the same study design. However, heterogeneity of the alleles genotyped, phenotypes derived, patients included and outcomes measured (see Results, below) precluded any planned meta-analyses.
Given the absence of either clinical utility studies or meta-analyses of the clinical validity data, attempts were made in an exploratory analysis to measure the clinical sensitivity and specificity of testing for particular alleles, as recommended by Flockhart et al. 54 in an American College of Medical Genetics statement. Data to calculate sensitivity and specificity were derived from the number of events reported in studies in the text/tables.
Results
Number of studies identified and included
The literature search yielded 1186 citations after duplicates had been removed. Of the titles and abstracts screened at screening stage one, 57 were assessed in detail at screening stage two. At this stage, 27 citations55–81 (reporting on 23 studies) were excluded (see Appendix 2), leaving 30 citations to be included (22 studies41,73,82–101 reporting on clinical outcomes by CYP2D6 status and eight studies49,73,89,102–106 reporting on endoxifen plasma concentrations by CYP2D6 status). No studies were found that met the criteria for clinical utility.
Following completion of the search in June 2009, a further nine citations were identified that met the inclusion criteria for the review (Figure 2): five studies82,107–110 reported clinical outcomes by CYP2D6 status, another111 presented additional data for one of these studies,109 two112,113 reported endoxifen plasma concentrations by CYP2D6 status, and one114 included data on clinical outcomes (which also included patients from a study88 previously identified by the literature search) and endoxifen plasma concentrations by CYP2D6 status, in two separate studies. 114
FIGURE 2.
Identification of eligible studies.
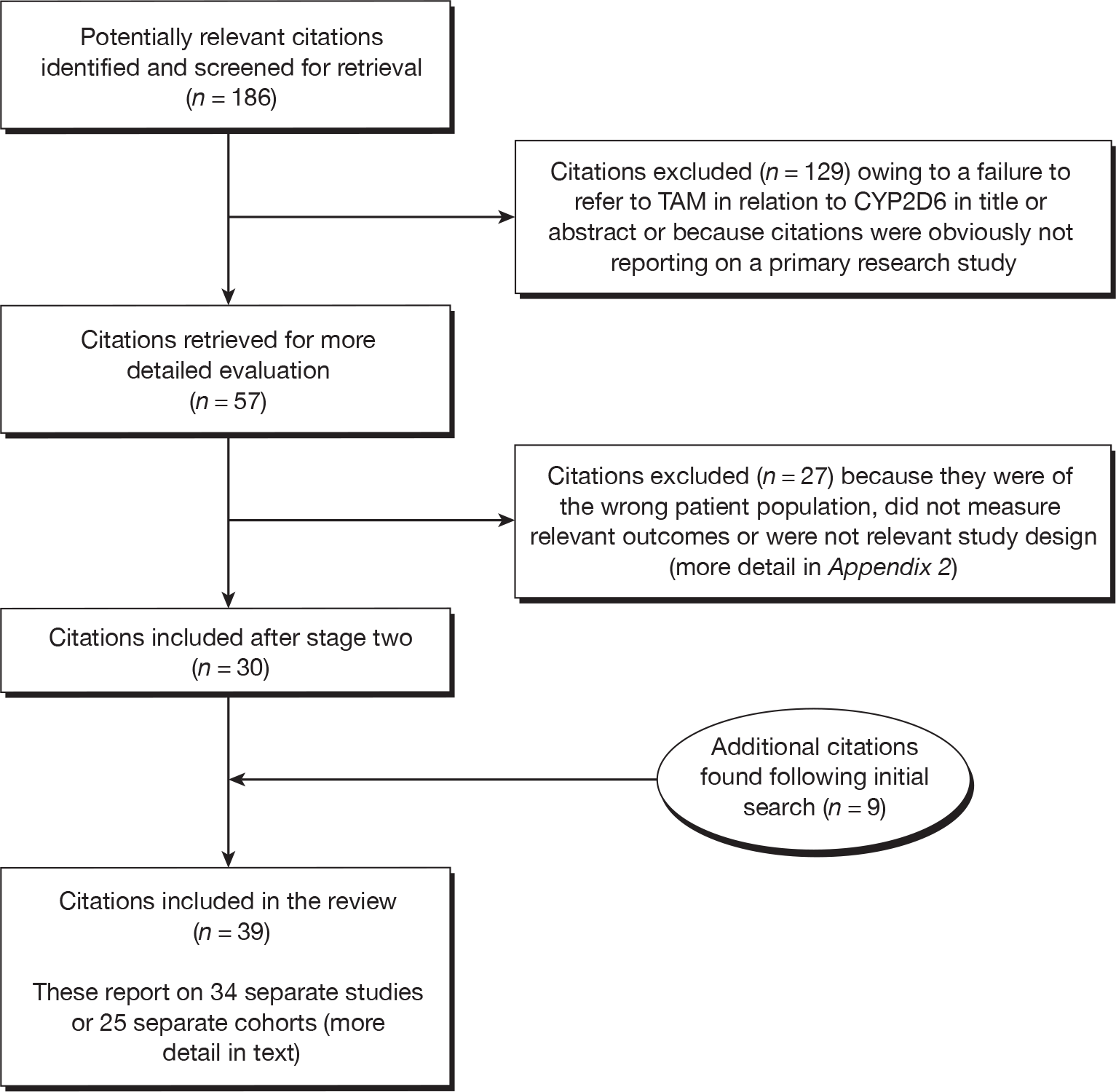
The separate search for the link between endoxifen concentrations and clinical outcomes yielded 4998 citations after duplicates had been removed. Of these, none met the criteria for inclusion into the review.
Two ongoing studies69,115 which are of some relevance to clinical utility but which do not meet the inclusion criteria have also since been identified. Both of these studies have been presented as conference posters. Details of these ongoing studies are provided in Appendix 2.
It is further apparent that many of the different studies included in the review are in fact reporting on the same cohort of patients but with a few subtle differences, such as using only a specific subgroup of patients, considering different genotypes, taking into account concomitant medication that inhibits CYP2D6 or analysing different alleles and genotype classifications. As these cohorts share the same patients and study characteristics, it is preferable to consider the quantity and quality of research available by cohort rather than individual study or paper. Thus, in total there are 25 cohorts (and where reference is made to the cohorts as a whole, rather than specific studies, the latest fully published study in the table is used to derive the name of the cohort, e.g. the cohort including the studies by Jin et al. ,105 Borges et al. ,102 Henry et al. 87,107 and Rae et al. 95 is referred to as the ‘Henry et al. cohort87’).
Quality assessment of included studies
Given that there are 25 distinct cohorts, the quality of each cohort as opposed to individual study is summarised here (see also Appendix 3). While the majority of the studies of these cohorts were published as full papers in peer-reviewed journals, it is important to note that six82,86,90,98,109,113 cohorts have reported findings only at conferences.
All but seven41,49,86,90,93,98,113 of the cohorts were explicit about both the source population from which the study population was derived, and the definition of the study population itself. Six82,86,90,98,109,113 cohorts had reported their studies only as abstracts, not full papers, and so space was limited to present this information. Compared with the typical sample sizes required to provide sufficient power to detect a range of typical genetic effect sizes for various minor allele frequencies,51 the majority of the samples in this review are small.
The majority (n = 12) of cohorts41,73,83,87,93,96,97,99–101,113,114 presented the rationale for the alleles tested for, with all but two49,112 providing rationale for CYP2D6 per se. All described the test used for genotyping and/or the specific procedure, with TaqMan or AmpliChip being the most commonly used in 1283,86,87,91–94,96,97,104,108,114 and six41,82,87,90,109,113 cohorts, respectively. Three cohorts91,97,104 reported quality control methods and seven cohorts87,91,96,97,104,114 reported on the Hardy–Weinberg equilibrium (two of these cohorts were reported in the same paper114).
In around half (n = 12) of the cohorts it was clear there were missing genotype data,41,82,83,87,96–101,108,112 reasons being provided in seven of these. 82,83,87,97,99,108,112 Only three of the cohorts, all abstracts,86,90,113 failed to present the number of patients contributing to each analysis.
Characteristics of included cohorts
The cohort characteristics are summarised in Table 6, where it is clearly evident that the size of the cohorts varied, with the smallest containing 12 subjects49 compared with the largest of 288082 [the International Tamoxifen Pharmacogenomics Consortium (ITPC) cohort that included patients from three published studies: Matthew Goetz, Mayo Clinic, Minnesota, USA, 2010, personal communication]. Generally, however, cohorts included between 60 and 300 patients, two other exceptions being two larger cohorts of 67799 and 1361108 patients, this last cohort itself including patients from two previously published cohorts. 83,96
| Cohort (and studies); number of patients genotyped for CYP2D6 | Study design; country of origin; length of patient follow-up | TAM dose; duration | Types of patients and key characteristics | Concomitant CYP2D6 inhibitors/chemotherapy accounted for? | Outcomes measured |
|---|---|---|---|---|---|
| Stearns et al. 2003;49 n = 12 | Prospective cohort; USA; 4 weeks | 20 mg/day; 4 weeks |
Women with breast cancer receiving TAM adjuvant therapy and taking paroxetine for hot flushes Postmenopausal: not reported ER+: not reported LN+: not reported Tumour size: not reported Metastatic disease: not reported |
Women were not permitted any concomitant medications known to inhibit CYP2D6 activity except for paroxetine 10 mg/day | Endoxifen concentrations |
|
Goetz et al. cohort: 83 Goetz et al. 2004116 (conference abstract), Goetz et al. 2005,84 n = 223 Goetz et al. 200783 (re-analysis); n = 180 Goetz et al. 2009110 (longer-term follow-up); n = 210 |
Retrospective analysis of samples from RCT, TAM-only arm); USA; mean 11.4 (range 5.7–14.1) years and median 14.5 years (longer-term follow-up) | 20 mg/day; 5 years |
Women with breast cancer receiving TAM adjuvant therapy (95% Caucasian) Postmenopausal: 100% ER+: 100% LN+: 36% Tumour ≥ 3 cm: 22% Metastatic disease: 0% |
Co-administration of CYP2D6 inhibitors was not accounted for in the initial analysis, hence the re-analysis No concomitant chemotherapy |
Efficacy AEs (Goetz et al. 200584 only) |
|
Henry et al. cohort: 87 Jin et al. 2005;105 n = 50 Borges et al. 2006;102 n = 158 Henry et al. 2009;87 n = 276 Henry et al. 2009;107 n = 276 Rae et al. 2009;95 n = 280 |
Prospective, observational, open-label, registry study (analysed retrospectively by Henry et al. 2009,87,107 Rae et al. 200995); USA; 12 months (4 months in Henry et al. 2009107) | 20 mg/day; 5 years (planned) |
Women with breast cancer starting TAM adjuvant therapy, extended in the 2009 analysis to include chemoprevention (91% ‘white’ in Jin et al. 2005105 and Borges et al. 2006102 ) Postmenopausal: 52% ER+ and/or PgR+: 100% LN+: not reported Tumour size: not reported Metastatic disease: 0% |
Co-administration of SSRIs was permitted and accounted for in the analysis No concomitant chemotherapy |
AEs (Henry et al. 2009,87 Henry et al. 2009107 and Rae et al. 200995 only) Endoxifen concentrations |
| Nowell et al. 2005;92 n = 337 (165 TAM and 172 no TAM) | Retrospective study of archived paraffin blocks; USA; median 5.4 years | Not reported |
Women with breast cancer receiving TAM adjuvant therapy and women with breast cancer receiving no TAM as controls (81% Caucasian, 19% African American) Postmenopausal: 59%a ER+: 67% LN+: 48% Tumour size: not reported Metastatic disease: 5% |
No information was available concerning concomitant medications Concomitant chemotherapy is allowed |
Efficacy |
| Wegman et al. 2005;100 n = 226 (112 TAM and 114 no TAM) | Retrospective analysis of frozen tumour tissues; Sweden; mean 10.7 (range 0.24–18.6) years | 40 mg/day; 2 years |
Women with breast cancer receiving TAM adjuvant therapy and women with breast cancer receiving no TAM as controls Postmenopausal: 100% ER+: 69% LN+: 89% Tumour > 2 cm: 61% Metastatic disease: not reported |
Adjuvant chemotherapy was allowed | Efficacy |
|
Gonzalez-Santiago et al. cohort: 86 Gonzalez-Santiago et al. 2006;85 n = 85 Gonzalez-Santiago et al. 2007;86 n = 84 |
Not reported; Spain; median 4.03 years in Gonzalez-Santiago et al. 200685 and mean 5.5 years in Gonzalez-Santiago et al. 200786 | Not reported |
Women with breast cancer receiving TAM adjuvant therapy Postmenopausal: not reported ER+ and/or PgR+: 99% LN+: 62%b Tumour size: not reported Metastatic disease: 0% |
Co-administration of CYP2D6 inhibitors was not accounted for in the initial analysis but was in the 2007 analysis | Efficacy |
| Gjerde et al. 2005103 (conference abstract), 2007;104 n = 151 | Prospective cohort; Norway; not reported | 20 mg/day; ≥ 80 days |
Women with breast cancer receiving TAM adjuvant therapy (100% Caucasian) Postmenopausal: not reported ER+ and/or PgR+: 100% LN+: not reported Tumour size: not reported Metastatic disease: not reported |
No information was available concerning concomitant medications (it is noted that SSRIs are not approved for hot flushes in Norway) | Endoxifen concentrations |
| Lim et al. 2006,89 2006106 (conference abstracts); Lim et al. 2007;73 n = 211 | Prospective cohort (PK) Korea; not reported | 20 mg/day; ≥ 8 weeks |
Women with early or metastatic breast cancer taking TAM in PK study (only patients with metastatic cancer were permitted in the efficacy study (100% South Korean) Postmenopausal: not reportedb ER+: not reported LN+: not reported Tumour size: not reported Metastatic disease: not reported |
Patients taking SSRIs were excluded | Endoxifen concentrations |
| Schroth et al. 2007;96 n = 486 (206 TAM and 280 no TAM) | Retrospective analysis of paraffin-embedded tumour samples from a single centre; Germany; median (range) 71 (4–227) months | Not reported |
Women with breast cancer receiving TAM adjuvant therapy and women with breast cancer receiving no TAM as controls Postmenopausal: 100% (TAM) ER+: 100% (TAM) LN+: 31% (TAM) Tumour ≥ 2 cm: 55% (TAM) Metastatic disease: 0% |
Information on SSRI use was incomplete No concomitant chemotherapy for patients taking TAM |
Efficacy |
| Wang et al. 2007;98 n = 58 | Not reported; USA; not reported | Not reported |
Women with breast cancer receiving TAM adjuvant therapy who were described as ‘ethnically diverse’ Postmenopausal: not reported ER+: not reported LN+: not reported Tumour size: not reported Metastatic disease: not reported |
Not reported | AEs |
| Wegman et al. 2007;99 n = 677 (of which 238 were randomised to either 2 or 5 years of adjuvant TAM) | Retrospective analysis of frozen tumour tissues; Sweden; mean (range) 7.3 (0.04–17.9) years (median 7.08) | 20 or 40 mg/day; 2–5 years |
Women with breast cancer receiving TAM adjuvant therapy Postmenopausal: 100% ER+: 100% LN+: 69% Tumour ≥ 2 cm: 72% Metastatic disease: 0% |
SSRIs were rarely used | Efficacy |
|
Kiyotani et al. cohort: 114 Kiyotani et al. 2008;88 n = 67 Kiyotani et al. 2010;114 n = 282 |
Retrospective analysis of samples of patients who were pathologically diagnosed and received surgical treatment; Japan; median (range) follow-up 8 years (1.6 to 21.6) years in Kiyotani et al. 200888 and median (range) follow-up 7.1 years (0.8 to 23.5 years) in Kiyotani et al. 2010114 | 20 mg/day; 5 years |
Women with breast cancer starting TAM adjuvant therapy (100% Japanese) Postmenopausal: 53%c ER+: 74%c LN+: 17%c Tumour > 2 cm: 38%c Metastatic disease: 0% |
Co-administration of SSRIs was not permitted No concomitant chemotherapy |
Efficacy |
| Madlensky et al. 2008;90 n = 1411 | Retrospective analysis of samples from RCT; USA; mean (range) 7.3 (6 to 11) years | Not reported |
Women with breast cancer receiving TAM adjuvant therapy Postmenopausal: not reportedb ER+: not reported LN+: not reported Tumour size: not reported Metastatic disease: 0% |
Not reported | AEs |
| Newman et al. 2008;91 n = 115 | Retrospective analysis of germline DNA samples from a single centre; UK; median 10 years | 20 mg/day; median > 4 years |
Women with familial breast cancer and BRCA1 or BRCA2 mutations receiving TAM adjuvant therapy (100% Caucasian) Postmenopausal: not reportedb ER+: 77% LN+: 39% Tumour > 3 cm: 23% Metastatic disease: 0% |
Four patients were co-prescribed drugs reported to inhibit CYP2D6, concomitant with TAM treatment | Efficacy |
| Xu et al. 2008;101 n = 293 (152 TAM and 141 no TAM) | Retrospective cohort; China; median (range) follow-up TAM = 63 (4 to 122) months and no TAM = 120 (4 to 193) months | 20 mg/day; 5 years |
Women with breast cancer receiving TAM adjuvant therapy and women with breast cancer receiving no TAM adjuvant therapy as controls Postmenopausal: 76%a (TAM) ER+: 82% (TAM) LN+: 7% (TAM) Tumour ≥ 2 cm: 27% (TAM) Metastatic disease: 0% |
Medication known to inhibit CYP2D6 was not permitted No concomitant chemotherapy for patients taking TAM |
Efficacy |
| Bonnanni et al. 2009;112 n = 75 (25 TAM, 25 ANA + TAM, 25 ANA) | Prospective randomised, open-label phase IIb trial; Italy; 12 months |
TAM: 10 mg/week; 1 year ANA + TAM: 10 mg/week; 1 year + ANA: 1 mg/day; 1 year ANA, no TAM, ANA: 10mg/week; 1 year |
Women with breast cancer receiving TAM Postmenopausal: not reported ER+ and/or PgR+: 100% LN+: not reported Tumour size: not reported Metastatic disease: not reported |
Not reported | Endoxifen concentrations |
| de Duenas et al. 2009;113 n = 115 | Prospective clinical study; Spain; not reported | Not reported |
Women with breast cancer receiving TAM adjuvant therapy Postmenopausal: not reported ER+: not reported LN+: not reported Tumour size: not reported Metastatic disease: 0% |
Concomitant use of CYP2D6 inhibitors not permitted | Endoxifen concentrations |
| dGoetz et al. 200982 on behalf of the ITPC; n = 2880 | Requested patient data from 12 ITPC project sites; not reported | Any dose permitted but the majority (2151/2880) given 20 mg/day |
Women with breast cancer receiving TAM adjuvant therapy Postmenopausal: 100% ER+: 100% LN+: 48% Tumour > 2 cm: 48% Metastatic disease: 0% |
Data on co-administration of CYP2D6 inhibitors was missing for 61% of patients | Efficacy |
| Okishiro et al. 2009;93 n = 173 | Retrospective cohort; Japan; median (range) 56 (8–109) months | 20 mg; median (range) 52 (9 to 60) months |
Women with breast cancer receiving TAM adjuvant therapy (100% Asian) Postmenopausal: 22% ER+: 91% LN+: not reported Tumour > 2 cm: 43% Metastatic disease: 0% |
Patients who received paroxetine concomitantly with TAM were excluded |
Efficacy AEs |
| Onitilo et al. 2009;94 n = 220 | Restrospective analysis of samples held in a population based repository, USA; up to 12.68 years | Not reported | All patients were Caucasian (and one patient was a man) | Not reported | AEs |
| Ramon et al. 2010;41 n = 91 | Retrospective analysis of samples from a single centre; Spain; mean (range) 108 (91 to 133) months | Not reported |
Women with breast cancer receiving TAM adjuvant therapy Postmenopausal: 40% ER+: 100% LN+: 50% Tumour: not reported Metastatic disease: 0% |
Information on SSRI use was incomplete Concomitant chemotherapy is allowed |
Efficacy AEs |
| Schroth et al. 2009;108 n = 1361e | Retrospective analysis of German (see Schroth et al. 200796) and US (see Goetz et al. cohort83) cohorts of patients treated with adjuvant TAM for early-stage breast cancer (retrospectively and prospectively collected); Germany and USA; median (range) 76.1 (2.1 to 243.6) months | Not reported |
Women with breast cancer receiving TAM adjuvant therapy Postmenopausal: 96% ER+: 97% LN+: 34% Tumour > 2 cm: 47% Metastatic disease: 0% |
Information on SSRI use was incomplete No concomitant chemotherapy |
Efficacy |
| Thompson et al. 2009;109 n = 618 | Retrospective analysis of samples from two separate sites; UK; median follow-up 9.4 and 4.9 years in each respective cohort | 20 mg/day; 5 years |
Women with breast cancer receiving TAM adjuvant therapy (100% Caucasian) Postmenopausal: 85% ER+: 100% LN+: 45% Tumour > 2 cm: 64% Metastatic disease: 0% |
The CYP2D6 metabolism status of patients was adjusted for co-medication Some patients received concomitant chemotherapy |
Efficacy |
| Toyama et al. 2009;97 n = 154 | Retrospective analysis of frozen tumours from single centre; Japan; median (range) 7.9 years (25 to 249 months) | 20 mg/day; 2–5 years (average 3.2 years) |
Women with ER+ breast cancer receiving TAM adjuvant therapy (no metastatic breast cancer) (100% Asian) Postmenopausal: not reported ER+: 96% LN+: 0% Tumour > 2 cm: 48% Metastatic disease: not reported |
Concomitant use of SSRIs was permitted (Tatsuya Toyama, Nagoya City University Hospital, Nagoya, Japan, 2010, personal communication) It is assumed that no patient received chemotherapy, as patients are ER+ and LN–, who, as stated, are usually recommended for hormone therapy alone |
Efficacy |
| Kiyotani et al. 2010;114 n = 98 | Not reported; Japan; not reported | 20 mg/day; not reported |
Women with breast cancer receiving TAM adjuvant therapy Postmenopausal: not reported ER+: not reported LN+: not reported Tumour size: not reported Metastatic disease: not reported |
Not reported |
Efficacy Endoxifen concentrations |
Unsurprisingly, all seven cohorts that measured endoxifen plasma concentrations were followed up prospectively. 49,73,87,104,112–114 All other studies were analysed retrospectively, using archived samples. In five cohorts,92,96,100,101,112 alongside data on patients receiving TAM, additional comparative data were provided for patients not receiving TAM.
Seven cohorts49,83,87,90,92,94,98 were solely from the USA, 1041,86,91,96,99,100,104,109,112,113 solely from Europe (including two from the UK91,109), six73,88,93,97,101,114 from Asia, one108 a combination of US and German patients and one82 from 12 unspecified ITPC project sites in the USA, Europe and Asia. The average duration of the studies varied considerably, from 4 weeks49 to 11.4 years,83 although, where average duration data were provided, all retrospective analyses were of at least 5 years’ duration.
Where cohorts provided data on TAM dose, this was 20 mg/day, the exceptions being two Swedish cohorts,99,100 in which average doses were higher, and the pharmacokinetic study of plasma concentrations by Bonanni et al. 112 in which they were lower. The majority of patients in these cohorts also received their dose for 5 years, the Swedish cohorts99,100 again being notable exceptions where 1–2 years’ dose duration was not uncommon and three other cohorts where it varied from a matter of weeks49,73,104 in pharmacokinetic studies to an average of 3 years. 97
Five cohorts83,87,96,108,114 were explicit in prohibiting adjuvant chemotherapy, while four41,92,100,109 were explicit in stating that this was permitted, concomitant chemotherapy in the other studies being uncertain. Data on CYP2D6 inhibitor use were also often either lacking or incomplete, with four93,101,113,114 cohorts explicitly prohibiting their use and five83,86,87,91,109 accounting for these in their analysis (noticeably, where there was more than one study for any given cohort this account was made in the more recent studies83,86,95,107).
It is also known that the study of endoxifen plasma concentrations by Lim et al. 73 also included patients with metastatic disease. No other study appears to include patients with metastatic breast cancer except Nowell et al. ,92 in which 5% of patients have metastatic disease and possibly also Wegman et al. 100 in which the inclusion criteria state that patients were required to have either histological verified lymph node metastases or a tumour diameter > 3 cm. It is also impossible to be sure in five of the pharmacokinetic studies49,102,104,112,114 and three of the efficacy studies whether or not the studies included patients with metastatic breast cancer. 94,98,100
Very few cohorts provided information on ethnicity, this being mentioned in just under half (n = 12) of the cohorts. 73,83,87,91–94,97,98,104,109,114 When this information was provided, Caucasians or Asians were represented in the study by at least 90% of all participants in all studies except Nowell et al. 92 in which there were 81% Caucasians and 19% African Americans, and Wang et al. 98 who simply stated that their population was ‘ethnically diverse’.
Not all cohorts provided data on the hormonal status of their patients. In six cohorts, all83,96,99,100,116 or nearly all (96%)108 of the women were known to be postmenopausal. In 10 others, there was a mix, although in only two instances41,93 was there a minority of postmenopausal women (40% and 22%, respectively). Similarly, not all cohorts provided data on hormone receptor status. Where these data were provided, all, or nearly all (> 90%), of the women were ER+ (or ER+ and/or progesterone receptor positive) in nine41,83,87,96,99,104,109,112,116 and five73,86,93,97,108 cohorts, respectively. In the remaining five cohorts91,92,100,101,114 in which this information was known, the proportion of ER+ women varied between 67% and 82%.
Less than half of the cohorts provided data on nodal status (n = 13)83,86,91,92,96,97,99–101,108,109,114,116 or tumour size (n = 12). 83,91,93,96,97,99–101,108,109,114,116 It was noticeable that the proportion of LN+ patients varied considerably across the cohorts, from no such patients97 to 89%,100 with a minority of patients being node positive in the majority of the studies. The proportion of patients with a tumour size ≥ 2 cm also appeared to vary significantly across the studies, from 27%101 to 72%,99 with a further two cohorts reporting only tumour sizes ≥ 3 cm in just under one-quarter of patients. 83,91
Fifteen cohorts measured efficacy. 41,82,83,86,91–93,96,97,99–101,108,109,114 Standard breast cancer study outcome measures, such as OS and DFS, were utilised; however, the definitions of these same outcomes often differed from study to study. In addition, depending on the cohort, the analysis was adjusted for in 14 cohorts. 82,83,86,91–93,96,99–101,104,108,109,114 Six cohorts41,83,87,90,94,98 reported on AEs and seven cohorts49,73,87,104,112–114 reported on endoxifen plasma concentrations. No cohort reported on health-related quality of life.
Derivation and classification of phenotypes
An important finding from our review was that currently there is no consensus about how CYP2D6 phenotypes should be derived from their genotypes and how they should thus be compared. In the current review, a large number of classifications and comparisons were utilised by different cohorts, and in some instances within cohorts. The different classifications used are summarised in Table 7, where it is evident that 10 cohorts82,83,87,90,91,96,104,108,112,113 used standard phenotypes (PM, IM, EM and UM), even though these were not always classified in the same manner from study to study, while others considered enzymatic function or simply compared genotypes. This heterogeneity makes comparisons across studies problematic, which is compounded further when one considers each cohort genotyped for different alleles, which may also be summarised as follows:
-
Number of cohorts that:
-
– genotyped for *4 only = 5
-
– genotyped for *4 plus at least one other allele = 16
-
– genotyped for *10 only = 3
-
– genotyped for *10 plus at least one other allele = 12
-
– genotyped for both *4 and *10 = 9
-
– used the AmpliChip = 6.
