
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 09/101/01. The protocol was agreed in February 2010. The assessment report began editorial review in September 2010 and was accepted for publication in January 2011. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
In the past, Anne Armstrong has received consultancy fees, reimbursement for attending a conference and hospitality at a conference, from Roche. The North West Medicines Information Centre, where Helen Davis is the Assistant Director, has received a consultancy fee from GlaxoSmithKline in the past for participation in a group discussing various mechanisms for managing NHS medicines budgets.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2011. This work was produced by Fleeman et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2011 Queen’s Printer and Controller of HMSO
Chapter 1 Background
Description of health problem
Breast cancer is the uncontrolled, abnormal growth of malignant breast tissue affecting predominantly women. Though frequently referred to as a homogeneous disease, breast cancer has been recognised as a biologically heterogeneous disease1 with several subgroups, including those with different stages and types of the disease. Metastatic breast cancer (mBC) is an advanced stage of the disease when the disease has spread beyond the original organ. Common sites of metastasis include bone, the liver, lung and brain.
Aetiology
After gender, the strongest risk factor for breast cancer is age. The incidence of breast cancer increases with age, doubling every 10 years until menopause, after which the rate of increase slows. Breast cancer is rare under the age of 20 years.
Genetic and hormonal risk factors have also been identified in the aetiology of breast cancer,2,3 and women with a family history of breast cancer have an increased risk of developing the disease. 4 Mutations in some genes can increase the risk of developing breast cancer. Mutations of the breast cancer 1 (BRAC1) gene, which belongs to a class of genes known as tumour suppressor genes, account for 2% of breast cancers, where the risk is as high as 85% by the age of 35 years. 5 Breast cancer 2 (BRCA2) mutations account for up to 1% of breast cancers, with a 60% chance of breast cancer. Other gene mutations contributing less frequently to familial breast cancer include mutations in tumour protein 53 (p53), MutS Homolog1 (MSH1), MutS Homolog2 (MSH2) and phosphatase tensin homologue (PTEN).
Higher concentrations of some endogenous hormones appear to increase breast cancer risk. 6 Early age at menarche, late natural menopause, later age at first full-term pregnancy and never breastfeeding are all associated with an increased risk of breast cancer,6 whereas childbearing and higher numbers of full-term pregnancies increase protection. 6 Use of exogenous hormones such as oral contraception, oestrogen replacement therapy and combined endocrine therapy increase the risk of breast cancer, as do other factors such as breast density (a risk factor independent of endogenous hormones), a body mass index of 25+ in post-menopausal women, moderate-to-heavy alcohol intake and a sedentary lifestyle. 6
Pathology and prognosis
There are several prognostic factors that are taken into account by clinicians when deciding on treatment options and making a clinical prognosis. 7 These include age, tumour size, histological type, nuclear grade, histological grade, number of metastatic axillary lymph nodes and clinical stage. Patients with stage IV disease are classified as having mBC according to the tumour/nodes/metastasis staging system developed and maintained by the American Joint Committee on Cancer8 and the Union for International Cancer Control. 9
Hormone receptor status and human epidermal growth factor 2 (HER2) status are two other predictive factors that are taken into consideration in estimating the prognosis of patients with breast cancer. As noted above, many breast cancer tumours are stimulated to grow and change by ERs and PgRs. Tumours that express either oestrogen receptor-positive (ER+) or progesterone receptor-positive (PgR+) are commonly referred to as being hormone receptor positive (HR+), and patients with HR+ breast cancer generally have an improved prognosis compared with those who are hormone receptor negative (HR–). Overviews of hormonal therapy, at least in the adjuvant therapy of early breast cancer,10,11 suggest that, where such evidence is sufficiently well defined, it is the ER rather than the PgR activity that is the most useful prognostic factor. More recently it has been discovered that over-expression of ErbB2 protein (also known as HER2), which is a member of the epidermal growth factor receptor family, and/or amplification of the HER2 gene results in an abnormally high number of HER2 genes per cancer cell, which results in cancer cells growing and dividing more quickly. Thus, human epidermal growth factor 2-positive (HER2+) breast cancer is considered to be an aggressive disease and there is growing evidence that the prognosis of HER2+ patients is generally poor, whether or not they are HR– or HR+. It should be emphasised that prior to this understanding of the role of HER2, trials did not routinely present data on this subgroup of patients.
Both HR+ tumours and HER2+ tumours are determined by immunohistochemistry (IHC). 5 Fluorescence in situ hybridisation (FISH) can also be used to measure HER2 expression by measuring the number of gene copies present. An IHC score of ≥ 3 or a FISH amplification of > 2.1 confirms a HER2+ status. An IHC of > 2 is usually confirmed by FISH. 5 Biological markers such as HER2 are also used as a predictor of prognosis and as a guide to therapy.
In England and Wales, 80%, 72% and 64% of people diagnosed with breast cancer live for at least 5 years, 10 years and 20 years after diagnosis, respectively. 12 Although therapeutic innovations have provided modest improvements in survival rates over the past two decades, mBC remains an incurable disease and the aim of treatment is to prolong survival and palliation. 13 Following a diagnosis of mBC, the average length of survival has been reported to be 12 months for those receiving no treatment,14 compared with 18–24 months for those receiving chemotherapy, a figure reduced by up to 50% for patients who are HER2+. 15
Epidemiology
Breast cancer is the most common cancer in the UK with 48,034 new cases diagnosed in 2008, 99% (47,693) being in women. 16 Accounting for almost one-third (31%) of all new cases of cancer in women in the UK, the lifetime risk of breast cancer for a woman is one in nine. 16
There is little regional variation in breast cancer rates in the UK,17 although there appears to be geographical variation within Europe. Breast cancer is one of the few cancers to show a clear trend of increasing rates from the most to least deprived groups,16 with rates in the most deprived groups around 20% lower than in the most affluent. 16 The European age-standardised incidence rate (EASR) for women has increased by 5% from 114 per 100,000 in 1998 to 120 per 100,000 in 2007, with the number of cases rising from 40,377 to 45,695, an increase of 13%. The EASR has been projected to increase from 119 per 100,000 in 2000–4 to 124 per 100,000 in 2020–4, with the average number of new cases per year rising from 41,900 to 55,700 over the same time period. Analysis of breast cancer survival by level of deprivation has, however, consistently shown higher survival for more affluent women. 18
UK data on breast cancer by stage of disease are not routinely collected and so neither the incidence nor the prevalence of mBC in the UK is known. However, assuming that 25% of all women diagnosed with breast cancer have mBC, of whom 6% are HR+/HER2+ [based on 2008 and 2009 data obtained from the Clatterbridge Centre for Oncology NHS Foundation Trust (Nicky Thorp, Clatterbridge Centre for Oncology NHS Foundation Trust, June 2010, personal communication)], approximately 700 patients each year may be diagnosed with HR+/HER2+ mBC in the UK. An independent estimate from 2008 data derived from the IMS Oncology Analyzer™ (IMS Health®, Plymouth Meeting, PA, USA) obtained by GlaxoSmithKline19 has estimated there may be around 450 new cases of HR+/HER2+ mBC each year in the UK, whereas Roche20 has estimated the number to be nearer to 1000 based on data from Dybdal et al. 21 and their own market research.
Impact of health problem
The impact of a diagnosis of mBC breast cancer on a patient is both physiological and psychological,22 affecting not only the patients but also their families and wider social network. Physical ill-health can stem from both the disease and disease treatment. The National Institute for Health and Clinical Excellence (NICE) Clinical Guideline No. 81 (CG81)23 gives guidance for the management of complications such as lymphoedema, fatigue and metastases. Adequate rehabilitation is vital as women may be less productive after treatment for the disease. 24 The psychological impact on the patient can be debilitating, including depression and fear of loss of autonomy,25 sexuality and body image. 26
Description of technologies under assessment
Lapatinib
Lapatinib (LAP) (Tyverb®/Tykerb®, GlaxoSmithKline) inhibits the tyrosine kinase components of the epidermal growth factor receptors (ErbB1 and ErbB2), implicated in the growth of various tumours. 27 LAP belongs to a group of medicines called protein kinase inhibitors that work by blocking enzymes known as protein kinases. Protein kinases can be found in some receptors on the surface of cancer cells including HER2. HER2, a receptor for epidermal growth factor, is involved in stimulating the cells to divide uncontrollably. By blocking these receptors, LAP helps to control cell division.
The most common side effects of LAP are loss of appetite, diarrhoea, nausea, vomiting, rash and fatigue. Monitoring of left ventricular function and for pulmonary toxicity should be carried out regularly. Monitoring of liver function should be performed before treatment and at monthly intervals. 28 The manufacturer has advised caution in the use of LAP in patients with moderate-to-severe hepatic impairment and severe renal impairment. Pregnancy should be avoided and breastfeeding discontinued during treatment with LAP.
Lapatinib is an orally active drug given once per day and is available as 250-mg tablets.
Currently, LAP is licensed in combination with capecitabine (Xeloda®, Roche) for patients with advanced or metastatic disease with progression following prior therapy, which must have included anthracyclines and taxanes and therapy with trastuzumab (TRA) (Herceptin®, Roche) in the metastatic setting. 29 It is not, however, recommended for first-line treatment by NICE.
In June 2010, the European Medicines Agency (EMA) granted conditional approval for the use of LAP in combination with an aromatase inhibitor (AI) for the first-line treatment of post-menopausal women with HR+/HER2+ mBC. 30,31
Trastuzumab
Trastuzumab is a recombinant humanised immunoglobulin G1 (IgG1) monoclonal antibody directed against HER2. It is administered by intravenous (i.v.) infusion, the regimen and dose is dependent on several clinical factors including the patient’s weight, other medications and stage of disease. It is commonly administered every 3 weeks, with the infusion taking approximately 30–90 minutes each time.
The most common side effects of TRA are fatigue and diarrhoea. Recent clinical trial data suggest that patients require a left ventricular ejection fraction (LVEF) of > 55% for treatment with TRA and the summary of product characteristics for TRA states that cardiac monitoring is required every 12 weeks during treatment. However, the optimal frequency of cardiac monitoring in the clinical practice setting is not universally agreed. 32
Trastuzumab should be used with caution in patients with symptomatic heart failure, a history of hypertension, coronary artery disease and uncontrolled arrhythmias. Pregnancy should be avoided during treatment and breastfeeding should be avoided during treatment and for 6 months after. 28
Trastuzumab is currently licensed in the UK for the following indications:33
-
the treatment of early breast cancer which over-expresses HER2
-
in combination with paclitaxel or docetaxel (Taxotere®, Sanofi-Aventis), for mBC in patients with HER2+ tumours who have not received chemotherapy for mBC and in whom anthracycline treatment is inappropriate
-
in combination with an AI, for mBC in post-menopausal patients with HR+/HER2+ tumours not previously treated with TRA
-
as a monotherapy for mBC in patients with HER2+ tumours who have received at least two chemotherapy regimens including, where appropriate, an anthracycline and a taxane; women with ER+ breast cancer should also have received endocrine therapy.
The AG contacted the EMA for clarification about point 3 above, as the interpretation of this licence varied among NHS clinicians. In particular, it was not clear whether or not TRA was indicated for a woman who had been given TRA during the treatment of early breast cancer, who subsequently progressed to mBC. The EMA responded by stating that TRA was licensed for use in mBC in TRA-naive patients. Of note, at the time the trials were recruiting, the use of TRA for patients with early or advanced breast cancer was relatively rare. In its submission, Roche also state that the majority (76%) of patients who present for mBC have not previously received TRA for adjuvant therapy based on its own market research,20 although it is not clear over which time period this research was conducted.
Aromatase inhibitors
Aromatase inhibitors are not, per se, one of the technologies under assessment in this appraisal. However, they are being assessed in combination with LAP and TRA and are one of the comparators. NICE issued guidance regarding the use of AIs in 2006. 34 During this appraisal, the Appraisal Committee ‘agreed that there is insufficient evidence to conclude that any one AI (used within the licensed indications) or treatment strategy is more clinically effective than another’. As such, the AIs considered in this technology appraisal are assumed to be equally clinically effective. However, in practice only letrozole (LET) (Femara®, Novartis) and anastrozole (ANA) (Arimidex®, AstraZeneca) are commonly used as first-line treatments for women with mBC (and they may also be offered as a second-line treatment), with exemestane (EXE) (Aromasin®, Pharmacia & Upjohn, Kalamazoo, MI, USA) being mostly used as a second-line treatment.
Aromatase inhibitors are a form of endocrine therapy and act predominantly by blocking the conversion of androgens to oestrogens in the peripheral tissues. As such, they are classified as antioestrogen therapies. AIs are classified into irreversible steroidal inhibitors (e.g. EXE) and non-steroidal inhibitors (e.g. ANA and LET), the latter inhibiting the enzyme by reversible competition.
Letrozole is indicated as adjuvant treatment of HR+ early breast cancer in post-menopausal women, advanced breast cancer in post-menopausal women (including those in whom other endocrine therapy has failed), early invasive breast cancer in post-menopausal women after standard adjuvant tamoxifen (TAM) therapy and pre-operative treatment in post-menopausal women with localised HR+ breast cancer to allow subsequent breast-conserving surgery.
Cautions and contraindication include the avoidance of use during pregnancy and breastfeeding. Avoidance has also been advised in severe hepatic impairment, whereas caution has been advised if creatinine clearance is < 10 ml/minute.
Anastrozole is indicated as adjuvant treatment of ER+ early invasive breast cancer in post-menopausal women, adjuvant treatment of ER+ early breast cancer in post-menopausal women following 2–3 years of TAM therapy and in advanced breast cancer in post-menopausal women which are ER+ or responsive to TAM. 28
Caution has been advised for the use of ANA in patients susceptible to osteoporosis; bone mineral density should be measured before treatment and at regular intervals during treatment. ANA is contraindicated in pre-menopausal women. Its use should also be avoided in patients with moderate-to-severe hepatic impairment and renal impairment where creatinine clearance is < 20 ml/minute. As with LET, it should also be avoided in pregnancy and breastfeeding.
Common side effects of ANA include hot flushes, vaginal dryness, vaginal bleeding, hair thinning, anorexia, nausea, vomiting, diarrhoea, headache, arthralgia, bone fractures and rash (including Stevens–Johnson syndrome). 28
Exemestane is indicated as adjuvant treatment of ER+ early breast cancer in post-menopausal women following 2–3 years of TAM therapy and in advanced breast cancer in post-menopausal women where endocrine therapy has failed.
As with other AIs, EXE is contraindicated in pre-menopausal women and should be avoided in pregnant and breastfeeding women. Caution in its use is advised in patients with renal and hepatic impairment. Common side effects include nausea, vomiting, abdominal pain, dyspepsia, constipation, anorexia, dizziness, fatigue, headache, depression, insomnia, hot flushes, sweating, alopecia and rash.
Current service provision
The aim of current treatments for mBC is to palliate symptoms, prolong survival and maintain a good quality of life (QoL) with minimal adverse events (AEs). Choice of treatment depends on previous therapy, hormone receptor status, HER2 status and the extent of the disease.
The National Institute for Health and Clinical Excellence23 recommends that endocrine therapy (such as TAM or an AI) is offered as a first-line treatment to the majority of women with ER+ advanced breast cancer. However, providing patients understand and are prepared to accept the toxicity of chemotherapy, this is also recommended as first-line treatment when the ER+ mBC is life-threatening or requires early relief of symptoms because of significant visceral organ involvement.
For patients who are receiving treatment with TRA for advanced breast cancer, NICE recommends15 that treatment with TRA is discontinued at the time of disease progression outside the central nervous system, but that TRA is continued if disease progression is within the central nervous system alone.
In practice, for patients with HR+/HER2+, TRA is commonly given in combination with chemotherapy (paclitaxel or docetaxel). However, variation in management of patients by age has also been reported. 35,36 Variation in practice regarding continued use of TRA at the time of disease progression also exists,37 partly because of uncertainty about mechanisms of resistance and whether or not this is partial or absolute.
As already noted, TRA, in combination with an AI, has been licensed for the treatment of post-menopausal patients with HR+/HER2+ mBC not previously treated with TRA. 33 Given the growing number of patients who are treated with TRA in the early breast cancer setting, this may result in a decreasing number of patients being eligible for treatment with either LAP or TRA in combination with an AI. According to the estimates from the manufacturers of these drugs, the number of patients eligible for LAP or TRA in combination with an AI is currently expected to be < 100 women each year.
Chapter 2 Definition of the decision problem
Decision problem
Interventions
The following two interventions are being considered:
-
LAP + AI
-
TRA + AI.
Population including subgroups
The population of interest is patients with mBC receiving first-line treatment who must:
-
have HR+ tumours, and
-
have tumours over-expressing the ErbB2 receptor, i.e. HER2+.
Relevant comparators
For LAP + AI, the relevant comparators are:
-
AIs alone
-
TRA + AI.
For TRA + AI, the relevant comparators are:
-
AIs alone
-
LAP + AI.
Outcomes
The NICE scope identified the following relevant outcomes:
-
overall survival (OS)
-
progression-free survival (PFS)
-
time to progression (TTP)
-
response rate, which (although not specified in the scope) may further be broken down to:
-
– overall response rate (ORR)
-
– complete response (CR)
-
– partial response (PR)
-
-
AEs
-
clinical benefit rate (CBR)
-
health-related QoL.
Key issues
It is important to note that the following criteria were to be fulfilled a priori.
-
Only trials that measure clinical effectiveness in the population of interest were to be included in the systematic review, i.e. women must have mBC, have tumours that are HR+/HER2+ and have had no prior treatment for mBC.
-
– Women were to be considered to have HR+ breast cancer if they had ER+ or PgR+ tumours.
-
-
Where head-to-head comparisons do not exist, indirect comparisons were to be attempted.
-
Cost-effectiveness of treatments was to be expressed in terms of incremental cost per quality-adjusted life-year (QALY) gained.
-
The time horizon for estimating clinical effectiveness and cost-effectiveness was to be sufficiently long to reflect any differences in costs or outcomes between the technologies being compared.
-
Costs were to be considered from an NHS and Personal Social Services (PSS) perspective.
Overall aims and objectives of assessment
The remit of this appraisal is to review the clinical effectiveness and cost-effectiveness evidence base for LAP in combination with an AI (LAP + AI) and TRA in combination with an AI (TRA + AI) within their licensed indications for the first-line treatment of patients who have HR+/HER2+ mBC.
Chapter 3 Assessment of clinical effectiveness
Methods for reviewing effectiveness
Evidence for the clinical effectiveness of LAP + AI and TRA + AI for the first-line treatment of patients with HR+/HER2+ mBC was assessed by conducting a systematic review of published research evidence. The review was undertaken following the general principles published by the Centre for Reviews and Dissemination’s (CRD’s) guidance for undertaking reviews in health care. 38
Identification of studies
Randomised controlled trials (RCTs) were identified by searching major electronic medical databases including MEDLINE, EMBASE and the Cochrane Library. The search strategy was broad and not limited to RCTs. Information on studies in progress, unpublished research or research reported in the grey literature were sought by searching a range of relevant databases including the National Research Register and Controlled Clinical Trials. In addition, bibliographies of previous reviews and retrieved articles were searched for further studies. The search strategy used for MEDLINE is presented in Appendix 1. The same search strategies were used to identify economic evaluations.
Further attempts to identify studies were made by contacting clinical experts and examining the reference lists of all retrieved articles. The manufacturers’ submissions (MS) were assessed for unpublished data.
Inclusion and exclusion criteria
Two reviewers (NF/MM) independently screened all titles and abstracts. Full-paper manuscripts of any titles/abstracts that were considered relevant by either reviewer were obtained where possible. The relevance of each study was assessed (NF/MM) according to the criteria in Table 1. Studies that did not meet the criteria were excluded and their bibliographic details were listed alongside reasons for their exclusion. These are listed in Appendix 2. Any discrepancies were resolved by consensus.
| Study design | RCTs |
|---|---|
| Population(s) | Post-menopausal women with HR+/HER2+ mBC who have not previously received treatment for metastatic disease and for whom treatment with an AI is suitable. The following broad subgroups are considered if data permit:
|
| Intervention(s) |
LAP in combination with an AI TRA in combination with an AI |
| Comparators |
The two interventions will be compared with each other The interventions will also be compared with AIs |
| Outcomes | The outcome measures to be considered include:
|
Data abstraction strategy
Data were extracted by one reviewer (MM) using a standardised data extraction form in Microsoft Word 2007 (Microsoft Corporation, Redmond, WA, USA) and checked independently by a second reviewer (NF). Disagreements were resolved by discussion.
Critical appraisal strategy
The quality of the individual clinical effectiveness studies was assessed according to criteria based on the CRD’s guidance for undertaking reviews in health care. 38 The assessment of risk of bias was conducted independently by both reviewers (MM/NF). Disagreements were resolved through discussion.
Methods of data synthesis
The results of the data extraction and quality assessment for each study are presented in structured tables and as a narrative summary. The possible effects of study quality on the effectiveness data and review findings are discussed.
It was intended by the Assessment Group (AG) that meta-analyses would be conducted in which direct evidence would be pooled using a standard meta-analysis39 and, where a direct comparison between LAP + AI and TRA + AI was not possible, by indirect comparisons. 40 However, the AG considered it inappropriate to conduct either of these analyses, as discussed further in the next section.
Results
Quantity and quality of research available
Identification of studies
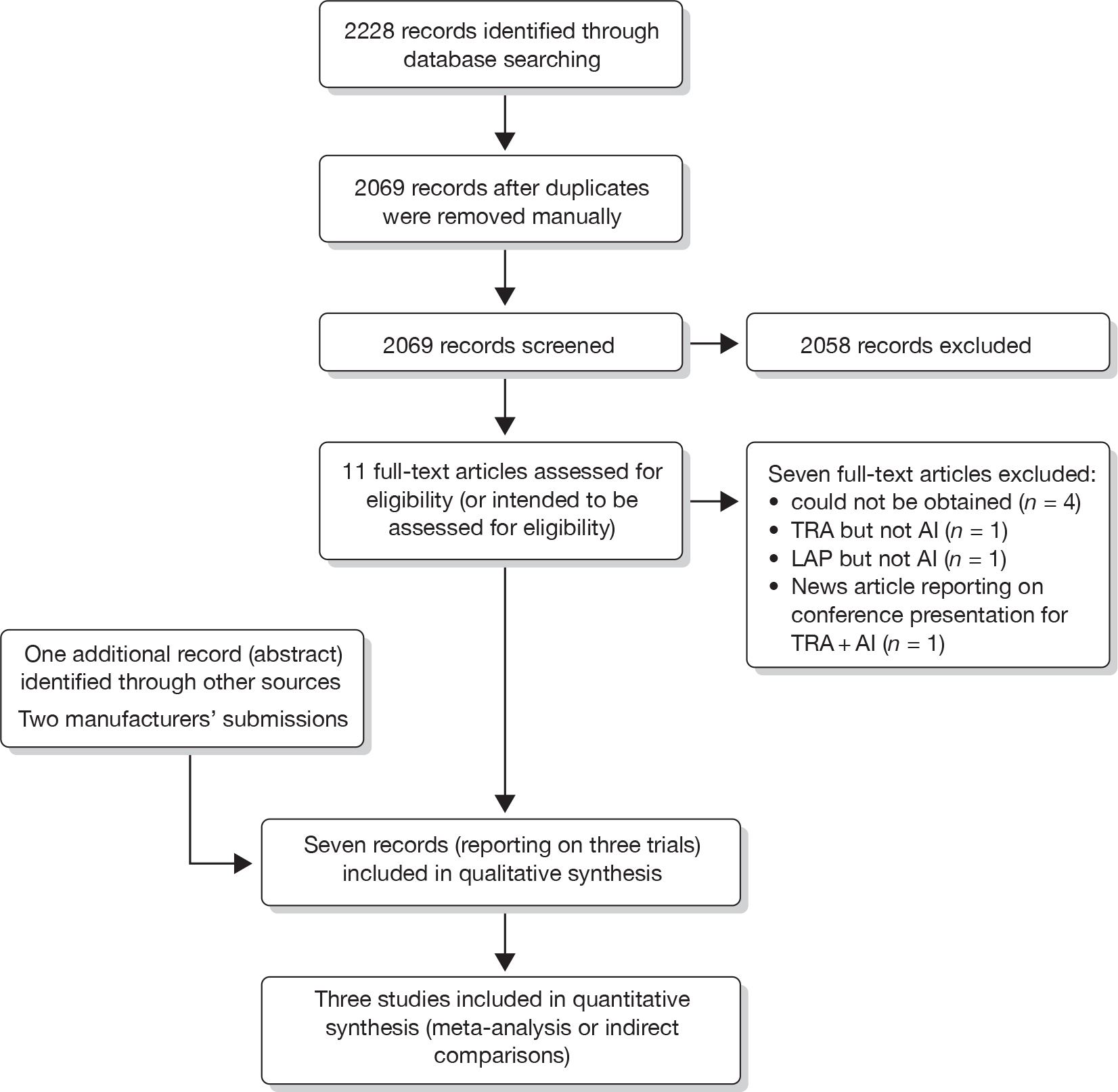
Once duplicates were removed, a total of 2069 references were identified (Figure 1); a scan of the titles and abstracts resulted in 11 potential records. 41–51 Four of these citations48–51 reporting on two trials [the efficacy and safety of lapatinib combined with letrozole (EGF30008)49 trial and the efficacy and safety of trastuzumab combined with anastrozole (TAnDEM)50 trial] met the inclusion criteria and a further trial [efficacy and safety of letrozole combined with trastuzumab (eLEcTRA)52 trial], reported only as a conference abstract, was also suitable for inclusion following information passed on to the AG by Roche at the NICE consultation meeting in February 2010. Thus, three trials were included in the systematic review. Additional data on these trials were submitted to NICE from the manufacturer of LAP (GlaxoSmithKline19) and the manufacturer of TRA (Roche20), including the relevant clinical study report (CSR) for the EGF3000849 and the TAnDEM. 50 Of the seven excluded citations, three45–47 were excluded either because they did not examine LAP or TRA in combination with an AI or because it was a conference report in relation to the TAnDEM. 50 Four citations41–44 were excluded because they could not be obtained. Each was a Physician Data Query (identified through The Cochrane Library) relating to the three included trials and indexed prior to the final study publication dates. In their submissions, both Roche and GlaxoSmithKline identified additional studies that they utilised as indirect evidence. The majority of these trials had also been identified by the AG’s search, but as none were limited to the HR+/HER2+ mBC population (or at least did not include subgroup analysis on the HR+/HER2+ population), they did not meet the review inclusion criteria. Reasons outlining all of the excluded citations, including those identified and included by the manufacturers, are given in Appendix 2.
FIGURE 1.
Identification of eligible studies.
Included trials
Key characteristics of the included trials are summarised in Table 2.
| Study and principal citation | Type of study and years of recruitment | Population | Interventions, dose and duration | Size of study | Notes |
|---|---|---|---|---|---|
| EGF3000849 | Double-blind multicentre trial conducted internationally: 212 sites in 29 countries, 2003–6 | First-line post-menopausal HR+/HER2+ mBC |
LAP + LET vs LET + placebo LAP = 1500 mg/day (oral) LET = 2.5 mg/day (oral) Placebo = pill (oral) Treatment was planned to continue until disease progression or study withdrawal |
n = 219a (LAP + LET = 111; LET = 108) |
The trial was funded by GlaxoSmithKline and excluded patients with extensive symptomatic visceral disease, including hepatic involvement and pulmonary lymphangitic spread of tumour, or the disease was considered by the investigator to be rapidly progressing or life-threatening Second-line treatment was permitted following disease progression |
| TAnDEM50 | Open-label multicentre trial conducted internationally: 77 sites in 22 countries (including eight sites in the UK), 2001–4 | First-line post-menopausal HR+/HER2+ mBC |
TRA + ANA vs ANA TRA = 4 mg/kg loading dose (i.v.) followed by 2 mg/kg/week (i.v.) or 8 mg/kg on day 1 followed by 6 mg/kg 3-weekly ANA = 1 mg/day (oral) Treatment was planned to continue until disease progression |
n = 208b (TRA + ANA = 103; ANA = 104) |
The trial was funded and conducted by Roche and permitted patients in the ANA group to cross over to TRA + ANA following disease progression and patients in both groups were permitted chemotherapy following disease progression, i.e. patients were permitted second-line treatment A greater proportion of patients in the ANA group received second-line treatment |
| eLEcTRA52 | Open-label multicentre trial conducted internationally: 32 sites in seven countries, 2003–7 | First-line post-menopausal HR+/HER2+ mBC |
TRA + LET vs LET TRA = 4 mg/kg loading dose (i.v.) followed by 2 mg/kg/week (i.v.) or 8 mg/kg on day 1 followed by 6 mg/kg 3-weekly LET = 2.5 mg/day (oral) Treatment was planned to continue until disease progression |
n = 57c (TRA + LET = 26; LET = 31c) |
The trial was funded by Novartis, with Roche described as a collaborator, and halted prematurely because of slow recruitment Patients were permitted to receive second-line TRA following disease progression A greater proportion of patients in the LET group received second-line treatment |
All three trials (the EGF30008,49 the TAnDEM50 and the eLEcTRA52) were multicentre and multinational trials (between 7 and 29 countries) enrolling post-menopausal patients receiving first-line treatment for mBC; all three trials included patients who had HR+/HER2+ mBC, although the EGF3000849 and the eLEcTRA52 also included patients who were HR+/ human epidermal growth factor receptor 2 negative (HER2–). The trials were designed to evaluate the efficacy and safety of the addition of LAP + LET to LET (EGF3000849), TRA + ANA to ANA (TAnDEM50) and TRA + LET to LET (eLeCTRA52). In all trials, treatment was administered at licensed doses until disease progression, at which point patients received second-line therapy which included chemotherapy; for patients in the ANA group, TRA + ANA was also a second-line treatment option. Both chemotherapy and TRA were second-line treatment options in the EGF30008 trial. 49
Clinical end points, including OS, PFS and TTP, which are commonly used in trials of breast cancer, were utilised in at least one of the trials included in this appraisal. However, the only efficacy end points common to all three, and reported on by all three, were the secondary end points, CBR and ORR. All three trials also reported on AEs. The eLEcTRA52 trial intended to report on OS, but, to date, no findings for OS have been reported, possibly because this trial was halted prematurely because of slow recruitment. Only the EGF3000849 trial reported on QoL.
