
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 06/01/02. The contractual start date was in September 2007. The draft report began editorial review in January 2013 and was accepted for publication in July 2013. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2014. This work was produced by Goodacre et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Asthma is a chronic inflammatory disease of the airways characterised by reversible airflow obstruction and bronchospasm. An acute asthma exacerbation (commonly referred to as an asthma attack) is characterised by shortness of breath, wheezing and chest tightness. Acute asthma was responsible for 55,259 emergency admissions and 153,877 bed-days in England in 2011–12,1 and many more emergency department (ED) attendances.
Management of acute asthma
The management of acute asthma in the UK NHS is subject to guidance issued by the British Thoracic Society (BTS) and Scottish Intercollegiate Guidelines Network (SIGN). 2 The severity of an acute asthma attack is categorised on the basis of presenting clinical characteristics into near-fatal, life-threatening, severe, moderate and brittle asthma. Patients with any features of life-threatening or severe asthma are referred to hospital for emergency treatment.
Prehospital and ED treatment of acute asthma in adults includes supplemental oxygen therapy, oral or parenteral steroids (prednisolone or hydrocortisone), nebulised β2-agonist bronchodilators (salbutamol or terbutaline) and nebulised ipratropium bromide. Patients are admitted to hospital if they have any feature of a life-threatening or near-fatal attack or any feature of a severe attack persisting after initial treatment. Patients whose peak flow is greater than 75% best or predicted 1 hour after initial treatment may be discharged from the ED unless they meet any of the following criteria, when admission may be appropriate: persistent significant symptoms; concerns about compliance; living alone/socially isolated; psychological problems; physical disability or learning difficulties; previous near-fatal or brittle asthma; exacerbation despite adequate dose steroid tablets prepresentation; presentation at night; or pregnancy.
Magnesium sulphate in acute asthma
Magnesium is an essential mineral nutrient that is present in every cell of every organism. Magnesium-dependent enzymes appear in virtually every metabolic pathway but notably, magnesium ions block calcium channels and, thus, affect nerve and muscle activity. Magnesium sulphate has an established therapeutic role in pre-eclampsia,3 torsade de pointes4 and hypomagnesaemia. 5 Its use has also been explored in ventricular arrhythmias other than torsade de pointes,6 cardiac arrest,7 myocardial infarction,8 atrial fibrillation9,10 and acute asthma. 11
The use of magnesium sulphate in acute asthma is based on possible smooth muscle relaxation and anti-inflammatory action. It can be given via the intravenous (i.v.) or nebulised route and may have a role augmenting treatment in a therapeutic ‘gap’ between the immediate action of nebulised bronchodilators and the delayed action of steroids. Doses of 1.2–2 g have been evaluated in acute severe asthma,11 although doses of 4–6 g can be used in other conditions. 3 The dose of nebulised magnesium sulphate is limited by the need to avoid administering a hypertonic nebulised solution and concurrent therapeutic need for nebulised β2-agonist. The maximum dose is therefore 500 mg per nebuliser.
Evidence for intravenous magnesium sulphate in acute asthma
Intravenous magnesium sulphate has been compared with placebo in five meta-analyses,11–15 two of which analysed adults separately from children. 11,14 The meta-analyses included a total of 15 randomised trials,16–30 nine of which were undertaken in adults. 16–24 The trials of adults used a bolus dose of either 1.2 g or 2.0 g of magnesium sulphate, given over 20–30 minutes. Only one trial followed the bolus dose with an infusion. 18
The most recent meta-analysis11 included all nine adult trials. 16–24 A variety of methods were used to measure pulmonary function, therefore these outcomes were pooled by calculating a standardised mean difference (SMD). The pooled relative risk (RR) for hospital admission after treatment with i.v. magnesium sulphate was 0.91 [95% confidence Interval (CI) 0.78 to 1.07; p = 0.27] and the pooled SMD in pulmonary function was 0.15 (95% CI 0.01 to 0.29; p = 0.035). The authors concluded that treatment with i.v. magnesium sulphate was associated with a modest improvement in pulmonary function, but the clinical significance of this effect was uncertain. Although there was no significant effect on hospital admission, the summary estimate included a potentially important reduction in admissions of up to 22%. Existing evidence was therefore insufficient to either recommend i.v. magnesium sulphate as standard treatment for acute severe asthma or rule out a potentially valuable role.
One further trial (n = 63) of i.v. magnesium sulphate has been published since the most recent meta-analysis was undertaken. 31 The trial reported a significant effect on predicted forced expiratory volume in 1 second (FEV1) at 120 minutes (62.84% vs. 56.7%; mean difference = 6.07; 95% CI 1.87 to 10.62; p < 0.01) and fewer patients admitted to hospital in the intervention group (2/30 vs. 9/30). Addition of these data to the most recent meta-analysis resulted in a pooled SMD of 0.35 (95% CI 0.06 to 0.64; p = 0.02) and a pooled RR for hospital admission of 0.85 (95% CI 0.68 to 1.06; p = 0.14).
Evidence for nebulised magnesium sulphate in acute asthma
Nebulised magnesium sulphate has been compared with placebo in three meta-analyses11,32,33 and eight randomised trials,34–41 of which five were undertaken in adults,34–38 two in children39–40 and one in a mixed population. 41 The most recent meta-analysis11 was the only one to report trials of adults and children separately. The trial with a mixed population was analysed with the trials of adults. The dose of magnesium sulphate used ranged from 95 to 500 mg, given up to four times, with doses every 20 to 30 minutes. The pooled RR for hospital admission after treatment with nebulised magnesium sulphate was 0.66 (95% CI 0.44 to 1.00; p = 0.048) and the pooled SMD in pulmonary function was 0.20 (95% CI –0.02 to 0.42; p = 0.076). Although the effect of nebulised magnesium sulphate on hospital admissions just reached significance, most of the admissions in this analysis were in one trial,36 and the effect was not consistent across the other trials. The authors concluded that the existing evidence was inadequate to either support nebulised magnesium sulphate as standard treatment for acute severe asthma or rule out a potentially valuable role.
Comparison between intravenous and nebulised magnesium sulphate
No previous trials have compared i.v. with nebulised magnesium sulphate.
Limitations of the existing evidence for intravenous or nebulised magnesium sulphate
The existing evidence for both i.v. and nebulised magnesium sulphate suggests a potentially worthwhile effect on pulmonary function and hospital admissions, but estimates of effect are imprecise and include the possibility of either no clinically worthwhile effect or a substantial effect. Furthermore, the existing evidence is subject to the following limitations:
-
Most previous trials were relatively small and powered to detect changes in pulmonary function rather than hospital admission.
-
Even if meta-analysis suggests a statistically significant difference in pulmonary function it is not clear whether such changes are important to patients or affect their clinical outcome.
-
Factors such as publication bias may influence selection of studies into meta-analysis, leading to overestimates of effectiveness. It has been noted that 35% of subsequent large trials conflict with the results of a previous meta-analysis. 42
-
The clinically important change in admission rate in patients with severe asthma identified in the meta-analysis by Rowe et al. 12 was based on post-hoc subgroup analysis.
-
No previous trials have included a head-to-head comparison of nebulised with i.v. magnesium sulphate.
Current use of intravenous and nebulised magnesium sulphate in the National Health Service
Current BTS/SIGN guidelines for the management of acute asthma2 state that there is limited evidence that, in adults, magnesium sulphate has bronchodilator effects and, although experience suggests that magnesium sulphate is safe when given by the i.v. or nebulised route, trials comparing these routes of administration are awaited. The guidelines suggest considering giving a single dose of i.v. magnesium sulphate to patients with acute severe asthma who have not had a good initial response to inhaled bronchodilator therapy or who have life-threatening or near-fatal asthma. Similar advice is provided by guidelines used in the USA. 43
A postal survey of the use of magnesium sulphate in the treatment of acute asthma in the ED was undertaken in the UK in 2009. 44 The lead clinician of each ED was mailed a survey asking about the use of magnesium sulphate in his or her department and 180 out of 251 responded (72%). Magnesium sulphate was reportedly used in 93% of the EDs, mostly because it was expected to relieve breathlessness (70%) or reduce critical care admissions (51%). Most departments used magnesium sulphate for patients with acute severe asthma (84%) and life-threatening exacerbations (87%), with 68% stating they would give the drug if there was no response to repeated nebulisers. In comparison, nebulised magnesium sulphate was used in only two EDs (1%). The main reason given for not administering via a nebuliser was insufficient evidence (51%). The authors commented that the reported use of i.v. magnesium sulphate was more extensive than current guidelines or available evidence appeared to support.
Research objectives
We aimed to measure the clinical effectiveness and cost-effectiveness of i.v. and nebulised magnesium sulphate in acute severe asthma and, thus, determine whether or not either should be standard first-line treatment for patients presenting to the ED with acute severe asthma.
We planned to test the following specific hypotheses:
-
intravenous or nebulised magnesium sulphate will reduce the proportion of patients who require admission at initial presentation or during the following week
-
intravenous or nebulised magnesium sulphate will improve patient assessment of their breathlessness over 2 hours after initiation of treatment.
We also planned to measure the effect of i.v. or nebulised magnesium sulphate on:
-
length of hospital stay and use of high-dependency unit (HDU) or intensive care unit (ICU)
-
mortality, adverse events and use of respiratory support
-
change in peak expiratory flow rate (PEFR) and physiological variables after initial treatment
-
patient-reported health utility
-
patient satisfaction with care
-
use of health and social services over the following month
-
time taken by patients off work
-
health and social care costs and productivity losses.
Chapter 2 Methods
The 3Mg trial was a multicentre, double-blind, placebo-controlled, three-arm, randomised trial and economic analysis of i.v. or nebulised magnesium sulphate in acute severe asthma. The trial took place in 34 EDs in England and Scotland.
Recruitment and allocation of participants
Adults (age over 16 years) attending the ED with acute severe asthma were recruited to the trial. Acute severe asthma was defined as acute asthma with one or more of the following: PEFR < 50% of best or predicted; respiratory rate > 25 breaths per minute; heart rate > 110 beats per minute; or inability to complete sentences in one breath. The percentage of best or predicted PEFR was calculated using the patient’s recent best PEFR (within 2 years) if it was known. If the recent best PEFR was not known, the predicted PEFR from age and height charts was used. This approach is recommended in BTS/SIGN guidance2 and was used to calculate all estimates of the percentage of best or predicted PEFR used in the trial. For convenience we use the term ‘% predicted PEFR’ to encompass all such estimates.
The following individuals were excluded:
-
patients with life-threatening features, defined as one or more of the following: oxygen saturation < 92% despite supplemental oxygen; silent chest; cyanosis; poor respiratory effort; bradycardia; arrhythmia; hypotension; exhaustion; coma; or confusion
-
patients with a contraindication to either nebulised or i.v. magnesium sulphate: pregnancy; hepatic or renal failure; heart block; or known hypermagnesaemia
-
patients who were unable to provide written or verbal consent
-
previous participants in the 3Mg trial
-
patients who had received i.v. or nebulised magnesium sulphate in the 24 hours prior to attendance at the ED.
The final exclusion criterion was added as a protocol amendment during the trial.
Anonymised basic details (age, sex, time and date of ED attendance) were collected from all potentially eligible patients to allow completion of a CONsolidated Standards Of Reporting Trials (CONSORT) flow chart. Patients were identified by ED medical staff, who completed a patient recruitment form (see Appendix 1) to verify that the patient met the eligibility criteria and to record that consent was taken prior to randomisation. Eligible patients were initially given a brief information sheet with details of the study design, trial treatments and potential side effects (see Appendix 2). When their condition permitted, they were given a full information sheet with further details on trial processes and requirements (see Appendix 3). All patients were required to give consent before being recruited to the trial. If the patient’s condition permitted, full written consent was taken before recruitment using the Research Ethics Committee (REC)-approved consent form (see Appendix 4). If not, verbal consent was obtained from the patient, recorded on the consent form and written consent requested as soon as the patient’s condition improved. No provisions were made for personal or professional legal representation or for recruitment before consent. Therefore, any patient unable to provide written or verbal consent was excluded from the trial. Both oral and written consent were taken in the presence of a witness who also signed the consent form, in addition to the person taking consent.
Once eligibility had been confirmed and consent acquired, the participants were randomly allocated to a treatment group. The recruiting clinician accessed a web-based randomisation system or automated telephone hotline provided by the Sheffield Clinical Trials Research Unit (CTRU) in partnership with epiGenesys (a University of Sheffield subsidiary software development company) and participants were allocated to a numbered treatment pack kept in the ED. The randomisation system only revealed the allocated pack number after patient details had been recorded and the patient irreversibly entered into the trial. A simple randomisation sequence was used in the first 20 hospitals open to recruitment, as planned in the protocol. 45 However, recruitment rates were lower than anticipated and, therefore, additional hospitals were opened to recruitment. To reduce the risk of random imbalances in the number allocated to each arm of the trial, blocked randomisation (block sizes of four or six), stratified by hospital, was used for subsequent centres. To avoid the risk of subversion of the randomisation process in these hospitals, decisions regarding the randomisation sequence were made independently by the CTRU and were not communicated to the investigators.
Each treatment pack contained an i.v. infusion and nebuliser solutions, either of which could be active treatment or placebo. Participants, hospital staff and research staff were all blind to allocated treatment, unless the formal unblinding procedure was undertaken. An emergency unblinding (code-break) procedure was in place to enable hospital staff to reveal the allocation of treatment when it was essential to know for their on-going clinical care whether or not the patient had received magnesium sulphate. A 24-hour unblinding service was available via the randomisation system (online or telephone), which immediately provided treatment allocation to the site and automatically alerted the study team and local principal investigator (PI) by e-mail that a participant had been unblinded. In case the online and telephone systems were unavailable, emergency unblinding envelopes were also prepared by the pharmacy production unit according to the randomisation schedule and stored with the investigational medicinal products (IMPs) at site. Tamper stickers were checked regularly to ensure that envelopes had not been opened and were returned, still sealed, to the central study team to ensure full accountability. If an envelope was opened it was reported to the study team and recorded as a participant unblinding.
Interventions
Patients were randomised to one of three treatment arms. Each patient received one i.v. and one nebulised treatment (consisting of three nebuliser vials given consecutively). The i.v. infusions and nebuliser vials were prepared as apparently identical solutions, with identical primary packaging and labelling to ensure blinding. Blinded treatment packs were assembled and labelled with a participant number in accordance with a randomisation schedule supplied by the CTRU. The three treatment arms are shown in Table 1.
| Treatment arm | Intravenous infusion | Nebulisers |
|---|---|---|
| 1 | i.v. magnesium sulphate, 8 mmol (2 g) in 100 ml of water for injections, adjusted to isotonicity with sodium chloride, given over 20 minutes | 7.5-ml vial of 0.9% saline, given three times 20 minutes apart |
| 2 | i.v. 0.9% saline, 100 ml given over 20 minutes | 7.5-ml vial of 2 mmol (500 mg) magnesium sulphate, given three times 20 minutes apart |
| 3 | i.v. 0.9% saline, 100 ml given over 20 minutes | 7.5-ml vial of 0.9% saline, given three times 20 minutes apart |
All three groups received standard therapy at the discretion of the treating physician, but guided by BTS/SIGN guidelines2 and the 3Mg Clinical Protocol (see Appendix 5). Recommended standard therapy included supplemental oxygen, nebulised salbutamol, nebulised ipratropium bromide and oral prednisolone, administered during recruitment, followed by up to 5 mg of salbutamol added to each trial nebuliser. The BTS/SIGN guidelines2 do not recommend magnesium sulphate, but suggest that i.v. use should be considered in patients with life-threatening features or those with severe asthma who do not respond to treatment. We, therefore, did not prohibit the use of magnesium sulphate outside the trial or recommend its use in patients unresponsive to treatment. During the trial we identified the occasional use of magnesium sulphate at an early stage of treatment of patients without life-threatening features. We therefore added an additional exclusion criteria that patients must not have received magnesium sulphate in the previous 24 hours before entry into the study.
Patients were managed in the ED and data collected until 2 hours after randomisation. At this point, if not already undertaken, a final disposition decision (hospital admission or discharge) was made.
Outcome measures
Two primary outcomes were specified in the trial protocol:
-
The health service primary outcome – the proportion of patients admitted to hospital, either after ED treatment or at any time over the subsequent week.
-
The patient-centred primary outcome – the patient’s visual analogue scale (VAS) for breathlessness over 2 hours after initiation of treatment.
Secondary outcomes included mortality; adverse events; the use of ventilation or respiratory support; length of hospital stay; use of HDU or ICU; change in PEFR and physiological variables (oxygen saturation, heart rate, respiratory rate, blood pressure) over 2 hours; quality of life at baseline and at 1 month [measured by European Quality of Life 5-Dimensions (EQ-5D)]; number of unscheduled health-care contacts (ED, walk-in centre or general practitioner attendances) over the subsequent month; and satisfaction with care.
The two primary outcome measures were selected to identify important changes in patient management and symptoms of asthma. The primary outcome of hospital admission included any admission over the following week, because this time period would encompass the expected duration of an asthma exacerbation and a typical course of associated treatment. Admission during this time therefore represented an overall failure of treatment, whereas admission later than 1 week was considered a separate episode.
The VAS and the Borg scale have both been used to measure breathlessness during exercise,46 but have only recently been tested in acute asthma. Kendrick et al. 47 showed that the Borg scale correlated with measures of respiratory function in a cohort of patients with asthma or chronic obstructive pulmonary disease, whereas Karras et al. 48 and Gupta et al. 49 showed correlation between the VAS and measures of respiratory function in cohorts with acute asthma. The study by Karras48 also showed that the mean VAS change among patients who reported their asthma to be ‘a little better’ after treatment was 22 mm on a 100-mm VAS and concluded that this represented a minimum clinically significant change. On the basis of these studies we concluded that the VAS was the best validated measure, offering a simple and reliable means of measuring symptomatic breathlessness in people with acute asthma, with an estimate of the minimum clinically significant change in VAS.
Outcomes were measured in two phases: (1) over 2 hours after randomisation and (2) at 1 month after attendance. During the first phase we measured variables that reflect patient response to emergency treatment, such as VAS, PEFR and physiological variables. During the second phase we measured variables that reflect the overall patient experience of an asthma attack and its subsequent treatment, such as adverse events, use of health services, satisfaction with care and quality of life.
Data collection and management
A recruitment form was completed for all patients attending the ED with acute severe asthma at participating sites. These data were used to complete the CONSORT flow chart and generate reports on non-recruited patients for discussion at management group meetings.
Clinical staff recorded baseline data, details of co-interventions and outcome data up to 2 hours after randomisation on the paper case report form (CRF) (see Appendix 6). Further data were collected up to 1 month after recruitment by research nurses using routine data sources and by patient self-completion questionnaire. A postal survey consisting of the EQ-5D, a health-care resource use questionnaire, and a patient satisfaction questionnaire was mailed to all participants who were alive and had not withdrawn from the trial 1 month after recruitment (see Appendices 7 and 8). A repeat mailing was undertaken for all non-responders at 2 weeks, with telephone completion of the EQ-5D attempted 2 weeks later for non-responders to either mailings. Patients who had not responded to mail or telephone contact by 8 weeks after study entry were recorded as lost to follow-up for questionnaire data.
Trial data were entered via web-based interface to a database developed in-house by the CTRU. The system, and its underlying database, resided on a server in Corporate Information and Computing Services at the University of Sheffield. Automated backups were made nightly. Prospect was accessed remotely via a secure web browser and all data transmissions were encrypted. Access was restricted by username and password (issued by the CTRU) and an automated audit trail recorded when (and by which user) records were created, updated or deleted. The profile of each user was set to allow only the appropriate information to be viewed and edited, e.g. site data inputters could only enter and view data about patients from their own sites. Quality control procedures were applied to validate the trial data. Error reports were generated where data clarification was required and data queries resolved by research nurses at sites. All activities were performed in accordance with Sheffield CTRU standard operating procedures (SOPs).
Proposed sample size
We planned to recruit 1200 participants, divided equally between the three trial arms (400 participants per arm). We anticipated that the hospital admission would be recorded for all participants, but that a proportion of cases would not have VAS measured. The sample size would therefore provide the following statistical power:
-
Proportion of patients admitted – assuming that 80% of patients with severe asthma are admitted after ED management, the study would have 90% power to detect a 10% absolute reduction in the proportion admitted (i.e. to 70%) for any pair of treatment groups compared (α = 0.05).
-
VAS breathlessness – assuming that the standard deviation on a 100-mm VAS is 3 cm, that 2.2 cm represents a minimum clinically significant difference48 and that 20% of participants will not have their VAS measured, the study would have 90% power to detect a 8-mm difference in a 100-mm VAS at 2 hours after treatment initiation (α = 0.05).
Statistical analysis
Analysis of coprimary outcomes
For the health service primary outcome, patients were considered to have been admitted if either (i) they had not been discharged within 4 hours and/or (ii) if they were recorded as having been readmitted at any point within 7 days following randomisation. The proportion admitted was analysed using logistic regression. We anticipated that data would be missing from only a very small proportion of the trial population (those withdrawing within a few hours of recruitment) and, therefore, planned only a complete case analysis.
For the patient-centred primary outcome, breathlessness was defined as the change in VAS from baseline to 2 hours and was analysed using linear regression. As both missing data and measurements outside the allotted time window were anticipated, the analysis plan proposed three analyses:
-
‘As is’ – complete case analysis, no adjustment is made for timing.
-
‘Imputed’– measurements more than ± 15 minutes from their scheduled time were adjusted by linear interpolation or extrapolation.
-
Multiple imputation by chained estimation. Missing 2-hour VAS scores (n = 108; 10%) were imputed based on age, sex, smoking status, previous admission for asthma, previous admission to ICU or HDU for asthma, time since last admission for asthma, baseline and 1-hour VAS, PEFR and heart rate over 2 hours’ observations, and status at 4 hours (i.e. discharged or admitted/awaiting decision).
Analysis was undertaken on an intention-to-treat basis. Patients were removed from analysis post randomisation only if recruitment was an unequivocal protocol violation (i.e. no consent had been recorded or if they had previously been recruited) or if the patient withdrew from the trial prior to any treatments having been administered. In all other cases, participants were analysed in accordance with the groups they were allocated to regardless of whether or not they actually completed their allocated treatment. A secondary, per-protocol analysis excluded participants who did not receive treatment, defined as a minimum nebulised dosage of 7.5 ml (the equivalent of one nebuliser) or 50 ml of i.v. volume (50% of the i.v. dose).
The primary and most of the secondary analyses included two covariates: treatment group and centre (hospital). For the purposes of the analysis, hospitals recruiting fewer than 10 patients in total were combined into one group for analyses in which centre was used as a covariate. The robustness of the findings to potential differences in baseline characteristics was assessed, in particular the initial breathlessness (VAS) and age.
We used Simes’ method,50 which is a modification of the Bonferroni method but has better power to adjust for multiplicity arising from having two primary outcomes. However, we did not adjust the CIs associated with the estimate of the treatment effect with each outcome. We tested the two hypotheses simultaneously through the analysis of variance. With three groups (A = nebuliser, B = i.v. and C = control) we had two degrees of freedom for analysis, which we split into two orthogonal contrasts (–2, +1, +1) to contrast both active treatments compared with control and (0, –1, +1) to contrast the active treatments.
Secondary outcomes
The length of hospital stay was analysed using a log-normal distribution, which allowed for interval censoring of non-admitted patients and right censoring of hospital duration among those still in hospital 30 days after randomisation. PEFR and physiological measures were analysed in the same manner as breathlessness. All secondary analyses were undertaken as complete case, intention-to-treat analyses.
Subgroup analyses
We prospectively defined three subgroup analyses, within which patients were stratified on the basis of:
-
Asthma severity, according to whether baseline PEFR (pretreatment PEFR as a percentage of predicted value) was above or below median baseline PEFR. A previous meta-analysis12 suggested that i.v. magnesium sulphate is more effective in patients with severe asthma.
-
Age, above or below 50 years. Older patients with a diagnosis of asthma are more likely to have chronic respiratory disease that may be less responsive to treatment with magnesium sulphate.
-
Treatment before arrival. We recruited patients on arrival at hospital, thus testing magnesium sulphate as a first-line treatment. However, some patients received prehospital treatment with nebulisers, thus making magnesium sulphate, in effect, a second-line treatment. Patients with severe asthma after receiving prehospital treatment are likely to have more severe asthma than those presenting without prehospital treatment.
Economic evaluation
The economic evaluation took an approach consistent with the National Institute for Health and Care Excellence (NICE) reference case analysis. 51 The perspective taken was that of the NHS and personal social services. Health benefits were measured in quality-adjusted life-years (QALYs) using the EQ-5D and the time horizon over which health benefits were derived was 30 days. Health benefit was also measured by assessing breathlessness on a 100-mm VAS at 1 and 2 hours after the initiation of study treatment. The data collected 2 hours after the initiation of treatment were used in the cost-effectiveness analysis.
The following resources were identified as being important and collected using either the CRF or patient-completed questionnaire:
-
trial medication
-
ICU – asthma related (days)
-
HDU – asthma related (days)
-
ward – asthma related (days)
-
ICU – non-asthma related (days)
-
HDU – non-asthma related (days)
-
ward – non-asthma related (days)
-
telephone health advice [e.g. general practitioner (GP), NHS Direct] (number of times used)
-
GP surgery consultations (number of times used)
-
GP home visits (number of times used)
-
nurse home visits (number of times used)
-
social worker visits (number of times used)
-
ED attendances (number of times used)
-
attendance at hospital as an outpatient (number of times used)
-
asthma-related concomitant medications: salbutamol, prednisolone, ipratropium bromide, salmeterol xinafoate/fluticasone propionate (Seretide®, GlaxoSmithKline), budesonide (Pulmicort®, AstraZeneca), montelukast, beclometasone dipropionate (Clenil® Modulite®, Chiesi Ltd), aminophylline, theophylline, hydrocortisone, tiotropium (Spiriva®, Boehringer Ingelheim Ltd), magnesium sulphate, salmeterol, ipratropium bromide/salbutamol (Combivent®, Boehringer Ingelheim) and terbutaline (Bricanyl®, AstraZeneca UK Ltd).
Inpatient stays and medications were collected via CRFs, with the remainder collected by patient questionnaire. Productivity loss as a consequence of the number of days patients took off work during the study was collected using the patient questionnaire and separate analyses were conducted excluding and including productivity loss.
Unit costs for medication were taken from the British National Formulary (BNF),5 with the implied dose being that of the most common recorded within the trial (Table 2). Other unit costs were derived primarily from NHS reference costs52 and the Personal Social Service Research Unit annual unit costs publication. 53 NHS reference costs were inflated to 2011/12 prices using the gross domestic product deflator (as the relevant Hospital and Community Health Services Index figure is not yet available). The costs of NHS Direct advice were estimated from an evaluation of this service54 and the costs of production losses were estimated using data from the Office for National Statistics. 55 These unit costs are shown in Table 3.
| Item of resource (medicationsa) | Route | Dose | Cost per doseb (£) |
|---|---|---|---|
| Treatment arm 1 (2 mg of i.v. magnesium sulphate) | – | – | 7.39 |
| Treatment arm 2 (2 mg of nebulised magnesium sulphate) | – | – | 7.39c |
| Treatment arm 3 (saline) | – | – | 0 |
| Aminophylline | Oral | 1 × 225 mg | 0.04 |
| Aminophylline | i.v. | 2 × 10-ml ampoule | 1.56 |
| Beclometasone dipropionate | Inhaled | 2 × 100 µg | 0.07 |
| Combivent | Nebulised | 1 × 2.5 ml | 0.40 |
| Hydrocortisone | Oral | 1 × 20 mg | 1.53 |
| Hydrocortisone | i.v. | 2 × 100 mg | 2.16 |
| Ipratropium bromide | Nebulised | 1 × 500 µg | 0.37 |
| Ipratropium bromide | Inhaled | 2 × 20 µg | 0.05 |
| Magnesium sulphate | i.v. | 1 × 2 g | 7.39 |
| Methylprednisolone | Oral | 2 × 16 mg | 1.14 |
| Montelukast | Oral | 1 × 10 mg | 0.96 |
| Prednisolone | Oral | 8 × 5 mg | 0.35 |
| Pulmicort | Nebulised | 1 × 1 mg | 30.30 |
| Pulmicort | Inhaled | 1 × 200 µg | 0.12 |
| Salbutamol | i.v. | 1 × 5 mg | 0.19 |
| Salbutamol | Nebulised | 1 × 5 mg | 0.19 |
| Salbutamol | Inhaled | 2 × 100 mg | 0.02 |
| Salmeterol (Serevent®, GlaxoSmithKline) | Inhaled | 2 × 25 µg | 0.46 |
| Seretide | Inhaled | 2 × 250 µg | 0.60 |
| Terbutaline | Inhaled | 1 × 500 µg | 0.07 |
| Theophylline | Oral | 1 × 200 mg | 0.05 |
| Tiotropium | Inhaled | 1 × 18 µg | 1.06 |
| Item of resource | Cost per unit (£) | Year | Citation |
|---|---|---|---|
| Telephone advice from NHS Direct | 25 | 2011/12 | 54 |
| GP surgery consultations | 37 | 2011/12 | 53 |
| GP home visits | 124 | 2011/12 | 53 |
| Nurse home visits | 37 | 2011/12 | 53 |
| Social worker visits | 108 | 2011/12 | 53 |
| ED attendance | 96 | 2011/12 | 52 |
| Outpatient visits – asthma related | 133 | 2011/12 | 52 |
| Inpatient days – asthma related | 358 | 2011/12 | 52 |
| Inpatient days – asthma and other related | 261 | 2011/12 | 52 |
| ICU days | 868 | 2011/12 | 52 |
| HDU days | 623 | 2011/12 | 52 |
| Days off work | 101 | 2011/12 | 55 |
Quality-adjusted life-years were calculated for each patient over the duration of the study using the EuroQoL tariff56 and by applying the trapezoidal rule based on data at baseline and at 30 days. The change from baseline in breathlessness 2 hours after the initiation of study treatment, as measured using a 100-mm VAS, was calculated for each patient. Reduction from baseline is defined as minus one times the change from baseline to reflect the fact that a reduction in breathlessness is a positive outcome.
The primary analysis was a cost-effectiveness analysis using the QALYs associated with treatment. Additionally, the change from baseline in breathlessness 2 hours after the initiation of study treatment, as measured using a 100-mm VAS, was used as a secondary cost-effectiveness analysis. The focus of the analysis is on the probability that the intervention arms are cost-effective at funding thresholds of £20,000 and £30,000 per QALY. In addition, cost-effectiveness acceptability frontiers are presented over the range £0–100,000 per QALY. As with all cost-effectiveness analyses, it is necessary for the measure of effectiveness to be linear in the sense that the value of an incremental increase of E units of effectiveness is EK, where K is the value to the decision-maker of increasing effectiveness by one unit. Unlike QALYs, it is not clear how much value a decision-maker may give to a unit reduction in breathlessness on the VAS scale, but is likely to be much less than that for a QALY. Cost-effectiveness acceptability frontiers are presented over the range £0–1000 per unit reduction in breathlessness. Sensitivity analyses were performed incorporating production losses (i.e. time taken from paid employment).
Missing data were imputed using a single-value imputation approach. A multiple imputation algorithm was attempted but it failed to converge, possibly as a consequence of the large number of missing data associated with some outcome measures and the assumptions being made about their underlying probability distributions in the multiple imputation algorithm.
The analysis was initially planned to use bootstrapping, but this was replaced by an analysis using a Bayesian approach using a bivariate normal likelihood function for the effectiveness and total cost. 57 The bivariate normal likelihood function is justified by appealing to the central limit theorem, which says that for data from any underlying probability distribution with finite mean and variance, the distribution of the sample means will tend to be a normal distribution with sufficiently large sample sizes. This change was undertaken in advance of the analysis commencing.
The analysis was conducted using the freely available software WinBUGS 1.4.3 (Imperial College and Medical Research Council, UK). 58 Weak prior information was included in the analysis. The model converged very quickly but for accuracy and precision purposes the results are based on 100,000 Markov chain Monte Carlo iterations after a burn-in of 25,000 iterations.
With sample sizes as large as those in this study, the results are expected to be similar to what would have been generated using bootstrap or a more exact approach that specifically models the underlying distribution of the derived effectiveness and total cost data. Nevertheless, a benefit of this approach is that it allows a more direct interpretation about the probability associated with the parameters of interest (i.e. population mean incremental cost and effectiveness) and the probability of positive net benefit.
Predictors of unsuccessful treatment
To maximise the value of this project, we planned to undertake an additional analysis of trial data to identify factors that predicted unsuccessful treatment for acute severe asthma. Predicting unsuccessful treatment would be helpful for deciding which patients need asthma nurse review after discharge,59 which need hospital admission and which need HDU or ICU support. Currently these decisions are made largely on PEFR recordings,2 although it is not clear how useful these are as predictors of relapse.
Data collection for the trial included variables that may be potentially useful predictors of unsuccessful treatment, such as baseline and post-treatment PEFR, physiological variables, age, sex, smoking status, and previous hospital, HDU and ICU admissions. We examined the ability of these factors to predict unsuccessful treatment, defined at two levels: (1) need for critical care, i.e. HDU or ICU care, airway management, respiratory support or cardiopulmonary resuscitation or respiratory arrest, cardiac arrhythmia or death within 7 days of initial attendance; and (2) need for emergency medical treatment (including critical care) within 7 days of presentation, either by attendance at the ED or unscheduled inpatient review.
Models were fitted separately for the two definitions. For each, an initial screening phase assessed potential covariates, with those achieving a minimum significance level of p < 0.15 retained for the multivariable modelling. Outcomes were modelled using logistic regression. Continuous covariates (physiological variables, PEFR and age) were modelled using fractional polynomials,60 but the resultant model compared with two alternative functional forms (linear and quadratic) to assess whether or not a simpler model could achieve an adequate fit. Predictive ability was assessed by calculating the area under the receiver operator characteristic (AUROC) curve of the model. Internal validation was performed by two methods: (1) bootstrap validation, assessing the robustness of model covariates and (2) cross-validation. The model was fitted separately to the following subgroups defined by (i) seasonality (October–March vs. April–September), (ii) time period (2008–10 vs. 2011–12) and (iii) type of hospital (teaching vs. non-teaching). In each case the resultant models were assessed for consistency between the subgroups.
For each definition, three models were incrementally compared:
-
a model based solely on %PEFR at admission
-
a model using the best-fitting combination of baseline (pretreatment) physiological covariates
-
a model incorporating change in physiological measures over 2 hours, in addition to those used in model 2.