-
| Cohort (and studies); no. of patients genotyped for CYP2D6 | CYP2D6 alleles tested | Derivation of phenotype from CYP2D6 | Comparisons (in paper) | ‘Standardised comparisons’ |
|---|---|---|---|---|
| Stearns et al. 2003;49 n = 12 | *4, *6, *8 | No specific phenotypes defined in initial study | wt genotype vs vt genotype | Other |
|
Goetz et al. cohort: 83 Goetz et al. 2004116 (conference abstract), Goetz et al. 2005;84 n = 223 Goetz et al. 200783 (re-analysis); n = 180 Goetz et al. 2009110 (longer-term follow-up); n = 210 |
*4; *6 (*4 only in Goetz et al. 200783) Also genotyped for CYP3A5 in Goetz et al. 200584 |
No specific phenotypes defined in initial study In re-analysis: |
*4/*4 vs wt/wt + wt/*4 (no *6 variants were detected) | |
|
PM vs EM IM vs EM PM + IM vs EM |
PM vs EM + IM PM vs EM IM vs EM PM + IM vs EM |
|||
|
Henry et al. cohort 87 Jin et al. 2005;105 n = 50 Borges et al. 2006;102 n = 158 Henry et al. 2009;87 n = 276 Henry et al. 2009;107 n = 276 Rae et al. 2009;95 n = 280 |
*1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29 to *31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N Fewer alleles were tested in the earliest study by Jin et al. 2005:105 *3, *4, *5, *6 Also genotyped for ESR1 and ESR2 in Henry et al. 200987 |
No specific phenotypes defined in original study | vt/vt vs wt/vt vs wt/wt | Other |
|
In Borges et al. 2006102 PM = *3, *4, *5, *6/*3, *4, *5, *6 IM = *9, *10, *17, *29, *41, *41 × N/*3, *4, *5, *6 or *9, *10, *17, *29, *41, *41 × N/*9, *10, *17, *29, *41, *41 × N or *1, *2, *35/*3, *4, *5, *6 or *1, *2, *35/*9, *10, *17, *29, *41, *41 × N EM = *1, *1 × N, *2, *2 × N, *35/*1, *1 × N, *2, *2 × N, *35 |
PM vs IM vs EM |
PM vs EM PM vs IM IM vs EM |
||
| No specific phenotypes defined in Henry et al. 200987 | Other | |||
|
In Henry et al. 2009107 and Rae et al. 200995 Each CYP2D6 allele was assigned a value from 0 (for non-functional alleles) to 1 (for fully functional alleles) based on its relative activity for dextromethorphan O-demethylation. For each subject, the two allele scores were summed. Patients were classified as PM if score < 1, IM if 1 to < 2, and EM if ≥ 2, i.e. per allele: |
PM vs EM + IM | PM vs EM + IM | ||
|
In Rae et al. 2009 95 For concomitant medication that inhibits CYP2D6, two points were deducted from each patient’s CYP2D6 metabolism score for strong inhibitors, one point for moderate inhibitors and zero points for the weak inhibitor/no inhibitors |
||||
| Nowell et al. 2005;92 n = 337 (165 TAM and 172 no TAM) |
*3, *4, *6 Also genotyped for SULT1A1 and UGT2B15 |
No specific phenotypes defined | *4/*4 + wt/*4 vs wt/wt | PM + IM vs EM |
| Wegman et al. 2005;100 n = 226 (112 TAM and 114 no TAM) |
*4 Also genotyped for SULT1A1 |
No specific phenotypes defined | *4/*4 + wt/*4 vs wt/wt | PM + IM vs EM |
|
Gonzalez-Santiago et al. cohort: 86 Gonzalez-Santiago et al. 2006;85 n = 85 Gonzalez-Santiago et al. 2007;86 n = 84 |
*4 | No specific phenotypes defined | *4/*4 + wt/*4 vs wt/wt | PM + IM vs EM |
| Gjerde et al. 2005103 (conference abstract), Gjerde et al. 2007;104 n = 151 |
*3, *4, *5, *6, *2 × 2 Also genotyped for SULT1A1 |
PM: vt/vt IM = wt/vt EM = wt/wt UM = *2 × 2 |
PM vs IM vs EM vs UM | Other |
| Lim et al. 2006,89 2006106 (conference abstracts), Lim et al. 2007;73 n = 212 | *5, *10, *2 × N | No specific phenotypes defined | *10/*10 vs wt/wt vs *10/*10 and vt/vt vs wt/wt vs vt/vt | Other |
| Schroth et al. 2007;96 n = 486 (206 TAM and 280 no TAM) |
*4, *5, *10, *41 *3, *6, *7 and *8 were also genotyped for but excluded because PCR amplification rates were poor Also genotyped for CYP2C19, CYP3A5, CYP2B6 and CYP2C9 |
EM = wt/wt hetEM = 4,*5, *10 or *41/wt IM = *4,*5, *10 or *41/*10 or *41 PM = *4 or *5/*4 or *5 Decreased = hetEM + IM + PM |
Decreased vs EM PM vs EM IM + PM vs EM IM vs EM hetEM vs EM |
PM + IM vs EM PM vs EM IM vs EM |
| Wang et al. 2007;98 n = 58 | *4 | No specific phenotypes defined | 4/*4 + wt/*4 vs wt/wt | PM + IM vs EM |
| Wegman et al. 2007;99 n = 677 (of which 238 were randomised to either 2 or 5 years of adjuvant TAM) |
*4 Also genotyped for CYP3A5, SULT1A1 and UGT2B15 |
No specific phenotypes defined | 4/*4 + wt/*4 vs wt/wt | PM + IM vs EM |
|
Kiyotani et al. cohort: 114 Kiyotani et al. 2008;88 n = 67 Kiyotani et al. 2010;114 n = 282 |
*4, *5, *6, *10, *14, *18, *21, *41 In Kiyotani et al. 2010114 also genotyped for *36 |
No specific phenotypes defined in original study |
*10/*10 vs wt/wt wt/*10 vs wt/wt *10/*10 vs wt/wt + wt/*10 |
Other |
|
wt = *1 vt = *4, *5, *10, *14, *21, *36, *41 |
vt/vt vs wt/vt vs wt/wt | Other | ||
| Madlensky et al. 2008;90 n = 1411 | *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1xN, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N | Not reported how phenotypes are derived | EM vs hetEM vs IM vs PM vs UM | Other |
| Newman et al. 2008;91 n = 115 | *3, *4, *5, *41 |
PM1 = *3, *4, *5/*3, *4, *5 PM2 = concomitant use of a potent CYP2D6 inhibitor in wt/wt individuals or moderate inhibitor use in patients heterozygous for *3, *4, *5 or *41 IM = *3, *4, *5 or *41/*41 and no use of CYP2D6 inhibitors EM = wt, *3, *4, *5 or *41/wt and no use of CYP2D6 inhibitors |
PM1 vs EM PM2 vs EM PM1 + PM2 vs EM + IM |
PM vs EM PM vs IM |
| Xu et al. 2008;101 n = 293 (152 TAM and 141 no TAM) | *10 | No specific phenotypes defined | 10/*10 vs wt/wt + wt/*10 | Other |
| Bonnanni et al. 2009;112 n = 75 (25 TAM, 25 ANA + TAM, 25 ANA) | *2, *3, *4, *5, *6, *9, *29, *41A |
PM = *3, *4, *5, *6/*3, *4, *5, *6 EM = *2, *9, *29, *41/*2, *3, *4, *5, *6, *9, *29, *41 |
PM vs EM | PM vs EM |
| de Duenas et al. 2009;113 n = 115 | *1– *10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29 to *31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N | Not reported | PM vs EM | PM vs EM |
| Goetz et al. 200982 on behalf of the ITPC; n = 2880a |
Varies by ITPC site but all genotyped for *4 and the majority genotyped for *3, *5, *6, *10, *17 and *41 1168/2880 used AmpliChip, i.e. *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29 to *31, *35, *36, *40, *41, *1xN, *2xN, *4xN, *10xN, *17xN, *35xN, *41xN |
EM = wt/wt or wt/*10, *17, *41 IM = *10, *17, *41/*10, *17, *41 or wt/*3, *4, *5, *6 or *10, *17, *41/*3, *4, *5, *6 PM = *3, *4, *5, *6/*3, *4, *5, *6 |
PM vs EM | PM vs EM + IM |
| Okishiro et al. 2009;93 n = 173 |
*10 Also genotyped for CYP2C19 |
No specific phenotypes defined | *10/*10 vs wt/wt + wt/*10 | Other |
| Onitilo et al. 2009;94 n = 220 |
*4 Also genotyped for ESR1, ESR2 and CYP19 |
No specific phenotypes defined | *4/ *4 + wt/*4 vs wt/wt | PM + IM vs EM |
| Ramon et al. 2010;41 n = 91 | *1– *10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N | Three-group analysis:
|
1 vs 2 vs 3 A vs B |
Other |
| Schroth et al. 2009;108 n = 1361 | *3, *4, *5, *10, *41, wt × 2, *2 × 2 |
PM = 3, *4, *5/*3, *4, *5 IM = *10, *41/*3, *4, *5, *10, *41 hetEM = *1, *2, *35/*3, *4, *5, *10, *41 EM = *1, *2, *35/*1, *2, *35 UM = *1, *2, *35/*1 × 2, *2 × 2 Decreased = PM + IM + hetEM |
PM vs EM + UM hetEM + IM vs EM + UM decreased v EM + UM |
PM vs EM IM vs EM PM + IM vs EM |
| Thompson et al. 2009;109 n = 618 | *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N |
Normal = *1, *2, *35/*1, *2, *35 Decreased = any other genotype Re-assignment of phenotypes using only 4 alleles: |
Decreased vs normal (as determined by AmpliChip using many SNPs and then repeated just using four common alleles) | PM + IM vs EM |
| Toyama et al. 2009;97 n = 154 | *10 | No specific phenotypes defined | *10/*10 vs wt/*10 vs wt/wt | Other |
| Kiyotani et al. 2010;114 n = 98 | *4, *5, *6, *10, *14, *18, *21, *36, *41 |
wt = *1 vt = *4, *5, *10, *14, *21, *36, *41 |
vt/vt vs wt/vt vs wt/wt | Other |
For the purposes of this review, when considering study outcomes, ‘standardised comparisons’ are made in the tables and text in which alleles are simply considered to be wt (i.e. normal function), null (i.e. loss of function) or vt (any allele that is not wt, which includes null), and phenotypes are considered to be EM (wt/wt), IM (wt/vt) or PM (null/null) [this classification is the same as classification 1 for Test C in Table 2 (where null = *3, *4, *5 and vt = *10)]. It should be noted that for the purposes of these comparisons, UMs are likely to be classified as EMs. This is because not all genotyping methods are able to detect UMs and where studies have used methods that can, UMs appear to be classified with EMs, for example in Schroth et al. 108 Thus, the following comparisons can be considered:
-
PM versus EM
-
IM versus EM
-
PM + IM versus EM
-
PM versus EM + IM
-
Asian patients genotyped for the *10 allele
-
other comparisons that do not fit these categorisations.
Differences in cohort characteristics by genotype or phenotype
The cohort characteristics in Table 6 are for all patients in the cohort, regardless of CYP2D6 status. Only eight cohorts83,91,93,97,99–101,108 provided any of these data by genotype or phenotype, which is perhaps unsurprising given the retrospective nature of these studies (these data possibly not being originally collected).
It was noticeable in the Goetz et al. cohort83 that, compared with the cohort as a whole genotyped by paraffin tissue, more patients in the *4/*4 group had a tumour size ≥ 3 cm (38% compared with 22%) and were LN+ (69% compared with 38%). 84 Patients with this genotype were also more likely to be older (median age 73 years compared with 68 years) and have had a mastectomy (92% compared with 83%). It should be noted, however, that the number of patients with the *4/*4 genotype was small (n = 13) compared with those with other genotypes (n = 177).
Similarly, in Newman et al. ,91 compared with patients with other phenotypes, patients with the PM phenotype had a larger tumour size (42% compared with 21% had tumour size > 3 cm) and were more likely to have one or more positive lymph nodes at diagnosis (55% compared with 39%). These patients were also more likely to have had a mastectomy (67% compared with 49%) and be ER+ (92% vs 76%), but the median age in both groups of patients was similar (43 years compared with 45 years). Again, the number of patients with the PM phenotype was very small (n = 12) compared with those with other genotypes (n = 103).
Wegman et al. 100 also presented demographic data but these were for the group of patients as a whole, i.e. including both the patients who received TAM and the control group who did not. For each of the groups compared, *4/*4, wt/*4 and wt/wt, the proportion of patients who were node negative but with a tumour > 3 cm was similar in each group (11%, 9% and 12%, respectively). However, some differences were evident in terms of patients who were node positive with a tumour > 2 cm (33%, 58% and 44%, respectively) and ER+ (44%, 22% and 31%, respectively). Once again, the number of patients with the *4/*4 genotype was extremely small (n = 9) compared with those with other genotypes (n = 217).
In their later (separate) study, in which all patients received TAM, Wegman et al. 99 reported no real differences in tumour size or nodal status (tumour ≥ 2 cm being 71% for *4/*4, 75% for wt/*4 and 71% for wt/wt; patients with node > 0 cm being 71%, 69% and 69%, respectively). As with the previously mentioned studies, the number of patients with the *4/*4 genotype was small (n = 34) compared with those with other genotypes (n = 643).
The only study to compare patients with decreased activity as a whole (n = 716, of whom 79 were PM) with EMs (n = 609) was by Schroth et al.,108 who reported no real differences in tumour size > 2 cm (48% compared with 47%), lymph nodal status (36% compared with 31%) or age at diagnosis (median age was 66 years in both groups) between groups. No real differences were evident for any other cohort characteristics presented by the authors.
Xu et al. 101 reported differences in cohort characteristics in Asian women across genotype groups, namely *10/*10, wt/*10 and wt/wt. While lymph nodal status appeared similar across the groups (6%, 8% and 7%, respectively, were node positive), more patients with the *10/*10 genotype appeared to have larger tumours (32% > 2 cm compared with 23% and 21%, respectively) and fewer were ER+ (86% compared with 92% and 96%, respectively). In this cohort, there were almost as many women with the *10/*10 genotype (n = 72) as with other genotypes (n = 80).
Toyama et al. 97 reported few differences between the three groups of Asian female patients, with the notable exception that more women with the wt/wt genotype had a tumour > 2 cm (59% compared with 44% in each of the other genotype groups). Data were not presented on nodal status. Age differed only slightly (median 56 years in the*10/*10 group compared with 60 years in the other two groups). The vast majority (> 96%) of patients were ER+ in all groups. The proportion of patients with the *10/*10 genotype was relatively small (n = 28) compared with other genotypes (n = 126).
In Okishiro et al. ,93 fewer differences were apparent when patients with the *10/*10 genotype were compared with patients with all other genotypes, the number of patients with tumour size > 2 cm and LN+ in each group being comparable (29% vs 28% and 43% vs 45%, respectively) and median age being similar (47 years compared with 46 years). However, it was noticeable that there were more patients with the *10/*10 genotype with ER+ breast cancer (92% compared with 60%). The number of patients with the *10/*10 genotype was again relatively small (n = 40) compared with other genotypes (n = 133).
While other studies neither presented cohort characteristics by genotype/phenotype nor reported any differences between groups, seven other cohorts did adjust for prognostic factors. 82,86,92,96,104,109,114
Efficacy
The efficacy of TAM treatment was considered by genotype/phenotype, using the following comparisons as described above (see Derivation and classification of phenotypes):
-
PM vs EM
-
IM vs EM
-
PM + IM vs EM
-
PM vs EM + IM
-
Asian patients genotyped for the *10 allele
-
other comparisons that do not fit these categorisations.
Unfortunately, not all clinical end points measured by the cohorts were defined. Where end points were defined, it was apparent that different cohorts used different definitions for the same end points. Given these inconsistencies and/or a lack of information to correctly classify a clinical end point [e.g. information on censoring would enable recurrence outcomes to be classified as DFS, RFS or time to recurrence (TTR)], the efficacy outcomes are presented below in terms of OS, breast cancer mortality (i.e. mortality attributed only to breast cancer and not from any cause as with OS) and outcomes such as DFS, RFS and TTR, which can be considered relating to relapse/recurrence.
Overall survival is considered the least ambiguous and most clinically relevant clinical end point. 117 Seven cohorts82,83,91,92,96,97,108 examined OS by CYP2D6 status. One108 of these studies also includes 350 patients from two of the other included cohorts,83,96 and the large ITPC cohort82 also contains data from three published data sets (Matthew Goetz, personal communication). Two cohorts considered breast cancer mortality97,101 and 13 cohorts41,82,83,86,91,93,96,97,99,100,108,109,114 reported outcomes relevant to relapse/recurrence.
Poor metaboliser versus extensive metaboliser
Five studies83,91,96,108,110 from four cohorts83,91,96,108 compared PMs with EMs. One cohort83 genotyped for the *4 PM allele only, while the other three all genotyped for three or more alleles, all including *4 and *5. 91,96,108 CYP2D6 inhibitors were accounted for in two cohorts83,91 by altering an EM patient’s phenotype to PM when a potent CYP2D6-inhibiting drug was taken concomitantly and an IM patient’s phenotype to PM when a moderate CYP2D6 inhibitor was used.
Neither of the two cohorts reporting on OS83,91 reported a significant difference between PMs and EMs. However, three cohorts83,91,108 reported improved outcomes in terms of relapse/recurrence for EMs compared with PMs.
Overall survival
Two cohorts83,91 considered differences in OS between PMs and EMs. One of these cohorts83 genotyped for the *4 allele, whereas the other genotyped for *3, *4 and *5. 91 Both of these cohorts83,91 appeared similar in terms of the tumour size and nodal status of patients; in terms of other cohort characteristics, there were differences in the proportions of postmenopausal women and women with ER+ breast cancer. While in some regards these cohorts appeared similar, in one cohort83 it was known that, compared with all patients, a greater proportion of patients with the PM phenotype had a tumour size ≥ 3 cm and were LN+.
In these two cohorts,83,91 OS appeared to be improved in EMs compared with PMs, with unadjusted hazard ratios (HRs) of between 1.9 and 3.5,83,91 although confidence intervals (CIs) were wide and no significant differences in OS were reported (Table 8). It is unclear whether or not the unadjusted findings were influenced by differences in terms of cohort characteristics in the phenotype groups [for comparisons of other phenotype groups (PM + IM vs EM and PM vs EM + IM) in these cohorts, these HRs were adjusted for tumour size83 and/or nodal status83,91]. Furthermore, while concomitant use of CYP2D6 inhibitors was taken into consideration in both cohorts, adjuvant chemotherapy use was unknown (in the Goetz et al. cohort,83 it was prohibited).
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
|
Goetz et al. cohort: 83 Goetz et al. 200783 |
*4 | Time from registration to death from any cause |
Cox HR (unadjusted) HR 2.00; 95% CI 0.92 to 4.17; p = 0.077 |
| Newman et al. 200891 | *3, *4, *5, *41 | Not reported |
Cox HR (unadjusted) PM1 vs EM: HR 3.5; 95% CI 0.8 to 15.4; p = 0.079 PM2 vs EM: HR 3.4; 95% CI 0.77 to 14.9; p = 0.084 |
Relapse/recurrence
Two studies83,110 from the Goetz et al. cohort83 compared DFS, and one study83 compared RFS for PMs compared with EMs. This cohort genotyped for *4 and accounted for CYP2D6 inhibitors in one study83 by altering an EM patient’s phenotype to PM when a potent inhibitor was used and an IM patient’s phenotype to PM when a moderate inhibitor was used. The Goetz et al. cohort83 and two others91,96 also measured TTR. These other two cohorts genotyped for a wider range of alleles (*3, *4, *591 and *4, *5, *10, *4196). There were also variations in the proportion of postmenopausal women and those with ER+ breast cancer across the three cohorts, although the majority (> 75%) of patients in all cohorts were ER+. The proportion of LN+ patients across the cohorts was also similar and, in two cohorts83,91 the proportion of patients with a tumour ≥ 3 cm was also similar (it was not possible to compare with the other cohort108 as this reported tumour size > 2 cm). In two of these cohorts,83,91 there were differences between PMs and EMs in terms of tumour size and nodal size.
In the Goetz et al. cohort,83 both DFS and RFS were reported to be significantly worse for PMs (Table 9). All three cohorts83,91,108 that measured TTR also reported significant differences between PMs and EMs, although this applied only when patients whose phenotype was modified to PM were included in the group of PMs in one cohort. 91
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
|
Goetz et al. cohort: 83 |
*4 | DFS: time from randomisation to documentation of the first of the following events: any recurrence (local, regional or distant) of breast cancer, a contralateral breast cancer, a second primary cancer or death from any cause |
Cox HR (unadjusted), DFS: HR 2.44; 95% CI 1.27 to 4.69; p = 0.008 Cox HR (adjusted for tumour size and nodal status), DFS: HR 2.00; p = 0.02 (longer-term follow-up) |
| RFS: time from randomisation to documentation of the first of the following events: any recurrence (local, regional or distant) of breast cancer, a contralateral breast cancer or death |
Cox HR (unadjusted), RFS: HR 2.69; 95% CI 1.34 to 5.37; p = 0.005 |
||
| TTR: the time from randomisation to documentation of a breast event, where a breast event is any recurrence (local, regional or distant) of breast cancer or the documentation of contralateral breast cancer (including ductal carcinoma in situ) |
Cox HR (unadjusted), TTR: HR 3.20; 95% CI 1.37 to 7.55; p = 0.007 Cox HR (adjusted for tumour size and nodal status), TTR: HR 4.00; p = 0.01 (longer-term follow-up) |
||
| Newman et al. 200891 | *3, *4, *5, *41 | TTR: time to tumour recurrence with contralateral, ipsilateral or metastatic disease |
Cox HR (unadjusted), TTR: PM1 vs EM: HR 2.9; 95% CI 0.9 to 9.4; p = 0.076 PM2 vs EM : HR 3.2; 95% CI 0.98 to 10.4; p = 0.044 |
| Schroth et al. 2009108 | *3, *4, *5, *10, *41, wt × 2, *2 × 2 | TTR: time from diagnosis or randomisation to documentation of a breast event, any local, regional or distant recurrence of breast cancer or a contralateral breast cancer |
Cox HR (unadjusted): HR 2.12; 95% CI 1.28 to 3.50; p = 0.003 Cox HR (adjusted for tumour size, nodal status and histological grade, and stratified by menopause status and mode of patient recruitment): HR 1.90; 95% CI 1.10 to 3.28; p = 0.02 |
Intermediate metaboliser versus extensive metaboliser
Two studies83,110 from one cohort83 genotyped for *4 only when IMs were considered to be wt/*4. This cohort also accounted for CYP2D6 inhibitors by classifying patients who were wt/wt (but taking a moderate CYP2D6 inhibitor) to be IMs.
There was no evidence of a difference in OS or relapse/recurrence between IMs and EMs in this cohort. 83
Overall survival
One cohort83 considered differences in OS between IMs and EMs (Table 10). This cohort accounted for CYP2D6 inhibitors, but it was unclear whether or not adjuvant chemotherapy was permitted. No difference in OS or relapse/recurrence between IMs and EMs was reported. 83
Relapse/recurrence
Only one cohort83 reported on DFS, RFS and TTR between IMs and EMs (Table 11). This cohort adjusted for CYP2D6 inhibitors by altering phenotypes accordingly and also prohibited the use of adjuvant chemotherapy. No difference in DFS, RFS or TTR between IMs and EMs was reported.
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
|
Goetz et al. cohort:83 |
*4 | DFS: time from randomisation to documentation of the first of the following events: any recurrence (local, regional or distant) of breast cancer, a contralateral breast cancer, a second primary cancer or death from any cause |
Cox HR (unadjusted), DFS: HR 1.52; 95% CI 0.93 to 2.49; p = 0.097 Cox HR (adjusted for tumour size and nodal status), DFS: HR 1.40; p = 0.10 (longer-term follow-up) |
| RFS: time from randomisation to documentation of the first of the following events: any recurrence (local, regional or distant) of breast cancer, a contralateral breast cancer or death |
Cox HR (unadjusted), RFS: HR 1.63; 95% CI 0.95 to 2.78; p = 0.075 |
||
| TTR: the time from randomisation to documentation of a breast event, where a breast event is any recurrence (local, regional or distant) of breast cancer or the documentation of contralateral breast cancer (including ductal carcinoma in situ) |
Cox HR (unadjusted), TTR: HR 1.40; 95% CI 0.68 to 3.05; p = 0.337 Cox HR (adjusted for tumour size and nodal status), TTR: HR 1.80; p = 0.08 (longer-term follow-up) |
Poor metaboliser plus intermediate metaboliser versus extensive metaboliser
Nine studies83,85,86,92,96,99,100,108,109 from eight cohorts83,86,92,96,99,100,108,109 compared PMs combined with IMs with EMs. Four of these cohorts83,86,92,100 genotyped only for (or at least utilised in the analysis) *4, whereas the other four96,99,108,109 genotyped for a wider range of alleles. Three83,86,109 of these cohorts considered the impact of CYP2D6 inhibitors on phenotype.
Four cohorts83,92,96,108 reported on OS but not one reported significant differences between the PM + IM or EM groups. Seven cohorts83,86,92,96,99,100,108 assessed relapse/recurrence, four83,86,96,108 of these reporting significantly worse outcomes for the PM + IM group. An important finding was that, in one of these cohorts,104 the significant differences were found only when using the AmpliChip to genotype for an extensive number of alleles and not when only four common alleles were genotyped. In the three cohorts in which there were no significant differences, the data suggested, if anything, that the PM + IM group had better outcomes than EMs. Reasons for these contradictory findings are unknown but may be attributable to cohort characteristics.
Overall survival
Four cohorts83,92,96,108 compared OS between PM + IMs and EMs. Three83,96,108 of these cohorts appeared relatively similar in terms of the proportions of postmenopausal women with ER+ breast cancer and nodal status. It was not possible to compare tumour size across all three cohorts, as one cohort83 reported > 3 cm and the other two96,108 > 2 cm (where the cohorts did appear similar). It should be noted that patients from two83,96 of these cohorts were actually included in the other. 108 The fourth cohort92 appeared to differ from these other cohorts in a number of ways: nearly one-fifth (19%) of all patients were African American; only three-fifths (59%) of women were postmenopausal and two-thirds (67%) had ER+ breast cancer. Over twice as many women were LN+ [four-fifths (79%) compared with no more than one-third in the other cohorts (31–36%)] and 5% had stage IV breast cancer (metastatic disease) compared with no patients in the other cohorts. Finally, this cohort permitted adjuvant chemotherapy, unlike the other three cohorts. However, these cohort characteristics do include data on patients in a control group who did not receive anti-oestrogen therapy.