As patients received second-line treatment once their disease had progressed, data on OS should be treated with caution as clearly this extra treatment could potentially impact on OS. Differences in second-line therapies received were clearly evident across the trials, with a greater proportion of patients receiving chemotherapy in the EGF3000849 trial (Table 3).
Data on PFS, TTP, CBR and ORR in all trials should be treated with caution because of the way these end points were measured. This is discussed in more detail below.
Overall, the risk of bias assessment conducted by the AG (Table 4) found the EGF3000849 to be of a good standard. Some small imbalances in baseline characteristics between the LAP + LET vs LET arms in the HR+/HER2+ population were noted (ECOG performance status ≥ 1: 46% vs 53%; Diseases stage, lymph node: 51% vs 40%; Previous therapy, chemotherapy: 55% vs 40%, other: 10% vs 4%), which did not exist between groups in the population as a whole. The imbalances were not however deemed to be of clinical significance by the study authors or the AG. However, while the study was a double-blind study, because of the significantly increased incidences of diarrhoea and rash in the LAP + LET group (see below), the effectiveness of blinding may be questioned.
| Criteria | EGF3000849 | TAnDEM50 | eLEcTRA52 |
|---|---|---|---|
| Was the method used to assign participants to the treatment groups really random? | ✓ | ✓ | ? |
| Was the allocation of treatment concealed? | ✓ | NA (open label) | NA (open label) |
| Was the number of participants who were randomised stated? | ✓ | ✓ | ✓ |
| Were details of baseline comparability presented in terms of prognostic factors? | ✓ | ✓ | ✓ |
| Was baseline comparability achieved in terms of prognostic factors? | ✓ | ✓ | ✓ |
| Were the eligibility criteria for study entry specified? | ✓ | ✓ | ✓ |
| Were any co-interventions identified that may influence the outcomes for each group? | ✓ | ✓ | ✓ |
| Were the outcome assessors blinded to the treatment allocation? | ✓/✗ | ✓/✗ | ✗ |
| Were the individuals who administered the intervention blinded to the treatment allocation? | ✓/✗ | ✗ | ✗ |
| Were the participants who received the intervention blinded to the treatment allocation? | ✓ | ✗ | ✗ |
| Was the success of the blinding procedure assessed? | ✗ | NA (open label) | NA (open label) |
| Were at least 80% of the participants originally included in the randomisation process followed up in the final analysis? | ✓ | ✓ | ✗ |
| Were the reasons for withdrawals stated? | ✓ | ✓ | Trial was stopped prematurely |
| Is there any evidence to suggest that the authors measured more outcomes than they reported? | ✓/✗a | ✓ | ✓ |
| Was an ITT analysis included? | ✓ | ✓ | ✗ |
The TAnDEM50 was similarly considered to be of good standard, the weakest aspect being the lack of blinding. As LAP is administered orally, the EGF3000849 was able to blind treatment by also administering a placebo pill with LET. To have blinded treatment for TRA, however, a placebo i.v. therapy would have been required, which may arguably have been difficult to justify from an ethical perspective. Generally, baseline characteristics were well balanced across the TRA + ANA and ANA arms although there were a small number of imbalances (Site of therapy, lung: 41.7% vs 46.2%, bone: 62.1% vs 51.0%, other: 69.9% vs 62.5%; Previous therapy, hormonal: 60.2% vs 66.3%, chemotherapy: 53.4% vs 59.6%, anthracycline: 44.7% vs 51.0%). Clinical advice received by the AG was that these were not a cause for concern in terms of biasing any results.
The eLEcTRA52 trial was deemed to be of poorer quality compared with the EGF3000848 and the TAnDEM trials. 50 This may be a reflection of poor-quality reporting rather than trial design, as this trial was only published as an abstract, with limited additional data subsequently available from Roche. 20 However, the fact that the trial ended prematurely because of slow recruitment did affect quality. Firstly, slow recruitment is attributed in the Roche submission20 to the fact that investigators believed TRA + LET was superior to LET (although no evidence is presented to support this claim) and investigators were reluctant to continue randomising patients into the LET group. This could have introduced selection bias. Secondly, because < 25% of the intended patients were recruited, the trial lacked statistical power and finally, there were large differences in baseline comparability between the TRA + LET and LET groups in the HER2+ population [ECOG performance status ≥ 1: 69% vs 45%; median (range) time from primary diagnosis to randomisation, months: 3 (0–486) vs 30 (0–75); Site of metastases, locoregional: 46% vs 29%, liver: 19% vs 39%, soft tissue: 31% vs 36%; Previous therapy, any adjuvant: 42% vs 71%, adjuvant endocrine therapy: 31% vs 65%].
Comparing baseline characteristics across the three trials was problematic because of differences in how measures were defined and/or reported and so the characteristics have not been presented in this report. However, it was noticeable that the median age of patients in the TAnDEM50 differs to that of the other two trials, the median age being around 55 years compared with around 60 years in the other two trials. There also appears to be more patients with soft tissue metastases in the TAnDEM50 than the EGF30008. 49
Arguably, of greater significance were the aforementioned differences in second-line treatment and the choice of exclusion criteria in the EGF3000848 trial. According to the MS,19 the EGF3000849 excluded patients with symptomatic visceral disease and patients in whom the disease was considered by the investigator to be rapidly progressing or life-threatening. 19 The potential importance of second-line treatment and/or this criterion became apparent when analysing median OS, which was reported to be ≤ 23.9 months [unadjusted intention-to-treat (ITT) population] or 28.6 months (centrally confirmed hormone receptor status) in the TAnDEM50 trial compared with 32.3 months in the EGF3000849 trial. If it is assumed that there is a ‘class effect’ (and certainly for early breast cancer where NICE guidance34 exists on the use of AIs, it is indeed assumed that LET and ANA are equally effective), then if the populations were truly similar, a similar median OS would be expected for patients in the LET and ANA arms of the different trials.
The generalisability of the trials to the UK population may also be questioned as it is unclear whether or not patients at imminent risk of death, as some patients may have been in the TAnDEM trial,50 would be eligible for treatment with LAP + AI or TRA + AI. However, clinical advice received by the AG was that some clinicians would offer TRA or LAP with an AI if patients were deemed to be too unfit for chemotherapy, even if they were at risk of imminent death. Equally, those who were not at risk of imminent of death, as in the EGF30008 trial,49 may also be offered TRA or LAP with an AI. Hence, both study populations appear generalisable to the UK.
In summary, although study designs appear appropriate for the comparison of LAP + AI versus AI or TRA + AI versus AI, key differences in the trials led the AG to the conclusion that it would not be appropriate to pool data or make meaningful comparisons, directly or indirectly, across the two completed trials. This decision was primarily based on apparent differences in patient populations – the key factors being the differences in second-line treatment received and the explicit exclusion of patients with symptomatic visceral disease and/or in whom the disease was considered by the investigator to be rapidly progressing or life-threatening from the EGF3000848 trial (but not the other trials). As the eLEcTRA52 trial was halted prematurely some data were not available/reported and data which were reported should be treated with extreme caution. For these reasons, the AG decided to focus on discussing the trials individually.
Assessment of effectiveness
EGF30008: lapatinib in combination with letrozole versus letrozole alone
Patients were recruited into the EGF3000849 trial between December 2003 and December 2006, during which time there were amendments to the original protocol. One amendment led to increased target enrolment, from 760 to 1280 subjects, in order to ensure adequate statistical power in the HR+/HER2+ subgroup (October 2005). The decision to focus on the HR+/HER2 subgroup was made as a result of pre-clinical studies and clinical studies suggesting that LAP modulates its effect in breast cancer primarily via ErbB2. 53,54 Another significant amendment was the definition of the HR+/HER2+ population as the primary population of interest, at which time the primary end point was changed from TTP to PFS (October 2007); PFS was defined as the time from randomisation until the earliest date of disease progression or death from any cause, if sooner. The decision to change the primary end point was made because, unlike TTP, PFS includes death (and is thus a better correlate with OS) and is, therefore, preferred by the US Food and Drug Administration (FDA)55 and the Committee for Medicinal Products for Human Use. 56 All of the EGF3000849 trial amendments were carried out prior to un-blinding and are less likely to increase risk of bias than if amendments had occurred after blinding.
The findings from the EGF3000849 trial are summarised in Table 5, where it can be seen that data were available for the HR+/HER2+ population as well as the wider population of patients recruited, which included patients who were HR+/HER2–. The wider population in the study was referred to as the ITT population.
| Outcome | HR+/HER2+ populationa | All patients (i.e. including those who are HR+/HER2–b) | ||||
|---|---|---|---|---|---|---|
| LAP + LET (n = 111) | LET (n = 108) | HR/OR (95% CI); p-value | LAP + LET (n = 644) | LET (n = 642) | HR/OR (95% CI); p-value | |
| OS (months)c | 33.3 | 32.3 | HR = 0.74 (0.5 to 1.1); p = 0.113 | Not reported | Not reported | Not reported |
| PFS (months)c | 8.2 | 3.0 |
HR = 0.71 (0.53 to 0.96); p = 0.019 Cox regression analysis (adjusting for known baseline prognostic factors) HR = 0.65 (0.47 to 0.89); p = 0.008 |
11.9 | 10.8 | HR = 0.86 (0.76 to 0.98); p = 026 |
| TTP (months)c | 8.2d | 3.0d | HR = 0.71 (0.53 to 0.96); p = 0.019 | Not reported | Not reported | Not reported |
| ORR (%)e | 28 | 15 | OR = 0.4 (0.2 to 0.9); p = 0.021 | 33 | 32 | OR not reported; p = 0.726 |
| CR (%) | 5 | 4 | 5 | 4 | ||
| PR (%) | 23 | 11 | 28 | 27 | ||
| SD ≥ 6 months (%)e | 20 | 14 | Not reported | 26 | 25 | Not reported |
| CBR (%)f | 48 | 29 | OR = 0.4 (0.2 to 0.8); p = 0.003 | 58 | 56 | OR not reported; p = 0.761 |
No significant differences were reported in terms of OS between the groups, although there was a possible trend in favour of LAP + LET compared with LET. 19 A pre-planned analysis within known prognostic factor subpopulations reported consistently improved OS with LAP + LET compared with LET in the following groups: the European Cooperative Oncology Group (ECOG) performance status score < 1 and patients with fewer than three metastatic sites; site of disease (non-visceral/visceral) did not significantly affect OS. 19 It should be noted that OS data were not mature at the time of data cut-off, as only 104 subjects (47%) had died and 41% were still being followed for survival. It should also be reiterated that once study medication had been discontinued, 153 patients (76%) in the HR+/HER2+ population received second-line treatment, mostly chemotherapy (around 60% of all patients), which may have affected OS.
For the HR+/HER2+ population, the EGF3000849 trial reported significant improvements in PFS in the LAP + LET group when compared with the LET group. 49 When adjusted for baseline prognostic factors, the stepwise Cox regression analysis for PFS confirmed the benefit of LAP + LET compared with LET. 51 A pre-planned analysis within known prognostic factor subpopulations reported consistently improved PFS with LAP + LET compared with LET in the following groups: patients with an ECOG performance status score > 0, patients without bone as the only site of metastasis, patients with and without liver metastases, patients with fewer than three metastatic sites and patients having received prior endocrine therapy for < 6 months. 51 Significant differences were also reported for differences in PFS in the ITT population,49 but here the differences between the groups were less pronounced. In particular, it was noticeable that the PFS was greater in the ITT population than in the HR+/HER2+ population, particularly for patients receiving LET (the difference in PFS between the ITT and HR/HER2+ populations here was 7.8 months compared with 3.7 months between the same two populations among patients receiving LAP + LET).
Because only one subject of the HR+/HER2+ population died from a cause other than breast cancer in this study, the TTP findings were almost identical to those reported for PFS. 19 In the same population, ORR was significantly improved for patients treated with LAP + LET compared with LET, as was CBR. However, the differences in ORR and CBR between treatment groups were not significant in the ITT population.
Because assessment of disease progression is liable to subjectivity, introducing an observation bias, which needs to be considered when interpreting PFS, TPP, ORR and CBR, blinded independent review has been recommended in order to circumvent such problems. 57,58 In the EGF30008 trial,49 investigator assessment and a blinded Independent Radiological Review Committee (IRC) were employed. The main reasons for differences in PFS assessments between the investigators and IRC, as noted in the CSR, were primarily because of differences in the censoring methods used. The differences were, however, constant across both treatment arms, which reduces the risk of bias.
Patients who received LAP + LET were more likely to experience AEs, with nearly all patients in the HR+/HER2+ population experiencing an AE compared with around three-quarters of patients who received LET (Table 6). Serious adverse events (SAEs), however, were relatively rare in both groups. Only three patient deaths were attributed to treatment, one of these taking LAP + LET in the HR+/HER2+ population.
| Adverse events | HR+/HER2+ population | All patients (i.e. including those who are HR+/HER2–) | ||
|---|---|---|---|---|
| LAP + LET (n = 111) | LET (n = 108) | LAP + LET (n = 644) | LET (n = 642) | |
| Any AE (%) | 96a,b | 77a,b | Not reported | Not reported |
| SAEs (%) | Not reported | Not reported | 8b,c | 4b,c |
| Discontinued treatment because of AE (%) | Not reported | Not reported | 2b,c,d | 1b,c,d |
| Treatment-related deaths, n | 1 (< 1%) | 0 | 1 (< 1%) | 2 (< 1%) |
In patients with HR+/HER2+ mBC, and in all patients as a whole (i.e. including HR+/HER2– mBC), the most common AEs were diarrhoea, rash, nausea, arthralgia and fatigue, of which the majority were grade 1 or 2 (Tables 7 and 8). In particular, incidences of diarrhoea, rash and nausea were significantly higher in patients receiving LAP + LET. It was reported by Johnston et al. 49 that 15% of all 60 patients with grades 3 or 4 diarrhoea discontinued LAP + LET as a result, i.e. around 1% of all patients. For the remainder of patients, diarrhoea was managed by dose reduction (19%), dose interruption (36%) or supportive intervention without treatment dose adjustments (31%).
| AEs | LAP+LET (n=111) | LET (n=108) | ||
|---|---|---|---|---|
| Total (any grade) (%) | Grade 3 or higher (%) | Total (any grade) (%) | Grade 3 or higher (%) | |
| Diarrhoea | 68 | 7 | 8 | 0 |
| Rash | 46 | 0 | 8 | 0 |
| Nausea | 27 | 0 | 18 | < 1 |
| Fatigue | 22 | 4 | 14 | 0 |
| Arthralgia | 18 | 4 | 20 | < 1 |
| Back pain | 17 | 2 | 9 | < 1 |
| Vomiting | 17 | < 1 | 7 | 0 |
| Headache | 14 | 0 | 11 | < 1 |
| Asthenia | 14 | 2 | 9 | 0 |
| Pruritus | 13 | 0 | 5 | < 1 |
| Dizziness | 12 | 0 | 8 | 0 |
| Cough | 11 | 0 | 9 | 0 |
| Alopecia | 11 | 0 | 4 | 0 |
| Musculoskeletal pain | 10 | < 1 | 5 | 0 |
| Epistaxis | 10 | < 1 | 2 | 0 |
| Dyspnoea | 9 | < 1 | 10 | 4 |
| Hot flush | 6 | 0 | 12 | 0 |
| Alanine aminotransferase increase | 11 | < 1 | 6 | < 1 |
| Aspartate aminotransferase increase | 10 | < 1 | 5 | 2 |
| AEs | LAP+LET (n=111) | LET (n=108) | ||
|---|---|---|---|---|
| Total (any grade) (%) | Grade 3 or higher (%) | Total (any grade) (%) | Grade 3 or higher (%) | |
| Diarrhoeaa | 64 | 9 | 20 | 1 |
| Rasha | 45 | 1 | 13 | 0 |
| Nauseaa | 31 | 1 | 21 | 1 |
| Arthralgia | 19 | 1 | 23 | 1 |
| Fatigue | 20 | 2 | 17 | 0 |
| Back pain | 16 | 2 | 16 | < 1 |
| Vomitinga | 17 | 1 | 11 | < 1 |
| Headache | 14 | < 1 | 13 | 0 |
| Cough | 12 | < 1 | 14 | 0 |
| Hot flusha | 11 | 0 | 15 | 0 |
| Asthenia | 12 | 1 | 11 | 1 |
| Pain in extremity | 10 | < 1 | 11 | 1 |
| Dyspnoea | 10 | 1 | 12 | < 1 |
| Pruritusa | 12 | < 1 | 9 | 0 |
| Alopeciaa | 13 | < 1 | 7 | 0 |
| Constipation | 9 | 0 | 11 | < 1 |
| Anorexia | 11 | 1 | 9 | < 1 |
| Dry skin | 13 | < 1 | 4 | 0 |
| Epistaxis | 11 | < 1 | 2 | 0 |
| Nail disorder | 11 | < 1 | 1 | 0 |
An additional 8 months of data beyond trial reporting had been collected (through to 3 February 2009) and presented in the GlaxoSmithKline submission. 19 These data remain consistent with the initial study results, although more patients in the LET group reported AEs than before [629 (96%) patients reported an AE in the LAP + LET group compared with 537 (86%) in the LET group].
Overall, therefore, no new safety issues were identified, the safety profile of LAP + LET being consistent with the safety profiles of both drugs when given as single agents and with safety data from previously reported LAP studies.
Finally, QoL was also assessed in the EGF30008 trial,49 utilising the Functional Assessment of Cancer Therapy-Breast (FACT-B) questionnaire. 19 Within the HR+/HER2+ population, QoL scores and changes from baseline were reported to be generally stable over time for subjects who stayed in the study in both the LAP + LET and LET groups, suggesting maintenance of QoL. The Quality-Adjusted Time Without Symptoms of Disease or Toxicity of Treatment (Q-TWIST) difference between treatment groups for the HR+/HER2+ population ranged from 8 weeks to 9.5 weeks, favouring LAP + LET over LET for all hypothetical utility levels, although none of the findings were reported to be statistically significant. 59
TAnDEM: trastuzumab in combination with anastrozole versus anastrozole alone
Between March 2001 and May 2006, the TAnDEM50 trial enrolled 207 HR+/HER2+ patients, of whom 103 were randomly assigned to TRA + ANA and 104 to ANA. According to the CSR, prior to any patient being recruited into the trial, there had already been amendments to the protocol, including a request at the behest of the FDA that the primary end point was changed from TTP to PFS. PFS was defined as the time between random assignment and the date of progressive disease (PD), clinical or radiographic, or death. There were also amendments following recruitment of the first patient. The most significant was perhaps the amendment that allowed for the crossover of patients from ANA to TRA + ANA following disease progression, thus, impacting on the size of the OS results. No statistical methods were described to address this issue of crossover a priori, the trial simply being separated into two treatment phases: main and extension. The main phase was defined as the first 24 months of treatment or until disease progression, and the extension phase was defined as the treatment period after 24 months or the treatment period after disease progression, whichever came earliest. Patients had a safety follow-up assessment 28 days after their last dose of treatment. Subsequently, post-hoc analyses were performed by Roche which attempted to take into account the effects of crossover, as described further below.
The findings from the TAnDEM50 trial are summarised in Table 9. No significant differences in terms of OS were found between the groups. However, it should be noted that 70% of the patients randomised to ANA subsequently received TRA + ANA in the extension phase of the trial and this may have impacted on the findings. In addition, around one-third (32%) of ANA patients went on to receive chemotherapy compared with a minority of patients who had been randomised to TRA + ANA (8%). With some legitimacy, the manufacturer of TRA argued that this could impact on the size of the OS estimates because, in this situation, the ITT results will be significantly compromised and will either underestimate or overestimate the treatment effect between groups. Roche contended that this would underestimate the treatment effect and show a reduced incremental gain from TRA + ANA over ANA. Thus, unplanned exploratory post-hoc analyses were performed by Roche to investigate the impact of this crossover from the control group of the trial on OS.
| Outcome | TRA + ANA (n = 103) | ANA (n = 104) | HR (95% CI); p-value |
|---|---|---|---|
| OS (range) months,a unadjusted ITT populationb | 28.5 (22.8–42.4) | 23.9 (18.2–37.4) | HR = 0.84 (0.59 to 1.20); p = 0.325 |
| OS (range) months,a centrally confirmed hormone receptor statusb | 34.1 (23.9–52.0) | 28.6 (17.4–40.0) | HR = 0.85 (no CIs); p = 0.451 |
| OS (range) months,a adjusted for crossover by RPSFT | 28.52c | 21.98c | HR = 0.73 (0.51 to 1.04); p = not reported |
| OS (range) months,d PP analysis (patients who did not cross over) | 28.5 (22.8–42.4) | 17.2e | p = 0.218;f p = 0.048g |
| PFS (range) months,a ITT populationb | 4.8 (3.7–7.0) | 2.4 (2.0–4.6) | HR=0.63 (0.47 to 0.84); p = 0.0016 |
| PFS (range) months,a centrally confirmed hormone receptor statusb | 5.6 (3.8–8.3) | 3.8 (2.0–6.3) | HR=0.62 (no CIs); p = 0.006 |
| PFS (range) months,a updatedd | 5.8 (4.6–8.3) | 2.9 (2.1–4.5) | HR= 0.55 (0.41 to 0.74); p < 0.0001 |
| TTP (range) months,a ITT populationb | 4.8 (3.7–7.7) | 2.4 (2.0–4.6) | HR not reported; p = 0.0007 |
| TTP (range) months,a centrally confirmed hormone receptor statusb | 5.6 (3.8–8.3) | 3.9 (2.1–6.3) | HR=0.62 (no CIs); p = 0.0007 |
| TRA + ANA (n = 74) | ANA (n = 73) | OR (95% CI); p-value | |
| ORR centrally confirmed hormone receptor status (%)b | 20 | 5 | OR not reported; p = 0.018 |
| CR (%) | 0 | 0 | |
| PR (%) | 20 | 5 | |
| SD ≥ 6 months (%) | 38 | 38 | Not reported |
| CBR (range) %,h ITT populationb | 43 (33–53) | 28 (20–38) | OR not reported; p = 0.026 |
Currently there is no uniform agreement about which is the best method(s) to use for adjusting for crossover. In a study by Kaufman et al. ,50 an attempt to highlight the impact of crossover on OS was explored using a per protocol (PP) analysis approach, in which the median OS for patients receiving TRA + ANA (28.5 months, n = 103) from randomisation was compared with the subgroup of patients who initially received ANA and did not cross over to receive TRA (17.2 months, n = 31). By log-rank testing, there was no significant difference in the OS analysis (p = 0.218). However, because of the small number of patients with long survival times available for analysis, the Wilcoxon test was also used as this gives more weight to early time points than the log-rank test. The analysis using the Wilcoxon test resulted in a modestly statistically significant difference (p = 0.048). Similarly, comparing patients in the ANA group who crossed over to receive TRA + ANA (n = 73) with those who did not (n = 31) resulted in an OS estimate of 25.1 months and 17.2 months, respectively. These differences were reported to be not statistically significant using the log rank test (p = 0.358), but were statistically significant using the Wilcoxon test (p = 0.040). The AG notes that where there is a relatively large proportion of patients who cross over, these PP approaches are prone to selection bias.
In their submission, the crossover adjustment employed by Roche20 was based on a rank-preserving structural failure time (RPSFT) approach initially proposed by Robins and Tsiatis60 and later modified by Mark and Robins. 61 Using the RPFST approach (ITT population), median OS in the TRA + ANA group becomes 28.52 months and the median OS in the ANA group becomes 21.98 months. The RPSFT method is an accelerated failure time model, a form of randomisation-based analysis that more effectively preserves the integrity of randomisation than do PP analyses. However, its validity has been questioned when imbalances occur post randomisation, e.g. when there is an unequal distribution of patients receiving second-line treatment across the arms. 62
The use of the RPSFT approach in the current appraisal was justified by the manufacturer as it has also been used for two other NICE appraisals: sunitinib for the treatment of gastrointestinal stromal tumours63 and everolimus for the second-line treatment of advanced renal cell carcinoma. 64 However, in the former appraisal, only 7% of patients crossed over to receive sunitinib whereas, in the latter, 81% of patients crossed over to receive everolimus. In the sunitinib appraisal,63 because so few patients crossed over from the control arm to sunitinib, the Appraisal Committee had confidence in the results from the RPSFT as well as the PP analysis, which was also performed. For everolimus,64 two different methods were employed to adjust for crossover by the manufacturer, the inverse probability censoring weight (IPCW) approach65 and the RPSFT approach. Because 81% of people had crossed over to receive everolimus, the Appraisal Committee agreed that it was appropriate to adjust the results utilising statistical methods such as these to control for crossover. However, as both methods gave different OS estimates,64 it was unclear which method, if either, was most suitable.
A recent paper by Morden et al. 62 explored various approaches to adjusting for crossover using a simulation exercise. Methods tested included PP approaches and accelerated failure time model methods. The authors found that that when there is crossover from the control group, commonly adopted approaches such as censoring at the time of crossover, or considering treatment as a time-dependent covariate, may be associated with biased estimates of the true treatment effect, where the reasons for the crossover are strongly related to their underlying prognosis. Where patients who cross over are excluded from the analysis altogether (i.e. a PP analysis), biases were reported to be small in situations with a low proportion of switchers (as was the case, for example, with sunitinib). However, as the number of patients who switch increases, the risk of bias was also reported to increase.
Specifically, with regard to accelerated failure time model methods, three methods were considered by Morden et al. :62 the RPSFT developed by Robins and Tsiatis,60 the iterative parameter estimation algorithm approach66 (which is a modification of the RPSFT method in which the test-based estimation is replaced with a likelihood-based analysis) and a parametric randomisation-based method (which as an extension to the previous two methods). 67 The findings from their simulation exercise suggested that the RPSFT60 and the iterative parameter estimation algorithm66 gave estimates close to the true treatment effect, whereas the parametric randomisation-based method67 overestimated the true treatment effect. The iterative parameter estimation algorithm66 appeared to be the most accurate method when the proportion of patients who crossed over was relatively high.
In the TAnDEM trial,50 the AG notes that some imbalances appear to have occurred post-randomisation, e.g. four times as many patients in the ANA arm received chemotherapy as a second-line treatment compared with TRA + ANA (32% compared with 8%). The AG also notes that the proportion of patients who crossed over was relatively high, being 70%, which, as Morden et al. 62 report, increases the likelihood of bias. However, the AG does agree that attempts to adjust for crossover are worthwhile – ideally different randomisation-based methods should be used to compute and compare a range of OS estimates to assess sensitivity of treatment effects, the applicability of each individual method employed depending on the trial circumstances and characteristics – in the MS such sensitivity is not investigated. It should be noted that in order to undertake such analyses, individual patient data are required. Such data were not available to the AG and thus the AG was unable to employ any of the aforementioned approaches. Thus, the AG has utilised its own method for adjusting for crossover for the purposes of conducting its economic analysis. This is described further in Chapter 4, Specific model features and parameters: trastuzumab in combination with anastrozole versus anastrozole alone.
As the assessment of disease progression (and therefore PFS, TTP, ORR and CBR) may be prone to subjectivity, and thus to observation bias, three universally accepted methods57,58 and procedures for assessing disease progression were employed in the TAnDEM50 trial to minimise the risk: an investigator assessment (ITT), a centrally confirmed assessment by a Response Evaluation Committee (REC) and, in situations in which the investigator assessment was different from the REC assessment, an independent oncologist was appointed to make a reconciliation assessment. Patients in the TRA + ANA group experienced significant improvement in PFS and TTP. Significant differences were also reported in terms of ORR and CBR in the ITT population, although, interestingly, no CR was recorded for any patient, the difference occurring as a result of improvements in PR in the TRA + ANA group.
Patients who received TRA + ANA were more likely to experience AEs, with nearly 90% experiencing an AE compared with 65% of patients who received ANA (Table 10). SAEs were also more common in the TRA + ANA group, nearly 25% experiencing an SAE compared with < 10% of patients receiving ANA. There were no treatment-related deaths in either group.
| Adverse events | TRA + ANA (n = 103) | ANA (n = 104) |
|---|---|---|
| Any AE (%) | 87 | 65 |
| SAEs (%) | 23 | 6 |
| Discontinued treatment because of AEs (%) | 9 | 1 |
| Treatment-related deaths (%) | 0 | 0 |
The most frequently reported AEs in both groups were fatigue, diarrhoea, vomiting and arthralgia, of which the majority were grades 1 or 2 (Table 11). AEs were more common in the TRA + ANA group than in the ANA group, although it should also be noted that duration of treatment was longer in the TRA + ANA group and that the open-label design of the study meant that AEs in the ANA group were only reported until the patients crossed over to TRA + ANA.
| AEs | TRA+ANA (n=103) | ANA (n=104) | ||
|---|---|---|---|---|
| Total (any grade) (%) | Grade 3 or higher (%) | Total (any grade) (%) | Grade 3 or higher (%) | |
| Fatigue | 21 | 1 | 10 | 0 |
| Diarrhea | 20 | 1 | 8 | 0 |
| Vomiting | 21 | 3 | 5 | 1 |
| Arthralgia | 15 | 0 | 10 | 1 |
| Pyrexia | 18 | 0 | 7 | 0 |
| Back pain | 15 | 2 | 7 | 2 |
| Dyspnoea | 13 | 2 | 9 | 0 |
| Nausea | 17 | 1 | 5 | 0 |
| Cough | 14 | 0 | 6 | 0 |
| Headache | 14 | 0 | 6 | 0 |
| Nasopharyngitis | 17 | 0 | 2 | 0 |
| Bone pain | 11 | 2 | 6 | 0 |
| Constipation | 12 | 0 | 5 | 0 |
| Chills | 15 | 1 | 0 | 0 |
| Hypertension | 7 | 2 | 4 | 4 |
Overall, therefore, no new safety issues were identified, the safety profile of TRA + ANA being consistent with the safety profiles of both drugs when given as single agents and with safety data from previously reported TRA studies.
eLEcTRA: trastuzumab in combination with letrozole versus letrozole alone
The eLEcTRA52 trial planned to enrol 370 patients with HR+ mBC, but between 2003 and 2007 enrolled only 92 patients, at which point the study was halted because of slow recruitment. The slow recruitment is attributed in the Roche submission20 to the fact that investigators believed that TRA + LET was superior to LET (although no evidence is presented to support this claim). When the trial was halted, patients who were HR+/HER2+ had been randomly assigned to TRA + LET (n = 26) or LET (n = 31) and patients who were HR+/HER2– had been assigned to receive LET (n = 35).
