
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 08/226/01. The protocol was agreed in February 2011. The assessment report began editorial review in September 2011 and was accepted for publication in February 2012. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2012. This work was produced by Pavey et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to NETSCC. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2012 Queen’s Printer and Controller of HMSO
Chapter 1 Background
This report contains reference to confidential information provided as part of the NICE appraisal process. This information has been removed from the report and the results, discussions and conclusions of the report do not include the confidential information. These sections are clearly marked in the report.
Description of health problem
Chronic myeloid leukaemia (CML) is one of the blood cancers in which there is an overproduction of one type of white blood cell (WBC), the granulocytes, by the bone marrow. The typical CML progression course is triphasic: the chronic phase (CP), the accelerated phase (AP) and the blast-crisis (BC) phase, with the last two being grouped together as ‘advanced phase’. 1
Molecular mechanism
The molecular characteristic of CML is the presence of an acquired BCR–ABL fusion gene [oncogene fusion protein consisting of breakpoint cluster region (BCR) and Abelson oncogene (ABL)] in multipotent stem cells. More than 90% of people who are diagnosed with CML have an acquired (non-inherited) chromosomal abnormality caused by a reciprocal translocation between chromosomes 9 and 22 in an individual stem cell. The result is a shortened 22q, which is called the Philadelphia chromosome. 2,3 More specifically, the Abelson oncogene (ABL1), which is located on chromosome 9, translocates to the BCR gene on chromosome 22. The result is a fusion gene, BCR–ABL, and its corresponding protein, a constitutively active BCR–ABL tyrosine kinase. BCR–ABL tyrosine kinase is not controlled by normal cellular mechanisms and its presence leads to enhanced cell proliferation, resistance to apoptosis (programmed cell death) and genomic instability. These are key features in the pathophysiology of CML. 4,5 Within the CML population, approximately 10% of people do not have a demonstrable Philadelphia chromosome but have a complex of different translocations that still results in the formation of the BCR–ABL gene and its product. 6
Diagnosis
Chronic myeloid leukaemia is diagnosed by the presence of a characteristic pattern of cells in the blood and bone marrow in conjunction with specific cytogenetic and molecular abnormalities.
At presentation, patients typically have an enlarged spleen and a raised white cell count, with higher than normal numbers of immature WBCs. Bone marrow biopsy typically shows very little fat present and the bone marrow space occupied entirely by large numbers of leukaemia cells. 7
The presence of the Philadelphia chromosome is important both in terms of diagnosis and for monitoring responses to treatment. It is usually demonstrated by cytogenetic techniques that involve examining bone marrow cells in mitosis under a microscope to allow visualisation of metaphase chromosomes. 8 This test can also identify additional clonal chromosomal abnormalities in Philadelphia chromosome-positive (Ph+) cells (clonal cytogenetic evolution), which may be important indicators of prognosis. The technique requires at least 20–30 bone marrow cells in mitosis, which can be difficult to achieve. 9 There are considerable sampling errors because of the relatively small numbers of cells examined and the infrequency of measurement because bone marrow examination is a relatively invasive, although minor, procedure. The sensitivity is approximately 5% if 20 metaphase chromosomes are examined. 6
Fluorescence in situ hybridisation (FISH) is a sensitive and quantitative method that is used to detect specific chromosomal aberrations not only in cells undergoing metaphase, but also in interphase nuclei. 6,10 It uses specific fluorescent probes to map the chromosomal location of genes and identify other genetic abnormalities. In the case of CML, the probe looks for the BCR–ABL fusion gene in bone marrow or peripheral blood cells. 10 This test is usually performed in addition to the conventional cytogenetic test and uses approximately 200 bone marrow or blood cells for interphase FISH. 6 The limit of detection is between 1% and 5% abnormal cells. 6
Reverse transcriptase-polymerase chain reaction (RT-PCR) to detect BCR–ABL transcripts is also sometimes used to provide confirmation of diagnosis in CML. Through this technique, the level of BCR–ABL transcripts in peripheral blood or bone marrow is measured and one CML cell in 100,000 normal cells can be detected. 6 This qualitative technique is a simplified version of real-time quantitative polymerase chain reaction (qPCR), which is used to detect and quantify the level of BCR–ABL transcripts in a sample, and can be used to monitor disease progression and molecular response to treatment more closely. All of the above diagnostic techniques are currently recommended in the UK for the confirmation of CML diagnosis. 8
Natural history and clinical presentation
With the advent of a new class of drugs called tyrosine kinase inhibitors (TKIs) for the treatment of CML, imatinib being the first (see Imatinib, below), the natural history of the disease has been markedly changed. Current evidence suggests that patients whose disease responds favourably to treatment with imatinib may remain essentially symptom free for at least 10 years. 7 The following paragraphs describe the natural history of the disease in the absence of imatinib treatment.
Traditionally, CML has been regarded as a progressive disease that evolves through three phases. The initial chronic phase (CP) during which the disease is stable and slow to progress is followed after a variable interval by progression through an accelerated phase (AP) to a rapidly fatal blast crisis (BC). In approximately, one-third of patients there is no demonstrable AP, with the disease progressing directly from CP to BC. Transition between the phases may be gradual or rapid.
Chronic phase
Most people (approximately 90%) with CML are diagnosed during the CP. 1 Symptoms tend to be mild and non-specific and may include tiredness, anaemia, a feeling of ‘fullness’ or a tender lump on the left side of the abdomen caused by enlargement of the spleen, night sweats and weight loss. Approximately half of patients in the CP are asymptomatic and are diagnosed as a result of a routine blood test. 7
Hydroxycarbamide can be used to control the white blood count but does not alter the natural history of the disease. 11 In patients who are treated with hydroxycarbamide, the CP, although variable in length, typically lasts between 3 and 5 years, during which time the patient may be well, with stable WBC counts.
Accelerated phase
The AP lasts for 6–24 months, during which time progression is more rapid. The AP is associated with increases in the percentage of immature blast cells seen in blood and bone marrow rather than fully differentiated cells. 7 Evidence of cytogenetic abnormalities in addition to the Philadelphia chromosome (clonal evolution; see Table 1 for definition) is also an indication of disease progression. 12 New symptoms, such as bruising or bleeding and infections, may become apparent together with a worsening of additional symptoms. 13
| WHO criteria17 | Criteria used in recent trials21 |
|---|---|
| AP | |
| Blast cells in blood or bone marrow 10–19% | Blast cells in blood or bone marrow 15–29%; blast cells plus promyelocytes in blood or bone marrow > 30%, with blast cells < 30% |
| Basophils in blood 20% or more | Basophils in blood ≥ 20% |
| Persistent thrombocytopenia (platelet count < 100 × 109/l) uncontrolled by therapy | Persistent thrombocytopenia (platelet count < 100 × 109/l) unrelated to therapy |
| Thrombocytosis (platelet count > 1000 × 109/l) unrelated to therapy | Not included |
| Increasing spleen size and increasing WBC count unresponsive to therapy | Not included |
| Cytogenetic evidence of clonal evolution (the appearance of additional genetic abnormalities that were not present at the time of diagnosis) | |
| BC | |
| Percentage of blast cells in blood or bone marrow (≥ 20%) | Percentage of blast cells in blood or bone marrow (≥ 30%) |
| Extramedullary blast proliferation or large foci or clusters of blasts in the bone marrow biopsy | Extramedullary blast involvement excluding the liver and spleen |
Blast crisis
Also known as blastic phase, the BC is usually fatal within 3–6 months of onset. 7 This phase is characterised by the rapid expansion of a population of differentiation-arrested blast cells (immature and non-functioning cells). So much of the bone marrow becomes replaced with immature cells that the other blood cells are prevented from functioning. An increased proportion of blast cells are found in blood and bone marrow, and blast cells may also spread to tissues and organs beyond the bone marrow (extramedullary blast involvement). The BC may be associated with significant symptoms, including fever, sweats, pain, weight loss, hepatosplenomegaly, enlarged lymph nodes and extramedullary disease. 13–15
Although the three phases of CML are well recognised clinically, there are several descriptions of defining criteria available in the literature. Varying definitions have been used in clinical trials. In 2001, the World Health Organization (WHO) proposed a new classification system with the intention to refine the criteria for AP and BC. 16 The fourth edition of this document was released in October 2008. 17 Table 1 describes the criteria used to define the AP and BC recommended by the WHO and those used in a recent single-arm clinical study of nilotinib; however, the trials in this report and other current single-arm studies do not report their criteria. 18–21 The implication is that more stringent criteria may be used in current trials.
Epidemiology of chronic myeloid leukaemia
Incidence
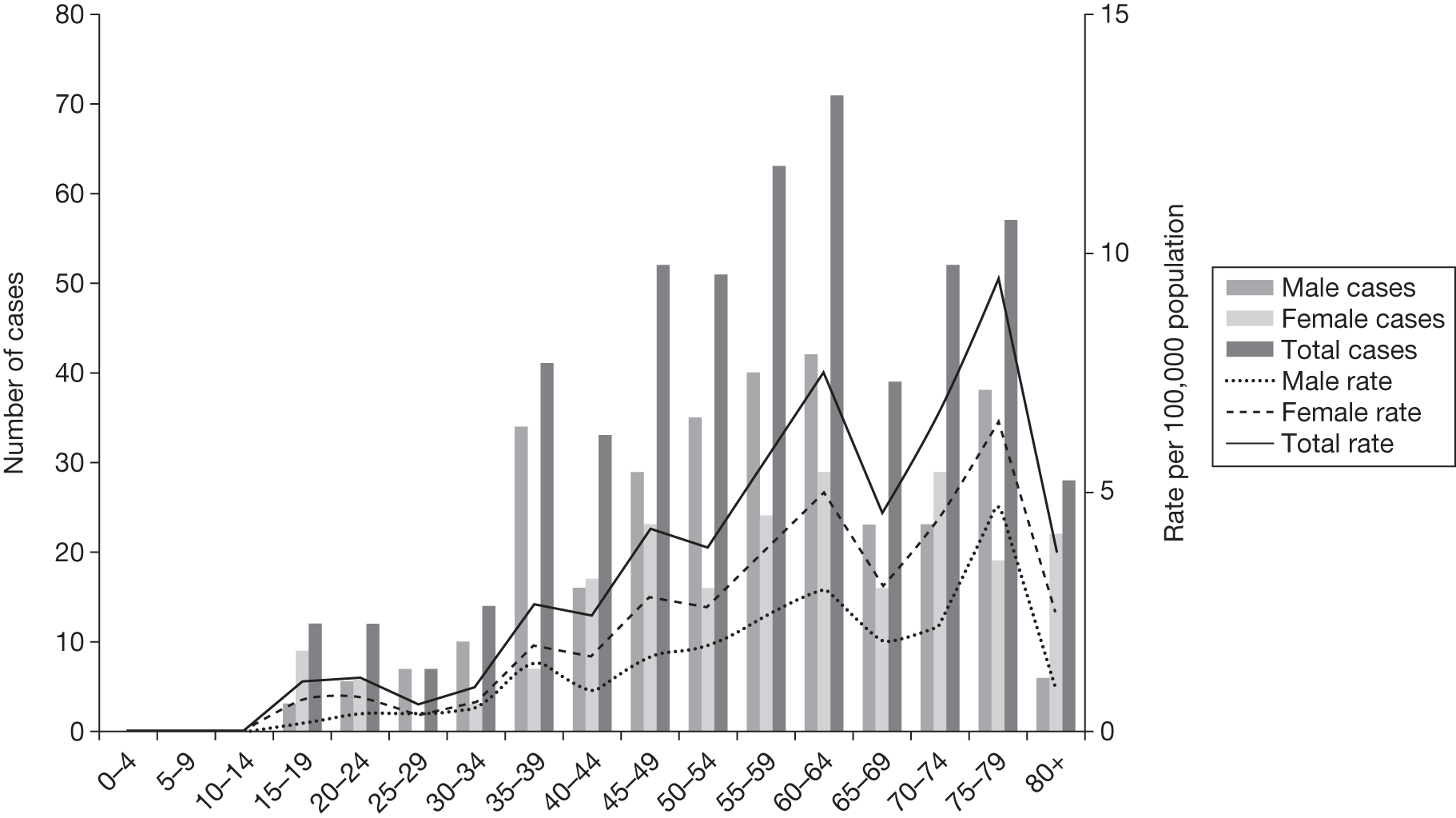
The Haematological Malignancy Research Network (HMRN), based in Yorkshire, estimates that 530 cases of CML are newly diagnosed in the UK each year, an annual age-standardised rate of 1.1 per 100,000 for men and 0.7 per 100,000 for women. 22
Figure 1 shows the annual estimated incidence of CML in the UK with age and sex distributions. The data are extrapolated from those collected within the HMRN region, whose population of 3.7 million is broadly representative of the UK as a whole. Approximately 60% of those diagnosed with CML are male. CML occurs in all age groups, although it is uncommon in those aged < 30 years; the median age at diagnosis is 58 years (this includes all phases). 22
FIGURE 1.
Annual estimated incidence in the UK, by age and sex. Source: Haematological Malignancy Research Network (www.hmrn.org).
Prognosis
There are two prognostic staging scores for CML in common use: the Sokal score23 and the Euro or Hasford score. 24 Details of how the scores are calculated are shown in Table 2. Both scores are used to determine if a patient is at low, intermediate or high risk of death and may also predict response to treatment. Both must be applied at diagnosis, prior to any treatment. The Sokal score is based on age, spleen size, and platelet and peripheral blood blast count. The Hasford score also includes data on eosinophil and basophil counts. The level and timing of haematological, cytogenetic and molecular responses provides important prognostic information and it is a widely accepted goal for patients to achieve a complete cytogenetic response (CCyR) within 18 months of CML therapy. 1,6 Both scores were developed prior to the introduction of tyrosine kinase inhibitors (TKIs) [the Hasford score in response to improvements in survival seen with interferon (IFN) treatment] but they appear to have some value in predicting response to treatment with TKIs.
| Patient characteristics | Calculation using the Sokal score23 | Calculation using the Hasford score24 |
|---|---|---|
| Age | 0.116 × (age – 43.4) | 0.666 when age ≥ 50 years |
| Spleena | 0.0345 × (spleen – 7.51) | 0.042 × spleen |
| Platelet count, × 109/l | 0.188 × [platelet count/700)2 – 0.563] | 1.0956 when platelet count ≥ 1500 × 109/l |
| Blood myeloblasts, % | 0.0887 × (myeloblasts – 2.10) | 0.0584 × myeloblasts |
| Blood basophils, % | NA | 0.20399 when basophils > 3% |
| Blood eosinophils, % | NA | 0.0413 × eosinophils |
| RRb | ||
| Low | < 0.8 | ≤ 780 |
| Intermediate | 0.8 to 1.2 | 781 to 1480 |
| High | > 1.2 | > 1480 |
At the 18-month follow-up of the IRIS trial26 (International Randomised Study of Interferon versus STI571) of imatinib and IFN, 49%, 67% and 76% of people with high-, intermediate- and low-risk Sokal scores, respectively, had achieved a CCyR. This relationship was maintained at the 48-month update with patients with a high Sokal score having a 69% probability of achieving a complete cytological response compared with 84% and 91% for patients with intermediate- and low-risk scores, respectively. 27 A similar relationship was seen with molecular response at 12 months; 38% of those in the high-risk group had a reduction from baseline of at least 3-log in BCR–ABL transcripts compared with 45% in the intermediate-risk group and 66% of those in the low-risk group (p = 0.007). 28
The trials in this assessment, ENESTnd (Evaluating Nilotinib Efficacy and Safety in clinical Trials – Newly Diagnosed patients) and DASISION (Dasatinib vs Imatinib in Patients With Newly Diagnosed Chronic Phase CML), use the Sokal and Hasford staging scores, respectively. 20,29 The ENESTnd study reported at 12 months that rates of CCyR for study arms nilotinib (300 mg), nilotinib (400 mg) and imatinib (400 mg) were 74%, 63% and 49%, respectively, for patients at high risk (Sokal). Rates of major molecular response (MMR) for study arms nilotinib (300 mg), nilotinib (400 mg) and imatinib (400 mg) were 41%, 32% and 17%, respectively, for patients at high risk (Sokal).
The DASISION study29 reported at 12 months that rates of CCyR for study arms dasatinib (100 mg) and imatinib (400 mg) were 78% and 64%, respectively, for patients at high risk (Hasford). Rates of MMR for study arms dasatinib (100 mg) and imatinib (400 mg) were 31% and 16%, respectively, for patients at high risk (Hasford) (see Chapter 3, Complete cytogenetic response and Major molecular response for full results). Comparability between ENESTnd20 and DASISION29 risk score responses should be treated with caution, with between-trial differences potentially resulting from the different risk group scoring systems adopted.
Survival
The most recently available survival statistics for all leukaemia in the UK are based on data collected from 2001–7. 30 The 5-year relative survival (survival of patients taking into account other causes of death) rate was 39.7% for men and 41.0% for women up to 2006, with a predicted rate of 42.7% for 2007. 30 The predicted 10-year survival rate for 2007 was 33.8% for men and 35.3% for women. 30 With fewer survival statistics available for CML, the IRIS trial26 of imatinib (see Imatinib, below) reports overall survival (OS) at 8 years of 85% for patients receiving imatinib.
Recent analysis of survival among patients with CML in the USA, derived from the 1973–2006 limited-use database of the Surveillance, Epidemiology and End Results Program of the United States National Cancer Institute suggests a dramatic recent increase in long-term survival for people with CML since the introduction of imatinib into routine clinical practice. 31 For all age groups combined, 5-year relative survival increased from 32.5% in 1990–2 to 54.6% in 1999–2006 (p < 0.05). For the period 1999–2006, 5-year relative survival was approximately 78.0% for age groups 15–44 year and 45–54 years, 63% for 55–64 years, 39.5% for 65–74 years and 24.7% for the ≥ 75 years age group. 31 There were indications from the data of improvements in long-term survival in the older age groups but long-term prognosis remained poor and essentially unchanged for the oldest patients (≥ 75 years age group). 31
Disease monitoring and treatment response
Disease monitoring plays a key role in assessing response to therapy and detecting early relapse. Several measures of disease status are used for monitoring: blood counts (haematological response), the proportion of Philadelphia chromosomes in bone marrow aspirate [cytogenetic response (CyR)] and the presence or absence (qualitative molecular response) and number (quantitative molecular response) of BCR–ABL transcripts in peripheral blood and bone marrow using PCR technology. In clinical trials, CyRs are variously defined as complete, partial, overall, major and minor, and the definitions vary according to the phase of the disease in which a patient is diagnosed (Table 3).
| Response | Percentage of Ph+ chromosomes in metaphase in bone marrow (%) |
|---|---|
| Complete (major) | None |
| Partial (major) | 1–35 |
| Minor | 36–65 |
| Minimal | 66–95 |
| None | > 95 |
The following definitions are commonly used to describe response in chronic disease.
Haematological response
Classification of haematological response varies widely among trials. Hochhaus and colleagues32 provide a definition of a complete haematological response (CHR) as:
-
WBC count no more than the upper limit of normal
-
absolute neutrophil count of at least 1 × 109/l
-
platelet count of < 450 × 109/l and no more than the institutional upper limit of normal
-
no blasts or promyelocytes in peripheral blood
-
< 2% basophils in peripheral blood
-
no extramedullary involvement, with all of these being maintained for 4 weeks.
Other trials have used variations of this definition, including some or all of the elements. The trials in this assessment do not report haematological response.
Cytogenetic response
The definition of CyR appears to be fairly standard across most trials and is split into complete, partial, minor, minimal and none (see Table 3). A CCyR is defined as absence of the Philadelphia chromosome among at least 20 cells in metaphase in a bone marrow aspirate. 32 A commonly used additional term is major CyR, which encompasses complete and partial.
Molecular response
In people with a CCyR, quantitative PCR techniques can be used to monitor the level of BCR–ABL transcripts in peripheral blood (and sometimes bone marrow). A complete molecular response (CMR) has been defined as undetectable levels of BCR–ABL transcripts in an assay that can detect a reduction from baseline of at least 4.5 logs. A MMR is a standardised BCR–ABL : ABL ratio of < 0.1%, which is equivalent to a 3-log reduction from the 100% baseline for untreated patients. 28,33
Surrogate outcomes
In the absence of long-term follow-up, the above measures of treatment response may be regarded as ‘surrogate outcomes’ for patient-relevant outcomes [OS, disease progression, quality of life (QoL)], with CyR and molecular response used as the primary outcomes in current trials. 18–21 The use of surrogate outcomes rather than more patient-relevant outcomes may be easier, more economical and provide earlier results. 34 This can lead to faster licensing time and dissemination of new treatments. 35 The use of surrogate outcomes is essential in Phase II and Phase III trials aiming to establish a drug’s potential benefit. 36 However, the use of surrogate outcomes can also be harmful when there is a lack of an independent causal association between a change in the surrogate outcomes and a change in the patient-relevant outcomes, thus the evaluation and validation of using a surrogate outcome is warranted. 34–36 The value of surrogate outcomes can be judged against a hierarchy of evidence, which ranges from biologically plausible relationships (weak evidence) to changes in the surrogate corresponding to equal changes in the patient-relevant outcome, assessed by clinical trials (strong evidence). 34,35
Schrover et al. 37 reported on the development of a predictive survival model for patients with chronic myeloid leukaemia in chronic phase (CP-CML), according to CyR rates in seven IFN-based RCTs. 37 They estimated a weighted odds ratio – for the survival of patients who achieved a major cytogenetic response (MCyR) when compared with those who did not – of 7 (95% CI 5 to 11) at 2 years and 5 (95% CI 3 to 8) at 4 years. Median survival was increased by 1.8 years for every 25 percentage point increase in MCyR rate. The predictive model reported by Schrover et al. 37 provides support for CyR predicting long-term survival within IFN class treatments for CP-CML. The evidence for the use of surrogate outcomes within the TKI (imatinib, dasatinib and nilotinib) class of CP-CML treatment is unclear, and may not be available particularly for the newer second-generation TKIs. Therefore, only imatinib may provide evidence for the use of surrogate outcomes within the TKI drug class (see Chapter 4, Assessment of evidence to support the use of complete cytogenetic response and major molecular response as surrogate outcomes).
Disease progression
Typically, disease progression describes the process in which the disease develops into the AP or to BC. Differences in the definition of AP have resulted in the use of more specific definitions of disease progression. The definition of progression used in a trial of this assessment relies on participants meeting any one of the four criteria:20
-
development of AP or BC CML
-
loss of CHR
-
increase in Ph+ bone marrow metaphases to more than 35%
-
increasing WBC count (a doubling of white cell count to > 20 × 109/l) in the absence of complete haematological response.
Treatment
Allogeneic stem cell transplant
Currently, the only known curative treatment for CML is allogeneic haematopoietic stem cell transplantation (alloHSCT), either from a matched related or unrelated donor. 38,39 Patient age, disease phase and duration, the degree of mismatch between patient and donor and therapy before transplantation all influence outcome. Younger patients in CP receiving a transplant from a matched sibling donor soon after diagnosis have the best prognosis. 40 Two studies have shown similar outcomes for transplantation in patients with CP-CML using either a fully matched related or unrelated donor, with 5-year survival rates of > 70% for people aged ≤ 50 years who undergo transplantation within a year of diagnosis. 41,42 Results are less promising for those in AP and BC. 39
The morbidity and mortality of alloHSCT is considerable; transplant-related mortality ranges from 15% to 40%. 43
Allogeneic haematopoietic stem cell transplantation is not a treatment option for many people, either for reasons related to age at diagnosis (the median age for diagnosis of CML is 59 years, and many patients are considered to be unsuitable for a transplant at diagnosis) or because of lack of a suitable donor. 6 UK recommendations propose the use of alloHSCT with failure of imatinib and or second-generation TKIs, or where younger patients have progressed to the AP. 8
Medical treatment
UK guidelines (see Chapter 1, Current service provision) recommend imatinib as a first-line treatment for CML in the CP.
Imatinib
Imatinib [originally STI571; trade name Gleevec® (USA) or Glivec® (Europe/Australia/Latin America), Novartis] is an orally administered TKI.
Pharmacology
Imatinib is a first-generation TKI, specifically designed to inhibit the BCR–ABL fusion protein by occupying the ATP-binding pocket of the ABL–kinase domain. This prevents a change in conformation of the protein to the active form of the molecule. By blocking the ATP-binding site, imatinib reduces cell proliferation and stops disease progression. 5
Licensing
In the UK, imatinib is licensed (since 7 November 2001) for the treatment of adults with CP, AP or BC CML. Imatinib has also received approval for this indication by the Food and Drug Administration (FDA) and EMA. Imatinib has orphan drug status.
Adverse events
The adverse events (AEs) of imatinib treatment are reported in detail in Chapter 3 (see Adverse events). The most common serious side effects (seen in more than 1 in 10 patients) are weight increase, headache, nausea, vomiting, diarrhoea, dyspepsia, abdominal pain, oedema, rash, muscle cramps and spasms, fatigue, neutropenia (low WBC counts), thrombocytopenia (low blood platelet counts) and anaemia (low red blood cell counts). 44
Dose
For patients with CP, the recommended dose for adults is 400 mg taken once a day, increased if required to 800 mg daily, in divided doses. For AP or BC, the recommended dose is 600 mg once daily, increased if required to 800 mg daily, in divided doses. The dose can be altered based on patient response. 44
Cost
According to the current edition of the Monthly Index of Medical Specialities (MIMS) (July 2011), the cost of treatment with imatinib at doses of 400 mg, 600 mg and 800 mg per day is £57.48, £86.22 and £114.96, respectively. 45 These prices reflect the 7% increase as of April 2011.
Efficacy
The efficacy data for imatinib is based on a large open-label, randomised controlled trial (RCT; IRIS)26 in which a total of 1106 people with newly diagnosed, CP-CML received either imatinib or interferon alpha (IFN-α) plus low-dose cytarabine. 26 After a median follow-up of 19 months, the estimated rate of a MCyR at 18 months was 87.1% in the imatinib group and 34.7% in the control group (p < 0.001). Corresponding figures for a CCyR were 76.2% and 14.5%, respectively (p < 0.001). 26
Patients who received imatinib continue to be followed up; after a median follow-up of 60 months, Kaplan–Meier estimates of cumulative CCyR rates were 87.0%. An estimated 7% of patients had progressed to AP CML or BC, and the estimated OS of patients who received imatinib as initial therapy was 89.0%. 27
The most recent data from key imatinib trials, at 8 years’ follow-up, show that 55.0% of patients randomised to imatinib remained on treatment. Event-free survival (EFS; prespecified event while on therapy, e.g. loss of CHR or CCyR, discontinuation due to toxicity, progression to accelerated/blast phase, death) was 81%, no disease progression to AP or BC was 92% and OS was 85% (93% for CML-related deaths only and patients prior to SCT). 46 The annual rates of progression to AP or BC in years 4–8 after initiation of therapy were 0.9%, 0.5%, 0%, 0% and 0.4%, respectively. However, with the high crossover rate of the IFN arm, comparison results were not reported.
There are serious limitations in the interpretation of these results, as 45% of the patients had abandoned the study by 8 years and patients were censored at the moment of discontinuing imatinib. This particularly affects those patients who discontinued imatinib because of intolerance and patients who failed to achieve a CyR and abandoned the study to receive other therapies before having an ‘event’. Consequently, the OS and EFS reported in the IRIS study26 are likely to be substantial overestimates. Marin et al. 47 report an intention-to-treat (ITT) analysis in 204 patients treated with imatinib 400 mg/day as first-line therapy. 47 In the study, the 5-year probabilities of CCyR, MMR, OS, progression-free survival (PFS), and EFS were similar to the ones reported in the IRIS study. 26 For example, the EFS (defined as in the IRIS study26) was 81.3% [confidence interval (CI) 73.0% to 87.5%], which is similar to the 83% rate in the IRIS study. 26 However, with EFS redefined to include as ‘event’ those patients who had discontinued imatinib due to toxicity or lack of a CyR, the recalculated EFS was 62.7%. In other words, the probability of having abandoned the imatinib therapy at 5 years due to toxicity, progression or unsatisfactory response was 37.3%.
Notwithstanding this, it has been recently shown that patients taking imatinib who achieve a durable CMR can potentially stop treatment without molecular relapse. Mahon et al. 48 showed patients with a median of 50 months’ imatinib therapy had molecular-free relapse rates of 41% at 12 months and 38% at 24 months after discontinuation of imatinib. 48
The definition of longer-term treatment end points used by TKI studies will have an impact on perceived differences between trials. Based on 435 patients with early CP-CML, Kantarjian et al. 49 recently showed PFS/EFS rates of 96%, 90%, 89% and 81% when applying different definitions in the research literature. The definitions are drawn from the researchers’ own centre, IRIS26 and the two studies included in this review, DASISION29 and ENESTnd. 20 It was concluded that uniform definitions of PFS and EFS are needed.
Description of new interventions
Nilotinib and dasatinib were initially developed for the treatment of patients who are resistant or intolerant to imatinib, and were selected due to their potency and activity against mutated forms of BCR–ABL1. 50 Nilotinib and dasatinib are now being considered as alternative treatments to imatinib as a first-line treatment.
Two Phase II trials18,21 report efficacy data for nilotinib. Rosti et al. 21 reported on 73 CP untreated patients with Ph+ CML (nilotinib, 400 mg twice daily): 97% showed complete haematological response, 96% achieved CCyR and 85% achieved a MMR, at 12 months. At 3 months, 78% achieved CCyR and 52% MMR. Cortes et al. 18 reported that of 51 patients with early CP-CML, observed for at least 3 months (nilotinib, 400 mg twice daily), 98% achieved a CCyR, and 76% achieved a MMR. Responses occurred rapidly, with 96% of patients achieving CCyR by 3 months and 98% achieving CCyR by 6 months.
A similar study of dasatinib by Cortes et al. 19 reported on 50 patients with early CP-CML who were observed for at least 3 months (dasatinib, 100 mg once daily or 50 mg twice daily): 98% achieved a CCyR and 82% achieved a MMR, with 94% of patients achieving CCyR by 6 months.
Dasatinib
Dasatinib (BMS-354825; trade name Sprycel®, Bristol-Myers Squibb) is a second-generation TKI.
Pharmacology
Dasatinib is a highly potent, orally active TKI, which can bind to both the active and inactive conformation of the ABL kinase domain. 6,51 In vitro, dasatinib is shown to be active against almost all imatinib-resistant BCR–ABL mutations and is 350 times more potent than imatinib. 52,53
Licensing
Since 2006, the EMA has approved dasatinib for the treatment of adults with CP, AP or BC CML with resistance or intolerance to prior therapy including imatinib. In December 2010, the EMA extended the licence for its use as a first-line treatment for adults newly diagnosed with CP-CML. Dasatinib has also received approval for this indication by the FDA (October 2010). Dasatinib has orphan drug status.
Adverse events
The AEs of dasatinib treatment are reported in detail in Chapter 3 (see Adverse events). The most common (seen in more than 1 in 10 patients) reported side effects in the trials are headache, pleural effusion, shortness of breath, cough, diarrhoea, nausea, vomiting, abdominal pain, skin rash, musculoskeletal pain, infections, haemorrhage, superficial oedema (swelling), fatigue, fever, neutropenia (low WBC counts) and thrombocytopenia (low blood platelet counts) and anaemia (low red blood cell counts). 54 Grade 3 and 4 haematological AEs in recent trials were approximately 21%, 10–19% and 6–10% for neutropenia, thrombocytopenia and anaemia, respectively. 19,20
Dose
For patients in CP, the recommended dose for adults > 18 years is 100 mg taken once a day, increased if required to 140 mg once a day. For AP or BC the recommended dose is 140 mg once daily, increased if required to 180 mg once a day. The dose can be altered based on patient response. 54
Cost
According to BNF 61 (March 2011), the cost of treatment with dasatinib at a dose of 100 mg once a day is £83.50 per day (140 mg – £116.90; 180 mg – £150.30), and is available as 20-, 50-, 70- and 100-mg tablets. 55
Nilotinib
Nilotinib (AMN107; trade name Tasigna®, Novartis) is a second-generation TKI.
Pharmacology
Nilotinib is an orally active phenylaminopyrimidine derivative of imatinib and is approximately 10–50 times more potent than imatinib at inhibiting BCR–ABL. 56 Studies performed in vitro suggest that nilotinib inhibits 32 of 33 mutant BCR–ABL forms resistant to imatinib at physiologically relevant concentrations. 57,58 Nilotinib, like imatinib, binds to the inactive conformation of ABL, but with a slightly better topographical fit. 15
Licensing
Since 2007, the EMA has approved nilotinib for the treatment of adults with CP and AP Ph+ CML with resistance or intolerance to prior therapy including imatinib. Nilotinib has not been approved for use in the BC. In September 2010, the EMA extended the licence for its use as a first-line treatment for adults who were newly diagnosed with CP-CML. Nilotinib has also received approval for this indication by the FDA (June 2010). Nilotinib has orphan drug status.
Adverse events
The AEs of nilotinib treatment are reported in detail in Chapter 3 (see Adverse events). The most common side effects with nilotinib (reported by > 1 patient in 10) are headache, nausea (feeling sick), constipation, diarrhoea, rash, pruritus (itching), fatigue (tiredness) and increased blood levels of lipase (an enzyme produced by the pancreas) and bilirubin, thrombocytopenia (low blood platelet counts), neutropenia (low WBC counts) and anaemia (low red blood cell counts). 59 Grade 3 and 4 haematological AEs in recent trials were approximately 12%, 12% and 5% for neutropenia, thrombocytopenia and anaemia, respectively. 18,20 The FDA has stipulated that nilotinib carry a ‘black box’ warning for possible heart problems due to QTc (the time between the start of the Q wave and the end of the T wave of a heart’s electrical cycle, corrected for the person’s heart rate) prolongation, that may lead to an irregular heart beat and possibly sudden death. Nilotinib has been shown to prolong cardiac ventricular repolarisation, which can result in ventricular tachycardia and death. Nilotinib should not be used in patients who have hypokalaemia, hypomagnesaemia or long QT syndrome. 60
Dose
In newly diagnosed patients with CP-CML, the recommended dose is 300 mg twice a day. The recommended starting dose for patients with CP or AP CML who do not respond to, or tolerate, other treatments is 400 mg twice daily. 59
Quality of life
Assessment of health-related quality of life (HRQoL) has become an important feature of cancer trials, enabling evaluation of treatment effectiveness from the perspective of the person with the condition and facilitating improved clinical decision-making.
There are several general HRQoL instruments for people with cancer that can be used to assess quality of life both in research studies and in clinical practice, for example the Functional Assessment of Cancer Therapy scale and the European Organisation for Research and Treatment of Cancer quality-of-life questionnaire QLQC-30. Disease-specific instruments for CML appear not to have been widely used in clinical trials.
A recent systematic review of HRQoL in CML highlighted the relative paucity of research and methodological shortcomings in this area. 61 Only one study identified addressed the effect of a TKI on QoL, with imatinib shown to be superior to IFN in terms of HRQoL, but this was measured only in the first year of treatment. 62 The review concluded that monitoring of HRQoL and side effects of CML treatment from the patient’s perspective will be of importance to determine the net clinical benefit of new therapies. 61 Assessment of QoL in CML is further discussed in Chapter 3 (see Health-related quality of life).
Current service provision
In 2009, the European LeukaemiaNet recommend imatinib 400 mg daily as a first-line treatment for all patients in the CP, with dasatinib, nilotinib or higher-dose imatinib as second-line treatment. Third-line treatment is continued dasatinib or nilotinib, with an option for alloHSCT, and alloHSCT after dasatinib or nilotinib failure. 50 In 2007, the British Committee for Standards in Haematology also recommended imatinib daily as a first-line treatment for all patients, with higher-dose imatinib or dasatinib and potentially nilotinib as second-line treatments. 8 These guidelines are due to be updated in July 2012. The National Institute for Health and Clinical Excellence (NICE) guidance on CML (TA70–2003) recommends imatinib for the first-line treatment of adults with the Philadelphia-chromosome type of CML in the CP.
Current use of new interventions in the National Health Service
Anecdotal evidence suggests that dasatinib and nilotinib are currently widely used in the NHS in England and Wales following failure of treatment with imatinib. NICE has recently provided guidance on the use of nilotinib or dasatinib as second-line treatment of CML. In the draft guidance on 18 August 2011, NICE recommended nilotinib for the treatment of the CP and AP of CML that is resistant or intolerant to standard-dose imatinib. Dasatinib and high-dose imatinib are not recommended in the draft guidance. Consultees have the opportunity to appeal against the draft guidance. Until NICE issues final guidance, NHS bodies should make decisions locally on the funding of specific treatments. This draft guidance does not mean that people currently taking dasatinib or high-dose imatinib will stop receiving them. They have the option to continue treatment until they and their clinicians consider it appropriate to stop.
Chapter 2 Definition of the decision problem
The purpose of this technology assessment report is to assess the clinical effectiveness and cost-effectiveness of dasatinib, nilotinib and imatinib (standard dose) for the first-line treatment of CML. Decision modelling to estimate the cost-effectiveness of alternative ways of using health technologies should start with a clearly defined decision problem.
Decision problem
A decision problem comprises a clear definition of (1) the targeted patient population and health problem, (2) the alternative treatment pathways to which they might be exposed, and (3) the main outcomes against which those pathways will be compared.
Interventions and comparators
Table 4 shows the three treatment pathways that will be evaluated using the decision model, assuming that second-line use of TKIs is or is not available within the NHS. For a description of how these admittedly simplified treatment sequences were arrived at, please see the cost-effectiveness analysis methods (see Chapter 7, Approaches to modelling treatments for chronic myeloid leukaemia).
| Treatment pathways to be compared by the decision model, without second-line use of TKIs (scenarios 1 and 2) | ||||
|---|---|---|---|---|
| No. | Initial (‘first-line’) treatment | Second-line treatment in CP (if first-line fails/intolerant) | Treatment in AP or BC | |
| 1 | Dasatinib, 100 mg (or 140 mg if required) once daily | Either stem cell transplant or hydroxycarbamide | Hydroxycarbamide + medical management | |
| 2 | Nilotinib, 300 mg twice daily | |||
| 3 | Imatinib, 400 mg once daily | |||
| Treatment pathways to be compared by the decision model, with nilotinib available as second-line treatment (scenarios 3 and 4) | ||||
| No. | Initial (‘first-line’) treatment | Second-line treatment in CP (if first-line fails/intolerant) | Third-line treatment in CP (if second-line fails) | Treatment in AP or BC |
| 1 | Dasatinib, 100 mg (or 140 mg if required) once daily | Nilotinib 400 mg twice daily | Either stem cell transplant or hydroxycarbamide | Hydroxycarbamide + medical management |
| 2 | Nilotinib, 300 mg twice daily | Either stem cell transplant or hydroxycarbamide | Not applicable | |
| 3 | Imatinib, 400 mg once daily | Nilotinib 400 mg twice daily | Either stem cell transplant or hydroxycarbamide | |
Apart from those relating to cytogenetic or molecular response at 12 months, no important and statistically significant subgroup differences emerged in the clinical effectiveness evidence.
Population
Adults with newly diagnosed CP, Ph+ CML. If possible, newly diagnosed CP-CML without genetic mutation (non-Philadelphia chromosome) will also be considered. In reality, for consistency, the patient population modelled will have to closely mirror the populations in the main trials from which the effectiveness estimates are derived.
Outcomes
The main outcomes that will determine the development of the decision model are:
-
lifetime quality-adjusted life-years (QALYs)
-
lifetime care costs [NHS and Personal Social Services (PSS)].
However, the modelling may also usefully estimate the following outcomes in the short or long term:
-
PCR
-
time to progression
-
OS
-
response rates: cytogenetic, molecular and haematological
-
time to treatment failure
-
adverse effects of treatment.
Overall aims and objectives of assessment
This technology assessment reviews the available evidence for the clinical effectiveness and cost-effectiveness of dasatinib, nilotinib and imatinib (standard dose) for the first-line treatment of Ph+ CML according to their marketing authorisation. The assessment draws on relevant evidence to determine what, if any, is the clinical effectiveness and cost-effectiveness of the interventions compared with each other in the CP. The policy questions addressed are as follows.
In CP:
-
What is the clinical effectiveness of first-line treatment for newly diagnosed Ph+ CML with dasatinib or with nilotinib or with imatinib (standard dose), using each of the three treatments as comparators?
-
What is the cost-effectiveness of first-line treatment for newly diagnosed Ph+ CML with dasatinib or with nilotinib or with imatinib (standard dose), using each of the three treatments as comparators?
Chapter 3 Assessment of clinical effectiveness
The clinical effectiveness of dasatinib, nilotinib and imatinib was assessed by a systematic review of published evidence. The review was undertaken following the general principles published by the NHS Centre for Reviews and Dissemination (CRD) and the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines. 63,64
Methods for reviewing effectiveness
Identification of studies
The search strategy comprised of the following main elements:
-
searching of electronic databases
-
contact with experts in the field
-
scrutiny of bibliographies of retrieved papers and manufacturer submissions
-
follow-up on mentions of potentially relevant ongoing trials noted in previous National Institute for Health and Clinical Excellence (NICE) guidance on imatinib for CML.
The main electronic databases of interest were MEDLINE (Ovid); EMBASE; The Cochrane Library including the Cochrane Database of Systematic Reviews (CDSR), Cochrane Central Register of Controlled Trials (CCRCT), Database of Abstracts of Reviews of Effects (DARE), NHS Economic Evaluation Database (NHS EED) and Health Technology Assessment (HTA) databases; National Research Register (NRR); Web of Science [including Conference Proceedings Citation Index (CPCI)]; Current Controlled Trials (CCT); ClinicalTrials.gov; FDA website; and the European Medicines Agency (EMA) website. These were searched from search end date of the last technology appraisal report on this topic, October 2002. 65
The searches were developed and implemented by a trained information specialist (CC) using the search strategy detailed in the technology appraisal by Thompson-Coon et al. 66 as the starting point (see Appendix 1 for full search strategy). This strategy was reviewed by PenTAG, including a clinical expert (CR).
Relevant studies were identified in two stages using predefined inclusion and exclusion criteria (see Appendix 2 for full research protocol). One reviewer (TP) examined all titles and abstracts, with two reviewers (TJ-H and LC) each examining approximately 50% each of all titles and abstracts (therefore all titles and abstracts were examined by at least two reviewers). Full texts of any potentially relevant studies were obtained. The relevance of each paper was assessed independently by two reviewers (TP and TJ-H) and any discrepancies resolved by discussion.
Inclusion and exclusion criteria
Inclusion criteria
For the review of clinical effectiveness, in the first instance, only systematic reviews of randomised controlled trials and RCTs were considered. However, if key outcomes of interest were not measured at all in the included RCTs, we discussed extending the range of included studies to other study designs. Other study designs were not required after scrutiny of the included RCTs. The systematic reviews were used as a source for finding further studies and to compare with our systematic review. Systematic reviews provided as part of manufacturers’ submissions were treated in a similar manner.
Population
Adults with CP-CML, naive to any treatment specifically directed against CML.
Interventions
-
dasatinib
-
nilotinib
-
imatinib (400-mg standard dose).
Each should be used in accordance with the marketing authorisation and in the populations indicated in Chapter 1 (see Description of new interventions), noting that CML without genetic mutation is outside the existing marketing authorisations.
Comparators
Imatinib or nilotinib when the intervention is dasatinib; imatinib or dasatinib when the intervention is nilotinib; dasatinib or nilotinib when the intervention is standard-dose imatinib.
Outcomes
All potentially relevant outcomes in the included studies were considered, particularly those capturing:
-
response rates – cytogenetic, molecular and haematological
-
EFS
-
PCR
-
time to progression
-
OS
-
time to treatment failure
-
adverse effects of treatment
-
HRQoL.
Exclusion criteria
Studies were excluded if they did not match the inclusion criteria, particularly:
-
non-randomised studies (except if agreed by PenTAG, in the absence of RCTs)
-
animal models
-
preclinical and biological studies
-
narrative reviews, editorials, opinions
-
non-English-language papers
-
reports published as meeting abstracts only, for which insufficient methodological details are reported to allow critical appraisal of study quality.
Data abstraction strategy
Data were extracted by one reviewer (TP) using a standardised data extraction form and checked independently by a second (TJ-H). Disagreements were resolved by discussion, with involvement of a third reviewer if necessary. Data extraction forms for each included study are included in Appendix 3.
Quality assessment strategy
The methodological quality of randomised controlled studies was assessed according to criteria specified by the CRD. 64 Quality was assessed by one reviewer (TP) and judgements were checked by a second (TJH or LC). Any disagreement was resolved by discussion, with involvement of a third reviewer if necessary.
Internal validity
The instrument sought to assess the following considerations:
-
Was the assignment to the treatment groups really random?
-
Was the treatment allocation concealed?
-
Were the groups similar at baseline in terms of prognostic factors?
-
Were the eligibility criteria specified?
-
Were outcome assessors blinded to the treatment allocation?
-
Was the care-provider blinded?
-
Was the patient blinded?
-
Were point estimates and a measure of variability presented for the primary outcome measure?
-
Did the analyses include an ITT analysis?
-
Were withdrawals and dropouts completely described?
In addition, methodological notes were made for each included study, with the reviewer’s observation on sample size and power calculations; participant attrition; methods of data analysis; and conflicts of interest.
External validity
External validity was judged according to the ability of a reader to consider the applicability of findings to a patient group and service setting. Study findings can only be generalisable if they provide enough information to consider whether or not a cohort is representative of the affected population at large. Therefore, studies that appeared to be typical of the UK CML population with regard to these considerations were judged to be externally valid.
Methods of data synthesis
Data were tabulated and discussed in a narrative review. Given the paucity of data, a meta-analysis was not conducted.
Mixed-treatment indirect comparisons were used as far as data allowed to facilitate comparison between the drugs for which there are no head-to-head data for dasatinib and nilotinib. From the data provided from the included trials, indirect comparisons are based on raw unadjusted results in the form of unadjusted odds ratios. The indirect log-odds ratio and corresponding variance were calculated using standard formulae presented in the appendix of Bucher et al. 67 Assuming the sampling distribution of the log-odds ratio to be normally distributed, the Wald method was used to construct 95% CIs for the odds ratio and calculate the p-value. A fixed-effect approach was used, which assumes that the relative effect of the interventions is the same across the two study populations. 67 To check this assumption we compared the baseline characteristics between trials. The participants were similar with respect to median age, the percentage of males, median time between diagnosis and randomisation, median white cell count and median platelet count. It was not possible to use more sophisticated methods (e.g. sensitivity analyses and subgroup analyses) to validate the assumption of similar relative effects, as we did not have access to the original data.
Handling company submissions to the National Institute for Health and Clinical Excellence
All clinical effectiveness data included in the pharmaceutical company submissions to NICE were assessed to see if they met the inclusion criteria and had not already been identified from published sources.
Results of clinical effectiveness
Identification of evidence
The electronic searches retrieved a total of 3227 titles and abstracts. Two additional papers were found by hand-searching of reference lists, with two papers retrieved from updated searches. No additional papers were found by searching the bibliographies of included studies. A total of 2510 papers were excluded on title and abstract. Full text of the remaining 35 papers was requested for more in-depth screening. The process is illustrated in detail in Figure 2.
FIGURE 2.
Flow diagram study of inclusion process for clinical effectiveness. EconLit, American Economic Association’s electronic bibliography.

Two clinical randomised controlled trials were included, one each studying dasatinib and nilotinib compared with imatinib (Table 5), with any additional abstracts or presentations related to the trials also included. 20,29 A further trial was identified, but was published only as a conference abstract. As sufficient detail was not available to make assessments of methodological quality, this was not formally included in the systematic review, with a summary of results available in Appendix 6. 68 Kantarjian et al. (dasatinib) provided an additional seven conference abstracts/presentations. 29,69–75 Saglio et al. (nilotinib) provided an additional 24-month follow-up paper and five conference abstracts/presentations. 20,76–81 One conference abstract of a systematic review assessing CML as a first-line treatment was identified and provided indirect comparison analysis of dasatinib and nilotinib. 82 Another paper also provided indirect comparison analysis of dasatinib and nilotinib. 83 Additional data were also retrieved from the industry submissions of Bristol-Myers Squibb (BMS, 2011, unpublished; dasatinib) and Novartis (2011, unpublished; nilotinib). 84,85
| Study | Year published | Study type | n | Intervention | Comparator | Supplementary publications |
|---|---|---|---|---|---|---|
| Kantarjian et al.,29 DASISION | 2010 | RCT, two-arm | 519 | Dasatinib | Imatinib | Saglio et al.69 (cardiovascular comorbidities) |
| Guilhot et al.70 (baseline medications) | ||||||
| Schiffer et al.71 (lymphocytosis) | ||||||
| Khoury et al.72 (baseline comorbidities) | ||||||
| Shah et al.73 (18-month follow-up data) | ||||||
| Kantarjian et al.74 (18-month follow-up data) | ||||||
| Kantarjian et al.75 (24-month follow-up data) | ||||||
| BMS84 (industry submission) | ||||||
| Saglio et al.,20 ENESTnd | 2010 | RCT, three-arm | 561 |
Nilotinib (300 mg) Nilotinib (400 mg) |
Imatinib | Beaumont et al.76 (hospitalisation) |
| Hochhaus et al.77 (MMR by Sokal group, EFS) | ||||||
| Larson et al.78 (18-month cardiac safety profile) | ||||||
| Hughes et al.79 (18-month follow-up data) | ||||||
| Hughes et al.80 (24-month follow-up data) | ||||||
| Kantarjian et al.81 (24-month follow-up data) | ||||||
| Novartis85 (industry submission) |
The details of studies retrieved as full papers and subsequently excluded, along with the reasons for their exclusion, are detailed in Appendix 5.
Assessment of effectiveness
Study characteristics
Dasatinib compared with imatinib
Kantarjian et al. 29 report on the DASISION trial, a multinational, open-label Phase III randomised controlled trial. Patients with newly diagnosed CP were randomised to either dasatinib (100 mg, n = 259) or imatinib (400 mg, n = 260). The trial has been reported in one full publication, with seven conference abstract/presentations providing additional data. Inclusion and exclusion criteria are detailed in Table 6. The aim of the study was to assess the efficacy and safety of dasatinib (100 mg) compared with imatinib (400 mg). The primary outcome was confirmed CCyR within 12 months, with a secondary outcome of MMR (at any time). Other secondary outcomes are detailed in Table 6.
| Study | Inclusion criteria | Exclusion criteria | Primary outcomes | Secondary outcomes |
|---|---|---|---|---|
| DASISION29 |
Newly diagnosed (≤ 3 months) ECOG score at least 0–2 No prior TKI treatment Adequate hepatic and renal function |
Serious or uncontrolled medical disorders or cardiovascular disease History of serious bleeding disorder, concurrent cancer, previous chemotherapy, pleural effusion at baseline |
CCyR (within 12 months) |
MMR (at any time) Time to confirmed CCyR and MMR response Rates of CCyR and MMR response by 12 months PCR OS |
Participants were randomly assigned in a 1 : 1 ratio stratified by Hasford score (see Chapter 1, Prognosis, for definition) to receive either dasatinib (100 mg daily) or imatinib (400 mg daily). All participants had a minimum follow-up of 12 months, with a median duration of 14 months’ treatment for dasatinib and 14.3 months for imatinib. The median dose of dasatinib was 99 mg per day and of imatinib was 400 mg per day.
Conference abstracts/presentations with additional data assessed:
-
whether or not baseline CV conditions, baseline comorbidities and medications impacted the efficacy and safety of the drugs (see Supplementary publications, below)69,70,72
-
whether or not the safety profile, responses and outcomes in patients with sustained lymphocytosis was determined (see Supplementary publications, below)71
Nilotinib compared with imatinib
Saglio et al. 20 report on the ENESTnd trial, a multicentre, open-label Phase III randomised controlled trial. Patients with newly diagnosed CP were randomised to either nilotinib (300 mg; n = 282) or nilotinib (400 mg; n = 281) or imatinib (400 mg; n = 283). The trial has been reported in one full publication and six conference abstracts/presentations providing additional data. Inclusion and exclusion criteria are detailed in Table 7. The aim of the study was to assess the efficacy and safety of nilotinib (300 mg or 400 mg) compared with imatinib (400 mg). Nilotinib 300 mg is licensed for first-line treatment of CML, with nilotinib 400 mg licensed for second-line treatment of CML. At the time of writing the current British NationalFormulary (BNF 61) provided indication for the use of nilotinib only for second-line treatment of CML (i.e. 400 mg). The primary outcome was MMR at 12 months, with a secondary outcome of CCyR by 12 months; other secondary outcomes are detailed in Table 7.
| Study | Inclusion criteria | Exclusion criteria | Primary outcomes | Secondary outcomes |
|---|---|---|---|---|
| ENESTnd20 |
Newly diagnosed (≤ 6 months) ECOG score 0–2 No prior TKI treatment (except imatinib ≤ 2-weeks) Adequate organ function |
Impaired cardiac function Medication affecting liver enzymes or QT interval prohibited |
MMR (at 12 months) |
Complete cytogenetic response (CCyR) (by 12 months) Rate of MMR and CCyR over time Time to and duration of MMR and CCyR Rate of BCR–ABL : ABL ratio of ≤ 0.01% and ≤ 0.0032% at 12 months EFS PFS Progression to AP/BC OS Safety Dose intensity Pharmacokinetics |
Participants were randomly assigned in a 1 : 1 : 1 ratio stratified by Sokal score (see Chapter 1, Prognosis, for definition) to receive either nilotinib (300 mg twice daily) or nilotinib (400 mg twice daily) or imatinib (400 mg daily). All participants had a minimum follow-up of 12 months, with a median duration of 14 months’ treatment for all study groups. The median dose of nilotinib was 592 mg per day (nilotinib 300 mg twice daily) or 779 mg per day (nilotinib 400 mg twice daily) and of imatinib was 400 mg per day.
Papers and conference abstracts/presentations with additional data assessed:
-
hospitalisation of patients (see Adverse events, below)76
-
cardiac safety profile of the study drugs (see Adverse events, below)78
-
MMR stratified by Sokal score at 12 months77
Population characteristics: baseline
Dasatinib compared with imatinib
For the DASISION trial,29 the population demographic, disease status and use of previous therapies were well matched (details are shown in Table 8).
| Study | DASISION29 | ENESTnd20 | ||||
|---|---|---|---|---|---|---|
| Intervention | Dasatinib (100 mg) | Imatinib (400 mg) | Nilotinib (300 mg) | Nilotinib (400 mg) | Imatinib (400 mg) | |
| n | 259 | 260 | 282 | 281 | 283 | |
| Age, median years (range) | 46 (18–24) | 49 (18–78) | 47 (18–85) | 47 (18–81) | 46 (18–80) | |
| Male (%) | 144 (56) | 163 (63) | 158 (56) | 175 (62) | 158 (56) | |
| Race or ethnic group (%) | ||||||
| Asian | 76 (27) | 66 (23) | 71 (25) | |||
| Black | 12 (4) | 11 (4) | 7 (2) | |||
| White | 170 (60) | 185 (66) | 187 (66) | |||
| Other | 24 (9) | 19 (7) | 18 (6) | |||
| ECOG performance score (%) | ||||||
| 0 | 213 (82) | 205 (79) | ||||
| 1 | 46 (18) | 53 (20) | ||||
| 2 | 0 | 2 (1) | ||||
| Risk groupa | ||||||
| Low | 86 (33) | 87 (33) | 103 (37) | 103 (37) | 104 (37) | |
| Intermediate | 124 (48) | 123 (47) | 101 (36) | 100 (36) | 101 (36) | |
| High | 49 (19) | 50 (19) | 78 (28) | 78 (28) | 78 (28) | |
| Time since diagnosis, median days (range) | 31 (0–296) | 31 (0–244) | 31 (0–182) | 31 (3–189) | 28 (1–183) | |
| White cell count (× 10−9/l), median (range) | 25.1 (2.5–493) | 23.5 (1.4–475) | 23 (2–247) | 23 (2–435) | 26 (3–482) | |
| Platelet count (× 10−9/l), median (range) | 448 (58–1880) | 390 (29–2930) | 424 (90–3880) | 374 (103–1819) | 375 (66–2232) | |
| Peripheral blood blasts (%), median (range) | 1 (0–10) | 1 (0–11) | ||||
| Peripheral blood basophils (%), median (range) | 4 (0–27.8) | 4 (0–19.5) | ||||
| Bone marrow blasts (%), median (range) | 2 (0–14) | 2 (0–12) | ||||
| Haemoglobin (g/dl), median (range) | 12 (5.5–17.6) | 12 (6.2–17.6) | 12.2 (6.4 – 17.1) | |||
| Spleen size ≥ 10 cm below costal margin (%) | 31 (11) | 34 (12) | 40 (14) | |||
| Atypical BCR–ABL transcripts (%) | 3 (1) | 1 (< 1) | 5 (2) | 1 (< 1) | 2 (1) | |
| Previous therapy for CML (%) | Hydroxycarbamide | 189 (73) | 190 (73) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| Anagrelide | 8 (3) | 3 (1) | ||||
| Imatinib (≤ 2 weeks) | 3 (1) | 4 (2) | ||||
Nilotinib compared with imatinib
For the ENESTnd trial,20 the population demographic, disease status and use of previous therapies were well matched (see Table 8 for details).
Comparability of baseline population characteristics between trials
With no head-to-head trial of dasatinib and nilotinib and an indirect comparison analysis conducted (see Indirect comparison of dasatinib and nilotinib, below), comparability between the trials is discussed. Participants in the DASISION29 and ENESTnd20 trials were of a similar age and gender distribution. However, the median age (46–49 years) was younger than that of the general population in which the median age at diagnosis is 58 years (this includes AP/BC patients). Risk group scores were measured by the Hasford risk score for the DASISION trial29 and the Sokal risk score for the ENESTnd trial. 20 However, risk distribution was fairly similar between trials, with ENESTnd20 reporting a slightly lower percentage of patients with intermediate risk and a slightly higher percentage with a high risk, compared with DASISION. 29 The European Cooperative Oncology Group (ECOG) performance status for both trials included patients within a score of 0–2. As shown in Tables 6 and 7, the exclusion criteria were slightly different for the two trials and based on the known AEs of the drugs (e.g. pleural effusion for dasatinib and QT prolongation for nilotinib). Furthermore, the two trials had different responses as primary outcomes for the trials, namely CCyR and MMR for DASISION29 and ENESTnd,20 respectively. However, both trials reported the other response as a secondary outcome.
Assessment of study quality
Dasatinib compared with imatinib
The DASISION trial29 is a good-quality international, multicentre, open-label, Phase III randomised controlled trial. There is no discussion regarding how patients were randomised. The trial was reported as open label, therefore allocation concealment of the patients, outcome or carer blinding was not possible. These criteria have been demonstrated to potentially bias results of RCTs; however, this is unlikely to have an impact, as the outcomes of the trial are objective. Baseline groups are similar and well reported. The statistical analysis and handling of data are also well reported. Although a sample size calculation is not reported, the groups are of a similar size to the ENESTnd trial,20 which does report a sample size calculation (Table 9). The large contribution from BMS to the study and manuscript construction would provide a strong conflict of interest. The study population is not wholly representative of a UK CML population, as a result of the lower median age and the large contribution of Asian patients to the study population.
| Assessment | DASISION29 | ENESTnd20 |
|---|---|---|
| Study design | RCT | RCT |
| Is a power calculation provided? | No | Yes |
| Is the sample size adequate? | Not reported | Yes |
| Was ethical approval obtained? | Yes | Yes |
| Were the study eligibility criteria specified? | Yes | Yes |
| Were the eligibility criteria appropriate? | Yes | Yes |
| Were patients recruited prospectively? | Yes | Yes |
| Was assignment to the treatment groups really random? | Not reported | Not reported |
| Were groups stratified? | Yes | Yes |
| Was the treatment allocation concealed? | No | No |
| Were adequate baseline details presented? | Yes | Yes |
| Were the participants representative of the population in question? | Yes | Yes |
| Were the groups similar at baseline? | Yes | Yes |
| Were baseline differences adequately adjusted for in the analysis? | Yes | Yes |
| Were the outcome assessors blind? | No | No |
| Was the care-provider blind? | No | No |
| Are the outcome measures relevant to the research question? | Yes | Yes |
| Is compliance with treatment adequate? | Yes | Yes |
| Are withdrawals/dropouts adequately described? | Yes | Yes |
| Are all patients accounted for? | Yes | Yes |
| Is the number randomised reported? | Yes | Yes |
| Are protocol violations specified? | Yes | Yes |
| Are data analyses appropriate? | Yes | Yes |
| Is analysis conducted on an ITT basis? | Yes | Yes |
| Are missing data appropriately accounted for? | Yes | Yes |
| Were any subgroup analyses justified? | Not reported | NA |
| Are the conclusions supported by the results? | Yes | Yes |
| Conflict of interest declared? | Yes | Yes |
Nilotinib compared with imatinib
The ENESTnd trial20 is a good-quality international, multicentre, open-label, Phase III randomised controlled trial. 20 There is no discussion regarding how patients were randomised. The trial was reported as open label, therefore allocation concealment of the patients, outcome or carer blinding was not possible. These criteria have been demonstrated to potentially bias results of RCTs; however, this is unlikely to have an impact as the outcomes of the trial are objective. Baseline groups are similar and well reported. The statistical analysis and handling of data are also well reported (see Table 9). The large contribution from Novartis to the study and manuscript construction would provide a strong conflict of interest. The study population is not wholly representative of a UK CML population, as a result of the lower median age and the unknown ethnicity of the patients.
Treatment status
Dasatinib compared with imatinib
The DASISION trial29 reports (Table 10) that at 12 months’ follow-up 85% and 81% of patients still continued to receive treatment with dasatinib and imatinib, respectively. Reported discontinuation rates for dasatinib and imatinib were drug-related AEs (5% vs 4%), disease progression (4% vs 5%) and treatment failure (2% vs 4%). At 18 months’ follow-up, 81% and 80% of patients still continued to receive treatment with dasatinib and imatinib, respectively. 73 At 24 months’ follow-up, 77% and 75% still continued to receive treatment with dasatinib and imatinib, respectively. 75 Reported discontinuation rates for dasatinib and imatinib were drug-related AEs (7% vs 5%), disease progression (5% vs 7%) and treatment failure (3% vs 4%). Significant differences were not reported.
| AEs | 12 months’ follow-up29 | 18 months’ follow-up73 | 24 months’ follow-up75 | |||
|---|---|---|---|---|---|---|
| Dasatinib (n = 258) | Imatinib (n = 258) | Dasatinib (n = 258) | Imatinib (n = 258) | Dasatinib (n = 258) | Imatinib (n = 258) | |
| No. of patients (%) | ||||||
| Received treatment | 258 (100.0) | 258 (100.0) | 258 (100.0) | 258 (100.0) | 258 (100.0) | 258 (100.0) |
| Continue to receive treatment | 218 (85.0) | 210 (81.0) | 209 (81.0) | 206 (80.0) | 199 (77.0) | 194 (75.0) |
| Discontinued treatment | 40 (15.0) | 48 (19.0) | 49 (19.0) | 52 (20.0) | 59 (23.0) | 64 (25.0) |
| Had drug-related AEs | 13 (5.0) | 11 (4.3) | 15 (6.0) | 10 (4.0) | 18 (7.0) | 12 (5.0) |
| Haematological, including cytopenia | 4 (1.6) | 3 (1.2) | 6 (2.3) | 3 (1.2) | 6 (2.3) | 4 (1.6) |
| Non-haematological | 9 (3.5) | 8 (3.1) | 12 (5) | 8 (3.0) | ||
| Diseased progressed | 11 (4.3) | 14 (5.4) | 14 (5) | 17 (7.0) | ||
| Increased white-cell count | 1 (0.4) | 0 | ||||
| Loss of CHR | 0 | 0 | ||||
| Loss of MCyR | 1 (0.4) | 4 (1.6) | ||||
| Progression to accelerated or blastic phase | 5 (1.9) | 9 (3.5) | 6 (2.3) | 9 (3.5) | 9 (3.5) | 15 (5.8) |
| Death | 4 (1.6) | 1 (0.4) | 16 (6.0) | 14 (5.0) | ||
| Treatment failed | 6 (2.3) | 10 (3.9) | 8 (3.0) | 11 (4.0) | ||
| Did not have complete haematological or CyR at 6 months | 2 (0.8) | 4 (1.6) | ||||
| Had less than partial CyR at 12 months | 3 (1.2) | 6 (2.3) | ||||
| Did not have a CCyR at 18 months | 1 (0.4) | 0 | ||||
| Had AE unrelated to drug | 3 (1.2) | 1 (0.4) | ||||
| Withdrew consent | 2 (0.8) | 3 (1.2) | ||||
| Became pregnant | 2 (0.8) | 0 | ||||
| Did not adhere to therapy | 0 | 2 (0.8) | ||||
| Was lost to follow-up | 0 | 3 (1.2) | ||||
| Requested to discontinue | 2 (0.8) | 1 (0.4) | ||||
| Had other reason | 1 (.04) | 3 (1.2) | ||||
Nilotinib compared with imatinib
The ENESTnd trial20 reports (Table 11) at 12 months’ follow-up that 84% and 79% of patients still continued to receive treatment with nilotinib 300 mg (licensed for first-line treatment of CML) and imatinib, respectively. 20 Discontinuation rates for nilotinib 300 mg and imatinib were drug-related AEs (5% vs 7%), disease progression (< 1% vs 4%) and suboptimal response/treatment failure (2% vs 4%). At 24 months’ follow-up 75% and 68% of patients still continued to receive treatment with nilotinib 300 mg and imatinib, respectively. 81 Discontinuation rates for nilotinib 300 mg and imatinib were drug-related AEs (6% vs 9%), disease progression (< 1% vs 4%) and suboptimal response/treatment failure (9% vs 13%) (Novartis, 2011). 85 Significant differences were not reported.
| AE | 12 months’ follow-up20 | 24 months’ follow-up81,85 | ||||
|---|---|---|---|---|---|---|
| Nilotinib 300 mg (n = 282) | Nilotinib 400 mg (n = 281) | Imatinib 400 mg (n = 283) | Nilotinib 300 mg (n = 282) | Nilotinib 400 mg (n = 281) | Imatinib 400 mg (n = 283) | |
| No. of patients (%) | ||||||
| Received treatment | 279 (99) | 278(99) | 279 (99) | 279 (99) | 278 (99) | 279 (99) |
| Still on study | 268 (95) | 271 (96) | 274 (97) | 262 (93) | 267 (95) | 260 (92) |
| Continue to receive treatment | 236 (84) | 230(82) | 224 (79) | 210 (75) | 220 (78) | 191 (68) |
| Discontinued treatment | 46 (16) | 51 (18) | 59 (21) | 72 (25) | 61 (22) | 92 (32) |
| AE(s) | 13 (5) | 26 (9) | 21 (7) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| Abnormal laboratory value(s) | 6 (2) | 5 (2) | 3 (1) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| Abnormal test procedure result(s) | 0 (0) | 1 (< 1) | 1 (< 1) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| Subject’s condition no longer requires drug | 1 (< 1) | 0 (0) | 0 (0) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| Withdrew consent | 6 (2) | 5 (2) | 3 (1) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| Was lost to follow-up | 2 (< 1) | 2 (< 1) | 1 (< 1) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| Death | 2 (< 1) | 0 (0) | 0 (0) | 3 (1) | 1 (< 1) | 0 (0) |
| Diseased progressed | 2 (< 1) | 2 (< 1) | 10 (4) | 2 (< 1) | 4 (1) | 12 (4) |
| Protocol deviation | 4 (1) | 5 (2) | 4 (1) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| Suboptimal response/treatment failure | 10 (4) | 5 (2) | 16 (6) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
At 12 months, only a small percentage, approximately double the number of imatinib patients in ENESTnd20 (21), had to discontinue due to AEs compared with imatinib patients in DASISION29 (11). However, it is unknown whether this is due to different measurement techniques of AEs, difference in the population characteristics between trials, or chance.
Assessment of clinical effectiveness
Complete cytogenetic response
Cytogenetic responses are shown in Table 12. DASISION29 and ENESTnd20 report CCyR by 12, 18 and 24 months’ follow-up. DASISION29 reports confirmed CCyR (i.e. two assessments 28 days apart) for 12, 18 and 24 months’ follow-up, which ENESTnd20 does not. Both trials report CCyR by risk group categorisation by 12 months. CCyR is the primary outcome in the DASISION trial. 29
| Study | DASISION29,73–75 | ENESTnd20,79,81,85 | ||||||||||
| Intervention | Dasatinib (100 mg) | Imatinib (400 mg) | p-value | RR (95% CI)a | Nilotinib (300 mg) | p-value | RR (95% CI)a,b | Nilotinib (400 mg) | p-value | RR (95% CI)a,b | Imatinib (400 mg) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCyR rates 12 monthsc (%) | 216/259 (83) | 186/260 (72) | 0.001 | 1.17 (1.06 to 1.28) | 226/282 (80) | 0.001 | 1.23 (1.11 to 1.36) | 220/281 (78) | 0.001 | 1.20 (1.08 to 1.34) | 184/283 (65) | |
| CCyR rates 18 monthsc (%) | 218/259 (84) | 203/260 (78) | 0.093 | 1.08 (0.98 to 1.17) | 240/282 (85) | < 0.001 | 1.15 (1.09 to 1.25) | 230/281 (82) | 0.017 | 1.11 (1.01 to 1.21) | 209/283 (74) | |
| CCyR rates 24 monthsc (%) | 223/259 (86) | 213/260 (82) | 0.23 | 1.05 (0.97 to 1.13) | 245/282 (87) | 0.0018 | 1.13 1.04 to 1.22) | 238/281 (85) | 0.016 | 1.10 (1.01 to 1.19) | 218/283 (77) | |
| CCyR rates 12 months confirmedd | 199/259 (77) | 172/260 (66) | 0.007 | 1.16 (1.04 to 1.30) | ||||||||
| CCyR rates 18 months confirmedd (%) | 202/259 (78) | 182/260 (70) | 0.037 | 1.11 (1.00 to 1.24) | ||||||||
| CCyR rates 24 months confirmedd (%) | 207/259 (80) | 192/260 (74) | 0.12 | 1.08 (0.98 to 1.19) | ||||||||
| Risk group CCyR rates 12 monthse (%) | Low | 81/86 (94) | 66/87 (76) | (CiC information has been removed) | (CiC information has been removed. | (CiC information has been removed) | ||||||
| Intermediate | 97/124 (78) | 88/123 (72) | ||||||||||
| High | 38/49 (78) | 32/50 (64) | 58/78 (74) | 49/78 (63) | 38/78 (49) | |||||||
| Risk group CCyR rates 18 months confirmedd,e (%) | Low | 79/86 (92) | 63/87 (72) | |||||||||
| Intermediate | 88/124 (71) | 87/123 (71) | ||||||||||
| High | 36/49 (73) | 32/50 (64) | ||||||||||
| Risk group CCyR rates 24 months confirmede (%) | Low | 94/103 (91) | 97/103 (94) | 94/104 (90) | ||||||||
| Intermediate | 88/101 (87) | 85/100 (85) | 78/101 (77) | |||||||||
| High | 63/78 (81) | 56/78 (72) | 46/78 (59) | |||||||||
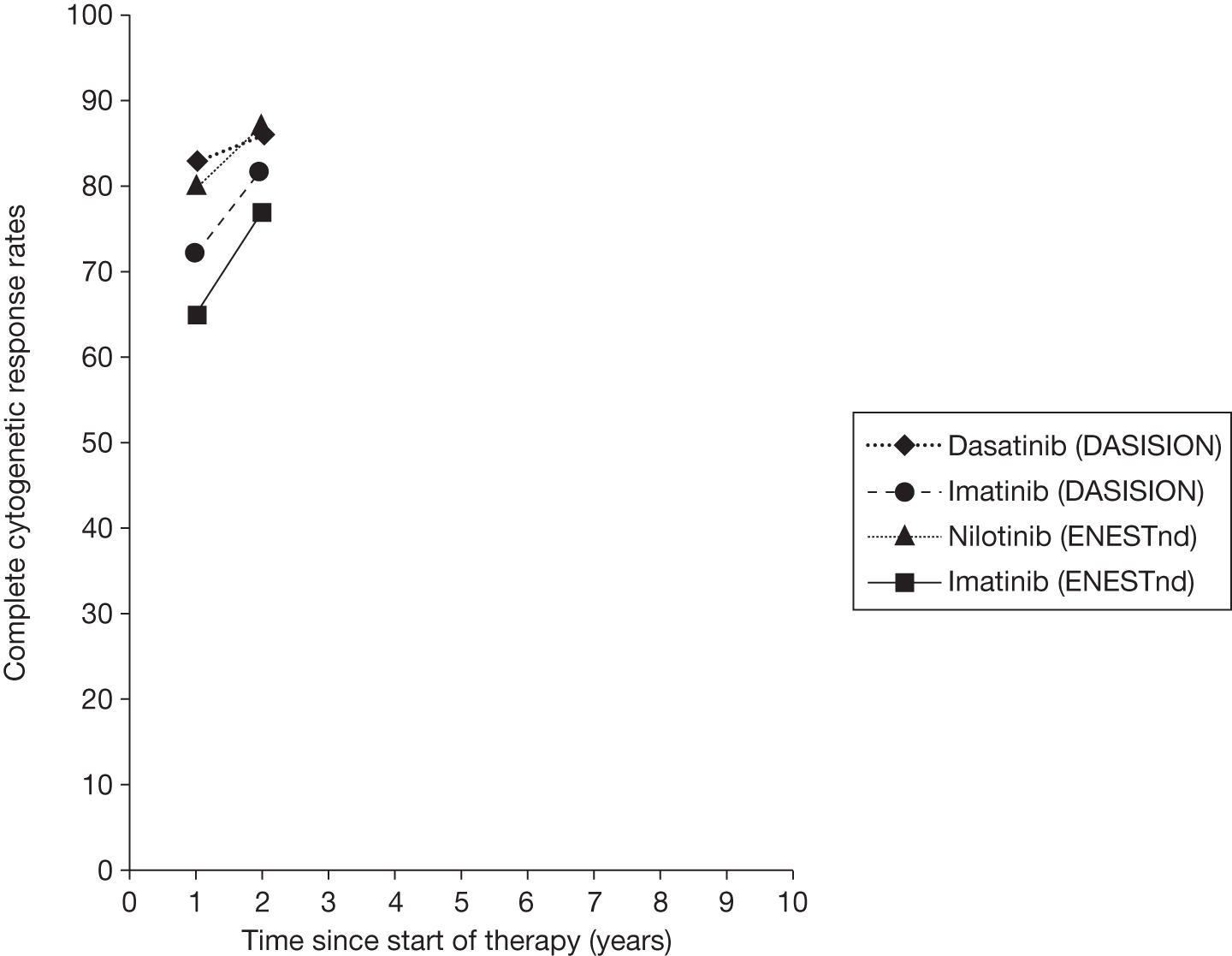
Figures 3 and 4 summarise the CCyR data. We present these on two axes: available follow-up data (see Figure 3) and potential long-term survival (see Figure 4).
FIGURE 3.
Complete cytogenetic response (24 months) all patients.
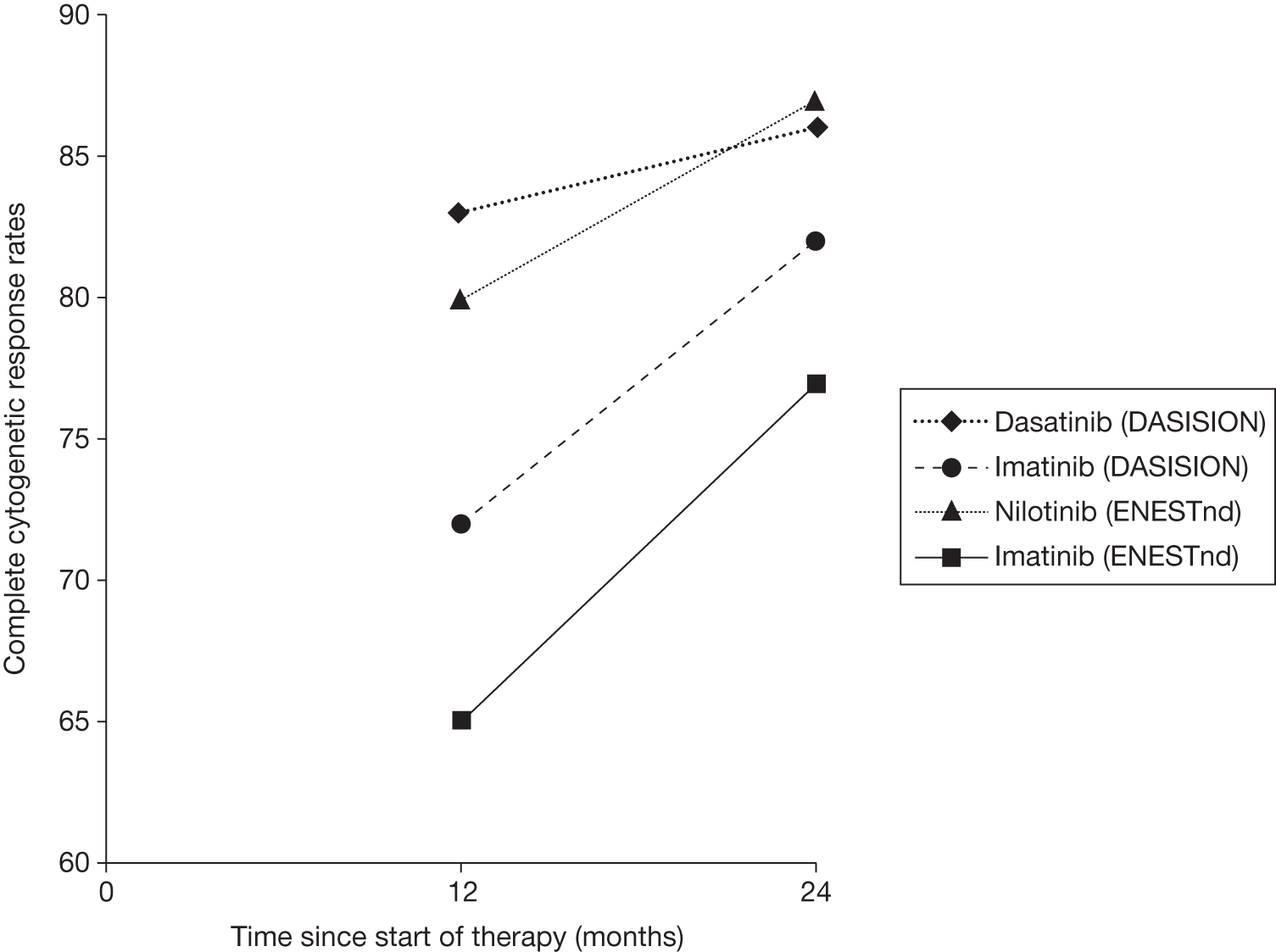
FIGURE 4.
Complete cytogenetic response (10 years) all patients.
Dasatinib compared with imatinib
The DASISION trial29 reports that significantly more patients taking dasatinib (83%) achieved a CCyR than patients taking imatinib (72%) by 12 months’ follow-up [p = 0.001; relative risk (RR) 1.17, 95% CI 1.06 to 1.28]. 29 This difference was not significant by 18 months (84% vs 78%, p = 0.093; RR 1.08, 95% CI 0.98 to 1.17) or 24 months (86% vs 82%, p = 0.23; RR 1.05, 95% CI 0.97 to 1.13). 74,75 There was a significant difference for patients with a confirmed CCyR (i.e. two assessments, 28 days apart) by 12 months’ (77% vs 66%, p = 0.007; RR 1.16, 95% CI 1.04 to 1.30) and 18 months’ (78% vs 70%, p = 0.037; RR 1.11, 95% CI 1.00 to 1.24) follow-up. 29,73
By 24 months’ follow-up there was no significant difference for patients with a confirmed CCyR (80% vs 74%, p = 0.12; RR 1.08, 95% CI 0.98 to 1.19). 75 Differences between confirmed and non-confirmed CCyR suggest that more transitory responses may be seen with imatinib.
By 12 months’ follow-up, CCyR rates were higher for patients receiving dasatinib across all Hasford risk categories than with imatinib, with rates among those categorised as high risk of 78% and 64% for dasatinib and imatinib, respectively. 29 By 18 months’ follow-up, confirmed CCyR rates remained higher for patients receiving dasatinib across all Hasford risk categories compared with imatinib. 73
Nilotinib compared with imatinib (nilotinib 300 mg licensed for first-line treatment of chronic myeloid leukaemia)
The ENESTnd trial20 reports that significantly more patients taking nilotinib 300 mg (80%) achieved a CCyR than patients taking imatinib (65%) by 12 months’ follow-up (p = 0.001; RR 1.23, 95% CI 1.11 to 1.36). 20 By 18 months’ follow-up, rates of CCyR for nilotinib 300 mg and imatinib were 85% and 74%, respectively (p < 0.001; RR 1.15, 95% CI 1.09 to 1.25). 79 By 24 months, nilotinib 300 mg (87%) continued to be significantly superior to imatinib (77%) (p = 0.0018; RR 1.13, 95% CI 1.04 to 1.22). 81 For patients receiving nilotinib 300 mg, CCyR rates were higher across all Sokal risk categories than with imatinib by 12 and 24 months’ follow-up, with high-risk CCyR rates of 74% compared with 49% (12 months) and 81% compared with 59% (24 months) for nilotinib 300 mg and imatinib, respectively. 20,81,85
Major molecular response
Table 13 shows MMR in the two key trials. DASISION29 and ENESTnd20 report MMR at 12, 18 and 24 months’ follow-up. ENESTnd20 reports MMR at any time (12- and 24-month cumulative, MMR may be lost at specific time point). DASISION29 reports MMR at any time (12- and 18-month cumulative). Both trials report MMR by risk group categorisation at 12, 18 and 24 months. MMR is the primary outcome in the ENESTnd trial. 20
| Study | DASISION29,73–75 | ENESTnd20,79–81 | ||||||||||
| Intervention | Dasatinib (100 mg) | Imatinib (400 mg) | p-value | RR (95% CI)a | Nilotinib (300 mg) | p-value | RR (95% CI)a,b | Nilotinib (400 mg) | p-value | RR (95% CI)a,b | Imatinib (400 mg) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MMR 12 monthsc (%) | 119/259 (46) | 73/260 (28) | < 0.001 | 1.63 (1.29 to 2.09) | 125/282 (44) | 0.001 | 2.02 (1.56 to 2.65) | 121/281 (43) | 0.001 | 1.97 (1.51 to 2.58) | 62/283 (22) | |
| MMR 18 monthsc (%) | 145/259 (56) | 96/260 (37) | < 0.001 | 1.52 (1.25 to 1.85) | ||||||||
| MMR 24 monthsa (%) | 175/282 (62) | < 0.001 | 1.67 (1.40 to 2.00) | 165/281 (59) | < 0.001 | 1.58 (1.32 to 1.90) | 105/283 (37) | |||||
| MMR at any time (12 months)c,d (%) | 135/259 (52) | 88/260 (34) | < 0.001 | 1.54 (1.25 to 1.91) | (CiC information has been removed) (57) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) (54) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) (30) | |
| MMR at any time (18-month)c,d(%) | 148/259 (57) | 107/260 (41) | < 0.001 | 1.39 (1.15 to 1.67) | 186/282 (66) | < 0.001 | 1.65 (1.40 to 1.95) | 174/281 (62) | < 0.001 | 1.55 (1.31 to 1.84) | 113/283 (40) | |
| MMR at any time (24-month)c,d (%) | 166/259 (64) | 120/260 (46) | < 0.001 | 1.39 (1.18 to 1.64) | 201/282 (71) | < 0.001 | 1.63 (1.37 to 1.84) | 187/281 (67) | < 0.001 | 1.52 (1.30 to 1.78) | 124/283 (44) | |
| Risk group MMR rates 12 monthse (%) | Low | 48/86 (56) | 31/87 (36) | (CiC information has been removed) (41) | (CiC information has been removed) (32) | (CiC information has been removed) (17) | ||||||
| Intermediate | 56/124 (45) | 34/123 (28) | ||||||||||
| High | 15/49 (31 | 8/50 (16) | ||||||||||
| Risk group MMR rates 18 monthse (%) | Low | 54/86 (63) | 42/87 (48) | 71/103 (69) | 71/103 (69) | 53/104 (51) | ||||||
| Intermediate | 69/124 (56) | 49/123 (40) | 69/101 (68) | 63/100 (63) | 39/101 (39) | |||||||
| High | 25/49 (51) | 15/50 (30) | 46/78 (59) | 40/78 (51) | 22/78 (28) | |||||||
| Risk group MMR rates 24 monthse (%) | Low | 63/86 (73) | 49/87 (56) | 75/103 (73) | 76/103 (74) | 68/104 (65) | ||||||
| Intermediate | 76/124 (61) | 62/123 (50) | 75/101 (74) | 67/100 (67) | 44/101 (44) | |||||||
| High | 28/49 (57) | 19/50 (38) | 51/78 (65) | 44/78 (56) | 25/78 (32) | |||||||
Figures 5 and 6 summarise the MMR data. We present these on two axes: available follow-up data (see Figure 5) and potential long-term survival (see Figure 6).
FIGURE 5.
Major molecular response (24 months), all patients.
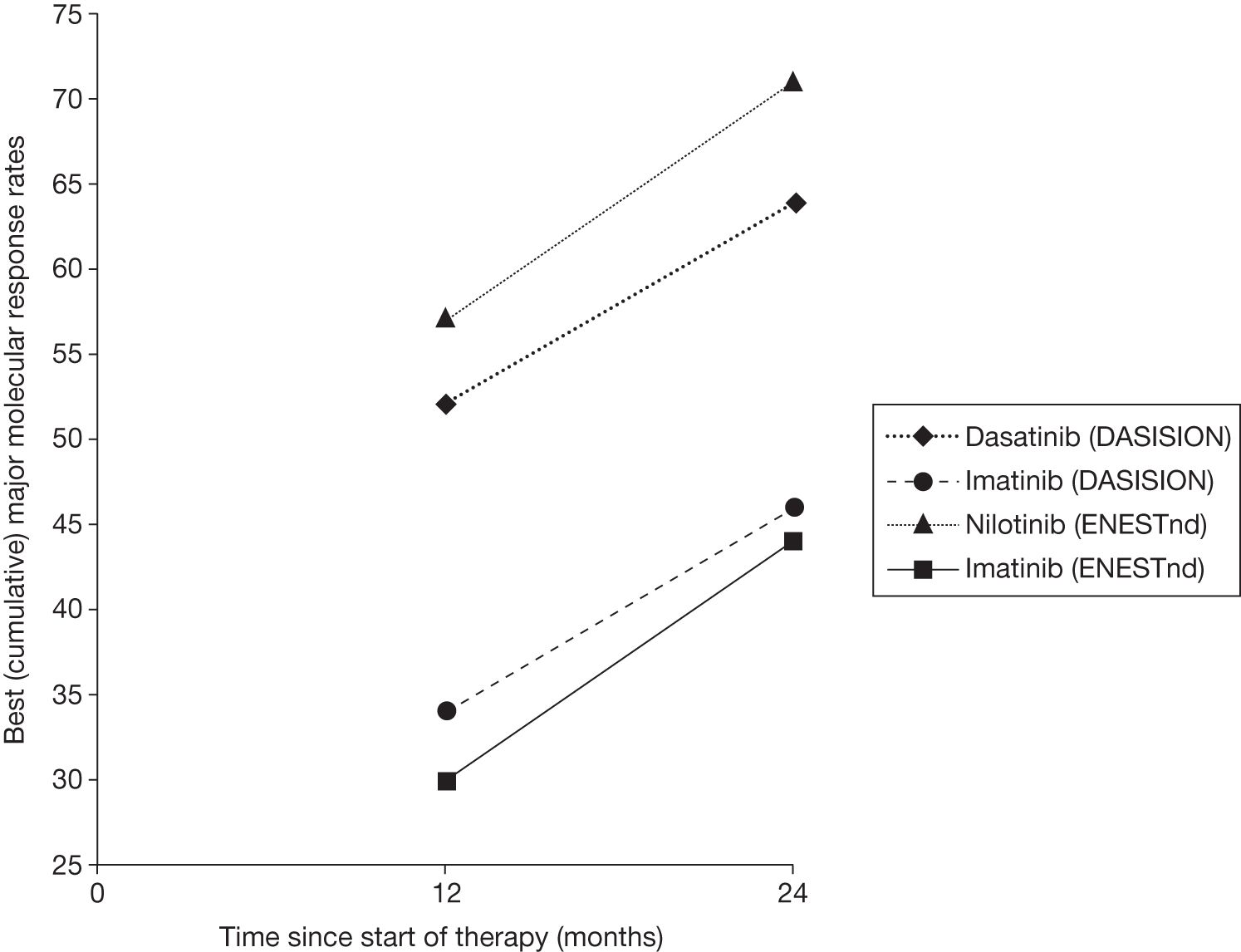
FIGURE 6.
Major molecular response (10 years), all patients.
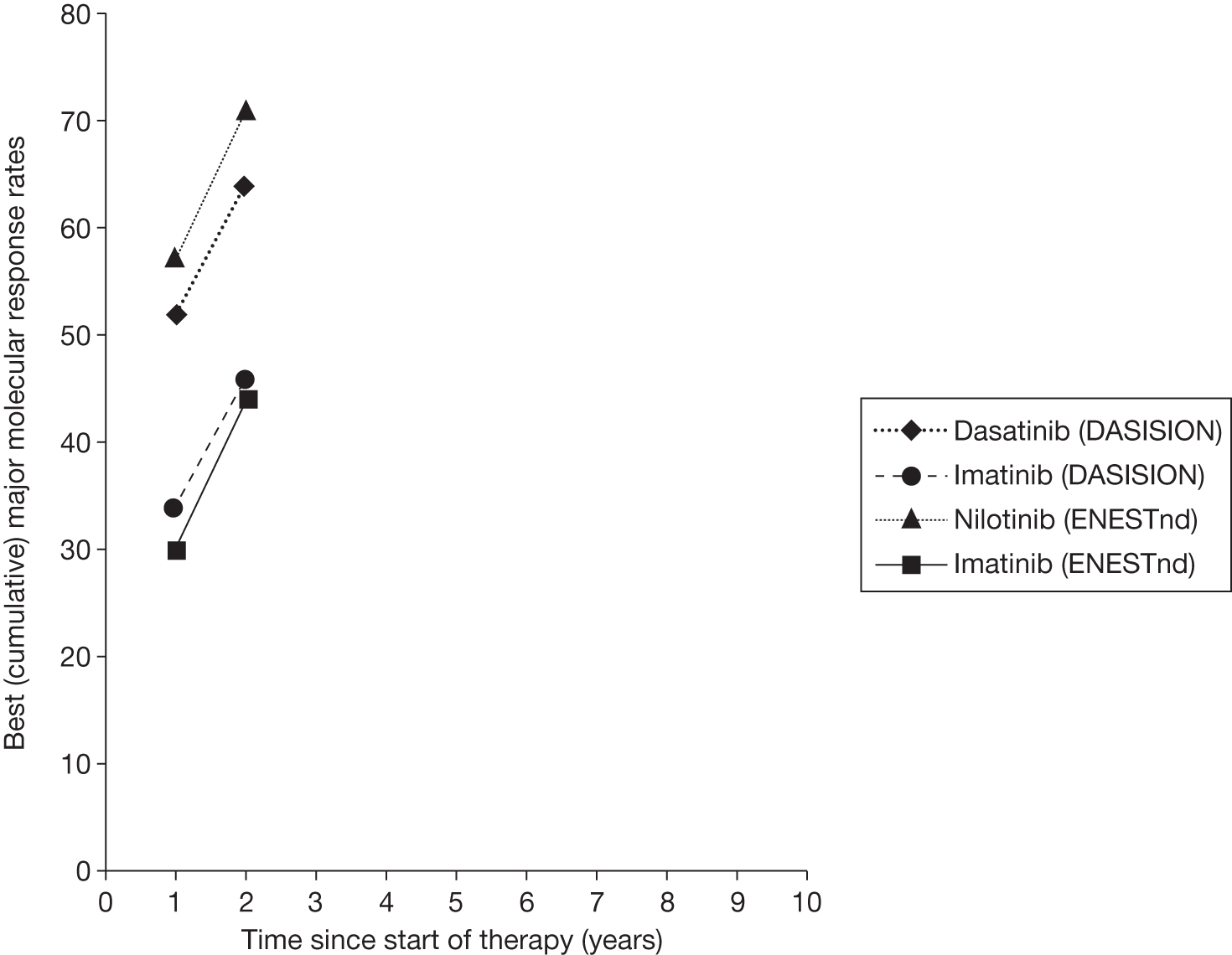
Dasatinib compared with imatinib
The DASISION trial29 reports that significantly more patients taking dasatinib (46%) achieved a MMR than those taking imatinib (28%) at 12 months’ follow-up (p < 0.0001; RR 1.63, 95% CI 1.29 to 2.09) and 18 months’ follow-up (56% vs 37%, p = 0.001; RR 1.52, 95% CI 1.25 to 1.85), and at 24 months’ follow-up. 29,74 A significant difference also seen for a MMR at any time at 12 months’ (52% vs 34%, p < 0.001; RR 1.54, 95% CI 1.25 to 1.91), 18 months’ (57% vs 41%, p = 0.001; RR 1.39, 95% CI 1.15 to 1.67) and 24 months’ follow-up (64% vs 46%, p = 0.001; RR 1.39, 95% CI 1.18 to 1.64)29,73,75
At 12 months’ follow-up, MMR rates were higher for patients receiving dasatinib across all Hasford risk categories than for patients receiving imatinib. 29 At 18 months’ follow-up, MMR rates remained higher for patients receiving dasatinib across all Hasford risk categories than for patients receiving imatinib, with MMR rates of 51% and imatinib 30% for dasatinib and imatinib, respectively, among those categorised as high risk. 73 At 24 months’ follow-up, MMR rates remained higher for patients receiving dasatinib across all Hasford risk categories than for patients receiving imatinib, with MMR rates of 57% and imatinib 38% for dasatinib and imatinib, respectively, among those categorised as high risk. 75
Nilotinib compared with imatinib (nilotinib 300 mg licensed for first-line treatment of chronic myeloid leukaemia)
The ENESTnd trial20 reports that significantly more patients receiving nilotinib 300 mg (44%) achieved a MMR than patients taking imatinib (22%) at 12 months’ follow-up (p = 0.001; RR 2.02, 95% CI 1.56 to 2.65). 20 At 24 months’ follow-up, MMR rates continued to be significantly higher for patients receiving nilotinib 300 mg (62%) than patients receiving imatinib (37%) (p = 0.001; RR 1.67, 95% CI 1.40 to 2.00). 80 A significant difference was also seen for a MMR at any time between nilotinib 300 mg and imatinib at 12 months’ [57% vs 30%, (CiC information has been removed)], 18 months’ (66% vs 40%, p < 0.001; RR 1.65, 95% CI 1.40 to 1.95) and 24 months’ follow-up (71% vs 44%, p = 0.001; RR 1.63, 95% CI 1.37 to 1.84). 79,81,85
(CiC information has been removed.) At 18 months’ follow-up, MMR rates were higher for patients receiving nilotinib 300 mg across all Sokal risk categories than for those receiving imatinib, with MMR rates of 59% and 28% for nilotinib 300 mg and imatinib, respectively, among those categorised as high risk. 79 At 24 months’ follow-up, MMR rates remained higher for patients receiving nilotinib 300 mg across all Sokal risk categories than for patients receiving imatinib, with MMR rates of 65% and 32% for nilotinib and imatinib, respectively, among those categorised as high risk. 81
Complete molecular response
Results for CMR from the two key trials are shown in Table 14. ENESTnd20 reports CMR by 12, 18 and 24 months. DASISION29 reports CMR by 18 and 24 months.
| Study | DASISION73,75 | ENESTnd20,79,81 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | Dasatinib (100 mg) | Imatinib (400 mg) | p-value | RR (95% CI)a | Nilotinib (300 mg) | p-value | RR (95% CI)a,b | Nilotinib (400 mg) | p-value | RR (95% CI)a,b | Imatinib (400 mg) | |
| CMR 12 monthsa (BCR–ABL 0.0032%) (%) | 37/282 (13) | < 0.001 | 3.38 (1.70 to 6.93) | 34/281 (12) | < 0.001 | 3.11 (1.56 to 6.43) | 11/283 (4) | |||||
| CMR 18 monthsc (BCR–ABL 0.0032%) (%) | 34/259 (13) | 18/260 (7) | 0.04 | 1.90 (1.00 to 3.24) | 59/282 (21) | < 0.001 | 3.48 (2.04 to 6.09) | 48/281 (17) | < 0.001 | 2.84 (1.64 to 5.04) | 17/283 (6) | |
| CMR 24 monthsc (BCR–ABL 0.0032%) (%) | 44/259 (17) | 21/260 (8) | 0.002 | 2.10 (1.26 to 3.57) | 73/282 (26) | < 0.001 | 2.62 (1.72 to 4.03) | 59/281 (21) | < 0.001 | 2.12 (1.37 to 3.32) | 28/283 (10) | |
| Risk group CMR rates 24 monthsd (%) | Low | 25/103 (24) | 30/103 (29) | 10/104 (10) | ||||||||
| Intermediate | 33/101 (33) | 13/100 (13) | 15/101 (15) | |||||||||
| High | 16/78 (21) | 16/78 (21) | 4/78 (5) | |||||||||
Dasatinib compared with imatinib
The DASISION trial29 reports that by 18 months CMR (BCR–ABL 0.0032%) rates were significantly higher for patients receiving dasatinib (13%, p = 0.04; RR 1.90, 95% CI 1.00 to 3.24) than for patients receiving imatinib (7%). 73 This difference was maintained by 24 months’ follow-up for dasatinib (17%, p = 0.002; RR 2.10, 95% CI 1.26 to 3.57) compared with imatinib (8%). 75
Nilotinib compared with imatinib (nilotinib 300 mg licensed for first-line treatment of chronic myeloid leukaemia)
The ENESTnd trial20 reports that by 12 months, CMR (BCR–ABL 0.0032%) rates were significantly higher for patients receiving nilotinib 300 mg (13%, p < 0.001; RR 3.38, 95% CI 1.70 to 6.93) than for patients receiving imatinib (4%). 20 By 18 months, CMR (BCR–ABL 0.0032%) rates were significantly higher for patients receiving nilotinib 300 mg (21%, p < 0.001; RR 3.48, 95% CI 2.04 to 6.09) than for patients receiving imatinib (6%). 79 By 24 months, CMR (BCR–ABL 0.0032%) rates continued to be significantly higher for patients receiving nilotinib 300 mg (26%, p < 0.001; RR 2.62, 95% CI 1.72 to 4.03) than for patients receiving imatinib (10%). 81
By 24 months’ follow-up, CMR rates were higher for patients receiving nilotinib 300 mg across all Sokal risk categories than for patients receiving imatinib, with CMR rates of 21% and 5% for nilotinib and imatinib, respectively, among those categorised as high risk. 81
Time to complete cytogenetic response and major molecular response
Dasatinib compared with imatinib
The DASISION trial29 reports that at 12, 18 and 24 months’ follow-up the time to a CCyR and a confirmed CCyR was significantly shorter for patients receiving dasatinib than for those receiving imatinib [both hazard ratios (HRs) 1.5, p < 0.0001]. 29,75,87 The median time to a confirmed CCyR was 3.1 and 5.6 months for dasatinib and imatinib, respectively (BMS 2011). 84
The time to a MMR was also significantly shorter for patients receiving dasatinib (HR 2.0, p < 0.0001) than for those receiving imatinib at 12 months’ follow-up (HR 2.0, p < 0001). 29 The median time to MMR was 6.3 and 9.2 months for dasatinib and imatinib, respectively (BMS 2011). 84 At 18 and 24 months’ follow-up, patients receiving dasatinib were significantly still more likely to achieve a MMR (HR 1.84, p < 0001; HR 1.69, p < 0001). 73,75
Durability of major molecular response
Dasatinib compared with imatinib
No information about durability of MMR was available for dasatinib.
