
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 10/108/01. The protocol was agreed in November 2012. The assessment report began editorial review in July 2013 and was accepted for publication in January 2014. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2015. This work was produced by Edwards et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Background
Description of health problem
Ovarian cancer is the fifth most common cancer in the UK, and is the fourth most common cause of cancer death. 1 Ovarian tumours are classified based on the cell type from which the tumour originates: surface epithelium, germ or stroma. Most ovarian malignancies are epithelial in origin, accounting for 80–90% of ovarian cancers. 1 Today, it is widely accepted that fallopian tube carcinoma and primary peritoneal carcinoma are, in general, histologically serous, and are considered to arise from the same pathophysiology as epithelial ovarian cancer. 2 Epithelial tumours can be further divided based on their histology (high-grade serous, low-grade serous, mucinous, endometrioid, clear cell, and undifferentiated or unclassifiable). The most common type of ovarian cancer in the UK is high-grade serous carcinoma. Other, rarer subtypes include germ cell tumours, which tend to occur in premenopausal women and are highly sensitive to chemotherapy (and therefore treatable), or borderline ovarian cancer. 1,3 Borderline ovarian cancers have low malignant potential and are usually considered separately as they do not usually require treatment with chemotherapy. It is thought that most histologies share common risk factors, with the probable exception of mucinous carcinomas. 1
Epidemiology
Incidence and prevalence
Ovarian cancer is predominantly a disease of older, postmenopausal women, with > 80% of cases being diagnosed in women of > 50 years of age. 1 The highest age-specific incidence rates are seen for women aged 80–84 years at diagnosis, with an incidence of 69 per 100,000, which drops to 64 per 100,000 in women aged ≥ 85 years. 1 However, for women with BRCA-deficient tumours, the age of diagnosis can be about 10 years earlier.
In 2008, around 6500 women were diagnosed with ovarian cancer in the UK, making it the second most common gynaecological cancer and the fifth most common cancer in women. 1 Focusing on England and Wales, in 2008, there were 5304 new cases in England and 400 in Wales, giving age-standardised rates per 100,000 of 15.8 [95% confidence Interval (CI) 15.4 to 16.2] and 19.6 (95% CI 17.7 to 21.5), respectively. 1 In 2010, 4295 deaths were attributed to ovarian cancer, accounting for 5.7% of all female deaths from cancer. 1 It has been estimated that the lifetime risk (adjusting for multiple primaries) of developing ovarian cancer is 1 in 54 for women in the UK (based on data from 2008). 1
Aetiology and pathology
Diagnosing ovarian cancer can be difficult. Patients typically present with subtle symptoms, such as difficulty eating, abdominal bloating and feeling ‘full’ quickly, all of which are suggestive of other, more minor conditions. As a result, many people (≈60%) are diagnosed with ovarian cancer when their disease is in an advanced stage. 4 Stage of ovarian cancer at diagnosis is based on the International Federation of Gynecology and Obstetrics (FIGO) classification system. 2 The FIGO system is a scale of I–IV, where stage I represents early stage disease and stages III and IV represent advanced disease (summarised in Table 1).
| Stage | Criteria |
|---|---|
| I | Tumour confined to the ovaries |
| IIA |
|
| IIB | As for 1A, but tumour limited to both ovaries |
| IIC | Tumour limited to one or both ovaries, with any of the following:
|
| II | Tumour involves one or both ovaries with pelvic extension |
| IIA | Extension and/or metastases in the uterus and/or fallopian tubes but with no malignant cells in ascites or peritoneal washings |
| IIB | Extension to other pelvic organs but with no malignant cells in ascites or peritoneal washings |
| IIC | Tumour staged either 2A or 2B with malignant cells in ascites or peritoneal washings |
| III | Tumour involves one or both ovaries with peritoneal metastasis outside the pelvis and/or regional lymph node metastasis Liver capsule metastasis equals stage 3 |
| IIIA | Microscopic peritoneal metastasis beyond the pelvis |
| IIIB | Macroscopic peritoneal metastasis beyond the pelvis, none of which exceed 2 cm in greatest dimension |
| IIIC | Peritoneal metastasis beyond the pelvis, larger than 2 cm in greatest dimension and/or regional lymph node metastasis |
| IV | Distant metastasis (beyond the peritoneal cavity) |
The aetiology of ovarian cancer is not yet fully understood. Various factors have been linked with an increased risk of developing ovarian cancer, and, conversely, others have been proposed as having a ‘protective’ effect and reducing ovarian cancer risk. The strongest known risk factors associated with a higher risk of ovarian cancer are increasing age and the presence of a mutation in the BRCA1 and BRCA2 genes, with the latter accounting for around 10% of cases. 1 The BRCA1 and BRCA2 genes are also associated with risk of breast cancer, and studies have shown a doubling in ovarian cancer risk for women with a previous breast cancer. Women who have a first-degree relative (i.e. parent, sibling or offspring) diagnosed with ovarian cancer have a three- to fourfold increased risk of developing the disease compared with women with no family history, although about only 10% of ovarian cancer cases occur in women with a family history. 1
Ovarian cancer risk tends to be reduced by factors that interrupt ovulation, such as pregnancy (with a dose–response relationship between increasing risk and a lower number of children), breastfeeding and oral contraceptive use. 1 Conversely, factors that prolong exposure to ovulation, such as nulliparity and infertility, increase risk. 1 It has been reported that 5 years’ use of oestrogen-only hormone replacement treatment (HRT) is associated with a 22% increase in the risk of ovarian cancer, which is considerably larger than the 10% risk increase identified with use of oestrogen–progestin HRT over the same time period. 1 It is estimated that about 50 cases of ovarian cancer in the UK in 2010 were linked with HRT, which is equivalent to about 1% of all ovarian cancers. 1 Past or short-term use of HRT is thought unlikely to increase the risk of ovarian cancer.
Risk of ovarian cancer seems to be higher in people who have some other gynaecological medical conditions. For example, studies have found that women with endometriosis have a 30–66% increased risk. 1 In addition, young women (15–29 years old) with ovarian cysts and functional cysts (harmless, short-lived cysts that are formed as a part of the menstrual cycle) have been found to have double the usual risk of ovarian cancer later in life, and women who had cysts surgically removed, or unilateral oophorectomy, have a ninefold risk increase. 1 Hysterectomy may reduce ovarian cancer risk, with case–control studies reporting a 30–40% risk reduction regardless of age at time of surgery, and a 50% risk reduction for women whose hysterectomy was 15 or more years before the study. 1
Lifestyle and environmental factors also affect risk of ovarian cancer, with both current and past smoking and high body mass index (BMI) being linked with increased risk. 1
Prognosis
Treatments for newly diagnosed ovarian cancer are given with curative intent. Primary treatment is determined by the stage and risk of disease at diagnosis. 1 Treatment options are surgery, or surgery followed by adjuvant chemotherapy (most likely platinum based) or chemotherapy alone. Alternatively, if it is thought that removal of all the cancer during the initial surgery could be problematic because of tumour size, chemotherapy may be administered before surgery (neoadjuvant chemotherapy) to shrink the tumour, with additional adjuvant chemotherapy after surgery. Clinically complete remission is achieved in most newly diagnosed patients through a combination of cytoreductive surgery and chemotherapy.
Considering chemotherapy, up to 10% of patients might not respond to first-line chemotherapeutic treatment and, of those who do respond, between 55% and 75% of people will relapse within 2 years. 5 It is these latter populations, more specifically those people who have received prior platinum-based treatment, that are the focus of this systematic review. Diagnosis of recurrent disease varies in UK clinical practice, with diagnosis based on clinical examination, biochemical markers (CA125) or radiological confirmation, or any combination of these three. Clinical expert advice is that, typically, a patient is diagnosed as relapsed if they have a serial rise in CA125 or have developed clinical signs, such as ascites. Diagnosis is typically confirmed with radiological scans. If a patient has no clinical symptoms but does have a rise in CA125, although possibly classified as relapse, the patient might not start a new chemotherapeutic regimen until they go on to develop symptoms. Date of relapse by CA125 is likely to be about 4 months earlier than date of relapse based on radiological scans. A patient is considered to have relapsed if they have progressed after achieving complete response (CR) or partial response (PR), or after their disease has been stable for some time (typically 8–12 weeks).
Prognostic factors thought to influence outcome (i.e. response to treatment and survival) are:
-
the stage of the disease at diagnosis (FIGO stage)
-
age
-
patient’s general health (typically referred to as performance status) at the time of presentation
-
extent of residual disease after debulking surgery
-
tumour grade
-
tumour histology.
Of the prognostic factors listed, the stage of disease at diagnosis and extent of residual disease after debulking surgery are considered to be strong predictors of survival. Relative 5-year survival rate is > 90% for early stage disease but falls markedly to < 10% for later stages. 1,3
Based on age-standardised relative survival rates during 2005–9 in England, data indicate that 72.3% of women are expected to survive for at least 1 year, falling to 42.9% surviving for ≥ 5 years, and to 35.4% surviving for ≥ 10 years. 1 Relative survival for ovarian cancer is higher in younger women, even after taking account of the higher background mortality in older people;1 5-year relative survival rates for ovarian cancer in England during 2005–9 ranged from 87% in people aged 15–39 years to 16% in those aged 80–99 years. The higher survival rate in younger women is likely to be attributable to a combination of better general health, more effective response to treatment and earlier diagnosis in younger people. 1
As with most cancers, relative survival for ovarian cancer is improving. 1 Much of the increase occurred during the 1980s and 1990s, and appears to be levelling off in the 2000s (Table 2). 1 Increased use of platinum-based chemotherapy, wider access to optimal primary treatment and greater determination to treat recurrent disease are all thought to have contributed to the observed improvements in overall survival (OS) at 1 and 5 years. 1
| Time period | 1 year | 5 years |
|---|---|---|
| 1971–5 | 42.0 | 21.0 |
| 2005–9 | 72.3 | 42.9 |
Measurement of disease
Initially, an elevated level of CA125 (determined by a blood test) is used as an indicator in the diagnosis of ovarian cancer. About 90% of people who have later stages of ovarian cancer have an elevated CA125 level, whereas about 50% of people with early-stage ovarian cancers have an elevated CA125 level; normal CA125 level is 0–35 U/ml. 6 However, CA125 is not specific to ovarian tumours, and other benign conditions of the womb and ovaries also result in elevated CA125 (e.g. endometriosis, fibroids and pelvic inflammatory disease). 1 Other non-gynaecological conditions that are associated with increased CA125 are liver cirrhosis and pleural effusions. If a person is found to have ovarian cancer that produces CA125 then this blood test can be used to monitor the clinical effectiveness of treatment. 1
As CA125 elevation is not specific to ovarian cancer, it is recommended that diagnosis of ovarian cancer be confirmed by an ultrasound scan of the abdomen and pelvis. 3 If the ultrasound, serum CA125 and clinical status suggest ovarian cancer, a computed tomography (CT) scan of the pelvis and abdomen is carried out to establish the extent of disease. Expert advice is that the ratio of CA125 to carcinoembryonic antigen (CEA) may be a useful guide in assessing ovarian cancer. Research has suggested that a CA125–CEA ratio of < 25 may be suggestive of a non-ovarian malignancy. 7
Impact of health problem
Significance for patients in terms of ill-health (burden of disease)
As a result of the difficulties of diagnosing ovarian cancer, many women present with advanced disease (e.g. 60% of women are diagnosed with stage III or IV disease), having had subtle symptoms for months before presentation. 1,3 Only around 29% of women are diagnosed at FIGO stage I, 4% at stage II and 6% are unstaged. 1
Treatments for newly diagnosed ovarian cancer are given with curative intent; however, for women with advanced, recurrent disease, second- and subsequent-line chemotherapies are typically given with palliative rather than curative intent, with the aim of alleviating symptoms and prolonging survival. Thus, key considerations in the choice of treatment at these stages in the pathway are maintaining the patient’s quality of life (QoL) and adverse effects associated with the individual treatments.
A recent study by Hess and Stehman8 investigated health-related quality of life (HRQoL) for women with ovarian cancer before, during and after chemotherapy, via a systematic review. The review resulted in identification of a total of 139 unique studies of patients with ovarian cancer in which QoL data were collected. Within these studies, > 90 different measures of QoL were administered. The authors found that there was limited longitudinal data beyond the initial treatment and immediate follow-up which limited the understanding of the long-term impact upon QoL for ovarian cancer survivors.
Significance for the UK NHS
Patients with ovarian cancer require significant amounts of hospital resources, including surgery and multiple courses of chemotherapy. In 2011–12, ovarian cancer accounted for 36,690 finished consultant episodes, 34,376 admissions and totalling 66,003 bed-days, in England alone. 9
Current service provision
National Institute for Health and Care Excellence (NICE) guidance is available on the initial recognition and management of ovarian cancer,3 first-line chemotherapeutic treatments for ovarian cancer,5 and on the use of topotecan [Hycamtin®, GlaxoSmithKline (GSK)]; paclitaxel [Taxol®, Bristol-Myers Squibb (BMS)]; and pegylated liposomal doxorubicin hydrochloride (PLDH; Caelyx®, Schering-Plough) as second-line or subsequent treatments of advanced ovarian cancer. 10
Initial management of ovarian cancer
After confirmation of a diagnosis of ovarian cancer, primary treatment is determined by the patient’s age and general health, in addition to the histology and grade of their cancer. Typically, surgery is the preferred initial treatment, the goal of which is to excise all macroscopic disease, irrespective of stage of disease.
For suspected early (stage I) ovarian cancer, NICE recommends optimal surgical staging, with no adjuvant chemotherapy for cancers identified as low-risk disease (grade 1 or 2, stage IA or IB). 3 For suspected early-stage disease that is considered high risk (grade 3 or stage IC), NICE recommends that surgery be followed by chemotherapy treatment comprising six cycles of carboplatin. 3
As noted earlier, most people are diagnosed with ovarian cancer when their disease has reached an advanced stage (stages II–IV). In such cases, complete excision of the tumour during surgery may be difficult and patients will typically require additional chemotherapeutic treatment. Chemotherapy may be administered prior to surgery (typically three cycles), with the objective of shrinking the tumour to facilitate excision and improve the probability of removal of all macroscopic disease. First-line chemotherapy is the first round of chemotherapeutic treatment a patient receives, whether it is as a neoadjuvant treatment before surgery, an adjuvant treatment to surgery or at some time in the longer term after surgery. Second- and subsequent-line treatment is for those who have either relapsed after first-line chemotherapeutic treatment or experienced progression of their disease while receiving chemotherapy.
Prior to offering cytotoxic chemotherapy to women with advanced ovarian cancer (stages II–IV), NICE recommends confirmation of tissue diagnosis with histology (or by cytology if histology is not appropriate). 3 For first-line chemotherapy, NICE recommends paclitaxel in combination with a platinum-based compound or platinum-based therapy alone (cisplatin or carboplatin). 5 NICE does not recommend the use of bevacizumab (Avastin®, Genentech) in combination with paclitaxel and carboplatin as a first-line chemotherapeutic treatment. 11
The NICE pathway for the management of advanced ovarian cancer is outlined in Figure 1.
FIGURE 1.
Treatment pathway recommended by NICE for the management of patients with advanced (stages II–IV) ovarian cancer. 12 a, Do not offer intraperitoneal chemotherapy to women with ovarian cancer, except as part of a clinical trial.

Second- and subsequent-line chemotherapeutic treatment
Although first-line chemotherapeutic treatment achieves a response in approximately 70–80% of patients, most patients will eventually relapse and require second-line therapy. 13 Between 55% and 75% of those who respond to first-line therapy will relapse within 2 years of completing treatment. Second- and subsequent-line therapies are typically given with palliative rather than curative intent, with the aim of alleviating symptoms and prolonging survival. Thus, key considerations in the choice of treatment at these stages in the pathway are maintaining the patient’s QoL and adverse effects associated with the individual treatments.
A patient’s response to first-line platinum-based therapy is indicative of their response to second and subsequent lines of platinum-based treatment, with the length of the platinum-free interval (PFI) and the extent of relapse (site and number of tumours) particularly prognostic of response. However, most patients will develop resistance to platinum-based therapy over time, with decreasing length of PFI with increasing rounds of treatment. Platinum-resistant ovarian cancer (defined in Table 3) has a particularly poor prognosis, with a reported median OS of < 12 months. 14
| Categorisation | Definition |
|---|---|
| Platinum sensitive | Disease that responds to first-line platinum-based therapy but relapses at ≥ 6 months after completion of initial platinum-based chemotherapy |
| PPS | Relapses between 6 and 12 months after completion of initial platinum-based chemotherapy |
| FPS | Relapses at ≥ 12 months after completion of initial platinum-based chemotherapy |
| Platinum resistant | Disease that relapses within 6 months of completion of initial platinum-based chemotherapy |
| Platinum refractory | Disease that does not respond to initial platinum-based chemotherapy |
The choice of second and subsequent lines of treatment has long been based on a patient’s PFI, i.e. the period of time between the last treatment of one regimen and the first treatment of the next regimen. Current NICE guidance on second-line or subsequent treatment of advanced ovarian cancer is based on the duration of time since last platinum-based therapy, with treatment options of paclitaxel, either as a monotherapy or in combination with platinum-based (carboplatin or cisplatin) therapy, PLDH monotherapy and topotecan monotherapy. 10 Treatments options as recommended by NICE, based on degree of platinum sensitivity, are depicted in Figure 2. In recently completed technology appraisals (TAs), NICE did not recommend bevacizumab in combination with gemcitabine (Gemzar®, Eli Lilly and Company) and carboplatin16 or trabectedin (Yondelis®, PharmaMar) plus PLDH17 for the treatment of recurrent ovarian cancer.

An important consideration in the choice of second-line treatment is the adverse effect of neurotoxicity, which is commonly associated with paclitaxel and also with carboplatin. Neurotoxicity can persist for up to 2 years after the end of treatment. 18 Patients who relapse after first-line treatment with paclitaxel–platinum combination therapy and are subsequently rechallenged with the same regimen within 12 months [i.e. those who are partially platinum sensitive (PPS)] are at an increased risk of developing neurotoxicity. 19 However, despite the associated increased risk of neurotoxicity, paclitaxel plus carboplatin is generally the preferred second-line treatment in UK practice in recurrent platinum-sensitive cancer, particularly for patients who relapse at > 12 months after completion of first-line chemotherapy. Carboplatin is chosen over cisplatin because of its more favourable adverse effect profile.
Current service cost
An analysis of Hospital Episode Statistics for 2006–8 of patients dying from prostate, breast, lung, upper gastrointestinal, colorectal or ovarian cancer indicates that patients with ovarian cancer and in their last year of life required 53,700 elective bed-days (at a cost of £14,274,623) and 216,723 emergency bed-days (at a cost of £58,606,527). 20 On a per-person basis, ovarian cancer had a longer elective stay and a longer emergency length of stay than the other cancers. 20 Ovarian cancer also had the highest overall cost, at £8000 per person. 20
Description of technologies under assessment
Topotecan
Topotecan is a semisynthetic, water-soluble derivative of camptothecin, a natural product isolated from the tree Camptotheca acuminate. 21 Topotecan elicits a chemotherapeutic effect through inhibition of the topoisomerase I enzyme, which has a crucial role in cell replication. Topoisomerase enzymes are involved in DNA replication, acting to relieve strain in the double-stranded DNA helix by ‘cutting’ one strand to release tension followed by reconnection of the two separate strands. Topotecan binds to the topoisomerase I–DNA complex, thus blocking the action of topoisomerase I and preventing re-formation of the DNA double helix.
Topotecan is licensed for the treatment of patients with metastatic carcinoma of the ovary after failure of at least one other treatment (i.e. topotecan is licensed as a second- and subsequent-line treatment). 22 The initial recommended dose of topotecan is 1.5 mg/m2 of body surface area (BSA), to be administered by intravenous (i.v.) infusion over 30 minutes for five consecutive days, with a 3-week interval between the start of each course. 22 It is recommended that topotecan be given for a minimum of four cycles. If well tolerated, treatment can be continued until disease progression. Topotecan should be administered under the supervision of a clinician experienced in the use of chemotherapy. Topotecan has also been evaluated in randomised controlled trials (RCTs) at an i.v. dose of 4.0 mg/m2 weekly,23 and as an oral treatment (dose of 2.3 mg/m2/day). 24 A dose for oral administration of topotecan has not been recommended for ovarian cancer.
Topotecan is contraindicated in patients who:22
-
have a history of severe hypersensitivity to the active substance or to any of the excipients
-
are breastfeeding
-
already have severe bone marrow depression before starting the first course, as evidenced by baseline neutrophil count of < 1.5 × 109/l and/or a platelet count of < 100 × 109/l.
Special warnings and precautions for use of topotecan include haematological toxicity, severe myelosuppression, topotecan-induced neutropenia, development of interstitial lung disease and thrombocytopenia. 22
The most common adverse events (AEs) associated with topotecan (reported by at least 1 out of 10 patients) are infection; febrile neutropenia; neutropenia; thrombocytopenia; anaemia; leucopenia; anorexia (which may be severe); nausea; vomiting; diarrhoea; constipation; mucositis; abdominal pain; alopecia; pyrexia; asthenia; and fatigue. 22
Pegylated liposomal doxorubicin hydrochloride
The active component in PLDH is doxorubicin hydrochloride, which is a member of the anthracycline class of antibiotics. Anthracyclines act by inhibiting synthesis, transcription and replication of DNA, and have potent antineoplastic (inhibits the growth and spread of cancerous cells) activity. 25 However, anthracyclines are also highly destructive to cellular membranes and are known to generate chemical species (oxygen-derived free radicals) that, as well as directly damaging DNA, are thought to damage the membranes of the heart, which may lead to congestive heart failure. 25 Cardiotoxic adverse effects of anthracyclines are irreversible and accumulative, and limit the clinical usefulness of this class of antibiotics.
Liposomes are minuscule spheres comprising a lipid bilayer, which can be used as vehicles for the administration of drugs. Coating the liposomes with methoxypolyethylene glycol (MPEG), a process known as pegylation, protects the liposome from detection by the body’s immune system. Encapsulation of doxorubicin hydrochloride in pegylated liposomes seems to increase the localisation and concentration of doxorubicin hydrochloride in cancerous cells, while simultaneously reducing the toxicity of doxorubicin hydrochloride to non-cancer tissues and cells, and, thereby, reducing the risk of severe adverse effects. 26
Pegylated liposomal doxorubicin hydrochloride (2 mg doxorubicin hydrochloride in a pegylated liposomal formulation) is licensed for the treatment of advanced ovarian cancer in women who have failed a first-line platinum-based chemotherapy regimen. 27 The licensed dose of PLDH when given as a monotherapy is 50 mg/m2 given intravenously once every 4 weeks for as long as disease does not progress and the patient continues to tolerate treatment;27 clinical expert advice is that typical UK clinical practice is to administer PLDH at a dose of 40 mg/m2. It should not be administered as a bolus injection or undiluted solution. PLDH should be given under the supervision of a clinician who is qualified in the use of cytotoxic medicines. 27 Importantly, PLDH cannot be interchanged with other medicines containing doxorubicin hydrochloride.
Randomised controlled trials have also evaluated PLDH in combination with other agents, both platinum-based and non-platinum based. 28–31 No dose has been recommended for PLDH when used in combination treatment. Doses of PLDH evaluated in doublet chemotherapy were 30 mg/m2 in combination with trabectedin30 and with carboplatin,28,31 and 45 mg/m2 in combination with carboplatin. 29 In all RCTs, the interval between cycles was 4 weeks. Clinical experts fed back that PLDH would most likely be used at a dose of 30 mg/m2 in combination regimens.
Pegylated liposomal doxorubicin hydrochloride is contraindicated in people with hypersensitivity to the active substance or to any of the excipients. 27 Special warnings and precautions for use of PLDH include cardiac toxicity, myelosuppression, and infusion-associated reactions. 27 It is recommended that all patients receiving PLDH routinely undergo frequent electrocardiogram (ECG) monitoring. 27
The most common undesirable adverse effect associated with PLDH (50 mg/m2 every 4 weeks) treatment in breast cancer and ovarian cancer RCTs was palmar–plantar erythrodysaesthesia (PPE), which is characterised by painful, macular, reddening skin eruptions. 27 The overall incidence of PPE was 44.0–46.1%. 27 These effects were reported to be predominantly mild, with severe (grade III) cases reported in 17–19.5% of patients. 27 The reported incidence of life-threatening (grade IV) cases was < 1%. 27
In patients with ovarian cancer, the most common adverse effects (reported by at least 1 out of 10 patients) associated with PLDH treatment were leucopenia; anaemia; neutropenia; thrombocytopenia; anorexia; constipation; diarrhoea; nausea; stomatitis; vomiting; PPE; alopecia; rash; asthenia; and mucous membrane disorder. Clinically significant laboratory abnormalities associated with PLDH included increases in total bilirubin (usually in patients with liver metastases) (5%) and serum creatinine levels (5%). 27
Paclitaxel
Paclitaxel is a taxane, a class of drugs isolated from the Pacific yew tree (Taxus brevifolia). 32 Paclitaxel targets a protein that is a key component of microtubules. Microtubules are important in various cellular processes, including the initiation of DNA synthesis. Unlike other taxanes, which inhibit microtubule assembly, paclitaxel stabilises the microtubule polymer, protecting the microtubule from disassembly and, therefore, further involvement in cellular processes.
In the UK, paclitaxel is licensed as first-line chemotherapy in combination with cisplatin or carboplatin for ovarian cancer patients with advanced carcinoma of the ovary or with residual disease (> 1 cm) after initial laparotomy. 33 Paclitaxel is also licensed as a second-line chemotherapy for ovarian cancer after failure of standard, platinum-containing therapy. 33 The recommended dose for paclitaxel when used as a second- and subsequent-line treatment is 175 mg/m2 administered over a period of 3 hours, followed by a platinum-based compound, with a 3-week interval between courses of treatment. 33 Prior to treatment with paclitaxel, patients should undergo pre-treatment with corticosteroids, antihistamines and H2-receptor antagonists. 33
Paclitaxel is contraindicated during lactation and should not be used in patients with a baseline neutrophil count of < 1500/mm3. 33 Special warnings and precautions for use of paclitaxel include hypersensitivity reactions and bone marrow suppression (primarily neutropenia). 33 Patients with hepatic impairment may be at increased risk of toxicity, particularly grade 3 and grade 4 myelosuppression. 33
The most common adverse effects associated with paclitaxel (reported by at least 1 out of 10 patients) are infection (mainly urinary tract and upper respiratory tract infections); myelosuppression; neutropenia; anaemia; thrombocytopenia; leucopenia; bleeding; minor hypersensitivity reactions (mainly flushing and rash); neurotoxicity (mainly peripheral neuropathy); hypotension; diarrhoea; vomiting; nausea; mucosal inflammation; alopecia; arthralgia; and myalgia. 33
Trabectedin
Trabectedin is a synthetic antineoplastic drug, the structure of which is derived from a natural product originally extracted from the marine Caribbean tunicate (‘sea squirt’, a marine animal) Ecteinascidia turbinata. 34 Trabectedin binds to the minor groove of DNA, a process that triggers various events that affect multiple transcription factors, DNA binding proteins and DNA repair pathways, and ultimately results in disruption of the cell cycle.
Trabectedin in combination with PLDH is licensed for the treatment of patients with relapsed platinum-sensitive ovarian cancer. 35 PLDH is administered first at a dose of 30 mg/m2, immediately followed by administration of trabectedin as a 3-hour infusion at a dose of 1.1 mg/m2. The recommended interval between treatment cycles is 3 weeks. To minimise the risk of PLDH infusion reactions, the initial dose of PLDH is administered at a rate of no greater than 1 mg/minute. 35 If no infusion reaction is observed, subsequent PLDH infusions may be administered over a 1-hour period.
All patients should be treated with corticosteroids 30 minutes before administration of PLDH (in combination therapy) or trabectedin (when used as a monotherapy). 35 Corticosteroids not only act as antiemetic prophylaxis, but also seem to afford hepatoprotective effects. 35
Trabectedin is contraindicated in:35
-
people who are hypersensitive to trabectedin or to any of the excipients
-
people who have concurrent serious or uncontrolled infection
-
people who are breastfeeding
-
concomitant combination with yellow fever vaccine.
Patients must meet specific criteria on hepatic function parameters before treatment (or re-treatment) with trabectedin can commence. 35 If patients do not meet the criteria listed below, treatment must be delayed for up to 3 weeks until the required levels are reached. Patients must have:
-
absolute neutrophil count of ≥ 1500/mm3
-
platelet count of ≥ 100,000/mm3
-
bilirubin level of less than or equal to the upper limit of normal (ULN)
-
alkaline phosphatase level of ≤ 2.5 × ULN
-
albumin level of ≥ 5 g/l
-
alanine transaminase and aspartate transaminase levels of ≤ 2.5 × ULN
-
creatinine clearance rate of ≥ 30 ml/minute (monotherapy), serum creatinine level of ≤ 1.5 mg/dl (≤ 132.6 µmol/l) or creatinine clearance rate of ≥ 60 ml/minute (combination therapy)
-
creatine phosphokinase level of ≤ 2.5 × ULN
-
haemoglobin level of ≥ 9 g/dl.
Additional special warnings and precautions for use of trabectedin include hepatic impairment; renal impairment; neutropenia; thrombocytopenia; nausea; vomiting; rhabdomyolysis; severe elevations of creatine phosphokinase level (> 5 × ULN); liver function test abnormalities; and injection site reactions. 35
The most common adverse effects associated with trabectedin (reported by at least 1 out of 10 patients) are neutropenia; leucopenia; anaemia; thrombocytopenia; anorexia; nausea; vomiting; constipation; stomatitis; diarrhoea; hyperbilirubinaemia; increase in alanine aminotransferase; increase in aspartate aminotransferase; increase in blood alkaline phosphatase; PPE syndrome; alopecia; fatigue; asthenia; mucosal inflammation; and pyrexia. 35
Gemcitabine
Gemcitabine is an analogue of the nucleoside deoxycytidine; in cells, nucleosides are modified enzymatically to produce nucleotides, which are the building blocks of RNA and DNA. As a nucleoside analogue, gemcitabine is a prodrug and, as such, once transported into a cell undergoes modification to produce the active form. 36 The activated form of gemcitabine replaces one of the nucleosides essential for DNA replication. Incorporation of the modified form of gemcitabine onto the growing DNA strand blocks further DNA synthesis and leads to apoptosis (cell death). 36
Gemcitabine is licensed for the treatment of patients with locally advanced or metastatic epithelial ovarian carcinoma, in combination with carboplatin, in patients with relapsed disease after a recurrence-free interval of at least 6 months after platinum-based, first-line therapy. 37 Gemcitabine in combination with carboplatin for the treatment of recurrent ovarian cancer has not as yet been evaluated by NICE as part of the single technology appraisal (STA) process. When used as a treatment for recurrent ovarian cancer, it is recommended that gemcitabine be administered at a dose of 1000 mg/m2 as a 30-minute i.v. infusion on days 1 and 8 of each 21-day cycle. 37 Carboplatin should be administered after gemcitabine on day 1 of the cycle, and at a dose consistent with a target area under the curve (AUC) of 4.0 mg/ml/minute. Dosage reduction with each cycle or within a cycle may be applied based on the grade of toxicity experienced by the patient. 37
Gemcitabine is contraindicated in people who are hypersensitive to the active substance or to any of the excipients and in those who are breastfeeding. 37 Prolongation of the infusion time of gemcitabine and increased dosing frequency have been shown to increase toxicity. 37 Additional special warnings and precautions for use of gemcitabine include haematological toxicity, hepatic insufficiency, concomitant radiotherapy, use with concomitant live vaccinations (e.g. yellow fever), risk of cardiac and/or vascular disorders, pulmonary effects, renal effects, and effects on sodium levels.
The most common adverse effects (reported by at least 1 out of 10 patients) associated with gemcitabine treatment are leucopenia; bone marrow suppression (typically mild to moderate); thrombocytopenia; anaemia; dyspnoea (usually mild and passes rapidly without treatment); vomiting; nausea; elevation of liver transaminases and alkaline phosphatase; allergic skin rash; haematuria; mild proteinuria; influenza-like symptoms; and oedema/peripheral oedema. 37
Chapter 2 Definition of the decision problem
Decision problem
Population including subgroups
The population of interest is people with ovarian cancer that has recurred after first-line (or subsequent) platinum-based chemotherapy or that is refractory to platinum-based chemotherapy.
Subgroups of particular interest are:
-
people with platinum-sensitive recurrent ovarian cancer (i.e. relapse at ≥ 6 months after completion of initial platinum-based chemotherapy), who will be divided further, evidence permitting, into those with partial (i.e. relapse within 6–12 months) and those with full platinum sensitivity (i.e. relapse at ≥ 12 months)
-
people with platinum-resistant (i.e. relapse within 6 months of completion of initial platinum-based chemotherapy) or platinum-refractory (i.e. disease that does not respond to initial platinum-based chemotherapy) recurrent ovarian cancer
-
those who are allergic to platinum-based treatment.
Interventions
The technology assessment report considers five interventions used within their licensed indication:
-
paclitaxel alone or in combination with platinum chemotherapy
-
PLDH alone or in combination with platinum chemotherapy
-
gemcitabine in combination with carboplatin
-
trabectedin in combination with PLDH
-
topotecan.
As per the final protocol,38 the clinical effectiveness and cost-effectiveness of the five interventions of interest have been evaluated in the prespecified subgroups listed above (see Population including subgroups). Interventions of interest for the individual subgroups are presented in Table 4.
| Population | Interventions of interest |
|---|---|
| Platinum sensitive |
|
| Platinum resistant or platinum refractory |
|
| People who are allergic to platinum-based compounds |
|
Relevant comparators
As per the final protocol,38 the relevant comparators have been evaluated based on the prespecified subgroups listed above (see Population including subgroups). Comparators of interest listed by individual subgroup are presented in Table 5.
| Population | Comparators of interest |
|---|---|
| Platinum sensitive |
|
| Platinum resistant or platinum refractory |
|
| People who are allergic to platinum-based compounds |
|
In the final protocol, bevacizumab in platinum-containing chemotherapy was listed as a potential comparator of interest for platinum-sensitive patients subject to appraisal by NICE. 38 Subsequent to finalisation of the protocol, the outcome of the NICE STA was not to recommend bevacizumab in combination with gemcitabine and carboplatin for the treatment of first recurrence of platinum-sensitive ovarian cancer. 16 Therefore, bevacizumab in platinum-containing chemotherapy has not been evaluated as a comparator in this group of patients.
Outcomes
The outcomes of interest considered for this review included:
-
OS
-
progression-free survival (PFS)
-
overall response rate (ORR)
-
adverse effects of treatment
-
HRQoL.
In addition to PFS, although not listed in the final protocol, time to progression (TTP) was also analysed in the evaluation of clinical effectiveness.
Overall aims and objectives of assessment
The purpose of this report is to assess the clinical effectiveness and cost-effectiveness of paclitaxel (monotherapy or in combination with platinum-based chemotherapy), PLDH (monotherapy or in combination with platinum-based chemotherapy), gemcitabine in combination with carboplatin, trabectedin in combination with PLDH, and topotecan as a monotherapy within their licensed indications for the treatment of advanced ovarian cancer that has relapsed after first-line treatment with a platinum-based regimen.
This report contains reference to confidential information provided as part of the NICE appraisal process. This information has been removed from the report and the results, discussions and conclusions of the report do not include the confidential information. These sections are clearly marked in the report.
Chapter 3 Assessment of clinical effectiveness
Methods for reviewing effectiveness
Evidence for the clinical effectiveness of topotecan, PLDH, paclitaxel, trabectedin and gemcitabine was assessed by conducting a systematic review of published research evidence. The review was undertaken following the general principles published by the Centre for Reviews and Dissemination (CRD). 39 The protocol for the systematic review was registered on PROSPERO (registration number CRD42013003555).
Identification of studies
The literature search for this review was designed to update and expand the systematic search carried out in TA91, which evaluated the clinical effectiveness and cost-effectiveness of topotecan, PLDH and paclitaxel. 13 Medical subject heading (MeSH) and text terms for ovarian cancer, topotecan, PLDH and paclitaxel were taken from the search strategy presented in TA91, and text terms added for the interventions trabectedin and gemcitabine. To ensure the capture of all potentially relevant studies to inform a network meta-analysis (NMA), the decision was taken not to restrict the start date of the update search to the end date of the search (2004) reported in TA91.
As a result of the large number of studies retrieved from the scoping search, the decision was taken to implement search filters for RCT. Filters developed and validated by Scottish Intercollegiate Guidelines Network were used. 40 The identified RCTs facilitated construction of three distinct networks for the outcomes of OS and PFS for both the platinum-sensitive (two networks) and platinum-resistant/-refractory (PRR) (one network) subgroups. In an attempt to identify a study to link the discrete networks for the platinum-sensitive subgroup, the retrieved abstracts were re-examined to consider interventions that were outside the scope of this review. Owing to time constraints, the decision was taken not to search for non-randomised trials. Bibliographies of previous reviews and retrieved articles were searched for additional studies. Clinical trial registries were also searched to identify planned, ongoing and finalised clinical trials of interest. In addition, clinical experts were contacted with a request for information on any additional studies of which they had knowledge. The manufacturer submissions (MSs) were assessed for unpublished data. Electronic databases searched were EMBASE, MEDLINE® and Cochrane Central Register of Controlled Trials (CENTRAL). Although the protocol stipulates that the Index of Scientific and Technical Proceedings would be searched to identify relevant conference proceedings, owing to time constraints this was not undertaken. However, based on the conference abstracts retrieved from the search of the prespecified electronic databases, the Technology Assessment Group (TAG) considers it likely that the key conference abstracts have been identified. Conference abstracts that were reviewed and found not to report additional results to those presented in the relevant full publication were excluded.
Electronic databases were initially searched on 18 January 2013 and results uploaded into Reference Manager version 11.0 (Thomson Research Soft, San Francisco, CA, USA) and deduplicated. An update search was carried out on 23 May 2013. No papers or abstracts published after this date were included in the review. Full details of the strategies are presented in Appendix 1.
Titles and abstracts returned by the search strategy were examined independently by two researchers (SB and TK) and screened for possible inclusion. Disagreements were resolved by discussion, or involvement of a third reviewer (SJE) in cases for which consensus could not be achieved. Full texts of potentially relevant studies were ordered. Full publications were assessed independently by two reviewers (SB and TK/AS) for inclusion or exclusion against prespecified criteria, with disagreements resolved by discussion or input from a third reviewer when consensus could not be achieved.
Inclusion and exclusion criteria
For the review of clinical effectiveness, only RCTs were considered for inclusion in the review. Systematic reviews and non-randomised studies were excluded, as were studies that considered drugs administered as ‘maintenance therapy’ following directly on from first-line therapy without evidence of disease progression. Inclusion criteria were based on the decision problem outlined in Chapter 2 (see Decision problem) (presented as a whole in Table 6). No restrictions were imposed on language or date of publication. Reference lists of identified systematic reviews were used as a source of potential additional RCTs, as well as a resource to compare studies retrieved from the systematic literature search.
As in TA91,13 second-line chemotherapy was defined as the second chemotherapy regimen, administered either as a result of relapse after first-line therapy or immediately following on from first-line therapy in patients with progressive disease (PD) or stable disease (SD). The definition applied in cases where the second-line regimen comprised the same treatments as the first-line regimen.
For the purposes of this review, based on expert opinion, supportive care was defined as treatment for recurrent ovarian cancer that does not have an anti-tumour mode of action.
| PICO | Inclusion criteria |
|---|---|
| Study design | RCTs |
| Population | People with ovarian cancer that has recurred after first-line (or subsequent) platinum-based chemotherapy or is refractory to platinum-based chemotherapy |
| Interventions | For people with platinum-sensitive ovarian cancer:
|
| Comparators | For people with platinum-sensitive ovarian cancer:
|
Data abstraction strategy
Data pertaining to study design, methodology, baseline characteristics, and clinical outcomes efficacy were extracted by two reviewers (TK/AS) into a standardised data extraction form and validated by a second (SB). Discrepancies were resolved by discussion when necessary. Authors of reports published as meeting abstracts only, where insufficient methodological details were reported to allow critical appraisal of study quality were contacted with a request for additional information. If no additional information was obtained, the studies were excluded. Data abstraction forms for the included studies are provided in Appendix 2.
Critical appraisal strategy
The quality of the clinical effectiveness data was assessed by two independent reviewers (TK and SB) and checked for agreement. The study quality was assessed according to recommendations by the NHS CRD39 and Cochrane Handbook for Systematic Reviews of Interventions41 and recorded using the Cochrane risk of bias tool. 41
Methods of data synthesis
Details of results on clinical effectiveness and quality assessment for each included study are presented in structured tables and as a narrative summary. The possible effects of study quality on the clinical effectiveness data and review findings are discussed. The 16 RCTs identified evaluated 14 different pairwise comparisons. Therefore, there were insufficient data for most comparisons to carry out a standard pairwise meta-analysis. However, the TAG determined that the data identified were sufficiently homogeneous to investigate comparative effectiveness of interventions via a NMA. The methods used for the NMA followed the guidance described in the NICE Decisions Support Units (DSUs) Technical Support Documents (TSDs) for Evidence Synthesis. In essence, a NMA assumes that each trial included in the network could have potentially included all treatments of interest but that some of these treatments are missing completely at random (MCAR). To illustrate this further, in a simple indirect comparison of three treatments A, B and C, the trials of A compared with B and of B compared with C are assumed to have been potentially trials of A compared with B compared with C but where one arm from each trial is MCAR. In this example, an estimate of the relative treatment effect of A compared with C can be inferred using treatment B as a common comparator.
The TAG conducted a NMA for each network using a Bayesian Markov chain Monte Carlo simulation in WinBUGS version 1.43 (MRC Biostatistics Unit, Cambridge, UK). The following were implemented for each analysis:
-
Uninformed priors (also called ‘flat’ priors) were used.
-
All outcomes were considered independent. For example, although OS and PFS might be correlated in advanced ovarian cancer, the degree of correlation is unlikely to be derived from summary trial estimates provided in published papers. 42 As such, in the absence of individual patient data (IPD), the TAG took the pragmatic approach of assuming all efficacy and safety outcomes were independent.
-
Results for all efficacy outcomes analysed were based on 50,000 iterations after a ‘burn-in’ of 30,000 iterations. For safety outcomes all analyses had a ‘burn-in’ of 30,000 iterations, with results based on 100,000 iterations.
-
Summary effect estimates for OS and PFS were hazard ratios (HRs), whereas ORR and all safety outcomes used odds ratios (ORs) as summary effect estimates.
-
As a result of disparity in HRs reported in the identified trials, in terms of unadjusted HRs compared with adjusted HRs, together with variation in adjustment factors, for consistency the TAG used only unadjusted HRs in the NMA.
-
Any results taken forward into the economic model (see Chapter 4, Treatment effectiveness) used the posterior sampling to retain the correlation between parameter estimates caused by their joint estimation from a single data set. 43
However, the ability of the TAG to conduct NMAs was limited by the low number of trials identified (typically only one trial per treatment comparison). The constraints imposed by the limited number of trials available for analysis were:
-
Implementation of a fixed-effects model for all analyses. Although it was planned that fixed- and random-effects models would be explored and the model with the lowest deviance information criterion selected as the preferred model, the sparse number of trials available necessitated the use of a fixed-effects model. Using an uninformed prior for the between trial heterogeneity in a random-effects model ‘overwhelmed’ the influence of the available data for analysis with the posterior estimation of tau approximating the prior value used. Identification of an alternative source for the prior, for example from an existing systematic review, was explored but no suitable review was identified. 43 As such, despite the potential clinical heterogeneity from two studies, which are discussed in detail later (see Comparability of baseline characteristics), the TAG made the pragmatic decision to use a fixed-effects model in the absence of any reliable estimate available.
-
Disconnected networks identified for each outcome assessed. The trials identified in the clinical systematic review were unable to populate a single network for any of the outcomes assessed. A wider selection of treatments was assessed, as the systematic review was conducted in such a way as to identify all trials with at least one intervention of interest present. Unfortunately this did not uncover trials that could link the disconnected networks together. 44 In addition, the TAG’s clinical advisors did not consider any of the suggested assumptions to link the disconnected networks together to have face validity.
-
Heterogeneity and inconsistency. The networks constructed, typically ‘linear’ in nature, and the sparse number of trials available, typically only one per pairwise comparison, prevented the TAG from exploring any potential heterogeneity or inconsistency in each analysis.
The potential impact of these limitations is discussed where the results are reported.
Manufacturer submissions
All data submitted by the manufacturers were assessed. Data presented that met the inclusion criteria, and had not been identified in another published source, were extracted and quality assessed in accordance with the procedures outlined in this protocol. Economic evaluation included in the MSs, which complied with NICE’s advice on presentation, was assessed for clinical validity, reasonableness of assumptions and appropriateness of the data used in the economic model (see Description and critique of manufacturer submitted evidence).
Interpreting the results from the clinical trials
Clinical effectiveness
For the outcomes of OS and PFS/TTP, which are time-to-event outcomes, most trials identified evaluated comparative clinical effectiveness using a HR, which is the ratio of the hazard (e.g. death or progression) rate between two groups. Typically, a reported HR of < 1 indicates that the event of interest is occurring more slowly in the experimental group compared with the control group. In some trials identified, HR of > 1 (i.e. event occurs more frequently in the experimental group) is reported to favour a treatment. In these instances, the event recorded is not the hazard but the opposite event, i.e. survival or no progression over time. For the purposes of this review, PFS and TTP have been reported and evaluated under the outcome heading of PFS. Many trials identified also assess the extent to which a tumour shrinks compared with initial size, which is the response rate. Response rate is a dichotomous event (i.e. patients either respond or do not respond) and is reported as the proportion of patients achieving a response according to prespecified criteria.
Adverse effects
Many trials evaluating chemotherapeutic treatments categorise the severity of adverse effects based on criteria developed by the National Cancer Institute (NCI), one aim of which was to standardise reporting of adverse effects. 45 According to the National Cancer Institute Common Toxicity Criteria (NCI-CTC),46 adverse effects are graded from 0 to 5, with increasing grade indicating more severe adverse effects (Table 7). The NCI-CTC also provides a detailed list of adverse effects commonly occurring in oncology trials, together with clinical descriptions on grade of severity that are specific for each adverse reaction.
| Grade | Degree of severity |
|---|---|
| 1 | Mild; with no or mild symptoms; no interventions required |
| 2 | Moderate; minimal intervention indicated; some limitation of activities |
| 3 | Severe but not life-threatening; hospitalisation required; limitation of patient’s ability to care for him/herself |
| 4 | Life-threatening; urgent intervention required |
| 5 | Death related to AE |
Results
The RCTs meeting the inclusion criteria are discussed in the sections that follow. Initially, a summary of the quantity and quality of the evidence is provided, together with a table presenting an overview of the included trials. Additionally, a more detailed narrative description, together with an overview of trial quality, for each included trial is presented, including those trials previously identified in TA91. 13 A narrative description of population baseline characteristics and potential imbalances are discussed for each trial. Owing to the number of trials identified, baseline characteristics are not tabulated within the main body of the report but are provided within the data abstraction forms in Appendix 2. Instead, baseline characteristics for key prognostic factors in recurrent ovarian cancer (age, number of prior lines of chemotherapy, interval since last chemotherapy, and performance score) are presented for included trials in a summary table (see Table 10).
Clinical effectiveness results are reported by outcome (OS, PFS, ORR, QoL and adverse effects). Within the efficacy outcomes of OS, PFS and ORR, results are presented separately based on platinum sensitivity. Results by population are ordered: platinum sensitive, which is broken down further to fully platinum sensitive (FPS) and PPS, when data are available; PRR; and the overall population (when trial includes patients with platinum-sensitive disease or PRR disease). Results for QoL and adverse effects are presented for the overall population, irrespective of sensitivity to platinum. Within the outcome, results are initially presented separately for each trial reporting data, and are supplemented with the findings from the NMA, including a description of assumptions made and potential bias across the trials included in the network.
