
Notes
Article history
The research reported in this issue of the journal was commissioned by the HTA programme as project number 05/04/01. The contractual start date was in May 2007. The draft report began editorial review in April 2012 and was accepted for publication in July 2012. As the funder, by devising a commissioning brief, the HTA programme specified the research question and study design.The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Dr Derralynn Hughes has received funding for research and travel expenses, honoraria for lectures and consultancy projects or advisory boards from Shire Human Genetic Therapies (HGT ), Genzyme, Amicus, Actelion and Biomarin. Consultancy and advisory board work is administered via University College London (UCL ) business and used in part to fund research. Dr Stephen Waldek is a member of the Fabry Registry Board and the Fabry Expert Group and has received funding for research, and honoraria for lectures and consultancy on research studies from Shire HGT and Genzyme. He has also received research grants from Biomarin, Synageva and Amicus. Dr Atul Mehta has received research grants, honoraria for speaking at educational meetings, travel expenses and consultancy from Genzyme, Shire HGT and Actelion. Dr Robin Lachmann has received honoraria and consultancy fees from Genzyme, Shire and Actelion, and unrestricted educational grant funding from Genzyme and Shire HGT.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2012. This work was produced by Wyatt et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to NETSCC. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2012 Queen’s Printer and Controller of HMSO
Chapter 1 Background
Lysosomal storage disorders
Lysosomal storage disorders (LSDs) are a heterogeneous group of inherited metabolic disorders characterised by the accumulation of undigested macromolecules owing to lysosomal dysfunction. There are more than 70 LSDs, whose common feature is an error in the metabolism of lipids, glycoproteins or glycosaminoglycans (GAGs), usually due to a deficiency of a lysosomal enzyme or transport protein. Most of these disorders are autosomal recessively inherited while three, Fabry disease, Hunter disease [mucopolysaccharidosis type II (MPS II)] and Dannon disease, are X-linked.
As the function of the lysosomal enzymes is to remove unwanted material such as GAGs and sphingolipids from the cell, a deficit in any of these enzymes will result in progressive accumulation of material in affected organs and tissues. This results in an increase in the size and number of these organelles and ultimately in cellular dysfunction and organ failure. 1 Some forms of storage disorders are associated with defects of membrane proteins, errors in enzyme targeting or defective function of enzyme activators. 2 In others, such as Pompe disease, the accumulation in lysosomes and within cells has ‘downstream’ effects including a disturbance in autophagy. 3,4
The majority of these disorders are characterised by substantial neurological involvement, with developmental regression, seizures and learning difficulties being relatively common features. There is a wide variety of clinical manifestations, depending on the particular disorder. Most affected individuals have a reduced life expectancy and suffer considerable morbidity.
Individually, most LSDs occur with a birth prevalence of less than 1 : 100,000, with the estimated combined prevalence of approximately 1 in every 7000 to 8000 births. 5,6 Higher prevalence of specific lysosomal storage diseases is encountered in some populations, for example Gaucher disease and Tay–Sachs disease among Ashkenazi Jews and aspartylglucosaminuria, Salla disease and infantile neuronal ceroid lipofuscinosis in Finland. 6,7
The clinical picture of most LSDs is heterogeneous with age at onset and type and progression of symptoms varying substantially among individual patients suffering from the same disorder. The clinical course of these disorders is not easily predictable in an individual, especially in the later-onset disorders. 8 However, although there is some correlation between the specific mutation and the severity of the problems experienced by an individual, the genotype/phenotype relationship is variable. 1 There are a great number of mutations responsible for most LSDs and, although these have been largely elucidated, the molecular pathways through which the storage material causes cellular and organ pathology are still largely unknown, making phenotype prediction difficult.
In general, a correlation exists between residual enzyme activity and severity of disease manifestation. This correlation is loose, however, and does not allow prediction of the clinical course on an individual basis. In some LSDs external genetic or environmental factors have a marked influence on the flux through the defective pathway and therefore also have a major impact on disease manifestation. 9 The likelihood that a particular cell type is involved in storage accumulation is determined by the flux of the substrate (the metabolic demand) and the residual capacity of that cell type to carry out the catabolic reaction. Mutations causing a complete loss of enzyme activity result mostly in a severe phenotype of early onset. In contrast, alleles that still allow the expression of low amounts of residual enzyme activity are frequently associated with attenuated forms of disease. For patients with a missense mutation in a lysosomal enzyme gene, and therefore showing a relatively high residual enzyme activity, storage is likely to occur in fewer tissue types. Accumulation of storage compounds also depends critically on the origin of the substrate; it is difficult to predict which cell may be affected just from the enzyme activity. For example, some Fabry missense mutations affect the kidney whereas others affect the heart.
It is the heterogeneity in individuals’ residual degradative capacity that accounts for some LSDs manifesting as relatively benign non-neuropathic variants and others as devastating neuropathic variants. In the latter case storage is not restricted to cells in visceral tissues but also involves cells inside the brain. Many LSDs have traditionally been classified into subtypes, although it is increasingly recognised that most LSDs have a broad continuum of clinical severity and age at presentation5 rather than falling into clinically discrete forms.
The symptoms arising from these disorders are generally progressive and clinical diagnosis becomes easier with time for the severe forms,10 but attenuated forms can be undiagnosed until late adulthood. For the most part, diagnosis relies on observation of clinical features which raises clinical suspicion and can be confirmed by formal testing.
Summary of available treatments for lysosomal storage disorders
No definitive, curative treatment is yet available for any LSD. For most of the disorders, symptomatic management for specific problems is currently the only therapeutic option. For some LSDs it is possible to either augment the deficient enzyme, for example by enzyme replacement therapy (ERT), bone marrow transplantation (BMT), or enzyme enhancement therapy, or partially inhibit synthesis of the parent substrates by substrate reduction therapy (SRT). Treatment options are summarised in Table 1.
| Disease | Approximate prevalence (Australian data5) | ERT | SRT | Median age at diagnosis (range) |
|---|---|---|---|---|
| Gaucher disease | 1 : 57,000 |
Alglucerase Ceredase® Genzyme Corporation Licensed USA 1991 EU 1994 Imiglucerase Cerezyme® Genzyme Corporation Licensed USA 1994 EU 1997 Dose: i.v.; varies according to disease severity Velaglucerase alpha VPRIV® Shire licensed USA 2010 EU 2010 Dose: 60 U/kg i.v. every 2 weeks |
Miglustat Zavesca® Actelion Pharmaceuticals Licensed USA 2003 EU 2003 Dose: 100 mg t.i.d. orally For patients in whom ERT is not appropriate |
9.5 (0–73.2) years |
| Fabry disease | 1 : 117,000 |
Agalsidase beta Fabrazyme® Genzyme Corporation Licensed USA 2003 EU 2001 Dose: 1 mg/kg i.v. Agalsidase alpha Replagal® Shire Human Genetic Therapies Inc. Licensed EU 2001 Dose: 0.2 mg/kg i.v. |
28.6 (0–55.7) years | |
| MPS IH/MPS IHS/MPS IS | 1 : 88,000 |
Laronidase Aldurazyme® Genzyme Corporation Licensed USA 2003 EU 2003 Dose: 0.58 mg/kg i.v. weekly Prescribed for Hurler and Hurler–Scheie forms of MPS I and for people with the Scheie form who have moderate to severe symptoms. Risks and benefits of treating mildly affected patients with the Scheie form have not been established |
1.0 (0.3–29.1) years | |
| MPS II Hunter disease | 1 : 136,000 |
Idursulfase Elaprase® Shire Human Genetic Therapies Inc. Licensed USA 2006 EU 2007 Dose: 0.5 mg/kg i.v. weekly |
2.8 (0.0–22.0) years | |
| MPS VI Maroteaux-Lamy disease | 1 : 235,000 |
Galsulfase Naglazyme® Genzyme Corporation Licensed USA 2005 EU 2006 Dose: 1 mg/kg i.v. weekly |
1.4 (0–43.4) years | |
| Pompe disease (early and late onset) | 1 : 146,000 |
Alglucosidase alpha Myozyme® Genzyme Corporation Licensed USA 2006 EU 2006 Dose: 20 mg/kg i.v. every 2 weeks |
0.5 (0.1–55.0) years | |
| NPC | 1 : 211,000 |
Miglustat Zavesca® Actelion Pharmaceuticals Licensed EU 2009 Dose: 200 mg t.i.d. orally |
9.3 (0.1–37.7) years | |
| Late onset | 1 : 201,000 | SRT shows some promise in mouse models |
Miglustat Zavesca® Actelion Pharmaceuticals Currently unlicensed – used in clinical trials |
27.0 years |
| Tay–Sachs disease/Sandhoff disease | 1 : 384,000 |
Enzyme replacement therapy
Enzyme replacement therapy provides exogenous, recombinant enzymes to replace defective lysosomal enzymes. Patients receive the therapy via intravenous infusions either weekly or biweekly, and often in their own home.
In the mid-1960s de Duve11 and Brady and colleagues12 speculated that injection of exogenously purified enzyme into an affected person might provide therapeutic benefit. The first reported clinical investigation of ERT was the intravenous injection of hexosaminidase A that had been isolated from human urine, into an infant with Sandhoff disease, the O-variant form of Tay–Sachs disease. 13 A significant reduction in globoside occurred in the circulation shortly after infusing the enzyme, but none of the injected enzyme reached the brain and the patient experienced pyrexia following infusion. The authors reported that there was no change in the patient’s clinical condition.
The first administration of lysosomal enzyme in two patients with Fabry disease consisted of a single intravenous injection that caused brief, but significant, reductions in the globotriaosylceramide (Gb3) substrate in the blood. 14 The level of Gb3 in the circulation fell rapidly, but within 48–72 hours it had returned to the pre-infusion value in both patients. Further investigations of ERT for Fabry disease were delayed for many years until improved procedures were developed for the production and purification of larger quantities of α-galactosidase A (α-Gal A). 15
The first large-scale attempt at ERT occurred in patients with Gaucher disease, in the 1980s, using modified human enzyme purified from human placentas. Alglucerase (Ceredase®, Genzyme Corporation) was subsequently approved by the Food and Drug Administration (FDA) for the treatment of patients with Gaucher disease type 1 (GD1) in 1991 and subsequently by the European Medicines Agency (EMA) in 1994. Owing to supply issues and safety concerns around using human tissue, Genzyme Corporation then developed an alternative, recombinant form of the enzyme in Chinese hamster ovary (CHO) cells called imiglucerase (Cerezyme®, Genzyme Corporation). 16,17
During the last two decades, recombinant DNA techniques for manufacturing highly purified therapeutic enzymes have led to the practical application of ERT to other disorders that do not primarily affect the central nervous system (CNS). There are currently nine licensed ERTs in the UK for six LSDs:
-
Alglucerase (licensed in 1994), imiglucerase (licensed in 1997) and velaglucerase alpha (VPRIV®, Shire HGT Inc.; licensed in 2010) for non-neuropathic Gaucher disease (GD1).
-
Agalsidase beta (Fabrazyme®, Genzyme Corporation; licensed in 2001) and agalsidase alpha (Replagal®, Shire HGT Inc.; licensed in 2001) for Fabry disease.
-
Laronidase (Aldurazyme®, Genzyme Corporation; licensed in 2003) for mucopolysaccharidosis type I (MPS I).
-
Idursulfase (Elaprase®, Shire HGT Inc.; licensed in 2007) for MPS II.
-
Galsulfase (Naglazyme®, Genzyme Corporation; licensed in 2006) for mucopolysaccharidosis type VI (MPS VI).
-
Alglucosidase alpha (Myozyme®, Genzyme Corporation; licensed in 2006) for Pompe disease.
Enzyme replacement therapies for Niemann–Pick types A and B, alpha-mannosidosis, mucopolysaccharidosis IVA (Morquio A disease) and Wolman’s disease (cholesterol ester storage disease), as well as an alternative ERT for Pompe disease, are currently being developed and undergoing clinical trials.
In 2005, the HTA commissioned an examination of the evidence for the effectiveness and cost-effectiveness of ERTs for Gaucher disease, Fabry disease and MPS I. For all three conditions the reports suggested, on the basis of the limited data available, that there may be beneficial effects of ERT on symptom-related markers. 18,19 However, even assuming that ERT restores people with Gaucher disease to full health for their remaining lives, the incremental cost-effectiveness of ERT was calculated at more than 10 times above the usually accepted threshold of £20,000–30,000 for what constitutes ‘good value for money’ when using NHS resources to improve health. 20
Shortage of enzyme replacement therapies
In 2009, Genzyme Corporation became aware of problems in the bioreactors used to produce ERTs imiglucerase and agalsidase beta at their production site in Allston Landing in the USA. The bioreactors were contaminated with a calicivirus that is not known to cause disease in humans, but can attack the cells used to produce these medicines. The contamination had an impact on cell growth, affecting the quantity of the enzymes produced by the cells.
In June 2009 the company stopped production of new batches of imiglucerase and agalsidase beta for an extended period of time in order to sanitise the production facilities and conduct an investigation to prevent the contamination from recurring. A shortage of both medicines followed, and the company, in agreement with the EMA, recommended some temporary changes to the way imiglucerase and agalsidase beta were prescribed and used in Europe. A paper supporting these temporary guidelines was subsequently written by a European multidisciplinary expert panel. 21
The following recommendations were made:
-
For imiglucerase, patients at greatest need of treatment were recommended to receive this medicine but at a reduced dosage or at a reduced infusion frequency. Priority was given to infants, children and adolescents, and adults with severe, life-threatening disease progression. It was recommended that no patient should be treated at a dose less than 15 units per kilogram body weight every 2 weeks, or alternative treatment should be considered.
-
For agalsidase beta, priority was given to children and adolescents, and adult male patients, who were recommended to continue to receive agalsidase beta as one infusion every 2 weeks. Adult female patients, in whom the disease is less severe, would receive agalsidase beta at a reduced dose.
In reality, in the UK, some less severely affected patients had their imiglucerase treatment stopped during this period. After some time, and with permission from the EMA, velaglucerase alpha was prescribed for these patients in replacement of imiglucerase. In the UK, it was agreed that some centres would reduce use of agalsidase beta to help ease the situation. Patients with the more severe disease were maintained on agalsidase beta at full dose, although others were changed to agalsidase alpha. All patients were closely monitored while they received reduced or altered doses of imiglucerase or agalsidase beta, and reporting of side effects continued as normal.
In August 2010, Genzyme Corporation announced that there was insufficient agalsidase beta to meet the needs of the nearly 600 Fabry disease patients receiving the treatment in Europe, and the EMA recommended the use of alternate Fabry disease treatments, such as agalsidase alpha.
Finally, in February 2011, Genzyme Corporation announced the end of the imiglucerase shortage, but the company conceded that supplies of agalsidase beta are not likely to be restored until a new production facility in the USA gains FDA approval. At the time of writing, there continues to be a shortage of agalsidase beta.
Treatment and the blood brain barrier
Although there is evidence of the efficacy of ERT to treat non-neurological symptoms of LSDs, treatment of the neurological aspects of many LSDs requires the delivery of therapeutic proteins across the blood–brain barrier (BBB). Although substrate reduction therapies do appear to cross the BBB in small amounts (approximately 10%), the large recombinant enzymes used for treatment of LSDs do not appear to cross the BBB in sufficient amounts to be effective, thus limiting their use for treating disorders with neurological symptoms. 22
The BBB maintains a homeostatic environment in the CNS. 23 Attempts have been made to use proteins capable of being transported across the BBB as carriers. When such carrier proteins or peptides are joined to a therapeutic protein (such as ERT), they may ferry the attached protein cargo across the BBB. 24–26
Other options for bypassing the BBB are being explored, including the direct infusion of enzymes via an intrathecal route. 27,28
Substrate reduction therapy
Substrate reduction therapy attempts to address the failure of the lysosomal metabolic pathway by reducing the level of the substrate to a point where residual degradative activity is sufficient to prevent substrate accumulation in the lysosomes. At present, miglustat (N-butyldeoxynojirimycin, Zavesca®, Actelion Pharmaceuticals Ltd) is the only licensed SRT in the UK. Miglustat is an orally administered drug that inhibits ceramide glucosyltransferase, the enzyme that synthesises glucosylceramide. Glucosylceramide is the storage compound in Gaucher disease, and is also the first step in the synthesis of most glycosphingolipids that accumulate in Niemann–Pick disease. 29 Miglustat is currently a treatment option for mild to moderate GD1 and Niemann–Pick disease type C (NPC).
The efficacy and safety of miglustat has also been evaluated in other disorders. Miglustat therapy concomitant with ERT was evaluated in a randomised controlled trial (RCT) in patients with Gaucher disease type 3 (GD3), but there were no significant benefits on the neurological primary end points of vertical saccadic eye movement velocity. 30 Similarly, miglustat treatment did not lead to any measurable benefits in a cohort of patients with late-onset Tay–Sachs disease in a randomised, open-label, 12-month study with a 24-month extension. 31 More recently, evaluation of miglustat treatment in patients with mucopolysaccharidosis type III (MPS III) was conducted in a randomised, double-blind, placebo-controlled study. Again, miglustat treatment was not associated with any improvement or stabilisation in behaviour problems in patients with MPS III. 32
Many other potential substrate inhibitors have been identified, and some are currently being considered for their possible use as therapeutic options for Gaucher disease33,34 and MPS. 35–37
Haematopoietic stem cell transplantation
Allogenic BMTs and, more recently, transplantation of stem cells sourced from umbilical cord blood have been used as therapy for many LSDs that have significant brain involvement, although therapeutic efficacy has been variable. The principle of haematopoietic stem cell transplantation (HSCT) is the replacement of multipotent haematopoietic stem cell or blood with that of a donor. Following successful transplantation the donor-derived stem cells populate the bone marrow and provide a continuous endogenous source of the deficient enzyme. Haematopoietic cells, including activated lymphocytes, monocytes and precursors of microglia, have the potential to cross the BBB. 38,39 Because a proportion of lysosomal enzymes synthesised intracellularly are secreted and then internalised and directed to lysosomes, cross-correction of neighbouring cells lacking a normal lysosomal enzyme is possible. The release of enzymes into plasma, with subsequent uptake by enzyme-deficient cells throughout the body, may also occur, following a BMT. 40 There are, however, some major limitations, including the development of graft-versus-host disease, graft rejection and high mortality and morbidity rates owing to infection or regimen associated toxicity.
The first BMTs were done on patients with mucopolysaccharidosis type I subtype Hurler disease (MPS IH) and reported in the early 1980s41 and stem cell transplantation has since been carried out for at least 20 different LSDs. 23 Although transplantation has been relatively successful in some LSDs such as MPS I, other disorders, including MPS II (Hunter disease) and MPS III (Sanfilippo disease), have not responded well to such treatment. Results in other LSDs have been extremely variable and despite initial promising results in people with Gaucher disease, HSCT is no longer recommended in this disease because of the high associated mortality and morbidity and the effectiveness of ERT in treating this condition.
In general terms, tissues least susceptible to correction by a BMT are brain and bone. For conditions that do not primarily affect the CNS, transplantation can influence the natural history of the disorder. 42 In those disorders that primarily affect the CNS, such as infantile Tay–Sachs disease, Sandhoff disease or MPS III (Sanfilippo disease), transplantation does not appear to be effective in slowing down the progression of the disease. When carried out in individuals with some neurological involvement, BMT is reported to be least effective in addressing the skeletal component and best at addressing the neurological component of these disorders. Similarly, where there is significant skeletal impact on the disorder, such as in MPS IV (Morquio disease), stem cell transplantation does not lead to an improvement in growth or other skeletal features. 8,43
In MPS I, a transplant performed early enough in the disease process, generally before the age of 2 years, before extensive cerebral damage has occurred, can lead to improvement or stabilisation in neurocognitive development for most children, preventing progressive mental deterioration. In MPS IH, however, transplantation after the onset of significant neurological signs does not lead to an improvement of neurological function, and in most patients a steady loss of skills continues. 8 Furthermore, some clinical manifestations appear to be irreversible or continue to progress despite successful stem cell transplantation, causing residual disease burden in long-term survivors.
Pharmacological chaperone therapy
Some missense mutations and small in-frame deletions may lead to a misfolded protein without significantly affecting the active site. Mutations that affect accurate folding of the lysosomal enzymes prevent these proteins from reaching their final destination, directing them instead to the endoplasmic reticulum where they undergo degradation.
Pharmacological chaperone therapy is designed to enhance innate enzyme activity by stabilising the misfolded lysosomal enzyme, allowing it to pass through the Golgi apparatus onto the lysosomes. 22 Once appropriately folded, the enzymes can leave the endoplasmic reticulum and go to the Golgi apparatus, where they undergo further maturation before trafficking to the lysosomes. Once the mature enzymes reach the lysosomes, the chaperones dissociate and full or partial catalytic activity is restored. Phase I trials have reported elevation of plasma alpha galactosidase levels in healthy volunteers. 44 Phase III trials are now in progress in males and females with Fabry disease with a pharmacological chaperone (1-deoxygalactonojirimycin; DGJ; AmigalTM, Amicus Therapeutics). DGJ has been demonstrated to enhance trafficking of mutant α-Gal A to lysosomes of fibroblasts derived from persons with Fabry disease and increase enzyme activity while reducing Gb3 substrate in tissues of a transgenic/knockout animal model of Fabry disease.
Unfortunately, the use of chaperone therapy is restricted to patients with missense mutations who are shown to be responsive to the drug. For Pompe disease, such mutations are believed to occur in only 10–15% of the patient population. 45 These patients would, in theory, be candidates for a chaperone approach.
Chaperones are still in the early stages of clinical development, but have shown promising results in cell culture and animal models for a number of lysosomal disorders, including Gaucher disease, Fabry disease, Pompe disease, and MPS I36,46–49 and MPS III. 50
Gene therapy
Lysosomal storage disorders should be excellent candidates for therapy by gene transfer as they are generally well characterised, single-gene disorders that are not subject to complex regulation mechanisms. In addition, it has been demonstrated that restoration of enzyme activity to just 15–20% of the normal level provides restored clinical efficiency. 51 Moreover, because lysosomal enzymes are secreted and internalised by mannose-6-phosphate (M6P)-mediated endocytosis, any corrected cells should be able to have an effect on neighbouring tissue. Some progress has been made in developing gene therapy methods for treating CNS disease. A gene can be delivered into an organism via one of two ways – using the in vivo or the ex vivo techniques. The in vivo approach inserts genetically altered genes directly into the patient, while the ex vivo approach removes tissue from the patient, extracts the cells in question and genetically alters them before returning them to the patient.
Both in vivo and ex vivo gene therapy have been carried out successfully in many animal models. 52–54 However, although gene therapy studies performed in animal models are rather promising, many important issues regarding safety and efficacy of these strategies need to be addressed before large-scale clinical trials using viral vectors can be undertaken. Although gene therapy may allow constant delivery of a therapeutic protein to targeted organs and thus be able to overcome some of the problems seen in ERT, there is still a long way to go until gene therapy becomes a realistic therapeutic option for LSD patients. Clinical trials are currently underway for gene therapy in Pompe disease and metachromatic leucodystrophy.
Future prospects for lysosomal storage disorder therapy
Each therapeutic modality described above targets only one aspect of the complex pathophysiology of a LSD, and each has inherent strengths and weaknesses. It has been suggested, therefore, that the future of successful LSD therapy may lie in the combining of different types of treatment. 55 Potential strategies include the enhancement of enzyme activity by ex vivo gene therapy prior to cell transplantation, simultaneous use of stem cells and small molecule substrate inhibitors or HSCT, or ERT combined with agents that have the potential to transport across the BBB.
Summary of lysosomal storage disorders investigated in this study
Gaucher disease
Aetiology
Gaucher disease (OMIM 230800, 230900, 231000) is caused by deficient activity of the lysosomal enzyme glucosylceramidase, which is responsible for hydrolysing glucosylceramide to glucose and ceramide. The result of this deficiency is the accumulation of its undegraded substrate, glucosylceramide, and other glycolipids. The major substrate source is the breakdown of senescent blood cells and tissue debris; the incompletely metabolised glucosylceramide is subsequently stored in cells of monocyte/macrophage lineage of the reticuloendothelial system. In the CNS, glucosylceramide is believed to originate from the turnover of membrane gangliosides, although neuronal cell death may be the basis of neuropathic involvement. 56
Gaucher disease arises as a result of a mutation in the GBA gene. The abnormal alleles include missense and nonsense mutations, splice junction mutations, deletions and insertions of one or more nucleotides, and complex alleles resulting from gene conversion or recombination with the downstream pseudogene. 57,58 At least 200 GBA mutations have been identified.
Clinical features
Gaucher disease comprises a continuum of clinical findings from a perinatal lethal disorder to an asymptomatic form. The identification of three major clinical types (1, 2 and 3) and two other subtypes (perinatal-lethal and cardiovascular) is useful in determining prognosis and management. The major clinical types are delineated by the absence (type 1) or presence (types 2 and 3) of primary CNS involvement.
Gaucher disease type 1
Gaucher disease type 1 is further characterised by the presence of clinical or radiographic evidence of bone disease (osteopenia, focal lytic or sclerotic lesions, and osteonecrosis), hepatosplenomegaly, anaemia and thrombocytopenia.
Clinical or radiographic evidence of bone disease occurs in 70–100% of individuals with GD1. Bone disease ranges from asymptomatic osteopenia to focal lytic or sclerotic lesions and osteonecrosis. 59 Bone involvement, which may lead to acute or chronic bone pain, pathological fractures and subchondral joint collapse with secondary degenerative arthritis, is often the most debilitating aspect of GD1. 60 Acute bone pain manifests as ‘bone crises’ or episodes of deep bone pain that are usually confined to one extremity or joint61 and are often accompanied by fever and leucocytosis. Bone involvement in Gaucher disease may not correlate with the severity of haematological or visceral problems.
Although individuals with GD1 do not have primary neurological disease, neurological complications (spinal cord or nerve root compression) may occur secondary to bone disease (e.g. severe osteopenia with vertebral compression, emboli following long bone fracture) or coagulopathy (e.g. haematomyelia). 62
The spleen is enlarged (i.e. 1500–3000 cm3 in size, compared with 50–200 cm3 in the average adult) with resultant hypersplenism associated with pancytopenia (i.e. anaemia, leucopenia and thrombocytopenia). Infarction of the spleen can result in acute abdominal pain. Rarely, acute surgical emergencies may arise because of splenic rupture. 63 Liver enlargement is common, although cirrhosis and hepatic failure are rare.
Cytopenia is almost universal in untreated Gaucher disease. Anaemia, thrombocytopenia and leucopenia may be present simultaneously or independently. 64 The pattern of cytopenia in Gaucher disease is dependent on spleen status. Low platelet count may result from hypersplenism, splenic pooling of platelets or marrow infiltration or infarction. Anaemia may result from hypersplenism, haemodilution (e.g. pregnancy), iron deficiency or B12 deficiency and, in advanced disease, decreased erythropoiesis as a result of bone marrow failure from Gaucher cell infiltration or medullary infarction.
Acquired coagulation factor deficiencies include low-grade disseminated intravascular coagulation and specific inherited coagulation factor deficiencies (e.g. factor XI deficiency among Ashkenazi Jews). An investigation of Egyptian individuals with GD1 revealed a wide variety of coagulation factor abnormalities (fibrinogen, factors II, VII, VIII, X and XII). 65
The following pulmonary problems are associated with Gaucher disease:
-
interstitial lung disease
-
alveolar/lobar consolidation
-
pulmonary hypertension; this is well documented in individuals with liver disease and presumably the result of inability to detoxify gut-derived factors, which adversely affect the pulmonary endothelium with resultant pulmonary hypertension. Pulmonary hypertension can also occur in individuals with Gaucher disease without liver disease. 66
Gaucher disease types 2 and 3
In the past, Gaucher disease types 2 and 3 (GD2 and GD3) were distinguished by age at onset and rate of disease progression, but these distinctions are not absolute. Gaucher disease type 2 is classified by onset before age 2 years, limited psychomotor development and a rapidly progressive course with death by age 2–4 years. Individuals with GD3 may have onset before age 2 years, but often have a more slowly progressive course and may live into their third or fourth decade.
It is increasingly recognised that neuropathic Gaucher disease represents a phenotypic continuum, ranging from abnormalities of horizontal ocular saccades at the mild end to hydrops fetalis at the severe end. 67 Bulbar signs include stridor, squint and swallowing difficulty whereas pyramidal signs include opisthotonus, head retroflexion, spasticity and trismus. Oculomotor apraxia, saccadic initiation failure and opticokinetic nystagmus are common. 68 Oculomotor involvement may be found as an isolated sign of neurological disease in individuals with a chronic progressive course and severe systemic involvement (e.g. massive hepatosplenomegaly). Generalised tonic–clonic seizures and progressive myoclonic epilepsy have been observed in some individuals. 69,70 Dementia and ataxia have been observed in the later stages of chronic neurological disease.
The perinatal lethal form
The most severe type of Gaucher disease, the perinatal lethal form, causes severe or life-threatening complications starting before birth or in infancy. Patients may present clinically with ichthyosiform or collodion skin abnormalities or hydrops fetalis. 71 Other features of this form are hepatosplenomegaly, pancytopenia, arthrogryposis and distinctive facial features. 72 Most infants with this form of Gaucher disease survive for only a few days.
The cardiovascular form
The cardiovascular form occurs in individuals homozygous for the D409H mutation. These patients present with an atypical phenotype dominated by cardiovascular disease with calcification of the mitral and aortic valves. 73 Additional findings include mild splenomegaly, corneal opacities and supranuclear ophthalmoplegia. 74 Cardiopulmonary complications have been described with all the clinical subtypes, although varying in frequency and severity.
Epidemiology
A study from Australia reported a disease frequency of 1 : 57,000 (1.75 per 100,000);5 a similar study from the Netherlands reported 1.16 per 100,000. 6
Non-neuropathic GD1 is prevalent in the Ashkenazi Jewish population, with a disease prevalence of 1 : 855 and an estimated carrier frequency of 1 : 18. 75 There is now a very effective screening programme for this high-risk group and the actual birth prevalence may be a lot lower.
The prevalence of neuropathic Gaucher disease (GD2 and GD3) varies across ethnic groups, but appears to be higher among those who are not of European origin.
Gaucher disease is inherited in an autosomal recessive manner. The variants N370S, 84GG, IVS2 + 1G → A and L444P account for 90% of the Gaucher disease-causing alleles in Ashkenazi Jewish individuals with GD1 and for 50–60% of mutant alleles in non-Jewish individuals with GD1. Non-Jewish individuals with Gaucher disease tend to be compound heterozygotes with one common and one ‘rare’ mutation or a unique mutation.
Diagnosis
Gaucher disease is suspected in individuals with characteristic bone lesions, hepatosplenomegaly and haematological changes, or signs of CNS involvement. 76 Clinical findings alone are not diagnostic – diagnosis relies on demonstration of deficient glucosylceramidase enzyme activity in peripheral blood leucocytes or other nucleated cells. In affected individuals, glucosylceramidase activity is 0–15% of normal activity. Testing for the four common GBA alleles (N370S, L444P, 84GG, IVS2 + 1) has been included in panels specifically designed for carrier screening in the Ashkenazi Jewish population,77 but clearly such screening does not lead to 100% detection.
Affected individuals may first be suspected of having Gaucher disease following bone marrow examination for Gaucher disease-related manifestations (e.g. anaemia, thrombocytopenia and/or splenomegaly). 78
Management
Treatment of manifestations
Partial or total splenectomy for massive splenomegaly and thrombocytopenia was carried out on many patients in the pre-ERT era. Although useful in alleviating some of the problems in Gaucher disease, splenectomy may increase the likelihood of bone problems and other complications and, hence, is now carried out only very rarely. Supportive care for all affected individuals may include transfusion of blood products for severe anaemia and bleeding, analgesics for bone pain, joint replacement surgery for relief from chronic pain and restoration of function, oral bisphosphonates and calcium for osteopenia, and vitamin D if the patient is deficient.
Haematopoietic stem cell transplantation
Individuals with severe Gaucher disease, primarily those with chronic neurological involvement (GD3), have been shown to benefit from HSCT. 79 Successful engraftment can correct the metabolic defect, improve blood counts and reduce increased liver volume. However, the availability of ERT and the morbidity and mortality associated with transplantation have limited its use in recent years. 80
Substrate reduction therapy
Substrate reduction therapy may be indicated in symptomatic individuals with mild to moderate GD1 for whom ERT is not a therapeutic option (e.g. because of constraints such as allergy, hypersensitivity or poor venous access). SRT aims to restore metabolic homeostasis by limiting the amount of substrate precursors synthesised (and eventually subject to catabolism) to a level that can be effectively cleared by the mutant enzyme with residual hydrolytic activity. 81 A potential concern regarding the use of SRT is its non-specificity, i.e. the substrate whose production is blocked or limited is also a precursor in the formation of other glycosphingolipids (ganglio- and lacto-series).
Miglustat, an imino sugar that reversibly inhibits glucosylceramide synthase and reduces intracellular storage of glucosylceramide, is the first oral agent for the treatment of individuals with Gaucher disease.
In a 1-year, open-label study, 28 adults with GD1 (seven with previous splenectomies) from four international clinics, who were unable or unwilling to receive ERT, received 100–300 mg oral N-butyldeoxynojirimycin (miglustat) three times daily (t.i.d.) for 12 months. 82 The authors reported reduced organomegaly and small haematological improvements after 12 months’ therapy.
In an extension of the above open-label study,82 18 patients from four centres who had completed 12 months of treatment with miglustat continued on treatment for a further 2 years. 83 After 36 months on treatment, statistically significant improvements were reported in all the major efficacy end points (organ volume, haematological parameters), and no new cases of peripheral neuropathy were reported. Diarrhoea and weight loss, which were frequently reported during the initial 12-month study, decreased in magnitude and prevalence during the second and third years.
In a non-comparative, open-label study in adult patients with GD1 who were unwilling or unable to receive ERT, or who had discontinued ERT for at least 3 months, 10 patients (seven men and three women, mean age 46.3 years) received miglustat (100 mg t.i.d.) for 12 months, with the option of continuing treatment for a further 12 months. 84 The authors reported that in the seven patients who completed 24 months of treatment with miglustat, there was a significant decrease in liver and spleen volume at 6 and 18 months, with clinical improvement noted at 24 months. Bone involvement and platelet and haemoglobin (Hb) values remained stable, with no significant changes noted during the observation period. This study reports that adverse effects such as diarrhoea, abdominal pain, distension, weight loss, tremors and paraesthesiae decreased over the course of the trial.
In a separate study by Elstein and colleagues,85 36 clinically stable patients on ERT were enrolled in an initial 6-month, open-label trial that compared the effects of combined enzyme and miglustat therapy with miglustat therapy alone. Following the open-label trial, patients were given the choice of receiving either miglustat alone or in combination with ERT for a further 18 months. In practice, all patients received miglustat monotherapy. At the end of the extension period, all clinical end points indicated disease stabilisation.
Pastores and colleagues86 evaluated the effects of miglustat on bone manifestations and bone mineral density (BMD). They recruited 72 patients with GD1 who had previously participated in three multinational, open-label, clinical trials of miglustat. The data were collected over 2 years. Although 63% of patients reported bone pain when entering the study, 83% of patients reported no bone pain at 6, 12 and 24 months. The authors suggested the beneficial effect of miglustat on bone symptoms might be explained by its wide tissue distribution and by a direct effect on bone cells.
Giraldo and colleagues87 reported the results of an open-label 12-month study in which 25 patients with mild to moderate GD1 were treated with miglustat. Ten patients were therapy naive, whereas 15 had previously been treated with ERT. At 6 months, the previously untreated group showed improvements in Hb, platelet counts and chitotriosidase levels that were comparable with those in the ERT-treated group.
A more recent study evaluated the efficacy and safety of miglustat, concomitant with ERT, in patients with type GD3. 30 In the 24-month, Phase II, open-label, clinical trial, 30 patients were enrolled and were randomised 2 : 1 to receive miglustat or ‘no miglustat treatment’ for 12 months. The randomised phase was followed by an optional 12-month extension phase in which all patients received miglustat. All patients received ERT for the duration of the study. The primary efficacy end point was change from baseline in vertical saccadic eye movement velocity, and secondary end points included changes in neurological and neuropsychological assessments, pulmonary function, liver and spleen volumes, haematological and clinical assessments. No significant between-group differences in any of the neurological or neuropsychological evaluations were observed, but improvement in pulmonary function and a decrease in chitotriosidase levels were observed with miglustat plus ERT compared with ERT alone.
Enzyme replacement therapy
There are currently three licensed ERTs in the UK for non-neuropathic Gaucher disease (GD1) – alglucerase, imiglucerase and velaglucerase alpha. Imiglucerase is a modified form of human acid β-glucosidase and is produced by recombinant DNA technology using a mammalian CHO cell culture. Dosage is usually between 15 U/kg and 60 U/kg of body weight once every 2 weeks, but is individualised for each patient based on evaluation of all clinical manifestations of the disease. 88 Velaglucerase alpha is produced in a human fibroblast cell line by recombinant DNA technology, and the recommended dose is 60 U/kg administered every other week. 89 Imiglucerase was approved for use by the US FDA in 1994 and by the European Agency for Evaluation of Medicinal Products in 1997. Velaglucerase alpha was approved in both the USA and Europe in 2010.
There is a lack of good-quality randomised trials demonstrating the effectiveness of ERT in Gaucher disease. Individuals with GD1 report improved health-related quality of life (HRQoL) of life after 24–48 months of ERT. 90–92 After prolonged treatment, ERT reduces the rate of bone loss in a dose-dependent manner,93 improves bone pain and reduces bone crises. 94,95
Individuals with GD2 disease and pyramidal tract signs are not likely to respond to ERT, perhaps because the underlying neuropathology is cell death rather than lysosomal storage of glucosylceramide. 96 It has been suggested that people with GD2 and those with hydrops fetalis are not appropriate candidates for a BMT, ERT or SRT. 97,98
There is evidence suggesting that individuals with GD3 disease appear to derive some benefit from ERT, although long-term prognosis remains to be defined for this heterogeneous group. 99 Onset of progressive myoclonic seizures while on ERT appears to indicate a poor prognosis. 70 Brain stem auditory evoked responses have been reported to deteriorate in individuals with GD3 disease on ERT. 97
A Health Technology Assessment (HTA)-commissioned systematic review of the clinical effectiveness and cost-effectiveness of ERT for Gaucher disease18 identified 63 studies that fulfilled their inclusion criteria. Of these, only two were RCTs. 100,101 Grabowski and colleagues100 compared the effectiveness of the placental enzyme alglucerase with the recombinant enzyme imiglucerase, whereas the study by Schiffmann and colleagues101 was the only study identified that compared ERT with a concurrent, randomised control arm with no ERT.
Grabowski and colleagues100 conducted a double-blind, parallel-group randomised trial of 30 patients with GD1, randomised to receive alglucerase or imiglucerase, infused every 2 weeks for 9 months, both at a dose of 60 U/kg. Hb levels, platelet counts,and serum acid phosphatase and angiotensin-converting enzyme activities were monitored routinely, and liver and spleen volumes were assessed at baseline and after 6 and 9 months on treatment. No significant differences were found in the rate or extent of improvement in outcome measures in either treatment group, and no major immunological adverse events (AEs) occurred in either group.
Schiffmann and colleagues101 randomised 29 splenectomised Gaucher patients into three groups receiving vitamin D analogue (calcitriol), ERT (alglucerase or imiglucerase) or calcitriol plus ERT for 6 months. 101 All patients received alglucerase/imiglucerase for a further 18 months, such that the cumulative dose at 24 months was the same in each arm. The primary outcome measure was BMD of the lumbar spine measured by single-energy quantitative computerised tomography; the group hypothesised that ERT would increase bone density and that calcitriol might further enhance the skeletal response to this therapy. Other outcomes measured included bone marrow fat fraction, liver volume and Hb and platelet levels. The group reported that bone density declined in all groups with no significant difference between the groups. Conversely, Hb, platelet counts and liver volume significantly improved in all groups.
All the other studies identified by Connock and colleagues18 were considered to be of moderate quality at best and none had reliable comparator data. Although data were suggestive of benefit from ERT, the overall conclusion was that there was a paucity of high-quality evidence and that it was therefore difficult to reliably estimate whether or not any reported effects translated into improved patient well-being and survival, or an altered need for health services. The studies reported improvements in haematological parameters and in hepatomegaly and splenomegaly, with most parameters tending to return towards normal ranges in the majority of patients after a year or more of treatment. For organomegaly and Hb, the rates and extent of response were reported to have been greater the more severe the pre-ERT condition. Platelet levels were reported to have improved more slowly. For most people liver size was reduced to near 1.2 times that expected and the spleen was reduced by 5- to 10-fold. ERT was also reported to have a beneficial effect on bone crises and fracture rate, as well as on pain, although the quantitative evidence for these benefits was described by the authors of the HTA report as being ‘extremely weak’.
An analysis by Weinreb and colleagues,102 of physician-reported data from 1028 patients on the International Collaborative Gaucher Group Gaucher Registry, attempted to overcome the problems associated with small numbers of patients in previously reported studies. The authors describe the effects of 2–5 years of treatment with alglucerase or imiglucerase on specific manifestations of GD1, and reported short-term efficacy of this treatment. Among anaemic patients, Hb concentration was reported to increase to normal or near normal within 6–12 months, with a sustained response through to 5 years. In thrombocytopenic patients with intact spleens, the most rapid response occurred during the first 2 years, with slower improvement thereafter. The likelihood of achieving a normal platelet count decreased with increasing severity of baseline thrombocytopenia. In patients who had undergone splenectomy, platelet counts returned to normal within 6–12 months. In non-splenectomised patients, splenomegaly decreased 50–60%, but rarely to volumes below five times normal size, while hepatomegaly decreased by 30–40% during follow-up. In patients with pre-treatment bone pain or bone crises, 52% (67/128) were pain free after 2 years and 94% (48/51) reported no additional crises. The authors argued that ERT prevents progressive manifestations of Gaucher disease, and ameliorates Gaucher disease-associated anaemia, thrombocytopenia, organomegaly, bone pain and bone crises.
A similar analysis of data from 884 children with GD1 on the Gaucher Registry sought to determine the effects of long-term ERT with alglucerase or imiglucerase on haematological and visceral manifestations, linear growth and skeletal disease. 103 The authors reported that within 8 years of ERT, most clinical parameters studied became normal or nearly normal.
A 9-month, Phase I/II, open-label, single-centre trial and ongoing extension study was conducted to evaluate safety and efficacy of velaglucerase alpha. 104–106 Twelve symptomatic adult GD1 patients with intact spleens received velaglucerase alpha (60 U/kg) every other week for a total of 20 doses. An extension study was offered to patients completing the trial where step-wise dose reduction to 30 U/kg per infusion was instituted. Statistically significant improvements were reported in mean percentage change from baseline to 9 months and baseline to 48 months, for Hb, platelet counts, normalised liver volume and normalised spleen volume. 104 The effects of velaglucerase alpha on BMD were reported separately106 and indicated statistically significant improvements in lumbar spine and femoral neck BMD. No drug-related serious AEs and no antibodies were observed.
Regular intravenous infusions of the recombinant enzyme have been demonstrated to be safe and effective in reversing those features resulting from haematological and visceral (liver/spleen) involvement. 102,104 It is likely that end-stage histological changes (e.g. fibrosis, infarction) influence the response to ERT. Thrombocytopenia may persist in individuals with residual spleneomegaly and/or the presence of splenic nodules. 107 ERT appears to be well tolerated. Approximately 10–15% of individuals develop antibodies to infused imiglucerase, whereas antibody formation has been reported in 1% of persons receiving velaglucerase. In most cases these individuals remain asymptomatic. 108,109 Adverse effects (e.g. pruritus, hives) are relatively well controlled with premedication using antihistamines.
The only cost-effectiveness analysis performed to date is by Connock and colleagues,18 who conducted a cost-effectiveness analysis based on UK costs in 2006. In this analysis, even assuming that ERT restores people with Gaucher disease to full health for their remaining lives, the incremental cost-effectiveness of ERT is more than 10 times above the usually accepted threshold for what constitutes ‘good value for money’ when using NHS resources to improve health. The authors emphasise that owing to the weak research evidence base, extreme uncertainty surrounds these cost-effectiveness estimates. However, even with the most favourable possible assumptions their analysis concluded that the incremental cost-effectiveness of ERT appears prohibitive given current drug costs.
A summary of the main evidence from RCTs and other studies on the efficacy and safety of ERT and SRT for Gaucher disease is provided in Tables 2 and 3, respectively.
| Study | Population (size, age, gender, country) | Specified aims | Study design | Outcomes/measures | Follow-up | Findings |
|---|---|---|---|---|---|---|
| Grabowski and colleagues (1995)100 |
N = 30 patients with moderate or severe GD1 Alglucerase: 15 patients (four children and 11 adults) Imiglucerase: 15 patients (three children and 12 adults) Age range: 12–69 years 17 males, 13 females Ohio, USA |
To examine the therapeutic effects of placental enzyme alglucerase with the recombinant enzyme imiglucerase |
Double-blind, parallel-group randomised trial (Dose 60 U/kg body weight every 2 weeks for 9 months) |
Hb levels, platelet counts, serum acid phosphatase and ACE activities Liver and spleen volumes IgG antibody levels |
Hb levels, platelet counts, serum acid phosphate and ACE measured every 2 weeks for 9 months Liver and spleen volumes after 6 and 9 months IgG antibody levels every 3 months |
Significant improvements were reported in Hb levels, platelet counts, serum acid phosphatase, ACE activities and liver and spleen volumes in both treatment groups No significant differences were found in the rate or extent of change between the treatment groups although the incidence of IgG antibody formation was greater in the alglucerase group (40%) than in the imiglucerase group (20%) |
| Damiano and colleagues (1998)90 |
N = 212 GD1 patients on ERT Mean age: 45.1 ± 17 years 87 males, 125 females USA |
To estimate:
|
Cross-sectional, retrospective study |
SF-36 Health Survey Changes in physical, mental and general HRQoL since starting ERT |
N/A |
Patients with Gaucher disease on ERT reported an improvement in HRQoL that was greater than that reported by patients with other chronic diseases Those patients treated for up to 51 months scored below equivalent adults in the general population |
| Masek and colleagues (1999)91 |
N = 25 GD1 patients Mean age: 41.7 ± 11.5 years Age range: 26–73 years 11 males, 14 females USA |
To examine the effect of ERT, alglucerase, on HRQoL |
Prospective study (Information about Alglucerase dose was individualised by physicians and dose changes were unknown) |
QoL: SF-36 Psychological functioning: Symptom Checklist-90-Revised |
Every 6 months for 24 months |
Significant improvement in seven of eight SF-36 scale scores beginning at 18 months of therapy (p < 0.05–0.001) was reported. The SF-36 scale that showed improvement first was vitality (energy level and fatigue) at 6 months of therapy (p < 0.01) The SF-36 scales that showed the largest improvements were role-physical and social functioning (p < 0.001) after 24 months Within the study population it was reported that there was significant improvement in mood and global functioning and fewer psychological symptoms reported at 24 months of therapy |
| Schiffmann and colleagues (2002)101 |
N = 29 splenectomised Gaucher disease patients Age range: 27.7–48.5 years 16 males, 13 females USA |
To examine the effect of vitamin D supplementation on bone density in splenectomised Gaucher disease patients | RCT: three groups receiving (1) vitamin D analogue (calcitriol), (2) ERT or (3) calcitriol and ERT for 6 months (dose 60 IU/kg body weight every 2 weeks). The group with only calcitriol had alglucerase every 2 weeks during months 7–12 |
BMD of lumbar spine Bone marrow fat fraction, liver volume, Hb and platelet levels Biochemical skeletal markers: alkaline phosphatise and osteocalcin levels |
24 months |
Over the 2-year period, from the three groups, the total mean reduction of bone density was 8.76 ± 17.7 mg/cm3 (p = 0.06). In Group 1, the onset of bone loss was delayed by 6 months A significant increase in fat fraction after 6 months. The improvement in fat fraction was delayed by 6 months, pending inclusion of ERT into the treatment regimen Significant increase in bone-specific alkaline phosphatases and serum osteocalcin over time was reported. Hb and platelets significantly increased in all three treatment groups and liver volume decreased. The onset of improvement was delayed by 6 months in Group 1 |
| Weinreb and colleagues (2002)102 |
N = 1028 GD1 patients from the ICGG Gaucher Registry Mean age at first enzyme infusion: 30 ± 19 years 493 males, 535 females Worldwide |
To assess the effects of 2–5 years of treatment with alglucerase or imiglucerase on specific manifestations of GD1 | Analysis of physician-reported data within the Gaucher Register |
Hb levels, platelet counts, and liver and spleen volumes Symptoms and signs of skeletal involvement |
N/A |
It was reported that patients on ERT for as long as 5 years had completely or partially improved Gaucher disease-associated anaemia, thrombocytopenia, organomegaly, bone pain and bone crises However, some patients continue to have signs or symptoms of the disease |
| Wenstrup and colleagues (2007)93 |
N = 502 (160 untreated and 342 ERT-treated GD1 patients from the ICGG Gaucher Registry) Mean age at diagnosis: no ERT: 27 ± 15 years; with ERT: 21 ± 15 years 234 males, 268 females International |
To determine the effect of ERT on BMD | Analysis of physician-reported data within the Gaucher Register | BMD of lumbar spine | N/A | The BMD of patients with Gaucher disease treated with ERT increased to –0.12 (60 U/kg/2 weeks), –0.48 (30 U/kg/2 weeks) and –0.66 (15 U/kg/2 weeks) SD of the mean of the reference population after 8 years of ERT, approaching the reference population |
| Charrow and colleagues (2007)94 |
N = 2153 GD1 patients on the ICGG Gaucher Registry. Patients subsets: with bone pain (n = 244) or with bone crisis (n = 219) during the year prior to ERT Age at first infusions: less than 5 years, n = 227; 5–17 years, n = 480; 18+ years, n = 1446 998 males, 1155 females International |
To evaluate the effect of ERT in reducing bone crises and bone pain in GD1 patients | Retro analysis of longitudinal data for single cohort | Bone pain (in last month) and bone crisis (since last submission of data to registry) as recorded from Gaucher Registry CRFs (i.e. physician reported) | Follow-up 1, 2 and 3 years after ERT treatment |
It was reported that there were statistically significant and clinically meaningful reductions in both bone pain and bone crisis following ERT This reduction was maintained for at least the first 3 years of treatment |
| Weinreb and colleagues (2007)92 |
N = 32 previously untreated adult GD1 patients Median age: 42.5 years Age range: 12.0–70.0 years USA (seven centres) |
To determine if HRQoL (SF-36) is decreased in GD1 patients with skeletal manifestations and investigate the impact of ERT on perceived HRQoL |
4-year multisite, prospective, open-label, single cohort study (Dose 60 U/kg every 2 weeks for 2 years, then dose reduction to 45 or 30 U/kg if therapeutic goals were met) |
SF-36 (PCS and MCS calculated) | Baseline and after 1, 2, 3 and 4 years of ERT |
Baseline PCS scores lowest for patients with highest pain ratings and most advanced bone involvement; however, even this group improved on PCS after ERT Reductions in reported bone pain over the study, PCS and MCS increased significantly after 2 years of ERT and mean values within the normal range Statistically significant improvements in all eight SF-36 subscales after 2 years of treatments |
| Sims and colleagues (2008)95 |
N = 33 imiglucerase-naive patients with at least one skeletal manifestation Median age: 43.0 years Age range: 12.0–70.0 years 19 males, 14 females USA |
To evaluate the effectiveness of imiglucerase in treating skeletal manifestations of GD1 patients who had not previously received ERT |
Prospective multicenter non-randomised, open-label, single cohort study (Imiglucerase i.v. dose 60 U/kg/2 weeks) |
BMD z-scores (lumbar and femoral) bone pain, bone crisis (physician reported) Self-report bone pain (6-point scale), bone crisis outcomes Markers of bone metabolism: osteocalcin, bone-specific alkaline phosphatase, N–telopeptide crosslinks and D-PYD |
48 months |
Mean lumbar and femoral neck DXA z-scores and t-score improved progressively with treatment Reduction in patient-reported bone pain levels was reported by month 3. The number of patients reporting bone crisis decreased in the first 12 months and remained below baseline for the remainder of the study Biochemical markers for bone formation (osteocalcin and bone-specific alkaline phosphatase) increased in the first 12 months of the study and consistently remained above baseline values throughout; markers for bone resorption (N–telopeptide crosslinks and D-PYD) decreased Decreases the risk of skeletal events and increases lumbar spine and femoral neck BMD during the first 4 years of treatment |
| Andersson and colleagues (2008)103 |
N = 884 GD1 child patients on the ICGG Gaucher Registry that have intact spleens and are on ERT (imiglucerase and alglucerase) Aged less than 18 years 463 males, 421 females International |
To analyse clinical responses to ERT (alglucerase or imiglucerase) in a large international cohort of children with GD1 | Observational longitudinal analysis of registry data |
Height (z-scores) Hb levels (standardised) Platelet count, liver and spleen volumes, lumbar BMD z-scores and reported bone crises |
Follow-up monitoring until 8 years after first ERT infusion or 18 years of age for all measures except BMD which was analysed until 12 years after first infusion |
Height z-scores, normalised Hb levels, platelet counts, liver and spleen volumes and BMD z-scores all improved significantly over the 8-year treatment period to normal or near-normal 15/90 (16.7%) patients who had bone crises prior to ERT reported a bone crisis during follow-up monitoring; however; there were no further events after 2 years of ERT |
| Zimran and colleagues (2010)104 |
N = 12 symptomatic adult GD1 patients (treatment-naive or not having imiglucerase within previous 12 months) treated in single centre Mean age: 41.7 ± 17.3 years Age range: 19–70 years Five males, seven females Israel |
To evaluate the safety and efficacy of velaglucerase alpha |
A 9-month, Phase I/II open-label, single-centre trial and ongoing (39 month) extension study (Dose 60 U/kg per infusion every other week for a total of 20 doses: extension study dose step-wise reduction to 30 U/kg) |
Hb level Platelet count Liver and spleen volumes |
Hb level and platelet count at baseline and every 3 months Liver and spleen volumes at baseline, 6 and 9 months and 24, 33 and 45 months for the extension study |
Statistically significant improvements were reported in the mean percentage change from baseline to 9 months for Hb concentration (+19.2%), platelet counts (+67.6%), normalised liver volume (–18.2%) and normalised spleen volume (–49.5%) with statistically significant improvements from baseline in both Hb concentration and platelet counts achieved within the first 3 months Continuous improvement in clinical parameters was not throughout the extension study and normalisation of Hb was observed in all patients by 24 months |
| Elstein and colleagues (2011)106 |
N = 10 treatment-naive symptomatic GD1 patients Median age: 35 years Age range: 18–62 years Four males, six females Jerusalem, Israel |
Evaluation of BMD in patients with GD1 |
Long-term extension prospective clinical study (Dose 60 U/kg infusion every other week; extension after 1 year. Patients who achieved two or more therapeutic goals began step-wise reduction from 60 to 45 then 30 U/kg every other week) |
BMD for lumbar spine and femoral neck representing a therapeutic goal for skeletal pathology Clinical parameters (Hb concentration, platelet count, liver volume, spleen volume) representing therapeutic goals for anemia, thrombocytopenia, hepatomegaly and splenomegaly |
Assessments were taken at baseline, 9 months, 24 months and yearly thereafter, for a total of 69 months |
Velaglucerase alpha was associated with clinically meaningful and statistically significant lumbar spine and femoral neck BMD Improvements as early as month 24 (lumbar spine) and month 33 (femoral neck) despite dose reduction and significant baseline skeletal pathology All patients receiving ERT achieved all five long-term therapeutic goals within 4 years of starting treatment and after ≥ 2 years dose reduction |
| Elstein and colleagues (2011)105 |
N = 12 adults with symptomatic GD1 and intact spleens Eight patients went through to extension study Three males, nine females Israel |
To examine the impact of velaglucerase alpha for 9 months |
Open-label, Phase I/II study and extension study (Dose 60 U/kg infusion every other week; extension after 1 year. Patients who achieved two or more therapeutic goals began step-wise reduction from 60 to 45 then 30 U/kg every other week) |
Five clinical parameters (Hb concentration, platelet count, liver volume, spleen volume and BMD) were used to represent therapeutic goals for anaemia, thrombocytopenia, hepatomegaly, splenomegaly and skeletal pathology | Assessments were taken at baseline, 9, 24, 36, 48 and 57 months | All patients receiving ERT achieved all five long-term therapeutic goals within 4 years of starting treatment and after ≥ 2 years dose reduction |
| Study | Population (size, age, gender, country) | Specified aims | Study design | Outcomes/measures | Follow-up | Findings |
|---|---|---|---|---|---|---|
| Elstein and colleagues (2004)83 |
N = 18 GD1 patients not receiving ERT Age range: 22–69 years Four international clinics |
Examines the longer-term efficacy and safety data of miglustat |
Extension of the open-label study (Cox and colleagues82) for a further 2 years (Dose 100 mg t.i.d.) |
Liver and spleen volume Hb level, platelet count and chitotriosidase activity Safety data |
Liver and spleen volumes – every 6 months for 36 months Biochemistry markers and safety data – every 3 months for 36 months |
A statistically significant, progressive reduction in mean liver and spleen organ volumes were reported over 36 months of treatment (18% and 30%, respectively). Hb level and platelet counts became significantly increased from baseline. Plasma chitotriosidase activity decreased throughout the 3-year period during which it was measured No new cases of peripheral neuropathy were reported |
| Pastores and colleagues (2005)84 |
N = 10 adult GD1 symptomatic patients not on ERT Mean age: 46.3 ± 10.1 years Age range 32–62 years Three males, seven females USA |
To further assess previous observations of the effects of miglustat in adult patients with mild to moderate GD1 and to evaluate the tolerability and safety profile of miglustat |
Phase II, open-label, non-comparative study with a 12-month treatment period and an optional 12-month extension period (Dose miglustat 100 mg t.i.d. for 12 months with the option of continuing on treatment for another 12 months) |
Percentage change in liver volume and spleen volume Hb levels, platelet count, chitotriosidase activity and bone assessments Neurological assessment including MMSE |
Liver and spleen volumes, bone assessments: baseline and every 6 months for 12 months (possibly 24 months) Biochemical assays performed at baseline and every 3 months MMSE every 3 months for 12 months and then 6-month intervals during extension |
Significant decrease in liver and spleen volume at 6 and 18 months with clinical improvement noted over 24 months Hb levels remained stable throughout the study and the mean platelet counts were increased from baseline starting at 6 months, with a significant mean change in absolute platelet count at 12 months Reduction in chitotriosidase levels (N/S) No clinically significant deterioration in MMSE total score |
| Giraldo and colleagues (2006)87 |
N = 25 GD1 patients (10 therapy-naive) Mean age: 46.4 years Nine males, 16 females Spain |
To evaluate the efficacy, safety and tolerability of SRT (miglustat) in mild to moderate GD1 patients |
Prospective, open-label, 12-month study (Dose 100 mg t.i.d. miglustat orally) |
Hb levels, platelet count, chitotriosidase levels and CCL-18/PARC Bone marrow infiltration |
Baseline, 6 and 12 months Bone marrow infiltration was re-evaluated after 12 months |
At 6 months: previously untreated group showed improvements in Hb levels, platelet counts and chitotriosidase levels that were comparable with the ERT-treated group After 12 months, it was reported that there was more clearance of vertebral infiltration in the treatment-naive group |
| Pastores and colleagues (2007)86 |
N = 72 GD1 patients: 41 previous ERT and 31 treatment-naive Mean age: 41.2 ± 13.1 years 37 males, 35 females Three independent multinational centres |
Evaluation of the effects of miglustat on bone manifestations and BMD in GD1 patients |
Pooled analysis of data collected prospectively over 2 years from patients from three multinational, open-label, clinical trials [Dosing regimen 100 mg t.i.d. although dose increase was allowed (up to a maximum of 300 mg t.i.d.) and dose decreases were implemented in the case of AE] |
Skeletal manifestations of GD: bone pain, bone crisis, new fractures, AVN and BMD (lumbar and/or hip) |
Skeletal manifestations were assessed over a 2-year observation period All patients who had a baseline assessment and at least one follow-up measurement were included in the BMD analysis |
Lumbar spine and femoral BMD z-scores increased significantly from baseline at each time point assessed; improvement as early as 6 months after the initiation of miglustat therapy Femoral BMD significant increased in splenectomised Patients and femoral and lumbar BMD significantly improved in osteoporotic patients Reduction in bone pain (from 63% to 17%) after 2 years of treatment. These findings were comparable among all subgroups, including high-risk splenectomised patients No new bone crises, AVN or pathological fractures reported during 2 years of follow-up |
| Elstein and colleagues (2007)85 |
N = 36 clinically stable GD1 patients receiving ERT for a minimum of 2 years Mean age: 37.2 ± 13.4 years Age range: 17–69 years 16 males, 20 females Single centre in Israel |
To assess the tolerability and pharmacokinetic profile of miglustat in patients clinically stable on ERT and to compare their results with those patients who were taking medication either alone or in combination |
Randomised, open-label, parallel-group, Phase II, single-centre study, 6 months [Miglustat dose 100 mg capsule t.i.d. Extension phase of study (18 months) whereby patients chose to receive either SRT alone or in combination with ERT. In practice, all patients received SRT alone] |
Spleen and liver volumes, Hb level, platelet count, chitotriosidase QoL: SF-36, a modified Medical Outcomes Study Health Distress scores for assessing frustration, distress and anxiety Two surveys assessing symptoms and treatment-related issues |
Organ volumes: baseline and after 6 months Physical examination and biochemical tests performed monthly QoL questionnaires: 3 and 6 months following start of randomised therapy Extension study: six patients from each group provided pharmacokinetic profiles at month 1 and were monitored every 3 months for an additional 18 months |
At 6 months: no statistically significant treatment differences in organ volume or Hb between treatment groups Statistically significant differences in platelet count between miglustat and imiglucerase groups Statistically significant improvements in SF-36 mental component scores in patients receiving miglustat compared with those receiving imiglucerase or combination Miglustat group reported greater treatment convenience compared with the imiglucerase and combination groups 18-month extension study: all clinical end points indicated disease stabilisation |
| Schiffmann and colleagues (2008)30 |
N = 30 Gaucher disease type 3 patients Mean age: miglustat group, 10.4 ± 5.1 years; no treatment group, 9.9 ± 4.0 years 12 males, 18 females USA and England |
To evaluate the efficacy and safety of miglustat, concomitant with ERT, in patients with Gaucher disease type 3 patients |
Initial 12-month randomised controlled phase followed by an optional 12-month non-comparative extension phase Randomised 2 : 1 to receive miglustat or ‘no miglustat treatment’: all patients received ERT during the 24-month period Miglustat dose: if patient aged ≥ 12 years 200 mg t.i.d. or younger patients received a lower dosage adjusted to their body surface area |
Ophthalmological assessment: horizontal and vertical saccadic eye movements (Other neurological assessments including Purdue Pegboard Test and Wechsler Scale Trail Making Test) Liver and spleen organ volumes, pulmonary functioning and chitotriosidase activity |
Baseline, 12 months and optional 12-month extension |
There were no significant differences in vertical saccadic eye movement-β, horizontal saccadic eye movement-α and horizontal saccadic eye movement-β between the two treatment groups at any time point No significant between-group differences in any of the neurological or neuropsychological evaluations Organ volumes and haematological parameters remained stable in both treatment groups Improvements in pulmonary function and decrease of chitotriosidase levels were observed with miglustat plus ERT compared with ERT alone |
Fabry disease
Aetiology
Fabry disease (also known as Anderson–Fabry disease; OMIM 301500) results from absolute or partial deficiency of the enzyme α-Gal A and the subsequent progressive accumulation of Gb3 [also known as ceramide trihexoside (CTH)] in the lysosomes of cells in almost all organs of the body,110 particularly the heart, kidneys and nerve tissue. The exact mechanisms by which Gb3 storage leads to clinical features including organ dysfunction are unknown.
Clinical features
Clinical presentation of Fabry disease can be broadly divided into a classical form (occurring in males with less than 1% α-Gal A enzyme activity) a later-onset variant in males with greater than 1% α-Gal A activity, and heterozygous females.
Classical males
The classical form usually has its onset in childhood or adolescence with crises of severe pain in the extremities (acroparaesthesias), vascular cutaneous lesions (angiokeratomas), sweating abnormalities (hypohidrosis or anhidrosis), characteristic corneal and lenticular opacities, abdominal pain and proteinuria. Gradual deterioration of renal function to end-stage renal disease (ESRD) usually occurs in men in the third to fifth decade of life. In middle age, most males successfully treated for ESRD develop cardiovascular and/or cerebrovascular disease.
Angiokeratomas appear as clusters of individual punctate, dark red to blue-black angiectases in the superficial layers of the skin, present especially around the mouth, genitals and umbilicus. 111
Pain (acroparaesthesias) occurring as episodic crises of agonising, burning pain in the distal extremities most often begins in childhood or early adolescence and signal clinical onset of the disease. These crises are of variable length and are usually triggered by exercise, fatigue, emotional stress or rapid changes in temperature and humidity.
Anhidrosis, or hypohidrosis, is an early and almost constant finding. Hyperhidrosis may also occur. 112
Ocular involvement can include the cornea, lens, conjunctiva and retina. A characteristic corneal opacity, termed cornea verticillata and observed only by slit-lamp microscopy, is found in affected males and most heterozygous females. Aneurysmal dilatation and tortuosity of conjuctival and retinal vessels also occur. 113
Cardiovascular disease is present in most males with the classic phenotype by middle age. Left ventricular hypertrophy, valvular involvement and conduction abnormalities are early symptoms. Arrhythmias are common and range from a short PR interval through heart block to atrial fibrillation, supraventricular and ventricular tachycardias. Echocardiography demonstrates an increased thickness of the interventricular septum and the left ventricular posterior wall. 114 Angina, palpitations/arrhythmia and exertional dyspnoea have been found in 23–27% of males and 22–25% of females. 115 Hypertension, angina pectoris, myocardial ischaemia and infarction, congestive heart failure and severe mitral regurgitation are late signs.
Cerebrovascular manifestations result from multifocal small vessel involvement and can include thrombosis, transient ischaemic attacks (TIAs), basilar artery ischaemia and aneurysm, seizures, hemiplegia, hemianesthesia, aphasia, labyrinthine disorders or frank cerebral haemorrhage. 116 Rolfs and colleagues117 reported that in Germany, a GLA mutation was identified in 21 of 432 males (4.9%) and 7 of 289 females (2.4%) aged 18–55 years suffering cryptogenic stroke, while others have challenged this figure. 118
Gradual deterioration of renal function usually occurs in the third to fifth decade of life, although ESRD has been reported in the second decade. Glycosphingolipid deposition in intestinal small vessels and in the autonomic ganglia of the bowel may cause episodic diarrhoea, nausea, vomiting, bloating, cramping abdominal pain and/or intestinal malabsorption. 119
Other common clinical manifestations include high-frequency hearing loss, tinnitus and dizziness as a result of cranial nerve VIII involvement. 120 Progressive glycosphingolipid deposition in the lymphatic vessels and lymph nodes results in irreversible lymphoedema requiring treatment with compression hosiery, whereas several affected individuals have had pulmonary involvement, manifesting clinically as chronic bronchitis, wheezing or dyspnoea. 121 Depression, anxiety, severe fatigue and other psychosocial manifestations lead to decreased quality of life (QoL) in many affected individuals. 122
Late-onset Fabry disease
Individuals who present at a later age often lack the characteristic skin lesions and acroparaesthesias, but may have ESRD or cardiac manifestations, and are at risk of neurological complications such as stroke/TIA. Two organ-specific variants have been described where one organ is predominantly affected and there is mild involvement in other organs.
Cardiac variant males with cardiac disease are asymptomatic during most of their lives and present in the sixth to eighth decade of life with left ventricular hypertrophy, mitral insufficiency and/or cardiomyopathy, and mild to moderate proteinuria with normal renal function for age. Many have been diagnosed as a result of having hypertrophic cardiomyopathy (HCM). Screening of males with ‘late-onset’ HCM found that 6.3% who were diagnosed at or after 40 years of age and 1.4% of males who were diagnosed before 40 years of age had low α-Gal A enzyme activity and GLA gene mutations. 123 Cardiac variants may thus be underdiagnosed among affected individuals with cardiomyopathies.
Renal variants were identified among Japanese individuals on chronic haemodialysis in whom ESRD had been misdiagnosed as chronic glomerulonephritis. 124 The early symptoms of classic Fabry disease may not occur in individuals with the renal variant who develop renal insufficiency.
Heterozygous females
Heterozygous females typically have milder symptoms and a later age at onset than males. Severity may range from relatively asymptomatic throughout a normal lifespan to symptoms as severe as those observed in males with the classic phenotype. Manifestations include the characteristic cornea verticillata (70–90%) and lenticular opacities that do not impair vision; pain/tingling in the extremities (acroparethesias) (50–90%); angiokeratomas (10–50%) that are usually isolated or sparse; and hypohidrosis. In addition, females may have chronic abdominal pain and diarrhoea (Gupta and colleagues125). With advancing age, females may develop mild to moderate enlargement of the left heart (left ventricular hypertrophy) and valvular disease, myocardial ischaemia and infarction, cardiac arrhythmias, TIA, strokes and ESRD. 126–128 Excessive guilt, fatigue, occupational difficulty, suicidal ideation and depression have been noted. 129
Epidemiology
The cumulative prevalence of Fabry disease is estimated to be approximately 1 : 50,000 (2 per 100,000) males. 130 Recent reports have estimated population prevalences ranging from 1 : 80,000 (1.25 per 100,000) to 1 : 117,000 (0.85 per 100,000). 5,130 However, milder forms of the disease that present later in life and primarily affect the cardiovascular, cerebrovascular or renal system may be more common and may be underdiagnosed, suggesting that the true prevalence is likely to be higher.
Fabry disease is found among all ethnic, racial and demographic groups.
Diagnosis
The vast array of symptoms seen in Fabry disease can mean that it is difficult to diagnose. In males, the most reliable method for the diagnosis of Fabry disease is the demonstration of deficient α-Gal A enzyme activity in plasma, isolated leucocytes and/or cultured cells. However, in females, measurement of α-Gal A enzyme activity is unreliable. Although demonstration of decreased α-Gal A enzyme activity is diagnostic of the heterozygote state, many females have normal α-Gal A enzyme activity. Molecular genetic testing is therefore the most reliable method for the diagnosis of carrier females.
Inheritance
Unlike most other LSDs, Fabry disease is inherited in an X-linked manner. A female heterozygote has a 50% chance of transmitting the GLA mutation in each pregnancy. An affected male transmits his mutation to all of his daughters.
Management
Symptomatic management
Carbamazepine or gabapentin may be used to reduce the pain caused by the acroparaesthesias, whereas the addition of angiotensin-converting enzyme (ACE) inhibitors and angiotensin-receptor blockers is reported to slow the decline of renal function. 131
Enzyme replacement therapy
Two enzyme replacement products using recombinant or gene-activated human α-galactosidase A enzyme are available: agalsidase alpha and agalsidase beta. Agalsidase alpha is produced using a genetically engineered human fibroblast cell line and has a recommended dose of 0.2 mg/kg body weight administered once every 2 weeks as an intravenous infusion. 132 Agalsidase beta is produced using a CHO cell line and has a recommended dose of 1 mg/kg body weight administered once every 2 weeks as an intravenous infusion. 133 Both therapies were approved in 2001 by the European Agency for Evaluation of Medicinal Products and agalsidase beta was approved by the FDA for use in the USA in 2003.
A 2006 HTA-commissioned systematic review of effectiveness and cost-effectiveness,19 which included studies of both forms of ERT, identified three randomised placebo-controlled trials, all described below,134–136 (n = 70; duration 5–6 months) and 11 observational, non-comparative, before-and-after studies (n = 493; duration up to 24 months). The three controlled trials134–136 included a total of 27 patients on agalsidase beta and 21 patients on agalsidase alpha. The studies, summarised below,134–136 were small, of short duration and looked at different outcome measures, making direct comparisons difficult. There was no convincing evidence for an effect on neurological events including the risk of TIA or stroke. The review concluded that overall results suggested some beneficial effect of ERT on measures of pain and cardiovascular function and an apparent stabilisation of renal function.
Eng and colleagues137 evaluated the safety and effectiveness of agalsidase beta in a multicentre, randomised, placebo-controlled, double-blind study. Fifty-eight, predominantly male, patients were treated biweekly for 20 weeks with agalsidase beta (11 doses of 1 mg/kg). The primary efficacy end point in this study was the percentage of patients in each group who were free of microvascular endothelial deposits of Gb3 in renal-biopsy specimens after 20 weeks of treatment. This end point was reached by 20 of the 29 patients (69%) in the treatment arm, compared with none of the 29 patients in the placebo group (p < 0.001). Individuals in the treated group also had decreased microvascular endothelial deposits of Gb3 in the skin (p < 0.001) and heart (p < 0.001) compared with those receiving the placebo. Similar results were reported for patients enrolled in the open-label study extension; renal scores were maintained or further decreased for 95% of those who received 6 months of open-label treatment. The authors reported that mild to moderate infusion reactions (i.e. rigors and fever) were more common in the agalsidase beta treated group than in the placebo group.
Another trial using agalsidase beta134 measured the effect of therapy on neuropathic pain in the absence of neuropathic pain medications. Twenty-six hemizygous male patients aged ≥ 18 years were treated with 0.2 mg/kg biweekly for 6 months (12 doses in total). Mean neuropathic Pain Severity Score [measured using question three of the Brief Pain Inventory (BPI)] declined in patients treated with agalsidase beta, whereas no significant change was measured in the placebo group. The authors also reported a reduction in plasma glycosphingolipid levels, a significant improvement in cardiac conduction and a significant increase in body weight in patients treated with agalsidase beta.
In a RCT that was published after the publication of the Connock and colleagues review,19 the safety and efficacy of ERT on the cardiac manifestations of Fabry disease was assessed. Fifteen hemizygous adult male patients (median age 37 years) with Fabry disease were randomised to receive placebo (n = 8) or ERT with agalsidase alpha (n = 7). 138 Patients in the treatment arm were given 0.2 mg/kg every 2 weeks for 6 months and all subjects subsequently received open-label ERT for an additional 24 months. Left ventricular mass (LVM), as measured by magnetic resonance imaging (MRI), was significantly reduced following 6 months of treatment with agalsidase alpha compared with placebo (p = 0.041). A mean 20% reduction in myocardial Gb3 content, as assessed by serial transvenous endomyocardial biopsies, was demonstrated over the 6 months of ERT compared with a mean 10% increase in patients receiving placebo (p = 0.42).
A separate analysis of the effect of agalsidase alpha on hearing loss in the Fabry disease patients enrolled in the above study was conducted135 and was included in the Connock and colleagues review. 19 The effect of hearing loss was measured by pure-tone audiometry, impedance audiometry and otoacoustic emission. In patients who received treatment for a further 24 months, ERT with agalsidase alpha was reported to reverse the hearing deterioration above baseline. This improvement occurred in 15 out of 20 ears and did not appear to be dependent on the initial severity of hearing loss.
More recently, a Phase IV, randomised, double-blind, placebo-controlled trial of agalsidase beta reported a reduction in clinical progression of the disease in treated patients with respect to renal, cardiac and CNS events. 139 Eighty-two patients attending 41 referral centres in nine countries were randomised 2 : 1 to receive agalsidase beta compared with placebo and the primary end point was the time to first clinical event (renal, cardiac or cerebrovascular event, or death). Thirteen (42%) of the 31 patients in the placebo group and 14 (27%) of the 51 patients in the agalsidase beta group experienced clinical events during the treatment period.
These findings are consistent with studies that have reported that agalsidase beta improves or stabilises cardiac function140 and renal function. 141
In other uncontrolled studies, treatment with agalsidase alpha has also been reported to stabilise renal function, cardiac abnormalities and pain,142 improve QoL,143 reduce abnormal cerebral perfusion and resolve abnormally increased cerebrovascular blood flow. 144–146
There is little evidence for the efficacy of agalsidase beta or agalsidase alpha in children as there are only a few published studies and no RCTs. An open-label study of 14 male and two female paediatric patients (aged 8–16 years) treated with agalsidase beta at 1 mg/kg every 2 weeks for 49 weeks reported a reduction in Gb3 accumulation in dermal endothelium. 147 Schiffmann and colleagues148 reported the results of a 4-year prospective, open-label, clinical trial of agalsidase alpha in one female and 16 male patients (aged 7.3–18.4 years) with Fabry disease. The authors reported improvement of Fabry disease-related features including reduced plasma Gb3 levels, improved pain (as assessed by the BPI) and improved heart rate variability.
The efficacy and tolerability of the two agalsidase preparations administered at identical protein dose was compared in a open-label RCT in the Netherlands and Norway. Thirty-four Fabry patients aged 24–76 years with severe renal impairment proteinuria greater than 1 g/l, treated with either agalsidase alpha or agalsidase beta at 0.2 mg/kg biweekly, showed no difference in the reduction of LVM (the primary end point) or other disease parameters after 12 or 24 months of either treatment. 149 The authors reported that treatment failure occurred frequently in both groups and seemed to be related to age and severity of disease pre treatment.
Enzyme replacement therapy appears to be well tolerated by patients with Fabry disease. For example, a European study150 indicated that ERT was safe and well tolerated in 15 severely affected female heterozygotes that were treated for up to 55 weeks. In addition, the safety of both agalsidase alpha and beta in young children has been demonstrated in several open-label, clinical trials. 147,148,151,152 The development of antibodies to treatment has been reported with both preparations, although there is no clear evidence that this affects clinical efficacy. 153 Deegan154 recently published a summary of the immune response of Fabry patients to ERT.
The more common side effects of both agalsidase preparations are infusion reactions including headache, flushing (redness), nausea, chills, fever, general pain or discomfort and tiredness. Serious anaphylactic and allergic reactions are less common.
Connock and colleagues19 conducted a cost-effectiveness analysis of ERT in Fabry disease. The conclusions were similar to those found in the review of ERT in Gaucher disease. The efficacy data were acknowledged to be poor, resulting in considerable uncertainty around all estimates. Even where the model was based on the most favourable possible assumptions, applying conventional thresholds of societal willingness to pay for health gains for the UK NHS [£30,000 per quality-adjusted life-year (QALY)] and current treatment prices, the authors concluded that ERT (either agalsidase alpha or agalsidase beta) for Fabry disease was highly unlikely to be considered to be cost-effective.
A summary of the main evidence from RCTs and other studies on the efficacy and safety of agalsidase alpha and agalsidase beta for Fabry disease is presented in Table 4.
| Study | Population (size, age, gender, country) | Specified aims | Study design | Outcomes/measures | Follow-up | Findings |
|---|---|---|---|---|---|---|
| Eng and colleagues (2001)136 |
N = 15 All males Age range: 18–45 years USA |
To evaluate the safety and pharmacokinetics of agalsidase beta infusions |
Multidose, open-label, dose escalation study Patients were sequentially enrolled into one of five agalsidase beta dosing regimens for five doses with three patients per group: Group A, 0.3 mg/kg biweekly; Group B, 1.0 mg/kg biweekly; Group C, 3.0 mg/kg biweekly; Group D, 1.0 mg/kg every 48 hours; and Group E, 3.0 mg/kg every 48 hours |
Medical and safety evaluations: medical history, physical examinations, vital signs, routine serum and urine chemistries, haematology indices and ECG Plasma and tissue α-Gal A activities, and plasma and tissue Gb3 levels Echocardiograms and renal MRIs Collection of biopsies of liver and skin Pain assessment using the Short Form McGill Pain Questionnaire |
At baseline, prior to each infusion and after infusion five Plasma levels measured at baseline, before each infusion, and either 14 days (for the every-48-hour groups) or 21–28 days (for the biweekly groups) after infusion five |
The primary finding was that infused agalsidase β safely and effectively cleared the accumulated Gb3 from endothelium of the liver, skin, heart and kidney – major sites of pathology in classical Fabry disease Pre- and post-treatment ECGs, echocardiograms and renal MRIs were unchanged Compared with the pre-treatment baseline results, the ‘overall pain’ and ‘present pain intensity’ scores were significantly improved after five infusions at all doses No significant changes in blood indices or blood and urine chemistries |
| Schiffmann and colleagues (2001)134 |
N = 26 hemizygous males with AFD Age range: 16–56 years USA |
To evaluate the safety and efficacy of i.v. agalsidase beta for Fabry disease |
RCT (double-blinded, placebo-controlled) Placebo or agalsidase beta at 0.2 mg/kg, twice a week managed i.v. for 6 months (12 doses in total) |
Effect of therapy on neuropathic pain while without neuropathic pain medications, measured by question 3 of the BPI Kidney function: glomeruli mesangial widening Inulin clearance, creatinine clearance, plasma glycosphingolipid levels, cardiac conduction and body weight |
6 months |
Mean (SE) BPI neuropathic Pain Severity Score declined from 6.2 (0.46) to 4.3 (0.73) in patients treated with α-Gal A compared with no significant change in the placebo group (p = 0.02) Mean (SE) pain-related QoL declined from 3.2 (0.55) to 2.1 (0.56) for patients receiving α-Gal A compared with 4.8 (0.59) to 4.2 (0.74) for placebo (p = 0.05) In the kidney, glomeruli with mesangial widening decreased by a mean of 12.5% for patients receiving α-Gal A compared with a 16.5% increase for placebo (p = 0.01) Mean inulin clearance decreased by 6.2 ml/minute for patients receiving α-Gal A compared with 19.5 ml/minute for placebo (p = 0.19) Mean creatinine clearance increased by 2.1 ml/minute (0.4 ml/second) for patients receiving α-Gal A compared with a decrease of 16.1 ml/minute (0.3 ml/second) for placebo (p = 0.02) In patients treated with α-Gal A, there was an approximately 50% reduction in plasma glycosphingolipid levels, a significant improvement in cardiac conduction and a significant increase in body weight |
| Eng and colleagues (2001)137 |
N = 58 Mean age: placebo group: 28.4 years; ERT group: 32.0 years 56 males, two females International, multicentre |
To evaluate the safety and effectiveness of agalsidase beta |
Phase III, double-blind RCT Patients received placebo or agalsidase beta 1 mg/kg i.v. every 2 weeks for 20 weeks Thereafter, all patients received agalsidase beta in an open-label extension study for 6 months |
Primary end point was percentage of patients in whom renal microvascular endothelial deposits of Gb3 were cleared Evaluation of the histological clearance of microvascular endothelial deposits of Gb3 in the endomyocardium and skin Changes in the level of pain and the QoL |
Follow-up at 14 and 20 weeks Individual scores for the kidney-, heart- and skin-biopsy specimens and composite scores for all three compared at baseline and after the week-20 infusion Extension study: 6 months |
It was reported that 20 of the 29 patients in the agalsidase beta group had no microvascular endothelial deposits of Gb3 after 20 weeks compared with none of the 20 patients in the placebo group (p < 0.001) Patients in the agalsidase beta-treated group also had significantly decreased microvascular endothelial deposits of Gb3 in skin and heart These treated patients also had significant improvements in two components of the SF-36 (physical role and emotional role), whereas patients in the placebo group had significant improvement in the physical role and body-pain components of the SF-36 After 6 months of open-label therapy, all patients in the former placebo group and 98% of patients in the former agalsidase beta group who had biopsies had clearance of microvascular endothelial deposits of Gb3 |
| Moore and colleagues (2002)146 |
N = 26 male hemizygous patients Age range: 19–47 years Mean age: 33.7 ± 8.1years ERT: n = 14 Placebo: n = 12 USA |
To examine the functional blood flow response of the brain after visual stimulation (reversing checkerboard pattern) |
RCT (double-blind, placebo-controlled) (i.v. dose of agalsidase alpha every 2 weeks for 6 months) |
Functional blood flow response of the brain after visual stimulation by assessing change in CBF | 6 months |
ERT was reported to significantly lower the absolute rCBF during visual stimulation in the treatment group compared with the placebo (p < 0.001) ERT reverses the exaggerated cerebrovascular response in Fabry disease |
| Waldek (2003)140 |
N = 1 female Age: 46 years UK |
To examine response to agalsidase beta |
Case study from Phase III and Phase III extension study (Dose 1 mg/kg of body weight every 2 weeks for 6 or 12 months) |
Electrocardiographic abnormalities using PR intervals Cardiac Gb3 levels |
12 months | Within the first 6 months of treatment the patient’s PR interval had returned to normal value and the Gb3 level in cardiac tissue had declined |
| Hajioff and colleagues (2003)135 |
N = 15 hemizygous male Fabry patients Median age: 37 years Age range: 25–49 years |
To examine the effect of agalsidase alpha on hearing loss in Fabry patients |
RCT Randomised to receive placebo (n = 8) or ERT (n = 7) for 6 months; all patients received open-label ERT for an additional 24 months (Dose 0.2 mg/kg over 40 minutes every 2 weeks) |
Pure-tone audiometry, impedance audiometry and otoacoustic emissions Cardiac (LVM, electrocardiography and myocardial Gb3 content), renal (glomerular filtration rate) and metabolic (plasma and urine Gb3 content) functions were assessed |
Performed at baseline, 6, 18 and 30 months |
Deterioration in the high-frequency sensorineural hearing loss occurred in the placebo and treatment groups during the first 6 months of the study However, for the patients who received treatment for a further 24 months it was reported that the hearing deterioration was reversed above baseline level This improvement occurred in 15 out of 20 ears and did not appear to be dependent on the initial severity of hearing loss |
| Hoffmann and colleagues (2004)143 |
N = 1 female Age: 34 years Germany |
To examine the effect of ERT on cardiomyopathy, renal function and autonomous nervous function | Case report |
Gastrointestinal symptoms severity, including diarrhoea, constipation, abdominal pain and appetite Body weight and BMI |
Remarkable improvement of gastrointestinal symptoms, body weight, BMI, physical activity and overall well-being | |
| Beck and colleagues (2004)142 |
N = 545 (n = 314 receiving ERT) 281 males, 264 females 60 centres from 11 European countries |
To monitor the long-term efficacy and safety of ERT with agalsidase alpha on renal function, heart size, pain and QoL | Cohort study of the Fabry Outcome Survey, which is a European outcomes database for patients with Fabry disease who are receiving, or are candidates for, ERT with agalsidase alpha | Renal function (assessed by eGFR), heart size (assessed by echocardiography), pain (assessed by the BPI) and QoL (assessed by the EQ-5D) | Every 3 months for the first year of ERT treatment and every 6 months thereafter |
It was reported that treatment with agalsidase alpha stabilised renal function in patients with a mild or moderate deterioration in renal function at baseline, reduced left ventricular size in patients who had an enlarged heart at baseline, and improved pain scores and QoL These improvements were similar in hemizygous men and heterozygous women with Fabry disease |
| Banikazemi and colleagues (2007)139 |
N = 82 adults with kidney dysfunction from Fabry disease Mean age: agalsidase beta group 46.9 years; placebo group 44.3 years 72 males, 10 females 41 referral centres in 9 countries in North America and Europe |
To see whether or not agalsidase beta delays the onset of a composite clinical outcome of renal, cardiovascular and cerebrovascular events, and death in patients with advanced Fabry disease |
Randomised (2 : 1 treatment-to-placebo randomisation) double-blind, placebo-controlled trial [Dose i.v. infusion of agalsidase beta (1 mg/kg of body weight) (n = 51) or placebo every 2 weeks (n = 31) for up to 35 months] |
Primary end point: the time to first clinical event (renal, cardiac or cerebrovascular event, or death) Serum creatinine level, proteinuria, ration of urinary albumin to urinary creatinine, eGFRs |
Serum measured every 4 months All baseline measures were repeated every 12 weeks, except for echocardiography, head MRI and exercise tolerance which were repeated every 24 weeks for up to 35 months |
Thirteen (42%) of the 31 patients in the placebo group and 14 (27%) of the 51 patients in the agalsidase beta group experienced clinical events with deteriorating renal function being the most frequent The authors reported that compared with placebo, agalsidase beta reduced the frequency and delayed the time to clinical events Subgroup analyses reported larger treatment effects in patients with baseline eGFRs greater than 55 ml/minute/1.73 m2 [HR 0.19 (95% CI 0.05 to 0.82); p = 0.025] compared with 55 ml/minute/1.73 m2 or less [HR 0.85 (95% CI 0.32 to 2.3); p = 0.75] |
| Germain and colleagues (2007)141 |
N = 58 patients that had previously participated in placebo-controlled, double-blind trial of agalsidase beta (1 mg/kg every 2 weeks) or placebo for 20 weeks Age range: 17–62 years Mean age: 31.3 years 56 males, two females |
To examine the long-term effect of agalsidase beta for Fabry disease | Open-label, Phase III extension trial with no placebo treatment (agalsidase beta dose 1 mg/kg every 2 weeks for up to additional 54 months) |
Gb3 clearance levels from kidney, heart and skin biopsies. Plasma Gb3, eGFR, proteinuria and serum creatinine levels Pain scores assessed by the Short Form McGill Pain Questionnaire SF-36 IgG antibody levels |
Follow-up: every 6 months after entry into extension study for 54 months |
After 54 months of treatment, median eGFR, protetinuria and serum creatinine remained stable and normal indicating a stabilisation of renal disease progression Sustained Gb3 clearance from kidney, skin and heart capillary endothelium, multiple renal cell types and plasma had been reported after 54 months At month 54 pain scores had significantly improved for those who had reported pain pre treatment (p = 0.016). A statistically significant improvement in mean visual analogue scale scores at month 54 was also reported for these patients (p = 0.007) For most SF-36 components, patients experienced a mean improvement after long-term treatment with agalsidase beta Overall, 52 of the 58 patients seroconverted with the majority of patients seroconverted within the first 3 months of receiving agalsidase beta |
| Vedder and colleagues (2007)149 |
N = 34 Age range: 19–60 years 18 males, 16 females Netherlands and Norway |
To compare the efficacy of tolerability towards the two agalsidase preparations | Comparative trial (patients treated either with agalsidase alpha or agalsidase beta at equal dose of 0.2 mg/kg biweekly) | Primary end point was reduction in LVM, plasma Gb3 levels, glomerular filtration rate and pain scores | 12 or 24 months |
Both treatment groups showed statistically significant declines in plasma Gb3 levels after 12 and 24 months After 12 and 24 months of treatment, no statistically significant reduction in LVM was reported in either group or between the treatment groups No significant differences in glomerular filtration rate or pain scores for either treatment group |
| Hughes and colleagues (2008)138 |
N = 15 male patients Mean age: 37 years |
To assess the safety and efficacy of ERT with agalsidase alpha on the cardiac manifestations of Anderson–Fabry disease |
RCT (double-blind, placebo-controlled) Participants received placebo (n = 8) or agalsidase alpha (n = 7) 0.2 mg/kg every 2 weeks After 6 months of the randomised trial patients were enrolled in a 2-year, open-label extension study |
The primary end point was myocardial Gb3 content, LVM QRS duration, urine sediment and plasma Gb3 levels | Baseline and 6 months |
A mean 20% reduction in myocardial Gb3 content in cardiac tissue was demonstrated over the 6 months of ERT compared with a mean 10% increase in patients receiving placebo LVM was significantly reduced following 6 months of treatment compared with placebo group Mean reduction in QRS duration of 12.9 ± 11.8 ms over the 6 months compared with an increase (+4 ms) in the placebo group, although this was reported to be driven by one patient Mean decrease of plasma Gb3 of 45% in the group given agalsidase alpha compared with no change in the placebo group between baseline and 6 months (p < 0.001 compared with placebo) In the open-label phase, the original placebo group demonstrated a similar statistically significant decrease in plasma and urine Gb3 levels |
| Wraith and colleagues (2008)147 |
N = 16 Age range: 8–16 years 14 males, two females International: seven sites in four countries (France, UK, USA and Poland) |
To evaluate the safety and explore the efficacy of ERT with agalsidase beta |
Open-label study 48-week treatment period: agalsidase beta dose (1 mg/kg) infused i.v. every 2 weeks |
Skin biopsies to measure Gb3 levels Gastrointestinal symptoms Renal function: serum creatinine levels Cardiac function: ECG sinus arrhythmias or conduction abnormalities BMI |
Plasma Gb3 samples and gastrointestinal parameters obtained at week 0 and every 4 weeks thereafter Skin Gb3, cardiac function measured at baseline and at 24 and 48 weeks |
It was reported that agalsidase beta safely and effectively reduced the Gb3 accumulation in dermal endothelium already evident in children with Fabry disease With treatment, reports of gastrointestinal symptoms declined steadily Serum creatinine levels did not vary significantly during the course of treatment No changes occurred in the ECG in the 48 weeks of treatment During the 48 weeks of treatment, BMI fluctuated slightly but only two individual changes were notable |
| Schiffmann and colleagues (2010)148 |
N = 17 Age range: 7.3–18.4 years 16 males, one female Four centres |
To investigate the safety and potential efficacy of long-term treatment with agalsidase alpha |
3.5-year extension Patients had already completed a 6-month, open-label agalsidase alpha study |
Plasma and urine sediment Gb3 Kidney function using eGFR Pain levels assessed by the BPI Cardiac structure and function, including LVM and heart rate variability |
Baseline and every 6 months for 3.5 years |
In treated males, there were sustained and statistically significant improvements in the clinical features, including reduced plasma Gb3 levels, reduced pain severity and improved heart rate variability. Mean urine Gb3 levels were reduced to normal range (p < 0.5 compared with baseline during years 1.5–4) Kidney function and LVM indexed to height remained stable throughout |
Mucopolysaccharidoses
Aetiology
The mucopolysaccharidoses are a family of disorders caused by inherited defects in the catabolism of sulphated components of connective tissue known as GAGs. Different mucopolysaccharidoses are caused by different enzyme deficiencies leading to the accumulation of biochemically different GAG degradation products.
Clinical features
Seven distinct clinical types and numerous subtypes of the mucopolysaccharidoses have been identified and are commonly referred to by the name of the clinician who first described the condition. They include MPS IH (Hurler), mucopolysaccharidosis type I subtype Scheie disease (MPS IS) (Scheie), MPS II (Hunter), MPS III (Sanfilippo), MPS IV (Morquio), MPS VI (Maroteaux–Lamy), MPS VII (Sly) and MPS IX (Natowicz).
Mucopolysaccharidosis type I: Hurler, Hurler–Scheie and Scheie disease
Mucopolysaccharidosis type I is caused by deficient activity of the enzyme α-l-iduronidase (IDUA) leading to the accumulation of GAGs, dermatan sulphate (DS) and heparan sulphate (HS). The accumulation of GAGs in many tissues including connective tissue, brain, heart and liver, in turn leads to skeletal abnormalities, respiratory problems, joint problems, developmental delay and other issues such as corneal cloudiness, enlarged liver and spleen, recurrent hernias and heart disease.
There are three subtypes of MPS I: type IH (Hurler disease; OMIM 607014) that presents in the first year of life, has severe neurological symptoms and a life expectancy of only one decade; MPS IHS (Hurler–Scheie disease; OMIM 607015) is an intermediate form with a life expectancy of only 2–3 decades; and MPS IS (Scheie; OMIM 607016) is an attenuated form with later presentation and longer life expectancy than MPS IH and MPS IHS.
In its severe, classical form, Hurler disease exhibits the full range of features seen in a LSD. Infants are usually normal at birth but a spinal gibbus may be noted by the parents during the first 6 months of life. 5 Umbilical and inguinal herniae are common, as is the early onset of recurrent upper respiratory tract infections. Between 6 and 9 months the facial appearance changes, commonly described as ‘coarse’, with mid-facial hypoplasia, frontal bossing and an enlarged tongue. Airway involvement leads to upper airway obstruction and, with time, enlargement of the liver and spleen, cardiac valve disease and dysostosis multiplex lead to a very characteristic clinical phenotype. Progressive cognitive impairment occurs and with no treatment, most infants die in the first decade of life from cardiorespiratory disease.
Clinical features in patients with MPS IH/S and MPS IS are extremely heterogeneous and there is considerable overlap in phenotypes. Generally cognitive impairment is slight or absent and the main abnormalities are progressive joint, heart valve, respiratory disease and corneal disease. Hepatosplenomegaly and dysostosis multiplex are very variable. Patients with severe MPS IH/S die in their second or third decade from cardiac or respiratory disease, while the most attenuated MPS IS patients can have a normal life expectancy albeit with significant disability. 155
Mucopolysaccharidosis type II: Hunter disease
Mucopolysaccharidosis type II (MPS II or Hunter syndrome; OMIM 309900) is an X-linked recessive disease caused by the deficiency of the lysosomal enzyme iduronate-2-sulphatase (I2S), resulting in an accumulation of DS and HS. In most parts of the world the disorder is not as prevalent as MPS I (1 : 162,0005) but has the same degree of heterogeneity. The mean age at onset of symptoms is a little later than in MPS I (1.5 years) and diagnosis is usually made later as well (3.5 years156). Although X-linked and primarily affecting boys, a number of affected females have been reported. 157
Patients with MPS II are usually divided into ‘severe’ or ‘attenuated’ variants depending on the presence or absence of cognitive impairment. There is often considerable overlap between these two groups. Severe patients with CNS disease have organ involvement similar to MPS IH, but the cornea generally remain clear in contrast to the progressive clouding that is a feature of both MPS I and MPS VI. Generally the dyostosis is milder and most patients will survive without treatment up to their mid-teenage years.
Patients with ‘mild’ or attenuated disease are at risk from progressive cardiac valve disease, restrictive respiratory disease and cervical myelopathy due to ligamentous and dural hyperplasia. Despite these complications many patients with the attenuated form will survive into middle age and a number have gone on to have their own families.
Epidemiology
The estimated birth prevalence for all types of MPS is around 3.5 in 100,000 live births. 158 In the UK, Connock and colleagues19 calculated the birth prevalence of MPS I (from data collected between 1981 and 2003) as 1.07 per 100,000 births. 19 The birth prevalence of MPS II among UK males has been reported as approximately 1 in 130,000 (0.8 per 100,000) male live births. 159
Diagnosis
A clinical diagnosis of the mucopolysaccharidoses can often be made through examination and measurement of urinary GAGs. Enzyme assays must be used to provide a definitive diagnosis. Prenatal diagnosis using amniocentesis and chorionic villus sampling can verify if a fetus either carries a copy of the defective gene or is affected with the disorder.
Management
All MPS disorders demonstrate significant clinical heterogeneity with severe and attenuated phenotypes. Disease-modifying treatments are available for MPS I (both mild and attenuated phenotypes), MPS II (non-neurological phenotype) and MPS VI (both severe and attenuated phenotypes).
Treatment of mucopolysaccharidosis
Haematopoietic stem cell transplantation
Haematopoietic stem cell transplantation has been tried in all MPS disorders but is currently recommended only in MPS IH. Although it has some effect in MPS VI, almost all patients are now offered recombinant ERT. For reasons that are not clearly understood the results in MPS II have been very disappointing and so in most centres this disorder is no longer considered as appropriate for HSCT.
Haematopoietic stem cell transplantation is now regarded as the standard therapy for MPS IH. 160 Patients are commonly given a period of ERT prior to the transplant. Increasingly, umbilical cord blood is being used as the source of donor cells. In early-treated patients there is rapid resolution of hepatosplenomegaly, decline in urine GAG excretion and improvement in respiratory function and cardiomyopathy, while cardiac valve disease often progresses. The coarse hair and facial features soften and become more normal. Importantly, HSCT can stabilise neurological decline assuming the development quotient (DQ) before transplantation is reasonable (greater than 70). However, skeletal disease is resistant to treatment and kyphoscoliosis, hip dysplasia and genu valgum deformities often require complex surgery. The role and results of HSCT in MPS IH have been reviewed recently. 161
Enzyme replacement therapy in mucopolysaccharidosis type I
Recombinant ERT (recombinant human IDUA, laronidase) is used to treat patients with attenuated MPS I. Laronidase is licensed for intravenous administration for symptomatic MPS IS and MPS HS patients. Approval was granted by both the FDA and the European Agency for the Evaluation of Medicinal Products in 2003, with a recommended dose of 0.58 mg/kg body weight every week.
There is a paucity of high-quality evidence for the efficacy of ERT in MPS I. The HTA-commissioned systematic review of effectiveness and cost-effectiveness of ERT in MPS I19 identified a single placebo-controlled RCT162 and one Phase I/II observational study. 163 Both studies, described below, reported some evidence suggesting efficacy.
Kakkis and colleagues163 undertook an observational study of 10 patients aged 5–22 years, treated with laronidase weekly for 52 weeks. The authors reported decreased hepatosplenogmegaly in all patients. They also reported a significant increase in the range of motion of shoulder flexion and elbow extension, and a 61% decrease in the number episodes of apnoea and hypopnoea. In the six prepubertal patients, the rate of growth in height and weight increased by a mean of 85% and 131%, respectively, at 52 weeks of treatment.
Wraith and colleagues160 reported a double-blind, placebo-controlled, randomised clinical trial of laronidase in patients (age range 6–43 years) with attenuated MPS I. Twenty-two of 45 patients were randomised to receive 0.58 mg/kg laronidase intravenously for 26 weeks. The efficacy end points compared the median change from baseline to week 26 between groups in percentage of predicted normal forced vital capacity (FVC) and in 6-minute walk test distance. After 26 weeks of treatment, patients receiving laronidase showed a mean 5.6% increase in the per cent of predicted normal FVC compared with the placebo group. The treatment group also showed a mean 38.1 m increase in the distance walked in the 6-minute walk test compared with the placebo group. In addition, laronidase was reported to reduce urine GAG excretion, reduce mean liver volume and, in more severely affected patients, improve sleep apnoea/hypopnoea and shoulder flexion.
The 45 patients who completed the 26-week, placebo-controlled trial, described above, were subsequently enrolled in a 3.5-year, open-label, extension study. 164 The per cent predicted FVC remained stable over the course of treatment, and the 6-minute walk distance increased by an average of 31.7 m in the first 2 years, with a final gain of 17.1 m. Improvements in the sleep apnoea/hypopnoea and shoulder flexion were most rapid during the first 2 years, whereas urinary glycosaminoglacan levels declined rapidly (60–70% reduction in most patients) during the first 12 weeks of treatment and then plateaued thereafter but did not reach normal levels.
Clarke and colleagues164 reported laronidase to be well tolerated in all but one patient, who experienced an anaphylactic reaction. A two-centre study of 17 MPS I patients aged between 1 and 35 years reported that ERT could be safely given within the patient’s home. 165 Serious and sometimes fatal allergic reactions have occurred with use of laronidase, and some patients experience hives, difficulty breathing and/or swelling of the face, lips, tongue or throat.
The authors of the HTA-commissioned review19 concluded that the lack of data related to natural history, efficacy and the highly heterogeneous nature of the conditions meant that it would not be appropriate to attempt a cost-effectiveness analysis. They nonetheless argue that the extremely high costs of ERT in this condition mean that it is unlikely that, even if the treatment is highly effective, it would meet the current thresholds for cost-effectiveness. As with Gaucher disease and Fabry disease, this argument is crucially dependent on current drug costs.
A summary of the main evidence from RCTs and other studies on the efficacy and safety of laronidase for MPS I is presented in Table 5.
| Study | Population (size, age, gender, country) | Specified aims | Study design | Outcomes/measures | Follow-up | Findings |
|---|---|---|---|---|---|---|
| Kakkis and colleagues (2001)163 |
N = 10 MPS I patients (One Hurler syndrome, eight Hurler–Scheie syndrome and one Scheie syndrome) Age range: 5–22 years Six males, four females USA |
To examine the efficacy of ERT in patients with MPS I |
Observational study Laronidase dose 125,000 U/kg i.v. weekly for 52 weeks |
Height and weight Liver and spleen size and volume Range of motion: shoulder flexion, elbow and knee extension Airway function Cardiac function using NYHA score Symptomatic and ophthalmic changes Enzyme activity in leucocytes and toxicity screening |
6, 12, 26 and 52 weeks |
It was reported that the rate of growth in height and weight had increased by a mean of 85% and 131%, respectively, at 52 weeks in the six prepubertal patients Hepatosplenomegaly decreased significantly in all patients The mean maximal range of motion of shoulder flexion and elbow extension increased significantly The number of episodes of apnoea and hypopnoea during sleep decreased 61% NYHA functional class improved by one or two classes in all patients During treatment all patients had improved endurance and fewer limitations in their ability to perform daily activities The extent of corneal clouding did not change in any of the eight patients with this problem Urinary GAG excretion decreased after 3–4 weeks of treatment |
| Wraith and colleagues (2004)162 |
N = 45 MPS I patients (One Hurler syndrome, 37 Hurler–Scheie syndrome and seven Scheie syndrome) Age range: 6–43 years Mean age: 15.5 ± 8 years Multinational: five centres |
To examine the efficacy and safety of laronidase in patients with MPS I |
Randomised, double-blind placebo-controlled, Phase III clinical trial of laronidase with attenuated MPS I patients N = 22 patients randomised to treatment group Dose 100 U/kg laronidase or placebo i.v. weekly for 26 weeks |
Primary outcomes: respiratory function, FVC Physical capacity: 6-minute walk test distance Additional outcomes: liver volume, urinary GAG excretion, sleep study AHI, active shoulder flexion and the disability score index of the CHAQ for patients aged ≤ 18 years and of the HAQ |
26 weeks |
It was reported that after 26 weeks, patients receiving laronidase compared with placebo showed mean improvements of 5.6 percentage points in per cent of predicted normal FVC (median, 3.0; p = 0.009) and 38.1 m in 6-minute walk test distance (median, 38.5; p = 0.066) Over the study, mean liver volume decreased by 18.9% in the laronidase group and increased by 1.3% in the placebo group, for a between-group difference of 20.0% (p = 0.001) Furthermore, the laronidase group showed a mean reduction of 54.1% in urinary GAG excretion whereas the placebo group had a mean increase of 47.3% (p < 0.001) Laronidase-treated patients had a mean decrease of 6.0 ADI events per hour of sleep whereas placebo-treated patients had a mean increase of 0.3 ADI events per hour. The 11.4 events per hour treatment benefit was statistically significant p = 0.014 For the total study population the mean change in shoulder flexion and changes in the disability index did not differ significantly between treatment groups |
| Clarke and colleagues (2009)164 |
N = 45 attenuated MPS I patients (38 Hurler–Scheie syndrome and seven Scheie syndrome) Age range: 6–43 years Mean age: 16 years 22 males, 23 females Europe and USA |
To evaluate the long-term safety and efficacy of recombinant human laronidase in patients with MPS I |
Open-label, extension study of Wraith and colleagues 2004160 3.5 years Weekly infusions of laronidase at dose 100 U/kg body weight |
Primary outcomes: respiratory function, FVC Physical capacity: 6-minute walk test distance Additional outcomes: liver volume, urinary GAG excretion, sleep study AHI, active shoulder flexion and the disability score index of the CHAQ for patients aged less than or equal to 18 years and of the HAQ |
Biochemical measures every 12 weeks Visual acuity and sleep study weeks 24, 72 and 120 Physical exam and standard laboratory safety evaluations on weeks 4 and 12 then every 12 weeks until week 120 then week 144 |
After 3.5 or 4 years of treatment, responses to treatment in individuals: Per cent predicted normal FVC: 18% improved, 55% stable and 28% declined Distance walked in the 6-minute walk test: 50% improved, 28% stable and 23% declined Liver volumes: 95% normal values and 92% of patients with abnormal values attained normal values Urinary GAG levels: 100% of patients improved, 15% attained normal values and 72% were within twice the upper limit of normal AHI events: 31% improved, 63% stable and 6% declined Shoulder range of motion: 46% improved, 49% stable and 5% declined Corrected visual acuity: 24% improved, 66% stable and 10% declined CHAQ/HAQ disability index: 57% improved, 20% stable and 23% declined |
| Giugliani and colleagues (2009)166 |
N = 33 (10 Hurler syndrome, 16 Hurler–Scheie syndrome and seven Scheie syndrome) Age range: 1.4–20.7 years Mean age: 7.1 years 45% male 63% Caucasian Weight greater than 12.5 kg, with clinical symptoms of MPS I less than 10% normal IDUA activity and normal renal function 30 from Brazil, 4 from Canada |
To assess whether or not alternate dosing regimens might provide a better reduction in lysosomal storage |
Randomised, open-label, multicentre, international dose optimisation trial Patients were stratified by urinary GAG level (greater than or equal to 300 or less than 300 µg/mg creatinine) and then randomised to one of four treatment groups Four treatments: 0.58 mg/kg weekly (n = 8); 1.2 mg/kg (200 U/kg) every 2 weeks (n = 8); 1.2 mg/kg (200 U/kg) weekly (n = 8); 1.8 mg/kg (300 U/kg) every 2 weeks (n = 9) |
Primary efficacy end point: the difference in the percentage change from baseline to week 26/27 in mean urinary GAG levels Secondary end points: the differences between groups in mean percentage change in liver organ volume and mean absolute change in the 6-minute walk test distance Safety and immunogenicity were evaluated |
Urinary GAG collected at baseline and then every 3–4 weeks until week 26/27 Liver volume assessed by CT and 6-minute walk test distance were assessed at baseline, week 11 and week 26/27 |
No significant differences in the reduction of urinary glycosaminoglaycan excretion or liver volume were reported for the four treatment regimens Laronidase had an acceptable safety profile in all dose regimen groups. The approved 0.58 mg/kg/week regimen provided near maximal reductions in GAG storage and best benefit to risk ratio In the 0.58 mg/kg/week group mean reduction in urinary GAG excretion was 63% and mean reduction in liver volume was 30% (95% CI 27% to 33%) Estimated mean improvement in 6-minute walk test distance (in all treatment groups) was 20 m (95% CI 10 m to 49 m) |
Enzyme replacement therapy in mucopolysaccharidosis type II
Idursulfase is licensed for treatment of patients with MPS II in the USA and the European Union (EU), gaining marketing approval by the FDA in 2006 and EMA in 2007. There have been few reports of the long-term efficacy of this treatment.
In a randomised, double-blind, placebo-controlled trial, 12 patients were randomised to receive idursulfase at 0.15, 0.5 or 1.5 mg/kg infused every other week, or placebo, for 24 weeks, followed by an open-label extension. 167 The primary end point was a change in urinary excretion of GAGs from baseline. Urinary GAGs decreased within 2 weeks of initiating treatment, and further decreased by 59% after 48 weeks on treatment. Both liver and spleen volume were decreased at 24 weeks and 48 weeks. The distance walked in 6 minutes increased an average of 48 metres after 48 weeks.
In a multinational, Phase II/III, clinical study of idursulfase in MPS II, Muenzer and colleagues167 reported a double-blind, placebo-controlled, clinical trial in which 96 patients with MPS II were randomised to receive placebo infusions, weekly infusions of idursulfase at 0.5 mg/kg or fortnightly infusions of idursulfase at 0.5 mg/kg. The primary end point used was a composite of distance walked in 6 minutes and the per cent of predicted FVC based on the sum of the ranks of change from baseline. Patients in the weekly and fortnightly idursulfase groups exhibited significant improvement in the composite end point compared with placebo after 1 year (p < 0.005 and p < 0.05, respectively). The weekly dosing group experienced a 37 m increase in the 6-minute walk distance, a 2.7% increase in per cent of predicted FVC and a 160 ml increase in the absolute FVC compared with placebo group at 53 weeks.
In the Muenzer and colleagues study,167 treatment with idursulfase was generally well tolerated in both groups and most infusion-related complications were mild. Antibodies to the infused proteins developed in many patients but these were often transient and their long-term impact remains unclear. Some of the more common side effects of idursulfase include anxiety, headache, irritability, pain, swelling or redness at the infusion site and stomach upset.
A summary of the main evidence from RCTs and other studies on the efficacy and safety of idursulfase is presented in Table 6.
| Study | Population (size, age, gender, country) | Specified aims | Study design | Outcomes/measures | Follow-up | Findings |
|---|---|---|---|---|---|---|
| Muenzer and colleagues (2006)167 |
N = 96 male patients Age range: 5–31 years Mean ages (years) at enrolment: 13. ± 1.22 (placebo); 14.4 ± 1.2 (idursulfase every other week); 15.1 ± 1.11 (idursulfase weekly) Multicentre, multinational |
To evaluate the safety and efficacy of recombinant human iduronate-2-sulfatase (idursulfase) in the treatment of MPS II |
A randomised, double-blind, placebo-controlled trial Patients were randomised to placebo infusions (n = 32) Weekly idursulfase (0.5 mg/kg) infusions (n = 32) or every-other-week infusions of idursulfase (0.5 mg/kg) (n = 32) |
Efficacy Efficacy was evaluated using a composite end point consisting of distance walked in 6 minutes and the percentage of predicted FVC based on the sum of the ranks of change from baseline Safety A clinical assessment was done at every infusion visit. A physical examination, which included measurement of vital signs, height, weight, serum chemistry, CBC, urinalysis and an electrocardiogram, was performed every 4 months. AEs were monitored and recorded throughout the study Blood samples were taken at baseline and periodically thereafter for measurement of serum idursulfase antibodies |
All measurements were made at baseline and at weeks 18, 36 and 53 |
Patients in the weekly and every-other-week idursulfase groups exhibited statistically significant improvement in the composite end point compared with placebo (p = 0.0049 for weekly and p = 0.0416 for every-other-week) after 1 year The weekly dosing group experienced a 37 m increase in the 6-minute walk test distance (p = 0.013), a 2.7% increase in per cent of predicted FVC (p = 0.065) and a 160 ml increase in absolute FVC (p = 0.001) compared with placebo group at 53 weeks Idursulfase was generally well tolerated, but infusion reactions did occur. Idursulfase antibodies were detected in 46.9% of patients during the study |
| Muenzer and colleagues (2007)168 |
N = 12 male patients with attenuated MPS II Mean age at enrolment was 14 years Age range: 6–20 years USA, single centre |
To evaluate the safety and explore the efficacy of idursulfase (recombinant human iduronate-2-sulfatase) treatment for MPS II |
A randomised, double-blind, placebo-controlled trial for 24 weeks followed by an open-label extension study Three groups of four patients were enrolled sequentially, with three patients in each group receiving idursulfase and one patient receiving placebo The first group received idursulfase at 0.15 mg/kg infused every other week with the second and third groups receiving 0.5 and 1.5 mg/kg, respectively After 24 weeks the placebo-treated patients were changed to idursulfase at the dose of their group |
Efficacy The primary end point was a change from baseline in urinary GAG excretion Safety Safety evaluations were performed at every visit and included a physical examination, serum chemistry, complete blood count, urinalysis, measurement of vital signs, height, weight and an electrocardiogram AEs were monitored and recorded throughout the study, either at study visits or by telephone contact by a study coordinator during the weeks without a scheduled visit. Anti-idursulfase antibodies in plasma were detected by an ELISA and positive results were confirmed by RIP |
The primary end point was the extent of reduction in urinary GAG excretion Secondary end points included liver and spleen size, 6-minute walk test, pulmonary function, joint mobility, heart size and function and a sleep study. All clinical assessments were performed the week prior to receiving the first infusion of idursulfase Repeat evaluations were performed at weeks 13 and 24 during the double-blinded phase and at weeks 25 and 51 of the open-label phase |
During the double-blind phase, the mean decrease in urinary GAG for all dose groups as well as the per cent changes from baseline at each visit were statistically significant (mean change, p = 0.0092; per cent change p = 0.0007) From pooled results of all three groups, it was reported that idursulfase significantly reduced urinary GAG excretion at 6 and 12 months (p < 0.0001) They reported significant decreases in liver and spleen volumes (p ≤ 0.0001), compared with placebo group 6-minute walk test distance significantly increased compared with placebo An improvement in elbow mobility between the weekly idursulfase group compared with the placebo was observed Absolute FVC was significantly increased from baseline in the weekly dosing group (0.22 ± 0.051) compared with both placebo (0.06 ± 0.031; p = 0.0011) and EOW groups (0.07 ± 0.03; p = 0.0176) after 53 weeks |
| Okuyama and colleagues (2010)169 |
10 adult males with attenuated MPS II Mean age at enrolment: 30.1 years Age range: 21–53 years Japan |
To evaluate the safety and explore the efficacy of idursulfase (recombinant human iduronate-2-sulfatase) treatment for MPS II | A multicenter, open-label clinical study in 10 male patients, aged 21–53 years, each received weekly i.v. infusions of 0.5 mg/kg idursulfase for 12 months |
Efficacy Urinary GAG level Liver and spleen volumes were quantitated by CT Per cent predicted FVC 6-minute walk test distance Cardiac structure and function Active joint range of motion was measured by goniometry and included the shoulder (flexion, extension and abduction), elbow (flexion and extension), hip (flexion and extension) and knee (flexion and extension) The sleep study ODI Safety Safety evaluation included continuous monitoring of AEs and periodic clinical laboratory and physical examination evaluations AEs were reported by severity (mild, moderate, severe, life-threatening) and by relatedness to idursulfase Antibodies to idursulfase |
Assessments were done at baseline and repeated at months 3, 6, 9 and 12 Nine patients completed the 12-month study; one patient died of causes unrelated to idursulfase after receiving 41 of 44 scheduled infusions |
Idursulfase was well tolerated over the course of the study AEs were mainly mild, unrelated and attributable to expected symptoms of MPS II disease Idursulfase reduced GAG storage, as evidenced by the statistically significant reductions in urinary GAG levels (p = 0.004) and hepatosplenomegaly (p = 0.002) There were trends towards improvement in functional capacity (mean 54.5 m increase in 6-minute walk test distance), respiratory function (mean 15.0% relative increase in per cent predicted FVC), joint range of motion (mean increases ranging from 8.1–19.0 degrees for several joints), and LVMI (mean 12.4% decrease) IgG antibodies were detected in 60% (6/10) of patients treated with idursulfase |
| Muenzer and colleagues (2011)170 |
Extension study of Muenzer and colleagues167 94 male patients enrolled (from original 96 patients in Muenzer and colleagues167) |
The primary objective of the study was to assess the long-term safety and clinical outcome in patients treated with idursulfase | This study was a multicentered, multinational, open-labelled, clinical trial |
Primary outcome measures 6-minute walk test distance and FVC Secondary measures Measurement of liver and spleen volume (measured by MRI), urine GAG excretion, cardiac mass (measured by echocardiography), JROM (measured by goniometry), linear growth velocity, and functional status (CHAQ DIS) Safety AEs were recorded continuously throughout the study. A clinical assessment was done at each study visit The presence of antiidursulfase antibodies in plasma were detected by an enzyme-linked immunosorbent assay; samples testing positive for IgG antibodies were further tested for neutralising activity by an in vitro enzyme inhibition assay and by inhibition of cell uptake of idursulfase |
Safety and clinical outcomes were assessed every 4 months during the first year of the study and every 6 months thereafter. |
No change in per cent of predicted FVC was seen, but absolute FVC demonstrated sustained improvement and was increased 25.1% at the end of the study Statistically significant increases in 6-minute walk test distance were observed at most time points Mean liver and spleen volumes remained reduced throughout the 2-year extension study Mean JROM improved for the shoulder and remained stable in other joints. Both the parent- and child-assessed CHAQ DIS demonstrated significant improvement Infusion-related AEs occurred in 53% of patients and peaked at month 3 of treatment and declined thereafter. Neutralising IgG antibodies were detected in 23% of patients and seemed to attenuate the improvement in pulmonary function |
Pompe disease
Aetiology and classification
Pompe disease (otherwise known as glycogen storage disease type II or acid maltase deficiency; OMIM 232300) is caused by a deficiency in the lysosomal enzyme acid α-glucosidase. This leads to an accumulation of glycogen within the cells, particularly within striated muscle. Although lysosomal dysfunction is at the heart of the disease, the exact pathophysiology is as yet unclear. The clinical manifestations are believed to be due to secondary effects of lysosomal dysfunction such as mitochondrial malfunction and autophagy. 171
There is a spectrum of disease severity. Traditionally the disease is classified according to age at onset172 and, at present, is generally divided into infantile onset and late onset. 173,174 Complete deficiency of α-glucosidase causes the progressively lethal infantile Pompe disease whereas partial deficiency leads to a milder late-onset phenotype. The latter condition may present at any age and is subdivided in non-classical infantile, childhood, juvenile and adult Pompe disease. 173,175,176 What distinguishes the slower-onset form from the classical infantile form is the presence of cardiomyopathy in the infantile form. 174
Clinical features
Adult onset
In both juvenile- and adult-onset disease the two main characteristics are respiratory compromise and a symmetrical proximal myopathy. These are both progressive but are independent of each other, with patients presenting with either or both the respiratory or the skeletal manifestations. In addition, some people may have swallowing difficulties, including involvement of the tongue, and skeletal deformities such as kyphoscoliosis.
As well as spinal deformities, there is an increased incidence of bone disease in the form of reduced bone density and mass. 177,178
Although not a mode of presentation, abnormalities of the cerebral circulation have been described and these can cause cerebral haemorrhage. 179
Infantile onset
The infantile form of Pompe disease represents the severe end of the spectrum of enzyme deficiency with an estimated incidence of 1 : 138,000. Infants with the classic form of infantile-onset Pompe disease typically experience muscle weakness, hypotonia, hepatomegaly, early cardiac enlargement because of HCM, cardiac failure and respiratory failure. Infants usually have a slightly enlarged tongue with associated drooling and ‘wood hard’ character of lower limb muscles owing to glycogen accumulation. Cardiac involvement includes cardiomegaly, cardiomyopathy, left ventricular hypertrophy or electrocardiographic changes. 174
Natural history studies suggest median ages of symptom onset at 1.6–2 months, ventilator dependency at 4.7 months and, if untreated, death from cardiorespiratory failure at 6–9 months. 174,180
The non-classic form of infantile-onset Pompe disease is associated with slightly longer survival than classic disease, and usually appears by age 1 year. It is characterised by delayed motor skills and progressive muscle weakness. The heart may be abnormally large, but affected individuals usually do not experience heart failure. The muscle weakness in this disorder leads to serious breathing problems, and most children with non-classic infantile-onset Pompe disease live only into early childhood.
Epidemiology
The worldwide incidence of Pompe disease in adults is reported as between 1 in 40,000 (2.5 per 100,000) and 1 in 60,000 (1.7 per 100,000). 181,182 Although the disease seems to occur in all adult ethnic groups, the estimated frequency of Pompe disease may vary among different ethnic groups and nationalities and reported estimates range from 1 in 14,000 (7.1 per 100,000) to 1 in 300,000 (0.3 per 100,000). 172
The infantile form of this disease is rarer with an incidence of 1 in 138 000 (0.72 per 100,000). This disorder has a pan-ethnic distribution. In infants, the disease appears to be more common among African Americans and in southern China and Taiwan, whereas the prevalence of adults with Pompe disease may be comparatively high in the Netherlands. 181
These estimates may actually be underestimates because of misdiagnosis and the presentation of some cases very late in life. Newborn screening in Taiwan has reported a prevalence of 1 in 4000 (25 per 100,000), with many of those probably having the later-onset juvenile or adult forms of the disease. 183
Diagnosis
Diagnosis of Pompe disease is usually based on natural history, decreased or absent α-glucosidase activity in muscle or skin biopsies, histopathology and GAA gene mutation analysis. Measuring creatine phosphokinase can be a marker as this is classically elevated in Pompe disease. Confirmation of the diagnosis is by measurement of α-glucosidase in leucocytes or, more recently, measuring the enzyme activity in blood spots. 184 The gene encoding α-glucosidase has been identified and confirmation can now be done using gene mutation analysis.
Inheritance and genetics
Pompe disease is inherited as an autosomal recessive trait. The gene is found at 17q25.2–q25.3 and is 20 exons long. Although over 200 mutations are described, the IVS1 – 3T → G accounts for about 50% of adult cases in Caucasians. 185
Cross-reactive immunological material (CRIM) status affects treatment outcomes in Pompe disease infants. 186 The presence of CRIM-negative disease seems to be higher in the infantile population and seems to be associated with greater immunological reaction to ERT and poorer outcome.
Management
Symptomatic management
Prior to the development of ERT there was no disease-specific treatment for Pompe disease and patients were managed with supportive treatment and palliative care. Although these multidisciplinary approaches cannot generally alter the disease course, they may impact on QoL.
Enzyme replacement therapy
Adult Pompe disease
Human recombinant α-glucosidase (alglucosidase alpha) was licensed in Europe in 2006. It is administered in a dose of 20 mg/kg body weight intravenously every 2 weeks. In adults there has only been one placebo-controlled, clinical trial reporting modest improvement in respiratory function and muscle strength in Pompe patients. 187 In this multicentre study with eight centres in the USA and Europe, 90 patients aged ≥ 8 years were randomly assigned 2 : 1 to receive biweekly intravenous alglucosidase alpha at 20 mg/kg body weight or placebo, for 78 weeks. All patients were ambulatory and free of invasive ventilation. At 78 weeks, significant differences were observed between the alglucosidase alpha and placebo groups in the two primary end points – distance walked during a 6-minute walk test and per cent of predicted FVC.
Several case reports and single cohort studies have been published and recently reviewed. 188 In addition, ERT has been reported to lead to improvements in nutrition and body composition. 189 This is reported to be coupled with significant improvement in gastrointestinal function seen after 6 months of ERT. 190
Infantile Pompe disease
The outcomes for the infantile forms of this disorder on ERT seem to be affected by age at diagnosis, CRIM status and immunological response towards the biological therapy. 191
Enzyme replacement therapy for infantile Pompe disease has been reported to improve survival as well as cardiac, respiratory and motor functions. 192,193 An open trial of 18 patients, aged ≤ 7 months, with infantile Pompe disease, randomised to receive alglucosidase alpha at either 20 or 40 mg/kg every other week for 52 weeks, reported an invasive ventilator-free survival of 88.9% at 18 months compared with a historical control group of 61 infants who had infantile Pompe disease, only one of whom survived to 18 months. 192 In an extension of this study, the children continued to receive alglucosidase alpha at the original dose for up to 3 years. These children continued to exhibit the benefits of ERT at age 36 months, and over the entire study period it was reported that ERT reduced the risk of death by 95%, risk of invasive ventilation or death by 91% and the risk of any type of ventilation or death by 87% compared with historical controls. However, the dramatic improvement in ventilator-free survival reported at 52 weeks on ERT was not sustained on longer follow-up, with reported ventilator-free survival rates of 66.7% at 24 months and 49.4% at 36 months. 193
A review of the treatment of infantile Pompe disease with alglucosidase alpha in the UK was published in 2010. 194 At the time of reporting, a total of 20 infants had been treated with ERT since 2000. The results of the review suggested that the UK experience for infantile Pompe disease is not as good as that reported previously,192 with ventilator-free survival of 35%. The authors considered age at clinical presentation rather than age at commencement of treatment to be the overriding factor determining outcome in the UK cohort, and implied that ERT does not improve the outcome of the most severe cases.
Safety
Enzyme replacement therapy is well tolerated in both juveniles and adults, although hypersensitivity reactions do occur in 5–8% but are easily managed. 187
New therapies
Alternative ERTs are currently under investigation (Biomarin BMN-701: IGF2-GAA). Therapy with a small molecule chaperone has also been developed (Amicus therapeutics) and trials are underway. There has been a review of pre-clinical work of gene therapy in Pompe disease195 and one clinical trial has recently commenced.
A summary of the main evidence from RCTs and other studies on the efficacy and safety of alglucosidase alpha for adult and infantile Pompe disease is presented in Table 7.
| Study | Population (size, age, gender, country) | Specified aims | Study design | Outcomes/measures | Follow-up | Findings |
|---|---|---|---|---|---|---|
| Banugaria and colleagues (2011)191 |
N = 34 infants with Pompe disease 11 CRIM-negative patients, nine high-titre CRIM-positive patients, 14 low-titre CRIM-positive patients USA |
To examine the impact of antibodies on clinical outcomes in diseases treated with therapeutic protein | Retrospective analysis | Clinical outcome measures were overall survival, ventilator-free survival, LVMI, Alberta Infant Motor Scale score and urine Glc4 levels | Not applicable |
Clinical outcomes in the high-titre CRIM-positive group were poor across all areas evaluated relative to the low-titre CRIM-positive group For the CRIM-negative and high-titre CRIM-positive groups, no statistically significant differences were observed for any outcome measures and both patient groups did poorly Irrespective of CRIM status, patients with infantile Pompe disease with high sustained antibody titre have an attenuated therapeutic response to ERT |
| Bernstein and colleagues (2010)190 |
N = 3 later-onset Pompe disease patients Age at ERT initiation: 16–52 years One male; two female USA |
To examine the effect of ERT on QoL symptoms of later-onset Pompe disease patients | Alglucosidase alpha infusions at 20 mg/kg every other week |
Self-reported gastrointestinal symptoms Height and weight |
Contacted by telephone twice per month for 6 months to 2 years, were examined annually and were extensively interviewed |
All gastrointestinal symptoms resolved within the first 6 months of ERT with rhGAA All three patients gained weight and remain symptom-free, two for over 4 years |
| Chakrapani and colleagues (2010)194 |
N = 20 patients that have been treated with ERT Median age at diagnosis: 5.75 months Age range: 0.25–31 months Median age at commencement of ERT: 6.5 months UK: four centres |
To review the treatment of infantile Pompe disease | Clinical review | Survival and ventilator use (invasive and non-invasive) | Not applicable | Ventilator-free survival in 7/20 cases (35%), although 7/20 cases died (35%) and 6/20 (30%) were ventilator-dependent in 2010 |
| Kishnani and colleagues (2007)192 |
N = 18 patients with rapidly progressing infantile-onset Pompe disease Age ≤ 7 months 11 males, seven females 13 sites (six in the USA, five in Europe, one in Taiwan and one in Israel) |
To examine the safety and efficacy of i.v. rhGAA |
52-week, open-label, RCT (Randomised in a 1 : 1 ratio to receive an i.v. infusion of either 20 mg/kg or 40 mg/kg of rhGAA every other week – dose could be adjusted every 4 weeks) |
Safety data Echocardiographic measurements Muscle GAA activity and glycogen content in quadriceps muscle biopsy |
Survival and ventilator use (invasive and non-invasive) were analysed at ages 24 months and 36 months Echocardiograms performed at baseline and at 4, 8, 12, 26, 38 and 52 weeks Muscle biopsy at baseline, 12 and 52 weeks |
rhGAA treatment markedly improved cardiomyopathy (LVM reduction) in all patients with 52-week follow-up data rhGAA treatment greatly increased skeletal muscle GAA activity in all patients, but to a greater degree in patients receiving 40 mg/kg rhGAA than in patients receiving 20 mg/kg rhGAA rhGAA treatment clearly prolongs ventilator-free survival and overall survival in patients with infantile-onset Pompe disease compared with an untreated historical control population |
| Kishnani and colleagues (2009)193 |
N = 16 patients Age: less than 19 months old 13 sites (six in the USA, five in Europe, one in Taiwan and one in Israel) |
To examine the long-term safety and efficacy of i.v. rhGAA |
Extension study of Kishnani and colleagues192 Dose of alglucosidase alpha at either 20 mg/kg biweekly (n = 8) or 40 mg/kg biweekly (n = 8), for up to a total of 3 years |
Survival and ventilator use (invasive and non-invasive) |
ERT reduced the risk of death by 95%, reduced the risk of invasive ventilation or death by 91% and reduced the risk of any type of ventilation or death by 87%, compared with an untreated historical control group Cardiomyopathy continued to improve and 11 patients learned and sustained substantial motor skills No significant differences in either safety or efficacy parameters were observed between the 20 and 40 mg/kg biweekly doses |
|
| Ravaglia and colleagues (2010)189 |
N = 17 patients with late-onset Pompe disease Aged: 52.6 ± 16.8 years Eight males, nine females Italy, single centre |
To evaluate changes in body composition and nutritional status of patients in different stages of disease, baseline and during ERT | Non-comparative clinical trial |
Plasma albumin and prealbumin Body composition: BMI, fat mass and lean mass 6-minute walking test and FVC |
Outcome parameters were assessed every 3 months for 18–24 months |
Plasma albumin and prealbumin levels significantly increased, as early as in the first 6 months of ERT After a 18–24 month treatment with ERT, the authors observed an increase in BMI and fat mass and a small, non-significant increase in lean mass Results for the 6-minute walking test and FVC were not given |
| van der Ploeg and colleagues (2010)187 |
N = 90 patients with late-onset Pompe disease who were ambulatory and free of invasive ventilation Age range: 10–70 years 45 males, 45 females Eight centres in the USA and Europe |
To assess the safety and efficacy of alglucosidase alpha in patients with late-onset Pompe disease |
Randomised, double-blind, placebo-controlled 2 : 1 to receive biweekly infusions of alglucosidase alpha (20 mg/kg of body weight) or placebo |
Distance walked during a 6-minute walk test Per cent of predicted FVC Medical Outcomes Study SF-36 |
78 weeks: serum samples every 4 weeks for first year and again at weeks 64 and 78 | At 78 weeks significant differences were observed between the alglucosidase alpha and placebo groups in the two primary end points, the 6-minute walk test and the FVC |
Niemann–Pick disease type C
Niemann–Pick disease describes a heterogeneous group of lipid storage disorders, with common features of hepatosplenomegaly and sphingomyelin storage in reticuloendothelial and parenchymal tissues, with or without neurological involvement. 196
Today, there are three commonly recognised forms of Niemann–Pick disease: Niemann–Pick types A (NPA), B (NPB) and C (NPC) was clinically differentiated from Niemann–Pick A and B by Crocker and Mays. 197 Brady and colleagues12 subsequently demonstrated that NPA and NPB were caused by sphingomyelinase deficiency, whereas this enzyme was not deficient in NPC disease.
Aetiology
Niemann–Pick type C disease (OMIM 257220, 607625) is an atypical LSD in that it is caused not by an enzyme deficiency, but by problems in intracellular cholesterol transport, which leads to too much cholesterol in the liver and spleen and excessive amounts of other lipids in the brain, although sphingomyelin accumulation is not prominent. 196 In NPC, the brain and other organs are affected, leading to progressive intellectual decline, loss of motor skills, seizures and dementia. Speech can become slurred and swallowing problems may develop. The rate at which the disease progresses varies greatly between patients; children who develop neurological symptoms in early childhood are thought to have a more aggressive form of the disease, others may remain symptom free for many years. NPC has been reported in all ethnic groups but it is most common among Puerto Ricans of Spanish descent.
Niemann–Pick type C disease is caused by mutations of either the NPC1 or NPC2 genes. Mutations of the NPC1 gene, which codes for an integral transmembrane protein,198 are responsible for about 95% of cases of NPC disease. The NPC2 gene codes for a soluble lysosomal protein,199 but there is no evidence to date that this is an acid hydrolase.
In patients with NPC disease, the pattern of accumulating lipids is different in brain and in non-neural organs. In the liver and spleen, there is a complex pattern of accumulating products, with no predominating compound. Studies using cultured skin fibroblasts suggested that NPC disease was caused by a defect in esterification and export of cholesterol from the lysosome. 200,201 The characterisation of the NPC1 and NPC2 genes has since given limited support to this hypothesis, and observations on the brains of patients and animal models of NPC disease have shown that in nervous tissue, storage of glycosphingolipids is much greater than that of cholesterol. The underlying cellular defect appears to be in lipid trafficking, but the precise pathogenic mechanisms are not yet fully understood. 196
Clinical features
Like many other LSDs, NPC disease shows a spectrum of disease with an age at onset ranging from the perinatal period until the seventh decade of life. 196 NPC disease tends to be a neurovisceral condition, but visceral and neurological manifestations arise at different stages, and follow completely independent courses. A small number of patients will die during their first 6 months of life, usually from hepatic or respiratory failure, but most patients will develop a progressive and fatal neurological disease.
Presenting symptoms are variable, but clumsiness and cognitive decline are common. Cerebellar signs and dystonia are common findings and vertical supranuclear gaze palsy is also characteristic. A degree of splenomegaly is almost universal even if not clinically detectable, but liver disease is not evident outside the neonatal period. The clinical course is progressive with both cortical and deep brain structures becoming involved often with seizures developing. 202 Cerebellar involvement is prominent, resulting in ataxia and dysarthria, whereas dysphagia often necessitates tube feeding. Dementia is universal but can be early and aggressive or slowly progressive, manifesting as psychiatric disease in adults. Patients end up bedbound, mute and entirely dependent on 24-hour nursing care. Death is normally due to aspiration pneumonia.
Atypical presentations are seen, particularly in older patients. Some patients first present with isolated splenomegaly and it may be many years before neurological disease develops. In general, prognosis is related to age at first neurological presentation, with the disease being more indolent and slowly progressive the later it manifests.
Epidemiology
Niemann–Pick type C disease is inherited in an autosomal recessive manner. The prevalence of NPC disease has been estimated at between 0.66 and 0.83 per 100,000 in Western Europe. 201,203 The true prevalence of NPC disease in early life is probably underestimated, owing to its varied clinical presentation and high infant mortality rate. Although it is generally considered that NPC disease occurs with similar frequency across the world, it has been reported to be more common in some genetic isolates such as the French Arcadian population of Nova Scotia. 204
Diagnosis
For many patients, it is only years after birth that the first clinical signs are observed and diagnosis is made. A clinical diagnosis of NPC disease requires in-depth screening for characteristic neurological and systemic features and must be confirmed by biochemical and/or genetic testing. The key diagnostic test for NPC disease is filipin staining of cultured fibroblasts, which demonstrates impaired cholesterol esterification.
Management
Until recently, treatment for NPC disease has been limited to symptomatic therapy for relief of specific manifestations of the disease.
Treatment
Miglustat was approved in the EU in 2009 for the treatment of NPC disease and is currently prescribed off-label in the USA and is under consideration for approval by the FDA.
Recent advances in understanding the pathogenesis of NPC disease led to trials of SRT. NPC disease results from mutations in genes that encode proteins responsible for trafficking of unesterified cholesterol between several cell compartments. A defect of one of these genes leads to accumulation of cholesterol, sphingosine, sphingomyelin and glycosphingolipids. Although the complex mechanisms that lead to the accumulation of these substrates in NPC disease have been poorly understood, it is known that miglustat inhibits the synthesis of glucosylceramide, the precursor of glycosphingolipids, and this substance has therefore been considered as a SRT option for NPC disease patients.
Observations of ganglioside storage in NPC disease neurons and neuropathological findings in NPC disease brains that are common to other ganglioside storage disorders (neuroaxonal dystrophy) led Zervas and colleagues to attempt to treat NPC diseased mice with miglustat. 205 In these murine studies, treatment with miglustat led to a delay in the onset of symptoms and prolonged lifespan. 205
As miglustat was already licensed for the treatment of GD1, after the results of the animal studies were published, a number of patients with NPC disease were treated on a named-patient basis. The first patient with NPC disease treated with miglustat was reported in 2004. 206 In this individual, miglustat was reported to reverse lysosomal storage and correct the abnormal lipid trafficking seen in peripheral blood lymphocytes. The patient’s clinical condition remained stable, and no progression of the disease was observed over the 6-month treatment period.
In 2007, Chien and colleagues207 described two children, aged 9–14 years, with NPC disease who were treated with miglustat for 1 year. Improvements in swallowing, mobility and cognitive function were reported for both children. Santos and colleagues208 reported an apparently remarkable response to miglustat in a 10-year-old girl with significant improvements in ataxia, dysarthria, mobility and seizure control, which were maintained for at least 1 year. However, Paciorkowski and colleagues209 reported the use of miglustat in a 3-year-old girl whose motor and cognitive function continued to decline despite treatment. Galanaud and colleagues210 treated three adult patients with miglustat and reported that the clinical condition of the patients had stabilised or shown slight improvement after 24 months on treatment. Magnetic resonance spectroscopy was reported to show a sustained decrease in choline–creatinine ratio in all three patients, suggesting that SRT may have a beneficial effect on the brain over time in patients with NPC. The same effect was observed in a patient with 22 years of symptomatic disease as that seen in two other patients with 4 years of symptomatic disease, suggesting that even in advanced stages of disease, some brain dysfunction may still be reversible.
Fecarotta and colleagues211 treated three children with significant dysphagia with miglustat for 3 years and reported sustained improvements in swallowing based on videofluoroscopy. ≥
Patterson and colleagues212 reported a trial of 29 patients with NPC aged ≥ 12 years, randomised in a 2 : 1 ratio to miglustat or standard care. The primary end point was measurement of the velocity of visual saccades at 12 months, which increased in the standard care group and decreased in the treated group over 12 months. This difference was statistically significant, but only when the six patients taking benzodiazepines (which are known to slow saccades) were excluded. Clinically and statistically significant improvements were also seen in swallowing and ambulation. The non-controlled, open-label extension reported that the majority of adult and paediatric patients showed stabilisation of symptoms, based on assessments of horizontal saccadic eye movements, swallowing and ambulation. 213,214
In Spain, Pineda and colleagues215 have reported long-term follow-up data on a cohort of 16 children with NPC disease who have been treated with miglustat. They report that in patients who had symptom onset in childhood, there was a stabilisation of disease, but that patients with the more severe infantile-onset forms did not respond to therapy.
Safety
Miglustat is well tolerated in NPC patients in clinical practice, although the incidence of diarrhoea, flatulence and weight loss is quite high. The safety and tolerability of miglustat at 200 mg t.i.d. in study participants was found to be consistent with previous trials in GD1 in which half this dose was used, and the safety and tolerability in children has been found to be comparable with that observed in adults and juveniles. 213 Although changes to diet, involving the exclusion of sucrose and similar sugars, may help some individuals with gastrointestinal side effects, there are still significant numbers of patients who have to stop taking miglustat because of these side effects.
A summary of the main evidence from RCTs and other prospective studies on the efficacy and safety of miglustat for NPC is presented in Table 8.
| Study | Population (size, age, gender, country) | Specified aims | Study design | Outcomes/measures | Follow-up | Findings |
|---|---|---|---|---|---|---|
| Lachmann and colleagues (2004)206 |
N = 1 female NPC patient Age: 36 years UK |
Examining the biochemical response of a NPC patient starting miglustat |
Clinical case study Miglustat dose: 100 mg once daily |
Clinical monitoring of the function of the endosomal-lysosomal system in peripheral blood cells | Monthly for 6 months |
Reduced pathological lipid storage, improved endosomal uptake and normalised lipid trafficking in peripheral blood B lymphocytes cells was reported It was also reported that the clinical condition of the patient remained stable and that no progression of the disease was observed over the 6-month treatment period |
| Patterson and colleagues (2007)211 |
N = 29 NPC patients Miglustat treatment group (n = 20) Mean age: 25.4 ± 9.8 years Age range: 12–42 years Standard care group (n = 9) Mean age 22.9 ± 7.5 years Age range 13–32 years Miglustat treatment group: 9 males, 11 females Standard care: 5 males, 4 females USA and UK |
To establish the effect of miglustat treatment on several clinical markers of NPC severity |
RCT Randomised 2 : 1 ratio to either miglustat dose 200 mg taken orally t.i.d. for 12 months or standard symptomatic care |
HSEM velocity Neurological assessment including assessment of swallowing, auditory acuity, ambulatory ability and MMSE |
HSEM velocity was assessed at screening and at 12 months Swallowing assessment was performed at screening, 6 and 12 months Auditory acuity and ambulatory ability measured at screening and months 3, 6, 9 and 12 MMSE assessed at visit 1 or 2 and months 3, 6, 9 and 12 |
HSEM velocity was reported to have improved in patients treated with miglustat compared with those receiving standard care. Results were significant when patients taking benzodiazepines were excluded Improvements in swallowing capacity, stable auditory acuity and a slower deterioration in ambulatory index was reported for the treated group compared with the standard care group Improvement in MMSE was reported for the miglustat group |
| Chien and colleagues (2007)207 |
N = 2 (one patient with infantile-onset NPC and one patient with childhood-onset NPC) Ages: 9 and 14 years Taiwan, Province of China |
To examine the initial response and the maintenance of effects over 1 year of SRT | 12-month case reports | Liver and spleen volumes, plasma chitotriosidase activity, swallowing ability, ambulation index measurements and MMSE | 6 and 12 months |
Liver and spleen volumes and plasma chitotriosidase activities were stabilised in both cases Substantial improvement in swallowing and walking ability by month 6 from one patient The other patient was reported to have shown cognitive improvement in MMSE by 6 months which was sustained up to 12 months |
| Paciorkowski and colleagues (2008)209 |
N = 1 female NPC patient Age: 3.3 years USA |
To investigate the use of motion analysis system, among other standardised measures, to provide quantitative outcome data |
Case reports (Miglustat dose 40 mg t.i.d.) |
General and neurological examination, including growth parameters Lab analyses including AST levels 3-dimensional motion analysis of patient’s gait Developmental assessment: BDI-2 and Peabody Development of Fine Motor Scale |
3-monthly intervals for 12 months Motion analysis was performed at 6-month intervals, BDI-2 at 3 month intervals and the Peabody Development of Fine Motor Scale at 6-month intervals |
Dementia and motor dysfunction progressed despite the miglustat with the patient losing the ability to walk between 9 and 12 months of the study Mild elevation of AST level throughout, but other lab measurements were stable Decline in gait velocity and increased knee hyperextension at 6 months. Unable to walk at 12 months It was reported by month 3, the patient experienced some improvement in the adaptive and personal-social domains. This was not sustained, however, and at the end of the study abilities in these domains had declined to < 0.1 percentile rank |
| Santos and colleagues (2008)208 |
N = 1 female Age at start of miglustat: 10 years Brazil |
To assess the initial impact of miglustat on a NPC patient |
Case report (Miglustat dose 100 mg t.i.d. and after 10 days, dosage increased to 200 mg t.i.d.) |
Clinical disability score Parent-reported CBCL to assess psychopathological, behavioural and social problems |
12 months |
Reduced disability score due to significant improvement in ataxia, dysarthria, better balance and good seizure control observed within 40 days of miglustat and were sustained at 12 months It was reported that miglustat had a positive impact on cognitive function as well as on psychopathological symptoms of depression and of affective and thought problems as assessed by the CBCL |
| Galanaud and colleagues (2009)210 |
N = 3 Ages: 21, 23 and 38 years Two males, one female France |
To assess the efficacy of miglustat |
Case studies (Miglustat initial dose of 600 mg/day; that was decreased to 300 mg after 3–6 months in two patients) |
Clinical assessment of swallowing, dysarthria, awareness and ambulation Brain magnetic resonance spectroscopy including Cho/Cr ratio |
Assessed at baseline, and after 12,18 and 24 months |
All patients reported mild clinical improvement or stabilisation after 24 months on treatment Sustained decrease in the Cho/Cr ratio was observed in all three patients over time |
| Patterson and colleagues (2010)213 |
N = 12 Age: 7.2 ± 2.5 years Age range: 4–11 years 5 males, 7 females USA and UK |
To examine the long-term effects of miglustat on the disease progression of NPC patients |
Initial 12-month, open-label, non-controlled treatment period plus a prospective 12-month, open-label, extended phase extension Miglustat dose 200 mg orally t.i.d. according to patient body surface |
Change in HSEM Neurological and neuropsychological assessments Assessment of swallowing and ambulation index |
Baseline, 12 and 24 months |
It was reported that a decrease in mean HSEM asymptotic peak velocity suggested an improvement at month 24, although mean HSEM initial peak velocity-amplitude slope values increased (i.e. worsened) from baseline to month 12 but did not deteriorate further to month 24 Mean SAI showed a small increase (slight worsening of ambulation) Swallowing ability appeared stable at 24 months |
| Wraith and colleagues (2010)214 |
N = 25 At recruitment to original study: Mean age: 24.6 ± 9.1 years Age range: 12–42 years USA and UK |
To examine the long-term efficiency and safety of miglustat in juvenile and adult patients participating in a subsequent non-controlled, open-label extension phase | Non-controlled, open-label extension of the RCT reported by Patterson and colleagues212 | HSEM-α, HSEM-β, swallowing evaluations, SAI and MMSE | Screening, baseline and months 12 and 24 |
Mean absolute HSEM-α value decreased (indicating improvement) from baseline among patients completing 12 months of miglustat therapy, and was stable (compared with baseline) among patients completing 24 months on miglustat Mean HSEM-β values increased (indicating mild deterioration) from baseline among patients completing 12 months of miglustat therapy as well as those completing 24 months on miglustat Swallowing was improved or stable (compared with baseline) in 86% of patients completing 12 months of miglustat therapy Minimal change in mean SAI scores and slight increase in MMSE for those who completed 12 months and 24 months of miglustat treatment |
| Pineda and colleagues (2010)215 |
N = 16 symptomatic NPC patients (5 early infantile, 4 late infantile and 7 juvenile) Age range: 1.3–15.6 years Nine males, seven females Spain and Portugal |
To evaluate the efficacy and tolerability of miglustat |
Case series study Various dosages based on body surface area (30–100 mg t.i.d. and 50–200 mg b.i.d.) |
Clinical assessment [neurological examination, modified functional disability scale and cognitive development evaluation (DDST and WIS-R)], biochemical analyses (plasma ChT and PARC activities) and imagining studies | Full assessment battery at baseline, months 6 and 12 and every year thereafter. Neurological examinations and biochemical analyses at screening and months 4 and 8. Disability scale assessment were performed every 4 months |
Cerebral hypometabolism and neurological symptoms were stabilised during treatment in juvenile-onset NPC Early-infantile and late-infantile NPC patients, who had higher disease severity at baseline, showed increased disability scores and progressive cerebral hypometabolism during follow-up Although cognitive scale scores remained relatively stable in patients with juvenile NPC, cognition deteriorated in early-infantile and late-infantile patients |
| Fecarotta and colleagues (2011)211 |
N = 3 female NPC patients with significant dysphagia (One late infantile patient and two juvenile patients) Ages at start of treatment: 9.6, 9.4 and 12 years Italy |
To examine the long-term effects of SRT on swallowing ability and dysphagia |
Observational long-term study [SRT dose ranging from 250 to 300 mg/mq/day divided in three doses for 3 years or longer (36–48 months)] |
Clinical evaluation of swallowing using radiological videofluoroscopic techniques Neurological involvement |
Periodically assessed (either every 3 or 6 months) for 48 months |
All patients with abnormalities at baseline showed improvements in swallowing ability after treatment The authors also reported that improvement of swallowing co-ordination occurred in parallel with improvement or stabilisation of neurological conditions |
Patterns of treatment in England
Treatment centres
Services for patients with LSDs, including treatments such as ERT and SRT, are overseen by the National Specialised Commissioning Group (NSCG) [formerly the National Commissioning Group (NCG)], an arm of the Department of Health, which plans and funds the provision of care for very rare conditions (www.ncg.nhs.uk/).
When this study commenced in 2006, six hospitals in England had been nationally designated and funded by the NSCG, to provide a service for patients with LSDs.
Centres for children
-
Central Manchester and Manchester Children’s University Hospitals NHS Trust.
-
Great Ormond Street Hospital for Children NHS Trust.
Centres for adults
-
Salford Royal NHS Foundation Trust.
-
Royal Free Hampstead NHS Trust.
-
University College London Hospitals NHS Foundation Trust, National Hospital for Nervous Diseases, Queen Square.
Centres for adults and children
-
Addenbrooke’s NHS Trust.
Birmingham Children’s Hospital NHS Foundation Trust received NSCG-designated status shortly afterwards in 2007.
It was estimated that the total number of patients with a LSD known to these centres was 1455 in 2006. As would be expected from prevalence data, the most common LSDs are Gaucher disease and Fabry disease in adults and the mucoploysaccharidosis disorders (in particular MPS I and MPS III) in children.
At present, Wales, Scotland and Northern Ireland have separate prescribing arrangements and there are no precise data as to the numbers of patients with LSDs living in these regions, although some do receive care at the designated centres.
Rationale for the cohort study
Most people who have LSDs suffer substantial morbidity and most have a reduced lifespan. All of these conditions are extremely rare. It has proved difficult to conduct large-scale clinical trials, and current evidence of efficacy of ERT is based primarily on small, relatively short duration trials, often with surrogate outcomes and open-label studies. All of the recombinant enzymes marketed thus far are extremely expensive.
Currently, there are several lysosomal condition-specific databases that are held by the pharmaceutical companies which manufacture the ERTs and the SRT currently licensed in the UK. This has led to the development of two registries for Fabry disease, which do not appear to be compatible with each other, hindering comparisons of treatment efficacy. The Society for Mucopolysaccharide Diseases (UK) also has a registry of all UK people diagnosed with an MPS disorder since 1981.
It was felt necessary to establish this UK cohort study independent of the pharmaceutical industry, not least because the intention was to collect data on some LSDs for which treatments are not yet available. In addition, given that there are currently three pharmaceutical companies that manufacture these treatments, to conduct the study with any one of the companies might have led to potential conflicts of interest.
It has been argued18,19 that there is currently little point in conducting further studies of effectiveness or cost-effectiveness of ERT in Gaucher disease, Fabry disease or MPS I. This is based on the argument that the costs of the drugs are currently so high that however effective these treatments are there is no possibility that they can cross currently accepted thresholds for willingness to pay. The authors of these reviews argue that, if society has decided that because of the particular rarity and severity of these conditions it is willing to pay for therapy, then further information is not required, whereas if society is to apply the thresholds generally used to make such decisions, no amount of information will move the decision across this threshold.
In our view this stance, although arguable, is mistaken. Better estimates of the effectiveness of the interventions, of the relative effectiveness of treatment depending on when in the course of the condition treatment begins and of different treatment regimens are important for patients and their families as well as for clinicians. The costs of the drugs may well change substantially in the future with changes in technology and the possible entry into the market of other providers. In these circumstances evidence of effectiveness will be needed to underpin decisions on cost-effectiveness. This study aimed to provide at least partial answers to these questions in addition to providing better data on NHS costs to inform future estimates of cost-effectiveness.
Background and objectives
The National Collaborative Study (NCS) LSD study is a longitudinal, cohort study of adults and children with LSDs, who are treated within the seven NSCG (formerly the NCG)-designated treatment centres in England. The study aimed to determine the natural history of the LSDs under investigation, and to estimate effectiveness and cost-effectiveness of treatment strategies.
Lysosomal storage disorders represent a group of less than 40 genetically distinct diseases. This study included patients with six of these conditions for which ERT or SRT is currently available or is being developed:
-
Gaucher disease
-
Fabry disease
-
MPS I
-
MPS II
-
Pompe disease
-
NPC.
The limitations of an observational cohort study design are acknowledged. A prospective RCT of the appropriate treatment in each condition with clinical end points and long-term follow-up would clearly provide the highest quality evidence of effectiveness. Such trials are currently lacking and it is believed that recruitment is unlikely to be feasible, particularly for therapies already in widespread use and given the relatively small number of people with these conditions.
Primary objectives
-
To compare the natural history of treated and untreated LSDs for those disorders where a specific therapy is currently available.
-
To estimate the effectiveness of ERT and SRT.
-
To estimate the cost-effectiveness of specific therapies for LSDs.
-
To describe the natural history of LSDs where ERT is likely to become available.
Secondary objective
-
To compare the effectiveness of agalsidase alpha with agalsidase beta in children and adults with Fabry disease.
-
To estimate the lifetime health-care cost and other economic impacts on people with LSDs and their families.
-
To provide the basis for future research to develop treatment–responsive measures in adults and children.
Chapter 2 Methods
Study procedures
Local collaborators
Clinical consultants at each of the seven study sites acted as local principal investigators for the study. A formal participation agreement was signed for each centre between the local principal investigators, the participating NHS Trust Research and Development Directors and the Peninsula College of Medicine & Dentistry (PCMD). Where possible, a research nurse was employed to recruit patients into the study, collect patient clinical, QoL and service-use data, according to the study protocol. Where it was not possible to recruit a nurse into this role, a researcher was employed. The research nurse or researcher was responsible for all aspects of the study conduct at their site, and for ensuring that the study was run ethically, and in accordance with good clinical practice. The number of hours each nurse or researcher was employed to work on the study was based on the number of LSD patients treated at their clinic.
At the outset, regular meetings and teleconferences were held with the management team in order to design and agree processes and systems. Following the commencement of data collection, both nurse/researcher meetings and management meetings were held every 6 months. In addition, monthly teleconference meetings were held with the nurses and researchers.
Study Steering Committee
The running of the project was under the supervision of a Study Steering Committee, chaired by Professor Richard Hobbs (Professor and Head of the Department of Primary Health Care, University of Oxford). The study Steering Committee met on four occasions during the project and provided advice throughout the course of the study (see Appendix 4).
Ethics and research governance
Overall ethical approval for this study was obtained from the South West Multicentre Research Ethics Committee (MREC) in February 2007 (REC reference 06/MRE06/70). In addition to the overall MREC approval, each local centre obtained their Local Research Ethics Committee (LREC) site-specific approval and local NHS Trust Research and Development approval prior to starting the study.
Subsequent approvals for amendments to the protocol were sought and obtained from the MREC. Details of these amendments are provided in Changes to original protocol.
Patient and public involvement
From the outset, several of the LSD patient advocate groups in the UK were invited to be part of this observational study and to co-write the original funding bid. The Gauchers Association accepted this invitation and agreed to assist in the initial proposal for funding, as a representative organisation for all UK LSD support groups. The management team arranged regular meetings with the support groups (collectively and at their individual meetings) and a representative from these groups was always invited to attend each of the management group meetings.
The support groups advised the research team on all aspects of study design. In particular, they reviewed the content of materials such as the Patient Information Sheet (PIS) and Patient Consent Form, and made recommendations regarding the choice of outcome measures and on ethical and feasibility questions. They also provided invaluable support in assisting with the process of sending a final set of questionnaires to the participants, and wrote a letter to their support group members urging them to participate.
Participant identification and consent
The process of identification and consent for patients is summarised in Figures 1 and 2.
FIGURE 1.
Flow diagram for identification and consent for adults with LSDs.
FIGURE 2.
Flow diagram for identification and consent for children aged < 16 years with LSDs.
Inclusion/exclusion criteria
All patients with Gaucher disease, Fabry disease, MPS I, MPS II, Pompe disease or NPC, living in the UK and attending one of the designated treatment centres, were considered for inclusion in this study.
Patients were deemed ineligible for inclusion if their treating clinician felt they would be distressed in any way by being approached or by participating in this study.
Identification
Identification of eligible patients was aided by the way in which LSD patients are cared for in the UK. The majority of people with LSDs are under the care of one of the NSCG specialist centres, regardless of whether or not there is a therapy available. Only these centres are able to prescribe ERT or SRT, and therefore most patients diagnosed with these conditions are referred to one of these centres.
The research nurse or researcher identified all eligible patients from the hospital department database or department patient lists. The patient’s initials and date of birth were entered into a condition-specific recruitment spreadsheet and a study ID was assigned to each patient. The LSD consultant was asked to confirm the patient’s eligibility to participate in the study as per the inclusion/exclusion criteria. Eligibility status was entered into the spreadsheet and eligible patients and/or their carers were sent a letter introducing the study (see Appendix 6).
Invitation letters and PISs (see Appendix 8, Patient Information Sheet for participants and Patient Information Sheet for carer) were sent to patients and/or their carers at least 1 month before they were due for their clinical review appointment. This ensured the patient had sufficient time to read and absorb the information, and also have the opportunity to discuss the study with relatives, general practitioners (GPs), research staff, etc.
Some patients were missed and hence not recruited by the researcher and/or the LSD consultant at their first clinical review appointment after receiving their invitation to participate in the study. This might have been due to patients not attending, or conflicting researcher and/or consultant commitments on the day of their attendance. In such cases a second invitation letter and PIS was sent 1 month before the next clinical review appointment (see Appendices 7 and 8).
Explanation of the study
The LSD consultant or research nurse explained the study to the patient and/or their carers at their hospital visit, using the appropriate PIS. The QoL, fatigue and service-use questionnaires were explained to the patient and/or their carer allowing sufficient time for each patient and/or their carer to ask questions and have them answered to their satisfaction.
Consent
The LSD consultant or research nurse sought written, informed consent to participate in the study from each participant [see Appendix 9, Consent form for participants and Consent form for participants (notes only)], parent or carer [for children aged < 16 years, see Appendix 9, Consent form for parents/carers and Consent for parents/carer (notes only)].
Two-tier consent: full versus ‘notes only’ consent
The study operated a two-tier consent process whereby if a patient (or his or her parent/carer) did not wish to complete the QoL, fatigue or resource use questionnaires, they were asked if they agreed to their data being extracted from their medical notes for the purposes of the study [see Appendix 9, Consent form for participants (notes only) and Consent form for parents/carers (notes only)] – this was referred to as a ‘notes only’ consent.
Consenting patients who lack capacity
Following consultation with the MREC and in accordance with the Mental Capacity Act 2005, the research team made an initial assumption that each patient had the capacity to give informed consent and, as such, every effort was made to support patients to make their own decision regarding participation in the study. Information about the study was provided to each individual in a way that was deemed most appropriate to help them understand the study and make their own decision.
If the treating clinician or another member of the health-care team believed, on the balance of probabilities, that the individual lacked capacity to give informed consent, then steps were taken in order to identify someone to consult (and thereby consent on behalf of the patient – a ‘personal consultee’) before the patient could be included in the study. It was essential that the consultee was involved in the patient’s care, interested in their welfare and willing to help. In accordance with the Mental Capacity Act, this person could not be a professional or paid care worker.
Where there was no willing ‘personal consultee’, the researcher/LSD consultant nominated a person to be the consultee. The nominated person must have had no connection with the research project.
The consultee was given information about the research project and asked:
-
for advice about whether or not the person who lacked capacity should take part in the study; and
-
what they thought the person’s feelings and wishes would be if they had the capacity to decide whether or not to take part.
Once a willing consultee had been identified, they were asked to provide written, informed consent on behalf of the patient [see Consent for consultees or Consent form for consultees (notes only)].
Reconsenting 16-year-olds
When a study participant reached 16 years of age, and the LSD consultant confirmed their continued eligibility to take part in the study, they were approached for reconsenting using adult (self-) consent forms.
In situations where the parent/carer had not consented to their child’s participation in the study, it was agreed with the MREC that the nurse or LSD consultant was free to approach the patient directly for consent when the patient turned 16 years old.
Similarly, when the parent/carer had given ‘notes only’ consent for their child’s participation in the study, it was agreed with the MREC that the researcher was free to approach the patient directly for full consent when the patient turned 16 years old.
Informing the patient’s general practitioner of participation in study
Once consent had been obtained from the patient or carer, a PIS and letter notifying them of the patient’s involvement in the study was sent to the patient’s GP (see Appendix 8, Patient Information Sheet for participants and Appendix 10).
Data collection
Data were collected on all consenting patients and entered onto the condition-specific database by research nurses at each centre. Each database followed the same structure with (a) a standard set of data fields common to all conditions and (b) condition-specific data fields. Clinical, QoL and service-use data were collected prospectively and clinical data were collected retrospectively. Historical, retrospective data were ascertained from patients’ notes and from Hospital Information Systems (HISs). Patient data were entered anonymously, using the patient’s study ID.
Following a tendering process, macro electronic data capture from MACRO™ Electronic Data Capture, version 3 (InferMed, London, UK) was chosen as the platform to build the database for this study. The secure, web-based electronic data collection (EDC) system allowed each site to enter data remotely, while data were held on a server within the PCMD at Plymouth University. A condition-specific database, with a common front-end, was designed for each condition by the study co-ordinator and database manager. The database allowed an unlimited number of prospective and retrospective data points to be collected, provided an audit trail of all data entries or amendments and allowed discrepancies or queries to be raised by the co-ordinating centre.
Identification of outcome measures
Our aim was to identify for each condition a limited set of outcome measures that would capture key areas of function and symptoms. Although we were able to ask patients to provide some data during the course of the study (e.g. QoL scales) we were not in a position to add new clinical investigations, and where possible we wished to be able to collect equivalent historical clinical data from hospital records. Criteria for selection of measures therefore included the clinical significance of the target area of function, reliability of the specific test suggested, ability of the measure to reflect change and clinical co-applicants’ views of likely availability of data across the sites.
Selection of clinical outcome data
Clinical data fields were agreed within the management team prior to the commencement of the study. The choice of outcomes was guided by the principle that only data which would clearly contribute to determining the extent of disease severity and progression would be collected. The process of identifying the clinical data fields was initially piloted with Gaucher disease.
Working within disease-specific groups, the clinicians from the seven sites identified the key organ(s) or system(s) or area(s) of function affected by each disorder. They were then asked to identify what measures they believed would most reliably assess each aspect of the condition, reflect disease progression and were likely to be assessed for most patients as part of routine clinical appointments. They were also asked to consider whether common or equivalent approaches to assessment of each measure were carried out at the seven participating centres, and whether or not the assessments were different for children and adults.
For each condition, the agreed data fields included biochemical and clinical investigations that are routinely collected at the treating centres, as recommended in relevant UK National Guidelines. 217–221 These condition-specific guidelines are written by a multidisciplinary group of clinical experts, a non-clinical expert chairman and patient representatives, at the invitation of the NCG of the UK.
The final process of refining the final data set for each condition was hampered by a difference of opinions between the clinical co-applicants. Although all UK treating centres practise in accordance with the National Guidelines, it became apparent that the exact series of tests and investigations carried out differs from centre to centre. Further communications within the working groups via email, teleconference and face-to-face meetings eventually generated a data set to be collected for each disorder. It was recognised at this stage, however, that the data sets contained some tests and investigations that were not conducted at all sites. Nonetheless, it was accepted that such fields should remain in the database and data would be captured where available.
A series of Case Report Forms (CRFs) was then created for each condition with each page pertaining to a specific organ, system or function (see Appendix 11). Where appropriate, forms were adapted to capture data from children. These CRFs were subsequently used to model the electronic CRFs (eCRFs) of the database. The final analysis focused on key fields that were identified by the clinical co-applicants after data collection was complete. These fields are discussed in the condition-specific results chapters.
Retrospective data fields
Although it was evident that it would not be possible to obtain the historical data for all clinical fields for every patient, it was agreed that all data fields should remain ‘active’ in the retrospective databases and that nurses and researchers at each site would endeavour to collect all information available.
Clearly, retrospective QoL and service-use data were not available as these had not been routinely collected.
Patient-reported outcomes
As with clinical data, the aim of including patient-reported outcomes was to ensure the assessment of the key impacts these disorders have on patients and their families and carers. Our choice of measures was based on consultation both with clinicians involved in the care of patients with LSDs (including members of our external steering group) and with the relevant patient support groups and associations. We based our initial ideas about possible measures to include on existing research evidence. These were summarised in a ‘non-clinical data collection plan’ that formed the basis for discussion and agreement with all co-investigators and the patient support groups involved.
Those consulted were asked firstly to consider what the most important impacts of the conditions were and secondly what measures might be employed to assess these aspects. They were also asked to keep in mind the need to limit the burden of data collection on patients and carers.
Quality of life in adults
We aimed to assess the HRQoL of LSD patients. The two best-established generic HRQoL measures are the Short Form questionnaire-36 items (SF-36) and the European Quality of Life-5 Dimensions (EQ-5D or EuroQol instrument). 221 Both have associated preference-based scoring systems that allow the calculation of ‘social preference weights’ (utilities) for each defined health state, and such weights are available from valuations by the UK general population. For the SF-36, this is done via the Short Form questionnaire-6 Dimensions (SF-6D) health state classification system. 222 The EQ-5D and SF-36 are also the preferred sources of utility estimates for health technology assessments conducted for the National Institute for Health and Clinical Excellence (NICE). 223 We chose to use both the SF-36 and the EQ-5D because there is ongoing debate about the validity, reliability and responsiveness of the two instruments in different disease areas, and also because the additional respondent burden of completing the EQ-5D is small. 224
Quality of life in children
We considered three candidate questionnaire-based instruments for measuring HRQoL in children that were available in mid-2007 – the Pediatric Quality of Life Inventory (PedsQL™), the Child Health Questionnaire and the KIDSCREEN instrument. Both the PedsQL and Child Health Questionnaire are recommended by the 2001 HTA monograph on Quality-of-life Measures in Chronic Diseases of Childhood. 225
After examining the research literature and consultation with the patient support groups, it was decided that the PedsQL instrument was the most appropriate for this study. The PedsQL was originally developed for assessing the QoL of children with cancer, but has now been used and validated in a wide number of disease areas. 226 The PedsQL’s 15 questions cover the three main domains of physical functioning, psychological functioning and social functioning. Importantly, it includes both child- and parent-reported QoL in all versions except for the youngest children.
Fatigue
Patient support groups were concerned that the chosen generic measures of HRQoL might fail to capture fatigue, which they regard as a prominent feature of many LSDs. In addition, anecdotally, reduced fatigue and improved vitality were reported by some patients to be one of the perceived benefits of being on treatment. This led us to include the Fatigue Severity Scale227 among the suite of instruments that adult patients were asked to complete (see Appendix 14).
The FSS is a nine-item self-report questionnaire, in which patients are asked to respond using a seven-point Likert scale. Responses are added together and averaged to determine the overall score. Scores > 4 are considered to be indicative of significant fatigue. 228,229
Assessment of impact on carers
The health, cost and other impacts on the parents or carers of people with chronic and serious conditions is well documented and increasingly recognised as an important aspect of economic evaluations. 230 We initially considered assessing the QoL of parents/carers and the partners of adult patients as part of this study, but after consultation these measures were omitted on the basis of the burden and complexity of data collection. Instead, we elected to include a well-established measure for assessing the impact of patients on their main carers, the Carer Strain Index (CSI)231 (see Appendix 12).
Cost data collection methods
The funded research protocol included a cost–utility analysis and, therefore, required the collection of cost data as part of its aim to describe the potential economic and non-clinical outcomes of ERT and SRT.
Discussion with the clinician co-investigators suggested that hospital clinical records would only reliably collect information about those treatments, appointments or tests that took place in their hospital or unit. Furthermore, each specialist treatment centre has different arrangements for ‘shared care’, further complicating the possibility of collating NHS patient records for the LSD study patients. We therefore concluded that our primary mode of data collection for NHS and other costs should be to directly ask people with a LSD, or their parents or carers, about their level of use of health care and other services. This questionnaire-based data collection took place at their annual review appointment at the specialist LSD units.
Service-use and costs questionnaire
The Client Services Receipt Inventory (CSRI) is a comprehensive set of questions that has become used in many large UK-based cost and cost-effectiveness studies since the early 1990s, especially where the client group’s needs typically span both health services and social care (e.g. older people or people with mental health problems). 232,233 The questions cover use of hospital services (whether inpatient, outpatient, day care or accident and emergency), general practice and primary care, as well as a wide range of other community-based health- and social-care services or professionals from whom people may receive care or other help. As the names and types of available services have changed over time, and because of variation in the purpose of different costing studies, the actual detailed content of the questionnaire is not highly standardised.
An up-to-date version of the CSRI was kindly supplied by one of its developers (Professor Martin Knapp, Personal Social Services Research Unit, London School of Economics). In consultation with both clinicians and the patient and family support associations, we piloted and adapted the questionnaire in a number of ways. In particular we:
-
removed a question about contact with police and judicial services
-
included a new question on the out-of-pocket costs incurred by patients or their families
-
amended the list of other services outside of hospital that people might have used
-
included a question on the number of short-term absences from work (or school) owing to health problems, and the number of days involved in these
-
adapted the question to assess the amount of help (in hours per week) ‘from friends or relatives as a consequence of your health problems’ (e.g. for child care, personal care, help around the house, help outside the home, transport).
Following a suggestion from the patients/family associations, we also asked an open question about any ways in which people felt their LSD ‘has or may have constrained your career (such as, missing a promotion; having to choose a less stressful job; having to cut your usual number of hours)’. There were two versions of the final questionnaire, one for self-completion by adult patients and the other for proxy completion by the carers or parents of children or adults unable to complete the questionnaires themselves. The final resource and service-use questionnaires (known as Service Use and Costs Questionnaires) are shown in Appendix 13.
Summary of health-related quality of life and carer impact measures used
Table 9 shows the full range of QoL and carer impact questionnaires that were used, and the ages of patients to whom they were given.
| Instrument | Version | Age of patients (years) |
|---|---|---|
| SF-36 | Version 1.0 | ≥ 16 |
| EQ-5D | UK/Eire | ≥ 13 |
| PedsQL | Version 4.0 – UK English | |
| PedsQL 2–4 | 2–4 | |
| PedsQL 5–7 | 5–7 | |
| PedsQL 8–12 | 8–12 | |
| PedsQL 13–16 | 13–16 | |
| Age-relevant child- and parent-completed questionnaires | ||
| NCS-LSD Service Use and Costs Questionnaire | Child proxy | 0–16 |
| Adult | ≥ 16 | |
| CSI (if applicable) | All ages | |
| FSS | ≥ 16 |
Data collection time points
Prospective data
The Department of Health has issued UK National Guidelines for each condition under investigation. 216–220 These guidelines advise a follow-up assessment protocol for each condition. Although clinical practice differs at each of the seven treatment centres, it was evident that most patients are invited to attend their clinic for an annual review when they would undergo a set of routine clinical investigations. It was agreed that patients should be invited to consent to participate in the study at their annual appointment, at which time clinical data would be captured from the hospital records of consenting patients.
Follow-up data were collected at each subsequent annual visit. Each patient was asked to complete the relevant pack of QoL and service-use questionnaires at study entry and at each subsequent annual review.
Retrospective data
For patients on ERT or SRT, we aimed to capture clinical data from the following retrospective time points:
-
at diagnosis (in some situations this would coincide with the start of treatment)
-
12 months before the start of treatment.
-
at the start of treatment.
In addition, for people who have been on treatment for several years, data were collected at:
-
1 year post commencement of treatment
-
2 years post commencement of treatment
-
4 years post commencement of treatment
-
every 2 years thereafter until start of prospective data collection.
For patients not on ERT or SRT we aimed to record yearly data points going as far back as possible.
It was recognised that these were ideal scenarios and it was not always possible to collect data at these exact time points. The research nurses and researchers were asked to use reasonable judgement in determining the most useful time point at which to extract data.
Data collection
Clinical data
The research nurses and researchers extracted the required clinical data from the patient’s medical records and/or the HIS and entered it into a CRF. For each time point (see Retrospective data), results from the most recent test carried out in the previous 12 months were used.
For both prospective and retrospective data collection, the date of each test was entered into the database. If the exact date of the test was unknown, the ‘15th’ of the month was entered. If the month was unknown, then ‘June’ was entered.
Quality-of-life and service-use data
Questionnaires were given to patients (or their carer/parent) who had consented to have the additional questionnaires at their annual check-up appointment with their clinician (see Two-tier consent). They were also asked to complete additional questionnaires at any additional monitoring appointment they attended. The patient had the right to refuse to complete the questionnaires at all times.
Although it was intended that these should be completed by patients (or their parents/main carer) during their clinic visits, in practice many people were unable to complete them during this time. The research nurse or researcher at each centre therefore put systems in place that enabled patients to take home some or all of their questionnaires and return them by post. In certain circumstances, for instance if there was a problem with the provision of ERT, and where it would assist in our understanding of the effectiveness of these treatments, additional questionnaires were posted to patients who were attending clinic less often. Similarly, in circumstances in which study patients attended for routine clinical follow-up but were not seen for study purposes, follow-up questionnaires were posted to them. In all such situations it was made clear in an accompanying letter that patients were under no obligation to complete these or to remain in the study.
The patient ID was always written on the top of each sheet in the pack either before or after completion. This was done before the questionnaires were removed from the treating centre if they were taken home by the patient.
Where questionnaires were not completed during the clinic visit we contacted the patient or their carer up to a maximum of two times at 2-week intervals. Each time a patient or their carer was contacted by the research nurse or researcher, a dated record of the conversation or message left was entered in the database.
Data entry
Data collected at the patient’s annual review were entered into a CRF and then entered into the database by the research nurse or researcher. Similarly, retrospective data extracted from patients’ notes and/or HIS were collected into a CRF and entered as a retrospective time point in the database.
Once returned, the QoL and service-use questionnaire answers were entered into the database and the paper copies kept in study folders.
Collection of additional data owing to shortage of enzyme replacement therapies
For reasons explained in Chapter 1, there was a worldwide shortage of imiglucerase and agalsidase beta during the study period and, as a result, some Fabry disease and Gaucher disease patients had their treatment regimen altered during this time. The clinical experts agreed that additional clinical data should be captured during this period in order to assess disease progression on reduced or altered treatment. It was anticipated that patients on altered regimen would be monitored more closely during this period, and would visit their clinicians on a more regular basis than usual. Clinical data were entered on the study database each time patients visited their LSD clinic. In addition, approval was gained from the South West MREC to ask these patients to complete additional QoL and service-use data questionnaires each time they attended their clinic.
However, in reality, patients receiving home infusions did not visit their clinic for additional monitoring visits unless they experienced additional complications. Therefore, while some additional data were received from patients on altered regimen, this was not as many as was initially anticipated.
Data quality assurance
In order to minimise the level of risk of data errors, data checks such as valid ranges, filter checks and logical checks were built into the eCRFs within the database. At least 10% of all data were checked using source data verification (SDV).
Monitoring visits and source data verification
Each site received a monitoring visit approximately 1 year following commencement of data collection, with additional visits arranged where there was a change in personnel or if there was any concern over the conduct of the study for any reason. The study site file was checked for completeness at these visits and consent forms of all participating patients were checked for conformity.
Using an online random number generator, approximately 10% of patients at each site were randomly selected to be verified against source data at the monitoring visits. Source data included patient medical records, any letters held within these records and any clinical data held on the HIS. For QoL and service-use data, the database entry was checked against the paper copy of the questionnaire completed by the patient and/or their carer. Some site monitoring visits were carried out in-house by the study co-ordinators, whereas others were subcontracted to a Clinical Research Organisation.
Methods used in analysis
Effectiveness of treatment
Mixed-effects models
The database contains longitudinal individual-level patient data for all consenting patients attending the participating treatment centres. In the core analysis, linear and generalised linear mixed models were developed to study individual dynamics for the selected continuous and binary outcomes, respectively. 234 Separate models were fitted for each condition owing to the heterogeneity in age at diagnosis, clinical presentation and rate of disease progression for the six LSDs. Each model related the relevant outcome (or suitable transformation of the outcome) to a linear combination of the patient’s age at the time the outcome was measured, their gender and the time from commencing treatment on ERT or SRT, to the current visit. Treated patients contributed data (where available) from the period before they were first treated as well as during treatment, providing aspects of a before-and-after design. Untreated patients contributed natural history data of relevance to estimating the effect of age at the time of the measurement. All analyses were based on complete records without imputation of missing data. This approach provides valid estimates of treatment effects under the assumption that the missing data mechanism is ‘missing at random’. 235
In the base models, it was assumed that the temporal components had independent additive linear effects on outcomes. Non-linear effects of time on treatment were explored by categorising time on ERT or SRT for Gaucher disease, Fabry disease and Pompe disease and by fitting generalised additive mixed models with smoothing splines. 236 Patient heterogeneity in disease severity at diagnosis and the rate of disease progression was addressed by fitting random intercept and random slope models. Likelihood ratio tests were used to compare the random intercept and random slope models. Separate models were fitted for adults and children where appropriate.
For each condition, there are differences in the demographic profile and underlying severity of patients seen by each of the centres, and clearly for many of the outcomes there is significant variability in the way these are assessed by individual observers across the centres. Clustering of patients within centres was explored by fitting hierarchical models with a random centre effect. Choice of a random effect for centre made it possible to assess the relative contribution to variance of centre- and individual-level effects. A sensitivity analysis was conducted in which all longitudinal models were fitted with a fixed-centre effect. The results for the effect of time on ERT were very similar (please contact the authors for a full tabulation of the results of modelling centre as a fixed effect).
For each condition, treatment efficacy was assessed based on the estimated effects of time since first infusion from the mixed-effects models described above. Where data were not available for significant numbers of untreated patients, model estimation of treatment efficacy takes advantage of the fact that the age and stage of their condition at which patients have begun taking ERT was dependent on the time when the treatment first became available. Historical data are available for many of these patients on their clinical condition at the time of beginning treatment while for others we have data only on their current clinical situation.
Potential sources of confounding bias were addressed through adjustment of the regression models for age and sex, and, where appropriate, through stratification (e.g. fitting separate models for splenectomised and non-splenectomised patients when assessing the effect of ERT on platelet counts in Fabry disease). Issues with data quality and low sample size made it impractical to consider more comprehensive adjustment for potential confounding factors. We prioritised the development of Bayesian predictive models for Fabry disease and Gaucher disease, as these conditions had sufficient data to compare the effects of starting ERT at different points in the disease process. Given space limitations in the report, we only presented illustrative results for outcomes which showed evidence of a beneficial effect of ERT in frequentist analyses.
Cox regression models
Kaplan–Meier survival curves were estimated to illustrate differences in the age at first recorded occurrence of binary events that could be considered progressive (e.g. restricted mobility or hearing loss). Treatment group differences in survival function were tested using Cox regression models (ERT compared with not on ERT). Individual patients entered the risk set on recruitment to the study and could contribute intervals of time at risk to more than one of the treatment categories; patients for whom there was outcome data prior to starting treatment contributed time at risk to both the ERT and untreated curves.
Bayesian longitudinal models
Given the rare nature of these disorders and the corresponding modest sample sizes, conventional analyses may not have the power to detect or exclude clinically worthwhile treatment benefits. Consequently, additional assessments of treatment efficacy were conducted in a Bayesian framework for selected outcomes to supplement analyses using classical methods. Although definitive answers are not always possible, taking a Bayesian approach can provide a clearer guide by quantifying the probabilities that clinical effects lie in a particular range. 237 These probabilities, calculated by combining study data with a prior distribution, apply directly to future patients and can be used explicitly in formal decision analysis.
Bayesian versions of the mixed-effects models were also used to make predictions about the expected trajectories for patients starting ERT at different ages. This was achieved by specifying the effect of time on ERT as a quadratic function, with parameters specific to the age interval during which patients started on ERT. Means of the posterior predictive distribution were plotted against age to illustrate the model findings. Non-informative priors were chosen for the fixed and random effects in each model.
Natural history
The mixed-effects models described above provided the basis for characterising the natural history of treated and untreated LSDs for individual outcome measures. The estimated age effects summarise the expected age-related change in outcome for untreated patients. Given the constraints on the overall sample size and the number of untreated data points per patient, the mean outcome trajectory for each condition was approximated by a linear function of age. Further analysis of natural history was conducted by exploring dynamic linear growth curve models in a Bayesian framework with patient-specific random effects, and non-informative priors for the mean and slope parameters. 238
Non-linear effects of age were considered by adding a quadratic age term to the Bayesian models. Models with linear age effects are presented except where strong evidence for non-linearity was identified.
Comparison of the effectiveness of agalsidase alpha and agalsidase beta in Fabry disease
Both agalsidase alpha and agalsidase beta are licensed for use in the UK for the treatment of Fabry disease. Both treatments received their licence in 2002. There is a fivefold difference in the licensed dosing regimen, although costs per patient are broadly similar. It appears that, although all centres use both drugs, there has been tendency for each centre to use one or other as their initial drug of choice. This appears to have been determined mainly by historical reasons, based partly on which drugs trials the centres were initially involved in. Some patients subsequently switch to the alternative treatment, for clinical reasons, and it has been suggested that more recently there may be more variety in initial drug choice. There are National Guidelines216–220 for the initiation of therapy to which all centres adhere that suggest that the populations receiving either treatment are likely to be broadly similar.
We compared the outcome of treatment depending on which of the two drugs patients were initially assigned (the equivalent of an intention-to-treat analysis) in a multivariate model allowing for potential confounding variables. The model partitioned the effect of time on ERT into two components: a common effect of being treated with an ERT (either agalsidase alpha or agalsidase beta) and a separate component for the incremental effect of receiving agalsidase beta rather than agalsidase alpha. To simplify the comparisons, the effect of time on ERT was considered as a linear function in all models (for both components). In addition, we compared recorded side effects and frequency of switching treatments.
Key for longitudinal model outputs
Models for the continuous outcomes have two outputs: (1) a table of estimated effects of gender, age at the time the outcome was measured and time on ERT on outcome (Table 10); and (2) a graph showing the shape of the estimated relationship between outcome and time on ERT (Figure 3). For selected models, we have also fitted a Bayesian version of the model to make predictions about how starting ERT at different ages impacts on age-related outcomes. More details of the generic templates used for the model outputs are given below. Throughout the remainder of the report, we use the term ‘age’ to refer to the age at measurement of the outcome unless specified otherwise.
| N Data a | Estimate of change in outcomeb | Standard errorc | 95% CId | p-valuee | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 413 | 0.00 | |||
| Female | 563 | –1.77 | 0.16 | –2.08 to –1.45 | < 0.001 |
| Age | |||||
| Linear effect/year | 0.01 | 0.005 | 0.001 to 0.02 | 0.04 | |
| Time on ERTf | < 0.001 | ||||
| Not on ERT | 41 | 0.00 | |||
| < 12 months | 68 | 0.86 | 0.18 | 0.51 to 1.21 | < 0.001 |
| 12–36 months | 155 | 1.11 | 0.15 | 0.81 to 1.40 | < 0.001 |
| > 36 months | 712 | 1.18 | 0.15 | 0.88 to 1.47 | < 0.001 |
| Varianceg components | |||||
| Individual | 1.08 | ||||
| Centre | 0.12 | ||||
| Residual | 0.66 | ||||
FIGURE 3.
Example graph of estimated relationship between age- and gender-adjusted outcome and time on ERT.
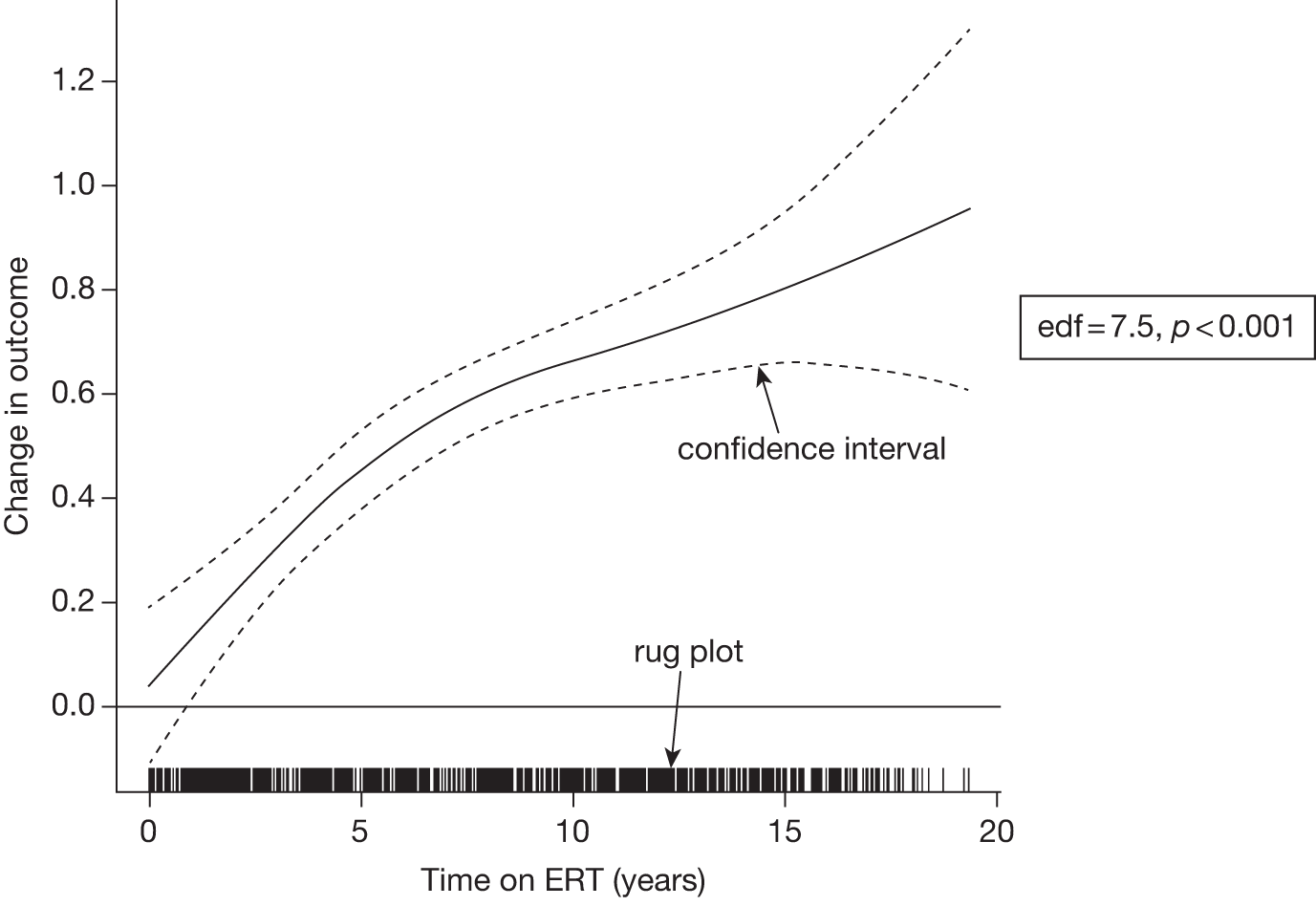
Figure 3 illustrates the shape of the relationship between outcome and time on ERT after adjustment for age and gender. The vertical axis measures the expected change in outcome as patients accumulate time spent on ERT, relative to the point at which patients start on ERT (expressed as an absolute difference). The solid line provides estimates of mean change in outcome and the dashed lines provide a 95% confidence interval (CI) around the model estimates. For example, Figure 3 suggests that treatment with ERT for 5 years is associated with a change in outcome of approximately 0.4 units. This effect is incremental to the underlying age-related change in outcome.
The extent to which the estimated relationship departs from linearity can be assessed by considering the estimated degrees of freedom (labelled in the text as ‘edf’). Values of the edf close to 1 indicate that the relationship is linear or close to linear.
The rug plot provides a visual representation of the frequency distribution for time on ERT. Each individual data point is represented by a single tick mark at the appropriate location on the chosen time scale (years).
Cost data analysis methods
Cost-effectiveness of enzyme replacement therapy
Because of the mixed and weak evidence of the effectiveness of ERT, and because any significant effects relate to biological markers of disease severity, we have decided that the planned model-based cost-effectiveness analysis would not be worthwhile. Instead, we have conducted a cost of illness analysis for each LSD to assess comprehensively the financial burden of each disorder on the NHS, social care and other publicly funded care and support services.
Cost of illness analysis methods
The self-reported or parent-reported retrospective data on health service and social care use were collated in a single database for all six disorders. For hospital-care usage, the reference (recall) period was the last 12 months, and recorded:
-
the number of days in hospital and reasons for inpatient stays
-
the number of and reasons for outpatient attendances, day hospital admissions and accident and emergency attendances.
For services and care professionals used outside of hospital, the data for 20 different types of service provider (GP, district nurse, occupational therapist, social worker, etc.) were recorded: whether or not that service/professional was used in the past 12 months (yes/no); the number of contacts; the typical length of each contact (in minutes); whether or not the visit/care was provided at home; and, if the service was paid for privately, the amount paid per attendance/use. It is important to note that, although ERT infusion sessions were reported in the questionnaire data, and are costed in the analyses, these costs do not include the actual ERT drug acquisition costs that are borne by the National Specialised Commissioning Team (NSCT) (and the NHS). The per patient annual costs of providing ERT are calculated and shown separately in each section based on data supplied by the NSCT.
Total costs for each service type were calculated by multiplying the number of episodes of each type of service use (e.g. inpatient stay days, outpatient appointments or GP visits) either by the NHS reference cost for that type of episode of care or by the mean number of contact hours reported per episode and the hourly cost to the public sector for the different types of health- and social-care professional. The unit costs used for each type of health- or social-care service or professional and the sources of the unit costs are shown for each condition. The main source of unit costs for hospital care was the NHS reference costs 2009–2010239 (for primary care trusts and NHS trusts combined), and the main source of the cost per hour of client/patient contact time for carers and other support workers normally based outside hospitals was the Unit Costs of Health and Social Care 2010. 240 Therefore the base year for the analysis is 2010. Table 11 shows the unit costs used to calculate the total NHS and social-care costs per LSD patient.
| Question number | Resource type and unit | Unit cost (£) | Source |
|---|---|---|---|
| 2a | Inpatient stays – elective | 940 per day | NSRC2009–10 (sheet TPCTEI) |
| 2a | Inpatient stays – non-elective short-stay (1 day/night) | 535 per day | NSRC2009–10 (sheet TPCTNEI_S) |
| 2a | Inpatient stays – non-elective long-stay (> 1 day/night) | 403 per day | NSRC2009–10 (sheet TPCTNEI_L) |
| 2b, 2c or 3 | *ERT Infusions in hospital | 297 per episode | NSRC2009–10 (sheet: TPCTCHEMTHPY_DEL_oth code SB14Z = deliver complex chemotherapy, including prolonged infusional treatment at first attendance) |
| 2b or 3 | *LSD annual/6-monthly check-up | 470 per episode | NSRC2009–10 (sheet: TPCCLFUSFF consultant-led face to face outpatients 261 = paediatric metabolic disease) |
| Various | *Kidney dialysis | 157 episode session | NSRC2009–10 renal dialysis outpatient (sheet: TPCTRENAL_OP) |
| 2b |
Hospital outpatient clinics Unless were for * (above) |
99 per appointment | NSRC 2009–10 face to face outpatient appointments (weighted average, consultant and non-consultant-led, first attendance and follow-ups) |
| 2c |
Day hospital stays Unless were for * (above) |
668 per admission | NSRC2009–10 (sheet: TPCTDC) |
| 2d | A&E attendances | 103 per attendance | NSRC2009–10 A&E services not leading to admitted (Sheet: TPCTAandEMSNA) |
| 2b | Hospital outpatient clinics | 99 per appointment | NSRC2009–10 face-to-face outpatient appointments (weighted average, consultant and non-consultant led, first attendance and follow-ups) |
| 1 | Visit to GP | 163 | UC2010 Section 10.8b (per hour of patient contact)a |
| 1 | GP home visit | 270 | UC2010 Section 10.8b (home visit at £4.50 per minute)a |
| 2 | GP practice nurse consultation | 10 | UC2010 Section 10.6 (GP practice nurse, per consultation) |
| 3 | District nurse | 64 | UC2010 (community nurse, per hour with patient) |
| 4 | Community mental health nurse | 48 | UC2010 (mental health nurse, per hour with patient) |
| 5 | Other nurse or health visitor | 88 | UC2010 Section 10.3 (health visitor, per hour with patient) |
| 6 | Counsellor | 44 | UC2010 Section 2.14 (counselling services in primary medical care, per hour with patient or per contact hour) |
| 7 | Other therapist – physiotherapistb | 37 (at home 39) | UC2010 Section 9.1 (physiotherapist, per hour with patient) |
| 7 | Other therapist – occupational therapistb | 38 (at home 39) | UC2010 Section 9.2 (NHS occupational therapist, per hour with patient) |
| 7 | Other therapist – speech/languageb | 37 (at home 39) | UC2010 Section 9.3 (community speech and language therapist) |
| 7 | Other therapist – chiropodist/podiatristb | 22 | UC2010 Section 9.4 (community chiropodist/podiatrist) |
| 8 | ‘Alternative’ medicine or therapyb | 0 | Note: highly unlikely to be NHS or publicly-funded or subsidised |
| 9 | Psychologist | 81 | UC2010 Section 9.5 (clinical psychologist, per hour with patient) |
| 10 | Psychiatrist | 283 | UC2010 Section 15.7 (consultant: psychiatric, per hour with patient) |
| 11 | Other community-based doctorb | As GP if not clearly stated | See GP above |
| 12 | Occupational therapist | 38 (at home 39) | UC2010 Section 9.2 (NHS occupational therapist, per hour with patient). Note: local authority occupational therapists more costly per hour of client contact: £77 |
| 13 | Social worker – children | 147 | UC2010 Section 11.3 [social worker (children), per hour with client] |
| 13 | Social worker – adult | 158 | UC2010 Section 11.2 [social worker (adult), per hour with client] |
| 14 | Home help/home care worker | 25 | UC2010 Section 11.6 (local authority home care worker, per hour face-to-face weekday contact) |
| 15 | Care attendant | 25 | Assumed same as above |
| 16 | Community support worker | 25 | Assumed same as above |
| 17 | Housing worker | 25 | Assumed same as above |
| 18 | Voluntary workerb | 0 | Voluntary – unpaid (no cost to NHS or public sector) |
| 19 | Day centre/drop-in/social clubb | 0 | If publicly funded/subsidised, zero marginal cost of modest changes in usage by a numerically small patient group |
| 20 | Self-help groupb | 0 |
To test for linear or non-linear associations between costs and time on ERT we also fitted regression models (generalised estimated equations with a gamma distribution and a log-link function) to total costs, hospital costs and community costs. The use of such gamma models allows for covariates to be estimated in terms of their multiplicative effect, allowing the expression of findings as a percentage reduction or increase in mean costs. 241 For these analyses we used the same demographic covariates as those used for analysing the potential impact of ERT on continuous measures of clinical effectiveness.
The cost and resource use data were analysed using PASW® Statistics 18 (SPSS Inc., Chicago, IL, USA). To avoid impressions of spurious accuracy, all mean and median costs are reported to the nearest £100 if > £1000, to the nearest £10 if < £1000 and to the nearest £1 if < £100.
Changes to original protocol
Changes to the original aims
The original aim of the NCS-LSD study was to collect data on all LSDs for which ERT is currently available or being developed, with the intention of including all children and adults diagnosed with other LSDs (for which there is currently no treatment) at a later date.
However, the LSDs represent a group of > 70 genetically distinct diseases, and it became apparent early on that it would not be possible to include all conditions in this study. The initial choice of disorders to be investigated was consequently based on the availability of drug therapies, and it was agreed to collect data on the following conditions, for which therapies were already licensed or under development:
-
Gaucher disease
-
Fabry disease
-
MPS types I, II, IV and VI
-
Pompe disease
-
NPB and NPC
-
mannosidosis.
However, it emerged that an industry-funded, multicentre, clinical trial of ERT in MPS IV patients was commencing, and the ensuing confidentiality clause with the drug manufacturer would preclude the collection of clinical data from a large cohort of MPS IV patients until March 2011. For this reason it was decided not include MPS IV in this study.
Meanwhile, the prevalence of mannosidosis, NPB and MPS VI is known to be very low within the UK – the worldwide incidence of alpha-mannosidosis is in the range of 1 per 500,000242 to 1 per 1,000,000; the incidence of NPB in the UK is estimated to be in the order of 1 : 10,000,000 (Niemann–Pick Disease Group UK website243), while the worldwide estimates for MPS VI vary from 1 : 248,0005 to 1 : 1,300,000,244 or 1100 patients currently worldwide. The study steering committee therefore recommended that the cost and time implications for creating a condition-specific database for each of these diseases were unjustified, and that investigation of more than six LSDs would be beyond the scope of the study.
Finally, it was agreed that data would be collected for the following conditions:
-
Gaucher disease
-
Fabry disease
-
MPS I
-
MPS II
-
Pompe disease
-
NPC.
Our decision to limit our data collection to these conditions meant that it was not possible to fulfil our fourth primary objective, i.e. to describe the natural history of LSDs where ERT is likely to become available.
Amendments to the protocol
Approvals for amendments to the study protocol and documentation sought and obtained from the South West MREC (Table 12).
| Amendment number/date | Reason for amendment |
|---|---|
|
Substantial amendment 1 Approved 29 April 2008 |
The protocol and initial documentation were amended in response to MREC request |
|
Substantial amendment 2 Approved 10 December 2008 |
The protocol was amended to meet the requirements of the Mental Capacity Act and address the process of consenting adults who lack understanding into the study. Two additional consent forms for people who lacked capacity were created |
|
Substantial amendment 3 Approved 7 August 2009 |
The protocol was revised to allow the follow-up letter and PIS to be resent to patients who were missed at their annual appointment |
|
Substantial amendment 4 Approved 25 September 2009 |
The protocol was amended to allow the collection of additional QoL and service-use information from patients who were on reduced or altered treatment regimen owing to a world shortage in their ERT drugs |
|
Substantial amendment 5 Approved 28 October 2009 |
The protocol was amended to include the FSS in the suite of questionnaires that were given to participants Letters were approved to explain to patients with MPS IV and MPS VI that data collection had been deferred for these conditions |
|
Substantial amendment 6 Approved 8 December 2009 |
The protocol was amended to post additional sets of questionnaires to patients who are on an altered treatment regime owing to a world shortage in their ERT |
|
Substantial amendment 7 Approved 22 April 2010 |
The protocol was amended to allow patients who were previously incorrectly consented to be reconsented into the study |
|
Substantial amendment 8 Approved 29 April 2010 |
The protocol was amended to allow follow-up QoL questionnaires to be posted to patients missed at their annual appointment |
|
Substantial amendment 9 Approved 10 September 2010 |
The protocol was amended to allow a final set of follow-up QoL questionnaires to be posted to patients who had not completed such questionnaires during the previous 4 months |
Chapter 3 Results – Gaucher disease
Patient characteristics
At the start of the study, 272 patients were identified by the treating centres as having Gaucher disease. Of these, 223 were deemed eligible for inclusion and 185 patients (83% of those deemed eligible) were approached in clinic and invited to participate. One hundred and seventy-five patients (95% of those approached), comprising 75 males and 100 females, agreed to participate in the study. Patient demographic characteristics are presented in Tables 13 and 14.
| Patient characteristic | |
|---|---|
| Gender | |
| Male, n | 65 |
| Female, n | 85 |
| Age at recruitment (years) | |
| Mean (SD), n | 46.4 (14.7) |
| Median (min.–max.) | 46.6 (16.8–83.1) |
| Age at diagnosis (years) | |
| Mean (SD), n | 24.8 (16.8) |
| Median (min.–max.) | 21.9 (0–72.2) |
| Type of Gaucher disease | |
| GD1, n | 145 |
| GD3, n | 5 |
| Splenectomised | |
| Yes, n | 48 |
| No, n | 102 |
| Initial treatment | |
| Not on ERT, n | 11 |
| ERT, n | 135 |
| Clinical triala, n | 4 |
| Initial type of ERT | |
| Imiglucerase, n | 78 |
| Alglucerase, n | 55 |
| Velaglucerase alpha, n | 2 |
| Treatment at recruitment | |
| Not on ERT, n | 11 |
| ERT, n | 131 |
| SRT, n | 8 |
| Type of ERT at recruitment | |
| Imiglucerase, n | 120 |
| Alglucerase, n | 1 |
| Taliglucerase alpha, n | 1 |
| Velaglucerase alpha, n | 8 |
| Unknown, n | 1 |
| Age at first infusion (years) | |
| Mean (SD) | 35.2 (15.2) |
| Median (range) | 35.9 (1.68–80.5) |
| Time on ERT (years) at recruitment | |
| Mean (SD) | 10.8 (4.7) |
| Median (min.–max.) | 11.7 (0–17.8) |
| Patient characteristic | |
|---|---|
| Gender | |
| Male, n | 10 |
| Female, n | 15 |
| Age at recruitment (years) | |
| Mean (SD) | 9.74 (4.1) |
| Median (min.–max.) | 9.98 (1.14–15.6) |
| Age at diagnosis (years) | |
| Mean (SD) | 3.25 (2.8) |
| Median (min.–max.) | 2.17 (0–11.1) |
| Type of Gaucher disease | |
| GD1, n | 14 |
| GD3, n | 11 |
| Splenectomised | |
| Yes, n | 2 |
| No, n | 23 |
| Initial treatment | |
| Not on ERT, n | 0 |
| ERT, n | 25 |
| Clinical triala, n | 0 |
| Initial type of ERT | |
| Imiglucerase, n | 22 |
| Alglucerase, n | 2 |
| Missing, n | 1 |
| Treatment at recruitment | |
| Not on ERT, n | 0 |
| ERT, n | 24 |
| SRT, n | 1 |
| Type of ERT at recruitment | |
| Imiglucerase, n | 22 |
| Alglucerase, n | 0 |
| Taliglucerase alpha, n | 0 |
| Velaglucerase alpha, n | 2 |
| Age at first infusion (years) | |
| Mean (SD) | 3.49 (2.8) |
| Median (range) | 2.57 (0.24–11.4) |
| Time on ERT (years) at recruitment | |
| Mean (SD) | 6.27 (4.3) |
| Median (min.–max.) | 5.57 (0–13.7) |
Twenty-five of the participants were children and 150 were adults, with the average age of patients with Gaucher disease at recruitment being 46.4 (range 16.8–83.1) years for adults and 9.74 (range 1.14–15.6) years for children.
Owing to the world shortage of some ERTs (as discussed in Chapter 1), patients with Gaucher disease attended their treating clinic more regularly during the later period of data collection, and clinical data were collected at each visit. We collected data from 156 patients at a second data point, and from 74 patients at a third data point. For six patients, we had five prospective data points. The number of retrospective data points per patient ranged from 1 to 15.
At recruitment to the study, 155 patients were on ERT (131 adults and 24 children), with the average time on ERT being 10.8 (range 0–17.8) years for adults and 6.27 (range 0–13.7) years for children. Of those on ERT, 142 patients (120 adults and 22 children) were recorded as receiving imiglucerase, one adult was receiving alglucerase, one adult was receiving taliglucerase alpha and 10 patients (eight adults and two children) received velaglucerase. The type of ERT for one adult patient at recruitment remained unknown. A further nine patients (eight adults and one child) received SRT (miglustat), whereas 11 adults were not on ERT or SRT. Of the 142 patients on imiglucerase at recruitment, 14 patients stopped treatment owing to shortage of this therapy.
The sample was stratified by age [≥ 16 years (adult) compared with < 16 years (child)] and by splenectomy status for modelling of platelet counts and bone pain. Once patients began receiving SRT their data were excluded from further analyses. With the exception of neurological measures, the patients with GD1 and GD3 disease were included together in all the models.
Key markers of Gaucher disease progression
The following measures were identified as key markers of disease progression:
-
platelet count
-
Hb
-
absence/presence of bone pain
-
spleen volume/size
-
liver volume/size
-
liver function tests.
In addition, adults completed the SF-36, the EQ-5D, the FSS and the Service Use and Costs Questionnaire, while children or their carers completed the age-appropriate PedsQL questionnaire. Carers of children or adults were asked to complete the Service Use and Costs Questionnaire and the CSI.
Longitudinal models were fitted to assess relationships between continuous measures of function and length of time on ERT, after adjustment for age and clustering by centre. In the base models, the effect of time on ERT was categorised as (1) not on ERT; (2) < 12 months on ERT; (3) 12–36 months on ERT; and (4) > 36 months on ERT. Further analysis was conducted to explore the possibility that time spent on ERT would have a non-linear effect on function. Patients contributed data points to the model both before and after starting ERT. A Bayesian version of the model was used for generating expected trajectories to illustrate model predictions about the effect of starting ERT treatment at different ages.
Summary of Gaucher disease results
These data provide strong evidence for an association between time on ERT and a clinically significant improvement in platelet count and Hb in adults, irrespective of whether or not they have undergone splenectomy, and in children. There is also a strong, statistically significant association between time on ERT and a clinically important decrease in the likelihood of having an enlarged spleen or liver based on estimated spleen volumes from scans or on palpation. In all of these analyses the data appear to suggest very substantial improvements over the first years of treatment (lasting perhaps 5–10 years) and then a plateauing of the effect. The shape of these relationships needs to be interpreted with caution owing to the wider CIs around the effect size associated with longer periods on ERT.
Data for liver function tests were difficult to interpret because of differences in methods of analysis and normal ranges across centres. When considering aspartate transaminase (AST) results in adults, we found a strong association between time on ERT and reduced AST levels, as well as a lower risk of a having an ‘abnormal’ AST level. No association was found in children, possibly as a result of sparse data.
The data provide some evidence that a longer duration of ERT may be associated with a reduced risk of bone pain in adults and children.
The data provide no evidence of an association between duration of ERT and either subscale of the SF-36, or either the EQ-5D or visual analogue scale (VAS) score or the FSS. Similarly, no association was seen between duration of ERT and the total PedsQL score, although a significant reduction was seen in the social functioning score with time on ERT. No evidence was found for a relationship between the time on ERT and the burden of carers, as assessed by the CSI.
Platelet count
Adults
We have platelet counts for 138 adult patients. A total of 925 measurements for platelet count were recorded for these patients across all time points. Platelet counts ranged from 11 × 109/l to 798 × 109/l. A linear mixed model for platelet count in adults (not shown) confirmed that there was a significant relationship between splenectomy status and estimated platelet count, with the model suggesting that splenectomised patients had a mean platelet count 81.7 × 109/l higher than non-splenectomised patients (95% CI 61.3 to 102.1 × 109/l; p < 0.001). In order to examine the differences in treatment effects in the two groups of patients, the data for platelet count were stratified by splenectomy status. Five patients were splenectomised after their first recorded data point and therefore their data contributed to the analyses for splenectomised and non-splenectomised patients.
Splenectomised patients
We have platelet counts for 47 splenectomised adult patients. A total of 375 measurements for platelet count were recorded for these patients across all time points. Platelet counts ranged from 28 × 109/l to 798 × 109/l.
The results presented in Table 15 show a statistically significant association between age and a decline in platelet count (p < 0.001) in splenectomised adults with Gaucher disease. There is a statistically significant association between platelet count and time on ERT (p = 0.002) with the model suggesting a platelet count 68.2 × 109/l higher in those treated for ≥ 3 years compared with the untreated (95% CI 31.1 to 105.2 × 109/l). In this initial analysis, we considered the response to treatment with all patients on ERT for > 36 months considered in a single group despite the considerable within-group heterogeneity in time on ERT.
| N Data | Estimate of change in platelet count (× 109/l) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 127 | 0.00 | |||
| Female | 248 | –0.32 | 25.4 | –50.1 to 49.5 | 0.99 |
| Age | |||||
| Linear effect/year | –4.40 | 0.98 | –6.32 to –2.48 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 14 | 0.0 | 0.002 | ||
| < 12 months | 26 | 38.0 | 20.8 | –2.76 to 78.7 | 0.06 |
| 12–36 months | 58 | 57.5 | 18.5 | 21.2 to 93.7 | 0.002 |
| > 36 months | 277 | 68.2 | 18.9 | 31.1 to 105.2 | < 0.001 |
| Variance components | |||||
| Individual | 15,462 | ||||
| Centre | 611 | ||||
| Residual | 3099 | ||||
Further analysis was conducted to explore the overall shape of the relationship between platelet count and time on ERT (considering actual time of ERT rather than categorising into four groups as in the previous analysis). Figure 4 illustrates the significant non-linear effect of time on ERT (edf = 2.33; p = 0.01). This graph appears to suggest that the effect of ERT peaks at around 7 years after commencement of ERT. The apparent decline seen in the graph should be viewed with caution, as demonstrated by the wide CIs around the line.
FIGURE 4.
The age-adjusted association between time on ERT and platelet count in splenectomised adults with Gaucher disease (time on ERT treated as a continuous variable).
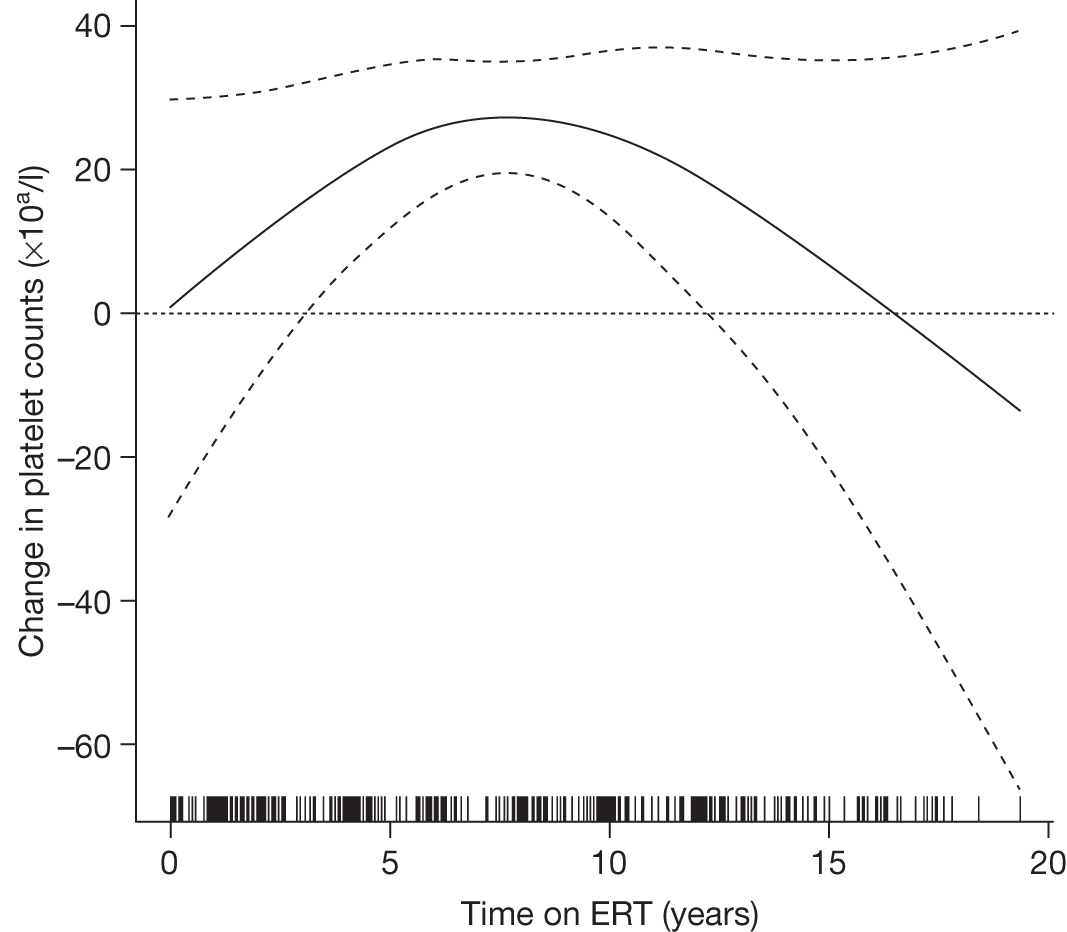
Non-splenectomised patients
A total of 550 measurements for platelet count were recorded for the 96 non-splenectomised adult patients across all time points. The measurements range from 11 × 109/l to 670 × 109/l.
The results presented in Table 16 show a statistically significant association between age and a decline in platelet count (p = 0.008). There is a statistically significant association between platelet count and time on ERT (p < 0.001) with the model suggesting a platelet count 74.3 × 109/l higher in those treated for ≥ 3 years compared with the untreated (95% CI 59.2 to 89.4 × 109/l). In this initial analysis, we considered the response to treatment with all patients on ERT for > 36 months considered in a single group despite the considerable within-group heterogeneity in time on ERT.
| N Data | Estimate of change in platelet count (× 109/l) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 260 | 0.00 | |||
| Female | 290 | 15.6 | 12.8 | –9.48 to 40.7 | 0.22 |
| Age | |||||
| Linear effect/year | –0.90 | 0.34 | –1.56 to –0.23 | 0.008 | |
| Time on ERT | |||||
| Not on ERT | 21 | 0.00 | < 0.001 | ||
| < 12 months | 35 | 24.1 | 8.1 | 8.22 to 39.9 | 0.003 |
| 12–36 months | 81 | 47.7 | 7.26 | 33.5 to 61.9 | < 0.001 |
| > 36 months | 413 | 74.3 | 7.69 | 59.2 to 89.4 | < 0.001 |
| Variance components | |||||
| Individual | 8069.2 | ||||
| Centre | 0.00 | ||||
| Residual | 632 | ||||
Further analysis was conducted to explore the overall shape of the relationship between platelet count and time on ERT (considering actual time of ERT rather than categorising into four groups as in the previous analysis). Figure 5 illustrates the significant non-linear effect of time on ERT (edf = 2.92; p < 0.001). This graph appears to suggest that the effect of ERT plateaus at around 8 years after commencement of ERT. The exact shape of the line in the later years should be viewed with caution as demonstrated by the wide CIs around the line.
FIGURE 5.
The age-adjusted association between time on ERT and platelet count in non-splenectomised adults with Gaucher disease (time on ERT treated as a continuous variable).
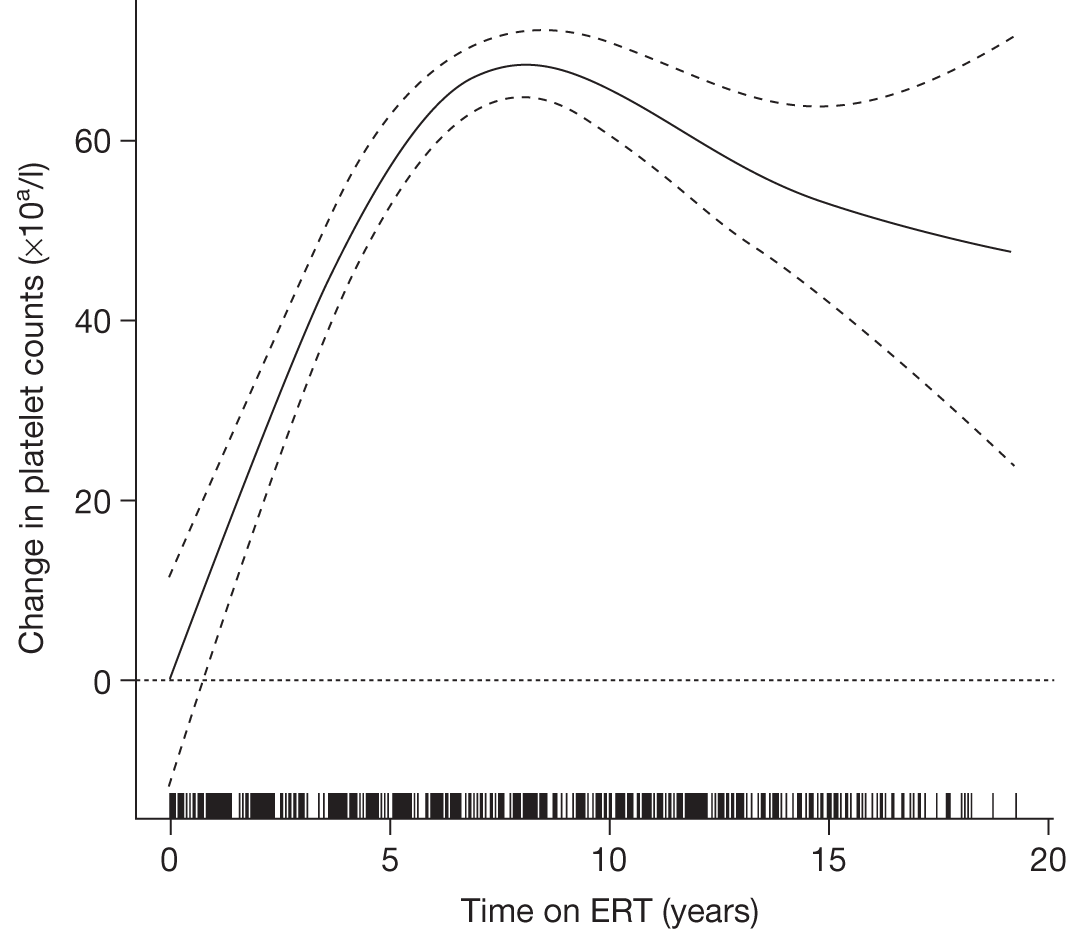
Bayesian projections
As an aid to illustrating the findings of the longitudinal model for platelet counts, we used a Bayesian version of the model to make predictions about the outcomes for untreated patients and the expected trajectories for patients starting ERT at different ages (Figure 6). Three different treatment scenarios were considered: (1) the patient remains untreated through adulthood; (2) the patient starts ERT at the age of 18 years (close to the mean age at diagnosis); and (3) the patient commences ERT at the age of 45 years. It should be noted that these are mean trajectories and in the interest of clarity the credible intervals around these trajectories are not shown.
FIGURE 6.
Platelet counts in non-splenectomised adults in different treatment scenarios.
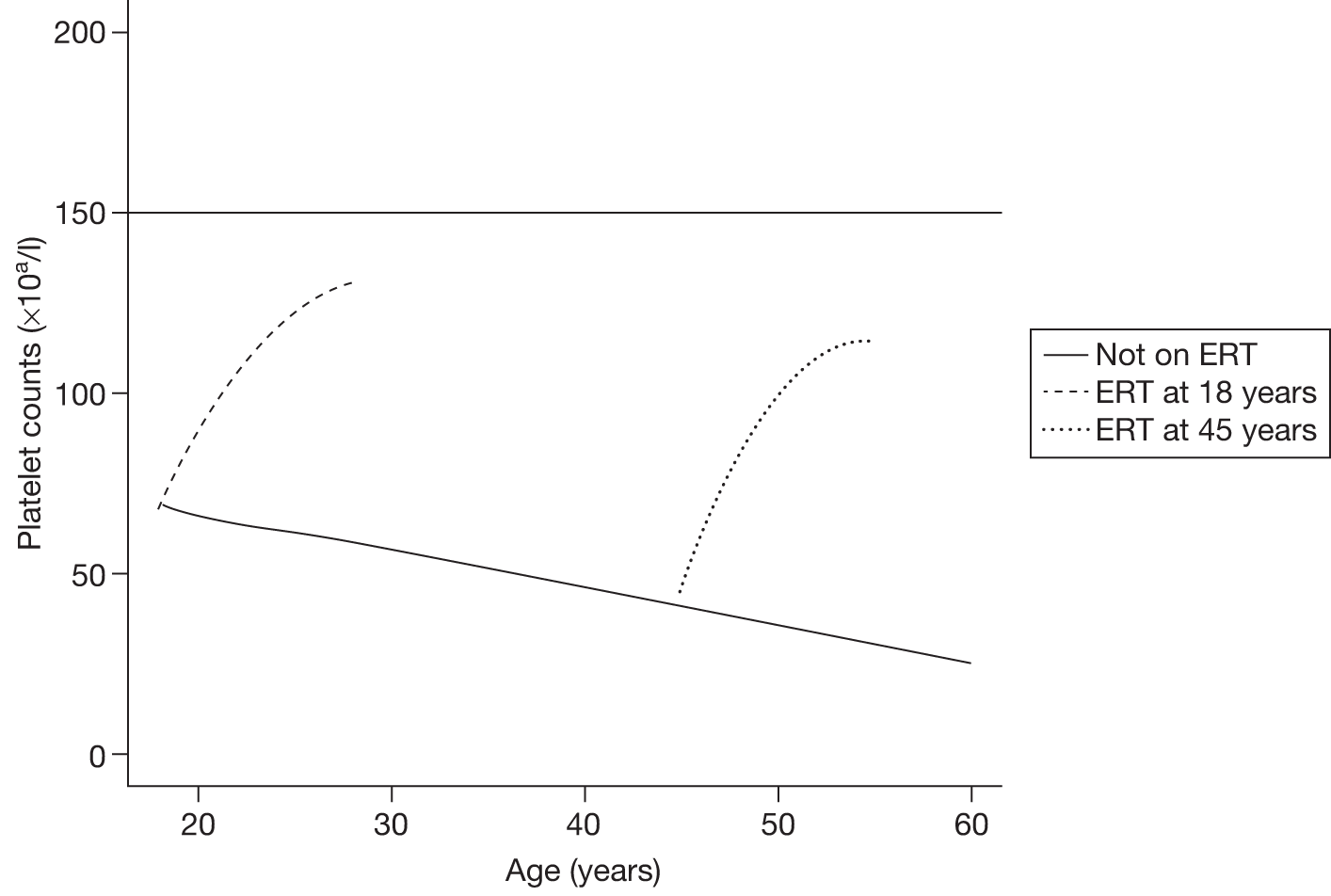
The model suggests that, in the absence of treatment, platelet counts in non-splenectomised patients decline at a rate of approximately 1 × 109/l/year through adulthood. Figure 6 suggests that for patients who commence treatment at 18 years of age, platelet counts recover to levels approaching the normal range (150 × 109/l) after approximately 5 years of being on ERT. For patients starting treatment aged 45 years, although the trajectory of their platelet count is of greater magnitude, they are unable to attain the same platelet count, because their pre-treatment count was greatly reduced.
We specified the Bayesian model to allow the shape of the post-treatment trajectories to depend on the age at which ERT is started, to explore the possibility that capacity to respond depends on the age at which treatment is commenced or that patients observed in late middle age or old age have less aggressive forms of the disease and so may respond more successfully to treatment. Although the graph does appear to suggest that patients diagnosed and treated later in life respond more quickly, this observation should be interpreted with caution. Estimates from the Bayesian model indicate that there is a 55% probability that first infusion at age 45 years will result in greater increases in platelet count after 10 years on ERT than first infusion at age 18 years. This is not far removed from the 50% probability that would result if age at starting ERT had no impact on response to treatment. Our data suggest that, even in patients diagnosed later, treatment with ERT can achieve a clinically relevant improvement in platelet count and that there is little evidence that age of commencement of treatment affects the patient’s likelihood of benefit.
Children
Splenectomised
We have platelet counts for only two of the splenectomised children; there are insufficient data to perform an analysis.
Non-splenectomised
A total of 119 measurements for platelet count were recorded for the 23 non-splenectomised children across all time points. The range of these measurements was 14 to 462 × 109/l.
The results presented in Table 17 show no statistically significant association between age and platelet count (p = 0.73). There is a statistically significant association between platelet count and time on ERT (p < 0.001) with the model suggesting a platelet count 94.2 × 109/l higher in those treated for ≥ 3 years compared with the untreated (95% CI 50.3 to 138.1 × 109/l). In this initial analysis, we considered the response to treatment with all patients on ERT for > 36 months considered in a single group despite the considerable within-group heterogeneity in time on ERT.
| N Data | Estimate of change in platelet count (× 109/l) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 55 | 0.00 | |||
| Female | 64 | 9.11 | 15.2 | –20.7 to 38.9 | 0.55 |
| Age | |||||
| Linear effect/year | 0.78 | 2.3 | –3.73 to 5.28 | 0.73 | |
| Time on ERT | |||||
| Not on ERT | 12 | 0.0 | < 0.001 | ||
| < 12 months | 24 | 46.1 | 21.5 | 3.91 to 88.2 | 0.03 |
| 12–36 months | 35 | 83.4 | 19.0 | 46.2 to 120.6 | < 0.001 |
| > 36 months | 48 | 94.2 | 22.4 | 50.3 to 138.1 | < 0.001 |
| Variance components | |||||
| Individual | 3660 | ||||
| Centre | 0 | ||||
| Residual | 2730 | ||||
Further analysis was conducted to explore the overall shape of the relationship between platelet count and time on ERT (considering actual time of ERT rather than categorising into four groups as in the previous analysis). Figure 7 illustrates the significant non-linear effect of time on ERT (edf = 2.44; p < 0.001). This graph appears to suggest that the effect of ERT plateaus at around 5 years after commencement of ERT. The exact shape of the line in the later years should be viewed with caution as this is based on sparse data, as demonstrated by the wide CIs around the line.
FIGURE 7.
The age-adjusted association between time on ERT and platelet counts in non-splenectomised children with Gaucher disease (time on ERT treated as a continuous variable).
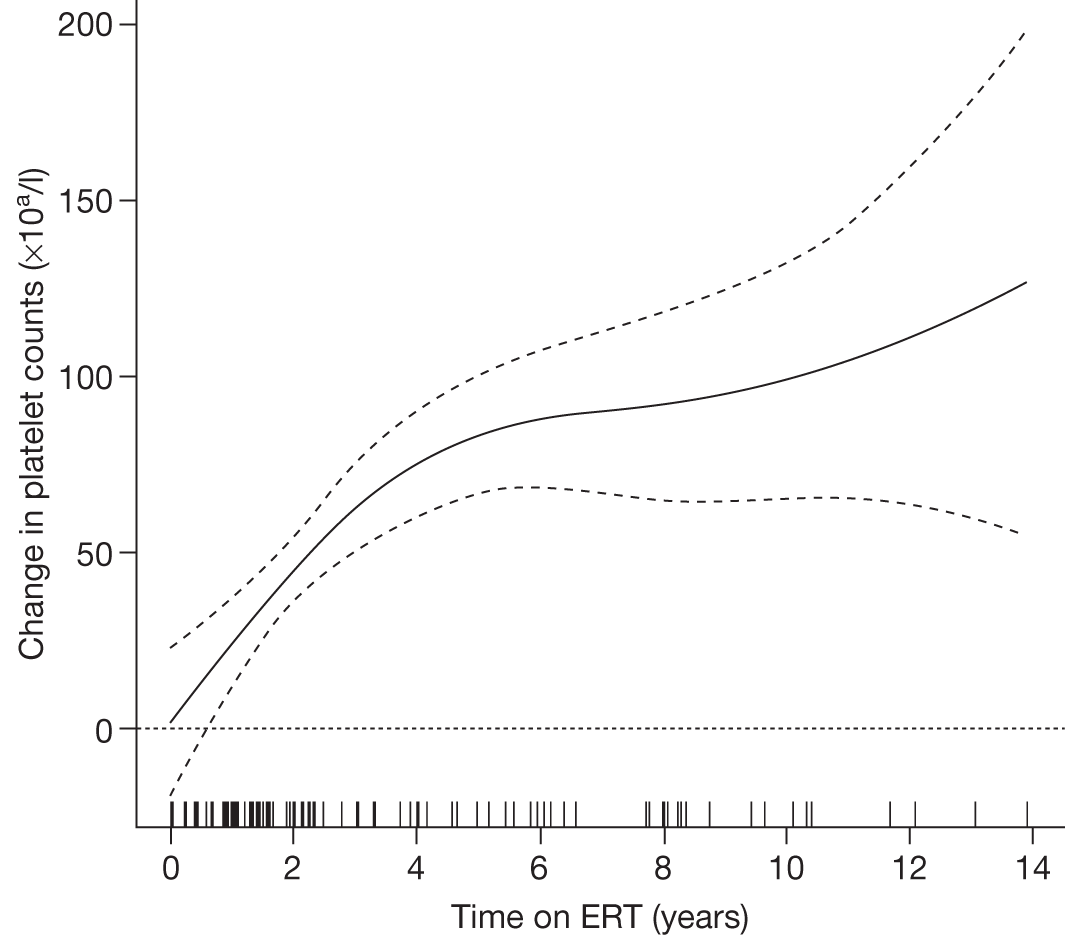
Haemoglobin
We have Hb values for 146 patients (127 adults and 19 children). A total of 955 measurements for Hb were recorded for the 127 adult patients across all time points. The Hb values range from 9 to 17 g/dl. As Hb levels are primarily determined by bone marrow and not the spleen, data were not stratified by spleen status.
Adults
The results presented in Table 18 show there is no significant association between age and increase in Hb (p = 0.20). There is a statistically significant association between increasing Hb levels and time on ERT (p < 0.001) with the model suggesting a Hb 1.19 g/dl higher in those treated for ≥ 3 years compared with the untreated (95% CI 0.87 to 1.50 g/dl). In this initial analysis, we considered the response to treatment with all patients on ERT for > 36 months considered in a single group despite the considerable within-group heterogeneity in time on ERT.
| N Data | Estimate of change in Hb (g/dl) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 407 | 0.00 | |||
| Female | 548 | –1.78 | 0.16 | –2.09 to –1.46 | < 0.001 |
| Age | |||||
| Linear effect/year | 0.007 | 0.005 | –0.003 to 0.02 | 0.20 | |
| Time on ERT | |||||
| Not on ERT | 39 | 0.00 | < 0.001 | ||
| < 12 months | 61 | 0.89 | 0.18 | 0.54 to 1.24 | < 0.001 |
| 12–36 months | 147 | 1.12 | 0.16 | 0.81 to 1.43 | < 0.001 |
| > 36 months | 708 | 1.19 | 0.16 | 0.87 to 1.50 | < 0.001 |
| Variance components | |||||
| Individual | 0.96 | ||||
| Centre | 0.09 | ||||
| Residual | 0.66 | ||||
Further analysis was conducted to explore the overall shape of the relationship between Hb and time on ERT (considering actual time on ERT rather than categorising into four groups as in the previous analysis). Figure 8 illustrates the significant non-linear effect of time on ERT (edf = 2.14; p < 0.001). This graph appears to suggest that ERT continues to have an incremental effect for the first 20 years of treatment, although the magnitude of the effect decreases after about 7 years. The exact shape of the line in the later years should be viewed with caution, as demonstrated by the wide CIs around the line.
FIGURE 8.
The age-adjusted association between time on ERT and Hb in adults with Gaucher disease (time on ERT treated as a continuous variable).
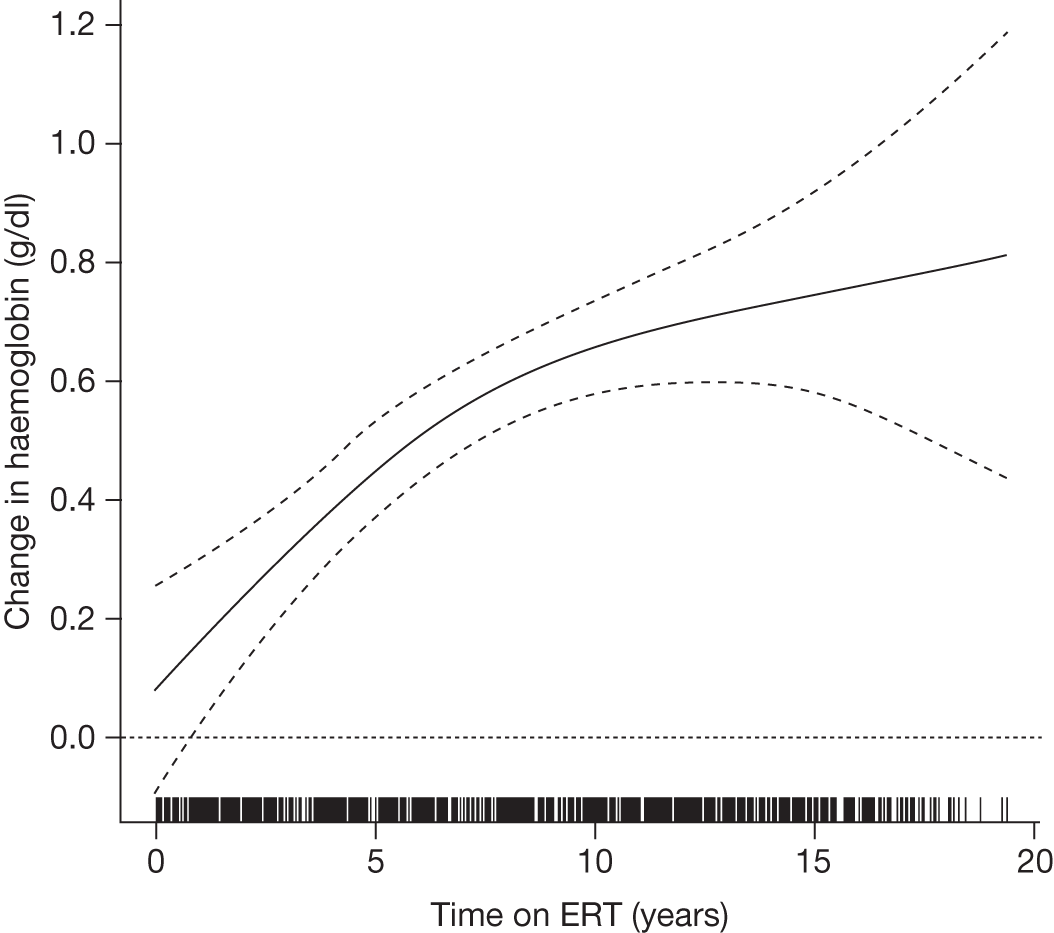
Bayesian projections
Again, the longitudinal model for Hb levels was fitted in a Bayesian framework and used to make predictions about how starting ERT at different ages impacts on age-related Hb levels (Figure 9). Three different treatment scenarios were considered: (1) the patient remains untreated; (2) the patient starts ERT at the age of 18 years; and (3) the patient commences ERT at the age of 45 years. It should be noted that these are mean trajectories and in the interest of clarity the credible intervals around these trajectories are not shown.
FIGURE 9.
Haemoglobin levels in adults with different treatment scenarios.
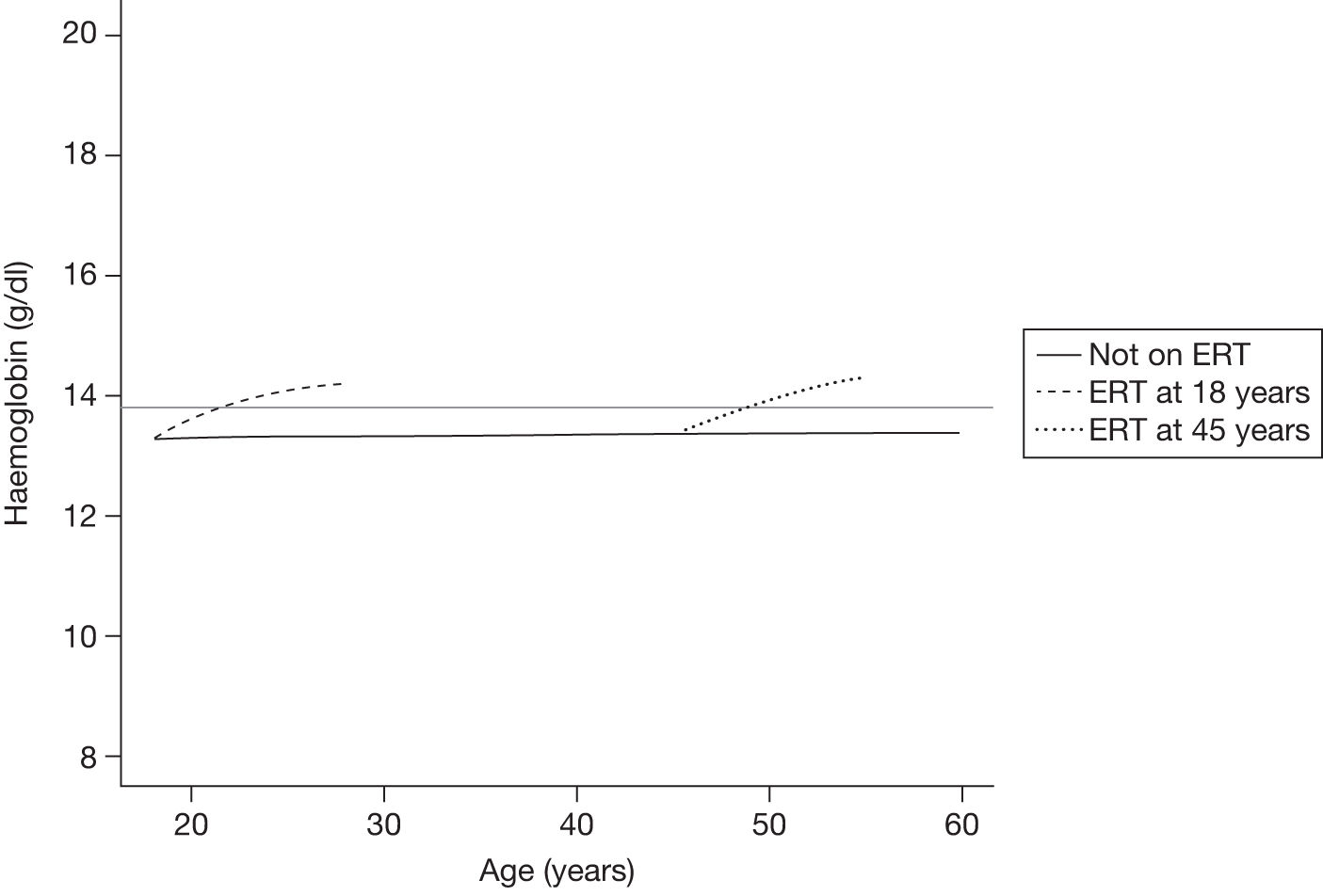
This model provides no strong evidence that the age at commencing ERT affects the degree of increase in Hb achieved. The Bayesian probability that first infusion at age 45 years will result in greater increases in Hb after 10 years on ERT than first infusion at age 18 years = 52%.
Children
A total of 145 measurements for Hb were recorded for the 19 children across all time points. The measurements for Hb in children ranged from 7 to 15 g/dl.
The results presented in Table 19 show a statistically significant association between age and increase in Hb (p < 0.001). There is a statistically significant association between increasing Hb levels and time on ERT (p < 0.001) with the model suggesting a Hb 1.14 g/dl higher in those treated for ≥ 3 years compared with the untreated (95% CI 0.47 to 1.80 g/dl). In this initial analysis, we considered the response to treatment with all patients on ERT for > 36 months considered in a single group despite the considerable within-group heterogeneity in time on ERT.
| N Data | Estimate of change in Hb (g/dl) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 58 | 0.00 | |||
| Female | 87 | –0.32 | 0.24 | –0.79 to 0.15 | 0.17 |
| Age | |||||
| Linear effect/year | 0.14 | 0.03 | 0.08 to 0.19 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 13 | 0.00 | < 0.001 | ||
| < 12 months | 26 | 1.07 | 0.34 | 0.40 to 1.74 | 0.002 |
| 12–36 months | 42 | 1.41 | 0.30 | 0.82 to 1.99 | < 0.001 |
| > 36 months | 64 | 1.14 | 0.34 | 0.47 to 1.80 | 0.001 |
| Variance components | |||||
| Individual | 0.26 | ||||
| Centre | 0.41 | ||||
| Residual | 0.82 | ||||
Further analysis was conducted to explore the overall shape of the relationship between Hb and time on ERT (considering actual time on ERT rather than categorising into four groups as in the previous analysis). Figure 10 illustrates a non-linear statistically significant effect of time on ERT with increased Hb levels (edf = 2.36; p = 0.02) with the effect appearing to peak at approximately 4 years. However, the shape of the line should be viewed with caution, as demonstrated by the wide CIs around the line.
FIGURE 10.
The age-adjusted association between time on ERT and Hb levels in children with Gaucher disease (time on ERT treated as a continuous variable).
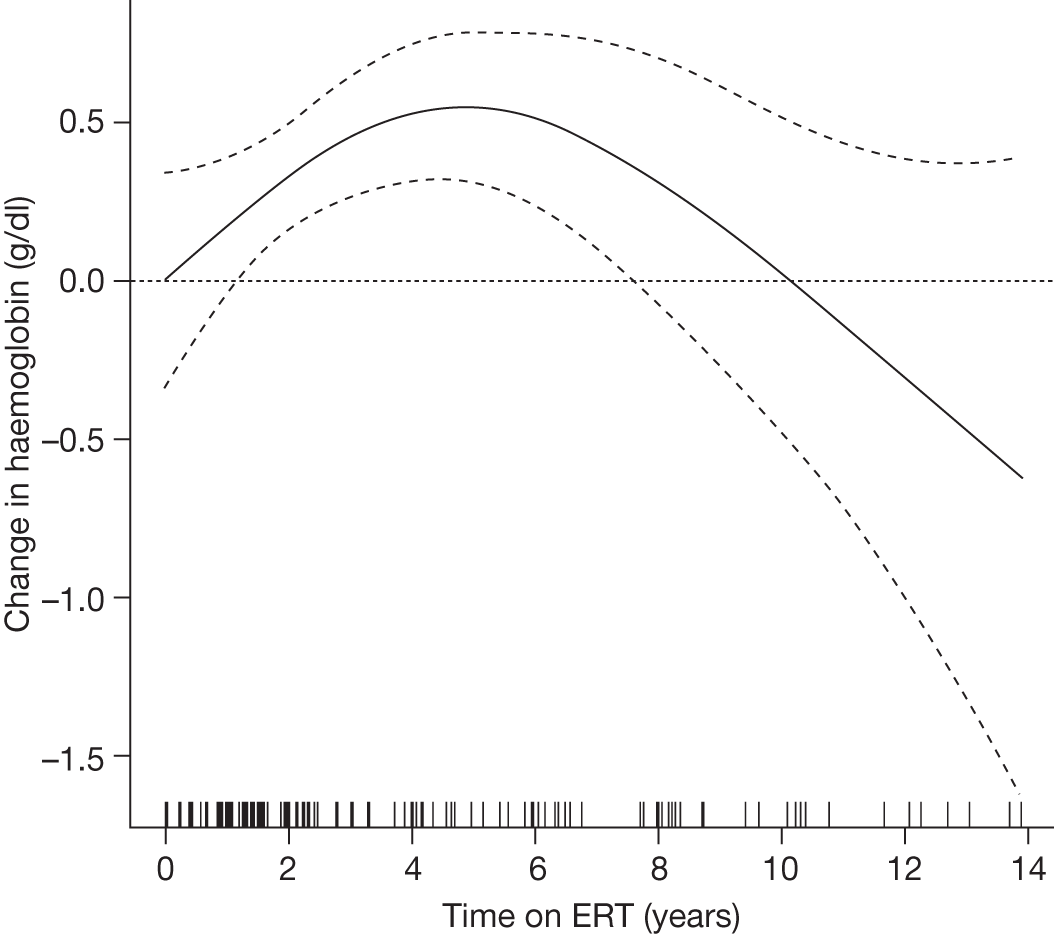
Bone pain
Adults
Reports of bone pain in the previous 12 months were recorded. As splenectomised patients have been reported to be at increased risk of skeletal complications,245,246 we have adjusted for this in the model. We have bone pain data on 175 patients (150 adults and 25 children) and a total of 636 recordings for bone pain (pain or no pain) were obtained for the 150 adults across all time points.
The data in Table 20 suggest there was no significant association between bone pain and age (p = 0.14) or gender (p = 0.75).
| N Pain | N No pain | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 148 | 109 | 1.00 | ||
| Female | 240 | 139 | 1.13 | 0.53 to 2.37 | 0.75 |
| Splenectomy | |||||
| No | 199 | 197 | 1.00 | ||
| Yes | 189 | 51 | 3.45 | 1.52 to 7.82 | 0.003 |
| Age | |||||
| Linear effect/year | 1.02 | 0.99 to 1.04 | 0.14 | ||
| Time on ERT | |||||
| Not on ERT | 18 | 5 | 1.00 | 0.35 | |
| < 12 months | 24 | 15 | 0.42 | 0.09 to 1.89 | 0.26 |
| 1 2–36 months | 65 | 36 | 0.44 | 0.11 to 1.70 | 0.23 |
| > 36 months | 281 | 192 | 0.33 | 0.09 to 1.20 | 0.09 |
Splenectomised adults had significantly higher odds of bone pain than non-splenectomised adults [odds ratio (OR) 3.45, 95% CI 1.52 to 7.82; p = 0.003]. After adjusting for splenectomy status, the risk of having bone pain is reduced with time since starting on ERT, although the relationship is not statistically significant in this model where time on ERT is categorised into four groups (p = 0.35).
Further analysis was conducted to explore the overall shape of the relationship between bone pain and time on ERT, treating time as a continuous variable. Figure 11 shows this relationship to be highly significant (edf = 1; p = 0.001). The latter formulation of the model is able to take account of the shape of the relationship within the large heterogeneous group of patients who have been on ERT for > 36 months. This suggests a strong statistically significant relationship between time on ERT and reduced risk of bone pain.
FIGURE 11.
The age-adjusted association between time on ERT and bone pain in adults with Gaucher disease (time on ERT treated as a continuous variable).
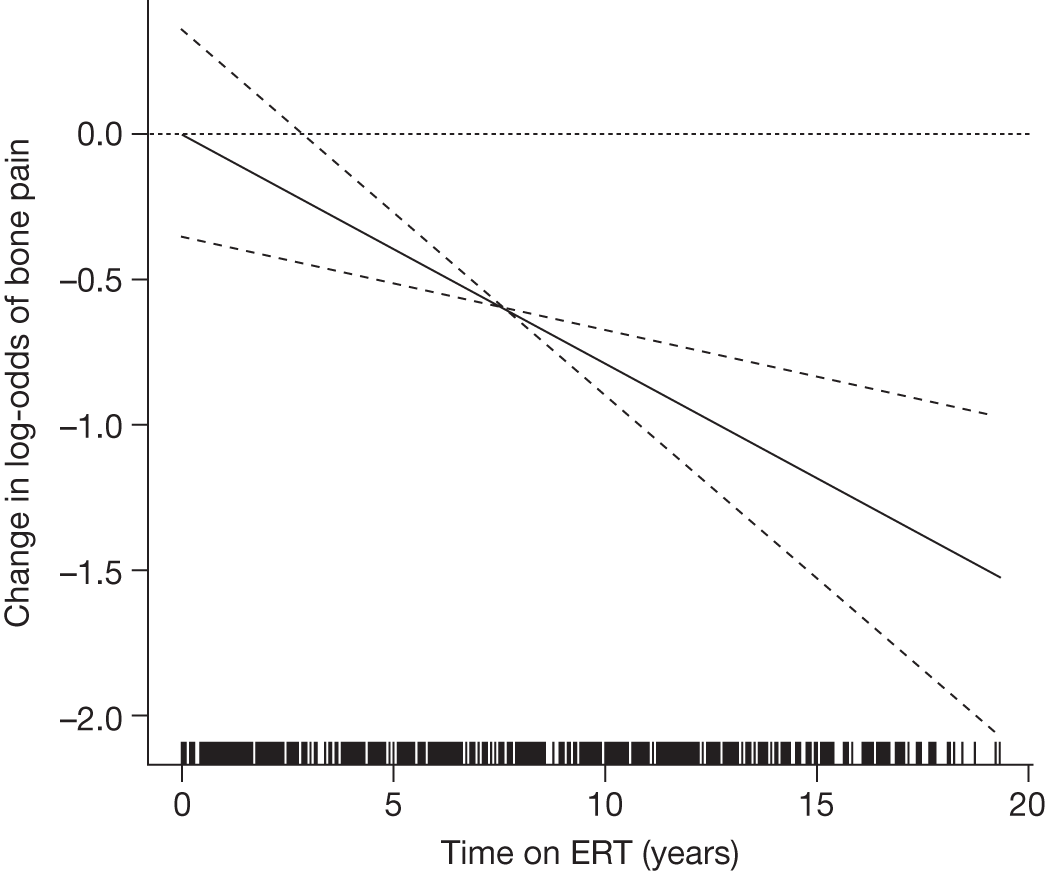
Children
A total of 108 recordings for bone pain (pain or no pain) were obtained for the 25 children across all time points (Table 21).
| N Pain | N No pain | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 12 | 38 | 1.00 | ||
| Female | 21 | 37 | 0.57 | 0.11 to 2.90 | 0.49 |
| Splenectomy | |||||
| No | 22 | 74 | 1.00 | ||
| Yes | 11 | 1 | 46.5 | 1.98 to 1091.8 | 0.02 |
| Age | |||||
| Linear effect/year | 1.26 | 1.04 to 1.54 | 0.02 | ||
| Time on ERT | |||||
| Not on ERT | 4 | 8 | 1.00 | 0.09 | |
| < 12 months | 3 | 13 | 0.06 | 0.005 to 0.88 | 0.04 |
| 12–36 months | 10 | 19 | 0.33 | 0.04 to 2.71 | 0.31 |
| > 36 months | 16 | 35 | 0.08 | 0.007 to 0.98 | 0.05 |
As with adults, splenectomised children had higher odds of bone pain (OR 46.5, 95% CI 1.98 to 1091.8; p = 0.02) than non-splenectomised children, although we only had data on two splenectomised children. Overall, the risk of bone pain increased significantly with each year of age (OR 1.26, 95% CI 1.04 to 1.54; p = 0.02).
As with adults, after adjusting for splenectomy status and age, the risk of having bone pain is reduced with time since starting on ERT, although, again, this does not reach conventional levels of statistical significance in this model where time on ERT is categorised into four groups (p = 0.09).
Again, further analysis was conducted to explore the overall shape of the relationship between bone pain and time on ERT, treating time as a continuous variable. Figure 12 suggests a statistically significant linear relationship between time on ERT and reduced risk of bone pain in children (edf = 1; p = 0.02).
FIGURE 12.
The age-adjusted association between time on ERT and bone pain in children with Gaucher disease (time on ERT treated as a continuous variable).
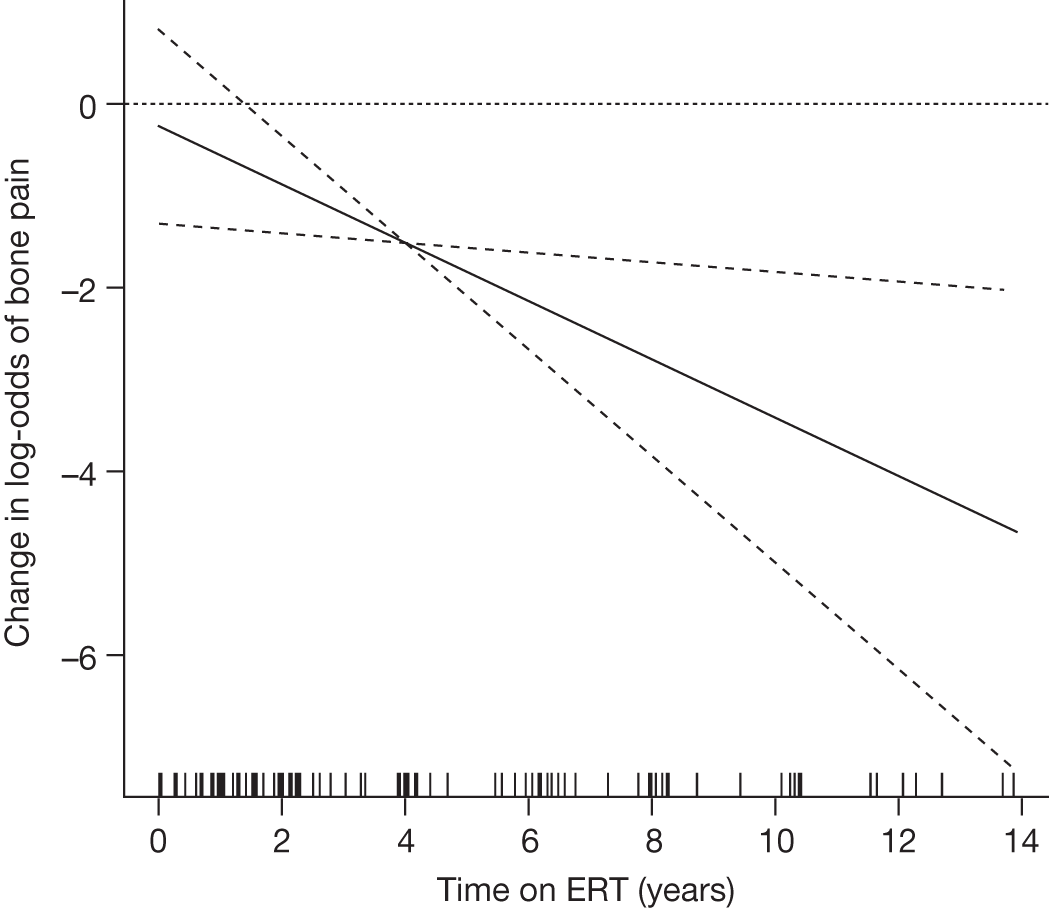
Spleen volume
Adults
We have spleen volume from 63 adult patients, with 298 measurements across all time points. The range of spleen volumes was 113–3170 ml (Table 22).
| N Data | Estimate of change in spleen volume (ml) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 133 | 0.00 | |||
| Female | 165 | –172.6 | 50.8 | –272.2 to –73.03 | < 0.001 |
| Age | |||||
| Linear effect/year | –0.36 | 1.65 | –3.59 to 2.87 | 0.83 | |
| Time on ERT | |||||
| Not on ERT | 8 | 0.00 | < 0.001 | ||
| < 12 months | 21 | –500.8 | 91.2 | –679.5 to –322.05 | < 0.001 |
| 12–36 months | 44 | –734.7 | 87.8 | –906.7 to –562.6 | < 0.001 |
| > 36 months | 225 | –972.2 | 91.6 | –1151.7 to –792.6 | < 0.001 |
| Variance components | |||||
| Individual | 276,920 | ||||
| Centre | 2678 | ||||
| Residual | 29,286 | ||||
There was no association between spleen volume and age (p = 0.83). We found a statistically significant association between time on ERT and decrease in spleen volume in adults (p < 0.001).
Figure 13 shows the statistically significant non-linear association between spleen volume and time on ERT (edf = 2.9; p < 0.001). This analysis suggests a rapid decrease in spleen volume following the start of treatment with ERT, with the maximum effect obtained by about 8 years, before an apparent plateauing of effect.
FIGURE 13.
The age-adjusted association between time on ERT and spleen volume in adults with Gaucher disease (time on ERT treated as a continuous variable).
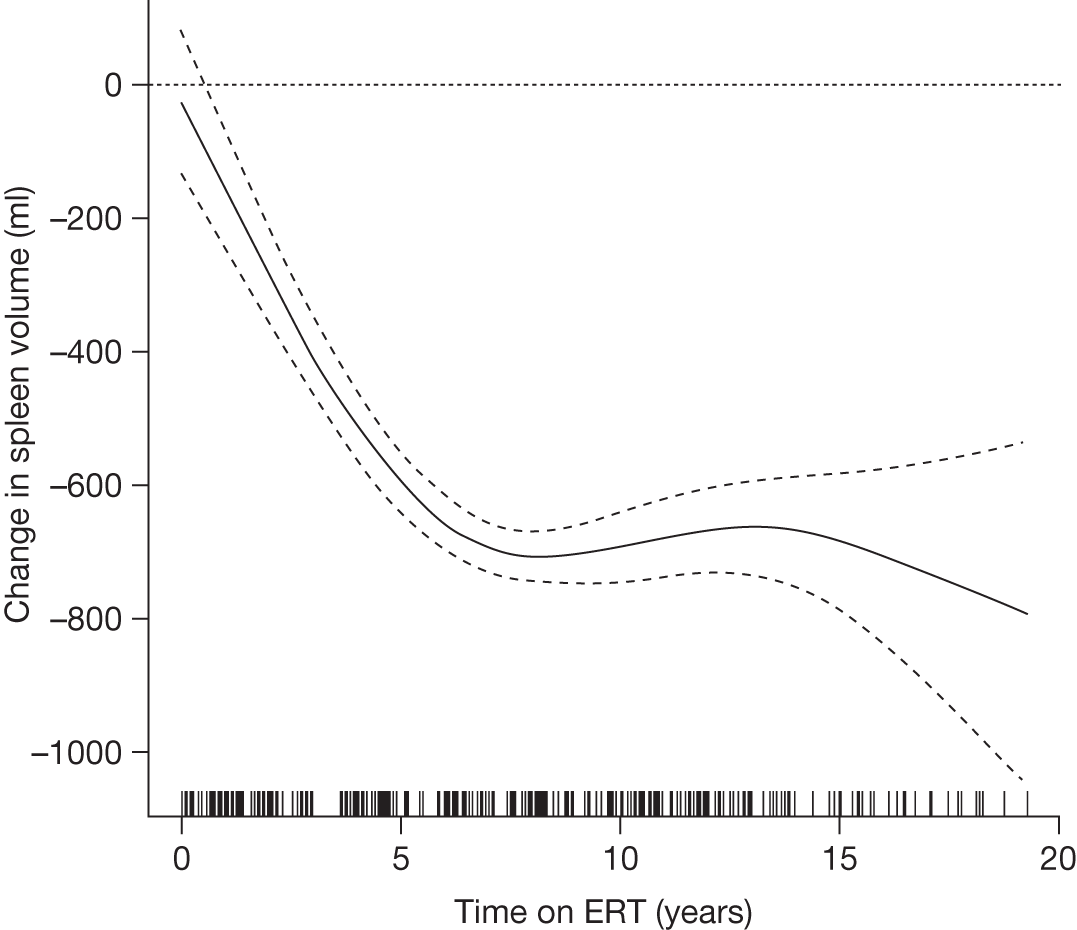
Bayesian projections
Again, the longitudinal model for spleen volume was fitted in a Bayesian framework and used to make predictions about how starting ERT at different ages impacts on age-related spleen volume (Figure 14). Three different treatment scenarios were considered: (1) the patient remains untreated; (2) the patient starts ERT at the age of 18 years; and (3) the patient commences ERT at the age of 45 years. It should be noted that these are mean trajectories and in the interest of clarity the credible intervals around these trajectories are not shown.
FIGURE 14.
Spleen volumes in patients with different treatment scenarios.
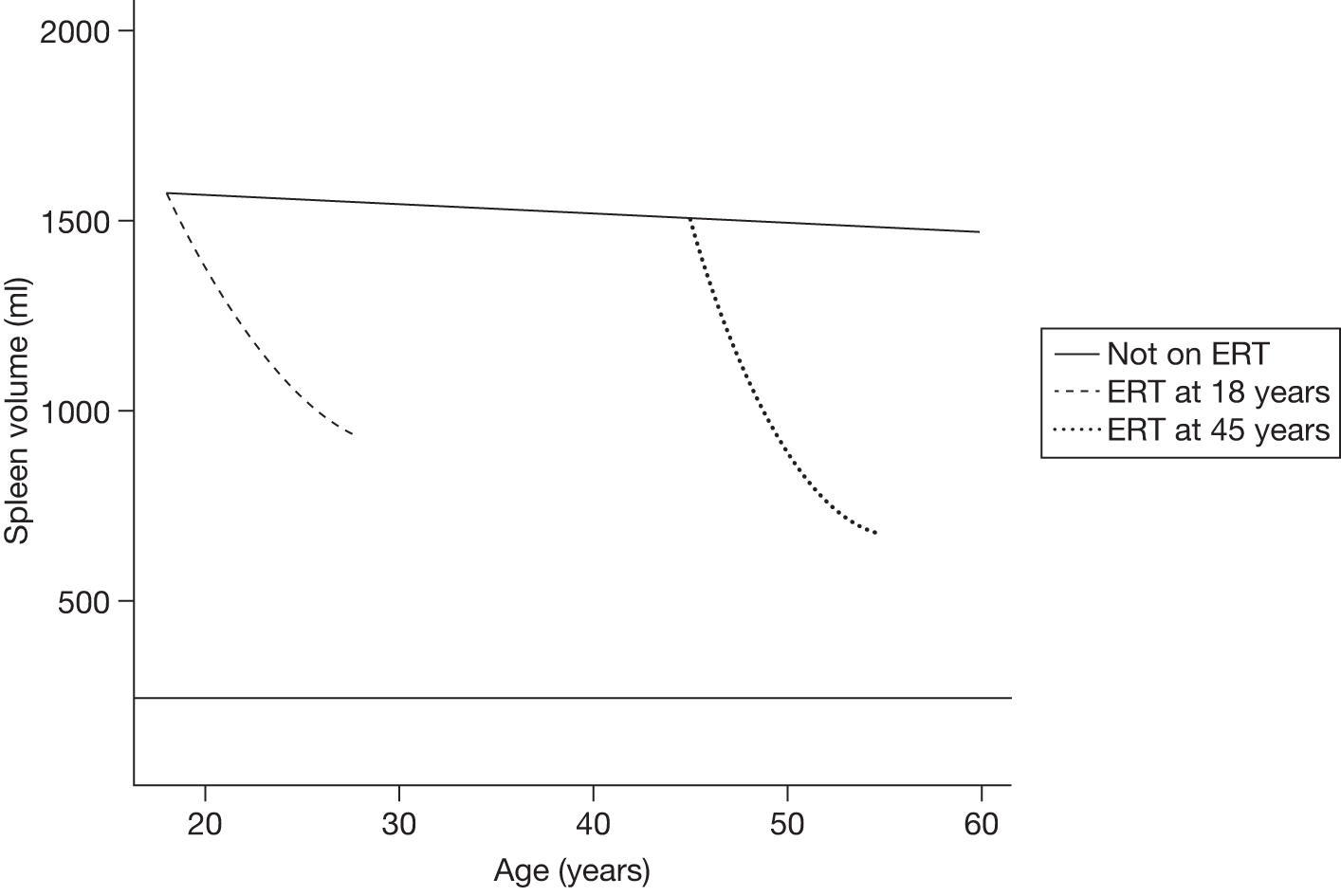
Patients who are not on ERT had a mean spleen volume of about 1500 ml, approximately three to four times the normal spleen volume in a healthy adult. The data suggest that spleen volume remains essentially unchanged over time in untreated adults. This, however, is likely to reflect, at least in part, bias owing to an increased likelihood of splenectomy (and hence removal from the model) of those patients with substantially enlarged spleens, as well as the heterogeneity of disease severity in the older patient population. The model predicts that patients who went on ERT at the ages of 18 and 45 years show a similar trajectory, with an initial reduction in spleen volume for 7 years, followed by a plateauing off of treatment effect. Estimates from the Bayesian model indicate that there is a 83% probability that first infusion at age 45 years will result in a greater reduction in spleen volume after 10 years on ERT than first infusion at age 18 years. A possible explanation for this finding is that the patients observed in late middle age have less aggressive forms of the disease and so may respond more successfully to treatment.
Clinical enlargement of spleen (adults)
Enlargement of the spleen was determined either from a scan or from palpation, with a palpable spleen at rest or on inspiration indicating organ enlargement. In this section, for patients for whom no palpation data are available, we have included data from their scan volumes, and categorised patients whose volume is > 250 ml as ‘enlarged’ (Table 23).
| N Enlarged | N Normal | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 131 | 38 | 1.00 | ||
| Female | 138 | 56 | 0.67 | 0.22 to 2.03 | 0.48 |
| Age | |||||
| Linear effect/year | 0.99 | 0.96 to 1.03 | 0.84 | ||
| Time on ERT | |||||
| Not on ERT | 15 | 1 | 1.0 | < 0.001 | |
| < 12 months | 26 | 3 | 0.24 | 0.009 to 6.26 | 0.39 |
| 12–36 months | 47 | 10 | 0.08 | 0.004 to 1.43 | 0.09 |
| > 36 months | 181 | 80 | 0.02 | 0.001 to 0.34 | 0.007 |
There was no significant association between the risk of an enlarged spleen and age (p = 0.84). There was a strong statistically significant association between time on ERT and a reduced risk of having an enlarged spleen (p < 0.001) with the model suggesting that the OR for having an enlarged spleen after ≥ 3 years of treatment compared with no treatment was 0.02 (95% CI 0.001 to 0.34). In this initial analysis, we considered the response to treatment with all patients on ERT for > 36 months in a single group despite the considerable within-group heterogeneity in time on ERT.
Further analysis was conducted to explore the overall shape of the relationship between time on ERT (considering actual time on ERT rather than categorising into four groups as in the previous analysis) and the log-odds of having an enlarged spleen. Figure 15 illustrates an apparently linear association between the log-odds of enlarged spleen and time on ERT in people with Gaucher disease (edf = 1; p < 0.001).
FIGURE 15.
The age-adjusted association between time on ERT and the risk of having an enlarged spleen in adults with Gaucher disease (time on ERT treated as a continuous variable).
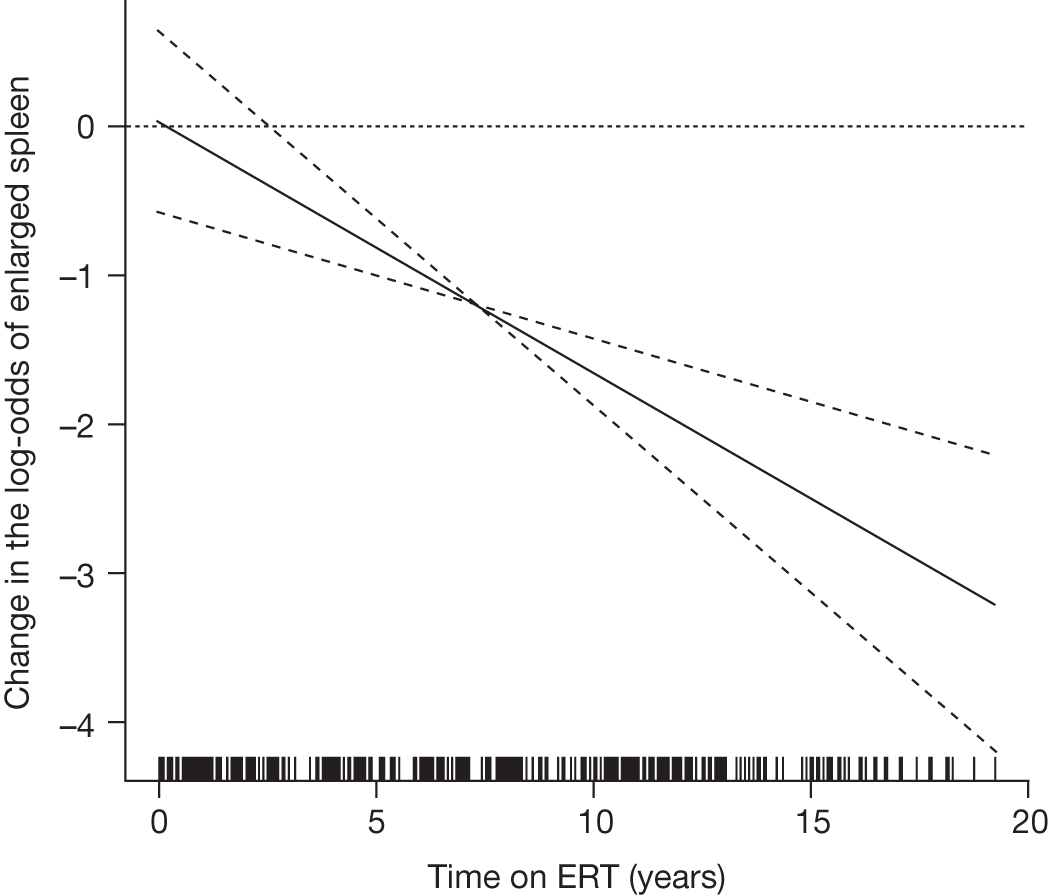
Liver size
We have liver volume data for 67 adult patients, and a total of 529 measurements for liver volume were recorded for these patients across all time points. The range of these measurements was 204–4500 ml (Table 24).
| N Data | Estimate of change in liver volume (ml) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 204 | 0.0 | |||
| Female | 325 | –222.1 | 76.2 | –371.4 to –72.7 | 0.03 |
| Age | |||||
| Linear effect/year | –10.4 | 2.52 | –15.3 to –5.46 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 15 | 0.0 | < 0.001 | ||
| < 12 months | 31 | –392.0 | 74.3 | –537.6 to –246.4 | < 0.001 |
| 12–36 months | 82 | –658.7 | 66.4 | –788.8 to –528.5 | < 0.001 |
| > 36 months | 401 | –801.9 | 69.2 | –937.5 to –666.3 | < 0.001 |
| Variance components | |||||
| Individual | 323,911 | ||||
| Centre | 11,203 | ||||
| Residual | 37,862 | ||||
There was a significant association between age and reduced liver volume (p < 0.001). There was a statistically significant association between time on ERT and decline in liver volume (p < 0.001). It is likely that the apparent decrease in the volume with age reflects, at least in part, differences in severity by age at diagnosis in our data set.
Further analysis was conducted to explore the overall shape of the relationship between liver volume and time on ERT (considering actual time on ERT rather than categorising into four groups as in the previous analysis). Figure 16 illustrates a statistically significant non-linear effect of time on ERT (edf = 2.92; p < 0.001). This suggests a rapid effect of ERT over the first 5 years of treatment followed by an apparent plateauing of the effect.
FIGURE 16.
The age-adjusted association between time on ERT and liver volume in adults with Gaucher disease (time on ERT treated as a continuous variable).
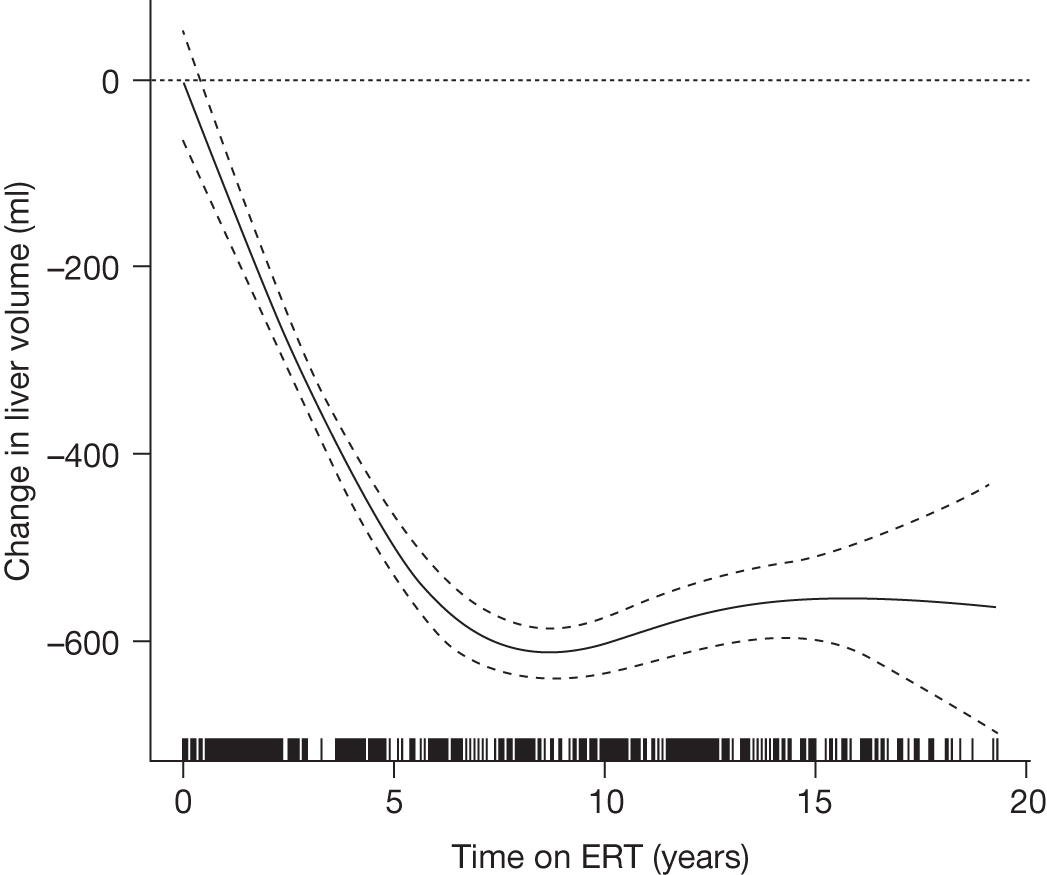
Liver from scan and palpation
As for the spleen, enlargement of the liver was determined either from a scan or from palpation, with a palpable liver at rest or on inhalation indicating organ enlargement. In this section, for patients for whom no palpation data are available, we have included data from their scan volumes, and categorised patients whose liver volume is > 1660 ml as ‘enlarged’ (Table 25).
| N Enlargd | N Normal | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 143 | 88 | 1.00 | ||
| Female | 179 | 180 | 0.58 | 0.25 to 1.35 | 0.21 |
| Age | |||||
| Linear effect/year | 0.96 | 0.94 to 0.98 | 0.008 | ||
| Time on ERT | |||||
| Not on ERT | 21 | 4 | 1.0 | < 0.001 | |
| < 12 months | 33 | 9 | 0.51 | 0.09 to 2.67 | 0.42 |
| 12–36 months | 59 | 32 | 0.17 | 0.04 to 0.78 | 0.02 |
| > 36 months | 209 | 223 | 0.06 | 0.01 to 0.25 | < 0.001 |
There was a significant reduction in the odds of enlarged liver with age (p = 0.008). There was a strong, statistically significant, association between time on ERT and a reduced risk of having an enlarged liver (p < 0.001), with the model suggesting that the OR for having an enlarged liver after ≥ 3 years of treatment compared with no treatment was 0.06 (95% CI 0.01 to 0.25; p < 0.001). In this initial analysis, we considered the response to treatment of all patients on ERT for > 36 months in a single group despite the considerable within-group heterogeneity in time on ERT.
Further analysis was conducted to explore the overall shape of the relationship between time on ERT (considering actual time on ERT rather than categorising into four groups as in the previous analysis) and the log-odds of having an enlarged liver. Figure 17 illustrates the relationship between liver size and time on ERT. The risk of liver enlargement appears to decrease during the first 10 years of treatment and then plateau (edf = 2.7; p < 0.001) although again interpretation of the shape of the relationship over time needs to take account of the wide confidence limits, particularly with longer duration of treatment, when the data become more sparse.
FIGURE 17.
The age-adjusted association between time on ERT and the risk of having an enlarged liver in adults with Gaucher disease (time on ERT treated as a continuous variable).
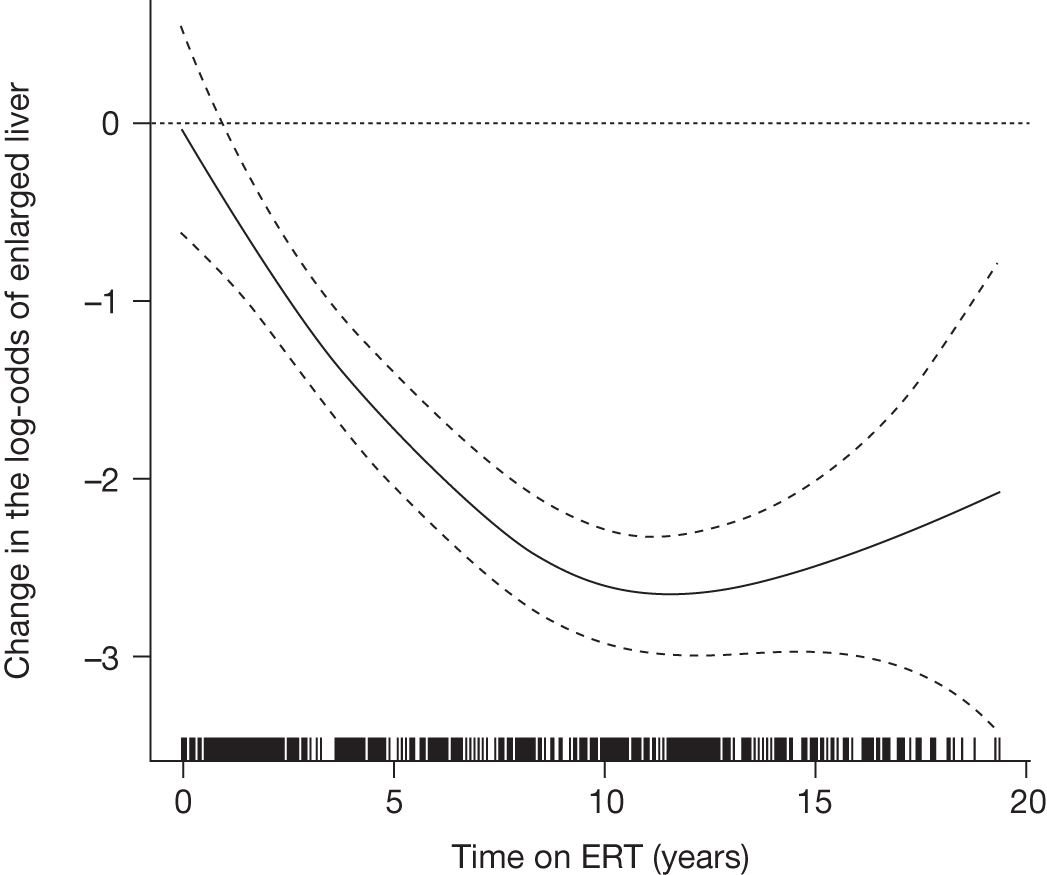
Bayesian projections
The longitudinal model for liver volume was fitted in a Bayesian framework and used to make predictions about how starting ERT at different ages impacts on age-related liver volume (Figure 18). Three different treatment scenarios were considered: (1) the patient remains untreated; (2) the patient starts ERT at the age of 18 years; and (3) the patient commences ERT at the age of 45 years. It should be noted that these are mean trajectories and in the interest of clarity the credible intervals around these trajectories are not shown.
FIGURE 18.
Liver volume in patients with different treatment scenarios.
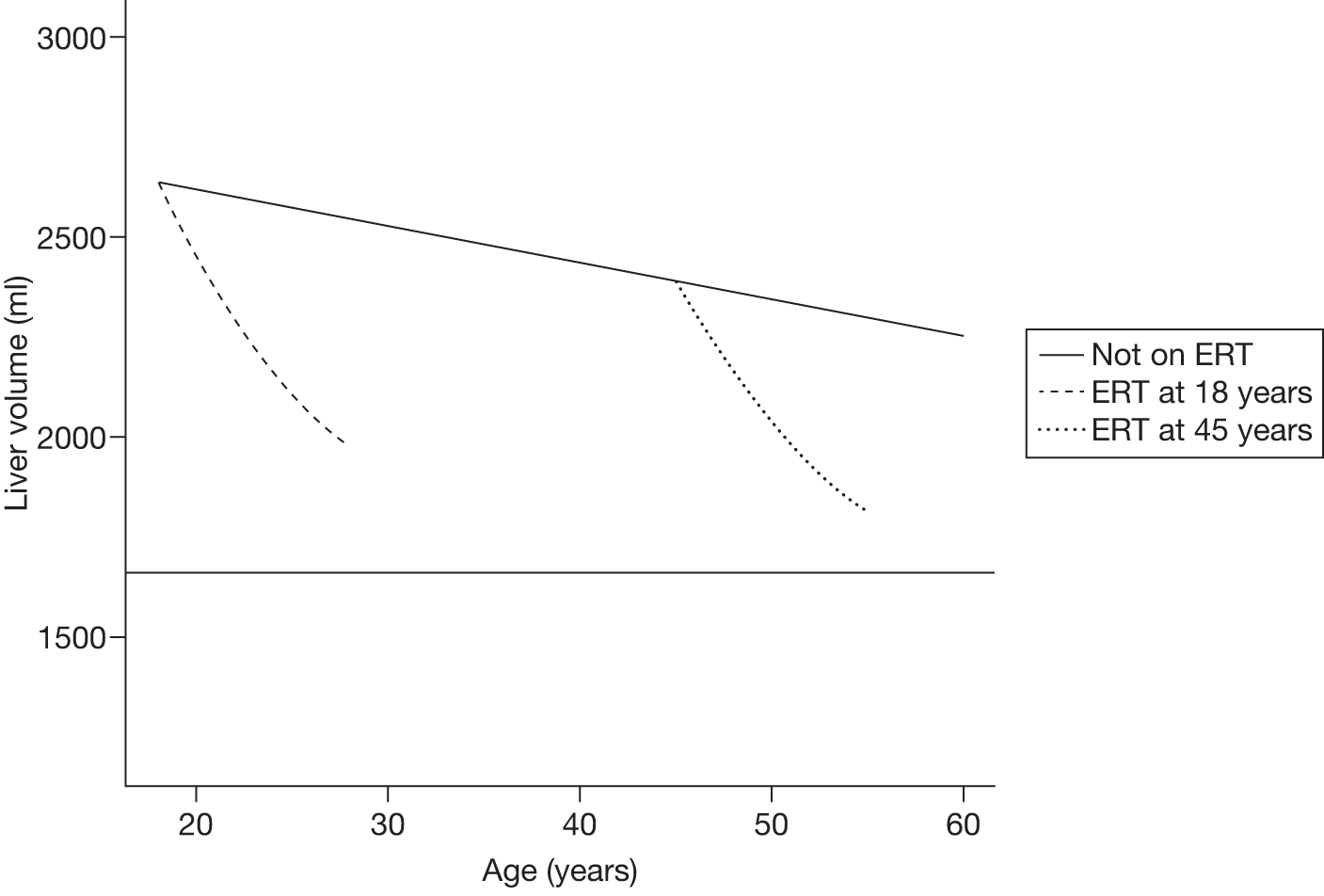
The model predicts that patients who are not on ERT had a mean liver volume of > 2500 ml, approximately 125% that of the normal liver volume in a healthy adult. Initiation of treatment with ERT results in a reduction in liver volume for the first 10 years of treatment followed by a plateau. Estimates from the Bayesian model indicate that there is a 48% probability that first infusion at age 45 years will result in a greater reduction in liver volume after 10 years on ERT than first infusion at age 18 years.
Liver function tests
Alanine transaminase
Although data were collected on alanine transaminase (ALT) levels there was substantial variation between sites in the methods used and ‘normal’ ranges and so further analyses were not carried out.
Aspartate transaminase
Adults
AST values for 67 patients were collected from one site. A total of 534 measurements for AST were recorded across all time points. Values ranged from 3 to 176 IU/l (Table 26).
| N Data | Estimate of change of AST (IU/l) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 188 | 0.00 | |||
| Female | 346 | –9.84 | 4.86 | –19.4 to –0.31 | 0.04 |
| Age | |||||
| Linear effect/year | –0.78 | 0.15 | –1.07 to –0.48 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 23 | 0.00 | 0.02 | ||
| < 12 months | 41 | –5.19 | 4.83 | –14.6 to 4.27 | 0.28 |
| 12–36 months | 90 | –6.26 | 4.33 | –14.7 to 2.22 | 0.15 |
| > 36 months | 380 | –11.5 | 4.22 | –19.7 to –3.23 | 0.007 |
| Variance components | |||||
| Individual | 315.6 | ||||
| Residual | 318.2 | ||||
There was a statistically significant association between age and lower AST levels in adults (p < 0.001) and a statistically significant association between time on ERT and reduced AST levels (p = 0.02). In this initial analysis, we considered the response to treatment with all patients on ERT for > 36 months considered in a single group despite the considerable within-group heterogeneity in time on ERT.
Further analysis was conducted to explore the overall shape of the relationship between time on ERT (considering actual time on ERT rather than categorising into four groups as in the previous analysis) and AST. Figure 19 shows the non-linear association between AST levels in adults and time on ERT (edf = 2.01; p < 0.001). AST levels decline over the first 15 years before an apparent plateauing, although this should be viewed with caution owing to the widening CIs.
FIGURE 19.
The age-adjusted association between time on ERT and AST levels in adults with Gaucher disease (time on ERT treated as a continuous variable).
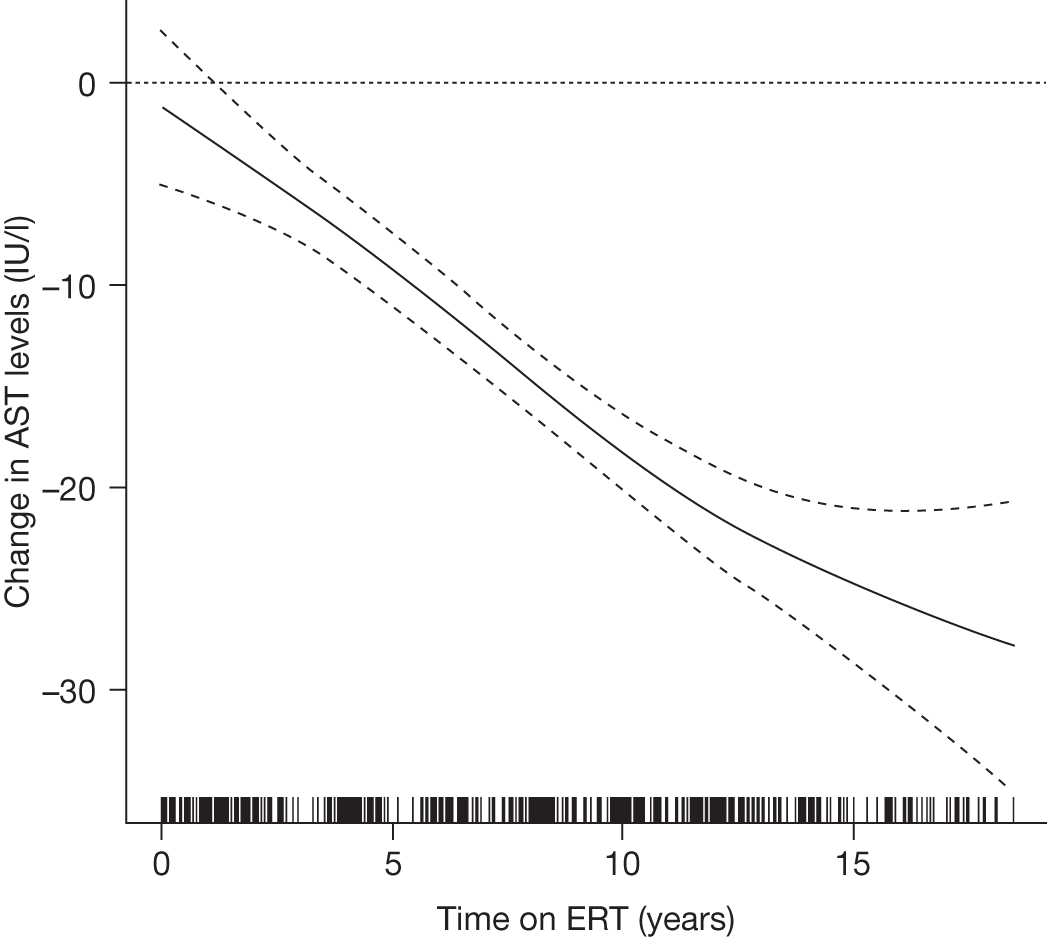
In addition to the previous analysis, in a secondary analysis AST data were dichotomised as normal/abnormal, and the risk of having a raised AST level (defined as above 40 IU/l according to the unit ‘normal’ values) was assessed (Table 27).
| N Abnormal | N Normal | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 83 | 105 | 1.00 | ||
| Female | 94 | 252 | 0.30 | 0.11 to 0.81 | 0.02 |
| Age | |||||
| Linear effect/year | 0.94 | 0.91 to 0.97 | 0.001 | ||
| Time on ERT | |||||
| Not on ERT | 12 | 11 | 1.00 | 0.007 | |
| < 12 months | 24 | 17 | 1.09 | 0.31 to 3.85 | 0.88 |
| 12–36 months | 40 | 50 | 0.57 | 0.18 to 1.74 | 0.32 |
| > 36 months | 101 | 279 | 0.27 | 0.09 to 0.81 | 0.02 |
The odds of abnormal AST levels in adults were significantly reduced with age (p = 0.001) and time on ERT (p = 0.007).
There was a statistically significant association between time on ERT and a reduced risk of having raised AST levels (p = 0.007), with the model suggesting that the odds of patients having raised AST levels fell by > 70% after ≥ 3 years of treatment (95% CI 0.09 to 0.81; p = 0.02) compared with untreated patients.
Children
We have 18 values for AST from six children across all time points, with values ranging from 11 to 107 IU/l (Table 28).
| N Data | Estimate | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 2 | 0.00 | |||
| Female | 16 | –49.5 | 21.8 | –92.2 to –6.77 | 0.04 |
| Age | |||||
| Linear effect/year | –1.99 | 1.24 | –4.42 to 0.44 | 0.12 | |
| Time on ERT | |||||
| Not on ERT | 2 | 0.00 | 0.34 | ||
| < 12 months | 8 | 2.44 | 15.5 | –27.8 to 32.8 | 0.87 |
| 12–36 months | 3 | –15.0 | 17.9 | –50.1 to 20.1 | 0.41 |
| > 36 months | 5 | –9.04 | 17.3 | –42.9 to 24.8 | 0.61 |
| Variance components | |||||
| Individual | 133.2 | ||||
| Residual | 328.8 | ||||
There was no statistically significant association between AST levels in children and age (p = 0.12) or time on ERT (p = 0.34).
There are insufficient data to fit a model for the dichotomous variable in children.
Neurological involvement
Peripheral neuropathy
At the time of entry into the study, seven GD1 patients were reported to have neurological involvement, of whom six were on ERT. Four of the seven with neurological involvement are reported to have a peripheral neuropathy, of whom three were on ERT at the time. There were insufficient data for further analysis.
Parkinsonian features
Three patients with GD1 were reported to have Parkinsonian features at the time of entry into the study. All three of these patients were on ERT at this time. There were insufficient data for further analysis.
Quality-of-life assessments
SF-36
One hundred and eighty-seven SF-36 questionnaires were completed across all prospective time points by 117 patients.
Data are presented separately below for the physical component score (PCS) and the mental component score (MCS) (Tables 29–31).
| PCS | MCS | |
|---|---|---|
| Overall | ||
| Mean (SD) | 42.6 (12.4) | 50.8 (9.97) |
| N Data | 187 | 187 |
| ≤ 3 years on ERT | ||
| Mean (SD) | 44.2 (12.6) | 52.3 (8.3) |
| N Data | 18 | 18 |
| > 3 years on ERT | ||
| Mean (SD) | 42.3(12.5) | 50.8 (10.0) |
| N Data | 168 | 168 |
| N Data | Estimate of change of PCS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 79 | 0.00 | |||
| Female | 108 | –0.81 | 2.18 | –5.08 to 3.46 | 0.71 |
| Age | |||||
| Linear effect/year | –0.33 | 0.07 | –0.46 to –0.19 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 1 | 0.00 | 0.86 | ||
| < 12 months | 7 | –3.53 | 6.89 | –17.03 to 9.97 | 0.61 |
| 12–36 months | 11 | –0.30 | 5.77 | –11.6 to 11.0 | 0.96 |
| > 36 months | 168 | –1.27 | 6.49 | –13.9 to 11.4 | 0.85 |
| Variance components | |||||
| Individual | 120.2 | ||||
| Centre | 0.0 | ||||
| Residual | 22.8 | ||||
| N Data | Estimate of change of MCS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 79 | 0.00 | |||
| Female | 108 | –0.66 | 1.77 | –4.13 to 2.81 | 0.71 |
| Age | |||||
| Linear effect/year | –0.011 | 0.06 | –0.13 to 0.11 | 0.86 | |
| Time on ERT | |||||
| Not on ERT | 1 | 0.00 | 0.84 | ||
| < 12 months | 7 | 5.28 | 8.25 | –10.8 to 21.4 | 0.52 |
| 12–36 months | 11 | 4.58 | 7.32 | –9.76 to 18.9 | 0.54 |
| > 36 months | 168 | 3.14 | 7.67 | –11.8 to 18.2 | 0.68 |
| Variance components | |||||
| Individual | 62.4 | ||||
| Centre | 0.00 | ||||
| Residual | 38.4 | ||||
There was a statistically significant association between lower PCS as age increased (p < 0.001). There was no significant association between these scores and time on ERT (p = 0.86). It is important to note that because these data were not available in the retrospective data points this analysis essentially compares people with different durations of ERT rather than comparing those not on ERT with those on ERT (Table 30).
Further analysis was conducted to explore the overall shape of the relationship between time on ERT (considering actual time on ERT rather than categorising into four groups as in the previous analysis) and PCS. Figure 20 suggests no evidence for a non-linear association between PCS and time on ERT (edf = 1.34; p = 0.66)
FIGURE 20.
The age-adjusted association between time on ERT and PCS in adults with Gaucher disease.
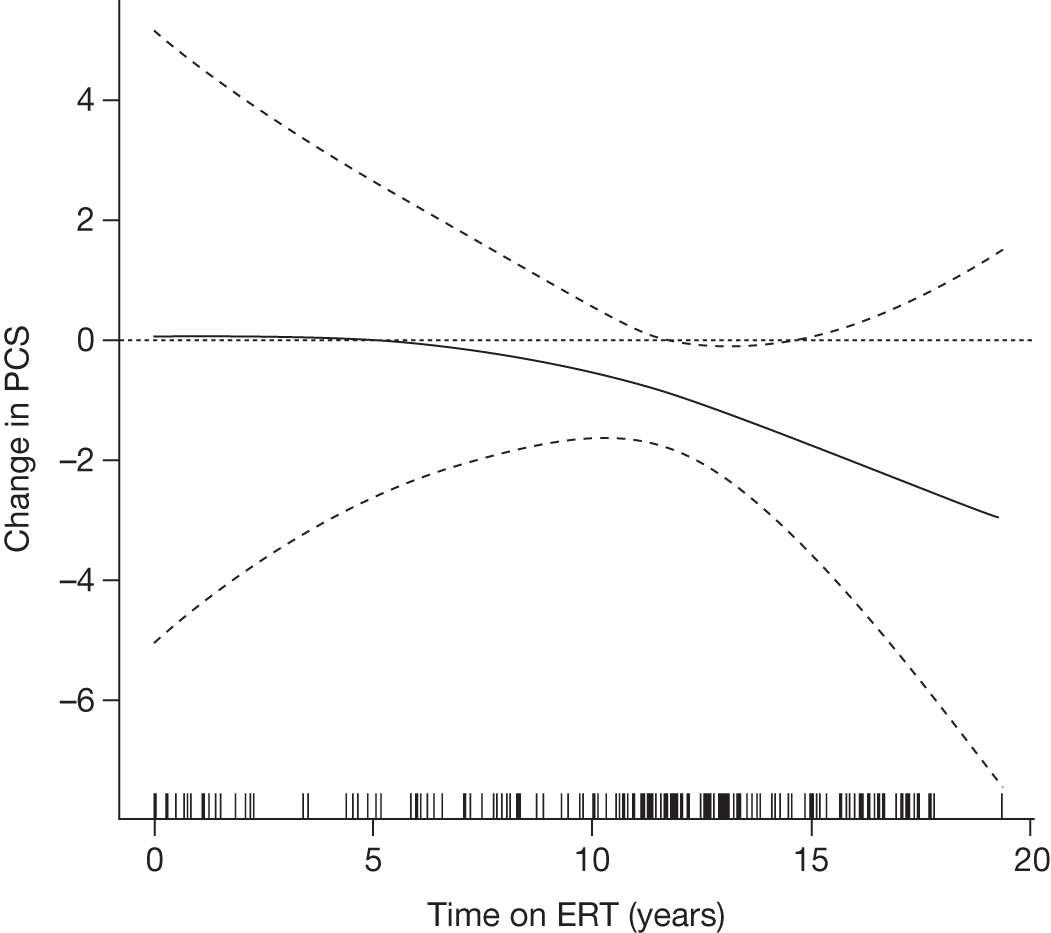
There was no significant association between MCS and age (p = 0.86) or time on ERT (p = 0.84). It is important to note that because these data were not available in the retrospective data points this analysis essentially compares people with different durations of ERT rather than comparing those not on ERT with those on ERT (Table 31).
Figure 21 suggests that MCS may decrease with the time on ERT, but this effect did not reach conventional levels of statistical significance (edf = 1.62; p = 0.19).
FIGURE 21.
The age-adjusted association between time on ERT and MCS in adults with Gaucher disease.
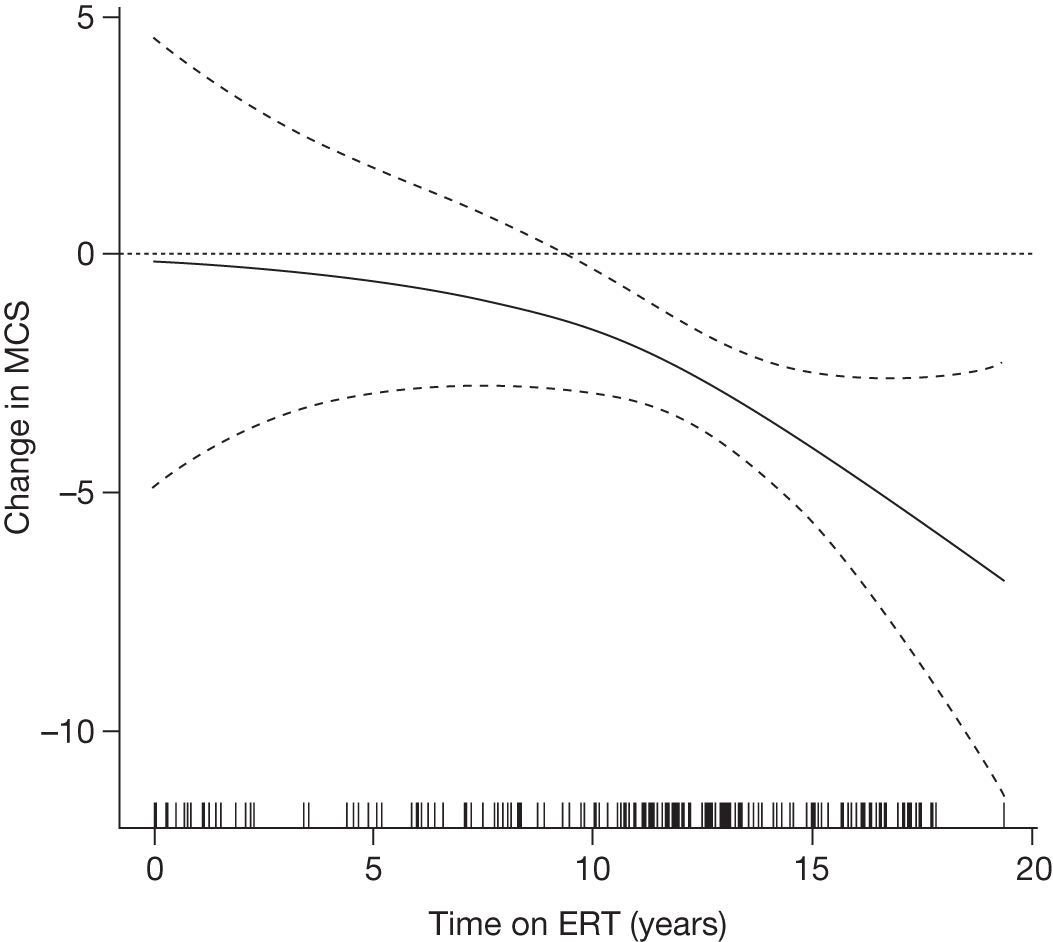
EQ-5D
In addition to the SF-36, participants aged > 13 years were invited to complete the EQ-5D. Two hundred and fourteen EQ-5D index scores were generated across all prospective time points. Data are presented below for the EQ-5D score (range from –0.32 to 1.0), with 1.0 being ‘perfect health’.
A longitudinal model was fitted to assess the linear relationship between EQ-5D and time on ERT, after adjusting for age (Table 32).
| N Data | Estimate of change of EQ-5D | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 90 | 0.00 | |||
| Female | 124 | –0.02 | 0.04 | –0.11 to 0.06 | 0.62 |
| Age | |||||
| Linear effect/year | –0.003 | 0.001 | –0.006 to –0.0005 | 0.02 | |
| Time on ERT | |||||
| Not on ERT | 2 | 0.00 | 0.92 | ||
| < 12 months | 7 | –0.02 | 0.12 | –0.26 to 0.23 | 0.88 |
| 12–36 months | 12 | 0.02 | 0.11 | –0.19 to 0.23 | 0.87 |
| > 36 months | 193 | –0.02 | 0.11 | –0.23 to 0.18 | 0.83 |
| Variance components | |||||
| Individual | 0.04 | ||||
| Centre | 0.0008 | ||||
| Residual | 0.02 | ||||
The linear model showed a small, but significant, reduction in EQ-5D score with age (p = 0.02), but no significant association was observed between EQ-5D and time on ERT (p = 0.92). No evidence was found for a non-linear association between the EQ-5D score and time on ERT (edf = 1; p = 0.43) (Figure 22).
FIGURE 22.
The age-adjusted association between time on ERT and EQ-5D index score in adults with Gaucher disease.
Equivalent modelling analyses were also conducted using SF-6D (SF-36-derived) utility weights, but no statistically significant associations (at α = 0.05 level) were found with time on ERT. The tabulated results of the SF-6D longitudinal modelling analyses are available on request from the study authors.
EQ-5D visual analogue scale
In addition to scoring on the five domains of the EQ-5D, participants were asked to rate their health on a VAS. The EQ-5D VAS asks people to rate their health state on a 10-cm line from 0, ‘worst imaginable health state’, to 100, ‘best imaginable health state’. The 209 VAS scores completed across all prospective time points ranged from 5 to 100. A longitudinal model was fitted to assess the linear relationship between the visual analogue score and time on ERT after adjusting for age.
The linear model shown in Table 33 shows a significant association between EQ-5D VAS with age (p = 0.03), but no significant association between the EQ-5D VAS and time on ERT was found (p = 0.71). No evidence was found for a non-linear association with EQ-5D VAS and time on ERT (edf = 1; p = 0.64) (Figure 23).
| N Data | Estimate of change of EQ-5D VAS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 88 | 0.00 | |||
| Female | 121 | 2.22 | 3.36 | –4.37 to 8.81 | 0.51 |
| Age | |||||
| Linear effect/year | –0.24 | 0.11 | –0.45 to –0.03 | 0.03 | |
| Time on ERT | |||||
| Not on ERT | 2 | 0.00 | 0.71 | ||
| < 12 months | 7 | 2.63 | 12.9 | –22.6 to 27.9 | 0.84 |
| 12–36 months | 12 | –6.65 | 11.5 | –29.2 to 15.9 | 0.56 |
| > 36 months | 188 | –1.85 | 11.2 | –23.8 to 20.2 | 0.87 |
| Variance components | |||||
| Individual | 208.9 | ||||
| Centre | 0.00 | ||||
| Residual | 196.3 | ||||
FIGURE 23.
The age-adjusted association between time on ERT and EQ-5D VAS score in adults with Gaucher disease (time on ERT treated as continuous variable).
PedsQL
Forty-one PedsQL questionnaires were completed by children with Gaucher disease or their carers. Table 34 shows a descriptive summary of the total score and the scores from the component scale scores by type of treatment. The scale ranges from 0 to 100, where a higher score indicates better HRQoL.
| Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Physical health summary score | Total score | |
|---|---|---|---|---|---|---|---|
| Overall | |||||||
| Mean (SD) | 61.9 (33.9) | 65.38 (19.4) | 73.7 (24.1) | 56.9 (16.8) | 64.06 (16.7) | 61.9 (33.9) | 62.5 (20.6) |
| n | 39 | 40 | 41 | 38 | 38 | 39 | 36 |
| ≤ 3 years on ERT | |||||||
| Mean (SD) | 66.4 (27.5) | 60.25 (15.9) | 85.0 (18.4) | 40.1 (7.21) | 50.9 (5.89) | 66.4 (27.5) | 46.9 (4.52) |
| n | 5 | 4 | 5 | 2 | 2 | 5 | 2 |
| > 3 years on ERT | |||||||
| Mean (SD) | 69.4 (33.8) | 69.3 (19.3) | 75.1 (24.4) | 61.8 (18.7) | 68.7 (17.4) | 69.4 (33.8) | 70.5 (19.3) |
| n | 10 | 12 | 12 | 12 | 12 | 10 | 10 |
After adjusting for age, a statistically significant reduction was seen in the social functioning subscale of PedsQL with time on ERT (p = 0.03), but not for the overall score (Table 35).
| Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Physical health summary score | Total score | |
|---|---|---|---|---|---|---|---|
| Gender | |||||||
| Male | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Female | –10.8 | 11.7 | 14.9 | 14.5 | 10.03 | –10.8 | –5.96 |
| 95% CI | –43.8 to 22.1 | –7.64 to 31.1 | –5.57 to 35.5 | –1.53 to 30.6 | –7.0 to 27.1 | –43.8 to 22.1 | –29.5 to 17.6 |
| p-value | 0.53 | 0.25 | 0.17 | 0.09 | 0.26 | 0.53 | 0.63 |
| Current age | |||||||
| Mean increment/year | 2.02 | 1.85 | 2.36 | 1.95 | 3.84 | 2.02 | 6.73 |
| 95% CI | –3.27 to 7.32 | –1.61 to 5.32 | –1.08 to 5.8 | –1.48 to 5.37 | 0.42 to 7.25 | –3.27 to 7.32 | 2.31 to 11.1 |
| p-value | 0.46 | 0.31 | 0.19 | 0.28 | 0.04 | 0.46 | 0.009 |
| Time on ERT | |||||||
| Mean increment/year | –1.65 | –0.95 | –4.25 | –0.35 | –1.50 | –1.65 | –0.89 |
| 95% CI | –6.89 to 3.58 | –4.22 to 2.31 | –7.72 to –0.77 | –2.89 to 2.19 | –4.19 to 1.18 | –6.89 to 3.58 | –4.44 to 2.64 |
| p-value | 0.54 | 0.57 | 0.03 | 0.79 | 0.29 | 0.54 | 0.63 |
| Variance components | |||||||
| Individual | 895.1 | 182.3 | 151.1 | 0.00 | 229.1 | 895.1 | 404.9 |
| Centre | 299.5 | 0.00 | 0.00 | 0.00 | 0.00 | 299.5 | 0.00 |
| Residual | 14.53 | 225.7 | 324.4 | 271.4 | 23.3 | 14.53 | 6.78 |
Fatigue Severity Scale
One hundred and thirty-four FSS questionnaires were collected. The scores ranged from 1 to 7 [where a high score (≥ 4) is indicative of significant fatigue]. 228,229 Exploratory modelling suggested no evidence for an association between fatigue and time on ERT (edf = 1; p = 0.57). This is well illustrated by Figure 24.
FIGURE 24.
The age-adjusted association between time on ERT and FSS in adults with Gaucher disease (time on ERT treated as a continuous variable).
No further longitudinal analysis was conducted because FSSs are only available for prospective data points for patients who have been on ERT for > 36 months, and we have no data for patients who are not on ERT.
Carer Strain Index
Sixteen CSI questionnaires were completed across all prospective time points. Data for the CSI total score ranged from 2 to 23; the maximum possible score of 26 signifies the greatest degree of caregiver burden. A longitudinal model was fitted to assess the linear relationship between the CSI and time on ERT, after adjusting for age (Table 36).
| N Data | Estimate of change of CSI | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 9 | 0.00 | |||
| Female | 7 | –7.62 | 3.69 | –14.8 to –0.38 | 0.06 |
| Age | |||||
| Linear effect/year | –0.23 | 0.11 | –0.44 to –0.01 | 0.05 | |
| Time on ERT | |||||
| Not on ERT | 0 | 0.00 | 0.40 | ||
| < 12 months | 3 | ||||
| 12–36 months | 2 | –3.29 | 5.99 | –15.03 to 8.45 | 0.59 |
| > 36 months | 11 | 1.85 | 4.43 | –6.83 to 10.5 | 0.68 |
| Variance components | |||||
| Individual | 14.8 | ||||
| Centre | 0.00 | ||||
| Residual | 10.1 | ||||
The linear model shows a significant reduction in the CSI score with the age of the person they are caring for (p = 0.05). No evidence was found for an association between CSI score and time on ERT (p = 0.40).
No evidence was found for a non-linear association between the CSI and time on ERT (edf = 1.54; p = 0.53) (Figure 25).
FIGURE 25.
The age-adjusted association between time on ERT and CSI score for carers of Gaucher disease patients.
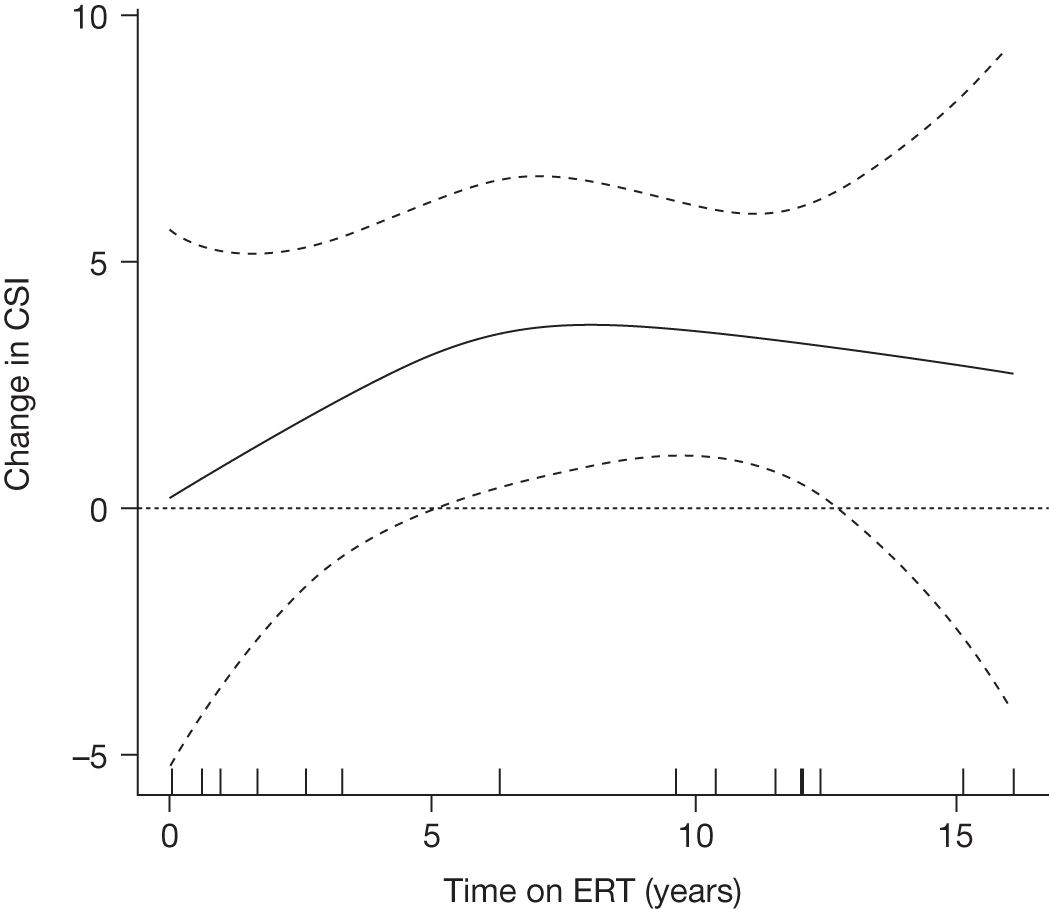
Safety and complications
Of the 175 participants with Gaucher disease, no patients reported that they had experienced any anaphylactic reactions or febrile reactions or had pre medication. One patient was reported to have a positive antibody status to the infused product but did not stop treatment.
Twenty-six patients stopped ERT during the period of data collection. Of these, five patients stopped treatment as they were pregnant or breastfeeding, 14 patients stopped because of treatment shortages and one stopped because of possible side effects. One patient decided to cease treatment, one patient stopped at the end of a clinical trial and no reason for stopping treatment was cited for the remaining four patients.
During the course of the study, no patient died from disease-related complications.
Cost of ERT in people with Gaucher disease
Table 37 shows the current purchase cost to the NHS of the ERTs imiglucerase and velaglucerase, and the SRT miglustat. Note that the drug costs for velaglucerase include the nursing cost for providing the infusion sessions (‘home care’).
| Drug full name | Proprietary name and unit | 2011 base price per unit (£) |
|---|---|---|
| Imiglucerase | Cerezyme®, 200 IU | 495.65 |
| Imiglucerase | Cerezyme®, 400 IU | 991.29 |
| Velaglucerase | Velaglucerase®, 200 IU | 495.65 |
| Velaglucerase | Velaglucerase®, 400 IU | 991.29 |
| Miglustat (SRT) | Zavesca®, 100 mg | 46.84 |
Table 38 shows the NSCT-estimated annual NHS per patient cost of providing these drugs. Note that these costs include both the drug costs and home-care costs where the NSCT funds them.
| Drug | Adults | Children |
|---|---|---|
| Imiglucerase – 1 unit (unbundled) | £126,261 | £107,404 |
| Velaglucerase – 1 unit | £144,868 | £187,841 |
| Miglustat | £54,320 |
Cost of care for adults with Gaucher disease
Total care cost – financial burden of Gaucher disease
Table 39 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for an adult with Gaucher disease. Of the estimated mean per patient annual cost of £3000, four-fifths is as a result of NHS hospital services used; approximately half of this (£1200 per patient per year) is from outpatient stays, whereas about a third (£830) is from inpatient visits (see Table 40). Of the £750 per patient/year from using services outside hospital, < £90 per patient is from GP visits whereas ≥ £500 is due to regular visits from either district nurses, health visitors or other nurses (and many of these will be for regular ERT infusions) (see Table 41).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. who used this type of service | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 132 | 87 | 107 | 2400 | 5166 | 1100 | 940–4500 |
| Services outside hospital | 132 | 87 | 102 | 750 | 1815 | 120 | 54–330 |
| Total health- (NHS) and social-care cost | 132 | 87 | 118 | 3000 | 5577 | 1400 | 650–2800 |
| Type of hospital care | No. who used this type of service (%) | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| Inpatient stays | 23 (17) | 830 | 3999 | 1900 | 940–3800 |
| Outpatient visits | 102 (77) | 1200 | 1953 | 940 | 570–1400 |
| Day cases | 10 (8) | 410 | 2424 | 1400 | 670–7700 |
| Accident and emergency visits | 7 (5) | 12 | 72 | 100 | 100–310 |
| Total hospital (NHS) care cost | 107 (81) | 2400 | 5166 | 1100 | 940–4500 |
| Care provider | No. who used this provider (%) | Mean cost (£) | Standard deviation | Median cost (£) | Interquartile range (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 90 (68) | 84 | 116 | 82 | 55–140 |
| GP nurse appointments | 49 (37) | 5 | 15 | 8 | 5–10 |
| District nurses | 9 (7) | 120 | 1072 | 64 | 21–4000 |
| Community mental health nurse | 0 | 0 | 0 | 0 | 0–0 |
| Other nurse or health visitor | 19 (14) | 420 | 1428 | 2100 | 530–4600 |
| Counsellor | 1 (1) | < 1 | 3 | 29 | N/A |
| Other therapist | 5 (4) | 3 | 19 | 37 | 25–170 |
| ‘Alternative’ medicine or therapy | 1 (1) | < 1 | 6 | 74 | N/A |
| Psychologist | 1 (1) | 1 | 14 | 160 | N/A |
| Psychiatrist | 0 | 0 | 0 | 0 | 0–0 |
| Other community-based doctor | 0 | 0 | 0 | 0 | 0–0 |
| Occupational therapist | 5 (4) | < 1 | 3 | 10 | 6–26 |
| Social worker | 3 (2) | 5 | 41 | 160 | 53–440 |
| Home help | 4 (3) | 110 | 890 | 2800 | 25–9100 |
| Care attendant | 0 | 0 | 0 | 0 | 0–0 |
| Community support worker | 0 | 0 | 0 | 0 | 0–0 |
| Housing worker | 1 (1) | < 1 | 7 | 75 | N/A |
| All non-hospital NHS and social-care providers | 102 (77) | 750 | 1815 | 120 | 54–330 |
Cost breakdown by hospital- and community-based services
Tables 40 and 41 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by adults with Gaucher disease. Only 23 patients, or about one-sixth of those adults who provided valid service-use data, had hospital stays as inpatients, but this accounted for over one-third of the NHS hospital costs in Gaucher adults. Eight patients had annual hospital costs of > £10,000 and three of these had estimated hospital costs of between £25,000 and £35,000 (due to treatment for cancer, leg ulcers and kidney dialysis). Over three-quarters (77%) of patients reported having at least one hospital outpatient attendance during the previous 12 months.
The majority of costs related to using community-based services were due to the relatively small minority of adults with Gaucher disease who used district nurses (nine patients, 7%) or who used health visitors or other nurses regularly (19 patients, 14%). They accounted for £540 of the £750 yearly per patient cost of services used outside of hospital. Although over two-thirds of adults with Gaucher disease had seen their GP at least once during the past year, and over one-third reported seeing a practice nurse at their GP surgery, these accounted for < £90 of the £3000 annual cost of NHS and publicly funded social-care services consumed. Other support providers used by smaller numbers of adults with Gaucher disease were occupational therapists, social workers and home help/home-care workers (see Table 41).
Cost of care for children with Gaucher disease
Total care cost – financial burden of Gaucher disease
Table 42 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for a child with Gaucher disease, based on the 20 children whose parents or carers supplied service-use data at study entry (mean age 10 years; age range 1.1–15.6 years; eight males). Of the estimated mean per patient annual cost of £2900, approximately three-quarters is as a result of NHS hospital services used, and of this two-thirds (£1400 per patient per year) is from inpatient stays, whereas about 20% (£440) is from outpatient visits (see Table 43). Of the £800 per patient/year from using services outside hospital, only £120 per patient is from GP visits, whereas > £400 is from regular visits from health visitors or other nurses (see Table 44).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. who used this type of service | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 20 | 83 | 11 | 2100 | 2748 | 2500 | 1500–6300 |
| Services outside hospital | 20 | 83 | 20 | 800 | 1348 | 150 | 110–990 |
| Total health- (NHS) and social-care cost | 20 | 83 | 20 | 2900 | 2822 | 2700 | 130–4500 |
| Type of hospital care | No. who used this type of service | Mean cost (£) | Standard deviation | Median cost (£) | Interquartile range (£) |
|---|---|---|---|---|---|
| Inpatient stays | 8 | 1400 | 2344 | 2500 | 1500–6600 |
| Outpatient visits | 6 | 440 | 747 | 1400 | 1200–1800 |
| Day cases | 2 | 190 | 596 | 1900 | 1300–2400 |
| Accident and emergency visits | 2 | 15 | 50 | 160 | 100–210 |
| Total hospital (NHS) care cost | 11 | 2100 | 2748 | 2500 | 1500–6300 |
| Care provider | No. who used this provider | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 20 | 120 | 141 | 120 | 44–130 |
| GP nurse appointments | 5 | 3 | 9 | 5 | 2.6–41 |
| District nurses | 0 | 0 | 0 | 0 | 0–0 |
| Community mental health nurse | 1 | < 1 | 1.8 | 8 | N/A |
| Other nurse or health visitor | 4 | 470 | 1349 | 210 | 88–5300 |
| Counsellor | 1 | 5 | 25 | 110 | N/A |
| Other therapist | 2 | 6 | 20 | 120 | 37–83 |
| ‘Alternative’ medicine or therapy | 0 | 0 | 0 | 0 | 0–0 |
| Psychologist | 3 | 73 | 200 | 320 | 320–810 |
| Psychiatrist | 0 | 0 | 0 | 0 | 0–0 |
| Other community-based doctor | 1 | 8 | 37 | 170 | N/A |
| Occupational therapist | 4 | 17 | 53 | 38 | 25–230 |
| Social worker | 1 | 66 | 296 | 1300 | N/A |
| Home help | 1 | 22 | 97 | 430 | N/A |
| Care attendant | 0 | 0 | 0 | 0 | 0–0 |
| Community support worker | 0 | 0 | 0 | 0 | 0–0 |
| Housing worker | 0 | 0 | 0 | 0 | 0–0 |
| All non-hospital NHS and social-care providers | 20 | 800 | 1348 | 150 | 110–990 |
Cost breakdown by hospital- and community-based services
Tables 43 and 44 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by children with Gaucher disease. Only eight patients, or three-fifths, of those children who provided valid service-use data had hospital stays as inpatients during the year, but this accounted for over three quarters of the NHS hospital costs in Gaucher children. Three children accumulated inpatient care costs of between £5000 and £7000 each (for a range of reasons such as foot surgery, portacath insertions, influenza and chest problems). Only six of the children had at least one hospital outpatient attendance during the previous 12 months.
The majority of costs related to using community-based services were as a result of the relatively small minority of children with Gaucher disease who used health visitors or other nurses regularly (four patients). They accounted for £470 of the £800 yearly per patient cost of services used outside of hospital. Although all 20 children with Gaucher disease had seen their GP at least once during the past year, and five also reported seeing a practice nurse at their GP surgery, together these accounted for < £120 of the £2900 annual cost of NHS and publicly funded social-care services consumed. Other support providers used by smaller numbers of children with Gaucher disease were occupational therapists, psychologists and physiotherapists (see Table 44).
Association between time on ERT and cost of care for people with Gaucher disease
From the longitudinal regression modelling of costs, in adult patients with Gaucher disease, there was no statistically significant association between time on ERT and either total NHS and social-care costs, hospital-care costs, or non-hospital-care costs. In child patients with Gaucher disease, although there was a statistically significant association between costs and time on ERT (17% higher, 95% CI 2% to 34%; p = 0.03), this was based on only 29 data points from 17 children (only eight with data from more than one time point) and in the opposite direction to what would be expected, and so is probably a spurious result due mainly to two or three high-cost patients who happen to also have been on ERT for longer. The tabulated results of these analyses are available on request from the study authors.
Discussion of Gaucher disease results
Since the licensing of alglucerase in the early 1990s, there has been a multitude of studies examining the efficacy of ERT in people with Gaucher disease (see Chapter 1 and review by Connock and colleagues18). Only two of these studies were RCTs;100,101 others analysed longitudinal data held on the Genzyme-sponsored Gaucher Registry,93,102,103 whereas others were open-label cohort studies. Only one (Schiffmann and colleagues101) compared ERT with a concurrent randomised control group.
Among the outcomes assessed in these studies were Hb levels, platelet count, chiotriosidase, skeletal manifestations, liver and spleen size, immunoglobulin G (IgG) antibody levels and HRQoL. Improvement in nearly all measures at all time points was reported in these studies although, given the nature of the studies, interpretation is complex.
In this study, we examined potential associations between treatment and platelet count, Hb levels, absence/presence of bone pain, spleen volume/size, liver volume/size and liver function tests. We also examined the relationship between treatment and QoL assessed by either SF-36 or PedsQL, depending on age. These analyses depend on the fact that patients began treatment with ERT at different ages, dependent on the time alglucerase became available.
Platelet count
A significant improvement in platelet count associated with the use of ERT has been consistently reported. 82,100–104 It has been suggested that a greater degree of improvement is likely in individuals with a lower pre-treatment platelet count at the start of treatment. 103 Andersson and colleagues103 reported that response to treatment for the patients with the most-severe conditions at baseline continued in a nearly linear basis over the 8 years of follow-up monitoring, whereas patients with the least severe conditions at baseline achieved most of the treatment effect within the first 12 months of treatment.
Splenectomy is followed by rapid increases in erythrocytes and platelets, and is therefore a confounding treatment variable in the analysis of platelet count, and so in this data set analyses were stratified by splenectomy status.
In splenectomised patients, we found a statistically significant association between platelet count and time on ERT. These increases are clinically meaningful, with patients who have been treated for ≥ 3 years having adjusted platelet counts a mean of 68.2 × 109/l higher than untreated patients (95% CI 31.1 to 105.2 × 109/l; p < 0.001). Further analyses suggested a plateauing of effect at around 7 years after commencement of ERT.
We found a similar magnitude of an effect of time on treatment on platelet count among those patients who had not been splenectomised. Once again, the model suggested that this effect plateaus, in this case after about 8 years of treatment.
Estimates from the Bayesian model indicate that there is a 55% probability that first infusion at age 45 years will result in greater increases in platelet count after 10 years on ERT than first infusion at age 18 years, suggesting little evidence that age at commencement of treatment affects the patient’s likelihood of benefit.
Although data were more limited we saw similar evidence of increases in platelet counts among children with duration of ERT.
Haemoglobin
A significant improvement in Hb levels in patients on ERT was reported by many studies. 92,100,103–105 Again, most of the studies demonstrated a greater degree of improvement in individuals with a lower pre-treatment Hb level than in patients in whom Hb is normal at the start of treatment; Andersson and colleagues103 reported that, after 8 years of ERT, the median Hb level was not substantially different from that of the normal population, and that the mean increases in Hb values after 8 years of treatment were 2.9 and 1.0 g/dl for patients in the 5th and 95th percentiles at baseline, respectively.
We found a statistically significant association between increasing Hb levels and time on ERT, with the model suggesting a mean Hb 1.19 g/dl higher in those treated for ≥ 3 years than in the untreated (p < 0.001). These data appear to suggest that ERT continues to have an incremental effect on Hb levels for the first 20 years of treatment although the magnitude of the effect decreases after about 7 years.
As with platelet count, the Bayesian model provides no strong evidence that the age at commencing ERT affects the degree of increase in Hb achieved. The Bayesian probability that first infusion at age 45 years will result in greater increases in Hb after 10 years on ERT than first infusion at age 18 years is 52%.
As seen with platelet counts, our data suggest a significant association between duration of treatment with ERT and improved Hb levels in children as well as in adults.
Bone pain
Skeletal manifestations including bone pain, bone crises and bone marrow density were examined in a number of previous studies. 93–95,101–103,106 Substantial and significant improvements in bone pain were reported by Weinreb and colleagues,92 Charrow and colleagues94 and Sims and colleagues95 but, in all, the interpretation is complicated by study design.
As splenectomised patients have been reported to be at increased risk of skeletal complications,245, 246 all analyses of bone pain in these data have been stratified by splenectomy status. As expected, splenectomised adults had significantly higher odds of bone pain than non-splenectomised adults (p = 0.003). After adjusting for splenectomy status, although the central estimate suggested a decreased risk of bone pain associated with duration of ERT (treated as a categorical variable), these findings were not statistically significant. In further analyses in which time on ERT was treated as a continuous variable, we found a strong statistically significant relationship between time on ERT and a reduced risk of bone pain. This analysis is able to take account of the large heterogeneous group of patients who have been on ERT for > 36 months.
As with adults, splenectomised children had higher odds of bone pain than non-splenectomised children. The risk of bone pain increased significantly with age. After adjusting for splenectomy status, although the central estimate suggested a decreased risk of bone pain associated with duration of ERT (treated as a categorical variable), these findings were not statistically significant. However, as with adults, when time on ERT was treated as a continuous variable, we found a strong statistically significant relationship between time on ERT and a reduced risk of bone pain (edf = 1; p = 0.001).
Spleen volume/size
A reduction in spleen size with ERT has been consistently reported in previous studies. 82,100,102–105
We found a statistically significant association between time on ERT and decrease in spleen volume in adults, irrespective of whether categorised on the basis of clinical palpation or measured by scan. The data suggest a rapid decrease in spleen volume following the start of treatment with ERT, with the maximum effect obtained by about 8 years, before an apparent plateauing of effect.
Patients who are not on ERT had a mean spleen volume of about 1500 ml, approximately three to four times the normal spleen volume in a healthy adult. The data suggest that spleen volume remains essentially unchanged over time in untreated adults. This, however, is likely to reflect, at least in part, bias due to an increased likelihood of splenectomy (and hence removal from the model) of those patients with substantially enlarged spleens, as well as the heterogeneity of disease severity in the older patient population. The model predicts that patients who went on ERT at the age of 18 and 45 years show a similar trajectory, with an initial reduction in spleen volume for 7 years, followed by a plateauing off of treatment effect.
Estimates from the Bayesian model indicate that there is a 83% probability that first infusion at age 45 years will result in a greater reduction in spleen volume after 10 years on ERT than first infusion at age 18 years. A possible explanation for this finding is that the patients observed in late middle age have less aggressive forms of the disease and so may respond more successfully to treatment. The model predicts that patients who went on ERT at the age of 18 and 45 years show a similar trajectory, with an initial reduction in spleen volume for 7 years, followed by a levelling off of treatment effect.
Liver volume/size
Liver volume has been consistently reported to decrease after commencement of treatment with ERT. 82,100–105
We found a strong statistically significant association between duration of treatment and liver size whether this was categorised on the basis of palpation or measurement by scan. Dependent on the method of estimating liver volume, the data suggest that these effects plateau somewhere between 5 and 10 years after commencing treatment.
Estimates from the Bayesian model provide little reason to believe that the effects of treatment depend on age at commencement, indicating that there is a 48% probability that first infusion at age 45 years will result in a greater reduction in liver volume after 10 years on ERT than first infusion at age 18 years.
Liver function tests
Although the effects of ERT on liver size are well documented, relatively few studies have reported on biochemical markers of liver function. El-Beshlawy and colleagues247 reported that liver enzymes (ALT and AST) were elevated in 1 of 22 Egyptian paediatric patients prior to starting treatment, and that after 5 months of ERT these levels returned to normal. Hsu and colleagues248 similarly reported that ERT was effective in improving liver function in six Taiwanese patients with GD1.
Although we attempted to collect data on ALT levels, in this study data there was substantial variation between sites in the methods used and ‘normal’ ranges, and so no further analyses were conducted.
We examined the association between treatment with ERT and AST levels in adults in analyses that treated AST as a continuous measures and that estimated the risk of having an ‘abnormal’ value. Both sets of analyses suggested that ERT was associated with a reduction in AST levels. No statistically significant association was seen in children but data available were limited.
Neurological involvement
The majority of people with Gaucher disease recruited in this study have a diagnosis of GD1. Although considered non-neuronopathic, and with the majority of patients not having primary CNS involvement, there is increasing evidence that patients with GD1 may exhibit various neurological complications. 62 Many of these complications are secondary to mechanical trauma due to disease-related complications (such as bone destruction), or may develop as a consequence of nerve root or spinal cord compression following vertebral body collapse. To date, there has been limited investigation of primary neurological involvement in GD1. In their epidemiological survey, Pastores and colleagues62 reported that 73% of respondents with GD1 had experienced a neurological complaint in the previous 3 months.
A recent multinational, observational study reported on the prevalence and incidence of peripheral neuropathy and associated conditions in patients with GD1, either untreated or receiving ERT. 249 Of 103 patients at baseline, 11 patients were diagnosed with sensory motor axonal polyneuropathy and two had a mononeuropathy of the ulnar nerve. The 2-year follow-up period revealed another six cases of polyneuropathy. The prevalence and incidence of polyneuropathy in patients with GD1 was increased compared with the general population (estimated between 0.09% and 1.3%).
At the time of entry into this study, seven GD1 patients were reported to have neurological involvement, of whom six where on ERT. Four of the seven with neurological involvement were reported to have a peripheral neuropathy, of whom three were on ERT at the time.
There have been reports of the association of Parkinsonian features and GD1,250 and the carrier status for the some genotypes (specifically N370S allele) has been shown to be over-represented in patients with Parkinson’s disease. 251
In our study, three patients with GD1 were reported to have Parkinsonian features at the time of entry into the study. All three of these patients were on ERT at this time. There were insufficient data for further analysis. This is consistent with a recent survey by Pastores and colleagues,62 which reported that 3 of 55 respondents had Parkinson’s disease.
The data collected in this study did not allow meaningful analysis of the associations between ERT and risk of neurological involvement.
Fatigue and quality of life
Health-related quality of life can be diminished in patients with Gaucher disease because of its many debilitating clinical manifestations. Masek and colleagues91 reported that treatment with ERT was associated with improvements in QoL in adults with GD1, as assessed by the SF-36 over a 24-month period. Prior to starting therapy, the study population’s health profile was reported to be significantly reduced compared with the general US adult population, but by 24 months of therapy there were no differences between the two.
Similarly, a study by Weinreb and colleagues92 reported improvement in SF-36 scores in a cohort of 32 patients between baseline scores at the commencement of treatment and after 2 years of treatment. Mean PCS and MCS increased to within the normal range after 2 years of treatment and were maintained until year 4. Large HRQoL gains were observed even in patients with the most advanced disease and lowest baseline PCS.
In our study, we found no significant association between the PCS of the SF-36 and time on ERT. The data suggested an association between a decrease in the MCS of the SF-36, but this was not statistically significant.
We included a measure of fatigue in the data set as patient groups report that this is an important but under-researched area. We found no evidence of an association between fatigue and time on ERT (edf = 1; p = 0.57) (see Figure 24). However, these analyses are hampered by a lack of data on people not on treatment.
Costs associated with Gaucher disease
Much of the past discussion regarding the costs associated with having a LSD has concentrated on the costs associated with ERT. In this study we were also keen to capture the wider costs of care falling on the public sector.
Based on patients’ self-reported health- and social-care service use, the annual cost of caring for people with Gaucher disease, excluding the purchase cost of ERT or SRT, was estimated at £3000 for an adult and £2900 for a child. These costs, however, are dwarfed by the cost of the therapies; the mean annual cost of ERT for adults with Gaucher disease is either £126,300 or £144,900, depending on which ERT drug is used, and for those on SRT the mean annual cost is £54,320. For children, the mean annual cost of ERT is £107,400 or £187,800.
From the longitudinal regression modelling of costs, there was no statistically significant association (i.e. p < 0.05) between time on ERT and either total NHS and social-care costs, hospital-care costs, or non-hospital-care costs for patients with Gaucher disease. The tabulated results of these analyses are available on request from the study authors.
Owing to these high associated costs, and the lack of measureable effect of ERT on either clinical outcomes or HRQoL measures, it was infeasible to conduct either a cost-effectiveness or cost–utility analysis. As they apply to all six LSDs, the limitations of these cost estimates are summarised and discussed in Chapter 9.
Chapter 4 Results – Fabry disease
Patient characteristics
At the start of the study 499 patients were identified by the treating centres as having Fabry disease. Of these, 442 were deemed eligible for inclusion and 333 patients (75% of those deemed eligible) were approached in clinics and invited to participate. Three hundred and eleven patients (94% of those approached), comprising 131 males and 180 females, agreed to participate in the study. Patient demographic characteristics are presented in Tables 45 and 46.
| Patient characteristic | Male, n = 120 | Female, n = 169 |
|---|---|---|
| Age (years) | ||
| Mean (SD) | 44.9 (14.5) | 43.9 (15.1) |
| Median (min.–max.) | 44.5 (16.4–78.6) | 43.7 (16.2–75.4) |
| Age at diagnosis (years) | ||
| Mean (SD) | 35.2 (18.2) | 35.8 (18.2) |
| Median (min.–max.) | 35.6 (0–76.3) | 34.9 (0–72.2) |
| Initial treatment | ||
| Not on ERT, n | 11 | 65 |
| ERT, n | 94 | 101 |
| Clinical trial, n | 15 | 2 |
| Missing, n | 0 | 1 |
| Initial type of ERT | ||
| Agalsidase beta, n | 62 | 53 |
| Agalsidase alpha, n | 32 | 48 |
| Treatment at recruitment | ||
| Not on ERT, n | 11 | 66 |
| ERT, n | 108 | 103 |
| Clinical trial, na | 1 | 0 |
| Type of ERT at recruitment | ||
| Agalsidase beta, n | 69 | 54 |
| Agalsidase alpha, n | 39 | 49 |
| Age at first infusion (years) | ||
| Mean (SD) | 40.9 (15.1) | 44.4 (14.6) |
| Median (min.–max.) | 42.0 (11.5–75.9) | 46.1 (13.6–72.6) |
| Time on ERT (years) | ||
| Mean (SD) | 3.74 (2.66) | 3.34 (2.25) |
| Median (min.–max.) | 3.51 (0–9.72) | 3.55 (0–8.77) |
| Patient characteristic | Male, n = 11 | Female, n = 11 |
|---|---|---|
| Age (years) | ||
| Mean (SD) | 8.9 (4.1) | 11.6 (3.3) |
| Median (min.–max.) | 9.1 (1.8–14.6) | 12.1 (4.6–15.9) |
| Age at diagnosis (years) | ||
| Mean (SD) | 4.6 (4.6) | 7.8 (4.4) |
| Median (min.–max.) | 3.4 (0–12.6) | 6.7 (1.3–13.2) |
| Initial treatment | ||
| Not on ERT, n | 7 | 8 |
| ERT, n | 4 | 3 |
| Initial type of ERT | ||
| Agalsidase beta, n | 2 | 1 |
| Agalsidase alpha, n | 2 | 2 |
| Treatment at recruitment | ||
| Not on ERT, n | 7 | 8 |
| ERT, n | 4 | 3 |
| Type of ERT at recruitment | ||
| Agalsidase beta, n | 3 | 1 |
| Agalsidase alpha, n | 1 | 2 |
| Age at first infusion (years) | ||
| Mean (SD) | 9.9 (2.3) | 9.8 (1.4) |
| Median (min.–max.) | 8.9 (8.6–14.7) | 9.7 (8.5–11.3) |
| Time on ERT (years) | ||
| Mean (SD) | 1.6 (1.8) | 2.3 (0.7) |
| Median (min.–max.) | 1.2 (0–4.2) | 2.4 (1.7–3.0) |
The majority of participants were adults (289), with only 22 children with Fabry disease enrolled in the study. At recruitment, the average age of adult male patients with Fabry disease was 44.9 (range 16.4–78.6) years, whereas that of adult female patients was 43.9 (range 16.2–75.4) years. The average age at recruitment for boys was 8.9 (range 1.8–14.6) years and for girls 11.6 (range 4.6–15.9) years. The average age at diagnosis was 35.2 (range 0–76.3) years for adult males and 34.9 (range 0–72.1) years for adult females. The average age at diagnosis for boys was 4.6 (range 0–12.6) years and for girls was 7.8 (range 1.3–13.2) years.
As with Gaucher disease, owing to the world shortage of agalsidase beta therapies, patients with Fabry disease attended their treating clinic more regularly during the later period of data collection, and clinical data were collected at each visit. Of the 311 patients enrolled in the study, we have a second data point from 290 patients and a third data point for 192 patients. For 19 patients, we have data from five prospective time points. Retrospective clinical data were available for 270 patients and the number of retrospective data points ranges from 1 to 11.
At recruitment 219 patients (212 adults and seven children) were on ERT with 123 adult patients (69 male and 54 female) and four children (three boys and one girl) receiving agalsidase beta, 88 adult patients (39 male and 49 female) and three children (one boy and two girls) receiving agalsidase alpha, and one unknown. The average time on ERT was 3.74 years (mean 3.62 years, range 0–9.72 years, for males and mean 3.31 years, range 0–8.77 years, for females). Of the 91 patients not on treatment at recruitment, 18 were male and 73 were female. Of the 127 patients on agalsidase beta at recruitment, 66 patients switched to agalsidase alpha during the shortage period (from June 2009).
Patient demography reported by gender (see Tables 45 and 46), demonstrates that most Fabry patients who are not on treatment are females, reflecting the X-linked inheritance of this disease.
Key markers of Fabry disease progression
The following measures were identified by the clinical principal investigators as key markers of disease progression in Fabry disease:
-
left ventricular mass index (LVMI)
-
estimated glomerular filtration rate (eGFR)
-
proteinuria
-
BPI
-
hearing
-
TIA/stroke
-
Fatigue Severity Scale (FSS).
In addition, adults completed the SF-36, EQ-5D, FSS and the Service Use and Costs Questionnaire, whereas children or their carers completed the age-appropriate PedsQL questionnaire. Carers of children or adults were asked to complete the Service Use and Costs Questionnaire and the CSI. In addition, we attempted to ascertain DQ if this had been assessed for children.
Summary of analysis
Longitudinal models were fitted to assess relationships between continuous measures of function and length of time on ERT, after adjustment for age and clustering by centre. In the base models, the effect of time on ERT was categorised as (1) not on ERT; (2) < 12 months on ERT; (3) 12–36 months on ERT; and (4) > 36 months on ERT. Further analysis was conducted to explore the possibility that time spent on ERT would have a non-linear effect on function. Patients contributed data points to the model both before and after starting ERT. A Bayesian version of the model was used for generating expected trajectories to illustrate model predictions about the effect of starting ERT treatment at different ages.
Kaplan–Meier survival curves were estimated to illustrate differences in the age at first recorded occurrence of binary events that could be considered progressive (i.e. abnormal hearing, TIA/stroke). Treatment group differences in survival function were tested using Cox regression models. Individual patients contributed intervals of time at risk to more than one of the treatment categories.
There are currently two ERTs licensed for use in the UK – agalsidase beta and agalsidase alpha. For the purposes of these analyses, unless otherwise stated, patients on both therapies have been analysed together, and ERT refers to either agalsidase beta or agalsidase alpha. The analyses at the end of this section compare the association between each of the two therapies and key outcome measures.
Summary of results
These data provide evidence for an association between time on ERT and a decrease in LVMI as well as an increase in the eGFR in adults after adjustment for age, although the magnitude of the differences is fairly small. The effect of treatment on eGFR appears to plateau after 5 or 6 years on treatment although these results should be interpreted with caution because of the wide CIs in the later years. In analyses that separated males and females, the association between time on ERT and increase in age adjusted eGFR remained statistically significant for women but not for men.
After adjusting for use of an ACE inhibitor, there was a significant reduction in the risk of proteinuria and time on ERT. However, the same analyses on children did not suggest a significant association although the numbers involved are small hence the analyses have little power to detect an effect.
We found no statistically significant association between time on ERT and Pain Severity Scores, but did see a statistically significant association with decrease in the scale measuring interference by pain on QoL in adults.
The risk of having a stroke or TIA increased significantly after this group of patients reached around 40 years of age. We found no association between the risk of stroke or TIA and the use of ERT. We similarly found no statistically significant association between use of ERT and the risk of needing a hearing aid.
A statistically significant association was found between duration of ERT and decrease in the SF-36 PCS. There was a significant, non-linear relationship between time on ERT and lower (i.e. worse) scores on the SF-36 MCS. Although no association was seen between the EQ-5D score and time on ERT, a significant reduction in patient-reported health status was reported using the EQ-5D VAS. Finally, after adjusting for age we found a small but statistically significant increase in fatigue score with time on ERT, suggesting worsening fatigue with time on ERT.
We had planned to examine the association between use of ERT and developmental quotient in children, but almost no data on this outcome were recorded. Similarly, there were insufficient data to analyse the effect of time on ERT with the CSI.
For each outcome we examined the relative effects in those patients initially treated with agalsidase beta with those initially treated with agalsidase alpha. There were no statistically significant differences in any of the outcomes for adults or children.
Heart size – left ventricular mass index
Left ventricular mass index was determined by standard echocardiography and adjusted for body surface area.
Adults
Left ventricular mass index data were collected for 227 adults. The range of LVMI for adults was 37–371 g/m2.
A longitudinal model was fitted to assess the relationship between LVMI and time on ERT, after adjusting for age. As can be seen in Table 47, there is a statistically significant increase in LVMI with age (p < 0.001). After adjusting for age, there was no statistically significant association between LVMI and time on ERT (p = 0.11) when time on ERT was categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months.
| N Data | Estimate of change in LVMI (g/m2) | Standard error | 95% CI | p-value | ||
|---|---|---|---|---|---|---|
| Gender | ||||||
| Male | 210 | 0.00 | ||||
| Female | 374 | –51.4 | 5.45 | –62.1 to –40.7 | < 0.001 | |
| Age | ||||||
| Linear effect/year | 1.52 | 0.18 | 1.16 to 1.87 | < 0.001 | ||
| Time on ERT | ||||||
| Not on ERT | 194 | 0.00 | 0.11 | |||
| < 12 months | 51 | 2.59 | 4.87 | –6.95 to 12.2 | 0.59 | |
| 12–36 months | 129 | –2.33 | 4.02 | –10.2 to 5.56 | 0.56 | |
| > 36 months | 210 | –8.17 | 4.17 | –16.4 to 0.006 | 0.05 | |
| Variance components | ||||||
| Individual | 1223 | |||||
| Centre | 168 | |||||
| Residual | 540 | |||||
However, further analysis was conducted to explore the relationship of time on ERT with LVMI when time is treated as a continuous variable. Figure 26 suggests that ERT has a significant linear effect of reducing LVMI (edf = 1; p = 0.01).
FIGURE 26.
The age-adjusted association between time on ERT and LVMI in adults with Fabry disease (time on ERT treated as a continuous variable).
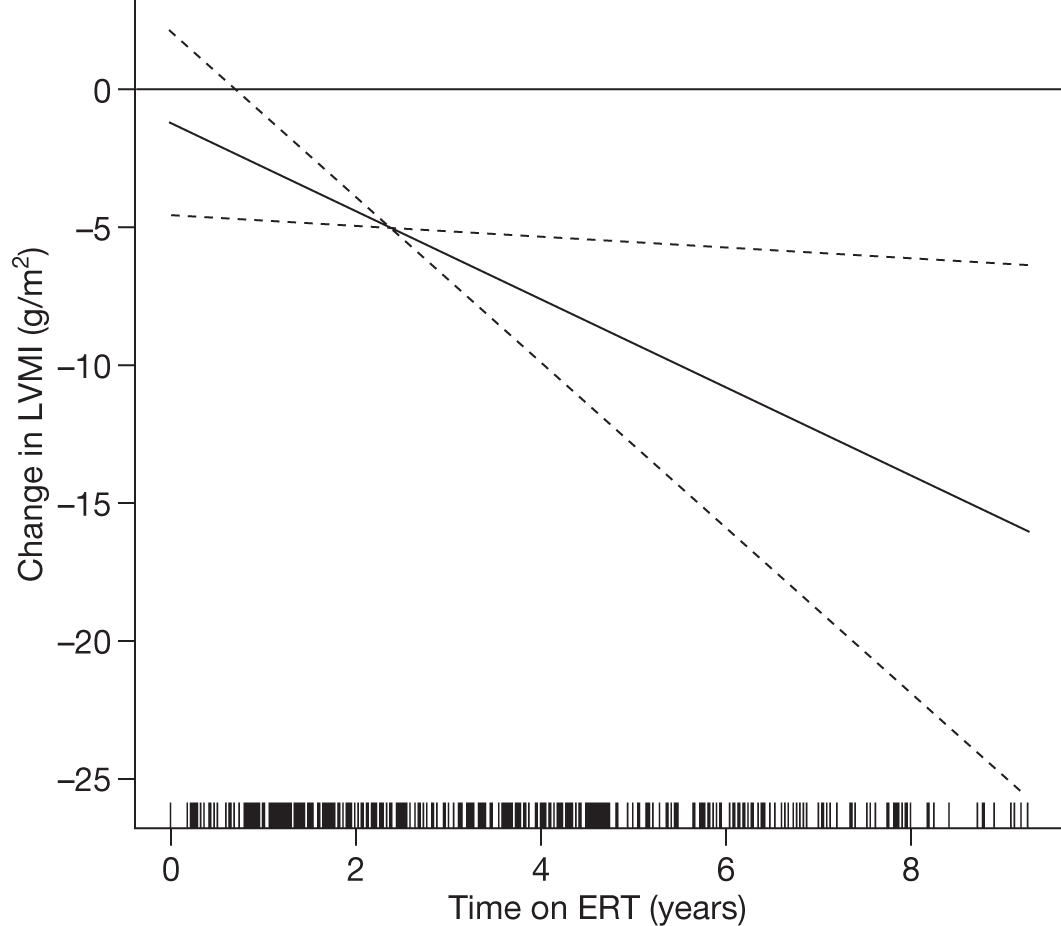
Children
Heart volume data were available for nine children. The range of LVMI for children was 41–101 g/m2.
A longitudinal model was fitted to assess the linear relationship between LVMI in children and time on ERT, after adjusting for age (Table 48). As can be seen in Table 48, LVMI was not significantly associated with age (p = 0.63). After adjusting for age, time on ERT was not associated with a significant change in LVMI (p = 0.92) when time on ERT was categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months.
| N Data | Estimate of change in LVMI (g/m2) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 16 | 0.00 | |||
| Female | 30 | –3.53 | 6.20 | –15.7 to 8.61 | 0.57 |
| Age | |||||
| Linear effect/year | 0.37 | 0.76 | –1.12 to 1.86 | 0.63 | |
| Time on ERT | |||||
| Not on ERT | 35 | 0.00 | 0.92 | ||
| < 12 months | 2 | –2.12 | 8.74 | –19.2 to 15.0 | 0.81 |
| 12–36 months | 7 | –1.87 | 6.16 | –13.9 to 10.2 | 0.76 |
| > 36 months | 2 | –4.64 | 10.0 | –24.2 to 14.9 | 0.65 |
| Variance components | |||||
| Individual | 77 | ||||
| Centre | 17 | ||||
| Residual | 119 | ||||
Further analysis was conducted to explore the shape of the relationship between LVMI and time on ERT treated as a continuous variable. This further modelling provided no evidence of a non-linear association between LVMI levels in children and time on ERT (edf = 1.0; p = 0.78) (Figure 27). The small numbers of children for whom data were available means that, inevitably, these results have wide CIs.
FIGURE 27.
The age-adjusted association between time on ERT and LVMI in children with Fabry disease (time on ERT treated as a continuous variable).
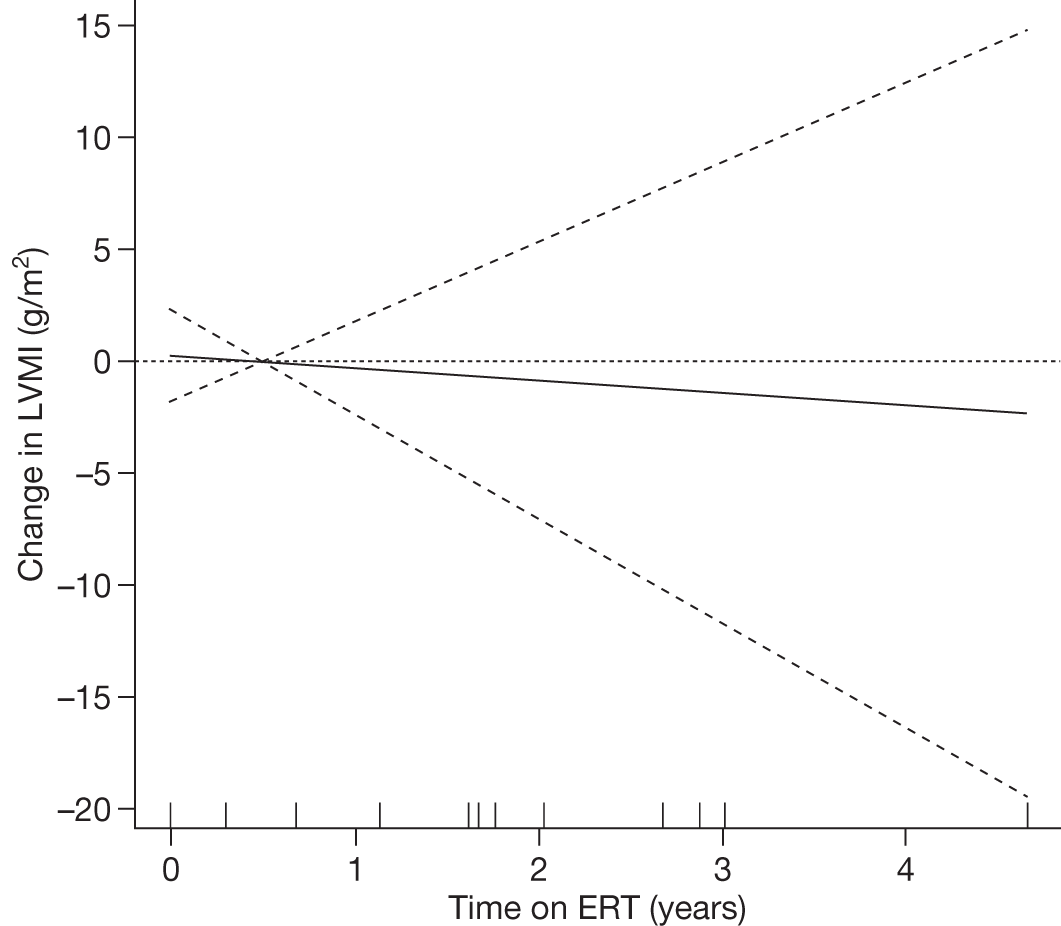
Renal function – estimated glomerular filtration rate
Adults
Renal function in adults was assessed using eGFR calculated by the Modification of Diet in Renal Disease formula. 252 eGFR data were collected for 266 adults (112 males and 154 females) including a total of 1360 data points. The values range from 4.9 to 203.2 ml/minute/1.73 m2 after exclusion of eight outliers (eGFR > 400 ml/minute/1.73 m2). Nineteen patients had an eGFR of < 15 ml/minute/1.73 m2, all of whom were on dialysis at recruitment and 16 of whom were on dialysis across all time points.
A longitudinal model was fitted to assess the relationship between eGFR in people with Fabry disease and time on ERT, after adjusting for age. As can be seen in Table 49, there is a significant decline in eGFR levels in adults with Fabry disease with age. The slope of the decline is –0.95 ml/minute/1.73 m2/year (p < 0.001). After adjusting for age, there was a statistically significant association between increased eGFR and time on ERT (p = 0.002) when time on ERT was categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months.
| N Data | Estimate of change in eGFR ml/minute/1.73 m2 | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 538 | 0.00 | |||
| Female | 822 | 4.19 | 2.56 | –0.83 to 9.22 | 0.10 |
| Age | |||||
| Linear effect/year | –0.95 | 0.08 | –1.11 to –0.79 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 535 | 0.00 | 0.002 | ||
| < 12 months | 141 | 1.01 | 1.30 | –1.54 to 3.57 | 0.44 |
| 12–36 months | 295 | 2.14 | 1.06 | 0.05 to 4.23 | 0.04 |
| > 36 months | 389 | 4.35 | 1.14 | 2.10 to 6.59 | < 0.001 |
| Variance components | |||||
| Individual | 394 | ||||
| Centre | 6 | ||||
| Residual | 138 | ||||
Further analysis was conducted to explore the shape of the relationship between eGFR and time on ERT treated as a continuous variable. This further modelling of the data suggested a non-linear relationship between eGFR levels and time on ERT (edf = 2.05; p < 0.001). Figure 28 suggests that age adjusted eGFR in people with Fabry disease increases after commencing ERT, and reaches a plateau after approximately 6 years.
FIGURE 28.
The age-adjusted association between time on ERT and eGFR in adults with Fabry disease (time on ERT treated as a continuous variable).
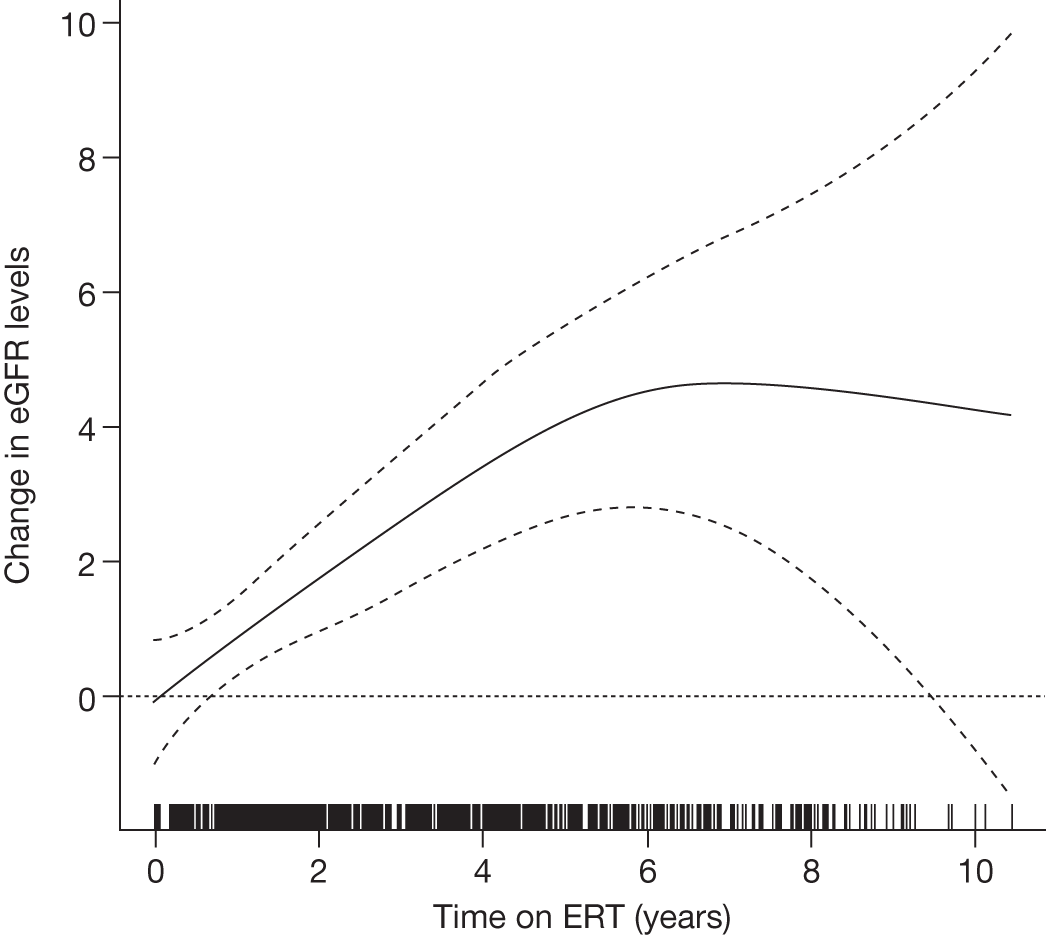
Table 49 shows that eGFR was higher in females than in males, although this difference was not statistically significant. While it is believed that the biology of disease in females is fundamentally the same as in males, it is understood that renal manifestations differ between the genders. In order to examine any potential differences in the treatment effect between the genders, separate longitudinal models were fitted to assess the linear relationship between eGFR and time on ERT, in male and female adults, after adjusting for age (Tables 50 and 51).
| N Data | Estimate of change in eGFR ml/minute/1.73 m2 | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Age | |||||
| Linear effect/year | –1.40 | 0.14 | –1.68 to –1.12 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 123 | 0.00 | 0.11 | ||
| < 12 months | 64 | –1.99 | 2.06 | –6.03 to 2.05 | 0.34 |
| 12–36 months | 150 | 2.38 | 1.64 | –0.84 to 5.60 | 0.15 |
| > 36 months | 201 | 2.29 | 1.80 | –1.24 to 5.82 | 0.21 |
| Variance components | |||||
| Individual | 495 | ||||
| Centre | 11 | ||||
| Residual | 145 | ||||
| N Data | Estimate of change in eGFR ml/minute/1.73 m2 |
Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Age | |||||
| Linear effect/year | –0.69 | 0.09 | –0.86 to –0.51 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 412 | 0.00 | < 0.001 | ||
| < 12 months | 77 | 2.73 | 1.64 | –0.48 to 5.94 | 0.09 |
| 12–36 months | 145 | 1.00 | 1.38 | –1.70 to 3.70 | 0.47 |
| > 36 months | 188 | 6.02 | 1.45 | 3.17 to 8.86 | < 0.001 |
| Variance components | |||||
| Individual | 319.6 | ||||
| Centre | 0.0 | ||||
| Residual | 127.7 | ||||
As can be seen in Tables 50 and 51, there is a significant decline in eGFR levels in male and female adults with Fabry disease with age, although the slope of decline is greater for males (–1.40 ml/minute/1.73 m2/year; p < 0.001) than for females (–0.69 ml/minute/1.73 m2/year; p < 0.001). After adjusting for age, there was no statistically significant association between increased eGFR and time on ERT in male patients (p = 0.11) when time on ERT was categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months. However, in female patients there was a significant increase in the slope of the eGFR with time on ERT (p < 0.001).
Further analysis was conducted to explore the shape of the relationship between eGFR and time on ERT treated as a continuous variable. In these analyses we found no statistically significant association between eGFR in adult males and time on ERT (slope = 0.36 ml/minute/m2/year, edf = 1; p = 0.21) (Figure 29). However, for adult female patients there was a statistically significant linear increase in eGFR with time on ERT after adjusting for age (slope = 1.15 ml/minute/m2/year, edf = 1; p < 0.001) (Figure 30).
FIGURE 29.
The age-adjusted association between time on ERT and eGFR in male adults with Fabry disease (time on ERT treated as a continuous variable).
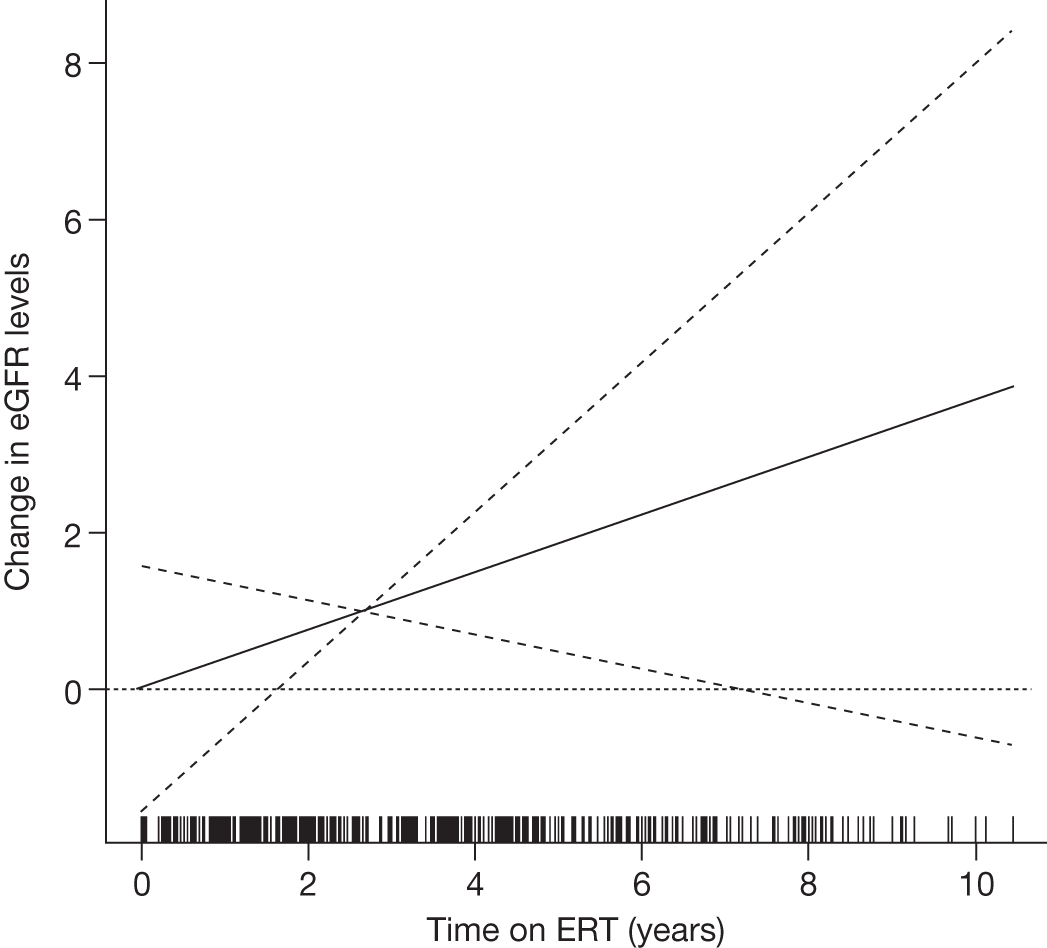
FIGURE 30.
The age-adjusted association between time on ERT and eGFR in femaleadults with Fabry disease (time on ERT treated as a continuous variable).
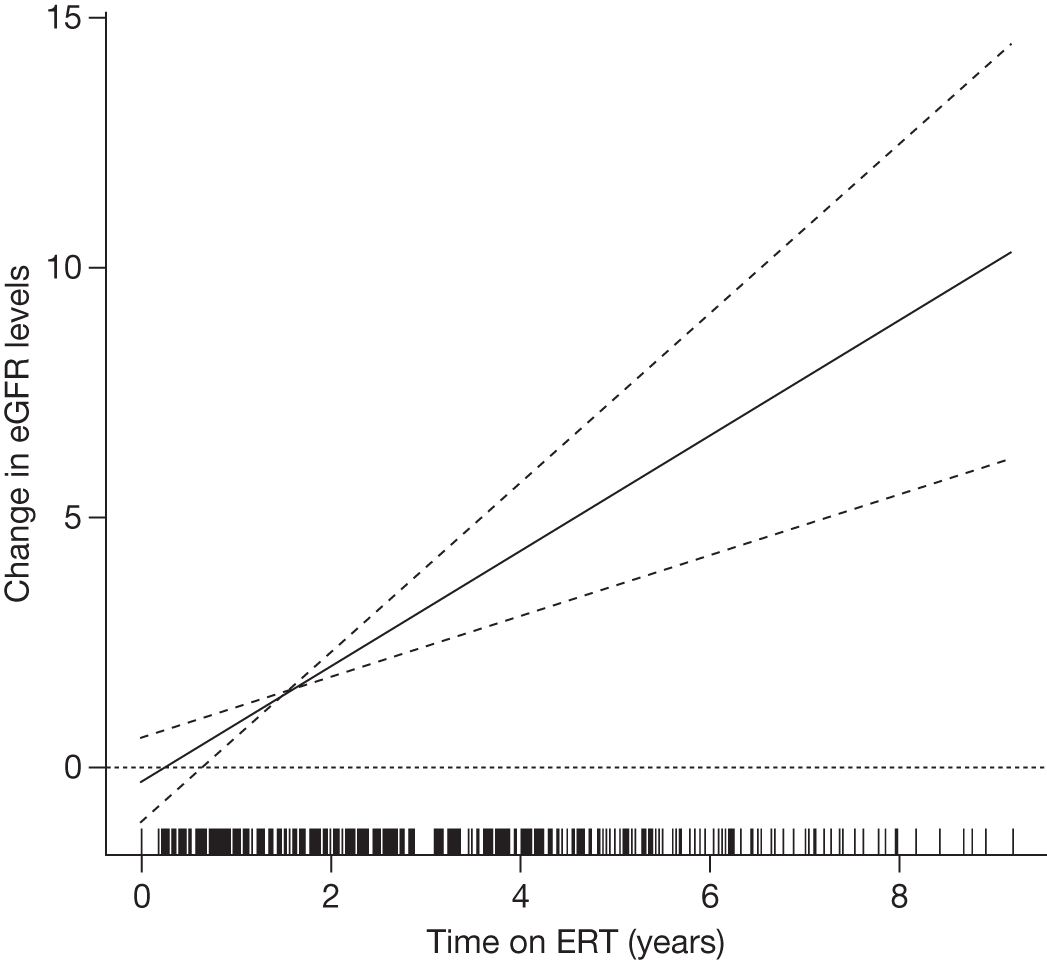
Bayesian predictions
The longitudinal model for eGFR levels in adults was fitted in a Bayesian framework and used to make predictions about how starting ERT at different ages impacts on age-related trajectories (Figure 31). Three different treatment scenarios were considered: (1) the patient remains untreated; (2) the patient starts ERT at the age of 18 years; and (3) the patient starts ERT at age 45 years. It should be noted that these are mean trajectories and in the interest of clarity the credible intervals around these trajectories are not shown.
FIGURE 31.
Estimated glomerular filtration rate in adults with different treatment scenarios.
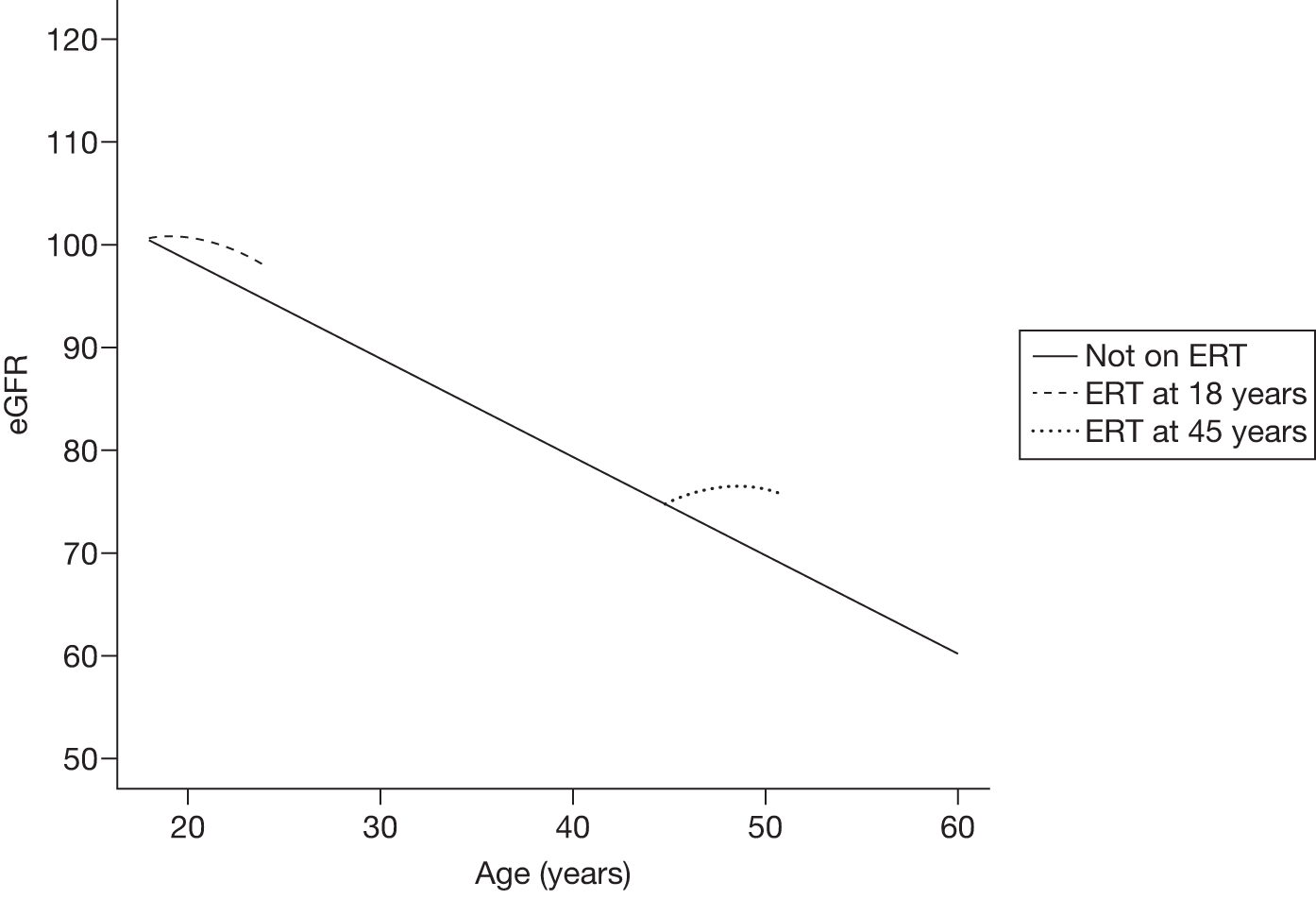
We specified the Bayesian model to allow the shape of the post-treatment trajectories to depend on the age at which ERT is started, in order to explore the possibility that patients observed in late middle age or old age have less aggressive forms of the disease and so may respond differently to treatment. As can be seen from Figure 31, the eGFR of patients starting on ERT at either age is predicted to increase relative to untreated patients, before levelling off. The treatment effect appears to be of slightly greater magnitude for patients who commenced treatment at age 45 years than for those who commenced treatment at 18 years of age; the Bayesian probability that first infusion at age 45 years will result in greater increase in eGFR after 5 years on ERT than first infusion at age 18 years = 55%.
When data from male and female patients were fitted separately to the Bayesian model, the expected improvements in eGFR on treatment (relative to remaining untreated) were of a larger magnitude in female patients than in males (Figures 32 and 33). There was little evidence to suggest that patients who commence treatment later in life respond better to treatment than patients treated at a younger age. The Bayesian probability that first infusion at age 45 years will result in a greater increase in eGFR after 5 years on ERT than first infusion 18 years of age is 54% for both males and females.
FIGURE 32.
Estimated glomerular filtration rate in male adults with different treatment scenarios.
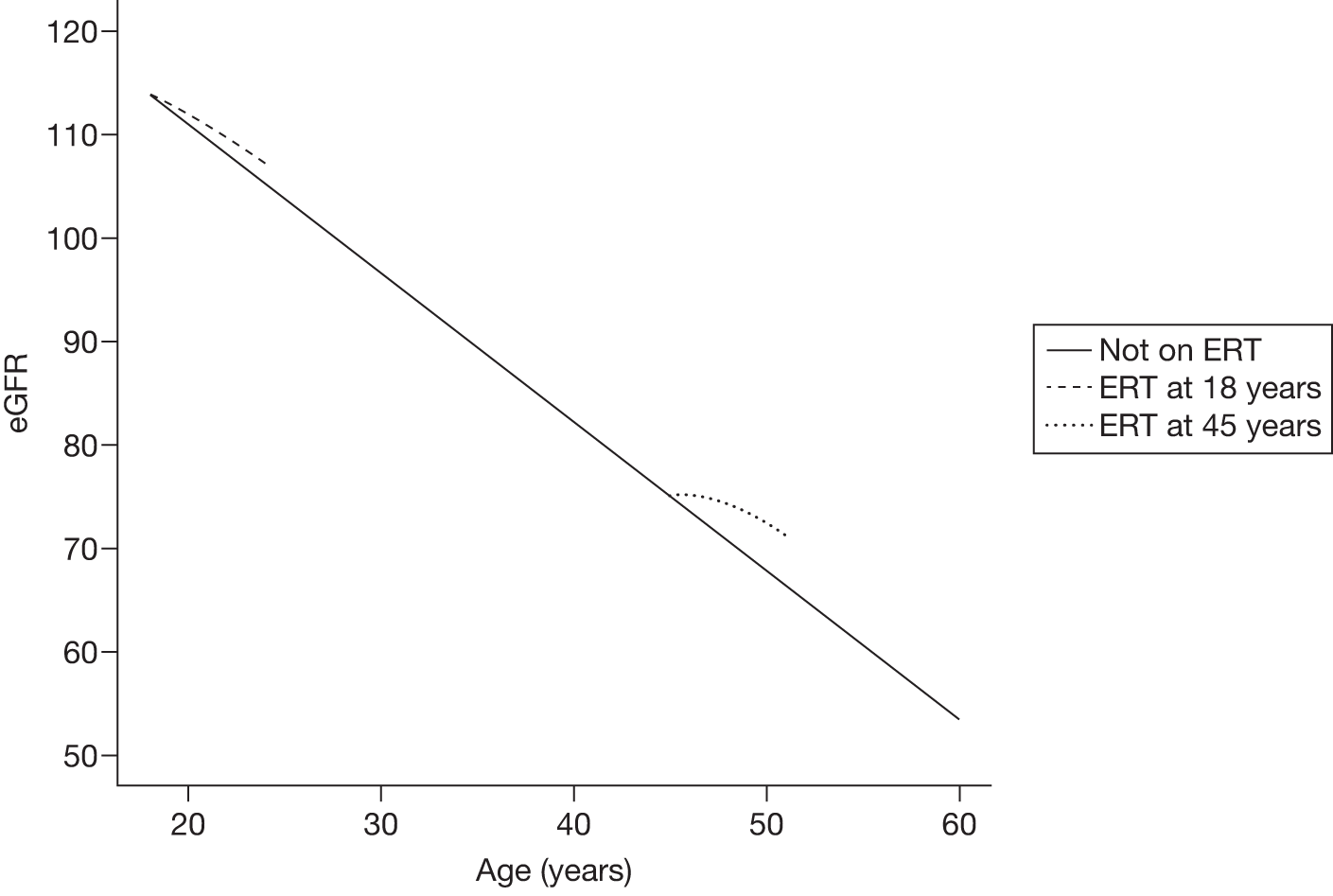
FIGURE 33.
Estimated glomerular filtration rate in female adults with different treatment scenarios.
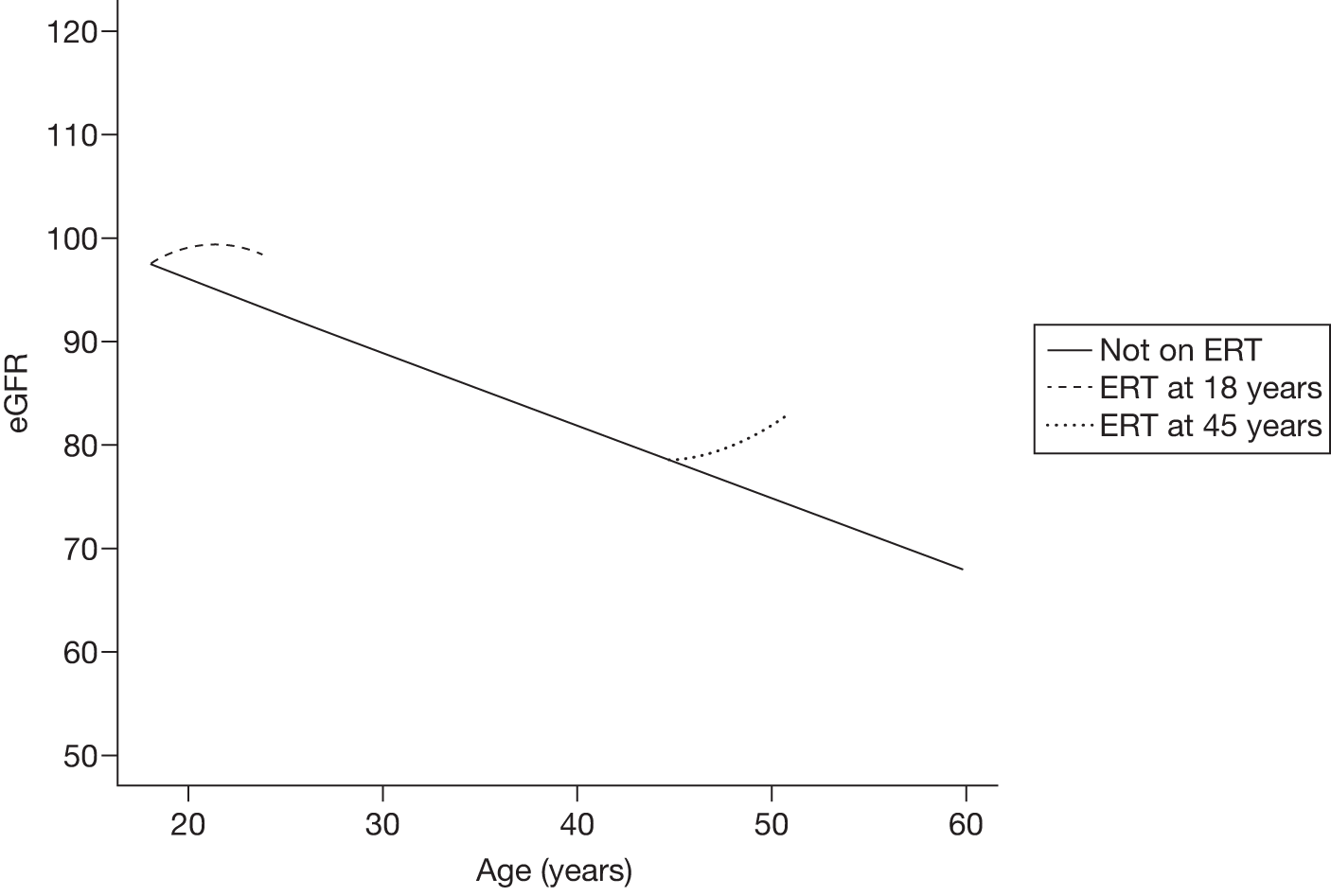
Children
Renal function in children was assessed using eGFR calculated by the new abbreviated Schwartz formula. 253 eGFR data were collected for 17 children (8 boys and 9 girls) with Fabry disease. There were 78 data points for these children across all time points and values ranged from 18.8 to 169.3 ml/minute/1.73 m2.
A longitudinal model was fitted to assess the linear relationship between eGFR in children with Fabry disease and time on ERT, after adjusting for age. As can be seen in Table 52, a statistically significant decrease in the slope of eGFR was seen with age (–2.24 ml/minute/1.73 m2, 95% CI –4.27 to –0.22; p = 0.04). After adjusting for age, there was an increase in the slope of eGFR with time on ERT, but this did not reach conventional levels of statistical significance (p = 0.06).
| N Data | Estimate of change in eGFR ml/minute/1.73 m2 | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 36 | 0.00 | |||
| Female | 42 | 14.4 | 9.67 | –4.61 to 33.3 | 0.15 |
| Age | |||||
| Linear effect/year | –2.24 | 1.03 | –4.27 to –0.22 | 0.04 | |
| Time on ERT | |||||
| Not on ERT | 55 | 0.00 | 0.06 | ||
| < 12 months | 5 | 0.25 | 8.02 | –15.5 to 16.0 | 0.98 |
| 12–36 months | 13 | 15.1 | 6.37 | 2.62 to 27.6 | 0.02 |
| > 36 months | 5 | 0.11 | 9.66 | –18.8 to 19.1 | 0.99 |
| Variance components | |||||
| Individual | 360 | ||||
| Centre | 218 | ||||
| Residual | 196 | ||||
In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and eGFR. Further analysis was conducted to explore the shape of the relationship between eGFR and time on ERT treated as a continuous variable. This analysis provided no statistically significant evidence of an association between age adjusted eGFR levels and time on ERT (edf = 2.2; p = 0.14) (Figure 34).
FIGURE 34.
The age-adjusted association between time on ERT and eGFR in children with Fabry disease (time on ERT treated as a continuous variable).
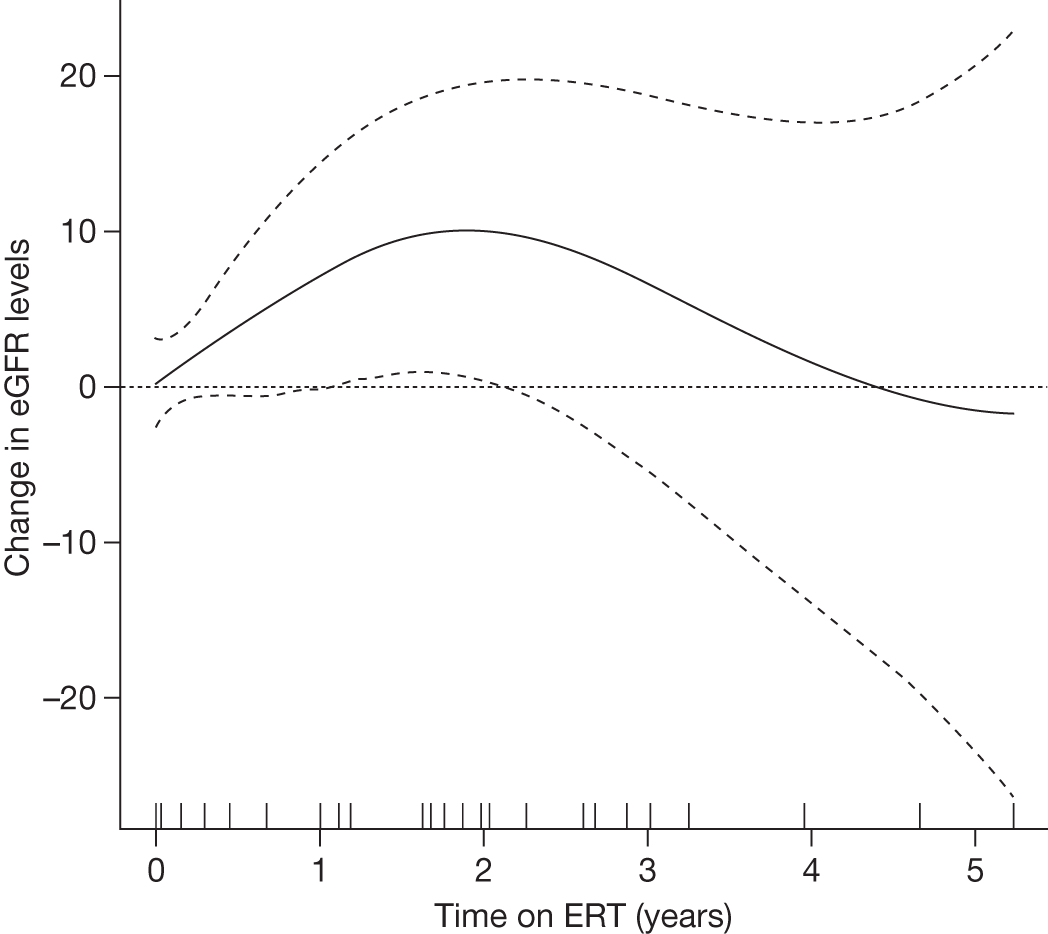
In order to examine any potential differences in the treatment effect between the genders, separate longitudinal models were fitted to assess the linear relationship between eGFR and time on ERT, in male and female children, after adjusting for age (Tables 53 and 54).
| N Data | Estimate of change in eGFR ml/minute/1.73 m2 | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Age | |||||
| Linear effect/year | –1.66 | 1.64 | –4.87 to 1.55 | 0.32 | |
| Time on ERT | |||||
| Not on ERT | 22 | 0.00 | 0.21 | ||
| < 12 months | 4 | 6.17 | 9.22 | –11.9 to 24.2 | 0.51 |
| 12–36 months | 7 | 18.1 | 9.17 | 0.13 to 36.07 | 0.06 |
| > 36 months | 3 | 9.02 | 14.30 | –19.0 to 37.05 | 0.53 |
| Variance components | |||||
| Individual | 650 | ||||
| Centre | 129 | ||||
| Residual | 168 | ||||
| N Data | Estimate of change in eGFR ml/minute/1.73 m2 |
Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Age | |||||
| Linear effect/year | –2.68 | 1.27 | –5.17 to –0.19 | 0.04 | |
| Time on ERT | |||||
| Not on ERT | 33 | 0.00 | 0.15 | ||
| < 12 months | 1 | –12.1 | 16.9 | –45.2 to 21.0 | 0.47 |
| 12–36 months | 6 | 15.6 | 8.86 | –1.76 to 32.9 | 0.08 |
| > 36 months | 2 | –1.10 | 13.8 | –28.1 to 25.9 | 0.94 |
| Variance components | |||||
| Individual | 187 | ||||
| Centre | 241 | ||||
| Residual | 227 | ||||
As can be seen in Tables 53 and 54, there is a significant decline in eGFR levels in girls with age (–2.68 ml/minute/1.73 m2/year, 95% CI –5.17 to –0.19; p = 0.04), whereas no significant association between age and eGFR was observed for boys (p = 0.32). After adjusting for age, there was no statistically significant association between eGFR and time on ERT in either boys (p = 0.21) or girls (p = 0.15). These analyses should be treated with considerable caution owing to the paucity of data.
Proteinuria
Proteinuria was measured using either urinary protein:creatinine ratio or urinary albumin:creatinine ratio. We have proteinuria data for 264 patients (106 males and 158 females). Results were categorised as high or within normal range by centre, according to centre reference ranges. Table 55 shows the effect of ERT and age on the log-odds of having proteinuria.
| N High | N Normal | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 271 | 148 | 1.00 | ||
| Female | 323 | 295 | 0.05 | 0.01 to 0.20 | < 0.001 |
| Age | |||||
| Linear effect/year | 1.06 | 1.02 to 1.12 | 0.008 | ||
| ACE inhibitors | |||||
| No | 510 | 428 | 1.00 | ||
| Yes | 84 | 15 | 2.18 | 0.74 to 6.45 | 0.16 |
| Time on ERT | |||||
| Not on ERT | 166 | 182 | 1.00 | < 0.001 | |
| < 12 months | 58 | 47 | 0.59 | 0.24 to 1.45 | 0.25 |
| 12–36 months | 142 | 95 | 0.48 | 0.22 to 1.05 | 0.07 |
| > 36 months | 228 | 119 | 0.59 | 0.25 to 1.38 | 0.22 |
A longitudinal model was fitted to assess the linear relationship between the risk of having proteinuria and time on ERT, after adjusting for age and whether or not the patient was taking an ACE inhibitor (see Table 55). As can be seen in Table 55, there was a statistically significant association between risk of proteinuria and age (p < 0.001). After adjusting for use of an ACE inhibitor, there was also a statistically significant association between the risk of proteinuria and time on ERT (p < 0.001). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and proteinuria.
The apparent discrepancy between the significant p-value for the overall effect of time on ERT and the lack of significance for the individual category coefficients is an example of the Hauck–Donner effect. 254 This phenomenon can arise in generalised linear models when one or more coefficients has a large absolute value and results in inflated standard errors. The more reliable likelihood ratio test provides evidence for a significant reduction in risk of proteinuria with time on ERT.
Further analysis was conducted to explore the shape of the relationship between proteinuria and time on ERT treated as a continuous variable. This analysis of the shape of the relationship between risk of proteinuria and time on ERT provided no statistically significant evidence of a non-linear association (edf = 1.0; p = 0.16) (Figure 35).
FIGURE 35.
The age-adjusted association between time on ERT and the risk of proteinuria in adults with Fabry disease (time on ERT treated as a continuous variable).
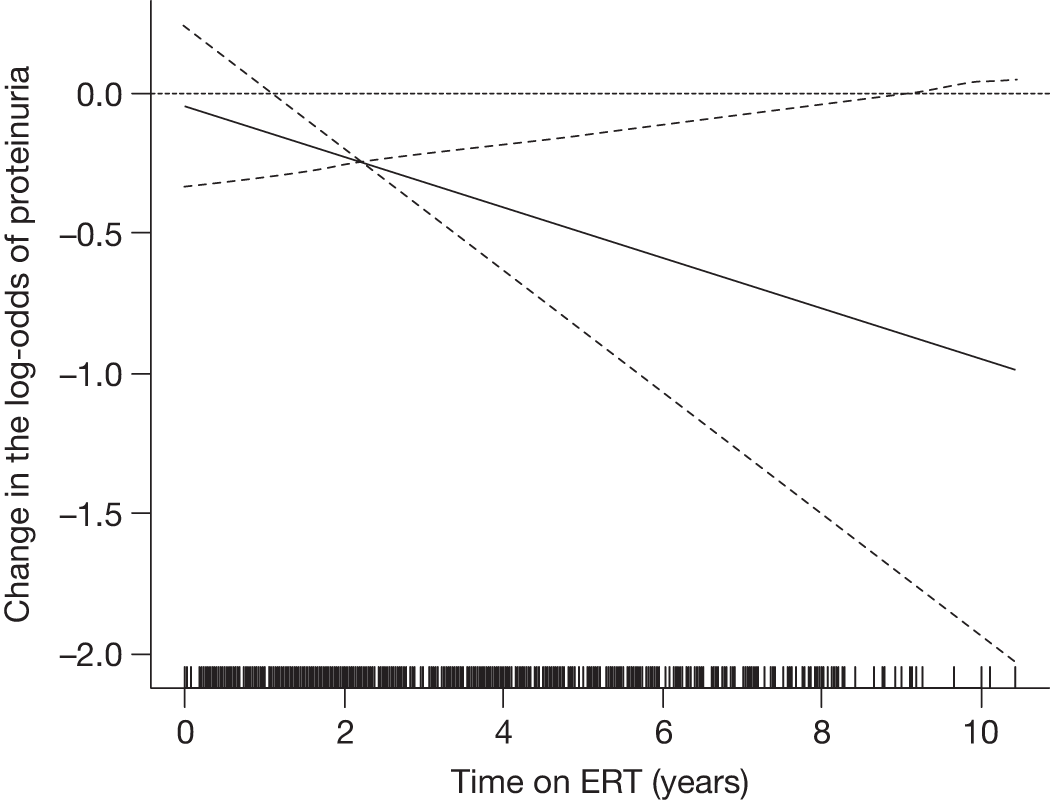
Further examination of any potential differences in the treatment effect between the genders showed no statistically significant linear relationship between proteinuria and time on ERT in male or female adults (p = 0.09 and p = 0.46, respectively), after adjusting for age and for use of an ACE inhibitor (Tables 56 and 57).
| N High | N Normal | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Age | |||||
| Linear effect/year | 1.10 | 1.03 to 1.19 | 0.009 | ||
| ACE inhibitors | |||||
| No | 184 | 199 | 1.00 | ||
| Yes | 34 | 6 | 9.20 | 0.39 to 217.2 | 0.17 |
| Time on ERT | |||||
| Not on ERT | 36 | 39 | 1.00 | 0.09 | |
| < 12 months | 20 | 29 | 0.78 | 0.18 to 3.37 | 0.74 |
| 12–36 months | 58 | 60 | 0.69 | 0.19 to 2.52 | 0.57 |
| > 36 months | 104 | 77 | 0.98 | 0.25 to 3.88 | 0.98 |
| N High | N Normal | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Age | |||||
| Linear effect/year | 1.05 | 1.004 to 1.10 | 0.03 | ||
| ACE inhibitors | |||||
| No | 220 | 339 | 1.00 | ||
| Yes | 43 | 18 | 1.68 | 0.56 to 5.04 | 0.36 |
| Time on ERT | |||||
| Not on ERT | 92 | 183 | 1.00 | 0.46 | |
| < 12 months | 28 | 32 | 0.55 | 0.19 to 1.57 | 0.27 |
| 12–36 months | 56 | 64 | 0.49 | 0.20 to 1.19 | 0.12 |
| > 36 months | 87 | 78 | 0.49 | 0.19 to 1.28 | 0.14 |
Brief Pain Inventory
The BPI provides two scores: the Pain Severity Score (ranging from 0 to 10, where 0 is ‘no pain’ and 10 is ‘pain as bad as you can imagine’); and the Pain Interference Score (ranging from 0 to 10, where 0 is ‘pain does not interfere’ and 10 is ‘pain completely interferes’).
Pain severity
Pain Severity Score data were collected for 149 adults with Fabry disease across all time points.
A longitudinal model was fitted to assess the relationship between Pain Severity Score and time on ERT, after adjusting for age (Table 58). As can be seen in Table 58, the Pain Severity Score was not significantly associated with age (p = 0.95). After adjusting for age, time on ERT was not associated with Pain Severity Score (p = 0.46). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and Pain Severity Score.
| N Data | Estimate of change in Pain Severity Score | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 218 | 0.00 | |||
| Female | 256 | 0.12 | 0.36 | –0.58 to 0.82 | 0.74 |
| Age | |||||
| Linear effect/year | –0.0008 | 0.01 | –0.02 to 0.02 | 0.95 | |
| Time on ERT | |||||
| Not on ERT | 161 | 0.00 | 0.46 | ||
| < 12 months | 38 | 0.07 | 0.29 | –0.49 to 0.64 | 0.79 |
| 12–36 months | 120 | –0.29 | 0.21 | –0.70 to 0.12 | 0.17 |
| > 36 months | 155 | –0.26 | 0.22 | –0.69 to 0.17 | 0.24 |
| Variance components | |||||
| Individual | 4.11 | ||||
| Centre | 0.00 | ||||
| Residual | 1.61 | ||||
Further analysis was conducted to explore the shape of the relationship between the Pain Severity Score and time on ERT treated as a continuous variable. This further modelling provided no evidence of a non-linear association between Pain Severity Score and time on ERT (edf = 1; p = 0.07) (Figure 36).
FIGURE 36.
The age-adjusted association between time on ERT and Pain Severity Score in adults with Fabry disease (time on ERT treated as a continuous variable).
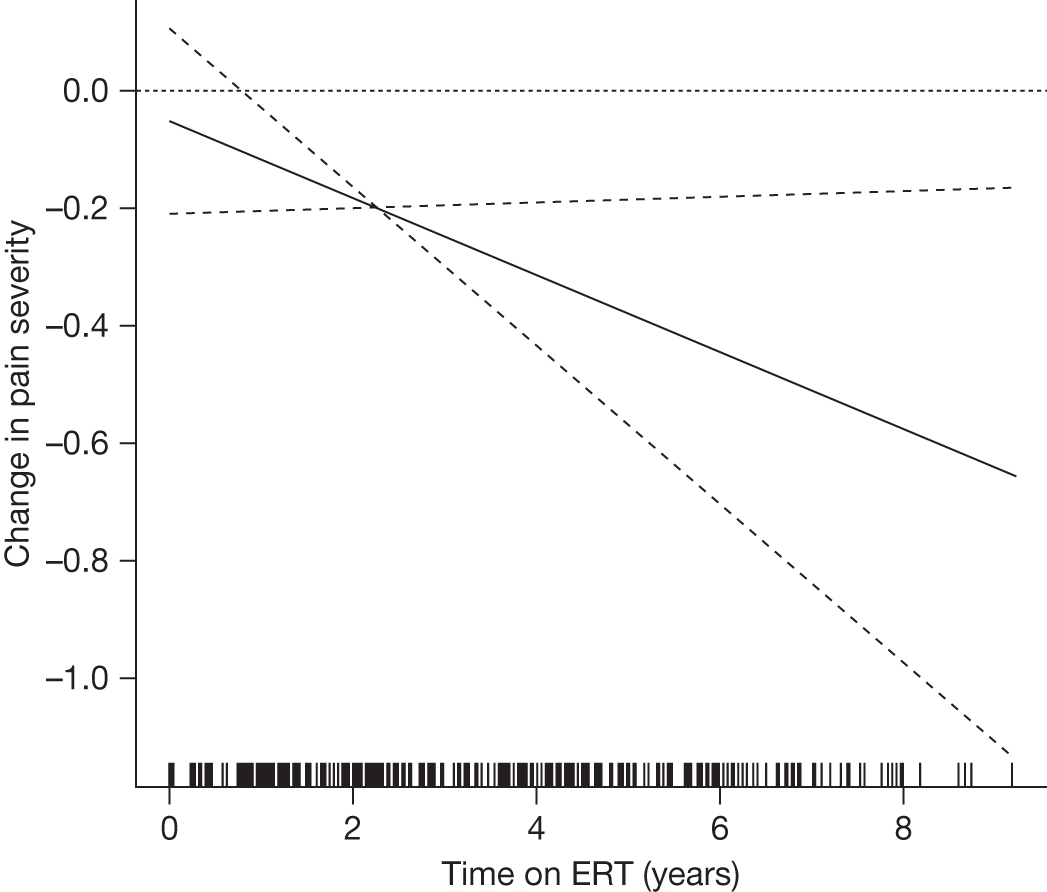
Pain interference
Pain Interference Score data were collected for 148 adult patients with Fabry disease across all time points.
A longitudinal model was fitted to assess the relationship between Pain Interference Score and time on ERT, after adjusting for age. As can be seen in Table 59, as with Pain Severity Score, the Pain Interference Score was not significantly associated with age (p = 0.62). However, after adjusting for age, time on ERT was associated with a significant reduction in Pain Interference Score (p < 0.001).
| N Data | Estimate of change in Pain Interference Score | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 215 | 0.00 | |||
| Female | 248 | –0.21 | 0.42 | –1.03 to 0.61 | 0.61 |
| Age | |||||
| Linear effect/year | 0.007 | 0.01 | –0.01 to 0.03 | 0.62 | |
| Time on ERT | |||||
| Not on ERT | 156 | 0.00 | < 0.001 | ||
| < 12 months | 35 | 0.57 | 0.38 | –0.17 to 1.31 | 0.14 |
| 12–36 months | 121 | –0.19 | 0.27 | –0.72 to 0.34 | 0.47 |
| > 36 months | 151 | –0.86 | 0.28 | –1.41 to –0.31 | 0.003 |
| Variance components | |||||
| Individual | 5.09 | ||||
| Centre | 0.00 | ||||
| Residual | 2.73 | ||||
In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and Pain Interference Score.
Further analysis was conducted to explore the shape of the relationship between Pain Interference Score and time on ERT treated as a continuous variable. Figure 37 illustrates the significant non-linear effect of time on ERT (edf = 1.66; p < 0.001) and Pain Interference Score. This graph suggests a significant reduction in Pain Interference Score, which plateaus at around 5 years after commencement of ERT, although the shape of this relationship should be viewed with caution because of the wide CIs around the line, particularly in later years.
FIGURE 37.
The age-adjusted association between time on ERT and Pain Interference Score in adults with Fabry disease (linear mixed-effects model).
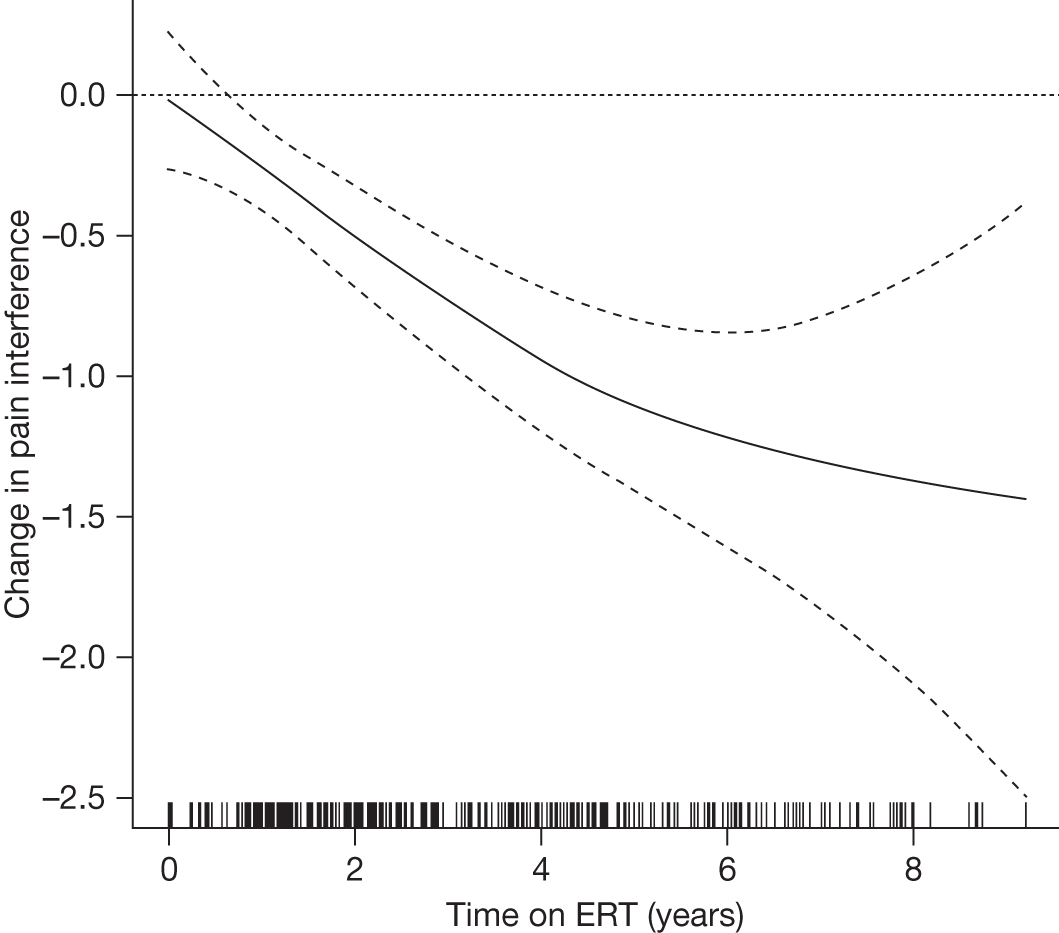
Audiology – use of a hearing aid
Patients were categorised according to whether or not they required a hearing aid. Nineteen out of 231 patients were reported as using a hearing aid at some point during the data collection period.
Figure 38 shows Kaplan–Meier curves for the age at the first recorded instance of the patient using a hearing aid. The curves illustrate the risk of moving from not requiring a hearing aid to requiring a hearing aid at different ages, on and off ERT.
FIGURE 38.
Risk of requiring a hearing aid by age and treatment status for people with Fabry disease (Kaplan–Meier curve).
The model shows a similar profile for patients on and off ERT, and indicates that, by the age of 60 years, approximately 40% of patients with Fabry disease require a hearing aid.
There was no significant association between treatment status and the probability of requiring a hearing aid – the hazard ratio (HR) for ERT relative to not on ERT is 0.96 (95% CI 0.28 to 3.18; p = 0.95).
Neurology – transient ischaemic attack/stroke
Patients were categorised according to whether or not they had experienced one or more TIAs or strokes. At recruitment, 30 patients (18 males and 12 females) were reported as having had a TIA or stroke. No further strokes were reported during the study period.
Figure 39 shows Kaplan–Meier curves for the age at first experiencing a TIA or stroke. The curves illustrate the risk of having a TIA or stroke at different ages, on and off ERT.
FIGURE 39.
Risk of TIA/stroke by age and treatment status for people with Fabry disease (Kaplan–Meier curve).
The model indicates that people with Fabry disease on or off ERT have a low probability of having a TIA or stroke before 40 years of age, but the risk of having a TIA or stroke increases after 40 years of age. Apparent differences in the survival curve should be interpreted with caution, as these do not reach conventional levels of statistical significance.
There was no statistically significant association between treatment status and the probability of having a TIA or stroke (HR = 2.08; 95% CI 0.42 to 10.20; p = 0.36).
Quality-of-life assessments
SF-36
Four hundred and seventy-seven SF-36 questionnaires were completed by 242 patients. Data are presented separately below for the PCS and the MCS (Tables 60–62, Figures 40 and 41).
| PCS | MCS | |
|---|---|---|
| Overall | ||
| Mean (SD) | 41.7 (12.7) | 48.7 (10.7) |
| N Data | 477 | 477 |
| ≤ 3 years on ERT | ||
| Mean (SD) | 41.5 (12.5) | 48.0 (10.5) |
| N Data | 124 | 124 |
| > 3 years on ERT | ||
| Mean (SD) | 39.5 (12.7) | 47.5 (11.1) |
| N Data | 266 | 266 |
| N Data | Estimate of change of PCS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 206 | 0.0 | |||
| Female | 271 | 2.68 | 1.59 | –0.44 to 5.79 | 0.09 |
| Age | |||||
| Linear effect/year | –0.22 | 0.05 | –0.32 to –0.12 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 87 | 0.00 | 0.41 | ||
| < 12 months | 32 | –0.76 | 1.65 | –3.99 to 2.47 | 0.64 |
| 12–36 months | 92 | –1.36 | 1.46 | –4.22 to 1.50 | 0.35 |
| > 36 months | 266 | –2.49 | 1.52 | –5.47 to 0.48 | 0.10 |
| Variance components | |||||
| Individual | 113.6 | ||||
| Centre | 1.8 | ||||
| Residual | 30.0 | ||||
| N Data | Estimate of change of MCS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 206 | 0.0 | |||
| Female | 271 | 2.45 | 1.35 | –0.19 to 5.09 | 0.07 |
| Age | |||||
| Linear effect/year | 0.009 | 0.04 | –0.07 to 0.08 | 0.85 | |
| Time on ERT | |||||
| Not on ERT | 87 | 0.00 | 0.04 | ||
| < 12 months | 32 | –4.39 | 1.78 | –7.87 to –0.90 | 0.01 |
| 12–36 months | 92 | –0.72 | 1.50 | –3.66 to 2.22 | 0.63 |
| > 36 months | 266 | –2.08 | 1.47 | –4.96 to 0.80 | 0.15 |
| Variance components | |||||
| Individual | 76.6 | ||||
| Centre | 4.1 | ||||
| Residual | 31.4 | ||||
FIGURE 40.
The age-adjusted association between time on ERT and SF-36 PCS in adults with Fabry disease.
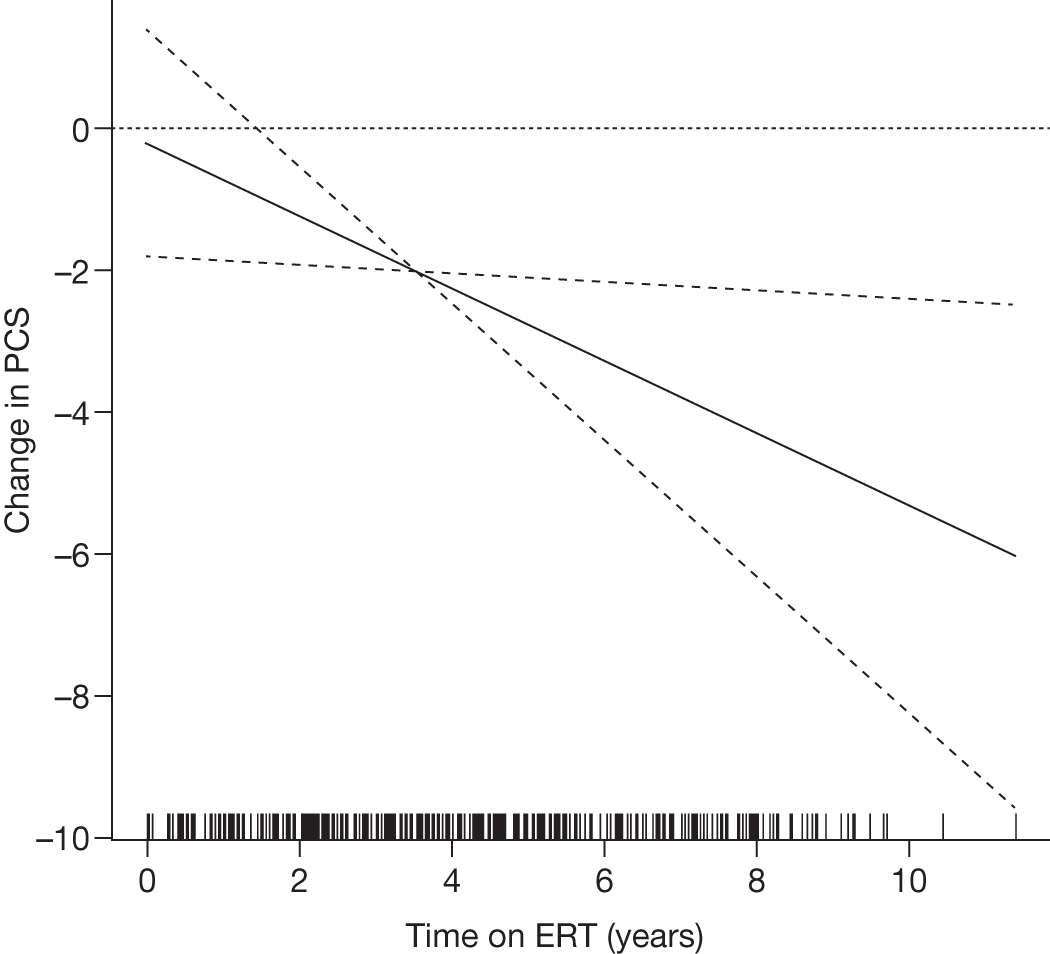
FIGURE 41.
The age-adjusted association between time on ERT and SF-36 MCS in adults with Fabry disease.
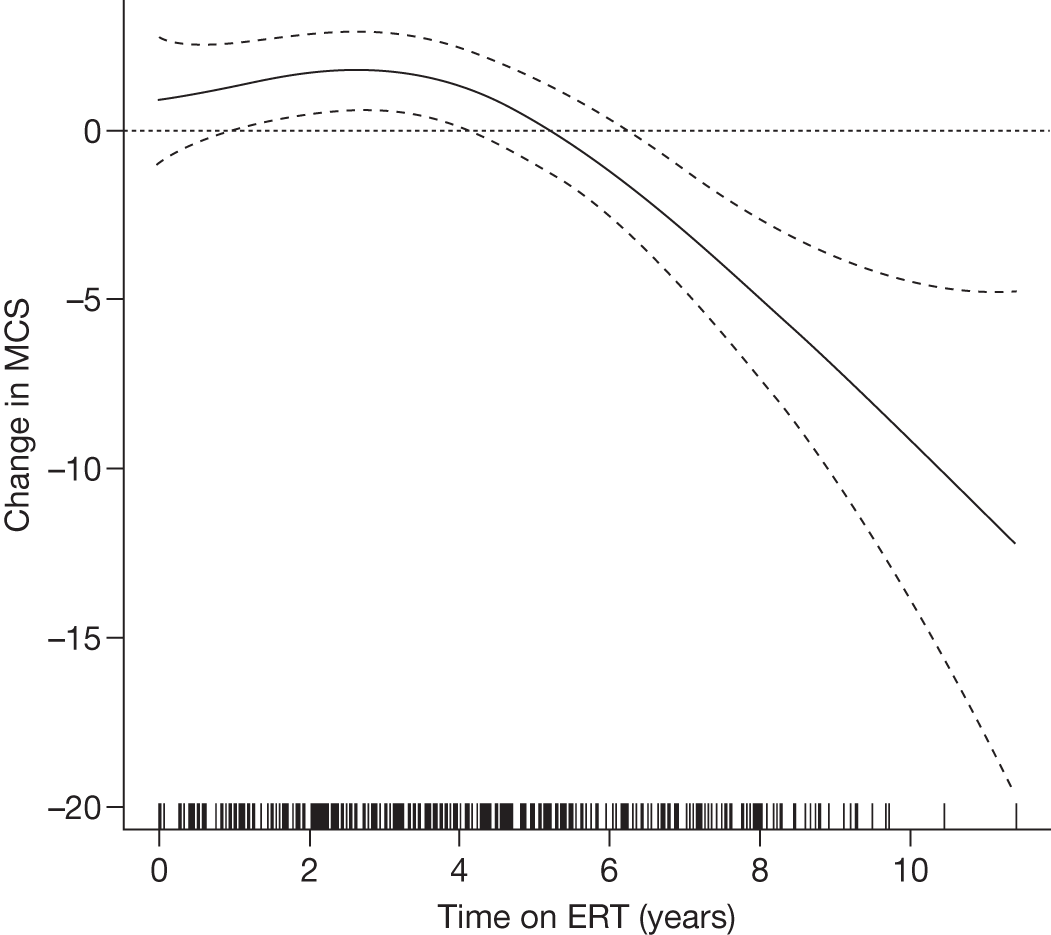
The mean PCS was 41.7 ± 12.7 and the mean MCS was 48.7 ± 10.7.
A longitudinal model was fitted to assess the linear relationship between the PCS and time on ERT, after adjusting for age (see Table 59). As can be seen in Table 61, there was an association between lower PCS and age (p < 0.001), but not with time on ERT (p = 0.41). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and PCS.
Further analysis was conducted to explore the shape of the relationship between PCS and time on ERT treated as a continuous variable. This analysis suggested a statistically significant association between a decrease in the PCS and time on ERT that was well approximated by a linear function (edf = 1; p = 0.02) (see Figure 40), suggesting a decrease in self-reported physical health associated with longer time on ERT.
A longitudinal model was fitted to assess the linear relationship between the MCS and time on ERT, after adjusting for age (see Table 62 and Figure 41). After adjusting for age, time on ERT was significantly associated with a lower MCS (p = 0.04). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and MCS. Further analysis was conducted to explore the shape of the relationship between MCS and time on ERT treated as a continuous variable. This analysis suggested a non-linear association between MCS and time on ERT (edf = 2.82; p = 0.0001) with a decline in scores seen associated with being on treatment for > 5 years. These results, which suggest a decrease in self-reported mental health with time on ERT, should be interpreted in the light of the wide CIs seen associated with longer periods of treatment.
EQ-5D
In addition to the SF-36, participants > 13 years of age were invited to complete the EQ-5D. Four hundred and ninety-seven EQ-5D index scores were generated across all prospective time points. Data are presented in Table 63 for the EQ-5D score (range from –0.24 to 1.0, with 1.0 being ‘perfect health’).
| N Data | Estimate of change of EQ-5D | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 210 | 0.00 | |||
| Female | 287 | 0.05 | 0.03 | –0.01 to 0.12 | 0.11 |
| Age | |||||
| Linear effect/year | –0.002 | 0.001 | –0.004 to –0.0001 | 0.03 | |
| Time on ERT | |||||
| Not on ERT | 95 | 0.00 | 0.09 | ||
| < 12 months | 30 | –0.02 | 0.04 | –0.10 to 0.06 | 0.65 |
| 12–36 months | 99 | –0.06 | 0.03 | –0.13 to 0.004 | 0.06 |
| > 36 months | 273 | –0.007 | 0.03 | –0.07 to 0.06 | 0.83 |
| Variance components | |||||
| Individual | 0.05 | ||||
| Centre | 0.00 | ||||
| Residual | 0.02 | ||||
A longitudinal model was fitted to assess the linear relationship between EQ-5D and time on ERT, after adjusting for age.
The linear model shows a very small but significant association between EQ-5D and age (p = 0.03) and no significant association between EQ-5D and time on ERT (p = 0.09).
Equivalent modelling analyses were also conducted using SF-6D (SF-36-derived) utility weights, but no statistically significant associations (at α = 0.05 level) were found with time on ERT. The tabulated results of the SF-6D longitudinal modelling analyses are available on request from the study authors.
No evidence was found for a non-linear association between the EQ-5D score and time on ERT (edf = 1.23; p = 0.44) (Figure 42).
FIGURE 42.
The age-adjusted association between time on ERT and EQ-5D in adults with Fabry disease (time on ERT treated as a continuous variable).
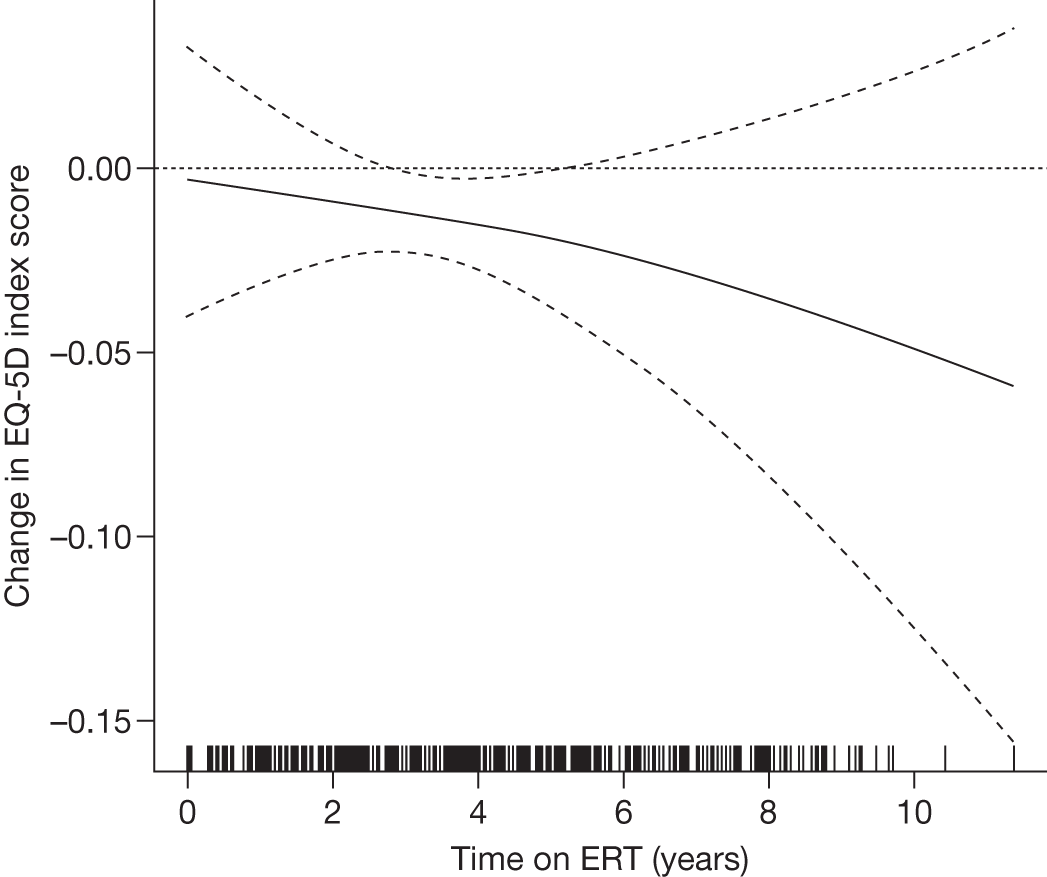
EQ-5D visual analogue scale
In addition to scoring on the five domains of the EQ-5D, participants were asked to rate their health on a VAS. The EQ-5D VAS asks people to rate their health state on a 10-cm line from 0, ‘worst imaginable health state’, to 100, ‘best imaginable health state’.
Four hundred and seventy EQ-5D questionnaires were completed across all prospective time points with range 4–100. A longitudinal model was fitted to assess the linear relationship between the visual analogue score and time on ERT after adjusting for age.
The linear model in Table 64 shows a non-significant association between EQ-5D VAS score and age (p = 0.35). There is, however, a statistically significant association between a reduction in the EQ-5D VAS score with time on ERT (p = 0.005). When time on ERT was treated as a continuous variable, there is strong evidence of a non-linear association of a reduction in the EQ-5D VAS and time on ERT (edf = 1.28; p < 0.001) (Figure 43).
| N Data | Estimate of change of EQ-5D VAS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 202 | 0.00 | |||
| Female | 268 | 2.98 | 2.55 | –2.02 to 7.98 | 0.24 |
| Age | |||||
| Linear effect/year | –0.08 | 0.08 | –0.23 to 0.08 | 0.35 | |
| Time on ERT | |||||
| Not on ERT | 95 | 0.00 | 0.005 | ||
| < 12 months | 29 | –9.87 | 3.99 | –17.7 to –2.05 | 0.01 |
| 12–36 months | 89 | –8.89 | 3.11 | –14.9 to –2.79 | 0.004 |
| > 36 months | 257 | –10.1 | 2.89 | –15.7 to –4.43 | < 0.001 |
| Variance components | |||||
| Individual | 251.5 | ||||
| Centre | 0.6 | ||||
| Residual | 177.5 | ||||
FIGURE 43.
The age-adjusted association between time on ERT and EQ-5D in adults with Fabry disease (time on ERT treated as a continuous variable).
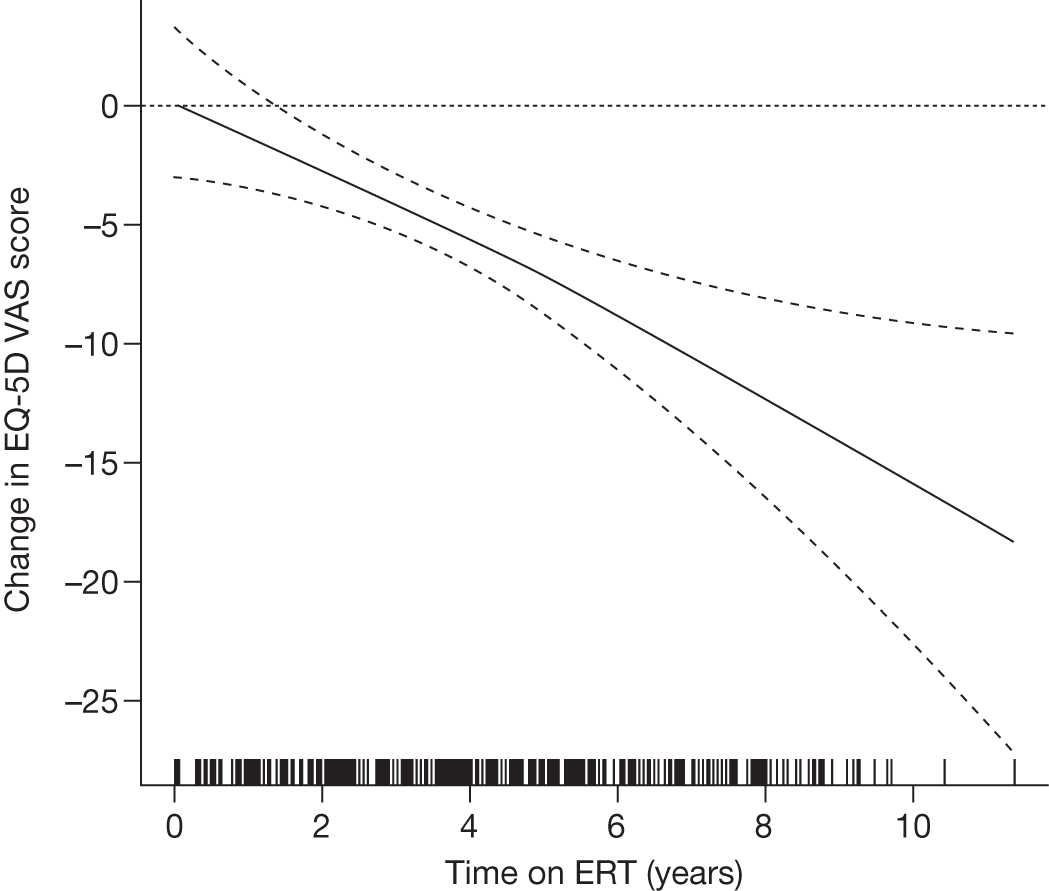
PedsQL
We have 22 PedsQL questionnaires completed by children or their carers. Table 65 shows a descriptive summary of the total score and the scores from the component scale scores by type of treatment. The scale ranges from 0 to 100, where higher scores indicate better HRQoL.
| Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Physical health summary score | Total score | |
|---|---|---|---|---|---|---|---|
| Overall | |||||||
| Mean (SD) | 75.5 (25.2) | 68.05 (23.9) | 76.8 (28.2) | 60.5 (20.5) | 66.1 (19.8) | 75.5 (25.2) | 68.1 (21.2) |
| n | 21 | 22 | 22 | 19 | 19 | 21 | 18 |
| Not on ERT | |||||||
| Mean (SD) | 83.9 (18.5) | 75.5 (18.7) | 83.0 (26.4) | 65.1 (19.0) | 71.7 (18.7) | 83.9 (18.5) | 74.5 (17.8) |
| n | 14 | 14 | 14 | 11 | 11 | 14 | 11 |
| ≤ 3 years on ERT | |||||||
| Mean (SD) | 57.3 (33.4) | 66.6 (28.9) | 72.6 (33.8) | 52.5 (17.1) | 63.9 (24.1) | 57.3 (33.4) | 61.6 (27.3) |
| n | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| > 3 years on ERT | |||||||
| Mean (SD) | 54.9 (41.6) | 36.2 (23.8) | 65.3 (30.8) | 52.47 (34.8) | 51.3 (18.7) | 54.9 (41.6) | 48.9 (29.3) |
| n | 2 | 3 | 3 | 3 | 3 | 2 | 2 |
The linear model shows a reduction in all PedsQL scores with age. These associations were significant in the total score (p = 0.03), the physical functioning (p = 0.006) and school functioning (p = 0.03) subscales. After adjusting for age, no relationship between time on ERT and any PedsQL subscale was seen (Table 66).
| Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Physical health summary score | Total score | |
|---|---|---|---|---|---|---|---|
| Gender | |||||||
| Male | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Female | 10.4 | 11.6 | 2.73 | 3.01 | 6.23 | 10.4 | 10.1 |
| 95% CI | –12.4 to 33.2 | –11.2 to 34.5 | –28.5 to 33.9 | –16.9 to 23.0 | –14.3 to 26.7 | –12.4 to 33.2 | –11.8 to 32.0 |
| p-value | 0.38 | 0.33 | 0.86 | 0.77 | 0.56 | 0.38 | 0.38 |
| Current age | |||||||
| Mean increment/year | –4.82 | –2.66 | –3.36 | –4.34 | –3.47 | –4.82 | –4.48 |
| 95% CI | –7.88 to –1.75 | –5.91 to 0.59 | –7.77 to 1.04 | –8.24 to –0.63 | –7.28 to 0.34 | –7.88 to –1.75 | –8.32 to –0.66 |
| p-value | 0.006 | 0.12 | 0.15 | 0.03 | 0.09 | 0.006 | 0.03 |
| Time on ERT | |||||||
| Mean increment/year | –3.77 | –7.91 | –0.17 | –1.10 | –2.96 | –3.77 | –4.17 |
| 95% CI | –12.8 to 5.25 | –16.3 to 0.47 | –11.4 to 11.1 | –8.52 to 6.32 | –10.4 to 4.52 | –12.8 to 5.25 | –12.8 to 4.45 |
| p-value | 0.42 | 0.08 | 0.97 | 0.77 | 0.45 | 0.42 | 0.35 |
| Variance components | |||||||
| Individual | 219.7 | 232.6 | 587.5 | 68.5 | 202.1 | 219.7 | 210.1 |
| Centre | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Residual | 146.1 | 191.2 | 186.7 | 274.5 | 134.9 | 146.1 | 122.7 |
Fatigue Severity Scale
Two hundred and forty-three FSS questionnaires completed by 173 patients were collected. The scores ranged from 1 to 7 [where a high score (≥ 4) is indicative of significant fatigue]. 228,229
A longitudinal model was fitted to assess the linear relationship between FSS and time on ERT, after adjusting for age (Table 67). As can be seen in Table 67, the FSS was not significantly associated with age (p = 0.49). There was, however, a significant association between time on ERT and an increase in the age-adjusted FSS (p = 0.02), suggesting an increase in fatigue with time on ERT. In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and fatigue severity.
| N Data | Estimate of change of mean FSS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 116 | 0.0 | |||
| Female | 127 | –0.47 | 0.27 | –0.99 to 0.06 | 0.09 |
| Age | |||||
| Linear effect/year | 0.007 | 0.01 | –0.01 to 0.03 | 0.49 | |
| Time on ERT | |||||
| Not on ERT | 38 | 0.00 | 0.02 | ||
| < 12 months | 10 | 0.21 | 0.52 | –0.81 to 1.23 | 0.67 |
| 12–36 months | 52 | 0.66 | 0.36 | –0.04 to 1.46 | 0.07 |
| > 36 months | 143 | 1.00 | 0.32 | 0.37 to 1.63 | 0.002 |
| Variance components | |||||
| Individual | 2.61 | ||||
| Centre | 0.00 | ||||
| Residual | 0.61 | ||||
Further analysis was conducted to explore the shape of the relationship between fatigue severity and time on ERT treated as a continuous variable. This analysis suggested that the relationship between fatigue severity and time on ERT was curvilinear (edf = 2.19; p = 0.02) (Figure 44), with self-reported fatigue increasing with duration on ERT.
FIGURE 44.
The age-adjusted association between time on ERT and FSS in adults with Fabry disease (time on ERT treated as a continuous variable).
