
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 05/503/04. The contractual start date was in April 2009. The draft report began editorial review in October 2011 and was accepted for publication in May 2012. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors' report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen's Printer and Controller of HMSO 2013. This work was produced by Lenney et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Background
Respiratory disease has recently been declared a target for improved management by the Department of Health. Although a major focus of this initiative is adult chest disease, especially chronic obstructive pulmonary disease, it has been recognised that asthma is an important unresolved burden, especially in childhood where much asthma originates. The longitudinal studies from Aberdeen1 have shown that 60% of adults with asthma suffered their first symptoms in early childhood, continuing with these through later childhood and into adult life.
Rationale
Asthma remains a common medical condition seen in children in primary care and a frequent cause of medical paediatric hospital admission. It affects one in eight children nationwide, of whom many are prescribed low-dose inhaled corticosteroids (ICSs). 1 When treatment with low-dose ICSs fails to control asthma symptoms or adequately prevent exacerbations, national guidelines2 suggest ensuring compliance, including giving appropriate information about the disease to children and their families, maximising inhaler technique and treating comorbidities such as rhinitis. Once these measures have been established and if asthma remains uncontrolled, step 3 of the national guidelines recommends increasing the treatment. The evidence base at this step of the guidelines is much more limited in children than it is in adults. The reasons for this are that few studies have been undertaken in children and most that have taken place have used inappropriate adult-based outcomes such as lung function measurements, which suffer from assay insensitivity and fail to capture the episodic nature of much of childhood asthma. Pharmaceutical company studies have generally been conducted only as part of their requirements to obtain a licence to market their product. These studies have generally been of short duration and have not added to clinicians' understanding of how and where to use the medications. 3,4 In addition, they have not necessarily selected a representative population because of their tight entry criteria and their intensive study requirements. Such requirements mean that ‘real-life’ compliance and hence outcomes do not occur. In the independent national Dutch study,5 which enrolled patients uncontrolled on low-dose ICSs, three treatment groups were employed: ICSs alone, ICSs in double the dose and ICSs plus a long-acting beta2-agonist. There was essentially no difference in outcome measures between the three treatment groups, probably because the primary outcome measure was lung function (forced expiratory volume in 1 second or FEV1). Comparing this study with a similar adult study,6 which also used lung function as the primary outcome measure, the mean FEV1 on entry into the Dutch paediatric study was approximately 89% of that expected for the children's heights and the mean FEV1 on entry into the adult study was 74% of that expected. It is therefore not surprising that the paediatric study was unable to show any differences between the treatment groups.
There is little scientific evidence on how to treat children with asthma who are not well controlled on low-dose ICS therapy, apart from the limited value of increasing the ICS dose. 7 There is no clinical study evidence showing that, when control is poor in children with asthma, the dose of the inhaled steroid should be increased. We therefore decided not to introduce into this study a treatment limb with a higher ICS dosage. There is anecdotal information, however, from many studies undertaken within the pharmaceutical industry that, when children enter a study that is controlled and double blind in nature, up to 30% of them improve, their symptoms reduce and their lung function increases (Bisgaard H, Professor of Respiratory Paediatrics at the University Hospital in Copenhagen, personal correspondence – during development of paediatric asthma studies). It is therefore surprising that approximately one-third of children receiving ICSs are prescribed high-dose inhaled steroid therapy (≥ 800 μg beclometasone dipropionate or equivalent) or are commenced on ‘add-on’ therapies such as long-acting beta-2 agonists or leukotriene receptor antagonists in addition to low-dose ICSs. Concerns about the safety of high-dose ICSs have been raised in relation to growth impairment,8 hypoglycaemia9 and suppression of the adrenal cortex,10 resulting in warnings on prescribing from the Medicines and Healthcare products Regulatory Agency (MHRA) in the UK. 11 Asthma is a very common condition and the worth of these regimens has not been proven by appropriately devised paediatric studies. The UK guidelines on asthma management2 have been developed in a ‘stepwise’ manner, the amount of medication increasing at each step if symptoms are not controlled; however, as stated above, it may be that childhood asthma differs from that in adults. It seems that children with relatively poorly controlled asthma who exhibit frequent symptoms do not necessarily show abnormal lung function between their periods of symptoms. It is for this reason that our study focused on outcome measures such as exacerbations and quality of life, although spirometric values at the first (T0) and last (T48) visits were measured and recorded.
A study is needed that is simple, is pragmatic (but placebo controlled and double blind), has outcomes that will be of practical benefit to children and will provide evidence for the use of add-on medications in the most cost-effective and efficient way. Since the MASCOT study was commenced the Best Add-on Therapy Giving Effective Response (BADGER) trial has been completed and reported. 12 This study, however, required reversibility or hyper-responsiveness as an entry criterion, which excluded many patients, was short term in nature and focused primarily on symptomatic control as measured by the Childhood Asthma Control Test as opposed to exacerbations.
Potential risks and benefits
The medications used in this study are subject to marketing authorisations and prescribed in accordance with their licensed indications. The management of any symptoms or exacerbations was in accordance with usual clinical practice, and a research worker, either the local principal investigator or research nurse, was available throughout the study to discuss specific issues with individuals concerned. Patients were free to withdraw from the study at any time with no detriment to their future care. All ethical aspects of the study were discussed when informed written consent was obtained. Appropriate patient and family information leaflets (Appendix 1) were developed and were discussed at the screening consultation. Patients and their families were provided with copies of the information sheets and their signed consent/assent forms.
All of the medications have been shown to be efficacious for children with chronic asthma when used appropriately as preventative therapy. The ultimate aims of preventative asthma treatment are the prevention of chronic symptoms and the maintenance of near-normal lung function and normal activity levels with prevention of recurrent acute episodes in order to maximise quality of life. The potential benefit for participants of taking these medications as part of the trial is that they will improve control of their asthma, reducing symptoms and exacerbations and meeting the goals above.
Objective
Primary objective
To determine whether or not, in children aged 6–14 years with asthma uncontrolled on low-dose ICSs, their control can be improved by adding in a long-acting beta-2 agonist [salmeterol, Seretide®, GlaxoSmithKline (GSK)] or a leukotriene receptor antagonist [montelukast, Singulair®, Merck Sharp & Dohme (MSD)], as measured by a reduced number of exacerbations requiring treatment with oral corticosteroids over the 48-week study period.
Chapter 2 Methods
MASCOT was designed as a prospective, controlled, double-blind, multicentre, randomised clinical trial to determine whether or not control of asthma symptoms in children aged 6–14 years with asthma uncontrolled on low-dose ICSs could be improved by adding in a long-acting beta-2 agonist (salmeterol) or a leukotriene receptor antagonist (montelukast).
Trial design
At the time that consent was obtained, the children commenced the run-in part of the study. This was an open run-in for 4 weeks commencing at T–4 (time minus 4 weeks) and continuing until T0 (time zero). If they continued with sufficient symptoms they were then eligible to enter the double-blind randomised study lasting 48 weeks (i.e. to T48).
Participants
Inclusion and exclusion criteria at registration (T–4 weeks)
Inclusion criteria
-
Children with physician-diagnosed asthma aged from 6 years to 14 years, 11 months.
-
Children who required frequent short-acting beta-2 agonist relief therapy: seven or more puffs in the past 7 days.
-
Children with symptoms of asthma (i.e. wheeze, shortness of breath but not cough alone) that resulted in:
-
nocturnal wakening in the last week and/or
-
interference with usual activities in the last week and/or
-
those who had had exacerbations, defined as a short course of oral corticosteroids, an unscheduled general practitioner (GP) or accident and emergency (A&E) department visit or a hospital admission within the previous 6 months.
-
-
Fully informed written (proxy) consent and assent, where appropriate.
Exclusion criteria
-
Children who received long-acting beta-2 agonists, leukotriene receptor antagonists, regular theophylline therapy or high-dose ICSs (> 1000 μg) and unlicensed beclometasone dipropionate or equivalent (at the discretion of the investigator).
-
Children with other respiratory diseases, cystic fibrosis, cardiac disease or immunological disorders.
Inclusion and exclusion criteria at randomisation (T0)
Inclusion criteria
-
Children with asthma aged 6–14 years 11 months.
-
Children who required frequent short-acting beta-2 agonist relief therapy: seven or more puffs in the past 7 days.
-
Children with symptoms of asthma (i.e. wheeze, shortness of breath but not cough alone) that resulted in:
-
nocturnal wakening in the last week and/or
-
interference with usual activities in the last week.
-
-
Continuing consent/assent (where appropriate).
Exclusion criteria
-
Children whose asthma was controlled after the 4-week run-in, in which control was defined as the absence of any symptoms of asthma (except cough alone) or when the symptoms of asthma had not interfered with usual activities in the last week.
-
Children who received long-acting beta-2 agonists, leukotriene receptor antagonists, regular theophylline therapy or high-dose ICSs (> 1000 μg) and unlicensed beclometasone dipropionate or equivalent (at the discretion of the investigator).
-
Children with other respiratory diseases, cystic fibrosis, cardiac disease or immunological disorders.
Interventions
During the 4-week run-in period all patients were commenced on fluticasone propionate inhalers (Flixotide®, GSK) at 200 μg per day (100 μg twice daily). Children who remained symptomatic at the end of the run-in period were randomised into one of three double-blind treatment regimens:
-
inhaled fluticasone propionate 100 μg twice daily plus placebo tablet once daily
-
inhaled fluticasone propionate 100 μg and salmeterol 50 μg twice daily (combination inhaler) plus placebo tablet once daily
-
inhaled fluticasone propionate 100 μg twice daily plus montelukast 5-mg tablet once daily.
Study procedure
Run-in period (T–4 to T0)
Patients were screened for eligibility in GP surgeries, primary care and paediatric clinics within secondary care. Following full informed written (proxy) consent, those eligible were registered into the study, had their inhaler technique checked (with additional training if necessary) and were provided with information about asthma and its management. All research centres taking part were centrally trained and instructed in appropriate strategies of approaching patients and their families in an attempt to obtain uniformity. Each registered patient was dispensed the same low-dose ICSs (fluticasone propionate, 100 μg twice daily). The patients then participated in an open 4-week ‘run-in’ period, completing a hand-held patient record providing information to aid assessment of asthma control.
The same criteria were used in all centres to determine control of symptoms. Poorly controlled patients were described as those:
-
requiring frequent short-acting beta-2 agonist relief therapy: seven or more puffs in the last week and/ or
-
with asthma symptoms affecting sleeping and/or usual activities in the last week and/or
-
who had had exacerbations (defined as a short course of oral corticosteroids, an unscheduled GP or A&E department visit or a hospital admission within the previous 6 months).
The purpose of the run-in period was to ensure that recruitment was limited to patients for whom control of their asthma presented a problem, rather than patients for whom only inhaler technique and management advice was sufficient to provide good control. Most run-ins lose approximately 25% of patients; it was anticipated that improved inhaler technique, education and attention to compliance as well as patients all using the same ICS may well make up to 50% ineligible for entry into the randomised part of the study. All patients registered but not randomised had some information collected (exacerbations, hospital appointments, medication changes and use) from their GP data approximately 1 year after registration.
At the T–4 review, families were also issued with a copy of a leaflet requesting the collection of a DNA sample for storage and investigation at a later date. They were invited to consent and provide a DNA sample (saliva) at their T0 clinic visit. Consent to provide a DNA sample was documented separately from consent for the main trial. It was hoped that at the end of the MASCOT study it might be possible to analyse whether or not there were any genetic implications for patients in terms of response or not to either of the ‘add-on’ medications.
The next study visit (T0) was organised with the participant and his or her carer/s within the ideal visit window of ≥ 24 days and ≤ 30 days from the T–4 date.
Entry to the full study (T0)
Following the run-in period patients were reassessed for study entry based upon the inclusion and exclusion criteria outlined earlier. Those achieving the threshold criteria for T0 were entered into the randomised part of the study.
Symptoms, exacerbations and beta-2 agonist use were ascertained by reviewing the hand-held record with the patient and his or her carer/s. Baseline Paediatric Asthma Quality of Life Questionnaire with Standardised Activities [(PAQLQ(S)]13 (sometimes referred to as the Juniper scale; interviewer administered if the child was ≤ 10 years, patient administered if the child was ≥ 11 years) and Paediatric Asthma Caregiver's Quality of Life Questionnaire (PACQLQ) (carer/parent administered) assessments were conducted and a full examination was performed, including height and weight measurements. Spirometry was carried out to measure the patient's FEV1, forced vital capacity (FVC) and FEV1 : FVC ratio (best of three before and after bronchodilator).
Each child was then randomised by pharmacy using a centrally supplied list and dispensed the first 3-month treatment pack accordingly. Treatment was to continue for 8 weeks and was double-blind using identical inhalers and placebo tablets, with patients receiving active and/or placebo medications:
-
inhaled fluticasone propionate 100 μg twice daily plus placebo tablet once daily
-
inhaled fluticasone propionate 100 μg and salmeterol 50 μg twice daily (combination inhaler) plus placebo tablet once daily
-
inhaled fluticasone propionate 100 μg twice daily plus montelukast 5-mg tablet once daily.
Following a separate consent process a DNA specimen was obtained. Individuals declining to provide DNA were not precluded from entry into main study.
Randomisation +8 weeks (T8, study visit) and randomisation +24 weeks (T24, study visit)
Symptoms, exacerbations and beta-2 agonist use were ascertained by reviewing the hand-held record with patients and carer/s. The health economics questionnaire completed during the period since the last clinic visit was checked for completeness and stored locally. A new blank questionnaire was inserted into the hand-held record to be used until the next appointment. Repeat quality of life assessments were administered as above and a symptom-directed physical examination was performed if appropriate. Adverse events were reported and recorded. Those who had achieved control of their asthma symptoms continued on the same treatment for the next 16 weeks. In the case of those whose symptoms had not improved but were no worse, the researcher discussed the family willingness to continue in the study.
Those who were clinically worse could be withdrawn from the study and given alternative treatment according to clinician advice, as in routine practice. The decision to withdraw a patient from the study was based on the child's health that day and review of clinical information written in the hand-held record over the preceding weeks. The decision was made at the clinical discretion of the investigator. The reason for discontinuation was documented in the case report form (CRF). Follow-up was continued until the end of the trial as per the study visit schedule, unless consent was withdrawn.
Randomisation +36 weeks (T36, telephone call)
Symptoms, exacerbations and beta-2 agonist use were ascertained by telephone. The research nurse confirmed that the hand-held record and the health economics questionnaire were being completed. Participants were asked to remove the completed questionnaire and place it at the back of the record to avoid any confusion. The research nurse reminded participants to begin a new health economics questionnaire to be used until their next appointment.
Adverse events were reported and recorded. Those who had achieved control of their asthma symptoms were requested to continue in the study. For those whose symptoms had not improved but were no worse, the RN discussed their willingness to continue in the study. Those whose asthma symptoms appeared to be worse were either offered an unscheduled study appointment with a member of the research team (if possible) or advised to visit their GP for further medical advice. If the practitioner believed they were clinically worse, the patient could be withdrawn from the study and given alternative treatment according to routine practice. The reason for discontinuation was documented in the CRF. Follow-up was continued until the end of the study as per the study visit schedule, unless consent was withdrawn.
Randomisation +48 weeks (T48, study visit)
Symptoms, exacerbations and beta-2 agonist use were ascertained by reviewing the hand-held record with patients and carer/s. The two health economics questionnaires completed since the last clinic visit were checked for completeness and were stored locally. Final quality of life assessments were administered as previously and a physical examination performed, including height and weight measurements. Spirometry was carried out to measure the patient's FEV1, FVC and FEV1 : FVC ratio (best of three before and after bronchodilator). Adverse events were reported and recorded.
Patients were asked to provide current details for their GP. They were told that within 7 days their GP would be informed which treatment they had been taking. If the patient was under the care of another clinician for asthma management before entering the study, that clinician would also be provided with details of the treatment. The PI/RN discussed future management with patients and their carer/s.
Table 1 provides a schedule of the study procedures.
| Procedure | Screening (T-4) | Baseline [T0 (clinic)]a | Follow-up schedule (weeks) | Premature discontinuation | |||
|---|---|---|---|---|---|---|---|
| T8 (clinic) | T24 (clinic) | T36 (telephone) | T48 (clinic) (study completion) | ||||
| Signed consent form | ✗ | ||||||
| Assessment of eligibility criteria | ✗ | ✗ | |||||
| Quality of life questionnaires administered | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Health economics questionnaire completed | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| FEV1, FVC, FEV1: FVC ratio | ✗ | ✗ | (✗) | ||||
| Review of patient-held record | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Review of medical history | ✗ | ✗ | |||||
| Review of concomitant medications | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Study intervention | ✗ | ✗ | ✗ | ✗ | |||
| Physical examination | |||||||
| Complete | ✗ | ✗ | |||||
| Symptom directed | (✗) | (✗) | (✗) | (✗) | (✗) | ||
| Vital signs | (✗) | (✗) | (✗) | (✗) | (✗) | (✗) | |
| Assessment of adverse events | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Special assay or procedure | |||||||
| Consent and obtain saliva sample for later DNA analysis | ✗ | ||||||
Soon after the study opened in all recruitment centres, it became clear that there were difficulties in recruiting the numbers of patients required. Even those centres achieving better recruitment rates than others were failing to increase at a satisfactory rate. After individual discussions with each centre, the chief investigator and trial co-ordinator arranged two meetings centrally where all recruitment nurses met to plan other strategies to increase recruitment. The chief investigator and trial co-ordinator also visited all sites between October 2009 and January 2010 to determine which of the new recruitment strategies would best suit each centre. It became clear that all strategies could not be undertaken by all sites and so agreement was reached as to which would be suitable for individual centres. All 12 new strategies are shown in Appendix 3 (three for secondary and nine for primary care).
Data collection
The data were recorded on to standardised CRFs designed collaboratively by the Trial Management Group (TMG). These were returned to the Medicines for Children Research Network Clinical Trials Unit (MCRN CTU) and the data entered onto a validated study database (MACROĸ™ v3.0, InferMed, London, UK) by trained staff. Confirmation of patient existence was by receipt of a fully signed consent form. Each CRF was checked for adherence to the trial protocol and for missing and/or erroneous values. Discrepancies were raised and queried with study sites to enable the correct data to be obtained or reasons, where possible, for missing data/errors.
Outcomes
Primary outcome
The primary outcome was number of asthma exacerbations requiring treatment with oral corticosteroids over the planned 48-week study period. Data on number of courses of oral corticosteroids prescribed for asthma symptoms since last visit were collected at each follow-up visit (8, 24, 36 and 48 weeks). The total number of courses of oral corticosteroids prescribed for asthma symptoms between randomisation and last follow-up visit was calculated for each patient. For the primary outcome a window of 5 weeks was chosen around the 48-week time point. In the case of children followed up for less than 43 weeks, data were excluded from the primary analysis. Data for children followed up for more than 53 weeks were truncated at 53 weeks.
Secondary outcomes
-
Quality of life as measured by the PAQLQ(S) and the PACQLQ.
-
Time from randomisation to first exacerbation requiring treatment with a short course of oral corticosteroids.
-
Number of school days missed due to respiratory problems.
-
Number of hospital admissions due to respiratory problems.
-
Amount of rescue beta-2 agonist therapy prescribed for asthma symptoms.
-
Time from randomisation to treatment withdrawal (because of lack of efficacy or side effects).
-
Lung function at 48 weeks assessed by spirometry.
-
Adverse events assessed during the trial.
-
Cost-effectiveness.
Sample size calculation
Assuming that the number of asthma exacerbations requiring treatment with oral corticosteroids was a Poisson random variable, the sample size for the trial was estimated using the following formula as described by Friede and Schmidli:14
where nc is the number of patients in the control arm, λc is the control group rate, t is the length of follow-up and θ* is the RR. The equation does not allow for overdispersion, which would lead to an inflated sample size.
Data from the UK General Practice Research Database for 1032 children who had at least one course of oral steroids in the previous 12 months were used to estimate a mean rate of 1.5 per year with variance 1.02 and dispersion parameter 0.68. 15 This was the best available estimate of exacerbation rate and dispersion parameter at the time of designing the MASCOT trial. However, as the MASCOT randomised population would have had their inhaler technique corrected, and other population differences may alter the exacerbation rate, an initial ‘target’ sample size was estimated with the intention of undertaking an internal blinded pilot after the first 75 children had been randomised and completed their 24-week follow-up assessment to check parameter assumptions and adjust the sample size if required. Recruitment rates and percentage of children randomised after the 4-week run-in were also to be closely monitored.
Bonferroni's adjustment was used in the sample size calculation (1.7% two-sided significance level) to allow for the three primary treatment comparisons of interest. To have 80% power to detect, as significant, at least a 30% reduction in exacerbation rate (from 1.5 per year to 1.05 per year, equivalent to a RR of 0.7) and allowing for a loss to follow up of 10%, 147 patients per group were required. Our preliminary target number to be randomised was 150 children per treatment group (450 in total). This number would also give > 99% power to detect, as significant, a difference of 0.5 points between treatment groups on the PAQLQ(S), with assumed standard deviation (SD) 0.71.
Randomisation and blinding
Randomisation code lists were generated (by an individual at the MCRN CTU who was not involved with the MASCOT trial) with the software package Stata (Release 9, StataCorp LP, College Station, TX, USA) using block randomisation with variable block length, stratified by secondary care centre, with allocation to the three treatment arms in the ratio of 1 : 1 : 1. The pharmacy at each secondary care centre held the randomisation list for that centre with treatment allocations labelled A, B or C. After determining a patient's treatment allocation from the list, the pharmacist selected an appropriate treatment pack and removed a serrated label showing A, B or C before dispensing to the patient. Study drugs were identical in appearance and identically packaged, with all patients, clinicians and trial personnel blinded to treatment allocation throughout. Patients could be unblinded at any time if clinically required using emergency contact details for a 24-hour pharmacy. All patients were routinely unblinded when they completed the 48-week study or when they withdrew prematurely. Routine unblinding was undertaken by a member of the MCRN CTU, who was not involved with the MASCOT study, who then contacted the child's GP. The MASCOT trial clinician and research nurse were not aware of the unblinding information to minimise the potential of unblinding other patients still in the study.
Statistical methods
Interim monitoring
It was planned to check the estimates of the overall exacerbation rate (not split by treatment group) and the dispersion parameter after the first 75 children had been randomised and completed their 24-week follow-up assessment. This figure was chosen to provide adequate data for the sample size review but to ensure that the review was undertaken before the recruitment period ended. This blinded internal pilot would be reviewed by the Independent Data and Safety Monitoring Committee (IDSMC) prior to reviewing any unblinded comparative data.
Two equally spaced interim analyses to estimate relative treatment effects for the primary efficacy outcome were also intended, planned after one-third and two-thirds of the target total number of children had been randomised and followed for 48 weeks. The Haybittle-Peto stopping guideline16,17 would be used and 99.9% confidence intervals (CIs) calculated for the difference between each pair of drugs at each interim analysis. All interim analysis results of primary and secondary outcomes would be confidential to the IDSMC.
Because of the early closure of the trial the internal pilot and planned interim analyses were not undertaken.
Analysis plan
All analyses were conducted according to the statistical analysis plan (SAP) (see Appendix 2), which provides a detailed and comprehensive description of the main, preplanned analyses for the trial. The main features are summarised below. Analyses were performed with standard statistical software (SAS version 14; SAS Institute Inc., Cary, NC, USA).
The protocol intended to follow patients to 48 weeks after randomisation and analyses were to include all patients with data up to 48 weeks. However, because of the unplanned early closure of the study a number of patients had < 48 weeks' follow-up data. The SAP (see Appendix 2) discussed this in detail and outlined a plan to present primary analyses using all available data up to 48 weeks but also to include secondary analyses using all available data up to 24 weeks. This was not included in the original protocol but was identified in the SAP before undertaking any unblinded analyses.
The CONSORT (CONsolidated Standards of Reporting Trials) flow diagram18,19 was used to summarise representativeness of the study sample and patient throughput. Baseline characteristics were presented by treatment group and overall with continuous variables presented with means and SDs [or median and interquartile range (IQR)] and categorical variables with numbers and percentages.
All primary analyses are based on an intention-to-treat (ITT) principle as far as practically possible. Secondary per-protocol analyses are presented as sensitivity analyses to examine the robustness of the results. Patients were included in the per-protocol analysis if they had taken at least 70% of their inhalers and 70% of their tablets up until the time point of interest. Patients with major protocol deviations or those withdrawn from treatment or the trial before the time point of interest were excluded from the per-protocol analysis set. This per-protocol analysis set definition differed slightly to that described in the SAP as it was felt to be too stringent. This amendment was made while treatment groups were still blinded. For the analysis of adverse events, all patients who received at least one dose of the trial medication were included. Patients were included in the treatment group for the treatment that they actually received, meaning that if a patient crossed over to another arm for some reason they would contribute safety data to this group instead of, or in addition to, their randomised group.
For the primary outcome a p-value of < 0.017 was used to declare statistical significance, with 98.3% CIs reported to allow for the multiplicity of primary treatment comparisons. For all other analyses a p-value of < 0.05 was used to declare statistical significance, with 95% CIs reported throughout. The relative effects of treatment were estimated for each pair-wise treatment comparison (fluticasone vs fluticasone plus salmeterol; fluticasone vs fluticasone plus montelukast; fluticasone plus salmeterol vs fluticasone plus montelukast).
All count data (number of exacerbations, number of school days missed and number of hospital admissions) were analysed using Poisson regression with adjustment of standard errors [multiplying by the square root of the scale parameter estimated as the Pearson's chi-squared statistic divided by its degrees of freedom (df)] to account for overdispersion. Adjustment for centre in the regression model was originally planned in the protocol but, as recognised in the SAP (see Appendix 2), was felt to be impracticable because of the limited number of patients randomised.
Time-to-event data (time to first exacerbation and time to withdrawal) were analysed using Kaplan-Meier curves and log-rank test with relative effects of treatments summarised using hazard ratios (HRs) and 95% CIs. The seasonality of having an asthma exacerbation episode was to be explored by fitting season as a time-dependent covariate in the model for time to first exacerbation. However, this analysis was not undertaken because of concerns about limited data available to estimate parameters of the model.
Continuous data (quality of life and lung function) collected at baseline and follow-up were analysed using analysis of covariance (ANCOVA), which adjusts for the baseline value and treatment group. The adjusted mean differences from ANCOVA are presented with 95% CIs as well as means and SDs for the baseline and relevant time points for each treatment group. For each type of beta-2 agonist inhaler recorded during follow-up, the total amount prescribed was calculated as dose per actuation multiplied by the number of doses. This continuous outcome was analysed using analysis of variance (ANOVA).
Health economics
The MASCOT economic analysis focused on determining the differences in the patient pathways between the three groups in terms of their costs and benefits. The intention was therefore that the analysis would take a number of different forms. It was intended to compute incremental cost-effectiveness ratios (ICERs) for (B) inhaled fluticasone propionate 100 μg and salmeterol 50 μg twice daily (combination inhaler) plus placebo tablet once daily and (C) inhaled fluticasone propionate 100 μg twice daily plus montelukast 5-mg tablet once daily compared with the base case of (A) inhaled fluticasone propionate 100 μg twice daily plus placebo tablet once daily, and for (B) compared with (C).
Methods
The intention in the economic analysis was to build up as full a picture as possible of the resource costs and outcomes for the children in each of the three groups.
The intervention used and hence cost for each of the three groups was collected over the study period. In addition, data were collected on primary and secondary health-care contacts and medications. As there was no direct access to GP or hospital records the intention was to collect resource information directly from the patients using a structured diary that included questions on visits to the GP, medications prescribed in addition to the study medication, visits to hospital and any out-of-pocket expenses. All questions referred to the child's asthma to avoid spurious events being recorded. Although not the primary focus of the study, the study further aimed to incorporate patient and societal costs in terms of time lost from school and time lost from work. The school time would not be valued per se but results reported by group.
The child or his or her carer already completed a regular asthma diary that was discussed with the RN at each clinic visit and so the health economics information could be placed in this diary without putting undue pressure on the child or family. The diary was collected at T8, T24, T36 and T48. It was felt that periods longer than 3 months would not be acceptable as patient memory is poor after this length of time. To improve completion each family received a telephone call from the RN prior to their visit to remind them to complete and bring the record and questionnaire to their next appointment.
Although the intention was to collect medicine usage from the economic diary, which related only to any additional medication prescribed as a result of the child's asthma, in practice a further concomitant medication form was collected at each visit that asked the child/carer about all of the medications he or she had taken since the last visit and included a specific question regarding rescue medications (inhalers). In practice, this form took precedence over the medication section in the health economics booklet and medications were not collected in the booklet. Medicine usage was taken from these data. In economic terms we further sought to split the medicines into preventers, relievers and over-the-counter (OTC) drugs.
It is important to determine differences in the patient pathways and resource use as a result of the different regimens. The 4-week run-in period was ignored in terms of the economics as the costs will be common across all patients.
All data were used for each of the four periods. Because the numbers were so small in the study the decision was made to use all available data for each time period, but not to correct for missing data. The smaller number of completions at T36, for example, could have been handled by imputing an average cost for this period but this was not carried out. Given that the data set was so small these data may have been unrepresentative and therefore inappropriate for substitution.
Monetary costs were developed and applied to each of the trial arms. The resources were valued using national data sets (see Appendix 4) at 2010–11 costings.
A key cost driver in each of the three groups was the assigned study medication. Table 2 shows the monthly and 48-week costs for each of the three intervention medications.
| Medication | Monthly cost (£) | 48-week cost (£) |
|---|---|---|
| Inhaled fluticasone propionate 100 µg twice daily (group A) | 8.93 | 107.16 |
| Inhaled fluticasone propionate 100 µg and salmeterol 50 µg twice daily (combination inhaler) (group B) | 31.19 | 374.28 |
| Inhaled fluticasone propionate 100 µg twice daily and montelukast 5-mg tablet once daily (group C) | 34.62 | 415.44 |
Given that cost data are often positively skewed, and the sample size was so low in this study, the non-parametric bootstrap was used to obtain CIs for the mean differences in cost. Bootstrapping is a resampling procedure: 2000 independent samples were generated for each treatment group by sampling with replacements from the study data, with each bootstrap sample being the same size as the original sample. The mean of each of these samples was calculated and the bias-corrected bootstrap method used to calculate 95% CIs for the mean differences in cost. Bootstrapping was performed using Stata statistical software.
Outcome
The outcome used in the study was the PAQLQ(S). The economic analysis used this measure for asthma in children. No additional outcome measures were used. The PAQLQ(S) has 23 questions in three domains – symptoms, actions and emotional function – which are scored on a Likert scale (1–7, where 1 = severe and 7 = no symptom); the PAQLQ(S) score is then the mean of those scores. To convert this measure into a useful composite measure for the economic analysis, we created an indexed PAQLQ(S) score to proxy for quality-adjusted life-years (QALYs) using the transformation (PAQLQ – 1)/6, which generates a score within the range (0,1). PAQLQ(S) data were used from each of the four time points to accurately plot the changes in quality of life over the period of the trial.
There were very minor differences at baseline (insignificant in t-tests and Wilcoxon rank-sum tests); however, there was some variation in performance at different time points between the treatment groups.
We used two methods of calculating QALYs over 1 year:21 the standard area under the curve approach22 as well as unadjusted patient-specific changes in QALYs. To calculate differential QALYs attributable to the intervention, we also used regression-based adjusted QALYs. The regression model used was ordinary least squares estimation of area under the curve QALYs, controlling for baseline indexed PAQLQ(S) score and treatment group; the coefficient for treatment group then represented the baseline-adjusted treatment effect of the intervention received by patients. There were no statistically significant differences in incremental QALYs using any of the methods. We elected to base our results on the incremental approach to estimating QALYs of each treatment compared with a baseline treatment.
The study measured incremental cost-effectiveness using the ICERs for fluticasone plus salmeterol compared with fluticasone and fluticasone plus montelukast compared with fluticasone. ICERs show the ratio of the difference in costs to the difference in outcomes between the two groups. In terms of outcome, the ICER was to be based on the PAQLQ(S) results. Fluticasone plus montelukast was also to be compared directly with fluticasone plus salmeterol for completeness. Cost-effectiveness acceptability curves (CEACs) calculated for each of the three comparative regimens showed the probability that each option was cost-effective at different willingness-to-pay thresholds.
Protocol amendments
All protocol amendments are summarised in Appendix 5.
Chapter 3 Results
Early trial closure
Recruitment rates were closely monitored throughout the study, which highlighted within 8 months of the first patient being registered that additional strategies and additional centres would be required to improve recruitment and achieve the target of 450 patients randomised. Site visits were undertaken from October 2009 and a number of new strategies were developed through discussion with sites (see Appendix 3). A funding extension application was submitted to the NIHR HTA programme in February 2010 requesting an extension of recruitment time together with funding for new sites. One of the differences noted about recruitment centres that were more successful than others was how well integration had occurred between staff within the newly formed Medicines for Children Research Network (MCRN), Primary Care Research Network (PCRN) and Comprehensive Local Research Network (CLRN). If the study was to succeed it would also have been necessary to open other recruitment centres and these had been carefully selected based on the knowledge we had accrued through discussions and particularly the ability to liaise and work effectively between secondary and primary care. It was recognised that some of the initial recruitment sites would have to be closed. Funding was not granted, however, and the study was closed down prematurely on 24 June 2010. For those patients who were already randomised, follow-up was to continue to T48 or the end of January 2011, whichever was the earliest, as January 2011 was the date that the medications expired. Data cleaning and site close-down visits took place between February and July 2011, with final analyses undertaken during August and September 2011.
Participant flow and recruitment
The first patient was registered on 27 January 2009, the first patient was randomised on 19 May 2009, the last patient was registered on 25 June 2010 and the last patient was randomised on 24 June 2010. Table 3 shows all 13 recruiting centres, the date each site was initiated, the original target recruitment figure, the number of participants registered, the number of participants randomised and the dates of the first and last randomisation. All centres registered at least one patient and 12 centres randomised at least one patient.
A total of 166 patients were registered at T–4 and 63 (38%) of these were eligible and consented to be randomised at T0. The percentage randomised at each centre ranged between 20% and 58% for those that randomised at least one patient.
Figure 1 provides a CONSORT flow diagram showing the numbers of participants recruited and randomly assigned to the three trial arms.
| Centre code | Hospital | Date site initiated | Original target recruitment | Date of first registration (T-4) | Date of last registration | Number registered | Date of first randomisation (T0) | Date of last randomisation | Date of last follow-up visit | Number randomised |
|---|---|---|---|---|---|---|---|---|---|---|
| 002 | Royal Devon & Exeter NHS Foundation Trust | 19 November 2008 | 23 | 16 February 2009 | 25 May 2010 | 10 | 13 October 2009 | 9 March 2010 | 7 February 2011 | 3 |
| 009 | Arrowe Park Hospital, Wirral | 20 January 2009 | 13 | 27 January 2009 | 23 June 2010 | 26 | 28 July 2009 | 15 June 2010 | 26 January 2011 | 12 |
| 031 | Leicester Royal Infrmary | 11 February 2009 | 45 | 28 April 2009 | 8 June 2010 | 21 | 2 June 2009 | 24 June 2010 | 28 January 2011 | 7 |
| 036 | Norfolk and Norwich University Hospitalsa | 01 April 2009 | 45 | 2 July 2009 | 28 April 2010 | 8 | 4 November 2009 | 26 May 2010 | 24 November 2010 | 3 |
| 116 | Bristol Royal Hospital for Children | 08 December 2008 | 23 | 26 June 2009 | 28 May 2010 | 7 | 24 July 2009 | 18 November 2009 | 22 October 2010 | 2 |
| 134 | Royal London Hospital | 23 December 2008 | 45 | 9 February 2009 | 17 May 2010 | 17 | 26 May 2009 | 25 May 2010 | 18 October 2010 | 8 |
| 168 | Derbyshire Children's Hospital | 18 February 2009 | 23 | 21 April 2009 | 3 June 2010 | 10 | 19 May 2009 | 3 June 2010 | 18 November 2010 | 3 |
| 213 | Queen's Medical Centre, Nottingham | 06 April 2009 | 23 | 4 June 2009 | 21 June 2010 | 10 | 29 June 2009 | 11 May 2010 | 8 February 2011 | 4 |
| 243 | Alder Hey Children's NHS Foundation Trust | 26 February 2009 | 32 | 23 June 2009 | 21 June 2010 | 14 | 21 July 2009 | 22 June 2010 | 27 January 2011 | 6 |
| 246 | Royal Manchester Children's Hospital | 23 December 2008 | 45 | 5 June 2009 | 25 June 2010 | 7 | 9 December 2009 | 29 January 2010 | 12 January 2011 | 2 |
| 451 | Royal Aberdeen Children's Hospital | 13 January 2009 | 45 | 20 August 2009 | 9 June 2010 | 10 | 24 February 2010 | 24 February 2010 | 24 January 2011 | 2 |
| 511 | University Hospitals Coventry and Warwickshire NHS Trust | 26 January 2009 | 45 | 4 February 2009 | 21 June 2010 | 7 | N/A | N/A | N/A | 0 |
| 522 | University Hospital of North Staffordshire | 06 April 2009 | 45 | 26 June 2009 | 21 May 2010 | 19 | 24 July 2009 | 16 June 2010 | 11 January 2011 | 11 |
| Total | 452 | 166 | 63 |
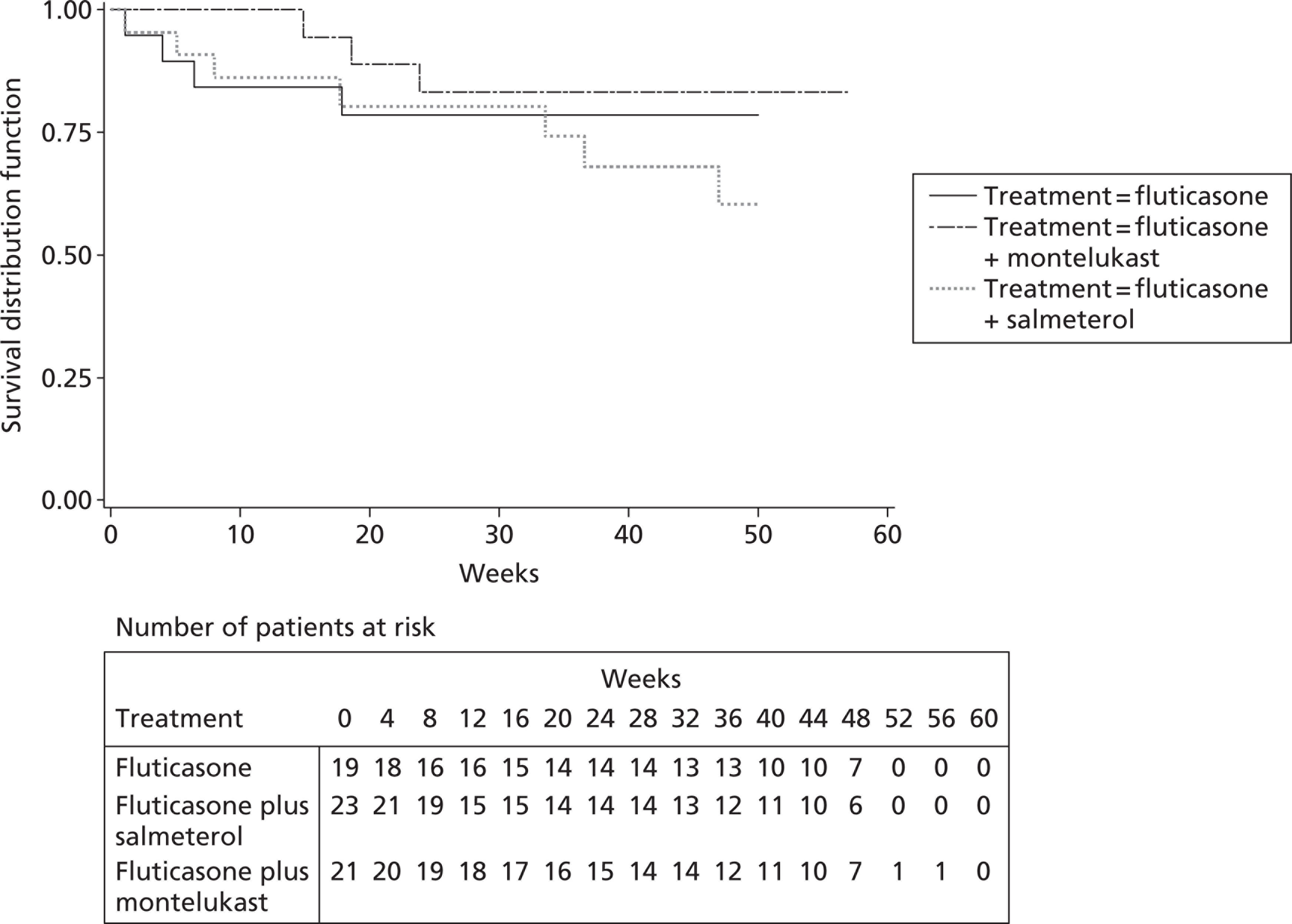
FIGURE 1.
CONSORT flow diagram. SABA, short-acting beta-2 agonist. All patients who did not reach the minimum of 43 weeks because of early closure of the trial have primary outcome data up to at least T24.

Baseline comparability of randomised groups
A summary of the baseline characteristics for all randomised participants is given in Table 4. Overall there was a preponderance of males with the major comorbid condition being eczema, neither of which was unexpected. The male-to-female ratio in children with asthma is approximately 2 : 1 and there is a strong association between eczema and asthma in children. The age of the children ranged from 6.50 to 14.67 years (average 10.39 years).
The distribution of characteristics is similar across treatment groups but a higher percentage of males were randomised to fluticasone alone and a higher percentage of children with respiratory and dermatological abnormalities were randomised to fluticasone plus salmeterol. Because of the small number of children recruited this probably occurred by chance but could suggest that patients randomised to fluticasone plus salmeterol had more severe asthma.
| Baseline characteristic | Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) |
|---|---|---|---|---|
| General | ||||
| Age (years), mean (SD), range (n) | 10.37 (1.82), 7.33 to 13.58 (19) | 10.46 (2.33), 6.50 to 13.75 (23) | 10.33 (2.37), 6.50 to 14.67 (21) | 10.39 (2.17), 6.50 to 14.67 (63) |
| Sex, n / N (%) | ||||
| Male | 17/19 (89.5) | 13/23 (56.5) | 10/21 (47.6) | 40/63 (63.5) |
| Female | 2/19 (10.5) | 10/23 (43.5) | 11/21 (52.4) | 23/63 (36.5) |
| Physical examination | ||||
| Height (cm), mean (SD), range (n) | 143.18 (15.63), 121.00 to 184.40 (19) | 144.60 (12.11), 128.00 to 166.00 (23) | 141.56 (15.98), 117.60 to 167.30 (21) | 143.16 (14.38), 117.60 to 184.40 (63) |
| Weight (kg), mean (SD), range (n) | 39.88 (12.44), 22.00 to 59.50 (19) | 42.28 (11.49), 22.90 to 64.80 (23) | 38.56 (12.41), 20.50 to 64.80 (21) | 40.31 (12.00), 20.50 to 64.80 (63) |
| General appearance, n/N (%) | ||||
| Normal | 19/19 (100.0) | 23/23 (100.0) | 21/21 (100.0) | 63/63 (100.0) |
| Abnormal | 0/19 (0.0) | 0/23 (0.0) | 0/21 (0.0) | 0/63 (0.0) |
| Ear, nose, throat, n/N (%) | ||||
| Normal | 17/19 (89.5) | 19/23 (82.6) | 19/20 (95.0) | 55/62 (88.7) |
| Abnormala | 2/19 (10.5) | 4/23 (17.4) | 1/20 (5.0) | 7/62 (11.3) |
| Cardiovascular, n/N (%) | ||||
| Normal | 19/19 (100.0) | 23/23 (100.0) | 20/20 (100.0) | 62/62 (100.0) |
| Abnormal | 0/19 (0.0) | 0/23 (0.0) | 0/20 (0.0) | 0/62 (0.0) |
| Respiratory, n/N (%) | ||||
| Normal | 18/19 (94.7) | 17/23 (73.9) | 18/20 (90.0) | 53/62 (85.5) |
| Abnormalb | 1/19 (5.3) | 6/23 (26.1) | 2/20 (10.0) | 9/62 (14.5) |
| Gastrointestinal, n/N (%) | ||||
| Normal | 19/19 (100.0) | 22/23 (95.7) | 18/20 (90.0) | 59/62 (95.2) |
| Abnormalc | 0/19 (0.0) | 1/23 (4.3) | 2/20 (10.0) | 3/62 (4.8) |
| Endocrine, n/N (%) | ||||
| Normal | 19/19 (100.0) | 23/23 (100.0) | 20/20 (100.0) | 62/62 (100.0) |
| Abnormal | 0/19 (0.0) | 0/23 (0.0) | 0/20 (0.0) | 0/62 (0.0) |
| Dermatological, n/N (%) | ||||
| Normal | 18/19 (94.7) | 18/23 (78.3) | 17/20 (85.0) | 53/62 (85.5) |
| Abnormald | 1/19 (5.3) | 5/23 (21.7) | 3/20 (15.0) | 9/62 (14.5) |
| Musculoskeletal, n/N (%) | ||||
| Normal | 18/19 (94.7) | 23/23 (100.0) | 20/20 (100.0) | 61/62 (98.4) |
| Abnormale | 1/19 (5.3) | 0/23 (0.0) | 0/20 (0.0) | 1/62 (1.6) |
| Other site,f n/N (%) | ||||
| Normal | 10/10 (100.0) | 7/7 (100.0) | 10/10 (100.0) | 27/27 (100.0) |
| Abnormal | 0/10 (0.0) | 0/7 (0.0) | 0/10 (0.0) | 0/27 (0.0) |
| Systolic blood pressure (mmHg), mean (SD), range (n) | 103.89 (13.11), 80.00 to 135.00 (19) | 106.96 (10.12), 90.00 to 134.00 (23) | 102.24 (10.86), 84.00 to 119.00 (21) | 104.46 (11.33), 80.00 to 135.00 (63) |
| Diastolic blood pressure (mmHg), mean (SD), range (n) | 62.84 (12.11), 46.00 to 89.00 (19) | 63.87 (8.38), 49.00 to 82.00 (23) | 60.00 (8.63), 44.00 to 77.00 (21) | 2.27 (9.71), 44.00 to 89.00 (63) |
| Pulse rate (beats/minute), mean (SD), range (n) | 80.58 (17.81), 57.00 to 118.00 (19) | 85.65 (14.96), 54.00 to 125.00 (23) | 85.48 (15.35), 55.00 to 114.00 (21) | 84.06 (15.90), 54.00 to 125.00 (63) |
| Respiratory rate (breaths/ minute), mean (SD), range (n) | 21.89 (4.84), 16.00 to 36.00 (19) | 20.48 (2.85), 16.00 to 25.00 (23) | 21.33 (3.37), 14.00 to 30.00 (21) | 21.19 (3.70), 14.00 to 36.00 (63) |
| Spirometryg | ||||
| FEV1 (l), mean (SD), range (n) | 1.98 (0.89), 0.90 to 4.70 (18) | 1.83 (0.68), 0.80 to 3.10 (19) | 1.82 (0.77), 0.20 to 3.20 (19) | 1.88 (0.77), 0.20 to 4.70 (56) |
| FVC (l), mean (SD), range (n) | 2.29 (0.92), 1.20 to 4.70 (18) | 2.20 (0.60), 1.30 to 3.20 (19) | 2.35 (0.77), 0.80 to 3.50 (19) | 2.28 (0.76), 0.80 to 4.70 (56) |
| FEV1 (%), mean (SD), range (n) | 88.29 (17.55), 48.0 to 116.0 (17) | 79.79 (19.90), 47.0 to 106.0 (19) | 86.47 (13.32), 56.0 to 104.0 (19) | 84.73 (17.21), 47.0 to 116.0 (55) |
| FVC (%), mean (SD), range (n) | 92.06 (15.09), 57.0 to 112.0 (17) | 87.00 (16.91), 49.0 to 112.0 (19) | 94.05 (15.32), 53.0 to 128.0 (19) | 91.00 (15.82), 49.0 to 128.0 (55) |
| FEV1 (l) after 400 µg salbutamol (GSK) administered, mean (SD), range (n) | 2.19 (0.84), 0.80 to 4.30 (15) | 1.99 (0.67), 0.80 to 3.10 (20) | 2.04 (0.68), 0.80 to 3.20 (18) | 2.06 (0.72), 0.80 to 4.30 (53) |
| FVC (l) after 400 µg salbutamol administered, mean (SD), range (n) | 2.60 (0.94), 1.30 to 4.80 (15) | 2.38 (0.62), 1.30 to 3.70 (20) | 2.34 (0.80), 0.90 to 3.60 (18) | 2.43 (0.78), 0.90 to 4.80 (53) |
| FEV1 (%)after 400 µg salbutamol administered, mean (SD), range (n) | 90.64 (17.95), 46.0 to 123.0 (14) | 86.30 (19.75), 49.0 to 111.0 (20) | 92.28 (13.94), 56.0 to 110.0 (18) | 89.54 (17.30), 46.0 to 123.0 (52) |
| FVC (%) after 400 µg salbutamol administered, mean (SD), range (n) | 94.21 (17.17), 58.0 to 124.0 (14) | 91.50 (15.83), 55.0 to 117.0 (20) | 95.06 (15.51), 54.0 to 118.0 (18) | 93.46 (15.85), 54.0 to 124.0 (52) |
No ineligible patients were randomised.
Unblinding of randomised treatments
No patient required unblinding from randomised treatment other than at the end of the study. The process was described in Chapter 2, Randomisation and blinding.