The findings from the eLEcTRA52 trial are summarised in Table 12: a large difference in TTP was observed between the two treatment groups [although this difference was not statistically significant (HR = 0.67; p = 0.23)]. Interestingly, however, significant differences were reported in TTP between the two cohorts of patients that received LET (median 3.3 months vs 15.2 months for HR+/HER2+ mBC vs HR+/HER2– mBC, respectively; HR = 0.71; p = 0.03). Large differences were also observed between TRA + LET and LET for ORR and CBR, but again these differences were not statistically significant (p = 0.3124 and p = 0.0636, respectively).
| Outcome | TRA + LET (n = 26) | LET (n = 31) |
|---|---|---|
| TTP (months)a | 14.1 | 3.3 |
| ORR (%) | 27 | 13 |
| CBR (%)b | 65 | 39 |
Patients who received TRA + LET were slightly more likely to experience SAEs and/or ‘clinically significant AEs’ (which were not defined) (Table 13). The most common AEs for patients in either group were musculoskeletal and connective tissue disorders and gastrointestinal disorders, whereas infections were also relatively common in both groups, particularly the TRA + LET group (Table 14). Fatigue was a problem particular to TRA + LET patients and around 15% experienced hepatobiliary disorders, whereas no patient in the LET group experienced these AEs.
| Adverse events | TRA + LET (n = 26) | LET (n = 31) |
|---|---|---|
| SAEs (%) | 27 | 23 |
| SAEs and/or clinically significant AEs (%)a | 39 | 36 |
| Discontinued treatment because of SAEs and/or clinically significant AEs (%)a | 4 | 0 |
| Death during treatment (%) | 0 | 3.2 |
| AEs | TRA + LET (n = 26) (%) | LET (n = 31) (%) |
|---|---|---|
| Musculoskeletal and connective tissue disorders | 57.7 | 38.7 |
| Gastrointestinal disorders | 57.7 | 32.2 |
| Infections | 30.8 | 16.1 |
| Fatigue | 26.9 | 0.0 |
| Metabolism disorders | 20.0 | 3.2 |
| Headache | 19.2 | 9.7 |
| Hepatobiliary disorders | 15.4 | 0.0 |
| Bone fractures | 7.7 | 6.5 |
| Psychiatric disorders | 3.8 | 16.1 |
| Hot flushes | 7.7 | 3.2 |
| Cardiac events | 7.7 | 9.7 |
Notwithstanding the caveats raised by the AG in comparing data across trials, it is also impossible to compare the AE profiles of TRA + LET in the eLeCTRA52 trial with TRA + ANA in the TAnDEM50 trial or LET in the EGF3000849 trial because of the different ways in which AEs have been categorised, with the possible exceptions of fatigue and headaches. However, comparisons with the eLEcTRA52 trial are arguably still inappropriate given the small number of patients in this trial and the fact that the trial was halted early.
As noted above, there are known concerns about cardiac safety associated with TRA, but there were fewer cardiac events recorded in the TRA + LET group than in the LET group.
Overall, therefore, no new safety issues were identified, the safety profile of TRA + LET being consistent with the safety profiles of both drugs when given as single agents and with the safety data from previously reported TRA studies.
Although there were three trials49,50,52 identified that compared the interventions of interest with a comparator of interest in the relevant population, only two of these trials49,50 were completed as intended. These two trials were primarily sponsored by the manufacturers of LAP (GlaxoSmithKline) and TRA (Roche) and it was from these two manufacturers that the MS19,20 were received. In both of the MS, the manufacturers reported and appraised each of the pivotal trials individually, an approach also undertaken by the AG. Unlike the AG, however, Roche also performed a meta-analysis and both manufacturers also conducted indirect comparison analyses in order to compare LAP + AI with TRA + AI.
Meta-analysis (Roche)
The fixed-effect standard meta-analyses undertaken by Roche20 examined PFS and were conducted for ANA versus TAM (two trials68,69) and ANA versus megestrol acetate (Magace®, Bristol-Myers Squibb) (two trials70,71). There were insufficient trials to conduct meta-analyses for any other comparisons, such as LAP + LET versus LET or TRA + AI versus AI.
For the meta-analysis, forest plots for HR for individual studies and pooled studies were presented. The I2 statistic was calculated to assess the potential heterogeneity between studies. The studies68–71 included in these meta-analyses appeared to be associated with statistical and clinical heterogeneity. No significant differences were found for PFS between treatment groups in either meta-analysis.
Given that ANA was being compared with TAM or megestrol acetate, and given that it was unclear how many patients were HR+/HER2+, the AG believes that the relevance of these analyses to the current appraisal is limited. They were, however, relevant to the Roche submission20 because the results from these meta-analyses were used in its indirect comparison analyses.
Indirect comparisons analyses
Both manufacturers performed indirect comparisons analyses, although different approaches were employed, as summarised in Table 15.
| Approach taken | GlaxoSmithKline | Roche |
|---|---|---|
| Population included | Post-menopausal women with HR+ mBC who have not received prior therapy for advanced or mBC, i.e. patients for whom treatment with endocrine therapy was considered appropriate | Post-menopausal women with HR+ mBC |
| Outcomes analysed |
PFS/TTP, OS TTP has been used where possible and, when TTP was not reported, PFS has been used assuming this was similar to TTP. Used Cox results not log-rank results |
PFS, OS PFS = TTP, if TTP was explicitly defined as the time from randomisation to disease progression or death from any cause (if the reason for death was not reported, it was assumed that the death was from any cause) Where HRs were unavailable, summary statistics were used based on Parmar et al. 72 |
| Included studies | Five studies were included in both the PFS and OS analyses | Seven studies were included in PFS analysis, 11 studies in the PFS/TTP analysis and 12 studies in the OS analysis |
| Synthesis methodology |
No direct meta-analysis Series of the adjusted indirect comparisons, using the methods and principles as described by Bucher et al. 73 |
For indirect comparisons, analyses were performed using Bayesian network meta-analyses (also known as mixed-treatment comparisons), as described by Sutton and Higgins74 |
| Assessment of homogeneity and similarity between included studies | No assessment was reported, although the manufacturer stated in the methods that it anticipated systematic differences between studies (i.e. heterogeneity). Thus, a random-effects model was used for the calculation of RR. Heterogeneity was intended to be assessed by measuring the degree of inconsistency in the studies’ results (I2 statistic). However, neither the I2 statistic nor measures of RR were calculated; HRs were calculated and utilised in the analysis instead | For indirect comparisons, the manufacturer discussed with clinical experts and assessed statistically (from the posterior median variance of the random effects) the suitability of including particular trials in the analyses. A series of sensitivity analysis were performed to explore the nature of heterogeneity |
| Manufacturers’ quality assessment | The manufacturer discussed the limitations of its indirect comparisons. These included failure to fulfil basic assumptions of homogeneity, similarity and consistency for the indirect comparisons | The manufacturer utilised clinical experts to assess the suitability of trials to be included in their analyses. The manufacturer discussed the limitations of their direct and network meta-analyses and sensitivity analyses were performed |
| AG comment | The studies included in the indirect comparisons analysis included trials in which the HR+/HER2+ status was unknown; only two trials included analyses of this specific population – the EGF3000849 and the TAnDEM50 trials | The manufacturer also utilised clinical experts to assess the suitability of trials to be included in their analyses. However, there were only three trials in which the HR+/HER2+ status of patients analyses was known – the EGF30008,49 the TAnDEM50 and the eLEcTRA52 trials |
A complex network meta-analysis using the methods described by Puhan et al. 75 was planned by GlaxoSmithKline,19 but was not possible because of the lack of data for the outcomes of interest: OS and PFS/TTP. Thus, adjusted indirect comparisons analyses were performed for single outcomes as available, using the methods and principles as described by Bucher et al. 73 and incorporating data from five studies; the EGF3000849 and the TAnDEM50 trials were included as well as one study comparing LET (2.5 mg/day) with TAM (20 mg/day)76 and two studies comparing ANA (1 mg/day) with TAM (20 mg/day). 68,69 The eLEcTRA52 study was not included in the GlaxoSmithKline analyses as it was published only as an abstract and the AG agrees with the manufacturer’s argument that a lack of data from this trial justifies its exclusion.
The findings for both OS and PFS/TTP presented by the manufacturer suggest that there are no significant differences between any of the interventions for OS. Both LAP + LET and TRA + LET result in significantly improved outcomes for PFS/TTP when compared with ANA, LET and TAM. For reasons discussed below, the AG believes these findings should be treated with extreme caution and so the findings are not presented in this report.
Roche20 employed an indirect network meta-analyses based on a Bayesian approach,74 in which a number of different analyses were performed for OS (base case of 12 trials49,50,68–71,77–82) and PFS (base case of seven trials49,50,68–70,78,81). A number of assumptions were made and tested by sensitivity analyses. These included an assumption that PFS = TTP (which enabled four additional trials52,77,79,80 to be considered) and that OS findings for the TAnDEM50 trial based on the RPSFT adjustment should be used in the base case. In addition, for every outcome, the assumption that AIs hold a ‘class effect’ (i.e. LET = ANA, as suggested by clinical experts and as found in a head-to-head trial of second-line ANA vs LET82) was tested. This assumption related to the mixed HER2 status population (i.e. the population in which the proportion of patients with HER2+ breast cancer was unknown, as in the aforementioned ANA vs LET trial82). The mixed HER2 population was chosen because the HR+/HER2+ population was too specific to allow the inclusion of any trials other than the EGF30008,49 the TAnDEM50 and the eLEcTRa trials. 52
The findings presented by the manufacturer in which a ‘class effect’ was assumed for AIs and which were derived from the EGF3000849 and the TAnDEM50 for OS and PFS and from the EGF30008,49 the TAnDEM50 and the eLEcTRa52 for PFS/TTP suggest that there are no significant differences between LAP + LET and TRA + ANA for OS, PFS or PFS/TTP. For reasons discussed below, the AG believes these findings should be treated with extreme caution and so findings are not presented in this report.
Aside from the fact that the EGF3000849 and the TAnDEM50 trials were too dissimilar in terms of patient populations, the AG believes that both the manufacturers’ indirect comparisons analyses had one other major limitation, namely that the basic requirement for indirect comparisons with regard to exchangeability of relative treatment effect between trials in the two MS could not be assumed. This is a limitation recognised by the manufacturers themselves19,20 and is amplified when patient population characteristics are considered. Crucially, it was unknown how many patients with HR+/HER2+ mBC were included in the trials. Only three trials49,50,52 presented data for patients with HR+/HER2+ mBC. The other trials68–71,76,77,78,82–90 included patients of mixed/unknown status. The importance of missing data on HR+/HER2+ status is twofold. Firstly, patients with HR+/HER2+ mBC are the population of interest to this review and, as Roche has acknowledged (MS, p. 18):20 ‘All results should be treated with extreme caution when applied to the HR+/HER2+ population as there is no evidence base capable of informing this analysis in the population specified by the decision problem’. Secondly, both the EGF3000849 and the eLEcTRA52 trials suggest that the effects of LET in patients with HR+/HER2+ mBC tumours are significantly compromised when compared with those with HR+/HER2– mBC. Thus, the indirect comparisons analyses may be overstating the benefit of AIs and, if so, there is a need to adjust for the results based on HER2+ status. However, given that the proportion of such patients is unknown, such adjustments are currently impossible. It is important to note, as Roche has also stated (MS, p. 18):20 ‘…understanding of HER2 was not fully developed at the period when most of the evidence base identified was formed as many of the trials conducted were not stratified for HER2 positivity and it is clearly plausible that an imbalance in this strong indicator of extremely poor prognosis could have biased the estimates of relative efficacy generated’.
Roche20 also acknowledges a number of additional limitations to its indirect comparisons analyses, namely ‘the low number of trials by pairwise comparison, the heterogeneity in the length of follow-up observed in the selected studies and the different methods used to adjust for cross-over in the individual studies’ (MS, p. 17). A final limitation is the fact that not all trials included patients receiving first-line treatment. In fact, two trials78,82 were second line, including the trial by Rose et al. ,82 which was a key trial for suggesting a ‘class effect’ for AIs. The AG believes that pooling trials with different lines of treatment is inappropriate and misleading; thus, these results should be interpreted with caution.
In summary, given the limitations described above, the AG believes that conducting indirect comparisons analyses with the limited data available is inappropriate. Therefore, any findings generated from these analyses should be treated with caution.
Summary
The findings from the three main trials examining the efficacy of LAP + LET (EGF3000849), TRA + ANA (TAnDEM50) and TRA + LET (eLEcTRA52) all suggest that LAP + AI or TRA + AI results in improved outcomes when compared with AIs alone (LET or ANA). In the EGF30008 and the TAnDEM trials,49,50 although these differences were not significant for OS, statistically significantly different outcomes were reported for PFS and TTP and large differences were reported between TRA + LET and LET patients in the eLEcTRA trial;52 this trial lacked statistical power to adequately test for significant differences. In addition, both ORR and CBR appeared to be improved for patients taking LAP + AI or TRA + AI. No new safety concerns were identified from the trials, although both AEs and SAEs were more common in the LAP + LET and TRA + AI groups than in the groups treated with AIs alone. For LAP + LET, the most significant AE was diarrhoea, experienced by around two-thirds of all patients. The impact this may have had on patient QoL is difficult to estimate, as to date the findings have been presented only as a conference abstract. 59 However, it would appear there are no significant differences between patients in either treatment group. Indeed, the majority of cases of AEs (including diarrhoea) were of grades 1 or 2 severity. Nevertheless, diarrhoea did result in around 1% of all patients who received LAP + LET discontinuing their treatment as a result; all other patients were managed by dose reduction, dose interruption or supportive intervention without treatment dose adjustments. For TRA + ANA patients, the most frequently reported AEs were fatigue, diarrhoea and vomiting, experienced by around one-fifth of all patients, of which the majority were grades 1 or 2 severity. Fatigue was also a problem for around one-quarter of patients who received TRA + LET, but infections, gastrointestinal disorders and musculoskeletal and connective tissue disorders were even more common; over half of TRA + LET patients experienced these last two AEs. Around one-third of LET patients also reported gastrointestinal disorders and musculoskeletal and connective tissue disorders.
However, extreme caution must be exercised in comparing the aforementioned findings across trials. Thus, for example, it would be wrong to assume that because TRA + ANA appears to have comparable efficacy and a more preferable side-effects profile than LAP + LET or TRA + LET, it is therefore superior to other treatments, for a number of reasons.
-
Such a comparison would be considered to be too simplistic and naive as it breaks the randomisation procedure and would not account for differences in baseline characteristics in treatment groups across the trials. To compare the outcomes more accurately, a direct comparison or indirect comparisons analyses would need to be considered.
-
The ORR hides the fact that none of the patients in the TAnDEM50 trial examining TRA + ANA were CRs, unlike in the EGF30008 trial,49 in which 5% of LAP + LET and 4% of LET patients were CRs. It was not known if any patients taking TRA + LET or LET were CRs in the eLEcTRA52 trial because this trial did not report ORR by CR and PR.
-
Trials did not always report data in the same way, so it is unclear, for example, if a greater proportion of patients in the eLEcTRA52 trial experienced gastrointestinal disorders than in other trials because such a category of AE did not exist (instead there were data on diarrohea and vomitting, etc.). A similar problem was encountered in trying to compare baseline characteristics across trials.
-
It is difficult to compare the results of the eLEcTRA52 trial with those of the other two trials because this trial was halted prematurely because of slow recruitment.
-
Most crucially, it was apparent from the exclusion criteria that there may have been differences in the patients included in the EGF30008 and the TAnDEM trials. 49,50 This appeared to be supported by data reported by the two trials, which suggested large differences in median OS in the AI arms (LET and ANA, respectively). Notwithstanding the dangers of crude comparisons across trials just highlighted, if patients were similar in terms of their baseline characteristics, we would expect similar estimates for OS in the AI-only arms trials given that the evidence to date suggests that there is no difference between LET and ANA in terms of efficacy, albeit in early HR+ breast cancer. 34,91 However, depending on which estimate for OS was used in the TAnDEM trial,50 differences of between 3.7 and 10.4 months were observed in the AI arms of the EGF3000849 and TAnDEM trials. 50
Alternative explanations for differences in OS are that there are real differences between LET and ANA, or that differences between the AI groups occurred as a result of differences in second-line treatment received following progression. In relation to the first alternative, there appears to be a broad consensus within clinical practice that there is little to choose between LET and ANA, certainly in terms of efficacy, and there is also evidence of a ‘class effect’ (i.e. LET = ANA), albeit from studies of early HR+ breast cancer. 34,91 In relation to the second possible explanation, patients in both the EGF3000849 and the TAnDEM50 trials were permitted to receive second-line treatment and this appears to differ between the trials, with chemotherapy being more common in the EGF30008 trial. 49
The fact that patients were able to cross over in the TAnDEM50 trial and the fact that in both trials second-line therapy was permitted has added an extra complication in interpreting and comparing the findings, namely how much of the benefit in OS is attributable to the first-line treatment and how much of the benefit is attributable to subsequent treatment following disease progression? Post-hoc attempts have been made by both Kaufman et al. 50 and Roche20 to address this issue in the TAnDEM trial,50 but no such attempts were made for the EGF30008 trial. 49
Kaufman et al. 50 compared the median OS between those receiving TRA + ANA with those who initially received ANA but did not cross over to receive TRA, and those in the AI group who crossed over to receive TRA + ANA with those who did not. Significant differences, when the Wilcoxon test was employed, were reported in favour of TRA + ANA and those in the ANA group who crossed over. The AG believes this was an inappropriate method as it is prone to selection bias. A different method was employed by Roche,20 namely the RPSFT method, which allowed for a comparisons between the TRA + ANA and ANA groups. Using this method OS gains were found to be greater than when no adjustment was made. However, the main justification for employing the RPSFT approach appears to be that it has been used in previous submissions to NICE. 63,64 The AG notes that other, possibly more appropriate, methods exist and believes that different randomisation-based methods should ideally be used to compute and compare a range of OS estimates to assess sensitivity of treatment effects. Therefore, the AG believes that the findings from the RPSFT approach should be treated with caution.
Because crude comparisons of OS across trials are too simplistic, both manufacturers conducted adjusted indirect comparisons analyses,19,20 with Roche employing a network meta-analysis based on a Bayesian approach,74 in which a number of different analyses and sensitivity analyses were performed and in which it was assumed, and tested, that there was a ‘class effect’ for AIs. The findings from both the manufacturers’ approaches appeared to support the trial findings suggesting that LAP + LET and TRA + AI were better than AIs alone in terms of PFS and/or TTP, but not OS. In addition, their analyses suggested that there was little difference between LAP + LET, TRA + ANA and TRA + AI. However, the AG believes that these indirect comparisons must also be treated with caution for a number of important reasons. First and foremost, as discussed above, the AG believes that the patient populations are not sufficiently similar in the EGF3000849 and the TAnDEM trials. 50 Hence, these trials should not be compared with each other at all. On the other hand, if there are differences in efficacy between LET and ANA, then there may be grounds to conduct an indirect comparisons analysis if the other trials are sufficiently similar. However, both indirect comparisons analyses had one other major limitation, recognised by the manufacturers themselves,19,20 namely that the basic requirement for indirect comparisons with regard to exchangeability of relative treatment effect between trials in the two MS could not be assumed. Crucially, it was unknown how many patients with HR+/HER2+ mBC were included. Both the EGF3000849 and the eLEcTRA52 trials suggest that the effects of LET in patients with HR+/HER2+ mBC tumours are significantly compromised when compared with those with HR+/HER2– mBC. As has been acknowledged by Roche,20 ‘it is clearly plausible that an imbalance in this strong indicator [HER2+] of extremely poor prognosis could have biased the estimates of relative efficacy generated’ (MS, p. 18). Other areas of heterogeneity include the proportion of patients with advanced breast cancer, length of follow-up and proportion of patients receiving first-line treatment.
Thus, overall, the AG believes comparisons across trials cannot be made and that only the individual findings from each trial should be considered.
Chapter 4 Assessment of cost-effectiveness
Systematic review of existing cost-effectiveness evidence
In this section, firstly, a critical appraisal of the available economic evidence describing (1) LAP + LET and (2) TRA + ANA is described. Secondly, the AG’s critique of the two economic evaluations submitted by the manufacturers is presented.
Review of published cost-effectiveness studies
Full details of the search strategy conducted by the AG and the methods used for selecting evidence are presented in Chapter 3, Quantity and quality of research available. The AG concluded that none of the 107 economics studies identified from the electronic searches were eligible for inclusion in the literature review as they did not include any of the relevant interventions (LAP + AI or TRA + AI). The authors of the GlaxoSmithKline MS noted that ‘no economic evaluations of lapatinib plus an AI were identified’ (MS, p. 100). The authors of the Roche MS stated that, although they summarised the characteristics and results of five studies,92–96 ‘four were of poor relevance to the decision problem as they were not in the population of relevance and were not set in the UK’ (MS, p. 209). The only study that Roche deemed to be relevant to the review was the poster by Hastings et al. ,96 which is discussed below.
The AG notes that the poster presented at American Society of Clinical Oncology (ASCO) conference (June 2010) by Hastings et al. 96 is relevant to the technologies under assessment. It is noted that the authors of this poster are employees of GlaxoSmithKline, yet the poster was only discussed in the MS submitted by Roche.
Summary and critique of Hastings poster
The Hastings et al. 96 poster describes an indirect comparison of the cost-effectiveness of LAP + LET versus TRA + ANA in post-menopausal women with HR+/HER2+ mBC who have not received prior treatment. The perspective of the economic analysis is the UK NHS. The evidence network used to estimate treatment effectiveness appears to be the same as that described in the MS submitted by GlaxoSmithKline and includes both the EGF3000849 and the TAnDEM trials;50 the utility values for PFS and post-progression survival (PPS) health states are also the same. Base-case results are shown in Table 16 and are different to the estimates provided in the MS by GlaxoSmithKline. Hastings et al. 96 conclude that LAP + LET is cheaper and more clinically effective than TRA + ANA and is therefore dominant. The AG is of the opinion that the results of the indirect analysis performed by Hastings et al. 96 are unreliable as the studies which make up the evidence network are inappropriate (for more details see Chapter 3). In addition, the AG notes that without access to more detailed information on costs, it is difficult to comment on the reliability of the cost-effectiveness results in this study.
| Measure | LAP + LET | TRA + ANA | Difference |
|---|---|---|---|
| Total QALYs (discounted) | 2.626 | 2.330 | 0.296 |
| Total costs (£) (discounted) | 60,614 | 64,003 | –3.389 |
| Cost per QALY (£) | Dominant |
Conclusions of the review of existing cost-effectiveness evidence
There is no relevant, currently available, published cost-effectiveness evidence to describe the use of LAP + LET or TRA + ANA in women who are HR+/HER2+ with mBC.
Overview and critique of GlaxoSmithKline economic evaluation
Overview of submitted economic evaluation and economic model
The purpose of the manufacturer’s model is to assess the cost-effectiveness of first-line treatment with LAP + LET in HR+/HER2+ patients with mBC. In the MS, the combination of LAP + LET is compared with the following interventions: LET monotherapy, TRA + ANA and ANA monotherapy. A decision-analytic model was developed by the manufacturer to estimate PFS, OS, lifetime costs of treatment of mBC and QALYs. The model schema is presented in Figure 2. The model features three health states: alive and no progression, alive with progression and dead. The manufacturer estimates costs from the perspective of the NHS and PSS and health outcomes in terms of life-years, progression-free life-years (PFLYs), post-progression life-years (PPLY) and QALYs; variables are estimated daily for 10 years. The economic evaluation has a time horizon of 10 years and both costs and benefits are discounted at 3.5% per annum. The manufacturer’s reference case adequately reflects the NICE reference case97 (Table 17).
FIGURE 2.
The structure of the model submitted by GlaxoSmithKline.
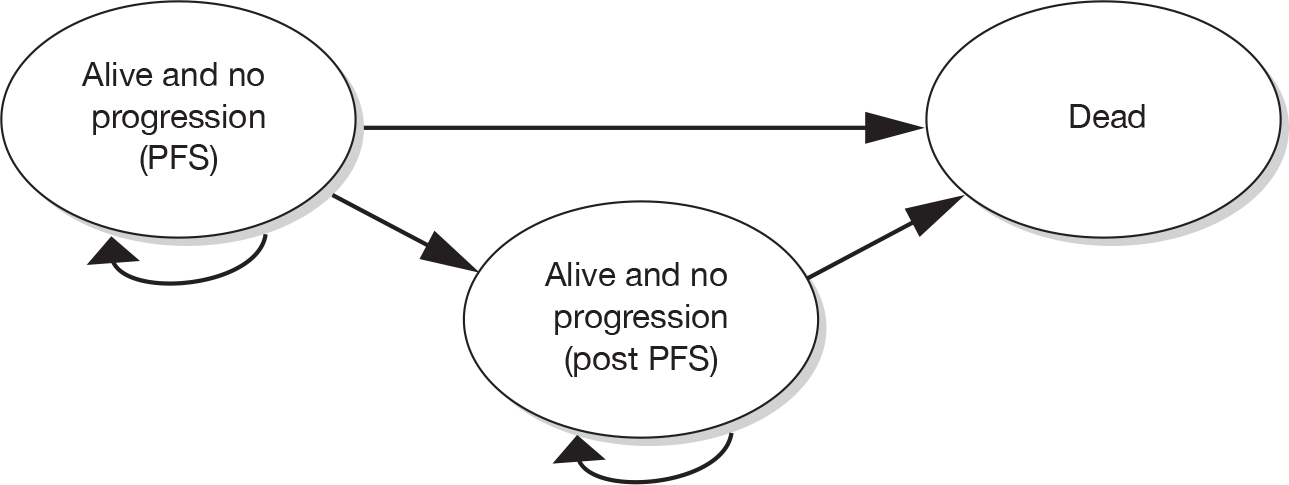
| NICE reference case requirements | Reference case97 | Does the de novo economic evaluation match the reference case? |
|---|---|---|
| Defining the decision problem | The scope developed by the institute | Yes |
| Comparators | Therapies routinely used in the NHS, including technologies currently regarded as best practice |
Yes. Intervention is LAP + LET Best practice: AIs. New intervention also under appraisal: TRA + ANA |
| Perspective on costs | NHS and PSS | NHS and PSS |
| Perspective on outcomes | All health effects on individuals | All health effects on individuals |
| Type of economic evaluation | Cost-effectiveness analysis | Cost–utility analysis |
| Synthesis of evidence on outcomes | Based on a systematic review | Based on a systematic review and an adjusted indirect comparisons exercise |
| Measure of health benefits | QALYs | QALYs |
| Source of data for measurement of QoL | Reported directly by patients and/or carers | (1) Utilities are reported directly by patients from the EGF3000849 trial for PFS health states (2) published utilities are used for PPS health states |
| Source of preference data for valuation of changes in QoL | Representative sample of general public | Algorithm used to map FACT-G values into EQ-5D values; EQ-5D valuations are based on values from representative values of general public |
| Discount rate | An annual rate of 3.5% on both costs and QALYs | An annual rate of 3.5% on both costs and QALYs |
| Equity weighting | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit |
Summary and critique: clinical effectiveness data
Direct clinical evidence: lapatinib in combination with letrozole versus letrozole alone
The key clinical data (PFS, OS and AEs) used in the manufacturer’s economic model comparing LAP + LET versus LET are taken directly from the EGF30008 trial. 49 The AG’s description and critique of the EGF3000849 trial is presented in Chapter 3 of this report.
Progression-free survival and OS estimates for patients receiving LET were estimated by fitting Weibull survival functions to patient-level failure time data for HR+/HER2+ patients in the EGF30008 trial. 49 PFS and OS estimates for patients receiving LAP + LET were obtained by applying the HRs for LAP + LET versus LET to the PFS and OS curves for LET for HR+/HER2+ patients in the EGF30008 trial. 49 In general, the AG’s preferred approach to projective modelling is to assess PFS and PPS separately and then combine them in order to get a more reliable estimate of OS, rather than simply modelling OS as a single entity.
Indirect clinical evidence: lapatinib in combination with letrozole versus trastuzumab in combination with anastrozole/anastrozole alone
An indirect comparisons analysis was carried out by the manufacturer to compare LAP + LET with other drugs. In order to perform the indirect comparison and derive (PFS, OS and AEs) parameter estimates for TRA + ANA and ANA monotherapy, data from the TAnDEM50 and other published AI trials (with or without an anti-HER2 therapy and mixed HER2 populations) were used. A summary and critique of the evidence network employed by the manufacturer is presented in Chapter 3 of this report.