While three83,96,108 of the cohorts (all with similar cohort characteristics) presented a HR suggesting a slight increase in OS and the other92 suggested an improved outcome for PMs; none of the differences was statistically significant (Table 12). This cohort92 also compared HRs between patients taking TAM and those who were not; the HRs were similar in both groups, suggesting that genotype is not associated with disease and *4 is not associated with response in this TAM-treated cohort.
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
| Goetz et al. cohort,83 Goetz et al. 200783 | *4 | Time from registration to death from any cause |
Cox HR (adjusted for tumour size and nodal status): HR 1.34; 95% CI 0.83 to 2.16; p = 0.223 |
| Nowell et al. 200592 |
*3, *4, *6 Only *4 used for the analysis |
Time from diagnosis to death or last contact |
Cox HR (adjusted for the age, stage with nodal status at diagnosis, race, ER status and PgR status): TAM: HR 0.77; 95% CI 0.32 to 1.81; p = 0.51 No TAM: HR 0.79; 95% CI 0.42 to 1.26; p = 0.26 |
| Schroth et al. 200796 |
*4, *5, *10, *41 *3, *6, *7 and *8 were also genotyped for but excluded because PCR amplification rates were poor |
Time from surgery to death from any cause |
Cox HR (adjusted for tumour size and nodal status): HR 1.73 95% CI 0.88 to 3.41; p = 0.11 |
| Schroth et al. 2009108 | *3, *4, *5, *10, *41, wt × 2, *2 × 2 | Time from registration to death from any cause |
Cox HR (unadjusted): HR 1.13; 95% CI 0.88 to 1.47; p = 0.34 Cox HR (adjusted for tumour size, nodal status and histological grade and stratified by menopause status and mode of patient recruitment): HR 1.15; 95% CI 0.88 to 1.51; p = 0.32 |
Relapse/recurrence
Eight cohorts83,86,92,96,99,100,108,109 comparing PM + IM with EM reported on a number of relapse/recurrence outcomes. Ostensibly, two cohorts83,108 reported on DFS, two96,108 on event-free survival (EFS), two92,109 on RFS, one100 on distant RFS, one on recurrence-free time (RFT)108 and one on TTR. 108 However, it is apparent from Table 13 that definitions of the same end point varied from study to study and, moreover, in some instances, definitions of one end point in one cohort seemed to match those of another end point in another. For example, the definitions of EFS and RFT used in Schroth et al. 96 appear similar to those of DFS and RFS (respectively) used in the Goetz et al. cohort. 83
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
| Nowell et al. 200592 |
*3, *4, *6 Only *4 used for the analysis |
RFS: not reported |
Cox HR (adjusted for the age, stage with nodal status at diagnosis, race, ER status and PgR status), RFS: TAM: HR 0.67; 95% CI 0.33 to 1.35; p = 0.19 No TAM: HR 0.69; 95% CI 0.40 to 1.18; p = 0.19 |
|
Goetz et al. cohort:83 Goetz et al. 200783 |
*4 | DFS: time from randomisation to documentation of the first of the following events: any recurrence (local, regional or distant) of breast cancer, a contralateral breast cancer, a second primary cancer or death from any cause |
Cox HR (adjusted for tumour size and nodal status), DFS: HR 1.60; 95% CI 1.06 to 2.43; p = 0.027 |
| RFS: time from randomisation to documentation of the first of the following events: any recurrence (local, regional or distant) of breast cancer, a contralateral breast cancer or death |
Cox HR (adjusted for tumour size and nodal status), RFS: HR 1.74; 95% CI 1.10 to 2.74; p = 0.017 |
||
| TTR: the time from randomisation to documentation of a breast event, where a breast event is any recurrence (local, regional or distant) of breast cancer or the documentation of contralateral breast cancer (including ductal carcinoma in situ) |
Cox HR (adjusted for tumour size and nodal status), TTR: TTR: HR 1.91; 95% CI 1.05 to 3.45; p = 0.034 |
||
| Wegman et al. 2005100 | *4 | Distant RFS: not reported |
Distant recurrence rate ratio adjusted for age, tumour size, LN status), ER+ patients, TAM vs no TAM: wt/wt: RR 0.91; 95% CI 0.53 to 1.57; p = 0.75 *4/*4 + wt/*4: RR 0.28; 95% CI 0.11 to 0.74; p = 0.008 |
| Gonzalez-Santiago et al. cohort: 86 | *4 | ||
| Gonzalez-Santiago et al. 200685 | Relapse: not reported |
Cox HR (adjusted for unspecified variables), relapse: HR 3.48; 95% CI 1.1 to 10.7; p = 0.029 |
|
| Gonzalez-Santiago et al. 200786 | Recurrence: not reported |
Cox HR (adjusted for disease stage), RFS: HR 2.82, 95% CI 1.0 to 7.9; p = 0.049 |
|
| Schroth et al. 200796 |
*4, *5, *10, *41 *3, *6, *7 and *8 were also genotyped for but excluded because PCR amplification rates were poor |
EFS: time from surgery to the occurrence of either local or distant recurrence, contralateral breast cancer or death from any cause |
Cox HR (adjusted for tumour size and nodal status), EFS: HR 1.89; 95% CI 1.10 to 3.25; p = 0.02 |
| RFT: time from surgery to the occurrence of a breast event (i.e., local or distant recurrence or contralateral breast cancer) |
Cox HR (adjusted for tumour size and nodal status), RFT: HR 2.24; 95% CI 1.16 to 4.33; p = 0.02 Differences in RFT were not observed in the control group |
||
| Wegman et al. 200799 | *4 | RFS: not reported |
Cox HR (unadjusted), RFS: 2 years of TAM (n = 103): HR 0.87; 95% CI 0.38 to 1.97; p = 0.74 5 years of TAM (n = 108): HR 0.33; 95% CI 0.08 to 1.43; p = 0.14 Cox HR (adjusted for TAM duration, tumour stage, tumour size and LN status): ‘No differences could be seen’ |
| Schroth et al. 2009108 | *3, *4, *5, *10, *41, wt × 2, *2 × 2 |
DFS: time to first occurrence of a breast event, a second non-breast primary cancer or death from any cause EFS: time to the first occurrence of a breast event or death from any cause TTR: time from diagnosis or randomisation to documentation of a breast event, any local, regional or distant recurrence of breast cancer or a contralateral breast cancer |
Cox HR (unadjusted), DFS: HR 1.31; 95% CI 1.06 to 1.61; p = 0.02 Cox HR (adjusted for tumour size, nodal status and histological grade and stratified by menopause status and mode of patient recruitment), DFS: HR 1.29; 95% CI 1.03 to 1.61; p = 0.02 Cox HR (unadjusted), EFS: HR 1.35; 95% CI 1.08 to 1.68; p = 0.07 Cox HR (adjusted for tumour size, nodal status and histological grade and stratified by menopause status and mode of patient recruitment), EFS: HR 1.33; 95% CI 1.06 to 1.68; p = 0.01 Cox HR (unadjusted), TTR: HR 1.57; 95% CI 1.18 to 2.08; p = 0.02 |
| Thompson et al. 2009;109 n = 618 |
Comprehensive testing: *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N Limited testing: *4,*5,*10,*41 |
RFS: locoregional recurrence, DCIS, distant metastases, contralateral DCIS or death due to breast cancer |
Cox HR (adjusted for tumour size and nodal status), RFS: All women Comprehensive testing:a HR 1.52; 95% CI 0.98 to 2.36; p = 0.06 Limited testing:a HR 1.03; 95% CI 0.67 to 1.58; p = 0.88 Postmenopausal Comprehensive testing:a HR 1.96; 95% CI 1.05 to 3.66; p = 0.04 Limited testing:a HR 1.26; 95% CI 0.74 to 2.16; p = 0.88 Taking into account adherence:b HR 3.02; 95% CI 1.07 to 8.47; p = 0.04 |
The majority of these studies include a majority (> 85%) of postmenopausal women and the majority include a majority (> 97%) of women with ER+ breast cancer, the exceptions being Nowell et al. ,92 in which a significant minority of patients in this cohort were neither postmenopausal (41%) nor had ER+ breast cancer (33%) and the Wegman et al. 100 study, in which 69% were ER+ (although analysis in this study was confined to those who were ER+). Alongside two other cohorts,83,86 Wegman et al. 100 also had a majority (> 62%) of patients who were LN+, unlike the other cohorts, in which these were a minority (31–45%). Another noticeable difference about the Wegman et al. cohort100 was that patients received 40 mg/day TAM for 2 years instead of the standard 20 mg/day for 5 years, and patients were permitted adjuvant chemotherapy. One other cohort99 also reported a dose that was different to the standard (20 or 40 mg/day for 2 or 5 years) and two other cohorts92,109 also permitted adjuvant chemotherapy. It should be noted that three cohorts86,96,108 did not provide data on drug dose/duration and two86,99 did not present information about adjuvant chemotherapy. Three cohorts83,86,109 explicitly stated that they adjusted for CYP2D6 inhibitors in derivation of phenotype. Comparison of tumour size was possible only for those cohorts presenting data on tumour size ≥ 2 cm, where the proportion of women varied from 47%108 to 72%. 99
Five of the cohorts83,86,96,108,109 reported statistically significantly more favourable outcomes for EMs than for PM + IMs in terms of relapse/recurrence (see Table 13), although the magnitude of the difference in terms of the HR was modest (between 1.6 and 3.5). Interestingly, a significant difference for RFS was found only in Thompson et al. 109 when comprehensive testing (i.e. genotyping using the AmpliChip) was conducted instead of just testing for a limited number of common alleles. This study was also able to consider the impact of adherence by reclassifying those with poor adherence as PMs. The difference between the two groups remained statistically significant, with the HR increasing to 3. Three cohorts92,99,100 suggested PMs to have equal or improved outcomes in terms of RFS and distant RFS, although the findings were not statistically significant in any of the cohorts. As noted above, in two of these cohorts99,100 the dose of TAM was known to be greater and duration of treatment shorter in some if not all of these patients than is standard, and a greater proportion of patients were also LN+ than in the other cohorts (although the authors did adjust for these as well as presenting unadjusted results).
Three cohorts92,96,100 also included a control group of patients not taking TAM (but receiving adjuvant chemotherapy and/or radiotherapy or no drug treatment). Wegman et al. 100 reported that patients with the *4 allele taking TAM still had a better distant RFS than those in the control group (relative risk 0.28, 95% CI 0.11 to 0.74; p = 0.0089). In Nowell et al. ,92 the HRs in the control group were similar to those in the TAM group, whereas, in Schroth et al. ,96 it was stated that significant differences between PM + IMs and EMs found in the TAM group were not found in the control group.
Poor metaboliser versus extensive metaboliser plus intermediate metaboliser
Three cohorts82,83,91 examined differences between PMs and EM + IMs, including the large ITPC study. 82 Different alleles were genotyped for in each cohort (*4,83 *3, *4 and *591) and different alleles at different study sites in the ITPC study. 82 Only one of the studies91 adjusted for CYP2D6 inhibitors in deriving the phenotype.
There was no evidence of a difference in OS or relapse/recurrence between PMs and EMs + IMs in any of the three cohorts. 82,83,91
Overall survival
Three cohorts82,83,91 reported on OS (Table 14). Two of these cohorts included only postmenopausal women with ER+ breast cancer,82,83 the other cohort91 included women of any menopausal status and in which 77% had ER+ breast cancer. Differences in cohort characteristics between PMs and the other phenotypes were noted in two83,91 of these cohorts, where a greater proportion of PMs had tumour size ≥ 3 cm and were LN+.
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
|
Goetz et al. cohort:83 Goetz et al. 200584 |
*4, *6 Only *4 used for the analysis |
Time from registration to death from any cause |
Cox HR (unadjusted): HR 1.73; 95% CI 0.79 to 3.76; p = 0.169 Cox HR (adjusted for tumour size and nodal status): HR 1.12; 95% CI 0.50 to 2.50; p = 0.78 |
| Newman et al. 200891 | *3, *4, *5, *41 | Not reported |
Cox HR (adjusted for nodal status): HR 2.5; 95% CI 0.8 to 8.2; p = 0.17 BRCA1 HR 0; 95% CI NA; p = 0.18 BRCA2 HR 9.7; 95% CI 2.3 to 41.0; p = 0.008 |
| Goetz et al. 200982 on behalf of the ITPC; n = 2880a |
Varies by ITPC site but all genotyped for *4 and the majority genotyped for *3, *5, *6, *10, *17, *41 1168/2880 tested for *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N |
Not reported |
Not known Cox HR adjusted for positive nodes and project sites: HR 0.92 (p = 0.50) |
No significant differences were found in any cohort when the HR between groups was adjusted for nodal status. The only study84 reporting an unadjusted analysis also reported no significant differences between the two groups. However, a subgroup analysis of BRCA status by Newman et al. 91 reported a significantly worse median OS in patients with BRCA2 mutations and low CYP2D6 activity (adjusted HR 9.7; 95% CI 2.3 to 41.0; p = 0.008). A formal test of the interaction showed that this difference between BRCA1 and BRCA2 patients was significant between the two groups for survival (p = 0.022 after adjustment for nodal status) and remained significant when also adjusted for ER status. Importantly, when the entire ER+ group was considered, CYP2D6 status was not associated with outcome, but the positive association persisted in ER+ patients in the BRCA2 tumour group.
Relapse/recurrence
One cohort82 reported on DFS, two cohorts83,91 reported on RFS and one91 of these also reported on TTR (Table 15). Two82,83 of these cohorts included only postmenopausal women with ER+ breast cancer, the other cohort91 included 77% with ER+ breast cancer and women of any menopausal status. In the Goetz et al. cohort83 there was a greater proportion of PMs than EMs + IMs with tumour size ≥ 3 cm and who were LN+.
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
|
Goetz et al. cohort:83 Goetz et al. 200584 |
*4, *6 Only *4 used for the analysis |
RFS: time from randomisation to documentation of the first of the following events: any recurrence (local, regional or distant) of breast cancer, a contralateral breast cancer or death |
Cox HR (unadjusted), RFS: HR 2.71; 95% CI 1.15 to 6.41; p = 0.023 Cox HR (adjusted for tumour size and nodal status): HR 1.85; 95% CI 0.76 to 4.52; p = 0.176 |
| Newman et al. 200891 | *3, *4, *5, *41 | RFS: time from surgery to first of the following events: any recurrence (local, regional or distant) of breast cancer, a contralateral breast cancer or death |
Cox HR (adjusted for nodal status), RFS: All patients: HR 1.9; 95% CI 0.8 to 4.8; p = 0.19 BRCA1: HR 1.1; 95% CI 0.2 to 5.5; p = 0.90 BRCA2: HR 3.6; 95% CI 0.9 to 13.4; p = 0.094 |
| TTR: time to tumour recurrence with contralateral, ipsilateral or metastatic disease |
Cox HR (adjusted for nodal status), TTR: All patients: HR 2.1; 95% CI 0.84 to 5.4; p = 0.14 BRCA1: HR 1.3; 95% CI 0.3 to 6.2; p = 0.73 BRCA2: HR 3.8; 95% CI 1.0 to 14.5; p = 0.083 |
||
| Goetz et al. 2009 on behalf of the ITPC;82 n = 2880a |
Varies by ITPC site but all genotyped for *4 and the majority genotyped for *3, *5, *6, *10, *17, *41 1168/2880 tested for *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N |
Not reported |
Cox HR adjusted for positive nodes and project sites, DFS HR 1.07 (p = 0.51) |
When an unadjusted HR was presented, the Goetz et al. cohort83 reported significantly worse RFS for PMs than for EMs + IMs. However, when the HR was adjusted for tumour size and nodal status, no significant differences were reported. The large ITPC cohort82 reported no significant differences in DFS, whereas in the other cohort91 significant differences in RFS and TTR were apparent only in the subgroup of patients with BRCA2 mutations.
Asian patients genotyped for the *10 allele
Four cohorts93,97,101,114 studied associations between efficacy and CYP2D6 status in which only the *10 allele was genotyped for in three cohorts93,97,101 and additional common alleles in the other. 114 These four cohorts93,97,101,114 prohibited the concomitant use of CYP2D6 inhibitors.
There was no convincing evidence of differences by genotypes for OS, breast cancer mortality or relapse/recurrence in any of the four cohorts of Asian patients genotyped for the *10 allele. 93,97,101,114
Overall survival
One cohort97 examining only OS genotyped for *10 (Table 16). The Kaplan–Meier curve suggests that *10/*10 may have had a slightly improved OS compared with wt/*10 or wt/wt, but differences were not statistically significant. In this cohort, the majority (96%) of patients had ER+ breast cancer and 48% had a tumour size > 2 cm, but it was reported that a greater proportion (59%) of patients with the wt/wt genotype had a tumour > 2 cm. No patients in this cohort were LN+. It was not known how many, if any, women were postmenopausal or received adjuvant chemotherapy or CYP2D6 inhibitors.
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
| Toyama et al. 200997 | *10 | Interval from the date of primary surgery to death from any cause | It is stated that no associations were found between the *10 genotype and OS. No HRs have yet been calculated (Tatsuya Toyama, personal communication) |
Breast cancer mortality
Two97,101 cohorts of Asian patients were examined for breast cancer mortality by *10 genotypes (Table 17). The majority of women had ER+ breast cancer in both cohorts, although this varied from 82% to 96%. Three-quarters of women in Xu et al. 101 were postmenopausal; these data were not reported in the other cohort. 97 While the proportion of patients with tumour size ≥ 2 cm varied in the cohorts (from 27% to 48%), few if any patients were LN+ (0–7%). Adjuvant chemotherapy was not permitted in Xu et al. ;101 it was unclear if patients received this in the other cohort. 97 Interestingly, in one cohort a greater proportion (32%) of patients with the *10/*10 genotype than other genotypes had tumours ≥ 2 cm, whereas in the other cohort97 a greater proportion (59%) of patients with the wt/wt genotype had a tumour size > 2 cm.
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
| Xu et al. 2008101 | *10 | Time from date of diagnosis to death where breast cancer was the primary or underlying cause of death |
Cox HR (adjusted for age, clinical stage, LN status, tumour size, adjuvant therapy, surgery, C-erbB2 status, and ER or progesterone receptor) *10/*10 vs wt/wt + wt/*10: HR 2.7; 95% CI 0.4 to 17.3; p = 0.28 No TAM (n = 141) CYP2D6 *10 genotype was not significantly associated with breast cancer mortality (p = 0.78) |
| Toyama et al. 200997 | *10 | Time from the date of primary surgery to death from breast cancer recurrence | It is stated that no associations were found between the *10 genotype and breast cancer mortality. No HRs have yet been calculated (Tatsuya Toyama, personal communication) |
The findings are summarised in Table 17. While Xu et al. 101 suggested that, compared with wt/wt + wt/*10, breast cancer mortality was higher for women with the *10/*10 genotype, the CIs were wide and the finding was not statistically significant. From the Kaplan–Meier curve, the other cohort suggested that *10/*10 patients may actually have improved mortality from breast cancer when compared with patients with either the wt/wt or wt/*10 genotypes, but again the findings were not statistically significant.
One cohort also compared breast cancer mortality in patients taking TAM with a control group of patients receiving chemotherapy, the majority of whom had ER– breast cancer. As with patients taking TAM, there was no significant association between patients with *10 alleles and DFS in this group, suggesting that *10 is not a prognostic factor for breast cancer independent of TAM.
Relapse/recurrence
Five studies88,93,97,101,114 from four cohorts93,97,101,114 reported on relapse/recurrence outcomes. Ostensibly two cohorts97,101 reported on DFS and two93,114 reported on RFS, although, as can be seen in Table 18, the outcome definitions were very similar for both DFS and RFS.
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
|
Kiyotani et al. cohort:114 Kiyotani et al. 200888 |
*4, *5, *6, *10, *14, *18, *21, *41 | RFS: the period between surgical treatment to the recurrence of a breast cancer (i.e. local or distant recurrence or contralateral breast cancer) |
Cox HR (unadjusted), RFS: *10/*10 vs wt/*10: HR 2.19; 95% CI 0.24 to 19.79; p = 0.49 *10/*10 vs wt/wt: HR 8.67; 95% CI 1.06 to 71.09; p = 0.036 Cox HR (adjusted for tumour size), RFS: *10/*10 vs wt/wt: HR 10.04; 95% CI 1.17 to 86.27; p = 0.044 |
| Kiyotani et al. 2010114 | In Kiyotani et al. 2010114 also genotyped for *36 |
Cox HR (adjusted for tumour size and nodal status), RFS: wt/*10 vs wt/wt: HR 4.44; 95% CI 1.31 to 15.00 *10/*10 vs wt/wt: HR 9.52; 95% CI 2.79 to 32.45; p < 0.001 |
|
| Xu et al. 2008101 | *10 | DFS: time from date of diagnosis to first distant metastasis or death from breast cancer without a recorded relapse |
Cox HR (adjusted for age, clinical stage, LN status, tumour size, adjuvant therapy, surgery, C-erbB2 status, and ER or PgR): TAM: *10/*10 vs wt/wt + wt/*10: HR 4.7; 95% CI 1.1 to 20.0; p = 0.04 No TAM: CYP2D6 *10 genotype was not significantly associated with DFS (p = 0.99) |
| Okishiro et al. 200993 | *10 | RFS: distant recurrences, locoregional recurrences, ipsilateral in-breast recurrences, and contralateral breast cancers were included |
Cox HR (unadjusted): *10/*10 vs. wt/wt + wt/*10: HR 0.94; 95% CI 0.34 to 2.60; p = 0.95 Cox HR (adjusted for tumour size, LN status, histological grade, PgR status, HER2 status and adjuvant therapy) *10/*10 vs. wt/wt + wt/*10: HR 0.6; 95% CI 0.18 to 1.92; p = 0.39 |
| Toyama et al. 200997 | *10 | DFS: time from the date of primary surgery to the first locoregional recurrence, distant metastasis, ipsilateral breast recurrence or contralateral breast cancers | It is stated that no associations were found between the *10 genotype and DFS. No HRs have yet been calculated (Tatsuya Toyama, personal communication) |
The proportion of postmenopausal women in each cohort differed, varying from 22% to 76% in the three93,101,114 cohorts that reported these data. In all cohorts, the majority of women had ER+ breast cancer, although this varied from 74% to 82% in two101,114 cohorts to 91–96% in the other two. 93,97 Differences also existed for tumour size and nodal status, the proportion of women with tumours ≥ 2 cm varying from 27% to 48% and LN+ patients varying from 0% to 17%, although nodal status was not reported in one cohort. 93 Three93,101,114 of the studies explicitly excluded patients with CYP2D6 inhibitors, and two101,114 did not allow adjuvant chemotherapy, these data not being reported in the other cohort(s). There were, however, some differences within cohorts by genotype in the three93,97,101 cohorts that reported these data. In one cohort, a greater proportion (32%) of patients with the *10/*10 genotype had tumours ≥ 2 cm, whereas, in the other,97 a greater proportion (59%) of patients with the wt/wt genotype had a tumour > 2 cm. No such differences were apparent in the third cohort,93 although 90% of patients with the *10/*10 genotype had ER+ cancer compared with 60% with the other genotypes.
Compared with other genotypes, all four cohorts93,97,101,114 suggest that there may be modestly poorer outcomes in terms of DFS and RFS for patients with the *10/*10 genotype, but CIs are extremely wide and a significant difference was reported in only one114 of these cohorts. This cohort still reports wide CIs, suggesting that the finding should be treated with caution.
One cohort also compared DFS in patients taking TAM with a control group of patients receiving chemotherapy, the majority of whom had ER– breast cancer. As with patients taking TAM, there was no significant association between patients with *10 alleles and DFS in this group, suggesting that *10 is not a prognostic factor for breast cancer independent of TAM.
Other genotype/phenotype/functional classification comparisons
Three cohorts41,96,108 reported outcomes by phenotypes that do not fit the ‘standard comparisons’ explored above. Unlike all of the other studies that genotyped for multiple alleles, two96,108 of these cohorts were unique in differentiating between patients who could be classified as IMs and those who could be classified as heterozygous EMs (hetEMs). Thus, in these cohorts, both the IMs and hetEMs would be considered subsets of patients who fit the IM phenotype in other studies. The other cohort41 conducted two comparisons, the first, which could equate to PMs, IMs and EMs, but then a secondary analysis in which the rationale for combining particular genotypes into two groups (A and B) was less clear.