Ethical issues
The trial was undertaken in accordance with the Medicine for Human Use (Clinical Trials) Regulations 200461 and subsequent amendments, and was approved by the Scotland A REC. The main ethical issue was that patients with acute severe asthma may lack capacity to provide informed consent or the ability to complete a written consent form, yet the very nature of the trial required that recruitment should take place quickly in an emergency and include acutely ill patients. 62,63 We initially planned to use personal or professional legal representatives to provide proxy consent for patients lacking capacity. However, on the advice of the ethics committee this provision was dropped and only patients able to provide some form of consent were recruited to the trial. In addition to the person taking consent, a witness also had to sign the consent form to verify that the patient had capacity to give informed consent. It was felt that patients who were too ill to consent were likely to have life-threatening asthma and that excluding patients lacking capacity would not compromise validity.
Participants were therefore only recruited into the trial if they could provide written or verbal-informed consent. We used the following process for seeking consent, based on the Medicine for Human Use (Clinical Trials) Regulations 2004 and subsequent amendments, and taking into account the ethics committee review:
-
All patients were given emergency treatment with supplemental oxygen, salbutamol nebuliser and ipratropium bromide nebuliser while consent was being sought. Initial investigations, such as arterial blood gas sampling and chest radiography, continued simultaneously.
-
Potential participants were given a brief initial information sheet (see Appendix 2) and asked if they wished to consider participation in the trial.
-
Those patients that would consider participation were given further verbal information.
-
Potential participants who were able to express their consent and were able to complete the consent form (see Appendix 4) were asked to provide written consent.
-
Potential participants who were able to express their consent but unable to complete the consent form as a result of their acute illness were recorded on the consent form as having provided verbal consent.
-
If the potential participant was not competent to give written or verbal consent, then he or she was not recruited into the trial.
-
Every recruited participant was reviewed at regular intervals during their treatment. As soon as their condition improved, they were provided with the full information sheet (see Appendix 3). Those participants who had completed a written consent form were asked if they were happy to remain in the trial. Those participants who had not completed a written consent form were asked to do so.
The risks to participants in this trial were considered to be low. Magnesium sulphate has been used by i.v. and nebulised routes in a number of trials and, although unlicensed, has frequently been used in the treatment of acute severe asthma. It is also included as a possible treatment for acute asthma in BTS/SIGN guidelines. 2 Although minor side effects such as nausea or flushing are common, serious side effects (arrhythmias and coma) are uncommon. Potential participants were advised of these risks when they were invited to participate.
Research governance
The trial was conducted in accordance with Medical Research Council (MRC) Guidelines for Good Clinical Practice (GCP)64 and the Medicines for Human Use (Clinical Trials) Regulations 2004. Sheffield Teaching Hospitals (STH) NHS Foundation Trust was the trial sponsor. The trial was an IMP trial covered by clinical trial regulations from the Medicines and Healthcare products Regulatory Agency (MHRA). A clinical trial authorisation was applied for and received from the MHRA. A site agreement between the sponsor, participating site, CTRU and University of Sheffield outlined responsibilities of all parties and was signed prior to commencement of recruitment at sites.
All clinicians responsible for recruiting patients to the trial were required to complete training in GCP. This presented substantial logistical barriers as, at the time the trial commenced, there was no requirement for GCP training as part of emergency medicine specialist training and there was a rapid turnover of doctors in emergency medicine. To address this we established GCP training on the emergency medicine specialist training curriculum, developed a GCP training package and made it available through the College of Emergency Medicine website, and promoted GCP training in the Emergency Medicine Journal. 65
Blinded treatment packs were manufactured in conjunction with the CTRU, initially by STH NHS Foundation Trust Pharmacy Production Unit at the Royal Hallamshire Hospital (RHH) and subsequently by Tayside Pharmaceuticals. All products were checked by a qualified person (QP) prior to release. The change to Tayside Pharmaceuticals was necessary as RHH did not maintain its manufacturer’s authorisation for IMPs, the manufacturer’s/importer’s licence (MIA), after the production of the first batch of IMPs. However, RHH continued to label the kits with a randomisation code in accordance with a randomisation schedule supplied by the CTRU and distribute kits to sites. This assembly service is permitted under the exemption for hospitals (regulation 37, UK SI 2004/1031). The pharmacy production units maintained an investigational medical products dossier (IMPD) (see Appendix 9) and relevant documentation.
Blinded treatment kits were manufactured, assembled and labelled as per European Commission Good Manufacturing Practice annex 13 requirements66 to enable the treatment to be identified and the batch source of the materials traced. Treatment kits consisted of a box containing an infusion bag and three nebuliser solution vials. Boxes carried an outer label identifying the trial and kit number. An unblinded kit number list and randomisation schedule (accessed via the online randomisation system using a pharmacy production unique username) allowed the RHH production unit to identify which arm of the trial each kit belonged to, and label the kits with a randomisation code. IMPs were supplied on a demand basis to the participating sites with minimal waste of materials. Treatment kit accountability logs were maintained by all parties (production units, CTRU, sites, hospital pharmacies), to allow full reconciliation of IMPs including assignment to patients.
Three committees were established to govern the conduct of the trial: the Trial Steering Committee (TSC), the independent Data Monitoring and Ethics Committee (DMEC) and the Project Management Group (PMG). These committees functioned in accordance with Sheffield CTRU SOPs.
The TSC consisted of four independent members [Professor of Respiratory Medicine (as chairperson), statistician, consultant in emergency medicine, patient representative] and three members of the trial team (chief investigator, emergency medicine co-applicant on the grant and the project manager). The TSC supervised the trial and in particular, the progress of the trial, adherence to protocol, patient safety and consideration of new information. The TSC made the decisions on how to proceed with the trial following recommendations from the DMEC.
The DMEC consisted of an independent statistician, a respiratory consultant and an emergency medicine consultant. The principal duty of the DMEC was patient safety. The DMEC agreed a charter stating the intended interim analyses and stopping rules for the study, and made recommendations to the TSC and PMG. The DMEC could make recommendations to:
-
continue recruiting
-
stop the trial
-
continue, with modification to the protocol.
The PMG consisted of the chief investigator, co-applicants, project manager and co-ordinators, statistician, research nurses and sponsor representative. The role of this group was to oversee the day-to-day management of the trial.
Reporting of serious adverse events
Serious adverse events (SAEs) were reported in accordance with the 3Mg trial SAE reporting protocol and the sponsor’s (STH) SOP67 for reporting, managing and recording adverse events for STH studies. All SAEs occurring within 30 days of recruitment to the trial were reported immediately to the sponsor on learning of their occurrence. Site trial staff and delegated ED staff were responsible for recording all adverse events that were reported by the participant and making them known to the PI. An investigators’ brochure (IB) was maintained by the trial team as the reference safety information for reporting SAEs (see Appendix 10).
Magnesium sulphate is a naturally occurring compound that is a normal constituent of the human body and, since the trial involved administering magnesium sulphate over a single 1-hour period, it was expected that any effect on other medications would be limited to the first few hours after administration. Thus, the SAE reporting procedure for the 3Mg trial only recorded those concomitant medications given in the 48-hour period after the trial drug was administered.
Reporting of protocol violations and deviations
Protocol violations and deviations were reported in accordance with the 3Mg and STH protocol violation and deviation SOPs. The site research nurse was responsible for reviewing the participant CRF and ED notes after entry into the trial to determine if treatment was given in accordance with the protocol, consent was obtained correctly and by a suitably trained and delegated doctor, and that the patient met the eligibility criteria. Any suspected protocol violation or deviation was reported to the local PI and to the central 3Mg team. The chief investigator/CTRU reviewed and confirmed if the incident was a violation/deviation and reported to the sponsor. Participants continued to participate in the trial except if the patient had given no informed consent or if they have requested to be withdrawn from the study. Fully consented patients enrolled on the trial were followed up and analysed as per intention-to-treat analysis outlined in the 3Mg protocol (see Appendix 11).
Trial monitoring
Throughout the trial there was ongoing management and monitoring to ensure that the integrity of the data and the rights and well-being of participants were protected. Monitoring was completed both at site and at a central level, and regular reports were submitted to relevant parties.
Reporting
The trial team were required to submit annual reports on trial progress, data completion rates, and safety and protocol compliance to the MHRA and MREC; and 6-monthly reports to the funding body [National Institute for Health Research (NIHR) Health Technology Assessment programme]. Reports were also prepared for all-trial oversight committees.
Site monitoring
On-site monitoring was performed before (prior to recruitment commencing at site), during (after third patient recruited and then annually) and after recruitment ended at a trial site. Monitors checked the following during site visits:
-
source data verification – data recorded on the CRFs against available source documents
-
SAEs/suspected unexpected serious adverse reactions – reported to the sponsor and followed up to resolution
-
resolution of data queries
-
investigator site file maintenance
-
training records for site staff (3Mg trial specific and GCP) and appropriate delegation of duties
-
IMP accountability and storage of IMPs in both ED and pharmacy
-
patient consent procedures
-
reporting of protocol deviations/violations.
Central data validation
Validation checks were built into the database which enabled validation reports to be generated monthly. Any missing values, values out of range or inconsistent with the data set were flagged. Queries were sent to sites and followed up to resolution prior to data lock. Data entry validation was completed on 5% of CRFs and questionnaires, queries resolved, and database entry error rates reviewed to assess if they were within acceptable limits.
Production of investigational medicinal product
Monitors independent of the study team checked QP-release certificates for all batches of product and verified that labelling with randomisation number had been done correctly according to the randomisation number and unblinded kit list.
During the trial we had to change the supplier of the IMPs. Initially the IMPs were supplied by the pharmacy production unit at RHH. However, this unit did not maintain a MIA licence and subsequently stopped fully supporting IMP trials during the trial, so we moved production to Tayside Pharmaceuticals. Labelling and distribution services remained the responsibility of the RHH pharmacy production unit.
Changes to the trial protocol
All changes to the trial protocol and study conduct were reviewed by the sponsor and submitted to the REC and MHRA for approval as appropriate. In summary, the following amendments were made:
-
prior to first participant recruitment:
-
change of sponsor from the University of Sheffield to STH NHS Foundation Trust
-
option for consent from a legal representative removed, written or oral informed consent must be obtained, as required by the REC
-
changes to statistical analysis plan to clarify that the primary analysis incorporated an adjustment for hospital; clarification that covariate adjusted analysis would be performed; subgroup analysis for asthma severity to be based on PEFR instead of VAS score (prior to recruitment commencing).
-
-
during trial recruitment:
-
changes to the number of recruiting sites and list of participating sites/PIs
-
minor changes to study documents for clarity or administrative purposes
-
changes to the storage requirements for the IMP, to allow storage at temperatures up to 30 °C
-
research alert page introduced as a study document to use at sites
-
IMPD and IB updates
-
option to telephone patients to collect EQ-5D data if no response from initial and reminder postal questionnaire
-
pharmacy production unit changed to Tayside Pharmaceuticals
-
addition of extra exclusion criteria – patients who have received i.v. or nebulised magnesium sulphate in the previous 24 hours prior to attendance at the ED
-
clarification that concomitant medications in SAE reports to be reported only for the period up to 48 hours post IMP administration
-
extension of recruitment period to 30 June 2012
-
change from ‘doctors will consent the patient’ to include the option for other health-care professionals to take consent.
-
Archiving
The site files and all study documentation were sent to the CTRU and archived according to the sponsor SOP for a period of 15 years. A log of all documents archived and a list of named individuals who can access the archive is kept by the CTRU.
Chapter 3 Results
Trial progress
The project started on 1 September 2007. Original recruitment predictions were based on the assumption of 12 hospitals recruiting one patient per centre per week for 2 years, with recruitment complete by the end of February 2010. The process of setting up the trial took much longer than anticipated and, after the start of recruitment at the first site, it took a further year until the original target of 12 recruiting sites was achieved. The main reasons for the delays were (a) slow progress in drawing up contracts between the sponsor and participating sites; (b) slow progress in securing NHS research governance approval at the participating sites; and (c) the time taken to provide GCP training for recruiting clinicians. Ethical approval, by contrast, was secured quickly and efficiently. The need to provide GCP training for emergency physicians was a specific problem for this trial. Owing to the acuity of the condition, patients could present to the ED at any time and we aimed to train as many emergency physicians as possible. There appeared to be no existing NHS infrastructure for supporting trials in emergency medicine, so we developed a GCP training package that was proportionate to the limited trial activities that the recruiting doctors were involved with and promoted it through the College of Emergency Medicine.
Patient recruitment
The trial opened to recruitment on 30 July 2008 and closed on 30 June 2012. A total of 1109 participants were recruited to the trial, 92% of the intended sample size of 1200. Figure 1 shows the number of patients recruited and the number of recruiting centres open per month, alongside the cumulative recruitment to the trial. Figure 2 shows the actual recruitment compared with the initial planned recruitment and subsequent revised recruitment plan.
FIGURE 1.
Number of recruiting centres and actual recruitment rates (30 July 2008–30 June 2012).
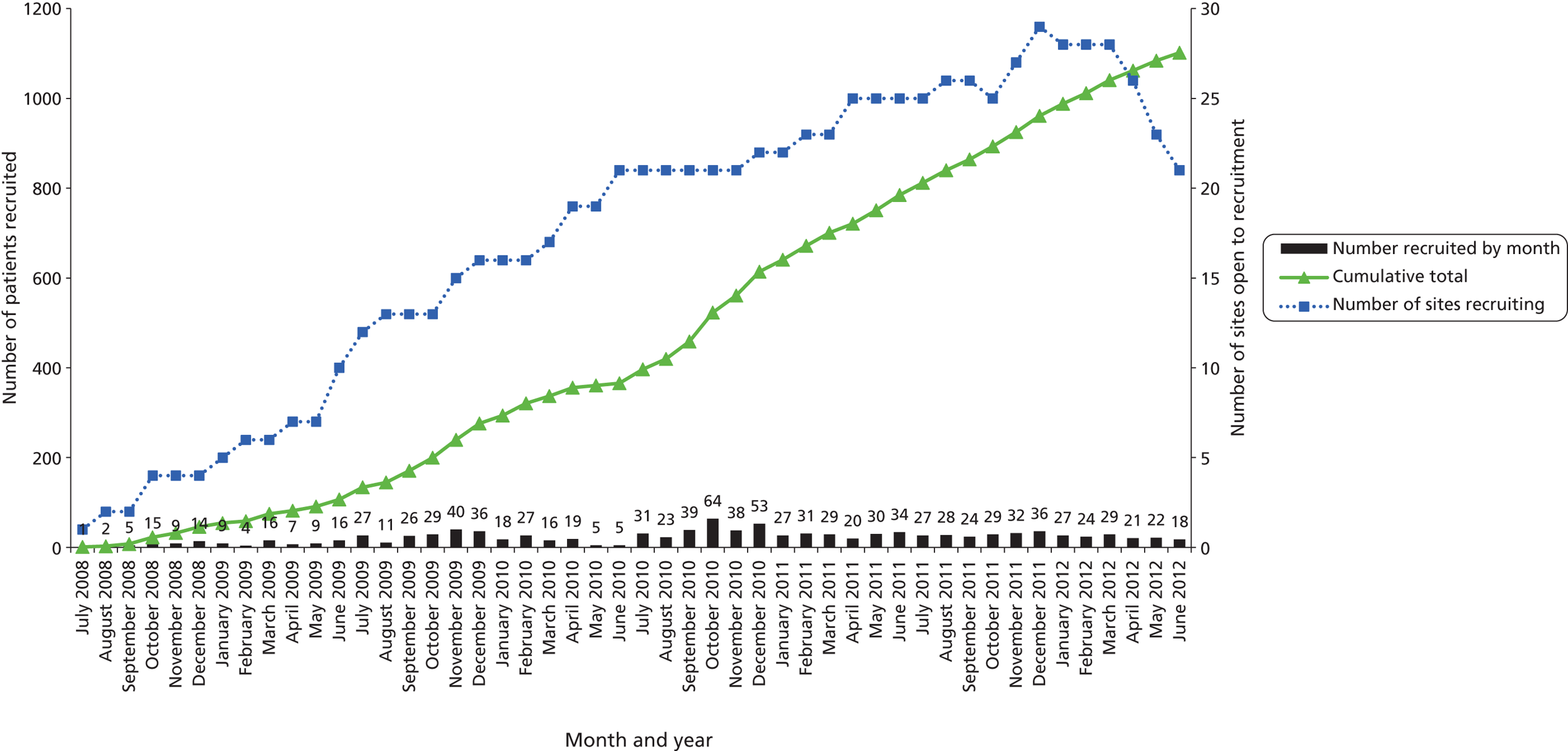
FIGURE 2.
Predicted vs. actual recruitment of participants.
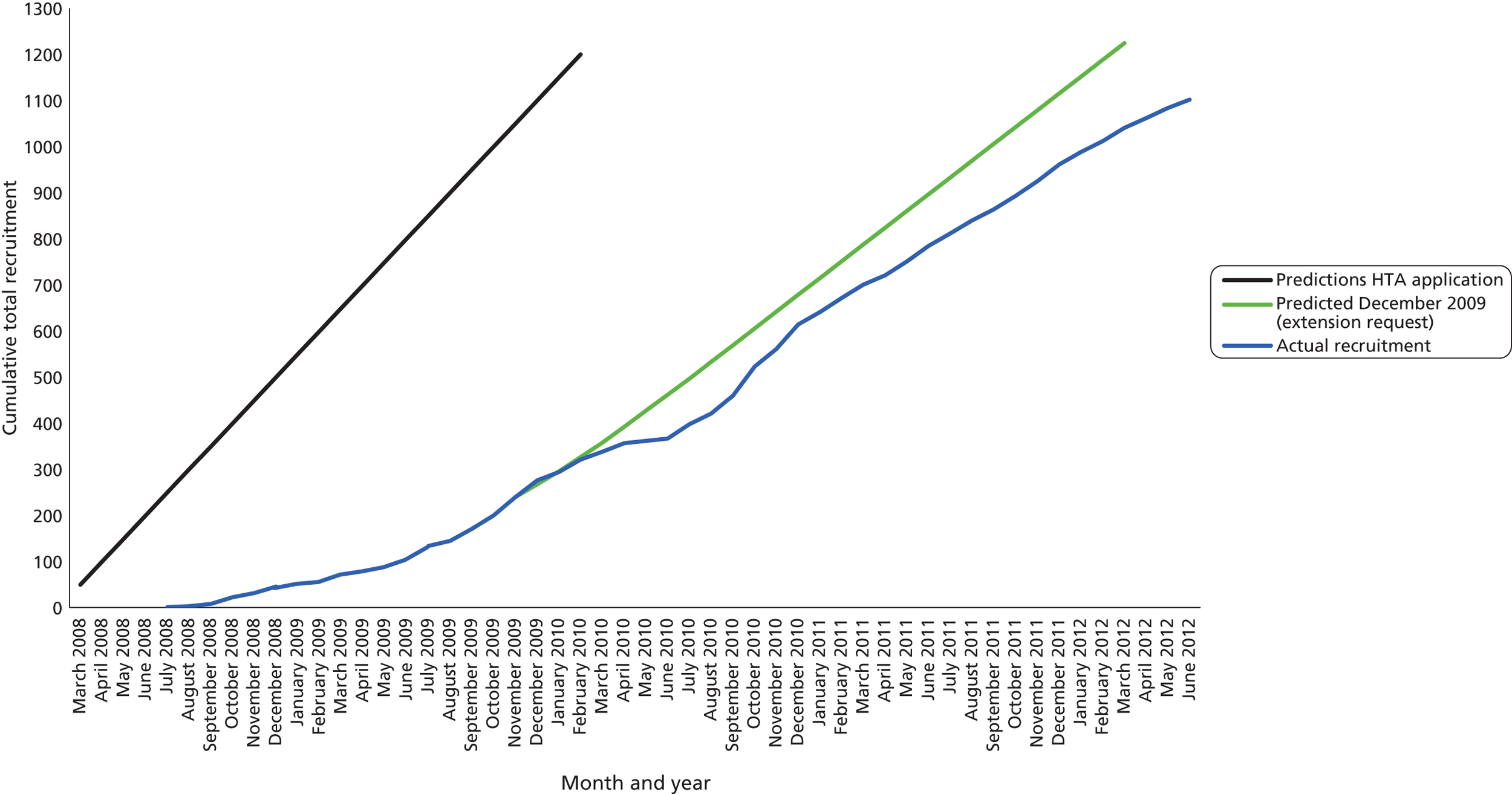
Recruitment at the participating centres was slower than expected as a result of a combination of a lower than anticipated availability of eligible patients and difficulties in ensuring that GCP-trained staff were available to recruit. To address the shortfall in recruitment, we increased the number of sites and promotional activities. Later in the trial we replaced existing sites that were experiencing recruitment fatigue or had small numbers of eligible patients with new sites that we identified via the NIHR Injuries and Emergencies National Priority Group. We were granted a funded extension to the trial to continue recruitment until 30 June 2012.
Using data available at the time of the funded extension request (after 276 patients were recruited), we revised the recruitment predictions assuming that we would recruit, on average, 0.4 patients per site per week and adjusted this for the predicted number of sites that would be recruiting each month. At this stage, we predicted that we would reach the target number by March 2012. Mid-trial we had to change the IMP manufacturer, and recruitment to the trial had to be suspended between May and June 2010 as the new IMPs were not delivered as a result of a problem with the sterile production process. The seasonality of asthma usually meant that recruitment increased in the winter months, but the final winter season saw generally lower numbers of presenting patients. We kept a large proportion of the sites open to recruitment for an additional 3 months compared with the revised recruitment plan, staggering the close of sites to assist with scheduling close-out monitoring.
Non-recruited patients
During the trial we asked participating centres to record basic anonymised details of patients attending ED aged 16 years or over with acute severe asthma who were not recruited. These data were captured by a mixture of prospective screening by recruiting doctors and retrospective case note review by research nurses. Unfortunately, limited research nurse availability and service pressure in the ED prevented reliable collection of these data at some centres. Nevertheless, data were collected from a total of 3948 non-recruited patients attending at recruiting centres during the trial. Of these, 1165 patients were not identified by recruiting doctors because of a variety of administrative reasons (the ED was too busy, no GCP-trained doctors were available or no treatment packs were available) but were retrospectively identified by research nurses on the basis of information in the ED record. The remaining 2783 patients were identified prospectively, but excluded on the basis of ineligibility (n = 847), declining to participate (n = 200), inability to give consent (n = 21), administrative reasons as outlined above (n = 306) and other reasons (n = 201), while no reason was recorded for 89 patients. The patient characteristics recorded (age and sex) were not obviously different between those who did not take part and those who did (Table 4), with the possible exception of the subset who were eligible but unable to give consent.
| Patient classification | Age (years), mean (SD) | Sex, n (%) male |
|---|---|---|
| Recruited (N = 1109) | 36 (14) | 321 (30) |
| Not identified (N = 1165) | 36 (16) | 359 (31) |
| Ineligible (N = 847) | 37 (15) | 217 (26) |
| Declined to participate (N = 200) | 37 (16) | 62 (31) |
| Unable to give consent (N = 31) | 44 (19) | 11 (36) |
| Administrative reasons (N = 306) | 37 (15) | 94 (31) |
| Other (N = 201) | 38 (17) | 50 (25) |
| Not recorded (N = 89) | 35 (18) | 30 (33) |
The trial population
The CONSORT flow chart (Figure 3) shows the flow of participants through the trial. Of the 1109 participants randomised, 25 were excluded from the analysis. Eleven patients received no medication and no data were collected after randomisation. These patients either withdrew consent prior to any medication being delivered or recruiting doctors had randomised them before taking consent and patients subsequently refused consent. Two patients self-discharged without being treated. There were nine occasions where the numbered medication pack was not available in the ED and no treatment was subsequently given. The remaining three patients received treatment but there were protocol violations and these patients should not have been recruited: two were subsequently found to be previous participants and one was a prisoner. All remaining 1084 patients were included in analyses according to intention-to-treat principles, regardless of whether or not they received any medication.
FIGURE 3.
The CONSORT flow chart.
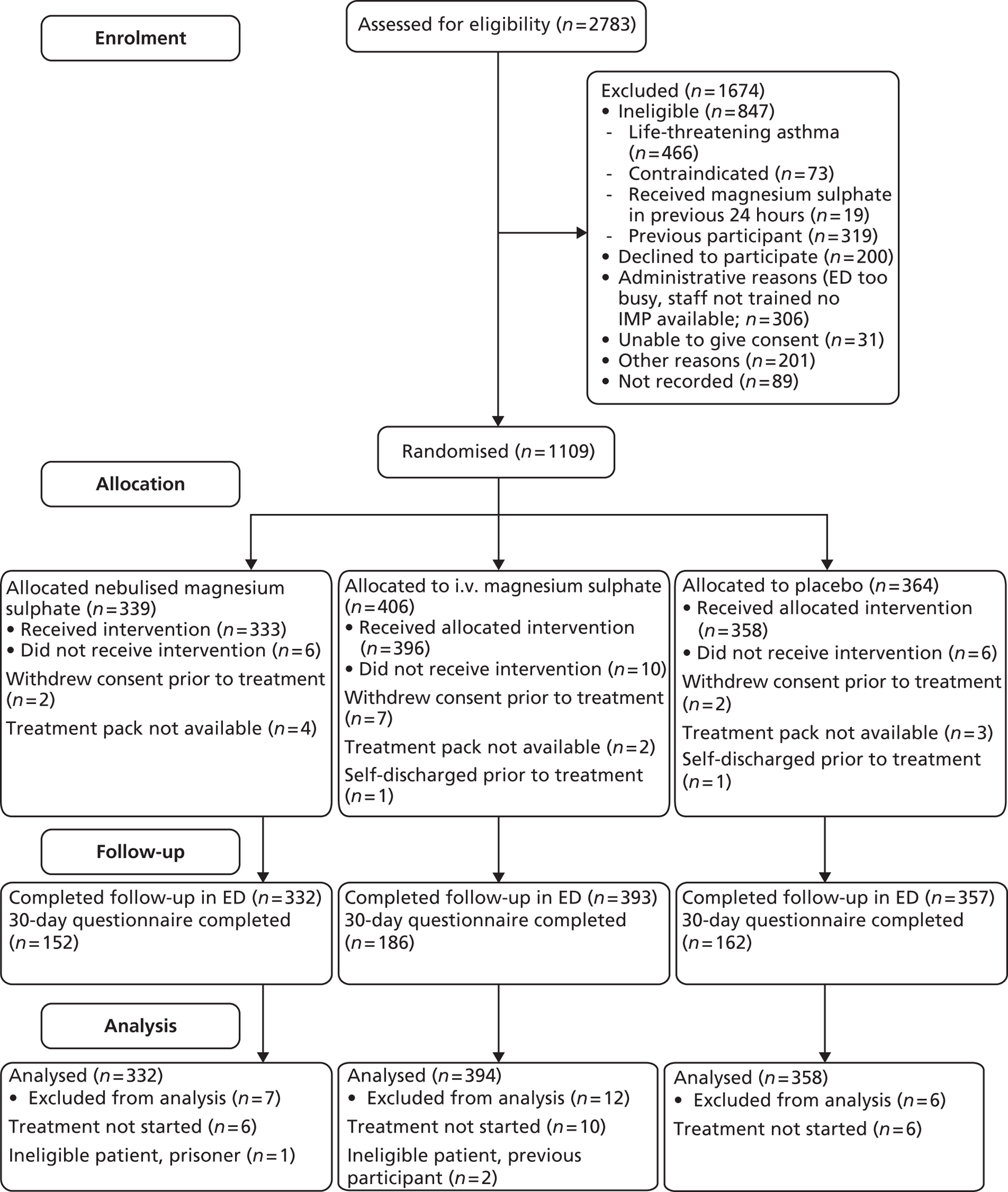
Protocol deviations which did not result in the participants being removed from analysis were also reported and reviewed at PMG meetings. A total of 203 protocol deviations were reported, which can be broadly categorised as follows: 42 deviations from the trial treatment protocol (randomised but trial treatment not commenced, treatment started but not completed or incorrect treatment), 143 consent not fully documented (no witness signature, verbal but not written consent, consent taken by a doctor without GCP training and/or not on delegation, log or tick boxes not completed), 15 administrative errors (prescription incorrectly completed, patient details recorded incorrectly, randomisation after treatment commenced or pack opened in error), and three screening assessment queries. The apparently high number of deviations can be attributed to the complexities of recruiting in an emergency setting and our requirement for sites to report any deviation from the protocol, however minor. Deviations from the consent procedure were reviewed to ensure that there was sufficient evidence that consent had been obtained before deciding if the participant should remain in the trial.
Table 5 summarises the recruitment and allocation of patients across the centres. Overall, 34 centres recruited to the study, of which 24 recruited 10 or more patients. The remaining centres were combined into one group for the purposes of all analyses. Table 6 shows the demographics and characteristics of the trial population. PEFR values reported in this table are predicted values. The actual baseline values are reported alongside the 1- and 2-hour values in Table 25. Patients were generally female (70%), white (90%) and relatively young (82% were below the age of 50 years). Overall, 64% of patients had previously been admitted to hospital for asthma, with the percentage being higher in the active two arms (68% nebulised, 66% i.v. and 59% placebo) and 14% of patients had previously been admitted to ITU. Where previous admissions had been recorded, around half had been admitted in the past year.
| Centre | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Royal Infirmary of Edinburgh | 56 (17%) | 67 (17%) | 56 (16%) | 179 (17%) |
| Sheffield – Northern General Hospital | 30 (9%) | 41 (10%) | 35 (10%) | 106 (10%) |
| Royal United Hospital Bath | 25 (8%) | 29 (7%) | 23 (6%) | 77 (7%) |
| Aberdeen Royal Infirmary | 17 (5%) | 23 (6%) | 17 (5%) | 57 (5%) |
| University Hospital of N. Staffordshire | 20 (6%) | 17 (4%) | 20 (6%) | 57 (5%) |
| Crosshouse Hospital | 18 (5%) | 15 (4%) | 19 (5%) | 52 (5%) |
| Bristol Frenchay Hospital | 13 (4%) | 17 (4%) | 18 (5%) | 48 (4%) |
| Barnsley Hospital | 14 (4%) | 14 (4%) | 18 (5%) | 46 (4%) |
| Plymouth – Derriford Hospital | 12 (4%) | 14 (4%) | 12 (3%) | 38 (4%) |
| York Hospital | 10 (3%) | 15 (4%) | 13 (4%) | 38 (4%) |
| Ayr Hospital | 9 (3%) | 12 (3%) | 10 (3%) | 31 (3%) |
| Royal Devon and Exeter Hospital | 12 (4%) | 8 (2%) | 11 (3%) | 31 (3%) |
| Bristol Royal Infirmary | 10 (3%) | 11 (3%) | 9 (3%) | 30 (3%) |
| Leicester Royal Infirmary | 9 (3%) | 7 (2%) | 14 (4%) | 30 (3%) |
| Kettering General Hospital | 9 (3%) | 10 (3%) | 10 (3%) | 29 (3%) |
| Lancaster Royal Infirmary | 8 (2%) | 16 (4%) | 5 (1%) | 29 (3%) |
| Derbyshire Royal Infirmary | 6 (2%) | 11 (3%) | 11 (3%) | 28 (3%) |
| Fife – Queen Margaret Hospital | 5 (2%) | 11 (3%) | 10 (3%) | 26 (2%) |
| Hull Royal Infirmary | 10 (3%) | 9 (2%) | 4 (1%) | 23 (2%) |
| Royal Alexandra Hospital – Paisley | 7 (2%) | 8 (2%) | 6 (2%) | 21 (2%) |
| University Hospital Coventry | 5 (2%) | 7 (2%) | 7 (2%) | 19 (2%) |
| Fife – Victoria Hospital | 5 (2%) | 5 (1%) | 5 (1%) | 15 (1%) |
| Addenbrookes Hospital, Cambridge | 3 (1%) | 6 (2%) | 4 (1%) | 13 (1%) |
| The Royal London Hospital | 3 (1%) | 4 (1%) | 4 (1%) | 11 (1%) |
| Southend University Hospital | 4 (1%) | 2 (1%) | 3 (1%) | 9 (1%) |
| James Cook University Hospital – Middlesbrough | 3 (1%) | 3 (1%) | 1 (< 1%) | 7 (1%) |
| Hairmyres Hospital | 2 (1%) | 3 (1%) | 2 (1%) | 7 (1%) |
| Leeds Teaching Hospitals | 1 (< 1%) | 1 (< 1%) | 3 (1%) | 5 (< 1%) |
| Northampton General Hospital | 1 (< 1%) | 1 (< 1%) | 3 (1%) | 5 (< 1%) |
| Rotherham General Hospital | 2 (1%) | 3 (1%) | 0 | 5 (< 1%) |
| Doncaster Royal Infirmary | 0 | 3 (1%) | 1 (< 1%) | 4 (< 1%) |
| Bradford Royal Infirmary | 1 (< 1%) | 0 | 2 (1%) | 3 (< 1%) |
| Queens Medical Centre, Nottingham | 1 (< 1%) | 1 (< 1%) | 1 (< 1%) | 3 (< 1%) |
| Pinderfields Hospital | 1 (< 1%) | 0 | 1 (< 1%) | 2 (< 1%) |
| Patient characteristic | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Age (years) | ||||
| Mean (SD) | 36.5 (14.8) | 35.6 (13.1) | 36.4 (14.1) | 36.1 (14.0) |
| Median (IQR) | 35.0 (23–47) | 34.0 (25–44) | 34.5 (24–47) | 34.0 (24–46) |
| Min., max. | 16, 85 | 16, 84 | 16, 88 | 16, 88 |
| Sexa | ||||
| Male | 100 (30%) | 115 (29%) | 106 (30%) | 321 (30%) |
| Female | 232 (70%) | 279 (71%) | 252 (70%) | 763 (70%) |
| Ethnicitya | ||||
| White | 286 (86%) | 369 (94%) | 319 (89%) | 974 (90%) |
| Mixed | 2 (1%) | 1 (< 1%) | 5 (1%) | 8 (1%) |
| Asian or Asian British | 14 (4%) | 8 (2%) | 16 (4%) | 38 (4%) |
| Black or black British | 2 (1%) | 5 (1%) | 4 (1%) | 11 (1%) |
| Other | 2 (1%) | 0 | 0 | 2 (< 1%) |
| Not stated | 22 (7%) | 8 (2%) | 11 (3%) | 41 (4%) |
| Missing | 4 (1%) | 3 (1%) | 3 (1%) | 10 (1%) |
| Smoking statusa | ||||
| Never | 151 (45%) | 156 (40%) | 143 (40%) | 450 (42%) |
| Current | 98 (30%) | 138 (35%) | 127 (35%) | 363 (33%) |
| Previous | 72 (22%) | 95 (24%) | 81 (23%) | 248 (23%) |
| Missing | 11 (3%) | 5 (1%) | 7 (2%) | 23 (2%) |
| Predicted PEFR | ||||
| n | 324 | 389 | 346 | 1059 |
| Mean (SD) | 430.0 (118.8) | 431.8 (116.9) | 435.0 (110.8) | 432.3 (115.4) |
| Median (IQR) | 425.0 (350–500) | 435.0 (350–500) | 425.0 (350–500) | 425.0 (350–500) |
| Min., max. | 100, 700 | 140, 800 | 150, 790 | 100, 800 |
| Previous admissions with asthma | ||||
| At least one previous ITU admissiona | 56 (17%) | 61 (15%) | 39 (11%) | 156 (14%) |
| At least one previous admissiona | 226 (68%) | 260 (66%) | 213 (59%) | 699 (64%) |
| If yes, time since last admission with asthma (months) | ||||
| n | 221 | 256 | 208 | 688 |
| Mean (SD) | 42.0 (72.2) | 38.5 (69.5) | 40.8 (61.9) | 40.1 (68.0) |
| Median (IQR) | 12.0 (4–47) | 12.0 (4–37) | 17.0 (6–48) | 12.5 (4–47) |
| Min., max. | 3 days, 32 years | 1 day, 50 years | 1 day, 40 years | 1 day, 50 years |
| Entry criterion for acute severe asthmaab | ||||
| PEFR < 50% of best or predicted | 179 (54%) | 205 (52%) | 192 (53%) | 576 (53%) |
| Heart rate > 110 beats per minute | 213 (64%) | 251 (64%) | 218 (61%) | 682 (63%) |
| Respiratory rate > 25 breaths per minute | 178 (54%) | 227 (58%) | 204 (57%) | 609 (67%) |
| Unable to complete sentences in one breath | 138 (42%) | 159 (40%) | 139 (39%) | 436 (40%) |
| Baseline PEFRa | ||||
| < 33% predicted | 53 (16%) | 50 (13%) | 56 (16%) | 156 (15%) |
| 33–50% predicted | 112 (34%) | 116 (29%) | 116 (32%) | 344 (32%) |
| 50–75% predicted | 107 (32%) | 148 (38%) | 118 (33%) | 373 (34%) |
| ≥ 75% predicted | 36 (11%) | 61 (15%) | 37 (10%) | 134 (12%) |
| Not recorded | 24 (7%) | 19 (5%) | 31 (9%) | 74 (7%) |
Trial and other treatments given
Table 7 shows the concurrent medications given in the 24 hours prior to hospital attendance, in the ambulance and ED immediately prior to randomisation, and alongside trial treatments in the 4 hours immediately after randomisation. Most patients (88%) had used salbutamol in the 24 hours prior to attendance and one-third had taken prednisolone. Use of salbutamol (95%) and ipratropium bromide (72%) was typical in the ambulance or ED prior to randomisation, whereas 41% were given prednisolone and 21% hydrocortisone. Salbutamol, ipratropium bromide, prednisolone and hydrocortisone were also commonly given alongside trial treatments.