Protocol deviations
Protocol deviations were classified as major or minor according to a preplanned classification system outlined in the SAP (see Appendix 2). There were 18 minor protocol deviations reported for 17 patients (three randomised to fluticasone, five randomised to fluticasone plus salmeterol, two randomised to fluticasone plus montelukast and seven who were not randomised) (Table 5). There were no major protocol deviations. Eleven of the protocol deviations were due to visits that occurred outside the predefined time interval, one of which led to missing data. One protocol deviation occurred because two different parents completed the quality of life questionnaire at two different visits. Six protocol deviations were related to the spirometry test undertaken at T0 or T48.
| Patient registration number | Date of deviation | Nature of deviation | Impact (minor/major) |
|---|---|---|---|
| 10009 | 2 November 2009 | The T0 visit occurred on the 31st day following the T–4 visit. The family were unable to make the T0 visit within the 30-day time frame because of other commitmentsa | Minor |
| 11006 | 13 November 2009 | Patient's T0 visit date occurred 32 days following the T–4 visit. Site are unable to explain the reason for this as the member of staff has now left the trusta | Minor |
| 13010 | 18 October 2010 | Patient was unable to attend the scheduled T48 visit (22 November 2010) because of religious commitments and holiday out of the country. Visit was carried out 5 weeks after the T36 visit on 18 October 2010 (42 weeks after randomisation) | Minor |
| 22001 | 10 August 2009 | Time period between the T–4 and T0 visits exceeded 30 daysa | Minor |
| 30004 | 11 February 2010 | The patient was seen for their T0 visit 22 days after the T–4 visit. This was checked with the CTU prior to the visit as Dr Turner was unable to see the patient the following weeka | Minor |
| 30007 | 7 April 2010 | Site provided a file note to explain that the patient was due to come in for her T0 visit on 1 April 2010; however, the PI was on annual leave that week. An attempt was made to sign up a co-investigator to see the patient; however, this was unsuccessful because of their Good Clinical Practice training being inadequate and the delegation log not being signed prior to the PI going on leave. The CTU was contacted and it was agreed that the patient could extend her run-in phase to 34 days and be seen for her T0 visit on 7 April 2010. The patient was therefore reviewed by a member of child health's medical team on 1 April 2010 and prescribed another fluticasone propionate Accuhaler 100 µg twice daily (GSK) to ensure that she had an adequate supply until her rescheduled appointment on 7 April 2010a | Minor |
| 60008 | 3 June 2010 | Patient started the run-in period on 22 April 2010 but it was arranged at site that the child would not begin taking the medications until 3 May 2010. The T0 visit was scheduled for 28 days following 3 May 2010. This is a deviation because the run-in period for this child was 6 weeks (although only 4 weeks were spent on run-in trial medications). T0 visit occurred on 3 June 2010a | Minor |
| 70008 | 7 February 2011 | Patient's final T48 visit fell outside of the visit window allocated for the closure of the trial because patient failed to attend. Patient attended on 7 February 2011, which is a protocol deviation because of trial closure and shortened follow-up dates; however, 7 February 2011 is exactly 48 weeks from randomisation for this patient | Minor |
| 70011 | 15 June 2010 | Patient's T0 visit fell 3 days outside of the visit window because parents were unable to attend initial appointmenta | Minor |
| 12008 | 14 July 2010 | Patient's T0 visit fell 9 days outside of the visit window; however, the trial had already been discontinued and so this patient could not be randomiseda | Minor |
| 12002 | 8 February 2011 | Patient's final T48 visit fell outside of the visit window allocated for the closure of the trial because of illness. Parent then attended the final visit without the child. This meant that spirometry, physical assessments and child's quality of life assessment could not be completed (research nurse mailed them to parents but did not receive back) | Minor |
| 13007 | 2 February 2010 | Different caregiver completed the quality of life questionnaire at the T24 visit from the T8 visit (mother at T24, father at T8). Father was not present at T24 | Minor |
| 10010 | 15 January 2010 | Pre-reversibility test not saved; therefore, initial spirometry data are not available for T0. Also, at T48, long-acting and short-acting bronchodilators had not been withheld for the appropriate time | Minor |
| 13006 | 15 June 2010 | Long-acting bronchodilators had not been withheld for the appropriate time at the T48 spirometry assessment – patient had taken study medication that morning | Minor |
| 13010 | 21 December 2009 | At T0 short-acting bronchodilator taken approximately 2.5 hours prior to spirometry testing | Minor |
| 13014 | 25 May 2010 | At T0 short-acting bronchodilator not withheld for 4 hours prior to spirometry testing | Minor |
| 13015 | 25 May 2010 | At T0 short-acting bronchodilator not withheld for 4 hours prior to spirometry testing | Minor |
| 60001 | 19 May 2009 | Required salbutamol 2 hours before spirometry testing at T0 | Minor |
Efficacy outcomes
Primary outcome
Number of asthma exacerbations requiring treatment with oral corticosteroids
In total, 38 (60%) randomised patients completed their 48-week visit and 54 (86%) patients completed their 24-week visit and could be included in the ITT analyses (Tables 6 and 7 respectively). Reasons for exclusions are provided in Appendix 6. For the primary analysis (ITT at 48 weeks), seven (18%) patients had at least one exacerbation requiring oral steroids, with most having only one episode. The effect of treatment overall (p = 0.98) and for each pair-wise comparison was not significant (Table 8). Because of limited data, the CI around the RR for each pair-wise comparison is extremely wide and includes values of RR that could indicate beneficial, harmful or equivalent treatment effects (see Table 8). The secondary ITT analysis at 24 weeks includes data for 54 patients (see Table 7) of whom 10 (18.5%) had experienced at least one exacerbation. Similarly to the 48-week analysis, all treatment effects are not significant with very wide CIs (see Table 8). The per-protocol analyses show similar results and uncertainty to the ITT analyses (see Table 8). Because of the limited number of patients and events available for parameter estimation, secondary analyses adjusting for prognostic factors were not undertaken as planned in the SAP.
| Fluticasone (n = 11) | Fluticasone plus salmeterol (n = 15) | Fluticasone plus montelukast (n = 12) | Total (n = 38) | |
|---|---|---|---|---|
| Children with at least one exacerbation, n (%) | 1 (9.1) | 5 (33.3) | 1 (8.3) | 7 (18.4) |
| Total no. of exacerbations over 48 weeks,a n (%) | ||||
| 0 | 10 (90.9) | 10 (66.7) | 11 (91.7) | 31 (81.6) |
| 1 | 0 (0.0) | 4 (26.7) | 0 (0.0) | 4 (10.5) |
| 2 | 0 (0.0) | 1 (6.7) | 0 (0.0) | 1 (2.6) |
| 3 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 4 | 1 (9.1) | 0 (0.0) | 1 (8.3) | 2 (5.3) |
| Exposure (weeks) | ||||
| Total | 528.4 | 721.3 | 580.1 | 1829.8 |
| Mean (SD) | 48.03 (1.06) | 48.09 (1.96) | 48.35 (2.35) | 48.15 (1.85) |
| Range | 46.14 to 50.00 | 43.00 to 50.00 | 43.00 to 53.00 | 43.00 to 53.00 |
| Fluticasone (n = 18) | Fluticasone plus salmeterol (n = 7) | Fluticasone plus montelukast (n = 19) | Total (n = 54) | |
|---|---|---|---|---|
| Children with at least one exacerbation, n (%) | 4 (22.2) | 3 (17.6) | 3 (15.8) | 10 (18.5) |
| Total no. of exacerbations over 24 weeks,a n (%) | ||||
| 0 | 14 (77.8) | 14 (82.4) | 16 (84.2) | 44 (81.5) |
| 1 | 1 (5.6) | 2 (11.8) | 3 (15.8) | 6 (11.1) |
| 2 | 2 (11.1) | 1 (5.9) | 0 (0.0) | 3 (5.6) |
| 3 | 1 (5.6) | 0 (0.0) | 0 (0.0) | 1 (1.9) |
| Exposure (weeks) | ||||
| Total | 436.3 | 421.3 | 464.6 | 1322.2 |
| Mean (SD) | 24.24 (1.03) | 24.78 (1.43) | 24.45 (1.28) | 24.48 (1.25) |
| Range | 22.00 to 26.00 | 22.29 to 29.00 | 22.00 to 28.14 | 22.00 to 29.00 |
| Analysis set | Number of patients | Time point | Scale parametera | Fluticasone vs fluticasone plus salmeterol | Fluticasone vs fluticasone plus montelukast | Fluticasone plus salmeterol vs futicasone plus montelukast | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RR | CI | p-valueb | RR | CI | p-valueb | RR | CI | p-valueb | ||||
| ITT | nA = 11, nB = 15, nC = 12 | 48 weeks | 1.68 | 0.91 | 0.07 to 12.05c | 0.93 | 1.10 | 0.06 to 18.6c | 0.94 | 1.21 | 0.09 to 15.97c | 0.86 |
| 0.11 to 7.59d | 0.11 to 11.21d | 0.14 to 10.07d | ||||||||||
| nA = 18, nB = 17, nC = 19 | 24 weeks | 1.17 | 1.93 | 0.35 to 10.67c | 0.36 | 2.84 | 0.43 to 18.79c | 0.19 | 1.47 | 0.17 to 12.39c | 0.67 | |
| 0.47 to 7.86d | 0.60 to 13.40d | 0.26 to 8.47d | ||||||||||
| Per protocol | nA = 9, >nB = 11, nC = 8 | 48 weeks | 1.27 | 1.64 | 0.16 to 16.54c | 0.61 | –e | –e | –e | –e | –e | –e |
| 0.25 to 10.94d | ||||||||||||
| nA = 15, NB = 13, >nC = 16 | 24 weeks | 1.18 | 2.36 | 0.35 to 15.93c | 0.28 | 2.87 | 0.42 to 19.35c | 0.19 | 1.22 | 0.12 to 12.15c | 0.84 | |
| 0.49 to 11.32d | 0.60 to 13.76d | 0.18 to 8.05d | ||||||||||
Secondary outcomes
Quality of life of children as measured by the Paediatric Asthma Quality of Life Questionnaire with Standardised Activities
A total of 37 children completed the quality of life questionnaire at baseline and at 48 weeks' follow-up. The results show an improvement in mean scores for activity limitations, symptoms and emotional function across all treatment groups at 48 weeks, with the largest improvement in mean score for fluticasone plus montelukast across all domains (Table 9). However, the pair-wise treatment comparison results from ANCOVA show that none of the mean differences adjusted for baseline score is statistically significant. The 24-week analysis results based on 50 patients show an improvement in mean scores for activity limitations, symptoms and emotional function across all treatment groups (Table 10). The largest improvement was seen for fluticasone plus salmeterol across all domains but none of the mean differences adjusted for baseline score is statistically significant.
| Domain | Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | Fluticasone vs futicasone plus salmeterol | Fluticasone vs futicasone plus montelukast | Fluticasone plus salmeterol vs futicasone plus montelukast | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline mean (SD), range | T48 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T48 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T48 mean (SD), range | Change mean (SD), range | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | |
| Activity limitations (nA = 10, nB = 14,anC = 12) | 4.76 (0.91), 3.60 to 6.00 | 6.20 (1.07), 3.40 to 7.00 | 1.44 (1.00), −0.20 to 3.40 | 4.39 (1.30), 2.60 to 6.60 | 5.60 (1.47), 3.00 to 7.00 | 1.21 (2.12), −2.80 to 4.40 | 4.25 (1.47), 1.80 to 6.80 | 6.30 (0.85), 4.40 to 7.00 | 2.05 (1.61), −0.60 to 4.20 | 0.59 (−0.43 to 1.61), p = 0.25 | −0.12 (−1.18 to 0.94), p = 0.82 | −0.70 (−1.67 to 0.26), p = 0.16 |
| Symptoms (nA = 10, nB = 15, nC = 12) | 4.90 (0.92), 3.70 to 6.30 | 6.28 (0.84), 4.40 to 7.00 | 1.38 (0.93), 0.00 to 3.10 | 4.35 (1.51), 1.30 to 6.80 | 5.30 (1.77), 1.50 to 7.00 | 0.95 (2.17), −2.20 to 5.70 | 4.32 (1.63), 1.50 to 6.70 | 6.23 (0.92), 4.70 to 7.00 | 1.91 (1.79), −0.80 to 4.40 | 0.90 (−0.22 to 2.02), p = 0.11 | −0.03 (−1.20 to 1.15), p = 0.96 | −0.93 (−1.98 to 0.12), p = 0.08 |
| Emotional function (nA = 10, nB = 15, nC = 12) | 5.31 (0.82), 3.50 to 6.25 | 6.40 (0.86), 4.25 to 7.00 | 1.09 (0.62), 0.13 to 2.13 | 4.33 (1.68), 1.75 to 7.00 | 5.58 (1.62), 2.13 to 7.00 | 1.25 (1.72), −1.50 to 4.38 | 4.79 (1.54), 2.00 to 6.75 | 6.42 (0.83), 4.88 to 7.00 | 1.63 (1.66), −0.63 to 4.13 | 0.50 (−0.48 to 1.49), p = 0.31 | −0.19 (−1.19 to 0.82), p = 0.71 | −0.69 (−1.60 to 0.22), p = 0.13 |
| Total (nA = 10, nB = 15, nC = 12) | 5.01 (0.64), 3.91 to 5.78 | 6.30 (0.88), 4.13 to 7.00 | 1.29 (0.61), 0.22 to 2.52 | 4.34 (1.39), 2.04 to 6.83 | 5.44 (1.56), 2.26 to 7.00 | 1.10 (1.86), −2.09 to 4.96 | 4.47 (1.50), 1.74 to 6.65 | 6.31 (0.85), 4.78 to 7.00 | 1.84 (1.66), −0.70 to 4.26 | 0.73 (−0.29 to 1.74), p = 0.15 | −0.12 (−1.17 to 0.94), p = 0.82 | −0.84 (−1.78 to 0.10), p = 0.08 |
| Domain | Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | Fluticasone vs futicasone plus salmeterol | Fluticasone vs futicasone plus montelukast | Fluticasone plus salmeterol vs futicasone plus montelukast | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline mean (SD), range | T24 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T24 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T24 mean (SD), range | Change mean (SD), range | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | |
| Activity limitations (nA = 17, nB = 15,anC = 17) | 4.52 (1.18), 2.20 to 6.00 | 5.60 (1.50), 2.20 to 7.00 | 1.08 (1.02), −0.40 to 3.60 | 4.40 (1.26), 2.60 to 6.60 | 6.00 (0.91), 4.40 to 7.00 | 1.60 (1.39), −0.80 to 4.20 | 4.21 (1.32), 1.80 to 6.80 | 5.75 (1.48), 2.40 to 7.00 | 1.54 (1.33), −0.60 to 3.80 | −0.47 (−1.28 to 0.35), p = 0.26 | −0.33 (−1.12 to 0.47), p = 0.41 | 0.14 (−0.68 to 0.96), p = 0.73 |
| Symptoms (nA = 17, nB = 16, nC = 17) | 4.64 (1.15), 2.60 to 6.60 | 5.49 (1.31), 2.70 to 7.00 | 0.85 (0.97), −1.00 to 2.90 | 4.45 (1.51), 1.30 to 6.80 | 5.91 (1.18), 2.70 to 7.00 | 1.46 (1.42), −1.10 to 3.60 | 4.24 (1.42), 1.50 to 6.70 | 5.46 (1.67), 1.50 to 7.00 | 1.22 (1.42), −1.40 to 3.20 | −0.52 (−1.34 to 0.29), p = 0.20 | −0.20 (−1.01 to 0.61), p = 0.63 | 0.33 (−0.49 to 1.14), p = 0.43 |
| Emotional function (nA = 17, nB = 16, nC = 17) | 5.21 (1.13), 3.38 to 6.88 | 5.86 (1.38), 1.50 to 7.00 | 0.65 (1.11), −1.88 to 2.63 | 4.45 (1.70), 1.75 to 7.00 | 6.22 (1.04), 3.75 to 7.00 | 1.77 (1.65), 0 to 4.88 | 4.79 (1.32), 2.00 to 6.75 | 5.93 (1.66), 1.38 to 7.00 | 1.14 (1.41), −1.50 to 3.63 | −0.73 (−1.61 to 0.15), p = 0.10 | −0.28 (−1.13 to 0.57), p = 0.51 | 0.45 (−0.41 to 1.31), p = 0.30 |
| Total (nA = 17, nB = 16, nC = 17) | 4.81 (1.01), 2.96 to 6.52 | 5.64 (1.29), 2.17 to 7.00 | 0.83 (0.90), −1.09 to 2.96 | 4.43 (1.39), 2.04 to 6.83 | 6.05 (1.01), 3.43 to 7.00 | 1.62 (1.38), −0.52 to 3.74 | 4.43 (1.29), 1.74 to 6.65 | 5.69 (1.59), 1.65 to 7.00 | 1.26 (1.30), −0.83 to 3.13 | −0.63 (−1.42 to 0.15), p = 0.11 | −0.27 (−1.05 to 0.50), p = 0.48 | 0.36 (−0.42 to 1.14), p = 0.36 |
Quality of life of caregivers as measured by the Paediatric Asthma Caregiver's Quality of Life Questionnaire
A total of 38 caregivers completed the quality of life questionnaire at baseline and at 48 weeks. The results show an improvement in mean scores for activity limitations and emotional function across all treatment groups at 48 weeks (Table 11). In support of the PAQLQ(S) analysis, the largest improvement in mean score at 48 weeks is seen for fluticasone plus montelukast across all domains. The pair-wise treatment comparison results from ANCOVA, however, show that none of the mean differences adjusted for baseline score is statistically significant. Similar results were obtained using data up to 24 weeks (Table 12).
| Domain | Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | Fluticasone vs futicasone plus salmeterol | Fluticasone vs futicasone plus montelukast | Fluticasone plus salmeterol vs futicasone plus montelukast | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline mean (SD), range | T48 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T48 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T48 mean (SD), range | Change mean (SD), range | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | |
| Activity limitations (nA = 11, nB = 15, nC = 12) | 4.75 (1.34), 2.50 to 7.00 | 6.07 (1.16), 3.50 to 7.00 | 1.32 (1.41), −1.25 to 3.50 | 5.08 (1.43), 1.25 to 6.75 | 6.30 (0.99), 4.25 to 7.00 | 1.22 (1.30), −1.00 to 3.75 | 3.85 (1.53), 1.75 to 6.50 | 5.81 (1.41), 3.50 to 7.00 | 1.96 (1.21), 0.50 to 4.75 | −0.09 (−0.92 to 0.75), p = 0.83 | −0.13 (−1.03 to 0.77), p = 0.77 | −0.04 (−0.90 to 0.83), p = 0.93 |
| Emotional function (nA = 11, nB = 15, nC = 12) | 4.44 (1.09), 2.22 to 6.22 | 5.80 (1.56), 1.67 to 7.00 | 1.36 (1.05), −0.56 to 3.00 | 4.35 (1.35), 1.00 to 6.00 | 5.73 (1.27), 2.33 to 7.00 | 1.38 (1.45), −0.56 to 4.44 | 4.05 (1.49), 2.00 to 6.00 | 5.67 (1.27), 3.78 to 7.00 | 1.62 (1.48), −1.11 to 4.11 | 0.02 (−0.95 to 0.99), p = 0.97 | −0.07 (−1.10 to 0.96), p = 0.89 | −0.08 (−1.04 to 0.87), p = 0.86 |
| Total (nA = 11, nB = 15, nC = 12) | 4.54 (1.08), 2.46 to 6.46 | 5.88 (1.42), 2.23 to 7.00 | 1.34 (1.07), −0.23 to 3.15 | 4.57 (1.28), 1.08 to 6.15 | 5.91 (1.13), 3.08 to 7.00 | 1.34 (1.25), −0.38 to 4.00 | 3.99 (1.43), 2.00 to 6.15 | 5.71 (1.25), 3.77 to 7.00 | 1.72 (1.27), −0.38 to 4.08 | −0.01 (−0.87 to 0.85), p = 0.99 | −0.12 (−1.04 to 0.80), p = 0.79 | −0.12 (−0.97 to 0.74), p = 0.78 |
| Domain | Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | Fluticasone vs fluticasone plus salmeterol | Fluticasone vs fluticasone plus montelukast | Fluticasone plus salmeterol vs fluticasone plus montelukast | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline mean (SD), range | T24 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T24 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T24 mean (SD), range | Change mean (SD), range | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | |
| Activity limitations (nA = 17, nB = 14, nC = 16) | 4.49 (1.49), 2.00 to 7.00 | 6.00 (1.31), 3.25 to 7.00 | 1.51 (1.29), −1.25 to 3.50 | 5.36 (0.99), 3.25 to 6.75 | 6.04 (1.24), 3.25 to 7.00 | 0.68 (1.33), −2.00 to 3.25 | 4.09 (1.64), 1.75 to 7.00 | 5.92 (1.07), 4.00 to 7.00 | 1.83 (1.55), −2.25 to 4.25 | 0.30 (−0.53 to 1.12), p = 0.47 | −0.07 (−0.85 to 0.70), p = 0.85 | −0.37 (−1.23 to 0.49), p = 0.39 |
| Emotional function (nA = 17, nB = 16, nC = 16) | 4.22 (1.36), 1.67 to 6.78 | 5.66 (1.42), 2.33 to 7.00 | 1.44 (1.27), −0.56 to 4.44 | 4.35 (1.31), 1.00 to 6.00 | 5.42 (1.82), 1.78 to 7.00 | 1.07 (1.22), −1.78 to 2.89 | 4.32 (1.43), 2.00 to 6.33 | 5.63 (1.18), 3.00 to 7.00 | 1.31 (1.57), −3.33 to 4.11 | 0.32 (−0.57 to 1.20), p = 0.47 | 0.09 (−0.79 to 0.98), p = 0.83 | −0.23 (−1.12 to 0.67), p = 0.62 |
| Total (nA = 17, nB = 16, nC = 16) | 4.30 (1.33), 2.00 to 6.85 | 5.76 (1.36), 2.62 to 7.00 | 1.46 (1.18), −0.77 to 4.15 | 4.54 (1.24), 1.08 to 6.15 | 5.57 (1.66), 2.28 to 7.00 | 1.03 (1.21), −1.85 to 2.85 | 4.25 (1.41), 2.00 to 6.54 | 5.72 (1.08), 3.54 to 7.00 | 1.47 (1.51), −3.00 to 4.08 | 0.32 (−0.51 to 1.15), p = 0.44 | 0.02 (−0.81 to 0.85), p = 0.96 | −0.30 (−1.15 to 0.54), p = 0.47 |
Time from randomisation to first exacerbation requiring treatment with a short course of oral corticosteroids
In total, 63 randomised patients could be included in the analysis of time to first exacerbation (Table 13) [median length of follow-up 47 weeks (range 0 to 57 weeks, IQR 33 to 49 weeks) for the 49 censored patients]. Overall, there were 14 (22.2%) patients who had at least one exacerbation event, with most events in the fluticasone plus salmeterol group [seven events (30.4%)] and least events in the fluticasone plus montelukast group [three events (14.3%)]. No statistically significant difference in time to first exacerbation could be detected between the three treatment groups with the log-rank test (Figure 2, χ2 = 1.90, 2df, p = 0.39). Because of the limited data, wide CIs for pair-wise HRs (Table 14) make it impossible to draw any meaningful conclusions.
| Treatment | No. of patients | No. of events (%) | Median exacerbation-free time | 24-week exacerbation-free probability (95% CI) |
|---|---|---|---|---|
| Fluticasone | 19 | 4 (21.1) | Not estimable | 0.79 (0.52 to 0.91) |
| Fluticasone plus salmeterol | 23 | 7 (30.4) | 0.80 (0.55 to 0.92) | |
| Fluticasone plus montelukast | 21 | 3 (14.3) | 0.83 (0.57 to 0.94) | |
| Total | 63 | 14 (22.2) |
FIGURE 2.
Kaplan–Meier survival curves for time to first exacerbation by treatment.
| Comparison | HR (95% CI) |
|---|---|
| Fluticasone vs fluticasone plus salmeterol | 0.63 (0.19 to 2.08) |
| Fluticasone vs fluticasone plus montelukast | 1.52 (0.34 to 6.70) |
| Fluticasone plus salmeterol vs fluticasone plus montelukast | 2.37 (0.68 to 8.20) |
Number of school days missed due to respiratory problems
A total of 37 children with data available up to 48 weeks were included in the ITT analysis. Fewer children missed at least 1 day of school over 48 weeks on fluticasone plus montelukast (18.2%) than on fluticasone (63.6%) and fluticasone plus salmeterol (60%) (Table 15); however, this pattern is not supported by the 24-week data, based on 54 patients, which show that a similar percentage of patients missed at least 1 day across treatment groups (Table 16). The RRs for number of school days missed, estimated from Poisson regression, are not statistically significant (Table 17) and wide CIs prevent useful conclusions for the 48-week and 24-week analyses.
| Fluticasone (n = 11) | Fluticasone plus salmeterol (n = 15) | Fluticasone plus montelukast (n = 11) | Total (n = 37) | |
|---|---|---|---|---|
| Children with at least 1 school day missed, n (%) | 7 (63.6) | 9 (60.0) | 2 (18.2) | 18 (48.6) |
| Total no. of school days missed over 48 weeks,a n (%) | ||||
| 0 | 4 (36.4) | 6 (40.0) | 9 (81.8) | 19 (51.4) |
| 1 | 2 (18.2) | 1 (6.7) | 0 (0.0) | 3 (8.1) |
| 2 | 1 (9.1) | 3 (20.0) | 0 (0.0) | 4 (10.8) |
| 3 | 1 (9.1) | 1 (6.7) | 0 (0.0) | 2 (5.4) |
| 4 | 1 (9.1) | 0 (0.0) | 0 (0.0) | 1 (2.7) |
| 5 | 1 (9.1) | 0 (0) | 1 (9.1) | 2 (5.4) |
| 6 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 7 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 8 | 1 (9.1) | 0 (0.0) | 0 (0.0) | 1 (2.7) |
| 9 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 10 | 0 (0.0) | 3 (20.0) | 0 (0.0) | 3 (8.1) |
| 11 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 12 | 0 (0.0) | 0 (0.0) | 1 (9.1) | 1 (2.7) |
| 13 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 14 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 15 | 0 (0.0) | 1 (6.7) | 0 (0.0) | 1 (2.7) |
| Median (IQR) | 1 (0 to 4) | 2 (0 to 10) | 0 (0 to 0) | 0 (0 to 3) |
| Range | 0 to 8 | 0 to 15 | 0 to 12 | 0 to 15 |
| Total exposure time (weeks) | ||||
| Mean (SD) | 48.04 (1.06) | 48.09 (1.96) | 48.38 (2.46) | 48.16 (1.87) |
| Range | 46.14 to 50.00 | 43.00 to 50.00 | 43.00 to 53.00 | 43.00 to 53.00 |
| Fluticasone (n = 18) | Fluticasone plus salmeterol (n = 17) | Fluticasone plus montelukast (n = 19) | Total (n = 54) | |
|---|---|---|---|---|
| Children with at least 1 school day missed, n (%) | 6 (33.3) | 8 (47.1) | 6 (31.6) | 20 (37.0) |
| Total no. of school days missed over 24 weeks,a n (%) | ||||
| 0 | 12 (66.7) | 9 (52.9) | 13 (68.4) | 34 (63.0) |
| 1 | 0 (0.0) | 3 (17.6) | 0 (0.0) | 3 (5.6) |
| 2 | 2 (11.1) | 2 (11.8) | 3 (15.8) | 7 (13.0) |
| 3 | 2 (11.1) | 2 (11.8) | 0 (0.0) | 4 (7.4) |
| 4 | 1 (5.6) | 0 (0.0) | 0 (0.0) | 1 (1.9) |
| 5 | 0 (0.0) | 1 (5.9) | 1 (5.3) | 2 (3.7) |
| 6 | 1 (5.6) | 0 (0.0) | 0 (0.0) | 1 (1.9) |
| 7 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 8 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 9 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 10 | 0 (0.0) | 0 (0.0) | 2 (10.5) | 2 (3.7) |
| Median (IQR) | 0 (0 to 2) | 0 (0 to 2) | 0 (0 to 2) | 0 (0 to 2) |
| Range | 0 to 6 | 0 to 5 | 0 to 10 | 0 to 10 |
| Total exposure time (weeks) | ||||
| Mean (SD) | 24.24 (1.03) | 24.78 (1.43) | 24.45 (1.27) | 24.48 (1.25) |
| Range | 22.00 to 26.00 | 22.29 to 29.00 | 22.00 to 28.14 | 22.00 to 29.00 |
| Analysis set | No. of patients | Time point (weeks) | Scale parametera | Fluticasone vs fluticasone plus salmeterol | Fluticasone vs fluticasone plus montelukast | Fluticasone plus salmeterol vs futicasone plus montelukast | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RR | 95% CI | p-valueb | RR | 95% CI | p-valueb | RR | 95% CI | p-valueb | ||||
| ITT | nA = 11, nB = 15, nC = 11 | 48 | 2.58 | 0.60 | 0.17 to 2.05 | 0.41 | 1.42 | 0.29 to 7.07 | 0.67 | 2.39 | 0.59 to 9.71 | 0.22 |
| nA = 18, nB = 17, nC = 19 | 24 | 2.00 | 1.07 | 0.30 to 3.84 | 0.91 | 0.69 | 0.22 to 2.12 | 0.51 | 0.64 | 0.20 to 2.05 | 0.45 | |
| Per protocol | nA = 9, nB = 11, nC = 8 | 48 | 2.72 | 0.69 | 0.14 to 3.33 | 0.65 | 0.93 | 0.15 to 5.66 | 0.94 | 1.35 | 0.27 to 6.68 | 0.72 |
| nA = 15, nB = 13, nC = 16 | 24 | 2.00 | 1.97 | 0.41 to 9.46 | 0.40 | 0.74 | 0.24 to 2.31 | 0.61 | 0.38 | 0.08 to 1.68 | 0.20 | |
Number of hospital admissions due to respiratory problems
Tables 18 and 19 show the number of hospital admissions over 48 weeks and 24 weeks respectively. Two hospital admissions occurred in two patients on fluticasone plus salmeterol over 48 weeks (see Table 18). Analysis of the number of admissions with Poisson regression was not possible because of sparse data and no conclusions can be drawn (Table 20). For the patients with data up to 24 weeks, four (7.4%) patients required at least one hospital admission, with a similar frequency across treatment groups (see Table 19). The CIs for the 24-week RRs estimated from Poisson regression (see Table 20) are very wide and include relative effects in both directions.
| Fluticasone (n = 11) | Fluticasone plus salmeterol (n = 15) | Fluticasone plus montelukast (n = 11) | Total (n = 37) | |
|---|---|---|---|---|
| Children with at least one hospital admission, n (%) | 0 (0.0) | 2 (13.3) | 0 (0.0) | 2 (5.4) |
| Total no. of hospital admissions over 48 weeks,a n (%) | ||||
| 0 | 11 (100.0) | 13 (86.7) | 11 (100.0) | 35 (94.6) |
| 1 | 0 (0.0) | 2 (13.3) | 0 (0.0) | 2 (5.4) |
| Total exposure time (weeks) | ||||
| Mean (SD) | 48.04 (1.06) | 8.09 (1.96) | 48.38 (2.46) | 48.16 (1.87) |
| Range | 46.14 to 50.00 | 43.00 to 50.00 | 43.00 to 53.00 | 43.00 to 53.00 |
| Fluticasone (n = 18) | Fluticasone plus salmeterol (n = 17) | Fluticasone plus montelukast (n = 19) | Total (n = 54) | |
|---|---|---|---|---|
| Children with at least one school day missed, n (%) | 2 (11.1) | 1 (5.9) | 1 (5.3) | 4 (7.4) |
| Total no. of hospital admissions over 48 weeks,a n (%) | ||||
| 0 | 16 (88.9) | 16 (94.1) | 18 (94.7) | 50 (92.6) |
| 1 | 1 (5.6) | 1 (5.9) | 1 (5.3) | 3 (5.6) |
| 2 | 1 (5.6) | 0 (0.0) | 0 (0.0) | 1 (1.9) |
| Total exposure time (weeks) | ||||
| Mean (SD) | 24.24 (1.03) | 24.78 (1.43) | 24.45 (1.27) | 24.48 (1.25) |
| Range | 22.00 to 26.00 | 22.29 to 29.00 | 22.00 to 28.14 | 22.00 to 29.00 |
| Analysis set | No. of patients | Time point | Scale parametera | Fluticasone vs fluticasone plus salmeterol | Fluticasone vs fluticasone plus montelukast | Fluticasone plus salmeterol vs fluticasone plus montelukast | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RR | 95% CI | p-valueb | RR | 95% CI | p-valueb | RR | 95% CI | p-valueb | ||||
| ITT | nA = 11, nB = 15, nC = 11 | 48 weeksc | – | – | – | – | – | – | – | – | – | – |
| nA = 18, nB = 17, nC = 19 | 24 weeks | 1.08 | 2.90 | 0.25 to 33.11 | 0.39 | 3.19 | 0.28 to 36.51 | 0.35 | 1.10 | 0.06 to 21.79 | 0.95 | |
| Per protocol | nA = 9, nB = 11, nC = 8 | 48 weeksc | – | – | – | – | – | – | – | – | – | – |
| nA = 15, nB = 13, nC = 16 | 24 weeks | 1.14 | 1.77 | 0.12 to 27.21 | 0.68 | 2.15 | 0.14 to 33.06 | 0.58 | 1.22 | 0.05 to 28.50 | 0.90 | |
Amount of rescue beta-2 agonist therapy prescribed for asthma symptoms
A total of 23 out of 37 (62.2%) children were prescribed at least one beta-2 agonist over 48 weeks of follow-up (Table 21), with a median total dose per patient of 24,000 μg. In total, 28 out of 53 (52.8%) children were prescribed at least one beta-2 agonist over 24 weeks of follow-up (Table 22), with a median total dose per patient of 20,000 μg. There was no significant difference in the mean amount prescribed between the three groups from ANOVA at 48 weeks (F-value = 0.96, 2df, p = 0.39) or 24 weeks (F-value = 1.2, 2df, p = 0.31) (Table 23).
| Fluticasone (n = 11) | Fluticasone plus salmeterol (n = 14a) | Fluticasone plus montelukast (n = 12) | Total (n = 37) | |
|---|---|---|---|---|
| Children with at least one beta-2 agonist prescribed, n (%) | 6 (54.5) | 10 (71.4) | 7 (58.3) | 23 (62.2) |
| Total no. of beta-2 agonists prescribed over 48 weeks,a n (%) | ||||
| 0 | 5 (45.5) | 4 (28.6) | 5 (41.7) | 14 (37.8) |
| 1 | 0 (0.0) | 3 (21.4) | 1 (8.3) | 4 (10.8) |
| 2 | 0 (0.0) | 1 (7.1) | 1 (8.3) | 2 (5.4) |
| 3 | 2 (18.2) | 1 (7.1) | 3 (25.0) | 6 (16.2) |
| 4 | 2 (18.2) | 0 (0.0) | 0 (0.0) | 2 (5.4) |
| 5 | 1 (9.1) | 1 (7.1) | 0 (0.0) | 2 (5.4) |
| 6 | 0 (0.0) | 2 (14.3) | 1 (8.3) | 3 (8.1) |
| 7 | 1 (9.1) | 0 (0.0) | 1 (8.3) | 2 (5.4) |
| 8 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 9 | 0 (0.0) | 1 (7.1) | 0 (0.0) | 1 (2.7) |
| 40 | 0 (0.0) | 1 (7.1) | 0 (0.0) | 1 (2.7) |
| Median (IQR) | 3 (0 to 4) | 1.5 (0 to 6) | 1.5 (0 to 3) | 2 (0 to 4) |
| Range | 0 to 7 | 0 to 40 | 0 to 7 | 0 to 40 |
| Amount of beta-2 agonists prescribed over 48 weeksa (µg) | ||||
| Median (IQR) | 36,000 (0 to 80,000) | 22,000 (0 to 120,000) | 22,000 (0 to 60,000) | 24,000 (0 to 80,000) |
| Range | 0 to 140,000 | 0 to 800,000 | 0 to 140,000 | 0 to 800,000 |
| Fluticasone (n = 18) | Fluticasone plus salmeterol (n = 16a) | Fluticasone plus montelukast (n = 19) | Total (n = 53) | |
|---|---|---|---|---|
| Children with at least one beta-2 agonist prescribed, n (%) | 9 (50.0) | 10 (62.5) | 9 (47.4) | 28 (52.8) |
| Total no. of beta-2 agonists prescribed over 24 weeks,a n (%) | ||||
| 0 | 9 (50.0) | 6 (37.5) | 10 (52.6) | 25 (47.2) |
| 1 | 2 (11.1) | 4 (25.0) | 4 (21.1) | 10 (18.9) |
| 2 | 4 (22.2) | 4 (25.0) | 4 (21.1) | 12 (22.6) |
| 3 | 1 (5.6) | 0 (0.0) | 1 (5.3) | 2 (3.8) |
| 4 | 2 (11.1) | 1 (6.3) | 0 (0.0) | 3 (5.7) |
| 31 | 0 (0.0) | 1 (6.3) | 0 (0.0) | 1 (1.9) |
| Median (IQR) | 0.5 (0 to 2) | 1 (0 to 2) | 0 (0 to 2) | 1 (0 to 2) |
| Range | 0 to 4 | 0 to 31 | 0 to 3 | 0 to 31 |
| Amount of beta-2 agonists prescribed over 24 weeksa (µg) | ||||
| Median (IQR) | 6000 (0 to 40,000) | 20,000 (0 to 40,000) | 0 (0 to 24,000) | 20,000 (0 to 40,000) |
| Range | 0 to 80,000 | 0 to 620,000 | 0 to 60,000 | 0 to 620,000 |
| Comparison of means | Fluticasone vs fluticasone plus salmeterol, mean difference (µg) (95% CIa) | Fluticasone vs fluticasone plus montelukast, mean difference (µg) (95% CIa) | Fluticasone plus salmeterol vs fluticasone plus montelukast, mean difference (µg) (95% CIa) |
|---|---|---|---|
| 48 weeks | −59,481 (−195,859 to 76,897) | 6758 (−134,532 to 148,048) | 66,238 (−66,920 to 199,396) |
| 24 weeks | −34,861 (−107,549 to 37,827) | 7942 (−61,642 to 77,525) | 42,803 (−28,980 to 114,585) |
Time from randomisation to treatment withdrawal (because of lack of efficacy or side effects)
Although 13 patients were withdrawn from treatment or the study, only one (on fluticasone) withdrew from treatment because of poor asthma control (patient also recorded intercurrent illness as reason) during follow-up (see Table 34). The planned analysis of time to treatment withdrawal was therefore not possible.
Lung function at 48 weeks assessed by spirometry
A total of 30 children had lung function assessed by spirometry at baseline and at 48 weeks and could be included in ANCOVA (Table 24). There were no statistically significant adjusted mean differences between the three treatment groups, with wide CIs that include clinically significant differences in both directions.
| Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | Fluticasone vs futicasone plus salmeterol | Fluticasone vs futicasone plus montelukast | Fluticasone plus salmeterol vs fluticasone plus montelukast | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline mean (SD), range | T48 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T48 mean (SD), range | Change mean (SD), range | Baseline mean (SD), range | T48 mean (SD), range | Change mean (SD), range | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | Adjusted mean difference (95% CI), p-value | |
| FEV1% predicted (nA = 8, nB = 13, nC = 9) | 86.13 (22.56), 48.00 to 116.00 | 86.25 (17.77), 49.00 to 106.00 | 0.12 (12.74), −16.00 to 19.00 | 74.46 (19.70), 47.00 to 103.00 | 90.00 (13.91), 64.00 to 107.00 | 15.54 (19.77), −17.00 to 56.00 | 84.56 (16.94), 56.00 to 104.00 | 89.11 (13.63), 73.00 to 120.00 | 4.55 (15.83), −14.00 to 30.00 | −8.58 (−20.71 to 3.56), p = 0.16 | −3.51 (−16.24 to 9.22), p = 0.58 | 5.07 (−6.57 to 16.71), p = 0.38 |
| FVC% predicted (nA = 8, nB = 13, nC = 9) | 87.50 (19.93), 57.00 to 112.00 | 92.00 (12.22), 74.00 to 111.00 | 4.50 (10.88), −7.00 to 23.00 | 84.08 (18.29), 49.00 to 111.00 | 95.15 (16.35), 67.00 to 128.00 | 11.07 (24.59), −12.00 to 79.00 | 91.89 (20.19), 53.00 to 128.00 | 91.67 (13.52), 68.00 to 113.00 | −0.22 (14.59), −15.00 to 28.00 | −4.17 (−16.79 to 8.45), p = 0.50 | 1.64 (−12.03 to 15.30), p = 0.81 | 5.81 (−6.53 to 18.15), p = 0.34 |
Safety outcomes
Adverse events were assessed at each visit between randomisation and the final visit. Adverse events were classified according to the Medical Dictionary for Regulatory Activities (MedRA) (www.meddramsso.com) System Organ Class by the MCRN CTU Data Manager, with all classifications approved by the chief investigator after discussion with the trial working group.
The number (and percentage) of patients experiencing each adverse event is shown for each treatment in Table 25; this is categorised further by severity in Table 26. For each patient who experienced more than one episode of the same adverse event, the maximum severity is shown. No formal statistical testing was undertaken on these data.
| Adverse event | Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patients (n = 19) | % | Events | Patients (n = 23) | % | Events | Patients (n = 21) | % | Events | |
| Ear and labyrinth disorders | 1 | 4.3 | 1 | ||||||
| Eye disorders | 1 | 4.8 | 1 | ||||||
| Gastrointestinal disorders | 5 | 26.3 | 6 | 5 | 21.7 | 7 | 5 | 23.8 | 9 |
| General disorders/administration site conditions | 4 | 21.1 | 5 | 6 | 26.1 | 10 | 5 | 23.8 | 6 |
| Immune system disorders | 2 | 10.5 | 2 | 3 | 13.0 | 4 | 1 | 4.8 | 2 |
| Infections and infestations | 7 | 36.8 | 11 | 9 | 39.1 | 13 | 7 | 33.3 | 13 |
| Injury, poisoning and procedural complications | 3 | 15.8 | 3 | 1 | 4.3 | 1 | 3 | 14.3 | 3 |
| Musculoskeletal and connective tissue disorders | 1 | 5.3 | 1 | 2 | 9.5 | 3 | |||
| Nervous system disorders | 5 | 26.3 | 13 | 1 | 4.3 | 2 | 7 | 33.3 | 37 |
| Psychiatric disorders | 2 | 8.7 | 2 | ||||||
| Reproductive system and breast disorders | 1 | 4.3 | 2 | ||||||
| Respiratory, thoracic and mediastinal disorders | 13 | 68.4 | 28 | 15 | 65.2 | 31 | 13 | 61.9 | 28 |
| Skin and subcutaneous tissue disorders | 2 | 10.5 | 2 | 3 | 13.0 | 3 | 2 | 9.5 | 5 |
| Total | 42 | 71 | 47 | 76 | 46 | 107 | |||
| Adverse event | Severity | No. of events | No. of patients | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | Total | Fluticasone (%) | Fluticasone plus salmeterol (%) | Fluticasone plus montelukast (%) | Total (%) | ||
| Ear and labyrinth disorders | Mild | 1 | 1 | 1 (4.3) | 1 (1.6) | ||||
| Total | 1 | 1 | 1 (4.3) | 1 (1.6) | |||||
| Eye disorders | Mild | 1 | 1 | 1 (4.8) | 1 (1.6) | ||||
| Total | 1 | 1 | 1 (4.8) | 1 (1.6) | |||||
| Gastrointestinal disorders | Mild | 6 | 7 | 9 | 22 | 5 (26.3) | 5 (21.7) | 5 (23.8) | 15 (23.8) |
| Total | 6 | 7 | 9c | 22 | 5 (26.3) | 5 (21.7) | 5c (23.8) | 15 (23.8) | |
| General disorders/administration site conditions | Mild | 5 | 10 | 6 | 21 | 4 (21.1) | 6 (26.1) | 5 (23.8) | 15 (23.8) |
| Total | 5 | 10 | 6 | 21 | 4 (21.1) | 6 (26.1) | 5 (23.8) | 15 (23.8) | |
| Immune system disorders | Mild | 4 | 2 | 6 | 3 (13.0) | 1 (4.8) | 4 (6.3) | ||
| Moderate | 2 | 2 | 2 (10.5) | 2 (3.2) | |||||
| Total | 2 | 4 | 2 | 8 | 2 (10.5) | 3 (13.0) | 1 (4.8) | 6 (9.5) | |
| Infections and infestations | Mild | 9 | 6 | 10 | 25 | 6 (31.6) | 5 (21.7) | 5 (23.8) | 16 (25.4) |
| Moderate | 2 | 7 | 3 | 12 | 1 (5.3) | 4 (17.4) | 2 (9.5) | 7 (11.1) | |
| Total | 11 | 13 | 13 | 37 | 7 (36.8) | 9 (39.1) | 7 (33.3) | 23 (36.5) | |
| Injury, poisoning and procedural complications | Mild | 3 | 3 | 6 | 3 (15.8) | 3 (14.3) | 6 (9.5) | ||
| Moderate | 1 | 1 | 1 (4.3) | 1 (1.6) | |||||
| Total | 3 | 1 | 3 | 7 | 3 (15.8) | 1 (4.3) | 3 (14.3) | 7 (11.1) | |
| Musculoskeletal and connective tissue | Mild | 1 | 2 | 3 | 1 (5.3) | 1 (4.8) | 2 (3.2) | ||
| Moderate | 1 | 1 | 1 (4.8) | 1 (1.6) | |||||
| Total | 1 | 3 | 4 | 1 (5.3) | 2 (9.5) | 3 (4.8) | |||
| Nervous system disorders | Mild | 13 | 2 | 16 | 31 | 5 (26.3) | 1 (4.3) | 5 (23.8) | 11 (11.1) |
| Moderate | 21 | 21 | 2 (9.5) | 2 (3.2) | |||||
| Total | 13 | 2 | 37 | 52 | 5 (26.3) | 1 (4.3) | 7 (33.3) | 13 (20.6) | |
| Psychiatric disorders | Mild | 1 | 1 | 1 (4.3) | 1 (1.6) | ||||
| Moderate | 1 | 1 | 1 (4.3) | 1 (1.6) | |||||
| Total | 2 | 2 | 2 (8.7) | 2 (3.2) | |||||
| Reproductive system and breast disorders | Mild | 2 | 2 | 1 (4.3) | 1 (1.6) | ||||
| Total | 2 | 2 | 1 (4.3) | 1 (1.6) | |||||
| Respiratory, thoracic and mediastinal disorders | Mild | 24 | 23 | 21 | 68 | 11 (57.9) | 9 (39.1) | 10 (47.6) | 30 (47.6) |
| Moderate | 4 | 8 | 7 | 19 | 2 (10.5) | 6 (26.1) | 3 (14.3) | 11 (11.1) | |
| Total | 28 | 31c | 28c | 87 | 13 (68.4) | 15c (65.2) | 13c (61.9) | 41 (65.1) | |
| Skin and subcutaneous tissue disorders | Mild | 2 | 2 | 3 | 7 | 2 (10.5) | 2 (8.7) | 1 (4.8) | 5 (7.9) |
| Moderate | 1 | 2 | 3 | 1 (4.3) | 1 (4.8) | 2 (3.2) | |||
| Total | 2 | 3 | 5 | 10 | 2 (10.5) | 3 (13.0) | 2 (9.5) | 7 (11.1) | |
| Total | 71 | 76 | 107 | 254 | 42 | 47 | 46 | 135 | |
In total, 53 out of 63 (84%) randomised patients experienced at least one adverse event between randomisation and last follow-up, with a similar percentage of patients across treatment groups [fluticasone 17 (89%); fluticasone plus salmeterol 18 (78%); fluticasone plus montelukast 18 (86%)]. The most common events were respiratory and nervous system disorders, infections and infestations, and general and gastrointestinal disorders. The percentage of patients reporting each adverse event was similar across treatment groups (see Table 25) except for nervous system disorders. Fewer patients reported nervous system disorders on fluticasone plus salmeterol [one patient (4.3%)] than on fluticasone plus montelukast [seven patients (33.3%)] and fluticasone [five patients (26.3%)], with a much greater number of events on fluticasone plus montelukast (37 events compared with 13 on fluticasone and two on fluticasone plus salmeterol) because of one patient experiencing 20 events. All adverse events were graded as mild or moderate (see Table 26) with a similar distribution of severity across treatment groups except for the nervous system disorders described previously.
Serious adverse events and suspected unexpected serious adverse reactions
Between randomisation and trial closure, six patients reported a total of seven serious adverse events (SAEs) (Table 27), with similar frequency across treatment groups (two patients reported three events on fluticasone; three patients reported three events on fluticasone plus salmeterol; one patient reported one event on fluticasone plus montelukast). All SAEs were mild or moderate in severity and all were classified as unrelated to treatment. There were no suspected unexpected serious adverse reactions (SUSARs) and no deaths reported during the trial.
| Patient registration number | Treatment allocation | Time occurred | Description | MedRA SOC | Seriousness | Severity | Expectedness | Relationship | Cause | Outcome | Patient status | Unblinded |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 134005 | Fluticasone | T0–T8 | History of cough requiring regular salbutamol use (4 days out of the previous 7 days). Temperature up to 39°C. Low oral intake. Contact with H1N1. Infective exacerbation with O2 requirement | Respiratory, thoracic and mediastinal disorders | Required hospitalisation | Moderate | Unexpected | Unrelated | Disease under study | Not resolved/ongoing | Continuing in trial | No |
| 134005 | Fluticasone | T8–T24 | Exacerbation of asthma? Viral-induced wheeze | Infections and infestations | Required hospitalisation | Mild | Expected | Unrelated | Disease under study | Resolved | Continuing in trial | No |
| 009010 | Fluticasone | T0–T8 | Patient admitted to children's emergency department following anaphylaxis? as a result of eating ice cream | Immune system disorders | Required hospitalisation | Moderate | Unexpected | Unrelated | Other illness | Resolved | Continuing in trial | No |
| 009003 | Fluticasone plus salmeterol | T36–T48 | Patient complained of episodes of chest pain after swimming. Diagnosis of muscular sprain from swimming | General disorders/ administration site conditions | Required hospitalisation | Mild | Unexpected | Unrelated | Other illness | Resolved | Continuing in trial | No |
| 036001 | Fluticasone plus salmeterol | T24–T36 | Been having ‘hayfever’ symptoms past few days. Started multidosing. Came to children's assessment unit 1230. Peak expiratory flow rate 100 l/minute | Respiratory, thoracic and mediastinal disorders | Required hospitalisation | Mild | Expected | Unrelated | Disease under study | Resolved | Continuing in trial | No |
| 522010 | Fluticasone plus salmeterol | T8–T24 | Admitted as a result of asthma symptoms. Patient reported shortness of breath. Received one nebuliser (salbutamol) and one dose of prednisolone | Respiratory, thoracic and mediastinal disorders | Medically significant/important | Mild | Unexpected | Unrelated | Disease under study | Resolved | Continuing in trial | No |
| 522011 | Fluticasone plus montelukast | T8–T24 | Diagnosis: exacerbation of asthma. Treatment: salbutamol inhalers and oral prednisolone | Respiratory, thoracic and mediastinal disorders | Required hospitalisation | Moderate | Expected | Unrelated | Disease under study | Resolved | Continuing in trial | No |
Health economics
The sample size is extremely small in each group; therefore, although every endeavour has been made to fully analyse and report the economic results and to take necessary statistical safeguards, the results should be interpreted with extreme caution.