Summarising the data from the three cohorts is problematic owing to the different genotype/phenotype/functional classifications used, although there was some suggestive evidence that EMs have better relapse/recurrence outcomes than patients with other phenotypes in one of these cohorts. 108
Relapse/recurrence
One cohort41 considered DFS by CYP2D6 status by comparing patients in two separate analyses. First, patients were split into three groups. These groups could broadly be considered to be PM, IM and EM in which no significant differences were reported when all were compared with each other, although the authors highlighted the gradual improvement in DFS in function of CYP2D6. The second analysis did report a significant difference between two groups (Table 19), although, as discussed above, there appears to be no obvious rationale behind the groupings of genotypes into these two groups. In the other two cohorts,96,108 RFT was significantly worse for IM + PMs (in effect, a subset of PM + IMs using the ‘standardised comparisons’ above) and also worse (but not statistically significant) for hetEMs96 and TTR was significantly worse for hetEMs + IMs than for EMs. 108 As reported above, using the ‘standardised comparisons’ of PM + IM versus EM, the HRs were actually greater for both RFT (adjusted HR 2.24; 95% CI 1.16 to 4.33; p = 0.02)96 and TTR (unadjusted HR 1.57; 95% CI 1.18 to 2.08; p = 0.02). 108
| Cohort/study | Alleles tested | Outcome definition | Summary of findings |
|---|---|---|---|
| Schroth et al. 200796 |
*4, *5, *10, *41 *3, *6, *7 and *8 were also genotyped for but excluded because PCR amplification rates were poor |
RFT was defined as the time from surgery to the occurrence of a breast event (i.e. local or distant recurrence or contralateral breast cancer) |
Cox HR (adjusted for tumour size and nodal status), RFT: hetEM vs EM:a HR 1.88; 95% CI 0.89 to 4.02; p = 0.09 IM + PM vs EM:a HR 1.63; 95% CI 1.07 to 2.46; p = 0.02 |
| Ramon et al. 201041 | *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N | DFS: calculated from the beginning of therapy to (a) the time of relapse, (b) the appearance of a contralateral breast cancer or (c) death |
Mean DFS (months): three-group analysis b 1: 98 2: 114 3: 118 p = 0.413 Mean DFS (months): two-group analysis b A: 95 B: 119 p = 0.016 |
| Schroth et al. 2009108 | *3, *4, *5, *10, *41, wt × 2, *2 × 2 | TTR: time from diagnosis or randomisation to documentation of a breast event, any local, regional or distant recurrence of breast cancer or a contralateral breast cancer |
Cox HR (unadjusted), TTR: hetEM + IM vs EM: HR 1.49; 95% CI 1.12 to 2.00; p = 0.06 Cox HR (adjusted for tumour size, nodal status and histological grade and stratified by menopause status and mode of patient recruitment), TTR: HR = 1.40; 95% CI 1.04 to 1.90; p = 0.03 |
Adverse events
Nine studies41,84,87,90,93–95,98,107 from seven cohorts41,83,87,90,93,94,98 have considered any AEs in relation to CYP2D6 status (Table 20).
| Cohort/study | Alleles tested | Summary of findings |
|---|---|---|
|
Goetz et al. cohort:83 Goetz et al. 200584 |
*4, *6 Only *4 used for the analysis |
Incidence of moderate (grade 2) or severe (grade 3) hot flushes, n (%): ■ *4/*4: 0/13 (0) ■ wt/*4: 9/40 (23) ■ wt/wt: 27/137 (20) For CYP2D6 *4/*4 patients, 0 (0%) of 13 patients experienced grade 2 or 3 hot flushes compared with 36 (20%) of 177 patients with the wt/wt or wt/*4 genotypes (p = 0.064) |
|
Henry et al. cohort:87 |
*1*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N |
The authors did not observe a statistically significant association between the CYP2D6 genotype and either baseline BMD or percentage change in BMD (data not presented in the paper) Change in hot flush score: ■ EM: 26.9 ± 8.8 ■ IM: 44.3 ± 10.2 ■ PM: 20.6 ± 16.9 IM significantly higher than EM (p = 0.011) and PM (p = 0.038) Change in hot flush score,a ITT analysis: ■ EM: 25.3 ± 4.7 ■ IM: 41.8 ± 6.2 ■ p = 0.040 Trend suggesting that EMs and PMs were more likely to remain free of hot flushes during TAM therapy than IMs (p = 0.100 and p = 0.089, respectively) Severity, n (%): EM ■ no hot flushes: 50/208 (24.0) ■ mild/moderate: 96/208 (46.2) ■ severe/very severe: 62/208 (29.8) PM + IM ■ no hot flushes: 9/21 (42.9) ■ mild/moderate: 10/21 (47.6) ■ severe/very severe: 2/21 (9.5) |
| Rae et al. 200995 |
Significant correlation between increasing CYP2D6 score and drug discontinuation due to side effects (r2 = 0.935, p = 0.018) Adjustment of scores for concomitant medications that alter CYP2D6 activity eliminated the relationship between CYP2D6 score and treatment discontinuation rates |
|
| Wang et al. 200798 | *4 |
Risk of AE by *4 status ■ OR = 1.9 to 8.0; p = 0.1 to 0.6 |
| Madlensky et al. 200890 | *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N |
Hot flushes: ■ EM: 79.8% ■ hetEM: 76.3 ■ IM: 80.1 ■ PM: 63.9 ■ UM: 75% ■ χ2 = 11.3; p = 0.02) After controlling for age, menopausal status and time since diagnosis, the PM group was half as likely to report hot flushes as the referent EM group (OR 0.46; 95% CI 0.28 to 0.78; p = 0.003) |
| Okishiro et al. 200993 | *10 | There was no significant difference in the extent of changes in BMD and total cholesterol concentrations between patients with the *10/*10 genotype and those with the wt/w + wt/*10 genotype |
| Onitilo et al. 200994 | *4 | *4/ + *4 + wt/*4 was compared with wt/wt. No significant difference in time to deep-venous thrombosis was noted (p = 0.3) |
| Ramon et al. 201041 | *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N |
No toxicity n (%): ■ A (*4/*4, *4/*41, *1/*5, *2/*5): 6/16 (37.5) ■ B (all other genotypes): 40/75 (53.3) Mild toxicity n (%): ■ A (*4/*4, *4/*41, *1/*5, *2/*5): 7/16 (43.8) ■ B (all other genotypes): 27/75 (36.0) Severe toxicity n (%): ■ A (*4/*4, *4/*41, *1/*5, *2/*5): 3/16 (18.8) ■ B (all other genotypes): 8/75 (10.7) ■ p = 0.2 |
The only AEs that appeared to differ by CYP2D6 status in any of the cohorts were hot flushes. 83,87,90 Here EMs and/or IMs appeared to be more prone to suffer hot flushes than PMs,83,87,90 more prone to experience severe hot flushes83,87 and more likely to discontinue treatment because of hot flushes. 87
Adverse event frequency
One cohort98 did not find a significant difference in the risk of AEs based on presence or absence of the *4 allele. 98 However, the number of patients with the *4/*4 genotype was very small, making comparisons difficult.
Toxicity
Severe, mild and no toxicity were considered in one cohort90 with regard to CYP2D6 status using an aggregation of genotypes that had produced significant results in relation to DFS, the rationale behind this grouping of genotypes in terms of enzyme function being unclear. No significant relationship between genotype and toxicity was found.
Deep-venous thrombosis
One cohort94 considered deep-venous thrombosis by CYP2D6 status. Patients were genotyped for *4 and it was reported that there was no difference in time to deep-venous thrombosis between patients with the *4/*4 + wt/wt genotypes and those with the wt/wt (EM).
Bone mineral density
Two cohorts87,93 examined the association between bone mineral density (BMD) and genotype. Whether patients were simply genotyped for *1093 (and those with the *10/*10 genotype were compared with those with the wt/wt + wt/*10 genotype) or whether a greater number of polymorphisms were tested,87 both studies suggest that change in BMD is not related to CYP2D6 status.
Hot flushes
Three cohorts83,87,90 examined hot flushes. It was found in one cohort that, after controlling for age, menopausal status and time since diagnosis, PMs were half as likely as EMs to report hot flushes. 90 Another cohort107 suggested that IMs (as defined using an ‘activity score’118) may be more prone to hot flushes than either EMs or PMs. This cohort also found that PMs were less likely than EMs + IMs to develop severe or very severe hot flushes. An earlier cohort84 found that no patients with the PM phenotype (*4/*4) developed moderate or severe hot flushes, compared with 20% of those with the EM + IM phenotypes (wt/wt + wt/*4), although the number of patients with the PM phenotype was small.
Discontinuation of treatment because of adverse events
In one cohort,87 it was reported that after 4 months 41/280 (14.6%) patients had discontinued treatment, 28/41 (68.3%) of these because of TAM side effects, most commonly as a result of hot flushes [13/28 (46.4%)]. None of these patients was found to be a PM. In fact, this study suggests that the greater the CYP2D6 activity, the greater the chance of withdrawal because of AEs, i.e. EMs are the phenotype at greatest risk.
Endoxifen concentrations
There were eight studies49,73,102,104,105,112–114 from seven cohorts49,73,87,104,112–114 that examined endoxifen concentrations in relation to CYP2D6. Four studies reported mean plasma concentrations102,105,112,113 and three reported median plasma concentrations. 104,112,114 Two studies reported decreases in endoxifen following the administration of an SSRI (paroxetine). 49,102 In five cohorts49,73,87,104,114 the TAM dose was known to be 20 mg/day, whereas in Bonanni et al. 112 the drug dose was only 10 mg/day as half of the patients receiving TAM in this cohort also received ANA 1 mg/day. In the other cohort113 the TAM dose was not stated.
The findings from each individual cohort appear to suggest differences in concentrations between PMs87,104 or those with the *10/*10 genotype73,114 and EMs (or those with the wt/wt genotype). However, in the Caucasian population, one cohort104 reported the levels for the IMs to be closer to those of EMs than PMs. The other cohort87 reported wt/vt genotypes (which may be equated as IM) to have levels closer to vt/vt genotypes. Reduced decreases in mean endoxifen plasma concentrations were also evident in patients taking potent CYP2D6 inhibitors in two cohorts. 49,102
Genotype studies
Four49,73,105,114 of the studies examined endoxifen levels in relation to genotype (Table 21). The earliest study49 included in this review examined the decrease in endoxifen concentrations in patients who were also taking the CYP2D6 inhibitor paroxetine. Decreases in endoxifen levels were greatest in patients with the wt/wt genotype, which suggests that TAM metabolism is vulnerable to drug interactions with this particular SSRI.
| Cohort/study | Alleles tested | Summary of findings |
|---|---|---|
| Stearns et al. 200349 | *4, *6, *8 |
Decrease in endoxifen concentrations after taking the CYP2D6 inhibitor paroxetine: wt/wt genotype: 64% (95% CI 39% to 89%) wt/vt or vt/vt genotype: 24% (95% CI 23% to 71%) p = 0.03 Where vt = *4, *6, *8 Baseline plasma endoxifen concentrations were lower in women with *4, *6 or *8 alleles than in those with wt (p = 0.002) |
|
Henry et al. cohort: 87 Jin et al. 2005105 |
*3, *4, *5, *6 |
Mean (range) endoxifen plasma concentrations, ng/ml: wt/wt: 78.0 (65.9 to 90.1) wt/vt: 43.1 (33.3 to 52.9) vt/vt: 20.0 (11.1 to 28.9) p < 0.01 vt = *3, *4, *5, *6 |
| Lim et al. 200773 | *5, *10, *2 × N |
Mean (range) endoxifen plasma concentrations, ng/ml: wt/wt (n = 64): 19.9 (18.0 to 21.9) wt/*10 (n = 89): 18.1 (16.8 to 19.5) *10/*10 (n = 49): 7.9 (7.1 to 8.8) p < 0.0001 Where wt = allele not containing *10 Mean (range) endoxifen plasma concentrations, ng/ml: wt/wt: (n = 55): 20.7 (18.5 to 22.9) wt/vt (n = 96): 18.0 (16.7 to 19.2) vt/vt (n = 51): 8.1 (7.2 to 9.0) p < 0.0001 Where wt = allele not containing *5 and *10 |
|
Kiyotani et al. cohort: 114 Kiyotani et al. 2010114 |
*4, *5, *6, *10, *14, *18, *21, *36, *41 |
Median endoxifen plasma concentrations, ng/ml: wt/wt (n = 24): 35.4 wt/vt (n = 45): 27.2 vt/vt (n = 29): 15.5 |
From the other three studies,73,105,114 it is evident that endoxifen concentrations in patients with the vt/vt genotype were lower than in those with the wt/wt genotype, regardless of which alleles were tested. In the two cohorts73,114 of Asian patients, in which *10 was one of the alleles tested and was thus the most common vt allele, endoxifen levels in those with the wt/vt genotype were nearer to those reported in patients with the wt/wt genotype. In US patients, in whom only *3, *4, *5 and *6 were tested, the mean endoxifen concentration in those with the wt/vt genotype was nearer to that of patients with the vt/vt genotype. 87
Phenotype studies
Four102,104,112,113 of the studies examined endoxifen levels in relation to phenotype (Table 22). As with the early study by Stearns et al.,49 decreases in endoxifen levels were greatest in patients with the wt/wt genotype (i.e. EMs), with potent CYP2D6 inhibitors (such as paroxetine) reducing the amount by which the levels decreased to levels similar to PMs. An interesting finding from this study was that the group of patients with alleles associated with UMs were the only ones who were not converted into PM status by CYP2D6 potent inhibitors. 102
| Cohort/study | Alleles tested | Summary of findings |
|---|---|---|
|
Henry et al. cohort:87 Borges et al. 2006102 |
*1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N |
Decrease in mean (SD) endoxifen plasma concentrations, nmol/l, 12 months: EM: 84.1 (39.4) PM: 19.4 (6.1) Potent CYP2D6 inhibitors: 24.6 (16.6) Weak CYP2D6 inhibitors: 50.1 (30.4) |
| Gjerde et al. 2007104 | *3, *4, *5, *6, *2 × 2 |
Median (range) endoxifen plasma concentrations, ng/ml: UM: 46.3 (37.6 to 141.4) EM: 52.3 (24.3 to 184.8) IM: 49.6 (27.3 to 108.2) PM: 36.7 (30.7 to 68.6) p = 0.003 (based on logistic regression analysis in which each variable was adjusted for age) |
| Bonnanni et al. 2009112 | *2, *3, *4, *5, *6, *9, *29, *41A |
Median (Q1, Q3) endoxifen concentrations, ng/ml, 6 months: EM (n = 43): 4.62 (3.52, 5.62) PM (n = 7): 5.00 (4.18, 6.79) Median (Q1, Q3) endoxifen concentrations, ng/ml, 12 months: EM (n = 42): 4.63 (2.98, 6.62) PM (n = 7): 3.67 (3.03, 5.13) |
| de Duenas et al. 2009113 | *1–*10AB, *11, *14A, *14B, *15, *17, *19, *20, *25, *26, *29–*31, *35, *36, *40, *41, *1 × N, *2 × N, *4 × N, *10 × N, *17 × N, *35 × N, *41 × N |
Mean (SD) plasma concentrations of endoxifen, nmol/l: EM = 21.0 (13.6) PM = 7.7 (1.5) p = 0.029 |
Two studies103,113 reported mean or median endoxifen levels to be significantly different between EMs and PMs, with one103 of these suggesting levels in IMs to be nearer those of EMs than of PMs. The other study112 did not report significant differences between EMs and PMs; however, in this study, one-third of patients received TAM alone, one-third received ANA alone and one-third received both TAM and ANA. Thus, the TAM dose in this study was lower than is standard (10 mg/week instead of 20 mg/day), which may explain this lack of difference. In addition, this study also considered effects on ANA concentrations from patients taking TAM and this aromatase inhibitor (data not presented in table). While ANA concentrations were not affected by the combination with low-dose TAM, endoxifen levels were lower in patients taking TAM and ANA [median (range) 3.18 (2.22 to 4.32) ng/ml] than those taking TAM alone [4.83 (3.65 to 6.19) ng/ml]. In a further analysis based on the CYP2D6 genotype, the differences for endoxifen were no longer significant after excluding PMs [median (range) 4.65 (2.07 to 6.49) ng/ml and 4.6 (3.98 to 6.85) ng/ml, respectively).
Exploratory analysis: clinical sensitivity and specificity
Because of the lack of definitive clinical validity evidence, in particular evidence for differences between groups of patients apparently being complicated depending on which alleles were tested, attempts were made to measure the clinical sensitivity and specificity of testing for particular alleles. This was possible for only nine studies85,86,88,92,96,99,100,108,114 from seven cohorts86,92,96,99,100,108,114 that presented data on events in the text or tables.
Exploring the clinical sensitivity and specificity is an approach recommended by Flockhart et al. 54 in an American College of Medical Genetics statement that defined clinical sensitivity as ‘the proportion of individuals with an event that have a genotype other than wt/wt (true positives) and clinical specificity is defined as the proportion of individuals that do not have the event who possess the wt/wt genotype (true negatives)’. The same definitions of true positives and true negatives were thus utilised for this exploratory analysis, although it should be noted that this previous paper by Flockhart et al. 54 referred to a test for CYP2C9 (cytochrome P450 2C9) and VKORC1 (Vitamin K epoxide reductase complex subunit 1) in relation to treatment with warfarin, and so the assumptions about what constitutes a true positive or true negative may not be directly applicable. However, given that there appears to be the greatest amount of suggestive evidence from our review for differences in clinical outcomes (if not endoxifen concentrations) between EMs (i.e. wt/wt) and those with other genotypes, it could be argued that these same assumptions may be appropriate.
Aside from testing for different alleles, there are a number of other notable differences in terms of cohort characteristics across the eight cohorts included in our exploratory analysis. The Kiyotani et al. cohort114 is perhaps the most notably different in that it is a study of Asian patients who appear to have notable differences in terms of nodal status (17% compared with > 60% in the majority of the other cohorts). However, other cohorts also stand out as differing from the rest: Nowell et al. 92 includes fewer postmenopausal women (59%), allows adjuvant chemotherapy and also includes 5% of women with metastatic disease. Wegman et al. 100 also allows adjuvant chemotherapy and has a large proportion of patients who are LN+ (89%) and the TAM dose was 40 mg/day for 2 years. In Wegman et al.,99 the TAM dose was either 20 or 40 mg/day and for some patients again only lasted for 2 years. Schroth et al. 96,108 reported the smallest proportion (34%) of women who were LN+ aside from the Kiyotani et al. cohort. 114
Estimates of clinical sensitivity and specificity were made in relation to each of the different outcomes for which we had these data, namely OS, breast cancer mortality and recurrence/relapse. Overall, our exploratory analysis suggested that testing for a greater number of alleles increased specificity but that sensitivity was generally low, no matter how many alleles were genotyped.
Overall survival
Taking into account the differences between the cohorts, the data suggest that the sensitivity and specificity of testing simply for *4 in the adjuvant setting may be 15% and 73%, respectively, for OS92 (Table 23). A more comprehensive genotype test (in terms of the number of alleles tested) appears to increase sensitivity and specificity to 18% and 83%, respectively. 108
| Cohort/study; number of patients | Alleles tested | Length of follow-up (years) | Eventsa | Sensitivity (%) | Specificity (%) | |
|---|---|---|---|---|---|---|
| wt/wt | Other | |||||
| Nowell et al. 2005;92 165 | *4 | Median 5.4 | 27/101 | 7/48 | 15 | 73 |
| Schroth et al. 2009;108 206 | *3, *4, *5, *10, *41, wt × 2, *2 × 2 | Median 6.3 | 132/716 | 102/609 | 18 | 83 |
Relapse/recurrence
The data from four cohorts86,92,99,100 suggest that the sensitivity of testing simply for *4 in the adjuvant setting may be between 21% and 37% for relapse/recurrence and specificity may be between 52% and 86% for relapse/recurrence (Table 24). If, in testing for *4, phenotype status is altered based on concomitant CYP2D6 use, based on a small number of patients (n = 84), for relapse/recurrence, sensitivity may be 50% and specificity 73%. 86 Utilising data from the only cohort to test simply for *10 suggests a sensitivity of between 19% and 50% and specificity of between 95% and 96%. If a more comprehensive genotyping strategy is used, data from the largest cohort of Schroth et al. 108 and Schroth et al. 96 (whose patients are actually included in the Schroth et al. 108 cohort) suggest a sensitivity of between 18% and 30% and a specificity of between 86% and 88%.
| Cohort/study; number of patients | Alleles tested | Length of follow-up (years) | Eventsa | Sensitivity (%) | Specificity (%) | |
|---|---|---|---|---|---|---|
| wt/wt | Other | |||||
| Nowell et al. 2005;92 165 | *4 | Median 5.4 | 38/112 | 10/48 | 21 | 66 |
| Wegman et al. 2005;100 112 | *4 | Mean 10.7 | 25/52 | 6/24 | 25 | 52 |
| Gonzalez-Santiago et al. cohort:86 | ||||||
| Gonzalez-Santiago et al. 2006;85 85 | *4 | Median 4 | 10/49 | 13/35 | 37 | 80 |
| Gonzalez-Santiago et al. 2007;86 84 | *4 accounting for CYP2D6 inhibitors | Mean 5.5 | 13/48 | 18/36 | 50 | 73 |
| Wegman et al. 2007;99 677 | *4 | Mean 7 | 103/480 | 45/137 | 33 | 79 |
| Kiyotani et al. cohort114 | ||||||
| Kiyotani et al. 2008;88 67 | *10 | Median 8 | 1/20 | 27/54 | 50 | 95 |
| Kiyotani et al. 2010;114 282 | *10 | Median 7.1 | 3/84 | 38/198 | 19 | 96 |
| Schroth et al. 2007;96 486 | *4, *5, *10, *41 | Median 5.9 | 17/118 | 24/79 | 30 | 86 |
| Schroth et al. 2009;108 1361 | *3, *4, *5, *10, *41, wt × 2, *2 × 2 | Median 6.3 | 135/609 | 202/716 | 18 | 88 |
Summary of clinical effectiveness evidence
No studies were found that explored the relationship between endoxifen levels and clinical outcomes or which considered the clinical utility of CYP2D6 testing. The clinical validity evidence was thus limited to studies which examined differences in clinical outcome (OS, breast cancer survival, relapse/recurrence and AEs) and endoxifen levels by genotype. Unfortunately, the heterogeneity between studies in terms of patient characteristics, alleles studied, comparisons made and clinical end points defined and measured has made meta-analyses inappropriate and comparisons difficult.
Taking into account these caveats, there is suggestive evidence from six cohorts83,86,91,96,108,109 that patients with the wt/wt genotype (EM phenotype) may have better outcomes in terms of relapse/recurrence than patients with other genotypes. However, three cohorts83,87,90 suggest that, alongside IMs, these EMs may also be more prone to hot flushes. The findings are confused when endoxifen plasma concentrations in Caucasians are considered. While there are differences in concentrations between patients with wt/wt and vt/vt genotypes87 or EM and PM phenotypes,104 one of these cohorts104 reports that those with the wt/vt genotype have levels closer to those with the wt/wt genotype and not the vt/vt genotype, as would arguably be expected from the suggestive evidence for relapse/recurrence from the aforementioned six cohorts83,86,91,96,108,109 in which relapse/recurrence outcomes were improved in EMs or, more specifically, compared with PMs + IMs. 83,86,96,108,109 There is no convincing evidence that Asian patients with the *10/*10 genotype have different outcomes to EMs in terms of efficacy or AEs, although there are clear differences in terms of mean and median endoxifen concentrations.
Given the absence of clinical utility studies and our inability to conduct meta-analyses of the clinical validity data, we carried out exploratory analyses of sensitivity and specificity. These were based on data from only a limited number of the cohorts and on the assumption that EMs should be considered separately to all other phenotypes in determining true negatives and true positives. Thus, these data should only be considered as exploratory, highlighting the type of data that may be useful for future studies. Based on the limited data presented here, the cohorts suggest that testing for a greater number of alleles increases specificity but that sensitivity is generally low no matter how many alleles are tested for.
Chapter 4 Assessment of cost-effectiveness
Systematic review of existing cost-effectiveness evidence
A systematic review of the economic literature was conducted to identify the existing evidence that assesses the cost-effectiveness of genotyping for CYP2D6 for the management of women with breast cancer. It followed the same principles stated in Chapter 3 (see Methods for reviewing effectiveness). The search strategies are listed in Appendix 1.
Identification of studies
A total of 63 studies were identified from the literature search for evidence relating to the costs and benefits of CYP2D6 for the management of women with breast cancer. None of these papers met the inclusion criteria of being an economic evaluation comparing TAM with any aromatase inhibitor and genotyped for CYP2D6. All excluded studies are listed in Appendix 2. However, two of these studies79,119 conducted a modelling exercise of the pharmacogenetic variation of CYP2D6 and considered the choice of optimal adjuvant endocrine therapy in women with early ER+ breast cancer (thus conducting a partial evaluation of the research question). Owing to the lack of any other published evidence, we have described both studies below as these studies offer a good starting point for the development of an economic evaluation; it is noted that one of these studies79 has been presented only as an abstract.
Study characteristics and model overview
Punglia et al. 119 undertook a modelling analysis to determine whether TAM or aromatase inhibitor monotherapy maximises DFS after 5 years of treatment. In this model, patients could be genotyped for only the *4 allele and treated with TAM or not genotypically selected and treated with TAM or aromatase inhibitors.
Veenstra et al. 79 developed a decision-analytic lifetime Markov model to evaluate pharmacogenetic testing for CYP2D6 variants to identify postmenopausal women who would be good candidates for alternative therapies. This paper classified women as PMs or EMs; in other words, the authors used the phenotype instead of the genotype to classify patients. The study by Punglia et al. 119 classified patients by genotype.
Model inputs and data sources
The Punglia et al. 119 model simulates the transition between two states: being well with no evidence of any cancer recurrence (‘being well’) and having a local or regional recurrence or a new primary breast cancer. Women starting in the ‘being well’ state face a monthly probability of experiencing a recurrence derived from the annual HRs from the BIG 1-98 trial. 120 The model estimates recurrence probabilities only for each CYP2D6*4 genotype: wt/wt, wt/*4 and *4/*4.
The frequencies for any of the genotypes in the population were derived from a study by Goetz et al. 84 and so the model used recurrence probabilities weighted for the genotypic frequency. The authors also re-ran the model using new data from a re-analysis of the same cohort of patients,83 where patients who had received SSRIs were reclassified to allow for a more accurate assessment of the effect of CYP2D6 on outcomes. A two-way sensitivity analysis was performed to test the uncertainty around the model, varying the HR for patients with the *4/*4 genotype and the HR for patients with the wt/*4 genotype.
The model briefly defined in the abstract by Veenstra et al. 79 consists of six health states and assesses a hypothetical cohort of 64-year-old women with ER+ breast cancer receiving TAM. The incidences of local regional relapse, metastasis and breast cancer death was obtained from the ATAC trial. 21 The HR for disease recurrence in PMs versus EMs was derived from a study by Goetz et al. (this study was not referenced in the abstract). Costs, utilities and background mortality rates were obtained from the published literature or publicly available sources.
Results and sensitivity analysis
In the Punglia et al. study,119 the base-case results reported 5-year DFS rates to be 84.0% for patients receiving aromatase inhibitors and 81.3% for those receiving TAM. DFS rates for wt/wt patients treated with TAM were 83.9%, i.e. similar to those unselected and treated with aromatase inhibitors. A two-way sensitivity analysis was performed, varying the increased HR for recurrence for *4/*4 patients relative to wt/wt patients from 1.0 to 3.0 and the increased HR for recurrence for wt/*4 patients relative to wt/wt patients from 0 to 1.0. The sensitivity analysis found that when a greater HR for *4/*4 patients was used, DFS rates for wt/wt patients treated with TAM exceeded those of patients treated with aromatase inhibitors. Thus, the authors concluded that this modelling exercise suggests that CYP2D6 testing could be considered for women newly diagnosed with breast cancer.
The results presented in the abstract by Veenstra et al. 79 are related to the projected DFS at 5 years, which is 81.4% for TAM and 83.3% for ANA compared with 81.0% (TAM) and 83.8% (ANA) from the ATAC trial. 21 These results are confused because it is not stated whether or not the first pair of data is in the CYP2D6-guided therapy. In terms of utility, treatment with TAM resulted in 11.95 QALYs, ANA in 12.15 QALYs and CYP2D6-guided therapy in 12.19 QALYs. The abstract states that a one-way sensitivity analysis and scenario analyses were conducted to evaluate uncertainty, but the results of these analyses have not been reported.
Critique of published models
Although the two examples are not full economic evaluations, it was felt that it would be beneficial to critique the approaches.
The Punglia et al. 119 modelling exercise can be critiqued as follows:
-
Methods of deriving the effectiveness data Data were collected from the BIG 1-98 trial120 but take account of only the direct effects of either TAM or aromatase inhibitors on DFS and do not account for any AEs. Women taking SSRIs are also excluded from the model.
-
Measurement of resource data No resources have been measured in the study.
-
Valuation of resource data As stated above, no resources have been described in the study.
-
Measurement and valuation of health benefits (utilities) There are no utility measures described in the study; in terms of clinical effectiveness, DFS was measured.
-
Method of synthesising the costs and effects Only effects have been measured, using HR on DFS between strategies.
-
Analysis of uncertainty Two-way sensitivity analysis is used to explore uncertainty, and the effect of varying the HR on the results are described. A probabilistic sensitivity analysis could be used to explore all of the parameters together, but, owing to the simple structure of the model and the lack of costs and utility data, it would not be very useful.
-
Generalisability of the results Finally, and arguably most crucially, the model considers testing only for the CYP2D6*4 allele. Although *4 is the most frequent allele with ‘loss of function’ (PM) enzymatic activity in the Caucasian population (see Table 5), other alleles should also be considered given the findings emerging from recent studies suggesting that differences in outcomes depend on which alleles are tested for109 and that up to one-third of patients are misclassified based on testing for *4 only. 121
The model described by Veenstra et al. 79 can be critiqued as follows:
-
Methods of deriving the effectiveness data Effectiveness data have been taken from one trial21 and from a ‘recent study’ (unreferenced). From the limited data available, the accuracy/reliability of the effectiveness data is unknown.
-
Measurement of resource data Insufficient details of resource measurement are presented; the abstract states that data were obtained from the published literature or publicly available sources.