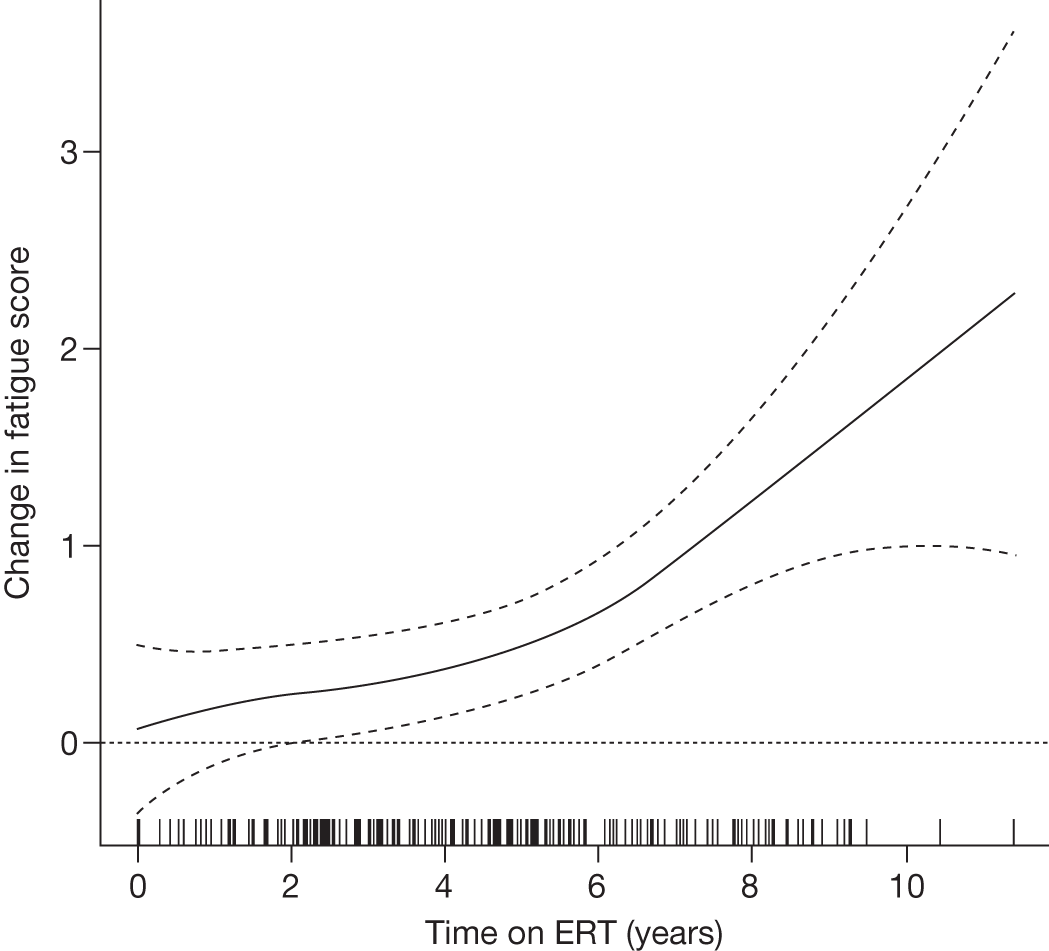
Carer Strain Index
Five CSI questionnaires were completed across all prospective time points. Data for the CSI total score ranged from 3 to 24. There were insufficient data for further analysis.
Comparison of the effectiveness of agalsidase alpha and agalsidase beta in Fabry disease
The longitudinal models presented above were extended to assess whether or not the effect of time on ERT varied according to the initial treatment patients received (agalsidase alpha or agalsidase beta). Each model partitioned the effect of time on ERT into two components: a common effect of being treated with an ERT (either agalsidase alpha or agalsidase beta) and a separate component for the incremental effect of receiving agalsidase beta rather than agalsidase alpha. To simplify the comparisons, the effect of time on ERT was considered as a linear function in all models (for both components). Patients who have never been treated with ERT were excluded from these models.
Heart size – left ventricular mass index
Adults
Left ventricular mass index data were collected for 172 adults, with 68 adults on agalsidase alpha and 104 adults on agalsidase beta (Table 68).
| N Data | Estimate of change in LVMI (g/m2) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 203 | 0.00 | |||
| Female | 255 | –42.50 | 6.26 | –54.77 to –30.24 | < 0.001 |
| Age | |||||
| Linear effect/year | 1.60 | 0.22 | 1.17 to 2.03 | < 0.001 | |
| Time on ERT | |||||
| Linear effect/year | –1.06 | 1.19 | –3.40 to 1.28 | 0.38 | |
| Comparison of treatments (per year) | |||||
| Agalsidase alpha | 0.00 | ||||
| Agalsidase beta | –1.31 | 1.43 | –4.11 to 1.48 | 0.36 | |
| Variance components | |||||
| Individual | 1264 | ||||
| Centre | 307 | ||||
| Residual | 608 | ||||
Treatment with agalsidase beta had a similar effect on adult LVMI levels to treatment with agalsidase alpha (incremental effect of agalsidase beta = –1.31 g/m2/year; 95% CI –4.11 to 1.48; p = 0.36).
Children
Left ventricular mass index data were collected for nine children, with five children on agalsidase alpha and four children on agalsidase beta.
No statistically significant evidence was found for a difference between the effects of agalsidase beta and agalsidase alpha on LVMI levels in children (incremental effect of agalsidase beta = 0.98 g/year, 95% CI –6.67 to 8.60; p = 0.80) (Table 69).
| N Data | Estimate of change in LVMI (g/m2) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 12 | 0.00 | |||
| Female | 11 | –10.48 | 9.28 | –28.70 to 7.71 | 0.27 |
| Age | |||||
| Linear effect/year | 2.12 | 1.39 | –0.61 to 4.85 | 0.14 | |
| Time on ERT | |||||
| Linear effect/year | –3.38 | 2.41 | –8.09 to 1.33 | 0.17 | |
| Comparison of treatments (per year) | |||||
| Agalsidase alpha | 0.00 | ||||
| Agalsidase beta | 0.98 | 3.90 | –6.67 to 8.60 | 0.80 | |
| Variance components | |||||
| Individual | 92 | ||||
| Centre | 0 | ||||
| Residual | 78 | ||||
Renal function – estimated glomerular filtration rate
Adults
Estimated glomerular filtration rate data were collected for 203 adults, with 86 adults on agalsidase alpha and 117 adults on agalsidase beta.
Overall, treatment with agalsidase beta had a similar effect on adult eGFR levels to treatment with agalsidase alpha (incremental effect of agalsidase beta = 0.36 ml/minute/1.73 m2/year; 95% CI –0.34 to 1.06; p = 0.32) (Table 70). No difference between treatments was found when the analysis was repeated in adult males only (incremental effect of agalsidase beta = –0.37 ml/minute/1.73 m2/year; 95% CI –1.38 to 0.64; p = 0.48) (Table 71). However, there was evidence to suggest that treatment with agalsidase beta increased eGFR levels at a faster rate than treatment with agalsidase alpha in adult females (incremental effect of agalsidase beta = 0.99 ml/minute/1.73 m2/year; 95% CI 0.06 to 1.91; p = 0.04) (Table 72).
| N Data | Estimate of change in eGFR ml/minute/1.73 m2 |
Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 514 | 0.00 | |||
| Female | 566 | 5.08 | 3.17 | –1.14 to 11.31 | 0.11 |
| Age | |||||
| Linear effect/year | –1.12 | 0.11 | –1.32 to –0.91 | < 0.001 | |
| Time on ERT | |||||
| Linear effect/year | 0.66 | 0.29 | 0.10 to 1.23 | 0.02 | |
| Comparison of treatments (per year) | |||||
| galsidase alpha | 0.00 | ||||
| Agalsidase beta | 0.36 | 0.36 | –0.34 to 1.06 | 0.32 | |
| Variance components | |||||
| Individual | 453 | ||||
| Centre | 18 | ||||
| Residual | 135 | ||||
| Estimate of change in eGFR ml/minute/1.73 m2 |
Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Age | ||||
| Linear effect/year | –1.39 | 0.16 | –1.70 to –1.08 | < 0.001 |
| Time on ERT | ||||
| Linear effect/year | 0.58 | 0.42 | –0.26 to 1.41 | 0.18 |
| Comparison of treatments (per year) | ||||
| Agalsidase alpha | 0.00 | |||
| Agalsidase beta | –0.37 | 0.51 | –1.38 to 0.64 | 0.48 |
| Variance components | ||||
| Individual | 523 | |||
| Centre | 0 | |||
| Residual | 149 | |||
| Estimate of change in eGFR ml/minute/1.73 m2 |
Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Age | ||||
| Linear effect/year | –0.84 | 0.14 | –1.11 to –0.57 | < 0.001 |
| Time on ERT | ||||
| Linear effect/year | 0.82 | 0.38 | 0.07 to 1.56 | 0.03 |
| Comparison of treatments (per year) | ||||
| Agalsidase alpha | 0.00 | |||
| Agalsidase beta | 0.99 | 0.47 | 0.06 to 1.91 | 0.04 |
| Variance components | ||||
| Individual | 393 | |||
| Centre | 20 | |||
| Residual | 116 | |||
Children
Estimated glomerular filtration rate data were collected for 15 children, with 5 children on agalsidase alpha and 10 children on agalsidase beta.
In children, treatment with agalsidase beta had a similar effect on adult eGFR levels to treatment with agalsidase alpha (incremental effect of agalsidase beta = 3.45 ml/minute/1.73 m2/year; 95% CI –4.14 to 11.05; p = 0.38) (Table 73).
| N Data | Estimate of change in eGFR ml/minute/1.73 m2 |
Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 25 | 0.00 | |||
| Female | 17 | 7.21 | 15.70 | –23.6 to 38.01 | 0.65 |
| Age | |||||
| Linear effect/year | –3.64 | 2.20 | –7.95 to 0.67 | 0.10 | |
| Time on ERT | |||||
| Linear effect/year | 1.70 | 3.76 | –5.67 to 9.07 | 0.65 | |
| Comparison of treatments (per year) | |||||
| Agalsidase alpha | 0.00 | ||||
| Agalsidase beta | 3.45 | 3.87 | –4.14 to 11.05 | 0.38 | |
| Variance components | |||||
| Individual | 692 | ||||
| Centre | 43 | ||||
| Residual | 238 | ||||
Proteinuria
Adults
Proteinuria data were collected for 197 adults, with 85 adults on agalsidase alpha and 112 adults on agalsidase beta.
Treatment with agalsidase beta did not change the risk of proteinuria in adults compared with treatment with agalsidase alpha (p = 0.90) (Table 74).
| N High | N Low | OR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 212 | 197 | 1.00 | ||
| Female | 231 | 213 | 0.26 | 0.07 to 1.00 | 0.05 |
| Age | |||||
| Linear effect/year | 1.07 | 1.02 to 1.12 | 0.003 | ||
| ACE inhibitors | |||||
| No | 374 | 392 | 1.00 | ||
| Yes | 69 | 18 | 1.96 | 0.65 to 5.88 | 0.23 |
| Time on ERT | |||||
| Linear effect/year | 0.88 | 0.72 to 1.07 | 0.19 | ||
| Comparison of treatments (per year) | |||||
| Agalsidase alpha | 1.00 | ||||
| Agalsidase beta | 0.98 | 0.76 to 1.28 | 0.90 | ||
Brief Pain Inventory
Pain severity
Brief Pain Inventory questionnaires were completed by 120 adults with Fabry disease on ERT (with 29 adults on agalsidase alpha and 91 adults on agalsidase beta) across all time points.
People with Fabry disease treated with agalsidase beta had similar levels of Pain Severity Score to patients treated with agalsidase alpha (p = 0.20) (Table 75).
| N Data | Estimate of change in Pain Severity Score | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 206 | 0.00 | |||
| Female | 166 | 0.54 | 0.40 | –0.25 to 1.33 | 0.18 |
| Age | |||||
| Linear effect/year | –0.004 | 0.01 | –0.03 to 0.02 | 0.76 | |
| Time on ERT | |||||
| Linear effect/year | –0.20 | 0.09 | –0.38 to –0.01 | 0.04 | |
| Comparison of treatments (per year) | |||||
| Agalsidase alpha | 0.00 | ||||
| Agalsidase beta | 0.12 | 0.10 | –0.07 to 0.31 | 0.20 | |
| Variance components | |||||
| Individual | 3.96 | ||||
| Centre | 0.00 | ||||
| Residual | 1.66 | ||||
Pain interference
People with Fabry disease treated with agalsidase beta had similar levels of Pain Interference Score to patients treated with agalsidase alpha (p = 0.42) (Table 76).
| N Data | Estimate of change in Pain Interference Score | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 205 | 0.00 | |||
| Female | 161 | 0.25 | 0.48 | –0.68 to 1.18 | 0.60 |
| Age | |||||
| Linear effect/year | 0.0004 | 0.02 | –0.03 to 0.03 | 0.98 | |
| Time on ERT | |||||
| Linear effect/year | –0.14 | 0.12 | –0.38 to 0.10 | 0.25 | |
| Comparison of treatments (per year) | |||||
| Agalsidase alpha | 0.00 | ||||
| Agalsidase beta | –0.10 | 0.13 | –0.36 to 0.15 | 0.42 | |
| Variance components | |||||
| Individual | 4.99 | ||||
| Centre | 0.53 | ||||
| Residual | 2.94 | ||||
Audiology – use of hearing aid
Hearing data were collected for 206 patients on ERT, with 89 patients on agalsidase alpha and 117 patients on agalsidase beta.
Figure 45 shows Kaplan–Meier curves for the age at the first recorded instance of the patient using a hearing aid. The curves illustrate the risk of moving from not requiring a hearing aid to requiring a hearing aid at different ages, for patients on agalsidase alpha and agalsidase beta.
FIGURE 45.
Risk of requiring a hearing aid by age and treatment type – agalsidase alpha and agalsidase beta (Kaplan–Meier curve).
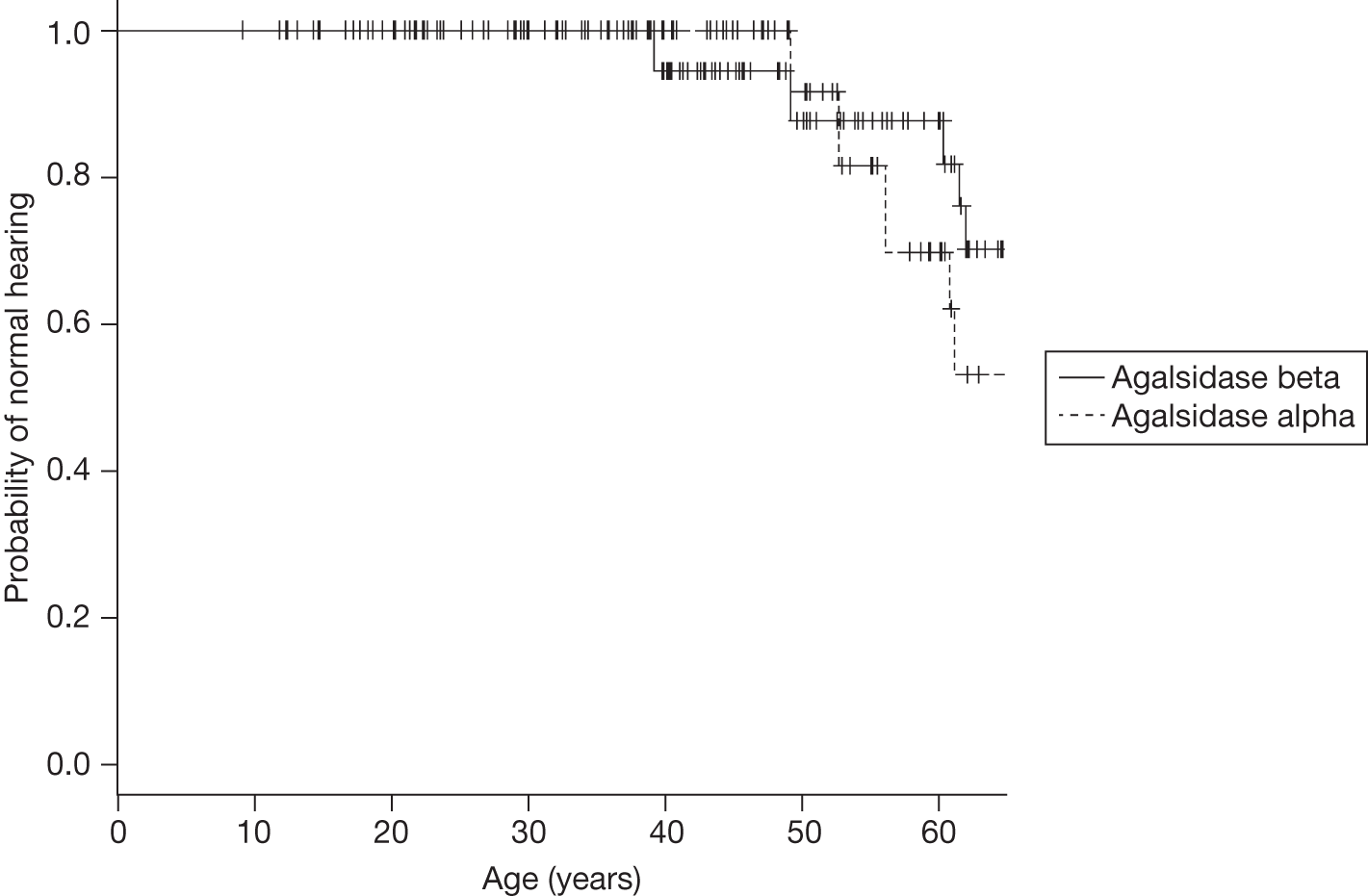
The HR comparison for the probability of requiring a hearing aid for the patients receiving agalsidase alpha or agalsidase beta was 1.87 (95% CI 0.56 to 6.22; p = 0.31).
Neurology – transient ischaemic attack/stroke
Transient ischaemic attack/stroke data were collected for 201 patients, with 90 patients on agalsidase alpha and 121 patients on agalsidase beta.
Figure 46 shows Kaplan–Meier curves for the age at first experiencing a TIA or stroke. The curves illustrate the risk of having a TIA or stroke at different ages, for patients on agalsidase alpha and agalsidase beta.
FIGURE 46.
Risk of TIA/stroke by age and treatment type – agalsidase alpha and agalsidase beta (Kaplan–Meier curve).
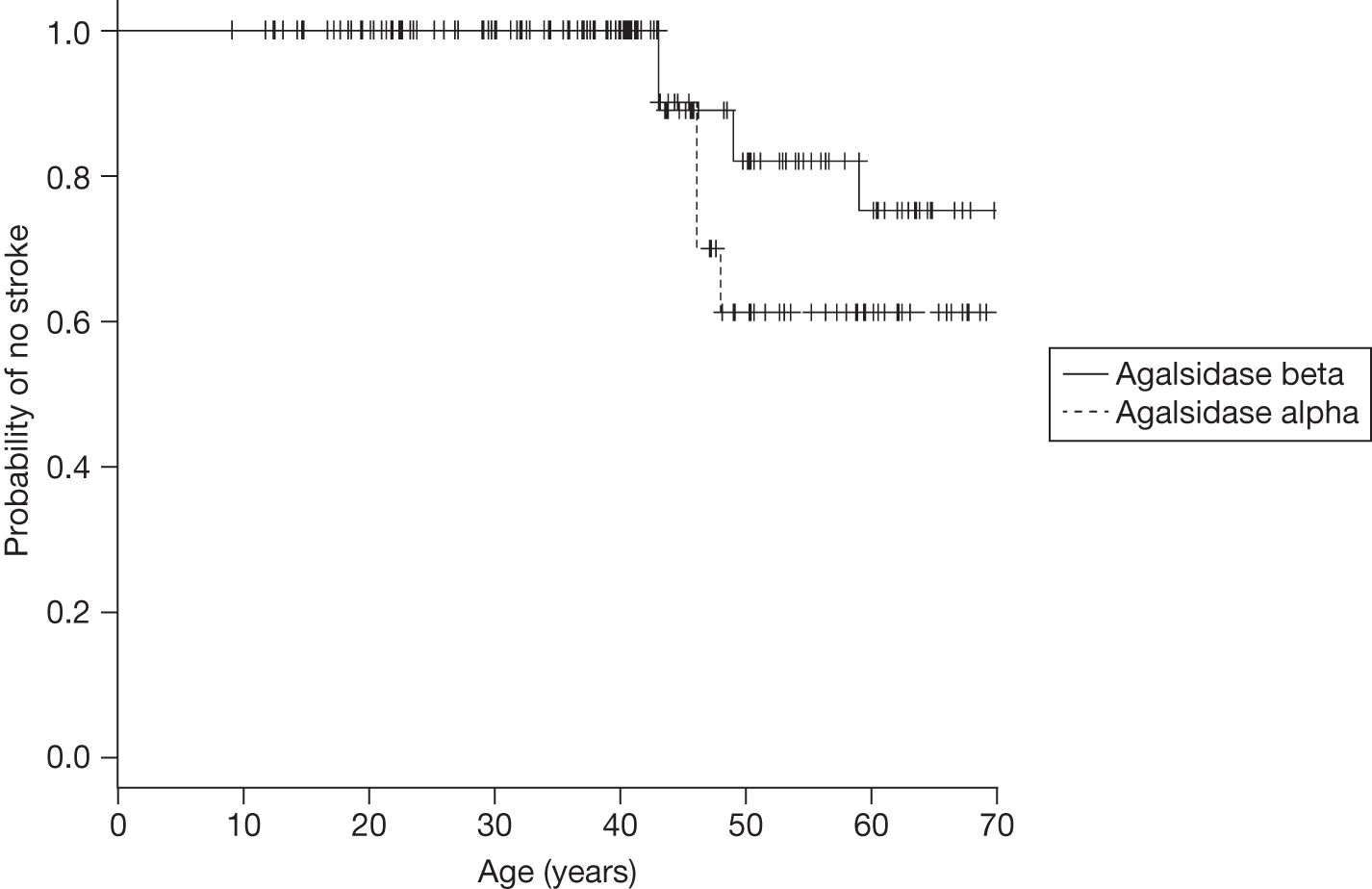
The model shows a similar profile for patients on both ERT formulations. There was no significant association between treatment status and the probability of requiring a hearing aid (the HR for being on agalsidase alpha relative to agalsidase beta = 1.41; 95% CI 0.38 to 5.25; p = 0.61).
Safety and complications
Of the 311 participants with Fabry disease in this study, six patients experienced anaphylactic reactions, five patients experienced febrile reactions and seven patients were reported to have a positive antibody status to infused product. None of the above patients had their treatment stopped following these events.
A further seven patients stopped ERT during the period of data collection. Of these, four patients were on agalsidase beta and three patients were on agalsidase alpha. One patient failed to comply with the regime, one patient stopped at the end of a clinical trial and one patient stopped during pregnancy. No reason for stopping treatment was cited for the remaining four patients.
There were no study-related adverse events among people with Fabry disease, although one patient refused to continue completing the study questionnaires 1 year after consenting to participate. Two patients died during the course of the study (one from disease-related complications and one from a non-related condition).
Cost of enzyme replacement therapy in people with Fabry disease
Table 77 shows the current purchase cost to the NHS of the ERTs agalsidase alpha and agalsidase beta. Note that the drug costs for agalsidase alpha include the nursing cost for providing the infusion sessions (‘home care’).
| Drug full name | Proprietary name and unit | 2011 base price per unit (£) |
|---|---|---|
| Agalsidase alpha | Replagal®, 3.5 mg | 1049.94 |
| Agalsidase beta | Fabrazyme®, 35 mg | 2086.76 |
| Agalsidase beta | Fabrazyme®, 5 mg | 298.11 |
Table 78 shows the NSCT-estimated annual NHS per patient cost of providing these drugs. Note that these costs include both the drug costs and home-care costs where the NSCT fund them.
| Drug | Adults | Children |
|---|---|---|
| Agalsidase alpha 3.5 mg | £120,840 | £89,199 |
| Agalsidase beta 35 mg and/or 5 mga | £106,394 | £79,478 |
Cost of care for adults with Fabry disease
Total care cost – financial burden of Fabry disease
Table 79 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for an adult with Fabry disease. Of the estimated mean per patient annual cost of £3300, 70% is as a result of NHS hospital services used, with just under a half of this (£1000 per patient per year) from inpatient stays, whereas about 40% (£940 per patient) is from outpatient visits (see Table 80). Of the £1000 per patient per year from using services outside of hospital, about one-tenth (£110 per patient) is as a result of GP visits and over four-fifths (£840) is as a result of regular visits from either district nurses, health visitors or other nurses (many of these will be for regular ERT infusions) (see Table 81).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. (%) who used this type of service | Mean cost (£) | Standard deviation | Median cost (£) | Interquartile range (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 257 | 89 | 192 (75) | 2300 | 4679 | 940 | 0–1880 |
| Services outside hospital | 257 | 89 | 205 (80) | 1000 | 2702 | 81.5 | 16–340 |
| Total health- (NHS) and social-care cost | 257 | 89 | 237 (92) | 3300 | 5958 | 1000 | 200–3200 |
| Type of hospital care | No. (%) who used this type of service | Mean cost (£) | Standard deviation | Median cost (£) | Interquartile range (£) |
|---|---|---|---|---|---|
| Inpatient stays | 60 (23) | 1000 | 3399 | 0 | 0–0 |
| Outpatient visits | 161 (63) | 940 | 2447 | 0 | 0–940 |
| Day cases | 41 (16) | 290 | 978 | 0 | 0–0 |
| Accident and emergency visits | 31 (12) | 21 | 83 | 0 | 0–0 |
| Total hospital (NHS) care cost | 192 (75) | 2300 | 4679 | 0 | 0–0 |
| Care provider | No. (%) who used this provider | Mean cost (£) | Standard deviation | Median cost (£) | Interquartile range (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 186 (72) | 110 | 149 | 54 | 0–140 |
| GP nurse appointments | 9 (4) | 7 | 15 | 0 | 0–8 |
| District nurses | 6 (2) | 31 | 345 | 0 | 0–0 |
| Community mental health nurse | 0 | 0 | 0 | 0 | 0–0 |
| Other nurse or health visitor | 48 (19) | 710 | 2088 | 0 | 0–0 |
| Counsellor | 6 (2) | < 1 | 6 | 0 | 0–0 |
| Other therapist | 8 (3) | 33 | 447 | 0 | 0–0 |
| ‘Alternative’ medicine or therapy | 2 (1) | 120 | 1589 | 0 | 0–0 |
| Psychologist | 6 (2) | 7 | 55 | 0 | 0–0 |
| Psychiatrist | 0 | 0 | 0 | 0 | 0–0 |
| Other community-based doctor | 2 (1) | 1 | 17 | 0 | 0–0 |
| Occupational therapist | 2 (1) | 2 | 24 | 0 | 0–0 |
| Social worker | 1 (0.4) | < 1 | 10 | 0 | 0–0 |
| Home help | 2 (1) | 8 | 91 | 0 | 0–0 |
| Care attendant | 0 | 0 | 0 | 0 | 0–0 |
| Community support worker | 0 | 0 | 0 | 0 | 0–0 |
| Housing worker | 1 (0.4) | < 1 | 3 | 0 | 0–0 |
| All non-hospital NHS and social-care providers | 205 (80) | 1000 | 2696 | 0 | 0–0 |
Cost breakdown by hospital- and community-based services
Tables 80 and 81 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by adults with Fabry disease. Sixty patients, or just under one-quarter of those adults who provided valid service-use data, had hospital stays as inpatients, and this accounted for > 40% of the NHS hospital costs in Fabry adults. In contrast, over three-fifths (63%) of patients reported having at least one hospital outpatient attendance during the previous 12 months.
The majority of costs related to using community-based services were as a result of the relatively small minority of adults with Fabry disease who used health visitors or other nurses regularly (48 patients, 19%). They accounted for £710 of the £1000 yearly per patient cost of services used outside hospital. Although nearly three-quarters of adults with Fabry disease had seen their GP at least once during the past year, these accounted for only £110 of the £3000 annual cost of NHS and publicly funded social-care services consumed. Other support providers used by smaller numbers of adults with Fabry disease were ‘other therapists’ (e.g. physiotherapists) and district nurses, psychologists and counsellors (see Table 81).
Cost of care for children with Fabry disease
Total care cost – financial burden of Fabry disease
Table 82 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for a child with Fabry disease, based on the 18 children whose parents or carers supplied service-use data at study entry (mean age 10 years, range 1.8–16 years, 10 male). Of the estimated mean per patient annual cost of £1300, approximately half is as a result of NHS hospital services used, approximately three-fifths (£380) is from inpatient stays, whereas about one-fifth (£130 per patient) is from outpatient visits (see Table 83). Of the £710 per patient per year from using services outside hospital, about one-fifth (£140 per patient) is as a result of GP visits while almost two-thirds (£460) is as a result of regular visits from either district nurses, health visitors or other nurses (note some of these may be for regular ERT infusions) (see Table 84).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. (%) who used this type of service | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 18 | 82 | 7 | 630 | 1007 | 1500 | 940–2700 |
| Services outside hospital | 18 | 82 | 17 | 710 | 1378 | 130 | 130–330 |
| Total health (NHS) and social-care cost | 18 | 82 | 17 | 1300 | 1600 | 240 | 130–3200 |
| Type of hospital care | No. (%) who used this type of service | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| Inpatient stays | 4 | 380 | 765 | 1900 | 940–2100 |
| Outpatient visits | 2 | 130 | 384 | 1200 | 1100–1200 |
| Day cases | 1 | 83 | 350 | 1500 | N/A |
| Accident and emergency visits | 2 | 34 | 122 | 310 | 100–520 |
| Total hospital (NHS) care cost | 7 | 630 | 1007 | 1500 | 940–2700 |
| Care provider | No. (%) who used this provider | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 17 | 140 | 98 | 130 | 82–130 |
| GP nurse appointments | 6 | 7 | 3 | 5 | 8–10 |
| District nurses | 2 | 220 | 905 | 1900 | 43–3800 |
| Community mental health nurse | 0 | 0 | 0 | 0 | 0–0 |
| Other nurse or health visitor | 2 | 240 | 996 | 2100 | 7–4200 |
| Counsellor | 0 | 0 | 0 | 0 | 0–0 |
| Other therapist | 1 | 1 | 4 | 19 | N/A |
| ‘Alternative’ medicine or therapy | 0 | 0 | 0 | 0 | 0–0 |
| Psychologist | 1 | 27 | 115 | 490 | N/A |
| Psychiatrist | 1 | 94 | 400 | 1700 | N/A |
| Other community-based doctor | 0 | 0 | 0 | 0 | 0–0 |
| Occupational therapist | 0 | 0 | 0 | 0 | 0–0 |
| Social worker | 0 | 0 | 0 | 0 | 0–0 |
| Home help | 0 | 0 | 0 | 0 | 0–0 |
| Care attendant | 0 | 0 | 0 | 0 | 0–0 |
| Community support worker | 0 | 0 | 0 | 0 | 0–0 |
| Housing worker | 0 | 0 | 0 | 0 | 0–0 |
| All non-hospital NHS and social-care providers | 18 | 710 | 1378 | 130 | 130–330 |
Cost breakdown by hospital- and community-based services
Tables 83 and 84 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by children with Fabry disease. Four patients who had valid service-use data had hospital stays as inpatients, and this accounted for > 40% of the NHS hospital costs for children with Fabry disease. In contrast, over three-fifths (63%) of patients reported having at least one hospital outpatient attendance during the previous 12 months.
The majority of costs related to using community-based services were due to the relatively small minority of children with Fabry disease who used district nurses, health visitors or other nurses regularly. They accounted for £460 of the £710 yearly per patient cost of services used outside hospital. Although almost all children with Fabry disease had seen their GP at least once during the past year, these visits accounted for only £140 of the £1300 annual cost of NHS and publicly funded social-care services consumed. Other support providers used by smaller numbers of children with Fabry disease were physiotherapists, psychologists and psychiatrists (see Table 84).
Association between time on enzyme replacement therapy and cost of caring for patients with Fabry disease
From the longitudinal regression modelling of costs for patients with Fabry disease, there was no statistically significant association (i.e. no p-values < 0.05 for the regression coefficient) in either adults or children between time on ERT and either total NHS and social-care costs, hospital-care costs, or non-hospital-care costs. The tabulated results of these analyses are available on request from the study authors.
Discussion of Fabry disease results
Evidence for the effectiveness of ERT in treating Fabry disease has come from three randomised, placebo-controlled trials,134–136 (n = 70, duration 5–6 months) including a total of 27 patients on agalsidase beta and 21 patients on agalsidase alpha, and 11 observational, non-comparative, before-and-after studies (n = 493, duration up to 24 months). These studies are summarised in a 2006 HTA-commissioned systematic review19 of effectiveness and cost-effectiveness. The review concluded that overall, the results suggest some beneficial effect of ERT on measures of pain and cardiovascular function with an apparent stabilisation of renal function.
In this study, we examined potential associations between treatment and LVMI, kidney function, pain, hearing, and TIA or strokes. We also examined the relationship between treatment and QoL assessed by either SF-36 or PedsQL depending on age. We had planned to examine the association between use of ERT and developmental quotient in children but almost no data on this outcome were recorded.
These analyses depend on the fact that patients began treatment with algalsidase alpha or algalsidase beta, at different ages dependent on the time that the drug became available. However, it should be noted that, of the 127 patients on agalsidase beta at recruitment, 66 patients switched to agalsidase alpha during the shortage period (from June 2009). Of these, two patients were recorded as switching back to agalsidase beta, whereas the remaining 64 patients remained on agalsidase alpha. Conversely, two patients who were on agalsidase alpha at recruitment switched to agalsidase beta during the same period. As our analysis compared the outcome of treatment depending on which of the two drugs patients were initially assigned (the equivalent of an intention-to-treat analysis), these changes in treatment regimen have not been taken into account in our models.
Overall, our results provide evidence to suggest positive effects of ERT on left ventricular hypertrophy and renal function (as assessed by age-adjusted eGFR and the risk of proteinuria). We also have some evidence suggesting that ERT is associated with a decrease in the extent to which pain interferes with QoL although we found no statistically significant association with the severity of pain.
We found no statistically significant evidence of an effect on the risk of hearing impairment or of TIA and stroke.
The duration of treatment with ERT was associated with worse scores on both SF-36 PCS and SF-36 MCS after adjusting for age. Finally, after adjusting for age we found a statistically significant relationship between higher (i.e. worse) fatigue score and the duration of treatment.
For each outcome we compared the relative effects in those patients initially treated with agalsidase beta with those initially treated with agalsidase alpha. There were no statistically significant differences in any of the outcomes for adults or children. Again, results will have been confounded by patients switching to agalsidase alpha during the shortage of agalsidase beta.
Left ventricular mass index
Some previous studies have suggested that ERT is associated with a decrease in left ventricular hypertrophy. In a cohort study of 545 patients, the Fabry Outcome Survey, treatment with agalsidase alpha was reported to reduce left ventricular size in patients who had an enlarged heart at baseline. 142 Hughes and colleagues138 reported that in a RCT of 15 male patients with Fabry disease, left ventricular mass (LVM) was significantly reduced following 6 months of treatment with agalsidase alpha compared with the placebo group. However, Eng and colleagues136 reported that echocardiograms of 15 patients in an open-label, dose escalation study remained unchanged after commencing treatment on agalsidase beta and no reduction in LVM was reported in either of the two treatment groups in a comparative trial of agalsidase alpha and agalsidase beta149 (n = 34).
Although in our initial analyses we found no statistically significant association between age adjusted LVMI in adults and time on ERT (p = 0.11), when time on ERT was categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, further analysis where time on ERT was treated as a continuous variable suggested that treatment with ERT has a significant linear effect of reducing LVMI (edf = 1; p = 0.01). We found no such association in children but this is unsurprising given the small numbers involved.
Renal function
Beck and colleagues142 reported that in their cohort study of 545 patients on agalsidase alpha, renal function stabilised in patients with a mild or moderate deterioration in renal function at baseline. Similarly, in an open-label, extension trial of a RCT of 58 patients, stabilisation of renal disease progression was reported after 54 months on agalsidase beta. 141 Meanwhile, Eng and colleagues136 reported that renal magnetic resonance images of patients in an open-label, dose-escalation study remained unchanged after commencing treatment on agalsidase beta and no change was seen in the glomerular filtration rate of 34 patients treated with either agalsidase alpha or beta in a comparative trial. 149
Germain and colleagues141 examined the long-term effects of agalsidase beta, in an open-label, Phase III, extension trial of 58 patients over 54 months. After 54 months of treatment, median proteinuria remained stable which they interpreted as a stabilisation of renal disease progression. They also suggested that significant renal involvement at pre-treatment increased the likelihood for renal progression over time.
In this study, we found a statistically significant association between time on ERT and an increase in age-adjusted eGFR in adults (p = 0.002) when time on ERT was categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months. Further analysis treating time on ERT as a continuous variable suggested a non-linear relationship between eGFR levels and time on ERT (edf = 2.05; p = 0.001) with the effect apparently plateauing after approximately 6 years. When data from men and women were analysed separately the association remained significant for women but not for men (although the association was in the same direction).
Bayesian models predicted that the eGFR of patients starting on ERT at either age 18 or 45 years would increase relative to untreated patients, before levelling off. The treatment effect appears to be of greater magnitude for patients who commenced treatment at 45 years than for those who commenced treatment at 18 years; the Bayesian probability that first infusion at age 45 years will result in greater increase in eGFR after 5 years on ERT than first infusion at age 18 years = 55%.
When data from male and female patients were fitted separately to the Bayesian model, the trajectories for the genders were very different. Both male and female patients with Fabry disease have improved eGFR on treatment relative to untreated patients, but the model suggests that effect in male patients is of a smaller magnitude than in females. Also, female patients who commence treatment later in life are predicted to continue to have improved eGFR for at least 5 years, whereas a plateauing of treatment effect is predicted in all other cases. The Bayesian probability that first infusion at age 45 years will result in greater increase in eGFR after 5 years on ERT than first infusion at age 18 years, is 54% in both males and females.
The paucity of data available means that estimates of the effect of ERT in children have very wide CIs and little power to detect an effect.
We were also able to explore the association between time on ERT and the risk of proteinuria. After adjusting for age and whether or not the patient was taking an ACE inhibitor, there was a statistically significant association between a decreased risk of proteinuria in adults and time on ERT when time on ERT was treated as a categorical (p < 0.001) variable. However, when time was treated as a continuous variable, no significant relationship was seen (edf = 1.0; p = 0.16).
Hearing
Hajioff and colleagues135 examined the effect of agalsidase alpha on hearing loss in 15 male patients with Fabry disease in a 6-month randomised trial, followed by an open-label extension for a further 24 months. After 24 months, they reported that hearing deterioration at baseline was reversed in 15 out of 20 ears.
In this study, we categorised hearing loss according to whether or not patients required a hearing aid. A Kaplan–Meier model shows a similar profile for patients on and off ERT, and indicates that by the age of 60 years, approximately 40% of patients with Fabry disease require a hearing aid. There was no statistically significant association between treatment status and the probability of requiring a hearing aid (the HR for ERT relative to not on ERT = 0.96; 95% CI 0.28 to 3.18; p = 0.95).
Transient ischaemic attack/stroke
Patients with Fabry disease are reported to have an increased risk of strokes and TIAs at an early age. 128,255 These can be seen in some patients as the first disease event. 256
Several studies have estimated the incidence of stroke in various small cohorts of patients with Fabry disease. Vedder and colleagues257 reported that 12 of 25 males (48%) and 13 of 41 females (32%) had experienced a cerebrovascular accident or lacunar stroke, at a median age of 46 and 52 years, respectively. Gupta and colleagues125 reported that 4 of 54 female patients with Fabry disease (7%) had experienced strokes, at a median age of 51 years, and Grewal and colleagues258 reviewed various types of imaging data and reported that 8 of 33 people with Fabry disease (24%) had experienced strokes at a median age as low as 26.5 years. Using data from the Fabry Outcome Survey (FOS) Registry, Mehta and Ginsberg259 reported that the overall prevalence of ischaemic stroke or TIAs was 13%, with events tending to occur at an early age. Prevalence of ischaemic strokes among male Fabry patients aged 25–44 years was 12 times that experienced in the general population. Clearly, the cumulative incidence in any cohort will depend on the age distribution.
In this study, patients were categorised according to whether or not they had had one or more TIAs or strokes. At recruitment, 30 patients (almost 10% of participants, 18 males and 12 females) were reported as having had a TIA or stroke. No further strokes were reported during the study period.
Similar to Gupta and colleagues,125 our data suggests that patients with Fabry disease, on or off ERT, have a low probability of having a TIA or stroke before the age of 40 years but that the risk increases thereafter.
We found no statistically significant association between treatment status and the probability of having a TIA or stroke (HR = 2.08; 95% CI 0.42 to 10.2; p = 36).
Pain
A number of previous studies have reported improvements in pain related to the use of ERT. In a cohort study of 545 patients on the Fabry Outcome Survey, treatment with agalsidase alpha was reported to improve pain scores and QoL. 142 Eng and colleagues136 also reported that ‘overall pain’ and ‘present pain intensity’ scores significantly improved after five infusions at all doses in a dose escalation study. In a double-blinded, placebo-controlled RCT of 26 males with Fabry disease, statistically significant reduction in mean neuropathic Pain Severity Score was reported in children treated with agalsidase alpha, whereas no significant change was measured in the placebo group. 134 The reduction in pain severity was reported to be sustained in the 3.5-year extension of this study. 148
We assessed pain using scores on the BPI. This produces scores for both severity of pain and the degree to which in interferes with activities. We found that, after adjustment, there was no statistically significant association between time on ERT and Pain Severity Scores. However, there was a statistically significant reduction in the extent to which pain interfered with activity, with duration on ERT (p < 0.001).
Fatigue
Fatigue has been reported as an important problem in people with Fabry disease. Ramaswami and colleagues152 reported that over 40% of patients aged under 18 years said that fatigue was a significant problem.
Guffon and colleagues260 reported results from a retrospective survey of 17 patients (mean age 34.7 years) treated with agalsidase beta for a mean of 18.76 months. An eight-item retrospective questionnaire was developed to assess the effect of ERT on pain severity, heat tolerance, physical activity, fatigue and psychological status. The change in mean score for fatigue reported for patients was from 5.53 to 3.71 (p = 0.046), suggesting an improvement in fatigue experienced by this group of patients, although the design of the study makes the results difficult to interpret.
In contrast, we found that, after adjustment for age, increasing duration of ERT was statistically significantly associated with a worsening of scores on the FSS. Further analysis suggested that the relationship between fatigue severity and time on ERT was curvilinear (edf = 2.19; p = 0.02) (see Figure 44), with a gradual increase in fatigue scores during the first 4 years after commencing treatment, but becoming more rapid thereafter.
Quality of life
In the previously discussed cohort study, Beck and colleagues142 reported that treatment with agalsidase alpha improved quality life as assessed by the EQ-5D in patients with Fabry disease. Eng and colleagues136 reported that patients who received agalsidase beta in a double-blind RCT experienced significant improvements in two components of the SF-36 (physical role and emotion role), whereas the patients in the placebo group had significant improvement in the physical role and body pain components of the SF-36.
We found statistically significant associations between duration of treatment and worse scores for both PCS and MCS of the SF-36.
One of the limitations of this study is the lack of historical QoL data. Virtually all patients with Fabry disease are on treatment and so we have few data from untreated patients. Analyses can therefore compare only the effects on QoL of different durations of treatment rather than being able to assess any change in QoL when patients first go on ERT, or the difference in QoL of patients on treatment, compared with those not on treatment.
Comparison of agalsidase alpha and agalsidase beta
There has been considerable controversy about the relative effectiveness of the two different agalsidase preparations available.
A trial by Vedder and colleagues149 compared the efficacy and tolerability of the two agalsidase preparations administered at identical protein dose in a open-label RCT. Thirty-four patients with Fabry disease were treated with either agalsidase alpha or agalsidase beta at equal dose of 0.2 mg/kg biweekly. The primary end point was reduction in LVM after 12 and 24 months of treatment. After 12 and 24 months of treatment, no reduction in LVM was seen in either group. Similarly, no changes in glomerular filtration rate, pain or Gb3 levels were found for either group following treatment. Treatment failure (defined as progression of renal disease, progression of cardiac disease, occurrence of a new cardiovascular attack, or new lacunar infarctions on MRI) within 24 months of therapy was seen in 8 of the 34 patients: six male patients (three in each treatment group) and two female patients (both agalsidase alpha). The occurrence of treatment failures did not differ between the two treatment groups.
We were able to compare the outcome in patients who were recorded as being on one or other of these preparations. Patients were analysed in a group according to which of the two preparations they were receiving at the earliest point recorded in our study data. We were not able to take account of the effect of changes in treatment allocation over time. None of these analyses suggested a statistically significant difference in effect between the two preparations.
Costs associated with Fabry disease
As with all other conditions investigated in this study, we were keen to capture the wider costs of care falling on the public sector in addition to the costs associated with ERT.
Based on patients’ self-reported health- and social-care service use, the annual cost of caring for people with Fabry disease, excluding the purchase cost of ERT, was estimated at £3300 for an adult and £1300 for a child. These costs, however, are dwarfed by the cost of the therapies; the mean annual cost of ERT for adults with Fabry disease is either £108,242 or £120,840, depending on which ERT drug is used. For children, the mean annual cost of ERT is £79,478 or £89,199.
From the longitudinal regression modelling of costs, there was no statistically significant association (i.e. p-value < 0.05) between time on ERT and either total NHS and social-care costs, hospital-care costs, or non-hospital-care costs for patients with Fabry disease. The tabulated results of these analyses are available on request from the study authors.
Owing to these high associated costs, and the lack of measureable effect of ERT on either clinical outcomes or HRQoL measures, it was infeasible to conduct either a cost-effectiveness or cost–utility analysis. As they apply to all six LSDs, the limitations of these cost estimates are summarised and discussed in Chapter 9.
Chapter 5 Results – mucopolysaccharidosis type I (MPS I)
Patient characteristics
At the start of the study 126 patients were identified by the treating centres as having MPS I. Of these, 111 patients were deemed eligible for inclusion and 72 patients (65% of those deemed eligible) were approached in clinics and invited to participate. Sixty-eight patients (94% of those approached), comprising 34 males and 34 females, consented to participate in the study. Patient demographic characteristics are presented in Tables 85 and 86.
| Patient characteristic | |
|---|---|
| Gender | |
| Male, n | 7 |
| Female, n | 13 |
| Type of MPS I | |
| Hurler, n | 8 |
| Hurler–Scheie, n | 10 |
| Scheie, n | 2 |
| Age at recruitment (years) | |
| Mean (SD) | 24.7 (6.2) |
| Median (min.–max.) | 24.7 (16.4–37.1) |
| Age at diagnosis (years) | |
| Mean (SD) | 6.74 (8.8) |
| Median (min.–max.) | 3.64 (0–35.1) |
| Initial treatment | |
| ERT (laronidase), n | 7 |
| HSCT, n | 8 |
| Clinical trial of ERT,a n | 5 |
| Type of MPS I on ERT | |
| Hurler, n | 0 |
| Hurler–Scheie, n | 10 |
| Scheie, n | 2 |
| Type of MPS I with HSCT | |
| Hurler, n | 8 |
| Hurler–Scheie, n | 0 |
| Scheie, n | 0 |
| Age at starting ERT (years) | |
| Mean (SD) | 20.86 (8.56) |
| Median (min.–max.) | 18.7 (10.1–34.9) |
| Age at HSCT (years) | |
| Mean (SD) | 1.15 (0.75) |
| Median (min.–max.) | 1.05 (0.45–2.87) |
| Time on ERT at recruitment (years) (n = 12) | |
| Mean (SD) | 4.79 (2.5) |
| Median (min.–max.) | 4.68 (1.38–7.8) |
| Patient characteristic | |
|---|---|
| Gender | |
| Male, n | 27 |
| Female, n | 21 |
| Type of MPS I | |
| Hurler, n | 35 |
| Hurler–Scheie, n | 12 |
| Scheie, n | 1 |
| Age at recruitment (years) | |
| Mean (SD) | 7.26 (4.1) |
| Median (min.–max.) | 6.41 (0.58–5.6) |
| Age at diagnosis (years) | |
| Mean (SD) | 1.09 (0.90) |
| Median (min.–max.) | 0.77 (0.05–3.71) |
| Initial treatment | |
| ERT (laronidase), n | 10 |
| HSCT, n | 36 |
| Clinical trial of ERT,a n | 2 |
| Type of MPS I on ERT | |
| Hurler, n | 0 |
| Hurler–Scheie, n | 11 |
| Scheie, n | 1 |
| Type of MPS I with HSCT | |
| Hurler, n | 35 |
| Hurler–Scheie, n | 1 |
| Scheie, n | 0 |
| Age at starting ERT (years) | |
| Mean (SD) | 5.36 (4.21) |
| Median (min.–max.) | 3.38 (0.48–13.3) |
| Age at HSCT (years) | |
| Mean (SD) | 1.32 (0.59) |
| Median (min.–max.) | 1.23 (0.61–3.52) |
| Time on ERT at recruitment (years) (n = 12) | |
| Mean (SD) | 3.32 (1.9) |
| Median (min.–max.) | 4.07 (0.09–6.1) |
At recruitment, 48 of the participants were children (aged ≤ 16 years) and 20 were adults. The average age of children at recruitment to the study was 7.3 (range 0.58–15.6) years and that of adults was 24.7 (range 16.4–37.1) years.
We collected data from all patients at the time of recruitment. Data were also collected from 55 patients at their 12-month appointment, 20 patients at their 24-month appointment and 2 patients at their 36-month appointment. We collected retrospective data from 59 patients at up to 12 time points.
As seen in Tables 85 and 86, 43 patients (8 adults and 35 children) had the severe Hurler phenotype (MPS IH), which is characterised by early and progressive CNS involvement. Three patients (two adults and one child) had the attenuated Scheie phenotype (MPS IS) with no CNS involvement, and 22 patients (10 adults and 12 children) had the intermediate Hurler–Scheie phenotype (MPS IHS).
Of those with the severe MPS IH, 35 children had received a HSCT at an average age of 1.32 (range 0.61–3.52) years and eight of the adults had received a HSCT at an average age of 1.15 (range 0.45–2.87) years. Two of these patients had received ERT for a short duration prior to the transplant.
One child with MPS IHS received a HSCT, while the remaining MPS IHS patients all received ERT (laronidase), as did all three MPS IS patients. At recruitment, the average time on ERT was 3.32 (range 0.09–6.1) years for children and 4.79 (1.38–7.8) years for adults.
Key markers of disease progression for mucopolysaccharidosis type I
The following measures were identified as key markers of disease progression:
-
FVC
-
mobility and 6-minute walk test
-
stature (height and weight)
-
hearing
-
heart valve disease
-
carpal tunnel syndrome (CTS)
-
cervical cord compression
-
spleen and liver size.
In addition, adults completed the SF-36, EQ-5D, FSS and the Service Use and Costs Questionnaire, whereas children or their carers completed the age-appropriate PedsQL questionnaire. Carers of children or adults were asked to complete the Service Use and Costs Questionnaire and the CSI.
Longitudinal models were fitted to assess relationships between continuous measures of function and length of time on ERT, after adjustment for age and clustering by centre. In the basic models, the effect of time on ERT was treated as a linear effect owing to the small number of data points. Further analysis was conducted to explore the possibility that time spent on ERT could have a non-linear effect on function. The models were extended to describe the trajectories of patients receiving a HSCT. Patients contributed data points to the model both before and after receiving a HSCT or starting ERT (where applicable). Clearly, a simplistic comparison of the effects of ERT and receiving a HSCT would be inappropriate given the underlying differences in the populations involved. An alternative model was examined in which the type of MPS I was added as a covariate, but this did not change the conclusions about the effect of treatment with ERT (results not shown).
Kaplan–Meier survival curves were estimated to illustrate differences in the age at first recorded occurrence of binary events that could be considered progressive (e.g. restricted mobility, abnormal hearing, valve disease, CTS, enlarged liver and enlarged spleen). Treatment group differences in survival function were tested using Cox regression models. Individual patients contributed intervals of time at risk to more than one of the treatment categories (no treatment, ERT, HSCT) as appropriate.
One would anticipate that the effect of time since receiving a HSCT might be non-linear. It is unclear what relation there should be and we have insufficient data to clearly establish the shape of the relationship. In the basic models we have assumed this is a linear relationship and as will be seen from the spline plots there is no strong evidence that this is inappropriate. However, we do recognise that if more data were available we might be led to a different set of assumptions.
For all outcomes, we restricted our analyses to models where treatment groups contain five patients or more.
Summary of results
All analyses in MPS I were hampered by a paucity of data related to the low number of patients recruited and lack of data recorded on key outcomes for a substantial proportion of these patients. This results in low power to detect any effects. The power is further diminished by the necessity of dealing with the heterogeneity within MPS I reflected by the different subtypes and the strong association between subtype and mode of treatment; of the 44 patients who received a HSCT, 43 had a more severe form of MPS I (i.e. Hurler subtype), whereas none of the 24 who had received ERT as their initial treatment had this phenotype.
We examined potential associations between treatment and FVC, mobility and 6-minute walk test, stature (height and weight), hearing, the presence/absence of heart valve disease and presence of CTS. In addition we examined the relationship between treatment and QoL assessed by either SF-36, EQ-5D or PedsQL depending on age, with DQ in children, and with the CSI.
We found no statistically significant relationship between time on ERT and any of these outcomes with the exception of an improvement (increase) in the social functioning subscale of the PedsQL.
Similarly, we found a statistically significant association between HSCT and an improvement of two subscales of the PedsQL, although not with the overall score. No other statistically significant associations with time since HSCT were observed.
Per cent of predicted forced vital capacity
Fourteen patients (12 on ERT and two HSCT recipients) were able to complete an upright FVC measurement. Results are reported as a per cent of the predicted forced vital capacity volume (FVC%). A total of 52 FVC measurements were recorded for these patients across all time points and the range of these measurements was 11–117%.
A longitudinal model was fitted to assess the linear relationship between FVC and time on ERT, after adjusting for age (Table 87). The model included a linear effect of time since HSCT making it possible to describe the trajectories of patients after HSCT.
| N Data | Estimate of change in FVC% | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 19 | 0.00 | |||
| Female | 33 | –12.19 | 14.3 | –40.2 to 15.8 | 0.39 |
| Current age | |||||
| Linear effect/year | –0.78 | 0.70 | –2.15 to 0.59 | 0.26 | |
| Time on ERT | |||||
| Linear effect/year | 1.69 | 1.16 | –0.58 to 3.96 | 0.15 | |
| Time since HSCT | |||||
| Linear effect/year | –0.78 | 1.21 | –3.15 to 1.59 | 0.52 | |
| Variance components | |||||
| Individual | 513.4 | ||||
| Centre | 0.0 | ||||
| Residual | 60.1 | ||||
As can be seen in Table 87, FVC was not significantly associated with age (p = 0.26), time on ERT (p = 0.15) or time since HSCT (p = 0.52). Females had a reduced FVC compared with males, but this difference was not statistically significant (p = 0.39).
No evidence was found for a non-linear association between FVC and time on ERT (edf = 1.0; p = 0.15) (Figure 47) or time after HSCT (edf = 1.0; p = 0.52) (Figure 48), although it is important to note that the data are very sparse, making clear interpretation difficult.
FIGURE 47.
The age-adjusted association between time on ERT and FVC% in adults and children with MPS I (time on ERT treated as a continuous variable).
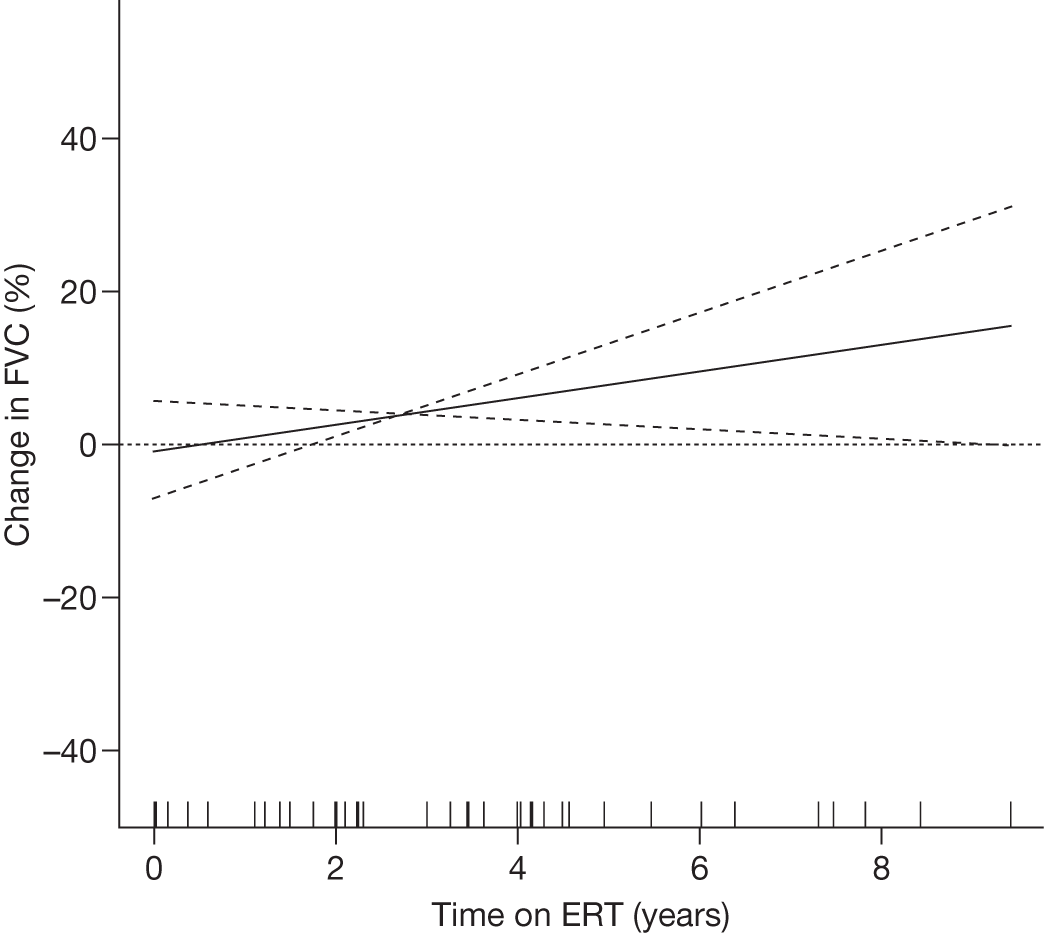
FIGURE 48.
The age-adjusted association between time since HSCT and adults and children with MPS I (time since HSCT treated as a continuous variable).
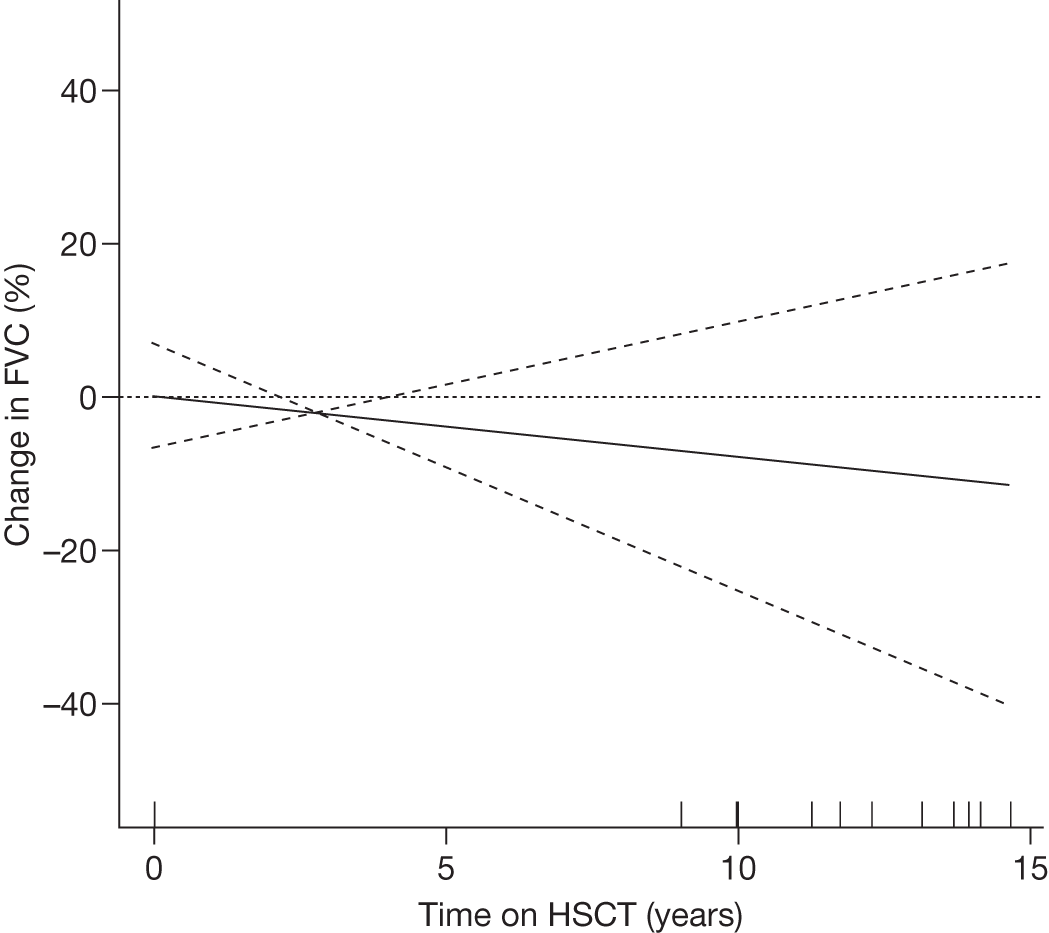
Mobility
Patients were categorised as being mobile (i.e. they could walk for 5 metres or stand for 6 minutes unaided), as having restricted mobility (i.e. can walk aided with one or two sticks) or as being immobile. Figure 49 shows Kaplan–Meier curves for the age at the first recorded instance of the patient having restricted mobility (or being immobile). The curves illustrate the risk of moving from being mobile to having restricted mobility at different ages, by treatment type.
FIGURE 49.
Risk of having restricted mobility by age and treatment status (ERT or HSCT) for people with MPS I (Kaplan–Meier curve). The curves were truncated at 20 years of age owing to fewer than five patients being at risk of restricted mobility in each treatment group.
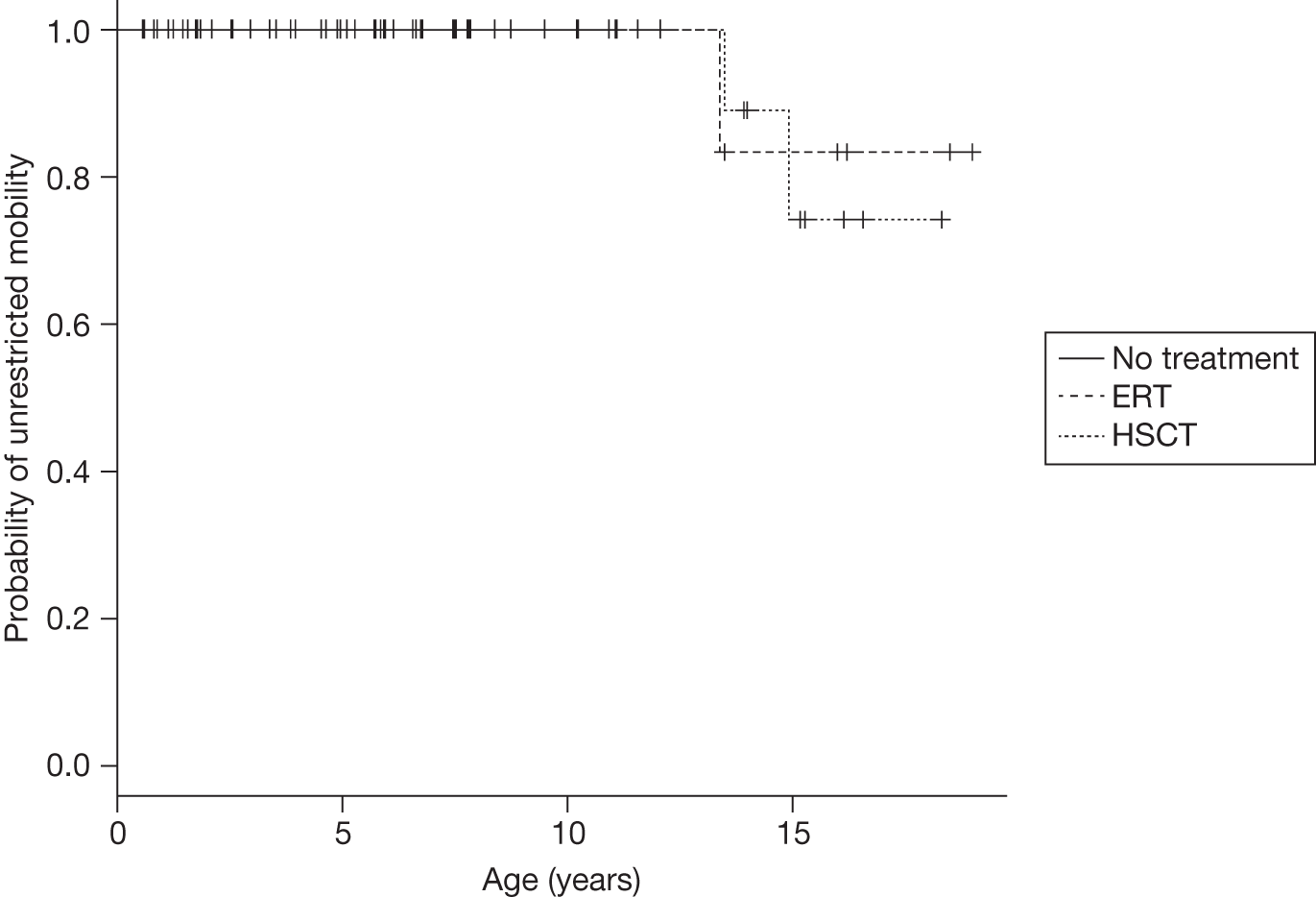
All MPS I patients for whom there was mobility data contributed pre-treatment data points; those patients who then had a HSCT or started ERT contributed data to the respective treatment group curves. The two patients who received ERT prior to having a HSCT contribute to both curves at the appropriate time points. The tick marks correspond to ages at which individual patients drop out of the risk set for one of the treatment groups and so are considered censored. For ease of interpretation, the survival curves were truncated at ages for which fewer than five patients were in the risk set to avoid large imprecise jumps in the curves.
As noted in Summary of results it is important to be clear that HSCT is primarily used in patients with Hurler disease, whereas the majority of those receiving ERT are classified as having Hurler–Scheie disease or Scheie disease.
The model indicates that approximately 20% of patients who have either had a HSCT or are on ERT have restricted mobility by the time they are 15 years old. There was no significant difference in the risk of having restricted mobility for patients on ERT compared with HSCT (the HR for ERT relative to HSCT = 0.51, 95% CI 0.04 to 5.88; p = 0.58); of course any such comparison would need to be treated with extreme caution owing to the differences in phenotype.
Beyond the model, three patients commenced treatment on ERT after the age of 18 years. One of the three was immobile at the time of commencing treatment (aged 25 years), whereas the other two were mobile. There was no change seen in the mobility status of the three patients during the data collection period.
6-minute walk test
Estimates of 6-minute walk tests were collected for 22 patients (14 patients were on ERT and 8 patients were HSCT recipients). Eighty-two estimates of distance walked in 6 minutes were recorded across all time points, and these estimates ranged from 150 to 547 m (Table 88).
| N Data | Estimate of change in distance walked (m) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 38 | 0.00 | |||
| Female | 44 | 4.41 | 44.9 | –83.6 to 92.4 | 0.93 |
| Current age | |||||
| Linear effect/year | 3.04 | 3.02 | –2.87 to 8.96 | 0.32 | |
| Time on ERT | |||||
| Linear effect/year | –7.05 | 5.86 | –18.5 to 4.43 | 0.23 | |
| Time since HSCT | |||||
| Linear effect/year | 1.18 | 4.87 | –8.36 to 10.72 | 0.81 | |
| Variance components | |||||
| Individual | 15,420.3 | ||||
| Centre | 0.00 | ||||
| Residual | 2384.2 | ||||
The linear model provided no evidence for an association between the distance walked and current age (p = 0.32). Distance walked was not significantly associated with either time on ERT or time since HSCT (p = 0.23 and p = 0.81, respectively).
Figures 50 and 51 show no evidence for a non-linear association between distance walked and time on ERT (edf = 1.0; p = 0.23) (see Figure 50) or time since HSCT (edf = 1.0; p = 0.81) (see Figure 51).
FIGURE 50.
The age-adjusted association between time on ERT and distance walked in people with MPS I (time on ERT treated as a continuous variable).
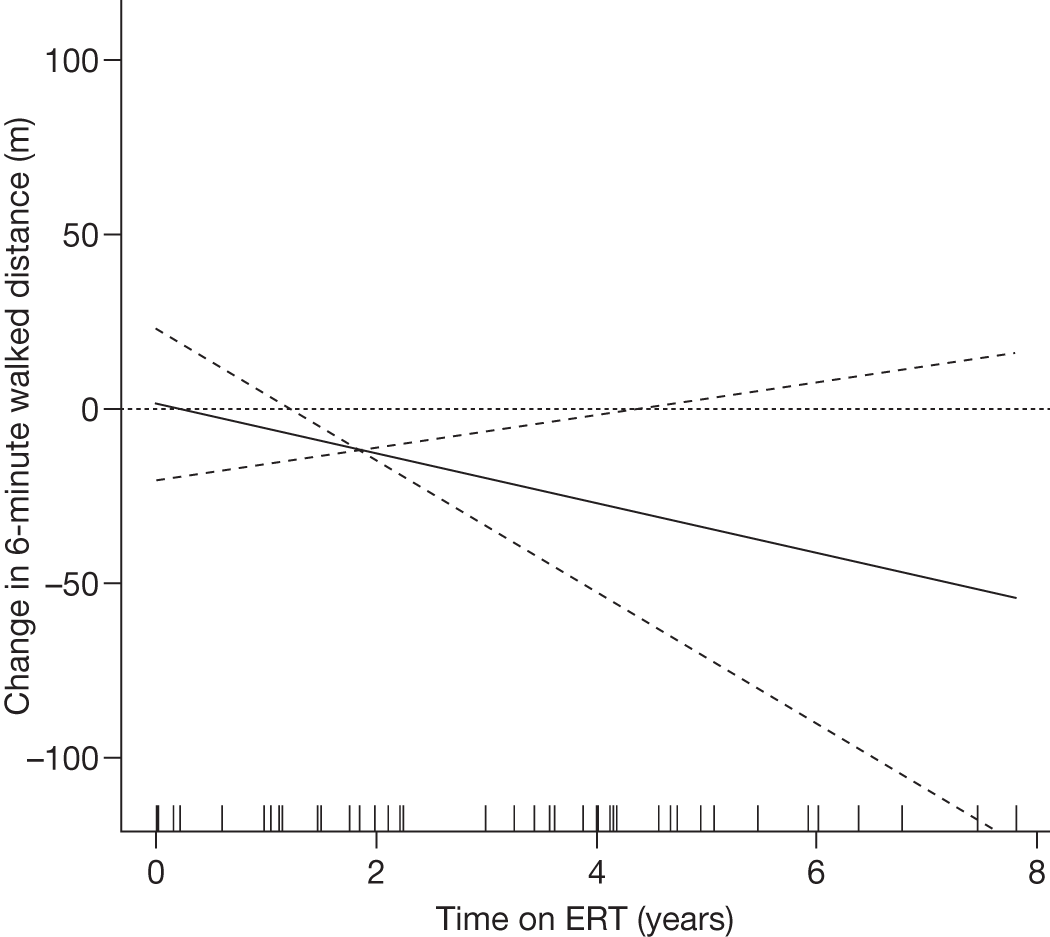
FIGURE 51.
The age-adjusted association between time since HSCT and distance walked in people with MPS I (time since HSCT treated as a continuous variable).
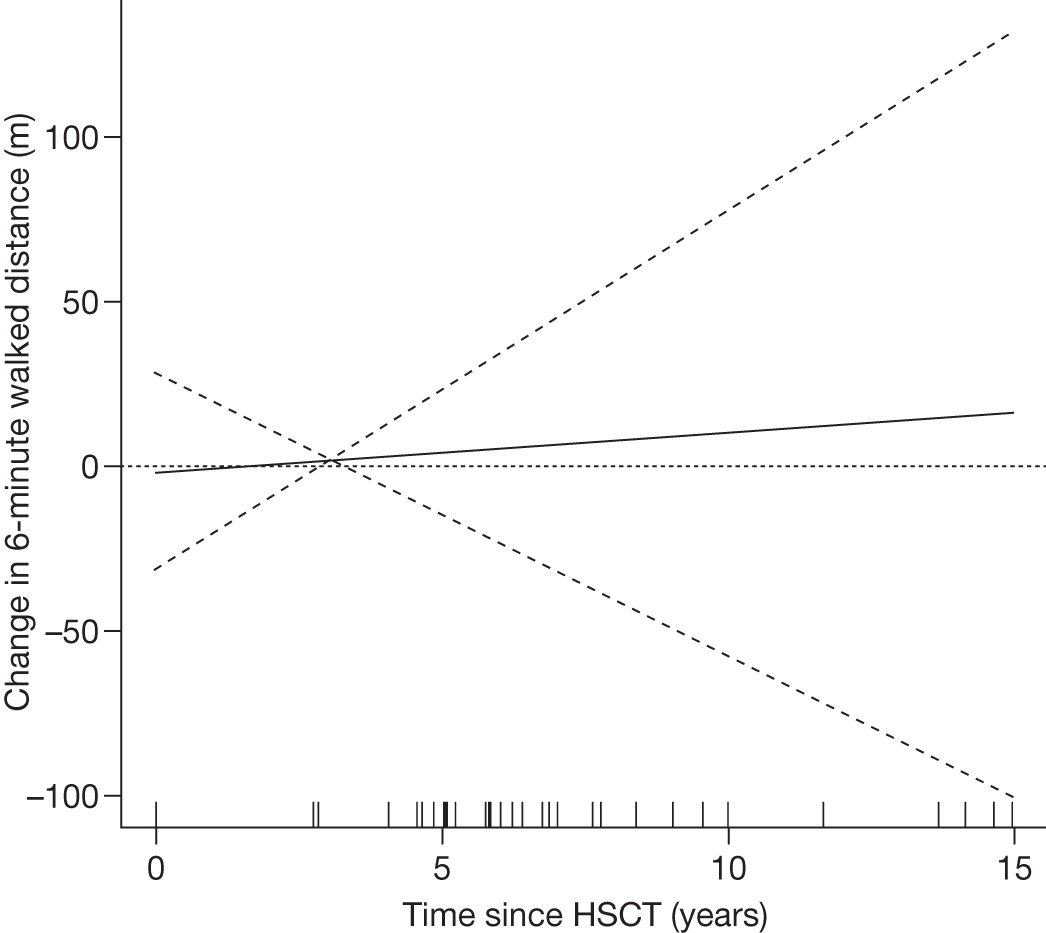
Hearing
The MPS I patients were classified as having normal or impaired hearing according to standard definitions. Hearing data were collected for 56 patients, with 40 patients experiencing impaired hearing across all time points.
Kaplan–Meier curves were not estimated for patients on ERT or for patients not yet treated, because the risk sets for these treatment groups had fewer than five patients at all ages. Figure 52 shows individual timelines for patients who received ERT and the age at which they first reported impaired hearing.
FIGURE 52.
Timelines of patients at risk of hearing loss who received ERT.
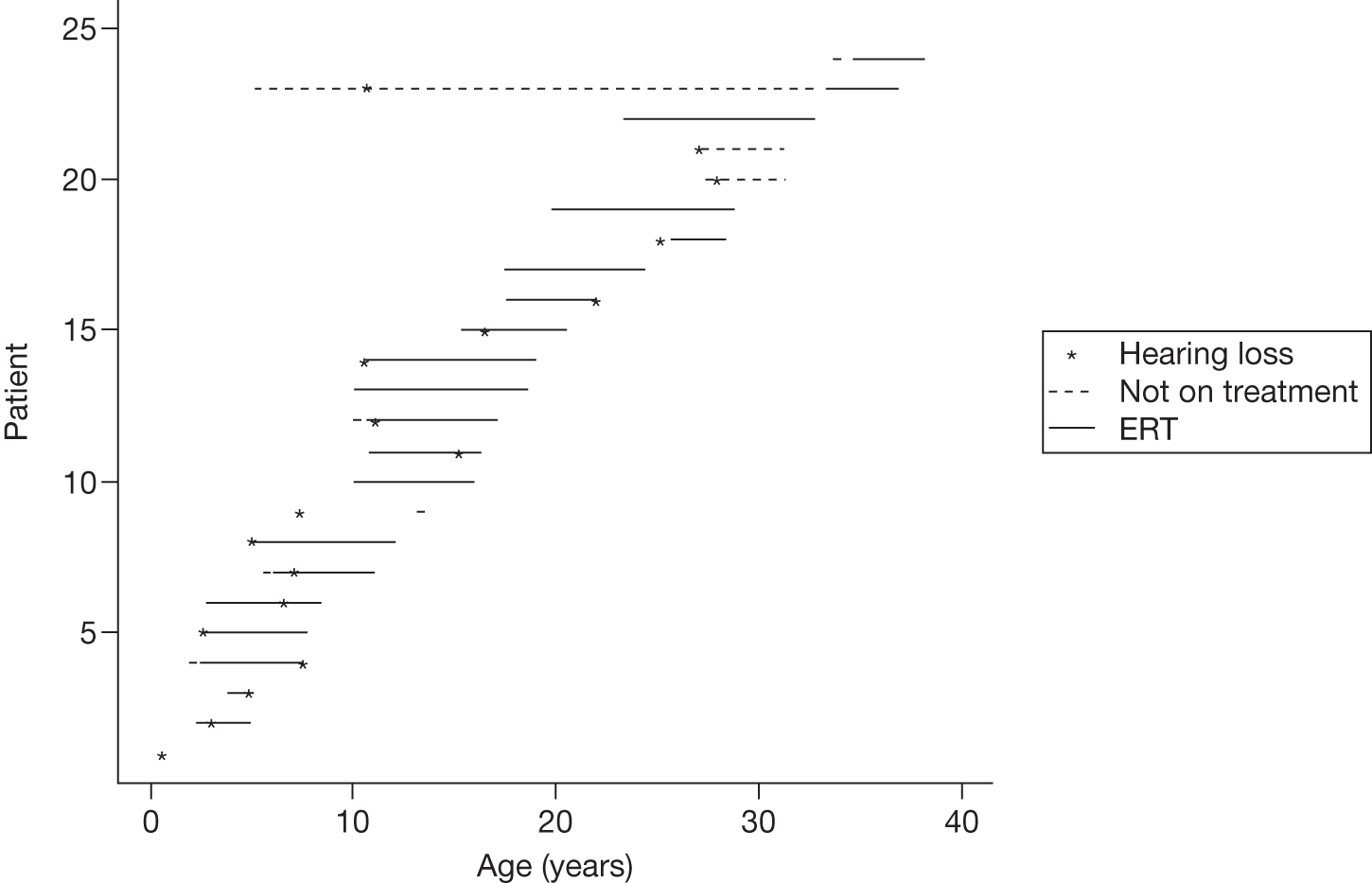
Figure 53 shows a Kaplan–Meier survival curve (and 95% CI) illustrating the probability of having impaired hearing by age for HSCT patients only.
FIGURE 53.
Risk of abnormal hearing by age and treatment status for children aged < 15 years old with MPS I (Kaplan–Meier curve). The Kaplan–Meier curve was truncated at 15 years of age because fewer than five HSCT patients were at risk of impaired hearing.
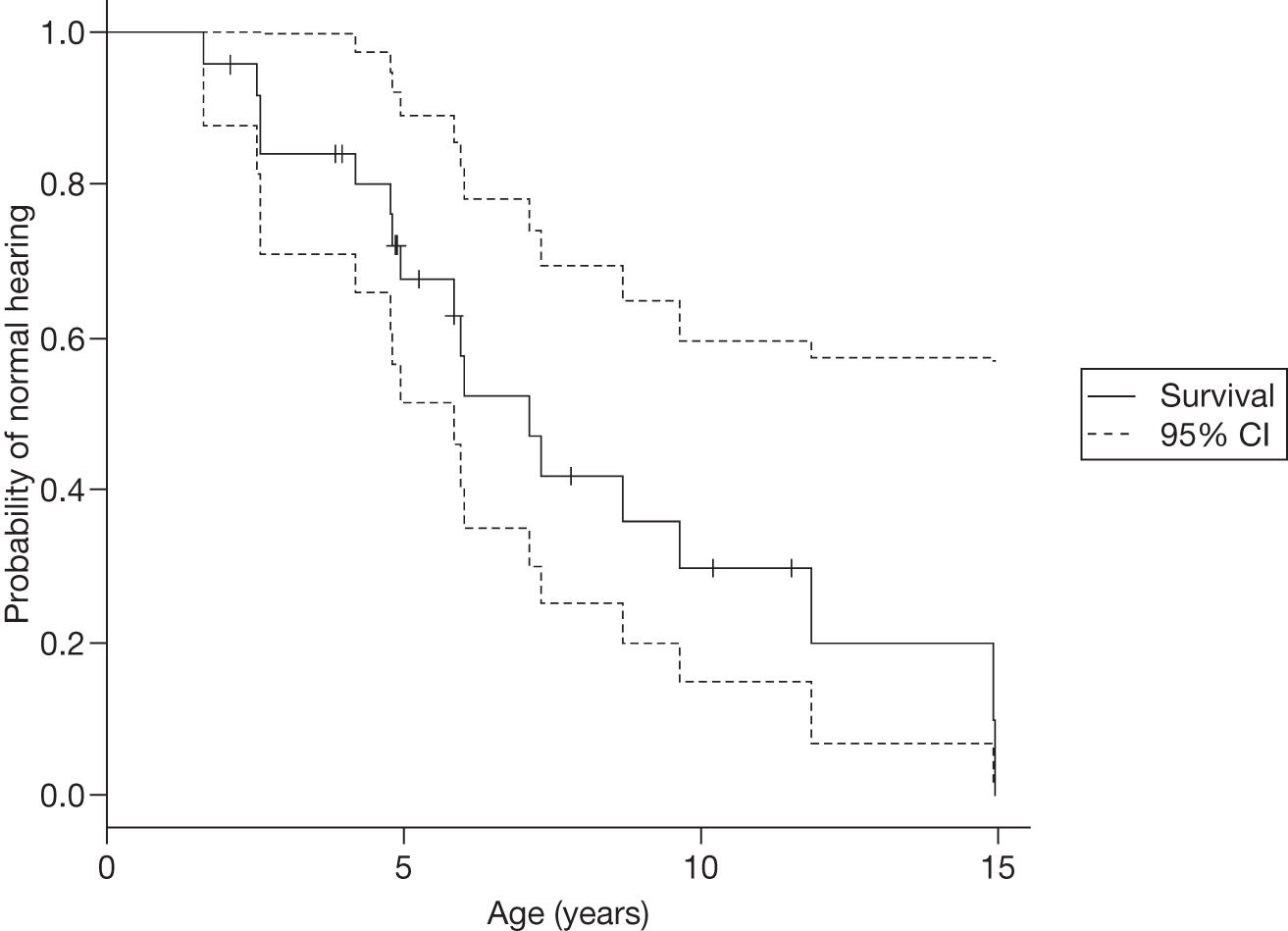
The model indicates that approximately 80% of HSCT patients have impaired hearing by the time they are 12 years old. The graph was truncated at the age 15 years, as the risk set dropped to five patients. Two of these patients went on to have impaired hearing before 18 years of age.
Height in children
Children’s height measurements were converted to z-scores using the British 1990 Growth Reference,261 with the Stata command ‘zanthro’ (StataCorp LP, College Station, TX, USA). This transformation allows one to examine the changes in height relative to the expected growth patterns for a child of the same age. Two hundred and twenty-two height measurements were recorded for 46 children (11 on ERT and 35 HSCT recipients), and these range from 48 to 158 cm, with a mean z-score of –1.62. This implies that these children are on average substantially shorter than their peers.
The model suggests that children’s height drops through the centiles over time (p < 0.001) (Table 89). After the adjustment for age, there was no statistically significant association between time on ERT and children’s height centiles (p = 0.16), or between time since HSCT (p = 0.25).
| N Data | Estimate of increment in height | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 122 | 0.00 | |||
| Female | 100 | –0.14 | 0.35 | –0.83 to 0.55 | 0.68 |
| Current age | |||||
| Linear effect/year | –0.17 | 0.05 | –0.27 to –0.07 | < 0.001 | |
| Time on ERT | |||||
| Linear effect/year | 0.14 | 0.09 | –0.04 to 0.32 | 0.16 | |
| Time since HSCT | |||||
| Linear effect/year | –0.06 | 0.05 | –0.16 to 0.04 | 0.25 | |
| Variance components | |||||
| Individual | 1.22 | ||||
| Centre | 0.12 | ||||
| Residual | 0.55 | ||||
Figure 54 shows no evidence for a non-linear relationship between children’s age-adjusted height and time on ERT (edf = 1; p = 0.18). However, data contributing to this model are sparse, as demonstrated by the wide CIs and the small number of tick marks on the rug plot. Similarly, Figure 55 suggests no evidence for a non-linear age-adjusted relationship between children’s height and time since HSCT (edf = 1; p = 0.17).
FIGURE 54.
The age-adjusted association between time on ERT and height z-scores in children with MPS I (time on ERT treated as a continuous variable).
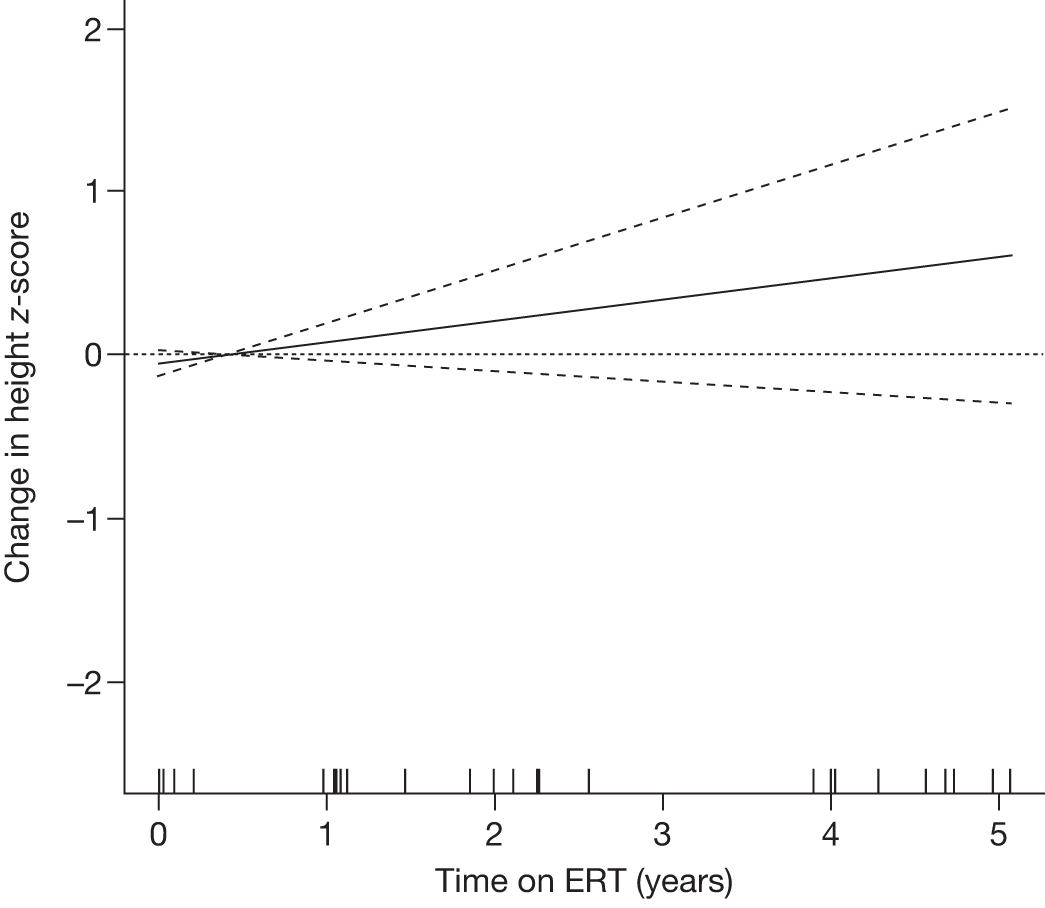
FIGURE 55.
The age-adjusted association between time since HSCT and height z-scores in children with MPS I (time since HSCT treated as a continuous variable).
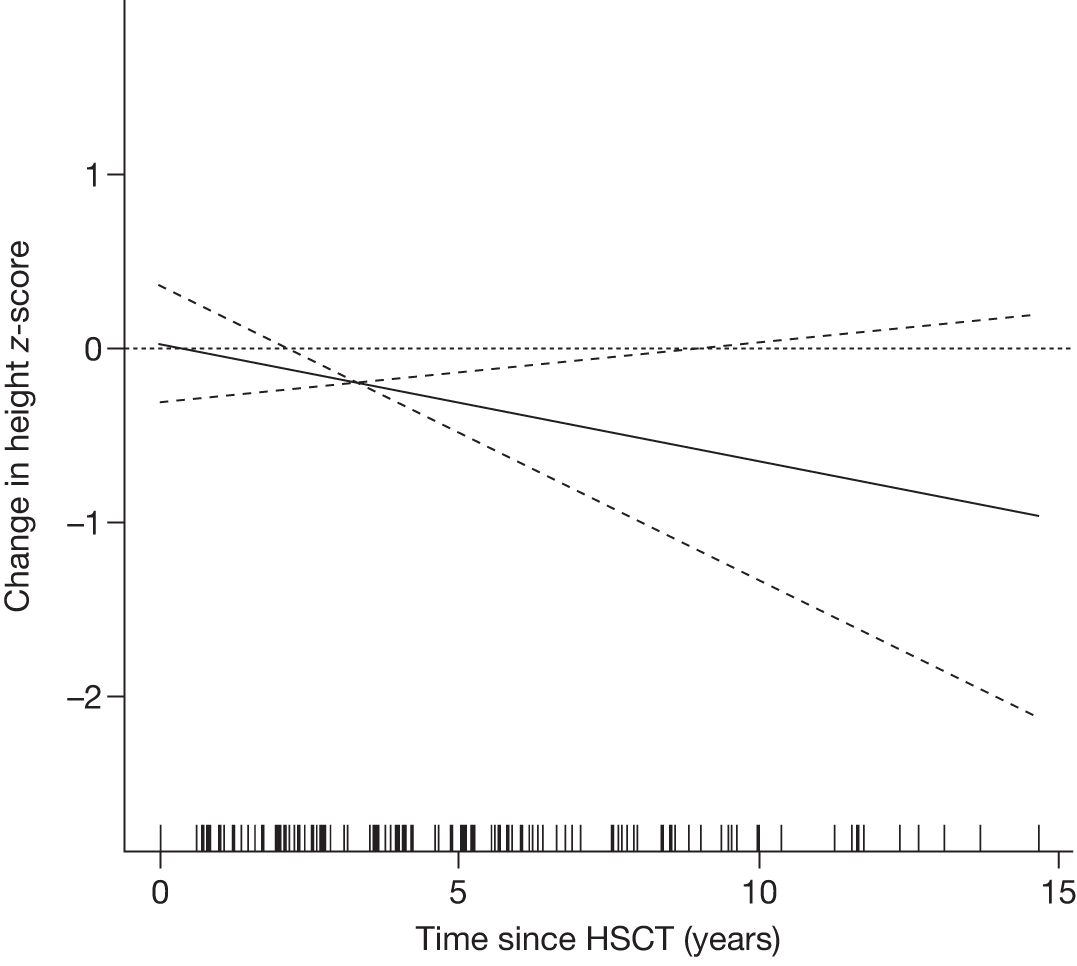
Weight
Children’s weights were converted to z-scores against 1990 UK norms261 using the ‘zanthro’ programme. Two hundred and forty-three weight measurements were recorded from 47 children (11 on ERT and 36 HSCT recipients) and these ranged from 6.9 to 60 kg, mean z-score –0.58. Although this suggests that these children weigh substantially less than their peers, on average, the discrepancy is considerably smaller than for height.
Overall, children’s weight z-scores reduced by an average of 0.12 standard deviations per year (p = 0.02) (Table 90). After adjusting for age, time on ERT was not associated with a significant change in weight centile (p = 0.77). Similarly, time since HSCT was not associated with a significant change in weight centile (p = 0.16).
| N Data | Estimate of increment in weight | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 138 | 0.00 | |||
| Female | 105 | –0.44 | 0.38 | –1.18 to 0.30 | 0.26 |
| Current age | |||||
| Linear effect/year | –0.12 | 0.05 | –0.22 to –0.02 | 0.02 | |
| Time on ERT | |||||
| Linear effect/year | 0.03 | 0.10 | –0.17 to 0.23 | 0.77 | |
| Time since HSCT | |||||
| Linear effect/year | –0.07 | 0.05 | –0.17 to 0.03 | 0.16 | |
| Variance components | |||||
| Individual | 1.54 | ||||
| Centre | 0.00 | ||||
| Residual | 0.72 | ||||
Further exploration of the data provided no evidence for a non-linear association between weight-for-age z-score and time on ERT (edf = 1; p = 0.77) (Figure 56) or time since HSCT (edf = 1; p = 0.16) (Figure 57) although again these findings need to be interpreted in the light of sparse data as demonstrated by the wide CIs.
FIGURE 56.
The age-adjusted association between time on ERT and weight z-scores in children with MPS I (time on ERT treated as a continuous variable).
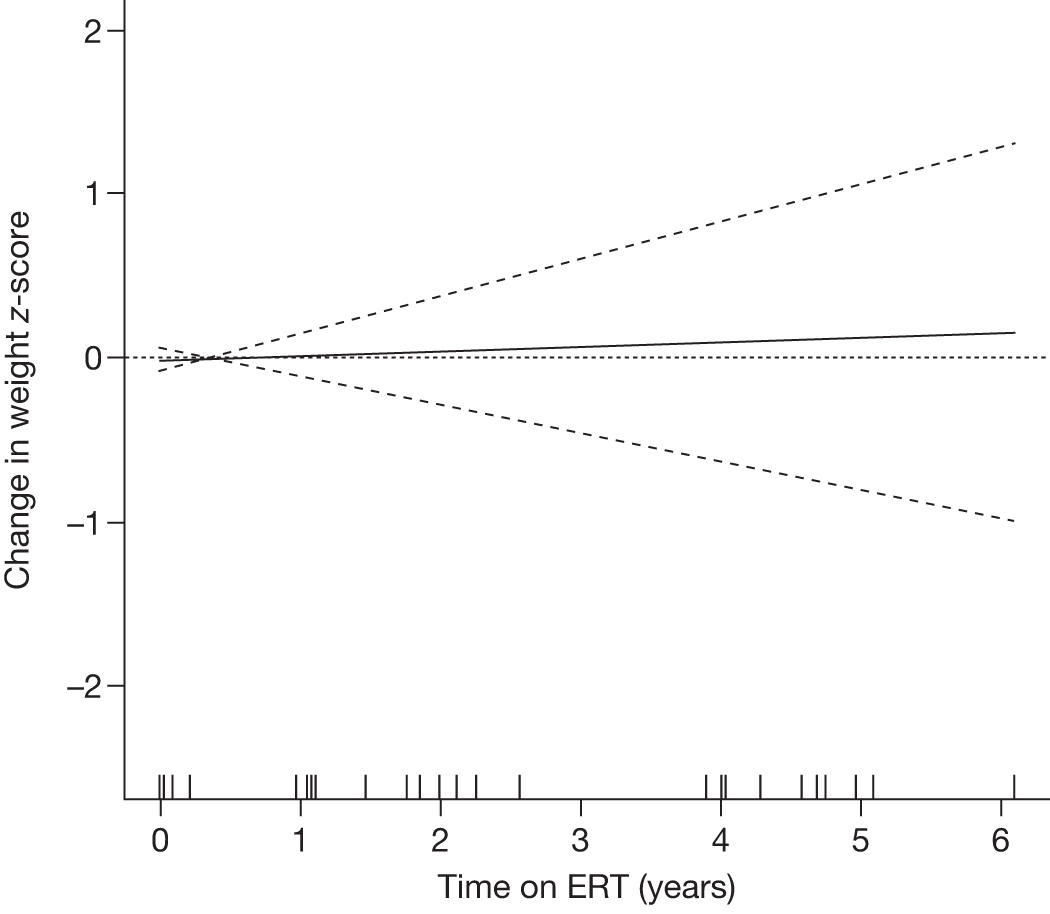
FIGURE 57.
The age-adjusted association between time since HSCT and weight z-scores in children with MPS I (time since HSCT treated as a continuous variable).
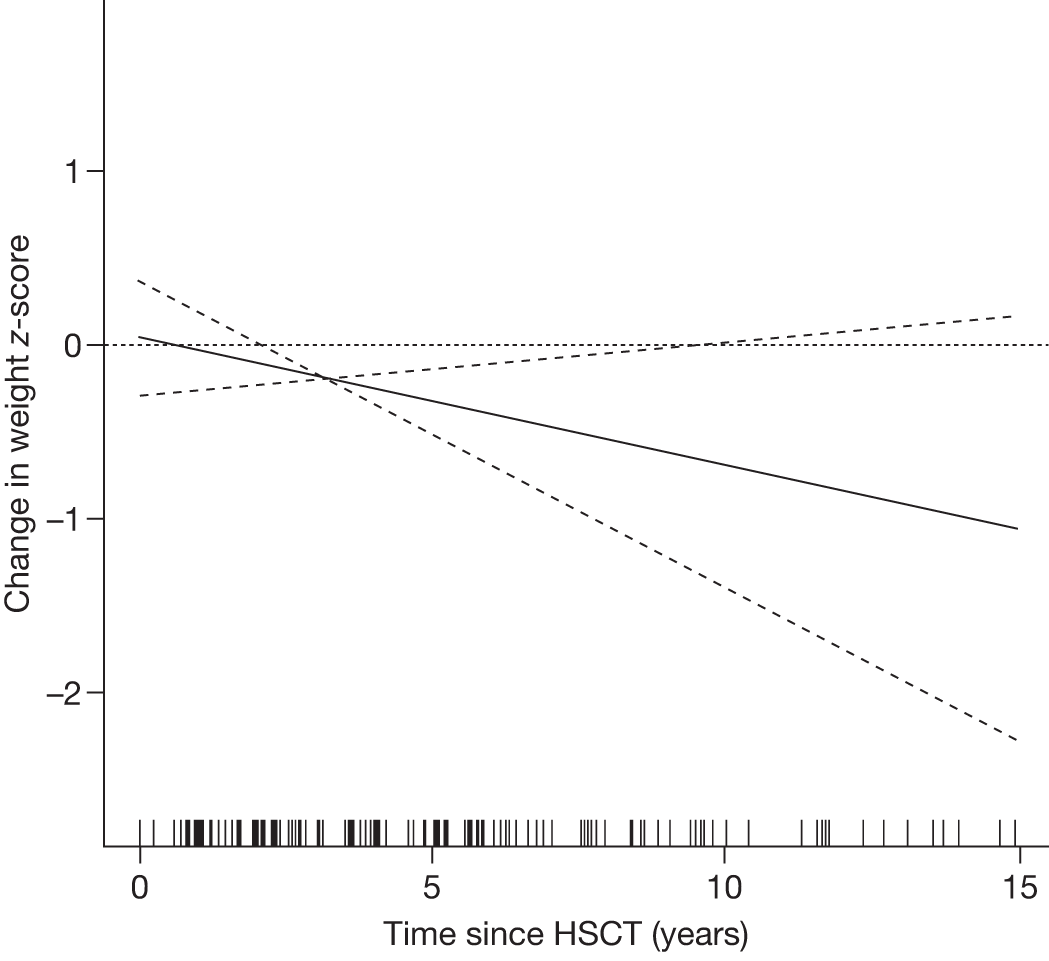
Cardiac valve involvement
The MPS I patients were categorised according to whether or not they were recorded as having cardiac valve involvement at a particular age. Figure 58 displays the timelines for the patients on ERT, showing the age at which they were first recorded as having valve disease. Nineteen of the 24 patients on ERT were classified as having valve disease across all time points. Kaplan–Meier curves were not estimated for patients on ERT or for patients not yet treated because the risk sets for these treatment groups had fewer than five patients at all ages.
FIGURE 58.
Age at first recording of valve disease among patients with MPS I who received ERT.
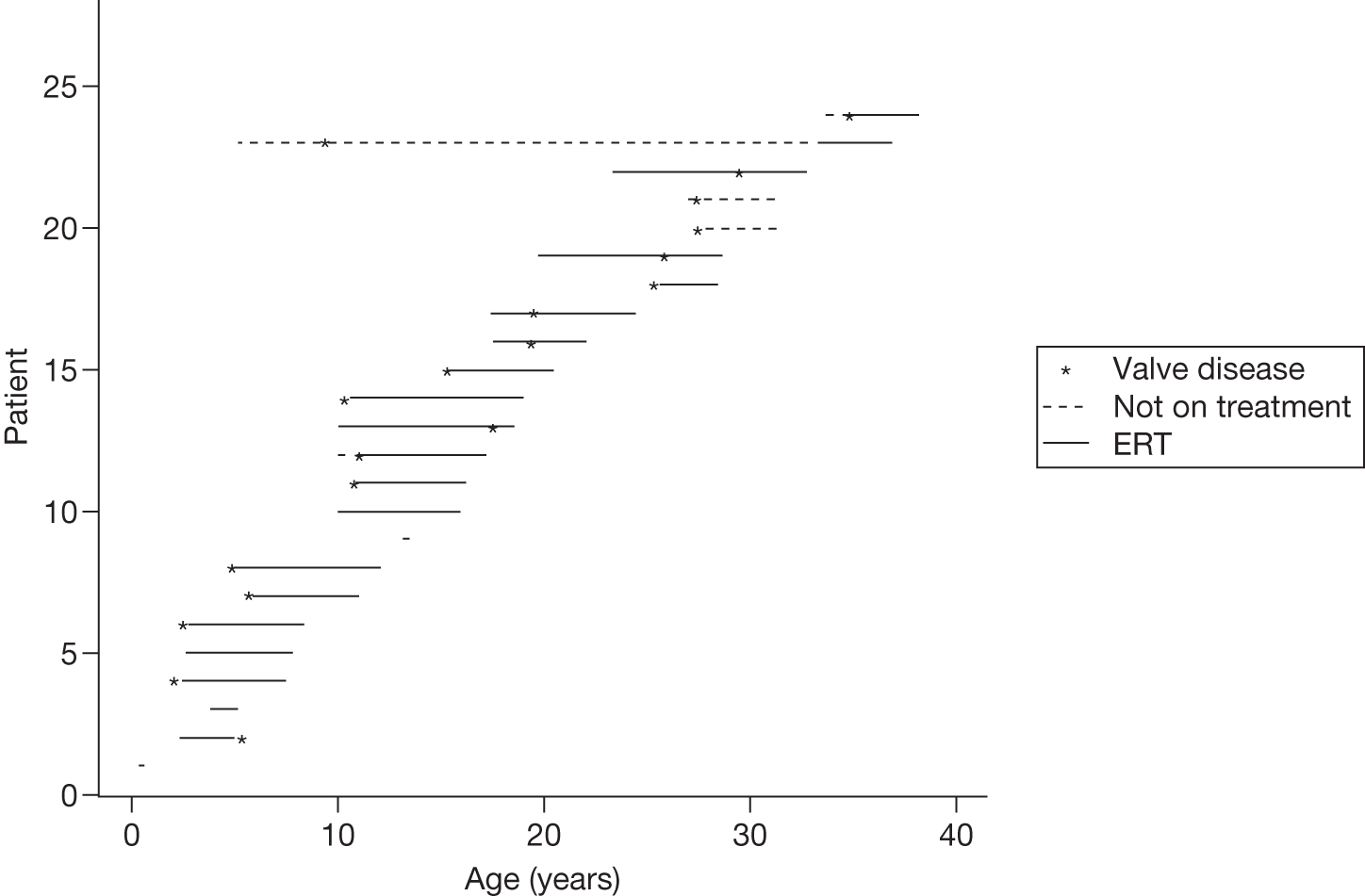
Figure 59 shows a Kaplan–Meier survival curve (and 95% CI) illustrating the probability of developing valve disease by age for HSCT patients only. The curve suggests approximately 80% of MPS I patients who have had a HSCT develop some form of valve disease by the time they are 9 years old. It is important to interpret these data in the light of the wide CIs around the survival curve.
FIGURE 59.
Risk of having valve disease by age in people with MPS I who have received a HSCT (Kaplan–Meier curve). The Kaplan–Meier curve was truncated at 10 years of age owing to fewer than five patients being at risk of developing valve disease.
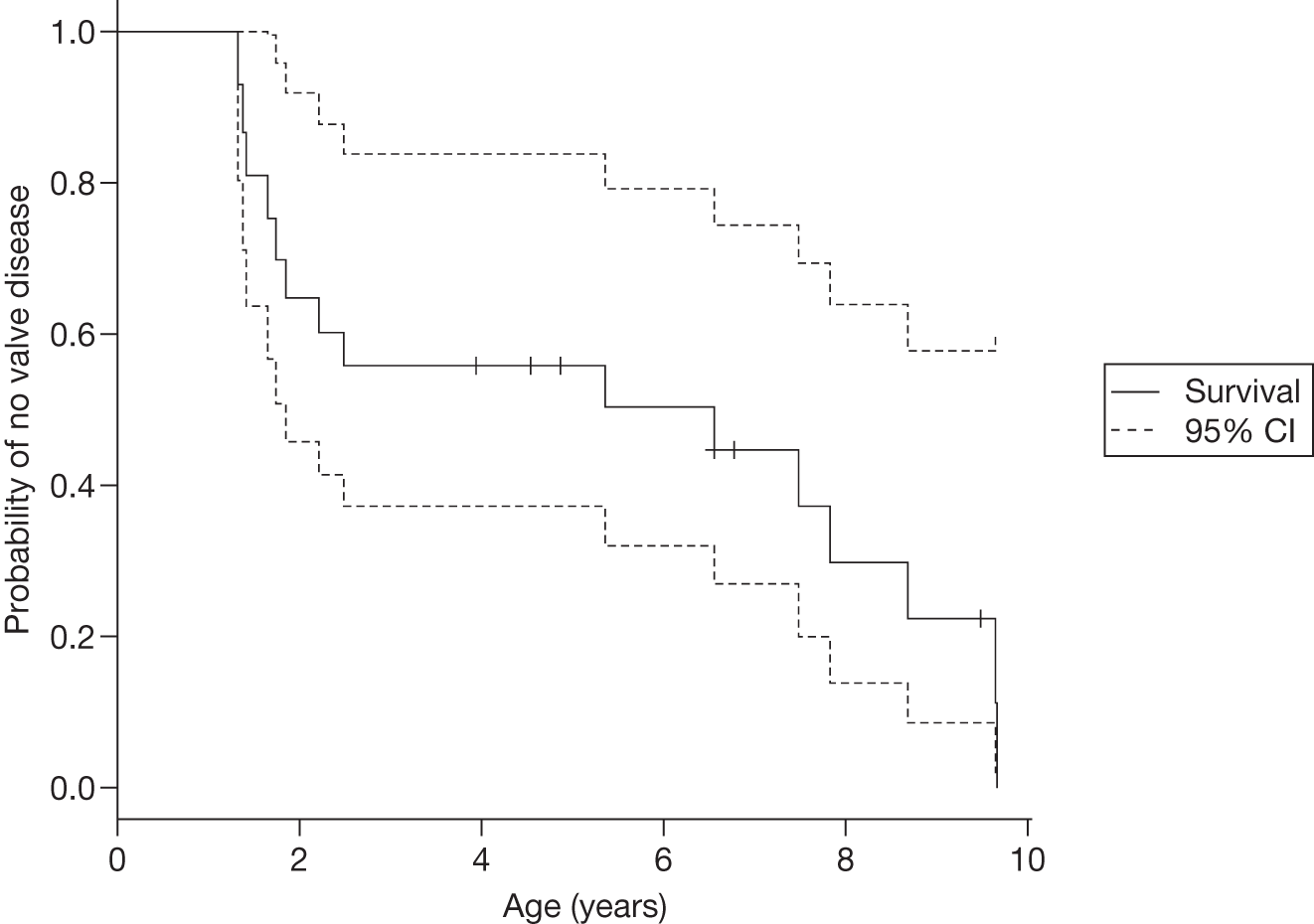
Carpal tunnel syndrome
Carpal tunnel syndrome was categorised in MPS I patients as absent or present. Figure 60 displays the timelines for patients on ERT, showing the age at which they were first reported as having CTS. Five of the 24 patients on ERT were classified as having CTS across all time points. Kaplan–Meier curves were not estimated for patients on ERT or for patients not yet treated because the risk sets for these treatment groups had fewer than five patients at all ages.
FIGURE 60.
Age at first recording of CTS among patients with MPS I who received ERT.
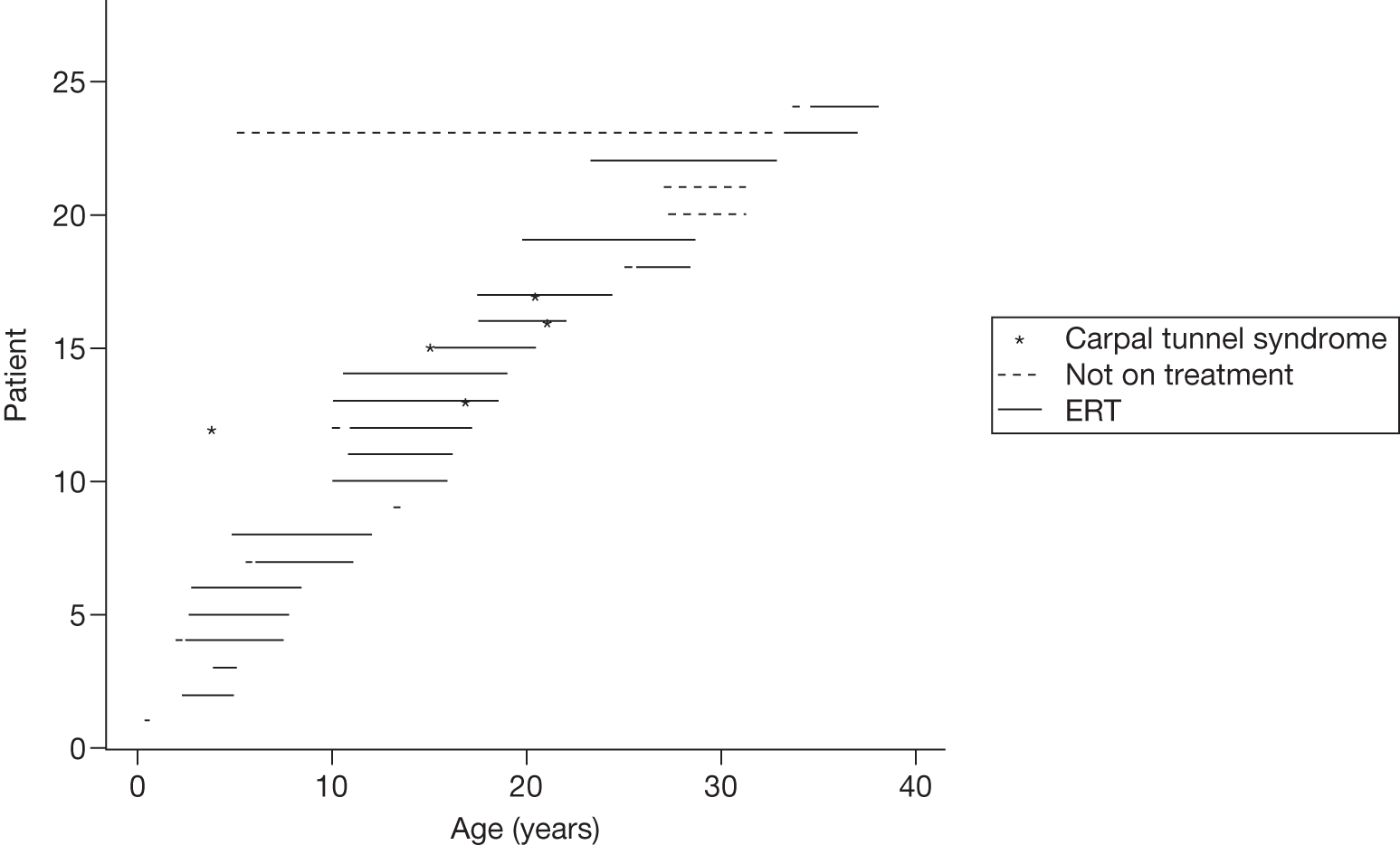
Figure 61 shows a Kaplan–Meier survival curve (and 95% CI) illustrating the probability of developing CTS by age for HSCT patients only. The model suggests that approximately 50% of MPS I patients who have received a HSCT develop CTS by the time they are 9 years old. Again, it is important to interpret these data in the light of the wide CIs around the survival curve.
FIGURE 61.
Risk of having CTS by age and treatment status for people with MPS I. The Kaplan–Meier curve was truncated at 11 years of age because fewer than five HSCT patients were at risk of developing CTS.
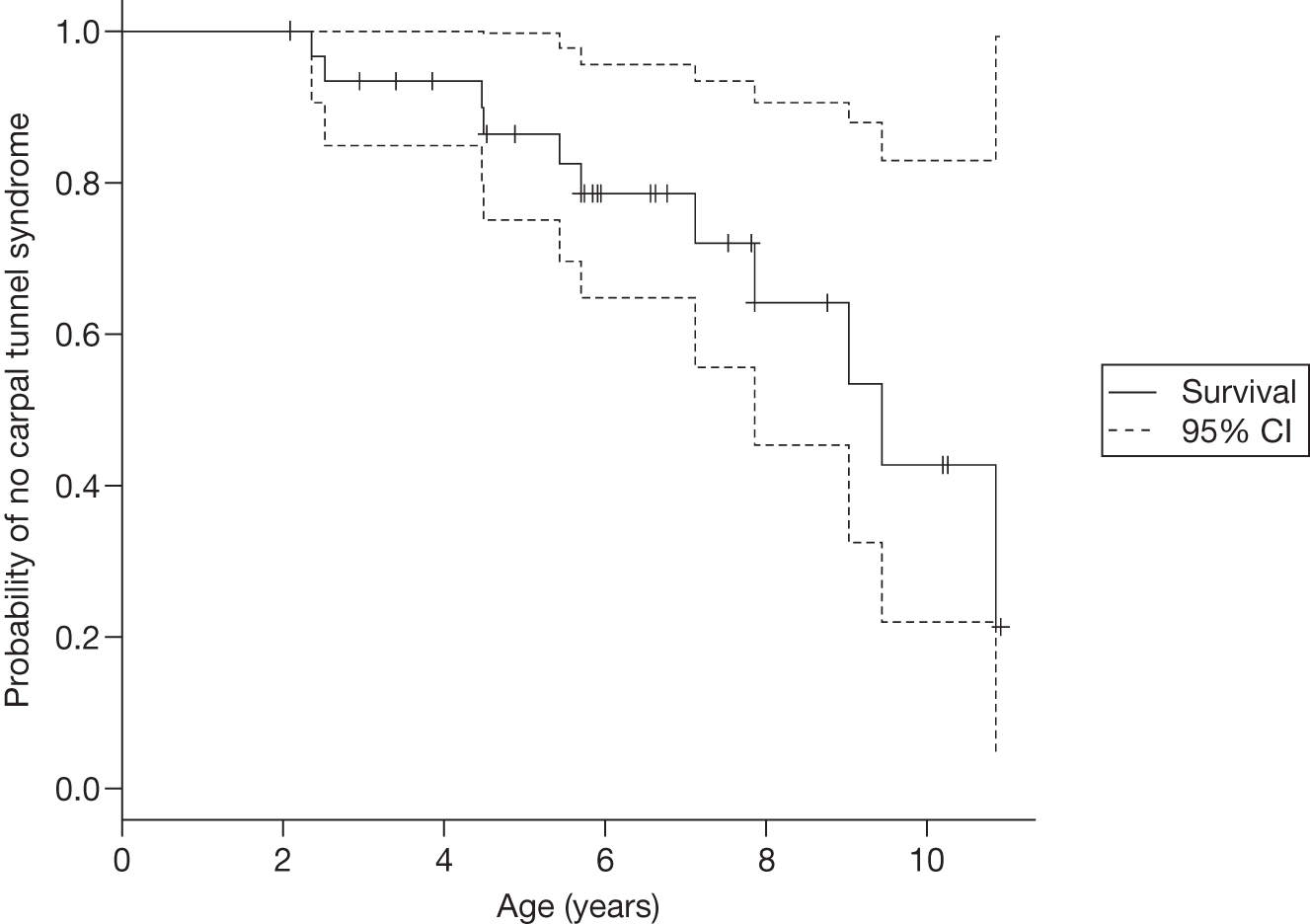
Developmental quotient score (children)
The DQ is a numerical expression of a child’s developmental level. Seventy-two DQ score measurements were available for 19 children (3 on ERT, 16 HSCT recipients). These scores ranged from 57 to 108, where a score of 100 indicates the child is exactly on target developmentally for their age (Table 91).
| N Data | Estimate of change in DQ | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 39 | 0.00 | |||
| Female | 33 | –0.88 | 5.85 | –12.3 to 10.58 | 0.88 |
| Current age | |||||
| Linear effect/year | –1.94 | 1.32 | –4.52 to 0.64 | 0.15 | |
| Time on ERT | |||||
| Linear effect/year | 2.40 | 2.71 | –2.91 to 7.71 | 0.38 | |
| Time since HSCT | |||||
| Linear effect/year | 0.78 | 1.29 | –1.74 to 3.31 | 0.54 | |
| Variance components | |||||
| Individual | 176.49 | ||||
| Centre | 78.96 | ||||
| Residual | 97.04 | ||||
Developmental quotient score was not significantly associated with age (p = 0.15) within this group of patients.
There was no significant association between DQ score and time on ERT (p = 0.38) or time since HSCT (p = 0.54). It is important to interpret these findings in the light of selection of patients for these two treatment modalities. Children selected for HSCT are those at risk of neurological involvement in whom a decline in DQ might be anticipated although we cannot draw such a conclusion from the data we have available.
No evidence was found for a non-linear association between DQ and time since HSCT (edf = 1.0; p = 0.72) (Figure 62). Owing to the lack of DQ score data for patients on ERT (n = 3), no further analyses were conducted.
FIGURE 62.
The age-adjusted association between time since HSCT and DQ score in children with MPS I (time since HSCT treated as a continuous variable).
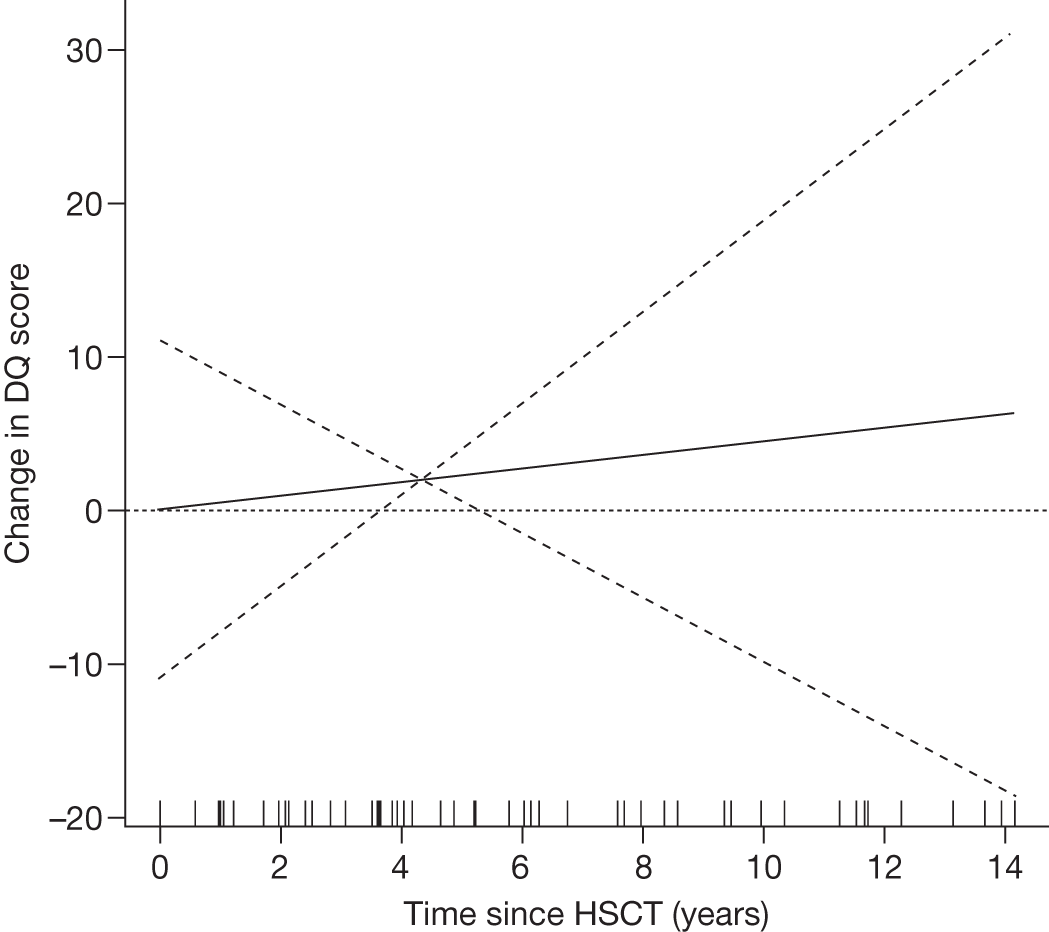
Cervical cord compression
Six MPS I patients had documented cervical cord compression and a further six patients had received surgery for cervical cord compression. Figure 63 shows a Kaplan–Meier survival curve illustrating the probability of having cervical cord compression by age, for patients following a HSCT or on ERT.
FIGURE 63.
Risk of cervical cord compression by age and treatment status for adults < 30 years old with MPS I (Kaplan–Meier curve). The number of patients who are > 30 years of age and are at risk of cervical cord compression is fewer than five.
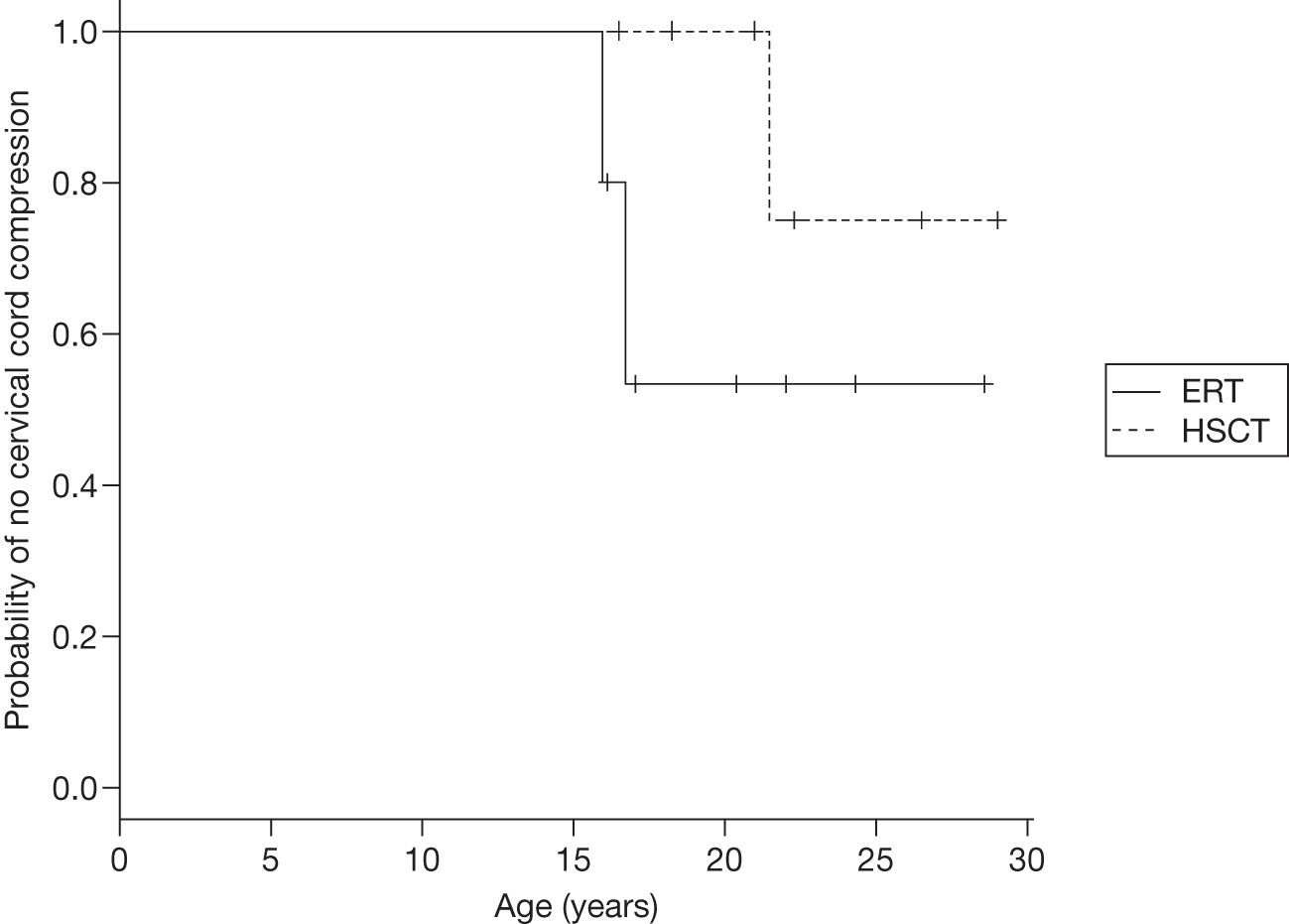
The model suggests that until the age of 15 years, both patient groups are at low risk of having cervical cord compression. Cases of cervical compression were observed in both treatment groups during early adulthood. There was no significant difference in risk between the groups (HR for ERT relative to HSCT = 3.7, 95% CI 0.33 to 41.1; p = 0.27). This comparison needs to be interpreted with caution owing to differences in underlying phenotype between groups.
Palpable splenic enlargement
The presence or absence of an enlarged spleen was recorded for 36 patients. Of these, 16 patients were reported as having an enlarged spleen on palpation. Figure 64 shows a Kaplan–Meier survival curve illustrating the probability of not having an enlarged spleen by age, for patients following a HSCT or on ERT.
FIGURE 64.
Risk of having an enlarged spleen by age and treatment status for people with MPS I (Kaplan–Meier curve).
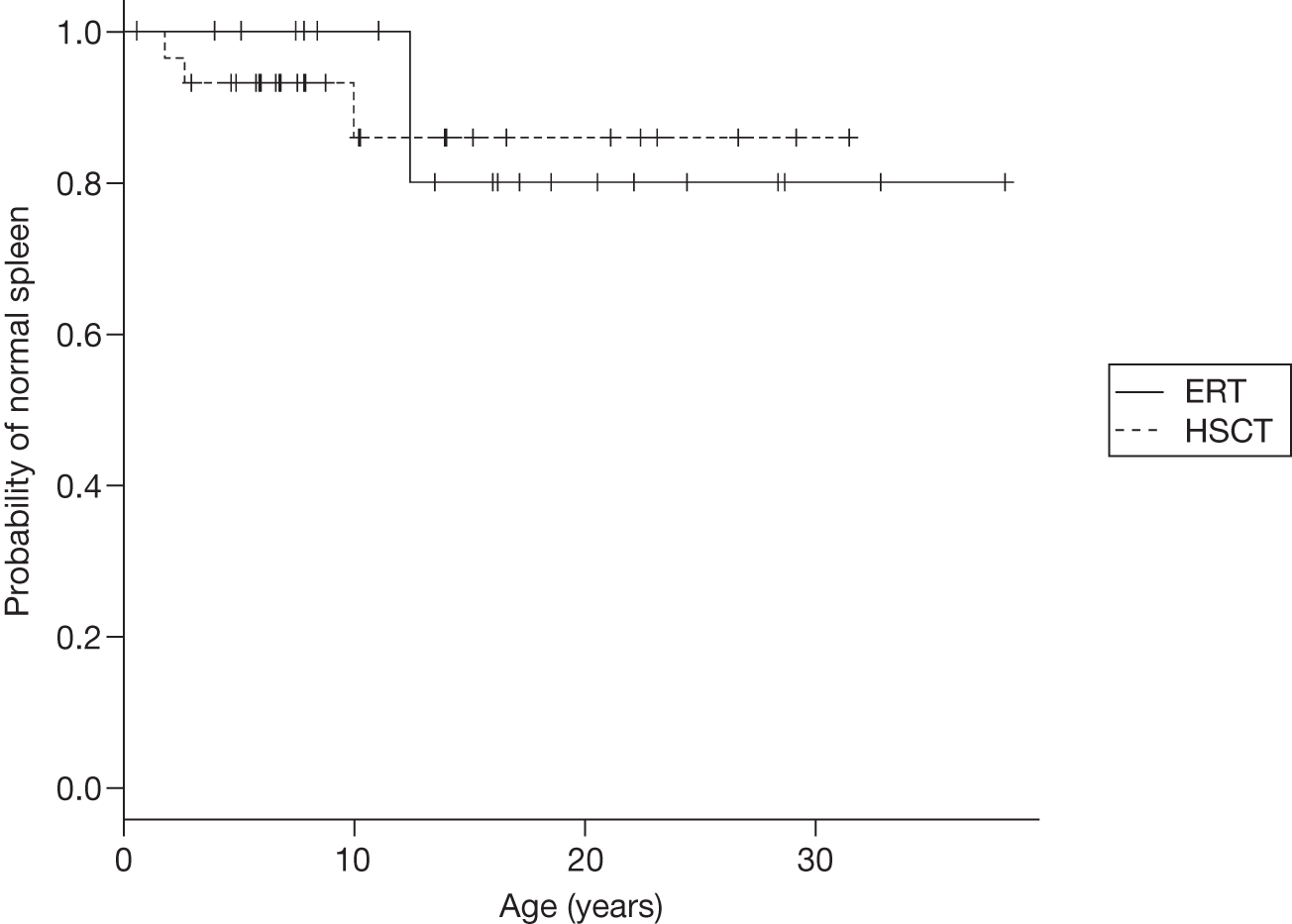
Although the curves appear to suggest that HSCT recipients are at increased risk of having an enlarged spleen at an earlier age than those receiving ERT (which would be compatible with the different underlying phenotypes), these differences are not statistically significant (HR for ERT relative to HSCT = 2.48; 95% CI 0.20 to 29.9; p = 0.47).
Palpable liver enlargement
The presence or absence of an enlarged liver was reported for 31 patients. Of these, 24 patients were reported as having an enlarged liver on palpation. Figure 65 shows timelines for MPS I patients, illustrating the age at which they were first reported as having an enlarged liver.
FIGURE 65.
Age at first recording of liver enlargement among patients with MPS I who received ERT.
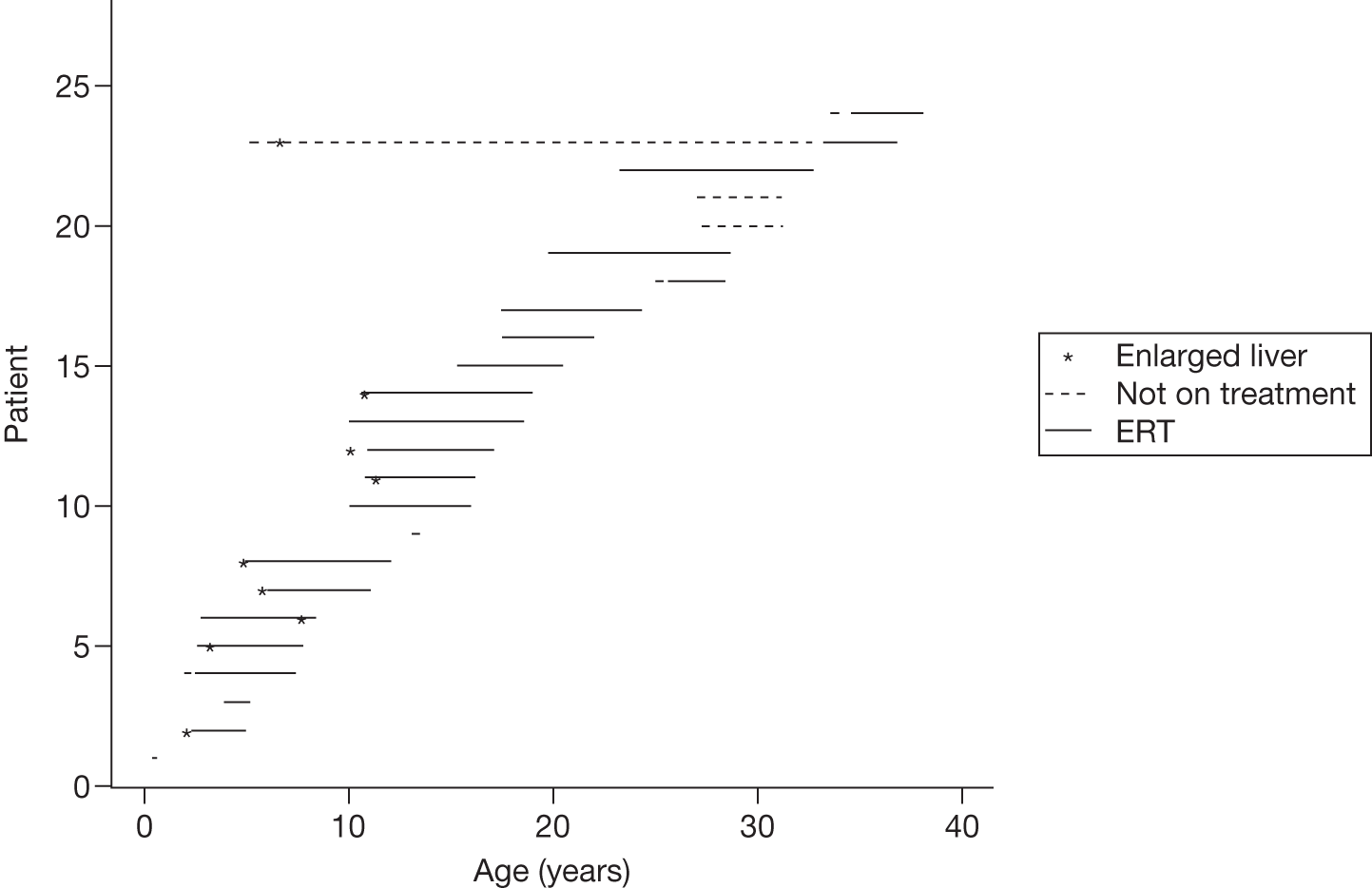
Figure 66 shows a Kaplan–Meier survival curve (and 95% CI) illustrating the probability of having an enlarged liver by age for HSCT patients only. The analysis is restricted to HSCT patients because the risk sets for untreated and ERT patients had fewer than five patients at all ages. The model suggests that by the age of 10 years, HSCT patients have a 30% chance of having an enlarged liver. It is important to interpret these data in light of the wide CIs around the survival curve.
FIGURE 66.
Risk of having an enlarged liver by age for people with MPS I who have received a HSCT (Kaplan–Meier curve).
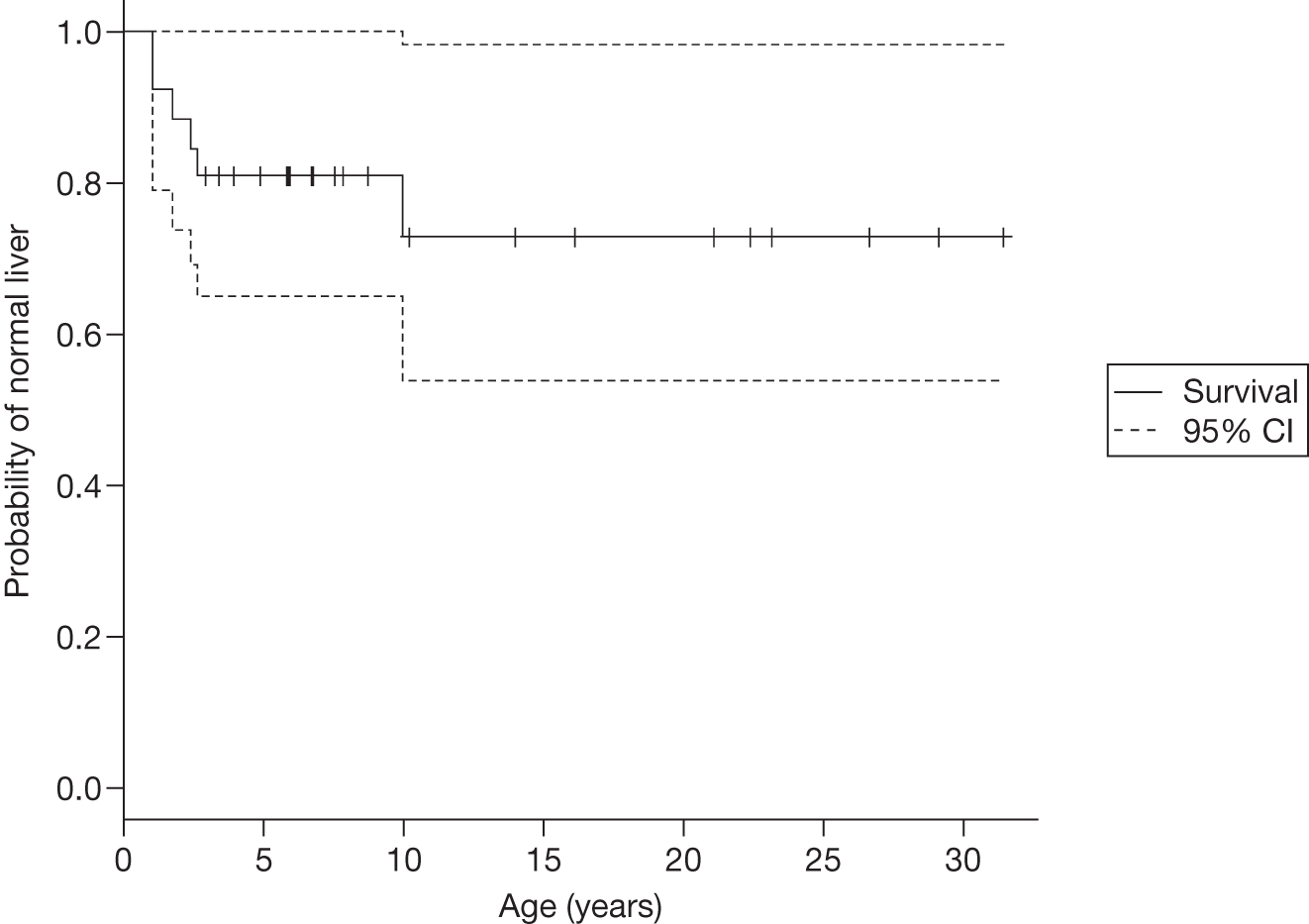
Quality-of-life assessments
SF-36
Thirty-three SF-36 questionnaires were collected from adult MPS I patients. Table 92 shows a descriptive summary of the PCS and MCS by type of treatment.
| Time | PCS | MCS |
|---|---|---|
| Overall | ||
| Mean (SD) | 33.15 (10.9) | 56.25 (9.8) |
| n | 33 | 33 |
| ERT patients | ||
| Mean (SD) | 34.99 (9.74) | 56.62 (9.25) |
| n | 19 | 19 |
| HSCT patients | ||
| Mean (SD) | 29.05 (8.42) | 55.13 (12.7) |
| n | 14 | 14 |
As can be seen in Table 93, the PCS was not significantly associated with age (p = 0.58), time on ERT (p = 0.68) or time since HSCT (p = 0.95).
| N Data | Estimate of increment in PCS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 12 | 0.00 | |||
| Female | 21 | –10.80 | 7.44 | –25.4 to 3.78 | 0.16 |
| Current age | |||||
| Linear effect/year | –0.27 | 0.48 | –1.21 to 0.67 | 0.58 | |
| Time on ERT | |||||
| Linear effect/year | 0.32 | 0.78 | –1.21 to 1.85 | 0.68 | |
| Time since HSCT | |||||
| Linear effect/year | 0.01 | 0.17 | –0.32 to 0.34 | 0.95 | |
| Variance components | |||||
| Individual | 63.9 | ||||
| Centre | 39.4 | ||||
| Residual | 14.8 | ||||
There was no evidence for a non-linear association between the PCS and time on ERT (edf = 1.90; p = 0.35) (Figure 67).
FIGURE 67.
The age-adjusted association between time on ERT and SF-36 PCS in adults with MPS I (time on ERT treated as a continuous variable).
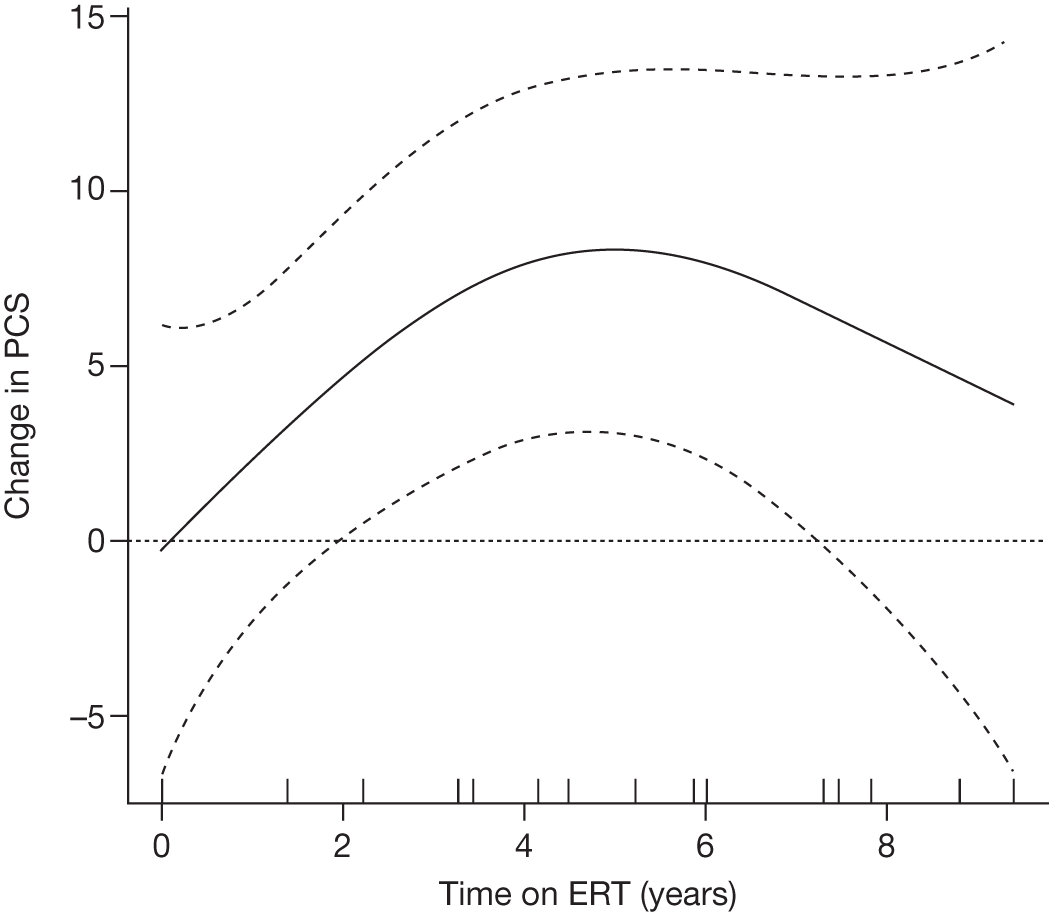
There were insufficient data to explore the shape of the relationship between PCS and time since HSCT.
It is important to note that because the SF-36 data were not available retrospectively, this analysis essentially compares patients with different durations of time on treatment rather than comparing those not on treatment with those on treatment (either HSCT or ERT).
As it can be seen in Table 94, the MCS was not significantly associated with age (p = 0.61), time on ERT (p = 0.14) or time since HSCT (p = 0.36).
| N Data | Estimate of increment in MCS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 12 | 0.00 | |||
| Female | 21 | –7.75 | 6.90 | –21.3 to 5.77 | 0.27 |
| Current age | |||||
| Linear effect/year | –0.23 | 0.46 | –1.13 to 0.67 | 0.61 | |
| Time on ERT | |||||
| Linear effect/year | –1.54 | 0.99 | –3.48 to 0.40 | 0.14 | |
| Time since HSCT | |||||
| Linear effect/year | –0.29 | 0.32 | –0.92 to 0.34 | 0.36 | |
| Variance components | |||||
| Individual | 0.0 | ||||
| Centre | 0.0 | ||||
| Residual | 114.9 | ||||
Figure 68 suggests that there was no evidence for a non-linear association between MCS and time on ERT (edf = 1.91; p = 0.19). There were insufficient data points to explore the shape of the relationship between MCS and time since HSCT.
FIGURE 68.
The age-adjusted association between time on ERT and SF-36 MCS in adults with MPS I (time on ERT treated as a continuous variable).
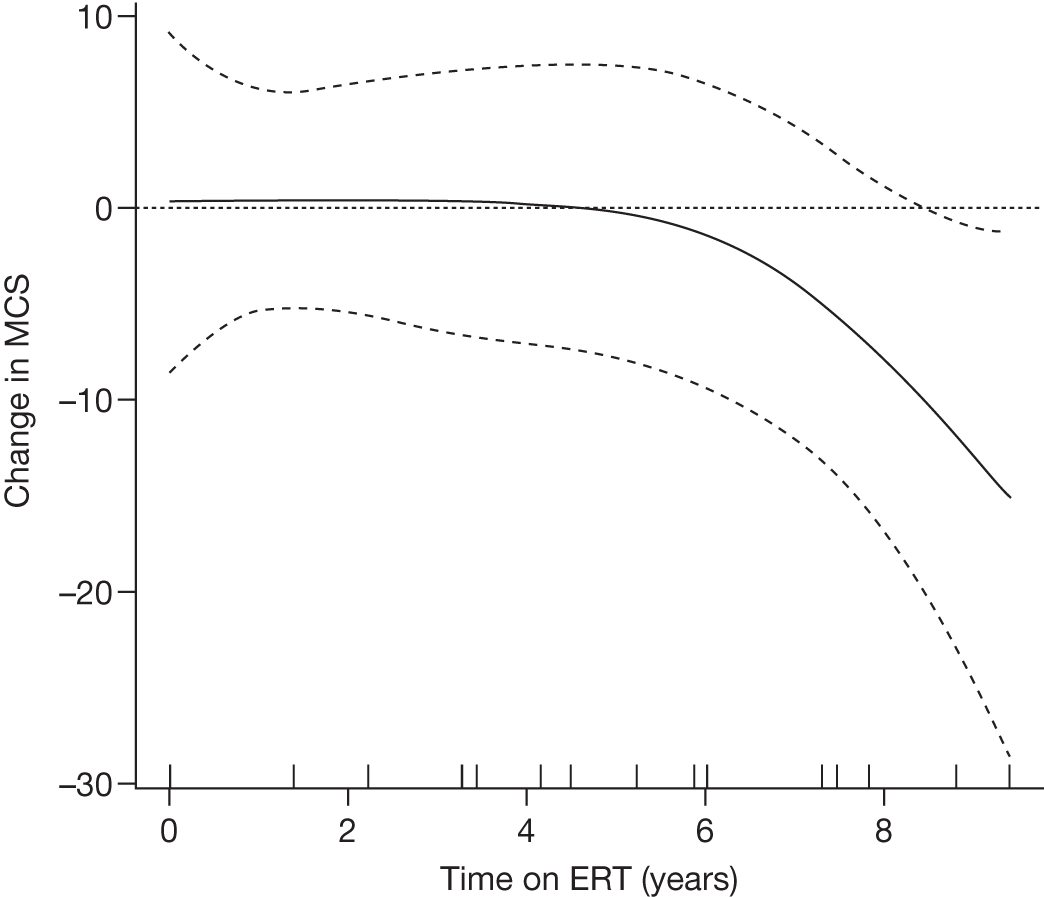
EQ-5D
In addition to the SF-36, participants aged > 13 years were invited to complete the EQ-5D. Forty-three EQ-5D questionnaires were completed across all prospective time points. Data are presented in Table 95 for EQ-5D score (ranging from –0.43 to 1.0, with 1.0 being ‘perfect health’).
| N Data | Estimate of increment in EQ-5D | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 16 | 0.00 | |||
| Female | 27 | –0.05 | 0.11 | –0.26 to 0.16 | 0.63 |
| Current age | |||||
| Linear effect/year | –0.0008 | 0.008 | –0.02 to 0.01 | 0.93 | |
| Time on ERT | |||||
| Linear effect/year | –0.02 | 0.024 | –0.07 to 0.03 | 0.38 | |
| Time since HSCT | |||||
| Linear effect/year | –0.004 | 0.008 | –0.02 to 0.01 | 0.61 | |
| Variance components | |||||
| Individual | 0.004 | ||||
| Centre | 0.014 | ||||
| Residual | 0.064 | ||||
A longitudinal model was fitted to assess the linear relationship between EQ-5D and time on ERT, and times since HSCT after adjusting for age.
The linear model showed no significant association between EQ-5D and age (p = 0.93), time on ERT (p = 0.38) or time since HSCT (p = 0.61).
No evidence was found for a non-linear association between the EQ-5D score and time on ERT (edf = 1.0; p = 0.38) (Figure 69) or time since HSCT (edf = 1.58; p = 0.51) (Figure 70), although it is important to note that the data are sparse.
FIGURE 69.
The age-adjusted association between time on ERT and EQ-5D score in adults with MPS I (time on ERT treated as a continuous variable).
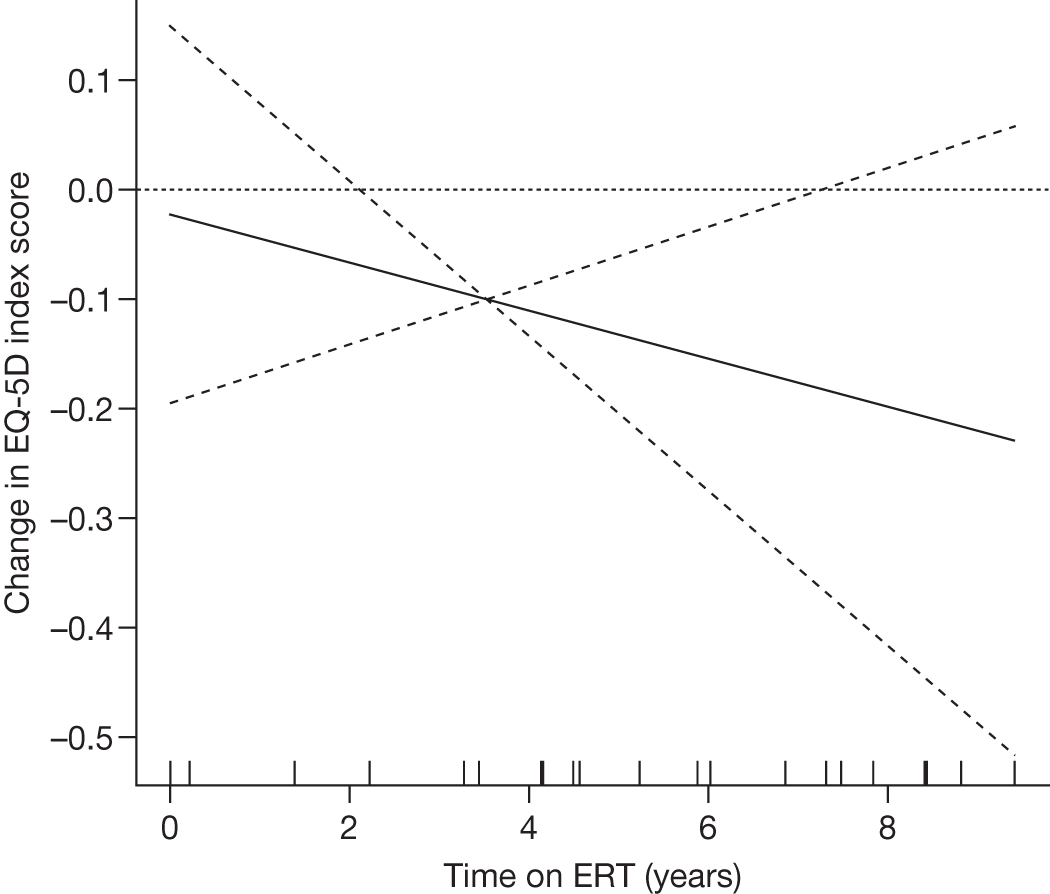
FIGURE 70.
The age-adjusted association between time since HSCT and EQ-5D score in adults with MPS I (time since HSCT treated as a continuous variable).
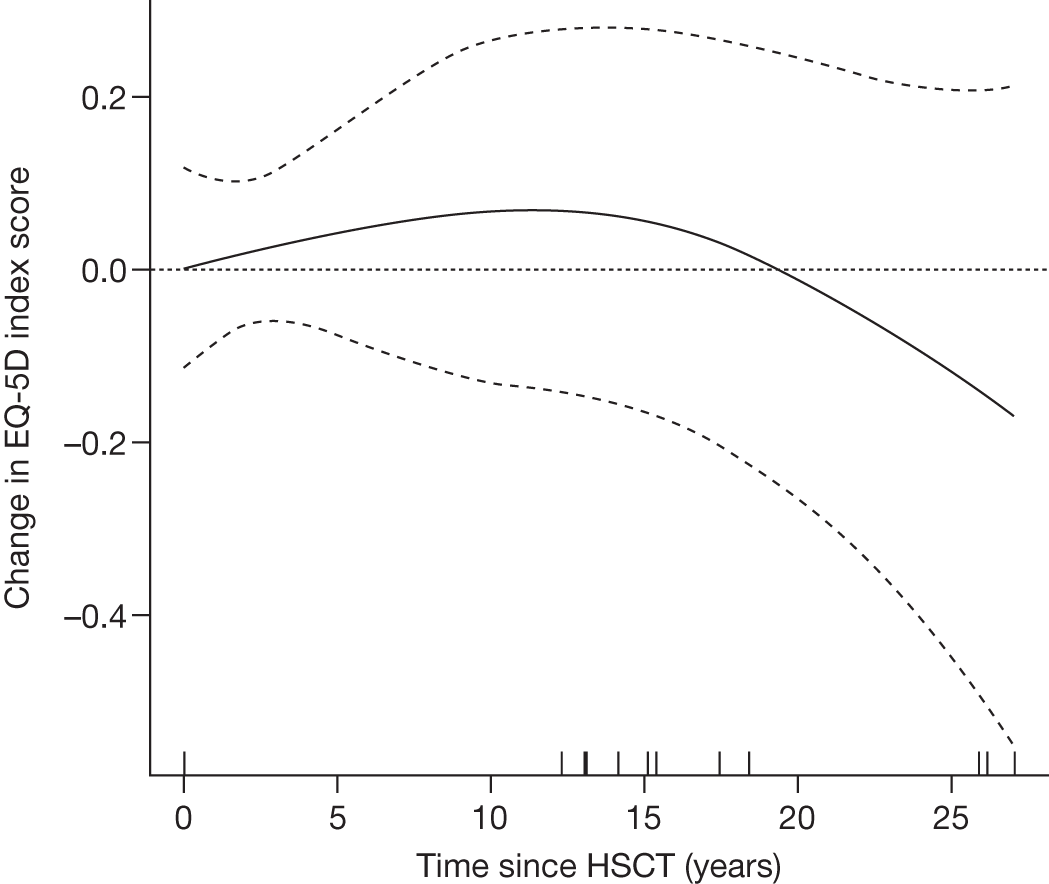
Equivalent modelling analyses were also conducted using SF-6D (SF-36-derived) utility weights, but no statistically significant associations (at α = 0.05 level) were found with time on ERT. The tabulated results of the SF-6D longitudinal modelling analyses are available on request from the study authors.
EQ-5D visual analogue scale
In addition to scoring on the five domains of the EQ-5D, participants were asked to rate their health on a VAS. The EQ-5D VAS asks people to rate their health state on a 10-cm line from 100, ‘best imaginable health state’, to 0, ‘worst imaginable health state’. The 43 VAS scores completed for this study ranged from 9 to 100. A longitudinal model was fitted to assess the linear relationship between the visual analogue score and time on ERT after adjusting for age.
The linear model shown in Table 96 shows no significant association in EQ-5D VAS with age (p = 0.87), time on ERT (p = 0.46) or time since HSCT (p = 0.41)
| N Data | Estimate of increment in EQ-5D VAS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 17 | 0.00 | |||
| Female | 26 | –3.64 | 9.87 | –22.9 to 15.7 | 0.71 |
| Current age | |||||
| Linear effect/year | 0.14 | 0.82 | –1.47 to 1.74 | 0.87 | |
| Time on ERT | |||||
| Linear effect/year | –1.49 | 1.97 | –5.35 to 2.37 | 0.46 | |
| Time since HSCT | |||||
| Linear effect/year | 0.63 | 0.75 | –0.84 to 2.1 | 0.41 | |
| Variance components | |||||
| Individual | 84.3 | ||||
| Centre | 227.3 | ||||
| Residual | 397.1 | ||||
No evidence was found for a non-linear association with EQ-5D VAS and time on ERT (edf = 1.84; p = 0.24) (Figure 71) or time since HSCT (edf = 1.59; p = 0.68) (Figure 72).
FIGURE 71.
The age-adjusted association between time on ERT and EQ-5D VAS in adults with MPS I (time on ERT treated as a continuous variable).
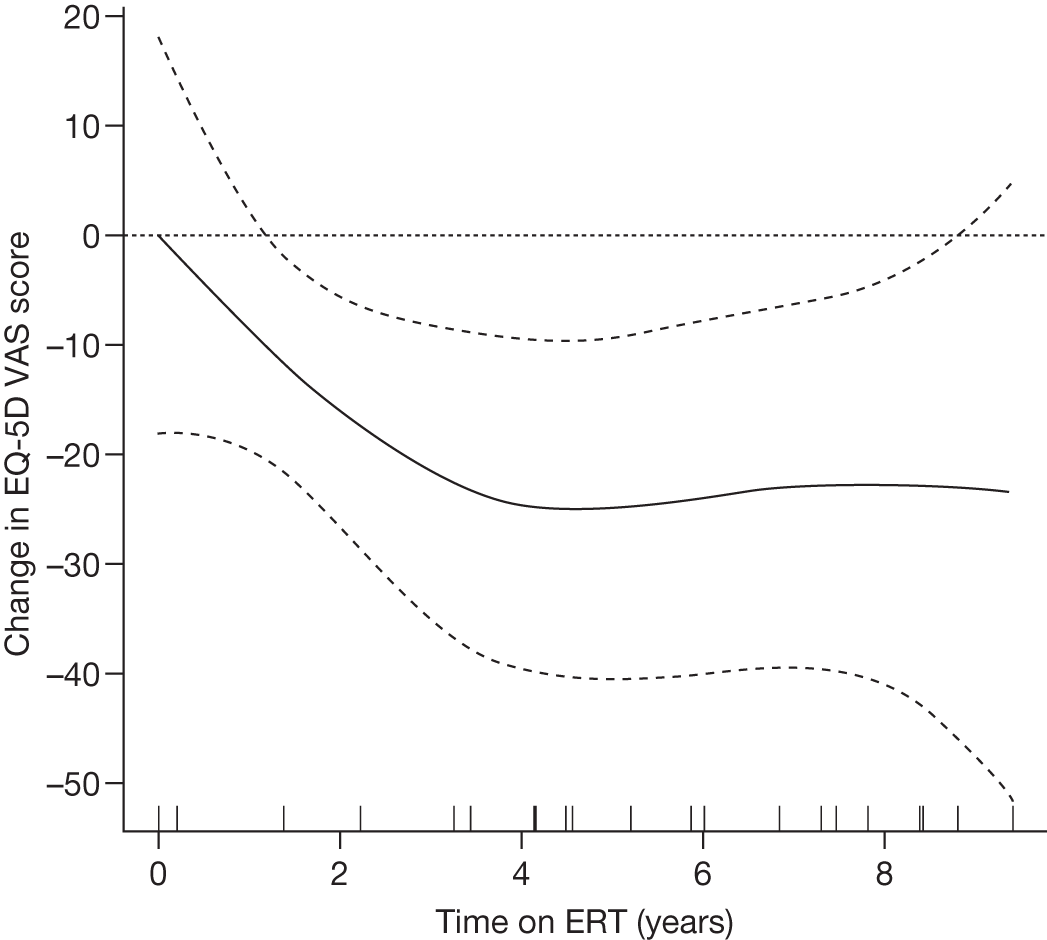
FIGURE 72.
The age-adjusted association between time since HSCT and EQ-5D VAS in adults with MPS I (time since HSCT treated as a continuous variable).
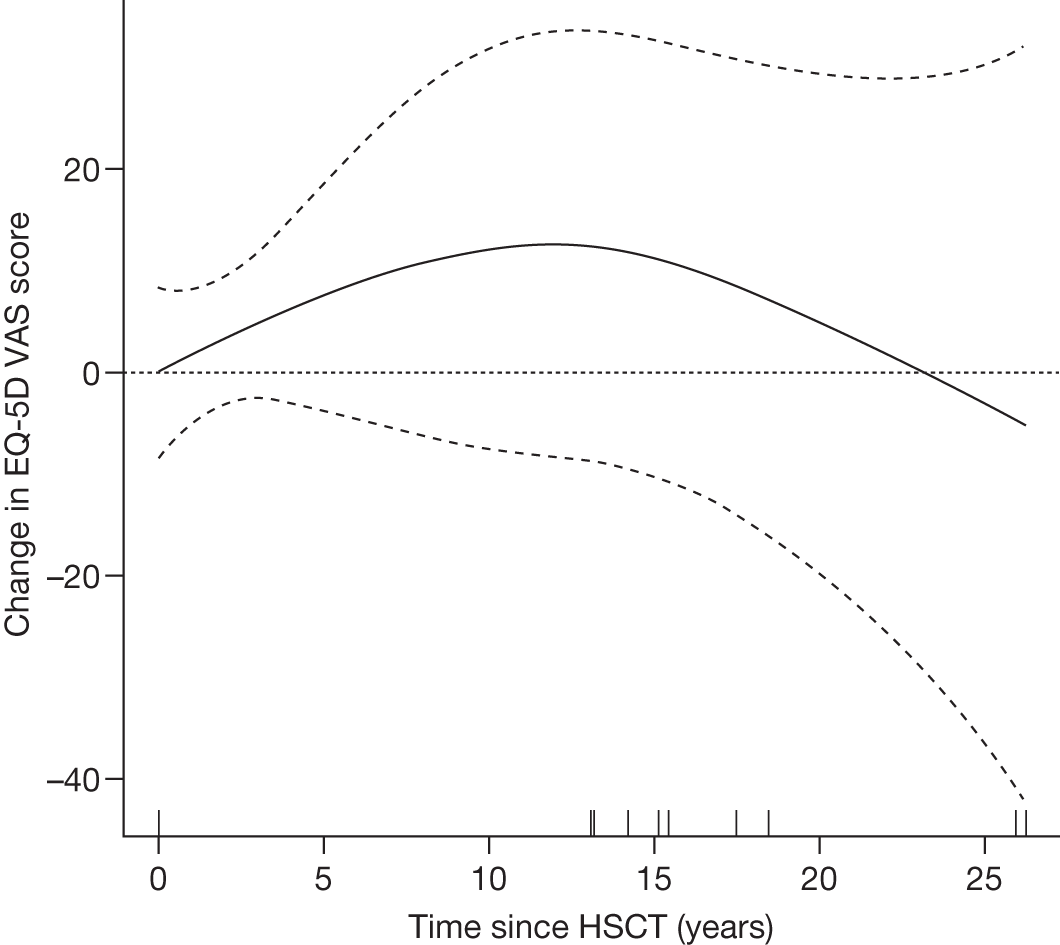
PedsQL
Fifty-six PedsQL questionnaires were completed by children or their carers. Table 97 shows a descriptive summary of the total score and the component summary scores by type of treatment. The scale ranges from 0 to 100 where higher scores indicate better HRQoL.
| Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Physical health summary score | Total score | |
|---|---|---|---|---|---|---|---|
| Overall | |||||||
| Mean (SD) | 44.04 (21.8) | 54.56 (21.0) | 55.23 (18.6) | 45.86 (16.1) | 50.49 (14.4) | 44.04 (21.8) | 45.2 (14.6) |
| n | 55 | 56 | 56 | 35 | 34 | 55 | 32 |
| ERT patients | |||||||
| Mean (SD) | 45.95 (29.0) | 54.57 (20.4) | 44.53 (30.9) | 52.2 (13.3) | 49.69 (20.8) | 45.95 (29.0) | 43.9 (24.4) |
| n | 5 | 6 | 6 | 5 | 5 | 5 | 4 |
| HSCT patients | |||||||
| Mean (SD) | 43.99 (21.7) | 54.61 (21.6) | 56.46 (16.8) | 45.25 (16.4) | 50.69 (13.9) | 43.99 (21.7) | 45.3 (13.8) |
| n | 48 | 48 | 48 | 28 | 27 | 48 | 26 |
Table 98 shows a descriptive summary of the PedsQL data, with time on ERT or since HSCT, after adjusting for age.
| Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Physical health summary score | Total score | |
|---|---|---|---|---|---|---|---|
| Gender | |||||||
| Male | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Female | –4.42 | 2.43 | 5.33 | –1.99 | –0.52 | –4.42 | 3.89 |
| 95% CI | –21.5 to 12.6 | –13.7 to 18.6 | –7.38 to 18.0 | –19.4 to 15.4 | –17.7 to 17.7 | –21.5 to 12.6 | –17.6 to 25.4 |
| p-value | 0.61 | 0.77 | 0.42 | 0.83 | 0.95 | 0.61 | 0.73 |
| Current age | |||||||
| Mean increment/year | –3.60 | –1.02 | –4.28 | 1.26 | –1.14 | –3.60 | –0.83 |
| 95% CI | –7.22 to 0.03 | –4.25 to 2.21 | –6.67 to –1.88 | –1.31 to 3.84 | –3.56 to 1.28 | –7.22 to 0.03 | –4.18 to 2.54 |
| p-value | 0.06 | 0.54 | 0.001 | 0.34 | 0.37 | 0.06 | 0.64 |
| Time on ERT | |||||||
| Mean increment/year | 11.7 | 3.84 | 9.65 | 2.73 | 7.18 | 11.7 | 6.75 |
| 95% CI | –0.63 to 24.1 | –5.89 to 13.6 | 2.23 to 17.1 | –5.0 to 10.5 | 0.03 to 14.3 | –0.63 to 24.1 | –4.28 to 17.8 |
| p-value | 0.07 | 0.44 | 0.01 | 0.49 | 0.06 | 0.07 | 0.24 |
| Time since HSCT | |||||||
| Mean increment/year | 1.85 | 0.82 | 4.69 | 0.86 | 2.76 | 1.85 | 2.52 |
| 95% CI | –1.88 to 5.57 | –2.59 to 4.22 | 2.14 to 7.25 | –1.59 to 3.32 | 0.48 to 5.05 | –1.88 to 5.57 | –0.18 to 5.23 |
| p-value | 0.34 | 0.64 | <0.001 | 0.49 | 0.02 | 0.34 | 0.08 |
| Variance components | |||||||
| Individual | 333.1 | 284.9 | 173.5 | 0.00 | 81.6 | 333.1 | 124.1 |
| Centre | 25.5 | 43.8 | 0.00 | 0.00 | 0.00 | 25.5 | 0.00 |
| Residual | 143.2 | 190.6 | 122.2 | 250.68 | 125.5 | 143.2 | 111.1 |
Although there is some suggestion of an improvement in PedsQL score with time on ERT, statistically significant effects are only seen in the social functioning subscale (mean increment/year = 9.65, 95% CI 2.23 to 17.1; p = 0.01) and not for the overall score.
Similarly, total PedsQL score and all subscales improved with time since HSCT, but the associations were statistically significant only for the social functioning and psychosocial health summary scales (p < 0.001 and p = 0.02, respectively) and not for the overall score.
Fatigue Severity Scale
Thirteen FSS questionnaires were completed by 10 patients prospectively. The scores ranged from 1.44 to 6.44. There were not enough data to carry out further analysis.
Carer Strain Index
Fifty-nine CSI questionnaires were completed across all prospective time points. Data for the CSI total score ranged from 1 to 24; the maximum possible score of 26 signifies the greatest degree of caregiver burden. A longitudinal model was fitted to assess the linear relationship between the CSI and time on ERT, after adjusting for age (Table 99).
| N Data | Estimate of increment in CSI | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 39 | 0.00 | |||
| Female | 20 | 1.28 | 2.09 | –2.82 to 5.37 | 0.54 |
| Current age | |||||
| Linear effect/year | –0.07 | 0.24 | –0.54 to 0.40 | 0.78 | |
| Time on ERT | |||||
| Linear effect/year | –1.37 | 1.29 | –3.89 to 1.16 | 0.29 | |
| Time since HSCT | |||||
| Linear effect/year | 0.12 | 0.27 | –0.41 to 0.65 | 0.67 | |
| Variance components | |||||
| Individual | 21.5 | ||||
| Centre | 0.0 | ||||
| Residual | 14.8 | ||||
The linear model shows no significant association in score on the CSI with age of person they are caring for (p = 0.78), with time on ERT (p = 0.29) or time since HSCT (p = 0.67).
No evidence was found for a non-linear association between the CSI score and time since HSCT (edf = 1; p = 0.69) (Figure 73).
FIGURE 73.
The age-adjusted association between time since HSCT and CSI for carers of people with MPS I (time since HSCT treated as a continuous variable).
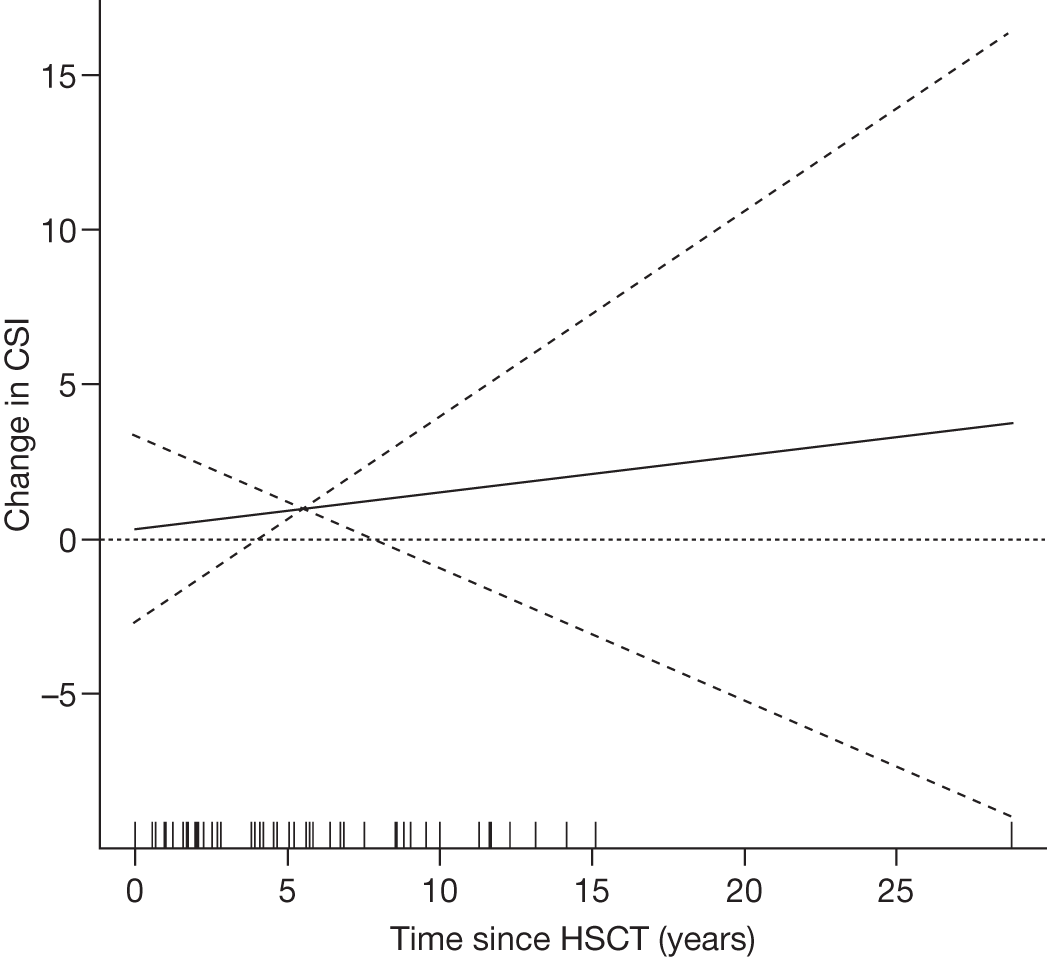
Safety and complications
Of the 68 participants with MPS I in this study, one patient was reported as experiencing anaphylactic reactions, seven patients required pre-medication and 10 patients were reported as having a positive antibody status to infused products. No MPS I patients were reported to experience any febrile reactions.
One patient stopped ERT during the period of data collection although no reason for stopping treatment was cited. This patient had required pre-medication.
During the period of data collection, no MPS I patients participating in the study died from disease-related complications.
Cost of enzyme replacement therapy in people with MPS I
Table 100 shows the current purchase cost to the NHS of the ERT laronidase.
| Drug full name | Proprietary name and unit | 2011 base price per unit (£) |
|---|---|---|
| Laronidase | Aldurazyme®, 500 IU | 444.70 |
Table 101 shows the NSCT-estimated annual NHS per patient cost of providing laronidase. Note that these costs include both the drug costs and home-care costs where the NSCT fund them.
| Drug | Adults | Infants | Children |
|---|---|---|---|
| Laronidase 1 unit | £258,201 | £27,844 | £139,563 |
Cost of care for adults with MPS I
Total care cost – financial burden of MPS I
Table 102 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for an adult with MPS I. Of the estimated mean per patient annual cost of £8500, just two-fifths is as a result of NHS hospital services used; of this over two-thirds (£2300 per patient per year) is from inpatient stays and only one-quarter (£880) is from outpatient visits (see Table 103). Of the £5100 per patient per year from using services outside hospital, < £90 per patient is as a result of GP or GP nurse appointments and > £3400 is as a result of regular visits from ‘health visitors or other nurses’, or home help/home-care workers (and some of these will be for regular ERT infusions) (see Table 104).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. (%) who used this type of service | Mean cost (£) | Standard deviation | Median cost (£) | Interquartile range (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 16 | 76 | 13 (81) | 3400 | 5209 | 1700 | 500–9200 |
| Services outside hospital | 16 | 76 | 13 (81) | 5100 | 6346 | 210 | 27–15,600 |
| Total health (NHS) and social-care cost | 16 | 76 | 14 (88) | 8500 | 9860 | 2000 | 1100–9200 |
| Type of hospital care | No. who used this type of service | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| Inpatient stays | 5 (31) | 2300 | 4803 | 8500 | 1300–16,400 |
| Outpatient visits | 13 (81) | 880 | 876 | 790 | 400–1700 |
| Day cases | 1 (6) | 170 | 668 | 2700 | N/A |
| Accident and emergency visits | 4 (25) | 26 | 46 | 100 | 100–100 |
| Total hospital (NHS) care cost | 13 (81) | 3400 | 5209 | 1700 | 500–9200 |
| Service provider | No. (%) who used this provider | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 11 (69) | 86 | 160 | 65 | 27–160 |
| GP nurse appointments | 6 (38) | 4 | 7 | 5 | 5–8 |
| District nurses | 0 | 0 | 0 | 0 | 0–0 |
| Community mental health nurse | 0 | 0 | 0 | 0 | 0–0 |
| Other nurse or health visitor | 3 (19) | 1300 | 3952 | 4600 | 44–15,400 |
| Counsellor | 0 | 0 | 0 | 0 | 0–0 |
| Other therapist | 1 (6) | 960 | 3861 | 15,400 | N/A |
| ‘Alternative’ medicine or therapy | 0 | 0 | 0 | 0 | 0–0 |
| Psychologist | 0 | 0 | 0 | 0 | 0–0 |
| Psychiatrist | 0 | 0 | 0 | 0 | 0–0 |
| Other community-based doctor | 0 | 0 | 0 | 0 | 0–0 |
| Occupational therapist | 1 (6) | 4 | 14 | 57 | N/A |
| Social worker | 5 (31) | 51 | 92 | 160 | 79–290 |
| Home help | 2 (13) | 2100 | 8449 | N/A | 75–33,800 |
| Care attendant | 1 (6) | 570 | 2281 | 9100 | N/A |
| Community support worker | 0 | 0 | 0 | 0 | 0–0 |
| Housing worker | 0 | 0 | 0 | 0 | 0–0 |
| All non-hospital NHS and social-care providers | 13 (81) | 5100 | 6346 | 210 | 27–15,600 |
Cost breakdown by hospital- and community-based services
Tables 103 and 104 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by adults with MPS I. Although only five patients (or just under one-third of those adults who provided valid service-use data) had hospital stays as inpatients, these hospital stays accounted for over two-thirds of the NHS hospital costs in MPS I adults. In contrast, 13 (four-fifths) of patients reported having at least one hospital outpatient attendance during the 12 months prior to entering the study.
The majority of costs related to using community-based services were due to the relatively small minority of adults with MPS I who used home help/home-care workers (two patients, 13%) or who used health visitors or other nurses regularly (three patients, 19%). They accounted for £3400 of the £5100 yearly per patient cost of services used outside hospital. Although 11 of the 16 adults with MPS I had seen their GP at least once during the past year, and six reported seeing a practice nurse at their GP surgery, these accounted for < £90 of the £8500 annual cost of NHS and publicly funded social-care services consumed. Other support providers used by smaller numbers of adults with MPS I were occupational therapists and social workers (see Table 104).
All of the resource use and cost findings relating to adults with MPS I should be treated with considerable caution because of the small number of patients for whom we have data.
Cost of care for children with MPS I
Total care cost – financial burden of MPS I
Table 105 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for a child with MPS I from the 39 patients for whom we had cost data at baseline (23 males, mean age 7 years, range 6 months to 15.7 years). Of the estimated mean per patient annual cost of £18,600, almost all is as a result of NHS hospital services used and £16,000 of this is from inpatient stays (see Table 106). Of the £1300 per patient per year from using services outside hospital, £171 is for GP or GP nurse appointments, £550 is for regular visits from ‘health visitors or other nurses’ for 12 children and £260 is for care attendants (for three children) (see Table 107).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. (%) who used this type of service | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 39 | 83 | 33 (85) | 17,300 | 34,488 | 4400 | 1900–23,500 |
| Services outside hospital | 39 | 83 | 39 (100) | 1300 | 2679 | 450 | 150–910 |
| Total health (NHS) and social-care cost | 39 | 83 | 39 (100) | 18,600 | 34,437 | 5300 | 2200–14,100 |
| Type of hospital care | No. who used this type of service | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| Inpatient stays | 28 (72) | 16,000 | 34,436 | 5500 | 1900–29,100 |
| Outpatient visits | 13 (33) | 500 | 1009 | 1100 | 690–2100 |
| Day cases | 12 (31) | 700 | 2209 | 1300 | 670–2100 |
| Accident and emergency visits | 5 (13) | 16 | 44 | 100 | 100–210 |
| Total hospital (NHS) care cost | 33 (85) | 17,300 | 34,488 | 4400 | 1900–23,500 |
| Service provider | No. (%) who used this provider | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 39 (100) | 160 | 90 | 130 | 110–200 |
| GP nurse appointments | 10 (16) | 11 | 34 | 16 | 6–63 |
| District nurses | 3 (8) | 13 | 57 | 160 | 32–320 |
| Community mental health nurse | 2 (5) | 2 | 7 | 24 | 24–36 |
| Other nurse or health visitor | 12 (31) | 550 | 2300 | 180 | 88–2300 |
| Counsellor | 0 | 0 | 0 | 0 | 0–0 |
| Other therapist | 15 (39) | 80 | 238 | 74 | 37–130 |
| ‘Alternative’ medicine or therapy | 0 | 0 | 0 | 0 | 0–0 |
| Psychologist | 5 (13) | 15 | 48 | 81 | 41–240 |
| Psychiatrist | 0 | 0 | 0 | 0 | 0–0 |
| Other community-based doctor | 6 (15) | 24 | 65 | 140 | 110–200 |
| Occupational therapist | 14 (36) | 32 | 73 | 76 | 20–110 |
| Social worker | 10 (26) | 130 | 409 | 150 | 74–440 |
| Home help | 1 (3) | 0.15 | 0.93 | 6 | 6–6 |
| Care attendant | 3 (8) | 260 | 1460 | 900 | 6–9100 |
| Community support worker | 1 (3) | 0.85 | 5.3 | 33 | 33–33 |
| Housing worker | 0 | 0 | 0 | 0 | 0 – 0 |
| All non-hospital NHS and social-care providers | 39 (100) | 1300 | 2679 | 450 | 150–910 |
Cost breakdown by hospital- and community-based services
Tables 106 and 107 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by children with MPS I. Nearly three-quarters (28) of those children who provided valid service-use data had inpatient hospital stays, and these accounted for over two-thirds of the NHS hospital costs in MPS I children. This included two children who had inpatient stays of 150 days for BMTs (> £140,000 each) and three other children who had received between £44,000 and £56,000 worth of inpatient hospital treatment during the year (again mainly for BMTs, but also for a spinal operation and fitting a ventricular-peritoneal shunt).
In contrast, just under one-third of children were reported as having at least one hospital outpatient attendance (13 children) or day case admission (12 children) during the 12 months prior to entering the study. These were for a wide range of check-ups and multiple consultant appointments for cardiac, orthopaedic, audiology, ear, nose and throat (ENT), ophthalmic and dental reasons. Overall, the mean per patient cost of these two types of hospital services was £500 and £700, respectively.
The majority of costs related to using community-based services were as a result of those children with MPS I who used home help/home-care workers (three patients, 8%) or who used health visitors or other nurses regularly (12 patients, 31%). They accounted for £260 and £550 of the yearly per patient cost of services used outside of hospital. A significant proportion of the children also saw social workers (10 children), occupational therapists (14 children) or ‘other therapists’ (15 children, mainly physiotherapists). Although all of the 39 children with MPS I had seen their GP at least once during the past year, and 10 reported seeing a practice nurse at their GP surgery, these accounted for only £160 and £11 of the estimated annual cost of NHS and social-care services consumed. Other care professionals used by smaller numbers of children with MPS I were psychologists and ‘other community-based doctors’ (typically paediatricians) (Table 107).
The resource use and cost findings relating to children with MPS I should be treated with considerable caution because of the relatively small number of patients for whom we have data.
Association of time on enzyme replacement surgery and the costs of caring for patients with MPS I
From the longitudinal regression modelling of costs, in child patients with MPS I there was no statistically significant association between time on ERT and either total NHS and social-care costs, hospital-care costs or non-hospital-care costs. In adult patients with MPS I, although there was a statistically significant association between time on ERT and total NHS and social-care costs (21% higher costs, 95% CI 1% to 45%; p = 0.04) and with non-hospital costs (38% higher costs, 95% CI 6% to 78%; p = 0.01), this was based on only 37 data points from 16 adults (only 10 adults with data from more than one time point) and is in the opposite direction to what would be expected. This result should therefore be interpreted with considerable caution, and may be mainly because of two high-cost patients who happen to also have been on ERT for longer. Restricting this analysis to only those patients who had not had an HSCT resulted in unreliable models, with only five patients with more than one data point. The tabulated results of these analyses are available on request from the study authors.
Discussion of MPS I results
There is considerable heterogeneity among people with MPS I and it is important to consider the distribution of people with different subtypes in study populations when interpreting results. Although three classic subtypes are described it has been more recently suggested that these should be seen more as a continuum rather than completely distinct conditions. The most severe form, Hurler syndrome, is characterised by the presence of neurological involvement, which is of particular relevance as laronidase does not cross the BBB.
Existing estimates of the effectiveness of ERT in MPS I, summarised in Table 6, are based on the results of one RCT,162 a before-and-after observational study,161 a dose optimisation study166 and two open-label, extension studies. 164,262 The strongest evidence is provided by the trial reported by Wraith and colleagues,162 a randomised, placebo-controlled trial including 45 patients (one with Hurler syndrome, 37 with Hurler–Scheie and seven with Scheie) with a 26-week follow-up. The treated group showed statistically significant improvement in FVC and an improvement in physical capacity as measured by the 6-minute walk test, although this did not reach conventional levels of clinical significance. Secondary outcomes including liver volume, urinary GAG excretion and the apnoea/hyponoea index also showed statistically significant effects favouring the intervention group. Only changes in shoulder flexion and a disability score index did not show significant effects favouring the intervention group. The other studies all suggest improvements across a number of outcomes over time in patients treated with laronidase although these are less straightforward to interpret given the lack of an untreated comparator.
In this study, we examined potential associations between treatment and FVC, mobility and 6-minute walk test, growth, hearing, heart valve disease and CTS. We also examined the relationship between treatment and QoL assessed by SF-36, EQ-5D or PedsQL depending on age and with DQ in children. These analyses depend on the fact that patients began treatment with laronidase at different ages dependent on the time that the drug became available.
It is important to note that, compared with the studies described above, our study population has more patients at the more severe end of the MPS I spectrum, with 43 patients having MPS IH, 22 patients with MPS IHS and only 3 with MPS IS. Clearly there is considerable potential for confounding, particularly by severity with those with more severe disease being diagnosed and beginning treatment earlier.
We found no statistically significant relationship between time on ERT and any outcome, with the exception of the social functioning subscale of the PedsQL. Similarly, no statistically significant association was observed with time since HSCT and any of the outcome measures, apart from two subscales of the PedsQL.
As previously described, all analyses in MPS I were hampered by a paucity of data. Not only were we able to recruit only a relatively small number of patients, but for many of those recruited data were lacking for key outcomes. This occurred despite the outcomes chosen being those that the clinical collaborators believed to be the best measures of disease progression and measures which they believed would be recorded for the vast majority of patients at each regular clinical contact. It is not clear to what extent this reflects the tests not being carried out or being performed but not recorded.
The result of this lack of data is that we have relatively low power to detect treatment effects. The power is further diminished by the necessity of dealing with the heterogeneity within MPS I reflected by the different subtypes. This heterogeneity and the strong association between subtype and mode of treatment makes comparison between HSCT and ERT uninterpretable [of the 44 patients who received a HSCT, 43 had a more severe form of MPS I (i.e. Hurler subtype), whereas of the 24 who had received ERT as their initial treatment, none had this phenotype].
Pulmonary function
Although a RCT by Wraith263 reported that FVC improved by 5.6% compared with controls (p = 0.009), we found no significant association between FVC and time on ERT in analyses adjusted for age, subtype, gender and centre. These analyses suggested an increase in FVC% of 1.69%/year but with a 95% CI including zero (–0.58% to 3.96%).
Liver and spleen size
A reduction in liver and spleen size was reported in all the previous studies with Wraith and colleagues162 reporting that mean liver volume decreased by 18.9% in the laronidase-treated group and increased by 1.3% in the placebo group (p = 0.001). Insufficient data were available in this study to assess the effects of ERT on liver and spleen volume.
In these data, HSCT recipients were at increased risk of having an enlarged spleen at an earlier age than those receiving ERT although this difference did not reach conventional levels of statistical significance. These differences would be compatible with underlying differences in phenotypes between treatment groups.
Mobility
Previous studies have suggested that the use of ERT is associated with statistically significant improvements in mobility as demonstrated by improvements in the 6-minute walk test. No effect was seen in our data but interpretation is severely hampered by the small number of patients for whom these results were recorded.
In line with current understanding, our data indicate that approximately 20% of patients who have had either a HSCT or are on ERT have restricted mobility by the time they are 15 years old.
Height and weight
Kakkis and colleagues163 reported that the rate of growth in height and weight of six pre-pubertal patients in their observational study increased by a mean of 85% and 131%, respectively, at 52 weeks after starting ERT. In this study, the authors compared the mean slopes of the best-fit lines for the 1–2 years before treatment for each patient, with the best-fit lines for the 1 year on treatment, as opposed to comparing with expected growth patterns for a child of the same age which makes interpretation difficult. In our data, after adjustment, time on ERT was not associated with a significant change in children’s weight centile (p = 0.77) or height centile (p = 0.16). Similarly, time since HSCT was not associated with a significant change in weight centile (p = 0.16) or height centile (p = 0.25).
Hearing
Some degree of hearing loss, which may be made worse by frequent ear infections, is common in people with MPS I who have either severe or more attenuated subtypes. Our data suggest that approximately 80% of patients who have had a HSCT (i.e. those with more severe subtypes) have impaired hearing by the time they are 12 years old. No analysis was conducted in untreated patients or patients on ERT owing to small numbers. None of the previous studies have reported the effect of ERT on hearing. A single case study has reported that mild hearing loss in a young boy with MPS I progressed to severe sensorineural hearing loss in the mild and high frequency range after 13 months on ERT. 264
Valve involvement
Most people with MPS I, including people those with the milder Scheie subtype, develop problems with their heart valves and may need mitral or aortic valve replacement in their teens and twenties. 265 Previous studies have suggested that while progression of cardiac valve disease may be stabilised, it does not seem to be reversed by ERT161,164,266 although the data on which these conclusions are based are difficult to interpret.
We were not able to examine the effects of ERT on cardiac valve disease owing to a paucity of data. Of those people with MPS I who have had a HSCT, the data suggest that 80% will develop some form of valve disease by the time they are 9 years old.
Carpal tunnel syndrome
Carpal tunnel syndrome is a very common complication of the mucopolysaccharidoses, but none of the previous studies have examined the effects of ERT on this outcome. Current experience suggests that almost all MPS patients have had surgery for CTS by the time they reach adulthood, irrespective of treatment (Stephen Waldek, Retired Metabolic Physician, formerly of Salford Royal NHS Foundation Trust, 2012, personal communication). In this study, five of the 24 MPS I patients on ERT and approximately 50% of patients who received a HSCT had developed CTS by the time they were 9 years old.
Development quotient
Neurological involvement is the defining characteristic of the more severe forms of MPS I and is associated with the development of cognitive impairment. Because the recombinant enzyme is not thought to cross the BBB, this is not likely to be effective in arresting this feature of the condition. For those at risk of cognitive impairment, current thinking is that the best option is an early HSCT where engrafted donor cells deliver enzyme to the host. Neuropsychological responses to HSCT are dependent on the age and intellectual capacity of the child at the time of the transplant. If the HSCT is undertaken prior to signs of significant developmental delay (usually < 2 years), there is a significant chance of decreasing the degree and rate of cognitive decline,267 whereas children showing significant cognitive impairment prior to undergoing HSCT do not show correction of existing impairment. 268
Among patients included in this study, those recognised to have neurological involvement MPS I patients who did not receive a HSCT would not have had neurological disease, and thus DQ in this group of patients would be expected to be reasonable. Meanwhile, there will be a few patients in the HSCT group who were transplanted too late and thus have some brain disease, and some patients in the ERT group who should have been transplanted but were not.
Development quotient score was not significantly associated with age (p = 0.15) within this group of patients. There was no significant association between DQ score and time on ERT (p = 0.38) or time since HSCT (p = 0.54).
Cervical cord compression
The model suggests that until the age of 15 years, both patient groups are at low risk of having cervical cord compression. Cases of cervical compression were observed in both treatment groups during early adulthood. There was no significant difference in risk between the groups (HR for ERT relative to HSCT = 0.37, 95% CI 0.33 to 41.1; p = 0.27).
Previous studies have not clearly documented an effect of HSCT on the prevention of spinal cord compression and spinal cord compression although myelopathy has been reported to develop and progress after HSCT. 269
Costs associated with MPS I
As with all other conditions investigated in this study, we were keen to capture the wider costs of care falling on the public sector in addition to the costs associated with ERT.
Based on patients’ self-reported health- and social-care service use, the annual cost of caring for people with MPS I, excluding the purchase cost of ERT, was estimated at £8500 for an adult and £18,600 for a child. These costs, however, are dwarfed by the cost of the therapies: the mean annual cost of ERT for adults with MPS I is £258,201 and for children with MPS I is £139,563.
From the longitudinal regression modelling of costs, there was no statistically significant association (i.e. p-value < 0.05) between time on ERT and either total NHS and social-care costs, hospital-care costs, or non-hospital-care costs for patients with MPS I. The tabulated results of these analyses are available on request from the study authors.
Owing to these high associated costs and the lack of measureable effect of ERT on either clinical outcomes or HRQoL measures, it was infeasible to conduct either a cost-effectiveness or cost–utility analysis. As they apply to all six LSDs, the limitations of these cost estimates are summarised and discussed in Chapter 9.
Chapter 6 Results – mucopolysaccharidosis type II (MPS II)
Patient characteristics
At the start of the study, 58 patients were identified by the treating centres as having MPS II. Of these, 51 patients were deemed eligible for inclusion and 43 patients (84% of those eligible for inclusion) were invited to participate. Thirty-nine patients (91% of those approached) agreed to participate. All consenting patients were male. 1 Patient characteristics are given in Tables 108 and 109.
| Patient characteristic | |
|---|---|
| Gendera | |
| Male, n | 3 |
| Female, n | 0 |
| Type of MPS II | |
| Attenuated (without neurological involvement), n | 0 |
| Severe (with neurological involvement), n | 3 |
| Age at diagnosis (years) | |
| Mean (SD) | 3.4 (1.7) |
| Median (min.–max.) | 4.1 (1.45–4.7) |
| Age at recruitment (years) | |
| Mean (SD) | 19.5 (1.9) |
| Median (min.–max.) | 19.6 (17.6–21.4) |
| Initial treatment | |
| Not on ERT, n | 0 |
| ERT (idursulfase), n | 2 |
| Clinical trial of ERT,b n | 1 |
| Age at starting ERT (years) | |
| Mean (SD) | 16.8 (1.1) |
| Median (min.–max.) | 16.6 (15.9–18.0) |
| Time on ERT at recruitment (years) | |
| Mean (SD) | 2.68 (0.90) |
| Median (min.–max.) | 3.03 (1.64–3.35) |
| Patient characteristic | |
|---|---|
| Gendera | |
| Male, n | 36 |
| Female, n | 0 |
| Type of MPS II | |
| Attenuated (without neurological involvement), n | 18 |
| Severe (with neurological involvement), n | 18 |
| Age at diagnosis (years) | |
| Mean (SD) | 3.3 (2.0) |
| Median (min.–max.) | 3.0 (0–7.8) |
| Age at recruitment (years) | |
| Mean (SD) | 9.62 (4.2) |
| Median (min.–max.) | 9.76 (2.3–15.6) |
| Initial treatment | |
| Not on ERT, n | 2 |
| ERT (idursulfase), n | 26 |
| Clinical trial of ERT,b n | 8 |
| Age at starting ERT (years) | |
| Mean (SD) | 6.47 (3.2) |
| Median (min.–max.) | 6.96 (1.1–12.0) |
| Time on ERT at recruitment (years) | |
| Mean (SD) | 2.27 (1.47) |
| Median (min.–max.) | 1.86 (0.11–5.91) |
At recruitment, 36 of the participants were children (aged ≤ 16 years) and three were adults. The average age of children at recruitment was 9.62 (range 2.3–15.6) years and that of adults was 19.5 (range 17.6–21.4 years). The average age at diagnosis of MPS II was 3.3 (range 0–7.8 years). It should be noted, however, that the age of this study population is not reflective of the disease population as a whole. Of the 14 patients who were either missed in clinic or declined to participate, seven were adults at the start of the study and two were teenagers. We have no treatment data on these patients, but it is anticipated that these people may possibly have more attenuated subtypes and therefore attend clinic less frequently.
We collected data from all consenting patients at the time of recruitment, from 29 patients at their 12-month appointment and five patients at their 24-month appointment. We also collected retrospective data from 30 patients at up to seven time points.
At recruitment, all but two of the patients (both children) were on ERT (idursulfase) and the average time on ERT was 2.31 (range 0.1–5.9) years. Of the 37 patients on ERT, nine patients were initially on a clinical trial prior to being prescribed open-label ERT.
Key markers of disease progression
The following measures were identified as key markers of MPS II disease progression:
-
FVC
-
mobility
-
6-minute walk test
-
stature (height and weight)
-
hearing
-
heart valve disease
-
CTS
-
spleen and liver size.
In addition, adults completed the SF-36, EQ-5D, FSS and the Service Use and Costs Questionnaire, while children or their carers completed the age-appropriate PedsQL questionnaire. Carers of children or adults were asked to complete the Service Use and Costs Questionnaire and the CSI.
Longitudinal models were fitted to assess relationships between continuous measures of function and length of time on ERT, after adjustment for age and clustering by centre. In the base models, the effect of time on ERT was treated as a linear effect because of the small number of data points. Further analysis was conducted to explore the possibility that time spent on ERT would have a non-linear effect on function. Patients contributed data points to the model both before and after starting ERT.
Kaplan–Meier survival curves were estimated to illustrate differences in the age at first recorded occurrence of binary events including restricted mobility, abnormal hearing, valve disease, CTS, enlarged liver and enlarged spleen that could be considered progressive. Treatment group differences in survival function were tested using Cox regression models. Individual patients contributed intervals of time at risk to more than one of the treatment categories (no treatment, ERT) as appropriate.
Summary of results
All analyses in MPS II were hampered by a paucity of data related to both the small number of affected patients recruited (39 of 58 patients deemed eligible) and lack of recording of data on key outcomes for a substantial proportion of these patients. This results in low power to detect any effects.
We examined potential associations between treatment and FVC, spleen and liver enlargement, mobility and 6-minute walk test, stature (height and weight), hearing, the presence/absence of heart valve disease and presence of CTS. We found a statistically significant association between duration of ERT and increasing height z-scores. The association between duration of ERT and weight z-scores was not statistically significant.
In addition we examined the relationship between treatment and QoL assessed by SF-36 and EQ-5D or PedsQL, depending on age. However, with only three adults in the study, we had insufficient completed SF-36 questionnaires to allow further analyses. We found a statistically significant relationship between time on ERT and an increase (improvement) in overall PedsQL score.
We found no statistically significant relationships between use of ERT and any other outcome.
Per cent of predicted forced vital capacity
Fourteen patients (four attenuated and 10 patients with the more severe form of MPS II) were able to complete an upright FVC measurement. Results are reported as a per cent of the predicted volume (FVC%). A total of 34 FVC (15 from attenuated MPS II patients) measurements were recorded for these patients across all time points and the range of these measurements was 13–108%. A longitudinal model was fitted to assess the linear relationship between FVC (adjusted for age) and time on ERT (Table 110).
| Estimate of change in FVC% | Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Current age | ||||
| Linear effect/year | 1.94 | 2.31 | –2.58 to 6.46 | 0.41 |
| Time on ERT | ||||
| Linear effect/year | –0.28 | 2.47 | –5.12 to 4.56 | 0.91 |
| Variance components | ||||
| Individual | 203.7 | |||
| Centre | 424.0 | |||
| Residual | 48.8 | |||
As can be seen in Table 110, FVC was not significantly associated with age (p = 0.41) or time on ERT (p = 0.91).
No evidence was found for a non-linear association between FVC and time on ERT (edf = 1.11; p = 0.86) (Figure 74) although it is important to note that the data are sparse.
FIGURE 74.
The age-adjusted association between time on ERT and FVC% in people with MPS II (time on ERT treated as a continuous variable).
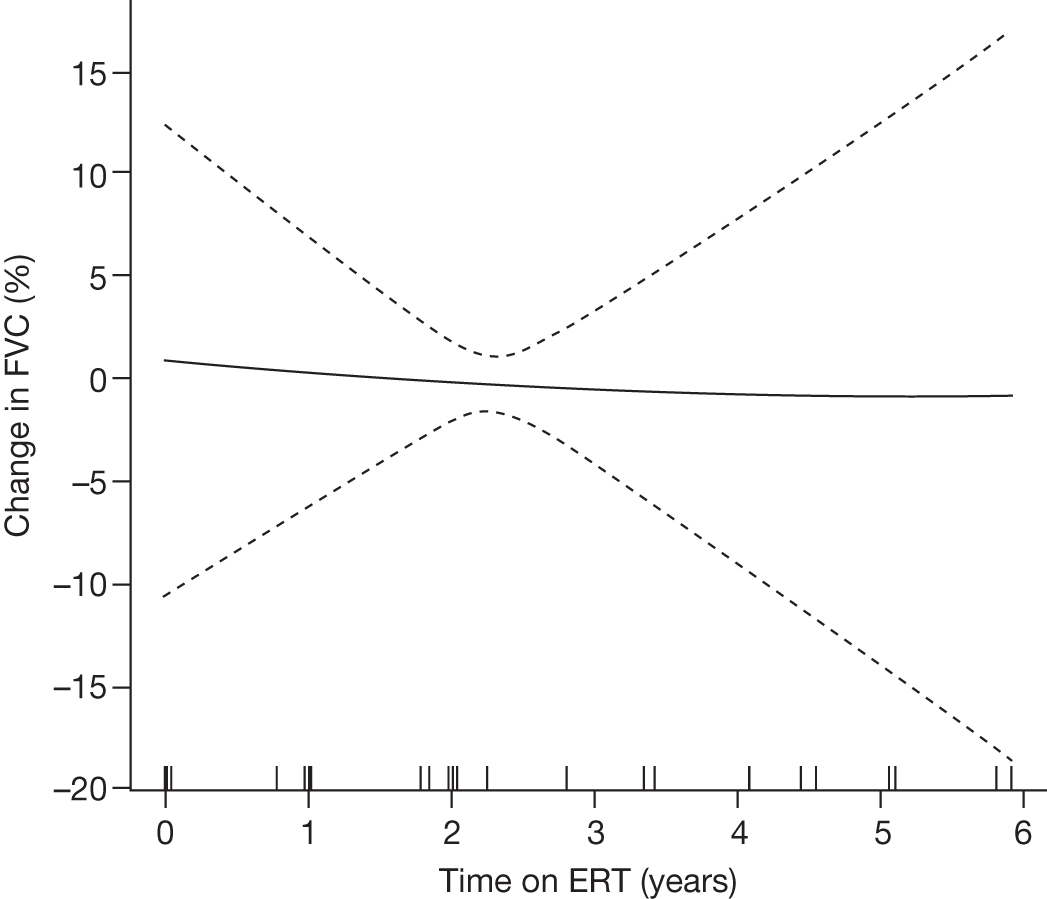
Mobility
Patients were categorised as being mobile (i.e. they could walk for five metres and stand for 6 minutes unaided) or as having restricted mobility (i.e. can walk aided with one or two sticks or immobile).
All MPS II patients for whom there are mobility data contribute pre-treatment data points; those patients who then start ERT contribute data to the treatment group curve. The tick marks correspond to ages at which individual patients drop out of the risk set for the treatment group and so are considered censored. For ease of interpretation, the survival curves were truncated at ages for which fewer than five patients were in the risk set to avoid large imprecise jumps in the curves.
Figure 75 shows Kaplan–Meier curves illustrating the risk of patients having restricted mobility by age for treated and untreated patients. The model suggests that by the time they are 10 years old, approximately 10–20% of patients have restricted mobility. Overall there was no statistically significant difference in the risk of restricted mobility between those on ERT and those not on ERT [HR (ERT/not on ERT) = 0.55; 95% CI 0.03 to 8.78; p = 0.67).
FIGURE 75.
Risk of having restricted mobility by age and treatment status for people with MPS II (Kaplan–Meier curve).
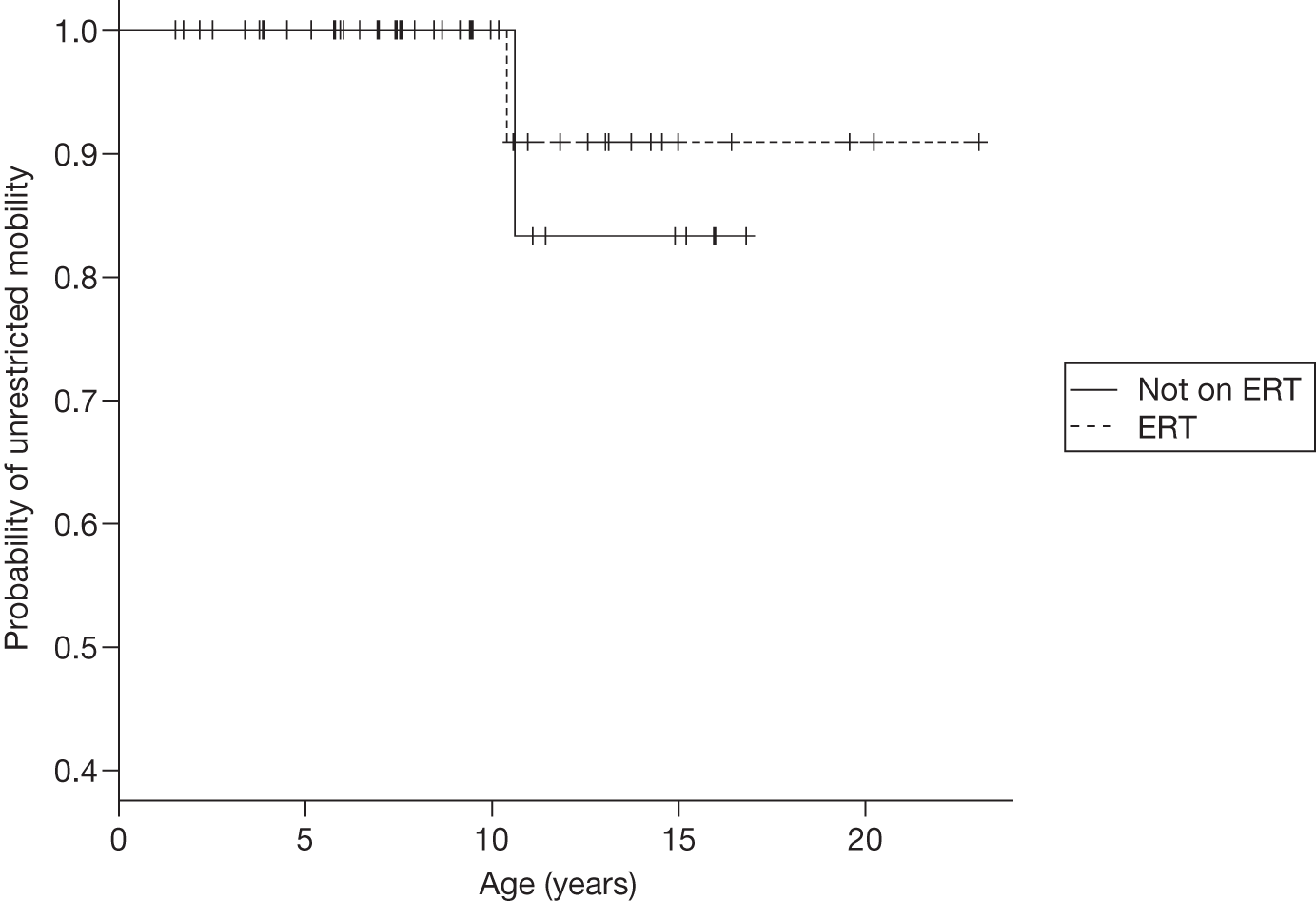
6-minute walk test
Estimates of 6-minute walk tests were collected for 23 patients (10 of these are attenuated MPS II patients). A total of 83 estimates (34 from attenuated MPS II) were recorded for these patients across all time points and these ranged from 45 to 604 m.
A longitudinal model was fitted to assess the linear relationship between the 6-minute walk test and time on ERT, after adjusting for age (Table 111).
| Estimate of change in distance walked (m) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Current age | ||||
| Linear effect/year | 2.07 | 5.85 | –9.39 to 13.5 | 0.72 |
| Time on ERT | ||||
| Linear effect/year | 10.4 | 8.03 | –5.34 to 26.1 | 0.19 |
| Variance components | ||||
| Individual | 9337.5 | |||
| Centre | 8492.3 | |||
| Residual | 2924.9 | |||
The linear model provided no evidence for an association between the distance walked and current age (p = 0.72) or time on ERT (p = 0.19).
Figure 76 suggests no evidence for a non-linear association between distance walked and time on ERT (edf = 1.0; p = 0.19)
FIGURE 76.
The age-adjusted association between time on ERT and distance walked (m) in people with MPS II (time on ERT treated as a continuous variable).
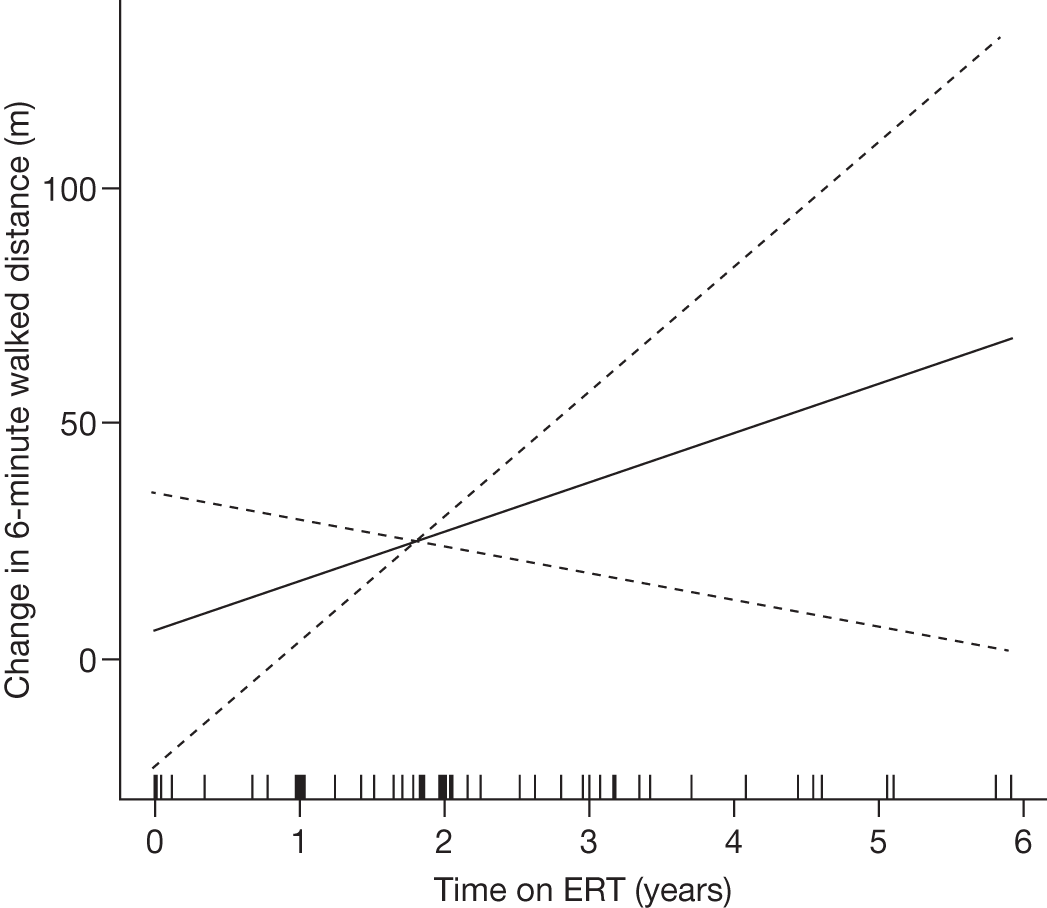
Height
Heights for 33 children were collected, with 132 height measurements recorded across all time points, ranging from 77 to 157 cm. Children’s height measurements were converted to standardised z-scores against 1990 UK norms261 and adjusted for age and gender using the Stata command ‘zanthro’. This transformation allows one to examine the changes in height relative to the expected growth patterns for a child of the same age. The mean z-score was –1.52. This implies that these children are, on average, substantially shorter than their peers (Table 112).
| Estimate of change in height | Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Current age | ||||
| Linear effect/year | –0.53 | 0.05 | –0.63 to –0.43 | < 0.001 |
| Time on ERT | ||||
| Linear effect/year | 0.28 | 0.07 | 0.14 to 0.42 | < 0.001 |
| Variance components | ||||
| Individual | 1.26 | |||
| Centre | 0.27 | |||
| Residual | 0.34 | |||
The model suggests that children’s height decreases through the centiles with age (p < 0.001), although the magnitude of the estimated decline (0.5 standard deviations per year) should be viewed with caution and may reflect selection effects in this small cohort.
After adjusting for age, time on ERT was associated with an improvement in height centile (p < 0.001), as seen in Table 111. Figure 77 suggests the relationship between the height z-score is well approximated by the linear function (edf 1.24; p < 0.001).
FIGURE 77.
The age-adjusted association between time on ERT and height z-scores for children with MPS II (time on ERT treated as a continuous variable).
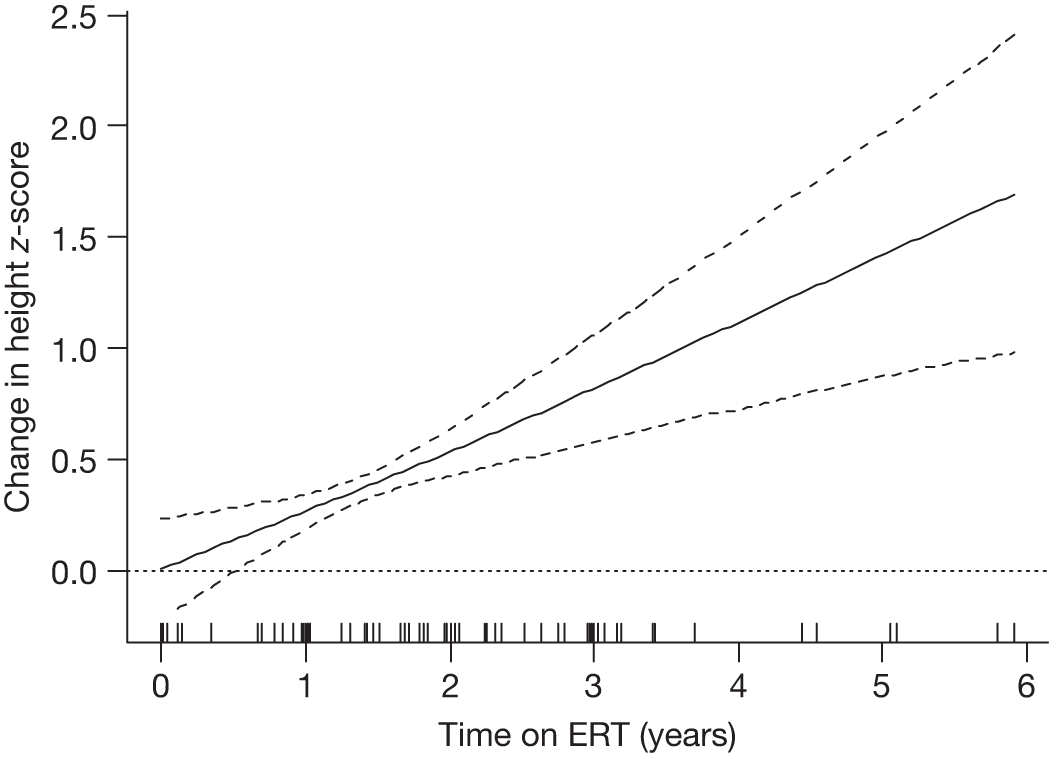
Weight
Weights for 35 children were collected, with 146 weight measurements recorded across all time points, ranging from 11 to 58.8 kg. Children’s weight measurements were converted to standardised z-scores against 1990 UK norms261 and adjusted for age and gender using the Stata command ‘zanthro’. This transformation allows one to examine the changes in weight relative to the expected growth patterns for a child of the same age. The mean z-score was 0.13. This suggests that mean weight in these children was slightly higher than their peers, unlike height which was substantially lower.
Children’s weight decreased through the centiles with age, decreasing by 0.26 standard deviations per year (p < 0.001). After adjusting for age, time on ERT was not associated with a significant change in weight centile (p = 0.37) (Table 113).
| Estimate of change in weight | Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Current age | ||||
| Linear effect/year | –0.26 | 0.03 | –0.32 to –0.20 | < 0.001 |
| Time on ERT | ||||
| Linear effect/year | 0.05 | 0.05 | –0.05 to 0.15 | 0.37 |
| Variance components | ||||
| Individual | 1.34 | |||
| Centre | 0.00 | |||
| Residual | 0.24 | |||
No evidence was found for a non-linear association between weight and time on ERT (edf = 1; p = 0.37) (Figure 78), although it is important to note that the data are sparse beyond three years of ERT.
FIGURE 78.
The age-adjusted association between time on ERT and weight z-scores for children with MPS II (time on ERT treated as a continuous variable).
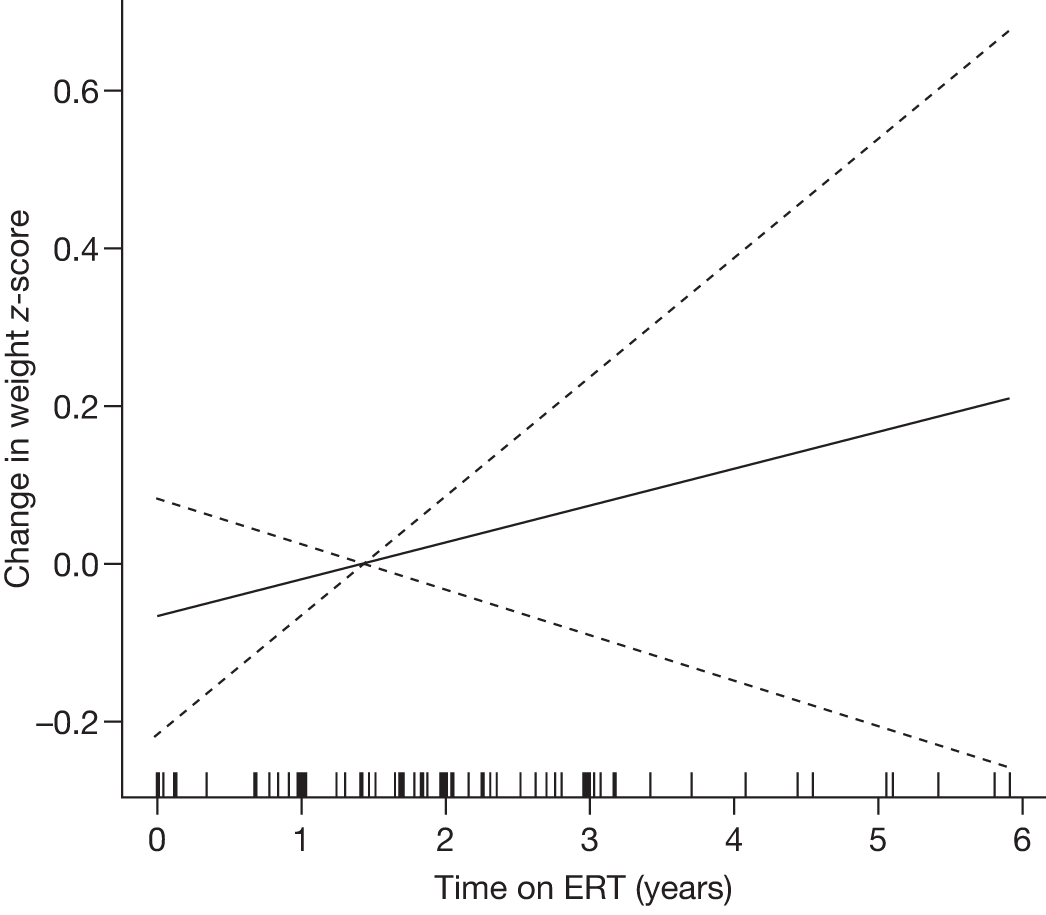
Hearing
The MPS II patients were classified as having normal or impaired hearing according to standard definitions. We have hearing data on 20 MPS II patients recorded across all time points, 18 of whom were classified as having impaired hearing.
Figure 79 shows a Kaplan–Meier survival curve illustrating the probability of having abnormal hearing by age for patients on ERT. Kaplan–Meier curves were not estimated for untreated patients because the risk sets had fewer than five patients at all ages. The graph suggests that about 60% of patients had normal hearing at 1 year of age. This rate dropped to about 8% by 5 years of age. By the age of 10 years, only 2% of patients with MPS II had normal hearing.
FIGURE 79.
Risk of having a hearing impairment by age for people with MPS II (Kaplan–Meier curve).
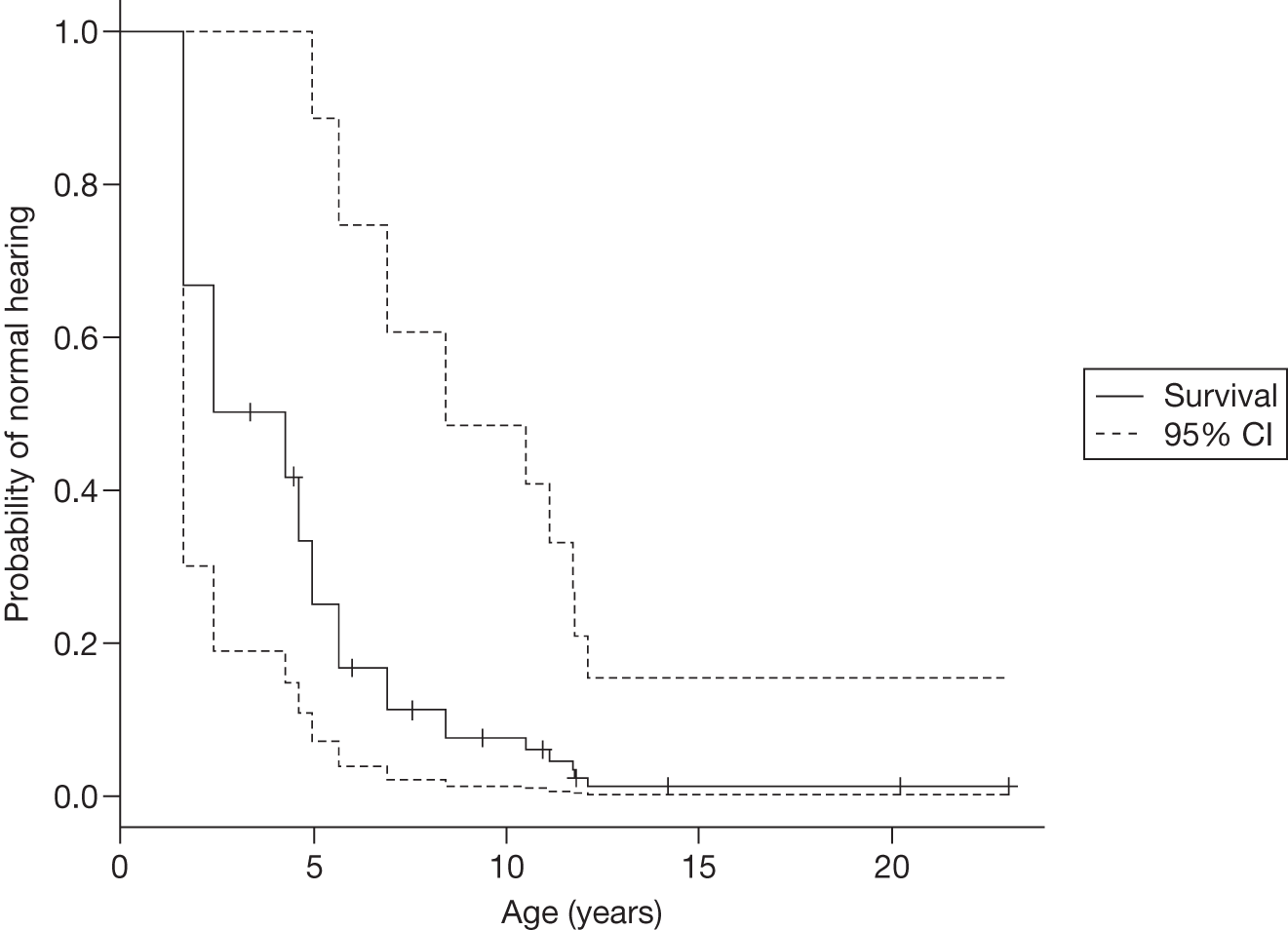
Cardiac valve involvement
Participants were categorised according to whether or not they were recorded as having cardiac valve involvement at a particular age. The age at first report of heart valve disease was available for 19 patients. Figure 80 shows Kaplan–Meier curves illustrating the likelihood of patients being reported as having no valve disease by age for treated and untreated patients.
FIGURE 80.
Risk of having valve disease by age and treatment status for people with MPS II (Kaplan–Meier curve).
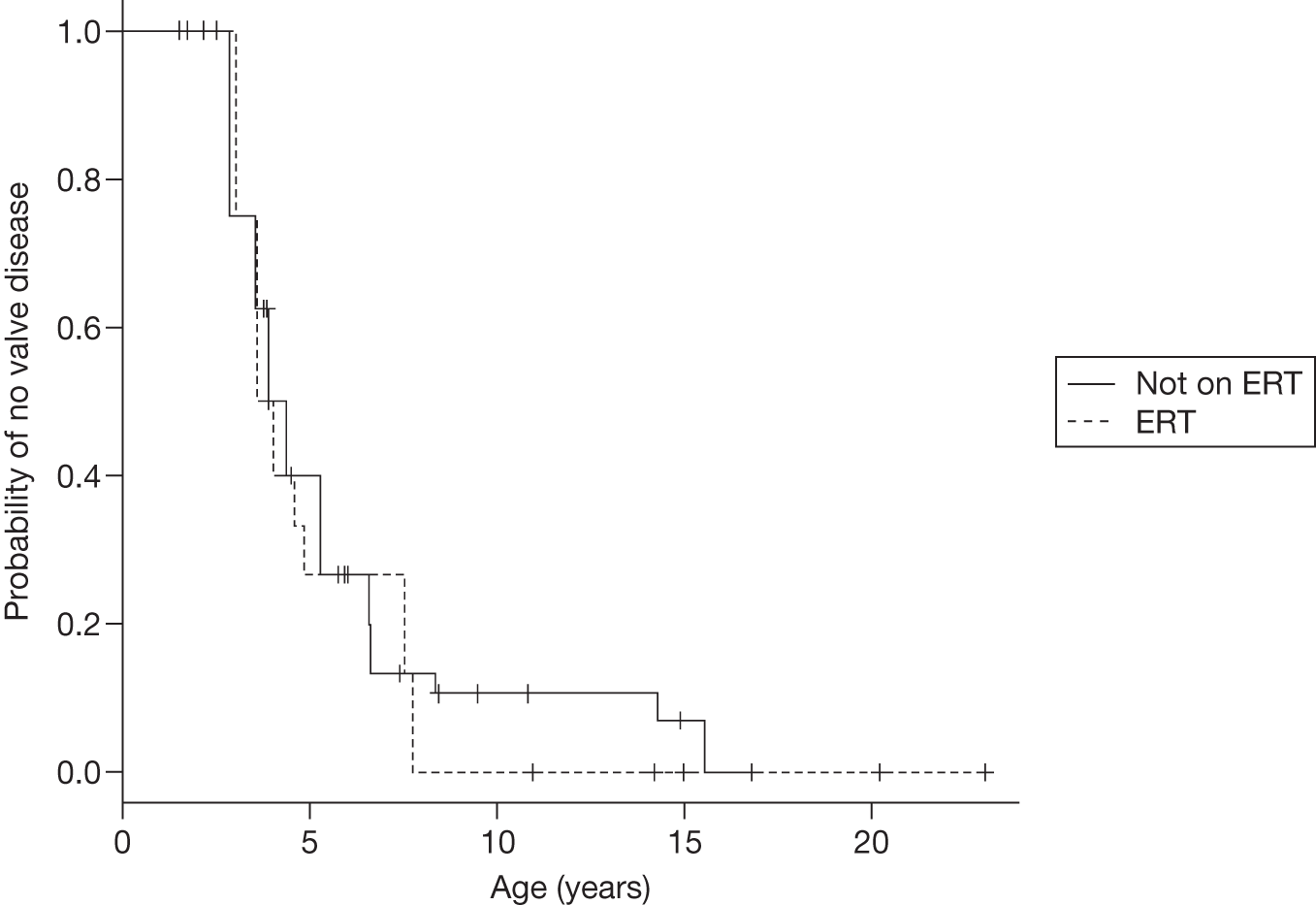
The risk of first reported valve disease by age did not differ between those on ERT and those not on ERT [HR (ERT/not on ERT) = 1.39; 95% CI 0.56 to 3.46; p = 0.48].
Carpal tunnel syndrome
Carpal tunnel syndrome was categorised in MPS II patients as present or absent. Data were collected for 19 patients who have CTS. Figure 81 shows Kaplan–Meier curves illustrating the risk of patients having CTS by age for treated and untreated patients. Overall, the HR was not significantly different between those on ERT and those not on ERT (HR = 1.44; 95% CI 0.36 to 5.76; p = 0.60).
FIGURE 81.
Risk of having CTS by age and treatment status for people with MPS II (Kaplan–Meier curve).
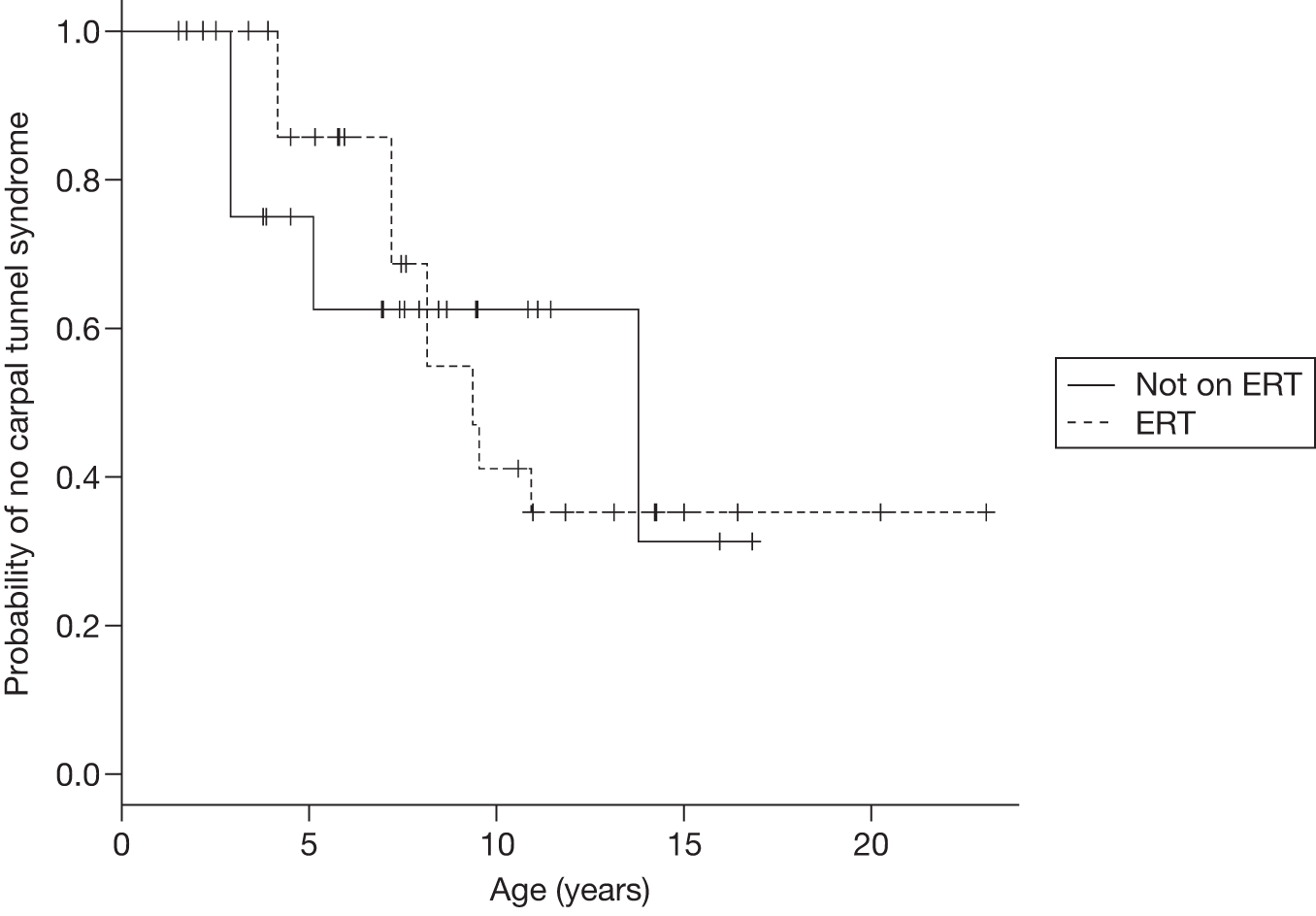
Palpable splenic enlargement
The presence or absence of an enlarged spleen was recorded for 20 patients, six of whom were reported as having an enlarged spleen on palpation. Figure 82 shows Kaplan–Meier curves illustrating the probability of not having a palpably enlarged spleen by age for treated and untreated patients.
FIGURE 82.
Risk of having an enlarged spleen by age and treatment status for people with MPS II (Kaplan–Meier curve).
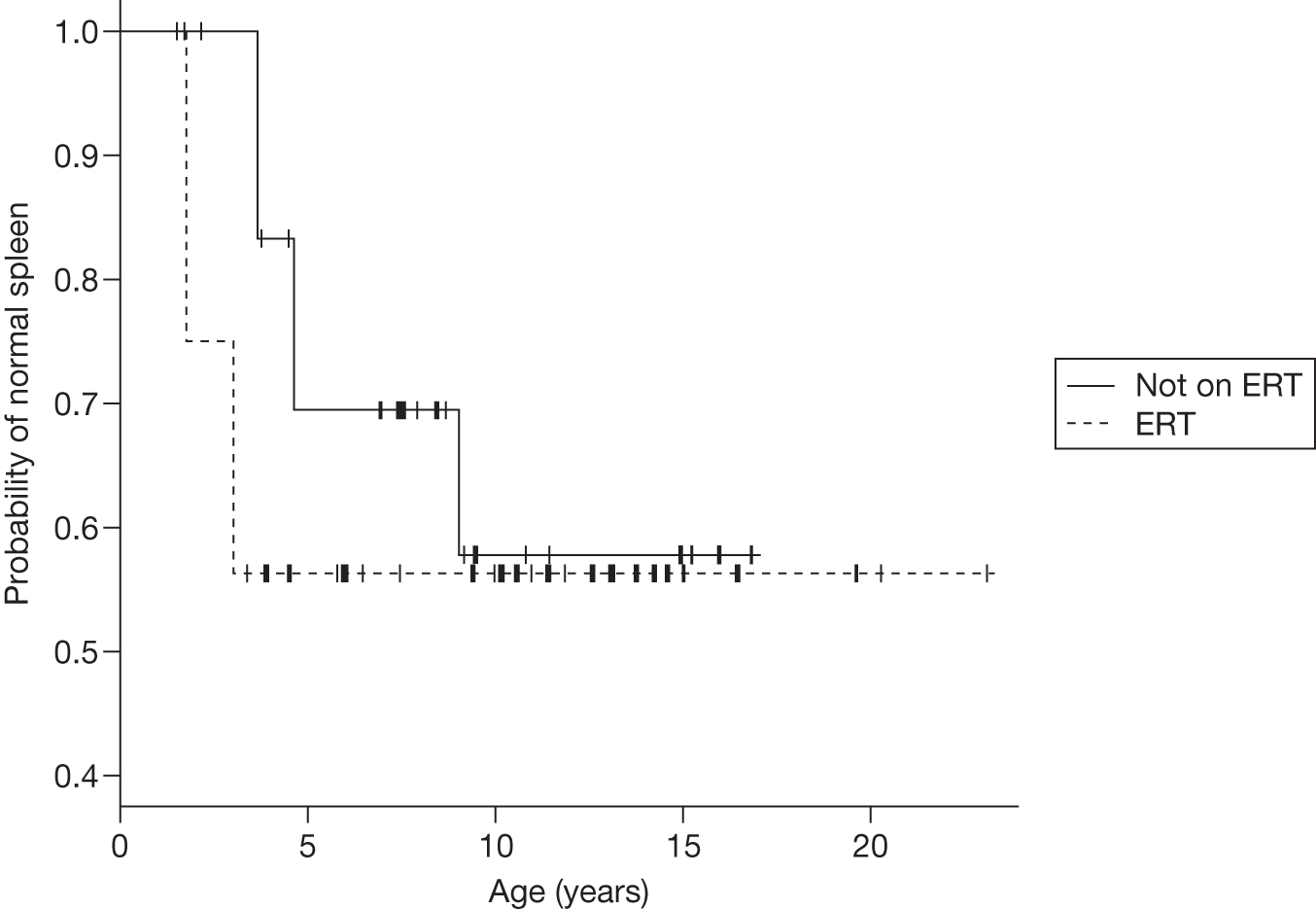
The model provides no evidence of a difference in the proportion of patients with an enlarged spleen associated with the use of ERT (HR for being on ERT relative to not on treatment = 0.62; 95% CI 0.09 to 3.98; p = 0.61).
Palpable liver enlargement
The presence or absence of having an enlarged liver was reported for 23 patients, 13 of whom were reported as having an enlarged liver on palpation. Figure 83 shows Kaplan–Meier survival curves illustrating the probability of not having a palpably enlarged liver by age for treated and untreated patients.
FIGURE 83.
Risk of having an enlarged liver by age and treatment status for people with MPS II (Kaplan–Meier curve).
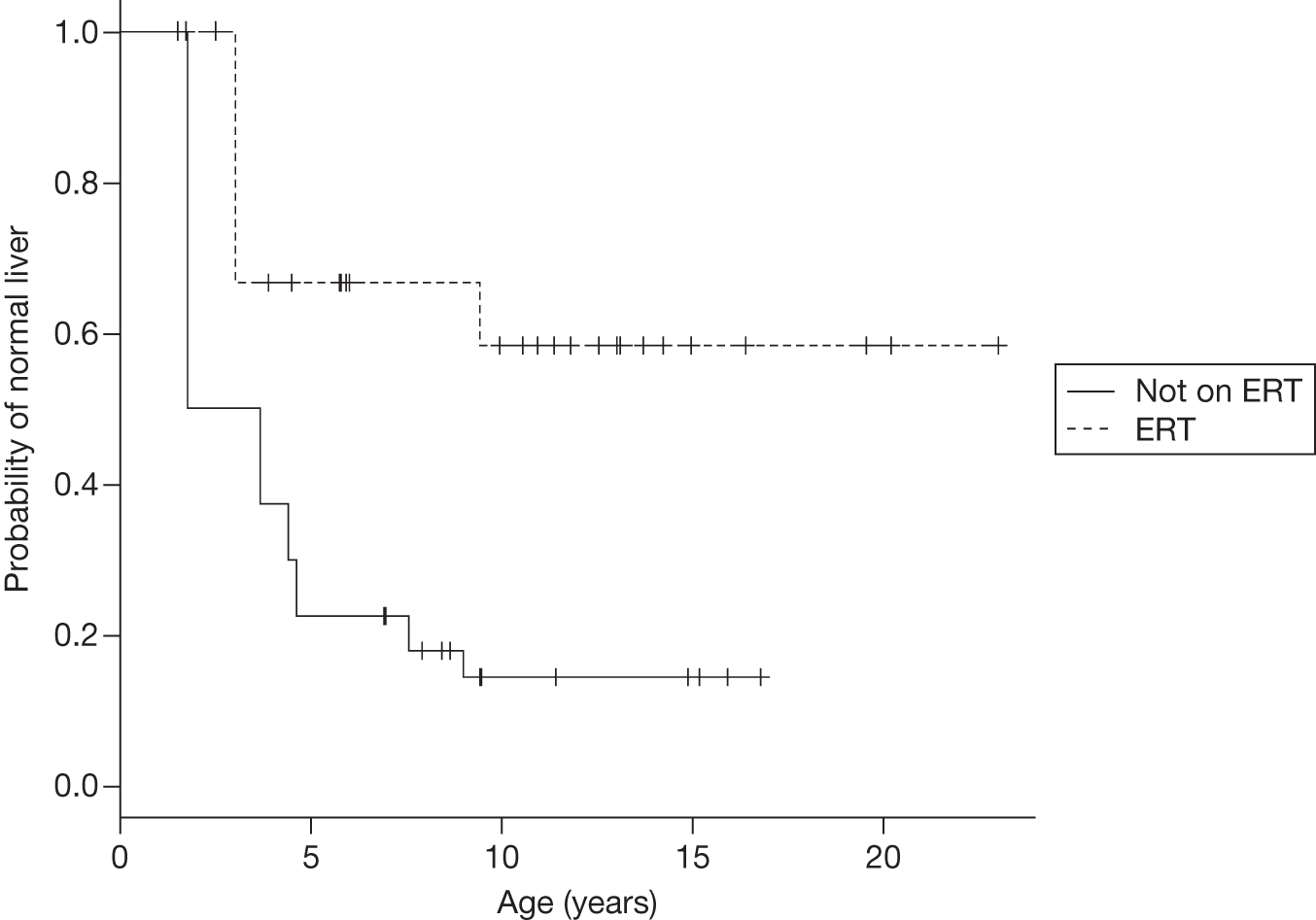
The model provides no evidence of a difference in the proportion of patients with an enlarged liver associated with the use of ERT (HR for being on ERT relative to not on treatment = 0.27; 95% CI 0.05 to 1.36; p = 0.11).
Quality-of-life assessments
SF-36
With only three adults with MPS II in this study, we had insufficient SF-36 data to analyse.
EQ-5D
Participants aged ≥ 13 years were invited to complete the EQ-5D questionnaire. Seventeen EQ-5D questionnaires were completed across all prospective time points. Data are presented in Table 114 for the EQ-5D index score (range from –0.095 to 1.0) with 1.0 being ‘perfect health’.
| Estimate of increment in EQ-5D | Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Current age | ||||
| Linear effect/year | –0.02 | 0.025 | –0.07 to 0.03 | 0.47 |
| Time on ERT | ||||
| Linear effect/year | 0.11 | 0.07 | –0.03 to 0.25 | 0.16 |
| Variance components | ||||
| Individual | 0.00 | |||
| Centre | 0.00 | |||
| Residual | 0.085 | |||
A longitudinal model was fitted to assess the linear relationship between EQ-5D and time on ERT after adjusting for age.
The linear model showed no significant association in EQ-5D with age (p = 0.47) or time on ERT (p = 0.16). No evidence was found for a non-linear association between the EQ-5D score and time on ERT (edf = 1.0; p = 0.17) (Figure 84), although it is important to note that these data are very sparse.
FIGURE 84.
The age-adjusted association between time on ERT and EQ-5D score in people with MPS II (time on ERT treated as a continuous variable).
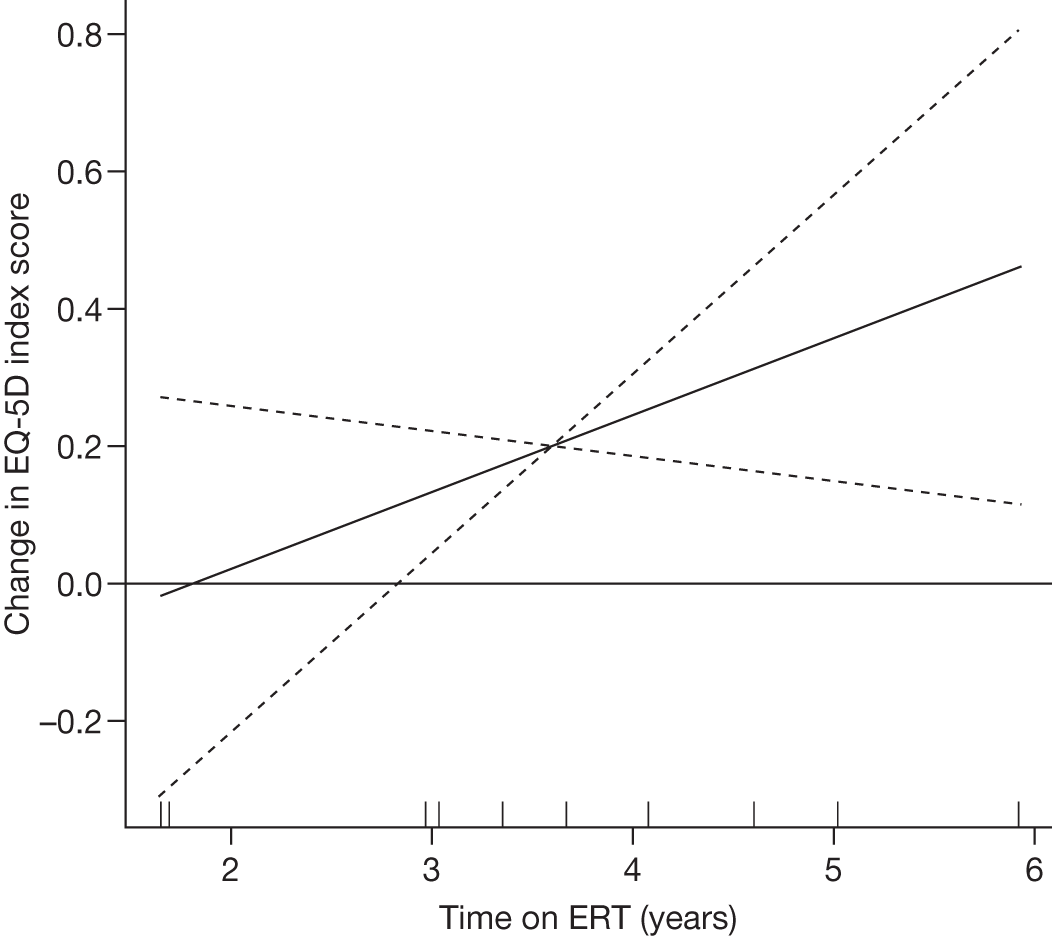
Equivalent modelling analyses were also conducted using SF-6D (SF-36-derived) utility weights, but no statistically significant associations (at α = 0.05 level) were found with time on ERT. The tabulated results of the SF-6D longitudinal modelling analyses are available on request from the study authors.
Visual analogue scale
In addition to scoring on the five domains of the EQ-5D participants were asked to rate their health on a VAS. The EQ-5D VAS asks people to rate their health state on a 10-cm line from 0, ‘worst imaginable health state’, to 100, ‘best imaginable health state’. The 17 VAS scores completed for this study ranged from 3 to 100. A longitudinal model was fitted to assess the linear relationship between the visual analogue score and time on ERT after adjusting for age.
The linear model shown in Table 115 shows no significant association in EQ-5D with age (p = 0.85) or with time on ERT (p = 0.89).
| Estimate of increment in EQ-5D VAS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Current age | ||||
| Linear effect/year | –0.48 | 2.40 | –5.18 to 4.22 | 0.85 |
| Time on ERT | ||||
| Linear effect/year | –0.93 | 7.32 | –15.3 to 13.4 | 0.89 |
| Variance components | ||||
| Individual | 0.00 | |||
| Centre | 0.00 | |||
| Residual | 674 | |||
PedsQL
Twenty-eight MPS II patients, or their parents or carers, completed the age-appropriate PedsQL questionnaire on at least one occasion, and 10 further questionnaires were completed at one of their annual check-ups.
Table 116 shows a descriptive summary of the PedsQL data, dichotomised by length of time (> 3 years or ≤ 3 years) on ERT.
| Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Physical health summary score | Total score | |
|---|---|---|---|---|---|---|---|
| Overall | |||||||
| Mean (SD) | 39.9 (31.3) | 46.3 (19.4) | 53.6 (31.2) | 47.8 (23.2) | 47.6 (18.2) | 39.9 (31.3) | 42.1 (17.2) |
| n | 36 | 36 | 36 | 26 | 26 | 36 | 25 |
| ≤ 3 years on ERT | |||||||
| Mean (SD) | 39.8 (30.9) | 45.1 (20.2) | 53.3 (30.1) | 42.5 (18.8) | 44.9 (16.1) | 39.8 (30.9) | 39.3 (13.1) |
| n | 25 | 25 | 25 | 16 | 16 | 25 | 15 |
| > 3 years on ERT | |||||||
| Mean (SD) | 46.02 (45.0) | 52.5 (22.4) | 78.2 (35.7) | 61.2 (22.5) | 62.7 (24.4) | 46.02 (45.0) | 53.6 (27.3) |
| n | 5 | 5 | 5 | 4 | 4 | 5 | 4 |
Table 117 shows the age-adjusted effects of duration of treatment on each of the PedsQL measures.
| Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Total score | |
|---|---|---|---|---|---|---|
| Current age | ||||||
| Mean increment (SD)/year | –6.67 (1.86) | –2.41 (1.21) | –0.69 (2.19) | 0.87 (2.02) | 1.53 (1.78) | 0.01 (1.56) |
| 95% CI | –10.3 to –3.03 | –4.79 to –0.02 | –5.0 to 3.61 | –3.09 to 4.85 | –1.96 to 5.03 | –3.04 to 3.07 |
| p-value | < 0.001 | 0.06 | 0.75 | 0.67 | 0.40 | 0.99 |
| Time on ERT | ||||||
| Mean increment (SD)/year | 10.08 (5.08) | 6.61 (3.76) | 1.78 (5.86) | 6.31 (4.58) | 4.19 (4.42) | 8.94 (3.81) |
| 95% CI | 0.13 to 20.0 | –0.75 to 13.9 | –9.72 to 13.3 | –2.67 to 15.3 | –4.47 to 12.8 | 1.47 to 16.4 |
| p-value | 0.06 | 0.09 | 0.76 | 0.18 | 0.35 | 0.03 |
| Variance components | ||||||
| Individual | 294.4 | 0.00 | 680.4 | 292.7 | 117.9 | 0.00 |
| Centre | 199.6 | 113.2 | 0.00 | 0.00 | 0.00 | 0.00 |
| Residual | 382.4 | 340.9 | 325.4 | 108.05 | 197.7 | 225.5 |
The linear model shows no significant association between total PedsQL score with age (p = 0.99). Although all the subscales showed some improvement with time on ERT (after adjusting for age), the differences did not achieve conventional levels of statistical significance for any of the subscales. However, the model does suggest a statistically significant association between time on ERT and an improvement in overall QoL score (p = 0.03).
Fatigue Severity Scale
Four FSS questionnaires were completed by two patients prospectively. The scores ranged from 3.22 to 6.0. There was not enough data to carry out further analysis.
Carer Strain Index
Thirty-seven CSI questionnaires were completed across all prospective time points. Data for the CSI total score ranged from 3 to 26; the maximum possible of 26 signifies the greatest degree of caregiver burden. A longitudinal model was fitted to assess the linear relationship between the CSI and time on ERT, after adjusting for age (Table 118).
| Estimate of increment in CSI | Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Current age | ||||
| Linear effect/year | 0.86 | 0.45 | –0.02 to 1.74 | 0.07 |
| Time on ERT | ||||
| Linear effect/year | –1.68 | 1.07 | –3.77 to 0.42 | 0.12 |
| Variance components | ||||
| Individual | 31.8 | |||
| Centre | 5.2 | |||
| Residual | 7.6 | |||
The linear model shows no significant association between score on the CSI with age of person they are caring for (p = 0.07) or with time on ERT (p = 0.12).
No evidence was found for a non-linear association between the CSI and time on ERT (edf = 1; p = 0.12) (Figure 85).
FIGURE 85.
The age-adjusted association between time on ERT and the CSI for carers of people with MPS II (time on ERT treated as a continuous variable).
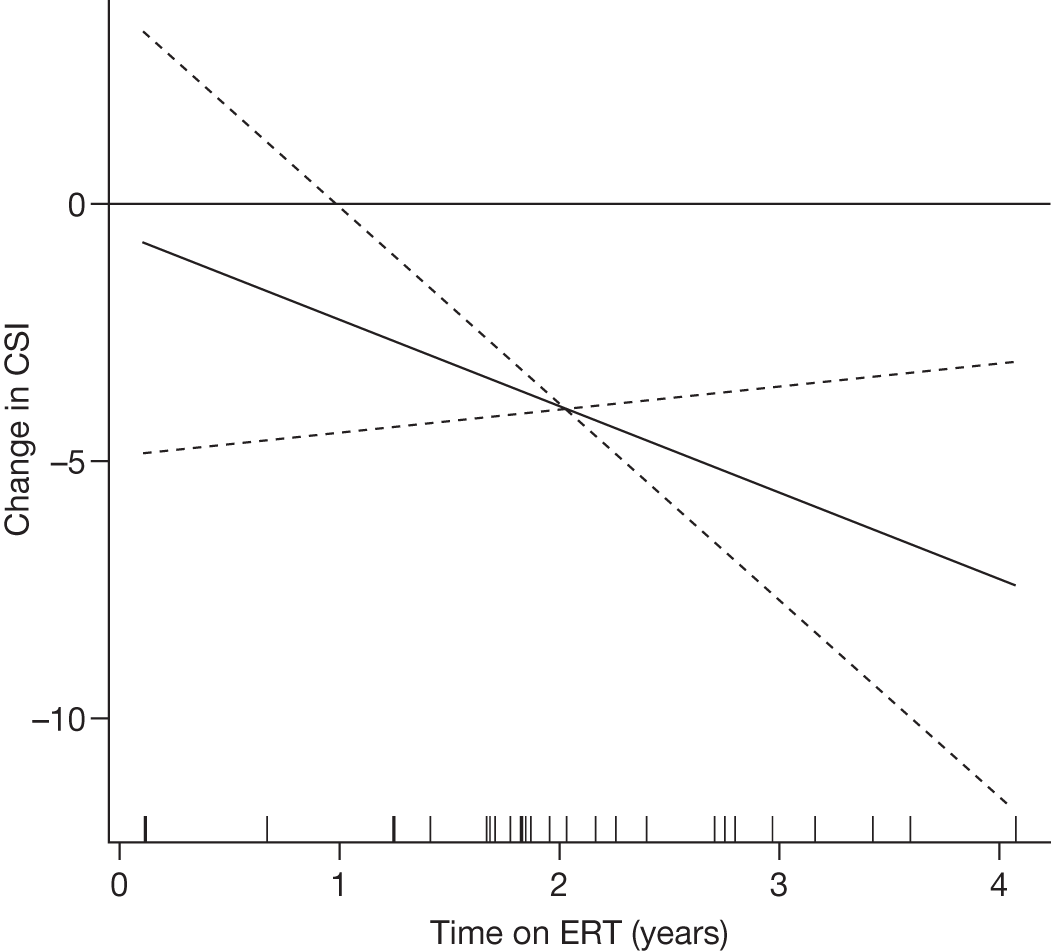
Safety and complications
Of the 39 participants with MPS II in this study, 1 patient reported that they had experienced anaphylactic reactions, 10 patients required pre-medication and 13 patients had reported positive antibody status to infused products. No MPS II patients reported experiencing any febrile reactions.
There were no reports of any patients stopping ERT during the period of data collection and no patients died from disease-related complications.
Cost of enzyme replacement therapy in people with MPS II
Table 119 shows the current purchase cost to the NHS of the ERT idursulfase.
| Drug full name | Proprietary name and unit | 2011 base price per unit (£) |
|---|---|---|
| Idursulfase | Elaprase®, 6 mg | 1985.00 |
Table 120 shows the NSCT-estimated annual NHS per patient cost of providing these drugs. Note that these costs include both the drug costs and home-care costs where the NSCT fund them.
| Drug | Adults | Children |
|---|---|---|
| Idursulfase 6 mg | £537,605 | £314,004 |
Cost of care for adults with MPS II
Total care cost – financial burden of MPS II
Table 121 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for an adult with MPS II, although this is only based on seven annual data points from four patients. Of the estimated mean per patient annual cost of £11,900, nearly two-thirds is as a result of NHS hospital services used, about half (£3800 per patient per year) from inpatient stays and about one-third (£2600) from outpatient visits (see Table 122). Of the £4400 per patient per year from using services outside hospital, < £100 per patient is as a result of GP visits and the remainder is almost entirely as a result of regular visits from either health visitors or other nurses, or other therapists for three of the patients (note that many of these will be for regular ERT infusions) (see Table 123).
| Type of service | No. with valid resource use dataa | Per cent of all at study entry | No. who used this type of service | Mean cost (£) | Standard deviation | Medianb cost (£) | Interquartile rangeb (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 7a | 100 | 6 | 7500 | 8535 | 1900 | 470–15,400 |
| Services outside hospital | 7a | 100 | 7 | 4400 | 7333 | 170 | 54–14,900 |
| Total health (NHS) and social-care cost | 7a | 100 | 7 | 11,900 | 11,180 | 15,500 | 520–20,400 |
| Type of hospital care | No. who used this type of service | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| Inpatient stays | 2 | 3800 | 7488 | N/A | 7000–19,700 |
| Outpatient visits | 6 | 2600 | 5659 | 570 | 470–1200 |
| Day cases | 2 | 1000 | 2244 | N/A | 1300–6000 |
| Accident and emergency visits | 0 | 0 | 0 | 0 | 0–0 |
| Total hospital (NHS) care cost | 6 | 7500 | 8535 | 1900 | 470–15,400 |
| Service provider | No. who used this provider | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 5 | 92 | 114 | 82 | 27–320 |
| GP nurse appointments | 5 | 12 | 18 | 10 | 5–52 |
| District nurses | 0 | 0 | 0 | 0 | 0–0 |
| Community mental health nurse | 0 | 0 | 0 | 0 | 0–0 |
| Other nurse or health visitor | 1 | 2200 | 5837 | 15,400 | 0–0 |
| Counsellor | 0 | 0 | 0 | 0 | 0–0 |
| Other therapist | 2 | 2100 | 5610 | N/A | 37–14,900 |
| ‘Alternative’ medicine or therapy | 0 | 0 | 0 | 0 | 0–0 |
| Psychologist | 0 | 0 | 0 | 0 | 0–0 |
| Psychiatrist | 0 | 0 | 0 | 0 | 0–0 |
| Other community-based doctor | 0 | 0 | 0 | 0 | 0–0 |
| Occupational therapist | 0 | 0 | 0 | 0 | 0–0 |
| Social worker | 0 | 0 | 0 | 0 | 0–0 |
| Home help | 0 | 0 | 0 | 0 | 0–0 |
| Care attendant | 0 | 0 | 0 | 0 | 0–0 |
| Community support worker | 0 | 0 | 0 | 0 | 0–0 |
| Housing worker | 0 | 0 | 0 | 0 | 0–0 |
| All non-hospital NHS and social-care providers | 7 | 4400 | 7333 | 170 | 54–14,900 |
Cost breakdown by hospital- and community-based services
Tables 122 and 123 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by adults with MPS II. Only two of the seven patient-years for which there were valid service-use data involved hospital stays as inpatients, but this accounted for over half of the NHS hospital costs in MPS II adults. In contrast, all but one of the patient-years involved having at least one hospital outpatient attendance during the previous 12 months.
The vast majority of costs related to using community-based services were because of the two or three adults with MPS II who used home helps or who used health visitors or other nurses regularly. They accounted for £4300 of the £4400 estimated annual per patient cost of services used outside hospital. Although over five of the annual data periods involved the adults with MPS II seeing their GP at least once during the year, as well as five seeing a practice nurse at their GP surgery, these accounted for < £110 of the £11,900 annual cost of NHS and publicly funded social-care services consumed. There was no evidence of using other types of care or support providers, but this probably mostly reflects the very small sample of patients and responses available for analysis (Table 123).
All of the resource use and cost findings relating to adults with MPS II should be treated with considerable caution because of the small number of patients for whom we have data.
Cost of care for children with MPS II
Total care cost – financial burden of MPS II
Table 124 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for a child with MPS II, based on the 29 children whose parents/carers provided service-use data at baseline (all male, mean age 9 years, age range 2–15 years). Of the estimated mean per patient annual cost of £7600 nearly two-thirds is as a result of NHS hospital services used, and of this almost two-thirds (£2900 per patient per year) is from inpatient stays, whereas over half (£1960) is from outpatient visits or day case treatment (see Table 125). Of the £2800 per patient per year from using services outside hospital, the majority (£2000) is as a result of regular visits from health visitors or other nurses (see Table 126).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. who used this type of service | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 29 | 81 | 23 | 4800 | 10,000 | 2700 | 1300–4600 |
| Services outside hospital | 29 | 81 | 29 | 2800 | 5310 | 500 | 130–1800 |
| Total health (NHS) and social-care cost | 29 | 81 | 29 | 7600 | 11,806 | 3500 | 1200–5900 |
| Type of hospital care | No. who used this type of service | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| Inpatient stays | 11 | 2900 | 7926 | 1900 | 1200–11,100 |
| Outpatient visits | 12 | 660 | 1260 | 1100 | 600–1900 |
| Day cases | 14 | 1300 | 2781 | 1300 | 940–3300 |
| Accident and emergency visits | 8 | 50 | 94 | 100 | 100–310 |
| Total hospital (NHS) care cost | 23 | 4800 | 10,000 | 2700 | 1300–4600 |
| Care provider | No. who used this provider | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 29 | 260 | 466 | 130 | 82–240 |
| GP nurse appointments | 11 | 3 | 5 | 5 | 5–10 |
| District nurses | 2 | 2 | 9 | N/A | 3–48 |
| Community mental health nurse | 0 | 0 | 0 | 0 | 0–0 |
| Other nurse or health visitor | 14 | 2000 | 5115 | 59 | 44–4200 |
| Counsellor | 1 | 6 | 33 | 180 | N/A |
| Other therapist | 4 | 38 | 165 | 74 | 39–890 |
| ‘Alternative’ medicine or therapy | 0 | 0 | 0 | 0 | 0–0 |
| Psychologist | 3 | 20 | 67 | 160 | 81–320 |
| Psychiatrist | 0 | 0 | 0 | 0 | 0–0 |
| Other community-based doctor | 6 | 95 | 277 | 190 | 160–750 |
| Occupational therapist | 13 | 49 | 118 | 51 | 38–120 |
| Social worker | 11 | 200 | 653 | 290 | 150–290 |
| Home help | 1 | 166 | 891 | 4800 | N/A |
| Care attendant | 0 | 0 | 0 | 0 | 0–0 |
| Community support worker | 1 | 0.29 | 1.5 | 8 | N/A |
| Housing worker | 0 | 0 | 0 | 0 | 0–0 |
| All non-hospital NHS and social-care providers | 29 | 2800 | 5310 | 500 | 130–1800 |
Cost breakdown by hospital- and community-based services
Tables 125 and 126 show the cost breakdown of the hospital and community (non-hospital) services and care professionals used by children with MPS II. Over one-third (11) of the children had hospital stays as inpatients, and this accounted for £2900 of the mean NHS hospital cost in MPS II children. The most costly inpatient stays were for gastrostomy operations for two children (40- and 15-day hospital stays costing an estimated £37,600 and £14,000, respectively), for ‘back problems’ (£9400) and for changing a tracheostomy (£4700). Just over one-third of the children with MPS II had hospital outpatient attendances or day case treatment during the year, mainly for a range of ENT, audiology, CTS, paediatrician, MRI scans and metabolic reasons. Four children had regular hospital-based ERT infusions which contributed to higher mean outpatient costs.
The majority of costs related to using community-based services were due to the relatively costly 14 children with MPS II who were seen by health visitors or other nurses regularly. They accounted for £2000 of the £2800 estimated annual per patient cost of services used outside of hospital. All 29 of the children with MPS II saw their GP at least once during the year, as well as over one-third (11) of the children seeing a practice nurse at their GP surgery, but together these accounted for only £263 of the £2800 annual cost of NHS and publicly funded social-care services consumed. Thirteen and four children, respectively, saw occupational therapists or physiotherapists during the previous year, and over one-third (11) of the children saw a social worker (see Table 123).
All of the resource use and cost findings relating to children with MPS II should be treated with considerable caution because of the small number of patients for whom we have data.
Association of time on enzyme replacement therapy and cost of caring for patients with MPS II
From the longitudinal regression modelling of costs for people with MPS II, in child patients there was no statistically significant association (i.e. p-value < 0.05) between time on ERT and total NHS and social-care costs or non-hospital-care costs. However, there was a statistically significant association between hospital costs and time on ERT (costs 3.78 times higher, 95% CI 2.7 to 5.3; p < 0.001), but this was based on only 33 data points from 24 children (only nine with data from more than one time point) and is in the opposite direction to what would be expected, and so should be interpreted with considerable caution. The tabulated results of these analyses are available on request from the study authors.
Discussion of MPS II results
Mucopolysaccharidosis type II is an X-linked recessive LSD in which there is a deficiency of the enzyme iduonate-2-sulfatase, resulting in an accumulation of GAGs. Idursulfase has been available and licensed in England since 2007.
Evidence for the effectiveness of ERT in MPS II depends primarily on two randomised trials,167,168 an open-label, before-and-after study169 and an open-label extension of the trial reported by Muenzer and colleagues in 2011. 170 The results are summarised in Table 7 in Chapter 1. Muenzer and colleagues167 randomised 96 patients with the attenuated form of MPS II to placebo or to one of two different dosing regimens of idursulfase and followed for 1 year. Both dosing groups were reported to show improvements in a composite outcome measure (including the 6-minute walk test and FVC) compared with placebo. These improvements were reported to be sustained over the 2 years of the extension study. 170 The much smaller study from the same group (12 patients randomised in equal proportions to placebo and two different dosing schedules of idursulfase) also reported significant reductions in GAG excretion and improvements in clinical outcomes, including liver and spleen size, 6-minute walk test, mobility at some joints and FVC. The small before-and-after study reported by Okuyama and colleagues169 of 10 patients reported improvements across a number of clinical outcomes over the course of 12 months.
Patient characteristics
Of the 51 MPS II patients deemed eligible for inclusion, 43 patients were invited to participate and 39 patients (36 children and 3 adults) agreed to participate. Of the 14 patients who were not approached or declined to participate, seven were adults and two were teenagers when first approached. Over half (21/39) of the MPS II patients who participated had the more severe form of the disease.
Height
In the first years of life the height of most patients with MPS II is above the 50th percentile; however, their rate of growth decreases with age and by the age of 8 years, height is below the third percentile. 270
Our findings suggest that relative to their peers the height of children with MPS II decreases with age. We found a statistically significant association between duration of ERT and increasing height z-scores (p < 0.001), suggesting a significant positive effect of treatment on growth. These findings reinforce those reported by Schulze-Frenking and colleagues271 who studied 18 children with attenuated MPS II, nine of whom had started ERT before 10 years of age and nine who had started ERT after 10 years of age. This group reported that ERT increased the rate of growth in both groups but the effect was greater in the group who started ERT before 10 years old.
Forced vital capacity
Respiratory disease and upper airway symptoms are characteristic in both forms of MPS II and become more severe with age. Previous studies (notably the relatively large trial reported by Muenzer and colleagues167) have reported improvements in FVC with treatment with ERT.
We were hampered in this study by the lack of recording of FVC for most included patients. Analysis of the 34 recorded measures of FVC from 14 patients suggested no evidence of an association between per cent predicted FVC and age or time on ERT. The lack of data inevitably means that these analyses are underpowered.
Mobility
Patients with MPS II can have severe skeletal deformities leading to limited joint mobility. Previous studies, notably Muenzer and colleagues,167 have reported substantial improvements in the distance that patients are able to walk (6-minute walk tests at 1 year reported improvement in the ERT group of 44.3 m compared with 7.3 m in the placebo group; p = 0.0131) when treated with ERT compared with placebo. The reports of effects on joint mobility are more mixed with differences reported in mobility at the elbow but not in other joints.
We were again hampered in our analyses by a paucity of data recorded for the majority of included patients with 83 estimates of distance walked in 6 minutes from 23 patients recorded. There was no association between the distance walked and age or time on ERT. Nor did we find any difference in the risk of having restricted mobility between those on ERT and those not receiving ERT. In interpreting these analyses it is important to note that the group of patients for whom we have data on these outcomes includes a disproportionately large number who have the more severe form of the condition while the trials were conducted among those with the more attenuated form.
Hearing
Hearing loss is common in patients with severe MPS II, with most patients having severe combined hearing loss once the disease is fully established. 272
Of the 20 participants with MPS II for whom hearing data were recorded, 18 were classified as having impaired hearing, with our model suggesting 40% of infants have impaired hearing at 1 year of age. The likelihood of unimpaired hearing dropped to about 8% by 5 years of age. By the age of 10 years, only 2% of patients with MPS II had normal hearing. Insufficient data were available to estimate whether or not the use of ERT reduced the likelihood of hearing loss and no protective effect has been reported from other studies.
Cardiac valve involvement
Cardiac abnormalities are common in patients with MPS II with the median age at onset of valve disease reported in a cohort of 82 patients as 6.2 (range 2.9–13.8) years. In a recent study of 24 MPS patients (eight patients with MPS I, six patients with MPS II and 10 patients with MPS IV) that looked at the effect of ERT on cardiological aspects of the conditions, the authors reported no positive effect of ERT on cardiac valves, concluding that ‘these valves are only slightly accessible to ERT’. 273 Our data provide no evidence for an effect of ERT on the timing of onset of valve disease in the 19 patients for whom these data were recorded.
Carpal tunnel syndrome
Carpal tunnel syndrome is common in MPS II patients. A study of seven MPS II patients (aged 4–16 years) with musculoskeletal abnormalities reported no effect of ERT and the risk of having or developing CTS. 274 Similarly, of the 19 patients for whom there are data on the presence or absence of CTS in this study, we found no difference in the risk of having CTS between those on ERT and those not receiving ERT.
Palpable splenic and liver enlargement
The accumulation of GAGs leads to enlargement of spleen and liver, although this does not appear to impair function. Previous studies and clinical experience (Stephen Waldek, Retired Metabolic Physician, formerly of Salford Royal NHS Foundation Trust, 2012, personal communication) have suggested that use of ERT leads to substantial reductions in liver and spleen volume.
Disappointingly, data on spleen size were available for only 20 patients in this study, with six patients reported as having a clinically enlarged spleen. Data on liver size were available for 23 patients, 13 of whom were reported as having palpably enlarged livers. We did not find a statistically significant association with use of ERT for either spleen or liver enlargement. The paucity of data makes these results difficult to interpret.
Costs associated with MPS II
As with all other conditions investigated in this study, we were keen to capture the wider costs of care falling on the public sector in addition to the costs associated with ERT.
Based on patients’ self-reported health- and social-care service use, the annual cost of caring for people with MPS II, excluding the purchase cost of ERT, was estimated at £11,900 for an adult and £7600 for a child. These costs, however, are dwarfed by the cost of the therapies; the mean annual cost of ERT for adults with MPS II is £537,605 and for children with MPS II is £314,004.
From the longitudinal regression modelling of costs, there was no statistically significant association (i.e. p-value < 0.05) between time on ERT and either total NHS and social-care costs, hospital-care costs, or non-hospital-care costs for patients with MPS II. The tabulated results of these analyses are available on request from the study authors.
Owing to these high associated costs, and the lack of measureable effect of ERT on either clinical outcomes or HRQoL measures, it was infeasible to conduct either a cost-effectiveness or cost–utility analysis. As they apply to all six LSDs, the limitations of these cost estimates are summarised and discussed in Chapter 9.
Chapter 7 Results – Pompe disease
Patient characteristics
At the start of the study, 93 patients were identified by the treating centres as having Pompe disease. Of these, 89 were deemed eligible for inclusion and 81 patients (91% of those deemed eligible) were approached in clinic and invited to participate in the study. Seventy-seven patients (95% of those approached) consented to participate. Patient demographic characteristics are presented in Tables 127 and 128.
| Patient characteristic | Late onset (N = 62) |
|---|---|
| Gender | |
| Male, n | 37 |
| Female, n | 25 |
| Age at recruitment (years) | |
| Mean (SD) | 46.5 (13.8) |
| Median (min.–max.) | 45.6 (16.3–76.7) |
| Age at diagnosis (years) | |
| Mean (SD) | 39.7 (15.2) |
| Median (min.–max.) | 37.7 (1.45–67.7) |
| Initial treatment | |
| Not on ERT, n | 3 |
| ERT (alglucosidase alpha), n | 59 |
| Age at first infusion (years) | |
| Mean (SD) | 45.5 (14.1) |
| Median (min.–max.) | 44.6 (16.4–74.7) |
| Time on ERT (years) | |
| Mean (SD) | 1.27 (0.8) |
| Median (min.–max.) | 1.26 (0–3.12) |
| Patient characteristic | Early onset (N = 12) | Late onset (N = 3) |
|---|---|---|
| Gender | ||
| Male, n | 6 | 3 |
| Female, n | 6 | 0 |
| Age at recruitment (years) | ||
| Mean (SD) | 3.1 (2.9) | 8.6 (6.4) |
| Median (min.–max.) | 2.6 (0.4–10.2) | 12.1 (1.28–12.5) |
| Age at diagnosis (years) | ||
| Mean (SD) | 0.69 (0.64) | 2.31 (2.1) |
| Median (min.–max.) | 0.49 (0.02–2.32) | 2.75 (0–4.2) |
| Initial treatment | ||
| Not on ERT, n | 0 | 0 |
| ERT (alglucosidase alpha), n | 10 | 3 |
| Clinical trial of ERT,a n | 2 | 0 |
| Age at first infusion (years) | ||
| Mean (SD) | 0.90 (0.81) | 6.49 (5.6) |
| Median (min.–max.) | 0.73 (0.05–2.65) | 8.97 (0.04–10.5) |
| Time on ERT (years) | ||
| Mean (SD) | 2.19 (2.9) | 2.14 (0.9) |
| Median (min.–max.) | 0.88 (0.15–9.7) | 1.34 (1.24–3.12) |
Twelve patients had a diagnosis of infantile-onset Pompe disease (characterised by the presence of cardiomyopathy) and 65 patients had a diagnosis of adult-onset Pompe disease. At recruitment, 15 of the participants were children (aged < 16 years) and 62 were adults. The average age of the infantile-onset patients at recruitment was 3.1 (range 0.4–10.2) years and the average age of children with adult-onset Pompe disease was 8.6 (range 1.28–12.5) years. The average age of adults (all with adult-onset Pompe disease) at recruitment was 46.5 (range 16.3–76.7) years. The average age at diagnosis of infantile-onset Pompe disease was 0.69 (range 0.02–2.32) years and that of adult-onset Pompe disease was 38.01 (range 0–67.7) years.
At the time of recruitment into the study, all the infantile-onset patients and almost all adult-onset patients (62 out of 65) received alglucosidase alpha. The average time on ERT was 2.19 (range 0.15–9.7) years for infantile-onset patients and 1.31 (range 0–3.12) years for adult-onset patients.
We collected data from all patients at the time of recruitment. Data were also collected from 71 patients at a second data point, and from 55 patients at a third data point. The number of retrospective data points per patient ranged from 1 to 4.
Separate results are presented for infantile and adult onset owing to the differing patterns of morbidity associated with these two forms of Pompe disease.
Key markers of disease progression
The following measures were identified as key markers of Pompe disease progression:
-
FVC
-
ventilation dependency
-
mobility
-
6-minute walk test
-
muscle strength
-
body mass index (BMI).
In addition, adults completed the SF-36, EQ-5D, FSS and the Service Use and Costs Questionnaire, whereas children or their carers completed the age-appropriate PedsQL questionnaire. Carers of children or adults were asked to complete the Service Use and Costs Questionnaire and the CSI.
Longitudinal models were fitted to assess relationships between continuous measures of function and length of time on ERT, after adjustment for age and clustering by centre. In the basic models, the effect of time on ERT was treated as a linear effect because of the small number of data points. Further analysis was conducted to explore the possibility that time spent on ERT would have a non-linear effect on function. Patients contributed data points to the model both before and after starting ERT.
Kaplan–Meier survival curves were estimated to illustrate differences in the age at first recorded occurrence of binary events that could be considered progressive including restricted mobility and ventilator dependency. Treatment group differences in survival function were tested using Cox regression models. Individual patients contributed intervals of time at risk to more than one of the treatment categories (no treatment, ERT) as appropriate.
Summary of results
Patients with infant-onset Pompe disease were analysed separately from the 65 patients with adult-onset Pompe disease. Only 12 patients with infantile-onset Pompe disease were included in this study and all started treatment at the time of diagnosis providing insufficient data to reliably estimate associations with ERT. Although we did not find evidence of an association with time on ERT and the CSI in the linear model, when time on ERT was treated as a continuous variable there was a significant association with time on ERT and a reduction in carer burden.
The remainder of this summary section refers to results of analyses confined to the patients with adult-onset Pompe disease.
These data provide evidence for an association between time on ERT and increased distance walked in the 6-minute walk test for patients with adult-onset Pompe disease. There is also evidence of a statistically significant association between time on ERT and increased muscle strength scores. However, improvements in both these measures are only seen over the first 2 years of treatment with ERT, although these results should be interpreted with caution because of the wide CIs. There were insufficient data to analyse the relationship between time on ERT and the risk of developing restricted mobility.
We found no evidence of a statistically significant association between the use of ERT and respiratory function as assessed by FVC or risk of becoming ventilator dependent among patients with adult-onset Pompe disease. There was no evidence of an association between BMI and time on ERT.
There was no statistically significant association between duration of treatment with ERT and QoL assessed by the SF-36, the EQ-5D or with fatigue assessed by the FSS. It is important to note that because the QoL data were not available retrospectively, this analysis essentially compares patients with different durations of time on treatment rather than comparing those not on treatment with those on ERT. Consequently, our analyses are not able to detect how QoL outcomes respond to initiation of treatment with ERT.
Per cent of predicted forced vital capacity
Adult-onset patients
Fifty-eight patients with adult-onset Pompe disease were able to complete an upright FVC test. Results are reported as a per cent of the predicted volume (FVC%). A total of 190 FVC measurements were recorded for these patients across all time points and the range of these measurements was 10–130%.
A longitudinal model was fitted to assess the linear relationship between FVC and time on ERT, after adjusting for age. As can be seen in Table 129, FVC was not significantly associated with age (p = 0.62). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and FVC. After adjusting for age, time on ERT was not significantly associated with change in FVC (p = 0.18). Further analysis was conducted to explore the shape of the relationship between FVC and time on ERT treated as a continuous variable. This analysis provided no evidence for a non-linear association between FVC and time on ERT (edf = 1.52; p = 0.22) (Figure 86).
| N Data | Estimate of change in FVC% | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 120 | 0.00 | |||
| Female | 70 | 12.7 | 7.2 | –1.41 to 26.8 | 0.08 |
| Age | |||||
| Linear effect/year | 0.12 | 0.24 | –0.35 to 0.59 | 0.62 | |
| Time on ERT | |||||
| Not on ERT | 52 | 0.00 | 0.18 | ||
| < 12 months | 37 | 1.91 | 1.27 | –0.58 to 4.39 | 0.14 |
| 12–36 months | 79 | 0.11 | 1.17 | –2.18 to 2.40 | 0.93 |
| > 36 months | 22 | –1.56 | 1.77 | –5.03 to 1.90 | 0.38 |
| Variance components | |||||
| Individual | 713.9 | ||||
| Centre | 0.0 | ||||
| Residual | 28.2 | ||||
FIGURE 86.
The age-adjusted association between time on ERT and FVC% in people with adult-onset Pompe disease (time on ERT treated as a continuous variable).
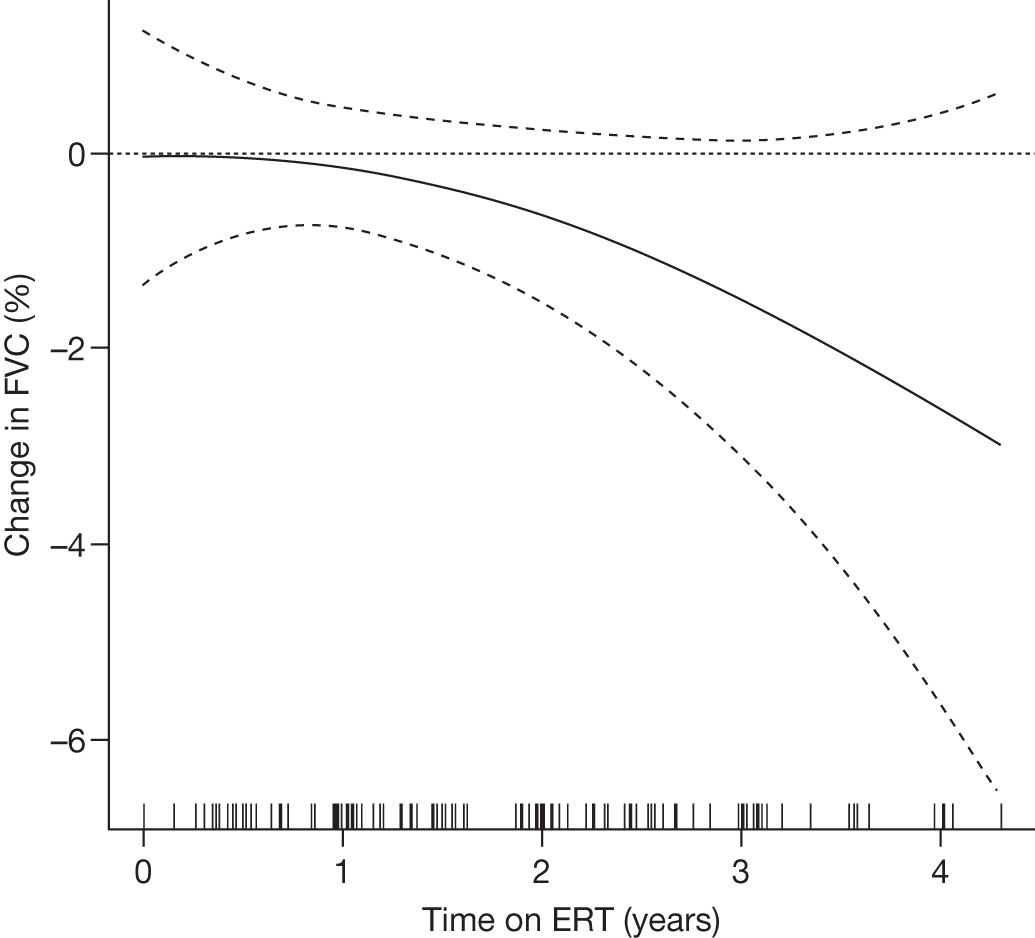
Ventilation
Adult-onset patients
We have ventilation data on 61 people with adult onset Pompe disease (58 on ERT and three not on treatment). We have 241 observations related to ventilation across all time points, where patients were categorised as being ventilator-free or ventilator-dependent.
Thirty-one patients (29 on ERT and two not on treatment) remained ventilator-free throughout the data collection period. By the age of 30, seven patients were reported as being ventilator-dependent (six on ERT and one not on treatment). By 50 years of age, a further 10 patients were ventilator-dependent (all on ERT) and a further 13 patients became ventilator-dependent after the age of 50 (all on ERT).
Infantile-onset patients
All of the infantile-onset patients were on ERT. Of the 12 patients, four were ventilator dependent at the time of being studied. There were insufficient data for further analyses.
Swallowing
Infantile-onset patients
One out of the 12 infantile-onset patients was reported as having swallowing difficulties by the fifth month of age. By the age of 4 years, 7 of the 12 infantile-onset patients reported having a swallowing difficulty. There were insufficient data for further analyses.
Mobility
Infantile-onset patients
Mobility data were collected for 11 infantile-onset Pompe disease patients. Of those, four were able to sit unsupported, three were able to stand and walk independently, two were able to walk upstairs one step, and two infants were unable to sit or stand unsupported. By the age of 5 years, 4 of the 11 early-onset patients were reported as having restricted mobility. Owing to the small numbers no further analyses were undertaken.
Adult-onset patients
We have mobility data on 64 adult onset Pompe patients, (61 on ERT and three not on treatment). We have 303 observations for mobility across all time points, where patients were categorised as being mobile if they could walk for 5 metres or stand for 6 minutes unaided, as having restricted mobility if they could walk aided with one or two sticks, or as being wheelchair dependent (i.e. immobile).
Thirty-seven patients (34 on ERT and three not on treatment) reported having no mobility problems at any point during data collection. Of the 27 patients with mobility problems (all on ERT), four reported having restricted mobility by the age of 30. By the age of 35, one additional patient reported having restricted mobility. By 60 years of age, 15 additional patients reported having restricted mobility, and seven more patients reported having restricted mobility after the age of 60. Nine of these patients (all on ERT) were reported as being wheelchair dependent by 60 years of age.
6-minute walk test
Adult-onset patients
We have a 6-minute walk test results for 22 adult-onset patients, with 71 measurements across all time points. The distances walked ranged from 88 to 581 m.
As can be seen in Table 130, the distance walked in 6 minutes by patients with adult-onset Pompe disease was significantly, negatively, associated with age (p < 0.001). After adjusting for age, time on ERT was associated with a significant positive change in distance walked (p = 0.0001). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and distance walked. Further analysis was conducted to explore the shape of the relationship between distance walked and time on ERT treated as a continuous variable. This analysis (illustrated in Figure 87) suggested a significant non-linear effect of time on ERT (edf = 2.8; p < 0.001). The graph suggests the distance walked by patients with adult-onset Pompe disease improves for the first 2 years after commencing ERT, when the treatment effect peaks before appearing to decline. However, the wide CIs mean that this result should be interpreted with caution as it may reflect selection effects in this small cohort.
| N Data | Estimate of change in distance walked (m) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 54 | 0.00 | |||
| Female | 17 | –65.5 | 36.4 | –137.2 to 5.44 | 0.07 |
| Current age | |||||
| Linear effect/year | –4.6 | 1.0 | –6.56 to –2.64 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 18 | 0.00 | < 0.001 | ||
| < 12 months | 10 | 36.3 | 14.4 | 8.07 to 64.5 | 0.014 |
| 12–36 months | 33 | 45.0 | 10.7 | 23.9 to 66.3 | < 0.001 |
| > 36 months | 10 | 4.16 | 16.6 | –28.4 to 36.7 | 0.80 |
| Variance components | |||||
| Individual | 5008.3 | ||||
| Centre | 3741.2 | ||||
| Residual | 1153.4 | ||||
FIGURE 87.
The age-adjusted association between time on ERT and distance walked in people with adult-onset Pompe disease (time on ERT treated as a continuous variable).
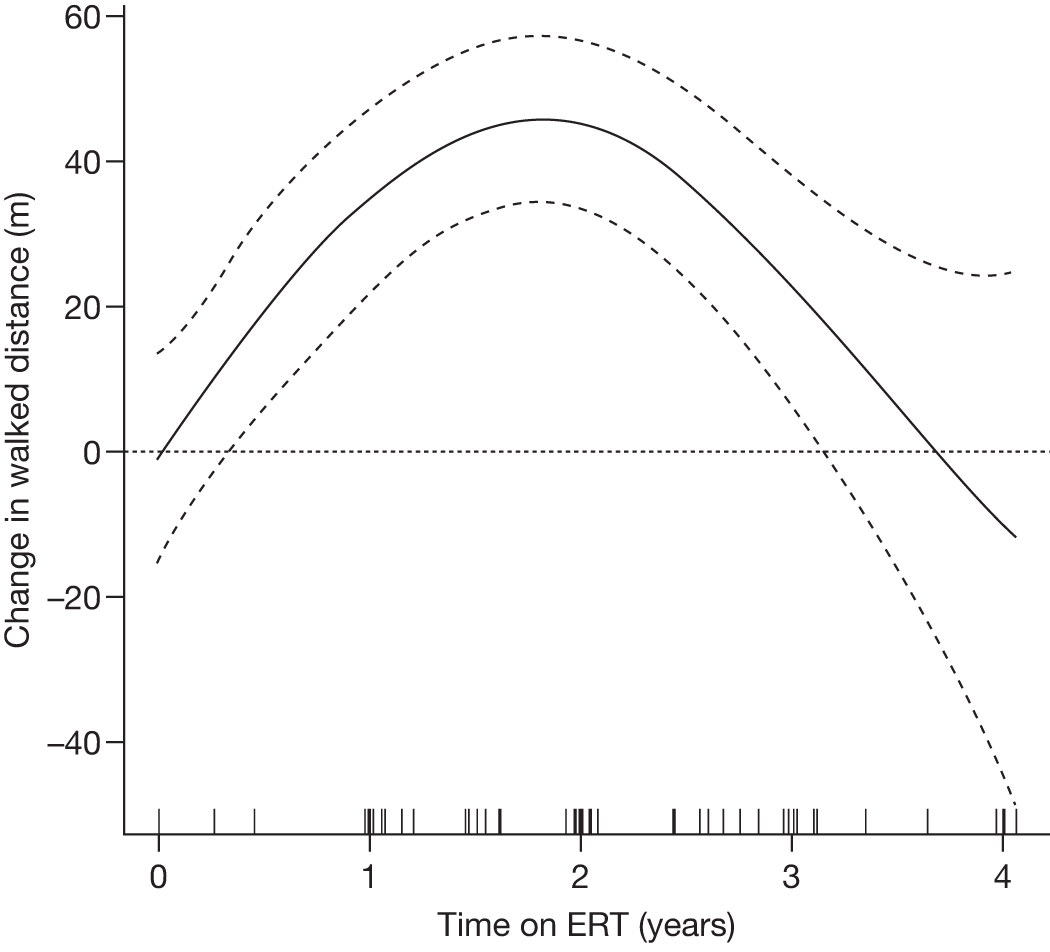
Muscle strength test
Adult-onset patients
We have muscle strength test data for 54 patients with adult-onset Pompe disease, with 176 scores for these patients across all time points. Scores ranged from 62 to 120.
As can be seen in Table 131, muscle strength score was not significantly associated with age in adult-onset patients (p = 0.56). After adjusting for age, time on ERT was associated with a significant positive change in muscle test score (p < 0.001). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and muscle test scores. Further analysis was conducted to explore the shape of the relationship between muscle test scores and time on ERT treated as a continuous variable. This analysis suggested that there was a significant non-linear association between muscle test score and time on ERT (edf = 2.71; p < 0.001) (Figure 88). This analysis appears to suggest that the scores improve for the first 2 years of treatment and then plateau. However, the wide CIs around the line mean that this result should be interpreted with caution.
| N Data | Estimate of change in muscle test score | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 113 | 0.00 | |||
| Female | 63 | 0.76 | 3.45 | –6.0 to 7.5 | 0.83 |
| Current age | |||||
| Linear effect/year | 0.07 | 0.12 | –0.16 to 0.30 | 0.56 | |
| Time on ERT | |||||
| Not on ERT | 56 | 0.00 | < 0.001 | ||
| < 12 months | 34 | 3.49 | 1.08 | 1.37 to 5.61 | 0.001 |
| 12–36 months | 73 | 4.02 | 0.89 | 2.27 to 5.76 | < 0.001 |
| > 36 months | 13 | 1.44 | 1.64 | –1.77 to 4.65 | 0.38 |
| Variance components | |||||
| Individual | 132.2 | ||||
| Centre | 16.4 | ||||
| Residual | 20.1 | ||||
FIGURE 88.
The age-adjusted association between time on ERT and muscle strength score in people with adult-onset Pompe disease (time on ERT treated as a continuous variable).
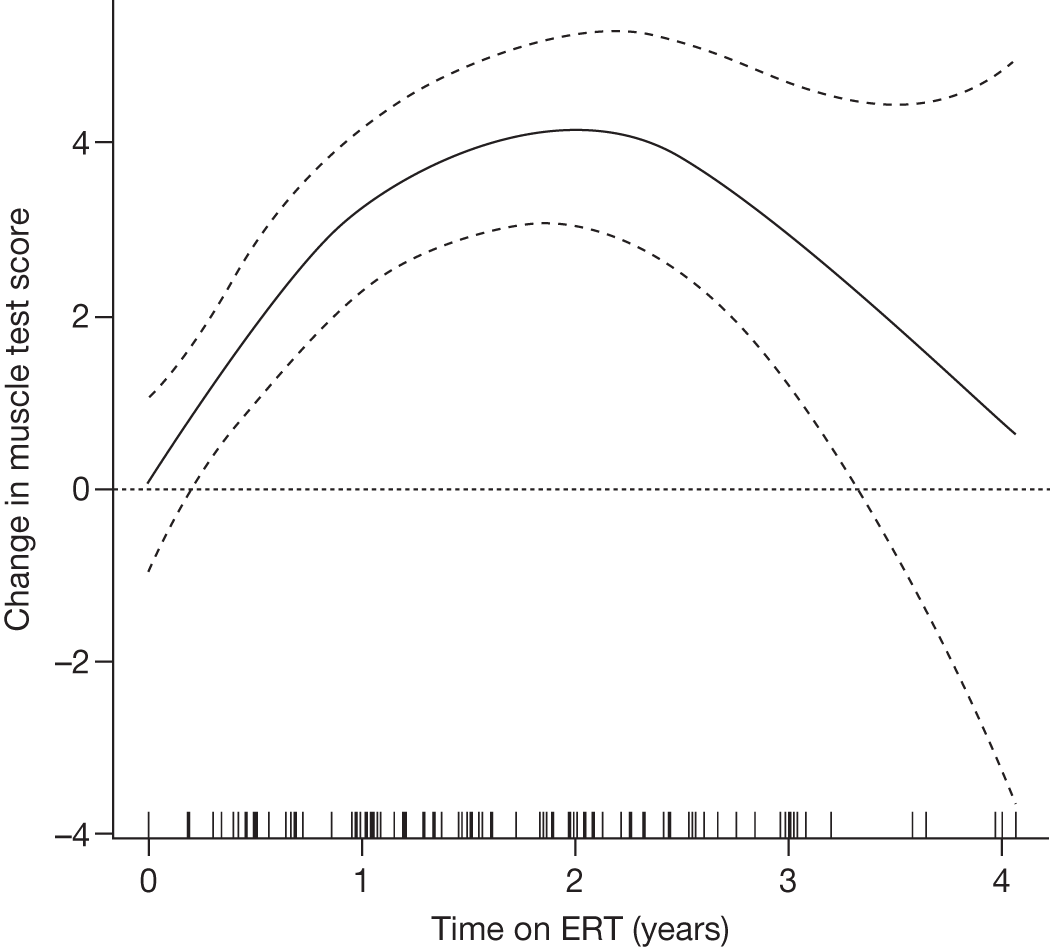
Body mass index
Adult-onset patients
We have 208 BMI measurements for 63 patients with adult-onset Pompe disease, across all time points. Values range from 13.31 to 46.17 kg/m2.
As seen in Table 132, there was a significant increase in BMI with age (p < 0.001). However, after adjusting for age there was no significant association between BMI and time on ERT (p = 0.51). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and BMI. Further analysis was conducted to explore the shape of the relationship between BMI and time on ERT treated as a continuous variable. This analysis provided no evidence for a non-linear association between BMI and time on ERT (edf = 1.0; p = 0.77) (Figure 89).
| N Data | Estimate of change in BMI (kg/m2) | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 131 | 0.00 | |||
| Female | 77 | 2.09 | 1.35 | –0.56 to 4.74 | 0.12 |
| Age | |||||
| Linear effect/year | 0.19 | 0.04 | 0.11 to 0.27 | < 0.001 | |
| Time on ERT | |||||
| Not on ERT | 75 | 0.00 | 0.51 | ||
| < 12 months | 42 | 0.42 | 0.29 | –0.15 to 0.98 | 0.16 |
| 12–36 months | 78 | 0.19 | 0.26 | –0.32 to 0.69 | 0.45 |
| > 36 months | 13 | 0.50 | 0.52 | –0.52 to 1.52 | 0.33 |
| Variance components | |||||
| Individual | 25.9 | ||||
| Centre | 0.00 | ||||
| Residual | 1.89 | ||||
FIGURE 89.
The age-adjusted association between time on ERT and BMI in people with adult-onset Pompe disease (time on ERT treated as a continuous variable).
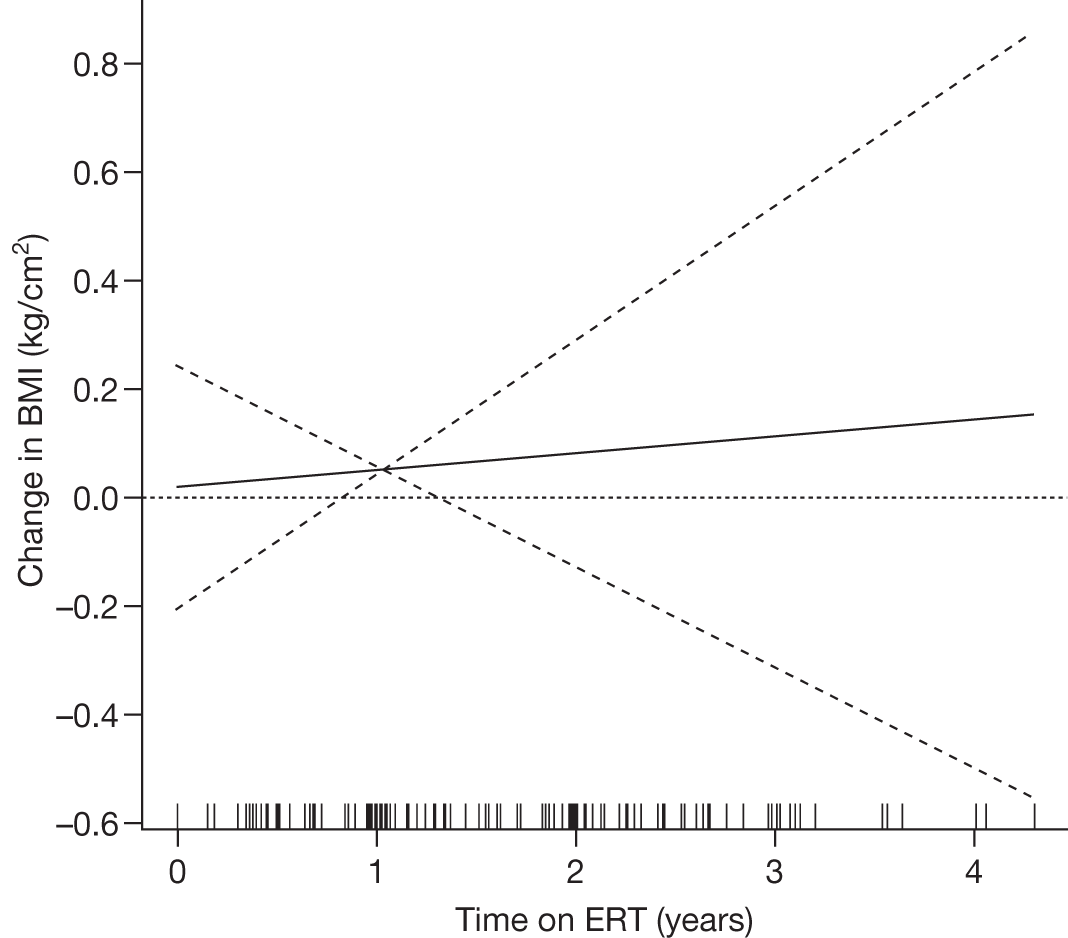
Height
Infantile-onset patients
Twenty-nine height measurements were collected for 11 patients with infantile Pompe disease, across all time points. Children’s height measurements were converted to z-scores using 1990 UK norms261 with the Stata command ‘zanthro’. This transformation allows one to examine the changes in height relative to the expected growth patterns for a child of the same age. Values range from 60.2 to 138.0 cm, with a mean z-score of 0.17 (Table 133).
| N Data | Estimate of increment in height | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 10 | 0.00 | |||
| Female | 19 | –1.75 | 0.57 | –2.88 to –0.62 | 0.005 |
| Age | |||||
| Linear effect/year | –0.57 | 0.38 | –1.31 to 0.17 | 0.14 | |
| Time on ERT | |||||
| Linear effect/year | 0.51 | 0.38 | –0.24 to 1.27 | 0.19 | |
| Variance components | |||||
| Individual | 0.66 | ||||
| Centre | 1.33 | ||||
| Residual | 0.35 | ||||
After the adjustment for age, there was no statistically significant association between time on ERT and infantile height (p = 0.19). Further analysis was conducted to explore the shape of the relationship between height centiles and time on ERT. This analysis provides no evidence for a non-linear association between height z-scores and time on ERT (edf = 1; p = 0.19) (Figure 90).
FIGURE 90.
The age-adjusted association between time on ERT and height z-scores in children with infantile Pompe disease (time on ERT treated as a continuous variable).
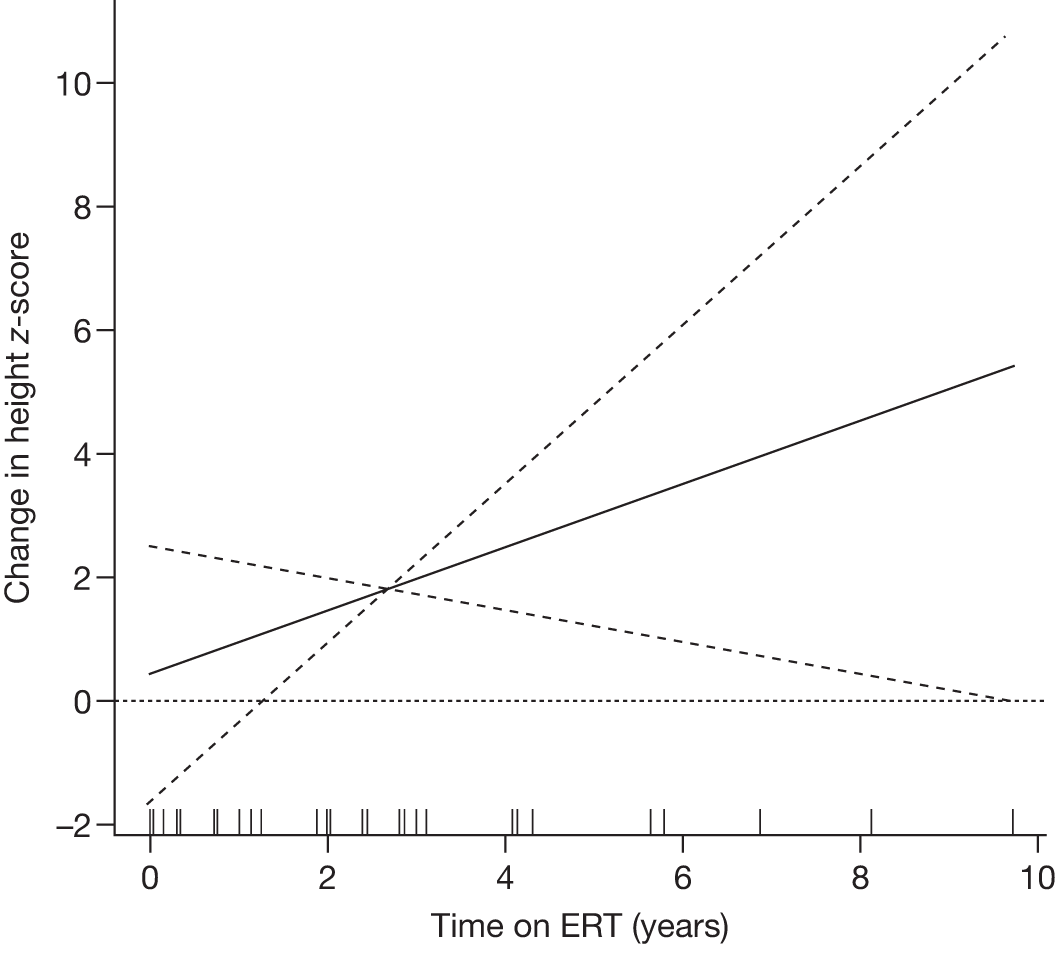
Weight
Infantile-onset patients
Thirty-one weight measurements were recorded for 12 patients with infantile Pompe disease, across all time points. Children’s weight measurements were converted to standardised z-scores against 1990 UK norms,261 adjusted for age and gender, using the Stata command ‘zanthro’. Values range from 5.7 to 32.2 kg, with a mean z-score of –0.15 (Table 134).
| N Data | Estimate of increment in weight | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 13 | 0.00 | |||
| Female | 18 | –0.13 | 0.73 | –1.56 to 1.31 | 0.86 |
| Age | |||||
| Linear effect/year | 0.32 | 0.49 | –0.64 to 1.28 | 0.52 | |
| Time on ERT | |||||
| Linear effect/year | –0.19 | 0.50 | –1.17 to 0.78 | 0.70 | |
| Variance components | |||||
| Individual | 1.03 | ||||
| Centre | 0.00 | ||||
| Residual | 0.99 | ||||
After the adjustment for age, there was no statistically significant association between time on ERT and infantile weight (p = 0.70). Further analysis was conducted to explore the shape of the relationship between weight centiles and time on ERT. This analysis provided no evidence for a non-linear association between infantile weight and time on ERT (edf = 1.87; p = 0.36) (Figure 91).
FIGURE 91.
The age-adjusted association between time on ERT and weight z-scores in children with infantile Pompe disease (time on ERT treated as a continuous variable).
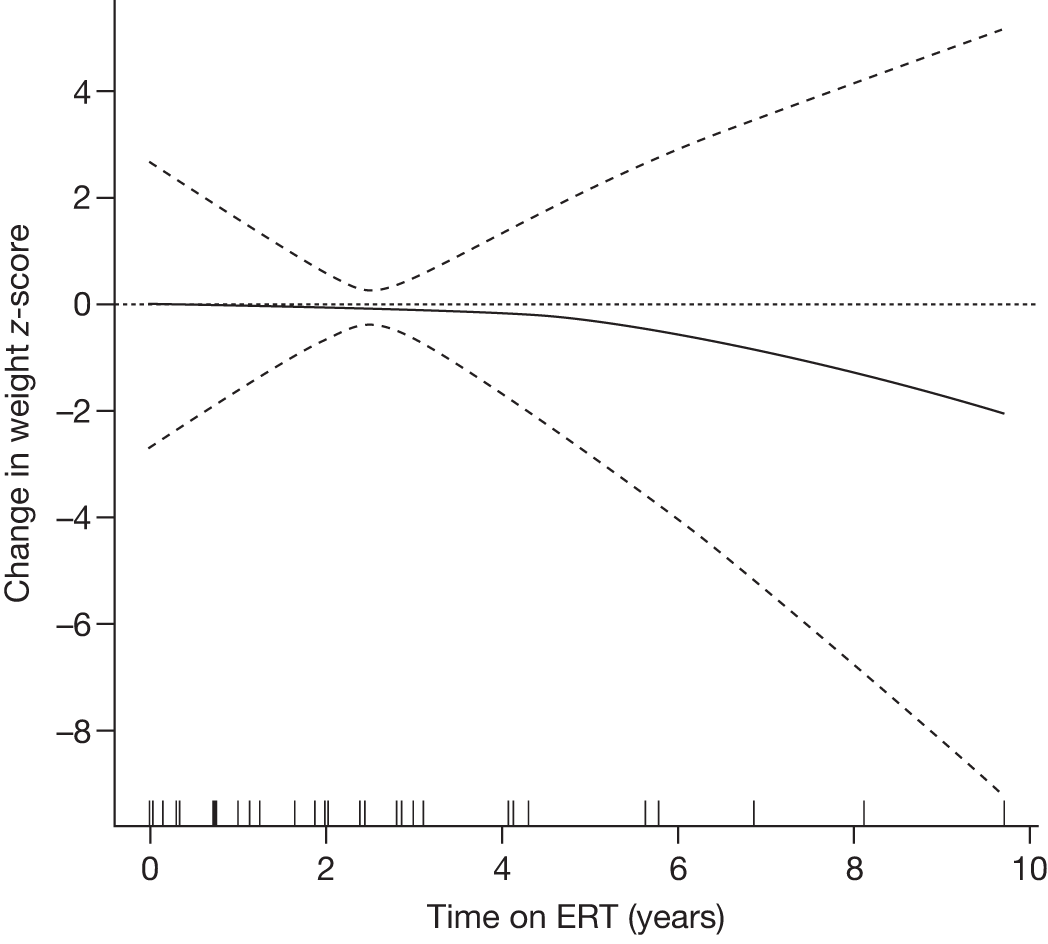
Quality-of-life assessments
SF-36
One hundred and thirty-four SF-36 questionnaires were completed across all prospective time points. Data are presented separately for PCS and MCS. The higher the SF-36 score, the better health the person believes themselves to be in (Tables 135–137).
| PCS | MCS | |
|---|---|---|
| Overall | ||
| Mean (SD) | 29.8 (8.73) | 50.07 (12.5) |
| N Data | 134 | 134 |
| ≤ 3 years on ERT | ||
| Mean (SD) | 29.8 (8.9) | 49.7 (12.8) |
| N Data | 93 | 93 |
| > 3 years on ERT | ||
| Mean (SD) | 28.4 (7.7) | 52.1 (12.5) |
| N Data | 35 | 35 |
| N Data | Estimate | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 85 | 0.00 | |||
| Female | 49 | 0.29 | 2.05 | –3.71 to 4.39 | 0.88 |
| Age | |||||
| Linear effect/year | –0.13 | 0.07 | –0.28 to 0.03 | 0.08 | |
| Time on ERT | |||||
| Not on ERT | 6 | 0.00 | 0.16 | ||
| < 12 months | 22 | –6.64 | 3.31 | –13.1 to –0.17 | 0.04 |
| 12–36 months | 71 | –6.32 | 3.03 | –12.2 to –0.34 | 0.04 |
| > 36 months | 35 | –7.24 | 3.27 | –13.9 to –1.12 | 0.03 |
| Variance components | |||||
| Individual | 40.3 | ||||
| Centre | 3.4 | ||||
| Residual | 32.7 | ||||
| N Data | Estimate | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 85 | 0.00 | |||
| Female | 49 | –1.58 | 3.20 | –7.85 to 4.69 | 0.62 |
| Age | |||||
| Linear effect/year | 0.007 | 0.12 | –0.23 to 0.24 | 0.95 | |
| Time on ERT | |||||
| Not on ERT | 6 | 0.00 | 0.88 | ||
| < 12 months | 22 | –3.29 | 4.49 | –12.1 to 5.51 | 0.46 |
| 12–36 months | 71 | –2.88 | 4.13 | –10.9 to 5.21 | 0.49 |
| > 36 months | 35 | –2.40 | 4.47 | –11.2 to 6.36 | 0.59 |
| Variance components | |||||
| Individual | 109.5 | ||||
| Centre | 17.8 | ||||
| Residual | 55.7 | ||||
The PCS was not significantly associated with age (p = 0.08). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and PCS. After adjusting for age, the PCS was not significantly associated with time on ERT (p = 0.16). Further analysis was conducted to explore the shape of the relationship between PCS and time on ERT treated as a continuous variable. This analysis provided no evidence of a non-linear association between PCS and time on ERT (edf = 1; p = 0.55) (Figure 92).
FIGURE 92.
The age-adjusted association between time on ERT and SF-36 PCS in people with adult-onset Pompe disease (time on ERT treated as a continuous variable).
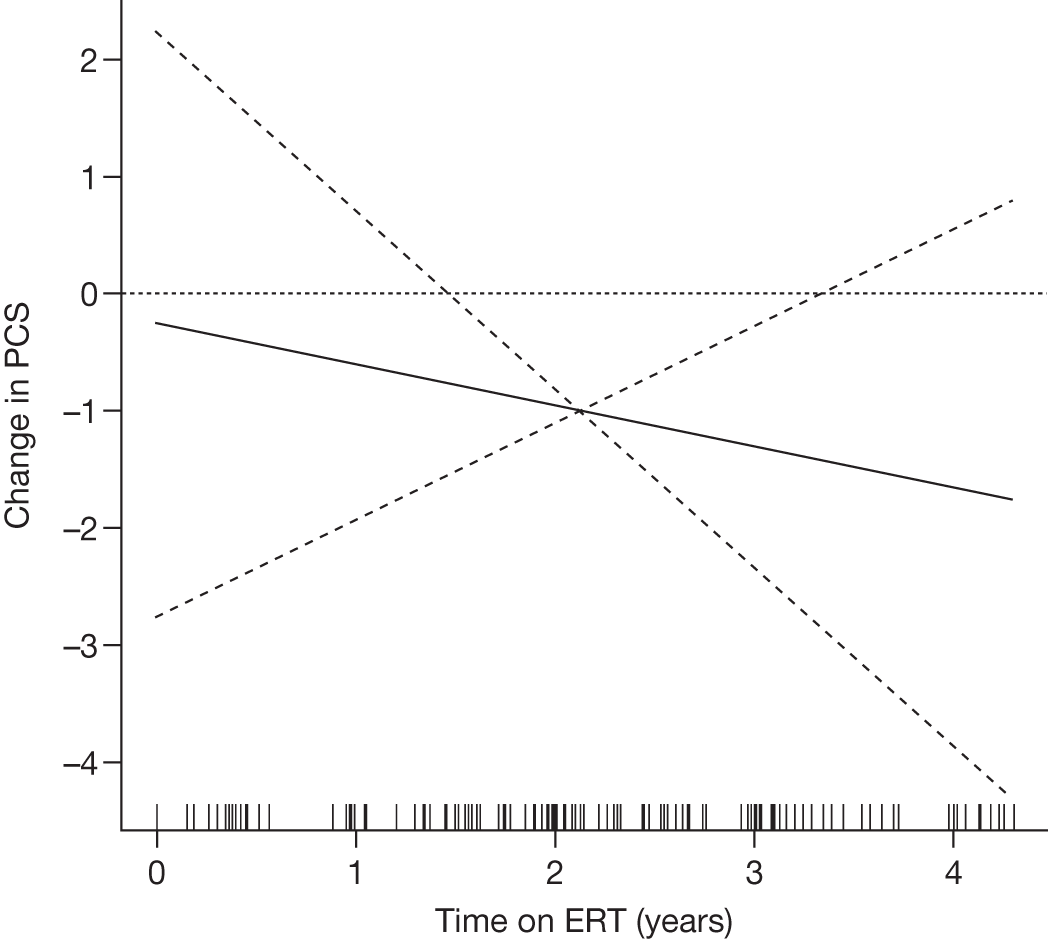
The likelihood ratio test for the overall effect of time on ERT did not reach statistical significance even though the coefficients for the individual categories were significant using the Wald test. The potential for different results can arise in small to moderate samples because of skewed sampling distributions of the model parameters. 275 The likelihood ratio test makes no assumption about the shape of the sampling distribution and so is generally superior to the Wald test in small samples. We conclude that the apparent association between PCS and time on ERT failed to reach conventional levels of statistical significance.
It is also important to note that because the SF-36 data were not available retrospectively, this analysis essentially compares patients with different durations of time on treatment rather than comparing those not on treatment with those on ERT. Consequently, our analysis is not able to detect how the physical component summary score responds to initiation of treatment with ERT (Table 137).
As with the PCS, the MCS was not significantly associated with age (p = 0.95). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and MCS. Further analysis was conducted to explore the shape of the relationship between MCS and time on ERT treated as a continuous variable. After adjusting for age, the MCS was not significantly associated with time on ERT (p = 0.88). This analysis provided no evidence of a non-linear association between MCS and time on ERT (edf = 1; p = 0.87) (Figure 93).
FIGURE 93.
The age-adjusted association between time on ERT and SF-36 MCS in people with adult-onset Pompe disease (time on ERT treated as a continuous variable).
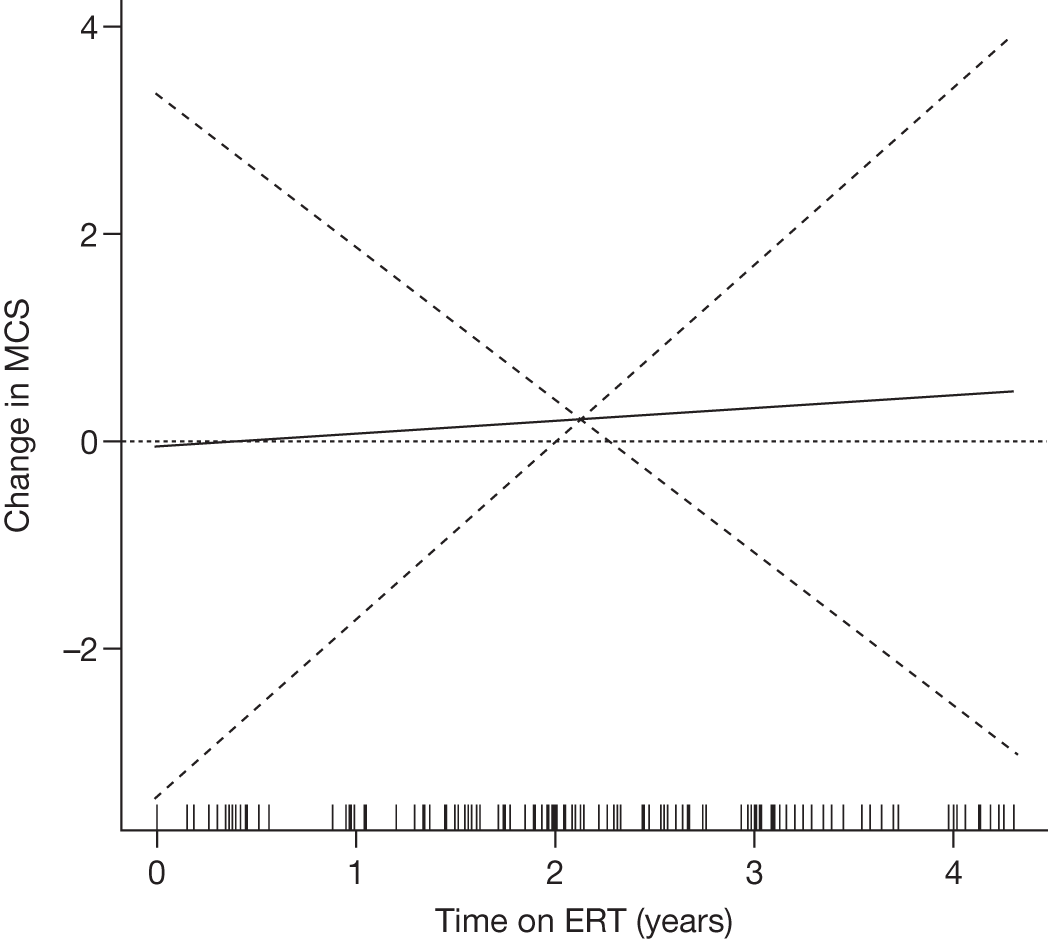
Again, it is important to note that because the SF-36 data were not available retrospectively, this analysis essentially compares patients with different durations of time on treatment rather than comparing those not on treatment with those on ERT. Consequently, our analysis is not able to detect how the MCS responds to initiation of treatment with ERT.
EQ-5D
In addition to the SF-36, participants aged ≥ 13 years were invited to complete the EQ-5D. One hundred and thirty-two EQ-5D questionnaires were completed across all prospective time points. Data are presented in Table 138 for the EQ-5D score (range from –0.35 to 1.0, with 1.0 being ‘perfect health’).
| N Data | Estimate of change of EQ-5D | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 81 | 0.00 | |||
| Female | 51 | –0.0005 | 0.07 | –0.14 to 0.14 | 0.99 |
| Age | |||||
| Linear effect/year | 0.002 | 0.003 | –0.004 to 0.008 | 0.46 | |
| Time on ERT | |||||
| Not on ERT | 7 | 0.00 | 0.06 | ||
| < 12 months | 23 | –0.19 | 0.09 | –0.36 to –0.01 | 0.03 |
| 12–36 months | 71 | –0.20 | 0.08 | –0.35 to –0.04 | 0.02 |
| > 36 months | 31 | –0.25 | 0.09 | –0.43 to –0.07 | 0.008 |
| Variance components | |||||
| Individual | 0.06 | ||||
| Centre | 0.002 | ||||
| Residual | 0.023 | ||||
A longitudinal model was fitted to assess the linear relationship between EQ-5D and time on ERT after adjusting for age.
The linear model showed no significant association between EQ-5D and age (p = 0.46). When time on ERT was categorised as < 12 months, 12–36 months or > 36 months, there appears to be a worsening in reduction in EQ-5D with time on ERT; however, this does not reach conventional levels of statistical significance (p = 0.06).
Further modelling of the data provided no evidence for a non-linear association between the EQ-5D and time on ERT (edf = 1.0; p = 0.27) (Figure 94).
FIGURE 94.
The age-adjusted association between time on ERT and EQ-5D score in people with adult-onset Pompe disease (time on ERT treated as a continuous variable).
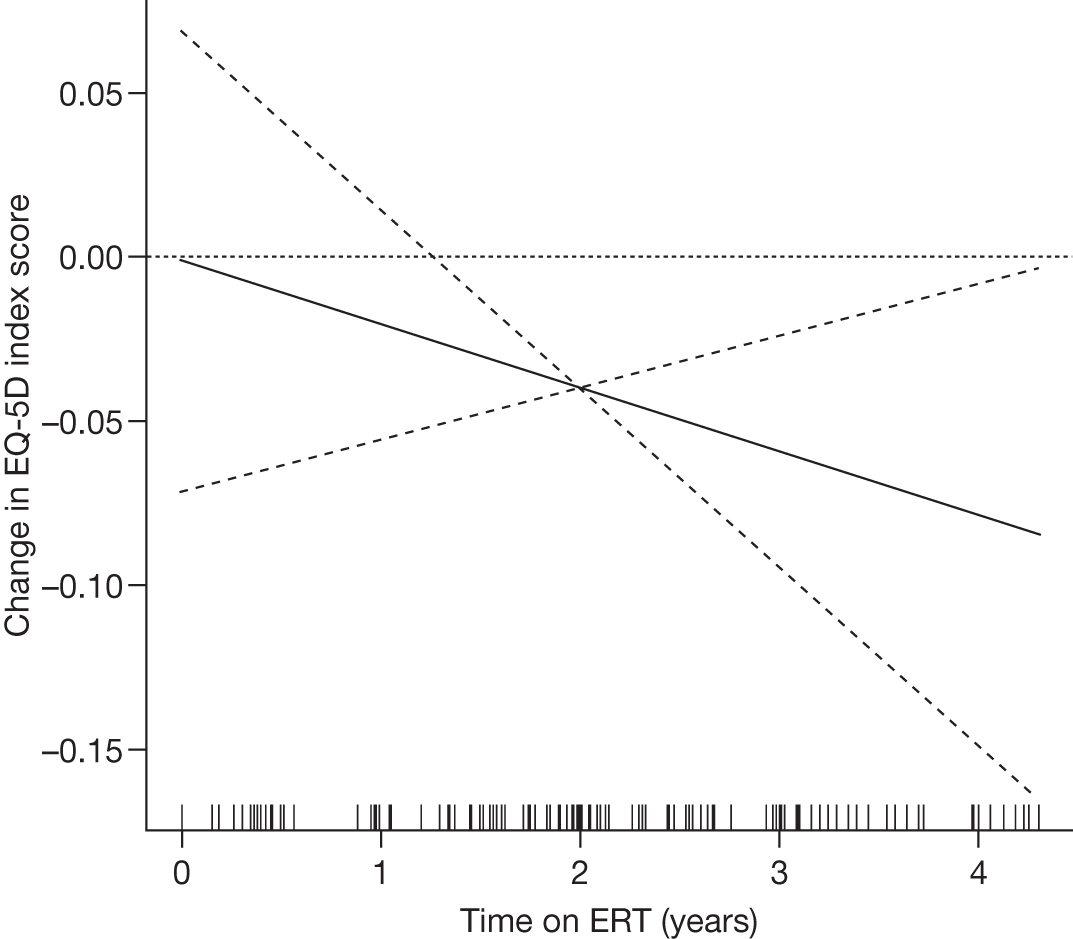
Equivalent modelling analyses were also conducted using SF-6D (SF-36-derived) utility weights, but no statistically significant associations (at α = 0.05 level) were found with time on ERT. The tabulated results of the SF-6D longitudinal modelling analyses are available on request from the study authors.
EQ-5D visual analogue scale
In addition to scoring on the five domains of the EQ-5D, participants were asked to rate their health on a VAS. The EQ-5D VAS asks people to rate their health state on a 10-cm line from 0, ‘worst imaginable health state’, to 100, ‘best imaginable health state’. The 123 VAS scores completed for this study ranged from 0 to 100. A longitudinal model was fitted to assess the linear relationship between the visual analogue score and time on ERT after adjusting for age.
The linear model shown in Table 139 shows no significant association in EQ-5D VAS score with age (p = 0.26) or with time on ERT (p = 0.80).
| N Data | Estimate of change of EQ-5D VAS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 79 | 0.00 | |||
| Female | 44 | 14.8 | 5.05 | 4.90 to 24.7 | 0.004 |
| Age | |||||
| Linear effect/year | 0.20 | 0.18 | –0.15 to 0.55 | 0.26 | |
| Time on ERT | |||||
| Not on ERT | 7 | 0.00 | 0.80 | ||
| < 12 months | 15 | –7.66 | 8.91 | –25.1 to 9.80 | 0.39 |
| 12–36 months | 65 | –6.13 | 7.66 | –21.1 to 8.88 | 0.42 |
| > 36 months | 36 | –7.54 | 8.33 | –23.8 to 8.78 | 0.37 |
| Variance components | |||||
| Individual | 216.0 | ||||
| Centre | 54.5 | ||||
| Residual | 229.8 | ||||
No evidence was found for a non-linear association with EQ-5D VAS score and time on ERT (edf = 1; p = 0.76) (Figure 95).
FIGURE 95.
The age-adjusted association between time on ERT and the EQ-5D VAS in people with adult-onset Pompe disease (time on ERT treated as a continuous variable).
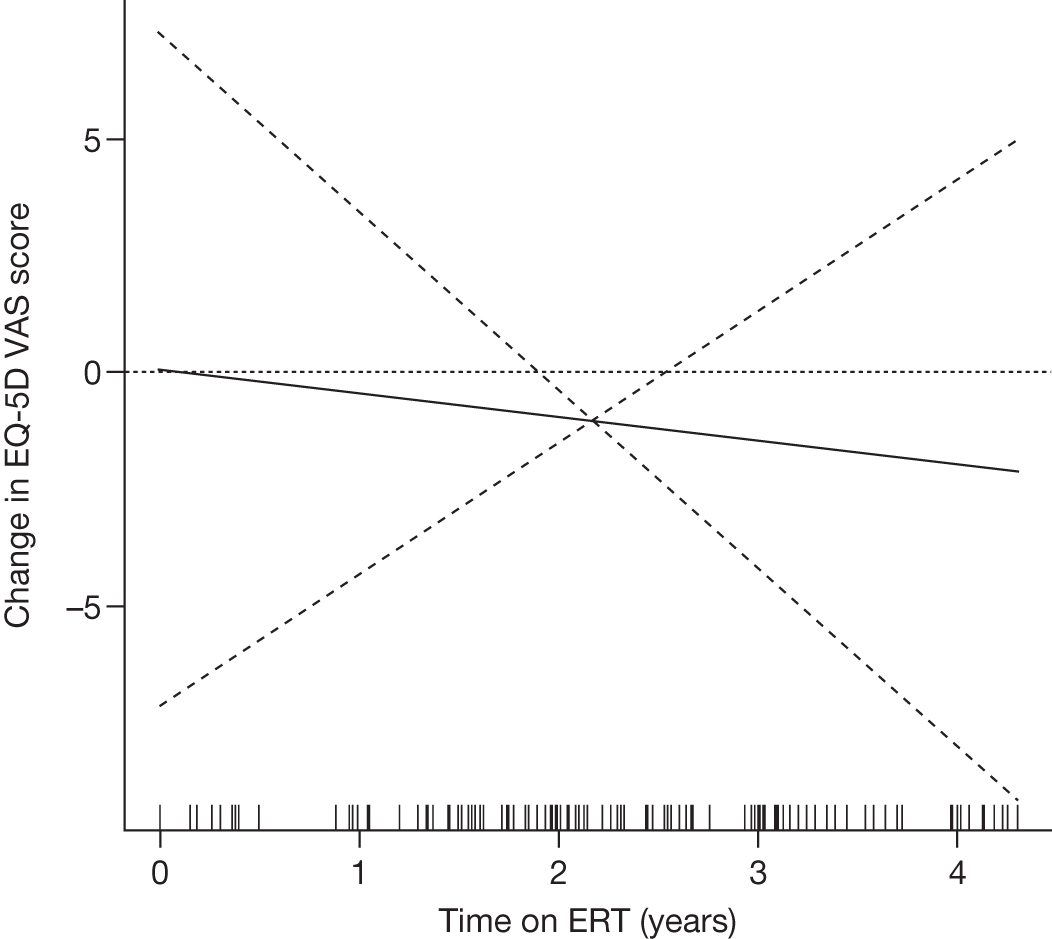
PedsQL
Thirteen PedsQL questionnaires were completed by children with infantile-onset Pompe disease, or their carers. Table 140 shows a descriptive summary of the total score and the scores from the component summary scores by type of treatment. The scale ranges from 0 to 100, where higher scores indicate better HRQoL.
| Time | Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Physical health summary score | Total score |
|---|---|---|---|---|---|---|---|
| Overall | |||||||
| Mean (SD) | 34.02 (26.8) | 64.9 (25.4) | 44.7 (19.2) | 85.0 (10.0) | 80.6 (3.5) | 34.02 (26.8) | 66.8 (17.7) |
| n | 12 | 12 | 13 | 3 | 3 | 12 | 2 |
| ≤ 3 years on ERT | |||||||
| Mean (SD) | 29.5 (25.7) | 51.3 (23.03) | 38.7 (19.7) | N/A | N/A | 29.5 (25.7) | N/A |
| n | 8 | 7 | 8 | 0 | 0 | 8 | 0 |
| > 3 years on ERT | |||||||
| Mean (SD) | 42.9 (30.4) | 84.0 (14.3) | 54.2 (15.4) | 85.0 (10.0) | 80.6 (3.5) | 42.9 (30.4) | 66.8 (17.7) |
| n | 4 | 5 | 5 | 3 | 3 | 4 | 2 |
Table 140 shows descriptive statistics of the PedsQL data, with time on ERT, after adjusting for age. As can be seen from the table there were insufficient data points to explore the relationship between PedsQL scores and time on ERT.
Fatigue Severity Scale
We collected 63 FSS questionnaires for patients with adult-onset Pompe disease. The scores ranged from 3.22 to 7 [where a high score (≥ 4) is indicative of significant fatigue228,229].
As can be seen in Table 141, there was no statistically significant difference in fatigue scores between males and females (p = 0.06) or with age (p = 0.98). After adjusting for age, there was no statistically significant association between fatigue scores and time on ERT (p = 0.17). In this initial analysis, we examined the association between time on ERT, categorised as ‘not treated’, < 12 months, 12–36 months and > 36 months, and fatigue scores.
| N Data | Estimate | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 41 | 0.00 | |||
| Female | 22 | 0.62 | 0.33 | –0.03 to 1.27 | 0.06 |
| Age | |||||
| Linear effect/year | –0.0001 | 0.01 | –0.02 to 0.02 | 0.98 | |
| Time on ERT | |||||
| Not on ERT | 2 | 0.00 | 0.17 | ||
| < 12 months | 2 | –0.44 | 1.05 | –2.49 to 1.62 | 0.67 |
| 12–36 months | 27 | 1.06 | 0.82 | –0.54 to 2.66 | 0.19 |
| > 36 months | 32 | 0.83 | 0.81 | –0.76 to 2.42 | 0.31 |
| Variance components | |||||
| Individual | 0.71 | ||||
| Centre | 0.09 | ||||
| Residual | 0.36 | ||||
Further analysis was conducted to explore the shape of the relationship between fatigue scores and time on ERT treated as a continuous variable. This analysis provided no strong evidence for a non-linear association between fatigue and time on ERT (edf = 2.44; p = 0.15) (Figure 96).
FIGURE 96.
The age-adjusted association between time on ERT and FSS in people with adult-onset Pompe disease (time on ERT treated as a continuous variable).
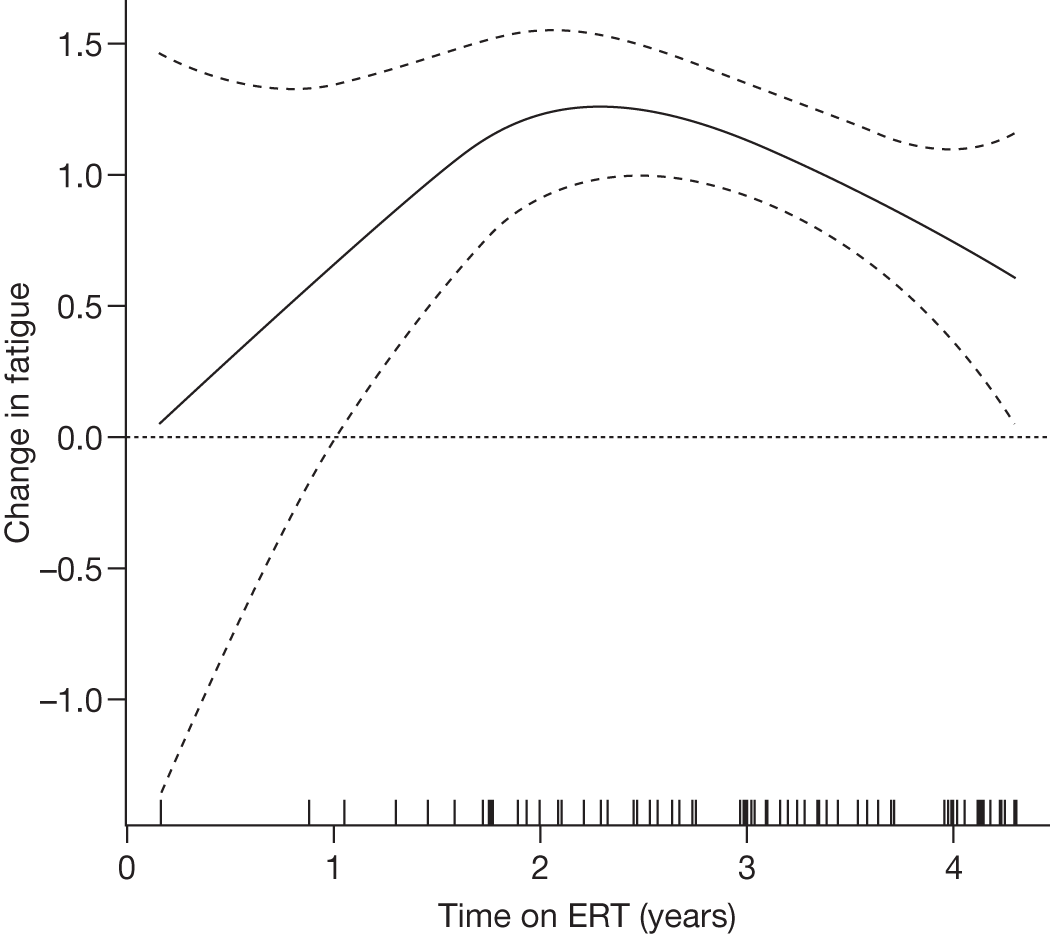
Carer Strain Index
Thirteen CSI questionnaires were completed across all prospective time points by carers of children with infantile-onset Pompe disease. Only one form was completed by the carer of an adult-onset patient, and therefore no analysis was possible. CSI total score ranged from 1 to 24; the maximum possible score of 26 signifies the greatest degree of caregiver burden. A longitudinal model was fitted to assess the linear relationship between the CSI and time on ERT, after adjusting for age (Table 142).
| N Data | Estimate of change of CSI | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 5 | 0.00 | |||
| Female | 8 | –7.82 | 6.37 | –20.3 to 4.66 | 0.24 |
| Age | |||||
| Linear effect/year | 0.31 | 1.18 | –2.0 to 2.62 | 0.79 | |
| Time on ERT | |||||
| Not on ERT | 0 | 0.00 | 0.08 | ||
| < 12 months | 3 | 0.00 | 0.00 | 0.00 | 0.00 |
| 12–36 months | 5 | 6.49 | 3.22 | 0.17 to 12.8 | 0.07 |
| > 36 months | 5 | –3.02 | 9.21 | –21.1 to 15.0 | 0.75 |
| Variance components | |||||
| Individual | 66 | ||||
| Centre | 0.0 | ||||
| Residual | 6 | ||||
The linear model shows no significant association in score on the CSI with age of person they are caring for (p = 0.79). When time on ERT was categorised as < 12 months, 12–36 months or > 36 months, no significant association in CSI was seen with time on ERT (p = 0.08).
However, further modelling of the data provided evidence for a non-linear association between the CSI and time on ERT treated as a continuous variable (edf = 3.1; p = 0.02) (see Figure 97). This relationship, seen in Figure 97, suggests that the burden of care for carers of patients with infantile-onset Pompe disease reduces with time on ERT, although these results should be treated with caution because of the sparse data.
FIGURE 97.
The age-adjusted association between time on ERT and the CSI for carers of children with infantile-onset Pompe disease (time on ERT treated as a continuous variable).
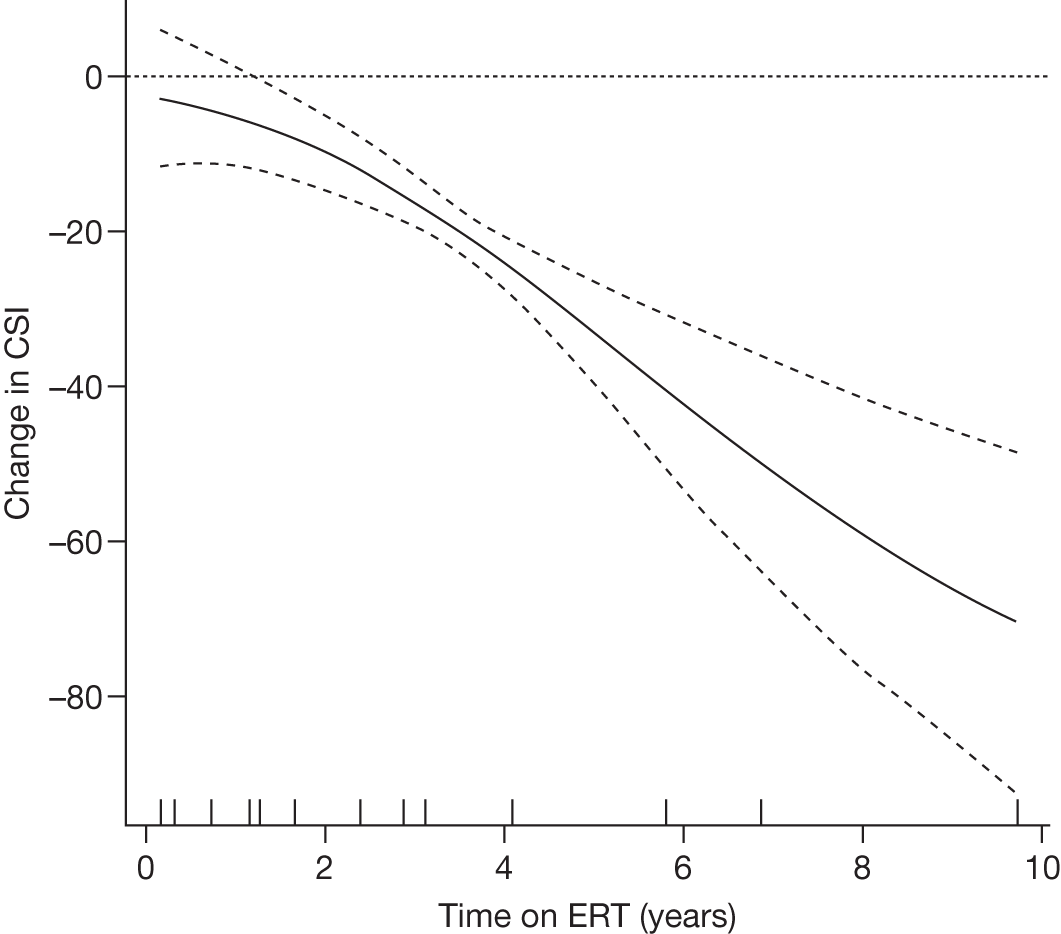
Safety and complications
Of the 77 participants with Pompe disease in this study, one patient reported that they had experienced febrile reactions, two patients reported anaphylactic reactions, five patients required pre-medication and 15 patients were reported as having positive antibody status to infused products.
Two patients stopped ERT during the period of data collection. One patient stopped treatment owing to pregnancy and no reason for stopping treatment was cited for the other patient.
During the course of the study, one patient with Pompe disease died from disease-related complications after 4 months.
Cost of enzyme replacement therapy in people with Pompe disease
Table 143 shows the current unit purchase cost to the NHS of alglucosidase alpha.
| Drug full name | Proprietary name and unit | 2011 base price per unit (£) |
|---|---|---|
| Aglucosidase alpha | Myozyme®, 50 mg | 356.06 |
Table 144 shows the NSCT-estimated annual NHS per patient cost of providing these drugs. Note that these costs include both the drug costs and home-care costs where the NSCT funds them.
| Drug | Adults | Infants | Children |
|---|---|---|---|
| Aglucosidase alpha 50 mg | £282,798 | £26,025 | £121,780 |
Cost of care for adults with Pompe disease
Total care cost – financial burden of Pompe disease
Table 145 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for an adult with Pompe disease. Of the estimated mean per patient annual cost of £6300, almost half is as a result of NHS hospital services used; of this two-fifths (£1200 per patient per year) is from inpatient stays, whereas just over two-fifths (£1300) is from outpatient visits (see Table 146). Of the £3300 per patient per year from using services outside hospital, < £80 per patient is from GP visits, whereas > £2400 is from regular visits from either home helps, health visitors or other nurses (see Table 147).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. who used this type of service (%) | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 54 | 87 | 50 (93) | 3000 | 4291 | 1500 | 940–4100 |
| Services outside hospital | 54 | 87 | 50 (93) | 3300 | 5584 | 230 | 54–3000 |
| Total health (NHS) and social-care cost | 54 | 87 | 53 (98) | 6300 | 6915 | 3200 | 1000–7900 |
| Type of hospital care | No. who used this type of service (%) | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| Inpatient stays | 23 (43) | 1200 | 3876 | 940 | 940–2400 |
| Outpatient visits | 43 (80) | 1300 | 1819 | 940 | 470–1900 |
| Day cases | 10 (19) | 510 | 1265 | 3300 | 940–3600 |
| Accident and emergency visits | 5 (9) | 11 | 38 | 100 | 100–210 |
| Total hospital (NHS) care cost | 50 (93) | 3000 | 4291 | 1500 | 940–4100 |
| Care provider | No. who used this provider (%) | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 39 (72) | 74 | 113 | 54 | 41–110 |
| GP nurse appointments | 19 (35) | 5 | 13 | 5 | 5–16 |
| District nurses | 5 (9) | 5 | 21 | 32 | 5–130 |
| Community mental health nurse | 2 (4) | 5 | 39 | 150 | 8–290 |
| Other nurse or health visitor | 14 (26) | 1300 | 2712 | 7100 | 1900–7700 |
| Counsellor | 1 (2) | 42 | 311 | 2300 | N/A |
| Other therapist | 8 (15) | 100 | 578 | 47 | 20–960 |
| ‘Alternative’ medicine or therapy | 0 | 0 | 0 | 0 | 0–0 |
| Psychologist | 1 (2) | 3 | 22 | 160 | N/A |
| Psychiatrist | 0 | 0 | 0 | 0 | 0–0 |
| Other community-based doctor | 0 | 0 | 0 | 0 | 0–0 |
| Occupational therapist | 8 (15) | 8 | 33 | 38 | 20–57 |
| Social worker | 4 (7) | 18 | 69 | 280 | 110–320 |
| Home help | 4 (7) | 1100 | 5272 | 9000 | 2700–36,500 |
| Care attendant | 2 (4) | 680 | 4967 | 18,300 | 170–36,500 |
| Community support worker | 0 | 0 | 0 | 0 | 0–0 |
| Housing worker | 1 (2) | < 1 | 3 | 25 | N/A |
| All non-hospital NHS and social-care providers | 50 (93%) | 3300 | 5584 | 230 | 54–3000 |
Cost breakdown by hospital- and community-based services
Tables 146 and 147 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by adults with Pompe disease. Only 23 patients, or just over two-fifths, of those adults who provided valid service-use data had hospital stays as inpatients, and this accounted for about the same proportion of the NHS hospital costs for adults with Pompe disease. In contrast, 80% of patients reported having at least one hospital outpatient attendance during the previous 12 months, and these accounted for 43% of the overall hospital costs.
The majority of costs related to using community-based services were due to the relatively small minority of adults with Pompe disease who used home helps (four patients, including one using £36,500 worth of care, e.g. 4 hours a day 365 days a year) or who used health visitors or other nurses regularly (14 patients). They accounted for £2400 of the £3300 annual per patient cost of services used outside hospital. Although over two-thirds of adults with Pompe disease had seen their GP at least once during the past year, and over one-third reported seeing a practice nurse at their GP surgery, these accounted for < £80 of the £3300 annual cost of NHS and publicly funded social-care services consumed. Other support providers used by smaller numbers of adults with Pompe disease were occupational therapists, social workers, district nurses and care attendants (see Table 147).
Cost of care for children with Pompe disease
Total care cost – financial burden of Pompe disease
Table 148 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for a child with Pompe disease, based on the 13 children who provided service-use data when they entered the study (7 males, mean age 4.7 years, age range 5 months to 12 years). Of the estimated mean per patient annual cost of £10,100, > £9000 is from NHS hospital services used, and of this £7500 per patient is from inpatient stays (see Table 149). Of the £780 per patient per year from using services outside hospital, GP visits, district nurse visits and other community-based doctors account for the majority of these costs (see Table 150).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. who used this type of service (%) | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 13 | 87 | 10 (77) | 9300 | 17,428 | 2300 | 1900–11,700 |
| Services outside hospital | 13 | 87 | 13 (100) | 780 | 795 | 560 | 130–1800 |
| Total health (NHS) and social-care cost | 13 | 87 | 13 (100) | 10,100 | 17,469 | 2700 | 1100–10,600 |
| Type of hospital care | No. who used this type of service | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| Inpatient stays | 7 | 7500 | 17,022 | 1900 | 940–25,300 |
| Outpatient visits | 2 | 300 | 753 | 2000 | 1700–2300 |
| Day cases | 4 | 1400 | 2643 | 5300 | 940–7100 |
| Accident and emergency visits | 2 | 87 | 237 | 570 | 310–820 |
| Total hospital (NHS) care cost | 10 | 9300 | 17,400 | 2300 | 1900–11,700 |
| Care provider | No. who used this provider | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 13 | 200 | 190 | 130 | 54–260 |
| GP nurse appointments | 7 | 8.5 | 11 | 12 | 10–23 |
| District nurses | 2 | 140 | 402 | 880 | 320–1400 |
| Community mental health nurse | 0 | 0 | 0 | 0 | 0–0 |
| Other nurse or health visitor | 5 | 49 | 82 | 77 | 53–88 |
| Counsellor | 0 | 0 | 0 | 0 | 0–0 |
| Other therapist | 1 | 58 | 131 | 39 | 19–440 |
| ‘Alternative’ medicine or therapy | 1 | 7 | 24 | 85 | N/A |
| Psychologist | 1 | 28 | 101 | 365 | N/A |
| Psychiatrist | 0 | 0 | 0 | 0 | 0–0 |
| Other community-based doctor | 3 | 210 | 585 | 280 | 280–2100 |
| Occupational therapist | 3 | 30 | 57 | 120 | 120–150 |
| Social worker | 1 | 57 | 204 | 740 | N/A |
| Home help | 0 | 0 | 0 | 0 | 0–0 |
| Care attendant | 0 | 0 | 0 | 0 | 0–0 |
| Community support worker | 0 | 0 | 0 | 0 | 0–0 |
| Housing worker | 0 | 0 | 0 | 0 | 0–0 |
| All non-hospital NHS and social-care providers | 13 | 780 | 795 | 560 | 130–1800 |
Cost breakdown by hospital- and community-based services
Tables 149 and 150 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by children with Pompe disease. Seven of the 13 children who provided valid service-use data had hospital stays as inpatients, but these were sometimes very costly inpatient episodes. This included one child whose inpatient stays cost an estimated £25,300 (mainly treatment for aspiration pneumonia and a chest infection with collapsed lung), and another whose inpatient stay cost £59,200 (for a reported 63 days in hospital owing to their Pompe disease). In contrast, fewer children were reported as having hospital outpatient attendances or day case treatment during the previous 12 months, and these accounted < 20% of the overall hospital costs for the 16 children.
The majority of costs related to using community-based services was as a result of all children using some GP services, plus the few children who used district nurses (four children) or other community-based doctors (three patients, typically seeing paediatricians). Together these three accounted for £550 of the £780 annual per patient cost of services used outside of hospital. Other care professionals and support providers typically seen by children with Pompe disease were GP practice nurses, occupational therapists, health visitors and other nurses (see Table 150), but these accounted for a relatively small proportion of the estimated mean annual costs.
Association between time on enzyme replacement therapy and costs for patients with Pompe disease
In patients with early- or late-onset Pompe disease, there was no statistically significant association (i.e. no p-values < 0.05 for the regression coefficient) in either adults or children between time on ERT and either total NHS and social-care costs, hospital-care costs or non-hospital-care costs. The tabulated results of these analyses are available on request from the study authors.
Discussion of Pompe disease results
Pompe disease is caused by glycogen accumulation due to a deficiency of the lysosomal acid alpha-glucosidase enzyme by which it is degraded. A total or partial deficiency of this enzyme causes lysosomal glycogen storage, leading to a systemic disorder characterised by cardiomyopathy, muscle weakness, hypotonia and respiratory disorders. Three forms of presentation have been described according to the age at which clinical signs appear: in adulthood, adolescence or infancy. The last, characterised by very severe or even total enzyme deficiency, usually manifests in the first months of life.
Enzyme replacement therapy with recombinant human alglucosidase alpha (rhGAA) was approved for treating patients with Pompe disease in 2006.
Infantile Pompe disease
Kishnani and colleagues192 reported the largest study thus far involving patients with infantile-onset Pompe disease. They studied 18 infants with severe infantile-onset Pompe disease treated with ERT, comparing this group with a historical control group of 62 patients meeting the inclusion criteria for the study. They reported improvements in respiratory function, cardiomyopathy and, among a subset of patients, motor function after 52 weeks of treatment with alglucosidase alpha. They also suggested increased survival in the treated group compared with the historical controls. They also reported on an extension of this study which suggested that the benefits compared with historical controls were maintained. 193 Chakrapani and colleagues194 reported the results of a study of 20 children with the infantile-onset form treated with ERT and suggested signs of benefit compared with historical experience.
Twelve patients with infantile Pompe disease were recruited to this study; the average age of the participants at the start of the study was 3 years and the mean age at diagnosis was 9 months. Inevitably, this cohort excludes those infants with the most severe problems who will have died before recruitment.
All participants in the study were on treatment at recruitment and one patient required ventilation. Of the 12 patients, four were ventilator dependent by the conclusion of the study. There were insufficient data for further analyses.
One out of the 12 infantile-onset patients was reported as having swallowing difficulties by the fifth month of age. By the age of 4 years, 7 of the 12 infantile-onset patients were reported as having a swallowing difficulty. There were insufficient data for further analyses.
Clearly, the nature of this sample (all on ERT as soon as diagnosed) means that the ability to investigate the effects of ERT is severely limited.
Late-onset Pompe disease
The central trial of the effectiveness of ERT in late-onset Pompe disease involved 90 patients aged between 10 to 70 years, 60 of whom were randomised to receive alglucosidase alpha and 30 to placebo for 78 weeks. 187 Compared with placebo, the treated group showed a statistically significant improvement in the 6-minute walk test and per cent predicted FVC. There were no significant differences reported in muscle test score or SF-36 PCS.
In this cohort study, 65 patients with late-onset Pompe disease agreed to participate; their mean age at recruitment was 45 years and mean age at diagnosis was 38.6 years. All apart from three were receiving alglucosidase alpha and the mean duration of treatment (at recruitment) was 15 months.
Respiratory outcome measures
Unlike the trial reported by Kishnani and colleagues,192,193 we found no statistically significant association between the use of ERT and per cent predicted FVC after adjustment. We also examined whether or not the use of ERT was associated with a decreased risk of becoming ventilator dependent. By the age of 55 years, almost 80% of patients were dependent on ventilation regardless of treatment status.
These differences in our results and those reported by van der Ploeg and colleagues187 might reflect a variety of causes. It is important to note that the van der Ploeg and colleagues study excluded patients who had a per cent predicted FVC outside the range of 30–80% or who required invasive ventilation or non-invasive ventilation when awake and upright. Within the cohort reported here, the range of per cent predicted FVC was 10–130% and 30 patients were receiving ventilation at some point during our observations.
6-minute walk test
The trial by van der Ploeg and colleagues reported a 25.1 m increase in distance walked in the group who had received ERT for 78 weeks compared with the controls whose distance walked decreased by an estimated 3 m. 187 An observational study276 involving 38 patients with late-onset Pompe disease who received alglucosidase alpha over 36 months reported an increase in 32 m after 12 months and a further increase of 12 m after 24 months, although the distance fell back after 36 months’ treatment to 13 m compared with baseline.
We were limited in our analyses by having data for the 6-minute walk test data on only 22 patients (71 measures across all time points). Nonetheless, our results appear to reinforce previous findings, showing a significant decline in distance walked with age but a statistically significant association between time on ERT and an increase in distance walked (p < 0.001). Further analysis suggests that the distance walked increased for the first 2 years and then appeared to plateau before declining again.
Muscle test
One of the defining features of late-onset Pompe disease is the progressive deterioration in proximal arm and leg strength. Previous studies have provided only inconclusive evidence of an effect of ERT on muscle strength, with the trial by van der Ploeg and colleagues reporting an improvement in proximal arm and leg muscle strength which did not reach statistical significance. 187 The observational study reported by Regnery and colleagues276 suggested no improvement in muscle strength compared with baseline after 3 years of treatment with ERT.
Muscle strength data were collected for 54 patients, including 176 scores across all time points. We did not find a significant association with age. There was a statistically significant association between duration of ERT and improved muscle strength (p < 0.001). Similarly to the 6-minute walk test, this effect appears to plateau after about 2 years of treatment before declining again.
Body mass index
Bernstein and colleagues190 reported a series of three patients with late-onset Pompe disease who were treated for 6 months and apparently gained weight. In this study we found no statistically significant association between ERT and changes in BMI.
Quality-of-life data
Quality of life was not assessed in the trial by van der Ploeg and colleagues. 187 Strothotte and colleagues277 reported an observational study looking at late-onset patients pre and 12 months post ERT and suggested there was no association between SF-36 QoL scores and treatment.
In this cohort we found no statistically significant association between QoL scores (EQ-5D or SF-36) and duration of treatment with alglucosidase alpha.
Costs associated with Pompe disease
As with all other conditions investigated in this study, we were keen to capture the wider costs of care falling on the public sector in addition to the costs associated with ERT.
Based on patients’ self-reported health- and social-care service use, the annual cost of caring for people with Pompe disease, excluding the purchase cost of ERT, was estimated at £6300 for an adult and £10,000 for a child. These costs, however, are dwarfed by the cost of the therapies; the mean annual cost of ERT for adults with Pompe disease is £282,798 and £121,780 for children with Pompe disease.
From the longitudinal regression modelling of costs, there was no statistically significant association (i.e. p-value < 0.05) between time on ERT and either total NHS and social-care costs, hospital-care costs, or non-hospital-care costs for patients with Pompe disease. The tabulated results of these analyses are available on request from the study authors.
Owing to these high associated costs, and the lack of measureable effect of ERT on either clinical outcomes or HRQoL measures, it was infeasible to conduct either a cost-effectiveness or cost–utility analysis. As they apply to all six LSDs, the limitations of these cost estimates are summarised and discussed in Chapter 9.
Chapter 8 Results – Niemann–Pick disease type C (NPC)
Patient characteristics
At the start of the study 58 patients were identified by the treating centres as having NPC. Of these, 56 patients were deemed eligible for inclusion and 36 patients (64% of those deemed eligible) were approached in clinics and invited to participate. All thirty-six patients (14 males and 22 females) agreed to participate. Patient demographic characteristics are presented in Tables 151 and 152.
| Patient characteristic | |
|---|---|
| Gender | |
| Male, n | 4 |
| Female, n | 9 |
| Age at recruitment (years) | |
| Mean (SD) | 28.9 (9.4) |
| Median (min.–max.) | 26.7 (17.3–45.2) |
| Age at diagnosis (years) | |
| Mean (SD) | 18.6 (11.5) |
| Median (min.–max.) | 17.2 (4.0–32.5) |
| Initial treatment | |
| Not on SRT, n | 7 |
| SRT (miglustat), n | 5 |
| Clinical trial,a n | 1 |
| Age at starting SRT | |
| Mean (SD) | 27.5 (11.5) |
| Median (min.–max.) | 26.9 (11.8–42.6) |
| Time on SRT (years) | |
| Mean (SD) | 3.74 (2.6) |
| Median (min.–max.) | 4.05 (0–6.92) |
| Patient characteristic | |
|---|---|
| Gender | |
| Male, n | 10 |
| Female, n | 13 |
| Age at recruitment (years) | |
| Mean (SD) | 6.46 (4.2) |
| Median (min.–max.) | 5.44 (1.19–15.4) |
| Age at diagnosis (years) | |
| Mean (SD) | 2.67 (3.3) |
| Median (min.–max.) | 1.30 (0.04–10.9) |
| Initial treatment | |
| Not on SRT, n | 15 |
| SRT (miglustat), n | 6 |
| Clinical trial,a n | 1 |
| Missing, n | 1 |
| Age at starting SRT | |
| Mean (SD) | 7.64 (4.4) |
| Median (min.–max.) | 7.93 (0.47–12.9) |
| Time on SRT (years) | |
| Mean (SD) | 0.84 (1.02) |
| Median (min.–max.) | 0.36 (0–2.49) |
At recruitment, 23 of the participants were children (aged < 16 years) and 13 were adults. The average age of children at recruitment was 6.46 (range 1.19–15.4) years and that of adults was 28.9 (range 17.3–45.2) years. We collected data from all patients at the time of recruitment. Data were collected from 29 patients at their 12-month appointment and 5 patients at their 24-month appointment. We collected retrospective data from 25 patients at up to 12 time points.
At recruitment, 13 patients (seven children) were on SRT (miglustat), and the average time on SRT was 2.08 (range 0.1–6.92) years. Of the 13 patients on SRT, two patients were initially on a clinical trial prior to being prescribed open-label SRT.
Key markers of disease progression
The following measures were identified as key markers of disease progression:
-
measures of CNS involvement
-
– seizures/epilepsy
-
– vertical supranuclear gaze palsy
-
– ataxia
-
– dystonia
-
– dysarthia
-
– swallowing difficulties
-
– cataplexy
-
-
height
-
weight.
In addition, adults completed the SF-36, EQ-5D, FSS and the Service Use and Costs Questionnaire, whereas children or their carers completed the age-appropriate PedsQL questionnaire. Carers of children or adults were asked to complete the Service Use and Costs Questionnaire and the CSI.
Longitudinal models were fitted to assess relationships between continuous measures of function and length of time on SRT, after adjustment for age and clustering by centre. In the base models, the effect of time on SRT was treated as a linear effect because of the small number of data points. Further analysis was conducted to explore the possibility that time spent on SRT would have a non-linear effect on function. Patients contributed data points to the model both before and after starting SRT.
Summary of results
All analyses in NPC were hampered by a paucity of data related to both the small number of affected patients recruited and the lack of data recorded for key outcomes for a substantial proportion of these patients. This results in low power to detect any effects, and, for this reason, all analyses of NPC outcomes, with the exception of height and weight, were conducted using combined data from children and adults.
We examined potential associations between treatment and stature (height and weight) and several CNS measures. In addition, we examined the relationship between treatment and QoL assessed by either SF-36, EQ-5D or PedsQL depending on age. The effect of caring for someone with NPC was assessed using the CSI.
We found no statistically significant relationship between SRT and height or weight. Longitudinal effects of time on SRT could not be explored with the CNS measures as there were insufficient data. There were no statistically significant associations between any of the CNS measures and receiving SRT, apart from a statistically significant association with an increase in the number of cataplexic episodes on SRT.
Small but significant improvements were seen in SF-36 MCS and in the EQ-5D scale, with time on SRT. However, we found no statistically significant association between SRT and SF-36 PCS or with the CSI. There were insufficient data to explore the relationship between time on SRT and PedsQL or the FSS.
Height in children
Children’s height measurements were converted to z-scores using 1990 UK norms261 with the Stata command ‘zanthro’. Fifty-four height measurements were recorded for 21 children (seven of whom were receiving SRT). The mean z-score was –0.79 (Table 153).
| N Data | Estimate of change in height | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 19 | 0.00 | |||
| Female | 35 | 0.67 | 0.72 | –0.74 to 2.08 | 0.35 |
| Current age | |||||
| Linear effect/year | –0.07 | 0.07 | –0.21 to 0.07 | 0.32 | |
| Time on SRT | |||||
| Linear effect/year | –0.005 | 0.22 | –0.44 to 0.43 | 0.98 | |
| Variance components | |||||
| Individual | 2.02 | ||||
| Centre | 0.00 | ||||
| Residual | 0.49 | ||||
After adjusting for age and centre, time on SRT was not associated with a significant change in height centile (p = 0.98). No evidence was found for a non-linear association between height and time on SRT (edf = 1.13; p = 0.92) (Figure 98) although it is important to note that the data are sparse.
FIGURE 98.
The age-adjusted association between time on SRT and height z-scores in children with NPC (time on SRT treated as a continuous variable).
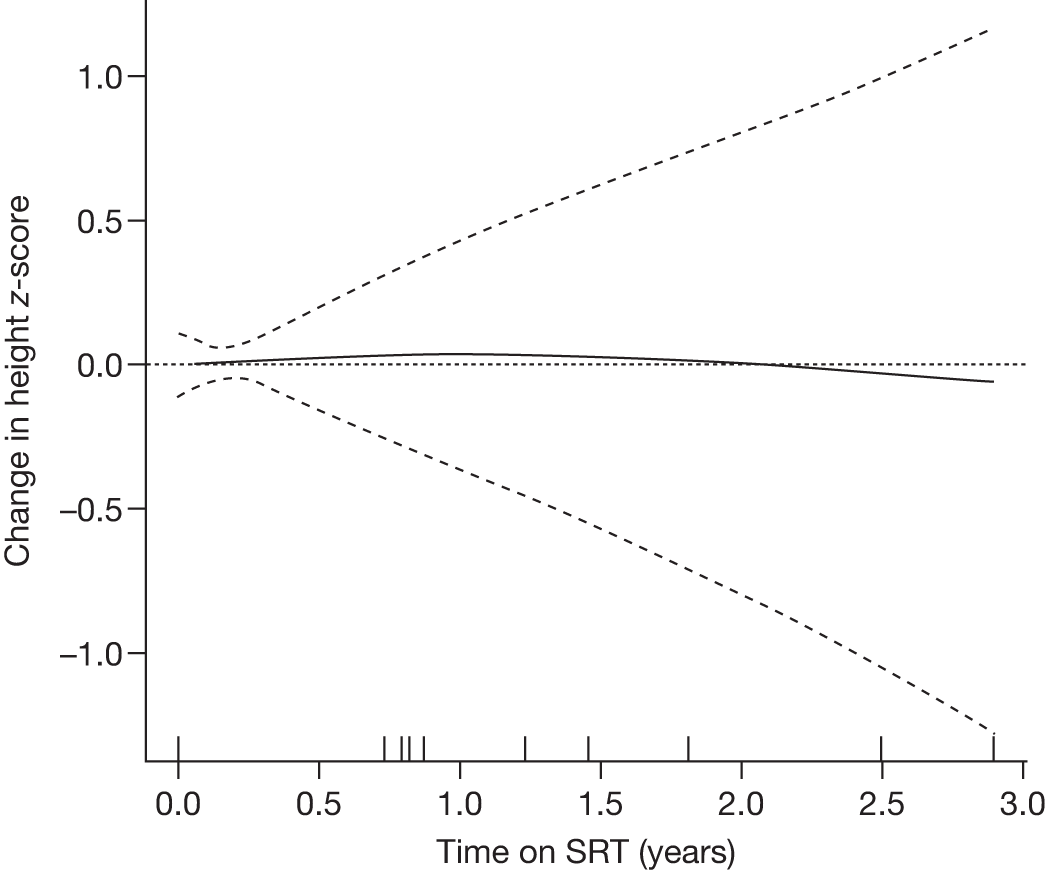
Weight
We have weight measures for 21 children (seven of whom were receiving SRT), with 55 weight measurements recorded across all time points, ranging from 3 to 65.6 kg. Children’s weight measurements were converted to standardised z-scores against 1990 UK norms,261 adjusted for age and gender, using the Stata command ‘zanthro’. The mean z-score was –0.19 (Table 154).
| N Data | Estimate of change in weight | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 19 | 0.00 | |||
| Female | 36 | 0.61 | 0.68 | –0.72 to 1.94 | 0.38 |
| Current age | |||||
| Linear effect/year | –0.04 | 0.06 | –0.16 to 0.08 | 0.49 | |
| Time on SRT | |||||
| Linear effect/year | –0.12 | 0.20 | –0.51 to 0.27 | 0.57 | |
| Variance components | |||||
| Individual | 1.85 | ||||
| Centre | 0.00 | ||||
| Residual | 0.38 | ||||
After adjusting for age, time on SRT was not associated with a significant change in weight centile (p = 0.57). No evidence was found for a non-linear association between weight and time on SRT (edf = 1; p = 0.57) (Figure 99) although it is again important to note that the data are sparse.
FIGURE 99.
The age-adjusted association between time on SRT and weight z-scores in children with NPC (time on SRT treated as a continuous variable).
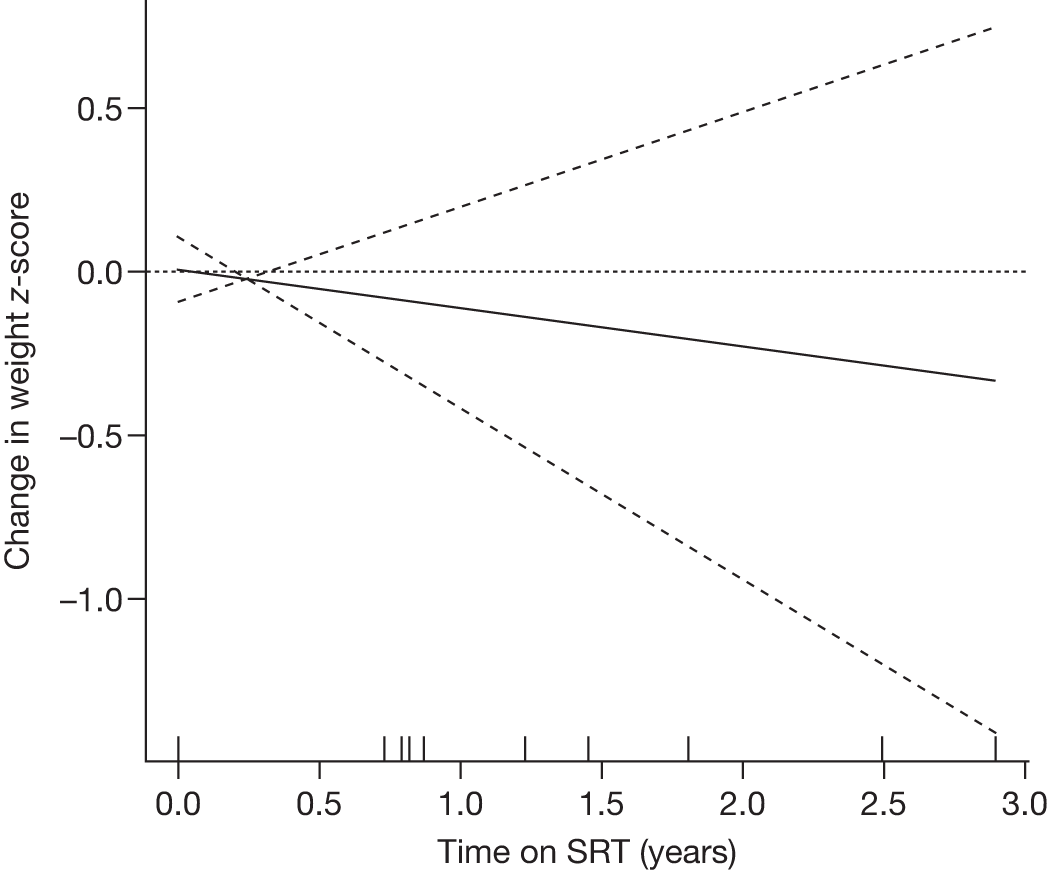
Central nervous system measures
There were insufficient data to conduct analyses on the individual CNS measures, nor were we able to produce a composite measure. Below are the descriptive details for each outcome measure. In all cases, severity was scored on a scale of 0–4, where 0 represents normal and 4 represents most severe symptoms (see Appendix 11, Clinical Record Form for NPC patients for further details). A longitudinal model for each of these CNS measures is shown in Table 155.
| Seizures/epilepsy | Vertical supranuclear gaze palsy | Ataxia | Dystonia | Dysarthia | Swallowing difficulties | Cataplexy | |
|---|---|---|---|---|---|---|---|
| Male | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Female | 0.85 | 1.27 | 1.04 | 1.87 | 0.89 | 0.85 | 0.95 |
| Standard error | 0.8 | 0.51 | 0.52 | 0.62 | 0.43 | 0.51 | 0.46 |
| 95% CI | –0.69 to 2.41 | 0.28 to 2.27 | 0.01 to 2.07 | 0.66 to 3.08 | 0.04 to 1.75 | –0.15 to 1.85 | 0.04 to 1.87 |
| p-value | 0.28 | 0.02 | 0.053 | 0.005 | 0.04 | 0.10 | 0.04 |
| Current age | |||||||
| Mean increment/year | –0.05 | 0.07 | 0.04 | –0.03 | 0.04 | 0.03 | –0.05 |
| Standard error | 0.03 | 0.02 | 0.02 | 0.04 | 0.01 | 0.02 | 0.03 |
| 95% CI | –0.12 to 0.007 | 0.02 to 0.12 | –0.002 to 0.08 | –0.11 to 0.04 | 0.01 to 0.07 | –0.01 to 0.06 | –0.11 to 0.01 |
| p-value | 0.09 | 0.01 | 0.07 | 0.34 | 0.009 | 0.18 | 0.13 |
| Time on SRT | |||||||
| Mean increment/year | 0.41 | –0.10 | –0.10 | 0.15 | –0.054 | –0.09 | 0.44 |
| Standard error | 0.25 | 0.13 | 0.11 | 0.15 | 0.07 | 0.13 | 0.20 |
| 95% CI | –0.09 to 0.91 | –0.35 to 0.15 | –0.33 to 0.11 | –0.14 to 0.45 | –0.19 to 0.09 | –0.35 to 0.17 | 0.05 to 0.84 |
| p-value | 0.12 | 0.43 | 0.35 | 0.33 | 0.46 | 0.51 | 0.03 |
| Variance component | |||||||
| Individual | 1.98 | 0.77 | 1.10 | 0.98 | 0.84 | 1.05 | 0.19 |
| Centre | 0.00 | 0.23 | 0.00 | 0.73 | 0.00 | 0.00 | 0.34 |
| Residual | 1.00 | 0.59 | 0.49 | 0.82 | 0.19 | 0.74 | 0.67 |
Seizures/epilepsy
Thirty-four recordings were collected for the occurrence of seizures/epilepsy across all time points. The mean number of seizures or epileptic fits was 1.26, range 0–4.
Sixteen of these data points are from patients taking SRT and the mean number of seizures was 1.0, range 0–4. Eighteen data points are from patients not on SRT and the mean number of seizures in this group was 1.5, range 0–4.
Vertical supranuclear gaze palsy
Thirty-nine measures of severity of vertical supranuclear gaze palsy were recorded across all time points (mean = 2.33, range 0–4). Sixteen of these data points were from patients taking SRT (mean = 2.5; range 0–4) across all time points. Twenty-three data points were from patients not on SRT and the mean number of vertical supranuclear gaze palsy noted in this group was 2.2 (range 0–4) across all time points.
Ataxia
Fifty-five measures of severity of ataxia were collected across all time points. The mean score was 1.98 (range 0–4). There were 23 recordings for ataxia for patients taking SRT (mean = 1.35; range 0–3) across all time points and 32 recordings for ataxia from patients not receiving SRT; the mean score for ataxia in this group was 2.44 (range 0–4).
Dystonia
Thirty-six measures of severity of dystonia were collected across all time points. The mean score for dystonia was 1.5 (range 0–4). There were 17 measures of dystonia for patients taking SRT. The mean score in this group was 0.88 (range 0–3) across all time points. There were 19 recordings for dystonia from patients not receiving SRT. The mean score in this group was 2.05 (range 0–4).
Dysarthia
Forty-eight measures of severity of dysarthia were collected across all time points; the mean score for speech problems was 1.18 (range 0–4). Sixteen of these data points were from patients taking SRT (mean = 0.87; range 0–2) across all time points. Thirty-two data points were from patients not on SRT and the mean score for dysarthia was 1.32 (range 0–4) across all time points.
Swallowing difficulties
Fifty-nine measures of severity of swallowing difficulties were collected across all time points (mean = 1.18; range 0–4). Twenty-two of these data points were from patients taking SRT (mean = 0.64; range 0–2). Thirty-seven data points were from patients not on SRT, with a mean score of 1.35 (range 0–4).
Cataplexy
Thirty-three measures of severity of cataplexy were collected across all time points. The mean score was 1.39 (range 0–4). Sixteen of these data points were from patients taking SRT and the mean score was 1.5, range 0–3, across all time points. Seventeen data points were from patients not on SRT and the mean score was 1.29 (range 0–4) across all time points.
After adjusting for age, the only significant change associated with time on SRT was an increase in the number of cataplexic episodes (p = 0.03).
Quality-of-life assessments
SF-36
Twelve SF-36 questionnaires were collected from adult NPC patients. Table 156 shows a descriptive summary of PCS and MCS by type of treatment.
| Time | PCS | MCS |
|---|---|---|
| Overall | ||
| Mean (SD) | 36.7 (12.4) | 49.7 (13.4) |
| n | 12 | 12 |
| SRT patients | ||
| Mean (SD) | 35.12 (11.1) | 48.0 (16.4) |
| n | 8 | 8 |
| Untreated | ||
| Mean (SD) | 37.9 (14.8) | 47.4 (7.72) |
| n | 4 | 4 |
As can be seen Table 157, the PCS was not significantly associated with age (p = 0.57) or time on SRT (p = 0.47). The MCS was not significantly associated with age (p = 0.41) but was significantly associated with time on SRT (p = 0.02), suggesting an improvement in mental functioning for patients on SRT.
| PCS | MCS | |
|---|---|---|
| Male | 0.00 | 0.00 |
| Female | –17.5 | –8.23 |
| Standard error | 7.14 | 10.2 |
| 95% CI | –31.5 to –3.53 | –28.3 to 11.8 |
| p-value | 0.03 | 0.44 |
| Current age | ||
| Mean increment/year | 0.23 | –0.49 |
| Standard error | 0.40 | 0.58 |
| 95% CI | –0.55 to 1.03 | –1.65 to 0.65 |
| p-value | 0.57 | 0.41 |
| Time on SRT | ||
| Mean increment/year | –0.95 | 2.18 |
| Standard error | 1.29 | 0.82 |
| 95% CI | –3.48 to 1.57 | 0.57 to 3.80 |
| p-value | 0.47 | 0.02 |
| Variance component | ||
| Individual | 68.6 | 194.7 |
| Centre | 0.00 | 0.00 |
| Residual | 22.4 | 0.71 |
EQ-5D
In addition to the SF-36, participants aged ≥ 13 years were invited to complete the EQ-5D. Twenty-two EQ-5D questionnaires were completed across all prospective time points. Data are presented in Table 158 for the EQ-5D score (range –0.59 to 1.0) with 1.0 being ‘perfect health’.
| N Data | Estimate of increment in EQ-5D | Standard error | 95% CI | p-value | |
|---|---|---|---|---|---|
| Gender | |||||
| Male | 13 | 0.00 | |||
| Female | 9 | –0.84 | 0.23 | –1.29 to –0.38 | 0.002 |
| Current age | |||||
| Linear effect/year | –0.02 | 0.01 | –0.04 to –0.0004 | 0.05 | |
| Time on SRT | |||||
| Linear effect/year | 0.09 | 0.04 | 0.01 to 0.17 | 0.02 | |
| Variance components | |||||
| Individual | 0.11 | 0.11 | |||
| Centre | 0.00 | 0.00 | |||
| Residual | 0.012 | 0.03 | |||
A longitudinal model was fitted to assess the linear relationship between EQ-5D and time on SRT, after adjusting for age.
The linear model showed a small but significant reduction in EQ-5D with age (p = 0.05) and a significant increase in EQ-5D with time on SRT (p = 0.02)
There was some evidence for a non-linear association between the EQ-5D score and time on SRT (edf = 1.0; p = 0.02) (Figure 100), although it is important to note that the data were very sparse.
FIGURE 100.
The age-adjusted association between time on SRT and EQ-5D scores in adults with NPC (time on SRT treated as a continuous variable).
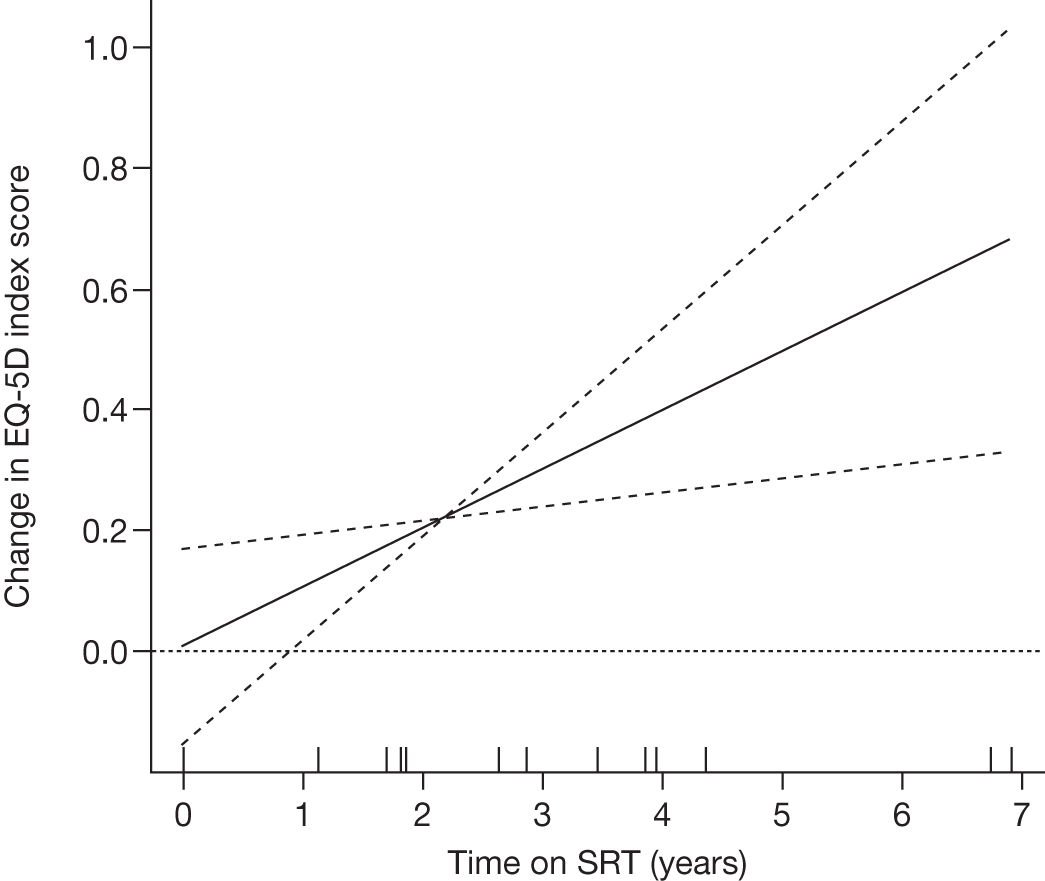
Equivalent modelling analyses were also conducted using SF-6D (SF-36-derived) utility weights, but no statistically significant associations (at α = 0.05 level) were found with time on ERT although, again, the data were very sparse. The tabulated results of the SF-6D longitudinal modelling analyses are available on request from the study authors.
EQ-5D visual analogue scale
In addition to scoring on the five domains of the EQ-5D, participants were asked to rate their health on a VAS. The EQ-5D VAS asks people to rate their health state on a 10-cm line from 0, ‘worst imaginable health state’, to 100, ‘best imaginable health state’. The 20 VAS scores completed for this study ranged from 4 to 98. A longitudinal model was fitted to assess the linear relationship between the visual analogue score and time on SRT after adjusting for age.
The linear model shown in Table 159 shows no significant association in EQ-5D with age (p = 0.31) or with time on SRT (p = 0.75).
| Estimate of increment in EQ-5D VAS | Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Gender | ||||
| Male | 0.00 | |||
| Female | –38.6 | 14.1 | –66.2 to –10.9 | 0.01 |
| Current age | ||||
| Linear effect/year | –0.75 | 0.71 | –2.14 to 0.64 | 0.31 |
| Time on SRT | ||||
| Linear effect/year | 0.78 | 2.44 | –4.0 to 5.56 | 0.75 |
| Variance components | ||||
| Individual | 286.6 | |||
| Centre | 83.5 | |||
| Residual | 41.3 | |||
No evidence was found for a non-linear association with EQ-5D VAS and time on SRT (edf = 1; p = 0.75) (Figure 101), although it is important to note that the data are very sparse.
FIGURE 101.
The age-adjusted association between time on SRT and EQ-5D VAS in adults with NPC (time on SRT treated as a continuous variable)
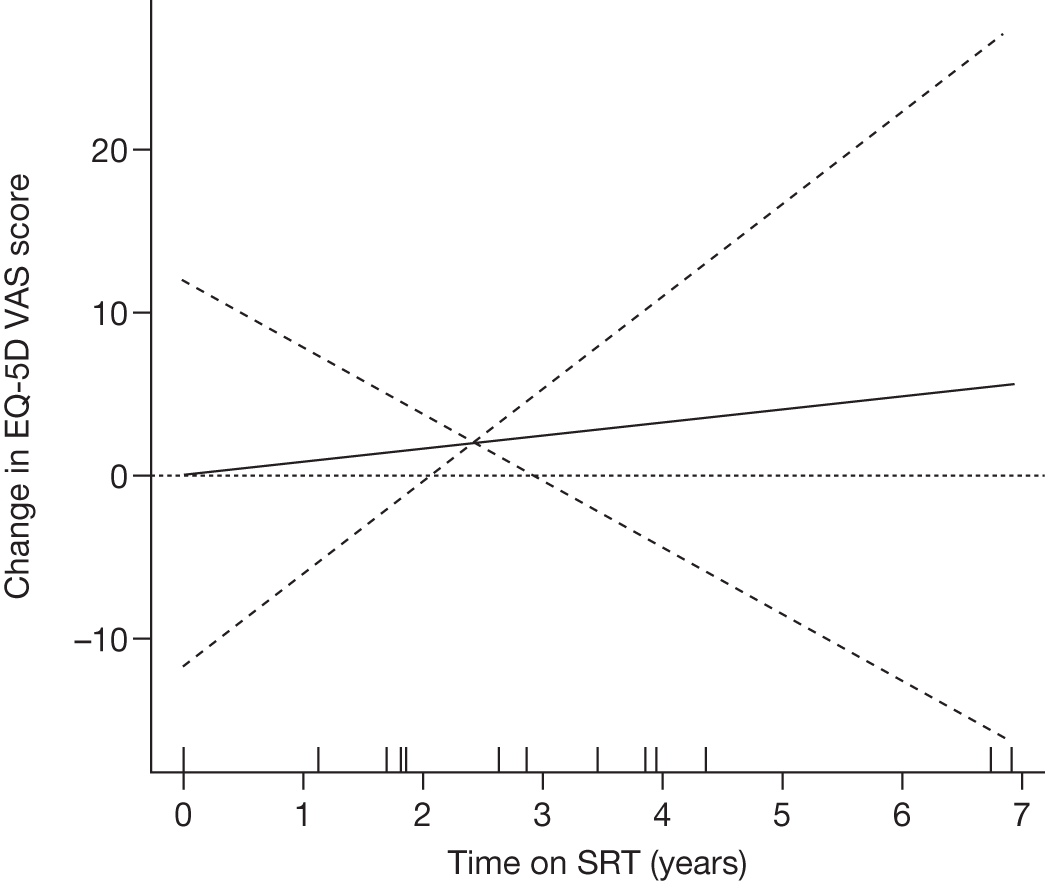
PedsQL
Fifteen PedsQL questionnaires completed by children or their carers were collected. Table 160 shows a descriptive summary of the total score and the scores from the component scale scores by type of treatment.
| Physical functioning | Emotional functioning | Social functioning | School functioning | Psychosocial health summary score | Physical health summary score | Total score | |
|---|---|---|---|---|---|---|---|
| Overall | |||||||
| Mean (SD) | 49.9 (37.4) | 58.7 (27.2) | 51.9 (32.4) | 39.2 (22.9) | 43.1 (23.1) | 49.9 (37.4) | 39.9 (24.5) |
| n | 15 | 15 | 14 | 8 | 8 | 15 | 8 |
| Not on treatment | |||||||
| Mean (SD) | 62.6 (43.1) | 66.1 (31.4) | 72.4 (28.3) | 46.6 (33.1) | 53.4 (30.0) | 62.6 (43.1) | 49.0 (33.8) |
| n | 9 | 9 | 9 | 4 | 4 | 9 | 4 |
| SRT | |||||||
| Mean (SD) | 40.7 (27.2) | 51.5 (19.4) | 28.6 (12.3) | 34.1 (13.0) | 36.8 (15.6) | 40.7 (27.2) | 35.2 (14.5) |
| n | 6 | 6 | 5 | 4 | 4 | 6 | 4 |
There were insufficient data across time points to fit longitudinal models for the PedsQL data.
Fatigue Severity Scale
Eight FSS questionnaires were completed by seven patients prospectively. The scores ranged from 1.22 to 7.0. There were not enough data to carry out further analysis.
Carer Strain Index
Seventeen CSI questionnaires were completed across all prospective time points. Data for the CSI total score ranged from 4 to 23; the maximum possible score is 26. A longitudinal model was fitted to assess the linear relationship between the CSI and time on SRT, after adjusting for age (Table 161).
| Estimate of increment in CSI | Standard error | 95% CI | p-value | |
|---|---|---|---|---|
| Gender | ||||
| Male | 0.00 | |||
| Female | 2.99 | 4.03 | –4.91 to 10.8 | 0.47 |
| Current age | ||||
| Linear effect/year | 0.00 | 0.00 | ||
| Time on SRT | ||||
| Linear effect/year | 1.38 | 1.99 | –2.52 to 5.28 | 0.50 |
| Variance components | ||||
| Individual | 33.3 | |||
| Centre | 0.0 | |||
| Residual | 0.0 | |||
The linear model shows no significant association in score on the CSI with gender of the person they are caring for (p = 0.47) or with time on SRT (p = 0.50).
Safety and complications
Of the 36 participants with NPC in this study, no patients reported that they had experienced AEs (febrile reactions, anaphylactic reactions, pre-medication or positive antibody status).
During the period of data collection one patient stopped SRT with no specific reason given and no NPC patients died from disease-related complications.
Cost of substrate reduction therapy in people with NPC
Table 162 shows the current unit purchase cost to the NHS of the SRT miglustat.
| Drug full name | Proprietary name and unit | 2011 base price per unit (£) |
|---|---|---|
| Miglustat (SRT) | Zavesca®, 100 mg | 46.84 |
Table 163 shows the NSCT-estimated annual NHS per patient cost of providing these drugs. Note that these costs include both the drug costs and home-care costs where the NSCT fund them.
| Drug | Adults | Children |
|---|---|---|
| Miglustat | £94,108 | £48,627 |
Cost of care for adults with NPC
Total care cost – financial burden of NPC
Table 164 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for an adult with NPC, although this is only based on 21 annual data points from 12 patients. Of the estimated mean per patient annual cost of £3800, only about one-quarter is as a result of NHS hospital services used. Of this, about two-fifths (£410 per patient per year) is from inpatient stays, whereas almost all of the remainder costs (£590) are from outpatient visits (see Table 165). Of the £2800 per patient per year from using services outside of hospital, < £60 is owing to GP visits and > £2400 is as a result of regular visits from ‘care attendants’ (see Table 166).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. who used this type of service (%) | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 21b | 52 | 16 (76) | 1000 | 1402 | 1000 | 300–1400 |
| Services outside hospital | 21b | 52 | 18 (86) | 2800 | 7825 | 380 | 150–1700 |
| Total health (NHS) and social-care cost | 21b | 52 | 19 (90) | 3800 | 7877 | 1100 | 530–3100 |
| Type of hospital care | No. who used this type of service (%) | Mean cost (£) | Standard deviation | Mediana cost (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| Inpatient stays | 3 (14) | 410 | 1284 | 1900 | 1100–5600 |
| Outpatient visits | 15 (71) | 590 | 540 | 940 | 300–1000 |
| Day cases | 0 | 0 | 0 | 0 | N/A |
| Accident and emergency visits | 4 (19) | 29 | 74 | 100 | 100–310 |
| Total hospital (NHS) care cost | 16 (76) | 1000 | 1402 | 1000 | 300–1400 |
| Care provider | No. who used this provider (%) | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 11 (52) | 51 | 73 | 110 | 14–180 |
| GP nurse appointments | 8 (38) | 3 | 5 | 5 | 3–10 |
| District nurses | 3 (14) | 24 | 64 | 192 | 96–220 |
| Community mental health nurse | 3 (14) | 14 | 36 | 72 | 72–140 |
| Other nurse or health visitor | 3 (14) | 45 | 137 | 260 | 88–590 |
| Counsellor | 0 | 0 | 0 | 0 | 0–0 |
| Other therapist | 2 (10) | 14 | 54 | 0 | 37–250 |
| ‘Alternative’ medicine or therapy | 0 | 0 | 0 | 0 | N/A |
| Psychologist | 1 (5) | 1 | 6 | 27 | N/A |
| Psychiatrist | 2 (10) | 55 | 231 | 580 | 94–1100 |
| Other community-based doctor | 1 (5) | 5 | 22 | 99 | N/A |
| Occupational therapist | 9 (43) | 26 | 49 | 39 | 19–160 |
| Social worker | 6 (29) | 150 | 350 | 400 | 110–950 |
| Home help | 4 (19) | 270 | 764 | 1450 | 100–2600 |
| Care attendant | 3 (14) | 2400 | 7863 | 18,300 | 65–32,000b |
| Community support worker | 2 (10) | 20 | 69 | 210 | 110–300 |
| Housing worker | 0 | 0 | 0 | 0 | 0–0 |
| All non-hospital NHS and social-care providers | 18 (86) | 2800 | 7825 | 380 | 150–1700 |
Cost breakdown by hospital- and community-based services
Tables 165 and 166 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by adults with NPC. Only 3 of the 21 patient-years, or about 14% of the years for which we had valid service-use data, had hospital stays as inpatients, and this accounted for about two-fifths of the NHS hospital costs in NPC adults. In contrast, for over two-thirds (71%) of patient-years there was at least one hospital outpatient attendance during the previous 12 months.
The majority of costs related to using community-based services were as a result of the two or three adults with NPC who reported using care attendants. They accounted for £2400 of the £2800 annual per patient cost of services used outside of hospital. Although over half of the adult NPC patient-years had involved seeing a GP at least once during the past year, and over one-third involved seeing a practice nurse at a GP surgery, these accounted for < £60 of the £3800 annual per patient cost of NHS and publicly funded social-care services consumed. Other support providers used by smaller proportions of adults with NPC were district nurses, psychiatrists, occupational therapists, community support workers, social workers and community mental health nurses (see Table 165).
All of the resource use and cost findings relating to adults with NPC should be treated with considerable caution because of the small number of patients for whom we have data.
Cost of care for children with NPC
Total care cost – financial burden of NPC
Table 167 shows the estimated annual cost to the NHS and publicly funded social-care services of caring for a child with NPC, based on the 19 patients whose parents or carers provided service-use data at study entry (8 males, mean age 6.6 years, age range 1.2–15.4 years). Of the estimated mean per patient annual cost of £4200, over three-quarters is due to NHS hospital services used. Of this, > 80% (£2600 per patient per year) is from inpatient stays, whereas almost all of the remainder costs (£550) are from outpatient visits (see Table 168). Of the £940 per patient per year from using services outside of hospital, only £200 per patient is as a result of GP visits and a wide range of other health- and social-care professionals are used by some children with NPC, for both physical and mental health problems (see Table 169).
| Type of service | No. with valid resource use data | Per cent of all at study entry | No. who used this type of service | Mean cost (£) | Standard deviation | Median costa (£) | Interquartile rangea (£) |
|---|---|---|---|---|---|---|---|
| Hospital services | 19 | 82 | 12 | 3300 | 4926 | 5700 | 990–7500 |
| Services outside hospital | 19 | 82 | 19 | 940 | 1240 | 210 | 130–1300 |
| Total health (NHS) and social-care cost | 19 | 82 | 19 | 4200 | 5351 | 1100 | 300–5400 |
| Type of hospital care | No. who used this type of service | Mean cost (£) | Standard deviation | Median cost (£) | Interquartile range (£) |
|---|---|---|---|---|---|
| Inpatient stays | 8 | 2600 | 4678 | 5900 | 940–10,300 |
| Outpatient visits | 6 | 550 | 970 | 1600 | 990–2000 |
| Day cases | 2 | 70 | 210 | 670 | 670–670 |
| Accident and emergency visits | 1 | 5 | 24 | 100 | N/A |
| Total hospital (NHS) care cost | 12 | 3300 | 4926 | 5700 | 990–7500 |
| Care provider | No. who used this provider | Mean cost (£) | Standard deviation | Median cost (£) | Interquartile range (£) |
|---|---|---|---|---|---|
| GP visits (including home visits) | 19 | 200 | 227 | 130 | 68–220 |
| GP nurse appointments | 7 | 3 | 7 | 5 | 3–18 |
| District nurses | 2 | 16 | 48 | 150 | 130–170 |
| Community mental health nurse | 0 | 0 | 0 | 0 | N/A |
| Other nurse or health visitor | 6 | 93 | 160 | 310 | 180–440 |
| Counsellor | 0 | 0 | 0 | 0 | N/A |
| Other therapist | 6 | 100 | 280 | 130 | 50–440 |
| ‘Alternative’ medicine or therapy | 0 | 0 | 0 | 0 | N/A |
| Psychologist | 6 | 58 | 123 | 120 | 81–240 |
| Psychiatrist | 1 | 30 | 130 | 570 | N/A |
| Other community-based doctor | 3 | 54 | 159 | 280 | 82–650 |
| Occupational therapist | 10 | 71 | 117 | 110 | 29–200 |
| Social worker | 4 | 110 | 244 | 590 | 150–740 |
| Home help | 2 | 14 | 48 | 140 | 75–200 |
| Care attendant | 0 | 0 | 0 | 0 | N/A |
| Community support worker | 1 | 190 | 826 | 3600 | N/A |
| Housing worker | 0 | 0 | 0 | 0 | N/A |
| All non-hospital NHS and social-care providers | 19 | 940 | 1240 | 210 | 130–1300 |
Cost breakdown by hospital- and community-based services
Tables 168 and 169 show the cost breakdown of the hospital and community (non-hospital) services and professionals used by children with NPC. Eight of the 19 patients had hospital stays as inpatients, and this accounted for 80% of the NHS hospital costs in NPC children. Five of these children received inpatient care during the year costing > £5000 and one child incurred inpatient costs of > £16,000 (due to respite care and a gastrostomy operation). Six of the NPC child patients reported having at least one hospital outpatient attendance during the previous 12 months, with a median annual outpatient cost of £1600 for these children.
The costs related to using community-based services or care professionals were due to a wide range of services used, including GPs, general practice nurses, health visitors, social workers, other therapists (mostly physiotherapists), other doctors (paediatricians), psychologists and psychiatrists (see Table 169). Although all of the child NPC patients had seen a GP at least once during the past year, and over one-third reported seeing a practice nurse at a GP surgery, these two services accounted for only £200 of the £4200 annual per patient cost of NHS and publicly funded social-care services consumed.
Association of time on enzyme replacement therapy and costs of caring for patients with NPC
From the longitudinal regression modelling of costs in child patients with NPC, the models did not converge, and so no reliable estimation of the association between time on SRT and costs could be made. In adult patients with NPC, although there was a statistically significant association between hospital costs and time on SRT (costs 27% lower; 95% CI 12% to 41%; p = 0.002), this was based on only 21 data points from 12 patients (only six with data from more than one time point). This result should therefore be interpreted with considerable caution, as it may be mainly because of the three high-cost patients (hospital costs > £2000 per year) who were not on SRT during the study. The tabulated results of these analyses are available on request from the study authors.
Discussion of NPC results
Niemann–Pick disease type C is an autosomal recessive disorder characterised by progressive neurological deterioration leading to premature death. The disease is characterised by impaired intracellular lipid transport and build-up of lipids in various tissues, particularly the brain. Miglustat is the only approved therapy for patients with NPC. It has been approved in the EU for the treatment of progressive neurological manifestations in adult patients and paediatric patients with NPC.
Existing estimates of the effectiveness of SRT in NPC, summarised in Table 9, are based on the results of one RCT,212 two open-label extension studies of that RCT213,214 and several case reports. 206–211,215 The strongest evidence is provided by the trial reported by Patterson and colleagues,212 a randomised, placebo-controlled trial comprising 29 patients aged ≥ 12 years (20 on SRT and 9 control subjects) with 52 weeks’ follow-up. Twelve children (< 12 years old) were also included but all received miglustat. The treated group in the RCT showed an improvement in horizontal saccadic eye movements compared with patients receiving standard care; results were significant when patients taking benzodiazepines were excluded. Improvements in swallowing ability and stabilisation of auditory acuity and ambulation were reported in treated patients ≥ 12 years old. The additional child cohort also reported an improvement (compared with baseline) in horizontal saccadic eye movement. The other studies all suggest improvements (or stabilising of outcome) across a number of outcomes over time in patients treated with miglustat, although these are less straightforward to interpret given the lack of an untreated comparator.
In this study, we examined potential associations between SRT and stature (height and weight), several CNS measures and QoL. These analyses depend on the fact that patients began treatment with miglustat at different ages dependent on the time that the drug became available. Clearly there is considerable potential for confounding, particularly by severity with those with more severe disease being diagnosed and beginning treatment earlier.
We found no statistically significant relationship between time on SRT and height or weight. Of the CNS measures studied the only statistically significant association with treatment was an increase in the cataplexy score. There were small but statistically significant associations with time on SRT and the EQ-5D score and MCS of the SF-36. No association was seen in PCS of the SF-36 or the CSI. There were insufficient data to analyse the effect of SRT on the PedsQL.
As previously described, all analyses in NPC were hampered by a paucity of data. Not only were we able to recruit only a relatively small number of patients but for many of those recruited, data were lacking for key outcomes. This occurred despite the outcomes chosen being those that the clinical collaborators believed to be the best measures of disease progression and measures which they believed would be recorded for the vast majority of patients at each regular clinical contact. It is not clear to what extent this reflects the tests not being carried out or being performed but not recorded. This lack of data means that we have low power to detect treatment effects.
Costs associated with NPC
As with all other conditions investigated in this study, we were keen to capture the wider costs of care falling on the public sector in addition to the costs associated with SRT.
Based on patients’ self-reported health- and social-care service use, the annual cost of caring for people with NPC, excluding the purchase cost of SRT, was estimated at £3800 for an adult and £4200 for a child. These costs, however, are dwarfed by the cost of the therapies; the mean annual cost of SRT for adults with NPC is £94,108 and £48,627 for children with NPC.
From the longitudinal regression modelling of costs, there was no statistically significant association (i.e. p-value < 0.05) between time on SRT and either total NHS and social-care costs, hospital-care costs, or non-hospital-care costs for patients with NPC. The tabulated results of these analyses are available on request from the study authors.
Owing to these high associated costs, and the lack of measureable effect of SRT on either clinical outcomes or HRQoL measures, it was infeasible to conduct either a cost-effectiveness or cost–utility analysis. As they apply to all six LSDs, the limitations of these cost estimates are summarised and discussed in Chapter 9.
Chapter 9 Discussion
Lysosomal storage disorders are all individually rare and, although the effects vary greatly between and within conditions, the consequences can be extremely severe and life-shortening for many of those affected. Against this background it is perhaps unsurprising that where treatments become available it has generally proven difficult to mount long-term, high-quality, placebo-controlled RCTs.
Over the last two decades, exogenous ERT has become available for a number of these conditions. Although the safety of these treatments has been reasonably well documented,102,104,147,148,164,167,187 the evidence of efficacy is much weaker. Most trials conducted to date have included relatively small numbers of patients and have relied primarily on biochemical markers or other surrogate outcome measures or have relied on data from uncontrolled studies. The evidence on efficacy of ERT in Gaucher disease, Fabry disease and MPS I was well summarised by Connock and colleagues in 2006. 18,19 These treatments are expensive and place a considerable burden on patients and carers, but current evidence provides limited information on the relative costs and benefits, or on questions such as the effects of starting (or stopping) therapy at different points in the disease course.
This study aimed to improve our estimates of effectiveness and costs by conducting an observational, multicentre, cohort study among patients with LSDs receiving treatment in the seven NSCG-designated centres for the treatment of LSDs (Addenbrooke’s Hospital Cambridge, Birmingham Children’s Hospital, University College London Hospitals, Great Ormond Street Hospital for Children, Royal Free Hospital London, Salford Royal Hospital and Royal Manchester Children’s Hospital) in England. We collected clinical data retrospectively and prospectively from patient records and QoL, carer burden and service-use data directly from patients and their families.
The results of our analyses for each condition are discussed in the relevant chapters. The degree to which our data can provide useful information regarding the effects of ERT varies between the six conditions studied, primarily reflecting the numbers of patients with each condition.
The data strongly reinforce the conclusions of previous research that ERT is effective in mitigating the effects of Gaucher disease as assessed by surrogate outcomes such as Hb and platelet counts and liver and spleen size. The estimates of the effects provided for clinically important outcomes such as bone pain, fatigue and QoL are less precise because of the paucity of data.
For Fabry disease, the commonest of the conditions we assessed, we were able to conclude with considerable confidence that the use of ERT is associated with significant improvements in both cardiac and renal manifestations of the condition and found some evidence that it is associated with a decreased impact of pain on QoL. We found no evidence that ERT is associated with a decrease in the risk of strokes or TIA. Our data suggested that the duration of ERT use was associated with a worsening of Qol scores and of fatigue. It is unclear whether this association is causal or whether it reflects confounding in a heterogeneous condition in which age at diagnosis (and hence possibly treatment initiation) may be associated with intrinsic severity.
There are two preparations available for the treatment of Fabry disease, agalsidase alpha and agalsidase beta, and there has been considerable debate regarding their relative effectiveness. The decision as to which of these was prescribed for an individual patient was largely determined by the consultant who was responsible for commencing treatment. We found no statistically significant difference in the magnitude of the association between ERT and any outcome dependent on which preparation was prescribed.
For the other conditions studied we were able to recruit only relatively small numbers of patients and consequently have little power to estimate clinical effects. We recruited only 12 patients with infantile-onset Pompe disease, all of whom received treatment with ERT from the time of diagnosis. This meant that we could not reliably estimate the associations between the use of ERT and clinical outcome. The magnitude of the effect of ERT reported in a previous comparison of these patients with historical controls193 provides strong evidence of efficacy, although the long-term prognosis remains poor, reflected in the finding here that 4 of 11 patients had restricted mobility and 4 of 11 were ventilator dependent by the age of 5 years. In patients with adult-onset Pompe disease, we did find evidence of an association between use of ERT and the distance patients could walk and their muscle test scores, but not with a range of other clinical outcomes, nor with QoL or fatigue scores. The estimates of all of these associations were imprecise owing to the limited data available.
Our sample included only 24 patients with MPS I who received ERT, providing little power to estimate its effects on clinical outcomes or QoL. Although we found no evidence of an association between ERT and any outcome measures, the estimates of effect are imprecise therefore limiting interpretation. Similarly, among patients with MPS II, the small number of patients recruited and limited availability of data on key outcomes for those included provided little power. A statistically significant association between use of ERT and children’s height was seen but we found no other statistically significant associations. For both conditions the apparent lack of association between ERT and positive clinical outcomes needs to be interpreted with considerable caution owing to the paucity of data.
Analyses of the association between the use of SRT and clinical outcome in people with NPC were similarly hampered by both the small numbers recruited and the lack of data regarding key outcomes for those who did participate. No statistically significant associations were found between use of SRT and clinical or QoL outcomes, but these findings need to be interpreted in light of the lack of power available.
Costs associated with lysosomal storage disorders
Much of the past discussion regarding the costs associated with having a LSD has concentrated on the substantial costs associated with ERT. In this study we aimed to capture, in addition, the wider costs within the public sector. Based on self-reported health- and social-care service use within this study, the public sector annual cost of caring for people with LSDs – but excluding the purchase cost of ERT or SRTs – varies from just > £3000 to nearly £12,000 for adults and from £1300 to £18,600 for children. Although the care of patients with Gaucher disease, Fabry disease and NPC costs ≤ £4000 per year, care costs are > £10,000 for adults with MPS II and children with Pompe disease, and > £18,000 for children with MPS I. For all LSDs, on average, hospital care accounted for a higher proportion of all care costs for children than for adults.
It is important to acknowledge that these costs are dwarfed by the costs of ERT. For adults with LSDs the annual cost of ERT is between 30 and 45 times the estimated annual cost of other treatments and care, whereas for children the annual cost of ERT is between 37 and 66 times the estimated annual cost of other treatments and care.
In this study we were unable to clearly demonstrate the effectiveness of ERT consistently across both clinical domains and HRQoL in any of the LSDs. It is, however, possible to conduct a crude threshold analysis based on the annual cost of providing ERT to adults or children with each LSD – which is known with reasonable certainty. In MPS I, MPS II or Pompe disease, the mean annual per patient NHS cost of receiving ERT is between £258,000 and £538,000 for adults, and between £122,000 and £314,000 for children. In Gaucher disease and Fabry disease the annual NHS cost of ERT varied between £79,000 and £188,000, depending on child or adult status and on specific ERT (see Appendix 18).
Currently in England and Wales, there is a maximum willingness-to-pay threshold for a QALY which is conventionally used by NICE for informing its guidance on health technologies. When judging the cost-effectiveness or value for money to the NHS of drugs or medical devices, anything with an implied cost of new QALYs of > £30,000 per QALY is generally deemed to not be cost-effective.
Combined with the current annual price to the NHS of the different ERTs, 3.6 and 17.9 discounted QALYs would need to be generated for each year of being on treatment in order for them to be considered cost-effective (or between 2.6 and 10.5 discounted QALYs for child patients). The effectiveness of ERT in adults and children with MPS-II would have to be particularly high in order to justify the very high current costs of ERT in this LSD. These estimates are shown for all LSDs in Appendix 19.
Study strengths and limitations
Intrinsic limitations of observational data
There are inevitable weaknesses in the use of observational data to assess efficacy. Key among these is the difficulty in controlling for confounding by variables related to the intrinsic severity of the conditions. This is a particular issue in conditions such as LSDs which are heterogeneous in their manifestations. In this study we aimed to make use of the fact that the primary determinant of when during the course of their disease older patients began the use of ERT was the time when the therapy became available. There is however scope for this to be confounded both by the severity of the condition leading to differences in the age at recognition and in differences in the age at which clinical decisions regarding initiation of treatment are made.
It is also important to recognise the potential impact in the analyses of differential attendance at participating centres. Although all those receiving ERT are invited to appointments at least annually, actual attendance is lower. It is likely that this attendance will be related to differences in outcome. This may be further complicated by the lack of completeness we found in data collection and recording of key outcomes among those who did attend which again may be related to clinical condition. Finally, although the collection of retrospective data from the patients’ medical records increased our ability to examine the natural history of treated and untreated conditions, there may well have been changes over time in both which tests are carried out and how they are performed.
Difficulties in recruitment
At the start of the study we identified 1106 potentially eligible patients across the six conditions included. Of these, the consultants responsible for their care decided that 133 (12%) were in some way too vulnerable to be approached, leaving 973 potential participants. Of these, 758 (77.9%) were approached and asked whether or not they would be prepared to participate.
We recognised that there was considerable suspicion within the patient community that the findings of this study might be used to withdraw treatment which they valued. This suspicion had been exacerbated by the findings of a previous review funded by the HTA which concluded that whatever assumptions were made regarding effectiveness, the high costs of the drugs meant that ERT was unlikely to meet conventional thresholds for cost-effectiveness. The research team made strenuous efforts to work with the patient support groups in the design of the study to reassure them about the nature of the study. These efforts were largely successful and these groups made a major contribution to the design of the study, including the design of information leaflets, and to publicise the study among their members. In large part because of this assistance the number of eligible people asked to participate who refused consent was small (47/758, 6.2%).
As seen from these figures, a major problem with recruitment was the failure to ask potentially eligible patients whether or not they would be prepared to take part. The clinical co-applicants were clear that consent for participation be obtained by either a doctor or nurse. We had considerable difficulty recruiting and retaining research nurses in the study and for about 50% of the time, we had non-clinical research analysts rather than nurses based in the treatment centres. During these periods consent had to be obtained by the consultants. This process was variably successful as it was dependent on the consultant concerned both being aware that a particular patient had not yet been recruited and having sufficient time available during clinic to go through the consent process. The problem of recruitment was further compounded by one centre where the research nurse left part way through the study and the centre was unable to provide accommodation for a replacement nurse thereafter leading to the cessation of further recruitment in that location.
Recruitment was also hindered by the fact that, although all patients are offered appointments at least once per year, they are drawn from very large geographical areas and actual clinic attendance is less frequent for many patients. This meant that for many patients if one opportunity for recruitment was missed, no further opportunities would present themselves during the course of the study.
Data quality
Clinical data fields were agreed within the management team working within disease-specific groups. The clinicians from the seven sites were asked to identify which measures they believed would most reliably reflect disease progression and would be available and recorded in the notes for most patients as part of routine clinical assessment. They were also asked to consider whether common or equivalent approaches to assessment of each measure were carried out at the seven participating centres.
The process of refining a universal minimal data set for each condition was hampered by differences of opinions between the clinical co-applicants, and different working practices at each of the treatment centres. While each centre practised in accordance with the national guidelines,216–220 the exact series of tests and investigations carried out differed from centre to centre, such that the final set of clinical measures contained some tests and investigations which were not conducted at all sites.
The nurses and researchers at each site were required to collect a broad spectrum of clinical information from medical notes, much of which was not available within the patient’s main set of hospital records but held in differing clinical departments within the Trust. This made the data collection process (and in particular the retrospective data collection process) much slower and more prone to error than had been anticipated.
It became clear that for many of the agreed clinical outcome measures these were in fact either not assessed, or were assessed but not recorded in the clinical notes in a fashion which could easily be retrieved. This included not merely the results of the more complex tests or investigations but even in some cases simple clinical observations such as height in children or the presence or absence of hepatosplenomegaly. Our inability to extract data on key outcomes was a particular problem for retrospective data points where very large amounts of data were missing. The consequence is that we had far less data available than we would have anticipated from the numbers of patients recruited.
Strengths and limitations of cost data and analysis
The health- and social-care service-use data have been collected using a well-established self-completion questionnaire, the CSRI,233 which was used across all the LSDs in the study, and which had been amended in various ways to be more suitable for people with LSDs and the spectrum of services and professionals that they might use (on advice from the family associations/patient support groups). The data collected also included self-reported costs and resource use falling on patients and their families or informal carers, and other wider ‘economic impacts’ such as lost days of school or work owing to health problems (although these costs and impacts have not been reported in this report). Hospital and other service-use data have been costed using the most reliable and up-to-date national sources of unit costs available in the UK (e.g. NHS reference costs 2009–2010239 and the PSSRU’s Unit Costs of Health and Social Care 2010240), with the data carefully cleaned and checked for anomalies in SPSS.
The cost of care results should be interpreted in light of the self-reported (i.e. recalled) nature of the service-use data; the often small samples of patients or parents who provided service-use data at study entry for some LSDs; and that those patients who supplied health- and social-care service-use data were a smaller, and possibly atypical, subset of all patients recruited to the study in each LSD.
No self-report service-use data can be assumed as entirely valid and reliable, especially if there is no other routine or administrative data source against which to check the accuracy. However, research in adults suggests that patient self-report agrees closely with service or provider records for hospital use, with recall periods of up to 6 months, but that for medication and other care products patient recall can be quite incomplete. 278 Furthermore, for people with LSDs, the types of health/care service use that accounted for the majority of the mean care costs were either long inpatient hospital stays or regular (e.g. 26 or 52 times a year) social-care or nursing visits; and so recall of such service use should hopefully be more reliable than for more intermittent and shorter (and therefore less costly) episodes of care.
In addition, there were some specific shortcomings of the questionnaire which its piloting did not reveal, and which may have affected the reliability of some aspects of the results. Most notably, there was no specific question about the number and duration of ERT or other infusion sessions received outside hospital. Consequently, patients appeared to report these sessions under a variety of service headings (e.g. as multiple visits by ‘other nurse or health visitor’ or by ‘care attendants’, or even occasionally as ‘alternative medicine or therapy’). Because of the similarity of hourly pay rates attached to these different types of nurse or carer, the overall impact on the estimated care costs should not be great. However, not knowing for certain whether or not they were for ERT infusions (or other regular infusions or treatments) means that it is difficult to estimate a care cost excluding ERT infusion or home care costs. This is important because for some drugs the ERT infusion sessions are paid for by the drug manufacturer (and are nominally included in the drug price; Julie Partridge, NSCT, January 2012, personal communication from the NSCT, 10 January 2012). So, without complete data on which regular nursing visits were definitely for ERT infusions, summing the estimated costs of care in a given LSD with the drug costs of ERT for that LSD might be double-counting the cost of providing the ERT sessions.
Similarly, the questionnaire did not separately ask how many annual or biannual LSD consultant checks/monitoring days had been attended; nevertheless, this should have still been captured in their reporting under either outpatient appointments or day cases. Also, the questionnaire responses of study participants indicated that the standard types of hospital visit (inpatient admission, outpatient appointments or day case admissions) are not easily distinguishable for patients regardless of how well the question is worded. Yet these are the categories of patient care episodes which all hospitals use and which are the basis of national data sets of unit costs.
Chapter 10 Conclusion
This study provides further evidence on the effectiveness of ERT for people with Gaucher disease, Fabry disease and Pompe disease and in people with MPS I and MPS II. However, the estimates of effectiveness (and hence cost-effectiveness) remain imprecise, particularly for the less common conditions. There is little strong evidence on the effectiveness of starting or stopping therapy at different points in the course of the condition. This information is needed both to guide clinical policies and so that people with the condition and their carers can make rational decisions about treatment options.
It is in our view unlikely that either for conditions where treatment is currently available or for those where ERT is likely to become available in the future, large randomised, placebo-controlled trials powered on clinical outcomes will be conducted. We have shown that observational data from a cohort study which collects both retrospective and prospective data can be used to provide estimates of treatment effectiveness. We have also shown that these analyses can be extended using Bayesian methods to provide estimates of the likely effects of therapy commenced at different points in the course of the condition. Our study was, however, severely weakened by the lack of consistency in assessing and clearly recording key outcomes that reflect disease progression, which led to substantial amounts of missing data. This not only reduced the power of our analyses but also increased the possibility of bias because of unmeasured confounding factors which may have influenced the likelihood of the recording of particular outcomes for individuals.
If future research is to more effectively address the unanswered question regarding effectiveness and cost-effectiveness the following steps will be required:
-
Agreement regarding appropriate outcome measures can be used to assess disease progression for each condition.
-
Agreement between designated UK treatment centres to collect these measures in a common data set for all patients with these conditions receiving ERT or SRT.
-
For the less common conditions, to attempt to extend this approach to include centres in other countries.
Acknowledgements
We would like to thank Marie Meehan, Andrea Hill, Debbie Hugh, Sarah West, Sandhya Maddukuri, Kate Blackler, Hannah Russon, Navneeta Reddy, Jennifer Hutchinson, Nike Aina and Noura Hamdi for their hard work in the recruitment of patients to this study, and in managing the patients’ data at each of the seven sites. We would like to thank Tim Cox, Patrick Deegan, Chris Hendriksz, Philip Lee, Uma Ramaswami, Ed Wraith, Simon Jones and Tanya Collin-Histed for their contribution to the design and conduct of the study. We would also like to thank the nursing and support staff at each of the sites who assisted with the administering processes of the study and in sending out patient questionnaires, with special thanks to Elizabeth Morris, Jackie Imrie and Gill Moss.
We are extremely grateful to all members of the LSDs patient support groups for the advice and support at each step of this study. They helped us appreciate the difficulties experienced by this group of patients, and assisted in the design of our questionnaires to ensure that we captured all aspects of the conditions we were studying. We also thank all members of the Trial Steering Committee for their advice along the way.
We would like to thank Sheena Oxer for her hard work in the initial set-up, and co-ordination of the study in the early days of data collection, and Laura Cocking for her help in the design and the management of the condition-specific databases throughout the study.
We are very grateful to Dr Gregory Pastores for allowing us to use his online gene reviews as a source for the background section of this report. We are also very grateful to Alison Hurst for her assistance in editing and formatting the final manuscript.
We would like to thank Julie Partridge and Steve Morgan, from NSCT, for supplying the ERT cost data.
Finally, this study would not have been possible without the patients and their families who allowed us to collect information from their hospital records and gave their time to complete the questionnaires. We thank them for their invaluable contribution.
The co-ordinating team based at the PCMD in Exeter comprised Professor Stuart Logan (Chief Investigator), Professor Ken Stein (Methodological Input and probabilistic modelling), Katrina Wyatt (Project Manager), Louise Klinger, Sheena Oxer and Lindsey Anderson (Study Co-ordinators), Professor William Henley and Vasilis Nikolaou (Study Statisticians), and Rob Anderson (Study Health Economist). In addition there were 12 other co-applicants, including 11 clinicians, with representatives from each of the seven NCSG-designated LSD treating centres (Professor Timothy Cox, Dr Patrick Deegan, Dr Chris Hendriksz, Dr Derralynn Hughes, Dr Robin Lachmann, Dr Philip Lee, Dr Atul Mehta, Dr Uma Ramaswami, Dr Ashok Vellodi, Dr Edmond Wraith, Dr Stephen Waldek), as well as Tanya Collin-Histed from the Gauchers Association, acting as representative of all the LSD patient support groups. Together, the co-ordinating team and the co-applicants comprised the Management Team. Additional clinical support was given throughout the study by Dr Simon Jones.
Contribution of authors
Katrina Wyatt (Project Manager) contributed to the design of the study, data collection forms and questionnaires, carried out the evidence literature review, contributed to interpretation of the data and assisted in writing all sections of the report in conjunction with William Henley, Lindsey Anderson, Rob Anderson and Stuart Logan.
William Henley (Study Statistician) contributed to the design of the study and was responsible for the analysis and interpretation of the data, and wrote the statistics section of the methods chapter.
Lindsey Anderson (Study Co-ordinator) managed the conduct of the study including administration relating to the collaborating centres, and assisted in writing all sections of the report.
Rob Anderson (Study Health Economist) contributed to the design of the study, data collection forms and questionnaires and was responsible for the cost analysis and interpretation of the data.
Vasilis Nikolaou (Statistician) conducted the clinical data and HRQoL analyses.
Ken Stein contributed to the design of the study and assisted in the editing of the report.
Louise Klinger (Study Co-ordinator) managed the conduct of the study including administration relating to the collaborating centres.
Derralynn Hughes contributed to the design of the study and clinical data collection forms, and assisted in the writing of the background and editing of the Fabry disease, Gaucher disease and Pompe disease chapters for the final report.
Stephen Waldek contributed to the design of the study and clinical data collection forms, and assisted in the writing of the background chapter and editing of all chapters in the final report.
Robin Lachmann contributed to the design of the study and clinical data collection forms, and assisted in the writing of the background chapter and editing of all chapters in the final report.
Atul Mehta contributed to the design of the study and clinical data collection forms, and assisted in the writing of the background chapter and editing of all chapters in the final report.
Ashok Vellodi contributed to the design of the study and clinical data collection forms, and assisted in the writing of the background chapter and editing of the MPS II and Gaucher disease chapters in the final report.
Stuart Logan (Principal Investigator) contributed to the design of the study, contributed to interpretation of the data and was primarily responsible for writing all sections of the report in conjunction with Katrina Wyatt, William Henley, Lindsey Anderson and Rob Anderson.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Gieselmann V. What can cell biology tell us about heterogeneity in lysosomal storage diseases?. Acta Paediatr Suppl 2005;94:80-6.
- Platt FM, Walkley SU, Platt FM, Walkley SU. Lysosomal disorders of the brain: recent advances in molecular and cellular pathogenesis and treatment. New York, NY: Oxford University Press; 2004.
- Raben N, Shea L, Hill V, Plotz P. Monitoring autophagy in lysosomal storage disorders. Methods Enzymol 2009;453:417-49.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008;132:27-42.
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281:249-54.
- Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, et al. The frequency of lysosomal storage diseases in the Netherlands. Hum Genet 1999;105:151-6.
- Peltonen L. Molecular background of the Finnish disease heritage. Ann Med 1997;29:553-6.
- Wenger DA, Coppola S, Liu SL. Insights into the diagnosis and treatment of lysosomal storage diseases. Arch Neurol 2003;60:322-8.
- Aerts JM, van Breemen MJ, Bussink AP, Brinkman J, Hollak CEM, Langeveld M, et al. Drug transport(ers) and the diseased brain proceedings of the Esteve Foundation Symposium 11. Amsterdam: Elsevier; 2005.
- Wraith JE. The management of lysosomal disorders. Current Paediatrics 2004;14.
- de Duve C. From cytases to lysosomes. Fed Proc 1964;23:1045-9.
- Brady RN, Ruis H, McCormick DB, Wright LD. Bacterial degradation of biotin. Catabolism of 14C-biotin and its sulfoxides. J Biol Chem 1966;241:4717-21.
- Johnson WG, Desnick RJ, Long DM, Sharp HL, Krivit W, Brady B, et al. Intravenous injection of purified hexosaminidase A into a patient with Tay–Sachs disease. Birth Defects Orig Artic Ser 9:120-4.
- Brady RO, Tallman JF, Johnson WG, Gal AE, Leahy WR, Quirk JM, et al. Replacement therapy for inherited enzyme deficiency. Use of purified ceramidetrihexosidase in Fabry’s disease. N Engl J Med 1973;289:9-14.
- Schiffmann R, Brady RO, Mehta A, Beck M, Sunder-Plassmann G. Fabry Disease: perspectives from 5 years of FOS. Oxford: Oxford Pharma Genesis; 2006.
- Brady RO. Enzyme replacement for lysosomal diseases. Annu Rev Med 2006;57:283-96.
- Pastores GM, Weinreb NJ, Aerts H, Andria G, Cox TM, Giralt M, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol 2004;41:4-14.
- Connock M, Burls A, Frew E, Fry-Smith A, Juarez-Garcia A, McCabe C, et al. The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Gaucher’s disease: a systematic review. Health Technol Assess 2006;10.
- Connock M, Juarez-Garcia A, Frew E, Mans A, Dretzke J, Fry-Smith A, et al. A systematic review of the clinical effectiveness and cost-effectiveness of enzyme replacement therapies for Fabry’s disease and mucopolysaccharidosis type 1. Health Technol Assess 2006;10.
- Rawlins M, Barnett D, Stevens A. Pharmacoeconomics: NICE’s approach to decision-making. Br J Clin Pharmacol 2010;70:346-9.
- Linthorst GE, Germain DP, Hollak CE, Hughes D, Rolfs A, Wanner C, et al. Expert opinion on temporary treatment recommendations for Fabry disease during the shortage of enzyme replacement therapy (ERT). Mol Genet Metab 2011;102:99-102.
- Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet 2002;3:954-66.
- Vellodi A. Lysosomal storage disorders. Br J Haematol 2004;128:413-31.
- Bickel U, Yoshikawa T, Pardridge WM. Delivery of peptides and proteins through the blood-brain barrier. Adv Drug Deliv Rev 2001;46:247-79.
- Demeule M, Poirier J, Jodoin J, Bertrand Y, Desrosiers RR, Dagenais C, et al. High transcytosis of melanotransferrin (P97) across the blood-brain barrier. J Neurochem 2002;83:924-33.
- Zhang Y, Pardridge WM. Delivery of beta-galactosidase to mouse brain via the blood-brain barrier transferrin receptor. J Pharmacol Exp Ther 2005;313:1075-81.
- von Specht BU, Geiger B, Arnon R, Passwell J, Keren G, Goldman B, et al. Enzyme replacement in Tay–Sachs disease. Neurology 1979;29:848-54.
- Hemsley KM, King B, Hopwood JJ. Injection of recombinant human sulfamidase into the CSF via the cerebellomedullary cistern in MPS IIIA mice. Mol Genet Metab 2007;90:313-28.
- Platt FM, Neises GR, Dwek RA, Butters TD. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J Biol Chem 1994;269:8362-5.
- Schiffmann R, Fitzgibbon EJ, Harris C, DeVile C, Davies EH, Abel L, et al. Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol 2008;64:514-22.
- Shapiro BE, Pastores GM, Gianutsos J, Luzy C, Kolodny EH. Miglustat in late-onset Tay–Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet Med 2009;11:425-33.
- Guffon N, Bin-Dorel S, Decullier E, Paillet C, Guitton J, Fouilhoux A. Evaluation of miglustat treatment in patients with Type III Mucopolysaccharidosis: a randomized, double-blind, placebo-controlled study. J Pediatr 2011;159:838-44.
- McEachern KA, Fung J, Komarnitsky S, Siegel CS, Chuang WL, Hutto E, et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol Genet Metab 2007;91:259-67.
- Lukina E, Watman N, Arreguin EA, Banikazemi M, Dragosky M, Iastrebner M, et al. A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood 2010;116:893-9.
- Roberts AL, Rees MH, Klebe S, Fletcher JM, Byers S. Improvement in behaviour after substrate deprivation therapy with rhodamine B in a mouse model of MPS IIIA. Mol Genet Metab 2007;92:115-21.
- Piotrowska E, Jakobkiewicz-Banecka J, Baranska S, Tylki-Szymanska A, Czartoryska B, Wegrzyn A, et al. Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur J Hum Genet 2006;14:846-52.
- Piotrowska E, Jakobkiewicz-Banecka J, Maryniak A, Tylki-Szymanska A, Puk E, Liberek A, et al. Two-year follow-up of Sanfilippo Disease patients treated with a genistein-rich isoflavone extract: assessment of effects on cognitive functions and general status of patients. Med Sci Monit 2011;17:CR196-202.
- Priller J, Flugel A, Wehner T, Boentert M, Haas CA, Prinz M, et al. Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat Med 2001;7:1356-61.
- Asheuer M, Pflumio F, Benhamida S, Dubart-Kupperschmitt A, Fouquet F, Imai Y, et al. Human CD34+ cells differentiate into microglia and express recombinant therapeutic protein. Proc Natl Acad Sci U S A 2004;101:3557-62.
- Hoogerbrugge PM, Brouwer OF, Bordigoni P, Ringden O, Kapaun P, Ortega JJ, et al. Allogeneic bone marrow transplantation for lysosomal storage diseases. The European Group for Bone Marrow Transplantation. Lancet 1995;345:1398-402.
- Hobbs JR, Hugh-Jones K, Barrett AJ, Byrom N, Chambers D, Henry K, et al. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone marrow transplantation. Lancet 1981;2:709-12.
- Wraith JE. The management of lysosomal disorders. Current Paediatrics 2004;14.
- Tomatsu S, Montano AM, Oikawa H, Smith M, Barrera L, Chinen Y, et al. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol 2011;12:931-45.
- Fan JQ, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med 1999;5:112-15.
- Porto C, Cardone M, Fontana F, Rossi B, Tuzzi MR, Tarallo A, et al. The pharmacological rone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts. Mol Ther 2009;17:964-71.
- Zheng W, Padia J, Urban DJ, Jadhav A, Goker-Alpan O, Simeonov A, et al. Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease. Proc Natl Acad Sci U S A 2007;104:13-7.
- Yam GH, Bosshard N, Zuber C, Steinmann B, Roth J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. Am J Physiol Cell Physiol 2006;290:C1076-82.
- Ishii S, Chang HH, Kawasaki K, Yasuda K, Wu HL, Garman SC, et al. Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem J 2007;406:285-95.
- Okumiya T, Kroos MA, Vliet LV, Takeuchi H, Van der Ploeg AT, Reuser AJ. Chemical chaperones improve transport and enhance stability of mutant alpha-glucosidases in glycogen storage disease type II. Mol Genet Metab 2007;90:49-57.
- Delgadillo V, O’Callaghan Mdel M, Artuch R, Montero R, Pineda M. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis 2011;34:1039-44.
- Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther 2006;13:839-49.
- Mango RL, Xu L, Sands MS, Vogler C, Seiler G, Schwarz T, et al. Neonatal retroviral vector-mediated hepatic gene therapy reduces bone, joint, and cartilage disease in mucopolysaccharidosis VII mice and dogs. Mol Genet Metab 2004;82:4-19.
- Li C, Ziegler RJ, Cherry M, Lukason M, Desnick RJ, Yew NS, et al. Adenovirus-transduced lung as a portal for delivering alpha-galactosidase A into systemic circulation for Fabry disease. Mol Ther 2002;5:745-54.
- Biffi A, Capotondo A, Fasano S, del Carro U, Marchesini S, Azuma H, et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J Clin Invest 2006;116:3070-82.
- Beck M. Therapy for lysosomal storage disorders. IUBMB Life 2010;62:33-40.
- Aerts JM, Hollak C, Boot R, Groener A. Biochemistry of glycosphingolipid storage disorders: implications for therapeutic intervention. Philos Trans R Soc Lond B Biol Sci 2003;358:905-14.
- Rozenberg R, Fox DC, Sobreira E, Pereira LV. Detection of 12 new mutations in Gaucher disease Brazilian patients. Blood Cells Mol Dis 2006;37:204-9.
- Alfonso P, Aznarez S, Giralt M, Pocovi M, Giraldo P. Mutation analysis and genotype/phenotype relationships of Gaucher disease patients in Spain. J Hum Genet 2007;52:391-6.
- Wenstrup RJ, Roca-Espiau M, Weinreb NJ, Bembi B. Skeletal aspects of Gaucher disease: a review. Br J Radiol 2002;75:A2-1.
- Pastores GM, Patel MJ, Firooznia H. Bone and joint complications related to Gaucher disease. Curr Rheumatol Rep 2000;2:175-80.
- Cohen IJ. Bone crises in Gaucher disease. Isr Med Assoc J 2003;5:838-9.
- Pastores GM, Barnett NL, Bathan P, Kolodny EH. A neurological symptom survey of patients with type I Gaucher disease. J Inherit Metab Dis 2003;26:641-5.
- Stone DL, Ginns EI, Krasnewich D, Sidransky E. Life-threatening splenic hemorrhage in two patients with Gaucher disease. Am J Hematol 2000;64:140-2.
- Zimran A, Altarescu G, Rudensky B, Abrahamov A, Elstein D. Survey of hematological aspects of Gaucher disease. Hematology 2005;10:151-6.
- Deghady A, Marzouk I, El-Shayeb A, Wali Y. Coagulation abnormalities in type 1 Gaucher disease in children. Pediatr Hematol Oncol 2006;23:411-17.
- Mistry PK, Sirrs S, Chan A, Pritzker MR, Duffy TP, Grace ME, et al. Pulmonary hypertension in type 1 Gaucher’s disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab 2002;77:91-8.
- Goker-Alpan O, Schiffmann R, Park JK, Stubblefield BK, Tayebi N, Sidransky E. Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J Pediatr 2003;143:273-6.
- Harris CM, Taylor DS, Vellodi A. Ocular motor abnormalities in Gaucher disease. Neuropediatrics 1999;30:289-93.
- Verghese J, Goldberg RF, Desnick RJ, Grace ME, Goldman JE, Lee SC, et al. Myoclonus from selective dentate nucleus degeneration in type 3 Gaucher disease. Arch Neurol 2000;57:389-95.
- Frei KP, Schiffmann R. Myoclonus in Gaucher disease. Adv Neurol 2002;89:41-8.
- Orvisky E, Park JK, LaMarca ME, Ginns EI, Martin BM, Tayebi N, et al. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: correlation with phenotype and genotype. Mol Genet Metab 2002;76:262-70.
- Mignot C, Gelot A, Bessieres B, Daffos F, Voyer M, Menez F, et al. Perinatal-lethal Gaucher disease. Am J Med Genet A 2003;120A:338-44.
- Bohlega S, Kambouris M, Shahid M, Al Homsi M, Al Sous W. Gaucher disease with oculomotor apraxia and cardiovascular calcification (Gaucher type IIIC). Neurology 2000;54:261-3.
- George R, McMahon J, Lytle B, Clark B, Lichtin A. Severe valvular and aortic arch calcification in a patient with Gaucher’s disease homozygous for the D409H mutation. Clin Genet 2001;59:360-3.
- Grabowski GA, Petsko GA, Kolody EH, Scriver CR, Beaudet AL, Sly WS. The online metabolic and molecular bases of inherited disease. New York, NY: McGraw-Hill; 2010.
- Mistry PK, Cappellini MD, Lukina E, Ozsan H, Mach Pascual S, Rosenbaum H, et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am J Hematol 2011;86:110-15.
- Zuckerman S, Lahad A, Shmueli A, Zimran A, Peleg L, Orr-Urtreger A, et al. Carrier screening for Gaucher disease: lessons for low-penetrance, treatable diseases. JAMA 2007;298:1281-90.
- Beutler E. Gaucher disease: multiple lessons from a single gene disorder. Acta Paediatr Suppl 2006;95:103-9.
- Somaraju UR, Tadepalli K. Hematopoietic stem cell transplantation for Gaucher disease. Cochrane Database Syst Rev 2008;4.
- Vellodi A, Tylki-Szymanska A, Davies EH, Kolodny E, Bembi B, Collin-Histed T, et al. Management of neuronopathic Gaucher disease: revised recommendations. J Inherit Metab Dis 2009;32:660-4.
- Dwek RA, Butters TD, Platt FM, Zitzmann N. Targeting glycosylation as a therapeutic approach. Nat Rev Drug Discov 2002;1:65-7.
- Cox T, Lachmann R, Hollak C, Aerts J, van Weely S, Hrebicek M, et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000;355:1481-5.
- Elstein D, Hollak C, Aerts JM, van Weely S, Maas M, Cox TM, et al. Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J Inherit Metab Dis 2004;27:757-66.
- Pastores GM, Barnett NL, Kolodny EH. An open-label, noncomparative study of miglustat in type I Gaucher disease: efficacy and tolerability over 24 months of treatment. Clin Ther 2005;27:1215-27.
- Elstein D, Dweck A, Attias D, Hadas-Halpern I, Zevin S, Altarescu G, et al. Oral maintenance clinical trial with miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood 2007;110:2296-301.
- Pastores GM, Elstein D, Hrebicek M, Zimran A. Effect of miglustat on bone disease in adults with type 1 Gaucher disease: a pooled analysis of three multinational, open-label studies. Clin Ther 2007;29:1645-54.
- Giraldo P, Latre P, Alfonso P, Acedo A, Alonso D, Barez A, et al. Short-term effect of miglustat in every day clinical use in treatment-naive or previously treated patients with type 1 Gaucher’s disease. Haematologica 2006;91:703-6.
- Genzyme Therapeutics . Summary of Product Characteristics for Cerezyme 2010. www.medicines.org.uk/emc/medicine/18403 (accessed February 2012).
- Electronic Medicines Compendium . Summary of Product characteristics,VPRIV 2010. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001249/WC500096382.pdf (accessed November 2010).
- Damiano AM, Pastores GM, Ware JE. The health-related quality of life of adults with Gaucher’s disease receiving enzyme replacement therapy: results from a retrospective study. Qual Life Res 1998;7:373-86.
- Masek BJ, Sims KB, Bove CM, Korson MS, Short P, Norman DK. Quality of life assessment in adults with type 1 Gaucher disease. Qual Life Res 1999;8:263-8.
- Weinreb N, Barranger J, Packman S, Prakash-Cheng A, Rosenbloom B, Sims K, et al. Imiglucerase (Cerezyme) improves quality of life in patients with skeletal manifestations of Gaucher disease. Clin Genet 2007;71:576-88.
- Wenstrup RJ, Kacena KA, Kaplan P, Pastores GM, Prakash-Cheng A, Zimran A, et al. Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res 2007;22:119-26.
- Charrow J, Dulisse B, Grabowski GA, Weinreb NJ. The effect of enzyme replacement therapy on bone crisis and bone pain in patients with type 1 Gaucher disease. Clin Genet 2007;71:205-11.
- Sims KB, Pastores GM, Weinreb NJ, Barranger J, Rosenbloom BE, Packman S, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet 2008;73:430-40.
- Takahashi T, Yoshida Y, Sato W, Yano T, Shoji Y, Sawaishi Y, et al. Enzyme therapy in Gaucher disease type 2: an autopsy case. Tohoku J Exp Med 1998;186:143-9.
- Campbell PE, Harris CM, Sirimanna T, Vellodi A. A model of neuronopathic Gaucher disease. J Inherit Metab Dis 2003;26:629-39.
- Migita M, Hamada H, Fujimura J, Watanabe A, Shimada T, Fukunaga Y. Glucocerebrosidase level in the cerebrospinal fluid during enzyme replacement therapy--unsuccessful treatment of the neurological abnormality in type 2 Gaucher disease. Eur J Pediatr 2003;162:524-5.
- Vellodi A, Bembi B, de Villemeur TB, Collin-Histed T, Erikson A, Mengel E, et al. Management of neuronopathic Gaucher disease: a European consensus. J Inherit Metab Dis 2001;24:319-27.
- Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA, et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med 1995;122:33-9.
- Schiffmann R, Mankin H, Dambrosia JM, Xavier RJ, Kreps C, Hill SC, et al. Decreased bone density in splenectomized Gaucher patients receiving enzyme replacement therapy. Blood Cells Mol Dis 2002;28:288-96.
- Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med 2002;113:112-19.
- Andersson H, Kaplan P, Kacena K, Yee J. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 2008;122:1182-90.
- Zimran A, Altarescu G, Philips M, Attias D, Jmoudiak M, Deeb M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood 2010;115:4651-6.
- Elstein D, Cohn GM, Wang N, Djordjevic M, Brutaru C, Zimran A. Early achievement and maintenance of the therapeutic goals using velaglucerase alfa in type 1 Gaucher disease. Blood Cells Mol Dis 2011;46:119-23.
- Elstein D, Foldes AJ, Zahrieh D, Cohn GM, Djordjevic M, Brutaru C, et al. Significant and continuous improvement in bone mineral density among type 1 Gaucher disease patients treated with velaglucerase alfa: 69-month experience, including dose reduction. Blood Cells Mol Dis 2011;47:56-61.
- Stein P, Malhotra A, Haims A, Pastores GM, Mistry PK. Focal splenic lesions in type I Gaucher disease are associated with poor platelet and splenic response to macrophage-targeted enzyme replacement therapy. J Inherit Metab Dis 2010;33:769-74.
- Rosenberg M, Kingma W, Fitzpatrick MA, Richards SM. Immunosurveillance of alglucerase enzyme therapy for Gaucher patients: induction of humoral tolerance in seroconverted patients after repeat administration. Blood 1999;93:2081-8.
- Starzyk K, Richards S, Yee J, Smith SE, Kingma W. The long-term international safety experience of imiglucerase therapy for Gaucher disease. Mol Genet Metab 2007;90:157-63.
- Desnick RJ, Wasserstein MP. Fabry disease: clinical features and recent advances in enzyme replacement therapy. Adv Nephrol Necker Hosp 2001;31:317-39.
- Orteu CH, Jansen T, Lidove O, Jaussaud R, Hughes DA, Pintos-Morell G, et al. Fabry disease and the skin: data from FOS, the Fabry outcome survey. Br J Dermatol 2007;157:331-7.
- Lidove O, Jaussaud R, Aractingi S, Mehta A, Beck M, Sunder-Plassmann G. FabRY Disease: perspectives from 5 years of FOS. Oxford: Oxford Pharma Genesis; 2006.
- Sodi A, Ioannidis AS, Mehta A, Davey C, Beck M, Pitz S. Ocular manifestations of Fabry’s disease: data from the Fabry Outcome Survey. Br J Ophthalmol 2007;91:210-14.
- Pieroni M, Chimenti C, De Cobelli F, Morgante E, Del Maschio A, Gaudio C, et al. Fabry’s disease cardiomyopathy: echocardiographic detection of endomyocardial glycosphingolipid compartmentalization. J Am Coll Cardiol 2006;47:1663-71.
- Linhart A, Kampmann C, Zamorano JL, Sunder-Plassmann G, Beck M, Mehta A, et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J 2007;28:1228-35.
- Politei JM, Capizzano AA. Magnetic resonance image findings in 5 young patients with Fabry disease. Neurologist 2006;12:103-5.
- Rolfs A, Bottcher T, Zschiesche M, Morris P, Winchester B, Bauer P, et al. Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet 2005;366:1794-6.
- Brouns R, Sheorajpanday R, Braxel E, Eyskens F, Baker R, Hughes D, et al. Middelheim Fabry Study (MiFaS): a retrospective Belgian study on the prevalence of Fabry disease in young patients with cryptogenic stroke. Clin Neurol Neurosurg 2007;109:479-84.
- Hoffmann B, Beck M, Sunder-Plassmann G, Borsini W, Ricci R, Mehta A. Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy – a retrospective analysis from the Fabry Outcome Survey. Clin J Pain 2007;23:535-42.
- Hegemann S, Hajioff D, Conti G, Beck M, Sunder-Plassmann G, Widmer U, et al. Hearing loss in Fabry disease: data from the Fabry Outcome Survey. Eur J Clin Invest 2006;36:654-62.
- Magage S, Lubanda JC, Susa Z, Bultas J, Karetova D, Dobrovolny R, et al. Natural history of the respiratory involvement in Anderson-Fabry disease. J Inherit Metab Dis 2007;30:790-9.
- Cole AL, Lee PJ, Hughes DA, Deegan PB, Waldek S, Lachmann RH. Depression in adults with Fabry disease: a common and under-diagnosed problem. J Inherit Metab Dis 2007;30:943-51.
- Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ, et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002;105:1407-11.
- Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a ‘renal variant’ phenotype. Kidney Int 2003;64:801-7.
- Gupta S, Ries M, Kotsopoulos S, Schiffmann R. The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. Medicine (Baltimore) 2005;84:261-8.
- Shah JS, Elliott PM. Fabry disease and the heart: an overview of the natural history and the effect of enzyme replacement therapy. Acta Paediatrica 2005;94:11-4.
- Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet 2006;43:347-52.
- Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab 2008;93:112-28.
- Sadek J, Shellhaas R, Camfield CS, Camfield PR, Burley J. Psychiatric findings in four female carriers of Fabry disease. Psychiatr Genet 2004;14:199-201.
- Desnick RJ, Ioannou YA, Eng CM, Scriver CR, Beaudet AL, Sly WS, et al. The metabolic and molecular bases of inherited disease. New York, NY: McGraw-Hill; 2001.
- Tahir H, Jackson LL, Warnock DG. Antiproteinuric therapy and fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol 2007;18:2609-17.
- Electronic Medicines Compendium . Summary of Product Characteristics for Replagal 2012. www.medicines.org.uk/EMC/medicine/19760/SPC/Replagal+1mg+ml+concentrate+for+solution+for+infusion/ (accessed accessed April 2012).
- Electronic Medicines Compendium . Summary of Product Characteristics for Fabrazyme 2011. www.medicines.org.uk/EMC/medicine/18664/XPIL/Fabrazyme+35+mg,+powder+for+concentrate+for+solution+for+infusion/ (accessed December 2011).
- Schiffmann R, Kopp JB, Austin HA, Sabnis S, Moore DF, Weibel T, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001;285:2743-9.
- Hajioff D, Enever Y, Quiney R, Zuckerman J, Mackermot K, Mehta A. Hearing loss in Fabry disease: the effect of agalsidase alfa replacement therapy. J Inherit Metab Dis 2003;26:787-94.
- Eng CM, Banikazemi M, Gordon RE, Goldman M, Phelps R, Kim L, et al. A phase 1/2 clinical trial of enzyme replacement in fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet 2001;68:711-22.
- Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. Safety and efficacy of recombinant human alpha-galactosidase A--replacement therapy in Fabry’s disease. N Engl J Med 2001;345:9-16.
- Hughes DA, Elliott PM, Shah J, Zuckerman J, Coghlan G, Brookes J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart 2008;94:153-8.
- Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 2007;146:77-86.
- Waldek S. PR interval and the response to enzyme-replacement therapy for Fabry’s disease. N Engl J Med 2003;348:1186-7.
- Germain DP, Waldek S, Banikazemi M, Bushinsky DA, Charrow J, Desnick RJ, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol 2007;18:1547-57.
- Beck M, Ricci R, Widmer U, Dehout F, de Lorenzo AG, Kampmann C, et al. Fabry disease: overall effects of agalsidase alfa treatment. Eur J Clin Invest 2004;34:838-44.
- Hoffmann B, Reinhardt D, Koletzko B. Effect of enzyme-replacement therapy on gastrointestinal symptoms in Fabry disease. Eur J Gastroenterol Hepatol 2004;16:1067-9.
- Moore DF, Scott LT, Gladwin MT, Altarescu G, Kaneski C, Suzuki K, et al. Regional cerebral hyperperfusion and nitric oxide pathway dysregulation in Fabry disease: reversal by enzyme replacement therapy. Circulation 2001;104:1506-12.
- Moore DF, Herscovitch P, Schiffmann R. Selective arterial distribution of cerebral hyperperfusion in Fabry disease. J Neuroimaging 2001;11:303-7.
- Moore DF, Altarescu G, Herscovitch P, Schiffmann R. Enzyme replacement reverses abnormal cerebrovascular responses in Fabry disease. BMC Neurol 2002;2.
- Wraith JE, Tylki-Szymanska A, Guffon N, Lien YH, Tsimaratos M, Vellodi A, et al. Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr 2008;152:563-70.
- Schiffmann R, Martin RA, Reimschisel T, Johnson K, Castaneda V, Lien YH, et al. Four-year prospective clinical trial of agalsidase alfa in children with Fabry disease. J Pediatr 2010;156:832-7.
- Vedder AC, Linthorst GE, Houge G, Groener JE, Ormel EE, Bouma BJ, et al. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS One 2007;2.
- Baehner F, Kampmann C, Whybra C, Miebach E, Wiethoff CM, Beck M. Enzyme replacement therapy in heterozygous females with Fabry disease: results of a phase IIIB study. J Inherit Metab Dis 2003;26:617-27.
- Ries M, Gupta S, Moore DF, Sachdev V, Quirk JM, Murray GJ, et al. Pediatric Fabry disease. Pediatrics 2005;115:e344-55.
- Ramaswami U, Wendt S, Pintos-Morell G, Parini R, Whybra C, Leon Leal JA, et al. Enzyme replacement therapy with agalsidase alfa in children with Fabry disease. Acta Paediatr 2007;96:122-7.
- Linthorst GE, Hollak CE, Donker-Koopman WE, Strijland A, Aerts JM. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int 2004;66:1589-95.
- Deegan PB. Fabry disease, enzyme replacement therapy and the significance of antibody responses. J Inherit Metab Dis 2012;35:227-43.
- Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis 2008;3.
- Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J. Initial report from the Hunter Outcome Survey. Genet Med 2008;10:508-16.
- Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol 2005;32:270-2.
- Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis 2005;28:1011-17.
- Young ID, Harper PS. Incidence of Hunter’s syndrome. Hum Genet 1982;60:391-2.
- Miebach E, Church H, Cooper A, Mercer J, Tylee K, Wynn RF, et al. The craniocervical junction following successful haematopoietic stem cell transplantation for mucopolysaccharidosis type I H (Hurler syndrome). J Inherit Metab Dis 2011;34:755-61.
- Aldenhoven M, Boelens JJ, de Koning TJ. The clinical outcome of Hurler syndrome after stem cell transplantation. Biol Blood Marrow Transplant 2008;14:485-98.
- Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr 2004;144:581-8.
- Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M, et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 2001;344:182-8.
- Clarke LA, Wraith JE, Beck M, Kolodny EH, Pastores GM, Muenzer J, et al. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics 2009;123:229-40.
- Cox-Brinkman J, Timmermans RG, Wijburg FA, Donker WE, van de Ploeg AT, Aerts JM, et al. Home treatment with enzyme replacement therapy for mucopolysaccharidosis type I is feasible and safe. J Inherit Metab Dis 2007;30.
- Giugliani R, Rojas VM, Martins AM, Valadares ER, Clarke JT, Goes JE, et al. A dose-optimization trial of laronidase (Aldurazyme) in patients with mucopolysaccharidosis I. Mol Genet Metab 2009;96:13-9.
- Muenzer J, Wraith JE, Beck M, Giugliani R, Harmatz P, Eng CM, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med 2006;8:465-73.
- Muenzer J, Gucsavas-Calikoglu M, McCandless SE, Schuetz TJ, Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab 2007;90:329-37.
- Okuyama T, Tanaka A, Suzuki Y, Ida H, Tanaka T, Cox GF, et al. Japan Elaprase Treatment (JET) study: idursulfase enzyme replacement therapy in adult patients with attenuated Hunter syndrome (Mucopolysaccharidosis II, MPS II). Mol Genet Metab 2010;99:18-25.
- Muenzer J, Beck M, Eng CM, Giugliani R, Harmatz P, Martin R, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med 2011;13:95-101.
- Parenti G, Andria G. Pompe disease: from new views on pathophysiology to innovative therapeutic strategies. Curr Pharm Biotechnol 2011;12:902-15.
- Hirschhorn R, Arnold JJR, Scriver C, Beaudet A, Sly W, Valle D. The metabolic and molecular bases of inherited disease 8th edition. New York, NY: McGraw-Hill; 2001.
- Slonim AE, Bulone L, Ritz S, Goldberg T, Chen A, Martiniuk F. Identification of two subtypes of infantile acid maltase deficiency. J Pediatr 2000;137:283-5.
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. Pompe disease diagnosis and management guideline. Genet Med 2006;8:267-88.
- Engel AG, Gomez MR, Seybold ME, Lambert EH. The spectrum and diagnosis of acid maltase deficiency. Neurology 1973;23:95-106.
- Wokke JH, Ausems MG, van den Boogaard MJ, Ippel EF, van Diggelene O, Kroos MA, et al. Genotype-phenotype correlation in adult-onset acid maltase deficiency. Ann Neurol 1995;38:450-4.
- van den Berg LE, Zandbergen AA, van Capelle CI, de Vries JM, Hop WC, van den Hout JM, et al. Low bone mass in Pompe disease: muscular strength as a predictor of bone mineral density. Bone 2010;47:643-9.
- Papadimas GK, Terzis G, Spengos K, Methenitis S, Papadopoulos C, Vassilopoulou S, et al. Bone mineral density in adult patients with Pompe disease. Bone 2011;48.
- Sacconi S, Bocquet JD, Chanalet S, Tanant V, Salviati L, Desnuelle C. Abnormalities of cerebral arteries are frequent in patients with late-onset Pompe disease. J Neurol 2010;257:1730-3.
- van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003;112:332-40.
- Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet 1999;7:713-16.
- Martiniuk F, Chen A, Mack A, Arvanitopoulos E, Chen Y, Rom WN, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet 1998;79:69-72.
- Chien YH, Lee NC, Huang HJ, Thurberg BL, Tsai FJ, Hwu WL. Later-onset Pompe disease: early detection and early treatment initiation enabled by newborn screening. J Pediatr 2011;158:1023-7.
- Kallwass H, Carr C, Gerrein J, Titlow M, Pomponio R, Bali D, et al. Rapid diagnosis of late-onset Pompe disease by fluorometric assay of alpha-glucosidase activities in dried blood spots. Mol Genet Metab 2007;90:449-52.
- Huie ML, Chen AS, Tsujino S, Shanske S, DiMauro S, Engel AG, et al. Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (–13T → G) mutation in a majority of patients and a novel IVS10 (+1GT → CT) mutation. Hum Mol Genet 1994;3:2231-6.
- Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab 2010;99:26-33.
- van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 2010;362:1396-406.
- Angelini C, Semplicini C. Enzyme replacement therapy for Pompe Disease. Curr Neurol Neurosci Rep 2011;12:70-5.
- Ravaglia S, Danesino C, Moglia A, Costa A, Cena H, Maccarini L, et al. Changes in nutritional status and body composition during enzyme replacement therapy in adult-onset type II glycogenosis. Eur J Neurol 2010;17:957-62.
- Bernstein DL, Bialer MG, Mehta L, Desnick RJ. Pompe disease: dramatic improvement in gastrointestinal function following enzyme replacement therapy. A report of three later-onset patients. Mol Genet Metab 2010;101:130-3.
- Banugaria SG, Prater SN, Ng YK, Kobori JA, Finkel RS, Ladda RL, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med 2011;13:729-36.
- Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 2007;68:99-109.
- Kishnani PS, Corzo D, Leslie ND, Gruskin D, Van der Ploeg A, Clancy JP, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res 2009;66:329-35.
- Chakrapani A, Vellodi A, Robinson P, Jones S, Wraith JE. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. J Inherit Metab Dis 2010;33:747-50.
- Byrne BJ, Falk DJ, Pacak CA, Nayak S, Herzog RW, Elder ME, et al. Pompe disease gene therapy. Hum Mol Genet 2011;20:R61-8.
- Vanier MT. Niemann–Pick disease type C. Orphanet J Rare Dis 2010;5.
- Crocker AC, Mays VB. Sphingomyelin synthesis in Niemann–Pick disease. Am J Clin Nutr 1961;9:63-7.
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, et al. Niemann–Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 1997;277:228-31.
- Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann–Pick C disease. Science 2000;290:2298-301.
- Pentchev P, Vanier MT, Suzuki K, Patterson MC, Scriver CR, Beaudet AL, et al. The metabolic and molecular bases of inherited disease 7th edition. New York, NY: McGraw Hill; 1995.
- Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea ED, Neufeld EB, et al. The metabolic and molecular bases of inherited disease 8th edition. New York, NY: McGraw Hill; 2001.
- Sevin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, et al. The adult form of Niemann–Pick disease type C. Brain 2007;130:120-33.
- Vanier MT, Millat G. Niemann–Pick disease type C. Clin Genet 2003;64:269-81.
- Imrie J, Dasgupta S, Besley GT, Harris C, Heptinstall L, Knight S, et al. The natural history of Niemann–Pick disease type C in the UK. J Inherit Metab Dis 2007;30:51-9.
- Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann–Pick disease type C. Curr Biol 2001;11:1283-7.
- Lachmann RH, te Vruchte D, Lloyd-Evans E, Reinkensmeier G, Sillence DJ, Fernandez-Guillen L, et al. Treatment with miglustat reverses the lipid-trafficking defect in Niemann–Pick disease type C. Neurobiol Dis 2004;16:654-8.
- Chien YH, Lee NC, Tsai LK, Huang AC, Peng SF, Chen SJ, et al. Treatment of Niemann–Pick disease type C in two children with miglustat: initial responses and maintenance of effects over 1 year. J Inherit Metab Dis 2007;30.
- Santos ML, Raskin S, Telles DS, Lohr Junior A, Liberalesso PB, Vieira SC, et al. Treatment of a child diagnosed with Niemann–Pick disease type C with miglustat: a case report in Brazil. J Inherit Metab Dis 2008;31:457-61.
- Paciorkowski AR, Westwell M, Ounpuu S, Bell K, Kagan J, Mazzarella C, et al. Motion analysis of a child with Niemann–Pick disease type C treated with miglustat. Mov Disord 2008;23:124-8.
- Galanaud D, Tourbah A, Lehericy S, Leveque N, Heron B, Billette de Villemeur T, et al. 24 month-treatment with miglustat of three patients with Niemann–Pick disease type C: follow up using brain spectroscopy. Mol Genet Metab 2009;96:55-8.
- Fecarotta S, Amitrano M, Romano A, Della Casa R, Bruschini D, Astarita L, et al. The videofluoroscopic swallowing study shows a sustained improvement of dysphagia in children with Niemann–Pick disease type C after therapy with miglustat. Am J Med Genet A 2011;155A:540-7.
- Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann–Pick C disease: a randomised controlled study. Lancet Neurol 2007;6:765-72.
- Patterson MC, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, et al. Long-term miglustat therapy in children with Niemann–Pick disease type C. J Child Neurol 2010;25:300-5.
- Wraith JE, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, et al. Miglustat in adult and juvenile patients with Niemann–Pick disease type C: long-term data from a clinical trial. Mol Genet Metab 2010;99:351-7.
- Pineda M, Perez-Poyato MS, O’Callaghan M, Vilaseca MA, Pocovi M, Domingo R, et al. Clinical experience with miglustat therapy in pediatric patients with Niemann–Pick disease type C: a case series. Mol Genet Metab 2010;99:358-66.
- Deegan P, Hughes D, Mehta A, Cox T. UK National guideline for adult Gaucher Disease. London: Department of Health; 2005.
- Hughes D, Ramaswami U, Elliot P, Deegan P, Waldek S, Apperley G, et al. Guidelines for the Diagnosis and Management of Anderson-Fabry Disease 2005. www.specialisedservices.nhs.uk/library/23/Guidelines_for_Anderson_Fabry_Disease.pdf.
- Wraith E, Lee P, Vellodi A, Cleary MA, Ramaswami U, Jessop E. Guidelines for the Investigation and Management of Infantile Pompe Disease 2005. http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_4137655.pdf.
- Wraith JE, Vellodi A, Cleary M, Ramaswami U, Lavery C, Jessop E. Guidelines for the Investagation and Management of Mucopolysaccharidosis Type I 2005. www.specialisedservices.nhs.uk/library/23/Guidelines_for_the_Investigation_and_Management_of_Mucopolysaccharidosis_type_I.pdf.
- Vellodi A, Wraith JE, Chakrapani A, Hendriksz C, Jones S. Mucopolysaccharidosis Type II: guidelines for assessment, monitoring and enzyme replacement therapy (ERT) 2005. www.specialisedservices.NHS.uk/library/23/Guidelines_for_Mucopolysaccharidosis_Type)II.pdf.
- Drummond MF, Sculpher MJ, Torrance GW, O’Brien BJ, Stoddart GL. Methods for the Economic Evaluation of Health Care Programmes. Oxford: Oxford University Press; 2005.
- Brazier J, Roberts J, Deverill M. The estimation of a preference-based measure of health from the SF-36. J Health Econ 2002;21:271-92.
- National Institute for Health and Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2005.
- Brazier J, Roberts J, Tsuchiya A, Busschbach J. A comparison of the EQ-5D and SF-6D across seven patient groups. Health Econ 2004;13:873-84.
- Eiser C, Morse R. Quality-of-life measures in chronic diseases of childhood. Health Technol Assess 2001;5.
- Varni JW, Burwinkle TM, Katz ER. The PedsQL in pediatric cancer: reliability and validity of the pediatric quality of life inventory generic core scales, multidimensional fatigue scale, and cancer module. Cancer 2002;94:2090-106.
- Krupp LB, LaRocca NG, Muir-Nash J, Steinberg AD. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol 1989;46:1121-3.
- Valko PO, Bassetti CL, Bloch KE, Held U, Baumann CR. Validation of the fatigue severity scale in a Swiss cohort. Sleep 2008;31:1601-7.
- Egner A, Phillips VL, Vora R, Wiggers E. Depression, fatigue, and health-related quality of life among people with advanced multiple sclerosis: results from an exploratory telerehabilitation study. NeuroRehabilitation 2003;18:125-33.
- Dixon SWM, Salek S. Incorporating carer effects into economic evaluation. Pharmacoeconomics 2006;24:43-5.
- Thornton M, Travis SS. Analysis of the reliability of the modified caregiver strain index. J Gerontol B Psychol Sci Soc Sci 2003;58:S127-32.
- Sleed M, Beecham J, Knapp M, McAuley C, McCurry N. Assessing services, supports and costs for young families under stress. Child Care Health Dev 2006;32:101-10.
- Beecham J, Knapp M, Thornicroft G. Measuring mental health needs, 2nd edition. London: Gaskell; 2001.
- Diggle PJ, Heagerty PJ, Liang K, Zeger SL. Analysis of longitudinal data. Second edition. Oxford: Statistical Science Series; 2002.
- Little RJA, Rubin DB. Statistical analysis with missing data, 2nd Edition. New York, NY: John Wiley; 2002.
- Wood SN. Generalized additive models: an introduction with R. Boca Raton, FL: Chapman & Hall/CRC Press; 2006.
- Lilford RJ, Thornton JG, Braunholtz D. Clinical trials and rare diseases: a way out of a conundrum. BMJ 1995;311:1621-5.
- Congdon P. Bayesian statistical modelling. Chichester: Wiley; 2002.
- Department of Health (DoH) . NHS Reference Costs 2009–2010 (for NHS Trust and PCT Combined) 2011.
- Curtis L. Unit Costs of Health and Social Care 2010. Canterbury: Personal Social Services Research Unit, University of Kent; 2011.
- Mihaylova B, Briggs A, O’Hagan A, Thompson SG. Review of statistical methods for analysing healthcare resources and costs. Health Econ 2011;20:897-916.
- Malm G, Gustafsson B, Berglund G, Lindstrom M, Naess K, Borgstrom B, et al. Outcome in six children with mucopolysaccharidosis type IH, Hurler syndrome, after haematopoietic stem cell transplantation (HSCT). Acta Paediatr 2008;97:1108-12.
- Niemann–Pick Disease Group UK website n.d. www.niemannpick.org.uk.
- Lowry RB, Applegarth DA, Toone JR, MacDonald E, Thunem NY. An update on the frequency of mucopolysaccharide syndromes in British Columbia. Hum Genet 1990;85:389-90.
- Rodrigue SW, Rosenthal DI, Barton NW, Zurakowski D, Mankin HJ. Risk factors for osteonecrosis in patients with type 1 Gaucher’s disease. Clin Orthop Relat Res 1999;362:201-7.
- Deegan PB, Pavlova E, Tindall J, Stein PE, Bearcroft P, Mehta A, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine 2011;90:52-60.
- El-Beshlawy A, Ragab L, Youssry I, Yakout K, El-Kiki H, Eid K, et al. Enzyme replacement therapy and bony changes in Egyptian paediatric Gaucher disease patients. J Inherit Metab Dis 2006;29:92-8.
- Hsu CC, Chien YH, Lai MY, Hwu WL. Enzyme replacement therapy with imiglucerase in Taiwanese patients with type I Gaucher disease. J Formos Med Assoc 2002;101:627-31.
- Biegstraaten M, Mengel E, Marodi L, Petakov M, Niederau C, Giraldo P, et al. Peripheral neuropathy in adult type 1 Gaucher disease: a 2-year prospective observational study. Brain 2010;133:2909-19.
- Tayebi N, Callahan M, Madike V, Stubblefield BK, Orvisky E, Krasnewich D, et al. Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol Genet Metab 2001;73:313-21.
- Clark LN, Nicolai A, Afridi S, Harris J, Mejia-Santana H, Strug L, et al. Pilot association study of the beta-glucocerebrosidase N370S allele and Parkinson’s disease in subjects of Jewish ethnicity. Mov Disord 2005;20:100-3.
- Levey AS, Coresh J, Greene T, Stevens LA, Zhang YL, Hendriksen S, et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med 2006;145:247-54.
- Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol 2009;20:629-37.
- Hauck WW, Donner A. Wald’s test as applied to hypotheses in logit analysis. J Am Stat Assoc 1977;72:851-3.
- Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004;34:236-42.
- Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke 2009;40:788-94.
- Vedder AC, Linthorst GE, van Breemen MJ, Groener JE, Bemelman FJ, Strijland A, et al. The Dutch Fabry cohort: diversity of clinical manifestations and Gb3 levels. J Inherit Metab Dis 2007;30:68-7.
- Grewal RP. Stroke in Fabry’s disease. J Neurol 1994;241:153-6.
- Mehta A, Ginsberg L. Natural history of the cerebrovascular complications of Fabry disease. Acta Paediatrica 2005;94:24-7.
- Guffon N, Fouilhoux A. Clinical benefit in Fabry patients given enzyme replacement therapy--a case series. J Inherit Metab Dis 2004;27:221-7.
- Cole TJ, Freeman JV, Preece MA. British 1990 growth reference centiles for weight, height, body mass index and head circumference fitted by maximum penalized likelihood. Stat Med 1998;17:407-29.
- Sifuentes M, Doroshow R, Hoft R, Mason G, Walot I, Diament M, et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab 2007;90:171-80.
- Wraith JE. The clinical presentation of lysosomal storage disorders. Acta Neurol Taiwan 2004;13:101-6.
- Mercimek-Mahmutoglu S, Reilly C, Human D, Waters PJ, Stoeckler-Ipsiroglu S. Progression of organ manifestations upon enzyme replacement therapy in a patient with mucopolysaccharidosis type I/Hurler. World J Pediatr 2009;5:319-21.
- Neufeld EF, Muenzer J, Scriver CR, Beaudet AL, Sly WS, Valle D, et al. The metabolic and molecular bases of inherited disease 8th edition. New York, NY: McGraw-Hill, Medical Publishing Division; 2001.
- Braunlin EA, Berry JM, Whitley CB. Cardiac findings after enzyme replacement therapy for mucopolysaccharidosis type I. Am J Cardiol 2006;98:416-18.
- Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123:19-2.
- Clarke LA, Heppner J, Pagon RA, Bird TD, Dolan CR, Stephens K, et al. GeneReviews. Seattle, WA: University of Washington; 1993.
- Kachur E, Del Maestro R. Mucopolysaccharidoses and spinal cord compression: case report and review of the literature with implications of bone marrow transplantation. Neurosurgery 2000;47:223-8.
- Schwartz IV, Ribeiro MG, Mota JG, Toralles MB, Correia P, Horovitz D, et al. A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr Suppl 2007;96:63-70.
- Schulze-Frenking G, Jones SA, Roberts J, Beck M, Wraith JE. Effects of enzyme replacement therapy on growth in patients with mucopolysaccharidosis type II. J Inherit Metab Dis 2011;34:203-8.
- Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr 2008;167:267-77.
- Brands MM, Frohn-Mulder IM, Hagemans ML, Hop WC, Oussoren E, Helbing WA, et al. Mucopolysaccharidosis: cardiologic features and effects of enzyme-replacement therapy in 24 children with MPS I, II and VI [published online ahead of print 26 January 2012]. J Inherit Metab Dis 2012.
- White KK, Hale S, Goldberg MJ. Musculoskeletal health in Hunter disease (MPS II): ERT improves functional outcomes. J Pediatr Rehabil Med 2010;3:101-7.
- Enders CG. Applied Missing Data Analysis. New York, NY: Guilford Press; 2010.
- Regnery C, Kornblum C, Hanisch F, Vielhaber S, Strigl-Pill N, Grunert B, et al. 36 months observational clinical study of 38 adult Pompe disease patients under alglucosidase alfa enzyme replacement therapy [published online ahead of print 31 January 2012]. J Inherit Metab Dis 2012.
- Strothotte S, Strigl-Pill N, Grunert B, Kornblum C, Eger K, Wessig C, et al. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J Neurol 2010;257:91-7.
- van den Brink M, van den Hout WB, Stiggelbout AM, van de Velde CJ, Kievit J. Cost measurement in economic evaluations of health care: whom to ask?. Med Care 2004;42:740-6.
Appendix 1 List of collaborators
| Name | Organisation | Post |
|---|---|---|
| Professor Stuart Logan | PCMD | Director of Institute for Health and Social Care Research |
| Dr Katrina Wyatt | PCMD | Research Fellow |
| Dr Rob Anderson | PCMD | Senior Lecturer in Health Economics |
| Dr Ken Stein | PCMD | Senior Lecturer in Public Health |
| Professor Timothy Cox | Addenbrookes Hospital/Cambridge University, Department of Medicine | Professor of Medicine |
| Dr Patrick Deegan | Addenbrookes Hospital/Cambridge University, Department of Medicine | Senior Research Associate and Consultant |
| Dr Uma Ramaswami | The Willink Biochemical Genetics Unit, Manchester | Consultant Metabolic Paediatrician |
| Dr Atul Mehta | Royal Free and University College Medical School, Department of Haematology | Clinical Director and Consultant in Metabolic Disorders |
| Dr Derralyn Hughes | Royal Free and University College Medical School, Department of Haematology | Clinical Lecturer in Haematology |
| Dr Edmond Wraith | Royal Manchester Children’s Hospital, The Willink Biochemical Genetics Unit | Consultant Paediatrician |
| Dr Stephen Waldek | Formerly of Salford Royal Hospitals NHS Trust, Department of Lysosomal Storage Disorder | Clinical Director |
| Dr Ashok Vellodi | Great Ormond Street Hospital for Children, Metabolic Unit | Consultant Metabolic Paediatrician |
| Ms Tanya Collin-Histed | Gauchers Association | Chief Executive Gauchers Association |
| Dr Robin Lachmann | National Hospital for Neurology and Neurosurgery, Charles Dent Metabolic Unit | Consultant in Metabolic Medicine |
Appendix 2 Project Management Team
The co-ordinating team responsible for overall project management were based at the PCMD in Exeter, Devon. The team comprised Professor Stuart Logan (Chief Investigator), Professor Ken Stein (Methodological Input and probabilistic modelling), Katrina Wyatt (Project Manager), Louise Klinger, Sheena Oxer and Lindsey Anderson (Study Co-ordinators), Professor William Henley and Vasilis Nikolaou (Study Statisticians), and Rob Anderson (Study Health Economist).
In addition to the PCMD research staff, there were 12 other co-applicants, including 11 clinicians, with representatives from each of the seven NCSG-designated LSD treating centres (Professor Timothy Cox, Dr Patrick Deegan, Dr Chris Hendriksz, Dr Derralynn Hughes, Dr Robin Lachmann, Dr Philip Lee, Dr Atul Mehta, Dr Uma Ramaswami, Dr Ashok Vellodi, Dr Edmond Wraith, Dr Stephen Waldek), as well as Tanya Collin-Histed from the Gauchers Association, acting as representative of all the LSD patient support groups. Together, the co-ordinating team and the co-applicants comprised the Management Team. Additional clinical support was given throughout the study by Dr Simon Jones.
Appendix 3 Study Support Group members
| Support Group | Contact |
|---|---|
| Gauchers Association | Ms Tanya Collin-Histed |
| Society for Mucopolysaccharide Diseases | Mrs Christine Lavery |
| Niemann–Pick Disease Group | Ms Toni Mathieson |
| Association for Glycogen Storage Disease UK | Mr Allan Muir |
| Batten Disease Family Association | Ms Jan Sablitzky |
Appendix 4 Study Steering Committee members
| Member | Role |
|---|---|
| Professor Richard Hobbs | Head of Primary Health Care, University of Oxford |
| Professor Michael Beck | Metabolic Clinician, University of Mainz |
| Dr Fiona Stewart | Consultant Clinical Geneticist, Belfast City Hospital |
| Professor Carla Hollak | Professor of Internal Medicine, University of Amsterdam |
| Dr Angie Wade | Senior Lecturer in Medical Statistics, Institute of Child Health, University College London |
Appendix 5 Protocol
A long-term cohort study of people with lysosomal storage disorders
Main Applicant:
Professor Stuart Logan1
Co-applicants:
Professor Tim Cox2
Dr Patrick Deegan2
Dr Derralynn Hughes3
Dr Philip Lee4
Dr Robin Lachmann4
Dr Atul Mehta3
Dr Uma Ramaswami5
Dr Ed Wraith6
Dr Stephen Waldek7
Dr Ashok Vellodi8
Dr Chris Hendriksz9
Ms Tanya Collin-Histed on behalf of the Gaucher Association10
Dr Ken Stein11
Dr Rob Anderson11
Dr Katrina Wyatt1
1Peninsula Medical School, St Lukes Campus, Exeter EX1 2LU
2Department of Medicine, University of Cambridge, Addenbrooke’s Hospital, Cambridge
3Department of Academic Haematology, Royal Free and University College Medical School, London
4Charles Dent Metabolic Unit, Post Box 92, National Hospital for Neurology & Neurosurgery, Queen Square, London WC1N 3BG
5Department of Paediatrics, University of Cambridge, Addenbrooke’s Hospital, Cambridge
6Willink Biochemical Genetics Unit, Royal Manchester Children’s Hospital, Manchester
7Hope Hospital Manchester M6 8HD
8Metabolic Unit, Great Ormond Street Hospital for Children NHS Trust, London
9Birmingham Children’s Hospital, Steelhouse lane, Birmingham, B4 6NH
10Gaucher Association, 3 Bull Pitch, Dursley, Gloucestershire GL11 4NG
11Peninsula Technology Assessment Group, Peninsula Medical School, Exeter
A long-term cohort study of people with lysosomal storage disorders
Aim: To conduct a prospective cohort study of people in England with a lysosomal storage disorder to determine natural history and estimate effectiveness and cost-effectiveness of current and potential treatment strategies.
Objectives: Primary objectives:
-
To determine the natural history of treated and untreated lysosomal storage disorders for those disorders where enzyme replacement therapy is currently available;
-
To estimate the effectiveness of enzyme replacement therapy;
-
To estimate the cost-effectiveness of enzyme replacement therapy for lysosomal storage disorders;
-
To determine the natural history of lysosomal storage disorders where enzyme replacement therapy is likely to become available.
Secondary objectives include:
-
To compare the effectiveness of Replagal and Fabryzme in children and adults with Fabry disease
-
To estimate the lifetime health care cost and other economic impacts on people with lysosomal storage disorders and their families
-
To provide the basis for future research to develop treatment-responsive measures in adults and children.
Lay summary
Lysosomal storage disorders are a group of rare, inherited diseases. In total they affect fewer than 1 : 7000 people. Traditionally, the therapeutic options for lysosomal storage disorders have focussed on managing the symptoms of the disease rather than treating the disease itself. However, in recent years, treatments which address the cause of the disease, the enzyme deficiency, are being developed for these disorders. Enzyme replacement therapies are now available for the treatment of Gaucher, Fabry and MPS I, and several more are being developed.
People with these disorders are treated at one of seven designated treatment centres in England. The Peninsula Medical School, in collaboration with the treatment centres and the support groups, would like to look at how effective and cost effective these therapies are. However, because these conditions are so rare, usual ways of testing how effective a treatment is, such as a randomised, controlled trial are much harder to conduct. Therefore, we hope to carry out a long term cohort study, whereby we collect data, at each centre, from all consenting adults and children with these conditions. By following people with these conditions over a period of time we will better understand how effective treatments are, when the best time to start giving these treatments is, what the appropriate dosing schedules are, and which symptoms led to the diagnosis of the disorder. Another aspect of the study will be to estimate the value for money of these treatments. In order to do this we will look at how frequently people use the NHS, the cost of their treatment, related costs to their family, and compare these for people who are receiving treatment with those people who are not, or for whom no treatment is currently available. This study is intended to last three years in the first instance and, in addition to addressing specific questions, will create a valuable research resource for patients and clinicians.
Background
Lysosomal storage disorders (LSDs) are a heterogeneous group of disorders with a combined prevalence of between 1 : 5,000 and 1 : 10,000. The prevalence of the more common individual lysosomal diseases is between 1 : 20,000 and 1 : 100,000 [1,2]. Higher prevalences of specific lysosomal storage diseases are encountered in some populations, for example Gaucher and Tay–Sachs disease among Ashkenazim Jews and aspartylglucosaminuria, and Salla disease and infantile neuronal ceroid lipofuscinosis in Finland [3]. The clinical picture of most lysosomal storage disorders is heterogeneous with age at onset, and type and progression of symptoms varying substantially among individual patients suffering from the same disorder. Within each condition, there is considerable variation in the underlying genetic mutation. There is a correlation between the specific mutation and the severity of the problems experienced by an individual but the genotype/phenotype relation is variable [4]. In general, a correlation exists between residual enzyme activity and severity of disease manifestation. In some lysosomal storage disorders external genetic or environmental factors markedly influence the flux through the defective pathway and therefore also have a major impact on disease manifestation.
There are > 40 LSDs whose common feature is the deficiency of a lysosomal enzyme or transport protein. This deficiency results in a progressive intracellular accumulation of glycophospholipids, causing tissue damage and ultimately organ failure [4]. The likelihood that a particular cell type is involved in storage accumulation is determined by the flux of the substrate (the metabolic demand) and the residual capacity of that cell type to carry out the catabolic reaction. In general, the more severe the mutation the more cell types accumulate the storage material. For patients with a missense lysosomal enzyme gene, and therefore showing a relatively high residual enzyme activity, storage is likely to occur in fewer cell types. It is the heterogeneity in individuals’ residual degradative capacity which accounts for some lysosomal storage disorders manifesting as relatively benign non-neuropathic variants and others as devastating neuropathic variants. In the latter case storage is not restricted to cells in visceral tissues but also involves cells inside the brain. Many LSDs have traditionally been classified into subtypes, although it is increasingly recognised that most LSDs have a broad continuum of clinical severity and age of presentation [5] rather than falling into clinically discrete forms.
The symptoms arising from these disorders are generally progressive and clinical diagnosis becomes easier with time [6]. For the most part diagnosis relies on observation of clinical features raising a clinical suspicion resulting in formal testing.
The clinical course of these disorders is not easily predictable in an individual, especially in the later-onset disorders [7]. Although mutation analysis can predict the likelihood of neurological involvement for some LSDs, as mentioned, there is often variability in the genotype/phenotype relationships. The situation is further complicated by the large number of mutations identified, which, coupled with the fact that most patients are compound heterozygotes, makes phenotype prediction difficult. In addition, the relative frequency of different patterns of mutation varies between ethnic groups making comparisons between outcomes in different countries problematic.
Treatments for lysosomal storage disorders
No definitive, curative treatment is yet available for any LSD. For most of the disorders, symptomatic treatment for specific problems is currently the only therapeutic option. For some LSDs it is possible to either augment the deficient enzyme (eg. by enzyme replacement therapy (ERT) or enzyme enhancement therapy - such as bone marrow transplant) or partially inhibit synthesis of the parent substrates by substrate reduction therapy. Treatment options are summarised in Table 1.
Bone marrow transplant
The first bone marrow transplants (haematopoietic stem cell transplant) were done on patients with Hurler’s disease and reported in the early 1980s [8]. Since then bone marrow transplant [BMT] has been carried out for at least 20 different LSDs [9]. The results of BMT are variable but it appears that for the most part it is in the disorders which do not affect the central nervous system to any great extent where BMT has the greatest effect. [6] When carried out in individuals with CNS involvement, BMT is reported to be least effective in addressing the skeletal and neurological component of these disorders. For disorders which primarily affect the CNS, such as infantile Tay Sachs, Sandhoff or MPS III (Sanfilippo disease), BMT does not appear to be effective in slowing down the disease progression. Similarly, where there is significant skeletal impact on the disorder such as MPS IV (Morquio disease), BMT has not been reported to lead to an improvement in growth or other skeletal features [7]. In MPS I and VI (Maroteaux Lamy disease) a transplant early on in the course of the condition has been reported to be associated with some improvements although in MPS I, BMT after the onset of significant neurological signs does not lead to an improvement of neurological function, and in most patients a steady loss of skills continues [7]. Furthermore, it appears that bone and cartilage cells remain MPS cells. [8,9]
Substrate reduction therapy
At present, Miglustat (Zavesca), is the only licensed substrate reduction therapy in the UK. Miglustat inhibits glucsylceramide synthetase which is the first step in the synthesis of most glycosphingolipids. It is currently licensed in the UK for treatment of mild to moderate type I Gaucher, in patients for whom enzyme replacement therapy is unsuitable.
A one year open label study involving 28 adults (seven with previous splenectomies) from four international Gauchers referral clinics, who were unable or unwilling to receive ERT reported reduced organomegaly and small haematological improvements after 12 months’ therapy [10]. An extension study to 36 months was conducted with 18 of the 22 eligible patients (14 completed the 36 month study) which reported a further reduction in liver and spleen volume, as well as haematological parameters with a reduction in the incidence of side effects (as experienced in the first 12 months) [11].
Other disorders where the effectiveness of Miglustat is currently being assessed are, Gaucher type 3, Niemann–Pick type C and late onset Tay Sachs [9].
Enzyme replacement therapy (ERT)
There are currently four licensed enzyme replacement therapies in the UK for three LSDs; imiglucerase (Cerezyme®) for non neuropathic Gaucher disease (type I); agalsidase beta (Fabrazyme®) and agalsidase alpha (Replagal®) for Fabry and laronidase (Aldurazyme®) for mucopolysaccharidosis (MPS) type I.
Other enzyme replacement therapies are currently being developed for Pompe [13], and MPS type II [14] and VI (Maroteaux -Lamy) [15]. Enzyme replacement therapy for Niemann – Pick Type B is at the pre-clinical stage [9].
Treatment and the blood brain barrier
Whereas substrate reduction therapies do appear to cross the blood brain barrier in small amounts (approx 10%), currently available enzyme replacement therapies do not appear to cross the blood brain barrier in sufficient amounts to be effective. This inevitably limits their potential effectiveness in those conditions in which CNS involvement is an important feature. There is some evidence that if patients are given sufficiently high doses of immunosuppressant drugs there may be better penetration of the enzyme into the CNS. It has been established that injecting the replacement therapy directly into the CNS is not an effective means of crossing the blood brain barrier [personal communication].
HTA commissioned reviews of effectiveness and cost-effectiveness
An examination of the evidence for the effectiveness and cost effectiveness of enzyme replacement therapies for Gauchers, Fabry and MPS type I was commissioned by the HTA. For all three conditions the reports suggested on the basis of the limited data available that there are beneficial effects of ERT on symptom-related markers. [16, 17]. The following sections summarise key points from these reports.
ERT for Gaucher Disease
Gaucher disease is classified into three subtypes by clinical features. Type I can present at any age and has predominantly visceral symptoms without neurological effects. Type II presents in childhood and has neurological and visceral symptoms. It causes severe progressive brain disease and death occurs in infancy. Type III presents in early childhood with the presence of visceral and/or neurological symptoms. Imiglucerase is licensed for use in symptomatic Type I disease and to treat the visceral symptoms of Type III disease.
Effectiveness
The systematic review identified 63 studies (involving 10 patients or more) [16]. These included one RCT which compared ERT to usual treatment and one RCT which compared two different derivations of ERT but provided only before and after data on the effectiveness of ERT. The other studies were considered to be of moderate quality at best and none had reliable comparator data.
All studies were suggestive of benefit from ERT. The RCT comparing ERT to usual treatment reported a potentially beneficial effect in haemoglobin and platelet levels and, to a lesser extent, on hepatomegaly. The other studies reported improvements in haematological parameters and in hepatomegaly and splenomegaly, with most parameters tending to return towards normal in the majority of patients after a year or more of treatment. For organomegaly and haemoglobin, the rates and extent of response are reported to have been greater the more abnormal the pre-ERT condition. Platelet levels are reported to improve more slowly. For most people liver size was reduced to near 1.2 times that expected for normal weight and the spleen was reduced by 5–10 fold. ERT was also reported to have a beneficial effect on bone crises and fracture rate, as well as on pain, although the quantitative evidence for these benefits was described by the authors of the HTA report as being ‘extremely weak’.
The overall conclusion was that there was a paucity of high quality evidence and that it was therefore difficult to reliably estimate whether these reported effects translate into improved patient wellbeing and survival, or an altered need for health services.
Cost effectiveness
All published cost-effectiveness studies are over nine years old and conducted outside the UK. The authors of the report described above conducted a new cost-effectiveness analysis based on UK costs [17]. In this analysis, even assuming that ERT restores people with Gaucher to full health for their remaining lives, the incremental cost-effectiveness of ERT is > 10 times above the usually accepted threshold for what constitutes ‘good value for money’ when using NHS resources to improve health. The authors emphasise that due to the weak research evidence base, extreme uncertainty surrounds these cost-effectiveness estimates. However, even with the most favourable possible assumptions the incremental cost-effectiveness of ERT appears prohibitive given current drug costs.
ERT in Fabry’s disease
Fabry’s disease is an x-linked lysosomal storage disorder caused by a deficiency of the enzyme α-galactosidase A, an enzyme involved in the breakdown of lipids. As a result of this deficiency glycosphingolipids accumulate in the body’s tissues, particularly the heart, kidneys and nerve tissue. Symptoms usually appear during childhood and adolescence and affect many organ systems such as heart, CNS, kidney, bowel, pancreas and lung [19]. It is a clinically heterogeneous disease and is usually slowly progressive with symptoms changing with age [20]. A substantial proportion of patients will develop cerebrovascular disease (transient ischaemic attacks and stroke). There are two ERTs licensed for use in the UK for Fabry’s disease, agalsidase alpha (Replagal®) and agalsidase beta (Fabryzyme®). Both are given intravenously, with the recommended dose being 0.2mg and 1mg/ kg body weight bi-weekly, respectively.
Effectiveness
Considering studies of either form of ERT, the systematic review identified three randomised placebo-controlled trials (n = 70, duration 5–6 months) and 11 uncontrolled before and after studies (n = 493, duration up to 24 months) [18]. Of the three controlled trials, 27 patients received Fabryzyme and 21 received Replagal. The studies are small, of short duration and use different outcome measures which made direct comparisons difficult. Overall their results suggest some beneficial effect of ERT on measures of pain and cardiovascular function, and an apparent stabilisation of renal function based on measures of creatinine clearance. The studies were unable to demonstrate any effect on neurological effects including the risk of transient ischaemic attacks or stroke. However, this is unsurprising given the lack of power to detect such effects as well as the short duration of treatment, and a beneficial effect cannot be excluded on the basis of current data.
There is currently a trial going on in Holland comparing Fabryzme and Replagal, however no results have been published as yet [personal communication].
Cost effectiveness
The authors of the report conducted a cost-effectiveness analysis of ERT in Fabry disease. The conclusions are similar to those reviewing ERT in Gaucher disease. The data are acknowledged to be poor, resulting in considerable uncertainty around all estimates. However, even where the model is based on the most favourable possible assumptions, applying conventional thresholds of societal willingness-to-pay for health gains for the UK NHS (£30,000 per QALY), and current treatment prices, the authors conclude that ERT (either Replagal® or Fabrazyme®) for Fabry’s was highly unlikely to be cost-effective. These conclusions are crucially dependent on current drug costs.
ERT for MPS I
MPS I is an inherited autosomal recessive disorder caused by deficient activity of the enzyme IDUA which results in an accumulation of glycoaminoglycans (GAGs) in many tissues including connective tissue, brain, heart and liver. This in turn leads to skeletal abnormalities, respiratory problems, joint problems, developmental delay and other issues such as corneal cloudiness, enlarged liver and spleen, recurrent hernias and heart disease. There are three subtypes: type IH (Hurler disease) which presents in the first year of life, has severe neurological symptoms and a life expectancy of only one decade; MPS IHS (Hurler-Scheie disease) is an intermediate form with a life expectancy of only two to three decades; and MPS IS (Scheie), is an attenuated form with later presentation and longer life expectancy than IH and IHS. Laronidase is licensed for IV administration for symptomatic MPS IS and HIS patients. The recommended does is 0.58 mg/kg body weight every week.
Effectiveness
The systematic review identified one placebo-controlled RCT and a phase I/II observational study provides evidence of effectiveness. In the RCT 45 people with moderate to mild disease (predominantly HS) took part in a 26 week duration trial, with an open label extension for an additional 72 weeks. The Phase I/ II study included 10 patients (8 patients had the HS subtype with one patient each with H and S subtypes). The duration was 26 weeks with a subsequent extension to 52 weeks and beyond. Both studies reported positive effects on functional ability (specifically performance on the 6-minute walking distance), markers of lysosomal storage and markers measuring change in specific disease symptoms.
Cost effectiveness
The authors of the review concluded that the lack of basic data related to natural history, in particular a lack of quality-of-life data, lack of efficacy data and the highly heterogeneous nature of the conditions meant that it would not be appropriate to attempt a cost-effectiveness analysis. They nonetheless argue that the extremely high costs of ERT in this condition mean that it is unlikely that, even if the treatment is highly effective, it would meet the current thresholds for cost-effectiveness. Again this argument is crucially dependent on current drug costs.
Table 1 shows the conditions for which there are treatments available. Please note the data on which symptoms are responsive and those which do not appear to respond to ERT are taken from a review article, publication date 2004 and are not taken from primary research studies.
Patterns of treatment in England
Treatment Centres
Services for patients with Lysosomal Storage Disorders (LSDs) including treatments such as Enzyme Replacement Therapy and Substrate Reduction Therapy are being nationally commissioned by the National Commissioning Group (NCG – formerly the National Specialist Commissioning Advisory Group, NSCAG) until March 2011. In England, seven hospitals have been nationally designated and funded, to provide a service for patients with lysosomal storage disorders (LSDs).
Centres for children
-
Central Manchester and Manchester Children’s University Hospitals NHS Trust (estimated 279 child patients)
-
Great Ormond Street Hospital for Children NHS Trust (estimated 148 patients)
-
Birmingham Children’s Hospital NHS Foundation Trust (estimated 137 patients)
Centres for adults
-
Salford Royal Hospital NHS Trust (estimated 311 adult patients)
-
Royal Free Hampstead NHS Trust (estimated 231 adult patients)
-
University College London Hospitals NHS Foundation Trust, National Hospital for Nervous Diseases, Queen Square (estimated 145 patients)
Centre for adults and children
-
Addenbrooke’s NHS Trust (estimated 204 patients; 159 adults 45 children)
| Disease | Approx. Prevalence (Australian data [5]) |
Enzyme Replacement Therapy | Substrate Reduction Therapy | Median Age of diagnosis* (range) | Symptoms responsive to ERT. | Symptoms which appear largely unresponsive to ERT (taken from [12] |
|---|---|---|---|---|---|---|
| Fabry | 1 : 117,000 |
Fabrazyme (agalsidase beta) 1 mg/kg IV Replagal (agalsidase alfa) 0.2 mg/kg IV |
28.6 years (0–55.7) |
Hypohidrosis, neuropathic pain, decreased cold and warm sensing, GI disturbance | Progressive renal, cerebrovascular (stroke – no evidence of effect, rather than evidence of no effect), cardiac disease | |
| Gaucher | 1 : 57,000 |
Initially Ceredase (licensed USA 1994, Europe 1998) Imiglucerase - Cerezyme licensed 2003 |
Miglustat (Zavesca) licensed for patients in whom ERT is not appropriate. |
9.5 years (0 -73.2) |
Anaemia, thrombocytopenia, bone crises, bone fractures | Neurological abnormalities, interstitial lung disease |
| MPSI Hurler’s disease/ Hurler-Scheie/ | 1 : 88,000 |
Iduronidase / Aldurazyme – 0.5 mg/kg Prescribed for Hurler and Hurler-Scheie forms of MPS I and for people with the Scheie form who have moderate to severe symptoms. Risks and benefits of treating mildly affected patients with the Scheie form have not been established. |
1.0 years (0.3 – 29.1) |
Hepatospenomegaly, decreased joint range of motion, restrictive pulmornary disease |
Macroephaly, hydrocephalus, Coarse facial features, bone deformities, mental retardation, cardiac disease, cornea clouding Aldurazyme has not been evaluated for symptoms of the central nervous system. |
|
| MPS VI Maroteaux-Lamy Disease | 1 : 235,000 | Naglazyme (Galsulfase) |
1.4 years (0 – 43.4) |
Decreased joint range of motion, gait difficulties | Coarse facial features, bone deformities | |
| MPS II Hunter syndrome | 1 : 136,000 | Iduronate-2-sulfatase Idursulfase (Elaprase) |
2.8 years (0.0 – 22.0) |
|||
|
Pompe’s disease (Early and Late onset) |
1 : 146,000 | Myozyme® (alglucosidase alfa) – 4 infants in an open label study suggest that Rx should begin as early as possible – 15-40 mg/kg. further study with 3 patients with late onset Pompe showed good results. |
0.5 years (0.1 -55.0) early and late - onset |
Cardiac hypertrophy, heart failure, skeletal muscle weakness, respiratory failure | Lower motor neuron disease | |
| Late onset | 1 : 201,000 | Substrate reduction therapy shows some promise in mouse models | Miglustat used in clinical trials | |||
| Tay–Sachs disease / Sndhoff disease | 1 : 384,000 | |||||
| Niemann–Pick type C | 1 : 211,000 | Miglustat used in clinical trials |
9.3 years (0.1 -37.7) |
From these figures there would appear to be 1455 patients with an LSD who are seen at one of the treatment centres.
As would be expected from prevalence data the most common LSDs among patients seen in these centres are Gaucher and Fabry in adults and the mucoploysaccharidosis disorders (in particular MPS I and MPS III), in children [Treatment Centres, personal communication].
At present, Wales, Scotland and Northern Ireland have separate prescribing arrangements and there is no precise data as to the numbers of patients with lysosomal storage disorders living in these regions, although some do receive care at the designated centres.
Rationale for study
It has been argued [16, 17] that there is currently little point in conducting further studies of effectiveness or cost-effectiveness of ERT in Gaucher disease, Fabry disease or MPSI. This is based on the argument that the costs of the drugs are currently so high that however effective these treatments are there is no possibility that they can cross currently accepted thresholds for willingness to pay. The authors of these reviews argue that, if society has decided that because of the particular rarity and severity of these conditions it is willing to pay for therapy, then further information is not required while, if society is to apply the thresholds generally used to make such decisions, no amount of information will move the decision across this threshold.
In our view this stance, while arguable, is mistaken. Better estimates of the effectiveness of the interventions, of the relative effectiveness of treatment depending on when in the course of the condition treatment begins and of different treatment regimens are important for patients and their families as well as for clinicians. The costs of the drugs may well change substantially in the future with changes in technology and the possible entry into the market of other providers. In these circumstances evidence of effectiveness will be needed to underpin decisions on cost effectiveness. The proposed study will provide at least partial answers to these questions in addition to providing better data on NHS costs to inform future estimates of cost effectiveness.
In addition, the proposed study offers the opportunity to assemble a cohort of patients with other LSDs for which ERT may become available in the future. We anticipate that the same difficulty in carrying out long term randomised controlled trials will apply in these conditions and that better estimates of the natural history of untreated UK patients will make possible later estimates of cost-effectiveness of therapy based on observational data.
Currently there are several lysosomal condition-specific databases which are held by the pharmaceutical companies which manufacture the enzyme replacement therapies and the substrate reduction therapy currently lisenced in the UK. This has led to the development of two registries for Fabry’s, which do not appear to be compatible with each other, hindering comparison treatment efficacy. The MPS society (UK) also has a registry of all UK people diagnosed with an MPS disorder since 1981. In addition there is a national Gaucher registry held at Addenbrookes which is part of the Gauchers Disease – diagnosis and management advice service.
It was felt to be necessary to establish this UK cohort study independent of the pharmaceutical industry, not least because the intention is to collect data on all lysosomal storage disorders and not solely ones where there are treatments. In addition, given that there are currently three pharmaceutical companies that manufacture these treatments, to conduct the study with any one of the companies might lead to potential conflicts of interest.
Background
We aim to conduct a longitudinal, prospective cohort study involving all adults and children with lysosomal storage disorders, living in the UK who are treated within the seven designated treatment centres in England. As new therapies and treatment modalities are being proposed and developed for these disorders, issues around diagnosis, when to start treatment, and valid and reliable outcome measures to assess treatment effectiveness are raised. With lysosomal storage disorders, early diagnosis is important to allow treatment before irreversible organ damage occurs. Furthermore, it is possible that given the progressive nature of these disorders, there might be clinical markers within each condition which indicate the optimum point at which enzyme replacement therapy should initiated.
The study will initially collect data on conditions for which ERT is currently available or being developed, although it is intended that eventually all children and adults diagnosed with an LSD will form part of the study. We believe that the majority of people with lysosomal storage disorders will be referred to these centres, regardless of whether there is a specific treatment available; where there is no specific treatment, people receive palliative care from these centres.
Methods
Identification/Recruitment of Eligible Patients
All patients with lysosomal storage disorders, living in the UK and attending the treatment centres will be identified and consent will be sought for their participation in the study.
1. Identification
The research assistant/nurse will identify eligible patients from the department database, or department patient lists and will enter the patient’s initials and date of birth into an ‘Initial table for Recruiting’ spreadsheet and assign a study ID. There will be one spreadsheet per condition.
The LSD consultant will be asked to confirm the patient’s eligibility to take part in the study i.e. that they will not be distressed by being approached to take part. Eligibility status will be entered into the spreadsheet and eligible patients and / or their carer will be sent an introductory letter (Appendix 1, 2, 2b or 2c).
To ensure patients / patients’ carers have sufficient time to read and absorb the information and have the time and opportunity to discuss the study with relatives, GPs, research staff etc, an invitation letter and patient information sheet will be posted to the patient / patient’s carer at least one month before they are due at their clinical review appointment (Appendices 3-6).
It is anticipated that some patients will be missed by the researcher and / or the LSD consultant at their first clinical review appointment after receiving their invitation to participate in the study. This might be due to the patient not attending the clinic, or due to other commitments for the researcher and/or consultant on the day of their attendance. In such cases, the patient will be sent an additional invitation letter and patient information sheet one month before they are next due at their clinical review appointment (Appendices 3-6).
2. Explanation of the Trial
The LSD consultant or research nurse will meet patients when they attend their clinical review appointment. The study will be verbally explained to the patient / patient’s carer using the appropriate Information Sheet and the use and timing of the questionnaires will be explained to the patient/patient’s carer. Sufficient time will be allocated for the patient / patient’s carer to ask questions and have them answered to their satisfaction.
3. Consent
Written informed consent (Appendix 7 or 8) will be obtained from each participant. For people under 16, written parental or guardian consent will be obtained (Appendix 8).
3.1 Two-tier consent
The study will operate a two-tier consent process whereby if a patient or their carer does not wish to complete the quality of life, fatigue or resource use questionnaires, they will be asked if they agree to their data being extracted from their medical notes for the purposes of the study (Appendix 7B or 8B).
3.2 Consenting patients who lack capacity
The research team will initially assume that each patient has capacity and every effort will be made to support them to help them make their own decision regarding participation in the study. Information about the study will be provided to each individual in a way that is most appropriate to help them understand the study and make their own decision.
If the treating clinician or another member of the healthcare team believes on the balance of probabilities, that the individual lacks capacity to give informed consent, then they must take reasonable steps to identify someone to consult, before they are included in the research. That person (the consultee) must be involved in the person’s care, interested in their welfare and must be willing to help. They must not be a professional or paid care worker.
Where there is no willing ‘personal consultee’, the researcher will identify an appropriate adult (such as a psychologist or social worker) involved in their care but unconnected to the study and ask them to assist in explaining the study.
The consultee will be given information about the research project and be asked:
-
for advice about whether the person who lacks capacity should take part in the study, and
-
what they think the person’s feelings and wishes would be, if they had capacity to decide whether to take part.
Once a willing consultee has been identified, they will be asked to provide written informed consent on behalf of the patient (Appendix 25 or 25B).
3.3 Re-consenting 16 year olds
When a patient who is in the study turns 16 they must be approached for re-consenting. They will be consented using adult forms.
-
When the parent/carer has not given any consent for their child’s participation in the study, the researcher can approach the patient directly for consent when the patient turns 16.
-
When the parent/carer has given ‘notes only’ consent for their child’s participation in the study, the researcher can approach the patient directly for full consent when the patient turns 16.
-
Patients can only be re-consented when the LSD consultant has confirmed their eligibility to take part in the study. That is, that they will not be distressed by being approached to take part.
4. Informing the patient’s GP of participation in study
Once consent has been received from the patient or their carer, a letter will be sent to the patient’s GP (Appendices 23 – 24) notifying them of the patient’s involvement in the study, along with a PIS.
Data collection
Data will be collected on all consenting patients onto a condition-specific database. Each database will follow the same structure with a set of data common to all conditions and condition-specific data. Data collected will include both prospective data and limited historical data. Historical data is available for a number of conditions contained within the variety of existing registers and in the patients’ notes. Data fields will be agreed within the group but will be guided by the principle that only data which will clearly contribute to answering a specific question will be included.
Identification of data fields
Procedures and data collection will be piloted on the following three disorders – Gaucher, Pompe and MPS I. Clinicians from the seven sites have identified, individually and in working groups, which data fields are important to collect for these disorders. Initially, the team determined basic information relating to which key organs are affected in each disorder, and then the primary tests which demonstrate the functioning of that organ. Further communications within the working groups clarified the data fields to be collected for each disorder.
Questionnaires
Questionnaires are to be handed out to patients (or carer/parent) consenting to have the additional questionnaires at their annual check up appointment with their clinician. They may also be asked to complete additional questionnaires at any additional monitoring appointment they attend. The patient will obviously have the right to refuse to complete the questionnaires at all times. Ideally questionnaires will be completed during the hospital visit, but if this is not possible the patient/carer will be given a Stamped Addressed Envelope and asked to return the questionnaires to the Research Nurse. In certain circumstances, for instance if there is a problem with the provision of ERT, and where it would assist in our understanding of the effectiveness of these treatments, additional questionnaires may be posted to patients who are attending clinic less often. Similarly, in circumstances where study patients attend for routine clinical follow up but are not seen for study purposes, follow up questionnaires may be posted to them. In all such situations it will be made clear in an accompanying letter that patients are under no obligation to complete these or to remain in the study. A stamped addressed envelope will be provided with the questionnaires for their return. The patient ID will be written on the top of each sheet in the pack either before or after completion. When the questionnaires are taken home, this will be done before they are removed from the treating centre. The Patient/Carer will be contacted up to a maximum of three times at two week intervals to chase if the questionnaires have not been returned. A brief record of the conversation/message left each time will be made on the notes page (Appendix 25) and kept in the study file. Dates will be recorded in the database.
Once returned the questionnaire answers will be entered into the database and the paper copies will be kept in study folders.
The age-appropriate questionnaires to be given to the patient and carer (if applicable) are detailed in Table 2.
For those conditions where the senses are impaired the HUI (Health Utilities Index) will also be given to patients over 5 years of age.
-
Pompe – where there is cardiomyopathy
-
Gaucher – Type III
-
MPS I – all
-
MPS II – all
-
Fabry – all
-
Niemann Pick C – all
| Age | Senses affected by condition? | Questionnaires |
|---|---|---|
| 0-1 | Not applicable | Service use and cost – child proxy (app 16B) |
| Caregiver Strain Index (app 18) | ||
| 2–4 | Not applicable | PedsQL toddler– parent (app 20) |
| Service use and cost – child proxy (app 16B) | ||
| Caregiver Strain Index (app 18) | ||
| 5–7 | No | PedsQL 5-7 – child (app 12) |
| PedsQL 5-7 – parent (app 11) | ||
| Service use and cost – child proxy (app 16B) | ||
| Caregiver Strain Index (app 18) | ||
| Yes | As above plus: HUI - proxy (app 19B) | |
| 8–12 | No | PedsQL 8-12 – child (app14) |
| PedsQL 8-12 – parent (app13) | ||
| Service use and cost – child proxy (app 16B) | ||
| Caregiver Strain Index (app 18) | ||
| Yes | As above plus: HUI – proxy (app 19B) | |
| 13–15 | No | PedsQL 13-18 – child (app 21) |
| PedsQL 13-18 – parent (app 22) | ||
| Service use and cost – Child proxy (app 16B) | ||
| EQ-5D (app 10) | ||
| Caregiver Strain Index (app 18) | ||
| Yes | As above plus: HUI – proxy (app 19B) | |
| 16-or over | No | EQ-5D (app 10) |
| SF-36 (app 9) | ||
| Service use and cost – adult (app 16) | ||
| Caregiver Strain Index (if applicable) (app 18) | ||
| FSS (app 26) | ||
| Yes | As above plus: HUI – self (app 19) |
Database
We will use MACRO, a web-based EDC system from InferMed. A secure, condition-specific database will be designed for all conditions. Data will be collected at the patients’ annual or six-monthly review and entered onto the database by the Research Nurse / Analyst at each study site.
Data quality assurance
Data accuracy is a requirement under the Data Protection Act, and together with data completeness is essential for maximising validity and reliability research outputs. To ensure data consistency between centres a data code-book or definitions manual will be developed. Data checks (valid ranges, filter checks, logical checks) will also be conducted as part of data entry processes and will be built into the database system. Key data will be ‘double entered’ using SDV.
Analysis
The database will contain longitudinal individual-level patient data for all consenting patients attending the participating treatment centres.
Natural History
Data will be analysed to describe the natural history of treated and untreated LSDs. Key outcome measures relevant to each disorder will be analysed by genotype where this information is available and where there are sufficient numbers of patients with a specific genotype. Exploratory analysis of patient trajectories will be conducted using graphical methods. For conditions where sufficient data are available, formal statistical modelling which exploits the longitudinal nature of the data (both prospective and retrospective) will be used to study individual dynamics. Important issues to be addressed will include accounting for non-linearity in the rate of disease progression, and patient heterogeneity in age of presentation and clinical severity. This will be achieved by exploring dynamic linear growth curve models in a Bayesian framework with patient-specific random effects, and random walk priors for the mean and slope parameters [21]. Such models can be extended to allow for dependency among the set of outcome measures for each condition by inclusion of patient-specific latent variables [22]. The latent variables represent unobserved constructs and provide a means of identifying the main elements of the underlying structure of the disease process. The flexible framework makes it possible to combine patient characteristics measured on different scales and to make adjustments for outcome-specific measurement errors. Given the likely sparsity of data for individual LSDs, careful attention will need to be given to specification of prior distributions to ensure model identification.
Effectiveness of treatment
For each condition, different approaches will be needed to estimate the effectiveness of ERT. The approach will depend largely on the amount of data available on untreated patients. Where data are not available for significant numbers of untreated patients, treatment efficacy will be estimated by taking advantage of the fact that the age and stage of their condition at which patients have begun taking ERT was dependent on the time when the treatment first became available. Historical data are available for many of these patients on their clinical condition at the time of beginning treatment while for others we will have data only on current clinical situation. The analysis of the available data will require (a) longitudinal analyses of changes in outcome measures and resource use before and after treatment, taking account of a range of covariates (e.g. baseline severity, demographic characteristics) and (b) extrapolation of pre- and post-treatment data to estimate the likely lifetime costs and effects in untreated and treated cohorts of patients.
Given the rare nature of these disorders and the corresponding modest sample sizes, conventional analyses may not have the power to detect or exclude clinically worthwhile treatment benefits. Consequently, we propose making assessments of treatment efficacy in a Bayesian framework to supplement analyses using classical methods. Although definitive answers may not always be possible, with frequentist confidence limits unlikely to exclude a null result, taking a Bayesian approach can provide a clearer guide by quantifying the probabilities that clinical effects lie in a particular range [23]. These probabilities, calculated by combining study data with a prior distribution, apply directly to future patients and can be used explicitly in formal decision analysis. A key component of our approach will be obtaining credible data on priors through incorporation of information from previous trials and elicitation of opinions from clinicians. We will conduct extensive sensitivity analyses to assess the effect of uncertainty in the choice of model specification and prior assumptions.
Comparison of the effectiveness of agalsidase alpha and agalsidase beta in Fabry disease
Both agalsidase alpha and agalsidase beta are licensed for use in the UK for the treatment of Fabry disease. Both treatments received their license in 2002. There is a five fold difference in the licensed dosing regimen although costs per patient are broadly similar. It appears that, although all centres use both drugs, there has been tendency for each centre to use one or other as their initial drug of choice. This it appears has been determined mainly by historical reasons based partly upon which drugs trials they were involved in. Some patients subsequently switch to the alternative treatment, for clinical reasons, and it has been suggested that more recently there may be more variety in initial drug choice. There are national guidelines for the initiation of therapy to which all centres adhere which suggests that the populations receiving either treatment are likely to be broadly similar. There are currently approximately 185 adults and 45 children with Fabry disease receiving treatment with one or other of these drugs.
We will compare the outcome of treatment depending on which of the two drugs patients were initially assigned (the equivalent of an intention to treat analysis) in a multivariate model allowing for potential confounding variables. We will in addition compare recorded side-effects and frequency of switching treatments.
Costs of care
Data will be collected on health care resource use using the Service use and cost questionnaire (Appendix 16). Information such as numbers of hospitalisations, outpatient and GP appointments, medication use, and other therapies will be collected according to disorder, patient age and severity for all patients. Additional data will be collected on associated family/carer costs and on family/carer related quality-of-life impacts using the Caregiver Strain Index (App 18). These data will be used to estimate lifetime health care costs according to disorder and severity.
Cost-effectiveness
These data will be used to help develop and populate a number of decision-modelling based cost–utility analyses for the main policy comparisons that might be relevant and feasible. Wherever possible these analyses will make use of the models previously developed by the West Midlands Health Technology Assessment Collaboration.
Given that the numbers involved will be relatively small (relative to typical epidemiological cohort studies) and also, for evidence relating both to treatment effects and economic impacts, subject to considerable uncertainty it will be essential to investigate the cost-effectiveness through modelling. Decision modelling in particular provides an explicit framework for integrating (a) disease natural history data (b) evidence and/or assumptions about treatment effectiveness and cost, (c) extrapolating these data over time, and (d) quantifying uncertainty surrounding all of the model inputs, so that a wide range of policy scenarios can be explored (23,24). The models will be used to establish the level at which the costs of the ERT would meet conventional thresholds of cost effectiveness taking account of NHS and societal costs.
Development of condition-specific rating scales
Currently there are condition-specific rating scales for Gauchers (the Severity Score Index [26, 27]) and Fabry (the Mainz severity Score Index [28]). There is no condition-specific severity scoring system for MPS I (although such a scale is under development [26]). However these scales have been developed from adult data and their relevance to children has not been established, nor do they appear to be particularly sensitive to treatment [personal communication]. There is an urgent need for the development of better severity scoring systems. The development of such scales is not part of the current application, however, the natural history data and the carer/family data collected as part of this study can be used to inform the development of such disorder-specific treatment responsive measures. The availability of a whole population sample for these conditions will provide the basis for further development and testing of such systems.
Ethical considerations
Multi-centre ethics agreement has been obtained from the South West Research Ethics Committee, and site-specific ethics approval has been granted by the relevant local ethics committee for each site. Research Governance approval has also been granted for the data collection and analysis of this data for the seven treatment centres to collect the data and for the Peninsula Medical School to undertake data analysis with the centres.
Benefits to the NHS
A longitudinal cohort study collecting individual patient data from people with lysosomal storage disorders will provide benefits to the NHS, designated treatment centres and patients. As suggested by the HTA-commissioned assessments of enzyme replacement therapy for Gaucher, Fabry and MPS I [16,18], in order for an evaluation of the clinical and cost-effectiveness of emerging enzyme replacement therapies to be conducted, comprehensive and valid data of sufficient quality needs to be collected, prior to the therapy being licensed. This study proposes to collect data from all people with lysosomal storage disorders in the UK who attend the seven treatment centres in England, thereby minimising selection bias. A similar type of study, which established a Cystic Fibrosis database, has reported benefits to clinicians and patients [30]. Similar to LSDs, many patients with CF are seen at specialised clinics, where care is tailored to the individual. Their data capture and reporting system has been customised to allow for individual patient reports regarding their disorder. The system also allows the participating clinicians to compare care programmes between centres.
Staffing implications
Professor Stuart Logan will have overall responsibility for the project. Dr Katrina Wyatt and Professor Logan will have day to day responsibility for the project which will be coordinated by Sheena Oxer. Dr Rob Anderson and Dr Ken Stein will supervise the data modelling and health economic analyses and Dr William Henley will manage the statistical analyses. The clinical applicants will ensure appropriate design of data gathering and clinical relevance of analyses. The patient support groups will provide input to ensure that appropriate account is taken of patient and family views.
Data collection will require considerable clinical expertise. There are in total 1127 patients with LSDs seen at the participating centres and we anticipate very high rates of agreement to participate. We estimate that initial data entry will take approximately 2 hours per patient and each follow-up visit approximately half an hour per patient. We are currently funding a full time research analyst in Cambridge, two 0.75WTE research analysts in Manchester, three 0.7WTE research analysts in London, and one 0.5WTE research analyst in Birmingham. Given the amount of data which will also need to be collected retrospectively we propose to fund a data entry research assistant at each site for 12 months. The study also requires a fulltime research fellow with modelling experience to develop and analyse the models, with additional support from the Peninsula Technology Assessment Group (PenTAG).
Additional support has been requested to allow for travel between the sites, conference attendance, computers and printers (for all sites for data collection) and recruitment.
NHS support and Treatment costs
Following extensive discussion with each of the treatment centres regarding additional treatment and NHS costs, it has been agreed that while there are no additional treatment costs (ie drug costs or investigations) associated with this study there are time implications for the consultants who manage patients with LSDs. In order to collect the necessary information from each patient, it will be necessary to spend additional time with each patient to explain the study, gain consent and collect and record additional information, more frequently than would otherwise be required in a routine consultation. As each patient is seen by a consultant for their management and treatment, this will does carry a time implication for each treatment centre. Two hours consultant time has been agreed per week per treatment centre for the duration of the study.
References
-
B.J.H.M. Poorthuis et al. , The frequency of lysosomal storage diseases in The Netherlands, Hum. Genet. 105 (1999), pp. 151–156.
-
P.F. Meikle, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders, JAMA 281 (1999), pp. 249–254.
-
L. Peltonen, Molecular background of Finnish disease heritage, Ann. Med. 29 (1997), pp. 553–556.
-
Acta Pediatrica 2005; 94 (suppl 447): 79
-
Meikle PJ, Hopwood JJ, Clague AE, Clarey WF. Prevalence of Lysosomal Storage Disorders. JAMA 1999 281(3) 249-254
-
Wraith JE. The management of lysosomal storage disorders. Current Peadiatrics 2004: 14; 389-393
-
Wenger DA, Coppola S, Liu S-L. Insights into the diagnosis and treatment of lysosomal storage disease. Arch Neurol 2003: 60; 322-328
-
Hobbs JR, Hugh-Jones K, Barrett AJ, Byrom N, Chambers D, Henry K, James DC, Lucas CF, Rogers TR, Benson PF, Tansley LR, Patrick AD, Mossman J, Young EP. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone marrow transplantation. Lancet. 1981 Oct 3;2(8249):709-12
-
Vellodi A. Lysosomal Storage Disorders. Br. J. Haem. 2004 128; 413-431
-
Cox T, Lachmann R, Hollak C, Aerts J, van Weely S, Hrebicek M, Platt F, Butters T, Dwek R, Moyses C, Gow I, Elstein D, Zimran A. Novel oral treatment of Gauchers disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000 355(9214):1481-5.
-
Elstein D, Hollak C, Aerts JM, van Weely S, Maas M, Cox TM, Lachmann RH, Hrebicek M, Platt FM, Butters TD, Dwek RA, Zimran A. Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease J Inherit Metab Dis. 2004; 27(6): 757-66.
-
Brady R, Schiffmann R. Enzyme-replacement therapy for metabolic storage disorders The Lancet 2004; 3: 752-56
-
L. Klinge, V. Straub, U. Neudorf, J. Schaper, T. Bosbach, K. Görlinger, M. Wallot, S. Richards and T. Voit. 2005 Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscular disorders 15 (1) 24-32
-
Muenzer J, Lamsa JC, Garcia A, Dacosta J, Garcia J, Treco DA 2002 Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr Suppl. 2002;91(439):98-9.
-
Harmatz, P, Whitley CB, Waber L, et al 2004.Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J Pediatrics 144(5) 574-580
-
Connock M, Burls A, Frew E et al The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Gauchers disease NICE Technology Assessment Report 2005
-
Connock M, Burls A, Frew E, Juarez- Garcia A et al. The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Gaucher disease: a systematic review. Health Technol Assess. 2006;10 (24): 1-152
-
Connock M, Frew E, Juarez- Garcia A et al. The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Fabry’s disease and MPS I. NICE Technology Assessment Report 2005
-
Peters FPJ, Vermeulen A, Kho TL. Anderson- Fabry’s disease: α – galactosidase deficiency. Lancet 2001; 357: 138-40
-
Mehta A, Ricci R, Widner U et al. On behalf of the FOS investigators. Fabry disease defined: baseline clinical manifestations of 366 patients in FOS – the Fabry Outcome Survey. Eur J Clin Invest 2004 34: 236-42
-
Congdon, P. Bayesian Statistical Modelling. Wiley. 2002
-
Dunson, DB. Bayesian methods for latent trait modelling of longitudinal data. Statistical Methods in Medical Research 2007 16(5): 399 - 415
-
Lilford, RJ, Thornton, JG, Braunholtz, D. Clinical trials and rare diseases: a way out of a conundrum. BMJ 1995; 311: 1621-25
-
M. Buxton, M. F. Drummond, B. A. van Hout, R. L. Prince, T. A. Sheldon, T. Szucs, and M. Vray. Modelling in economic evaluation: an unavoidable fact of life. Health Economics 6:217-227, 1997.
-
K Kuntz, M Weinstein. Modelling in economic evaluations. In: Economic evaluation in health care: merging theory with practice, edited by Drummond M and McGuire A, Oxford:Oxford University Press, 2005, p. 141-171.
-
Zimran A, Gross E, West C, Sorge J, Kubitz M, Beutler E. Prediction of severity of Gauchers disease by identification of mutations at DNA level. The Lancet 1989; 334(8659): 349 -352
-
Zimran A, Kay A, Gelbart T, Garver P, Thurston D, Saven A et al. Guacher disease Clinical, laboratory, radiology and genetic features of 53 patients. Medicine 1992; 71(6): 337-353
-
Whybra C, Kampmann C, Krummenauer F, Ries M, Mengel E, Miebach E et al. The Mainz Severity Score Index: a new instrument for quantifying the Anderson Fabry disease phenotype, and the response of new patients to enzyme replacement therapy. Clinical Genetics 2004; 65(4): 299-307
-
Haley SM, Fragala MA, Aseltine R, Ni P, Skrinar AM. Development of a disease-specific disability instrument for Pompe disease. Pediatr Rehabil 2003: 6(2); 77-84
-
Mehta G, Sims EJ, Culross F, McCormick JD. Potential benefits of the UK cystic fibrosis database 2004. Suppl 44 vol 97 pages 60-71
Appendix 6 Initial letters
Initial contact letter to potential participants