| Medication Usage | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Used medication 24 hours prior to attendance | 304 (92%) | 370 (94%) | 320 (89%) | 994 (92%) |
| Salbutamol | 293 (88%) | 350 (89%) | 309 (86%) | 952 (88%) |
| Prednisolone | 115 (35%) | 140 (36%) | 106 (30%) | 361 (33%) |
| Seretide | 58 (17%) | 54 (14%) | 62 (17%) | 174 (16%) |
| Ipratropium bromide | 42 (13%) | 58 (15%) | 47 (13%) | 147 (14%) |
| Pulmicort | 32 (10%) | 33 (8%) | 24 (7%) | 89 (8%) |
| Beclometasone (Clenil) | 25 (8%) | 23 (6%) | 22 (6%) | 70 (6%) |
| Montelukast | 15 (5%) | 12 (3%) | 14 (4%) | 41 (4%) |
| Amoxicillin | 9 (3%) | 13 (3%) | 12 (3%) | 34 (3%) |
| Salmeterol (Serevent) | 12 (4%) | 10 (3%) | 7 (2%) | 29 (3%) |
| Terbutaline (Bricanyl) | 8 (2%) | 9 (2%) | 8 (2%) | 25 (2%) |
| Theophylline | 8 (2%) | 7 (2%) | 7 (2%) | 22 (2%) |
| Tiotropium (Spiriva) | 7 (2%) | 5 (1%) | 7 (2%) | 19 (2%) |
| Hydrocortisone | 6 (2%) | 5 (1%) | 4 (1%) | 15 (1%) |
| Aminophylline | 7 (2%) | 4 (1%) | 3 (1%) | 14 (1%) |
| Formoterol (Oxis, AstraZeneca UK Ltd) | 5 (2%) | 1 (< 1%) | 0 | 6 (1%) |
| Clarithromycin | 2 (1%) | 1 (< 1%) | 0 | 3 (< 1%) |
| Combivent | 0 | 2 (1%) | 1 (< 1%) | 3 (< 1%) |
| Zarfirlukast | 2 (1%) | 0 | 1 (< 1%) | 3 (< 1%) |
| Other | 5 (2%) | 4 (1%) | 6 (2%) | 15 (1%) |
| Given medication in ambulance or ED prerandomisation | 325 (98%) | 375 (95%) | 344 (96%) | 1044 (96%) |
| Salbutamol | 320 (96%) | 367 (93%) | 338 (94%) | 1025 (95%) |
| Ipratropium bromide | 241 (73%) | 279 (71%) | 259 (72%) | 779 (72%) |
| Prednisolone | 126 (38%) | 154 (39%) | 168 (47%) | 448 (41%) |
| Hydrocortisone | 71 (21%) | 86 (22%) | 69 (19%) | 226 (21%) |
| Combivent | 9 (3%) | 19 (5%) | 10 (3%) | 38 (4%) |
| Amoxicillin | 7 (2%) | 3 (1%) | 3 (1%) | 13 (1%) |
| Amoxicillin trihydrate/potassium clavulanate (Augmentin, GlaxoSmithKline UK) | 2 (1%) | 5 (1%) | 2 (1%) | 9 (1%) |
| Clarithromycin | 1 (<1%) | 2 (1%) | 4 (1%) | 7 (1%) |
| Aminophylline | 2 (1%) | 1 (< 1%) | 0 | 3 (< 1%) |
| Magnesium sulphate | 0 | 0 | 3 (1%) | 3 (< 1%) |
| Theophylline | 0 | 1 (< 1%) | 1 (< 1%) | 2 (< 1%) |
| Montelukast | 0 | 1 (< 1%) | 0 | 1 (< 1%) |
| Pulmicort | 0 | 0 | 1 (< 1%) | 1 (< 1%) |
| Other | 2 (1%) | 0 | 0 | 2 (< 1%) |
| Given medication 0–4 hours post randomisation | 180 (54%) | 195 (49%) | 182 (51%) | 557 (51%) |
| Salbutamol | 107 (32%) | 101 (26%) | 93 (26%) | 301 (28%) |
| Prednisolone | 64 (19%) | 62 (16%) | 54 (15%) | 180 (17%) |
| Ipratropium bromide | 59 (18%) | 50 (13%) | 53 (15%) | 162 (15%) |
| Hydrocortisone | 16 (5%) | 25 (6%) | 19 (5%) | 60 (6%) |
| Magnesium sulphate | 21 (6%) | 16 (4%) | 21 (6%) | 58 (5%) |
| Amoxicillin | 9 (3%) | 16 (4%) | 15 (4%) | 40 (4%) |
| Augmentin | 11 (3%) | 12 (3%) | 7 (2%) | 30 (3%) |
| Combivent | 7 (2%) | 13 (3%) | 7 (2%) | 27 (2%) |
| Clarithromycin | 7 (2%) | 9 (2%) | 9 (3%) | 25 (2%) |
| Aminophylline | 8 (2%) | 7 (2%) | 9 (3%) | 24 (2%) |
| Pulmicort | 2 (1%) | 3 (1%) | 1 (< 1%) | 6 (1%) |
| Seretide | 0 | 1 (< 1%) | 1 (< 1%) | 2 (< 1%) |
| Theophylline | 1 (< 1%) | 1 (< 1%) | 0 | 2 (< 1%) |
| Other | 9 (3%) | 3 (1%) | 3 (1%) | 15 (1%) |
Table 8 shows the proportion of patients receiving prednisolone or hydrocortisone at any point from 24 hours before attendance to 4 hours after randomisation. Around one-third of patients had taken corticosteroids in the 24 hours before attendance, 61% were given corticosteroids before randomisation and 21% after. Some patients were given additional corticosteroids in the ambulance or ED despite having taken corticosteroids in the previous 24 hours and, therefore, overall 95% of the trial population received corticosteroid therapy at some point from 24 hours prior to hospital attendance to 4 hours after randomisation. Table 9 shows the total dose of salbutamol given in the ambulance or ED prior to randomisation or up to 4 hours after randomisation. All but 10 patients (1%) received salbutamol at some point and 95% received salbutamol prior to randomisation, with a mean dose of 4.9 mg. Overall, it appears that there was adherence to BTS/SIGN guidance and substantial use of standard treatments that are known to be effective. 2
| Usage | Nebulised magnesium (n = 332) | Intravenous magnesium (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Any usage in or before ED | 316 (95%) | 372 (94%) | 344 (96%) | 1032 (95%) |
| Last 24 hours before attendance | 119 (36%) | 143 (36%) | 110 (31%) | 372 (34%) |
| Ambulance/ED pre randomisation | 191 (58%) | 236 (60%) | 231 (65%) | 658 (61%) |
| After randomisation 0–4 hours | 77 (23%) | 83 (21%) | 69 (19%) | 229 (21%) |
| Usage | Nebulised magnesium (n = 332) | Intravenous magnesium (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Usage in ambulance or ED | ||||
| Any usagea | 329 (99%) | 391 (99%) | 354 (99%) | 1074 (99%) |
| Mean (SD) dose given (mg) | 8.7 (3.4) | 8.0 (3.4) | 8.2 (3.4) | 8.3 (3.4) |
| Prerandomisation usage (ambulance or ED) | ||||
| Any usagea | 320 (96%) | 367 (93%) | 338 (94%) | 1025 (95%) |
| Mean (SD) dose given (mg) | 5.0 (1.3) | 4.8 (1.5) | 4.9 (1.4) | 4.9 (1.4) |
| Post-randomisation usage in ED | ||||
| Any usagea | 232 (70%) | 243 (62%) | 232 (65%) | 707 (65%) |
| Mean (SD) dose given (mg) | 3.8 (3.4) | 3.3 (3.3) | 3.4 (3.2) | 3.4 (3.3) |
Three patients (all in the placebo group) were given i.v. magnesium sulphate in the ED or ambulance prior to randomisation. After these cases were identified, the protocol was amended to exclude patients who had received magnesium sulphate in the 24 hours prior to randomisation. A further 58 patients (5%) received i.v. magnesium sulphate after randomisation as a result of the treating physician deciding that the patient’s response to initial treatment suggested that they were no longer in equipoise. These cases were evenly distributed across the three groups.
Table 10 shows the trial medications received by the three groups. Most patients (89%) received the full i.v. infusion and only 2% received less than half. Similarly, most patients (99%) received all three nebulisers and a substantial proportion of the nebuliser solution.
| Medication received | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Total volume of i.v. infusion (ml) | ||||
| Mean (SD) | 97.2 (14.8) | 96.5 (16.1) | 97.9 (12.7) | 97.1 (14.6) |
| 100a | 299 (90%) | 349 (89%) | 320 (89%) | 968 (89%) |
| 90–99.9a | 11 (3%) | 18 (5%) | 16 (4%) | 45 (4%) |
| 70–89.9a | 8 (2%) | 8 (2%) | 4 (1%) | 20 (2%) |
| 50–69.9a | 0 | 2 (1%) | 1 (< 1%) | 3 (< 1%) |
| 0–49.9a | 7 (2%) | 11 (3%) | 6 (2%) | 24 (2%) |
| Missinga | 7 (2%) | 6 (2%) | 11 (3%) | 24 (2%) |
| Number of nebulisers given | ||||
| 0 | 0 | 0 | 0 | 0 |
| 1a | 2 (1%) | 4 (1%) | 0 | 6 (1%) |
| 2a | 5 (2%) | 1 (< 1%) | 2 (1%) | 8 (1%) |
| 3a | 323 (98%) | 387 (99%) | 355 (99%) | 1065 (99%) |
| Missinga | 2 | 2 | 1 | 5 |
| Total volume of nebuliser excluding salbutamol (ml) | ||||
| Mean (SD) | 21.0 (4.1) | 21.5 (3.9) | 21.5 (3.6) | 21.3 (3.9) |
| 22.5a | 261 (79%) | 349 (89%) | 307 (86%) | 917 (85%) |
| 20–22.4a | 11 (3%) | 7 (2%) | 8 (2%) | 26 (2%) |
| 15–19.9a | 21 (6%) | 12 (3%) | 18 (5%) | 51 (5%) |
| 7.5–14.9a | 18 (5%) | 9 (2%) | 9 (3%) | 36 (3%) |
| 0–7.4a | 5 (2%) | 9 (2%) | 6 (2%) | 20 (2%) |
| Missinga | 16 (5%) | 8 (2%) | 10 (3%) | 34 (3%) |
Primary outcome analysis
Table 11 shows the primary, service-orientated outcome, namely admission to hospital at presentation or within 1 week of presentation. Overall, 827 out of 1084 (76%) of patients were admitted to hospital within 1 week: 811 were admitted at initial attendance, 14 were initially discharged but were admitted within the next week, and the status of two patients was unknown and, therefore, the patients were analysed as having been admitted. The percentage admitted was lowest in the i.v. magnesium sulphate group (72%) but similar in the placebo (78%) and nebulised magnesium sulphate (79%) groups. None of the contrasts or pairwise comparisons was statistically significant at the 5% level. These findings were also borne out in the per-protocol subset, which showed very similar results (Table 12).
| Classification | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Status at 4 hours | ||||
| Admitteda | 254 (77%) | 279 (71%) | 278 (78%) | 811 (75%) |
| Dischargeda | 77 (23%) | 114 (29%) | 80 (22%) | 271 (25%) |
| Dead | 0 | 0 | 0 | 0 |
| Unknowna | 1 (< 1%) | 1 (< 1%) | 0 | 2 (< 1%) |
| Subsequent hospital admission within 7 daysa | 15 (5%) | 10 (3%) | 7 (2%) | 32 (3%) |
| Subsequent hospital admission following discharge at initial attendancea | 6 (2%) | 5 (1%) | 3 (1%) | 14 (1%) |
| Admitted to hospital at any time within 7 daysa | 261 (79%) | 285 (72%) | 281 (78%) | 827 (76%) |
| Comparisons | Odds ratio (95% CI) | p-value | ||
| Active vs. placebo | 0.84 (0.61 to 1.15) | 0.276 | ||
| i.v. vs. nebuliser | 0.76 (0.53 to 1.10) | 0.146 | ||
| i.v. vs. placebo | 0.73 (0.51 to 1.04) | 0.083 | ||
| Nebuliser vs. placebo | 0.96 (0.65 to 1.40) | 0.819 | ||
| Classification | Nebulised magnesium sulphate (n = 305) | Intravenous magnesium sulphate (n = 368) | Placebo (n = 333) | Total (n = 1006) |
|---|---|---|---|---|
| Status at 4 hours | ||||
| Admitteda | 236 (77%) | 263 (71%) | 258 (77%) | 757 (75%) |
| Dischargeda | 69 (23%) | 105 (29%) | 75 (23%) | 249 (25%) |
| Dead | 0 | 0 | 0 | 0 |
| Unknown | 0 | 0 | 0 | 0 |
| Subsequent hospital admission within 7 daysa | 14 (5%) | 8 (2%) | 7 (2%) | 29 (3%) |
| Subsequent hospital admission following discharge at initial attendancea | 6 (2%) | 4 (1%) | 3 (1%) | 13 (1%) |
| Admitted to hospital at any time within 7 daysa | 242 (79%) | 267 (73%) | 261 (78%) | 770 (77%) |
| Comparisons | Odds ratio (95% CI) | p-value | ||
| Active vs. placebo | 0.85 (0.61 to 1.19) | 0.348 | ||
| i.v. vs. nebuliser | 0.78 (0.54 to 1.15) | 0.210 | ||
| i.v. vs. placebo | 0.75 (0.52 to 1.09) | 0.133 | ||
| Nebuliser vs. placebo | 0.96 (0.64 to 1.44) | 0.848 | ||
Patient breathlessness is summarised in Tables 13 (complete case), 14 (complete case, per-protocol) and 15 (imputed). The complete case analysis is also presented graphically in Figure 4. The change in VAS at 2 hours was recorded in 976 out of 1084 (90%) members of the cohort. Improvements in breathlessness were observed in all three groups. In the complete case data, the mean change from baseline was largest [34.3 mm; standard deviation (SD) 27.7 mm] in the i.v. group and smallest (28.2 mm; SD 27.4 mm) in the nebulised group, whereas the change in the placebo group was between the two (31.3 mm; SD 29.4 mm). Overall, magnesium sulphate did not reduce breathlessness (mean difference = 0 mm, 95% CI –1.9 to 1.9 mm; p = 0.999). Although i.v. magnesium sulphate appeared superior to nebulised magnesium sulphate (mean difference = 5.1 mm; 95% CI 0.8 to 9.4 mm; p = 0.019), the magnitude of the difference in clinical terms is small. The post-hoc comparison of i.v. magnesium sulphate against placebo yielded no significant difference (mean difference 2.6 mm; 95% CI –1.6 to 6.8 mm; p = 0.231), although this may be partly due to a small imbalance at baseline, as the pretreatment VAS scores were slightly higher in the placebo group than in the i.v. group. Nonetheless, the magnitude of difference between i.v. and placebo is minor when considered in the context that the minimum clinically significant difference for VAS breathlessness has been estimated to be 22 mm. 48
| Measurement | Nebulised magnesium (n = 332) | Intravenous magnesium (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| VAS at baseline | ||||
| Number of observations | 326 | 386 | 349 | 1061 |
| Mean [mm (SD)] | 61.6 (23.3) | 61.9 (22.8) | 63.1 (23.5) | 62.2 (23.2) |
| Median [mm (IQR)] | 65.0 (46–80) | 65.0 (46–80) | 69.0 (46–82) | 67.0 (46–80) |
| Min., max. (mm) | 2, 100 | 4, 100 | 0, 100 | 0, 100 |
| VAS at 1 hour | ||||
| Number of observations | 314 | 372 | 344 | 1030 |
| Mean [mm (SD)] | 42.9 (25.7) | 37.7 (25.4) | 41.9 (25.0) | 40.7 (25.4) |
| Median [mm (IQR)] | 42.5 (23–63) | 34.5 (16–58) | 41.0 (20–62) | 39.0 (19–60) |
| Min., max. (mm) | 0, 100 | 0, 98 | 0, 99 | 0, 100 |
| Change in VAS at 1 hour | ||||
| Number of observations | 314 | 372 | 344 | 1030 |
| Mean [mm (SD)] | –18.4 (22.8) | –24.2 (24.4) | –21.5 (24.7) | –21.5 (24.1) |
| Median [mm (IQR)] | –15.0 (–32 to –4) | –20.0 (–40 to –7) | –17.0 (–37 to –5) | –18.0 (–35 to –5) |
| Min., max. (mm) | –86, 63 | –96, 39 | –93, 60 | –96, 63 |
| VAS at 2 hours | ||||
| Number of observations | 296 | 357 | 323 | 976 |
| Mean [mm (SD)] | 32.9 (27.8) | 27.7 (26.4) | 32.4 (27.5) | 30.8 (27.3) |
| Median [mm (IQR)] | 27.5 (8–53) | 18.0 (5–45) | 25.0 (9–53) | 23.0 (7–52) |
| Min., max. (mm) | 0, 99 | 0, 100 | 0, 100 | 0, 100 |
| Change in VAS at 2 hours | ||||
| Number of observations | 296 | 357 | 323 | 976 |
| Mean [mm (SD)] | –28.2 (27.4) | –34.3 (27.7) | –31.3 (29.4) | –31.5 (28.2) |
| Median [mm (IQR)] | –28.0 (–47 to –9) | –33.0 (–53 to –14) | –29.0 (–53 to –10) | –30.0 (–52 to –11) |
| Min., max. (mm) | –94, 90 | –99, 45 | –98, 63 | –99, 90 |
| Comparisons | Mean difference (95% CI) | p-value | ||
| Active vs. placebo | 0.0 (–1.9 to 1.9) | 0.999 | ||
| i.v. vs. nebuliser | –5.1 (–9.4 to –0.8) | 0.019 | ||
| i.v. vs. placebo | –2.6 (–6.8 to 1.6) | 0.231 | ||
| Nebuliser vs. placebo | 2.6 (–1.8 to 7.0) | 0.253 | ||
| Measurement | Nebulised magnesium (n = 305) | Intravenous magnesium (n = 368) | Placebo (n = 333) | Overall (n = 1006) |
|---|---|---|---|---|
| VAS at baseline | ||||
| Number of observations | 303 | 362 | 326 | 991 |
| Mean [mm (SD)] | 61.7 (23.3) | 61.6 (22.7) | 63.0 (23.5) | 62.1 (23.1) |
| Median [mm (IQR)] | 65.0 (46–80) | 65.0 (47–80) | 69.0 (45–82) | 66.0 (46–80) |
| Min., max. (mm) | 2, 100 | 4, 100 | 0, 100 | 0, 100 |
| VAS at 1 hour | ||||
| Number of observations | 297 | 354 | 322 | 973 |
| Mean [mm (SD)] | 43.6 (25.7) | 37.9 (25.7) | 42.0 (25.0) | 41.0 (25.5) |
| Median [mm (IQR)] | 43.0 (24–64) | 34.5 (16–59) | 41.0 (20–62) | 39.0 (19–61) |
| Min., max. (mm) | 0, 100 | 0, 98 | 0, 99 | 0, 100 |
| Change in VAS at 1 hour | ||||
| Number of observations | 297 | 354 | 322 | 973 |
| Mean [mm (SD)] | –17.9 (22.2) | –23.8 (24.2) | –21.3 (24.5) | –21.2 (23.8) |
| Median [mm (IQR)] | –15.0 (–30 to –3) | –19.5 (–39 to –7) | –17.0 (–37 to –5) | –17.0 (–34 to –5) |
| Min., max. (mm) | –86, 63 | –96, 39 | –93, 60 | –96, 63 |
| VAS at 2 hours | ||||
| Number of observations | 280 | 341 | 304 | 925 |
| Mean [mm (SD)] | 33.7 (27.8) | 28.0 (26.6) | 32.7 (27.7) | 31.3 (27.4) |
| Median [mm (IQR)] | 28.5 (10–54) | 18.0 (5–45) | 25.5 (10–53) | 24.0 (7–52) |
| Min., max. (mm) | 0, 99 | 0, 100 | 0, 100 | 0, 100 |
| Change in VAS at 2 hours | ||||
| Number of observations | 280 | 341 | 304 | 925 |
| Mean [mm (SD)] | –27.7 (27.1) | –34.0 (27.7) | –31.1 (29.4) | –31.1 (28.2) |
| Median [mm (IQR)] | –27.5 (–46 to –9) | –33.0 (–52 to –14) | –28.5 (–53 to –10) | –30.0 (–51 to –11) |
| Min., max. (mm) | –88, 90 | –99, 45 | –98, 63 | –99, 90 |
| Comparisons | Mean difference (95% CI) | p-value | ||
| Active vs. placebo | 0.1 (–1.8 to 2.0) | 0.950 | ||
| i.v. vs. nebuliser | –5.2 (–9.6 to –0.8) | 0.021 | ||
| i.v. vs. placebo | –2.5 (–6.8 to 1.8) | 0.261 | ||
| Nebuliser vs. placebo | 2.6 (–1.8 to 7.0) | 0.253 | ||
| Imputation strategy | Number of participants included | Mean difference (95% CI) | p-value |
|---|---|---|---|
| Active vs. placebo | |||
| ITT, no imputation | 976 | 0.0 (–1.9 to 1.9) | 0.999 |
| ITT, linear interpolation | 981 | 0.1 (–1.7 to 1.9) | 0.956 |
| ITT, multiple imputation | 1084 | –1.0 (–2.3 to 4.3) | 0.549 |
| PP, no imputation | 925 | 0.1 (–1.8 to 2.0) | 0.950 |
| i.v. vs. nebuliser | |||
| ITT, no imputation | 976 | –5.1 (–9.4 to –0.8) | 0.019 |
| ITT, linear interpolation | 981 | –4.8 (–9.0 to –0.5) | 0.027 |
| ITT, multiple imputation | 1084 | –5.2 (–9.1 to 1.3) | 0.007 |
| PP, no imputation | 925 | –5.2 (–9.6 to –0.8) | 0.021 |
| i.v. vs. placebo | |||
| ITT, no imputation | 976 | –2.6 (–6.8 to 1.6) | 0.231 |
| ITT, linear interpolation | 981 | –2.3 (–6.4 to 1.9) | 0.279 |
| ITT, multiple imputation | 1084 | –3.6 (–7.3 to 0.1) | 0.059 |
| PP, no imputation | 925 | –2.5 (–6.8 to 1.8) | 0.261 |
| Nebuliser vs. placebo | |||
| ITT, no imputation | 976 | 2.6 (–1.8 to 7.0) | 0.253 |
| ITT, linear interpolation | 981 | 2.5 (–1.9 to 6.9) | 0.261 |
| ITT, multiple imputation | 1084 | 1.6 (–2.2 to 5.5) | 0.401 |
| PP, no imputation | 925 | 2.6 (–1.8 to 7.0) | 0.253 |
FIGURE 4.
Change in VAS breathlessness (complete case analysis). Neb, nebuliser; Pla, placebo.
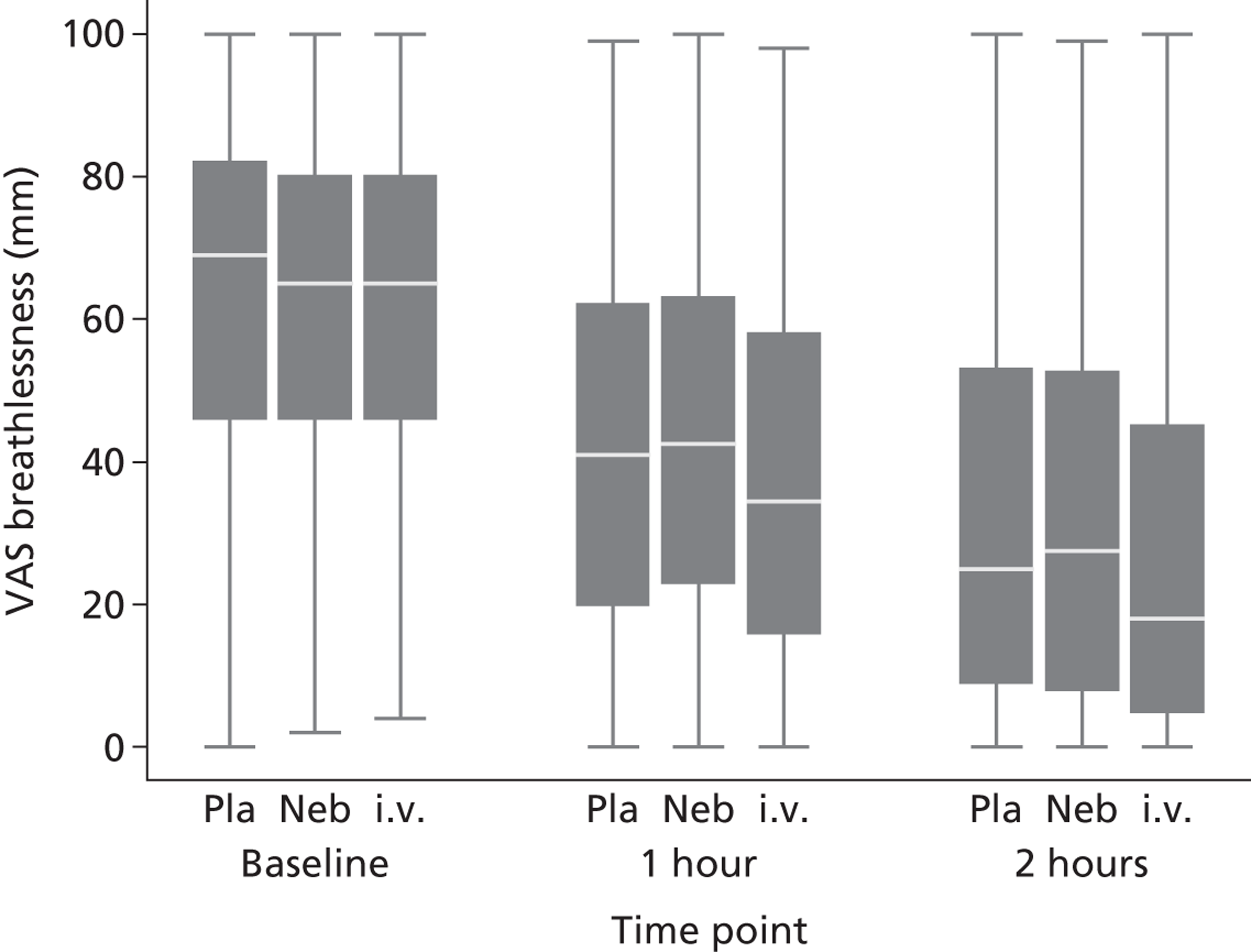
Secondary outcome analysis
Table 16 and Figure 5 compare the hospital length of stay after initial attendance for the three groups. The length of stay was not recorded unless the patients had been admitted; these patients are represented by the ‘spike’ at 3.9 hours in the Kaplan–Meier graph. The length of stay was shorter in the i.v. group, although not significantly so.
| Measurement/classification | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Time to dischargea | ||||
| Time not known | 3 (1%) | 6 (2%) | 5 (1%) | 14 (1%) |
| Patient died | 1 (< 1%) | 1 (< 1%) | 0 | 2 (< 1%) |
| Patient admitted, duration not known | 1 (< 1%) | 2 (1%) | 4 (1%) | 7 (1%) |
| Nothing recorded | 1 (< 1%) | 3 (1%) | 1 (< 1%) | 5 (< 1%) |
| Time recorded | ||||
| n | 329 | 388 | 353 | 1070 |
| Mean [hours (SD)] | 63.2 (79.7) | 57.0 (75.1) | 63.3 (84.3) | 61.0 (79.6) |
| Median [hours (IQR)] | 35.1 (5–88) | 31.5 (4–78) | 36.4 (5–87) | 34.1 (4–84) |
| Min., max. (hours) | 3, 623 | 4, 723 | 1, 694 | 1, 723 |
| Not admitted/discharged within 4 hoursa | 80 (24%) | 120 (30%) | 83 (23%) | 283 (26%) |
| 4–6 hoursa | 4 (1%) | 11 (3%) | 8 (2%) | 23 (2%) |
| 6–12 hoursa | 16 (5%) | 10 (3%) | 15 (4%) | 41 (4%) |
| 12–24 hoursa | 33 (10%) | 34 (9%) | 38 (11%) | 105 (10%) |
| > 24 hoursa | 196 (59%) | 213 (54%) | 209 (58%) | 618 (57%) |
| Comparisons | Time ratio (95% CI) | p-value | ||
| Active vs. placebo | 0.92 (0.76 to 1.13) | 0.432 | ||
| i.v. vs. nebuliser | 0.87 (0.69 to 1.09) | 0.230 | ||
| i.v. vs. placebo | 0.86 (0.69 to 1.08) | 0.192 | ||
| Nebuliser vs. placebo | 0.99 (0.78 to 1.25) | 0.936 | ||
FIGURE 5.
Length of stay following initial attendance.
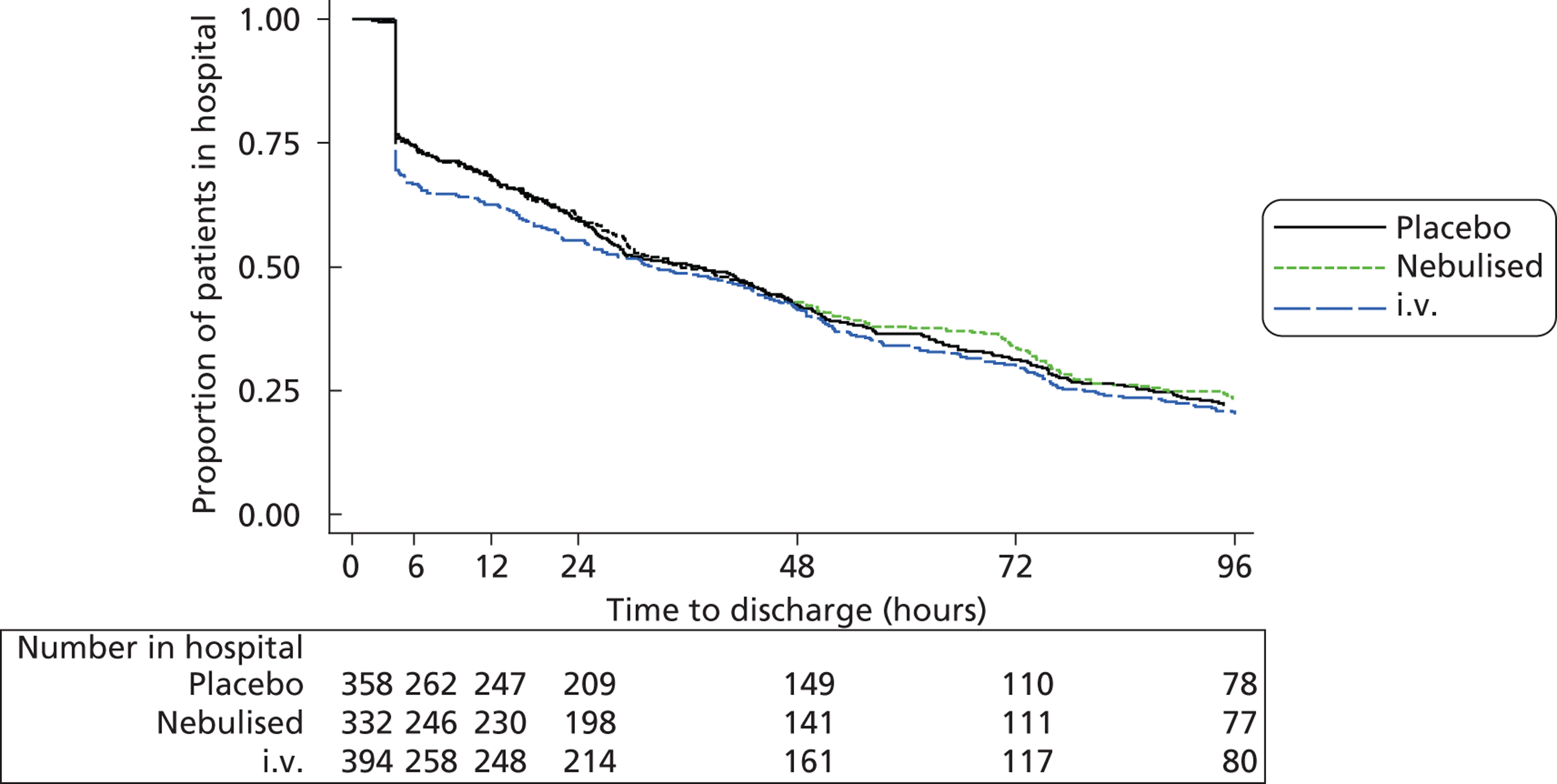
Table 17 shows the number of days spent on ICU, HDU and a general medical ward up to 30 days after recruitment. Only 2% of the cohort spent any days on ICU and only 6% spent any days on HDU. There were no significant differences between the three groups in the number of days spent in any location.