-
Valuation of resource data Insufficient details of resource valuation are available from the abstract.
-
Measurement and valuation of health benefits (utilities) The utilities have been obtained from published literature but the abstract does not give more information.
-
Method of synthesising the costs and effects Results have been reported as QALYs gained for each strategy, but there are no cost data or cost-effectiveness ratios reported. Results on projected DFS have been inadequately reported.
-
Analysis of uncertainty The results of the sensitivity analysis are not reported.
-
Generalisability of the results Owing to the limited information included in the abstract, it is difficult to assess the generalisability of the results. The authors have not stated the methods used to phenotype women, in other words data describing the alleles that have been tested to categorise women as PMs and EMs are missing; this makes it difficult to determine to what extent the study is able to accomplish its primary objective.
Independent economic assessment
Given the lack of studies relevant to the research question and UK clinical practice, we aimed to structure and populate an economic model to evaluate the incremental costs and benefits of CYP2D6 testing for the management of women with breast cancer potentially eligible for management with TAM.
Requirements for a de novo economic evaluation
In order to undertake an economic evaluation of pharmacogenetic testing within the framework of the wider model of breast cancer care, the following clinical data requirements are considered to be most important:
-
Epidemiological data Data related to allele frequencies for selected genetic variants and how these are distributed in populations; in particular, the data should indicate which groups of patients (as defined by genotype and/or phenotype) would need to be identified by the pharmacogenetic test.
-
Clinical effectiveness Evidence of a link between phenotypes and drug metabolism and data describing clinical outcomes and AEs, including long-term effects of the drugs. It will be necessary to pay special attention to the choice of the time horizon of the economic evaluation which will be related to the length of the treatment (e.g. as stated in guidelines) and ‘carry-over’ effects of the treatment (i.e. delayed effects of the treatment after discontinuation).
-
Test accuracy Data are required on both the sensitivity and specificity of the test and how accurate the test is in linking genotypes to phenotypes and then to clinical events and predictive value of the test.
-
Uptake of the test The degree of test uptake, by patients or clinicians, will have an impact on cost-effectiveness.
-
The impact that pharmacogenetic test results will have on clinicians’ behaviour Data are required including the impact that test results have on prescribing decisions, and how this affects the overall delivery of care.
From a health economics perspective, the key elements that need to be considered when undertaking a de novo economic evaluation of CYP2D6 testing as a management option for women with breast cancer after surgery are discussed below.
Study question
The economic question of interest: what is the relative cost-effectiveness of CYP2D6 testing as a management option for women with breast cancer after surgery? Both the costs and benefits (utility) of the alternatives being compared require identification, measurement and valuation.
Selection of alternatives
The current standard of care for women with ER+ breast cancer after surgery is 5 years of TAM for women with ER+ breast cancer. This could be compared with the following potential comparators:
-
Five years of TAM or aromatase inhibitors based on the results of genotyping for CYP2D6 This strategy represents the three different pathways of care as a result of the genotyping for CYP2D6 as explained in Chapter 2 (see Current service provision):*
-
– poor metaboliser pathway – five years of aromatase inhibitors
-
– intermediate pathway – five years of aromatase inhibitors
-
– extensive metaboliser pathway – five years of TAM.
-
-
Five years of aromatase inhibitors This strategy represents people taking aromatase inhibitors for 5 years.
-
Two years of TAM then aromatase inhibitors As stated in Chapter 2 (see Current service provision), this strategy is recommended for postmenopausal women who have received 2 years of TAM and who are not considered to be at low risk of recurrence, who are intolerant to TAM or for whom TAM is contraindicated because of toxicities.
*Unfortunately, there is no consensus about how to define each of the phenotypes from genotypes.
Effectiveness data
Every economic evaluation is reliant on good-quality clinical effectiveness data. Currently, the quality and quantity of the data available from published clinical studies are limited and so the data are not easily incorporated into any form of economic evaluation. As mentioned previously, there are 34 relevant published clinical studies describing 25 cohorts. However, owing to the heterogeneity of the studies, data from these studies cannot be synthesised for use in an economic evaluation.
Outcome measurement and valuation
Our clinical review has identified a number of outcomes that have been used to measure the efficacy of genotyping for CYP2D6. Of these, it is likely that a relapse/recurrence outcome, such as DFS, would be most appropriate for a condition such as breast cancer. These data would then need to be extrapolated and the life-years gained estimated. It then needs to be considered whether we adjust these adjusted life-years on a quality basis with appropriate utility data for the health states relevant to breast cancer. Owing to the treatments under evaluation, it is also necessary to include AEs in the model. These should include the following AEs:
-
hot flushes: presented in women treated with either TAM or with aromatase inhibitors21,22
-
endometrial cancer: more likely to occur in women treated with TAM21,22
-
hip fractures: more likely to occur in women treated with aromatase inhibitors than those treated with TAM21,22
-
spine fractures: more likely to occur in women treated with aromatase inhibitors than those treated with TAM21,22
-
vaginal bleeding: more likely in women treated with TAM21
-
ischaemic cerebral events: more likely in women treated with TAM21
-
cardiovascular events: increased risk in women treated with aromatase inhibitors22,122
-
deep-venous thrombosis: more likely in women treated with TAM21,22
-
arthralgia: more likely in women treated with aromatase inhibitors. 22
Costing
No CYP2D6 genotyping is currently provided by the NHS and so there is no national price list for these tests. This means that the cost of the test used in the economic evaluation should be varied in sensitivity analyses.
Modelling
A simple decision tree could be used to model the sensitivity and specificity of the genotyping test (Figure 3). Beyond this point, a de novo Markov model would be more appropriate. A Markov model structure is considered appropriate because it is assumed that breast cancer is a condition that causes patients to move between a limited number of relevant health states during their lives. This type of model allows a large number of cycles to be simulated without the need to create a new decision tree in each cycle. Figure 4 depicts the schematic model that includes the possible health states and possible transitions between these states.
FIGURE 3.
Decision tree for genotyping for CYP2D6. The Markov model referenced in this figure is depicted in Figure 4.
FIGURE 4.
Markov model for genotyping for CYP2D6.
As can be seen from Figure 4, the Markov model reflects seven health states:
-
DFS without AEs: women at risk of an AE owing to the medication received or at risk of any relapse/recurrence
-
DFS with AEs: women at risk of any relapse/recurrence with AEs
-
contralateral disease: those women with a new primary tumour in the contralateral breast
-
locoregional disease: women suffering a locoregional recurrence or ipsilateral second primary tumour
-
metastatic disease: women with metastases (detail relating to different sites of metastases could be incorporated if relevant data are available)
-
breast cancer death: death from metastatic disease only, as death from either contralateral disease or locoregional disease is unlikely
-
non-breast cancer death: death from any cause apart from breast cancer.
Trying to model the cost-effectiveness of this technology seems to be premature given the quantity and quality of the clinical effectiveness and cost-effectiveness evidence available. It is particularly challenging because there are problems with identifying the alleles to test for, derivation of phenotypes and the lack of cost information available.
Adjustments for timing of costs and outcomes
The model should be developed with a cycle length of 1 year (a 1-year cycle has been used in previous economic evaluations of TAM versus aromatase inhibitors123–126) and be simulated for the remaining lifetime of all patients. The starting age should be derived from the nature of the evidence available. The mean age of the patients in the reviewed cohorts ranged from 43 to 73 years; such wide variation makes it difficult to establish a starting age.
Costs and QALYs should be discounted using a 3.5% annual rate.
Is it appropriate to develop a de novo model with the data available?
In trying to develop a de novo model, it is apparent there are too many uncertainties around the data from the clinical review, as well as other important parameters, to produce even a very simple early economic model. Thus, instead of being able to present findings from a de novo Markov model, our economic assessment has only been able to identify a number of important parameters, and relevant data, which we believe need to be included in a cost-effectiveness model. These important parameters and data requirements are discussed in detail below.
Patient population
As TAM is considered the standard of care for premenopausal women with ER+ breast cancer, and as anti-oestrogen therapy is not used for patients with ER– breast cancer, the only situation in which genotyping for CYP2D6 might prove to be useful is if alternative treatment options to TAM, namely aromatase inhibitors, are considered to be at least as appropriate. Currently, aromatase inhibitors are recommended for women with ER+ early breast cancer deemed not to be at low risk of disease recurrence, and for women with ER+ advanced breast cancer with no prior history of anti-oestrogen therapy. 20 The decision to offer aromatase inhibitors to higher risk patients is largely based on the results of published RCTs21–27 and systematic reviews28–31 of women with ER+ early breast cancer suggesting a modest benefit for patients taking aromatase inhibitors over those taking TAM. Thus, the population of interest is likely to be postmenopausal women with ER+ breast cancer.
It has been proposed that patients be given TAM or an aromatase inhibitor depending on the test results. This decision may be based on their genotype or phenotype. Thus, it is important to know the distribution of postmenopausal patients with early breast cancer with each of these genotypes or phenotypes. This distribution could be inferred from the studies included in the review.
Concomitant CYP2D6 inhibitor medication
While NICE18 states that neither paroxetine nor fluoxetine should be given to women taking TAM, a few studies included in our review state that women on TAM are taking these SSRIs. 49,86,87,91,97,99,109 To accurately reflect this situation in the model, it is necessary to know how SSRIs change the phenotype of these women, as well as the extent to which women with breast cancer are taking paroxetine or fluoxetine in clinical practice. Ferraldeschi et al. 127 conducted a survey to assess the current practice of breast oncologists in the UK with respect to CYP2D6 testing and SSRI co-treatment. The authors reported that 22% of respondents stated that there was enough evidence to routinely use CYP2D6 testing, 37% required more evidence and 41% were unsure. There was general agreement (80%) that concomitant medications might affect the clinical efficacy of TAM, and 86% indicated that they discuss drug interactions with their patients. Importantly, 93% would not prescribe a potent CYP2D6 inhibitor concomitant with TAM and would prescribe alternative treatments, where available.
Adherence to treatment
Two separate studies from the USA128 and the UK129 have reported that non-adherence to TAM ranges from 13% to 22% after the first year of treatment to 50% in years 4 and 5 of treatment. Data on aromatase inhibitor non-adherence suggests that this rises from 14–22% in year 1 to 21–38% in year 3. 130 However, crucially, there is a lack of data with regard to TAM adherence by genotype. Data on adherence are important because what few data we have suggest that the most common reason for discontinuing treatment is experience of hot flushes,95 which are most common in EMs and IMs. 84,90,107
Timing of test
In the absence of any clinical guidelines or prospective clinical studies, it is difficult to know where one would model the CYP2D6 test along the treatment pathway. Currently, the decision to prescribe TAM or an aromatase inhibitor (or indeed, a switching strategy) is made depending on the risk of disease recurrence. However, other factors are also taken into account, for example in relation to AEs from each drug. Thus, would CYP2D6 testing be required only after risk and these other factors been taken into account or would the results from CYP2D6 be another of these other factors to consider? From a practical point of view, as patients are already tested for ER and HER2 status before they are treated, it may be surmised that CYP2D6 testing would occur at the same time, even if the CYP2D6 test results are considered only at a later stage. Finally, in the absence of any clinical guidelines or prospective clinical studies, it is also unclear which pathways patients would follow after CYP2D6 testing, although current treatment pathways for those treated with TAM and aromatase inhibitors would seem logical.
CYP2D6 test availability, cost and accuracy
A number of genotyping tests exist. Many are designed ‘in house’ (using techniques such as TaqMan) to test for specific alleles. Others are offered commercially, such as the AmpliChip. No CYP2D6 genotyping is currently provided by the NHS and so there is no national price list for these tests. Many of these tests are designed specifically for research studies; therefore, the test performance in clinical practice is unknown, although generally genotyping has been shown to have high analytical validity. 45 The greatest uncertainty is in determining clinical sensitivity and specificity and predictive value, i.e. how accurate the test is in linking phenotypes to clinical events. Although we have conducted some exploratory post hoc analyses of sensitivity and specificity for some tests used in the studies included within our systematic review (see Chapter 3, Exploratory analysis: clinical sensitivity and specificity), it should be noted that these are from only a selected sample of studies reporting the necessary data to calculate these values. It is important to note that these are not tests that may be used in clinical practice. They are indicative, however, of the types of data that would be required for an economic model.
Penetrance, the degree of phenotypic expression of genetic variation, is a key parameter for an economic evaluation of pharmacogenetic genetic testing. 131 It is possible to design a test with almost perfect characteristics in terms of test sensitivity and specificity, but the test can still have poor positive predictive value because of the impact of low penetrance, which will affect the cost-effectiveness of the test. We were not able to estimate the degree of gene penetrance and associated positive predictive value for the CYP2D6 test because the clinical data were not able to inform which alleles should be tested. This information is a prerequisite before gene penetrance and test positive predictive value can be established.
Anticipating the importance of the types of tests, alleles tested and their cost to the model, we undertook a survey of several laboratories with regard to their current practices in relation to CYP2D6 testing for patients to be treated with TAM. We chose five laboratories that are currently testing patients for several genes, not only CYP2D6, and, to gain greater insight, chose three from the UK, one from the Netherlands and one from the USA. These laboratories were chosen because they were known to us as laboratories offering the services, often in relation to research studies, and we therefore knew that they were likely to be using the most up-to-date techniques. The survey was conducted between January 2010 and June 2010 and the questions were submitted via e-mail.
The findings from this brief consultation exercise are summarised in Table 25, where it can be seen that there is wide variation regarding the number of alleles tested. The costs also vary considerably by laboratory, from as little as £30 to as much as £500. This wide variation in alleles tested is concordant with the wide variation in the number of alleles tested across the studies included in our review. The wide variation in cost is likely to be due in part to the wide variation in the number of alleles tested, as most assays (e.g. TaqMan) require each allele to be tested individually, so increasing the materials required, time taken, etc. It is perhaps worth noting here that the AmpliChip, which tests for 33 alleles, has been quoted as costing US$500 per test in the USA132 and £300 per test in the UK. 45 The AmpliChip is, to date, the only test that is licensed for use by the FDA and, unlike many tests, is able to test multiple alleles simultaneously.
| Question | Laboratory | ||||
|---|---|---|---|---|---|
| LAB21 (Cambridge, UK) | Mayo Clinic (Rochester, MN, USA) | DxS (Manchester, UK) | LGC (Middlesex, UK) | Erasmus University Medical Centre (Rotterdam) | |
| How many requests per year do you get for CYP2D6 testing for TAM? | Overall number is small but increasing, last 12 months: 12 requests | 1500 tests per year | Two per month | No tests | 300 tests per year |
| When you do clinical testing for CYP2D6 which alleles do you test? | *2, *2A, *3 *4, *6, *7 *8, *9, *10, *11, *12, *17 and N | *2 through *12, *14, *15, *17 and *41 | NS | No tests | *3, *4, *5, *6, *9, *10, *41 |
| Do you use TaqMan? | Yes, along with a kit from Luminex® and sequencing | No, use a kit from Luminex Molecular Diagnostics | No, use Amplification-Refractory Mutation System and Scorpions technology® (DxS Surrey, UK) | No, use a fluorescent probe called HyBeacon® (LGC Middlesex, UK) | Yes |
| Do you offer AmpliChip testing? | No | No, it is too costly | No | No | Yes, and TaqMan analysis as duplicate to confirm the eight most prevalent alleles and the gene duplication |
| How much do you charge for a CYP2D6 test? | £500 | US$439.30 | £30 | NS | €382 |
The number of alleles tested is important to correctly classify patients into their correct phenotype. A recent paper by Schroth et al.,121 re-analysed data from German patients in the large Schroth et al. cohort108 and reported that one-third of patients identified as PMs by the AmpliChip were also identified as PMs by testing for only *4. This proportion rose to 62% when testing for three alleles (*3–*5) and 100% when testing for five (*3–*7). If replicated, the findings from this analysis could suggest that only five alleles need to be tested, if the treatment decision is made on whether or not a patient is a PM. Unfortunately, we do not know this to be true. The clinical evidence from our review suggests that it may be more important to correctly identify EMs (although the evidence relating to endoxifen levels does not seem to support this). Thus, a wider range of alleles would need to be incorporated, in particular those associated with the IM phenotype, such as *10.
Test uptake
The degree of test uptake is an important parameter to consider when evaluating the incremental costs and benefits of pharmacogenetic testing. Low test uptake could affect the cost-effectiveness of the testing, i.e. low uptake reduces the cost-effectiveness. The uptake of pharmacogenetic tests, and whether uptake is driven by the patient or the clinician, are not known and are topics for future research. There are some data that have reported test uptake for breast cancer chemoprevention. 133,134 Uptake rates range between 11.7% in populations with poor genetic counselling and 31% in populations informed by genetic counselling. 133
Time horizon
The time horizon of the model must reflect, at the very least, the duration of the treatment and differences in resource use and/or changes in patient outcomes. Where the time frame of the model exceeds the duration of treatment, it is therefore also important to know how long patients are likely to live for, how long they are likely to be disease free, what other treatments they will receive subsequently, etc. Given that 75% of patients have a life expectancy > 5 years,5 it would seem appropriate to model beyond treatment and until death. As noted above, this means taking into account ‘carry-over’ effects from treatment but (also noted above) may also be problematic given the current lack of established pathways of care. Nevertheless, it would seem feasible to populate a model that followed patients up to death.
Uncertainty
Understanding the impact of uncertainty is a key aspect of any economic model. A model of CYP2D6 testing that is subsequently structured and populated is likely to be an early economic model that in part will aim to inform the need for further research to collect data on key parameters. As a minimum, two key types of uncertainty should be addressed: parameter and structural uncertainty. Parameter uncertainty would be addressed using probabilistic sensitivity analysis, which would allow the analyst to estimate the expected value of perfect information for future research and types of future research required. Structural uncertainty reflecting different possible care pathways should also be explored and included in the model in such a way that allows the analyst to explore the impact of structural uncertainty on whether or not additional evidence is needed. 135
Summary
It is not known if testing for CYP2D6 is cost-effective because no economic evaluations relevant to the UK addressing this question were identified by our review. Two studies79,119 did conduct a modelling exercise but crucially they did not include any data on costs. Other notable weaknesses of these models include the limitation of genotyping to only *4, and the omission of data on AEs.
We have also been unable to produce our own de novo economic model. To a large extent, this is because there is a lack of convincing evidence from the clinical review suggesting that genotyping for CYP2D6 would have any clinical benefit. In addition, there are a number of other important parameters where data would be required for modelling, which are currently lacking. Thus, we have outlined the structure and information requirements appropriate to developing such a Markov model, highlighting the important parameters where more data are required.
Chapter 5 Discussion
From the 25 cohorts included in our clinical review, the evidence is arguably at best suggestive, but not convincing, that genotyping for CYP2D6 may have a role to play in the management of breast cancer. Given that six cohorts83,86,91,96,108,109 suggest that EMs appear to have better outcomes than either PMs or PMs + IMs in terms of relapse/recurrence, this could translate to EMs being suitable candidates for TAM and PMs (and possibly IMs) being offered aromatase inhibitors instead, assuming that the differences in relapse/recurrence outcomes between the two phenotypes are similar in magnitude to the differences found in studies comparing aromatase inhibitors with TAM. However, the suggestive evidence is taken from cohorts that, with two exceptions,108,109 are relatively small in number (≤ 500 patients). In addition, three cohorts92,99,100 have failed to report a similar association. Thus, the evidence must be treated with caution.
Uncertainty in the clinical evidence is compounded further from heterogeneity across the cohorts in terms of patient populations, alleles tested and the manner in which phenotypes are defined. Even within the cohorts there appear to be differences between patients with different genotypes/phenotypes in the few cohorts (n = 8)83,91,93,97,99–101,108 that report these data. To illustrate, two cohorts83,91 appear to show that PMs have poorer outcomes in terms of relapse/recurrence than EMs, and that PMs are more likely to have larger tumours and a greater number of positive lymph nodes, whereas a different cohort100 (which suggests that PMs may have better outcomes than EMs) reported PMs to be less likely to have larger tumours and be LN+. Although the findings were adjusted for these factors, there is still a concern that CYP2D6 status may not be related to outcomes – or at least not directly.
Not only are there differences in terms of patient characteristics, but there are also, just as crucially, differences in outcome definitions. The most unambiguous end point to define is OS, but the only consistent finding across all studies is that there is no relationship between OS and genotype or phenotype. This lack of effect may be because there are indeed no differences or may indicate that longer-term studies are required. Interestingly, evidence published to date comparing OS in patients taking TAM versus aromatase inhibitors has failed to find any significant differences in OS. 28–31
However, perhaps most important of all, there are also differences in terms of the alleles that are being tested. At the very least, all the US and European cohorts41,49,83,86,87,90–92,94,96,98–100,104,108,109,112,113 have genotyped for *4 and all the Asian cohorts73,88,93,97,101,114 have genotyped for *10, the only two alleles for which there appears to be consensus about their importance in these respective populations. However, additional alleles tested for vary from study to study, and only nine cohorts41,82,87,90,96,108,109,113,114 have tested for both *4 and *10. Furthermore, the derivation of phenotypes from these tests also varies from cohort to cohort. An important finding from our review, therefore, is that there does not appear to be any consensus about what alleles to test for or how to derive phenotypes and make meaningful comparisons.
The AmpliChip is an approved CYP2D6 test that could be used in clinical practice and which includes *4 and *10 alongside another 31 alleles. However, only six cohorts41,82,87,90,109,113 in our review utilised the AmpliChip and only two41,87 of these have published findings in full papers. The only other relevant evidence published on the AmpliChip to date has focused on its analytical sensitivity and specificity. 43,136–141
Perhaps the most significant finding from the cohorts using the AmpliChip is that one cohort109 has reported that differences in RFS between PMs and EMs are significant only when the AmpliChip is used and not when testing for just four common alleles (*4, *5, *10 and *41). More recently, a paper by Schroth et al. 121 re-analysed data from German patients in the large Schroth et al. cohort108 and reported that one-third of patients identified as PMs by the AmpliChip were also identified as PMs by testing for only *4 instead. This proportion rose to 62% when testing for three alleles (*3–*5) and to 100% when testing for five (*3–*7). Alongside this evidence, which suggests that a greater number of alleles are required to accurately classify patients, we have undertaken exploratory analysis that also seems to confirm that sensitivity and specificity are increased when the number of alleles tested is increased. However, despite the increase, sensitivity is generally low no matter how many alleles are tested for.
Each of the seven cohorts49,73,87,104,112–114 examining endoxifen concentrations show evidence of an association between these and CYP2D6 status in Caucasians and Asians. Endoxifen levels were reported to be markedly different between both PMs and EMs49,87,104,112,113 and those with the *10/*10 and the wt/wt (EM phenotype) genotypes. 73,114 However, there is conflicting evidence from the two cohorts87,104 regarding IMs (or those with the wt/vt genotype), with one cohort suggesting that IMs have levels closer to EMs104 and the other suggesting that they are closer to PMs. 87
Our review intended to examine the evidence base for an association between endoxifen levels and clinical outcomes, but no study was found which examined this relationship. Assuming that endoxifen concentrations translate into improved outcomes, based on the evidence from the six cohorts83,86,91,96,108,109 that suggest that EMs appear to have better outcomes than either PMs or PMs + IMs in terms of recurrence/relapse, we would probably expect endoxifen concentrations of IMs to be closer to those of PMs. As noted above, in Caucasians, one cohort104 suggested that, on the contrary, endoxifen levels for IMs are closer to EMs than PMs, whereas the other cohort87 did indeed suggest that the levels of IMs are closer to PMs. It is difficult to reconcile these apparently contradictory findings without conducting further studies but reasons for this may be because of the number of patients taking CYP2D6 inhibitors in these studies and/or adherence to TAM or because other enzymes are playing a more important role. It should also be noted that the number of patients included in these two cohorts was small (between 5087 and 151 patients104).
We also intended to review the evidence for clinical utility but, again, our research failed to identify any such studies. Given the lack of convincing evidence for clinical validity, however, this is unsurprising as it would be inappropriate to conduct any clinical utility studies until such evidence becomes available.
Similarly, given the lack of convincing evidence for clinical validity, it is also unsurprising that we identified no full economic evaluations. However, we did identify two decision models that may be informative to later work. 79,119 The findings from these evaluations must be treated with caution, however, because models assume testing only for *4 and include no data on costs. It should also be emphasised that the actual cost of the pharmacogenetic test itself would form only a very small proportion of the overall costs of implementing pharmacogenetic testing into patient care pathways.
Given these deficiencies in the evidence base, we thus encountered a number of problems in attempting to develop a Markov model to address the cost-effectiveness of CYP2D6 testing. Instead, we have been able to identify the important parameters for which additional data are needed to populate an economic model. As we have discussed above, we believe, crucially, that there is too much uncertainty as to which alleles to test for, how to derive phenotypes, which patients would subsequently be considered appropriate to receive TAM and which patients would be considered suitable for aromatase inhibitors. Uncertainty about the type and quantity of alleles to test for makes it difficult to comment on the sensitivity/specificity requirements of any pharmacogenetic test that might be considered for routine use in UK clinical practice; estimating what the cost of such a test might be is also impossible. We also lack evidence of any impact on OS or convincing evidence for other outcomes such as DFS and we need more robust clinical utility data. Crucially, in the absence of any clinical utility studies about CYP2D6 testing, it is impossible to predict how prescribing behaviour may change as a result of genotyping for CYP2D6 and to model future pathways of care, including costs and benefits.
Chapter 6 Conclusions
Our review aimed to answer a number of questions, namely:
-
In patients treated with TAM, do women with breast cancer identified as EMs for CYP2D6 have similar or different clinical outcomes to those identified as PMs, IMs or UMs?
-
Is there a relationship between CYP2D6 status and endoxifen concentrations?
-
Are endoxifen concentrations related to clinical outcomes?
-
– Do women with breast cancer who are identified as EMs for CYP2D6 have similar or different clinical outcomes with TAM compared with aromatase inhibitors?
-
-
What is the relative cost-effectiveness of CYP2D6 testing as a management option for women with breast cancer?
This is a relatively new area of research that is evolving rapidly and, although international consortia are collaborating, the data are limited and conflicting, which limited the ability of the review to answer the questions above.
Six individual cohorts83,86,91,96,108,109 suggest that EMs may have different outcomes in terms of relapse/recurrence (but not OS) to PMs and PMs + IMs. However, this evidence is far from conclusive, based on typically small numbers of patients and heterogeneous patient populations, clinical outcomes and alleles tested, not to mention differences in how both phenotypes and clinical outcomes are defined. In addition, three other cohorts have failed to find an association. There also appears to be evidence of a link between endoxifen concentrations and CYP2D6 status, from even smaller cohorts than have been used to assess efficacy, but this seems to suggest a contrary finding – that PMs are different to EMs + IMs – not supported from the evidence to date in terms of clinical outcomes. Unfortunately, we found no studies that measured the association between endoxifen levels and clinical outcomes, and we found no studies that directly assess whether patients who are identified as EMs for CYP2D6 have similar or different clinical outcomes with TAM when compared with aromatase inhibitors. It might be inferred, however, that if patients who are EMs have better outcomes than PMs and if the magnitude of this difference was similar to that identified in studies comparing aromatase inhibitors with TAM, then EMs might be suitable candidates for TAM, whereas aromatase inhibitors might be more suitable for the PMs. However, as just stated, there is no convincing evidence to support this. Finally, given the lack of data available to address the previous clinical questions and given additional uncertainties surrounding costs and pathways of care, it has been impossible to assess the cost-effectiveness of testing for CYP2D6.