Progression to accelerated phase or blast crisis
Dasatinib compared with imatinib
The DASISION trial29 reports that at 12 months, progression to AP or BC was not significantly different for patients receiving imatinib (n = 9) compared with patients receiving dasatinib (n = 5). At 18 months there was only one extra progression, in a patient treated with imatinib. 74 At 24 months’ rates were imatinib (n = 15) compared with dasatinib (n = 9).
Nilotinib compared with imatinib
The ENESTnd trial20 reports that rates of progression to AP or BC were significantly higher at 12 months for imatinib (n = 11) than with nilotinib 300 mg (n = 2, p = 0.01) and at 24 months (2 vs 12, p = 0.005). 20,81 Of note, the rate of progression to AP/BC in the ENESTnd study20 for imatinib is considerably higher than that previously reported for imatinib in the IRIS study. 26
Time to progression
Dasatinib compared with imatinib
Time to progression was not reported for dasatinib.
Time to treatment failure
Time to treatment failure was not reported in the DASISION29 or ENESTnd trials. 20
Survival
This section reports on OS, PFS and EFS. PFS is usually defined as all cause death or progression to AP/BC, but definition may be subjective to the trial. EFS is defined by the researchers of the trials and usually includes all cause death, progression to AP/BC and loss of response. Results and details of the trial survival definitions are shown in Table 15. Figures 7 and 8 summarise the OS data.
| Study | DASISION29,73,75 | ENESTnd77,79,81 | ||||||
|---|---|---|---|---|---|---|---|---|
| Intervention | Dasatinib (100 mg) | Imatinib (400 mg) | p-value | Nilotinib (300 mg) | p-value | Nilotinib (400 mg) | p-value | Imatinib (400 mg) |
| EFSa 12 months | 97.6% | 0.09 | 99.6 | 0.001 | 95.7% | |||
| EFSa 24 months | 96.4% | 0.12 | 97.8 | 0.01 | 93.6% | |||
| EFSb 24 months | 84.8% | 83.8% | ||||||
| PFSc 12 months | 96% | 97% | ||||||
| PFSc 18 months | 94.9% | 93.7% | ||||||
| PFSc,d 24 months | 93.7% | 92.1% | 98% | 0.07 | 97.7% | 0.04 | 95.2% | |
| OS 12 months | 97% | 99% | ||||||
| OS 18 months | 96% | 97.9% | 98.5% | 0.28 | 99.3% | 0.03 | 96.9% | |
| OS 24 months | 95.3% | 95.2% | 97.4% | 0.64 | 97.8% | 0.21 | 96.3% | |
FIGURE 7.
Overall survival (24-month axis).
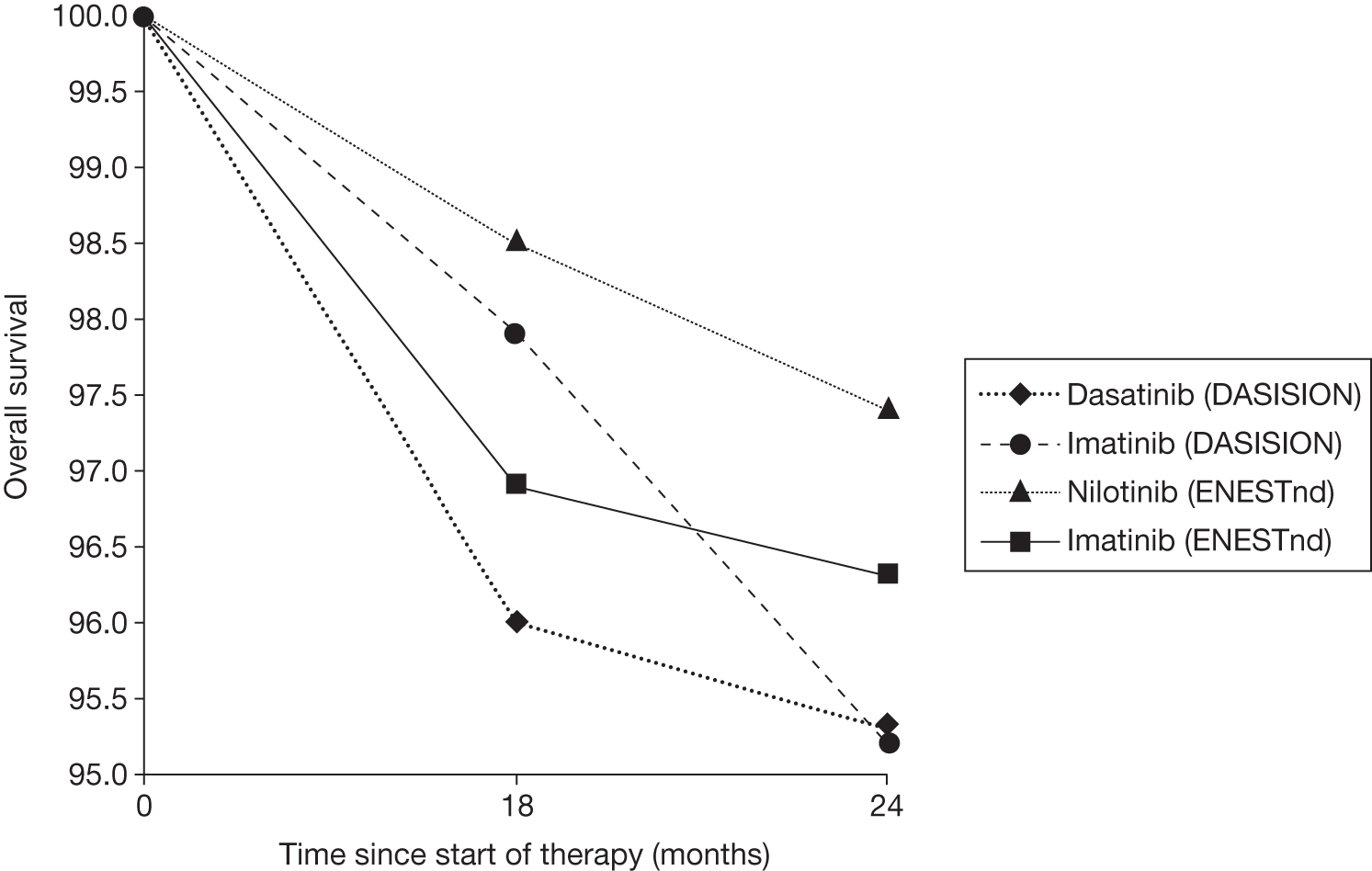
FIGURE 8.
Overall survival (10-year axis).
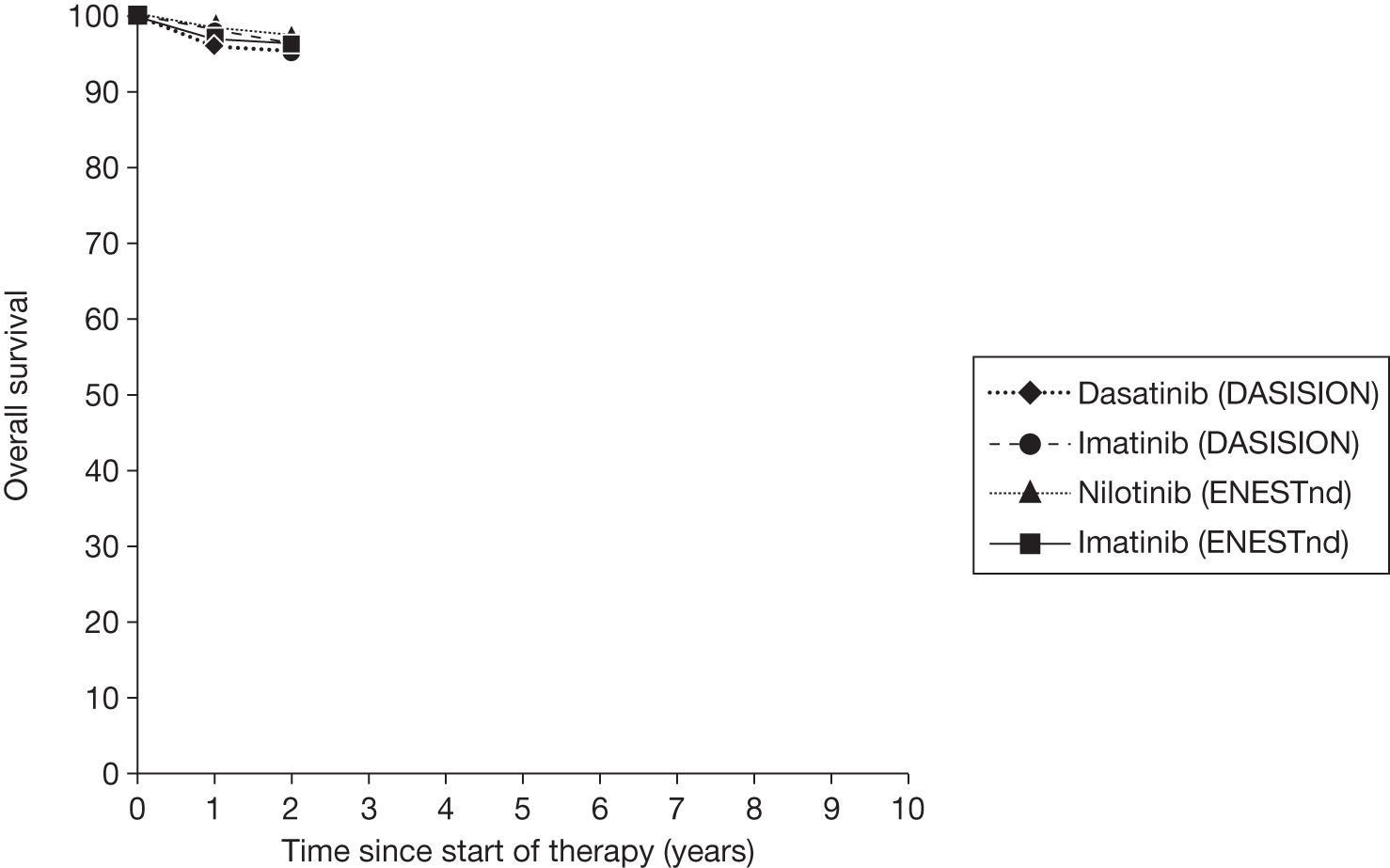
We present these on two axes, available follow-up data (see Figure 7) and potential long-term survival (see Figure 8), as an indication of the immaturity of these data in relation to expected long-term survival.
Dasatinib compared with imatinib
The DASISION trial29 reports (see Table 15) that PFS and OS were not statistically different between dasatinib and imatinib at 12 months (PFS 96% vs 97%; OS 97% vs 99%) 18 months (PFS 94.9% vs 93.7; OS 96 vs 97.9%) and 24 months’ follow-up (PFS 93.7% vs 92.1%; OS 95.3% vs 95.2%), as calculated by PenTAG. 29,73,75
Nilotinib compared with imatinib (nilotinib 300 mg licensed for first-line treatment of chronic myeloid leukaemia, nilotinib 400 mg licensed for second-line treatment of chronic myeloid leukaemia)
At 12 months’ follow-up, the ENESTnd trial20 reports (see Table 15) no significant difference in EFS compared with imatinib (95.7%) for nilotinib 300 mg (97.6%; p = 0.09) but significantly higher EFS for nilotinib 400 mg (99.6%,; p = 0.001), with differences maintained at 24 months’ follow-up. 77,81 PFS at 24 months was also not significantly different for nilotinib 300 mg (98%; p = 0.07) but significantly higher for nilotinib 400 mg (97.7%; p = 0.04) compared with imatinib (95.2%). 81
At 18 months, OS was not significantly different for nilotinib 300 mg (98.5%; p = 0.28) and significantly higher for nilotinib 400 mg (99.3%; p = 0.03) than for imatinib (96.9%). 79 At 24 months OS was not significantly different for either dose of nilotinib compared with imatinib (97.4%, p = 0.64; 97.8%, p = 0.21; 96.3%, respectively). 81
Supplementary publications
As well as the main trial reports, supplementary publications present a number of additional analyses, which are reported in this section.
Dasatinib compared with imatinib (DASISION)
Four supplementary publications were identified:
-
Saglio et al. 69 report on the efficacy and safety of dasatinib and imatinib by baseline cardiovascular comorbidities.
-
Guilhot et al. 70 report on the efficacy and safety of dasatinib and imatinib by use of baseline medications.
-
Khoury et al. 72 report on the efficacy and safety of dasatinib and imatinib by baseline comorbidities.
-
Schiffer et al. 71 report on the responses of patients experiencing lymphocytosis.
Baseline cardiovascular condition, medication or comorbidities generally had no impact on efficacy and safety of dasatinib or imatinib as a first-line treatment for CML. 69,70,72 Schiffer et al. 71 reported on the responses of patients with lymphocytosis (an increase in thymus and natural killer WBCs) compared with those without at 14 months’ follow-up. For patients taking dasatinib, CCyR rates were slightly higher for patients with lymphocytosis (84% of n = 61) compared with those without (75% of n = 197). For patients taking imatinib, CCyR rates were lower for patients with lymphocytosis (50% of n = 14) than for those without (70% of 244).
Adverse events
Results for AEs are shown in Tables 16 and 17 for the DASISION29 and ENESTnd20 trials, respectively. Both trials report the measurement of similar haematological and non-haematological events, with ENESTnd20 also reporting biochemical abnormalities.
| AEs | 12 months’ follow-up29 | 18 months’ follow-up73 | 24 months’ follow-up75 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dasatinib (n = 258) | Imatinib (n = 258) | Dasatinib (n = 258) | Imatinib (n = 258) | Dasatinib (n = 258) | Imatinib (n = 258) | |||||||
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | |
| No. of patients (%)a | ||||||||||||
| Haematological | ||||||||||||
| Neutropenia | 168 (65) | 54 (21) | 150 (58) | 52 (20) | 57 (22) | 52 (20) | 62 (24) | 54 (21) | ||||
| Thrombocytopenia | 181 (70) | 49 (19) | 160 (62) | 26 (10) | 49 (19) | 26 (10) | 52 (20) | 28 (11) | ||||
| Anaemia | 232 (90) | 26 (10) | 217 (84) | 18 (7) | 28 (11) | 18 (7) | 28 (11) | 21 (8) | ||||
| Bleeding | < 1 | 1 | < 1 | 1 | ||||||||
| Non-haematological AE | ||||||||||||
| Fluid retention | 49 (19) | 1 | 108 (42) | 1 | 59 (23) | 111 (43) | 65 (25) | 111 (43) | ||||
| Superficial oedema | 23 (9) | 0 | 93 (36) | < 1 | 26 (10) | 93 (36) | 28 (11) | 93 (36) | ||||
| Pleural effusion | 26 (10) | 0 | 0 | 0 | 31 (12) | 0 | 36 (14) | 0 | ||||
| Other | 13 (5) | 1 | 21 (8) | < 1 | ||||||||
| Diarrhoea | 44 (17) | < 1 | 44 (17) | 1 | 46 (18) | 49 (19) | 49 (19) | 54 (21) | ||||
| Nausea | 21 (8) | 0 | 52 (20) | 0 | 23 (9) | 54 (21) | 26 (10) | 59 (23) | ||||
| Vomiting | 13 (5) | 0 | 26 (10) | 0 | 13 (5) | 26 (10) | 13 (5) | 26 (10) | ||||
| Myalgia | 15 (6) | 0 | 31 (12) | 0 | 15 (6) | 31 (12) | ||||||
| Muscle inflammation | 10 (4) | 0 | 44 (17) | < 1 | 10 (4) | 49 (19) | 10 (4) | 49 (19) | ||||
| Musculoskeletal pain | 28 (11) | 0 | 36 (14) | < 1 | 31 (12) | 41 (16) | 31 (12) | 41 (16) | ||||
| Rash | 28 (11) | 0 | 44 (17) | 1 | 28 (11) | 49 (19) | 28 (11) | 49 (19) | ||||
| Headache | 31 (12) | 0 | 26 (10) | 0 | ||||||||
| Fatigue | 21 (8) | < 1 | 26 (10) | 0 | 21 (8) | 28 (11) | 23 (9) | 28 (11) | ||||
| AE | 12 months’ follow-up20 | 24 months’ follow-up80,81,85 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nilotinib 300 mg (n = 279) | Nilotinib 400 mg (n = 277) | Imatinib 400 mg (n = 280) | Nilotinib 300 mg (n = 279) | Nilotinib 400 mg (n = 277) | Imatinib 400 mg (n = 280) | |||||||
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | |
| No. of patients (%)a | ||||||||||||
| Haematological | ||||||||||||
| Neutropenia | 120 (43) | 33 (12) | 106 (38) | 27 (10) | 189 (68) | 56 (20) | 41 (15) | 33 (12) | 30 (11) | 31 (11) | 57 (20) | 59 (21) |
| Thrombocytopenia | 133 (48) | 28 (10) | 136 (49) | 33 (12) | 156 (56) | 24 (9) | 48 (17) | 28 (10) | 54 (20) | 33 (12) | 48 (17) | 25 (9) |
| Anaemia | 105 (38) | 9 (3) | 105 (38) | 9 (3) | 132 (47) | 14 (5) | 18 (6) | 11 (4) | 24 (9) | 11 (4) | 46 (16) | 14 (5) |
| Non-haematological | ||||||||||||
| Rash | 86 (31) | < 1 | 100 (36) | 7 (3) | 32 (11) | 1 | 89 (32) | < 1 | 103 (37) | 8 (3) | 36 (13) | 6 (2) |
| Headache | 39 (14) | 1 | 58 (21) | 1 | 23 (8) | 0 | 39 (14) | 1 | 61 (22) | 1 | 25 (9) | < 1 |
| Nausea | 32 (11) | < 1 | 54 (19) | 1 | 86 (31) | 0 | 39 (14) | < 1 | 59 (21) | 1 | 95 (34) | 0 |
| Alopecia | 22 (8) | 0 | 36 (13) | 0 | 11(4) | 0 | 25 (9) | 0 | 36 (13) | 0 | 14 (5) | 0 |
| Pruritus | 41 (15) | < 1 | 36 (13) | < 1 | 15 (5) | 0 | 45 (16) | < 1 | 36 (13) | < 1 | 17 (6) | 0 |
| Myalgia | 27 (10) | < 1 | 28 (10) | 0 | 28 (10) | 0 | 28 (10) | < 1 | 28 (10) | 0 | 31 (11) | 0 |
| Fatigue | 30 (11) | 0 | 25 (9) | 1 | 22 (8) | < 1 | 31 (11) | 0 | 25 (9) | < 1 | 28 (10) | < 1 |
| Vomiting | 13 (5) | 0 | 24 (9) | 1 | 40 (14) | 0 | 14 (5) | 0 | 25 (9) | 1 | 50 (18) | 0 |
| Diarrhoea | 22 (8) | 1 | 18 (6) | 0 | 60 (21) | 1 | 22 (8) | < 1 | 20 (7) | 0 | 73 (26) | 1 |
| Muscle spasm | 20 (7) | 0 | 17 (6) | 1 | 67 (24) | 1 | 22 (8) | 0 | 20 (7) | < 1 | 75 (27) | < 1 |
| Peripheral oedema | 14 (5) | 0 | 15 (5) | 0 | 38 (14) | 0 | 14 (5) | 0 | 17 (6) | 0 | 42 (15) | 0 |
| Eyelid oedema | 2 (1) | 0 | 5 (2) | < 1 | 37 (13) | < 1 | < 1 | 0 | 6 (2) | < 1 | 45 (16) | < 1 |
| Periorbital oedema | 1 (< 1) | 0 | 2 (1) | 0 | 34 (12) | 0 | < 1 | 0 | 1 | 0 | 39 (14) | 0 |
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | |
| Biochemical abnormality | ||||||||||||
| Increased total bilirubin | 149 (53) | 10 (4) | 171 (62) | 21 (8) | 27 (10) | < 1 | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) |
| Increased alkaline phosphate | 59 (21) | 0 | 76 (27) | 0 | 92 (33) | < 1 | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) |
| Decreased phosphate | 88 (32) | 13 (5) | 94 (34) | 13 (5) | 126 (45) | 21 (8) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) |
| Increased glucose | 100 (36) | 17 (6) | 113 (41) | 10 (4) | 57 (20) | 0 | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) |
| Increased lipase | 67 (24) | 16 (6) | 80 (29) | 16 (6) | 30 (11) | 9 (3) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) |
| Increase amylase | 42 (15) | 1 (< 1) | 51 (18) | 1 | 35 (12) | 1 | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) |
| Increased creatinine | 13 (5) | 0 | 15 (5) | 0 | 36 (13) | < 1 | ||||||
| Increased ALT | 184 (66) | 11 (4) | 203 (73) | 25 (9) | 57 (20) | 7 (2) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) |
| Increased AST | 112 (40) | 4 (1) | 134 (48) | 8 (3) | 65 (23) | 1 | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) | (AiC information has been removed) |
| Hospitalisation 76 | ||||||||||||
| Nilotinib 300 mg (n = 279) | p-value | Nilotinib 400 mg (n = 277) | p-value | Imatinib 400 mg (n = 280) | ||||||||
| No. of hospitalisations | 48 | 74 | 57 | |||||||||
| Total hospital days | 434 | 591 | 642 | |||||||||
| Length of stay, days: median (range) | 4 (1–64) | 4 (1–101) | 5 (1–86) | |||||||||
| Hospital days per 1000 patient-days | 2.72 | 0.057 | 3.69 | 0.681 | 3.99 | |||||||
Dasatinib compared with imatinib
The DASISION trial29 reports that both of the drugs were well tolerated, with discontinuation due to AEs at 5% and 4% for dasatinib and imatinib, respectively (12 months). 29 At 12, 18 and 24 months’ follow-up, rates of haematological events were similar between dasatinib and imatinib (all grades and grades 3–4), except grade 3 or 4 thrombocytopenia, for which there were nearly twice as many events in the dasatinib arm (19–20%) compared with the imatinib arm (10–11%). 29,73,75 An increased frequency of fluid retention and superficial oedema was displayed for patients receiving imatinib across all grades at 12, 18 and 24 months’ follow-up. 29,75,87 Rates of pleural effusion were higher for patients receiving dasatinib (10–14%) than for patients receiving imatinib (0%). 29,75,87 Other non-haematological events – including rash, vomiting, nausea and myalgia – generally appeared lower across time points for dasatinib than for imatinib.
Nilotinib compared with imatinib (nilotinib 300 mg licensed for first-line treatment of chronic myeloid leukaemia, nilotinib 400 mg licensed for second-line treatment of chronic myeloid leukaemia)
The ENESTnd trial20 reports that both drugs were well tolerated with discontinuation due to AEs at 5%, 9% and 7% for nilotinib 300 mg, 400 mg and imatinib, respectively, at 12 months, and 6%, 10% and 9% at 24 months (Novartis 2011). 20,85 At 12 months’ follow-up, haematological events across all grades were lower for patients receiving either dose of nilotinib than for those receiving imatinib. Most grade 3/4 haematological events were also lower, with neutropenia events approximately double for patients receiving imatinib (20%) compared with nilotinib 300 mg (12%) and 400 mg (10%). 20 For non-haematological events, nausea, diarrhoea, vomiting and muscle spasm events were approximately three times higher for patients receiving imatinib than for those receiving both doses of nilotinib across all grades. Across all grades, oedema events were also higher for patients taking imatinib compared with both doses of nilotinib, particularly eyelid and periorbital oedema. 20 Conversely, rash, headache, pruritus and alopecia events were up to three times higher for both doses of nilotinib than for imatinib across all grades. 20
(CiC information has been removed.)
Biochemical abnormalities of grade 3/4 were uncommon in any study arm. Across all grades, increased bilirubin, glucose, ALT and AST were more common for patients receiving nilotinib 300 mg and 400 mg. Biochemical abnormalities are normally manageable and not clinically important. 20 As previously stated, nilotinib carries a ‘black box’ warning for possible heart problems due to QTc prolongation, where prolonged cardiac ventricular repolarisation can result in ventricular tachycardia and death. No patient in the ENESTnd study20 had an increased QTc of more than 500 milliseconds (where complexities may arise) at 12, 18 or 24 months’ follow-up. 20,78,81
The number of hospitalisations, hospital days and length of stay were lower for nilotinib 300 mg than for imatinib. There were more hospitalisations for patients receiving nilotinib 400 mg than for those receiving imatinib; the length of stay and hospital days were lower. 76
Health-related quality of life
Health-related quality of life was not reported in the DASISION29 or ENESTnd20 trials.
Indirect comparison of dasatinib and nilotinib
No trials compared dasatinib and nilotinib head to head. However, an indirect comparison of nilotinib to dasatinib was carried out using results from the DASISION29 and ENESTnd trials. 20
The primary outcomes reported are MMR at 12 months and CCyR by 12 months. Because the DASISION trial29 reported CCyR as well as confirmed CCyR, two sets of results are reported for the CCyR outcome. 29 As shown in Table 18, there was no difference between dasatinib and nilotinib for CCyR, MMR or CMR rates for 12 months’ or 24 months’ follow-up.
| Outcome | aComparison20,29,75,88 | PenTAG (current review) | Oxford Outcomes (2010) | (CiC information has been removed.) | |||
|---|---|---|---|---|---|---|---|
| Odds ratio | 95% CI | Odds ratio | 95% CI | (CiC information has been removed) | (CiC information has been removed.) | ||
| MMR at 12 months | Nilotinib (300 mg) vs dasatinib | 1.28 | 0.77 to 2.16 | 1.33 | 0.77 to 2.15 | (CiC information has been removed) | (CiC information has been removed) |
| Nilotinib (400 mg) vs dasatinib | 1.24 | 0.74 to 2.08 | 1.28 | 0.74 to 2.06 | (CiC information has been removed) | (CiC information has been removed) | |
| Best MMR by 24 months | Nilotinib (300 mg) vs dasatinib | 1.53 | 0.93 to 2.51 | (CiC information has been removed) | (CiC information has been removed) | ||
| Nilotinib (400 mg) vs dasatinib | 1.22 | 0.75 to 2.00 | (CiC information has been removed) | (CiC information has been removed) | |||
| CCyR by 12 months | Nilotinib (300 mg) vs dasatinib | 1.09 | 0.61 to 1.92 | 1.13 | 0.61 to 1.93 | (CiC information has been removed) | (CiC information has been removed) |
| Nilotinib (400 mg) vs dasatinib | 0.95 | 0.54 to 1.67 | 0.99 | 0.54 to 1.67 | (CiC information has been removed) | (CiC information has been removed) | |
| Complete confirmed CyR by 12 monthsb | Nilotinib (300 mg) vs dasatinib | 1.28 | 0.74 to 2.20 | (CiC information has been removed) | (CiC information has been removed) | ||
| Nilotinib (400 mg) vs dasatinib | 1.12 | 0.65 to 1.92 | (CiC information has been removed) | (CiC information has been removed) | |||
| CCyR by 18 months | Nilotinib (300 mg) vs dasatinib | (CiC information has been removed) | (CiC information has been removed) | ||||
| Nilotinib (400 mg) vs dasatinib | (CiC information has been removed) | (CiC information has been removed) | |||||
| CCyR by 24 months | Nilotinib (300 mg) vs dasatinib | 1.44 | 0.76 to 2.76 | ||||
| Nilotinib (400 mg) vs dasatinib | 1.21 | 0.64 to 2.28 | |||||
| Complete confirmed CyR by 24 monthsb | Nilotinib (300 mg) vs dasatinib | 1.40 | 0.77 to 2.56 | ||||
| Nilotinib (400 mg) vs dasatinib | 1.17 | 0.65 to 2.12 | |||||
| Complete molecular response by 24 months | Nilotinib (300 mg) vs dasatinib | 1.37 | 0.66 to 2.82 | ||||
| Nilotinib (400 mg) vs dasatinib | 1.04 | 0.50 to 2.17 | |||||
Oxford Outcomes conducted an indirect comparison of dasatinib and nilotinib based on the data from the DASISION and ENESTnd trials. 20,29,82 The indirect results for 12 months’ follow-up showed no statistical difference between dasatinib and nilotinib for CCyR or MMR data.
(CiC information has been removed.)
Signorovitch et al. 85 report on the indirect comparison of the DASISION29 and ENESTnd20 trials, with individual patient data for patients receiving nilotinib (ENESTnd20). 85 Individual patient data for patients receiving 300 mg nilotinib were weighted to match the baseline characteristics reported for patients receiving dasatinib including age, gender, ECOG performance and haematology laboratory values. After matching patients receiving 300 mg, nilotinib, compared with dasatinib, had significantly higher rates of MMR (56.8% vs 45.9%; p = 0.001) and OS (99.5 vs 97.3; p = 0.046). CCyR was not assessed due to different measurement procedures of the trials. We have analysed CCyR as we believe they are sufficiently similar to warrant comparison.
Overall clinical effectiveness conclusions
From the two trials available, both the second-generation TKIs dasatinib 100 mg (once daily; DASISION trial29) and nilotinib 300 mg (twice daily; ENESTnd trial20) have a statistically significant advantage compared with the first-generation TKI imatinib 400 mg (once daily) as measured by surrogate outcomes; however, there are insufficient data to assess longer-term patient-relevant outcomes (e.g. PFS, OS, HRQoL). Rates of CCyR and MMR for the second-generation TKI were higher, more rapidly attained, and deeper (MMR) compared with imatinib.
All three drugs were well tolerated with discontinuation due to AEs of < 10%.
With no head-to-head data available, an indirect comparison analysis was conducted between dasatinib and nilotinib. There was no difference between dasatinib and nilotinib for the primary outcomes of CCyR or MMR at 12 or 24 months’ follow-up. The results of the DASISION29 and ENESTnd20 trials are summarised in Tables 19 and 20, respectively.
| DASISION29 (dasatinib 100 mg vs imatinib 400 mg) | |
|---|---|
| CCyR |
Rates of CCyR and confirmed CCyR were significantly higher (11%) for patients receiving dasatinib compared with imatinib by 12 months (p < 0.008), but not by 24 months (4%; p > 0.1) CCyR rates were higher across all Hasford risk groups The difference in confirmed CCyR rates were maintained by 18 months. Time to a CCyR was shorter for patients receiving dasatinib at 12, 18 and 24 months (HR 1.5; p < 0.0001) |
| MMR |
Rates of MMR were significantly higher (18%) for patients receiving dasatinib compared with imatinib at 12-month (p < 0.001), which was maintained at 18 months (19%; p < 0.001) MMR rates were higher across all Hasford risk groups Time to a MMR was also shorter for patients receiving dasatinib at 12 months (HR 2.0, p < 0.0001), 18 months (HR 1.84; p < 0.0001) and 24 months (HR 1.69; p < 0.0001) |
| Survival | PFS and OS were similar between dasatinib and imatinib at 12 months’ follow-up (PFS 96% vs 97%; OS 97% vs 99%), 18 months’ follow-up (PFS 94.9% vs 93.7; OS 96 vs 97.9%) and 24 months’ follow-up (PFS 93.7% vs 92.1; OS 95.3 vs 95.2%) |
| AEs |
Rates of haematological events were similar between dasatinib and imatinib Except grade 3 or 4 thrombocytopenia, where there were nearly twice as many events in the dasatinib arm (19%) compared with the imatinib arm (10%) Pleural effusion rates were higher for patients receiving dasatinib (12%) compared with patients receiving imatinib (0%) Rates of non-haematological events demonstrated higher rates of fluid retention and superficial oedema for patients receiving imatinib across all grades Other non-haematological events generally appeared lower for dasatinib compared with imatinib, including rash, vomiting, nausea and myalgia |
| ENESTnd20 (nilotinib 300 mg vs imatinib 400 mg) | |
|---|---|
| CCyR |
Rates of CCyR were significantly higher (15%) for patients receiving nilotinib 300 mg compared with imatinib by 12 months (p < 0.001) CCyR rates were higher across all Sokal risk groups The difference in CCyR rates were maintained by 18 and 24 months (p < 0.002) |
| MMR |
Rates of MMR were significantly higher (22%) for patients receiving dasatinib compared with imatinib at 12-month (p < 0.001) MMR rates were higher across all Sokal risk groups The difference in MMR rates were maintained at 18 and 24 months. (CiC information has been removed) Median time to MMR was significantly shorter (p < 0.0001) for patients receiving nilotinib 300 mg (8.6 months; 95% CI 8.3 to 11.1 months) compared with patients receiving imatinib (22.1 months; 95% CI 19.5 to 27.6 months) For patients with an MMR at 12 months, 93% receiving nilotinib 300 mg and 92% receiving imatinib were still in MMR at 24 months |
| Survival |
PFS at 24 months was not statistically different for nilotinib 300 mg (98%) compared with imatinib (95.2%) At 18 and 24 months, OS was not statistically different for nilotinib 300 mg (98.5%; 97.4%) compared with imatinib (96.9%; 96.3%) |
| AEs |
Haematological events across all grades were lower for patients receiving nilotinib 300 mg than those receiving imatinib For non-haematological events, nausea, diarrhoea, vomiting and muscle spasms events were up to three times higher for patients receiving imatinib compared with nilotinib 300 mg across all grades Conversely rash, headache, pruritus and alopecia events were up to three times higher for nilotinib 300 mg compared with imatinib across all grades |
Chapter 4 Assessment of evidence to support the use of complete cytogenetic response and major molecular response as surrogate outcomes
Owing to short-term follow-up, DASISION29 and ENESTnd20 trials both provide surrogate outcomes as indicators of potential patient benefit. For a biomarker to be accepted as an appropriate surrogate measure of the final outcome, the following criteria should be met:
-
evidence of biological plausibility of relationship between the surrogate outcome and the final patient-relevant outcome (from pathophysiological studies and/or understanding of the disease process)
-
evidence demonstrating consistent association between surrogate outcome and final patient-relevant outcome (from observational studies)
-
evidence demonstrating treatment effects on the surrogate correspond to treatment effects on the patient-relevant outcome (from RCTs). 35
As discussed in Chapter 1 (see Surrogate outcomes), two published trials have both presented evidence supporting (major or complete) CyR as a surrogate outcome in the prediction of all-cause survival for patients with CML in CP receiving first-line IFN treatment. 37,89 Our initial literature searches (see Chapter 3, Identification of evidence) failed to identify an assessment of the evidence for the use of CyR or molecular response as acceptable surrogate outcome for long-term (≥ 1 year) OS within the TKI class of therapies (i.e. imatinib, dasatinib and nilotinib) for the first-line treatment of CP-CML.
We therefore undertook this systematic review to assess the evidence base for the use of CyR and molecular response as surrogate measures for survival or HRQoL with dasatinib, nilotinib and imatinib.
Methods for reviewing effectiveness of surrogate outcome measures
This systematic review was undertaken following the general principles published by the NHS CRD and the PRISMA guidelines. 63,64
Identification of studies
The search strategy comprised of the following main elements:
-
searching of electronic databases
-
scrutiny of bibliographies of retrieved papers and manufacturer submissions.
The following databases were searched: MEDLINE (Ovid), EMBASE, The Cochrane Library (including the CDSR, CCTR, DARE, NHS EED and HTA databases), NRR, Web of Science (including Conference Proceedings); CCT; ClinicalTrials.gov, FDA website and the EMA website. These were searched from search end date of the last technology appraisal report on this topic of October 2002. 65
The searches were written by CC with advice from TP, RA, RT, OC and RG. The surrogate terms circulated were cross-checked against a previous review of surrogate outcomes and CR for sensitivity and inclusion. 90
Inclusion and exclusion criteria
Inclusion criteria
Studies were included if they met the following criteria:
Population
Adults with CP-CML, naive to any IFN or TKI treatment.
Interventions
Dasatinib or nilotinib or imatinib in accordance with the marketing authorisation.
Comparators
Any or none.
Outcomes
Final patient-relevant outcomes:
-
PCR
-
overall all-cause survival
-
HRQoL.
Potential surrogate outcomes:
-
CCyR
-
MMR.
Study design
Any observational or experimental study that reported the association between CCyR and/or MMR and any one of the above final patient-relevant outcomes.
We excluded conference abstracts, narrative reviews, editorials, opinion pieces, non-English-language papers and individual case studies.
Studies were selected in two stages. First, two reviewers (TP and OC) examined all titles and abstracts. Second, full texts of any potentially relevant studies were obtained and the relevance of each paper was assessed independently by the same two reviewers according to the inclusion and exclusion criteria, and any discrepancies were resolved by discussion.
Data extraction strategy
Study characteristics and surrogate/final outcome data were extracted by one reviewer (OC) using a standardised data extraction form and independently checked by a second (TP or RT). Data digitalisation software (WinDIG, version 2.5 WinDIG, Geneva, Switzerland) was used to extract data from Kaplan–Meier survival curves. Disagreements were resolved by discussion, with involvement of a third reviewer if necessary. Data extraction forms for each included study are included in Appendix 4.
Quality assessment strategy
The methodological quality of included studies was assessed according to a modified list of criteria specified by the CRD. Quality was assessed by one reviewer (OC) and judgements were checked by a second (TP or RT).
Internal validity
The instrument sought to assess the following considerations:
-
Is the hypothesis/aim/objective of the study clearly described?
-
Were the case series collected at more than one centre?
-
Are patient characteristics adequately described?
-
Are inclusion and exclusion criteria clearly reported?
-
Were data collected prospectively?
-
Were patients recruited consecutively?
-
Did all of the participants receive the same intervention?
-
Is the use of any concurrent therapies adequately described?
-
Was an ITT analysis performed?
-
Were dropouts from the trial adequately described?
In addition, data about population, treatment discontinuation and subsequent therapies, surrogate end points response and patient-relevant outcomes were recorded (see Appendix 4).
External validity
External validity was judged according to the ability of a reader to consider the applicability of findings to the UK CML population.
Methods of data synthesis
An initial review of included studies revealed two key limitations. First, there was a lack of data reported to assess the trial level association between TKI treatment effects on CCyR and TKI treatment effect on patient-relevant outcome. This would be needed for high-level evidence of surrogacy. Second, there was no presentation of data of the association of CCyR or MMR and HRQoL. It was therefore decided to focus on studies that reported OS and/or PFS stratified by either CCyR or MMR. 37,89
For each study, levels of OS and PFS were extracted by response stratum at each year following trial recruitment (or randomisation) up to the latest follow-up point reported. In most studies OS and PFS data were reported in Kaplan–Meier curves using landmark analysis to evaluate differences in the final patient-relevant outcomes between responder and non-responders. The landmark method determines each patient’s response at a fixed time point, with survival estimates calculated from that time point and associated statistical tests being conditional on patients’ landmark responses. (In the included papers, the survival probabilities referred to the starting point of the treatment rather than to the time of response.) Note that in this method, patients who die before the landmark time point are excluded from the analysis. 91
We selected 12 months after the start of first-line TKI therapy as the landmark for our analysis, as the DASISION29 and ENESTnd20 trials consider, respectively, the rate of MMR and confirmed CCyR at 12 months after randomisation as primary end points. 20,29 A weighted average of the OS and PFS at different yearly intervals was estimated for both the responders and non-responders by taking into account the initial number of patients in the two groups. Wilson 95% CIs were derived for each point estimate assuming binomial distributed variables and no censoring of data. 92 Analyses were carried out using Stata© version 11.2 (StataCorp, TX, USA).
Results
Identification of evidence
The electronic searches retrieved a total of 5033 titles and abstracts. Two papers were found by updated databases searches, and three were identified by reviewers through hand-searching and/or referenced in industry submissions. These papers were then excluded based on title and abstract because the population was treated with IFN or they were conference abstracts. 93–95
After de-duplication, 3555 papers were screened and the majority of them were excluded on title and abstract. Full text of the remaining 63 papers was requested for more in-depth screening. The process is illustrated in detail in Figure 9. The last step of the process was the inclusion of selected papers in the quantitative analysis for the assessment of both CCyR and MMR as surrogate measure for OS and PFS. Where a study had been reported in several publications, it was considered only once according to the type of relationship reported (CCyR and/or MMR vs OS and/or PFS) and the paper reporting maximum follow-up was used. One study from India was deemed not externally valid in portraying the UK patients with CML population and treatment response. 96 Although reporting on OS and PFS stratified by level of CyR, this study was excluded from the quantitative analysis. The details of studies excluded on full review, along with the reasons for their exclusion, are detailed in Appendix 5.
FIGURE 9.
Flow diagram study inclusion process surrogate outcomes.
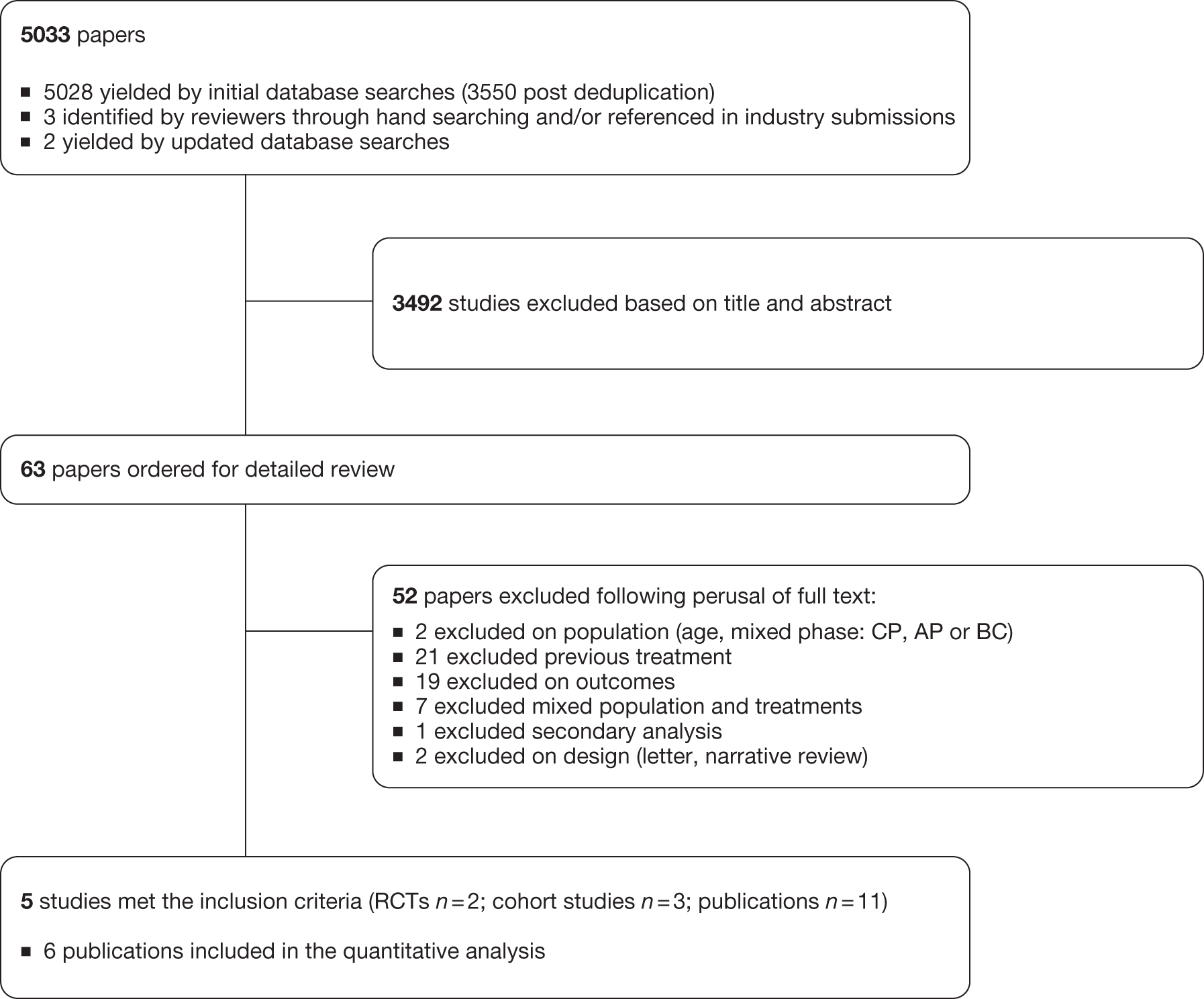
Assessment of surrogate evidence
Study and population characteristics
Eleven publications were included, all related to imatinib, reporting on five separate studies (Table 21). They are five reports of two cohort/single-arm studies, a single report of a RCT and five reports of the IRIS RCT. 26 Differences in details about the same study extracted from different papers are due to different follow-up and different analyses carried on. 27,28,96–104 No studies were identified considering patients with CML who were treated by dasatinib or nilotinib.
| Study (country) | Authors | Year published | Study type | n (imatinib arm) | Median age: years (range) | Intervention | Comparator | Follow-up (months) |
|---|---|---|---|---|---|---|---|---|
| (UK) | De Lavallade et al.97 | 2008 | Cohort/single arm | 204 | 46 (18–79) | Imatinib | None | 38 |
| Marin et al.103 | 2008 | Cohort/single arm | 224 | 46 (18–79) | Imatinib | None | 46 | |
| (India) | Rajappa et al.96 | 2008 | Cohort/single arm | 201 | 32 (18–72) | Imatinib | None | 29 |
| (US) | Kantarjian et al.101 | 2006 | Cohort/single arm | 279 | 48 (15–84) | Imatinib | None | 42 |
| Kantarjian et al.102 | 2008 | Cohort/single arm | 276 | 48 (15–84) | Imatinib | None | 48 | |
| (Germany) | Hehlmann et al.98 | 2011 | RCT | 324 | 54 (16–88) | Imatinib | Imatinib 400 mg/day combined with IFN imatinib 800 mg/day | 43 |
| IRIS26 (international) | Druker et al.27 | 2006 | RCT | 553 | 50 (18–70) | Imatinib | IFN-α plus cytarabine | 60 |
| Hochhaus et al.99 | 2009 | RCT | 551 | 50 (18–70) | Imatinib | IFN-α plus cytarabine | 70 | |
| Hughes et al.28 | 2003 | RCT | 333 | 51 (18–70) | Imatinib | IFN-α plus cytarabine | 25 | |
| Hughes et al.100 | 2010 | RCT | 476 | 50 (20–69) | Imatinib | IFN-α plus cytarabine | 77 | |
| Roy et al.104 | 2006 | RCT (retrospective comparison) | 551 | 50 (18–70) | Imatinib | IFN-α plus cytarabine | 42 |
Only the arm receiving imatinib standard-dose first-line therapy was considered from each RCT study, because the IRIS trial26 was inadequate to demonstrate a survival benefit for imatinib compared with IFN-α therapy in newly diagnosed Ph+ CP CML due to the high rate of crossover (65% at 72-month follow-up) from IFN-alpha to imatinib. 99 Hehlmann et al. ,105 on the other hand, compare the 400 mg/day imatinib with the high-dose therapy (i.e. 800 mg/day) or combined therapy with IFN.
The number of patients in the imatinib arm varied from 201 up to 553, with a median age between 32 and 54 years (overall range 15–88 years). The median follow-up ranged from 25 to 77 months, thus some evidence on the treatment effect on survival at 6 or 7 years after the initiation of imatinib is available. Two publications are UK studies, as many as are US studies; one publication is set in Germany, one in India, while the IRIS trial26 is a multicentre international study.
The inclusion criteria for the studies were similar: patients with newly diagnosed (within 6 months of study entry) Ph+ CML in CP, previously untreated with the exception of hydroxycarbamide and anagrelide. (Kantarjian et al. 102 include five (2%) patients with a CML duration of < 12 months.)
Assessment of study quality
Table 22 illustrates the results of the quality assessment performed on the 11 included publications.
As a number of publications reported different analyses based on the same study population we individually assessed a number of quality features associated with each of these studies separately, such as whether or not the ITT principle was applied.
| Assessment | DeLavallade et al.97 | Marin et al.103 | Rajappa et al.96 | Kantarjian et al.101 | Kantarjian et al.102 | Hehlmann et al.105 | Druker et al.27 | Hochhaus et al.99 | Hughes et al.28 | Hughes et al.100 | Roy et al.104 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Is the hypothesis/aim/objective of the study clearly described? | Yes | Yes | Yes | Yes | Yes | ||||||
| Were the case series collected at more than one centre? | No | No | No | No | Yes | ||||||
| Are patient characteristics adequately described? | Yes | Yes | Yes | Yes | Yes | ||||||
| Are inclusion and exclusion criteria clearly reported? | Yes | Yes | Yes | Yes | Yes | ||||||
| Is the use of any concurrent therapies adequately described? | Yes | No | Unclear | Yes | Yes | ||||||
| Were patients recruited consecutively? | Yes | Unclear | Unclear | Unclear | Unclear | ||||||
| Were data collected prospectively? | Yes | No | Yes | Unclear | Unclear | Yes | Yes | Yes | Yes | Yesa | No |
| Did all the participants receive the same intervention? | Yes | Yes | Yes | Unclear | No | Yes | Yesb | Yesa | Yesc | Yesd | Yesa |
| Was an ITT analysis performed? | No | No | No | No | No | No | No | No | No | No | No |
| Were dropouts from the trial adequately described? | Yes | Yes | No | No | No | Yes | Yes | Yes | No | No | Yes |
Analysis of overall survival and progression-free survival by cytogenetic and molecular response
For the purpose of this analysis, we focused on four main outcomes:
-
Final patient-relevant outcomes:
-
– OS, calculated since the start of imatinib therapy (or diagnosis) until death from any cause or date of last visit;
-
– PCR, described as survival without evidence of progression to accelerated or blast phase disease;27,99,100,103 or survival without evidence of AP or BC disease, white-cell count increasing, loss of complete haematological or CyR or death from any cause during therapy. 28,96,98,101,102,104
-
-
Potential surrogate outcomes:
-
– CCyR, defined as absence of the Philadelphia chromosome among at least 20 cells in metaphase in a bone marrow aspirate (see Chapter 1, Disease Monitoring and treatment response), as opposed to no CCyR;
-
– MMR, a standardised BCR–ABL : ABL ratio of less than 0.1% which is equivalent to a 3-log reduction from the 100% baseline for untreated patients (see Chapter 1, Disease monitoring and treatment response), as opposed to no MMR.
-
To prevent double counting, patient cohorts presented in more than one paper were included only once in the analysis. Selection was based on the study reporting the longest follow-up and an appropriate comparison between responder (complete cytogenetic responders vs not complete cytogenetic responders, or major molecular responders vs not major molecular responders) (Table 23). This choice is based on the primary end points assessed in the key trials assessing the clinical effectiveness of dasatinib and nilotinib, which consider, respectively, the rate of MMR and confirmed CCyR at/by 12 months after randomisation. 20,29 Kantarjian et al. compare patients showing a major CyR (≤ 35% Ph+ chromosomes in bone marrow aspirates) with patients without a MCyR at 12 months [the group of people achieving a minor CyR at 12 months after the first-line treatment initiation (n = 5) in Kantarjian et al. 102 study report was excluded from the pooled OS average estimate], whereas other studies compare patients with a CCyR with patients with minor CyR, no MMR or no CyR at all. 99,101–103 Molecular response is often assessed after a certain degree of CyR has been reached, so four out of seven papers present the final outcomes by a conjoint assessment of complete cytogenetic and MMR.
| Authors | Final outcome by level of CR | Final outcome by level of MMR | ||
|---|---|---|---|---|
| OS | PFS | OS | PFS | |
| De Lavallade et al.97 | CCyR vs no CCyR | CCyR vs no CCyR | CCyR + MMR vs CCyR + no MMR | CCyR + MMR vs CCyR + no MMR |
| Marin et al.103 | CCyR vs failurea | CCyR vs no CCyR | – | – |
| Kantarjian et al.101 | MCyR vs no MCyR | – | CCyR + MMR vs CCyR + no MMR | – |
| Kantarjian et al.102 | CCyR vs minor CyR | CCyR vs minor CyR | MMR vs no MMR | MMR vs no MMR |
| Hehlmann et al.98 | – | – | MMR vs no MMR | MMR vs no MMR |
| Druker et al.27 | – | CCyR vs no MCyR | – | CCyR + MMR vs no CCyR + no MMR |
| Hochhaus et al.99 | – | CCyR vs no CyR | – | – |
| Hughes et al.28 | – | CCyR + MMR vs no CCyR | – | CCyR + MMR vs CCyR + no MMR |
| Hughes et al.100 | – | – | MMR vs no MMR | MMR vs no MMR |
| Roy et al.104 | CCyR vs no CCyR | – | – | – |
As previously described (see Methods of data synthesis, above), 12-month landmark analysis after the starting of the imatinib therapy was selected for this analysis. Although this method should consider the survival of patients starting from the date when the event (CCyR or MMR) presents itself, survival data in the studies refer to the beginning of the first-line therapy; hence, the realignment of the year points’ survival probabilities towards a common time reference was not required.
Survival by level of cytogenetic response
Figure 10 shows the weighted pooled OS (95% CI) at yearly intervals after the initiation of imatinib treatment by CyR. Three publications provided data for the estimates. 97,102,104 The impact of failing to achieve a CCyR at 12 months becomes increasingly apparent over time, with increasing differences in OS between those who respond and those who do not. No non-responder group data at 48 months are reported. It was decided not to include the non-responder group data from Kantarjian et al. ,102 because they included five patients who developed a minor CyR at 12 months.
FIGURE 10.
Pooled weighted average (95% CI) of OS by level of CyR at yearly intervals after first-line imatinib initiation.
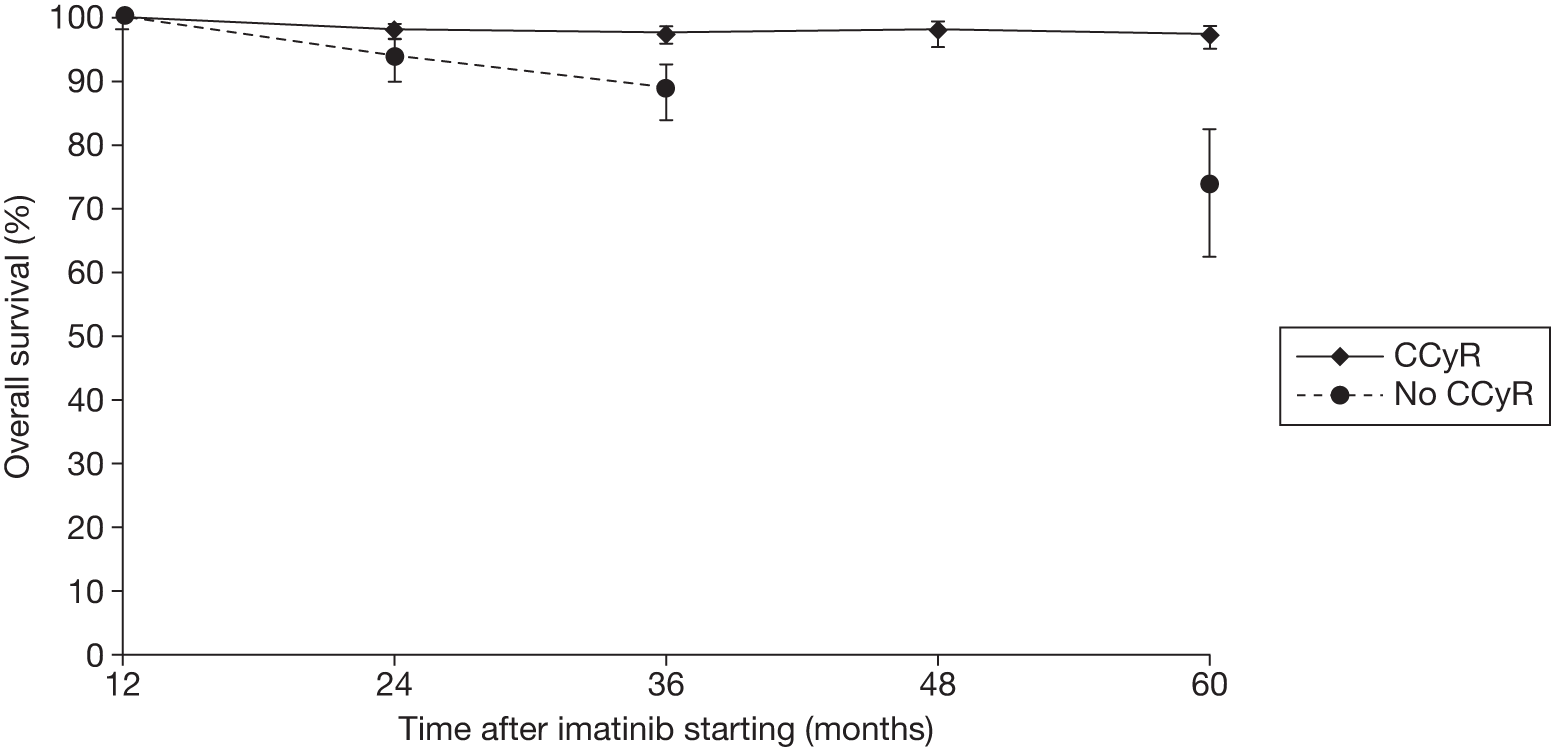
The weighted average of the PFS by CCyR at 12 months at yearly intervals after the initiation of imatinib therapy is shown in Figure 11. The estimates were obtained by the three papers that reported PFS across groups with different level of CyR. 97,99,102 The plotted values and the uncertainty around the estimates of OS and PFS by CyR are given in Table 24.
| Time (months) | OS% (95% CI) | PFS% (95% CI) | ||
|---|---|---|---|---|
| CCyR | No CCyR | CCyR | No CCyR | |
| 12 | 100 (100 to 99.3) | 100 (100 to 98.1) | 100 (100 to 99.3) | 98.9 (99.8 to 94) |
| 24 | 98.1 (98.9 to 96.5) | 94 (96.5 to 89.7) | 98.8 (99.4 to 97.4) | 94.3 (97.6 to 87.7) |
| 36 | 97.5 (98.5 to 95.9) | 89 (92.6 to 83.8) | 97.6 (98.5 to 95.9) | 85.5 (91.4 to 77.1) |
| 48 | 98 (99.3 to 95.3) | Not reported | 97.6 (98.5 to 95.9) | 85.5 (91.4 to 77.1) |
| 60 | 97.4 (98.6 to 94.9) | 74.1 (82.4 to 62.4) | 96.8 (97.9 to 95) | 75.2 (82.5 to 64.9) |
| 72 | Not reported | Not reported | 95.5 (97.0 to 93.1) | 80 (91.5 to 56.7) |
FIGURE 11.
Pooled weighted average (95% CI) of PFS by level of CyR at yearly intervals after first-line imatinib initiation.
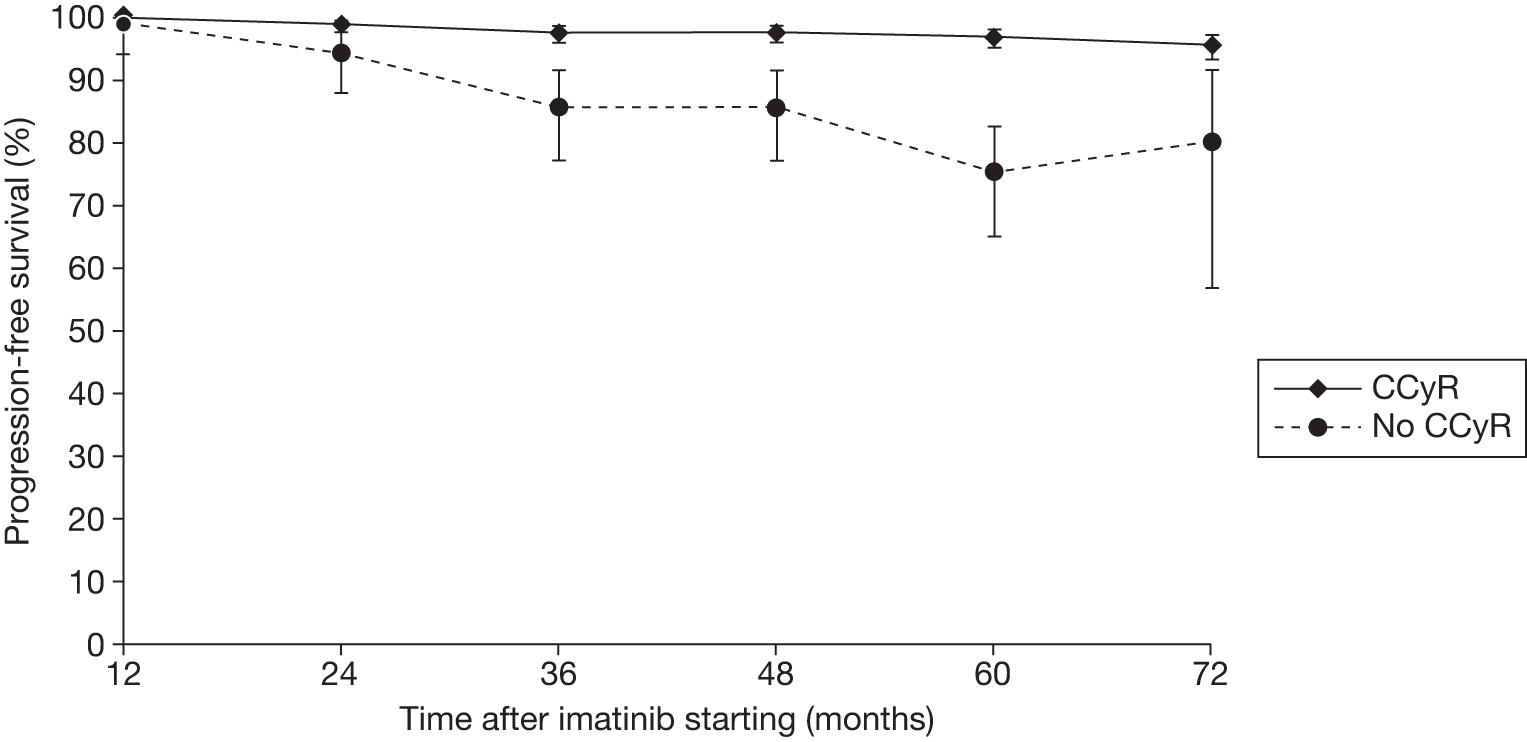
Survival by level of molecular response
Figure 12 shows the weighted average OS at yearly intervals after the start of the first-line imatinib therapy for CP-CML by level of molecular response. Three publications provided data for the estimates. 98,100,102 It is worth highlighting Hehlmenn et al. ,105 considered in the OS curves by landmark analysis of MMR at 12 months for the whole study population (n = 848) because, independent of the treatment approach (imatinib 400 mg/day, imatinib 800 mg/day, imatinib 400 mg/day + IFN-α), they found that MMR compared with no MMR at 12 months was associated with better PFS (99% vs 95%, p = 0.0143 at 3 years) and OS (99% vs 95%, p = 0.0156 at 3 years). Consistent with the weighting approach used in this report, the number of units involved in the construction of Kaplan–Meier curves, the overall sample size population in this case, was considered.
FIGURE 12.
Pooled weighted average (95% CI) of OS by level of molecular response at yearly intervals after first-line imatinib initiation.
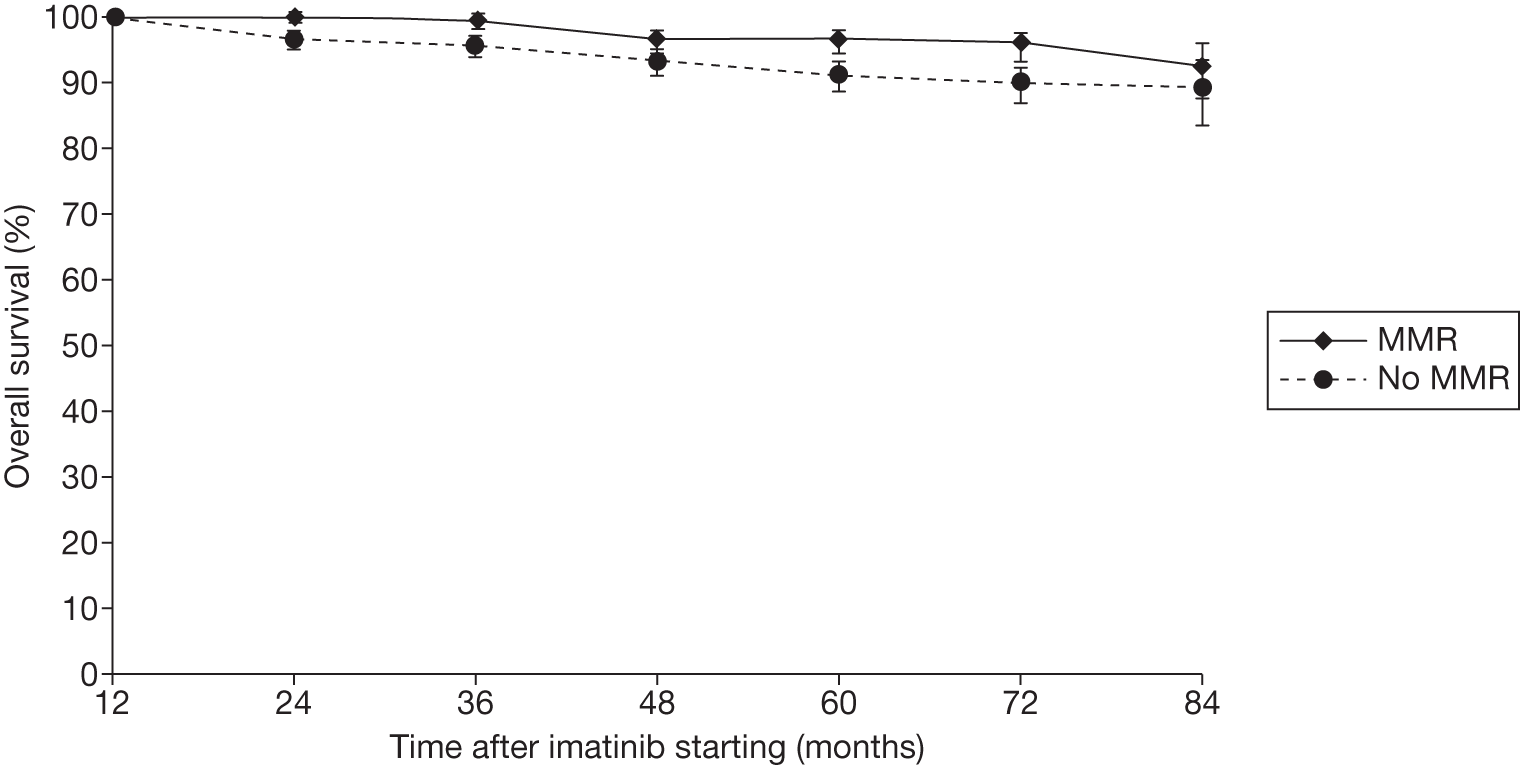
The pooled PFS by MMR at 12 months after the starting of imatinib therapy for CP-CML is shown in Figure 13. The estimates are derived from three publications that reported on PFS for groups of patients presenting different levels of molecular response. 98,100,102
FIGURE 13.
Pooled weighted average (95% CI) of PFS by level of molecular response at yearly intervals after first-line imatinib initiation.
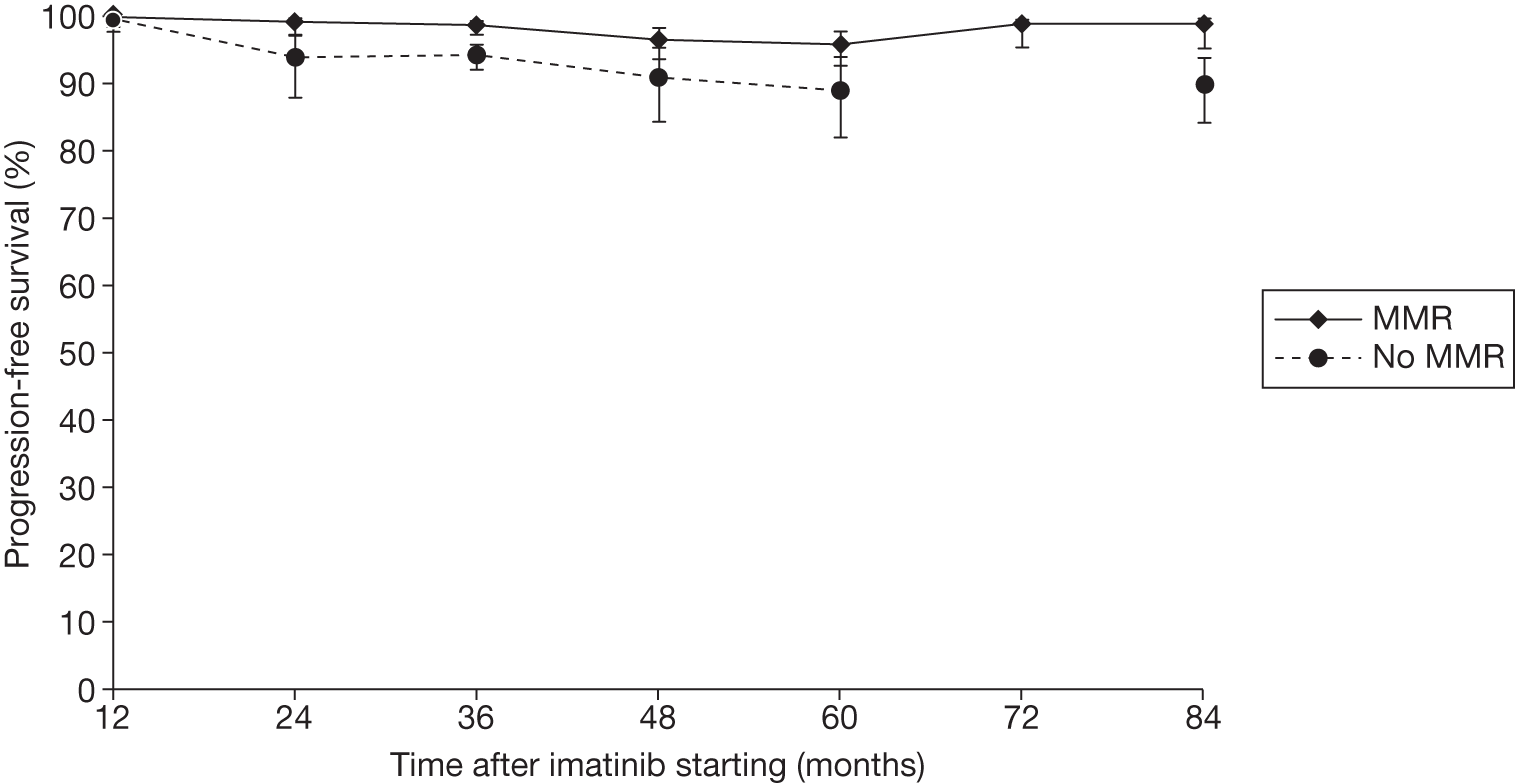
No non-responder PFS estimate at 72 months after therapy initiation was reported. The IRIS report26 by Hughes et al. 100 shows PCR curves by BCR–ABL transcript levels at 12 months converted to the International Scale (IS) (i.e. ≤ 0.1%, > 0.1% to ≤ 1%, > 1% to ≤ 10%, >10%) up to 84 months’ follow-up. These curves provide data for the PFS for patients achieving MMR at 12 months, defined as ≤ 0.1% IS, but not for the cumulative group of patients who do not achieve MMR at 12 months. The same authors give a tabulated value for the 7-year PFS in patients with no MMR at 12 months’ landmark time. The plotted values and the uncertainty around the estimates of OS and PFS by level of molecular response are given in Table 25.
| Time (months) | OS% (95% CI) | PFS% (95% CI) | ||
|---|---|---|---|---|
| MMR | No MMR | MMR | No MMR | |
| 12 | 100 (100 to 99.1) | 100 (100 to 99.4) | 100 (100 to 98.5) | 99.6 (99.9 to 97.8) |
| 24 | 100 (100 to 99.1) | 96.7 (97.9 to 95) | 99.2 (99.8 to 97.1) | 94 (97.3 to 87.9) |
| 36 | 99.2 (99.8 to 97.9) | 95.7 (97.1 to 93.8) | 98.6 (99.3 to 97.3) | 94.3 (95.8 to 92.1) |
| 48 | 96.7 (97.9 to 94.4) | 93.3 (95.0 to 91.0) | 96.6 (98.3 to 93.7) | 91 (95.4 to 84.4) |
| 60 | 96.6 (97.9 to 94.9) | 91.2 (93.2 to 88.6) | 95.8 (97.8 to 92.7) | 89 (93.9 to 82.0) |
| 72 | 92.5 (95.9 to 87.6) | 90 (92.3 to 87.0) | 99 (99.6 to 95.3) | Not reported |
| 84 | 96 (97.5 to 93.2) | 89.2 (93.4 to 83.5) | 99 (99.6 to 95.3) | 89.9 (93.9 to 84.2) |
Overall surrogate outcome conclusions
The end points assessed as surrogates for the target clinical outcomes are CCyR and MMR, in the 12 months after the first-line treatment (imatinib) initiation for CP chronic myelogenous leukaemia. A plausible biological rationale for the adoption of the two end points is clear after the disease mechanism and the definition of CCyR and MMR have been explained (see Chapter 1, Description of health problem and Disease monitoring and treatment response). Although biological plausibility is a basic step towards the identification of a surrogate end point, it alone is not sufficient for an end point to be accepted as a surrogate outcome. Evidence of an association between the end point and final patient-related outcome is also needed. Ideally, evidence should be in the form of multiple randomised controlled trials that have assessed the effects of the treatment on both the end point marker and final patient-relevant outcome. 35,90 However, this systematic review only identified evidence of the association between CyR and molecular response in patients and survival treated with TKI for CP-CML from the imatinib arms of three cohort studies and two randomised controlled trials. This observational comparison is considered level 2 evidence, rather than the best-quality evidence of a comparison of surrogate response according to randomised treatment allocation (level 1 evidence). 106 In addition, evidence is not available for dasatinib and nilotinib.
Nevertheless these studies do consistently show that patients who experience either a CCyR or MMR following 12 months’ imatinib treatment have better long-term (up to 7 years) OS and PFS than patients who do not respond at 12 months. Our inability to further explore the validation of the surrogate outcomes is limited by the amount and quality of data available (i.e. aggregate data instead of individual patient data). Other limitations include:
-
The reliance on the landmark analysis (patients who die before the landmark time point are excluded from analysis and response may, confoundingly, act as a surrogate marker for patients with favourable prognosis). 91
-
The pooling of subpopulations from different trials (although the exclusion criteria applied yielded very similar groups).
-
The assumption of no censoring for the estimation of 95% CI for the weighted average OS and PFS. 91
A strength is that we chose to approach the problem of deriving survival curves for patients in CP-CML conditioning to their achievement of either a CCyR or MMR at 12 months after the first-line treatment initiation, in a systematic way, using all of the available evidence to obtain weighted average estimates for the OS and PFS to inform the cost-effectiveness model discussed in this report [see Chapter 7, Surrogate-predicted overall survival (for surrogate survival only)].
Summary of surrogate outcomes
In summary, there is observational association evidence supporting the use of CCyR and MMR at 12 months as surrogates for OS and PFS in patients with CML in CP. This is based entirely on imatinib treatment studies. In the absence of evidence of adequacy of these surrogates for dasatinib and nilotinib as first-line therapies for CP-CML, assuming a TKIs class-specific relationship between the surrogate outcomes and the patient-relevant outcomes, these results can be potentially applied to other drugs in the same class.
Chapter 5 Cost-effectiveness: systematic review
Methods
We undertook a systematic literature search to identify economic evaluations of the therapies under investigation, which were carried out in line with the scope of the current assessment. Appendix 1 outlines in detail the search strategy used and databases searched. Manufacturer submissions to NICE were reviewed to identify additional studies.
All titles and abstracts were examined. The relevance of each paper was assessed according to the inclusion and exclusion criteria. The review was carried out by two researchers (RA and LC).
Results
Our literature search did not identify any published full economic evaluations meeting the inclusion criteria. However, we identified five conference abstracts that met the specified inclusion criteria. Three evaluated resource utilisation and costs associated with the use of TKIs for the management of CML;107–109 one examined long-term survival outcomes following treatment with dasatinib, imatinib and nilotinib;110 and one estimated lifetime QALYs and costs of Ph+ patients with CP-CML who were initiating therapy with nilotinib or imatinib using a literature-based Markov model. 111
There is insufficient detail in the abstracts or reports to undertake a detailed critical appraisal of the methods used or to rule out that some of them may relate to second-line treatment with nilotinib or dasatinib. The corresponding authors were contacted but no additional information was received; however, a summary of study characteristics and results is given below (Table 26).
| Study characteristics | Ovanfors et al. (2011)111 | Simons et al. (2009)107 | Szabo et al.(2010)109 | Taylor et al.(2010)110 | Wu et al.(2010)108 |
|---|---|---|---|---|---|
| Intervention | Nilotinib 300 mg b.i.d. | Dasatinib and nilotinib | TKIs (dasatinib, imatinib) | Dasatinib, nilotinib, imatinib | TKIs with pleural effusion |
| Comparator | Imatinib 400 mg q.d. | No comparator | No comparator (not head to head) | No comparator (not head to head) | TKIs with no pleural effusion |
| Patient population | Newly diagnosed patients with Ph+ CML | Patients with CML | Patients with CML | Newly diagnosed patients with CML | Patients with CML |
| Analysis by CML stage | Chronic | Unknown | Chronic; accelerated; blast | Chronic | Unknown |
| Model type | Literature-based Markov model | Not relevantb | Not relevantc | Disease model | Not relevanta |
| Time horizon | Lifetime | Unknown | Unknown | Unknown | Unknown |
| Perspective | Sweden | Unknown | UK | Unknown | Unknown |
| Discounting | Unknown | Unknown | Unknown | Unknown | Unknown |
| Effectiveness data | ENESTnd20 and IRIS26 | Unknown | Unknown | DASISION29 (dasatinib and imatinib) and ENESTnd20 (nilotinib) | MarketScan and Ingenix Impact databases (2001–9) |
| Base-case results | Discounted incremental cost per life-year and cost per QALY are estimated at US$21,028 and US$22,914, respectively | Total costs are US$2721.29 and US$426.44 for monitoring parameters for nilotinib and dasatinib, respectively | Higher costs were associated with patients not responding to treatment in each CML phase | QALYs and life-years were 12.238 and 14.727 for dasatinib; 11.506 and 13.822 for imatinib; and 12.016 and 14.426 for nilotinib, respectively | Compared with pleural effusion-free patients. Pleural-effusion patients have a substantial economic burden, with higher pleural effusion-related costs, CML-related costs, and total medical cost |
| Source of funding | Unknown; although author list suggests industry linked (Novartis) | Unknown; although author list suggests industry linked (BMS) | Unknown; although author list suggests industry linked (BMS) | Unknown; although author list suggests industry linked (BMS) | Unknown |
Chapter 6 Assessment of industry submissions
Introduction
Two manufacturer submissions were received for this MTA. BMS provided a full economic model for dasatinib and Novartis provided a full economic model for nilotinib. In this section, a summary of the critique of these two economic models is presented. The full critique of the two models is available in Appendix 7. There are two major sources of uncertainty in estimating the cost-effectiveness of dasatinib and nilotinib for first-line treatment of CML. First, the clinical effectiveness evidence from the DASISION29 RCT of dasatinib compared with imatinib and the ENESTnd20 RCT of nilotinib compared with imatinib is extremely immature, with current follow-up of only 2 years. Therefore, given that CML is a chronic disease, with current survival from diagnosis of around 15–20 years, it is necessary to extrapolate clinical effectiveness over many years, thus introducing substantial uncertainty.
Bristol-Myers Squibb submission
Scope of the submission
The submission from BMS considers the use of dasatinib for the first-line treatment of people with CML as an alternative to the standard dose of imatinib (400 mg daily) or nilotinib (600 mg daily).
The clinical effectiveness outcomes considered are:
-
OS
-
PFS
-
response rates
-
adverse effects of treatment
-
HRQoL.
The outcomes for the economic analysis are:
-
incremental cost per QALY
-
incremental cost per life-year gained.
In order to derive these outcomes, the following costs have been considered:
-
cost of first- and second-line TKIs
-
cost of second- or third-line treatment post TKI failure
-
the cost of treating serious AEs.
The time horizon for the economic analysis is between 46 and 86 years, and costs are considered from an NHS perspective. No subgroup analysis is conducted for the economic evaluation.
Summary of submitted cost-effectiveness evidence
The manufacturer uses a ‘time in state’ (area under the curve) model extrapolating CML-related survival and PCR data. The health states represent the CP and AP/blast phases, as well as death. Within the CP, patients may also be in first-, second- or third-line treatment, whereas in the AP/blast phase they may be receiving either third-line treatments or palliative care. Time is modelled in blocks of 1 month (Figure 14).
FIGURE 14.
Bristol-Myers Squibb model structure. Source: Figure 5, p. 40 of BMS submission.
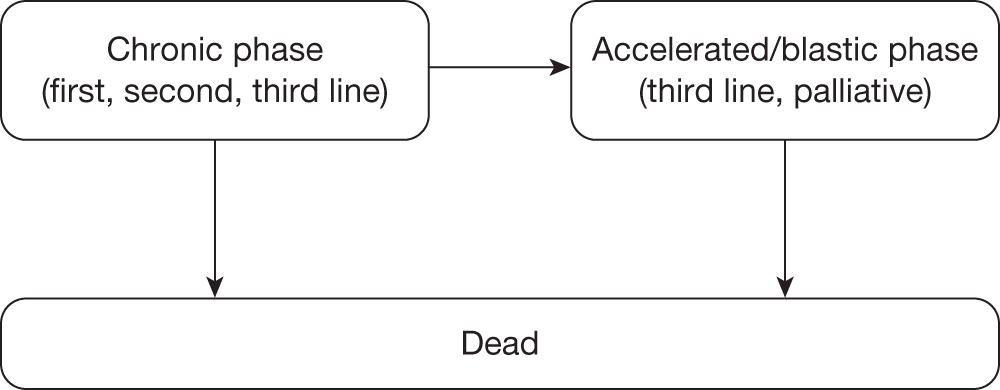
Bristol-Myers Squibb have modelled one scenario with three different comparators. The interventions and sequence of treatments are summarised in Table 27.
| Line of treatment | Intervention | Comparator 1 | Comparator 2 |
|---|---|---|---|
| First line | Dasatinib (100 mg) | Imatinib (400 mg) | Nilotinib (600 mg) |
| Second line | Nilotinib (800 mg) |
Dasatinib (100 mg) or nilotinib (800 mg) (50 : 50 spilt) |
Dasatinib (100 mg) |
| Third line | SCT or chemo/combination therapy or in-hospital palliative care | SCT or chemo/combination therapy or in-hospital palliative care | SCT or chemo/combination therapy or in-hospital palliative care |
The BMS base-case analysis produces incremental cost-effectiveness ratios (ICERs) of (Table 28):
-
£26,000 per QALY for dasatinib in comparison with imatinib as first-line TKI, and
-
£145,000 per QALY for nilotinib compared with dasatinib (nilotinib provides more benefit at greater cost than dasatinib) as a first-line TKI.
| Dasatinib | Imatinib | Nilotinib | ||
|---|---|---|---|---|
|
PFS (years, undiscounted) PYs (years, undiscounted) |
Mean | 19.16 | 17.14 | 19.28 |
| PYs (years, undiscounted) | Mean | 1.30 | 1.69 | 1.31 |
| Life-years (undiscounted) | Mean | 20.46 | 18.83 | 20.59 |
| QALYs (discounted) | PFS | 9.50 | 7.97 | 9.66 |
| PY | 1.14 | 1.92 | 1.04 | |
| Total | 10.64 | 9.89 | 10.70 | |
| First-line drug cost, £ (discounted) | 283,209 | 84,836 | 282,887 | |
| Second-line drug acquisition cost, £ (discounted) | 60,336 | 164,690 | 77,350 | |
| Third-line treatment cost, £ (discounted) | 82,324 | 145,215 | 75,619 | |
| AEs first-line, £ (discounted) | 2321 | 818 | 1291 | |
| AEs second-line, £ (discounted) | 412 | 1159 | 562 | |
| AEs third-line, £ (discounted) | 310 | 616 | 265 | |
| SCT,a £ (discounted) | 5350 | 10,093 | 4954 | |
| Other, £ | 63,955 | 70,864 | 63,685 | |
| Total costs, £ (discounted) | 498,217 | 478,293 | 506,613 | |
| ICERs | ||||
| Cost/life-year gained, £ (dasatinib vs imatinib) | 32,785 | |||
| Cost/life-year gained, £ (dasatinib vs nilotinib) | 116,447 | |||
| Cost/QALY, £ (dasatinib vs imatinib) | 26,305 | |||
| Cost/QALY, £ (dasatinib vs nilotinib) | 144,778 | |||
The sensitivity analysis shows the key parameters to which the model is sensitive:
-
drug costs
-
OS
-
the cost of SCT.
The BMS model contained a number of formula errors. After correcting for these errors the model predicts ICERs of:
-
£36,000 per QALY for first-line dasatinib compared with first-line imatinib, and
-
£103,000 per QALY for dasatinib compared with nilotinib (dasatinib provides more benefit at greater cost than nilotinib).
In the original model, the cost of nilotinib used by BMS does not account for the Patient Access Scheme (PAS) discount applied to nilotinib.
[Academic-in-confidence (AiC) information has been removed.]
Including this change, the BMS model predicts an ICER of £45,600 per QALY for dasatinib compared with imatinib. When comparing dasatinib with nilotinib, the model predicts that nilotinib is more effective and less costly.
Furthermore, BMS assume that dasatinib is taken as a third-line treatment in all treatment arms. However, in the NICE draft guidance final appraisal determination, dasatinib was not recommended (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). (In the draft guidance on 18 August 2011, NICE has recommended nilotinib for the treatment of the chronic and APs of CML that is resistant or intolerant to standard-dose imatinib. Dasatinib and high-dose imatinib are not recommended in the draft guidance. Consultees have the opportunity to appeal against the draft guidance. Until NICE issues final guidance, NHS bodies should make decisions locally on the funding of specific treatments. This draft guidance does not mean that people currently taking dasatinib or high-dose imatinib will stop receiving them. They have the option to continue treatment until they and their clinicians consider it appropriate to stop.) When the BMS model is adjusted so that dasatinib is not taken third line, the ICER of dasatinib compared with imatinib increases further, from £45,600 to £64,000 per QALY, and nilotinib is still more effective and less costly than dasatinib.
Finally, BMS assumes that half of all patients in the imatinib and nilotinib treatment arms who are eligible for second-line treatment take dasatinib. Again, in the NICE draft guidance final appraisal determination, dasatinib was not recommended (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). When the BMS model is adjusted so that dasatinib is not taken second line, and instead when we assume that all second-line patients in the imatinib arm take nilotinib second line, the ICER of dasatinib compared with imatinib increases further, from £64,000 to £96,000 per QALY. There appears to be no simple way to adjust the BMS model to disallow for patients taking dasatinib second line.
In summary, the BMS adjusted model yields an ICER for dasatinib compared with imatinib of £96,000 per QALY. Furthermore, nilotinib is more effective and less costly than dasatinib.
Commentary on the robustness of the submitted evidence
Strengths
-
The approach taken to modelling is reasonable although quite complex.
-
The sources and justification of estimates are also generally reasonable.
-
Resource use is largely based on a survey of six UK clinicians who manage patients with CML.
Weaknesses
-
There are a number of formula errors in the BMS model. When corrected, the base case ICER changes from £26,000 to £36,000 per QALY for dasatinib in comparison with imatinib, and from £145,000 to £103,000 per QALY for dasatinib in comparison with nilotinib.
-
Bristol-Myers Squibb does not account for the reduced price of nilotinib due to the PAS discount. In addition to the formula errors, if the (CiC information has been removed)discount in the price of nilotinib in first line and second line is accounted for, the best-case ICER for the BMS model is £45,600 per QALY for dasatinib compared with imatinib. When comparing dasatinib with nilotinib, the model predicts that nilotinib is more effective and less costly. However, it is acknowledged that BMS was unable to account for the discount as it did not have knowledge of the PAS discount at the time of its submission.
-
The starting age of the simulated cohort, 46 years, is considerably lower than the mean age of newly diagnosed patients with CML in the UK (56 years).
-
The model does not adopt a lifetime time horizon. Instead the model is run until the cohort is 86 years old, at which point 20% of the cohort is still alive. If the model is extended to the age of 100 years, 10% of the population is still alive. Assuming an equal distribution of males and females, data from the ONS predict that 2% of those alive at 46 years will be alive at the age of 100 years. This suggests that BMS overestimates the period that those with CML will survive.
-
Bristol-Myers Squibb uses 42-month follow-up data from a RCT to predict OS for those with a complete, partial and ‘less than partial’ CyR to treatment at 12 months. 104 Survival data are digitally extracted from published Kaplan–Meier curves and fitted to a Weibull distribution. There is no use of MMR response rates; the model uses only CyR rates.
-
Bristol-Myers Squibb outlines the clinical effectiveness of second-line TKIs in its submission. However, these data are not used to model the cost-effectiveness of second-line therapy.
-
With the BMS model, there are a number of assumptions that are not defined in detail. In addition, several parameters within the manufacturer submission do not reflect the data that is used in the model. For example, the data used to estimate the PFS curves (explained in table 19, p. 47 of the manufacturer submission) do not match the data in the model. Also, the source quoted for PFS data in the submission is Hochhaus et al. 99 However, the model appears to be using data from Druker et al. ,27 which is a study with a shorter follow-up period. If the model is updated to use data from Hochhaus et al. 99 then the ICER changes as follows:
-
– dasatinib compared with imatinib: from £36,052 to £42,556 per QALY
-
– dasatinib compared with nilotinib: from £103,483 to £103,593 per QALY.
-
-
Bristol-Myers Squibb assumes that dasatinib is taken second and third line. Given that BMS prepared its submission before NICE’s recent draft guidance FAD on second-line TKIs, the BMS assumption on the use of dasatinib second and third line was reasonable. However, in the NICE draft guidance FAD, dasatinib second and third line was not recommended (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). When the BMS model is adjusted to remove dasatinib second and third line, the cost-effectiveness of dasatinib worsens substantially, as quantified above.
-
Bristol-Myers Squibb developed a highly complex model in an area in which data are not of high quality. We believe the cost-effectiveness model could have been developed in a simpler way.
-
It is not clear how BMS calculated the cost of SCT. (CiC information has been removed.)
-
On several occasions, the BMS report of the modelling differs from the actual model.
Areas of uncertainty
The BMS model does not provide the raw data that were used to fit the OS and time to treatment discontinuation curves. However, the choice of distribution and coefficients of the distribution appear to be correct on the basis of graphs showing the observed data and the fitted curves.
A considerable area of uncertainty is the chosen sequence of second-line TKI treatments that might follow failure of different first-line TKIs. This is partly because the submission was prepared before NICE’s draft guidance FAD on the use of dasatinib, nilotinib or high-dose imatinib as second-line treatments (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). However, uncertainty also results from the fact that data on the effectiveness of second-line TKI treatments are only available following the use of imatinib as first-line treatment.
Key issues
-
The BMS model does not use the cost of nilotinib agreed under the PAS in the submission. However, it is acknowledged that BMS was unable to account for the discount, as it did not have knowledge of the PAS discount at the time of its submission.
-
The BMS model is structured in such a way that it would require significant changes to run it without second-line treatment, should this be required by NICE.
-
The time horizon chosen by the BMS model does not reflect the lifetime of a patient with CML. In the model, nearly 20% of the population is still alive in the last cycle (86 years old), suggesting that the model overestimates the period that those with CML will survive.
-
The BMS model has a number of formula errors, correcting for which impacts on the ICER.
-
The cost and proportions of patients who receive SCT have a significant impact on ICERs, but the source of the BMS estimates of these parameters is unclear. Clinical opinion is required to assess whether or not the BMS assumption on the provision and costing of SCT is appropriate.
Novartis submission
Scope of the submissions
The submission from Novartis considers the use of nilotinib for the first-line treatment of people with CML as an alternative to the standard dose of imatinib (400 mg daily). In one analysis, dasatinib is used in the cost-effectiveness model as second-line treatment when first-line treatment with imatinib or nilotinib fails. In the other analysis, no second-line TKIs are assumed.