Quantity and quality of research available
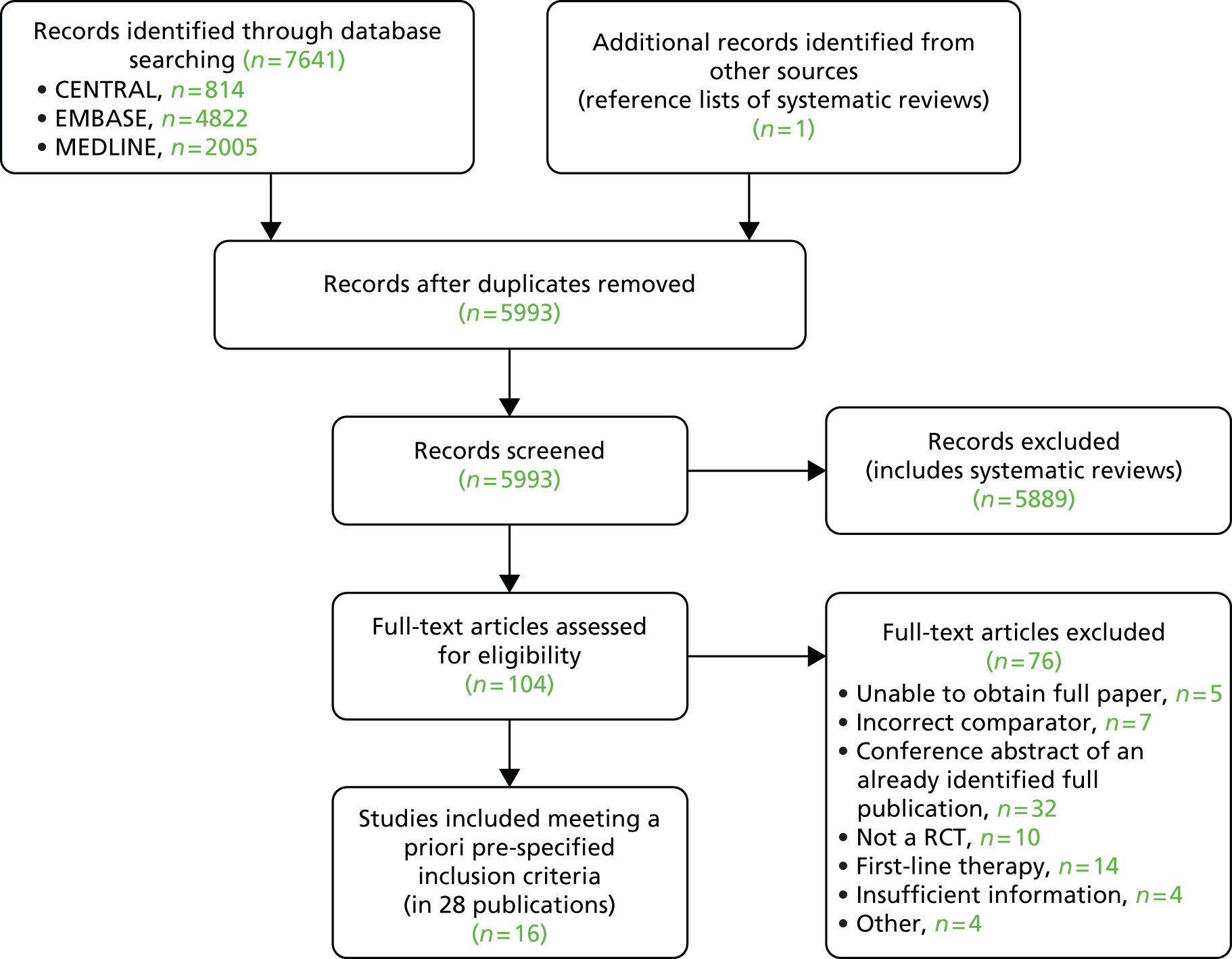
The searches retrieved a total of 5993 records (post deduplication) that were of possible relevance to the review (Figure 3). These were screened and 104 full references were ordered. Of these, five had to be cancelled because they were unobtainable. Of the full references evaluated, 28 papers describing 16 studies were included in the review.
FIGURE 3.
A PRISMA flow diagram for studies included and excluded from the clinical effectiveness review.
The full list of studies included in the review is given below (see Table 8), whereas a list of the papers screened but subsequently excluded (with reasons for exclusion) from the review is presented in Appendix 3.
Included studies
Sixteen RCTs reported in 15 primary publications, with 13 accompanying publications, were included in the review. One RCT from TA9113 was included, which was identified in the literature search only as an abstract and the results of which have not been published in full elsewhere (referred to hereafter as Trial 30–57). 47 An overview of the identified trials is provided in Table 8. Of the 16 RCTs identified, five evaluated the intervention and comparator within their licensed indication, and dose and route of administration. 13,21,48–50 The remaining 11 RCTs evaluated the intervention or comparator outside the parameters specified in the licence, in terms of, for example, dose or route of administration. No RCT identified evaluated interventions specifically in a population who were allergic or intolerant to platinum-based treatments. Of the nine RCTs identified in TA91, only one RCT51 has been excluded from this update. Cantu et al. 51 evaluated paclitaxel alone compared with a combination of cyclophosphamide, doxorubicin and cisplatin (CAP). Doxorubicin administered in the trial is the non-pegylated formulation and is outside the scope of this review, which specifies PLDH as the intervention of interest.
Two manufacturers [Eli Lilly and Company (gemcitabine); PharmaMar (trabectedin)] submitted clinical evidence for consideration for this multiple technology appraisal (MTA).
Eli Lilly (gemcitabine) did not carry out a systematic review of the literature; instead, the manufacturer reported clinical data from three studies:
-
a Phase III study comparing gemcitabine plus carboplatin with carboplatin monotherapy in patients with platinum-sensitive, recurrent ovarian cancer (study JHQJ)
-
a single-arm, Phase II study of gemcitabine plus carboplatin in platinum-sensitive, recurrent ovarian cancer (study JHRW)
-
a single-arm, Phase I/II dose-finding study of gemcitabine plus carboplatin in platinum-sensitive, recurrent ovarian cancer (study SO026).
The data provided by the manufacturer for JHQJ, the Phase III study comparing gemcitabine plus carboplatin with carboplatin monotherapy, are reported in the full publication of the trial,50 which was identified and included as part of the systematic review of the literature on clinical effectiveness (see Results, above).
The two additional studies (JHRW and SO026) are single-arm trials and as such do not meet the criteria for inclusion in the review (see Results, above).
PharmaMar (trabectedin) carried out a systematic search of the literature. Specifically, the manufacturer updated the review carried out for the STA TA222,15 which evaluated the use of trabectedin plus PLDH in the treatment of platinum-sensitive ovarian cancer. The manufacturer searched the following databases: EMBASE; MEDLINE; MEDLINE® In-Process & Other Non-Indexed Citations; and The Cochrane Library. Studies were included if:
-
the study type was a RCT
-
the population of interest was relapsed platinum-sensitive ovarian cancer
-
outcome data for PFS, OS or AEs were included
-
the interventions and comparators of interest included at least one of trabectedin, PLDH, paclitaxel, topotecan, etoposide, or best supportive care.
The manufacturer limited the comparators within searches to the comparators outlined in the NICE pathway for patients who are unsuitable for platinum-based chemotherapy; this represents the target population for the MS.
The manufacturer identified two additional relevant studies relating to OVA-301,30 which were identified as part of the review of the clinical effectiveness literature and are discussed in a subsequent section (see subsequent text).
| Study and principal citation | Trial design | Population (n) | PFI | Randomised treatments | Supplementary publications | |
|---|---|---|---|---|---|---|
| Intervention | Comparator | |||||
| Both intervention and comparator used within licensed indication and at licensed dose | ||||||
| ten Bokkel Huinink et al.21 | Phase III, multicentre, open-label RCT | 235 | Disease that recurred or progressed after first-line platinum based therapy (no minimum PFI specified) | Topotecan (1.5 mg/m2 as a 30-minute i.v. infusion) for five consecutive days every 21 days | Paclitaxel (175 mg/m2 as a 3-hour i.v. infusion) every 21 days | ten Bokkel Huinink et al.52 Gore et al.53 |
| Gordon et al.49 | Phase III, multicentre, open-label RCT | 474 | Disease that recurred after, or failed, first-line platinum-based chemotherapy (no minimum PFI specified) | PLDH (50 mg/m2 as a 1-hour i.v. infusion) every 28 days | Topotecan (1.5 mg/m2 as a 30-minute infusion) for five consecutive days every 21 days | Gordon et al.54 |
| Trial 30–57; data taken from TA9113 | Phase III, multicentre, open-label RCT | 216 | Disease that recurred after, or failed, one platinum-based first-line regimen (no minimum PFI specified) | PLDH (50 mg/m2) every 28 days | Paclitaxel (175 mg/m2) every 21 days | One conference abstract (O’Byrne et al.47) |
| Gonzalez-Martin et al.48 | Phase II, ‘pick the winner’ design, multicentre RCT; level of masking unclear | 81 | Progression > 6 months after completion of platinum-based chemotherapy | Paclitaxel (175 mg/m2 as a 3-hour i.v. infusion) plus carboplatin (AUC 5) every 3 weeks | Carboplatin alone (AUC 5) every 3 weeks | None identified |
| Pfisterer et al.50 | Phase III, multicentre, international, open-label RCT | 356 | Disease recurrence at least 6 months after completion of first-line, platinum-based therapy | Gemcitabine (1000 mg/m2) plus carboplatin (AUC 4) every 21 days | Carboplatin alone (AUC 5) every 21 days | None identified |
| Intervention or comparator used outside licensed indication or dose | ||||||
| Gore et al.24 | Multicentre, open-label RCT (phase not clear) | 266 | Disease progression on first-line platinum-based chemotherapy or relapse within 12 months of completion of first-line platinum-based treatment | Oral topotecan (2.3 mg/m2) given daily | Intravenous topotecan (1.5 mg/m2) for five consecutive days every 21 days | None identified |
| Sehouli et al.23 | Phase II, multicentre RCT | 194 | Disease that had recurred after radical surgery and at least one platinum-based chemotherapy with recurrence at < 6 months after cessation of platinum-based treatment | Topotecan (4.0 mg/m2 as a 30-minute i.v. infusion on days 1, 8 and 15) weekly every 28 days | Topotecan (1.25 mg/m2 as a 30-minute i.v. infusion) for five consecutive days every 21 days | None identified |
| Alberts et al.28 | Phase II RCT; level of masking unclear | 61 | Disease that recurred within 6–24 months of completing platinum-based chemotherapy | PLDH (30 mg/m2 as a 1-hour i.v. infusion) plus carboplatin (AUC 5) every 4 weeks | Carboplatin alone (AUC 5) every 4 weeks | Markman et al.55 |
| Bafaloukos et al.29 | Phase II RCT; level of masking unclear | 204 | Recurrence at > 6 months after completion of platinum-based chemotherapy | PLDH (45 mg/m2 as a 90-minute i.v. infusion) plus carboplatin (AUC 5) every 4 weeks | Paclitaxel (175 mg/m2 as a 3-hour i.v. infusion) plus carboplatin (AUC 5) every 21 days | None identified |
| CALYPSO ; Pujade-Lauraine et al.31 | Phase III, non-inferiority, multicentre, international, open-label RCT | 976 | Disease that had recurred/progressed > 6 months after first- or second-line platinum-based chemotherapy | PLDH (30 mg/m2 as an i.v. infusion) plus carboplatin (AUC 5) every 28 days | Paclitaxel (175 mg/m2 as an i.v. infusion) plus carboplatin (AUC 5) every 21 days | Wagner et al.,56 Gladieff et al.,57 Kurtz et al.,58 Brundage et al.59 |
| Rosenberg et al.60 | Multicentre RCT (phase not clear) | 208 | Disease that recurred or progressed after first-line platinum-based therapy (no minimum PFI specified) | Paclitaxel (67 mg/m2) weekly | Paclitaxel (200 mg/m2) every 21 days | None identified |
| ICON4/AGO-OVAR 2.2; Parmar et al.61 | Phase III, multicentre, international RCT (three parallel RCTs, each with its own protocol) | 802 | Disease that had been treatment free for > 6 months | Paclitaxel (175 or 185 mg/m2) plus carboplatin or cisplatin every 21 days | Carboplatin or cisplatin alone every 21 days | None identified |
| CARTAXHY; Lortholary et al.62 | Phase II, multicentre, open-label, three-armed RCTa | 165 | Disease progression during or relapse within 6 months of completing platinum-based chemotherapy | Paclitaxel (80 mg/m2 on days 1, 8 and 15) weekly plus carboplatin (AUC 5) every 28 days | Paclitaxel (80 mg/m2 on days 1, 8 and 15) weekly every 28 days | None identified |
| Piccart et al.63 | Phase II, open-label, multicentre RCT | 86 | Disease that progressed or stabilised after prior platinum-based treatment; for those experiencing relapse, relapse was to have occurred within 12 months of last platinum-based therapy | Paclitaxel (175 mg/m2 as a 3-hour infusion) every 21 days | Oxaliplatin (130 mg/m2 as a 2-hour infusion) every 21 days | None identified |
| OVA-301; Monk et al.30 | Phase III, open-label, multicentre, international RCT | 672 | Disease that was persistent, recurrent or progressing on current treatment | Trabectedin (1.1 mg/m2 as a 3-hour infusion) plus PLDH (30 mg/m2 as a 90-minute infusion) every 21 days | PLDH (50 mg/m2 as a 90-minute infusion) every 28 days | Monk et al.,64 Poveda et al.,65 Kaye et al.,66 Krasner et al.67 |
| Omura et al.68 | Phase III, multicentre RCT | 372 | Histologically confirmed ovarian cancer treated with no more than one prior platinum-based regimen and no prior taxane | Paclitaxel 250 mg/m2 (24-hour infusion) every 21 days (patients in this group also randomised to filgrastim (Neupogen®, Amgen) 5 or 10 µg/kg subcutaneously) | Paclitaxel 175 mg/m2 (24-hour infusion) every 21 days | None identified |
Study characteristics
Pegylated liposomal doxorubicin hydrochloride plus carboplatin compared with paclitaxel plus carboplatin
Two RCTs29,31 were identified for this comparison. The RCTs were of similar design but one was a Phase II RCT29 and one was a Phase III RCT. 31 In addition, the dose of PLDH evaluated differed between the trials, with 45 mg/m2 and 30 mg/m2 used by Bafaloukos et al. 29 and Pujade-Lauraine et al. ,31 respectively. The licence for PLDH does not recommend a dose of PLDH for use in combination with platinum-based chemotherapy. Bafaloukos et al. 29 note in the discussion that, at the time of initiation of the trial, limited information was available on the optimal dose for PLDH in combination with carboplatin. As highlighted by Bafaloukos et al. ,29 retrospective analyses suggest that lower dose intensities of PLDH (30–40 mg/m2) are as clinically effective but with improved tolerance. Clinical experts have fed back that, in UK clinical practice, PLDH would most likely be used at a dose of 30 mg/m2 when combined with carboplatin.
Bafaloukos et al. 29 report the results of a randomised study in which 204 patients with histologically confirmed recurrent ovarian cancer were randomised to either PLDH (45 mg/m2) plus carboplatin (AUC 5) every 28 days or paclitaxel (175 mg/m2) plus carboplatin (AUC 5) every 21 days. Patients recruited had disease that had recurred at least 6 months after platinum-based chemotherapy, i.e. women with platinum-sensitive disease. Women with only elevated CA125 levels (twice the ULN or more) as an indicator of disease were also included.
The primary aim of the study was to evaluate the comparative clinical effectiveness of the two treatment regimens in terms of response rate and toxicity in women with platinum-sensitive ovarian cancer relapsing after first-line platinum-based therapy. OS and TTP were analysed as secondary outcomes. Subsequent to randomisation, 15 patients were found to be ineligible (reasons provided). Therefore, analyses presented are based on data from 189 eligible patients (96 in the paclitaxel plus carboplatin group vs. 93 in the PLDH plus carboplatin group). The reported power calculation indicates that 201 patients were needed to identify a 20% difference in response rate between the groups. The study might have been underpowered to detect a difference between groups in response rate.
Randomisation (1 : 1) was performed at the central Hellenic Cooperative Oncology Group (HeCOG) Data Office in Athens but details on the method of randomisation were not reported. Stratification criteria were not applied at randomisation. Tumour response was evaluated using World Health Organization (WHO) criteria for patients with measurable disease and CA125 based on Rustin’s criteria for patients without measurable disease. Median duration of follow-up was reported as 43.6 months (95% CI 0.1 to 74.8 months), but the range of follow-up was not reported either for the full trial population or for the individual treatment groups.
All patients received standard premedication of dexamethasone, diphenhydramine and ranitidine (Zantac®, GSK) prior to paclitaxel. In the group receiving paclitaxel, premedication was administered twice, orally 12 hours before and again intravenously 30 minutes before paclitaxel infusion. In the group receiving PLDH, premedication was administered only intravenously prior to PLDH infusion. Six cycles of chemotherapy were administered, unless disease progression or unacceptable toxicity occurred. A maximum of 2 weeks’ delay was allowed for toxicity and treatment was discontinued if longer toxicity-related delays occurred. For grade 3 and grade 4 thrombocytopenia, a 25% and a 50% dose reduction, respectively, was recommended for all drugs.
A median of six cycles of paclitaxel plus carboplatin (range 1–9) and six cycles of PLDH plus carboplatin (range 1–8) were administered. Most patients in each group completed the planned treatment (68% in paclitaxel plus carboplatin and 70% in PLDH plus carboplatin).
In the second RCT identified for this comparison, Pujade-Lauraine et al. 31 report the results of a randomised international, multicentre, open-label, Phase III non-inferiority trial (CALYPSO) in which 976 patients with platinum-sensitive (disease progression > 6 months after prior treatment) relapsed/recurrent ovarian cancer received a combination of PLDH plus carboplatin (n = 467) or carboplatin plus paclitaxel (n = 509). Prior treatment must have included a taxane and no more than two previous platinum-based regimens (i.e. patients had failed first- or second-line treatment). Patients with measurable [according to Response Evaluation Criteria in Solid Tumours (RECIST)] and CA125 assessable [according to Gynecologic Cancer Intergroup (GCIG)] criteria were eligible.
The primary publication presents results on PFS. Accompanying publications were identified that present results on mature OS data,56 clinical effectiveness results in the subgroup of patients with PPS ovarian cancer (relapse at between 6 and 12 months since receipt of last cycle of chemotherapy)57 and QoL. 59
The trial was of a non-inferiority design with the aim of determining whether PLDH (30 mg/m2) plus carboplatin (AUC 5) every 4 weeks was non-inferior to the standard treatment of paclitaxel (175 mg/m2) plus carboplatin every 3 weeks. 31 The goal was to evaluate the comparative effectiveness of the treatments in terms of efficacy and toxicity. The primary outcome of the trial was PFS, with OS, QoL and toxicity as prespecified secondary outcome measures. Determination of disease progression was based on RECIST and GCIG criteria modifications, and included any of the following: occurrence (clinically or by imaging) of any new lesion; increase in measurable and/or non-measurable tumour defined by RECIST; CA125 elevation defined by GCIG criteria; health status deterioration attributable to disease; and death from any cause before progression was diagnosed. Assessments were independently reviewed. All patients were observed for at least 5 years from random assignment to assess OS.
Randomisation was in permuted blocks of six in a 1 : 1 ratio, and patients were stratified based on therapy-free interval from last chemotherapy (6–12 months vs. 12 months), measurable disease (yes vs. no) and centre. Despite randomisation, an imbalance in treatment allocation was noted (467 randomised to PLDH plus carboplatin vs. 509 randomised to paclitaxel plus carboplatin).
All patients received antiemetics, including a serotonin antagonist and corticosteroid. Patients randomly assigned to paclitaxel plus carboplatin received premedication to prevent hypersensitivity reactions. Dose delay and dose reduction were allowed for haematological and non-haematological toxicity. In the absence of unacceptable toxicity or disease progression, patients were treated for a total of six courses of therapy; if SD or PR was achieved after six courses of therapy, patients were allowed to remain on therapy until progression.
To assert non-inferiority of PLDH plus carboplatin, it was estimated that a sample size of 898 evaluable patients (estimate of 745 progression) would be required. 31 The calculation was based on non-inferiority margin with a HR of 1.23 at 15 months or a 7.9% absolute difference at 12 months (90% power and a one-sided CI of 95%).
Median follow-up was 22 months; median follow-up in the individual treatment groups not reported. 31 The median number of cycles was six in each treatment group, with a range of cycles from 1 to 14 in the PLDH plus carboplatin group and 1 to 12 in the paclitaxel plus carboplatin group. A significantly larger proportion of patients in the PLDH plus carboplatin group completed at least six cycles of treatment (85% vs. 77%; p < 0.001).
Pegylated liposomal doxorubicin hydrochloride plus carboplatin compared with carboplatin alone
Alberts et al. 28 reported the results of a randomised study in which 61 patients from the USA with recurrent stage III or i.v. epithelial or peritoneal ovarian carcinoma were randomised to PLDH (i.v. infusion of 30 mg/m2) plus carboplatin (AUC 5) once every 4 weeks (31 patients) or carboplatin (AUC 5) alone once every 4 weeks (30 patients). A follow-up study reporting final OS results was also identified. 55
To be eligible for enrolment, patients had to have histologically diagnosed stage III or IV disease that was determined to be progressive based on RECIST or GCIG CA125 criteria. Patients also had to have a progression-free interval and a PFI of 6–24 months after first-line platinum-based chemotherapy, which indicates that the study focused on women with platinum-sensitive disease. Patients were excluded if Zubrod performance status was > 1. Prior treatment with up to 12 courses of a non-platinum containing consolidation treatment during the 6- to 24-month PFI was allowed on the proviso that treatment had been completed at least 28 days prior to registration.
The primary aim of the study was to evaluate the comparative clinical effectiveness of the two treatment regimens in terms of OS in women with platinum-sensitive ovarian cancer. PFS, confirmed CR rate and time to treatment failure were analysed as secondary outcomes. Objective response and disease progression were defined according to standard RECIST criteria. 69 GCIG CA125 progression criteria were also implemented in defining disease progression. 70
Details on the method of randomisation were not reported, but randomisation was 1 : 1 to each group and was reported to be equal between the groups. Randomisation was stratified by disease measurability, number of disease sites and serous histology. The power calculation reported indicates that the study had initially planned to recruit 900 patients over a period of 4.5 years. However, as a result of slow patient accrual, the study closed early with only 61 patients enrolled. Initially designed as a Phase III RCT, results were reported as for a Phase II RCT. Median duration of follow-up was reported as 22.4 months, but the range of follow-up was not reported either for the full trial population or for the individual treatment groups. Markman et al. 55 reported a longer follow-up of the same trial. However, the duration of follow-up in this study is unclear.
Each treatment was given until progression, intolerable toxicity or a request from either the clinician or the patient to be removed from the study. Dose modifications were allowed, based on toxicity to PLDH. The maximum cumulative dose of PLDH was 600 mg/m2. Any patient with a compromised left ventricular ejection fraction (LVEF) (< 45% or decreases by a relative 20% from baseline) was removed from PLDH and continued on the carboplatin treatment. Carboplatin dose modifications were allowed for gastrointestinal and neurological toxicity. Patients with persistently greater than or equal to grade 2 peripheral neuropathy, despite dose reduction, were permanently taken off carboplatin treatments. The median number of treatment cycles given was 7 (range 1–18) for patients in the PLDH plus carboplatin group and 6 (range 2–16) for those in the carboplatin alone group. No major protocol violations were reported.
Trabectedin plus pegylated liposomal doxorubicin hydrochloride compared with pegylated liposomal doxorubicin hydrochloride alone
Monk et al. 30 report the results of an open-label, randomised, multicentre (124 centres in 21 countries), Phase III trial involving 672 women with recurrent ovarian cancer after failure of first-line platinum-based chemotherapy (OVA-301). Patients with platinum-resistant ovarian cancer (PFI < 6 months) or platinum-sensitive ovarian cancer (PFI ≥ 6 months) were eligible, but those who experienced progression during first-line therapy (platinum refractory) were excluded. Measurable disease by RECIST criteria69 was also an inclusion criterion. Related publications identified were a follow-up study reporting mature OS analysis,64 clinical efficacy results for the subgroup of patients with PPS65 and full results for QoL. 67
The aim of OVA-30130 was to compare the efficacy and safety of PLDH (30 mg/m2) plus trabectedin (1.1 mg/m2) every 21 days (n = 337) compared with PLDH (50 mg/m2) alone every 28 days (n = 335). The primary outcome was PFS, which was defined as time from randomisation to disease progression or death. Primary analysis of PFS was based on independent radiology review (radiological evaluation alone) by radiologists who were masked to treatment allocation. Secondary end points included OS, ORR (response maintained ≥ 4 weeks by RECIST69), and duration of response (calculated from date of first documentation of response to date of PD or death from PD). QoL was a tertiary outcome and was evaluated using the European Organisation for Research and Treatment of Cancer (EORTC) quality of life questionnaire C30 (QLQ-C30) and ovarian cancer-specific QLQ-OV28. 71 All efficacy analyses were based on the intention-to-treat (ITT) principle.
Randomisation was by a permuted block method (1 : 1 ratio) and patients were stratified by performance status [Eastern Cooperative Oncology Group (ECOG) score of 0 or 1 vs. 2] and platinum sensitivity (sensitive vs. resistant). After enrolment of 440 patients, and before central radiology review, the study was amended, changing the two primary efficacy end points, OS and PFS, to a single primary end point, PFS. OS became a secondary end point; the sample size remained unchanged. The sample size calculation indicated that 415 PFS events were required to test statistical difference between treatment groups with at least 90% power; it is reported that approximately 650 patients were to be randomised over 2 years.
Treatment was continued until disease progression or confirmation of CR and could be continued for two or more cycles beyond confirmed CR. A maximum of two dose reductions for each drug was allowed (in the trabectedin plus PLDH group, trabectedin could be reduced to 0.9 mg/m2 and subsequently to 0.75 mg/m2, and PLDH to 25 mg/m2, then to 20 mg/m2; in the PLDH group, PLDH could be reduced to 37.5 mg/m2 and then to 28 mg/m2). Median cumulative trabectedin dose was 5.6 mg/m2 (range 1–23 mg/m2). For PLDH, median cumulative PLDH dose was 154.4 mg/m2 (range 15–630 mg/m2) and 216 mg/m2 (range 3–1061 mg/m2) when administered in combination with trabectedin and as a monotherapy, respectively. Incidence of dose reductions was similar between groups, whereas cycle delays were less frequent with PLDH alone than trabectedin plus PLDH.
Median duration of follow-up in the initial publication was not reported30 but median follow-up in the longer-term study was 47 months. 64
The authors report that, despite stratification before randomisation, there was an imbalance between groups in mean baseline PFI that favoured PLDH alone (13.3 months with PLDH alone vs. 10.6 months with trabectedin plus PLDH; p = 0.009). Post hoc hypothesis-generating analyses on the influence of PFI on OS were carried out (discussed in Assessment of effectiveness).
It should be noted that use of trabectedin plus PLDH as an intervention in patients with PRR is not covered by the scope of this review. Clinical effectiveness data for only platinum-sensitive patients are presented.
Pegylated liposomal doxorubicin hydrochloride compared with topotecan
Gordon et al. 49 report the results of a Phase III randomised study comparing PLDH compared with topotecan in 474 women with histologically proven recurrent epithelial ovarian carcinoma that recurred after or did not respond to first-line platinum-based chemotherapy. The RCT was open label in design and was carried out at multiple centres (104 sites) in the USA and Europe. Patients with either measurable or assessable disease were included, where measurable disease was defined as presence of bidimensionally measurable lesions with clearly defined margins based on imaging scans and assessable disease was defined as unidimensionally measurable lesions by imaging scan in conjunction with serum CA125 levels of > 100 U/ml. A follow-up publication reported data on more mature OS, together with subgroup analyses based on platinum sensitivity. 54
Patients were randomised to receive either PLDH 50 mg/m2 as a 1-hour infusion every 28 days (239 patients) or topotecan 1.5 mg/m2 daily for five consecutive days every 21 days (235 patients). 49 In the absence of disease progression, treatment in each group could be continued for up to 1 year. Treatment could also continue if the patient demonstrated sustained clinical benefit. Patients who discontinued treatment after 6 months (six cycles of PLDH or eight cycles of topotecan) were considered protocol completed.
The study was described as randomised, but details on the method of randomisation were not reported. Patients were stratified for platinum sensitivity and for the presence or absence of bulky disease (tumour mass > 5 cm). Patients were classified as platinum sensitive if they had a PFI of > 6 months after first-line platinum-based chemotherapy and platinum refractory if they had SD, progressed during initial platinum-based therapy or relapsed within 6 months after completion of therapy. In the subsequent publication,54 analyses for OS and PFS for the subgroups of patients with PPS disease (PFI > 6 to ≤ 12 months) and FPS disease (PFI > 12 months) are presented. The authors report that the main outcome measures of efficacy were PFS and OS. Overall response rate (confirmed CR plus PR), time to response, duration of response, QoL, and safety and toxicity were also assessed. The study was designed with 80% power to demonstrate statistical equivalence between the two treatment groups. The initial sample size calculation found that a total of 350 assessable patients, 175 patients in each treatment group, would need to be randomised. To accommodate two interim analyses (necessitating 5% more patients) and anticipated loss of 20% of randomised patients who might not be assessable for efficacy end points, the sample size was increased to 460.
Protocol deviations included: (1) failure to meet entry criteria (seven patients receiving PLDH, two patients receiving topotecan); (2) patients who continued on study after first clinically significant change in LVEF (13 patients receiving PLDH); (3) patients who continued treatment after documented disease progression (40 patients receiving PLDH, 42 patients receiving topotecan); and (4) patients who completed fewer than eight cycles of treatment but were deemed protocol completed by the investigator (20 patients receiving topotecan).
Dose modifications were permitted. Reasons for reduction in PLDH dose included PPE, haematological toxicity, elevated bilirubin, stomatitis, or all other grade 3 and grade 4 events until resolution to grade 2 or lower. In the event of severe neutropenia during any cycle with topotecan, the dose was reduced by 0.25 mg/m2 for subsequent courses. Treatment with either drug was temporarily suspended or discontinued in cases of: disease progression; serious or intolerable AEs precluding further treatment; inability to tolerate study drug despite dose modification; LVEF of < 45% or a 20% decrease from baseline; and patient’s decision to withdraw participation or patients requiring radiation.
Median duration of follow-up was not reported in either publication. 49,54 In addition, information on mean or median number of cycles received in each treatment group was not provided. However, the mean cycle dose and cycle length for each treatment group were reported to be close to those specified in the protocol, indicating good compliance in following the dosing guidelines.
Pegylated liposomal doxorubicin hydrochloride compared with paclitaxel
In a publication available as only a conference abstract, O’Byrne et al. 47 provided a brief overview of a trial comparing PLDH compared with paclitaxel. The search did not retrieve a full publication of this study. However, as part of TA91,13 the manufacturer of PLDH (Schering-Plough) provided a full trial report as part of the industry submission. 72 The description of trial methodology and results for OS and adverse effects have been adapted from TA91. 13
The trial by Schering-Plough was a Phase III, randomised, open-label study involving 216 women with epithelial ovarian carcinoma after failure of first-line platinum-based chemotherapy. Additionally, to be eligible, women had to have measurable disease and be taxane naive. The trial was designed to compare the clinical effectiveness and safety of PLDH (50 mg/m2) every 28 days compared with paclitaxel (175 mg/m2) every 21 days.
Randomisation was carried out in a 1 : 1 ratio, with patient stratification by platinum sensitivity and bulky disease. No details on the method of randomisation are reported.
TA9113 reports that the planned enrolment was for 438 patients but only 216 were randomised (108 in each treatment arm), with the trial closing early due to poor accrual. It is thought that poor accrual was associated with the approval of Taxol for use in combination with platinum-based therapy for the first-line treatment of ovarian cancer by the European Agency for the Evaluation of Medicinal Products.
Patients were assessed weekly for haematological toxicities, and radiological imaging was repeated every 7–8 weeks to assess disease status. Patients achieving either a CR or PR were re-evaluated 4 weeks later to confirm the initial observation of response. All participants were to have been followed for a minimum of 1 year for survival and disease progression.
At baseline, the two treatment groups were balanced in terms of age, treatment-free interval (TFI), disease bulk, the number of previous chemotherapy regimens, the type of previous chemotherapy agents received, histology and performance status.
As a result of the low recruitment rate, efficacy analysis in TA9113 was limited to OS. AEs were also described.
Topotecan compared with paclitaxel
ten Bokkel Huinink et al. 21 report the results of an open-label, Phase III randomised study involving 235 patients with stage III/IV ovarian cancer, who had progressed during or after treatment with one platinum-based chemotherapy. The study was designed to compare the effectiveness and toxicity of topotecan (1.5 mg/m2) for five consecutive days every 21 days compared with paclitaxel (175 mg/m2) every 21 days. Enrolled patients had at least one bidimensionally measurable lesion as evidenced by CT or magnetic resonance imaging (MRI) scan, ultrasound or physical examination. Patients who had received more than one prior chemotherapy, or who had been previously treated with topotecan or paclitaxel, were ineligible. A second publication reporting more mature OS data was also identified. 52 A related study reports results from an analysis of patients who received third-line treatment during the trial, and specifically crossover therapy with the treatment received in the other group. 53
The primary outcome measures were response rate, duration of response and TTP. Response rate included CR or PR as a best response as determined by WHO criteria, with all responses independently reviewed by a radiologist who was masked to treatment allocation. Secondary outcome measures were time to response and OS. Of the 235 patients randomised, nine patients did not receive treatment and were excluded from analyses. An additional 24 patients were not evaluated for response, but were included in the calculation of response rate.
Randomisation was reported to be carried out by telephone, but details on the method of randomisation were not available. Patients were stratified by age (< 65 vs. ≥ 65 years), ascites (present vs. absent) and prior response to platinum-based therapy (resistant vs. early vs. interim vs. late response). Resistant disease was defined as no response to initial chemotherapy or having an initial PR or CR with subsequent progression while still receiving treatment. Early, interim and late response were defined as initial CR or PR with subsequent relapse within 3 months (early), 3–6 months (interim) or > 6 months (late) after cessation of chemotherapy.
Patients with a CR or PR continued treatment until either progression or 6 months past the maximal response; those who progressed were removed from the study. Those whose best response was SD after six cycles could be removed from the study or switched to the alternative regimen.
Patients on paclitaxel received premedication with dexamethasone and H1- and H2-receptor antagonists to prevent hypersensitivity. No premedication was initially given to those on topotecan but was allowed in subsequent cycles if nausea or vomiting occurred. Dose reductions in each group were permitted for toxicity. The minimum dose allowed was 1.0 mg/m2 per day for topotecan and 135 mg/m2 for paclitaxel; the dose of topotecan could also be escalated to a maximum of 2 mg/m2 per day. Patients were withdrawn from treatment if there was a > 2-week delay in treatment at the minimum dose of either medication because of toxicity. The target dose was achieved in 90% of cycles of topotecan and 98% of cycles of paclitaxel. Median number of cycles received was five in each group, with patients treated with topotecan receiving between 1 and 17 cycles compared with between 1 and 12 cycles for patients treated with paclitaxel.
A sample size calculation was not reported. Median duration of follow-up at the time of the first publication was unclear. 21 Median follow-up at the time of the publication reporting more mature OS data was reported in TA91 to be 58.5 weeks in the topotecan group (range 0–86 weeks) and 52.6 weeks in the paclitaxel group (range 0–117 weeks). 13
Gemcitabine plus carboplatin compared with carboplatin alone
Pfisterer et al. 50 report the results of a Phase III international, open-label randomised study assessing the comparative clinical effectiveness of gemcitabine (1000 mg/m2) plus carboplatin (AUC 4) (n = 178) compared with carboplatin alone (AUC 5; n = 178) in patients with platinum-sensitive recurrent ovarian cancer, with recurrence occurring at least 6 months after completion of first-line platinum-based therapy. Patients were enrolled with measurable or assessable lesions according to Southwest Oncology Group (SWOG) criteria. Exclusion criteria included an ECOG score of > 2, inadequate bone marrow or kidney function or serious concomitant conditions, or life expectancy of < 12 weeks.
The primary outcome of the trial was PFS, with OS, response rate, duration of response, QoL and toxicity measured as secondary outcomes. It should be noted that the study was not powered to detect a difference between treatments in OS. Randomisation was carried out through the central Arbeitsgemeinschaft Gynaekologische Onkologie (AGO)-Ovarian Cancer Study Group (OVAR) office (method of randomisation not reported), with patients randomised at a 1 : 1 ratio. Patients were stratified by PFI (6–12 months vs. ≥ 12 months), first-line therapy (platinum plus paclitaxel vs. other platinum-based therapy) and bidimensionally measurable disease (yes vs. no).
Median duration of follow-up was reported as 17 months, but the range of follow-up was not reported either for the full trial population or the individual treatment groups. Treatment cycles in each group were repeated every 21 days for six cycles, in the absence of PD or unacceptable toxicity. At the investigator’s discretion, benefiting patients could receive a maximum of 10 cycles of therapy. The median number of cycles administered was six cycles in each group. Cycles could be postponed up to 2 weeks owing to toxicity, and longer toxicity-related delays led to treatment discontinuation. For grade 3 non-haematological toxicities (excluding nausea/vomiting), dose modifications and/or study discontinuation were at the investigator’s discretion. Patients in the gemcitabine plus carboplatin arm received 75.6% of the planned mean dose of gemcitabine (92.8% on day 1 and 63.4% on day 8) and 96.2% of the planned dose of carboplatin. Patients in the carboplatin arm received 98.2% of the planned dose.
Paclitaxel plus carboplatin compared with platinum-based therapy alone
Two RCTs were identified for this comparison. 48,61 One RCT was a collaboration between the International Collaborative Ovarian Neoplasm (ICON) group and the AGO group and hereafter is referred to as ICON4/AGO-OVAR 2.2 (Parmar et al. 61). The RCTs identified were of similar design, but one was a Phase II RCT (Gonzalez-Martin et al. 48) and the other was a Phase III RCT (ICON4/AGO-OVAR 2.261).
The ICON4/AGO-OVAR 2.2 trial61 comprised results from two randomised trials that were run in parallel. ICON4/AGO-OVAR 2.261 was an international multicentre trial enrolling 802 patients in 119 hospitals across five countries. ICON4 was co-ordinated by the Istituto di Recerche farmacologiche Mario Negri (IRFMN), and the Medical Research Council’s Clinical Trials Unit (MRC CTU), and AGO-OVAR 2.2 was co-ordinated by AGO. Each co-ordinating unit had its own protocol, with minor differences in eligibility criteria.
All centres enrolled patients with relapsed epithelial ovarian cancer who had previously received platinum-based chemotherapy and had been treatment free for at least 6 months; patients in IRFMN were required to have been treatment free for a minimum of 12 months. The IRFMN and AGO-OVAR 2.2 protocols specified that women were to have received only one prior chemotherapy treatment to be eligible for enrolment, whereas the MRC-CTU protocol permitted women to have received more than one previous line of chemotherapy. Measurable disease at baseline was an entry criteria for patients randomised in centres co-ordinated by the IRFMN, but not MRC CTU or AGO co-ordinated centres. The IRFMN and MRC CTU protocols required that patients have had previous platinum-based chemotherapy, with or without paclitaxel. By contrast, the AGO protocol specified that patients must have previously received cisplatin plus paclitaxel or carboplatin plus paclitaxel. Patients with concomitant or previous malignant disease were ineligible.
The trial compared the clinical effectiveness of paclitaxel plus platinum-based chemotherapy with platinum-based chemotherapy alone. Patients were randomised to receive paclitaxel [175 (ICON4) or 185 (AGO-OVAR 2.2) mg/m2 as a 3-hour infusion] plus platinum chemotherapy (392 patients) or conventional platinum-based therapy (410 patients). Platinum-based therapy comprised carboplatin (AUC 5) or cisplatin (minimum 75 mg/m2 as monotherapy or 50 mg/m2 in combination therapy). In all protocols, cycles were administered every 21 days.
The aim of the study was to evaluate whether paclitaxel should be given in addition to platinum-based chemotherapy in patients with platinum-sensitive disease, who would otherwise be treated with conventional platinum-containing regimens. Randomisation used a computer minimisation method (1 : 1 ratio) and patients were stratified by multiple factors that were determined by the protocol of the assigned centre. In ICON4 protocols, patients were stratified by age, centre, last chemotherapy received, time since last chemotherapy completed and intended platinum treatment. In AGO-OVAR 2.2, patients were stratified by whether the patient had undergone secondary debulking surgery and time since completion of last chemotherapy.
The primary outcome measure of all protocols was OS; secondary outcomes were PFS and QoL. Progression required clinical or radiological evidence of disease (not only raised CA125 level). The sample size calculation found that 800 patients would be sufficient to detect an 11% difference between the groups if the control group survival was 50% at a power of 90% and a 5% significance level.
Median follow-up was 42 months. Of the full trial population, 72% of patients received a minimum of six cycles of assigned chemotherapy; reasons for not completing six cycles included disease progression or death, toxicity or patient preference.
Gonzalez-Martin et al. 48 reported the results of a Phase II study, in which 81 patients with platinum-sensitive recurrent ovarian cancer, who had received no more than two previous chemotherapy lines, were randomised to receive carboplatin alone (AUC 5; 40 patients) every 21 days or paclitaxel (125 mg/m2 of > 3 hours) plus carboplatin (AUC 5; 41 patients) every 21 days. Patients had to have measurable disease as measured by CT or clinically evident but non-measurable disease that was evaluable by CA125 level, based on Rustin’s criteria. Patients who had an ECOG performance status of > 2, life expectancy of < 12 weeks or inadequate bone marrow, liver or kidney function were ineligible.
The primary outcome measure was ORR (CR or PR), which was evaluated using WHO criteria in those with measurable disease, or by CA125 level, according to Rustin’s criteria, in those without measurable disease. OS, TTP and QoL were reported as secondary outcome measures.
Both treatments were administered for a minimum of six cycles unless there was progression, unacceptable toxicity or a patient refused treatment. After six cycles, patients could continue for three further cycles if clinical benefit could be expected. All patients randomised to receive paclitaxel were treated with standard premedication 30 minutes before infusion, which comprised dexamethasone, diphenhydramine and ranitidine. In cases of grade 4 neutropenia or thrombocytopenia, doses were reduced to carboplatin AUC 4 (both groups) and paclitaxel 150 mg/m2.
Randomisation was reported to have been carried out by a central data centre (no further details reported). Patients were stratified by PFI [6–12 months (PPS) compared with > 12 months (FPS)] and number of previous chemotherapy lines (one vs. two). The trial was an unusual ‘pick up the winner’ design. The authors of the trial comment that this type of design has a ‘90% chance of selecting the better treatment if the difference is at least 15% and the smaller response rate is assumed to be 30%’. A sample size calculation is not presented, but the authors state that the trial was not designed or powered to detect differences in survival. The authors go on to comment that ‘no formal statistical comparison between the two arms was planned’, but a selection of statistical comparisons are reported ‘for exploratory purposes only’.
Patients in both treatment groups received a median of six cycles of treatment, with between two and nine cycles of carboplatin alone administered and between one and eight cycles of paclitaxel plus carboplatin administered. Three patients in the paclitaxel plus carboplatin arm did not receive one cycle of treatment. The proportion of patients requiring a dose reduction was small and was similar between the groups (4.7% with carboplatin alone vs. 6.6% with paclitaxel plus carboplatin). By contrast, a significantly larger proportion of patients required a dose delay in the carboplatin alone group (34.4%) compared with the paclitaxel plus carboplatin group (21%; p-value for difference < 0.006). The difference was attributed to the absence of haematological recovery by day 21 in the group receiving carboplatin alone.
The three patients who received no treatment in the paclitaxel plus carboplatin group were included in the ITT analysis of overall response but were excluded from other analyses. Median duration of follow-up was 67.7 weeks. At this time point, 32 patients had died and median OS has not been reached in the paclitaxel plus carboplatin group. The range of follow-up was not reported either for the full trial population or the individual treatment groups.
Paclitaxel plus carboplatin compared with paclitaxel alone
Lortholary et al. 62 reported the results of a Phase II, multicentre, open-label, three-armed randomised trial (CARTAXHY) in patients with PRR recurrent ovarian cancer. Eligible patients were those who had received at least one prior therapy, with the most recent regimen combining platinum with a taxane agent. In addition, patients were required to have either measurable (according to RECIST criteria) or CA125 assessable disease, an ECOG performance status of ≤ 2 and a life expectancy of > 12 weeks. Patients with measurable disease (according to RECIST criteria) or evaluable disease (CA125) were enrolled. Patients who had previously been treated with weekly paclitaxel were excluded.
In total, 165 patients were randomised (1 : 1 : 1 ratio) to treatment with weekly paclitaxel (80 mg/m2 administered on days 1, 8 and 15 of a 4-week cycle; 57 patients), weekly paclitaxel plus carboplatin (AUC 5 administered on day 1 of a 4-week cycle; 51 patients) or weekly paclitaxel plus weekly topotecan (3 mg/m2 administered on days 1, 8 and 15 of a 4-week cycle; 57 patients). The combination of paclitaxel plus topotecan in the treatment of patients with PRR ovarian cancer is not covered by the scope of this review, and the efficacy results for this group are not presented.
The primary outcome was PFS. Secondary end points were response rate, OS, QoL and toxicity. QoL was assessed using the EORTC Quality of Life Questionnaire (EORTC-QLQ) and toxicity was assessed according to NCI-CTC. The efficacy analyses were based on the ITT principle.
Randomisation was carried out at the Group d‘Investigateurs Nationaux pour I’Etude des Cancers Ovariens (GINECO) data centre but details on the method of randomisation are not available. Patients were stratified according to centre, TFIs (progression during treatment vs. relapse between 0 and 3 months vs. relapse at > 3 months and ≤ 6 months), and presence of a measurable lesion at baseline.
Treatments were administered for six to nine cycles or until progression or unacceptable toxicity. On progression, patients treated with weekly paclitaxel or weekly paclitaxel plus weekly topotecan received carboplatin (AUC 5) and patients treated with weekly paclitaxel plus carboplatin went on to receive treatment of physician’s choice. One patient in the weekly paclitaxel group did not receive any treatment. Patients received a median three cycles in each group.
Dose reductions for toxicity of one level were to paclitaxel 65 mg/m2, carboplatin AUC 4 mg/ml/minute, and topotecan 2.4 mg/m2. Dose reductions of two levels were to paclitaxel 5 mg/m2, carboplatin AUC 3.5 mg/ml/minute, and topotecan 2 mg/m2. In cases in which there was a treatment delay of > 2 weeks, patients were discontinued from the study.
The sample size calculation indicated that 165 patients would be required for adequate power to detect a difference among groups with 80% power. Median duration of follow-up was 15 months.
Paclitaxel compared with oxaliplatin
Piccart et al. 63 report the results of a multicentre (17 European centres across six countries), open-label, randomised, Phase II study. Patients were enrolled who had histologically or cytologically proven advanced ovarian cancer that had progressed or stabilised after prior treatment, with relapse occurring within 12 months of the last platinum-based chemotherapy regimen. No more than two prior cisplatin- and/or carboplatin-containing chemotherapy regimens were permitted. Patients were also ineligible if they had prior treatment with platinum derivatives other than cisplatin and carboplatin or with paclitaxel, docetaxel, or high-dose chemotherapy with haematopoietic stem cell support.
The primary aim of the trial was to evaluate the clinical effectiveness of oxaliplatin (Eloxatin®, Sanofi) (130 mg/m2 over 2 hours) every 21 days (n = 45) compared with paclitaxel (175 mg/m2 over 3 hours) every 21 days (n = 41).
Patients were assigned to their study group by the EORTC. No details on the method of randomisation are reported in the full publication. Patients were stratified by centre, performance status (0 vs. 1 vs. 2), PFI (0–6 months vs. 6–12 months) and number of prior platinum-based regimens (1 vs. 2). The primary outcome measure was the objective confirmed response rate, which was assigned as per WHO criteria and verified by two independent radiologists. Secondary outcome measures were TTP, OS, time to treatment failure and QoL.