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) | p-values | |
|---|---|---|---|---|---|---|
| Active vs. placebo | Intravenous vs. nebuliser | |||||
| Days in hospital at any location | ||||||
| Number (%) with any staya | 253 (76) | 278 (71) | 267 (75) | 798 (74) | 0.613 | 0.087 |
| Mean [days (SD)] | 3.3 (4.8) | 3.1 (5.0) | 2.9 (3.9) | 3.1 (4.6) | ||
| Median [days (IQR)]b | 2.0 (1–4) | 2.0 (0–4) | 2.0 (0–4) | 2.0 (0–4) | 0.598 | 0.218 |
| Days on ICU | ||||||
| Number (%) with any ICU staya | 9 (3) | 11 (3) | 5 (1) | 25 (2) | 0.161 | 0.947 |
| Mean [days (SD)] | 3.3 (4.8) | 3.1 (5.0) | 2.9 (3.9) | 3.1 (4.6) | ||
| Median [days (IQR)]b | 2.0 (1–4) | 2.0 (0–4) | 2.0 (0–4) | 2.0 (0–4) | 0.159 | 0.941 |
| Days on HDU | ||||||
| Number (%) with any HDU staya | 22 (7) | 23 (6) | 20 (6) | 65 (6) | 0.690 | 0.661 |
| Mean [days (SD)] | 3.3 (4.8) | 3.1 (5.0) | 2.9 (3.9) | 3.1 (4.6) | ||
| Median [days (IQR)]b | 2.0 (1–4) | 2.0 (0–4) | 2.0 (0–4) | 2.0 (0–4) | 0.715 | 0.630 |
| Days on ward | ||||||
| Number (%) with any ward staya | 247 (74) | 275 (70) | 258 (72) | 780 (72) | 0.954 | 0.169 |
| Mean [days (SD)] | 3.3 (4.8) | 3.1 (5.0) | 2.9 (3.9) | 3.1 (4.6) | ||
| Median [days (IQR)]b | 2.0 (1–4) | 2.0 (0–4) | 2.0 (0–4) | 2.0 (0–4) | 0.612 | 0.323 |
Table 18 shows the number of patients requiring invasive or non-invasive ventilatory support. Only 1% of the cohort required ventilatory support and there were no significant differences between the three groups.
| Classification | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Required ventilationa | 3 (1%) | 6 (2%) | 4 (1%) | 13 (1%) |
| Non-invasive ventilationa | 2 (1%) | 2 (1%) | 3 (1%) | 7 (1%) |
| Emergency intubationa | 2 (1%) | 4 (1%) | 1 (< 1%) | 7 (1%) |
| Comparisons | Odds ratio (95% CI) | p-value | ||
| Active vs. placebo | 1.05 (0.31 to 3.51) | 0.936 | ||
| i.v. vs. nebuliser | 1.70 (0.42 to 6.83) | 0.458 | ||
| i.v. vs. placebo | 1.37 (0.38 to 4.89) | 0.629 | ||
| Nebuliser vs. placebo | 0.81 (0.18 to 3.63) | 0.780 | ||
Tables 19–22 show the change in heart rate, respiratory rate and blood pressure (systolic and diastolic) over the first 2 hours after initiation of treatment. These changes are also shown in Figures 6–9. Mean values of all four parameters fell during treatment, but there were no significant differences between the treatment groups in the magnitude of change.
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Pulse at baseline | ||||
| Number of observations | 331 | 394 | 356 | 1081 |
| Mean [beats per minute (SD)] | 111.1 (19.8) | 112.0 (19.1) | 110.4 (18.9) | 111.2 (19.2) |
| Median [beats per minute (IQR)] | 112.0 (97–123) | 112.0 (98–124) | 110.5 (98–123) | 112.0 (98–123) |
| Min., Max. (beats per minute) | 52, 177 | 60, 168 | 54, 180 | 52, 180 |
| Pulse at 1 hour | ||||
| Number of observations | 326 | 387 | 353 | 1066 |
| Mean [beats per minute (SD)] | 106.8 (18.0) | 106.2 (18.6) | 106.5 (18.1) | 106.5 (18.2) |
| Median [beats per minute (IQR)] | 107.0 (95–119) | 106.0 (94–119) | 106.0 (94–119) | 106.0 (94–119) |
| Min., max. (beats per minute) | 61, 162 | 59, 162 | 52, 164 | 52, 164 |
| Change in pulse at 1 hour | ||||
| Number of observations | 326 | 387 | 353 | 1066 |
| Mean [beats per minute (SD)] | –4.4 (13.0) | –5.7 (12.7) | –3.9 (12.3) | –4.7 (12.7) |
| Median [beats per minute (IQR)] | –4.0 (–12 to 3) | –6.0 (–13 to 1) | –2.0 (–10 to 3) | –4.0 (–12 to 3) |
| Min., max. (beats per minute) | –50, 54 | –42, 46 | –45, 44 | –50, 54 |
| Pulse at 2 hours | ||||
| Number of observations | 311 | 379 | 340 | 1030 |
| Mean [beats per minute (SD)] | 104.9 (17.3) | 105.7 (18.1) | 105.9 (17.5) | 105.5 (17.7) |
| Median [beats per minute (IQR)] | 104.0 (93–118) | 105.0 (92–117) | 105.0 (95–118) | 105.0 (93–117) |
| Min., max. (beats per minute) | 62, 166 | 57, 164 | 52, 160 | 52, 166 |
| Change in pulse at 2 hours | ||||
| Number of observations | 311 | 379 | 340 | 1030 |
| Mean [beats per minute (SD)] | –6.3 (15.1) | –6.3 (14.7) | –4.5 (14.4) | –5.7 (14.7) |
| Median [beats per minute (IQR)] | –5.0 (–16 to 3) | –6.0 (–15 to 2) | –4.0 (–14 to 4) | –5.0 (–15 to 3) |
| Min., max. (beats per minute) | –60, 34 | –50, 53 | –52, 40 | –60, 53 |
| Comparisons | Mean difference [beats per minute (95% CI)] | p-value | ||
| Active vs. placebo | –1.8 (–3.7 to 0.1) | 0.067 | ||
| i.v. vs. nebuliser | –0.1 (–2.3 to 2.1) | 0.940 | ||
| i.v. vs. placebo | –1.8 (–4.0 to 0.3) | 0.096 | ||
| Nebuliser vs. placebo | –1.8 (–4.0 to 0.5) | 0.130 | ||
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Respiratory rate at baseline | ||||
| Number of observations | 330 | 392 | 356 | 1078 |
| Mean [breaths per minute (SD)] | 25.7 (7.2) | 25.4 (6.4) | 25.2 (6.3) | 25.4 (6.6) |
| Median [breaths per minute (IQR)] | 24.0 (20–29) | 25.0 (21–28) | 24.0 (20–29) | 24.0 (20–28) |
| Min., max. (breaths per minute) | 12, 58 | 10, 48 | 13, 45 | 10, 58 |
| Respiratory rate at 1 hour | ||||
| Number of observations | 323 | 383 | 350 | 1056 |
| Mean [breaths per minute (SD)] | 22.7 (6.3) | 22.1 (5.9) | 21.8 (5.3) | 22.2 (5.8) |
| Median [breaths per minute (IQR)] | 21.0 (19–25) | 20.0 (18–24) | 20.0 (18–24) | 20.0 (18–24) |
| Min., max. (breaths per minute) | 8, 50 | 11, 48 | 11, 40 | 8, 50 |
| Change in respiratory rate at 1 hour | ||||
| Number of observations | 323 | 381 | 350 | 1054 |
| Mean [breaths per minute (SD)] | −3.1 (5.9) | −3.4 (5.6) | −3.3 (5.4) | −3.3 (5.6) |
| Median [breaths per minute (IQR)] | −2.0 (–6 to 0) | −3.0 (−6 to 0) | −2.0 (−6 to 0) | −2.0 (−6 to 0) |
| Min., max. (breaths per minute) | −24, 20 | −24, 21 | −20, 11 | −24, 21 |
| Respiratory rate at 2 hours | ||||
| Number of observations | 307 | 376 | 336 | 1019 |
| Mean [breaths per minute (SD)] | 21.3 (5.7) | 20.6 (5.3) | 21.0 (5.4) | 21.0 (5.5) |
| Median [breaths per minute (IQR)] | 20.0 (18–24) | 20.0 (18–23) | 20.0 (18–24) | 20.0 (18–24) |
| Min., max. (breaths per minute) | 10, 60 | 11, 48 | 9, 48 | 9, 60 |
| Change in respiratory rate at 2 hours | ||||
| Number of observations | 307 | 374 | 336 | 1017 |
| Mean [breaths per minute (SD)] | −4.3 (7.0) | −4.8 (5.9) | −4.2 (6.3) | −4.5 (6.4) |
| Median [breaths per minute (IQR)] | −4.0 (−8 to 0) | −4.0 (−8 to −1) | −4.0 (–8 to 0) | −4.0 (–8 to 0) |
| Min., max. (breaths per minute) | −38, 16 | −30, 12 | −27, 14 | −38, 16 |
| Comparisons | Mean difference [breaths per minute (95% CI)] | p-value | ||
| Active vs. placebo | –0.5 (–1.3 to 0.4) | 0.264 | ||
| i.v. vs. nebuliser | –0.4 (–1.4 to 0.5) | 0.396 | ||
| i.v. vs. placebo | –0.7 (–1.6 to 0.3) | 0.154 | ||
| Nebuliser vs. placebo | –0.3 (–1.3 to 0.7) | 0.594 | ||
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Systolic BP at baseline | ||||
| Number of observations | 330 | 392 | 356 | 1078 |
| Mean [mmHg (SD)] | 132.5 (20.4) | 133.1 (20.6) | 130.6 (19.7) | 132.1 (20.2) |
| Median [mmHg (IQR)] | 129.5 (118–142) | 130.0 (118–145) | 129.0 (116–141) | 130.0 (118–143) |
| Min., max. | 84, 230 | 93, 216 | 86, 235 | 84, 235 |
| Systolic BP at 1 hour | ||||
| Number of observations | 323 | 383 | 351 | 1057 |
| Mean [mmHg (SD)] | 128.2 (19.4) | 125.2 (15.8) | 126.6 (17.2) | 126.6 (17.4) |
| Median [mmHg (IQR)] | 125.0 (116–140) | 124.0 (115–133) | 125.0 (114–137) | 124.0 (115–137) |
| Min., max. (mmHg) | 80, 215 | 79, 186 | 85, 203 | 79, 215 |
| Change in systolic BP at 1 hour | ||||
| Number of observations | 322 | 381 | 351 | 1054 |
| Mean [mmHg (SD)] | −4.5 (17.8) | −7.8 (19.4) | −4.2 (18.1) | −5.6 (18.6) |
| Median [mmHg (IQR)] | −4.5 (–14 to 5) | −6.0 (–17 to 4) | −3.0 (–14 to 6) | −4.0 (–15 to 5) |
| Min., max. (mmHg) | −65, 74 | −82, 92 | −71, 48 | −82, 92 |
| Systolic BP at 2 hours | ||||
| Number of observations | 309 | 373 | 338 | 1020 |
| Mean [mmHg (SD)] | 127.0 (19.3) | 125.5 (16.5) | 124.9 (18.5) | 125.7 (18.1) |
| Median [mmHg (IQR)] | 124.0 (114–137) | 124.0 (114–134) | 122.0 (112–134) | 123.0 (114–135) |
| Min., max. (mmHg) | 85, 194 | 88, 187 | 76, 212 | 76, 212 |
| Change in systolic BP at 2 hours | ||||
| Number of observations | 309 | 371 | 338 | 1018 |
| Mean [mmHg (SD)] | −5.6 (19.9) | −7.7 (18.2) | −6.1 (19.5) | –6.5 (19.2) |
| Median [mmHg (IQR)] | −5.0 (–17 to 6) | −7.0 (–16 to 4) | −5.0 (–17 to 6) | −5.5 (–17 to 5) |
| Min., max. (mmHg) | −82, 51 | −82, 60 | −78, 47 | −82, 60 |
| Comparisons | Mean difference [mmHg (95% CI)] | p-value | ||
| Active vs. placebo | –0.6 (–3.1 to 2.0) | 0.664 | ||
| i.v. vs. nebuliser | –1.8 (–4.7 to 1.1) | 0.214 | ||
| i.v. vs. placebo | –1.5 (–4.3 to 1.4) | 0.308 | ||
| Nebuliser vs. placebo | 0.4 (–2.6 to 3.3) | 0.810 | ||
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Diastolic BP at baseline | ||||
| Number of observations | 330 | 391 | 356 | 1077 |
| Mean [mmHg (SD)] | 76.3 (15.5) | 75.7 (14.8) | 75.0 (15.1) | 75.6 (15.1) |
| Median [mmHg (IQR)] | 75.0 (66–86) | 74.0 (66–85) | 74.0 (65–84) | 74.0 (65–85) |
| Min., max. (mmHg) | 30, 151 | 38, 135 | 33, 145 | 30, 151 |
| Diastolic BP at 1 hour | ||||
| Number of observations | 323 | 383 | 351 | 1057 |
| Mean [mmHg (SD)] | 73.2 (14.4) | 69.3 (11.5) | 70.8 (13.5) | 71.0 (13.2) |
| Median [mmHg (IQR)] | 73.0 (64–81) | 69.0 (61–77) | 70.0 (62–79) | 71.0 (62–78) |
| Min., max. (mmHg) | 36, 135 | 30, 115 | 32, 133 | 30, 135 |
| Change in diastolic BP at 1 hour | ||||
| Number of observations | 322 | 380 | 351 | 1053 |
| Mean [mmHg (SD)] | –3.4 (14.1) | –6.3 (13.8) | –4.2 (13.6) | –4.7 (13.9) |
| Median [mmHg (IQR)] | –2.0 (–11 to 5) | –5.0 (–13 to 2) | –4.0 (–12 to 3) | –4.0 (–12 to 3) |
| Min., max. (mmHg) | –65, 49 | –67, 41 | –72, 69 | –72, 69 |
| Diastolic BP at 2 hours | ||||
| Number of observations | 309 | 372 | 337 | 1018 |
| Mean [mmHg (SD)] | 70.6 (13.9) | 68.3 (12.3) | 69.7 (14.3) | 69.4 (13.5) |
| Median [mmHg (IQR)] | 69.0 (62–80) | 68.0 (61–76) | 69.0 (61–78) | 69.0 (61–78) |
| Min., max. (mmHg) | 35, 117 | 32, 113 | 37, 148 | 32, 148 |
| Change in diastolic BP at 2 hours | ||||
| Number of observations | 309 | 369 | 337 | 1015 |
| Mean [mmHg (SD)] | –5.8 (14.4) | –7.5 (14.9) | –5.4 (14.6) | –6.3 (14.7) |
| Median [mmHg (IQR)] | –5.0 (–15 to 4) | –7.0 (–16 to 2) | –5.0 (–14 to 4) | –6.0 (–15 to 3) |
| Min., max. (mmHg) | –53, 31 | –81, 40 | –72, 35 | –81, 40 |
| Comparisons | Mean difference [mmHg (95% CI)] | p-value | ||
| Active vs. placebo | –1.1 (–3.0 to 0.8) | 0.248 | ||
| i.v. vs. nebuliser | –1.6 (–3.8 to 0.6) | 0.151 | ||
| i.v. vs. placebo | –1.9 (–4.1 to 0.2) | 0.080 | ||
| Nebuliser vs. placebo | –0.3 (–2.6 to 1.9) | 0.782 | ||
FIGURE 6.
Heart rate (beats per minute) during and after trial treatment. Neb, nebuliser; Pla, placebo.
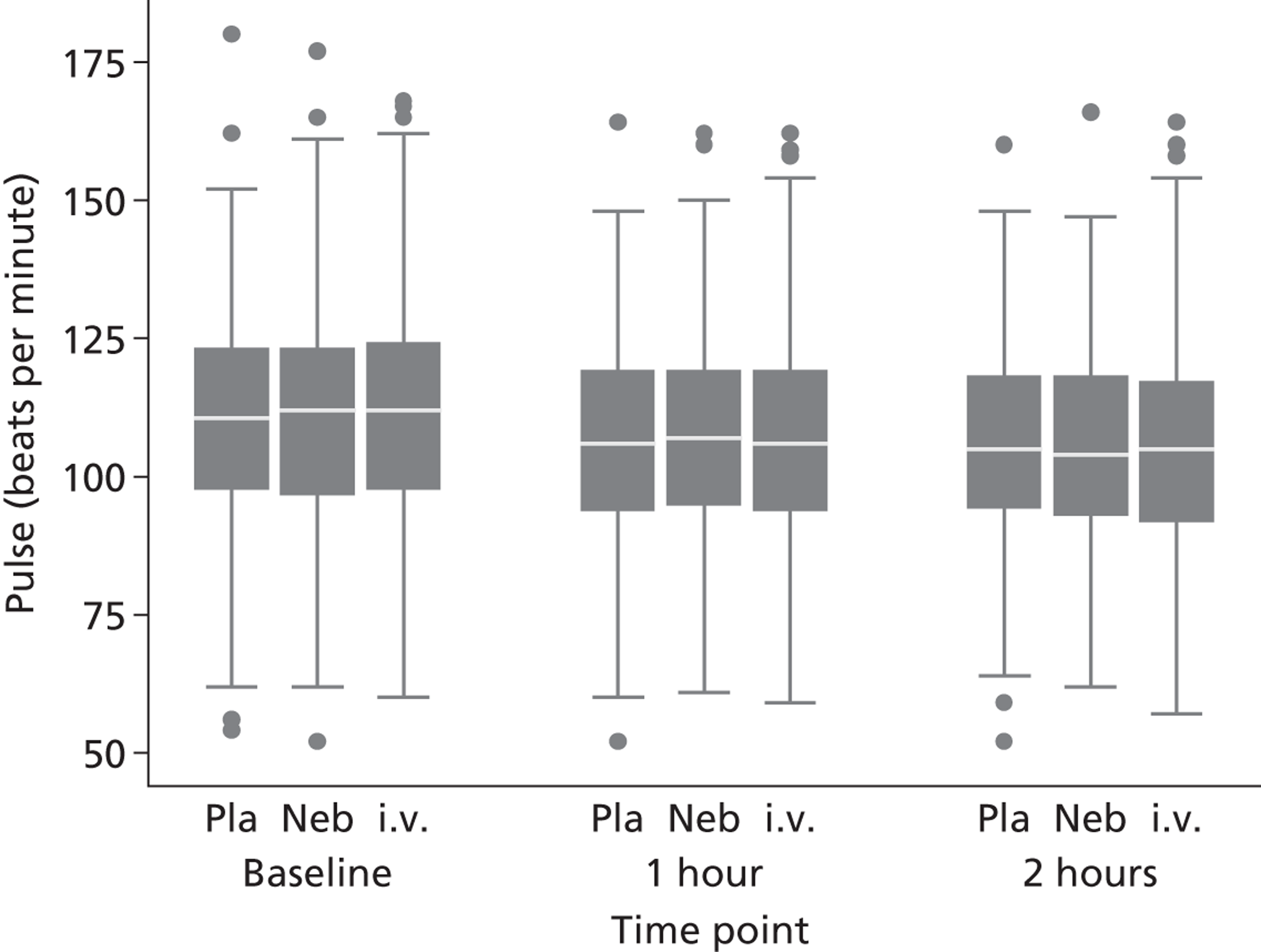
FIGURE 7.
Respiratory rate (breaths per minute) during and after trial treatment. Neb, nebuliser; Pla, placebo.
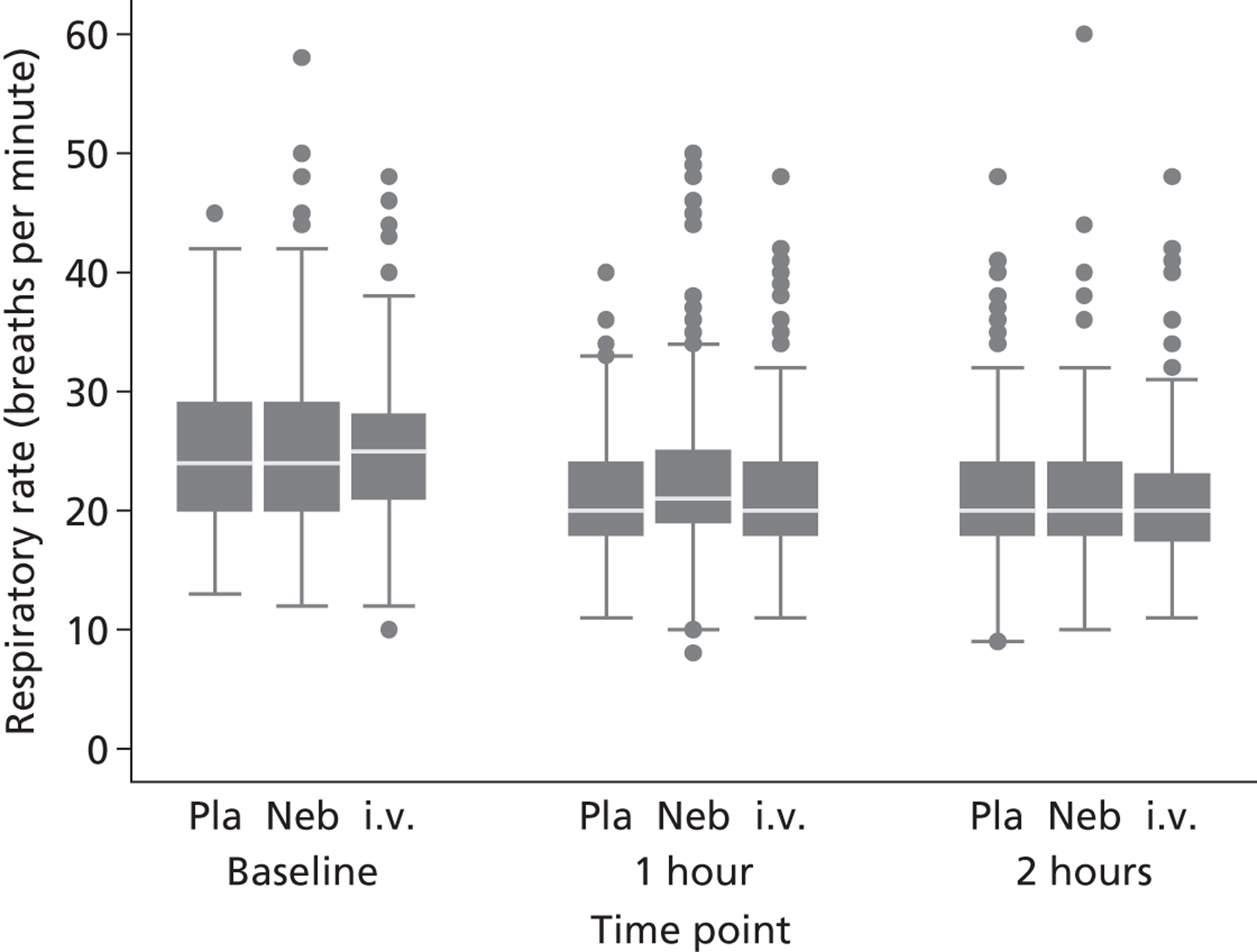
FIGURE 8.
Systolic blood pressure (mmHg) during and after trial treatment. Neb, nebuliser; Pla, placebo.
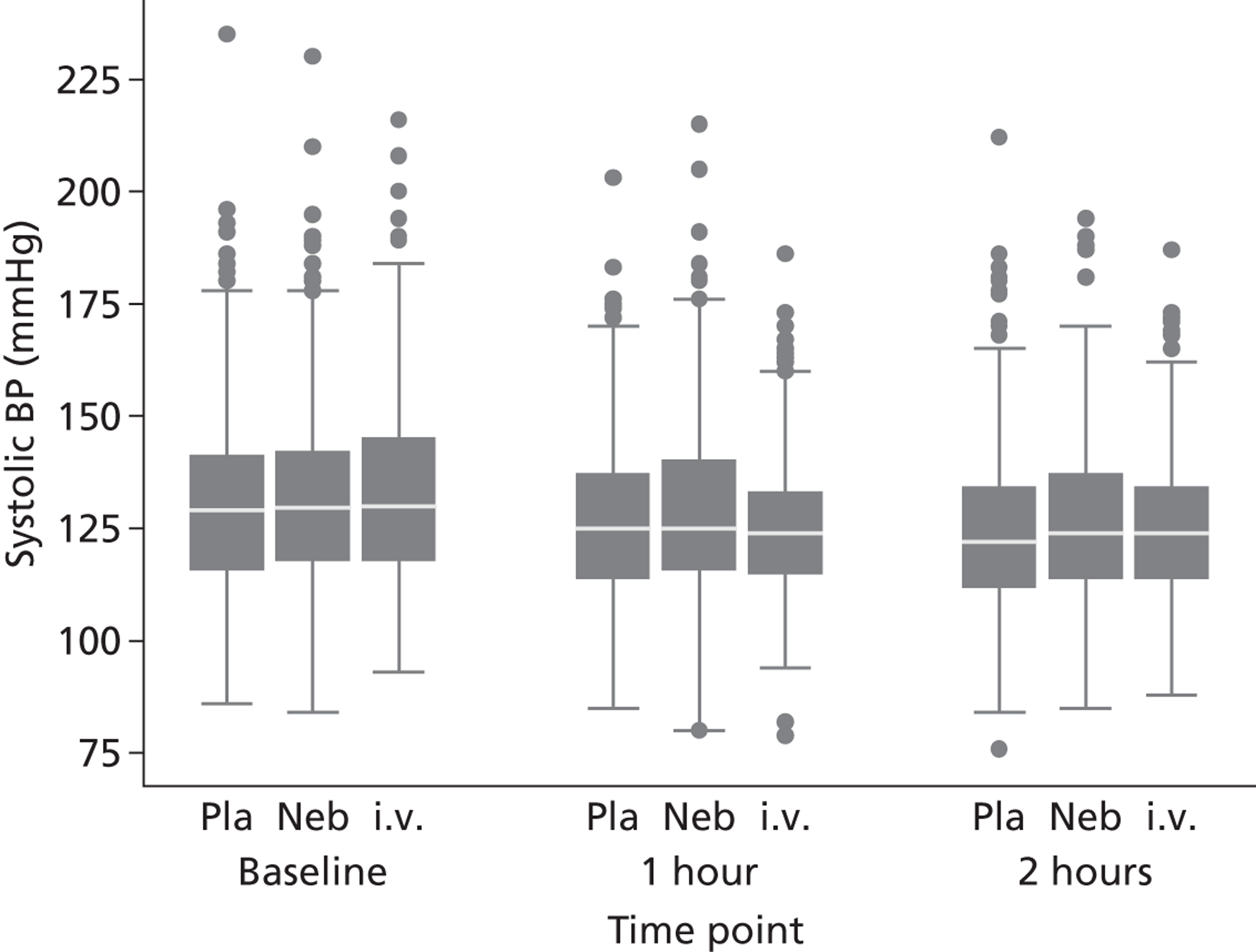
FIGURE 9.
Diastolic blood pressure (mmHg) during and after trial treatment. Neb, nebuliser; Pla, placebo.
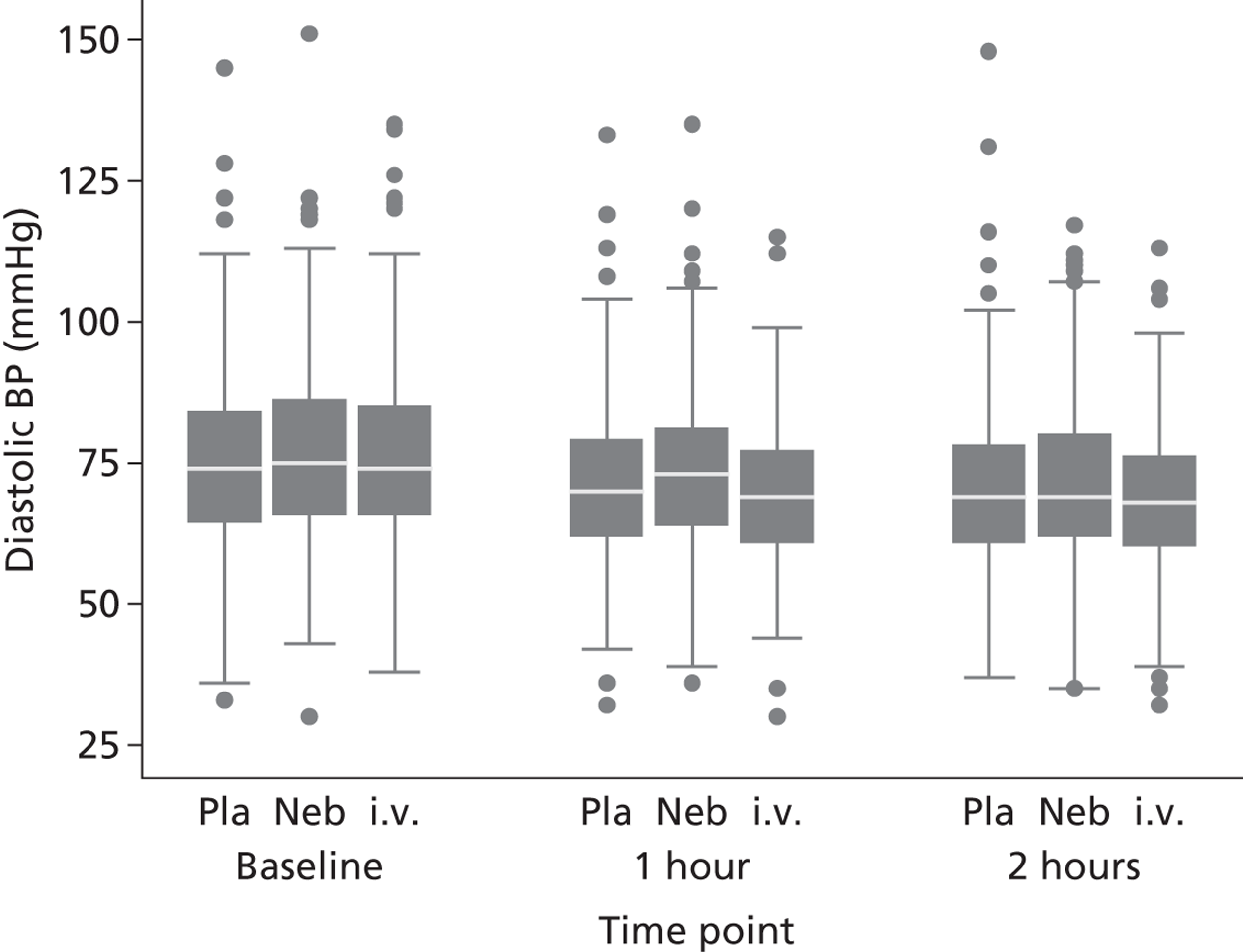
Table 23 shows the change in peripheral oxygen saturation over the first 2 hours after initiation of treatment. Three-quarters of the cohort were receiving supplemental oxygen at baseline. The proportion of participants receiving supplemental oxygen increased at 1 hour and then decreased at 2 hours. This means that the analysis of peripheral oxygen saturation, shown separately for those receiving and not receiving supplemental oxygen, includes different patients at different times. Unsurprisingly, there is little change in the peripheral oxygen saturation over time and little difference between groups, since administration of oxygen is likely to be titrated in response to demand and/or peripheral oxygen saturation. Patients receiving supplemental oxygen had a mean peripheral oxygen saturation of around 98%, whereas those patients not receiving supplemental oxygen had a peripheral oxygen saturation of 96–97%.