The clinical effectiveness outcomes considered are:
-
PFS
-
time to discontinuation
-
adverse effects of treatment
-
HRQoL.
The outcomes for the economic analysis were:
-
incremental cost per QALY
-
incremental cost per life-year gained.
In order to derive these outcomes the following costs were estimated in the model:
-
cost of first- and second-line TKIs,
-
cost of post-TKI failure second- or third-line treatment
-
the cost of treating AEs.
The time horizon for the economic analysis is lifetime and costs are considered from the NHS perspective.
The Novartis cost-effectiveness modelling reflects a cost discount (PAS for the cost of first-line nilotinib) (CiC information has been removed). Its cost of second-line nilotinib also reflects this cost discount (also a PAS). No subgroup analyses are conducted for the economic evaluation, although a policy scenario without the use of second-generation TKIs is simulated.
Summary of submitted cost-effectiveness evidence
The manufacturer uses a Markov approach to model the cost-effectiveness of nilotinib compared with the current standard of care (imatinib 400 mg daily). This model has nine states. Patients enter the model in the CP. The model estimates when one treatment fails and hence the patient is switched to an alternative treatment. At the end of each cycle, patients have a probability of remaining on current treatment, progressing to an alternative treatment or dying (Figure 15).
FIGURE 15.
Novartis model structure. Source: Novartis industry submission (p. 82).
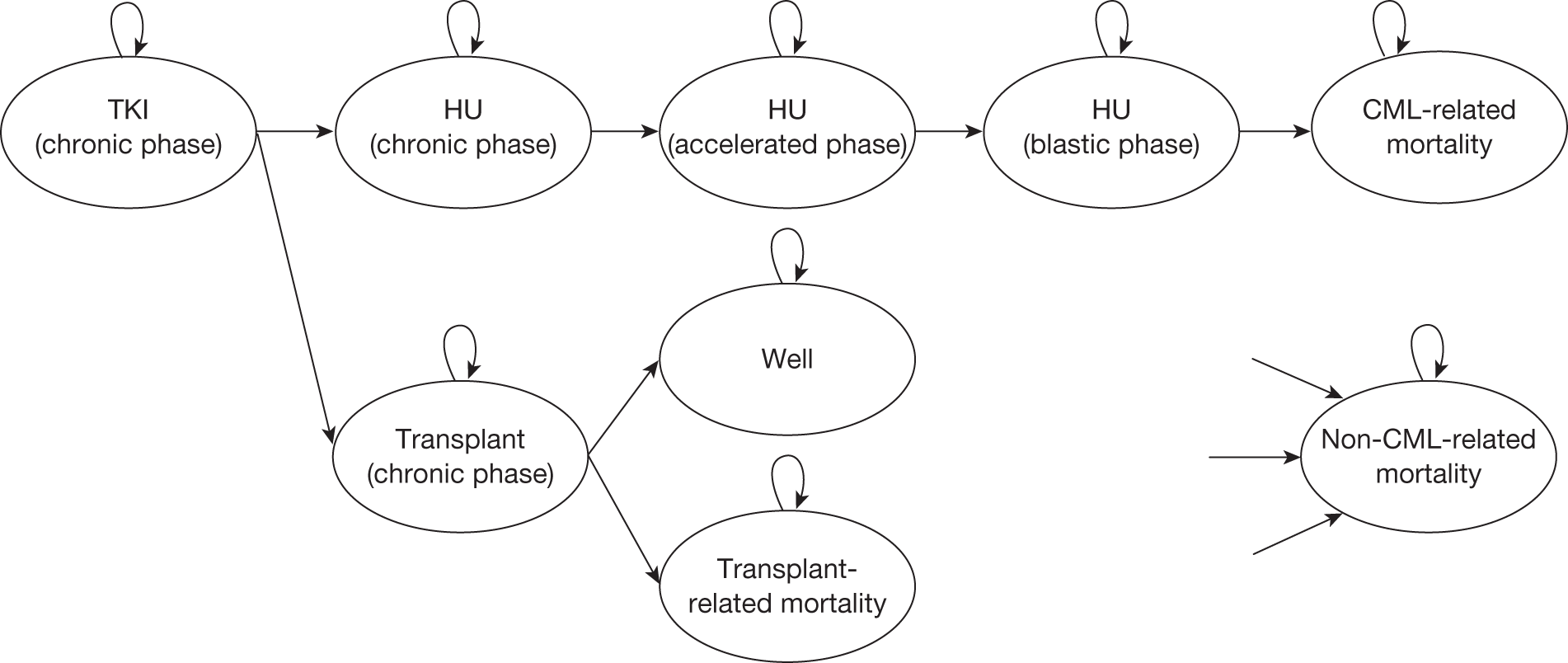
Novartis modelled two different scenarios to reflect the availability or not of second-generation TKIs as second-line treatment. The interventions and sequence of treatment are summarised in Table 29.
| Line of treatment | Scenario 1 | Scenario 2 | ||
|---|---|---|---|---|
| Nilotinib | Imatinib | Nilotinib | Imatinib | |
| First line | Nilotinib (600 mg) | Imatinib (400 mg) | Nilotinib (600 mg) | Imatinib (400 mg) |
| Second line | Dasatinib (100 mg) | Dasatinib (100 mg) | SCT or hydroxycarbamide | SCT or hydroxycarbamide |
| Third line | SCT or hydroxycarbamide | SCT or hydroxycarbamide | NA | NA |
The Novartis model predicts that nilotinib is both more effective and less costly than imatinib (dominates) when followed by dasatinib as second-line treatment. In a scenario analysis without dasatinib as second-line treatment, the model predicts an ICER of £5908 per QALY for nilotinib in comparison with imatinib (Table 30).
| Model output | Nilotinib/dasatinib | Imatinib/dasatinib | Nilotinib | Imatinib | ||
|---|---|---|---|---|---|---|
| PFS (years, undiscounted) | Mean | 12.66 | 11.94 | 10.64 | 9.30 | |
| PY (years, undiscounted) | Mean | 0.88 | 0.90 | 0.74 | 0.68 | |
| Life-years (undiscounted) | Mean | 13.54 | 12.83 | 11.38 | 9.97 | |
| QALYs (discounted) | PFS | 9.93 | 9.38 | 8.31 | 7.25 | |
| PY | 0.47 | 0.48 | 0.40 | 0.37 | ||
| Total | 10.40 | 9.85 | 8.71 | 7.62 | ||
| First-line drug cost, £ (discounted) | 114,771 | 104,038 | 114,771 | 104,038 | ||
| Second-line drug acquisition cost, £ (discounted) | 57,532 | 77,284 | Refer to SCT | Refer to SCT | ||
| Third-line treatment cost, £ (discounted) | 170 | 175 | 411 | 147 | ||
| AEs first line, £ (discounted) | 111 | 178 | 111 | 178 | ||
| AEs second line, £ (discounted) | 37 | 51 | NA | NA | ||
| AEs third line, £ (discounted) | NA | NA | NA | NA | ||
| SCT, £ (discounted) | 28,772 | 31,183 | 42,383 | 49,986 | ||
| Other | 15,979 | 14,835 | 12,966 | 11,667 | ||
| Total costs, £ (discounted) | 217,373 | 227,744 | 170,643 | 166,015 | ||
| ICERs | ||||||
| Cost/life-year gained, £ (nilotinib vs imatinib, with second line) | –(27,739) | |||||
| Cost/life-year gained, £ (nilotinib vs imatinib, without second line) | 4701 | |||||
| Cost/QALY, £ (nilotinib vs imatinib, with second line) | –(34,889) | |||||
| Cost/QALY, £ (nilotinib vs imatinib, without second line) | 5908 | |||||
The sensitivity analysis shows the key parameters to which the cost-effectiveness results are sensitive to are:
-
drug costs (i.e. without PAS)
-
time to discontinuation of first-line TKI.
No major formula errors have been identified in the Novartis model.
Commentary on the robustness of submitted evidence
Strengths
-
The approach taken to modelling is reasonable.
-
The sources and justification of estimates are also generally reasonable.
Weaknesses
-
Novartis makes no use of the major molecular and CCyR rates from the RCT of nilotinib compared with imatinib, both of which are important indicators of clinical effectiveness.
-
We believe that the Novartis method of estimating the time on hydroxycarbamide in CP is flawed.
Areas of uncertainty
The Novartis model does not provide the raw data which were used to fit the OS and time-to-treatment discontinuation curves. However, the choice of distribution and coefficients of the distribution appear to be correct on the basis of graphs showing the observed data and the fitted curves.
Another area of uncertainty is the chosen sequence of second-line TKI treatments that might follow the failure of different first-line TKIs. This is partly because this submission was prepared before NICE’s draft guidance FAD on the use of dasatinib, nilotinib or high-dose imatinib as second-line treatments (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). However, uncertainty also results from the fact that data on the effectiveness of second-line TKI treatments are only available following the use of imatinib as first-line treatment.
Another area of uncertainty is regarding the cost and utility of stem cell patients. Assumptions around SCT have a significant impact on the model. Novartis uses a one-off cost of £99,224 for each transplant with a post-transplant utility for survivors of 0.813.
Key issues
-
Novartis uses a PAS for pricing nilotinib as first-line treatment. This has a significant impact on the results.
-
Novartis makes no use of the major molecular and CCyR rates from the RCT of nilotinib compared with imatinib, both of which are important indicators of clinical effectiveness.
-
The cost and the proportions of patients who receive SCT differ between the Novartis and BMS models and they have a significant impact on ICERs. Clinical opinion is required to assess whether or not the BMS assumption on the provision and costing of SCT is appropriate.
Summary of manufacturers’ cost-effectiveness submissions
A summary of the two cost-effectiveness submissions is displayed below:
-
Novartis uses PAS for pricing nilotinib as first-line treatment. This has significant impact on cost-effectiveness, and BMS was unable to reflect this in its model.
-
BMS and Novartis assume different second- and third-line treatments. BMS assumes that both dasatinib and nilotinib are available second line. In one analysis, Novartis assumes that only dasatinib is available second line, and in its other analysis it assumes that neither dasatinib nor nilotinib is available second line. However, in the NICE draft guidance FAD, nilotinib, but not dasatinib, was recommended to be used second line. We have adjusted the BMS model to reflect NICE’s guidance (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99).
-
The time horizon chosen by the BMS model does not reflect the lifetime of a patient with CML. In the model, nearly 20% of the population is still alive in the last cycle (86 years old).
-
The BMS model has a number of formulae errors, correcting for which has an impact on ICERs.
-
The cost and the proportions of patients who receive SCT differ between the models and have a significant impact on ICERs.
Chapter 7 Peninsula Technology Assessment Group cost-effectiveness analyses
There are various approaches to modelling the costs and effectiveness of treatments for CML, as exemplified by the quite different approaches taken by the two manufacturers of nilotinib and dasatinib for this MTA. In the following sections we describe:
-
an overview of the main alternative modelling approaches, given key sources of uncertainty
-
the choice of approaches (scenarios) for which we produce cost-effectiveness results
-
the methods for estimating or extrapolating survival and occupancy of key health/treatment states.
We then describe the value, source and justification for all utility, cost and other input parameters used in the model.
Approaches to modelling treatments for chronic myeloid leukaemia
There are two major sources of uncertainty in estimating the cost-effectiveness of dasatinib and nilotinib for first-line treatment of CML. First, the clinical effectiveness evidence from the DASISION RCT29 of dasatinib compared with imatinib and the ENESTnd RCT20 of nilotinib compared with imatinib is extremely immature, with current follow-up of only 2 years. Therefore, given that CML is a chronic disease, with current survival from diagnosis of around 15–20 years, it is necessary to extrapolate clinical effectiveness over many years, thus introducing substantial uncertainty. Second, cost-effectiveness is heavily influenced by our assumptions for subsequent lines of treatment, and there is much uncertainty about the nature and the cost of such treatment.
Given this extensive structural uncertainty, we believe that it is useful to present a range of deterministic scenario analyses, depending on key structural assumptions, which we believe cover the main plausible structural assumptions. Furthermore, given that it is not possible to designate any one scenario as the most plausible, we do not present a single base-case analysis.
Our scenario analyses are presented in Table 31. It shows two alternative assumptions relating to possible treatment sequences following the failure of first-line TKIs (table rows), three alternative approaches to estimating survival (right-hand columns), and also some scenarios in which only costs and benefits during first-line treatment are compared (the simplified method). The three alternative methods for estimating cost-effectiveness are:
-
the cumulative survival method, in which OS is estimated as the cumulative result of the duration of successive treatments
-
the surrogate survival method, in which OS is estimated from the 12-month treatment response, using either CCyR or MMR
-
the simplified method, in which the per-patient costs and benefits occurring after treatment with TKIs are assumed equal between treatment arms.
| Treatments | Method | |||||
|---|---|---|---|---|---|---|
| First line | Second line | Third line | Simplified? | Cumulative survival | MMR surrogate survival | CCyR surrogate survival |
| Imatinib | Hydroxycarbamide or SCT | None | No | 1 | 1a | 1b |
| Dasatinib | ||||||
| Nilotinib | ||||||
| Imatinib | Hydroxycarbamide or SCT | None | Yes | 2 | 2a | 2b |
| Dasatinib | ||||||
| Nilotinib | ||||||
| Imatinib | Nilotinib | Hydroxycarbamide or SCT | No | 3 | 3a | 3b |
| Dasatinib | ||||||
| Nilotinib | Hydroxycarbamide or SCT | None | ||||
| Imatinib | Nilotinib | Hydroxycarbamide or SCT | Yes | 4 | 4a | 4b |
| Dasatinib | ||||||
| Nilotinib | Hydroxycarbamide or SCT | None | ||||
Each of these methods is described in the following sections, together with their advantages and disadvantages for evaluation different first- and second-line TKI treatment sequences.
We did not model scenarios 3a, 3b, 4a and 4b (see Table 31). This is because the historical OS data used to estimate the surrogate relationships did not reflect the use of second-line TKIs. Therefore, although these analyses include the use of a TKI as second-line treatment, the relative effectiveness of this treatment compared with those having hydroxycarbamide or SCT second line would not be captured in any survival modelling based solely on the surrogate relationship.
Peninsula Technology Assessment Group model structure
The PenTAG cost-effectiveness model is a state-transition model with states for the main disease phases, and for the different possible treatments within the CP. It is very similar to the Novartis model in terms of states and allowable transitions.
Patients enter the model in the CP. During each model cycle, a patient is assumed to be in one of the health states. In Figure 16, arrows represent possible transitions between health states. At the end of each cycle, patients have a probability of remaining on their health state (shown by circular arrows), progressing to an alternative state or dying (see Figure 16). In scenarios 3 and 4, after first-line treatment failure, patients in the imatinib and dasatinib treatment arms progress to second-line nilotinib, as shown in Figure 16 by the dotted ellipse. Patients in the nilotinib arm progress directly to hydroxycarbamide or SCT. In scenarios 1 and 2, all patients progress directly to hydroxycarbamide or SCT after first-line TKI.
FIGURE 16.
Structure of PenTAG cost-effectiveness model.
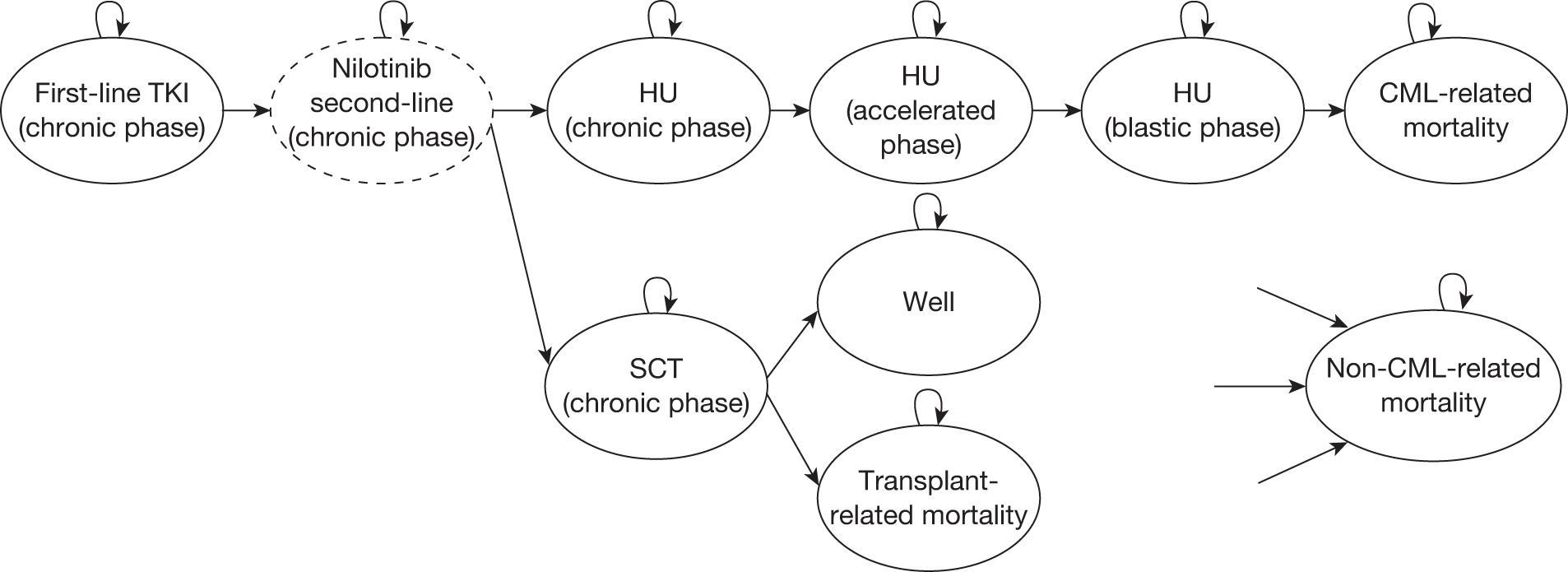
Cumulative survival approach
In this approach, OS for each treatment arm is estimated in scenarios 1 and 2 (see Table 31) as the sum of time on first-line treatment and OS following either hydroxycarbamide or SCT. For scenarios 3 and 4, OS for the nilotinib comparator is as for scenarios 1 and 2, whereas OS for the imatinib and dasatinib arms equals the sum of time on first-line treatment, time on second-line nilotinib and OS following either hydroxycarbamide or SCT. This general approach is the same as that used by Novartis (see Comparison of PenTAG model with Novartis model, below). The method ignores the CCyR and MMR response rates from the two main RCTs of first-line dasatinib compared with imatinib and first-line nilotinib compared with imatinib.
An important assumption in this approach is that OS after second-line nilotinib and OS after hydroxycarbamide or SCT are independent of previous treatment. In the cumulative survival method, the time-independent annual transition probability from CP to AP, while people take hydroxycarbamide, was assumed to be the same for all three treatment arms.
Surrogate-predicted survival approach
In this approach, OS for all three treatment comparators is estimated using a surrogate relationship based on MMR at 12 months on first-line TKI (scenarios 1a and 2a), and in a separate analysis, on CCyR at 12 months (scenarios 1b and 2b). The methods of estimating OS based on the surrogate relationships with MMR and CCyR are described below [see Surrogate-predicted overall survival (for surrogate survival only)] and are based on the results of our clinical effectiveness systematic review and meta-analysis of surrogate outcomes (see Chapter 4, Analysis of overall survival and progression-free survival by cytogenetic and molecular response).
Modelling for these scenarios uses only the proportion of patients with or without a response at 12 months. We also assume that, for a given response rate, OS is independent of first-line treatment comparator.
The model does not reflect possible differences in the depth, speed of achieving or duration of a response. Given that (1) dasatinib and nilotinib are believed to be superior to imatinib in all these respects (see Novartis and RCP submissions and our clinical effectiveness systematic review, see Chapter 3, Overall clinical effectiveness conclusions) and (2) the historical surrogate data are all based on OS for patients taking imatinib, it is possible that this method underestimates OS for dasatinib and nilotinib but the extent of this is unquantifiable.
The BMS modelling also predicts OS by a surrogate relationship based on CCyR, but not on MMR. Novartis does not model OS by a surrogate method, instead using only the cumulative survival method (see Comparison of PenTAG model with Novartis model and Comparison of PenTAG model with BMS model, below, comparing the PenTAG with the manufacturers’ economic analyses). Our scenario analyses, which make use of the surrogate-based survival relationships, do so by adjusting the analyses based on the cumulative survival approach. In the following paragraphs, we describe the adjustments to the cumulative survival model that are needed to reflect the surrogate OS for the three treatment arms estimated below [see Surrogate-predicted overall survival (for surrogate survival only)].
It is not surprising that OS for each treatment arm under the cumulative survival method is different to OS as predicted by the MMR and CCyR surrogate relationships, given that the cumulative survival method relies on numerous assumptions that have a cumulative impact. Specifically, in the Results section (see PenTAG cost-effectiveness results, below) we show that OS under the cumulative survival method is far shorter, at approximately 16–18 years, than under the surrogate survival methods, at approximately 21–23 years. We are then faced with the decision of how to adjust the model, which is based on the cumulative survival method, so that it predicts OS specific to each treatment as estimated by the surrogate survival method. BMS achieved this by leaving unaltered the transition probabilities under the cumulative survival method, but setting the transition probabilities that determine the times in AP and BC as the ‘balancing items’, so as to achieve the surrogate OS experienced in historical trials of imatinib.
We ruled out this approach because this would result in unrealistically long mean times in AP plus BC of approximately 5–8 years, the difference between typical OS predicted under the surrogate relationship and typical OS under the cumulative survival method. In practice, typical times in these advanced disease states are believed to be 6 months to 1 year (see Time on hydroxycarbamide in chronic phase, and time in accelerated phase and blast crisis, below).
For the transition probabilities in the PenTAG model, the mean times corresponding to first-line TKIs and second-line nilotinib were not altered (from their cumulative survival model values) because they are informed by good evidence from high-quality trials. This left three choices:
-
adjust the annual transition probability from CP to AP while people take hydroxycarbamide
-
adjust mortality after the SCT operation
-
or some combination of the above.
These choices seemed plausible given that the corresponding transition probabilities are informed by poorer-quality evidence. The third option was ruled out as too complex. The second option was ruled out because even if we assumed that all patients are completely cured after SCT then the modelled OS is still shorter than OS predicted from the surrogate relationships. The first option was selected as it was possible to model OS from the surrogate relationship. Under the surrogate survival method, this probability was unique for each treatment arm.
A pair of analyses was performed for each of scenarios 1a, 1b, 2a and 2b (see Table 31). First, the transition probability from CP to AP while people take hydroxycarbamide, unique to each treatment arm, was set to precisely match the mean OS from the appropriate surrogate relationship. Second, the transition probability from CP to AP for the imatinib treatment arm was left unadjusted (as in the cumulative survival method) but the transition probabilities from CP to AP for the nilotinib and dasatinib treatment arms were adjusted so as to model the differences in OS between treatment arms from the surrogate OS. These adjustments are shown graphically in the Results section (see Figure 42). The purpose of the second analysis was to capture the essence of OS estimated by the historical surrogate data, which is the magnitude of the difference in OS according to response (see Chapter 4).
Simplified method
In this simplified approach (used in scenarios 2, 2a, 2b and 4 in Table 31), the post-TKI (first-line TKIs and second-line nilotinib) per-patient costs and QALYs are set to be equal across treatment arms. The costs and QALYs while patients are on TKIs are modelled specific to each treatment arm, exactly as normal. However, because slightly different proportions of patients will have died during the time when they are taking first- or second-line TKIs, there will still be small differences in the total costs and QALYs accrued after this time point between the treatments compared.
Specifically, suppose the total discounted per-patient post-TKI treatment cost in the imatinib treatment arm is given as Cim, and suppose the proportion of patients who are still alive and start second- or third-line treatment on hydroxycarbamide or SCT in the imatinib and nilotinib treatment arms are Pim and Pnil, respectively (which are calculated from scenarios 1 or 3; see Table 31). Then we estimate the total discounted per-patient post-TKI treatment cost in the nilotinib treatment arm as:
and similarly for the dasatinib treatment arm. The total discounted per-patient post-TKI treatment QALYs in the nilotinib and dasatinib treatment arms are calculated similarly. In the Results section, we show that the proportions still alive and starting second- or third-line treatment on hydroxycarbamide or SCT are similar across the three treatment arms, as the durations of TKI treatments are similar across treatments. Therefore, this method largely equalises all post-TKI costs and QALYs between treatment arms.
One further adjustment is performed when using the simplified analysis method in combination with the surrogate survival method (scenarios 1a, 1b, 2a, 2b; see Table 31). In these scenarios, relative survival between treatment arms is modelled by setting the time on hydroxycarbamide in CP as a function of treatment arm in order to recreate the modelled OS based on surrogate data (see Surrogate-predicted survival approach, above). In this case, if denoting Tim and Tnil as the mean times on hydroxycarbamide in CP for those patients who receive this treatment in the imatinib and nilotinib treatment arms, respectively, then we estimate the total discounted per-patient post-TKI treatment cost in the nilotinib treatment arm as:
and similarly for the dasatinib arm, and for the QALYs in the dasatinib and nilotinib treatment arms.
This simplified method clearly does not represent our best estimate of the courses of treatments after resistance or intolerance to TKIs. However, we include this scenario to represent largely the cost-effectiveness of the treatment arms allowing for the ‘pure’ cost-effectiveness of first-line TKIs and second-line nilotinib. Also, we believe this analysis may be useful given the substantial uncertainty in the nature and costs of subsequent lines of treatment. This is especially true given that we predict that patients will take first-line TKIs for many years (between 7 and 9 years, see Results). Therefore, in the simplified method analysis the results should not reflect the treatments post TKIs (which remain uncertain, and also start about 8 years from diagnosis) and their associated costs and QALYs.
Perspective, discounting and time horizon
The model cycle length is 3 months, and the model time horizon is 50 years, or age 107 years, at which time all people have died. A model half-cycle correction is applied.
Future costs and benefits (QALYs) are discounted at 3.5% per year, and the perspective is that of the NHS and PSS, in accordance with the NICE reference case.
Modelled treatment sequences post first-line treatment
Treatment sequences in chronic-phase chronic myeloid leukaemia
As presented in Table 31, in scenarios 1 (‘a’ and ‘b’), 2 (‘a’ and ‘b’), 3 and 4, we assumed that patients with CP-CML received either SCT or hydroxycarbamide for second- or third -line treatment, and no further lines of treatment before reaching AP or BC. The people with CML who receive SCT are deemed to receive it immediately following TKI failure.
Scenarios 3 and 4 represent our best estimate of the probable future lines of treatment, and reflect NICE’s draft guidance FAD to recommend nilotinib – but neither dasatinib nor high-dose imatinib – as second-line treatment after imatinib in CML (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). (In the draft guidance on 18 August 2011, NICE has recommended nilotinib for the treatment of the CPs and APs of CML that is resistant or intolerant to standard-dose imatinib. Dasatinib and high-dose imatinib are not recommended in the draft guidance. Consultees have the opportunity to appeal against the draft guidance. Until NICE issues final guidance, NHS bodies should make decisions locally on the funding of specific treatments. This draft guidance does not mean that people currently taking dasatinib or high-dose imatinib will stop receiving them. They have the option to continue treatment until they and their clinicians consider it appropriate to stop.) Here, for the nilotinib as first-line treatment comparator, we again assume a mixture of SCT and hydroxycarbamide for second-line treatment, but no further lines of treatment. In contrast, in the imatinib and dasatinib comparators, we assume that all patients will receive nilotinib second-line, and a mixture of SCT and hydroxycarbamide for third-line treatment.
Treatments in accelerated phase and blast crisis
The range of treatments that CML patients may receive in the advanced stages of disease is wide, and quite variable between patients. We are aware from our clinical experts that these might include the use of TKIs, various chemotherapies, reconsideration for SCT; however, for simplicity we assume that patients take only hydroxycarbamide in AP and BC. We believe this is justified mainly because of a lack of evidence relating to the effectiveness of the these treatments in the advanced stages of CML, and believe it would be inconsistent to include their costs but not their effects. Further, in common with the manufacturers’ analyses, we felt it would be too difficult to create a well-evidenced submodel for the advanced phases of disease, which included SCT or the possibility of second or third CPs.
Treatment pathways not modelled
When we assume treatment with hydroxycarbamide in CP-CML, it is likely that in reality a wider mixture of treatments would be offered, including other chemotherapies and IFN-α. Although we have costed for the use of hydroxycarbamide only, our survival data following hydroxycarbamide rely on data for which a mixture of post-TKI treatments has been used (see Overall survival on hydroxycarbamide following tyrosine kinase inhibitor failure).
As discussed in the previous section, we also chose not to model SCT, other forms of chemotherapy or the possibility of second or subsequent CPs after entering either of the advanced phases of disease.
There is some limited evidence that some patients on TKIs with a deep and durable response may be taken off treatment as they are effectively cured. 48 We have not modelled this possibility.
Summary of scenario analyses
The relative merits of our scenario analyses are presented in Table 32.
| Advantages/disadvantages | Scenario 1 | Scenario 1a | Scenario 1b | Scenario 2 | Scenario 2a | Scenario 2b | Scenario 3 | Scenario 4 |
|---|---|---|---|---|---|---|---|---|
| Advantages | ||||||||
| Equity across treatment arms because same number of lines of therapy | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Cost-effectiveness of first-line drugs not affected by cost-effectiveness of second-line nilotinib | ✓ | ✓ | ||||||
| MMR from two RCTs used, which is a known predictor of survival | ✓ | ✓ | ||||||
| CCyR from two RCTs used, which is a known predictor of survival | ✓ | ✓ | ||||||
| Nature of subsequent lines of treatment uncertain; this issue is bypassed | ✓ | ✓ | ✓ | ✓ | ||||
| Cost-effectiveness of first-line drugs only marginally affected by cost-effectiveness of subsequent treatment | ✓ | ✓ | ✓ | ✓ | ||||
| Subsequent lines of treatment are our best estimate of future treatments on NHS given NICE’s draft guidance FADa recommendations on second-line drugs; such related medical costs should be modelled | ✓ | |||||||
| Allows treatment with second-line nilotinib, which NICE’s draft guidance FADa has recently recommended | ✓ | ✓ | ||||||
| Disadvantages | ||||||||
| Does not use any response rates from RCTs | ✓ | ✓ | ✓ | ✓ | ||||
| Second-line nilotinib not modelled, although recently recommended in the NICE draft guidance FADa | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Survival does not reflect exact nature of second-line treatment | ✓ | ✓ | ||||||
| Only marginally affected by subsequent lines of treatment, and their related medical costs | ✓ | ✓ | ✓ | ✓ | ||||
| Cost-effectiveness of first-line drugs affected by cost-effectiveness of second-line nilotinib | ✓ | ✓ | ||||||
In the context of a MTA, a secondary purpose of the economic model produced by the independent technology assessment/review group is to enable consideration and comparison of the similarities and implications of the modelling approaches used by the manufacturers.
Table 33 shows the scenarios analysed with the PenTAG model and how they relate to the model-based cost-effectiveness analyses provided by Novartis and BMS.
| First line | Second line | Third line | Model all costs? | Cumulative survival method | MMR surrogate survival method | CCyR surrogate survival method |
|---|---|---|---|---|---|---|
| Imatinib/nilotinib | Hydroxycarbamide or SCT | None | Yes | Novartis 1 | 1a | 1b |
| Imatinib/nilotinib | Hydroxycarbamide or SCT | None | No, only costs while on first-line drugs | 2 | 2a | 2b |
| Imatinib | Nilotinib | Hydroxycarbamide or SCT | Yes | 3 | 3a | 3b |
| Dasatinib | Nilotinib | Hydroxycarbamide or SCT | ||||
| Nilotinib | Hydroxycarbamide or SCT | None | ||||
| Imatinib | Nilotinib | Hydroxycarbamide or SCT | No, only costs while on first- or second-line TKIs | 4 | 4a | 4b |
| Dasatinib | Nilotinib | Hydroxycarbamide or SCT | ||||
| Nilotinib | Hydroxycarbamide or SCT | None | ||||
| Imatinib | 50% nilotinib, 50% dasatinib | SCT or dasatinib-based therapy | BMS | |||
| Dasatinib | Nilotinib | |||||
| Nilotinib | Dasatinib | |||||
| Imatinib/nilotinib | Dasatinib | Hydroxycarbamide or SCT | Novartis 2 | NA | NA |
Note that the Novartis analysis in the last row of this table (their ‘base-case scenario’) is probably no longer valid, given that in the NICE draft guidance FAD, dasatinib as second-line treatment for CML was not recommended (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). It is acknowledged that this was unknown to Novartis at the time of their submission. However, the Novartis ‘scenario A’ analysis in the first row of this table, where no TKI is assumed second line, is still valid and therefore of interest (see the comparison of Novartis results with PenTAG scenario 1 analysis at the end of the cost-effectiveness section Comparison of PenTAG model with the Novartis model). BMS model only a single scenario.
Effectiveness parameters and assumptions
Surrogate-predicted overall survival (for surrogate survival only)
Overall survival for all three treatment arms was estimated using a surrogate relationship based on CCyR at 12 months and, in a separate analysis, on MMR at 12 months. In each case, OS was estimated in four stages.
Stage 1 Overall survival for responders, and separately for non-responders, was estimated as a function of time using imatinib-arm data from a meta-analysis of trials of imatinib first-line (including the IRIS RCT26). The estimates of OS for responders and non-responders, separately for CyR and molecular response, using a meta-analysis are given in Chapter 4 (see Analysis of overall survival and progression-free survival by cytogenetic and molecular response).
Stage 2 Mortality due to CML was estimated from these historical imatinib trial data. Mortality was assumed to occur due to CML-related causes and non-CML causes. Given limited historical data, the probability of CML-related death was assumed to be constant over time. Non-CML mortality was taken from UK life tables,112 and the age at diagnosis was estimated as the average age at diagnosis across all historical trials, weighted by the number of responders or non-responders in each trial, as appropriate. The probability of CML-related death was estimated using the Microsoft Excel ‘Solver’ function (Microsoft Corporation, Redmond, WA, USA) in such as way that the sum of squares of differences between the actual historical OS and modelled OS at each year was minimised. The actual historical OS and fitted OS are shown in Figure 17.
FIGURE 17.
Historical vs fitted OS for patients (a) with and without a CCyR and (b) with and without a MMR.
Stage 3 Overall survival was estimated separately for responders and non-responders given a cohort of patients starting first-line treatment at the age of 57 years (the mean age at diagnosis for our modelling, and at present in the UK). OS was estimated by applying mortality from the general population with starting age of 57 years and the appropriate estimate of CML-related mortality from Stage 2.
Stage 4 Overall survival was estimated for each treatment arm (imatinib, dasatinib, nilotinib) by averaging the responder and non-responder OS, estimated in Stage 3, weighted by the proportion of patients who did and did not achieve a response to first-line treatment at 12 months (Figure 18). Our estimates of mean OS based on these estimation methods are given in Figure 19.
FIGURE 18.
Overall survival for each treatment arm estimated by surrogate relationship based on CCyR and, separately, MMR.
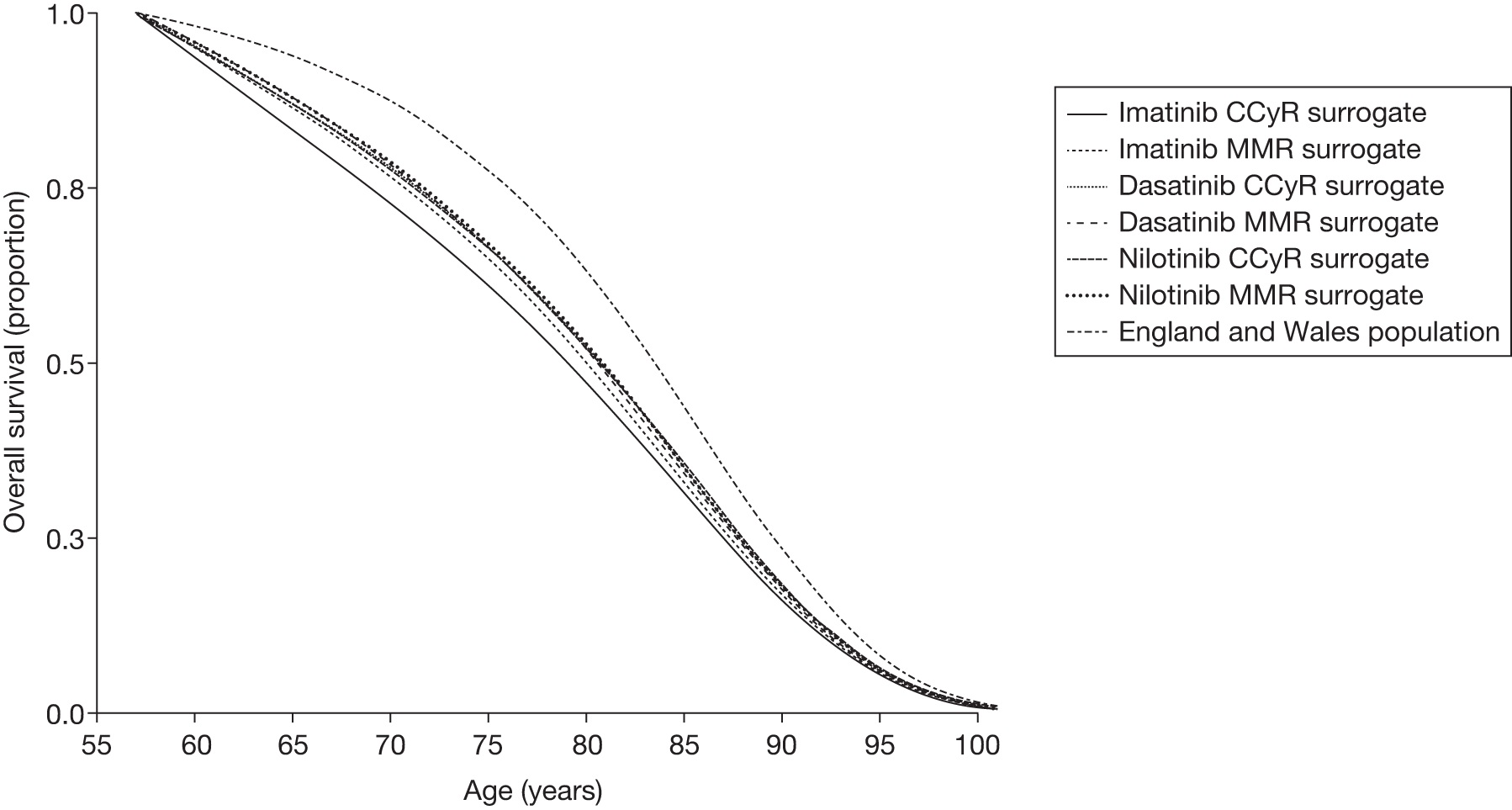
FIGURE 19.
Estimated mean OS as a function of surrogate measure used and treatment arm; treatment started at age 57 years.
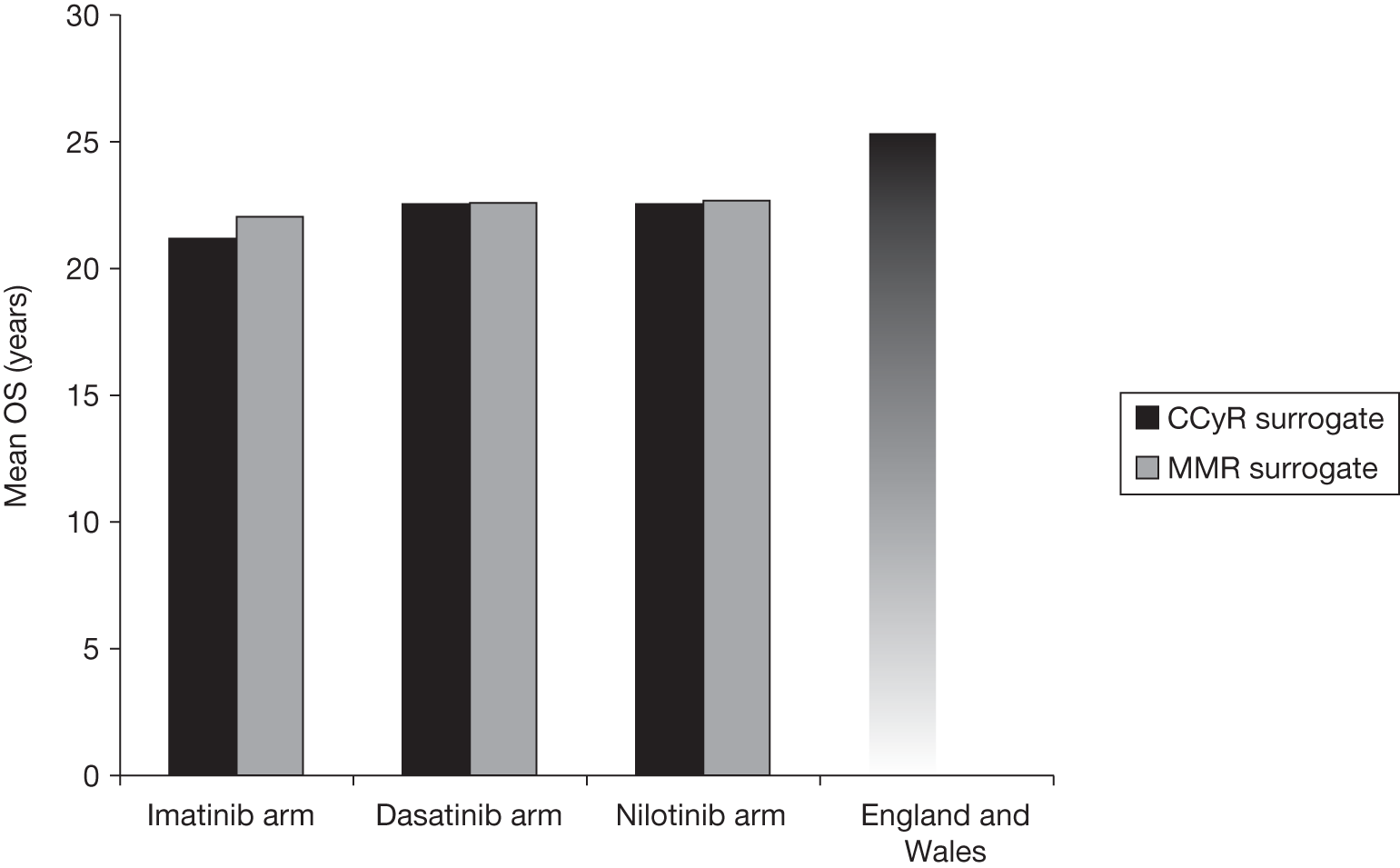
Finally, we compare our estimates of expected OS with the actual 24 month OS from the RCT ENESTnd,20 the RCT DASISION,29 and with the longer-term imatinib survival data from the IRIS trial26 of imatinib compared with IFN-α.
Comparison of actual and expected overall survival
Given that the OS from the RCTs of first-line dasatinib and nilotinib is very immature, it is difficult to gauge the accuracy of the modelled OS shown in Figure 18. Nonetheless, it appears that the modelled OS is consistent with these data. At 2 years’ follow-up:
-
dasatinib empirical OS was 95% based on DASISION trial data,29 compared with 97% in the model based on either the CCyR or MMR surrogate relationships75
-
nilotinib empirical OS was 97% based on ENESTnd trial data,20 compared with 97% in the model based on either the CCyR or MMR surrogate relationships85
-
imatinib empirical OS was 95% and 96% in the dasatinib and nilotinib RCTs, respectively, compared with 96% in the model based on the CCyR surrogate relationship and 97% based on the MMR surrogate relationship. 75,85
In addition, the estimated OS for the imatinib arm closely predicts the actual OS in the imatinib arm of the IRIS RCT26 (Figure 20). This is not a completely independent verification of OS by this method, because some of the data on OS for imatinib responders and non-responders from the IRIS RCT26 were also used to estimate the OS surrogate relationships. However, other historical data also heavily influenced the surrogate OS estimates so it is a useful calibration of the model’s survival outputs using this method (see Chapter 4, Study and population characteristics).
FIGURE 20.
Overall survival for the imatinib treatment arm estimated by surrogate relationship compared with actual OS from the IRIS RCT. 26
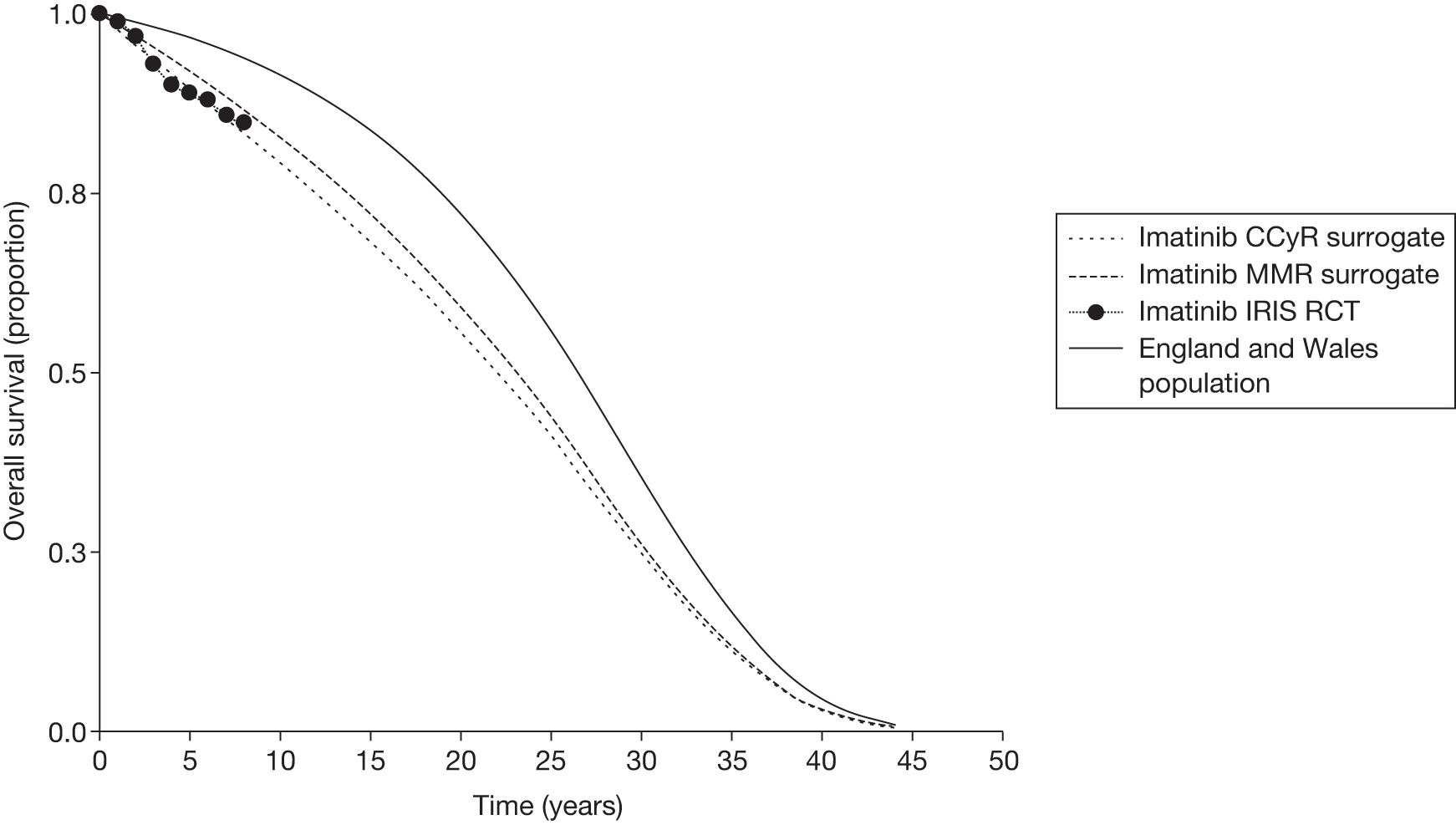
Estimated complete cytogenetic response and major molecular response at 12 months by first-line treatment
Estimates of response rates for CCyR and MMR are available for imatinib and dasatinib from DASISION29 and for imatinib and nilotinib (300 mg) from ENESTnd20 (Tables 34 and 35).
| Study | Imatinib | Dasatinib | Nilotinib (300 mg) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of responders | Total participants | Response rate | SE | No. of responders | Total participants | Response rate | SE | No. of responders | Total participants | Response rate | SE | |
| Kantarjian et al.29 | 186 | 260 | 0.715 | 0.028 | 216 | 259 | 0.834 | 0.023 | – | – | – | – |
| Saglio et al.20 | 184 | 283 | 0.650 | 0.028 | – | – | – | – | 226 | 282 | 0.801 | 0.024 |
| Study | Imatinib | Dasatinib | Nilotinib (300 mg) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of responders | Total participants | Response rate | SE | No. of responders | Total participants | Response rate | SE | No. of responders | Total participants | Response rate | SE | |
| Kantarjian et al.29 | 73 | 260 | 0.281 | 0.028 | 119 | 259 | 0.459 | 0.031 | – | – | – | – |
| Saglio et al.20 | 63 | 283 | 0.219 | 0.025 | – | – | – | – | 125 | 282 | 0.443 | 0.03 |
As estimates of response rates are required for all three treatments in the cost-effectiveness model, a mixed-treatment (MTC) approach was taken using the data above. The method is described in Appendix 7. The imputed and overall estimated response rates for CCyR and MMR are shown in Tables 36 and 37, respectively. Note that, as required, the overall response rate estimates for CCyR and MMR are weighted in favour of the trial report estimates rather than the imputed estimates.
| Source | Imatinib | Dasatinib | Nilotinib (300 mg) | |||
|---|---|---|---|---|---|---|
| Response rate | SE | Response rate | SE | Response rate | SE | |
| Kantarjian et al.29 | 0.715 | 0.028 | 0.834 | 0.023 | 0.844 | 0.031 |
| Saglio et al.20 | 0.650 | 0.028 | 0.786 | 0.042 | 0.801 | 0.024 |
| Meta-analysed for model | 0.683 | 0.020 | 0.823 | 0.020 | 0.817 | 0.019 |
| Source | Imatinib | Dasatinib | Nilotinib (300 mg) | |||
|---|---|---|---|---|---|---|
| Response rate | SE | Response rate | SE | Response rate | SE | |
| Kantarjian et al.29 | 0.281 | 0.028 | 0.459 | 0.031 | 0.525 | 0.057 |
| Saglio et al.20 | 0.219 | 0.025 | 0.380 | 0.055 | 0.443 | 0.030 |
| Meta-analysed for model | 0.246 | 0.018 | 0.440 | 0.027 | 0.460 | 0.026 |
Overall survival by subsequent treatment and disease phase
Duration of first-line tyrosine kinase inhibitor treatment
The mean times on first-line treatment for imatinib, dasatinib and nilotinib are very important quantities in the estimation of the cost-effectiveness of first-line nilotinib and dasatinib compared with imatinib. We used the following sources of data:
-
For nilotinib, we used treatment duration data up to 2.5 years’ follow-up from the RCT ENESTnd. 20
-
For dasatinib, we used treatment duration data up to 2 years’ follow-up from the RCT DASISION. 29
-
For imatinib, we used three sources of data from ENESTnd,20 DASISION,29 and the IRIS RCT,26 which has up to 8 years’ follow-up.
Follow-up was limited to just 2 or 2.5 years in the RCTs of first-line nilotinib and dasatinib. Given that a large proportion of patients were still on treatment at this time (0.65–0.80), it was necessary to extrapolate these proportions. This was achieved by using data from the IRIS RCT26 of first-line imatinib compared with IFN-α, with follow-up extending to 8 years. We deemed this trial as appropriate because these are the longest follow-up TKI treatment duration data that currently exist.
Treatment duration for all three drugs was estimated in the following three stages, described below, and which assumes that temporal pattern of treatment discontinuation in the new drugs would be broadly similar.
-
In Stage 1, we modelled the treatment duration of imatinib in the IRIS RCT. 26
-
In Stage 2, we modelled the treatment duration of dasatinib from DASISION,29 the treatment duration of nilotinib from ENESTnd,20 and the treatment duration of imatinib from DASISION29 and ENESTnd. 20
-
In Stage 3, we synthesised these quantities to estimate the modelled treatment durations for imatinib, dasatinib and nilotinib.
Stage 1
First, we fitted a curve to the proportion of patients on imatinib treatment in the IRIS RCT. 26 The empirical data were taken from four publications. 27,46,99,113 Treatment cessation due to non-CML mortality was modelled independently of treatment cessation due to any other causes. Non-CML mortality was modelled by using mortality of the general population in England and Wales, with starting age of 50 years, the median starting age in the IRIS RCT. 26 We modelled treatment cessation due to any other causes as a Weibull distribution, which is most commonly used in survival analysis (Figure 21). Fitting was achieved by minimising the sums of squares of differences between actual and modelled treatment duration. The resulting parameters of the Weibull distribution, which modelled treatment cessation due to any causes except non-CML mortality, were lambda = 0.093, gamma = 0.861. Including all causes of treatment cessation (‘fitted with general mortality’ in Figure 21), the mean treatment duration was 13.0 years.
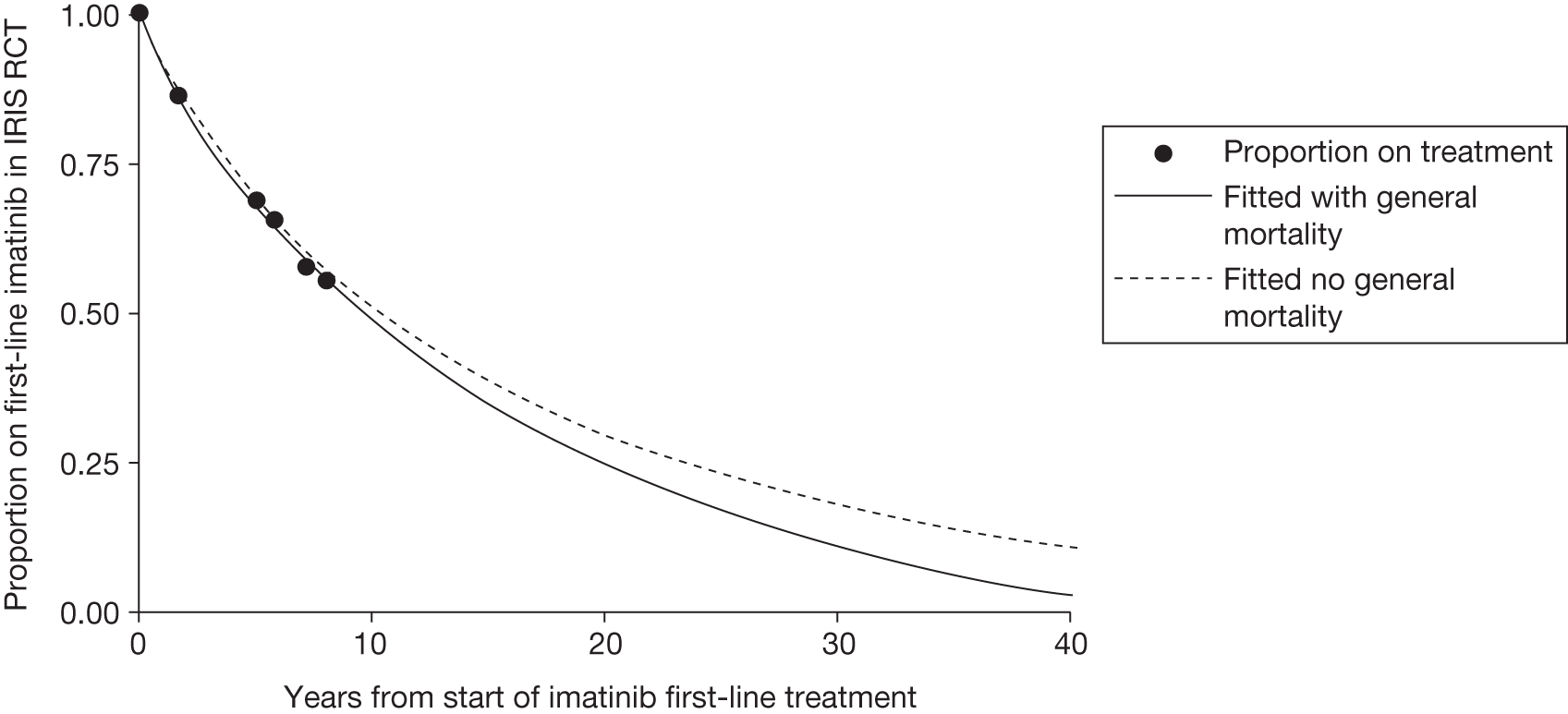
Stage 2
Next, the proportion of patients on nilotinib treatment in the nilotinib compared with imatinib (ENESTnd20) RCT was modelled, again splitting out non-CML mortality and modelling the remaining causes of treatment cessation as a Weibull distribution. Given such short follow-up in the nilotinib compared with imatinib RCT, it was not reasonable to estimate a shape parameter from the data from this trial. Instead, we assumed the same shape parameter of the Weibull distribution (of gamma = 0.861) as estimated in the longer-duration IRIS RCT for imatinib (Stage 1). This strongly impacts the extrapolated nilotinib treatment duration. When modelling non-CML mortality, a starting age of 57 years for first-line treatment of CML was assumed, our estimate for general age at diagnosis of the patient population in the UK [see Cohort starting age (age at diagnosis with chronic-phase chronic myeloid leukaemia)].
In the ENESTnd RCT20 of nilotinib compared with imatinib, 12-month data on nilotinib and imatinib treatment discontinuation is given in the online appendix of the paper describing this RCT. However, the most up-to-date data on treatment discontinuation are the 24-month data, which is provided by Novartis in its report to NICE and used in its model. Therefore, in common with Novartis, we based our estimate of nilotinib and imatinib treatment duration in this RCT on the Kaplan–Meier data provided by Novartis (Figure 22). This yielded parameter lambda = 0.144, and a mean treatment duration of 8.5 years (Figure 23). If, instead, we had modelled treatment cessation due to causes other than non-CML mortality as an exponential distribution, i.e. not using the shape parameter from the IRIS RCT,26 then the mean nilotinib treatment duration would have been 6.9 years. However, we repeat that we believe this method is less sound because it does not use the valuable information on treatment duration at longer follow-up from the IRIS RCT. 26
FIGURE 22.
Kaplan–Meier estimates of time on treatment in first-line nilotinib vs imatinib RCT. b.i.d., twice a day; q.d., once a day. Source: Figure 5 of Novartis’s manufacturer’s submission to NICE.
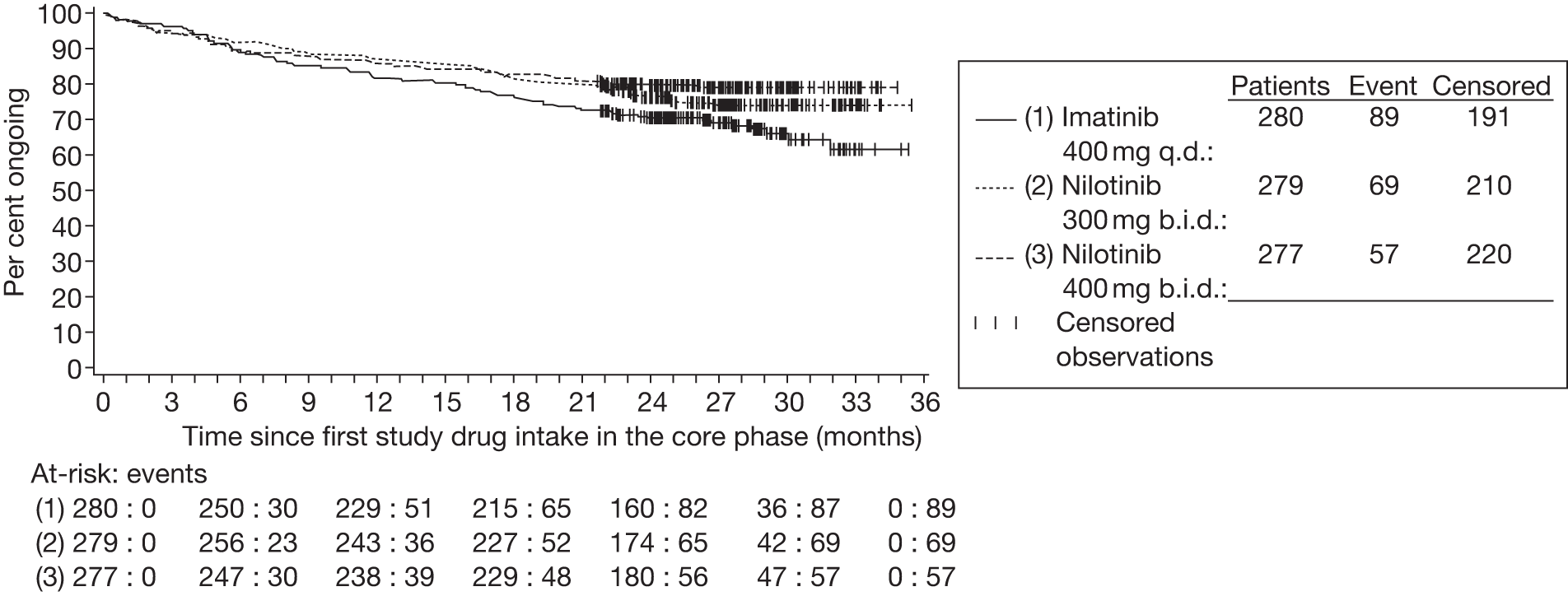
FIGURE 23.
Actual vs modelled nilotinib treatment duration in nilotinib vs imatinib RCT.
Data for treatment duration in the RCT of dasatinib compared with imatinib were taken from Kantarjian et al. 29 for 12 months’ data, Shah et al. 73 for 18 months’ data, and from conference slides by Kantarjian et al.75 for 24 months’ data.
Exactly the same procedure was then followed for dasatinib, imatinib in the dasatinib RCT and imatinib in the nilotinib RCT (Figures 24–26), with the corresponding lambda parameters equal to 0.150, 0.166 and 0.190, and corresponding mean treatment duration values of 8.2, 7.5 and 6.6 years. If instead we had modelled treatment cessation due to causes other than non-CML mortality as an exponential distribution – i.e. not using the shape parameter from the IRIS RCT26 – then the mean treatment durations would have been much shorter: 6.6, 6.0 and 5.4 years.
FIGURE 24.
Actual vs modelled dasatinib treatment duration in dasatinib vs imatinib RCT.
FIGURE 25.
Actual vs modelled imatinib treatment duration in dasatinib vs imatinib RCT.
FIGURE 26.
Actual vs modelled imatinib treatment duration in nilotinib vs imatinib RCT.
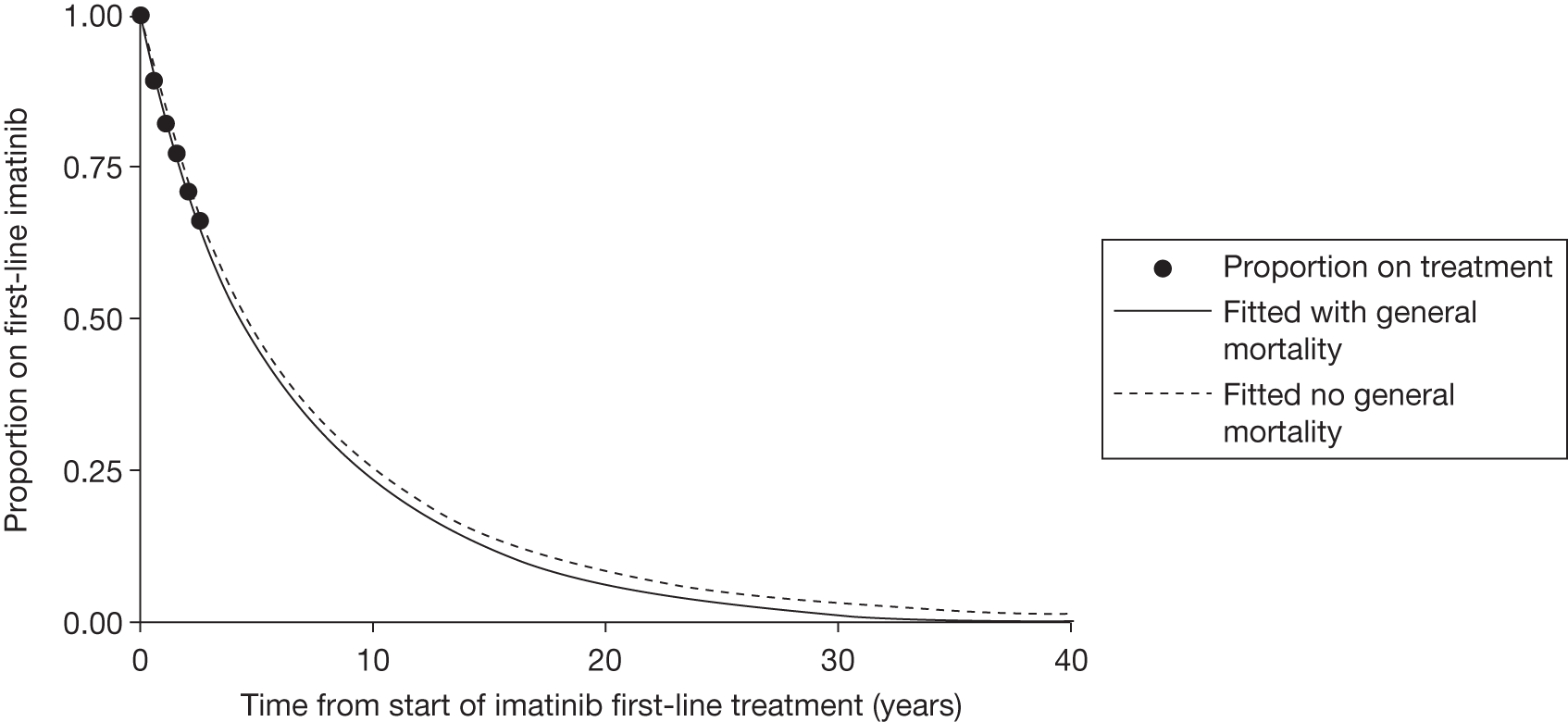
All the fitted curves are shown together in Figure 27. This clearly shows the model’s prediction that the treatment duration of dasatinib and nilotinib will be considerably shorter than treatment duration extrapolated from the IRIS RCT. 26 One possible reason may be the lack of any second-line TKI treatment following imatinib during the IRIS trial,26 so participants stayed on imatinib longer.
FIGURE 27.
First-line treatment durations before adjustment for indirect comparison.
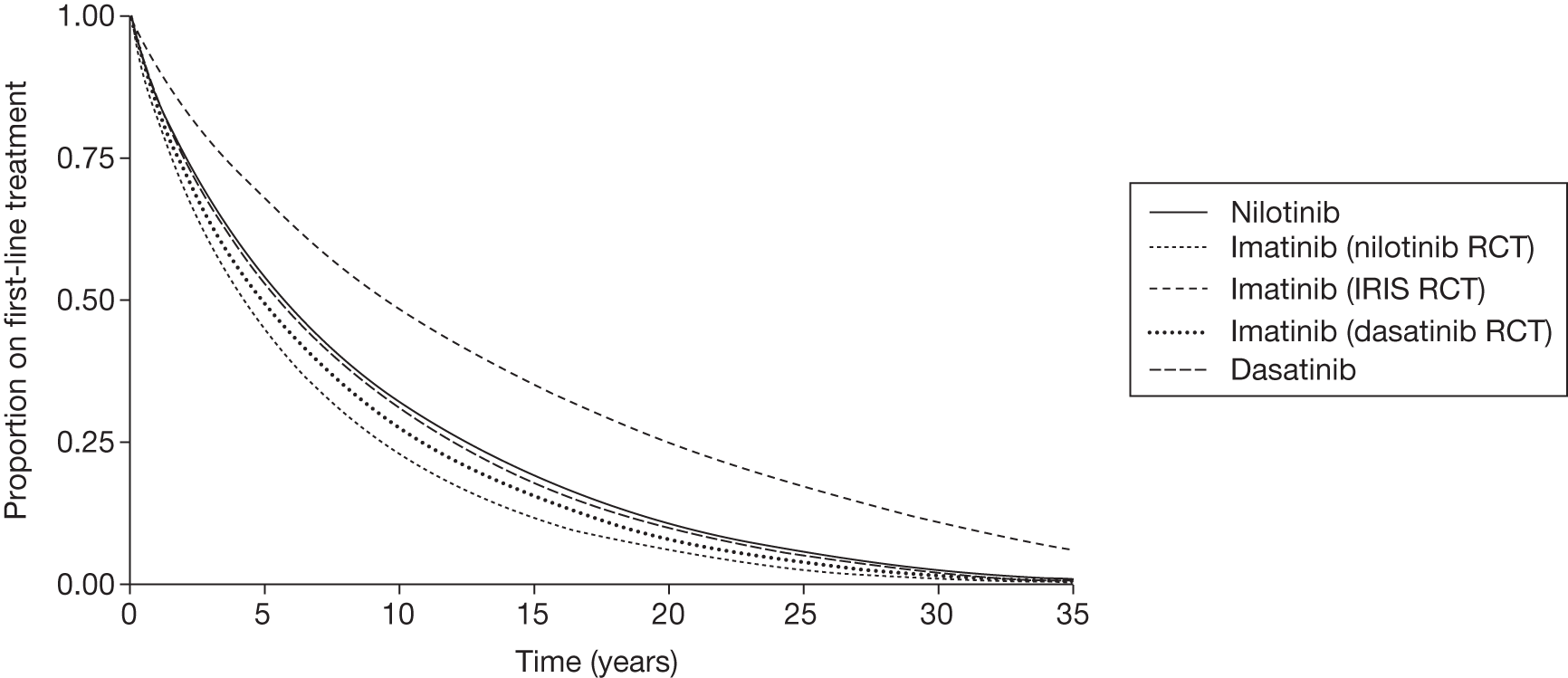
Stage 3
In this final stage, the treatment duration curves were adjusted for the purposes of the indirect comparison between the three first-line treatments (Figure 28 and Table 38). The treatment duration for imatinib was estimated in such a way that the mean treatment duration (excluding non-CML mortality) was set to the average of the mean imatinib treatment duration (excluding non-CML mortality) in the RCT DASISION29 and the mean imatinib treatment duration (excluding non-CML mortality) in the RCT ENESTnd. 20 It was appropriate to take the average duration from the two RCTs, as this is consistent with our estimate of average response rates for patients taking all first-line drugs in our MTC (see Estimated complete cytogenetic response and major molecular response at 12 months by first-line treatment, above). In addition, the shape parameter gamma was unchanged at 0.861, the value estimated from the IRIS RCT. 26 Given that treatment cessation due to non-CML mortality is a relatively minor cause of cessation, the modelled mean duration for imatinib is approximately equal to the modelled mean duration for imatinib in the ENESTnd20 and DASISION29 RCTs (see Figure 28 and Table 38).
| Trial data used for estimation | Imatinib | Nilotinib | Dasatinib |
|---|---|---|---|
| DASISION29 | 7.5 | – | 8.2 |
| ENESTnd20 | 6.6 | 8.5 | – |
| IRIS RCT26 | 13.0 | – | – |
| Modelled for indirect comparison | 7.1 | 9.0 | 7.8 |
FIGURE 28.
First-line treatment durations after adjustment for indirect comparison.
Next, parameter lambda for the treatment duration of nilotinib was adjusted by the ratio of lambda for imatinib for the indirect comparison, and lambda for imatinib from the nilotinib compared with imatinib RCT. The shape parameter gamma was unchanged at 0.861. This adjustment follows that suggested by Bucher et al. 67 Treatment duration for dasatinib was similarly adjusted for the indirect comparison.
Overall survival on hydroxycarbamide following tyrosine kinase inhibitor failure
Given a lack of evidence for survival on hydroxycarbamide following TKI therapy, we have adopted the same strategy as the Novartis (2009) submission to NICE for second-line nilotinib for patients with CP-CML who are resistant or intolerant to imatinib (Novartis 2009 submission, p. 36) to estimate survival on hydroxycarbamide following TKI failure. 114 This is based on a cohort study by Kantarjian et al. ,115 who present combined results for a subgroup of ‘other’ patients who are resistant or intolerant to imatinib. The 61 patients of this subgroup received a range of treatments including tipifarnib, lonafarnib, decitabine, cytarabine, homoharringtonine and IFN, with 12 receiving hydroxycarbamide. Survival when taking hydroxycarbamide is assumed to be the same as that of the ‘other’ treatment arm for imatinib-intolerant patients, even though, as acknowledged by Novartis in its 2009 report, some of the non-hydroxycarbamide treatments in this treatment group may prolong survival compared with hydroxycarbamide. However, the Novartis consultation with clinical experts for its second-line submission also suggested that this was a reasonable assumption given the lack of available relevant data on hydroxycarbamide in this setting. 114 Given the lack of relevant data, and in common with Novartis in its current submission, we assume that OS on hydroxycarbamide is independent of previous treatment. Given these limitations, our estimate of OS hydroxycarbamide following TKI failure is uncertain.
The Kaplan–Meier estimates of OS on hydroxycarbamide were read off at yearly intervals from figure 2a of Kantarjian et al. 115 As previously, two sources of mortality were modelled: CML-specific and non-CML. Non-CML mortality was modelled assuming the median initial age in the Kantarjian et al. 115 study of 54 years. The probability of CML-specific mortality was assumed to be constant over time. This probability was adjusted in such a way as to minimise the sums of squares of differences between the actual and modelled survival (Figure 29). This yielded a mean OS of 7.0 years for this trial, and a 5-year survival of 50%. Note that OS on hydroxycarbamide is lower in our model, because we assume that patients start first-line treatment at the age of 57 years, and these first-line treatments are taken for 7–9 years, so patients start second-line hydroxycarbamide aged approximately 65 years.
FIGURE 29.
Actual vs modelled OS for hydroxycarbamide following TKI failure. Kaplan–Meier data are from Kantarjian et al. 115
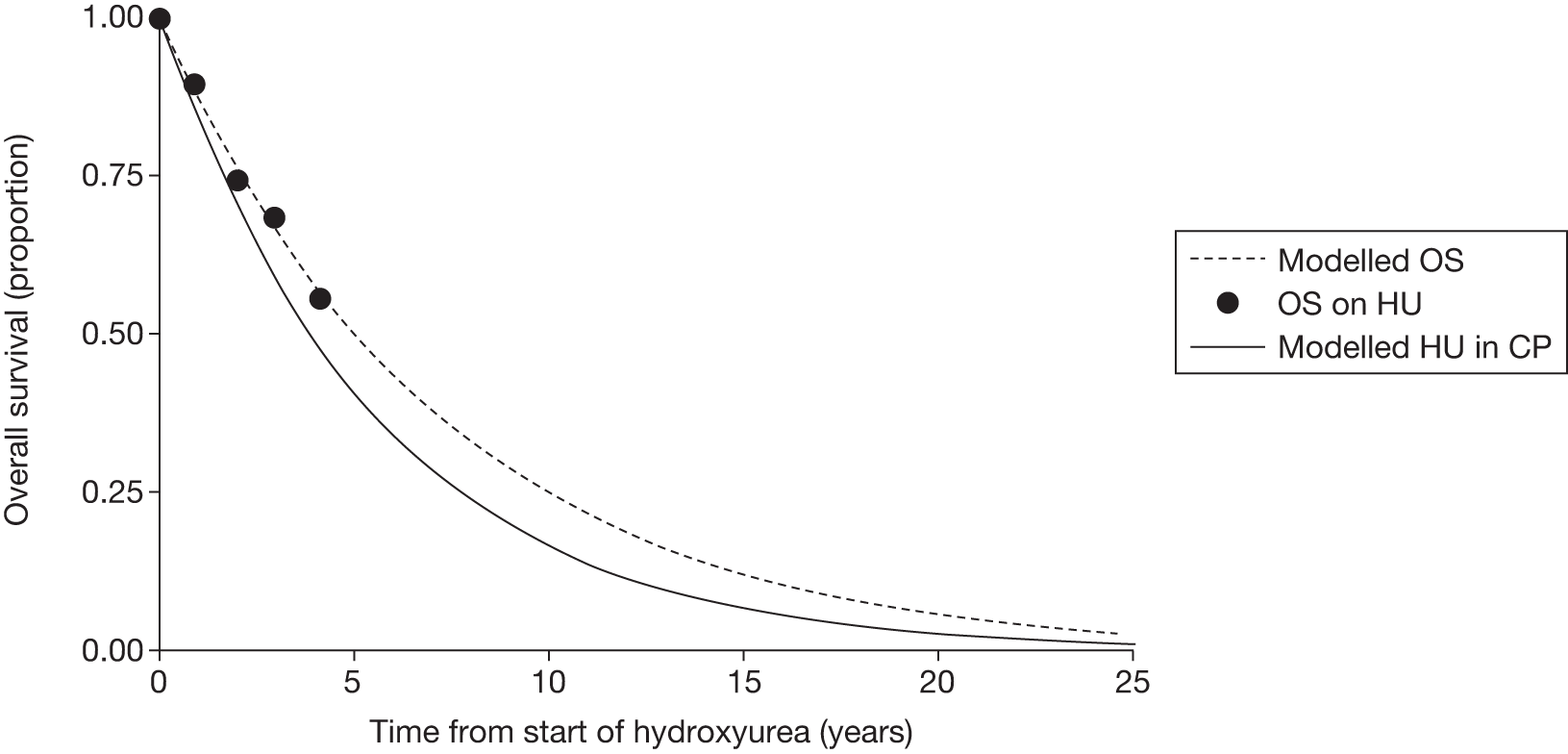
The derivation of our estimated time on hydroxycarbamide in CP, shown in Figure 29, is explained in the next section.
Time on hydroxycarbamide in chronic phase, and time in accelerated phase and blast crisis
In common with the current Novartis analysis, the previous CML disease and treatment model developed by PenTAG in 2009 (of second-line treatment for patients with CP-CML who are resistant or intolerant to imatinib) assumed that time spent in AP and BC is independent of treatment arm. The mean time in AP was 9.6 months and in BC 13.1 months using this source. These values were in turn taken from a previous cost-effectiveness analysis in CML, in which the time in AP and BC was calculated from published survival curves. 116–118
Given the similarity in the estimates, but also the uncertainty around the time spent in the advanced phases, we have adopted the same estimate as the PenTAG (2009) model for the duration of AP (9.6 months). However, for the BC phase, current clinical opinion suggests a considerably shorter duration than the previous estimates used, with life expectancy about 3–6 months (see stated considerations in NICE’s draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML; this is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99. The committee recommendations are draft – consultees have the opportunity to appeal against them and final guidance has not been issued on this appraisal topic). Also in common with Novartis, we applied constant probabilities of transition from AP to BC and from BC to death.
We now explain our derivation of the time on hydroxycarbamide in CP, as shown in Figure 29. First, as explained in the previous section (see Overall survival on hydroxycarbamide following tyrosine kinase inhibitor failure), we model general population mortality while people are on hydroxycarbamide in CP. We also specify a constant annual probability of 0.71 of transition from AP to BC, corresponding to our estimate of mean time in AP of 0.8 years (= 9.6 months). Similarly, we specify a constant annual probability of 0.87 of transition from BC to death, corresponding to our revised estimate of mean time in BC of 0.5 years (= 6 months). In addition, we specified a constant probability of transition from CP to AP. Given that we have modelled OS on hydroxycarbamide as explained in the previous section, this then specifies the constant probability of transition from CP to AP on hydroxycarbamide. In the model, this was achieved by using the Solver function in Excel. This yields the constant quarterly probability of 0.043 of transition from CP to AP while on hydroxycarbamide.
Proportion of patients receiving stem cell transplant post tyrosine kinase inhibitor failure
In all scenarios, some patients are assumed to receive SCT and some to receive hydroxycarbamide, as second- or third-line treatment after TKI failure (see Table 31). We did not identify any published evidence on the proportion of patients receiving SCT after first-line TKI failure. We therefore asked three of our clinical experts the proportion of patients that they believed would receive SCT, specifically after failure of TKIs, and at different ages. The similarity of their responses was notable, particularly the steep drop in the estimated percentage of patients who would receive a SCT in the CP after the age of 65 years. Over the age of 75 years, all three clinicians said that no patients with CP-CML would be likely to receive SCT. To approximate the responses that we received from our clinical experts, we first estimated the percentage of patients who receive a SCT for each of a range of ages (Table 39). Because of both the high cost of SCT and its important impact on life expectancy for those that survive to 5 years or more, these key assumptions will be varied in sensitivity analysis. For ease of modelling, we then estimated the proportion of patients receiving SCT as a simple linear function of time by least squares (Figure 30).
| Age (years) at which TKIs fail | Percentage of patients who get an SCT |
|---|---|
| 55–59 | 60 |
| 60–64 | 40 |
| 65–69 | 15 |
| 70–74 | 5 |
| 75+ | 0 |
FIGURE 30.
Proportion of patients receiving SCT as a function of age in the PenTAG model.
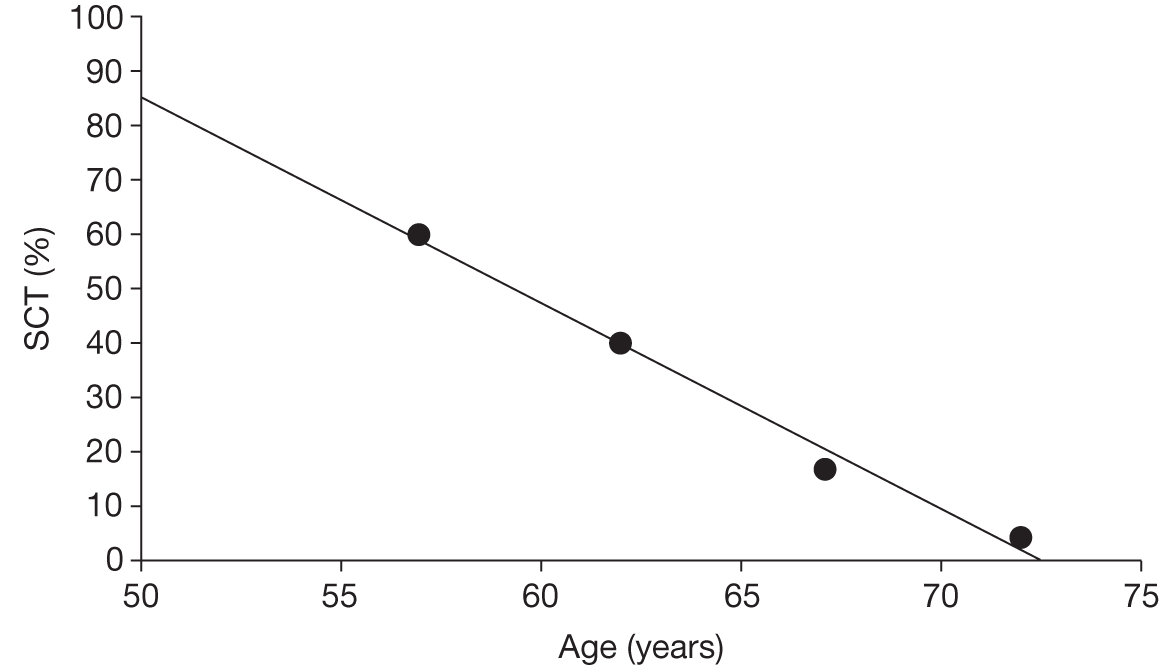
The corresponding equation is:
Overall survival following stem cell transplant
We reviewed a number of potential published sources for estimating OS following SCT, including those used in the manufacturers’ models. The Novartis source relates to a cohort of European patients in which only 30% were patients with CML, and the data manipulation to estimate the likely survival of the CML subgroup involves the assumption that the mean pre-transplant eligibility/European Group for Blood and Marrow Transplantation (EBMT) risk score for patients with CML is 4. 119 The whole data set, as used by Novartis, is also from patients transplanted between 1980 and 2005. Graphs in the Gratwohl paper (their figure 2A and B) further indicate that patients with CML, and patients transplanted in the most recent period (2001–5), have the greatest survival compared with other diseases and the whole cohort as used by Novartis. 119
This produces an estimate of survival at 5 years (34% for those with a risk score of 4), which seems far lower than other published estimates in patients with CML we identified and reviewed. 120–122 EBMT registry data were as cited in Pavlu et al. 122 – 61% survival at 2 years, and higher (66–70%) in those patients with CML transplanted in the first CP. 123
We therefore used an alternative UK-based source. This is a review and analysis of 173 patients with CML who received SCTs in CP at Hammersmith Hospital, London, between 2000 and 2010. 124 Of these patients, 74% survived to 3 years and 72% to 6 years. This is also very similar to the survival estimate from the US Center for Bone Marrow & Transplant Research, which analysed data from 1309 patients transplanted between 1999 and 2004; its 3-year survival estimate for patients with CP-CML undergoing transplant post imatinib was 72%. 120
Similar to Novartis, we modelled OS following SCT by assuming that patients fall in to one of two distinct groups – those who have high mortality soon after transplant and those who have low mortality. Investigation of possible fits to the two Hammersmith Hospital data points revealed a plausible solution that:
-
The high-risk group has constant probability of death of 0.55 (lower i.e. dashed line in Figure 31).
-
The low-risk group has mortality equal to that of the general England and Wales population (upper dashed line in Figure 31).
-
Twenty-five per cent of patients are in the high-risk group.
The survival of the resulting total population then closely matched the empirical survival data (continuous line in Figure 31). Note that it is not important whether or not these two groups are clinically plausible. Instead, it is the survival function for all patients combined (continuous line in Figure 31) that should appear plausible compared with available empirical estimates. (A further substantial advantage of modelling survival according to the weighted average of two cohorts is that it greatly simplifies modelling, and bypasses the need for transition probabilities related to the time spent in the SCT health state.)
FIGURE 31.
Modelled OS following SCT vs actual OS from Hammersmith Hospital, 2000–10.
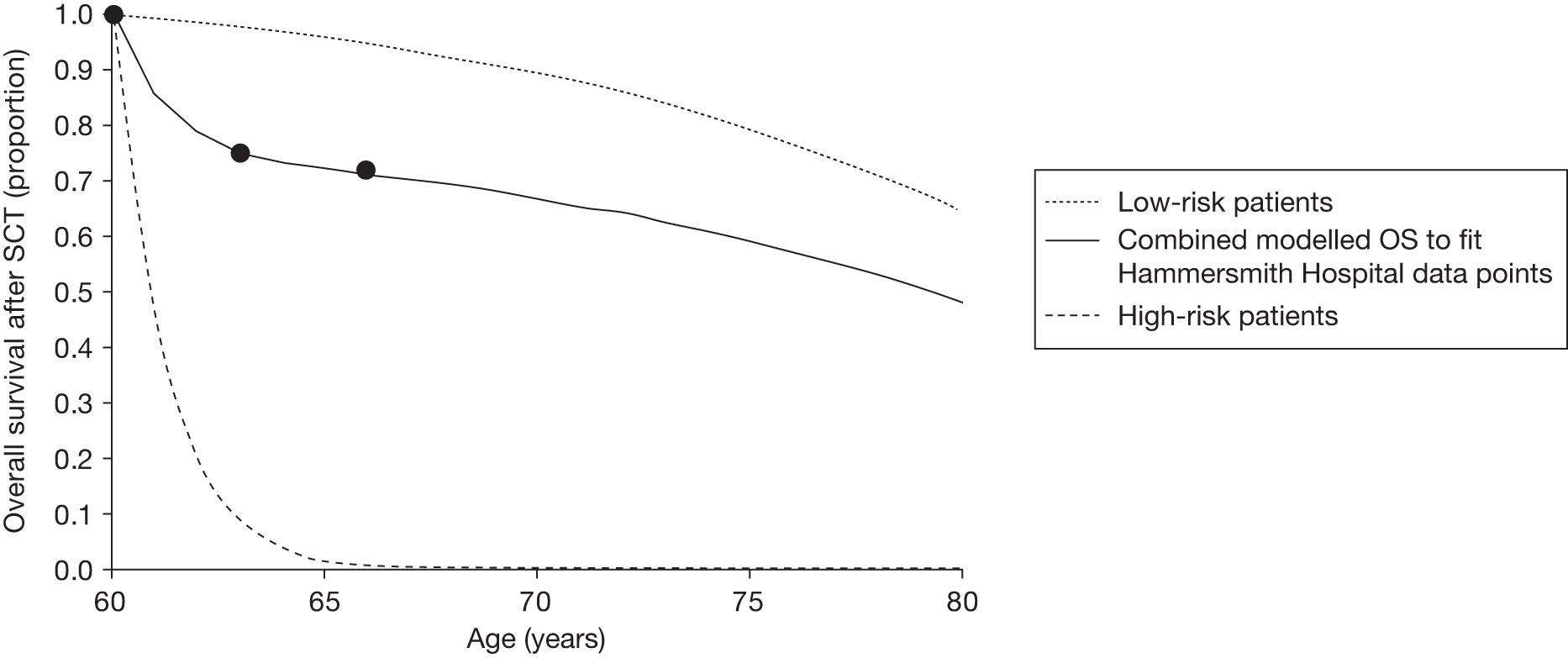
The life expectancy for patients having SCT at the age of 60 years is then 17.4 years, compared with 22.8 years in the general England and Wales population.
Duration of second-line nilotinib treatment
Second-line nilotinib is modelled in scenarios 3 and 4 only (see Table 31). In these scenarios, we assume that all patients initially randomised to imatinib or dasatinib take nilotinib after first-line treatment failure. Patients randomised to nilotinib take either hydroxycarbamide or SCT after nilotinib failure. We are aware of no clinical evidence of nilotinib after dasatinib failure. However, there has been a Phase II single-arm trial of nilotinib after imatinib failure. 124 In the absence of further data, we assume the duration of second-line nilotinib is independent of whether dasatinib or imatinib was taken first line.
The proportion of all patients (70% imatinib resistant and 30% imatinib intolerant) still on second-line nilotinib treatment from the single-arm trial is reported as 0.47 at 2 years. 125 But the proportion is most recently reported as 0.39 at 2 years in an update publication of Kantarjian et al. 126 Also, in their model, Novartis presents the Kaplan–Meier data for the proportion of imatinib-resistant (not imatinib-intolerant) patients still on second-line nilotinib treatment from 0 to 2 years. We assume that it is merely coincidental that its proportion at 2 years is the same as the published value of 0.47 from the 2008 abstract for both imatinib-resistant and imatinib-intolerant patients combined. Therefore, we used the Kaplan–Meier data presented by Novartis for imatinib-resistant patients only. We fitted an exponential curve to these data, and this yielded a mean time on treatment of 2.4 years. Given that the duration of treatment was short, it was not necessary to model treatment cessation due to non-CML mortality.
Cohort starting age (age at diagnosis with chronic-phase chronic myeloid leukaemia)
As stated in our background section (see Chapter 1, Incidence) the estimated mean age at diagnosis of CML patients in the UK is 58 years, according to data reported by the HMRN. We corresponded with the epidemiologist at the HMRN to obtain a current estimate of the age at diagnosis of those patients with CML who are diagnosed in the CP (the population relevant to this MTA).
Using data for 192 patients diagnosed with CML in CP between September 2004 and August 2010, the mean age at diagnosis was 57 years [standard deviation (SD) = 17; data kindly analysed and supplied by Dr Alex Smith, HMRN, Department of Health Sciences, University of York]. These data are not from UK-wide sources but actually from 14 hospitals mainly in the Yorkshire and Humber region of England.
Our cost-effectiveness modelling uses a starting age at diagnosis of 57 years.
Utility parameters and assumptions
Health-related quality-of-life literature
We undertook a systematic literature search to identify studies which had either directly or indirectly elicited social preference weights or ‘utilities’ for different CML health states. Appendix 1 shows the full search strategy used and databases searched. Manufacturer submissions to NICE were also reviewed to identify any additional studies.
Because this was to update the search previously conducted in 2009 for our technology assessment of the same drugs for second-line treatment of CML, titles and abstracts from the bibliographic searches were examined only for the years 2009–11. The review was carried out by one researcher (RA).
Our searches identified only one new study of relevance, the 2010 study by Szabo et al. , which used the time trade-off (TTO) technique to elicit valuations of seven CML health state descriptions from 339 members of the public in the USA, Canada, UK (UK n = 97) and Australia. 127 This study, and the EQ-5D-derived utility values from the IRIS trial26 reported by Reed et al. 116 and previously supplied in a Novartis submission, appear to be the only two research-based sources of utility values for HRQoL in people with CML (excluding those based on clinicians’ estimates). 114
Utilities in Peninsula Technology Assessment Group model
Our choice of utilities is given in Table 40.
| Treatment | Mean (SE) | Source |
|---|---|---|
| First line (CP) | ||
| Dasatinib, nilotinib, imatinib | 0.83 (0.004) at diagnosis, age 57 years, decreasing with age | Based on Reed et al.116 from IRIS RCT26 |
| Second/third line (CP) | ||
| SCT |
75% patients (low-risk group) utility equal to general population minus 0.041 25% (high-risk) utility general population minus 0.079a |
Decrement value from Lee et al.128 |
| Hydroxycarbamide | As dasatinib, nilotinib, imatinib first line | Based on Reed et al.116 from IRIS RCT26 |
| AP | ||
| Hydroxycarbamide | 0.73 (0.06) | Dalziela et al.65 |
| Blast phase | ||
| Hydroxycarbamide | 0.52 (0.08) | Dalziela et al.65 |
In our cost-effectiveness model we chose to use the EQ-5D-based valuations of CML health states previously reported by Reed et al. ,116 and supplemented by the unpublished data from the same trial (IRIS) for the AP and BC phases of the disease (Dr Shelby Reed, Duke University Medical Center, Durham, NC, USA, 5 July 2011, personal communication; Novartis data cited in Dalziel et al. 65). These data were collected for patients taking imatinib during the IRIS trial,26 as reported by Reed et al. 116 and used by Dalziel et al. 65 in a previous HTA of imatinib for CML. These data are drawn from a large sample of patients, using the EQ-5D, which is preferred in the NICE reference case. 129
It was necessary to estimate utility values for people taking dasatinib and nilotinib in CP, because no values are cited in the literature. In common with BMS and Novartis, we set these values equal to the value for imatinib in CP, based on clinical opinion and the similarity of the incidence of AEs by treatment.
The utilities for AP and blast phase reported by Reed et al. 116 are slightly different from those quoted by Dalziel et al. ,65 although both are taken from the IRIS trial originally. 26,116,130 In the Reed et al. analysis,116 no difference was assumed between AP and blast phase, as the observed difference in values was not statistically significant. We have therefore used the utility values cited by Dalziel et al. 65 in our model.
Utilities decrease with increasing age. 131 In common with Novartis (but not BMS), we adjusted the utilities in the model for age using the following equation (Ara and Brazier 2010):131 utility = 0.951 + 0.021 × male – 0.000259 × age – 0.000033 × age2.
The estimated utility of people taking imatinib in CP-CML of 0.854 from Reed et al. 116 is for patients of mean age of 50 years in the IRIS trial. 116 Given the formula from Ara and Brazier,131 the mean utility of a member of the general population aged 50 years, assuming that 58% are male (our assumption for CML population), is 0.867. This implies a disutility of 0.867 – 0.854 = 0.013 for patients taking imatinib in CP-CML compared with the general population. Therefore, for patients taking either imatinib, dasatinib or nilotinib in CP, we assumed that general population utilities as a decreasing function of age with a disutility of 0.013 at all ages.
We assume that the utility for patients taking hydroxycarbamide in CP equals that for the TKIs, and we further assume the same decrease in utility over time.
We do not model additional utility decrements associated with AEs in the base case.