For patients randomised to receive paclitaxel infusion, premedication included oral dexamethasone (20 mg) 12 and 6 hours before infusion, and diphenhydramine (50 mg intravenously) plus cimetidine (300 mg; Tagamet®, GSK) or ranitidine (50 mg intravenously) 30 minutes before the infusion. Antiemetic therapy before oxaliplatin infusion was a serotonin antagonist (5-HT3), with a single dose of corticosteroid (e.g. dexamethasone 20 mg).
Treatment in each group was continued until disease progression, unacceptable toxicity or patient refusal. The initial paclitaxel and oxaliplatin doses could be reduced in subsequent cycles, or the cycles could be delayed by 1 or 2 weeks, depending on toxicity. Dose reduction of below 90 mg/m2 for paclitaxel and 75 mg/m2 oxaliplatin per cycle was not permitted, and patients requiring these or lower doses went off study. The median number of cycles of treatment was six (range 1–8) in the paclitaxel group and four (range 1–8) in the oxaliplatin group. Most patients had a delivered relative dose intensity of at least 95%.
Median duration of follow-up was not reported. A total of five patients were not assessable for response (two in the paclitaxel arm and three in the oxaliplatin arm): four were ineligible because of eligibility deviations and one died 6 days after the first oxaliplatin cycle, as a result of a massive pulmonary thromboembolism (unrelated to treatment).
A sample size calculation was not reported. The authors comment that, despite the use of several centres, as a result of wider use of paclitaxel as a first-line treatment at the time the trial was initiated, accrual of paclitaxel-naive patients became slow in the later stages of the trial. It is unclear whether the trial was adequately powered to detect a difference between treatments.
Topotecan oral compared with topotecan intravenous
Gore et al. 24 report the results of a multicentre, international (Europe, South Africa and North America) randomised trial of open-label design that compared topotecan administered orally (2.3 mg/m2 daily for five consecutive days; n = 135) with intravenously (1.5 mg/m2 daily for five consecutive days; n = 131). Both treatment regimens were given on a 21-day cycle. Patients were enrolled who had relapsed epithelial ovarian cancer (histological diagnosis) that was measurable at baseline and was of FIGO stage III or IV (266 patients randomised). To be eligible, patients were also required to have an ECOG score of ≤ 2. Patients had either progressed during or relapsed within 12 months of completing first-line chemotherapy, and only one prior chemotherapy regimen was permitted. Initial treatment must have been platinum based and could have been given in conjunction with a taxane.
The aim of the study was to compare the efficacy, safety and tolerability of oral topotecan compared with standard i.v. topotecan in patients with relapsed ovarian cancer. Randomisation (1 : 1 ratio) was carried out by telephone (no further details reported) and stratified by prior taxane exposure, interval from previous platinum therapy and tumour diameter (< 5 cm vs. ≥ 5 cm). Three categorisations of response to first-line chemotherapy were defined: platinum refractory (PD or SD during initial chemotherapy); platinum resistant (initial response followed by relapse within 6 months); and platinum sensitive (initial response with subsequent relapse at > 6 months).
Outcomes assessed included response rate (as per WHO criteria), time to response, TTP, survival and toxicity. Median follow-up was not stated. Although open label in design, all responses that were claimed to be confirmed or partial were subject to independent, blinded radiological review. The only outcome evaluated for the subgroups categorised by extent of sensitivity to platinum was response rate.
Duration of treatment with topotecan was determined by response to therapy and was at the discretion of the clinician. It was recommended that patients with SD receive a minimum of four cycles of treatment and that patients responding to treatment receive at least two cycles of treatment beyond response. Patients assigned to oral topotecan received a median of four (range 1–23) cycles and those assigned i.v. topotecan received a median of six (range 1–26) cycles. Dose reductions were permitted for grade 3 or 4 AEs, with about 10% of patients in each group requiring a reduction in dose.
Topotecan administered on five consecutive days (conventional regimen) compared with topotecan administered weekly
Sehouli et al. 23 report the results of a randomised, multicentre, Phase II trial in Germany involving 194 patients with platinum-resistant recurrent epithelial ovarian or primary peritoneal cancer after radical surgery and at least one platinum-containing chemotherapy. Patients with disease measurable by CT or MRI, or disease evaluable by CA125 according to GCIG criteria, were eligible. Platinum resistance was defined as clinical disease progression after a TFI of < 6 months after a platinum-based regimen. Inclusion criteria with regards to number of previous lines of chemotherapy were not specified.
The primary goal of the trial was to compare weekly administration of topotecan at a dose of 4.0 mg/m2 each week, applied on days 1, 8 and 15 of a 28-day cycle (n = 97) compared with the conventional regimen of 1.25 mg/m2 for five consecutive days (n = 97). The rationale for the trial was that weekly administration of topotecan is considered to be less toxic and is widely used in clinical practice, despite the lack of an evidence base of effectiveness. It should be noted that the dose used in the ‘conventional’ 5-day regimen is lower than the dose recommended in the Summary of Product Characteristics (SmPC) for topotecan (1.5 mg/m2).
Randomisation was central with permutated blocks in a 1 : 1 ratio and was carried out by phone and facsimile. However, the level of masking in the trial is unclear. The primary outcome evaluated was the clinical benefit rate, which was defined as the composite of CR, PR and SD. Response was determined according to RECIST for measurable disease or GCIG criteria for serum CA125 levels. Use of the CA125 marker or scans to evaluate response was at investigators’ discretion, with all responses confirmed by a second examination. Secondary end points were toxicity, PFS and OS; QoL was also explored. All analyses were based on the ITT principle. No sample size calculation was reported but it is stated that the study was not powered for a direct comparison between the dosing schedules or to reveal differences in response rates.
Treatment in each group was continued until intolerable toxicity or disease progression or until the patient refused further therapy, with maximum treatment duration of 12 months. Dose of topotecan could be reduced by 25% for any grade 3 or 4 adverse effects according to the NCI-CTC.
Median follow-up was 23.4 months (range 12.7–41.4 months). Patients in the weekly topotecan group received statistically significantly fewer cycles of chemotherapy than the group receiving topotecan at the conventional dosing regimen (3.5 with weekly topotecan vs. 4.8 with conventional topotecan; p = 0.002).
Paclitaxel high dose (250 mg/m2) compared with paclitaxel standard dose (175 mg/m2)
Omura et al. 68 conducted a Phase III, randomised, multicentre trial comparing two doses of paclitaxel (250 mg/m2 vs. 175 mg/m2) involving patients with recurrent or persistent histologically confirmed epithelial ovarian cancer despite prior platinum therapy. A third group, paclitaxel 135 mg/m2, was closed early because of inadequate patient accrual. Eligible patients had received not more than one prior platinum-based regimen, had adequate bone marrow, kidney and liver function; and a Gynecologic Oncology Group (GOG) performance status of 0, 1 or 2.
The aim of the trial was to evaluate whether increasing dose of paclitaxel was associated with an increase in response. The primary outcome measures were PFS and OS. Objective response (CR or PR) rates were recorded in patients with measurable disease (pleural effusion or elevated CA125 level were not regarded as measurable disease). The study also assessed whether prophylactic filgrastim 10 µg/kg was more effective than filgrastim 5 µg/kg at reducing the incidence of febrile neutropenia in patients receiving paclitaxel 250 mg/m2. The TAG considers that the administration of filgrastim is unlikely to influence comparative clinical effectiveness.
Sequential, permuted block randomisation was used to assign patients to paclitaxel 175 or 250 mg/m2 by 24-hour i.v. infusion every 3 weeks. Both treatments were administered for a minimum of six cycles. Patients could continue treatment indefinitely if there was no clinical progression or excessive toxicity after six cycles. Paclitaxel dose intensity could be reduced for some grade 3 or greater toxicities (not otherwise specified). Patients experiencing neutropenic fever while receiving paclitaxel 175 mg/m2 were allowed filgrastim during subsequent therapy cycles.
Based on the sample size calculation, it was estimated that 540 patients, followed until approximately 80% had died, would provide an 80% chance of detecting a true HR of 1.4 between paclitaxel 135 mg/m2 and either of the more intense regimens (type I error p = 0.025 for one-tailed test). However, the study failed to enrol sufficient patients in the paclitaxel 135 mg/m2 arm and a decision was made to ‘allocate all of the type I error to the comparison of the two higher-dose regimens’. Initially designed to evaluate effects of the two paclitaxel regimens in platinum-resistant clinically measurable disease, owing to slow accrual, after commencement of the trial, the eligibility criteria were expanded to include patients with platinum-sensitive disease and without clinically measurable disease.
Of the 184 women randomly assigned to paclitaxel 175 mg/m2 and the 188 to paclitaxel 250 mg/m2, 164 (89%) and 166 (88%), respectively, were eligible. Ten eligible women (three in the paclitaxel 175 mg/m2 group and seven in the paclitaxel 250 mg/m2 group) were not assessed for tumour response because of death, toxicity or withdrawal but were classified as not responding for an ITT analysis among eligible patients. The primary survival outcomes were restricted to eligible patients.
Median duration of follow-up is not reported. The proportion of women receiving six or more cycles of therapy was similar between the two groups, with 58% and 55% of patients in the paclitaxel 175 mg/m2 and paclitaxel 250 mg/m2 group, respectively, receiving six or more cycles. One patient refused to take any dose of the allocated treatment.
Paclitaxel weekly compared with paclitaxel every 3 weeks
Rosenberg et al. 60 report the results of a randomised bifactorial multicentre study carried out at sites in Sweden and Finland. The aim of the study was to assess the efficacy and toxicity of paclitaxel given at the same dose intensity administered either weekly or every 21 days. Patients were randomised to paclitaxel 67 mg/m2 every 7 days or paclitaxel 200 mg/m2 every 21 days. Enrolled patients (n = 208) had advanced ovarian cancer (histologically proven) that had progressed during or relapsed after administration of a platinum-based regimen. To be eligible, patients had to have measurable disease that had been documented clinically and/or radiologically. Only one prior platinum-containing regimen was permitted. In addition, all patients were taxane naive.
The RCT was of a bifactorial design. In addition to randomisation to either paclitaxel weekly or every 21 days, patients were also randomised to oral dexamethasone (20 mg) taken 12 hours and 6 hours before paclitaxel infusion or administration of i.v. dexamethasone (20 mg) 30 minutes before paclitaxel infusion. Results in the full publication cited here focus on treatment with paclitaxel. Premedication with clemastine 2 mg (Tavegel®, Novartis) and cimetidine 300 mg (or ranitidine 50 mg) was given intravenously to all patients 30 minutes prior to paclitaxel infusion.
The primary endpoint of the study was clinical response rate as per WHO criteria, with TTP and OS evaluated as secondary outcomes. Randomisation was reported to have been carried out at the BMS office in Stockholm and patients were randomised in a 1 : 1 ratio. Patients were stratified by platinum resistance, with a differentiation at 6 months (randomisation strata: relapse at ≤ 6 months vs. > 6 months after primary platinum-based treatment). No further details on the method of randomisation are reported. The level of masking in the trial is unclear.
Median duration of follow-up was 27 months (range 7–47+ months). Patients to whom paclitaxel was administered weekly at a dose of 67 mg/m2 received a median of 5.7 courses of treatment (range 1–16 courses) compared with a median of seven courses in the group receiving paclitaxel 200 mg/m2 every 21 days (range 1–17 courses). More patients in the paclitaxel weekly arm (32 vs. 20) were taken off the study early (within 9 weeks) owing to either early progression or for administrative reasons. The difference in early progressions could be because of a low initial weekly dose or some patients may have had a more aggressive tumour biology.
The sample size calculation estimated that 318 patients would be required to detect the prespecified relative difference between groups of 54% with 80% power. To ensure a sufficient number of evaluable patients, it had been planned to recruit a total of 350 patients. Owing to slow recruitment of taxane-naive patients with recurrent disease, the study closed early after inclusion of 208 patients. The study may therefore have been underpowered to detect a difference between the two regimens.
Quality assessment of studies included in clinical effectiveness review
Pegylated liposomal doxorubicin hydrochloride plus carboplatin compared with paclitaxel plus carboplatin
The trial carried out by Bafaloukos et al. 29 is generally well designed with the primary analysis based on the ITT population. However, limited details on trial methodology are provided in the full publication. Randomisation is reported to have been carried out at the central HeCOG Data Office in Athens but a description of the method of randomisation is not reported. The level of masking within the trial is unclear. The primary outcome is response rate, which is determined by radiological scan or CA125 level. Assessment of response is associated with disparity in interpretation of scan results, both across different assessors and within categorisation (CR or PR) by an individual assessor. It is unclear whether radiological scans were evaluated by an independent review panel. In addition, TTP was measured from date of treatment initiation rather than date of randomisation, which is a more commonly used definition for TTP in clinical trials. The evaluation of the quality of the trial is presented in Table 9.
The CALYPSO trial31 is a well-designed and well-conducted trial. Progression and response were reviewed independently. Although the methods indicate that analyses are based on the ITT principle, three randomised patients (one in the PLDH plus carboplatin group and two in the paclitaxel plus carboplatin group) were judged to be ineligible because of absence of evidence of ovarian cancer post randomisation and were excluded from analyses of clinical effectiveness. Thus, the analyses are not strict ITT analyses. The evaluation of the quality of the trial is presented in Table 9.
Pegylated liposomal doxorubicin hydrochloride plus carboplatin compared with carboplatin alone
The Alberts et al. 28 trial seems to be generally well designed, although limited details on the methodology of the trial are provided in the full publication. The method of randomisation and level of masking are unclear. As the primary outcome is OS, masking, or lack of masking, is unlikely to introduce bias into the evaluation of treatment effect. The key issue associated with trial design is that the study is likely to have been underpowered as a result of early closure owing to slow patient accrual (61 patients recruited out of a planned 900 patients). The authors identify several factors that could have contributed to slow accrual, including dissolution of the SWOG Gynecologic Cancer Committee after initiation of the trial and publication of results from the larger ICON4/AGO-OVAR 2.2 trial. 61 The evaluation of the quality of the trial is presented in Table 9.
Trabectedin plus pegylated liposomal doxorubicin hydrochloride compared with pegylated liposomal doxorubicin hydrochloride alone
The OVA-301 trial30 was a well-conducted trial. Methodologically, the design of the trial was robust, with clinical effectiveness analyses based on the ITT population, and progression and response reviewed by an independent radiologist who was masked to treatment allocation. A secondary analysis of the primary outcome of PFS was carried out based on review by an independent oncologist (radiological assessment in conjunction with prespecified clinical data) who was also masked to treatment allocation. The methods of the trial are well reported. As noted in the Final Appraisal Determination (FAD) for the assessment of trabectedin plus PLDH as part of the Technology Appraisal process (TA222),73 one potential area that affects the external validity of the trial is the omission of a platinum-based chemotherapy as a comparator, particularly as a large proportion of patients enrolled had platinum-sensitive disease. The authors commented that the inclusion of platinum-resistant patients contributed to the decision against use of a platinum-based control, as platinum-based therapy would have been inappropriate in this setting. The evaluation of the quality of the trial is presented in Table 9.
Pegylated liposomal doxorubicin hydrochloride compared with topotecan
The trial carried out by Gordon et al. 49 was generally a well-designed trial. Although open label in design, scans for assessment of disease response and progression underwent independent radiological review. Although the methods state that analyses are based on the ITT principle, in the first publication, results are based on patients who received at least a partial dose of study drug (474 patients out of 481 randomised), which is a modified ITT analysis. However, in the publication describing longer-term follow-up of OS, analysis of OS is based on the ‘all randomised’ population and, as such, is a true ITT analysis. The evaluation of the quality of the trial is presented in Table 9.
Pegylated liposomal doxorubicin hydrochloride compared with paclitaxel
Technology appraisal no. 9113 reports that the study carried out by Schering-Plough (Trial 30–57) was a reasonably good-quality, randomised, open-label comparative trial. The key issue noted was that approximately 50% of the planned number of patients were recruited (216 recruited out of planned 438 patients). It is therefore likely that the trial is underpowered to detect a difference between PLDH and paclitaxel in treatment effect. TA91 also notes that the results of the trial ‘are likely to be preliminary and the longer term implications of any differences observed in the treatment effect at the time of data analysis are unclear’. The evaluation of the quality of the trial is presented in Table 9.
Topotecan compared with paclitaxel
A key strength of the trial evaluating topotecan compared with paclitaxel (ten Bokkel Huinink et al. 21) is that, for the primary outcome of response rate, all claimed responses were evaluated by an independent radiologist who was masked to treatment allocation. As a sample size calculation was not reported, there is uncertainty whether the trial was adequately powered to detect a difference between treatments. Furthermore, results are not based on the ITT principle, with only patients who received at least one dose of study drug being included in the final analysis. The trial design allowed patients to cross over to the alternative treatment should they fail to respond to their allocated treatment. The switch in treatment during the trial generates confounding in the final analysis of OS. The evaluation of the quality of the trial is presented in Table 9.
Gemcitabine plus carboplatin compared with carboplatin alone
The trial carried out by Pfisterer et al. 50 is generally a well-designed and well-conducted trial, with efficacy analyses based on the ITT principle. With PFS as a primary outcome and an open-label design, there is potential for bias. It is unclear from the full publication whether radiological assessment of progression was reviewed by an independent panel. The evaluation of the quality of the trial is presented in Table 9.
Paclitaxel plus carboplatin compared with platinum-based therapy alone
ICON4 and AGO-OVAR 2.2 are well-conducted parallel trials. 61 Comprehensive details on most aspects of trial methodology are provided in the full publication. 61 The level of masking is unclear but OS is the primary outcome and therefore awareness of treatment allocation is unlikely to influence results of this outcome. Analyses of clinical effectiveness are based on the ITT population. The evaluation of the quality of the trial is presented in Table 9.
The trial carried out by Gonzalez-Martin et al. 48 was a Phase II trial of a ‘pick-the-winner’ design, which the authors state has a ‘90% chance of selecting the better treatment if the difference is at least 15% and the smaller response rate is assumed to be 30%’. Therefore, no sample size calculation was carried out. A ‘pick-the-winner’ trial is designed as a screening trial to facilitate a selection between promising experimental regimens in a Phase II setting, and, as such, do not typically include the standard of care. Trials with a ‘pick the winner’ design are underpowered for hypothesis testing or comparisons of treatment effect on the outcomes of interest, such as survival. 74 Therefore, as the authors comment, all reported statistical analyses are exploratory and reported p-values should be interpreted with caution. Limited details on trial methodology are reported and the level of masking in the trial is unclear. Although it is reported that randomisation was carried out in a central data centre, the method of randomisation is not described. The evaluation of the quality of the trial is presented in Table 9.
Paclitaxel plus carboplatin compared with paclitaxel alone
Limited details of the methodology of the CARTAXHY trial62 are available in the publication presenting the results of the trial. A key strength of the trial is that clinical efficacy analyses were based on the ITT principle. Although it is stated that the study is randomised, details on the method of randomisation are not reported. As an open-label trial, there is potential for bias in the assessment of progression and response. It is unclear whether radiological scans underwent independent radiological review. The evaluation of the quality of the trial is presented in Table 9.
Paclitaxel compared oxaliplatin
The trial reported by Piccart et al. 63 is generally a well-designed trial. The primary outcome was objective confirmed response. As an open-label design, the outcome of confirmed response could potentially be open to bias. The descriptions of the methods states that response was verified by two independent radiologists. However, it is unclear whether the independent radiologists were truly independent and masked to treatment allocation. Although limited details are reported on the method of randomisation, it is reported that the treatment allocation was assigned by the EORTC. The key issue associated with the trial is the uncertainty around the power of the trial. The evaluation of the quality of the trial is presented in Table 9.
Topotecan oral compared with topotecan intravenous
The trial reported by Gore et al. 24 is generally well designed, with analysis based on the ITT population. In addition, although open label in design, assessments of CR and PR were validated by masked independent radiological review.
It is stated that randomisation was carried out by telephone but no details on the method of randomisation are reported. No power calculation is reported and thus it is unclear whether the study is adequately powered. The population is clinically homogeneous in that all patients randomised had measurable disease at baseline and also had received only one prior chemotherapeutic treatment. The evaluation of the quality of the trial is presented in Table 9.
Topotecan administered on five consecutive days (conventional regimen) compared with topotecan administered weekly
There are several factors that impact on the quality of the design and conduct of the trial carried out by Sehouli et al. 23 Although it is stated that all analyses are carried out based on the ITT principle, the analysis of clinical benefit does not include all patients randomised. There is no discussion of the omission of patients from this analysis. The dose used for the ‘conventional’ regimen for topotecan is lower than that recommended in the SmPC. The authors comment that the reduced dose is widely accepted by many international cancer societies but go on to highlight that there are no RCTs evaluating the comparative effectiveness of 1.25 mg/m2 compared with 1.5 mg/m2 of topotecan. In addition, use of radiological scans or CA125 level to determine response was at the discretion of the investigator. It is widely accepted that CA125 level is not sufficient to confirm response to treatment. Examination of the results for response indicates that a large proportion of patients were evaluated by CA125 level alone (80.1%). Moreover, it is unclear whether the investigator was masked to treatment allocation. Although responses had to be confirmed by a second examination, it is unclear whether response was confirmed by the same investigator or by independent review. The trial was not adequately powered to detect a difference between groups. These factors potentially limit the comparison of the results from this trial with similar trials in ovarian cancer. The evaluation of the quality of the trial is presented in Table 9.
Paclitaxel high dose (250 mg/m2) compared with paclitaxel standard dose (175 mg/m2)
Limited methodological details were reported in Omura et al. 68 The method of randomisation was robust, with treatment regimens sequentially assigned from stratified, permuted blocks. The level of masking in the trial is unclear. Although the methods state that the analyses are based on the ITT principle, patients identified post randomisation to be ineligible for participation in the trial were excluded from all analyses, and, therefore, analyses are not based on the ITT population. A key issue with the trial is the sample size, with only 265 patients recruited from a planned 540, even after expansion of the protocol to include platinum-sensitive patients and those with measurable disease. Thus, the study is likely to be underpowered to detect a true difference between the treatment regimens for which results are reported.
Paclitaxel weekly compared with paclitaxel every 3 weeks
The trial carried out by Rosenberg et al. 60 is of reasonable quality. Efficacy analyses are based on the ITT principle. Limited details are reported on trial methodology in terms of method of randomisation and level of masking. The key issue with the trial is that it is potentially underpowered to detect a difference in the primary outcome of response rate between the paclitaxel regimens evaluated. The evaluation of the quality of the trial is presented in Table 9.
| Study | Potential bias affecting trial methodology | Potential bias affecting outcome | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Random sequence generation | Allocation concealment | Selective reporting | Other bias | Masking of personnel | Masking of outcome assessment | Incomplete outcome data | |||||||||||||||
| Low | Unclear | High | Low | Unclear | High | Low | Unclear | High | Low | Unclear | High | Low | Unclear | High | Low | Unclear | High | Low | Unclear | High | |
| Bafaloukos et al.29 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| TTP | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| CALYPSO Pujade-Lauraine et al.31 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Alberts et al.28 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| OVA-301 Monk et al.30 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| QoL | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Gordon et al.49 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| QoL | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Trial 30–57; data taken from TA9113 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| ten Bokkel Huinink et al.21 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| TTP | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| QoL | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Pfisterer et al.50 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| QoL | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| ICON4/AGO-OVAR 2.2 Parmar et al.61 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| QoL | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Gonzalez-Martin et al.48 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| QoL | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| CARTAXHY Lortholary et al.62 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| QoL | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Piccart et al.63 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| QoL | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Gore et al.24 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| TTP | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Sehouli et al.23 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| QoL | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Omura et al.68 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| PFS | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
| Rosenberg et al.60 | ✓ | ✓ | ✓ | ✓ | |||||||||||||||||
| OS | ✓ | ✓ | ✓ | ||||||||||||||||||
| TTP | ✓ | ✓ | ✓ | ||||||||||||||||||
| Response rate | ✓ | ✓ | ✓ | ||||||||||||||||||
| AEs | ✓ | ✓ | ✓ | ||||||||||||||||||
Comparability of baseline characteristics
Within most of the trials identified, the treatment groups were well matched in terms of population baseline characteristics, including age, TFI, the number of previous chemotherapy agents received, disease measurability (for those trials including patients with measurable and non-measurable disease), and performance status. Differences between groups that were reported to be significant are described below; imbalances that were reported to be non-significant or for which the significance of the difference was not assessed in the trial are not discussed. Detailed baseline characteristics of the individual trials are available in the data abstraction forms presented in Appendix 2.
An unanticipated imbalance in PFI was noted in a retrospective analysis of OVA-301. 30 Patients in the PLDH monotherapy group had a significantly longer mean PFI than patients in the trabectedin plus PLDH group (mean PFI: 13.3 months with PLDH alone vs. 10.6 months with trabectedin plus PLDH; p = 0.009). Longer PFI is correlated with increased likelihood of response to treatment. Therefore, the potential direction of bias in analysis of treatment effect is against trabectedin plus PLDH. To account for this imbalance, the authors carried out additional exploratory analyses based on PFI as a continuous covariate (discussed in Chapter 4, Trabectedin for the treatment of patients with relapsed platinum-sensitive ovarian cancer). The analyses were not prespecified and as such were hypothesis generating.
Baseline characteristics of key prognostic factors (based on expert advice) are summarised in Table 10. Also, based on expert advice, the TAG has focused on the subgroups of platinum-sensitive ovarian cancer and PRR ovarian cancer rather than the full trial population. Baseline characteristics are considered in terms of comparability within platinum-sensitive patients and PRR patients.
Considering patients with platinum-sensitive disease, a potential source of heterogeneity within the trials is the proportion of patients with FPS (relapse at > 12 months after last platinum-based treatment) ovarian cancer compared with PPS (relapse at ≥ 6–12 months after last platinum-based treatment) at baseline. The greater the duration of PFI, the more favourable the prognosis. In trials involving patients with only platinum-sensitive disease,28,29,31,48,50,61 the proportion of patients with PPS ovarian cancer ranges from 28.6% to 43.0%. Considering the large trial ICON4/AGO-OVAR 2.2,61 the proportion of patients with PPS compared with FPS is 74.7% and 25.3%, respectively. ICON4/AGO-OVAR 2.261 has been reported to have longer median PFS and OS for both groups compared with other trials involving platinum-sensitive patients, which is thought to be attributable to the comparatively larger proportion of patients with FPS ovarian cancer who have an improved prognosis compared with those who are PPS. Given that the NMA is based on relative treatment effects (HR), and that most trials are well balanced between groups in FPS ovarian cancer compared with PPS, the TAG considered the trials sufficiently clinically homogeneous to compare treatments in a NMA.
Number of prior lines of chemotherapy is another source of potential heterogeneity. Increasing number of previous chemotherapy regimens is associated with a decrease in response to treatment. Of the 16 trials identified, seven included patients who had received two or more prior lines of chemotherapeutic treatment. In trials involving only patients with platinum-sensitive disease, the proportion of patients with more than one line of prior chemotherapy in each trial is generally small, ranging from 4% to 15.5%. By contrast, as could be expected, in trials involving patients with PRR ovarian cancer, the proportion of patients with two or more chemotherapy regimens is larger, at about 30% in all trials. In all trials, the number of patients with multiple lines of prior chemotherapy is well balanced within the trial. It is possible that inclusion of trials in which patients received two or more chemotherapy regimens is likely to underestimate the effects of the evaluated treatments in patients with first recurrence of disease, and thus potentially bias the results of an indirect comparison towards treatments that are given as second line. Again, as the HR used in the NMA is a relative treatment effect, the impact of these trials on the overall result could be minimal.
Scales evaluating performance status are used to assess disease progression and how a patient’s daily living abilities are affected by their disease. On the ECOG scoring system (also referred to as the Zubrod or WHO score), the lower a patient’s performance score, the greater their capacity for physical activity; a score of ‘0’ or ‘1’ indicates that the patient is ambulatory. In the Karnofsky scale, which scores from ‘100 to 0’, higher performance score is favourable: a score of > 80 indicates that a patient is able to carry on normal activity and to work with no special care required. Good performance status has been shown to be an important prognostic factor in several types of cancer. 75
In the identified trials, the proportion of patients with unfavourable baseline performance score (ECOG/Zubrod/WHO score of ≥ 2; Karnofsky score of < 80) is small, ranging from 0% to 16% across the trials. Including patients with less favourable performance scores is likely to underestimate the effect of the treatments. For example, in Gonzalez-Martin et al. ,48 the 12.3% increase in proportion of people with ECOG score 2 in the platinum treatment group may limit the benefit received by people receiving platinum monotherapy when compared with paclitaxel plus platinum (i.e. paclitaxel plus platinum may have less benefit over platinum monotherapy). In addition, in Rosenberg et al. 60 the 9% increase in WHO score 2 in the three-weekly paclitaxel group may limit the benefit received by three-weekly paclitaxel monotherapy compared with weekly paclitaxel monotherapy (i.e. the benefit of paclitaxel weekly may have less benefit over three-weekly paclitaxel). However, in those trials that include patients with a less favourable performance score, the proportion of patients in each treatment group is well balanced and thus the impact on the overall result could be minimal.
Diagnosis of recurrent disease based on raised CA125 levels alone has been found to predate evidence of disease progression from clinical examinations or radiological scans by a median of 4 months in 70% of patients with ovarian cancer. 76 Thus, there is uncertainty whether patients diagnosed as having recurrent disease by only CA125 level would have the same diagnosis on radiological scan. In addition, it is also possible that the degree of sensitivity to platinum could differ. For example, based on CA125 level alone, a patient could be categorised as PPS at baseline but as FPS 4 months later with radiological confirmation. Of the trials identified, seven RCTs23,28,29,31,48,62,68 reported that patients with only CA125 level as an indicator of recurrent disease were enrolled. In trials in patients with platinum-sensitive disease, there was considerable variation across the trials in the proportion of patients with non-measurable disease at baseline, ranging from 8.5% to 38.2%. In some trials, patients with non-measurable disease were not included in analyses of response rate. Despite the identified disparity in methods used to diagnose recurrent disease at baseline, as the proportion of patients in each group within the individual trials was well balanced, the TAG considered that the heterogeneity could have a minimal impact on the NMA.
Considering heterogeneity among treatments evaluated, it is important to note that ICON461 evaluated the efficacy of adding paclitaxel to ‘conventional’ platinum-based chemotherapy compared with platinum-based therapy alone. A large proportion of patients in each treatment group received carboplatin as the platinum component of their regimen (80% in the paclitaxel plus platinum-based therapy group vs. 29% in the platinum-based chemotherapy alone group). Of the remaining 20% of patients in the paclitaxel plus platinum group, 10% were administered cisplatin, and 5% received paclitaxel plus carboplatin or cisplatin, switching between the two platinum monotherapies. In the conventional platinum-based monotherapy group, 4% of patients received cisplatin alone, and a further 2% received either carboplatin or cisplatin monotherapy, switching between the two platinum monotherapies. Moreover, 17% of patients in the conventional chemotherapy group received the triple therapy of cyclophosphamide, doxorubicin and cisplatin, which the ICON investigators had compared against carboplatin in an earlier trial and found no statistically significant difference between the treatments in effect on OS. 61 Although a small proportion of patients received platinum treatment other than carboplatin, there is evidence that the regimens received have similar efficacy.
Although differences in key prognostic factors across the trials have been identified, when considering the trials that would inform the NMA for platinum-sensitive disease and for PRR disease, the TAG considers the trials sufficiently clinically homogeneous to compare clinical effectiveness of treatments.
| Trial name | Intervention | Age (median,a years) | Performance score | Proportion of patients with two or more lines of previous chemotherapy | Platinum sensitivity (interval since last chemotherapy) | Measure of ovarian cancer at baseline |
|---|---|---|---|---|---|---|
| Bafaloukos et al.29 | PLDH plus carboplatin | 62 (38–89) | ECOG 0: 55/93 (59%) | 4/93 (4%) | 6–12 months: 22/93 (23%) | Elevated CA125 level only: 9/93 (10%) |
| ECOG 1: 30/93 (32%) | 12.1–24 months: 38/93 (41%) | |||||
| ECOG 2: 1/93 (1%) | > 24 months: 29/93 (31%) | |||||
| Paclitaxel plus carboplatin | 63 (37–81) | ECOG 0: 62/96 (65%) | 4/96 (4%) | 6–12 months: 32/96 (33%) | Elevated CA125 level only: 7/96 (7%) | |
| ECOG 1: 27/96 (28%) | 12.1–24 months: 32/96 (33%) | |||||
| ECOG 2: 0/96 (0%) | > 24 months: 23/96 (24%) | |||||
| CALYPSO (Pujade-Lauraine et al.31) | PLDH plus carboplatin | 60.5 (24–82) | ECOG 0: 286/466 (61.4%) | 58/466 (12.4%) | 6–12 months: 161/466 (35.0%) | Measurable disease: 281/466 (60.3%) |
| ECOG 1: 158/466 (33.9%) | > 12 months: 305/466 (65.0%) | |||||
| ECOG 2: 13/466 (2.8) | ||||||
| Paclitaxel plus carboplatin | 61 (27–82) | ECOG 0: 317/466 (62.5%) | 88/466 (17.3%) | 6–12 months: 183/466 (36.1%) | Measurable disease: 321/466 (63.3%) | |
| ECOG 1: 164/466 (32.3%) | > 12 months: 324/466 (63.9%) | |||||
| ECOG 2: 15/466 (3.0%) | ||||||
| Alberts et al.28 | PLDH plus carboplatin | 66.9 (43–87) | Zubrod 0: 20/31 (65%) | 0/31 (0%) | 6–12 months: 13/31 (43%) | Measurable disease: 19 (61%) |
| Zubrod 1: 11/31 (35%) | 12–24 months: 18/31 (57%) | Elevated CA125 level: 4 (13%) | ||||
| Other non-measurable disease: 8 (26%) | ||||||
| Carboplatin alone | 62.5 (31–80) | Zubrod 0: 16/30 (53%) | 0/30 (0%) | 6–12 months: 13/30 (43%) | Measurable disease: 20 (67%) | |
| Zubrod 1: 14/30 (47%) | 12–24 months: 17/30 (57%) | Elevated CA125 level: 2 (7%) | ||||
| Other non-measurable disease: 8 (27%) | ||||||
| OVA-301 (Monk et al.30) | Trabectedin plus PLDH | 56 (26–82) | ECOG 0: 230/337 (68%) | 0/337 (0%) | < 6 months: 115/333 (35%) | All patients had measurable disease at baseline |
| ECOG 1: 98/337 (29%) | 6–12 months: 123/333 (37%) | |||||
| ECOG 2: 9/337 (3%) | > 12 months: 95/333 (28%) | |||||
| PLDH alone | 58 (27–87) | ECOG 0: 192/335 (57%) | 0/335 (0%) | < 6 months: 117/330 (35%) | All patients had measurable disease at baseline | |
| ECOG 1: 132/335 (39%) | 6–12 months: 91/330 (28%) | |||||
| ECOG 2: 11/335 (3%) | > 12 months: 122/330 (37%) | |||||
| Gordon et al.49 | PLDH | 60 (27–87) | Karnofsky < 80: 39/239 (16.3%) | 0/239 (0%) | < 6 months: 130/239 (54.4%) | NR |
| Karnofsky ≥ 80: 199/239 (83.3%) | ≥ 6 months: 109/239 (45.6%) | |||||
| Topotecan | 60 (25–85) | Karnofsky < 80: 37/235 (15.7%) | 0/235 (0%) | < 6 months: 130/239 (54.4%) | NR | |
| Karnofsky ≥ 80: 195/235 (83.0%) | ≥ 6 months: 109/239 (45.6%) | |||||
| Trial 30–5713 | PLDH | 60.5 (27–80) | Karnofsky < 80: 11/108 (10.2%) | 0/108 (0%) | NR | All patients had measurable disease at baseline |
| Karnofsky ≥ 80: 95/108 (88%) | ||||||
| Not available: 2/108 (1.9%) | ||||||
| Paclitaxel | 61 (20–78) | Karnofsky < 80: 12/108 (11.1%) | 0/108 (0%) | < 6 months: 67/108 (62%) | All patients had measurable disease at baseline | |
| Karnofsky ≥ 80: 90/108 (83.3%) | ≥ 6 months: 41/108 (38%) | |||||
| Not available: 6/108 (5.6%) | ||||||
| ten Bokkel Huinink et al.21 | Topotecan | Mean: 59.2 (29–85) | ECOG 0: 41 (36.6%) | 0/117 (0%) | < 6 months: 52/112 (46.4%) | All patients had measurable disease at baseline |
| ECOG 1: 51 (45.5%) | ≥ 6 months: 60/112 (53.6%) | |||||
| ECOG 2: 20 (17.9%) | ||||||
| Paclitaxel | Mean: 58.3 (29–79) | ECOG 0: 42 (36.8%) | 0/118 (0%) | < 6 months: 55/114 (48.4%) | All patients had measurable disease at baseline | |
| ECOG 1: 53 (46.5%) | ≥ 6 months: 59/114 (51.8%) | |||||
| ECOG 2: 17 (14.9%) | ||||||
| Pfisterer et al.50 | Gemcitabine plus carboplatin | 59 (36–78) | ECOG 0: 83/178 (46.6%) | 0/178 (0%) | 6–12 months: 71/178 (39.9%) | NR |
| ECOG 1: 79/178 (44.3%) | > 12 months: 106/178 (59.6%) | |||||
| ECOG 2: 11/178 (6.2%) | ||||||
| Carboplatin alone | 58 (21–81) | ECOG 0: 93/178 (52.2%) | 0/178 (0%) | 6–12 months: 71/178 (39.9%) | NR | |
| ECOG 1: 72/178 (40.4%) | > 12 months: 107/178 (60.1%) | |||||
| ECOG 2: 9/178 (5.1%) | ||||||
| ICON4/AGO-OVAR 2.2 (Parmar et al.61) | Paclitaxel plus platinum | 60.0 | WHO 0: 246/392 (62.8%) | 37/392 (9.4%) | 6–12 months: 92/392 (35.0%) | NR |
| WHO 1: 121/392 (30.9%) | > 12 months: 300/392 (65.0%) | |||||
| WHO 2–3: 25/392 (6.4%) | ||||||
| Platinum monotherapy | 59.2 | WHO 0: 262/410 (63.9%) | 30/410 (7.3%) | 6–12 months: 111/410 (27.1%) | NR | |
| WHO 1: 122/410 (29.7%) | > 12 months: 299/410 (72.9%) | |||||
| WHO 2–3: 26/410 (6.3%) | ||||||
| Gonzalez-Martin et al.48 | Paclitaxel plus carboplatin | 59 (40–77) | ECOG 0: 17/41 (47.2%) | 7/41 (18.4%) | 6–12 months: 17/41 (45%) | WHO criteria: 27 (71%) |
| ECOG 1: 17/41 (47.2%) | > 12 months: 21/41 (55%) | CA125 criteria: 11 (28.9%) | ||||
| ECOG 2: 2/41 (5.6%) | ||||||
| Carboplatin alone | 61 (35–77) | ECOG 0: 14/40 (35.9%) | 5/40 (12.5%) | 6–12 months: 16/40 (40%) | WHO criteria: 25 (62.5%) | |
| ECOG 1: 18/40 (46.2%) | > 12 months: 24/40 (60%) | CA125 criteria: 15 (37.5%) | ||||
| ECOG 2: 7/40 (17.9%) | ||||||
| CARTAXHY (Lortholary et al.62) | Weekly paclitaxel plus carboplatin | 60 (43–77) | 0–1: 47/51 (92%) 2: 4/51 (8%) | 15/51 (29%) | All patients platinum resistant | Measurable (RECIST): 35/51 (68%) Elevated CA125 level only: 14/51 (28%) |
| Weekly paclitaxel | 60 (30–80) | 0–1: 54/57 (95%) 2: 3/57 (5%) | 15/57 (26%) | All patients platinum resistant | Measurable (RECIST): 32/57 (57%) Elevated CA125 level only: 21/57 (37%) | |
| Piccart et al.63 | Paclitaxel | 62 (37–81) | WHO 0–1: 35/41 (85%) | 11/41 (27%) | < 6 months: 31/41 (76%) | NR |
| WHO 2: 6/41 (15%) | 6–12 months: 10/41 (24%) | |||||
| Oxaliplatin | 59 (28–71) | WHO 0–1: 38/45 (84%) | 16/45 (36%) | < 6 months: 32/45 (71%) | NR | |
| WHO 2: 7/45 (16%) | 6–12 months: 13/45 (29%) | |||||
| Gore et al.24 | Oral topotecan | 60 (23–80) | ECOG 0: 59/135 (45%) | 0/135 (0%) | Platinum sensitive: 58 (43%) | All patients had measurable disease at baseline |
| ECOG 1: 60/135 (46%) | Platinum resistant: 37 (27%) | |||||
| ECOG 2: 12/135 (9%) | Platinum refractory: 40 (30%) | |||||
| Intravenous topotecan | 60 (27–80) | ECOG 0: 47/131 (35%) | 0/131 (0%) | Platinum sensitive: 56 (43%) | All patients had measurable disease at baseline | |
| ECOG 1: 77/131 (57%) | Platinum resistant: 36 (27%) | |||||
| ECOG 2: 11/131 (8%) | Platinum refractory: 39 (30%) | |||||
| Sehouli et al.23 | Weekly topotecan | 65 (41–82) | ECOG 0: 33/97 (34%) | 28/97 (29%) | All patients platinum resistant | Measurable disease: 86/97 (89%) |
| ECOG 1: 48/97 (49%) | ||||||
| ECOG 2: 12/97 (12%) | ||||||
| Conventional topotecan | 65 (36–85) | ECOG 0: 34/97 (35%) | 31/97 (32%) | All patients platinum resistant | Measurable disease: 90/97 (93%) | |
| ECOG 1: 50/97 (52%) | ||||||
| ECOG 2: 11/97 (11%) | ||||||
| Omura et al.68 | Paclitaxel 250 mg/m2 | 62 (24–80) | GOG 0: 88/166 (53%) | 0/166 (0%) | < 6 months: 132/166 (79%) | 134/166 (81%) |
| GOG 1: 63/166 (38%) | > 6 months: 34/166 (21%) | |||||
| GOG 2: 15/166 (9%) | ||||||
| Paclitaxel 175 mg/m2 | 60 (23–88) | GOG 0: 89/164 (54%) | 0/164 (0%) | < 6 months: 125/164 (76%) | 131/164 (80%) | |
| GOG 1: 65/164 (40%) | > 6 months: 39/164 (24%) | |||||
| GOG 2: 10/164 (6%) | ||||||
| Rosenberg et al.60 | Weekly paclitaxel | 59 (37–74) | WHO 0: 57/105 (54%) | 0/105 (0%) | < 6 months: 57/105 (54%) | All patients had measurable disease at baseline |
| WHO 1: 40/105 (38%) | > 6 months: 48/105 (46%) | |||||
| WHO 2: 8/105 (7%) | ||||||
| Three-weekly paclitaxel | 60 (40–76) | WHO 0: 56/103 (54%) | 0/103 (0%) | < 6 months: 51/103 (50%) | All patients had measurable disease at baseline | |
| WHO 1: 33/103 (32%) | > 6 months: 52/103 (50%) | |||||
| WHO 2: 14/103 (16%) |
Assessment of effectiveness
Based on clinical expert advice, the TAG has focused on the clinical effectiveness of interventions in populations defined by degree of platinum sensitivity [i.e. platinum sensitive (i.e. recurrence ≥ 6 months after last platinum-based treatment) and platinum resistant (i.e. recurrence < 6 months after last platinum-based treatment) or refractory (progression during platinum-based treatment)]. When it was not possible to extract data for the prespecified populations, for completeness, the TAG presents data for the full population of the study.
Overall survival
Overall survival is universally accepted as a measure of benefit in trials evaluating treatments for cancer, and is generally considered to be the most reliable end point. 77 However, the large number of patients required to ensure adequate power to detect a difference between treatments and long follow-up periods can hinder the collection and analysis of survival data. The FDA and other regulatory authorities define OS as the time from randomisation until death from any cause. 77 It should be noted that some of the trials reported here define OS as the time from administration of first cycle of study drug until death from any cause. As the event recorded is all-cause mortality, there is no bias associated with measurement of the end point.
A potential area of confounding with measurement of OS derives from the use of post-progression therapies. It has been proposed that subsequent lines of therapy are likely to be more effective in the less clinically effective group than in the treatment group, and is more likely to be considered when there is no significant difference in OS between the treatment and the control. Confounding from post-progression therapy is most likely to be an issue in trials in which most patients cross over to the alternative group after progression or in trials in which the ‘new’ therapy is available as a post-progression treatment in the control group. 78
Summary of results for overall survival
Most trials identified reported results for the outcome of OS. No trial was identified evaluating treatments in a population solely comprising patients who were allergic or intolerant to platinum-based chemotherapy. Here, results for patients with platinum-sensitive or PRR disease are summarised. For trials not limited to either platinum-sensitive or PRR patients (i.e. includes a mix of PFI), results for the full trial population are presented in the main body of the text.
Ten RCTs evaluating eight different head-to-head comparisons of interventions and comparators of interest were identified (Table 11).
| Trial name | Intervention | Comparator | HR (95% CI) |
|---|---|---|---|
| CALYPSO (Pujade-Lauraine et al.56) | PLDH (30 mg/m2) plus carboplatin every 21 days | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | 0.99a (0.85 to 1.16) |
| Bafaloukos et al.29 | PLDH (45 mg/m2) plus carboplatin every 28 days | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | 1.15 (0.78 to 1.66) |
| ICON4/AGO-OVAR 2.2 (Parmar et al.61) | Paclitaxel plus platinum | Conventional platinum treatment | 0.82 (0.69 to 0.97) |
| Gonzalez-Martin et al.48 | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | 0.31 (0.14 to 0.68) |
| ten Bokkel Huinink et al.52 | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 1.01 (0.66 to 1.54) |
| Trial 30–57 (taken from TA91)13 | PLDH (50 mg/m2) every 28 days | Paclitaxel (175 mg/m2) every 21 days | 1.05 (0.66 to 1.66) |
| Gordon et al.54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 1.43b (1.06 to 1.92) |
| Alberts et al.28 | PLDH (30 mg/m2) plus carboplatin every 4 weeks | Carboplatin alone every 4 weeks | 0.70 (0.40 to 1.21) |
| OVA-301 (Monk et al.30) | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 0.83 (0.67 to 1.04) |
| Pfisterer et al.50 | Gemcitabine (1000 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | 0.96 (0.75 to 1.23) |
To inform the decision problem, a NMA was carried out. Based on trials identified, it was not possible to construct a complete network. Two discrete networks were generated: one evaluating platinum-based therapies and the second comparing non-platinum-based regimens. It should be stressed that results from the two discrete networks are not directly comparable.