It can be seen from Table 28 that there was some loss of patients completing the health economics diary over each time period but completion of the health economics booklet was very similar across the groups. This included patients whose 48-week visit took place within 48 weeks because of the early closure of the trial. We removed patients from the 48-week visit analysis when the final visit took place inside 43 weeks from baseline.
| Group | Baseline | T8 | T24 | T36 | T48 |
|---|---|---|---|---|---|
| Fluticasone | 19 | 19 | 17 | 12 | 11 |
| Fluticasone plus salmeterol | 23 | 22 | 17 | 14 | 15 |
| Fluticasone plus montelukast | 21 | 19 | 19 | 13 | 12 |
Table 29 indicates the total events of care (resource use) for each of the three treatment groups during the study period.
| Resource | Resource use | ||
|---|---|---|---|
| Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | |
| Intervention | 68 | 73 | 72 |
| GP visit | 5 | 9 | 6 |
| GP nurse visit | 3 | ||
| Walk-in doctor visit | 3 | 2 | |
| GP other visit | 2 | 2 | 3 |
| Home doctor visit | 1 | ||
| Out-of-hours GP visit | 1 | ||
| A&E visit | 0 | 5 | 2 |
| Prescribed inhalers | 6 | 12 | 8 |
| Prescribed medicines | 5 | 13 | 7 |
| OTC medicines | 0 | 2 | 2 |
| ‘Rescue’ medication (salbutamol) | 11 | 28 | 6 |
| Total | 100 | 151 | 106 |
Table 30 shows the main cost drivers in the treatment of childhood asthma. The main cost driver, not surprisingly, is the cost of the intervention itself, followed by the costs of GP visits, prescribed inhalers and prescribed medications. With larger patient numbers these results could change.
| Resource | Total resource cost (£) | Total cost (£) | ||
|---|---|---|---|---|
| Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | ||
| Intervention | 1536 | 6300 | 6439 | 14,275 |
| GP visit | 140 | 252 | 168 | 560 |
| GP nurse visit | 0 | 30 | 0 | 30 |
| Walk-in doctor visit | 120 | 80 | 0 | 200 |
| GP other visit | 80 | 113.70 | 290.10 | 483.80 |
| Home doctor visit | 0 | 94 | 0 | 94 |
| Out-of-hours GP visit | 0 | 120 | 0 | 120 |
| A&E visit | 0 | 483.50 | 193.40 | 676.90 |
| Prescribed inhalers | 89 | 280.08 | 100.90 | 469.98 |
| Prescribed medicines | 18.20 | 159.54 | 22.46 | 200.20 |
| OTC medicines | 0 | 3.04 | 0.38 | 3.42 |
| ‘Rescue’ medication (salbutamol) | 7.94 | 85.76 | 6.26 | 99.96 |
| Total | 1991.14 | 8001.62 | 7220.50 | 17,213.26 |
| Average cost per person | 104.80 | 347.90 | 343.83 | 273.23 |
Given that the cost of the trial intervention is the greatest cost driver it is not surprising that the fluticasone plus salmeterol group and the fluticasone plus montelukast group have the greatest overall costs. That said, even though the intervention cost is higher in the fluticasone plus montelukast group, once the overall costs have been factored in it is the fluticasone plus salmeterol group that has the greatest overall cost, although the difference is small. This shows the importance of calculating the full economic cost of an intervention, not just the cost of the intervention itself.
Outcome
The QALY gains shown in Table 31 are based upon independent bootstrapped samples of n = 2000.
| Mean | SD | |
|---|---|---|
| Incremental QALYs | ||
| Fluticasone | 0.09 | 0.04 |
| Fluticasone plus salmeterol | 0.12 | 0.14 |
| Fluticasone plus montelukast | 0.13 | 0.12 |
| Intervention + treatment costs | ||
| Fluticasone | 144.75 | 82.13 |
| Fluticasone plus salmeterol | 458.80 | 95.24 |
| Fluticasone plus montelukast | 447.99 | 128.46 |
Bootstrapped t-tests and Wilcoxon rank-sum tests did not reveal any statistically significant differences in mean QALYs between baseline and 48 weeks when comparing any of the treatment groups in two-way tests. Figure 3 provides a graphical representation of quality of life over time.
FIGURE 3.
Quality-adjusted life-years over time in the trial. A, fluticasone; B, fluticasone plus salmeterol; C, fluticasone plus montelukast.
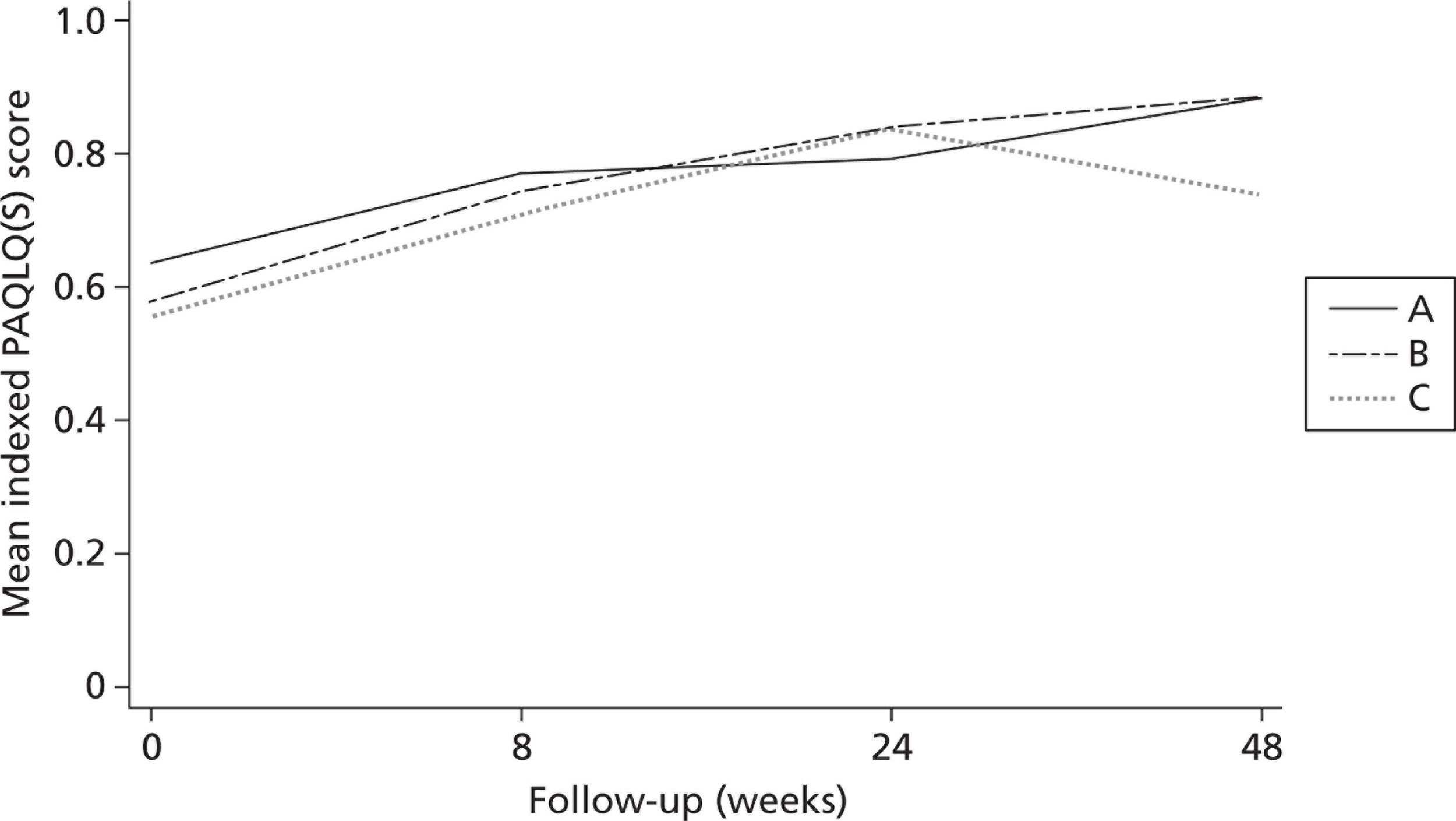
Cost-effectiveness analysis
In terms of economics the variables of interest between the groups are the differences in costs and effects. Table 32 shows the mean differences in incremental costs and QALY gains between the three groups.
| Model | Difference in costs (£) | Difference in QALYs | ||||
|---|---|---|---|---|---|---|
| Mean | SD | p-valuea | Mean | SD | p-valuea | |
| Fluticasone plus salmeterol vs futicasone | 314.05 | 34.90 | 0.48 | 0.03 | 0.04 | 0.46 |
| Fluticasone plus montelukast vs futicasone | 303.24 | 44.59 | 0.35 | 0.04 | 0.04 | 0.43 |
| Fluticasone plus montelukast vs futicasone plus salmeterol | −10.81 | 44.50 | 0.19 | 0.02 | 0.05 | 0.89 |
The measure of interest from these data is incremental cost-effectiveness, captured by the ICER, which shows the difference in the sum of the costs over effects for each individual patient between each of the treatment arms. Table 31 shows that there is very little difference in QALYs before and after the interventions. Similarly, cost differences are not great between the groups, with fluticasone plus salmeterol and fluticasone plus montelukast incurring approximately three times the cost per person as fluticasone. The difference in outcomes was greater for fluticasone plus salmeterol and fluticasone plus montelukast even though the cost was greater; hence, it can be seen that a positive ICER is generated for fluticasone plus salmeterol compared with fluticasone and fluticasone plus montelukast compared with fluticasone (Table 33).
| ICER (£/QALY) | |
|---|---|
| Fluticasone plus salmeterol vs fluticasone | 12,054 |
| Fluticasone plus montelukast vs fluticasone | 6827 |
| Fluticasone plus montelukast vs fluticasone plus salmeterol | –588 |
It can be seen from the ICER results that the treatment of choice is inhaled fluticasone propionate 100 μg twice daily plus montelukast 5-mg tablet once daily, as this produces 1 additional QALY for £6827, whereas it would cost £12,054 to produce an additional QALY from inhaled fluticasone propionate 100 μg and salmeterol 50 μg twice daily (combination inhaler) plus placebo tablet once daily. Both are by comparison with inhaled fluticasone propionate 100 μg. To further aid interpretation of the results the CEACs are presented for fluticasone plus salmeterol and fluticasone plus montelukast compared with fluticasone, and fluticasone plus montelukast compared with fluticasone plus salmeterol (Figure 4).
FIGURE 4.
Cost-effectiveness acceptability curves. A, futicasone; B, futicasone plus salmeterol; C, futicasone plus montelukast.
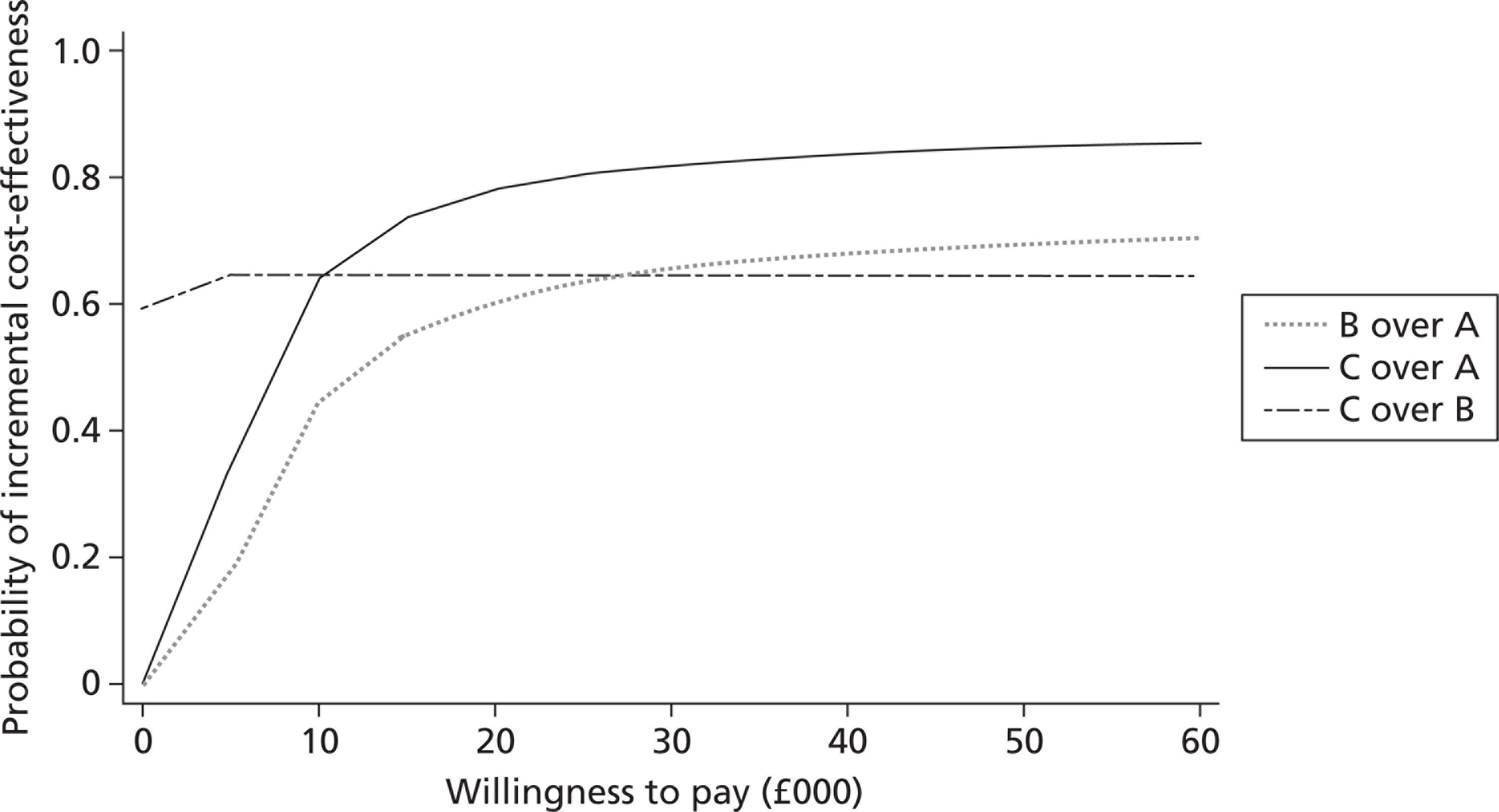
Figure 4 shows the probabilities that fluticasone plus salmeterol and fluticasone plus montelukast are under the £30,000 per QALY cost-effectiveness threshold as required by NICE. The probability of cost-effectiveness at this level is around 80% for fluticasone plus montelukast and 60% for fluticasone plus salmeterol. Moreover, the comparison between fluticasone plus montelukast and fluticasone plus salmeterol shows that fluticasone plus montelukast dominates fluticasone plus salmeterol; however, the CEAC for fluticasone plus montelukast compared with fluticasone plus salmeterol declines after the threshold ICER because of increasing uncertainty, reflecting the lack of statistical power in the incremental QALYs. In fact, there is very little evidence supporting fluticasone plus salmeterol or fluticasone plus montelukast over the other.
Figure 5 illustrates the relative spread of incremental costs and QALYs for fluticasone plus salmeterol and fluticasone plus montelukast compared with fluticasone on a cost-effectiveness plane. This is useful because it shows that, although fluticasone plus montelukast has a lower average incremental cost and higher average incremental QALY gain, it also has greater uncertainty, which is why fluticasone plus salmeterol and fluticasone plus montelukast cannot easily be compared directly.
FIGURE 5.
The cost-effectiveness plane.
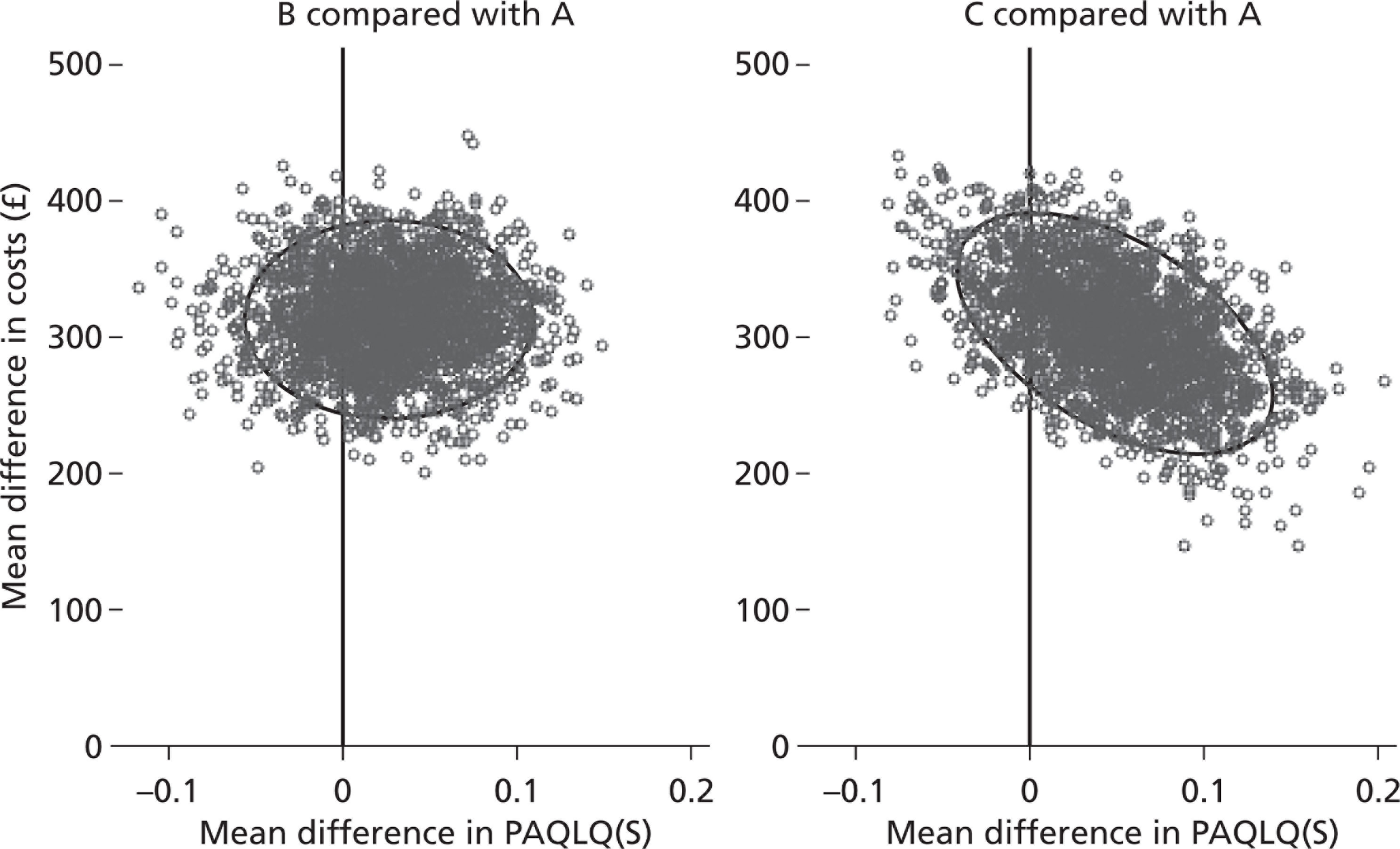
Withdrawals
In total, 13 (20.6%) patients withdrew from the study before the 48-week visit or before early study closure. The numbers of withdrawals and reasons for withdrawal are similar across the three treatment groups (Table 34). Six of the 13 patients had indicated a withdrawal from treatment but no further follow-up data were available. There were no crossovers to another treatment arm.
| Time point of withdrawal | Reason | Fluticasone (n = 19), n (%) | Fluticasone plus salmeterol (n = 23), n (%) | Fluticasone plus montelukast (n = 21), n (%) | Total (n = 63), n (%) |
|---|---|---|---|---|---|
| T0–T8 | Total | 0 (0) | 3 (13.0) | 2 (9.5) | 5 (7.9) |
| Spiritual reasons | 0 | 1 | 1 | 2 | |
| Intercurrent illness | 0 | 0 | 1 | 1 | |
| Patient decision | 0 | 2 | 0 | 2 | |
| T8–24 | Total | 2 (10.5) | 3 (13.0) | 2 (9.5) | 7 (11.1) |
| Lost to follow-up | 1 | 2 | 0 | 3 | |
| Study schedule too intrusive | 0 | 1 | 0 | 1 | |
| Non-compliance | 0 | 0 | 1 | 1 | |
| Unknown | 0 | 0 | 1 | 1 | |
| Poor asthma control and intercurrent illness | 1 | 0 | 0 | 1 | |
| T24–T36 | Total | 1 (5.3) | 0 (0) | 0 (0) | 1 (1.6) |
| Lost to follow-up | 1 | 0 | 0 | 1 | |
| Total | 3 (15.8) | 6 (26.1) | 4 (19.0) | 13 (20.6) |
Compliance
The number of doses missed (inhaler and tablets) since the previous visit was captured on the follow-up CRF. The expected number of tablet doses for each visit was calculated as the number of days since the last visit (i.e. one per day) and the expected number of inhaler doses was calculated as twice the number of days since the last visit (i.e. two per day).
For each visit, each patient had the proportion of doses missed (number of doses missed/number of expected doses) calculated separately for inhalers and tablets. These proportions were averaged over all patients per treatment group separately for inhalers and tablets and are displayed as summary measures in Table 35. The proportion of treatments missed is similar across the groups and time points, except for fluticasone plus montelukast at the 48-week visit.
| Visit | Fluticasone (n) | Fluticasone plus salmeterol (n) | Fluticasone plus montelukast (n) | Total (n) | Cumulative withdrawals (n) | Proportion of inhaler doses missed (patients with missing information) | Proportion of tablet doses missed (patients with missing information) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | Total | Fluticasone | Fluticasone plus salmeterol | Fluticasone plus montelukast | Total | ||||||
| T0 | 19 | 23 | 21 | 63 | 0 | – | – | – | – | – | – | – | – |
| T8 | 19 | 22 | 20 | 61 | 2 | 0.07 (2/19) | 0.04 (3/22) | 0.04 (2/20) | 0.05 (5/61) | 0.05 (2/19) | 0.07 (3/22) | 0.09 (2/20) | 0.07 (5/61) |
| T24 | 18 | 17 | 19 | 54 | 9 | 0.04 (3/18) | 0.05 (4/17) | 0.05 (3/19) | 0.04 (8/54) | 0.04 (2/18) | 0.04 (4/17) | 0.06 (3/19) | 0.04 (7/54) |
| T36a | 12 | 14 | 13 | 39 | 13 | 0.02 (1/12) | 0.04 (1/14) | 0.04 (3/13) | 0.03 (5/39) | 0.02 (1/12) | 0.03 (1/14) | 0.03 (3/13) | 0.03 (5/39) |
| No T36a | 4 | 3 | 4 | 11 | – | – | – | – | – | – | – | – | – |
| T48 | 16 | 17 | 17 | 50 | 13 | 0.07 (1/16) | 0.04 (2/17) | 0.28 (1/17) | 0.13 (4/50) | 0.08 (1/16) | 0.04 (2/17) | 0.35 (1/17) | 0.16 (4/50) |
Outcome of non-randomised patients
The GPs of all 103 children who were registered at T–4 but who were not randomised at T0 were contacted towards the end of the study and asked to complete a questionnaire about each of their patients, as described in the protocol. As the exposure time since date of registration varied, data are summarised according to the time intervals for which the data were collected. The data presented for ‘up to 1 year’ are likely to be an underestimate of events because only the cumulative data over the period from registration until completion of the form were collected. Some patients with data reported over periods > 1 year may have had a number of their events during the first year from registration.
The questionnaire was kept very simple and short to try and maximise response. Nine (14.5%) children had at least one exacerbation that required a course of oral corticosteroids (Table 36). A total of 50 (80.6%) required at least one prescription of beta2 agonist after registration (Table 37). Unfortunately it was not possible to summarise the amount of beta2 agonist prescribed as the reporting of type of inhaler and amount prescribed was inconsistent and incomplete. In total, 18 (29.0%) patients were prescribed at least one additional treatment for asthma control (Table 38), with the majority (61%) of these being combination therapy (ICS plus long-acting beta2 agonist). Four (6.5%) children required an A&E visit (Table 39) and one (1.6%) child needed a hospital admission for their asthma (Table 40).
| Length of follow-up | |||||
|---|---|---|---|---|---|
| Up to 1 year (n = 32) | 1–1.5 years (n = 19) | 1.5–2 years (n = 10) | 2–2.5 years (n = 1) | Total (n = 62) | |
| Children with at least one exacerbation, n (%) | 2 (6.3) | 6 (31.6) | 1 (10.0) | 0 (0.0) | 9 (14.5) |
| Total no. of exacerbations since registration (T–4), n (%) | |||||
| 0 | 30 (93.8) | 13 (68.4) | 9 (90.0) | 1 (100.0) | 53 (85.5) |
| 1 | 2 (6.3) | 3 (15.8) | 1 (10.0) | 0 (0.0) | 5 (8.1) |
| 2 | 0 (0.0) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 2 (3.2) |
| 3 | 0 (0.0) | 2 (10.5) | 0 (0.0) | 0 (0.0) | 2 (3.2) |
| Median (IQR) | 0 (0 to 0) | 0 (0 to 1) | 0 (0 to 0) | 1 | 0 (0 to 0) |
| Range | 0 to 1 | 0 to 3 | 0 to 2 | – | 0 to 3 |
| Total exposure time (weeks) | |||||
| Median (IQR) | 41.57 (33.57 to 45.86) | 65.14 (61.57 to 69.14) | 86.29 (82.07 to 89.93) | 108.14 | 51.93 (41.57 to 68.43) |
| Range (n) | 28.29 to 52.00 (30) | 55.0 to 78.14 (19a) | 78.43 to 102.14 (8a) | 28.29 to 108.14 | |
| Length of follow-up | |||||
|---|---|---|---|---|---|
| Up to 1 year (n = 32) | 1–1.5 years (n = 19) | 1.5–2 years (n = 10) | 2–2.5 years (n = 1) | Total (n = 62) | |
| Children with at least one beta-2 agonist prescribed, n (%) | 22 (68.8) | 18 (94.7) | 9 (90.0) | 1 (100.0) | 50 (80.6) |
| Total no. of beta-2 agonists prescribed since T–4,a n (%) | |||||
| 0 | 10 (31.3) | 1 (5.3) | 1 (10.0) | 0 (0.0) | 12 (19.4) |
| 1 | 2 (6.3) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 3 (4.8) |
| 2 | 4 (12.5) | 2 (10.5) | 2 (20.0) | 0 (0.0) | 8 (12.9) |
| 3 | 2 (6.3) | 2 (10.5) | 2 (20.0) | 0 (0.0) | 6 (9.7) |
| 4 | 5 (15.6) | 3 (15.8) | 0 (0.0) | 0 (0.0) | 8 (12.9) |
| 5 | 4 (12.5) | 1 (5.3) | 2 (20.0) | 0 (0.0) | 7 (11.3) |
| 6 | 2 (6.3) | 2 (10.5) | 0 (0.0) | 0 (0.0) | 4 (6.5) |
| 7 | 0 (0.0) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 1 (1.6) |
| 8 | 0 (0.0) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 1 (1.6) |
| 9 | 0 (0.0) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 1 (1.6) |
| 10 | 1 (3.1) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 2 (3.2) |
| 11 | 0 (0.0) | 2 (10.5) | 0 (0.0) | 0 (0.0) | 2 (3.2) |
| 12 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (100.0) | 1 (1.6) |
| 13 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 14 | 1 (3.1) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 2 (3.2) |
| 15 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 16 | 0 (0.0) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 1 (1.6) |
| 17 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 18 | 1 (3.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.6) |
| 19 | 0 (0.0) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 1 (1.6) |
| 28 | 0 (0.0) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 1 (1.6) |
| Median (IQR) | 2.5 (0 to 5) | 5 (3 to 10) | 4 (2 to 9) | – | 4 (2 to 6) |
| Range | 0 to 18 | 0 to 28 | 0 to 16 | – | 0 to 28 |
| Prescription | Length of follow-up | ||||
|---|---|---|---|---|---|
| Up to 1 year (n = 32) | 1–1.5 years (n = 19) | 1.5–2 years (n = 10) | 2–2.5 years (n = 1) | Total (n = 62) | |
| None prescribed, n (%) | 24 (75.0) | 11 (57.9) | 8 (80.0) | 1 (100.0) | 44 (71.0) |
| At least one prescription, n (%) | 8 (25.0) | 8 (42.1) | 2 (20.0) | 0 (0.0) | 18 (29.0) |
| Montelukast, n (%) | 2 (6.3) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 3 (4.8) |
| Combination therapy (ICS and long-acting beta-2 agonist), n (%) | 6 (18.8) | 4 (21.1) | 1 (10.0) | 0 (0.0) | 11 (17.7) |
| Montelukast and combination therapy (ICS and long-acting beta-2 agonist), n (%) | 0 (0.0) | 2 (10.5) | 0 (0.0) | 0 (0.0) | 2 (3.2) |
| Montelukast and long-acting beta-2 agonist and combination therapy (ICS and long-acting beta-2 agonist), n (%) | 0 (0.0) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 1 (1.6) |
| Long-acting beta-2 agonist and combination therapy (ICS and long-acting beta-2 agonist), n (%) | 0 (0.0) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 1 (1.6) |
| Number of admissions | Length of follow-up | ||||
|---|---|---|---|---|---|
| Up to 1 year (n = 32) | 1–1.5 years (n = 19) | 1.5–2 years (n = 10) | 2–2.5 years (n = 1) | Total (n = 62) | |
| Had at least one admission, n (%) | 1 (3.1) | 2 (10.5) | 1 (10.0) | 0 (0.0) | 4 (6.5) |
| 0, n (%) | 31 (96.9) | 16 (84.2) | 9 (90.0) | 1 (100.0) | 57 (91.9) |
| 1, n (%) | 1 (3.1) | 2 (10.5) | 0 (0.0) | 0 (0.0) | 3 (4.8) |
| 2, n (%) | 0 (0.0) | 0 (0.0) | 1 (10.0) | 0 (0.0) | 1 (1.6) |
| Number of admissions | Length of follow-up | ||||
|---|---|---|---|---|---|
| Up to 1 year (n = 32) | 1–1.5 years (n = 19) | 1.5–2 years (n = 10) | 2–2.5 years (n = 1) | Total (n = 62) | |
| 0, n (%) | 32 (100.0) | 18 (94.7) | 10 (100.0) | 1 (100.0) | 61 (98.4) |
| 1, n (%) | 0 (0.0) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 1 (1.6) |
Chapter 4 Discussion
Main findings
The MASCOT study faced a number of challenges from the outset. Issues relating to study set-up, the pharmaceutical companies and the lack of a NHS facility for packaging and distribution are dealt with in the section on strengths and weaknesses. The most significant issue was the difficulty in recruitment of children with asthma into the study, particularly from primary care. A number of novel strategies were developed to improve recruitment; however, although recruitment did increase towards the end of the study, the percentage of children eligible for randomisation after the run-in fell and an application for additional funding for the study was rejected.
The strategies are clearly outlined in Appendix 3 and should be considered in any study in the future that attempts to recruit children from both primary and secondary care. Had these strategies been considered from the outset of the study the recruitment issue may have been less of a problem. Unfortunately, although all centres agreed that they would institute the strategies appropriate to them from January 2010 onwards, it often takes time before the rewards of such strategies take hold. Had the study been able to progress into and through the 2010 autumn peak seen in children with asthma there was a feeling among the MASCOT staff that we would have shown an upturn in both registration and randomisation of suitable patients. We still have no doubt that such patients exist in the UK.
Because of the early study closure and resulting small sample size there was insufficient power to detect any statistically significant differences between the three groups for the efficacy analyses. At 48 weeks the RR of exacerbations requiring treatment with oral corticosteroids was 0.91 (98.3% CI 0.07 to 12.05, p = 0.93) for fluticasone compared with fluticasone plus salmeterol; 1.10 (98.3% CI 0.06 to 18.6, p = 0.94) for fluticasone compared with fluticasone plus montelukast; and 1.21 (98.3% CI 0.09 to 15.97, p = 0.86) for fluticasone plus salmeterol compared with fluticasone plus montelukast. The CIs are extremely wide and include clinically important RRs that could favour any of the treatments. The results for the 24-week analysis were similarly inconclusive as were the results for the time to first exacerbation comparing fluticasone with fluticasone plus salmeterol (HR 0.63, 95% CI 0.19 to 2.08); time to first exacerbation comparing fluticasone with fluticasone plus montelukast (HR 1.52, 95% CI 0.34 to 6.7); and time to first exacerbation comparing fluticasone plus salmeterol with fluticasone plus montelukast (HR 2.37, 95% CI 0.68 to 8.2).
Although there were no statistically significant differences in mean quality of life scores adjusted for baseline values for any of the pair-wise treatment comparisons, the mean quality of life score had improved at 48 weeks and at 24 weeks for all treatment groups across all domains, both for the child and for the caregiver. Fewer children missed at least 1 day of school over 48 weeks on fluticasone plus montelukast (18.2%) than on fluticasone (63.6%) and fluticasone plus salmeterol (60%), whereas more children on fluticasone plus salmeterol (71.4%) required at least one beta-2 agonist than children on fluticasone (54.5%) or fluticasone plus montelukast (58.3%) over 48 weeks. These patterns were not supported by the 24-week data and wide CIs for pair-wise comparisons of relative treatment effects make conclusions difficult to draw. Only a few children required a hospital admission during the study, with relative treatment effects difficult to estimate.
The majority of patients (84%) experienced at least one mild or moderate adverse event during follow-up. The most common events reported were respiratory disorders, nervous system disorders, infections and infestations, general disorders and gastrointestinal disorders. The percentage of patients reporting each adverse event type was similar across treatment groups except for nervous system disorders, which were reported by fewer patients on fluticasone plus salmeterol [one patient (4.3%)] than patients on fluticasone plus montelukast [eight patients (38.1%)] and fluticasone [five patients (26.3%)], with a much greater number of events on fluticasone plus montelukast because of one patient experiencing 20 events.
Health economic analyses were extremely limited by the small sample size and very small non-significant differences in quality of life outcomes between the three treatment groups, which mean that very little confidence can be placed in the ICER and CEAC results. Nevertheless, interpreting the results with great caution indicates that fluticasone plus montelukast could be the economic treatment of choice. A key component of this treatment is montelukast, which lost patent status in August 2012. Historically, prices of drugs that lose patent status have fallen between 30% and 70%, particularly within 4–5 years. 23,24 This will further improve the cost-effectiveness profile of fluticasone plus montelukast relative to fluticasone plus salmeterol; however, this would be based solely on intervention cost.
The limited data available for children who were registered at T–4 but who were not randomised at T0 suggest that their regimens over the subsequent 12 months were not entirely successful in spite of the control achieved over the 4-week run-in, with 14.5% having an exacerbation, 80.6% requiring at least one short-acting beta-2 agonist prescription and 29% requiring a prescription for at least one further asthma treatment. These results also require careful interpretation as the data represent only 60.2% of the registered non-randomised patients.
Strengths and weaknesses
MASCOT was planned as the largest long-term paediatric asthma study ever. Although UK guidelines on asthma management2 have advocated that, when control is poor on low-dose ICSs alone, combination therapy is the first-line step-up treatment, the evidence base for this remains poor. Before the development of the MCRN in 2005, clinical studies in children were often inadequate in both number and design. MASCOT, it was hoped, would address this by being designed by a paediatric CTU in Liverpool, supported by key respiratory paediatric doctors, nurses and patients. As in all studies the key to success is appropriate study design, good working relations between all concerned and successful recruitment.
MASCOT was in the first wave of MCRN studies funded by the HTA programme. Funding for MASCOT was approved in January 2006 pending the agreement of GSK and MSD to supply the medications, including the necessary placebos. Reaching this agreement took a considerable length of time. It was then discovered that there was no NHS facility large enough to package and supply the medicines for all of the participating research sites. An application was made to the HTA for additional funding for a commercial company to undertake this procedure. The HTA supported the application.
The protocol indicated that metered dose inhalers (MDIs) would be used for the study. Unfortunately, we were then informed that GSK was closing down its factory which was the only worldwide manufacturer for the supply of their MDIs for research purposes. With the approval of all of the principal investigators the study protocol was amended to allow the GSK medications to be in the form of dry powder through Accuhalers. When the montelukast tablets from MSD arrived from America it was noted that their expiration date was only 5 months later. These challenges delayed the study opening and have been well documented in a recent publication in Thorax. 25
The study finally opened fully in all centres in May 2009, some 3 years and 4 months after the initial funding had been approved. When the study was being developed in 2005 there was clinical belief that it would not be difficult to recruit up to 90 children over a 12-month period from each of the major recruitment centres. The reality was very different. Prescribing habits in both primary and secondary care changed between these dates, with increasing numbers of children with asthma being commenced on long-acting beta-2 agonists or montelukast, rendering them ineligible for the MASCOT study. The chief investigator and the trial co-ordinator visited all of the research centres from October 2009 to January 2010 to try to discover ways to improve recruitment. A number of novel ideas were developed and these were put in place by January/February 2010.
The TMG requested a meeting with the HTA to discuss recruitment challenges, the development of novel additional recruitment strategies and the possibility of opening new recruitment sites. The TMG considered closing sites that were failing to recruit successfully and the chief investigator and trial co-ordinator had communicated with other sites where there were good working relationships between the research networks and where the prospective principal investigator believed that it would be practically possible to recruit ≥ 30 patients within the allocated time. The HTA met with the TMG but wanted to see a doubling of patients recruited and randomised over the subsequent 3 months before considering the possibility of opening any new potential sites. Over those 3 months patient recruitment into the study did double but there was little increase in patients with sufficient symptoms to be randomised into the double-blind arm of the study. The HTA therefore closed the study to recruitment in June 2010. Patients who had been randomised up to that date were allowed to continue in the study either for a full 48 weeks or until the end of January 2011, whichever occurred first.
As indicated in the results section, the small number of children recruited and randomised was insufficient to show clinically or statistically significant differences in asthma exacerbations between the three treatment groups. Interestingly, however, quality of life measurements improved from baseline in all three groups for the duration of the study. This phenomenon is well reported in clinical trials and is thought to be the result of participating in the study itself rather than a specific effect of the individual medications. Differences were seen between the results obtained at 24 weeks and those at 48 weeks but these differences were not consistent and were probably once again related to the small number of patients randomised.
Because of previous safety concerns regarding long-acting beta-2 agonists, particularly in adult male US asthma sufferers, it was encouraging that we saw no safety issues related to any of the treatment groups in this study, but our patient numbers were too small to draw any conclusions. Other recently published papers26,27 have, to a certain degree, also allayed previous concerns.
Our health economic data suggest a possible benefit of fluticasone plus montelukast over fluticasone therapy alone but caution is needed because of the small number of children participating in the MASCOT study.
Chapter 5 Conclusions
Implications for health care
Unfortunately, the early cessation of this study leaves the question of how best to treat children uncontrolled on ICSs unanswered. Since commencing the MASCOT study an American asthma study, the BADGER trial,12 has been published which concluded that long-acting beta-2 agonist step-up was more likely to provide a better response than either fluticasone alone or fluticasone plus montelukast. This is in keeping with the present national UK guidelines for asthma therapy. 2 Further studies that have compared add-on long-acting beta-2 agonist with doubling the dose of fluticasone in children28,29 have favoured long-acting beta-2 agonist step-up. However, these studies have major limitations, such as short duration, required reversibility to be recruited and inappropriate outcome measures for children, which make it difficult to generalise the results. Now that MASCOT has been prematurely terminated, the question may never be adequately addressed.
Recommendations for research
We believe that there are two major priorities arising from this work. The first is to examine whether or not patients treated in primary care for a chronic condition can be approached by someone from outside their current care team to consider study participation.
Second, it is important to look at alternative study designs to answer the key research question of what is the most appropriate treatment for children uncontrolled on low-dose ICSs. This might be carried out using routine data included in primary care databases, possibly supplemented with patient-reported outcome data.
Setting the study in the context of existing research
Compared with the adult literature, the results of studies of asthma in children have been inconsistent, and there are far fewer studies available. The earlier studies, in particular, produced negative results5 and there was little information available for the Cochrane systematic review. 30 The MASCOT study, therefore, had the potential to be the definitive paper to state what is the appropriate treatment at step 3 of the national and international guidelines for asthma when low-dose ICSs are not successfully controlling symptoms and exacerbations. Failure to recruit sufficient numbers has prevented this study goal.
Acknowledgements
The MASCOT TMG are very grateful to all of the principal investigators, research practitioners, site pharmacists, GPs, children and their carers and families for their contribution, commitment and enthusiasm. The MASCOT TMG is also grateful to the following: GlaxoSmithKline plc for the manufacture of the Flixotide Accuhaler and the Seretide Accuhaler and Merck Sharp and Dohme Ltd for the manufacture of Singulair (montelukast) and the matching placebo; the Local Research Networks in England; Sophie Perry and Heather Granville, MASCOT trial co-ordinators; Casey Quinn and Lisa Webster, health economic analysis support; Sadie Clayton, respiratory nurse; the Trial Steering Committee (chaired by Dr Anne Thomson) and the Data Monitoring and Ethics Committee (chaired by Dr David Spencer) for their support and work throughout the study; Mr and Mrs Hilton for their invaluable contributions; and the Guy Hilton Asthma Trust for financial support to produce and distribute hand-held records for patients.
The MASCOT research team acknowledges the support of the NIHR through the MRCN. It also acknowledges the funding supplied from the HTA programme.
Contribution of authors
Warren Lenney was the chief investigator, liaised with all the of the clinical centres involved, worked closely with the trial co-ordinators and headed up the team that drew up the interim and final reports.
David Price was a member of the TMG and contributed to the design and conduct of the study, provided expertise in primary care and prepared the report for publication.
Andrew McKay (Trial Statistician) was a member of the TMG, performed the statistical analyses and report preparation during the study and prepared the report for publication.
Catrin Tudur Smith (Senior Lecturer in Biostatistics) was a member of the TMG, led the statistical team, contributed to the design of the study, its conduct and analysis and prepared the report for publication.
Paula Williamson (Director of the CTU and Professor of Medical Statistics) was a member of the TMG, contributed to the design and conduct of the study and reviewed a draft of the report.
Marilyn James (Professor of the Economics of Health and Social Policy) led the health economics team and contributed to the design and analysis of the study and prepared the report for publication.
| Name | Role | Hospital |
|---|---|---|
| Dr. Andrew Collinson | Principal investigator | Royal and Exeter, NHS Foundation Trust |
| Dr Patrick Oades | Principal investigator | |
| Michelle Curtis | Research nurse | |
| Celia Charlton | Research nurse | |
| Tania Crabb | South West PCRN | |
| Dr David Lacy | Principal investigator | Arrowe Park Hospital, Wirral |
| Lucy Meredith | Research nurse | |
| Becky Bryson | Paediatric respiratory nurse | |
| Dr Hitesh Pandya | Principal investigator | Leicester Royal Infirmary |
| Samantha Hunt | Research nurse | |
| Dr Chris Upton | Principal investigator | Norfolk and Norwich University Hospitals |
| Alison Betteridge | Research nurse | |
| Dr Tom Hilliard | Principal investigator | Bristol Royal Hospital for Children |
| Myriam Kanchanatheera | Research nurse | |
| Dr Caroline Pao | Principal investigator | Royal London Hospital |
| Pauleen Waithe | Research nurse | |
| Gemma Barry | Research nurse | |
| Frances Ling | Research nurse | |
| Shelley Mieres | Research nurse | |
| Dr Will Carroll | Principal investigator | Derbyshire Children's Hospital |
| Vanessa Unsworth | Research nurse | |
| Coral Smith | Research nurse | |
| Samantha Jones | Research nurse | |
| Professor Harish Vyas | Principal investigator | University Hospital Nottingham |
| Caroline Youle | Research nurse | |
| Dr Jon Couriel | Principal investigator | Alder Hey Children's NHS Foundation Trust |
| Gillian Webster | Research nurse | |
| Janet Hall | Research nurse | |
| Carol Stanton | North West PCRN | |
| Dr Clare Murray | Principal investigator | Royal Manchester Children's Hospital |
| Helena Prady | Research nurse | |
| Claire Fish | Research nurse | |
| Dr Stephen Turner | Principal investigator | Royal Aberdeen Children's Hospital |
| Richard Leece | Research nurse | |
| Lindsay Cameron | Research nurse | |
| Amanda Cardy | Scottish PCRN | |
| Dr Edward Simmonds | Principal investigator | University Hospitals Coventry and Warwickshire NHS Trust |
| Karen Miles | Research nurse | |
| Pauline Derbyshire | West Midlands PCRN | |
| Professor Warren Lenney | Principal investigator | University Hospital of North Staffordshire |
| Hilary Shepley | Research nurse | |
| Katharine Beamond | Research nurse | |
| Margo Brady | Research nurse | |
| Marie Phipps | Research nurse | |
| Dr Mark Porcheret | GP |
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
Publications
-
Lenney W, Perry S, Price D. Clinical trials and tribulations: the MASCOT study. Thorax 2011;66:457–8.
References
- Turner S, Thomas M, von Ziegenweidt J, Price D. Prescribing trends in asthma: a longitudinal observational study. Arch Dis Child 2008;941:16-22.
- British Thoracic Society . Scottish Intercollegiate Guidelines Network. British guideline on the management of asthma. Thorax 2003;58:1-94.
- Van den Berg NJ, Ossip MS, Hederos CA, Anttila H, Ribeiro BL, Davies PI. Salmeterol/fluticasone propionate (50/100 microg) in combination in a Diskus inhaler (Seretide) is effective and safe in children with asthma. Pediatr Pulmonol 2000;30:97-105.
- Knorr B, Matz J, Bernstein JA, Nguyen H, Seidenberg BC, Reiss TF, et al. Montelukast for chronic asthma in 6- to 14-year-old children: a randomized, double-blind trial. Pediatric Montelukast Study Group. JAMA 1998;279:1181-6.
- Verberne AA, Frost C, Duiverman EJ, Grol MH, Kerrebijn KF. Addition of salmeterol versus doubling the dose of beclomethasone in children with asthma. The Dutch Asthma Study Group. Am J Respir Crit Care Med 1998;158:213-19.
- Greening AP, Ind PW, Northfield M, Shaw G. Added salmeterol versus higher-dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Allen & Hanburys Limited UK Study Group. Lancet 1994;344:219-24.
- Garrett J, Williams S, Wong C, Holdaway D. Treatment of acute asthmatic exacerbations with an increased dose of inhaled steroid. Arch Dis Child 1998;79:12-7.
- Allen D, Mullen M, Mullen B. A meta-analysis of the effect of oral and inhaled corticosteriods on growth. J Allergy Clin Immunol 1994;93:967-76.
- Russell G. Inhaled corticosteroids and adrenal insufficiency. Arch Dis Child 2002;87:455-6.
- Crowley S. Inhaled glucocorticoids and adrenal function: an update. Paediatr Respir Rev 2003;42:153-61.
- Current Problems in Pharmacovigilance. 2002.
- Lemanske RF, Mauger DT, Sorkness CA, Jackson DJ, Boehmer SJ, Martinez FD, et al. Step-up therapy for children with uncontrolled asthma receiving inhaled corticosteroids. N Engl J Med 2010;362:975-85.
- Juniper E, O’Byrne P, Guyatt G, Ferrie P, King D. Development and validation of a questionnaire to measure asthma control. Eur Respir J 1999;144:902-7.
- Friede T, Schmidli H. Blinded sample size reestimation with count data: methods and applications in multiple sclerosis. Stat Med 2010;29:1145-56.
- The Clinical Practice Research Datalink n.d. www.cprd.com (accessed 2005).
- Haybittle JL. Repeated assessment of results in clinical trials of cancer treatment. Br J Radiol 1971;44:793-7.
- Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. I. Introduction and design. Br J Cancer 1976;34:585-612.
- Schulz K, Altman D, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340.
- Moher D, Hopwell S, Schulz K, Gøtzsche P, Devereaux P, Elbourne D, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340.
- BNF for Children, 2011–2012. London: BMJ Group, Pharmaceutical Press and RCPCH Publications; 2011.
- Manca A, Hawkins N, Sculpher M. Estimating mean QALYs in trial based cost-effectiveness analysis: the importance of controlling for baseline utility. Health Econ 2005;14:487-96.
- Drummond M, McGuire A. Economic evaluation in health care: merging theory with practice. Oxford: Oxford University Press; 2001.
- Suh D, Manning W, Schondelmeyer S, Hadsall R. Effect of multiple-source entry on price competition after patent expiration in the pharmaceutical industry. Health Services Res 2000;35:529-47.
- Engström A, Jacob J, Lundin D. Sharp drop in prices after the introduction of generic substitution. Solna, Sweden: LFN-Swedish Pharmaceutical Benefits Board; 2006.
- Lenney W, Perry S, Price D. Clinical trials and tribulations: the MASCOT study. Thorax 2011;66:457-8.
- Carroll W, Jones P, Boit P, Clayton S, Cliff I, Lenney W. Childhood evaluation of salmeterol tolerance – a double-blind randomized controlled trial. Pediatr Allergy Immunol 2010;21:366-44.
- Maspero J, Guerra F, Cuevas F, Gutierrez J, Soto-Ramos M, Anderton S, et al. Efficacy and tolerability of salmeterol/fluticasone propionate versus montelukast in childhood asthma: a prospective, randomized, double-blind, double-dummy, parallel-group study. Clin Ther 2008;308:1492-504.
- de Blic J, Ogorodova L, Klink R, Sidorenko I, Valiulis A, Hofman J, et al. Salmeterol/fluticasone propionate vs. double dose fluticasone propionate on lung function and asthma control in children. Paediatr Allergy Immunol 2011;208:763-71.
- Gappa M, Zachgo W, von Berg A, Kamin W, Stern-Strater C, Steinkamp G, et al. Add-on salmeterol compared to double dose fluticasone in pediatric asthma: a double-blind, randomized trial (VIAPAED). Pediatric Pulmonol 2009;4411:1132-42.
- Ni Chroinin M, Lasserson T, Greenstone I, Ducharme F. Addition of long-acting beta-agonists to inhaled corticosteroids for chronic asthma in children. Cochrane Database Syst Rev 2009;3.
- Curtis L. Unit costs of health and social care 2010. Canterbury: PPSRU, University of Kent; 2010.
- NHS reference costs 2009–10. London: DoH; 2011.