As noted earlier, the AG is not confident that the results of the indirect comparisons analysis conducted by the manufacturer are reliable. Firstly, the TAnDEM50 trial is included in the network and this trial does not explicitly exclude patients who have extensive symptomatic visceral or rapidly progressing or life-threatening disease, which means that the patient populations of the studies in the network may be different; in particular, the patients in the TAnDEM50 and the EGF3000849 trials are not comparable. Secondly, there were large differences in the second-line treatment received in these trials. A further criticism is that in the indirect comparisons analysis, it is likely that the AI trials include patients who are both HER2+ and HER2–; the AG is of the opinion that the inclusion of HER2– patients is inappropriate given that the decision problem is focused on treating women who are HER2+ and it is becoming apparent49,52 that an AI as monotherapy is less effective in women who are HER2+ than in women who are HER2–. Finally, it should be noted that GlaxoSmithKline uses the TAnDEM50 trial data published in 2009 that does not use the RPFST method to adjust (for crossover) OS estimates for patients receiving ANA in the TAnDEM trial. 50
In summary, the AG considers that the submitted model results for the comparison of LAP + LET versus LET is the only comparison that is wholly valid to inform decision-making in this area because of the limited comparative clinical effectiveness data available.
Summary and critique: costs and resource use
The manufacturer estimates the following costs for each treatment strategy: acquisition and administration of medications, patient monitoring, treatment of AEs, other costs during PFS and PPS and total costs. The key cost parameters used in the model are summarised in Table 18.
| Item | Most (£) | Source |
|---|---|---|
| LAP (250 mg) 70 pack | 804.30/pack; 11.49 per tablet | BNF 5928 |
| LET (2.5 mg) 28 pack | 66.50/pack; 2.38 per tablet | BNF 5928 |
| TRA (150-mg vial) | 407.40 | BNF 5928 |
| ANA (1 mg) 28 pack | 68.56/pack; 2.45 per tablet | BNF 5928 |
| Dispensing costs | 8.50 | PSSRU (15 minutes of community pharmacist time, £34.00/hour)98 |
| ECHO/MUGA monitoring costs | 46.50/month | NHS reference costs 2008–200999 (50% : 50%, testing every 3 months) |
| Total pre-progression cost | 562.00/month of PFS | Remak and Brazil100/PSSRU98 |
| Total post-progression cost | 803.92/month of PPS | Remak and Brazil100/PSSRU98 |
| Non-severe AE (e.g. chills, constipation, cough, epistaxis, hot flushes, nasopharyngitis) | 99 | Probability for hospitalisation for grade 3 or higher AEs was based on data from the EGF30008 trial.49 Visit and hospitalisation costs for grade 3 or higher AEs based on NHS reference costs 2008–200999 |
| Alopecia | 158 | |
| Dyspnoea | 722 | |
| Headache | 255 | |
| Nausea | 420 | |
| Vomiting | 1398 |
The economic model makes use of pre- and post-progression cost data from the Remak and Brazil study;100 however, the manufacturer does not comment on the relevance and/or generalisability of the assumptions employed in this study to HR+/HER2+patients with mBC in England and Wales.
In summary, the AG notes that in the MS, the methods used by the manufacturer to identify, measure and value cost items are not fully described. The AG notes that further information is provided in the manufacturer’s accompanying cost-effectiveness report. Table 19 identifies key costs which could have been discussed in further detail by the manufacturer.
| Assumption made | Limitation |
|---|---|
| Economic evaluation uses a 4 mg/kg loading dose of TRA followed by subsequent doses of 6 mg/kg in a 3-weekly scenario | SPC states that a 8 mg/kg loading dose be used then followed by subsequent doses of 6 mg/kg in a 3-weekly scenario. In the model a 10 mg/kg loading dose is costed (4 mg/kg + 6 mg/kg) |
| MS describes costs of PFS and PPS using cost categories described in Remak and Brazil100 paper | Remak and Brazil100 paper also describes cost categories related to end-of-life treatment which are not included in the economic evaluation |
| MS appears to assume that patients did not receive second-line chemotherapy or post-progression treatments as further treatments are not discussed | Inappropriate assumption – (1) not a valid assumption for clinical practice in England and Wales as often patients go on to receive additional treatments and (2) high proportion of patients received second-line chemotherapy treatment in the TAnDEM trial,50 which gives first-line treatment in a similar setting to patients with mBC |
| MS assumes a 14-day wastage of oral tablets | GlaxoSmithKline model uses drug costs on a per tablet basis. As drugs can only be bought in packs (and any unused drugs cannot be shared) this is inappropriate. The full (rather than half) pharmacy dispensing cost should also be included in the cost associated with wastage |
GlaxoSmithKline estimates drug costs per tablet which leads to inaccuracies:
|
Pack prices from BNF 59:28
|
| GlaxoSmithKline assumes that delivery of TRA is always an outpatient procedure. Delivery of simple parenteral chemotherapy [SB12Z Deliver parenteral chemotherapy at first attendance (£272) and SB15Z Deliver subsequent elements of a chemotherapy cycle (£272)] | Depending on patient condition and local circumstance, delivery could be on a day-case basis. A weighted average of outpatient and day-case costs would be more meaningful |
| Monitoring costs in the economic model are £46.50 per month for both LAP + LET and TRA + ANA patients | In the base case, GlaxoSmithKline assumes that cardiac monitoring occurs every 3 months and that both MUGA and ECHO scans are used in equal proportions. AG clinical advisors have stated that MUGA scans are used less frequently (30%) than ECHOs (70%) |
| AEs | No real information is presented in MS to explain methods used to cost concurrent events; all AEs appear to be costed as individual episodes, which is unrepresentative of clinical practice |
Summary and critique: utilities
Utility values for PFS without AEs were estimated using data from the EGF3000849 trial on the Functional Assessment of Cancer Therapy-General Scale (FACT-G)101 plus Breast Cancer subscale (FACT-B)19 and an algorithm was used to map from the FACT-G to patient preference-based utilities. 102,103 The pre-progression utility value used in the model was 0.86.
In the EGF0008 trial,49 FACT assessments were routinely completed by patients only until withdrawal of study medications (i.e. typically at disease progression). This means that post-progression utility values for patients are largely unavailable, and the manufacturer states that the generalisability of the values that are available is uncertain. In order to identify a utility decrement for PD to apply to patients with PD, the manufacturer used the results of a study by Lloyd et al. 104 of societal preferences for different stages of mBC in the UK. The absolute reduction in utility compared with no progression used in the model was 0.23; this means that PP utility value can be no higher than 0.62.
Disutility values from grades 3 or higher AEs were obtained from published and unpublished sources,104–106 and where no data were available, assumptions were made. The utility decrements employed in the economic model include nausea (0.1), vomiting (0.1), diarrhoea (0.1), alopecia (0.11), asthenia/fatigue/lethargy (0.12), and skin and nail disorders (0.15).
The AG notes that the manufacturer does not sufficiently describe the results of the FACT assessments from the EGF30008 trial,49 nor does the manufacturer adequately describe (or test the sensitivity of) the mapping exercise undertaken. Therefore, it is difficult to comment on the usefulness of the PFS utility values.
For PPS, the AG agrees with the manufacturer that the paper by Lloyd et al. 104 describing UK-based societal preferences is relevant to health-care decision-making in the UK. However, as (1) the health states described in the Lloyd et al. 104 paper were derived from literature reviews, exploratory interviews with physicians and an oncology focus group made up of specialist nurses and (2) the health states were gender neutral and there was no mention of ‘cancer’ in the health-state descriptions, the AG is also very aware that health-state descriptions and the valuations of the general public may not fully reflect the experiences of patients with cancer nor the true preferences of the general public.
Summary and critique: results
The manufacturer presents detailed summaries of costs and outcomes (PFLYs, PPLYs, Life-years and QALYs) for the following regimens: LAP + LET, LET monotherapy, TRA + ANA and ANA monotherapy. Base-case results for the pair-wise comparisons are reported in Table 20. The results show that LAP + LET is not cost-effective compared with any of the AIs. LAP + LET appears to be cost-effective compared with TRA + ANA; however, as there is much uncertainty about the reliability of the indirect comparison results, these results are not considered by the AG to be meaningful.
| Outcomes, costs and ICER | LAP + LET | LET | TRA + ANA | ANA | Incremental comparisons: LAP + LET vs | ||
|---|---|---|---|---|---|---|---|
| LETa | TRA + ANA | ANA | |||||
| Outcomes | |||||||
| PFLYs | 1.181 | 0.738 | 1.042 | 0.592 | 0.444 | 0.139 | 0.589 |
| PPLYs | 2.218 | 2.079 | 2.004 | 2.065 | 0.138 | 0.214 | 0.153 |
| Life-years | 3.399 | 2.817 | 3.045 | 2.657 | 0.582 | 0.354 | 0.742 |
| QALYs | 2.389 | 1.923 | 2.137 | 1.788 | 0.467 | 0.252 | 0.601 |
| Costs | |||||||
| Acquisition costs (£) | 30,219 | 688 | 23,818 | 576 | 29,531 | 6401 | 29,643 |
| Administration costs (£) | 260 | 83 | 4236 | 66 | 177 | –3976 | 194 |
| Monitoring costs (£) | 659 | 0 | 581 | 0 | 659 | 78 | 659 |
| Treatment-specific AE costs (£) | 113 | 71 | 109 | 67 | 42 | 4 | 46 |
| Other progression-free costs (£) | 7966 | 4975 | 7026 | 3991 | 2991 | 940 | 3975 |
| Other post-progression costs (£) | 21,396 | 20,060 | 19,330 | 19,919 | 1336 | 2066 | 1447 |
| Total costs (£) | 60,614 | 25,878 | 55,101 | 24,620 | 34,737 | 5513 | 35,995 |
| ICER | |||||||
| Cost per LYG (£) | 59,684 | 15,590 | 48,478 | ||||
| Cost per PFLYG (£) | 78,317 | 39,532 | 61,074 | ||||
| Cost per QALY gained (£) | 74,448 | 21,836 | 59,895 | ||||
Summary and critique: sensitivity analysis and probabilistic sensitivity analysis
The AG notes that 51 scenarios were examined by the manufacturer using sensitivity analysis. The results of the sensitivity analysis are presented in table 41 of the MS. For the comparison with LET monotherapy, the incremental cost per QALY is in the range of £41,877 to LAP + LET being dominated by LET monotherapy. The cost per QALY gained versus ANA monotherapy ranges from £38,170 to £378,674. For the comparison with TRA + ANA, the range is LAP + LET dominating the comparator to a cost per QALY estimate of £45,106. The approach to sensitivity analysis adopted by the manufacturer makes it is difficult for the AG to acquire any real insight into the true drivers affecting the size of the incremental cost-effectiveness ratios (ICERs).
A summary of the probabilistic sensitivity analysis (PSA) is presented in Figure 3 and the cost-effectiveness acceptability curve (CEAC) is shown in Figure 4. The CEAC shows that, at a cost-effectiveness threshold of £30,000 per QALY gained, the probability of LAP + LET being cost-effective is very low (< 25%) compared with any AI and low (approximately 50%) compared with TRA + ANA.
FIGURE 3.
Incremental cost-effectiveness ratios for LAP + LET versus comparators.
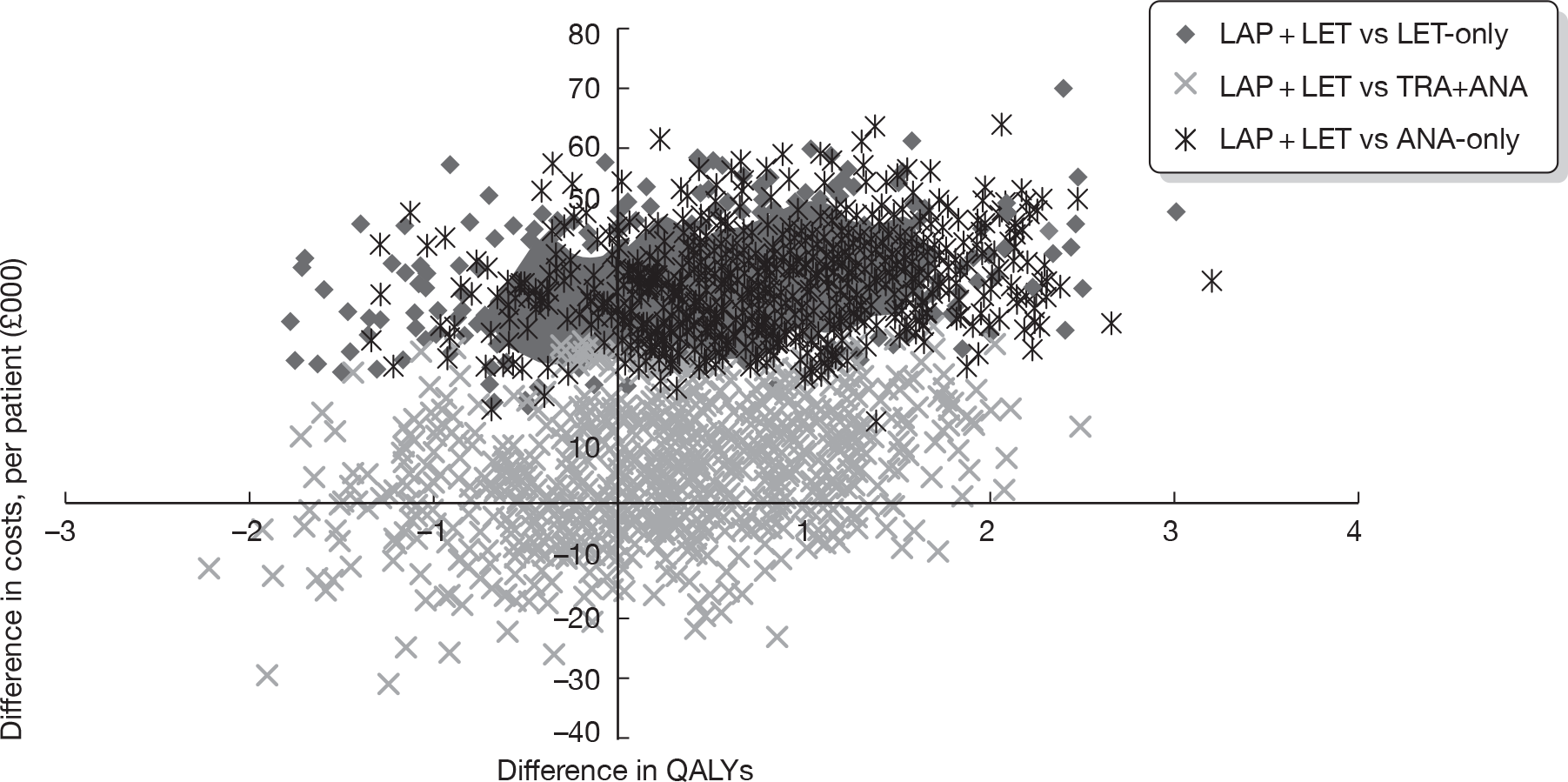
FIGURE 4.
Cost-effectiveness acceptability curve for LAP + LET versus LET, TRA + ANA and ANA.
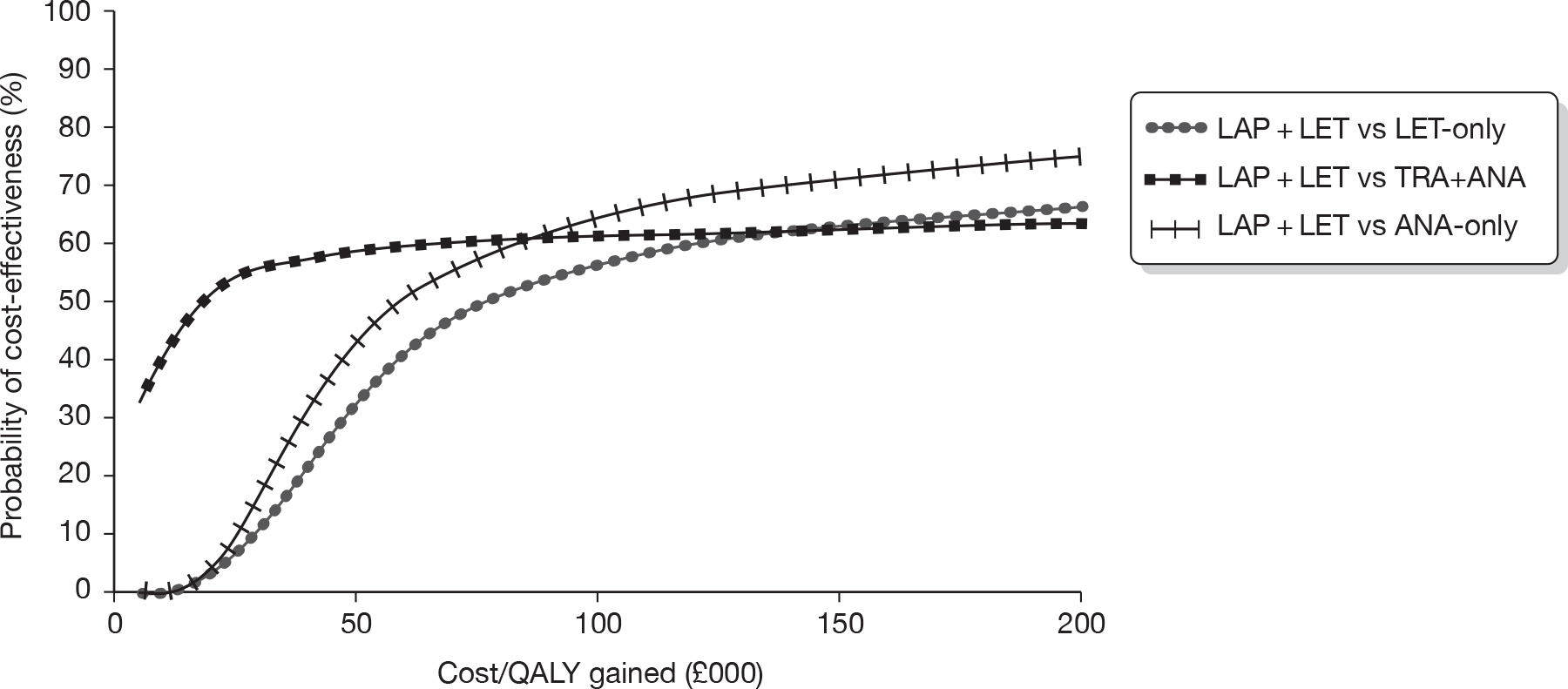
Summary and critique: end-of-life treatment criteria
The AG notes that the manufacturer has not requested that LAP + LET be considered by NICE as an end-of-life treatment.
Conclusions of the Assessment Group
From the information presented in the MS, the AG agrees with the manufacturer that LAP + LET is not cost-effective when compared with LET.
The AG also considers that the methods used in the indirect analysis undertaken by the manufacturer are unreliable and concludes that the ICERs derived from the remaining comparisons (LAP + LET vs TRA + ANA; LAP + LET vs ANA) are not meaningful.
GlaxoSmithKline did not make a case for LAP to be considered as an end-of-life treatment.
Overview and critique of Roche economic evaluation
Overview of submitted economic evaluation and economic model
The purpose of the manufacturer’s model is to assess the cost-effectiveness of first-line treatment with TRA + ANA in HR+/HER2+ patients with mBC. In the MS, the combination of TRA + ANA is compared with the following interventions: ANA monotherapy, LAP + LET and LET monotherapy. An area under the curve (AUC) model was designed to calculate the present value of the health outcomes and NHS/PSS costs attributable to each possible treatment option calculated. The model schema is presented in Figure 5. The model features three health states (PFS, PD and death) and has a cycle length of 1 month. The manufacturer estimates costs from the perspective of the NHS/PSS and health outcomes in terms of life-years gained (LYG) and QALYs. The economic evaluation has a time horizon of 15 years and both costs and benefits are discounted at 3.5% per annum (implemented monthly). The manufacturer’s economic evaluation adequately reflects the NICE reference case97 (Table 21).
FIGURE 5.
The structure of the model submitted by Roche.
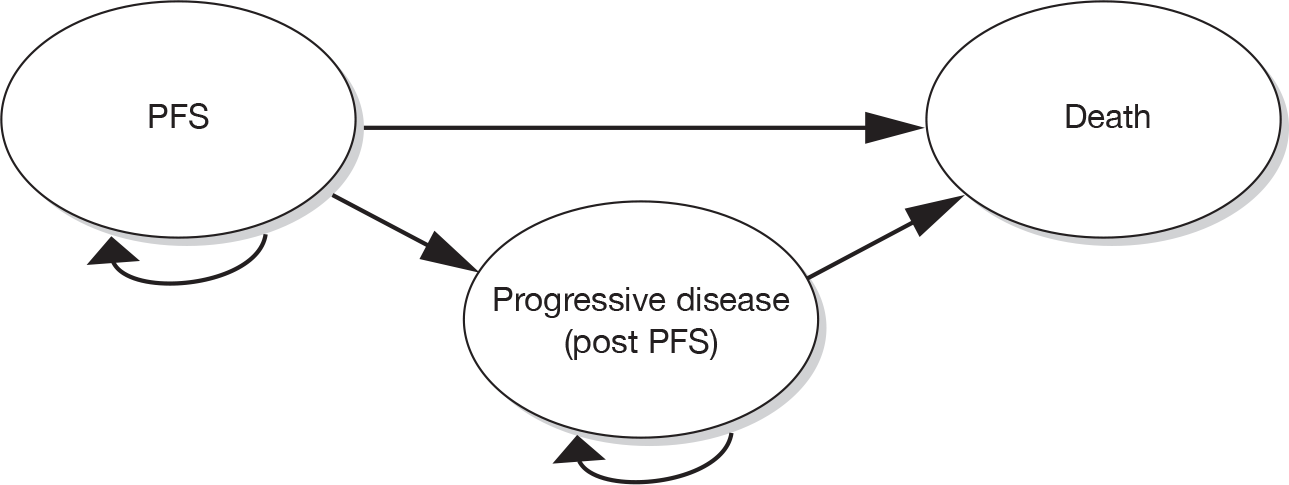
| NICE reference case requirements | Reference case97 | Does the de novo economic evaluation match the reference case? |
|---|---|---|
| Defining the decision problem | The scope developed by the institute | Yes |
| Comparators | Therapies routinely used in the NHS, including technologies currently regarded as best practice | Yes. Best practice: AIs. New intervention also under appraisal: LAP + LET |
| Perspective on costs | NHS and PSS | NHS and PSS |
| Perspective on outcomes | All health effects on individuals | All health effects on individuals |
| Type of economic evaluation | Cost-effectiveness analysis | Cost–utility analysis |
| Synthesis of evidence on outcomes | Based on a systematic review | Based on a systematic review and indirect analysis exercise |
| Measure of health benefits | QALYs | QALYs |
| Source of data for measurement of QoL | Reported directly by patients and/or carers | Health-state descriptions derived from reviews of the literature and lay and professional focus groups |
| Source of preference data for valuation of changes in QoL | Representative sample of general public | Representative sample of general population |
| Discount rate | An annual rate of 3.5% on both costs and QALYs | An annual rate of 3.5% on both costs and QALYs (implemented monthly) |
| Equity weighting | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit |
Summary and critique: clinical effectiveness data
Direct clinical evidence
The key clinical data (PFS and OS) used in the MS are taken directly from the TAnDEM50 trial and utilise some previously unpublished data as the model inputs were taken from an April 2008 data cut (the published paper from the trial uses an older data cut). As the PFS curves from the TAnDEM50 trial were practically complete, the Kaplan–Meier PFS curves for the two regimens were used directly to model the majority of disease progression of patients within the economic model (uncertainty in the Kaplan–Meier PFS curves was addressed in the sensitivity analysis). In general, the AG’s preferred approach to projective modelling would be to assess PFS and PPS separately and then combine them in order to get a more reliable estimate of OS, rather than simply modelling OS as a single entity.
In the TAnDEM50 trial estimates of OS were affected by (1) high rates of crossover of patients from ANA to TRA + ANA and (2) second-line chemotherapy imbalance by trial group. Both of these factors have been considered by the AG in Chapter 3, Quantity and quality of research available. The AG is aware that the OS estimates for TRA + ANA used in the base case are the adjusted values; use of the RPSFT approach reduced the OS HR of TRA + ANA versus ANA. To date, the manufacturer has been unable to account for the second-line therapy imbalance using quantitative methods. As the Kaplan–Meier OS curves were not complete, parametric fitting of the curves was carried out to allow extrapolation beyond the follow-up period. The manufacturer concluded that the exponential distribution most accurately portrayed the OS curves of the two regimens for the time period beyond the availability of the Kaplan–Meier OS curves.
The AG considers that the manufacturer’s approach to adjusting for cross-over in the TAnDEM50 is limited and requires further exploration/justification before confidence can be placed in the OS results generated.
Indirect clinical evidence
As the manufacturer’s systematic review found no network capable of linking all of the regimens in the population of interest, the manufacturer assumed that LET and ANA hold an ‘AI class effect’ in terms of PFS and OS and that the PFS and OS curves observed for ANA patients in the TAnDEM50 trial would therefore hold for LET patients. In order to integrate the combination therapies into the evidence network, the manufacturer also assumed that HER2 status is independent of the relative treatment effect of the AI therapies. The AG’s critique of the manufacturer’s evidence approach to indirect analysis is discussed fully in Chapter 3, Quantity and quality of research available.
In summary, the AG is not confident that the results of the indirect comparisons analyses conducted by the manufacturer are reliable. Firstly, the EGF3000849 trial is included in the network and this trial explicitly excludes patients who have extensive symptomatic visceral or rapidly progressing or life-threatening disease, which means that the patient populations in the EGF3000849 and the TAnDEM50 trials may not be comparable. Secondly, the AG agrees with the manufacturer that ‘given … mixing of heterogeneous populations, the results produced by the [indirect] analysis should be treated with caution’ (MS, p. 216). Specifically, the AG is of the opinion that inclusion of evidence derived from HER2– patients is inappropriate given that the decision problem is focused on treating women who are HER2+ and it is becoming apparent49,52 that an AI as monotherapy is less effective in women who are HER2+ than in women who are HER2–.
In summary, the AG considers that the submitted model results for the comparison of TRA + ANA versus ANA is the only comparison that is wholly valid to inform decision-making in this area.
Summary and critique: resource use and costs
The manufacturer presents a detailed and comprehensive description of resource use and costs used in the economic model. The cost categories are presented as follows: monthly drug costs, treatment duration, administration and monitoring costs, pharmacy preparation, response assessment, cardiac monitoring, PFS best supportive care, AEs, progressed disease costs and end-of-life costs. The key cost parameters used in the economic model are summarised in Table 22. In terms of costs, the key difference between the treatment regimens is the acquisition costs of the drugs (LAP and TRA are much more expensive than the AIs), followed by administration costs (i.v. TRA is much more expensive to administer than the other oral drugs) and finally, the costs of ‘PD, BSC and second-line treatment’ (post-PFS costs are higher for the patients taking LAP and TRA).
| Item | Cost (£) | Source |
|---|---|---|
| LAPa (250 mg) | 11.49 per tablet purchased; 12.32 per tablet taken; 2249.53/month | BNF 5928 |
| LETa (2.5 mg) | 2.38 per tablet purchased; 2.55 per tablet taken; 77.50/month | BNF 5928 |
| TRAb (150-mg vial) | 1956.33/month (vial sharing used in model); 2230.16/month (full wastage) | BNF 5928 |
| ANAa (1 mg) | 2.45 per tablet purchased; 2.62 per tablet taken; 79.90/month | BNF 5928 |
| ECHO/MUGA monitoring costs | 46.31/month | Ward et al.;107 NHS reference costs 2008–200999 (70% : 30%, testing every 4 months) |
| Total subsequent monthly cost (administration, cardiac monitoring, pharmacy preparation) |
92.99 (ANA/LET) 273.58 (LAP + LET) 297.87 (TRA + ANA) |
NHS reference costs 2008–2009;99 PSSRU108 and Clinician Advisory Board |
| Progressive disease costs (second-line treatment with EXE monotherapy) | 92.88 | Roche Advisory Board; BNF 5928 |
| PFS BSC costs | 192.83/month | NICE CG No. 81;23 PSSRU 2009108 |
| Post-progression BSC costs | 542/month | NICE CG No. 81;23 PSSRU 2009108 |
| End-of-life costs | 3418/last 14 days of life | NICE CG No. 8123 |
| Examples of AE costs | ||
| Back pain | 194 | NHS reference costs 2008–200999 (PS05A) |
| Cardiac failure | 370 | NHS reference costs 2008–200999 (EB05Z) |
| Chest pain | 400 | NHS reference costs 2008–200999 (PA22Z) |
| Hypertension | 560 | NHS reference costs 2008–200999 (EB041) |
| Vomiting | 553 | NHS reference costs 2008–200999 (PA28B) |
| Utility values | ||
| Cooper et al.109/NICE23 (base case) | PFS = 0.73; PD = 0.45; SD = 0.65 | |
| Hastings et al.96 (sensitivity analysis) | PFS = 0.86; PD = 0.62 | |
The AG notes that the economic evaluation in the MS uses a 3-weekly schedule for TRA; although this is not the weekly schedule used in the TAnDEM trial,50 the AG agrees that the 3-weekly schedule is typically used in clinical practice in England and Wales.