In the network evaluating platinum-based chemotherapies, PLDH plus carboplatin and paclitaxel plus carboplatin were found to significantly improve OS compared with platinum monotherapy (Table 12). However, no statistically significant differences in OS were identified between the remaining treatments considered in the network.
| Comparator | Intervention | |||
|---|---|---|---|---|
| Paclitaxel plus carboplatin | Gemcitabine plus carboplatin: HR (95% CrI) | PLDH plus carboplatin: HR (95% CrI) | Platinum monotherapy: HR (95% CrI) | |
| Paclitaxel plus carboplatin | – | 1.247 (0.921 to 1.652) | 1.023 (0.889 to1.172) | 1.290 (1.096 to1.509) |
| Gemcitabine plus carboplatin | – | – | 0.839 (0.602 to1.135) | 1.051 (0.815 to1.335) |
| PLDH plus carboplatin | – | – | – | 1.267 (1.030 to 1.545) |
| Platinum monotherapy | – | – | – | – |
Analysis of non-platinum-based regimens indicates that PLDH monotherapy and trabectedin plus PLDH are both significantly more effective at prolonging OS than topotecan monotherapy (Table 13). No other significant OS differences were identified.
| Comparator | Intervention | |||
|---|---|---|---|---|
| PLDH monotherapy | Trabectedin plus PLDH: HR (95% CrI) | Paclitaxel monotherapy: HR (95% CrI) | Topotecan monotherapy: HR (95% CrI) | |
| PLDH monotherapy | – | 0.835 (0.667 to 1.032) | 1.219 (0.850 to 1.690) | 1.367 (1.035 to 1.770) |
| Trabectedin plus PLDH | – | – | 1.479 (0.962 to 2.176) | 1.658 (1.157 to 2.307) |
| Paclitaxel monotherapy | – | – | – | 1.145 (0.808 to 1.576) |
| Topotecan monotherapy | – | – | – | |
Platinum-free interval is a prognostic factor for response. To investigate any potential differences in clinical efficacy between treatments with PFI, when data were available, OS was analysed for the subgroups of patients with FPS (relapse at > 12 months after last platinum-based treatment) and PPS (relapse at ≥ 6 to ≤ 12 months after last platinum-based treatment) ovarian cancer. Few trials involving platinum-sensitive patients evaluated treatment effect in these two subgroups: four trials54,56,61,64 afforded data on both FPS and PPS ovarian cancer; two trials56,61 evaluated platinum-based regimens and two trials54,64 non-platinum-based regimens.
Three of the four trials15,54,56 reported a HR as a measure of treatment effect (Table 14). The difference between treatment groups was not statistically significant in any trial. The fourth trial61 did not report a HR, but the proportion of people having an event was similar in each treatment group.
| Trial | Intervention | Comparator | HR (95% CI) |
|---|---|---|---|
| CALYPSO56 | PLDH (30 mg/m2) plus carboplatin every 21 days | Paclitaxel (175 mg/m2) plus carboplatin every 3 weeks | 0.99 (0.81 to 1.21) |
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | NR |
| Gordon et al.54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) daily for 5 days every 21 days | 1.15a (0.714 to 1.852) |
| OVA-30130,64 | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 21 days | PLDH (50 mg/m2) every 4 weeks | 0.89b (0.58 to 1.35) |
HR for OS was not available from ICON4/AGO-OVAR 2.261 and so it was not possible to carry out a NMA.
In patients with PPS ovarian cancer, PLDH monotherapy has been found to significantly prolong OS compared with topotecan (Table 15). Furthermore, trabectedin plus PLDH has been found to be significantly more effective than PLDH alone at increasing OS. The trial comparing platinum-based regimens did not report a HR for OS in this subgroup of patients. However, a similar proportion of patients in each group had had an event at the time of analysis.
| Trial | Intervention | Comparator | HR (95% CI) |
|---|---|---|---|
| CALYPSO56 | PLDH (30 mg/m2) plus carboplatin every 21 days | Paclitaxel (175 mg/m2) plus carboplatin every 3 weeks | 1.01 (0.80 to 1.28) |
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | NR |
| Gordon et al.54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) daily for 5 days every 21 days | 1.58a (1.07 to 2.33) |
| OVA-30130,64 | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 21 days | PLDH (50 mg/m2) every 4 weeks | 0.64 (0.47 to 0.86) |
The results of the NMA are in agreement with the results of the individual trials (Table 16). Trabectedin plus PLDH was found to be significantly more effective at increasing OS than PLDH monotherapy and topotecan monotherapy. The difference between PLDH monotherapy and topotecan monotherapy remained significant and favoured PLDH monotherapy.
| Comparator | Intervention | ||
|---|---|---|---|
| PLDH monotherapy | Trabectedin plus PLDH: HR (95% CrI) | Topotecan monotherapy: HR (95% CrI) | |
| PLDH monotherapy | – | 0.621 (0.493 to 0.771) | 1.610 (1.072 to 2.334) |
| Trabectedin plus PLDH | – | – | 2.628 (1.636 to 4.011) |
| Topotecan monotherapy | – | – | – |
Platinum-resistant disease has been defined as disease that initially responds followed by relapse at < 6 months after last platinum-based chemotherapy. Platinum-refractory indicates disease does not respond to or progresses during first-line platinum-based chemotherapy.
Five RCTs13,23,52,54,62 reporting results for five different head-to-head comparisons involving PRR patients were identified (Table 17). Two RCTs enrolled only patients with PRR, with the remaining three RCTs reporting results from a subgroup of patients within the trial. None of the trials identified a significant difference in OS between the two treatment groups evaluated.
| Trial name | Intervention | Comparator | HR (95% CI) |
|---|---|---|---|
| ten Bokkel Huinink et al.52 | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 0.74 (0.5 to 1.09) |
| Trial 30–57 (taken from TA9113) | PLDH (50 mg/m2) every 28 days | Paclitaxel (175 mg/m2) every 21 days | 0.87 (0.61 to 1.24) |
| Gordon et al.54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 1.07a (0.82 to 1.39) |
| Sehouli et al.23 | Topotecan (4.0 mg/m2) (weekly; days 1, 8 and 15) every 28 days | Topotecan (1.25 mg/m2) for five consecutive days every 21 days | 1.04 (0.74 to 1.44) |
| Lortholary et al.62 | Weekly paclitaxel (80 mg/m2) plus carboplatin | Weekly paclitaxel (80 mg/m2) on 4-week cycle | 1.07 (0.86 to 1.34) |
Four of the five identified trials were included in the network13,23,52,54 (Table 18); the treatment regimens evaluated in the trial reported by Lortholary et al. 62 did not inform the network. Trabectedin plus PLDH is outside of the scope for this review for the population of PRR patients; data have been included within the network to capture all the available evidence but are not included in the economic analysis. The results of the NMA are in alignment with the results of the individual trials, with no statistically significant differences in OS among the treatments evaluated.
| Comparator | Intervention | ||||
|---|---|---|---|---|---|
| PLDH monotherapy | Trabectedin plus PLDH | Paclitaxel monotherapy: HR (95% CrI) | Topotecan monotherapy: HR (95% CrI) | Topotecan monotherapy (weekly): HR (95% CrI) | |
| PLDH monotherapy | – | 0.928 (0.699 to 1.208) | 1.053 (0.783 to 1.382) | 0.973 (0.764 to 1.221) | 1.026 (0.669 to 1.505) |
| Trabectedin plus PLDH | – | – | 1.155 (0.763 to 1.681) | 1.069 (0.734 to 1.508) | 1.127 (0.666 to 1.775) |
| Paclitaxel monotherapy | – | – | – | 0.939 (0.694 to 1.244) | 0.989 (0.619 to 1.499) |
| Topotecan monotherapy | – | – | – | – | 1.054 (0.744 to 1.447) |
Platinum sensitive
In the trial carried out by Bafaloukos et al. 29 OS was calculated from the initiation of treatment until the date of last follow-up or the patient’s death. Analysis of OS was carried out on the ITT population when 122 patients were known to have died. It is important to note that the study was not powered to detect differences in OS. Median OS was 24.7 months in the PLDH plus carboplatin group and 29.4 months in the paclitaxel plus carboplatin group, with no statistically significant difference between the groups (HR 1.15, 95% CI 0.78 to 1.66; p = 0.455; see Table 23). The proportion of patients receiving post-progression therapy was similar between the groups [61/93 (65.6%) patients in the PLDH plus carboplatin group vs. 61/96 (63.5%) patients in the paclitaxel plus carboplatin group].
The authors carried out a univariate and multivariate analysis based on the Cox proportional hazards model to evaluate the influence of prespecified prognostic factors on survival. Results indicated that performance status score of zero and longer PFI (> 12 months) were important independent prognostic factors for survival (Table 19).
| Variable | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI for HR | p-value | HR | 95% CI for HR | p-value | |
| Age (years) | ||||||
| ≤ 65 years | 1 | – | – | – | – | – |
| > 65 years | 0.83 | 0.57 to 1.21 | 0.329 | – | – | – |
| Performance status | ||||||
| 0 | 1 | – | – | 1 | – | – |
| 1–2 | 1.96 | 1.32 to 2.90 | 0.001 | 1.89 | 1.25 to 2.88 | 0.003 |
| Previous exposure to taxanes | ||||||
| No | 1 | – | – | – | – | – |
| Yes | 1.18 | 0.62 to 2.27 | 0.610 | – | – | – |
| Disease status | ||||||
| Non-measurable | 1 | – | – | – | – | – |
| Measurable | 1.49 | 0.88 to 2.55 | 0.141 | – | – | – |
| PFI (months) | ||||||
| 6–12 | 1 | – | – | 1 | – | – |
| 12.1–24 | 0.58 | 0.37 to 0.89 | 0.013 | 0.54 | 0.34 to 0.86 | 0.009 |
Wagner et al. 56 report mature OS data from CALYPSO. 31 Based on a median follow-up of 49 months (range 0–68 months) and a total of 663 deaths, median OS was 30.7 months in the PLDH plus carboplatin group and 33.0 months in the paclitaxel plus carboplatin group. The accompanying HR of 0.99 (95% CI 0.85 to 1.16; p = 0.94; see Table 23) indicates that there was no statistically significant difference between treatments in OS; HR reported is for paclitaxel plus carboplatin compared with PLDH plus carboplatin. It should be noted that OS was not defined. Analysis of crossover treatment identified an imbalance between treatment groups, with a significantly larger proportion of patients randomised to paclitaxel plus carboplatin receiving PLDH (68%) compared with the alternative scenario of patients randomised to PLDH plus carboplatin receiving subsequent paclitaxel (43%; p < 0.001).
In a multivariate analysis, TFI of ≥ 12 months, ECOG performance status of 0, CA125 level of < 100 U/ml, non-measurable disease and one involved disease site were identified as factors significantly correlated with OS (Table 20).
| Variable | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI for HR | p-value | HR | 95% CI for HR | p-value | |
| Age (years) | ||||||
| < 70 | 0.98 | 0.83 to 1.16 | 0.80 | – | – | – |
| ≥ 70 years | 1.10 | 0.76 to 1.58 | 0.62 | – | – | – |
| BMI (kg/m2) | ||||||
| < 30 | 1.00 | 0.85 to 1.19 | 0.98 | 1 | – | – |
| ≥ 30 | 0.95 | 0.67 to 1.35 | 0.76 | 1.89 | 1.25 to 2.88 | 0.003 |
| TFI (months) | ||||||
| 6–12 | 1.01 | 0.80 to 1.28 | 0.92 | – | – | – |
| ≥ 12 | 0.99 | 0.81 to 1.21 | 0.90 | 0.50 | 0.43 to 0.59 | < 0.001 |
| Measurable disease/longest lesion (mm) | ||||||
| No | 0.88 | 0.65 to 1.21 | 0.56 | – | – | – |
| Yes | 1.07 | 0.90 to 1.27 | 0.47 | – | – | – |
| ≤ 50 | – | – | – | 1.28 | 1.04 to 1.57 | 0.02 |
| > 50 | – | – | – | 1.78 | 1.40 to 2.26 | < 0.001 |
| CA125 (U/ml) | ||||||
| < 100 | – | – | – | – | – | – |
| ≥ 100 | – | – | – | 1.78 | 1.49 to 2.14 | < 0.001 |
| No. of prior lines of chemotherapy | ||||||
| 1 | 0.99 | 0.84 to 1.17 | 0.92 | 1 | – | – |
| ≥ 2 | 0.97 | 0.65 to 1.46 | 0.74 | 0.54 | 0.34 to 0.86 | 0.009 |
| ECOG performance status | ||||||
| 0 | 0.99 | 0.81 to 1.20 | 0.92 | – | – | – |
| ≥ 1 | 0.99 | 0.78 to 1.27 | 0.95 | 1.37 | 1.17 to 1.60 | < 0.001 |
| Involved disease sites | ||||||
| 1 | – | – | – | – | – | – |
| > 1 | – | – | – | 1.26 | 1.05 to 1.52 | 0.014 |
Data from Alberts et al. 28 were immature in terms of OS (based on data for 32 patients who had died). Longer-term data (evaluating 50 patients who had died) reported by Markman et al. 55 found a median OS of 31 months in the PLDH plus carboplatin group and 18 months in the carboplatin alone group, giving a median OS gain of 8 months with PLDH plus carboplatin (p = 0.20). Markman et al. 55 did not report the HR for the comparison between groups. Using the methods presented by Tierney et al. ,79 the TAG calculated a HR of 0.70 (95% CI 0.40 to 1.21; see Table 23), for which HR < 1 favours PLDH plus carboplatin.
At the time of first publication of analysis of PFS from OVA-301,30 OS data were immature. Final OS analysis was reported in a follow-up study,64 in which OS analysis was based on 522 events (analysis planned once 520 deaths had occurred). Various subgroup analyses of OS are reported, including platinum-sensitive disease compared with platinum-resistant disease.
In the subgroup of patients with platinum-sensitive disease (relapse at > 6 months after last platinum-based treatment), of 430 patients randomised, 316 had died (156 in the trabectedin plus PLDH group vs. 160 in the PLDH alone group). Median OS in the trabectedin plus PLDH group was 27.0 months compared with 24.1 months in the PLDH alone group. The difference between groups did not reach statistical significance (HR 0.83, 95% CI 0.67 to 1.04; p = 0.106; see Table 23).
Observation of an unexpected, and statistically significant, difference in mean baseline PFI that favoured the PLDH group prompted the authors to carry out a post hoc analysis based on three categorisations of PFI (6 months vs. 6–12 months vs. > 12 months). The analysis suggested that patients with a longer PFI have longer OS, with median OS in each category of:
-
< 6 months PFI: 13.6 months (95% CI 11.7 to 14.8)
-
6–12 months PFI: 20.3 months (95% CI 17.7 to 21.7)
-
> 12 months PFI: 32.5 months (95% CI 28.4 to 38.5).
It should be noted that the analysis carried out (log-rank) stratified by dichotomous PFI and could not account for the observed imbalance between treatment groups in baseline PFI.
In the MS, PharmaMar present the results of a multivariate analysis Cox regression performed to provide a result for treatment effect adjusting for prespecified key prognostic factors (including PFI). The HR for OS from this analysis for the platinum-sensitive population was 0.78 (95% CI 0.62 to 0.98; p = 0.0319; taken from the PharmaMar submission), which suggests a 22% reduction for death in patients randomised to trabectedin plus PLDH. In this analysis, median OS was 28.4 months with trabectedin plus PLDH compared with 24.1 months with PLDH monotherapy. As noted earlier, as a result of variation in the reporting of adjusted and unadjusted HRs, the TAG has used unadjusted HRs in the NMA.
Data for OS for patients with platinum-sensitive disease from Gordon et al. 49 are based on 46% of the full trial population. OS results are based on a modified ITT population, and OS was defined as the time from the start of study drug administration to death. In the longer-term follow-up study (Gordon et al. 54), for the full population, OS results are also reported based on the ITT population and the more commonly used definition of OS of time from date of randomisation until date of death (presented in Interpreting the results from clinical trials, Clinical effectiveness). At the time of analysis, 87% of patients had died and 13% of observations were censored.
In platinum-sensitive patients, Gordon et al. 54 found a median OS of 107.9 weeks in the PLDH group compared with 70.1 weeks in the topotecan group. The difference between groups was statistically significant and favoured PLDH (HR 1.432, 95% CI 1.066 to 1.923; p = 0.017; see Table 23); in this analysis, HR of > 1 favours PLDH. The gain in OS corresponded to a 30% reduction in the risk of death for patients treated with PLDH. Survival rates at 1, 2 and 3 years are presented in Table 21. Results for PPS, FPS and PRR patients are discussed in subsequent sections.
| Treatment | Survival rate (%) | ||
|---|---|---|---|
| 1 year | 2 years | 3 years | |
| PLDH | 74.1 (95% CI 65.8 to 82.4) | 51.2 (95% CI 41.6 to 60.7) | 28.4 (95% CI 19.6 to 37.1) |
| Topotecan | 66.2 (95% CI 57.4 to 75.1) | 31.0 (95% CI 22.2 to 39.7) | 17.5 (95% CI 10.2 to 24.7) |
Trial 30–5713 evaluated OS as the primary outcome. TA9113 presents results for the subgroup of patients with platinum-sensitive disease (44 patients in the PLDH group vs. 41 patients in the paclitaxel group). Median OS was 65.4 weeks (range 3.9–263.7+ weeks) with PLDH and 57.0 weeks (range 14–172.3 weeks) with paclitaxel. The corresponding HR of 1.051 (95% CI 0.663 to 1.667; see Table 23) indicates that the difference between treatments is not statistically significant; HR of > 1 favours PLDH.
ten Bokkel Huinink et al. 21 defined OS as time from initial drug administration to death. Analysis of OS for the subgroup of patients with platinum-sensitive (late relapse) disease is not reported in either publication by ten Bokkel Huinink et al. 21,52 reporting OS data. TA9113 found no statistically significant difference between topotecan and paclitaxel in OS, reporting an unadjusted HR of 1.010 (95% CI 0.663 to 1.541; see Table 23) in platinum-sensitive patients, for which HR < 1 favours topotecan. It should be noted that interpretation of OS results are potentially confounded by the permitted crossover to the alternative treatment should a patient not respond to their allocated treatment. In the full population, 43.8% (49/112) and 53.5% (61/114) in the topotecan and paclitaxel groups, respectively, crossed over to the alternative treatment during the trial.
In the trial carried out by Pfisterer et al. ,50 OS was measured from the date of randomisation to the date of death from any cause. It should be noted that the trial was not powered to detect a difference between treatments in OS. At the time of analysis, 71% of patients had died. The RCT found no statistically significant difference between gemcitabine plus carboplatin and carboplatin alone in median OS (HR 0.96, 95% CI 0.75 to 1.23; p = 0.7349). Median OS was 18.0 months in the gemcitabine plus carboplatin group and 17.3 months in the carboplatin alone group.
ICON4/AGO-OVAR 2.261 defined OS as the time from randomisation to death from any cause. Patients known to be alive at the time of analysis were censored at the time of their last follow-up. At the time of analysis (median follow-up of 42 months), 530 patients (66%) had died. Median OS was significantly prolonged in the paclitaxel plus platinum-based chemotherapy compared with platinum-based chemotherapy alone (HR 0.82, 95% CI 0.69 to 0.97; p = 0.02; see Table 23). The difference between groups translates into an absolute difference in 2-year survival of 7% in favour of adding paclitaxel to platinum-based chemotherapy (57% vs. 50%). Paclitaxel plus platinum-based chemotherapy was associated with a gain in median OS of 5 months (median OS: 29 months with paclitaxel plus platinum-based chemotherapy vs. 24 months with platinum-based therapy alone).
The authors of ICON4/AGO-OVAR 2.261 also carried out an exploratory analysis to investigate the effect of randomisation strata on OS (summarised in Table 22). 61 No statistically significant difference between treatment groups was identified for any of the subgroups analysed but, as the authors noted, many of the subgroups were small and may have lacked the power to detect any real differences between the groups. A non-significant trend was noted within the subgroups of age (< 55 years vs. 55–65 years vs. > 65 years) and the number of previous lines of chemotherapy (1 vs. 2 vs. > 2).
| Randomisation strata | No. of events per number of patients | p-value (interaction or trend) | |
|---|---|---|---|
| Paclitaxel plus platinum | Platinum alone | ||
| Randomisation group | |||
| ICON4 MRC CTU | 169/266 | 176/270 | 0.84 (interaction) |
| ICON4 Italy | 67/100 | 80/113 | |
| AGO | 19/26 | 19/27 | |
| Age (years) | |||
| < 55 | 77/127 | 77/123 | 0.84 (trend) |
| 55–65 | 100/151 | 106/162 | |
| > 65 | 78/114 | 92/125 | |
| WHO performance | |||
| 0 | 146/246 | 161/262 | 0.53 (interaction) |
| > 0 | 109/146 | 114/148 | |
| Intended platinum treatment | |||
| Carboplatin | 206/332 | 215/341 | 0.16 (interaction) |
| Cisplatin | 49/60 | 60/69 | |
| Previous lines of chemotherapy | |||
| 1 | 227/354 | 260/380 | 0.08 (trend) |
| 2 | 18/22 | 12/24 | |
| > 2 | 10/15 | 3/6 | |
| Time since completion of last chemotherapy cycle (months) | |||
| ≤ 12 | 75/92 | 88/111 | 0.21 (interaction) |
| ≥ 12 | 180/300 | 187/299 | |
| Previous exposure to taxane | |||
| No | 154/223 | 176/235 | 0.49 (interaction) |
| Yes | 101/169 | 99/175 | |
The data reported by Gonzalez-Martin et al. 48 for OS are immature. At the time of analysis, median OS had not been reached in the paclitaxel plus carboplatin group. Of the 81 patients randomised, 32 patients had died, 23 in the carboplatin-alone group and nine in the paclitaxel plus carboplatin group. Analysis of available OS data found that median OS was prolonged in the paclitaxel plus carboplatin group, being significantly longer than the median OS of 72.7 weeks in the carboplatin alone group (HR 0.31, 95% CI 0.14 to 0.68; p = 0.0021; Table 23). OS was defined as time from date of randomisation to death. It should be noted that the study was not powered to identify a difference between groups in OS and that the statistical comparative analysis was exploratory.
| Study | Intervention | Comparison | Median OS (events/n) | HR | 95% CI | p-value | |
|---|---|---|---|---|---|---|---|
| Intervention | Comparator | ||||||
| Alberts et al.28 | PLDH (30 mg/m2) plus carboplatin (AUC 5) every 4 weeks | Carboplatin (AUC 5) every 4 weeks | 31 months (26/31) | 18 months (24/30) | 0.70a | 0.40 to 1.21 | 0.20 |
| Bafaloukos et al.29 | PLDH (45 mg/m2) plus carboplatin (AUC 5) on day 1 every 28 days | Paclitaxel 175 mg/m2 plus carboplatin (AUC 5) on day 1 every 21 days | 24.7 months (events NR) | 29.4 months (events NR) | 1.15 | 0.78 to 1.66 | 0.455b |
| Gordon et al.54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2 per day) for 5 days every 21 days | 107.9 weeks (n = 109) | 70.1 weeks (n = 111) | 1.43c | 1.07 to 1.92 | 0.017 |
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | 29 months | 24 months | 0.82 | 0.69 to 0.97 | 0.02 |
| OVA-30130,64 | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 27.0 months (156/218) | 24.1 months (160/211) | 0.83 | 0.67 to 1.04 | 0.106b |
| CALYPSO56 | PLDH (30 mg/m2) plus carboplatin (AUC 5) every 21 days | Paclitaxel (175 mg/m2) plus carboplatin (AUC 5) every 21 days | 33.0 months (346/509) | 30.7 months (317/467) | 0.99d | 0.85 to 1.16 | 0.94b |
| Gonzalez-Martin et al.48 | Paclitaxel (175 mg/m2) plus carboplatin (AUC 5) every 21 days | Carboplatin (AUC 5) alone every 21 days | Not yet reached (9/41) | 72.7 weeks (23/40) | 0.31 | 0.14 to 0.68 | 0.0021b |
| Pfisterer et al.50 | Gemcitabine plus carboplatin every 21 days | Carboplatin alone every 21 days | 18.0 months | 17.3 months | 0.96 | 0.75 to 1.23 | 0.7349 |
| ten Bokkel Huinink et al.52 | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 1.01e | 0.66 to 1.54 | |||
| Trial 30–5713 | PLDH (50 mg/m2) every 28 days | Paclitaxel (175 mg/m2) every 21 days | 65.4 weeks | 57.0 weeks | 1.05f | 0.66 to 1.67 | 0.833 |
The RCTs available for inclusion in the NMA evaluating OS in patients with platinum-sensitive recurrent ovarian cancer are summarised in Table 23. Unfortunately, as described earlier, a single network could not be constructed out of the available trials. The two networks constructed for this outcome are depicted in Figure 4.
Network 1 (see Figure 4a) consisted of the following comparators:
-
paclitaxel plus carboplatin
-
gemcitabine plus carboplatin
-
PLDH plus carboplatin
-
platinum as a monotherapy.
Paclitaxel plus carboplatin was chosen as the baseline treatment as this would best help inform the economic evaluation conducted by the TAG (see Chapter 4, Treatment effectiveness). However, results are reported in Table 24, sequentially covering all possible comparisons. Overall, there was no significant difference (at the 5% level) for any of the doublet chemotherapies assessed compared with paclitaxel plus carboplatin. Platinum monotherapy was associated with a significant reduction in OS compared with all doublet chemotherapies, with the exception of gemcitabine plus carboplatin, for which no significant difference was found.
Network 2 (see Figure 4b) consisted of the following comparators:
-
PLDH monotherapy
-
trabectedin plus PLDH
-
paclitaxel monotherapy
-
topotecan monotherapy.
Pegylated liposomal doxorubicin hydrochloride monotherapy was chosen as the baseline treatment, as this would best help inform the economic evaluation conducted by the TAG (see Chapter 4, Treatment effectiveness). However, results are reported in Table 24, sequentially covering all possible comparisons. Overall, there was no significant difference (at the 5% level) for trabectedin plus PLDH or paclitaxel monotherapy compared with PLDH monotherapy. Topotecan monotherapy was associated with a significant reduction in OS compared with all other chemotherapy regimens assessed, with the exception of paclitaxel monotherapy, where no significant difference was found (albeit with a non-significant trend in favour of paclitaxel monotherapy).
FIGURE 4.
Networks for OS for people with platinum-sensitive recurrent ovarian cancer. (a) Network 1; and (b) network 2.
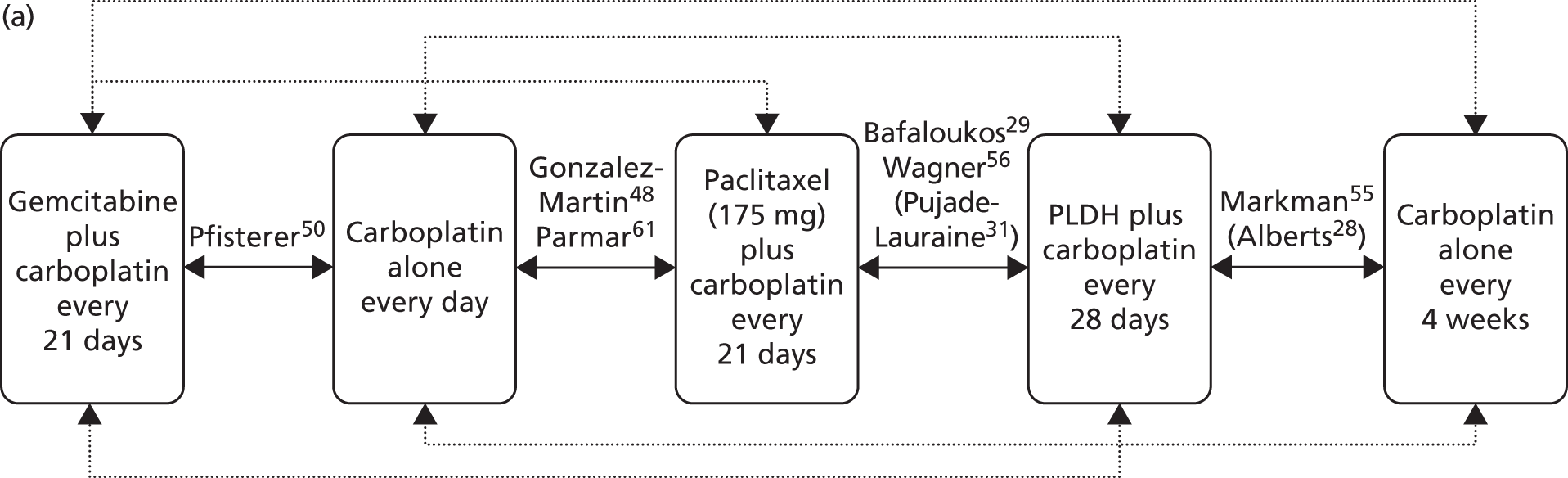
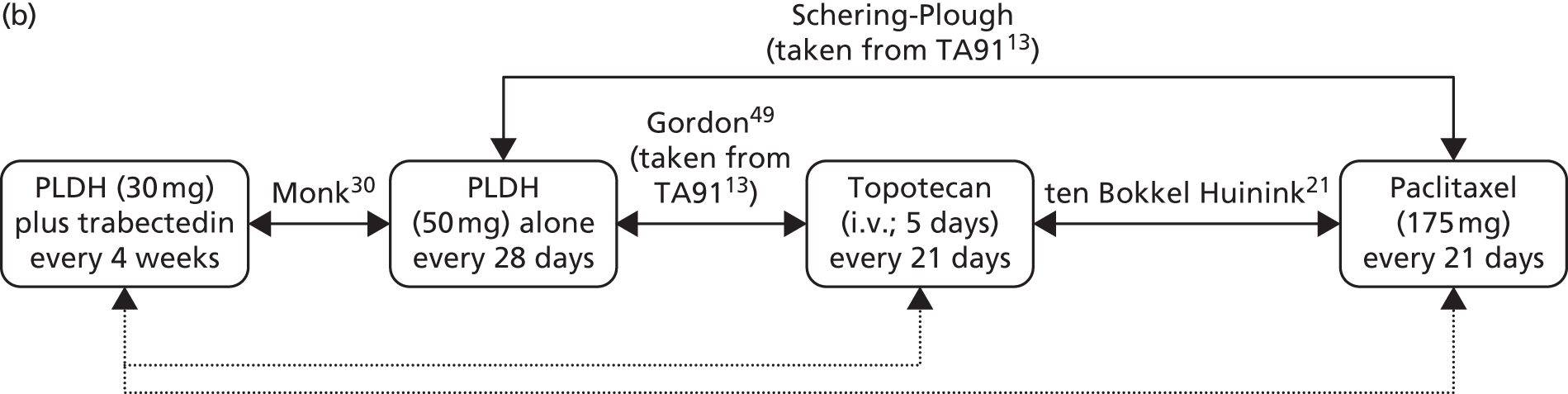
| Comparison | HR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| Network 1 | |||
| vs. paclitaxel plus carboplatin (HR < 1 favours comparator, HR > 1 favours paclitaxel plus carboplatin) | |||
| Gemcitabine plus carboplatin | 1.247 | 0.921 | 1.652 |
| PLDH plus carboplatin | 1.023 | 0.889 | 1.172 |
| Platinum as a monotherapy | 1.290 | 1.096 | 1.509 |
| vs. gemcitabine plus carboplatin (HR < 1 favours comparator, HR > 1 favours gemcitabine plus carboplatin) | |||
| PLDH plus carboplatin | 0.839 | 0.602 | 1.135 |
| Platinum as a monotherapy | 1.051 | 0.815 | 1.335 |
| vs. PLDH plus carboplatin (HR < 1 favours comparator, HR > 1 favours PLDH plus carboplatin) | |||
| Platinum as a monotherapy | 1.267 | 1.030 | 1.545 |
| Network 2 | |||
| vs. PLDH monotherapy (HR < 1 favours comparator, HR > 1 favours PLDH monotherapy) | |||
| Trabectedin plus PLDH | 0.835 | 0.667 | 1.032 |
| Paclitaxel monotherapy | 1.219 | 0.850 | 1.690 |
| Topotecan monotherapy | 1.367 | 1.035 | 1.770 |
| vs. trabectedin plus PLDH (HR < 1 favours comparator, HR > 1 favours trabectedin plus PLDH) | |||
| Paclitaxel monotherapy | 1.479 | 0.962 | 2.176 |
| Topotecan monotherapy | 1.658 | 1.157 | 2.307 |
| vs. paclitaxel monotherapy (HR < 1 favours comparator, HR > 1 favours paclitaxel monotherapy) | |||
| Topotecan monotherapy | 1.145 | 0.808 | 1.576 |
Fully platinum sensitive
Mature OS data from CALYPSO are reported in a follow-up publication to that of Pujade-Lauraine et al. 31,56 A univariate Cox regression analysis was carried out in prespecified patient subgroups, one of which was based on TFI of 6–12 months (PPS) compared with ≥ 12 months (FPS). A total of 631 patients (305 patients in the PLDH plus carboplatin group and 326 patients in the paclitaxel plus carboplatin group) had a TFI of ≥ 12 months. The univariate analysis identified no statistically significant difference between PLDH plus carboplatin and paclitaxel plus carboplatin in OS in this subgroup of patients (HR 0.99, 95% CI 0.81 to 1.21; p = 0.90). It should be noted that OS was not defined.
Neither the long-term follow-up study of OVA-30164 nor the accompanying publication presenting results for the subgroup of patients with PPS disease report data on OS in the FPS subgroup. 65 Although TA22215 reports OS data for patients with FPS disease, data are based 81% of the planned 520 deaths for the full trial population and are therefore immature. Data are reported here for completeness but have not been included in the NMA. In TA222,15 median OS in the FPS subgroup is reported as 31.7 months in the PLDH alone group. Median OS had not been reached in the trabectedin plus PLDH group. Accompanying HR of 0.89 (95% CI 0.58 to 1.35; p = 0.5746) indicates that there is no statistically significant difference between treatments in OS in this subgroup of patients. In addition to being based on immature data, this is a post hoc analysis, and as such is exploratory and hypothesis generating.
In the subgroup of patients with FPS ovarian cancer (PFI of > 12 months; 97 patients), Gordon et al. 53 found no statistically significant difference between PLDH and topotecan in OS, with a HR of 1.15 (95% CI 0.71 to 1.85; p = 0.057; see Table 25), for which HR of > 1 favours PLDH. The median OS in each group was not reported. It should be noted that the number of patients with FPS ovarian cancer in each treatment group was not reported. Furthermore, although randomisation was stratified by platinum sensitivity (sensitive vs. resistant/refractory), patients were not stratified based on PPS compared with FPS, and these subgroup analyses were not prespecified. As subgroup analyses, the results should be interpreted with caution.
ICON4/AGO-OVAR 2.261 carried out a subgroup analysis to determine the effect of paclitaxel plus platinum chemotherapy on OS in various subgroups, including time since completion of last chemotherapy regimen (≤ 12 months vs. > 12 months). Most patients had received only one prior regimen of chemotherapy (92%) and therefore TFI is akin to PFI. In the subgroup of patients with FPS ovarian cancer (599 patients), a similar proportion of people in each treatment group had died at the time of analysis [180/300 (60.0%) with paclitaxel plus carboplatin vs. 187/299 (62.5%) with carboplatin alone]. Median OS in each group for this population, or an accompanying HR or p-value for the difference between groups was not reported.
| Study | Notes | Intervention | Comparison | Intervention | Comparator | HR | 95% CI | p-value |
|---|---|---|---|---|---|---|---|---|
| Median OS (events/n) | Median OS (events/n) | |||||||
| Gordon et al.54 | Drug-free interval > 12 months n = 97 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) daily for 5 days every 21 days | NR | NR | 1.15a | 0.71 to 1.85 | 0.057 |
| CALYPSO56 | Prespecified subgroup of FPS patients | PLDH (30 mg/m2) plus carboplatin (AUC 5) every 21 days | Paclitaxel (175 mg/m2) plus carboplatin (AUC 5) every 3 weeks | 0.99 | 0.81 to 1.21 | 0.90 | ||
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | 180/300 | 187/299 | NR | NR | NR | |
| OVA-30115,30,64 | FPS subgroup | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 21 days | PLDH (50 mg/m2) every 4 weeks | Not reached | 31.7 months | 0.89 | 0.58 to 1.35b | 0.5746 |
The trials identified for potential inclusion in the NMA for OS in patients with FPS recurrent ovarian cancer are detailed in Table 25. Of the three RCTs identified, only two trials reported the required data for analysis49,56 and as they did not contain a common comparator it was not possible perform an indirect comparison.
Partially platinum sensitive
A univariate Cox regression analysis of data from CALYPSO based on TFI of 6–12 months (PPS) included 344 patients (161 patients in the PLDH plus carboplatin group and 183 patients in the paclitaxel plus carboplatin group). 56 The univariate analysis identified no statistically significant difference between PLDH plus carboplatin and paclitaxel plus carboplatin in OS in this subgroup of patients (HR 1.01, 95% CI 0.80 to 1.28; p = 0.92; see Table 26). It should be noted that OS was not defined.
An accompanying publication to OVA-30130 reports results for the subgroup of patients with PPS ovarian cancer (relapse within 6–12 months of completion of platinum-based chemotherapy). OS data presented by Poveda et al. 65 (419 deaths) are not as mature those in the long-term study reported by Monk et al. 64 (522 deaths) and therefore are not reported here.
In the subgroup of patients with PPS ovarian cancer (relapse at 6–12 months after last platinum-based treatment), trabectedin plus PLDH significantly prolonged OS compared with PLDH alone (22.4 months with trabectedin plus PLDH vs. 16.4 months with PLDH alone; HR 0.64, 95% CI 0.47 to 0.86; p = 0.0027; see Table 26). 64 The authors highlight that this is a post hoc analysis, and as such is exploratory and hypothesis generating.
In the subgroup of patients with PPS ovarian cancer (PFI of > 6–≤ 12 months; 122 patients), Gordon et al. 54 found that PLDH significantly prolonged OS compared with topotecan (HR 1.58, 95% CI 1.07 to 2.34; p = 0.021; see Table 26), for which HR of > 1 favours PLDH. The median OS in each group was not reported. It should be noted that the number of patients with PPS ovarian cancer in each treatment group was not reported. Furthermore, although randomisation was stratified by platinum sensitivity (sensitive vs. resistant/refractory), patients were not stratified based on PPS ovarian cancer compared with FPS ovarian cancer and these subgroup analyses were not prespecified.
In ICON4/AGO-OVAR 2.2,61 to be eligible for randomisation in the MRC CTU and AGO-OVAR protocols, patients had to have been treatment free for > 6 months. Thus, the subgroup of patients with a TFI of ≤ 12 months are, by the definition used in this review, PPS (213 patients). A similar proportion of people in each treatment group had died at the time of analysis [75/92 (81.5%) with paclitaxel plus carboplatin vs. 88/111 (79.3%) with carboplatin alone]. Median OS in each group for this population, or an accompanying HR or p-value for the difference between groups were not reported.
| Study | Notes | Intervention | Comparison | Intervention | Comparator | HR | 95% CI | p-value |
|---|---|---|---|---|---|---|---|---|
| Median OS (events/n) | Median OS (events/n) | |||||||
| Gordon et al.54 | Drug free interval 6–12 months n = 122 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | NR | NR | 1.58a | 1.07 to 2.34 | 0.021 |
| OVA-30130,64 | PPS subgroup (6–12 months PFI) | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 21 days | PLDH (50 mg/m2) every 4 weeks | 22.4 months | 16.4 months | 0.64 | 0.47 to 0.86 | 0.0027b |
| CALYPSO56 | Prespecified subgroup of partially sensitive patients (6–12 months) | PLDH (30 mg/m2) plus carboplatin every 21 days | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | (n = 161) | (n = 183) | 1.01 | 0.80 to 1.28 | 0.92 |
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | 75/92 | 88/111 | NR | NR |
The RCTs available for inclusion in the NMA evaluating OS in patients with platinum-sensitive recurrent ovarian cancer are summarised in Table 26. Unfortunately, as described earlier, a single network could not be constructed out of the available trials. The two networks constructed for this outcome are depicted in Figure 5.
Only Wagner et al. 56 were able to provide data for network 1 (see Figure 5) and the results are presented in Table 27. The trial demonstrated no significant difference in OS for PLDH plus carboplatin compared with paclitaxel plus carboplatin.
Network 2 (see Figure 5) consisted of the following comparators:
-
PLDH monotherapy
-
trabectedin plus PLDH
-
topotecan monotherapy.
The results of this NMA are presented in Table 27. Trabectedin plus PLDH was associated with significantly greater OS than PLDH monotherapy or topotecan monotherapy. Topotecan monotherapy was associated with a significant reduction in OS compared with all other chemotherapy regimens assessed.
FIGURE 5.
Networks for OS for people with PPS recurrent ovarian cancer. (a) Network 1; and (b) network 2.

| Comparison | HR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| vs. PLDH monotherapy (HR < 1 favours comparator, HR > 1 favours PLDH monotherapy) | |||
| Trabectedin plus PLDH | 0.621 | 0.493 | 0.771 |
| Topotecan monotherapy | 1.610 | 1.072 | 2.334 |
| vs. trabectedin plus PLDH (HR < 1 favours comparator, HR > 1 favours trabectedin plus PLDH) | |||
| Topotecan monotherapy | 2.628 | 1.636 | 4.011 |
Platinum resistant/refractory
In the subgroup of patients with PRR ovarian cancer (254 patients), Gordon et al. 54 found a median OS of 38.3 weeks in the PLDH group and 42.1 weeks in the topotecan group (median OS taken from TA9113). There was no statistically significant difference between the groups in OS (HR 1.07, 95% CI 0.82 to 1.39; p = 0.618; see Table 29); HR of > 1 favours PLDH. Survival rates at 1, 2 and 3 years are presented in Table 28.
| Treatment | Survival rate (%) | ||
|---|---|---|---|
| 1 year | 2 years | 3 years | |
| PLDH | 41.5 (95% CI 32.8 to 50.1) | 21.1 (95% CI 14.1 to 28.2) | 13.8 (95% CI 7.6 to 20.0) |
| Topotecan | 43.2 (95% CI 34.5 to 51.9) | 17.2 (95% CI 10.5 to 23.8) | 9.5 (95% CI 4.2 to 14.7) |
TA9113 presents results for the subgroup of patients with PRR disease (64 patients in the PLDH group vs. 67 patients in the paclitaxel group). 11 There was no statistically significant difference between PLDH and paclitaxel in this subgroup of patients, with a HR of 0.87 (95% CI 0.61 to 1.24), for which HR of > 1 favours PLDH. Median OS was 36.7 weeks [range 2.3–241.1 weeks (upper limit includes a censored observation)] for PLDH and 54.3 weeks [range 1.7–211.4 weeks (upper limit includes a censored observation); see Table 29] for paclitaxel.
Analysis of OS for the subgroup of patients with PRR (refractory, early and interim relapse) disease is not reported in the publications by ten Bokkel Huinink et al. 21,52 TA9113 found no statistically significant difference between topotecan and paclitaxel in OS, reporting an unadjusted HR of 0.74 (95% CI 0.5 to 1.09; see Table 29) in PRR patients, for which HR < 1 favours topotecan. 13 It should be noted that interpretation of OS results are potentially confounded by the permitted crossover to the alternative treatment should a patient not respond to their allocated treatment.
OS (not defined) was evaluated by Lortholary et al. 62 as a secondary outcome and was reported not to differ among treatment groups, with median OS of 19.9 months, 15.2 months and 18.6 months for weekly paclitaxel, weekly paclitaxel plus carboplatin and weekly paclitaxel plus weekly topotecan, respectively. The number of events at the time of analysis is unclear. As discussed earlier, results from the weekly paclitaxel plus topotecan group are not of interest to this systematic review. The authors of the study were contacted with a request for the HR for the comparison of weekly paclitaxel compared with weekly paclitaxel plus carboplatin. The authors helpfully provided the requested information, which indicates that there is no significant difference between the two treatment groups in median OS (HR 1.07, 95% CI 0.86 to 1.34; p = 0.53; see Table 29).
OS was not defined by Sehouli et al. 23 After a median duration of follow-up of 23.4 months, 55 (28.4%) patients remained alive. Median OS in the weekly topotecan group was 9.6 months compared with 9.3 months in the conventional topotecan group. The difference between groups did not reach statistical significance, with a HR of 1.04 (95% CI 0.74 to 1.44; p = 0.83; see Table 29). The authors carried out a multivariate regression analysis that identified the factors listed below as independent predictors of OS:
-
duration of chemotherapy (HR 0.99, 95% CI 0.99 to 1.00; p < 0.001)
-
baseline ECOG score (HR 1.47, 95% CI 1.16 to 1.86; p = 0.001)
-
administration of follow-up chemotherapy (HR 0.53, 95% CI 0.37 to 0.76; p = 0.001).
| Study | Notes | Intervention | Comparison | Intervention | Comparator | HR | 95% CI | p-value |
|---|---|---|---|---|---|---|---|---|
| Median OS (events/n) | Median OS (events/n) | |||||||
| Gordon et al.54 | Platinum refractory/resistant | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 38.3 weeks (n = 130) | 42.1 weeks (n = 125) | 1.07a | 0.82 to 1.39 | 0.618 |
| Sehouli et al.23 | Full population recurrent platinum-resistant-patients | Topotecan (4.0 mg/m2) (weekly; days 1, 8 and 15) every 28 days | Topotecan (1.25 mg/m2) for five consecutive days every 21 days | 9.6 months (95% CI 6.3 to 14.2) | 9.3 months (95% CI 7.5 to 11.4) | 1.04 | 0.74 to 1.44 | 0.83 |
| ten Bokkel Huinink et al.52 | HR taken from TA91;13 data not presented in the published paper | Topotecan (1.5 mg/m2/day) for 5 days | Paclitaxel (175 mg/m2/day) as 3-hour infusion every 21 days | 0.74b | 0.5 to 1.09 | |||
| Trial 30–5713 | Trial was terminated prematurely | PLDH (50 mg/m2) every 28 days | Paclitaxel (175 mg/m2) every 21 days | 36.7 weeks (range 2.3–241.1 weeks) | 54.3 weeks (range 1.7–211.4 weeks) | 0.87 | 0.61 to 1.24 | 0.427 |
| Lortholary et al.62 | Full population relapsed within 6 months | Weekly paclitaxel (80 mg/m2 on days 1, 8 and 15) plus carboplatin (AUC 5) every 4 weeks | Weekly paclitaxel (80 mg/m2 on days 1, 8 and 15) every 4 weeks | 15.2 months | 19.9 months | 1.07c | 0.86 to 1.34 | 0.53 |
The RCTs available for inclusion in the NMA evaluating OS in patients with PRR recurrent ovarian cancer are summarised in Table 29. The network of trials constructed for this outcome is depicted in Figure 6 and contains the following comparators:
-
PLDH monotherapy
-
trabectedin plus PLDH
-
paclitaxel monotherapy
-
topotecan monotherapy, i.e. topotecan 1.25 or 1.5 mg/m2 daily for 5 days every 21 days
-
topotecan monotherapy (weekly); i.e. topotecan 4.0 mg/m2 (weekly) on days 1, 8 and 15 of a 28-day cycle.
The results from this NMA are presented in Table 30. Overall, there was no significant difference in OS (at the 5% level) for any of the chemotherapies assessed compared with PLDH monotherapy (or with each other).
A RCT that provided results for this population but which did not share a common comparator within the network compared low-dose paclitaxel (80 mg/m2) with low-dose paclitaxel (80 mg/m2) plus carboplatin. 62 However, Lortholary et al. 62 identified no significant difference in OS between the two different treatment regimens (see Table 29). Trabectedin plus PLDH is outside of the scope for this review for the population of PRR patients; data have been included within the network to capture all of the available evidence but are not included in the economic analysis.
FIGURE 6.
Networks for OS for people with PRR recurrent ovarian cancer.