Appendix 1 Patient information sheets (children aged 6–10 years, children aged 11–15 years, adults)
Patient information sheets (children aged 6–10 years, children aged 11–15 years, adults) (PDF download)
Patient information sheets (children aged 6–10 years, children aged 11–15 years, adults) (PDF download)
Patient information sheets (children aged 6–10 years, children aged 11–15 years, adults) (PDF download)
Appendix 2 Statistical analysis plan
1 Introduction
The Statistical Analysis Plan (SAP) provides a detailed and comprehensive description of the main, pre-planned analyses for the study ‘MASCOT: Management of asthma in school-age Children On Therapy: a prospective, multi-centre, randomised, double blind, controlled, trial comparing inhaled fluticasone/salmeterol (combination inhaler) + placebo tablet, and inhaled fluticasone + montelukast tablet with inhaled fluticasone propionate + placebo tablet for improved asthma control’. This study is carried out in accordance with the World Medical Association Declaration of Helsinki (1964) and the Tokyo (1975), Venice (1983), Hong Kong (1989) and South Africa (1996) amendments and will be conducted in compliance with the protocol, MCRN CTU Standard Operating Procedures and EU Directive 2001/20/EC, transposed into UK law as the UK Statutory Instrument 2004 No 1031: Medicines for Human Use (Clinical Trials) Regulations 2004.
This statistical analysis plan details the intended analyses and should be clear and detailed enough to be followed by any statistician. This will prevent the introduction of bias or data dredging.
These planned analyses will be performed by the trial statistician under the supervision of the lead statistician. The analysis results will be described in a statistical analysis report, to be used as the basis of the primary research publications according to the study publication plan.
All analyses are to be performed with Standard Statistical Software (SAS). The final analysis datasets, programs and outputs will be archived following good clinical practice guidelines (ICH E9). The testing and validation of the statistical analysis programs will be performed following the relevant Standard Operation Procedure.
Due to the early trial closure and lack of data some analyses described in protocol v8.1 are not planned now.
2 Design
2.1 Study design
This study is a prospective, multi-centre, randomised, double blind, controlled trial involving 13 secondary care and associated general practices throughout the United Kingdom.
It is planned that 900 patients, that satisfy the entry criteria at screening, will be recruited and registered following consent at T–4 for the 4-week run-in period. It is anticipated that 50% of the patients will still satisfy the entry criteria at T0 after completing the run-in period so therefore 450 patients (sample size) will be eligible to be randomised and continue with the trial after obtaining further consent, 150 in each of the three study arms:
-
Inhaled Afluticasone/salmeterol (combination inhaler) + placebo tablet
-
Inhaled fluticasone + montelukast tablet
-
Inhaled fluticasone propionate + placebo tablet.
Patients will be allocated to one of the three treatment groups based on a 1 : 1 : 1 randomisation procedure stratified by Secondary Care Centre.
Randomisation
Randomisation lists will be generated in STATA using simple block randomisation with variable block length. Pharmacy holds the MASCOT randomisation list for the site with treatment allocations labelled A, B or C. The randomising pharmacy staff must check with the research team that the patient meets the eligibility criteria for randomisation prior to dispensing at T0 and record that this check has been made.
Dispensing to Randomised Patients
The individual three-month patient packs will be ready-labelled with the trial label (space for patient randomisation number blank for completion by pharmacy). The label will have a serrated section on the side listing the treatment allocation contained (either A, B or C) along with the batch number of that pack. There is also space on the serrated section to complete the patient randomisation number and date of dispensing, prior to dispensing of the pack.
After determining the patient's treatment allocation (A, B or C) as described above, the pharmacist will select an appropriate pack. Before giving the pack to the nurse/patient, the pharmacist will complete and remove the serrated label section showing the allocation of that pack. Pharmacy will retain the serrated labels containing the treatment arm allocation and batch number for that patient pack and affix these to the appropriate accountability log for that patient.
Schedule of Study Procedures
| Procedures | Screening (T-4) | Baseline (T0 [clinic])* | Follow-Up Schedule (weeks) | Premature Discontinuation | ||||
|---|---|---|---|---|---|---|---|---|
| T + 8 Weeks (clinic) | T + 24 Weeks (clinic) | T + 36 Weeks (telephone) | T + 48 Weeks (clinic) Study Completion | |||||
| Signed Consent Form | ✗ | |||||||
| Assessment of Eligibility Criteria | ✗ | ✗ | ||||||
| Quality of Life Questionnaires Administered | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Health Economics Questionnaire Completed | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| FEV1, FVC, FEV1/FVC Ratio | ✗ | ✗ | (✗) | |||||
| Review Patient Held Record | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Review of Medical History | ✗ | ✗ | ||||||
| Review of Concomitant Medications | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Study Intervention | ✗ | ✗ | ✗ | ✗ | ||||
| Physical Exam | Complete | ✗ | ✗ | |||||
| Symptom-Directed | (✗) | (✗) | (✗) | (✗) | ||||
| Vital Signs | ✗ | (✗) | (✗) | (✗) | (✗) | (✗) | ||
| Assessment of Adverse Events | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Special Assay or Procedure | Consent and obtain saliva sample for later DNA analysis | ✗ | ||||||
2.2 Study objectives
The primary objective is to determine, in 6–14 year old children with asthma uncontrolled on low-dose ICS, whether their asthma control can be improved by adding in a long-acting beta2 agonist (salmeterol) or a leukotriene receptor antagonist (montelukast) as measured by a reduced number of exacerbations requiring treatment with oral corticosteroids over the 48 week study period.
2.2.1 Primary outcome
The primary outcome is the number of asthma exacerbations requiring treatment with oral corticosteroids over the 48 week study period.
2.2.2 Secondary outcomes
-
Quality of Life as measured by the Paediatric Asthma Quality of Life Questionnaire with Standardised Activities (PAQLQ(S)) and the Paediatric Asthma Caregivers Quality of Life Questionnaire (PACQLQ)
-
Time from randomisation to first exacerbation requiring treatment with a short course of oral corticosteroids
-
School attendance
-
Hospital admissions
-
Amount of rescue beta2 agonist therapy prescribed
-
Time from randomisation to treatment withdrawal (due to lack of efficacy or side effects)
-
Lung function at 48 weeks (as assessed by spirometry)
-
Cost effectiveness
-
Adverse events
2.3 Inclusion/exclusion criteria for registration at T–4 weeks
2.3.1 Inclusion criteria
-
Children with physician diagnosed asthma aged 6 years–14 years, 11months.
-
Those requiring frequent short-acting beta2 agonist relief therapy ≥ 7 puffs in the past seven days.
-
Those with symptoms of asthma (i.e. wheeze, shortness of breath but not cough alone) resulting in:
-
Nocturnal wakening in the last week because of asthma symptoms and/or
-
Asthma has interfered with usual activities in the last week and/or
-
Those who have had exacerbations, defined as a short course of oral corticosteroids, an unscheduled GP or A&E Department visit or a hospital admission within the previous 6 months.
-
Fully informed written (proxy) consent and assent, where appropriate.
2.3.2 Exclusion criteria
-
Children receiving long acting beta-2 agonists, leukotriene receptor antagonists, regular theophylline therapy or high dose ICS > 1000 micrograms and unlicensed beclometasone dipropionate or equivalent (at the discretion of the investigator).
-
Children with other respiratory diseases, cystic fibrosis, cardiac disease or immunological disorders.
2.4 Inclusion/exclusion criteria for randomisation at T0 weeks
2.4.1 Inclusion criteria
-
Children with asthma aged 6 years–14 years.
-
Those requiring frequent short-acting beta2 agonist relief therapy ≥ 7 puffs in the past seven days.
-
Those with symptoms of asthma (i.e. wheeze, shortness of breath but not cough alone) resulting in:
-
Nocturnal wakening in the last week because of asthma symptoms and/or
-
Asthma has interfered with usual activities in the last week.
-
-
Continuing consent/assent (where appropriate).
2.4.2 Exclusion criteria
-
Children whose asthma is controlled after the 4 week run-in, where control is defined as the absence of any symptoms of asthma (except cough alone) or where the symptoms of asthma have not interfered with usual activities in the last week.
-
Children receiving long acting beta-2 agonists, leukotriene receptor antagonists, regular theophylline therapy or high dose ICS > 1000 micrograms and unlicensed beclometasone dipropionate or equivalent (at the discretion of the investigator).
-
Children with other respiratory diseases, cystic fibrosis, cardiac disease or immunological disorders.
2.5 Sample size
The primary outcome ‘Number of asthma exacerbations requiring treatment with short courses of oral corticosteroids over 48 weeks’ will be modelled as a Poisson random variable. The sample size for the primary outcome is estimated using the following formula as described by Friede and Schmidli (personal communication):
where nc is the number of patients in the control arm, λc is the control group rate, t is the length of follow-up and θ* the rate ratio. The formula above does not allow for overdispersion which would read to an inflated sample size.
For 1032 children who have had at least one course of oral steroid in the previous 12 months, the mean exacerbation rate per year is estimated from the UK General Practice Research Database as 1.5 per year with variance 1.02 and dispersion parameter 0.68. This is our current best estimate of exacerbation rate and dispersion parameter but may not be entirely representative of the MASCOT randomised population who will have had inhaler technique corrected. Therefore, a target sample size is estimated here with the intention of undertaking an internal blinded pilot to check parameter assumptions and adjust sample size if required. As described by Friede and Schmidli (personal communication), analogous formulae to those above based on the overall event rate across groups can be used to undertake a blinded sample size review (see section 9.5 of MASCOT trial protocol) which was planned after the first 75 children had been randomised and completed their 24 week follow-up assessment.
As there are three primary treatment comparisons, each will be tested at the two-sided significance level of 1.7% to adjust for the multiplicity and to give a study-wise 5% two-tailed significance level. To have power of 80% to detect, as significant, at least a 30% reduction in exacerbation rate (from 1.5 per year to 1.05 per year, equivalent to a rate ratio of 0.7) and allowing for a loss to follow up of 10%, 147 patients per group are required.
Thirteen main centres will participate in this study, with an anticipated total accrual for screening of around 75 patients per month across these sites, enabling recruitment of approximately 900 children over 12 months. Assuming either 50% or 75% of children to be eligible for randomisation after the 4 week run-in period would enable either 150 or 225 children to be randomised to each treatment group. The exact percentage of children who will be eligible for randomisation following the 4 week run-in period cannot be estimated yet but will be monitored closely to assess the likely impact on recruitment figures (see interim analysis section).
In summary, our preliminary target number to be randomised is 150 children per treatment group (450 in total) with recruitment rates, percentage of children randomised and parameters for sample size calculations closely monitored.
Secondary outcome ‘Quality of Life’: With 150 children in each group, the power to detect, as significant, a difference of 0.5 points between treatment groups on the Juniper Paediatric Asthma Quality of Life Questionnaire, with assumed standard deviation 0.71[2] is greater than 99%.
The MASCOT trial suffered from a very low recruitment rate and due to this it was evident that the trial would not reach the target number of patients. An extension application was submitted to the HTA which was rejected. The HTA agreed, with some additional funding, that randomised patients could continue follow-up until they had either completed their full 48 week follow-up or up until approximately the end of January 2011, whichever was soonest for each individual patient. The reason why follow-up could not be continued beyond January 2011 was that the current batch of study medications would expire. This allowed for at least seven months of follow-up data for the last few patients who were randomised into the trial and allowed full follow-up to be collected for many.
The MASCOT trial did not reach the point of undertaking the internal pilot analysis.
2.6 Recruitment
Recruitment started in April 2009 and was due to end in April 2010 (12 months of recruitment). Follow-up was due to end in April 2011 (52 weeks after last patient recruited). However, due to a slow recruitment rate the trial was stopped early and the last patient was randomised on 24/06/2010 with last follow-up on 08/02/2011. All randomised patients have data collected up to at least T24.
3 Description of Study Population
3.1 Representativeness of study sample and patient throughput
Details on the number of patients who were assessed for eligibility at T–4, those who meet the study inclusion criteria at T–4, those who were eligible at T–4 but who subsequently dropped out prior to T0, those who were assessed for eligibility at T0, those who were eligible and randomised, those who were eligible but not randomised, those who withdraw from the study after randomisation and those who are lost to follow-up will be summarised in a CONSORT[1] flow diagram. Eligible patients who are randomised will be described with respect to demographics and history. The number of ineligible patients randomised will also be reported.
3.2 Baseline comparability of randomised groups
Patients in each treatment group (Inhaled fluticasone/salmeterol (combination inhaler) + placebo tablet, Inhaled fluticasone + montelukast tablet, Inhaled fluticasone propionate + placebo tablet) will be described with respect to gender, age, physical examination measurements (height, weight, general appearance, ENT, cardiovascular, respiratory, gastrointestinal, endocrine, dermatological, musculoskeletal, systolic blood pressure, diastolic blood pressure, pulse rate, respiratory rate) and spirometry measurements (FEV1, FVC, FEV1/FVC ratio actual and percentage predicted).Tests of statistical significance will not be undertaken for baseline characteristics; rather the clinical importance of any imbalance will be noted.
3.3 Definition of outcomes and losses to follow-up
3.3.1 Primary outcome
Number of asthma exacerbations requiring treatment with oral corticosteroids over the 48 week study period. Data on ‘Number of courses of oral corticosteroids prescribed for asthma symptoms since last visit’ are collected on the follow-up CRF at visits T+8, T+24, T+36 and T+48. For each patient, this data will be used to calculate the total number of courses of oral corticosteroids prescribed for asthma symptoms over 48 weeks since date of randomisation at T0.
If any of the trial patients are lost to follow up, contact will initially be attempted through the research nurse and lead investigator at each centre. If the lead investigator at the trial centre is not the patient's usual clinician responsible for their speciality care then follow-up will also be attempted through this latter clinician. Where these attempts are unsuccessful, the child's GP will be asked to contact the family and provide follow-up information to the recruiting centre. This information is included on the patient information sheet. Wherever possible, information on the reason for loss to follow-up will be recorded. The number of patients and reasons for loss to follow-up will be summarised.
3.3.2 Secondary outcomes
Quality of Life as measured by the Paediatric Asthma Quality of Life Questionnaire with Standardised Activities (PAQLQ(S)) and the Paediatric Asthma Caregivers Quality of Life Questionnaire (PACQLQ). The overall and domain specific scores (‘Activity limitations’, ‘Emotional function’ and additionally ‘Symptoms’ for the PAQLQ(S)) at T48 will be analysed (adjusted for baseline T0). A secondary analysis will be performed using the T24 questionnaires.
Time from randomisation to first exacerbation requiring treatment with a short course of oral corticosteroids – calculated as days from date of randomisation to date of first course of oral corticosteroids from the follow-up CRF. Days to last follow-up visit will be calculated for patients who were not prescribed a course of oral corticosteroids and these observations will be censored in analyses of time to first exacerbation requiring treatment with oral corticosteroid.
School attendance – number of school days missed due to respiratory problems collected on the follow-up CRF at visits T+8, T+24, T+36 and T+48 will be summed across the 48 weeks of follow-up with a secondary analysis using only data up to T24.
Hospital admissions – number of hospital admissions due to respiratory problems collected on the follow-up CRF at visits T+8, T+24, T+36 and T+48 will be summed across the 48 weeks of follow-up with a secondary analysis using only data up to T24.
Amount of rescue beta2 agonist therapy prescribed for asthma symptoms – the amount prescribed (mcg) will be calculated for each patient based on the type of inhaler, number of inhalers and dose prescribed. This data is collected on the follow-up CRF and concomitant medications CRF at visits T+8, T+24, T+36 and T+48. The primary analysis will include data up to T48 and a secondary analysis will consist of data up to T24.
Time from randomisation to treatment withdrawal (due to lack of efficacy or side effects) – calculated as days between randomisation and date randomised treatment last taken (from follow-up CRF). Days to last follow-up visit will be calculated for patients who did not withdraw from treatment and these observations will be censored in analyses of time to treatment withdrawal. If reason for treatment withdrawal is recorded as poor asthma control or unacceptable adverse events these observations will be counted as events in analyses. Observations recording any other reason for treatment withdrawal will be censored.
Lung function at 48 weeks (as assessed by spirometry) – measured as change in percent predicted for FEV1 & FVC – data collected on the assessment and randomisation at T0 and on the follow-up CRF at T+48.
Cost effectiveness – See Health Economic Analysis Plan.
Adverse events – data collected on the AE/SAE CRFs at visits T0, T+8, T+24, T+36 and T+48.
The number lost to follow-up within each treatment group will be reported and the reasons where known will be documented. Any deaths and their causes will be reported separately.
3.4 Description of adherence with therapy
Deviations from the intended treatment (withdrawals from randomised treatment), cross-over into another arm and withdrawal from study will be summarised for each treatment group along with reasons where available.
Information on the number of doses missed (inhaler and tablets) will be summarised by treatment group separately for data up to T24 and T48. At each study visit the number of doses missed and the number of expected doses is taken. For each patient these will be totalled and the proportion for each patient (number of doses missed/number of expected doses) calculated. These will be averaged over all patients per group and displayed as summary measures in a table.
4 Trial Monitoring
4.1 Interim analysis plan
We planned to check the estimate of overall exacerbation rate (not split by treatment group) and dispersion parameter after the first 75 children had been randomised and completed their 24 week follow-up assessment. We also planned to undertake interim analysis after 1/3 and 2/3 of the target total number of children had been randomised. However, due to the premature closure of the trial no internal pilot or interim analysis was undertaken.
5 Unblinding of Randomised Treatments
The number of patients who were unblinded will be reported for each group and the reason as to why they were unblinded will also be reported. Treatment packs were identically packaged; therefore the risk of unblinding was minimal. Checks were made on the order of patients being randomised and records were kept of any unblinding requests that were made by sites.
6 Patient Groups for Analysis
6.1 Intention to treat (ITT) analysis
To provide a pragmatic comparison of the different interventions, the principle of intention-to-treat, as far as is practically possible, will be the main strategy of the analysis adopted for the primary outcome and all secondary outcomes. These analyses will be conducted on all patients assigned to the three treatment groups as randomised, regardless of the treatment (study or otherwise) actually received.
No imputation methods will be used for any missing primary outcome data for the primary analyses. Due to the early closure of the trial primary analyses will focus on the pre-planned T48 time point with secondary sensitivity analyses presented using all patients' data up to T24. The early trial closure is not expected to introduce bias. However, as there may be a seasonality effect the distribution of follow up by month of year will be explored graphically and compared across treatment groups.
The membership of the analysis set will be determined and documented and reasons for participant exclusion will be given prior to the blind being broken and the randomisation lists being requested (the analysis set may be refined under review prior to the final statistical analysis). Reasons may include missing data, loss to follow up. The reason should be identified and potential relationship to treatment and the primary outcome should be given.
The number of patients with missing data will be compared across treatment arms to check for any imbalance.
6.2 Per protocol analysis
The membership of the analysis set will be determined and documented and reasons for participant exclusion will be given prior to the blind being broken and the randomisation lists being requested (the analysis set may be refined under review prior to the final statistical analysis). Patients who have not missed any randomised treatment up to the time point of interest (T48 and T24) will be included in the per protocol analysis set. Patients that withdrew from treatment or had a major protocol violation will be excluded. This is a sensitivity analysis and will be used to demonstrate the robustness of the results.
6.3 Safety analysis
For the analysis of safety outcomes, all patients who have received at least one dose of the trial medication will be included. Patients will be included in the treatment group they actually received meaning that if a patient crossed over to another arm for some reason they would contribute safety data to this group instead of, or in addition to, their randomised group.
7 Data Analysis
7.1 Analysis of primary efficacy outcome
The primary outcome of number of asthma exacerbations requiring treatment with short courses of oral corticosteroids over 48 weeks will be compared between treatment groups using a Poisson regression model with two dummy variables representing 3 treatment groups. We originally planned to adjust the primary analysis by centre as this was stratified for during randomisation. However, due to the early closure of the trial the limited number of patients randomised makes it impracticable to include the centre effects in the statistical model. Adjustment of standard errors (multiplying by the square root of the scale parameter estimated as the Pearson's chi-squared statistic divided by its degrees of freedom) to account for overdispersion will be made if the scale parameter is greater than one. Statistical tests will be nominally performed at the 1.7% two-sided significance level (with corresponding 98.3% confidence interval), to give a study-wise 5% two-sided significance level (see sample size calculation). The relative exacerbation rate and confidence interval will be calculated for each pair-wise treatment comparison: Fluticasone compared with Fluticasone plus salmeterol to evaluate the effect of adding in long acting beta-2 agonist (salmeterol 50 micrograms twice daily); Fluticasone compared with Fluticasone plus montelukast to evaluate the effect of adding in oral leukotriene receptor antagonist (montelukast 5 mg once daily); and Fluticasone plus salmeterol compared with Fluticasone plus montelukast to evaluate the effect of adding in long acting beta-2 agonist (salmeterol 50 micrograms twice daily) compared with adding in oral leukotriene receptor antagonist tablet (montelukast 5 mg once daily).
The primary analysis will be based on the 48 week data for patients with data available with a secondary analysis based only on data up to 24 weeks for all patients. A further secondary analysis adjusting the Poisson regression model for important prognostic factors (e.g. age, sex, duration of asthma, rhinitis, baseline PACQL(S)/PACQLQ, baseline number of courses of oral steroids) will be explored if sufficient data are available. Backward elimination will be used to find which variables are best associated with the outcome. A p-value of 0.15 will be used as variables which may not be strongly associated with the outcome may still have an effect on the associations of other variables with the outcome.
At each visit the primary outcome data are collected by asking for the Number of courses of oral corticosteroids prescribed for asthma symptoms since last visit. Therefore if a patient missed a visit during follow-up the primary outcome data would still be captured at the subsequent visit if one took place. Therefore, all children with T48 data should have a complete profile of Number of courses of oral corticosteroids since randomisation and this will be used for the primary analysis. Similarly for the T24 analysis the data should be complete for all patients with T24 data. For the small minority of children who do not have data available for the primary outcome at T48 and where reason for missingness is not due to the early closure of the trial sensitivity analyses using different imputation assumptions informed by data collected on reasons for missing data will be performed as follows:
-
If the reason for missing T48 data is related to the patient's poor condition (e.g. study withdrawal due to poor asthma control), the missing number of exacerbations requiring oral steroids since last visit will be replaced by the worst possible number (i.e. highest number) taken from all follow-up assessments for all patients estimated using interpolation to ensure that the number relates to a common period of time.
-
If the reason for missing T48 data is related to the patient's good condition (e.g. study withdrawal due to patient having good asthma control), the missing number of exacerbations requiring oral steroids since last visit will be replaced by 0.
-
If the reason for missing T48 data is not related to the patient's condition (e.g. patient withdrew from study as moved from the area) or if the reason is not available, two separate analyses will be undertaken replacing the missing number of exacerbations requiring oral steroids by the (i) worst possible number (i.e. highest number) taken from all follow-up assessments for all patients using interpolation, and (ii) value of zero indicating the best possible outcome.
7.2 Analysis of secondary efficacy outcomes
For secondary outcomes statistical tests will be performed at the 5% two-sided significance level (with corresponding 95% confidence interval) unless indicated otherwise. Analyses will not be adjusted by centre as originally planned due to the limited number of patients randomised which make it impracticable to include the centre effects in the statistical model. Secondary analyses adjusting for important prognostic factors (age, sex, duration of asthma, rhinitis, baseline AQLQ, baseline number of courses of oral steroids) will be explored if sufficient data are available.
7.2.1 Quality of life
This outcome is measured by the Paediatric Asthma Quality of Life Questionnaire with Standardised Activities (PAQLQ(S)) and the Paediatric Asthma Caregivers Quality of Life Questionnaire (PACQLQ). PAQLQ(S) was self-administered for older children and interviewer administered for younger children with consistency maintained throughout the trial duration for each child. Interviewer and self-administered questionnaires were combined in analyses.
Baseline characteristics of responders and non-responders will be compared and potential biases assessed.
SCORING THE PAQLQ(S)
The 23 questions (items) in the PAQLQ(S) are divided into three areas, or domains:
Domains Questions
-
Activity limitations: 1, 2, 3, 19, 22
-
Symptoms: 4, 6, 8, 10, 12, 14, 16, 18, 20, 23
-
Emotional function: 5, 7, 9, 11, 13, 15, 17, 21
To score each question a seven point scale is used (1 = extremely bothered, 2 = very bothered, 3 = quite bothered, 4 = somewhat bothered, 5 = bothered a bit, 6 = hardly bothered at all, 7 = not bothered).
Individual questions are equally weighted. The overall PAQLQ(S) score is the mean of the responses to each of the 23 questions (add all 23 responses together and divide the total by 23). The resultant overall score will be between 1 and 7. The domain scores are calculated by adding the responses for each of the items in the domain and dividing by the number of items in the domain. Therefore, the scores from a domain with five items and a domain with ten items will both be between 1 and 7.
SCORING THE PACQLQ
To score each question a seven point scale is used (1 = all of the time, 2 = most of the time, 3 = quite often, 4 = some of the time, 5 = once in a while, 6 = hardly any of the time, 7 = none of the time).
The scoring method for the PACQLQ is exactly the same as for the PAQLQ. All the questions are equally weighted and so the overall score is the mean of the responses to all 13 questions. The domain scores are also the mean values for the items in each domain:
Domains Questions
-
Activity Limitations: 2, 4, 6, 8
-
Emotional Function: 1, 3, 5, 7, 9, 10, 11, 12, 13
MISSING DATA
Whenever children fail to complete a questionnaire correctly, there is potential for bias to creep in and the more gaps there are, the greater the risk of a serious bias.
In children, the individual questions within the PAQLQ(S) are not highly correlated with each other (3, 7, 8). The PAQLQ(S) was developed to capture the breadth of functional diversity (physical, emotional, social and educational) and so the PAQLQ(S) does not have a high Cronbach's alpha. Therefore, one cannot assume that a missing item will be closely correlated with other items within the questionnaire. Neither can one assume that it will behave the same way as the other items in response to an intervention.
How much missing data is acceptable? The PAQLQ(S) scoring manual recommends only allowing up to 10% missing data in a single questionnaire or domain. The PAQLQ(S) has 23 questions and so for the overall score, we would only accept a maximum of two missing responses. For the symptom and emotional function domain scores, only one missing value would be accepted per domain. For the activity limitation domain score, missing responses would not be accepted. For the PACQLQ, which has 13 questions, only 1 missing response would be accepted from the overall score and none missing for each of the domains.
The optimum method, which is associated with the least risk of bias, is to interpolate (pro-rate) missing values using either previous or subsequent completions of the questionnaire. For instance:
| Visit 1 | Visit 2 | |
|---|---|---|
| Item 1 | 4 | 6 |
| Item 2 | 3 | 5 |
| Item 3 | 4 | 4 |
| Item 4 | 5 | 6 |
| Item 5 | 2 | Missing |
Total visit 1 score for items answered on both visits: 4 + 3 + 4 + 5 = 16 (A)
Total visit 2 score for items answered on both visits: 6 + 5 + 4 + 6 = 21 (B)
Item 5 score at visit 1 = 2
Item 5 score at visit 2 = B/A × 2 = 21/16 × 2 = 2.63
This would mean that the mean score for visit 1 was 3.6(4 + 3 + 4 + 5 + 2/5) and the mean score for visit 2 was 4.73(6 + 5 + 4 + 6 + 2.63/5)
When a child returns for follow-up, limitation is assessed in those patient-specific activities that were chosen at the baseline visit. The situation arises where a child has not performed one or more of the chosen activities within the time of interest. The child's response should be recorded by the interviewer as ‘9’ or some other non-valid score to indicate that the answer is ‘missing’. The answer sheet for the Activity domain for this hypothetical patient may look something like this:
| Activities | Baseline | Follow-up |
|---|---|---|
| Bicycling | 4 | 9* |
| Hurrying | 3 | 2 |
| Sleeping | 4 | 2 |
INTERPRETATION OF 7-POINT SCALE
All the questions in the PAQLQ(S) (and its various versions) ask about problems that occur as a result of asthma. Therefore the questionnaire cannot be completed by anyone who does not have asthma and so there can be no ‘normal’ values. The best score is 7.0, which means that the child has no impairments due to their asthma. However, once the score begins to drop below 7.0, this means that the child is experiencing some degree of impairment even if quite mild.
METHOD OF ANALYSIS
The analysis will use the method of analysis of covariance (ANCOVA) and the model will only contain two covariates, these being the baseline score at T0 and the treatment group (in the form of two dummy variables). Those participants that do not have any week 48 data will not be included in the analyses as these data were only collected at T48. The adjusted (from ANCOVA) mean differences will be presented with 95% CI as well as means and standard deviations for T0, T48 and the change (T48 – T0) for each treatment group. Alternative transformations will be explored if the data are found not to be normally distributed.
7.2.2 Time from randomisation to first exacerbation requiring treatment with a short course of oral corticosteroids
This will be calculated for each child and compared across treatment groups using Kaplan–Meier curves and log-rank test with relative effects of treatments summarised using hazard ratios and 95% confidence intervals. The seasonality of having an asthma exacerbation episode will be explored by fitting season as a time-dependent covariate in the model for time to first exacerbation.
7.2.3 School attendance
The number of school days missed from randomisation at T0 to up until T48 will be summarised and presented for each treatment group. Choice of summary measures will depend on the distribution of the data. The number of school days missed will be compared between treatment groups using a Poisson regression model with two dummy variables representing 3 treatment groups. Adjustment of standard errors (multiplying by the square root of the scale parameter estimated as the Pearson's chi-squared statistic divided by its degrees of freedom) to account for overdispersion will be made if the scale parameter is greater than one. Statistical tests will be performed at the 5% two-sided significance level. The relative number of school days missed and confidence interval will be calculated for each pair-wise treatment comparison: Fluticasone compared with Fluticasone plus salmeterol to evaluate the effect of adding in long acting beta-2 agonist (salmeterol 50 micrograms twice daily); Fluticasone compared with Fluticasone plus montelukast to evaluate the effect of adding in oral leukotriene receptor antagonist (montelukast 5 mg once daily); and Fluticasone plus salmeterol compared with Fluticasone plus montelukast to evaluate the effect of adding in long acting beta-2 agonist (salmeterol 50 micrograms twice daily) compared with adding in oral leukotriene receptor antagonist tablet (montelukast 5 mg once daily).
The primary analysis will be based on the 48 week data for patients with data available with a secondary analysis based only on data up to 24 weeks for all patients.
7.2.4 Hospital admissions
The number of hospital admissions from randomisation at T0 to up until T48 will be summarised and presented for each treatment group. Choice of summary measures will depend on the distribution of the data. The number of hospital admissions will be compared between treatment groups using a Poisson regression model with two dummy variables representing 3 treatment groups. Adjustment of standard errors (multiplying by the square root of the scale parameter estimated as the Pearson's chi-squared statistic divided by its degrees of freedom) to account for overdispersion will be made if the scale parameter is greater than one. Statistical tests will be performed at the 5% two-sided significance level. The relative number of hospital admissions and confidence interval will be calculated for each pair-wise treatment comparison: Fluticasone compared with Fluticasone plus salmeterol to evaluate the effect of adding in long acting beta-2 agonist (salmeterol 50 micrograms twice daily); Fluticasone compared with Fluticasone plus montelukast to evaluate the effect of adding in oral leukotriene receptor antagonist (montelukast 5 mg once daily); and Fluticasone plus salmeterol compared with Fluticasone plus montelukast to evaluate the effect of adding in long acting beta-2 agonist (salmeterol 50 micrograms twice daily) compared with adding in oral leukotriene receptor antagonist tablet (montelukast 5 mg once daily).
The primary analysis will be based on the 48 week data for patients with data available with a secondary analysis based only on data up to 24 weeks for all patients.
7.2.5 Amount of rescue beta2 agonist therapy prescribed
The total amount of rescue beta2 agonist therapy prescribed up to T48 (measured in mcg) will be summarised and presented for each treatment group along with the number of patients requiring at least one prescription. The total amount will be calculated as the sum of all the doses of each inhaler prescribed. This will be analysed (transformed if appropriate) and compared between treatment groups using analysis of variance. Blinded distribution data will be reviewed and the statistical analysis plan amended accordingly. The number of missing inhalers recorded will be compared across treatment arms to check they are approximately equal. Missing inhaler types will be assumed to be 100 mcg with 200 doses as this is the most commonly prescribed inhaler type. A secondary analysis using data up to T24 will also be undertaken.
7.2.6 Time from randomisation to treatment withdrawal (due to lack of efficacy or side effects)
This will be calculated for each child and compared across treatment groups using Kaplan–Meier curves and log-rank test with relative effects of treatments summarised using hazard ratios and 95% confidence intervals. To examine different reasons for treatment withdrawal (competing risks of unacceptable adverse effects and poor asthma control) a cumulative incidence analysis will be undertaken. This approach does not assume competing risks are independent and allows the assessment of cause-specific withdrawal in the presence of other competing risks.
All ‘other’ reasons for treatment withdrawal will be sent to the chief investigator to categorise as poor asthma control (lack of efficacy), unacceptable adverse events or some other unrelated reason (which would be censored). This will be carried out before any analyses have been undertaken and before any treatment allocations have been unblinded.
7.2.7 Lung function at 48 weeks (as assessed by spirometry)
The lung function variables of interest here are percent predicted FEV1 and percent predicted FVC.
The analysis will use the method of analysis of covariance (ANCOVA) and will not adjust for any missing data and the model will only contain two covariates, these being the baseline lung function value and the treatment group (in the form of two dummy variables). Those participants that do not have any week 48 data will not be included in the analyses as these data were only collected at T48. The adjusted (from ANCOVA) mean differences will be presented with 95% CI as well as means and standard deviations for T0, T48 and the change (T48 – T0) for each treatment group. Alternative transformations will be explored if the data are found not to be normally distributed.
Reasons for missing data will be documented and the results interpreted as appropriate.
7.2.8 Cost effectiveness
See Health Economic Analysis Plan.
7.3 Non-randomised patients
As a supplementary analysis to explore the value of having a run-in period, follow-up data for a number of outcomes have been obtained for the non-randomised patients from their GPs. The outcomes are as follows:
-
Total number of courses of oral corticosteroids prescribed since registration.
-
Amount of rescue beta2 agonist therapy prescribed since registration.
-
Time from registration to first exacerbation requiring treatment with a short course of oral corticosteroids.
-
Number of times admitted to AE due to respiratory problems since registration.
-
Number of times admitted as an in-patient due to respiratory problems since registration.
-
Whether the patients were prescribed montelukast, long-acting beta2 agonists (LABA) or combination therapy (inhaled corticosteroids & LABA).
Data for the non-randomised patients will be summarised using appropriate summary measures. Most patients would be expected to have approximately 1 year of follow-up. However, if there is too much variability in length of follow-up, summary measures will be presented grouped according to length of follow-up. For example,
-
Up to 1 year.
-
1–1.5 years.
-
1.5–2 years.
-
2–2.5 years, etc.
7.4 Safety analysis
All adverse events (AEs) and serious adverse events (SAEs) reported by the clinical investigator will be categorised and presented, identified by treatment group.
The AEs and SAEs will be grouped together by a member of the trial management team using the MedDRA classification system and entered into an Excel file. This Excel file will be sent to the chief investigators to review and approve.
The number (and percentage) of patients experiencing each AE/SAE will be presented for each treatment group categorised by severity and relationship to drug. For each patient, only the maximum severity experienced of each type of AE will be displayed. The number (and percentage) of occurrences of each AE/SAE will also be presented for each treatment arm. No formal statistical testing will be undertaken.
8 Reporting Protocol Deviations
The table (given in Appendix A) lists potential deviations of important protocol specifications, including eligibility criteria, treatment regimens and study assessments. Protocol deviations are classified according to this system prior to unblinding of treatment. The number (and percentage) of patients with major and minor protocol deviations will be summarised by treatment group with details of type of deviation provided. No formal statistical testing will be undertaken.
9 Setting Results in Context of Previous Research
Once the trial has been completed the results of the trial will be set in context of the existing evidence base.
References
- Moher D, Schulz KF, Altman D. CONSORT group . The CONSORT Statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. Lancet n.d.;357:1191-4.
- Juniper EF, O’Byrne PM, Guyatt GH, . Development and validation of a questionnaire to measure asthma control. Eur Respir J 1999;14:902-7.
Protocol Deviations Table
Note:
-
Impact refers to the impact of the potential protocol deviation on the risk of introducing bias in the defined en-points of the trial. This is generally graded as:
-
Major (in which case patients who experience this protocol deviation would generally be excluded from the ‘per protocol’ analysis set)
-
Minor (in which case patients who experience this protocol deviation would generally be included in the ‘per protocol’ analysis set)
-
-
Justification refers to the protocol-specific justification for the assessment of the impact of each potential protocol deviation.
| Protocol specification | Potential deviation(s) | Impact | Justification |
|---|---|---|---|
| Inclusion criteria | |||
| Children with physician diagnosed asthma | Child without asthma recruited | Major | Violation of this criteria would result in a different prognosis |
| Aged 6 years–14 years, 11 months | Child aged < 6 years, ≥ 15 years | Minor | Any violation of age criteria would be expected to be minimal (a few months rather than years) and there is no evidence to suggest that this would result in a different prognosis |
| Children requiring frequent short-acting beta2 agonist relief therapy ≥ 7 puffs in the past seven days | Child required < 7 puffs in the past seven days | Minor/Major | Violation of this criteria could result in a different prognosis but would depend on number of puffs required in the past seven days (0 puffs would be major) |
| Children with symptoms of asthma (i.e. Wheeze, shortness of breath but not cough alone) resulting in: | All of the specified inclusion criteria violated | Major | Violation of all criteria together would result in a different prognosis |
| i. Nocturnal wakening in the last week because of asthma symptoms and/or | |||
| ii. Asthma has interfered with usual activities in the last week and/or | |||
| iii. Those who have had exacerbations, defined as a short course of oral corticosteroids, an unscheduled GP or A&E department visit or a hospital admission within the previous 6 months (T–4 only) | |||
| Fully informed written (proxy) consent and assent, where appropriate | Fully informed written consent not provided or provided with inaccuracies | Minor | Although major impact for patient, would only be expected to have minor impact on defined end-points and would be very unlikely to happen |
| Exclusion criteria | |||
| Children receiving long acting beta-2 agonists, leukotriene receptor antagonists, regular theophylline therapy or high dose ICS > 1000 micrograms and unlicensed beclometasone dipropionate or equivalent (at the discretion of the investigator) | Child receiving one or more of the prohibited treatments directly prior to entry into the trial | Major | Violation of this criterion could result in a different prognosis |
| Children with other respiratory diseases, cystic fibrosis, cardiac disease or immunological disorders | Child has another respiratory disease | Major | Violation of this criterion could result in a different prognosis |
| Non-English speaking | Both parents/guardians non-English speaking | Minor | Violation of this criterion could result in missing or inaccurate outcome data but graded as minor since unlikely to happen |
| Treatment regime | |||
| Run-in fluticasone propionate inhalers at 100 micrograms twice daily | Incorrect dose (including zero) taken during run-in | Minor/Major | Could lead to randomising children with a different prognosis. Approximately one third of children regularly do not take their medication as prescribed |
| Inhaled fluticasone propionate 100 micrograms twice daily plus placebo tablet once daily | Incorrect treatment pack provided to patient | Major | May influence effectiveness |
| Inhaled fluticasone propionate 100 micrograms and salmeterol 50 micrograms twice daily (combination inhaler) plus placebo tablet once daily | Incorrect dose (including zero) taken or prescribed (e.g. no medication prescribed at a visit) | Minor/Major | High dose may be expected to result in increase in adverse event rate. Low dose (or zero dose) may influence effectiveness of drug. Degree of impact depends on magnitude of discrepancy between correct and actual dose given and length of time on incorrect dose |
| Inhaled fluticasone propionate 100 micrograms twice daily plus montelukast 5-mg tablet once daily | |||
| Administration of an excluded concomitant medication | Child administered one of the excluded concomitant medications listed | Minor/Major | Degree of impact depends on which concomitant medication administered e.g. CYP3A inhibitors may affect the efficacy of montelukast (see Singulair SPC Section 4.5 ‘Interactions with other medicinal products and other forms of interaction’) |
| The following are not permitted for the duration of the trial period: | |||
| Inhaled corticosteroids (other than the trial treatment) | |||
| Long-acting beta2 agonists (other than trial treatment) | |||
| Leukotriene receptor antagonists (other than trial treatment) | |||
| All beta-blockers | |||
| Theophylline | |||
| Cyp3a inhibitors (e.g. Ketaconazole, itraconazole) | |||
| T0 visit timing | |||
| T0 visit should be 4 weeks after T–4 visit to allow 4 weeks run-in for each child | T0 visit occurs outside the visit schedule | Minor/Major | Could lead to randomising children with a different prognosis. Degree of impact depends on number of days outside the visit schedule |
| Assessment of end-points | |||
| Primary: | Child misses 48 week visit leading to missing data for primary end-point | Minor | Data for primary outcome can be collected from GP database at end of trial |
| Number of asthma exacerbations requiring treatment with short courses of oral corticosteroids over 48 weeks from date of randomisation | |||
Secondary:
|
Deviation in the method of assessment e.g. QoL caregiver questionnaire completed by different caregiver, child cannot complete spirometry test | Minor/Major | Introduce bias if reason for missing data is related to asthma and prognosis. Power and integrity of results will diminish as degree of missing data increases |
Appendix 3 MASCOT revised recruitment strategies
The strategies listed below were new methods that would be tested to boost recruitment to MASCOT in addition to existing recruitment approaches.
| Strategy | Cost and funding source |
|---|---|
| Secondary care | |
| Use of CLRN-funded/employed nurses working in A&E to review admissions logs and departmental databases weekly for potentially eligible patients. These patients would then be sent a participant invitation letter by the A&E team and advised to contact the local MASCOT nurse directly if interested | Supported by CLRN – no direct costs, application was for actual nurse time |
| Use of CLRN-funded nurses to work clinical shifts in A&E and identify/approach potentially eligible patients as they present at the hospital | Supported by CLRN – no direct costs, application was for actual nurse time |
| The MASCOT poster to be displayed on the plasma screens in patient waiting areas (mainly A&E and outpatients). This would hopefully capture interest from families and prompt them to ask the clinician they see about the study (or contact the research team themselves directly). Where trusts did not have these facilities, posters would be printed and displayed | No associated costs for the plasma screens |
| Primary care | |
| Recruitment of community pharmacists to help identify potentially eligible patients. In conjunction with a Local Pharmaceutical Committee, pharmacies within an area would be identified who would be paid to conduct mini asthma medication reviews for children who might be eligible. Following the review, if a child met the trial eligibility criteria, their contact details would be passed to the local research nurse (with the family's permission) who would then contact them directly and arrange initial study contact | CLRN – pharmacists were paid £25 per review (we have received funding for 15 pharmacies to conduct up to 10 reviews each – total of £3750). Funding to be secured on a per site basis |
| Pharmacists would be approached to assist in identifying patients. They would be provided with a supply of MASCOT leaflets or information sheets (depending on their preference) and asked to insert one into the medication bag each time a prescription of salbutamol/ICS/oral corticosteroid was dispensed to a patient in the 6–14 years age group with asthma. MASCOT posters to be displayed in these pharmacies. Patients indicating their interest immediately to a pharmacist would have their details passed on to the local research nurse. Alternatively, patients would be encouraged to contact the research team directly once they had considered the information they had received | Leaflets/information sheets printed at the CTU or by MCRN administrative staff |
| To provide reminders to GPs about the trial and the eligibility criteria, for all patients identified during the database searches (existing search strategy), reminders that they could be eligible for MASCOT would be added to the notes of their previous consultation. When the GP reviewed these during the next appointment it should prompt them to remember the study and mention it to the patient. They could make a referral via fax (referral form provided) if the patient was happy for them to do so | CLRN – the usual funding paid to a practice for conducting the database search would be increased when this was also undertaken |
| To attempt to capture the large percentage of patients who are sent participant invitation letters following the database search but who did not respond, practice staff would be funded to make a follow-up telephone call to these families. The family would simply be asked if they received the letter, a brief assessment of eligibility would be made and, if they agree, the family's details would be passed on to the research nurse to contact the family to discuss the trial further | CLRN – time taken for this activity depends on the number of patients identified in the search |
| Practices who cannot spare staff time to make follow-up telephone calls, despite funding being available, could be asked to carry out a second mail-out to the patients who were contacted but who did not respond. The participant follow-up invitation letter would be used, which directs interested patients to contact the local research team. The plan would be for the second mail-out to be 1 calendar month after the first | CLRN – estimated at 1–2 hours' administration time per practice, depending on number of potentially eligible patients found during the initial search |
| MASCOT posters to be placed in walk-in centres, which should generate some interest directly from patients. Walk-in centre staff to be encouraged to refer in to the study team if they saw any patients who may be eligible and they agreed for their contact details to be passed on | No specific cost to the centres, although walk-in centres would be paid the usual £25 per patient registered into the study from the main MASCOT grant |
| Paediatric respiratory clinics held at community hospitals to be approached as they would be more likely than secondary care clinics to treat the type of patients being looked for. The methods used above for walk-in centres would be rolled out to community clinics | No specific costs to the centres, although centres paid the usual £25 per patient registered from the main MASCOT grant |
| A system has been developed for MASCOT in which a study reminder would ‘pop up’ on the GP's computer screen when coding during a consultation. The pop-up is based on asthma and medication codes to trigger eligible patients, prompting the GP (or practice nurse) to mention the study to the family and seek consent for their details to be passed to the team. A download will be undertaken fortnightly and each patient who consented will be sent an invitation letter | Funding to develop the system for MASCOT and install it at practices within the West Midlands was gained from the West Midlands North CLRN |
| School health teams to be utilised to identify potentially eligible patients. Each local authority has different processes and procedures and so various strategies needed depending on how each team operates (and what capacity each team has to support MASCOT). We have worked within the protocol but adapted existing processes for different areas | No associated costs |
Appendix 4 Resource components and unit costs
Resource components sources and their unit costs
| Resource | Resource cost per episode (£) | Source |
|---|---|---|
| GP visit | 28.00 | Department of Health31 |
| GP nurse visit | 10.00 | Department of Health31 |
| Walk-in doctor visit | 40.00 | Curtis32 |
| Walk-in nurse visit | 40.00 | Curtis32 |
| GP other visit | 69.11 | Curtis32 |
| Home doctor visit | 94.00 | Department of Health31 |
| PM doctor visit | 120.00 | Department of Health31 |
| A&E visit | 96.70 | Curtis32 |
| Hospital admission | 1150.00 | Curtis32 |
| Prescribed inhalersa | 15.47 | BNF for Children 20 |
| Prescribed medicinesa | 12.92 | BNF for Children 20 |
| OTC medicinesa | 1.28 | www.boots.com/en/Pharmacy-Health/; www.tesco.com/superstore |
| ‘Rescue’ medicationa (salbutamol) | 2.22 | BNF for Children 20 |
Unit cost for prescribed medicines
| Generic name | Product name | Strength | Unit | Unit cost (£) |
|---|---|---|---|---|
| Adrenaline | Anapen® (Lincoln Medical) | 500 | mg | 30.67 |
| Amoxicillin | Amoxicillin | 125 | mg | 0.06 |
| Amoxicillin | Amoxicillin | 250 | mg | 0.05 |
| Amoxicillin | Amoxicillin | 500 | mg | 0.06 |
| Hydroxyzine hydrochloride | Atarax® (Alliance) | 25 | mg | 0.06 |
| Beclometasone diproprionate | Asmabec Clikhaler® (UCB Pharma) | 50 | μg | 0.13 |
| Beclometasone diproprionate | Beclometasone | 100 | μg | 0.05 |
| Beclometasone diproprionate | Beclometasone | 200 | μg | 0.05 |
| Beclametasone diproprionate | Clenil Modulite® (Chiesi) | 100 | μg | 0.07 |
| Beclometasone diproprionate | Clenil Modulite® (Chiesi) | 200 | μg | 0.08 |
| Cetrizine hydrochloride | Ceterizine hydrochloride | 10 | mg | 0.05 |
| Chlorphenamine maleate | Piriton® (GSK Consumer Healthcare) | 2 | mg | 0.08 |
| Chlorphenamine maleate | Chlorphenamine | 2 | mg | 0.08 |
| Chlorphenamine maleate | Chlorphenamine | 5 | mg | 0.20 |
| Chlorphenamine maleate | Chlorphenamine | 4 | mg | 0.04 |
| Clarithromycin | Clarithromycin | 250 | mg | 0.23 |
| Co-amoxiclav | Co-amoxiclav | 250 | mg | 0.01 |
| Desloratadine | Neoclarityn® (Schering-Plough) | 5 | mg | 1.35 |
| Fexofenadine hydrochloride | Fexofenadine | 30 | mg | 0.18 |
| Fluticasone propionate | Flixotide Accuhaler® (GlaxoSmithKline) | 100 | μg | 0.09 |
| Flucloxacillin | Flucloxacillin | 250 | mg | 0.01 |
| Hydrocortisone ointment | Hydrocortisone | 1 | mg | 1.92 |
| buprofen | Buprofen | 200 | mg | 0.01 |
| Loratadine | Loratadine | 10 | mg | 0.12 |
| Oxymetazoline (drops) | Oxytetracycline | 1 | 0.5% | 1.91 |
| Oxytetracycline | Oxytetracycline | 250 | mg | 0.01 |
| Paracetamol (acetaminophen) paediatric soluble | Calpol® [McNeil Healthcare (UK)] | 1 | mg | 1.78 |
| Paracetamol (acetaminophen) paediatric soluble | Calpol® | 10 | mg | 0.18 |
| Paracetamol (acetaminophen) | Calpol® | 240 | mg | 0.07 |
| Paracetamol (acetaminophen) | Paracetamol | 240 | mg | 0.01 |
| Paracetamol (acetaminophen) | Paracetamol | 300 | mg | 0.09 |
| Paracetamol (acetaminophen) | Paracetamol | 360 | mg | 0.01 |
| Paracetamol (acetaminophen) | Paracetamol | 480 | mg | 0.01 |
| Paracetamol (acetaminophen) (32) | Paracetamol | 500 | mg | 0.03 |
| Paracetamol (acetaminophen) (16) | Paracetamol | 500 | mg | 0.01 |
| Paracetamol (acetominophen) (soluble) | Paracetamol | 250 | mg | 0.01 |
| Paracetamol (acetominophen) (soluble) | Paracetamol | 500 | mg | 0.15 |
| Pholcodine | Pholcodine Linctus | 10 | mg | 0.04 |
| Prednisolone | Prednisolone | 50 | mg | 1.20 |
| Prednisolone | Prednisolone | 40 | mg | 0.21 |
| Prednisolone | Prednisolone | 30 | mg | 0.21 |
| Prednisolone | Prednisolone | 20 | mg | 0.21 |
| Prednisolone | Prednisolone | 10 | mg | 0.21 |
| Salbutamol (albuterol) | Salbutamol | 200 | mg | 0.02 |
| Salbutamol (albuterol) | Ventolin® (GlaxoSmithKline) | 100 | mg | 0.08 |
| Salbutamol (albuterol) | Ventolin® (GlaxoSmithKline) | 200 | μg | 0.08 |
| Salbutamol (albuterol) | Salamol® (IVAX) | 100 | μg | 0.03 |
| Salbutamol (albuterol) | Salamol® (IVAX) | 200 | μg | 0.03 |
| Salmeterol | Serevent® (GlaxoSmithKline) | 50 | μg | 0.59 |
| Terbutaline sulphate | Bricanyl® (AstraZeneca) | 500 | μg | 0.01 |
| Trimethoprim | Trimethoprim | 100 | mg | 0.01 |
| Xylometazoline hydrochloride | Otrivine® | 1 | 0.5% | 1.91 |
Unit cost for over-the-counter medicines
| Product name | Description | Strength | Unit | Unit cost (£) |
|---|---|---|---|---|
| Dextromethorphan hydrobromide | Average price cough medicine | 5 | ml | 3.99 |
| Dextromethorphan hydrobromide | Average price cough medicine | 10 | ml | 3.99 |
| Honey and lemon linctus | Average price honey and lemon linctus | 5 | ml | 0.16 |
| Paracetamol, phenylephrine hydrochloride | Lemsip | 1 | mg | 3.80 |
| Menthol eucalyptus | Average price lozenges | 2 | Pack | 1.35 |
| Cold relief capsulesa | Tesco | 1 | Pack | 0.68 |
| Guaifenesin | Tixylix | 5 | ml | 0.16 |
| Bells Simple Linctus | Average price cough medicine | 5 | ml | 0.16 |
| Buttercup cough syrup | Buttercup | 10 | ml | 0.25 |
Appendix 5 Details of protocol amendments
Version 2.0 (19 March 2008)
Amendments and clarifications prior to full ethical approval (v1.0 22 January 2008 to v2.0 19 March 2008).