Utility: after stem cell transplant
Although patients who survive SCT can, in most cases, be regarded as ‘cured’, in the early years following the transplant many patients will experience complications such as graft-versus-host disease (GvHD) and serious infections (due to the after-effects of myeloablation and the use of immunosuppressive agents). 132 As well as increasing mortality risk, these complications have inevitable quality-of-life impacts. 133,134
Novartis assumes that 52% of those receiving SCT experience a 0.079 utility decrement compared with patients in CP taking dasatinib, nilotinib or imatinib for the rest of their lives, while the other 48% experience the same utilities as patients in CP taking TKIs. This disutility is based on the quality-of-life impact of chronic GvHD in a 1997 health state preference elicitation study by Lee et al. (conducted with 12 US clinicians familiar with bone marrow transplant patients). 128
In the absence of other research evidence, our assumption for the utility of survivors after SCT is similar to that of Novartis. We modelled OS following SCT by assuming that patients are in one of two groups: a high-risk group with a constant high probability of death and a low-risk group with mortality equal to that of the general England and Wales population (see Overall survival following stem cell transplant, above). In the low-risk group, we assume that 52% of patients, those with chronic GvHD, have a disutility of 0.079 compared with the general population of England and Wales (not versus people in CP on TKIs as Novartis assumes). The remaining 48% of patients are effectively cured of CML, and therefore experience the age-related utility of the general population of England and Wales. Hence, on average, patients in the low-risk group have a disutility of 52% × 0.079 = 0.041 compared with the general population of England and Wales. We further assume that patients in the high-risk group have a disutility of 0.079 compared with patients taking TKIs in CP. This reflects the earlier impact of many of the post-transplant complications, and that chronic GvHD is one of a number of possible serious complications.
Cost parameters and assumptions
We model the following costs, which are inflated to 2011–12 values where appropriate:
-
drug acquisition
-
treatment for AEs
-
a range of medical management costs, including nurse treatment, consultant outpatient visits, bone marrow tests, and hospitalisation.
In addition to the cost of drug acquisition, mean drug costs per person allow for treatment duration (see Duration of first-line TKI treatment and Duration of second-line nilotinib treatment, above) and dose intensity (see Dose intensities, below).
Drug acquisition costs
Table 41 and Figure 32 present the drug prices. The prices of dasatinib and hydroxycarbamide were taken from BNF 61 (2010), (CiC information has been removed) and the price of imatinib was taken from MIMS, which is more up to date than the lower price in BNF 61 (2010). 45,55 NICE have agreed these sources of price information.
| Dose and frequency | Price (£) | Cost per 3-month model cycle (£) |
|---|---|---|
| Dasatinib (Sprycel) | ||
| 100 mg per day |
2504.96 per 50 mg, 60-tablet pack, 2504.96 per 100 mg, 30-tablet pack |
7624 |
| Nilotinib (Tasigna) | ||
|
First line: 300 mg per day (150 mg twice a day) Second line: 400 mg per day (200 mg twice a day) |
(CiC information has been removed) | (CiC information has been removed) |
| Imatinib (Glivec) | ||
| 400 mg once daily |
862.19 per 100 mg, 60-tablet pack, 1724.39 per 400 mg, 30-tablet pack |
5249 |
| Hydroxycarbamide | ||
| 20–30 mg/kg daily or 80 mg/kg every third day | 10.47 per 500 mg, 100-capsule pack | 36a |
FIGURE 32.
(CiC information has been removed).
In common with Novartis, we assume that the main alternative treatment to SCT after TKI failure is hydroxycarbamide. This drug costs only £36 per 3 months (see Table 41). However, hydroxycarbamide may not be the only, or even the main, treatment for patients post TKI failure who do not receive SCT (and we have taken our survival estimates for patients on hydroxycarbamide for second- or third-line treatment from a study population in which patients were taking several other treatments such as IFN-α, homoharringtonine and cytarabine). Therefore, to reflect the broader mix of drugs that such patients might receive, the cost of treatment with hydroxycarbamide is varied widely in the sensitivity analyses.
Dose intensities
For consistency between the costs of the drugs and the clinical outcomes, it is necessary to model the amounts of the drugs actually taken in the relevant clinical trials. The dose intensity of a drug is defined as the amount of drug administered in a trial as a proportion of the amount that would have been administered if there had been no dose reductions or dose interruptions. This does not include people who withdraw from treatment due to AEs. Mean dose intensities per person used in our model are given in Table 42.
| Drug | Treatment line | Mean dose intensity (%) | Source |
|---|---|---|---|
| Dasatinib | First line | 99 | BMS submission, p. 27 |
| Nilotinib | First line | (CiC information has been removed) | Novartis submission, p. 75 |
| Nilotinib | Second line | 99 | Kantarjian et al.,126 blood paper |
| Imatinib | First line | (CiC information has been removed) | Novartis submission, p. 75 |
| Hydroxycarbamide | Second and third line | 100 | PenTAG assumption |
In the absence of a published estimate of the mean dose intensity for first-line dasatinib, this was estimated as 99% which is the median dose intensity cited by BMS (p. 27 of BMS report). The mean dose intensity for first-line nilotinib of (CiC information has been removed) was provided by Novartis, which states that this came from its analysis at 12 months (p. 75 of Novartis report). The mean dose intensity for imatinib of (CiC information has been removed) was provided by Novartis, which states that this came from its analysis at 12 months (p. 75 of Novartis report) of the nilotinib compared with imatinib RCT, with mean dose of (CiC information has been removed) compared with the 400-mg planned dose. However, Novartis actually used a dose intensity of imatinib of 106%, which it cites on p. 105 of its report, which it states came from its analysis at 24 months. 85 However, it does not state that this is a mean dose intensity. Given that this mean actually represents a median dose intensity, we have chosen the mean dose intensity of (CiC information has been removed) from its 12-month analysis. 20 Our estimate of (CiC information has been removed) is consistent with the median dose intensity of imatinib of 100% from the dasatinib compared with imatinib RCT (p. 27 BMS report). Our estimate is also consistent with the mean dose intensity at 6 years for imatinib in the IRIS RCT of 100% for the 364 patients who remained on imatinib at 6 years. 99
Our estimate of the mean dose intensity of second-line nilotinib of 99% is taken from the single-arm trial of nilotinib for people resistant to or intolerant of imatinib. 124 The mean dose intensity is not reported; however, we used the median dose intensity of 789 mg/day, out of a planned dose of 800 mg/day. Given that hydroxycarbamide is extremely cheap compared with the other drugs, we have not searched the literature for a mean dose intensity, rather we have simply assumed 100%.
We understand that imatinib will come off European patent in 2016. 135 It is likely that its price will then come down considerably. In a sensitivity analysis, we model setting the price reduction on patent expiry to 25% for all drugs, and setting the price reduction to 25% for imatinib and dasatinib and 0% for nilotinib (see Sensitivity analyses: patient expiry).
Cost of serious adverse events
We included an estimate of the cost of treating selected serious (grade 3 or 4) AEs while on first-line or second-line TKIs. Based on the reported rates of different AEs during the first 12 months of treatment, we decided to include only the cost of treating neutropenia, thrombocytopenia and anaemia. No other types of grade 3 or 4 AE were experienced by > 1% of patients in either of the main RCTs (i.e. DASISION29 and ENESTnd20). Although there were very few patients experiencing grade 3 or 4 pleural effusions with dasatinib, because this complication is quite common at lower grades and specific to this TKI, we also estimated the cost of these. The number of additional AEs from months 13 to 24 was so small that we chose to model only AEs during the first year of treatment with TKIs. Rates of AEs costed in the model are shown in Table 43.
| Adverse event | Dasatinib | Nilotinib (300 mg) | Imatinib (DASISION29) | Imatinib (ENESTnd20) |
|---|---|---|---|---|
| Neutropenia (grades 3 and 4) | 20.9% | 11.8% | 20.2% | 20.0% |
| Thrombocytopenia (grades 3 and 4) | 19.0% | 10.0% | 10.1% | 8.6% |
| Anaemia (grades 3 and 4) | 10.1% | 3.2% | 7.0% | 5.0% |
| Pleural effusion (all grades) | 10.1% | 0.0% | 0.0% | 0.0% |
The cost of treating each of these four types of AE was taken from the Oxford Outcomes study, and used a weighted average of the cost of treating a patient experiencing these complications when hospitalised or not hospitalised. They were also inflated from the 2008 to the 2011 values (Table 44).
| Serious AE | Cost of treating if hospitalised (£) | Cost of treating if not hospitalised (£) | Percentage that would be hospitalised | £2008 cost per AE | £2011 cost per AE |
|---|---|---|---|---|---|
| Neutropenia (grades 3 and 4) | 1668 | 279 | 14.0 | 473 | 497 |
| Thrombocytopenia (grades 3 and 4) | 1234 | 467 | 0.5 | 471 | 494 |
| Anaemia (grades 3 and 4) | 324 | 324 | 0.7 | 324 | 340 |
| Pleural effusion (all grades) | 30 | 0 | 30 | 31 |
Table 45 shows the resulting annual cost of treating the main grade 3/4 AEs, and the cost of treating pleural effusions in those taking imatinib. The incidence rates of neutropenia, thrombocytopenia and anaemia in those taking nilotinib as second-line treatment are 29%, 29% and 3.2%, respectively (sourced from Kantarjian et al. ,124 except for anaemia, which is assumed to be the same as for first-line treatment with nilotinib).
| Serious AEs | Costs (£) | |||
|---|---|---|---|---|
| First-line treatment | Second-line treatment | |||
| Dasatinib | Nilotinib (300 mg) | Imatiniba | Nilotinib (400 mg) | |
| Neutropenia | 104 | 59 | 99 | 144 |
| Thrombocytopenia | 94 | 50 | 46 | 144 |
| Anaemia | 34 | 11 | 21 | 11 |
| Pleural effusion | 47 | – | – | – |
| Total annual cost | 280 | 119 | 166 | 299 |
Cost of other medical management and monitoring
We based our medical management and monitoring costs on the mean frequency of hospital outpatient appointments and tests reported by the Oxford Outcomes 2009 survey of six UK-based CML clinicians. As with the estimates used in the BMS cost-effectiveness model, these were based on the frequency of routine appointments and tests after the first 3 months of treatment, and separately for patients in the chronic and advanced phases. They were also inflated to 2011 prices and adjusted for some tests when our clinical expert believed the frequency from the Oxford Outcomes survey was unrealistic or illogical (e.g. having a frequency of mutation analysis, when only one such test per patient would be conducted; C Rudin, Royal Devon and Exeter Hospital, UK, 31 July 2011, personal communication). Following comments from Novartis, with which our clinical expert also agreed, we also took out those costs in CP for repeat bone marrow aspirations (original monthly frequency of 0.3) and hospital nurse consultations (monthly frequency of 0.4), and reduced the frequency of haematology consultant outpatient appointments to quarterly if on a TKI and every 6 weeks if on hydroxycarbamide. [Although there would, in reality, be a higher frequency of visits when patients start taking TKIs, these costs would be equal (i.e. cancel out) between treatment arms.]
Note that, unlike the BMS modelling analyses, we did not include different costs for patients responding and not responding to treatment. This is for simplicity, and because the time that most patients are not responding to treatment, and before they are switched to a new treatment, should be relatively small relative to overall time in first- or second-line treatment. Also, in the questions that distinguished patients as either responding or not responding to treatment in the Oxford Outcomes cost survey (used by BMS), response was not defined; so it is wholly unclear whether this related to cytogenetic, molecular or some other type or level of treatment response for those who answered this survey.
Table 46 shows the resulting estimates of the medical management costs per month for patients in the chronic and advanced phases (AP and BC).
| Procedure | Frequency (per month)a | Unit cost (£2009)c | Monthly cost (£2010) |
|---|---|---|---|
| CP on TKI | |||
| Haematologist/oncologist-led outpatient appointments | 0.33 | 127 | 41.91 |
| Tests (various)b | b | Various | 13.87 |
| Hospital in patient: ward-days | 0 | 246 | 0 |
| Hospital in patient: ICU-days | 0 | 1219 | 0 |
| CP total | 56 | ||
| CP on hydroxycarbamide | |||
| Haematologist/oncologist-led outpatient appointments | 0.72 | £127 | 91.44 |
| Tests (various)b | b | Various | 13.87 |
| Hospital in patient: ward-days | 0 | £246 | 0 |
| Hospital in patient: ICU-days | 0 | £1,219 | 0 |
| CP total | 106 | ||
| AP | |||
| Nurse-led outpatient appointments | 0.50 | £100 | 50.00 |
| Haematologist/oncologist-led outpatient appointments | 1.30 | £127 | 165.10 |
| Tests (various)b | b | Various | 352.45 |
| Hospital in patient: ward-days | 1.72 | £246 | 423.83 |
| Hospital in patient: ICU-days | 0.10 | £1,219 | 121.90 |
| Advanced phase total | 1113 | ||
Cost of care post tyrosine kinase inhibitor: failure
Cost of stem cell transplant (second or third line)
Estimating the NHS cost of adult SCT is complicated by a number of factors:
-
It is a complex multistage process (typically presented as eight phases, from decision to transplant to after 100 days’ follow-up). 137
-
The resource use and cost of many phases differs for related and unrelated donors, and also, for example, depending on whether or not related donor SCT recipients may have reduced-intensity chemotherapy (reduces transplant cost) or whether or not unrelated donor SCT recipients require more or less myeloablative therapy.
-
The cost categories and HRGs within the National Schedule of Reference Costs (NSRC) are relatively new and their use is still evolving. They do not appear to cover all phases of the SCT process. (There is anecdotal evidence from specialist commissioners that the HRGs may not yet be consistently used in cost submissions from NHS trusts and primary care trusts.)
-
The costs vary considerably in different parts of England and Wales, and from trust to trust, for example depending on overhead allocation rates, critical care costs and the prices paid for obtaining out-of-area unrelated donor cells.
As with the Novartis analysis, we therefore based our base-case, per-patient cost estimate for a SCT on an unpublished September 2009 report by the London Specialised Commissioning Group, which is the most comprehensive and UK-based cost and pricing analysis of adult bone marrow and SCT currently available (Mr Mike Millen, London Specialised Commissioning Group, 29 July 2011, personal communication). They report a mean cost of transplant for phases 1–6 [from ‘decision to transplant and donor selection’ (= phase 1) through ‘transplant inpatient admission’ (= phase 4), to day 100 post transplant (= phase 6)]of £47,500 (£2009) for related donor allografts and £79,600 for unrelated donor allografts.
For the cost of transplant phases 1–6, we took a weighted average of these two costs, based on assumed 25% : 75% split of related (usually sibling) compared with unrelated (volunteer) donor transplants (sources: Ashfaq et al. ;138 Jessica Whitton, Senior Commissioning Manager, SW Specialist Commissioning Manager, 14 July 2011, personal communication; note same split as that assumed by Novartis submission) and inflated to 2011 costs.
For the short-term cost of phases 7 and 8 (i.e. from 100+ days post transplant to approximately 2 years) we also estimated the cost of antifungal drugs used and the cost of repeat donor lymphocyte infusions. The mean costs for both of these are also taken from the 2009 London SCG analysis, but the mean per-patient cost of donor lymphocyte infusions has been based on 3 years of data relating to adult allogeneic stem cell transplants from University of Bristol Hospital (Jessica Whitton, Senior Commissioning Manager, SW Specialised Commissioning Group, 27 July 2011, personal communication).
Table 47 shows the estimation of our base-case cost of SCT.
| Related donor | Unrelated donor | Source and notes | |
|---|---|---|---|
| Cost for phases 1–6 (£2009) | 47,500 | 79,600 | London SCG137 |
| Inflated to 2011 (i.e. 2 years), £ | 49,115 | 82,306 | PSSRU, Curtis136 |
| Percentage split of related vs unrelated | 25 | 75 | Ashfaq et al.138 |
| Weighted average, £ | 74,008 | ||
| Plus cost of antifungal drugs | 5369 | London SCG137 (weighted average) | |
| Plus donor lymphocyte infusions | 2225 | London SCG137 (weighted average, also using UBH dataa on percentage of related and unrelated donor patients receiving different numbers of DLIs) | |
| Mean per-patient cost of SCT | 81,600b |
We also estimated the cost of SCT by an alternative method, starting with the NSRC HRG cost estimate for an inpatient stay for ‘peripheral blood STC in adults’ (code SA28A = national average cost of £34,783, just for phase 4 of transplant process) and then used a table in the LSCG report, which shows the percentage split of total costs across transplant phases (1–6) to estimate the total cost of phases 1–3, 5 and 6 (from decision to transplant to 100 days post transplant). The estimate from this method comes out as £81,300 – very close to our first method. The method above was used in the model.
Longer term following stem cell transplant
Unlike the Novartis submission, we chose to include an estimate of the cost of long-term care following SCT, especially to reflect the monitoring and treatment of longer-term complications such as chronic GvHD. Our estimate of £113 per month for those suffering cGvHD includes (1) the NHS cost of a quarterly specialist appointment with a clinical haematologist (£125 per appointment) plus (2) the estimated cost of immunosuppressive drug therapies (either ciclosporin with prednisolone or mycophenolate with prednisolone for the base-case assumptions). The calculation of these cost estimates is shown in Table 48. An estimate of the monthly cost of a more intensive immunosuppressive drug therapy regime, typically for treating more severe cGvHD, is also shown (ciclosporin, mycophenolate, methotrexate and prednisolone), and this higher estimate is used in sensitivity analysis.
| Immunosuppressive regime | Drug costs (£)a | Quarterly appointments costs (£) | Total quarterly cost (by regime) (£) | Percentage split | Total quarterly cost (weighted average) (£) |
|---|---|---|---|---|---|
| Ciclosporin (50 mg b.i.d.) plus prednisolone (20 mg q.d.) | 65.96 | 42 | 107.62 | 60 | 64.57 |
| Mycophenolate (1 g b.i.d.) plus prednisolone (20 mg q.d.) | 80.32 | 42 | 121.97 | 40 | 48.79 |
| Weighted mean cost per month | 113 |
Cost of care in advanced phases
Accelerated phase
In addition to the substantially higher costs of medical management (outpatient appointments and tests; see Cost of other medical management and monitoring, above) we assumed that patients in the AP would be treated with hydroxycarbamide. We acknowledge that this is a considerable simplification of the range of possible treatments that people in this heterogeneous group are likely to receive; CML patients within the AP or BC phase may receive SCT after chemotherapy or TKIs as an adjunct to chemotherapy. However, although the use of some TKIs in the AP is licensed, the evidence for their effectiveness in the APs of the disease is very limited (and the recent NICE draft guidance FAD on nilotinib, dasatinib and high-dose imatinib in second-line treatment of CML emphasised this; the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99).
Blast crisis
The quarterly care costs for patients in the BC phase were assumed to be the same as in the AP, but with the addition at death of an inpatient palliative care stay (£425) plus two non-medical specialist palliative care home visits (£72 each).
Peninsula Technology Assessment Group cost-effectiveness results
We first present and discuss the base-case results for the different scenarios and then the results of the sensitivity analyses. For the base-case analyses we first present the results based on scenario 1, the full results of the cumulative survival method without second-line nilotinib. Next, we present the results of scenario 2, which is the same as scenario 1 but using the simplified method (which equalises post-TKI costs and outcomes). Next, we present the results of scenario 3, which is the same as scenario 1, but allowing second-line nilotinib, and, finally, scenario 4, the same as scenario 2, but allowing for second-line nilotinib (as presented in Table 31).
The results for the scenarios based on surrogate survival methods (1a and 1b for MMR based, and 2a and 2b for CCyR based) are presented below (see Sensitivity analyses: surrogate overall survival).
Note that we have chosen not to conduct and present probabilistic sensitivity analyses (PSAs) because of the unusually large amount of structural uncertainty that is inherent in the present decision problem(s). This structural uncertainty relates to both the variety of ways in which long-term survival might be estimated and uncertainty surrounding the possible sequences and mixes of treatments post first-line TKI failure. As a result, we believe that structural uncertainty would dominate total (structural and parameter) uncertainty and, therefore, if we presented PSAs based just on parameter uncertainty, then this would be of little use to the committee. Furthermore, it might actually mislead users of our report who do not appreciate the substantial structural uncertainty.
Theoretically, it would have been possible to incorporate some of the structural uncertainty in to a PSA by some kind of model averaging. For example, we present scenario analyses with and without second-line nilotinib. To incorporate the uncertainty in whether we assume use of second-line nilotinib, we could have assigned a probability to the use of second-line nilotinib, and present just one analysis. However, we believe that it would be more helpful to the committee to present the two analyses separately, thus allowing the committee to decide for themselves which scenario they prefer, i.e. allowing them to use their expert judgement to estimate the probability of second-line nilotinib use for themselves.
Summary of cost-effectiveness results
Table 49 shows the cost-effectiveness results for scenarios 1–4, conventionally with comparators in order of increasing effectiveness, and the ICERs representing the incremental costs and QALYs gained by moving to the next most effective non-dominated. In the more detailed results tables in the rest of the chapter the ICERs are calculated relative to the current best clinical practice in the NHS (imatinib as first line) and then between nilotinib and dasatinib.
| Scenario | Discounted cost (£) | Undiscounted life-years | Discounted QALYs | Incremental cost (£) | Incremental QALYs | ICER (£ per QALY) |
|---|---|---|---|---|---|---|
| Scenario 1: cumulative survival without second-line nilotinib | ||||||
| Imatinib – then hydroxycarbamide/SCT | 159,000 | 16.5 | 9.0 | |||
| Nilotinib – then hydroxycarbamide/SCT | 170,000 | 17.4 | 9.4 | 11,000 | 0.4 | 25,000 |
| Dasatinib – then hydroxycarbamide/SCT | 224,000 | 16.8 | 9.2 | 54,000 | –0.3 |
Dasatinib dominated by nilotinib |
| Scenario 2: cumulative survival without second-line nilotinib – simplified method | ||||||
| Imatinib – then hydroxycarbamide/SCT | 159,000 | 9.0 | ||||
| Nilotinib – then hydroxycarbamide/SCT | 172,000 | 9.7 | 13,000 | 0.7 | 20,000 | |
| Dasatinib – then hydroxycarbamide/SCT | 225,000 | 9.3 | 53,000 | –0.4 | Dasatinib dominated by nilotinib | |
| Scenario 3: cumulative survival with second-line nilotinib | ||||||
| Nilotinib – then hydroxycarbamide/SCT | 170,000 | 17.4 | 9.4 | |||
| Imatinib – then nilotinib | 188,000 | 17.3 | 9.5 | 19,000 | 0.1 | 192,000a |
| Dasatinib – then nilotinib | 252,000 | 17.6 | 9.7 | 63,000 | 0.1 | 450,000 |
| Scenario 4: cumulative survival with second-line nilotinib – simplified method | ||||||
| Nilotinib – then hydroxycarbamide/SCT | 166,000 | 9.1 | ||||
| Imatinib – then nilotinib | 188,000 | 9.5 | 22,000 | 0.5 | 46,000a | |
| Dasatinib – then nilotinib | 253,000 | 9.7 | 65,000 | 0.2 | 301,000 | |
The variation in cost-effectiveness results across the four scenarios is considerable, with the ICERs for nilotinib compared with imatinib being either slightly above (scenario 1) or on (scenario 2) the £20,000 cost-effectiveness threshold, or – in scenarios 3 and 4 – generating slightly fewer lifetime QALYs than imatinib followed by nilotinib, but yielding significant cost savings. However, in all scenarios dasatinib is shown to be either dominated (by nilotinib) or have an ICER relative to imatinib of over £250,000 per QALY gained. These widely different cost-effectiveness results again reinforce the significance of structural uncertainty in the modelling of CML, including the substantial impact of assumptions regarding second- and third-line treatment sequences after first-line TKI failure.
The interpretation of the ICERs for scenarios 3 and 4 is unusual, because having nilotinib as second-line treatment after imatinib or dasatinib – but of course not after nilotinib failure – results in the nilotinib comparator being both less effective and more costly over patients’ lifetimes than the current best practice treatment of imatinib. Depending on which modelling assumptions are used, this means that adopting nilotinib as first-line treatment yields considerable cost savings for relatively modest QALY losses per patient (either £192,000 or £46,000 of savings yielded per QALY lost, in scenarios 3 and 4). This is discussed further in the following sections and in the Discussion (see Chapter 8).
Results: scenario 1 – cumulative survival method without second-line nilotinib
Table 50 presents the cost-effectiveness results for scenario 1.
| Treatment | Imatinib | Nilotinib | Dasatinib | Nilotinib–imatinib | Dasatinib–imatinib | Nilotinib–dasatinib |
|---|---|---|---|---|---|---|
| Life-years (undiscounted) | ||||||
| First-line TKI | 7.0 | 8.9 | 7.7 | 1.9 | 0.7 | 1.2 |
| Second-line nilotinib | – | – | – | – | – | – |
| SCT | 5.8 | 4.9 | 5.5 | –0.9 | –0.4 | –0.6 |
| Hydroxycarbamide CP | 2.9 | 2.8 | 2.9 | –0.1 | –0.0 | –0.1 |
| Hydroxycarbamide AP | 0.5 | 0.4 | 0.5 | –0.0 | –0.0 | –0.0 |
| Hydroxycarbamide BC | 0.3 | 0.3 | 0.3 | –0.0 | –0.0 | –0.0 |
| OS (mean) | 16.5 | 17.4 | 16.8 | 0.9 | 0.3 | 0.6 |
| OS (median) | 15.0 | 16.3 | 15.4 | 1.3 | 0.4 | 0.9 |
| Mean age start | ||||||
| First-line TKI | 57 | 57 | 57 | 0.0 | 0.0 | 0.0 |
| Second-line nilotinib | – | – | – | – | – | – |
| SCT | 64 | 66 | 65 | 1.9 | 0.7 | 1.2 |
| Hydroxycarbamide CP | 64 | 66 | 65 | 1.9 | 0.7 | 1.2 |
| Hydroxycarbamide AP | 69 | 71 | 70 | 1.8 | 0.7 | 1.1 |
| Hydroxycarbamide BC | 70 | 72 | 71 | 1.8 | 0.7 | 1.1 |
| Cohort splita | ||||||
| % starting second-line nilotinib | 0 | NA | 0 | NA | 0 | NA |
| % starting SCT/hydroxycarbamide | 90 | 84 | 88 | –6 | –2 | –4 |
| % SCT (whole cohort) | 33 | 28 | 32 | –5 | –2 | –3 |
| % SCT (eligible cohort) | 37 | 34 | 36 | –3 | –1 | –2 |
| % hydroxycarbamide (whole cohort) | 56 | 56 | 56 | –1 | 0 | –1 |
| % hydroxycarbamide (eligible cohort) | 63 | 66 | 64 | 3 | 1 | 2 |
| % AP (whole cohort) | 49 | 48 | 49 | –2 | 0 | –1 |
| % BC (whole cohort) | 49 | 48 | 49 | –2 | 0 | –1 |
| Life-years (undiscounted eligible cohorta) | ||||||
| First-line TKI | 7.0 | 8.9 | 7.7 | 1.9 | 0.7 | 1.2 |
| Second-line nilotinib | – | – | – | – | – | – |
| SCT | 17.4 | 17.2 | 17.3 | –0.2 | –0.1 | –0.1 |
| Hydroxycarbamide CP | 5.1 | 5.0 | 5.1 | –0.1 | –0.0 | –0.1 |
| Hydroxycarbamide AP | 0.9 | 0.9 | 0.9 | –0.0 | –0.0 | –0.0 |
| Hydroxycarbamide BC | 0.6 | 0.6 | 0.6 | –0.0 | –0.0 | –0.0 |
| QALYs (discounted) | ||||||
| First-line TKI | 4.5 | 5.5 | 4.9 | 1.0 | 0.4 | 0.6 |
| Second-line nilotinib | – | – | – | – | – | – |
| SCT | 2.6 | 2.2 | 2.4 | –0.4 | –0.2 | –0.3 |
| Hydroxycarbamide CP | 1.5 | 1.4 | 1.5 | –0.1 | –0.0 | –0.1 |
| Hydroxycarbamide AP | 0.2 | 0.2 | 0.2 | –0.0 | –0.0 | –0.0 |
| Hydroxycarbamide BC | 0.1 | 0.1 | 0.1 | –0.0 | –0.0 | –0.0 |
| Total | 9.0 | 9.4 | 9.2 | 0.4 | 0.2 | 0.3 |
| Costs (discounted), £ | ||||||
| First-line TKI | 118,635 | 133,386 | 184,774 | 14,751 | 66,139 | –51,388 |
| First-line AEs | 166 | 119 | 282 | –47 | 116 | –163 |
| First-line medical management | 3811 | 4658 | 4127 | 846 | 316 | 530 |
| Second-line nilotinib | – | – | – | – | – | – |
| Second-line nilotinib AEs | – | – | – | – | – | – |
| Second-line nilotinib medical management | – | – | – | – | – | – |
| SCT transplant | 24,486 | 20,646 | 23,005 | –3840 | –1482 | –2359 |
| SCT medical management | 2562 | 2148 | 2401 | –415 | –161 | –254 |
| Hydroxycarbamide acquisition in CP | 282 | 264 | 276 | –19 | –6 | –12 |
| Hydroxycarbamide CP medical management | 2494 | 2330 | 2439 | –164 | –55 | –109 |
| Hydroxycarbamide AP acquisition + medical management | 4098 | 3828 | 4007 | –270 | –91 | –179 |
| Hydroxycarbamide BC acquisition + medical management | 2735 | 2555 | 2675 | –180 | –61 | –120 |
| Total | 159,270 | 169,932 | 223,985 | 10,662 | 64,715 | –54,053 |
| Cost/life-years gained | 12,000 | 205,000 | –97,000 | |||
| Cost/QALY | 25,000 | 414,000 | –205,000 | |||
Scenario 1: survival results
The relative proportions of patients in each health state for each treatment over time are displayed in Figure 33. The mean duration in each health state for each treatment (as reported in Table 50, above) is represented in these graphs by the area under each curve. For example, mean survival after SCT is represented by the light shaded area. Virtually all patients are predicted to have died by age 97, 20 years from start of first-line treatment.
FIGURE 33.
Scenario 1 cohort composition over time by treatment arm.
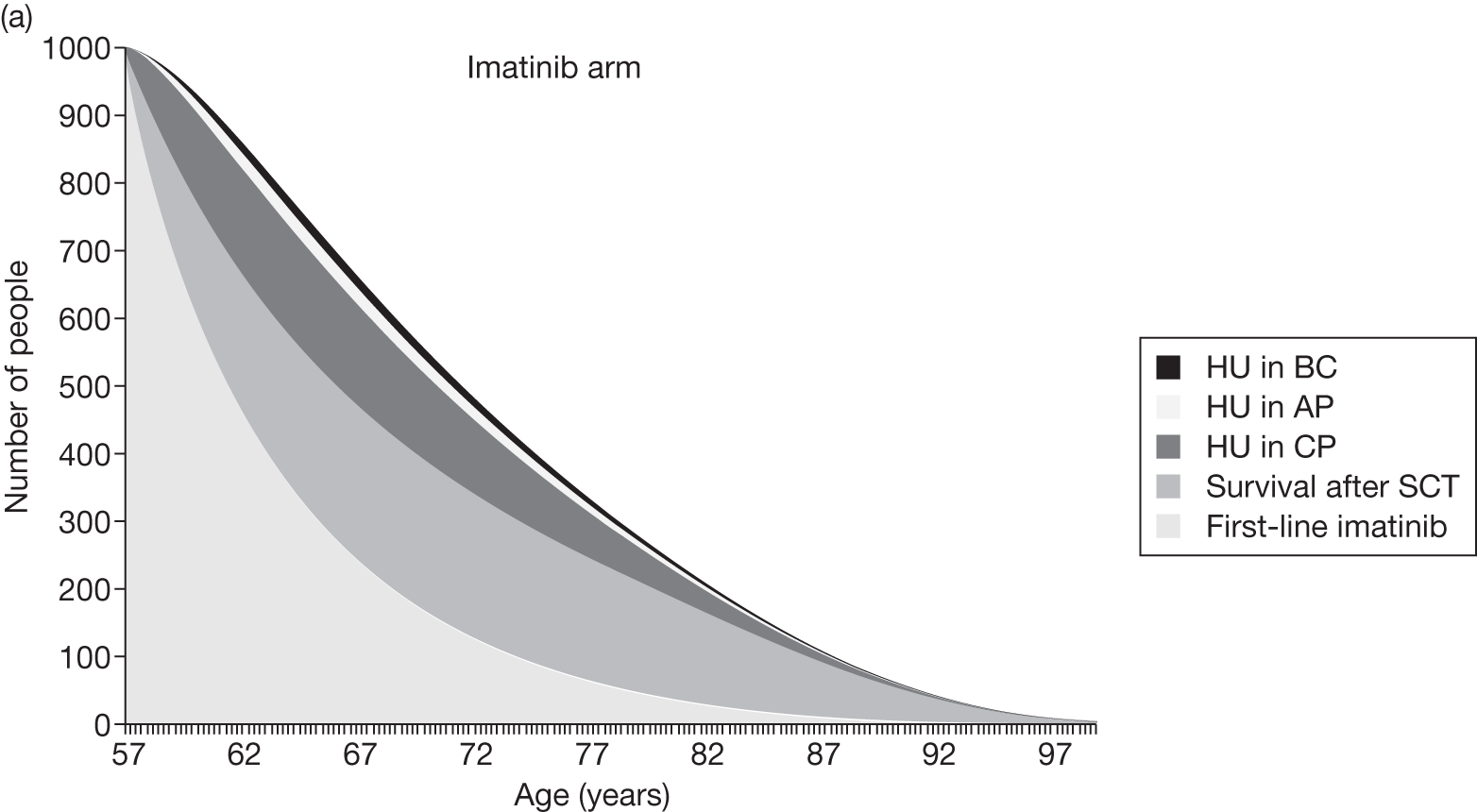
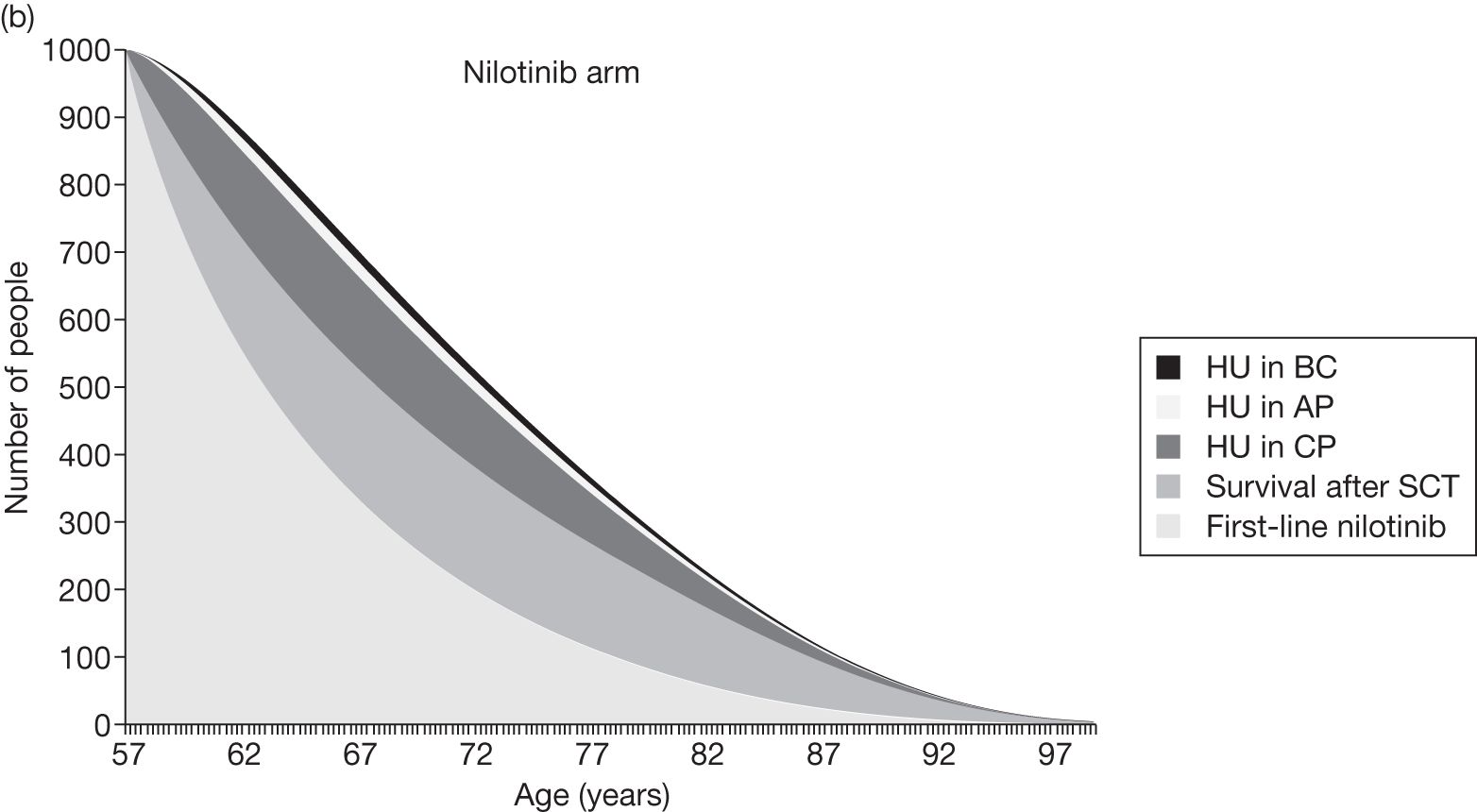
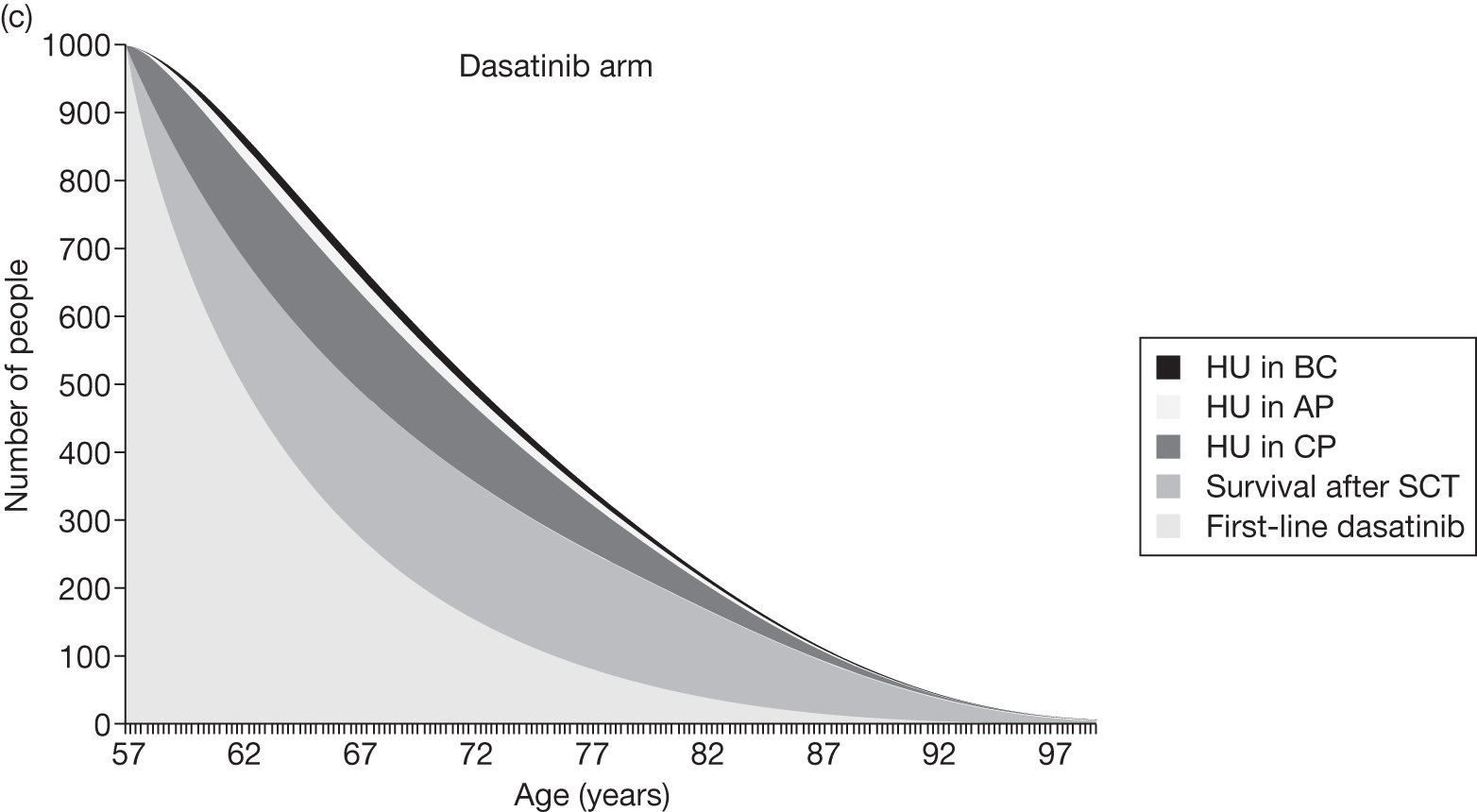
As previously explained (see Figure 28), we predict that the mean duration of first-line treatment is least for people on imatinib (7.0 years), greater for dasatinib (7.7 years) and greatest for nilotinib (8.9 years) (see Table 50 and Figure 33).
We predict similar mean survival times after SCT for all treatment arms, but with shortest duration in the nilotinib arm (4.9 years), longer in the dasatinib arm (5.5 years) and longest in the imatinib arm (5.8 years) (see Table 50). This order is explained by three factors. First, fewest people reach second-line treatment in the nilotinib arm (84%), and most reach second-line treatment in the imatinib arm (90%) (see Table 50), because this reflects the relative duration of first-line treatment, and the longer people spend on first-line treatment, the greater the mortality. Also, the lower the proportion reaching second-line treatment, the lower the mean survival time after SCT averaged over all patients starting first-line treatment. Second, we assume that the proportion of patients receiving SCT declines with increasing age (see Proportion of patients receiving stem cell transplant post TKI failure). Given that people are generally slightly older when they have SCT in the nilotinib arm (66 years old) than those in the imatinib arm (64 years old), this also reduces the proportion of patients having SCT in the nilotinib arm, relative to the other treatment arms. Combined, we predict that only 28% of patients in the nilotinib arm receive SCT, with similar proportions in the imatinib and dasatinib arms (32% and 33%). Third, the mean survival after SCT, for those patients who receive SCT (the ‘eligible’ cohort in Table 50), is marginally lower in the nilotinib arm (17.2 years) than in the imatinib arm (17.4 years). This is due to the fact that people typically receive SCT in the nilotinib arm when they are slightly older, as explained above, and therefore general mortality is greatest in the nilotinib arm.
The mean time on hydroxycarbamide in CP, averaged over all patients initially starting first-line treatment, is almost the same across treatment arms: 2.8 years for nilotinib and 2.9 years for imatinib and dasatinib. There are two treatment-dependent factors that operate in different directions here. First, of those patients who reach second-line treatment, the highest proportion received hydroxycarbamide in the nilotinib arm (66%, the ‘eligible patients’ in Table 50) and the lowest proportion received hydroxycarbamide in the imatinib arm (63%). The explanation is similar to that given in the previous paragraph for the proportion of eligible patients who receive SCT. In this case, a large proportion receive hydroxycarbamide in the nilotinib arm because patients are typically slightly older when they start second-line treatment, and the proportion of eligible patients who receive hydroxycarbamide increases with age (as the proportion who receive SCT decreases with age). Second, as stated in the previous paragraph, the proportion of patients who receive second-line treatment is least in the nilotinib arm. Together, these factors cancel out, resulting in the same proportion of all patients who start first-line treatment taking hydroxycarbamide in CP, 56% in all treatment arms (see Table 50).
The mean time on hydroxycarbamide in AP, averaged across all patients initially starting first-line treatment, is almost the same across treatment arms: 0.4 years for nilotinib and 0.5 years for imatinib and dasatinib. Similarly, the proportion of all patients randomised to first-line treatment who receive hydroxycarbamide in AP is almost the same across treatment arms, at 48–49%. Again, this is explained by two competing treatment-dependent influences that cancel out. First, we might expect the proportion of all patients who receive hydroxycarbamide in AP to be least for the nilotinib arm because the proportion of patients who reach second-line treatment is least in this arm (84% nilotinib arm vs 90% imatinib arm). Conversely, the greatest proportion of those patients who receive second-line treatment who receive hydroxycarbamide do so in the nilotinib arm (66% nilotinib arm vs 63% imatinib arm) and it is necessary to pass through the hydroxycarbamide in CP state in order to reach the hydroxycarbamide in AP state.
Finally, mean time on hydroxycarbamide in BC, averaged over all patients initially starting first-line treatment, is the same across treatment arms, at 0.3 years. This is explained in the previous paragraph for the mean time on hydroxycarbamide in AP, and the fact that we assume no mortality on hydroxycarbamide in AP, given such a short time in AP.
Notice that the mean undiscounted life-years for those patients who receive the relevant treatment, the eligible cohorts in Table 50, is very nearly independent of treatment arm. Any slight differences between arms is due to the slight differences in general mortality owing to the slight differences in mean ages at the start of each treatment. Also notice that the life expectancy of those patients who take SCT, at about 17 years, is far higher than for those patients who take hydroxycarbamide in AP, at approximately 6.5 years (the sum of the mean times on hydroxycarbamide in CP, AP and BC). We predict a long life expectancy after SCT because we assume that 75% of patients who have SCT subsequently experience the same mortality as the general population of England and Wales.
Overall survival from time of starting first-line treatment, which reflects the sum of the times on the component lines of treatments, is similar across treatment arms, but greatest in the nilotinib arm (17.4 years) and least in the imatinib arm (16.5 years). The difference in OS between the nilotinib and imatinib arms, at 0.9 years, is less than the difference in the time on first-line nilotinib and first-line imatinib, at 1.9 years. This is because a lower proportion of patients received SCT in the nilotinib arm (28%) than in the imatinib arm (33%), and life expectancy after SCT is high, at about 17 years.
We now turn to the estimated QALYs in Table 50. First, notice that the relative differences in discounted QALYs between treatment arms is consistent with the relative differences in undiscounted life-years. For example, both life-years and QALYs are 5% higher in the nilotinib arm than in the imatinib arm. Next, the ratio of discounted QALYs to undiscounted life-years is approximately 55% in all arms. This is accounted for by the rather substantial discounting, given high life expectancies of approximately 17 years, and by the application of utility values that are typically approximately 0.80, averaged over the entire cohort, over all time.
Scenario 1: cost results
We now turn to the expected costs per person. The expected costs of first-line drug acquisition are by far the largest single cost item (see Table 50) and account for the largest incremental costs compared with the imatinib arm (Figure 34). Notice, further, that the mean acquisition costs of imatinib and nilotinib are fairly similar (£119,000 and £133,000, respectively), whereas the cost of dasatinib is far higher, at about £185,000. The expected drug acquisition costs are calculated as the product of the mean drug acquisition cost per person per unit time and the discounted mean duration of drug treatment.
FIGURE 34.
Scenario 1 incremental costs vs imatinib treatment arm. Med man, medical management.
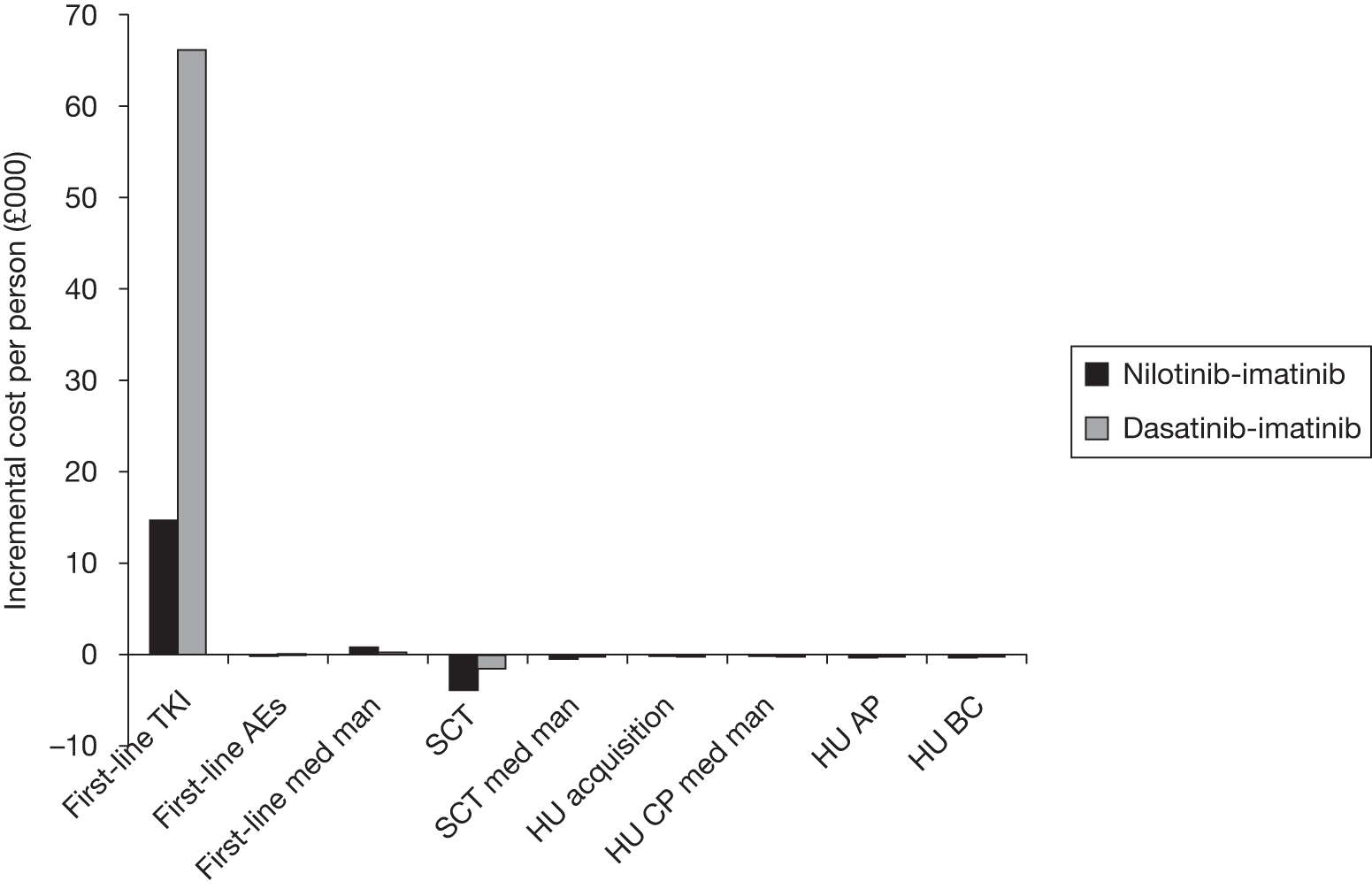
(CiC information has been removed.)
The expected incremental costs of medical management during first-line treatment and the expected incremental cost of the SCT operation are the next largest single cost items (see Table 50). The expected medical management costs during first-line treatment are greatest in the nilotinib arm and least in the imatinib arm, reflecting the order of duration of first-line treatments.
The expected cost of SCT, averaged over all patients at about £21,000, is least for nilotinib and greatest for imatinib because the proportion of all patients who have SCT is least for the nilotinib arm (28%) and greatest for the imatinib arm (33%).
All other costs contribute only marginally to the incremental costs. The per-patient medical management costs after SCT are least in the nilotinib arm, also because the proportion of all patients who have SCT is least for the nilotinib arm (28%). Although the absolute per-patient costs of medical management while taking hydroxycarbamide in CP are rather large, the incremental costs compared with imatinib are small because we predict similar mean per-patient duration of hydroxycarbamide in CP, at about 2.9 years. The incremental costs of hydroxycarbamide acquisition and medical management in AP and BC are very small for the same reason. The costs of AEs while on first-line treatment and the cost of hydroxycarbamide acquisition in CP are both extremely small.
Scenario 1: cost-effectiveness results
Combining all of the information on expected costs and QALYs per person, we estimate the following cost-effectiveness results (see Table 50 and Figure 35):
-
nilotinib compared with imatinib ICER of £25,000 per QALY
-
dasatinib compared with imatinib ICER of £414,000 per QALY
-
nilotinib dominates dasatinib.
Figure 35 displays the results from both scenarios 1 and 3 on the same cost-effectiveness plane. Filled symbols represent treatment arms, which include second-line treatment with nilotinib, and empty symbols represent treatment arms without second-line nilotinib. The top graph shows that the difference in QALYs between the arms is rather small, but the difference in total costs per person is large. In both graphs, the continuous line represents a willingness to pay of £30,000 per QALY compared with treatment with imatinib followed by nilotinib, and the dotted line represents a willingness to pay of £30,000 per QALY compared with treatment with imatinib without second-line nilotinib. Scenario 1 concerns the empty symbols only, and scenario 3 concerns the filled symbols plus the treatment arm with nilotinib first line.
FIGURE 35.
Cost-effectiveness results for scenarios 1 and 3 (wide axes top and narrow axes bottom). Straight lines represent £30,000 per QALY threshold anchored on comparators involving first-line imatinib (cheapest in each scenario).
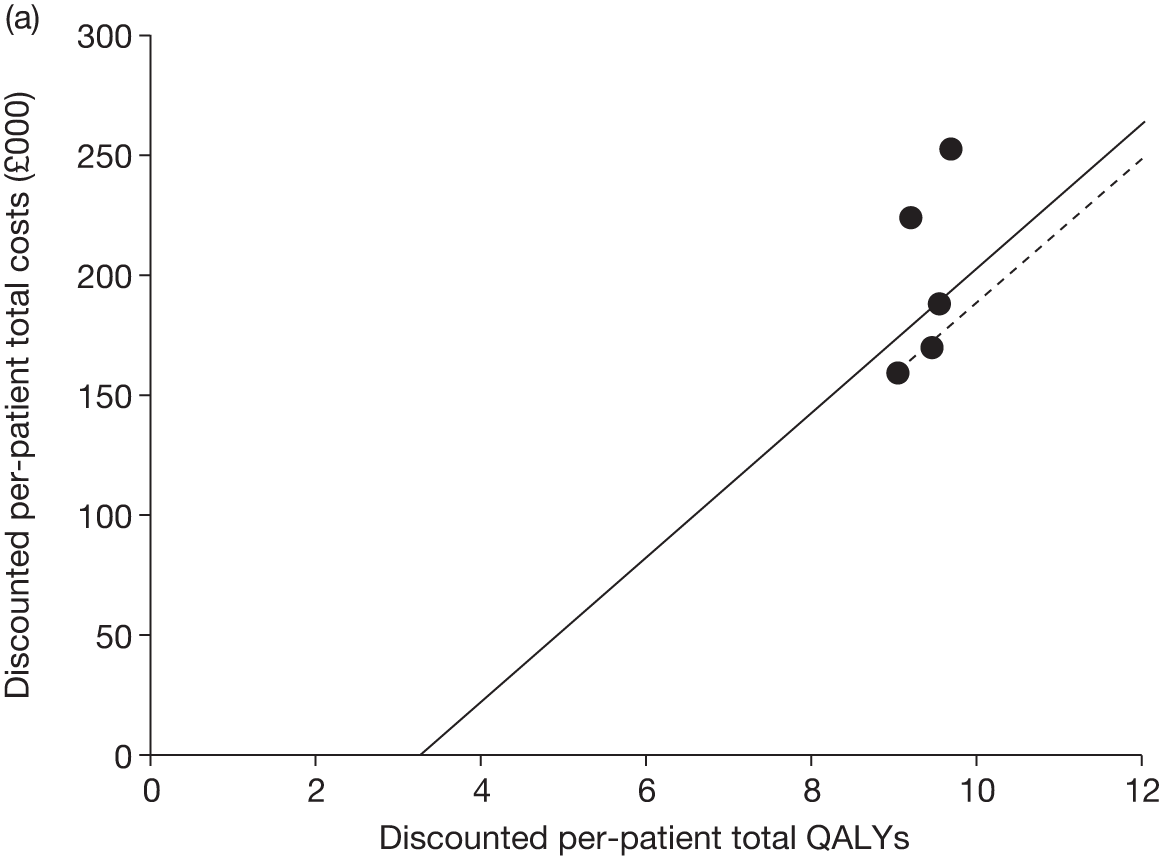
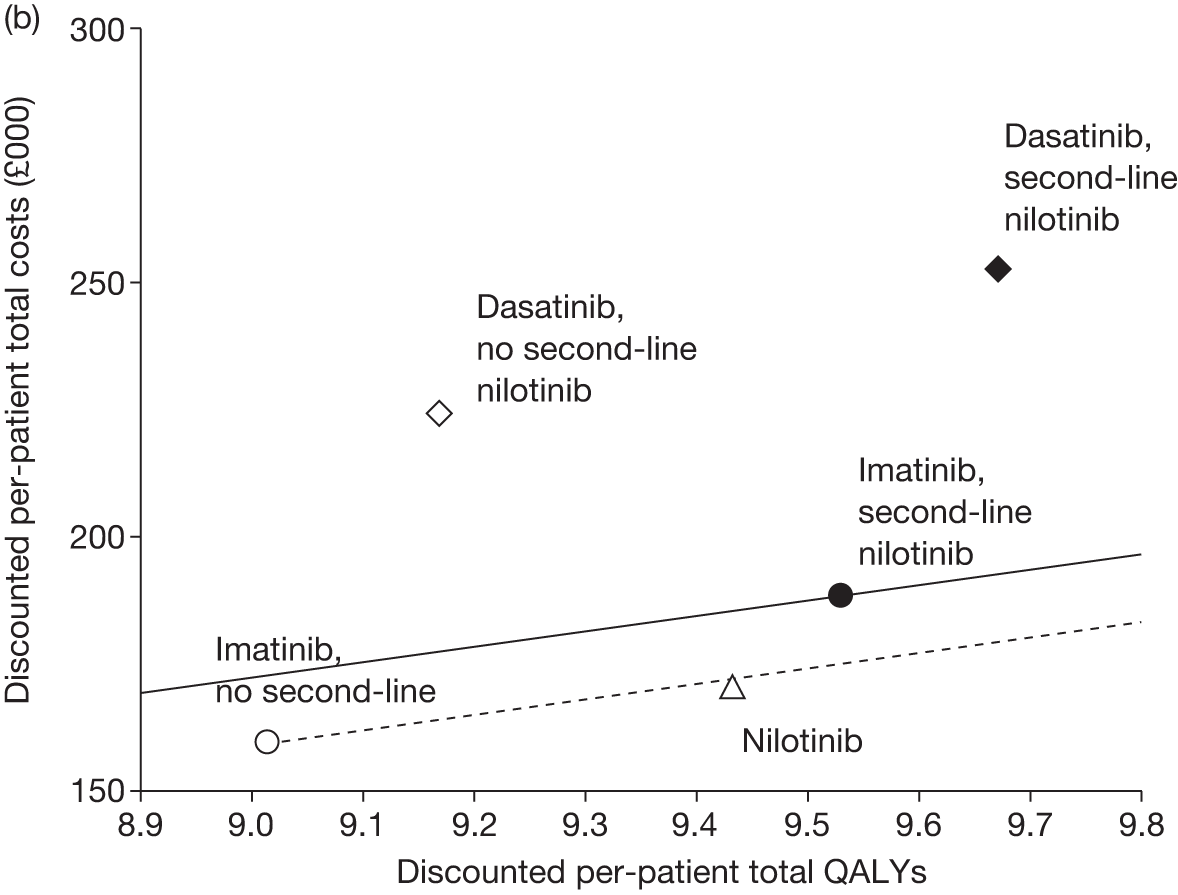
For scenario 1, the symbol for nilotinib lies just below the £30,000 willingness-to-pay line, based on imatinib first-line with no nilotinib second-line, which reflects the fact that the ICER of nilotinib compared with imatinib is £25,000 per QALY, only slightly < £30,000 per QALY.
For scenario 1, the symbol for dasatinib without second-line nilotinib lies well above the willingness-to-pay line, which reflects the very high ICER of dasatinib compared with imatinib of £414,000 per QALY.
We discuss the results from scenario 3 below (see Results: scenario 3 – cumulative survival method with second-line nilotinib).
Results: scenario 2 – cumulative survival, simplified method, without second-line nilotinib
In the simplified method, the post-TKI (first-line TKIs) per-patient costs and QALYs are set equal across treatment arms. The costs and QALYs while patients are on TKIs are modelled specifically to each treatment arm, i.e. exactly as normal (see Simplified method, above). The method substantially reduces the impact of the nature, costs and utilities associated with treatments post first-line TKIs. We believe that this is useful, given that these treatments will typically be taken many years in the future and the substantial uncertainty in the nature and costs of such treatments. Table 51 presents the cost-effectiveness results for scenario 2.
| Treatment | Imatinib | Nilotinib | Dasatinib | Nilotinib–Imatinib | Dasatinib–Imatinib | Nilotinib–Dasatinib |
|---|---|---|---|---|---|---|
| QALYs (discounted) | ||||||
| First-line TKI | 4.5 | 5.5 | 4.9 | 1.0 | 0.4 | 0.6 |
| Second-line nilotinib | – | – | – | – | – | – |
| SCT | 2.6 | 2.4 | 2.5 | –0.2 | –0.1 | –0.1 |
| Hydroxycarbamide CP | 1.5 | 1.4 | 1.5 | –0.1 | –0.0 | –0.1 |
| Hydroxycarbamide AP | 0.2 | 0.2 | 0.2 | –0.0 | –0.0 | –0.0 |
| Hydroxycarbamide BC | 0.1 | 0.1 | 0.1 | –0.0 | –0.0 | –0.0 |
| Total | 9.0 | 9.7 | 9.3 | 0.7 | 0.3 | 0.4 |
| Costs (discounted), £ | ||||||
| First-line TKI | 118,635 | 133,386 | 184,774 | 14,751 | 66,139 | –51,388 |
| First-line AEs | 166 | 119 | 282 | -47 | 116 | –163 |
| First-line medical management | 3811 | 4658 | 4127 | 846 | 316 | 530 |
| Second-line nilotinib | – | – | – | – | – | – |
| Second-line nilotinib AEs | – | – | – | – | – | – |
| Second-line nilotinib medical management | – | – | – | – | – | – |
| SCT transplant | 24,486 | 22,935 | 23,954 | –1551 | –532 | –1019 |
| SCT medical management | 2562 | 2400 | 2507 | –162 | –56 | –107 |
| Hydroxycarbamide acquisition in CP | 282 | 259 | 274 | –23 | –8 | –15 |
| Hydroxycarbamide CP medical management | 2494 | 2289 | 2421 | –205 | –73 | –132 |
| Hydroxycarbamide AP acquisition + medical management | 4098 | 3838 | 4009 | –260 | –89 | –171 |
| Hydroxycarbamide BC acquisition + medical management | 2735 | 2562 | 2676 | –173 | –59 | –114 |
| Total | 159,270 | 172,446 | 225,023 | 13,176 | 65,753 | –52,577 |
| Cost/QALY | 20,000 | 256,000 | –129,000 | |||
The proportions still alive and starting second- or third-line treatment on hydroxycarbamide or SCT are similar across the three treatment arms (from 84% for nilotinib to 90% for imatinib), since the durations of TKI treatments are similar across treatments. Therefore, this method largely nets off all post-TKI costs and QALYs between treatment arms. For example, the incremental QALYs associated with time after SCT is –0.2 for nilotinib–imatinib, which is smaller than the corresponding figure of –0.4 from scenario 1, and the incremental per-person cost of SCT operations is –£1551 for nilotinib–imatinib (Figure 36), compared with –£3840 in scenario 1 (see Table 50).
FIGURE 36.
Scenario 2 incremental costs vs imatinib treatment arm. Med man, medical management.
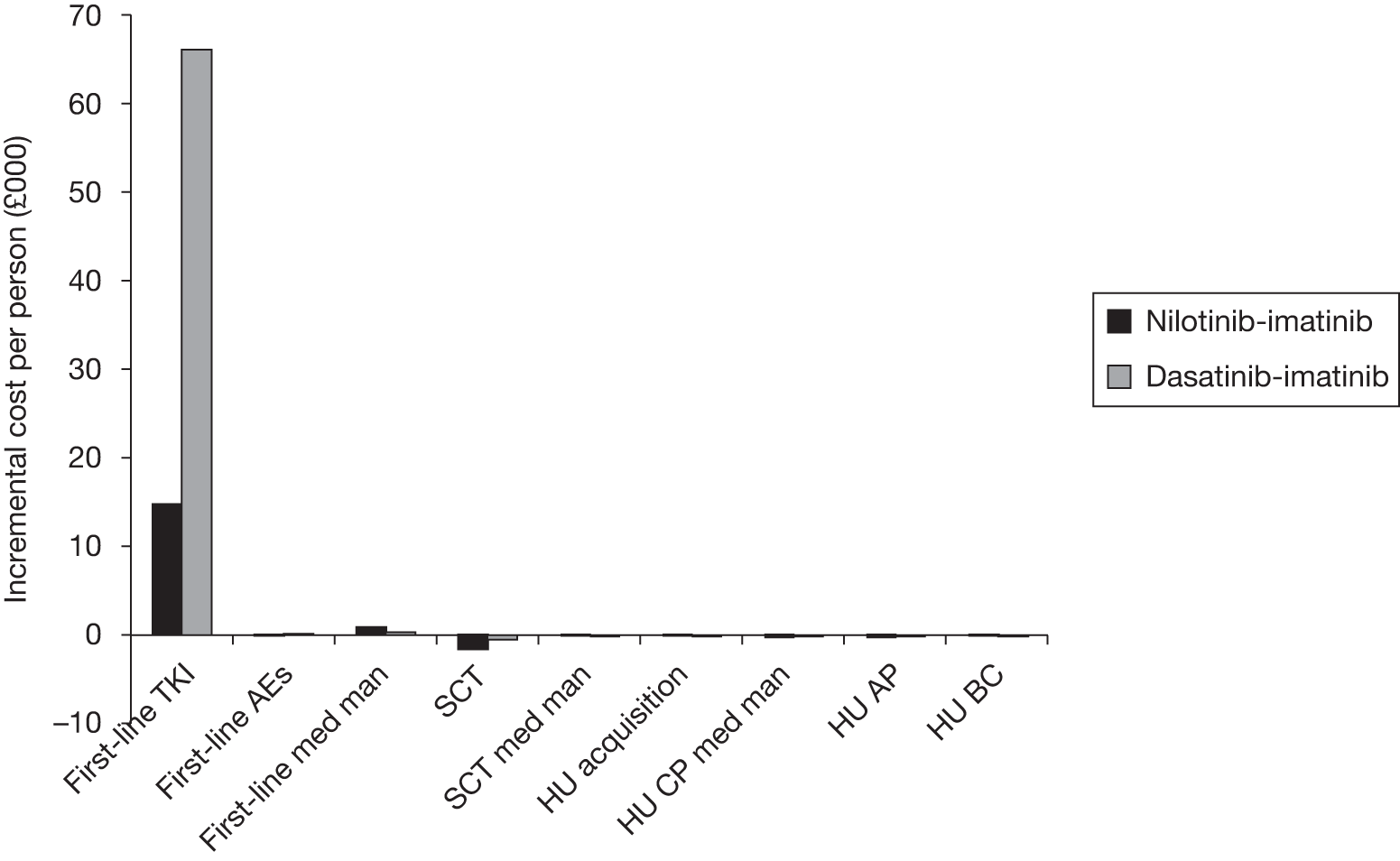
Combining all the information on expected costs and QALYs per person, we estimate the following cost-effectiveness results (see Table 51 and Figure 37):
-
nilotinib compared with imatinib ICER of £20,000 per QALY
-
dasatinib compared with imatinib ICER of £256,000 per QALY
-
nilotinib dominates dasatinib (dasatinib costs more and confers fewer benefits).
As in Figure 35, the dotted line in Figure 37 represents a willingness to pay of £30,000 per QALY compared with treatment with imatinib.
FIGURE 37.
Cost-effectiveness results for scenario 2 (wide axes top and narrow axes bottom). Straight lines represent £30,000 per QALY threshold anchored on comparators involving first-line imatinib (cheapest in each scenario).
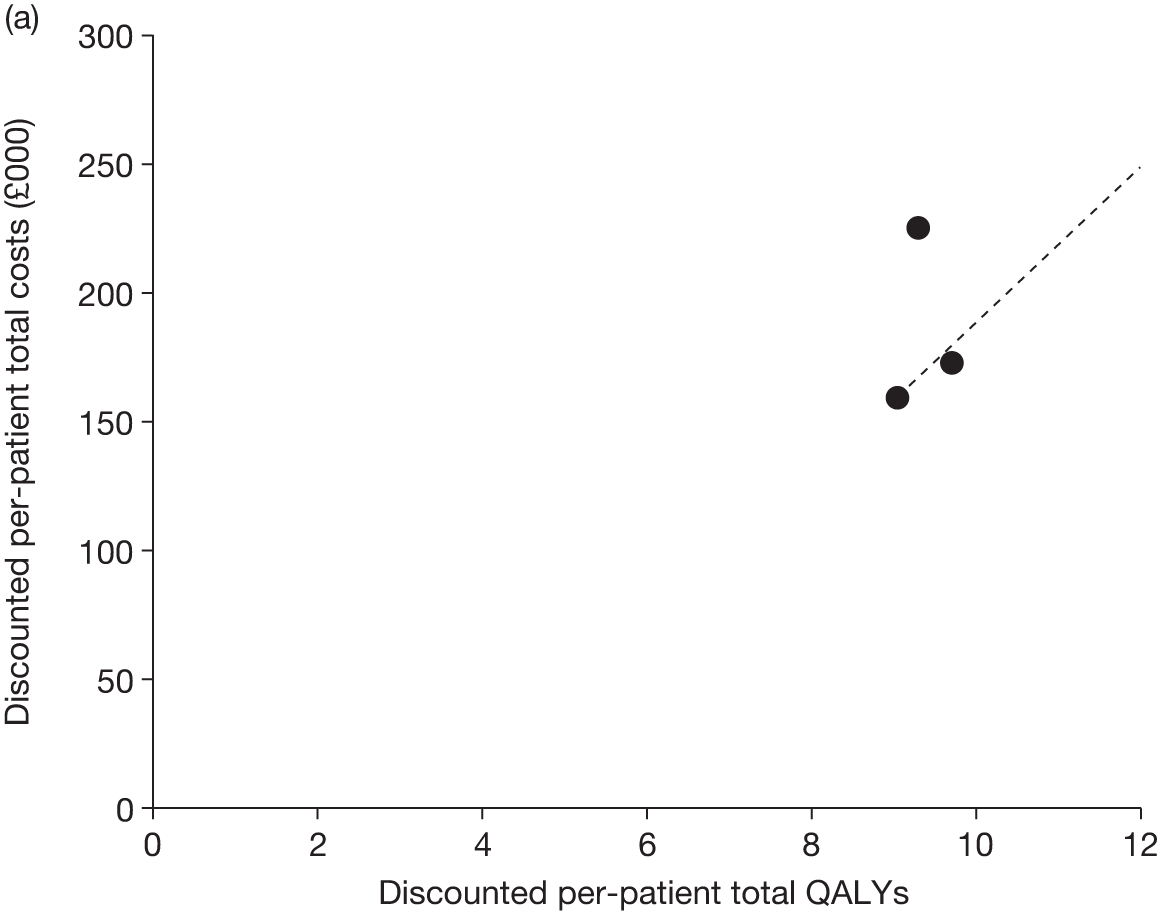
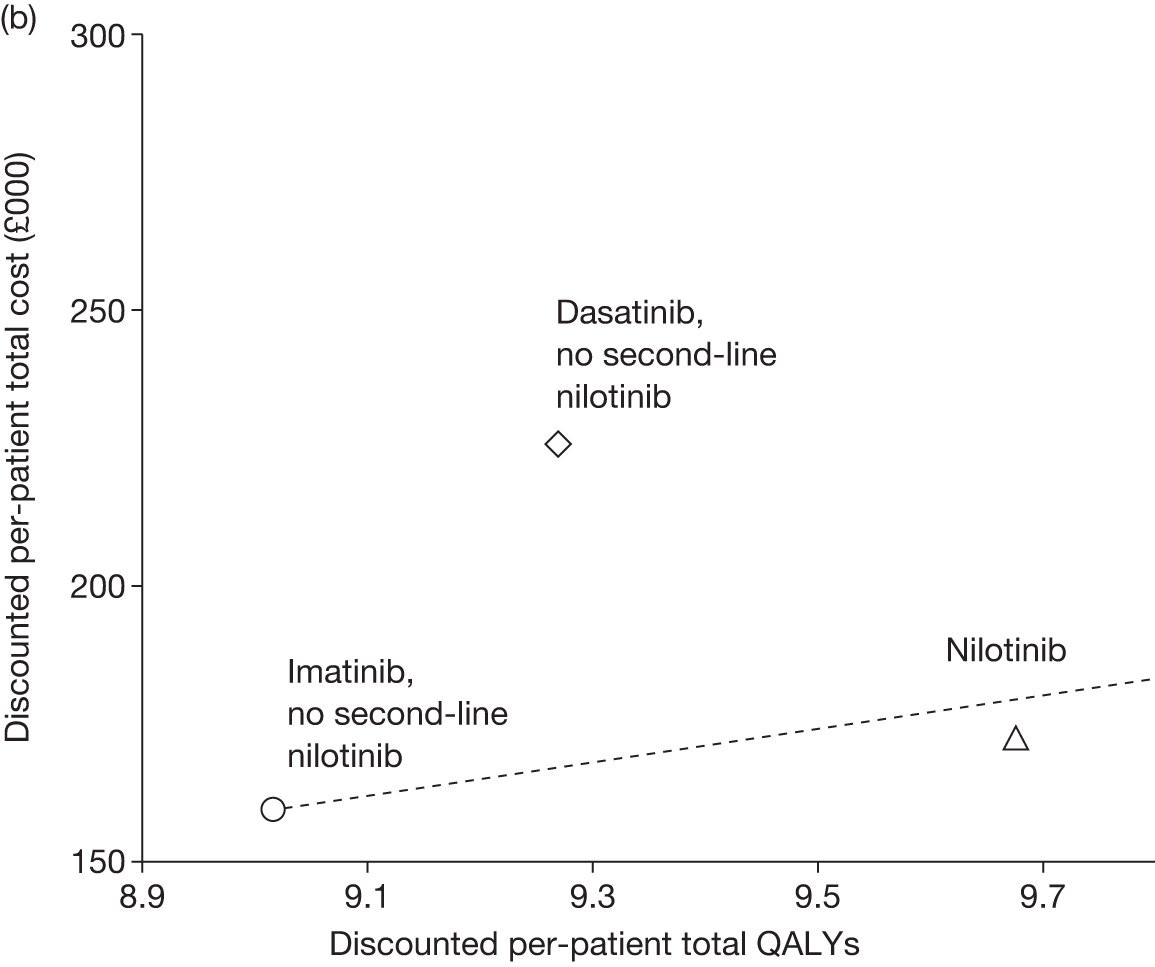
The ICER for nilotinib compared with imatinib falls from £25,000 per QALY under scenario 1 to £20,000 per QALY under the simplified method. This is a result of increasing the importance of the costs and QALYs associated with TKI treatment relative to the costs and QALYs of treatments after TKI failure.
Results: scenario 3 – cumulative survival method with second-line nilotinib
Table 52 presents the cost-effectiveness results for scenario 3.
| Treatment | Imatinib | Nilotinib | Dasatinib | Nilotinib–imatinib | Dasatinib–imatinib | Nilotinib–dasatinib |
|---|---|---|---|---|---|---|
| Life-years (undiscounted) | ||||||
| First-line TKI | 7.0 | 8.9 | 7.7 | 1.9 | 0.7 | 1.2 |
| Second-line nilotinib | 2.2 | – | 2.2 | –2.2 | –0.1 | –2.2 |
| SCT | 4.2 | 4.9 | 3.9 | 0.7 | –0.3 | 1.0 |
| Hydroxycarbamide CP | 3.0 | 2.8 | 3.0 | –0.3 | –0.0 | –0.2 |
| Hydroxycarbamide AP | 0.5 | 0.4 | 0.5 | –0.0 | –0.0 | –0.0 |
| Hydroxycarbamide BC | 0.3 | 0.3 | 0.3 | –0.0 | –0.0 | –0.0 |
| OS (mean) | 17.3 | 17.4 | 17.6 | 0.1 | 0.3 | –0.2 |
| OS (median) | 16.0 | 16.2 | 16.5 | 0.2 | 0.5 | –0.3 |
| Mean age start | ||||||
| First-line TKI | 57 | 57 | 57 | 0.0 | 0.0 | 0.0 |
| Second-line nilotinib | 64 | 66 | 65 | 1.9 | 0.7 | 1.2 |
| SCT | 66 | 66 | 67 | –0.3 | 0.6 | –0.9 |
| Hydroxycarbamide CP | 66 | 66 | 67 | –0.3 | 0.6 | –0.9 |
| Hydroxycarbamide AP | 71 | 71 | 72 | –0.3 | 0.6 | –0.9 |
| Hydroxycarbamide BC | 72 | 72 | 73 | –0.3 | 0.6 | –0.9 |
| Cohort splita | ||||||
| % starting second-line nilotinib | 90 | – | 88 | – | –2 | – |
| % starting SCT/hydroxycarbamide | 86 | 84 | 84 | –2 | –2 | 0 |
| % SCT (whole cohort) | 26 | 28 | 24 | 3 | –2 | 4 |
| % SCT (eligible cohort) | 30 | 34 | 29 | 4 | –1 | 5 |
| % hydroxycarbamide (whole cohort) | 61 | 56 | 60 | –5 | –1 | –5 |
| % hydroxycarbamide (eligible cohort) | 70 | 66 | 71 | –4 | 1 | –5 |
| % AP (whole cohort) | 52 | 48 | 51 | –4 | –1 | –3 |
| % BC (whole cohort) | 52 | 48 | 51 | –4 | –1 | –3 |
| Life-years (undisclosed eligible cohort)a | ||||||
| First-line TKI | 7.0 | 8.9 | 7.7 | 1.9 | 0.7 | 1.2 |
| Second-line nilotinib | 2.5 | – | 2.5 | –2.5 | –0.0 | –2.5 |
| SCT | 16.4 | 17.2 | 16.3 | 0.8 | –0.1 | 0.9 |
| Hydroxycarbamide CP | 5.0 | 5.0 | 5.0 | 0.0 | –0.0 | 0.0 |
| Hydroxycarbamide AP | 0.9 | 0.9 | 0.9 | –0.0 | –0.0 | –0.0 |
| Hydroxycarbamide BC | 0.6 | 0.6 | 0.6 | –0.0 | –0.0 | –0.0 |
| QALYs (discounted) | ||||||
| First-line TKI | 4.5 | 5.5 | 4.9 | 1.0 | 0.4 | 0.6 |
| Second-line nilotinib | 1.4 | – | 1.3 | –1.4 | –0.1 | –1.3 |
| SCT | 1.8 | 2.2 | 1.7 | 0.4 | –0.1 | 0.5 |
| Hydroxycarbamide CP | 1.5 | 1.4 | 1.5 | –0.1 | –0.0 | –0.0 |
| Hydroxycarbamide AP | 0.2 | 0.2 | 0.2 | –0.0 | –0.0 | –0.0 |
| Hydroxycarbamide BC | 0.1 | 0.1 | 0.1 | –0.0 | –0.0 | –0.0 |
| Total | 9.5 | 9.4 | 9.7 | –0.1 | 0.1 | –0.2 |
| Costs (discounted), £ | ||||||
| First-line TKI | 118,635 | 133,386 | 184,774 | 14,751 | 66,139 | –51,388 |
| First-line AEs | 166 | 119 | 282 | –47 | 116 | –163 |
| First-line medical management | 3811 | 4658 | 4127 | 846 | 316 | 530 |
| Second-line nilotinib | 35,393 | 0 | 34,096 | –35,393 | –1297 | –34,096 |
| Second-line nilotinib AEs | 299 | 0 | 299 | –299 | 0 | –299 |
| Second-line nilotinib medical management | 1148 | 0 | 1106 | –1148 | –42 | –1106 |
| SCT transplant | 17,724 | 20,646 | 16,601 | 2921 | –1123 | 4044 |
| SCT medical management | 1784 | 2148 | 1667 | 364 | –116 | 480 |
| Hydroxycarbamide acquisition in CP | 280 | 264 | 272 | –16 | –8 | –8 |
| Hydroxycarbamide CP medical management | 2471 | 2330 | 2402 | –141 | –69 | –72 |
| Hydroxycarbamide AP acquisition + medical management | 4060 | 3828 | 3947 | –232 | –113 | –119 |
| Hydroxycarbamide BC acquisition + medical management | 2710 | 2555 | 2634 | –155 | –75 | –79 |
| Total | 188,480 | 169,932 | 252,208 | –18,548 | 63,728 | –82,276 |
| Cost/LYG | –216,000 | 201,000 | 356,000 | |||
| Cost/QALY | 192,000b | 450,000 | 345,000b | |||
Scenario 3: survival results
The relative proportions of patients in each health state for each treatment over time are displayed in Figure 38. Virtually all patients are predicted to have died by the age of 97 years, 20 years from start of first-line treatment.
FIGURE 38.
Scenario 3 cohort composition over time by treatment arm.
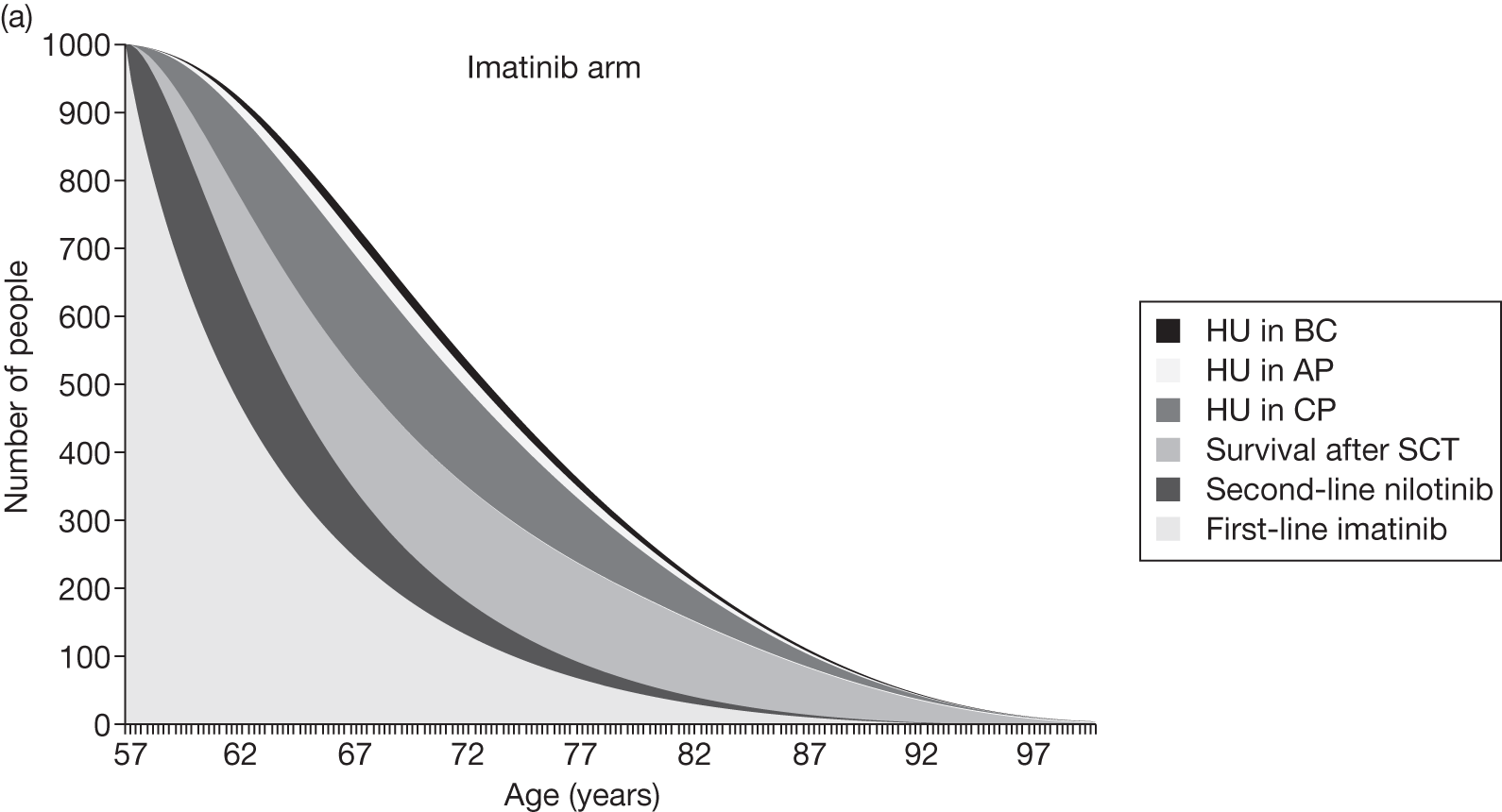
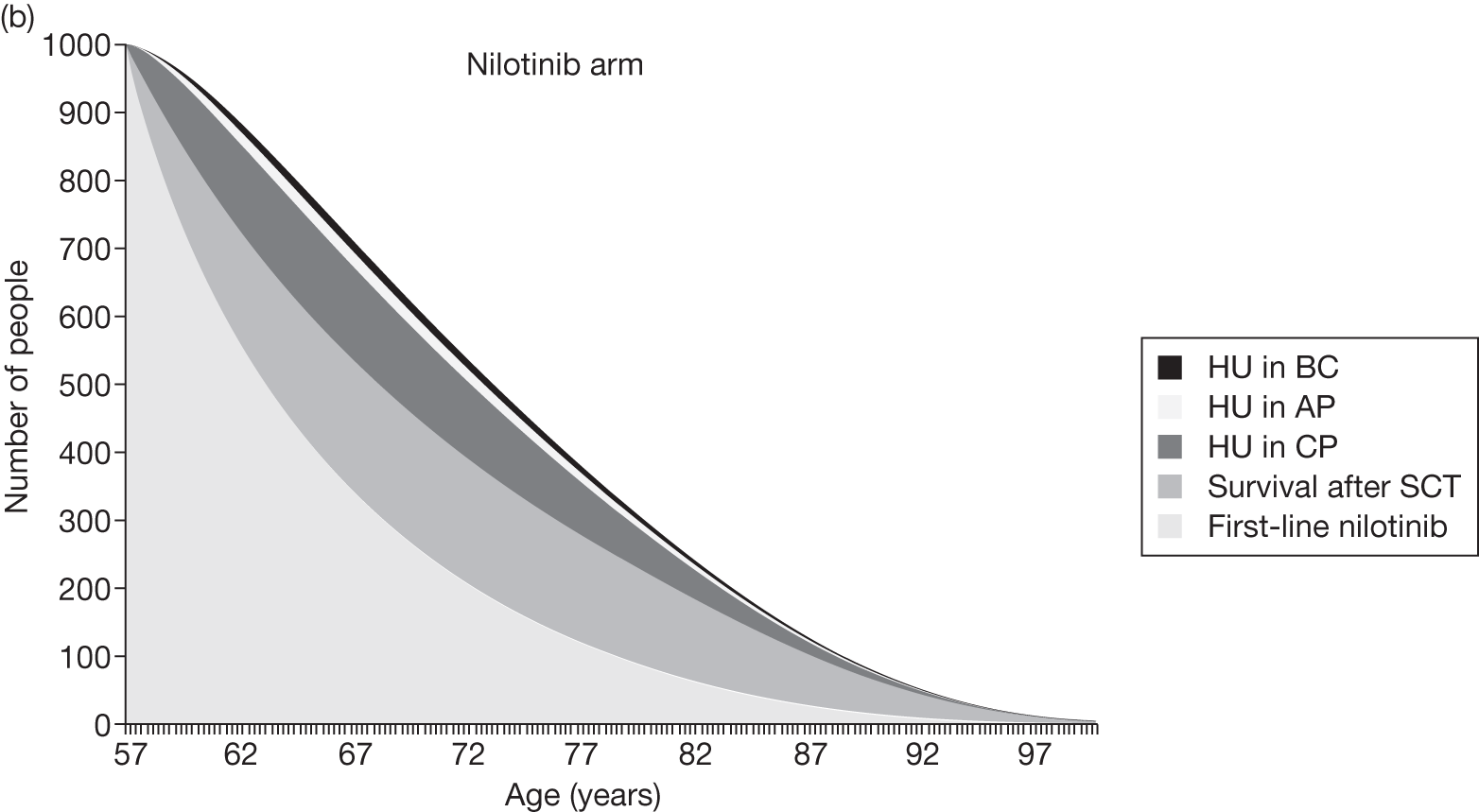
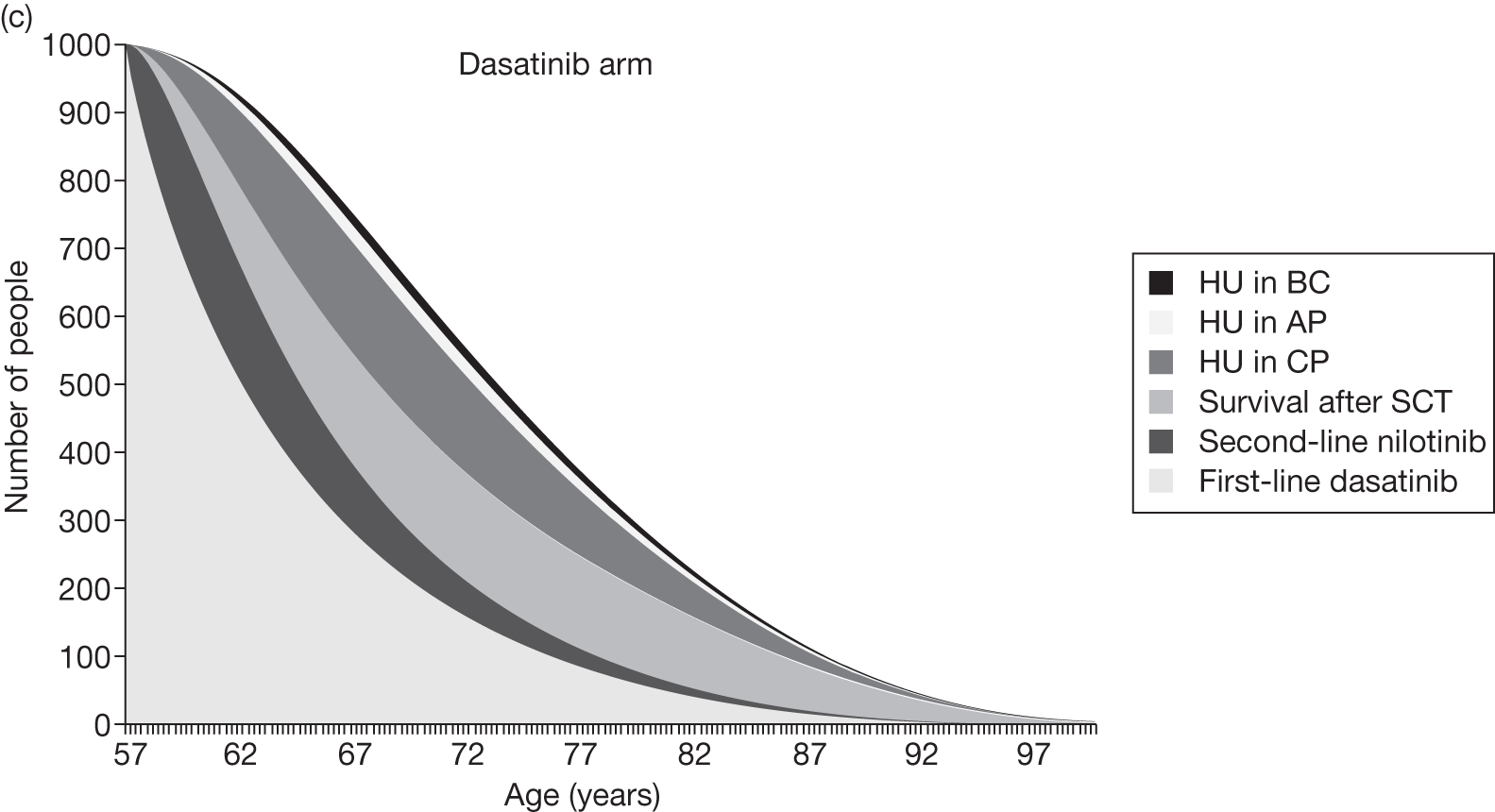
By design, the mean durations of first-line TKI treatment are the same in this scenario as in scenarios 1 and 2.
In the imatinib and dasatinib treatment arms, for those patients who take second-line nilotinib, we predict a mean time on second-line nilotinib of 2.5 years, which reflects the findings from the single-arm trial of second-line nilotinib. 115 Clearly, there is no second-line nilotinib in the nilotinib arm.
The proportion of patients who have SCT in both the imatinib and dasatinib arms is lower than in scenario 1 because people are typically older when they reach this treatment option, because of the extra line of treatment with nilotinib, and because the proportion receiving SCT declines with age. For example, in the imatinib arm, 26% of patients are predicted to receive SCT after second-line nilotinib, compared with 33% in scenario 1 (no second-line nilotinib). This explains why the expected survival time after SCT, averaged over the whole cohort, is lower in this scenario than in scenario 1. For example, for the imatinib arm, the mean survival is 4.2 years in scenario 3 compared with 5.8 years in scenario 1.
The proportion of patients who receive hydroxycarbamide in CP in the imatinib arm, at 61%, is higher than in scenario 1, at 56%. This is also because the second-line nilotinib delays the time when patients are eligible for SCT or hydroxycarbamide, and because the proportion receiving SCT declines with age, and therefore the proportion receiving hydroxycarbamide increases with age. Similarly, in the dasatinib arm, the corresponding proportions receiving hydroxycarbamide are 60% and 56%.
Scenario 3: cost results
The incremental per-patient drug costs are given in Figure 39. First, by design, the incremental costs of acquisition of first-line TKIs, of treatment for first-line AEs and of medical management while on first-line TKI are exactly as in scenario 1 (see Figure 34). Next, there are substantial cost savings, approximately £35,000 per patient, in the nilotinib arm by having no cost for second-line nilotinib acquisition, and having no cost of medical management while on second-line nilotinib (approximately £1100 per patient). Notice that the mean acquisition cost of second-line nilotinib, at approximately £35,000 per patient, is substantially lower than the mean acquisition cost of first-line nilotinib, at approximately £133,000 per patient. This is because we assume that nilotinib is taken for far less time as a second-line treatment (typically 2.5 years) than as a first-line treatment (typically 8.9 years).
FIGURE 39.
Incremental costs vs imatinib treatment arm. Med man, medical management.