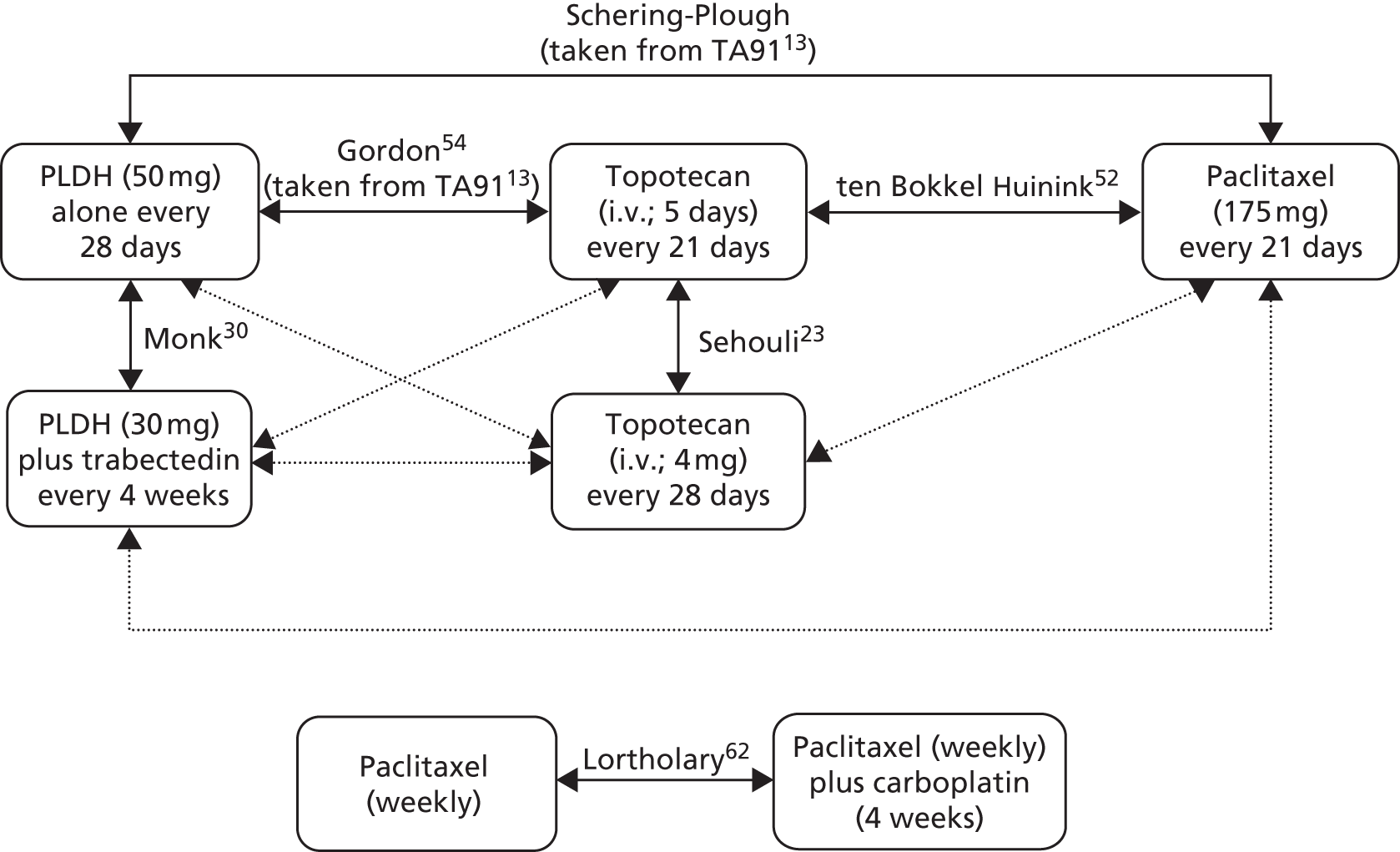
| Comparison | HR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| vs. PLDH monotherapy (HR < 1 favours comparator, HR > 1 favours PLDH monotherapy) | |||
| Trabectedin plus PLDH | 0.928 | 0.699 | 1.208 |
| Paclitaxel monotherapy | 1.053 | 0.783 | 1.382 |
| Topotecan monotherapy | 0.973 | 0.764 | 1.221 |
| Topotecan monotherapy (weekly) | 1.026 | 0.669 | 1.505 |
| vs. trabectedin plus PLDH (HR < 1 favours comparator, HR > 1 favours trabectedin plus PLDH) | |||
| Paclitaxel monotherapy | 1.155 | 0.763 | 1.681 |
| Topotecan monotherapy | 1.069 | 0.734 | 1.508 |
| Topotecan monotherapy (weekly) | 1.127 | 0.666 | 1.775 |
| vs. paclitaxel monotherapy (HR < 1 favours comparator, HR > 1 favours paclitaxel monotherapy) | |||
| Topotecan monotherapy | 0.939 | 0.694 | 1.244 |
| Topotecan monotherapy (weekly) | 0.989 | 0.619 | 1.499 |
| vs. topotecan monotherapy (HR < 1 favours comparator, HR > 1 favours topotecan monotherapy) | |||
| Topotecan monotherapy (weekly) | 1.054 | 0.744 | 1.447 |
Full population (mixed platinum-free intervals)
Based on 522 deaths (analysis planned at 520 deaths), OVA-301 found no significant difference in OS between the two treatments, with median OS of 22.2 months in the trabectedin plus PLDH group and 18.9 months in the PLDH alone group (HR 0.86, 95% CI 0.72 to 1.02; p = 0.084; see Table 34). 64 Survival rates in the two treatment groups at various time points are presented in Table 31.
| Treatment | Survival rate (%) | ||
|---|---|---|---|
| 12 months | 24 months | 30 months | |
| Trabectedin plus PLDH | 74 (95% CI 69 to 79) | 45 (95% CI to 40 to 51) | 37 (95% CI 14.9 to 15.5) |
| PLDH alone | 68 (95% CI 62 to 72) | 41 (95% CI 35 to 46) | 37 (95% CI 31 to 42) |
As noted earlier, data for OS from Gordon et al. 49 are based on a modified ITT population and OS was defined as the time from the start of study drug administration to death. In the longer-term study,54 additional analyses are presented in which OS results for the full trial population are based on the ITT population and the more commonly used definition of OS of time from date of randomisation until date of death. At the time of analysis, 87% of patients had died and 13% of observations were censored. For completeness, both results are reported here.
Based on the modified ITT population (n = 474) and the original definition of OS, Gordon et al. 54 found that PLDH significantly prolonged median OS compared with topotecan, with a median gain of 3.0 weeks (median OS 62.7 weeks with PLDH vs. 59.7 weeks with topotecan; HR 1.22, 95% CI 1.00 to 1.48; p = 0.05; see Table 34); in this analysis, HR of > 1 favours PLDH. The gain in OS associated with PLDH corresponded to an 18% reduction in the risk of death. Similar results were observed in the analysis of all patients randomised (n = 481), with a median gain of 6.6 weeks associated with PLDH (median OS 63.6 weeks with PLDH vs. 57.0 weeks with topotecan; HR 1.23, 95% CI 1.01 to 1.50; p = 0.038; see Table 34); in this analysis, HR of > 1 favours PLDH. Survival rates in the two treatment groups at various time points are presented in Table 32.
| Treatment | Survival rate (%) | ||
|---|---|---|---|
| 1 year | 2 years | 3 years | |
| PLDH | 56.3 (95% CI 50.0 to 62.6) | 34.7 (95% CI 28.6 to 40.8) | 20.2 (95% CI 14.9 to 15.5) |
| Topotecan | 54.0 (95% CI 47.6 to 60.3) | 23.6 (95% CI 18.1 to 29.2) | 13.2 (95% CI 8.8 to 17.7) |
To investigate the influence of multiple putative prognostic factors on OS, the authors carried out a multivariate Cox regression analysis. 54 Variables evaluated were treatment, platinum sensitivity (sensitive vs. resistant/refractory), bulky disease (yes vs. no), baseline Karnofsky performance status (KPS) (< 80 vs. ≥ 80). The adjusted HR for OS was similar to that of the primary analysis, which led the authors to conclude that the results were not affected by potential prognostic factors (summarised in Table 33). Results suggest that age of < 65 years, platinum-sensitive disease and absence of ascites at baseline are associated with improved survival.
| Variable | Group | n | HRa | 95% CI for HR |
|---|---|---|---|---|
| Age (years) | < 65 | 294 | 1.322 | 1.022 to 1.710 |
| ≥ 65 | 180 | 1.077 | 0.786 to 1.477 | |
| Baseline KPS | < 80 | 76 | 0.871 | 0.531 to 1.427 |
| ≥ 80 | 394 | 1.242 | 0.999 to 1.543 | |
| Drug-free interval (months) | ≤ 6b | 211 | 1.103 | 0.826 to 1.474 |
| < 12 | 367 | 1.224 | 0.983 to 1.523 | |
| > 18 | 107 | 1.088 | 0.687 to 1.726 | |
| Bulky disease | Present | 213 | 1.131 | 0.849 to 1.506 |
| Absent | 261 | 1.294 | 0.991 to 1.691 | |
| Platinum sensitivity | Sensitive | 219 | 1.432 | 1.066 to 1.923 |
| Refractory | 255 | 1.069 | 0.823 to 1.387 | |
| Baseline ascites | Present | 142 | 0.978 | 0.689 to 1.389 |
| Absent | 330 | 1.387 | 1.088 to 1.768 |
In the full trial population of Trial 30–5713 (216 patients), there was no statistically significant difference between PLDH and paclitaxel in OS, with a HR of 0.93 (95% CI 0.70 to 1.23; see Table 34); HR of > 1 favours PLDH. Median OS was 46.6 weeks [range 2.3–263.7 weeks (includes censored observation)] with PLDH compared with 56.3 weeks (range 1.4–211.4 weeks) with paclitaxel.
Data reported here are taken from the longer-term follow-up study reported by ten Bokkel Huinink et al. 52 in which data had been collected for > 4 years. For analysis of OS, 20.5% of patients in the topotecan group and 12.3% of patients in the paclitaxel group were censored. There was no statistically significant difference between topotecan and paclitaxel in median OS (63 weeks with topotecan vs. 53 weeks with paclitaxel; p = 0.44; see Table 34). 52 An accompanying HR was not reported in the full publication. However, TA9113 reported a HR of 0.91 (95% CI 0.68 to 1.23) for OS, for which HR < 1 favours topotecan. 13 The HR had been adjusted for stratification factors. It should be noted that interpretation of OS results are potentially confounded by the permitted crossover to the alternative treatment should a patient not respond to their allocated treatment.
Piccart et al. 63 evaluated OS as a secondary outcome measure, with OS defined as the time from day 1 of treatment to death. At the time of analysis, of the 86 patients randomised, 45 had died [52%; 25/41 (61.0%) in the paclitaxel group vs. 20/45 (44.4%) in the oxaliplatin group; see Table 34]. Median OS was 37 weeks in the paclitaxel group compared with 42 weeks in the oxaliplatin group. Statistical significance was not assessed in the full publication. Neither an accompanying HR nor a p-value for the difference between groups was reported.
In the full trial population, Gore et al. 24 found that median OS was significantly prolonged with i.v. topotecan compared with oral topotecan, with a median OS of 51 weeks with oral topotecan compared with 58 weeks with i.v. topotecan (risk ratio of death 1.36, 95% CI 1.00 to 1.85; p = 0.033; see Table 34). It should be noted that OS was not defined in the full publication.
Omura et al. 68 defined OS as the time from randomisation until the date of death, or last contact if the date of death was unknown. Estimated median OS for the paclitaxel 175 mg/m2 and the 250 mg/m2 regimens were 13.1 and 12.3 months, respectively. The accompanying HR of 0.97 (95% CI 0.77 to 1.22; ratio of 250 mg/m2 compared with 175 mg/m2; see Table 34) indicated that OS was not statistically significantly different between the two paclitaxel regimens. The HR was adjusted for initial performance score, cell type, response to prior platinum, cooperative group and measurable disease. An unadjusted HR was not reported.
Rosenberg et al. 60 defined OS as time from date of randomisation to death or censored observation. In the full trial population, there was no statistically significant difference between treatment regimens in median OS (p = 0.98). Median OS was 13.6 months (95% CI 10.5 to 18.7 months) in the group receiving paclitaxel every 7 days compared with 14.7 months (95% CI 12.3 to 19.1 months) in the group receiving paclitaxel every 21 days. It is unclear how many events had occurred at the time of analysis.
| Study | Notes | Intervention | Comparison | Median OS (events/n) | HR | 95% CI | p-value | |
|---|---|---|---|---|---|---|---|---|
| Intervention | Comparator | |||||||
| Gordon et al.54 | ‘Assessable population’–contains mix of platinum-sensitive patients and platinum-refractory patients | PLDH 50 mg/m2 every 28 days | Topotecan 1.5 mg/m2 per day for 5 days every 21 days | 62.7 weeks | 59.7 weeks | 1.22a | 1.00 to 1.48 | 0.05 |
| 63.6 weeks | 57.0 weeks | 1.23 | 1.01 to 1.50 | 0.038b | ||||
| Piccart et al.63 | Approximately 75% of population is platinum refractory, 25% is platinum sensitive | Paclitaxel 175 mg/m2 over 3 hours every 3 weeks | Oxaliplatin 130 mg/m2 over 2 hours every 3 weeks | 37 weeks (n = 25/41) | 42 weeks (n = 20/45) | NR | NR | NR |
| OVA-30164 | Full population contains platinum-sensitive patients and platinum-resistant | PLDH 30 mg/m2 i.v. plus trabectedin 1.1 mg/m2 every 3 weeks | PLDH 50 mg/m2 every 4 weeks | 22.2 months (95% CI 19.3 to 25 months) (258/337) | 18.9 months (95% CI 17.1 to 21.5 months) (264/335) | 0.86 | 0.72 to 1.02 | 0.084 |
| Gore et al.24 | Full population contains platinum refractory, platinum-resistant and platinum-sensitive patients | Oral topotecan 2.3 mg/m2/day | i.v. topotecan 1.5 mg/m2/day for 5 days every 21 days | 51 weeks (n = 135) | 58 weeks (n = 131) | 1.36 | 1.00 to 1.85 | 0.033 |
| ten Bokkel Huinink et al.52 | cHR taken from TA9113 | Topotecan 1.5 mg/m2/day for 5 days | Paclitaxel 175 mg/m2/day as 3-hour infusion every 21 days | 63 weeks (range < 1–238.4+ weeks) | 53 weeks (range < 1–226.3+ weeks) | 0.91; adjusted for stratification factors | 0.68 to 1.23 | 0.44 |
| p = 0.44 | ||||||||
| Trial 30–5713 | Trial was terminated prematurely | PLDH 50 mg/m2/day every 28 days | Paclitaxel 175 mg/m2/day every 21 days | 46.6 weeks (range 2.3–263.7+ weeks) | 56.3 weeks (range 1.4–211.4 weeks) | 0.93 | 0.70 to 1.23 | 0.0618 |
| Rosenberg et al.60 | Mixed population | Paclitaxel weekly | Paclitaxel 3 weekly | 13.6 months (range 10.5–18.7 months) (n = 105) | 14.7 months (range 12.3–19.1 months) (n = 103) | |||
| p = 0.98 | ||||||||
| Omura et al.68 | Paclitaxel 250 mg/m2 every 21 days | Paclitaxel 175 mg/m2 every 21 days | 12.3 months | 13.1 months | 0.97 | 0.774 to 1.22 | NR | |
The RCTs available for inclusion in the NMA evaluating OS in patients with mixed PFIs in recurrent ovarian cancer are summarised in Table 34. However, based on expert clinical opinion, the TAG decided not to evaluate this mixed patient population, as the results would not be considered clinically meaningful.
Progression-free survival
In oncology trials, progression of disease is typically assessed according to internationally recognised criteria, such as the RECIST criteria,69 which are based on clinical signs, ultrasound scans or X-rays. RECIST criteria encompass measurable and non-measurable disease. Increase in levels of CA125 biomarker is also used to determine disease progression, typically in patients with non-measurable lesions at baseline; according to criteria developed by Rustin et al. ,80 increase in CA125 level has been shown to predate evidence of disease progression from clinical examinations or radiological scans in 70% of patients with ovarian cancer by a median of 4 months. 76 There are two time-to-event measures of disease progression (definitions as reported in Food and Drug Administration guidance on conducting oncology trials):77
-
PFS, which is defined as time from randomisation to disease progression or death (includes all deaths)
-
TTP, which is defined as time from randomisation to disease progression (deaths before progression are censored).
The terms PFS and TTP are often used interchangeably. For example, a trial might refer to the outcome of PFS but the definition indicates that all-cause mortality has not been included in the analysis. For the purposes of the review, the TAG has considered PFS and TTP together and has reported the outcome as defined in the individual trials. As for OS, in some cases, PFS and TTP have been measured from the time of treatment initiation rather than randomisation.
Progressive events occur in a shorter timeframe and more frequently than OS events. Therefore, PFS data are available much sooner than OS data. Additionally, there is no confounding from postprogression therapy. However, because PFS is based on assessment of change in tumour size, there is a degree of subjective assessment, with associated potential for measurement errors. Assessment bias is more likely in an open-label trial. Differences in the timing of measurement between the groups may arise if the treatments under evaluation have different cycle lengths, which could lead to a difference in progression date. In clinical trials, it has been reported that an increase in CA125 level frequently triggers subsequent postprogression therapy before clinical or radiological confirmation of progression. The practice of using CA125 level alone also introduces disparity across trials in terms of the date of disease progression.
The criteria used to determine progression were initially developed for use in clinical trials using response rate as a primary end point (e.g. Phase II screening trials), with the goal of facilitating evaluation of changes in tumour burden during treatment rather than to associate the changes with a clinical benefit. 78 However, changes in tumour size are recognised as signals of a drug’s anti-tumour activity.
Summary of results for progression-free survival/time to progression
Results are presented for PFS or TTP, as reported in the trial. PFS and TTP are often used interchangeably and, for the purposes of the results presented here, TTP has been assumed to approximate to PFS. Definitions as reported in the trials are provided in the main text. No trial was identified evaluating treatments in a population solely comprising patients who were allergic or intolerant to platinum-based chemotherapy. Here, results for patients with platinum-sensitive or PRR disease are summarised. For trials not limited to either platinum-sensitive or PRR patients (i.e. includes a mix of PFI), results for the full trial population are presented in the main text.
Nine RCTs28–31,48,50,52,54,61 evaluating seven different head-to-head comparisons of interventions and comparators of interest reported on PFS/TTP (Table 35).
| Trial name | Intervention | Comparator | HR (95% CI) |
|---|---|---|---|
| CALYPSO31 | PLDH (30 mg/m2) plus carboplatin every 21 days | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | 0.82 (0.72 to 0.94) |
| Bafaloukos et al.29 | PLDH (45 mg/m2) plus carboplatin every 28 days | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | NR |
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | 0.76 (0.66 to 0.89) |
| Gonzalez-Martin et al.48 | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | 0.54 (0.32 to 0.92) |
| ten Bokkel Huinink et al.52 | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 0.82 (0.54 to 1.26) |
| Gordon et al.54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 1.29 (0.98 to 1.69)a |
| Alberts et al.28 | PLDH (30 mg/m2) plus carboplatin every 4 weeks | Carboplatin alone every 4 weeks | 0.54 (0.32 to 0.93) |
| OVA-30130 | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 0.73 (0.56 to 0.95) |
| Pfisterer et al.50 | Gemcitabine (1000 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | 0.72 (0.58 to 0.90) |
As for OS, based on trials identified, it was not possible to construct a complete network. Again, two discrete networks were generated, one evaluating platinum-based therapies and the second comparing non-platinum-based regimens. It should be stressed that results from the two discrete networks are not directly comparable.
In the network evaluating platinum-based chemotherapies, all combination chemotherapy regimens significantly improved PFS compared with platinum monotherapy (Table 36). In addition, PLDH plus carboplatin was found to be significantly more effective at prolonging PFS than paclitaxel plus carboplatin. No other statistically significant differences were identified between combination regimens.
| Comparator | Intervention | |||
|---|---|---|---|---|
| Paclitaxel plus carboplatin | Gemcitabine plus carboplatin: HR (95% CrI) | PLDH plus carboplatin: HR (95% CrI) | Platinum monotherapy: HR (95% CrI) | |
| Paclitaxel plus carboplatin | – | 0.985 (0.748 to 1.273) | 0.817 (0.717 to 0.927) | 1.361 (1.182 to 1.559) |
| Gemcitabine plus carboplatin | – | – | 0.845 (0.624 to 1.116) | 1.400 (1.106 to 1.749) |
| PLDH plus carboplatin | – | – | – | 1.672 (1.389 to 1.997) |
| Platinum monotherapy | – | – | – | – |
Analysis of non-platinum-based regimens indicates that trabectedin plus PLDH significantly improves PFS compared with PLDH, paclitaxel and topotecan when given as monotherapy (Table 37). No statistically significant differences were identified among the monotherapies evaluated (PLDH, topotecan and paclitaxel).
| Comparator | Intervention | |||
|---|---|---|---|---|
| PLDH monotherapy | Trabectedin plus PLDH: HR (95% CrI) | Paclitaxel monotherapy: HR (95% CrI) | Topotecan monotherapy: HR (95% CrI) | |
| PLDH monotherapy | – | 0.736 (0.560 to 0.949) | 1.615 (0.939 to 2.586) | 1.298 (0.979 to 1.688) |
| Trabectedin plus PLDH | – | – | 2.236 (1.209 to 3.795) | 1.797 (1.207 to 2.578) |
| Paclitaxel monotherapy | – | – | – | 0.842 (0.539 to 1.262) |
| Topotecan monotherapy | – | – | – | – |
Where available, PFS/TTP data were analysed for the subgroups of patients with FPS (relapse at > 12 months after last platinum-based treatment) and PPS (relapse at ≥ 6 to ≤ 12 months after last platinum-based treatment). As for OS, few trials involving platinum-sensitive patients evaluated treatment effect in these two subgroups: three trials afforded data on FPS and four trials on PPS. Two trials evaluated platinum-based regimens and two trials non-platinum-based regimens.
One65 of the three trials50,61,65 reported a HR as a measure of treatment effect (Table 38). The difference between trabectedin plus PLDH and PLDH monotherapy was not statistically significant. The two remaining trials50,61 did not report a HR for PFS but the proportion of people having an event was similar in each treatment group. The lack of HRs for two of the trials50,61 precluded carrying out a NMA.
| Trial | Intervention | Comparator | HR (95% CI) |
|---|---|---|---|
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | NR |
| OVA-30165 | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 0.70 (0.47 to 1.03) |
| Pfisterer et al.50 | Gemcitabine (1000 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | NR |
Two of the four trials56,65 evaluating treatments in the subgroup of patients with PPS ovarian cancer reported HR as a measure of effect. PLDH plus carboplatin was found to significantly prolong PFS compared with paclitaxel plus carboplatin (Table 39). 56 In addition, trabectedin plus PLDH significantly improved PFS compared with PLDH alone. 65 The two remaining trials50,61 did not report HRs. The proportion of patients experiencing an event was similar in the two treatment groups in each trial. The lack of HRs for two of the trials50,61 precluded carrying out a NMA.
| Trial | Intervention | Comparator | HR (95% CI) |
|---|---|---|---|
| CALYPSO56 | PLDH (30 mg/m2) plus carboplatin every 21 days | Paclitaxel (175 mg/m2) plus carboplatin every 3 weeks | 0.73 (0.58 to 0.90) |
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | NR |
| OVA-30165 | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 21 days | PLDH (50 mg/m2) every 4 weeks | 0.65 (0.45 to 0.92) |
| Pfisterer et al.50 | Gemcitabine (1000 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | NR |
Four RCTs23,52,54,62 reporting results for four different head-to-head comparisons involving PRR patients were identified. Two RCTs enrolled only patients with PRR23,62 with the remaining two RCTs52,54 reporting results from a subgroup of patients within the trial. None of the trials identified a significant difference in PFS/TTP between the two treatment groups evaluated (Table 40).
| Trial name | Intervention | Comparator | HR (95% CI) |
|---|---|---|---|
| ten Bokkel Huinink et al.52 | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 0.75 (0.50 to 1.12) |
| Gordon et al.54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 0.99a (0.77 to 1.28) |
| Sehouli et al.23 | Topotecan (4.0 mg/m2) (weekly; days 1, 8 and 15) every 28 days | Topotecan (1.25 mg/m2) for five consecutive days every 21 days | 1.29 (0.96 to 1.76) |
| Lortholary et al.62 | Weekly paclitaxel (80 mg/m2) plus carboplatin | Weekly paclitaxel (80 mg/m2) on 4-week cycle | 0.92 (0.76 to 1.2) |
Three23,52,54 of the four identified trials were included in the network; the treatment regimens evaluated in the trial reported by Lortholary et al. 62 did not inform the network. Trabectedin plus PLDH is outside of the scope for this review for the population of PRR patients; data have been included within the network to capture all of the available evidence but are not included in the economic analysis. The results of the NMA are in alignment with the results of the individual trials, with no statistically significant differences in PFS among PLDH, paclitaxel and topotecan monotherapy (Table 41).
| Comparator | Intervention | ||||
|---|---|---|---|---|---|
| PLDH monotherapy | Trabectedin plus PLDH | Paclitaxel monotherapy: HR (95% CrI) | Topotecan monotherapy: HR (95% CrI) | Topotecan monotherapy (weekly): HR (95% CrI) | |
| PLDH monotherapy | – | 0.961 (0.697 to 1.292) | 1.360 (0.817 to 2.123) | 0.998 (0.767 to 1.277) | 1.302 (0.859 to 1.894) |
| Trabectedin plus PLDH | – | – | 1.450 (0.791 to 2.454) | 1.064 (0.698 to 1.555) | 1.389 (0.811 to 2.216) |
| Paclitaxel monotherapy | – | – | 0.765 (0.502 to 1.122) | 0.999 (0.585 to 1.599) | |
| Topotecan monotherapy | – | – | – | 1.305 (0.951 to 1.744) | |
| Topotecan monotherapy (weekly) | – | – | – | – | |
Platinum sensitive
Bafaloukos et al. 29 evaluated TTP, which was defined as the time from the initiation of treatment to the first disease progression. Deaths as a result of disease without previous documentation of progression were considered events in TTP. Median TTP was 11.8 months in the PLDH plus carboplatin group compared with 10.8 months in the paclitaxel plus carboplatin group (see Table 47), with no statistically significant difference between treatments for this outcome (p = 0.904). It is important to note that the study was not powered to detect differences in TTP. An accompanying HR was not reported.
Progression-free survival was the primary outcome in the CALYPSO trial31 and primary analysis was based on the ITT population. Although a comprehensive description of criteria for categorisation of disease progression is provided, it is unclear when monitoring for progression began, that is, from randomisation or from first administration of study drug. Tumour assessment was carried out every 3 months while patients were receiving treatment.
After a median follow-up of 22 months, 832 PFS events had occurred. PLDH plus carboplatin significantly prolonged median PFS compared with carboplatin plus paclitaxel, with a median PFS gain of 1.9 months (median PFS 11.3 months with PLDH plus carboplatin vs. 9.4 months with paclitaxel plus carboplatin; HR 0.82, 95% CI 0.72 to 0.94; p = 0.005). The test for non-inferiority of PLDH plus carboplatin afforded a p-value of < 0.001. A similar proportion of patients in each group had disease progression based on RECIST criteria69 (Table 42).
| Disease progression measure | PLDH plus carboplatin | Paclitaxel plus carboplatin | ||
|---|---|---|---|---|
| No. of patients | % | No. of patients | % | |
| RECIST criteria69 | 301 | 79 | 363 | 80 |
| CA125 GCIG criteria70 | 79 | 21 | 89 | 20 |
Exploratory analysis of the effects of several baseline characteristics on PFS was carried out using Cox proportional hazards regression. Factors evaluated were age; number of previous lines of chemotherapy; TFI; surgery at relapse; measurability status of tumour; size of tumour (< 5 cm or ≥ 5 cm); number of tumour sites (1 or > 1); tumour grade; histological classification of tumour cells; CA125 level; ECOG performance score; and treatment arm. Limited results are available in the full publication (summarised in Table 43). TFI, measurable disease, CA125 level of ≥ 100 and PLDH plus carboplatin were found to be associated with a significant effect on PFS. It is unclear whether the remainder of the putative prognostic factors had no effect on PFS.
| Baseline factor | n | HR | 95% CI | p-value |
|---|---|---|---|---|
| TFI (months) | ||||
| 6–12 | 342 | 1.00 | 0.48 to 0.65 | < 0.001 |
| > 12 | 617 | 0.56 | – | – |
| Measurable disease | ||||
| No | 362 | 1.00 | 1.27 to 1.70 | < 0.001 |
| Yes | 597 | 1.47 | – | – |
| CA125 (U/ml) | ||||
| < 100 | 316 | 1.00 | 1.52 to 2.07 | < 0.001 |
| ≥ 100 | 643 | 1.77 | – | – |
| Treatment group | ||||
| Paclitaxel plus carboplatin | 499 | 1.00 | 0.71 to 0.93 | 0.003 |
| PLDH plus carboplatin | 460 | 0.80 | – | – |
Alberts et al. 28 reported that PFS was measured as a secondary outcome but a definition of PFS was not provided. Based on 55 out of 61 women having progressed or died, Alberts et al. 28 found a median PFS (unadjusted) of 12 months in the PLDH plus carboplatin group and 8 months in the carboplatin alone group (HR 0.54, 95% CI 0.32 to 0.93; p = 0.03; see Table 47). Longer-term data (all women had progressed or died) reported by Markman et al. 55 found similar results, with median PFS of 12 months and 8 months in the PLDH plus carboplatin group and carboplatin alone group, respectively (HR not reported; p = 0.02).
PFS was the primary outcome of the OVA-301 trial, and was defined as time from random assignment to disease progression or death. 30 Three analyses for PFS were performed, based on review by independent radiologists, independent oncologists and investigator. The primary analysis was based on review by independent radiologists who were masked to treatment allocation, with disease progression determined by radiological evaluation alone according to RECIST criteria. 69 The primary analysis included only those patients who had measurable disease at baseline. A secondary analysis was based on review by independent oncologists who were also masked to treatment and who categorised disease progression based on radiological assessments together with clinical data. The secondary analysis included all randomised patients.
The sample size calculation estimates that 415 progressive events would be needed to test statistical difference at a two-sided 5% significance level with at least 90% power, based on assumed median PFS of 16 weeks and 22 weeks for PLDH alone and trabectedin plus PLDH, respectively. At the time of analysis of PFS, in the full trial population (includes platinum-resistant patients), 389 events had occurred according to independent radiology review and 432 events based on independent oncologist review. Based on event rate, the primary analysis of PFS could be underpowered. In the FAD for TA222, the Committee concluded that ‘despite the technical difficulties, the analysis based on the independent radiologists’ assessment was the most robust’. 73 For this reason, the TAG has used results from the primary analysis of PFS in the NMA.
In the subgroup of patients with platinum-sensitive disease, all three analyses found that median PFS was significantly prolonged with trabectedin plus PLDH compared with PLDH alone (Table 44). Multivariate analysis of potential prognostic factors found that treatment with trabectedin plus PLDH remained significant after adjustment of prognostic factors; the multivariate analysis was based on the full trial population and is presented in the section outlining results in the full trial population.
| Review | Median PFS (months) | HR (95% CI) | p-value | |
|---|---|---|---|---|
| Trabectedin plus PLDH | PLDH alone | |||
| Independent radiologist | 9.2 | 7.5 | 0.73 (0.56 to 0.95) | 0.0170 |
| Independent oncologist | 9.7 | 7.2 | 0.66 (0.52 to 0.85) | 0.0010 |
| Investigator | 9.4 | 5.8 | 0.62 (0.50 to 0.78) | < 0.0001 |
In Gordon et al. ,49 PFS was defined as the time from the first day of study drug dosing to documented disease progression or death due to any cause while the patient was on the study drug or during the long-term follow-up period. In platinum-sensitive patients, Gordon et al. 49 found that PLDH significantly prolonged PFS compared with topotecan (p = 0.037; HR not reported). Median PFS was reported to be 28.9 weeks and 23.3 weeks in the PLDH and topotecan groups, respectively. However, results presented in TA91,13 which are based on data provided by the manufacturer as part of the appraisal process, indicate that there is no statistically significant difference between PLDH and topotecan in PFS in platinum-sensitive patients, with a median PFS of 27.3 weeks with PLDH, and 22.7 weeks with the topotecan-treated group (HR 1.29, 95% CI 0.98 to 1.69; HR of > 1 favours PLDH). As data reported in TA9113 are more mature, the TAG has used the HR reported in TA91 in its NMA.
ten Bokkel Huinink et al. 21 evaluated TTP as a secondary outcome, defining TTP as time from first study drug to documented progression or administration of third-line therapy. Analysis of TTP for the subgroup of patients with platinum-sensitive (late relapse) disease is not reported in either publication by ten Bokkel Huinink et al. 21,52 TA9113 found no statistically significant difference between topotecan and paclitaxel in TTP, reporting an unadjusted HR of 0.82 (95% CI 0.54 to 1.26; see Table 47) in platinum-sensitive patients, for which HR < 1 favours topotecan. There was no significant difference between topotecan and paclitaxel in TTP (p = 0.08), with a median TTP of 18.9 weeks in the topotecan group compared with 14.7 weeks in the paclitaxel group.
PFS was the primary outcome in the trial reported by Pfisterer et al. 50 and was defined as time from the date of randomisation to the date of disease progression or death from any cause. PD was based on clinical and/or radiological evaluation. CA125 elevation without accompanying clinical or radiological evidence was not sufficient to determine disease progression. Analysis occurred after observation of 325 events. Gemcitabine plus carboplatin was associated with a gain in median PFS of 2.8 months, with the difference between groups reaching statistical significance (HR 0.72, 95% CI 0.58 to 0.90; p = 0.0031). Median PFS was 8.6 months (95% CI 7.9 to 9.7 months) with gemcitabine plus carboplatin compared with 5.8 months (95% CI 5.2 to 7.1 months) with carboplatin alone.
Univariate analysis to investigate the effect of prespecified prognostic factors on PFS found PFI to be an important prognostic factor (p = 0.0015; Table 45).
| Covariate | Univariate analysis | ||
|---|---|---|---|
| HR | (95% CI) | Wald’s p-value | |
| Age (years) | |||
| 60 | 1 | ||
| > 60 | 1.04 | 0.83 to 1.29 | 0.7528 |
| ECOG performance | |||
| 0 | 1 | ||
| 1 or 2 | 1.16 | 0.93 to 1.44 | 0.1994 |
| Prior platinum treatment | |||
| Platinum plus non-paclitaxel | 1 | ||
| Platinum plus paclitaxel | 1.06 | 0.83 to 1.34 | 0.6575 |
| Disease status | |||
| Assessable | 1 | ||
| Bidimensionally measured | 0.81 | 0.48 to 1.36 | 0.4143 |
| PFI (months)a | |||
| 6–12 | 1 | ||
| > 12 | 0.70 | 0.56 to 0.87 | 0.0015 |
ICON4/AGO-OVAR 2.261 defined PFS as the time from randomisation to first appearance of PD or death from any cause, which is the definition most commonly used across trials. Raised CA125 level without clinical or radiological evidence of PD was not considered to demonstrate disease progression. As for OS, patients known to be alive and without PD at the time of analysis were censored at their last follow-up. At analysis (median follow-up of 42 months), 717 (89%) of patients had developed PD or died. Paclitaxel plus platinum-based chemotherapy was associated with a significantly improved PFS compared with platinum-based therapy alone (HR 0.76, 95% CI 0.66 to 0.89; p = 0.0004). The improvement translates into an estimated absolute difference in 1-year PFS of 10% (40% vs. 50%) and an absolute difference in median PFS of 3 months in favour of combination treatment (median PFS 12 months with paclitaxel plus platinum-based chemotherapy vs. 9 months with platinum-based chemotherapy alone).
The authors carried out an exploratory analysis to investigate the effect of randomisation strata on PFS (summarised in Table 46). 61 Again, as for OS, no statistically significant difference between treatment groups was identified for any of the subgroups analysed. A non-significant trend was observed within the subgroups of age (< 55 vs. 55–65 vs. > 65 years) and the number of previous lines of chemotherapy (1 vs. 2 vs. > 2).
| Randomisation strata | No. of events per number of patients | p-value (interaction or trend) | |
|---|---|---|---|
| Paclitaxel plus platinum | Platinum alone | ||
| Randomisation group | |||
| ICON4 MRC CTU | 243/266 | 253/270 | 0.93 (interaction) |
| ICON4 Italy | 80/100 | 94/113 | |
| AGO | 23/26 | 24/27 | |
| Age (years) | |||
| < 55 | 114/127 | 111/123 | 0.08 (trend) |
| 55–65 | 135/151 | 146/162 | |
| > 65 | 97/114 | 114/125 | |
| WHO performance | |||
| 0 | 212/246 | 232/262 | 0.53 (interaction) |
| > 0 | 134/146 | 139/148 | |
| Intended platinum treatment | |||
| Carboplatin | 294/332 | 303/341 | 0.66 (interaction) |
| Cisplatin | 52/60 | 68/69 | |
| Previous lines of chemotherapy | |||
| 1 | 310/354 | 343/380 | 0.19 (trend) |
| 2 | 22/22 | 22/24 | |
| > 2 | 14/15 | 6/6 | |
| Time since completion of last chemotherapy cycle (months) | |||
| ≤ 12 | 90/92 | 109/111 | 0.87 (interaction) |
| ≥ 12 | 256/300 | 262/299 | |
| Previous exposure to taxane | |||
| No | 195/223 | 214/235 | 0.49 (interaction) |
| Yes | 151/169 | 157/175 | |
Gonzalez-Martin et al. 48 reported that paclitaxel plus carboplatin was associated with a significantly prolonged TTP compared with carboplatin alone (median TTP: 33.7 weeks with carboplatin alone vs. 49.1 weeks with paclitaxel plus carboplatin; HR 0.54, 95% CI 0.32 to 0.92; p = 0.021; see Table 47). TTP was defined as the time from date of randomisation to date of documentation of tumour progression. It should be noted that the study was not powered to identify a difference between groups in TTP and that the statistical comparative analysis was exploratory.
| Study | Population notes | Intervention | Comparison | Median PFS | HR | 95% CI | p-value | |
|---|---|---|---|---|---|---|---|---|
| Intervention | Comparator | |||||||
| Gonzalez-Martin et al.,48 TTP | Platinum sensitive | Paclitaxel (175 mg/m2) plus carboplatin (AUC 5) every 21 days | Carboplatin (AUC 5) every 21 days | 49.1 weeks (95% CI 36.9 to 61.3 weeks) | 33.7 weeks (95% CI 25.8 to 41.5 weeks) | 0.54 | 0.32 to 0.92 | 0.03a |
| Gordon et al.49 | Platinum-sensitive subgroup HR from TA9113 (not reported in Gordon 200148) |
PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 27.3 weeks (n = 109) | 22.7 weeks (n = 111) | 1.29b | 0.98 to 1.69 | |
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | 12 months | 9 months | 0.76 | 0.66 to 0.89 | 0.0004 | |
| CALYPSO31 | Platinum-sensitive patients (overall population) | PLDH (30 mg/m2) plus carboplatin every 21 days | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | 11.3 months (n = 467) | 9.4 months (n = 507) n = 363 had disease progression according to RECIST criteria;69 n = 89 had progression according to CA125 GCIG criteria70 |
0.82 | 0.72 to 0.94 | 0.005a |
| OVA-30130 | Platinum-sensitive subgroup | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 9.2 months (115/218) | 7.5 months (111/213) | 0.73 | 0.56 to 0.95 | 0.017 |
| Alberts et al.28 | PFI of 6–24 months | PLDH (30 mg/m2) plus carboplatin (AUC 5) every 4 weeks | Carboplatin (AUC 5) every 4 weeks | 12 months (26/30) | 8 months (29/30) | 0.54 | 0.32 to 0.93 | 0.02 |
| Bafaloukos et al.,29 TTP | PLDH (45 mg/m2) plus carboplatin (AUC 5) every 28 days | Paclitaxel (175 mg/m2; 3-hour infusion) plus carboplatin (AUC 5) every 21 days | 11.8 months | 10.8 months | NR | NR | p = 0.904 | |
| NR | ||||||||
| Pfisterer et al.50 | Platinum-sensitive | Gemcitabine (1000 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | 8.6 months (95% CI 7.9 to 9.7 months) (n = 178) | 5.8 months (95% CI 5.2 to 7.1 months) (n = 178) | 0.72 | 0.58 to 0.90 | 0.0031a |
| ten Bokkel Huinink et al.,52 TTP | HR taken from TA91;13 data are not presented in the published paper | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 0.82 | 0.54 to 1.26 | |||
The RCTs available for inclusion in the NMA evaluating PFS in patients with platinum-sensitive recurrent ovarian cancer are summarised in Table 47. Unfortunately, as described earlier, a single network could not be constructed out of the available trials. The two networks constructed for this outcome are depicted in Figure 7.
Network 1 (see Figure 7a) consisted of the following comparators:
-
paclitaxel plus carboplatin
-
gemcitabine plus carboplatin
-
PLDH plus carboplatin
-
platinum as a monotherapy.
Paclitaxel plus carboplatin was chosen as the baseline treatment, as this would best help to inform the economic evaluation conducted by the TAG (see Chapter 4, Treatment effectiveness). However, results are reported in Table 48 sequentially covering all possible comparisons. Overall, only PLDH plus carboplatin had a significantly improved PFS (at the 5% level) compared with paclitaxel plus carboplatin. Platinum monotherapy was associated with a significant reduction in PFS compared with all doublet chemotherapies assessed.
Network 2 (see Figure 7b) consisted of the following comparators:
-
PLDH monotherapy
-
trabectedin plus PLDH
-
paclitaxel monotherapy
-
topotecan monotherapy.
Pegylated liposomal doxorubicin hydrochloride monotherapy was chosen as the baseline treatment, as this would best help to inform the economic evaluation conducted by the TAG (see Chapter 4, Treatment effectiveness). However, results are reported in Table 48 sequentially covering all possible comparisons. Overall, only trabectedin plus PLDH demonstrated a significant difference increase in PFS (at the 5% level) compared with PLDH monotherapy. Trabectedin plus PLDH would also be considered to have a statistically significant prolonged PFS when compared directly with paclitaxel monotherapy or topotecan monotherapy. None of the other comparisons of chemotherapies would be considered significantly different from one another.
FIGURE 7.
Networks for PFS for people with platinum-sensitive recurrent ovarian cancer. (a) Network 1; and (b) network 2.


| Comparison | HR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| Network 1 | |||
| vs. paclitaxel plus carboplatin (HR < 1 favours comparator, HR > 1 favours paclitaxel plus carboplatin) | |||
| Gemcitabine plus carboplatin | 0.985 | 0.748 | 1.273 |
| PLDH plus carboplatin | 0.817 | 0.717 | 0.927 |
| Platinum monotherapy | 1.361 | 1.182 | 1.559 |
| vs. gemcitabine plus carboplatin (HR < 1 favours comparator, HR > 1 favours gemcitabine plus carboplatin) | |||
| PLDH plus carboplatin | 0.845 | 0.624 | 1.116 |
| Platinum monotherapy | 1.400 | 1.106 | 1.749 |
| vs. PLDH plus carboplatin (HR < 1 favours comparator, HR > 1 favours PLDH plus carboplatin) | |||
| Platinum monotherapy | 1.672 | 1.389 | 1.997 |
| Network 2 | |||
| vs. PLDH monotherapy (HR < 1 favours comparator, HR > 1 favours PLDH monotherapy) | |||
| Trabectedin plus PLDH | 0.736 | 0.560 | 0.949 |
| Paclitaxel monotherapy | 1.615 | 0.939 | 2.586 |
| Topotecan monotherapy | 1.298 | 0.979 | 1.688 |
| vs. trabectedin plus PLDH (HR < 1 favours comparator, HR > 1 favours trabectedin plus PLDH) | |||
| Paclitaxel monotherapy | 2.236 | 1.209 | 3.795 |
| Topotecan monotherapy | 1.797 | 1.207 | 2.578 |
| vs. paclitaxel monotherapy (HR < 1 favours comparator, HR > 1 favours paclitaxel monotherapy) | |||
| Topotecan monotherapy | 0.842 | 0.539 | 1.262 |
Fully platinum sensitive
In the subgroup of patients with FPS disease, in the primary analysis of PFS (independent radiologist), OVA-301 found no statistically significant difference between treatment groups in median PFS, with median PFS of 11.1 months in the trabectedin plus PLDH group compared with 8.9 months in the PLDH alone group (HR 0.70, 95% CI 0.47 to 1.03; see Table 49). 65 Secondary analysis based on independent review by oncologists found the difference in PFS to be statistically significant and favouring trabectedin plus PLDH [median PFS: 11.1 months with trabectedin plus PLDH vs. 9.0 months with PLDH alone; HR 0.66, 95% CI 0.46 to 0.97; p = 0.0311 (log-rank)]. The FAD of the STA of trabectedin plus PLDH (TA22273) states that the primary analysis was thought to be the most robust analysis.
It is important to reiterate that, in the full trial population, fewer events had occurred than the planned event rate required to generate 90% power and, as a consequence, the analysis might have been underpowered. In a subgroup analysis, the power to detect a statistically significant difference is further reduced. In addition, analysis of results for FPS patients was not preplanned and is therefore hypothesis generating.
In the trial reported by Pfisterer et al. ,50 in the subgroup of patients with FPS ovarian cancer, at the time of analysis, a similar proportion in each treatment group had progressed or died [93/106 (87.7%) with gemcitabine plus carboplatin vs. 97/107 (90.7%) with carboplatin alone]. Median PFS in each treatment group for this population was not reported. An accompanying HR or p-value for the difference between treatment groups was not available in the full publication.
As for OS, ICON4/AGO-OVAR 2.261 carried out a subgroup analysis to determine the effect of paclitaxel plus platinum chemotherapy on PFS in various subgroups, including time since completion of last chemotherapy regimen (≤ 12 months vs. > 12 months). In the subgroup of patients with FPS ovarian cancer (599 patients), a similar proportion of people in each treatment group had progressed or died at the time of analysis [256/300 (85.3%) with paclitaxel plus carboplatin vs. 262/299 (87.3%) with carboplatin alone]. Median PFS in each treatment group for this population was not reported. An accompanying HR or p-value for the difference between treatment groups was not available in the full publication.
| Study | Intervention | Comparison | Median PFS, (events, n/N) | HR | 95% CI | p-value | |
|---|---|---|---|---|---|---|---|
| Intervention | Comparator | ||||||
| OVA-30130,65 | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 11.1 months (n = 94) | 8.9 months (n = 122) | 0.70 | 0.47 to 1.03 | 0.0152 |
| Pfisterer et al.50 | Gemcitabine (1000 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | 93/106 | 97/107 | |||
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | 256/300 | 262/299 | |||
The trials identified for potential inclusion in the NMA for PFS in patients with FPS recurrent ovarian cancer are detailed in Table 49. Of the three RCTs identified, only one trial30,65 reported the required data for analysis, and so it was not possible perform an indirect comparison.
Partially platinum sensitive
A separate publication of CALYPSO31 reported an analysis of PFS in the subgroup of patients with PPS ovarian cancer. 57 The PPS subgroup comprised 161 patients in the PLDH plus carboplatin group and 183 patients in the paclitaxel plus carboplatin group. Baseline characteristics were similar in the two treatment groups. Median follow-up was 23 months and 326 patients experienced an event (progression or death).
Pegylated liposomal doxorubicin hydrochloride plus carboplatin significantly prolonged median PFS compared with paclitaxel plus carboplatin in this subgroup of patients, with a gain of 0.6 months in PFS (median PFS: 9.4 months with PLDH plus carboplatin vs. 8.8 months with paclitaxel plus carboplatin; HR 0.73, 95% CI 0.58 to 0.90; p = 0.004 for superiority; see Table 50).
In the subgroup of patients with PPS ovarian cancer, in the primary analysis of PFS (independent radiologist), OVA-30164 found that trabectedin plus PLDH significantly prolonged median PFS compared with PLDH alone (HR 0.65, 95% CI 0.45 to 0.92; Table 50). Median PFS was 7.4 months in the trabectedin plus PLDH group compared with 5.5 months in the PLDH alone group. Results based on review by independent oncologist align with those of the primary analysis (HR 0.54, 95% CI 0.39 to 0.76). As noted above, analysis of results for PPS patients is potentially underpowered and was not preplanned.
At the time of analysis of PFS in Pfisterer et al. ,50 most patients categorised as having PPS ovarian cancer had progressed or died [69/71 (97.2%) with gemcitabine plus carboplatin vs. 65/71 (91.5%) with carboplatin alone]. Median PFS in each treatment group for this population was not reported. An accompanying HR or p-value for the difference between treatment groups was not available in the full publication.