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date; correction of typographical errors |
| 11 | The text ‘fully informed written (proxy) consent’ moved from T0 to T–4 visit in trial schematic. ‘Verbal consent/assent’ added to T0 visit activities |
| 20–1 | Exclusion criteria corrected to read ‘> 1000 micrograms and unlicensed beclometasone dipropionate’ |
| 23 | Clarification of procedures for recruiting via primary care to include opportunistic recruitment |
| 25 | Changed procedure for randomisation – patients are allocated to a treatment arm using a randomisation list rather than through allocation of next sequential randomised treatment pack; pharmacy team are unblinded to treatment allocation |
| 29–31 | Description of trial packaging altered and dispensing procedure updated to reflect this |
| 31 | Clarification that inhaler technique checked throughout trial; first drug shipment size changed from one-third to one-quarter of total allocation |
| 32 | Accountability procedures updated to reflect randomisation and dispensing procedures |
| 38 | Randomisation procedure updated |
| 38–9 | Expanded details relating to withdrawal of patients from trial treatment |
| 55 | Fluticasone was added to the list of active treatments |
| 62 | The text ‘CTA reference’ replaced by ‘EudraCT number’ |
Version 3.0 (16 May 2008)
Amendments and clarifications (v2.0 19 March 2008 to v3.0 16 May 2008).
| Page no. | Comment |
|---|---|
| 15 | Clarification of reference from Summary of Product Characteristics |
| 24–5 | Change in pharmacy contact at University Hospital of North Staffordshire (Susan Thomson to Cath Jackson), Royal Aberdeen Children's Hospital (from Valerie Macgregor to Martina Freeman) and the Royal Devon & Exeter NHS Foundation Trust (from Kate O'Connor to Fiona Hall). Contact details amended appropriately |
| 31 | Clarification on documentation of patient randomisation number |
| 32 | Long-acting beta-2 agonists and leukotriene receptor antagonists (other than trial medication) added to list of concomitant medications |
| 35 | Guidance added on the window for timing of patient study visits |
| 68 | The amount available for reimbursement of patient travel expenses was changed from £10 to £8 |
| 75 | Principal investigator at Norfolk and Norwich University Hospitals changed from Professor David Price to Dr Chris Upton |
| 77–8 | South Manchester University Hospital and Wirral Universities Teaching Hospital added as lead participating sites; six GP practices in the Norfolk area added as participating sites (Appendix A) |
| 79–80 | Five participating primary care sites added to Appendix B |
| 81 | T0 pharmacy dispensing procedure for Norfolk patients changed to allow patients to be randomised away from the Norfolk and Norwich University Hospitals |
| 112 | Search codes for GP databases added as an appendix (Appendix E) |
Version 4.0 (24 July 2008)
Amendments and clarifications (v3.0 16 May 2008 to v4.0 24 July 2008)
| Page no. | Comment |
|---|---|
| 21 | Explanation that patients who withdraw early from trial treatment but continue to allow data collection will discuss with the principal investigator whether and when to become unblended |
| 22 | MCRN CTU to unblind individual patients leaving the trial completely within 7 days of completion/withdrawal whenever possible |
| 26 | Addition of pharmacy contact (Neil Caldwell) at Arrowe Park Hospital, Wirral |
| 29–30 | Change in process of supplying the investigational medicinal product (IMP) to patients at T–4 and T0 (from IMP being dispensed prior to visit and supplied then if eligible to being dispensed after the visit and then transported to the patient) |
| 34 | CYP3A inhibitors changed from a prohibited concomitant medication group to limited use only whilst on trial treatment |
| 34–6 | Routine unblinding procedure changed from all patients being unblinded only at the end of the trial as a whole to each participant being unblinded when they end their individual participation in the trial. Process for this described (MCRN CTU to inform GP with 7 days) |
| 40 | Patients must now be asked for current GP details at the final visit |
| 41 | Clarification that European Respiratory Society standards for spirometry will be accepted as well as American Thoracic Society standards |
| 45 | Further guidance on completion of the PACQLQ added |
| 62 | Description of new unblinding procedure with regards to study discontinuation |
| 63 | Confirmation that MHRA authorisation has now been granted for MASCOT |
| 65 | Screening logs to be submitted monthly instead of weekly |
| 80–1 | Four GP practices in the Norfolk area added as participating sites (Appendix A) |
| 84–5 | Clarification on process of supplying IMP to Norfolk patients seen at outreach centres – to include change from IMP dispensed prior to the visit to dispensed after the visit and transported to the patient (Appendix C) |
Version 5.0 (30 January 2009)
Amendments and clarifications (v4.0 24 July 2008 to v5.0 30 January 2009)
| Page no. | Comment |
|---|---|
| Throughout | Removal of the word ‘confidential’ from the page headers |
| 3 | Change in contact details for Aptuit Ltd (from Ciaran Flanagan to Dr Ivan Langan) |
| 5 | Contact details for the MASCOT Data Manager added following the appointment of Emma Dyson |
| 29 | Correction of montelukast matching placebo tablet shelf life from 24 months to 5 years |
| 71 | Nemonie Marriott to replace Zahira Maqsood as the primary care representative on the TMG |
Version 6.0 (20 May 2009)
Amendments and clarifications (v5.0 30 January 2009 to v6.0 20 May 2009)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date; correction of typographical errors |
| 3–6 | Correction and updates to contact details of various individuals |
| 10 | Update in number of secondary care centres from 12 to 13 |
| 14–16 | Updates to ‘Potential risks’ section for all three study medications (information derived from Summary of Product Characteristics) |
| 18 | Addition of adverse events as a secondary end point |
| 19 | Clarification of inclusion criteria 1 at both T–4 and T0 (from ‘Those requiring frequent short-acting beta-2 agonist relief therapy ≥ 7 puffs per week’ to ‘Those requiring frequent short-acting beta-2 agonist relief therapy ≥ 7 puffs in the past seven days’) |
| 21–2 | Additions to recruitment section including recruitment through community pharmacists and school health professionals and promotion through the media. Expansion of existing strategies to include use of a follow-up letter or telephone call and recruitment through any appropriate primary care centre |
| 23 | Update to site name (from Royal Liverpool Children's Hospital to Alder Hey Children's NHS Foundation Trust) |
| 23 | Change in pharmacist at Royal Manchester Children's Hospital (from Judith Thornton to Carolyn Davies) and Derbyshire Children's Hospital (from Julie Vanes and Liz Bedford to Peter Fox) and updates to associated contact details |
| 27 | Clarification added that pharmacists must complete all sections on the trial medication labels prior to dispensing |
| 32 | Revision of text to show that unblinding information at completion of/withdrawal from trial treatment will be provided to the patient's GP only. Secondary care clinicians will now be instructed to contact the GP if they require this information |
| 34–5 | Addition of caveat that the research nurse/principal investigator can conduct the T8 and T24 study visits at the patient's home, instead of in clinic, in exceptional circumstances |
| 41–3 | Updates to sample size calculation formula and justifications (sample size remains unchanged) |
| 43 | Change in planned interim sample size review – now to be conducted after the first 75 children have completed 24 weeks' follow-up |
| 44–5 | Various updates and clarifications to the SAP. Discussion of how secondary outcomes will be analysed |
| 58–9 | Revision of the text detailing how data queries will be documented and resolved. All data queries will be actioned using specific data query forms; no changes will be made to the original CRF page/s |
| 62 | Change as to who the £25.00 participant identification payment can be made to – this now includes any appropriate health-care provider, not just GP practices |
| 67 | Removal of the Machin et al. (1997) reference. Addition of Metcalfe et al. (2003) reference |
| 74 | Removal of Appendix B, which listed ‘satellite’ GP centres in the Greater London area affiliated to the Royal London Hospital. These are now referenced in section 8.1 |
| 77 | Removal of Appendix D, Summary of Product Characteristics – now to be provided to sites separate from the protocol |
| 105 | Appendix E (now Appendix C) – updates to the GP Database Search Guidance |
Version 6.1 (18 January 2010)
Minor amendments and clarifications (v6.0 20 May 2009 to v6.1 18 January 2010)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date |
| 4 | Update to contact details for Professor Marilyn James, Head of Health Economics |
| 5 | Removal of Ms Elizabeth Stokes from the MASCOT Health Economist role, addition of Mr Andrew McKay as MASCOT Trial Statistician, update to email contact information for MASCOT Trial Coordinator and Data Manager |
| 6 | Update to email contact information for Dr Iolo Doull and Dr David Spencer |
| 23–4 | Change in pharmacist at University Hospital of North Staffordshire (from Catherine Jackson to Susan Brammer), Leicester Royal Infirmary (from David Harris to Judith Bwire), Norfolk and Norwich University Hospitals (from Nicola Rudge to Susan George) and all associated contact information |
| 70 | Removal of Appendix A (‘List of participating sites’) – now to be provided to sites separate to the protocol |
| Throughout | Removal of all associated references to ‘Appendix A: List of Participating Sites’ throughout protocol |
Version 7.0 (26 March 2010)
Amendments and clarifications (v6.1 18 January 2010 to v7.0 26 March 2010)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date |
| 19 | Removal of the exclusion criterion ‘non-English speaking’ from both the T–4 and the T0 visits |
| 21–2 | Slight amendments to the current recruitment strategies, including the addition of nurses visiting schools to discuss asthma and the study |
| 25–6 | Change in maximum storage temperature of Flixotide and Seretide from 30°C to 25°C |
| 28 | Removal of the requirement for a standard operating procedure to be in place prior to a nurse delivering the IMP to a patient's home |
| 29 | Removal of statement outlining size of the IMP shipments |
| 33 | Change in description of inclusion criterion from seven or more puffs (of SABA) ‘per week’ to seven or more puffs ‘in the past week’ |
| 33 | Clarification that ideal visit window between T–4 and T0 is ≥ 24 days and ≤ 30 days |
| 51 | Removal of the ethical consideration ‘ineligibility of non-English speakers’ |
| 53 | Statement added that translators (when used) will need to countersign the consent form used in the informed consent discussion |
Version 8.0
Amendments and clarifications (v7.0 26 March 2010 to v8.0 21 March 2011)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date |
| 2 | Detailed notice of substantial amendment to the MASCOT protocol as a result of funding being withdrawn from the study |
Version 8.1
Amendments and clarifications (v8.0 21 Mach 2011 to v8.1 13 June 2011)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date |
| 43–4 | Details of minor changes to the SAP and subsequent minor changes to some analysis methods for secondary outcomes |
Appendix 6 Reasons for exclusion of participants from outcome analyses
Primary outcome intention-to-treat analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 11 | 15 | 12 | 38 |
| Excluded – patient has primary outcome data past T24 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 4 | 2 | 4 | 10 |
| Excluded – patient has primary outcome data past T36 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 1 | 0 | 1 | 2 |
| Excluded – patient has primary outcome data only up to T24 as withdrew before T36 | 2 | 0 | 1 | 3 |
| Excluded – patient has primary outcome data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 so has no primary outcome data | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8; had no T8 visit but agreed to have data collected at T24 | 0 | 0 | 1 | 1 |
Primary outcome intention-to-treat analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 18 | 17 | 19 | 54 |
| Excluded – patient has primary outcome data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 so has no primary outcome data | 0 | 1 | 1 | 2 |
Primary outcome per-protocol analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 9 | 11 | 8 | 28 |
| Excluded – patient has primary outcome data past T24 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 4 | 2 | 4 | 10 |
| Excluded – patient has primary outcome data past T36 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 1 | 0 | 1 | 2 |
| Excluded – patient has primary outcome data up to T48 but did not take > 70% of tablets | 0 | 0 | 1 | 1 |
| Excluded – patient has primary outcome data up to T48 but has missing number of missed doses data for at least one visit | 2 | 4 | 3 | 9 |
| Excluded – patient has primary outcome data only up to T24 as withdrew before T36 | 2 | 0 | 1 | 3 |
| Excluded – patient has primary outcome data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 and has missing number of missed doses data for the T24 visit | 0 | 0 | 1 | 1 |
| Excluded – patient withdrew before T8 so has no primary outcome data | 0 | 1 | 1 | 2 |
Primary outcome per-protocol analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 15 | 13 | 16 | 44 |
| Excluded – patient has primary outcome data up to T24 but did not take > 70% of inhalers | 0 | 1 | 0 | 1 |
| Excluded – patient has primary outcome data up to T24 but did not take > 70% of tablets | 0 | 0 | 1 | 1 |
| Excluded – patient has primary outcome data up to T24 but has missing number of missed doses data for at least one visit | 3 | 3 | 1 | 7 |
| Excluded – patient has primary outcome data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 and has missing number of missed doses data for the T24 visit | 0 | 0 | 1 | 1 |
| Excluded – patient withdrew before T8 so has no primary outcome data | 0 | 1 | 1 | 2 |
PAQLQ(S) activity limitations domain score analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 10 | 14 | 12 | 36 |
| Excluded – patient completed up to T48 but has no PAQLQ(S) for T48 | 1 | 0 | 0 | 1 |
| Excluded – patient has a final PAQLQ(S) but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 5 | 2 | 5 | 12 |
| Excluded – patient has a T0 and T48 PAQLQ(S) but has a missing answer in the activity limitations domain at T0 | 0 | 1 | 0 | 1 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T36 and last PAQLQ(S) questionnaire does not reach the minimum of 43 weeks to be included in the T48 analysis | 2 | 0 | 1 | 3 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. Only has a T0 PAQLQ(S) | 0 | 0 | 1 | 1 |
PAQLQ(S) activity limitations domain score analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 17 | 15 | 17 | 49 |
| Excluded – patient completed up to T24 but has no PAQLQ(S) for T24 | 1 | 1 | 1 | 3 |
| Excluded – patient has a T0 and T24 PAQLQ(S) but has a missing answer in the activity limitations domain at T0 | 0 | 1 | 0 | 1 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. Only has a T0 PAQLQ(S) | 0 | 0 | 1 | 1 |
PAQLQ(S) activity limitations domain score analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 17 | 15 | 17 | 49 |
| Excluded – patient completed up to T24 but has no PAQLQ(S) for T24 | 1 | 1 | 1 | 3 |
| Excluded – patient has a T0 and T24 PAQLQ(S) but has a missing answer in the activity limitations domain at T0 | 0 | 1 | 0 | 1 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. No PAQLQ(S) was filled in at T24 | 0 | 0 | 1 | 1 |
PAQLQ(S) symptoms domain score analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 10 | 15 | 12 | 37 |
| Excluded – patient completed up to T48 but has no PAQLQ(S) for T48 | 1 | 0 | 0 | 1 |
| Excluded – patient has a final PAQLQ(S) but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 5 | 2 | 5 | 12 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T36 and last PAQLQ(S) does not reach the minimum of 43 weeks to be included in the T48 analysis | 2 | 0 | 1 | 3 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. Only has a T0 PAQLQ(S) | 0 | 0 | 1 | 1 |
PAQLQ(S) symptoms domain score analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 17 | 16 | 17 | 50 |
| Excluded – patient completed up to T24 but has no PAQLQ(S) for T24 | 1 | 1 | 1 | 3 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. No PAQLQ(S) was filled in at T24 | 0 | 0 | 1 | 1 |
PAQLQ(S) total score analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 10 | 15 | 12 | 37 |
| Excluded – patient completed up to T48 but has no PAQLQ(S) for T48 | 1 | 0 | 0 | 1 |
| Excluded – patient has a final PAQLQ(S) but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 5 | 2 | 5 | 12 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T36 and last PAQLQ(S) does not reach the minimum of 43 weeks to be included in the T48 analysis | 2 | 0 | 1 | 3 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. Only has a T0 PAQLQ(S) | 0 | 0 | 1 | 1 |
PAQLQ(S) total score analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 17 | 16 | 17 | 50 |
| Excluded – patient completed up to T24 but has no PAQLQ(S) for T24 | 1 | 1 | 1 | 3 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. No PAQLQ(S) was filled in at T24 | 0 | 0 | 1 | 1 |
PACQLQ activity limitations domain score analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 11 | 15 | 12 | 38 |
| Excluded – patient does not have a final PACQLQ and did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 0 | 0 | 1 | 1 |
| Excluded – patient has a final PACQLQ but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 5 | 2 | 4 | 11 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T36 and last PACQLQ does not reach the minimum of 43 weeks to be included in the T48 analysis | 2 | 0 | 1 | 3 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. Only has a T0 PACQLQ | 0 | 0 | 1 | 1 |
PACQLQ activity limitations domain score analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 17 | 14 | 16 | 47 |
| Excluded – patient completed up to T24 but has no PACQLQ for T24 | 1 | 1 | 2 | 4 |
| Excluded – patient has a T0 and T24 PACQLQ but has a missing answer in the activity limitations domain at the T0 visit | 0 | 2 | 0 | 2 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. No PACQLQ was filled in at T24 | 0 | 0 | 1 | 1 |
PACQLQ emotional functioning domain score analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 11 | 15 | 12 | 38 |
| Excluded – patient does not have a final PACQLQ and did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 0 | 0 | 1 | 1 |
| Excluded – patient has a final PACQLQ but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 5 | 2 | 4 | 11 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T36 and last PACQLQ does not reach the minimum of 43 weeks to be included in the T48 analysis | 2 | 0 | 1 | 3 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. Only has a T0 PACQLQ | 0 | 0 | 1 | 1 |
PACQLQ emotional functioning domain score analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 17 | 16 | 16 | 49 |
| Excluded – patient completed up to T24 but has no PACQLQ for T24 | 1 | 1 | 2 | 4 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24. No PACQLQ was filled in at T24 | 0 | 0 | 1 | 1 |
Time from randomisation to first exacerbation requiring treatment with a short course of oral corticosteroids analysis set inclusion/exclusions with reasons
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 19 | 23 | 21 | 63 |
Number of school days missed intention-to-treat analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 11 | 15 | 11 | 37 |
| Excluded – patient completed the trial up to T48 but has missing number of school days missed data at T48 visit | 0 | 0 | 1 | 1 |
| Excluded – patient has number of school days missed data past T24 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 4 | 2 | 4 | 10 |
| Excluded – patient has number of school days missed data past T36 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 1 | 0 | 1 | 2 |
| Excluded – patient has number of school days missed data only up to T24 as withdrew before T36 | 2 | 0 | 1 | 3 |
| Excluded – patient has number of school days missed data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 so has no number of school days missed data | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 | 0 | 0 | 1 | 1 |
Number of school days missed intention-to-treat analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 18 | 17 | 19 | 54 |
| Excluded – patient has number of school days missed data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 so has no number of school days missed data | 0 | 1 | 1 | 2 |
Number of school days missed per-protocol analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 9 | 11 | 8 | 28 |
| Excluded – patient has missing number of school days missed data at T48 visit and has missing number of missed doses data for at least one visit | 0 | 0 | 1 | 1 |
| Excluded – patient has number of school days missed data past T24 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 4 | 2 | 4 | 10 |
| Excluded – patient has number of school days missed data past T36 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 1 | 0 | 1 | 2 |
| Excluded – patient has number of school days missed data up to T48 but did not take > 70% of tablets | 0 | 0 | 1 | 1 |
| Excluded – patient has number of school days missed data up to T48 but has missing number of missed doses data for at least one visit | 2 | 4 | 2 | 8 |
| Excluded – patient has number of school days missed data only up to T24 as withdrew before T36 | 2 | 0 | 1 | 3 |
| Excluded – patient has number of school days missed data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 and has missing number of missed doses data for the T24 visit | 0 | 0 | 1 | 1 |
| Excluded – patient withdrew before T8 so has no number of school days missed data | 0 | 1 | 1 | 2 |
Number of school days missed per-protocol analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 15 | 13 | 16 | 44 |
| Excluded – patient has number of school days missed data up to T24 but did not take > 70% of inhalers | 0 | 1 | 0 | 1 |
| Excluded – patient has number of school days missed data up to T24 but did not take > 70% of tablets | 0 | 0 | 1 | 1 |
| Excluded – patient has number of school days missed data up to T24 but has missing number of missed doses data for at least one visit | 3 | 3 | 1 | 7 |
| Excluded – patient has number of school days missed data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 and has missing number of missed doses data for the T24 visit | 0 | 0 | 1 | 1 |
| Excluded – patient withdrew before T8 so has no number of school days missed data | 0 | 1 | 1 | 2 |
Number of hospital admissions missed intention-to-treat analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 11 | 15 | 11 | 37 |
| Excluded – patient completed the trial up to T48 but has missing number of hospital admissions data at T48 visit | 0 | 0 | 1 | 1 |
| Excluded – patient has number of hospital admissions data past T24 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 4 | 2 | 4 | 10 |
| Excluded – patient has number of hospital admissions data past T36 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 1 | 0 | 1 | 2 |
| Excluded – patient has number of hospital admissions data only up to T24 as withdrew before T36 | 2 | 0 | 1 | 3 |
| Excluded – patient has number of hospital admissions data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 so has no number of hospital admissions data | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 | 0 | 0 | 1 | 1 |
Number of hospital admissions missed intention-to-treat analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 18 | 17 | 19 | 54 |
| Excluded – patient has number of hospital admissions data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 so has no number of hospital admissions data | 0 | 1 | 1 | 2 |
Number of hospital admissions missed per-protocol analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 9 | 11 | 8 | 28 |
| Excluded – patient has number of hospital admissions data past T24 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 4 | 2 | 4 | 10 |
| Excluded – patient has number of hospital admissions data past T36 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 1 | 0 | 1 | 2 |
| Excluded – patient has number of hospital admissions data up to T48 but did not take > 70% of tablets | 0 | 0 | 1 | 1 |
| Excluded – patient has number of hospital admissions data up to T48 but has missing number of missed doses data for at least one visit | 2 | 4 | 3 | 9 |
| Excluded – patient has number of hospital admissions data only up to T24 as withdrew before T36 | 2 | 0 | 1 | 3 |
| Excluded – patient has number of hospital admissions data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 and has missing number of missed doses data for the T24 visit | 0 | 0 | 1 | 1 |
| Excluded – patient withdrew before T8 so has no number of hospital admissions data | 0 | 1 | 1 | 2 |
Number of hospital admissions missed per-protocol analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 15 | 13 | 16 | 44 |
| Excluded – patient has number of hospital admissions data up to T24 but did not take > 70% of inhalers | 0 | 1 | 0 | 1 |
| Excluded – patient has number of hospital admissions data up to T24 but did not take > 70% of tablets | 0 | 0 | 1 | 1 |
| Excluded – patient has number of hospital admissions data up to T24 but has missing number of missed doses data for at least one visit | 3 | 3 | 1 | 7 |
| Excluded – patient has number of hospital admissions data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 and has missing number of missed doses data for the T24 visit | 0 | 0 | 1 | 1 |
| Excluded – patient withdrew before T8 so has no number of hospital admissions data | 0 | 1 | 1 | 2 |
Amount of rescue beta-2 agonist therapy prescribed analysis set inclusion/exclusions with reasons: T48
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 11 | 14 | 12 | 37 |
| Excluded – patient completed trial up to T48 but has missing number of beta-2 agonists data for T24 visit | 0 | 1 | 0 | 1 |
| Excluded – patient has number of beta-2 agonists data past T24 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 4 | 2 | 4 | 10 |
| Excluded – patient has number of beta-2 agonists data past T36 but did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 1 | 0 | 1 | 2 |
| Excluded – patient has number of beta-2 agonists data only up to T24 as withdrew before T36 | 2 | 0 | 1 | 3 |
| Excluded – patient has number of beta-2 agonists data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 so has no number of beta-2 agonists data | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 | 0 | 0 | 1 | 1 |
Amount of rescue beta-2 agonist therapy prescribed analysis set inclusion/exclusions with reasons: T24
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 18 | 16 | 19 | 53 |
| Excluded – patient completed trial up to T48 but has missing number of beta-2 agonists data for T24 visit | 0 | 1 | 0 | 1 |
| Excluded – patient has number of beta-2 agonists data only up to T8 as withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T8 so has no number of beta-2 agonists data | 0 | 1 | 1 | 2 |
Forced expiratory volume in 1 second per cent predicted analysis set inclusion/exclusions with reasons
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 8 | 13 | 9 | 30 |
| Excluded – patient did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 5 | 2 | 5 | 12 |
| Excluded – patient had no spirometry at both the T0 and the T48 visits | 0 | 1 | 0 | 1 |
| Excluded – patient has FEV1(%) data at T0 but missing FEV1(%) data at T48 | 1 | 0 | 0 | 1 |
| Excluded – patient has FEV1(%) data at T0 but spirometry not carried out at T48 | 1 | 0 | 1 | 2 |
| Excluded – patient has FEV1(%) data at T48 but missing FEV1(%) data at T0 | 0 | 1 | 0 | 1 |
| Excluded – patient has FEV1(%) data at T48 but spirometry not carried out at T0 | 1 | 0 | 2 | 3 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T36 | 2 | 0 | 1 | 3 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 | 0 | 0 | 1 | 1 |
Forced vital capacity per cent predicted analysis set inclusion/exclusions with reasons
| Analysis status | Treatment | |||
|---|---|---|---|---|
| Fluticasone (n = 19) | Fluticasone plus salmeterol (n = 23) | Fluticasone plus montelukast (n = 21) | Total (n = 63) | |
| Included | 8 | 13 | 9 | 30 |
| Excluded – patient did not reach the minimum of 43 weeks for a T48 visit because of early closure of the trial | 5 | 2 | 5 | 12 |
| Excluded – patient had no spirometry at both the T0 and the T48 visits | 0 | 1 | 0 | 1 |
| Excluded – patient has FVC(%) data at T0 but missing FVC(%) data at T48 | 1 | 0 | 0 | 1 |
| Excluded – patient has FVC(%) data at T0 but spirometry not carried out at T48 | 1 | 0 | 1 | 2 |
| Excluded – patient has FVC(%) data at T48 but missing FVC(%) data at T0 | 0 | 1 | 0 | 1 |
| Excluded – patient has FVC(%) data at T48 but spirometry not carried out at T0 | 1 | 0 | 2 | 3 |
| Excluded – patient withdrew before T24 | 1 | 5 | 1 | 7 |
| Excluded – patient withdrew before T36 | 2 | 0 | 1 | 3 |
| Excluded – patient withdrew before T8 | 0 | 1 | 1 | 2 |
| Excluded – patient withdrew from trial treatment before T8, had no T8 visit but agreed to have data collected at T24 | 0 | 0 | 1 | 1 |
Appendix 7 Protocol
General Information
This document describes the MASCOT trial and provides information about procedures for entering patients into it. The protocol should not be used as an aide-memoir or guide for the treatment of other patients; every care was taken in its drafting, but corrections or amendments may be necessary. These will be circulated to the registered investigators in the trial, but centres entering patients for the first time are advised to contact the coordinating centre (Medicines for Children Research Network Clinical Trials Unit [MCRN CTU], Liverpool [mascot@mcrnctu.org.uk]) to confirm they have the most up to date version. Clinical problems relating to this trial should be referred to the relevant chief investigator via the MCRN CTU.
Statement of Compliance
This study will be carried out in accordance with the World Medical Association Declaration of Helsinki (1964) and the Tokyo (1975), Venice (1983), Hong Kong (1989) and South Africa (1996) amendments and will be conducted in compliance with the protocol, MCRN CTU Standard Operating Procedures and EU Directive 2001/20/EC, transposed into UK law as the UK Statutory Instrument 2004 No 1031: Medicines for Human Use (Clinical Trials) Regulations 2004 and amendments.
As per the MCRN CTU Standard Operating Procedures no waivers from the MASCOT protocol will be granted.
Contact Details: Institutions
| Co-Sponsors: | Clinical Trials Unit: | DNA Storage Facility: |
|---|---|---|
| Keele University Research Services | Medicines for Children Clinical Trials Unit | Prof Munir Pirmohamed |
| Room DH 1.13 | (Director: Professor Paula Williamson) | Dept. Of Pharmacology & Therapeutics |
| Dorothy Hodgkin Building | Institute of Child Health | The University of Liverpool |
| Keele University | Alder Hey Children's NHS Foundation Trust | Ashton Street |
| Keele | Eaton Road | Liverpool |
| Staffordshire | Liverpool | L69 3GE |
| Tel: 01782 733 374 | L1 2 2AP | Tel: 0151 794 5549 |
| Fax: 01782 733 740 | Tel: 0151 282 4729 | Fax: 0151 794 5540 |
| University Hospital of North Staffordshire NHS Trust Research & Development Department | Fax: 0151 282 4721 | Email: munirp@liverpool.ac.uk |
| North Staffordshire Medical Institute | Email: mascot@mcrnctu.org.uk | |
| Hartshill Road | ||
| Hartshil | ||
| Stoke-on-Trent | ||
| ST4 7QB | ||
| Tel: 01782 554 839 | ||
| Fax: 01782 554 610 | ||
| Clinical Trial Supplies Company | ||
| Aptuit Ltd | ||
| Unit 107 | ||
| Tenth Avenue | ||
| Deeside Industrial Park | ||
| Deeside | ||
| Flintshire | ||
| CH5 2UA | ||
| Tel: 01244 84 5723 | ||
| Fax: 01244 84 5701 | ||
| Email: ivan.langan@aptuit.com | ||
Details of participating sites can be found in Appendix A
Contact Details: Individuals
| Individual/s Authorised to Sign the Protocol and Protocol Amendments on behalf of the Co-Sponsors: | Chief investigator (CI): | Medical Expert who will Advise on Safety Reports in the Absence of the CI: |
|---|---|---|
| Dr Darren Clement | Professor Warren Lenney | Professor David Price |
| Research & Development Manager Research & Development Department | Consultant Paediatrician/Professor of Paediatrics | GPIAG Professor of Primary Care Respiratory Medicine |
| University Hospital of North Staffordshire NHS Trust | Research & Development Department | Dept of General Practice and Primary Care |
| Guy Hilton Research Centre | University Hospital of North Staffordshire | University of Aberdeen Foresterhill Health Centre |
| Thornburrow Drive | North Staffordshire Medical Institute | Westburn Road |
| Stoke-on-Trent | Hartshill Road | Aberdeen |
| ST4 7QB | Stoke-on-Trent | AB25 2AY |
| Tel: 01782 554 839 | ST4 7QB | Tel: 020 71931876 |
| Fax: 01782 747 319 | Tel: 01782 554839 | Aberdeen office fax: 01224 550683 |
| E-mail: Darren.clement@uhns.nhs.uk | Fax: 01782 412236 | Email: david@respiratoryresearch.org |
| Ms Judith Garside | Email: warren.lenney@uhns.nhs.uk | |
| Research Grants and Contracts Manager | ||
| Research Services | ||
| Room DH 1.13 | ||
| Dorothy Hodgkin Building | ||
| Keele University | ||
| Keele | ||
| Staffordshire | ||
| ST5 5BG | ||
| Tel: 01782 733 374 | ||
| Fax: 01782 733 740 | ||
| E-mail: j.m.garside@uso.keele.ac.uk | ||
| Head of Health Economics: | Head of Trial Management: | Head of Statistics: |
| Professor Marilyn James | Ms Helen Hickey | Dr Catrin Tudur-Smith |
| Professor of Applied Health Economics | Medicines for Children Clinical Trials Unit | Medicines for Children Clinical Trials Unit |
| Liverpool John Moores University and Liverpool Primary Care Trust Centre for Public Health | Institute of Child Health | Institute of Child Health |
| 5th Floor Kingsway House | Alder Hey Children's NHS Foundation Trust | Alder Hey Children's NHS Foundation Trust |
| Hatton Garden | Eaton Road | Eaton Road |
| Liverpool | Liverpool | Liverpool |
| L3 2AJ | L12 2AP | L12 2AP |
| Tel: 0151 231 8783 | Tel: 0151 252 5240 | Tel: 0151 794 4059 |
| Fax: 0151 231 8020 | Fax: 0151 282 4721 | Fax: 0151 282 4721 |
| Email: m.james@ljmu.ac.uk | Email: mascot@mcrnctu.org.uk | Email: mascot@mcrnctu.org.uk |
| Health Economist: | Trial Manager: | Trial Statistician: |
| Ms Elizabeth Stokes | Miss Sophie Perry | To be appointed |
| Research Assistant in Applied Health Economics | Medicines for Children Clinical Trials Unit | Medicines for Children Clinical Trials Unit |
| Liverpool John Moores University | Institute of Child Health | Institute of Child Health |
| Centre for Public Health | Alder Hey Children's NHS Foundation Trust | Alder Hey Children's NHS Foundation Trust |
| 5th Floor Kingsway House | Eaton Road | Eaton Road |
| Hatton Garden | Liverpool | Liverpool |
| Liverpool | L12 2AP | L12 2AP |
| L3 2AJ | Tel: 0151 282 4706 | Tel: 0151 282 4706 |
| Tel: 0151 231 8783 | Fax: 0151 282 4721 | Fax: 0151 282 4721 |
| Fax: 0151 231 8020 | Email: mascot@mcrnctu.org.uk | Email: mascot@mcrnctu.org.uk |
| Email: E.A.Stokes@ljmu.ac.uk | ||
| Data Manager: | ||
| Miss Emma Dyson | ||
| Medicines for Children Clinical Trials Unit | ||
| Institute of Child Health | ||
| Alder Hey Children's NHS Foundation Trust | ||
| Eaton Road | ||
| Liverpool | ||
| L12 2AP | ||
| Tel: 0151 282 4705 | ||
| Fax: 0151 282 4721 | ||
| Email: mascot@mcrnctu.org.uk |
Contact Details: Independent Oversight Committees
| Trial Steering Committee (TSC) | ||
|---|---|---|
| Chair | Medical Experts | Statistician |
| Dr Anne Thomson | Dr Gary Connett | Prof Chris Frost |
| Consultant in Paediatric Respiratory Medicine & General Paediatrics | Consultant in Paediatric Respiratory Medicine | Professor of Medical Statistics |
| Oxford Children's Hospital | Department of Paediatrics | Room 142b Keppel Street |
| The John Radcliffe | Southampton General Hospital | London |
| Headington | Tremona Road | WC1E 7HT |
| Oxford | Southampton | Tel: 020 7 927 2242 |
| OX3 9DU | SO16 6YD | E-mail: chris.frost@lshtm.ac.uk |
| Tel: 01865 234199 | Tel: 02380 798973 | |
| Email: anne.thomson@paediatrics.ox.ac.uk | Email: Gary.Connett@suht.swest.nhs.uk | |
| Dr Iolo Doull | ||
| Consultant Respiratory Paediatrician | ||
| Childrens Hospital for Wales | ||
| Heath Park | ||
| Cardiff | ||
| CF14 4XW | ||
| Tel: 029 2074 3530 | ||
| Email: doullij@cf.ac.uk | ||
| Lay Representatives | ||
| Mr & Mrs John Hilton | ||
| The Guy Hilton Asthma Trust | ||
| Lyme House | ||
| Devils Lane | ||
| Longsdon | ||
| Stoke-on-Trent | ||
| ST9 9QP | ||
| Tel: 01538 385479 | ||
| Email: guystrust@btopenworld.com | ||
| Independent Data & Safety Monitoring Committee (IDSMC) | ||
|---|---|---|
| Chair | Medical Expert | Statistician |
| Dr David Spencer | Dr John Alexander | Mr Andy Vail |
| Consultant in Paediatric Respiratory Medicine | Consultant Paediatrician | Senior Lecturer and Statistician |
| The Regional Cardiothoracic Centre | Paediatric Intensive Care Unit | Research and Development Department |
| The Freeman Hospital | City General Hospital | Clinical Sciences Building |
| High Heaton | Stoke-on-Trent | Salford Royal Hospitals NHS Trust |
| Newcastle upon Tyne Tyne and Wear | ST4 6QG | Stott Lane |
| NE7 7DN | Tel: 01782 552 751 | Salford |
| Email: david.spencer2@nuth.northy.nhs.uk | Email: John.Alexander@uhns.nhs.uk | M6 8HD |
| Tel: 0161 206 4262 | ||
| Email: Andy.Vail@manchester.ac.uk | ||
List of abbreviations
- AE
- Adverse Event
- AR
- Adverse Reaction
- CI
- Chief Investigator
- CRF
- Case Report Form
- CTU
- Clinical Trials Unit
- GP
- General Practitioner
- IB
- Investigator's Brochure
- ICS
- Inhaled Cortico-steroids
- IDSMC
- Independent Data and Safety and Monitoring Committee
- IEC
- Independent Ethical Committee
- IMP
- Investigational Medicinal Product
- LREC
- Local Research Ethics Committee
- MCRN CTU
- Medicines for Children Clinical Trials Unit
- MREC
- Main Research Ethics Committee
- PI
- Principal Investigator
- R&D
- Research & Development
- SAE
- Serious Adverse Event
- SAR
- Serious Adverse Reaction
- SPC
- Summary of product characteristics
- SUSAR
- Suspected Unexpected Serious Adverse Reaction
- TSC
- Trial Steering Committee
- UAR
- Unexpected Adverse Reaction
1 PROTOCOL SUMMARY
Title
Management of Asthma in School-age Children On Therapy
Phase
IV
Population
The target population will be children (aged 6–14 years) requiring frequent short-acting beta-2 agonist relief therapy ≥ 7 puffs per week and with asthma symptoms resulting in nocturnal awakening and/or compromised or reduced activity/exercise and/or those who have had exacerbations (defined as a short course of oral corticosteroids, an unscheduled GP or A&E Department visit or a hospital admission within the previous 6 months).
Number of Sites
13 secondary care and associated general practices throughout the United Kingdom. Site details are listed in Appendices A.
Study Duration
Total study duration for each randomised child is 52 weeks, comprising a 4-week run-in when all will be prescribed standard medication. Those continuing to fulfil eligibility criteria and giving informed consent after 4 weeks will be randomised (designated as time T0) and have follow-up reviews at T+8, T+24, T+36 and T+48 weeks.
Description of Agent/Intervention
All patients recruited into the study will undergo a 4-week run-in period when they will be provided with information about asthma and its management and prescribed the same low-dose inhaled corticosteroid, fluticasone propionate, in the dose of 100 micrograms twice daily. After 4 weeks, those that are eligible will be randomised to one of the following three regimen:
-
Inhaled fluticasone propionate 100 micrograms twice daily plus placebo tablet once daily
-
Inhaled fluticasone propionate 100 micrograms and salmeterol 50 micrograms twice daily (combination inhaler) plus placebo tablet once daily
-
Inhaled fluticasone propionate 100 micrograms twice daily plus montelukast 5-mg tablet once daily.
The allocated treatment will be double-blinded, achieved by using identical inhalers and placebo tablets.
Objectives
Primary
The main research objective is to determine, in 6–14 year old children with asthma, uncontrolled on low-dose ICS, whether their control can be improved by adding in a long-acting beta-2 agonist (salmeterol) or a leukotriene receptor antagonist (montelukast) as measured by a reduced number of exacerbations requiring treatment with oral corticosteroids over the 48 week study period.
Schematic of Study Design
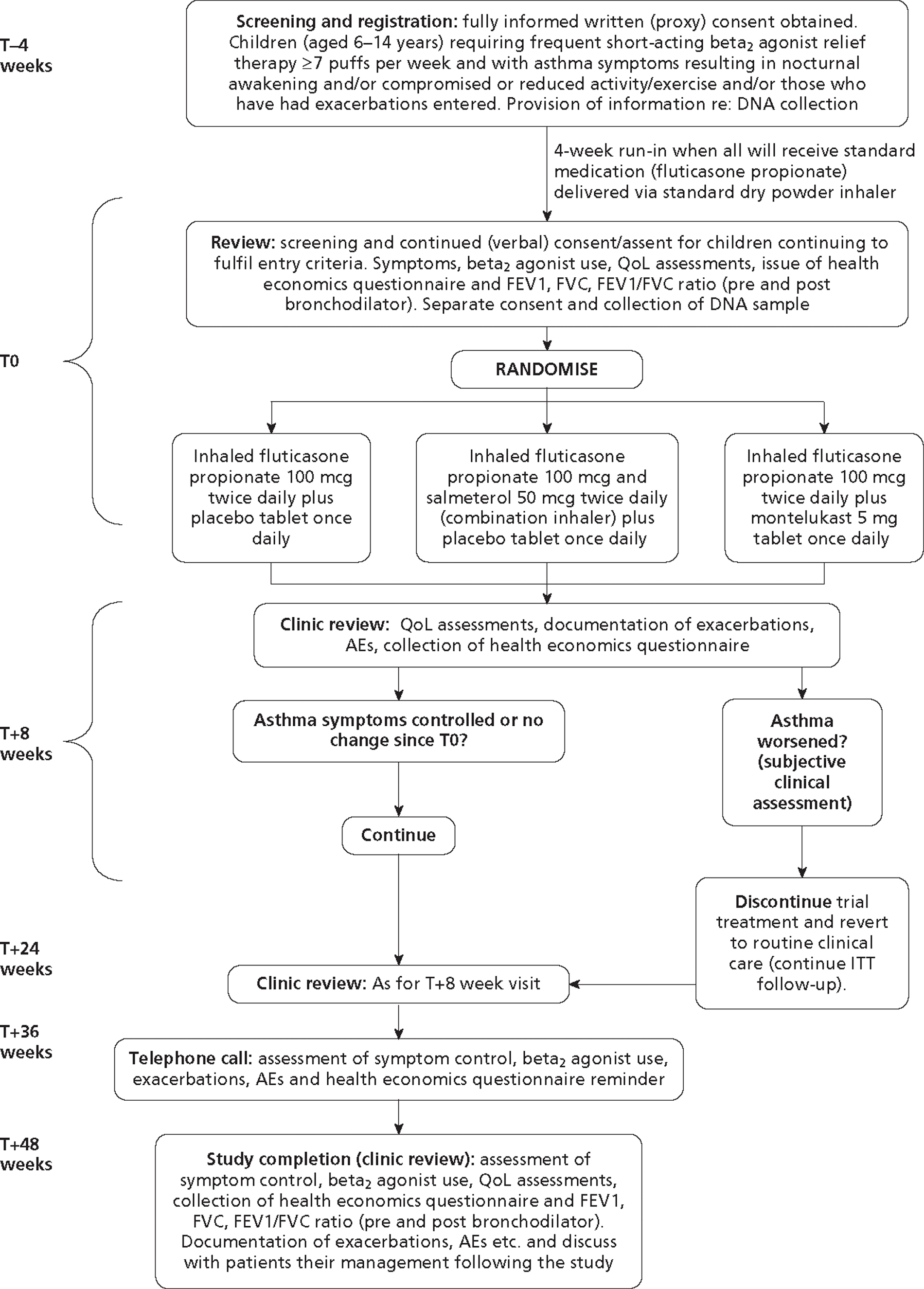
2 BACKGROUND INFORMATION
2.1 Introduction
Respiratory disease has recently been declared a target for improved management by the Department of Health. Although the major burden of chest diseases is in the adult population, it is accepted that the majority of adult chest diseases originates in childhood. The longitudinal studies from Aberdeen1 have shown that 60% of adults with asthma suffered their first symptoms in early childhood, continuing with these through later childhood and into adult life.
2.2 Rationale
Asthma remains the most common medical condition seen in children in primary care and the most frequent cause for medical paediatric hospital admission. It affects 1 in 8 children nationwide, approximately 50% of whom are prescribed low-dose inhaled corticosteroids (ICS). When treatment with low-dose ICS fails to control asthma symptoms, the National Guidelines2 suggest ensuring compliance, maximising inhaler technique and giving appropriate information about the disease to children and their families. Once these measures have been established and if asthma symptoms persist, the Guidelines recommend changing the treatment (Step 3 of the National Guidelines). The evidence at this step of the Guidelines is much weaker in children than it is in adults. The reasons for this are that few studies have been undertaken in children and most that have taken place have used adult-based outcomes such as lung function measurements. This is unsatisfactory because we know that as a chronic disease entity asthma in children is much more variable than in adults and between periods of symptoms, lung function is often normal. Pharmaceutical company studies have really only been conducted as part of their requirements to obtain a license to market their product. These studies have generally been of short-term duration. They have not added to clinicians' understanding of how and where to use the medications3,4. They have not necessarily selected a representative population due to their entry criteria and their intensive study requirements. Such requirements mean that ‘real-life’ compliance does not occur. In the independent National Dutch Study5 which attempted to enter patients uncontrolled on low-dose ICS, three treatment groups were employed: inhaled corticosteroids alone, inhaled corticosteroids in double the dose and inhaled corticosteroids + a long-acting beta2 agonist. There was essentially no difference in outcome measures between the three treatment groups as once again the primary outcome measure was that of lung function (FEV1). Comparing this study with a similar adult study6 both of which used lung function as the primary outcome measure, the mean FEV1 on entry into the paediatric study was approximately 89% expected for the children's heights. In the adult study the mean FEV1 on entry into the study was 74% expected. It is therefore not surprising that the paediatric study was unable to show any differences between the treatment groups.
We do not have the scientific information about how to treat children with asthma who are not well controlled on low-dose ICS therapy. It used to be recommended that when low-dose ICS were not effective their dose should be doubled. Studies in children, however, have investigated this statement and the results are not impressive7. There is no scientific evidence that when control is poor in children with asthma, the dose of the inhaled steroid should be increased. We have therefore decided not to introduce into this study a treatment limb with a higher ICS dosage. There is anecdotal information, however, from many studies undertaken within the pharmaceutical industry that when children enter a study which is controlled and double-blind in nature, up to 30% of them improve, their symptoms reduce and their lung function increases8. It is therefore surprising that approximately one third of children receiving ICS are prescribed high-dose inhaled steroid therapy (≥ 800 micrograms and unlicensed beclometasone dipropionate or equivalent) or they are commenced on ‘add-on’ therapies such as long-acting beta2 agonists (LABA) or leukotriene receptor antagonists (LTRA) in addition to low-dose ICS. Concerns about the safety of high-dose ICS have been raised in relation to growth impairment9, hypoglycaemia10 and suppression of the adrenal cortex11 resulting in warnings on prescribing from the Medicines & Healthcare Products Regulatory Agency (MHRA) in the UK12 and from the Food and Drug Administration (FDA) in the USA. It is therefore unacceptable that approximately one third of children with asthma are being treated with the above regimes. Asthma is a very common condition and the worth of these regimes has not been proven by appropriately devised paediatric studies. The National Guidelines have been developed in a ‘stepwise’ manner, the amount of medication increasing at each step if symptoms are not controlled.
However, as stated above, it may be that childhood asthma differs from that in adults. It seems that relatively poorly controlled asthma in children who exhibit frequent symptoms do not necessarily show abnormal lung function between their periods of symptoms. It is for this reason that in our study we will be concentrating on outcome measures such as exacerbations and quality of life although we will have the opportunity to measure spirometric values at the first (T0) and last (T48) visits in the randomised part of the study. It could be that an increase in medication may only be needed for a short time in children with asthma and there have been suggestions that once control is achieved children should have their add-on therapy reviewed. To incorporate such a step within the present study would make it excessively complicated and would have major implications on the number of patients included. The inclusion of such a step would make the study impractical within the UK. A study is needed which is simple, pragmatic (but placebo-controlled and double-blinded), has outcomes which will be of practical benefit to children and will provide evidence for the use of add-on medications in the most cost effective and efficient way.
Children dislike exacerbations. School attendance, daily activity and general well-being increase when asthma is well controlled. Once families understand sufficiently about asthma, inhaler technique has been evaluated and optimised, and compliance issues addressed, one of the reasons why a specific medication may be less effective could be related to the genetic make-up of the patient. In this study we will have the opportunity, through a separate consent process, to collect and store DNA specimens from saliva for later analysis of specific genetic polymorphisms in relation to asthma severity and outcome. This aspect will bring added value to the study.
2.3 Objectives
Primary Objective
To determine, in 6–14 year old children with asthma uncontrolled on low-dose ICS, whether their control can be improved by adding in a long-acting beta2 agonist (salmeterol) or a leukotriene receptor antagonist (montelukast) as measured by a reduced number of exacerbations requiring treatment with oral corticosteroids over the 48 week study period.
2.4 Potential Risks and Benefits
The medications used in this study are subject to Marketing Authorisations and are to be prescribed in accordance with their licensed indications. The management of any symptoms or exacerbations will be in accordance with usual clinical practice and a research worker, either the local principal investigator (PI) or research nurse (RN), will be available throughout the study to discuss specific issues with individuals concerned. Any concerns which cannot be satisfied at a local level will be forwarded to the chief investigator via the MASCOT Trial Coordinator based at the Medicines for Children Research Network Clinical Trials Unit (MCRN CTU). Any patient can withdraw from the study at any time with no detriment to their future care. All ethical aspects of the study will be discussed when informed written consent is obtained. Appropriate patient and family information leaflets have been developed and are discussed at the screening consultation. Patients and their families will be provided with a copy of the information sheets and their signed consent/assent forms.