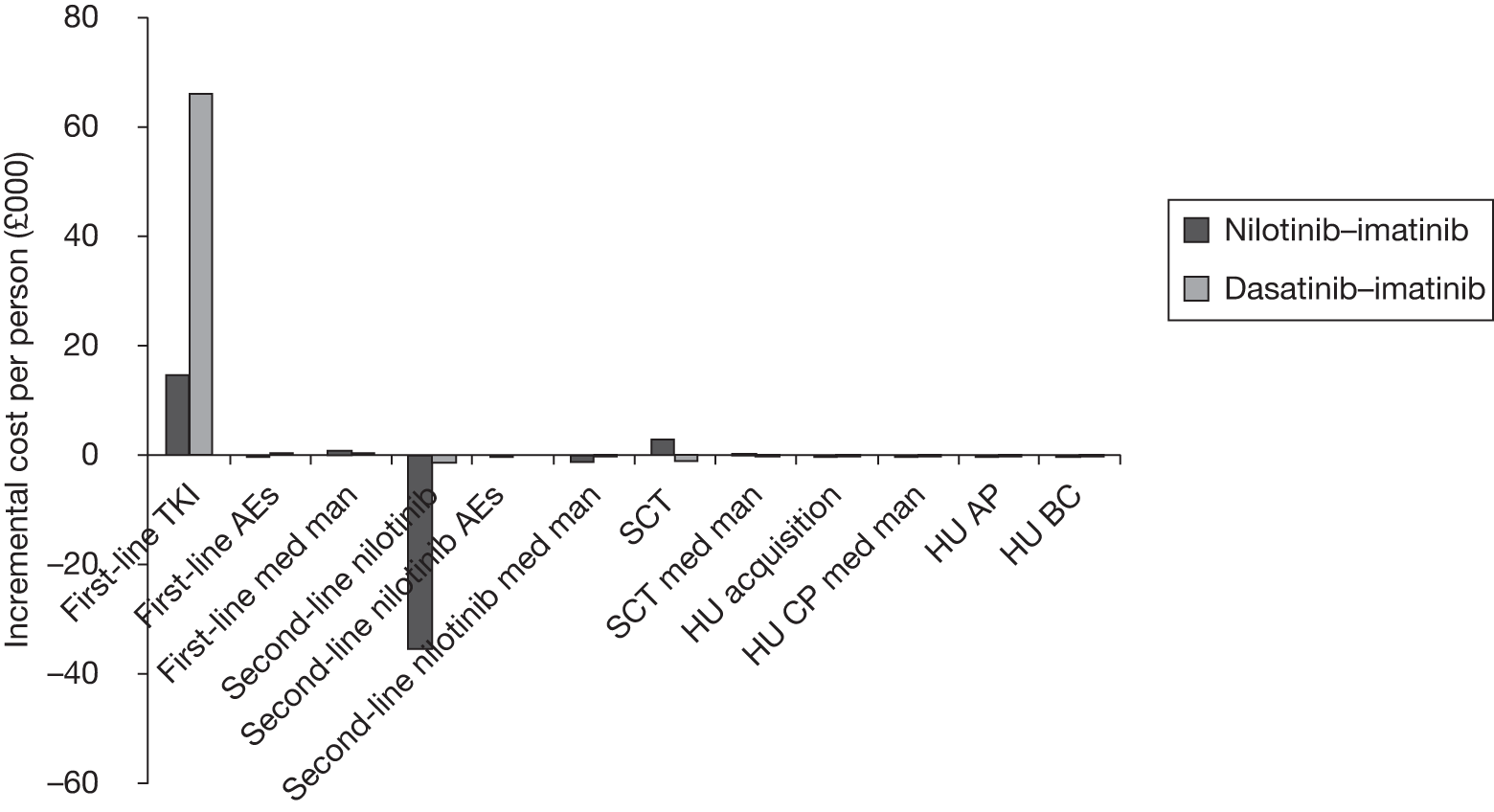
The incremental per-patient cost of SCT for nilotinib compared with imatinib is higher in scenario 3 than in scenario 1. This is because, as explained in the previous section, the proportion of patients who receive SCT falls when we allow for second-line nilotinib, because people are typically older when they receive SCT, and because nilotinib is taken second-line only in the imatinib and dasatinib arms. All other incremental costs are similar to those in scenario 1.
Scenario 3: cost-effectiveness results
Combining all of the information on expected costs and QALYs per person, we estimate the following cost-effectiveness results (see Table 52 and Figure 35):
-
Nilotinib compared with imatinib ICER of £192,000 per QALY, whereby the nilotinib arm provides slightly fewer QALYs (–0.1) at far less cost (–£19,000) than the imatinib arm. This implies that the nilotinib arm provides far better value for money than the imatinib arm at willingness-to-pay thresholds of £20,000 and £30,000 per QALY.
-
Dasatinib compared with imatinib ICER of £450,000 per QALY, whereby the dasatinib arm provides slightly more QALYs (0.1) at far more cost (£64,000) than the imatinib arm. This implies that the dasatinib arm provides far worse value for money than the imatinib arm at willingness-to-pay thresholds of £20,000 and £30,000 per QALY.
-
Nilotinib compared with dasatinib ICER of £345,000 per QALY, whereby the nilotinib arm provides slightly fewer QALYs (–0.2) at far less cost (–£8000) than the dasatinib arm. This implies that the nilotinib arm provides far better value for money than the imatinib arm at willingness-to-pay thresholds of £20,000 and £30,000 per QALY.
These results are displayed graphically in Figure 35, along with the results from scenario 1.
Results: scenario 4 – cumulative survival, simplified method, with second-line nilotinib
To reiterate, in the simplified method the post-TKI (first-line TKIs and second-line nilotinib) per-patient costs and QALYs are set equal across treatment arms. The costs and QALYs while patients are on TKIs are modelled specific to each treatment arm, i.e. exactly as in the previous scenario 3. Table 53 presents the cost-effectiveness results for scenario 4.
| Treatment | Imatinib | Nilotinib | Dasatinib | Nilotinib–imatinib | Dasatinib–imatinib | Nilotinib–dasatinib |
|---|---|---|---|---|---|---|
| QALYs (discounted) | ||||||
| First-line TKI | 4.5 | 5.5 | 4.9 | 1.0 | 0.4 | 0.6 |
| Second-line nilotinib | 1.4 | – | 1.3 | –1.4 | –0.1 | –1.3 |
| SCT | 1.8 | 1.8 | 1.8 | –0.0 | –0.0 | –0.0 |
| Hydroxycarbamide CP | 1.5 | 1.5 | 1.5 | –0.0 | –0.0 | 0.0 |
| Hydroxycarbamide AP | 0.2 | 0.2 | 0.2 | –0.0 | –0.0 | –0.0 |
| Hydroxycarbamide BC | 0.1 | 0.1 | 0.1 | –0.0 | –0.0 | –0.0 |
| Total | 9.5 | 9.1 | 9.7 | –0.5 | 0.2 | –0.7 |
| Costs (discounted) | ||||||
| First-line TKI | 118,635 | 133,386 | 184,774 | 14,751 | 66,139 | –51,388 |
| First-line AEs | 166 | 119 | 282 | –47 | 116 | –163 |
| First-line medical management | 3811 | 4,658 | 4127 | 846 | 316 | 530 |
| Second-line nilotinib | 35,393 | 0 | 34,096 | –35,393 | –1297 | –34,096 |
| Second-line nilotinib AEs | 299 | 0 | 299 | –299 | 0 | –299 |
| Second-line nilotinib medical management | 1148 | 0 | 1106 | –1148 | –42 | –1106 |
| SCT transplant | 17,724 | 17,267 | 17,293 | –458 | –432 | –26 |
| SCT medical management | 1784 | 1738 | 1740 | –46 | –43 | –3 |
| Hydroxycarbamide acquisition in CP | 280 | 273 | 271 | –7 | –9 | 2 |
| Hydroxycarbamide CP medical management | 2471 | 2408 | 2393 | –62 | –78 | 16 |
| Hydroxycarbamide AP acquisition + medical management | 4060 | 3955 | 3961 | –105 | –99 | –6 |
| Hydroxycarbamide BC acquisition + medical management | 2710 | 2640 | 2643 | –70 | –66 | –4 |
| Total | 188,480 | 166,443 | 252,985 | –22,037 | 64,505 | –86,542 |
| Cost/QALY | 46,000a | 301,000 | 125,000a | |||
As in scenario 2, the proportions still alive and starting second- or third-line treatment on hydroxycarbamide or undergoing SCT are similar across the three treatment arms (84% for nilotinib and dasatinib and 86% for imatinib), as the durations of first- and second-line TKI treatments are similar across treatment arms. Therefore, as in scenario 2, this method largely nets off all post-TKI costs and QALYs between treatment arms. For example, the incremental QALYs associated with time after SCT is 0.0 for nilotinib–imatinib, compared with the corresponding figure of 0.4 from scenario 3, and the incremental per-person cost of SCT operations is –£460 for nilotinib–imatinib (Figure 40) compared with £2900 in scenario 3 (see Table 52).
FIGURE 40.
Scenario 4 incremental costs vs imatinib treatment arm. Med man, medical management.
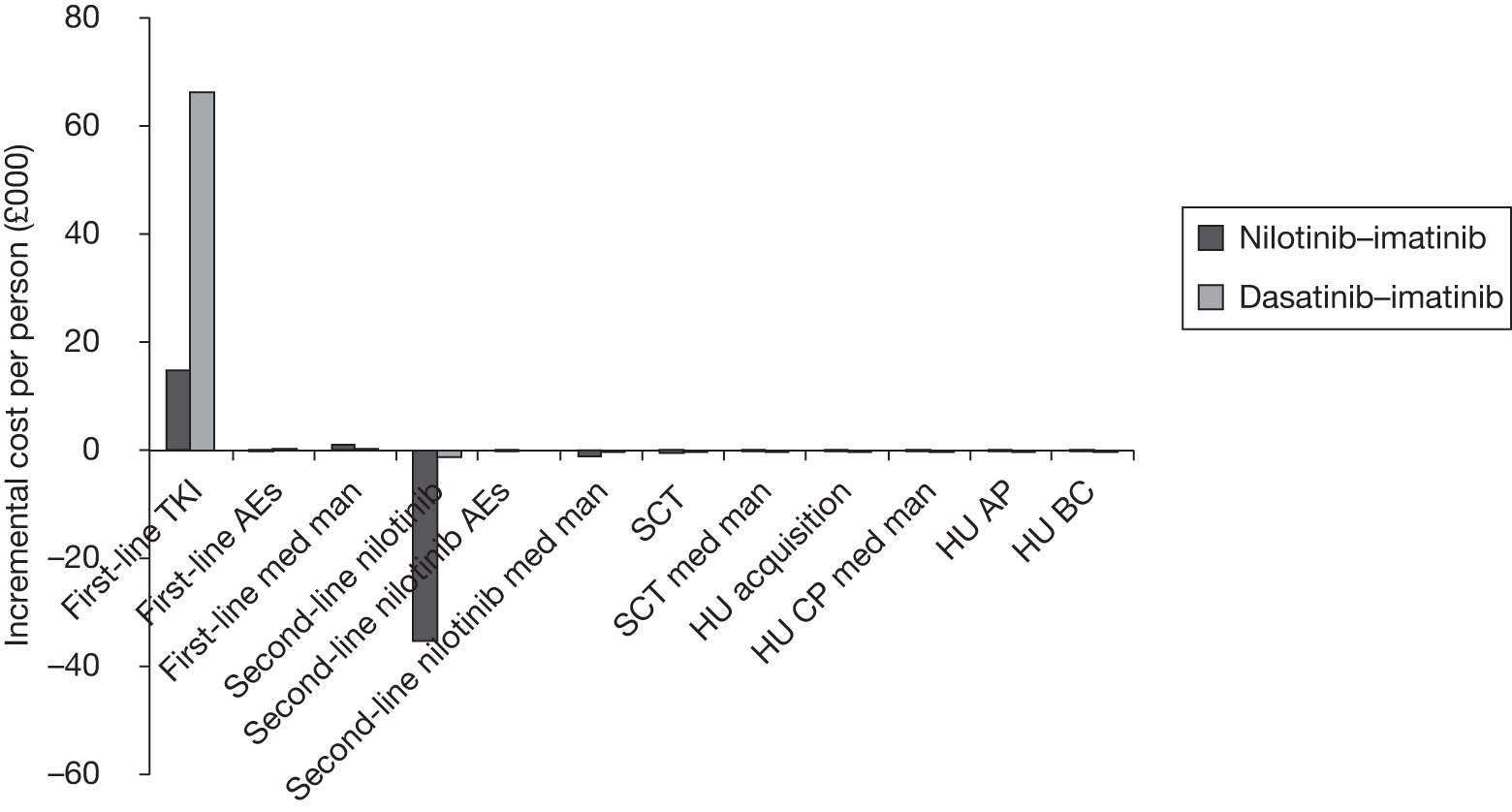
Combining all the information on expected costs and QALYs per person, we estimate the following cost-effectiveness results (see Table 51 and Figure 41):
-
Nilotinib compared with imatinib ICER of £46,000 per QALY, whereby the nilotinib arm provides fewer QALYs (–0.5) at less cost (–£22,000) than the imatinib arm. As in scenario 3, this implies that the nilotinib arm provides better value for money than the imatinib arm at willingness-to-pay thresholds of £20,000 and £30,000 per QALY.
-
Dasatinib compared with imatinib ICER of £301,000 per QALY, whereby the dasatinib arm provides slightly more QALYs (0.2) at far more cost (£65,000) than the imatinib arm. As in scenario 3, this implies that the dasatinib arm provides far worse value for money than the imatinib arm at willingness-to-pay thresholds of £20,000 and £30,000 per QALY.
-
Nilotinib compared with dasatinib ICER of £125,000 per QALY, whereby the nilotinib arm provides fewer QALYs (–0.7) at far less cost (–£87,000) than the dasatinib arm. As in scenario 3, this implies that the nilotinib arm provides far better value for money than the imatinib arm at willingness-to-pay thresholds of £20,000 and £30,000 per QALY.
The dotted line in Figure 41 represents a willingness-to-pay of £30,000 per QALY compared with treatment with imatinib followed by second-line nilotinib.
FIGURE 41.
Cost-effectiveness results for scenario 4 (wide axes top and narrow axes bottom). Straight lines represent £30,000 per QALY threshold anchored on comparators involving first-line imatinib (cheapest in each scenario).
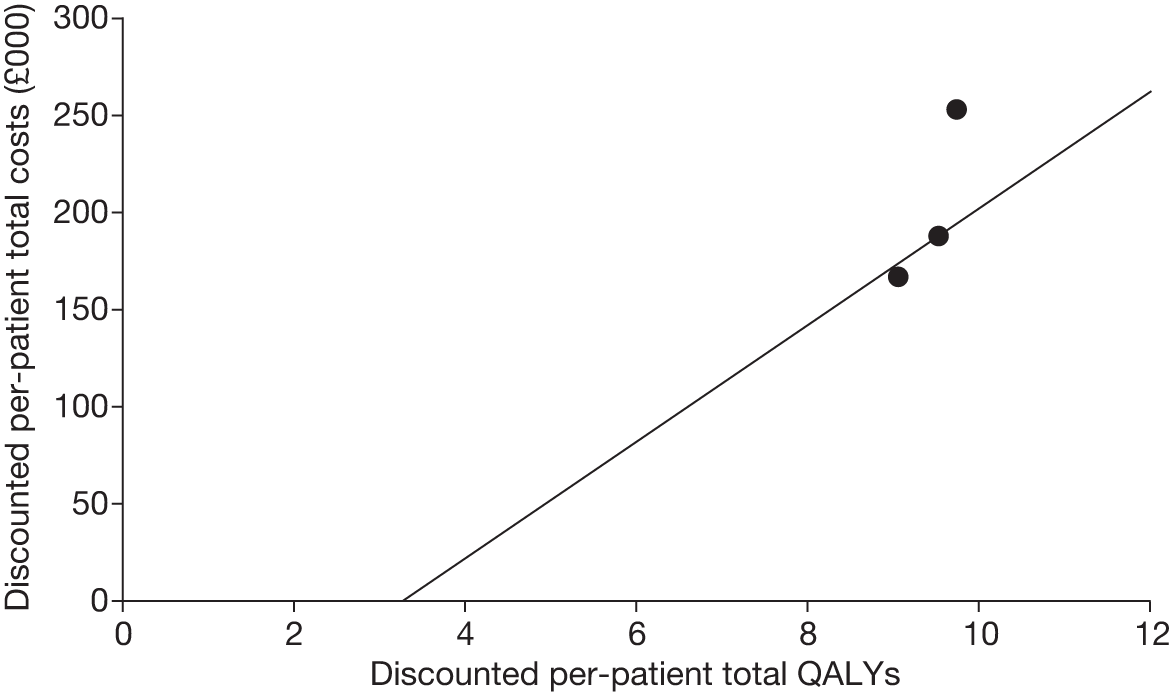
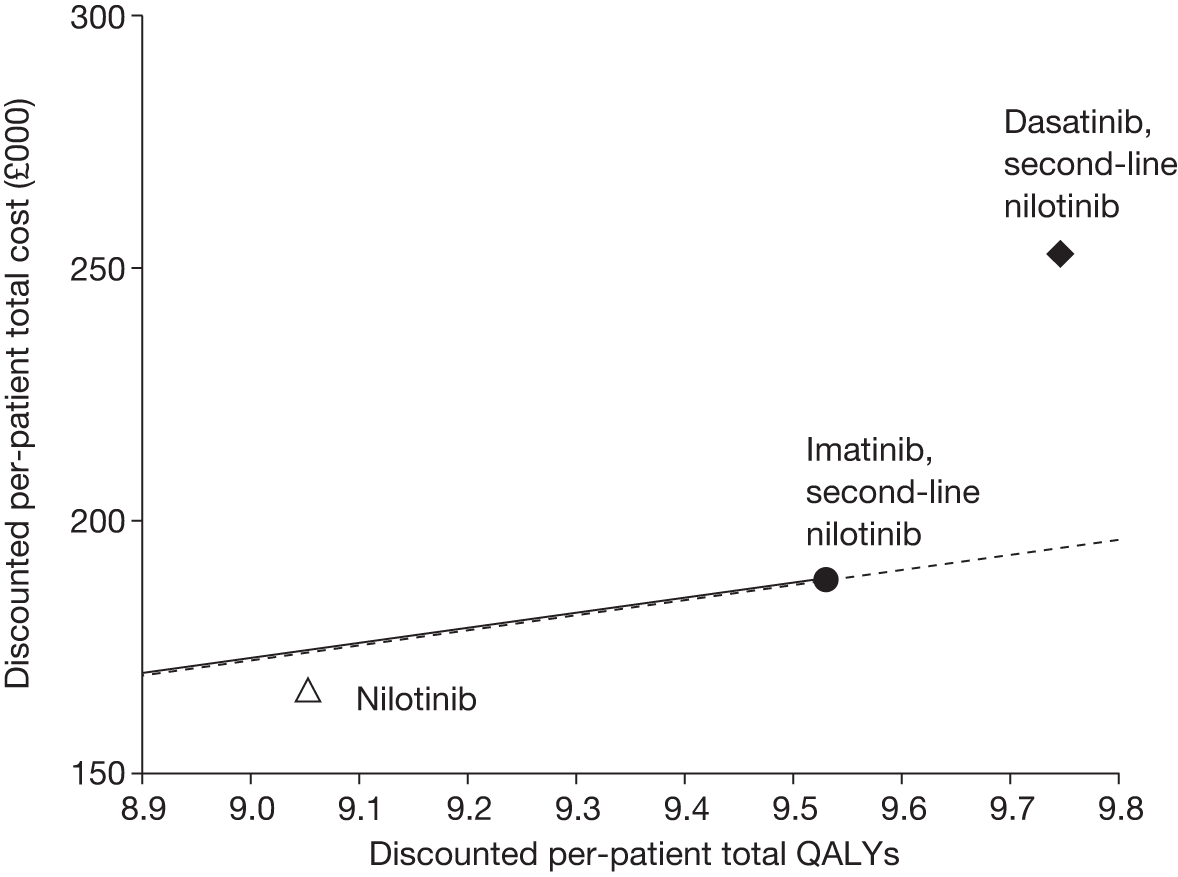
Deterministic sensitivity analyses
We now present the deterministic sensitivity analyses. Where relevant, the analyses are performed for each of the four modelling scenarios.
Sensitivity analyses for nilotinib compared with imatinib are reported in Table 54 and for dasatinib compared with imatinib in Table 55.
| Parameter | Base case | Sensitivity analysis | Scenario 1 (no second-line nilotinib) | Scenario 2 (no second-line nilotinib, simplified method) | Scenario 3 (second-line nilotinib) | Scenario 4 (second-line nilotinib, simplified method) |
|---|---|---|---|---|---|---|
| Base case | NA | NA | £25,000 | £20,000 | £192,000 a | £46,000 a |
| General | ||||||
| Discounting costs and benefits | 3.5% p.a. | 0% p.a. | £30,000 | £24,000 | Nilotinib dominates | £51,000a |
| Treatment pathways | ||||||
| Proportion receiving SCT | Mean 28% nilotinib, 33% imatinib, decreases with age | 31% at all ages (BMS assumption) | £24,000 | £20,000 | £86,000a | £48,000a |
| 75% if age < 65 years (Novartis) | £28,000 | £20,000 | £286,000a | £45,000a | ||
| Halve % at all ages | £23,000 | £20,000 | £98,000a | £48,000a | ||
| Effectiveness | ||||||
| Time on first-line TKI | 8.9 years nilotinib, 7.0 years imatinib | 7.0 years nilotinib, 7.0 years imatinib | Nilotinib dominates | Nilotinib dominates | £75,000a | £38,000a |
| 13.8 years nilotinib, 11.7 years imatinib (IRIS) | £14,000 | £13,000 | Nilotinib dominates | £79,000a | ||
| Time on second-line nilotinib | Mean 2.5 years | Same as mean time on first-line nilotinib = 8.9 years | NA | NA | £61,000a | £37,000a |
| Survival after SCT | Mean approximately 17 years | Mean 5.7 years (Novartis) | £16,000 | 17,000 | £54,000a | £49,000a |
| Time in CP on hydroxycarbamide | Mean 5 years | Mean 1.6 years (Novartis) | £22,000 | £18,000 | £341,000a | £49,000a |
| OS estimated by cumulative survival or surrogate survival | Cumulative survival | Cumulative survival means, MMR survival difference | £35,000 | £25,000 | NA | NA |
| Cumulative survival means, CCyR survival difference | £17,000 | £15,000 | NA | NA | ||
| Surrogate survival means, MMR survival difference | £40,000 | £29,000 | NA | NA | ||
| Surrogate survival means, CCyR survival difference | £19,000 | £17,000 | NA | NA | ||
| Costs | ||||||
| Drug price reduction on patent expiry | 0% nilotinib, 0% imatinib | 0% nilotinib, 25% imatinib | £60,000 | £42,000 | £42,000a | £16,000a |
| 25% nilotinib, 25% imatinib | £44,000 | £31,000 | £95,000a | £27,000a | ||
| Dose intensities |
(CiC information has been removed) first-line nilotinib (CiC information has been removed) imatinib 99% second-line nilotinib |
100% first-line nilotinib (CiC information has been removed) imatinib 99% second-line nilotinib |
£53,000 | £37,000 | £72,000a | £22,000a |
|
(CiC information has been removed) first-line nilotinib (CiC information has been removed) imatinib (Novartis) 99% second-line nilotinib |
£8000 | £9000 | £265,000a | £61,000a | ||
|
(CiC information has been removed) first-line nilotinib (CiC information has been removed) imatinib (CiC information has been removed) second-line nilotinib |
NA | NA | £166,000a | £41,000a | ||
| Cost SCT | £81,603 | £40,801 | £30,000 | £21,000 | £207,000a | £46,000a |
| £163,205 | £16,000 | £17,000 | £162,000a | £47,000a | ||
| Medical management costs after SCT | £113 per month | £57 per month | £26,000 | £20,000 | £194,000a | £46,000a |
| Medical management costs in CP | £56 per month TKIs, 106 per month hydroxycarbamide | £28 per month TKIs, 53 per month hydroxycarbamide | £25,000 | £19,000 | £189,000a | £46,000a |
| £112 per month TKIs, £211 per month hydroxycarbamide | £27,000 | £21,000 | £196,000a | £47,000a | ||
| Medical management costs in AP and BC | £1113 per month | £2227 per month | £24,000 | £19,000 | £196,000a | £47,000a |
| AEs costs | £166 per patient imatinib, 119 per patient nilotinib | £1660 per patient imatinib, £1190 per patient nilotinib | £24,000 | £19,000 | £196,000a | £47,000a |
| Utilities | ||||||
| Utilities | Equal to Novartis | £25,000 | £20,000 | £201,000a | £46,000a | |
| Reduce all utilities by 0.10 | £22,000 | £19,000 | £130,000a | £47,000a | ||
| Parameter | Base case | Sensitivity analysis | Scenario 1 (no second-line nilotinib) | Scenario 2 (no second-line nilotinib, simplified method) | Scenario 3 (second-line nilotinib) | Scenario 4 (second-line nilotinib, simplified method) |
|---|---|---|---|---|---|---|
| Base case | NA | NA | 414,000 | 256,000 | 450,000 | 301,000 |
| General | ||||||
| Discounting costs and benefits | 3.5% p.a. | 0% p.a. | £335,000 | £229,000 | £338,000 | £253,000 |
| Treatment pathways | ||||||
| Proportion receiving SCT | Mean 32% dasatinib, 33% imatinib, decreases with age | 31% at all ages (BMS assumption) | £338,000 | £247,000 | £397,000 | £294,000 |
| 75% if age < 65 years (Novartis) | £537,000 | £265,000 | £584,000 | £312,000 | ||
| Halve % at all ages | £331,000 | £246,000 | £378,000 | £290,000 | ||
| Effectiveness | ||||||
| Time on first-line TKI | 7.0 years dasatinib, 7.0 years imatinib | 7.0 years dasatinib, 7.0 years imatinib | Imatinib dominates | Imatinib dominates | Imatinib dominates | Imatinib dominates |
| 12.5 years dasatinib, 11.7 years imatinib (IRIS) | £565,000 | £427,000 | £641,000 | £508,000 | ||
| Time on second-line nilotinib | Mean 2.5 years | Same as mean time on first-line nilotinib = 8.9 years | NA | NA | £673,000 | £501,000 |
| Survival after SCT | Mean approximately 17 years | Mean 5.7 years (Novartis) | £246,000 | £224,000 | £292,000 | £266,000 |
| Time in CP on hydroxycarbamide | Mean 5 years | Mean 1.6 years (Novartis) | £356,000 | £229,000 | £373,000 | £263,000 |
| OS estimated by cumulative survival or surrogate survival | Cumulative survival | Cumulative survival means, MMR survival difference | £250,000 | £171,000 | NA | NA |
| Cumulative survival means, CCyR survival difference | £104,000 | £77,000 | NA | NA | ||
| Surrogate survival means, MMR survival difference | £303,000 | 196,000 | NA | NA | ||
| Surrogate survival means, CCyR survival difference | £124,000 | £86,000 | NA | NA | ||
| Costs | ||||||
| Drug price reduction on patent expiry | 0% dasatinib, 0% imatinib | 25% dasatinib, 25% imatinib | £425,000 | £262,000 | £462,000 | £308,000 |
| Dose intensities | (CiC information has been removed) imatinib, 99% dasatinib, 99% second-line nilotinib | (CiC information has been removed) imatinib (Novartis), 99% dasatinib, 99% second-line nilotinib | £369,000 | £228,000 | £400,000 | £268,000 |
| (CiC information has been removed) imatinib, 99% dasatinib, (CiC information has been removed) second-line nilotinib | NA | NA | £451,000 | £301,000 | ||
| Cost SCT | £81,603 | £40,801 | £419,000 | £257,000 | £454,000 | £302,000 |
| £163,205 | £405,000 | £254,000 | £442,000 | £299,000 | ||
| Medical management costs after SCT | £113 per month | £57 per month | £415,000 | £256,000 | £451,000 | £301,000 |
| Medical management costs in CP | £56 per month TKIs, £106 per month hydroxycarbamide | £28 per month TKIs, £53 per month hydroxycarbamide | £414,000 | £255,000 | £449,000 | £300,000 |
| 112 per month TKIs, 211 per month hydroxycarbamide | £416,000 | £257,000 | £452,000 | £302,000 | ||
| Medical management costs in AP and BC | £1113 per month | £2227 per month | £414,000 | £255,000 | £449,000 | £300,000 |
| AEs costs | £166 per patient imatinib, £282 per patient dasatinib | £1660 per patient imatinib, £2820 per patient dasatinib | £421,000 | £260,000 | £458,000 | £306,000 |
| Utilities | ||||||
| Utilities | Equal to Novartis | £413,000 | £255,000 | £448,000 | £299,000 | |
| Utilities | Reduce all utilities by 0.10 | £362,000 | £248,000 | £402,000 | £291,000 | |
The sensitivity analyses were chosen on the basis of either general interest (e.g. assuming no discounting), plausibility (e.g. modelling drug price falls on patent expiry) or using the Novartis assumptions.
Incremental cost-effectiveness ratios are shaded black if the drug is less cost-effective than imatinib at a willingness-to-pay threshold of £30,000 per QALY. The shading is grey if the drug is more cost-effective than imatinib at a threshold of £30,000 per QALY, but less cost-effective than imatinib at £20,000 per QALY. There is no shading if the drug is more cost-effective than imatinib at a threshold of £20,000 per QALY.
For scenarios 3 and 4 (imatinib is followed by second-line nilotinib) in Table 54, on nearly all occasions nilotinib is predicted to yield fewer QALYs and less cost than imatinib. Nilotinib then lies in the south-west quadrant of the cost-effectiveness plane relative to imatinib. In this case, the ICERs are denoted by table footnote ‘a’. ICERs above £30,000 per QALY imply that nilotinib is better value for money than imatinib at that threshold, contrary to the usual interpretation. When we assume that patients take second-line nilotinib after imatinib, nilotinib almost always provides good value for money compared with imatinib.
For scenarios 1 and 2 (no second-line nilotinib), nilotinib often lies close to the £20,000 and £30,000 per QALY willingness-to-pay thresholds.
We focus our discussion of the results on the comparison of nilotinib compared with imatinib rather than on dasatinib compared with imatinib. This is because the cost-effectiveness of nilotinib is often close to the threshold, and because dasatinib is always very poor value for money compared with imatinib.
Sensitivity analyses: discounting
Although CML is a chronic disease, discounting has little impact on the ICERs.
Sensitivity analyses: proportion receiving stem cell transplantation
First note that the ICERs for the simplified method (scenarios 2 and 4) are largely independent of our assumption for the proportion of patients receiving SCT. This is as intended, because the simplified method is designed to ensure that cost-effectiveness is insensitive to the nature, costs and QALYs of treatments post TKIs.
In scenario 1, the ICER of nilotinib compared with imatinib falls from £25,000 to £23,000 per QALY when we halve the proportion receiving SCT at all ages. This is because, now, a relatively smaller number of people receive SCT in the imatinib arm than in the nilotinib arm, and it is more cost-effective to be in the health state following SCT compared with the health state of receiving hydroxycarbamide treatment in CP, AP and then BC.
This assertion that it is more cost-effective for patients to receive SCT than to receive hydroxycarbamide is demonstrated as follows. The ICER between the treatment arm of first-line imatinib, followed by 100% patients taking SCT, compared with first-line imatinib, followed by 100% patients taking hydroxycarbamide, is £14,000 per QALY. Also, the corresponding ICER starting with first-line nilotinib is £15,000 per QALY.
In scenario 1, the ICER of nilotinib compared with imatinib increases from £25,000 to £28,000 per QALY with the Novartis assumption that 75% of patients have SCT if they are < 65 years old, and no patients receive SCT if they are older. This is because the difference in the proportion of people who receive SCT between imatinib and nilotinib increases from 5% to 8%, and, as we have just demonstrated, SCT is more cost-effective than treatment with hydroxycarbamide.
Dasatinib remains very poor value for money against imatinib regardless of our assumption for proportion receiving SCT.
Sensitivity analyses: time on first-line tyrosine kinase inhibitor
We consider two sensitivity analyses concerning duration of first-line TKI treatment. These parameters are worthy of sensitivity analysis because they strongly affect cost-effectiveness and because duration of all first-line treatments is uncertain, given that the two first-line RCTs are very immature.
First, we assume that all treatments have the same mean duration as for imatinib, at 7.0 years. (CiC information has been removed.) Imatinib dominates dasatinib because it is far less expensive per person per day.
Next, the absolute mean times on first-line TKIs were based on that for imatinib in the IRIS RCT. At the same time, the HRs were still taken from the RCT of first-line nilotinib compared with imatinib and dasatinib compared with imatinib. This yields mean times on treatment of 11.7 years for imatinib, 13.8 years for nilotinib and 12.5 years for dasatinib.
Sensitivity analyses: time on second-line nilotinib
The time on second-line nilotinib is relevant only in scenarios 3 and 4. Our estimate of the mean time on second-line nilotinib, at 2.5 years, is probably robust because it is taken from a single-arm, high-quality study. Nonetheless, when we increase the mean duration substantially to 8.9 years, which is our assumption for the duration on first-line nilotinib, the cost-effectiveness of nilotinib compared with imatinib deteriorates, but nilotinib still remains cost-effective at a willingness-to-pay threshold of £30,000 per QALY.
Sensitivity analyses: survival after stem cell transplantation
Our estimated mean survival after SCT of approximately 17 year is uncertain, given that our evidence is observational and we have no relevant evidence after failure of nilotinib or dasatinib. Novartis estimates a far shorter mean survival after SCT of 5.7 years. Assuming this shorter survival time, the ICER for nilotinib compared with imatinib under scenario 1 falls from £25,000 to £16,000 per QALY and under scenario 2 falls from £20,000 to £17,000 per QALY. In both cases, cost-effectiveness improves because being in the post-SCT health state is now less cost-effective, because patients still incur the initial cost of the operation, but live less long. In addition, more patients have SCT on imatinib (33%) than on nilotinib (28%).
Sensitivity analyses: time on hydroxycarbamide in chronic phase
Our estimated mean time on hydroxycarbamide in CP of 5 years is uncertain, given that our evidence is based on a study which included a mixture of treatments in addition to hydroxycarbamide, and because we have no relevant evidence after failure of nilotinib or dasatinib. Novartis estimates a far shorter time on hydroxycarbamide in CP of 1.6 years. Assuming this shorter survival time, the ICER for nilotinib compared with imatinib under scenario 1 falls from £25,000 to £22,000 per QALY and under scenario 2 falls from £20,000 to £18,000 per QALY.
Sensitivity analyses: surrogate overall survival
We now consider the sensitivity analyses whereby we retain the model structure under the cumulative survival method, but adjust the time on hydroxycarbamide in CP to reflect the OS experienced in historical trials. We believe that these sensitivity analyses are very important, because they are the only analyses which use the CCyR and MMR rates reported for first-line treatment with the three TKIs. The methods are explained above (see Surrogate-predicted survival approach). However, to summarise briefly, we present four sensitivity analyses (1a, 1b, 2a and 2b, as presented in Table 31) for each of scenarios 1 and 2. In the first analysis, the mean OS on imatinib is left unchanged, but the mean OS for nilotinib and dasatinib is adjusted to reflect the differences in OS between nilotinib, dasatinib and imatinib, which are estimated from the surrogate analysis based on MMR. The second analysis repeats the first analysis, but using the surrogate relationship based on CCyR (scenario 1b and 2b). In the third analysis, OS for all treatments is forced to equal OS estimated for each treatment based on the historical MMR surrogate. The final analysis is the same, but based on the historical CCyR surrogate. The resulting mean survival times are given in Figure 42. Figure 43 shows how OS as estimated by the cumulative survival method is far shorter than by the surrogate survival method (upper graph). In the third and fourth sensitivity analyses, the modelled OS is then adjusted to match the OS based on the surrogate experience (lower graph).
FIGURE 42.
Modelled OS by treatment arm as a function of method of estimating OS for methods related to MMR surrogate OS (upper graph) and CCyR surrogate OS (lower graph).
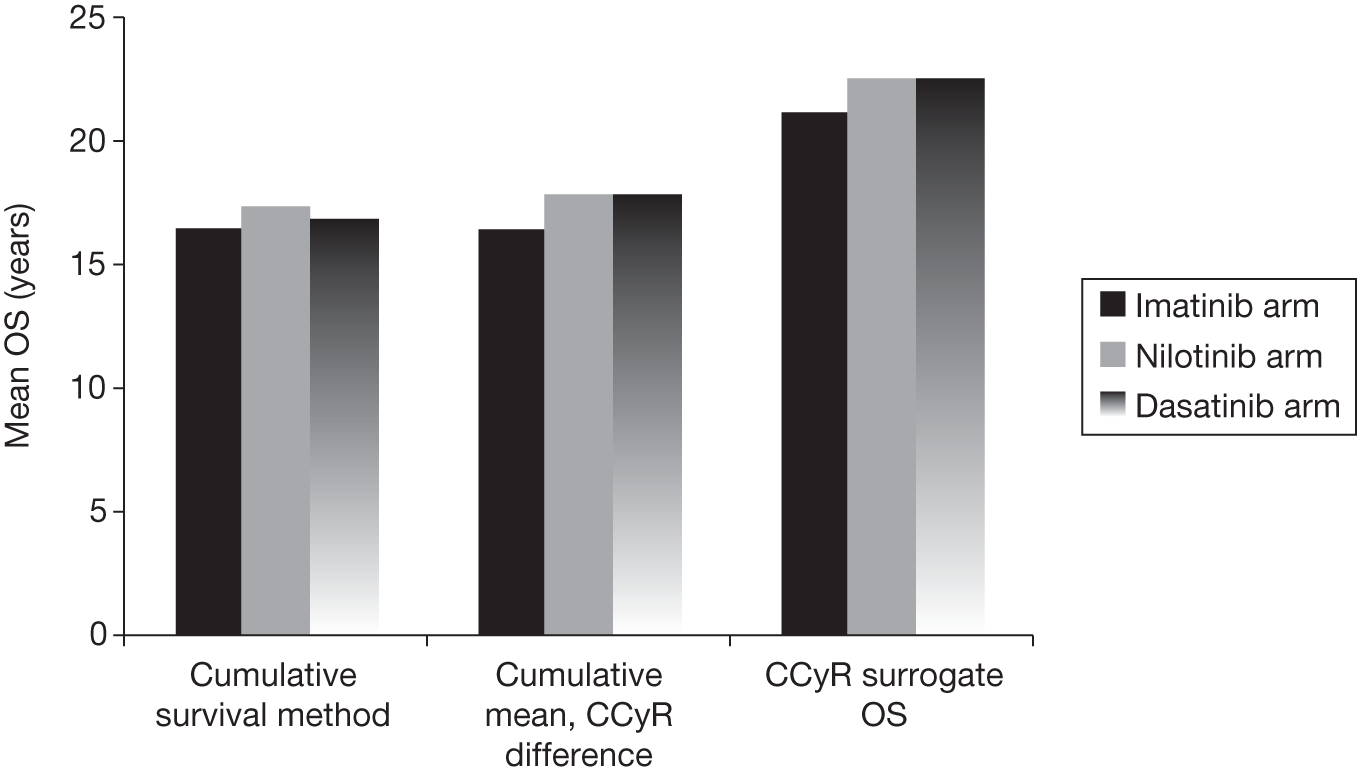
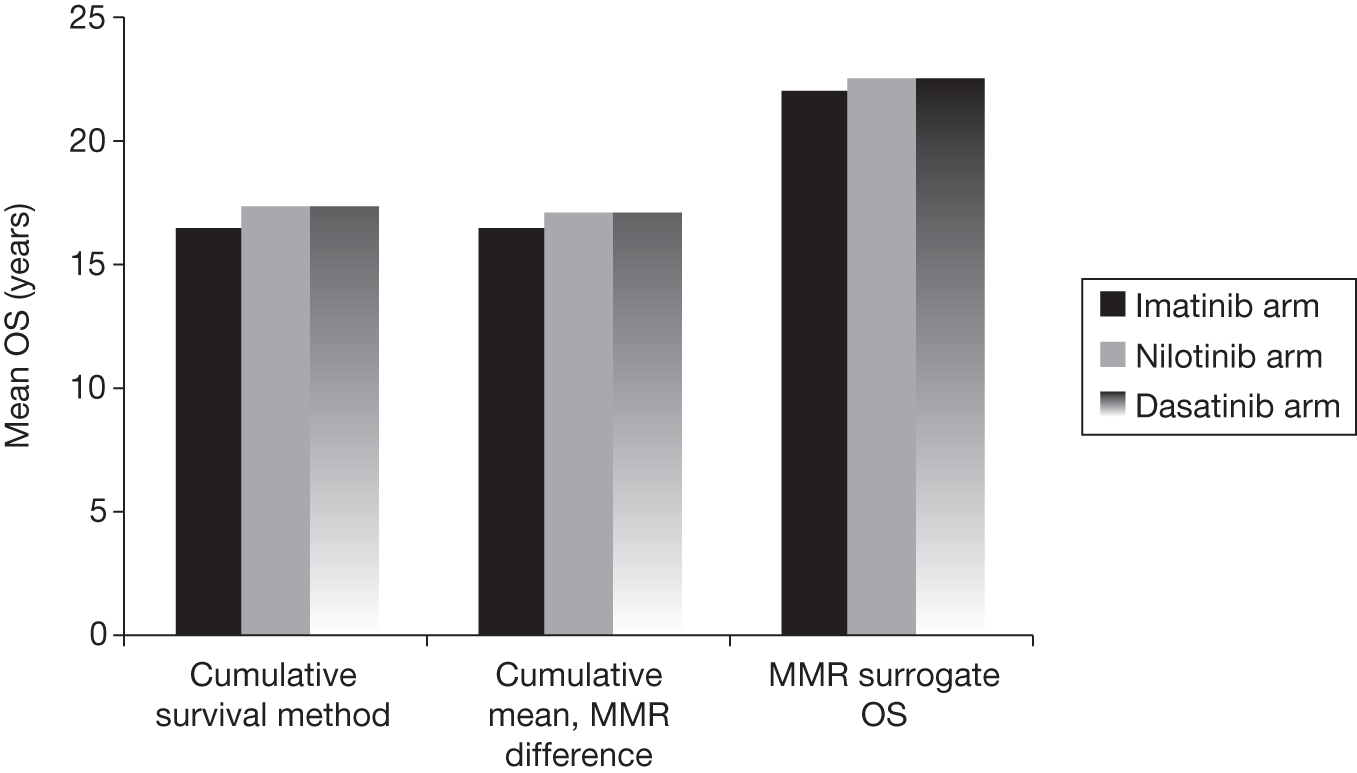
FIGURE 43.
Overall survival based on cumulative survival vs surrogate survival. (a) By treatment and response type, (b) when cumulative survival is adjusted to match surrogate survival. Filled circles represent actual OS from the imatinib arm of the IRIS trial.
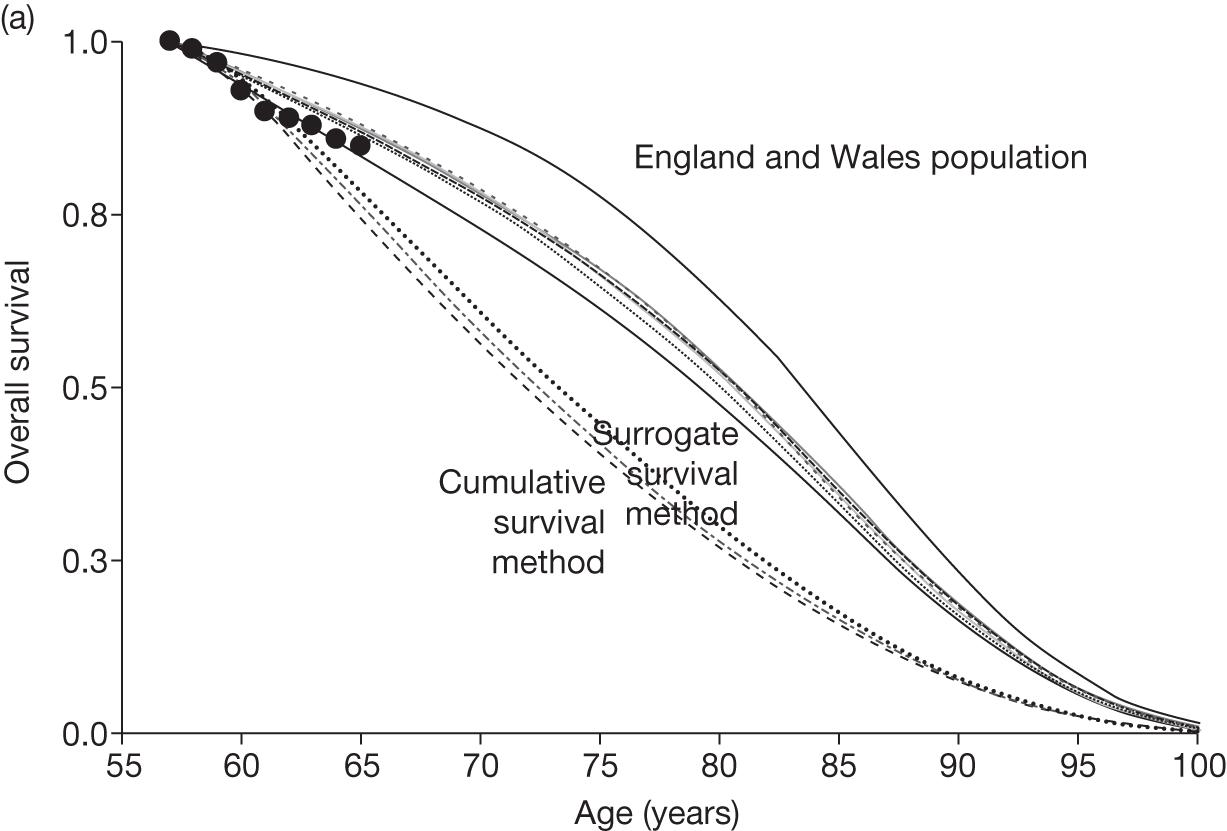
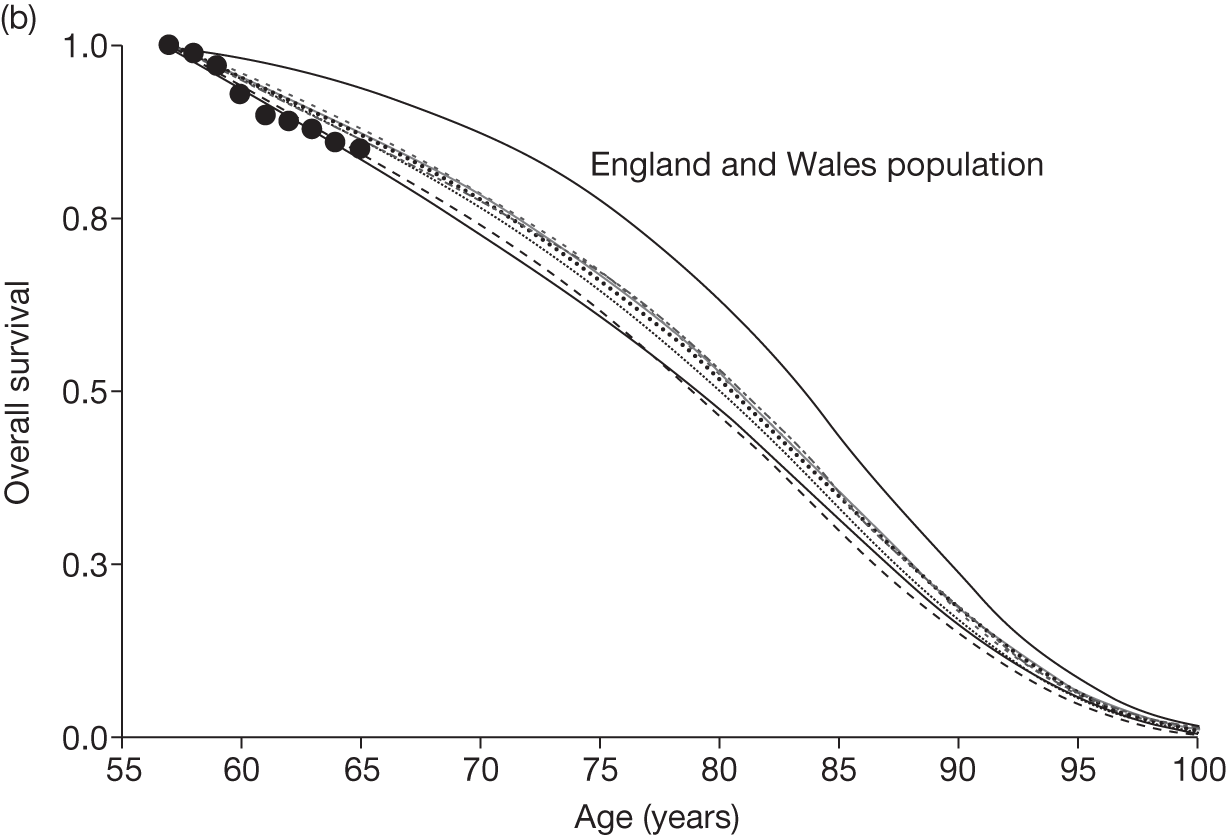
The sensitivity analyses reveal that the cost-effectiveness of nilotinib compared with imatinib worsens when we base OS on the MMR surrogate relationship, regardless of whether or not the OS of imatinib is adjusted to reflect that from the surrogate relationship (Table 56; and see Table 54, above). This is because we estimate only a slight advantage in OS, 0.6 years, for people taking nilotinib compared with people taking imatinib based on the MMR surrogate relationship, and this is less than the difference of 0.9 years based on the cumulative survival method. Conversely, the cost-effectiveness of nilotinib compared with imatinib improves when we base OS on the CCyR surrogate relationship, regardless of whether or not the OS of imatinib is adjusted to reflect that from the surrogate relationship. This is because we estimate a slightly greater advantage in OS, 1.3 years, for people taking nilotinib compared with people taking imatinib based on the CCyR surrogate relationship than the 0.9 years based on the cumulative survival method.
| Treatment | Discounted cost (£) | Undiscounted life-years | Discounted QALYs | Incremental cost (£) | Incremental QALYs | ICER vs imatinib (£ per QALY) |
|---|---|---|---|---|---|---|
| Cumulative survival means, MMR survival difference | ||||||
| Imatinib – then hydroxycarbamide/SCT | £159,000 | 16.5 | 9.0 | |||
| Nilotinib – then hydroxycarbamide/SCT | £170,000 | 17.1 | 9.3 | £11,000 | 0.3 | £35,000 |
| Dasatinib – then hydroxycarbamide/SCT | £224,000 | 17.1 | 9.3 | £65,000 | 0.3 | £250,000 |
| Cumulative survival means, CCyR survival difference | ||||||
| Imatinib – then hydroxycarbamide/SCT | £159,000 | 16.5 | 9.0 | |||
| Nilotinib – then hydroxycarbamide/SCT | £170,000 | 17.9 | 9.6 | £11,000 | 0.6 | £17,000 |
| Dasatinib – then hydroxycarbamide/SCT | £224,000 | 17.9 | 9.6 | £65,000 | 0.6 | £104,000 |
| Surrogate survival means, MMR survival difference | ||||||
| Imatinib – then hydroxycarbamide/SCT | £159,000 | 22.0 | 11.2 | |||
| Nilotinib – then hydroxycarbamide/SCT | £169,000 | 22.6 | 11.4 | £11,000 | 0.3 | £40,000 |
| Dasatinib – then hydroxycarbamide/SCT | £223,000 | 22.6 | 11.4 | £65,000 | 0.1 | £303,000 |
| Surrogate survival means, CCyR survival difference | ||||||
| Imatinib – then hydroxycarbamide/SCT | £159,000 | 21.2 | 10.9 | |||
| Nilotinib – then hydroxycarbamide/SCT | £169,000 | 22.5 | 11.4 | £10,000 | 0.5 | £19,000 |
| Dasatinib – then hydroxycarbamide/SCT | £223,000 | 22.6 | 11.4 | £64,000 | 0.1 | £124,000 |
Dasatinib remains very poor value for money when using OS based on the surrogate method.
Sensitivity analyses: patent expiry
Imatinib will lose patent protection in England and Wales in just a few years, in 2016 (note: this is after the currently tabled review date for this NICE guidance). 135 Also, dasatinib comes off patent in 2020 and nilotinib comes off patent in 2023. 139,140 Given that NICE’s recommendations from this HTA will come into force in 2012, this will be only 4 years before imatinib loses patent protection. Cost-effectiveness can be sensitive to the price fall when the drug patent expires. 141 Two sensitivity analyses were considered: first, setting the price reduction on patent expiry to 25% for all drugs and, second, setting the price reduction to 25% for imatinib and dasatinib and 0% for nilotinib. The reduction of 25% is not evidence based; however, we believe that this gives a guide to the possible changes in cost-effectiveness. In one sensitivity analysis, we model no price change for nilotinib, because this assumes that the price reduction on patent expiry will be relative to the list price of nilotinib, not to the price of nilotinib under the PAS.
(CiC information has been removed.)
In scenario 1, assuming a 25% reduction in the prices of nilotinib and imatinib, the ICER for nilotinib compared with imatinib increases from £25,000 to £44,000 per QALY. This is for two reasons. First and most importantly, imatinib is far closer to patent expiry than nilotinib. Second, we predict that patients take nilotinib for longer than imatinib. Also in scenario 1, assuming a 25% reduction in the price of imatinib only, with no change in the price of nilotinib, the ICER increases from £25,000 to £60,000 per QALY.
In scenario 4 (simplified method, with second-line nilotinib), nilotinib changes from being cost-effective compared with imatinib (although providing fewer QALYs) to being on the border of cost-effectiveness.
Dasatinib becomes even worse value for money compared with imatinib when we allow for price reduction on patent expiry.
These sensitivity analyses all assume patients starting TKI treatment in the year 2012. If instead we model patients starting treatment in the future, so-called ‘future incident cohorts’,142 the cost-effectiveness of drugs can be substantially altered. 143 In this case, all ICERs increase further. For example, modelling patients starting treatment in the year 2016, and assuming a 25% reduction in the prices of both nilotinib and imatinib, under scenario 1, the ICER for nilotinib compared with imatinib increases from £25,000 to £64,000 per QALY. Under scenario 2, the ICER for nilotinib compared with imatinib increases from £20,000 to £44,000 per QALY. In addition, under scenario 4 (with second-line nilotinib, simplified method), nilotinib changes from being good value for money (although less beneficial) to being poor value for money.
Sensitivity analyses: dose intensities
The ICERs of nilotinib compared with imatinib are very sensitive to even small changes in the dose intensities. Our estimate of the dose intensity of first-line nilotinib, at (CiC information has been removed), is taken from Novartis, and is evidence based. However, when we (CiC information has been removed) this to 100%, a value which is not evidence based, the ICER under scenario 1 increases from £25,000 to £53,000 per QALY, and the ICER under scenario 2 increases from £20,000 to £37,000 per QALY.
Conversely, leaving the dose intensity of first-line nilotinib unchanged at (CiC information has been removed), and increasing the dose intensity of imatinib from (CiC information has been removed) to (CiC information has been removed), which is the value used by Novartis, nilotinib becomes substantially better value. The ICER under scenario 1 decreases from £25,000 to £8,000 per QALY, and under scenario 2 decreases from £20,000 to £9000 per QALY.
These analyses highlight the crucial importance of the dose intensities in estimating the cost-effectiveness of nilotinib.
When the dose intensity of second-line nilotinib is changed from the evidence-based value of 99% to (CiC information has been removed), being the same as for first-line nilotinib, the cost-effectiveness of nilotinib in scenarios 3 and 4 worsens slightly.
Sensitivity analyses: cost of stem cell transplantation
The ICERs of nilotinib compared with imatinib are fairly sensitive to changes in the cost of SCT from our evidence-based estimate of £81,603. When the cost is increased, nilotinib becomes better value for money because a smaller proportion of the total cohort is predicted to have SCT in the nilotinib arm than in the imatinib arm.
Sensitivity analyses: medical management in chronic phase
The ICERs are fairly sensitive to the monthly cost of medical management in CP, whether on TKIs or hydroxycarbamide. For example, in scenarios 1 and 2 (no second-line nilotinib), the ICER of nilotinib compared with imatinib increases when the cost is increased. This is because we predict that patients will spend longer in CP taking TKIs or hydroxycarbamide in the nilotinib arm than in the imatinib arm.
Sensitivity analyses: other costs
All ICERs are insensitive to all other costs of medical management after SCT, medical management in AP and BC, and treatment of AEs.
Sensitivity analyses: utilities
All ICERs are virtually unchanged when we use the Novartis utilities. This is because we use the same age-dependent utilities while patients are taking TKIs or hydroxycarbamide in CP, and because the remaining utilities differ only slightly.
When all utilities are reduced by 0.10, the ICERs for nilotinib compared with imatinib in scenarios 1 and 2 decrease slightly. However, we caution that the reduction of 0.10 is not evidence based, but is arbitrary.
Comparison of Peninsula Technology Assessment Group model with industry submissions
Comparison of the Peninsula Technology Assessment Group model with the Novartis model
Scenario 1 in the PenTAG model uses the closest structural assumptions to the Novartis model in which no second-line TKIs are assumed. However, the models predict substantially different ICERs for nilotinib compared with imatinib, which span the usually accepted cost-effectiveness thresholds:
-
PenTAG ICER £25,000 per QALY
-
Novartis ICER £6000 per QALY.
Note that scenario 1 is only one of our four scenarios, all with their advantages and disadvantages.
First, we explain the causes of this difference in cost-effectiveness, and justify our choice of assumptions. Second, we describe some further key differences in model predictions. Third, in order to assess the impact of assumptions on cost-effectiveness, we adjust the Novartis model sequentially so that it becomes more like our model.
Causes of difference in cost-effectiveness of Novartis compared with the Peninsula Technology Assessment Group
Table 57 compares the results from the PenTAG scenario 1 and the Novartis analysis with no second-line TKI. The difference in cost-effectiveness is explained mostly by the following differences in the models. All of these differences act to make the cost-effectiveness of nilotinib compared with imatinib worse in the PenTAG model compared with the Novartis model:
-
Incremental QALYs in SCT: PenTAG –0.42 compared with –0.26 Novartis
-
Incremental QALYs on hydroxycarbamide in CP: PenTAG –0.11 compared with 0.01 Novartis
-
Incremental costs on first-line TKIs: PenTAG £14,751 compared with £10,733 Novartis
-
Incremental cost of SCT operation: PenTAG –£3840 compared with –£7603 Novartis.
These key differences are highlighted in Table 57. If just these incremental results from our model are used, then the Novartis ICER increases from £6000 to £25,000 per QALY, which matches the result from our model. This demonstrates that it is these incremental differences that drive the difference in cost-effectiveness estimates.
| Treatment | Imatinib | Nilotinib | Nilotinib–imatinib | |||
|---|---|---|---|---|---|---|
| PenTAG | Novartisa | PenTAG | Novartisa | PenTAG | Novartisa | |
| Life-years (undiscounted) | ||||||
| First-line TKI | 7.0 | 5.5 | 8.9 | 7.3 | 1.9 | 1.7 |
| SCT | 5.8 | 3.1 | 4.9 | 2.7 | –0.9 | –0.5 |
| Hydroxycarbamide CP | 2.9 | 0.6 | 2.8 | 0.7 | –0.1 | 0.1 |
| Hydroxycarbamide AP | 0.5 | 0.3 | 0.4 | 0.4 | 0.0 | 0.0 |
| Hydroxycarbamide BC | 0.3 | 0.3 | 0.3 | 0.4 | 0.0 | 0.0 |
| OS | 16.5 | 10.0 | 17.4 | 11.4 | 0.9 | 1.4 |
| Cohort splitb | ||||||
| % starting SCT/hydroxycarbamide | 90 | 94 | 84 | 90 | –6 | –4 |
| % SCT (whole cohort) | 33 | 55 | 28 | 47 | –5 | –8 |
| % SCT (eligible cohort) | 37 | 58 | 34 | 52 | –3 | –6 |
| % hydroxycarbamide (whole cohort) | 56 | 39 | 56 | 43 | –1 | 4 |
| % hydroxycarbamide (eligible cohort) | 63 | 42 | 66 | 48 | 3 | 6 |
| % AP (whole cohort) | 49 | 38 | 48 | 42 | –2 | 4 |
| % BC (whole cohort) | 49 | 38 | 48 | 42 | –2 | 4 |
| Life-years (undiscounted eligible cohort)b | ||||||
| First-line TKI | 7.0 | 5.5 | 8.9 | 7.3 | 1.9 | 1.7 |
| SCT | 17.4 | 5.7 | 17.2 | 5.7 | –0.2 | 0.0 |
| Hydroxycarbamide CP | 5.1 | 1.6 | 5.0 | 1.6 | –0.1 | 0.0 |
| Hydroxycarbamide AP | 0.9 | 0.8 | 0.9 | 0.8 | 0.0 | 0.0 |
| Hydroxycarbamide BC | 0.6 | 0.8 | 0.6 | 0.8 | 0.0 | 0.0 |
| QALYs (discounted) | ||||||
| First-line TKI | 4.54 | 3.77 | 5.52 | 4.75 | 0.98 | 0.98 |
| SCT | 2.61 | 1.66 | 2.18 | 1.40 | –0.42 | –0.26 |
| Hydroxycarbamide CP | 1.54 | 0.38 | 1.43 | 0.39 | –0.11 | 0.01 |
| Hydroxycarbamide AP | 0.22 | 0.13 | 0.21 | 0.14 | –0.01 | 0.00 |
| Hydroxycarbamide BC | 0.11 | 0.13 | 0.10 | 0.13 | –0.01 | 0.00 |
| Total | 9.01 | 6.07 | 9.43 | 6.81 | 0.42 | 0.74 |
| Costs, £ (discounted) | ||||||
| First-line TKI | 118,635 | 104,038 | 133,386 | 114,771 | 14,751 | 10,733 |
| First-line AEs | 166 | 178 | 119 | 111 | –47 | –67 |
| First-line medical management | 3811 | 5460 | 4658 | 6825 | 846 | 1365 |
| SCT transplant | 24,486 | 49,986 | 20,646 | 42,383 | 3840 | 7603 |
| SCT medical management | 2562 | 0 | 2148 | 0 | –415 | 0 |
| Hydroxycarbamide acquisition in CP | 282 | 73 | 264 | 76 | –19 | 3 |
| Hydroxycarbamide CP medical management | 2494 | 271 | 2330 | 279 | –164 | 8 |
| Hydroxycarbamide AP acquisition + medical management | 4098 | 844 | 3,828 | 874 | –270 | 30 |
| Hydroxycarbamide BC acquisition + medical management | 2735 | 1613 | 2555 | 1665 | –180 | 52 |
| End-of-life cost | 3541 | 3389 | –152 | |||
| Total costs | 159,270 | 166,003 | 169,932 | 170,373 | 10,662 | 4370 |
| Cost/LYG | 12,000 | 5000 | ||||
| Cost/QALY | 25,000 | 6000 | ||||
Difference in quality-adjusted life-years after stem cell transplantation
There are two important components to the QALYs after SCT, which apply to both models: first, the proportion of patients who receive SCT from all patients who start first-line treatment and, second, the mean time after SCT for those who have SCT.
The first component, the proportion of patients who receive SCT from all patients who start first-line treatment, actually works against the observation that incremental QALYs are lower in our model than in the Novartis model. In our model, 5% fewer patients have SCT on nilotinib than on imatinib, compared with 8% in the Novartis model.
However, the second component dominates. In our model, life expectancy after SCT is about 17.3 years, compared with 5.7 years in the Novartis model.
It is difficult to be certain whether we or Novartis have a better estimate for the life expectancy after SCT, for people having SCT after first-line imatinib or nilotinib, given that we both rely on observational evidence.
Difference in quality-adjusted life-years on hydroxycarbamide in chronic phase
We predict slightly lower QALYs on hydroxycarbamide in CP in the nilotinib arm compared with the imatinib arm, whereas Novartis predicts virtually the same QALYs. Initially, it appears surprising that we predict lower QALYs for the nilotinib arm compared with the imatinib arm, 1.54 compared with 1.43, given that we predict very similar mean times on hydroxycarbamide in CP, averaged over all patients starting first-line treatment (2.88 vs 2.79 years). The difference is due to discounting, given that hydroxycarbamide is taken in CP typically later in the nilotinib arm than in the imatinib arm.
Furthermore, the slight difference in discounted time on hydroxycarbamide is magnified in our model, because we assume that patients take hydroxycarbamide in CP for much longer than does Novartis – 5.0 years compared with 1.6 years. However, as stated (see Appendix 7), we believe that the Novartis method of calculating time on hydroxycarbamide in CP following TKI failure is flawed.
The difference between the models is not explained by utilities, because we and Novartis use the same utilities while on hydroxycarbamide in CP.
Difference in costs of first-line tyrosine kinase inhibitors
We predict that the mean acquisition cost of first-line nilotinib is £14,800 greater than the acquisition cost of first-line imatinib. Novartis assumes a smaller difference, at £10,700.
There are two factors that influence this difference between models. Most importantly, we assume a lower dose intensity for imatinib, at (CiC information has been removed), than Novartis, at (CiC information has been removed). Using the Novartis estimate in our model, we predict an incremental cost of £7600. Thus, changing the dose intensity overcompensates for the difference in costs.
As mentioned above, although both estimates of dose intensity are provided by Novartis, we favour 100% for the reasons given above (see Dose intensities, above). The mean acquisition cost of first-line TKIs is also a function of the mean time on first-line TKIs.
Difference in cost of stem cell transplantation operation
We predict that the mean cost of SCT operations, averaged over all patients starting first-line treatment, is lower, by approximately £3800, in the nilotinib arm than in the imatinib arm. Novartis estimates a greater difference, at approximately £7600.
The difference between models is mostly explained by the fact that we predict a smaller difference in the proportion of all patients who have SCT in the nilotinib arm compared with the imatinib arm: –5% for us compared with –8% for Novartis. In both models, fewer patients are predicted to have SCT in the nilotinib arm than in the imatinib arm. This, in turn, is a function of the differences in the assumed proportions of patients who have SCT as a function of age. We assume a linear decrease as a function of age, whereas Novartis assumes a flat rate of 75% up to the age of 65 years, and 0% thereafter.
The difference in the mean cost of SCT per patient is explained only to a small extent by the assumed cost of SCT. We assume £81,600, compared with Novartis’s £99,200. Specifically, changing our assumed cost to equal that of Novartis changes our incremental costs from £3800 to £4700.
It is difficult to be certain whether we or Novartis have more accurate estimates of the proportions of patients having SCT as a function of age and the cost of SCT because both assumptions are rather subjective.
Further key differences in model predictions
Time on first-line treatment
We predict longer expected times on first-line nilotinib and imatinib than Novartis. Specifically, the mean time on imatinib in the PenTAG model is 7.0 years compared with 5.5 years in the Novartis model, and the mean time on nilotinib in the PenTAG model is 8.9 years compared with 7.3 years in the Novartis model. Figure 44 shows these differences.
FIGURE 44.
Time on first-line treatment with imatinib (upper figure) and nilotinib (lower figure): PenTAG vs Novartis.
We and Novartis both fit Weibull distributions to the time on first-line treatment, and we both use the same empirical data from the trial of first-line imatinib compared with nilotinib. However, there are two reasons that explain the differences in time on treatment. First, we adjust our estimates of the time on treatment of both imatinib and nilotinib from the RCT of first-line imatinib compared with nilotinib to perform the indirect comparison of all three TKIs, imatinib, nilotinib and dasatinib, as explained in the Methods section. Novartis does not make this adjustment. Second, whereas we fit a curve to the Kaplan–Meier probabilities from the RCT of first-line imatinib compared with nilotinib, Novartis does not. Instead, it first adjusts the Kaplan–Meier probabilities. For example, at 12 months’ follow-up, the Kaplan–Meier estimate of the proportion of patients still on first-line nilotinib is 0.870, whereas Novartis adjusts this to 0.861 and then fits a Weibull curve to this figure. Novartis does not justify this adjustment, and the reason for the adjustment is not clear to us.
Overall survival
Novartis predicts much shorter OS than us for both treatment arms (Figures 45 and 46). This is because it predicts much shorter times on hydroxycarbamide in CP (5.0 years ‘us’ vs 1.6 years Novartis) and survival after SCT (17.3 years ‘us’ vs 5.7 years Novartis), and slightly shorter times on first-line nilotinib and imatinib than us, as mentioned above (see Causes of difference in cost-effectiveness Novartis vs PenTAG) and in the previous section.
FIGURE 45.
Overall survival (nilotinib), PenTAG vs Novartis.
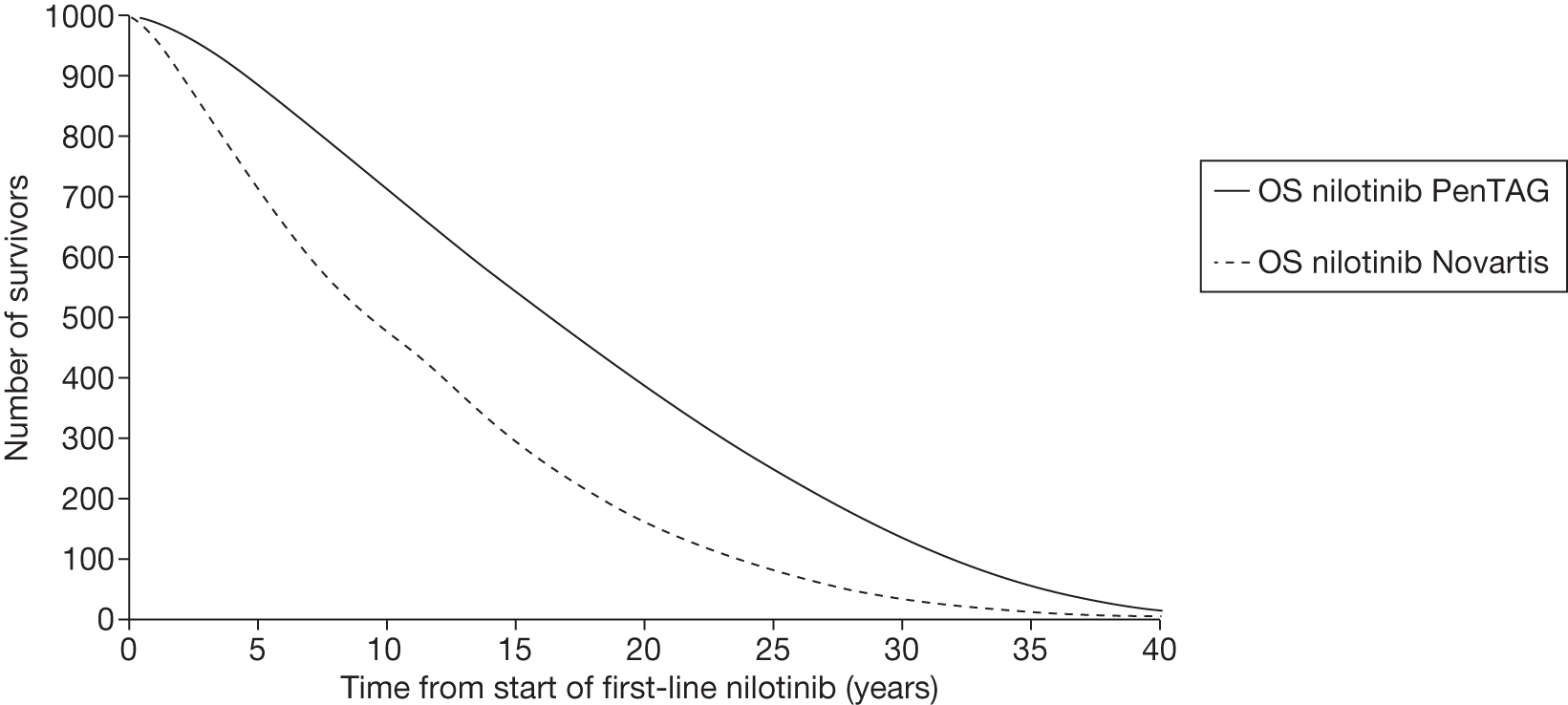
FIGURE 46.
Overall survival (imatinib), PenTAG vs Novartis.
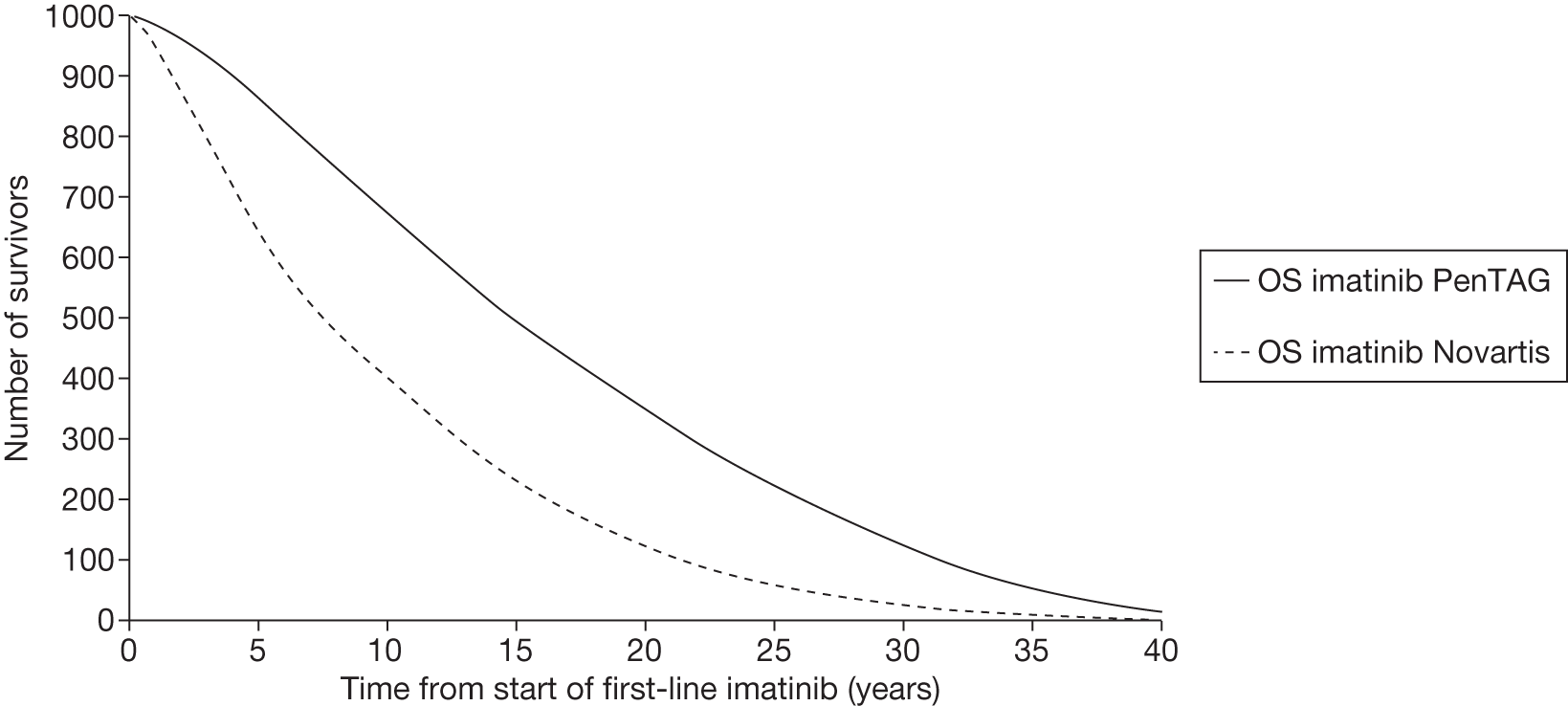
Adjustments to Novartis model
In order to further explore what is driving the difference in cost-effectiveness between the models, the following key parameters were identified:
-
time on first-line TKI treatment
-
SCT parameters
-
utility values.
Where differences between the key parameters were identified, the PenTAG values were input to the Novartis model and the resulting impact on the ICER was analysed.
Time on first-line treatment
As explained above, we predict longer expected times on first-line nilotinib and imatinib than Novartis. When the PenTAG treatment discontinuation rates for first-line treatment are input to the Novartis model, the ICER for nilotinib compared with imatinib decreases only slightly, from £6000 to £4000 per QALY.
Costs
Taking the Novartis ICER of £4000 per QALY updated for the PenTAG times on first-line treatment as the starting point, Table 58 summarises the difference in input costs between the two models, and the change in the Novartis ICER when PenTAG costs are used.
| Treatment | Cost (£, per person per 3 months) | ||
|---|---|---|---|
| PenTAG | Novartis | Updated ICER from Novartis model using PenTAG values | |
| First-line nilotinib | (CiC information has been removed) | (CiC information has been removed) | 12,000 |
| First-line imatinib | 5249 | 5547 | |
As shown in Table 58, imatinib is slightly more expensive in the Novartis model than in the PenTAG model. As stated above, this is because Novartis assumes a higher dose intensity for imatinib, 106%, than us (CiC information has been removed). Although this difference in dose intensities is small, it impacts strongly on cost-effectiveness given also that incremental QALYs are small. Using the PenTAG drug costs in the Novartis model causes the ICER to increase from £6000 to £12,000 per QALY.
Assumptions related to stem cell transplantation
There are considerable differences around the use of SCT between the PenTAG and Novartis models. First, Novartis assumes that 75% of patients who reach second-line treatment aged < 65 years have SCT, and no patients > 65 years receive SCT. Conversely, we assume a linear decrease in the proportion having SCT with increasing age. Further, we predict far longer survival after SCT (17.3 years) than Novartis (5.7 years) (see Table 57).
Out of all of the patients starting first-line treatment, fewer patients receive SCT in the PenTAG analysis than in the Novartis analysis. In the Novartis model, 47% of patients in the nilotinib arm receive SCT compared with 55% of patients in the imatinib arm (see Table 57, above). When we alter the Novartis model for our assumptions on the proportions having SCT and survival after SCT, the updated Novartis model matches the prediction from the PenTAG model that 28% of those in the nilotinib arm receive SCT compared with 33% of those in the imatinib arm. The ICER then increases further from £12,000 to £19,000 per QALY, for the reason stated above (see Difference in quality-adjusted life-years after stem cell transplantation), i.e. that we then predict substantially fewer QALYs after SCT in the nilotinib arm than in the imatinib arm. When we further change the Novartis assumption that SCT costs £99,225 to our value of £81,603, the ICER increases slightly, from £19,000 to £21,000 per QALY.
Utility values
There are only slight differences in the utility values used in the PenTAG and Novartis models. Both models vary utility by age in exactly the same way. Furthermore, the utilities while patients are taking TKIs and hydroxycarbamide in CP are equal in both models. Utility assumptions are slightly different between models for post-SCT and while in AP and BP. Indeed, the ICER remains at £21,000 per QALY when we update the Novartis model for our assumed utilities.
Time on hydroxycarbamide
We assume a much longer mean time on hydroxycarbamide in CP than Novartis, 5.0 compared with 1.6 years, where these values are averaged over people who receive hydroxycarbamide, rather than people starting first-line treatment. The mean time in AP is very similar between the models. When the Novartis model is further updated for our times on hydroxycarbamide in CP, AP and BC, the ICER increases only slightly, from £21,000 to £23,000 per QALY.
Comparison of Peninsula Technology Assessment Group model with the Bristol-Myer Squibbs model
Here, we compare our model and the BMS model for scenario 3, in which we model second-line nilotinib, using the BMS model corrected for errors and adjusted so that all patients receive nilotinib second line. We consider the dasatinib and imatinib treatment arms only.
This section is brief for the following reasons:
-
We present the results of the BMS model after we have made several corrections and adjustments.
-
Both models predict that dasatinib is very poor value compared with imatinib, with ICERs of £450,000 per QALY with our model and £95,000 per QALY with the BMS corrected and adjusted model.
-
We disagree with the BMS method of estimating OS via a historical surrogate relationship because this relationship does not reflect the use of second-line nilotinib, whereas BMS models second-line nilotinib. Indeed, it is for this reason that we did not attempt to model surrogate OS when we modelled second-line nilotinib (scenarios 3 and 4).
We estimate far longer OS than BMS of approximately 17.5 years compared with 12.5 years (Table 59). It is therefore surprising that we estimate similar discounted QALYs. This is largely because we assume that utilities decline with age, whereas BMS does not.
| Model output | Imatinib | Dasatinib | Dasatinib–imatinib | |||
|---|---|---|---|---|---|---|
| PenTAG | BMSa | PenTAG | BMSa | PenTAG | BMSa | |
| Life-years (undiscounted) | 17.3 | 12.3 | 17.6 | 12.9 | 0.3 | 0.6 |
| QALYs (discounted) | 9.5 | 9.8 | 9.7 | 10.6 | 0.1 | 0.8 |
| Costs, £ (discounted) | 188,000 | 378,000 | 252,000 | 457,000 | 64,000 | 79,000 |
| Cost/QALY | 450,000 | 95,000 | ||||
Although we estimate far lower total costs per patient than BMS, incremental total costs are similar, although this is probably purely coincidental.
Chapter 8 Discussion
Main findings
Clinical effectiveness
Both dasatinib 100 mg (once daily; DASISION trial29) and nilotinib 300 mg (twice daily; ENESTnd trial20) have a statistically significant advantage compared with the first-generation TKI imatinib 400 mg (once daily) with regard to surrogate outcomes (e.g. CCyR and MMR); however, there are insufficient data to assess longer-term patient-relevant outcomes (e.g. PFS, OS, HRQoL). Rates of CCyR and MMR for dasatinib and nilotinib were higher, more rapidly attained, and deeper (molecular response) compared with imatinib. All three drugs were well tolerated with discontinuation due to AEs of < 10%. Indirect comparison analysis showed no difference between dasatinib and nilotinib for the primary outcomes of CCyR or MMR at 12 months’ or 24 months’ follow-up.
There is observational association evidence supporting the use of CCyR and MMR at 12 months as surrogates for PFS and overall in patients with CP-CML. This is based entirely on imatinib treatment studies. In the absence of evidence of adequacy of these surrogates for dasatinib and nilotinib as first-line therapies, and assuming a TKI class-specific relationship between the surrogate outcomes and the patient-relevant outcomes, these results can be potentially applied to other drugs in the same class.
Cost-effectiveness
The whole of this technology assessment report has been prepared in the context of changing draft guidance about the use of the same drugs for second-line treatment of CML after imatinib as first-line treatment. In the draft guidance on 18 August 2011, NICE recommended nilotinib for the treatment of the CP and AP CML that is resistant or intolerant to standard-dose imatinib. Dasatinib and high-dose imatinib are not recommended in the draft guidance. Consultees have the opportunity to appeal against the draft guidance. Until NICE issues final guidance, NHS bodies should make decisions locally on the funding of specific treatments. This draft guidance does not mean that people currently taking dasatinib or high-dose imatinib will stop receiving them. They have the option to continue treatment until they and their clinicians consider it appropriate to stop.
We do not provide a single base case upon which to compare the cost-effectiveness of first-line nilotinib, dasatinib and imatinib because our model relies on numerous important assumptions. Furthermore, in many cases, there is no clear preference for one assumption over another. Instead, we present cost-effectiveness results for each of four main ‘scenarios’. In scenario 1, we do not model second-line nilotinib. In scenario 2, again, we do not model second-line nilotinib, but we use the simplified method, whereby the post-TKI per-patient costs and QALYs are set equal across treatment arms. We believe that this approach is appropriate owing to the substantial uncertainty in the nature, and associated costs and quality of life, of post-TKI treatments several years in the future, which is when patients will typically become eligible for such post-TKI treatments. Scenario 3 is the same as scenario 1, but allowing for second-line nilotinib, which has recently been recommended in the NICE draft guidance FAD (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). Similarly, scenario 4 is the same as scenario 2, but allowing for second-line nilotinib.
First-line dasatinib is predicted to provide very poor value for money compared with first-line imatinib regardless of the model structure, for example whether or not we allow for second-line treatment with nilotinib and regardless of when parameters are varied within plausible ranges.
Conversely, the findings for the cost-effectiveness of first-line nilotinib compared with first-line imatinib are rather complex.
Assuming first-line imatinib is followed by second-line nilotinib, on nearly all occasions, nilotinib is predicted to yield fewer QALYs at less cost than imatinib. This is because first-line imatinib, but not first-line nilotinib, is followed by second-line nilotinib, and the second-line nilotinib extends OS. Furthermore, assuming patients take second-line nilotinib after imatinib, first-line nilotinib almost always provides good value for money compared with imatinib. The only occasions when first-line nilotinib may represent worse value for money than first-line imatinib are when we allow for drug price decreases on patent expiry, and when the dose intensity of first-line nilotinib is (CiC information has been removed) to 100%.
Next, when we assume first-line imatinib is not followed by second-line nilotinib, first-line nilotinib often lies close to the £20,000 and £30,000 per QALY willingness-to-pay thresholds.
Still assuming that first-line imatinib is not followed by second-line nilotinib, the following parameters strongly influence the cost-effectiveness of first-line nilotinib and whether or not first-line nilotinib is cost-effective at a willingness-to-pay threshold of £30,000 per QALY:
-
proportion of patients receiving SCT on failure of first-line TKI imatinib and nilotinib
-
treatment duration of first-line imatinib and nilotinib
-
survival after SCT
-
time on hydroxycarbamide in CP after imatinib and nilotinib failure
-
whether we model CCyR and MMR response rates via surrogate relationships
-
reduction in the prices of imatinib and nilotinib on patent expiry
-
dose intensities of imatinib and nilotinib
-
cost of SCT operation.
Of special note are the analyses whereby OS is adjusted to match that experienced in historical trials of imatinib according to whether a CCyR or MMR is achieved. The findings differ according to whether the surrogate relationship is based on CCyR or MMR. Using CCyR substantially improves the cost-effectiveness of first-line nilotinib compared with imatinib, whereas the reverse is true with the MMR surrogate relationship.
Also of special note are the analyses whereby the prices of the TKIs are reduced on patent expiry. We believe this is highly relevant to this appraisal, especially given that imatinib will lose patent protection very soon, in 2016. We do not estimate the likely price cut on patent expiry, but even assuming a modest 25% reduction, the cost-effectiveness of first-line nilotinib worsens dramatically. Moreover, if we model patients who start first-line TKIs in the future, so-called ‘future incident cohorts’, the cost-effectiveness of nilotinib worsens still further.
Strengths and limitations of systematic review of clinical effectiveness
The strengths of this systematic review are that it was conducted by an independent research team using the latest evidence to a prespecified protocol.
The main limitation was lack of long-term evidence on dasatinib and nilotinib used first-line in the populations of interest, providing only immature data. Furthermore, there was only one trial for the each of the second-generation TKIs, namely dasatinib compared with imatinib and nilotinib compared with imatinib. This results in no head-to-head trials of dasatinib and nilotinib. With the immaturity of the data, primary end points of the trials are currently assessed using surrogate outcomes (i.e. CCyR and MMR). However, there is a lack of evidence for the use of surrogate outcomes for second-generation TKIs, with evidence available only for imatinib. It is assumed that the surrogate relationship exists for drugs of the same class.
Strength and limitations of systematic review of cost-effectiveness
The strengths of this systematic review are that it was conducted by an independent research team using the latest evidence to a prespecified protocol. However, we identified no studies reporting the cost-effectiveness of dasatinib and nilotinib.
Strengths and limitations of the appraisal of industry submissions
This was conducted by an independent research team using a number of established frameworks to identify strengths and weaknesses.
Strengths and limitations of the Peninsula Technology Assessment Group economic model
Strengths
-
Our assessment of the cost-effectiveness of drugs for CML is independent. We have carefully compared our model and the results of our analysis with those of Novartis and in so doing we have highlighted areas in common and those where there is disagreement.
-
Our model adheres to the NICE reference case methods and has been extensively checked. In addition to our four basic scenario analyses, we also present numerous one-way deterministic sensitivity analyses. We have chosen carefully for plausibility and to reflect the key areas of uncertainty and disagreements between ourselves and the Novartis modelling. This has involved developing a model that is capable of using either a surrogates-based estimation of OS or a cumulative treatment duration approach, or combinations of the two approaches. It is therefore also more capable of exploring the differences between Novartis and BMS model.
-
It is based on best available research evidence, from UK and recent patients wherever available and of reliable quality.
-
Where research evidence is lacking, we have checked key assumptions and parameter inputs with relevant clinical and other experts – for example, to inform our estimate cost of SCT, and the percentage who would get SCT at different ages.
-
Good calibration of model survival outputs against IRIS data (imatinib arm).
Limitations
Given that CML is a chronic condition, and that the main two RCTs provide very immature data on PFS, treatment duration and OS, our estimates of the cost-effectiveness of dasatinib and nilotinib are necessarily highly uncertain. They are also based on very small differences in clinical effectiveness outcomes between dasatinib and nilotinib. The following main sources of uncertainty exist in our modelling:
-
Immaturity of empirical trial data relative to life expectancy – forcing either reliance on surrogate relationships or cumulative survival/treatment duration approach. There is therefore considerable extrapolation from 12- to 30-month follow-up data using a variety of curve-fitting methods.
-
Overall great uncertainty over the very heterogeneous treatment and care pathways that patients with CML may follow – there are very many potential care and disease state paths which might be followed depending on how different people respond to treatment, their age, disease severity, availability of matched donors (for SCT), mutations that predict responsiveness to second-generation TKIs, etc. This includes not modelling complex treatment sequences in advanced disease (e.g. second and third CPs, and SCT following disease progression) and not modelling possible cessation of TKIs in those who experience a deep and durable initial response.
-
Some of the uncertainty regarding treatment sequences after first-line TKIs was because the NICE draft guidance FAD recommendation for second-line use of nilotinib, dasatinib or high-dose imatinib after standard-dose imatinib was not released until very recently (18 August 2011, the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). This meant that we could not choose the most plausible scenarios to model, or finalise exactly how to model them, until later than would normally be the case.
-
Uncertainty over both which treatment sequences of alternative TKIs are seen as clinically feasible and what clinical effectiveness (and treatment duration, and dose intensity) would be for some combinations (especially for dasatinib after nilotinib or nilotinib after dasatinib).
-
Uncertainty in evidence regarding treatments that would be received post TKI failure in CP: proportion getting SCT; also, using hydroxycarbamide as proxy for what in reality would be a range of treatments that might be offered (e.g. IFN and other chemotherapies).
-
Considerable uncertainty in survival and treatment costs either following SCT or with hydroxycarbamide.
-
Very limited sources of evidence for utility weights, and none available for post-TKI failure in CP. Also, no valid and reliable studies were available to reflect possible HRQoL decrement of being on TKIs but not responding to them. Single source for AP and BC based on very small numbers (n = 8 and 15).
-
The types and cost of care in AP and BC phases was uncertain. We may have underestimated these, but discounting, and the fact that we predict similar durations in these states across treatment arms, mean that this probably has only a minor impact on the ICERs. Also, with the widespread use of TKIs, the AP may in effect not exist for many patients now. Further, more effective treatment regimes in AP or BC may allow second or third CPs, or create sufficient recovery for SCT to be reconsidered. Our model does not capture these various treatment possibilities within advanced-phase CML.
-
An important assumption of the cumulative survival method is that OS after second-line nilotinib and OS after hydroxycarbamide or SCT are independent of previous treatment. There is very little research evidence to assess whether or not this assumption is plausible.
-
For the surrogate survival method, we consider only the proportion of patients with a response at 12 months. We do not consider the depth, speed of achieving, and duration of a MMR or CCyR. Given that dasatinib and nilotinib are superior to imatinib in all these respects (see Novartis report), and given that the historical surrogate data are based on OS for patients taking imatinib, it is likely that we underestimate OS for dasatinib and nilotinib. We also assume that, for a given response rate, OS is independent of treatment arm.
-
There is considerable current interest in being able to stop treatment, or reduce dose, in patients who respond very well to treatment and this might be where the benefit of the newer TKIs might be eventually demonstrated. 48 However, it is impossible to incorporate these ideas into the model without much more follow-up from the RCTs of dasatinib and nilotinib.
-
We have chosen not to conduct and present PSAs because of the unusually large amount of structural uncertainty that is inherent in the present decision problem(s). This structural uncertainty relates to both the variety of ways in which long-term survival might be estimated and uncertainty surrounding the possible sequences and mixes of treatments post first-line TKI failure. As a result, we believe that structural uncertainty would dominate total (structural and parameter) uncertainty, and therefore that if we presented PSAs based just on parameter uncertainty, this would be of little use to the committee. Furthermore, it might actually mislead users of our report who do not appreciate the substantial structural uncertainty.
-
Theoretically, it would have been possible to incorporate some of the structural uncertainty in to a PSA by some kind of model averaging. For example, we present scenario analyses with and without second-line nilotinib. To incorporate the uncertainty in whether or not we assume use of second-line nilotinib, we could have assigned a probability to the use of second-line nilotinib, and presented just one analysis. However, we believe that it would be more helpful to the committee to present the two analyses separately, thus allowing the committee to decide for themselves which scenario they prefer, i.e. allowing them to use their expert judgement to estimate the probability of second-line nilotinib use for themselves.
Chapter 9 Conclusions
Implications
From the two trials available, both the second-generation TKIs dasatinib and nilotinib have a statistically significant advantage compared with the first-generation TKI imatinib 400 mg as measured by surrogate outcomes. However, there are insufficient data to assess longer-term, patient-relevant outcomes (e.g. PFS, OS, HRQoL). All three drugs were well tolerated with discontinuation due to AEs of < 10%.
With no head-to-head data available, an indirect comparison analysis showed no difference between dasatinib and nilotinib for the primary outcomes of CCyR or MMR at 12 months’ or 24 months’ follow-up.
Based entirely on imatinib treatment, there is observational association evidence supporting the use of CCyR and MMR at 12 months as surrogates for OS and PFS in patients with CP-CML. In the absence of evidence of adequacy of these surrogates for dasatinib and nilotinib, and assuming a TKI class-specific relationship between the surrogate outcomes and the patient-relevant outcomes, these results can be potentially applied to other drugs in the same class.
Taking into account the treatment pathways for patients with CML, i.e. assuming the use of second-line nilotinib, first-line nilotinib appears to be more cost-effective than first-line imatinib for most scenarios. Dasatinib was not cost-effective if decision thresholds of £20,000 per QALY or £30,000 per QALY are used, compared with imatinib and nilotinib.
Suggested research priorities
-
Given the immature stage of trials assessing dasatinib or nilotinib compared with imatinib, longer-term follow-up data are required and will be available from the ongoing and currently recruiting trials. As well as the prespecified clinical outcomes (such as CCyR, MMR and survival) these should report both treatment duration and dose-intensity information for those treated if they are to be useful in estimating the long-term cost-effectiveness of the treatments.
-
With no current head-to-head data for dasatinib and nilotinib, a RCT assessing the two therapies directly or with an additional imatinib arm would be valuable.
-
More research-based data for assessing the predictive usefulness of surrogate outcomes (such as MMR and CCyR) within the CML population, especially for dasatinib and nilotinib.
-
Uncertainty in the cost-effectiveness analysis would be substantially reduced with better and more UK-specific data on the incidence and cost of SCT in patients with chronic CML.
-
Data on HRQoL for people in all stages of CML, and when on different treatments, are lacking. Studies should ideally use the EQ-5D or SF-36 generic HRQoL measures in order to allow social preference weights for the different states to be estimated.
-
Research to reflect the whole sequence of CML treatment, as opposed to ‘cross-sectionally’ at each line of treatment.
Acknowledgements
We would particularly like to thank the Expert Advisory Group for their help throughout the project. We would also like to thank Kevin Marsh (Chief Economist, Matrix Evidence) for providing overall direction for the critique of manufacturers’ submissions; Alex Smith (Research Fellow, Epidemiology and Genetics Unit, Department of Health Sciences, University of York) for data related to the Haematological Malignancy Research Network; Jessica Whitton (Senior Commissioning Manager, South West Specialised Commissioning Group, Bristol) for information related to SCT; Mike Millen (Senior Commissioning Manager, London Specialised Commissioning Group) for information related to the costing of SCT; Professor Rod Taylor (PenTAG, University of Exeter) for his guidance related to surrogate outcomes; Daniel Chalk (Associate Research Fellow, PenCHORD, University of Exeter) and Michael Allen (Senior Modeller, PenCHORD, University of Exeter) for checking the PenTAG economic model; and Jamie Peters for the WinBUGS analysis and methods. We would like to acknowledge the administrative support provided throughout the project by Sue Whiffin and Jenny Lowe.
Expert Advisory Group
Dr Claudius Rudin, Department of Haematology, Royal Devon and Exeter Hospital, Exeter, UK.
Competing interests: none.
Dr Jenny Byrne, Faculty of Medicine and Health Sciences, Nottingham City Hospital, Nottingham, UK.
Competing interests: none
Professor Stephen O’Brien, Northern Institute for Cancer Research, Newcastle University Medical School, Newcastle, UK.