In the subgroup of patients with PPS disease in ICON4/AGO-OVAR 2.2,61 almost all patients in each treatment group had progressed or died at the time of analysis [90/92 (97.8%) with paclitaxel plus carboplatin vs. 109/111 (98.2%) with carboplatin alone]. Median PFS in each treatment group for this population was not reported. An accompanying HR or p-value for the difference between treatment groups was not available in the full publication.
| Study | Population notes | Intervention | Comparison | Median PFS (events, n/N) | HR | 95% CI | p-value | |
|---|---|---|---|---|---|---|---|---|
| Intervention | Comparison | |||||||
| OVA-30130,64 | PFI 6–12 months | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 21 days | PLDH (50 mg/m2) every 4 weeks | 7.4 months (n = 123) Independent radiologist: (69/122) Independent oncologist: (68/91) |
5.5 months (n = 91) Independent radiologist: (55/86) Independent radiologist: (73/123) |
0.65 | 0.45 to 0.92 | 0.0152 |
| CALYPSO31,57 | Prespecified subgroup of partially sensitive patients | PLDH (30 mg/m2) plus carboplatin every 21 days | Paclitaxel (175 mg/m2) plus carboplatin every 3 weeks | 9.4 months (n = 161) | 8.8 months (n = 183) | 0.73 | 0.58 to 0.90 | 0.004 |
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | 90/92 | 109/111 | ||||
| Pfisterer et al.50 | Platinum sensitive | Gemcitabine plus carboplatin every 21 days | Carboplatin alone every 21 days | 69/71 | 65/71 | |||
The trials identified for potential inclusion in the NMA for PFS in patients with PPS recurrent ovarian cancer are detailed in Table 50. Of the four RCTs identified, only two trials30,31,50,63 reported the required data for analysis and as they did not contain a common comparator it was not possible to perform an indirect comparison.
Platinum resistant/refractory
In the subgroup of patients with PRR ovarian cancer, Gordon et al. 49 found no statistically significant difference in PFS between PLDH and topotecan (p = 0.733; HR not reported). Median PFS with PLDH was 9.1 weeks compared with 13.6 weeks with topotecan. Results presented in TA91 for this subgroup of patients are analogous to those reported in Gordon et al. ,49 with a HR reported of 0.99 (95% CI 0.77 to 1.28). 13
Analysis of TTP for the subgroup of patients with PRR (refractory, early and interim relapse) disease is not reported in either publication by ten Bokkel Huinink et al. 21,52 TA9113 found no statistically significant difference between topotecan and paclitaxel in TTP, reporting an unadjusted HR of 0.75 (95% CI 0.50 to 1.12; Table 51) in PRR patients, for which HR < 1 favours topotecan. There was no significant difference between topotecan and paclitaxel in TTP (p = 0.08), with a median TTP of 18.9 weeks in the topotecan group compared with 14.7 weeks in the paclitaxel group.
PFS was the primary outcome of the trial carried out by Lortholary et al. 62 and was determined according to criteria set out by GCIG. 70 Median PFS is based on 162 events occurring in a median follow-up of 15 months. No statistically significant differences in PFS were identified among the treatment arms, with a median PFS of 3.7, 4.8 and 5.4 months for weekly paclitaxel, weekly paclitaxel plus carboplatin, and weekly paclitaxel plus weekly topotecan, respectively. As discussed earlier, results from the weekly paclitaxel plus topotecan group are not of interest to this systematic review. The authors of the study were contacted with a request for the HR for the comparison of weekly paclitaxel compared with weekly paclitaxel plus carboplatin. The authors helpfully provided the requested information, which indicates that there is no significant difference between the two treatment groups in median PFS (HR 0.92, 95% CI 0.76 to 1.12; p = 0.42; see Table 51).
In addition, an exploratory analysis of PFS was carried out using a Cox model that adjusted for: age; number of metastatic sites; number of prior lines of chemotherapy (1 vs. ≥ 2); PFI (progression ≤ 1 month vs. > 1 month from last platinum dose); ECOG performance status (0 vs. 1 or 2); and tumour size (< 5 cm or ≥ 5 cm). The analysis found that (monotherapy vs. combination therapy) was not predictive of PFS. However, PFI and ECOG performance status were identified as independent predictors of PFS, with p-values of 0.03 and 0.01, respectively.
PFS was evaluated as a secondary outcome by Sehouli et al. 23 A definition for PFS was not provided in the full publication. There was no statistically significant difference between treatments in PFS (HR 1.29, 95% CI 0.96 to 1.76; p = 0.088). Median PFS was 3.0 months with conventional topotecan compared with 4.4 months with weekly topotecan.
| Study | Population notes | Intervention | Comparison | Median PFS (events, n/N) | HR | 95% CI | p-value | |
|---|---|---|---|---|---|---|---|---|
| Intervention | Comparator | |||||||
| Gordon et al.49 | Platinum-resistant patients (refractory term used in methods, resistant used in results) aHR from TA9113 (not calculated in Gordon et al.49) |
PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 9.1 weeks (n = 130) | 13.6 weeks (n = 124) | 0.99 | 0.77 to 1.28 | 0.733 |
| ten Bokkel Huinink et al.52 | HR taken from TA91;13 data are not presented in the published paper | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 0.75 | 0.50 to 1.12 | |||
| Lortholary et al.62 | Full population relapsed within 6 months | Weekly paclitaxel (80 mg/m2) plus carboplatin | Weekly paclitaxel (80 mg/m2) on 4-week cycle | 4.8 (95% CI 3.3 to 6.3)(n = 51) | 3.7 (95% CI 3.1 to 4.3) (n = 57) | 0.92 | 0.76 to 1.12 | 0.42 |
| Sehouli et al.23 | Full population recurrent platinum-resistant patients | Topotecan (4.0 mg/m2) (weekly; days 1, 8 and 15) every 28 days | Topotecan (1.25 mg/m2) for five consecutive days every 21 days | 4.4 months | 3.0 months | 1.29 | 0.96 to 1.76 | 0.088 |
| Mink et al.30 | Subgroup of women with PFI < 6 months | Trabectedin (1.1 mg/m2 as a 3-hour infusion) plus PLDH (30 mg/m2 as a 90-minute infusion) every 21 days | PLDH (50 mg/m2 as a 90-minute infusion) every 28 days | 4.0 months | 3.7 months | 0.95 | 0.7 to 1.3 | NR |
The RCTs available for inclusion in the NMA evaluating PFS in patients with PRR recurrent ovarian cancer are summarised in Table 51. The network of trials constructed for this outcome is depicted in Figure 8 and contains the following comparators:
-
PLDH monotherapy
-
trabectedin plus PLDH
-
paclitaxel monotherapy
-
topotecan monotherapy; i.e. topotecan 1.25 or 1.5 mg/m2 daily for 5 days every 21 days
-
topotecan monotherapy (weekly); i.e. topotecan 4.0 mg/m2 (weekly) on days 1, 8 and 15 of a 28-day cycle.
The results from this NMA are presented in Table 52. Overall, there was no significant difference in PFS (at the 5% level) for any of the chemotherapies assessed compared with PLDH monotherapy (or with each other).
A RCT that provided results for this population, but which did not share a common comparator within the network, compared low-dose paclitaxel (80 mg/m2) with low-dose paclitaxel (80 mg/m2) plus carboplatin. 62 However, Lortholary et al. 62 identified no significant difference in PFS between the two different treatment regimens (see Table 52). Trabectedin plus PLDH is outside of the scope for this review for the population of PRR patients; data have been included within the network to capture all of the available evidence but are not included in the economic analysis.
FIGURE 8.
Networks for PFS for people with PRR recurrent ovarian cancer.

| Comparison | HR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| vs. PLDH monotherapy (HR < 1 favours comparator, HR > 1 favours PLDH monotherapy) | |||
| Trabectedin plus PLDH | 0.961 | 0.697 | 1.292 |
| Paclitaxel monotherapy | 1.360 | 0.817 | 2.123 |
| Topotecan monotherapy | 0.998 | 0.767 | 1.277 |
| Topotecan monotherapy (weekly) | 1.302 | 0.859 | 1.894 |
| vs. trabectedin plus PLDH (HR < 1 favours comparator, HR > 1 favours trabectedin plus PLDH) | |||
| Paclitaxel monotherapy | 1.450 | 0.791 | 2.454 |
| Topotecan monotherapy | 1.064 | 0.698 | 1.555 |
| Topotecan monotherapy (weekly) | 1.389 | 0.811 | 2.216 |
| vs. paclitaxel monotherapy (HR < 1 favours comparator, HR > 1 favours paclitaxel monotherapy) | |||
| Topotecan monotherapy | 0.765 | 0.502 | 1.118 |
| Topotecan monotherapy (weekly) | 0.999 | 0.585 | 1.599 |
| vs. topotecan monotherapy (HR < 1 favours comparator, HR > 1 favours topotecan monotherapy) | |||
| Topotecan monotherapy (weekly) | 1.305 | 0.951 | 1.744 |
Full population (mixed platinum-free intervals)
In OVA-301 (all patients), after 389 events based on independent radiological review, trabectedin plus PLDH was found to significantly prolong PFS by 1.5 months compared with PLDH alone (median PFS: 7.3 months with trabectedin plus PLDH vs. 5.8 months with PLDH alone; HR 0.79, 95% CI 0.65 to 0.96; p = 0.019; see Table 54). 30
Multivariate analysis of baseline characteristics that are potential prognostic factors affecting PFS (based on independent radiology review) identified treatment with trabectedin plus PLDH and PFI (analysed as a continuum) as factors having a statistically significant effect on PFS (Table 53). 30
| Prognostic factor | PFS | p-value | |
|---|---|---|---|
| HR | 95% CI | ||
| Treatment arm (trabectedin/PLDH vs. PLDH alone) | 0.78 | 0.64 to 0.96 | 0.0195 |
| PFI, continuous | 0.97 | 0.96 to 0.98 | < 0.0001 |
| ECOG performance status (1–2 vs. 0) | 1.23 | 0.99 to 1.52 | 0.0591 |
| Race (non-white vs. white) | 1.23 | 0.97 to 1.56 | 0.0890 |
| Baseline CA125 (≥ 2 × ULN vs. < 2 × ULN) | 1.18 | 0.91 to 1.53 | 0.2245 |
| Age, continuous | 1.00 | 0.99 to 1.01 | 0.8542 |
| Baseline liver/lungs involvement (yes vs. no) | 1.21 | 0.98 to 1.49 | 0.0760 |
| Prior taxane (yes vs. no) | 1.00 | 0.77 to 1.29 | 0.9957 |
For the full trial population, Gordon et al. 49 observed a median PFS of 16.1 weeks with PLDH and of 17.0 weeks with topotecan, with no statistically significant difference between groups (HR 1.12, 95% CI 0.93 to 1.35; p = 0.095; see Table 54). The HR was not reported in the full publication and is as reported in TA91. 13
Data reported here are taken from the longer-term follow-up study reported by ten Bokkel Huinink et al. 52 in which data had been collected for > 4 years. For analysis of TTP, 25% of patients in the topotecan group and 12.3% of patients in the paclitaxel group were censored. There was no statistically significant difference between topotecan and paclitaxel in TTP (p = 0.08), with a median TTP of 18.9 weeks in the topotecan group compared with 14.7 weeks in the paclitaxel group (Table 54). 52 An accompanying HR was not reported in the full publication. 52 However, TA9113 reported a HR of 0.81 (95% CI 0.60 to 1.09) for TTP, for which HR of < 1 favours topotecan. 13 The HR was adjusted for stratification factors.
The methods section of Piccart et al. 63 indicates that TTP was a secondary outcome measure, with TTP defined as the time from day 1 of treatment to first observation of disease progression as per WHO criteria. However, results are presented for both TTP and PFS. At the time of analysis, of the 86 patients randomised, 69 had progressed (80.2%). Of the remaining 17 patients who had not progressed, nine were in the paclitaxel group and eight were in the oxaliplatin group.
Median TTP (the number of patients reported includes only those who have progressed) and PFS were the same and were reported as 14 weeks in the paclitaxel group compared with 12 weeks in the oxaliplatin group. Statistical significance was not assessed in the full publication.
Gore et al. 24 evaluated TTP; TTP was not defined in the full publication. In the full trial population, median TTP was 13 weeks with oral topotecan compared with17 weeks with i.v. topotecan (difference reported to be non-significant; p-value not reported; see Table 54). 24
In the trial carried out by Omura et al. ,68 PFS did not differ appreciably between treatment regimens. Patients assigned to paclitaxel 175 mg/m2 had an estimated median PFS of 4.8 months compared 5.5 months for patients receiving paclitaxel 250 mg/m2 (see Table 54). The statistical significance between the groups was not assessed.
Rosenberg et al. 60 evaluated TTP as a secondary outcome, and defined TTP as time from first day of study treatment to the date of documented tumour progression (as per WHO criteria) or censored observation. In the full trial population, median TTP was 6.1 months (95% CI 5.0 to 8.0 months) in the group receiving paclitaxel weekly compared with 8.1 months (95% CI 6.4 to 9.7 months) in the group receiving paclitaxel every 21 days. The difference between groups in TTP did not reach statistical significance (p = 0.85). It is unclear how many events had occurred at the time of analysis.
| Study | Population notes | Intervention | Comparison | Median PFS, (events, n/N) | HR | 95% CI | p-value | |
|---|---|---|---|---|---|---|---|---|
| Intervention | Comparator | |||||||
| Piccart et al.63 | Approximately 75% of the population is platinum refractory, 25% is platinum sensitive | Paclitaxel (175 mg/m2) over 3 hours every 3 weeks | Oxaliplatin (130 mg/m2) over 2 hours every 3 weeks | 14 weeks (n = 41) | 12 weeks (n = 45) | NR | NR | NR |
| OVA-30130 | Full population contains platinum-sensitive and platinum-resistant patients | PLDH (30 mg/m2 i.v.) plus trabectedin (1.1 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 7.3 months (n = 337) | 5.8 months (n = 335) | 0.79 | 0.65 to 0.96 | 0.019 |
| Gordon et al.49 | Combination of platinum-sensitive and platinum-refractory patients aHR from TA9113 (not calculated in Gordon et al.48) |
PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2 per day for 5 days) every 21 days | 16.1 weeks (n = 239) | 17.0 weeks (n = 235) | 1.12 | 0.93 to 1.35 | 0.095 |
| Gore et al.,24 TTP | Approximately 30% refractory; 27.5% resistant; 43% sensitive | Oral topotecan (2.3 mg/m2/day) | Intravenous topotecan (1.5 mg/m2/day for 5 days) every 21 days | 13 weeks (range 1.6–76.6 weeks) (n = 135) | 17 weeks (range 0.1–91.6 weeks) (n = 135) | NR | NR | NR |
| Rosenberg et al.,60 TTP | Platinum resistant: relapse at ≤ 6 months and > 6 months after primary platinum-based therapy | Paclitaxel weekly | Paclitaxel 3 weekly | 6.1 months (95% CI 5.0 to 8.0 months) (n = 105) | 8.1 months (95% CI 6.4 to 9.7 months) (n = 103) | NR | NR | p = 0.85 |
| ten Bokkel Huinink et al.,52 TTP | HR taken from TA91;13 data are not presented in the published paper | Topotecan (1.5 mg/m2/day) for 5 days | Paclitaxel (175 mg/m2/day) as 3 hour infusion every 21 days | 18.9 weeks (range < 1–92.6+ weeks) | 14.7 weeks (range < 1–137.3+ weeks) | 0.81 (adjusted for stratification factors) | 0.60 to 1.09 | 0.08 |
| Omura et al.68 | Paclitaxel (250 mg/m2) every 21 days | Paclitaxel (175 mg/m2) every 21 days | 5.5 months | 4.8 months | NR | NR | NR | |
The RCTs available for inclusion in the NMA evaluating PFS in patients with mixed PFIs in recurrent ovarian cancer are summarised in Table 54. However, based on expert clinical opinion, the TAG decided not to evaluate this mixed patient population, as the results would not be considered clinically meaningful.
Tumour response
As with PFS and TTP, for patients with measurable disease, assessment of tumour response is based on standard criteria, such as RECIST criteria. 69 In patients without measurable disease, changes in CA125 level are used to evaluate tumour response as per the algorithm outlined by Rustin et al. 76 There is some controversy over the use of CA125 level alone as an indicator for disease progression and for tumour response. However, an alternative opinion is that it is difficult to radiologically follow changes in measurable disease from baseline. ORR is typically reported as the combination of patients with a CR or those with a PR, as defined by the criteria implemented in the trial. ORR is considered to be a direct measure of the anti-tumour activity of a drug but not a direct measure of clinical benefit. 76,77 As for PFS and TTP, evaluation of CR and PR is open to assessment bias, particularly in an open-label trial. When CR and PR have been reported separately, for the purposes of the NMA, the TAG has combined CR and PR results. Results for SD and PD are also reported for completeness.
Summary of results for tumour response
Results are presented for ORR, which has been defined as the number of patients achieving CR or PR as their best response. Definitions of CR and PR as reported in the trials are provided in the main text. No trial was identified evaluating treatments in a population solely comprising patients who were allergic or intolerant to platinum-based chemotherapy. Here, results for patients with platinum-sensitive or PRR disease are summarised. For trials not limited to either platinum-sensitive or PRR patients (i.e. includes a mix of PFI), results for the full trial population are presented in the main text.
Twelve RCTs24,28–30,48,50,52,54,60,61,63,68 evaluating 11 different head-to-head comparisons of interventions and comparators of interest reported on ORR (Table 55). Of the 11 comparisons identified, only two trials30,50 reported a statistical significance in ORR. A larger proportion of patients treated with gemcitabine plus carboplatin achieved CR or PR than those treated with carboplatin alone. Trabectedin plus PLDH was also found to significantly improve the rate of CR or PR achieved compared with PLDH (50 mg/m2) alone.
| Trial name | Intervention | Comparator | OR (95%CI) |
|---|---|---|---|
| Bafaloukos et al.29 | PLDH (45 mg/m2) plus carboplatin every 28 days | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | 0.866 (0.535 to 1.402) |
| OVA-30130 | Trabectedin (1.1 mg/m2) plus PLDH (30 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 1.567 (1.043 to 2.354) |
| Gordon et al.54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 0.987 (0.563 to 1.727) |
| ten Bokkel Huinink et al.52 | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 1.442 (0.607 to 3.427) |
| Alberts et al.28 | PLDH (30 mg/m2) plus carboplatin every 4 weeks | Carboplatin alone every 4 weeks | 2.148 (0.792 to 5.825) |
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum | Conventional platinum treatment | 1.182 (0.831 to 1.682) |
| Gonzalez-Martin et al.48 | Paclitaxel (175 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | 0.661 (0.325 to 1.347) |
| Pfisterer et al.50 | Gemcitabine (1000 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | 1.527 (1.025 to 2.275) |
| Rosenberg et al.60 | Paclitaxel (67 mg/m2) weekly (one course = 3 weeks) | Paclitaxel (200 mg/m2) every 3 weeks | 1.127 (0.574 to 2.212) |
| Gore et al.24 | Oral topotecan (2.3 mg/m2) daily | Intravenous topotecan (1.5 mg/m2) for 5 days every 21 days | 0.531 (0.233 to 1.208) |
| Piccart et al.63 | Paclitaxel (175 mg/m2) every 3 weeks | Oxaliplatin (130 mg/m2) every 3 weeks | 0.520 (0.083 to 3.259) |
| Omura et al.68 | Paclitaxel 250 mg/m2 every 21 days | Paclitaxel 175 mg/m2 every 21 days | 0.748 (0.273 to 2.051) |
Based on the trials identified, it was not possible to construct a complete network. Again, two discrete networks were generated: one evaluating platinum-based therapies and the second comparing non-platinum-based regimens. It should be stressed that results from the two discrete networks are not directly comparable.
In the network evaluating platinum-based chemotherapies, paclitaxel plus carboplatin and gemcitabine plus carboplatin were found to have a significantly higher ORR than platinum monotherapy (Table 56). There was no significant difference between PLDH plus carboplatin and any of the chemotherapeutic treatments with which it was assessed.
| Comparator | Intervention | |||
|---|---|---|---|---|
| Paclitaxel plus carboplatin | PLDH plus carboplatin: HR (95% CrI) | Platinum monotherapy: HR (95% CrI) | Gemcitabine plus carboplatin: HR (95% CrI) | |
| Paclitaxel plus carboplatin | – | 0.994 (0.574 to 1.609) | 0.666 (0.474 to 0.908) | 1.370 (0.765 to 2.261) |
| PLDH plus carboplatin | – | – | 0.713 (0.386 to 1.208) | 1.467 (0.672 to 2.793) |
| Platinum monotherapy | – | 2.058 (1.305 to 3.108) | ||
| Gemcitabine plus carboplatin | – | – | – | – |
Analysis of non-platinum-based regimens indicates that trabectedin plus PLDH significantly improves ORR compared with PLDH, and oral topotecan (Table 57). Compared with oral topotecan, i.v. topotecan was found to be associated with a significant increase in the proportion of patients achieving CR or PR. No other statistically significant differences were identified.
| Comparator | Intervention | |||||
|---|---|---|---|---|---|---|
| PLDH monotherapy | Trabectedin plus PLDH: HR (95% CrI) | Topotecan monotherapy (i.v.): HR (95% CrI) | Paclitaxel monotherapy (every 3 weeks): HR (95% CrI) | Topotecan monotherapy (oral): HR (95% CrI) | Paclitaxel monotherapy (weekly): HR (95% CrI) | |
| PLDH monotherapy | – | 1.932 (1.231 to 2.905) | 1.072 (0.565 to 1.858) | 0.734 (0.207 to 1.871) | 0.483 (0.145 to 1.169) | 1.024 (0.204 to 3.097) |
| Trabectedin plus PLDH | – | – | 0.582 (0.260 to 1.122) | 0.399 (0.102 to 1.077) | 0.262 (0.071 to 0.674) | 0.556 (0.102 to 1.773) |
| Topotecan monotherapy (i.v.) | – | – | – | 0.683 (0.243 to 1.514) | 0.451 (0.170 to 0.951) | 0.953 (0.230 to 2.642) |
| Paclitaxel monotherapy (every 3 weeks) | – | – | – | – | 0.822 (0.191 to 2.337) | 1.393 (0.578 to 2.852) |
| Topotecan monotherapy (oral) | – | – | – | – | – | 2.554 (0.431 to 8.493) |
| Paclitaxel monotherapy (weekly) | – | – | – | – | – | – |
Most identified trials involving platinum-sensitive patients did not present data on tumour response separately for the subgroup of patients with FPS (relapse at > 12 months after last platinum-based treatment) and PPS (relapse at ≥ 6 to ≤ 12 months after last platinum-based treatment) ovarian cancer. No data were available for the subgroup of patients with FPS ovarian cancer.
Only the CALYPSO trial57 presented results (in an accompanying publication) for tumour response in patients with PPS ovarian cancer. There was no significant difference between PLDH plus carboplatin and paclitaxel plus carboplatin in the proportion of patients achieving CR or PR as their best response (OR 0.86, 95% CI 0.58 to 1.27).
Eight RCTs23,24,49,52,60,62,63,68 reporting results for eight different head-to-head comparisons involving PRR patients were identified (Table 58). Two RCTs enrolled only patients with PRR, with the remaining six RCTs reporting results from a subgroup of patients within the trial. None of the trials identified a significant difference in ORR between the two treatment groups evaluated.
| Trial name | Intervention | Comparator | OR (95% CI) |
|---|---|---|---|
| ten Bokkel Huinink et al.52 | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 1.967 (0.562 to 6.884) |
| Gordon et al.49 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 1.908 (0.788 to 4.616) |
| Sehouli et al.23 | Topotecan (4.0 mg/m2) (weekly; days 1, 8 and 15) every 28 days | Topotecan (1.25 mg/m2) for five consecutive days every 21 days | 0.491 (0.190 to 1.271) |
| Lortholary et al.62 | Weekly paclitaxel (80 mg/m2) plus carboplatin | Weekly paclitaxel (80 mg/m2) on a 4-week cycle | 1.060 (0.510 to 2.209) |
| Gore et al.24 | Oral topotecan (2.3 mg/m2) daily | Intravenous topotecan (1.5 mg/m2) for 5 days every 21 days | 0.974 (0.301 to 3.155) |
| Rosenberg et al.60 | Paclitaxel (67 mg/m2) weekly (one course = 3 weeks) | Paclitaxel (200 mg/m2) every 3 weeks | 0.757 (0.312 to 1.839) |
| Piccart et al.63 | Paclitaxel (175 mg/m2) every 3 weeks | Oxaliplatin (130 mg/m2) every 3 weeks | 2.581 (0.466 to 14.306) |
| Omura et al.68 | Paclitaxel 250 mg/m2 every 21 days | Paclitaxel 175 mg/m2 every 21 days | 1.659 (0.930 to 2.961) |
A NMA was carried out using five of the identified RCTs. Based on clinical expert advice, the decision was taken not to include the trial by Piccart et al. 63 comparing paclitaxel vs. oxaliplatin as oxaliplatin is not licensed for the treatment of ovarian cancer and is rarely used in UK clinical practice. In addition, the treatment regimens evaluated in the trial reported by Lortholary et al. 62 did not inform the network. In the NMA, PLDH was found to significantly increase ORR compared with paclitaxel (175 mg/m2) every 21 days and with an alternative regimen in which paclitaxel was given weekly at a dose of 67 mg/m2. PLDH monotherapy was also significantly more effective than an unconventional regimen of topotecan in which topotecan was administered weekly at a dose of 4 mg/m2.
No chemotherapeutic regimen was found to have a significantly higher ORR than PLDH monotherapy (Table 59). However, paclitaxel monotherapy, paclitaxel monotherapy (weekly) and topotecan monotherapy (i.v., weekly) were found to have significantly lower ORR than PLDH monotherapy. No other comparison of chemotherapies was found to have a statistically significant difference.
| Comparator | Intervention | |||||
|---|---|---|---|---|---|---|
| PLDH monotherapy | Topotecan monotherapy i.v. (conventional): HR (95% CrI) | Paclitaxel monotherapy (every 3 weeks): HR (95% CrI) | Topotecan monotherapy (oral): HR (95% CrI) | Paclitaxel monotherapy (weekly) | Topotecan monotherapy (unconventional i.v. regimen): HR (95% CrI) | |
| PLDH monotherapy | – | 0.529 (0.184 to 1.166) | 0.290 (0.040 to 0.982) | 0.622 (0.098 to 2.116) | 0.224 (0.022 to 0.884) | 0.253 (0.051 to 0.761) |
| Topotecan monotherapy i.v. (conventional) | – | – | 0.548 (0.111 to 1.553) | 1.176 (0.283 to 3.283) | 0.423 (0.059 to 1.470) | 0.478 (0.154 to 1.086) |
| Paclitaxel monotherapy (every 3 weeks) | – | – | – | 3.387 (0.379 to 13.810) | 0.771 (0.271 to 1.736) | 1.383 (0.191 to 5.216) |
| Topotecan monotherapy (oral) | – | – | – | – | 0.530 (0.041 to 2.321) | 0.601 (0.090 to 2.090) |
| Paclitaxel monotherapy (weekly) | – | – | – | – | – | 2.251 (0.215 to 9.439) |
| Topotecan monotherapy (unconventional i.v. regimen) | – | – | – | – | – | – |
Platinum sensitive
In Bafaloukos et al. ,29 tumour response was evaluated using either WHO criteria for patients with measurable disease at baseline or repetitive CA125 level measurements using the algorithm proposed by Rustin et al. ,80 and based on CA125 Rustin’s criteria for patients without measurable disease at baseline. 78 Bafaloukos et al. 29 included a small proportion of women with only CA125 level elevation at baseline as a marker of presence of disease [16/186 (8.6%)] for whom results were analysed both as part of the full trial population and as a subgroup. Tumour assessments for response were carried out every two cycles. A similar proportion of women achieved overall response (CR or PR) in the two treatment groups [47/93 (50.5%) with PLDH plus carboplatin vs. 56/96 (58.3%) with paclitaxel plus carboplatin; OR 0.886, 95% CI 0.535 to 1.402; see Table 60]. Bafaloukos et al. 29 found no statistically significant difference between PLDH plus carboplatin and paclitaxel plus carboplatin for overall response in any of the populations assessed: the full trial population (p = 0.309); patients with measurable disease at baseline (p = 0.427); and patients with evaluable disease (elevated CA125 level and/or effusions) (p = 0.713). It is unclear whether the clinicians evaluating response had been masked to treatment, or whether tumour response was evaluated by a central review panel.
Alberts et al. 28 based the primary analysis of response rate on confirmed response rates for CR or PR, with CR and PR assigned according to RECIST criteria. 69 Women with only CA125 level elevation at baseline as a marker of disease at study entry (six women) were excluded from the analysis of objective response. It is unclear whether the clinicians evaluating response had been masked to treatment, or whether tumour response was evaluated by a central review panel. Alberts et al. 28 found no statistically significant difference between PLDH plus carboplatin and carboplatin alone in confirmed response rate [14/27 (52%) with PLDH plus carboplatin vs. 8/28 (29%) with carboplatin alone; p = 0.10]. However, a follow-up publication by Markman et al. 55 reporting more mature data found the difference between groups to be statistically significant favouring PLDH plus carboplatin [16/27 (59%) with PLDH plus carboplatin vs. 8/29 (28%) with carboplatin alone; p = 0.10; OR 2.15, 95% CI 0.79 to 5.83; see Table 60]. As noted earlier, the duration of follow-up in the longer-term study is unclear. In addition, the follow-up publication does not discuss the inclusion of one additional patient in the analysis of the carboplatin group.
OVA-301. 30 evaluated tumour response as the ORR (CR or PR) with response maintained at ≥ 4 weeks, based on RECIST criteria. 69 The schedule for tumour assessment is unclear. The primary analysis was based on assessments by independent radiology review. In the subgroup of patients with platinum-sensitive ovarian cancer (218 patients in the trabectedin plus PLDH group vs. 213 patients in the PLDH alone group), trabectedin plus PLDH significantly improved ORR compared with PLDH alone [77/218 (35.3%) with trabectedin plus PLDH vs. 48/213 (22.5%) with PLDH alone; p = 0.042; OR 1.567, 95% CI 1.043 to 2.354; see Table 60]. It should be noted that most patients achieved PR. In the full trial population, only six patients achieved a CR: two in the trabectedin plus PLDH group and four in the PLDH alone group.
In Gordon et al. ,49 tumour response was determined by ORR, which comprised CR and PR. Patients achieving either a CR or PR underwent repeat radiological assessment at least 4 weeks later to confirm the response. CR was defined as complete disappearance of all measurable and assessable disease, no new lesions and no disease-related symptoms. PR was defined as a ≥ 50% reduction in the sum of products of the perpendicular diameters of all measurable lesions for at least 4 weeks. Although open-label in design, scans for assessment of disease response and progression underwent independent radiological review.
In the subgroup of patients with platinum-sensitive disease, a similar proportion in the PLDH and topotecan groups achieved either CR or PR as their best response [31/109 (28.4%) with PLDH vs. 32/111 (28.8%) with topotecan; p = 0.964; OR 0.987, 95% CI 0.563 to 1.727; see Table 60]. The difference between groups did not reach statistical significance. In addition, a similar proportion of patients in each group achieved SD as their best response [41/109 (37.6%) with PLDH vs. 42/111 (37.8%) with topotecan; see Table 60].
Response rate was a primary outcome evaluated by ten Bokkel Huinink et al. 21 Response included patients achieving either CR or PR as their best response, with CR or PR assigned as per WHO criteria. All claimed responses were independently reviewed and scans confirmed by a radiologist masked to treatment allocation. The timing of tumour assessment is unclear. Patients who were not fully assessed for efficacy or who were not evaluated for response were considered to be non-responders. Data for the subgroup of patients with platinum-sensitive disease (late relapse; relapse at > 6 months after cessation of chemotherapy) were reported separately. In this subgroup of patients, a larger proportion of patients in the topotecan group achieved either CR or PR compared with paclitaxel [15/52 (28.8%) with topotecan vs. 11/55 (20.0%) with paclitaxel; see Table 60] but the statistical significance of this result was not evaluated in the full publication. 21 The OR calculated by the TAG indicates the difference to be non-significant (OR 1.442, 95% CI 0.607 to 3.427; see Table 60).
Pfisterer et al. 50 implemented SWOG criteria to determine degree of tumour response. The outcome evaluated was overall response, which included patients achieving a CR or PR as their best response. SWOG defines a CR as complete disappearance of all measurable and evaluable disease and no evidence of non-evaluable disease and PR as sum of products of all lesions decreased by > 50% for at least 3–6 weeks, with no new lesions and no progression of evaluable lesions. Patients were assessed before random assignment, before every cycle during treatment, and every 2–3 months after treatment for at least 2 years. It is unclear from the full publication whether there was an independent review of claimed CR or PR.
Gemcitabine plus carboplatin significantly improved ORR compared with carboplatin alone, with 47.2% (84/178) of patients treated with gemcitabine plus carboplatin achieving CR or PR compared with 30.9% (55/178) of patients treated with carboplatin (p = 0.0016; OR 1.527, 95% CI 1.025 to 2.275; see Table 60).
The ICON4/AGO-OVAR 2.2 investigators defined response rate as patients achieving CR or PR. 61 It is unclear from the full publication which criteria (e.g. WHO, SWOG, or RECIST) were used to assign CR or PR. Timing of response assessment varied with protocol, with those in the AGO protocol assessed after the second and fourth cycles of treatment and those in the Italian ICON4 protocol assessed after three cycles. No further details are reported.
The authors reported that there was no statistically significant difference between treatment regimens in response rate, with 66% (78/119) patients in the paclitaxel plus platinum-based treatment achieving CR or PR compared with 54% (69/128) patients in the platinum chemotherapy alone group, which translates to a difference of 12% (95% CI –0.1% to 24%; p = 0.06). Although the methods state that all efficacy analyses are based on the ITT principle, it should be noted that the number of patients included in the analysis of response is not equal to the number of patients randomised to each group. One potential explanation of this potential discrepancy could be that the patients included in the analysis were those with measurable disease at baseline; number of patients with measurable disease was not reported in the table of baseline characteristics presented in the full publication.
Gonzalez-Martin et al. 48 used the WHO criteria to evaluate response in those with measurable disease at baseline, with tumour response assessed every three cycles. For patients without measurable disease at baseline, response was determined according to Rustin’s criteria. The RCT found that paclitaxel plus carboplatin significantly improved ORR (CR plus PR) compared with carboplatin alone [75.6% with paclitaxel plus carboplatin vs. 50.0% with carboplatin alone (p = 0.017; see Table 60)]. The authors commented that based on study design, paclitaxel plus carboplatin was the ‘winner’. Although analysis was based on the ITT population, it should be noted that the comparative statistical analysis was carried out as an exploratory exercise and the reported p-value should be interpreted with caution. In addition, overall response combines data for women with and without measurable disease at baseline.
Objective confirmed response rate was the primary efficacy end point in the trial carried out by Piccart et al. 63 Confirmed response was defined as CR or PR as per WHO criteria, and that was observed on at least two consecutive evaluations at least 4 weeks apart. Confirmed response was verified by two independent radiologists. ORR was defined by the total number of patients in each treatment group. Only patients receiving at least two treatment cycles were considered assessable for response. Of the 86 patients randomised, only five were not assessable: two in the paclitaxel group and three in the oxaliplatin group; four patients were deemed ineligible and one patient died 6 days after the first dose of oxaliplatin owing to causes unrelated to treatment.
In the subgroup of patients with platinum-sensitive disease (23 patients), 20% (2/10) of patients in the paclitaxel group achieved PR compared with 38% (5/13) of patients in the oxaliplatin group. The statistical significance of the difference was not assessed in the full publication. The TAG calculated the OR to be 0.520 (paclitaxel vs. oxaliplatin), with a 95% CI of 0.083 to 3.259 (non-significant difference). No patient achieved a CR. The authors caution that, because of the low number of patients in the analysis, conclusions cannot be drawn on the comparative effectiveness of treatments in this subgroup.
In Gore et al. ,24 tumour response was assessed based on WHO criteria such that a CR was the complete disappearance of all known measurable and evaluable disease determined by two measurements not < 4 weeks apart. A PR was defined as a > 50% decrease in measurable lesion size for at least 4 weeks, with no simultaneous increase in a known lesion or appearance of new lesions or increase in evaluable disease. Timing of assessment was determined by radiological method used to measure disease at baseline. Patients evaluated by CT or MRI at baseline were assessed for response at the end of alternate cycles, whereas those evaluated by chest radiograph or photography were assessed at the end of every cycle.
In the platinum-sensitive subgroup (relapse at > 6 months after initial response), although a larger proportion of patients in the i.v. topotecan group achieved a CR or PR as their best response, the difference between treatment groups did not reach statistical significance [11/58 (19%) with oral topotecan vs. 20/56 (36%) with i.v. topotecan; reported as not significant; p-value not reported; see Table 60].
Omura et al. 68 analysed ORR based on platinum sensitivity. A statistically significant treatment subgroup interaction was identified (p = 0.041). In the subgroup of patients with platinum-sensitive disease, there was no statistically significant difference between paclitaxel 250 mg/m2 and paclitaxel 175 mg/m2 in the proportion of patients achieving a CR or PR (OR 0.63, 95% CI 0.191 to 2.07). The OR was adjusted for histological cell type (papillary serous compared with clear cell or mucinous cell vs. other cell types), cooperative group, performance status and prior platinum sensitivity. The proportion of patients achieving either CR or PR in each group was 36.0% (9/25) and 48.1% (13/27) in the 250 mg/m2 and 175 mg/m2 groups, respectively. Unadjusted OR as calculated by the TAG was 0.748 (95% CI 0.273 to 2.051; see Table 60). For the purposes of the NMA, based on clinical expert advice, it has been assumed that doses of paclitaxel of 175 mg/m2 up to 250 mg/m2 are of equivalent clinical effectiveness and thus this trial has not been included in the NMA.
In the trial carried out by Rosenberg et al. ,60 patients were stratified at randomisation based on platinum resistance (relapse ≤ 6 months vs. > 6 months after primary platinum-based treatment). Results for the primary outcome of tumour response were reported separately for the subgroups categorised by platinum resistance. Evaluations of tumour size were carried out at baseline and subsequently every 6 weeks using the same imaging technique for all assessments. Tumour response was categorised as per WHO criteria, with overall response including CR or PR as a best response.
In the subgroup of patients with platinum-sensitive disease, a similar proportion of patients achieved either CR or PR in each treatment group [26/48 (54.2%) with paclitaxel every 7 days vs. 25/52 (48.1%) with paclitaxel every 21 days; see Table 60]. The statistical significance of the result in this subgroup of patients was not reported in the full publication. The OR calculated by the TAG indicates that the difference between groups did not reach statistical significance (OR 1.127, 95% CI 0.574 to 2.212; see Table 60). It should be noted that the results include patients with unconfirmed CR and PR. In the full trial population, 3 patients in each group had unconfirmed CR, and 7 and 6 patients in the paclitaxel every 7 days and paclitaxel every 21 days, respectively, had unconfirmed PR. The corresponding number of patients in the platinum-sensitive subgroup is not reported.
| Study | Intervention | Comparison | Overall response (OR, 95% CI)a | CR | PR | SD | PD | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | |||
| Gordon et al.49,54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 31/109 | 32/111 | 8/109 | 10/111 | 23/109 | 22/111 | 41/109 | 42/111 | ||
| OR 0.987 (0.563 to 1.727) | ||||||||||||
| ICON4/AGO-OVAR 2.261 | Paclitaxel plus platinum every 21 days | Conventional platinum-based treatment every 21 days | 78/392 | 69/410 | ||||||||
| OR 1.182 (0.831 to 1.682) | ||||||||||||
| Gonzalez-Martin et al.48 | Carboplatin alone (AUC 5) every 21 days | Paclitaxel (175 mg/m2) plus carboplatin (AUC 5) every 21 days | 20/40 (50.0%) | 31/41 (75.6%) | 8/40 (20.0%) | 11/41 (26.8%) | 12/40 (30.0%) | 20/41 (48.8%) | 5/40 (12.5%) | 2/41 (4.9%) | 13/40 (32.5%) | 2/41 (4.9%) |
| OR 0.661 (0.325 to 1.347) | ||||||||||||
| Bafaloukos et al.29 | PLDH (45 mg/m2) plus carboplatin (AUC 5) every 28 days | Paclitaxel (175 mg/m2) plus carboplatin (AUC 5) every 21 days | 47/93 (50.5%) | 56/96 (58.3%) | 21/93 (22.6%) | 33/96 (34.3%) | 26/93 (28.0%) | 23/96 (24.0%) | ||||
| OR 0.886 (0.535 to 1.402) | ||||||||||||
| Alberts et al.28 | PLDH (30 mg/m2) plus carboplatin (AUC 5) every 4 weeks | Carboplatin alone (AUC 5) every 4 weeks | 16/27 | 8/29 | ||||||||
| OR 2.15 (0.79 to 5.83) | ||||||||||||
| Rosenberg et al.60 | Paclitaxel (67 mg/m2) weekly | Paclitaxel (200 mg/m2) every 3 weeks | 26/48 | 25/52 | ||||||||
| OR 1.127 (0.574 to 2.212) | ||||||||||||
| Gore et al.24 | Oral topotecan 2.3 mg/m2/day | Intravenous topotecan 1.5 mg/m2/day for 5 days every 21 days | 11/58 | 20/56 | ||||||||
| OR 0.531 (0.233 to 1.208) | ||||||||||||
| ten Bokkel Huinink et al.21 | Topotecan (1.5 i.v. mg/m2/day) for 5 days | Paclitaxel (175 mg/m2/day) every 21 days | 15/52 | 11/55 | 4/52 | 3/55 | 11/52 | 8/55 | ||||
| OR 1.442 (0.607 to 3.427) | ||||||||||||
| Piccart et al.63 | Paclitaxel (175 mg/m2) every 3 weeks | Oxaliplatin (130 mg/m2) every 3 weeks | 2/10 | 5/13 | 0/10 | 0/13 | 2/10 | 5/13 | ||||
| OR 0.520 (0.083 to 3.259) | ||||||||||||
| Pfisterer et al.50 | Gemcitabine (1000 mg/m2) plus carboplatin every 21 days | Carboplatin alone every 21 days | 84/178 | 55/178 | 26/178 | 11/178 | 58/178 | 44/178 | 68/178 | 69/178 | 14/178 | 29/178 |
| OR 1.527 (1.025 to 2.275) | ||||||||||||
| OVA-30130 | PLDH (30 mg/2) plus trabectedin (1.1 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 77/218 | 48/213 | ||||||||
| OR 1.567 (1.043 to 2.354) | ||||||||||||
| Omura et al.68 | Paclitaxel (250 mg/m2) every 21 days | Paclitaxel (175 mg/m2) every 21 days | 9/25 | 13/27 | 4/25 | 4/27 | 5/25 | 9/27 | ||||
| OR 0.748 (0.273 to 2.051) | ||||||||||||
The RCTs available for inclusion in the NMA evaluating ORR in patients with platinum-sensitive recurrent ovarian cancer are summarised in Table 60. Unfortunately, as described earlier, a single network could not be constructed out of the available trials. The two networks constructed for this outcome are depicted in Figure 9.
Network 1 (see Figure 9a) consisted of the following comparators:
-
paclitaxel plus carboplatin
-
PLDH plus carboplatin
-
platinum as a monotherapy
-
gemcitabine plus carboplatin.
Although ORR does not inform the economic evaluation conducted by the TAG (see Chapter 4, Independent economic assessment), for consistency with OS and PFS, paclitaxel plus carboplatin was chosen as the baseline treatment. However, results are reported in Table 61 sequentially covering all possible comparisons. Overall, there was no significant difference (at the 5% level) for any of the doublet chemotherapies assessed compared with paclitaxel plus carboplatin (or with each other). Platinum monotherapy was associated with a significant reduction in ORR compared with all doublet chemotherapies, with the exception of PLDH plus carboplatin, where no significant difference was found.
Network 2 (see Figure 9b) consisted of the following comparators:
-
PLDH monotherapy
-
trabectedin plus PLDH
-
topotecan monotherapy (i.v.)
-
paclitaxel monotherapy, i.e. 175 mg/m2 or 200 mg/m2 every 21 days
-
topotecan (oral)
-
paclitaxel monotherapy (weekly); i.e. paclitaxel 67 mg/m2 every week for 21 days.
Pegylated liposomal doxorubicin hydrochloride monotherapy was chosen as the baseline treatment in order to maintain consistency with the results reported for the NMAs for OS and PFS. However, results are reported in Table 61 sequentially covering all possible comparisons. Overall, only trabectedin plus PLDH demonstrated a significant increase in ORR (at the 5% level) compared with PLDH monotherapy. Trabectedin plus PLDH would also be considered to have a statistically significant increased ORR when compared directly with topotecan monotherapy (oral) but to have no significant difference from any other treatment assessed. None of the other comparisons of chemotherapies would be considered significantly different from one another, with the exception of topotecan monotherapy (oral), which was found to have a significantly lower ORR than topotecan monotherapy (i.v.).
FIGURE 9.
Networks for ORR for people with platinum-sensitive recurrent ovarian cancer. (a) Network 1; and (b) network 2.