Potential Risks
The potential risks of the three products (fluticasone propionate, salmeterol and montelukast) used in MASCOT are summarised individually in the tables below. For more detailed information on the potential risks, special warning and precautions for use of these medications please refer to the Summary of Product Characteristics.
Adverse events are listed below by system organ class and frequency. Frequencies are defined as: very common (≥ 1/10), common (≥ 1/100 and < 1/10), uncommon (≥ 1/1000 and < 1/100), rare (≥ 1/10,000 and < 1/1000) and very rare (< 1/10,000) including isolated reports. Very common, common and uncommon events were generally determined from clinical trial data. Rare and very rare events were generally determined from spontaneous data.
Fluticasone propionate
| System Organ Class | Adverse Event | Frequency |
|---|---|---|
| Infections & Infestations | Candidiasis of the mouth and throat | Very Common |
| Immune System Disorders | Hypersensitivity reactions with the following manifestations: | |
| Cutaneous hypersensitivity reactions | Uncommon | |
| Angioedema (mainly facial and oropharyngeal oedema) | Very Rare | |
| Respiratory symptoms (dyspnoea and/or bronchospasm) | Very Rare | |
| Anaphylactic reactions | Very Rare | |
| Endocrine Disorders | Cushing's syndrome, Cushingoid features, adrenal suppression, growth retardation in children and adolescents, decreased bone mineral density, cataract, glaucoma | Very Rare |
| Metabolism & Nutrition Disorders | Hyperglycaemia (see Flixotide SPC Section 4.4 ‘Special Warnings and Precautions for Use’) | Very Rare |
| Gastrointestinal Disorders | Dyspepsia | Very Rare |
| Musculoskeletal & Connective Tissue Disorders | Arthralgia | Very Rare |
| Psychiatric Disorders | Anxiety, sleep disorders, behavioural changes, including hyperactivity and irritability (predominantly in children) | Very Rare |
| Respiratory, Thoracic & Mediastinal Disorders | Hoarseness/dysphonia | Common |
| Paradoxical bronchospasm | Very Rare | |
| Skin & Subcutaneous Tissue Disorders | Contusions | Common |
Salmeterol
| System Organ Class | Adverse Event | Frequency |
|---|---|---|
| Infections and Infestations | Candidiasis of the mouth and throat | Common |
| Pneumonia | *#Common | |
| Bronchitis | *#Common | |
| Immune System Disorders | Hypersensitivity reactions with the following manifestations: | |
| Cutaneous hypersensitivity reactions | Uncommon | |
| Angioedema (mainly facial and oropharyngeal oedema), Respiratory symptoms (dyspnoea and/or bronchospasm), Anaphylactic reactions including anaphylactic shock | Very Rare | |
| Endocrine Disorders | Cushing's syndrome, Cushingoid features, Adrenal suppression, Growth retardation in children and adolescents, Decreased bone mineral density | Very Rare |
| Metabolism and Nutrition Disorders | Hypokalaemia | #Common |
| Hyperglycaemia | Very Rare | |
| Psychiatric Disorders | Anxiety, sleep disorders and behavioural changes, including hyperactivity and irritability (predominantly in children) | Very Rare |
| Nervous System Disorders | Headache | *Very Common |
| Tremor | Common | |
| Cardiac Disorders | Palpitations | Common |
| Tachycardia | Uncommon | |
| Cardiac arrhythmias (including atrial fbrillation, supraventricular tachycardia and extrasystoles) | Very Rare | |
| Respiratory, Thoracic and Mediastinal Disorders | Nasopharyngitis | **#Very Common |
| Throat irritation | Common | |
| Hoarseness/dysphonia | Common | |
| Sinusitis | *#Common | |
| Paradoxical bronchospasm | Very Rare | |
| Skin and Subcutaneous Tissue Disorder | Contusions | *#Common |
| Musculoskeletal and Connective Tissue Disorders | Muscle cramps | Common |
| Traumatic fractures | *#Common | |
| Arthralgia | Very Rare | |
| Myalgia | Very Rare | |
| Eye Disorders | Cataract, glaucoma | Very rare |
Montelukast
(Please note no defnition of ‘very rare’ is available for montelukast)
| Body System Class | Adverse Event | Frequency |
|---|---|---|
| Body as a whole | Abdominal pain | Common |
| Asthenia/fatigue, malaise, oedema, | Very rare | |
| Digestive System Disorders | Thirst | Common |
| Diarrhoea, dry mouth, dyspepsia, nausea, vomiting | ||
| Nervous System Disorders | Headache | Common |
| Dizziness, drowsiness, paraesthesia/hypoesthesia, seizure | Very rare | |
| Psychiatric Disorders | Dream abnormalities including nightmares, hallucinations, insomnia, paraesthesia/hypoesthesia, irritability, agitation including aggressive behaviour, restlessness, anxiety, tremor, depression, suicidal thinking and behaviour (suicidality) | Very rare |
| Musculo-Skeletal Disorders | Arthralgia, myalgia including muscle cramps | Very rare |
| Hepato-Biliary Disorders | Elevated levels of serum transaminases (ALT, AST), cholestatic hepatitis | Very rare |
| Cardiovascular Disorders | Palpitations | Very rare |
| Skin & Subcutaneous Tissue Disorders | Angiooedema, urticaria, pruritus, rash, erythema nodosum, bruising | Very rare |
| Immune System Disorders | Hypersensitivity reactions including anaphylaxis, hepatic eosinophilic infiltration | Very rare |
Known Potential Benefits
All of the medications have Marketing Authorisations and have been shown to be efficacious for children with chronic asthma when used appropriately as preventative therapy. The ultimate aims of preventative asthma treatment are the prevention of chronic symptoms, maintenance of near normal lung function and normal activity levels and prevention of recurrent acute episodes in order to maximise quality of life. The potential benefit for participants of taking these medications as part of the trial is that they will improve control of their asthma, reducing symptoms and exacerbations and meeting the goals above.
3 SELECTION OF CENTRES/CLINICIANS
Hubs will be selected upon the basis of:
-
an institution with existing links with primary care teams or with the ability to engage and facilitate an effective relationship
-
having at least one lead clinician with a specific interest in, and responsibility for, supervising and managing children with asthma, who is enthusiastic about participating in the study
-
ensuring that sufficient time, staff and adequate facilities are available for the trial
-
providing information to all supporting staff members involved with the trial or with other elements of the patient's management
-
identifying that they will be able to recruit the required number of patients
-
acknowledging and agreeing to conform to the administrative and ethical requirements and responsibilities of the study, including signing-up to Good Clinical Practice and other regulatory documentation
General Practices will be selected upon the basis of:
-
Fully computerised prescribing records
-
Willingness to use GPIAG or equivalent asthma template for routine asthma consultations to ensure high quality data when patients reviewed
-
Willingness to allow MIQUEST or Apollo extraction tools to enable health resource utilisation (consultations and medications) to be collected electronically
3.1 Centre/Clinician Inclusion Criteria
-
Positive Site Specific Assessment (SSA) by LREC
-
Local R&D approval
-
Receipt of evidence of completion of (a) and (b) by MCRN CTU
-
Completion and return of ‘Signature and Delegation Log’ to MCRN CTU
3.2 Centre/Clinician Exclusion Criteria
-
Not meeting the inclusion criteria listed above
4 ENDPOINTS
4.1 Primary Endpoint
The primary outcome will be the number of asthma exacerbations requiring treatment with oral corticosteroids over the 48 week study period.
4.2 Secondary Endpoint(s)
-
Quality of Life as measured by the Paediatric Asthma Quality of Life Questionnaire (PAQLQ) and the Paediatric Asthma Caregivers Quality of Life Questionnaire (PACQLQ)13
-
Time from randomisation to first exacerbation requiring treatment with a short course of oral corticosteroids
-
School attendance
-
Hospital admissions
-
Amount of rescue beta2 agonist therapy prescribed
-
Time from randomisation to treatment withdrawal (due to lack of efficacy or side effects)
-
Lung function at 48 weeks (as assessed by spirometry)
-
Cost effectiveness
-
Adverse events
5 STUDY POPULATION
5.1 Inclusion and Exclusion Criteria at T–4 Weeks (Registration)
5.1.1 Inclusion Criteria
-
Children with physician diagnosed asthma aged 6 years–14 years, 11months
-
Those requiring frequent short-acting beta2 agonist relief therapy ≥ 7 puffs in the past seven days
-
Those with symptoms of asthma (i.e. wheeze, shortness of breath but not cough alone) resulting in:
-
Nocturnal wakening in the last week because of asthma symptoms and/or
-
Asthma has interfered with usual activities in the last week and/or
-
Those who have had exacerbations, defined as a short course of oral corticosteroids, an unscheduled GP or A&E Department visit or a hospital admission within the previous 6 months
-
-
Fully informed written (proxy) consent and assent, where appropriate
5.1.2 Exclusion Criteria
-
Children receiving long acting beta2-agonists, leukotriene receptor antagonists, regular theophylline therapy or high dose ICS > 1000 micrograms and unlicensed beclometasone dipropionate or equivalent (at the discretion of the investigator)
-
Children with other respiratory diseases, cystic fibrosis, cardiac disease or immunological disorders
-
Non-English speaking
5.2 Inclusion and Exclusion Criteria at T0 (Randomisation)
5.2.1 Inclusion Criteria
-
Children with asthma aged 6 years–14 years
-
Those requiring frequent short-acting beta2 agonist relief therapy ≥ 7 puffs in the past seven days
-
Those with symptoms of asthma (i.e. wheeze, shortness of breath but not cough alone) resulting in
-
Nocturnal wakening in the last week because of asthma symptoms and/or
-
Asthma has interfered with usual activities in the last week
-
-
Continuing consent/assent (where appropriate)
5.2.2 Exclusion Criteria
-
Children whose asthma is controlled after the 4 week run-in, where control is defined as the absence of any symptoms of asthma (except cough alone) or where the symptoms of asthma have not interfered with usual activities in the last week
-
Children receiving long acting beta2-agonists, leukotriene receptor antagonists, regular theophylline therapy or high dose ICS > 1000 micrograms and unlicensed beclometasone dipropionate or equivalent (at the discretion of the investigator)
-
Children with other respiratory diseases, cystic fibrosis, cardiac disease or immunological disorders.
-
Non-English speaking
5.3 Patient Transfer and Withdrawal
In consenting to the trial, patients are consented to trial treatment, follow-up and data collection. If voluntary withdrawal occurs, the patient and their parent/legal representative should be asked to allow continuation of scheduled evaluations, complete an end-of-study evaluation, and be given appropriate care under medical supervision until the symptoms of any adverse event resolve or the subject's condition becomes stable wherever relevant. Follow-up of these patients will be continued through the research nurse and lead investigator at each secondary care centre and, where this is unsuccessful, through the child's GP (if possible).
5.3.1 Patient Transfers
For patients moving from the area, every effort should be made for the patient to be followed up at another participating trial centre and for this trial centre to take over responsibility for the patient or for follow-up via GP.
A copy of the patient's Case Report Forms (CRFs) should be provided to the new site. The patient and their parent/legal representative will have to sign a new consent form at the new site and, until this occurs, the patient remains the responsibility of the original centre. The CTU should be notified in writing of patient transfers.
5.3.2 Withdrawal from Trial Intervention
Patients may be withdrawn from treatment for any of the following reasons:
-
Parent/legal representative (or, where applicable, the patient) withdraws consent for treatment.
-
Unacceptable adverse effects.
-
Intercurrent illness preventing further treatment.
-
Development of serious disease preventing further treatment or any change in the patient's condition that justifies the discontinuation of treatment in the clinician's opinion.
-
Lack of efficacy.
The patient should be asked if they are willing to still have data collected as per trial schedule or, failing this, to allow routine follow-up data to be used for trial purposes. Patients who withdraw from trial treatment but are willing to allow further data collection must have a discussion with the investigator as to whether to they will be unblinded at this point or remain blind to their randomised treatment allocation until the end of their 48 week follow-up period. The decision should be based on the patient and their carers' own preferences and whether the investigator feels they need to be aware of the patient's randomised treatment allocation in order to enable appropriate follow on care.
5.3.3 Withdrawal from Trial Completely
Patients are free to withdraw consent at any time without providing a reason. Patients who wish to withdraw consent for the trial will have anonymised data collected up to the point of that withdrawal of consent included in the analyses. The patient will not contribute further data to the study and the MCRN CTU should be informed in writing by the responsible physician and a withdrawal CRF should be completed. MCRN CTU will endeavour to unblind the patient and inform their GP of their randomised treatment allocation within seven days. Data up to the time of withdrawal will be included in the analyses unless the patient explicitly states that this is not their wish.
6 RECRUITMENT, REGISTRATION AND RANDOMISATION
6.1 Recruitment
Patients who are eligible for inclusion into the trial will be identified and recruited through both primary and secondary care. There are several strategies to support the recruitment of participants into MASCOT. Participating sites can adopt either one or a combination of these approaches, depending on their local arrangements and existing pathways for managing patients in the target population.
Recruitment Strategy #1
This strategy covers all primary care centres including (but not limited to) general practices and health centres, NHS walk-in centres, GP and nurse-led out of hours services and minor injury centres. Participants will be identified via General Practitioners, community-based specialist paediatric/respiratory nurses and other appropriate clinicians working within primary care. The primary care practitioners will be asked for an estimate on the number of their patients who meet the MASCOT eligibility criteria and, if they do see this patient population, they will be asked if they are willing to take part in helping to identify participants for the trial. This identification will be done by the primary care staff who will search their own patient database/s (either electronically – see Appendix C for search guidance – or manually) to find potentially eligible participants and then write to them (a standard letter will be provided), enclosing a Patient Information Sheet and instructions on how to proceed if they are interested in taking part or finding out more about the study. They will be asked either to send a reply slip back directly to the RN who would then contact them by telephone to ascertain potential eligibility and invite them to a T–4 visit OR to contact the RN by telephone/e-mail for further information. Following this initial mailout, the primary care practitioners may also follow up the letter with a telephone call or one subsequent letter.
Recruitment Strategy #2
In addition to the database searches outlined above, primary care practitioners will also conduct opportunistic recruitment and will ask any patients they identify if they are willing for their contact details to be passed to the research team. If they agree, the RN will then contact them directly to ascertain potential eligibility and invite them to a T–4 visit. Alternatively the practitioner can provide the family with a Patient Information Sheet, which gives the contact details of the research team, with advice to contact them directly if they want to find out more about the trial OR provide them with the approved MASCOT lay person poster (produced as a A5 sized handout) which also gives the contact details of the research team.
Recruitment Strategy #3
Participants will be identified via General Practitioners in primary care. The GPs will be asked for an estimate on the number of their patients who meet the MASCOT eligibility criteria. If they do see this patient population, they will be asked if they are willing to take part in the trial and to take on the role of PI. The practice staff will identify potentially eligible patients and write to them (a standard letter will be provided and printed on practice headed paper), enclosing a Patient Information Sheet and instructions on how to proceed if they are interested in taking part or finding out more about the study. They will be asked to either send a reply slip back directly to the GP who would contact them by telephone to ascertain potential eligibility and then invite them to the T–4 visit OR to contact the GP by telephone/e-mail for further information. Following this initial search, GPs will subsequently conduct opportunistic recruitment and will ask any patients they identify if they are willing for their contact details to be passed to the research team. If they agree, the RN will then contact them directly to ascertain potential eligibility and invite them to a T–4 visit.
Recruitment Strategy #4
Participants will be identified by community pharmacists. During the course of a patient's regular medication review, or whilst providing education on the use of asthma medications, pharmacists may identify patients they believe are potentially eligible for the trial. They will ask any patients who they think may be eligible if they are willing for their contact details to be passed to the research team. If they agree, the RN will then contact them to discuss the trial, ascertain potential eligibility and invite them to a T–4 visit (if appropriate). Alternatively the pharmacist can provide the family with a Patient Information Sheet, which gives the contact details of the research team, with advice to contact them directly if they want to find out more about the trial OR provide them with the approved MASCOT lay person poster (produced as a A5 sized handout) which also gives the contact details of the research team. All pharmacies agreeing to participate in this way will be fully briefed on the trial beforehand. Where it is more appropriate, pharmacists may flag a patient's potential eligibility for the trial to their GP instead of directly to the research team. The GP may then follow any of the methods listed in recruitment strategies #1 & #2 at their own discretion.
Recruitment Strategy #5
Participants will be identified via health professionals with a remit to work within schools (e.g. school nurses, health visitors). They may search their registers and databases to find potentially eligible patients. These patients will then be written to (a standard letter will be provided) enclosing a Patient Information Sheet and instructions on how to proceed if they are interested in taking part or in finding out more about the trial. Following this initial mailout, the school health team may also follow up the letter with a telephone call or one subsequent letter. Alternatively, they may identify patients who are potentially eligible during the course of their normal role. After gaining permission from the patient's parent/legal guardian, the health professional will pass their contact details to the research team. The RN will then contact the family directly to discuss the trial, ascertain potential eligibility and invite them to a T–4 visit (if appropriate).
Recruitment Strategy #6
Participants will be identified via secondary care (A&E admissions, routine OPD appointments, specialist nurse-led clinic appointments). Secondary care professionals will be approached by the MASCOT RN/LRN nurses and the PI and informed about the trial. If a patient presents who may be eligible for the trial, the medical staff treating them initially can follow one of two routes: 1) contact the local PI/RN and ask them to come and speak to the patient there and then about the trial, inviting them to attend a T–4 visit if they are potentially eligible and interested in participating OR 2) give the patient a PISC and ask for their permission to pass their contact details on to the RN who will call the patient later to discuss the trial and invite them for a T–4 visit if they are eligible.
Recruitment Strategy #7
Participants will be identified via secondary care by their usual NHS clinical team who will search the Trust databases to find potentially eligible participants. These patients will then be written to (a standard letter will be provided and printed on Trust headed paper), enclosing a PISC and instructions on how to proceed if they are interested in taking part or finding out more about the trial. They could either be asked to send a reply slip back directly to the RN who would contact them by telephone to ascertain the patient's eligibility and then invite them to the T–4 visit OR to contact the RN by telephone/e-mail for further information. Following this initial mailout, the secondary care team may also follow up the letter with a telephone call or one subsequent letter.
Recruitment Strategy #8
The trial may also be promoted via appropriate websites, magazines and newspapers to be accessed by both health professionals and the general public. Approval will be sought from the REC for any specific features or advertisements designed to promote the trial directly to the public, prior to submitting them for publication.
6.2 Screening (Registration)
A log of potential patients will be kept (the ‘Screening and Enrolment Log’), including individuals who decide not to participate in the study at the T–4 visit and ineligible referrals from primary care practitioners and secondary care clinicians. Screening will be performed of a patient's possible eligibility for the study and must be documented on the screening CRF at the T–4 visit.
Screening at T–4 (See Section 8 for T–4 assessments)
-
Confirm aged 6–14 years, 11 months
-
Explanation of the two different phases of the trial and understanding that eligibility for trial treatment will be reassessed at T0 visit
-
Fully informed written proxy consent (and assent, where appropriate) to participate in the trial
-
Assessments to determine eligibility (inc. review of medical history, symptoms, concomitant medications)
-
Fluticasone propionate dispensed (open label)
-
Submission of T–4 CRF to MCRN CTU within seven days of registration
-
Forward copy of consent/assent forms to MCRN CTU within seven days of registration
6.3 Enrolment/Baseline (Randomisation)
Screening at T0
-
A check of compliance with hand held asthma record completion
-
Review of symptoms and exacerbations
-
A check of concomitant medications prescribed/administered since T–4 visit
-
Complete physical examination performed
-
Verification that the eligibility criteria for randomisation is fulfilled
-
See Section 8 for T0 assessments
Randomisation Process
-
Continuing consent and assent (where appropriate) obtained verbally
-
Completion of randomisation CRF and trial prescription
-
Attend local pharmacy (see Table 1 for pharmacy contact details)
-
Participant's treatment allocation ascertained by pharmacy using the site randomisation list
-
Issue of treatment pack by pharmacy department (ensuring the patient and researcher are blinded to the allocation)
-
Submission of T0 CRF to MCRN CTU within seven days of randomisation
| For any queries relating to randomisation procedure, please contact: Trial Coordinator, Sophie Perry on 0151 252 4706 | |
|---|---|
| E-mail: mascot@mcrnctu.org.uk | |
| University Hospital of North Staffordshire | Alder Hey Children's NHS Foundation Trust |
| Cath Jackson | Catrin Barker |
| Pharmacy Department | Pharmacy Department |
| Royal Infirmary | Royal Liverpool Children's NHS Trust (Alder Hey) |
| University Hospital of North Staffordshire | Eaton Road |
| Stoke-on-Trent | Liverpool |
| Princes Road | L12 2AP |
| Stoke-on-Trent | Tel: 0151 252 5837 |
| ST4 7LN | Fax: 0151 220 3885 |
| Tel: 01782 555157 | Email: catrin.barker@alderhey.nhs.uk |
| Fax: 01782 555156 | |
| Email: Catherine.Jackson@uhns.nhs.uk | |
| Royal Manchester Children's Hospital | Derbyshire Children's Hospital |
| Carolyn Davies | Peter Fox |
| Pharmacy Department | Pharmacy Department |
| Manchester Royal Infirmary | Derbyshire Hospitals NHS Foundation Trust |
| Oxford Road | London Road |
| Manchester | Derby |
| M13 9WL | DE22 3NE |
| Tel: 0161 276 4623 | Tel: 01332 789101 |
| Email: carolyn.davies@CMFT.nhs.uk | Fax: 01332 789106 |
| Email: peter.fox@derbyhospitals.nhs.uk | |
| Queens Medical Centre, Nottingham | University Hospital (Walsgrave site) |
| Sheila Hodgson | Roger Cross |
| Pharmacy Department | Pharmacy Department |
| Queens Medical Centre Campus | University Hospitals Coventry & Warwickshire NHS Trust |
| Nottingham University Hospitals NHS Trust | Clifford Bridge Road |
| Derby Road | Coventry |
| Nottingham | CV2 2DX |
| NG7 2UH | Tel: 02476 966042 |
| Tel/Fax: 0115 9194450 ext 68450 | Fax: n/a |
| Email: sheila.hodgson@nuh.nhs.uk | Email: roger.cross@uhcw.nhs.uk |
| Royal Aberdeen Children's Hospital | Bristol Royal Hospital for Children |
| Martina Freeman | Lindsay Ball |
| Pharmacy Department (Clinical Trials) | Pharmacy Department |
| Aberdeen Royal Infirmary | Bristol Royal Infirmary |
| Foresterhil | Marlborough Street |
| Aberdeen | Bristol |
| AB25 2ZN | BS2 8HW |
| Tel: 01224 551733 | Tel: 0117 928 2053 |
| Fax: 01224 551061 | Fax: 0117 928 2683 |
| Email: martina.freeman@nhs.net | Email: Lindsay.Ball@ubht.nhs.uk |
| Royal Devon & Exeter NHS Foundation Trust | Royal London Hospital |
| Fiona Hall | Alexandra Farrell Outpatient Dispensary |
| Pharmacy Department | Outpatient Building |
| Royal Devon & Exeter Hospital (Wonford) | Royal London Hospital |
| Barrack Road | Whitechapel |
| Exeter | E1 1BB |
| EX2 5DW | Tel: 0207 377 7000 |
| Tel: 01392 402444 | Fax: 0207 377 7386 |
| Fax: 01392 406006 | Email: Alexandra.Farrell@bartsandthelondon.nhs.uk |
| Email: Fiona.Hall@rdeft.nhs.uk | |
| Leicester Royal Infirmary | Norfolk & Norwich University Hospital |
| David Harris | Nicola Rudge |
| Pharmacy Department | Pharmacy Department |
| Leicester Royal Infirmary | Norfolk & Norwich University Hospital |
| Leicester | Colney Lane |
| LE1 5WW | Norwich |
| Tel: 0116 258 5462 | NR4 7UY |
| Fax: 0116 258 6974 | Tel: 01603 289402 |
| Email: david.harris@uhl-tr.nhs.uk | Fax: 01603 287134 |
| Email: nicola.rudge@nnuh.nhs.uk | |
| Wirral University Teaching Hospitals NHS Foundation Trust | |
| Neil Caldwell | |
| Pharmacy Department | |
| Arrowe Park Hospital | |
| Arrowe Park Road | |
| Upton | |
| Wirra | |
| CH49 5PE | |
| Tel: 0151 678 5111 (ext. 2060) Fax: 0151 604 7066 | |
| Email: Neil.Caldwell@whnt.nhs.uk | |
7 TRIAL TREATMENT/S
7.1 Introduction
This study is designed as a prospective, controlled, double-blind, multicentre, randomised clinical trial comparing whether control of asthma symptoms can be improved by adding in a long-acting beta2 agonist (salmeterol) or a leukotriene receptor antagonist (montelukast) in 6–14 year olds with asthma uncontrolled on low-dose inhaled corticosteroids (ICS).
During the four week run-in period all patients will be commenced on fluticasone propionate inhalers at 200 micrograms per day (100 micrograms twice daily). Children who remain symptomatic at the end of the run-in period will be randomised into one of three double-blinded treatment regimen:
-
Inhaled fluticasone propionate 100 micrograms twice daily plus placebo tablet once daily
-
Inhaled fluticasone propionate 100 micrograms and salmeterol 50 micrograms twice daily (combination inhaler) plus placebo tablet once daily
-
Inhaled fluticasone propionate 100 micrograms twice daily plus montelukast 5 mg tablet once daily.
7.2 Formulation, Packaging, Labelling, Storage and Stability
A: Fluticasone propionate
Description and composition of the drug product
Product name – fluticasone propionate (brand name, Flixotide™).
Flixotide 100 micrograms Accuhaler is a multi-dose dry powder inhalation device delivering 100 micrograms of fluticasone propionate per inhalation. Participants will receive 100 micrograms twice daily, taken as one inhalation twice daily.
Flixotide has been manufactured and supplied by GlaxoSmithKline plc (GSK).
Type of container and closure system
The Flixotide Accuhaler is a moulded plastic device containing a foil strip with regularly placed blisters each containing a mixture of fluticasone propionate (100 micrograms) and lactose monohydrate. The blister strip consists of a formed foil base with a peelable foil laminate lid. Each inhaler contains 60 pre-dispensed doses of Flixotide. The inhaler device is packed in an individual cardboard container, which will be labelled for trial treatment (labels will include study acronym, EudraCT reference number, randomisation number, visit number, site number, instructions for use and storage, batch number and expiry date).
Stability and shelf life
The product should be stored at temperatures less than 30°C and should not be refrigerated or frozen. The product has a shelf life of 18 months.
B: Salmetrol + Fluticasone propionate
Description and composition of the drug product
Product name – salmeterol (brand name, Seretide™).
Seretide 50/100 micrograms Accuhaler is a multi-dose dry powder inhaler delivering 50 micrograms of salmeterol (as salmeterol xinafoate) and 100 micrograms of fluticasone propionate. Participants will receive 50 micrograms and 100 micrograms (respectively) twice daily, taken as one inhalation twice daily in a combined inhaler.
Seretide has been manufactured and supplied by GlaxoSmithKline plc.
Type of container and closure system
The Seretide Accuhaler is a moulded plastic device containing a foil strip with regularly placed blisters each containing a mixture of fluticasone propionate (100 micrograms), salmeterol (50 micrograms) and lactose monohydrate. The blister strip consists of a formed PVC base with a peelable foil laminate lid. Each inhaler contains 60 pre-dispensed doses of Seretide. The inhaler device is packed in an individual cardboard container, which will be labelled for trial treatment (labels will include study acronym, EudraCT reference number, randomisation number, visit number, site number, instructions for use and storage, batch number and expiry date).
Stability and shelf life
The product should be stored at temperatures less than 30°C and should not be refrigerated or frozen. The product has a shelf life of 18 months.
C: Montelukast
Description and composition of the drug product
Product name – montelukast (brand name, Singulair®).
Singulair Paediatric 5 mg Chewable Tablets contain montelukast sodium, which is equivalent to 5 mg montelukast. Participants will receive 5 mg once daily, taken as a single chewable tablet. The tablet is round, biconvex and 9.5 mm in diameter. Singulair also contains mannitol, microcrystalline cellulose, hydroxypropylcellulose, red ferric oxide (E172), croscarmellose, cherry flavour, aspartame and magnesium stearate.
Singulair has been manufactured and supplied by Merck Sharp and Dohme Ltd (MSD).
Type of container and closure system
The tablets will be packaged in monthly blister packs of 35 days supply, with an aluminium foil top. Three blister packs will be packaged in an outer cardboard carton and labelled for trial treatment (labels will include study acronym, EudraCT reference number, randomisation number, visit number, site number, instructions for use and storage, batch number and expiry date).
Stability and shelf life
The product should be stored at ambient temperature in the original packaging. The product has a shelf life of 24 months.
Placebo
Description and composition of the drug product
Product name – montelukast (brand name, Singulair®) matching placebo
The drug product (Singulair Paediatric 5 mg Chewable Tablets) placebo tablet is round, biconvex and 9.5 mm in diameter to match the active drug. The placebo contains mannitol, microcrystalline cellulose, hydroxypropylcellulose, red ferric oxide (E172), croscarmellose, cherry flavour, aspartame and magnesium stearate.
The drug product (Singulair) placebo has been manufactured and supplied by Merck Sharp and Dohme Ltd (MSD).
Type of container and closure system
The tablets will be packaged in monthly blister packs of 35 days supply, with an aluminium foil top. Three blister packs will be packaged in an outer cardboard carton and labelled for trial treatment (labels will include study acronym, EudraCT reference number, randomisation number, visit number, site number, instructions for use and storage, batch number and expiry date).
Stability and shelf life
The product should be stored at ambient temperature in the original packaging. The product has a shelf life of five years.
7.3 Preparation, Dosage and Administration of Study Treatment/s
Dispensing
For each randomised patient, treatment will continue for a maximum period of 48 weeks. Patients will be randomised by pharmacy using a randomisation list provided to the site by the coordinating centre. Pharmacy will ensure that the participant and the researcher are blinded to the treatment allocation. After randomisation patients will be dispensed their first treatment pack. Each treatment pack contains three months of trial medication, consisting of:
-
Three inhalers (each inhaler containing 60 inhalations or 30 days treatment)
-
Three blister packs (each blister pack containing 35 days treatment)
All treatments will be dispensed at the standard dose throughout the trial, unless interruption or discontinuation is warranted and agreed by the PI (see Section 7.4). The dose regimens are:
-
inhaled fluticasone propionate 100 micrograms twice daily + placebo tablet once daily
-
inhaled fluticasone propionate 100 micrograms and salmeterol 50 micrograms twice daily (combination inhaler) + placebo tablet once daily
-
inhaled fluticasone propionate 100 micrograms twice daily + montelukast 5 mg tablet once daily.
When pharmacy dispense the trial treatments they will add their own dispensing label, which will include information such as the name and address of the hospital, the patient's name or initials, date of dispensing and instructions for use. They will also complete the information specified on the medication trial labels (i.e. patient trial number, visit number etc.).
The medications will be dispensed upon production of a valid, signed trial prescription to either the RN or directly to the patient and their carer/s as detailed below*:
T–4 study visit
One inhaler of fluticasone propionate (open label) will be dispensed for a registered participant at T–4 on receipt of a valid trial prescription. If the T–4 visit is conducted at an outreach or community site (e.g. a GP practice) and the participant is registered during the visit the RN will arrange for them to receive the study medication once it has been dispensed from pharmacy. The dispensed medication will be collected from pharmacy and securely transported by a member of the research team, ensuring temperature monitoring is conducted until it is given to the patient. The RN will arrange with the patient and their carer/s to collect the medication from the outreach site, ensuring that it is collected by the family within an agreed timeframe (no longer than three days).
T0 study visit
The participant will be randomised to a treatment arm by pharmacy. Ensuring the RN and participant remain blinded as to the treatment allocation, one three month treatment pack containing three inhalers and three monthly blister cards will be dispensed on receipt of a valid trial prescription. If the T0 visit is conducted at an outreach or community site (e.g. a GP practice) and the participant is randomised during the visit the RN will arrange for them to receive the study medication once it has been dispensed from pharmacy. The dispensed medication will be collected from pharmacy and securely transported by a member of the research team, ensuring temperature monitoring is conducted until it is given to the patient. The RN will arrange with the patient and their carer/s to collect the medication from the outreach site, ensuring that it is collected by the family before the patient runs out of their current prescription.
T+8 study visit
The participant's randomised treatment allocation will be ascertained by pharmacy. Ensuring the RN and participant remain blinded as to the treatment allocation, one three month treatment pack each containing three inhalers and three monthly blister cards will be dispensed from pharmacy on receipt of a valid trial prescription.
Where the study visit is conducted at the lead local site and the patient has access to the trial's pharmacy department, the patient and their carer/s can collect the medication at the end of the study visit as per usual clinical practice. Where the patient is seen at an outreach or community site (e.g. a GP practice) for T+8, the RN will collect the appropriate medications from the pharmacy department prior to the visit. The RN will securely transport them to the visit site, ensuring temperature monitoring is conducted, where they will be given to the patient. If the patient is not eligible to continue in the trial or wishes to withdraw from trial treatment at that point, the dispensed medications will be returned to pharmacy for destruction.
T+24 study visit
The participant's randomised treatment allocation will be ascertained by pharmacy. Ensuring the RN and participant remain blinded as to the treatment allocation, two three month treatment packs each containing three inhalers and three monthly blister cards will be dispensed from pharmacy on receipt of a valid trial prescription.
Where the study visit is conducted at the lead local site and the patient has access to the trial's pharmacy department, the patient and their carer/s can collect the prescription at the end of the study visit as per usual clinical practice. Where the patient is seen at an outreach or community site (e.g. a GP practice) for T+24, the RN will collect the appropriate medications from the pharmacy department prior to the visit. The RN will securely transport them to the visit site, ensuring temperature monitoring is conducted, where they will be given to the patient. If the patient is not eligible to continue in the trial or wishes to withdraw from trial treatment at that point, the dispensed medications will be returned to pharmacy for destruction.
*In certain circumstances, the RN may deliver the study medications to the family home. A RN may only deliver medications to a patient's home if this procedure is authorised for their site by the coordinating centre and an appropriate SOP is in place. In addition, due to geographical constraints, Norfolk & Norwich University Hospital pharmacy department will follow a separate dispensing procedure for participants who cannot be seen at the lead local site (outlined in Appendix B).
Lost or Damaged Medications
In the event that a patient loses or damages the inhaler or tablet pack they are currently using, they will move straight onto the next month's medication from the pack they were dispensed with at their last visit. The patient should contact the RN who will bring forward the date of the next visit to ensure that the patient can be dispensed their next prescription (if applicable) in sufficient time to ensure there is no break in medication. However, if the patient has no more dispensed medication, they should contact the RN immediately to discuss how to manage their treatment. If no arrangements can be put in place for the patient to collect their next medication pack from the pharmacy within an acceptable period of time, the PI will withdraw the patient from the trial at that point. The patient will return to the care of their usual medical practitioner and will be followed up as per the protocol by the research team (see Section 5.3.2).
Administration
The patient and their carer/s will be instructed in the correct use of the medications dispensed. Patients will be instructed in the proper use and care of their inhaler by the RN at T–4 and will have their technique assessed. Further guidance will be provided throughout the remainder of the trial where necessary.
The trial treatments have two different routes of administration:
-
inhalation (fluticasone/salmeterol). One inhalation to be taken twice daily at regular intervals (e.g. once in the morning and once in the evening).
-
oral (montelukast/placebo). One tablet is to be administered daily, to be taken in the evening. If taken in conjunction with food, montelukast should be taken one hour before or two hours after eating.
7.4 Dose Modifications
The decision to interrupt or discontinue trial therapy is at the discretion of the treating physician. Doses may be interrupted or discontinued at any point during the trial period for reasons such as unacceptable adverse effects, intercurrent illness, development of serious disease or any change in the patient's condition that the physician believes warrants a change in medication (see Section 5.3.2). Any changes must be documented in the CRF along with the justification for those changes.
7.5 Accountability Procedures for Study Treatment/s
Clinical trial supplies will only be delivered to an investigator site once the site has been initiated. This can only be completed once full ethical and regulatory approvals have been granted. This must be confirmed by the Trial Coordinator acting on behalf of the study sponsor. The size of the shipments to each site will be pre-determined based on the patient recruitment target for that individual site. The first shipment will be approximately a quarter of the total trial supplies for the site and there will be a maximum of three subsequent shipments over the two year trial period. Recruitment will be monitored centrally and drug shipment dates will be tailored accordingly to ensure that pharmacies always hold adequate supplies of trial treatment. Pharmacies will document all shipment receipts and will provide copies of this documentation to the Trial Coordinator.
Following registration into the trial of an individual participant, the following accountability procedures for clinical trial supplies at pharmacy will apply:
At T–4 the RN/PI will collect an inhaler of fluticasone propionate (open label) from pharmacy and record the receipt of this by the patient on the T–4 CRF. Pharmacy will put their dispensing label (including information such as patient name or initials, pharmacy address, telephone number and date of dispensing) on to the inhaler carton and will complete, sign and date the accountability log. A second member of the pharmacy team will counter-sign the log to document the dispensing.
At T0 the patient will be randomised and a blinded treatment pack will be dispensed by pharmacy according to their treatment allocation. Pharmacy will ensure that the participant and the researcher remain blinded to the treatment allocation. Pharmacy will put their dispensing label (including information such as patient name or initials, pharmacy address, telephone number and date of dispensing) on to the pack and will complete, sign and date the accountability log. A second member of the pharmacy team will counter-sign and date the log to document the dispensing. The RN will record the participant's randomisation number in the CRF. Upon allocation, the patient's trial number will also be recorded in their individual hand held asthma record.
At each subsequent dispensing, the patient's randomised treatment allocation will be ascertained by pharmacy and a treatment pack/s dispensed on production of a valid trial prescription. Pharmacy will ensure that the participant and the researcher remain blinded to the treatment allocation. The accountability log will be updated and signed and dated by two members of the pharmacy team. Where the prescription is dispensed to the RN for transport to another site, the RN will transport the medications securely ensuring that the temperature of the products is controlled and recorded. At all study visits from T0–T+48, unused medications (including omitted doses from the previous prescription's regimen) will be collected by the RN and returned to pharmacy for destruction.
7.6 Assessment of Compliance with Study Treatment/s
Participants will be asked to retain all used and unused trial medications and packaging and bring them to each study visit from T0–T+48. The research doctor/nurse will collect the returned supplies in order to assess compliance with the trial treatment regimen. The inhalers supplied to the participants contain dose counters which show the number of doses remaining. Once the compliance checks have been completed and recorded, the RN will deliver all returned medications to pharmacy for destruction via their local procedures.
T0 study visit
All registered participants will be asked to bring the fluticasone inhaler they were issued at T–4 to the T0 study visit. The research doctor/nurse will ask the participant and their carer/s about compliance with the treatment regime and whether any doses have been missed. At the end of the study visit, after the patient has left, the researcher will use the dose counter on the inhaler to verify the information provided by the family. The number of inhalations reported by both the participant and the dose counter will be recorded on the CRF along with the reason given for any doses missed (if applicable).
T+8 study visit
The research nurse will collect all used medications and packaging from the participant. The participant will have been issued with 12 weeks of treatment at T0 so will retain one inhaler and one monthly blister pack for use over the next four weeks.
The research nurse will ask the participant and their carer/s about compliance with the treatment regime since their last visit and whether any inhalations or tablets have been missed. At the end of the study visit, after the patient has left, the researcher will use the dose counter on the inhaler and conduct a full pill count to verify the information provided by the family. The number of doses reported by the participant and from the medication counts will be recorded on the CRF along with the reason given for any doses missed (if applicable).
T+24 study visit
The research nurse will collect all used and unused medications and packaging from the participant. They will ask the participant and their carer/s about compliance with the treatment regime since their last visit and whether any inhalations or tablets have been missed. At the end of the study visit, after the patient has left, the researcher will use the dose counter on the inhaler and conduct a full pill count to verify the information provided by the family. The number of doses reported by the participant and from the medication counts will be recorded on the CRF along with the reason given for any doses missed (if applicable).
T+36 telephone call
The research nurse will ask the participant and/or their carer/s about compliance with the treatment regime since their last visit and whether any inhalations or tablets have been missed. The number of doses reported by the participant will be recorded on the CRF along with the reason given for any doses missed (if applicable). The RN will remind the participant to bring all used and unused medications and packaging to their next study visit.
T+48 study visit
The research nurse will collect all used and unused medications and packaging from the participant. They will ask the participant and their carer/s about compliance with the treatment regime since their last visit and whether any inhalations or tablets have been missed. At the end of the study visit, after the patient has left, the researcher will use the dose counter on the inhaler and conduct a full pill count to verify the information provided by the family. The number of doses reported by the participant and from the medication counts will be recorded on the CRF along with the reason given for any doses missed (if applicable).
Early withdrawal
If a patient wishes to prematurely withdraw from trial treatment, the research nurse will collect all used and unused medications and packaging from the participant. They will ask the participant and their carer/s about compliance with the treatment regime since their last visit and whether any inhalations or tablets have been missed. The researcher will use the dose counter on the inhaler and conduct a full pill count to verify the information provided by the family. The number of doses reported by the participant and from the medication counts will be recorded on the CRF along with the reason given for any doses missed (if applicable).
7.7 Concomitant Medications/Treatments
7.7.1 Medications Permitted
Details of concomitant medications will be collected at the T–4 visit and recorded on the CRF. They will be reviewed at all subsequent study visits (clinic visits, telephone call) until T+48. The trial treatments have very few adverse interactions with other medicinal products so concomitant medications, with the exception of those listed in Section 7.7.2, are permissible at the discretion of the investigator.
7.7.2 Medications Not Permitted/Precautions Required
The following are not permitted for the duration of the trial period:
-
Inhaled corticosteroids (other than the trial treatment)
-
Long-acting beta2 agonists (other than trial treatment)
-
Leukotriene receptor antagonists (other than trial treatment)
-
All beta-blockers
-
Theophylline
Caution should be exercised when prescribing CYP3A inhibitors as they may affect the efficacy of montelukast (see Singulair SPC Section 4.5 ‘Interactions with other medicinal products and other forms of interaction’). CYP3A inhibitors (e.g. ketaconazole, itraconazole) are not permitted for regular or frequent use during the trial treatment period. All prescribed CYP3A inhibitors should be documented on the Concomitant Medications CRF.
7.7.3 Data on Concomitant Medication
The dose and name of all concomitant medications should be documented on the CRF at T–4. This will be reassessed at each trial visit by the PI/RN. Any new medications introduced or any changes to current medications should be documented on the CRF.
7.8 Unblinding
Unblinding should be considered when knowledge of the treatment assignment is deemed essential for the child's care by their physician or a regulatory body. In general, unblinding of participants before they have completed their individual 48 week follow-up period should be considered when the participant has prematurely withdrawn from trial treatment or when there are compelling medical or safety reasons to do so.
N.B. If simply ceasing study treatment is a viable option for the patient's care, it should not be necessary for unblinding to occur.
7.8.1 Procedure
Emergency Unblinding
-
The decision to unblind a single case should be made when knowledge of an individual's allocated treatment is required to:
-
enable treatment of severe adverse event/s, or
-
enable administration of another therapy that is contraindicated by the trial treatment.
-
-
Where possible, requests for emergency or unplanned unblinding of individuals should be made via the Trial Coordinator at MCRN CTU. Agreement of the chief investigator (Professor Warren Lenney) will then be sought. Professor David Price will be consulted in the chief investigator's absence.
-
Contact the central pharmacy at University Hospital of North Staffordshire NHS Trust, where unblinding codes are held:
-
The central pharmacy will release the allocation details of an individual patient only, documenting:
-
Date information needed
-
Detailed reason for unblinding
-
Identity of recipient of the unblinding information
-
-
Ensure all necessary CRFs to time of unblinding are completed and submitted to MCRN CTU (if possible, completed before unblinding is performed)
-
All instances of unblinding should be recorded and reported in writing to the MCRN CTU by the local investigator, including the identity of all recipients of the unblinding information.
-
Allocation should not routinely be revealed to MCRN CTU personnel.
Accidental Unblinding
All instances of inadvertent unblinding should be recorded and reported in writing to the MCRN CTU by the local investigator. Reports to include:
-
Date of unblinding
-
Detailed explanation of circumstances
-
Recipients of the unblinding information
-
Action to prevent further occurrence
Allocation should not be routinely revealed to MCRN CTU personnel.
Routine Unblinding at the End of Follow-up
At the end of their individual 48 week follow-up period, participants will be instructed to contact their GP who will be informed which treatment allocation their patient had been randomised to. Upon receipt of a T+48 visit CRF, MCRN CTU will unblind that individual and write to the patient's GP to inform them which treatment they had been prescribed. MCRN CTU will endeavour to inform the patient's GP within seven days of the T+48 visit occurring. Where the patient was managed within secondary or tertiary care for their asthma treatment prior to entering the study, the relevant clinician or specialist nurse will be informed by the MCRN CTU that the patient has completed their involvement in the trial and advised to contact the GP if they need to know what randomised treatment the patient had been taking. The participant and their guardian will be made aware of this procedure in the patient information sheet and at their final study visit. In the interim period between the end of trial treatment at T+48 and the patient's GP being informed of their treatment allocation, patients will be treated as per local standard practice.
At Trial Closure
The end of the trial will be considered as the date of the final database lock. However the trial may be closed prematurely by the Trial Steering Committee, on the recommendation of the Independent Data and Safety Monitoring Committee.
Upon trial closure the central pharmacy department at University Hospital of North Staffordshire NHS Trust will return unblinding codes to the MCRN CTU. MCRN CTU will notify local investigators in writing of unblinding information for patients under their care. A copy of this notification should be placed in the medical records and a copy retained in the site file.
7.9 Co-enrolment Guidelines
To avoid potentially confounding issues, ideally patients should not be recruited into other trials. Individuals who have participated in a trial testing a medicinal product within one month preceding screening will be ineligible for the MASCOT study. Where recruitment into another trial is considered to be appropriate and without having any detrimental effect on the MASCOT trial this must first be discussed with the coordinating centre (MCRN CTU) who will contact the chief investigator (Professor Warren Lenney).
8 ASSESSMENTS AND PROCEDURES
8.1 Schedule of Study Visits
See schedule of study procedures, Table 2. Wherever possible, all study visits after randomisation (T0) should be conducted no earlier than one week before the appropriate date (e.g. no earlier than seven weeks after randomisation for T+8). When planning a visit which is later than the appropriate date, the research team should consider the participant's remaining trial medications and ensure they do not run out prior to the visit.
| Procedures | Follow-Up Schedule (weeks) | |||||||
|---|---|---|---|---|---|---|---|---|
| Screening (T–4) | Baseline (T0 [clinic])* | T+8 Weeks (clinic) | T+24 Weeks (clinic) | T+36 (telephone) | T+48 Weeks (clinic) Study Completion | Premature Discontinuation | ||
| Signed Consent Form | ✗ | |||||||
| Assessment of Eligibility Criteria | ✗ | ✗ | ||||||
| Quality of Life Questionnaires Administered | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Health Economics Questionnaire Completed | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| FEV1, FVC, FEV1/FVC Ratio | ✗ | ✗ | (✗) | |||||
| Review Patient Held Record | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Review of Medical History | ✗ | ✗ | ||||||
| Review of Concomitant Medications | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Study Intervention | ✗ | ✗ | ✗ | ✗ | ||||
| Physical Exam | Complete | ✗ | ✗ | |||||
| Symptom-Directed | (✗) | (✗) | (✗) | (✗) | ||||
| Vital Signs | ✗ | (✗) | (✗) | (✗) | (✗) | (✗) | ||
| Assessment of Adverse Events | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Special Assay or Procedure | Consent and obtain saliva sample for later DNA analysis | ✗ | ||||||
Due to geographical constraints, some participating centres sites may conduct certain study visits at ‘satellite’ sites (e.g. GP practices) in their region. Patients will be seen for study visits at these locations by the research team if they are unable to travel to their lead local centre. Patients will remain under the care of the local principal investigator for their region, who will be based at the lead site. Trial medications will be dispensed from the pharmacy department at the lead centre. For all satellite sites research governance approval will first be sought from the appropriate Trust and the main REC will also be notified.
Four Week Run-in Period (Study visit)
Patients will be screened in GP surgeries in primary care and in paediatric clinics in secondary care. Following full informed written (proxy) consent, those eligible will be registered into the study, have their inhaler technique checked and be provided with information about asthma and its management. All research centres taking part will be centrally trained and instructed in the approach to the patients and their families in an attempt to obtain uniformity. They will all be dispensed the same low-dose inhaled corticosteroid, fluticasone propionate, in the dose of 100 micrograms twice daily. They will participate in an open four week ‘run-in’ period and will complete a hand-held patient record that will provide information to aid assessment of ongoing control (see 8.2.1).
The same criteria will be used in all centres to determine whether the patient is effectively controlled or not. Poorly controlled, as defined in sections 5.1 and 5.2, will be those requiring frequent short-acting beta2 agonist relief therapy ≥ 7 puffs per week and with asthma symptoms affecting sleeping and/or usual activities in the last week and/or who have had exacerbations (defined as a short course of oral corticosteroids, an unscheduled GP or A&E Department visit or a hospital admission within the previous six months).
The purpose of this run-in period is to ensure that we are only recruiting those patients for whom control of their asthma presents a problem, rather than those for whom inhaler technique and management advice will be sufficient to provide symptomatic relief. Most run-ins lose approximately 25% patients but we anticipate that improved education and attention to compliance in this study may well make up to 50% ineligible for entry into the randomised part of the study. All patients registered will have GP data follow-up one year after registration, regardless of continuation into the main trial, which is detailed in the patient information sheet and consent form (PISC). We will collect data on things such as symptoms, exacerbations, hospital appointments, medication changes and use.
At the T–4 review, families will also be issued with a copy of the PISC requesting the collection of a DNA sample for storage and investigation at a later date (section 8.4.1). They will be invited to consent and provide a DNA sample (saliva) at their T0 clinic visit. Consent to provide a DNA sample is documented separately to that of consent for the main trial.
The next study visit (T0) will be organised with the participant and their carer/s to be in no less than 24 days time and no longer than 30 days time from T–4.
Time 0: Entry into full study (Study visit)
Following the run-in period patients will be re-assessed for study entry based upon inclusion and exclusion criteria (section 5.2). Those patients achieving the threshold criteria for T0 will be entered into the randomised part of the study.