Table 23 summarises the key costs that the AG believes the manufacturer could have considered in more detail in the MS.
| Assumption made | Limitation |
|---|---|
| Wastage (tablets) | Roche uses an average pill count per month to compare with a notional number of whole packs – this is not correct and underestimates wastage |
| Subsequent administration of TRA | Roche uses estimate from old interim local source instead of the correct NHS reference cost |
| Roche estimates drug costs per tablet purchased/taken which leads to inaccuracies | Only pack prices are available from the BNF 5928 and drug usage would be more accurately costed accordingly |
| Roche assumes that delivery of TRA is always a day-case procedure and uses day-case costs | Depending on patient condition and local circumstance, delivery could be on an outpatient basis. A weighted average of outpatient and day-case costs would be more meaningful |
| Cardiac monitoring costs used in the economic model | In the base case, Roche assumes that cardiac monitoring occurs every 4 months and that MUGA and ECHO scans are used in unequal proportions (30% : 70%). AG clinical advisors have stated that cardiac monitoring occurs every 3 months |
| MUGA cost = £316.64 based on uplifted cost from Ward et al.107 | NHS reference costs 2008–2009;99 aRA37Z OP = £203.05 is more appropriate |
| AEs are limited and poorly described and sourced AE costs described by Roche appear to be underestimates |
Examples: Back pain uses ‘paramedic attendance’ cost and ignores inpatient/outpatient/day-case episode costs In MS, hypercalcaemia cost is given hypertension cost (model); hypercalcaemia cost is not estimated in model |
Summary and critique: utilities and adverse events
The TAnDEM50 conducted by Roche did not collect data using a generic health-utility instrument. In order to estimate utility values for HR+/HER2+ patients, the manufacturer underook a focused review of the literature, identified 20 relevant studies (1996–2009) and presented utility values from six of these published studies (MS, p. 247). However, as no studies were identified as being relevant specifically to HR+/HER2+ patients, the manufacturer made a decison to use the assumptions of Winstanley and Murray in the recent breast cancer publication23 and to apply utilities as identified by Cooper et al. ;109 this decision was made to ensure alignment with the most recent relevant piece of research23 commissioned by NICE in breast cancer. In the sensitivity analysis, the manuacturer made use of the utility values cited by Hastings et al. 96 in the indirect comparison of LAP + LET versus TRA + ANA. The values used by Hastings et al. 96 were derived via mapping FACT-G101 data collected in the EGF3000849 trial to European Quality of Life-5 Dimensions (EQ-5D). Both sets of values are shown in Table 22. Disutility from AEs is not a feature of the economic model developed by Roche.
The Cooper et al. 109 paper pools utilities from many different sources (all derived from oncology nurses using the standard gamble technique). In contrast, the AG notes that the paper by Lloyd et al. ,104 identified by the manufacturer, asks 100 members of the general public to rank health states using the standard gamble technique to determine utility values. As the study by Lloyd et al. 104 is a large preference study designed to obtain UK-based societal preferences for distinct stages of mBC, the AG considers the paper by Lloyd et al. ,104 with caveats previously mentioned, to be the most useful evidence available that could help to inform the decision problem.
In the Roche model, only grade 3 or grade 4 AEs are considered. In the MS it is assumed that the AEs recorded for TRA + ANA are the same for LAP + LET and that the AEs recorded for ANA can be applied to LET. For comparison of TRA and LAP this seems unlikely as episodes of diarrhoea are reported more often for LAP patients. The AG also notes that the costs of several of the AEs listed in the MS (e.g. anaemia, cardiac failure, hypercalcaemia and hypertension) are not the cost inputs used in the economic model (‘AE cost data’). In summary, the AG is of the opinion that the AE costs used in the economic model are underestimated and require revision to make them reliable.
Results: summary and critique
The manufacturer presents detailed summaries of estimated costs and outcomes (time in PFS, time in PD, life-years, QALYs in PFS, QALYs in PD and total QALYs) for the following regimens: TRA + ANA, LAP + LET, ANA monotherapy and LET monotherapy (Table 24). As there are four regimens of interest, the manufacturer has chosen to represent the results of the economic evaluation in terms of the efficiency frontier. The efficiency frontier links the regimens that are not dominated. Figure 6 shows that, using this approach, LAP + LET does not lie on the efficiency frontier and that the key comparison is between TRA + ANA versus LET. When TRA + ANA is compared with LET, the ICER is estimated at approximately £54,336 per QALY gained. When TRA + ANA is compared with LAP + LET, it appears to be cost-effective.
| Total costs and QALYs | TRA + ANA | LAP + LET | ANA | LET |
|---|---|---|---|---|
| Total costs (£) | 54,748.92 | 51,882.53 | 23,340.88 | 23,327.52 |
| Total QALYs | 1.87 | 1.71 | 1.29 | 1.29 |
| ICER | ||||
| TRA + ANA vs LAP + LET | £17,914/QALY gained | |||
| TRA + ANA vs ANA | £54,151/QALY gained | |||
| TRA + ANA vs LET | £54,174/QALY gained | |||
FIGURE 6.
Cost-effectiveness plane.
Note: although ANA is, technically speaking, dominated by LET, it is only marginally more expensive, and in this diagram the separation between ANA and LET is indistinguishable.
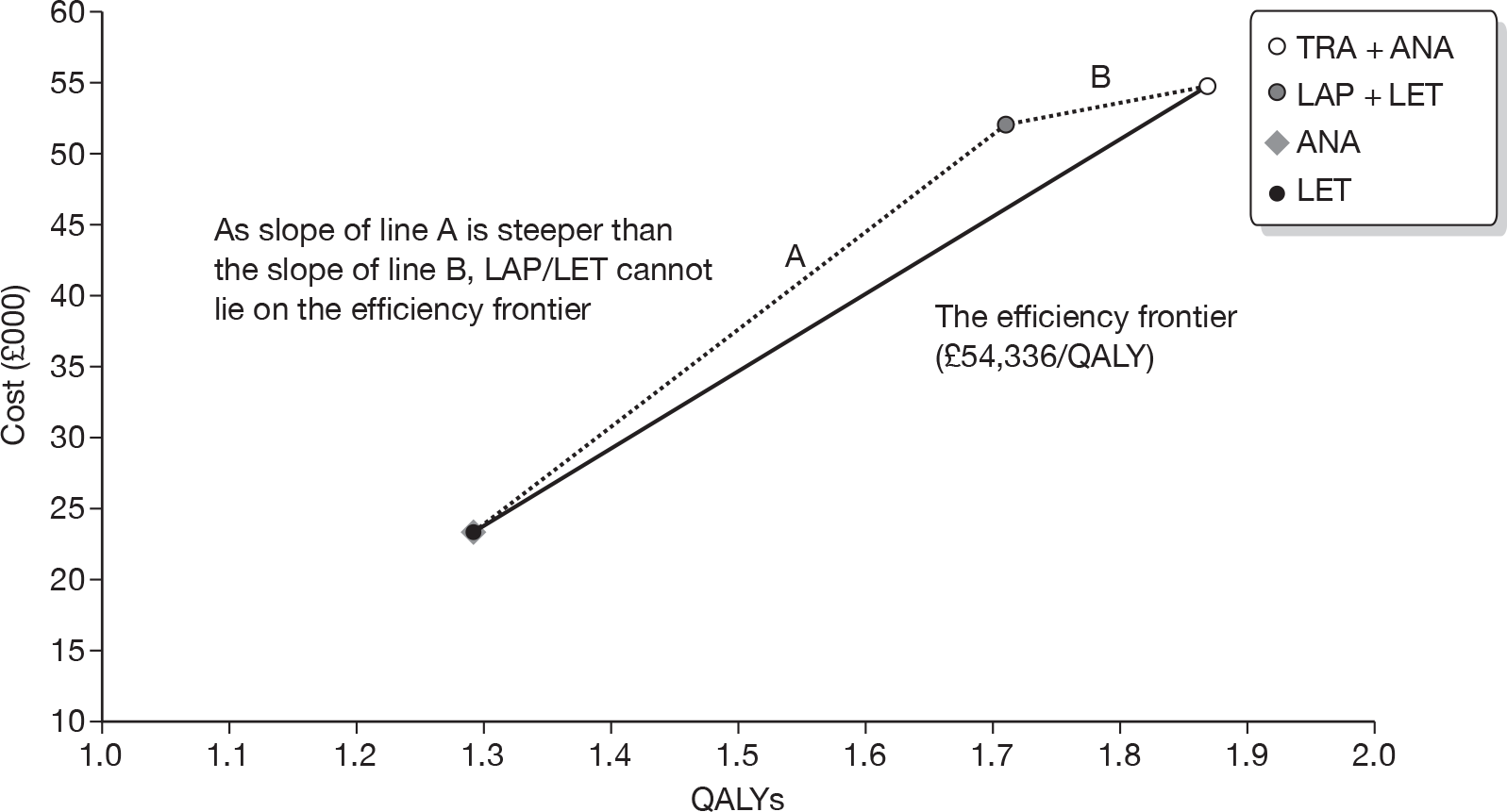
Summary and critique: sensitivity analysis and probabilistic sensitivity analysis
Sensitivity analysis
Twenty-three different parameters were modified in the univariate sensitivity analysis. The results are presented only for the comparison of TRA + ANA versus LET. The base-case ICER was most sensitive to variation in PFS utility values (£50,099–£59,355) and the rate used to discount health outcomes (£48,664–£58,400).
The manufacturer also described three multivariate/scenario analyses. The implementation of the HRS derived from the indirect comparisons analysis for PFS and OS into the model (using the ANA curve from the TAnDEM50 as a baseline; the Roche analysis shows LET is slightly preferred to ANA) leads to a change in the efficiency frontier and the results are as follows: ANA represents a cost-effective option up to a threshold of £3594; LET is the most cost-effective option from £3594 to £57,773; > £57,773 TRA + ANA represents the most cost-effective treatment.
When pessimistic (PFS = 0.65; PD = 0.35) and optimistic (PFS = 0.8; PD = 0.55) utility values are used in the multivariate analysis, the ICER ranges from £48,715 to £62,239. The manufacturer estimates that the base-case ICER would fall to £44,497 if the utility values used in the Hastings et al. 96 paper are employed (these values do not fall within the ± 10% of PFS and PD values used in the univariate sensitivity analysis).
Finally, the manufacturer attempts to account for the confounding influence of the imbalance in second-line chemotherapy in the TAnDEM50 trial and demonstrates that the base-case ICER could fall to around £49,426.
Probabilistic sensitivity analysis
The PSA carried out by the manufacturer is summarised in the MS (p. 278). The CEAC (MS, p. 279) shows that at a threshold of £30,000 per QALY gained, the combination therapies (LAP + LET or TRA + ANA) are never cost-effective. At a threshold of £55,000 per QALY gained, TRA + ANA was shown to be cost-effective in approximately 35% of simulations (i.e. had a low probability of cost-effectiveness).
End-of-life treatment criteria: summary and critique
This section provides an overview and critique of the manufacturer’s case for TRA + ANA as an end-of-life maintenance treatment for patients mBC. The NICE end-of-life treatment criteria have three key points:
-
the treatment is indicated for patients with a short life expectancy, normally < 24 months, and
-
there is sufficient evidence to indicate that the treatment offers an extension to life, normally of at least an additional 3 months, compared with current NHS treatment, and
-
the treatment is licensed or otherwise indicated for small patient populations.
Patient life expectancy of less than 24 months
The published literature110 on prognosis after a diagnosis of mBC, confirms that the disease is incurable and patient life expectancy is short. In a previous scope111 issued by NICE (LAP for the treatment of previously treated women with advanced, metastatic or recurrent breast cancer), it was stated that ‘…The average life expectancy after diagnosis of mBC is 18–24 months. This is reduced by up to 50% for patients with tumours over-expressing HER2’.
The manufacturer cites data from the comparator (ANA) group of the TAnDEM50 trial to support the argument that patients with mBC who are HR+/HER2+ have a very poor prognosis. Median OS is shown to range between 17.2 months (excluding all patients who crossed over) and be ≤ 32.1 months (excluding all patients with liver metastases); other OS estimates are also generated depending on the methodology used to undertake survival analysis (Table 25). The AG notes that although data from the EGF3000849 trial show that patients with mBC who are HR+/HER2+ have a median OS of 33.3 months, this trial explicitly excludes patients who have extensive symptomatic visceral or rapidly progressing or life-threatening disease. The AG acknowledges that use of second-line therapies may also influence estimates of OS.
Life extension of at least 3 months
The manufacturer attempts to demonstrate that there is sufficient evidence from the TAnDEM50 trial to indicate that TRA + ANA offers an extension to life of at least an additional 3 months, compared with current NHS treatment. In the TAnDEM trial,50 unadjusted median OS gained and RPFST-adjusted median OS gained were estimated to be 4.6 months and 6.54 months, respectively.
The AG is of the opinion that TRA + ANA compared with ANA yields a life extension of at least 3 months for patients who are HR+/HER2+ and who have had no prior treatment for mBC.
Licensed for a small patient population
The size of the patient population eligible for treatment with TRA + AI in England and Wales, i.e. women with HR+/HER2+ mBC, is estimated to be around 50 patients by both GlaxoSmithKline and Roche. However, TRA has indications in mBC, metastatic gastric cancer and early breast cancer. The manufacturer reports that, in England and Wales, across all the indications, 7158 patients are eligible to receive TRA each year (2333 from mBC, 506 from metastatic gastric cancer and 4319 from early breast cancer). It was difficult for the AG to verify these population figures as the references cited were not included in the references package as part of the MS; the data were from pharmaceutical company reports that the AG could not easily access.
Conclusions of the Assessment Group
From the information presented in the MS, the AG agrees with the manufacturer that TRA + ANA is not cost-effective when compared with ANA. The AG also considers that the methods used in the indirect analysis undertaken by the manufacturer are unreliable and concludes that the ICERs derived from the remaining comparisons (TRA + ANA vs LET; TRA + ANA vs LAP + LET) are not meaningful.
The manufacturer submitted a case for TRA + ANA to be considered as an end-of-life treatment for women with HR+/HER2+ mBC. The AG agrees that TRA + ANA meets the criteria as a treatment for patients with a short life expectancy and that it extends life by an additional 3 months when compared with current NHS treatment. However, the AG makes no comment on whether or not the criterion of a small patient population is met.
Independent economic assessment
Each of the novel treatment regimens considered in this AG report relies on clinical evidence derived from a single small RCT (the EGF3000849 or the TAnDEM50 trial). Moreover, the comparator treatments differ between these trials, albeit both drugs were drawn from the same class of compounds. These disparities alone suggest the need for caution when generalising these results of the RCTs. However, an even greater difficulty arises if the two study populations do not appear to match.
As discussed in earlier sections, there is reason to believe that, in some important respects, the protocol criteria governing the selection of subjects for these two trials were sufficiently dissimilar as to be likely to generate non-equivalent patient populations. In particular, the requirement in the EGF3000849 trial to exclude patients with extensive symptomatic visceral disease including rapidly progressing or life-threatening disease is not explicitly matched by a similar exclusion in the TAnDEM50 protocol.
As a result, it is reasonable to expect that patients in the EGF3000849 trial may have been somewhat fitter and with better prognoses than those recruited into the TAnDEM trial. 50 However, direct comparison of patient characteristics in the trials is restricted by differences in how measures were defined and/or reported in the two CSRs. Tables setting out the number and location of metastatic lesions at baseline in the two trials are available. However, they cannot be compared with full confidence as they are defined somewhat differently. There is strong evidence of a significant difference in the mean age of the populations. There is also evidence of a greater incidence of soft-tissue metastases in the TAnDEM50 trial patients (43.5% vs 30.14%, p = 0.004), although metastases at other sites are broadly comparable. Overall, the frequency of metastatic sites affected per patient (1.77 in the EGF3000849 vs 2.40 in the TAnDEM50 trial) also suggests more severe advanced disease in the TAnDEM50 patients, although this could be an artefact of differing reporting methods.
Coupled with the serious problems identified in earlier sections relating to the indirect comparison of treatment effects, these uncertainties led the AG to conclude that although two separate assessments of cost-effectiveness, each based on one of the principal RCTs, could be undertaken with some confidence, the evidence base is too insecure to allow a meaningful comparison of the two innovative compounds against each other. In this section, two separate cost-effectiveness analyses are reported, using a common modelling framework and common parameter values, but employing effectiveness data drawn from only a single RCT (either the TAnDEM50 or the EGF3000849 trial).
Methods: common model features and parameters
Model design
A common model structure has been adopted for both de novo cost-effectiveness analyses (Figure 7) and, wherever possible, has been implemented using the same parameter values. The de novo model employs outcome data derived from the relevant clinical trial in the form of Kaplan–Meier estimated survival values augmented by projected survival estimates calibrated against the observed data. The preferred approach uses PFS and PPS estimates directly as the basis for calculating expected OS in each group of the RCT. PFS and PPS values then furnish the information required to calculate all components of health service costs and also to estimate the expected future patient utility. These survival estimates are calculated separately for each date on which a resource is expected to be used (e.g. when prescriptions are dispensed or when a hospital visit or test takes place), avoiding the need for a general model cycle or for mid-cycle corrections.
FIGURE 7.
A schematic of the AG’s model structure for each treatment option.
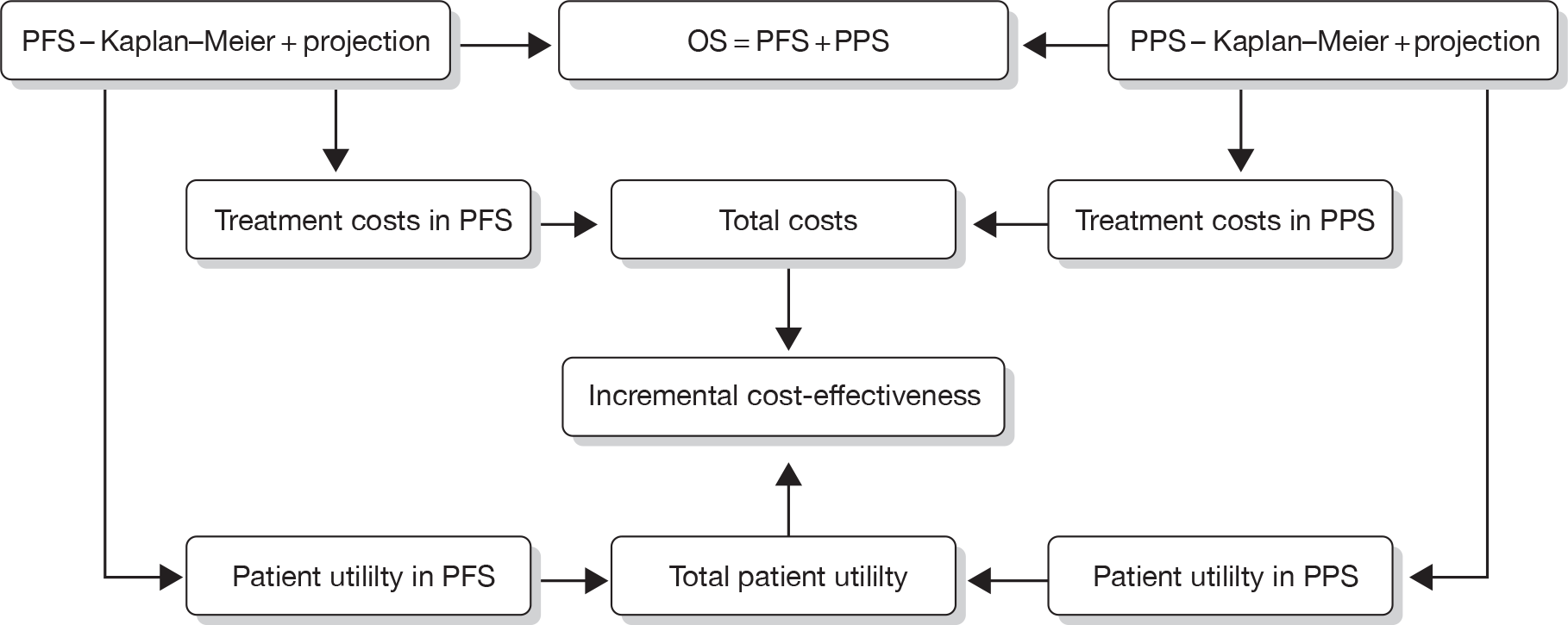
Both of the manufacturers’ models use PFS and OS as the primary sources for survival information, and derive time in PPS as the difference between OS and PFS. The AG finds this approach generally liable to generate substantial bias in OS estimates when projective parametric modelling is used. This is because recorded OS data are a result of combining patient experience in two distinct phases in which hazard rates would be expected to exhibit quite different dynamics (in PFS the patient is likely to have reduced event risks although the active drug continues to be effective, but in PPS event risks are more likely to revert to higher levels of uncontrolled disease progression). In most cases, standard parametric statistical models cannot accurately represent an outcome measure (such as OS) which is a compound of two very different processes, and modelled OS projected over several decades can result in very large cumulative errors. By contrast, in advanced disease the risk profile of patients entering PPS is usually quite stable and allows projective modelling with greater confidence [i.e. narrow confidence intervals (CIs)] and limits the risk of some of the more extreme estimates of long-term survival which can occur when modelling OS directly. At a pragmatic level, deriving PPS as the difference between OS and PFS can sometimes lead to modelling anomalies with negative estimates of PPS during projection, an error which cannot occur when PFS and PPS estimates are summed.
Undiscounted and discounted (3.5% per annum for costs and outcomes) deterministic results were generated for each year of remaining life up to 30 years. A PSA was carried out for all model variables for which sampling uncertainty could be estimated, and a range of univariate sensitivity analyses were performed for other variables and assumptions.
Cost parameter values
Model costing variables common to both models are listed in Table 26 with the parameter values used in the base-case analyses and the data sources employed.
| Cost item | Value | Source |
|---|---|---|
| Pharmacy dispensing costs (£) |
9.00 from hospital pharmacy 6.90 from community pharmacy |
Roche MS, based on hourly cost of pharmacist time108 and 12 minutes per script dispensed |
| Frequency of cardiac monitoring | Every 3 months | Clinical advisor opinion |
| ECHO : MUGA proportion of cardiac scans | 70 : 30 | Clinical advisor opinion |
| Unit cost per ECHO scan (£) | 74.37 | NHS reference costs 2008–2009;99 direct access diagnostics code DA02 |
| Unit cost per MUGA scan (£) | 203.05 | NHS reference costs 2008–2009;99 OP nuclear medicine code RA37Z |
| Frequency of OP follow-up (including CT scan) in PFS | Every 3 months | Clinical advisor opinion |
| Unit cost per OP follow-up visit (£) | 98.51 | NHS reference costs 2008–2009;99 consultant-led follow-up attendance, non-admitted, face to face, code 800 (clinical oncology) |
| Unit cost per CT scan (£) | 138.27 | NHS reference costs 2008–2009;99 code RA12Z – OP CT scan (two areas with contrast) |
| Annual cost of BSC in PFS (£) | 1831.54 | NICE guideline,23 updated for inflation108 |
| Annual cost of BSC in PPS (£) | 5597.82 | NICE guideline,23 updated for inflation108 |
| Terminal care costs (last 2 weeks of life) (£) | 1788.55 | Remak and Brazil,100 updated for inflation108 |
| Unit cost of EXE (per 30-tablet pack) (£) | 88.80 | BNF 5928 |
| Wastage per patient (half pack) (£) | 44.40 | BNF 5928 |
| Proportion of PPS patients receiving EXE (%) | 50 | Modelling assumption |
| Discounting rate (costs) | 3.5% per annum | NICE methods guide97 |
| Discounting rate (outcomes) | 3.5% per annum | NICE methods guide97 |
Patient utility valuation
The AG considered a number of sources for utility values for breast cancer patients referenced by the manufacturers and conducted an exploratory (but not systematic) search which failed to identify any useful additional material. Overall, there appears to be a particular dearth of relevant utility studies appropriate to this appraisal and conforming to the NICE recommended97 approach (UK data capturing population preferences using EQ-5D or either time trade-off or standard gamble methodology). Several standard gamble studies have been reported, but normally use a very small sample of health professionals to assess quite general health states, not particularly focused on advanced disease. The AG concluded that the best available option was the study reported by Lloyd et al. 104 published in 2006, which considered health states and a limited set of treatment-related AEs specific to mBC, and developed a mixed model using data collected from a sample of 100 UK residents broadly similar in age and gender to the general population. The values presented have face validity for both absolute values and interstate differences. The particular benefit is that they furnish an integrated system of utility estimates (rather than adopting values from disparate sources as is often the case).
The Lloyd et al. 104 model includes age and treatment response as model variables. However, it is important to note that the relevant age is not that of the patient, but of the study participant. To ensure consistency with the UK EQ-5D standard value scheme, the AG adopted the average age of respondents to the original multivessel disease study112 of just over 47 years in arriving at utility parameter values. For patients who are pre-progression it was necessary to calculate a weighted average of the model values for stable disease and treatment response, based on the reported response rate (CR + PR) in each group of each trial (see Specific model features and parameters: lapatinib in combination with letrozole versus letrozole alone and Specific model features and parameters: trastuzumab in combination with anastrozole versus anastrozole alone). A common health-state utility value was obtained for post-progression patients of 0.496 [standard error (SE) 0.160] for use in both models.
Specific model features and parameters: lapatinib in combination with letrozole versus letrozole alone
The manufacturer of LAP provided full details of survival analyses (PFS, PPS and OS) requested by the AG relating to data from the EGF30008 trial. 49 The AG employed these data to estimate the components of mean survival that could be expected over the lifetime of a patient, the results of which are presented below. Note that these may differ in detail from those generated by the AG model because of the approximations required to implement the continuous mathematical survival functions in a structure designed around discrete cycle periods.
Expected progression-free survival
Examination of the Kaplan–Meier estimates of PFS over time (data provided in confidence) shows an early advantage for the combination therapy (LAP + LET) compared with LET alone, but also indicates that this benefit steadily eroded over time until the two treatments were indistinguishable beyond about 16 months. The AG decided that the most reliable estimate of the mean expected PFS would be obtained by using the difference between the Kaplan–Meier AUC estimates up to the time of convergence (505 days). Thereafter, a single exponential model of PFS was applied to both the intervention and the comparator in the AG model, calibrated on pooled Kaplan–Meier data for the period > 500 days.
This approach yielded estimates of PFS up to 505 days:
-
198.5 (SE 17.6) PFS days for LET only
-
266.2 (SE 16.1) PFS days for LAP + LET,
i.e. a gain of 67.6 (SE 16.9) PFS days attributable to the use of LAP.
For the period beyond 505 days, a further 67.8 (SE 5.0) days PFS must be applied to all patients irrespective of treatment to obtain the overall mean PFS estimates per patient:
-
266.4 (SE 18.3) PFS days for LET only
-
334.0 (SE 16.9) PFS days for LAP + LET.
Expected post-progression survival
Examination of the Kaplan–Meier analysis of PPS in the EGF3000849 trial indicated that following disease progression, patients in both groups of the trial were at the same risk of death, which appears to be constant over time. Therefore, a single exponential model was calibrated from the pooled trial data for use in the AG model, yielding an estimated mean survival for patients in PPS of 764.8 (SE 5.0) days. This is applied only to patients whose progression event is non-fatal.
Expected overall survival
As no cross-over following disease progression was permitted in the EGF30008 trial,49 and there is no evidence of significant imbalance in post-progression therapies, no further adjustments to PFS and PPS estimates are necessary.
The best estimate of OS is obtained by summing PFS and PPS, after adjusting PPS to exclude patients dying at or before disease progression:
-
1009.6 (SE 18.9) OS days for LET only
-
1050.6 (SE 17.6) OS days for LAP + LET,
i.e. a gain of 41.0 (SE 25.8) OS days attributable to the use of LAP.
Patient utility values
Health-state utility values for patients in PFS, obtained from the Lloyd et al. 104 model, differ slightly between the EGF3000849 groups because of differential treatment response rates:
-
0.7749 (SE 0.1125) in the LET group
-
0.7794 (SE 0.1118) in the LAP + LET group.
Adverse events
Examining the incidence of the six grades 3/4 AEs featured in the Lloyd et al. 104 model (fatigue, diarrhoea/vomiting, stomatitis, febrile neutropenia, hand–foot syndrome and alopecia), showed generally low incidence in all categories with the exception of diarrhoea/vomiting, which was six times more common in the combination group than the LET group. However, the absolute difference in estimated utility per study patient is very small (< 0.01) with a wide CI, so the AG decided to examine the influence of the disutility of this AE through sensitivity analysis rather than through setting a value in the base case. A similar approach was taken to the differential cost per patient of AEs.
Cost of lapatinib: acquisition
The acquisition cost of LAP is £804.30 per pack of 70 tablets. The standard dose requires patients to take six tablets per day. In the AG model it is assumed that LAP is prescribed to non-progressed patients every 28 days, with sufficient packs to complete treatment for the next 4 weeks (taking account of any unused tablets from previous prescriptions). This requires two or three packs to be prescribed at each visit. Wastage is automatically included in this calculation as the dispensed tablets are unused at the time of progression (14 days’ supply on average), and no mid-cycle correction is necessary. It is assumed that prescriptions will be dispensed by a hospital pharmacist.
Cost of letrozole: acquisition
The acquisition cost of LET is £66.50 per pack of 28 tablets. It is assumed that one pack is dispensed every 28 days to all patients remaining in PFS on that day. This implies that wastage is limited to an average 14 days of treatment per patient. It is assumed that prescriptions will be dispensed by a community pharmacist, except for the first prescription, which is provided in the hospital.
Actual and expected delivery of treatment
Information provided by the manufacturer of LAP indicates that adjustments made to the dose intensity of treatments in the EGF3000849 trial were based on similar pill counts to those discussed in the next section in relation to ANA. The AG has no reason to consider these data are any more reliable than those for ANA, which were assessed at the individual patient level. The AG, therefore, decided not to make adjustments to calculations based on 100% compliance with the treatment protocol. This ensures a consistent approach in both appraisals.
Specific model features and parameters: trastuzumab in combination with anastrozole versus anastrozole alone
Expected progression-free survival
The manufacturer of TRA provided full details of the survival analyses (PFS, PPS and OS) requested by the AG relating to data from the TAnDEM trial. 50 The AG employed these data to estimate the components of mean survival that could be expected over the lifetime of a patient, the results of which are presented below. Note that these may differ in detail from those generated by the AG model because of the approximations required to implement the continuous mathematical survival functions in a structure designed around discrete cycle periods.
Examination of the cumulative hazard plots for PFS suggested an early relatively high risk of progression, followed by a more regular slowly reducing hazard trend in both trial groups. It was found that a two-parameter Weibull model offered an acceptable representation of the long-term trend in both groups (using data provided in confidence). The AG decided that the most reliable estimate of the mean expected PFS would be obtained by using the Kaplan–Meier AUC estimate up to the last recorded event in each group, and then adding the area under the projected long-term Weibull model curve at later times.