Version 4b (21.10.08)
Treatment centre name
Address
Telephone
Name participant
Address
Date
Dear [name]
The purpose of this letter is to briefly let you know about a study which is taking place at [name of treating hospital]. The study is a collaboration between this hospital, other hospitals in England which treat people with lysosomal storage disorders, the Peninsula College of Medicine and Dentistry, and associated patient support groups. We are asking everyone (adults and children) in England who has a lysosomal storage disorder, such as [insert person’s condition] to consider being part of our study.
This research study will involve collecting and entering data relating to the diagnosis and management of your condition on a specially designed, secure database. We believe that this information will help us understand more about these disorders and the effect of treatments on them. Please note that taking part in this study will not alter your medical care or your treatment in any way.
We will send you some more information about the study before your next clinical appointment and when you are next at the hospital, one of the nurses or doctors involved in your care will discuss the study with you in more detail and answer any questions that you may have. Only when you feel you have sufficient information about the study and what we are asking you to do, will you be asked to make a decision about whether or not you want to take part. Please note that if you decide not to take part your care will not be affected in any way.
Please be assured that your details have been added to this letter by hospital staff and have not been supplied to anyone outside the hospital.
Thank you for reading this letter and we look forward to meeting you.
Yours Sincerely
Chief Investigator and Name of treating consultant for that site.
Initial contact letter to young potential participants