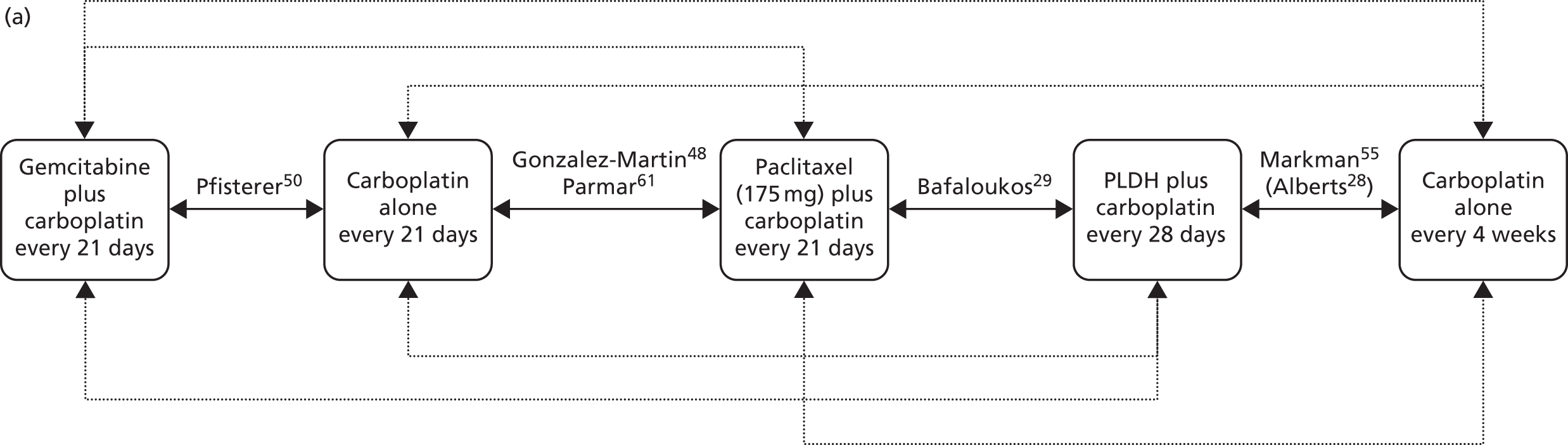
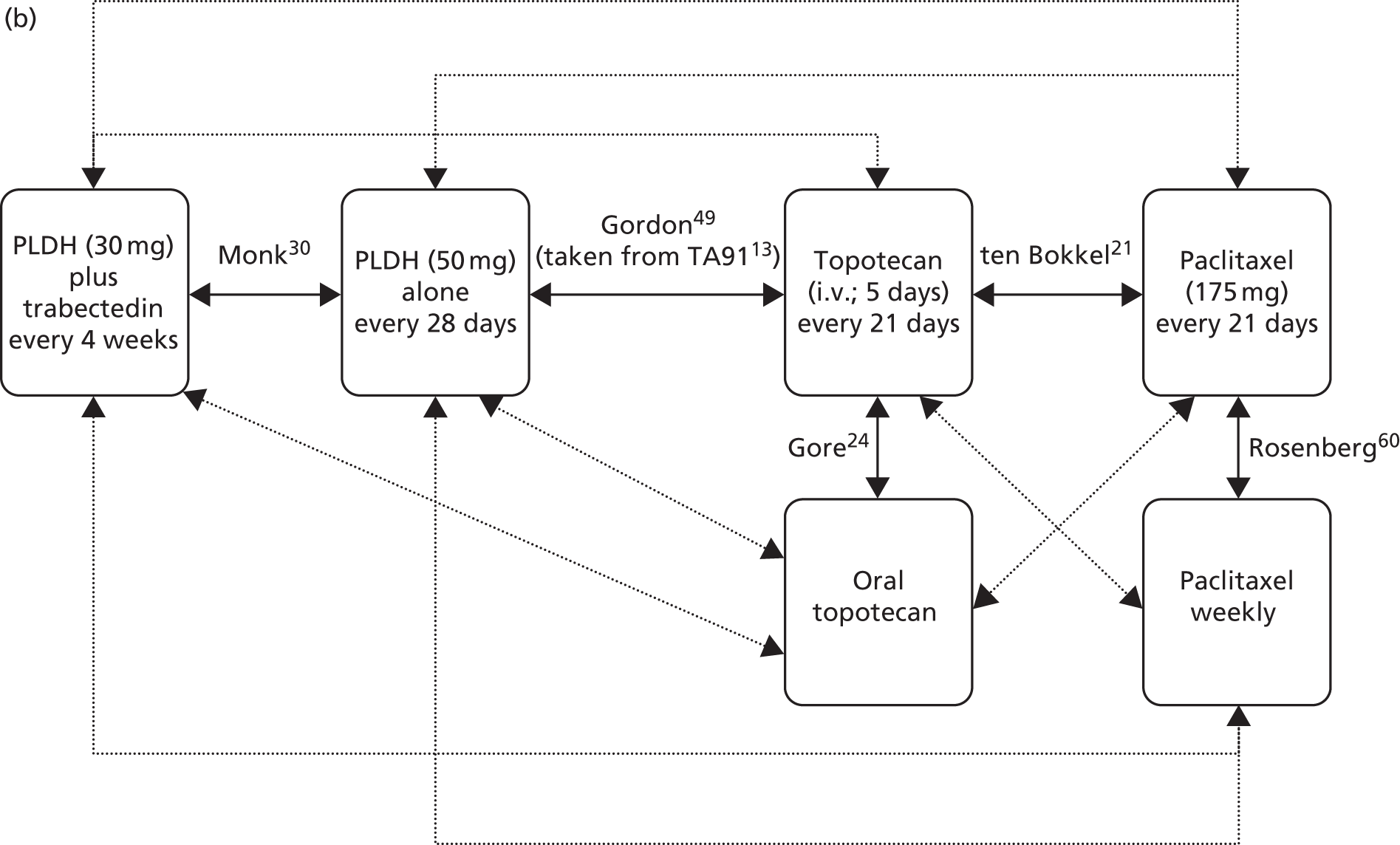
| Comparison | OR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| Network 1 | |||
| vs. paclitaxel plus carboplatin (OR > 1 favours comparator, OR < 1 favours paclitaxel plus carboplatin) | |||
| PLDH plus carboplatin | 0.994 | 0.574 | 1.609 |
| Platinum monotherapy | 0.666 | 0.474 | 0.908 |
| Gemcitabine plus carboplatin | 1.370 | 0.765 | 2.261 |
| vs. PLDH plus carboplatin (OR > 1 favours comparator, OR < 1 favours PLDH plus carboplatin) | |||
| Platinum monotherapy | 0.713 | 0.386 | 1.208 |
| Gemcitabine plus carboplatin | 1.467 | 0.672 | 2.793 |
| vs. platinum monotherapy (OR > 1 favours comparator, OR < 1 favours platinum monotherapy) | |||
| Gemcitabine plus carboplatin | 2.058 | 1.305 | 3.108 |
| Network 2 | |||
| vs. PLDH monotherapy (OR > 1 favours comparator, OR < 1 favours PLDH monotherapy) | |||
| Trabectedin plus PLDH | 1.932 | 1.231 | 2.905 |
| Topotecan monotherapy (i.v.) | 1.072 | 0.565 | 1.858 |
| Paclitaxel monotherapy | 0.734 | 0.207 | 1.871 |
| Topotecan monotherapy (oral) | 0.483 | 0.145 | 1.169 |
| Paclitaxel monotherapy (weekly) | 1.024 | 0.204 | 3.097 |
| vs. trabectedin plus PLDH (OR > 1 favours comparator, OR < 1 favours trabectedin plus PLDH) | |||
| Topotecan monotherapy (i.v.) | 0.582 | 0.260 | 1.122 |
| Paclitaxel monotherapy | 0.399 | 0.102 | 1.077 |
| Topotecan monotherapy (oral) | 0.262 | 0.071 | 0.674 |
| Paclitaxel monotherapy (weekly) | 0.556 | 0.102 | 1.773 |
| vs. topotecan monotherapy (i.v.) (OR > 1 favours comparator, OR < 1 favours topotecan monotherapy: i.v.) | |||
| Paclitaxel monotherapy | 0.683 | 0.243 | 1.514 |
| Topotecan monotherapy (oral) | 0.451 | 0.170 | 0.951 |
| Paclitaxel monotherapy (weekly) | 0.953 | 0.230 | 2.642 |
| vs. paclitaxel monotherapy (OR > 1 favours comparator, OR < 1 favours paclitaxel monotherapy) | |||
| Topotecan monotherapy (oral) | 0.822 | 0.191 | 2.337 |
| Paclitaxel monotherapy (weekly) | 1.393 | 0.578 | 2.852 |
| vs. topotecan monotherapy: oral (OR > 1 favours comparator, OR < 1 favours topotecan monotherapy: oral) | |||
| Paclitaxel monotherapy (weekly) | 2.554 | 0.431 | 8.493 |
Partially platinum sensitive
An accompanying publication to CALYPSO31 presents results for PFS and response rate (secondary outcome) for a subgroup of patients with PPS (TFI 6–12 months). 57 The principal publication provided a comprehensive description of the criteria for progression and indicated that tumour assessments occurred every 3 months and states that ORR was ‘response maintained ≥ 4 weeks by RECIST’. 31 Table 2 in the accompanying publication indicates that confirmed best responses are based on RECIST criteria,69 and ORR is the total of confirmed CR and PR. 57
There was no statistically significant difference between PLDH plus carboplatin and paclitaxel plus carboplatin in ORR [63/161 (39%) with PLDH plus carboplatin vs. 83/183 (45%) with paclitaxel plus carboplatin; p = 0.691]. The proportion of patients achieving CR, PR and SD, together with PD, are presented in Table 62.
| Study | Intervention | Comparison | Overall response (OR, 95% CI) | CR | PR | SD | PD | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | |||
| CALYPSO;57 prespecified subgroup of partially sensitive patients | PLDH (30 mg/m2) plus carboplatin every 21 days | Paclitaxel (175 mg/m2) plus carboplatin every 3 weeks | 63/161 | 83/183 | 19/161 | 14/183 | 44/161 | 69/183 | 64/161 | 61/183 | 17/161 | 19/183 |
| OR 0.86 (0.58 to 1.27) | ||||||||||||
As only a single trial was identified with data to inform ORR in patients with PPS recurrent ovarian cancer (see Table 62) no NMA was possible for this subgroup.
Platinum resistant/refractory
Gordon et al. 49 found no statistically significant difference between PLDH and topotecan in the proportion of patients with PRR ovarian cancer achieving either CR or PR as their best response [16/130 (12.3%) with PLDH vs. 8/124 (6.5%) with topotecan; p = 0.118; see Table 64]. However, a larger proportion of patients in the topotecan group achieved SD as their best response [36/130 (27.7%) with PLDH vs. 53/124 (42.7%) with topotecan; significance not assessed; see Table 64].
In the subgroup of patients with PRR disease (resistant, early relapse and interim relapse; 119 patients), ten Bokkel Huinink et al. 21 that 13.3% (8/60) of patients treated with topotecan and 6.8% (4/59) of patients treated with paclitaxel achieved either CR or PR as their best response (significance not assessed; see Table 64). Results for the individual categories that make up PRR are presented in Table 63.
| Platinum sensitivity | Resistant | Early relapse | Interim relapse | Total | ||||
|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | No. | % | |
| Topotecan | n = 34 | n = 6 | n = 20 | n = 60 | ||||
| CR | 0 | 0.0 | 0 | 0.0 | 1 | 5.0 | 1 | 1.67 |
| PR | 3 | 8.8 | 1 | 16.7 | 3 | 15.0 | 7 | 11.67 |
| Total (CR + PR) | 3 | 8.8 | 1 | 16.7 | 4 | 20.0 | 8 | 13.3 |
| Paclitaxel | n = 33 | n = 10 | n = 16 | n = 59 | ||||
| CR | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0 |
| PR | 1 | 3.0 | 1 | 10.0 | 2 | 12.5 | 4 | 6.8 |
| Total (CR + PR) | 1 | 3.0 | 1 | 10.0 | 2 | 12.5 | 4 | 6.8 |
Lortholary et al. 62 based response rate on the proportion of patients who achieved either a CR or PR as their best response. Patient response was determined according to RECIST criteria69 for patients with measurable disease and Rustin’s criteria for CA125 levels for patients with non-measurable disease. Chest CT and abdominopelvic or MRI were obtained every two cycles or as needed for assessment of duration of response. Objective response was to be confirmed radiologically at least 4 weeks after initial response. Lortholary et al. 62 found that a similar response rate was achieved in the weekly paclitaxel and weekly paclitaxel plus carboplatin groups [20/57 (35.1%) with weekly paclitaxel vs. 19/51 (37.3%) with weekly paclitaxel plus carboplatin; see Table 64]. The statistical significance of the difference between groups was not assessed by Lortholary et al. 62 The TAG calculated an OR of 1.06 (95% CI 0.510 to 2.209), which indicates that the difference between groups is not statistically significant.
In the subgroup of patients with PRR ovarian cancer (63 patients), Piccart et al. 63 found that 16% (5/31) of patients in the paclitaxel group achieved PR compared with 6% (2/32) of patients in the oxaliplatin group (see Table 64). No patient achieved a CR. The statistical significance of the difference was not assessed in the full publication. 63 The authors caution that, because of the low number of patients in the analysis, conclusions cannot be drawn on the comparative effectiveness of treatments in this subgroup.
In the subgroup of patients with PRR ovarian cancer (progression or SD during treatment or relapse at < 6 months after initial response), Gore et al. 24 found that a small proportion of patients in each group achieved a CR or PR as their best response, with no statistically significant difference between groups [6/77 (7.8%) with oral topotecan vs. 6/75 (8.0%) with i.v. topotecan; reported as not significant; p-value not reported; see Table 64].
Clinical benefit rate was the primary outcome in the trial carried out by Sehouli et al. 23 Clinical benefit rate comprised CR, PR and SD as best response. By contrast, most trials identified have evaluated ORR of CR or PR. In the trial, tumour response could be determined radiologically and categorised as per RECIST criteria69 or by change in CA125 level as per GCIG criteria,70 with choice of method of assessment at the discretion of the investigator. Schedule of assessment of response was not reported. It should be noted that, despite most patients having measurable disease at baseline, only a small proportion of women were evaluated radiologically for response (19.8%).
For the primary outcome of clinical benefit, 58% (46/80) of patients treated with the conventional dose of topotecan achieved CR, PR or SD compared with 47% (36/76) of patients receiving topotecan weekly. The statistical significance of the difference between groups was not reported. Considering ORR (CR or PR), the proportion of patients achieving CR or PR as best response was 18.8% (15/80) and 9.2% (7/76) in the conventional topotecan compared with weekly topotecan groups, respectively.
Of the 80 patients in the conventional topotecan group, response was evaluated by CA125 level alone in 62 patients (CR or PR = 13 patients). By comparison, 63 out of 76 patients were evaluated by CA125 level alone (CR or PR = five patients).
In the subgroup of patients with PRR disease, Omura et al. 68 found that paclitaxel 250 mg/m2 significantly increased the proportion of patients achieving a CR or PR compared with paclitaxel 175 mg/m2 (OR 2.59, 95% CI 1.36 to 4.95). The OR was adjusted for histological cell type (papillary serous compared with clear cell or mucinous cell vs. other cell types), cooperative group, performance status and prior platinum sensitivity. The proportion of patients achieving either CR or PR in each group was 36.7% (40/109) and 22.1% (23/104) in the 250 mg/m2 and 175 mg/m2 groups, respectively. Unadjusted OR as calculated by the TAG was 1.659 (95% CI 0.930 to 2.961; see Table 64), which is a non-statistically significant difference.
In the subgroup of patients with PRR, Rosenberg et al. 60 found that a similar proportion of patients achieved either CR or PR in each treatment group [11/57 (19.3%) with paclitaxel every 7 days vs. 13/51 (25.5%) with paclitaxel every 21 days; Table 64]. The statistical significance of the result in this subgroup of patients was not reported in the full publication. As noted earlier, unconfirmed CR and PR is not broken down by subgroup and it is unclear how many patients in the PRR analysis had unconfirmed CR or PR.
| Study | Intervention | Comparison | Overall response (OR, 95% CI)a | CR | PR | SD | PD | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | |||
| Gordon et al.49 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2) for 5 days every 21 days | 16/130 | 8/124 | 1/130 | 1/124 | 15/130 | 7/124 | 36/130 | 53/124 | ||
| 1.908 (0.788 to 4.616) | ||||||||||||
| Gore et al.24 | Oral topotecan (2.3 mg/m2) daily | Intravenous topotecan (1.5 mg/m2) for 5 days every 21 days | 6/77 | 6/75 | ||||||||
| 0.974 (0.301 to 3.155) | ||||||||||||
| Sehouli et al.23 | Topotecan (4.0 mg/m2) (weekly; days 1, 8 and 15) every 28 days | Topotecan (1.25 mg/m2) for five consecutive days every 21 days | 7/76 | 15/80 | 4/76 | 3/80 | 3/76 | 12/80 | 29/76 | 31/80 | 40/76 | 34/80 |
| 0.491 (0.190 to 1.271) | ||||||||||||
| Rosenberg et al.60 | Paclitaxel (67 mg/m2) weekly (one course = 3 weeks) | Paclitaxel (200 mg/m2) every 3 weeks | 11/57b | 13/51b | ||||||||
| 0.757 (0.312 to 1.839) | ||||||||||||
| Lortholary et al.62 | Weekly paclitaxel (80 mg/m2) plus carboplatin | Weekly paclitaxel (80 mg/m2) on 4-week cycle | 19/51 | 20/57 | 7/51 | 3/57 | 12/51 | 17/57 | 29/51 | 23/57 | 26/51 | 26/57 |
| 1.06 (0.510 to 2.209) | ||||||||||||
| Piccart et al.63 | Paclitaxel (175 mg/m2) over 3 hours every 3 weeks | Oxaliplatin (130 mg/m2) over 2 hours every 3 weeks | 5/31 | 2/32 | 0/31 | 0/32 | 5/31 | 2/32 | ||||
| 2.581 (0.466 to 14.306) | ||||||||||||
| cten Bokkel Huinink et al.21 | Topotecan (1.5 mg/m2) for 5 days every 21 days | Paclitaxel (175 mg/m2) every 21 days | 8/60 | 4/59 | 1/60 | 0/59 | 7/60 | 4/59 | ||||
| 1.967 (0.562 to 6.884) | ||||||||||||
| Omura et al.68 | Paclitaxel (250 mg/m2) every 21 days | Paclitaxel (175 mg/m2) every 21 days | 40/109 | 23/104 | 13/109 | 5/104 | 27/109 | 18/104 | ||||
| 1.659 (0.930 to 2.961) | ||||||||||||
The RCTs available for inclusion in the NMA evaluating ORR in patients with PRR recurrent ovarian cancer are summarised in Table 64. The network of trials constructed for this outcome is depicted in Figure 10 and contains the following comparators:
-
PLDH monotherapy
-
topotecan monotherapy (i.v.); i.e. topotecan 1.25 or 1.5 mg/m2 daily for 5 days every 21 days
-
paclitaxel monotherapy; i.e. 175 mg/m2 or 200 mg/m2 every 21 days
-
topotecan monotherapy (oral)
-
paclitaxel monotherapy (weekly); i.e. paclitaxel 67 mg/m2 every week for 21 days
-
topotecan monotherapy (i.v., weekly); i.e. topotecan 4.0 mg/m2 (weekly) on days 1, 8 and 15 of a 28-day cycle.
The results from this NMA are presented in Table 65. Overall, no chemotherapy was found to have a significantly higher ORR (at the 5% level) than PLDH monotherapy. However, paclitaxel monotherapy, paclitaxel monotherapy (weekly) and topotecan monotherapy (i.v., weekly) were found to have significantly lower ORR than PLDH monotherapy. No other comparison of chemotherapies was found to have a statistically significant difference.
A RCT that provided results for this population but which did not share a common comparator within the network compared low-dose paclitaxel (80 mg/m2) with low-dose paclitaxel (80 mg/m2) plus carboplatin. 62 However, Lortholary et al. 62 identified no significant difference in OS between the two different treatment regimens (OR 1.062, 95% CI 0.510 to 2.209).
FIGURE 10.
Networks for ORR in people with PRR recurrent ovarian cancer.

| Comparison | OR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| vs. PLDH monotherapy (OR > 1 favours comparator, OR < 1 favours PLDH monotherapy) | |||
| Topotecan monotherapy (i.v.) | 0.529 | 0.184 | 1.166 |
| Paclitaxel monotherapy | 0.290 | 0.040 | 0.982 |
| Topotecan monotherapy (oral) | 0.622 | 0.098 | 2.116 |
| Paclitaxel monotherapy (weekly) | 0.224 | 0.022 | 0.884 |
| Topotecan monotherapy (i.v., weekly) | 0.253 | 0.051 | 0.761 |
| vs. topotecan monotherapy: i.v. (OR > 1 favours comparator, OR < 1 favours topotecan monotherapy: i.v.) | |||
| Paclitaxel monotherapy | 0.548 | 0.111 | 1.553 |
| Topotecan monotherapy (oral) | 1.176 | 0.283 | 3.283 |
| Paclitaxel monotherapy (weekly) | 0.423 | 0.059 | 1.470 |
| Topotecan monotherapy (i.v., weekly) | 0.478 | 0.154 | 1.086 |
| vs. paclitaxel monotherapy (OR > 1 favours comparator, OR < 1 favours paclitaxel monotherapy) | |||
| Topotecan monotherapy (oral) | 3.387 | 0.379 | 13.810 |
| Paclitaxel monotherapy (weekly) | 0.771 | 0.271 | 1.736 |
| Topotecan monotherapy (i.v., weekly) | 1.383 | 0.191 | 5.216 |
| vs. topotecan monotherapy: oral (OR > 1 favours comparator, OR < 1 favours topotecan monotherapy: oral) | |||
| Paclitaxel monotherapy (weekly) | 0.530 | 0.041 | 2.321 |
| Topotecan monotherapy (i.v., weekly) | 0.601 | 0.090 | 2.090 |
| vs. paclitaxel monotherapy: weekly (OR > 1 favours comparator, OR < 1 favours paclitaxel monotherapy: weekly) | |||
| Topotecan monotherapy (i.v., weekly) | 2.251 | 0.215 | 9.439 |
Full population (mixed platinum-free intervals)
OVA-30130 evaluated tumour response as the ORR (CR or PR) with response maintained ≥ 4 weeks based on RECIST criteria. 69 In the full trial population, trabectedin plus PLDH significantly improved ORR compared with PLDH alone [93/337 (27.6%) with trabectedin plus PLDH vs. 63/335 (18.8%) with PLDH alone; p = 0.080; see Table 67). It should be noted that most patients achieved PR, with only six patients being assessed as CR: two in the trabectedin plus PLDH group and four in the PLDH alone group.
In the full trial population, Gordon et al. 49 found no statistically significant difference between PLDH and topotecan in the proportion of patients achieving either CR or PR as their best response [47/239 (19.7%) with PLDH vs. 40/235 (17.0%) with topotecan; p = 0.390; see Table 67]. In addition, a similar proportion of patients in each group achieved SD as their best response [77/239 (32.2%) with PLDH vs. 95/235 (40.4%) with topotecan; see Table 67].
In patients who received at least one dose of study drug (226 patients), ten Bokkel Huinink et al. 21 found no statistically significant difference between topotecan and paclitaxel in ORR [CR or PR; 23/112 (20.5%) with topotecan vs. 15/114 (13.2%) with paclitaxel; p = 0.138; see Table 67]. It should be noted that, of the 226 patients included in the analysis, only 202 were evaluated for response, with the remaining 24 patients considered to be non-responders.
The authors carried out an analysis of response rate relative to baseline disease characteristics. Higher response rates in both groups were observed in patients without ascites at baseline, with better performance status scores (lower score is better), with smaller tumour burden (< 5 cm), and in those who responded to first-line chemotherapy (summarised in Table 66).
| Baseline status | Topotecan response (%) | Paclitaxel response (%) |
|---|---|---|
| Age (years) | ||
| ≤ 40 | 0 | 0 |
| 41–64 | 19.7 | 12.0 |
| ≥ 65 | 23.7 | 16.7 |
| Ascites | ||
| Present | 18.9 | 7.5 |
| Absent | 21.3 | 16.2 |
| Performance status | ||
| 0 | 22.0 | 14.3 |
| 1 | 25.5 | 13.2 |
| 2 | 5.0 | 11.8 |
| Tumour burden (cm) | ||
| < 5 | 33.3 | 18.0 |
| 5 ≤ 10 | 10.9 | 12.5 |
| First-line response | ||
| Responders | 15.2 | 10.5 |
| Non-responders | 5.4 | 2.6 |
Piccart et al. 63 found that a similar proportion of patients achieved PR in the paclitaxel and oxaliplatin groups [7/41 (17.1%) with paclitaxel vs. 7/45 (15.6%) with oxaliplatin]. No patient achieved a CR. The statistical significance of the difference was not assessed in the full publication.
In the full trial population, Gore et al. 24 found no statistically significant difference between oral and i.v. topotecan in the proportion of patients achieving a CR or PR as their best response [17/135 (13%) with oral topotecan vs. 26/131 (20%) with i.v. topotecan; reported as not significant; p-value not reported; see Table 67].
Omura et al. 68 evaluated only patients with measurable disease for tumour response (131 patients treated with paclitaxel 175 mg/m2 vs. 134 patients treated with paclitaxel 250 mg/m2). ORR comprised patients with CR (disappearance of all gross evidence of disease for at least 4 weeks) or PR (≥ 50% reduction in the product of perpendicular measurements of each lesion for at least 4 weeks). Response was assessed before every other cycle of therapy. It is unclear from the methods whether the assessor was masked to treatment allocation.
In the full trial population, a significantly larger proportion of patients in the paclitaxel 250 mg/m2 group than in the 175 mg/m2 group achieved either CR or PR as their best response [49/134 (36%) with paclitaxel 250 mg/m2 vs. 36/131 (27%) with paclitaxel 175 mg/m2; see Table 67]. The accompanying OR was 1.89 (95% CI 1.07 to 3.31; p = 0.027). The OR had been adjusted for histological cell type (papillary serous compared with clear cell or mucinous cell vs. other cell types), cooperative group, performance status and prior platinum sensitivity.
In patients randomised to paclitaxel 250 mg/m2 and who were subsequently randomised to filgrastim 5 or 10 µg/kg, there was no statistically significant difference among the filgrastim groups in the proportion of patients achieving CR or PR [24/68 (35%) with 5 µg/kg filgrastim vs. 25/66 (37.9%) with 10 µg/kg filgrastim].
Rosenberg et al. 60 found no statistically significant difference between paclitaxel every 7 days and paclitaxel every 21 days in the proportion of patients achieving either CR or PR [37/105 (35.2%) with paclitaxel every 7 days vs. 38/103 (36.9%) with paclitaxel every 21 days; reported as not significant; p-value not reported]. As noted, patients with unconfirmed CR (six patients) and PR (13 patients) are included in this analysis and this should be borne in mind when interpreting the results.
| Study | Intervention | Comparison | Overall response (OR, 95% CI) | CR | PR | SD | PD | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | |||
| Gordon et al.49,54 | PLDH (50 mg/m2) every 28 days | Topotecan (1.5 mg/m2/day) for 5 days every 21 days | 47/239 | 40/235 | 38/239 | 29/235 | 9/239 | 11/235 | 77/239 | 95/235 | ||
| 1.155 (0.730 to 1.827) | ||||||||||||
| Gore et al.24 | Oral topotecan (2.3 mg/m2/day) | Intravenous topotecan (1.5 mg/m2/day) for 5 days every 21 days | 17/135 | 26/131 | 15/135 | 22/131 | 2/135 | 4/131 | 39/135 | 35/131 | 65/135 | 59/131 |
| 0.634 (0.329 to 1.224) | ||||||||||||
| ten Bokkel Huinink et al.21 | Topotecan (1.5 mg/m2/day) for 5 days | Paclitaxel (175 mg/m2/day) as 3 hour infusion every 21 days | 23/112 | 15/114 | 18/112 | 12/114 | 5/112 | 3/114 | ||||
| 1.561 (0.774 to 3.145) p = 0.138 |
||||||||||||
| aRosenberg et al.60 | Paclitaxel (67 mg/m2) every 7 days | Paclitaxel (200 mg/m2) every 21 days | 37/105 | 38/103 | 24/105 | 21/103 | 13/105 | 17/103 | 43/105 | 33/103 | 15/105 | 19/103 |
| 0.955 (0.563 to 1.620) | ||||||||||||
| Piccart et al.63 | Paclitaxel (175 mg/m2 over 3 hours) every 3 weeks | Oxaliplatin (130 mg/m2 over 2 hours) every 3 weeks | 7/41 | 7/45 | 7/41 | 7/45 | 0/41 | 0/45 | 14/41 | 15/45 | 18/41 | 20/45 |
| 1.098 (0.355 to 3.397) | ||||||||||||
| OVA-30130 | PLDH (30 mg/m2) plus trabectedin (1.1 mg/m2) every 3 weeks | PLDH (50 mg/m2) every 4 weeks | 93/337 | 63/335 | 2/337 | 4/335 | 91/337 | 59/335 | ||||
| 1.467 (1.030 to 2.090) | ||||||||||||
| Omura et al.68 | Paclitaxel (250 mg/m2) every 21 days | Paclitaxel (175 mg/m2) every 21 days | 49/134 | 36/131 | 32/134 | 27/131 | 17/134 | 9/131 | ||||
| 1.89 (1.07 to 3.31) | ||||||||||||
The RCTs available for inclusion in the NMA evaluating ORR in patients with mixed PFIs in recurrent ovarian cancer are summarised in Table 67. However, based on expert clinical opinion, the TAG decided not to evaluate this mixed-patient population, as the results would not be considered clinically meaningful.
Quality of life
Of the 16 RCTs identified, 10 reported some level of data on QoL. 21,23,30,31,48–50,61–63 A systematic review of health-related QoL reporting in ovarian cancer trials identified considerable disparity in the level of reporting of QoL results, the questionnaires used to evaluate QoL and the time points for evaluation. 8 Given the often palliative nature of second- and subsequent-line therapeutic treatments for ovarian cancer, there has been a move to place greater emphasis on assessment of QoL in this condition.
The most commonly used scale in the identified trials is the EORTC QLQ-C30 questionnaire,81 which was developed to assess the QoL of cancer patients and can be supplemented with disease-specific modules for individual cancers, including ovarian cancer. The QLQ-C30 questionnaire81 comprises six questions that address dyspnoea, sleep disturbance, appetite loss, constipation, diarrhoea and financial impact, in addition to one global QoL scale, five functional scales (physical, role, emotional, cognitive, and social) and three symptom scales (fatigue, pain and nausea/vomiting).
Here, a narrative description of QoL is presented for those trials providing data on this outcome.
Summary of results for quality of life
Owing to a paucity of data, results for individual trials assessing QoL are summarised here. It should be noted that, generally, reporting of results was limited, with few trials reporting scores generated from responses to the questionnaires.
Baseline QoL scores showed impaired global health scores and considerable symptom burden. At 3 months, PLDH plus carboplatin was associated with a significant improvement in global health compared with paclitaxel plus carboplatin. However, this benefit was not maintained at 6 months.
The QLQ-OV28 questionnaire indicated that paclitaxel plus carboplatin was associated with significantly worse peripheral neuropathy and other chemotherapy side effects at 3 months and 6 months compared with PLDH plus carboplatin. 71
Mean change in scores from baseline to end of treatment were similar between trabectedin plus PLDH and PLDH monotherapy, with no differences reaching statistical significance on any questionnaire. The difference between groups in mean scores for the QLQ-C30 global health status scale81 did not reach ≥ 5 at any time point, which indicated non-significance. Additional information on QoL in the subgroup of patients with PPS ovarian cancer provided in the MS indicates a difference in global health status score among responding patients beyond cycle 5, with patients in the trabectedin plus PLDH group having a higher score than those receiving PLDH monotherapy (higher score is favourable).
The EORTC QLQ-C3081 scores were similar between the groups and neither paclitaxel nor topotecan was associated with any compromise of QoL.
The ICON4/AGO-OVAR 2.261 investigators evaluated QoL using the EORTC QLQ-C3081 questionnaire. It was reported that, in the first 6 months after randomisation, patients receiving platinum monotherapy scored significantly worse on the nausea and vomiting symptom scale than did the paclitaxel plus platinum-based chemotherapy group. However, this difference seemed to be transient and was observed for only the first 15 weeks after randomisation. All other worst scores or AUCs were reported to be similar between treatment groups for the remaining eight symptom scales, the five functional scales, and global health status of the QLQ-C30. 81
Gonzalez-Martin et al. 48 also evaluated QoL using the QLQ-C3081 questionnaire. No differences between treatments in the five functional components of the QLQ-C30 were reported.
Quality of life was assessed using the EORTC QLQ-C3081 questionnaire. At week 12, no significant differences between the groups in any of the measured scores were noted. The proportion of patients who had a worsened global QoL score was also reported to be similar in the two treatment groups. Topotecan was associated with a significantly more favourable rating on the pain subscale of the EORTC QLQ-C30.
Based on responses to the EORTC QLQ-C3081 and QLQ-C28,71 no statistically significant differences between treatment groups for all scales/items at baseline or in changes in score from baseline to treatment discontinuation were noted.
Response to EORTC QLQ-C3081 indicated that global health scores were stable over time and similar across treatment arms. Among symptom and functional scales, patients receiving weekly paclitaxel plus carboplatin experienced improvements in constipation, abdominal/gastrointestinal symptoms, appetite loss, pain and emotional functioning. Patients treated with weekly paclitaxel alone experienced improvements in attitude to disease and insomnia, but worsening of dyspnoea and peripheral neuropathy.
Mean QoL score on the EORTC QLQ-C3081 increased by > 10 points between baseline and cycle 4 for patients in the paclitaxel group, irrespective of study withdrawal. By contrast, in the oxaliplatin group, the mean QoL score decreased through cycle 2, but by < 10 points, after which most patients’ mean scores returned to baseline levels.
It was reported that there were no differences between treatment groups in EORTC QLQ-OV28 scores. 71
Quality-of-life data were collected during CALYPSO31 using the EORTC QLQ-C3081 questionnaire and supplemented by the ovarian cancer-specific OV28 module. QoL was assessed at baseline and subsequently at the 3-, 6-, 9- and 12-month assessments. QoL was not assessed after progression of disease. Results for QoL are presented in full in an accompanying publication. 59
Analyses of QoL were restricted to those patients with both a completed baseline questionnaire and at least one QoL form completed during follow-up. At baseline, 90% of patients completed the questionnaires [421/467 (90.1%) with PLDH plus carboplatin vs. 458/509 (90.0%) with paclitaxel plus carboplatin]. Compliance remained high at 3 months’ follow-up (79.3% with PLDH plus carboplatin vs. 73.5% with paclitaxel plus carboplatin) but steadily declined over the remaining 9 months (completed questionnaires: 6 months – 68.3% with PLDH plus carboplatin vs. 60.3% with paclitaxel plus carboplatin; 12 months – 50.6% with PLDH plus carboplatin vs. 49.7% with paclitaxel plus carboplatin). Given that only 50% of patients were compliant at 12 months, the authors restricted reporting of results to data collected up to 9 months’ follow-up.
Baseline QoL scores showed impaired global health scores and considerable symptom burden (see Table 68).
At 3 months, PLDH plus carboplatin was associated with a significant improvement in global health compared with paclitaxel plus carboplatin (mean score at 3 months [standard deviation (sd) –2.2 (22.7) with paclitaxel plus carboplatin vs. 2.6 (26.0) with PLDH plus carboplatin; p = 0.01]. However, this benefit was not maintained at 6 months, at which time the difference between groups for this measure was not statistically significant [4.8 (24.4) with paclitaxel plus carboplatin vs. 2.4 (26.4) with PLDH plus carboplatin; p = 0.31]. It should be noted that the difference between groups is modest. Results from QoL analyses are presented in Table 68.
Other symptom scores for which there was a significant difference at 3 months, but which was not maintained at 6 months, are physical functioning; nausea and vomiting; pain; dyspnoea; and sexual functioning.
Assessment of QLQ-OV28 indicated that paclitaxel plus carboplatin was associated with significantly worse peripheral neuropathy and other chemotherapy side effects at 3 months and 6 months compared with PLDH plus carboplatin.
| Item/domain | Baseline scores | 3-month changea | 6-month changea | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CP | CD | CP | CD | p-value | CP | CD | p-value | |||||||
| n | Mean (sd) | n | Mean (sd) | n | Mean (sd) | n | Mean (sd) | n | Mean (sd) | n | Mean (sd) | |||
| Functional scales scores | ||||||||||||||
| Physical functioning | 452 | 79.5 (20.7) | 414 | 79.8 (20.1) | 313 | −7.4 (18.1) | 309 | −3.7 (18.8) | 0.01 | 228 | −1.1 (20.6) | 242 | −2.8 (19.1) | 0.36 |
| Role functioning | 447 | 72.6 (30.4) | 413 | 72.3 (31.9) | 310 | −9.2 (31.2) | 305 | −4.5 (33.8) | 0.07 | 225 | 2.4 (34.7) | 241 | −1.7 (31.5) | 0.18 |
| Emotional functioning | 447 | 63.4 (25.6) | 410 | 64.4 (25.2) | 308 | 6.5 (21.8) | 303 | 8.3 (22.6) | 0.31 | 225 | 6.8 (25.3) | 239 | 5.1 (23.7) | 0.46 |
| Cognitive functioning | 448 | 83.9 (20.2) | 412 | 83.8 (20.2) | 309 | −6.4 (20.3) | 305 | −3.6 (19.1) | 0.08 | 226 | −3.1 (21.3) | 242 | −5.2 (22.6) | 0.31 |
| Social functioning | 445 | 74.2 (28.2) | 411 | 78.4 (27.2) | 303 | −5.2 (28.3) | 304 | −5.1 (28.5) | 0.95 | 224 | 0.1 (31.2) | 242 | −4.3 (23.8) | 0.09 |
| Global health status score | ||||||||||||||
| Global health status/QoL | 447 | 62.2 (23.0) | 408 | 61.4 (24.2) | 307 | −2.2 (22.7) | 301 | 2.6 (26.0) | 0.01 | 227 | 4.8 (24.4) | 238 | 2.4 (26.4) | 0.31 |
| Symptoms scales scores | ||||||||||||||
| Fatigue | 450 | 34.7 (25.7) | 413 | 34.4 (27.5) | 310 | −9.4 (25.3) | 306 | −6.3 (27.0) | 0.13 | 225 | 1.2 (25.9) | 243 | −4.6 (27.6) | 0.02 |
| Nausea and vomiting | 449 | 8.0 (17.3) | 413 | 10.9 (20.7) | 309 | −3.5 (22.9) | 308 | −8.4 (26.1) | 0.01 | 225 | 3.2 (21.9) | 244 | −0.3 (24.9) | 0.11 |
| Pain | 450 | 27.1 (28.4) | 414 | 25.9 (28.2) | 310 | 1.3 (31.2) | 306 | 6.2 (28.7) | 0.04 | 227 | 6.4 (31.3) | 243 | 6.0 (29.3) | 0.86 |
| Dyspnoea | 444 | 17.9 (25.3) | 409 | 19.4 (27.8) | 307 | −11.8 (29.6) | 305 | −3.2 (30.0) | < 0.001 | 224 | −4.5 (30.1) | 244 | −5.7 (29.4) | 0.64 |
| Insomnia | 447 | 36.8 (32.9) | 413 | 36.6 (32.8) | 306 | 2.2 (31.7) | 304 | 5.7 (31.4) | 0.16 | 223 | 3.6 (30.2) | 242 | 4.6 (32.6) | 0.73 |
| Appetite loss | 445 | 18.7 (29.2) | 413 | 21.5 (30.8) | 305 | −2.3 (31.5) | 306 | 0.6 (30.1) | 0.24 | 225 | 7.3 (27.9) | 242 | 5.3 (31.3) | 0.47 |
| Constipation | 449 | 22.6 (31.1) | 409 | 23.6 (31.7) | 309 | −4.5 (33.2) | 301 | −5.5 (34.9) | 0.74 | 227 | 3.6 (34.2) | 239 | −2.7 (34.3) | 0.05 |
| Diarrhoea | 447 | 10.3 (21.1) | 409 | 13.5 (24.8) | 307 | 0.5 (24.0) | 300 | 3.4 (25.2) | 0.15 | 227 | −1.0 (24.5) | 238 | 3.8 (22.5) | 0.03 |
| Financial difficulties | 441 | 14.7 (27.3) | 405 | 12.4 (24.8) | 303 | −4.6 (26.8) | 297 | −2.4 (22.6) | 0.28 | 221 | −2.6 (30.5) | 239 | −2.0 (22.9) | 0.81 |
| QLQ–OV28 | ||||||||||||||
| Abdominal/gastrointestinal symptoms | 442 | 29.1 (22.6) | 411 | 30.3 (24.0) | 306 | 5.0 (22.0) | 305 | 4.7 (21.7) | 0.83 | 223 | 6.8 (23.1) | 238 | 5.1 (23.9) | 0.43 |
| Peripheral neuropathy | 434 | 17.7 (22.0) | 402 | 15.3 (20.6) | 300 | −27.4 (26.8) | 297 | −6.1 (18.9) | < 0.001 | 218 | −24.2 (30.5) | 233 | −9.8 (20.1) | < 0.001 |
| Other chemotherapy side effects | 435 | 15.0 (14.9) | 405 | 14.2 (15.1) | 301 | −24.7 (18.4) | 301 | −7.6 (16.8) | < 0.001 | 219 | −16.2 (19.9) | 236 | −9.5 (15.9) | < 0.001 |
| Hormonal/menopausal symptoms | 435 | 26.4 (28.0) | 405 | 24.2 (28.6) | 300 | −1.6 (24.2) | 301 | −0.6 (28.3) | 0.62 | 219 | −2.4 (28.4) | 235 | −2.9 (28.4) | 0.84 |
| Body image | 431 | 23.9 (27.6) | 401 | 24.3 (28.0) | 297 | −12.2 (29.3) | 292 | −1.2 (28.2) | < 0.001 | 212 | −10.4 (31.4) | 234 | −3.8 (27.0) | 0.02 |
| Attitude to disease and treatment | 432 | 57.2 (28.2) | 397 | 56.3 (28.5) | 295 | −0.4 (25.4) | 290 | 1.7 (25.8) | 0.32 | 216 | 0.0 (28.0) | 228 | 1.8 (24.3) | 0.48 |
| Sexual functioning | 385 | 20.4 (23.5) | 358 | 16.3 (21.7) | 241 | 4.5 (17.8) | 232 | 0.4 (18.5) | 0.02 | 173 | 0.8 (19.7) | 187 | 0.2 (18.5) | 0.78 |
OVA-30130 evaluated patient-reported outcomes as an exploratory end point using the cancer-specific EORTC QLQ-C3081 and QLQ-OV28 questionnaires, together with the generic European Quality of Life-5 Dimensions (EQ-5D) questionnaire, which is the utility measure preferred by NICE. Results from the analyses were reported in a follow-up publication by Krasner et al. 67
Patients completed questionnaires at baseline, on day 1 of each treatment cycle before administration of the allocated treatment, and at the end of treatment. Statistical analyses of QoL were based on all randomised patients. Non-random withdrawal from treatment across groups, most frequently as a result of disease progression or poor tolerability, is well recognised in trials evaluating treatments in cancer. To account for the potential imbalance in patients lost to follow-up between the groups, the authors implemented a pattern mixture model.
Compliance was high, with an overall rate of missing questionnaires of 15%, which was balanced across the groups (14.4% with trabectedin plus PLDH vs. 15.2% with PLDH alone). At most time points, the rate of missing questionnaires was < 10%, but at the end of treatment the rate rose to 34%.
Mean change in scores from baseline to end of treatment were similar between trabectedin plus PLDH and PLDH alone, with no differences reaching statistical significance on any questionnaire. The authors report that the difference between groups in mean scores for the QLQ-C3081 global health status scale did not reach ≥ 5 at any time point, which indicated non-significance. Mean change in QLQ-C30 global health status scale over time is presented in Figure 11. Minor, sporadic differences in the fatigue symptom scale were observed in cycles 3 and 9, with some worsening of fatigue for subjects with trabectedin plus PLDH.
In the submission received from PharmaMar, additional information on QoL in the subgroup of patients with PPS ovarian cancer is provided. The manufacturer notes that a difference in global health status score was observed among responding patients beyond cycle 5 in the PPS subgroup, with patients in the trabectedin plus PLDH group having a higher score than those receiving PLDH alone (higher score is favourable) (Figure 12). The manufacturer comments that the benefit associated with trabectedin plus PLDH is clinically meaningful. It should be noted that the analysis seems to be based on patients with PPS ovarian cancer who responded to treatment (n is reported to be 51) rather than the full PPS subgroup. In addition, all QoL analyses are exploratory.
FIGURE 11.
Mean QLQ-C30 global health status score over time (reproduced with permission from PharmaMar’s submission).
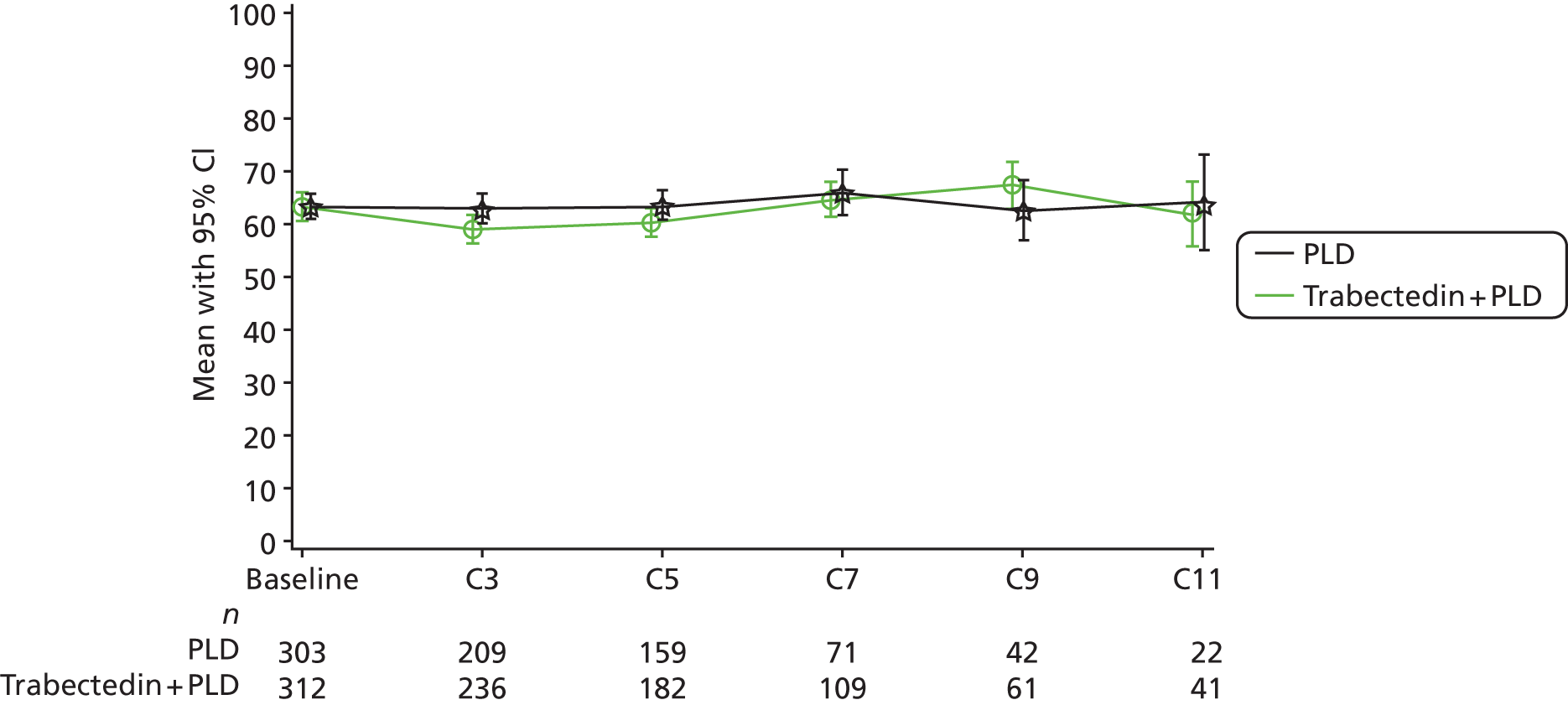
FIGURE 12.
Mean QLQ-C30 global health status score over time for the PPS subgroup (reproduced with permission from PharmaMar’s submission). SEM, standard error of the mean.

ten Bokkel Huinink et al. 21 evaluated QoL using the EORTC QLQ-C3081 questionnaire. It is reported that between 75% and 85% of patients enrolled in the study had evaluable QoL data. However, no results are reported in either of the two identified publications. 21,52 The authors comment that scores were similar between the groups and that neither paclitaxel nor topotecan was associated with any compromise of QoL.
Gordon et al. 49 report that QoL was assessed using the EORTC QLQ-C3081 questionnaire. All patients completed a QLQ-C30 questionnaire before study entry, during every cycle and 4 weeks after the last treatment dose. The full publication reports that about 82% of patients completed the questionnaire at baseline and that at study entry function and symptom scale scores were similar between the groups. At week 12, it is reported that there were no significant differences between the groups in any of the measured scores. No further details are reported in the full publication. Additional detail is reported in TA91,13 which is summarised here.
Technology appraisal no. 91 reports that only 50% of patients completed the questionnaire at 12 weeks. 13 At week 12, similar proportions of patients in the PLDH and topotecan groups had improved or stable global QoL scores, with no statistically significant difference identified between groups [68/239 (28.5%) with PLDH vs. 55/235 (23.4%) with topotecan; relative risk (RR) 0.82, 95% CI 0.61 to 1.12]. The proportion of patients who had a worsened global QoL score was also reported to be similar in the two treatment groups [49/239 (20.5%) with PLDH vs. 48/235 (20.4%) with topotecan; RR 0.97, 95% CI 0.70 to 1.42]. Considering the subscales of the QLQ-C30,79 a statistically significant difference between PLDH and topotecan was identified for only the pain subscale score (RR 1.26, 95% CI 1.08 to 1.50), which favoured topotecan (results from TA9113 summarised in Table 69).
| QoL subscale | PLDH, (n/N) | Topotecan, % (n/N) | RR (95 CI)a |
|---|---|---|---|
| Physical | 56 (66/118) | 56 (61/107) | 1.02 (0.81 to 1.28) |
| Role | 65 (77/118) | 58 (63/109) | 0.89 (0.72 to 1.10) |
| Emotional | 67 (80/119) | 74 (80/108) | 1.10 (0.93 to 1.31) |
| Cognitive | 73 (87/119) | 73 (79/108) | 1.00 (0.85 to 1.17) |
| Social | 69 (82/119) | 64 (69/108) | 0.93 (0.77 to 1.12) |
| Global QoL | 58 (68/117) | 52 (54/104) | 0.89 (0.70 to 1.13) |
| Fatigue | 57 (67/118) | 56 (61/109) | 0.99 (0.78 to 1.24) |
| Nausea/vomiting | 72 (86/119) | 71 (77/109) | 0.98 (0.83 to 1.15) |
| Pain | 64 (76/119) | 81 (88/109) | 1.26 (1.08 to 1.50) |
Pfisterer et al. 50 evaluated QoL using the EORTC QLQ-C3081 and QLQ-C28 (version 2) questionnaires. QoL was assessed 2 weeks before enrolment and before commencement of each treatment cycle. Questionnaire completion rate was high, with 85.4% (152/178) and 82.6% (147/178) of patients in the gemcitabine plus carboplatin and carboplatin alone groups, respectively, having completed a questionnaire at baseline and at least one postbaseline questionnaire. The authors report that there were no statistically significant differences between treatment groups for all scales/items at baseline or in changes in score from baseline to treatment discontinuation. No further details reported.