Thus, our review has raised more questions than answers, the most pertinent question being ‘what alleles would one test for in clinical practice?’. The evidence base around CYP2D6 testing to date is at too early a stage of development to be able to ascertain which alleles should be genotyped for and how phenotypes should then be derived. In the absence of any clinical utility studies, there are also too many uncertainties about expected future pathways of care, assuming that a CYP2D6 test were to be conducted. Thus, it is impossible to recommend routine CYP2D6 testing in clinical practice based on the evidence so far available.
Implications for service provision
Owing to a lack of relevant data, it has not been possible to ascertain whether testing for CYP2D6 is clinically effective or cost-effective. In particular, it is unclear which alleles would need to be tested and therefore which test, if any, should be used. Consequently, it is not possible to recommend CYP2D6 testing for routine clinical practice.
Suggested research priorities
There are many areas in which there is a need for further data and thus we have identified the following as research questions to be addressed:
-
How many and what type of alleles should be tested for? It is important that studies are able to determine the alleles which appear to be related to clinical outcomes and which would need to be included in any CYP2D6 test. To achieve this, studies need to include adequate numbers of patients, or at least samples that can be genotyped using techniques that can test for a number of different alleles. To date, there is also no evidence surrounding patients with multiple copies of an allele, i.e. UMs, and so testing to identify UMs may also be prudent.
-
What are acceptable levels of sensitivity and specificity for these tests when measuring different outcomes? To date we have found no literature assessing the sensitivity and specificity of CYP2D6 testing, and this is a matter that needs addressing. Although it is accepted that in the absence of a number of standard test alternatives it is difficult to assess sensitivity and specificity, given the high analytical validity of genotype tests these values may be calculated for testing for specific alleles. Acceptable values for sensitivity and specificity should also be agreed a priori.
-
How should phenotypes for the metabolism of TAM be defined? This is another research question that can be adequately addressed only once the relevant alleles have been determined. It may well be that there is no need to define phenotypes and it is enough to use only genotypes, although such classifications would arguably be of greater utility for patients as well as medical professionals who need to interpret tests.
-
What are the important health outcomes for women with breast cancer, and how should these be defined? The most unambiguous outcome to define is OS, but to date there has been no evidence of any difference in this outcome between genotypes or phenotypes, or indeed between TAM and aromatase inhibitors. This is no reason to dismiss it as an important outcome but, arguably, outcomes that measure relapse/recurrence, such as DFS, are more important for a condition such as breast cancer. While DFS is an outcome measure used in many studies, unfortunately, it is not a standardised outcome measure. For example, some definitions include death from any cause, whereas others include only breast cancer mortality. This is not a problem unique to pharmacogenetic studies, however, but occurs in all breast cancer studies.
-
Do different ethnic populations need to be tested using different tests? This is a research question that can really be addressed only once the relevant alleles have been determined. Initially, it might be useful to use standardised tests across different populations, with the possibility of refining the alleles tested for in different ethnic populations once these alleles have been established. The need to carry out such tests would largely be driven by costs of resources (materials, time, etc.) and, ultimately, it may be safer, simpler and no less cost-effective to use the same test in all populations.
-
What pathways of care would a patient follow if pharmacogenetic testing were to be introduced? Would testing be required for women of any menopausal status? To answer these questions, we need more evidence of differences (if they exist) in outcomes by CYP2D6 status in premenopausal women and a better understanding on current pathways of care. While NICE currently does have recommendations for the use of TAM or aromatase inhibitors based largely on risk of disease recurrence, data on the numbers of women using these are currently lacking; an estimate of the current use of TAM and aromatase inhibitors in the UK is required as the basis for calculating resource use data for an economic model.
-
If pharmacogenetic testing were to be introduced, what would be the uptake of pharmacogenetic testing and would uptake be driven mainly by clinicians or patients? A survey of clinicians’ intentions may be informative. Evidence from other areas in which pharmacogenetic testing has been introduced may also be useful.
Ideally, clinical studies will constitute companion studies to previously conducted RCTs of TAM. We are aware of such studies being undertaken by the ITPC, as well as of previously conducted trials of aromatase inhibitors versus TAM, and the results from these analyses are eagerly awaited.
Finally, TAM metabolism is complex and CYP2D6 does not appear to account for all variability in endoxifen levels. Studies examining the link between endoxifen levels and clinical outcomes are also needed, as are studies that examine polymorphisms in other TAM metabolic pathway enzymes.
Acknowledgements
The authors are pleased to acknowledge Janet Atkinson (Liverpool Reviews Implementation Group, University of Liverpool) who provided administrative support (including obtaining bibliographic sources), Adrian Bagust (Liverpool Reviews Implementation Group, University of Liverpool) for providing comments on the report, Juliet Hockenhull (Liverpool Reviews Implementation Group, University of Liverpool) who assisted with data extraction by cross-checking extracted data with that in the published papers, Nicky Thorp (Clatterbridge Centre for Oncology NHS Foundation Trust , Wirral) who provided additional advice regarding the use of tamoxifen, aromatase inhibitors and CYP2D6 inhibitors in clinical practice, Ayshe Latif (Genetic Medicine, University of Manchester) and Roberta Ferraldeschi (Genetic Medicine, University of Manchester) who provided invaluable insight into CYP2D6 genotyping, and Munir Pirmohamed (Biomedical Sciences, University of Liverpool) for information and advice throughout the project.
Contributions of authors
Nigel Fleeman Project lead, involved in all aspects of the clinical review and report writing.
Carlos Martin Saborido Involved in all aspects of the economics review and report writing.
Katherine Payne Involved in all aspects of the economics review and report writing.
Angela Boland Contributed to analysis and interpretation of data and commented on draft versions of the final report.
Rumona Dickson Participated in protocol development and the initial clinical study selection and commented on draft versions of the final report.
Yenal Dundar Conducted literature searches, assisted with quality assessment and commented on draft versions of the final report.
Ana Fernández Santander Contributed to analysis and interpretation of data and commented on draft versions of the final report.
Sacha Howell Contributed to analysis and interpretation of data and commented on draft versions of the final report.
William Newman Contributed to analysis and interpretation of data and commented on draft versions of the final report.
James Oyee Contributed to analysis and interpretation of data and commented on draft versions of the final report.
Tom Walley Contributed to analysis and interpretation of data and commented on draft versions of the final report.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Cancer Research UK. UK Breast Cancer – Incidence Statistics 2010. http://info.cancerresearchuk.org/cancerstats/types/breast/incidence/ (accessed 27 July 2010).
- Cancer Research UK. Breast Cancer – UK Mortality Statistics 2008. http://info.cancerresearchuk.org/cancerstats/types/breast/mortality/index.htm (accessed 25 January 2010).
- National Institute for Health and Clinical Excellence (NICE) . Hormonal Therapies for the Adjuvant Treatment of Early Oestrogen-Receptor-Positive Breast Cancer 2006.
- Micheli A, Mugno E, Krogh V, Quinn MJ, Coleman M, Hakulinen T, et al. Cancer prevalence in European registry areas. Ann Oncol 2002;13:840-65.
- Cancer Research UK . Breast Cancer – Survival Statistics n.d. http://info.cancerresearchuk.org/cancerstats/types/breast/survival/ (accessed 12 August 2011).
- Ward S, Simpson E, Davis S, Hind D, Rees A, Wilkinson A. Taxanes for the adjuvant treatment of early breast cancer: systematic review and economic evaluation. Health Technol Assess 2007;11.
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994;266:66-71.
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995;378:789-92.
- Pharoah PD, Antoniou A, Bobrow M, Zimmern RL, Easton DF, Ponder BA. Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet 2002;31:33-6.
- The Endogenous Hormones Breast Cancer Collaborative Group . Endogenous sex hormones and breast cancer in postmenopausal women: Reanalysis of nine prospective studies. J Natl Cancer Inst 2002;94:606-16.
- Travis R, Key T. Oestrogen exposure and breast cancer risk. Breast Cancer Res 2003;5:239-47.
- Collaborative Group on Hormonal Factors in Breast Cancer . Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet 2001;358:1389-99.
- John EM, Phipps AI, Knight JA, Milne RL, Dite GS, Hopper JL, et al. Medical radiation exposure and breast cancer risk: Findings from the Breast Cancer Family Registry. Int J Cancer 2007;121:386-94.
- American Joint Committee on Cancer . AJCC Cancer Staging Manual 2009.
- International Union Against Cancer . TNM Classification of Malignant Tumours 2009.
- Galea M, Blamey R, Elston C, Ellis I. The Nottingham Prognostic Index in primary breast cancer. Breast Cancer Res 1992;22:207-19.
- Adjuvant! Online n.d. www.adjuvantonline.com/index.jsp (accessed 27 July 2010).
- National Institute for Health and Clinical Excellence . Early and Locally Advanced Breast Cancer: Diagnosis and Treatment 2009. www.nice.org.uk/Guidance/CG80 (accessed 11 March 2009).
- Dowsett M, Haynes BP. Hormonal effects of aromatase inhibitors: focus on premenopausal effects and interaction with tamoxifen. J Steroid Biochem Mol Biol 2003;86:255-63.
- National Institute for Health and Clinical Excellence . Advanced Breast Cancer: Diagnosis and Treatment 2009. www.nice.org.uk/Guidance/CG81 (accessed 11 March 2009).
- Baum M, Budzar AU, Cuzick J, Forbes J, Houghton JH, Klijn JGM, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet 2002;359:2131-9.
- Breast International Group 1-98 Collaborative Group . A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med 2005;353:2747-57.
- Jakesz R, Samonigg H, Greil R, Gnant M, Schmid M, Kwasny W, et al. Extended adjuvant treatment with anastrozole: Results from the Austrian Breast and Colorectal Cancer Study Group Trial 6a (ABCSG-6a). J Clin Oncol 2005;23.
- Jakesz R, Jonat W, Gnant M, Mittlboeck M, Greil R, Tausch C, et al. Switching of postmenopausal women with endocrine-responsive early breast cancer to anastrozole after 2 years’ adjuvant tamoxifen: combined results of ABCSG trial 8 and ARNO 95 trial. Lancet 2005;366:455-62.
- Coombes RC, Kilburn LS, Snowdon CF, Paridaens R, Coleman RE, Jones SE, et al. Survival and safety of exemestane versus tamoxifen after 2-3 years’ tamoxifen treatment (Intergroup Exemestane Study): a randomised controlled trial. Lancet 2007;369:559-70.
- Boccardo F, Rubagotti A, Guglielmini P, Fini A, Paladini G, Mesiti M, et al. Switching to anastrozole versus continued tamoxifen treatment of early breast cancer. Updated results of the Italian tamoxifen anastrozole (ITA) trial. Ann Oncol 2006;17:vii10-14.
- Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Cancer Inst 2005;97:1262-71.
- Dowsett M, Cuzick J, Ingle J, Coates A, Forbes J, Bliss J, et al. Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J Clin Oncol 2010;28:509-18.
- Eisen A, Trudeau M, Shelley W, Messersmith H, Pritchard KI. Aromatase inhibitors in adjuvant therapy for hormone receptor positive breast cancer: a systematic review. Cancer Treat Rev 2008;34:157-74.
- Hind D, Ward S, De Nigris E, Simpson E, Carroll C, Wyld L. Hormonal therapies for early breast cancer: systematic review and economic evaluation. Health Technol Assess 2007;11.
- Jonat W, Gnant M, Boccardo F, Kaufmann M, Rubagotti A, Zuna I, et al. Effectiveness of switching from adjuvant tamoxifen to anastrozole in postmenopausal women with hormone-sensitive early-stage breast cancer: a meta-analysis. Lancet Oncol 2006;7:991-6.
- Cuppone F, Bria E, Verma S, Pritchard KI, Gandhi S, Carlini P, et al. Do adjuvant aromatase inhibitors increase the cardiovascular risk in postmenopausal women with early breast cancer? Meta-analysis of randomized trials. Cancer 2008;112:260-7.
- Aiello EJ, Buist DS, Wagner EH, Tuzzio L, Greene SM, Lamerato LE, et al. Diffusion of aromatase inhibitors for breast cancer therapy between 1996 and 2003 in the Cancer Research Network. Breast Cancer Res Treat 2008;107:397-403.
- Pirmohamed M, Park BK. Cytochrome P450 enzyme polymorphisms and adverse drug reactions. Toxicology 2003;192:23-32.
- US Food and Drug Administration . Sally’s Tamoxifen Review 2006. www.fda.gov/ohrms/dockets/ac/06/briefing/2006-4248B1-01-FDA-Tamoxifen%20Background%20Summary%20Final.pdf (accessed 12 August 2011).
- Stebbing J, Stearns V, Davidson NE. Role of CYP2D6 testing in selection of endocrine therapy for breast cancer. Pharmacogenomics 2007;8:1-3.
- Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet 1997;60:284-95.
- Alfaro CL, Lam YW, Simpson J, Ereshefsky L. CYP2D6 inhibition by fluoxetine, paroxetine, sertraline, and venlafaxine in a crossover study: intraindividual variability and plasma concentration correlations. J Clin Pharmacol 2000;40:58-66.
- Bradford LD. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002;3:229-43.
- Bernard S, Neville KA, Nguyen AT, Flockhart DA. Interethnic differences in genetic polymorphisms of CYP2D6 in the U.S. population: Clinical implications. Oncologist 2006;11:126-35.
- Ramon YCT, Altes A, Pare L, Del Rio E, Alonso C, Barnadas A, et al. Impact of CYP2D6 polymorphisms in tamoxifen adjuvant breast cancer treatment. Breast Cancer Res Treat 2010;119:33-8.
- US Food and Drug Administration . 510(k) Substantial Equivalence Determination Decision Summary 2004. www.accessdata.fda.gov/cdrh_docs/reviews/K043576.pdf (accessed 21 April 2008).
- Roche Molecular Systems Inc . US Food and Drug Administration. 510(k) Substantial Equivalence Determination Decision Summary for Roche AmpliChip CYP450 Microarray for Identifying CYP2D6 Genotype (510(k) Number k042259) 2004. www.accessdata.fda.gov/cdrh_docs/reviews/K042259.pdf (accessed 12 August 2011).
- Li X, Quigg RJ, Zhou J, Gu W, Nagesh Rao P, Reed EF. Clinical utility of microarrays: current status, existing challenges and future outlook. Curr Genomics 2008;9:466-74.
- Fleeman N, McLeod C, Bagust A, Beale S, Boland A, Dundar Y, et al. The clinical effectiveness and cost-effectiveness of testing for cytochrome P450 polymorphisms in patients with schizophrenia treated with antipsychotics: a systematic review and economic evaluation. Health Technol Assess 2010;14.
- Grossman I. Routine pharmacogenetic testing in clinical practice: dream or reality?. Pharmacogenomics 2007;8:1449-59.
- Piper MA. CYP2D6 pharmacogenomics of tamoxifen treatment. Washington, DC: Blue Cross Blue Shield Association; 2008.
- Centre for Reviews and Dissemination (CRD) . CRD’s Guidance for Undertaking Reviews in Healthcare: Systematic Reviews 2008.
- Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P, et al. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst 2003;95:1758-64.
- Hayden JA, Cote P, Bombardier C. Evaluation of the quality of prognosis studies in systematic reviews. Ann Intern Med 2006;144:427-37.
- Jorgensen AL, Williamson PR. Methodological quality of pharmacogenetic studies: issues of concern. Stat Med 2008;27:6547-69.
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analysis. BMJ 2003;327:557-60.
- Little J, Higgins J. The HuGENet HUGE Review Handbook 2006. www.genesens.net/_intranet/doc_nouvelles/HuGE%20Review%20Handbook%20v11.pdf (accessed 30 June 2008).
- Flockhart DA, O’Kane D, Williams MS, Watson MS, Gage B, Gandolfi R, et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med 2008;10:139-50.
- Bijl M, van Schaik R, Lammers L, Hofman A, Vulto A, van Gelder T, et al. Confirmation of Reduced Breast Cancer Survival in CYP2D6 Poor Metabolizers on Tamoxifen in a Population-Based Cohort Study n.d.
- Bijl MJ, van Schaik RH, Lammers LA, Hofman A, Vulto AG, van Gelder T, et al. The CYP2D6*4 polymorphism affects breast cancer survival in tamoxifen users. Breast Cancer Res Treat 2009;118:125-30.
- Boocock DJ, Brown K, Gibbs AH, Sanchez E, Turteltaub KW, White INH. Identification of human CYP forms involved in the activation of tamoxifen and irreversible binding to DNA. Carcinogenesis 2002;23:1897-901.
- Burton A. Cytochrome genotype affects outcome with tamoxifen. Lancet Oncol 2006;7.
- Chubak J, Buist DSM, Boudreau DM, Rossing MA, Lumley T, Weiss NS. Breast cancer recurrence risk in relation to antidepressant use after diagnosis. Breast Cancer Res Treat 2008;112:123-32.
- Coller JK. Oxidative metabolism of tamoxifen to Z-4-hydroxy-tamoxifen by cytochrome P450 isoforms: an appraisal of in vitro studies. Clin Exp Pharmacol Physiol 2003;30:845-8.
- Connolly RM, Barron TI, Feely J, Kennedy MJ. Co-Prescription Rates of Tamoxifen and CYP2D6 Inhibitors: Are We Compromising Breast Cancer Outcome? n.d.
- Crewe HK, Notley LM, Wunsch RM, Lennard MS, Gillam EMJ. Metabolism of tamoxifen by recombinant human cytochrome P450 enzymes: Formation of the 4-hydroxy, 4′ -hydroxy and N-desmethyl metabolites and isomerization of trans-4-hydroxytamoxifen. Drug Metab Dispos 2002;30:869-74.
- Desta Z, Ward BA, Soukhova NV, Flockhart DA. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: Prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther 2004;310:1062-75.
- Dezentje VO, Nortier JWR, Guchelaar HJ, van de Velde CJH, Gelderblom H. Clinical Implications of CYP2D6 Genotyping on Tamoxifen Treatment in Breast Cancer n.d.
- Dieudonne AS, Lambrechts D, Claes B, Vandorpe T, Wildiers H, Timmerman D, et al. Prevalent breast cancer patients with a homozygous mutant status for CYP2D6*4: response and biomarkers in tamoxifen users. Breast Cancer Res Treat 2009;118:531-8.
- Goetz MP, Suman V, Couch F, Ames M, Rae J, Erlander M, et al. An index based on HOXB13/IL17BR and CYP2D6 for determination of relapse and survival in tamoxifen-treated node negative breast cancer. Breast Cancer Res Treat 2006.
- Goetz MP, Suman VJ, Couch F, Ames MM, Rae JM, Erlander MG, et al. Cytochrome P450 2D6 and homeobox 13/interleukin-17B receptor: Combining inherited and tumor gene markers for prediction of tamoxifen resistance. Clin Cancer Res 2008;14:5864-8.
- Grabinski JL, Smith LS, Chisholm GB, Drengler R, Rodriguez GI, Lang AS, et al. Relationship Between CYP2D6 and Estrogen Receptor Alpha Polymorphisms on Tamoxifen Metabolism in Adjuvant Breast Cancer Treatment n.d.
- Irvin WJ, Peppercorn JM, Dees EC, Lange L, Chiu WK, Evans JP, et al. The Relationship Between CYP2D6 Genotype and Endocrine Symptoms in Women Taking Tamoxifen n.d.
- Johnson MD, Zuo H, Lee K-H, Trebley JP, Rae JM, Weatherman RV, et al. Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res Treat 2004;85:151-9.
- Lash TL, Pedersen L, Cronin-Fenton D, Ahern TP, Rosenberg CL, Lunetta KL, et al. Tamoxifen’s protection against breast cancer recurrence is not reduced by concurrent use of the SSRI citalopram. Br J Cancer 2008;99:616-21.
- Lim YC, Li L, Desta Z, Zhao Q, Rae JM, Flockhart DA, et al. Endoxifen, a secondary metabolite of tamoxifen, and 4-OH-tamoxifen induce similar changes in global gene expression patterns in MCF-7 breast cancer cells. J Pharmacol Exp Ther 2006;318:503-12.
- Lim HS, Lee HJ, Lee KS, Lee ES, Jang IJ, Ro J. Clinical implications of CYP2D6 genotypes predictive of tamoxifen pharmacokinetics in metastatic breast cancer. J Clin Oncol 2007;25:3837-45.
- Mortimer JE, Flatt SW, Parker BA, Gold EB, Wasserman L, Natarajan L, et al. Tamoxifen, hot flashes and recurrence in breast cancer. Breast Cancer Res Treat 2008;108:421-6.
- Ntukidem NI, Nguyen AT, Stearns V, Rehman M, Schott A, Skaar T, et al. Estrogen receptor genotypes, menopausal status, and the lipid effects of tamoxifen. Clin Pharmacol Ther 2008;83:702-10.
- Ro J, Lim HS, Han HS. Clinical outcomes in correlation with pharmacokinetics and CYP2D6 genotypes in metastatic breast cancer patients on tamoxifen. Am J Hematol Oncol 2008;7:65-7.
- Serrano D, Macis D, Gandini S, Johansson H, Guerrieri-Gonzaga A, Mellgren G, et al. Correlation of low dose tamoxifen and its metabolites plasma level with CYP 2D6, and SULT polymorphism; a pharmacogenetics study within a chemoprevention trial. J Clin Oncol 2009;25.
- Sridar C, Kent UM, Notley LM, Gillam EMJ, Hollenberg PF. Effect of tamoxifen on the enzymatic activity of human cytochrome CYP2B6. J Pharmacol Exp Ther 2002;301:945-52.
- Veenstra DL, Lin MP, Garrison LP, Burke W. An exploration of the potential clinical benefits and risks of CYP2D6 testing to guide tamoxifen therapy in breast cancer. Value Health 2009;12.
- Wu X, Hawse JR, Subramaniam M, Goetz MP, Ingle JN, Spelsberg TC. The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor alpha for degradation in breast cancer cells. Cancer Res 2009;69:1722-7.
- Hawse JR, Wu X, Subramaniam M, Goetz P, Spelsberg TC, Ingle TC. Endoxifen, But Not 4-Hydroxytamoxifen, Degrades the Estrogen Receptor in Breast Cancer Cells: A Differential Mechanism of Action Potentially Explaining CYP2D6 Effect n.d.
- Goetz MP, Berry DA, Klein TE. on Behalf of the International Tamoxifen Pharmacogenomics Consortium (ITPC). Adjuvant Tamoxifen Treatment Outcome According to Cytochrome P450 2D6 (CYP2D6) Phenotype in Early Stage Breast Cancer n.d.
- Goetz MP, Knox SK, Suman VJ, Rae JM, Safgren SL, Ames MM, et al. The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Res Treat 2007;101:113-21.
- Goetz MP, Rae JM, Suman VJ, Safgren SL, Ames MM, Visscher DW, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J Clin Oncol 2005;23:9312-18.
- Gonzalez-Santiago S, Zarate R, de la Haba-Rodriguez J, Gomez A, Bandres E, Borrega P, et al. Genetic polymorphism CYP2D6(*4) interaction in clinical outcomes of tamoxifen-treated breast cancer patients. Ann Oncol 2006;17:62-3.
- Gonzalez-Santiago S, Zárate R, Haba-Rodriguez J, Gomez A, Bandres E, Borrega P, et al. CYP2D6*4 polymorphism as blood predictive biomarker of breast cancer relapse in patients receiving adjuvant tamoxifen. J Clin Oncol 2007;25.
- Henry NL, Nguyen A, Azzouz F, Li L, Robarge J, Philips S, et al. Lack of association between oestrogen receptor polymorphisms and change in bone mineral density with tamoxifen therapy. Br J Cancer 2009;102:294-300.
- Kiyotani K, Mushiroda T, Sasa M, Bando Y, Sumitomo I, Hosono N, et al. Impact of CYP2D6*10 on recurrence-free survival in breast cancer patients receiving adjuvant tamoxifen therapy. Cancer Sci 2008;99:995-9.
- Lim HS, Lee HJ, Lee KS, Lee ES, Jang IJ, Ro J. Clinical Implications of CYP2D6 Genotypes Predictive of Tamoxifen Pharmacokinetics in Metastatic Breast Cancer n.d.
- Madlensky L, Flatt SW, Natarajan L, Lawrence HJ, Nikoloff DM, Fontecha M, et al. Hot Flashes Are Associated With CYP2D6 Genotype in Breast Cancer Survivors Taking Tamoxifen n.d.
- Newman WG, Hadfield KD, Latif A, Roberts SA, Shenton A, McHague C, et al. Impaired tamoxifen metabolism reduces survival in familial breast cancer patients. Clin Cancer Res 2008;14:5913-18.
- Nowell SA, Ahn JY, Rae JM, Scheys JO, Trovato A, Sweeney C, et al. Association of genetic variation in tamoxifen-metabolizing enzymes with overall survival and recurrence of disease in breast cancer patients. Breast Cancer Res Treat 2005;91:249-58.
- Okishiro M, Taguchi T, Kim SJ, Shimazu K, Tamaki Y, Noguchi S. Genetic polymorphisms of CYP2D6*10 and CYP2C19*2, *3 are not associated with prognosis, endometrial thickness, or bone mineral density in Japanese breast cancer patients treated with adjuvant tamoxifen. Cancer 2009;115:952-61.
- Onitilo AA, McCarty CA, Wilke RA, Glurich I, Engel JM, Flockhart DA, et al. Estrogen receptor genotype is associated with risk of venous thromboembolism during tamoxifen therapy. Breast Cancer Res Treat 2009;115:643-50.
- Rae JM, Sikora MJ, Henry NL, Li L, Kim S, Oesterreich S, et al. Cytochrome P450 2D6 activity predicts discontinuation of tamoxifen therapy in breast cancer patients. Pharmacogenomics J 2009;9:258-64.
- Schroth W, Antoniadou L, Fritz P, Schwab M, Muerdter T, Zanger UM, et al. Breast cancer treatment outcome with adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. J Clin Oncol 2007;25:5187-93.
- Toyama T, Yamashita H, Sugiura H, Kondo N, Iwase H, Fujii Y. No association between CYP2D6*10 genotype and survival of node-negative japanese breast cancer patients receiving adjuvant tamoxifen treatment. Jpn J Clin Oncol 2009;39:651-6.
- Wang P, Lorizio W, Ziv E, Wu AHB. An Analysis of the Association Between Cytochrome P-450 2D6 Polymorphism and Incidence of Adverse Effects Caused by Tamoxifen in Women With Breast Cancer n.d.
- Wegman P, Elingarami S, Carstensen J, Stal O, Nordenskjold B, Wingren S. Genetic variants of CYP3A5, CYP2D6, SULT1A1, UGT2B15 and tamoxifen response in postmenopausal patients with breast cancer. Breast Cancer Res 2007;9.
- Wegman P, Vainikka L, Stal O, Nordenskjold B, Skoog L, Rutqvist LE, et al. Genotype of metabolic enzymes and the benefit of tamoxifen in postmenopausal breast cancer patients. Breast Cancer Res 2005;7:R284-90.
- Xu Y, Sun Y, Yao L, Shi L, Wu Y, Ouyang T, et al. Association between CYP2D6 *10 genotype and survival of breast cancer patients receiving tamoxifen treatment. Ann Oncol 2008;19:1423-9.