Version 3b (21-10-08)
Treatment centre name
Address
Telephone
Name participant
Address
Date
Dear [name]
The purpose of this letter is to briefly let you know about a study which is taking place at [name of treating hospital]. The study is a partnership between this hospital, other hospitals in England which treat people with lysosomal storage disorders, as well as the Peninsula College of Medicine and Dentistry, and the patient support groups for people with lysosomal storage disorders. We are asking everyone (adults and children) in England who has a lysosomal storage disorder to consider whether they would like to be part of this study.
The study will involve collecting and entering information about the diagnosis and management of your condition onto a specially designed, secure database. We believe that this information will help us understand more about these disorders and the effect of treatments on them. Please note that taking part in this study will not alter your medical care or treatment in any way.
We will send you some more information about the study before your next clinical appointment and when you are next at the hospital, one of the nurses or doctors involved in your care will discuss the study with you in more detail and answer any questions that you may have. Only when you feel you have sufficient information about the study and what we are asking you to do, will you be asked to make a decision about whether or not you are happy to take part.
Please note that if you decide not to take part your care will not be affected in any way.
Please be assured that your details have been added to this letter by hospital staff and have not been supplied to anyone outside the hospital.
Thank you for reading this letter and we look forward to meeting you.
Yours Sincerely
Chief Investigator and Name of treating consultant for that site.
Initial contact letter to parents/carers of potential participants