This approach yielded estimates of:
-
189.6 (SE 21.4) PFS days for ANA only
-
514.8 (SE 64.1) PFS days for TRA + ANA,
i.e. a gain of 325.1 (SE 67.6) PFS days attributable to the use of TRA.
Expected post-progression survival
Examination of the cumulative hazard plots for PPS indicated that disease progression was not associated with any variation in risk away from a continuous long-term trend. However, analysis confirmed that simple exponential models (i.e. linear trends in cumulative hazard) were not adequate to describe the observed PPS data. Two-parameter Weibull models were fitted to data from both trial groups and offered an acceptable representation. The AG decided that the most reliable estimate of the mean expected PFS would be obtained by using the Kaplan–Meier AUC estimate up to the last recorded event in each group, and then adding the area under the projected long-term Weibull model curve at later times.
Using this approach generated estimates of:
-
869.6 (SE 46.3) PPS days for ANA only
-
649.6 (SE 63.1) PPS days for TRA + ANA,
i.e. a loss of 220.0 (SE 78.3) PPS days attributable to the use of TRA.
Expected overall survival
By combining estimates of mean PFS and mean PPS in each group and adjusting for the minority of patients who die at or before progression (5.77% in the ANA group and 5.83% in the TRA + ANA group), combined estimates for OS were obtained:
-
1009.0 (SE 50.5) OS days for ANA only
-
1126.5 (SE 85.6) OS days for TRA + ANA,
i.e. a gain of 117.5 (SE 100.9) OS days attributable to the use of TRA.
The manufacturer of TRA has drawn attention to two factors in the TAnDEM50 trial that are considered likely to distort the estimation of PPS in the comparator group:
-
a large number of patients in the comparator (ANA) group chose to ‘cross over’ to TRA + ANA therapy following disease progression and are likely to gain additional benefit in terms of extended PPS
-
a greater proportion of patients in the comparator group received second-line chemotherapy, also potentially extending PPS.
In the MS, the results of applying a statistical technique to attempt to counter the first of these confounding factors were presented, but no attempt was made to make any further adjustment to overcome the second-line chemotherapy imbalance.
As discussed in Chapter 3, Assessment of effectiveness, the suggested statistical adjustment is not universally accepted as the most suitable method to employ and may rely on restrictive assumptions that are not valid for use in the TAnDEM trial. 50 The AG asked for additional survival analyses to be undertaken in order to explore the sensitivity of OS estimates to other approaches to correcting PPS for crossover and imbalance in second-line chemotherapy; how the results of the survival analyses were used by the AG are discussed below.
Crossover: Separate Kaplan–Meier analyses of patients in the comparator group of the TAnDEM trial,50 split by whether or not they did or did not receive crossover TRA following progression, demonstrated a clear advantage for crossover patients. However, the data suggest that, after about 6 months has elapsed, this advantage diminishes, and it disappears altogether after about 3 years. The AG found that these complex trends in PPS could be well described by fitting bi-phase exponential models, from which it is possible to estimate the mean survival gain attributable to crossover for patients in the post-progression phase.
The net benefit of crossover (i.e. the area between the two modelled PPS lines) is estimated as 150.5 days. However, this advantage only accrues to those patients who do not die at or before progression (94% of the total), so that the mean PPS adjustment which may be subtracted in the calculation of OS in the control group is 141.8 (SE 12.8) days.
Second-line chemotherapy: Four Kaplan–Meier analyses of the TAnDEM50 patients, stratified by treatment group and post-progression use of second-line chemotherapy, were compared. No clear distinctions were apparent, though in both trial groups those receiving second-line therapy seemed to have a modest advantage. Generally, the hazard time profiles did not markedly differ from linearity indicating that an exponential parametric model would be appropriate. The AG chose to compare all patients receiving second-line chemotherapy to all patients who did not, recognising that this is necessarily only an exploratory analysis lacking a full standardisation. However, in view of the small numbers of patients in each stratum, more detailed analysis would most likely be unproductive. Exponential survival parameters were estimated suggesting a HR of 0.83 in favour of chemotherapy and a gain in PPS of 145.2 (SE 31.1) days. This figure must be adjusted for three factors:
-
the difference in the use of second-line chemotherapy between the trial groups is 24% (32% – 8%)
-
examination of the trial data indicated that the majority (82%) of chemotherapy patients also benefited from crossover TRA, and for these patients the effect of chemotherapy is already included in the crossover adjustment discussed above
-
the absolute difference in PPS only applies to patients who did not die at or before disease progression.
The net effect of these adjustments is an estimated additional second-line chemotherapy gain in PPS in the comparator group of 5.9 (SE 1.2) days.
When the adjustments for crossover and chemotherapy are included, the following final estimates for OS were obtained:
-
861.4 (SE 52.1) OS days for ANA only
-
1126.5 (SE 87.3) OS days for TRA + ANA,
i.e. a gain of 265.1 (SE 101.7) OS days attributable to the use of TRA.
Patient utility values
Health-state utility values for patients in PFS obtained from the Lloyd et al. 104 model, differ slightly been the TAnDEM50 groups because of differential treatment response rates:
-
0.7639 (SE 0.1139) in the ANA group
-
0.7687 (SE 0.1133) in the TRA + ANA group.
Adverse events
Examining the incidence of the six grades 3/4 AEs featured in the Lloyd et al. 104 model (fatigue, diarrhoea/vomiting, stomatitis, febrile neutropenia, hand–foot syndrome and alopecia) showed very low incidence in all categories and no significant differences on which to base any estimate of differential disutility from AEs for this comparison; the AG decided to examine the potential importance of this issue via sensitivity analysis. A similar approach was taken to the differential cost per patient of AEs.
Cost of trastuzumab: acquisition and administration
The cost of TRA treatment was estimated using the distribution of body weight recorded at baseline in the TAnDEM trial. 50 These data indicated that a log-normal distribution was appropriate and parameters were estimated by the method of moments (i.e. a weighted average of the individual doses and vials of TRA which would be required to treat the population of patients without vial sharing was estimated). This calculation automatically incorporated drug wastage. For the initial loading dose (8 mg/kg), the cost per dose was estimated as £1657.86 and for a regular dose (6 mg/kg) the cost per dose is £1292.88. These costs were applied to all patients remaining in PFS at the beginning of each 3-week period.
The costs of administering TRA are derived from the NHS reference costs 2008–2009,99 using average costs for day cases and outpatient weighted by national activity levels. For the loading dose, this uses Healthcare Resource Group code SB14Z, and for the regular dose, code SB12Z, as specified in clinical coders guidance. 113 The unit cost per treatment is £284.66 (loading dose) and £198.63 (regular doses).
Cost of anastrozole: acquisition
The acquisition cost of ANA is £68.56 per pack of 28 tablets. It is assumed that one pack is dispensed every 28 days to all patients remaining in PFS on that day. This implies that wastage is limited to an average of 14 days of treatment per patient. It is assumed that prescriptions will be dispensed by a community pharmacist, except for the first prescription which is provided in the hospital.
Actual and expected delivery of treatment
In their submitted model, the manufacturer of TRA adjusted the quantity of each treatment by a multiplier to represent the ratio of treatment actually received by patients and that expected during their time in PFS. This seems to be a compound of patient compliance, missed doses, dose adjustments and the lack of precision in the estimation of treatment volumes in their model. For TRA infusions this factor has a minor effect (× 0.987), but the effect is more pronounced for ANA. The AG considered this issue carefully on the basis of the detailed individual treatment records included in the TAnDEM CSR. 50 In the case of TRA infusions, there are almost no occasions when scheduled infusions were not administered on time and at the prescribed dose. A few instances of a missed appointment (e.g. at Christmas holiday) were generally followed by a double dose administered at the next scheduled visit. It appears, therefore, that the trial data do not support the notion that there is any serious systematic discrepancy between planned and administered delivery of TRA. There may be some merit in a minor adjustment in the submitted model to take account of the approximation involved in estimating PFS at monthly intervals, but this problem does not arise in the AG’s model which calculates PFS daily.
For ANA, the estimation of an adjustment factor appears to have been based on estimated compliance data using pill counts undertaken during the trial. The individual patient data track the issue of tablet packs at each patient visit and the number of tablets returned unused at the following visit. These data reveal that the method of calculating compliance is fundamentally unsound, as it takes no account of the occasional failure of patients to return unused tablets during the dosing period and a systematic failure to return unused tablets at the end of treatment. As a consequence, individual compliance figures ranging between 32% and 300% were estimated. Closer examination of individual patients’ drug issues and returns shows a generally exemplary adherence to schedule in all patients.
The AG is satisfied that there is no evidential basis for making any adjustments to the calculated expected use of either treatment on the grounds of deviation from treatment protocol, or to correct for approximations arising from the model structure, as the AG model is designed to avoid such problems.
Results
The results obtained from modelling the costs and outcomes of each of the trial-based appraisals are shown separately in this section. No attempt has been made to make any comparisons between the groups of the two trials, as the populations are not considered to be directly comparable and reliable indirect comparisons of treatment effects could not be undertaken (see Chapter 3, Quantity and quality of research available). Therefore, the only questions that may be addressed legitimately are as follows.
-
Can LAP + LET be considered a cost-effective alternative to LET alone?
-
Can TRA + ANA be considered a cost-effective alternative to ANA alone?
Base-case result: lapatinib in combination with letrozole versus letrozole alone
The base-case cost-effectiveness results based on the AG model are shown in Table 27. A small expected mean health gain per patient (2 months’ life extension and < 0.12 additional QALYs) is generated by an additional cost of > £25,000 per patient, most of which is incurred in the first 5 years. The cost-effectiveness ratio is stable over long time periods and exceeds £225,000 per QALY gained.
| Treatment | Cost per patient (£) | Outcomes per patient | ICER | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Time horizon (years) | Drugs | Monitoring | AEs | BSC | Terminal care | Total costs | Life-years | QALYs | £/QALY gained |
| LET | |||||||||
| 10 | 685 | 702 | 12,485 | 1633 | 15,506 | 2.539 | 1.238 | ||
| 20 | 686 | 702 | 12,620 | 1653 | 15,661 | 2.562 | 1.247 | ||
| 30 | 686 | 702 | 12,621 | 1653 | 15,662 | 2.562 | 1.247 | ||
| LAP + LET | |||||||||
| 10 | 26,051 | 1446 | 98 | 12,435 | 1620 | 41,650 | 2.704 | 1.354 | |
| 20 | 26,052 | 1446 | 98 | 12,574 | 1641 | 41,811 | 2.727 | 1.363 | |
| 30 | 26,052 | 1446 | 98 | 12,575 | 1641 | 41,812 | 2.728 | 1.363 | |
| Incremental | |||||||||
| 10 | 25,366 | 744 | 98 | –51 | –13 | 26,144 | 0.165 | 0.116 | 225,676 |
| 20 | 25,366 | 744 | 98 | –46 | –12 | 26,150 | 0.166 | 0.116 | 225,131 |
| 30 | 25,366 | 744 | 98 | –46 | –12 | 26,150 | 0.166 | 0.116 | 225,127 |
Univariate sensitivity analysis
Results from a sensitivity analysis covering the main model variables are shown in Table 28. The ICER is most sensitive to the health-state utility parameter values and to the cost of LAP, but is insensitive to most of the other variables. In all cases, the ICER remains > £141,000 per QALY, indicating that uncertainty in any single parameter value is unlikely to alter the cost-effectiveness of LAP + LET relative to conventional thresholds.
| Model variable | Variation in value | ICER (£) for | ||
|---|---|---|---|---|
| Low | High | Low | High | |
| Base case | 225,131 | |||
| Discount rate: costs | 0% | 6% | 228,880 | 222,701 |
| Discount rate: outcomes | 0% | 6% | 219,215 | 228,988 |
| Dispensing costs: community | £5 | £10 | 225,087 | 225,210 |
| Dispensing costs: hospital | £7 | £11 | 224,912 | 225,351 |
| Frequency of cardiac monitoring (per annum) | 3 | 6 | 224,331 | 227,037 |
| ECHO as % of scans | 50 | 100 | 226,136 | 223,625 |
| Frequency of PFS follow-up and CT scan (per annum) | 2 | 6 | 223,912 | 226,080 |
| Proportion of progressed patients on EXE | 0% | 100% | 225,666 | 224,596 |
| Net extra cost of AEs in LAP + LET group | £0 | £1000 | 224,284 | 232,761 |
| Net extra disutility of AEs in LAP + LET group | 0 | –0.01 | 211,246 | 229,503 |
| Utility in PFS: LET only | –10% | +10% | 157,265 | 396,037 |
| Utility in PFS: LAP + LET | –10% | +10% | 549,154 | 141,589 |
| Utility in PFS: both groups | –10% | +10% | 267,536 | 194,330 |
| Utility in PPS | –10% | +10% | 219,375 | 231,198 |
| Acquisition cost of LAP | –10% | +10% | 203,605 | 246,658 |
| Cost of cardiac scan | –10% | +10% | 224,710 | 225,553 |
| BSC annual costs | –10% | +10% | 225,118 | 225,145 |
| Terminal care costs | –10% | +10% | 225,142 | 225,121 |
Probabilistic sensitivity analysis
Probabilistic sensitivity was explored, running 1000 random iterations for all variables subject to measurable parameter uncertainty, using the base-case scenario over a 20-year time horizon. The PSA results are compared with the corresponding deterministic results in Table 29.
| Sensitivity analysis | Incremental cost (£) | Incremental QALYs | ICER (£) |
|---|---|---|---|
| Deterministic | 25,150 | 0.116 | 225,131 |
| Probabilistic | 25,034 | 0.109 | 228,913 |
The scatterplot of iteration results in the cost-effectiveness plane as shown in Figure 8, the scatterplot, indicates that all iterations lie substantially outside the region normally considered cost-effective. Figure 9 confirms that the probability of the combination therapy being cost-effective at a willingness-to-pay threshold of £50,000 per QALY gained is 0.1%, and does not reach 50% probability until £231,000 per QALY gained.
FIGURE 8.
Probabilistic sensitivity analysis of LAP + LET versus LET only: scatterplot of 1000 probabilistic iterations.
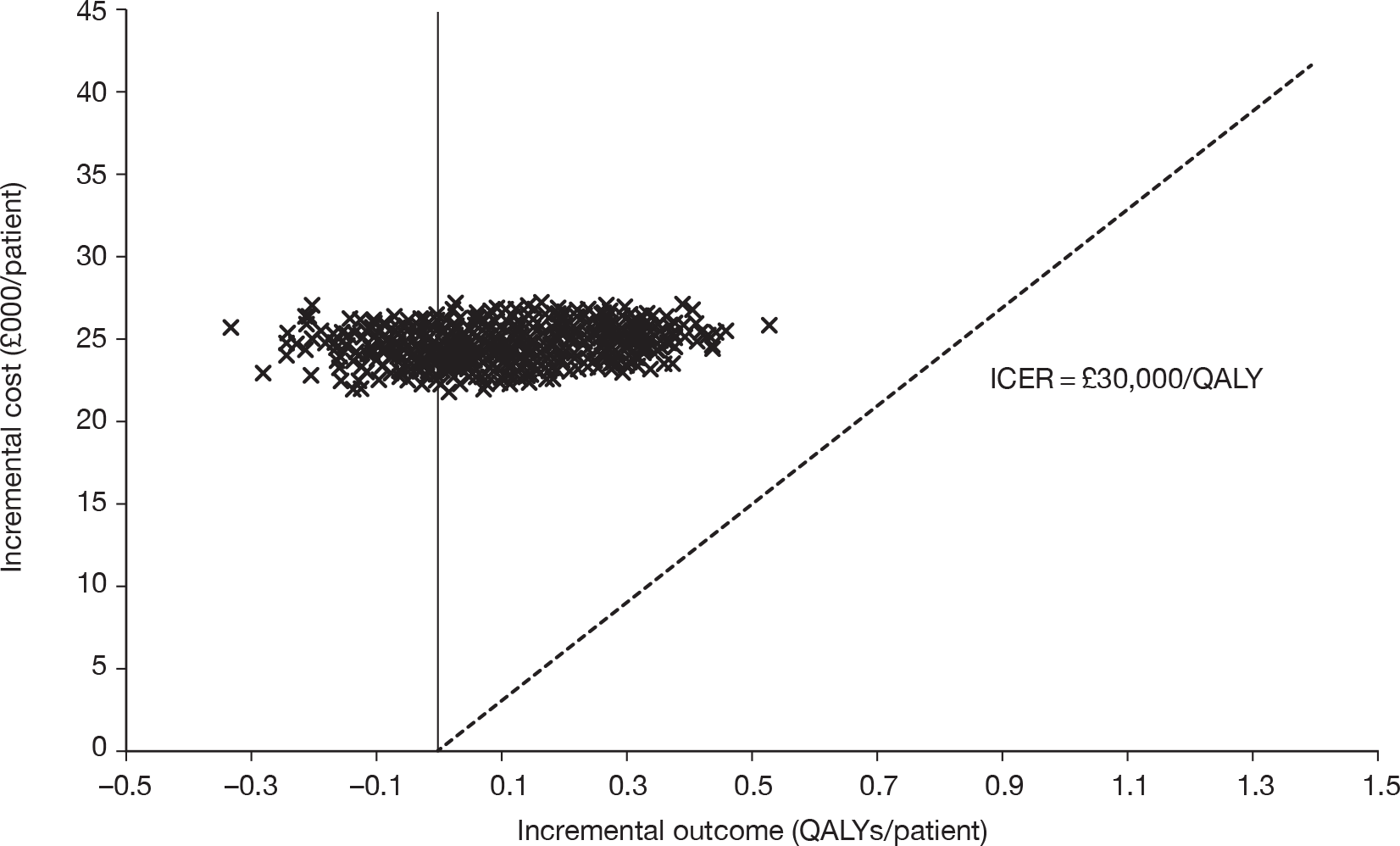
FIGURE 9.
Probabilistic sensitivity analysis of LAP + LET versus LET only: CEAC.
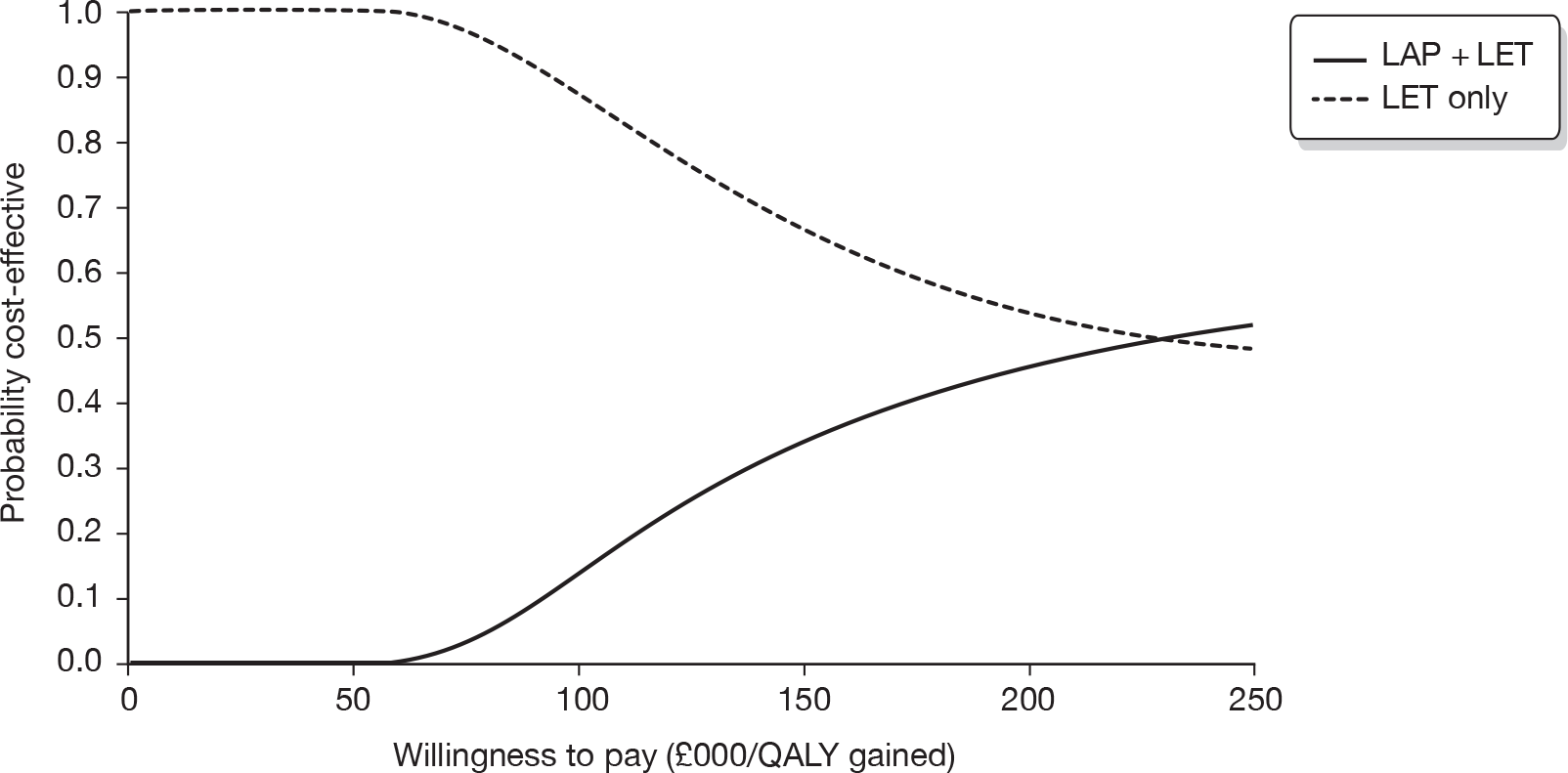
Base-case results: trastuzumab in combination with anastrozole versus anastrozole alone
The base-case cost-effectiveness results based on the AG model are shown in Table 30. A modest expected mean health gain per patient (< 8 months’ life extension and about 0.5 additional QALYs) is generated by a substantial additional cost of > £35,000 per patient, most of which is incurred in the first 5 years. The cost-effectiveness ratio is stable over long time periods and exceeds £69,000 per QALY gained.
| Treatment | Cost per patient (£) | Outcomes per patient | ICER | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Time horizon (years) | Drugs | Monitoring | AEs | BSC | Terminal care | Total costs | Life-years | QALYs | £/QALY gained |
| ANA | |||||||||
| 10 | 549 | 602 | 11,101 | 1632 | 13,884 | 2.204 | 1.235 | ||
| 20 | 549 | 602 | 11,194 | 1647 | 13,992 | 2.220 | 1.243 | ||
| 30 | 549 | 602 | 11,194 | 1648 | 13,993 | 2.220 | 1.243 | ||
| TRA + ANA | |||||||||
| 10 | 35,197 | 1843 | 90 | 11,875 | 1695 | 50,699 | 2.848 | 1.757 | |
| 20 | 36,251 | 1898 | 92 | 11,953 | 1696 | 51,891 | 2.888 | 1.788 | |
| 30 | 36,370 | 1905 | 93 | 11,961 | 1696 | 52,025 | 2.893 | 1.791 | |
| Incremental | |||||||||
| 10 | 34,648 | 1241 | 90 | 774 | 63 | 36,815 | 0.644 | 0.522 | 70,463 |
| 20 | 35,702 | 1297 | 92 | 759 | 49 | 37,899 | 0.669 | 0.545 | 69,514 |
| 30 | 35,821 | 1303 | 93 | 767 | 49 | 38,032 | 0.673 | 0.549 | 69,333 |
Univariate sensitivity analysis
Results from a sensitivity analysis covering the main model variables are shown in Table 31. The ICER is most sensitive to the health-state utility parameter values, to the cost of TRA and to discounting rates, but very insensitive to most of the other variables. In all cases, the ICER remains > £58,000 per QALY, indicating that uncertainty in any single parameter value is unlikely to alter the cost-effectiveness of TRA + ANA relative to conventional thresholds.
| Model variable | Variation in value | ICER (£) for | ||
|---|---|---|---|---|
| Low | High | Low | High | |
| Base case | 69,514 | |||
| Discount rate: costs | 0% | 6% | 76,076 | 66,870 |
| Discount rate: outcomes | 0% | 6% | 64,711 | 72,588 |
| Dispensing costs: community | £5 | £10 | 69,482 | 69,570 |
| Dispensing costs: hospital | £7 | £11 | 69,429 | 69,598 |
| Frequency of cardiac monitoring (per annum) | 3 | 6 | 69,249 | 70,051 |
| ECHO as % of scans | 50% | 100% | 69,778 | 69,117 |
| Frequency of PFS follow-up and CT scan (per annum) | 2 | 6 | 68,940 | 70.130 |
| Proportion of progressed patients on EXE | 0% | 100% | 69,628 | 69,399 |
| Net extra cost of AEs in TRA + ANA group | £0 | £1000 | 69,459 | 71,155 |
| Net extra disutility of AEs in TRA + ANA group | 0 | –0.01 | 69,514 | 70,713 |
| Utility in PFS: ANA only | –10% | +10% | 64,744 | 75,042 |
| Utility in PFS: TRA + ANA | –10% | +10% | 85,113 | 58,747 |
| Utility in PFS: both groups | –10% | +10% | 78,071 | 62,647 |
| Utility in PPS | –10% | +10% | 68,852 | 70,188 |
| Administration of TRA costs | –10% | +10% | 68,661 | 70,367 |
| Acquisition cost of TRA costs | –10% | +10% | 63,997 | 75,030 |
| Cost of cardiac scan | –10% | +10% | 69,397 | 69,630 |
| BSC annual costs | –10% | +10% | 69,363 | 69,664 |
| Terminal care costs | –10% | +10% | 69,505 | 69,523 |
Probabilistic sensitivity analysis
Probabilistic sensitivity was explored, running 1000 random iterations for all variables subject to measurable parameter uncertainty, using the base-case scenario over a 20-year time horizon. The PSA results are compared with the corresponding deterministic results in Table 32.
| Sensitivity analysis | Incremental cost (£) | Incremental QALYs | ICER (£) |
|---|---|---|---|
| Deterministic | 37,899 | 0.669 | 69,514 |
| Probabilistic | 33,489 | 0.513 | 65,284 |
The scatterplot of iteration results in the cost-effectiveness plane (Figure 10) indicates a strong positive correlation between incremental cost and incremental benefit. Figure 11 confirms that there is no measurable probability of the combination therapy being cost-effective at a willingness-to-pay threshold of £40,000 per QALY gained, and only a 6.3% probability at £50,000 per QALY gained.
FIGURE 10.
Probabilistic sensitivity analysis of TRA + ANA versus ANA only: scatterplot of 1000 probabilistic iterations.
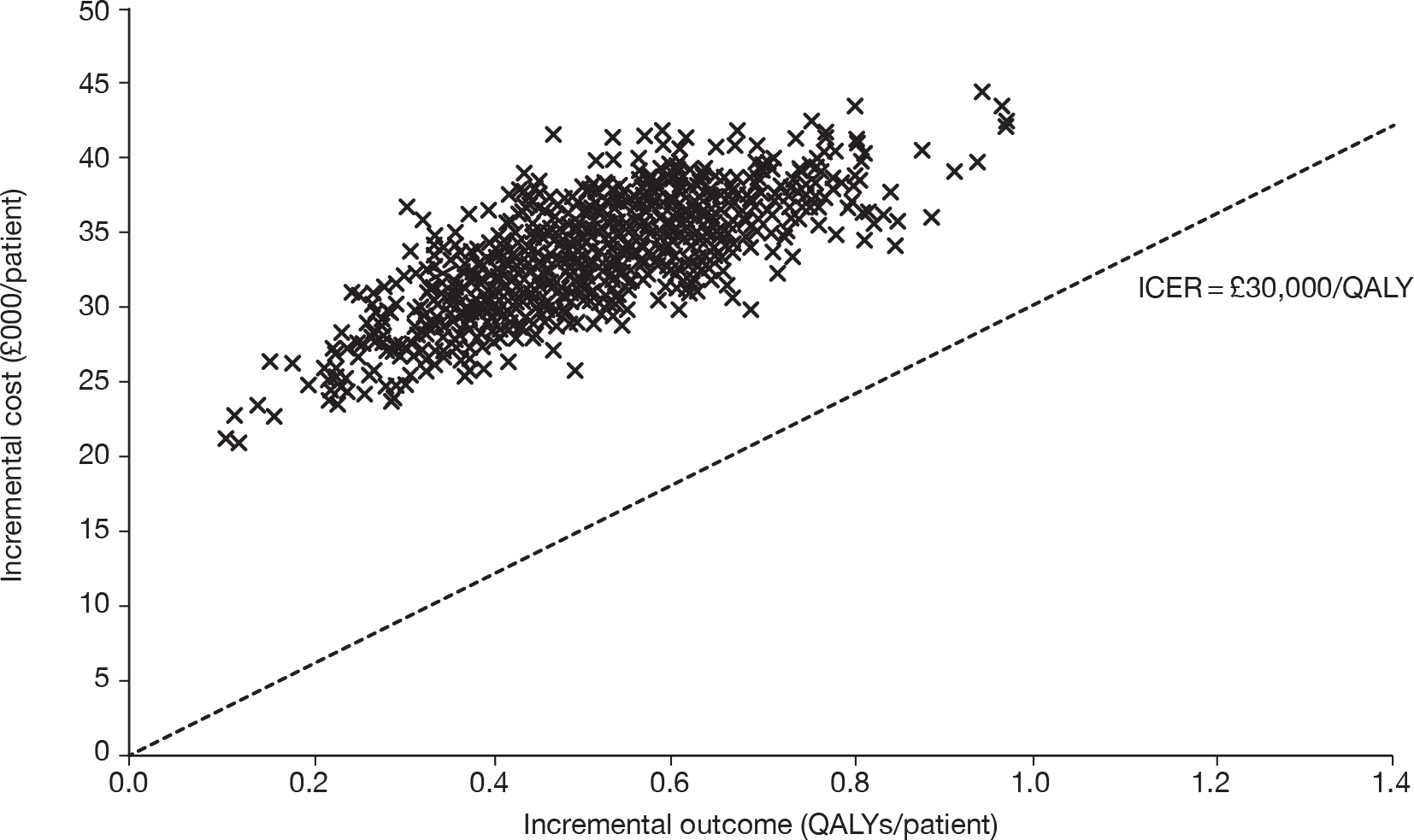
FIGURE 11.