Competing interests: Received fees for consulting, support for attendance at scientific meetings and/or honoraria for lectures from the following companies: Novartis, BMS, Pfizer (Wyeth), MSD, Chemgenex. No employment is held with these companies. Received research funding, mainly for clinical trial work, from the following companies: Novartis, Pfizer, BMS, Roche and Ariad.
Professor Jane Apperley, Chairperson of the Department of Haematology, Imperial College, London, UK.
Competing interests: Department has received research support from both Novartis and BMS. Participation in advisory boards for both Novartis and BMS. Speaking at satellite symposia for both Novartis and BMS.
Contribution of authors
-
Toby Pavey: Provided overall project management and led the systematic review of clinical effectiveness, including assessment of all abstracts and titles for possible inclusion. Also contributed to the review of surrogate outcomes, researched and justified model parameters, and drafted and/or edited all sections of the report.
-
Martin Hoyle: Led the design, development and execution of the economic model and wrote most of the sections on the design and results of the economic model, helped critique the two industry economic models and contributed to the review of surrogate outcomes.
-
Oriana Ciani: Assessed abstracts and titles for inclusion, led the systematic review of surrogate outcomes and contributed to the writing and editing of the report.
-
Louise Crathorne: Assessed abstracts and titles for inclusion, led the systematic review of cost-effectiveness and contributed to the writing and editing of the report.
-
Tracey Jones-Hughes: Assessed abstracts and titles for inclusion, researched model parameters and contributed to the writing and editing of the report.
-
Chris Cooper: Designed and carried out literature searches for the systematic reviews and identification of model parameters, and contributed to the writing and editing of the report.
-
Leeza Osipenko: Provided overall project management, critiqued the manufacturers’ economic models and contributed to the preparation of the report.
-
Meena Venkatachalam: Critiqued the economic analysis provided by the manufacturers and contributed to writing the report.
-
Claudius Rudin: Provided clinical input into the design of the model, advised on clinical matters and contributed to the editing of the report.
-
Obi Ukoumunne: Conducted indirect comparison analyses and wrote the accompanying section.
-
Ruth Garside: Contributed to the design of the assessment, the clinical effectiveness review and the surrogate outcomes review, and contributed to the writing and editing of the report.
-
Rob Anderson: Contributed to the systematic review of cost-effectiveness, contributed to the design of the model, helped critique the two industry economic models, contributed to the writing and editing of the report and was overall Director of the project and Guarantor of the report.
About the Peninsula Technology Assessment Group
PenTAG is part of the Institute of Health Service Research at the Peninsula College of Medicine and Dentistry. PenTAG was established in 2000 and currently has two major work streams: independent health technology assessments (HTAs) for NICE and the NIHR HTA programme, and evidence synthesis work in relation to the needs of the SW Peninsula Collaboration for Applied Health Research and Care (PenCLAHRC), as well as for other local and national decision-makers.
The group is multidisciplinary and draws on individuals’ backgrounds in public health, health services research, computing and decision analysis, systematic reviewing, statistics and health economics. The Peninsula College of Medicine and Dentistry is a school within the Universities of Plymouth and Exeter. The Institute of Health Research is made up of discrete methodologically related research groups, among which HTA is a strong and recurring theme.
Recent HTA projects include:
-
Bendamustine for the first-line treatment of chronic lymphocytic leukaemia (Binet stage B or C) in patients for whom fludarabine combination chemotherapy is not appropriate: a critique of the submission from Napp.
-
The effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer’s disease (review of TA111): a systematic review and economic model.
-
Ofatumumab (Arzerra®) for the treatment of chronic lymphocytic leukaemia in patients who are refractory to fludarabine and alemtuzumab: a critique of the submission from GSK.
-
Everolimus for the second-line treatment of advanced and/or metastatic renal cell carcinoma.
-
The clinical effectiveness and cost-effectiveness of sunitinib for the treatment of gastrointestinal (GI) stromal tumours: a critique of the submission from Pfizer.
-
The clinical- and cost-effectiveness of lenalidomide for multiple myeloma in people who have received at least one prior therapy: an evidence review of the submission from Celgene.
-
Bevacizumab, sorafenib tosylate, sunitinib and temsirolimus for renal cell carcinoma: a systematic review and economic model.
-
Machine perfusion systems and cold static storage of kidneys from deceased donors.
For more information about PenTAG and our other or previous work, please visit: www.sites.pcmd.ac.uk/pentag
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Baccarani M, Dreyling M. and on behalf of the ESMO Guidelines Working Group . Chronic myeloid leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010;21:v165-7.
- Nowell PC. A minute chromosome in human chronic granulocytic leukemia. Science 1960;132.
- Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973;243:290-3.
- Deininger MWN, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood 2000;96:3343-56.
- Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008;112:4808-17.
- O’Brien S, Berman E, Borghaei H, DeAngelo DJ, Devetten MP, Devine S, et al. Chronic myelogenous leukemia. J Natl Compr Canc Netw 2009;7:984-1023.
- Goldman JM. Chronic myeloid leukaemia. Medicine 2009;37:195-7.
- Goldman JM. British Committee for Standards in Haematology Recommendations for the Management of BCR-ABL-Positive Chronic Myeloid Leukaemia 2007. www.bcshguidelines.com/pdf/CML_guidelines_270707.pdf (accessed 23 June 2011).
- Seong D, Kantarjian H, Ro J, Talpaz M, Xu J, Robinson J, et al. Hypermetaphase fluorescence in situ hybridization for quantitative monitoring of Philadelphia chromosome-positive cells in patients with chronic myelogenous leukemia during treatment. Blood 1995;86:2343-9.
- Yanagi M, Shinjo K, Takeshita A, Tobita T, Yano K, Kobayashi M, et al. Simple and reliably sensitive diagnosis and monitoring of Philadelphia chromosome-positive cells in chronic myeloid leukemia by interphase fluorescence in situ hybridization of peripheral blood cells. Leukemia 1999;13:542-52.
- Gale RP, Hehlmann R, Zhang M-J, Hasford J, Goldman JM, Heimpel H, et al. Survival with bone marrow transplantation versus hydroxyurea or interferon for chronic myelogenous leukemia. Blood 1998;91:1810-9.
- O’Dwyer ME, Mauro MJ, Blasdel C, Farnsworth M, Kurilik G, Hsieh Y-C, et al. Clonal evolution and lack of cytogenetic response are adverse prognostic factors for hematologic relapse of chronic phase CML patients treated with imatinib mesylate. Blood 2004;103:451-5.
- Calabretta B, Perrotti D. The biology of CML blast crisis. Blood 2004;103:4010-22.
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med 1999;340:1330-40.
- Heaney N, Holyoake T. Therapeutic targets in chronic myeloid leukaemia. Hematol Oncol 2007;25:66-75.
- Jaffe E, Harris NL, Stein H, Vardiman JW. WHO classification. Tumours of haematopoietic and lymphoid tissues. Lyon: IARC; 2001.
- Swerdlow S, Campo E, Harris NL, Jaffe E, Pileri S, Stein H, et al. WHO classification. Tumours of haematopoietic and lymphoid tissues. Lyon: IARC; 2008.
- Cortes JE, Jones D, O’Brien S, Jabbour E, Konopleva M, Ferrajoli A, et al. Nilotinib as front-line treatment for patients with chronic myeloid leukemia in early chronic phase. J Clin Oncol 2010;28:392-7.
- Cortes JE, Jones D, O’Brien S, Jabbour E, Ravandi F, Koller C, et al. Results of dasatinib therapy in patients with early chronic-phase chronic myeloid leukemia. J Clin Oncol 2010;28:398-404.
- Saglio G, Kim D-W, Issaragrisil S, le Coutre P, Etienne G, Lobo C, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 2010;362:2251-9.
- Rosti G, Palandri F, Castagnetti F, Breccia M, Levato L, Gugliotta G, et al. Nilotinib for the frontline treatment of Ph(+) chronic myeloid leukemia. Blood 2009;114:4933-8.
- Haematological Malignancy Research Network . Chronic Myelogenous Leukaemia n.d. www.hmrn.org/Statistics/Cancer_Info.aspx?id=1 (accessed 2 March 2011).
- Sokal JE, Baccarani M, Russo D, Tura S. Staging and prognosis in chronic myelogenous leukemia. Semin Hematol 1988;25:49-61.
- Hasford J, Pfirrmann M, Hehlmann R, Allan NC, Baccarani M, Kluin-Nelemans JC, et al. A new prognostic score for survival of patients with chronic myeloid leukemia treated with interferon AlfaWriting Committee for the Collaborative CML Prognostic Factors Project Group. J Natl Cancer Inst 1998;90:850-9.
- Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2006;108:1809-20.
- O’Brien SG, Guilhot Fo, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003;348:994-1004.
- Druker BJ, Guilhot Fo, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006;355:2408-17.
- Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med 2003;349:1423-32.
- Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2010;362:2260-70.
- Rachet B, Maringe C, Nur U, Quaresma M, Shah A, Woods LM, et al. Population-based cancer survival trends in England and Wales up to 2007: an assessment of the NHS cancer plan for England. Lancet Oncol 2009;10:351-69.
- Surveillance Epidemiology and End Results . Cancer Statistics Review 1975–2007 n.d. seer.cancer.gov/csr/1975_2007/browse_csr.php?section=13%26page=sect_13_table.16.html (accessed 3 March 2011).
- Hochhaus A, Kantarjian HM, Baccarani M, Lipton JH, Apperley JF, Druker BJ, et al. Dasatinib induces notable hematologic and cytogenetic responses in chronic-phase chronic myeloid leukemia after failure of imatinib therapy. Blood 2007;109:2303-9.
- Cortes J, Rousselot P, Kim D-W, Ritchie E, Hamerschlak N, Coutre S, et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood 2007;109:3207-13.
- Lassere MN, Johnson KR, Boers M, Tugwell P, Brooks P, Simon L, et al. Definitions and validation criteria for biomarkers and surrogate endpoints: development and testing of a quantitative hierarchical levels of evidence schema. J Rheumatol 2007;34:607-15.
- Elston J, Taylor RS. Use of surrogate outcomes in cost-effectiveness models: A review of United Kingdom health technology assessment reports. Int J Technol Assess Health Care 2009;25:6-13.
- Bucher HC, Guyatt GH, Cook DJ, Holbrook A, McAlister FA. for the Evidence-Based Medicine Working Group . Users’ Guides to the Medical Literature. JAMA 1999;282:771-8.
- Schrover RJ, Adena MA, De Abreu Lourenco R, Miles Prince H, Seymour JF, Wonder MJ. Development of a predictive population survival model according to the cytogenetic response rate for patients with chronic myeloid leukemia in the chronic phase. Leuk Lymphoma 2006;47:1069-81.
- Goldman JM, Apperley JF, Jones L, Marcus R, Goolden AWG, Batchelor R, et al. Bone marrow transplantation for patients with chronic myeloid leukemia. N Engl J Medicine 1986;314:202-7.
- Clift R, Buckner C, Thomas E, Bryant E, Anasetti C, Bensinger W, et al. Marrow transplantation for patients in accelerated phase of chronic myeloid leukemia. Blood 1994;84:4368-73.
- Goldman J, Szydlo R, Horowitz M, Gale R, Ash R, Atkinson K, et al. Choice of pretransplant treatment and timing of transplants for chronic myelogenous leukemia in chronic phase. Blood 1993;82:2235-8.
- Davies SM, DeFor TE, McGlave PB, Miller JS, Verfaillie CM, Wagner JE, et al. Equivalent outcomes in patients with chronic myelogenous leukemia after early transplantation of phenotypically matched bone marrow from related or unrelated donors. Am J Med 2001;110:339-46.
- Hansen JA, Gooley TA, Martin PJ, Appelbaum F, Chauncey TR, Clift RA, et al. Bone marrow transplants from unrelated donors for patients with chronic myeloid leukemia. N Engl J Med 1998;338:962-8.
- Silver RT, Woolf SH, Hehlmann R, Appelbaum FR, Anderson J, Bennett C, et al. An evidence-based analysis of the effect of busulfan, hydroxyurea, interferon, and allogeneic bone marrow transplantation in treating the chronic phase of chronic myeloid leukemia: developed for the American Society of Hematology. Blood 1999;94:1517-36.
- European Medicines Agency (EMA) . EPARS for Authorised Medicinal Products for Human Use 2009. www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000406/human_med_000808.jsp%26murl=menus/medicines/medicines.jsp%26mid=WC0b01ac058001d125 (accessed 1 March 2011).
- MIMS . Drug Detail Glivec 2011. www.mims.co.uk/Drugs/cancer/antineoplastics/glivec/ (accessed 13 July 2011).
- Deininger M, O’Brien SG, Guilhot F, Goldman JM, Hochhaus A, Hughes TP, et al. International randomized study of interferon Vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. ASH Annual Meeting Abstracts 2009;114.
- Marin D, Khorashad JS, Foroni L, Milojkovic D, Szydlo R, Reid AG, et al. Does a rise in the BCR-ABL1 transcript level identify chronic phase CML patients responding to imatinib who have a high risk of cytogenetic relapse?. Br J Haematol 2009;145:373-5.
- Mahon F-X, Réa D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010;11:1029-35.
- Kantarjian H, O’Brien S, Jabbour E, Shan J, Ravandi F, Kadia T, et al. Impact of treatment end point definitions on perceived differences in long-term outcome with tyrosine kinase inhibitor therapy in chronic myeloid leukemia. J Clin Oncol 2011;29:3173-8.
- Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, et al. Chronic Myeloid Leukemia: An Update of Concepts and Management Recommendations of European LeukemiaNet. J Clin Oncol 2009;27:6041-51.
- Tokarski JS, Newitt JA, Chang CYJ, Cheng JD, Wittekind M, Kiefer SE, et al. The structure of dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res 2006;66:5790-7.
- Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004;305:399-401.
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-chloro-6-methyl- phenyl)–2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem 2004;47:6658-61.
- European Medicines Agency (EMA) . EPARS for Authorised Medicinal Products for Human Use 2010. www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000709/human_med_001062.jsp%26murl=menus/medicines/medicines.jsp%26jsenabled=true (accessed 1 March 2011).
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary n.d. bnf.org/bnf/bnf/current/129968.htm?q=dasatinib%26t=search%26ss=text%26p=3#_129968 (accessed 2 March 2011).
- Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant BCR-ABL. Cancer Cell 2005;7:129-41.
- O’Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan JR, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res 2005;65:4500-5.
- Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD. AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br J Cancer 2006;94:1765-9.
- European Medicines Agency (EMA) . EPARS for Authorised Medicinal Products for Human Use 2011. www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000798/human_med_001079.jsp%26murl=menus/medicines/medicines.jsp%26jsenabled=true (accessed 1 March 2011).
- US Food and Drug Administration (FDA) . Prescribing Information Tasigna (nilotinib) 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/022068s004s005lbl.pdf (accessed 17 June 2011).
- Efficace F, Cocks K, Breccia M, Sprangers M, Meyers CA, Vignetti M, et al. Time for a new era in the evaluation of targeted therapies for patients with chronic myeloid leukemia: Inclusion of quality of life and other patient-reported outcomes. Crit Rev Oncol Hematol 2011;81:123-35.
- Hahn EA, Glendenning GA, Sorensen MV, Hudgens SA, Druker BJ, Guilhot F, et al. quality of life in patients with newly diagnosed chronic phase chronic myeloid leukemia on imatinib versus interferon alfa plus low-dose cytarabine: results from the IRIS Study. J Clin Oncol 2003;21:2138-46.
- Moher D, Liberati A, Tetzlaff J, Altman DG. The PRISMA Group . Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol 2009;62:1006-12.
- NHS Centre for Reviews and Dissemination (CRD) . Systematic Reviews: CRD’s Guidance for Undertaking Reviews in Health Care 2009.
- Dalziel K, Round A, Stein K, Garside R, Price A. Effectiveness and cost-effectiveness of imatinib for first-line treatment of chronic myeloid leukaemia in chronic phase: a systematic review and economic analysis. Health Technol Assess 2004;8.
- Thompson-Coon J, Hoyle M, Pitt M, Rogers G, Moxham T, Liu Z, et al. Dasatinib and nilotinib for imatinib-resistant or -intolerant chronic myeloid leukaemia: a systematic review and economic evaluation. Health Technol Assess 2012;16.
- Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol 1997;50:683-91.
- Radich JP, Kopecky KJ, Kamel-Reid S, Stock W, Paietta E, Wadleigh M, et al. A randomized phase II trial of dasatinib 100 Mg vs imatinib 400 Mg in newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): The S0325 Intergroup Trial. Blood 2010;116:LBA-6.
- Saglio G, Hochhaus A, Cortes JE, Kantarjian H, Baccarani M, Bradley-Garelik MB, et al. Safety and Efficacy of Dasatinib Versus Imatinib by Baseline Cardiovascular Comorbidity In Patients with Chronic Myeloid Leukemia In Chronic Phase (CML-CP): Analysis of the DASISION Trial. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Guilhot F, Kantarjian H, Shah NP, Hochhaus A, Bradley-Garelik MB, Dejardin D, et al. Dasatinib (Versus Imatinib) In Patients (Pts) with Newly Diagnosed Chronic Myeloid Leukemia In Chronic Phase (CML-CP): Analysis of Safety and Efficacy by Use of Baseline Medications In the DASISION Trial. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Schiffer CA, Cortes JE, Saglio G, Coutre Pl, Guilhot F, Bahceci E, et al. Lymphocytosis Following First-Line Treatment for CML In Chronic Phase with Dasatinib Is Associated with Improved Responses: A Comparison with Imatinib. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Khoury HJ, Cortes JE, Kantarjian H, Baccarani M, Shah NP, Bradley-Garelik MB, et al. Safety and Efficacy of Dasatinib (DAS) Vs. Imatinib (IM) by Baseline Comorbidity In Patients with Chronic Myeloid Leukemia In Chronic Phase (CML-CP): Analysis of the DASISION Trial. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Shah N, Kantarjian H, Hochhaus A, Cortes JE, Bradley-Garelik MB, Zhu C, et al. Dasatinib Versus Imatinib In Patients with Newly Diagnosed Chronic Myeloid Leukemia In Chronic Phase (CML-CP) In the DASISION Trial: 18-Month Follow-up. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Kantarjian H, Shah NP, Cortes JE, Baccarani M, Bradley-Garelik MB, Zhu C, et al. Dasatinib or imatinib (IM) in newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): two-year follow-up from DASISION. ASCO Meeting Abstracts n.d.;29.
- Kantarjian H, Shah N, Cortes J, Baccarani M, Bradley-Garelik M, Zhu C, et al. Dasatinib or Imatinib (IM) in Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP): Two-Year Follow-up from DASISION n.d.
- Beaumont JL, Coombs J, Bollu A, Woodman RC, Cella D. Hospitalizations of patients with newly diagnosed CML-CP treated with nilotinib or imatinib: results from a randomized phase III trial. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Hochhaus A, Lobo C, Pasquini R, Clark R, Kim DW, Issaragrisil S, et al. Haematologica 2010.
- Larson RA, Hochhaus A, Saglio G, Rosti G, Lopez JL, Stenke L, et al. Cardiac safety profile of imatinib and nilotinib in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): results From ENESTnd. Blood 2010;116.
- Hughes TP, Hochhaus A, Saglio G, Kim D-W, Jootar S, le Coutre PD, et al. ENESTnd Update: continued superiority of nilotinib versus imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP). Blood (ASH Annual Meeting Abstracts) 2010;116.
- Hughes TP, Hochhaus A, Saglio G, Kim D-W, Jooter S, le Coutre PD, et al. ENESTnd 24-Month Update: Continued Superiority of Nilotinib Versus Imatinib in Patients With Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP) n.d.
- Kantarjian HM, Hochhaus A, Saglio G, Souza CD, Flinn IW, Stenke L, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol 2011;12:841-51.
- Mealing S, Barcena L, Hawkins N, Clark J, Davis C. Comparative efficacy of first line treatment of chronic myeloid leukaemia (CML): a systematic review and meta-analysis. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Signorovitch JE, Wu EQ, Betts KA, Parikh K, Kantor E, Guo A, et al. Comparative efficacy of nilotinib and dasatinib in newly diagnosed chronic myeloid leukemia: a matching-adjusted indirect comparison of randomized trials. Curr Med Res Opin n.d.:1263-71.
- Bristol-Myers Squibb . Dasatinib, Nilotinib and Standard-Dose Imatinib for the First-Line Treatment of Chronic Myeloid Leukaemia (CML) 2011.
- Novartis . Dasatinib, Nilotinib and Standard-Dose Imatinib for the First-Line Treatment of Chronic Myeloid Leukaemia (CML) 2011.
- Brixey AG, Light RW. Pleural effusions due to dasatinib. Curr Opin Pulm Med:351-6.
- Shah N, Kantarjian H, Hochhaus A, Cortes JE, Bradley-Garelik MB, Zhu C, et al. Dasatinib versus imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) in the DASISION trial: 18-month follow-up. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Larson RA, Kim D, Rosti G, Stenke L, Pasquini R, Hoenekopp A, et al. Comparison of nilotinib and imatinib in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): ENESTnd 24-month follow-up n.d.
- Anstrom KJ, Reed SD, Allen AS, Glendenning GA, Schulman KA. Long-term survival estimates for imatinib versus interferon-alpha plus low-dose cytarabine for patients with newly diagnosed chronic-phase chronic myeloid leukemia. Cancer 2004;101:2584-92.
- Lassere MN. The Biomarker-Surrogacy Evaluation Schema: a review of the biomarker-surrogate literature and a proposal for a criterion-based, quantitative, multidimensional hierarchical levels of evidence schema for evaluating the status of biomarkers as surrogate endpoints. Stat Methods Med Res 2008;17:303-40.
- Anderson JR, Cain KC, Gelber RD. Analysis of survival by tumor response and other comparisons of time-to-event by outcome variables. J Clin Oncol 2008;26:3913-15.
- Wilson EB. Probable Inference, the Law of Succession, and Statistical Inference. J Am Stat Assoc 1927;22.
- Braziel RM, Launder TM, Druker BJ, Olson SB, Magenis RE, Mauro MJ, et al. Hematopathologic and cytogenetic findings in imatinib mesylate-treated chronic myelogenous leukemia patients: 14 months’ experience. Blood 2002;100:435-41.
- Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 2002;346:645-52.
- Muller MC. Molecular response to first line imatinib therapy is predictive for long term event free survival in patients with chronic phase chronic myelogenous leukemia – an interim analysis of the randomized German CML Study IV. Blood (ASH Annual Meeting Abstracts) 2008.
- Rajappa S, Varadpande L, Paul T, Jacob R, Digumarti R. Imatinib mesylate in early chronic phase chronic myeloid leukemia: experience from a developing country. Leuk Lymphoma 2008;49:554-8.
- de Lavallade H, Apperley JF, Khorashad JS, Milojkovic D, Reid AG, Bua M, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: incidence of sustained responses in an intention-to-treat analysis. J Clin Oncol 2008;26:3358-63.
- Hehlmann R, Lauseker M, Jung-Munkwitz S, Leitner A, Müller MC, Pletsch N, et al. Tolerability-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon-α in newly diagnosed chronic myeloid leukemia. J Clin Oncol n.d.;29:1634-42.
- Hochhaus A, O’Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009;23:1054-61.
- Hughes TP, Hochhaus A, Branford S, Müller MC, Kaeda JS, Foroni L, et al. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: An analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood 2010;116:3758-65.
- Kantarjian HM, Talpaz M, O’Brien S, Jones D, Giles F, Garcia-Manero G, et al. Survival benefit with imatinib mesylate versus interferon-alpha-based regimens in newly diagnosed chronic-phase chronic myelogenous leukemia. Blood 2006;108:1835-40.
- Kantarjian H, O’Brien S, Shan J, Huang X, Garcia-Manero G, Faderl S, et al. Cytogenetic and molecular responses and outcome in chronic myelogenous leukemia: need for new response definitions?. Cancer 2008;112:837-45.
- Marin D, Milojkovic D, Olavarria E, Khorashad JS, de Lavallade H, Reid AG, et al. European LeukemiaNet criteria for failure or suboptimal response reliably identify patients with CML in early chronic phase treated with imatinib whose eventual outcome is poor. Blood 2008;112:4437-44.
- Roy L, Guilhot J, Krahnke T, Guerci-Bresler A, Druker BJ, Larson RA, et al. Survival advantage from imatinib compared with the combination interferon-alpha plus cytarabine in chronic-phase chronic myelogenous leukemia: historical comparison between two phase 3 trials. Blood 2006;108:1478-84.
- Hehlmann R, Lauseker M, Jung-Munkwitz S, Leitner A, Muller MC, Pletsch N, et al. Tolerability-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon in newly diagnosed chronic myeloid leukemia. J Clin Oncol 2011;29:1634-42.
- Elston J, Taylor RS. Use of surrogate outcomes in cost-effectiveness models: a review of United Kingdom health technology assessment reports. Int J Technol Assess Health Care 2009;25:6-13.
- Simons WR, Maclean R, Cairns J. Comparative cost for required monitoring to detect adverse events or laboratory abnormalities: dasatinib versus nilotinib. Blood (ASH Annual Meeting Abstracts) 2009.
- Wu EQ, Guerin A, Bollu V, Williams D, Guo A, Barido DPL, et al. The economic Burden of pleural effusions (PE) in patients with chronic myeloid leukemia (CML). Blood (ASH Annual Meeting Abstracts) 2010.
- Szabo SM, Davis C, Sidhu S, Levy AR. Characterizing resource use and treatment costs for chronic myelogenous leukaemia in the UK. Value Health 2007;12.
- Taylor M, Lewis L, Patton T, Hirji I, Davis C. A disease model to predict the long-term outcomes associated with treatments for chronic myelogenous leukemia (CML). Blood (ASH Annual Meeting Abstracts) 2010.
- Ovanfors A, Stephens J, Snedecor SJ, Patel D, Ji X, Carpiuc KT, et al. Cost-effectiveness of nilotinib versus imatinib as first-line treatment for newly diagnosed patients with Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia in the chronic phase (CML-CP): Swedish perspective. ASCO Meeting Abstracts 2011;29.
- The Government Actuary’s Department (GAD) . Life Tables n.d. www.gad.gov.uk/Demography%20Data/life%20Tables/index.html (accessed 29 May 2011).
- O’Brien SG, Guilhot F, Goldman JM, Hochhaus A, Hughes TP, Radich JP, et al. International randomized study of interferon versus STI571 (IRIS) 7-year follow-up: sustained survival, low rate of transformation and increased rate of major molecular response (MMR) in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CMLCP) treated with imatinib (IM). ASH Annual Meeting Abstracts 2008;112.
- Novartis . Nilotinib for the Treatment of Adults With Chronic Myeloid Leukaemia in the Chronic Phase or Accelerated Phase With Resistance or Intolerance As Well As Resistance to Prior Therapy, Including Imatinib. Industry Submission (unpublished) by Novartis to NICE for Technology Appraisal of Nilotinib and Dasatinib for Second-Line Treatment of CML n.d.
- Kantarjian H, O’Brien S, Talpaz M, Borthakur G, Ravandi F, Faderl S, et al. Outcome of patients with Philadelphia chromosome-positive chronic myelogenous leukemia post-imatinib mesylate failure. Cancer 2007:109-60.
- Reed SD, Anstrom KJ, Ludmer JA, Glendenning GA, Schulman KA. Cost-effectiveness of imatinib versus interferon-α plus low-dose cytarabine for patients with newly diagnosed chronic-phase chronic myeloid leukemia. Cancer 2004;101:2574-83.
- Kantarjian H, Cortes J. BCR-ABL tyrosine kinase inhibitors in chronic myeloid leukemia: using guidelines to make rational treatment choices. J Natl Compr Canc Netw 2008;6:37-42.
- Cervantes F, López-Guillermo A, Bosch F, Terol M-J, Rozman C, Montserrat E. An assessment of the clinicohematological criteria for the accelerated phase of chronic myeloid leukemia. Eur J Haematol 1996;57:286-91.
- Gratwohl A, Stern M, Brand R, Apperley J, Baldomero H, de Witte T, et al. Risk score for outcome after allogeneic hematopoietic stem cell transplantation. Cancer 2009;115:4715-26.
- Lee SJ, Kukreja M, Wang T, Giralt SA, Szer J, Arora M, et al. Impact of prior imatinib mesylate on the outcome of hematopoietic cell transplantation for chronic myeloid leukemia. Blood 2008;112:3500-7.
- Saussele S, Lauseker M, Gratwohl A, Beelen DW, Bunjes D, Schwerdtfeger R, et al. Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood 2010;115:1880-5.
- Pavlu J, Szydlo RM, Goldman JM, Apperley JF. Three decades of transplantation for chronic myeloid leukemia: what have we learned?. Blood 2011;117:755-63.
- Gratwohl A, Brand R, Apperley J, Crawley C, Ruutu T, Corradini P, et al. Allogeneic hematopoietic stem cell transplantation for chronic myeloid leukemia in Europe 2006: transplant activity, long-term data and current results. An analysis by the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Haematologica 2006;91:513-21.
- Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood 2007;110:3540-6.
- Kantarjian HM, Giles F, Bhalla KN, Larson RA, Gattermann N, Ottmann OG, et al. Nilotinib in chronic myeloid leukemia patients in chronic phase (CMLCP) with imatinib resistance or intolerance: 2-year follow-up results of a Phase 2 study. ASH Annual Meeting Abstracts 2008;112.
- Kantarjian HM, Giles FJ, Bhalla KN, Pinilla-Ibarz J, Larson RA, Gattermann N, et al. Nilotinib is effective in patients with chronic myeloid leukemia in chronic phase after imatinib resistance or intolerance: 24-month follow-up results. Blood 2011;117:1141-5.
- Szabo SM, Levy AR, Davis C, Holyoake TL, Cortes J. A multinational study of health state preference values associated with chronic myelogenous leukemia. Value Health 2010;13:103-11.
- Lee SJ, Kuntz KM, Horowitz MM, McGlave PB, Goldman JM, Sobocinski KA, et al. Unrelated donor bone marrow transplantation for chronic myelogenous leukemia: a decision analysis. Ann Intern Med 1997;127:1080-8.
- National Institute for Health and Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2008.
- Dalziel K, Round A, Garside R, Stein K. Cost effectiveness of imatinib compared with interferon-alpha or hydroxycarbamide for first-line treatment of chronic myeloid leukaemia. Pharmacoeconomics 2005;23:515-26.
- Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health 2010;13:509-18.
- Cadigan P. Dasatinib, nilotinib and standard-dose imatinib for the first-line treatment of chronic myeloid leukaemia (including part-review of NICE technology appraisal guidance 70) Statement. Industry Submission. London: Royal College of Physicians; 2011.
- Slovacek L, Slovackova B, Jebavy L. Global quality of life in patients who have undergone the hematopoietic stem cell transplantation: finding from transversal and retrospective study. Exp Oncol 2005;27:238-42.
- Hendriks MGJ, Schouten HC. Quality of life after stem cell transplantation: a patient, partner and physician perspective. Eur J Intern Med 2002;13:52-6.
- Rudzki J, Wolf D. Dose escalation of imatinib in chronic-phase chronic myeloid leukemia patients: is it still reasonable?. Expert Rev Hematol 2011;4:153-9.
- Curtis L. Unit Costs of Health and Social Care. 2010 n.d. www.pssru.ac.uk/pdf/uc/uc2010/uc2010_s09.pdf (accessed 2 July 2011).
- Millen M, Phennah D. Adult BMT costing and pricing model workshop. London Specialised Commissioning Group; 2009.
- Ashfaq K, Yahaya I, Hyde C, Andronis L, Barton P, Bayliss S, et al. Clinical effectiveness and cost-effectiveness of stem cell transplantation in the management of acute leukaemia: a systematic review. Health Technol Assess 2010;14.
- US Food and Drug Administration (FDA) . Orange Book: Approved Drug Products With Therapeutic Equivalence Evaluations – Nilotinib 2011. www.accessdata.fda.gov/scripts/cder/ob/docs/patexclnew.cfm?Appl_No=022068%26Product_No=001%26table1=OB_Rx (accessed 5 July 2011).
- US Food and Drug Administration (FDA) . Orange Book: Approved Drug Products With Therapeutic Equivalence Evaluations – Dasatinib 2011. www.accessdata.fda.gov/scripts/cder/ob/docs/patexclnew.cfm?Appl_No=021986%26Product_No=001%26table1=OB_Rx (accessed 5 July 2011).
- Hoyle M. Future drug prices and cost-effectiveness analyses. Pharmacoeconomics 2008;26:589-602.
- Hoyle M, Anderson R. Whose costs and benefits? Why economic evaluations should simulate both prevalent and all future incident patient cohorts. Med Decis Making 2010;30:426-37.
- Hoyle M. Allowing for the drug life cycle and future drug prices in cost-effectiveness analysis. Pharmacoeconomics 2011;29:1-15.
- Thompson-Coon J, Hoyle M, Pitt M, Rogers G, Moxham T, Liu Z, et al. Dasatinib and nilotinib for imatinib-resistant or -intolerant chronic myeloid leukaemia: a systematic review and economic evaluation. Health Technol Assess 2012;16.
- Lefebvre C, Manheimer E, Glanville J, Higgins JTP, Green S. Cochrane handbook for systematic reviews on interventions. Oxford: The Cochrane Collaboration; 2009.
- National Institute for Health and Clinical Excellence (NICE) . Imatinib for Chronic Myeloid Leukaemia: Technology Appraisal 70 2003.
- NHS Centre for Reviews and Dissemination . Systematic Reviews: CRD’s Guidance for Undertaking Reviews in Health Care 2009.
- Evers S, Goossens M, de Vet H, van Tulder M, . Criteria list for assessment of methodological quality of economic evaluations: consensus on health economic criteria. Int J Technol Assess Health Care 2005;21:240-5.
- Weinstein MC, O’Brien B, Hornberger J, Jackson J, Johannesson M, McCabe C, et al. Principles of good practice for decision analytic modeling in health-care evaluation: Report of the ISPOR Task Force on Good Research Practices–Modeling Studies. Value Health 2003;6:9-17.
- National Institute for Health and Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2008.
- Saglio G, Hochhaus A, Cortes JE, Kantarjian H, Baccarani M, Bradley-Garelik MB, et al. Safety and efficacy of dasatinib versus imatinib by baseline cardiovascular comorbidity in patients with chronic myeloid leukemia in chronic phase (CML-CP): analysis of the DASISION trial. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Guilhot F, Kantarjian H, Shah NP, Hochhaus A, Bradley-Garelik MB, Dejardin D, et al. Dasatinib (versus imatinib) in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): analysis of safety and efficacy by use of baseline medications in the DASISION trial. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Schiffer CA, Cortes JE, Saglio G, Coutre Pl, Guilhot F, Bahceci E, . Lymphocytosis following first-line treatment for cml in chronic phase with dasatinib is associated with improved responses: a comparison with imatinib. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Khoury HJ, Cortes JE, Kantarjian H, Baccarani M, Shah NP, Bradley-Garelik MB, et al. Safety and efficacy of dasatinib (DAS) vs. imatinib (IM) by baseline comorbidity in patients with chronic myeloid leukemia in chronic phase (CML-CP): analysis of the DASISION Trial. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Shah N, Kantarjian H, Hochhaus A, Cortes JE, Bradley-Garelik MB, Zhu C, et al. Dasatinib versus imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) in the DASISION trial: 18-month follow-up. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Kantarjian H, Shah NP, Cortes JE, Baccarani M, Bradley-Garelik MB, Zhu C, et al. Dasatinib or imatinib (IM) in newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): two-year follow-up from DASISION. ASCO Meeting Abstracts 2011;29.
- Kantarjian H, Shah N, Cortes J, Baccarani M, Bradley-Garelik M, Zhu C, et al. Dasatinib or Imatinib (IM) in Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP): Two-Year Follow-up from DASISION n.d.
- Hochhaus A, Lobo C, Pasquini R, Clark R, Kim DW, Issaragrisil S, et al. Continued superiority of nilotinib vs imatinib in patients with newly diagnosed chronic myeloid leukaemia in chronic phase (CML-CP): ENESTnd beyond 1 Year. 15th Congress of the European Hematology Association, Barcelona Spain 2011. Haematologica n.d.
- Beaumont JL, Coombs J, Bollu a, Woodman RC, Cella D. Hospitalizations of patients with newly diagnosed CML-CP treated with nilotinib or imatinib: results from a randomized phase III trial. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Larson RA, Hochhaus A, Saglio G, Rosti G, Lopez JL, Stenke L, et al. Cardiac safety profile of imatinib and nilotinib in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): results From ENESTnd. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Hughes TP, Hochhaus A, Saglio G, Kim D-W, Jootar S, Coutre PDl, et al. ENESTnd update: continued superiority of nilotinib versus imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP). Blood (ASH Annual Meeting Abstracts) 2010;116.
- Hughes T, Hochhaus A, Saglio G, Kim DW, Jooter S, Le Coutre P, et al. ENESTnd 24-Month Update: Continued Superiority of Nilotinib Versus Imatinib in Patients With Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP) n.d.
- Larson RA, Kim D, Rosti G, Stenke L, Pasquini R, Hoenekopp A, et al. Comparison of Nilotinib and Imatinib in Patients (pts) With Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP): ENESTnd 24-Month Follow-Up. (Conference Presentation) n.d.
- de Lavallade H, Apperley JF, Khorashad JS, Milojkovic D, Reid AG, Bua M, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: Incidence of sustained responses in an intention-to-treat analysis. J Clin Oncol 2008;26:3358-63.
- Hehlmann R, Lauseker M, Jung-Munkwitz S, Leitner A, Muller MC, Pletsch N, et al. Tolerability-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon in newly Diagnosed chronic myeloid leukemia. J Clin Oncol 2011;29:1634-42.
- Rajappa S, Varadpande L, Paul T, Jacob R, Digumarti R. Imatinib mesylate in early chronic phase chronic myeloid leukemia: Experience from a developing country. Leuk Lymphoma 2008;49:554-8.
- O’Brien S, Hedgley C, Apperley J, Clark R, . SPIRIT 2 STI571 Prospective International Randomised Trial n.d. www.spirit-cml.org/spirit-2-home/about.aspx (accessed 21 July 2011).
- National Institute for Health and Clinical Excellence (NICE) . Guidance on the Use of Imatinib for Chronic Myeloid Leukaemia (TA70) 2003. guidance.nice.org.uk/TA70/Guidance/Recommendation (accessed 16 May 2011).
- Briggs A, Sculpher M, Claxton K. Decision modelling for health economic evaluation. New York: Oxford University Press; 2006.
- Botteman F, Stephens J, Coombs J. Projecting the long-term survival of newly diagnosed patients with chronic myeloid leukaemia (CML) in chronic phase (CP) receiving nilotinib or imatinib. 15th Congress of the European Hematology Association (EHA), Barcelona, 2010. Haematologica 2010;95.
- Oxford Outcomes Ltd . Estimation of the Resources Used for Treating Patients With Chronic Myelogenous Leukemia in the United Kingdom: Final Report 2009.
- Mealing S, Barcena L, Hawkins N, Clark J, Davis C. Comparative efficacy of first line treatment of chronic myeloid leukaemia (CML): a systematic review and meta-analysis. Blood (ASH Annual Meeting Abstracts) 2010;116.
- Braziel RM, Launder TM, Druker BJ, Olson SB, Magenis RE, Mauro MJ, et al. Hematopathologic and cytogenetic findings in imatinib mesylate treated chronic myelogenous leukemia patients: 14 months’ experience. Blood 2002;100:435-41.
- Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med 2002;346:645-52.
- Ghatnekar O, Hjalte F, Taylor M. Cost-effectiveness of dasatinib versus high-dose imatinib in patients with chronic myeloid leukemia (CML), resistant to standard dose imatinib a Swedish model application. Acta Oncologica 2010;49:851-8.
- Allan NC, Shepherd PCA, Richards SM. UK Medical Research Council randomised, multicentre trial of interferon-[alpha]n1 for chronic myeloid leukaemia: improved survival irrespective of cytogenetic response. Lancet 1995;345:1392-7.
- Liverpool Reviews and Implementation Group (LRiG) . Erlotinib for the Treatment of Relapsed Non-Small Cell Lung Cancer: Evidence Review Group Report 2006. www.nice.org.uk/nicemedia/live/11714/35177/35177.pdf (accessed 3 August 2011).
- Doyle S, Lloyd A, Walker M. Health state utility scores in advanced non-small cell lung cancer. Lung Cancer 2008;62:374-80.
- Tabberer M, Stamuli E, Walker M, Summerhayes M, Lees M. Utilities Associated With Non-Small Cell Lung Cancer (NSCLC): A Community Study. International Society for Pharmacoeconomics and Outcomes (ISPOR) 2006.
- Curtis L. Unit Costs of Health and Social Care 2008 n.d. www/pssru.ac.uk/uc/uc2008contents.htm (accessed 3 August 2011).
- Bristol-Myers Squibb . Dasatinib, High Dose Imatinib and Nilotinib for the Treatment of Imatinib-Resistant Chronic Myeloid Leukaemia n.d.
- Drummond MF, Jefferson TO. Guidelines for authors and peer reviewers of economic submissions to the BMJ. BMJ 1996;313:275-83.
- Philips Z, Bojke L, Sculpher M, Claxton K, Golder S. Good practice guidelines for decision-analytic modelling in health technology assessment: a review and consolidation of quality assessment. Pharmacoeconomics 2006;24:355-71.
- Shah NP, Kim D-W, Kantarjian H, Rousselot P, Llacer PED, Enrico A, et al. Potent, transient inhibition of BCR-ABL with dasatinib 100 mg daily achieves rapid and durable cytogenetic responses and high transformation-free survival rates in chronic phase chronic myeloid leukemia patients with resistance, suboptimal response or intolerance to imatinib. Haematologica 2010;95:232-40.
- Garg RJ, Kantarjian H, O’Brien S, Quintás-Cardama A, Faderl S, Estrov Z, et al. The use of nilotinib or dasatinib after failure to 2 prior tyrosine kinase inhibitors: long-term follow-up. Blood 2009;114:4361-8.
- Kantarjian H, O’Brien S, Talpaz M, Borthakur G, Ravandi F, Faderl S, et al. Outcome of patients with Philadelphia chromosome-positive chronic myelogenous leukemia post-imatinib mesylate failure. Oxford: Wiley Subscription Services; 2007.
- Lee SJ, Kuntz KM, Horowitz MM, McGlave PB, Goldman JM, Sobocinski KA, et al. Unrelated donor bone marrow transplantation for chronic myelogenous leukemia: a decision analysis. Ann Inter Med 1997;127:1080-8.
- Varney SJ, Guest JF. The annual cost of blood transfusions in the UK. Transfus Med 2003;13:205-18.
- Department of Health . NHS Reference Costs 2008–9 n.d. www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_111591 (accessed 15 July 2011).
- Millen M, Phennah D. Adult BMT costing and pricing model workshop. London Specialised Commissioning Group; 2009.
- Lu G, Ades AE. Assessing evidence inconsistency in mixed treatment comparisons. J Am Stat Assoc 2006;101:447-59.
- Spiegelhalter D, Thomas A, Best N, Lunn D. WinBUGS User Manual, Version 1.4. Cambridge: Medical Research Council Biostatistics Unit; 2003.
- Sutton AJ, Abrams KR, Jones DR, Sheldon TA, Song F. Methods for Meta-Analysis in Medical Research. Chichester: Wiley; 2000.
Appendix 1 Literature search strategy
The strategy: notes
The strategy was based upon the previous PenTAG review on this population and for this set of interventions. 144
All controlled syntax and population/intervention terminology have been double-checked for currency, or for any form of update, as well as the possibility of entirely new terms or themes existing for this population and set of interventions.
Four additional lines have been incorporated for this review. With reference to the MEDLINE strategy (by way of example), lines 3 and 15 were incorporated in testing and have been retained for the sake of completeness. For line 3, it is noted as unlikely that references would appear using only the acronym CML as an expression of the population without referring to, or defining first, CML, but it is a common point of reference within title and abstract of texts and so a viable inclusion to the strategy in view of sensitivity. Similarly, with line 15, this is another way of referring to the Philadelphia chromosome (as reflected in Emtree’s controlled syntax) and has been incorporated for the sake of sensitivity. 145
Lines 10 and 11 were incorporated at the advice of our local clinical expert, Dr Claudius Rudin (Department of Haematology, Royal Devon and Exeter Hospital, Exeter, UK), who critically appraised this strategy. The lines reflect concepts usually defined in reference to our population and interventions and so have been incorporated into the search both at his advice and for the sake of overall completeness. We are grateful to him for his time and advice on this stage of the assessment.
Syntax and limits: notes
Population
We have searched explicitly for chronic-stage myeloid leukaemia [via controlled syntax (line 4 of the MEDLINE strategy) and free text (line 3 of the MEDLINE strategy)], as well as more broadly, and therefore with more sensitivity, using the controlled syntax (where available) and free text for the broader, overarching population group, myeloid leukaemia. This is to compensate for any unlikely deficiencies in indexing or referencing to the chronic stage of the broader myeloid population. Accordingly, any ‘rogue’ references that are implicitly chronic stage but are not explicitly defined as such can be picked up in the literature via screening.
Intervention
The interventions have been operationalised using both their formal and informal naming as well as their numerical drug forms. Over the Ovid platform this has been done using multiple placing (.mp.) (title, original title, abstract, name of substance word, subject heading word) for the syntax lines expressing the intervention (drug) names, to ensure that all theoretical bases have been covered, as well as expressing the numerical form via free text. In EMBASE, the relevant controlled syntax (Emtree) for the drugs has also been incorporated.
Limits
The relative youth of the interventions in question means that there are comparatively few data in the field when compared with other interventions for this population (i.e. imatinib). Accordingly, we ran our searching without recourse to methodological filters (RCTs, etc.), which opens a broader field of results for this review (e.g. observational studies) as mentioned in the protocol.
Limits have been applied (where the databases have allowed) to exclude studies carried out on animals as well as to limit returns to the date parameters of this assessment (2002 to current) and to English-language studies.
Results
All results were exported from the databases into a bibliographic tool (RefWorks, RS-RW, Bethesda MD, USA) [except ISI proceedings and EMBASE, which were imported directly into EndNote X4 (Thomas Reuters, CA, USA)] to manage the results before the aggregate volume was de-duplicated using the internal tool in EndNote X4. The result was passed to the review team in Research Information Systems (RIS) format. Copies of the result, a file of duplicates that have been removed, and a file containing the library before duplication, as well as individual files of each database search, have been retained and held in RIS format.
Surrogate outcomes
As the screening developed, the possibility of requiring deeper literature on surrogate outcomes was raised. One outcome, MMR, had been introduced to the search by our expert but an alternate measure, complete cytogenic response, was not explicitly defined within the search syntax.
A search of this term (and the acronym CCyR) was conducted in MEDLINE using the same project interventions that retrieved 15 results. These results were cross-checked and de-duplicated against the main review library which confirmed that all 15 results had been captured in the original search.
Although confident that this result suggested we had captured all relevant literature on these outcomes project-wide, we nevertheless repeated the search across the portfolio of resources used for the initial search. Of the 308 references retrieved in this search, every single reference was found to have already been retrieved and was, therefore, a duplicate record. Although this search retrieved no unique references, it does seek to confirm that saturation of these terms had already been achieved in the first search. The terms themselves appear well embedded within the relevant literature for this review.
As the surrogate terms for dasatinib and nilotinib had already been captured in the clinical effectiveness review, an additional search used the intervention imatinib. The alternative comparator, IFN, although not explicit as a comparator in this review, will have been captured in this search as it is the key comparator to imatinib, but data from the Schrover et al. has also been used to support this point. The same database sources were searched for this review as for the clinical effectiveness review.
As the search was operationalised without recourse to limits (other than the project timelines and limits to human-only references) these unfiltered results have a broad applicability for the project.
The results annex and the detailed search syntax for this search are at the bottom of this annex.
Notes on an additional search: The Cochrane Library
The Cochrane Library was in the process of updating from Issue 2 of 12, February 2011 to Issue 3 of 12, February 2011, when the initial searching was run. Rather than hold up the overall search delivery, we searched Issue 2 in the first instance.
A second search of The Cochrane Library was run on Thursday 17 March 2011 when the update to Issue 3 was complete and the results from this search were de-duplicated against the results found when the search of Issue 2 was conducted. Both searches yielded 51 hits and, accordingly, the de-duplication found no unique data in the new update. A record of the search is included below the first Cochrane search.
Citation alerts
We put citation alerts on the two papers identified as includable in the review process. The alerts were screened as they arose by way of updating searches.
Main search
Database: MEDLINE
-
Host: Ovid
-
Date parameters: 1948 to week 4 February 2011
-
Date searched: Monday 7 March 2011
-
Hits: 595
-
myeloid$ leuk?emia$.mp.
-
Leukemia, Myeloid/
-
(CML).tw.
-
leukemia, myeloid, chronic-phase/
-
leukemia, myeloid, chronic, atypical, bcr-abl negative/
-
exp leukemia, myelogenous, chronic, bcr-abl positive/
-
myelogenous$ leuk?emia$.mp.
-
myelocytic$ leuk?emia$.mp.
-
leukemia, myelomonocytic, chronic/
-
major cytogenetic response.ti,ab.
-
major molecular response.ti,ab.
-
Or/1- 11
-
Philadelphia Chromosome/
-
(Philadelphia adj1 Chromosome).mp.
-
(PH1 or PH 1 adj3 Chromosome).mp.
-
Or/13-15
-
12 or 16
-
nilotinib.mp.
-
“4-methyl-N-(3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-pyridin-3-ylpyrimidin-2-yl)amino)benzamide”.mp.
-
tasigna.mp.
-
((amn107 or amn-107 or amn) adj “107”).mp.
-
Or/18-21
-
dasatinib.mp.
-
sprycel.mp.
-
(BMS354825 or BMS 354825 or BMS-354825).mp.
-
Or/23-25
-
22 or 26
-
17 and 27
-
Animals/ not Humans/
-
28 NOT 29
-
limit 30 to English language
-
limit 31 to yr=“2002 -Current”
Database: MEDLINE In-Process & Other Non-Indexed Citations
-
Host: Ovid
-
Date parameters: 4 March 2011 to 7 March 2011
-
Date searched: Monday 7 March 2011
-
Hits: 66
-
myeloid$ leuk?emia$.mp.
-
(CML).tw.
-
myelogenous$ leuk?emia$.mp.
-
myelocytic$ leuk?emia$.mp.
-
major cytogenetic response.ti,ab.
-
major molecular response.ti,ab.
-
Or/1-6
-
(Philadelphia adj1 Chromosome).mp.
-
(PH1 or PH 1 adj3 Chromosome).mp.
-
Or/8-9
-
7 or 10
-
nilotinib.mp.
-
“4-methyl-N-(3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-pyridin-3-ylpyrimidin-2-yl)amino)benzamide”.mp.
-
tasigna.mp.
-
((amn107 or amn-107 or amn) adj “107”).mp.
-
Or/12-15
-
dasatinib.mp.
-
sprycel.mp.
-
(BMS354825 or BMS 354825 or BMS-354825).mp.
-
Or/17-19
-
16 or 20
-
11 and 21
-
limit 22 to English language
-
limit 23 to yr=“2002 -Current”
Database: PsycINFO
-
Host: Ovid
-
Date parameters: 1806 to March Week 1 2011
-
Date searched: Monday 7 March 2011
-
Hits: 3
-
myeloid$ leuk?emia$.mp.
-
(CML).tw.
-
myelogenous$ leuk?emia$.mp.
-
myelocytic$ leuk?emia$.mp.
-
major cytogenetic response.ti,ab.
-
major molecular response.ti,ab.
-
Or/1-6
-
(Philadelphia adj1 Chromosome).mp.
-
(PH1 or PH 1 adj3 Chromosome).mp.
-
Or/8-9
-
7 or 10
-
nilotinib.mp.
-
“4-methyl-N-(3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-pyridin-3-ylpyrimidin-2-yl)amino)benzamide”.mp.
-
tasigna.mp.
-
((amn107 or amn-107 or amn) adj “107”).mp.
-
Or/12-15
-
dasatinib.mp.
-
sprycel.mp.
-
(BMS354825 or BMS 354825 or BMS-354825).mp.
-
Or/17-19
-
16 or 20
-
11 and 21
-
Animals/ not Humans/
-
22 NOT 23
-
limit 24 to English language
-
limit 25 to yr=“2002-Current”
Database: EMBASE
-
Database host: Ovid
-
Date parameters: 1980 to 2011 Week 9
-
Date searched: Monday 7 March 2011
-
Hits: 2109
-
myeloid$ leuk?emia$.mp.
-
myelogenous$ leuk?emia$.mp.
-
myelocytic$ leuk?emia$.mp.
-
chronic myeloid leukemia/
-
(CML).tw.
-
myeloid leukemia/
-
major cytogenetic response.ti,ab.
-
major molecular response.ti,ab.
-
Or/1-8
-
Philadelphia 1 Chromosome/
-
(Philadelphia adj1 Chromosome).mp.
-
(PH1 or PH 1 adj3 Chromosome).mp.
-
Or/10-12
-
9 OR 13
-
Nilotinib/
-
nilotinib.mp.
-
tasigna.mp.
-
(amn107 or amn-107 or (amn adj “107”)).mp.
-
Or/15-18
-
dasatinib/
-
dasatinib.mp.
-
sprycel.mp.
-
(BMS354825 or BMS 354825 or BMS-354825).mp.
-
Or/20-23
-
19 OR 24
-
14 AND 25
-
limit 26 to English language
-
limit 27 to yr=“2002 -Current”
-
((animal$ or nonhumans) not human$).sh,hw.
-
28 NOT 29
Database: The Cochrane Library [Reviews, Database of Abstracts of Reviews of Effects (DARE), Cochrane Central Register of Controlled Trials (CENTRAL), Health Technology Assessment (HTA), NHS Economic Evaluation Database (NHS EED)]
-
Database host: Cochrane (www.thecochranelibrary.com/view/0/index.html)
-
Date parameters: Issue 2 of 12, February 2011 (updating)
-
Date searched: Monday 7 March 2011
-
Hits: 52 (CENTRAL = 45 + HTA = 6 + NHS EED = 1)
-
CML
-
myeloid* leukaemia*
-
myeloid* leukemia*
-
myelogenous* leukemia*
-
myelogenous* leukaemia*
-
myelocytic* leukemia*
-
myelocytic* leukaemia*
-
major cytogenetic response
-
major molecular response
-
#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9
-
Philadelphia Chromosome
-
#10 OR #11
-
nilotinib
-
tasigna
-
amn107
-
amn-107
-
#13 OR #14 OR #15 OR #16
-
dasatinib
-
sprycel
-
BMS354825
-
BMS 354825
-
BMS-354825
-
#18 OR #19 OR #20 OR #21 OR #22
-
#17 OR #23
-
#12 AND #24 Restrict YR 2002 -2011
Database: The Cochrane Library [Reviews, Database of Abstracts of Reviews of Effects (DARE), Cochrane Central Register of Controlled Trials (CENTRAL), Health Technology Assessment (HTA), NHS Economic Evaluation Database (NHS EED)]
-
Database host: Cochrane (www.thecochranelibrary.com/view/0/index.html)
-
Date parameters: Issue 3 of 12, February 2011
-
Date searched: Thursday 17 March 2011
-
Hits: 52 (CENTAL =45 + HTA =6 + NHS EED =1)
Note: This is the update search to the above search, undertaken when the data update from issue 2 to 3 had been completed. It incorporates the surrogate terms.
-
CML
-
myeloid* leukaemia*
-
myeloid* leukemia*
-
myelogenous* leukemia*
-
myelogenous* leukaemia*
-
myelocytic* leukemia*
-
myelocytic* leukaemia*
-
major cytogenetic response
-
major molecular response
-
Complete Cytogenic Response
-
CCyR
-
#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11
-
Philadelphia Chromosome
-
#12 OR #13
-
nilotinib
-
tasigna
-
amn107
-
amn-107
-
#15 OR #16 OR #17 OR #18
-
dasatinib
-
sprycel
-
BMS354825
-
BMS 354825
-
BMS-354825
-
#20 OR #21 OR #22 OR #23 OR #24
-
#19 OR #25
-
#14 AND #26 Restrict YR 2002-2011
Database: Centre for Reviews and Dissemination all [Database of Abstracts of Reviews of Effects (DARE), Health Technology Assessment (HTA) and NHS Economic Evaluation Database (NHS EED)]
-
Database host: CRD (www.crd.york.ac.uk/crdweb/)
-
Date searched: Monday 7 March 2011
-
Hits: 6 (HTA = 5 + NHS EED = 1)
-
CML
-
myeloid* leukaemia*
-
myeloid* leukemia*
-
myelogenous* leukemia*
-
myelogenous* leukaemia*
-
myelocytic* leukemia*
-
myelocytic* leukaemia*
-
major cytogenetic response
-
major molecular response
-
#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9
-
Philadelphia Chromosome
-
#10 OR #11
-
nilotinib
-
tasigna
-
amn107
-
amn-107
-
#13 OR #14 OR #15 OR #16
-
dasatinib
-
sprycel
-
BMS354825
-
BMS 354825
-
BMS-354825
-
#18 OR #19 OR #20 OR #21 OR #22
-
#17 OR #23
-
#12 AND #24 Restrict YR 2002-2011
Database: Science Citation Index Expanded (SCIE) plus Conference Proceedings Citation Index-Science (CPCI-S) plus Conference Proceedings Citation Index-Social Science & Humanities (CPCI-SSH)
-
Host: ISI
-
Date parameters: 1900 – present
-
Date searched: Monday 7 March 7 2011
-
Hits: 1021
-
TS=(myeloid* leukaemia*) OR TS=(myeloid* leukemia*)
-
TS=(myelogenous* leukemia*) or TS=(myelogenous* leukaemia*)
-
TS=(myelocytic* leukaemia*) OR TS=(myelocytic* leukemia*)
-
#1 OR #2 OR #3
-
(“Philadelphia Chromosome”)
-
#4 OR #5
-
TS=(nilotinib) OR TS=(tasigna) OR TS=(amn107) OR TS=(amn-107) OR TS=(amn adj “107”)
-
TS=(dasatinib) OR TS=( sprycel) OR TS=(BMS354825) OR TS=( BMS 354825) OR TS=( BMS-354825)
-
#7 OR #8
-
#6 and #9
Database: TRIP
-
Database host: www.tripdatabase.com/
-
Date searched: Monday 7 March 2011
-
Hits: 95
(CML or myeloid* leukaemia* or myeloid* leukemia* or myelogenous* leukemia* or myelogenous* leukaemia* or myelocytic* leukemia* or myelocytic* leukaemia* or Philadelphia Chromosome) AND (nilotinib or dasatinib)
Database: EconLit
-
Host: EBSCOhost
-
Date parameters: 1969 – present
-
Date searched: Tuesday 8 March 2011
-
Hits: 0
-
(Myeloid Leukaemia or Myeloid Leukemia)
-
(Myelogenous Leukaemia or Myelogenous Leukemia)
-
(Myelocytic Leukaemia or Myelocytic Leukemia)
-
(Philadelphia Chromosome)
-
S1 or S2 or S3 or S4
-
(dasatinib or nilotinib or tasigna or sprycel)
-
S5 AND S6
| Clinical trial | |
|---|---|
| Current Controlled Trials | Hand searched |
| ClinicalTrials.gov | (207) – Data not included in main review |
| NRR | Hand searched |
| EMA website | Hand searched |
| FDA website | Hand searched |
Surrogate outcomes search
Database: MEDLINE
-
Host: Ovid
-
Date parameters: 1948 to week 2 March 2011
-
Date searched: Thursday 17 March 2011
-
Hits: 44
-
Complete Cytogenetic Response.ti,ab.
-
Complete Cytogenic Response.ti,ab.
-
CCyR.tw.
-
Or/1-3
-
nilotinib.mp.
-
“4-methyl-N-(3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-pyridin-3-ylpyrimidin-2-yl)amino)benzamide”.mp.
-
tasigna.mp.
-
((amn107 or amn-107 or amn) adj “107”).mp.
-
Or/5-8
-
dasatinib.mp.
-
sprycel.mp.
-
(BMS354825 or BMS 354825 or BMS-354825).mp.
-
Or/10-12
-
9 or 13
-
4 and 14
-
limit 15 to english language
Database: MEDLINE In-Process & Other Non-Indexed Citations
-
Host: Ovid
-
Date parameters: Ovid MEDLINE(R) In-Process & Other Non-Indexed Citations, 16 March 2011
-
Date searched: Thursday 17 March 2011
-
Hits: 3
-
Complete Cytogenetic Response.ti,ab.
-
Complete Cytogenic Response.ti,ab.
-
CCyR.tw.
-
Or/1-3
-
nilotinib.mp.
-
“4-methyl-N-(3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-pyridin-3-ylpyrimidin-2-yl)amino)benzamide”.mp.
-
tasigna.mp.
-
((amn107 or amn-107 or amn) adj “107”).mp.
-
Or/5-8
-
dasatinib.mp.
-
sprycel.mp.
-
(BMS354825 or BMS 354825 or BMS-354825).mp.
-
Or/10-12
-
9 or 13
-
4 and 14
-
limit 15 to english language
Database: PsycINFO
-
Host: Ovid
-
Date parameters: 1806 to March Week 2 2011
-
Date searched: Thursday 17 March 2011
-
Hits: 0
-
Complete Cytogenetic Response.ti,ab.
-
Complete Cytogenic Response.ti,ab.
-
CCyR.tw.
-
Or/1-3
-
nilotinib.mp.
-
“4-methyl-N-(3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-pyridin-3-ylpyrimidin-2-yl)amino)benzamide”.mp.
-
tasigna.mp.
-
((amn107 or amn-107 or amn) adj “107”).mp.
-
Or/5-8
-
dasatinib.mp.
-
sprycel.mp.
-
(BMS354825 or BMS 354825 or BMS-354825).mp.
-
Or/10-12
-
9 or 13
-
4 and 14
-
limit 15 to english language
Database: EMBASE
-
Host: Ovid
-
Date parameters: 1980 to 2011 Week 10
-
Date searched: Thursday 17 March 2011
-
Hits: 199
-
Complete Cytogenetic Response.ti,ab.
-
Complete Cytogenic Response.ti,ab.
-
CCyR.tw.
-
Or/1-3
-
nilotinib.mp.
-
“4-methyl-N-(3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-pyridin-3-ylpyrimidin-2-yl)amino)benzamide”.mp.
-
tasigna.mp.
-
((amn107 or amn-107 or amn) adj “107”).mp.
-
Or/5-8
-
dasatinib.mp.
-
sprycel.mp.
-
(BMS354825 or BMS 354825 or BMS-354825).mp.
-
Or/10-12
-
9 or 13
-
4 and 14
-
limit 15 to english language
Database: The Cochrane Library [Reviews, Database of Abstracts of Reviews of Effects (DARE), Cochrane Central Register of Controlled Trials (CENTRAL), Health Technology Assessment (HTA) and NHS Economic Evaluation Database (NHS EED)]
-
Database host: Cochrane (www.thecochranelibrary.com/view/0/index.html)
-
Date parameters: Issue 3 of 12, February 2011
-
Date searched: Thursday 17 March 2011
-
Hits: 7 (CENTRAL = 7)
-
Complete Cytogenetic Response
-
Complete Cytogenic Response
-
CCyR
-
#1 or #2 or #3
-
nilotinib
-
tasigna
-
amn107
-
amn-107
-
#5 OR #6 OR #7 OR #8
-
dasatinib
-
sprycel
-
BMS354825
-
BMS 354825
-
BMS-354825
-
#10 OR #11 OR #12 OR #13 OR #14
-
#9 OR #15
-
#4 AND #16
Database: Centre for Reviews and Dissemination all [Database of Abstracts of Reviews of Effects (DARE), Health Technology Assessment (HTA) and NHS Economic Evaluation Database (NHS EED)]
-
Database host: CRD (www.crd.york.ac.uk/crdweb/)
-
Please contact authors for details of how date parameters were specified for this search
-
Date searched: Thursday 17 March 2011
-
Hits: 0
-
Complete Cytogenetic Response
-
Complete Cytogenic Response
-
CCyR
-
#1 or #2 or #3
-
nilotinib
-
tasigna
-
amn107
-
amn-107
-
#5 OR #6 OR #7 OR #8
-
dasatinib
-
sprycel
-
BMS354825
-
BMS 354825
-
BMS-354825
-
#10 OR #11 OR #12 OR #13 OR #14
-
#9 OR #15
-
#4 AND #16
Database: Science Citation Index Expanded plus Conference Proceedings Citation Index-Science plus Conference Proceedings Citation Index- Social Science & Humanities
-
Host: ISI
-
Date parameters: 1900 to Present
-
Date searched: Thursday 17 March 2011
-
Hits: 62
-
TS=( “Complete Cytogenetic Response”)
-
TS=( “Complete Cytogenic Response”)
-
TS=( “CCyR”)
-
#1 OR #2 OR #3
-
TS=(nilotinib) OR TS=(tasigna) OR TS=(amn107) OR TS=(amn-107) OR TS=(amn adj “107”)
-
TS=(dasatinib) OR TS=( sprycel) OR TS=(BMS354825) OR TS=( BMS 354825) OR TS=( BMS-354825)
-
#4 OR #5
-
#3 and #6
Surrogate outcomes additional search
-
Complete Cytogenetic Response.ti,ab.
-
Complete Cytogenic Response.ti,ab.
-
CCyR.tw.
-
major cytogenetic response.ti,ab.
-
major molecular response.ti,ab.
-
Surrogate adj3 outcome$1
-
Or/1-6
-
(Imatinib).mp.
-
(Gleevec or Glivec).mp.
-
(STI571 or STI-571 or (STI adj1 571)).mp.
-
Or/8-10
-
exp Interferon-alpha/
-
interferon.mp.
-
Or/12-13
-
11 OR 14
-
7 AND 15
-
limit 16 to english language
| Results additional surrogates search | |
|---|---|
| Database | Hits |
| MEDLINE | 390 |
| MEDLINE In-Process & Other Non-Indexed Citations | 20 |
| EMBASE | 828 |
| CRD | 13 |
| The Cochrane Library | 40 |
| SSCI and SCI | 510 |
| Total | 1801 |
| – EndNote deduplication | 592 |
| – Manual deduplication | 199 |
| n | 1010 |
Quality-of-life search
-
myeloid$ leuk?emia$.mp.
-
Leukemia, Myeloid/
-
(CML).tw.
-
leukemia, myeloid, chronic-phase/
-
leukemia, myeloid, chronic, atypical, bcr-abl negative/
-
exp leukemia, myelogenous, chronic, bcr-abl positive/
-
myelogenous$ leuk?emia$.mp.
-
myelocytic$ leuk?emia$.mp.
-
leukemia, myelomonocytic, chronic/
-
major cytogenetic response.ti,ab.
-
major molecular response.ti,ab.
-
Or/1-11
-
Philadelphia Chromosome/
-
(Philadelphia adj1 Chromosome).mp.
-
(PH1 or PH 1 adj3 Chromosome).mp.
-
Or/13-15
-
12 or 16
-
Quality of Life/
-
((quality adj3 life) or life quality or QOL).ti,ab.
-
(HRQL or HRQOL or HRQol).ti,ab.
-
(value adj2 life).ti,ab. or Value of Life/
-
(life adj2 qualit$3).tw.
-
(quality-adjusted life year$1 or QALY or QALYs).ti,ab. or Quality-Adjusted Life Years/
-
daly.ti,ab.
-
(disabilit$3 adj2 life).ti,ab.
-
Health Status Indicators/
-
(sf36 or sf 36 or short form 36 or shortform 36 or sf thirtysix or sf thirty six or shortform thirstysix or shortform thirty six or short form thirty six or short form thirtysix or short form thirty six).tw.
-
(sf6 or sf 6 or short form 6 or shortform 6 or sf six or sfsix or shortform six or short form six).tw.
-
(sf12 or sf 12 or short form 12 or shortform 12 or sf twelve of sftwelve or shortform twelve or short form twelve).tw.
-
(sf16 or sf 16 or short form 16 or shortform 16 or sf sixteen or sfsixteen or shortform sixteen or short form sixteen).tw.
-
(sf20 or sf 20 or short form 20 or shortform 20 or sf twenty of sftwenty or shortform twenty of short form twenty).tw.
-
(euroqol or euro qol or eq5d or eq 5d).tw.
-
(hye or hyes or health$ year$ equivalent$).tw.
-
hui$1.tw.
-
rosser.tw.
-
(willing$ adj2 pay).tw.
-
willing$ adj2 accept.tw.
-
standard gamble$.tw.
-
(health adj3 (utilit$3 or value$2 or preference$2)).tw.
-
(visual analog$3 scale or VAS).tw.
-
patient preference$2.tw.
-
(person$ trade-off or person$ trade off or (PTO)).ti,ab.
-
(Contingent value or contingent valuation).ti,ab.
-
(discrete choice).ti,ab.
-
(health status).ti,ab. or Health Status/
-
((quality adj3 (wellbeing index)) or QWB).ti,ab.
-
(health utilities index or (HUI)).ti,ab.
-
(time trade off or time tradeoff or (TTO)).ti,ab.
-
(utility or utilities).ti,ab.
-
(disutil$).ti,ab.
-
(disability).tw.
-
(wellbeing or well-being or well being or qwb).ti,ab.
-
quality of well being.tw.
-
quality of wellbeing.tw.
-
Or/18-54
-
17 and 55
-
Limit 56 to English Language
-
Limit 57 to “1990-Current”
| Results quality-of life search | |
|---|---|
| Database | Hits |
| MEDLINE | 540 |
| MEDLINE In-Process & Other Non-Indexed Citations | 22 |
| EMBASE | 1000 |
| NHS EED via CRD | 32 |
| NHS EED via The Cochrane Library | 16 |
| PsycINFO | 15 |
| EconLit | 21 |
| Total | 1646 |
| EndNote de-duplication | –436 |
| Manual de-duplication | –107 |
| Total hits for screening | 1103 |
Appendix 2 Protocol
Technology Assessment Report commissioned by the NETSCC HTA Programme on behalf of the National Institute for Health and Clinical ExcellenceHTA 08/226/01FINAL PROTOCOLFebruary 2011
Title of the project:
Dasatinib, nilotinib and standard dose imatinib for the first-line treatment of chronic myeloid leukaemia (including part-review of TA 70).
Name of TAR team and project ‘lead’
PenTAG, Peninsula College of Medicine and Dentistry, University of Exeter
Name: Chris Hyde
Post held: Professor of Public Health and Clinical Epidemiology
Official address: PenTAG, Peninsula Medical School, Veysey Building, Salmon Pool Lane, Exeter, EX2 4SG
Plain English summary
Chronic myeloid leukaemia is one of the blood cancers. Although it has serious consequences for the patient, the outlook with treatment is more favourable than might be expected. The typical age when chronic myeloid leukaemia becomes apparent is between 50 and 60 years and the average life expectancy is at least 15 years.
This project will examine the evidence on how good a number of drugs (dasatinib, nilotinib and standard dose imatinib) are for treating chronic myeloid leukaemia immediately after the disease has been diagnosed, as the first treatment that the patient receives. Concerning this use, the project will update the evidence previously presented to the National Institute of Health and Clinical Excellence in the case of imatinib and review for the first time evidence on dasatinib and nilotinib. The assessment will also assess whether the reviewed drugs are likely to be considered good value for money for the NHS.
Decision problem
Purpose
Chronic myeloid leukaemia (CML) is one of the blood cancers in which there is an overproduction of one type of white blood cell, the granulocytes, by the bone marrow. CML progresses slowly through three identifiable phases: the chronic phase, the accelerated phase and the blast crisis (transformation) phase, with the latter two being grouped together as advanced phase. In some cases categorisation can be difficult and there are various criteria for defining the three phases of CML.
The majority of people are diagnosed in the chronic phase. The course of the chronic phase is initially stable with most people remaining responsive to treatment; around 60% of people will remain in chronic phase and in complete cytogenic remission for at least 5 years. From the chronic phase, people with CML either go through the accelerated phase or move straight into blast crisis. The accelerated phase is a poorly defined period. Blast crisis generally lasts for between 3–6 months and is a terminal stage in which the disease transforms into a fatal acute leukaemia.
Ninety-five percent of people with CML have a specific chromosomal abnormality commonly known as the ‘Philadelphia chromosome’. This is caused by an exchange of genetic material between two chromosomes (known as reciprocal translocation); between parts of the long arms of chromosome 22 and chromosome 9. It is associated with fusion of the breakpoint cluster region (BCR) and Abelson (ABL) genes and the production of an abnormal tyrosine kinase oncoprotein. BCR-ABL is the only known cause of CML.
CML is a rare disease with an incidence of approximately 1 per 100,000 people every year. It accounts for about one in six cases of leukaemia in adults. Approximately 600 to 800 people are diagnosed with CML in England and Wales each year. It has been estimated that median life expectancy is at least 15 years. The median age at diagnosis is between 50 and 60 years.
NICE technology appraisal guidance 70 in 2003 recommends imatinib, a tyrosine-kinase inhibitor, as first-line treatment for people with Philadelphia chromosome positive CML in the chronic phase. 67,149 However, since then other tyrosine-kinase inhibitors have been developed and are being used in the initial treatment of CML. NICE is thus updating TAG 70 concerning the evidence on imatinib, and considering for the first time evidence on dasatinib and nilotinib as first-line treatment for people with Philadelphia chromosome positive CML in the chronic phase. The question referred to NICE is, “To appraise the clinical and cost effectiveness of dasatinib, nilotinib and standard-dose imatinib within their licensed indications for the first-line treatment of chronic myeloid leukaemia (including part-review of TA70).”
In addition, outside this appraisal, NICE is currently appraising dasatinib and nilotinib for imatinib-intolerant CML. An appraisal of dasatinib, nilotinib and high-dose imatinib for imatinib-resistant CML (part-review of TA70) is also underway.
Interventions
The technology assessment report (TAR) will consider three pharmaceutical interventions:
-
Dasatinib (Sprycel, Bristol Myers Squibb)
-
Nilotinib (Tasigna, Novartis Pharmaceuticals)
-
Imatinib (standard dose) (Glivec, Novartis Pharmaceuticals).
All of these are oral tyrosine kinase inhibitors (TKIs). These particular TKIs work by blocking specific signals in cells expressing the BCR-ABL protein, which reduces the uncontrolled proliferation of white blood cells. Imatinib and nilotinib have a high specificity for the BCR-ABL protein, whilst dasatinib acts on multiple targets.
Dasatinib (100 mg daily) has a marketing authorisation for the treatment of adult patients with newly diagnosed Philadelphia chromosome positive CML in the chronic phase. Nilotinib (400/300 mg twice daily) has a marketing authorisation for the treatment of adult patients with newly diagnosed Philadelphia chromosome positive CML in the chronic phase. Imatinib has a marketing authorisation for use in adult and paediatric patients with newly diagnosed Philadelphia chromosome positive CML for whom bone marrow transplantation is not considered as the first-line of treatment. The recommended starting dosage of imatinib is 400 mg/day for patients in chronic phase CML. This is the “standard dose” for the purposes of this appraisal.
Relevant comparators
The main comparators of interest are the alternative interventions particularly:
-
Dasatinib vs imatinib (standard dose)
-
Nilotinib vs imatinib (standard dose)
-
Dasatinib vs nilotinib.
Population and relevant sub-groups
Adults with newly diagnosed, chronic phase, Philadelphia chromosome positive CML. If possible newly diagnosed, chronic phase CML without genetic mutation will also be considered, clearly noting that this population is outside the marketing authorisation of the drugs of interest. No other sub-groups of interest have been identified.
Outcomes to be addressed
The following outcomes will be measured:
-
Event-free survival
-
Progression-free survival
-
Time to progression
-
Overall survival
-
Response rates – cytogenetic, molecular and haematological
-
Time to treatment failure
-
Adverse effects of treatment
-
Health-related quality of life.
Methods for synthesis of evidence of clinical effectiveness
The assessment report will include a systematic review of the evidence for clinical effectiveness of dasatinib, nilotinib and standard-dose imatinib for the first-line treatment of chronic myeloid leukaemia. The review will be undertaken following the general principles published by the NHS Centre for Reviews and Dissemination. 147 The components of the review question will be:
Population: Adults with chronic phase CML, naïve to any treatment specifically directed against CML.
Interventions: Dasatinib or nilotinib or imatinib (standard dose). Each should be employed in accordance with the marketing authorisation and in the populations indicated in the previous paragraph, noting that CML without genetic mutation is outside the existing marketing authorisations.
Comparators: The alternative interventions, particularly imatinib (standard dose) or nilotinib where the intervention is dasatinib, or imatinib (standard dose) or dasatinib where the intervention is nilotinib.
Outcomes: All potentially relevant outcomes in the included studies will be considered, particularly those capturing:
-
Event-free survival
-
Progression-free survival
-
Time to progression
-
Overall survival
-
Response rates – cytogenetic, molecular and haematological
-
Time to treatment failure
-
Adverse effects of treatment
-
Health-related quality of life.
Search strategy
The search strategy will comprise the following main elements:
-
Searching of electronic databases
-
Contact with experts in the field
-
Scrutiny of bibliographies of retrieved papers and manufacturer submissions
-
Follow-up on mentions of potentially relevant ongoing trials noted in previous NICE guidance on imatinib for CML.
The main electronic databases of interest will be:
MEDLINE (Ovid); PubMed; EMBASE; The Cochrane Library including the Cochrane Systematic Reviews Database, Cochrane Controlled Trials Register, DARE, NHS EED and HTA databases; NRR (National Research Register); Web of Science Proceedings; Current Controlled Trials; Clinical Trials.gov; FDA website; EMEA website. These will be searched from search end-date of the last technology appraisal report65 on this topic October 2002.
The searches will be developed and implemented by a trained information specialist using the search strategy detailed in the technology appraisal by Thomson Coon et al. as the starting point (see Appendix A for more information). 144
Inclusion criteria
For the review of clinical effectiveness, in the first instance, only systematic reviews of randomised controlled trials (RCTs) and RCTs will be considered. However, if key outcomes of interest are not measured at all in the included RCTs we will discuss whether or not extending the range of included study designs i.e. to controlled clinical trials could be of value and feasible in the time available with NICE. The systematic reviews will be used as a source for finding further included studies and to compare with our systematic review. Systematic reviews provided as part of manufacturer’s submissions will be treated in a similar manner. These criteria may be relaxed for consideration of adverse events, for which observational studies may be included.
Titles and abstracts will be examined for inclusion by two reviewers independently. Disagreement will be resolved by consensus.
Exclusion criteria
Studies will be excluded if they do not match the inclusion criteria, particularly:
-
Non-randomised studies (except if agreed, in the absence of RCTs)
-
Animal models
-
Preclinical and biological studies
-
Narrative reviews, editorials, opinions
-
Non-English-language papers
-
Reports published as meeting abstracts only, where insufficient methodological details are reported to allow critical appraisal of study quality.
Data extraction strategy
Data will be extracted independently by one reviewer using a standardised data extraction form and checked by another. Discrepancies will be resolved by discussion, with involvement of a third reviewer if necessary.
Quality assessment strategy
Consideration of study quality will be based on the guidelines set out by the NHS Centre for Reviews and Dissemination and include the following factors for RCTs:147
-
Timing, duration and location of the study
-
Method of randomisation
-
Allocation concealment
-
Blinding
-
Numbers of participants randomised, excluded and lost to follow up.
-
Whether intention-to-treat analysis is performed
-
Methods for handling missing data
-
Appropriateness of statistical analysis.
This framework will be adapted should other study designs subsequently be included. Quality will be assessed independently by one reviewer and checked by another, discrepancies again being resolved by discussion, with involvement of a third reviewer if necessary.
Methods of analysis/synthesis
Data will be tabulated and discussed in a narrative review. Where appropriate, meta-analysis will be employed to estimate a summary measure of effect on relevant outcomes based on intention-to-treat analyses.
Meta-analysis will be carried out using fixed and random effects models, using RevMAN supplemented with STATA or equivalent software as required. Heterogeneity will be explored through consideration of the study populations, methods and interventions, by visualisation of results and, in statistical terms, by the χ2 test for homogeneity and the I2 statistic. Mixed-treatment comparisons will be used as far as data allows to facilitate comparison between the drugs for which there is no direct comparison.
Methods for synthesising evidence of cost-effectiveness
Review question
For the interventions and populations indicated above, the existing evidence on cost-effectiveness will be systematically reviewed.
Search strategy
The searches will again be developed and implemented by a trained information specialist using the search strategy detailed in the technology appraisal by Thomson Coon et al. 144 as the starting point. The range of sources searched will include those for clinical effectiveness and extend to include NHS EED and EconLit. October 2002 will again be the starting point.
Study selection criteria and procedures
The inclusion and exclusion criteria for the systematic review of economic evaluations will be identical to those for the systematic review of clinical effectiveness, except:
Non-randomised studies will be included (e.g. decision model based analyses, or analyses of patient-level cost and effectiveness data alongside observational studies).
Full cost-effectiveness analyses, cost-utility analyses, cost–benefit analyses and cost consequence analyses will be included. (Economic evaluations which only report average cost-effectiveness ratios will only be included if the incremental ratios can be easily calculated from the published data.)
Stand alone cost analyses based in the UK NHS will also be sought and appraised.
Based on the above inclusion/exclusion criteria, study selection will be made by one reviewer. In addition, a random sample of the inclusion decisions will be checked by a second reviewer.
Study quality assessment
The methodological quality of the economic evaluations will be assessed by one reviewer according to internationally accepted criteria such as the Consensus on Health Economic Checklist (CHEC) questions developed by Evers et al. 148 Any studies based on decision models will also be assessed against the International Society for Pharmacoeconomics and Outcomes Research (ISPOR) guidelines for good practice in decision analytic modelling. 149
Data extraction strategy
Data will be extracted by one researcher into two summary tables: one to describe the study design and characteristics of each economic evaluation and the other to describe the main results. The tables may need to be split into a number of sub-tables if the number of included studies is large. The entries will be checked by a second reviewer.
In the study design table the main headings will include author and year; model type or trial based; study design [e.g. cost-effectiveness analysis (CEA), cost utility analysis (CUA) or cost-analysis]; service setting/country; study population; comparators; research question; perspective, time horizon and discounting; main costs included; main outcomes included; sensitivity analyses conducted; and other notable design features.
For modelling-based economic evaluations a supplementary study design table will record further descriptions of model structure (and note its consistency with the study perspective, and knowledge of disease/treatment processes); sources of transition and chance node probabilities; sources of utility values; sources of resource use and unit costs; handling of heterogeneity in populations; evidence of validation (e.g. debugging, calibration against external data, comparison with other models).
In the results table for each comparator we will show incremental cost, incremental effectiveness/utility and incremental cost-effectiveness ratio(s). Excluded comparators on the basis of dominance or extended dominance will also be noted. The original authors’ conclusions will be noted, and also any issues they raise concerning the generalisability of results. Finally the reviewers’ comments on study quality and generalisability (in relation to the TAR scope) of their results will be recorded.
Synthesis of extracted evidence
Narrative synthesis, supported by the data extraction tables, will be used to summarise the evidence base.
Economic modelling
The general approach will be consistent with the NICE reference standard. 150 A new cost-effectiveness analysis will be carried out from the perspective of the UK NHS and Personal Social Services (PSS) using a decision analytic model. This will build on the modelling approach used in a recent technology appraisal by PenTAG on a closely related topic and be informed by modelling approaches used in other related NICE appraisals and published cost-effectiveness literature reviewed (see Section 6). 144
Model structure will be determined on the basis of available research evidence and clinical expert opinion.
The sources of parameter values that determine the effectiveness of the interventions being compared will be obtained from our own systematic review of clinical effectiveness or other relevant research literature. Where required parameters are not available from good-quality published studies in the relevant patient group we may use data from manufacturer submissions to NICE.
Cost data will be identified from NHS and PSS reference costs or, where these are not relevant, will be extracted from published work and/or sponsor submissions to NICE. If insufficient data are retrieved from published sources, costs may be derived from individual NHS Trusts or groups of Trusts.
To reflect health related quality of life, utility values will be sought either directly from relevant research literature or indirectly from quality of life studies.
Analysis of uncertainty will focus on costs and utilities, assuming cost per QALY can be estimated. Uncertainty will be explored through one way sensitivity analysis and, if the data and modelling approach permit, probabilistic sensitivity analysis (PSA). The outputs of PSA will be presented using plots on the cost-effectiveness plane and cost-effectiveness acceptability curves.
A life-time time horizon will be taken for our analysis and both cost and outcomes (QALYs) will be discounted at 3.5%. 150
We will collate the available relevant material necessary to inform an assessment of the applicability of the End of Life Criteria.
The TAR team cannot guarantee to consider any data or information relating to the technologies if received after 03/06/11.
Handling the company submissions
All data submitted by the manufacturers will be considered if received by the TAR team no later than 03/06/11. Data arriving after this date will not be considered.
If the data meet the inclusion criteria for the review they will be extracted and quality assessed in accordance with the procedures outlined in this protocol. Any economic evaluations included in the company submission will be assessed against NICE’s guidance on the Methods of Technology Appraisal and will also be assessed for clinical validity, reasonableness of assumptions and appropriateness of the data used. 153
Where the TAR team have undertaken further analyses, using models submitted by manufacturers or via de novo modelling and cost-effectiveness analysis, a comparison will be made of the alternative models used for the analysis.
Expertise in this TAR team
| Name | Institution | Expertise |
|---|---|---|
| Toby Pavey | PenTAG, Peninsula Medical School, University of Exeter | Systematic reviewing, project management and overall lead for clinical effectiveness |
| Louise Crathorne | PenTAG, Peninsula Medical School, University of Exeter | Systematic reviewing |
| Tracey Jones-Hughes | PenTAG, Peninsula Medical School, University of Exeter | Systematic reviewing |
| Martin Hoyle | PenTAG, Peninsula Medical School, University of Exeter | Economic modelling and overall lead for cost-effectiveness |
| Kevin Marsh | Matrix Knowledge | Health economics (provisional, to be confirmed) |
| Chris Cooper | PenTAG, Peninsula Medical School, University of Exeter | Information science |
| Claudius Rudin | Royal Devon and Exeter Foundation Trust | Clinical expert |
| Ruth Garside | PenTAG, Peninsula Medical School, University of Exeter | Support for systematic reviews |
| Rob Anderson | PenTAG, Peninsula Medical School, University of Exeter | Overall project lead and project guarantor |
| Chris Hyde | PenTAG, Peninsula Medical School, University of Exeter | Protocol development |
TAR centre
About PenTAG
The Peninsula Technology Assessment Group (PenTAG) is part of the Institute of Health Service Research (IHSR) at the Peninsula Medical School. PenTAG was established in 2000 and carries out independent Health Technology Assessments (HTAs) for the UK HTA Programme, systematic reviews and economic analyses for the NICE (Technology Appraisal and Centre for Public Health Excellence) and systematic reviews as part of the Cochrane Collaboration Heart Group, as well as for other local and national decision-makers. The group is multi-disciplinary and draws on individuals’ backgrounds in public health, health services research, computing and decision analysis, systematic reviewing, statistics and health economics. The Peninsula Medical School is a school within the Universities of Plymouth and Exeter. The IHSR is made up of discrete but methodologically related research groups, among which HTA is a strong and recurring theme.
Recent projects include:
-
The effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer’s disease (review of TA111): a systematic review and economic model.
-
Dasatinib and nilotinib for imatinib-resistant or -intolerant chronic myeloid leukaemia: a systematic review and economic evaluation.
-
Systematic review of the effectiveness and cost-effectiveness of weight management schemes for the under fives.
-
Barriers to and facilitators for the effectiveness of multiple risk factor programmes aimed at reducing cardiovascular disease within a given population: a systematic review of qualitative research.
-
Population and community programmes addressing multiple risk factors to prevent cardiovascular disease: a qualitative study into how and why some programmes are more successful than others.
-
Barriers to and facilitators of conveying information to prevent first occurrence of skin cancer: a systematic review of qualitative research.
-
The harmful health effects of recreational ecstasy: a systematic review of observational evidence.
-
The use of surrogate outcomes in model-based cost-effectiveness analyses: a survey of UK health technology assessment reports.
-
The effectiveness and cost-effectiveness of cochlear implants for severe to profound deafness in children and adults: a systematic review and economic model.
-
The effectiveness and cost-effectiveness of methods of storing donated kidneys from deceased donors: a systematic review and economic model.
-
Bevacizumab, sorafenib tosylate, sunitinib and temsirolimus for renal cell carcinoma: a systematic review and economic model.
-
The effectiveness and cost-effectiveness of cinacalcet for secondary hyperparathyroidism in end stage renal disease patients on dialysis: systematic review and economic evaluation.
-
The effectiveness and cost-effectiveness of carmustine implants and temozolomide for the treatment of newly-diagnosed high grade glioma: systematic review and economic evaluation.
-
The effectiveness and cost-effectiveness of cardiac resynchronisation therapy for heart failure: systematic review and economic evaluation.
-
Inhaled corticosteroids and long-acting beta2-agonists for the treatment of chronic asthma in adults and children aged 12 years and over: a systematic review and economic analysis.
-
Inhaled corticosteroids and long-acting beta2-agonists for the treatment of chronic asthma in children under the age of 12 years: a systematic review and economic analysis.
Competing interests of authors
None.