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Mode of delivery at baselinea | ||||
| On oxygen | 256 (77%) | 294 (75%) | 259 (72%) | 809 (75%) |
| On air | 75 (23%) | 99 (25%) | 95 (27%) | 269 (25%) |
| Not recorded | 1 (< 1%) | 1 (< 1%) | 4 (1%) | 6 (1%) |
| Mode of delivery at 1 houra | ||||
| On oxygen | 285 (86%) | 338 (86%) | 299 (84%) | 922 (85%) |
| On air | 39 (12%) | 45 (11%) | 51 (14%) | 135 (12%) |
| Not recorded | 8 (2%) | 11 (3%) | 8 (2%) | 27 (2%) |
| Mode of delivery at 2 hoursa | ||||
| On oxygen | 213 (64%) | 224 (57%) | 199 (56%) | 636 (59%) |
| On air | 99 (30%) | 154 (39%) | 138 (39%) | 391 (36%) |
| Not recorded | 20 (6%) | 16 (4%) | 21 (6%) | 57 (5%) |
| Patients on oxygen | ||||
| Oxygen saturation at baseline (%) | ||||
| Number of observations | 256 | 294 | 259 | 809 |
| Mean [% (SD)] | 97.8 (2.1) | 97.9 (2.3) | 98.0 (2.2) | 97.9 (2.2) |
| Median [% (IQR)] | 98.0 (97–100) | 98.5 (97–100) | 99.0 (97–100) | 98.0 (97–100) |
| Min., max. (%) | 91, 100 | 88, 100 | 90, 100 | 88, 100 |
| Oxygen saturation at 1 hour (%) | ||||
| Number of observations | 285 | 338 | 299 | 922 |
| Mean [% (SD)] | 98.2 (1.9) | 98.3 (1.9) | 98.3 (1.9) | 98.2 (1.9) |
| Median [% (IQR)] | 99.0 (97–100) | 99.0 (97–100) | 99.0 (97–100) | 99.0 (97–100) |
| Min., max. (%) | 91, 100 | 89, 100 | 90, 100 | 89, 100 |
| Change in oxygen saturation at 1 hour (%) | ||||
| Number of observations | 235 | 266 | 234 | 735 |
| Mean [% (SD)] | 0.3 (2.0) | 0.3 (2.0) | 0.3 (1.9) | 0.3 (2.0) |
| Median [% (IQR)] | 0.0 (–1 to 1) | 0.0 (–1 to 1) | 0.0 (–1 to 1) | 0.0 (–1 to 1) |
| Min., max. (%) | –5, 6 | –5, 9 | –5, 7 | –5, 9 |
| Oxygen saturation at 2 hours (%) | ||||
| Number of observations | 213 | 224 | 199 | 636 |
| Mean [% (SD)] | 97.7 (2.3) | 97.7 (2.1) | 97.8 (2.2) | 97.7 (2.2) |
| Median [% (IQR)] | 98.0 (96–100) | 98.0 (97–99) | 98.0 (96–100) | 98.0 (96–100) |
| Min., max. (%) | 90, 100 | 90, 100 | 90, 100 | 90, 100 |
| Change in oxygen saturation at 2 hours (%) | ||||
| Number of observations | 176 | 181 | 164 | 521 |
| Mean [% (SD)] | –0.0 (2.3) | –0.1 (2.3) | –0.2 (2.1) | –0.1 (2.2) |
| Median [% (IQR)] | 0.0 (–1 to 1) | 0.0 (–1 to 1) | 0.0 (–1 to 1) | 0.0 (–1 to 1) |
| Min., max. (%) | –6, 6 | –8, 7 | –7, 7 | –8, 7 |
| Patients on air | ||||
| Oxygen saturation at baseline (%) | ||||
| Number of observations | 75 | 99 | 95 | 269 |
| Mean [% (SD)] | 96.3 (2.6) | 95.9 (2.5) | 95.9 (2.6) | 96.0 (2.6) |
| Median [% (IQR)] | 96.0 (95–98) | 96.0 (94–98) | 96.0 (94–98) | 96.0 (94–98) |
| Min., max. (%) | 88, 100 | 87, 100 | 85, 100 | 85, 100 |
| Oxygen saturation at 1 hour (%) | ||||
| Number of observations | 39 | 45 | 51 | 135 |
| Mean [% (SD)] | 96.6 (2.6) | 96.6 (2.9) | 97.1 (2.4) | 96.8 (2.7) |
| Median [% (IQR)] | 98.0 (95–98) | 97.0 (95–99) | 98.0 (95–99) | 98.0 (95–99) |
| Min., max. (%) | 92, 100 | 88, 100 | 92, 100 | 88, 100 |
| Change in oxygen saturation at 1 hour (%) | ||||
| Number of observations | 24 | 25 | 30 | 79 |
| Mean [% (SD)] | 0.0 (2.0) | 1.0 (2.1) | 1.5 (2.6) | 0.9 (2.3) |
| Median [% (IQR)] | 0.0 (–2 to 2) | 1.0 (0 to 3) | 1.0 (–1 to 4) | 1.0 (–1 to 3) |
| Min., max. (%) | –6, 3 | –4, 4 | –2, 7 | –6, 7 |
| Oxygen saturation at 2 hours (%) | ||||
| Number of observations | 99 | 154 | 138 | 391 |
| Mean [% (SD)] | 96.4 (2.6) | 96.3 (2.7) | 96.4 (2.4) | 96.4 (2.6) |
| Median [% (IQR)] | 96.0 (95–99) | 97.0 (95–98) | 96.0 (95–98) | 96.0 (95–98) |
| Min., max. (%) | 87, 100 | 86, 100 | 85, 100 | 85, 100 |
| Change in oxygen saturation at 2 hours (%) | ||||
| Number of observations | 34 | 52 | 56 | 142 |
| Mean [% (SD)] | –0.0 (2.6) | –0.1 (2.4) | 0.5 (2.6) | 0.2 (2.5) |
| Median [% (IQR)] | 0.0 (–1 to 1) | 0.0 (–1 to 1) | 0.0 (–1 to 2) | 0.0 (–1 to 1) |
| Min., max. (%) | –7, 6 | –9, 4 | –4, 10 | –9, 10 |
If the trial treatment had influenced oxygenation then this might have been apparent in terms of reduced requirement for inspired oxygen. Table 24 and Figure 10 show the change in inspired oxygen flow rate over the first 2 hours after initiation of treatment. Patients not receiving oxygen were analysed as having a flow rate of 0 l/minute. Patients allocated to i.v. magnesium sulphate had a slightly lower oxygen flow rate at 2 hours than patients allocated to nebulised magnesium sulphate, but there were no significant differences between either treatment arm and placebo.
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Oxygen flow at baseline | ||||
| Number of observations | 324 | 387 | 344 | 1055 |
| Mean [l/minute (SD)] | 5.1 (3.5) | 5.3 (4.0) | 4.8 (3.6) | 5.1 (3.7) |
| Median [l/minute (IQR)] | 6.0 (2–6) | 6.0 (0–6) | 6.0 (0–6) | 6.0 (0–6) |
| Min., max. (l/minute) | 0, 15 | 0, 15 | 0, 15 | 0, 15 |
| Oxygen flow at 1 hour | ||||
| Number of observations | 315 | 377 | 339 | 1031 |
| Mean [l/minute (SD)] | 5.6 (2.6) | 5.6 (2.6) | 5.3 (2.7) | 5.5 (2.6) |
| Median [l/minute (IQR)] | 6.0 (5–6) | 6.0 (6–6) | 6.0 (5–6) | 6.0 (5–6) |
| Min., max. (l/minute) | 0, 15 | 0, 15 | 0, 15 | 0, 15 |
| Change in oxygen flow at 1 hour | ||||
| Number of observations | 312 | 372 | 331 | 1015 |
| Mean [l/minute (SD)] | 0.5 (3.4) | 0.4 (3.9) | 0.5 (3.7) | 0.5 (3.7) |
| Median [l/minute (IQR)] | 0.0 (0–1) | 0.0 (0–2) | 0.0 (0–2) | 0.0 (0–2) |
| Min., max. (l/minute) | –10, 10 | –11, 10 | –15, 10 | –15, 10 |
| Oxygen flow at 2 hours (l/minute) | ||||
| Number of observations | 301 | 364 | 326 | 991 |
| Mean [l/minute (SD)] | 4.4 (3.7) | 3.6 (3.5) | 3.6 (3.5) | 3.8 (3.5) |
| Median [l/minute (IQR)] | 6.0 (0–6) | 5.0 (0–6) | 5.0 (0–6) | 5.0 (0–6) |
| Min., max. (l/minute) | 0, 15 | 0, 15 | 0, 15 | 0, 15 |
| Change in oxygen flow at 2 hours (l/minute) | ||||
| Number of observations | 299 | 361 | 320 | 980 |
| Mean [l/minute (SD)] | –0.7 (4.3) | –1.6 (4.8) | –1.3 (4.2) | –1.2 (4.5) |
| Median [l/minute (IQR)] | 0.0 (–4 to 0) | 0.0 (–6 to 0) | 0.0 (–5 to 0) | 0.0 (–5 to 0) |
| Min., max. (l/minute) | –15, 10 | –15, 10 | –15, 10 | –15, 10 |
| Comparisons | Mean difference [l/minute (95% CI)] | p-value | ||
| Active vs. placebo | 0.0 (–0.6 to 0.6) | 0.942 | ||
| i.v. vs. nebuliser | –0.9 (–1.6 to –0.2) | 0.008 | ||
| i.v. vs. placebo | –0.4 (–1.1 to 0.2) | 0.196 | ||
| Nebuliser vs. placebo | 0.5 (–0.2 to 1.2) | 0.173 | ||
FIGURE 10.
Oxygen flow rate (l/minute) during and after trial treatment. Neb, nebuliser; Pla, placebo.
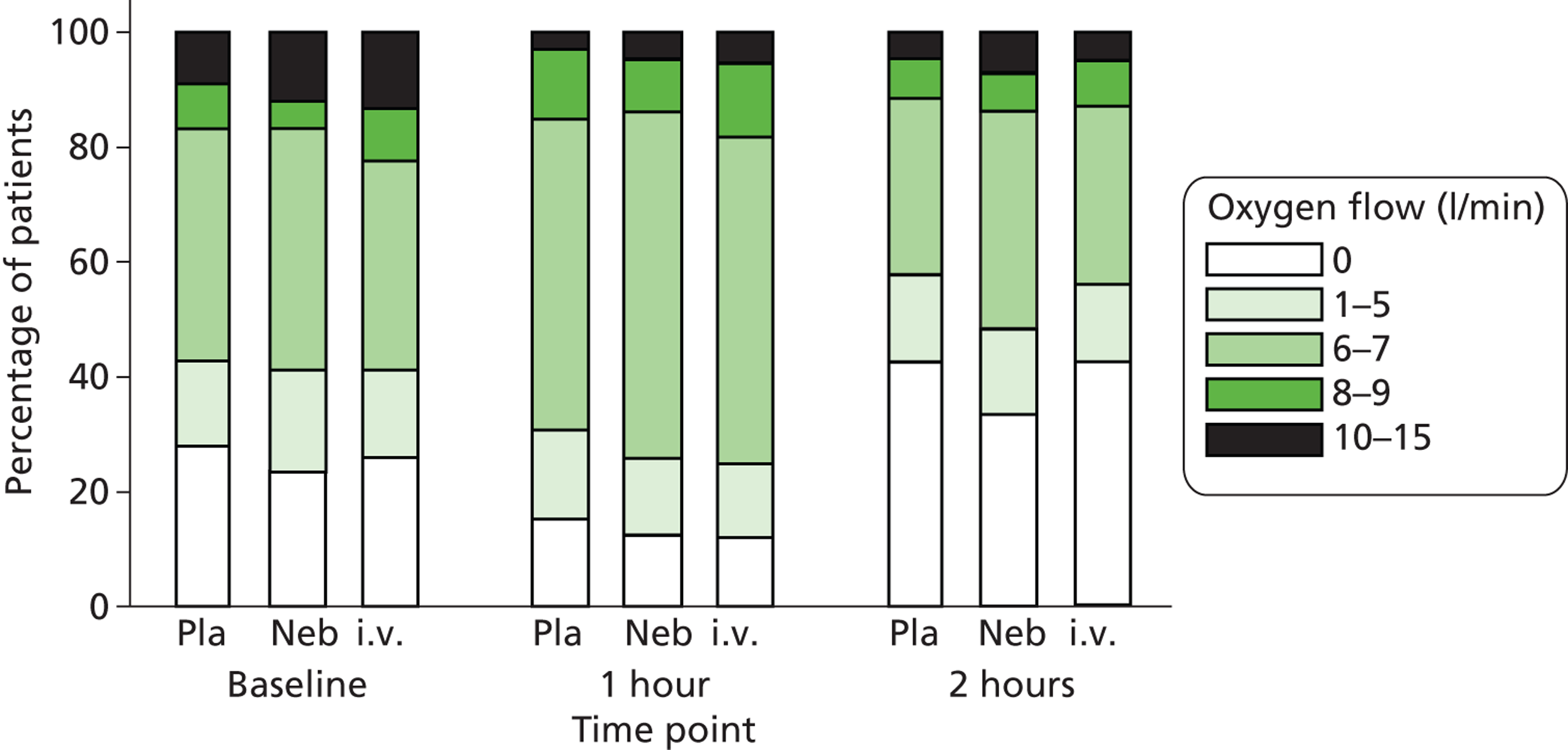
Table 25 and Figure 11 show the change in PEFR over the first 2 hours after initiation of treatment, and Table 26 and Figure 12 show these data as percentage of predicted PEFR. Mean PEFR and percentage predicted PEFR improved markedly in all three treatment groups over the 2 hours. The IQR for percentage predicted PEFR at 2 hours indicates that after treatment only around one-quarter of the cohort were recording a PEFR < 50% of predicted. Comparison of the three groups shows no evidence of any effect from i.v. or nebulised magnesium sulphate on PEFR.
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| PEFR at baseline | ||||
| Number of observations | 312 | 379 | 336 | 1027 |
| Mean [l/minute (SD)] | 209.1 (92.3) | 226.8 (92.3) | 214.5 (84.0) | 217.4 (89.9) |
| Median [l/minute (IQR)] | 200.0 (150–250) | 210.0 (150–270) | 200.0 (150–255) | 200.0 (150–260) |
| Min., max. (l/minute) | 50, 620 | 50, 530 | 50, 520 | 50, 620 |
| PEFR at 1 hour | ||||
| Number of observations | 293 | 358 | 324 | 975 |
| Mean [l/minute (SD)] | 255.2 (109.8) | 275.6 (104.9) | 259.0 (100.2) | 263.9 (105.2) |
| Median [l/minute (IQR)] | 240.0 (170–330) | 260.0 (190–350) | 250.0 (180–330) | 250.0 (180–340) |
| Min., max. (l/minute) | 50, 590 | 50, 650 | 60, 700 | 50, 700 |
| Change in PEFR at 1 hour | ||||
| Number of observations | 284 | 351 | 312 | 947 |
| Mean [l/minute (SD)] | 43.3 (63.9) | 48.2 (66.0) | 45.3 (65.1) | 45.8 (65.0) |
| Median [l/minute (IQR)] | 30.0 (0–70) | 30.0 (0–90) | 30.0 (3–70) | 30.0 (0–70) |
| Min., max. (l/minute) | –110, 330 | –100, 350 | –60, 580 | –110, 580 |
| PEFR at 2 hours | ||||
| Number of observations | 281 | 348 | 310 | 939 |
| Mean [l/minute (SD)] | 270.4 (117.4) | 288.1 (111.2) | 278.9 (105.5) | 279.8 (111.4) |
| Median [l/minute (IQR)] | 250.0 (190–350) | 280.0 (200–355) | 270.0 (200–350) | 270.0 (200–350) |
| Min., max. (l/minute) | 50, 800 | 50, 650 | 50, 600 | 50, 800 |
| Change in PEFR at 2 hours | ||||
| Number of observations | 272 | 339 | 298 | 909 |
| Mean [l/minute (SD)] | 58.3 (77.3) | 61.0 (73.6) | 62.5 (69.4) | 60.7 (73.3) |
| Median [l/minute (IQR)] | 40.0 (5–95) | 40.0 (10–90) | 50.0 (20–100) | 40.0 (10–100) |
| Min., max. (l/minute) | –89, 500 | −140, 410 | −80, 390 | –140, 500 |
| Comparisons | Mean difference [l/minute (95% CI)] | p-value | ||
| Active vs. placebo | –2.5 (–12.5 to 7.5) | 0.625 | ||
| i.v. vs. nebuliser | 0.3 (–11.2 to 11.7) | 0.964 | ||
| i.v. vs. placebo | –2.4 (–13.6 to 8.8) | 0.680 | ||
| Nebuliser vs. placebo | –2.6 (–14.5 to 9.2) | 0.664 | ||
FIGURE 11.
Peak expiratory flow rate (l/minute) during and after trial treatment. Neb, nebuliser; Pla, placebo.
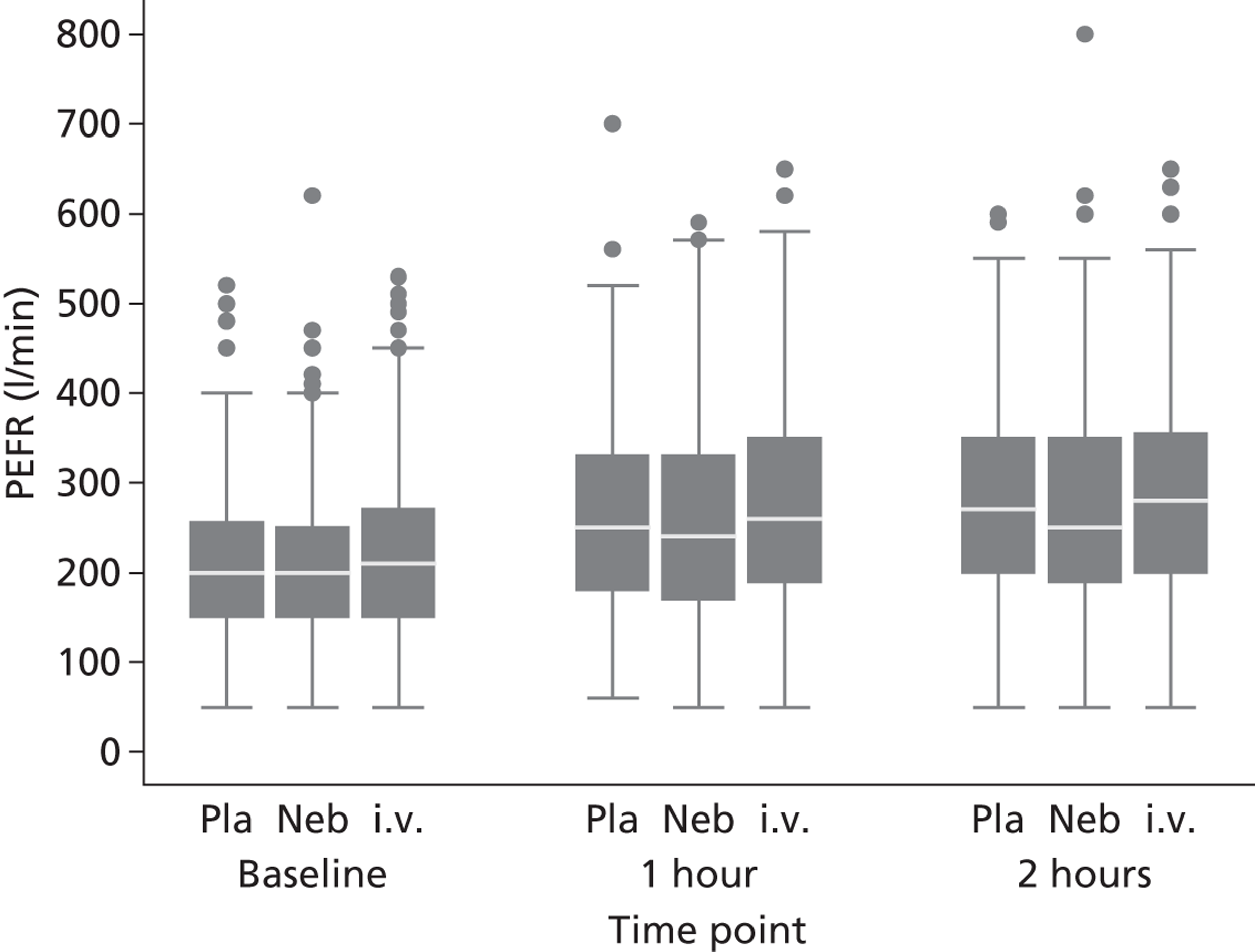
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| % PEFR at baseline | ||||
| Number of observations | 308 | 375 | 327 | 1010 |
| Mean [% of predicted (SD)] | 50.0 (19.6) | 54.3 (20.2) | 50.5 (19.1) | 51.7 (19.7) |
| Median [% of predicted (IQR)] | 47.5 (35–61) | 50.0 (40–67) | 48.0 (37–63) | 50.0 (38–63) |
| Min., max. (% of predicted) | 11, 116 | 12, 160 | 9, 112 | 9, 160 |
| % PEFR at 1 hour | ||||
| Number of observations | 290 | 356 | 315 | 961 |
| Mean [% of predicted (SD)] | 60.3 (22.8) | 65.5 (22.7) | 60.1 (20.9) | 62.1 (22.3) |
| Median [% of predicted (IQR)] | 57.1 (43–77) | 63.0 (50–80) | 60.0 (44–73) | 60.0 (45–78) |
| Min., max. (% of predicted) | 17, 125 | 13, 140 | 13, 125 | 13, 140 |
| % change in PEFR at 1 hour | ||||
| Number of observations | 282 | 349 | 304 | 935 |
| Mean [% of predicted (SD)] | 9.9 (15.0) | 11.4 (15.7) | 10.2 (14.7) | 10.6 (15.2) |
| Median [% of predicted (IQR)] | 7.0 (0–17) | 6.8 (0–20) | 7.5 (0–16) | 7.0 (0–17) |
| Min., max. (% of predicted) | –40, 63 | –24, 75 | –27, 97 | –40, 97 |
| % PEFR at 2 hours | ||||
| Number of observations | 278 | 346 | 301 | 925 |
| Mean [% of predicted (SD)] | 63.1 (24.0) | 68.6 (23.3) | 65.0 (22.7) | 65.8 (23.5) |
| Median [% of predicted (IQR)] | 60.0 (46–78) | 68.3 (52–86) | 64.9 (47–80) | 64.0 (49–81) |
| Min., max. (% of predicted) | 16, 160 | 13, 144 | 10, 164 | 10, 164 |
| % change in PEFR at 2 hours | ||||
| Number of observations | 270 | 337 | 291 | 898 |
| Mean [% of predicted (SD)] | 13.4 (18.0) | 14.4 (17.4) | 14.4 (16.3) | 14.1 (17.2) |
| Median [% of predicted (IQR)] | 9.8 (1–22) | 10.9 (3–23) | 10.9 (4–22) | 10.4 (3–22) |
| Min., max. (% of predicted) | –36, 100 | –30, 83 | –18, 103 | –36, 103 |
| Comparisons | Mean difference [% of predicted (95% CI)] | p-value | ||
| Active vs. placebo | –0.5 (–2.9 to 1.9) | 0.676 | ||
| i.v. vs. nebuliser | 0.3 (–2.4 to 3.0) | 0.841 | ||
| i.v. vs. placebo | –0.4 (–3.0 to 2.3) | 0.786 | ||
| Nebuliser vs. placebo | –0.6 (–3.4 to 2.1) | 0.652 | ||
FIGURE 12.
Peak expiratory flow rate (% of predicted) during and after trial treatment. Neb, nebuliser; Pla, placebo.
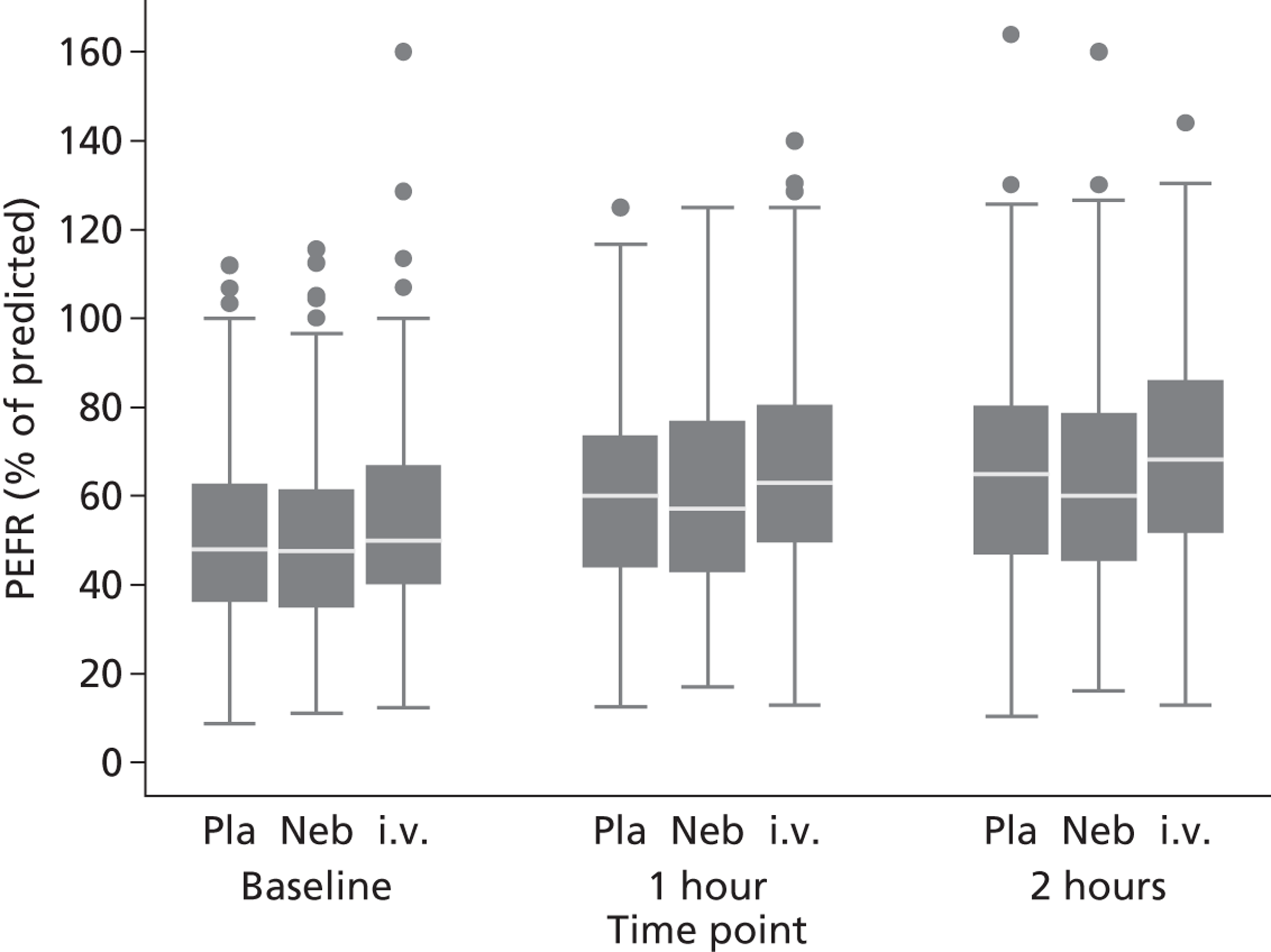
Adverse events and side effects
Table 27 shows the adverse events recorded up to 30 days and Table 28 shows those adverse events which were classified as being SAEs. There were very few adverse events in the prespecified categories: seven patients required intubation, seven patients required non-invasive ventilation, two patients suffered an arrhythmia, one patient suffered a cardiac arrest and two patients died. However, the definition of an adverse event used in the trial included any subsequent hospitalisation. A substantial number of patients were therefore recorded as having an adverse event by virtue of subsequent admission to hospital, either due to worsening of their asthma or other unrelated problems.
| Adverse event | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 395) | Placebo (n = 358) | Overall (n = 1085) |
|---|---|---|---|---|
| Any adverse event | 41 (12.3%) | 53 (13.4%) | 36 (10.1%) | 130 (12.0%) |
| Arrhythmia | 0 | 1 (0.3%) | 1 (0.3%) | 2 (0.2%) |
| Cardiac arrest | 0 | 1 (0.3%) | 0 | 1 (0.1%) |
| Death | 1 (0.3%) | 1 (0.3%) | 0 | 2 (0.2%) |
| Intubation | 2 (0.6%) | 4 (1.0%) | 1 (0.3%) | 7 (0.6%) |
| Non-invasive ventilation | 2 (0.6%) | 2 (0.5%) | 3 (0.8%) | 7 (0.6%) |
| Other asthma related | 26 (7.8%) | 25 (6.3%) | 22 (6.1%) | 73 (6.7%) |
| Other non-asthma related | 14 (4.2%) | 20 (5.1%) | 12 (3.4%) | 46 (4.2%) |
| Adverse event | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 395) | Placebo (n = 358) | Overall (n = 1085) |
|---|---|---|---|---|
| Any serious adverse event | 35 (10.5%) | 45 (11.4%) | 28 (7.8%) | 108 (10.0%) |
| Arrhythmia | 0 | 1 (0.3%) | 0 | 1 (0.1%) |
| Cardiac arrest | 0 | 1 (0.3%) | 0 | 1 (0.1%) |
| Death | 1 (0.3%) | 1 (0.3%) | 0 | 2 (0.2%) |
| Intubation | 2 (0.6%) | 4 (1.0%) | 1 (0.3%) | 7 (0.6%) |
| Non-invasive ventilation | 0 | 1 (0.3%) | 3 (0.8%) | 4 (0.4%) |
| Other asthma related | 24 (7.2%) | 23 (5.8%) | 21 (5.9%) | 68 (6.3%) |
| Other non-asthma related | 8 (2.4%) | 14 (3.6%) | 5 (1.4%) | 27 (2.5%) |
Table 29 shows the potential side effects recorded during or after trial treatment. The rate of side effects that have previously been reported in association with administration of magnesium sulphate (flushing, hypotension, nausea and vomiting) were generally low and were only slightly higher in the active treatment arms than the placebo arm. Overall, side effects were a little more common in the active treatment arms than the placebo arm [15.6% vs. 10.1%, odds ratio (OR) 1.68 (95% CI 1.11 to 2.52; p = 0.014)].
| Side effect | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1085) |
|---|---|---|---|---|
| Any side effecta | 52 (15.7%) | 61 (15.5%) | 36 (10.1%) | 149 (13.7%) |
| Flushinga | 3 (0.9%) | 7 (1.8%) | 2 (0.6%) | 12 (1.1%) |
| Hypotensiona | 31 (9.3%) | 31 (7.8%) | 22 (6.1%) | 84 (7.7%) |
| Nauseaa | 5 (1.5%) | 14 (3.5%) | 7 (2.0%) | 26 (2.4%) |
| Vomitinga | 6 (1.8%) | 6 (1.5%) | 3 (0.8%) | 15 (1.4%) |
| Othera | 12 (3.6%) | 15 (3.8%) | 5 (1.4%) | 32 (2.9%) |
| Comparisonsb | Odds ratio (95% CI) | p-value | ||
| Active vs. placebo | 1.68 (1.11 to 2.52) | 0.014 | ||
| i.v. vs. nebuliser | 1.00 (0.66 to 1.52) | 0.988 | ||
| i.v. vs. placebo | 1.68 (1.07 to 2.63) | 0.025 | ||
| Nebuliser vs. placebo | 1.67 (1.05 to 2.66) | 0.031 | ||
Thirty-day outcomes
Overall, 504 (47%) of the 30-day outcome questionnaires were returned. Table 30 shows the 30-day questionnaire response rate by age group and sex. Females were more likely to respond than males and the percentage response increased with participant age.
| Age group (years) | n (%) response | |
|---|---|---|
| Males | Females | |
| < 25 | 22 (28) | 87 (43) |
| 25–34 | 23 (30) | 91 (47) |
| 35–44 | 25 (35) | 79 (49) |
| 45–54 | 35 (59) | 73 (55) |
| 55–64 | 11 (55) | 34 (62) |
| 65–74 | 6 (55) | 12 (86) |
| ≥ 75 | 3 (60) | 3 (60) |
Figure 13 shows the overall distribution of EQ-5D scores and Table 31 shows patient-reported health utility, at baseline and 30 days, measured on the EQ-5D survey. The placebo group had a slightly higher baseline mean score than the nebulised group, which was in turn higher than the i.v. group. There were only small changes in EQ-5D score from baseline to 30 days and no significant differences between the groups. Health utility data for the normal UK population were used to calculate a regional age- and sex-adjusted expected normal value for each patient. 68 The results are shown in Figure 14. At 30 days, the responders had a significantly lower EQ-5D than age- or sex-matched UK norms (mean loss = –0.14, 95% CI –0.17 to –0.11; p < 0.001).
FIGURE 13.
Distribution of EQ-5D scores. EQ-5D values at 30 days.