- Borges S, Desta Z, Li L, Skaar TC, Ward BA, Nguyen A, et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: Implication for optimization of breast cancer treatment. Clin Pharmacol Ther 2006;80:61-74.
- Gjerde J, Hauglid M, Breilid H, Lundgren S, Varhaug JE, Kisanga ER, et al. Relationship Between CYP2D6 and SULT1A1 Genotypes and Serum Concentrations of Tamoxifen and Its Metabolites During Steady State Treatment in Breast Cancer Patients n.d.
- Gjerde J, Hauglid M, Breilid H, Lundgren S, Varhaug JE, Kisanga ER, et al. Effects of CYP2D6 and SULT1A1 genotypes including SULT1A1 gene copy number on tamoxifen metabolism. Ann Oncol 2007;19:56-61.
- Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee K-H, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst 2005;97:30-9.
- Lim H, Lee H, Lee K, Lee E, Jang I, Ro J. CYP2D6 Genotypes in Association With Steady State Plasma Concentrations of Active Metabolites of Tamoxifen in Patients With Breast Cancer n.d.
- Henry LN, Rae JM, Li L, Azzouz F, Skaar TC, Desta Z, et al. Association between CYP2D6 genotype and tamoxifen-induced hot flashes in a prospective cohort. Breast Cancer Res Treat 2009;117:571-5.
- Schroth W, Goetz MP, Hamann U, Fasching PA, Schmidt M, Winter S, et al. Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA 2009;302:1429-36.
- Thompson A, Quinlan P, Bray S, Johnson A, Nikoloff M, Fontecha M, et al. CYP2D6 Genotype Affects Outcome in Postmenopausal Breast Cancer Patients Treated With Tamoxifen Monotherapy n.d.
- Goetz M, Suman V, Ames M, Black J, Safgren S, Kuffel M, et al. Tamoxifen pharmacogenetics of CYP2D6, CYP2C19, and SULT1A1: long term follow-up of the North Central Cancer Treatment Group 89-30-52 adjuvant trial. Cancer Res 2009;69.
- Quill T, Abair T, Garcia K, Peck S, Oshaughnessy JA. Benefit from Adjuvant Paclitaxel According to Breast Cancer Subtype Assessed by Tissue Microarrays n.d.
- Bonanni B, Serrano D, Gandini S, Guerrieri-Gonzaga A, Johansson H, Macis D, et al. Randomized biomarker trial of anastrozole or low-dose tamoxifen or their combination in subjects with breast intraepithelial neoplasia. Clin Cancer Res 2009;15:7053-60.
- de Duenas EM, Ochoa E, Ferrer T, Ferrer C, Lisart R, Lopez A, et al. Tamoxifen Pharmacogenetics of CYP2D6: Effect of CYP2D6 Genotype on Tamoxifen Metabolism and Descriptive Analysis of CYP2D6 Variants in a Mediterranean Population of Early Breast Cancer Patients n.d.
- Kiyotani K, Mushiroda T, Imamura CK, Hosono N, Tsunoda T, Kubo M, et al. Significant effect of polymorphisms in CYP2D6 and ABCC2 on clinical outcomes of adjuvant tamoxifen therapy for breast cancer patients. J Clin Oncol 2010;28:1287-93.
- Lorizio W, Beattie M, Tchu S, Melese T, Melisko M, Wu A, et al. Effect of CYP2D6 Pharmacogenetic Testing on Selection of Therapy n.d.
- Goetz MP, Rae JM, Suman VJ, Safgren SL, Ames MM, Visscher DW, et al. Pharmacogenetics of Tamoxifen Biotransformation Is Associated With Clinical Outcomes of Efficacy and Hot Flashes n.d.
- Ferraldeschi R, Newman WG. The impact of CYP2D6 genotyping on tamoxifen treatment. Pharmaceuticals 2010;3:1122-38.
- Blake MJ, Gaedigk A, Pearce RE, Bomgaars LR, Christensen ML, Stowe C, et al. Ontogeny of dextromethorphan O- and N-demethylation in the first year of life. Clin Pharmacol Ther 2007;81:510-16.
- Punglia RS, Burstein HJ, Winer EP, Weeks JC. Pharmacogenomic variation of CYP2D6 and the choice of optimal adjuvant endocrine therapy for postmenopausal breast cancer: a modeling analysis. J Natl Cancer Inst 2008;100:642-8.
- Coates AS, Keshaviah A, Thurlimann B, Mouridsen H, Mauriac L, Forbes JF, et al. Five years of letrozole compared with tamoxifen as initial adjuvant therapy for postmenopausal women with endocrine-responsive early breast cancer: update of study BIG 1-98. J Clin Oncol 2007;25:486-92.
- Schroth W, Hamann U, Fasching PA, Dauser S, Winter S, Eichelbaum M, et al. CYP2D6 polymorphisms as predictors of outcome in breast cancer patients treated with tamoxifen: expanded polymorphism coverage improves risk stratification. Clin Cancer Res 2010;6:4468-77.
- Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med 2003;349:1793-802.
- Delea TE, El-Ouagari K, Karnon J, Sofrygin O. Cost-effectiveness of letrozole versus tamoxifen as initial adjuvant therapy in postmenopausal women with hormone-receptor positive early breast cancer from a Canadian perspective. Breast Cancer Res Treat 2008;108:375-87.
- Delea TE, Karnon J, Sofrygin O, Thomas SK, Papo NL, Barghout V. Cost-effectiveness of letrozole versus tamoxifen as initial adjuvant therapy in hormone receptor-positive postmenopausal women with early-stage breast cancer. Clin Breast Cancer 2007;7:608-18.
- El Ouagari K, Karnon J, Delea T, Talbot W, Brandman J. Cost-effectiveness of letrozole in the extended adjuvant treatment of women with early breast cancer. Breast Cancer Res Treat 2007;101:37-49.
- Karnon J. Aromatase inhibitors in breast cancer: a review of cost considerations and cost effectiveness. Pharmacoeconomics 2006;24:215-32.
- Ferraldeschi R, Howell S, Thompson A, Newman W. Avoidance of CYP2D6 inhibitors in patients receiving tamoxifen. J Clin Oncol 2010;28:e584-e585.
- Partridge AH, Wang PS, Winer EP, Avorn J. Nonadherence to adjuvant tamoxifen therapy in women with primary breast cancer. J Clin Oncol 2003;21:602-6.
- McCowan C, Shearer J, Donnan PT, Dewar JA, Crilly M, Thompson AM, et al. Cohort study examining tamoxifen adherence and its relationship to mortality in women with breast cancer. Br J Cancer 2008;99:1763-8.
- Partridge AH, LaFountain A, Mayer E, Taylor BS, Winer E, Asnis-Alibozek A. Adherence to initial adjuvant anastrozole therapy among women with early-stage breast cancer. J Clin Oncol 2008;26:556-62.
- Veenstra DL, Higashi MK, Phillips KA. Assessing the cost-effectiveness of pharmacogenomics. AAPS PharmSci 2000;2.
- Carlson B. First-in-class product seeks to promote better prescribing habits. Biotechnol Healthc 2009;6:7-9.
- Ropka ME, Keim J, Philbrick JT. Patient decisions about breast cancer chemoprevention: a systematic review and meta-analysis. J Clin Oncol 2010;28:3090-5.
- Matloff ET, Moyer A, Shannon KM, Niendorf KB, Col NF. Healthy women with a family history of breast cancer: impact of a tailored genetic counseling intervention on risk perception, knowledge, and menopausal therapy decision making. J Womens Health (Larchmt) 2006;15:843-56.
- Bojke L, Claxton K, Sculpher M, Palmer S. Characterizing structural uncertainty in decision analytic models: a review and application of methods. London: Blackwell Publishing; 2009.
- Dukek BA, O’Kane DJ. Comparison of Roche diagnostics AmpliChip CYP450IVD test with TM Bioscience Tag-It cytochrome P450 2D6 and 2C19 tests. Clin Chem 2006;52.
- Heller T, Kirchheiner J, Armstrong VW, Lathe H, Brockmoller J, Oellerich M. Assessment of Amplichip CYP450-based CYP2D6-genotyping and phenotype prediction compared to PCRRFLP- genotyping and phenotyping by metoprolol pharmacokinetics. Ther Drug Monit 2005;27.
- Heller T, Kirchheiner J, Armstrong VW, Luthe H, Tzvetkov M, Brockmöller J, et al. AmpliChip CYP450 GeneChip: a new gene chip that allows rapid and accurate CYP2D6 genotyping. Ther Drug Monit 2006;28:673-7.
- Lawrence HJ, Truong S, Patten N, Nakao A, Wu L. Detection of p53 Mutations in Cancer by the Amplichip p53 Test, a Microarray-Based Resequencing Assay n.d.
- Melis R, Lyon E, McMillin GA. Determination of CYP2D6, CYP2C9 and CYP2C19 genotypes with Tag-It mutation detection assays. Exp Rev Mol Diagn 2006;6:811-20.
- Rebsamen MC, Desmeules J, Daali Y, Chiappe A, Diemand A, Rey C, et al. The AmpliChip CYP450 test: cytochrome P450 2D6 genotype assessment and phenotype prediction. Pharmacogenomics J 2009;9:34-41.
- Dezentje VO, Guchelaar HJ, Nortier JWR, van de Velde CJH, Gelderblom H. Clinical implications of CYP2D6 genotyping in tamoxifen treatment for breast cancer. Clin Cancer Res 2009;15:15-21.
- Anderson K, Jacobson JS, Heitjan DF, Zivin JG, Hershman D, Neugut AI, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006;144:397-406.
- Annemans L. Methodological issues in evaluating cost effectiveness of adjuvant aromatase inhibitors in early breast cancer: a need for improved modelling to aid decision making. Pharmacoeconomics 2008;26:409-23.
- Armstrong K, Chen TM, Albert D, Randall TC, Schwartz JS. Cost-effectiveness of raloxifene and hormone replacement therapy in postmenopausal women: impact of breast cancer risk. Obstet Gynecol 2001;98:996-1003.
- Benedict A, Brown RE. Review of cost-effectiveness analyses in hormonal therapies in advanced breast cancer. Expert Opin Pharmacother 2005;6:1789-801.
- Preventive health program aims to reduce cost, incidence of breast cancer. Clin Resource Manag 2001;2:26-9.
- Borgstrom F, Johnell O, Kanis JA, Oden A, Sykes D, Jonsson B. Cost effectiveness of raloxifene in the treatment of osteoporosis in Sweden: an economic evaluation based on the MORE study. Pharmacoeconomics 2004;22:1153-65.
- Cuzick J, Sasieni P, Howell A. Should aromatase inhibitors be used as initial adjuvant treatment or sequenced after tamoxifen?. Br J Cancer 2006;94:460-4.
- Delea TE, Karnon J, Smith RE, Johnston SRD, Brandman J, Sung JCY, et al. Cost-effectiveness of extended adjuvant letrozole therapy after 5 years of adjuvant tamoxifen therapy in postmenopausal women with early-stage breast cancer. Am J Manag Care 2006;12:374-86.
- Dranitsaris G, Verma S, Trudeau M. Cost utility analysis of first-line hormonal therapy in advanced breast cancer: comparison of two aromatase inhibitors to tamoxifen. Am J Clin Oncol 2003;26:289-96.
- Duelge J, Hillner TJ. Is tamoxifen cost-effective for prevention of breast cancer?. J Clin Oncol 2000;18.
- Dunn C, Keam SJ. Letrozole: a pharmacoeconomic review of its use in postmenopausal women with breast cancer. Pharmacoeconomics 2006;24:495-517.
- Eckermann SD, Martin AJ, Stockler MR, Simes RJ. The benefits and costs of tamoxifen for breast cancer prevention. Aust NZ J Public Health 2003;27:34-40.
- Eisinger F. CYP2D6 and Ockham’s razor. J Clin Oncol 2008;26:686-7.
- Gil JM, Rubio-Terres C, Del Castillo A, Gonzalez P, Canorea F. Pharmacoeconomic analysis of adjuvant therapy with exemestane, anastrozole, letrozole or tamoxifen in postmenopausal women with operable and estrogen receptor-positive breast cancer. Clin Transl Oncol 2006;8:339-48.
- Goeree R, Blackhouse G, Adachi J. Cost-effectiveness of alternative treatments for women with osteoporosis in Canada. Curr Med Res Opin 2006;22:1425-36.
- Hershman D, Sundararajan V, Jacobson JS, Heitjan DF, Neugut AI, Grann VR. Outcomes of tamoxifen chemoprevention for breast cancer in very high-risk women: a cost-effectiveness analysis. J Clin Oncol 2002;20:9-16.
- Higa GM. New generation aromatase inhibitors in breast cancer. Weighing out potential costs and benefits. Pharmacoeconomics 2000;17:121-32.
- Higa GM. Aromatase inhibitors for breast cancer: pharmacoeconomic considerations. Expert Opin Pharmacother 2001;2:987-95.
- Hillner BE, Radice D. Cost-effectiveness analysis of exemestane compared with megestrol in patients with advanced breast carcinoma. Cancer 2001;91:484-9.
- Hillner BE. Benefit and projected cost-effectiveness of anastrozole versus tamoxifen as initial adjuvant therapy for patients with early-stage estrogen receptor-positive breast cancer. Cancer 2004;101:1311-22.
- Imai H, Kuroi K, Ohsumi S, Ono M, Shimozuma K. Economic evaluation of the prevention and treatment of breast cancer – present status and open issues. Breast Cancer 2007;14:81-7.
- Kanis JA, Borgstrom F, Johnell O, Oden A, Sykes D, Jonsson B. Cost-effectiveness of raloxifene in the UK: an economic evaluation based on the MORE study. Osteoporos Int 2005;16:15-2.
- Karnon J, Jones T. A stochastic economic evaluation of letrozole versus tamoxifen as a first-line hormonal therapy: for advanced breast cancer in postmenopausal patients. Pharmacoeconomics 2003;21:513-25.
- Karnon J, Johnston SRD, Jones T, Glendenning A. A trial-based cost-effectiveness analysis of letrozole followed by tamoxifen versus tamoxifen followed by letrozole for postmenopausal advanced breast cancer. Ann Oncol 2003;14:1629-33.
- Karnon J, Delea T, Johnston SRD, Smith R, Brandman J, Sung J, et al. Cost effectiveness of extended adjuvant letrozole in postmenopausal women after adjuvant tamoxifen therapy: the UK perspective. Pharmacoeconomics 2006;24:237-50.
- Karnon J, Delea T, Barghout V. Cost utility analysis of early adjuvant letrozole or anastrozole versus tamoxifen in postmenopausal women with early invasive breast cancer: the UK perspective. Eur J Health Econ 2008;9:171-83.
- Keyzer JF, Melnikow J, Kuppermann M, Birch S, Kuenneth C, Nuovo J, et al. Recruitment strategies for minority participation: challenges and cost lessons from the POWER interview. Ethn Dis 2005;15:395-406.
- Kellokumpu-Lehtinen P, Bergh J, Salminen E, Wiklund T, Lehtinen S, Aronen P, et al. Cost-effectiveness of intensive adjuvant chemotherapy for high-risk breast cancer: is tailored and dose-escalated chemotherapy with growth factor support (GFS) more costly and less effective than marrow-supported high-dose chemotherapy: results from a randomized study. Acta Oncol 2007;46:146-52.
- Kilian R, Porzsolt F. When to recommend and to pay for first-line adjuvant breast cancer treatment? A structured review of the literature. Breast 2005;14:636-42.
- Lindgren P, Jonsson B, Redaelli A, Radice D. Cost-effectiveness analysis of exemestane compared with megestrol in advanced breast cancer: a model for Europe and Australia. Pharmacoeconomics 2002;20:101-8.
- Locker GY, Mansel R, Cella D, Dobrez D, Sorensen S, Gandhi SK, et al. Cost-effectiveness analysis of anastrozole versus tamoxifen as primary adjuvant therapy for postmenopausal women with early breast cancer: a US healthcare system perspective. The 5-year completed treatment analysis of the ATAC (‘Arimidex’, Tamoxifen Alone or in Combination) trial. Breast Cancer Res Treat 2007;106:229-38.
- Lonning PE. Comparing cost/utility of giving an aromatase inhibitor as monotherapy for 5 years versus sequential administration following 2-3 or 5 years of tamoxifen as adjuvant treatment for postmenopausal breast cancer. Ann Oncol 2006;17:217-25.
- Lundkvist J, Wilking N, Holmberg S, Jonsson L. Cost-effectiveness of exemestane versus tamoxifen as adjuvant therapy for early-stage breast cancer after 2-3 years treatment with tamoxifen in Sweden. Breast Cancer Res Treat 2007;102:289-99.
- Mansel R, Locker G, Fallowfield L, Benedict A, Jones D. Cost-effectiveness analysis of anastrozole vs tamoxifen in adjuvant therapy for early stage breast cancer in the United Kingdom: the 5-year completed treatment analysis of the ATAC (‘Arimidex’, Tamoxifen alone or in combination) trial. Br J Cancer 2007;97:152-61.
- Marchetti M, Caruggi M, Colombo G. Cost utility and budget impact of third-generation aromatase inhibitors for advanced breast cancer: a literature-based model analysis of costs in the Italian National Health Service. Clin Ther 2004;26:1546-61.
- Meadows ES, Klein R, Rousculp MD, Smolen L, Ohsfeldt RL, Johnston JA. Cost-effectiveness of preventative therapies for postmenopausal women with osteopenia. BMC Womens Health 2007;7.
- Melnikow J, Birch S, Slee C, McCarthy TJ, Helms LJ, Kuppermann M. Tamoxifen for breast cancer risk reduction: impact of alternative approaches to quality-of-life adjustment on cost-effectiveness analysis. Med Care 2008;46:946-53.
- Miller L-AN, Roy A, Mody R, Higa GM. Comparative economic analysis of aromatase inhibitors and tamoxifen in the treatment of hormone-dependent breast cancer. Expert Opin Pharmacother 2007;8:1675-91.
- Moeremans K, Annemans L. Cost-effectiveness of anastrozole compared to tamoxifen in hormone receptor-positive early breast cancer. Analysis based on the ATAC trial. Int J Gyn Cancer 2006;16:S576-8.
- Mullins CD, Ohsfeldt RL. Modeling the annual costs of postmenopausal prevention therapy: raloxifene, alendronate, or estrogen-progestin therapy. J Manag Care Pharm 2003;9:150-8.
- Naeim A, Keeler EB. Is adjuvant therapy for older patients with node(–) early breast cancer cost-effective?. Crit Rev Oncol Hemat 2005;53:81-9.
- National Institute for Health and Clinical Excellence . Breast Cancer (early): Hormonal Treatments: Costing Template and Report 2006. http://guidance.nice.org.uk/TA112/CostingReport/xls/English (accessed 12 August 2011).
- Okubo I, Kondo M, Toi M, Ochiai T, Miki S. Cost-effectiveness of letrozole versus tamoxifen as first-line hormonal therapy in treating postmenopausal women with advanced breast cancer in Japan. Jpn J Cancer Chemo 2005;32:351-63.
- Ozanne EM, Esserman LJ. Evaluation of breast cancer risk assessment techniques: a cost-effectiveness analysis. Cancer Epidemiol Biomarkers Prev 2004;13:2043-52.
- Risebrough NA, Verma S, Trudeau M, Mittmann N. Cost-effectiveness of switching to exemestane versus continued tamoxifen as adjuvant therapy for postmenopausal women with primary breast cancer. Cancer 2007;110:499-508.
- Rocchi A, Verma S. Anastrozole is cost-effective vs tamoxifen as initial adjuvant therapy in early breast cancer: Canadian perspectives on the ATAC completed-treatment analysis. Support Care Cancer 2006;14:917-27.
- Rodriguez-Antona C, Gurwitz D, de Leon J, Llerena A, Kirchheiner J, de Mesa EG, et al. CYP2D6 genotyping for psychiatric patients treated with risperidone: Considerations for cost-effectiveness studies. Pharmacogenomics 2009;10:685-99.
- Sher DJ, Wittenberg E, Suh WW, Taghian AG, Punglia RS. Partial-breast irradiation versus whole-breast irradiation for early-stage breast cancer: a cost-effectiveness analysis. Int J Radiat Oncol Biol Phys 2009;74:440-6.
- Simons WR, Jones D, Buzdar A. Cost-effectiveness of anastrozole versus tamoxifen as first-line therapy for postmenopausal women with advanced breast cancer. Clin Ther 2003;25:2972-87.
- Skedgel C, Rayson D, Dewar R, Younis T. Cost–utility of adjuvant hormone therapies for breast cancer in post-menopausal women: sequential tamoxifen–exemestane and upfront anastrozole. Breast Cancer Res Treat 2007;101:325-33.
- Skedgel C, Rayson D, Dewar R, Younis T. Cost–utility of adjuvant hormone therapies with aromatase inhibitors in post-menopausal women with breast cancer: Upfront anastrozole, sequential tamoxifen–exemestane and extended tamoxifen–letrozole. Breast 2007;16:252-61.
- Smith TJ, Hillner BE. Tamoxifen should be cost-effective in reducing breast cancer risk in high-risk women. J Clin Oncol 2000;18:284-6.
- Thompson D, Taylor DCA, Montoya EL, Winer EP, Jones SE, Weinstein MC. Cost-effectiveness of switching to exemestane after 2 to 3 years of therapy with tamoxifen in postmenopausal women with early-stage breast cancer. Value Health 2007;10:367-76.
- Williams C, Brunskill S, Altman D, Briggs A, Campbell H, Clarke M, et al. Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy. Health Technol Assess 2006;10.
- Younis T, Rayson D, Dewar R, Skedgel C. Modeling for cost-effective-adjuvant aromatase inhibitor strategies for postmenopausal women with breast cancer. Ann Oncol 2007;18:293-8.
- Irvin WJ, Carey LA, Olajide O, Dees EC, Peppercorn J, Chiu WK, et al. Comprehensive CYP2D6 genotyping in a multiracial U.S. breast cancer population. J Clin Oncol 2009;27.
Appendix 1 Literature search strategies
Searches for studies linking outcomes to CYP2D6
Ovid MEDLINE, 2000 to June week 2 2009
| Hits | ||
|---|---|---|
| 1 | exp Genotype/ | 145,874 |
| 2 | exp Phenotype/ | 98,789 |
| 3 | (genotype$or phenotype$).tw. | 198,382 |
| 4 | exp Cytochrome P-450 Enzyme System/ | 31,587 |
| 5 | (CYP2D6 or CYP 2D6).mp. | 9297 |
| 6 | AmpliChip®$.tw. | 17 |
| 7 | or/1-6 | 349,228 |
| 8 | (tamoxifen or endoxifen or aromatase inhibitor$or anastrozole or arimidex or letrozole or femara or exemestane or aromasin or nolvadex or 4-hydroxy-N-desmethyl-tamoxifen).af. | 12,954 |
| 9 | exp Tamoxifen/ | 9297 |
| 10 | exp Aromatase Inhibitors/ | 2726 |
| 11 | or/8-10 | 14,339 |
| 12 | exp Breast Neoplasms/ | 91,430 |
| 13 | (breast$adj5 (neoplasm$or cancer$or tumo?r$or carcinoma$or adenocarcinoma$or sarcoma$or dcis or ductal or infiltrat$or intraductal$or lobular or medullary)).mp. | 112,215 |
| 14 | or/12-13 | 112,255 |
| 15 | 7 and 11 and 14 | 683 |
| 16 | animals/not (animals/and humans/) | 1,231,707 |
| 17 | 15 not 16 | 653 |
| 18 | limit 17 to (yr = “2000 - 2009”) | 544 |
EMBASE, 2000–9 week 24
| Hits | ||
|---|---|---|
| 1 | exp Genotype/ | 102,077 |
| 2 | exp Phenotype/ | 122,694 |
| 3 | (genotype$or phenotype$).tw. | 181,223 |
| 4 | exp Cytochrome P450/ | 16,955 |
| 5 | (CYP2D6 or CYP 2D6).mp. | 2854 |
| 6 | AmpliChip®$.tw. | 22 |
| 7 | or/1-6 | 275,475 |
| 8 | (tamoxifen or endoxifen or aromatase inhibitor$or anastrozole or arimidex or letrozole or femara or exemestane or aromasin or nolvadex or 4-hydroxy-N-desmethyl-tamoxifen).af. | 25,169 |
| 9 | exp Tamoxifen/ | 21,139 |
| 10 | exp Aromatase Inhibitor/ | 8089 |
| 11 | or/8-10 | 26,043 |
| 12 | exp Breast Tumour/ | 115,251 |
| 13 | (breast$adj5 (neoplasm$or cancer$or tumo?r$or carcinoma$or adenocarcinoma$or sarcoma$or dcis or ductal or infiltrat$or intraductal$or lobular or medullary)).mp. | 127,487 |
| 14 | or/12-13 | 128,366 |
| 15 | 7 and 11 and 14 | 656 |
| 16 | limit 15 to (human and yr = “2000 - 2009”) | 543 |
Web of Science, Science Citations Index and Conference Proceedings Science Index
| Hits | |
|---|---|
| Topic = ((genotype* or phenotype* or CYP2D6 or CYP 2D6 or Cytochrome P-450 or AmpliChip®*)) AND Topic = ((tamoxifen or endoxifen or aromatase inhibitor* or anastrozole or arimidex or letrozole or femara or exemestane or aromasin or nolvadex or 4-hydroxy-N-desmethyl-tamoxifen)) AND Topic = ((breast neoplasm* or breast cancer* or breast tumour* or breast tumour* or breast carcinoma* or breast adenocarcinoma* or breast sarcoma*)) | 365 |
The Cochrane Library, issue 2
| Hits | |
|---|---|
| (genotype* or phenotype* or CYP2D6 or CYP 2D6 or Cytochrome P-450 or AmpliChip®*) and (tamoxifen or endoxifen or aromatase inhibitor* or anastrozole or arimidex or letrozole or femara or exemestane or aromasin or nolvadex or 4-hydroxy-N-desmethyl-tamoxifen) and (breast neoplasm* or breast cancer* or breast tumour* or breast tumour* or breast carcinoma* or breast adenocarcinoma* or breast sarcoma*) | 7 |
Searches for studies linking outcomes to endoxifen
Ovid MEDLINE, 2000 to June Week 2 2009
| Hits | ||
|---|---|---|
| 1 | (tamoxifen or endoxifen or 4-hydroxy-N-desmethyl-tamoxifen).af. or exp Tamoxifen/ | 12,129 |
| 2 | exp Breast Neoplasms/or (breast$adj5 (neoplasm$or cancer$or tumo?r$or carcinoma$or adenocarcinoma$or sarcoma$or dcis or ductal or infiltrat$or intraductal$or lobular or medullary)).mp. | 113,203 |
| 3 | 1 and 2 | 7187 |
| 4 | animals/not (animals/and humans/) | 1,240,444 |
| 5 | 3 not 4 | 6968 |
| 6 | limit 5 to yr = “2000 - 2009” | 5403 |
| 7 | limit 6 to english language | 4974 |
The searches for the studies for the economics review were identified from the combined searches above.