Timetable/milestones
| Event | Expected due date |
|---|---|
| Final scope | 04/02/11 |
| Final protocol due | 11/02/11 |
| Consultee information meeting (CIM) (if applicable) | To be confirmed |
| Manufacturers’ submissions | 03/06/11 |
| ERG Appraisal Report due | 06/09/11 |
| 1st Appraisal Committee meeting | 08/11/11 |
| 2nd Appraisal Committee meeting | 08/02/12 |
Appendix 3 Clinical effectiveness data extraction forms
Data extraction: DASISION
| Study details29 | Population | Arms | Outcomes |
|---|---|---|---|
|
Study: Kantarjian et al. (2010) Design: RCT CML phase: Newly diagnosed chronic Country: Multinational No. of centres: Multilocation (109) Length of follow-up: 5-years (minimum) Notes The disease was considered to have progressed if any of the following occurred: a doubling of the white cell count to > 20 × 109/l in the absence of CHR; a loss of CHR; an increase in Ph+ bone marrow metaphases to more than 35%; progression to AP or blastic-phase CML, or death from any cause |
Inclusion criteria (total randomised n = 519): Newly diagnosed (≤ 3 months) ECOG score at least 0–2 No prior TKI treatment Adequate hepatic and renal function Exclusion criteria: Serious or uncontrolled medical disorders or cardiovascular disease History of serious bleeding disorder, concurrent cancer, previous chemotherapy, pleural effusion at baseline |
Arms n = 2 Arm 1: Dasatinib n: 259 Drug: Dasatinib Starting daily dose (mg): 100 mg Median dose: 99 mg Dosage details: Interruptions, reductions or escalations based on criteria (supplementary appendix) Concurrent treatment: Prior treatment with anagrelide or hydroxycarbamide allowed Duration of treatment: 14 months Arm 2: Imatinib n: 260 Drug: Imatinib Starting daily dose (mg): 400 mg Median dose: 400 mg Dosage details: Interruptions, reductions or escalations based on criteria (supplementary appendix) Concurrent treatment: Prior treatment with anagrelide or hydroxycarbamide allowed Duration of treatment: 14.3 months |
Primary outcome CCyR (within 12 months)Secondary outcomes MMR (at any time) |
| Baseline characteristics29 | ||
|---|---|---|
| Dasatinib (n = 259) | Imatinib (n = 260) | |
| Age | ||
| Median, years | 46 | 49 |
| Range, years | 18–84 | 18–78 |
| > 65 years (%) | 20 (8) | 24 (9) |
| Sex, no. (%) | ||
| Male | 144 (56) | 163 (63) |
| Female | 115 (44) | 97 (37) |
| ECOG status, no. (%) | ||
| 0 | 213 (82) | 205 (79) |
| 1 | 46 (18) | 53 (20) |
| 2 | 0 | 2 (1) |
| Hasford risk, no. (%) | ||
| Low | 86 (33) | 87 (33) |
| Intermediate | 124 (48) | 123 (47) |
| High | 49 (19) | 50 (19) |
| Time from diagnosis to randomisation, months | ||
| Median | 1 | 1 |
| Range | 0.03–9.7 | 0.1–8.0 |
| White cell count, × 10 −9 /l | ||
| Median | 25.1 | 23.5 |
| Range | 2.5–493.0 | 1.4–475.0 |
| Platelet count, × 10 −9 /l | ||
| Median | 448 | 390 |
| Range | 58–1880 | 29–2930 |
| Peripheral blood blasts, % | ||
| Median | 1.0 | 1.0 |
| Range | 0.0–10.0 | 0.0–11.0 |
| Peripheral blood basophils, % | ||
| Median | 4.0 | 4.0 |
| Range | 0.0–27.8 | 0.0–19.5 |
| Bone marrow blasts, % | ||
| Median | 2.0 | 2.0 |
| Range | 0.0–14.0 | 0.0–12.0 |
| BCR–ABL transcript type, no. (%) | ||
| b2a2 and b3a2 | 253 (98) | 255 (98) |
| b2a3 | 1 (< 1) | 1 (< 1) |
| b3a3 | 1 (< 1) | 1 (< 1) |
| Rare variant | 3 (1) | 1 (< 1) |
| Previous therapy for CML, no. (%) | ||
| Hydroxycarbamide | 189 (73) | 190 (73) |
| Anagrelide | 8 (3) | 3 (1) |
| Imatinib | 3 (1) | 4 (2) |
| Main results: 12 months9 | |||||||
|---|---|---|---|---|---|---|---|
| Dasatinib (n = 259) | Imatinib (n = 260) | ||||||
| No. | % | (95% CI) | No. | % | (95% CI) | p-value | |
| Response 12 months | |||||||
| Confirmed CCyR by 12 months (i.e. two assessments) | 199 | 77 | (71 to 82) | 172 | 66 | (60 to 72) | 0.007 |
| Complete CyR by 12 months (one assessment) | 216 | 83 | (78 to 88) | 186 | 72 | (66 to 72) | 0.001 |
| MMR at any time (12-month paper) | 135 | 52 | (46 to 58) | 88 | 34 | (28 to 40) | < 0.0001 |
| MMR response 12 months | 119 | 46 | (40 to 52) | 73 | 28 | (23 to 34) | < 0.0001 |
| Rates of CCyR at 12 months (Hasford risk) | |||||||
| Low | 81 | 94 | − | 66 | 76 | − | − |
| Intermediate | 97 | 78 | − | 88 | 72 | − | − |
| High | 38 | 78 | − | 32 | 64 | − | − |
| Rates of MMR at 12 months (Hasford risk) | |||||||
| Low | 48 | 56 | − | 31 | 36 | − | − |
| Intermediate | 56 | 45 | − | 34 | 28 | − | − |
| High | 15 | 31 | − | 8 | 16 | − | − |
| Dasatinib (n = 259): no. (%) | Imatinib (n = 260): no. (%) | |
|---|---|---|
| Treatment status 12 months | ||
| Received treatment | 258 (100.0) | 258 (100.0) |
| Continue to receive treatment | 218 (84.5) | 210 (81.4) |
| Discontinued to receive treatment | 40 (15.5) | 48 (18.6) |
| Had drug-related AEs (12 months) | 13 (5.0) | 11 (4.3) |
| Haematological, including cytopenia (12 months) | 4 (1.6) | 3 (1.2) |
| Non-haematological | 9 (3.5) | 8 (3.1) |
| Diseased progressed | 11 (4.3) | 14 (5.4) |
| Increased white cell count | 1 (0.4) | 0 |
| Loss of CHR | 0 | 0 |
| Loss of MCyR | 1 (0.4) | 4 (1.6) |
| Progression to accelerated or blastic phase (12 months) | 5 (1.9) | 9 (3.5) |
| Death | 4 (1.6) | 1 (0.4) |
| Treatment failed | 6 (2.3) | 10 (3.9) |
| Did not have complete haematological or cytogenetic response at 6 months | 2 (0.8) | 4 (1.6) |
| Had less than partial CyR at 12 months | 3 (1.2) | 6 (2.3) |
| Did not have a CCyR at 18 months | 1 (0.4) | 0 |
| Had AE unrelated to drug | 3 (1.2) | 1 (0.4) |
| Withdrew consent | 2 (0.8) | 3 (1.2) |
| Became pregnant | 2 (0.8) | 0 |
| Did not adhere to therapy | 0 | 2 (0.8) |
| Was lost to follow-up | 0 | 3 (1.2) |
| Requested to discontinue | 2 (0.8) | 1 (0.4) |
| Had other reason | 1 (0.04) | 3 (1.2) |
| Dasatinib (n = 258) | Imatinib (n = 258) | |||
|---|---|---|---|---|
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | |
| AEs 12 months | ||||
| Cytopenia, % | ||||
| Neutropenia (12 months) | 65 | 21 | 58 | 20 |
| Thrombocytopenia (12 months) | 70 | 19 | 62 | 10 |
| Anaemia (12 months) | 90 | 10 | 84 | 7 |
| Non-haematological AE, % | ||||
| Fluid retention (12 months) | 19 | 1 | 42 | 1 |
| Superficial oedema (12 months) | 9 | 0 | 36 | < 1 |
| Pleural effusion (12 months) | 10 | 0 | 0 | 0 |
| Other | 5 | 1 | 8 | < 1 |
| Diarrhoea (12 months) | 17 | < 1 | 17 | 1 |
| Nausea (12 months) | 8 | 0 | 20 | 0 |
| Vomiting (12 months) | 5 | 0 | 10 | 0 |
| Myalgia (12 months) | 6 | 0 | 12 | 0 |
| Muscle inflammation (12 months) | 4 | 0 | 17 | < 1 |
| Musculoskeletal pain (12 months) | 11 | 0 | 14 | < 1 |
| Rash (12 months) | 11 | 0 | 17 | 1 |
| Headache | 12 | 0 | 10 | 0 |
| Fatigue (12 months) | 8 | < 1 | 10 | 0 |
| Results: cardiovascular conditions, 12 months151 | ||||
|---|---|---|---|---|
| Dasatinib (n = 258) | Imatinib (n = 258) | |||
| Any cardiovascular condition | No cardiovascular condition | Any cardiovascular condition | No cardiovascular condition | |
| Response 12 months, % of patients | ||||
| CCyR | 86 | 83 | 76 | 71 |
| MMR | 63 | 43 | 26 | 28 |
| Dasatinib (n = 259) | Imatinib (n = 258) | |||
| Any cardiovascular condition | No cardiovascular condition | Any cardiovascular condition | No cardiovascular condition | |
| AEs cardiovascular 12 months | ||||
| Cytopenia, % of patients | ||||
| Neutropenia (12 months) | 5 | 24 | 17 | 21 |
| Thrombocytopenia (12 months) | 9 | 21 | 10 | 11 |
| Non-haematological AE, % of patients | ||||
| Fluid retention (12 months) | 35 | 16 | 57 | 39 |
| Superficial oedema (12 months) | 16 | 7 | 48 | 33 |
| Pleural effusion (12 months) | 23 | 7 | 0 | 0 |
| Vomiting (12 months) | 12 | 11 | 21 | 16 |
| Myalgia (12 months) | 9 | 11 | 14 | 18 |
| Rash (12 months) | 12 | 11 | 21 | 16 |
| Cardiac | 7 | 5 | 10 | 2 |
| Results: additional baseline medications, 12 months152 | ||||||
|---|---|---|---|---|---|---|
| Dasatinib (n = 259) | Imatinib (n = 260) | |||||
| No. of medications: | No. of medications: | |||||
| 0 | 1–3 | ≥ 4 | 0 | 1–3 | ≥ 4 | |
| Response 12 months, % of patients | ||||||
| CCyR | 79 | 85 | 87 | 76 | 70 | 71 |
| MMR | 43 | 49 | 42 | 35 | 26 | 23 |
| Dasatinib (n = 259) | Imatinib (n = 258) | |||||
| No. of medications | No. of medications | |||||
| 0 | 1–3 | ≥ 4 | 0 | 1–3 | ≥ 4 | |
| AEs 12 months | ||||||
| Cytopenia (grade 3/4), % of patients | ||||||
| Neutropenia | 29 | 13 | 31 | 31 | 18 | 11 |
| Thrombocytopenia | 28 | 17 | 13 | 9 | 9 | 17 |
| Non-haematological AE (all grades), % of patients | ||||||
| Diarrhoea | 17 | 19 | 13 | 13 | 19 | 17 |
| Fluid retention | 9 | 23 | 24 | 41 | 44 | 37 |
| Superficial oedema | 7 | 8 | 16 | 25 | 41 | 31 |
| Pleural effusion | 1 | 13 | 13 | 0 | 0 | 0 |
| Nausea/vomiting | 12 | 9 | 18 | 25 | 23 | 23 |
| Myalgia | 12 | 10 | 11 | 28 | 13 | 14 |
| Rash | 6 | 12 | 18 | 19 | 16 | 20 |
| Results: lymphocytosis, 14 months153 | ||||
|---|---|---|---|---|
| Lymphocytosis | ||||
| Dasatinib (n = 259) | Imatinib (n = 260) | |||
| Yes | No | Yes | No | |
| Response 12 months, % of patients | ||||
| CCyR | 83.6 | 75.1 | 50 | 69.7 |
| MCyR | 91.8 | 83.3 | 50 | 82.8 |
| Dasatinib (n = 259) | Imatinib (n = 258) | |||
| Yes | No | Yes | No | |
| AEs 12 months, % of patients | ||||
| Non-haematological AE (all grades) | ||||
| Fatigue | 16.4 | 9.1 | 7.1 | 11.9 |
| Pleural effusion | 18 | 7.6 | 0 | 1 |
| Myalgia | 11.5 | 18.8 | 7.1 | 24.2 |
| Results: baseline comorbidities, 12 months154 | ||||||
|---|---|---|---|---|---|---|
| Comorbidities | ||||||
| Dasatinib (n = 259) | Imatinib (n = 260) | |||||
| Diabetes | Hepatobiliary conditions | Hyperlipidaemia | Diabetes | Hepatobiliary conditions | Hyperlipidaemia | |
| Response condition 12 months, % of patients | ||||||
| CCyR | 67 | 78 | 96 | 69 | 75 | 79 |
| MMR | 44 | 56 | 59 | 15 | 29 | 32 |
| Response age 12 months, % of patients | ||||||
| < 46 | 46–65 | > 65 | < 46 | 46–65 | > 65 | |
| CCyR | 88 | 78 | 85 | 70 | 70 | 83 |
| MMR | 45 | 47 | 50 | 26 | 30 | 29 |
| Dasatinib (n = 259) | Imatinib (n = 258) | |||||
|---|---|---|---|---|---|---|
| No. of medications | No. of medications | |||||
| Any CB (n = 193) | No CB (n = 66) | Any CB (n = 192) | No CB (n = 68) | |||
| AEs 12 months, % of patients | ||||||
| Cytopenia (grade 3/4) | ||||||
| Neutropenia | 22 | 17 | 20 | 21 | ||
| Thrombocytopenia | 18 | 23 | 9 | 13 | ||
| Non-haematological AE (all grades) | ||||||
| Diarrhoea | 18 | 17 | 20 | 10 | ||
| Fluid retention | 19 | 20 | 47 | 28 | ||
| Pleural effusion | 11 | 8 | 0 | 0 | ||
| Nausea/vomiting | 14 | 5 | 24 | 22 | ||
| Myalgia | 12 | 8 | 16 | 19 | ||
| Rash | 14 | 5 | 15 | 21 | ||
| Main results: 18 months155,156 | |||||||
|---|---|---|---|---|---|---|---|
| Dasatinib (n = 259) | Imatinib (n = 260) | p-value | |||||
| No. | % | (95% CI) | No. | % | (95% CI) | ||
| Response 18 months | |||||||
| Confirmed CCyR by 18 months (i.e. two assessments) | 202 | 78 | – | 182 | 70 | – | 0.037 |
| CCyR 18 months (one assessment) | 218 | 84 | – | 203 | 78 | – | 0.093 |
| MMR at any time 18-month abstract | 148 | 57 | – | 107 | 41 | – | < 0.001 |
| MMR response 18 months | 145 | 56 | – | 96 | 37 | – | < 0.001 |
| CMR 18 months (BCR–ABL 0.0032%) | 34 | 13 | – | 18 | 7 | – | – |
| Rates of CCyR at 18 months (Hasford risk) | |||||||
| Low | 76 | 92 | – | 63 | 72 | – | – |
| Intermediate | 88 | 71 | – | 87 | 71 | – | – |
| High | 36 | 73 | – | 32 | 64 | – | – |
| Rates of MMR at 18 months (Hasford risk) | |||||||
| Low | 54 | 63 | – | 42 | 48 | – | – |
| Intermediate | 69 | 56 | – | 49 | 40 | – | – |
| High | 25 | 51 | – | 15 | 30 | – | – |
| PCR at 18 months | – | 94.9 | – | – | 93.7 | – | – |
| OS at 18 months | – | 96 | – | – | 97.9 | – | – |
| Dasatinib (n = 259) | Imatinib (n = 260) | |||
|---|---|---|---|---|
| No. (%) | ||||
| Treatment status 18 months | ||||
| Received treatment | 258 (100.0) | 258 (100.0) | ||
| Continue to receive treatment | 209 (81) | 206 (80) | ||
| Discontinued to receive treatment | 49 (19) | 52 (20) | ||
| Had drug-related AEs (18 months) | 15 (6) | 10 (4) | ||
| Haematological, including cytopenia (12 months) | 6 (2.3) | 3 (1.2) | ||
| Progression to accelerated or blastic phase (18 months) | 6 (2.3) | 9 (3.5) | ||
| Dasatinib (n = 258) | Imatinib (n = 258) | |||
|---|---|---|---|---|
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | |
| AEs 18 months, % of patients | ||||
| Cytopenia | ||||
| Neutropenia (18 months) | – | 22 | – | 20 |
| Thrombocytopenia (18 months) | – | 19 | – | 10 |
| Anaemia (18 months) | – | 11 | – | 7 |
| Bleeding | – | 0.8 | – | 1.2 |
| Non-haematological AEs | ||||
| Fluid retention (18 months) | 23 | – | 43 | – |
| Superficial oedema (18 months) | 10 | – | 36 | – |
| Pleural effusion (18 months) | 12 | – | 0 | – |
| Diarrhoea (18 months) | 18 | – | 19 | – |
| Nausea (18 months) | 9 | – | 21 | – |
| Vomiting (18 months) | 5 | – | 10 | – |
| Myalgia (18 months) | 6 | – | 12 | – |
| Muscle inflammation (18 months) | 4 | – | 19 | – |
| Musculoskeletal pain (18 months) | 12 | – | 16 | – |
| Rash (18 months) | 11 | – | 17 | – |
| Fatigue (18 months) | 8 | – | 11 | – |
| Main results: 24 months157 | |||||||
|---|---|---|---|---|---|---|---|
| Dasatinib (n = 259), no. % | Imatinib (n = 260), no. % | p-value | |||||
| no. | % | (95% CI) | no. | % | (95% CI) | ||
| Response 24 months | |||||||
| Confirmed CCyR by 24 months (i.e. two assessments) | 207 | 80 | – | 192 | 74 | – | 0.037 |
| CCyR 24 months (one assessment) | 223 | 86 | – | 213 | 82 | – | 0.23 |
| MMR at any time 24 month | 166 | 64 | – | 120 | 46 | – | < 0.001 |
| CMR 24 months (BCR–ABL 0.0032%) | 44 | 17 | – | 21 | 8 | – | – |
| Rates of MMR at 24 months (Hasford risk) | |||||||
| Low | 63 | 73 | – | 49 | 56 | – | – |
| Intermediate | 76 | 61 | – | 62 | 50 | – | – |
| High | 28 | 57 | – | 19 | 38 | – | – |
| PFS at 24 months | – | 93.7 | – | – | 92.1 | – | – |
| OS at 24 months | – | 95.3 | – | – | 95.2 | – | – |
| Dasatinib (n = 259) | Imatinib (n = 260) | |||
|---|---|---|---|---|
| Treatment status 24 months | ||||
| Received treatment | 258 (100.0) | 258 (100.0) | ||
| Continue to receive treatment | 199 (77) | 194 (75) | ||
| Discontinued to receive treatment | 59 (23) | 64 (25) | ||
| Had drug-related AEs (18 months) | 18 (7) | 12 (5) | ||
| Haematological, including cytopenia (12 months) | 6 (2.3) | 4 (1.6) | ||
| Non-haematological | 12 (5) | 8 (3) | ||
| Diseased progressed | 14 (5) | 17 (7) | ||
| Progression to accelerated or blastic phase (18 months) | 9 (3.5) | 15 (5.8) | ||
| Death | 16 (6) | 14 (5) | ||
| Treatment failed | 8 (3) | 11 (4) | ||
| Dasatinib (n = 258) | Imatinib (n = 258) | |||
|---|---|---|---|---|
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | |
| AEs 24 months, % of patients | ||||
| Cytopenia | ||||
| Neutropenia (18 months) | – | 24 | – | 21 |
| Thrombocytopenia (18 months) | – | 20 | – | 11 |
| Anaemia (18 months) | – | 11 | – | 8 |
| Bleeding | – | < 1 | – | 1 |
| Non-haematological AE | ||||
| Fluid retention (18 months) | 25 | – | 43 | – |
| Superficial oedema (18 months) | 11 | – | 36 | – |
| Pleural effusion (18 months) | 14 | – | 0 | – |
| Diarrhoea (18 months) | 49 (19) | – | 21 | – |
| Nausea (18 months) | 26 (10) | – | 23 | – |
| Vomiting (18 months) | 13 (5) | – | 10 | – |
| Myalgia (18 months) | – | – | 12 | – |
| Muscle inflammation (18 months) | 10 (4) | – | 19 | – |
| Musculoskeletal pain (18 months) | 31 (12) | – | 16 | – |
| Rash (18 months) | 28 (11) | – | 19 | – |
| Fatigue (18 months) | 23 (9) | – | 11 | – |
| Quality appraisal: DASISION | |
|---|---|
| Is a power calculation provided? | No |
| Is the sample size adequate? | Not reported |
| Was ethical approval obtained? | Yes |
| Were the study eligibility criteria specified? | Yes |
| Were the eligibility criteria appropriate? | Yes |
| Were patients recruited prospectively? | Yes |
| Was assignment to the treatment groups really random? | Not reported |
| Were groups stratified? | Yes |
| Was the treatment allocation concealed? | No |
| Are adequate baseline details presented? | Yes |
| Are the participants representative of the population in question? | Yes |
| Are groups similar at baseline? | Yes |
| Are any differences in baseline adequately adjusted for in the analysis? | Yes |
| Are outcome assessors blind? | No |
| Was the care-provider blinded? | No |
| Are outcome measures relevant to research question? | Yes |
| Are data collection tools shown or known to be valid for the outcome of interest? | Yes |
| Is compliance with treatment adequate? | Yes |
| Are withdrawals/dropouts adequately described? | Yes |
| Are all patients accounted for? | Yes |
| Is the number randomised reported? | Yes |
| Are protocol violations specified? | Yes |
| Are data analyses appropriate? | Yes |
| Is analysis conducted on an ITT basis? | Yes |
| Are missing data appropriately accounted for? | Yes |
| Were any subgroup analyses justified? | NA |
| Are the conclusions supported by the results? | Yes |
| Conflict of interest declared? | Yes |
Data extraction: ENESTnd
| Study details20 | Population | Arms | Outcomes |
|---|---|---|---|
|
Study: Saglio (2010) Design: RCT CML phase: Newly diagnosed chronic Countries: USA, UK No. of centres: Multilocation (63) Length of follow-up: 5 years (minimum) |
Inclusion criteria (total randomised n = 846): Newly diagnosed (≤ 6 months) ECOG score of at least 2 No prior TKI treatment (except imatinib ≤ 2 weeks) Adequate organ function Exclusion criteria: Impaired cardiac function Medication affecting liver enzymes or QT interval prohibited |
Arms n = 3 Arm 1: Nilotinib n: 282 Drug: Dasatinib Starting daily dose (mg): 300 mg twice daily Median dose: 592 mg Dosage details: Patients could discontinue therapy because of treatment failure (including progression), intolerable side effects, or other reasons. Dose escalation of nilotinib was not permitted Concurrent treatment: Prior treatment with anagrelide or hydroxycarbamide allowed Duration of treatment: 14 months Arm 2: Nilotinib n: 281 Drug: Nilotinib Starting daily dose (mg): 400 mg twice daily Median dose: 779 mg Dosage details: Patients could discontinue therapy because of treatment failure (including progression), intolerable side effects, or other reasons. Dose escalation of nilotinib was not permitted Concurrent treatment: Prior treatment with anagrelide or hydroxycarbamide allowed Duration of treatment: 14 months Arm 3: Imatinib n: 283 Drug: Nilotinib Starting daily dose (mg): 400 mg Median dose: 400 mg Dosage details: Patients could discontinue therapy because of treatment failure (including progression), intolerable side effects, or other reasons. An escalation in the imatinib dose to 400 mg twice daily was permitted in patients who had a suboptimal response or treatment failure, as defined by the European LeukemiaNet Concurrent treatment: Prior treatment with anagrelide or hydroxycarbamide allowed Duration of treatment: 14 months |
Primary outcome MMR (at 12 months)Secondary outcomes CCyR (by 12 months)Rate of MMR and CCyR over time Time to and duration of MMR and CCyR Rate of BCR–ABL : ABL ratio of ≤ 0.01% and ≤ 0.0032% by international scale at 12 months EFS (event defined as loss of CHR, loss of PCyR, loss of CCyR, progression to AP/BC or death from any cause during treatment) PFS (defined as progression to AP/BC or death from any cause during treatment) Progression to AP/BC (defined as progression to AP/BC or CML-related death) OS Safety Dose intensity Pharmacokinetics |
| Baseline characteristics20 | |||
|---|---|---|---|
| Nilotinib (300 mg; n = 282) | Nilotinib (400 mg; n = 281) | Imatinib (400 mg; n = 283) | |
| Age | |||
| Median (range) year | 47 (18–85) | 47 (18–81) | 46 (18–80) |
| Sex, no. (%) | |||
| Male | 158 (56) | 175 (62) | 158 (56) |
| Race or ethnic group, no. (%) | |||
| Asian20 | 76 (27) | 66 (23) | 71 (25) |
| Black | 12 (4) | 11 (4) | 7 (2) |
| White | 170 (60) | 185 (66) | 187 (66) |
| Other | 24 (9) | 19 (7) | 18 (6) |
| Sokal risk group, no. (%) | |||
| Low | 103 (37) | 103 (37) | 104 (37) |
| Intermediate | 101 (36) | 100 (36) | 101 (36) |
| High | 78 (28) | 78 (28) | 78 (28) |
| Time since diagnosis (range), days | |||
| Median | 31 (0–182) | 31 (3–189) | 28 (1–183) |
| White cell count, × 10 −3 /mm 3 | |||
| Median | 23 (2–247) | 23 (2–435) | 26 (3–482) |
| Platelet count, × 10 −3 /mm 3 | |||
| Median | 424 (90–3880) | 347 (103–1819) | 375 (66–2232) |
| Haemoglobin (range), g/dl | |||
| Median | 12.0 (5.5–17.6) | 12.0 (6.2–17.6) | 12.2 (6.4–17.1) |
| Spleen size ≥ 10 cm below costal margin, no. (%) | 31 (11) | 34 (12) | 40 (14) |
| Chromosomal abnormalities in addition to the Philadelphia chromosome | 34 (12) | 44 (16) | 31 (11) |
| Previous therapy for CML, no. (%) | 2 (1) | 1 (< 1) | 4 (1) |
| Main results: 12 months20,158 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nilotinib (300 mg) | Nilotinib (400 mg) | Imatinib (400 mg) | |||||||||
| No. | % | (95% CI) | p-value | no. | % | (95% CI) | p-value | No. | % | (95% CI) | |
| Response 12 months20,161 | |||||||||||
| Rates of MMR at 12 months (ITT) | 125/282 | 44 | − | 0.001 | 121/281 | 43 | − | 0.001 | 62/283 | 22 | − |
| Rates of MMR at 12 months (assessed) | 124/242 | 51 | − | − | 120/240 | 50 | − | − | 63/235 | 27 | − |
| Rates of MMR at 15 months (assessed) | 87/154 | 57 | − | − | 88/155 | 57 | − | − | 48145 | 33 | − |
| Rates of MMR at 18 months (assessed) | 50/83 | 60 | − | − | 44/78 | 56 | − | − | 23/89 | 26 | − |
| Rates of CCyR at 12 months (ITT) | 226/282 | 80 | − | 0.001 | 220/281 | 78 | − | 0.001 | 184/283 | 65 | − |
| Rates of CCyR at 12 months (assessed) | 226/244 | 93 | − | − | 219/236 | 93 | − | − | 184243 | 76 | − |
| Rates of CCyR at 12 months (high Sokal risk) | 58/78 | 74 | − | − | 49/78 | 63 | − | − | 38/78 | 49 | − |
| BCR–ABL ≤ 0.1% | − | 57 | − | − | – | 54 | – | – | – | 30 | – |
| BCR–ABL ≤ 0.01% | − | 24 | − | − | – | 21 | – | – | – | 10 | – |
| BCR–ABL ≤ 0.0032% | − | 13 | − | − | – | 12 | – | – | – | 4 | – |
| MMR by Sokal group | |||||||||||
| Low | − | 41 | − | 0.0238 | – | 53 | – | < .0001 | – | 26 | – |
| Intermediate | − | 51 | − | < .0001 | – | 40 | – | 0.0085 | – | 23 | – |
| High | − | 41 | − | 0.0008 | – | 32 | – | 0.0252 | – | 17 | – |
| EFS | − | 97.6 | − | 0.0898 | – | 99.6 | – | 0.0012 | – | 95.7 | – |
| Nilotinib (300 mg) | Nilotinib (400 mg) | Imatinib (400 mg) | |
|---|---|---|---|
| Treatment status 12 months,20 no. (%) | |||
| Received treatment | 279 (99) | 288 (99) | 279 (99) |
| Continue to receive treatment | 236 (84) | 230 (82) | 224 (79) |
| Discontinued to receive treatment | 46 (16) | 51 (18) | 59 (21) |
| AE(s) | 13 (5.0) | 26 (9) | 21 (7) |
| Abnormal laboratory value(s) | 6 (2) | 5 (2) | 3 (1) |
| Abnormal test procedure result(s) | 0 (0) | 1 (< 1) | 1 (< 1) |
| Subject’s condition no longer requires drug | 1 (< 1) | 0 (0) | 0 (0) |
| Withdrew consent | 6 (2) | 5 (2) | 3 (1) |
| Was lost to follow-up | 2 (< 1) | 2 (< 1) | 1 (< 1) |
| Death | 2 (< 1) | 0 (0) | 0 (0) |
| Diseased progressed | 2 (< 1) | 2 (< 1) | 10 (4) |
| Protocol deviation | 4 (1) | 5 (2) | 4 (1) |
| Suboptimal response/treatment failure | 10 (4) | 5 (2) | 16 (6) |
| Nilotinib (300 mg; n = 279) | Nilotinib (400 mg; n = 277) | Imatinib (400 mg; n = 280) | ||||
|---|---|---|---|---|---|---|
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | |
| AEs 12 months,20 no. of patients (%) | ||||||
| Haematological | ||||||
| Neutropenia | 120 (43) | 33 (12) | 106 (38) | 27 (10) | 189 (68) | 56 (20) |
| Thrombocytopenia | 133 (48) | 28 (10) | 136 (49) | 33 (12) | 156 (56) | 24 (9) |
| Anaemia | 105 (38) | 9 (3) | 105 (38) | 9 (3) | 132 (47) | 14 (5) |
| Non-haematological AE | ||||||
| Rash | 86 (31) | 1 (< 1) | 100 (36) | 7 (3) | 32 (11) | 4 (1) |
| Headache | 39 (14) | 3 (1) | 58 (21) | 3 (1) | 23 (8) | 0 |
| Nausea | 32 (11) | 1 (< 1) | 54 (19) | 3 (1) | 86 (31) | 0 |
| Alopecia | 22 (8) | 0 | 36 (13) | 0 | 11(4) | 0 |
| Pruritus | 41 (15) | 1 (< 1) | 36 (13) | 1 (< 1) | 15 (5) | 0 |
| Myalgia | 27 (10) | 1 (< 1) | 28 (10) | 0 | 28 (10) | 0 |
| Fatigue | 30 (11) | 0 | 25 (9) | 2 (1) | 22 (8) | 1 (< 1) |
| Vomiting | 13 (5) | 0 | 24 (9) | 3 (1) | 40 (14) | 0 |
| Diarrhoea | 22 (8) | 2 (1) | 18 (6) | 0 | 60 (21) | 3 (1) |
| Muscle spasm | 20 (7) | 0 | 17 (6) | 2 (1) | 67 (24) | 2 (1) |
| Peripheral oedema | 14 (5) | 0 | 15 (5) | 0 | 38 (14) | 0 |
| Eyelid oedema | 2 (1) | 0 | 5 (2) | 1(< 1) | 37 (13) | 1 (< 1) |
| Periorbital oedema | 1 (< 1) | 0 | 2 (1) | 0 | 34 (12) | 0 |
| Biochemical abnormality | ||||||
| Increased total bilirubin | 149 (53) | 10 (4) | 171 (62) | 21 (8) | 27 (10) | 1 (< 1) |
| Increased alkaline phosphate | 59 (21) | 0 | 76 (27) | 0 | 92 (33) | 1 (< 1) |
| Decreased phosphate | 88 (32) | 13 (5) | 94 (34) | 13 (5) | 126 (45) | 21 (8) |
| Increased glucose | 100 (36) | 17 (6) | 113 (41) | 10 (4) | 57 (20) | 0 |
| Increased lipase | 67 (24) | 16 (6) | 80 (29) | 16 (6) | 30 (11) | 9 (3) |
| Increase amylase | 42 (15) | 1 (< 1) | 51 (18) | 3 (1) | 35 (12) | 4 (1) |
| Increased creatinine | 13 (5) | 0 | 15 (5) | 0 | 36 (13) | 1 (< 1) |
| Increased ALT | 184 (66) | 11 (4) | 203 (73) | 25 (9) | 57 (20) | 7 (2) |
| Increased AST | 112 (40) | 4 (1) | 134 (48) | 8 (3) | 65 (23) | 3 (1) |
| Results: hospitalisation, 12 months159 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Nilotinib (300; n = 282) | p-value | Nilotinib (400; n = 281) | p-value | Imatinib (n = 283) | ||||
| No. of hospitalisations | 48 | 74 | 57 | |||||
| Total hospital days | 434 | 591 | 642 | |||||
| Length of stay, days | ||||||||
| Mean (SD) | 9.04 (23.95) | 7.99 (15.20) | 11.26 (15.98) | |||||
| Hospital days per 1000 patient-days | 2.72 | 0.057 | 3.69 | 0.61 | 3.99 | |||
| Nilotinib (300; n = 282) | p-value | Nilotinib (400; n = 281) | p-value | Imatinib (n = 283) | ||||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |||
| Time off from usual activities, average hours per week | ||||||||
| Baseline | 247 | 9.33 (18.40) | 0.882 | 240 | 9.20 (19.79) | 0.870 | 234 | 10.02 (22.08) |
| 3-month change for baseline | 225 | –5.03 (19.98) | 0.218 | 210 | –2.85 (19.78) | 0.544 | 206 | –5.83 (20.58 |
| 12-month change from baseline | 195 | –6.66 (20.57) | 0.799 | 190 | –6.17 (15.76) | 0.570 | 171 | –7.06 (26.63) |
| Results: cardiac safety, 12 months160 | |||
|---|---|---|---|
| Nilotinib (300; n = 279) | Nilotinib (400; n = 277) | Imatinib (n = 280) | |
| QT prolongation, % of patients | |||
| Absolute QTcF > 500 milliseconds | 0 | 0 | 0 |
| QTcF increase > 30 milliseconds | 26 | 26 | 18 |
| QTcF increase > 60 millisecond | 0.4 | 0.7 | 0 |
| Mean (%) LVEF change (SD) | |||
| 6 months | +1.2 (1.71) | +1.2 (1.77) | +1.2 (2.02) |
| 12 months | +1.3 (2.33) | +1.3 (1.99) | +1.3 (2.29) |
| Discontinued therapy | |||
| QT prolongation | 0 | 0 | 0 |
| LVEF | 0 | 0 | 0 |
| Ischaemic heart disease event | 1 | 2 | < 1 |
| Left ventricular dysfunction | 1 | 4 | < 1 |
| Response results: 18 month161 | |||||
|---|---|---|---|---|---|
| Nilotinib (300; n = 282) | p-value | Nilotinib (400; n = 281) | p-value | Imatinib (n = 283) | |
| MMR at any time, % of patients | 66 | < 0.0001 | 62 | < 0.0001 | 40 |
| Sokal group, % of patients | |||||
| Low | 70 | 69 | 51 | ||
| Intermediate | 67 | 63 | 39 | ||
| High | 59 | 51 | 28 | ||
| Complete molecular response (BCR–ABL ≤ 0.0032%), % of patients | 21 | < 0.0001 | 17 | < 0.0001 | 6 |
| CCyR, % of patients | 85 | < 0.001 | 82 | < 0.017 | 74 |
| Suboptimal response (12 months) | 2 | 2 | 11 | ||
| Treatment failure (12 months) | 3 | 2 | 8 | ||
| Estimated OS (at 18 months), % | 98.5 | 0.28 | 99.3 | 0.03 | 96.9 |
| Progression to AP/BC | |||||
| Excluding clonal evolution, n (%) | 2 (0.7) | < 0.006 | 1 (0.4) | < 0.003 | 12 (4.2) |
| Including clonal evolution, n (%) | 2 (0.7) | < 0.001 | 3 (1.2) | < 0.002 | 17 (6.9) |
| Total deaths, patient (n) | 5 | 2 | 9 | ||
| CML-related deaths | 2 | 1 | 8 | ||
| Main results: 24 months162,163 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nilotinib (300 mg) | Nilotinib (400 mg) | Imatinib (400 mg) | |||||||||
| No. | % | (95% CI) | p-value | No. | % | (95% CI) | p-value | No. | % | (95% CI) | |
| Response 24 months165,166 | |||||||||||
| Rates of MMR at 24 months (ITT) | 175/282 | 62– | – | < 0.001 | 165/281 | 59 | – | < 0.001 | 105/283 | 37 | – |
| MMR at any time 24 months (ITT) | 201/282 | 71 | – | < 0.001 | 187/281 | 67 | – | < 0.001 | 124/283 | 44 | – |
| Rates of CCyR at 24 months (ITT) | 245/282 | 87 | – | 0.001 | 238/281 | 85 | – | 0.017 | 218/283 | 74 | – |
| BCR–ABL ≤ 0.0032% | – | 26 | – | < 0.001 | – | 21 | – | – | – | 10 | 0.004 |
| MMR by Sokal group | |||||||||||
| Low | – | 73 | – | 0.0238 | – | 74 | – | < 0.0001 | – | 65 | – |
| Intermediate | – | 74 | – | < 0.0001 | – | 67 | – | 0.0085 | – | 44 | – |
| High | – | 65 | – | 0.0008 | – | 56 | – | 0.0252 | – | 32 | – |
| PFS | – | 98 | – | 0.07 | – | 97.7 | – | 0.04 | – | 95.2 | – |
| OS | – | 97.4 | – | 0.64 | – | 97.8 | – | 0.21 | – | 96.3 | – |
| Nilotinib (300 mg) | Nilotinib (400 mg) | Imatinib (400 mg) | ||||
|---|---|---|---|---|---|---|
| Treatment status 24 months,163 no. (%) | ||||||
| Received treatment | 279 (99) | 288 (99) | 279 (99) | |||
| Continue to receive treatment | 210 (75) | 220 (78) | 191 (68) | |||
| Discontinued to receive treatment | 72 (25) | 61 (22) | 92 (32) | |||
| Death | 3 (1) | 1 (< 1) | 0 (0) | |||
| Diseased progressed | 2 (< 1) | 4 (1) | 12 (4) | |||
| Protocol deviation | 4 (1) | 5 (2) | 4 (1) | |||
| Suboptimal response/treatment failure | 24 (9) | 5 (2) | 36 (13) | |||
| Nilotinib (300 mg; n = 279) | Nilotinib (400 mg; n = 277) | Imatinib (400 mg; n = 280) | ||||
|---|---|---|---|---|---|---|
| All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | All grades | Grade 3 or 4 | |
| AEs 24 months,162 no. of patients (%) | ||||||
| Haematological | ||||||
| Neutropenia | – | 33 (12) | – | 31 (11) | – | 59 (21) |
| Thrombocytopenia | – | 28 (10) | – | 33 (12) | – | 25 (9) |
| Anaemia | – | 11 (4) | – | 11 (4) | – | 14 (5) |
| Non-haematological AE | ||||||
| Rash | 86 (31) | 1 (< 1) | 89 (32) | < 1 | 103 (37) | 8 (3) |
| Headache | 39 (14) | 3 (1) | 39 (14) | 1 | 61 (22) | 1 |
| Nausea | 32 (11) | 1 (< 1) | 39 (14) | < 1 | 59 (21) | 1 |
| Alopecia | 22 (8) | 0 | 25 (9) | 0 | 36 (13) | 0 |
| Pruritus | 41 (15) | 1 (< 1) | 45 (16) | < 1 | 36 (13) | < 1 |
| Myalgia | 27 (10) | 1 (< 1) | 28 (10) | < 1 | 28 (10) | 0 |
| Fatigue | 30 (11) | 0 | 31 (11) | 0 | 25 (9) | < 1 |
| Vomiting | 13 (5) | 0 | 14 (5) | 0 | 25 (9) | 1 |
| Diarrhoea | 22 (8) | 2 (1) | 22 (8) | < 1 | 20 (7) | 0 |
| Muscle spasm | 20 (7) | 0 | 22 (8) | 0 | 20 (7) | < 1 |
| Peripheral oedema | 14 (5) | 0 | 14 (5) | 0 | 17 (6) | 0 |
| Eyelid oedema | 2 (1) | 0 | < 1 | 0 | 6 (2) | < 1 |
| Periorbital oedema | 1 (< 1) | 0 | < 1 | 0 | 1 | 0 |
| Quality appraisal: ENESTnd | |
|---|---|
| Is a power calculation provided? | Yes |
| Is the sample size adequate? | Not reported |
| Was ethical approval obtained? | Yes |
| Were the study eligibility criteria specified? | Yes |
| Were the eligibility criteria appropriate? | Yes |
| Were patients recruited prospectively? | Yes |
| Was assignment to the treatment groups really random? | Not reported |
| Were groups stratified? | Yes |
| Was the treatment allocation concealed? | No |
| Are adequate baseline details presented? | Yes |
| Are the participants representative of the population in question? | Yes |
| Are groups similar at baseline? | Yes |
| Are any differences in baseline adequately adjusted for in the analysis? | Yes |
| Are outcome assessors blind? | No |
| Was the care-provider blinded? | No |
| Are outcome measures relevant to research question? | Yes |
| Are data collection tools shown or known to be valid for the outcome of interest? | Yes |
| Is compliance with treatment adequate? | Yes |
| Are withdrawals/dropouts adequately described? | Yes |
| Are all patients accounted for? | Yes |
| Is the number randomised reported? | Yes |
| Are protocol violations specified? | Yes |
| Are data analyses appropriate? | Yes |
| Is analysis conducted on an ITT basis? | Yes |
| Are missing data appropriately accounted for? | Yes |
| Were any subgroup analyses justified? | NA |
| Are the conclusions supported by the results? | Yes |
| Conflict of interest declared? | Yes |
Appendix 4 Surrogate data extraction forms
De Lavallade et al.164 (2008)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| UK | 2000–6 | Cohort single arm | 1 | Imatinib 400 mg/day | 1 | Median: 38 months | |
| Range: 12 to 85 | |||||||
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Consecutive adult patients with BCR–ABL-positive CML in CP. Treatment started within 6 months of diagnosis | No prior treatment for leukaemia other than hydroxycarbamide | 204 | Seventeen of these patients included in IRIS trial | ||||
| Subsequent treatment and treatment duration | |||||||
| Criteria for interruption | Patients on treatment and subsequent therapy | ||||||
| ‘Dose was reduced in the presence of grades 3 to 4 toxicity with the aim of maintaining imatinib at or greater than 300 mg/d. Initially, the criteria for dose escalation were applied as in the IRIS Study, but as more evidence emerged, dose increases reflected those recommended by the European LeukemiaNet. Similarly, the criteria for discontinuing Imatinib varied as new tyrosine kinase inhibitors became available’ | ‘At the time of data analysis 54 patients (26%) had permanently discontinued imatinib after a median time of 15.5 months (range, 0.5 to 64 months). Reasons for discontinuation included AEs (n = 7), loss of CHR or progression to accelerated or blastic phase (n=26), loss of MCyR (n = 3), and failure to achieve MCyR while still in CHR (n = 18). After discontinuing imatinib, 18 patients underwent allogeneic stem-cell transplantation (four while still in CP) and the remaining 36 received one or more of hydroxycarbamide, interferon-α, dasatinib, nilotinib, or other agents. The dose of imatinib was increased in 75 patients (37%)’ | ||||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR | The failure to detect any Ph+ metaphases in two consecutive bone marrow examinations with a minimum of 30 metaphases examined |
1-year cumulative incidence: 57.4% 5-year cumulative incidence: 82.7% (95% CI 76.1% to 87.8%) 159 (77%) (median time, 7 months; range 3 to 55.4) but lost in 14 (8.8%) |
Bone marrow morphology and cytogenetics were assessed at diagnosis and every 3 months until patients achieved CCyR | Cumulative incidence of best CCyR according to CyR at 3 and 6 months reported | |||
| MCyR | Combination of complete and partial CyR (≤ 35% Ph+ metaphases) |
1-year cumulative incidence: 71.1% 5-year cumulative incidence: 85.1% (95% CI 82.8% to 93.0%) |
|||||
| Loss of CCyR | Detection of one or more Ph+ marrow metaphases, confirmed by a subsequent study | 14 (8.8%) | |||||
| MMR | A 3-log reduction in BCR–ABL transcript levels on the basis of two consecutive molecular studies |
1-year cumulative incidence: 12.3% 5-year cumulative incidence: 50.1% (95% CI 41.5% to 58.6%) 80 (39%) (median time, 15.7 months; range, 2 to 73) but lost in 8 (10%) |
BCR–ABL transcripts in the blood were measured at 6- to 12-week intervals | Samples obtained for quantitative real-time PCR were also analysed for kinase domain mutations | |||
| CMR | Two consecutive samples with no detectable transcripts |
1-year cumulative incidence: 0.5% 5-year cumulative incidence: 8.3% 10 (5%) (median time, 30.7 months; range, 12 to 67.4) but lost in 4 (40%) |
|||||
| PFS | Survival without evidence of accelerated or blastic phase disease | 5-year probability: 2.7% |
At 1 year 121 patients with CCyR PFS: 96% 72 patients failed to achieve CCyR PFS: 74% At 5 years 121 patients with CCyR PFS: 96% 72 patients failed to achieve CCyR PFS: 74% |
No significant difference in PFS or OS if patients achieving CCyR are subclassified by MMR | |||
| EFS | Death from any cause, progression resulting from CP, loss of CHR, loss of MCyR or increasing white cell count |
5-year probability: 81.3% (95% CI 73.0% to 87.5%) 5-year probability:* 62.7% (95% CI 55.0% to 70.2%) |
*Include in the definition that 18 patients were discontinuing imatinib because they failed to achieve a MCyR but did not lose CHR and that seven patients were intolerant to imatinib | ||||
| OS | 5-year probability: 83.2% |
At 1 year 121 patients with CCyR OS: 98% 72 patients failed to achieve CCyR OS: 74.1% |
|||||
Druker et al.27 (2006)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| International | 2000–1 | RCT | Multicentre | Imatinib 400 mg/day orally or subcutaneous IFN-α | Two |
Median: 60 months Mean: 50 ± 19 5-year follow up |
|
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Eligible patients had to be between 18 and 70 years of age and must have been diagnosed with Ph+ CML in CP within 6 months before study entry | No previous treatment except for hydroxycarbamide or anagrelide | 1106 (553 in each arm) |
359 (65%) patients had crossed over to imatinib, 14 (3%) had switched to the IFN therapy 382 patients continued imatinib first line, 18% of them with different dosage Focus on imatinib first-line-treated patients |
||||
| Subsequent treatment and treatment duration | |||||||
| Criteria for interruption | Patients on treatment and subsequent therapy | ||||||
| ‘Patients receiving imatinib who did not have a CHR within 3 months or whose bone marrow contained more than 65% Ph-positive cells at 12 months could have a stepwise increase in the dose of imatinib to 400 mg orally twice daily as long as there were no dose-limiting AEs’ |
‘382/553 (69%) in the imatinib group continued with their initial assigned treatment. 14/553 (3%) switched to IFN. Other 157/553 (28%) discontinued first-line treatment: 23 (4%) patients discontinued therapy for AE, 25 (5%) withdraw consent, 10 (2%) died, 15 (3%) violated the protocol, five (< 1%) loss to follow-up, 16 (3%) had SCT In patients remaining in first line therapy 6% received 600 mg/day, 4% received 800 mg/day, 8% received < 400 mg/day’ |
||||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR | No Ph+ metaphases on the basis of G-banding in at least 20 cells in metaphase per sample |
At 12 months 382/553 (69%) At 60 months 368/382 (96%) 481/553 (87%) |
|||||
| MCyR | Complete plus partial responses on the basis of G-banding in at least 20 cells in metaphase per sample |
At 12 months 470/553 (85%) At 60 months 509/553 (92%) |
|||||
| PCyR | 1–35% Ph+ metaphases on the basis of G-banding in at least 20 cells in metaphase per sample | ||||||
| MR | Results were expressed as ‘log reductions’ below a standardised baseline derived from a median ratio of BCR–ABL to BCR obtained from 30 untreated patients with CP-CML |
At 1 year 66/124 (53%) with ≥ –3-log 27/124 (22%) with ≥ –4-log At 4 years 99/124 (80%) with ≥ –3-log 51/124 (41%) with ≥ –4-log |
Signs of a molecular response were sought every 3 months after a CCyR was obtained | ||||
| Patient-relevant outcomes | |||||||
| Patient-relevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| PFS | Survival without evidence of accelerated or blastic phase disease |
At 60 months 93% (95% CI 90% to 96%) 35/553 (6%) progressed to AP or blastic phase Annual rate of progression 1.5% 2.8% 1.6% 0.9% 0.6% |
At 60 months 97% (95% CI 94% to 99%) among 350 patients with CCyR at 12 months 93% (95% CI 87% to 99%) among 86 patients with PCyR 81% (95% CI 70% to 92%) among 73 patients without MCyR 100% among 139 patients with CCyR and –3-log BCR–ABL transcripts at 12 or 18 months 98% among 54 patients with CCyR and less than –3-log BCR–ABL transcripts at 18 months 87% among 88 patients without CCyR Annual rate of progression for patients in CCyR 2.1% 0.8% 0.3% 0% |
Analyses of survival and EFS, using the Kaplan–Meier method according to the ITT principle and using all data available, regardless of whether or not crossover occurred Survival graphs (figures 2–4) |
|||
| EFS | Events were defined by the first occurrence of any of the following: death from any cause during treatment, progression to the AP or blastic phase of CML, or loss of a complete haematological or MCyR |
At 60 months 83% (95% CI 79% to 87%) |
|||||
| OS |
At 60 months 89% (95% CI 86% to 92%), 95% (95% CI 93% to 98%)* |
*After censoring for patients who had died for causes unrelated to CML or transplantation | |||||
Hehlmann et al.165 (2011)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| Germany | July 2002 to April 2009 | RCT (randomised treatment optimisation trial) | Multicentre | Monotherapy imatinib 400 mg/day vs imatinib 400 mg/day combined with IFN-α vs imatinib 800 mg/day | Three | Median follow-up was 28 months in the imatinib 800 mg/day arm, 43 months in the 400 mg/day arm and 48 months in the imatinib plus IFN-α arm | The 800 mg/day imatinib arm started later |
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Newly diagnosed patients with CP-CML | 1012 | Median age 54 years (range 16–88 years) for 325 people in imatinib 400 mg/day arm | |||||
| Subsequent treatment and treatment duration | |||||||
| Criteria for interruption | Patients on treatment and subsequent therapy | ||||||
| Treatment interruptions were discouraged and permitted only for grades 3 and 4 AEs. Simultaneous CYP3A4 inhibitors were avoided. If imatinib treatment failed, either SCT or risk-adapted drug treatment was recommended. After approval of second-generation TKIs for second-line treatment, either nilotinib or dasatinib was recommended. In older patients who were not eligible for transplantation, hydroxycarbamide was recommended if second-generation TKIs were not effective |
The data here refer to the 400 mg/day imatinib arm; 325 randomised patients, 43 months’ median follow-up At 1 year, the number of patients still receiving standard-dose imatinib were 271; 24 patients discontinued treatment at 12 months, four died, four underwent SCT, eight received second-generation TKI, five received hydroxycarbamide/IFN; 236 (73%) were under 400 mg/day imatinib at latest follow-up |
||||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR | Definitions followed the recommendations published by the European LeukemiaNet. | Tab. 2. CCyR at 6, 12, 18, 24 months for the three arms (400 mg/day n = 306, 800 mg/day n = 328, imatinib + IFN, n = 336) | |||||
| MMR | Definitions followed the recommendations published by the European LeukemiaNet. | Tab. 2. MMR at 6, 12, 18, 24 months for the three arms (400 mg/day n = 306, 800 mg/day n = 328, imatinib + IFN, n = 336) | Patients had to have an analysis within an interval of 9–15 months | ||||
| CMR | Definitions followed the recommendations published by the European LeukemiaNet. | Tab. 2. CMR at 6, 12, 18, 24 months for the 3 arms (400 mg/day n = 306, 800 mg/day n = 328, imatinib + IFN, n = 336) | |||||
| Patient-relevant outcomes | |||||||
| Patientrelevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| PFS | PFS was defined by survival free of AP and BC. Starting date for all time-to-event analyses was the date of diagnosis |
At 3 years PFS was 94% (95% CI, 92% to 95%) At 2 years Total 49 (4.8%) 800 mg/day 21 (6.2%) 400 mg/day 16 (4.9%) Imatinib + IFN 12 (3.4%) |
At 3 years Independent of treatment approach, MMR vs no MMR at 12 months was associated with better PFS [99% (95% CI 97% to 100%) vs 95% (95% CI 93% to 97%); p = 0.0143] MMR vs >1% on the international scale at 12 months showed better PFS [99% (95% CI 97% to 100%) vs 94% (95% CI, 90% to 97%); p = 0.0023] No difference was observed in the group with 0.1% to < 1% on the international scale, which is closely correlated with CCyR [PFS 97% (95% CI 94% to 99%)] |
PFS curves not reported. No stratification by CyR, but OS is given by 0.1%–1% IS transcripts level (which has been shown to closely correlate with complete cytogenic remission) | |||
| OS |
At 3 years OS 95% (95% CI 93% to 97%) with no differences between treatment arms At 2 years OS Total 96.6 800 mg/day 96.0 400 mg/day 96.9 Imatinib + IFN 96.8 Deaths Total 30 (3.0%) 800 mg/day 11 (3.3%) 400 mg/day 9 (2.8%) Imatinib + IFN 10 (2.9%) |
At 5 years CCyR vs no CCyR at 12 months was associated with better survival (96% vs 91%; p = 0.0154). At 3 years OS [99% (95% CI 97% to 100%) vs 95% (95% CI 93% to 97%); p = 0.0156] MMR vs >1% on the international scale at 12 months showed better OS [99% (95% CI 97% to 100%) vs 93% (95% CI 90% to 96%); p = 0.0011] No difference was observed in the group with 0.1% to < 1% on the international scale, which is closely correlated with CCyR OS, 98% (95% CI 95% to 100%) |
‘A possible advantage of high-dose therapy is supported by the higher rate of CCyR during the first 2 years, which is an accepted surrogate marker for OS, and by the high CMR rates, which demonstrate the depth of molecular remissions’ | ||||
Hochhaus et al.99 (2009)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| International | 2000–1 | RCT | Multicentre |
Imatinib 400 mg orally once a daily or IFN administered subcutaneously plus cytarabine |
Two |
Median: 70 months, range 0.2–78 months* 6-year follow-up for the last patient recruited |
*Duration of treatment with imatinib |
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Adult patients (aged 18–70 years) with previously untreated Ph+ CP-CML diagnosed within 6 months of study entry | Only prior therapy permitted with hydroxycarbamide or anagrelide | 1106 (553 in each arm) | 65% of patients crossed over to imatinib, 3% of patients crossed over to the alternative therapy. 364 (66%) patients initially assigned to imatinib were still receiving study treatment after 6-year follow-up; 239 of 359 patients who crossed over to imatinib were still receiving the treatment at 6-year follow-up. This study focuses on patients initially randomised to imatinib | ||||
| Subsequent treatment and treatment duration | |||||||
| Criteria for interruption | Patients on treatment and subsequent therapy | ||||||
|
Stepwise dose escalation of imatinib to 400 mg twice daily was permitted if there were no dose limiting AEs on imatinib 400 mg once daily and if any of the following criteria were met: failure to achieve a CHR within 3 months; bone marrow contained more than 65% Phþ metaphase cells at 12 months; or loss of MCyR |
In total, 364 of 553 (66%) patients randomised to imatinib were still receiving study treatment after 6 years of follow-up. 18 (3%) patients randomised to imatinib discontinued study treatment (six patients discontinued due to lack of efficacy, one patient due to unconfirmed loss of CCyR and 11 patients for other reasons including withdrawal of consent or loss to follow-up). The median (mean ± SD, range) last dose given at the time of discontinuation of imatinib study treatment was 400 mg (467 ± 179, 100–800 mg). Other reasons for study discontinuation included: SCT (3%), protocol violation (3%), death (2%) and loss to follow-up (1%). The last reported daily dose of imatinib in this group was 400 mg. Less than 400 mg was reported for 10% of patients [600 mg (6%) and 800 mg (4%)] | ||||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR | At least 20 metaphase cells were analysed to determine CyR. 0% Ph+ |
Last follow-up 349/553 (63%) At 6 months 228/529 (41%) |
Cytogenetic bone marrow assessments annually | ||||
| MCyR | At least 20 metaphase cells were analysed to determine CyR. Definition of MCyR previously reported |
Any time 490/553 (89%) [49/490 (10%) have documented loss MCyR] |
|||||
| PCyR | > 0–35% Ph+ |
At 6 months 92/529 (17%) |
|||||
| Minor/minimal CyR | > 35–95% Ph+ |
At 6 months 39/529 (7%) |
|||||
| No CyR | > 95% Ph+ |
At 6 months 19/529 (3%) |
|||||
| Patient-relevant outcomes | |||||||
| Patient-relevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| PFS | Progression to AP or blastic phase |
At 6 years 93% (95% CI 91% to 95%) Annual rate of progression 1.5% 2.8% 1.6% 0.9% 0.5% 0% |
Annual rate of progression in patients with CCyR (n = 456) 2.1% 0.7% 0.3% 0% 6 year rate without progression* 97% (CCyR) 94% (PCyR) 85% (minor/minimal CyR) 80% (no CyR) |
Survival graphs (Figure 3) *Landmark analysis on 529 patients divided according to their CyR status at 6 months |
|||
| EFS | An event was defined as loss of CHR or MCyR, progression to AP or BC, an increase in WBC count to >20x109/l or death from any cause during treatment |
At 6 years 83% (95% CI 80% to 86%) 86/553 (16%) experiencing an event at any time Annual event rate 3.3% 7.5% 4.8% 1.5% 0.8% 0.4% |
Annual event rate in patients with CCyR (n = 456) |
Survival graphs (Figure 3) *Landmark analysis on 529 patients divided according to their CyR status at 6 months |
|||
| OS |
At 6 years 88% (95% CI 85% to 92%) 95% (95% CI 92% to 97%)* |
*After censoring for patients who had died for causes unrelated to CML or transplantation | |||||
Hughes et al.28 (2003)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| International | 2000–1 | RCT | Multicentre |
Imatinib 400 mg/day or IFN- plus cytarabine |
2 |
Median: 25 months Max: 31 |
|
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Patients aged 18–70 years were enrolled within 6 months of receiving a diagnosis of CML in the CP. Patients could have received no previous treatment for the disease except HU and anagrelide |
1106 (553 in each arm) Median 51 years, range (18–70 years) in n = 408 patients with CCyR and n = 333 patients with CCyR and PCR |
||||||
| Subsequent treatment and treatment duration | |||||||
| Criteria for interruption | Patients on treatment and subsequent therapy | ||||||
| ‘Patients could cross over to the other group if strict definitions of treatment failure or intolerance were met. Details of the study design, conduct, and treatment plan have been reported previously. 26’ | ‘The remaining patients were not included in the analysis: 50 either had disease progression or had discontinued imatinib for other reasons before 12 months of treatment, and 135 had no quantitative PCR sample available’ | ||||||
| Surrogate Outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR |
At 6 months 50% imatinib 3% IFN At 12 months 240/553 (43%) 25/553 (4%) At 19 months 408/553 (74%) imatinib 47/553 (8%) IFN 12-months rate of remission 68% imatinib, 7% IFN |
Samples collected at the baseline, within 2 weeks of CCyR and every 3 months thereafter | |||||
| MR |
The primary BCR-ABL Values calculated as a percentage of BCR were converted to reflect the reduction in the value with use of a standardised logarithmic (base 10) scale |
At the time of CCyR* Median –2.5log (IQR 2.0–3.2) imatinib Median –2.2log (IQR 1.5–2.6) IFN At 15 months after CCyR Median –3.7log imatinib Median –2.5log IFN Proportion of patients with > –3log (MMR)** 39/120 (32%) imatinib 0/12 (0%) IFN After 6 months 50/120 (42%) imatinib 2/120 (13%) IFN After 12 months*** 137/240 (57%) imatinib 6/25 (24%) IFN |
*Median reduction in transcripts level by time after CCyR (Figure 1) **Patients with CCyR at the first assessment ***39% of all patients in the imatinib group and 2% in the IFN group |
||||
| Patient-relevant outcomes | |||||||
| Patient-relevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| PFS | Progression was defined as death, the development of AP or BC CML, an increasing white cell count, or the loss of complete haematologic or major cytogenetic response |
Progression rate 26/365 (7%) (Death 1/26, progression to AP or BP 8/26) |
At 24 months 100% (CCyR + MMR) 95% (CCyR + reduction less than 3-log) 85% (no CCyR) |
*Survival graph (Figure 3) | |||
Hughes et al.100 (2010)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| International | 2000–1 | RCT | Multicentre | Imatinib 400 mg/day or IFN-α plus cytarabine | 2 | Median: 77 months | |
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Patients enrolled on the imatinib arm of the IRIS trial with at least one BCR–ABL transcript measurement | 476 |
Median 50 years, range (20–69 years), IQR 39 to 59, SD 13.2, mean 48.2 For the substudy population ITT population (553 patients), median 50, range 39–58, IQR 39 to 58, SD 12.6, mean 48.2 |
|||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| MMR | MMR represents a 3-log reduction in BCR–ABL transcripts, and is defined as ≤ 0.1% international scale |
At 84 months 87% Proportion of patients in MMR with CCyR: At 3 months 33.3% (n = 51) At 6 months 48% (n = 127) At 9 months 47.1% (n = 138) At 12 months 62.1% (n = 177) At 18 months 77.9% (n = 163) |
Samples for RQ-PCR were collected after achievement of CCyR, at regular intervals or at physicians’ discretion | ||||
| MR | BCR–ABL transcript levels from individual laboratories converted to the international scale |
Proportion of patients with transcripts level > 0.1% and ≤ 1.0% with CCyR: At 3 months 41.2% (n = 51) At 6 months 41.7% (n = 127) At 9 months 39.9% (n = 138) At 12 months 32.8% (n = 177) At 18 months 16.6% (n = 163) |
|||||
| Patient-relevant outcomes | |||||||
| Patient-relevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| PFS | Survival without AP or BC progression |
7-year PFS rate MMR 6 months: 96.2% (95% CI 92% to 100%) MMR 12 months: 100% MMR 18 months: 100% No MMR 6 months: 93% (95% CI 89% to 97%) No MMR 12 months: 89.9% (95% CI 85% to 95%) No MMR 18 months: 90.1% (95% CI 84% to 97%) |
Landmark analyses were run to determine whether BCR–ABL (international scale) values at 6, 12 and 18 months were predictive of long-term outcomes Survival graphs (Figure 4) |
||||
| EFS | The time from treatment start until any of the following events that occur during study treatment: (1) loss of CHR; (2) loss of MCyR; (3) progression to AP/BC; or (4) death due to any cause |
7-year EFS MMR 6 months: 85.1% (95% CI 76% to 94%) 84.4% (95% CI 75% to 94%)* MMR 12 months: 91% (95% CI 85% to 97%) 86.6% (95% CI 80% to 94%)* MMR 18 months: 94.9% (95% CI 91% to 99%) 92.3% (95% CI 87% to 98%)* No MMR 6 months: 83.5% (95% CI 78% to 89%) 71.6% (95% CI 64% to 79%)* No MMR 12 months: 79.4% (95% CI 73% to 86%) 73.1% (95% CI 65% to 81%)* No MMR 18 months: 75.3% (95% CI 66% to 85%) 65.4% (95% CI 54% to 77%)* |
*Including loss of CCyR as an event Survival graphs (Figure 2) |
||||
| OS |
7-year OS rate MMR 6 months: 90.3% (95% CI 83% to 97%) MMR 12 months: 92.5% (95% CI 88% to 97%) MMR 18 months: 94.9% (95% CI 91% to 99%) No MMR 6 months: 89% (95% CI 85% to 94%) No MMR 12 months: 89.2% (95% CI 84% to 94%) No MMR 18 months: 89.8% (95% CI 84% to 96%) |
||||||
Kantarjian et al.101 (2006)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| USA | 2000–4 | Cohort study | 1 | Frontline therapies with 400 mg daily imatinib mesylate orally, 600 mg daily or 800 mg orally daily | 1 (+ a historic group of IFN-treated patients for comparison) | Median 42 months, range 12–66 in imatinib group | ‘The survival by imatinib mesylate dose was identical, justifying their inclusion as 1 treatment group for comparative survival analysis’ |
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Adults with newly diagnosed Ph+ early CP-CML (i.e. within 6 months from diagnosis) |
279 (73 at 400 mg/day dose, 12 at 600 mg/day, 194 at 800 mg/day) ‘Their survival by imatinib dose was identical justifying their inclusion as 1 treatment group’ |
Median 48 range (15–84) n = 279 | |||||
| Subsequent treatment and treatment duration | |||||||
| Criteria for interruption | Patients on treatment and subsequent therapy | ||||||
| – | Seven patients on imatinib mesylate (five from related donors, two from unrelated donors) and 97 patients on interferon (63 from related donors, 30 from unrelated donors, four from donors unspecified) underwent allogeneic SCT in CP. Five of the seven patients who received transplants after imatinib mesylate remain alive without evidence of disease after a median follow-up of 17 months (range 2–34 months) | ||||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR | Disappearance of Ph+ cells (0% Ph+) by routine cytogenetic analysis |
Latest follow-up: 243/279 (87%) Median time 3 months |
|||||
| PCyR | Reduction of Ph+ cells to 1–34% |
Latest follow-up: 17/279 (6%) Median time 3 months |
|||||
| MCyR | A MCyR referred to reduction of Ph+ cells to < 35% | ||||||
| MMR | BCR–ABL/ABL transcript levels < 0.05% | 163/267 (61%) | 267 patients had a follow-up molecular test performed | ||||
| Patient-relevant outcomes | |||||||
| Patient-relevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| TFS | Transformation-free survival was calculated from the date of start of therapy until progression to accelerated-blastic phase |
(Estimated) 3-year TFS 98% (MMR at 1 year) 95% (no MMR at 1 year) |
Therapy (imatinib mesylate vs IFN), entered into the model after accounting for the effect of the independent pre-treatment factors, remained a significant independent factor favouring imatinib mesylate therapy (HR 0.44; p < 0.01) Survival of patients in CCyR was not different by whether or not they achieved a MMR |
||||
| PFS | PFS was calculated from the date of start of therapy, until cytogenetic or haematological resistance or relapse, or progression to AP/blastic phase |
(Estimated) 3-year PFS 98% (MMR at 1 year) 94% (no MMR at 1 year) |
|||||
| OS | Survival was calculated from the date of start of therapy |
(Estimated) 3-year survival: 96% (Estimated) 5-year survival: 88% (Survival graph Figure 1) |
(Estimated) 3-year survival 98% (CCyR at 1 year, n = 210) 100% (PCyR at 1 year, n = 21) 75% (minor cytogenetic response at 1 year, n = 6) 84% (no MCyR at 1 year) 88% (no CR at 1 year, n = 11) (Survival graph Figure 3) (Estimated) 5-year survival 94% (CCyR at 1 year) 94% (PCyR at 1 year) |
||||
Kantarjian et al.102 (2008)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| USA | 1998–2004 | Cohort single arm | 1 | Imatinib mesylate | 1 | Median 48 months, range 4–78 months | Limitations of the study: heterogeneous group and imatinib doses; methodology of molecular studies |
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Adults with a diagnosis of Ph+ CML in early CP (diagnosis of CML for < 12 months) referred to our institution | 276 | Median age 48 years (15–84 years) | |||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR | Disappearance of Ph+ cells (0% Ph+) by routine cytogenetic analysis | The incidence of CCyR at 3, 6, 12, 18, 24, 36, 48 months, at last follow-up and overall, are shown in figure 1 | Response status was evaluated every 3 months in the first year and every 6 months in the subsequent 4 years | ||||
| Durable CCyR | CCyR lasting continuously for at least 3 months (documented twice), 6 months (documented three times) or 12 months (documented three to four times) |
Any 247/276 (89%) < 6 months 32/276 (12%) 6–11 months 18/276 (7%) 12–23 months 48/276 (17%) 24 months or more 149/276 (54%) CCyR durable for 12 months 76% in high-dose imatinib 59% with standard-dose imatinib |
|||||
| MMR | BCR–ABL/ABL transcript levels < 0.1% by real-time Taq human-based QPCR done on peripheral blood or marrow samples. This represents a 3-log reduction from the average baseline for untreated patients in our laboratory | The incidences of major (QPCR 0.1% or less) molecular and complete molecular responses at 3, 6, 12, 18, 24, 36, 48 months, at last follow-up and overall, are shown in Figure 1 | |||||
| Durable MMR | MMR lasting continuously for at least 3 months (documented twice), 6 months (documented three times) or 12 months (documented three to four times) |
Any 201/269 (75%) < 6 months 55/269 (20%) 6–11 months 30/269 (11%) 12–23 months 30/269 (11%) 24 months or more 86/269 (32%) MMR durable for 12 months 45% in high-dose imatinib 39% with standard-dose imatinib |
|||||
| Durable CMR | Undetectable BCR–ABL level lasting continuously for at least 3 months (documented twice), 6 months (documented three times) or 12 months (documented three to four times) |
Any 100/269 (37%) < 6 months 59/269 (22%) 6–11 months 16/269 (6%) 12–23 months 10/269 (4%) 24 months or more 15/269 (6%) |
|||||
| Patient-relevant outcomes | |||||||
| Patient-relevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| PFS | PFS was defined as being on therapy without any of the following: loss of a CyR (Ph positivity increase by at least 30% or to > 65%), loss of haematological response, progression to AP or blastic phase, or death from any cause during therapy. Loss of MMR in a patient who is still in at least a MCyR is not considered to define progression |
PFS is shown in figure 5A–D PFS at 6, 12, 18 and 24 months by molecular response only in patients who were in complete CyR are shown in Figure 7A–D PFS by whether patients achieved a durable CCyR (for at least 12 months) or major (for at least 12 months) and complete molecular response (for at least 6 months) are shown in Figure 8A–C |
Durable CCyR and durable MMR for at least 12 months predicted better PFS rates | ||||
| OS |
Survival from 6, 12, 18 and 24 months by CyR at these time points is shown in Figure 2A–D Survival from 6, 12, 18 and 24 months by molecular response in all patients is shown in Figure 4A–D Survival at 6, 12, 18 and 24 months by molecular response only in patients who were in complete CyR are shown in Figure 6A–D |
OS was not different whether or not patients had achieved these durable responses | |||||
Marin et al.103 (2008)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| UK | 2000–7 | Cohort single arm | 1 | Imatinib 400 mg/day | 1 | Median 46 months, range 13–43 months | |
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Consecutive adult patients with BCR–ABL-positive CP-CML who received imatinib as first-line therapy. Imatinib was started within 6 months of diagnosis and no patient had received any previous anti-leukaemia treatment other than hydroxycarbamide | 224 |
17 patients were included in the International Randomized Study of Interferon and STI571 (IRIS) study Same cohort as de Lavallade 2008 Median age 46.1 years (18–79 years) |
|||||
| Subsequent treatment and treatment duration | |||||||
| Criteria for interruption | Patients on treatment and subsequent therapy | ||||||
| – | A total of 29 patients discontinued the imatinib therapy, although still in CP, eight resulting from toxicity and 21 resulting from unsatisfactory response. The dose of imatinib was increased by more than 400 mg per day in 94 (42%) patients; 21 patients (9.4%) had the imatinib increased during the first year of therapy | ||||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR | Failure to detect any Ph+ metaphases in two consecutive bone marrow examinations with a minimum of 30 metaphases examined | 173/224 (77%) |
Bone marrow morphology and cytogenetics were assessed at diagnosis and then every 3 months until patients achieved CCyR Thereafter, patients were monitored by RQ-PCR and annual bone marrow examinations |
Probability of CCyR and loss of CCyR according to failure and suboptimal response at different time points available (Table 2) Probability of CCyR according to the criteria for failure also in survival graph (Figure 2) Loss of CCyR according to level of MR in Figure 4 |
|||
| MCyR | Combination of complete and partial CyRs (≤35% Ph+ metaphases) | 190/224 (85%) | |||||
| Loss of CCyR | Detection of one or more Ph+ marrow metaphases, also confirmed by a subsequent study, in a patient who had previously achieved CCyR | ||||||
| Failure | No CyR (Ph > 95%) at 6 months or < PCyR (Ph < 35%) at 12 months or < CCyR at 18 months or loss of CCyR at any time |
At 3 months: 8/224 At 6 months: 37/224 At 12 months: 50/224 At 18 months: 66/224 |
|||||
| Suboptimal response | Less than PCyR at 6 months or Less than complete CCyR at 12 months or less than MMR at 18 months or loss of MMR at any time |
At 6 months: 28/224 At 12 months: 45/224 At 18 months: 91/224 |
|||||
| MMR | MMR was defined as a 3-log reduction in transcript levels, 11 based on two consecutive molecular studies | 97/224 (43%) | Patients in CCyR who had failed to achieve MMR at 12 or 18 months were more likely to lose their CCyR than patients who did achieve MMR, 23.6% versus 2.6% (p < 0.04) and 24.6% versus 0% (p < 0.006), respectively | ||||
| CMR | CMR was defined as two consecutive samples with no detectable transcripts provided that control gene copy numbers were adequate | ||||||
| Patient-relevant outcomes | |||||||
| Patient-relevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| PFS | PFS was defined as survival without evidence of AP or blastic phase disease | 25/224 (11%) progressed to AP or blastic phase |
5-year survival: At 3 months 56.2 (95% CI 37.1 to 73.6) in Failure 84.6 (95% CI 77.8 to 89.6) in No Failure At 6 months 73.4 (95% CI 64.9 to 80.4) in Failure 87.1 (95% CI 81.4 to 91.2) in No Failure 87.1 (95% CI 85.0 to 88.9) in CyR (n = 185) 72.8 (95% CI 64.4 to 79.9) in No CyR (n = 34) 91.5 (95% CI 88.1 to 94.0) in MCyR (n = 157) 70.4 (95% CI 62.1 to 77.6) in No MCyR (n = 62) At 12 months 76.0 (95% CI 65.1 to 84.3) in Failure 90.0 (95% CI 85.4 to 93.3) in No Failure 90.0 (95% CI 86.9 to 92.4) in MCyR (n = 169) 76.3 (95% CI 67.7 to 83.2) in No MCyR (n = 46) 96.2 (95% CI 94.3 to 97.5) in CCyR (n = 127) 74.4 (95% CI 70.3 to 78.1) in No CCyR (n = 88) 94.4 (95% CI 86.5 to 97.8) in MMR (n = 32) 85.3 (95% CI 81.7 to 88.3) in No MMR (n = 183) At 18 months 76.4 (95% CI 67.8 to 83.3) in Failure 97.1 (95% CI 92.5 to 98.9) in No Failure 97.1 (95% CI 94.1 to 98.6) in CCyR (n = 132) 76.5 (95% CI 70.8 to 81.4) in No CCyR (n = 65) 94.5 (95% CI 89.2 to 97.3) in MMR (n = 41) 87.5 (95% CI 80.2 to 94.2) in No MMR (n = 156) At any time 6.95 (95% CI 2.2 to 21.7) in Loss of CCyR (n = 17) 0.04 (95% CI 0.0005 to 15654) in Loss of MMR (n = 10) 5-year PFS 72.8% more than 95% Ph+ (n = 34) 74.9%, 36% to 95% Ph+ (n = 28) 91.5% MCyR (n = –157) 76.3% No CyR (n = 46) 81.5% MCyR but No CCyR (n = 42) 96.2% CCyR (n = –127) |
PFS according to suboptimal response at different time points available (Table 2) PFS according to the criteria for failure in survival graph (Figure 2) At 6 months being in MCyR (RR = 3.3, p < 0.017) is independent predictor for PFS. At 12 months, the only independent predictors for PFS were: (1) being in CCyR (RR = 4.5, p < 0.02) and (2) prior loss of CCyR (RR = 24, p < 0.036) At 18 months, the only independent predictor for PFS was being in CCyR (RR = 6.9, p < 0.005) |
|||
| OS | 13/224 (6%) died |
5-year survival: At 3 months 60.2 (95% CI 40.2 to 79.8) in Failure 93.2 (95% CI 86.7 to 96.7) in No Failure At 6 months 81.8 (95% CI 70.2 to 89.6) in Failure 95.5 (95% CI 89.8 to 98.1) in No Failure 94.9 (95% CI 92.3 to 96.7) in CyR (n = 185) 84.6 (95% CI 72.5 to 92.0) in No CyR (n = 34) 93.2 (95% CI 83.7 to 97.3) in MCyR (n = 157) 74.2 (95% CI 58.8 to 85.3) in No MCyR (n = 62) At 12 months 87.1 (95% CI 81.7 to 91.1) in Failure 95.1 (95% CI 91.3 to 97.3) in No Failure 95.1 (95% CI 90.6 to 97.5) in MCyR (n = 169) 86.7 (95% CI 75.5 to 93.2) in No MCyR (n = 46) 98.4 (95% CI 95.9 to 99.4) in CCyR (n = 127) 86.0 (95% CI 79.1 to 90.9) in No CCyR (n = 88) 96.4 (95% CI 85.2 to 99.2) in MMR (n = 32) 93.4 (95% CI 88.3 to 96.4) in No MMR (n = 183) At 18 months 87.8 (95% CI 74.2 to 94.7) in Failure 98.5 (95% CI 95.0 to 99.6) in No Failure 98.5 (95% CI 93.9 to 99.6) in CCyR (n = 132) 87.6 (95% CI 80.5 to 92.3) in No CCyR (n = 65) 95.6 (95% CI 89.8 to 98.2) in MMR (n = 41) 94.5 (95% CI 85.4 to 98.1) in No MMR (n = 156) At any time 3.2 (95% CI 1.1 to 15.4) in Loss of CCyR (n = 17) 0.04 (95% CI.0003 to 21675) in Loss of MMR (n = 10) |
OS according to suboptimal response at different time points available (Table 2) | ||||
Rajappa et al.166 (2008)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
| India | 2003–6 | Cohort single arm | 1 | Imatinib standard oral dose 4000 mg/day | 1 | Median 29.5 months, range 3–58 months | |
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| All adult patients with newly diagnosed untreated CP-CML who were treated with imatinib | No other non-prescription drugs or indigenous medicines were allowed | 201 | Median age 32 years (18–72 years) | ||||
| Subsequent treatment and treatment duration | |||||||
| Criteria for interruption | Patients on treatment and subsequent therapy | ||||||
| Doses were escalated when patients showed clinical or laboratory evidence of progression | Among all patients, 43 (21%) needed temporary discontinuations in imatinib therapy due to AEs. Reasons for treatment discontinuations included myelosuppression in 26 (13%), 11 (5%) for skin reactions and unknown in 6 (3%). The mean daily dose was 346 mg or 86% of scheduled. No patient needed permanent discontinuation of imatinib therapy. The dose of imatinib was escalated to 600–800 mg for patients who had clinical or laboratory evidence of progression. None took dasatinib or underwent allogeneic SCT. At a median of 29 months, 94% patients are alive and on follow-up. Nine (4%) have died and 4 (2%) are lost to follow-up | ||||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR | Standard criteria for CHR, CCyR, partial CyR (PCR), minor response (Minor CR) and no response (NR) were applied (IRIS definition) | 113/201 (56%) | The bone marrow cytogenetics was repeated at least once every year | ||||
| PCyR | 45/201 (23%) | ||||||
| Minor CR | 35 (17%) | ||||||
| NR | 8/201 (4%) | ||||||
| Patient-relevant outcomes | |||||||
| Patient-relevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| PFS | PFS was defined by any of the following events, whichever occurred first: death from any cause, the development of AP-CML or BP-CML (defined by the presence of at least 20% blasts in the blood or bone marrow), loss of CHR, loss of CR (defined as an increase in Ph+ cells in metaphase by at least 30 percentage points) |
At 29 months 77% |
At 29 months 88% in CCyR 64% in other CR conditions |
Survival graph (Figures 1 and 2) | |||
| OS | Survival was calculated from initiation of treatment with imatinib to death from any cause or lost to follow-up |
At 29 months 94% 9/201 (4%) dead 4/201 (2%) lost to follow-up |
At 29 months 100% in CCyR 94% in other CR conditions |
Survival graph (Figures 3 and 4) | |||
Roy et al.104 (2006)
| General characteristics | |||||||
|---|---|---|---|---|---|---|---|
| Country | Year | Design | No. of centres | Treatment | No. of arms | Follow-up | Note |
|
France (CML91) International (IRIS) |
1991–6 CML91 2000–1 IRIS |
Retrospective comparison of two RCTs | Multicentre | Imatinib 400 mg/day vs IFN-α plus Ara-C | (2) |
IRIS: median 42 months, range (0.59–42 months) CML91: median 42 months, range (5.32–42 months) |
|
| Population | |||||||
| Inclusion criteria | Exclusion criteria | Sample size | Note | ||||
| Adults > 18 years of age with Ph+ CML in CP, diagnosed within the preceding 6 months, based on the date of the first cytogenetic analysis. Patients from the French CML91 trial were randomly assigned to the IFN-α plus Ara-C; the current comparison analysed only the 551 patients initially assigned to the imatinib arm, who actually received imatinib at the initial dose of 400 mg daily |
551 IRIS, median age 50 years, range (18–70 years) 325 CML91 |
||||||
| Subsequent treatment and treatment duration | |||||||
| Criteria for interruption | Patients on treatment and subsequent therapy | ||||||
|
A total of 130 patients (24%) in the imatinib group discontinued the treatment (P< .001). Time to discontinuation was 41.8 months (range, 0.16–42 months) The most common reason was lack of efficacy or intolerance, which occurred more frequently with the IFN-α plus Ara-C treatment. A few patients (14 of 551) in the IRIS trial assigned to the imatinib arm crossed over to IFN-α plus Ara–C combination. At the time of analysis, 38 patients (7%) had proceeded to bone marrow transplantation in the IRIS study. Nine patients died during treatment (8 receiving imatinib; 1, the IFN plus Ara–C combination) |
|||||||
| Surrogate outcomes | |||||||
| Surrogate outcomes | Surrogate outcomes (definition) | Results | Time points | Note | |||
| CCyR | Absence of Ph+ cells on karyotype analysis |
IRIS: The cytogenetic analyses were performed every 3 months for the first 12 months and every 6 months thereafter. CML91 study: Cytogenetics at 3 months was optional; however, they were performed at 6, 9, and 12 months for the first 12 months, and every 4 months thereafter |
|||||
| MCyR | The sum of complete and partial cytogenetic responses | ||||||
| Partial CR | Decrease of Ph+ marrow metaphase cells to 1–34% in CML91 or 1–35% in IRIS | ||||||
| Patient-relevant outcomes | |||||||
| Patient-relevant outcomes | Patient-relevant outcomes (definition) | Results | Results stratified by level of response | Note | |||
| PFS | The term ‘survival free of transformation’ (i.e. AP, BC patients, and death) will be used in this analysis. The definitions of AP and BC differed slightly between the two trials. The percentage of peripheral blasts was slightly lower in the CML91 study for the diagnosis of APs and blastic phases (15% and 30% for IRIS vs 10% and 20% for CML91, respectively) |
At 3 years For patients who achieved CCyR at 12 months, survival rates 96% (95% CI 94% to 98%) and 92% (95% CI 85% to 99%) for imatinib and IFN-α plus Ara-C groups, respectively |
|||||
Appendix 5 Excluded studies
| Excluded studies – clinical effectiveness | |
|---|---|
| Paper | Exclude (reason) |
| Abraham (2010) | Review article |
| Baccarani et al. (2010) | Duplication, full paper included |
| Botteman et al. (2010) | No relevant outcomes |
| Cortes (2009) | Review article |
| Giles et al. (2010) | Review article |
| Hughes et al. (2010b) | Not relevant populations |
| Jabbour, et al. (2007) | Review article |
| Kantarjian et al. (2010b) | Duplication, full paper included |
| Larson et al. (2010b) | Duplication, full paper included |
| Le Coutre et al. (2010) | Not relevant populations |
| MacNeil (2010) | Review article |
| Minami et al. (2010) | Not relevant populations |
| Ogura et al. (2010) | Duplication, full paper included |
| Quintas-Cardama et al. (2008) | No relevant outcomes |
| Research Report (2009) | Review article |
| Saglio et al. (2010b) | Duplication, full paper included |
| Shah (2007) | Review article |
| Wei et al. (2010) | Review article |
| Wendling (2010) | Review article |
| Excluded studies – cost-effectiveness | |
|---|---|
| Paper | Exclude (reason) |
| Bouwmans et al. (2009) | Previously treated population |
| Guerin et al. (2010) | Study design |
| Juarez-Garcia (2009) | Previously treated population |
| Ovanfors et al. (2011) | Insufficient information |
| Simons et al. (2009) | Insufficient information |
| Szabo et al. (2010) | Insufficient information |
| Taylor et al. (2010) | Insufficient information |
| Taylor et al. (2007) | Previously treated population |
| Wu et al. (2010) | Insufficient information |
| Excluded studies – surrogate outcomes | |
|---|---|
| Paper | Exclude (reason) |
| Al-Kali et al. (2010) | Final outcome not stratified by level of response |
| Alvarado et al. (2009) | Data derived from different trials |
| Anstrom et al. (2004) | Not relevant populations |
| Aziz et al. (2007) | Previously treated population |
| Aziz et al. (2010) | Previously treated population |
| Bee et al. (2006) | Previously treated population |
| Braziel et al. (2002) | Previously treated population |
| Cervantes et al. (2010) | Final outcome not stratified by level of response |
| Cortes et al. (2005) | Previously treated population |
| Cortes et al. (2006) | Previously treated population |
| Cortes et al. (2010 Das) | Final outcome not stratified by level of response |
| Cortes et al. (2010 Nil) | Final outcome not stratified by level of response |
| El-Zimaity et al. (2004) | Final outcome not stratified by level of response |
| Furukawa et al. (2011) | Previously treated population, not relevant populations |
| Guilhot et al. (2009) | Final outcome not stratified by level of response |
| Huntly et al. (2003) | Previously treated population |
| Jabbour et al. (2009) | Not relevant populations |
| Jiang et al. (2010) | Previously treated population |
| Kanda et al. (2008) | Previously treated population |
| Kantarjian et al. 2002 | Previously treated population |
| Kantarjian et al. (2003) | Final outcome not stratified by level of response |
| Khorashad et al. (2008) | Final outcome not stratified by level of response |
| Kim et al. (2010) | Previously treated population |
| Koffi et al. (2010) | Final outcome not stratified by level of response |
| Mahmoud et al. (2009) | No relevant outcomes |
| Marin et al. (2005) | No relevant final outcomes |
| Marin et al. (2009) | No relevant outcomes |
| Matsuo et al. (2007) | Previously treated population |
| Medhi et al. (2010) | Not relevant populations |
| Moran, Valia et al. (2011) | Not relevant populations |
| Muller et al. 2008 | Final outcome not stratified by level of response |
| Nagai et al. (2010) | Final outcome not stratified by level of response |
| Nannya et al. (2008) | Not relevant populations |
| O’Brien et Deininger (2003) | Final outcome not stratified by level of response |
| O’Brien et al. (2003) | Final outcome not stratified by level of response |
| O’Brien et al. (2008) | Final outcome not stratified by level of response |
| Palandri et al. (2007) | Letter to the editors |
| Palandri et al. (2009) | Previously treated population |
| Palandri et al. (2010) | Final outcome not stratified by level of response |
| Piazza et al. (2006) | Not relevant populations |
| Press et al. (2006) | Not relevant populations, previously treated population |
| Press et al. (2007) | Not relevant populations, previously treated population |
| Qin et al. (2009) | Previously treated population |
| Quintas-Cardama et al. (2009) | Previously treated population |
| Quintas-Cardama et al. (2009) | Review article |
| Rosti et al. (2009) | No relevant final outcomes |
| Santos et al. (2010) | Previously treated population |
| Schrover et al. (2006) | Results (survival by CyR) are estimated from IFN-α population |
| Sheehy et al. (2008) | Previously treated population |
| Shepherd et al. (2008) | Previously treated population |
| Sugita et al. (2008) | Previously treated population |
| Wang et al. (2003) | Previously treated population |
Appendix 6 Ongoing trials
| Study characteristics: S0325 | ||||
|---|---|---|---|---|
| Study | Drug therapy | Inclusion criteria | Exclusion criteria | Outcomes |
| S032568 | Dasatinib, imatinib | Newly diagnosed | Unknown | Haematological response |
| CCyR | ||||
| MMR | ||||
| OS | ||||
| PFS | ||||
| Summary of results: 12 months S032568 | |||||
|---|---|---|---|---|---|
| Response 12 months | |||||
| No. | Dasatinib (n = 123) | Imatinib (n = 123) | p-value | ||
| % | No. | % | |||
| Haematological response | 104 | 86 | 111 | 90 | 0.25 |
| Complete CyR 12 months | 55/67 | 82 | 40/58 | 69 | 0.097 |
| MMR | 39/90 | 59 | 39/90 | 59 | 0.042 |
| CMR | 21/99 | 21 | 13/90 | 14 | 0.26 |
| OS | 123 | 100 | 66 | 99 | 0.60 |
| PFS | 123 | 99 | 88 | 96 | 0.19 |
| Treatment status: 12 months | ||
|---|---|---|
| Dasatinib (n = 123) | Imatinib (n = 123) | |
| No. (%) | ||
| Discontinued to receive treatment | 38 | 47 |
| Had drug-related AEs (12 months) | 18 (15) | 13 (11) |
| Death | 5 | 2 |
| Withdrew consent | 3 (2) | 8 (7) |
| Had other reason | 12 (10) | 24 (20) |
| AEs: 12 months | ||||
|---|---|---|---|---|
| All grades | Dasatinib (n = 258) | Imatinib (n = 258) | ||
| Grade 3 or 4 | All grades | Grade 3 or 4 | ||
| Cytopenia, % of patients | ||||
| Thrombocytopenia (12 months) | 18 | 8 | ||
| Non-haematological AE | ||||
| Pleural effusion (12 months) | 11 | 2 | ||
| Study characteristics: SPIRIT 2167 | ||||
|---|---|---|---|---|
| Study | Drug therapy | Inclusion criteria | Exclusion criteria | Outcomes |
| SPIRIT 2167 | Dasatinib, imatinib |
Male or female patients ≥ 18 years of age Patients must have all of the following: |
|
Primary outcome EFS at 5 years Secondary outcomes CCyR 2 years Treatment failure rates 5 years CHR Levels of molecular response Quality of life OS 2 years and 5 years Broad comparison of costs |
Appendix 7 Full critique of manufacturer’s cost-effectiveness submission
Dasatinib, nilotinib and standard-dose imatinib for the first-line treatment of chronic myeloid leukaemia (CML)
Originally produced by: Matrix Evidence
Abridged version produced by: PenTAG
Original authors:
-
Kevin Marsh, Chief Economist, Matrix Eviden
ceLeeza Osipenko, Principal Economist, Matrix Evidence
Meena Venkatachalam, Economist, Matrix Evidence
Please note: In the draft guidance on 18 August 2011, NICE has recommended nilotinib, for the treatment of the CPs and APs of CML that is resistant or intolerant to standard-dose imatinib. Dasatinib and high-dose imatinib, are not recommended in the draft guidance. Consultees have the opportunity to appeal against the draft guidance. Until NICE issues final guidance, NHS bodies should make decisions locally on the funding of specific treatments. This draft guidance does not mean that people currently taking dasatinib or high-dose imatinib will stop receiving them. They have the option to continue treatment until they and their clinicians consider it appropriate to stop.
Bristol-Myers Squibb submission
Summary
Scope of the submissions
The submission from BMS considers the use of dasatinib (Sprycel®) for the first-line treatment of people with CML as an alternative to the standard dose of imatinib (400 mg daily) or nilotinib (600 mg daily).
The clinical effectiveness outcomes considered are OS, PFS, response rates, adverse effects of treatment and HRQoL.
The outcomes for the economic analysis are incremental cost per QALY, and incremental cost per life-year gained. In order to derive these outcomes the following costs have been considered: cost of first- and second-line TKIs, cost of post-TKI failure second- or third-line treatment, and the cost of treating serious AEs. The time horizon for the economic analysis is between 46 and 86 years old, and costs are considered from an NHS perspective. No subgroup analysis is conducted for the economic evaluation.
Summary of submitted cost-effectiveness evidence
The manufacturer uses a ‘time in state’ (area under the curve) model extrapolating CML-related survival and PCR data. The health states represent the CP, and AP/blast phase as well as death. Within the CP, patients may also be in first-, second or third-line treatment, whereas in the AP/blast phase they may be receiving either third-line treatments or palliative care. Time is modelled in blocks of 1 month.
The BMS base-case analysis produces ICERs of:
-
£26,305 per QALY for dasatinib in comparison with imatinib as a first-line TKI, and
-
£144,778 per QALY in comparison with nilotinib as a first-line TKI.
The sensitivity analysis shows the key parameters to which the model is sensitive: drug costs, OS and the cost of SCT.
The BMS model contained a number of formula errors. After correcting for these errors, the BMS model predicts ICERs of:
-
£36,052 per QALY for first-line dasatinib compared with first-line imatinib, and
-
£103,483 per QALY for dasatinib compared with nilotinib.
In the original model the cost of nilotinib used by BMS does not account for the PAS discount applied to nilotinib. If the cost of nilotinib is adjusted to reflect the (CiC information has been removed) decrease in the cost of nilotinib due to PAS, the cost of nilotinib in first line and second line is reduced from £2664 per month to (CiC information has been removed) per month. Including this change, the BMS model predicts an ICER of £45,600 per QALY for dasatinib compared with imatinib. When comparing dasatinib with nilotinib, the model predicts that nilotinib is more effective and less costly.
Further, BMS assumes that dasatinib is taken as a third-line treatment in all treatment arms. However, in the NICE draft guidance FAD, dasatinib was not recommended (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). When the BMS model is further adjusted so that dasatinib is not taken third line, the ICER of dasatinib compared with imatinib increases further, from £45,600 to £64,000 per QALY, and nilotinib is still more effective and less costly than dasatinib.
Finally, BMS assumes that half of all patients in the imatinib and nilotinib treatment arms who are eligible for second-line treatment take dasatinib. Again, in the NICE draft guidance FAD, dasatinib was not recommended (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). When the BMS model is further adjusted so that dasatinib is not taken second line, and instead when we assume that all second-line patients in the imatinib arm take nilotinib second line, the ICER of dasatinib compared with imatinib increases further, from £64,000 to £96,000 per QALY. There appears to be no simple way to adjust the BMS model to disallow patients taking dasatinib second line.
In summary, the BMS adjusted model yields an ICER for dasatinib compared with imatinib of £96,000 per QALY. Further, nilotinib is more effective and less costly than dasatinib.
Commentary on the robustness of the submitted evidence
Strengths
-
The approach taken to modelling is reasonable although quite complex.
-
The sources and justification of estimates are also generally reasonable.
-
Resource use is largely based on a survey of six UK clinicians who manage patients with CML.
Weaknesses
-
There are a number of formulae errors in the BMS model. When corrected, the base-case ICER changes from £26,305 to £36,052 per QALY for dasatinib in comparison with imatinib; and from £144,778 to £103,483 per QALY for dasatinib in comparison with nilotinib.
-
Unfortunately, due to BMS not having knowledge of the PAS, BMS does not account for the reduced price of nilotinib owing to the PAS discount. In addition to the formulae errors, if the (CiC information has been removed) discount in the price of nilotinib in first line and second line is accounted for, the best-case ICER for the BMS model is £45,600 per QALY for dasatinib compared with imatinib. When comparing dasatinib with nilotinib, the model predicts that nilotinib is more effective and less costly.
-
The starting age of the simulated cohort, 46 years, is considerably lower than the mean age of newly diagnosed patients with CML in the UK (56 years).
-
The model does not adopt a lifetime time horizon. Instead, the model is run until the cohort is 86 years old, at which point 20% of the cohort is still alive. If the model is extended to the age of 100 years, 10% of the population is still alive. Assuming an equal distribution of males and females, data from the ONS predict that 2% of those alive at 46 years will be alive at the age of 100 years. This suggests that BMS overestimates the period that those with CML will survive.
-
BMS use 42-month follow-up data from a RCT to predict OS for those with a complete, partial and ‘less than partial’ CyR to treatment at 12 months. Survival data are digitally extracted from published Kaplan–Meier curves and fitted to a Weibull distribution. There is no use of MMR response rates; the model utilises only CyR rates.
-
BMS outlines the effectiveness of second-line TKIs in their submission. However, these data are not used to model the effectiveness of second-line therapy.
-
There are a number of assumptions with the BMS model which are not defined in detail. In addition, several parameters within the manufacturer submission do not reflect the data that are used in the model. For example, the data used to estimate the PFS curves explained in Table 19 within the manufacturer submission do not match the data in the model. Also, the source quoted for PFS data in the submission is Hochhaus et al. However, the model appears to be using data from Drunker et al. , which is a study with a shorter follow-up period. If the model is updated to use data from Hochhaus et al. the ICER change as follows:
-
– dasatinib compared with imatinib, from £36,052 to £42,556 per QALY
-
– dasatinib compared with nilotinib, from £103,483 to £103,593 per QALY.
-
-
BMS assumes that dasatinib is taken second and third line. Given that BMS prepared their submission before NICE’s recent draft guidance FAD on second-line TKIs, the BMS assumption on the use of dasatinib second and third line was reasonable. However, in the NICE draft guidance FAD dasatinib second and third line was not recommended (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). When the BMS model is adjusted to remove dasatinib second and third line, the cost-effectiveness of dasatinib worsens substantially, as quantified above.
-
BMS developed a highly complex model in an area in which data are not of high quality. We believe the cost-effectiveness model could have been developed in a simpler way.
-
It is not clear how BMS calculated the cost of SCT. (AiC information has been removed.)
-
On several occasions, the BMS report of the modelling differs from the actual model.
Areas of uncertainty
The BMS model does not provide the raw data that were used to fit the OS and time to treatment discontinuation curves. However, the choice of distribution and coefficients of the distribution appear to be correct on the basis of graphs showing the observed data and the fitted curves.
A considerable area of uncertainty is the chosen sequence of second-line TKI treatments that might follow failure of different first-line TKIs. This is partly because the submission was prepared before NICE’s draft guidance FAD on the use of dasatinib, nilotinib or high-dose imatinib as second-line treatments (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). However, uncertainty also results from the fact that data on the effectiveness of second-line TKI treatments is available only following the use of imatinib as first-line treatment.
Key issues
Unfortunately, the BMS model does not use the cost of nilotinib agreed under the PAS in its submission.
The BMS model is structured in such a way that it would require significant changes to run it without second-line treatment, should this be required by NICE.
The time horizon chosen by the BMS model does not reflect the lifetime of a patient with CML. In the model, nearly 20% of the population is still alive in the last cycle (86 years old), suggesting that the model overestimates the period that those with CML will survive.
The BMS model has a number of formulae errors, correcting for which impacts ICER.
The cost and proportions of patients who receive SCT have a significant impact on ICERs, but the source of the BMS estimates of these parameters is unclear. Clinical opinion is required to assess whether or not the BMS assumption on the provision and costing of SCT is appropriate.
Background
Critique of manufacturer’s description of underlying health problem
In section 2 of its submission, BMS adequately describes the underlying health problem. BMS states the median age for disease onset to be 65 years, and the disease prevalence in England and Wales is ∼2660 patients (2003 data from NICE TA70). 168 BMS uses the following timeline to report phase duration of the disease:
-
CP 3–5 years
-
AP 2–15 months
-
BC 3–6 months.
Critique of manufacturer’s overview of current service provision
Bristol-Myers Squibb uses current treatment as counterfactual – imatinib 400 mg for first-line treatment of CML. However, the BMS cost-effectiveness analysis also compares their drug dasatinib with nilotinib. In its submission, BMS correctly uses the recently updated cost of £1724.39 per 30-tab pack for imatinib, which has not yet been published by BNF but is listed in MIMS.
Critique of manufacturer’s definition of decision problem
Population
The population in the BMS submission consists of the adults with newly diagnosed Ph+ CML in the CP. This is an adequate description of the population under consideration, and concurs with that defined in the NICE scope.
The BMS model uses an average age of 46 years old. This choice is based on the average age of patients in the DASISION trial. 29
Intervention sequences
Bristol-Myers Squibb has modelled one scenario with three different comparators. The interventions and sequence of treatments are summarised in Table 60.
| Line of treatment | Intervention | Comparator 1 | Comparator 2 |
|---|---|---|---|
| First line | Dasatinib (100 mg) | Imatinib (400 mg) | Nilotinib (600 mg) |
| Second line | Nilotinib (800 mg) | Dasatinib (100 mg) or nilotinib (800 mg) | Dasatinib (100 mg) |
| (50 : 50 spilt) | |||
| Third line | SCT or chemo/combination therapy or in-hospital palliative care | SCT or chemo/combination therapy or in-hospital palliative care | SCT or chemo/combination therapy or in-hospital palliative care |
Outcomes
In the BMS model, the outcomes for the economic analysis are incremental cost per QALY and incremental cost per life-year gained. There is no discussion of appropriate ways for measuring these outcomes in the decision problem section. However, these are the appropriate outcomes for this assessment.
Time frame
The BMS manufacturer submission state that a life time horizon is used, which is an appropriate timeline for modelling CML. However, in the BMS model nearly 20% of the population is alive at the end of the last cycle (86 years old).
Economic evaluation
Overview of manufacturer’s economic evaluation
The BMS base-case analysis produces an ICER of £26,305 per QALY for dasatinib compared with imatinib as first-line TKI, and £144,778 per QALY for dasatinib compared with nilotinib as a first-line TKI. Overall, we found the BMS economic model and evaluation to be based on plausible structural assumptions and input parameters, with the following exceptions:
The time horizon of the model does not follow a significant proportion of the population till death. Within the last cycle (86 years old) of the mode nearly 20% of the population remains alive.