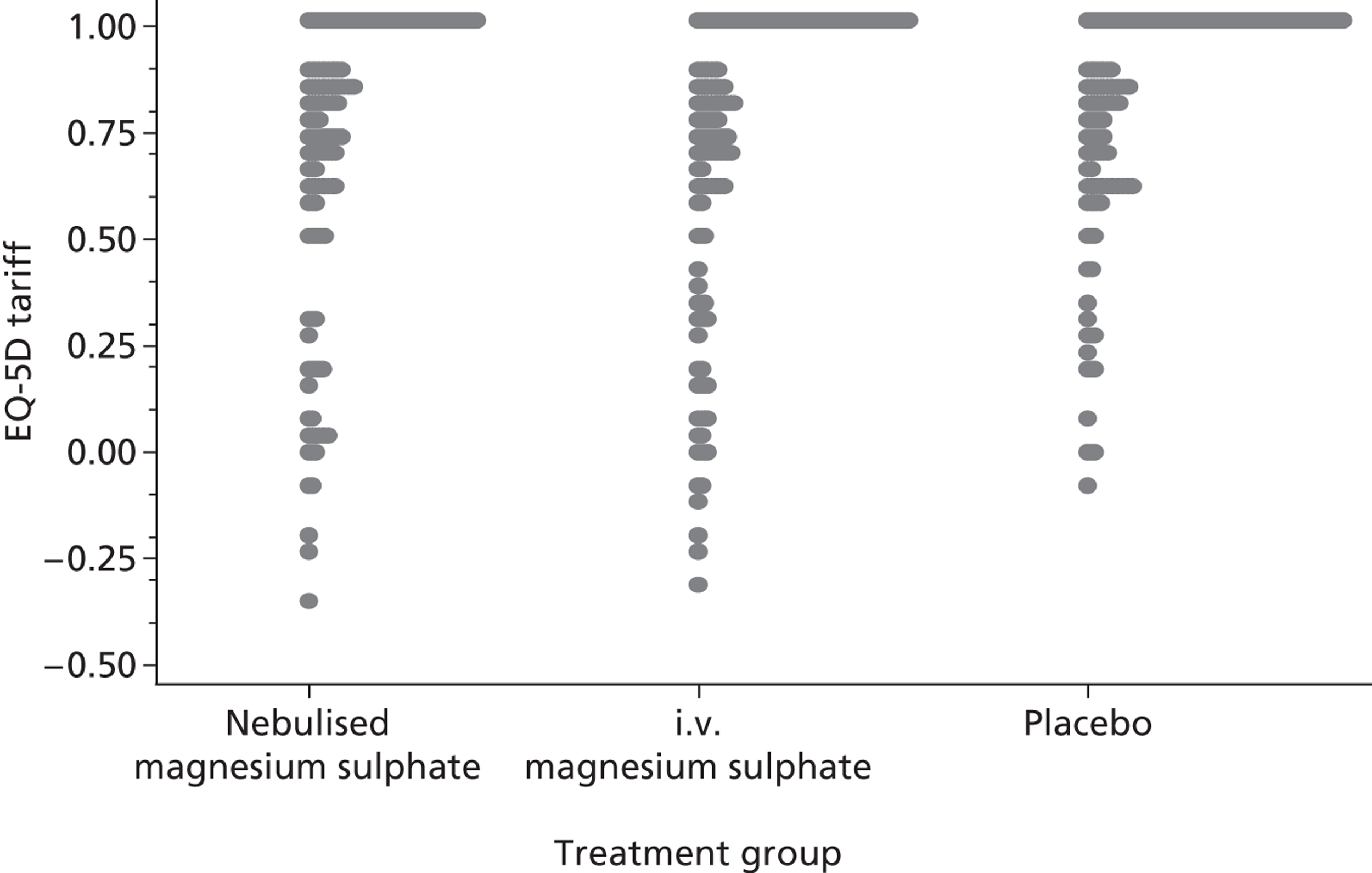
| Measurement | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Baseline | ||||
| Number (%) of responses | 282 (84.9) | 341 (86.5) | 309 (86.3) | 932 (86.0) |
| Mean (SD) | 0.734 (0.327) | 0.726 (0.354) | 0.746 (0.323) | 0.735 (0.336) |
| Median (IQR) | 0.814 (0.69–1.00) | 0.814 (0.69–1.00) | 0.883 (0.69–1.00) | 0.848 (0.64–1.00) |
| Min., max. | –0.35, 1.00 | –0.59, 1.00 | –0.35, 1.00 | –0.59, 1.00 |
| One month | ||||
| Number (%) of responses | 160 (48.2) | 169 (42.9) | 173 (48.3) | 502 (46.3) |
| Mean (SD) | 0.721 (0.326) | 0.731 (0.329) | 0.810 (0.250) | 0.755 (0.305) |
| Median (IQR) | 0.814 (0.62–1.00) | 0.814 (0.62–1.00) | 0.883 (0.69–1.00) | 0.848 (0.64–1.00) |
| Min., max. | –0.35, 1.00 | –0.32, 1.00 | –0.08, 1.00 | –0.35, 1.00 |
| Change in EQ-5D score at 1 month | ||||
| Number (%) of responses | 140 (42.2) | 146 (37.1) | 151 (42.2) | 437 (40.3) |
| Mean (SD) | 0.037 (0.377) | 0.035 (0.357) | 0.041 (0.341) | 0.038 (0.358) |
| Median (IQR) | 0.00 (–0.10 to 0.20) | 0.00 (–0.15 to 0.19) | 0.00 (–0.12 to 0.15) | 0.00 (–0.11 to 0.19) |
| Min., max. | –1.07, 1.09 | –1.02, 1.48 | –1.02, 1.32 | –1.07, 1.48 |
| Comparisons | Difference (95% CI) | p-value | ||
| Active vs. placebo | –0.01 (–0.08 to 0.06) | 0.842 | ||
| i.v. vs. nebuliser | –0.00 (–0.09 to 0.08) | 0.961 | ||
| i.v. vs. placebo | –0.01 (–0.09 to 0.07) | 0.884 | ||
| Nebuliser vs. placebo | –0.01 (–0.09 to 0.08) | 0.844 | ||
| Comparison of baseline scores (responders vs. non-responders) | ||||
| Difference | –0.10 | –0.05 | 0.03 | –0.04 |
| (95% CI) | –0.17 to –0.02 | –0.13 to 0.02 | –0.04 to 0.10 | –0.08 to 0.01 |
| p-value | 0.014 | 0.179 | 0.456 | 0.085 |
FIGURE 14.
EQ-5D score loss at 30 days compared with age- or sex-matched UK norms.
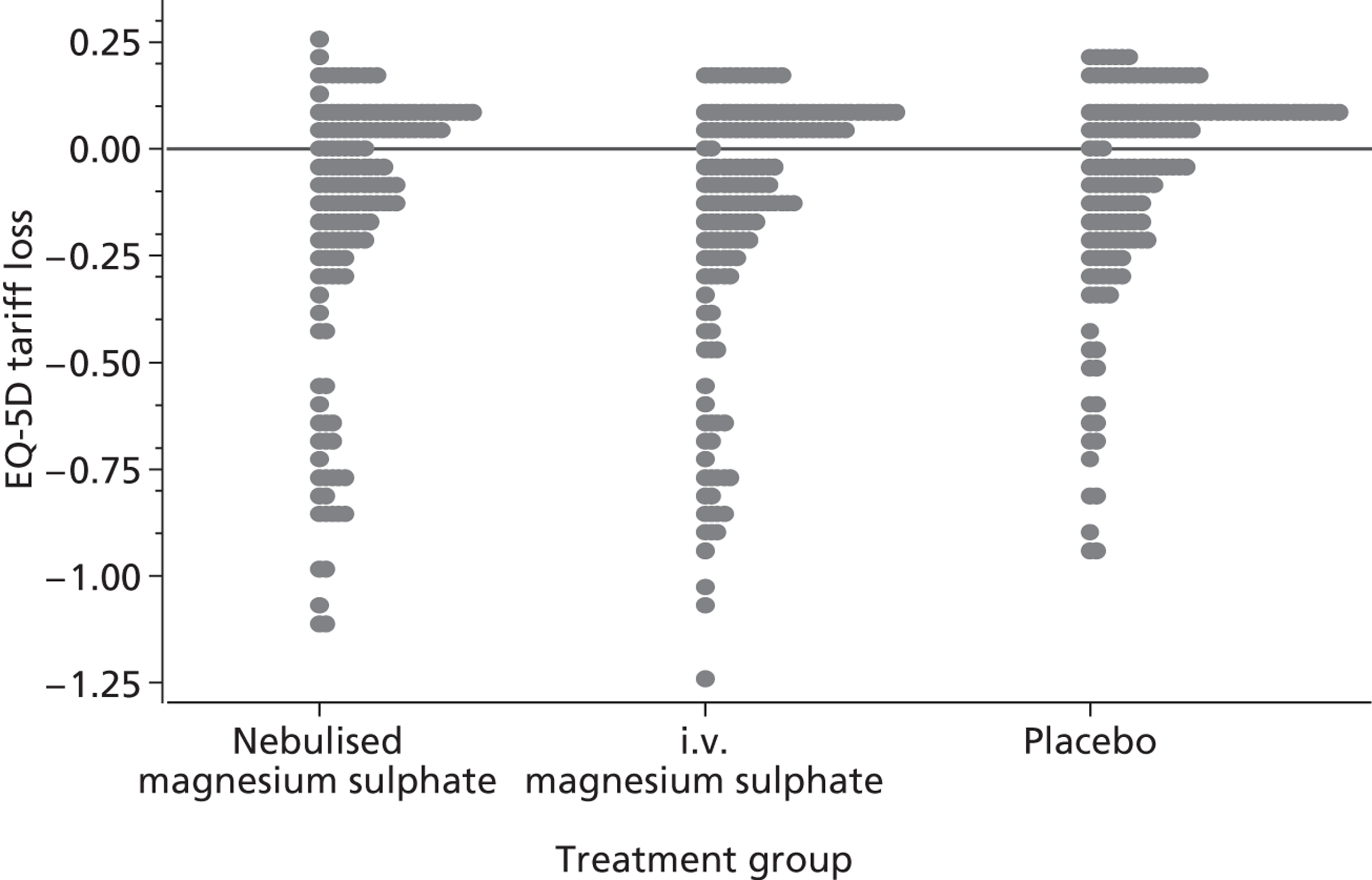
The 30-day questionnaire also asked patients to describe which NHS resources they had used in the 30 days following randomisation and these are described in Table 32. No obvious differences were noted between the groups, but most patients (74%) had at least one GP contact and reattendances at hospital were common, either as an outpatient (36%) or as an admission (29%).
| Health or social service | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Number (%) of responders | 138 (42%) | 151 (38%) | 144 (40%) | 433 (40%) |
| Use of telephone health advice | ||||
| None | 97 (70%) | 103 (68%) | 107 (74%) | 307 (71%) |
| 1 | 19 (14%) | 22 (15%) | 17 (12%) | 58 (13%) |
| 2 | 6 (4%) | 8 (5%) | 11 (8%) | 25 (6%) |
| 3 or more | 16 (12%) | 18 (12%) | 9 (6%) | 43 (10%) |
| Use of GP surgery consultations | ||||
| None | 39 (28%) | 43 (28%) | 39 (27%) | 121 (28%) |
| 1 | 37 (27%) | 33 (22%) | 31 (22%) | 101 (23%) |
| 2 | 24 (17%) | 28 (19%) | 35 (24%) | 87 (20%) |
| 3 | 16 (12%) | 17 (11%) | 18 (13%) | 51 (12%) |
| 4 or more | 22 (16%) | 30 (20%) | 21 (15%) | 73 (17%) |
| GP home visits | ||||
| None | 131 (95%) | 141 (93%) | 140 (97%) | 412 (95%) |
| 1 or more | 7 (5%) | 10 (7%) | 4 (3%) | 21 (5%) |
| Nurse home visits | ||||
| None | 131 (95%) | 145 (96%) | 141 (98%) | 417 (96%) |
| 1 or more | 7 (5%) | 6 (4%) | 3 (2%) | 16 (4%) |
| Social worker visits | ||||
| None | 130 (94%) | 145 (96%) | 143 (99%) | 418 (97%) |
| 1 or more | 8 (6%) | 6 (4%) | 1 (1%) | 15 (3%) |
| Outpatient attendances | ||||
| None | 83 (60%) | 90 (60%) | 103 (72%) | 276 (64%) |
| 1 | 35 (25%) | 38 (25%) | 28 (19%) | 101 (23%) |
| 2 | 14 (10%) | 15 (10%) | 6 (4%) | 35 (8%) |
| 3 or more | 6 (4%) | 8 (5%) | 7 (5%) | 21 (5%) |
| Inpatient nights | ||||
| None | 89 (64%) | 106 (70%) | 113 (78%) | 308 (71%) |
| 1 | 8 (6%) | 5 (3%) | 3 (2%) | 16 (4%) |
| 2 | 8 (6%) | 5 (3%) | 5 (3%) | 18 (4%) |
| 3 or more | 33 (24%) | 35 (23%) | 23 (16%) | 91 (21%) |
| Any self-reported medication use | ||||
| Yes | 131 (96%) | 144 (97%) | 138 (97%) | 413 (96%) |
Table 33 describes the time taken off work in the month following randomisation. Despite the groups being well matched in respect to age and sex, both at baseline and among responders, the placebo group contained a lower percentage of patients not in paid employment (31%) than the active groups (i.v. 43%; nebulised 45%). Restricting the analysis just to those in paid employment, 47% of responders in the placebo group took time off, compared with 55% in the i.v. magnesium sulphate group and 62% in the nebulised magnesium sulphate group, although this comparison was not statistically significant (comparison of active vs. placebo: χ2 = 3.1, p = 0.08). Likewise, the number of days taken off was not significantly different between groups (Mann–Whitney U-test, p = 0.21). Among patients who had taken time off work, typically this entailed more than 1 week (median 9 days).
| Classification | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Number (%) of responders | 138 (42%) | 151 (38%) | 144 (40%) | 433 (40%) |
| Any time taken off work | ||||
| Yes | 47 (34%) | 45 (31%) | 46 (32%) | 138 (33%) |
| No | 29 (21%) | 37 (26%) | 52 (37%) | 118 (28%) |
| Not in paid employment | 61 (45%) | 63 (43%) | 44 (31%) | 168 (40%) |
| Number of days if yes: | ||||
| ≤ 1 day | 3 (8%) | 0 | 4 (9%) | 7 (6%) |
| 2–3 days | 4 (10%) | 9 (22%) | 7 (15%) | 20 (16%) |
| 4–5 days | 3 (8%) | 5 (12%) | 7 (15%) | 15 (12%) |
| 6–10 days | 6 (15%) | 5 (12%) | 9 (20%) | 20 (16%) |
| 11–15 days | 11 (28%) | 6 (15%) | 6 (13%) | 23 (18%) |
| ≥ 16 days | 12 (31%) | 16 (39%) | 13 (28%) | 41 (33%) |
Table 34 shows the results of the 11 questions in the satisfaction with care questionnaire. In general, satisfaction with care was high across all three treatment groups and across most dimensions of care, with over 70% rating care as very good or excellent on 8 of the 11 dimensions. The dimensions of care relating to personal interest in the patient and their medical problems, the amount of time given by hospital staff and especially advice given about ways to avoid illness and stay healthy were generally rated lower. There were no significant differences in any of the primary contrasts between the treatment groups.
| Classification | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Overall (n = 1084) |
|---|---|---|---|---|
| Number (%) of questionnaires returned | 138 (42%) | 151 (38%) | 145 (41%) | 434 (40%) |
| The urgency with which you were assessed | ||||
| Poor | 1 (1%) | 4 (3%) | 1 (1%) | 6 (1%) |
| Fair | 8 (6%) | 5 (3%) | 10 (7%) | 23 (5%) |
| Good | 22 (16%) | 19 (13%) | 25 (17%) | 66 (15%) |
| Very good | 42 (30%) | 52 (34%) | 42 (29%) | 136 (31%) |
| Excellent | 65 (47%) | 71 (47%) | 67 (46%) | 203 (47%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.592 | |||
| i.v. vs. nebuliser | 0.781 | |||
| The thoroughness of your assessment | ||||
| Poor | 1 (1%) | 1 (1%) | 0 | 2 (< 1%) |
| Fair | 4 (3%) | 7 (5%) | 9 (6%) | 20 (5%) |
| Good | 24 (17%) | 20 (13%) | 25 (17%) | 69 (16%) |
| Very good | 52 (38%) | 51 (34%) | 51 (35%) | 154 (36%) |
| Excellent | 57 (41%) | 71 (47%) | 60 (41%) | 188 (43%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.400 | |||
| i.v. vs. nebuliser | 0.368 | |||
| Explanations given to you about medical procedures and tests | ||||
| Poor | 2 (1%) | 5 (3%) | 4 (3%) | 11 (3%) |
| Fair | 6 (4%) | 8 (5%) | 9 (6%) | 23 (5%) |
| Good | 23 (17%) | 15 (10%) | 23 (16%) | 61 (14%) |
| Very good | 58 (42%) | 55 (36%) | 46 (32%) | 159 (37%) |
| Excellent | 49 (36%) | 68 (45%) | 63 (43%) | 180 (41%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.960 | |||
| i.v. vs. nebuliser | 0.157 | |||
| Attention given to what you have to say | ||||
| Poor | 1 (1%) | 6 (4%) | 3 (2%) | 10 (2%) |
| Fair | 12 (9%) | 9 (6%) | 15 (10%) | 36 (8%) |
| Good | 26 (19%) | 26 (17%) | 27 (19%) | 79 (18%) |
| Very good | 45 (33%) | 58 (38%) | 53 (37%) | 156 (36%) |
| Excellent | 53 (39%) | 52 (34%) | 47 (32%) | 152 (35%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.342 | |||
| i.v. vs. nebuliser | 0.666 | |||
| Advice you got about ways to avoid illness and stay healthy | ||||
| Poor | 13 (10%) | 14 (9%) | 9 (6%) | 36 (8%) |
| Fair | 28 (21%) | 23 (15%) | 19 (13%) | 70 (16%) |
| Good | 38 (28%) | 36 (24%) | 39 (27%) | 113 (26%) |
| Very good | 30 (22%) | 38 (25%) | 39 (27%) | 107 (25%) |
| Excellent | 26 (19%) | 39 (26%) | 36 (25%) | 101 (24%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.143 | |||
| i.v. vs. nebuliser | 0.112 | |||
| Friendliness and courtesy shown to you by hospital staff | ||||
| Poor | 2 (1%) | 1 (1%) | 0 | 3 (1%) |
| Fair | 7 (5%) | 7 (5%) | 4 (3%) | 18 (4%) |
| Good | 21 (15%) | 25 (17%) | 26 (18%) | 72 (17%) |
| Very good | 41 (30%) | 43 (28%) | 38 (26%) | 122 (28%) |
| Excellent | 66 (48%) | 75 (50%) | 77 (53%) | 218 (50%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.407 | |||
| i.v. vs. nebuliser | 0.812 | |||
| Personal interest in you and your medical problems | ||||
| Poor | 1 (1%) | 4 (3%) | 4 (3%) | 9 (2%) |
| Fair | 12 (9%) | 19 (13%) | 11 (8%) | 42 (10%) |
| Good | 35 (25%) | 28 (19%) | 32 (22%) | 95 (22%) |
| Very good | 43 (31%) | 49 (33%) | 51 (35%) | 143 (33%) |
| Excellent | 47 (34%) | 50 (33%) | 47 (32%) | 144 (33%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.887 | |||
| i.v. vs. nebuliser | 0.733 | |||
| Respect shown to you and attention to your privacy | ||||
| Poor | 3 (2%) | 2 (1%) | 0 | 5 (1%) |
| Fair | 10 (7%) | 8 (5%) | 8 (6%) | 26 (6%) |
| Good | 28 (20%) | 30 (20%) | 32 (22%) | 90 (21%) |
| Very good | 45 (33%) | 47 (31%) | 41 (28%) | 133 (31%) |
| Excellent | 52 (38%) | 64 (42%) | 64 (44%) | 180 (41%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.484 | |||
| i.v. vs. nebuliser | 0.363 | |||
| Reassurance and support offered to you by hospital staff | ||||
| Poor | 5 (4%) | 7 (5%) | 4 (3%) | 16 (4%) |
| Fair | 14 (10%) | 13 (9%) | 13 (9%) | 40 (9%) |
| Good | 28 (20%) | 23 (15%) | 25 (17%) | 76 (18%) |
| Very good | 39 (28%) | 57 (38%) | 45 (31%) | 141 (33%) |
| Excellent | 51 (37%) | 51 (34%) | 58 (40%) | 160 (37%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.373 | |||
| i.v. vs. nebuliser | 0.922 | |||
| Amount of time the hospital staff gave you | ||||
| Poor | 4 (3%) | 4 (3%) | 3 (2%) | 11 (3%) |
| Fair | 20 (14%) | 22 (15%) | 15 (10%) | 57 (13%) |
| Good | 30 (22%) | 35 (23%) | 37 (26%) | 102 (24%) |
| Very good | 42 (30%) | 47 (31%) | 46 (32%) | 135 (31%) |
| Excellent | 42 (30%) | 43 (28%) | 44 (30%) | 129 (30%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.539 | |||
| i.v. vs. nebuliser | 0.790 | |||
| Overall, how satisfied are you with the service you received | ||||
| Poor | 4 (3%) | 1 (1%) | 1 (1%) | 6 (1%) |
| Fair | 8 (6%) | 13 (9%) | 6 (4%) | 27 (6%) |
| Good | 23 (17%) | 19 (13%) | 25 (17%) | 67 (16%) |
| Very good | 45 (33%) | 54 (36%) | 51 (35%) | 150 (35%) |
| Excellent | 57 (42%) | 63 (42%) | 62 (43%) | 182 (42%) |
| Comparisons | p-value | |||
| Active vs. placebo | 0.645 | |||
| i.v. vs. nebuliser | 0.741 | |||
Table 35 shows the results of questions asking the participants to guess whether they received active treatment or placebo. Most patients in the active treatment arms (61% of patients in the nebulised group and 60% of patients in the i.v. group) believed they had received active treatment, compared with 45% of patients in the placebo group. When asked how they thought they had received magnesium sulphate, 39% of patients in the nebuliser group and 26% of patients in the i.v. group correctly identified the route of administration. These findings suggest that participants had some ability to correctly identify whether or not they had received active treatment, but their guessing was only moderately better than that expected due to chance.
| Response | Nebulised magnesium sulphate (n = 332) | Intravenous magnesium sulphate (n = 394) | Placebo (n = 358) | Total (n = 1084) |
|---|---|---|---|---|
| Number of responses | 140 | 156 | 148 | 444 |
| Believed they had received magnesium sulphate | 85 (61%)a | 94 (60%)a | 66 (45%)a | 245 (55%) |
| How do you think magnesium sulphate was given? | ||||
| Nebuliser | 55 (39%)a | 35 (22%) | 28 (19%) | 118 (27%) |
| i.v. | 21 (15%) | 40 (26%)a | 28 (19%) | 89 (20%) |
| Both | 7 (5%) | 13 (8%) | 7 (5%) | 27 (6%) |
| Do not know/not answered | 2 (1%) | 6 (4%) | 3 (2%) | 11 (2%) |
| NA | 55 (39%) | 62 (40%) | 82 (55%) | 199 (45%) |
Subgroup analysis
The protocol prespecified three key factors for subgroup analyses: age, baseline PEFR and previous receipt of salbutamol. Table 36 shows the impact of these factors on the primary outcomes. In each case, the factor (age, PEFR or previous receipt of salbutamol) was added to a model that also contained treatment group and site. For change in VAS, the VAS at baseline was also included. Older age and lower PEFR at baseline were both highly associated with an increase in admissions and also a greater reduction from baseline in VAS. The latter is presumably a reflection of the greater severity (i.e. higher VAS at baseline) among those with lower percentage PEFR. The use of salbutamol in the ED or ambulance prior to randomisation was compromised by relatively small numbers without prior usage (n = 59), but those with previous use were more likely to be admitted than those who had not (77% vs. 56%; p = 0.001).
| Factor | Hospital admission | Change in VAS (mm) | ||
|---|---|---|---|---|
| OR (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| Age (per 10 year increase) | 1.27 (1.13 to 1.43) | < 0.001 | 2.3 (1.1 to 3.4) | < 0.001 |
| PEFR at baseline (per 10% of predicted increase) | 0.81 (0.75 to 0.88) | < 0.001 | –1.4 (–2.2 to –0.6) | 0.001 |
| Previous use of salbutamol (yes vs. no) | 2.83 (1.57 to 5.12) | 0.001 | –2.3 (–9.3 to 4.7) | 0.520 |
The results are displayed graphically in Figures 15 and 16. As the primary analysis showed no difference between the nebulised and placebo arms, the graphical displays focus on the comparison of i.v. and placebo. Figure 15 shows the ORs for admission to hospital for each subgroup and overall and, likewise, Figure 16 shows the mean differences in VAS. The results appear consistent across all subgroups. The subgroup of patients who had not previously received salbutamol appeared to have worse outcomes with active treatment, but this analysis is limited by small numbers and wide CIs.
FIGURE 15.
Hospital admission, i.v. vs. placebo, by subgroup.
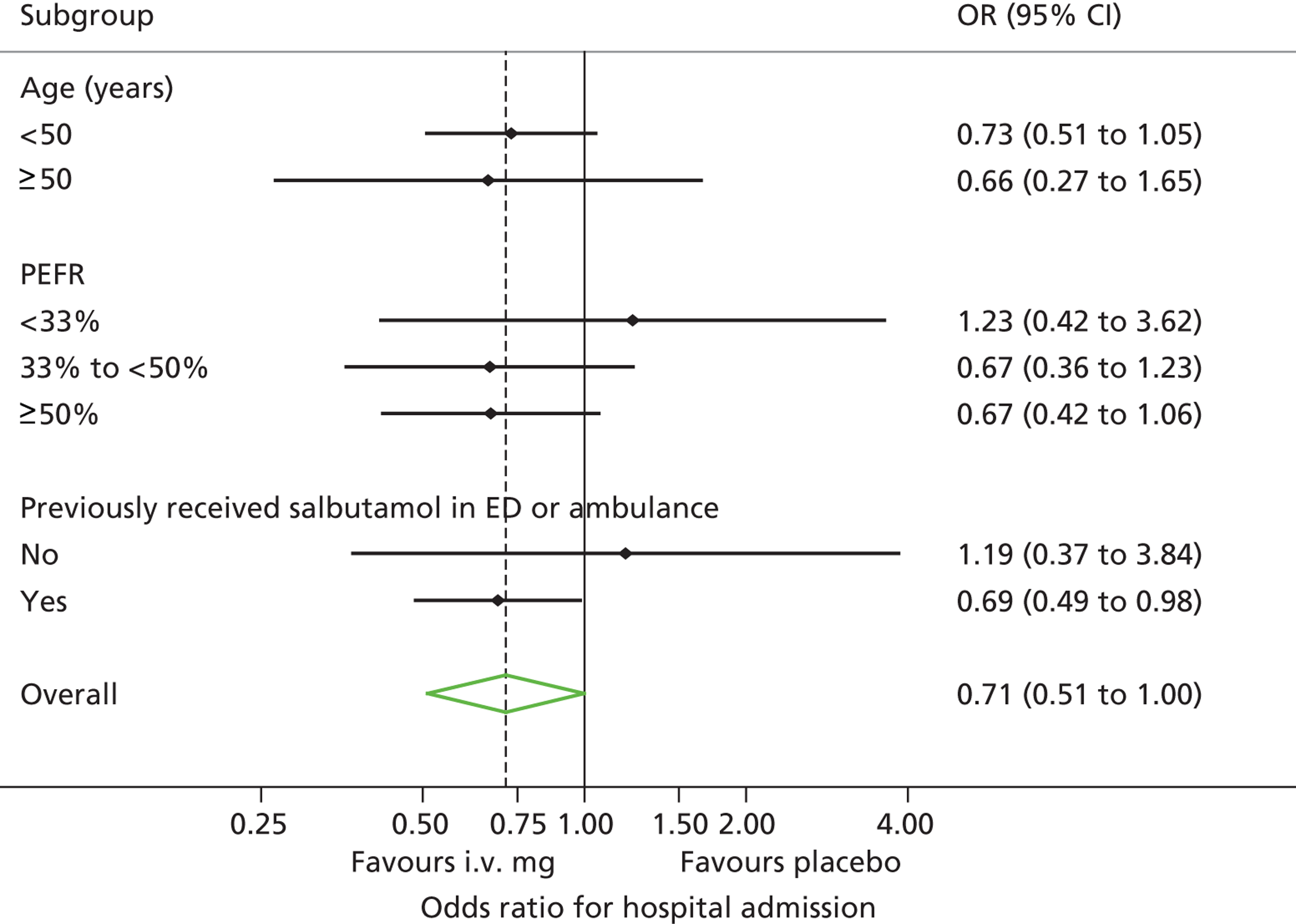
FIGURE 16.
Change in VAS by subgroup.
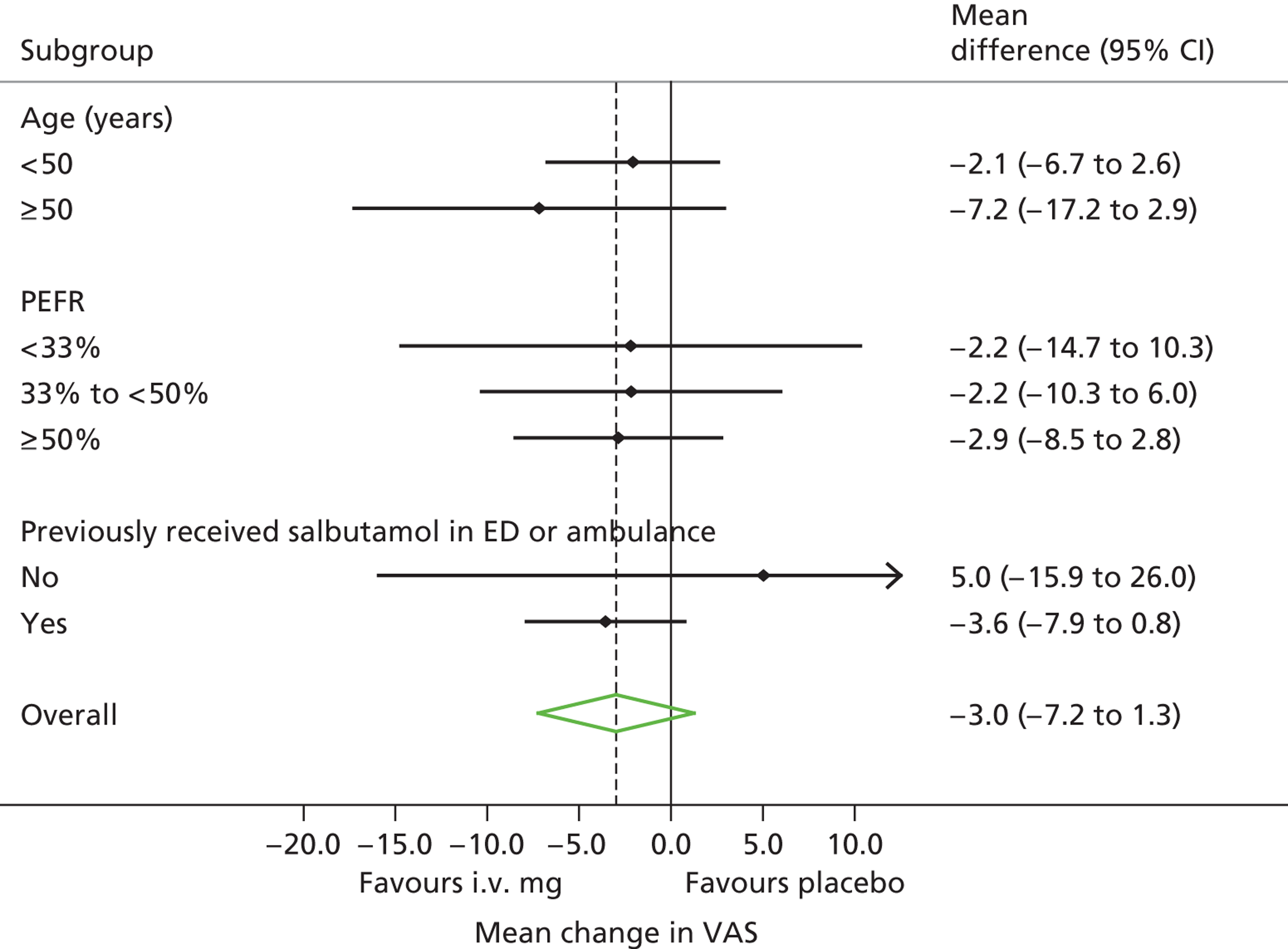
Economic analysis
Table 37 presents the results of the cost-effectiveness analysis of the QALY and cost data excluding productivity costs. The placebo-treated population have the highest population mean QALY and there was no apparent difference between the population mean QALYs associated with nebulised magnesium sulphate and i.v. magnesium sulphate; there is a 67% chance that placebo is associated with the highest population mean QALY. Nebulised magnesium sulphate has the highest population mean cost and placebo has the lowest population mean cost; there is a 95% chance that placebo is associated with the lowest population mean cost.
| Treatment | QALYs, mean (SD) | Incremental QALYs (95% CrI) | Cost, (£) mean (SD) | Incremental cost, £ (95% CrI) | ICER |
|---|---|---|---|---|---|
| Placebo | 0.063 (0.0030) | – | 1610 (89.7) | – | |
| Nebulised magnesium | 0.060 (0.0033) | –0.011 to 0.006 | 1974 (115.3) | 77 to 651 | Dominated |
| i.v. magnesium | 0.060 (0.0028) | –0.011 to 0.005 | 1870 (110.8) | –20 to 540 | Dominated |
Figure 17 presents the cost-effectiveness frontiers for willingness to pay in the range £0 to £100,000. There is a 97% and 96% chance that placebo has the highest net benefit at thresholds of £20,000 and £30,000 respectively.
FIGURE 17.
Cost-effectiveness frontiers for willingness to pay, main QALY analysis.
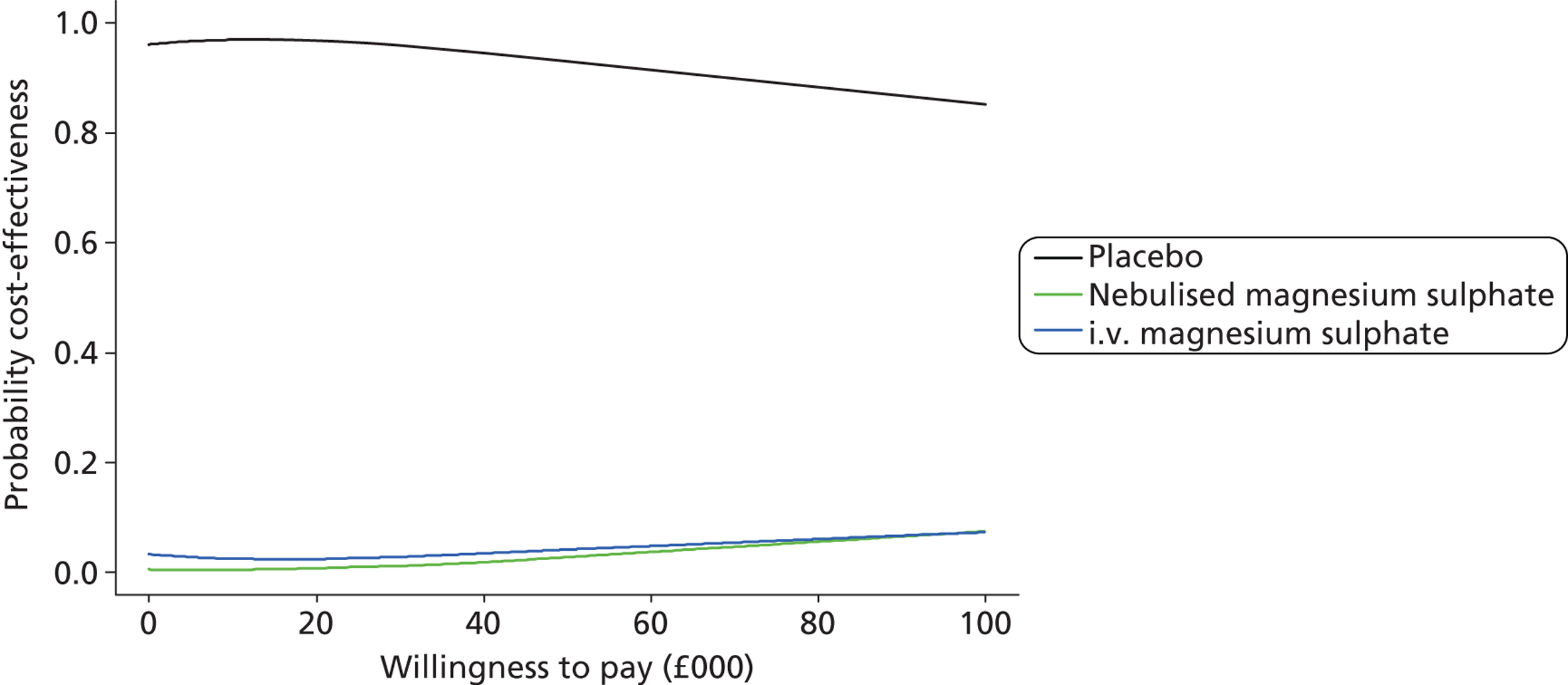
Table 38 presents the results of the cost-effectiveness analysis of the QALY and cost data, including productivity costs. Nebulised magnesium sulphate has the highest population mean cost and placebo has the lowest population mean cost; there is a 90% chance that placebo is associated with the lowest population mean cost.
| Treatment | QALYs, mean (SD) | Incremental QALYs (95% CrI) | Cost (£), mean (SD) | Incremental cost (95% CrI) | ICER |
|---|---|---|---|---|---|
| Placebo | 0.063 (0.0030) | – | 2007 (107.4) | – | |
| Nebulised magnesium sulphate | 0.060 (0.0033) | –0.011 to 0.006 | 2401 (120.9) | 78 to 712 | Dominated |
| i.v. magnesium sulphate | 0.060 (0.0028) | –0.011 to 0.005 | 2219 (120.5) | –104 to 530 | Dominated |
Figure 18 presents the cost-effectiveness frontiers for willingness-to-pay in the range £0 to £100,000. There is a 93% chance that placebo has the highest net benefit at thresholds of both £20,000 and £30,000.