Probabilistic sensitivity analysis of TRA + ANA versus ANA only: CEAC.
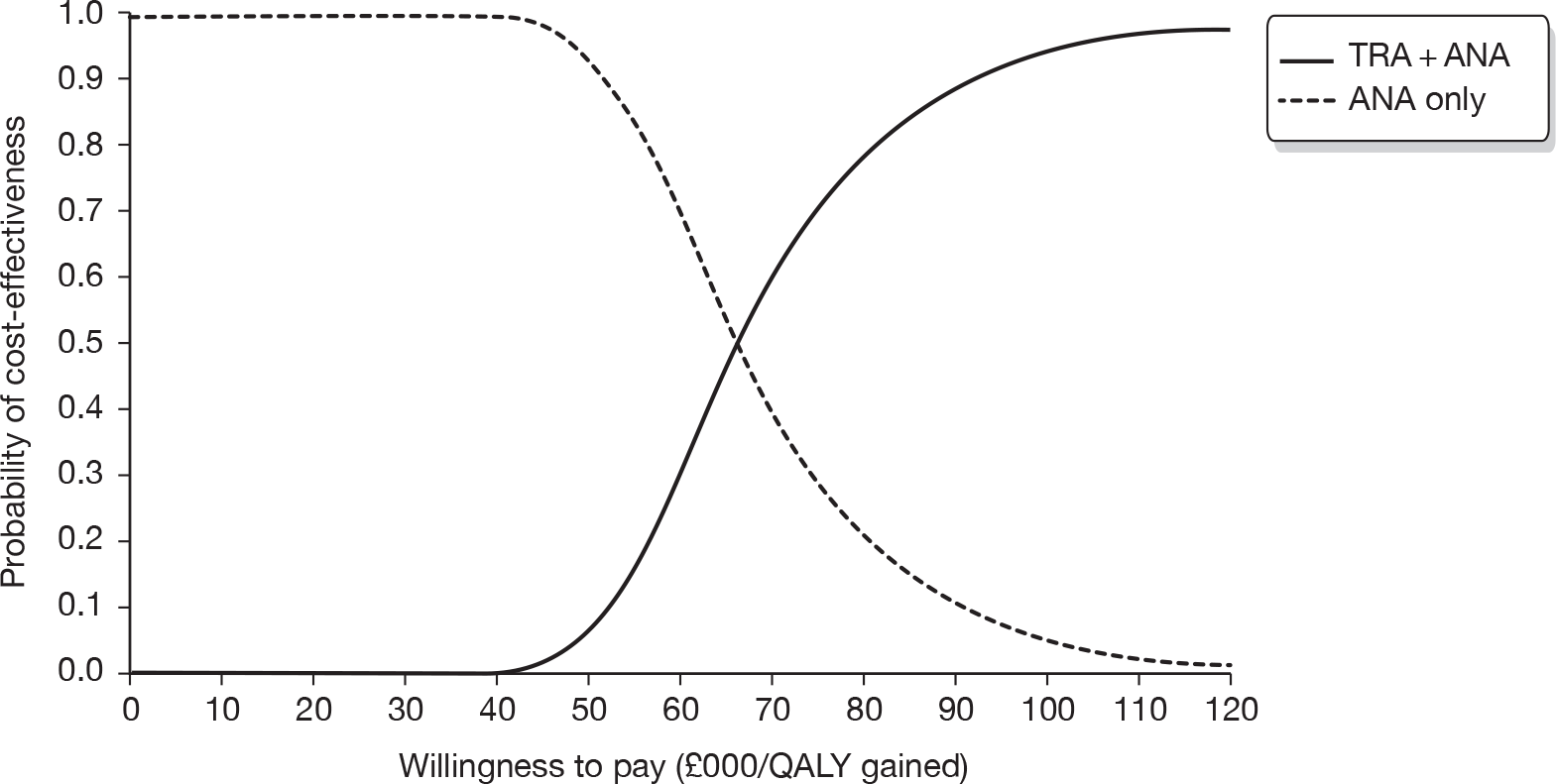
Summary of cost-effectiveness evidence
Cost-effectiveness review
In summary, the AG did not identify any relevant papers for inclusion in the cost-effectiveness review of LAP + AI or TRA + AI in patients who are HR+/HER2+ with mBC. The manufacturer of TRA identified a poster96 that was presented at the ASCO 2010 conference; the study described compared LAP + LET versus TRA + ANA, using an indirect comparisons analysis. The AG is of the opinion that the results of the indirect analysis performed by Hastings et al. 96 are unreliable as the studies that make up the evidence network are inappropriate. In addition, the AG notes that without access to more detailed information on costs, it is difficult to comment on the reliability of the cost-effectiveness results in this study.
Submitted economic evaluations by manufacturers
The two economic evaluations submitted by the manufacturers appear to meet the NICE reference case criteria. 97 However, the AG is critical of the approaches used by the manufacturers to estimate OS in each of their models; the AG is of the opinion that projective modelling in this group of patients can lead to substantial bias in OS estimates. In addition, the AG also identified several costing inaccuracies and inconsistencies in both of the economic evaluations submitted.
For the direct comparisons, GlaxoSmithKline demonstrated that LAP + LET is not cost-effective compared with LET and Roche demonstrated that TRA + ANA is not cost-effective compared with ANA.
Both of the manufacturers undertook indirect comparisons analyses in order to be able to compare LAP + LET versus TRA + ANA. GlaxoSmithKline demonstrated that LAP + LET is cost-effective compared with TRA + ANA. Roche demonstrated that TRA + ANA is cost-effective compared with LAP + LET. The AG concludes that the indirect comparisons analyses conducted by the manufacturers are unreliable and that only the ICERs estimated from the direct comparisons are valid.
Roche makes the case for TRA + ANA to be considered as an end-of-life treatment for women with HR+/HER2+ mBC. The AG does not have sufficient information to verify whether or not all three NICE criteria for consideration of end-of-life treatments are met.
Assessment Group’s cost-effectiveness results and sensitivity analysis
The AG reports the results of two separate de novo cost-effectiveness analyses using a common framework and common parameter values, but employing effectiveness data drawn only from a single RCT (the EGF3000849 or the TAnDEM53 trial). The AG model employs outcome data derived from the relevant clinical trial in the form of Kaplan–Meier estimated survival values augmented by projected survival estimates calibrated against the observed data. The AG uses PFS and PPS estimates directly as the basis for calculating expected OS in each group of the RCT.
As the AG is of the opinion that the evidence base is too unstable to allow meaningful comparison of LAP + AI versus TRA + ANA, the only questions that may be addressed legitimately are as follows.
-
Can LAP + LET be considered a cost-effective treatment compared with LET alone?
-
Can TRA + ANA be considered a cost-effective treatment compared with ANA alone?
Base-case result: lapatinib in combination with letrozole versus letrozole alone
The AG concludes that in HR+/HER2+ women with mBC, LAP + LET compared with LET is not cost-effective. Using a time horizon of 20 years, the AG estimates an ICER that exceeds £225,000 per QALY gained for the comparison of LAP + LET versus LET; the incremental total costs and QALYs per patient treated are estimated as £26,150 and 0.116, respectively.
Base-case result: trastuzumab in combination with anastrozole versus anastrozole alone
The AG concludes that in HR+/HER2+ women with mBC, TRA + ANA compared with ANA is not cost-effective. Using a time horizon of 20 years, the AG estimates an ICER that exceeds £69,000 per QALY gained for the comparison of TRA + ANA versus ANA; the incremental total costs and QALYs per patient treated are estimated as £37,899 and 0.545, respectively.
Lapatinib in combination with aromatase inhibitor versus trastuzumab in combination with aromatase inhibitor
The AG emphasises, again, that the currently available clinical evidence base is too unstable to allow meaningful comparison of LAP + AI versus TRA + AI.
Sensitivity analyses undertaken by the Assessment Group
For the comparison of LAP + LET versus LET, the univariate sensitivity analysis shows that the ICER is most sensitive to the choice of health-state utility parameter values and the cost of LAP, but is insensitive to most of the other variables. In all cases, the ICER remains > £141,000 per QALY gained. The PSA shows that the probability of LAP + LET being cost-effective is 0.1% at a willingness-to-pay threshold of £50,000 per QALY gained; to achieve a 50% probability of LAP + LET being cost-effective, the willingness-to-pay threshold needs to increase to £231,000 per QALY gained.
For the comparison of TRA + ANA versus ANA, the univariate sensitivity analysis shows that the ICER is most sensitive to the choice of health-state utility parameter values, the cost of TRA and discounting rates only. In all cases, the ICER exceeds £58,000 per QALY gained. The PSA shows that there is no measureable probability of TRA + ANA being cost-effective compared with ANA at a willingness-to-pay threshold of £40,000.
Chapter 5 Discussion
The size of the relevant study population of interest to this appraisal is small. The manufacturers of both LAP and TRA are in agreement that the population of post-menopausal women with HR+/HER2+ mBC eligible for LAP or TRA in combination with an AI in England and Wales is < 100 patients per year. From the limited data available, the AG agrees that these estimates appear to be reasonable.
Only three RCTs49,50,52 have been identified that present head-to-head comparisons of the interventions of interest to this appraisal. It was not possible to compare the data across the trials because of differences in the patient populations. Nevertheless, all three49,50,52 trials suggest that either LAP or TRA in combination with an AI improves efficacy, in terms of PFS and/or TTP, over an AI alone. These findings are only statistically significantly different in two trials,49,50 as the eLEcTRA52 trial lacks statistical power owing to being halted early because of slow recruitment. The trials49,50 that measured OS did not report any statistically significant differences between treatment groups, although the OS data in the HR+/HER2+ population of the EGF30008 trial had yet to reach maturity. AEs were more common in the groups in which either LAP or TRA was given in combination with an AI, but on the whole, were of grades 1 or 2 severity. However, around 1% of patients taking LAP + LET had to discontinue their treatment as a result of AEs related to diarrhoea. No new safety concerns were reported in any of the trials.
The comparison of LAP + AI versus TRA + AI is also of interest to this appraisal and, as there are no head-to-head trials of these interventions, the manufacturers used indirect comparisons analyses using mainly clinical data from the EGF3000849 and the TAnDEM50 trials to assess this comparison. The AG believes that the results of any indirect comparisons analyses of LAP + LET versus TRA + ANA using data from the EGF3000849 and the TAnDEM50 trials are unreliable because of heterogeneous patient populations. The AG considers that there are apparent differences in the study populations of these two key trials that prohibit comparison of patients and therefore results; these differences may be explained by the fact that patients were explicitly excluded from the EGF3000849 trial if they had extensive symptomatic visceral disease or their disease was rapidly progressing or life-threatening, or they may be explained by differences in second-line treatment received in the two trials.
In addition, to complete the evidence network in the indirect comparisons analyses presented in the submitted MS, the manufacturers had to use trials with mixed HER2– and HER2+ populations. The AG is of the opinion, that the use of clinical effectiveness evidence from a mixed population adds to the uncertainty regarding the results of the indirect analyses conducted by the manufacturers. To illustrate, in the EGF30008 trial,49 which included both HR+/HER2+ and HR+/HER2– populations, the clinical effectiveness of LET appears to be compromised in patients who are HR+/HER2+ compared with patients who are HR+/HER2–; this was also apparent for patients in the LAP + LET arm of the trial, but to a lesser extent. The significance of this finding is unclear but from a purely clinical viewpoint, could suggest that an AI alone is relatively less effective in patients with HR+/HER2+ mBC.
In summary, the AG is of the opinion that it is not useful to compare findings across the two trials49,50 because of the heterogeneous patient populations. In addition, reliance on clinical evidence from a mixed population adds to the uncertainty of the validity of the results for a HR+/HER2+ population. The AG, therefore, considers the results of the indirect comparisons analysis presented by the manufacturers to be unreliable.
A final issue that needs to be considered relates to the generalisability of these trials to the actual population of interest in the UK, namely post-menopausal women with HR+/HER2+ mBC who have not previously received treatment for mBC and for whom treatment with an AI is suitable. None of the patients in the EGF3000849 or the TAnDEM50 trials have received prior treatment with TRA; this is not surprising as, at the time the trials were recruiting, the use of TRA for patients with early or advanced breast cancer was relatively rare. This contrasts very much with what happens in clinical practice in the NHS today. Now, when a patient is diagnosed with early HER2+ breast cancer, TRA is the standard treatment of choice and in reality it is likely that only de novo patients with HR+/HER2+ mBC will be eligible for TRA + AI as per the wording of the EMA licence (i.e. TRA-naive), although Roche believes the majority (76%) of patients who present for mBC have not previously received TRA for adjuvant therapy based on its own market research. 20 Patients who have been treated with TRA previously are eligible for treatment with LAP + AI; however, whether or not the clinical effectiveness of LAP + AI is the same for patients who are and who are not TRA-naive is uncertain. An FDA post-approval commitment study, EGF114299, is currently being conducted by GlaxoSmithKline to compare the safety and efficacy of an AI in combination with LAP, TRA or both for the treatment of HR+/HER2+ mBC. However, the results from this study will not be available for many years as this study is not currently open for participant recruitment as of November 2011. 114
From a health economics perspective, the AG has confirmed by its independent analyses the assertion made by both manufacturers that LAP + LET and TRA + ANA are not cost-effective compared with AIs alone for women with HR+/HER2+mBC. However, the ICERs estimated by the AG for LAP + LET versus LET and TRA + ANA versus ANA are higher than those estimated by the manufacturer.
The AG is of the opinion that the protocol criteria governing the selection of patients for the EGF3000849 and the TAnDEM50 trials, and subsequent treatment received on progression in these trials, are sufficiently dissimilar to be likely to generate non-equivalent patient populations. This means that the results of any indirect comparisons analyses that include both of these trials in the evidence network are unreliable. Consequently, the AG did not address the cost-effectiveness of LAP + LET versus TRA + ANA as there were insufficient comparative clinical data available to allow estimation of meaningful ICERs.
Chapter 6 Conclusions and research recommendations
Clinical effectiveness evidence from two good-quality RCTs49,50 and a third trial that was halted prematurely58 demonstrates that LAP + LET or TRA + ANA improves median PFS and/or TTP compared with AI monotherapy in patients who are HR+/HER2+ mBC; it also appears that AIs alone may be less effective in patients with HR+/HER2+ tumours compared with HR+/HER2–. 49,52 To date, the trials49,50 do not show a statistically significant benefit in terms of OS for patients taking LAP + LET versus AI monotherapy or TRA + ANA versus AI monotherapy, although the OS data in the HR+/HER2+ population of the EGF3000849 trial had yet to reach maturity and no OS data were presented for the eLEcTRA trial,58 presumably because this trial was halted prematurely. The results of the economic evaluations conducted by the manufacturers and confirmed by the AG demonstrate that LAP + LET is not cost-effective compared with AI monotherapy, nor is TRA + ANA cost-effective compared with AI monotherapy.
As a result of differences in the patient populations of the EGF3000849 and the TAnDEM trials,50 the AG believes the results of the indirect comparisons analyses conducted by the manufacturers are inappropriate and for the same reason believes that it would be unsound to compare LAP + LET versus TRA + ANA in an economic evaluation.
Recommendations for further research
Given the uncertainties in the evidence base, the AG suggests that the following research priorities should be addressed (in order of priority).
-
Given that most patients who present for HR+/HER2+ mBC are now likely to have been previously treated for early breast cancer and given that this is almost certain to have included TRA (unlike at the time the pivotal trials in this appraisal were conducted), further research may be required into treating mBC in the HR+/HER2+ population who are not TRA (or LAP) naive. It is noted that such a study (EGF114299) is planned by GlaxoSmithKline.
-
As, increasingly, trials allow patients to cross over following disease progression, attempts should be made to consider how to adjust for crossover at the trial design stage, rather than as a post-hoc analysis.
-
As the EGF3000849 trial reports, there were large differences in PFS for HER2+ and HER2– patients receiving both LAP + LET and, in particular, LET. Further research may be warranted comparing the clinical effectiveness of AIs as monotherapy in patients with HER2+ and HER2– breast cancer.
Acknowledgements
The authors are pleased to acknowledge Janet Atkinson [Liverpool Reviews and Implementation Group (LRiG), University of Liverpool], who provided administrative support (including obtaining bibliographic sources) for this project. In addition, four referees considered and commented on the final version of this report post submission. The policy of the NIHR Evaluation, Trials and Studies Coordinating Centre (NETSCC) is not to name referees; however, individuals contributing peer review of HTA programme products are listed on the NETSCC website (www.netscc.ac.uk/). Finally, the authors would like to acknowledge the comments received by consultees prior to the first meeting of the NICE Appraisal Committee Meeting in November 2010, available from the NICE website (at http://guidance.nice.org.uk/TA/Wave0/167).
Contributions of authors
Nigel Fleeman Project lead, involved in all aspects of the clinical review and report writing.
Adrian Bagust Critical appraisal of manufacturers’ economic models, development of de novo model and report writing.
Angela Boland Involved in all aspects of the economics review and report writing.
Rumona Dickson Support of review process and commented on draft versions of the final report.
Yenal Dundar Conducted literature searches and contributed to the screening and selection process.
May Moonan Prepared the background section, contributed to analysis and interpretation of data and commented on draft versions of the final report.
James Oyee Contributed to analysis and interpretation of data, prepared sections of the report relating to statistical issues and commented on draft versions of the final report.
Michaela Blundell Additional statistical support.
Helen Davis Contributed to the background section and commented on draft versions of the final report.
Anne Armstrong Contributed to the analysis and interpretation of data and commented on draft versions of the final report.
Nicky Thorp Contributed to the background section, analysis and interpretation of data and commented on draft versions of the final report.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Germano S, O’Driscoll L. Breast cancer: understanding sensitivity and resistance to chemotherapy and targeted therapies to aid in personalised medicine. Curr Cancer Drug Targets 2009;9:398-41.
- Cancer Research UK . Breast Cancer – Risk Factors 2010. http://info.cancerresearchuk.org/cancerstats/types/breast/riskfactors/ (accessed 6 September 2010).
- Ward S, Simpson E, Davis S, Hind D, Rees A, Wilkinson A. Taxanes for the adjuvant treatment of early breast cancer: systematic review and economic evaluation. Health Technol Assess 2007;11.
- Pharoah PD, Antoniou A, Bobrow M, Zimmern RL, Easton DF, Ponder BA. Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet 2002;31:33-6.
- Hannah L, Crosby T, MacBeth F. Practical clinical oncology. Cambridge: Cambridge University Press; 2008.
- Endogenous Hormones Breast Cancer Collaborative Group . Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst 2002;94:606-16.
- Cassidy J, Bissett D, Spence R, Payne M. Oxford handbook of oncology. 2nd edn. Oxford: Oxford: Oxford University Press; 2006.
- American Joint Committee on Cancer . AJCC Cancer Staging Manual 2009.
- International Union Against Cancer . TNM Classification of Malignant Tumours 2009.
- Dowsett M, Cuzick J, Ingle J, Coates A, Forbes J, Bliss J, et al. Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J Clin Oncol 2010;28:509-18.
- Early Breast Cancer Trialists’ Collaborative Group . Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005;365:1687-717.
- Cancer Research UK. Predicted Improvements in Breast Cancer Survival Cancer Research UK 2010. www.cancerhelp.org.uk/about-cancer/cancer-questions/predicted-improvements-in-breast-cancer-survival (accessed 6 September 2010).
- Andreopoulou E, Hortobagyi GN. Prognostic factors in metastatic breast cancer: successes and challenges toward individualized therapy. J Clin Oncol 2008;26:3660-2.
- Cold S, Jensen NV, Brincker H, Rose C. The influence of chemotherapy on survival after recurrence in breast-cancer – a population-based study of patients treated in the 1950s, 1960s and 1970s. Eur J Cancer 1993;29:1146-52.
- National Institute for Health and Clinical Excellence (NICE) . Guidance on the Use of Trastuzumab for the Treatment of Advanced Breast Cancer 2002. www.nice.org.uk/nicemedia/pdf/TA107guidance.pdf (accessed 26 January 2010).
- Cancer Research UK . Breast Cancer – UK Incidence Statistics 2010. http://info.cancerresearchuk.org/cancerstats/types/breast/incidence/ (accessed 18 August 2011).
- Quinn M, Cooper N, Rowan S. Cancer Atlas of the United Kingdom and Ireland 1991–2000 2005. www.statistics.gov.uk/downloads/theme_health/caUKI91_00/Preliminary_pages.pdf (accessed 14 July 2010).
- Woods LM, Rachet B, Coleman MP. Origins of socio-economic inequalities in cancer survival: a review. Ann Oncol 2006;17:5-19.
- GlaxoSmithKline . Manufacturer Submission to the National Institute for Health and Clinical Excellence by GlaxoSmithKline UK. Multiple Technology Appraisal of Lapatinib (Tyverb®) for the Treatment of HR+ HER2+ Metastatic Breast Cancer 2010.
- Roche . Trastuzumab in Combination With Aromatase Inhibitors for the 1st Line Treatment of Metastatic Hormone Receptor Positive Breast Cancer Which over-Expressed HER2 2010.
- Dybdal N, Leiberman G, Anderson S, McCune B, Bajamonde A, Cohen R, et al. Determination of HER2 gene amplification by fluorescence in situ hybridization and concordance with the clinical trials immunohistochemical assay in women with metastatic breast cancer evaluated for treatment with trastuzumab. Breast Cancer Res Treat 2005;93:3-11.
- Turner J, Kelly B, Swanson C, Allison R, Wetzig N. Psychosocial impact of newly diagnosed advanced breast cancer. Psychooncology 2005;14:396-407.
- National Institute for Health and Clinical Excellence (NICE) . Advanced Breast Cancer: Diagnosis and Treatment 2009. www.nice.org.uk/Guidance/CG81 (accessed 26 January 2010).
- Quinlan E, Thomas-Maclean R, Hack T, Kwan W, Miedema B, Tatemichi S, et al. The impact of breast cancer among Canadian women: disability and productivity. Work 2009;34:285-96.
- Yoo GJ, Levine EG, Aviv C, Ewing C, Au A. Older women, breast cancer, and social support. Support Care Cancer 2010;18:1521-30.
- Montazeri A, Vahdaninia M, Harirchi I, Ebrahimi M, Khaleghi F, Jarvandi S. Quality of life in patients with breast cancer before and after diagnosis: an eighteen months follow-up study. BMC Cancer 2008;8.
- Kedrin D, Wyckoff J, Boimel PJ, Coniglio SJ, Hynes NE, Arteaga CL, et al. ERBB1 and ERBB2 have distinct functions in tumor cell invasion andi. Clin Cancer Res 2009;15:3733-9.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary 2010.
- European Medicines Agency (EMA) . Committee for Medicinal Products for Human Use Post-Authorisation Summary of Positive Opinion for Tyverb 2010. www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion/human/000795/WC500074419.pdf (accessed 24 November 2010).
- European Medicines Agency (EMA) . Committee for Medicinal Products for Human Use Summary of Positive Opinion for Tyverb 2008. www.ema.europa.eu/pdfs/human/opinion/Tyverb_38267507en.pdf (accessed 26 January 2010).
- Pharmaceutical Field . Further Indication for Tyverb Approved 2010. www.pharmafield.co.uk/news/2010/06/further-indication-for-tyverb-approved (accessed 14 July 2010).
- Jones AL, Barlow M, Barrett-Lee PJ, Canney PA, Gilmour IM, Robb SD, et al. Management of cardiac health in trastuzumab-treated patients with breast cancer: updated United Kingdom National Cancer Research Institute recommendations for monitoring. Br J Cancer 2009;100:684-92.
- European Medicines Agency (EMA) . Committee for Medicinal Products for Human Use Post-Authorisation Summary of Positive Opinion for Herceptin 2009. www.ema.europa.eu/pdfs/human/opinion/Herceptin_82246709en.pdf (accessed 26 January 2010).
- National Institute for Health and Clinical Excellence (NICE) . Hormonal Therapies for the Adjuvant Treatment of Early Oestrogen-Receptor-Positive Breast Cancer 2006. www.nice.org.uk/TA112 (accessed 26 January 2010).
- Lavelle K, Todd C, Moran A, Howell A, Bundred N, Campbell M. Non-standard management of breast cancer increases with age in the UK: a population based cohort of women greater than or equal to 65 years. Br J Cancer 2007;96:1197-203.
- Wyld L, Garg DK, Kumar ID, Brown H, Reed MWR. Stage and treatment variation with age in postmenopausal women with breast cancer: compliance with guidelines. Br J Cancer 2004;90:1486-91.
- Ladva S. Continued use of trastuzumab following disease progression in metastatic breast cancer. London: New Drugs Group; 2010.
- Centre for Reviews and Dissemination (CRD) . CRD’s Guidance for Undertaking Reviews in Healthcare: Systematic Reviews 2008.
- Egger M, Smith GD, Altman DG. Systematic reviews in health care – meta-analysis in context. London: BMJ books; 2001.
- Lu G, Ades AE. Combination of direct and indirect evidence in mixed treatment comparisons. Stat Med 2004;23:3105-24.
- Langer B. Phase II III Randomized Study of Anastrozole With or Without Trastuzumab (herceptin) in Postmenopausal Women With Hormone-Receptor Positive HER2-Overexpressing Metastatic Breast Cancer. Physician Data Query (PDQ) 2001. www.mrw.interscience.wiley.com/cochrane/clcentral/articles/021/CN-00362021/frame.html (accessed 30 June 2010).
- Piccart-Gebhart MJ, Coleman RE. Phase III Randomized Study of Trastuzumab (herceptin) in Women With HER2-Positive Primary Breast Cancer. Physician Data Query (PDQ) 2002. www.mrw.interscience.wiley.com/cochrane/clcentral/articles/007/CN-00404007/frame.html (accessed 30 June 2010).
- Stein S. Phase III Randomized Study of Letrozole With or Without GW572016 in Postmenopausal Women With Stage IIIB or IV Breast Cancer. Physician Data Query (PDQ) n.d. www.mrw.interscience.wiley.com/cochrane/clcentral/articles/289/CN-00548289/frame.html (accessed 30 June 2010).
- Novartis . Study of the Efficacy and Safety of Letrozole Combined With Trastuzumab in Patients With Metastatic Breast Ccancer. Physician Data Query (PDQ) 2006. www.mrw.interscience.wiley.com/cochrane/clcentral/articles/302/CN-00614302/frame.html (accessed 30 June 2010).
- Morris P, Modi S. Trastuzumab-refractory HER2-positive metastatic breast cancer – use of tanespimycin, a novel Hsp90 inhibitor. Am J Pediatr Hematol Oncol 2008;7.
- Maung K, O’Shaughnessy JA. Inhibition of ErbB1 and ErbB2 by lapatinib ditosylate, a dual kinase inhibitor: promising activity in pretreated advanced breast cancer. Clin Breast Cancer 2004;4:398-400.
- Ranganathan A, Moore Z, O’Shaughnessy JA. Trastuzumab prolongs progression-free survival in patients with hormone-sensitive and HER2-overexpressing metastatic breast cancer. Clin Breast Cancer 2007;7:450-1.
- Johnston S, Arteaga C. Lapatinib (tykerb) plus letrozole (femara) or letrozole alone for postmenopausal hormone receptor-positive metastatic breast cancer. P and T 2009;34.
- Johnston S, Pippen J, Pivot X, Lichinitser M, Sadeghi S, Dieras V, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor – positive metastatic breast cancer. J Clin Oncol 2009;27:5538-46.
- Kaufman B, Mackey JR, Clemens MR, Bapsy PP, Vaid A, Wardley A, et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol 2009;27:5529-37.
- Schwarzberg LS, Franco SX, Florance A, O’Rourke L, Maltzman J, Johnston SRD. Lapatinib plus letrozole as first-line therapy for HER-2+ hormone receptor-positive metastatic breast cancer. Oncologist 2010;15:122-9.
- Huober J, Fasching P, Paepke S, Kubista E, Barsoum M, Wallwiener D, et al. Letrozole in combination with trastuzumab is superior to letrozole monotherapy as first line treatment in patients with hormone-receptor-positive, HER2-positive metastatic breast cancer (MBC) – results of the eLEcTRA Trial. Cancer Res 2009;69.
- Gomez HL, Doval DC, Chavez MA, Ang PC-S, Aziz Z, Nag S, et al. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J Clin Oncol 2008;26:2999-3005.
- Cameron D, Casey M, Press M, Lindquist D, Pienkowski T, Romieu C, et al. A phase III randomized comparison of lapatinib plus capecitabine versus capecitabine alone in women with advanced breast cancer that has progressed on trastuzumab: updated efficacy and biomarker analyses. Breast Cancer Res Treat 2008;112:533-43.
- Food and Drug Administration . Guidance for Industry: Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics 2007. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071590.pdf (accessed 22 July 2010).
- Committee for Medicinal Products for Human Use . Guideline on the Evaluation of Anticancer Medicinal Products in Man 2005. www.tga.gov.au/docs/pdf/euguide/ewp/020595enrev3.pdf (accessed 22 July 2010).
- Fleming TR, Rothmann MD, Lu HL. Issues in using progression-free survival when evaluating oncology products. J Clin Oncol 2009;27:2874-80.
- Saad ED, Katz A. Progression-free survival and time to progression as primary end points in advanced breast cancer: often used, sometimes loosely defined. Ann Oncol 2009;20:460-4.
- Sherif B, Sherrill B, Amonkar M, Wu Y, Maltzman J, O’Rourke L, et al. Lapatinib plus letrozole compared with letrozole alone as first-line therapy in hormone receptor-positive HER2+ metastatic breast cancer (MBC): a quality-of-life (QOL) analysis. J Clin Oncol 2009;27.