Version 4b (21-10-08)
Treatment centre name
Address
Telephone
Name participant
Address
Date
Dear [name]
The purpose of this letter is to briefly let you know about a study which is taking place at [name of treating hospital]. The study is a collaboration between this hospital and the other hospitals in England which treat people with lysosomal storage disorders, as well as the Peninsula College of Medicine and Dentistry, and the associated patient support groups. We are asking everyone (adults and children) in England who has a lysosomal storage disorder to consider being part of our study. The study will involve collecting and entering data about the diagnosis and management of your child’s lysosomal storage disorder on a specially designed, secure database. We believe that this information will help us understand more about these disorders and the effect of treatments on them. Please note that taking part in this study will not alter their medical care or their treatment in any way.
We will send you some more information about the study before your child’s next clinical appointment and when you are both next at the hospital, one of the nurses or doctors involved in their care will discuss the study with you and your child in more detail and answer any questions that you may have. Only when you feel you have sufficient information about the study and what we are asking you to do, will you be asked to make a decision about whether or not you are happy for them to take part. Please note that if you decide not to take part your child’s care will not be affected in any way.
Please be assured that your details have been added to this letter by hospital staff and have not been supplied to anyone outside the hospital.
Thank you for reading this letter and we look forward to meeting you both.
Yours Sincerely
Chief Investigator and Name of treating consultant for that site.
Initial contact letter to carers of adult participants