FIGURE 18.
Cost-effectiveness frontiers for willingness to pay, QALY analysis including productivity costs.
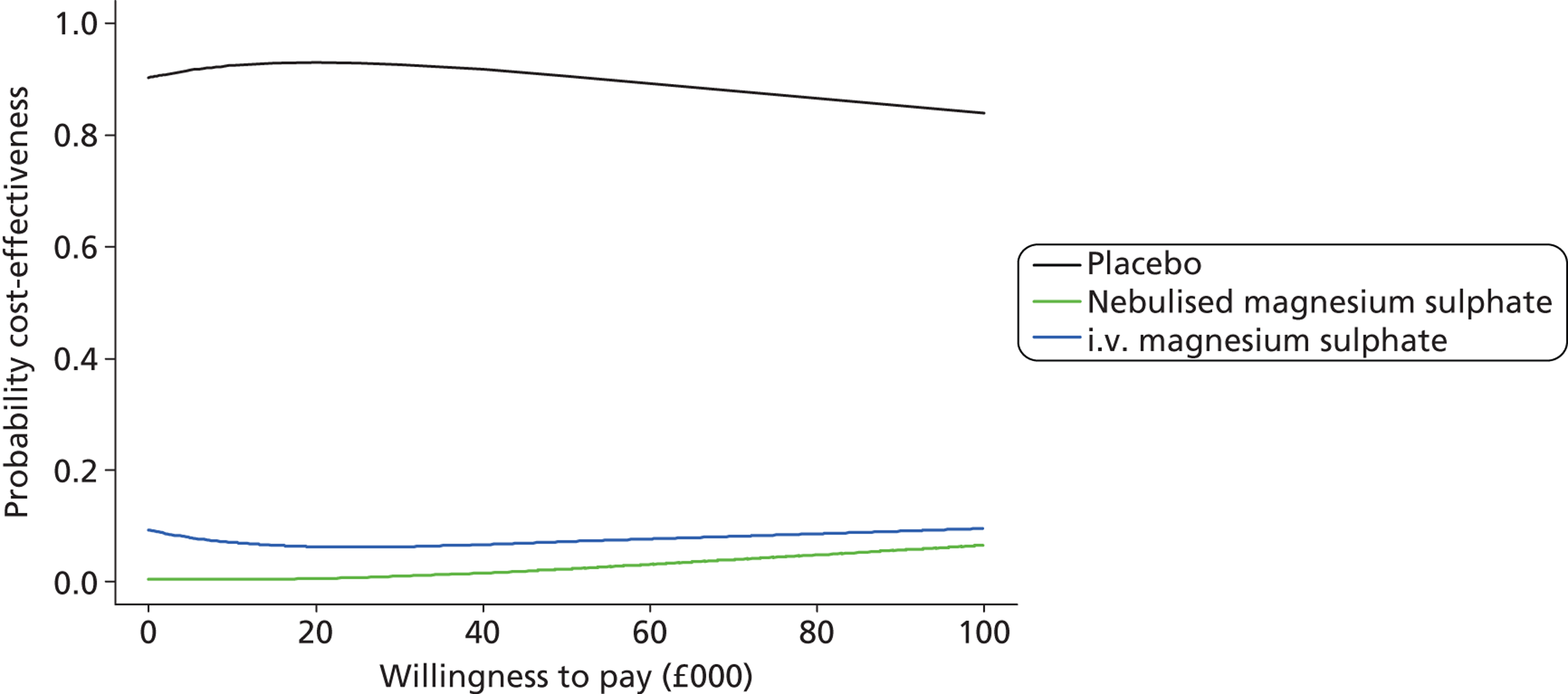
Table 39 presents the results of the cost-effectiveness analysis of the 2-hour breathlessness (change from baseline) and cost data excluding productivity costs. Intravenous magnesium sulphate has the greatest population mean change from baseline in 2-hour breathlessness, whereas nebulised magnesium sulphate has the smallest population mean change from baseline in 2-hour breathlessness; there is a 97% chance that i.v. magnesium sulphate is associated with the greatest population mean change from baseline 2-hour breathlessness score.
| Treatment | Breathlessness (VAS), mean (SD) | Incremental breathlessness (95% CrI) | Cost (£), mean (SD) | Incremental Cost (95% CrI) | ICER |
|---|---|---|---|---|---|
| Placebo | 29.5 (1.57) | – | 1610 (89.8) | – | |
| Nebulised magnesium sulphate | 27.8 (1.57) | –6.0 to 2.7 | 1974 (115.3) | 77 to 651 | Dominated |
| i.v. magnesium sulphate | 33.7 (1.43) | 0 to 8.4 | 1870 (110.9) | –20 to 540 | £61.7/mm |
Figure 19 presents the cost-effectiveness frontiers for willingness to pay in the range £0 to £1000. The incremental cost-effectiveness ratio (ICER) for i.v. magnesium sulphate relative to placebo was £61.70/mm.
FIGURE 19.
Cost-effectiveness frontiers for willingness to pay, main breathlessness analysis.
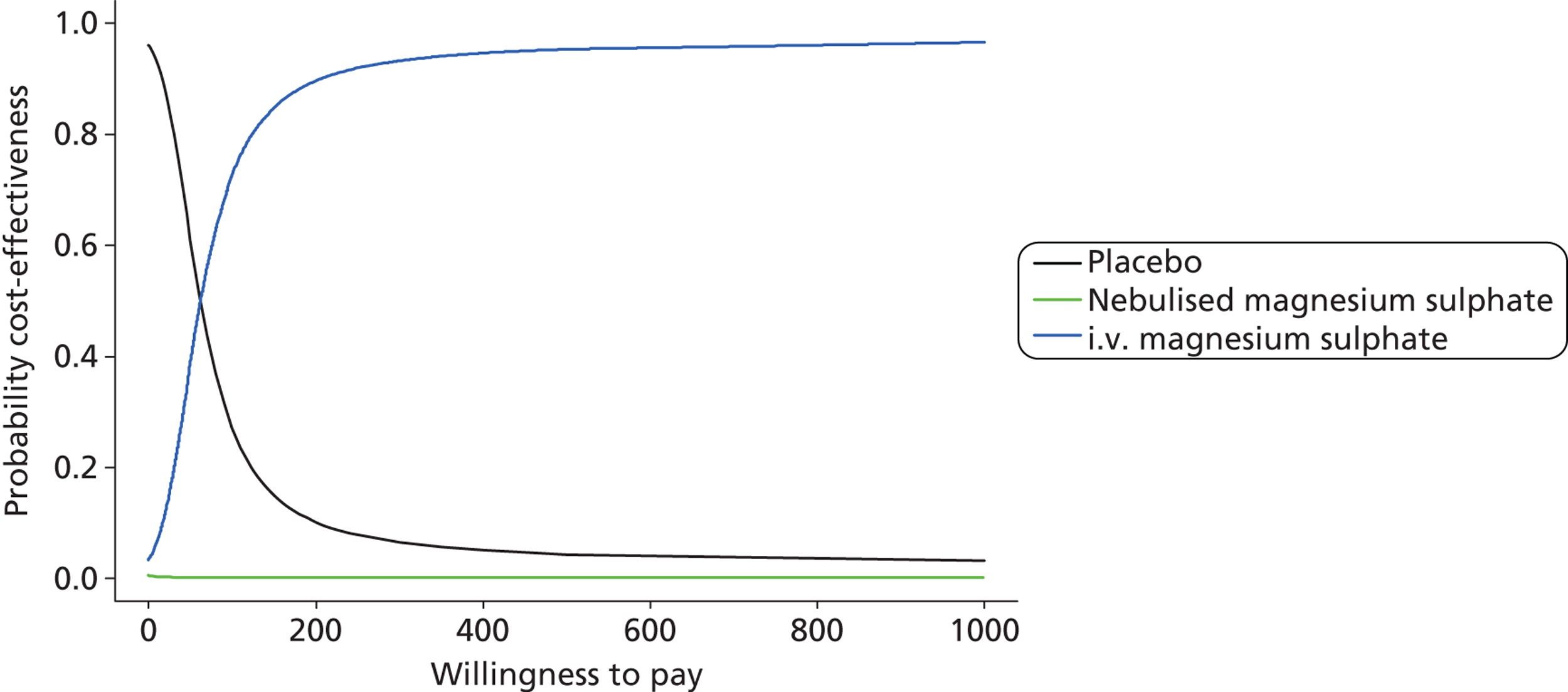
Table 40 presents the results of the cost-effectiveness analysis of the 2-hour breathlessness (change from baseline) and cost data including productivity costs. Figure 20 presents the cost-effectiveness frontiers for willingness to pay in the range of £0 to £1000. The ICER for i.v. magnesium sulphate relative to placebo was £50.48/mm.
| Treatment | Breathlessness (VAS), mean (SD) | Incremental breathlessness (95% CrI) | Mean cost, £ (SD) | Incremental cost, £ (95% CrI) | ICER |
|---|---|---|---|---|---|
| Placebo | 29.6 (1.57) | – | 2007 (107.4) | – | |
| Nebulised magnesium sulphate | 28.0 (1.57) | –6.0 to 2.7 | 2401 (120.9) | 78 to 712 | Dominated |
| i.v. magnesium sulphate | 33.7 (1.43) | 0.0 to 8.4 | 2219 (120.5) | –104 to 530 | £50.48/mm |
FIGURE 20.
Cost-effectiveness frontiers for willingness to pay, breathlessness analysis including productivity costs.
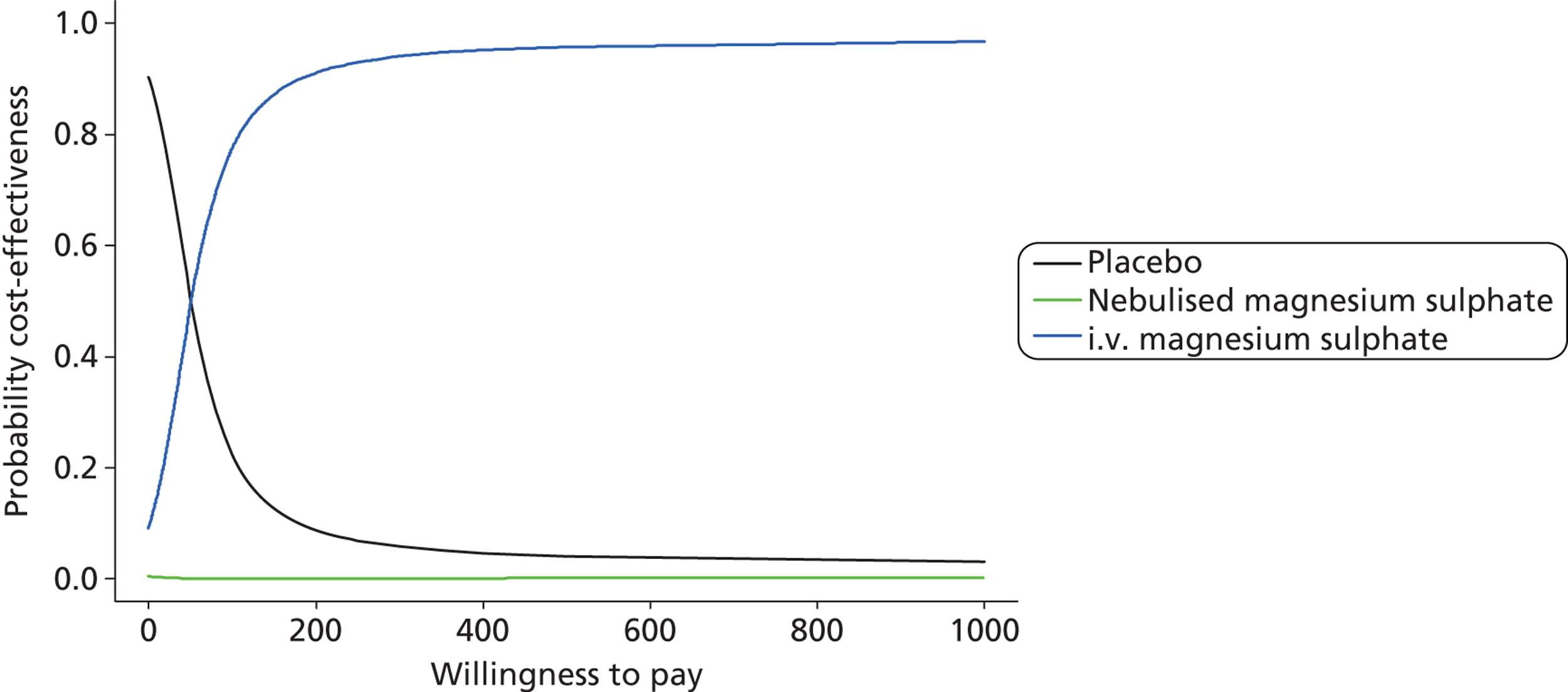
Additional analysis: predictors of unsuccessful treatment
Overall, 81 (7%) participants required critical care and 157 (14%) required emergency medical treatment (including critical care) within 7 days of initial attendance. Table 41 shows the criteria behind these classifications.
| Criterion | Number (%) of patients |
|---|---|
| Critical care | 81 (7.5) |
| Emergency medical treatment | 157 (14.5) |
| Reason | |
| Stay on HDU or ICU | 77 (7.1) |
| Arrhythmia | 2 (0.2) |
| Emergency intubation | 8 (0.7) |
| Non-invasive ventilation | 7 (0.6) |
| Other event | 4 (0.4) |
| Reattendance at ED/unscheduled inpatient reviewa | 111 (10.2) |
Tables 42 and 43 show the results of univariate analysis. Table 42 shows the association between baseline characteristics or categorised baseline physiological variables and each outcome. Table 43 shows the p-values for the associations between fractional polynomials of continuous variables (as used in the model) and for each outcome. There was some evidence that previous asthma admission, previous ITU admission, other serious illness and percentage predicted PEFR, heart rate, respiratory rate, oxygen flow rate and VAS breathlessness at baseline or after treatment were associated with unsuccessful treatment. Although percentage predicted PEFR at presentation was associated with unsuccessful treatment it did not provide very useful prognostic value (AUROC < 0.6).
| Factor | Requiring critical care | Requiring emergency medical treatment |
|---|---|---|
| Overall | 81 (7%) | 157 (14%) |
| Age group (years) | (p = 0.921) | (p = 0.722) |
| 16–24 | 21 (8%) | 43 (15%) |
| 25–34 | 20 (7%) | 42 (16%) |
| 35–44 | 16 (7%) | 27 (12%) |
| 45–54 | 13 (7%) | 26 (14%) |
| 55–64 | 7 (9%) | 13 (17%) |
| 65+ | 4 (11%) | 6 (17%) |
| Sex | (p = 0.798) | (p = 0.396) |
| Female | 56 (7%) | 115 (15%) |
| Male | 25 (8%) | 42 (13%) |
| Ethnic category | (p = 0.121) | (p = 0.180)a |
| White | 77 (8%) | 140 (14%) |
| Mixed | 2 (25%) | 4 (50%) |
| Asian or Asian British | 0 (0%) | 6 (16%) |
| Black or black British | 0 (0%) | 1 (9%) |
| Other | 0 (0%) | 0 (0%) |
| Not stated | 2 (4%) | 6 (12%) |
| Smoker | (p = 0.639) | (p = 0.822) |
| Never | 31 (7%) | 63 (14%) |
| Current | 28 (8%) | 54 (15%) |
| Previous | 22 (9%) | 39 (16%) |
| Missing | 0 (0%) | 1 (4%) |
| Previous asthma admission | (p = 0.061) | (p = 0.034) |
| No | 21 (5%) | 44 (11%) |
| Yes | 60 (9%) | 113 (16%) |
| Previous ITU asthma admission | (p = 0.002) | (p = 0.070) |
| No | 60 (6%) | 127 (14%) |
| Yes | 21 (13%) | 30 (19%) |
| Previous serious lung disease | (p = 0.348) | (p = 0.719) |
| No | 75 (8%) | 142 (15%) |
| Yes | 5 (5%) | 13 (13%) |
| Missing | 1 (7%) | 2 (14%) |
| Other serious illness | (p = 0.002) | (p = 0.001) |
| No | 55 (6%) | 111 (13%) |
| Yes | 26 (13%) | 44 (22%) |
| Missing | 0 (0%) | 2 (17%) |
| % of predicted PEFR at baseline | (p = 0.153) | (p = 0.042) |
| ≤ 35 | 22 (10%) | 42 (20%) |
| > 35–45 | 13 (6%) | 27 (13%) |
| > 45–54 | 13 (7%) | 24 (12%) |
| > 54–67 | 11 (6%) | 26 (14%) |
| > 67 | 9 (4%) | 20 (10%) |
| Missing | 13 (18%) | 18 (24%) |
| Pulse at baseline (beats per minute) | (p < 0.001) | (p < 0.001) |
| ≤ 95 | 6 (3%) | 26 (11%) |
| 95–107 | 15 (7%) | 22 (11%) |
| 107–115 | 18 (9%) | 31 (16%) |
| 115–125 | 11 (5%) | 23 (11%) |
| > 125 | 31 (14%) | 55 (25%) |
| Missing | 0 (0%) | 0 (0%) |
| Respiratory rate at baseline (breaths per minute) | (p < 0.001) | (p < 0.001) |
| ≤ 20 | 10 (4%) | 31 (11%) |
| > 20–23 | 12 (8%) | 21 (14%) |
| > 23–26 | 20 (8%) | 29 (11%) |
| > 26–30 | 11 (5%) | 27 (13%) |
| > 30 | 28 (15%) | 49 (26%) |
| Missing | 0 (0%) | 0 (0%) |
| Systolic BP at baseline (mmHg) | (p = 0.220) | (p = 0.634) |
| ≤ 114 | 14 (7%) | 31 (15%) |
| > 114–125 | 12 (5%) | 27 (11%) |
| > 125–135 | 16 (7%) | 35 (16%) |
| > 135–147 | 16 (8%) | 30 (15%) |
| > 147 | 23 (11%) | 34 (16%) |
| Missing | 0 (0%) | 0 (0%) |
| Diastolic BP at baseline (mmHg) | (p = 0.238) | (p = 0.192) |
| ≤ 63 | 17 (7%) | 30 (13%) |
| > 63–70 | 13 (7%) | 32 (16%) |
| > 70–78 | 11 (5%) | 23 (10%) |
| > 78–86 | 17 (8%) | 34 (16%) |
| > 86 | 23 (11%) | 38 (18%) |
| Missing | 0 (0%) | 0 (0%) |
| Oxygen flow at baseline (l/minute) | (p = 0.027) | (p = 0.054) |
| 0 | 11 (4%) | 34 (13%) |
| 1–5 | 15 (9%) | 26 (15%) |
| 6 | 38 (10%) | 62 (16%) |
| 7–10 | 10 (6%) | 21 (12%) |
| ≥ 10 | 7 (14%) | 14 (29%) |
| Missing | 0 (0%) | 0 (0%) |
| Variable | Requiring critical care | Requiring emergency medical treatment | ||
|---|---|---|---|---|
| Baseline | Post treatment | Baseline | Post treatment | |
| Heart rate | < 0.001 | < 0.001 | < 0.001 | < 0.001 |
| Respiratory rate | < 0.001 | < 0.001 | < 0.001 | < 0.001 |
| Systolic BP | 0.074 | 0.211 | 0.390 | 0.604 |
| Diastolic BP | 0.164 | 0.420 | 0.086 | 0.819 |
| Oxygen saturation (on oxygen) | 0.428 | 0.001 | 0.333 | 0.001 |
| Oxygen saturation (on air) | 0.549 | 0.945 | 0.283 | 0.272 |
| Oxygen flow rate | 0.009 | < 0.001 | 0.041 | < 0.001 |
| VAS breathlessness | 0.028 | < 0.001 | 0.003 | < 0.001 |
| % predicted PEFR | 0.008 | < 0.001 | 0.012 | < 0.001 |
Tables 44 and 45 show the results of multivariate analysis for prediction of need for critical care and need for any emergency medical treatment respectively. The effect of PEFR on hospitalisation remained similar in the three models and with both outcomes, but the addition of physiological features and the presence of serious comorbidity improved prognostic ability. The presence of other serious illness, raised heart rate and raised oxygen flow requirement were all associated with increased need for critical care. Change in physiology measures over the 2-hour observation period increased the predictive ability of the model further, with a decrease in heart rate or increase in PEFR predicting reduced need for critical care. The derived model was robust and produced comparable fits to the subgroups (temporal, season and teaching/non-teaching hospital). Nonetheless, the resultant model had limited predictive ability (model incorporating change in physiology: AUROC = 0.77), suggesting that there is limited means to predict events for individual patients.
| Factor | Model 1 (pretreatment PEFR) | Model 2 (pretreatment characteristics and physiology) | Model 3 (pretreatment characteristics and physiology, and 2-hour changes in physiology) | |||
|---|---|---|---|---|---|---|
| OR (95% CI) | p-value | OR (95% CI) | p-value | OR (95% CI) | p-value | |
| Number of patients with data | 1010 | 999 | 956 | |||
| PEFR (% of predicted)a | 0.82 (0.72 to 0.95) | 0.007 | 0.82 (0.71 to 0.95) | 0.008 | 0.83 (0.71 to 0.97) | 0.017 |
| Other serious illness | 2.15 (1.24 to 3.73) | 0.006 | 2.04 (1.12 to 3.70) | 0.019 | ||
| Heart rate (beats per minute)a | 1.27 (1.12 to 1.45) | < 0.001 | 1.45 (1.23 to 1.70) | < 0.001 | ||
| Oxygen flow (l/minute)a | 2.08 (1.08 to 4.00) | 0.028 | 2.11 (0.99 to 4.49) | 0.054 | ||
| Change in PEFR (% of predicted)a | 0.77 (0.63 to 0.95) | 0.015 | ||||
| Change in pulse (beats per minute)a | 1.70 (1.35 to 2.12) | < 0.001 | ||||
| Model AUROC | 0.60 | 0.70 | 0.77 | |||
| Factor | Model 1 (pretreatment PEFR) | Model 2 (pretreatment characteristics and physiology) | Model 3 (pretreatment characteristics and physiology, and 2-hour changes in physiology) | |||
|---|---|---|---|---|---|---|
| OR (95% CI) | p-value | OR (95% CI) | p-value | OR (95% CI) | p-value | |
| Number of patients with data | 1010 | 997 | 954 | |||
| PEFR (% of predicted)a | 0.87 (0.79 to 0.96) | 0.008 | 0.88 (0.79 to 0.97) | 0.010 | 0.87 (0.78 to 0.97) | 0.010 |
| Other serious illness | 1.77 (1.16 to 2.71) | 0.009 | 1.67 (1.07 to 2.61) | 0.023 | ||
| Heart rate (beats per minute)a | < 0.001 | < 0.001 | ||||
| Linear term | 0.48 (0.25 to 0.95) | 0.57 (0.28 to 1.17) | ||||
| Quadratic term | 1.04 (1.01 to 1.07) | 1.03 (1.00 to 1.07) | ||||
| Respiratory rate (breaths per minute)a | 1.34 (1.03 to 1.76) | 0.032 | 1.42 (1.06 to 1.88) | 0.017 | ||
| Change in PEFR (% of predicted)a | 0.82 (0.71 to 0.93) | 0.003 | ||||
| Change in pulse (beats per minute)a | 1.30 (1.12 to 1.52) | 0.001 | ||||
| Model AUROC | 0.57 | 0.64 | 0.69 | |||
A similar theme emerged from the analysis of prediction of need for any medical treatment, although more terms were identified. There was a quadratic relationship between pretreatment heart rate and needing emergency medical treatment, with the outcome being least likely when the heart rate was in the range 90–100 beats per minute. In addition, pretreatment respiratory rate was significantly associated with the need for emergency medical treatment (higher rate meaning a greater probability of the outcome). The derived model incorporating 2-hour change in physiology was robust and comparable in the subgroups, but again predictive ability remained low (AUROC = 0.69).
Chapter 4 Discussion
Main findings
Analysis of this three-arm trial involved two preplanned contrasts: (1) active treatment (i.v. and nebulised magnesium sulphate combined) compared with placebo and (2) i.v. compared with nebulised magnesium sulphate. This was based on a systematic review and meta-analysis of previous trials of magnesium sulphate in adults,11 which suggested that both i.v. and nebulised magnesium sulphate had a potential effect on respiratory function and hospital admission but found no direct comparisons between i.v. and nebulised magnesium sulphate. We found no evidence that active treatment was more effective than placebo, but we did find some evidence that active treatment was associated with a slightly higher risk of side effects [15.6% vs. 10.1%, OR 1.68 (95% CI 1.11 to 2.52; p = 0.014)].
Our choice of preplanned comparisons could be criticised if it was thought that one mode of administration was superior to the other, especially if the inferior mode of administration was associated with worse outcomes than placebo. We therefore reported comparisons between i.v. treatment and placebo, and between nebulised treatment and placebo, although it should be recognised that these comparisons were not those originally specified. We found no evidence that nebulised magnesium sulphate was more effective than placebo. In fact, any non-significant trends in the outcomes tended to favour placebo. The only significant effect of nebulised magnesium sulphate compared with placebo was a small increase in the risk of side effects [15.7% vs. 10.1%, OR 1.67 (95% CI 1.05 to 2.66; p = 0.031)]. Intravenous magnesium sulphate was associated with a lower rate of hospital admission than placebo, but the difference was not significant [OR 0.73 (95% CI 0.51 to 1.04, p = 0.083)], and there was no evidence of an effect on VAS breathlessness [mean difference 2.6 mm (95% CI –1.6 to 6.8 mm; p = 0.231)] compared with placebo. Although i.v. magnesium sulphate appeared superior to nebulised magnesium sulphate [mean difference = 5.1 mm, (95% CI 0.8 to 9.4 mm)], the magnitude of the difference in clinical terms is small when considered in the context of a minimum clinically significant difference for VAS breathlessness of 22 mm. 48 There was also no evidence of any clinically worthwhile effect from i.v. magnesium sulphate compared with placebo on secondary outcome measures, including change in PEFR [2.4 l/minute (95% CI –8.8 to 13.6 l/minute; p = 0.680)]. Overall, therefore, we were unable to demonstrate a clinically worthwhile benefit from i.v. magnesium sulphate.
Given the failure to demonstrate clinical effectiveness of magnesium sulphate it is not surprising that the economic analysis showed a corresponding lack of cost-effectiveness, with high probabilities that placebo is associated with the highest net benefit at conventionally used thresholds for willingness to pay on the cost per QALY analysis. The analysis of incremental cost per unit change in VAS breathlessness is more difficult to interpret as it showed that i.v. magnesium sulphate could provide a non-significant improvement in outcome at a higher cost, compared with placebo. The ICER was around £50–£60/mm improvement in VAS, but both cost and effectiveness estimates were surrounded by considerable uncertainty.
Our additional analysis to explore the predictive value of baseline characteristics and response to treatment showed that the percentage predicted PEFR at baseline predicted the need for critical care or emergency medical treatment, but that the predictive value was limited (AUROC 0.60 and 0.57 respectively). The presence of other serious illnesses, heart rate, the oxygen flow rate required, change in the percentage predicted PEFR and change in heart rate all predicted the need for critical care, and a predictive model based on these variables had a reasonable predictive value (AUROC 0.77). The presence of other serious illnesses, increased heart rate, increased respiratory rate, change in the percentage predicted PEFR and change in heart rate all predicted the need for emergency medical treatment within the next week (including critical care), but a predictive model based on these variables had only a limited predictive value (AUROC 0.69).
Current BTS/SIGN guidance2 uses PEFR and a number of other parameters to guide hospital admission decisions, but does not recommend the use of any decision rule or predictive model. Our findings confirm that PEFR is a useful, albeit limited, predictor of adverse outcome. Taking the heart rate, other serious illnesses and the required oxygen flow rate into account could improve prediction, but the predictive model based on these values was unlikely to be adequate for guiding practice. This was a secondary analysis, however, and the sample size was determined by the trial and recruitment constraints. A larger number of outcomes would be required to analyse the predictor variables with sufficient statistical power to ensure an optimal model.
Comparison with previous trials
The most recent meta-analysis of magnesium sulphate for acute asthma11 showed some heterogeneity among existing trials, but concluded that overall there was uncertain evidence suggesting that both i.v. and nebulised magnesium sulphate could have a clinically worthwhile effect on respiratory function and admission to hospital. It is not unusual for the results of meta-analyses of small trials to be negated or even overturned in large scale, robust trials and there are a number of potential reasons why the findings of the 3Mg trial and the meta-analysis are inconsistent. Meta-analyses may be subject to publication bias if positive trials are preferentially submitted and accepted for publication, and some previous trials may have been limited by inadequate allocation concealment or blinding that inflated estimates of treatment effects. All three arms of the 3Mg trial received treatment with nebulised beta-agonists, ipratropium bromide and corticosteroids in accordance with BTS/SIGN guidance,2 which may have limited the potential for magnesium sulphate to provide additional bronchodilatation, whereas it was not always clear that all patients received optimal standard treatment in previous trials. It is worth noting that patients in the placebo arm showed marked improvements in PEFR and VAS breathlessness, and few required respiratory support, indicating a good response to standard treatment alone.
One potential explanation that can probably be discounted is that the trial treatment was inadequate in the 3Mg trial. The protocol specified doses of i.v. and nebulised magnesium sulphate that were at the top end of those used in previous trials. Data presented in the supplemental tables show high adherence to the trial protocol, with most patients receiving the full dose of the relevant drugs. Pragmatic trials carry an inevitable risk that trial treatment will be delivered in a suboptimal manner, but we found no significant evidence of this in the 3Mg trial.
Strengths and weaknesses
The 3Mg trial was designed as a pragmatic trial to determine the effectiveness of using magnesium sulphate alongside other treatments as part of routine ED practice. As such it can tell us whether or not magnesium sulphate is an effective treatment for typical patients presenting to the ED with acute severe asthma. However, it cannot tell us whether magnesium sulphate may have benefits in a more narrowly defined patient population or a more specialised setting. The study population was pragmatically defined using information routinely available to ED staff. This means that the findings are generalisable to typical patients attending hospital with acute asthma, but also means that the study population could have included some patients with other diagnoses. In particular, the study population included a proportion of older patients and those who had smoked for a number of years in whom a diagnosis of chronic obstructive pulmonary disease may be more likely than asthma. We evaluated magnesium sulphate in combination with standard treatment rather than assessing it alone. This may have reduced the potential for magnesium sulphate to make a difference, but withholding standard treatment would have been unethical and would not have reflected typical practice. We selected primary outcomes that measured the effect of treatment on symptoms (VAS breathlessness) and clinical management (hospital admission). We also measured physiological parameters and PEFR as secondary outcomes. It is possible that other measures, such as FEV1, might have been more sensitive to changes in respiratory function, but these are not usually measured in the ED and are of uncertain clinical relevance.
We deliberately excluded patients with life-threatening asthma and were unable to power the study to detect differences in serious adverse outcomes (including death), therefore we are unable to determine whether or not magnesium sulphate may have an effect on serious adverse outcomes in life-threatening asthma.
Although 3Mg is the largest trial undertaken of magnesium sulphate in acute asthma it is still possible that i.v. magnesium sulphate has a modest effect on admission to hospital that was not proven in this trial. Some of the differences in outcome between i.v. magnesium sulphate and placebo showed trends towards benefit, most notably hospital admission, and the CI includes the possibility of a worthwhile effect (albeit also including the possibility of no effect or even a slightly increased admission rate). The recruitment rate was slower than expected and this meant that, despite additional funding, increasing the number of recruiting centres and prolonging recruitment, we fell short of our recruitment target of 1200 participants. This reduced the power of the trial from 90% to 87% to detect a 10% difference in admission rate. The effect on the power to detect other outcomes was minimal. The sample size was mainly determined by the primary health service outcome, so the trial was powered to detect an 8-mm change in VAS breathlessness despite the minimum clinically significant difference being estimated to be 22 mm. We also assumed a conservative attrition rate of 20% for VAS breathlessness, whereas it was actually only 10% and, therefore, power to detect differences in VAS breathlessness was maintained.
The trial was double blinded to reduce the potential for placebo effects and measurement bias. The responses to the questionnaire suggested that more patients in the active treatment arms than in the placebo arm thought that they had received active treatment, suggesting that blinding did not completely overcome the patient’s ability to detect that they had received active treatment. However, given the overall negative findings and pragmatic nature of the trial, this does not seem to be an important source of bias.
Although the attrition rates for the primary outcomes and physiological secondary outcomes were low, the response rate to the questionnaire was lower than anticipated. Previous trials in ED patients have achieved response rates of around 70%,69,70 but our response rate was 40%. The reasons for this are not clear but may reflect a younger age group, general population disengagement with traditional mailing over time or recognised associations between presentation with asthma at the ED and lower willingness to engage with health care. The low response rate means that the findings of the EQ-5D analysis, health-care resource use survey and patient satisfaction survey should be treated with caution. However, the lack of evidence of clinical effectiveness demonstrated in the primary outcomes and physiological secondary outcomes undermines the potential mechanism for changes in the questionnaire outcomes, while the lack of any important differences between the trial groups means that there is little to be gained from speculating about the potential effect of responder bias.