- Robins JM, Tsiatis AA. Correcting for non-compliance in randomized trials using rank preserving structural failure time models. Comm Stat Theor 1991;20:2609-31.
- Mark SD, Robins JM. A method for the analysis of randomized trials with compliance information: an application to the multiple risk factor intervention trial. Control Clin Trials 1993;14:79-97.
- Morden JP, Lambert PC, Latimer N, Abrams KR, Wailoo AJ. Assessing methods for dealing with treatment switching in randomised controlled trials. University of Leicester; 2010.
- National Institute for Health and Clinical Excellence (NICE) . Final Appriasal Determination: Sunitinib for the Treatment of Gastrointestinal Stromal Tumours 2009. www.nice.org.uk/nicemedia/live/11817/43174/43174.pdf (accessed 17 August 2010).
- National Institute for Health and Clinical Excellence (NICE) . Final Appraisal Determination: Everolimus for the Second-Line Treatment of Advanced Renal Cell Carcinoma 2010. http://guidance.nice.org.uk/TA/WaveCRS1/48 (accessed 17 August 2010).
- Robins J, Finkelstein D. Correcting for noncompliance and dependent censoring in an AIDS clinical trial with inverse probability of censoring weighted (IPCW) log-rank tests. Biometrics 2000;56:779-88.
- Branson M, Whitehead J. Estimating a treatment effect in survival studies in which patients switch treatment. Stat Med 2002;21:2449-63.
- Walker A, White I, Babiker A. Parametric randomization-based methods for correcting for treatment changes in the assessment of the causal effect of treatment. Stat Med 2004;23:571-90.
- Bonneterre J, Thürlimann B, Robertson JF, Krzakowski M, Mauriac L, Koralewski P, et al. Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: results of the tamoxifen or arimidex randomized group efficacy and tolerability study. J Clin Oncol 2000;18:3748-57.
- Nabholtz JM, Buzdar A, Pollak M, Harwin W, Burton G, Mangalik A, et al. Anastrozole is superior to tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: results of a North American multicenter randomized trial. J Clin Oncol 2000;18:3758-67.
- Buzdar AU, Jones SE, Vogel CL, Wolter J, Plourde P, Webster A. A phase III trial comparing anastrozole (1 and 10 milligrams), a potent and selective aromatase inhibitor, with megestrol acetate in postmenopausal women with advanced breast carcinoma. Arimidex Study Group. Cancer 1997;79:730-9.
- Jonat W, Howell A, Blomqvist C, Eiermann W, Winblad G, Tyrrell C, et al. A randomised trial comparing two doses of the new selective aromatase inhibitor anastrozole (Arimidex) with megestrol acetate in postmenopausal patients with advanced breast cancer. Eur J Cancer 1996;32:404-12.
- Parmar M, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Stat Med 1998;17:2815-34.
- Bucher H, Guyatt G, Griffith L, Walter S. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol 1997;50:683-91.
- Sutton AJ, Higgins JPT. Recent developments in meta-analysis. Oxford: John Wiley & Sons, Ltd; 2008.
- Puhan M, Bachmann L, Kleijnen J, ter Riet G, Kessels A. Inhaled drugs to reduce exacerbations in patients with chronic obstructive pulmonary disease: a network meta-analysis. BMC Med 2009;7.
- Mouridsen HT. Letrozole in advanced breast cancer: the PO25 trial. Breast Cancer Res Treat 2007;105:19-2.
- Buzdar A, Douma J, Davidson N, Elledge R, Morgan M, Smith R, et al. Phase III, multicenter, double-blind, randomized study of letrozole, an aromatase inhibitor, for advanced breast cancer versus megestrol acetate. J Clin Oncol 2001;19:3357-66.
- Dombernowsky P, Smith I, Falkson G, Leonard R, Panasci L, Bellmunt J, et al. Letrozole, a new oral aromatase inhibitor for advanced breast cancer: double-blind randomized trial showing a dose effect and improved efficacy and tolerability compared with megestrol acetate. J Clin Oncol 1998;16:453-61.
- Kaufmann M, Bajetta E, Dirix LY, Fein LE, Jones SE, Cervek J, et al. Exemestane improves survival compared with megoestrol acetate in postmenopausal patients with advanced breast cancer who have failed on tamoxifen. Results of a double-blind randomised phase III trial. Eur J Cancer 2000;36:S86-7.
- Mouridsen H, Sun Y, Gershanovich M, Perez-Carrion R, Becquart D, Chaudri-Ross HA, et al. Superiority of letrozole to tamoxifen in the first-line treatment of advanced breast cancer: evidence from metastatic subgroups and a test of functional ability. Oncologist 2004;9:489-96.
- Paridaens R, Dirix L, Beex L, Nooij M, Cufer T, Lohrisch C, et al. Promising results with exemestane in the first-line treatment of metastatic breast cancer: a randomized phase II EORTC trial with a tamoxifen control. Clin Breast Cancer 2000;1:S19-21.
- Rose C, Vtoraya O, Pluzanska A, Davidson N, Gershanovich M, Thomas R, et al. An open randomised trial of second-line endocrine therapy in advanced breast cancer. Comparison of the aromatase inhibitors letrozole and anastrozole. Eur J Cancer 2003;39:2318-27.
- Campos SM, Guastalla JP, Subar M, Abreu P, Winer EP, Cameron DA. A comparative study of exemestane versus anastrozole in patients with postmenopausal breast cancer with visceral metastases. Clin Breast Cancer 2009;9:39-44.
- Howell A, Robertson JF, Quaresma Albano J, Aschermannova A, Mauriac L, Kleeberg UR, et al. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol 2002;20:3396-403.
- Osborne CK, Pippen J, Jones SE, Parker LM, Ellis M, Come S, et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol 2002;20:3386-95.
- Robertson JF, Llombart-Cussac A, Rolski J, Feltl D, Dewar J, Macpherson E, et al. Activity of fulvestrant 500 mg versus anastrozole 1 mg as first-line treatment for advanced breast cancer: results from the FIRST study. J Clin Oncol 2009;27:4530-5.
- Paridaens RJ, Dirix LY, Beex LV, Nooij M, Cameron DA, Cufer T, et al. Phase III study comparing exemestane with tamoxifen as first-line hormonal treatment of metastatic breast cancer in postmenopausal women: the European Organisation for Research and Treatment of Cancer Breast Cancer Cooperative Group. J Clin Oncol 2008;26:4883-90.
- Chernozemsky I, Kalinov K, Tzekov H, Racheva M, Hristova S, Tomova A, et al. Randomized phase III trial of exemestane or tamoxifen in first-line hormonal treatment of postmenopausal women with metastatic breast cancer. Breast Cancer Res Treat 2007;106.
- Mauriac L, Romieu G, Bines J. Activity of fulvestrant versus exemestane in advanced breast cancer patients with or without visceral metastases: data from the EFECT trial. Breast Cancer Res Treat 2009;117:69-75.
- Kaufmann M, Bajetta E, Dirix LY, Fein LE, Jones SE, Zilembo N, et al. Exemestane is superior to megestrol acetate after tamoxifen failure in postmenopausal women with advanced breast cancer: results of a phase III randomized double-blind trial. J Clin Oncol 2000;18:1399-411.
- Hind D, Ward S, De Nigris E, Simpson E, Carroll C, Wyld L. Hormonal therapies for early breast cancer: systematic review and economic evaluation. Health Technol Assess 2007;11.
- Dranitsaris G, Leung P, Mather J, Oza A. Cost utility analysis of second line hormonal therapy in advanced breast cancer: a comparison of two aromatase inhibitors to megestrol acetate. Anticancer Drugs 2000;11:591-60.
- Dranitsaris G, Verma S, Trudeau M. Cost utility analysis of first-line hormonal therapy in advanced breast cancer. Am J Clin Oncol 2003;26:289-96.
- Marchetti M, Caruggi M, Colombo G. Cost utility and budget impact of third-generation aromatase inhibitors for advanced breast cancer: a literature based model analysis of cost in the Italian national health service. Clin Ther 2004;26:1546-61.
- Verma S, Rodchi A. Economic evaluation of antiaromatase agents in the second line treatment of metastatic breast cancer. Support Care Cancer 2003;11:728-34.
- Hastings V, Amonkar M, Lykopoulos K, Diaz J, Johnston S. Indirect comparison of the cost-effectiveness of letrozole plus lapatinib (LET+LAP) versus anastrozole plus trastuzumab (ANA + TZ) as first-line treatment for postmenopausal women with HER2+ and HR+ metastatic breast cancer (MBC) from the U.K. national health service (NHS) perspective. Chicago, IL, USA; 2010.
- National Institute for Health and Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2008. www.nice.org.uk/media/B52/A7/TAMethodsGuideUpdatedJune2008.pdf (accessed 26 January 2010).
- Curtis L. Unit costs of health and social care 2008. Kent: Personal Social Services Research Unit, University of Kent; 2008.
- Department of Health . NHS Reference Costs 2008–2009 2010. www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_111591 (accessed 25 August 2010).
- Remak E, Brazil L. Cost of managing women presenting with stage IV breast cancer in the United Kingdom. Br J Cancer 2004;91:77-83.
- Cella DF, Tulsky DS, Gray G, Sarafian B, Linn E, Bonomi A, et al. The functional assessment of cancer therapy scale: development and validation of the general measure. J Clin Oncol 1993;11:570-9.
- Dobrez D, Cella D, Pickard S, Lai J-S, Nickolov A. Estimation of patient preference-based utility weights from the functional assessment of cancer therapy-general. Value Health 2007;10:266-72.
- Delea TE, . Patient preference-based utility weights from the Functional Assessment of Cancer Therapy-general (FACT-G) in women with hormone receptor positive metastatic breast cancer receiving letrozole plus lapatinib or letrozole alone. Value Health 2010;3.
- Lloyd A, Nafees B, Narewska J, Dewilde S, Watkins J. Health state utilities for metastatic breast cancer. Br J Cancer 2006;95:683-90.
- Oxford Outcomes . Utility Measurement Study for Patients With Metastatic Breast Cancer Treated With Tyverb 2009.
- Doyle S, Lloyd A, Walker M. Health state utility scores in advanced non-small cell lung cancer. Lung Cancer 2009;62:374-80.
- Ward S, Pilgrim H, Hind D. Trastuzumab for the treatment of primary breast cancer in HER2-positive women: a single technology appraisal. Health Technol Assess 2009;13:1-6.
- Curtis L. Unit costs of health and social care 2009. Personal Social Services Research Unit, University of Kent; 2010.
- Cooper N, Abrams K, Sutton A, Turner D, Lambert P. A bayesian approach to markov modelling in cost effectiveness analyses: application to taxane use in advanced brest cancer. J Roy Statistical Society 2003;166:389-405.
- Regional Drugs and Therapeutics Centre . The Use of Bevacizumab in the Manaagment of Metastatic Breast Cancer 2007. www.nyrdtc.nhs.uk/docs/eva/Bevacizumab%20MBC%20RDTC%20FINAL.pdf (accessed 25 August 2010).
- National Institute for Health and Clinical Excellence (NICE) . Lapatinib for the Treatment of Previously Treated Women With Advanced, Metastatic or Recurrent Breast Cancer (final Scope) 2007. http://guidance.nice.org.uk/nicemedia/live/11731/35243/35243.pdf (accessed 25 August 2010).
- Dolan P, Gudex C, Kind P, Williams A. The time trade-off method: results from a general population survey. Health Econ 1996;5:141-54.
- Swain L. OPCS-4.5 Chemotherapy Regimens List & Guidance for Clinical Coders Version 2.0 Dated 10th March 2010 2010. www.wales.nhs.uk/sites3/Documents/920/chemoreglistandguideopcs45v2.pdf/ (accessed 25 August 2010).
- GlaxoSmithKline . A Study to Compare the Safety and Efficacy of an Aromatase Inhibitor in Combination With Lapatinib, Trastuzumab or Both for the Treatment of Hormone Receptor Positive, HER2+ Metastatic Breast Cancer n.d. http://clinicaltrials.gov/ct2/show/NCT01160211 (accessed 24 November 2010).
Appendix 1 Literature search strategies
MEDLINE 1950 to April week 4 2010
| Searches | Search terms | Results |
|---|---|---|
| 1 | (lapatinib or tykerb or tyverb or lapatinib ditosylate).af. | 456 |
| 2 | (trastuzumab or herceptin).af. | 340 |
| 3 | (letrozole or femara or anastrozole or arimidex or exemestane or aromasin).af. | 2106 |
| 4 | exp Aromatase Inhibitors/ | 4804 |
| 5 | aromatase inhibitor$.tw. | 3518 |
| 6 | 1 or 2 | 3627 |
| 7 | 3 or 5 | 4323 |
| 8 | 6 and 7 | 121 |
| 9 | exp Breast Neoplasms/ | 172,296 |
| 10 | (breast$adj5 (neoplasm$or cancer$or tumo?r$or carcinoma$or adenocarcinoma$or sarcoma$or dcis or ductal or infiltrat$or intraductal$or lobular or medullary)).mp. | 203,286 |
| 11 | 9 or 10 | 203,391 |
| 12 | 8 and 11 | 115 |
EMBASE 1980 to week 18 2010
| Searches | Search terms | Results |
|---|---|---|
| 1 | (lapatinib or tykerb or tyverb or lapatinib ditosylate).af. | 2435 |
| 2 | (trastuzumab or herceptin).af. | 10,741 |
| 3 | (letrozole or femara or anastrozole or arimidex or exemestane or aromasin).af. | 5962 |
| 4 | Aromatase Inhibitors.mp. or exp aromatase inhibitor/ | 11,914 |
| 5 | 1 or 2 | 11,728 |
| 6 | 3 or 4 | 11,959 |
| 7 | 5 and 6 | 1472 |
| 8 | (breast$adj5 (neoplasm$or cancer$or tumo?r$or carcinoma$or adenocarcinoma$or sarcoma$or dcis or ductal or infiltrat$or intraductal$or lobular or medullary)).mp. | 195,921 |
| 9 | exp breast cancer/ | 167,175 |
| 10 | 8 or 9 | 197,019 |
| 11 | 7 and 10 | 1378 |
| 12 | limit 11 to (human and english language) | 1195 |
The Cochrane Library Issue 4, 2010
| Searches | Search terms | Results |
|---|---|---|
| 1 | (lapatinib or tykerb or tyverb or lapatinib ditosylate or trastuzumab or herceptin or letrozole or femara or anastrozole or arimidex or exemestane or aromasin or aromatase inhibitor*) | 1196 |
| 2 | MeSH descriptor Breast Neoplasms explode all trees | 6865 |
| 3 | (breast cancer* or breast neoplasm* or breast tumor* or breast tumour* or breast carcinoma*) | 14,097 |
| 4 | (#2 OR #3) | 14,097 |
| 5 | (#1 AND #4) | 932 |
| Search Results by each database in The Cochrane Library | Results | |
| Cochrane Database of Systematic Reviews (CDSR) | 24 | |
| Database of Abstracts of Reviews of Effects (DARE) | 26 | |
| Cochrane Central Register of Controlled Trials (CENTRAL) | 757 | |
| Cochrane Methodology Register (CMR) | 11 | |
| Health Technology Assessment Database | 34 | |
| NHS Economic Evaluation Database (NHS EED) | 79 | |
The search strategy for The Cochrane Library is broader than MEDLINE or EMBASE, combining all the drug-related free-text words with breast cancer [both using medical subject heading (MeSH) Breast Neoplasms and free-text words] to identify relevant reviews and particularly economic evaluations in the area.
All databases
Total number of results from all databases: 2228.
After electronic removal of duplicates: 2202.
After manual removal of duplicates: 2069.
Appendix 2 Tables of excluded studies with rationale
Excluded studies from clinical review
The following citations were excluded by the AG at screening stage 2.
| Study | Reason for exclusion |
|---|---|
| Langer 200141 | Data Physicians Query apparently relating to the TAnDEM50 trial |
| Maung and O’Shaughnessy 200446 | LAP monotherapy, not LAP + AI |
| Morris and Modi 200845 | TRA + tanespimycin, not TRA + AI |
| Novartis 200644 | Data Physicians Query apparently relating to the eLEcTRA52 trial |
| Piccart-Gebhart and Coleman 200242 | Data Physicians Query apparently relating to the TAnDEM50 trial |
| Ranganathan et al. 200747 | News article reporting on conference presentation for the TAnDEM50 trial |
| Stein 200443 | Data Physicians Query apparently relating to the EGF3000849 trial |
Studies included in the indirect comparisons analyses performed by the manufacturers
The following studies were included in the GlaxoSmithKline19 and/or Roche20 submissions, but excluded by the AG.
| Study | Submission(s) included in | Reason for exclusion |
|---|---|---|
| TARGET (outside North America)68 | GlaxoSmithKline19 and Roche20 | ANA vs TAM, not HER2+ |
| PO2576 | GlaxoSmithKline19 and Roche20 | LET vs TAM, not limited to first-line, not HER2+ |
| TARGET (North America)69 | GlaxoSmithKline19 and Roche20 | ANA vs TAM, not HER2+ |
| Rose et al. 200382 | Roche20 | ANA vs LET, second-line, not HER2+ |
| Campos et al. 200983 | Roche20 | ANA vs EXE, not limited to first-line, not HER2+ |
| 02084 | Roche20 | ANA vs fulvestrant (Faslodex®, AstraZeneca) not limited to first-line, not HER2+ |
| 02185 | Roche20 | ANA vs fulvestrant, not limited to first-line, not HER2+ |
| FIRST86 | Roche20 | ANA vs fulvestrant, HER2 status not clear |
| Jonat et al. 199671 | Roche20 | ANA vs megestrol acetate, not clear if first-line, not HER2+ |
| Buzdar et al. 199770 | Roche20 | ANA vs megestrol acetate, not limited to first-line, not HER2+ |
| Dombernowsky et al. 199878 | Roche20 | LET vs megestrol acetate, second-line, not HER2+ |
| Buzdar et al. 200177 | Roche20 | LET vs megestrol acetate, not limited to first-line, not HER2+ |
| EORTC87 | Roche20 | EXE vs TAM, not HER2+ |
| Chernozemsky et al. 200788 | Roche20 | EXE vs TAM, not HER2+ |
| EFFECT89 | Roche20 | EXE vs fulvestrant, not clear if first-line, not HER2+ |
| Kaufmann et al. 200090 | Roche20 | EXE vs megestrol acetate, not clear if first-line, not HER2+ |
Appendix 3 Final protocol
Glossary
- Biological therapy
- Treatments that use natural substances from the body, or drugs made from these substances, to fight cancer or to lessen the side effects that may be caused by some cancer treatments, e.g. trastuzumab.
- Chemotherapy
- Treatment with drugs that kill cancer cells.
- Endocrine therapy
- Treatment that adds, blocks or removes hormones. Also commonly known as hormonal or antioestrogen therapy.
- Human epidermal growth factor receptor 2 positive (HER2+)
- Over-expression of the HER2 receptor (HER2 receptors present in cancer cells).
- Human epidermal growth factor receptor 2 negative (HER2–)
- HER2 receptors are not present in cancer cells.
- Heterogeneity
- In statistics this means that there is between-study variation. If heterogeneity exists, the pooled effect size in a meta-analysis has no meaning as the presence of heterogeneity indicates that there is more than one true effect size in the studies being combined.
- Hormone receptor
- A receptor that binds a hormone.
- Hormone receptor positive (HR+)
- A tumour consisting of cells that express receptors for certain hormones, usually the oestrogen receptor (ER), i.e. oestrogen receptor positive (ER+), or the progesterone receptor (PgR), i.e. progesterone receptor positive (PgR+).
- Meta-analysis
- A quantitative method for combining the results of many studies into one set of conclusions.
- Oestrogen
- A general term for female steroid sex hormones that are secreted by the ovaries and responsible for typical female sexual characteristics.
- Oestrogen receptor (ER)
- Proteins that bind oestrogens.
- Oestrogen receptor positive (ER+)
- Cells that contain a receptor (protein) to which oestrogens can bind (attach).
- Oestrogen receptor negative (ER–)
- Cells that do not have a receptor to which oestrogens will bind.
- Quality-adjusted life-year (QALY)
- An index of survival that is weighted or adjusted by a patient’s quality of life during the survival period. QALYs are calculated by multiplying the number of life-years by an appropriate utility or preference score.
- Receptor
- A protein molecule embedded in a membrane to which a signal molecule (ligand), such as a pharmaceutical drug, may attach itself and which usually initiates a cellular response (although some ligands merely block receptors without inducing any response).
List of abbreviations
- AE
- adverse event
- AG
- Assessment Group
- AI
- aromatase inhibitor
- ANA
- anastrozole
- ASCO
- American Society of Clinical Oncology
- AUC
- area under the curve
- CBR
- clinical benefit rate
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CR
- complete response
- CRD
- Centre for Reviews and Dissemination
- CSR
- clinical study report
- ECOG
- the European Cooperative Oncology Group
- EGF30008
- efficacy and safety of lapatinib combined with letrozole trial
- eLEcTRA
- efficacy and safety of letrozole combined with trastuzumab trial
- EMA
- European Medicines Agency
- EQ-5D
- European Quality of Life-5 Dimensions
- ER
- oestrogen receptor
- ER+
- oestrogen receptor positive
- EXE
- exemestane
- FACT
- Functional Assessment of Cancer Therapy
- FDA
- the US Food and Drug Administration
- FISH
- fluorescence in situ hybridisation
- HER2
- human epidermal growth factor receptor 2
- HER2+
- human epidermal growth factor receptor 2 positive
- HER2–
- human epidermal growth factor receptor 2 negative
- HR
- hazard ratio
- HR+
- hormone receptor positive
- HR–
- hormone receptor negative
- ICER
- incremental cost-effectiveness ratio
- IHC
- immunohistochemistry
- IPCW
- inverse probability censoring weight
- ITT
- intention to treat
- i.v.
- intravenous
- LAP
- lapatinib
- LET
- letrozole
- LVEF
- left ventricular ejection fraction
- LYG
- life-year gained
- mBC
- metastatic breast cancer
- MS
- manufacturer’s submission/manufacturers’ submissions
- NICE
- National Institute for Health and Clinical Excellence
- ORR
- overall response rate
- OS
- overall survival
- PD
- progressive disease
- PFLY
- progression-free life-year
- PFS
- progression-free survival
- PgR
- progesterone receptor
- PgR+
- progesterone receptor positive
- PP
- per protocol
- PPLY
- post-progression life-years
- PPS
- post-progression survival
- PR
- partial response
- PSA
- probabilistic sensitivity analysis
- PSS
- Personal Social Services
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- RPSFT
- rank-preserving structural failure time
- SAE
- serious adverse event
- SE
- standard error
- TAM
- tamoxifen
- TAnDEM
- efficacy and safety of trastuzumab combined with anastrozole trial
- TRA
- trastuzumab
- TTP
- time to progression
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Peter Brocklehurst, Professor of Women’s Health, Institute for Women’s Health, University College London
-
Professor Jenny Donovan, Professor of Social Medicine, University of Bristol
-
Professor Jonathan Green, Professor and Acting Head of Department, Child and Adolescent Psychiatry, University of Manchester Medical School
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Freddie Hamdy, Professor of Urology, Head of Nuffield Department of Surgery, University of Oxford
-
Professor Allan House, Professor of Liaison Psychiatry, University of Leeds
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Expert Advisory Network
-
Professor Douglas Altman, Professor of Statistics in Medicine, Centre for Statistics in Medicine, University of Oxford
-
Professor John Bond, Professor of Social Gerontology & Health Services Research, University of Newcastle upon Tyne
-
Professor Andrew Bradbury, Professor of Vascular Surgery, Solihull Hospital, Birmingham
-
Mr Shaun Brogan, Chief Executive, Ridgeway Primary Care Group, Aylesbury
-
Mrs Stella Burnside OBE, Chief Executive, Regulation and Improvement Authority, Belfast
-
Ms Tracy Bury, Project Manager, World Confederation of Physical Therapy, London
-
Professor Iain T Cameron, Professor of Obstetrics and Gynaecology and Head of the School of Medicine, University of Southampton
-
Professor Bruce Campbell, Consultant Vascular & General Surgeon, Royal Devon & Exeter Hospital, Wonford
-
Dr Christine Clark, Medical Writer and Consultant Pharmacist, Rossendale
-
Professor Collette Clifford, Professor of Nursing and Head of Research, The Medical School, University of Birmingham
-
Professor Barry Cookson, Director, Laboratory of Hospital Infection, Public Health Laboratory Service, London
-
Dr Carl Counsell, Clinical Senior Lecturer in Neurology, University of Aberdeen
-
Professor Howard Cuckle, Professor of Reproductive Epidemiology, Department of Paediatrics, Obstetrics & Gynaecology, University of Leeds
-
Professor Carol Dezateux, Professor of Paediatric Epidemiology, Institute of Child Health, London
-
Mr John Dunning, Consultant Cardiothoracic Surgeon, Papworth Hospital NHS Trust, Cambridge
-
Mr Jonothan Earnshaw, Consultant Vascular Surgeon, Gloucestershire Royal Hospital, Gloucester
-
Professor Martin Eccles, Professor of Clinical Effectiveness, Centre for Health Services Research, University of Newcastle upon Tyne
-
Professor Pam Enderby, Dean of Faculty of Medicine, Institute of General Practice and Primary Care, University of Sheffield
-
Professor Gene Feder, Professor of Primary Care Research & Development, Centre for Health Sciences, Barts and The London School of Medicine and Dentistry
-
Mr Leonard R Fenwick, Chief Executive, Freeman Hospital, Newcastle upon Tyne
-
Mrs Gillian Fletcher, Antenatal Teacher and Tutor and President, National Childbirth Trust, Henfield
-
Professor Jayne Franklyn, Professor of Medicine, University of Birmingham
-
Mr Tam Fry, Honorary Chairman, Child Growth Foundation, London
-
Professor Fiona Gilbert, Consultant Radiologist and NCRN Member, University of Aberdeen
-
Professor Paul Gregg, Professor of Orthopaedic Surgical Science, South Tees Hospital NHS Trust
-
Bec Hanley, Co-director, TwoCan Associates, West Sussex
-
Dr Maryann L Hardy, Senior Lecturer, University of Bradford
-
Mrs Sharon Hart, Healthcare Management Consultant, Reading
-
Professor Robert E Hawkins, CRC Professor and Director of Medical Oncology, Christie CRC Research Centre, Christie Hospital NHS Trust, Manchester
-
Professor Richard Hobbs, Head of Department of Primary Care & General Practice, University of Birmingham
-
Professor Alan Horwich, Dean and Section Chairman, The Institute of Cancer Research, London
-
Professor Allen Hutchinson, Director of Public Health and Deputy Dean of ScHARR, University of Sheffield
-
Professor Peter Jones, Professor of Psychiatry, University of Cambridge, Cambridge
-
Professor Stan Kaye, Cancer Research UK Professor of Medical Oncology, Royal Marsden Hospital and Institute of Cancer Research, Surrey
-
Dr Duncan Keeley, General Practitioner (Dr Burch & Ptnrs), The Health Centre, Thame
-
Dr Donna Lamping, Research Degrees Programme Director and Reader in Psychology, Health Services Research Unit, London School of Hygiene and Tropical Medicine, London
-
Professor James Lindesay, Professor of Psychiatry for the Elderly, University of Leicester
-
Professor Julian Little, Professor of Human Genome Epidemiology, University of Ottawa
-
Professor Alistaire McGuire, Professor of Health Economics, London School of Economics
-
Professor Neill McIntosh, Edward Clark Professor of Child Life and Health, University of Edinburgh
-
Professor Rajan Madhok, Consultant in Public Health, South Manchester Primary Care Trust
-
Professor Sir Alexander Markham, Director, Molecular Medicine Unit, St James’s University Hospital, Leeds
-
Dr Peter Moore, Freelance Science Writer, Ashtead
-
Dr Andrew Mortimore, Public Health Director, Southampton City Primary Care Trust
-
Dr Sue Moss, Associate Director, Cancer Screening Evaluation Unit, Institute of Cancer Research, Sutton
-
Professor Miranda Mugford, Professor of Health Economics and Group Co-ordinator, University of East Anglia
-
Professor Jim Neilson, Head of School of Reproductive & Developmental Medicine and Professor of Obstetrics and Gynaecology, University of Liverpool
-
Mrs Julietta Patnick, Director, NHS Cancer Screening Programmes, Sheffield
-
Professor Robert Peveler, Professor of Liaison Psychiatry, Royal South Hants Hospital, Southampton
-
Professor Chris Price, Director of Clinical Research, Bayer Diagnostics Europe, Stoke Poges
-
Professor William Rosenberg, Professor of Hepatology and Consultant Physician, University of Southampton
-
Professor Peter Sandercock, Professor of Medical Neurology, Department of Clinical Neurosciences, University of Edinburgh
-
Dr Philip Shackley, Senior Lecturer in Health Economics, Sheffield Vascular Institute, University of Sheffield
-
Dr Eamonn Sheridan, Consultant in Clinical Genetics, St James’s University Hospital, Leeds
-
Dr Margaret Somerville, Director of Public Health Learning, Peninsula Medical School, University of Plymouth
-
Professor Sarah Stewart-Brown, Professor of Public Health, Division of Health in the Community, University of Warwick, Coventry
-
Dr Nick Summerton, GP Appraiser and Codirector, Research Network, Yorkshire Clinical Consultant, Primary Care and Public Health, University of Oxford
-
Professor Ala Szczepura, Professor of Health Service Research, Centre for Health Services Studies, University of Warwick, Coventry
-
Dr Ross Taylor, Senior Lecturer, University of Aberdeen
-
Dr Richard Tiner, Medical Director, Medical Department, Association of the British Pharmaceutical Industry
-
Mrs Joan Webster, Consumer Member, Southern Derbyshire Community Health Council
-
Professor Martin Whittle, Clinical Co-director, National Co-ordinating Centre for Women’s and Children’s Health, Lymington