Symptoms, exacerbations and beta2 agonist use will be ascertained by reviewing the hand held record with the patient and their carer/s. Baseline Paediatric Asthma Quality of Life Questionnaire (interviewer administered if child is 10 years of age or younger, patient administered if aged 11 years or more) and Paediatric Asthma Caregivers Quality of Life Questionnaire (carer administered) assessments will be conducted and a complete physical examination will be performed, including height and weight measurements. Spirometry will be carried out to measure the patient's FEV1, FVC and FEV1/FVC ratio (best of three before and after bronchodilator).
Each patient is then randomised by pharmacy using a randomisation list supplied centrally and dispensed their first three month treatment pack according to their treatment allocation. Treatment is to continue for eight weeks and the allocated treatment will be double-blinded, achieved by using identical inhalers and placebo tablets, with patients allocated to receive either:
-
Inhaled fluticasone propionate 100 micrograms twice daily plus placebo tablet once daily
-
Inhaled fluticasone propionate 100 micrograms and salmeterol 50 micrograms twice daily (combination inhaler) plus placebo tablet once daily
-
Inhaled fluticasone propionate 100 micrograms twice daily plus montelukast 5 mg tablet once daily.
Following a separate consent process, a DNA specimen will be obtained (see Section 8.4.1). Individuals declining to provide DNA will not be precluded from entry into main trial.
Randomisation + 8 weeks (Study visit)
Symptoms, exacerbations and beta2 agonist use will be ascertained by reviewing the hand held record with the patient and their carer/s. The Health Economics questionnaire completed throughout the time period since the last clinic visit will be checked for completeness and removed. A new, blank questionnaire will be inserted into the hand held record to be used until the next appointment. Repeat Quality of Life Assessments will be administered (interviewer [if child is 10 years of age or younger], patient [if aged 11 years or more] and carer administered) and a symptom-directed physical examination will be performed as appropriate. Adverse events will be reported and recorded.
Those who have achieved control of their asthma symptoms will continue on the same treatment for the next 16 weeks. For those whose symptoms have not improved but are no worse, the PI/RN will discuss their willingness to continue with randomised treatment.
Those who are clinically worse may be withdrawn from randomised treatment and given alternative treatment according to clinician's advice as in routine practice. The decision to withdraw the patient from trial treatment is based on the patient's current clinical presentation and the review of information on symptoms/exacerbations etc collected in the hand held record over the preceding weeks. The decision is made at the discretion of the investigator using their informed clinical opinion. The reason for discontinuation of trial treatment must be documented on the CRF. Follow-up should be continued until the end of the trial as per the study visit schedule.
In certain circumstances, the PI/RN may conduct the T+8 study visit at the family home where it is appropriate and necessary to do so. The PI/RN must only conduct a study visit in the family home if this has been authorised for their site by the coordinating centre and appropriate lone working procedures are in place.
Randomisation + 24 weeks (Study visit)
Symptoms, exacerbations and beta2 agonist use will be ascertained by reviewing the hand held record with the patient and their carer/s. The Health Economics questionnaire completed throughout the time period since the last clinic visit will be checked for completeness and removed. Two new, blank questionnaires will be inserted into the hand held record to be used until the next study visit. Repeat Quality of Life Assessments will be administered (interviewer [if child is 10 years of age or younger], patient [if aged 11 years or more] and carer administered) and a symptom-directed physical examination will be performed as appropriate. Adverse events will be reported and recorded.
Those who have achieved control of their asthma symptoms will continue on the treatment. For those whose symptoms have not improved but are no worse, the PI/RN will discuss their willingness to continue with randomised treatment.
Those who are clinically worse may be withdrawn from randomised treatment and given alternative treatment according to clinician's advice as in routine practice. The decision to withdraw the patient from trial treatment is based on the patient's current clinical presentation and the review of information on symptoms/exacerbations etc collected in the hand held record over the preceding weeks. The decision is made at the discretion of the investigator using their informed clinical opinion. The reason for discontinuation of trial treatment must be documented on the CRF. Follow-up should be continued until the end of the trial as per the study visit schedule.
In certain circumstances, the PI/RN may conduct the T+24 study visit at the family home where it is appropriate and necessary to do so. The PI/RN must only conduct a study visit in the family home if this has been authorised for their site by the coordinating centre and appropriate lone working procedures are in place.
Randomisation + 36 weeks (Telephone call)
Symptoms, exacerbations and beta2 agonist use will be ascertained from the patient and/or their carer/s. The RN will check that they are continuing to complete the hand held record and that they have completed the inserted Health Economics questionnaire for the time period since their last clinic visit. They will be asked to remove the completed questionnaire and place it to the back of the record to avoid any confusion. The RN will remind them to begin a new Health Economics questionnaire to be used until their next appointment.
Adverse events will be reported and recorded. Those who have achieved control of their asthma symptoms will continue on the trial treatment. For those whose symptoms have not improved but are no worse, the RN will discuss their willingness to continue with randomised treatment. Those whose asthma symptoms appear to be worse will either be offered an unscheduled study appointment with a member of the research team (if possible) or advised to visit their General Practitioner to seek further medical advice. If the practitioner believes they are clinically worse the patient may be withdrawn from randomised treatment and given alternative treatment according to clinician's advice as in routine practice. The reason for discontinuation of trial treatment must be documented on the CRF. Follow-up should be continued until the end of the trial as per the study visit schedule.
Randomisation + 48 weeks (Clinic visit)
Symptoms, exacerbations and beta2 agonist use will be ascertained by reviewing the hand held record with the patient and their carer/s. The two Health Economics questionnaires completed throughout the time period since the last clinic visit will be checked for completeness and removed. Final Quality of Life assessments will be administered (interviewer [if child is 10 years of age or younger], patient [if aged 11 years or more] and carer administered) and a basic physical examination will be performed, including height and weight measurements. Further examination will be symptom led. Spirometry will be carried out to measure the patient's FEV1, FVC and FEV1/FVC ratio (best of three before and after bronchodilator). Adverse events will be reported and recorded.
Patients will be asked to provide current details for their General Practitioner (GP). They will be informed that their GP will be provided with details of which treatment they have been taking within seven days. If the patient was under the care of a different clinician for their asthma management prior to entering the study, that clinician will also be provided with details of the treatment wherever possible. The PI/RN will discuss future management with patients and their carer/s.
8.2 Procedures for assessing Efficacy
Efficacy of trial treatments will be assessed throughout the period of the study using both objective and subjective measures.
8.2.1 Hand Held Records
Hand held records will be used throughout by participants, from T–4 through to T0, and continuing until study completion (T+48) for those patients who proceed with randomisation at T0. The records are A6 sized folders, which are divided into sections relating to different aspects of a child's asthma and their management of it. They were developed by The Guy Hilton Asthma Trust and have been modified for the purposes of the MASCOT trial to collect data relevant to the trial outcomes.
There are sections in the records to capture information on daily symptoms, exacerbations, use of beta2 agonist relief therapy, management/treatment schedule and emergency contact numbers. The records can be completed by patients and/or their carer/s on a continuous basis so the information recorded will be current and accurate.
The records will be brought along to every study visit and reviewed by the PI/RN in conjunction with the patient and their carer/s. The information collected in the records will be used to assess how well participants' asthma is being controlled on the trial treatments and will be important in determining whether they should progress to the next stage of the trial. The information collected in the hand held records will be regarded as source data so at each clinic visit the completed pages will be removed from the record.
The original sheets will be sent to CTU, a copy retained in the Site File and the family will be provided with a copy for their own records (if requested).
The review of hand held records at each study visit, detailing patients' symptoms and exacerbations, will indicate how effectively their asthma is being controlled and so can be used to assess efficacy of the trial treatments.
8.2.2 Spirometry
Spirometry will be carried out at baseline (T0) and study completion (T+48) and will provide an objective measure of efficacy. Spirometry will be undertaken pre and post bronchodilator and will only be accepted if meeting American Thoracic Society (ATS)/European Respiratory Society (ERS) standards for acceptability and reproducibility. The following values will be recorded in the CRF:
-
Date and time of spirometry
-
Whether readings meet ATS standards for acceptability and reproducibility
-
Whether bronchodilators withheld for appropriate length of time – short-acting 4 hours and long-acting 24 hours
-
FEV1 pre and post 400 micrograms of salbutamol
-
FVC pre and post 400 micrograms of salbutamol
8.2.3 Paediatric Asthma Quality of Life Questionnaire (PAQLQ)
The Quality of Life (QoL) scores obtained at different timepoints (T0, T + 8, T + 24 and T + 48) throughout the trial can be used as a subjective measure of efficacy. The PAQLQ, devised by Elizabeth Juniper, will measure the physical, emotional, occupational and social effects of asthma in children. There are two versions of the PAQLQ, interviewer administered for children 10 years & younger and patient administered for children 11 years & older. The RN will select the version appropriate for the individual child's age and stage of development. Children will be asked to recall information from the previous seven days and to use this when selecting their responses. The child's asthma may also affect the parents' quality of life and so an additional scale, the Paediatric Asthma Caregivers Quality of Life Questionnaire (PACQLQ), will also be used. The PACQLQ has been designed by Elizabeth Juniper to complement the PAQLQ.
The PAQLQ has been selected for use in MASCOT due to its short recall requirement and ability to detect small but clinically important changes that children experience as a result of treatment or fluctuation in their asthma. (See Section 8.5 for procedure).
8.3 Procedures for Assessing Safety
An assessment of adverse events will be undertaken at each study visit from baseline (T0) to study completion (T + 48). These reviews will be carried out by the PI or RN conducting the visit or telephone call.
Adverse event reporting is detailed fully in Section 10 (Pharmacovigilance).
8.4 Substudies
8.4.1 Genetic Study
The study will include the collection of DNA (from saliva) which will be securely stored at the University of Liverpool for pharmacogenetic analysis at a later date.
A separate consent process will be undertaken for this purpose at the T0 visit. All participants registered in the trial will be asked to provide a sample, regardless of whether they are eligible for the randomised phase. If consent and assent (where applicable) is provided at T0, the genetic sample will be collected by asking the child to spit directly into a collection kit. The kit is specifically designed for the preservation, transportation and purification of DNA from saliva and is a non-invasive, highly reliable method of DNA collection. The amount of saliva required from the participant is 2 ml so the child may have to spit more than once in order to obtain a sufficient amount of material for DNA extraction. Once the saliva has been deposited, the RN will then seal and label the container with the child's unique trial identifier number. When the kit has been sealed the sample will remain stable at room temperature. It will be sent directly to a central facility at the University of Liverpool where it will be stored for future study.
The genetic samples collected will be used in the future to help determine whether specific polymorphisms affect severity or long term prognosis of asthma and its symptoms. For the patients who enter the randomised part of the trial, the samples will also be used to explore the possibility that a difference may be seen in those children responding to long acting beta2 agonists (LABAs) or leukotriene receptor antagonists (LTRAs) dependent on specific single nucleotide polymorphisms (SNPs) within the beta2 agonist receptor or the leukotriene receptor genes. However, no specific study is planned at this time. Approval from a Research Ethics Committee will be sought before any pharmacogenetic analyses are conducted.
8.4.2 RECRUIT
It is proposed that MASCOT will involve a qualitative substudy ‘Processes in recruitment to randomised controlled trials (RCTs) of medicines for children (RECRUIT)’. RECRUIT was approved in its own right by the North West REC on 2nd March 2007 (ref 07/MRE08/6).
RECRUIT will be examining communication processes in the recruitment of participants to MASCOT with the aim of identifying strategies for subsequent trials of medicines for children to improve trial recruitment and conduct. RECRUIT will involve:
-
Routine audio-recording of MASCOT discussions (consultations) between families and practitioners (trial recruiters)
-
Follow-up interviews with up to eight families (parents and children, where aged seven or over) who agree to participate in MASCOT
-
Follow-up interviews with up to eight families (parents and children, where aged seven or over) who decline to participate in MASCOT
-
Follow-up interviews with up to eight trial recruiters involved in approaching families to take part in MASCOT
Collection of data for 1 will be facilitated by MASCOT staff who will routinely seek permission to audio-record recruitment consultations from the families whom they approach for MASCOT. Data for 2, 3 and 4 will be collected by the Research Associates (RAs) employed on RECRUIT, who will be entirely independent of MASCOT.
If permission for audio-recording is declined by a family the recruitment consultation will not be recorded. If permission is given the recruiter will activate an audio-recorder. At the end of the MASCOT recruitment consultation the recruiter will discuss RECRUIT with the family and seek their permission to pass their details to one of the RAs employed on RECRUIT, who will then make contact with families and obtain written informed consent for participation in the RECRUIT study. Recordings from families who decline RECRUIT will be erased as soon as practicable. All families who express an interest in RECRUIT but are not selected for follow-up interview will be contacted by letter to thank them and inform them that their recordings have been erased. Audio-recordings of the recruitment consultations will only be released to the RECRUIT RAs after the consent of the participants has been obtained.
All interviews for RECRUIT will be conducted by experienced RAs with proven skills in the conduct of research in sensitive settings. Any distress during the interviews will be managed with care and compassion by the RAs and participants will be free to decline to answer any questions that they do not wish to answer or to stop the interview at any point. The RAs will receive appropriate training and follow a clear protocol for managing participants whose level of distress gives cause for concern. Any such families will be supported in obtaining appropriate help. If necessary, and after discussion with the participant, the lead clinician responsible for the child's care will be informed.
To allow MASCOT to become established and avoid the initial ‘teething’ phase that most trials experience, sampling for RECRUIT will not begin until the trial has been recruiting for approximately four months. Sampling to RECRUIT (and therefore audio-recording of trial consultations) will roll from trial site to trial site in blocks of up to three months' duration, with planned suspensions if accrual to RECRUIT allows. This will help to minimise the numbers of families who are approached but not selected for RECRUIT. Concentrated sampling at particular sites in time-limited blocks, with the possibility of planned suspensions, will minimise the impact of RECRUIT on MASCOT and the risk of overburdening particular trial sites. It will also facilitate liaison with the sites and assist recruiters in routine audio-recording of consultations.
8.5 Other Assessments
Childhood asthma represents a significant economic burden to the health service, in terms of treatment and hospital admissions, and to society (the child and their carers) in terms of quality of life; lost schooling and lost time from productive employment (that is time off work caring for a sick child)14.
An economic analysis will be conducted primarily from a health service perspective but extended to examine a societal perspective by including the child and his/her carer's viewpoint. Both the child and carer's quality of life perspective can be measured using the Paediatric Asthma Quality of Life and Paediatric Asthma Caregivers Quality of Life questionnaires devised by Elizabeth Juniper. Further effects on the carer can be measured by adding additional questions for the carer concerning time lost to paid employment as a result of caring for the child suffering an asthma exacerbation.
The three interventions will be evaluated in terms of their costs and benefits.
Key costs will be use of health service resources; this will include the drugs, visits to general practitioners, home visits from primary health care workers, hospital admissions and A&E attendances. Primary care costs will be a key cost driver in this evaluation. The analysis will be extended to measure costs as they accrue to the patient and their carer. Additional questions will be administered concerning time lost to paid employment due to caring for a child with an asthma related illness. The health economics questionnaire will be lodged in the hand held record at T0, and collected from there at all data collection time points from T+8 to T+48. The data required on resources requires actual recollection of events, so patients and carers will be encouraged to complete the questionnaire on an ongoing basis.
The important measures of outcome will be exacerbations, quality of life, time off school for the child and time off work for parents and carers. The study will seek to measure outcome from both an effectiveness perspective e.g. reduction in exacerbations and reduction in visits to primary care and hospital admissions and from a utility perspective the Juniper measure of quality of life in children. Key outcome measures will be considered alongside the costs.
8.5.1 Health Economics
Health economic data will be collected on primary and secondary health care contacts and medications prescribed. Although not the primary focus of the study, it will aim to incorporate patient and societal costs in terms of time lost to school by the children and time lost to work for the parents and carers. It will include any out of pocket expenses such as personal money spent on medications and aids and appliances. It is important to determine differences in the patient pathway as a result of different regimes.
The four week run-in period can be ignored in terms of the economics as the costs will be common across all patients.
For participants entering the randomised phase, a blank Health Economics (HE) questionnaire will be inserted into their hand held record at each visit for the patient and their carer/s to complete over the following weeks up until their next appointment. This should increase the reliability and accuracy of the data collected by decreasing the family's reliance on retrospective recall of information. The family will receive a telephone call from the RN prior to their visit to remind them to complete and bring along the record and questionnaire for review at their next study appointment. During the visit, the HE questionnaire pages will be removed from the record by the RN. The original will be sent to CTU within seven days and copies made for the Site File and provided to the family (if requested). A new, blank questionnaire will then be inserted into the record for use until the next contact with the study team. At T+24 weeks an extra copy of the HE questionnaire will be inserted into the back of handheld record. At the telephone contact at T+36 weeks, patients/carers will be asked to swap the two questionnaires over and enter the start date on the new questionnaire. At T+48 both questionnaires will be collected from the hand held record.
8.5.2 Quality of Life
The Paediatric Asthma Quality of Life Questionnaire (PAQLQ) should be the first questionnaire completed during the clinic visit and should precede any discussion with the research doctor or nurse. Ideally, children should be interviewed on their own and in a quiet room where there are no distractions. Parents/carers should be instructed to wait in another room if possible, where they can complete the Paediatric Asthma Caregiver's Quality of Life Questionnaire (PACQLQ) while the child completes the PAQLQ. The child should be made to feel comfortable and relaxed but be aware that they need to complete the questionnaire carefully. Where the PAQLQ is interviewer administered (i.e. for children ten years of age and younger) then the questionnaires should be read out exactly as they appear on the form. They should not be reworded or paraphrased for the child, even if they ask for clarification. Children will be asked to base their responses on the past seven days and adults on the previous two weeks.
Two coloured cards (green and blue) will be provided with the PAQLQ, which list sets of response options appropriate to the different questions. The appropriate response card is listed with each question. The interviewer should explain both cards to the child, reading through each of the response options with the younger children and asking the older ones to read each of the responses aloud to ensure they understand them. Children should be reminded that they are only able to choose one option. The PACQLQ is in a self-administered format and carers should be instructed to follow the instructions on the questionnaire.
The research nurse should ensure that all questions have been answered. Once they have collected the completed questionnaires from the participant and their carer, they will make a copy and send the originals to CTU within seven days. The copy will be retained in the Site File. The questionnaires should, wherever possible, only be completed during the study visit .They must always be completed by the same caregiver who completed the first questionnaire and this should be the child's main carer.
8.6 Loss to Follow-up
If any of the trial patients are lost to follow up, contact will initially be attempted through the research nurse and lead investigator at each centre. If the lead investigator at the trial centre is not the patient's usual clinician responsible for their speciality care then follow-up will also be attempted through this latter clinician. Where these attempts are unsuccessful, the child's GP will be asked to contact the family and provide follow-up information to the recruiting centre. This information will be included on the patient information sheet. Wherever possible, information on the reason for loss to follow-up will be recorded.
8.7 Trial Closure
The end of the trial will be considered as the date of the final database lock. However the trial may be closed prematurely by the Trial Steering Committee, on the recommendation of the Independent Data and Safety Monitoring Committee.
9 STATISTICAL CONSIDERATIONS
9.1 Introduction
A separate and full statistical analysis plan (SAP) will be developed prior to the final analysis of the trial. The SAP will be agreed by the trial steering committee before being sent to the independent data and safety monitoring committee for comment and approval. The main features of these planned statistical analyses are included here in the main protocol.
9.2 Method of Randomisation
The randomisation code list will be generated by an MCRN CTU (who is not involved with the MASCOT study) with the software package STATA using block randomisation with variable block length. Randomisation will be stratified by Secondary Care Centre with allocation to the three treatment arms in the ratio of 1 : 1 : 1.
9.3 Outcome Measures
9.3.1 Primary
Number of asthma exacerbations requiring treatment with short courses of oral corticosteroids over 48 weeks from date of randomisation.
9.3.2 Secondary
-
Quality of Life as measured by the Paediatric Asthma Quality of Life Questionnaire (PAQLQ) and the Paediatric Asthma Caregivers Quality of Life Questionnaire (PACQLQ)13
-
Time from randomisation to first exacerbation requiring treatment with a short course of oral corticosteroids
-
School attendance
-
Hospital admissions
-
Amount of rescue beta2 agonist therapy prescribed
-
Time from randomisation to treatment withdrawal (due to lack of efficacy or side effects)
-
Lung function at 48 weeks (as assessed by spirometry)
-
Cost effectiveness
-
Adverse events
9.4 Sample Size
The primary outcome ‘Number of asthma exacerbations requiring treatment with short courses of oral corticosteroids over 48 weeks’ will be modelled as a poisson random variable. The sample size for the primary outcome is estimated using the following formula as described by Friede and Schmidli (personal communication):
where nc is the number of patients in the control arm, λc is the control group rate, t is the length of follow-up and θ* the rate ratio. The formula above does not allow for overdispersion which would lead to an inflated sample size. A method of moments estimator for the dispersion parameter, where values greater than 1 indicates overdispersion, is given by:
For 1032 children who have had at least one course of oral steroid in the previous 12 months, the mean exacerbation rate per year is estimated from the UK General Practice Research Database as 1.5 per year with variance 1.02 and dispersion parameter 0.68. This is our current best estimate of exacerbation rate and dispersion parameter but may not be entirely representative of the MASCOT randomised population who will have had inhaler technique corrected. Therefore, a target sample size is estimated here with the intention of undertaking an internal blinded pilot to check parameter assumptions and adjust sample size if required. As described by Friede and Schmidli (personal communication), analogous formulae to those above based on the overall event rate across groups can be used to undertake a blinded sample size review (see section 9.5).
As there are three primary treatment comparisons, each will be tested at the two-sided significance level of 1.7% to adjust for the multiplicity and to give a study-wise 5% two-tailed significance level. To have power of 80% to detect, as significant, at least a 30% reduction in exacerbation rate (from 1.5 per year to 1.05 per year, equivalent to a rate ratio of 0.7) and allowing for a loss to follow up of 10%, 147 patients per group are required.
Thirteen main centres will participate in this study, with an anticipated total accrual for screening of around 75 patients per month across these sites, enabling recruitment of approximately 900 children over 12 months. Assuming either 50% or 75% of children to be eligible for randomisation after the 4 week run-in period would enable either 150 or 225 children to be randomised to each treatment group. The exact percentage of children who will be eligible for randomisation following the 4 week run-in period cannot be estimated yet but will be monitored closely to assess the likely impact on recruitment figures (see interim analysis section).
In summary, our preliminary target number to be randomised is 150 children per treatment group (450 total) with recruitment rates, percentage of children randomised and parameters for sample size calculations closely monitored.
Secondary outcome ‘Quality of Life’: With 150 children in each group, the power to detect, as significant, a difference of 0.5 points between treatment groups on the Juniper Paediatric Asthma Quality of Life Questionnaire, with assumed standard deviation 0.7113 is greater than 99%.
9.5 Interim Monitoring and Analyses
The estimate of overall exacerbation rate (not split by treatment group) and dispersion parameter will be checked after the first 75 children have been randomised and completed their 24 week follow-up assessment. This blinded internal pilot is anticipated to be undertaken at approximately 32 weeks after the first child is randomised. This figure has been chosen to provide adequate data for the sample size review but to ensure that the review is undertaken before the recruitment period ends. This blinded internal pilot will be reviewed by the Independent Data and Safety Monitoring Committee (IDSMC) prior to reviewing any unblinded comparative data. The initial analysis of trial data for IDSMC review is planned at 6 months after the first patient is randomised, to assess recruitment rates (including the percentage of children randomised after the 4 week run-in period) and toxicity.
Subsequent timing of the next analysis of the data will be determined on the basis of recruitment rates at the initial IDSMC meeting although it is anticipated that this will be approximately after a further 6 months. Additional interim analyses may also be triggered by a concern regarding Suspected Unexpected Serious Adverse Reactions (SUSARs).
It is noted that primary outcome data may not be available for the early interim analyses because of the time necessary (48 weeks) to follow-up participants to observe the end point. Any lack of important information will be taken into account by the IDSMC in their monitoring of the trial.
In order to estimate relative treatment effects for the primary efficacy outcome at each interim and final analysis, the Haybittle-Peto approach will be employed for 2 equally spaced interim analyses, planned after 1/3 and 2/3 of the target total number of children have been randomised and followed for 48 weeks, with 99.9% confidence intervals calculated for the difference between each pair of drugs. The final analysis will be undertaken after the final randomised child has completed 48 weeks follow-up and 95% confidence intervals will be calculated. This method has been chosen to ensure that interim efficacy results would have to be extreme before early termination is recommended in order to be convincing to the clinical community. The method also minimises controversy regarding interpretation of the results from estimation and hypothesis testing at the final analysis. No inflation factor needs to be applied to the sample size using this approach.
All interim analysis results of primary and secondary outcomes will be confidential to the IDSMC members and will not be for review by the trial management group (except the statistical team preparing the IDSMC report). The IDSMC members will make formal recommendations to the trial working group and TSC regarding the continuation of recruitment of patients into the study and will comply with a trial-specific IDSMC charter according to ICH GCP guidelines. The IDSMC may recommend to the TSC that the trial be stopped or amended if sufficient evidence emerges that one treatment is clearly indicated or contra-indicated, as considered by the IDSMC in light of the analyses presented, accounting for other emerging worldwide evidence and overall clinical relevance. The IDSMC will be asked to consider patient safety, particularly any Suspected Unexpected Serious Adverse Reactions (SUSARs) leading to death, alongside treatment efficacy when making their recommendation regarding continuation, amendment or discontinuation of the trial.
9.6 Analysis Plan
The study will be analysed and reported using the ‘Consolidated Standard of Reporting Trials’ (‘CONSORT’)15,16 and the ICH E9 guidelines.
A full and detailed statistical analysis plan will be written prior to the conduct of the final analysis according to the MCRN CTU Statistical Analysis Plan SOP. To provide a pragmatic comparison of the policies of the different treatments, primary analyses will be performed on an intention to treat basis, analysing all patients according to the treatment group originally allocated. A sensitivity analysis using the per protocol or complier average causal effect (CACE) population as appropriate will be explored to demonstrate robustness of the results. For the overall safety and tolerability assessment, the set of patients to be summarised will be defined as those subjects who received at least one dose of the study drug.
Missing data will be handled by considering the robustness of the complete case analysis to sensitivity analyses using different imputation assumptions informed by data collected on reasons for missing data. Data relating to the primary outcome (oral steroid usage) for those patients who fail to attend for the 48 week assessment visit can be obtained from their GP.
The primary outcome of number of asthma exacerbations requiring treatment with short courses of oral corticosteroids over 48 weeks will be compared between treatment groups using a Poisson regression model (adjusted for centre) with two dummy variables representing 3 treatment groups. Adjustment for overdispersion will be made if appropriate. A secondary analysis adjusting the model for other important prognostic factors (age, sex, duration of asthma, rhinitis, baseline AQLQ, baseline number of courses of oral steroids) will be explored. Statistical tests will be nominally performed at the 1.7% two-sided significance level (with corresponding 98.3% confidence interval), to give a study-wise 5% two-sided significance level (see sample size calculation). The relative exacerbation rate and confidence interval will be calculated for each pair-wise treatment comparison: A compared with B to evaluate the effect of adding in long acting beta-2 agonist (salmeterol 50 micrograms twice daily); A compared with C to evaluate the effect of adding in oral leukotriene receptor antagonist (montelukast 5 mg once daily); and B compared with C to evaluate the effect of adding in long acting beta-2 agonist (salmeterol 50 micrograms twice daily) compared with adding in oral leukotriene receptor antagonist tablet (montelukast 5 mg once daily).
For secondary outcomes statistical tests will be performed at the 5% two-sided significance level (with corresponding 95% confidence interval) unless indicated otherwise. Analyses will adjust by centre and secondary analyses adjusting for other important prognostic factors (age, sex, duration of asthma, rhinitis, baseline AQLQ, baseline number of courses of oral steroids) will be explored. For the secondary outcome quality of life, characteristics of responders and non-responders will be compared and potential biases assessed. The score on three domains of activity, symptoms and emotion measured by the Juniper Quality of Life scale, will be assessed over time and treatment groups compared using longitudinal analysis. A joint modelling approach where time to dropout is taken as the time to event outcome will also be used to explore relationships between dropout and outcome.
Time to first exacerbation requiring treatment with a short course of oral corticosteroids will be calculated for each child and compared across treatment groups using Kaplan-Meier curves and log-rank test with relative effects of treatments summarised using hazard ratios and 95% confidence intervals. The seasonality of having an asthma exacerbation episode will be explored by fitting season as a time-dependent covariate in the model for time to first exacerbation.
Time to withdrawal of randomized treatment will be calculated for each child and compared across treatment groups using Kaplan-Meier curves and log-rank test with relative effects of treatments summarised using hazard ratios and 95% confidence intervals. To examine different reasons for treatment withdrawal (competing risks of unacceptable adverse effects and inadequate asthma control) a cumulative incidence analysis will be undertaken. This approach does not assume competing risks are independent and allows the assessment of cause-specific withdrawal in the presence of other competing risks. All adverse events will be categorised appropriately and the number of patients with occurrences of each event summarised according to treatment group supplemented by calculation of confidence intervals wherever this aids interpretation.
The percentage of school days missed (transformed if appropriate) will be analysed and compared between treatment groups using analysis of variance. The number of hospital admissions will be summarised and compared between treatment groups using bootstrapping techniques which allow comparisons between groups to be based upon the mean difference while making allowance for the non-normal distribution in the calculation of confidence intervals and p-values (Metcalfe et al 200317). Lung function at 48 weeks will be compared between treatment groups using analysis of covariance (adjusted by baseline lung function). Number of doses of rescue beta2 agonist therapy prescribed over 48 weeks (transformed if appropriate) will be analysed and compared between treatment groups using analysis of variance. Blinded distribution data will be reviewed and the statistical analysis plan amended accordingly.
9.6.1 Economic analysis
Economic analysis will focus on determining the differences in the patient pathways between the three groups in terms of their costs and benefits. Analysis will therefore take a number of different forms.
Incremental Cost Effectiveness Ratios (ICER) will be calculated against the base case:
-
A Inhaled fluticasone propionate 100 micrograms twice daily plus placebo tablet once daily.
The other two scenarios:
-
B Inhaled fluticasone propionate 100 micrograms and salmeterol 50 micrograms twice daily (combination inhaler) plus placebo tablet once daily.
-
C Inhaled fluticasone propionate 100 micrograms twice daily plus montelukast 5mg tablet once daily will be compared against the base case.
ICERs calculate the ratio of the difference in cost to the difference in outcome between the two groups. In terms of outcome, the ICER will be based on the difference in the number of exacerbations between the groups. Further ICERs based on quality of life could also be calculated.
Cost effectiveness acceptability curves (CEACs) will be calculated for each of the three regimes showing the probability that each option is cost effective at different willingness to pay thresholds.
A subsidiary economic analysis may evaluate the cost of exacerbations in terms of the average cost per exacerbation and then cost the treatment alternatives in terms of exacerbations averted. This subsidiary analysis is a useful supplement to the main analysis. Cases averted are a useful tool for presentation of the data.
10 PHARMACOVIGILANCE
10.1 Terms and Definitions
The Medicines for Human Use (Clinical Trials) Regulations 2004 (SI 2004/1031) definitions:
Adverse Event (AE)
Any untoward medical occurrence in a subject to whom a medicinal product has been administered, including occurrences which are not necessarily caused by or related to that product.
Adverse Reaction (AR)
Any untoward and unintended response in a subject to an investigational medicinal product which is related to any dose administered to that subject.
Unexpected Adverse Reaction (UAR)
An adverse reaction the nature and severity of which is not consistent with the information about the medicinal product in question set out in:
-
In the case of a product with a marketing authorization, in the summary of product characteristics for that product
-
In the case of any other investigational medicinal product, in the investigator's brochure relating to the trial in question.
Serious Adverse Event (SAE), Serious Adverse Reaction (SAR) or Suspected Unexpected Serious Adverse Reaction (SUSAR)
Any adverse event, adverse reaction or unexpected adverse reaction, respectively, that:
-
results in death
-
is life-threatening* (subject at immediate risk of death)
-
requires in-patient hospitalisation or prolongation of existing hospitalisation**
-
results in persistent or significant disability or incapacity, or consists of a congenital anomaly or birth defect
-
Other important medical events
*‘life-threatening’ in the definition of ‘serious’ refers to an event in which the patient was at risk of death at the time of the event; it does not refer to an event which hypothetically might have caused death if it were more severe.
**Hospitalisation is defined as an inpatient admission, regardless of length of stay, even if the hospitalisation is a precautionary measure for continued observation. Hospitalisations for a pre-existing condition, including elective procedures that have not worsened, do not constitute an SAE.
***Other important medical events that may not result in death, be life-threatening, or require hospitalisation may be considered a serious adverse event/experience when, based upon appropriate medical judgment, they may jeopardise the subject and may require medical or surgical intervention to prevent one of the outcomes listed in this definition
10.2 Notes on Adverse Event Inclusions and Exclusions
10.2.1 Include
-
An exacerbation of a pre-existing illness
-
An increase in frequency or intensity of a pre-existing episodic event/condition
-
A condition (even though it may have been present prior to the start of the trial) detected after trial drug administration
-
Continuous persistent disease or symptoms present at baseline that worsens following the administration of the study/trial treatment
10.2.2 Do Not Include
-
Medical or surgical procedures – the condition which leads to the procedure is the adverse event
-
Pre-existing disease or conditions present before treatment that do not worsen
-
Situations where an untoward medical occurrence has occurred e.g. cosmetic elective surgery
-
Overdose of medication without signs or symptoms
-
The disease being treated or associated symptoms/signs unless more severe than expected for the patient's condition
10.2.3 Reporting of Pregnancy
Study participants will not routinely be tested for pregnancy as part of the trial screening process. Any pregnancy which does occur during the course of the study should be reported to the MCRN CTU immediately. The investigator should discuss the risks of continuing with the pregnancy with the patient and the possible effects on the foetus if they continue on trial treatment. It is at the investigator's discretion to decide whether the individual should be instructed to stop taking study drugs. All pregnancies that occur during trial treatment, or within seven days of finishing treatment, need to be followed up until completion and reported separately.
10.3 Notes Severity/Grading of Adverse Events
The assignment of the severity/grading should be made by the investigator responsible for the care of the participant using the definitions below.
Regardless of the classification of an AE as serious or not, its severity must be assessed according to medical criteria alone using the following categories:
-
Mild: does not interfere with routine activities
-
Moderate: interferes with routine activities
-
Severe: impossible to perform routine activities
A distinction is drawn between serious and severe AEs. Severity is a measure of intensity (see above) whereas seriousness is defined using the criteria in section 10.1, hence, a severe AE need not necessarily be a Serious Adverse Event.
10.4 Relationship to Trial Treatment
The assignment of the causality should be made by the investigator responsible for the care of the participant using the definitions in Table 3.
| Relationship | Description |
|---|---|
| Unrelated | There is no evidence of any causal relationship. N.B. An alternative cause for the AE should be given |
| Unlikely | There is little evidence to suggest there is a causal relationship (e.g. the event did not occur within a reasonable time after administration of the trial medication). There is another reasonable explanation for the event (e.g. the participant's clinical condition, other concomitant treatment) |
| Possibly | There is some evidence to suggest a causal relationship (e.g. because the event occurs within a reasonable time after administration of the trial medication). However, the influence of other factors may have contributed to the event (e.g. the participant's clinical condition, other concomitant treatments) |
| Probably | There is evidence to suggest a causal relationship and the influence of other factors is unlikely |
| Almost certainly | There is clear evidence to suggest a causal relationship and other possible contributing factors can be ruled out |
If any doubt about the causality exists the local investigator should inform the study coordinating centre who will notify the chief investigator. In the case of discrepant views on causality between the investigator and others, the MHRA will be informed of both points of view.
10.5 Expectedness
An AE whose causal relationship to the study drug is assessed by the investigator as ‘possible’, ‘probable’, or ‘almost certainly’ is an Adverse Drug Reaction.
All events judged by the investigator to be possibly, probably, or almost certainly related to the IMP, graded as serious and unexpected (see section 10.2 and SPCs for list of Expected Adverse Events) should be reported as a SUSAR.
10.6 Follow-up After Adverse Events
All adverse events should be followed until satisfactory resolution or until the investigator responsible for the care of the participant deems the event to be chronic or the patient to be stable.
When reporting SAEs and SUSARs the investigator responsible for the care of the participant should apply the following criteria to provide information relating to event outcomes: resolved; resolved with sequelae (specifying with additional narrative); not resolved/ongoing; ongoing at final follow-up; fatal or unknown.
10.7 Reporting Procedures
All adverse events should be reported. Depending on the nature of the event the reporting procedures below should be followed. All adverse events will be reported and recorded from the point that the participant provides informed consent and throughout the trial treatment period up until seven days after the patient has taken the final dose of investigational medicinal product. Any questions concerning adverse event reporting should be directed to the MCRN CTU in the first instance. A flowchart is given below to aid in determining reporting requirements.
10.7.1 Non serious ARs/AEs
All such events, whether expected or not, should be recorded on an Adverse Event Form, which should be transmitted to the MCRN CTU within seven days of the form being due.
10.7.2 Serious ARs/AEs/SUSARs
SARs, SAEs and SUSARs should be reported within 24 hours of the local site becoming aware of the event. The SAE form asks for the nature of event, date of onset, severity, corrective therapies given, outcome and causality. The responsible investigator should sign the causality of the event. Additional information should be sent within 5 days if the reaction has not resolved at the time of reporting.
The MCRN CTU will notify the MHRA and main REC of all SUSARs occurring during the study according to the following timelines; fatal and life-threatening within 7 days of notification and non-life threatening within 15 days. All investigators will be informed of all SUSARs occurring throughout the study. Local investigators should report any SUSARs and/or SAEs as required by their local Research & Development (R&D) Office.
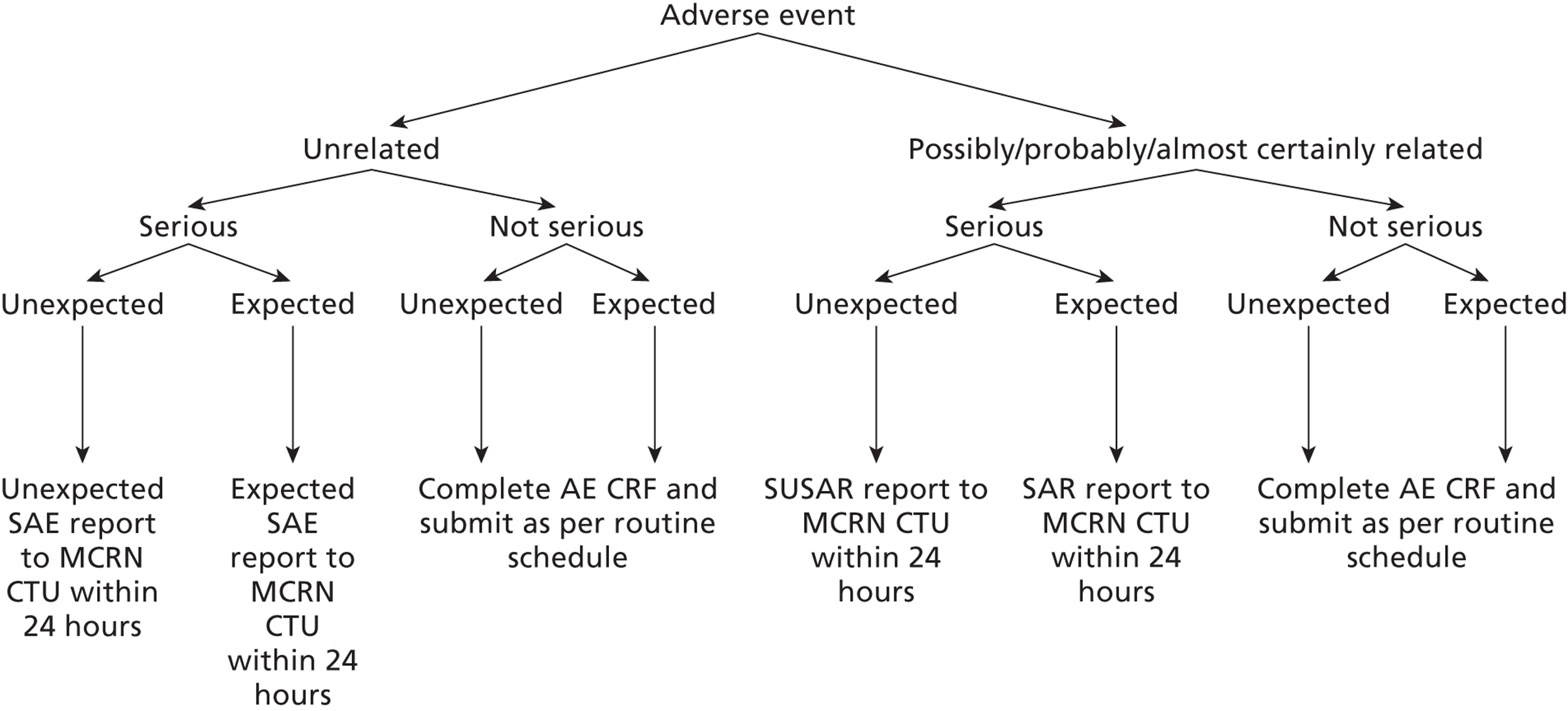
10.8 Responsibilities – Investigator
The Investigator is responsible for reporting all AEs that are observed or reported during the study, regardless of their relationship to study product.
All SAEs must be reported immediately by the investigator to the MCRN CTU on an SAE form unless the SAE is specified in the protocol or SPC as not requiring immediate reporting. All other adverse events should be reported on the regular progress/follow-up reports.
Minimum information required for reporting
-
Study identifier
-
Study centre
-
Patient number
-
A description of the event
-
Date of onset
-
Current status
-
Whether study treatment was discontinued
-
The reason why the event is classified as serious
-
Investigator assessment of the association between the event and study treatment
-
The SAE form should be completed by the responsible investigator i.e. the consultant named on the ‘signature list and delegation of responsibilities log’ who is responsible for the patient's care. The investigator should assess the SAE for the likelihood that that it is a response to an investigational medicine. In the absence of the responsible investigator the form should be completed and signed by a designated member of the site trial team and faxed to the MCRN CTU immediately. The responsible investigator should check the SAE form, make changes as appropriate, sign and then re-fax to the MCRN CTU as soon as possible. The initial report shall be followed by detailed, written reports.
-
Send the SAE form by fax (within 24 hours or next working day) to the MCRN CTU: Fax Number: 0151 282 4721
-
The responsible investigator must notify their local R&D department of the event (as per standard local procedure).
-
In the case of an SAE the subject must be followed-up until clinical recovery is complete and laboratory results have returned to normal, or until the event has stabilised. Follow-up may continue after completion of protocol treatment if necessary.
-
Follow-up information is noted on another SAE form by ticking the box marked ‘follow-up’ and faxing to the MCRN CTU as information becomes available. Extra, annotated information and/or copies of test results may be provided separately.
-
The patient must be identified by trial number, date of birth and initials only. The patient's name should not be used on any correspondence.
10.8.1 Maintenance of Blinding
Systems for SUSAR and SAR reporting should, as far as possible, maintain blinding of individual clinicians and of trials staff involved in the day-to-day running of the trial. Unblinding clinicians may be unavoidable if the information is necessary for the medical management of particular patients. The safety of patients in the trial always takes priority. In each report, seriousness, causality and expectedness should be evaluated for all of the trial treatments unless criteria have been fulfilled (section 7.8) and unblinding has taken place. Cases that are considered serious, unexpected and possibly, probably or almost certainly related to one of the trial therapies (i.e. possible SUSARs) would have to be unblinded at the clinical trials unit prior to reporting to the regulator.
10.9 Responsibilities – MCRN CTU
The MCRN CTU is undertaking duties delegated by the trial co-sponsors, Keele University and University Hospital of North Staffordshire NHS Trust, and is responsible for the reporting of SUSARs and other SARs to the regulatory authorities (MHRA and, if required, the research ethics committees) as follows:
-
SUSARs which are fatal or life-threatening must be reported not later than 7 days after the MCRN CTU is first aware of the reaction. Any additional relevant information must be reported within a further 8 days.
-
SUSARs that are not fatal or life-threatening must be reported within 15 days of the MCRN CTU first becoming aware of the reaction.
-
A list of all SARs (expected and unexpected) must be reported annually.
It is recommended that the following safety issues should also be reported in an expedited fashion:
-
An increase in the rate of occurrence or a qualitative change of an expected serious adverse reaction, which is judged to be clinically important;
-
Post-study SUSARs that occur after the patient has completed a clinical trial and are notified by the investigator to the sponsor;
-
New events related to the conduct of the trial or the development of the IMPs and likely to affect the safety of the subjects, such as:
-
A serious adverse event which could be associated with the trial procedures and which could modify the conduct of the trial;
-
A significant hazard to the subject population, such as lack of efficacy of an IMP used for the treatment of a life-threatening disease;
-
A major safety finding from a newly completed animal study (such as carcinogenicity).
-
Any anticipated end or temporary halt of a trial for safety reasons and conducted with the same IMP in another country by the same sponsor;
-
-
Recommendations of the Data Monitoring Committee, if any, where relevant for the safety of the subjects.
Staff at the MCRN CTU will liaise with the chief investigator (or, as specified in the protocol, Professor David Price) who will evaluate all SAEs received for seriousness, expectedness and causality. Investigator reports of suspected SARs will be reviewed immediately and those that are SUSARs identified and reported to regulatory authorities and MREC. The causality assessment given by the local investigator at the hospital cannot be overruled and in the case of disagreement, both opinions will be provided with the report.
The MCRN CTU will also send an annual safety report containing a list of all SARs to regulatory authorities and MREC. Copies of the report will be sent to the principal investigator at all institutions participating in the trial.
Patient safety incidents that take place in the course of research should be reported to the National Patient Safety Agency (NPSA) by each participating NHS Trust in accordance with local reporting procedures.
11 ETHICAL CONSIDERATIONS
11.1 Ethical Considerations
The study will abide by the principles of the World Medical Association Declaration of Helsinki (1964) and the Tokyo (1975), Venice (1983), Hong Kong (1989) and South Africa (1996).
We consider the specific ethical issues relating to participation in this trial to be:
-
Informed consent in a paediatric population: The parent or legal representative of the child will have an interview with the investigator, or a designated member of the investigating team, during which opportunity will be given to understand the objectives, risks and inconveniences of the trial and the conditions under which it is to be conducted. They will be provided with written information and contact details of the study nurse, from whom further information about the trial may be obtained, and will be made aware of their right to withdraw the child from the trial at any time without the child or family being subject to any detriment in the child's treatment. Children will receive information, according to their capacity of understanding, about the trial and its risks and benefits and their assent will be obtained, where appropriate.
-
The taking of a placebo treatment or active treatment (fluticasone, salmeterol or montelukast): The families will be informed about the National Guidelines and that there are various choices open to patients and carers with regard to asthma treatment. It is important that the family understands that no one will know which treatment the child is receiving due to the lack of scientific information as to which choice is better. All three treatment groups are potentially effective but it is possible that one may be better for one child than another.
-
DNA sampling: The samples will be labelled with the participant's trial identifier number. The purpose of undertaking genetic testing at some point in the future will be to determine whether those patients with specific polymorphisms differ in their response to particular treatments or whether specific polymorphisms affect severity or long term prognosis of asthma and its symptoms. The genetic study will be subject to a separate consent process (using a separate information sheet) to the main study and participants who refuse to take part will not be precluded from entry into MASCOT. The consent obtained will be related to genetic studies in asthma only. The DNA collected will not be used for any other genetic studies. Information about individual patients will not be available at any time but all participants will be informed of the outcome of the study and the outcome of any DNA analysis by publishing the final trial report on the public access area of the NIHR portal.
-
Ineligibility of non-English speakers: To include non-English speakers in the trial would require appropriate translations of all trial documentation and correspondence provided to participants, which would need to be fully checked and validated. Unfortunately there are not the resources within the trial to adequately provide this. The design of the trial also necessitates study visits to be held in primary care settings in some regions, which do not routinely have translation services available. In addition, the study specifies a telephone call at week 36 (T+36) between the RN and the patient and/or their carer/s, which would not be possible if they were unable to communicate in the same language. For these reasons, we must therefore limit trial entry to English speakers only.
11.2 Ethical Approval
The trial protocol will be submitted for the approval of the North West Research Ethics Committee (REC). Each participating centre must also undergo site specific assessment (SSA). A copy of local NHS Research & Development (R&D) approval and of the Patient Information sheet and Consent form (PISC) on local headed paper should be forwarded to MCRN CTU before patients are recruited into the study at that site. The CTU should receive notification of positive SSA for each new centre via the main REC. This will be through the chief investigator (Prof Warren Lenney) who is the main REC applicant. Details of any amendments to the original approved version of the protocol, which have subsequently been ethically approved, are indexed in Section 18.
Proxy consent from the parent or legally acceptable representative should be obtained prior to each patient registering in the trial after a full explanation has been given of the treatment options, including the conventional and generally accepted methods of treatment. Age and stage-of-development specific Patient Information and Consent leaflets should also be implemented and patient assent obtained (where appropriate). The right of the parent/legal representative to refuse consent for the minor to participate in the trial without giving reasons must be respected. After the patient has entered the trial, the clinician must remain free to give alternative treatment to that specified in the protocol at any stage if he/she feels it to be in the best interest of the patient. However, the reason for doing so should be recorded and the patient will remain within the trial for the purpose of follow-up and data analysis according to the treatment option to which they have been allocated. Similarly, the parent/legal representative of the patient remains free to withdraw the patient at any time from the protocol treatment and trial follow-up without giving reasons and without prejudicing the further treatment of the minor.
11.3 Informed Consent Process
11.3.1 General
Informed consent is a process initiated prior to an individual agreeing to participate in a trial and continues throughout the individual's participation. Informed consent is required for all patients participating in MCRN CTU coordinated trials. In obtaining and documenting informed consent, the investigator should comply with applicable regulatory requirements and should adhere to GCP and to the ethical principles that have their origin in the Declaration of Helsinki. Informed consent should only be taken by staff who are appropriately trained to do so and who have experience in providing information to minors.