Appendix 2 Table of excluded studies with rationale
Excluded studies from clinical review
The following citations were excluded at screening stage 2:
| Study | Reason for exclusion |
|---|---|
| Bijl et al. 200955,56 | Includes mostly patients with metastatic disease (≥ 75%) (Bijl et al. 200955 is a conference abstract) |
| Boocock et al. 200257 | Wrong outcome (N-desmethyl-TAM, not endoxifen) |
| Burton 200658 | Not a research study (description of Goetz et al. 200584) |
| Chubak et al. 200859 | Does not consider outcomes by CYP2D6 genotype |
| Coller 200360 | Not a primary research study (review) |
| Connolly et al. 200761 | Does not consider outcomes by CYP2D6 genotype |
| Crewe et al. 200262 | PK study that does not consider endoxifen |
| Desta et al. 200463 | PK study that does not consider endoxifen |
| Dezentje et al. 200864 | Not a primary research study (review, subsequently published in 2009142) |
| Dieudonne et al. 200965 | Wrong outcome (changes in follicle-stimulating hormone and sex hormone-binding globulin) |
| aGoetz et al. 2006,66 200867 | Does not consider outcomes by CYP2D6 genotype (index including HOXB13/IL17BR) (Goetz et al. 200666 is interim analysis presented as abstract) |
| Grabinski et al. 200668 | Wrong outcome (plasma levels of TAM and 4-hydroxytamoxifen, not endoxifen) |
| Johnson et al. 200470 | Does not link endoxifen to clinical outcomes |
| Does not consider endoxifen plasma levels by CYP2D6 genotype | |
| Lash et al. 200871 | Does not consider outcomes by CYP2D6 genotype |
| aLim et al. 200672 | Does not link endoxifen to clinical outcomesa |
| Does not consider endoxifen plasma levels by CYP2D6 genotypea | |
| aLim et al. 200773 | Includes only patients with metastatic disease in efficacy studya |
| aMortimer et al. 200874 | Does not consider outcomes by CYP2D6 genotype |
| aNtukidem et al. 200875 | Wrong outcome (serum total cholesterol) |
| Ro et al. 200876 | Single case reports |
| Serrano et al. 200977 | Wrong outcome (plasma levels of N-desmethyl-TAM, not endoxifen) |
| Wrong setting (chemoprevention) | |
| Sridar et al. 200278 | PK study that does not consider endoxifen or CYP2D6 |
| Veenstra et al. 200979 | Not a primary research study (economic analysis) |
| Wu et al. 200980 and Hawse et al. 200881 | Does not link endoxifen to clinical outcomes (considers metabolism of endoxifen in vitro) (Hawse et al. 200881 is interim analysis presented as conference abstract) |
In addition, one of the included citations by Lim et al. 73 also included data on an efficacy study containing only patients with metastatic disease. These data were excluded from the review, but as the citation also included data on a separate pharmacokinetic study of patients with early and metastatic breast cancer, this citation is included in the review.
Excluded studies from economics review
| Study | Reason for exclusion |
|---|---|
| Anderson et al. 2006143 | Does not consider CYP2D6 testing |
| Annemans 2008144 | Does not consider CYP2D6 testing |
| Armstrong et al. 2001145 | Does not consider CYP2D6 testing |
| Benedict and Brown 2005146 | Does not consider CYP2D6 testing |
| BlueCross BlueShield 2001147 | Does not consider CYP2D6 testing |
| Borgstrom et al. 2004148 | Does not consider CYP2D6 testing |
| Cuzick et al. 2006149 | Does not consider CYP2D6 testing |
| Delea et al. 2006150 | Does not consider CYP2D6 testing |
| Delea et al. 2007124 | Does not consider CYP2D6 testing |
| Delea et al. 2008123 | Does not consider CYP2D6 testing |
| Dranitsaris et al. 2003151 | Does not consider CYP2D6 testing |
| Duelge and Hillner 2000152 | Does not consider CYP2D6 testing |
| Dunn and Keam 2006153 | Does not consider CYP2D6 testing |
| Eckermann et al. 2003154 | Does not consider CYP2D6 testing |
| Eisinger 2008155 | Not an economic evaluation |
| El Ouagari et al. 2007125 | Does not consider CYP2D6 testing |
| aFleeman et al. 201045 | Not related to breast cancer |
| Gil et al. 2006156 | Does not consider CYP2D6 testing |
| Goeree et al. 2006157 | Does not consider CYP2D6 testing |
| Hershman et al. 2002158 | Does not consider CYP2D6 testing |
| Higa 2000159 | Does not consider CYP2D6 testing |
| Higa 2001160 | Does not consider CYP2D6 testing |
| Hillner and Radice 2001161 | Does not consider CYP2D6 testing |
| Hillner 2004162 | Does not consider CYP2D6 testing |
| Hind et al. 200730 | Does not consider CYP2D6 testing |
| Imai et al. 2007163 | Does not consider CYP2D6 testing |
| Kanis et al. 2005164 | Does not consider CYP2D6 testing |
| Karnon and Jones 2003165 | Does not consider CYP2D6 testing |
| Karnon et al. 2003166 | Does not consider CYP2D6 testing |
| Karnon 2006126 | Does not consider CYP2D6 testing |
| Karnon et al. 2006167 | Does not consider CYP2D6 testing |
| Karnon et al. 2008168 | Does not consider CYP2D6 testing |
| Keyzer et al. 2005169 | Does not consider CYP2D6 testing |
| Kellokumpu-Lehtinen et al. 2007170 | Does not consider CYP2D6 testing |
| Kilian and Porzsolt 2005171 | Does not consider CYP2D6 testing |
| Lindgren et al. 2002172 | Does not consider CYP2D6 testing |
| Locker et al. 2007173 | Does not consider CYP2D6 testing |
| Lonning 2006174 | Does not consider CYP2D6 testing |
| Lundkvist et al. 2007175 | Does not consider CYP2D6 testing |
| Mansel et al. 2007176 | Does not consider CYP2D6 testing |
| Marchetti et al. 2004177 | Does not consider CYP2D6 testing |
| Meadows et al. 2007178 | Does not consider CYP2D6 testing |
| Melnikow et al. 2008179 | Does not consider CYP2D6 testing |
| Miller et al. 2007180 | Does not consider CYP2D6 testing |
| Moeremans and Annemans 2006181 | Does not consider CYP2D6 testing |
| Mullins and Ohsfeldt 2003182 | Does not consider CYP2D6 testing |
| Naeim and Keeler 2005183 | Does not consider CYP2D6 testing |
| NICE 2006184 | Does not consider CYP2D6 testing |
| Okubo et al. 2005185 | Does not consider CYP2D6 testing |
| Ozanne and Esserman 2004186 | Does not consider CYP2D6 testing |
| Punglia et al. 2008119 | Includes CYP2D6 testing but does not include costs |
| Risebrough et al. 2007187 | Does not consider CYP2D6 testing |
| Rocchi and Verma 2006188 | Does not consider CYP2D6 testing |
| Rodriguez-Antona et al. 2009189 | Not related to breast cancer |
| Sher et al. 2009190 | Does not consider CYP2D6 testing |
| Simons et al. 2003191 | Does not consider CYP2D6 testing |
| Skedgel et al. 2007192 | Does not consider CYP2D6 testing |
| Skedgel et al. 2007193 | Does not consider CYP2D6 testing |
| Smith and Hillner 2000194 | Does not consider CYP2D6 testing |
| Thompson et al. 2007195 | Does not consider CYP2D6 testing |
| Veenstra et al. 200979 | Includes CYP2D6 testing but does not include costs |
| Williams et al. 2006196 | Does not consider CYP2D6 testing |
| Younis et al. 2007197 | Does not consider CYP2D6 testing |
Ongoing studies
One study appears to meet inclusion criteria of this review, but has not yet reported:
| Study | Outcomes to be measured |
|---|---|
| Irvin et al. 200969,198 |
Change in endoxifen levels after an increase in the TAM dose from 20 to 40 mg in patients with CYP2D6 IM genotypes Tolerability of increasing the dose of TAM from 20 to 40 mg per day in patients with CYP2D6 IM genotypes Feasibility of obtaining pharmacogenomic information from patients in the clinical setting and using it to guide changes in therapy CYP2D6 allele frequencies and endoxifen levels among African American women taking TAM Change in plasma endoxifen levels after an increase in TAM dose from 20 to 40 mg daily in patients with poor-metabolising genotypes |
Another ongoing study may be of interest regarding clinical utility:
| Study | Study details |
|---|---|
| Lorizio et al. 2009115 | Patients taking TAM, or for whom TAM was recommended, participate in a teaching session that discusses both positive and negative results regarding CYP2D6 genotype and breast cancer recurrence. CYP2D6 testing offered to all participants at the end of the session; results then released to their clinician. Clinicians informed of test results but no specific treatment recommendation provided. To determine whether or not a change in medication occurred, a follow-up phone call is conducted 4–6 months later. To date, 180 women have been enrolled, 100 have received the follow-up call, of which five were classified PM. Of these, four (80%) have had their treatment changed based on physician recommendation compared with 10 (11%) in IM or EM (p = 0.001) |
Appendix 3 Quality assessment
To assesses quality, the following questions were posed, based on elements of checklists developed to assess the methodological quality of prognostic factor studies50 and pharmacogenetic studies,51 with the corresponding responses presented in the table:
Patient sample (sample)
-
Is the source population clearly defined?
-
Is the study population clearly defined?
-
Does the study population clearly represent the source population or population of interest?
-
Are details given of how the sample size was calculated?
Choosing the genes/single nucleotide polymorphisms to genotype (see ‘SNP’, table below)
-
Are reasons given for choosing the genes and SNPs genotyped?
Reliability of genotypes (see ‘Test’, table below)
-
Is the genotyping procedure described?
-
Are the primers described?
-
Were quality control methods used and described?
-
Were findings from quality control methods, if used, described?
-
Are any genotype frequencies previously reported quoted?
Missing genotype data (see ‘Data’, table below)
-
Is it evident that there are any missing data?
-
Where missing data are evident, are reasons given?
-
Are checks for missingness at random reported?
-
Is missing genotype data imputed?
-
Does paper quote number of patients contributing to each analysis?
Confounding measurement and account (see ‘Confound’, table below)
-
Are potential confounders described?
-
Are potential confounders adjusted for?
Hardy–Weinberg Equilibrium (see ‘HWE’, table below)
-
Was a test presented to check for HWE?
Choice and definition of outcomes (see ‘Outcomes’, table below)
-
Does the paper clearly define the phenotypes?
-
Does the paper clearly define all outcomes investigated?
-
Is justification provided for the choice of phenotypes?
-
Is justification provided for the choice of outcomes?
-
Were the outcomes assessed blindly (i.e. did the assessor know the genotype/phenotype in relation to this?)
| Bonanni 2009112 | Henry 200987 | de Duenas 2009113 | Gjerde 2007104 | Goetz 200783 | Goetz 200982 on behalf of ITPC | Gonzalez-Santiago 200786 | Kiyotani 2010114 (efficacy) | Kiyotani 2010114 (metabolism) | Lim 200773 | Madlensky 200890 | Newman 200891 | Nowell 200592 | Okishiro 200993 | Onitilo 200994 | Ramon 201041 | Schroth 200796 | Schroth 2009108 | Stearns 200349 | Thompson 2009109 | Toyama 200997 | Wang 200798 | Wegman 2005100 | Wegman 200799 | Xu 2008101 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | 1 | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | / | ✓ | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ |
| 2 | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | / | / | / | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | |
| 3 | ✓ | ✓ | ✗ | ✓ | ✓ | / | ? | ✓ | ? | ✓ | ? | ✓ | ✓ | ? | ✓ | / | ✓ | ✓ | ? | ? | ✓ | ? | ? | ✓ | ✓ | |
| 4 | ✓ | / | ✗ | ✗ | / | – | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | α | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| SNP | 5 | ✗ | ✓ | ✓ | / | ✓ | / | / | ✓ | / | ✓ | / | / | / | ✓ | / | ✓ | ✓ | / | ✗ | / | ✓ | / | ✓ | ✓ | ✓ |
| Test | 6 | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | / | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| 7 | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | / | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | / | ✓ | |
| 8 | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | α | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | |
| 9 | – | – | – | ✓ | – | – | – | – | – | – | – | α | – | – | – | – | – | – | – | – | ✓ | – | – | – | – | |
| 10 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | |
| Data | 11 | ✓ | ✗ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✓ | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| 12 | ✓ | ✓ | – | – | ✓ | ✓ | ? | – | – | – | – | – | – | – | – | ✗ | ✗ | ✓ | – | – | ✓ | ✗ | ✗ | ✓ | / | |
| 13 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | – | ✗ | – | ✗ | ✗ | ✗ | ✗ | – | ✗ | ✗ | ✗ | – | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| 14 | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | – | ✗ | – | ✗ | – | – | – | – | ✗ | ✗ | ✗ | – | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| 15 | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Confound | 16 | ✗ | ✓ | ✗ | ✗ | ✓ | ✗ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | / | / | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | / | ✗ |
| 17 | ✗ | ✓ | ✗ | ✓ | ✓ | ✗ | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | |
| HWE | 18 | ✗ | ✓ | ✗ | ✓ | ✗ | ✗ | ✗ | ✓ | ✓ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ |
| Outcomes | 19 | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | – | – | – | – | ✗ | ✓ | – | – | – | ✓ | ✓ | ✓ | – | ✓ | ✓ | – | – | – | ✓ |
| 20 | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✗ | ✗ | ✗ | ✓ | |
| 21 | ✗ | ✓ | ✗ | ✗ | ✓ | ✗ | – | – | – | – | ✗ | ✓ | – | – | – | ✗ | ✓ | ✓ | – | ✗ | ✓ | – | – | – | ✗ | |
| 22 | ✗ | ✓ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| 23 | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | ? | |
List of abbreviations
- AE
- adverse event
- AJCC
- American Joint Committee on Cancer
- ANA
- anastrozole
- ATAC
- Arimidex, Tamoxifen, Alone or in Combination
- BIG 1-98
- Breast International Group 1-98
- BMD
- bone mineral density
- BRCA1
- breast cancer 1
- BRCA2
- breast cancer 2
- CI
- confidence interval
- CYP2D6
- cytochrome P450 2D6
- CYP450
- cytochrome P450
- DFS
- disease-free survival
- EFS
- event-free survival
- EM
- extensive metaboliser
- ER
- oestrogen receptor
- ER–
- oestrogen receptor negative
- ER+
- oestrogen receptor positive
- FDA
- Food and Drug Administration
- HER2
- human epidermal growth factor receptor 2
- hetEM
- heterozygous extensive metaboliser
- HR
- hazard ratio
- IES
- Intergroup Exemestane Study
- IM
- intermediate metaboliser
- ITA
- Italian tamoxifen anastrozole
- ITPC
- International Tamoxifen Pharmacogenomics Consortium
- LN+
- lymph node positive
- NICE
- National Institute for Health and Clinical Excellence
- NPI
- Nottingham Prognostic Index
- OS
- overall survival
- PM
- poor metaboliser
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- RFS
- recurrence-free survival
- RFT
- recurrence-free time
- SSRI
- selective serotonin reuptake inhibitor
- TAM
- tamoxifen
- TNM
- tumour/nodes/metastasis
- TTR
- time to recurrence
- UICC
- Union Internationale Contre le Cancer
- UM
- ultrarapid metaboliser
- vt
- variant type
- wt
- wild type
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Glossary
- Allele
- In humans, an allele is a member of a pair of different forms of a gene.
- AmpliChip®
- A type of assay used to detect CYP2D6 variants.
- Anti-oestrogen therapy
- Treatment that blocks the binding and actions of oestrogen.
- ARMS
- Genotyping method that uses two pairs of primers to amplify two alleles in one polymerase chain reaction.
- Biological therapy
- Treatments that use natural substances from the body, or drugs made from these substances, to fight cancer or to lessen the side effects that may be caused by some cancer treatments. An example includes trastuzumab (Herceptin®, Roche).
- Chemotherapy
- Treatment with drugs that kill cancer cells.
- Coronary arteries
- The arteries that supply the heart muscle with blood.
- Cost–benefit analysis
- A method of economic evaluation. An attempt to give the consequences of the alternative interventions a monetary value. In this way, the consequences can be more easily compared with the costs of the intervention. This involves measuring individuals’ ‘willingness to pay’ for given outcomes.
- Cost-effectiveness analysis
- A method of economic evaluation. The consequences of the alternatives are measured in natural units, such as years of life gained. The consequences are not given a monetary value.
- CYP2D6
- The enzyme belonging to the CYP450 enzyme system, also known as cytochrome P450 2D6. This is one of the most important enzymes involved in the metabolism of substances in the human body, mostly in the liver.
- CYP2D6
- The gene that encodes the CYP2D6 enzyme.
- DNA (deoxyribonucleic acid)
- A nucleic acid that contains the genetic instructions that make up living organisms.
- Debrisoquine
- A derivative of guanidine found in urine as a normal product of protein metabolism. It is frequently used for phenotyping the CYP2D6 enzyme (from the molar urinary metabolic ratio of debrisoquine to its metabolite, 4-hydroxydebrisoquine).
- Dextromethorphan
- A drug that is frequently used for phenotyping the CYP2D6 enzyme (from the molar urinary metabolic ratio of dextromethorphan to its metabolite, dextrorphan).
- Enzyme
- A protein molecule produced by living organisms that catalyses chemical reactions of substances (including drugs).
- Extensive metaboliser
- Somebody who metabolises tamoxifen normally at the normal therapeutic dose.
- Gene
- The basic biological unit of heredity – a segment of DNA that contributes to phenotype/function.
- Genotype
- The genetic constitution of an individual, i.e. the specific allelic make-up of an individual.
- Heterogeneity
- In statistics this means that there is between-study variation. If heterogeneity exists, the pooled effect size in a meta-analysis has no meaning, as the presence of heterogeneity indicates that there is more than one true effect size in the studies being combined.
- Heterozygote
- A person who has two copies of an allele that are different.
- Homozygote
- A person who has two copies of an allele that are the same.
- Intermediate metaboliser
- Somebody whose metabolism of tamoxifen lies somewhere between that of extensive metabolisers and poor metabolisers.
- Luminex
- A type of assay used to detect CYP2D6 variants.
- Metabolite
- A substance produced during metabolism (when it is drugs being metabolised, this usually refers to the end product that remains after metabolism).
- Nucleotide
- Small molecules that are the basic constituent of DNA.
- Oestrogen receptor negative
- Cancer cells that are oestrogen receptor negative do not need oestrogen to grow.
- Oestrogen receptor positive
- Cancer cells that may need oestrogen to grow (and can thus be treated with anti-oestrogen therapy).
- Pharmacogenetics
- A term used to define inherited variability in response to drug treatment.
- Phenotype
- The observable physical or behavioural traits of an organism, largely determined by the organism’s genotype but also influenced by environmental factors.
- Polymerase chain reaction
- A genotyping technique to amplify DNA for sequencing.
- Poor metaboliser
- Somebody with impaired metabolism of tamoxifen at the normal dose.
- Protein
- A complete biological molecule made of amino acids arranged in a linear chain defined by a gene and encoded in the genetic code. Types of proteins include enzymes and receptors.
- Quality-adjusted life-year
- An index of survival that is weighted or adjusted by a patient’s quality of life during the survival period. Quality-adjusted life-years are calculated by multiplying the number of life-years by an appropriate utility or preference score.
- Radiotherapy
- The use of high-energy radiation from X-rays, gamma rays, neutrons, protons and other sources to kill cancer cells and shrink tumours.
- Receptor protein
- A protein molecule embedded in a membrane, to which a signal molecule (ligand), such as a pharmaceutical drug, may attach itself to and which usually initiates a cellular response (although some ligands merely block receptors without inducing any response).
- Sensitivity
- The proportion of true-positive cases that are correctly identified by a test.
- Sequencing
- Method for determining the order of the nucleotide bases – adenine, guanine, cytosine and thymine – in a molecule of DNA.
- Single-nucleotide polymorphism
- The most common types of genetic variations in human beings that occur when a single nucleotide (adenosine, guanine, cytosine and thymine) in the genome sequence is changed.
- Specificity
- The proportion of true negative cases that are correctly identified by a test.
- TaqMan®
- A type of assay used to detect CYP2D6 variants.
- Ultrarapid metaboliser
- Somebody who metabolises tamoxifen more rapidly than extensive metabolisers at the normal dose.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Peter Brocklehurst, Professor of Women’s Health, Institute for Women’s Health, University College London
-
Professor Jenny Donovan, Professor of Social Medicine, University of Bristol
-
Professor Jonathan Green, Professor and Acting Head of Department, Child and Adolescent Psychiatry, University of Manchester Medical School
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Freddie Hamdy, Professor of Urology, Head of Nuffield Department of Surgery, University of Oxford
-
Professor Allan House, Professor of Liaison Psychiatry, University of Leeds
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Expert Advisory Network
-
Professor Douglas Altman, Professor of Statistics in Medicine, Centre for Statistics in Medicine, University of Oxford
-
Professor John Bond, Professor of Social Gerontology & Health Services Research, University of Newcastle upon Tyne
-
Professor Andrew Bradbury, Professor of Vascular Surgery, Solihull Hospital, Birmingham
-
Mr Shaun Brogan, Chief Executive, Ridgeway Primary Care Group, Aylesbury
-
Mrs Stella Burnside OBE, Chief Executive, Regulation and Improvement Authority, Belfast
-
Ms Tracy Bury, Project Manager, World Confederation of Physical Therapy, London
-
Professor Iain T Cameron, Professor of Obstetrics and Gynaecology and Head of the School of Medicine, University of Southampton
-
Professor Bruce Campbell, Consultant Vascular & General Surgeon, Royal Devon & Exeter Hospital, Wonford
-
Dr Christine Clark, Medical Writer and Consultant Pharmacist, Rossendale
-
Professor Collette Clifford, Professor of Nursing and Head of Research, The Medical School, University of Birmingham
-
Professor Barry Cookson, Director, Laboratory of Hospital Infection, Public Health Laboratory Service, London
-
Dr Carl Counsell, Clinical Senior Lecturer in Neurology, University of Aberdeen
-
Professor Howard Cuckle, Professor of Reproductive Epidemiology, Department of Paediatrics, Obstetrics & Gynaecology, University of Leeds
-
Professor Carol Dezateux, Professor of Paediatric Epidemiology, Institute of Child Health, London
-
Mr John Dunning, Consultant Cardiothoracic Surgeon, Papworth Hospital NHS Trust, Cambridge
-
Mr Jonothan Earnshaw, Consultant Vascular Surgeon, Gloucestershire Royal Hospital, Gloucester
-
Professor Martin Eccles, Professor of Clinical Effectiveness, Centre for Health Services Research, University of Newcastle upon Tyne
-
Professor Pam Enderby, Dean of Faculty of Medicine, Institute of General Practice and Primary Care, University of Sheffield
-
Professor Gene Feder, Professor of Primary Care Research & Development, Centre for Health Sciences, Barts and The London School of Medicine and Dentistry
-
Mr Leonard R Fenwick, Chief Executive, Freeman Hospital, Newcastle upon Tyne
-
Mrs Gillian Fletcher, Antenatal Teacher and Tutor and President, National Childbirth Trust, Henfield
-
Professor Jayne Franklyn, Professor of Medicine, University of Birmingham
-
Mr Tam Fry, Honorary Chairman, Child Growth Foundation, London
-
Professor Fiona Gilbert, Consultant Radiologist and NCRN Member, University of Aberdeen
-
Professor Paul Gregg, Professor of Orthopaedic Surgical Science, South Tees Hospital NHS Trust
-
Bec Hanley, Co-director, TwoCan Associates, West Sussex
-
Dr Maryann L Hardy, Senior Lecturer, University of Bradford
-
Mrs Sharon Hart, Healthcare Management Consultant, Reading
-
Professor Robert E Hawkins, CRC Professor and Director of Medical Oncology, Christie CRC Research Centre, Christie Hospital NHS Trust, Manchester
-
Professor Richard Hobbs, Head of Department of Primary Care & General Practice, University of Birmingham
-
Professor Alan Horwich, Dean and Section Chairman, The Institute of Cancer Research, London
-
Professor Allen Hutchinson, Director of Public Health and Deputy Dean of ScHARR, University of Sheffield
-
Professor Peter Jones, Professor of Psychiatry, University of Cambridge, Cambridge
-
Professor Stan Kaye, Cancer Research UK Professor of Medical Oncology, Royal Marsden Hospital and Institute of Cancer Research, Surrey
-
Dr Duncan Keeley, General Practitioner (Dr Burch & Ptnrs), The Health Centre, Thame
-
Dr Donna Lamping, Research Degrees Programme Director and Reader in Psychology, Health Services Research Unit, London School of Hygiene and Tropical Medicine, London
-
Professor James Lindesay, Professor of Psychiatry for the Elderly, University of Leicester
-
Professor Julian Little, Professor of Human Genome Epidemiology, University of Ottawa
-
Professor Alistaire McGuire, Professor of Health Economics, London School of Economics
-
Professor Neill McIntosh, Edward Clark Professor of Child Life and Health, University of Edinburgh
-
Professor Rajan Madhok, Consultant in Public Health, South Manchester Primary Care Trust
-
Professor Sir Alexander Markham, Director, Molecular Medicine Unit, St James’s University Hospital, Leeds
-
Dr Peter Moore, Freelance Science Writer, Ashtead
-
Dr Andrew Mortimore, Public Health Director, Southampton City Primary Care Trust
-
Dr Sue Moss, Associate Director, Cancer Screening Evaluation Unit, Institute of Cancer Research, Sutton
-
Professor Miranda Mugford, Professor of Health Economics and Group Co-ordinator, University of East Anglia
-
Professor Jim Neilson, Head of School of Reproductive & Developmental Medicine and Professor of Obstetrics and Gynaecology, University of Liverpool
-
Mrs Julietta Patnick, Director, NHS Cancer Screening Programmes, Sheffield
-
Professor Robert Peveler, Professor of Liaison Psychiatry, Royal South Hants Hospital, Southampton
-
Professor Chris Price, Director of Clinical Research, Bayer Diagnostics Europe, Stoke Poges
-
Professor William Rosenberg, Professor of Hepatology and Consultant Physician, University of Southampton
-
Professor Peter Sandercock, Professor of Medical Neurology, Department of Clinical Neurosciences, University of Edinburgh
-
Dr Philip Shackley, Senior Lecturer in Health Economics, Sheffield Vascular Institute, University of Sheffield
-
Dr Eamonn Sheridan, Consultant in Clinical Genetics, St James’s University Hospital, Leeds
-
Dr Margaret Somerville, Director of Public Health Learning, Peninsula Medical School, University of Plymouth
-
Professor Sarah Stewart-Brown, Professor of Public Health, Division of Health in the Community, University of Warwick, Coventry
-
Dr Nick Summerton, GP Appraiser and Codirector, Research Network, Yorkshire Clinical Consultant, Primary Care and Public Health, University of Oxford
-
Professor Ala Szczepura, Professor of Health Service Research, Centre for Health Services Studies, University of Warwick, Coventry
-
Dr Ross Taylor, Senior Lecturer, University of Aberdeen
-
Dr Richard Tiner, Medical Director, Medical Department, Association of the British Pharmaceutical Industry
-
Mrs Joan Webster, Consumer Member, Southern Derbyshire Community Health Council
-
Professor Martin Whittle, Clinical Co-director, National Co-ordinating Centre for Women’s and Children’s Health, Lymington