Chapter 5 Conclusions
Implications for health care
The findings of this trial suggest that there is no role for nebulised magnesium sulphate in the management of acute severe asthma in adults and only a limited potential role for i.v. magnesium sulphate. Patients receiving placebo alongside standard treatment showed marked improvements in breathlessness and PEFR, and few required respiratory support. Although most were admitted to hospital, we found no evidence that nebulised magnesium sulphate reduced the admission rate and the effect on admission rates from i.v. magnesium sulphate was not significant. The low rate of side effects and adverse events (other than those related to the underlying illness) suggests a low risk of harm from i.v. magnesium sulphate administration, but the corresponding evidence of benefit is modest and uncertain.
The findings of the trial do not apply to patients with life-threatening asthma, who were excluded from the trial. The low rate of adverse events associated with i.v. magnesium sulphate treatment may reassure clinicians who feel that in life-threatening asthma any weak evidence of benefit extrapolated from this trial or other sources provides a justification for administering i.v. magnesium sulphate. However, given the failure to demonstrate evidence of benefit, it is important to ensure that magnesium sulphate is not used as an alternative to effective treatments or as a way of delaying the provision of respiratory support.
The BTS/SIGN guidelines2 do not currently recommend routine use of i.v. or nebulised magnesium sulphate in the management of acute severe asthma, but recommend considering a single dose of i.v. magnesium sulphate (following consultation with senior medical staff) for patients with severe acute asthma who have not had a good initial response to inhaled bronchodilator therapy or patients with life-threatening or near fatal asthma. Our findings do not suggest any reliable expectation of clinical benefit from i.v. or nebulised magnesium sulphate.
The BTS/SIGN guidance recommends using PEFR to guide decision-making in acute asthma. Our secondary analysis suggests that PEFR predicts adverse outcome. Heart rate, other serious illnesses and the required oxygen flow rate could also be used to predict adverse outcome, but the predictive value of a model based on these parameters is not high.
Recommendations for research
Further clinical trials of magnesium sulphate in adults with acute asthma are very unlikely to be worthwhile. It is possible that i.v. treatment could have a modest effect on admission to hospital that was not detected by 3Mg, but a much larger trial would be needed to detect this. Despite having extensive experience of undertaking trials in the emergency setting, an excellent network of supporting hospitals and a simple, pragmatic trial protocol we failed to achieve our recruitment targets and took longer to complete the trial than expected. Undertaking a larger trial to obtain a more precise estimate of treatment effect would therefore require international collaboration and incur substantial costs. It is very unlikely that this could be justified given the low probability of detecting a worthwhile benefit.
The results of the 3Mg trial show that, although standard treatment for acute severe asthma produces marked improvements in breathlessness and PEFR, with a low rate of adverse outcome or requirement for respiratory support, most patients were admitted to hospital after ED treatment. This suggests there is still scope for new treatments or other changes in clinical practice, to improve the management of acute severe asthma.
Acknowledgements
Contributions
Membership of the 3Mg research team is outlined below. The co-applicants designed the trial and wrote the grant application. The Project Management Group and local investigators undertook the trial with independent oversight from the Trial Steering Committee and Data Monitoring Committee.
The 3Mg research team
Writing Group: Steve Goodacre, Judith Cohen, Mike Bradburn, John Stevens (University of Sheffield), Alasdair Gray (Royal Infirmary of Edinburgh), Jonathan Benger (University of the West of England) and Tim Coats (University of Leicester).
Project Management Group: Steve Goodacre, Judith Cohen, Mike Bradburn, Chin Maguire, Yvonne Meades (University of Sheffield), Alasdair Gray, Moyra Masson (Royal Infirmary of Edinburgh), Jonathan Benger (University of the West of England) and Tim Coats (University of Leicester).
Trial Steering Committee: Andrew Greening (Independent Chairperson, Royal Infirmary of Edinburgh), Magdy Sakr (Independent, University Hospitals of Coventry and Warwickshire), Roseanne McNamee (Independent, University of Manchester), Jenny Negus (Independent, Patient Representative), Steve Goodacre, Alasdair Gray and Judith Cohen.
Data Monitoring and Ethics Committee: Sandra Eldridge (Chairperson, Queen Mary University of London), Mark Elliott (St James University Hospital, Leeds) and Steve Crane (York Hospital).
Co-applicants: Steve Goodacre, Jon Nicholl, Mike Campbell, Julie Ratcliffe (University of Sheffield), Alasdair Gray (Royal Infirmary of Edinburgh), Jonathan Benger (University of the West of England), Tim Coats (University of Leicester), Stephen Holgate (University of Southampton) and Peter Jackson (Sheffield Teaching Hospitals NHS Foundation Trust).
Local investigators: Abdul Jalil (Doncaster Royal Infirmary), Alasdair Corfield (Royal Alexandra Hospital, Paisley), Alasdair Gray (Royal Infirmary of Edinburgh), Alastair Stevenson (Ayr Hospital), Angus Cooper (Aberdeen Royal Infirmary), Ann Marie Morris (University Hospital of North Staffordshire), Colin Dewar (Fife, Queen Margaret Hospital and Victoria Hospital), Crawford McGuffie (Crosshouse Hospital), David Robinson (Bradford Royal Infirmary), Barbara Madigan (Royal United Hospital Bath), Frank Coffey (Queen’s Medical Centre, Nottingham), Gary Kitching (York Hospital), Gavin Lloyd (Royal Devon and Exeter Hospital), Iain Lennon (Derbyshire Royal Infirmary), Jason Smith (Derriford Hospital, Plymouth), John Keaney (Hairmyres Hospital), Jonathan Benger (Bristol Royal Infirmary), Julian Humphrey (Barnsley Hospital), Khurram Iftikhar (Southend University Hospital), Matt Shepherd (Pinderfields Hospital), Matthew Pereira (Addenbrooke’s Hospital, Cambridge), Mehmood Chaudhry (University Hospital Coventry), Nathan Spencer (Kettering General Hospital), Patrick Dissmann (James Cook University Hospital, Middlesbrough), Samuel McBride (Lancaster Royal Infirmary), Simon Chapman (Bristol Frenchay Hospital), Steve Goodacre (Northern General Hospital, Sheffield), Suzanne Brady (Rotherham General Hospital), Taj Hassan (Leeds Teaching Hospitals), Tim Coats (Leicester Royal Infirmary), Tim Harris (The Royal London Hospital), Tristan Dyer (Northampton General Hospital) and William Townend (Hull Royal Infirmary).
We would like to acknowledge the following people who have not been included as members of the 3Mg research team but provided valuable input into the trial: Diana Papaioannou, Joseph Clark, Kathryn MacKellar, Katie Biggs and Martina Santarelli (3Mg central team); Andy Scott-Donkin, Beverley Page, Caroline Renton, Charlene Roe, Christine Harley, Deborah Hardy, Dominique Fairclough, Edelyn Danpiago, Elma Norwood, Emily Calton, Emma Beeston, Emma Gendall, Fiona Hudas, Geraldine Ward, Ian Massey, Isaac Narh, Janet Field, Jason Pott, Jayne Craig, Joanne Porter, Joseph Alade, Judith Edwards, Kerry Flahive, Kim Ray, Lee Northway, Lindsay Jenkinson, Lindsey Pearson, Loretta Barnett, Madaline Oboye, Margaret Clarke, Michelle McMillan, Natasha Barnes, Nicola Lancaster, Parizade Raymode, Paul Williams, Paula Welch, Philip Miller, Phillippa Smith, Rachel Lock, Rachel Walker, Racquel Carpio, Rebecca Coop, Rosalyn Squire, Rosanna Fong, Ruth Ballington, Sarah Day, Sarah Plant, Sarah Yusaf, Steve Harvey, Susan Brown, Vanessa Lawlor, Victoria Fowler and Victoria Katsande (site research nurses); Andrew Webster, David Watson, Jo Beahan, Mark Poulson, Moshood Olayinka, Paul Hunt and Steve Bush (site investigators); Amanda Loban, Chris Ellis, Cindy Cooper and Tim Chater (Sheffield CTRU); Carol Jacques, Gillian McVey, Jen Boston, Rodney Hughes, Roger Brookes and Tracy Jackson (Sheffield Teaching Hospitals); Chris Murray (EpiGenesys); and, Baxter Millar and Simon Bath (Tayside Pharmaceuticals).
Contributions of authors
Steve Goodacre, Judith Cohen, Mike Bradburn, John Stevens, Alasdair Gray, Jonathan Benger and Tim Coats contributed to drafting the report and approved the final draft.
Mike Bradburn undertook the statistical analysis.
John Stevens undertook the economic analysis.
All members of the writing group contributed to redrafting and approved the final draft.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
Publication
Goodacre S, Cohen J, Bradburn M, Gray A, Benger J, Coats T on behalf of the 3Mg research team. Intravenous or nebulised magnesium sulphate versus standard therapy for severe acute asthma (3Mg trial): a double-blind, randomised controlled trial. Lancet Respir Med 2013;1:293–300.
References
- The NHS Health and Social Care Information Centre . Hospital Episodes Statistics for England, 1998–2012 n.d. www.hesonline.nhs.uk/ (accessed 6 December 2012).
- British Thoracic Society/Scottish Intercollegiate Guidelines Network . British Guideline on the Management of Asthma: A National Clinical Guideline 2012. www.britthoracic.org.uk/Portals/0/Guidelines/AsthmaGuidelines/sign101%20Jan%202012.pdf (accessed 20 January 2014).
- The Magpie Trial Collaborative Group . Do women with pre-eclampsia, and their babies, benefit from magnesium sulphate? The Magpie Trial: a randomised placebo-controlled trial. Lancet 2002;359:1877-90.
- Tzivoni D, Banai S, Schuger C, Benhorin J, Keren A, Gottlieb S, et al. Treatment of torsade de pointes with magnesium sulfate. Circulation 1988;77:392-7. http://dx.doi.org/10.1161/01.CIR.77.2.392.
- British National Formulary. London: BMJ Publishing Group; 2012.
- Farouque HM, Sanders P, Young GD. Intravenous magnesium sulfate for acute termination of sustained monomorphic ventricular tachycardia associated with coronary artery disease. Am J Cardiol 2000;86:1270-2. http://dx.doi.org/10.1016/S0002-9149(00)01219-4.
- Thel MC, Armstrong AL, McNulty SE, Califf RM, O’Connor CM. Randomised trial of magnesium in in-hospital cardiac arrest. Duke Internal Medicine Housestaff. Lancet 1997;350:1272-6.
- Woods KL, Fletcher S. Long-term outcome after intravenous magnesium sulphate in suspected acute myocardial infarction: the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2). Lancet 1994;343:816-19. http://dx.doi.org/10.1016/S0140-6736(94)92024-9.
- Ho KM, Sheridan DJ, Paterson T. Use of intravenous magnesium to treat acute onset atrial fibrillation: a meta-analysis. Heart 2007;93:1433-40. http://dx.doi.org/10.1136/hrt.2006.111492.
- Onalan O, Crystal E, Daoulah A, Lau C, Crystal A, Lashevsky I. Meta-analysis of magnesium therapy for acute management of rapid atrial fibrillation. Am J Cardiol 2007;99:1726-32. http://dx.doi.org/10.1016/j.amjcard.2007.01.057.
- Mohammed S, Goodacre S. Intravenous and nebulised magnesium sulphate for acute asthma: systematic review and meta-analysis. Emerg Med J 2007;24:823-30. http://dx.doi.org/10.1136/emj.2007.052050.
- Rowe BH, Bretzlaff JA, Bourdon C, Bota GW, Camargo CA. Intravenous magnesium sulphate treatment for acute asthma in the emergency department: a systematic review of the literature. Ann Emerg Med 2000;36:181-90.
- Alter HJ, Koepsell TD, Hilty WM. Intravenous magnesium as an adjuvant in acute bronchospasm: a meta-analysis. Ann Emerg Med 2000;36:191-7. http://dx.doi.org/10.1067/mem.2000.109170.
- Rodrigo G, Rodrigo C, Burschtin O. Efficacy of magnesium sulfate in acute adult asthma: a meta-analysis of randomized trials. Am J Emerg Med 2000;18:216-21. http://dx.doi.org/10.1016/S0735-6757(00)90024-X.
- Cheuk DKL, Chau TCH, Lee SL. A meta-analysis on intravenous magnesium sulphate for treating acute asthma. Arch Dis Child 2005;90:74-7. http://dx.doi.org/10.1136/adc.2004.050005.
- Skobeloff EM, Spivey WH, McNamara RM, . Intravenous magnesium sulphate for the treatment of acute asthma in the emergency department. JAMA 1989;262:1210-13.
- Green SM, Rothrock SG. Intravenous magnesium for acute asthma: failure to decrease emergency treatment duration or need for hospitalization. Ann Emerg Med 1992;21:260-5. http://dx.doi.org/10.1016/S0196-0644(05)80885-6.
- Tiffany BR, Berk WA, Todd IK, White SR. Magnesium bolus or infusion fails to improve expiratory flow in acute asthma exacerbations. Chest 1993;104:831-4. http://dx.doi.org/10.1378/chest.104.3.831.
- Matusiewicz SP, Cusack S, Greening AP, Crompton GK. A double blind placebo controlled parallel group study of intravenous magnesium sulphate in acute severe asthma. Eur Res 1994;7.
- Skorodin MS, Tenholder MF, Yetter B, Owen KA, Waller RF, Khandelwahl S, et al. Magnesium sulfate in exacerbations of chronic obstructive pulmonary disease. Arch Intern Med 1995;155:496-500. http://dx.doi.org/10.1001/archinte.155.5.496.
- Bloch H, Silverman R, Mancherje N, Grant S, Jagminas L, Scharf SM. Intravenous magnesium sulfate as an adjunct in the treatment of acute asthma. Chest 1995;107:1576-81. http://dx.doi.org/10.1378/chest.107.6.1576.
- Silverman R, Osborn H, Runge J, Gallagher EJ, Chiang W, Feldman J, et al. IV magnesium sulfate in the treatment of acute severe asthma. A multicentre randomized controlled trial. Chest 2002;122:489-97. http://dx.doi.org/10.1378/chest.122.2.489.
- Boonyavorakul C, Thakkinstan A, Charonepan P. Intravenous magnesium sulfate in acute severe asthma. Respirology 2000;5:221-5. http://dx.doi.org/10.1046/j.1440-1843.2000.00252.x.
- Porter RS, Nester BA, Braitman LE, Geary U, Dalsey WC. Intravenous magnesium is ineffective in adult asthma, a randomized trial. Eur J Emerg Med 2001;8:9-15. http://dx.doi.org/10.1097/00063110-200103000-00003.
- Ciarallo L, Sauer AH, Shannon MW. Intravenous magnesium therapy for moderate to severe pediatric asthma: Results of a randomized, placebo-controlled trial. J Pediatr 1996;129:809-14. http://dx.doi.org/10.1016/S0022-3476(96)70023-9.
- Devi PR, Kumar L, Singhi SC, Prasad R, Singh M. Intravenous magnesium sulfate in acute severe asthma not responding to conventional therapy. Indian Pediatr 1997;34:389-97.
- Gurkan F, Haspolat K, Bosnak M, Dikici B, Derman O, Ece A. Intravenous magnesium sulphate in the management of moderate to severe acute asthmatic children nonresponding to conventional therapy. Eur J Emerg Med 1999;6:201-5. http://dx.doi.org/10.1097/00063110-199909000-00005.
- Scarfone RJ, Loiselle JM, Joffe MD, Mull CC, Stiller S, Thompson K, et al. A randomized trial of magnesium in the emergency department treatment of children with asthma. Ann Emerg Med 2000;36:572-8. http://dx.doi.org/10.1067/mem.2000.111060.
- Ciarallo L, Brousseau D, Reinert S. Higher-dose intravenous magnesium therapy for children with moderate to severe acute asthma. Arch Pediatr Adolesc Med 2000;154:979-83. http://dx.doi.org/10.1001/archpedi.154.10.979.
- Santana JC, Barreto SS, Piva JP, Garcia PC. Randomized clinical trial of intravenous magnesium sulfate versus salbutamol in early management of severe acute asthma in children. J Pediatr 2001;77:279-87.
- Singh AK, Gaur S, Kumar R. A randomised controlled trial of intravenous magnesium sulphate as an adjunct to standard therapy in acute severe asthma. Iran J Allergy Asthma Immunol 2008;7:221-9.
- Blitz M, Blitz S, Hughes R, Diner B, Beasley R, Knopp J, et al. Aerosolized magnesium sulfate in acute asthma: A systematic review. Chest 2005;128:337-44.
- Villeneuve EJ, Zed PJ. Nebulized Magnesium Sulfate in the Management of Acute Exacerbations of Asthma. Ann Pharmacother 2006;40:1118-24. http://dx.doi.org/10.1345/aph.1G496.
- Nannini LJ, Pendino JC, Corna RA, Mannarino S, Quispe R. Magnesium sulfate as a vehicle for nebulized salbutamol in acute asthma. Am J Med 2000;108:193-7. http://dx.doi.org/10.1016/S0002-9343(99)00463-5.
- Bessmertny O, DiGregorio RV, Cohen H, Becker E, Looney D, Golden J, et al. A randomized clinical trial of nebulized magnesium sulfate in addition to albuterol in the treatment of acute mild-to-moderate asthma exacerbations in adults. Ann Emerg Med 2002;39:585-91. http://dx.doi.org/10.1067/mem.2002.123300.
- Hughes R, Goldkorn A, Masoli M, Weatherall M, Burgess C, Beasley R. Use of isotonic nebulised magnesium sulphate as an adjuvant to salbutamol in treatment of severe asthma in adults: randomised placebo-controlled trial. Lancet 2003;361:2114-17. http://dx.doi.org/10.1016/S0140-6736(03)13721-X.
- Kokturk N, Turktas H, Kara P, Mullaoglu S, Yilmaz F, Karamercan A. A randomized clinical trial of magnesium sulphate as a vehicle for nebulized salbutamol in the treatment of moderate to severe asthma attack. Pulmon Pharmacol Therapeut 2005;18:416-21. http://dx.doi.org/10.1016/j.pupt.2005.03.003.
- Aggarwal P, Sharad S, Handa R, Dwiwedi SN, Irshad M. Comparison of nebulised magnesium sulphate and salbutamol combined with salbutamol alone in the treatment of acute bronchial asthma: a randomised study. Emerg Med J 2006;23:358-62. http://dx.doi.org/10.1136/emj.2005.026203.
- Mahajan P, Haritos D, Rosenberg N, Thomas R. Comparison of nebulized magnesium sulfate plus albuterol to nebulized albuterol plus saline in children with acute exacerbations of mild to moderate asthma. J Emerg Med 2004;27:21-5. http://dx.doi.org/10.1016/j.jemermed.2004.02.006.
- Meral A, Coker M, Tanac R. Inhalation therapy with magnesium sulfate and salbutamol sulfate in bronchial asthma. Turk J Pediatr 1996;38:169-75.
- Mangat HS, D’Souza GA, Jacob MS. Nebulized magnesium sulphate versus nebulized salbutamol in acute bronchial asthma: a clinical trial. Eur Respir J 1998;12:341-4. http://dx.doi.org/10.1183/09031936.98.12020341.
- LeLorier J, Gregoire G, Benhaddad A, Lapierre J, Derderian F. Discrepancies between meta-analyses and subsequent large randomised controlled trials. N Engl J Med 1997;337:536-42.
- National Heart, Lung, and Blood Institute . National Asthma Education and Prevention Program Expert Panel Report #3: Guidelines for the Diagnosis and Management of Asthma. US Department of Health and Human Services National Institutes of Health National Heart, Lung, and Blood Institute; 2007 n.d. www.nhlbi.nih.gov/guidelines/asthma/asthgdln.pdf (accessed 20 January 2014).
- Jones LA, Goodacre S. Magnesium sulphate in the treatment of acute asthma: evaluation of current practice in adult emergency departments. Emerg Med J 2009;26:783-5. http://dx.doi.org/10.1136/emj.2008.065938.
- Hewitt CE, Torgerson DJ. Is restricted randomisation necessary?. BMJ 2006;322:1506-8. http://dx.doi.org/10.1136/bmj.332.7556.1506.
- Wilson RC, Jones PW. Comparison of the visual analogue scale and Borg scale for measurement of dyspnoea during exercise. Clin Sci 1989;76:277-82.
- Kendrick KR, Baxi SC, Smith RM. Usefulness of the modified 0–10 Borg scale in assessing the degree of dyspnea in patients with COPD and asthma. J Emerg Nurs 2000;26.
- Karras DJ, Sammon ME, Terregino CA, Lopez BL, Griswold SK, Arnold GK. Clinically meaningful changes in quantitative measures of asthma severity. Acad Emerg Med 2000;7:327-34. http://dx.doi.org/10.1111/j.1553-2712.2000.tb02231.x.
- Gupta D, Aggarwal AN, Sublamaxi MV, Jindal SK. Assessing severity of asthma: spirometric correlates with visual analogue scale (VAS). Indian J Chest Dis Allied Sci 2000;42:95-100.
- Simes RJ. An improved Bonferroni procedure for multiple tests of significance. Biometrika 1986;73:751-4. http://dx.doi.org/10.2307/2336545.
- Guide to the Methods of Technology Appraisal. London: National Institute for Health and Clinical Excellence; 2008.
- Reference Costs 2012–12. London: Department of Health; 2012.
- Curtis L. Unit Costs of Health and Social Care 2011. Canterbury: Personal Social Services Research Unit; 2011.
- Turner J, O’Cathain A, Knowles E, Nicholl J, Tosh J, Sampson F, et al. Evaluation of NHS 111 Pilot Sites: Final Report. Sheffield: University of Sheffield; 2012.
- Office for National Statistics . Statistical Bulletin: 2012 Annual Survey of Hours and Earnings n.d. www.ons.gov.uk/ons/dcp171778_286243.pdf (accessed 16 January 2014).
- Dolan P. Modelling valuations for EurolQoL health states. Med Care 1997;35:1095-108. http://dx.doi.org/10.1097/00005650-199711000-00002.
- O’Hagan A, Stevens JW, Montmartin J. Bayesian cost-effectiveness analysis from clinical trial data. Stat Med 2001;20:733-53. http://dx.doi.org/10.1002/sim.861.
- Lunn DJ, Thomas A, Best N, Spiegelhalter D. WINBUGS – a Bayesian modelling framework: concepts, structure, and extensibility. Stat Comput 2000;10:325-37.
- Griffiths C, Foster G, Barnes N, Eldridge S, Tate H, Begum S, et al. A specialist nurse intervention to reduce unscheduled care for asthma: the East London controlled trial for high risk asthma (ELECTRA). BMJ 2004;328:144-7. http://dx.doi.org/10.1136/bmj.37950.784444.EE.
- Royston P, Altman DG. Approximating statistical functions by using fractional polynomial regression. J R Stat Soc 1997;46:411-42. http://dx.doi.org/10.1111/1467-9884.00093.
- The Medicines for Human Use (Clinical Trials) Regulations 2004 n.d. www.legislation.gov.uk/uksi/2004/1031/contents/made (accessed 20 January 2014).
- Coats TJ, Goodacre S. Eur J Emerg Med 2009;16:242-3. http://dx.doi.org/10.1097/MEJ.0b013e32830fe96a (accessed Consent in emergency and resuscitation research).
- Coats TJ, Ng G, Shakur H. Consent in emergency research. Emerg Med J 2006;23:489-90. http://dx.doi.org/10.1136/emj.2005.031005.
- Medical Research Council . Guidelines for Good Clinical Practice in Clinical Trials (1998) n.d. www.mrc.ac.uk/Utilities/Documentrecord/index.htm?d=MRC002416 (accessed 20 January 2014).
- Goodacre S, Coats T, Clancy M. Good clinical practice in clinical trials: Core knowledge for emergency physicians. Emerg Med J 2008;25. http://dx.doi.org/10.1136/emj.2008.063230.
- Eudralex (European Commission) . EU Guidelines for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use. Volume 4 (Annex 13) n.d. http://ec.europa.eu/health/files/eudralex/vol-4/2009_06_annex13.pdf (accessed 20 January 2014).
- Standard Operating Procedure for Recording, Managing and Recording Adverse Events for STH studies (SOP A123): Version 2.5. Sheffield: STH; 2009.
- Kind P, Hardman G, Macran S. UK Population Norms for EQ5D 1999. www.york.ac.uk/inst/che/pdf/DP172.pdf (accessed 16 January 2014).
- Goodacre SW, Bradburn M, Cross E, Collinson PO, Gray A, Hall AS. on behalf of the RATPAC research team . The RATPAC Trial (Randomised Assessment of Treatment using Panel Assay of Cardiac markers): A randomised controlled trial of point-of-care cardiac markers in the emergency department. Heart 2011;97:190-6. http://dx.doi.org/10.1136/hrt.2010.203166.
- Goodacre S, Nicholl J, Dixon S, Cross E, Angelini K, Arnold J, et al. Randomised controlled trial and economic evaluation of a chest pain observation unit compared with routine care. BMJ 2004;328:254-7. http://dx.doi.org/10.1136/bmj.37956.664236.EE.
Appendix 1 Patient recruitment form
Appendix 2 Brief information sheet
Initial patient information sheet for the 3Mg study
(Is magnesium sulphate, in addition to standard treatment, effective in patients with an asthma attack?)
You are being invited to take part in a research study. The purpose of the study is to find out whether treating an asthma attack with magnesium sulphate, in addition to standard treatment, relieves symptoms of breathlessness and reduces the chances that people with acute asthma will need to be admitted to hospital. The study will compare standard treatment for asthma to standard treatment and magnesium sulphate, given either into a vein or through a nebuliser.
Why have I been chosen?
You have come to hospital with an asthma attack. The doctors treating you think that magnesium sulphate, if it is effective, may ease your symptoms of breathlessness and improve your chances of avoiding hospital admission.
Do I have to take part?
No. It is up to you to decide whether or not to take part. If you do, you will be asked to sign a consent form. You will be free to withdraw at any time, without giving a reason. A decision to withdraw at any time or a decision not to take part, will not affect the standard of care you receive.
What will happen to me if I take part?
If you agree to take part you will be given the standard treatment for asthma. You will also be given an infusion into a vein in your arm and three nebulisers. Along with the standard treatments you will be given one of the following three alternatives:
-
magnesium sulphate added to the nebuliser but not the infusion
-
magnesium sulphate added to the infusion but not the nebuliser
-
nothing added to either the infusion or the nebuliser (just standard treatment).
We will then monitor your symptoms for up to 4 hours, after which the doctor will advise whether you should be admitted to hospital or go home. Neither you, nor the doctors will know which treatment you have been given.
What do I have to do?
All the treatments will be given by the doctors and nurses. You will to need answer some questions and do a Peak Flow recording (breathing test).
Are there any side effects to treatment?
Magnesium sulphate can cause feelings of nausea, vomiting, thirst or facial flushing, particularly when given through a vein. In rare cases overdose of magnesium sulphate can cause weakness, coma or heart problems. The doctors will monitor your heartbeat during treatment.
We will give you more information shortly, when you are feeling better.
Version 004: 16 June 2009
Appendix 3 Full information sheet
Headed notepaper from XXX Hospital
PATIENT INFORMATION SHEET FOR THE 3Mg STUDY
(Is magnesium sulphate, in addition to standard treatment, effective in patients with an asthma attack?)
You are being invited to take part in a research study that is organised by the University of Sheffield and undertaken at XXX Hospital. Before you decide it is important for you to understand why the research is being done and what it will involve. Please read the following information carefully. Ask us if there is anything that is not clear or if you would like more information.
What is the purpose of the study?
The purpose of the study is to find out whether treating an asthma attack with magnesium sulphate relieves symptoms of breathlessness and reduces the chances that people with acute asthma will need to be admitted to hospital.
Magnesium sulphate has been used to treat asthma attacks for several years. It can be given either as an infusion into a vein or by being inhaled using a nebuliser. We know that treatment with magnesium sulphate can improve the results of breathing tests, but we do not yet know whether it improves patient’s symptoms of breathlessness or reduces their chances of needing hospital admission. We also do not know whether it works better by infusion into a vein or inhaled through a nebuliser.
The study will compare standard treatment for asthma, with the addition of magnesium sulphate (given either into a vein or through a nebuliser), to standard treatment without magnesium sulphate.
Why have I been chosen?
You have come to hospital with an asthma attack. The doctors treating you think that magnesium sulphate, if it is effective, may ease your symptoms of breathlessness and improve your chances of avoiding hospital admission.
Do I have to take part?
No. It is up to you to decide whether or not to take part. If you do, you will be given this information sheet to keep and be asked to sign a consent form. You are still free to withdraw at any time and without giving a reason. A decision to withdraw at any time, or a decision not to take part, will not affect the standard of care you receive.
What will happen to me if I take part?
If you agree to take part you will be given standard treatment for asthma with oxygen, nebulisers and steroids (prednisolone or hydrocortisone). In addition you will also be given an infusion of saline (salt and water) into a vein in your arm. Along with the standard treatments you will be given one of the following three alternatives:
-
magnesium sulphate added to the nebuliser but not the infusion
-
magnesium sulphate added to the infusion but not the nebuliser
-
nothing added to either the infusion or the nebuliser (i.e. just standard treatment on its own).
After you have been given the treatment we will monitor your symptoms for up to 4 hours. Depending upon your response to treatment the doctor will then advise whether you should be admitted to hospital or go home.
One month from now we will examine all of your hospital records and then send you a questionnaire in the post or telephone you asking you about your health, your recent use of health services and what you thought about the care you received.
You will be put into one of the three treatment groups by chance (randomly). Neither you, the staff nor the researchers will know which treatment you have been given (this is known as a ‘blind trial’). At the end of the trial the researchers will compare patients in the three groups and then reveal which treatment was which to find out which treatment helped patients most.
If it may affect your care then the doctors treating you can find out which treatment you have been given.
What do I have to do?
All the treatments will be given by the doctors and nurses. You will to need answer some questions and do a peak flow recording (breathing test) to monitor your progress, and then complete a questionnaire in 1 month’s time.
Are there any side effects to treatment?
Magnesium sulphate can cause feelings of nausea, vomiting, thirst or facial flushing, particularly when given through a vein. In rare cases overdose of magnesium sulphate can cause weakness, coma or heart problems. The doctors will monitor your heartbeat during treatment.
What are the possible benefits of taking part?
We cannot promise that the study will help you but doing the study may help to improve the treatment of people with an asthma attack.
What if there is a problem?
If you have a concern about any aspect of this study, you should ask to speak with the researchers who will do their best to answer your questions (contact details below). If you remain unhappy and wish to complain formally, you can do this through the NHS Complaints Procedure. Details can be obtained from the hospital.
In the event that something does go wrong and you are harmed during the research study there are no special compensation arrangements. If you are harmed and this is due to someone’s negligence then you may have grounds for a legal action for compensation against the University of Sheffield or XXX Hospital but you may have to pay your legal costs. The normal National Health Service complaints mechanisms will still be available to you.
Will my taking part in the study be kept confidential?
We will inform your GP that you have taken part in this study. We will record information about your treatment over the next few hours and a member of the research team will record information from your hospital notes and computer records in 1 month’s time. All information that is collected about you during the course of this study will be kept strictly confidential. The information will be stored in a secure area of the hospital. A copy of the information will be sent to the University of Sheffield where it will be stored in a secure area and also kept as a password-protected computer file, both of which can only be accessed by the research team and regulatory authorities. We will destroy all identifiable information 5 years after the end of the study. An anonymised copy of the computer file (with any details that might identify you removed) will be retained and made available to other researchers for use in future studies.
What will happen if I don’t want to carry on with the study?
You can withdraw from the study at any time. We will need to keep the information you have given up to the time you withdraw but will not collect any new information or send you the questionnaire.
What will happen to the results of the research study?
We will publish the results in a scientific journal and produce a report that is freely available to anyone who wishes to read it. You will not be personally identified in any report or publication we produce. Please contact us using the details below if you would like to see a summary of the results when the trial is completed.
Who is organising and funding the research?
The research is organised by the University of Sheffield and funded by the Department of Health.
Who has reviewed the study?
This study was given a favourable ethical opinion for conduct in the NHS by the Scotland A Research Ethics Committee.
Further information can be obtained from: Prof SW Goodacre
Medical Care Research Unit
University of Sheffield
Regent Court, 30 Regent Street
Sheffield S1 4DA
0114 222 0842, s.goodacre@sheffield.ac.uk
Version 004: 16 June 2009
Appendix 4 Patient consent form
Appendix 5 Clinical protocol
Appendix 6 Case report form
Appendix 7 Health-care resource use questionnaire
Appendix 8 Satisfaction with care questionnaire
Appendix 9 Investigational Medical Products Dossier
Appendix 10 Investigator’s brochure
Appendix 11 Trial protocol
Appendix 12 Original project description
List of abbreviations
- AUROC
- area under the receiver operating characteristic
- BNF
- British National Formulary
- BTS
- British Thoracic Society
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- CTRU
- Clinical Trials Research Unit
- DMEC
- Data Monitoring and Ethics Committee
- ED
- emergency department
- EQ-5D
- European Quality of Life-5 Dimensions
- FEV1
- forced expiratory volume in 1 second
- GCP
- good clinical practice
- GP
- general practitioner
- HDU
- high-dependency unit
- IB
- investigator’s brochure
- ICER
- incremental cost-effectiveness ratio
- ICU
- intensive care unit
- IMP
- investigational medicinal product
- IMPD
- investigational medicinal product dossier
- i.v.
- intravenous
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MIA
- manufacturer’s/importer’s licence
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- OR
- odds ratio
- PEFR
- peak expiratory flow rate
- PI
- principal investigator
- PMG
- Project Management Group
- QALY
- quality-adjusted life-year
- QP
- qualified person
- REC
- Research Ethics Committee
- RHH
- Royal Hallamshire Hospital
- RR
- relative risk
- SAE
- serious adverse event
- SD
- standard deviation
- SIGN
- Scottish Intercollegiate Guidelines Network
- SMD
- standardised mean difference
- SOP
- standard operating procedure
- STH
- Sheffield Teaching Hospitals
- TSC
- Trial Steering Committee
- VAS
- visual analogue scale