Parental and age and stage-of-development appropriate Patient Information Sheet and Consent (PISC) forms, which will have been approved by an independent ethics committee (IEC), will be issued to potentially eligible patients and their families. The PISC will describe in detail the trial procedures (for both the run-in and randomised treatment phases), the trial interventions/products and potential risks and benefits of taking part in the study. All patients and their families will receive the appropriate version of the written information and be asked to read and review it. The PISC will emphasise that participation in the trial is voluntary and that the parent or legal representative* may, without the minor being subject to any resulting detriment, withdraw them from the trial at any time by revoking the informed consent. The rights and welfare of the patient will be protected by emphasising to them that the quality of medical care will not be adversely affected if they decline to participate in this study. All parents/legally acceptable representatives and patients will be given the opportunity to ask questions and will be given sufficient time to consider trial entry before consenting. They should have the opportunity to discuss the study with their usual medical practitioner and/or family/friends prior to agreeing to participate. The consent form will request permission for the patient's General Practitioner to be informed of their registration into the trial and also permission for personnel involved in the research or from regulatory authorities to have access to the individual's medical records. A copy of the informed consent/assent document will be given to the patient and their legally acceptable representative for their records.
*A mother always has legal responsibility for her child, however a father only has legal responsibility if he is married to the mother or has acquired legal responsibility for his child in the following ways:
-
For children born before 1 December 2003, unmarried fathers can get parental responsibility by:
-
marrying the mother of their child or by obtaining a parental responsibility order from the court
-
registering a parental responsibility agreement with the court or by an application to court
-
-
For children born after 1 December 2003, unmarried fathers can get parental responsibility by:
-
registering the child's birth jointly with the mother at the time of birth
-
re-registering the birth if you are the natural father
-
marrying the mother of their child or by obtaining a parental responsibility order from the court
-
registering with the court for parental responsibility
-
11.3.2 Obtaining Informed Consent
The consent process will be carried out by an appropriate researcher identified in the trial signature and delegation log. The consent process will be conducted by a researcher with training and experience with minors. The researcher delegated to obtain informed consent will be determined on a site by site basis, depending on the experience and knowledge of the individual staff at that site. Only personnel confident and competent to do so will be able to obtain informed consent. This can include PIs, other delegated investigators and research nurses. Where informed consent is being obtained by a research nurse, the patient and their family should have access to a clinician with expertise in paediatric respiratory medicine if they have any concerns about participation or any further questions that the nurse is unable to sufficiently answer.
Upon reviewing the PISC with the patient and their parent/s or legal representative, the researcher who is obtaining consent will fully explain the research study (both the run-in and randomised treatment phases) to the patient and their parent/legal representative and answer any questions they may have. They will discuss the objectives of the study and all potential benefits and inconveniences of taking part. They will clearly outline all of the responsibilities the patient and their family will be expected to meet if they agree to participate, including attendance at study visits and compliance with trial medications. A contact point where further information about the trial may be obtained will be provided. The patient and their family will be made aware that entry into the randomised phase of the trial is contingent on their continuing to meet the eligibility criteria following the four week run-in, as well as their continuing consent/assent to participate in the trial.
Both parental consent and the patient's assent, if appropriate, will be obtained prior to any study related procedures being carried out. The researcher and the parent/legal representative of the minor must personally sign and date the form. If capable, the patient should assent and personally sign and date the assent form. Assent forms do not substitute for the consent form signed by the patient's legally acceptable representative. Where the child is unable to provide assent, this should be documented in the patient's medical notes and recorded on a copy of the age and stage of development specific PISC. The original assent form should be filed in the medical notes and copies placed in the site file and forwarded to the MCRN CTU.
The original copy of the consent/assent form will be retained in the patient's medical notes and must be available for inspection. A copy will be returned to the MCRN CTU and one will also be placed in the Site File. A further copy of the signed consent/assent form will be provided to the child's parent/legally acceptable representative along with the PISC used during the recruitment consultation.
Following the four week run-in phase, any patients continuing to meet the eligibility criteria will progress to the randomised part of the trial. Once they have been found to be eligible, prior to randomisation, the researcher will reiterate previous written and verbal explanations about the trial and answer any further questions the family may have. The patient and their parent/s or legally acceptable representative will be asked to provide verbal consent (and assent, where appropriate) for their continued participation in the trial.
11.3.3 Informed Consent for DNA Collection
Consent for obtaining DNA in the form of saliva will be discussed initially at the T–4 study visit. All participants registered in the trial will be provided with a copy of a parental and age and stage of development specific PISC, approved by an IEC and developed specifically for the sub-study. They will have the opportunity to ask any questions and to discuss the sub-study with their usual medical practitioner and/or family/friends prior to making their decision.
Consent for participation in the genetic sub-study will be obtained at the T0 visit by the researcher identified in the site signature and delegation log. They will discuss the objectives of the study and all potential benefits and inconveniences of taking part. Participants and their families will be made aware that they are consenting for the DNA to be collected and stored for analysis at a later date. It will be made clear that the DNA specimen will be used only for analysis of specific genetic polymorphisms relating to asthma severity and outcomes and the steps that will be taken to maintain the confidentiality of the data.
Both parental consent and patient assent, if appropriate, will be obtained prior to the collection of the DNA sample. The research practitioner and the parent/legal representative of the minor must personally sign and date the form. If capable, the patient should assent and personally sign and date the assent form. Assent forms do not substitute for the consent form signed by the patient's legally acceptable representative.
The original copy of the consent/assent form will be retained in the patient's medical notes and must be available for inspection. A copy will be returned to the MCRN CTU and one will also be placed in the Site File. A further copy of the signed consent/assent form will be provided to the child's parent/legally acceptable representative along with the Patient Information Sheet relating to the trial.
11.4 Study Discontinuation
In the event that the study is discontinued, children will be treated according to usual standard clinical care. Patients withdrawing early from the trial as a whole will be unblinded within seven days of the local team becoming aware of their decision. Patients who withdraw early from trial treatment but continue to allow data collection and follow-up will have a discussion with the PI or investigator as to whether they will be unblinded at that point or wait until the end of their 48 week follow-up period. The decision will be based on the patient and their carers' own preferences and whether the investigator feels they need to be aware of the patient's randomised treatment allocation in order to provide appropriate follow on care.
12 REGULATORY APPROVAL
This trial is registered with the Medicines and Healthcare products Regulatory Agency (MHRA) and has been granted a Clinical Trial Authorisation (CTA). The EudraCT number is 2008–000511–16.
13 TRIAL MONITORING
Trial monitoring is carried out to ensure that the rights and well-being of human participants are protected during the course of a clinical trial. A risk assessment is performed for each trial co-ordinated by the MCRN CTU to determine the level and type of monitoring required for specific hazards. The type of trial monitoring should be specific to the individual trial and can take the form of on-site visits or central monitoring.
13.1 Risk Assessment
In accordance with the MCRN CTU Standard Operating Procedure (TM005) this trial has undergone a risk assessment, completed in partnership between the University of Liverpool, MCRN CTU, trial co-sponsors and the chief investigator, Prof Warren Lenney. In conducting this risk assessment, the contributors considered potential patient, organisational and study hazards, the likelihood of their occurrence and resulting impact should they occur.
The outcome of the risk assessment is expressed as a percentage, assigned according to the following categories:
-
Score ≤ 33% = Low risk
-
Score ≥ 34 to ≤ 67% = Moderate risk
-
Score ≥ 68 to ≤ 100% = High risk
The outcome of the MASCOT trial risk assessment was a score of 18.3% therefore it has been judged to be a low risk clinical trial. This level of risk has determined the approach to trial monitoring described in this section and additionally in Section 16.
13.2 Source Documents
-
Source data: All information in original records and certified copies of original records of clinical findings, observations, or other activities in a clinical trial necessary for the reconstruction and evaluation of the trial. Source data are contained in source documents (original records or certified copies). (ICH E6, 1.51).
-
Source document: Original documents, data, and records (e.g. hospital records, clinical and office charts, laboratory notes, memoranda, subjects diaries or evaluation checklists, pharmacy dispensing records, recorded data from automated instruments, copies or transcriptions certified after verification as being accurate copies, microfiches, photographic negatives, microfilm or magnetic media, x-rays, subject files, and records kept at the pharmacy, at the laboratories and at medico-technical departments involved in the clinical trial). (ICH E6, 1.52).
In order to resolve possible discrepancies between information appearing in the Case Report Form (CRF) and any other patient related documents, it is important to know what constitutes the source document and therefore the source data for all information in the CRF. The following data recorded in the CRF should be consistent and verifiable with source data in source documents other than the CRF (eg medical record, laboratory reports and nurses' notes).
The following parameters that will be documented in the CRF are not source data:
-
Relevant medical history and diagnosis (medical notes are source documents)
-
Data for evaluation of eligibility criteria (medical notes are source documents)
-
Physical examinations and assessments (medical notes are source documents)
-
Concomitant medications (including changes) and diagnoses (medical notes are source documents)
-
Dispensing of trial medication (pharmacy records are source documents)
-
Adverse events (medical notes are source documents)
For data where no prior record exists and which is recorded directly onto the trial Case Report Forms, e.g. quality of life evaluations, the Case Report Form will be considered the source document, unless otherwise indicated by the investigator. All such exemptions should be identified prior to the clinical phase of the trial.
In addition to the above, date of conducting informed consent and assent process including date of provision of patient information, individual screening study number, unique randomised trial number and the fact that the patient is participating in a clinical trial comprising three treatment arms of inhaled fluticasone propionate and placebo tablet versus inhaled fluticasone propionate and salmeterol (combination inhaler) plus placebo tablet versus inhaled fluticasone propionate and montelukast tablet should be added to the patient's medical record chronologically, i.e. when treatment is allocated to the patient. Further, study treatment allocation should also be noted in the patient's medical record after unblinding of the study (see Section 7.8).
13.3 Data Capture Methods
Trial data will be captured using paper based Case Report Forms (CRFs).
13.3.1 Case Report Forms
The study case report form (CRF) is the primary data collection instrument for the study. All data requested on the CRF must be recorded and all missing data must be explained. If a space on the CRF is left blank because the procedure was not done or the question was not asked, ‘N/D’ must be written. If the item is not applicable to the individual case, write ‘N/A’. All entries should be printed legibly in black ink. If any entry error has been made, to correct such an error, draw a single straight line through the incorrect entry and enter the correct data above it. All such changes must be initialled and dated. DO NOT ERASE OR WHITE OUT ERRORS. For clarification of illegible or uncertain entries, print the clarification above the item, then initial and date it. CRF pages will be provided in triplicate on No Carbon Required (NCR) paper and when complete, should be split into three collated sets. Originals should be sent to the MCRN CTU and the copies securely retained at site.
Screening logs should be maintained at site and submitted monthly to the MCRN CTU. The screening log will be used to record patients (in an anonymised format) who register in the trial and patients who:
-
Contact the research team after receiving information about MASCOT but decline to take part after talking to the research doctor/nurse about the trial
-
Were referred inappropriately by their GP, nurse or hospital clinician
-
Attended the T–4 visit but declined to consent
-
Consented to the trial at T–4 but were found to be ineligible
Registration and randomisation CRFs should be submitted to the MCRN CTU within seven days of patients being registered or randomised onto the study. All other routine CRFs should be completed and submitted to the CTU within seven days of the study visit occurring. SAEs, SARs and SUSARs should be reported as detailed in Section 10.
Health Economics questionnaires will be completed by the patient and their carer/s as paper records and will be collected by the RN at each study visit. They will check them with the family for completeness and query any omissions (where appropriate). The RN should retain a copy for their Site File and return the original to the CTU within seven days of the study visit occurring. A copy should also be provided to the family if requested. A Quality of Life (QoL) questionnaire will be administered at each study visit as a paper record. The above procedure and timelines should be followed (original to CTU within seven days, copies for patient site trial records and family if requested).
13.4 Data Monitoring at MCRN CTU
The MCRN CTU will review recruitment rates, withdrawals and losses to follow-up and identified problems will be reviewed by the TMG. Remedial action will be taken as necessary. Completed CRFs submitted to the CTU will be centrally monitored to ensure that data collected are consistent with adherence to the trial protocol. Data will be checked for missing or unusual values (range checks) and checked for consistency within participants over time. Discrepancies that have been raised will be queried with the RN/PI. They will be sent a data query form highlighting the discrepancy and asked to check the recorded information against the source data and either confirm or correct the recorded data as appropriate. All corrections and clarifications should be documented on the relevant data query form which must be signed off by a member of the research team who is authorised to do so on the site signature and delegation log. A copy of the completed data query form should be returned to CTU and the site's original copy should also be filed with the CRF it relates to. CTU will send reminders for any overdue or missing data queries.
13.5 Central and Clinical Site Monitoring
13.5.1 Central Monitoring
The MCRN CTU is to receive a copy of the PISC within a week of randomisation. If consent forms are not forwarded regularly by a participating centre, the Trial Coordinator will conduct a site visit to check the presence of a signed PISC in the medical notes of all registered patients.
Data submitted to the database will be centrally monitored by the CTU to ensure, as far as possible, that CRF data collected are consistent with adherence to the trial protocol. Data will be checked for missing or unusual values (range checks) and checked for consistency within participants over time. Discrepancies that have been raised will be queried. The MACRO data management system will automatically keep a log of what data has been changed, the time of each change, and the person who changed it.
The Trial Coordinator will review rates of recruitment, missing outcome data, SAEs, ADRs, study withdrawals and losses to follow-up across sites, and remedial action taken as necessary. The Trial Coordinator may arrange site visits to undertake source data verification.
Standardised paper Case Report Forms (CRFs) should be sent to the MCRN CTU promptly. The Trial Data Manager will conduct data entry checks and use automated validation checks at data entry. A site visit will be conducted if inconsistencies, unresolved queries, missing data are consistently noted at a given site.
Monthly recruitment reports will be provided by the Trial Coordinator, monitoring reasons cited for consent refusal and querying reasons for slow recruitment. The TMG is charged with providing solutions to problems where possible.
The Trial Coordinator will keep a central protocol deviation log which will be updated with all deviations reported from trial sites. If the Trial Coordinator identifies significant and/or persistent non-compliance on the part of the PI, this will be documented in the monitoring report and the MCRN CTU team will discuss any further action required. A site visit will be conducted if primary and secondary measures are consistently missing from a given site. The Trial Coordinator will be in regular contact with the PIs in order to monitor the impact that the study may have on the running of the service.
13.5.2 Site Monitoring
Site monitoring may be deemed to be necessary as a result of central data checks. In order to perform their role effectively, a member of the MCRN CTU staff (usually the Trial Coordinator) may need direct access to primary data, e.g. patient records, laboratory reports, appointment books, etc. Since this affects the patient's confidentiality, this fact is included in the Patient Information Sheet and Informed Consent Form.
13.5.3 Confidentiality
Individual participants' medical information obtained as a result of this study is considered confidential and disclosure to third parties is prohibited with the exceptions below.
Case report forms will be labelled with patient initials and a unique trial registration and/or randomisation number. DNA samples will be transferred to an external laboratory and will be identified by unique identifiers only. Medical information may be given to the participant's medical team and all appropriate medical personnel responsible for the participant's welfare.
Verification of appropriate informed consent will be enabled by the provision of copies of participants' signed informed consent/assent forms being supplied to the MCRN CTU by recruiting centres. This requires that name data will be transferred to the MCRN CTU, which is explained in the PISC. The MCRN CTU will preserve the confidentiality of participants taking part in the study and the University of Liverpool is a Data Controller registered with the Information Commissioners Office.
13.5.4 Quality Assurance and Quality Control of Data
QA includes all the planned and systematic actions established to ensure the trial is performed and data generated, documented/recorded and reported in compliance with applicable regulatory requirements. QC includes the operational techniques and activities done within the QA system to verify that the requirements for quality of the trial-related activities are fulfilled.
This trial has undergone a risk assessment, the outcome of which indicates that it is a low risk trial. As such, site visits will be conducted and source data verification performed only if indicated to be necessary as a result of central monitoring. To this end:
-
The PI, RN and designated pharmacist from each centre will attend the trial launch meeting, coordinated by the MCRN CTU in conjunction with the chief investigator, which will incorporate elements of trial specific training necessary to fulfil the requirements of the protocol
-
The Trial Coordinator is to verify appropriate approvals are in place prior to the initiation of a site and that relevant personnel have attended site specific training
-
The internal QA process of the MCRN CTU involves routine audit of certain activities across all trials, including random checking of adherence to informed consent procedure (monitoring receipt of signed consent forms)
-
The Trial Coordinator and Trial Statistician are to check safety reporting rates between centres
-
The Trial Coordinator and Trial Statistician are to monitor screening, recruitment and drop out rates between centres
-
The Trial Data Manager is to conduct data entry consistency checks and follow up data queries until resolved
-
Independent oversight of the trial will be provided by the Data Monitoring Committee and independent members of the Trial Steering Committee
13.6 Records Retention
The investigator at each investigational site must make arrangements to store the essential trial documents, including the Investigator Site File, until the MCRN CTU informs the investigator that the documents are no longer to be retained, or for a maximum of 15 years, whichever is soonest.
In addition, the PI is responsible for the archiving of all relevant source documents so that the trial data can be compared against source data after completion of the trial (e.g. in case of inspection from authorities).The PI is required to ensure the continued storage of the documents, even if they leave the clinic/practice or retires before the end of the required storage period. Delegation should be documented in writing.
The MCRN Clinical Trials Unit undertakes to store originally completed CRFs and separate copies of the above documents for the same period, except for source documents pertaining to the individual investigational site, which are kept by the PI only.
14 INDEMNITY
MASCOT is co-sponsored by Keele University and University Hospital of North Staffordshire NHS Trust and co-ordinated by the MCRN CTU in the University of Liverpool. As this is an investigator-initiated study, The Association of the British Pharmaceutical Industry (ABPI) guidelines for patient compensation by the pharmaceutical industry do not apply. However, in terms of liability NHS Trust and Non-Trust Hospitals have a duty of care to patients treated, whether or not the patient is taking part in a clinical trial, and they are legally liable for the negligent acts and omission of their employees. With regards to the MASCOT trial, University Hospital North Staffordshire NHS Trust will provide an indemnity in respect of clinical negligence to the extent that such an indemnity is permitted by the NHS Litigation Authority's Clinical Negligence Scheme for Trusts.
For General Practitioners participating in the MASCOT trial, indemnity in respect of clinical negligence will be provided through their Primary Care Trust (if they are employed through that PCT) following approval of the trial by their Research Office. Where an independent contractor (a GP or their practice staff working under a contract for services to a Primary Care Trust), undertakes research as part of their routine clinical services, their personal professional indemnity arrangements will provide them with adequate cover for this activity for their own practice patients. GPs involved in MASCOT, who are independent contractors, will inform their indemnity providers (e.g. MDU, MPS) of their participation in the study prior to the start of recruitment.
Clinical negligence is defined as
A breach of duty of care by members of the health care professions employed by NHS bodies or by others consequent on decisions or judgments made by members of those professions acting in their professional capacity in the course of their employment, and which are admitted as negligent by the employer or are determined as such through the legal process.
This study is funded by the Health Technology Assessment programme (HTA) of the Department of Health. Contractual agreements will be in place between sponsor and collaborating sites that will incorporate financial arrangements.
15 FINANCIAL ARRANGEMENTS
15.1 Participant Payments
Patients will be paid up to £8 per visit to cover any expenses incurred as a result of travelling to the centre for their study visit. This will be monitored by collecting tickets and receipts for those travelling via public transport and asking families who have travelled to the centre by private car to complete a mileage form.
15.2 Identification Payments
A nominal sum of £25 per patient registered for the four week run-in is allocated to be paid to the relevant healthcare provider (e.g. a GP practice) for provision of administration costs.
15.3 Pharmacy Departments
The dispensing service for the trial will be provided by the pharmacy department in each of the secondary care hubs. Provision of payment to support pharmacy costs (setup, storage, dispensing, reconciliation and GCP quality assurance), totalling £200 per participating site plus an additional fee of £10 per patient randomised has been allocated.
16 TRIAL COMMITTEES
16.1 Trial Management Group (TMG)
The Trial Management Group (TMG) will comprise Prof Warren Lenney, Prof Paula Williamson, Dr Catrin Tudur-Smith, Miss Sophie Perry, Prof David Price, Prof Marilyn James, Ms Sadie Clayton, Dr Jonathan Couriel and Ms Nemonie Marriott. The TMG will be responsible for the day-to-day running and management of the trial and will meet/teleconference approximately every two months during the first year and appropriately after that.
16.2 Trial Steering Committee (TSC)
The Trial Steering Committee (TSC) will consist of an independent chairperson, Dr Anne Thomson (Consultant in Paediatric Respiratory Medicine and General Paediatrics), two independent experts in the field of respiratory paediatrics, (Dr Gary Connett and Dr Iolo Doull), a biostatistician (Prof Chris Frost) and two lay representatives (Mr John Hilton and Mrs Ro Hilton) together with members from the TMG. The role of the TSC is to provide overall supervision for the trial and provide advice to the funder and the sponsor through its independent chairperson. The ultimate decision for the continuation of the trial lies with the TSC.
16.3 Independent Data and Safety Monitoring Committee (IDSMC)
The independent Data and Safety Monitoring Committee (IDSMC) consists of an independent chairperson, Dr David Spencer (Consultant in Respiratory Paediatrics), plus two independent members: Dr John Alexander, who is an expert in the field of paediatric medicine, and Mr Andy Vail, an expert in medical statistics.
The IDSMC will be responsible for reviewing and assessing recruitment, interim monitoring of safety and effectiveness, trial conduct and external data. The IDSMC will first convene before recruitment begins and will then define frequency of subsequent meetings (at least annually). Details of the interim analysis and monitoring plans are provided in section 9.
The IDSMC will provide a written recommendation to the Trial Steering Committee concerning the continuation of the study.
17 PUBLICATION
The results from different centres will be analysed together and published as soon as possible after the close of the trial. Individual clinicians must undertake not to submit any part of their individual data for publication without the prior consent of the Trial Management Group.
The Uniform Requirements for Manuscripts Submitted to Biomedical Journals (http://www.icmje.org/) and the CONSORT guidelines14,15 will be respected. The ISRCTN allocated to this trial should be attached to any publications resulting from this trial.
BMJ guidance on authorship and contributorship (see http://bmj.com/advice/3.html) will be use to acknowledge the level and nature of contribution of key individuals in publications arising from the trial. The publication strategy shall lie under the jurisdiction of the Trial Steering Committee.
18 PROTOCOL AMENDMENTS
18.1 Version 2.0 (19/Mar/2008)
Amendments and clarifications prior to full ethical approval (v1.0 22/Jan/2008 to v2.0 19/Mar/2008).
| Page No. | Comment |
|---|---|
| Throughout | Updated version and date; correction of typographical errors |
| 11 | The text ‘fully informed written (proxy) consent’ moved from T0 to T–4 visit in trial schematic. ‘Verbal consent/assent’ added to T0 visit activities |
| 20–21 | Exclusion criteria corrected to read ‘> 1000micrograms and unlicensed beclometasone dipropionate’ |
| 23 | Clarification of procedures for recruiting via primary care to include opportunistic recruitment |
| 25 | Changed procedure for randomisation – patients are allocated to a treatment arm using a randomisation list rather than through allocation of next sequential randomised treatment pack; pharmacy team are unblinded to treatment allocation |
| 29–31 | Description of trial packaging altered and dispensing procedure updated to reflect this |
| 31 | Clarification that inhaler technique checked throughout trial; first drug shipment size changed from one third to one quarter of total allocation |
| 32 | Accountability procedures updated to reflect randomisation and dispensing procedures |
| 38 | Randomisation procedure updated |
| 38–39 | Expanded details relating to withdrawal of patients from trial treatment |
| 55 | Fluticasone was added to the list of active treatments |
| 62 | The text ‘CTA reference’ replaced by ‘EudraCT number’ |
18.2 Version 3.0 (16/May/2008)
Amendments and Clarifications (v2.0 19/Mar/2008 to v3.0 16/May/2008).
| Page No. | Comment |
|---|---|
| 15 | Clarification of reference from Summary of Product Characteristics |
| 24–25 | Change in pharmacy contact at University Hospital of North Staffordshire (Susan Thomson to Cath Jackson), Royal Aberdeen Children's Hospital (from Valerie Macgregor to Martina Freeman) and the Royal Devon & Exeter (from Kate O'Connor to Fiona Hall). Contact details amended appropriately |
| 31 | Clarification on documentation of patient randomisation number |
| 32 | Long acting beta2 agonists and leukotriene receptor antagonists (other than trial medication) added to list of concomitant medications |
| 35 | Guidance added on the window for timing of patient study visits |
| 68 | The amount available for reimbursement of patient travel expenses was changed from £10 to £8 |
| 475 | Principal investigator at NNUH changed from Professor David Price to Dr Chris Upton |
| 77–78 | South Manchester University Hospital and Wirral Universities Teaching Hospital added as lead participating sites; six GP practices in the Norfolk area added as participating sites (Appendix A) |
| 79–80 | Five participating primary care sites added to Appendix B |
| 81 | T0 pharmacy dispensing procedure for Norfolk patients changed to allow patients to be randomised away from the NNUH |
| 112 | Search codes for GP databases added as an appendix (Appendix E) |
18.3 Version 4.0 (24/Jul/2008)
Amendments and Clarifications (v3.0 16/May/2008 to v4.0 24/Jul/2008)
| Page No. | Comment |
|---|---|
| 21 | Explanation that patients who withdraw early from trial treatment but continue to allow data collection will discuss with the PI whether and when to become unblinded |
| 22 | MCRN CTU to unblind individual patients leaving the trial completely within seven days of completion/withdrawal wherever possible |
| 26 | Addition of pharmacy contact (Neil Caldwell) at Arrowe Park Hospital |
| 29–30 | Change in process of supplying IMP to patients at T–4 and T0 (from IMP being dispensed prior to visit and supplied then if eligible to being dispensed after the visit and then transported to the patient) |
| 34 | CYP3A inhibitors changed from a prohibited concomitant medication group to limited use only whilst on trial treatment |
| 34–36 | Routine unblinding procedure changed from all patients only being unblinded at the end of the trial as a whole to each participant being unblinded when they end their individual participation in the trial. Process for this described (MCRN CTU to inform GP with seven days) |
| 40 | Patients must now be asked for current GP details at the final visit |
| 41 | Clarification that European Respiratory Society (ERS) standards for spirometry will be accepted as well as ATS |
| 45 | Further guidance on completion of the PACQLQ added |
| 62 | Description of new unblinding procedure with regards to study discontinuation |
| 63 | Confirmation that MHRA authorisation has now been granted for MASCOT |
| 65 | Screening logs to be submitted monthly instead of weekly |
| 80–81 | Four GP practices in the Norfolk area added as participating sites (Appendix A) |
| 84–85 | Clarification on process of supplying IMP to Norfolk patients seen at outreach centres – to include change from IMP dispensed prior to the visit to dispensed after the visit and transported to the patient (Appendix C) |
18.4 Version 5.0 (30/Jan/2009)
Amendments and Clarifications (v4.0 24/July/2008 to v5.0 30/Jan/2009)
| Page No. | Comment |
|---|---|
| Throughout | Removal of the word ‘CONFIDENTIAL’ from the page headers |
| 3 | Change in contact details for Aptuit Ltd (from Ciaran Flanagan to Dr Ivan Langan) |
| 5 | Contact details for the MASCOT Data Manager added following the appointment of Emma Dyson |
| 29 | Correction of montelukast matching placebo tablet shelf life from 24 months to fve years |
| 71 | Nemonie Marriott to replace Zahira Maqsood as the primary care representative on the Trial Management Group |
18.5 Version 6.0 (20/May/2009)
Amendments and Clarifications (v5.0 30/Jan/2009 to v6.0 20/May/2009)
| Page No. | Comment |
|---|---|
| Throughout | Updated version and date; correction of typographical errors |
| 3–6 | Correction and updates to contact details of various individuals |
| 10 | Update in number of secondary care centres from 12 to 13 |
| 14–16 | Updates to ‘Potential Risks’ section for all three study medications (information derived from SPCs) |
| 18 | Addition of ‘Adverse Events’ as a secondary endpoint |
| 19 | Clarification of inclusion criteria #1 at both T–4 and T0 (from ‘Those requiring frequent short-acting beta2 agonist relief therapy ≥ 7 puffs per week’ to ‘Those requiring frequent short-acting beta2 agonist relief therapy ≥ 7 puffs in the past seven days’ |
| 21–22 | Additions to recruitment section including recruitment via community pharmacists and school health professionals and promotion via the media. Expansion of existing strategies to include use of a follow-up letter or telephone call and recruitment via any appropriate primary care centre |
| 23 | Update to site name (from ‘Royal Liverpool Children's Hospital’ to ‘Alder Hey Children's NHS Foundation Trust’) |
| 23 | Change in pharmacist at Royal Manchester Children's Hospital (from Judith Thornton to Carolyn Davies) and Derbyshire Children's Hospital (from Julie Vanes and Liz Bedford to Peter Fox) and updates to associated contact details |
| 27 | Clarification added that pharmacists must complete all sections on the trial medication labels prior to dispensing |
| 32 | Revision of text to show that unblinding information at completion of/withdrawal from trial treatment will be provided to the patient's GP only. Secondary care clinicians will now be instructed to contact the GP if they require this information |
| 34–35 | Addition of caveat that the RN/PI can conduct the T+8 and T+24 study visits at the patient's home, instead of clinic, in exceptional circumstances |
| 41–43 | Updates to sample size calculation formula and justifications (sample size remains unchanged) |
| 43 | Change in planned interim sample size review – now to be conducted after the first 75 children have completed 24 week follow-up |
| 44–45 | Various updates and Clarifications to the statistical analysis plan. Discussion of how secondary outcomes will be analysed |
| 58–59 | Revision of the text detailing how data queries will be documented and resolved. All data queries will be actioned using specific Data Query Forms, no changes will be made to the original CRF page/s |
| 62 | Change as to who the £25.00 participant identification payment can be made to – this now includes any appropriate healthcare provider, not just GP practices |
| 67 | Removal of the Machin et al. (1997) reference. Addition of Metcalfe et al. (2003) reference |
| 74 | Removal of Appendix B which listed ‘Satellite’ GP centres in the Greater London area affiliated to the Royal London Hospital. These are now referenced in Section 8.1 |
| 77 | Removal of Appendix D, Summary of Product Characteristics – now to be provided to sites separate from the protocol |
| 105 | Appendix E (now Appendix C) – updates to the GP Database Search Guidance |
19 REFERENCES
- Srivastava P, Baird A, Mill A, . Risk factors for wheeze in transition from childhood to early adulthood. Am J Resp Crit Care Med 2000;161.
- SIGN . British Guideline on the Management of Asthma. Thorax 2003;58:i1-i94.
- Van den Berg NJ, Ossip MS, Hederos CA, . Salmeterol/fluticasone propionate in combination is effective and safe in children with asthma. Pediatr Pulmonol 2000;30:97-105.
- Knorr B, Matz J, Bernstein JA, . Montelukast for chronic asthma in 6–14 year old children. JAMA 1998;279:1181-6.
- Verberne AA, Frost C, Duiverman EJ, . Addition of salbutamol versus doubling the dose of beclomethasone in children with asthma. Am J Respir Crit Care Med 1998;158:213-19.
- Greening AP, Ind PW, Northfield M, . Added salmeterol versus higher-dose corticosteroid in asthma patients with symptoms on existing inhaled corticosteroid. Lancet 1994;344:219-24.
- Garrett J, Williams S, Wong C, . Treatment of acute asthma exacerbations with an increased dose of inhaled steroid. Arch Dis Child 1998;79:12-7.
- Allen D, Mullen M, Mullen B. A meta-analysis of the effect of oral and inhaled corticosteroids on growth. JACI 1994;93:967-76.
- Russell G. Inhaled corticosteroids and adrenal insufficiency. Arch Dis Child 2002;87:455-6.
- Crowley S. Inhaled glucocorticoids and adrenal function: an update. Paediatr Resp Rev 2003;4:153-61.
- Committee on Safety of Medicines and Medicines Control Agency . Current problems in pharmacovigilance. Drugs and Therapeutics Bulletin 2002;28.
- Juniper EF, O’Byrne PM, Guyatt GH, . Development and validation of a questionnaire to measure asthma control. Eur Respir J 1999;14:902-7.
- Newacheck PW, Halfon N. Prevalence, impact, and trends in childhood disability due to asthma. Arch Pediatr Adolesc Med 2000;154:287-93.
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA 1996;276:637-9.
- Moher D, Schulz KF, Altman D. CONSORT group . The CONSORT Statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. Lancet n.d.;357:1191-4.
- Metcalfe C, Thompson SG, Cowie MR, Sharples LD. The use of hospital admission data as a measure of outcome in clinical studies of heart failure. European Heart Journal 2003;24:105-12.
20 APPENDICES
Appendix A Lead Participating Sites
| Participating Site: | Professor Warren Lenney |
| Consultant Paediatrician/Professor of Paediatrics | |
| Medical Research Unit | |
| University Hospital of North Staffordshire | |
| Hartshill Road | |
| Hartshill | |
| Stoke on Trent | |
| ST4 7QB | |
| Tel: 01782 554610 | |
| Fax: 01782 412236 | |
| Email: warren.lenney@uhns.nhs.uk | |
| Participating Site: | Dr Chris Upton |
| Consultant Paediatrician | |
| Norfolk & Norwich University Hospital | |
| Colney Lane | |
| Norwich | |
| NR4 7UY | |
| Tel: 01603 287544 | |
| Fax: 01603 286318 | |
| Email: chris.upton@nnuh.nhs.uk | |
| Participating Site: | Dr Stephen Turner |
| Consultant Paediatrician & Clinical Senior Lecturer | |
| Royal Aberdeen Children's Hospital | |
| Westburn Road | |
| Foresterhill | |
| Aberdeen | |
| AB25 2ZG | |
| Tel: 01224 555195 | |
| Fax: 01224 551919 | |
| Email: s.w.turner@abdn.ac.uk | |
| Participating Site: | Dr Hitesh Pandya |
| Senior Lecturer & Honorary Consultant Paediatrician | |
| Robert Kilpatrick Clinical Sciences Building | |
| Leicester Royal Infirmary | |
| Infirmary Square | |
| Leicester | |
| LE2 7LX | |
| Tel: 0116 258 5881 | |
| Fax: 0116 252 3282 | |
| Email: hp28@le.ac.uk | |
| Participating Site: | Dr Tom Hilliard |
| Consultant Paediatrician | |
| Bristol Royal Hospital for Children | |
| Upper Maudlin Street | |
| Bristol | |
| BS2 8BJ | |
| Tel: 0117 342 8693 | |
| Fax: 0117 342 8494 | |
| Email: tom.hilliard@ubht.nhs.uk | |
| Participating Site: | Dr Will Carroll |
| Consultant Paediatrician | |
| Derbyshire Children's Hospital | |
| Uttoxeter Road | |
| Derby | |
| DE22 3NE | |
| Tel: 01332 786826 | |
| Fax: 01332 200857 | |
| Email: will.carroll@nhs.net | |
| Participating Site: | Dr Andrew Collinson |
| Consultant Paediatrician | |
| Royal Devon & Exeter NHS Foundation Trust | |
| Barrack Road | |
| Exeter | |
| EX2 5DW | |
| Tel: 01392 403694 | |
| Fax: 01392 406435 | |
| Email: Andrew.Collinson@rdeft.nhs.uk | |
| Participating Site: | Dr Edward Simmonds |
| Consultant Paediatrician | |
| University Hospitals of Coventry & Warwickshire NHS Trust | |
| Clifford Bridge Road | |
| Coventry | |
| CV2 2DX | |
| Tel: 02476 96 7232 | |
| Fax: 02476 967248 | |
| Email: edward.simmonds@uhcw.nhs.uk | |
| Participating Site: | Dr Clare Murray |
| Consultant Respiratory Paediatrician & Clinical Senior Lecturer | |
| Royal Manchester Children's Hospital | |
| Hospital Road | |
| Pendlebury | |
| Manchester | |
| M27 4HA | |
| Tel: 0161 922 2191 | |
| Fax: 0161 922 2191 | |
| E-mail: clare.murray@cmmc.nhs.uk | |
| Participating Site: | Dr Jonathan Couriel |
| Consultant Respiratory Paediatrician | |
| Alder Hey Children's NHS Foundation Trust | |
| Eaton Road | |
| West Derby | |
| Liverpool | |
| L12 2AP | |
| Tel: 0151 252 5911 | |
| Fax: 0151 252 5929 | |
| Email: jcouriel@alderhey.nhs.uk | |
| Participating Site: | Prof Harish Vyas |
| Consultant in PICU and Respiratory Medicine | |
| Queens Medical Centre Campus | |
| Nottingham University Hospital | |
| Derby Road | |
| Nottingham | |
| NG7 2UH | |
| Tel: 0115 9249924 (ext 64841) | |
| Fax: 0115 9420324 | |
| Email: harish.vyas@nuh.nhs.uk | |
| Participating Site: | Dr Caroline Pao |
| Consultant Paediatrician Respiratory and General Paediatrics | |
| Royal London Hospital | |
| Bart's & The London NHS Trust | |
| Whitechapel | |
| London | |
| E1 1BB | |
| Tel: 020 7733 7000 (ext. 3931) | |
| Fax: | |
| E-mail: Caroline.Pao@bartsandthelondon.nhs.uk | |
| Participating Site: | Dr Clare Murray |
| Consultant Respiratory Paediatrician & Clinical Senior Lecturer | |
| University Hospital of South Manchester | |
| Southmoor Road | |
| Wythenshawe | |
| Manchester | |
| M23 9LT | |
| Tel: 0161 291 5876 | |
| Fax: 0161 291 5730 | |
| E-mail: clare.murray@manchester.ac.uk | |
| Participating Site: | Dr David Lacy |
| Consultant Paediatrician | |
| Wirral Universities Teaching Hospital NHS Foundation Trust | |
| Arrowe Park Hospital | |
| Arrowe Park Road | |
| Upton | |
| Wirral | |
| CH49 5PE | |
| Tel: 0151 604 7498 | |
| Fax: 0151 604 7206 | |
| Email: David.Lacy@whnt.nhs.uk | |
| Participating Site: | Dr Hywel Jones |
| General Practitioner | |
| The Surgery | |
| 15 Dereham Road | |
| Mattishal | |
| Dereham | |
| NR20 3QA | |
| Tel: 01362 850227 | |
| Fax:01362 858466 | |
| Email: hywel.jones@nhs.net | |
| Participating Site: | Dr Rob Stone |
| General Practitioner | |
| Hellesdon Medical Practice | |
| 343 Reepham Road | |
| Hellesdon | |
| Norwich | |
| NR6 5QJ | |
| Tel: 01603 486602 | |
| Fax:01 603 401389 | |
| Email: rob.stone@nhs.net | |
| Participating Site: | Dr Stephen Thurston |
| General Practitioner | |
| Wymondham Postmill Close | |
| Wymondham | |
| Norfolk | |
| NR18 0RF | |
| Tel: 01953 602118 | |
| Fax: 01953 605313 | |
| Email: stephen.thurston@nhs.net | |
| Participating Site: | Dr Peter Franklin |
| General Practitioner | |
| Holt Medical Practice | |
| Kelling Hospital | |
| Old Cromer Road | |
| High Kelling | |
| Holt | |
| NR25 6QA | |
| Tel:01 263 712461 | |
| Fax: 01263 713211 | |
| Email: peter.franklin@nhs.net | |
| Participating Site: | Dr Alasdair Duthie |
| General Practitioner | |
| The Surgery | |
| Church Plain | |
| Loddon | |
| Norfolk | |
| NR14 6EX | |
| Tel: 01508 520222 | |
| Fax: 01508 528579 | |
| Email: alasdair.duthie@nhs.net | |
| Participating Site: | Dr Simon May |
| General Practitioner | |
| The Surgery | |
| Overstrand Road | |
| Cromer | |
| Norfolk | |
| NR27 0AJ | |
| Tel:01263 513148 | |
| Fax:01 263 515264 | |
| Email: simon.may@nhs.net | |
| Participating Site: | Dr Alistair Martin |
| General Practitioner | |
| Attleborough Surgery | |
| Station Road | |
| Attleborough | |
| Norfolk | |
| NR17 2AS | |
| Tel: 01953 452394 | |
| Fax: 01953 xxxx | |
| Email: alistair.martin@nhs.net | |
| Participating Site: | Dr Carsten Dernedde |
| General Practitioner | |
| Hoveton & Wroxham Medical Practice | |
| Stalham Road | |
| Hoveton | |
| Norfolk | |
| NR12 8DU | |
| Tel: 01603 782155 | |
| Fax: 01603 xxxx | |
| Email: carsten.dernedde@nhs.net | |
| Participating Site: | Dr Kevin Elsby |
| General Practitioner | |
| The Market Surgery | |
| 26 Norwich Road | |
| Aylsham | |
| Norfolk | |
| NR11 6BW | |
| Tel: 01263 733331 | |
| Fax: 01263 xxxx | |
| Email: Kevin.elsby@nhs.net | |
| Participating Site: | Dr Sanjay Gheyi |
| General Practitioner | |
| Coltishall Medical Practice | |
| St Johns Close | |
| Rectory Road | |
| Coltishall | |
| Norfolk | |
| NR12 7HL | |
| Tel: 01603 737593 | |
| Fax: 01603 xxxx | |
| Email: s.gheyi@nhs.net |
Appendix B Pharmacy Dispensing Procedure for Norfolk & Norwich University Hospital
Trial medications for all Norfolk participants, including those seen at the ten General Practitioners sites, will be stored at and dispensed from the pharmacy department at the Norfolk & Norwich University Hospital (NNUH). Trial medications will be dispensed from pharmacy at the NNUH using the following procedures:
T–4
Where the T-4 visit is conducted at an outreach centre (i.e. a GP practice), if the patient is registered in the trial the PI will write the T-4 prescription for that patient. The RN/PI will arrange with the patient to collect the study medication from the outreach centre on a day agreed with both the patient and the dispensing pharmacy department. On receipt of the prescription, pharmacy will prepare the medication for dispensing. It will be placed in a container with a temperature monitor provided centrally through the MASCOT trial. The medication will be transported via the hospital pharmacy service which delivers hospital dispensed prescriptions to GP practices within the community. Two members of the NNUH pharmacy team will sign and date the accountability log to confirm that the prescription has been dispensed. The medication will be securely transported to the outreach clinic where the patient will collect their study medication.
T0
Where the T0 visit is conducted at an outreach centre (i.e. a GP practice), if the patient is found to be eligible for randomisation and wishes to continue in the trial, the PI will write a trial prescription which will be faxed to pharmacy. The RN/PI will arrange with the patient to collect the study medication from the outreach centre on a day agreed with both the patient and the dispensing pharmacy department. The patient will be allocated their randomisation number by pharmacy as per the standard process detailed in Section 7 and an appropriate treatment pack will be prepared for dispensing. It will be placed in a container with a temperature monitor provided centrally through the MASCOT trial. The medication will be transported via the hospital pharmacy service which delivers hospital dispensed prescriptions to GP practices within the community. Two members of the NNUH pharmacy team will sign and date the accountability log to confirm that the prescription has been dispensed. The medication will be securely transported to the outreach clinic where the patient will collect their medication.
T+8 onwards
The PI/RN will ensure that pharmacy have received a signed valid prescription at least 48 hours before the medication needs to be dispensed. On receipt of the prescription, pharmacy will ascertain the patient's randomised treatment allocation and dispense the appropriate trial treatment pack/s for that patient. The pack/s will be placed in a container with a temperature monitor provided centrally through the MASCOT trial. The medication will be transported via the hospital pharmacy service which delivers hospital dispensed prescriptions to GP practices within the community. Two members of the NNUH pharmacy team will sign and date the accountability log to confirm that the prescription has been dispensed. The medication will be securely transported to the outreach clinic where the patient will be attending for their study visit that day. The medication will then be given to the patient by the RN at the end of the visit if they are eligible and willing to continue to the next part of the study. If the patient is not eligible to continue in the trial or wishes to withdraw from trial treatment at that point, the dispensed medications will be returned to pharmacy for destruction. The RN will collect any previously dispensed used/unused medications from the patient and return them to the hospital pharmacy within five working days for destruction.
Appendix C GP Database Search Guidance
As per ‘Recruitment Strategies – Primary Care #1 and #2’ (Section 6.1), General Practitioners' databases may be searched in order to identify potentially eligible patients for the study. Below are a set of guidelines for facilitating the electronic searches. However, this is not an exhaustive guide and should be adapted to fit the database systems and procedures in use by the individual practice.
ELIGIBILITY CRITERIA SEARCH
1. Age range – 6–14 yrs 11/12 months (SHARED) – shared by all in total group (not incl or excl)
2. Asthma (H33) Any code (incls past/active)
OR children on bronchodilators and/or shorting acting corticosteroids.
No date range incl (SHARED)
3. Bronchodilaters – current prescriptions.
Try 1 script (issue) in last year (SHARED)
4*. Not eligible (exclusion) if prescribed in the previous month and used currently Leukotriene Antagonists
Select drugs: Montelukast Zafilucast/Accolate
Issue (script) 1 in the last month
(EXCLUSION)
5*. Exclusion not shared
Exclude long acting beta agonists (select beta agonist) if prescribed in the last previous month and used currently
Select drugs: Formoteral/fumerate
Seretride (combination flixotide+) Symbicort
Current: 1 script (issue) in the last month
(EXCLUSION)
6. Not shared
Exclude theophyline
Current last year
7. EXCLUSION
Cystic Fibrosis (CF) – C370
Cardiac congenital P6
Exclude P68 congenital heart disease
*Please note that this criterion can be added on as an excluder when conducting the electronic search. However, depending on the time coding methods and the individual database, it may be easier to assess this during a manual notes search after the initial electronic search has been conducted to avoid any potentially eligible patients being prematurely excluded.
TO CREATE A SEARCH ON EMIS LV
Choose ST search & statistics
Select B – patient searches
Select A – build and perform a new search
Select A – search on today's practice population i.e. currently registered pop (SHARED)
Select A – add Feature
Select 2 – AGE upper 14, lower 6 (SHARED)
Select 2 – Classification Codes, type Read Code H33,
Select A – (Asthma**) press return (answer Y to all lower codes)
Select A – no date range, A (SHARED)
Select A – add Feature
Select 7 – Drugs, when prompted ‘Search on all drugs Y/N’ enter N for No
Select B – search by drug group
Select 3 – Respiratory System Drugs
Select 3 – (corticosteroids for inhalation), press return (Y for all variants of drug group)
Press return (C current), press (A – all)
Enter date range 01/01/2008, press return, (today's date)
Press return (blank frequency)
Enter Y to continue, set operator as (SHARED)
NOW ADD IN CRITERIA FOR DRUG EXCLUSIONS
Select A – add Feature
Select 7 – Drugs, when prompted ‘Search on all drugs Y/N’ enter N for No
Select I – Ingredient or brand, type in part of the name of the drug e.g. ‘Monte’ press return
Select 1 – (montelukart sodium (G) (T)) press return, Enter – (Y to all variants), press return
Enter – (C – current) press return
Enter – (A – all)
Enter date range – (6 months ago), press return, (today's date)
Press return (blank frequency)
Enter Y to continue, set operator as B (EXCLUDE)
Select A – add Feature
Select 7 – Drugs, when prompted ‘Search on all drugs Y/N’ enter N for No
Select I – Ingredient or brand, type in part of the name of the drug e.g. ‘Salmet’ press return
Select 1 – (salmeterol xinafoate (G) (T)) press return, Enter – (Y to all variants), press return
Enter – (C – current) press return
Enter – (A – all)
Enter date range – (6 months ago), press return, (today's date)
Press return (blank frequency)
Enter Y to continue, set operator as B (EXCLUDE)
Select A – add Feature
Select 7 – drugs, when prompted ‘Search on all drugs Y/N’ enter N for No
Select I – Ingredient or brand, type in part of the name of the drug e.g. ‘Formo’ press return
Select 3 – (Formoterol Fumarate (G) (T)) press return, Enter – (Y to all variants), press return
Enter – (C – current) press return
Enter – (A – all)
Enter date range – (6 months ago), press return, (today's date)
Press return (blank frequency)
Enter Y to continue, set operator as B (EXCLUDE)
When you have entered the appropriate criteria enter Y to confirm that the features are correct (N if they are not and edit the search) always check your date range & operator status.
Save the search with an appropriate name, prefixed by ‘MASCOT’, to the directory for one off searches. Enter Y to run the search. A message in a yellow band will appear at the top of the screen when the search has completed.
To view the results go to S ‘Search results’, type MASCOT and press return. Select your search
Choose A – A table showing the distribution by age and sex.
Once you are happy with the group of patients the search has generated, via WP Word Processing module mail merge the MASCOT participant invitation letter template onto practice letter headed paper, including the patient's name & address details.
The envelope to be mailed out to the patient and their family should include:
-
the participant invitation letter
-
parent/guardian MASCOT Patient Information Sheet
-
the age-specific MASCOT Patient information Sheet (optional)
-
a stamped SAE for use when returning the reply slip to the local MASCOT team
REMEMBER – only appropriate practice staff can view identifiable patient data. MASCOT research nurses should not be accessing patient names or any data which could easily identify a patient.
List of abbreviations
- A&E
- accident and emergency
- ANCOVA
- analysis of covariance
- ANOVA
- analysis of variance
- BADGER
- Best Add-on Therapy Giving Effective Response
- BNF
- British National Formulary
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CLRN
- Comprehensive Local Research Network
- CONSORT
- CONsolidated Standards of Reporting Trials
- CRF
- case report form
- CTU
- Clinical Trials Unit
- df
- degrees of freedom
- FEV1
- forced expiratory volume in 1 second
- FVC
- forced vital capacity
- GP
- general practitioner
- GSK
- GlaxoSmithKline
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- ICS
- inhaled corticosteroid
- IDSMC
- Independent Data and Safety Monitoring Committee
- IMP
- investigational medicinal product
- IQR
- interquartile range
- ITT
- intention to treat
- MASCOT
- Management of Asthma in School age Children On Therapy
- MCRN
- Medicines for Children Research Network
- MCRN CTU
- Medicines for Children Research Network Clinical Trials Unit
- MDI
- metered dose inhaler
- MedRA
- Medical Dictionary for Regulatory Activities
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MSD
- Merck Sharp & Dohme
- NIHR
- National Institute for Health Research
- OTC
- over the counter
- PACQLQ
- Paediatric Asthma Caregiver's Quality of Life Questionnaire
- PAQLQ
- Paediatric Asthma Quality of Life Questionnaire
- PAQLQ(S)
- Paediatric Asthma Quality of Life Questionnaire with Standardised Activities
- PCRN
- Primary Care Research Network
- QALY
- quality-adjusted life-year
- RR
- rate ratio
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SUSAR
- suspected unexpected serious adverse reaction
- TMG
- Trial Management Group
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.