Version 1 (24-04-09)
Treatment centre name
Address
Telephone
Name participant
Address
Date
Dear [carer’s name]
The purpose of this letter is to briefly let you know about a study which is taking place at [name of treating hospital]. The study is a collaboration between this hospital and the other hospitals in England which treat people with lysosomal storage disorders, as well as the Peninsula College of Medicine and Dentistry, and the associated patient support groups. We are asking everyone (adults and children) in England who has a lysosomal storage disorder to consider being part of our study. The study will involve collecting and entering data about the diagnosis and management of the patients’ lysosomal storage disorders on a specially designed, secure database. We believe that this information will help us understand more about these disorders and the effect of treatments on them. Please note that taking part in this study will not alter their medical care or their treatment in any way.
We will send you some more information about the study before [patient’s name]’s next clinical appointment and when you are both next at the hospital, one of the nurses or doctors involved in their care will discuss the study with you both in more detail and answer any questions that you may have. Only when you feel you have sufficient information about the study and what we are asking you to do, will you be asked to make a decision about whether or not you and [patient’s name] are happy for them to take part. Please note that if you decide not to take part, [patient’s name]’s care will not be affected in any way.
Please be assured that your details have been added to this letter by hospital staff and have not been supplied to anyone outside the hospital.
Thank you for reading this letter and we look forward to meeting you both.
Yours Sincerely
Chief Investigator and Name of treating consultant for that site.
Appendix 7 Follow-up letters
Follow-up letter to potential participants