In the context of the availability of second-generation TKIs for second-line treatment, the model ignores any additional effectiveness of second- and third-line treatments.
The PAS discount for first- and second-line nilotinib was not incorporated into the model.
A number of assumptions and parameters used within the model are not reflected in the manufacturer submission. In addition, there are discrepancies between the values stated in the manufacturer submission and the model.
A number of formulae errors have been identified in the model.
A full list of outputs from the original BMS model is presented below in Table 61.
| Dasatinib | Imatinib | Nilotinib | ||
|---|---|---|---|---|
| PFS (years, undiscounted) | Mean | 19.16 | 17.14 | 19.28 |
| PY (years, undiscounted) | Mean | 1.30 | 1.69 | 1.31 |
| Life-years (undiscounted) | Mean | 20.46 | 18.83 | 20.59 |
| QALYs (discounted) | PFS | 9.50 | 7.97 | 9.66 |
| PY | 1.14 | 1.92 | 1.04 | |
| Total | 10.64 | 9.89 | 10.70 | |
| First-line drug cost (discounted) | 283,209 | 84,836 | 282,887 | |
| Second-line FC drug acquisition cost (discounted) | 60,336 | 164,690 | 77,350 | |
| Third-line treatment cost (discounted) | 82,324 | 145,215 | 75,619 | |
| AEs first-line (discounted) | 2321 | 818 | 1291 | |
| AEs second line (discounted) | 412 | 1159 | 562 | |
| AEs third line (discounted) | 310 | 616 | 265 | |
| SCTa (discounted) | 5350 | 10,093 | 4954 | |
| Other | 63,955 | 70,864 | 63,685 | |
| Total costs (discounted) | 498,217 | 478,293 | 506,613 | |
| ICERs | ||||
| Cost/life-year gained (dasatinib vs imatinib) | 32,785 | |||
| Cost/life-year gained (dasatinib vs nilotinib) | 116,447 | |||
| Cost/QALY (dasatinib vs imatinib) | 26,305 | |||
| Cost/QALY (dasatinib vs nilotinib) | 144,778 | |||
Model structure
Bristol-Myers Squibb developed a ‘time in state’ (area under the curve) model, with the health states representing the early (CP) and advanced (AP/blast phase) stages as well as death. 169 This is based on extrapolating CML-related survival data and PCR data (Botteman et al. 170). Time is presented in blocks of 1 month, and patients were simulated from age 46 years until aged 86 years.
In the CP, patients can be on first-, second- or third-line treatment. Palliative care is only for patients in advanced phases (i.e. AP/blast phase). The model (Figure 47) distinguishes between disease stages (CP, AP/blast phase) and lines of treatment (first, second or third).
FIGURE 47.
Bristol-Myers Squibb model structure. Source: Figure 5, p. 40 of BMS submission.
The model is developed from the NHS perspective.
Natural history
The impact of TKIs on CML progression and survival is estimated using a combination of data on the effect of TKIs on CyR, and data on the impact of CyR on PFS and OS.
Effect data
Effect is defined as the probability that each TKI achieves a complete, partial and less-than-partial response. Full response is defined as CCYR, i.e. 0% Ph+ metastases at 12 months; partial response is defined as PCyR, i.e. ≤ 35% Ph+ metastases at 12 months; and less-than-partial response is defined as failed cytogenetic response, i.e. > 35% Ph+ metastases. The less-than-partial response is calculated as the residual of full and partial.
The clinical effectiveness data for those achieving a complete response in first-line therapy is taken from an unpublished systematic review commissioned by BMS. 171 This comprises a systematic review and network meta-analysis by Mealing et al. ,172 which pooled the effect estimates from the DASISION trial and another smaller trial by South West Oncology Group, updated to incorporate data presented at ASH 2010 and other peer-reviewed journals.
The clinical effectiveness data for those achieving a partial response in first-line therapy is taken directly from the respective RCTs – DASISION trial, for those receiving dasatinib and imatinib, and ENESTnd trial for those receiving nilotinib. Table 62 outlines the effectiveness of first-line therapy based on CyR category.
| Full (%) | Partial (%) | < Partial (%) | |
|---|---|---|---|
| Dasatinib 100 mg | 77.1 | 4.3 | 18.6 |
| Imatinib 400 mg | 62.4 | 14.6 | 23.0 |
| Nilotinib 600 mg | 77.7 | 4.3 | 18.0 |
The effectiveness of second-line TKI is assumed to be the same as second-line treatment post imatinib, as data for second-line treatment post dasatinib and nilotinib is not available. The data for second-line treatment is based on a report by PenTAG. 144 Table 63 outlines the effectiveness of second-line therapy based on response category.
| Full (%) | Partial (%) | Less than partial (%) | |
|---|---|---|---|
| Dasatinib 100 mg | 47.8 | 14.2 | 38.0 |
| Imatinib 800 mg | 16.3 | 16.3 | 67.4 |
| Nilotinib 800 mg | 35.1 | 15.3 | 49.6 |
Survival estimates
Both PFS and OS are modelled from CyR post first-line treatment. Data on the effectiveness of second-line therapy is not used to estimate either PFS or OS.
Surrogate outcome measures (e.g. level of CyR or molecular response) have been used in modelling CML as there is evidence that short-term response on these measures is predictive of longer-term survival or PCR. Also, the relationship between short-term CyR and long-term prognosis is believed (by BMS) to be similar for imatinib, dasatinib and nilotinib, although no references or research is cited to support this claim.
In the BMS model, CyR (and in particular a complete, partial or less-than-partial response at 12 months) is used as a predictor of both PFS and OS. This relationship has been demonstrated in the clinical literature and used in a recently published model of interventions for imatinib resistant CML patients. 104,173–175
The data for the OS curve and PFS curves are taken from a number of different sources. Table 64 summarises the sources used.
| Curve | Dasatinib | Imatinib | Nilotinib |
|---|---|---|---|
| Overall survival | |||
| Complete cytogenetic response | Roy et al. 2006 (IRIS study)104 | Roy et al. 2006 (IRIS study)104 | Roy et al. 2006 (IRIS study)104 |
| Partial cytogenetic response | Roy et al. 2006 (IRIS study)104 | Roy et al. 2006 (IRIS study)104 | Roy et al. 2006 (IRIS study)104 |
| < Partial cytogenetic response | Allen et al. 1995176 | Roy et al. 2006 (IRIS study)104 | Allen et al. 1995176 |
| PFS (all responses) | Hochhaus et al. 2009 (IRIS study)99 | Hochhaus et al. 2009 (IRIS study)99 | Hochhaus et al. 2009 (IRIS study)99 |
For patients receiving imatinib, long-term survival data are available from trial data. For patients receiving dasatinib and nilotinib, long-term survival information is unavailable since dasatinib and nilotinib have only recently been licensed for use in newly diagnosed CML patients (December 2010).
The Roy et al. (IRIS study) paper104 is a clinical trial focusing on the effectiveness of imatinib in comparison with interferon. Only data from patients in the imatinib arm of the IRIS study were used. It is assumed that the estimated OS for those on dasatinib and nilotinib with a CCyR and PCyR is the same as for those on imatinib, therefore data from Roy et al. 104 is used for all three comparators for complete and partial CyR. This assumption seems reasonable. It should be noted that the age group of the IRIS study is marginally older than the population which is modelled – 50 years old compared with 46 years old.
Data for the OS curve for a less-than-partial response for dasatinib and nilotinib is obtained from Allen et al. ,176 which is a clinical trial focusing on the effectiveness of interferon in comparison with cytotoxic drugs for the treatment of CML. It is assumed that the effectiveness of interferon for those with a less-than-partial CyR is similar to those with a less-than-partial CyR on dasatinib and nilotinib. In addition, the age group of the trial is significantly older than the population which is modelled – 57 years old compared with 46 years old.
The IRIS clinical trial data covers a period of 6 years. However, the majority of patients receiving imatinib in this trial were still both alive and on first-line therapy at the end of the trial (i.e. not progressed). 27,99 Therefore, the long-term trends of OS and PFS are not known. To extrapolate beyond the trial data, both the OS curves and PFS curves are based on Weibull distributions.
Health-related quality of life
Health-state utilities were taken from Szabo et al. 127 and are reproduced in Table 65. Szabo et al. 127 is a UK-, USA-, Australia- and Canada-based study, which derives utility values based on the TTO method. The utility values are based on interviewer-administered survey responses from a sample of the general population (n = 353, of which 97 were from the UK). Respondents were provided with descriptions of CML-related health states, which were derived in consultation with medical professionals, and which characterised the CP, AP and blast phase for both responding and non-responding states and for AEs.
| State | Value | Source |
|---|---|---|
| CP (responder) | 0.8500 | Szabo et al. 2010126 |
| CP (non-responder) | 0.6800 | Szabo et al. 2010126 |
| AP (responder) | 0.7900 | Szabo et al. 2010126 |
| AP (non-responder) | 0.5000 | Szabo et al. 2010126 |
| Blast phase (responder) | 0.5000 | Szabo et al. 2010126 |
| Blast phase (non-responder) | 0.3100 | Szabo et al. 2010126 |
| Progressed phasea (dasatinib) | 0.6346 | Calculated |
| Progressed phase (imatinib) | 0.5967 | Calculated |
| Progressed phase (nilotinib) | 0.6361 | Calculated |
| Post SCT | 0.7100 | (AiC information has been removed) |
The BMS model assumes that only patients with a full cytogenetic response receive the higher utility value and that those with either a partial or less-than-partial response receive the lower value.
Utility associated with the AP/blast phase health state was derived from the above values. The challenge in deriving these estimates is the lack of knowledge surrounding the proportion of time an individual can expect to spend in each health state. To derive the AP/blast phase health-state utility it is assumed that patients spend two-thirds of time in the AP, and one-third of time in the blast phase. These time proportions are then applied to the probability of responding and the associated utility values are outlined in Table 65 below.
For individuals who receive stem cell transplants, BMS uses a baseline utility value of 0.71. (AiC information has been removed.)
The AE decrements (Table 66) are derived primarily from the chemotherapy literature, and in particular previous NICE submissions. Where utility estimates for AEs were not available from the non-CML literature a 5% (–0.05) decrement was assumed as no reference has been identified.
| Event | Value | Source |
|---|---|---|
| Anaemia | –0.0730 | NICE 2006; LRIG178 |
| Diarrhoea | –0.0480 | NICE 2006; LRIG178 |
| Dyspnoea | –0.0500 | Doyle et al.179 |
| GI haemorrhage | –0.0500 | Assumption |
| Infection | –0.0500 | Assumption |
| Neutropenia | –0.1600 | Tabberer et al.180 |
| Pneumonia | –0.0500 | Assumption |
| Pyrexia | –0.0500 | Assumption |
| Rash | –0.0500 | Assumption |
| Thrombocytopenia | –0.0500 | Assumption |
We did not use the more recent TTO valuations of health states reported by Szabo et al. in our model because their methods do not meet the NICE reference case requirements, and because the study has a number of other notable weaknesses (Box 1, above). 127 (Being a TTO study in members of the public, the valuations produced by Szabo et al. do not reflect ‘changes in HRQoL as reported directly from patients’ and do not use the EQ-5D which is NICE’s preferred measure of HRQoL in adults. 150)
-
Although it is claimed that each health state description described the ‘typical patient experience’ of a person in that phase of CML (and either responding or not responding to treatment), at no point in the process of developing and testing these descriptions were patients with CML involved – only clinical experts and descriptions of symptoms in the literature were consulted.
-
The difference in the health-state descriptions for those responding and not responding to treatment is phrased entirely in terms being anxious and upset about the treatment not working and in terms of fear about the future: namely (for CP-CML) ‘My doctor has told me that my treatment is not working. This has made me anxious and upset’ and ‘I worry about my condition getting worse and I worry about my family. I understand that my health condition may get worse. I avoid making plans for the future’. Note that these distinctions are again based on how doctors perceive that CML patients are impacted when they are told they are not responding to treatment, and may bear little relation to the person’s wider health status and how it actually impacts on their quality of life.
-
It might be questioned whether or not a standard 10-year lifetime horizon for the TTO exercise may have biased responses, or at least whether or not they were compatible with assessing some of the states where life expectancy might nowadays be considerably longer than this.
Szabo et al. 127 also go on to make a number of misinformed criticisms of the EQ-5D based utilities from the IRIS study: first, they claim that IRIS did not collect EQ-5D data from patients in the accelerated or blast phase (they did, albeit in much smaller numbers); second, they claim that the pooling of data from different countries in the IRIS trial undermines the validity and applicability of the IRIS-based EQ-5D valuations (this seems flawed because the English-language EQ-5D was used in all four English-speaking countries, and UK-based valuations of the EQ-5D health states were used).
Resources and costs
Only direct medical costs incurred by the NHS (including staffing and primary care) are included in the model. All values have been inflated to 2010 using the Hospital and Community Health Services (HCHS) pay and prices inflation index. 181
Drug costs
Drug costs in the BMS model are identified from the BNF (2011). Where multiple options for achieving the same daily dose were available, BMS used a weighted average in the final calculation. BMS assumes the same BNF-derived cost for first- and second-line nilotinib (and therefore neither of these reflects the reduced price now available via the recently approved PASs). Table 67 presents the costs used in the model.
| Medication | Unit dose (mg) | Pack description | Pack price (£) |
|---|---|---|---|
| Dasatinib | 50 | 60-tab pack | 2504.96 |
| 100 | 30-tab pack | 2504.96 | |
| Imatinib | 100 | 60-tab pack | 862.19a |
| 400 | 30-tab pack | 1724.39a | |
| Nilotinib | 150 | 112-cap pack | 2432.85b |
| 200 | 112-cap pack | 2432.85 |
Adverse events costs
To cost AEs, BMS uses a number of sources:
-
Oxford Outcomes costing study (a survey of six UK-based clinicians who care for patients with CML)171
-
national UK databases
-
previous NICE oncology appraisals
-
expert opinion/assumption.
Where data from the NSRC are used, all information on elective and non-elective admissions has been identified and a weighted average was used in the model.
In deriving the cost estimate for each type of AE BMS have taken into account the proportion of people hospitalised for each AE and unit costs for an AE for those who were hospitalised and those who were not hospitalised. Separate values were specified for disease stage (CP or AP/blast phase). In deriving the final estimates used in the model BMS have assumed that two-thirds of time in the AP/blast phase state is spent in the AP stage and one-third in the blast phase stage. Table 68 below presents AE costs used in the BMS model.
| Event | Unit costs (£) | Sources/commentsa | |
|---|---|---|---|
| CP | AP/blast phase | ||
| Anaemia | 344.52 | 385.30 | |
| Diarrhoea | 82.17 | 82.17 | |
| Dyspnoea | 169.17 | 504.35 | |
| Fatigue | 21.90 | 21.90 | Derived from previous NICE appraisala |
| GI bleeding | 1082.61 | 1516.00 | |
| Headache | 809.16 | 809.16 | NHS SRC (currency code AA31Z)a |
| Infection | 574.88 | 1334.54 | |
| Leucopenia | 503.75 | 954.23 | Assumed same as neutropenia |
| Nausea | 270.90 | 270.90 | Derived from previous NICE appraisala |
| Neutropenia | 503.75 | 954.23 | |
| Pleural effusion | 184.43 | 286.01 | |
| Pneumonia | 949.09 | 1928.47 | |
| Pyrexia | 295.59 | 733.59 | |
| Skin rash | 152.62 | 188.02 | |
| Thrombocytopenia | 501.21 | 583.13 | |
| Vomiting | 0.00 | 0.00 | Assumed no additional cost of treatment |
Stem cell transplant cost
The BMS model uses an estimated monthly cost of (AiC information has been removed). This was regarded as an implausibly high level of ongoing costs by the NICE Appraisal Committee, which considered second-generation TKIs after resistance or intolerance to imatinib (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99. The committee recommendations are draft – consultees have the opportunity to appeal against them and final guidance has not been issued on this appraisal topic).
The overall cost of third-line treatment was adjusted so that only the proportion of patients who undergo SCT actually incur this one-off and additional ongoing cost.
Other costs
Other costs include outpatient visits, hospitalisation costs, various tests and scans. A full list of other costs used in the BMS model is presented in the Appendix (see Table 10).
Discounting
Costs and benefits were both discounted at annual rates of 3.5%, in line with the NICE reference case (NICE 2009).
Sensitivity analysis
Deterministic and PSAs have been conducted and presented. One-way statistical analysis was used to test the impact of disutility values (AEs). Additional parameters that BMS tested in the statistical analysis are presented in Table 69.
It was concluded that the model is sensitive to changes in the majority of parameters, and that the key drivers of cost-effectiveness are costs and QoL.
| Parameter | Set to | Dasatinib 100 mg vs nilotinib 600 mg | Dasatinib 100 mg vs imatinib 400 mg |
|---|---|---|---|
| Monthly cost of first-line dasatinib | 2000/3000 | ✓ | ✓ |
| Dose intensity (dasatinib, years 3+) | 1/0.75 | ✓ | ✓ |
| 12-month full response (first-line imatinib) | 0.5/0.8 | ✓ | |
| Monthly cost of first-line imatinib | 1500/2500 | ✓ | |
| 12-month no response (first-line imatinib) | 0.65/0.65 | ✓ | |
| Benefit discount rate (pa) | 0/0.06 | ✓ | |
| ICU ward-days (CP non-responding) | 0.5/1 | ✓ | |
| ICU ward-days (CP responding) | 0.5/1 | ✓ | |
| Monthly cost of second-line dasatinib | 2000/3000 | ✓ | ✓ |
| Monthly post-SCT cost | 1200/3600 | ✓ | |
| Dose intensity (imatinib, years 3+) | 1/0.75 | ✓ | |
| 12-month full response (first-line dasatinib) | 0.7/0.9 | ✓ | ✓ |
| Monthly cost of second-line nilotinib | 1500/4000 | ✓ | ✓ |
| Monthly cost of first-line nilotinib | 2000/3000 | ✓ | |
| Dose intensity (nilotinib, 3 years+) | 1/0.75 | ✓ | |
| 12-month full response (first-line nilotinib) | 0.7/0.9 | ✓ | |
| 12-month no response (first-line dasatinib) | 0.786/0.786 | ✓ | |
| 12-month switch rate (< partial, dasatinib) | 0.25/0.75 | ✓ | |
| 12-month no response (first-line nilotinib) | 0.8/0.8 | ✓ | |
| Dose intensity (nilotinib, year 1) | 0.9/0.8 | ✓ | |
| Dose intensity (dasatinib, year 1) | 0.9/0.8 | ✓ |
Model validation
In order to assess the clinical validity of the model results, a selection of key model outputs have been estimated (Table 44 in BMS submission) and presented. Given that all currently available long-term data, as well as clinical opinion, were used to construct the model, validation of these results is complex and largely indirect. However, BMS compared the results from this model with those from other models (Reed et al. ,116 (AiC information has been removed), PenTAG 2009, Ghatnekar et al. 175), and with additional short-term clinical data not used in model construction. 144,182
Uncertainty has been characterised through the use of statistical distributions. BMS presents the choice of distributions and the justification for each parameter category.
Major concerns with the Bristol-Myers Squibb model
Unfortunately, the BMS model does not use the cost of nilotinib agreed under the PAS in their submission.
The time horizon chosen by the BMS model does not reflect the lifetime of a CML patient. In the model, nearly 20% of the population is still alive in the last cycle (86 years old), suggesting that the model overestimates the period that those with CML will survive.
A number of assumptions and parameters used within the model are not reflected in the manufacturer submission. In addition, there are discrepancies between the values stated in the manufacturer submission and the model.
A number of formulae errors have been identified in the model.
Unfortunately, BMS assume that dasatinib is taken second line and third line. However, this has recently not been recommended in the NICE draft guidance FAD (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99).
The sources used to estimate the cost and proportions of patients who receive SCT are unclear.
Critical appraisal frameworks
This section summarises a critique of the BMS model. It is divided into the following two subsections:
-
appraisal of the BMS approach against general checklists
-
a critique of the BMS in light of the specific research problem.
Quality checklists
The BMS model has been appraised against the following commonly used quality checklists:
| NICE reference case requirement | Critical appraisal | Reviewer comment | |
|---|---|---|---|
| Defining the decision problem | The scope developed by the Institute | ✓ | |
| Comparator | Therapies routinely used in the NHS, including technologies regarded as current best practice | Comparator is either imatinib 400 mg daily and nilotinib 600 mg daily | |
| Perspective on costs | NHS and PSS | ✓ | |
| Perspective on outcomes | All health effects on individuals | ✓ | Disutility of AEs are included. Where disutility values could not be identified a value of –0.05 was assumed |
| Type of economic evaluation | Cost-effectiveness analysis | ✓ | |
| Synthesis of evidence on outcomes | Based on a systematic review | ✓ | Oxford Outcomes 2010: interventions used as first-line treatment for CML |
| Measure of health benefits | QALYs | ✓ | |
| Source of data for measurement of HRQoL | Reported directly by patients and/or carers | ✓ | Health state values based on Szabo et al.,127 which is an interviewer based survey of non-CML patients |
| Source of preference data for valuation of changes in HRQoL | Representative sample of the public | ✓ | |
| Discount rate | 3.5% per annum for costs and health effects | ✓ | |
| Equity weighting | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | ✓ | |
| Item | Critical Appraisal | Reviewer comment |
|---|---|---|
| Is there a well-defined question? | ✓ | – |
| Is there a clear description of alternatives (i.e. who did what to whom, where and how often)? | ✓ | – |
| Has the correct patient group/population of interest been clearly stated? | ✓ | No patient subgroups |
| Is the correct comparator used? | ✓ | Imatinib 400 mg daily and nilotinib 600 mg daily |
| Is the study type reasonable? | ✓ | Standard area under the curve model |
| Is the perspective of the analysis clearly stated? | ✓ | UK NHS and PSS |
| Is the perspective employed appropriate? | ✓ | – |
| Is effectiveness of the intervention established? | ✓ | |
| Has a lifetime horizon been used for analysis, and if not has a shorter time horizon been justified? | ✗ | At the last cycle of the model nearly 20% of the population is alive. The model needs to be extended to reflect a lifetime horizon |
| Are the costs and consequences consistent with the perspective employed? | ✓ | All costs from UK NHS and PSS perspective |
| Is differential timing considered? | ✓ | |
| Is incremental analysis performed? | ✓ | |
| Is sensitivity analysis undertaken and presented clearly? | ✓ | Univariate and PSAs clearly presented |
| Dimension of quality | Comments | ||
|---|---|---|---|
| Structure | |||
| S1 | Statement of decision problem/objective | ✓ | |
| S2 | Statement of scope/perspective | ✓ | NHS and PSS perspective. Cost and benefit inputs are consistent with the perspective. Scope of model stated |
| S3 | Rationale for structure | ✓ | Cohort model is appropriate |
| S4 | Structural assumptions | ? | Model assumptions are not explained clearly in the report. Model is highly complex |
| S5 | Strategies/comparators | ✓ | See S1 |
| S6 | Model type | ✓ | Cohort model is appropriate |
| S7 | Time horizon | ? | A lifetime horizon should have been adopted; however, nearly 20% of the population is alive at the last cycle |
| S8 | Disease states/pathways | ✓ | The disease states CP, AP, blast phase and death are commonly used for CML |
| S9 | Cycle length | ✓ | One month is appropriate |
| Data | |||
| D1 | Data identification | ✓ | Data identification methods are well described |
| D2 | Pre-model data analysis | ✓ | . |
| D2a | Baseline data | ✓ | Baseline data from Oxford Outcomes systematic review |
| D2b | Treatment effects | ✓ | |
| D2c | Quality-of-life weights (utilities) | ✓ | |
| D3 | Data incorporation | ? | Several explanations in the manufacturer submissions do not reflect the model |
| D4 | Assessment of uncertainty | ✓ | |
| D4a | Methodological | ✓ | |
| D4b | Structural | ✓ | |
| D4c | Heterogeneity | ✓ | No patient subgroups, as appropriate |
| D4d | Parameter | ✓ | Probabilistic and univariate sensitivity analyses performed |
| Consistency | |||
| C1 | Internal consistency | ? | Several logical errors identified within the cost-effectiveness model |
| C2 | External consistency | ✓ | |
Critique of the modelling approach and structure
The description of the BMS model (see Economic evaluation, above) identified a number of specific concerns with the BMS model. This section considers the implications of these concerns for the accuracy of the ICERs generated by the BMS model. Each concern is discussed in turn. The next section then concludes with a summary of the ICERs once relevant updates have been made to the model.
Formulae errors in the model
There were several formulae errors which were identified in the model calculations. Table 73 summarises these errors and Table 74 summarises the impact on the ICER.
| Description of error | Location (cells in Trace tab) |
|---|---|
| Major errors | |
| The QALY value of all those in a health state is based on the following formula: [(those in CP – those with SCT) × QALY] + [(those in AP/blast phase – those with SCT) × QALY)] + [(those with SCT) × QALY]. In the original formula there are two mistakes: (1) The SCT patients which are being subtracted are from the next cycle instead of the current cycle and (2) the number of SCT patients that are being subtracted is the cumulative value instead of the incremental value | Column IF, IM, and IT |
| For example, in cell IF75, based on the original calculation there are negative values of people in health states since the cumulative number of patients is being subtracted | |
| The probability of switching treatment from imatinib at 12 months when under < partial response: ‘PCT12MonthNCyRSwitchIMAT’ is input as 100% which contradicts Table 25 in the manufacturer submission where it clearly states this should be 58% | CX20 |
| The formula is using the wrong probability of switching, i.e. formula uses Pct18MonthPCyRSwitch but should be using Pct18MonthPCyRSwitchIMAT | CS26 |
| Minor errors | |
| The probability of switching at 18 months is applied to both cells where it should only be cell CU37 | CU37 and CU38 |
| The calculation of cost for third-line resource use for those who are new AP/blast phase patients (i.e. cell KP8) is using the population of new arrivals from next cycle instead of current cycle | Column GE and HL and HC |
| Formula is not using mortality adjusted population | Column GG and GH |
| Resource use cost was using dasatinib mortality unadjusted population for both CP and AP/blast phase | Column GS and HE |
| Formula is not using mortality adjusted population | Column GS and GT |
| ICER | Original values | After formula corrections | ||||
|---|---|---|---|---|---|---|
| ICER (£) | Incremental cost (£) | Incremental QALY | ICER (£) | Incremental cost (£) | Incremental QALY | |
| Dasatinib vs imatinib | 26,305 | 19,924 | 0.76 | 36,052 | 29,834 | 0.83 |
| Dasatinib vs nilotinib | 144,728 | (–8396) | –0.06 | 103,482 | (–8782) | –0.08 |
Application of Patient Access Scheme costs for nilotinib
The BMS model does not incorporate the new reduced price of nilotinib for first and second line under PAS. With (AiC information has been removed) discount in nilotinib, the best estimate ICER for dasatinib compared with imatinib is £45,600 per QALY. In the case of dasatinib compared with nilotinib, nilotinib is less costly and more effective.
Predicted survival
The structure of the BMS cohort-based cost-effectiveness model is appropriate. The use of the CP, AP and blast phase is appropriate and consistent with the clinical disease progression in trials.
However, a key concern with the model is that it does not adopt a lifetime time horizon, despite the submission stating that such a time horizon is adopted. The model runs for a cohort between 46 and 86 years old, at which point nearly 20% of the population remain alive. This suggests that the model overestimates the period that those with CML will survive.
This raises a number of questions of the BMS model. First, the model adopted a young onset age. Having stated that the average age at onset is 65 years old, BMS start the model at 46 years old. Furthermore, this onset age group is substantially younger than the population on which the trial data are established – 57 years old.
Second, the model seems to be overestimating the period of OS. Figure 48 shows cohort survival as predicted by the model until the age of 86 years, and as extrapolated beyond the model period by the review team (dashed line). At the end of the period modelled by BMS, nearly 20% of the cohort is still alive. Extending this survival trend beyond the model period demonstrates this implies that 10% of the cohort would be alive at age 100 years. This compares with 2% of the non-CML population alive at 46 years, who would be alive at 100 years (ONS).
FIGURE 48.
Overall survival predicted by BMS model. Graph produced by technology assessment group (i.e. not from BMS submission).
The impact on the ICERs of the extension to the period of the model to 100 years old is:
-
The ICER for dasatinib compared with imatinib reduces from £36,052 to £31,456 per QALY.
-
The ICER for dasatinib compared with nilotinib is reduced from £103,482 to £98,319 per QALY.
As summarised above (see Economic evaluation), estimates of survival are derived from the relationship between CyR and survival taken from the literature. Specifically, BMS state the source for the PFS curves as data from Hochhaus et al. ,99 which is a 6-year follow-up study of patients receiving imatinib in the first line. However, in the cost-effectiveness model, it appears that data from Druker et al. 27 is used instead, which is a 5-year follow-up study of patients receiving imatinib. The data extrapolated from Druker et al. estimate nearly identical PFS curves for those with a partial response and those with a less-than-partial response. 27 In comparison, data from Hochhaus et al. 99 estimate a higher PFS curve for those with a partial response than for those with a less-than-partial response. When the PFS coefficients estimated by Hochhaus et al. 99 are input into the model, the ICER for dasatinib compared with nilotinib increases from £45,600 to £52,574 per QALY. When comparing dasatinib with nilotinib, nilotinib continues to be dominant.
Updated Bristol-Myers Squibb results
Table 75 presents updated results after the formula error correction and in adjustment of nilotinib cost for first- and second-line therapy to equal PAS (AiC information has been removed). These results do not reflect the adjustments to the model to disallow dasatinib as second and third line.
| Dasatinib | Imatinib | Nilotinib | ||
|---|---|---|---|---|
| PFS (years, undiscounted) | Mean | 19.16 | 17.14 | 19.28 |
| PYs (years, undiscounted) | Mean | 1.30 | 1.69 | 1.31 |
| Life-years (undiscounted) | Mean | 20.46 | 18.83 | 20.59 |
| QALYs (discounted) | PFS | 10.16 | 9.17 | 10.24 |
| PY | 0.48 | 0.64 | 0.48 | |
| Total | 10.64 | 9.81 | 10.72 | |
| First-line drug cost (discounted), £ | 283,308 | 88,483 | (AiC information has been removed) | |
| Second-line drug acquisition cost (discounted), £ | 39,949 | 135,876 | 77,319 | |
| Third line treatment cost (discounted), £ | 82,062 | 135,775 | 75,416 | |
| AEs first line (discounted), £ | 2322 | 854 | 1292 | |
| AEs second line (discounted), £ | 412 | 1,156 | 562 | |
| AEs third line (discounted), £ | 309 | 565 | 264 | |
| SCT (discounted), £ | 5325 | 9281 | 4935 | |
| Other, £ | 63,899 | 67,861 | 63,971 | |
| Total costs (discounted), £ | 477,585 | 439,851 | 411,108 | |
| ICERs | ||||
| Cost/life-year gained, £ (dasatinib vs imatinib) | 62,093 | |||
| Cost/life-year gained, £ (dasatinib vs nilotinib) | (–922,003) | |||
| Cost/QALY, £ (dasatinib vs imatinib) | 45,600 | |||
| Cost/QALY, £ (dasatinib vs nilotinib) | (–783,367) | |||
Updated Bristol-Myers Squibb model to disallow dasatinib as second and third line
As explained above, BMS assume dasatinib is taken as second and third line. Technically, we adjusted the BMS model to disallow these options as follows.
-
First, to disallow dasatinib third line, in worksheet ‘thirdLineResUse’, cell D13 is changed from 0% to 100%, and cell D16 is changed from 80% to 0%. The ICER of dasatinib compared with imatinib then increases from £45,600 to £64,000 per QALY.
-
Next, to disallow dasatinib second line, in worksheet ‘Rx Sequence’, cell D12 changed from 50% to 0%, and cell D13 changed from 50% to 100%. The ICER of dasatinib compared with imatinib then increases from £64,000 to £96,000 per QALY.
Novartis submission
Summary
Scope of the submissions
The submission from Novartis considers the use of nilotinib (Tasigna®) for the first-line treatment of people with CML as an alternative to the standard dose of imatinib (400 mg daily). Dasatinib is used in the cost-effectiveness model as second-line treatment when first-line treatment with imatinib or nilotinib fails.
The clinical effectiveness outcomes considered are PFS, time to discontinuation, adverse effects of treatment and HRQoL.
The outcomes for the economic analysis were incremental cost per QALY, and incremental cost per life-year gained. In order to derive these outcomes the following costs were estimated in the model: cost of first- and second-line TKIs, cost of post-TKI failure second- or third-line treatment, and the cost of treating AEs. The time horizon for the economic analysis is lifetime and costs are considered from the NHS perspective.
The Novartis cost-effectiveness modelling reflects a cost discount (PAS) for the cost of first-line nilotinib (CiC information has been removed). Their cost of second-line nilotinib also reflects this cost discount (also a PAS). No subgroup analyses are conducted for the economic evaluation, although a policy scenario without the use of second-generation TKIs is simulated.
Summary of submitted cost-effectiveness evidence
The manufacturer uses a Markov approach to model the cost-effectiveness of nilotinib compared with the current standard of care (imatinib 400 mg daily). This model has nine states. Patients enter the model in the CP. The model estimates when one treatment fails and hence the patient is switched to an alternative treatment. At the end of each cycle, patients have a probability of remaining on current treatment, progressing to an alternative treatment or dying.
The Novartis model predicts that nilotinib is both more effective and less costly compared with imatinib, when followed by dasatinib as second-line treatment. In a scenario analysis without dasatinib as second-line treatment, the model predicts an ICER of £5908 per QALY for nilotinib in comparison with imatinib. The sensitivity analysis shows the key parameters which the cost-effectiveness results are sensitive to are drug costs (i.e. without PAS), and time to discontinuation of first-line TKI.
No major formula errors have been identified in the Novartis model.
Commentary on the robustness of submitted evidence
Strengths
-
The approach taken to modelling is reasonable.
-
The sources and justification of estimates are also generally reasonable.
Weaknesses
-
Novartis make no use of the major molecular and CCyR rates from the RCT of nilotinib compared with imatinib, both of which are important indicators of clinical effectiveness.
-
We believe that the Novartis method of estimating the time on hydroxycarbamide in CP is flawed.
Areas of uncertainty
The Novartis model does not provide the raw data that were used to fit the OS and time to treatment discontinuation curves. However, the choice of distribution and coefficients of the distribution appear to be correct on the basis of graphs showing the observed data and the fitted curves.
Another area of uncertainty is the chosen sequence of second-line TKI treatments that might follow the failure of different first-line TKIs. This is partly because this submission was prepared before NICE’s forthcoming draft guidance FAD on the use of dasatinib, nilotinib or high-dose imatinib as second-line treatments (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99). However, uncertainty also results from the fact that data on the effectiveness of second-line TKI treatments are available only for following the use of imatinib as first-line treatment.
Another area of uncertainty is regarding the cost and utility of stem cell patients. Assumptions around SCT significantly impact the model. Novartis uses a one-off cost of £99,224 for each transplant with a post-transplant utility for survivors of 0.813m, which decreases with age.
Key issues
Novartis use the PAS for pricing nilotinib as first-line treatment. This has significant impact on the results.
Novartis make no use of the major molecular and CCyR rates from the RCT of nilotinib compared with imatinib, both of which are important indicators of clinical effectiveness.
The cost and the proportions of patients who receive SCT differ between the Novartis and BMS models, and have a significant impact on ICERs. Clinical opinion is required on the BMS assumption that the provision and costing of SCT is appropriate.
Background
Critique of manufacturer’s description of underlying health problem
In section 1 of their submission, Novartis adequately describe the underlying health problem. Novartis state the median age for disease onset to be 55 years and disease prevalence in England and Wales as ∼2660 patients (2003 data from NICE TA70). Novartis use the following timeline to report phase duration of the disease:
-
CP: 3–5 years
-
AP: 1–2 years
-
BC: 3–12 months.
Critique of manufacturer’s overview of current service provision
Novartis use current treatment as the counterfactual (imatinib 400 mg for first-line treatment of CML) and the recently updated cost of £1724.39 per 30-tab pack for imatinib, which has not yet been published by BNF but is listed in MIMS.
Critique of manufacturer’s definition of decision problem
Population
The Novartis submission considers adult patients with Ph+ CML diagnosed in CP and who do not initially receive SCT. This is an adequate description of the population under consideration, and concurs with that defined in the NICE scope.
The Novartis model uses an average age of 57 years. This choice is based on the average age of patients in the ENESTnd trial. 20
Intervention sequences compared
Novartis modelled two different scenarios to reflect the availability or not of second-generation TKIs as second-line treatment. The interventions and sequence of treatment is summarised in Table 76.
| Line of treatment | Scenario 1 | Scenario 2 | ||
|---|---|---|---|---|
| Nilotinib | Imatinib | Nilotinib | Imatinib | |
| First line | Nilotinib (600 mg) | Imatinib (400 mg) | Nilotinib (600 mg) | Imatinib (400 mg) |
| Second line | Dasatinib (100 mg) | Dasatinib (100 mg) | SCT or hydroxycarbamide | SCT or hydroxycarbamide |
| Third line | SCT or hydroxycarbamide | SCT or hydroxycarbamide | NA | NA |
Outcomes
In the Novartis model, outcomes of the economic analysis were incremental cost per QALY and incremental cost per life-year gained. There was no discussion of appropriate ways for measuring these outcomes in the decision problem section. However, these are the appropriate outcomes for this assessment.
Time frame
Novartis used a lifetime horizon, which is an appropriate timeline for modelling CML.
Overview of manufacturer’s economic evaluation
The Novartis model estimates that nilotinib first line followed by dasatinib as second-line treatment would be both more effective (generating 0.55 extra discounted QALYs per patient) and less costly (£10,371 cheaper per patient) than imatinib followed by dasatinib. Without dasatinib as second-line treatment, the model predicts an ICER of £5908 per QALY for first-line nilotinib in comparison with imatinib.
Overall, we found the Novartis model to be robust. A full list of outputs from the original Novartis model is presented below in Table 77.
| Nilotinib/dasatinib | Imatinib/dasatinib | Nilotinib | Imatinib | |||
|---|---|---|---|---|---|---|
| PFS (years, undiscounted) | Mean | 12.66 | 11.94 | 10.64 | 9.30 | |
| PYs (years, undiscounted) | Mean | 0.88 | 0.90 | 0.74 | 0.68 | |
| Life-years (undiscounted) | Mean | 13.54 | 12.83 | 11.38 | 9.97 | |
| QALYs (discounted) | PFS | 9.93 | 9.38 | 8.31 | 7.25 | |
| PY | 0.47 | 0.48 | 0.40 | 0.37 | ||
| Total | 10.40 | 9.85 | 8.71 | 7.62 | ||
| First-line drug cost, £ (discounted) | (CiC information has been removed) | 104,038 | (CiC information has been removed) | 104,038 | ||
| Second-line drug acquisition cost, £ (discounted) | 57,532 | 77,284 | Refer to SCT | Refer to SCT | ||
| Third-line treatment cost, £ (discounted) | 170 | 175 | 411 | 147 | ||
| AEs first line, £ (discounted) | 111 | 178 | 111 | 178 | ||
| AEs second line, £ (discounted) | 37 | 51 | NA | NA | ||
| AEs third line, £ (discounted) | NA | NA | NA | NA | ||
| SCT, £ (discounted) | 28,772 | 31,183 | 42,383 | 49,986 | ||
| Other £ | 15,979 | 14,835 | 12,966 | 11,667 | ||
| Total costs, £ (discounted) | 217,373 | 227,744 | 170,643 | 166,015 | ||
| ICERs | ||||||
| Cost/life-year gained, £ (nilotinib vs imatinib, with second line) | –(27,739) | |||||
| Cost/life-year gained, £ (nilotinib vs imatinib, without second line) | 4701 | |||||
| Cost/QALY, £ (nilotinib vs imatinib, with second line) | –(34,889) | |||||
| Cost/QALY, £ (nilotinib vs imatinib, without second line) | 5908 | |||||
The base-case results in the Novartis report (p. 111) are different to those in the model. However, deterministic results in appendix (p. 132) agree with the model.
Model structure
A Markov model was developed in MS Excel 2007 for a hypothetical cohort of 1000 patients. The cycle length in the model is 1 month for the first 6 months, and then 3 months. A lifetime horizon is assumes in the model, with a final age of 100 years.
Equal numbers of male and female patients enter the model at the age of 57 years. Patients enter the model in CP. The model estimates when one treatment will fail and hence the patient is switched to an alternative treatment. At each cycle, patients have a probability of remaining on current treatment, progressing to an alternative treatment or dying (Figure 49). Patients are able to remain in CP, AP or blast phase for more than one cycle, and they may die from other causes at any time. Patients who receive a transplant may die from transplant-related mortality or remain well. Patients who are treated with hydroxycarbamide have a probability of progressing to AP. On progression to AP or blast phase, all patients are assumed to receive hydroxycarbamide therapy. Patients in AP have a probability of progressing to blast phase, and finally from blast phase to CML-related mortality. In blast phase, patients may only die as a result of CML. The Novartis model is developed from the NHS perspective.
FIGURE 49.
Novartis model structure.
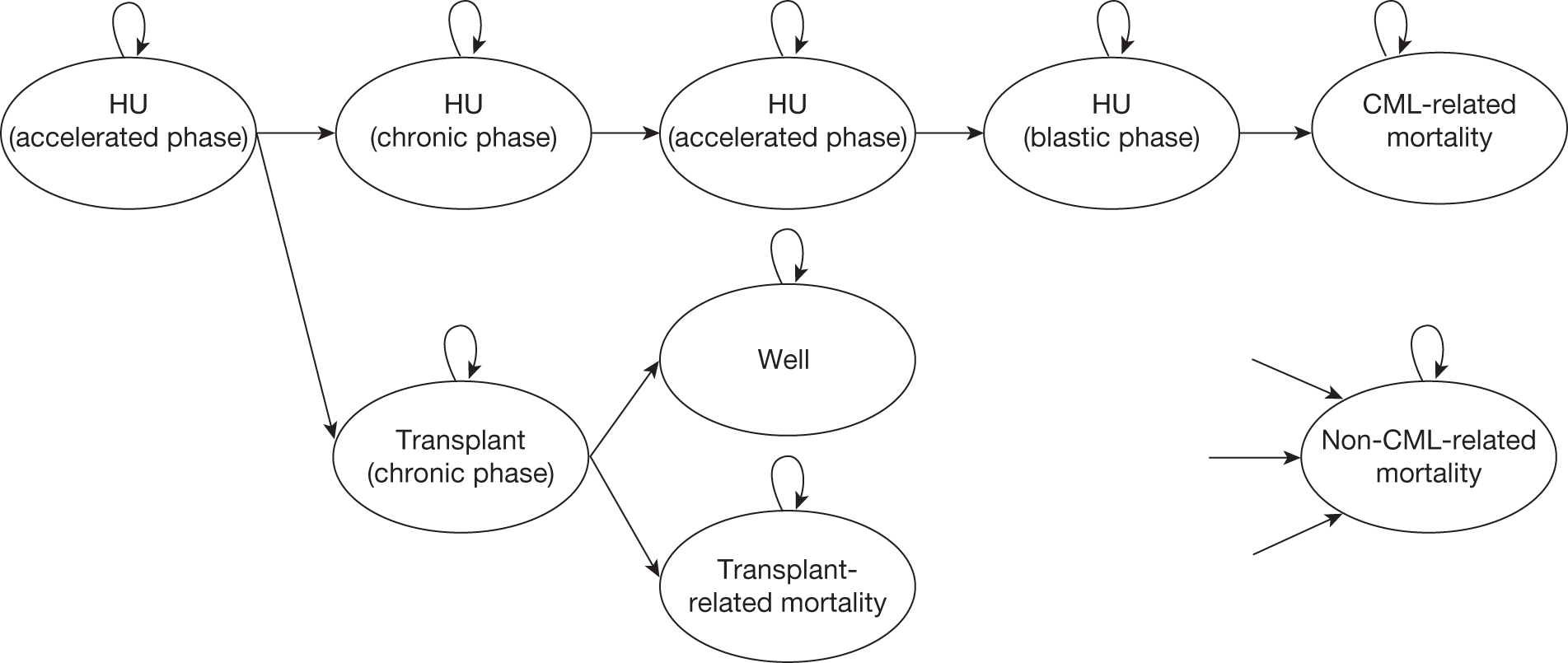
Natural history
The impact of TKIs on CML progression and survival is estimated using a combination of data on the effect of TKIs on discontinuity of treatment, and data on the relationship between discontinuity and PFS and OS.
Effect data
The Novartis model uses time to treatment discontinuation as the primary measure of the clinical effectiveness of the different treatments. The data for time to treatment discontinuation data used in the Novartis model are provided in Table 78. Table 79 summarises the multiple sources from which these data are taken.
| First-line treatment | First line | Second line (dasatinib) | Third-line CP (hydroxycarbamide) | Hydroxycarbamide | |||
|---|---|---|---|---|---|---|---|
| Imatinib | Nilotinib | Imatinib | Nilotinib | Imatinib and nilotinib | AP | Blast phase | |
| Per month for first 6 months in model | 0.05a | 0.13a | 0.28 | 0.22 | 0.052 | 0.104 | 0.101 |
| Per 3 months for > 6 months in model | 0.034a | 0.026a | 0.80 | 0.63 | 0.149 | 0.280 | 0.274 |
| Curve | First line | Second line | Hydroxycarbamide |
|---|---|---|---|
| Nilotinib | ENESTnd trial160 (24 Month Clinical Study Report) | Shah et al. (2010)185 and Garg et al. (2009)186 | CAMN107A 2101 trial |
| Imatinib | ENESTnd trial160 (24 Month Clinical Study Report) | Shah et al. (2010)185 | CAMN107A 2101 trial |
The data for first-line treatment is provided by the ENESTnd Trial (referred to as CAMN107A2303 in some parts of the industry submission). The trial assesses the clinical effectiveness of nilotinib in comparison with imatinib for newly diagnosed CP-CML patients, with a mean age of 47 years. The trial had a significantly younger starting population than the one included in the Novartis model (56 years old).
The data for second-line treatment is taken from two sources: Shah et al. 185 and Garg et al. 186 The Shah et al. 185 trial measured the effectiveness of dasatinib as second-line treatment after imatinib failure. Shah et al. 185 report the proportion remaining on treatment at 24 months. Based on this the monthly probability of discontinuing dasatinib post-imatinib is estimated as 0.22 and the quarterly probability is estimated as 0.63.
There is no study measuring the effectiveness of second-line dasatinib following nilotinib. It was assumed that second-line dasatinib following nilotinib would be less effective than when following first-line imatinib. In order to derive a lower effectiveness the effectiveness reported by Shah et al. 185 was averaged with the effectiveness reported by Garg et al. 186 who measure the effectiveness of dasatinib as third-line therapy. Similar to Shah et al. ,185 Garg et al. 186 report the proportion remaining on treatment at the end of the study. Based on these data the monthly probability of discontinuing dasatinib post nilotinib is estimated as 0.28 and the quarterly probability is estimate as 0.80. However, the median age for patients receiving dasatinib as third-line treatment in the study was 53 years old, which is slightly younger than the modelled population (56 years old).
Novartis state that the time spent in CP ( for data on time in state see Table 88) on hydroxycarbamide therapy is based on data reflecting the time in CP following second-line TKI treatment failure. The difference between the time to discontinuation and PFS curves is used to derive the number of years in CP on hydroxycarbamide. In order for this logic to be consistent, the PFS data should reflect progression only to the AP/blast phase. However, the data used to populate the PFS curve account for progression due to other reasons than progression to AP and blast phase, such as poor haematological response. Therefore, the estimated time spent in CP on hydroxycarbamide is not completely accurate. In addition, the source used to derive the PFS is not identified in the model. In the model it appears that the PFS data were fitted to an exponential curve. Based on the data, there is a 0.052 monthly probability of discontinuing hydroxycarbamide in CP and the quarterly probability is 0.149.
The time spent in the AP and blast phase on hydroxycarbamide is from the Kantarjian et al. study. 187 Novartis fit the data from the study to a number of different distributions to find the best way to extrapolate the data. Based on this an exponential curve was used. The exponential curve predicted a monthly probability of discontinuing hydroxycarbamide in the AP of 0.104, and a quarterly probability of 0.280. The monthly probability of discontinuing hydroxycarbamide in the blast phase, and ultimately leading to CML-related death, is 0.101, and the quarterly probability is 0.274.
Novartis assume that time on hydroxycarbamide and survival associated with SCT is independent of previous TKI treatments. For example, as can be seen in Table 78, it is assumed that the effectiveness of hydroxycarbamide is the same for both imatinib and nilotinib. In addition, these discontinuation rates are applied in both the scenario where second-line TKIs are available and the scenario where they are not available. Therefore it is assumed that hydroxycarbamide is equally effective following nilotinib and dasatinib failure, as it is with only nilotinib failure.
Survival data
Overall survival of patients is predicted based on the time to treatment discontinuation summarised in the previous section, which was used to determine transition probabilities within the Markov model. That is, survival is the cumulative result of the model’s assumptions about treatment discontinuation of first-, second- and third-line treatments (previous section). Figure 50 shows the OS predicted by the Novartis model. It demonstrates that at 100 years the entire population has died.
FIGURE 50.
Predicted OS: Novartis.
Time spent in AP and blast phase is based on data from Kantarjian et al. ,187 and is assumed to be the same independent of prior treatment. In order to model cost and QALY gains over a lifetime, the available evidence was extrapolated within the economic model.
Health-related quality of life
Novartis used evidence from a model-based cost-effectiveness analysis in which the utility estimates were based on responses to the EQ-5D preference-based measure of HRQoL of patients in the IRIS study who were receiving standard-dose imatinib. 116 Based on this paper, the modelled baseline utility of being in CP is assumed to be 0.854, while the baseline utility of being in AP or BC is 0.595. These utilities were assumed to be independent of drug therapy (in the Novartis model, utility is associated only with a given state as a person changes the state as he/she becomes a non-responder, thus it is reasonable for Novartis not to provide utility weight for non-responder as BMS has done).
Sensitivity analyses around the baseline utility values were conducted using utility values reported by Szabo et al. 127 Szabo et al. 127 is a UK-, USA-, Australia- and Canada-based study, which derives utility values based on the time trade-off method. The utility values are based on interviewer-administered survey responses from a sample of the general population (n = 353, of which 97 were from the UK). Respondents were provided with descriptions of CML-related health states that were derived in consultation with medical professionals, and which characterised the CP, AP and blast phase for both responding and non-responding states and for AEs.
The utilities are adjusted for age to take account of the fact that:
-
Patients in the modelled cohort (starting age 57 years) are older than patients in the IRIS study (mean age 50 years) from which the EQ-5D utility values were obtained.
-
The average utility of a given population decreases as age increases, for example the utility of patients remaining in good health in the CP will not remain constant but will decline gradually over time due to ageing. Assuming a constant utility by health state over time ignores the natural decline in quality of life associated with comorbidities, etc., potentially overestimating the benefits of treatment.
However, neither the Novartis report nor the model provide explanation about or further details on how the age adjustment calculation was undertaken.
The utility weights associated with SCT used in the Novartis model is 0.813. Further, in the base-case analysis, a decrement of 0.079 was applied to the long-term utility for 52% of patients following transplant to reflect common AEs associated with SCT. 188 Given that this relates to only one specific AE associated with allogeneic stem cell transplantation (alloSCT), it is likely to be an underestimate of the utility decrement experienced by patients following alloSCT. Table 80 presents utility weights that Novartis used in their model.
| State | Utility | Source |
|---|---|---|
| Health states | ||
| CP (first and second line) | 0.854 | Reed et al. 2004 (assumption for second line)116 |
| AP (first and second line) | 0.595 | Reed et al. 2004 (assumption for second line)116 |
| BC (first and second line) | 0.595 | Reed et al. 2004 (assumption for second line)116 |
| AEs | ||
| Disutility associated with AEs on nilotinib | 0.010 | Calculated |
| Disutility associated with AEs on imatinib | 0.016 | Calculated |
| Disutility associated with AEs on hydroxycarbamide | 0.000 | Assumption |
| Disutility associated with AEs on dasatinib | 0.019 | Calculated |
| Stem cell transplant (high-/low-risk groups) | 0.813 | Assumption |
| Utility decrement associated with SCT | 0.079 | Lee et al. 1997188 |
Only grade 3 and 4 AEs for TKI therapies were incorporated into the model because they are the most likely to impact upon quality of life and incur additional resource use beyond the routine appointments of these patients (see Table 82 for a list of AEs). It was assumed that grade 3 and 4 AEs would occur only within the first 18 months of treatment because the trial data suggest that very few grade 3 and 4 AEs occur beyond this time period. It was assumed that hydroxycarbamide therapy would not typically be associated with grade 3 and 4 AEs; hence disutility effects were applied only within the first 18 months for first-line treatment with nilotinib and imatinib, and second-line treatment with dasatinib.
Novartis searched the literature to identify utility values for common grade 3 and 4 AEs related to CML treatment with TKIs. AEs that were associated with substantial utility or cost impacts were included within the analysis. Where utilities were not available for these AEs related to CML, utilities associated with AEs for similar diseases were included. In general, these utilities were not based on EQ-5D data owing to the limited availability of this evidence. These utilities were used along with the duration of the AE and the probability of experiencing the AE to calculate the disutility of experiencing AEs resulting from first-line nilotinib or imatinib treatment and from second-line dasatinib treatment.
Resources and costs
Only direct medical costs are incorporated into the model. These include the costs associated with the different drug therapies, routine hospital appointments for administration and monitoring, and treatment for grade 3 and 4 AEs.
Drug costs
Drug costs used by Novartis are mainly taken from the BNF and are presented in Table 81.
| TKI treatment | Quarterly drug cost (£), including dose-intensity adjustments |
|---|---|
| First-line imatinib | 5547 |
| First-line nilotinib (with PAS) | (CiC information has been removed) |
| First-line nilotinib (without PAS) | 7319 |
| Second-line dasatinib | 7034 |
Novartis applies a cost discount (PAS) to nilotinib. (CiC information has been removed.)
For imatinib 400 mg, the cost of the 30-day pack is £1724, equivalent to a daily cost of £57.50. (CiC information has been removed.) The current NHS list price of 28-day pack of nilotinib is £2433 (600 mg); under the PAS, the cost per pack is (CiC information has been removed).
Adverse events costs
The costs of grade 3 and 4 AEs were considered because these were more likely to incur additional resource use beyond the regular intensive follow-up of these patients. The costs of grade 1 and 2 AEs were excluded as clinical expert opinion suggested that these would typically require minimal treatment and hence would have limited resource implications. Treatment for each AE was based on clinical expert opinion. The monthly costs of AEs associated with each therapy were weighted by their respective costs. AEs costs used by Novartis are presented in Table 82.
| AE | Cost (£)a | Assumption/source |
|---|---|---|
| Anaemia | 911 | One red blood cell transfusion [Varney and Guest (2003) inflated from 2000/1 to 2010/11]189 |
| Neutropenia | – | Minimal treatment |
| Thrombocytopenia | 537 | Weighted cost: grade 3 (64%) and grade 4 (36%) |
| Grade 3 | – | No treatment assumed for grade 3 |
| Grade 4 | 1493 | Three platelet transfusion [Varney and Guest (2003) inflated from 2000/1 to 2010/11]189 |
| GI bleed | 5233 | Five inpatient days (NHS Reference Costs 2008/9 inflated to 2010/11)190 |
| plus | ||
| cost of therapeutic endoscopic procedure (NHS Reference Costs 2008/09 inflated to 2010/11)190 | ||
| plus | ||
| three transfusions of platelet plus two transfusions of red blood cells [Varney and Guest (2003) inflated from 2000/1 to 2010/11]189 | ||
| CNS bleed | 4306 | Five inpatient days (NHS Reference Costs 2008/09 inflated to 2010/11)59,190 |
| plus | ||
| five transfusions of platelet [Varney and Guest (2003) inflated from 2000/1 to 2010/11]189 | ||
| plus | ||
| one CT scan (NHS Reference Costs 2008/9 inflated to 2010/11)190 | ||
| Pleural effusion | 2775 | Weighted cost: grade 3 (64%) and grade 4 (36%) |
| Grade 3 | 680 | Two inpatient days (NHS Reference Costs 2008/09 inflated to 2010/11)190 |
| Grade 4 | 6500 | One week intensive care ‘Adult Critical Care – 1 Organs Supported’ (NHS Reference Costs 2008/9 inflated to 2010/11)190 |
| Pericardial effusion | 1963 | Five inpatient days plus cost of two echocardiograms (NHS Reference Costs 2008/9 inflated to 2010/11)190 |
| CHF/cardiac dysfunction | 874 | Weighted cost: grade 3 (64%) and grade 4 (36%) |
| Grade 3 | 262 | Two echocardiograms (NHS Reference Costs 2008/9 inflated to 2010/11)190 |
| Grade 4 | 1963 | Five inpatient days plus cost of two echocardiograms (NHS Reference Costs 2008/9 inflated to 2010/11)190 |
Other costs
Based on clinical opinion, Novartis include the following appointments:
-
Patients in CP have a routine appointment at the start of treatment, with successive visits at intervals of 1, 2 and 4 weeks, and every 6 weeks thereafter.
-
Patients in AP are assumed to have six routine appointments per quarter.
-
Patients in BC are assumed to have 12 routine appointments per quarter.
Based on clinical advice, a routine appointment is assumed to be an outpatient visit, during which patients would receive a full blood chemistry test, plus a physical examination at every second appointment.
Patients are also likely to receive around three bone marrow tests during treatment. As these are low-cost tests, the model assumes that their cost is absorbed within the estimated cost of an outpatient visit.
The cost of each routine visit was therefore taken to be £138 (NHS reference costs 2008/9 – ‘Clinical Haematology: NHS Trusts Consultant Led Follow up Attendance Non-Admitted Face to Face’ inflated to 2010/11). 190
Novartis has also assumed based on clinical advice that patients will require, on average, a 2-week inpatient stay as end-of-life care.
The cost of alloSCT, in the first 100 days, is assumed to be £99,224 derived from a weighted average of the costs reported by the London Specialised Commissioning Group Workshop for related and unrelated donors, taking into account the cost of antifungal and donor lymphocyte infusion (DLI). 191
Table 83 summarises the other costs used in the model.
| Parameter | Value (£) | Source |
|---|---|---|
| Cost of routine appointment (outpatient visit) | 138 | NHS reference costs 2008/9 (inflated to 2010/11)190 |
| Cost of inpatient visits | 340 | NHS reference costs 2008/9 (inflated to 2010/11)190 |
| Cost of intensive care | 929 | NHS reference costs 2008/9 (inflated to 2010/11)190 |
| Cost of red blood cell transfusion | 911 | Varney and Guest 2003 (inflated from 2000/01 to 2010/11)189 |
| Cost of platelet transfusion | 498 | Varney and Guest 2003 (inflated from 2000/01 to 2010/11)189 |
| Therapeutic endoscopic procedure | 218 | NHS reference costs 2008/9 (inflated to 2010/11)190 |
| CT scan | 118 | NHS reference costs 2008/9 (inflated to 2010/11)190 |
| Echocardiogram | 131 | NHS reference costs 2008/9 (inflated to 2010/11)190 |
Discounting
All costs and QALYs are discounted by 3.5% as recommended by NICE.
Sensitivity analysis
A one-way sensitivity analysis is run to determine the impact of uncertainty in the following variables:
-
cost of alloSCT
-
cost of treating AEs
-
cost of first-line nilotinib treatment without the PAS
-
costs without dose adjustment
-
the impact of the disutility of alloSCT
-
baseline health state values (in CP and AP, BC)
-
disutility associated with AE.
Other parameters such as time horizon, age of patients and probability of receiving alloSCT have also been tested.
Probabilistic sensitivity analysis has been undertaken to explore the impact of joint uncertainty in all model parameters upon the cost-effectiveness results.
Model validation
No information on internal or external validation is presented by the manufacturer.
Major concerns with Novartis model
The Novartis model makes no use of cytogenetic or molecular response rates from the ENESTnd trial. 20
Critical appraisal frameworks
This section summarises a critique of the Novartis model. It is divided into the following two subsections:
-
appraisal of the Novartis approach against general checklists.
-
a critique of the Novartis in light of the specific research problem.
Quality checklists
The model was appraised against the following commonly used quality checklists:
| NICE reference case requirement | Critical appraisal | Reviewer comment | |
|---|---|---|---|
| Defining the decision problem | The scope developed by the Institute | ✓ | |
| Comparator | Therapies routinely used in the NHS, including technologies regarded as current best practice | Comparator is imatinib 400 mg daily. The model does not directly compare against first-line dasatinib which is the other current option available to patients | |
| Perspective on costs | NHS and PSS | ✓ | |
| Perspective on outcomes | All health effects on individuals | ✓ | Disutility of AEs are included. Where disutility values could not be identified a value of –0.05 was assumed |
| Type of economic evaluation | Cost−effectiveness analysis | ✓ | |
| Synthesis of evidence on outcomes | Based on a systematic review | ✓ | Oxford Outcomes 2010 – interventions used as first-line treatment for CML |
| Measure of health benefits | QALYs | ✓ | |
| Source of data for measurement of HRQoL | Reported directly by patients and/or carers | ✓ | Health state values based on Reed et al.118 based on the EQ-5D responses from patients within the IRIS study. The disutility values for AEs are mostly not based on EQ-5D data |
| Source of preference data for valuation of changes in HRQoL | Representative sample of the public | ✓ | |
| Discount rate | 3.5% p.a. for costs and health effects | ✓ | |
| Equity weighting | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | ✓ | |
| Item | Critical appraisal | Reviewer comment |
|---|---|---|
| Is there a well-defined question? | ✓ | – |
| Is there a clear description of alternatives (i.e. who did what to whom, where, and how often)? | ✓ | – |
| Has the correct patient group/population of interest been clearly stated? | ✓ | No patient subgroups |
| Is the correct comparator used? | ✓ | Imatinib 400 mg daily. Dasatinib is not included in the analysis, only as a second-line treatment |
| Is the study type reasonable? | ✓ | Standard Markov model |
| Is the perspective of the analysis clearly stated? | ✓ | UK NHS and PSS |
| Is the perspective employed appropriate? | ✓ | – |
| Is effectiveness of the intervention established? | ✓ | |
| Has a lifetime horizon been used for analysis, and if not has a shorter time horizon been justified? | ✓ | |
| Are the costs and consequences consistent with the perspective employed? | ✓ | All costs from UK NHS and PSS perspective |
| Is differential timing considered? | ✓ | |
| Is incremental analysis performed? | ✓ | – |
| Is sensitivity analysis undertaken and presented clearly? | ✓ | Univariate and PSAs clearly presented |
| Dimension of quality | Comments | ||
|---|---|---|---|
| Structure | |||
| S1 | Statement of decision problem/objective | ✓ | |
| S2 | Statement of scope/perspective | ✓ | NHS and PSS perspective. Cost and benefit inputs are consistent with the perspective. Scope of model stated |
| S3 | Rationale for structure | ✓ | Cohort model is appropriate |
| S4 | Structural assumptions | ✓ | Model assumptions are mostly explained clearly in the report. Overall, we are satisfied with the structural assumptions |
| S5 | Strategies/comparators | ✓ | See S1 |
| S6 | Model type | ✓ | Cohort model is appropriate |
| S7 | Time horizon | ✓ | |
| S8 | Disease states/pathways | ✓ | The disease states CP, AP, blast phase and death are commonly used for CML |
| S9 | Cycle length | ✓ | 3-month cycle is appropriate. The model accounts for a shorter cycle length in the beginning of the model to capture effect of AEs |
| Data | |||
| D1 | Data identification | ✓ | Data identification methods are well described |
| D2 | Pre-model data analysis | ✓ | |
| D2a | Baseline data | ✓ | Baseline data from RCT ENESTnd trial20 |
| D2b | Treatment effects | ✓ | |
| D2c | Quality of life weights (utilities) | ✓ | |
| D3 | Data incorporation | ✓ | Data incorporated in the model is referenced. See D2 |
| D4 | Assessment of uncertainty | ✓ | |
| D4a | Methodological | ✓ | |
| D4b | Structural | ✓ | |
| D4c | Heterogeneity | ✓ | No patient subgroups, as appropriate |
| D4d | Parameter | ✓ | Probabilistic and univariate sensitivity analyses performed |
| Consistency | |||
| C1 | Internal consistency | ✓ | |
| C2 | External consistency | ✓ | |
Critique of the modelling approach and structure
The approach adopted by Novartis was considered to be robust. Two issues were identified in the review of the model.
-
The Novartis model makes no use of cytogenetic or molecular response rates from the ENESTnd trial. 20
-
There are uncertainties around the cost and the proportions of patients who receive SCT.
At this stage no further analysis has been undertaken to investigate these issues.
Comparison of manufacturers’ models
Background
Both BMS (in section 2 of their report) and Novartis (in section 1 of their report) adequately describe underlying health problems in their reports. BMS state the median age at disease onset to be 65 years, whereas Novartis quotes median age as 55 years.
Table 87 shows that the duration of disease phases for those who are not treated differs slightly between the manufacturers’ descriptions of CML.
| CP (years) | AP | BC (months) | |
|---|---|---|---|
| BMS | 3–5 | 2–15 months | 3–6 |
| Novartis | 3–5 | 1–2 years | 3–12 |
Model outputs compared: state occupancy
This section describes and compares the main state occupancy and survival data predicted by each model. Table 88 presents time spent in each phase as predicted by the model. Table 89 presents the time spent in each line of treatment in two models.
| Phase | BMSa | Novartis | |||||
|---|---|---|---|---|---|---|---|
| Dasatinib | Imatinib | Nilotinib | Nilotinib/dasatinib | Imatinib/dasatinib | Imatinib | Nilotinib | |
| Chronic | 19.16 | 17.14 | 19.28 | 12.66 | 11.94 | 9.30 | 10.64 |
| Accelerated | 1.30 | 1.69 | 1.31 | 0.44 | 0.45 | 0.34 | 0.37 |
| Blast | 0.44 | 0.45 | 0.34 | 0.37 | |||
| Start age | 46 | 57 | |||||
| Mean age at death | 66.46 | 64.83 | 66.59 | 70.54 | 69.83 | 66.97 | 68.38 |
| Model | Treatment arm | First line | Second line | Third line | |||
|---|---|---|---|---|---|---|---|
| Description | Time | Description | Time | Description | Time | ||
| BMSa | Dasatinib | Dasatinib (100 mg) | 14.29 | Nilotinib (800 mg) | 3.16 | SCT or chemo/combination therapy or in-hospital palliative care | 3.01 |
| Imatinib | Imatinib (400 mg) | 5.09 | Dasatinib (100 mg) or Nilotinib (800 mg) | 9.02 | SCT or chemo/combination therapy or in-hospital palliative care | 4.72 | |
| Nilotinib | Nilotinib (600 mg) | 13.64 | Dasatinib (100 mg) | 4.29 | SCT or chemo/combination therapy or in-hospital palliative care | 2.67 | |
| Novartis | Nilotinib/dasatinib | Nilotinib (600 mg) | 7.28 | Dasatinib (100 mg) | 2.68 | SCT or hydroxycarbamide | 3.58 |
| Imatinib/dasatinib | Imatinib (400 mg) | 5.53 | Dasatinib (100 mg) | 3.55 | SCT or hydroxycarbamide | 3.75 | |
| Nilotinib | Nilotinib (600 mg) | 7.28 | SCT or hydroxycarbamide | 4.10 | NA | NA | |
| Imatinib | Imatinib (400 mg) | 5.53 | SCT or hydroxycarbamide | 4.44 | NA | NA | |
Table 88 demonstrates that the mean age of death in the two models is similar. This is partly explained by the different starting ages in the models. Given the earlier starting age in the BMS model, a similar age of death is produced by predicting much longer periods in each phase. This is the result of the different methods for predicting survival. In the BMS model the OS and PFS curves determine the proportion of the population in the AP/blast phase over time. In comparison in the Novartis model the proportion in AP/blast phase is determined by the discontinuation rate of hydroxycarbamide in the CP.
Drug costs
There is slight variation in the cost of treatment across the BMS and Novartis models. This is due to different dose intensity assumptions between BMS and Novartis, rather than listed costs used by the manufacturers. Table 90 outlines the drug costs that are used.
| TKI treatment | BMS | Novartis | ||
|---|---|---|---|---|
| Cost (£) per pack (see table 4.1.5.1) for details | Per day (£) | Quarterly drug cost (£), including dose intensity adjustments | Per day (£) | |
| First-line imatinib | 1724.39 | 57.48 | 5547.00 | 60.62 |
| First-line nilotinib (with PAS) | NA | NA | (CiC information has been removed) | (CiC information has been removed) |
| First-line nilotinib (without PAS) | 2432.85 | 86.89 | 7319.00 | 79.99 |
| Dasatinib | 2504.96 | 83.50 | 7034.00 | 76.87 |
Unfortunately, because of the timing when the industry submissions had to be supplied to NICE, the price of nilotinib used by BMS did not reflect the price discount recently approved under a PAS (two PASs: one for second line and one for first line). If the cost of nilotinib is adjusted to reflect the (CiC information has been removed) in the cost of nilotinib due to PAS, the cost of nilotinib in first line and second line is reduced from £2664 per month to (CiC information has been removed) per month. Based on this change, the BMS predicts an ICER of £45,600 for dasatinib compared with imatinib. When comparing dasatinib with nilotinib, the model predicts that nilotinib is more effective and less costly, and therefore nilotinib is the dominant comparator.
Other costs
In the BMS model there are three significant disease management costs: (1) costs associated with the management of patients in CP taking TKIs, and post-progression phase patients, (2) costs associated with third-line CP and AP/blast phase patients who do not receive SCT; and (3) costs of patients with SCT.
The resource use costs associated with each response category and costs associated with third-line therapy for non SCT patients are based on data from the Oxford Outcomes UK costing study. 171 This study identified the resource use and costs for treating patients with CML in the UK, as well as the frequency and length of hospital stay of patients with CML for managing serious (grade 3/4) treatment-related AEs and disease sequelae observed to occur in over 5% of CML patients enrolled in the large clinical trials. The Oxford Outcomes costing study incorporated a literature review, responses from six clinicians to the resource use questionnaire developed by Oxford Outcomes, and analysis of UK hospitalisation data from Hospital Episodes Statistics and the Cardiff Research Consortium. The results of the study are presented by type of CML patient.
(AiC information has been removed.)
(AiC information has been removed.)
Thus, (AiC information has been removed) references previous BMS submission, whereas current BMS submission references (AiC information has been removed) and, unfortunately, this approach provides no explanation for SCT cost derivation.
The Oxford Outcomes costing study quotes the cost of bone marrow transplant at £52,638 (2008). 171 It is unclear why BMS used other cost estimates from the Oxford Outcomes costing study and have not used this one. The original source used by BMS to cost SCT could not be traced.
In the Novartis model there are also three similar disease management costs: (1) costs associated with management of CP, AP and blast phase patients; (2) costs associated with treatment of patients who do not receive SCT (post-TKI failure); and (3) costs of STC patients. The management costs refer to the cost of routine appointments; each routine appointment costs £138. 190 The number of routine appointments varies by time, and is based on personal communication with medical experts. The cost of routine appointments over time can be found in Table 91. Patients who do not receive SCT are assumed to move to hydroxycarbamide therapy, which is £38 per 3 months (BNF, 2010). The cost of SCT is £99,224 based on data from the London Specialised Commissioning Group’s report on the cost of bone marrow transplant. 191
| Month | No. of routine appointments | Cost of routine appointments, first- and second-line therapy (£): nilotinib/imatinib |
|---|---|---|
| 1 | 3 | 414 |
| 2 | 1 | 138 |
| 3 | 0 | 0 |
| 4 | 1 | 138 |
| 5 | 0 | 0 |
| 6 | 1 | 138 |
| > 7 | 2 | 276a |
Table 92 provides a comparison of general resources costs across the two models, e.g. outpatient visits, tests, hospitals stays.
| Phase | BMS | Novartis | ||||
|---|---|---|---|---|---|---|
| Dasatinib | Imatinib | Nilotinib | Nilotinib | Imatinib | Dasatinib | |
| CP | 407 | 405 | 451 | (0, 414)a | (0, 414)a | (0, £414a) |
| AP | 490 | 488 | 539 | 92 | £92 | 92 |
| Blast phase | 182 | 182 | 182 | |||
It is evident from Table 92 that resource costs are different across the two models. Overall, BMS appear to have larger resource costs. This may imply that the Novartis model has underestimated disease management costs. In addition, the BMS model accounts for resource costs associated with patients who receive SCT. However, in the Novartis model there are no additional resource costs associated with SCT patients – only the cost of the transplant is considered. Table 93 shows the additional resource use associated with third-line therapy for non-SCT patients.
| Phase | BMS | Novartis | ||
|---|---|---|---|---|
| Probability of not receiving SCT (%) | Cost of care per month (£) | Probability of not receiving SCT (%) | Cost of care per month (£) | |
| CP | 69.2 | 2467 | 25a | 38 |
| AP/blast phase | 50.0 | 4,836 | NA | NA |
It is clear from Table 93 that BMS has substantially higher monthly costs associated with the treatment of patients without SCT. In the Novartis model, patients who do not receive SCT move to hydroxycarbamide therapy, which has a minimal cost of £38 per month. In the BMS model, patients who do not receive SCT are assumed to receive either chemotherapy care (in CP) or hospital care (in AP/blast phase), both of which are at a considerable cost. In addition, the BMS model assumes it is possible to receive SCT in the CP and the AP/blast phase in comparison with the Novartis model where SCT is available only to CP patients. Table 94 summarises the differences in costs associated with receiving SCT between both models.
| BMS | Novartis | ||||||
|---|---|---|---|---|---|---|---|
| Dasatinib | Imatinib | Nilotinib | Nilotinib/dasatinib | Imatinib/dasatinib | Imatinib | Nilotinib | |
| Percentage receiving SCT | 7.6 | 13.8 | 7.0 | 33.2 | 36.0 | 54.7 | 47.8 |
| Cost of SCT (£) | (AiC information has been removed) | 99,224 | |||||
In the Novartis model a more substantial percentage of the population receive SCT, even when including second-line TKIs. This is driven by the Novartis assumption that if TKI failure occurs, 75% of patients < 65 years old will receive SCT. This assumption is tested within the Novartis model and is shown not to impact the ICER greatly. In comparison, in the BMS model if TKI failure occurs there, is a 30.8% change of receiving SCT in the CP for any age, and a 50% chance of receiving SCT in the AP and blast phase at any age which is based on data from the Oxford Outcomes study. 171 The probability of receiving SCT used by BMS is tested in the additional sensitivity analysis performed by PenTAG.
(AiC information has been removed.) This cost is significantly higher than the cost of STC predicted by Novartis (£99,224 per transplant). When accounting for the formulae errors and discounted price of nilotinib in the first- and second line due to PAS, if the one-off cost of SCT per QALY from the Novartis model is input in the BMS model, while removing the (AiC information has been removed), there is a substantial increase in the ICER value, from £27,639 to £78,791 per QALY for dasatinib compared with imatinib. The inclusion of an (AiC information has been removed) is inflating the cost for all patients receiving SCT. The assumption made by BMS increases the total cost of imatinib as more patients in the imatinib arm receive SCT. The approach used by BMS is based on viewing SCT solely as a cost, since any treatment benefit is implicitly included in the ITT survival data used to inform the parametric survival analysis discussed previously (see Survival estimates, above).
Adverse event costs
Both the BMS and Novartis models account for AEs, but differ in the types of AEs that are included. The BMS model incorporates a wider range of AEs than Novartis. Table 95 summarises the types of AEs that are included in each model.
The estimated cost of AEs also differs between the models. Table 96 outlines the cost of AEs per month by phase of disease. As the incidence rates of AEs are very small, the difference in costs only has a small impact on the ICER and therefore costs were not tested with additional sensitivity analysis by PenTAG.
| Type of AE | BMS | Novartis |
|---|---|---|
| Anaemia | ✓ | ✓ |
| Diarrhoea | ✓ | – |
| Dyspnoea | ✓ | – |
| Fatigue | ✓ | – |
| Headache | ✓ | – |
| Infection and Infestations | ✓ | – |
| Leucopenia | ✓ | – |
| Nausea | ✓ | – |
| Neutropenia | ✓ | ✓ |
| Pleural effusion | ✓ | ✓ |
| Pyrexia | ✓ | – |
| Skin rash | ✓ | – |
| Thrombocytopenia | ✓ | ✓ |
| Vomiting | ✓ | – |
| GI bleed | – | ✓ |
| CNS bleed | – | ✓ |
| Dasatinib (£) | Imatinib (£) | Nilotinib (£) | ||
|---|---|---|---|---|
| BMS | CP | 20.83 | 16.88 | 12.07 |
| AP/blast phase | 0.00 | 28.29 | 26.19 | |
| Novartis | CP | 23.34 | 11.61 | 6.95 |
| AP/blast phasea | NA | NA | NA |
Another difference between the two models is the assumption about the duration of AEs. In the BMS model, AEs occur through the lifetime of the model and are based on the proportion of the cohort in CP and AP/blast phase. In the Novartis model, the incidence of AEs is assumed to last only up to 18 months. Therefore, after 18 months, there is no cost of AEs.
Health-related quality of life
The BMS and Novartis models use different sources for the utility associated within each disease state. Table 97 outlines the differences between the utility values.
| State | BMS | Novartis | ||
|---|---|---|---|---|
| Value | Source/notes | Value | Source/notes | |
| CP | 0.8500 | Responder (Szabo et al.)127 | 0.854a,c | (Reed et al.)116 Same value assumed for second line |
| 0.6800 | Non-responderb (Szabo et al.)127 | |||
| AP | 0.7900 | Responder (Szabo et al.)127 | 0.595a,c | (Reed et al.)116 Same value assumed for second line |
| 0.5000 | Non-responderb (Szabo et al.)127 | |||
| Blast phase | 0.5000 | Responder (Szabo et al.)127 | 0.595a,c | (Reed et al.)116 Same value assumed for second line |
| 0.3100 | Non-responderb (Szabo et al.)127 | |||
| SCT | 0.7100 | (AiC information has been removed) | 0.813c | Assumption. Disutility associated with SCT (0.079) is applied Lee et al.191 |
Szabo et al. 127 is a UK-, USA-, Australia- and Canada-based study that derives utility values based on the time trade-off method. The utility values are based on interviewer-administered survey responses from a sample of the general population (n = 353, of which 97 were from the UK). Respondents were provided with descriptions of CML-related health states that were derived in consultation with medical professionals.
Reed et al. 116 estimate utility values based on responses to EQ-5D questionnaires from patients in the IRIS study receiving standard dose imatinib. The average age of patients in the study was 50 years old. Owing to the younger age of the participants in the study compared with the cohort in the Novartis model, adjustments were made to the utility values to reflect age. The adjustment to utility values is not clearly described in either the manufacturer submission or the cost-effectiveness model.
The two biggest differences in utility values between the models appear to be within the AP and for SCT patients. To test the differences in utility values, the value for the AP estimated by Reed et al. 116 is input into the BMS model for AP responders. In addition, post-SCT value used by BMS is input into the Novartis model. Table 98 outlines the subsequent changes in ICERs due to changes in the utility values. The table shows the changes in the utility values have a minor impact on the ICER.
| Value | Novartis | BMS | ||
|---|---|---|---|---|
| Nilotinib vs imatinib with second-line dasatinib | Nilotinib vs imatinib without second-line dasatinib | Dasatinib vs imatinib | Dasatinib vs nilotinib | |
| Original unadjusted | (–£34,889) | £5908 | £36,052 | £103,483 |
| AP (0.595) | – | – | £35,538 | £104,451 |
| SCT (0.71) | –(£33,893) | £5658 | – | – |
The disutility of AEs also differs between the models. Table 99 outlines the disutility of AEs by phase of disease. As the disutility of AEs is minimal, the difference in values is likely to have only a small impact on the ICER and therefore values were not tested with additional sensitivity analysis.
| Model | Phase | Dasatinib | Imatinib | Nilotinib |
|---|---|---|---|---|
| BMS | CP | 0.005 | 0.004 | 0.002 |
| AP/blast phase | 0.004 | 0.004 | 0.004 | |
| Novartis | CP | 0.19 | 0.16 | 0.10 |
| AP/BCa | NA | NA | NA |
A major difference between the two models is the assumption about the duration of AEs. In the BMS model AEs occur through the lifetime of the model and are based on the proportion of the cohort in CP and AP/BC. In the Novartis model, the incidence of AEs is assumed to only last up to 18 months. Therefore, after 18 months there is no disutility of AEs.
Discussion
Summary of cost-effectiveness issues
Novartis use the PAS for pricing nilotinib as first-line treatment. This has significant impact on the results, and is unfortunately not reflected in the BMS model.
BMS and Novartis use different second- and third-line treatments. BMS assumes that dasatinib and nilotinib are both available as second-line treatments. In one scenario, Novartis assumes that only dasatinib is available second line, whereas in another scenario, it assumes no TKI second line. However, NICE’s draft guidance FAD has recently recommended nilotinib, but not dasatinib second line (the draft guidance FAD for second-line, high-dose imatinib, dasatinib and nilotinib for CML is available on the NICE website at http://guidance.nice.org.uk/TA/WaveR/99).
The time horizon chosen by the BMS model does not reflect the lifetime of a patient with CML. In the model, nearly 20% of the population is still alive in the last cycle (86 years old).
The BMS model has a number of formulae errors, correcting for which impacts ICER.
The cost and the proportions of patients who receive SCT differ between the models and has a significant impact on ICERs.
Appendix
| Event | Cost (£) | Source |
|---|---|---|
| Outpatient visits | ||
| Nurse | 25 | Curtis (2008) section 8.6. Value represents the hourly rate for a GP practice nurse181 |
| Haematologist/oncologist | 108 | Curtis (2008) section 13.5. Value represents the hourly rate for a general medical consultant181 |
| Tests | ||
| Complete blood count | 2.97 | NHS SRC. Currency code DAP823 |
| Cytogenetic analysis | 17.03 | NHS SRC. Currency code DAP838 |
| Bone marrow aspiration with biopsy | 637.10 | NHS SRC. (original value £565.26)a |
| FISH | 17.03 | NHS SRC. Currency code DAP838 |
| PCR | 1.34 | NHS SRC. Currency code DAP841 |
| Flow cytometry | 87.01 | NHS SRC. Currency code DA08 |
| Cytochemistry analysis | 17.03 | NHS SRC. Currency code DAP838 |
| Blood film examination | 2.97 | NHS SRC. Currency code DAP823 |
| Chest radiograph | ||
| CT scan chest | 116.72 | NHS SRC. Currency codes RA08Z – RA14Z |
| Blood chemistry | 1.34 | NHS SRC. Currency code DAP841 |
| Kinase domain mutation | 87.01 | NHS SRC. Currency code DA08 |
| C-reactive protein | 7.42 | NHS SRC. Currency code DAP831 |
| EKG | 131 | NHS SRC. Currency codes EA46Z, EA47SZ |
| Upper endoscopy | 221.14 | NHS SRC. Currency codes FZ26A, FZ27C |
| Hospitalisation | ||
| Day on a general ward | 246.41 | NHS SRC. Weighted average of all non-elective excess bed-day costs |
| Day in intensive care unit | 1219 | NHS SRC. Currency codes XC01Z-XC07Z (burns, spinal injuries and general critical care) |
| Day in hospice | 233 | Curtis (2008) section 1.5 Nursing-Led Inpatient Unit (NLIU) for intermediate care181 |
| Other | ||
| Blood transfusion | 57.07 | NHS SRC. Service code 821 |
| Donor lymphocyte infusion | 57.07 | Assumed same as blood transfusion |
| Platelet transfusion | 57.07 | Assumed same as blood transfusion |
| Lumbar puncture | 87 | NHS SRC. Currency code DA08 |
Appendix 8 WinBUGS mixed-treatment comparison analysis of complete cytogenetic response and major molecular response rates
The method of conducting the mixed-treatment comparison (MTC) for the CCyR and MMR in the two RCTs of first-line dasatinib and nilotinib involves two steps.
First, a fixed-effects MTC model (Lu and Ades 2006192) was used in WinBUGS (MRC Biostatistics Unit, Cambridge, UK)(Spiegelhalter et al. 2003193) to impute estimates of the response rates for CCyR and MMR for dasatinib from Saglio et al. 20 and for nilotinib (300 mg) from Kantarjian et al. 29 (the shaded cells in Tables 36 and 37). The MTC model allows estimation of the shaded cells using the precision of the available data. The WinBUGS code for this analysis is given below;
To fit this model, it was assumed that the total number of participants for the dasatinib arm of Saglio et al. 20 would have been 282 (as in the nilotinib arm), had dasatinib also been included in the trial. Similarly, it was assumed that the total number of participants for the nilotinib arm of Kantarjian et al. 29 would have been 259 (as in the dasatinib arm), had nilotinib also been included in the trial. Prior distributions, intended to be vague, were placed on the estimates of the trial baseline and treatment effects (e.g. and in the WinBUGS code). The impact of using different vague priors and different assumptions on the assumed total number of participants in the dasatinib arm of Saglio et al. 20 and the nilotinib arm of Kantarjian et al. 29 was assessed.
Second, all estimated response rates (those reported and those imputed from above) are assumed to follow a normal distribution. A fixed-effects meta-analysis (Sutton et al. 2000194) was then undertaken in WinBUGS to obtain an overall estimate of response rate for each treatment. Prior distributions, intended to be vague, were placed on the unknown parameters.
A burn-in of 20,000 iterations was used for both of the above steps, with estimates based on a sample of 200,000 iterations. Convergence of the analysis was checked using the trace, auto-correlation and density plots within WinBUGS.
The analyses were deemed to have been based on convergent samples and there was no impact on the results by assuming different prior distributions for the unknown parameters. There was no impact of assuming alternative total numbers of participants in the dasatinib arm of Saglio et al. 20 and the nilotinib arm of Kantarjian et al. 29
Glossary
- Allogeneic transplant
- A bone marrow or stem cell transplant using marrow from another person.
- Basophilia
- An excess number of basophils (a rare type of white cell) in the peripheral blood.
- Blast cells
- Immature cells found in, and produced by, the bone marrow. Not normally found in the peripheral blood.
- Bone marrow
- The soft substance that fills bone cavities. It is composed of mature and immature blood cells and fat. Red and white blood cells and platelets are formed in the bone marrow.
- Bone marrow transplant
- A procedure by which a patient’s bone marrow is replaced by healthy bone marrow. The bone marrow to be replaced may be deliberately destroyed by high doses of chemotherapy and/or radiation therapy. The replacement marrow may come from another person or it may be previously harvested from the patient’s own marrow.
- Chemotherapy
- The treatment of a disease by chemicals to destroy cancer cells. Chemotherapy can affect the whole body.
- Cytogenetic response
- A response to treatment at the level of chromosomal abnormalities. In the case of chronic myeloid leukaemia, it is assessed by counting the number of Philadelphia chromosome-positive cells (Ph+) in metaphase (usually 20 metaphases are analysed). A complete response generally means no Ph+ cells, a partial response leaves up to 35% Ph+ cells evident, and with a minor response from 35% to 95% Ph+ cells are still evident.
- Cytopenia
- A reduction in the number of cells circulating in the blood.
- EQ-5D
- A European quality-of-life questionnaire containing five physical and psychological dimensions.
- Extramedullary disease
- Disease occurring outside the bone marrow.
- Haematological response
- A haematological response refers to the normalisation of blood cell counts. Chronic myeloid leukaemia causes over-proliferation of white blood cells, which treatments aims to lower, and categories of response indicate the extent to which this occurs. Typically, the haematological response is classified as complete if the number of white blood cells is < 10 × 109/l, the number of platelets is < 450 × 109/l, there are no immature cells in the peripheral blood with normal differential count, and disappearance of symptoms and signs occurs.
- Hydroxycarbamide
- A drug that is used in the treatment of chronic myeloid leukaemia that inhibits DNA synthesis.
- Incremental cost-effectiveness ratio
- Demonstrates the total additional cost per quality-adjusted life-year gained of one alternative over another. There is no particular point at which an alternative is said to be ‘cost-effective’, as this will be a policy decision. The larger the incremental cost-effectiveness ratio, the less likely it is to be cost-effective.
- Interferon alpha
- Interferon is a protein derived from human cells. It has a role in fighting viral infections by preventing virus multiplication in cells. Interferon alpha is made by leucocytes. It is often used as first-line therapy for CML.
- Kaplan–Meier estimator
- Also known as the product limit estimator. This is an estimator for estimating the survival function from lifetime data. In medical research, it is often used to measure the fraction of patients living for a certain amount of time after treatment.
- Landmark analysis
- A form of survival analysis in which only patients who have survived a specified period of time are included.
- Leucocytes
- The white blood cells that are responsible for fighting infections.
- Leucopenia
- A reduced number of white cells in the blood – it may affect a single cell type or all white cells.
- Leukapheresis
- A process of removing excess white blood cells from the peripheral blood.
- Metaphase
- The second phase of mitosis (cell division). Cells in this phase of division are used for cytogenetic analysis in chronic myeloid leukaemia to identify the proportion of Philadelphia chromosome-positive chromosomes.
- Mitosis
- A division of cells, which consists of four phases: prophase, metaphase, anaphase and telophase.
- Myelocytes
- Committed progenitor cells produced by, and found in, the bone marrow, which develop into mature leucocytes.
- Neutropenia
- A decrease in neutrophils (white blood cells) circulating in the blood.
- Neutrophil
- The most common type of white blood cell in humans and mammals (also known as neutrophil granulocytes).
- Oncogene
- A gene that has the potential to cause cancer.
- Peripheral blood
- In this report, peripheral blood refers to blood in the circulatory system.
- Platelet
- Small fragments of cells found in the blood, which help to form clots and control bleeding (also called ‘thrombocytes’).
- Promyelocytes
- Committed progenitor cells produced by, and found in, the bone marrow, which develop into myelocytes.
- Stem cells
- Very early progenitor cells, which divide and mature to become all of the types of cells that make up the blood and immune system.
- Thrombocytes
- Platelets (fragments of bone marrow cells) found in the blood, which help to form clots and control bleeding.
- Thrombopenia
- A reduced number of thrombocytes (platelets) in the blood.
- Toxicity
- The quality of being poisonous. The National Cancer Institute grades the toxicity levels of treatments as 1 = mild, 2 = moderate, 3 = severe and 4 = life-threatening.
- Tyrosine kinase
- An enzymatic protein that adds phosphate residues to other proteins in the cell. In chronic myeloid leukaemia, the abnormal tyrosine kinase BCR–ABL (oncogene fusion protein consisting of BCR and ABL) phosphorylates proteins that cause cellular proliferation.
- Weibull distribution
- A continuous probability distribution, which is usually used in survival analysis.
List of abbreviations
- ABL
- Abelson oncogene
- AE
- adverse event
- AiC
- academic-in-confidence
- alloHSCT
- allogeneic haematopoietic stem cell transplant
- alloSCT
- allogeneic stem cell transplantation
- ALT
- alanine aminotransferase
- AP
- accelerated phase
- AST
- aspartate aminotransferase
- BC
- blast crisis
- BCR
- breakpoint cluster region
- BCR–ABL
- oncogene fusion protein consisting of BCR and ABL
- b.i.d.
- twice a day
- BMS
- Bristol-Myers Squibb
- BNF
- British National Formulary
- CB
- comorbities
- CCyR
- complete cytogenetic response
- CENTRAL
- Cochrane Central Register of Controlled Trials
- CHR
- complete haematological response
- CI
- confidence interval
- CiC
- commercial-in-confidence
- CML
- chronic myeloid leukaemia
- CMR
- complete molecular response
- CNS
- central nervous system
- CP
- chronic phase
- CP-CML
- chronic myeloid leukaemia in chronic phase
- CRD
- Centre for Reviews and Dissemination
- CyR
- cytogenetic response
- DARE
- Database of Abstracts of Reviews of Effects
- ECOG
- European Cooperative Oncology Group
- EFS
- event-free survival
- EMA
- European Medicines Agency
- EQ-5D
- European Quality of Life-5 Dimensions
- FAD
- final appraisal determination
- FDA
- US Food and Drug Administration
- FISH
- fluorescence in situ hybridisation
- GI
- gastrointestinal
- GvHD
- graft-versus-host disease
- HMRN
- Haematological Malignancy Research Network
- HR
- hazard ratio
- HRQoL
- heath-related quality of life
- ICER
- incremental cost-effectiveness ratio
- IFN
- interferon
- IFN-α
- interferon alpha
- IRIS
- International Randomised Study of Interferon versus STI571
- ITT
- intention to treat
- LVEF
- left ventricular ejection fraction
- MCyR
- major cytogenetic response
- mg
- milligram
- MHR
- major haematological response
- MIMS
- Monthly Index of Medical Specialities
- MMR
- major molecular response
- MTC
- mixed-treatment comparison
- NA
- not applicable
- NHS EED
- NHS Economic Evaluation Database
- NICE
- National Institute for Health and Clinical Excellence
- NSRC
- National Schedule of Reference Costs
- OS
- overall survival
- p.a.
- per annum
- PAS
- Patient Access Scheme
- PCR
- polymerase chain reaction
- PCyR
- partial cytogenetic response
- PFS
- progression-free survival
- Ph–
- Philadelphia chromosome-negative cell
- Ph+
- Philadelphia chromosome-positive cell
- PRISMA
- Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- PSA
- probabilistic sensitivity analysis
- PSS
- Personal Social Services
- PYs
- progressed-years (i.e. years in accelerated and blast phases)
- QALY
- quality-adjusted life-year
- q.d.
- once a day
- QoL
- quality of life
- qPCR
- real-time quantitative polymerase chain reaction
- QT
- In cardiology, the time between the start of the Q wave and the end of the T wave of a heart’s electrical cycle
- QTc
- Same as the QT interval (above), but corrected for the person’s heart rate
- RCT
- randomised controlled trial
- RR
- relative risk
- RQ-PCR
- real-time quantitative PCR
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- SCT
- stem cell transplantation
- SD
- standard deviation
- SE
- standard error
- SF-36
- Short Form questionnaire-36 items
- TKI
- tyrosine kinase inhibitor
- TTO
- time trade-off
- WBC
- white blood cell
- WHO
- World Health Organization
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Note
This monograph is based on the Technology Assessment Report produced for NICE. The full report contained a considerable number of data that were deemed commercial-in-confidence and academic-in-confidence. The full report was used by the Appraisal Committee at NICE in their deliberations. The full report with each piece of commercial-in-confidence and academic-in-confidence data removed and replaced by the statement ‘commercial-in-confidence and academic-in-confidence information (or data) removed’ is available on the NICE website: www.nice.org.uk.
The present monograph presents as full a version of the report as is possible while retaining readability, but some sections, sentences, tables and figures have been removed. Readers should bear in mind that the discussion, conclusions and implications for practice and research are based on all the data considered in the original full NICE report.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Professor of Bio-Statistics, Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor Zarko Alfirevic, Head of Department for Women’s and Children’s Health, Institute of Translational Medicine, University of Liverpool
-
Professor Judith Bliss, Director of ICR-Clinical Trials and Statistics Unit, The Institute of Cancer Research
-
Professor David Fitzmaurice, Professor of Primary Care Research, Department of Primary Care Clinical Sciences, University of Birmingham
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, Department of Specialist Radiology, University College Hospital, London
-
Professor Angela Harden, Professor of Community and Family Health, Institute for Health and Human Development, University of East London
-
Dr Joanne Lord, Reader, Health Economics Research Group, Brunel University
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Dion Morton, Professor of Surgery, Academic Department of Surgery, University of Birmingham
-
Professor Gail Mountain, Professor of Health Services Research, Rehabilitation and Assistive Technologies Group, University of Sheffield
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Director, Centre for Healthcare Randomised Trials, Health Services Research Unit, University of Aberdeen
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Peter Tyrer, Professor of Community Psychiatry, Centre for Mental Health, Imperial College London
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Caroline Watkins, Professor of Stroke and Older People’s Care, Chair of UK Forum for Stroke Training, Stroke Practice Research Unit, University of Central Lancashire
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health