Version 2b (08-04-08)
Treatment centre name
Address
Telephone
Name participant
Address
Date
Dear [name]
You may remember that we wrote to you a little while ago to tell you about a national study we are taking part in. We are writing again to enclose more information about the study and to remind you that someone will come and talk about the study and your possible involvement when you are next at the hospital.
Again, we would like to assure you that you do not have to take part in this study and if you decide to take part in this study you can withdraw at any time. If you do not want to take part or if you decide to leave the study, your treatment will not be affected in any way. You will be given as much time as you would like to decide whether or not to take part.
We hope you find the information sheet useful and would be very happy to answer any questions that you have about the study.
Yours sincerely
Name of treating consultant for that site
Follow-up letter to young participants

Version 2b (08-04-08)
Treatment centre name
Address
Telephone
Name participant
Address
Date
Dear [name]
You may remember that we wrote to you a little while ago to tell you about a national study we are taking part in. We are writing again to enclose more information about the study and to remind you that someone will come and talk to you and your parents/guardians about the study and your possible involvement when you are next at the hospital.
Again, we would like to assure you that you do not have to take part in this study and if you decide to take part in this study you can withdraw at any time. If you do not want to take part or if you decide to leave the study, your treatment will not be affected in any way. You will be given as much time as you need to decide whether or not to take part.
We hope you find the information sheet useful and would be very happy to answer any questions that you have about the study.
Yours sincerely
Name of treating consultant for that site
Follow-up letter to parents/carers

Version 3c (18-06-08)
Treatment centre name
Address
Telephone
Name participant
Address
Date
Dear [name]
You may remember that we wrote to you a little while ago to tell you about a national study we are running. We are writing again to enclose more information about the study and to remind you that someone will come and talk about the study and [name of person who is cared for] possible involvement when you are both next at the hospital.
Again, we would like to assure you that [name of person who is cared for] does not have to take part in this study and if you decide that they can take part in this study they are free to withdraw at any time. If you do not want them to take part or if you decide that they should then leave the study, their treatment will not be affected in any way. You will be given as much time as you need to decide whether or not [name of person who is cared for] should take part. Please also note that every effort will be made to explain the study to [name] and to gain their views about taking part.
We hope you both find the information sheet useful and would be very happy to answer any questions that either of you have about the study.
Yours sincerely
Name of treating consultant for that site
Follow-up letter to carers of adult participants

Version 1 (24-04-09)
Treatment centre name
Address
Telephone
Name participant
Address
Date
Dear [carer’s name]
You may remember that we wrote to you a little while ago to tell you about a national study we are running. We are writing again to enclose more information about the study and to remind you that someone will come and talk about the study and [patient name’s] possible involvement when you are both next at the hospital.
Again, we would like to assure you that [patient name] does not have to take part in this study and if you decide that they can take part in this study they are free to withdraw at any time. If you do not want them to take part or if you decide that they should then leave the study, their treatment will not be affected in any way. You will be given as much time as you need to decide whether or not [patient name] should take part. Please also note that every effort will be made to explain the study to [patient name] and to gain their views about taking part.
We hope you both find the information sheet useful and would be very happy to answer any questions that either of you have about the study.
Yours sincerely
Name of treating consultant for that site
Appendix 8 Patient Information Sheets
Patient Information Sheet for participants

PATIENT INFORMATION SHEET
Lysosomal Storage Disorders a research study
A study to investigate the natural history, effectiveness and cost effectiveness of current and emerging treatment options for people with a lysosomal storage disorder
1. What is this all about?
In England approximately 1100 people have been diagnosed with a lysosomal storage disorder, such as Gauchers, Fabrys, MPS1, Niemann Pick C or Pompe. The purpose of this study is to understand more about these disorders including how they have been diagnosed and how they are being treated, Enzyme replacement therapy and substrate reduction therapy are new treatments for some lysosomal storage disorders and new therapies are also being developed. In order to better understand the effects that these treatments are having, the researchers and doctors at your hospital and other hospitals which treat people with these disorders would like to gather together information from your hospital notes about the diagnosis and management of your lysosomal storage disorder. By collecting information on as many children and adults with these conditions as possible, it is hoped to understand these disorders and the effects of their treatments better.
You are invited to participate in this research study. Before you decide whether or not you are happy to be part of this study, it is important for you to understand why the research is being done and what it would mean to be involved. Please take time to read the following information carefully and discuss it with friends, relatives and your doctor if you wish. Please do feel free to ask us if there is anything that is not clear or if you would like more information. Please take as much time as you need to decide whether or not you wish to take part in this study.
Thank you for reading this
2. What is the purpose of the study?
Lysosomal storage disorders are a group of rare, inherited diseases. Traditionally, the treatment for lysosomal storage disorders has focussed on managing the symptoms of the disease rather than treating the disease itself. However, in recent years, treatments which seek to increase the level of enzymes in the body, known as enzyme replacement therapies are being developed for these disorders.
Enzyme replacement therapies are now available for the treatment of some of the disorders. The Peninsula Medical School, in partnership with a patient organisation and the treating hospitals would like to understand more about how effective and cost- effective these new treatments are. However, because these conditions are so rare, usual ways of testing how effective a treatment is are much harder to carry out. Therefore, we hope to carry out a long term research study, whereby we collect information, from all consenting adults and children with these conditions over a period of time. If we are able to collect this information over a period of time it should help us to understand
-
how well (or effective) these treatments are
-
when the best time to start giving these treatments is
-
what the appropriate doses are
-
which symptoms led to the diagnosis of the disorder
Another aspect of the study will be to estimate the value for money or cost-effectiveness of these treatments. In order to do this we will look at
-
how frequently people use the NHS
-
the cost of their treatment
-
other costs, related to your family
3. Why have I been chosen?
We intend to ask everyone with a diagnosis of a lysosomal storage disorder in the UK, who attends one of the 7 English NCG specialist centres, whether they would consider taking part in the research. The whole study will last for around three years and we would like everyone who agrees to participate to remain in the study until it is completed in 2011.
4. Do I have to take part?
It is entirely up to you to decide whether or not you would like to take part. If you decide that you are happy to be part of the study, you will be given a copy of this information sheet to keep and be asked to sign a consent form. If you do take part, you will be free to leave the study at any time, and will not have to give a reason. Please be assured that not taking part in the study or leaving the study will not affect the standard of care that you receive in any way.
5. What will happen to me if I decide to take part?
Once you have thought about the study and asked any questions that you might have, you will be asked whether or not you are happy to take part in the study.
If you decide that you are happy to take part you will be asked to agree to share relevant information about your condition with the research study team. The researchers will obtain information, such as when you were diagnosed, what treatments you are receiving, from your hospital notes. This information will be entered onto a separate and secure computer at your hospital and added to information collected at other hospitals to get a national picture of these conditions.
We would also like you to fill out two or three quality-of-life questionnaires about how you are feeling and what you feel able to do. We would also like you to fill out a questionnaire about the services you use. If someone cares for you on a regular basis we would like to ask them to fill out a questionnaire about the impact this has on them. This should take about 20–30 minutes. We will ask you to fill these out when you come to the hospital for your annual review, for the duration of the study.
6. What are the possible disadvantages of taking part?
We do not foresee any risk to you should you agree to take part in this study as being in this study will not affect your treatment or management of your condition in any way. The time it takes to complete the questionnaires may be an inconvenience for you, although we will ensure that this does not add too much extra time to your clinical appointment.
7. What are the possible benefits of taking part?
While we can not say that this study will be of direct benefit to you now, we do believe that it will help us to answer questions about lysosomal storage disorders and their treatments in the future. For example, we should have a better understanding of when treatments should be started and what symptoms could lead to earlier diagnosis of these disorders
8. Will my details be kept confidential?
Yes. If you consent to take part in the study, your medical records will be inspected and the relevant information entered on to a database designed for this study. Your name however, will not be entered on to the database; instead you will be given a unique identifying number and any information about you will be related to that number.
All information that is collected about you during the course of the study will be kept strictly confidential, and securely for 10 years, in accordance with good research practice guidelines. We do not expect to publish any individual information, rather we will look at the data from everyone in the study. However, should we publish any data about an individual we will ensure that is will not be possible to identify that person from the data.
9. What will happen to the results of the research study?
The results of the research are likely to be known by the end of 2011. The researchers will write an annual report updating everyone who has taken part to let them know how the study is going. The results will also be published in a medical journal so that other doctors and health professionals looking after people with lysosomal storage disorders can learn from it. These results should also be really useful for support groups.
No one who takes part in the study will be identified in any report/publication resulting from the research.
10. What if something goes wrong?
If you wish to complain or have any concerns about any aspect of the way you have been approached or have been treated during the course of this study, the normal National Health Service complaints mechanisms are available to you. The details are available on the website below:
http://www.nhs.uk/England/AboutTheNhs/ComplainCompliment.cmsx
Alternatively, you can contact the Patient Advice and Liaison Service (PALS) at your local hospital where you receive treatment and they will able to assist you with any complaint. The details are available below
The PALS desk is in the main reception area at: University College Hospital 235 Euston Road London NW1 2BU Monday to Friday 9am to 4pm:
PALS Tel: 020 7380 9975 email: pals@uclh.nhs.uk
<insert hospital logo here> UCL Hospitals is an NHS Foundation Trust incorporating the Eastman Dental Hospital, Elizabeth Garrett Anderson & Obstetric Hospital, The Heart Hospital, Hospital for Tropical Diseases, The Middlesex Hospital, National Hospital for Neurology & Neurosurgery, The Royal London Homoeopathic Hospital and University College Hospital.
Please see study website for a list and links to the major national support groups: http://www.pms.ac.uk/ncslsd
Who is funding and organising the study?
The research is funded by the Health Technology Assessment Programme, which is part of the Department of Health. The research is organised by a team from the Peninsula Medical School in collaboration with:
-
Manchester Children’s Hospital
-
Hope Hospital, Salford
-
Great Ormond Street Hospital
-
Royal Free Hospital, London
-
National Hospital for Neurology & Neurosurgery, London
-
Addenbrookes Hospital, Cambridge
-
Birmingham Children’s Hospital
-
Gauchers Association
Please note your consultant will not be paid for your participation in this project.
Who has reviewed the study?
The protocol for this study has been reviewed and approved by the Southwest Multi-Centre Research Ethics Committee.
Contact for Further Information
If you would like any further information about the research, please contact:
<NCS-LSD Study Coordinator>
Peninsula College of Medicine & Dentistry
St Luke’s Campus
Heavitree Road
Exeter
EX1 2LU
Please replace with local information i.e. the research nurses/analyst contact details
OR
<PI NCS-LSD Study>
Peninsula College of Medicine & Dentistry
St Luke’s Campus
Heavitree Road
Exeter
EX1 2LU
Please replace with local information i.e. the name and contact details of the clinician
Thank you for taking time to consider this study.
If you decide to take part, you will be given a signed consent form to keep for your records.
Patient Information Sheet for carer

PATIENT INFORMATION SHEET
Lysosomal Storage Disorders – a research study
A study to understand the natural history, effectiveness and cost effectiveness of current and emerging treatments for children with a lysosomal storage disorder and their families
Version 8 03-06-09
1. What is this all about?
In England approximately 1100 people have been diagnosed with a lysosomal storage disorder, such as Gauchers, Fabrys, MPS1, Niemann Pick C or Pompe. The purpose of this study is to understand more about these disorders including how they have been diagnosed and how they are being treated. Enzyme replacement therapy and substrate reduction therapy are new treatments for some lysosomal storage disorders and new therapies are also being developed. In order to better understand the effects that these treatments are having, the researchers and doctors at your hospital and other hospitals which treat people with these disorders would like to gather together information from your hospital notes about the diagnosis and management of your lysosomal storage disorder. By collecting information on as many children and adults with these conditions as possible, it is hoped to understand these disorders and the effects of their treatments better.
Your child is invited to participate in this research study. Before you decide whether or not you are happy to be part of this study, it is important for you to understand why the research is being done and what it would mean to be involved. Please take time to read the following information carefully and discuss it with friends, relatives and your doctor if you wish. Please do feel free to ask us if there is anything that is not clear or if you would like more information. Please take as much time as you need to decide whether or not you wish to take part in this study.
Thank you for reading this
2. What is the purpose of the study?
Lysosomal storage disorders are a group of rare, inherited diseases. Traditionally, the treatment for lysosomal storage disorders has focussed on managing the symptoms of the disease rather than treating the disease itself. However, in recent years, treatments which seek to increase the level of enzymes in the body, known as enzyme replacement therapies are being developed for these disorders.
Enzyme replacement therapies are now available for the treatment of some of the disorders. The Peninsula Medical School, in partnership with a patient organisation and the treating hospitals would like to understand more about how effective and cost- effective these new treatments are. However, because these conditions are so rare, usual ways of testing how effective a treatment is are much harder to carry out, we hope to carry out a long term research study, whereby we collect information, from all consenting children and adults with these conditions over a period of time. If we are able to collect this information over a period of time it should help us to understand
-
how well (or effective) these treatments are
-
when the best time to start giving these treatments is
-
what the appropriate doses are
-
which symptoms led to the diagnosis of the disorder
Another aspect of the study will be to estimate the value for money or cost-effectiveness of these treatments. In order to do this we will look at
-
how frequently people use the NHS
-
the cost of their treatment
-
other costs, related to your family
3. Why has my child been chosen?
We intend to ask everyone with a diagnosis of a lysosomal storage disorder in the UK, who attends one of the 7 English NCG specialist centres, whether they would consider taking part in the research. The whole study will last for around three years and we would like everyone who agrees to participate to remain in the study until it is completed in 2011.
4. Do I have to take part?
It is entirely up to you to decide whether or not you would like your child to take part. If you decide that you are happy for them to be part of the study, you will be given a copy of this information sheet to keep and be asked to sign a consent form. If they do take part, they will be free to leave the study at any time, and will not have to give a reason. Please be assured that not taking part in the study or leaving the study will not affect the standard of care that they receive in any way.
5. What will happen to my child if they take part?
Once you have thought about the study and asked any questions that you might have, you will be asked whether or not you are happy for your child to take part in the study.
If you decide that you are happy for your child to take part you will be asked to agree to share relevant information about your child’s condition with the research study team. The researchers will obtain information, such as when your child was diagnosed, what treatments they are receiving, from their hospital notes. This information will be entered onto a separate and secure computer at your child’s hospital and added to information collected at other hospitals to get a national picture of these conditions.
We would also like you to fill out two or three quality-of-life questionnaires about how you think your child is feeling and what you feel they are able to do. We will ask your child to complete these questionnaires if you think this appropriate. We would also like you to fill out two questionnaires about the services your child uses and the impact that your child’s condition has on you. This should take about 20-30 minutes. We will ask you to fill these out when your child comes to the hospital for their annual review, for the duration of the study.
6. What are the possible disadvantages of taking part?
We do not foresee any risk to your child being part of this study as being in this study will not affect their treatment or management of their condition in any way. The time it takes to complete the questionnaires may be an inconvenience for you, although we will ensure that this does not add too much extra time to their clinical appointment.
7. What are the possible benefits of taking part?
While we can not say that this study will be of direct benefit to you and your child now, we do believe that it will help us to answer questions about lysosomal storage disorders and their treatments in the future. For example, we should have a better understanding of when treatments should be started and what symptoms could lead to earlier diagnosis of these disorders
8. Will my child’s details be kept confidential?
Yes. If you consent for your child to take part in the study, their medical records will be inspected and the relevant information entered on to a database designed for this study. Their name however, will not be entered on to the database; instead they will be given a unique identifying number and any information about them will be related to that number.
All the information that is collected about your child during the course of the study will be kept strictly confidential, and securely for 10 years, in accordance with good research practice guidelines. We do not expect to publish any individual information; rather we will look at the data from everyone in the study. However, should we publish any data about an individual we will ensure that is will not be possible to identify that person from the data.
9. What will happen to the results of the research study?
The results of the research are likely to be known by the end of 2011. The researchers will write an annual report updating everyone who has taken part to let them know how the study is going.
The results will also be published in a medical journal so that other doctors and health professionals looking after people with lysosomal storage disorders can learn from it. These results should also be really useful for support groups.
No one who takes part in the study will be identified in any report/publication resulting from the research.
10. What if something goes wrong?
If you wish to complain or have any concerns about any aspect of the way you and your child have been approached or have been treated during the course of this study, the normal National Health Service complaints mechanisms are available to you. The details are available on the website below:
http://www.nhs.uk/England/AboutTheNhs/ComplainCompliment.cmsx
Alternatively, you can contact the Patient Advice and Liaison Service (PALS) at your local hospital where you receive treatment and they will able to assist you with any complaint. The details are available below
The PALS desk is in the main reception area at: <insert contact details here>
<insert hospital logo here> UCL Hospitals is an NHS Foundation Trust incorporating the Eastman Dental Hospital, Elizabeth Garrett Anderson & Obstetric Hospital, The Heart Hospital, Hospital for Tropical Diseases, The Middlesex Hospital, National Hospital for Neurology & Neurosurgery, The Royal London Homoeopathic Hospital and University College Hospital.
Please see study website for a list and links to the major national support groups: http://www.pms.ac.uk/ncslsd
Who is funding and organising the study?
The research is funded by the Health Technology Assessment Programme, which is part of the Department of Health. The research is organised by a team from the Peninsula Medical School in collaboration with:
-
Manchester Children’s Hospital
-
Hope Hospital, Salford
-
Great Ormond Street Hospital
-
Royal Free Hospital, London
-
National Hospital for Neurology & Neurosurgery, London
-
Addenbrookes Hospital, Cambridge
-
Birmingham Children’s Hospital
-
Gauchers Association
Please note your consultant will not be paid for your participation in this project.
Who has reviewed the study?
The protocol for this study has been reviewed and approved by the Southwest Multi-Centre Research Ethics Committee.
Contact for Further Information
If you would like any further information about the research, please contact:
<NCS-LSD Study Coordinator>
Peninsula College of Medicine & Dentistry
St Luke’s Campus
Heavitree Road
Exeter
EX1 2LU
Please replace with local information i.e. the research nurses/analyst contact details
OR
<PI NCS-LSD Study>
Peninsula College of Medicine & Dentistry
St Luke’s Campus
Heavitree Road
Exeter
EX1 2LU
Please replace with local information i.e. the name and contact details of the clinician
Thank you for taking time to consider this study.
If you decide to take part you will be a signed consent form to keep for your records.
Patient Information Sheet for consultees

PATIENT INFORMATION SHEET
Lysosomal Storage Disorders – a research study
A study to understand the natural history, effectiveness and cost effectiveness of current and emerging treatments for children with a lysosomal storage disorder and their families
Version 2 03-06-09
1. What is this all about?
In England approximately 1100 people have been diagnosed with a lysosomal storage disorder, such as Gauchers, Fabrys, MPS1, Niemann Pick C or Pompe. The purpose of this study is to understand more about these disorders including how they have been diagnosed and how they are being treated. Enzyme replacement therapy and substrate reduction therapy are new treatments for some lysosomal storage disorders and new therapies are also being developed. In order to better understand the effects that these treatments are having, the researchers and doctors at your hospital and other hospitals which treat people with these disorders would like to gather together information from your hospital notes about the diagnosis and management of your lysosomal storage disorder. By collecting information on as many children and adults with these conditions as possible, it is hoped to understand these disorders and the effects of their treatments better.
The person in your care is invited to participate in this research study. Before you decide whether or not you are happy for the person you care for to be part of this study, it is important for you to understand why the research is being done and what it would mean to be involved. Please take time to read the following information carefully and discuss it with friends, relatives and the patient’s doctor if you wish. Please do feel free to ask us if there is anything that is not clear or if you would like more information. Please take as much time as you need to decide whether or not the person in your care should take part in this study.
Thank you for reading this
2. What is the purpose of the study?
Lysosomal storage disorders are a group of rare, inherited diseases. Traditionally, the treatment for lysosomal storage disorders has focussed on managing the symptoms of the disease rather than treating the disease itself. However, in recent years, treatments which seek to increase the level of enzymes in the body, known as enzyme replacement therapies are being developed for these disorders.
Enzyme replacement therapies are now available for the treatment of some of the disorders. The Peninsula Medical School, in partnership with a patient organisation and the treating hospitals would like to understand more about how effective and cost- effective these new treatments are. However, because these conditions are so rare, usual ways of testing how effective a treatment is are much harder to carry out, we hope to carry out a long term research study, whereby we collect information, from all consenting children and adults with these conditions over a period of time. If we are able to collect this information over a period of time it should help us to understand
-
how well (or effective) these treatments are
-
when the best time to start giving these treatments is
-
what the appropriate doses are
-
which symptoms led to the diagnosis of the disorder
Another aspect of the study will be to estimate the value for money or cost-effectiveness of these treatments. In order to do this we will look at
-
how frequently people use the NHS
-
the cost of their treatment
-
other costs, related to your family
3. Who is taking part in this study?
We intend to ask everyone with a diagnosis of a lysosomal storage disorder in the UK, who attends one of the 7 NCG specialist centres in England, whether they would consider taking part in the research. The whole study will last for around three years and we would like everyone who agrees to participate to remain in the study until it is completed in 2011.
4. What is my role in this study?
As a carer of someone with a lysosomal storage disorder who lacks capacity, we ask you to act as their personal consultee. The role of personal consultee will mean that our researchers will:
-
Ask you for advice about whether the person in your care should take part in the project, and
-
What you think the person’s feelings and wishes would be, if they had capacity to decide whether to take part.
You are not obliged to undertake the role of consultee if they do not wish to do so. If you do not feel able to take on the role of consultee, then you may suggest that someone else takes on the role, or ask that a nominated consultee be appointed.
5. Does the person in my care have to take part?
It is important that we consider the wishes and feelings of the person in your care, when deciding whether or not they would like to take part in this study. As the patient’s personal consultee, if you decide that you are happy for them to be part of the study, you will be given a copy of this information sheet to keep and be asked to sign a consent form. If they do take part, they will be free to leave the study at any time, and will not have to give a reason. Please be assured that not taking part in the study or leaving the study will not affect the standard of care that they receive in any way.
6. What will happen to the person in my care if they take part?
Once you have thought about the study and asked any questions that you might have, you will be asked whether or not you are happy for the person in your care to take part in the study.
If you are happy for the person you care for to take part, you will be asked to agree to share relevant information about their condition with the research study team. The researchers will obtain information, such as date of diagnosis and treatments they are receiving, from their hospital notes. This information will be entered onto a separate and secure computer and added to information collected at other hospitals to get a national picture of these conditions.
We would also like you to fill out two or three quality-of-life questionnaires about how you think the person in your care is feeling and what you feel they are able to do. We would also like you to fill out two questionnaires about the services you and the patient use and the impact that the patient’s condition has on you. This should take about 20–30 minutes. We will ask you to fill these out when the person in your care comes to the hospital for their annual review, for the duration of the study.
7. What are the possible disadvantages of taking part?
We do not foresee any risk to the patient by taking part in this study as being in this study will not affect their treatment or management of their condition in any way. The time it takes to complete the questionnaires may be an inconvenience for you, although we will ensure that this does not add too much extra time to their clinical appointment.
8. What are the possible benefits of taking part?
While we can not say that this study will be of direct benefit to you and the person in your care now, we do believe that it will help us to answer questions about lysosomal storage disorders and their treatments in the future. For example, we should have a better understanding of when treatments should be started and what symptoms could lead to earlier diagnosis of these disorders
9. Will personal details be kept confidential?
Yes. If you consent for the person in your care to take part in the study, their medical records will be inspected and the relevant information entered on to a database designed for this study. Their name however, will not be entered on to the database; instead they will be given a unique identifying number and any information about them will be related to that number.
All the information that is collected about the patient during the course of the study will be kept strictly confidential, and securely for 10 years, in accordance with good research practice guidelines. We do not expect to publish any individual information; rather we will look at the data from everyone in the study. However, should we publish any data about an individual we will ensure that it will not be possible to identify that person from the data.
10. What will happen to the results of the study?
The results of the research are likely to be known by the end of 2011. The researchers will write an annual report updating everyone who has taken part to let them know how the study is going.
The results will also be published in a medical journal so that other doctors and health professionals looking after people with lysosomal storage disorders can learn from it. These results should also be really useful for support groups.
No one who takes part in the study will be identified in any report/publication resulting from the research.
11. What if something goes wrong?
If you wish to complain or have any concerns about any aspect of the way you or the person in your care have been approached or have been treated during the course of this study, the normal National Health Service complaints mechanisms are available to you. The details are available on the website below:
http://www.nhs.uk/England/AboutTheNhs/ComplainCompliment.cmsx
Alternatively, you can contact the Patient Advice and Liaison Service (PALS) at your local hospital where you receive treatment and they will able to assist you with any complaint. The details are available below
The PALS desk is in the main reception area at: <insert contact details here>
UCL Hospitals is an NHS Foundation Trust incorporating the Eastman Dental Hospital, Elizabeth Garrett Anderson & Obstetric Hospital, The Heart Hospital, Hospital for Tropical Diseases, The Middlesex Hospital, National Hospital for Neurology & Neurosurgery, The Royal London Homoeopathic Hospital and University College Hospital.
Please see study website for a list and links to the major national support groups: http://www.pms.ac.uk/ncslsd
Who is funding and organising the study?
The research is funded by the Health Technology Assessment Programme, which is part of the Department of Health. The research is organised by a team from the Peninsula Medical School in collaboration with:
-
Manchester Children’s Hospital
-
Hope Hospital, Salford
-
Great Ormond Street Hospital
-
Royal Free Hospital, London
-
National Hospital for Neurology & Neurosurgery, London
-
Addenbrookes Hospital, Cambridge
-
Birmingham Children’s Hospital
-
Gauchers Association.
Please note your consultant will not be paid for your participation in this project.
Who has reviewed the study?
The protocol for this study has been reviewed and approved by the Southwest Multi-Centre Research Ethics Committee.
Contact for Further Information
If you would like any further information about the research, please contact:
<NCS-LSD Study Coordinator>
Peninsula College of Medicine & Dentistry
St Luke’s Campus
Heavitree Road
Exeter
EX1 2LU
Please replace with local information i.e. the research nurses/analyst contact details
OR
<PI NCS-LSD Study>
Peninsula College of Medicine & Dentistry
St Luke’s Campus
Heavitree Road
Exeter
EX1 2LU
Please replace with local information i.e. the name and contact details of the clinician
Thank you for taking time to consider this study.
If you decide to take part you will be given a signed consent form to keep for your records.
Appendix 9 Consent Forms
Consent form for participants
Consent form for participants (notes only)
Additional consent form for participants who attend clinic for additional hospital visits
Consent form for parents/carers
Consent form for parents/carers (notes only)
Consent for parents (additional visits)
Consent form for consultees
Consent form for consultees (notes only)
Appendix 10 Letter to general practitioner
Appendix 11 Clinical Record Forms
Appendix 12 Caregiver Strain Index
If someone cares for you on a regular basis please can you ask them to fill this questionnaire in
Directions: Here is a list of things that other caregivers have found to be difficult. Please put a checkmark in the columns that apply to you. We have included some examples that are common caregiver experiences to help you think about each item. Your situation may be slightly different, but the item could still apply.
| Yes, On a Regular Basis = 2 | Yes, Sometimes = 1 | No = 0 | |
|---|---|---|---|
| My sleep is disturbed (For example: the person I care for is in and out of bed or wanders around at night) | ____________ | ____________ | ____________ |
| Caregiving is inconvenient (For example: helping takes so much time or it’s a long drive over to help) | ____________ | ____________ | ____________ |
| Caregiving is a physical strain (For example: lifting in or out of a chair; effort or concentration is required) | ____________ | ____________ | ____________ |
| Caregiving is confining (For example: helping restricts free time or I cannot go visiting) | ____________ | ____________ | ____________ |
| There have been family adjustments (For example: helping has disrupted my routine; there is no privacy) | ____________ | ____________ | ____________ |
| There have been changes in personal plans (For example: I had to turn down a job; I could not go on vacation) | ____________ | ____________ | ____________ |
| There have been other demands on my time (For example: other family members need me) | ____________ | ____________ | ____________ |
| There have been emotional adjustments (For example: severe arguments about caregiving) | ____________ | ____________ | ____________ |
| Some behaviour is upsetting (For example: incontinence; the person cared for has trouble remembering things; or the person I care for accuses people of taking things) | ____________ | ____________ | ____________ |
| It is upsetting to find the person I care for has changed so much from his/her former self (For example: he/she is a different person than he/she used to be) | ____________ | ____________ | ____________ |
| There have been work adjustments (For example: I have to take time off for caregiving duties) | ____________ | ____________ | ____________ |
| Caregiving is a financial strain | ____________ | ____________ | ____________ |
| I feel completely overwhelmed (For example: I worry about the person I care for; I have concerns about how I will manage) | ____________ | ____________ | ____________ |
| [Sum responses for ‘Yes, on a regular basis’ (2 pts each) and ‘yes, sometimes’ (1 pt each)] | |||
| Total Score = | |||
Appendix 13 Service-use and costs questionnaires
Service-use and cost questionnaire to patients and their families
Service-use and cost questionnaire for children (proxy)
Appendix 14 Fatigue Severity Scale of sleep disorders
The Fatigue Severity Scale is a method of evaluating the impact of fatigue on you. The Fatigue Severity Scale is a short questionnaire that requires you to rate your level of fatigue.
The Fatigue Severity Scale questionnaire contains nine statements that rate the severity of your fatigue symptoms.
Read each statement and circle a number from 1 to 7, based on how accurately it reflects your condition during the past week and the extent to which you agree or disagree that the statement applies to you.
A low value (e.g., 1) indicates strong disagreement with the statement, whereas a high value (e.g., 7) indicates strong agreement.
It is important that you circle a number (1 to 7) for every question.
| Fatigue Severity Scale Questionnaire | |||||||
|---|---|---|---|---|---|---|---|
| During the past week, I have found that: | Disagree | ⟷ | Agree | ||||
| My motivation is lower when I am fatigued. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Exercise brings on my fatigue. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| I am easily fatigued. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Fatigue interferes with my physical functioning. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Fatigue causes frequent problems for me. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| My fatigue prevents sustained physical functioning. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Fatigue interferes with carrying out certain duties and responsibilities. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Fatigue is among my three most disabling symptoms. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Fatigue interferes with my work, family, or social life. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Total Score: | |||||||
Arch Neurol 1989;46:1121–3. Copyright ©(1989) American Medical Association. All rights reserved. Reproduced with permission of the author.
Appendix 15 Postal questionnaire request letters
Patient enzyme replacement therapy reduction postal questionnaire letter

Dear
We are very grateful to you for consenting to participate in the National Collaborative Study for Lysosomal Storage Disorders. We had anticipated asking you to complete the questionnaires at each of your annual review visits until 2011.
We are aware however, that your clinician has changed your treatment regimen due to a current world shortage of your treatment drug, and we were wondering whether you would consider completing a further set of Quality of Life and Service Use Questionnaires as well as a fatigue severity assessment for the study.
The questionnaires are exactly the same as those you previously completed with the addition of a fatigue assessment. As these questionnaires are being given to you in addition to those at your annual clinical review, we are asking for your additional agreement.
We have enclosed a stamped addressed envelope for you to return the completed questionnaires, however please be assured that you are under no obligation to agree to this or to remain in the study.
If you have any questions or concerns regarding this, please free to contact one of the team.
If you would like to discuss any aspect of the study then please contact either of us at the address below, or talk to your clinician.
Best wishes
Name of treating consultant for that site and Chief Investigator
Parent enzyme reduction therapy reduction postal questionnaire letter

Dear
We are very grateful to you for allowing your child to participate in the National Collaborative Study for Lysosomal Storage Disorders. We had anticipated asking you to complete the questionnaires at each of your annual review visits until 2011.
We are aware however, that your child’s clinician has changed his/her treatment regimen due to a current world shortage of your treatment drug, and we were wondering whether you would consider completing a further set of Quality of Life and Service Use Questionnaires for the study.
The questionnaires are exactly the same as those you previously completed, however as this is not at your child’s annual review we are asking for your additional agreement.
We have enclosed a stamped addressed envelope for you to return the completed questionnaires, however please be assured that you are under no obligation to agree to this or for your child to remain in the study.
If you have any questions or concerns regarding this, please free to contact one of the team.
If you would like to discuss any aspect of the study then please contact either of us at the address below, or talk to your clinician.
Best wishes
Name of treating consultant for that site and Chief Investigator
Appendix 16 Questionnaire follow-up letters
Letter for follow-up postal questionnaire

Dear
We are very grateful to you for consenting to participate in the National Collaborative Study for Lysosomal Storage Disorders. We had anticipated asking you to complete the questionnaires at each of your annual review visits until 2011.
Unfortunately due to staffing issues we have been unable to meet with you to give you the follow up questionnaires in person however we have enclosed them with this letter.
The questionnaires are exactly the same as those you previously completed with the addition of a fatigue assessment.
We have enclosed a stamped addressed envelope for you to return the completed questionnaires, however please be assured that you are under no obligation to agree to this or to remain in the study.
If you have any questions or concerns regarding this, please free to contact one of the team.
If you would like to discuss any aspect of the study then please contact either of us at the address below, or talk to your clinician.
Best wishes
Name of treating consultant for that site and Chief Investigator
Letter for parent for follow–up postal questionnaires

Dear
We are very grateful to you for allowing your child to participate in the National Collaborative Study for Lysosomal Storage Disorders. We had anticipated asking you to complete the questionnaires at each of your annual review visits until 2011.
Unfortunately due to staffing issues we have been unable to meet with you and your child to give you the follow up questionnaires in person however we have enclosed them with this letter. The questionnaires are exactly the same as those you previously completed,
We have enclosed a stamped addressed envelope for you to return the completed questionnaires, however please be assured that you are under no obligation to agree to this or for your child to remain in the study.
If you have any questions or concerns regarding this, please free to contact one of the team.
If you would like to discuss any aspect of the study then please contact either of us at the address below, or talk to your clinician.
Best wishes
Name of treating consultant for that site and Chief Investigator
Letter for carer for follow-up postal questionnaires

Dear
We are very grateful to you for giving consent to participate in the National Collaborative Study for Lysosomal Storage Disorders on behalf of [patient name]. We had anticipated asking you to complete the questionnaires at each of [patient name]’s annual review visits until 2011.
Unfortunately due to staffing issues we have been unable to meet with you and [patient name] to give you the follow up questionnaires in person however we have enclosed them with this letter. The questionnaires are exactly the same as those you previously completed,
We have enclosed a stamped addressed envelope for you to return the completed questionnaires, however please be assured that [patient name] is under no obligation to agree to this or to remain in the study.
If you have any questions or concerns regarding this, please free to contact one of the team.
If you would like to discuss any aspect of the study then please contact either of us at the address below, or talk to your clinician.
Best wishes
Name of treating consultant for that site and Chief Investigator
Appendix 17 Final letters
Final letter for follow-up postal questionnaires

Dear
We are now coming towards the end of the National Collaborative Study for Lysosomal Storage Disorders and would like to take this opportunity to thank you for participating in this study.
We would also be extremely grateful if you would complete one final set of questionnaires which will help us compare your quality of life and health status now, with when you entered the study. The questionnaires are exactly the same as those you previously completed with the addition of a fatigue assessment. As these questionnaires are in addition to those given to you at your annual clinical review, we are asking for your additional agreement.
We have enclosed a stamped addressed envelope for you to return the completed questionnaires. However, please be assured that you are under no obligation to agree to this – your continuing treatment will not be affected in any way by your choice and no further communications regarding these questionnaires will be made if you do not return them to us.
We will shortly be analysing the data we have been collecting over the last couple of years, and we hope to send an abridged version of the study results to everyone who has participated in the study, by the end of next year.
Once again, we thank you for your time taken to complete these questionnaires and for your continued support of our research. If you would like to discuss any aspect of the study then please contact either of us at the address below, or talk to your clinician.
Best wishes
Name of treating consultant for that site and Chief Investigator
Final letter for follow-up postal questionnaires to parents

Dear
We are now coming towards the end of the National Collaborative Study for Lysosomal Storage Disorders and would like to take this opportunity to thank you and your child for participating in this study.
We would also be extremely grateful if you would complete one final set of questionnaires which will help us compare your child’s quality of life and health status now, with when they entered the study. The questionnaires are exactly the same as those you previously completed. As these questionnaires are in addition to those given to you at you’re your child’s annual clinical review, we are asking for your additional agreement.
We have enclosed a stamped addressed envelope for you to return the completed questionnaires. However, please be assured that you are under no obligation to agree to this – your child’s continuing treatment will not be affected in anyway by your choice and no further communications regarding these questionnaires will be made if you do not return them to us.
We will shortly be analysing the data we have been collecting over the last couple of years, and we hope to send an abridged version of the study results to everyone who has participated in the study, by the end of next year.
Once again, we thank you and your child for your time taken to complete these questionnaires and for your continued support of our research. If you would like to discuss any aspect of the study then please contact either of us at the address below, or talk to your child’s clinician.
Best wishes
Name of treating consultant for that site and Chief Investigator
Final letter for follow-up postal questionnaire for carers

Dear
We are now coming towards the end of the National Collaborative Study for Lysosomal Storage Disorders and would like to take this opportunity to thank you and [patient name] for participating in this study.
We would also be extremely grateful if you would complete one final set of questionnaires which will help us compare [patient name’s] quality of life and health status now, with when they entered the study. The questionnaires are exactly the same as those you previously completed with the addition of a fatigue assessment. As these questionnaires are in addition to those given to you at [patient name’s] annual clinical review, we are asking for your additional agreement.
We have enclosed a stamped addressed envelope for you to return the completed questionnaires. However, please be assured that you are under no obligation to agree to this – [patient name’s] continuing treatment will not be affected in any way by your choice and no further communications regarding these questionnaires will be made if you do not return them to us.
We will shortly be analysing the data we have been collecting over the last couple of years, and we hope to send an abridged version of the study results to everyone who has participated in the study, by the end of next year.
Once again, we thank you and [patient name] for your time taken to complete these questionnaires and for your continued support of our research. If you would like to discuss any aspect of the study then please contact either of us at the address below, or talk to [patient name’s] clinician.
Best wishes
Name of treating consultant for that site and Chief Investigator
Appendix 18 Mean annual drug cost to the NHS of enzyme replacement therapy by disorder and adult/child status
| LSD | Adults | Children | ||
|---|---|---|---|---|
| Annual cost (£) (ERT) | Annual cost (£) (alternative ERT drug) | Annual cost (£) (ERT) | Annual cost (£) (alternative ERT drug) | |
| Gaucher disease | 126,261 | 144,868 | 107,404 | 187,841 |
| Fabry disease | 120,840 | 108,242 | 89,199 | 79,478 |
| MPS I | 258,201 | 139,563 | ||
| MPS II | 537,605 | 314,004 | ||
| Pompe disease | 282,798 | 121,780 | ||
Appendix 19 Minimum additional discounted QALYs needed for each year on ERT for ERT to be judged as cost-effective
| LSD | Adults | Children | ||
|---|---|---|---|---|
| Annual cost (ERT) | Annual cost (alternative ERT drug) | Annual cost (ERT) | Annual cost (alternative ERT drug) | |
| Gaucher disease | 4.2 | 4.8 | 3.6 | 6.3 |
| Fabry disease | 4.0 | 3.6 | 3.0 | 2.6 |
| MPS I | 8.6 | 4.7 | ||
| MPS II | 17.9 | 10.5 | ||
| Pompe disease | 9.4 | 4.1 | ||
List of abbreviations
- α-Gal A
- α-galactosidase A
- ACE
- angiotensin-converting enzyme
- AE
- adverse event
- ALT
- alanine transaminase
- AST
- aspartate transaminase
- BBB
- blood–brain barrier
- BMD
- bone mineral density
- BMI
- body mass index
- BMT
- bone marrow transplant
- BPI
- Brief Pain Inventory
- CHO
- Chinese hamster ovary
- CI
- confidence interval
- CNS
- central nervous system
- CRF
- Case Report Form
- CRIM
- cross-reactive immunological material
- CSI
- Carer Strain Index
- CSRI
- Client Services Receipt Inventory
- CTS
- carpal tunnel syndrome
- DGJ
- 1-deoxygalactonojirimycin
- DQ
- development quotient
- DS
- dermatan sulphate
- EDC
- electronic data collection
- edf
- estimated degrees of freedom
- eGFR
- estimated glomerular filtration rate
- EMA
- European Medicines Agency
- ENT
- ear, nose and throat
- EQ-5D
- European Quality of Life-5 Dimensions
- ERT
- enzyme replacement therapy
- ESRD
- end-stage renal disease
- EU
- European Union
- FDA
- Food and Drug Administration
- FSS
- Fatigue Severity Scale
- FVC
- forced vital capacity
- GAG
- glycosaminoglycan
- Gb3
- globotriaosylceramide (also known as GL-3)
- GD1
- Gaucher disease type 1
- GD2
- Gaucher disease type 2
- GD3
- Gaucher disease type 3
- GP
- general practitioner
- Hb
- haemoglobin
- HCM
- hypertrophic cardiomyopathy
- HIS
- Hospital Information System
- HR
- hazard ratio
- HRQoL
- health-related quality of life
- HS
- heparan sulphate
- HSCT
- haematopoietic stem cell transplantation
- IDUA
- α-l-iduronidase
- IgG
- immunoglobulin G
- LSD
- lysosomal storage disorder
- LVM
- left ventricular mass
- LVMI
- left ventricular mass index
- MCS
- mental component score
- MPS I
- mucopolysaccharidosis type I
- MPS IH
- mucopolysaccharidosis type I subtype Hurler disease
- MPS IHS
- mucopolysaccharidosis type I subtype Hurler–Scheie syndrome
- MPS II
- mucopolysaccharidosis type II
- MPS IS
- mucopolysaccharidosis type I subtype Scheie disease
- MPS VI
- mucopolysaccharidosis type VI
- MREC
- Multicentre Research Ethics Committee
- MRI
- magnetic resonance imaging
- NCG
- National Commissioning Group
- NCS
- National Collaborative Study
- NICE
- National Institute for Health and Clinical Excellence
- NPC
- Niemann–Pick disease type C
- NSCAG
- National Specialist Commissioning Advisory Group
- NSCG
- National Specialised Commissioning Group
- NSCT
- National Specialised Commissioning Team
- OR
- odds ratio
- PCMD
- Peninsula College of Medicine & Dentistry
- PCS
- physical component score
- PedsQL
- Pediatric Quality of Life Inventory
- PIS
- Patient Information Sheet
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- SDV
- source data verification
- SF-36
- Short Form questionnaire-36 items
- SF-6D
- Short Form questionnaire-6 Dimensions
- SRT
- substrate reduction therapy
- TIA
- transient ischaemic attack
- t.i.d.
- three times daily
- VAS
- visual analogue scale
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Professor of Bio-Statistics, Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor Zarko Alfirevic, Head of Department for Women’s and Children’s Health, Institute of Translational Medicine, University of Liverpool
-
Professor Judith Bliss, Director of ICR-Clinical Trials and Statistics Unit, The Institute of Cancer Research
-
Professor David Fitzmaurice, Professor of Primary Care Research, Department of Primary Care Clinical Sciences, University of Birmingham
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, Department of Specialist Radiology, University College Hospital, London
-
Professor Angela Harden, Professor of Community and Family Health, Institute for Health and Human Development, University of East London
-
Dr Joanne Lord, Reader, Health Economics Research Group, Brunel University
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Dion Morton, Professor of Surgery, Academic Department of Surgery, University of Birmingham
-
Professor Gail Mountain, Professor of Health Services Research, Rehabilitation and Assistive Technologies Group, University of Sheffield
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Director, Centre for Healthcare Randomised Trials, Health Services Research Unit, University of Aberdeen
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Peter Tyrer, Professor of Community Psychiatry, Centre for Mental Health, Imperial College London
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Caroline Watkins, Professor of Stroke and Older People’s Care, Chair of UK Forum for Stroke Training, Stroke Practice Research Unit, University of Central Lancashire
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health