The ICON4/AGO-OVAR 2.2 investigators evaluated QoL using the EORTC QLQ-C3081 questionnaire. In total, 90% (482/536) of patients enrolled in centres following the MRC CTU ICON4 protocol completed the questionnaire at baseline, before receiving any study drug. The authors report that all scales were balanced across the two treatment groups at baseline and that most patients had little or no functional difficulties and few had moderate or severe symptoms at baseline (no further details reported). In the first 6 months after randomisation, patients receiving platinum monotherapy scored significantly worse on the nausea and vomiting symptom scale than did the paclitaxel plus platinum-based chemotherapy group (p = 0.0014 for worst score and p = 0.005 for AUC). However, this difference seemed to be transient and was observed for only the first 15 weeks after randomisation. All other worst scores or AUCs were reported to be similar between treatment groups for the remaining eight symptom scales, the five functional scales, and global health status of the QLQ-C3081 (no further details reported).
Gonzalez-Martin et al. 48 also evaluated QoL using the QLQ-C3081 questionnaire. The authors reported that there were no differences between treatments in the five functional components of the QLQ-C30. No other details were reported.
Lortholary et al. 62 explored QoL using the EORTC QLQ-C3081 and QLQ-OV28 questionnaires. Completion rate of questionnaires ranged between 40% and 70%, with questionnaires collected at baseline, and after two, four and six cycles of treatment. Global health scores were stable over time and similar across treatment arms. Among symptom and functional scales, patients receiving weekly paclitaxel plus carboplatin experienced improvements in constipation, abdominal/gastrointestinal symptoms, appetite loss, pain and emotional functioning. Patients treated with weekly paclitaxel experienced improvements in attitude to disease and insomnia, but worsening of dyspnoea and peripheral neuropathy. No further details reported.
Piccart et al. 63 used the EORTC QLQ-C3081 questionnaire and a specific checklist to evaluate QoL. Patients were to complete the questionnaires at least 8 days before their first treatment and, subsequent to start of treatment, every 6 weeks or every two visits. At baseline, completed questionnaires were available for 66 patients. However, at the end of the second treatment cycle (week 6) only 47 patients had completed their questionnaires, with a further drop to 31 completed questionnaires by the end of the fourth treatment cycle (12 weeks). The authors report that the mean QoL score increased by > 10 points between baseline and cycle 4 for patients in the paclitaxel group, irrespective of study withdrawal. By contrast, in the oxaliplatin group, the mean QoL score decreased through cycle 2, but by <10 points, after which most patients’ mean scores returned to baseline levels. The authors propose that the initial decrease in score in the oxaliplatin group is associated with peripheral neurotoxicity. No further details on scores are reported.
Sehouli et al. 23 explored disease-specific QoL using the EORTC QLQ-OV28 questionnaire. Details on schedule of completion of questionnaires were not reported. Baseline data were available for 120 patients (65 treated with conventional topotecan vs. 55 treated with weekly topotecan). A second assessment was available for considerably fewer patients (39 treated with conventional topotecan vs. 20 treated with weekly topotecan), but it is unclear at what point in the trial the second questionnaire was completed. Patients with at least a completed baseline and at least one follow-up assessment reported an improvement in neuropathy scales but a worsening in body image. The authors reported that there was no difference in scores between treatment groups. No further details were reported.
Adverse events
Summary of results for adverse effects
Data for adverse effects for individual trials are reported in the main text. Within each trial, the most frequently reported adverse effects were as expected for the individual treatments based on the SmPC. Commonly occurring adverse effects were alopecia, nausea and vomiting, haematological toxicities (neutropenia, anaemia, thrombocytopenia and leucopenia).
Based on expert clinical advice, the TAG restricted its comparison of AEs to those considered most problematic for patients or most likely to consume substantial health-care resource. The potential for a NMA was, therefore, investigated for the following severe (grades 3 and 4) AEs: allergic reaction; alopecia; anaemia; fatigue; febrile neutropenia; nausea and vomiting; and neuropathy. The results of each investigation are presented in the main text. The results were mixed, with most found to be non-significant or with chemotherapies having significant lower risk of one or more AEs but then being found to have significantly higher risks of others (e.g. PLDH plus carboplatin has significantly less risk of allergic reaction and alopecia but significantly higher risk of anaemia and nausea and vomiting when compared with paclitaxel plus carboplatin). In many cases, a NMA was not possible owing to the lack of available data in the trials assessed. In these instances, the individual trial results are reported with the ORs and 95% CIs calculated. Overall, no chemotherapy was consistently associated with either a lower risk or a higher risk of the severe AEs assessed.
Bafaloukos et al. 29 based the safety analysis on the 177 patients who received at least one cycle of allocated treatment (84 in the PLDH plus carboplatin group vs. 89 in the carboplatin plus paclitaxel group). A significantly larger proportion of patients in the paclitaxel plus carboplatin group discontinued treatment because of associated toxicity (13.5% with paclitaxel plus carboplatin vs. 3% with PLDH plus carboplatin; p = 0.016).
Neutropenia (grades 3 and 4) was the most commonly observed severe toxicity, with a similar proportion of people between groups experiencing this adverse effect (30% with paclitaxel plus carboplatin vs. 35% with PLDH plus carboplatin); the difference between groups did not reach statistical significance (p-value not reported).
Pegylated liposomal doxorubicin hydrochloride plus carboplatin was associated with a significantly higher rate of severe thrombocytopenia (grades 3 and 4: 11% with PLDH plus carboplatin vs. 2% with paclitaxel plus carboplatin; p = 0.016; Table 70) and PPE and skin toxicity (grades 1 and 2; 38% with PLDH plus carboplatin vs. 9% with paclitaxel plus carboplatin; p = 0.003). By contrast, paclitaxel plus carboplatin was associated with a significantly higher rate of severe neurotoxicity (7% with paclitaxel plus carboplatin vs. 0% with PLDH plus carboplatin; p = 0.029) and alopecia (20% with paclitaxel plus carboplatin vs. 5% with PLDH plus carboplatin; p = 0.003).
| Event | PLDH plus carboplatin (n = 84) | Paclitaxel plus carboplatin (n = 89) | ||||||
|---|---|---|---|---|---|---|---|---|
| Withdrawal owing to haematological events | 1a | 6b | ||||||
| Withdrawal owing to hypersensitivity | 2 | 3 | ||||||
| Withdrawal owing to grade 3 skin toxicity | 1 | 0 | ||||||
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| Neutropenia | 13 (15%) | 20 (24%) | 23 (27%) | 7 (8%) | 14 (16%) | 20 (22%) | 18 (20%) | 9 (10%) |
| Anaemia | 27 (32%) | 23 (27%) | 7 (8%) | 1 (1%) | 29 (33%) | 0 | 3 (3%) | 0 |
| Leucopenia | 25 (30%) | 30 | 4 (5%) | 1 (1%) | 24 (27%) | 23 (26%) | 5 (6%) | 1 (1%) |
| Thrombocytopeniac | 4 (5%) | 7 (8%) | 9 (10%) | 1 (1%) | 1 (1%) | 6 (7%) | 2 (2%) | 0 |
| Stomatitis | 7 (8%) | 5 (6%) | 3 (3%) | 0 | – | 1 (1%) | – | 0 |
| Nausea/vomiting | 16 (19%) | 12 (14%) | 4 (5%) | 0 | 18 (20%) | 10 (11%) | 1 (1%) | 0 |
| Diarrhoea | 5 (6%) | 1 (1%) | 0 | 0 | 5 (6%) | 1 (1%) | 1 (1%) | 0 |
| Infection | 3 (4%) | 1 (1%) | 1 (1%) | 1 (1%) | 1 (1%) | 3 (3%) | – | 0 |
| Neurotoxicityc | 19 (23%) | 1 (1%) | 0 | 0 | 24 (27%) | 27 (30%) | 5 (6%) | 1 (1%) |
| Alopeciac | 12 (14%) | 5 (6%) | 4 (5%) | 0 | 1 (1%) | 56 (63%) | 18 (20%) | 0 |
| Allergy | 4 (5%) | 2 (2%) | 1 (1%) | 0 | 18 (20%) | 9 (10%) | 1 (1%) | 0 |
| Skinc | 9 (11%) | 12 (14%) | 1 (1%) | 0 | 6 (7%) | 2 (2%) | 0 | 0 |
| Hand and foot | 2 (2%) | 8 (10%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Fatigue | 8 (10%) | 6 (7%) | 0 | 0 | 12 (13%) | 6 (7%) | 0 | 0 |
| Fever | 2 (2%) | 4 (5%) | 0 | 0 | – | 5 (6%) | 0 | 0 |
| Anorexia | 5 (6%) | – | 0 | 0 | 4 (4%) | 2 (2%) | 0 | 0 |
| Cardiac | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Arthralgias/myalgias | 6 (7%) | 0 | 0 | 0 | 18 (20%) | 8 (9%) | 0 | 0 |
In the CALYPSO trial,31 significantly fewer patients treated with PLDH plus carboplatin discontinued treatment early as a result of adverse effects compared with patients treated with paclitaxel plus carboplatin (6% with PLDH plus carboplatin vs. 15% with paclitaxel plus carboplatin; p < 0.001). There were two treatment-related deaths in the PLDH plus carboplatin group: one attributed to cerebral haemorrhage and one to acute myeloid leukaemia.
Overall, severe (grades 3 and 4) non-haematological toxicity occurred significantly more frequently in the paclitaxel plus carboplatin group (36.8% with paclitaxel plus carboplatin vs. 28.4% with PLDH plus carboplatin; p = 0.001). Incidence of anaemia and febrile neutropenia were similar between treatment groups. However, grade 3 and grade 4 neutropenia and thrombocytopenia were significantly more frequent in the paclitaxel-plus-carboplatin group (neutropenia: p < 0.01; thrombocytopenia: p < 0.001; Table 71).
Adverse events that occurred significantly more frequently in the paclitaxel plus carboplatin group than in the PLDH plus carboplatin group were grade 2 alopecia (complete or total hair loss) (p < 0.001), hypersensitivity reactions (p < 0.001), and sensory and motor neuropathy (sensory, p < 0.001; motor, p = 0.002; see Table 71). By contrast, PLDH plus carboplatin was associated with a significantly higher incidence of hand–foot syndrome (grades 2 and 3; p < 0.001), nausea (p < 0.001), vomiting (p < 0.001) and mucositis (p < 0.001; Table 71).
| Event | PLDH plus carboplatin (n = 466) | Paclitaxel plus carboplatin (n = 501) | p-value |
|---|---|---|---|
| Withdrawal due to hypersensitivity reaction | 1% | 6% | < 0.001 |
| Treatment-related fatalities | 3 | 1 | NR |
| Grades 3 and 4 | |||
| Neutropenia | 164 (35.2%) | 229 (45.7%) | < 0.01 |
| Febrile neutropenia | 12 (2.6%) | 21 (4.2%) | 0.171 |
| Infection | 12 (2.6%) | 16 (3.2%) | 0.723 |
| Thrombocytopenia | 74 (15.9%) | 31 (6.2%) | < 0.001 |
| Anaemia | 37 (7.9%) | 27 (5.4%) | 0.573 |
| Bleeding | 3 (0.6%) | 0 | 0.718 |
| Grade ≥ 2 | |||
| Alopecia | 31 (7%) | 419 (83.6%) | < 0.001 |
| Nausea | 164 (35.2%)a | 121 (24.2%)a | < 0.001 |
| Vomiting | 105 (22.5%)a | 78 (15.6%)a | < 0.001 |
| Constipation | 100 (21.5%) | 109 (21.8%) | 0.6 |
| Diarrhoea | 25 (5.4%)a | 41 (7.6%)a | < 0.001 |
| Fatigue | 172 (36.9%)a | 202 (40.3%)a | 0.220 |
| Mucositis | 65 (13.9%)a | 35 (7%)a | < 0.001 |
| Neuropathy (sensory) | 23 (4.9%)a | 135 (26.9%) | < 0.001 |
| Neuropathy (motor) | 7 (1.5%) | 22 (4.4%)a | 0.002 |
| Cardiovascular | 10 (2.1%)a | 17 (3.4%) | 0.616 |
| Allergic reaction | 26 (5.6%)a | 94 (18.8%) | < 0.001 |
| Hand–foot syndrome | 56 (12.0%)a | 11 (2.2%)a | < 0.001 |
| Arthralgia/myalgia | 19 (4.0%)a | 96 (19.2%)a | < 0.001 |
| Any grade | |||
| Alopeciab | 158 (34%) | 452 (90.2%) | – |
| Nausea | 365 (78.3%) | 354 (70.7%) | – |
| Vomiting | 228 (48.9%) | 181 (36.1%) | – |
| Constipation | 258 (55.4%) | 287 (57.5%) | – |
| Diarrhoea | 108 (23.2%) | 158 (31.6%) | – |
| Fatigue | 363 (77.9%) | 409 (81.6%) | – |
| Mucositis | 182 (39.1%) | 131 (26.1%) | – |
| Neuropathy (sensory) | 186 (39.9%) | 366 (73.1%) | – |
| Neuropathy (motor) | 34 (7.3%) | 67 (13.4%) | – |
| Cardiovascular | 49 (10.5%) | 57 (11.4%) | – |
| Allergic reaction | 72 (15.5%) | 165 (32.9%) | – |
| Hand–foot syndrome | 180 (38.6%) | 51 (10.2%) | – |
| Arthralgia/myalgia | 104 (22.3%) | 250 (49.9%) | – |
Alberts et al. 28 reported that the most common grade 3 and grade 4 AEs in the PLDH plus carboplatin group were haematological, with eight patients (26%) experiencing a grade 4 haematological AE (thrombocytopenia and neutropenia; Table 72). No patient in the PLDH plus carboplatin group had an allergic reaction compared with nine patients treated with carboplatin alone.
| Event | PLDH plus carboplatin (n = 31) | Carboplatin alone (n = 30) | ||||
|---|---|---|---|---|---|---|
| Withdrawal due to AEs | 15 (48%)a | 7 (23%)b | ||||
| Grade 4 haematological AEs | 8 (26%) | 0 | ||||
| Grade | ≤ 2 | 3 | 4 | ≤ 2 | 3 | 4 |
| Abdominal pain/cramping | 97% | 3% | 0% | 100% | 0% | 0% |
| Allergy/hypersensitivity | 100% | 0% | 0% | 83% | 13% | 3% |
| Anaemia | 84% | 16% | 0% | 100% | 0% | 0% |
| Catheter-related infection | 97% | 3% | 0% | 100% | 0% | 0% |
| Constipation/bowel obstruction | 94% | 6% | 0% | 97% | 3% | 0% |
| Depression | 100% | 0% | 0% | 97% | 3% | 0% |
| Dyspnoea | 94% | 3% | 0% | 93% | 3% | 3% |
| Fatigue/malaise/lethargy | 90% | 10% | 0% | 93% | 7% | 0% |
| Febrile neutropenia | 90% | 10% | 0% | 100% | 0% | 0% |
| Hand–foot skin reaction | 97% | 3% | 0% | 100% | 0% | 0% |
| Hypomagnesaemia | 97% | 3% | 0% | 100% | 0% | 0% |
| Hyponatraemia | 97% | 3% | 0% | 100% | 0% | 0% |
| Hypotension | 100% | 0% | 0% | 97% | 3% | 0% |
| Infection with grades 3 and 4 neutropenia | 94% | 6% | 0% | 100% | 0% | 0% |
| Leucopenia | 71% | 26% | 3% | 100% | 0% | 0% |
| Myalgia | 100% | 0% | 0% | 97% | 3% | 0% |
| Nausea | 94% | 6% | 0% | 100% | 0% | 0% |
| Neutropenia/granulocytopenia | 52% | 29% | 19% | 97% | 3% | 0% |
| PRBC transfusion | 90% | 10% | 0% | 100% | 0% | 0% |
| Platelet transfusion | 94% | 6% | 0% | 100% | 0% | 0% |
| Respiratory infection without neutropenia | 97% | 3% | 0% | 100% | 0% | 0% |
| Thrombocytopenia | 61% | 29% | 10% | 90% | 10% | 0% |
| Vomiting | 97% | 3% | 0% | 100% | 0% | 0% |
| Maximum grade any AE | 29% | 45% | 26% | 60% | 37% | 3% |
In OVA-301,30 safety was evaluated using NCI-CTC for AEs and the safety analysis population included all randomly assigned patients who received one or more doses of trabectedin or PLDH. Deaths were summarised by treatment and primary cause. Nineteen patients died during treatment (8 in the PLDH group vs. 11 in the trabectedin plus PLDH group). Twelve patients died (six in each group) as a result of disease progression. One patient in the PLDH group and five patients in the trabectedin plus PLDH group died as a result of an adverse effect. The full publication presented the most common grade 3 and grade 4 AEs, together with other AEs of interest that were potentially related to treatment, which are presented in Table 73. Grade 3 and grade 4 haematological adverse effects were more common in the trabectedin plus PLDH group than in the PLDH alone group. The incidence of known toxicities associated with PLDH, such as hand–foot syndrome, stomatitis and mucosal inflammation, was lower in the trabectedin plus PLDH arm than the PLDH monotherapy arm, although the number of events was low in the combination group.
| Event | Trabectedin plus PLDH (n = 333) | PLDH alone (n = 330) |
|---|---|---|
| Death due to AE | 5 | 1 |
| Grade 4 | ||
| Haematological | ||
| Neutropenia | 113 (33.9%) | 28 (8.5%) |
| Leucopenia | 28 (8.4%) | 8 (2.4%) |
| Thrombocytopenia | 27 (8.1%) | 2 (0.6%) |
| Anaemia | 10 (3.0%) | 1 (0.3%) |
| Febrile neutropenia | 8 (2.4%) | 1 (0.3%) |
| Non-haematological | ||
| Hand–foot syndrome | 0 | 4 (1.2%) |
| Mucosal inflammation | 0 | 0 |
| Stomatitis | 0 | 1 (0.3%) |
| Fatigue | 1 (0.3%) | 1 (0.3%) |
| Nausea | 0 | 0 |
| Vomiting | 1 (0.3%) | 0 |
| AST increase | 3 (0.9%) | 1 (0.3%) |
| ALT increase | 8 (2.4%) | 0 |
| Grade 3 | ||
| Haematological | ||
| Neutropenia | 96 (28.8%) | 46 (13.9%) |
| Leucopenia | 82 (24.6%) | 24 (7.3%) |
| Thrombocytopenia | 34 (10.2%) | 6 (1.8%) |
| Anaemia | 31 (9.3%) | 15 (4.5%) |
| Febrile neutropenia | 15 (4.5%) | 6 (1.8%) |
| Non-haematological | ||
| Hand–foot syndrome | 13 (3.9%) | 61 (18.5%) |
| Mucosal inflammation | 7 (2.1%) | 19 (5.8%) |
| Stomatitis | 3 (0.9%) | 16 (4.8%) |
| Fatigue | 19 (5.7%) | 8 (2.4%) |
| Nausea | 29 (8.7%) | 8 (2.4%) |
| Vomiting | 33 (9.9%) | 7 (2.1%) |
| AST increase | 21 (6.3%) | 1 (0.3%) |
| ALT increase | 95 (28.5%) | 1 (0.3%) |
| Other events of interest (grade not stated) | ||
| Alopecia | 40 (12%) | 44 (13%) |
| Alkaline phosphatase increase | 68 (20%) | 24 (7%) |
| Neuropathy | 34 (10%) | 24 (7%) |
| Bilirubin conjugated increase/hyperbilirubinaemia | 51 (15%) | 18 (5%) |
Gordon et al. 49 reported withdrawal rates due to adverse effects of 18% and 16% from the PLDH and topotecan groups, respectively. Almost all patients reported an adverse effect. The incidence of grade 1, 2 or 3 events was reported to be similar across the groups but grade 4 events occurred more frequently in the topotecan group. Gordon et al. 49 note that the toxicity profiles of topotecan and PLDH were different, with PLDH associated with adverse effects of mild to moderate severity. The most common adverse effect in the PLDH group was severe PPE, with the difference between PLDH and topotecan reaching statistical significance (p < 0.001; Table 74). By contrast, incidence of severe (grades 3 and 4) haematological toxicity was significantly higher with topotecan [neutropenia (p < 0.001) and leucopenia (p < 0.001); see Table 74].
Technology appraisal no. 91 presents additional data on adverse effects, reporting treatment-emergent AEs that occurred in at least 10% of patients (Table 75). 13 TA91 identified statistically significant differences between PLDH and topotecan for various grade 3 events. Adverse effects that were significantly higher in the PLDH compared with the topotecan group were:
-
mucous membrane disorder (RR 0.05, 95% CI 0.006 to 0.56)
-
stomatitis (RR 0.056, 95% CI 0.01 to 0.31)
-
PPE (RR 0.009, 95% CI 0.001 to 0.087)
-
rash (RR 0.11, 95% CI 0.017 to 0.61).
By contrast, adverse effects that were significantly higher in the topotecan group than the PLDH group were as follows:
-
fever (RR 4.07, 95% CI 1.00 to 16.82)
-
anaemia (RR 4.62, 95% CI 2.64 to 8.16)
-
leucopenia (RR 4.02, 95% CI 2.6 to 6.27)
-
neutropenia (RR 1.7, 95% CI 1.04 to 3.00)
-
thrombocytopenia (RR 13.56, 95% CI 4.54 to 40.99)
-
alopecia (RR 5.09, 95% CI 1.60 to 16.27).
Although a larger proportion of patients treated with PLDH experienced grade 4 pain, stomatitis and PPE, the difference between PLDH and topotecan did not reach statistical significance for these outcomes. 13 By contrast, incidence of grade 4 fever, anaemia, leucopenia, neutropenia and thrombocytopenia remained statistically significantly higher in the topotecan group than in the PLDH group.
| Event | PLDH (n = 239) | Topotecan (n = 235) | ||
|---|---|---|---|---|
| Withdrawal due to PPE | 9 (3.8%) | 0 | ||
| Withdrawal owing to sepsis | 0 | 2 (0.8%) | ||
| Withdrawal owing to any AE | 43 (18%) | 37 (16%) | ||
| Grade 4 AEs | 17.2% | 71.1% | ||
| All grades a | Grade 3 or 4 a | All grades a | Grade 3 or 4 a | |
| Neutropenia | 84 (35%) | 29 (12%) | 191 (81%) | 180 (77%) |
| Anaemia | 85 (36%) | 13 (5%) | 169 (72%) | 66 (28%) |
| Leucopenia | 31 (13%) | 24 (10%) | 152 (65%) | 117 (50%) |
| Thrombocytopenia | 87 (36%) | 3 (1%) | 148 (63%) | 80 (34%) |
| Alopecia | 38 (16%) | 3 (1%) | 114 (49%) | 14 (6%) |
| PPE | 117 (49%) | 55 (23%) | 2 (1%) | 0 |
| Stomatitis | 95 (40%) | 20 (8%) | 35 (15%) | 1 (0.4%) |
| Body system: AE | PLDH (N = 239) | Topotecan (N = 235) | ||||
|---|---|---|---|---|---|---|
| All grades, n (%) | Grade 3, n (%) | Grade 4, n (%) | All grades, n (%) | Grade 3, n (%) | Grade 4, n (%) | |
| Body as a whole | ||||||
| Asthenia | 96 (40.2) | 17 (7.1) | 0 | 121 (51.5) | 19 (8.1) | 0 |
| Abdominal pain | 80 (33.5) | 24 (10.0) | 1 (0.4) | 89 (37.9) | 19 (8.1) | 4 (1.7) |
| Fever | 51 (21.3) | 2 (0.8) | 0 | 72 (30.6) | 8 (3.4) | 5 (2.1) |
| Pain | 50 (20.9) | 4 (1.7) | 1 (0.4) | 40 (17.0) | 4 (1.7) | 0 |
| Mucous membrane disorder | 34 (14.2) | 9 (3.8) | 0 | 8 (3.4) | 0 | 0 |
| Back pain | 28 (11.7) | 4 (1.7) | 0 | 24 (10.2) | 2 (0.9) | 0 |
| Infection | 28 (11.7) | 5 (2.1) | 0 | 15 (6.4) | 2 (0.9) | 0 |
| Headache | 25 (10.5) | 2 (0.8) | 0 | 35 (14.9) | 0 | 0 |
| Digestive system | ||||||
| Nausea | 110 (46.0) | 12 (5.0) | 1 (0.4) | 148 (63.0) | 16 (6.8) | 3 (1.3) |
| Stomatitis | 99 (41.4) | 19 (7.9) | 1 (0.4) | 36 (15.3) | 1 (0.4) | 0 |
| Vomiting | 78 (32.6) | 17 (7.1) | 2 (0.8) | 103 (43.8) | 18 (7.7) | 5 (2.1) |
| Constipation | 72 (30.1) | 6 (2.5) | 0 | 107 (45.5) | 11 (4.7) | 2 (0.9) |
| Diarrhoea | 50 (20.9) | 5 (2.1) | 1 (0.4) | 82 (34.9) | 9 (3.8) | 1 (0.4) |
| Anorexia | 48 (20.1) | 6 (2.5) | 0 | 51 (21.7) | 3 (1.3) | 0 |
| Dyspepsia | 29 (12.1) | 2 (0.8) | 0 | 33 (14.0) | 0 | 0 |
| Intestinal obstruction | 27 (11.3) | 19 (7.9) | 4 (1.7) | 26 (11.1) | 14 (6.0) | 7 (3.0) |
| Haemic and lymphatic system | ||||||
| Anaemia | 96 (40.2) | 13 (5.4) | 1 (0.4) | 177 (75.3) | 59 (25.1) | 10 (4.3) |
| Leucopenia | 88 (36.8) | 21 (8.8) | 3 (1.3) | 151 (64.3) | 83 (35.3) | 36 (15.3) |
| Neutropenia | 84 (35.1) | 19 (7.9) | 10 (4.2) | 193 (82.1) | 33 (14.0) | 146 (62.1) |
| Thrombocytopenia | 31 (13.0) | 3 (1.3) | 0 | 153 (65.1) | 40 (17.0) | 40 (17.0) |
| Metabolic/nutritional disorder | ||||||
| Peripheral oedema | 27 (11.3) | 5 (2.1) | 0 | 41 (17.4) | 6 (2.6) | 0 |
| Nervous system | ||||||
| Paraesthesia | 24 (10.0) | 0 | 0 | 21 (8.9) | 0 | 0 |
| Dizziness | 10 (4.2) | 0 | 0 | 24 (10.2) | 0 | 0 |
| Respiratory system | ||||||
| Pharyngitis | 38 (15.9) | 0 | 0 | 42 (17.9) | 1 (0.4) | 0 |
| Dyspnoea | 36 (15.1) | 8 (3.3) | 2 (0.8) | 55 (23.4) | 7 (3.0) | 3 (1.3) |
| Cough increased | 23 (9.6) | 0 | 0 | 27 (11.5) | 0 | 0 |
| Skin and appendages | ||||||
| PPE | 121 (50.6) | 55 (23.0) | 2 (0.8) | 2 (0.9) | 0 | 0 |
| Rash | 68 (28.5) | 10 (4.2) | 0 | 29 (12.4) | 1 (0.4) | |
| Alopeciaa | 46 (19.2) | 3 (1.3) | 0 | 123 (52.3) | 15 (6.4) | 0 |
Technology appraisal no. 91 reports that 16.7% (18/108) of patients in the PLDH group and 6.5% (7/108) of patients in the paclitaxel group discontinued treatment because of adverse effects. 13 The five most commonly reported treatment emergent AEs associated with PLDH were nausea (51.9%), PPE (50.9%), stomatitis (48.1%), alopecia (43.5%), and asthenia (38.9%). In the paclitaxel group, the five most commonly reported AEs were alopecia (87.0%), nausea (43.5%), paraesthesia (43.5%), constipation (38.0%) and asthenia (33.3%).
The treatment-emergent AEs that occurred in at least 10% of participants in either treatment group for all grades, grades 3 and 4 are presented in Table 76. The incidence of grade 4 events was low in each group, with neutropenia being the only grade 4 event occurring in both the PLDH and paclitaxel groups (0.9% with PLDH vs. 2.8% with paclitaxel).
Technology appraisal no. 9113 presented forest plots to illustrate the significance of the difference between groups. Grade 3 events occurred in a significantly smaller proportion of people in the paclitaxel group compared with the PLDH group (RR of < 1 indicates paclitaxel was associated with a lower rate of AE):
-
PPE (0% with paclitaxel vs. 14.8% with PLDH); RR 0.031, 95% CI 0.003 to 0.297
-
stomatitis (0.9% with paclitaxel vs. 10.2% with PLDH); RR 0.091, 95% CI 0.02 to 0.53.
Alopecia was the only grade 3 adverse effect occurring significantly more frequently with paclitaxel than with PLDH (18.5% with paclitaxel vs. 2.8% PLDH; RR 6.67, 95% CI 2.20 to 20.66; see Table 76).
| AE classified by body system | PLDH | Paclitaxel | ||||
|---|---|---|---|---|---|---|
| All grades | Grade 3 | Grade 4 | All grades | Grade 3 | Grade 4 | |
| Asthenia | 42 (38.9%) | 4 (3.7%) | 0 | 36 (33.3%) | 6 (5.3%) | 1 (0.9%) |
| Abdominal pain | 34 (31.5%) | 12 (11.1%) | 0 | 35 (32.4%) | 7 (6.5%) | 0 |
| Fever | 28 (25.9%) | 7 (6.5%) | 0 | 8 (7.4%) | 3 (2.8%) | 0 |
| Pain | 24 (22.2%) | 1 (0.9%) | 0 | 24 (22.2%) | 3 (2.8%) | 0 |
| Infection | 23 (21.3%) | 2 (1.9%) | 1 (0.9%) | 10 (9.3%) | 1 (0.9%) | 0 |
| Headache | 12 (11.1%) | 1 (0.9%) | 0 | 13 (12.0%) | 2 (1.9%) | 0 |
| Ascites | 11 (10.2%) | 6 (5.6%) | 0 | 8 (7.4%) | 1 (0.9%) | 0 |
| Back pain | 11 (10.2%) | 1 (0.9%) | 0 | 14 (13.0%) | 1 (0.9%) | 0 |
| Cardiovascular system | ||||||
| Vasodilation | 5 (4.6%) | 1 (0.9%) | 0 | 13 (12.0%) | 1 (0.9%) | 0 |
| Digestive system | ||||||
| Nausea | 56 (51.9%) | 6 (5.6%) | 1 (0.9%) | 47 (43.5%) | 2 (1.9%) | 0 |
| Stomatitis | 52 (48.1%) | 11 (10.2%) | 0 | 12 (11.1%) | 1 (0.9%) | 0 |
| Vomiting | 37 (34.3%) | 10 (9.3%) | 2 (1.9%) | 34 (32.5%) | 4 (3.7%) | 0 |
| Constipation | 30 (27.8%) | 4 (3.7%) | 0 | 41 (38.0%) | 5 (4.6%) | 0 |
| Diarrhoea | 23 (21.3%) | 3 (2.8%) | 0 | 24 (22.2%) | 3 (2.8%) | 0 |
| Anorexia | 18 (16.7%) | 1 (0.9%) | 0 | 11 (10.2%) | 0 | 0 |
| Dyspepsia | 14 (13.0%) | 1 (0.9%) | 0 | 11 (10.2%) | 0 | 0 |
| Haemic and lymphatic system | ||||||
| Neutropenia | 18 (16.7%) | 6 (5.6%) | 1 (0.9%) | 23 (21.3%) | 10 (9.3%) | 3 (2.8%) |
| Anaemia | 17 (15.7%) | 3 (2.8%) | 0 | 23 (21.3%) | 5 (4.6%) | 0 |
| Leucopenia | 15 (13.9%) | 5 (4.6%) | 1 (0.9%) | 21 (19.4%) | 9 (8.3%) | 0 |
| Metabolic/nutritional disorder | ||||||
| Peripheral oedema | 14 (13.0%) | 0 | 0 | 15 (13.9%) | 1 (0.9%) | 0 |
| Musculoskeletal system | ||||||
| Myalgia | 4 (3.7%) | 1 (0.9%) | 0 | 31 (28.7%) | 7 (6.5%) | 0 |
| Arthralgia | 2 (1.9%) | 0 | 0 | 23 (21.3%) | 2 (1.9%) | 0 |
| Nervous system | ||||||
| Paraesthesia | 15 (13.9%) | 0 | 0 | 47 (43.5%) | 4 (3.7%) | 0 |
| Somnolence | 11 (10.3%) | 3 (2.8%) | 0 | 17 (15.7%) | 2 (1.9%) | 0 |
| Respiratory system | ||||||
| Dyspnoea | 18 (16.7%) | 6 (5.6%) | 1 (0.9%) | 15 (13.9%) | 1 (0.9%) | 0 |
| Pharyngitis | 8 (7.4%) | 0 | 0 | 18 (16.7%) | 0 | 0 |
| Skin and appendages | ||||||
| PPE | 55 (50.9%) | 16 (14.8%) | 1 (0.9%) | 13 (12.0%) | 0 | 0 |
| Alopecia | 47 (43.5%) | 3 (2.8%) | 0 | 94 (87.0%) | 20 (18.5%) | 1 (0.9%) |
| Rash | 15 (13.9%) | 2 (1.9%) | 0 | 19 (17.6%) | 1 (0.9%) | 0 |
ten Bokkel Huinink et al. 21 evaluated adverse effects according to the NCI-CTC. There were two treatment-related deaths in the topotecan group, which were attributed to topotecan-induced sepsis. There were no treatment-related deaths in the paclitaxel group. Ten patients (seven in the topotecan group vs. four in the paclitaxel group) discontinued treatment as a result of an adverse effect. Febrile neutropenia, infection and sepsis were the causes of withdrawal from the topotecan group, whereas discontinuations from the paclitaxel group were as a result of neurotoxicity. Severe (grades 3 and 4) haematological adverse effects predominantly occurred more frequently in the topotecan group than in the paclitaxel group, with differences between groups in grade 4 leucopenia, neutropenia, and thrombocytopenia reaching statistical significance (Table 77). The only haematological adverse effect that occurred more frequently in paclitaxel-treated patients was grade 3 neutropenia (see Table 77).
Most non-haematological adverse effects were mild to moderate in severity (grades 1 and 2). The most frequently reported adverse effects considered related or possibly related to treatment in both groups were alopecia and gastrointestinal disturbances, including nausea, vomiting, diarrhoea and constipation (see Table 77). A larger proportion of patients in the paclitaxel group experienced alopecia than in the topotecan group. Mild to moderate nausea, vomiting and constipation occurred more frequently in the topotecan group. By contrast, more patients in the paclitaxel group experienced mild to moderate diarrhoea.
| Event | Topotecan (n = 112) | Paclitaxel (n = 114) | ||
|---|---|---|---|---|
| Withdrawal for AE | 7 (7%) | 3 (4%) | ||
| Death due to sepsis/myelosuppression | 2 | 0 | ||
| Haematological, grades 3 and 4 | ||||
| Grade 3 (%) | Grade 4 (%) | Grade 3 (%) | Grade 4 (%) | |
| Leucopeniaa | 50.9 | 33.6 | 17.9 | 2.7 |
| Neutropeniaa | 15.3 | 79.3 | 28.6 | 23.2 |
| Thrombocytopeniaa | 24.3 | 25.2 | 0.9 | 1.8 |
| Anaemia | 36.9 | 3.6 | 3.6 | 2.7 |
| Non-haematologicalb | ||||
| Grade 1–2 (%) | Grades 3 and 4 (%) | Grade 1–2 (%) | Grades 3 and 4 (%) | |
| Alopecia | 75.9 | 0 | 92.1 | 0.9 |
| Nausea | 67.9 | 9.8 | 43.0 | 1.8 |
| Vomiting | 53.6 | 9.9 | 28.1 | 2.7 |
| Fatigue | 33.1 | 8.0 | 25.4 | 6.1 |
| Constipation | 37.5 | 5.4 | 30.7 | 0 |
| Diarrhoea | 33.9 | 6.3 | 37.8 | 0.9 |
| Abdominal pain | 21.5 | 5.4 | 36.0 | 3.5 |
| Fever (excludes febrile neutropenia) | 27.7 | 0.9 | 17.7 | 0 |
| Stomatitis | 23.2 | 0.9 | 14.0 | 0.9 |
| Dyspnoea | 17.8 | 6.3 | 13.2 | 5.3 |
| Asthenia | 17.0 | 5.4 | 9.6 | 3.5 |
| Arthralgia | 5.5 | 0.9 | 28.9 | 2.6 |
| Myalgia | 3.6 | 0 | 25.4 | 2.6 |
| Neuropathy | 0.9 | 0 | 15.8 | 0 |
| Skeletal pain | 4.5 | 0 | 11.4 | 5.3 |
| Flushing | 4.5 | 0 | 14.1 | 0 |
| Paraesthesia | 0.9 | 0 | 29.0 | 0 |
In the trial reported by Pfisterer et al. ,50 grade 3 and grade 4 haematological toxicities were significantly more frequent in the gemcitabine plus carboplatin group than in the carboplatin alone group, with neutropenia the predominant haematological toxicity (Table 78). The proportion of patients discontinuing treatment as a result of a haematological AE was small in each group (5.1% with gemcitabine plus carboplatin vs. 4.0% with carboplatin alone). Grade 3 and grade 4 non-haematological AEs were infrequent in each group, with < 5% of patients in each group experiencing a non-haematological toxicity reported in the full publication (see Table 78). Grade 2 alopecia occurred in 14.3% of patients treated with gemcitabine plus carboplatin compared with 2.3% of patients treated with of carboplatin alone (statistical significance of result not reported). AEs were graded according to the NCI-CTC guidance.
| Event | Gemcitabine plus carboplatin (n = 175) | Carboplatin alone (n = 174) | p-value (grades 3 and 4 together) | ||
|---|---|---|---|---|---|
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | ||
| Haematological | |||||
| Anaemia | 39 (22.3%) | 9 (5.1%) | 10 (5.7%) | 4 (2.3%) | < 0.001 |
| Neutropenia | 73 (41.7%) | 50 (28.6%) | 19 (10.9%) | 2 (1.1%) | < 0.001 |
| Thrombocytopenia | 53 (30.3%) | 8 (4.6%) | 18 (10.3%) | 2 (1.1%) | < 0.001 |
| Non-haematological | |||||
| Hypersensitivity | 3 (1.7%) | 1 (0.6%) | 3 (1.7%) | 2 (1.1%) | 0.7503 |
| Diarrhoea | 3 (1.7%) | 0 | 0 | 0 | 0.2479 |
| Dyspnoea | 2 (1.1%) | 0 | 2 (1.1%) | 1 (0.6%) | 0.6848 |
| Fatigue | 3 (1.7%) | 1 (0.6%) | 3 (1.7%) | 0 | 0.99 |
| Febrile neutropenia | 2 (1.1%) | 0 | 0 | 0 | 0.4986 |
| Infection without neutropenia | 0 | 1 (0.6%) | 0 | 0 | 0.99 |
| Infection with neutropenia | 0 | 0 | 0 | 0 | – |
| Neuropathy (motor) | 1 (0.6%) | 0 | 0 | 0 | 0.99 |
| Neuropathy (sensory) | 2 (1.1%) | 0 | 3 (1.7%) | 0 | 0.6848 |
| Vomiting | 5 (2.9%) | 0 | 2 (1.1%) | 1 (0.6%) | 0.7234 |
| Grade 1 | Grade 2 | Grade 1 | Grade 2 | ||
| Haematological | |||||
| Anaemia | 32 (18.3%) | 73 (41.7%) | 71 (40.8%) | 44 (25.3%) | – |
| Neutropenia | 9 (5.1%) | 27 (15.4%) | 44 (25.3%) | 33 (19.0%) | – |
| Thrombocytopenia | 41 (23.4%) | 36 (20.6%) | 66 (37.9%) | 14 (8.0%) | – |
| Non-haematological | |||||
| Hypersensitivity | 1 (0.6%) | 4 (2.3%) | 3 (1.7%) | 2 (1.1%) | – |
| Diarrhoea | 16 (9.1%) | 7 (4.0%) | 7 (4.0%)0 | 6 (3.4%) | – |
| Dyspnoea | 1 (0.6%) | 12 (6.9%)0 | 2 (1.1%) | 4 (2.3%) | – |
| Fatigue | 29 (16.6%) | 35 (20.0%) | 25 (14.4%) | 23 (13.2%) | – |
| Febrile neutropenia | 0 | 0 | 0 | 0 | – |
| Infection without neutropenia | 1 (0.6%) | 1 (0.6%) | 1 (0.6%) | 1 (0.6%) | – |
| Infection with neutropenia | 1 (0.6%) | 0 | 0 | 1 (0.6%) | – |
| Neuropathy (motor) | 9 (5.1%) | 1 (0.6%) | 6 (3.4%) | 1 (0.6%) | – |
| Neuropathy (sensory) | 43 (24.6%) | 7 (4.0%) | 38 (21.8%) | 6 (3.4%) | – |
| Vomiting | 41 (23.4%) | 28 (16.0%) | 32 (18.4%) | 1 (0.6%) | – |
| Alopeciaa | 61 (34.9%) | 25 (14.3%) | 27 (15.5%) | 23 (13.2%) | – |
In ICON4/AGO-OVAR 2.2,61 paclitaxel plus platinum-based therapy was associated with higher rates of alopecia compared with conventional platinum-based therapy alone [322/392 (86%) with paclitaxel plus platinum-based chemotherapy vs. 95/410 (25%) with conventional platinum-based therapy; Table 79). 61 Additionally, the proportion of patients experiencing a grade 2–4 neurological toxicity was higher in the paclitaxel plus platinum chemotherapy group [76/392 (20%)] than in the conventional platinum-based therapy group [4/410 (1%)]. By contrast, incidence of moderate or severe (grades 2–4) haematological adverse effects was higher in the conventional platinum-based therapy group.
| Event | Paclitaxel plus platinum chemotherapy (n = 392) | Conventional platinum-based chemotherapy (n = 410) |
|---|---|---|
| ‘Moderate or severe’: neurological (grades 2–4) | 76 (20%) | 4 (1%) |
| Not yet known | 15 | 31 |
| Haematological | 111 (29%) | 182 (46%) |
| Not yet known | 8 | 16 |
| Infection | 64 (17%) | 53 (14%) |
| Not yet known | 15 | 24 |
| Renal | 31 (8%) | 37 (9%) |
| Not yet known | 8 | 16 |
| Mucositis (grades 2 and 3) | 26 (7%) | 21 (6%) |
| Not yet known | 15 | 31 |
| Nausea and vomiting (grades 2–4) | 131 (35%) | 153 (40%) |
| Not yet known | 15 | 29 |
| Alopecia (grades 2–4) | 322 (86%) | 95 (25%) |
| Not yet known | 28 | 19 |
Gonzalez-Martin et al. 48 based the safety analysis on 78 patients who received at least one cycle of treatment. Adverse effects were graded according to NCI-CTC criteria. Grades 3 and 4 haematological toxicity was similar between the groups. Although severe neutropenia (grades 3 and 4) was more common in the paclitaxel plus carboplatin group, the difference between groups was not statistically significant (p = 0.24; see Table 80). Treatment with paclitaxel plus carboplatin was associated with a higher incidence of grade 2–4 non-haematological adverse effects and with significantly higher incidences of alopecia, mucositis, myalgia/arthralgia and peripheral neuropathy than treatment with carboplatin alone (Table 80).
| Event | Carboplatin (n = 40) | Paclitaxel plus carboplatin (n = 38) | p-value | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NCI-CTC grade | NCI-CTC grade | ||||||||||||
| Haematological | |||||||||||||
| 0 | 1 | 2 | 3 | 4 | 3 and 4 | 0 | 1 | 2 | 3 | 4 | 3 and 4 | ||
| Leucopenia | 17 | 16 | 6 | 1 | – | 1 (2.5) | 19 | 11 | 6 | 2 | – | 2 (5.3) | 0.93 |
| Neutropenia | 13 | 11 | 12 | 3 | 1 | 4 (10.0) | 16 | 7 | 8 | 6 | 1 | 7 (18.4) | 0.24 |
| Thrombocytopenia | 8 | 17 | 10 | 3 | 2 | 5 (12.5) | 20 | 12 | 5 | 1 | – | 1 (2.6) | 0.25 |
| Anaemia | 4 | 20 | 10 | 5 | 1 | 6 (15.0) | 8 | 20 | 8 | 2 | – | 2 (5.3) | 0.33 |
| Non-haematological | |||||||||||||
| 0 | 1 | 2 | 3 | 4 | 2–4 | 0 | 1 | 2 | 3 | 4 | 2–4 | ||
| Allergy | 33 | 4 | 3 | 1 | – | 4 (10) | 28 | 4 | 2 | 3 | 1 | 6 (15.8) | – |
| Alopecia | 30 | 3 | 7 | – | – | 7 (17.5) | 5 | – | 11 | 22 | – | 33 (86.8) | 0.001 |
| Fever | 36 | 4 | – | – | – | – | 34 | 2 | 2 | – | – | 2 (5.3) | |
| Infection | 39 | – | – | 1 | – | 1 (2.5) | 33 | 3 | 1 | 1 | – | 2 (5.3) | |
| Haemorrhage | 36 | 4 | – | – | – | – | 36 | 2 | – | – | – | – | |
| Nausea | 13 | 15 | 12 | – | – | 12 (30.0) | 17 | 15 | 6 | – | – | 6 (15.8) | |
| Vomiting | 21 | 9 | 6 | 4 | – | 10 (25.0) | 24 | 9 | 4 | 1 | – | 5 (13.2) | |
| Stomatitis/mucositis | 37 | 3 | – | – | – | – | 27 | 4 | 7 | – | – | 7 (18.4) | 0.004 |
| Diarrhoea | 34 | 5 | 1 | – | – | 1 (2.5) | 35 | 2 | 1 | – | – | 1 (2.6) | |
| Constipation | 27 | 10 | 3 | – | – | 3 (7.5) | 25 | 10 | 3 | – | – | 3 (7.9) | |
| Creatinine | 35 | 4 | 1 | – | – | 1 (2.5) | 36 | 1 | 1 | – | – | 1 (2.6) | |
| Pulmonary (dyspnoea) | 38 | 1 | 1 | – | – | 1 (2.5) | 35 | 1 | 1 | 1 | – | 2 (5.3) | |
| Neurosensory | 34 | 6 | – | – | – | – | 17 | 12 | 9 | – | – | 9 (23.7) | 0.009 |
| Myalgias/arthralgias | 39 | 1 | – | – | – | – | 15 | 9 | 12 | 2 | – | 14 (36.8) | 0.001 |
| Mood depression | 39 | – | 1 | – | – | 1 (2.5) | 36 | 1 | – | 1 | – | 1 (2.6) | |
| Asthenia | 20 | 10 | 10 | – | – | 10 (25.0) | 16 | 11 | 9 | 2 | – | 11 (28.9) | |
| Anorexia | 35 | 1 | 3 | 1 | – | 4 (10.0) | 35 | 2 | 1 | – | – | 1 (2.6) | |
In the trial reported by Lortholary et al. ,62 one patient randomised to treatment with weekly paclitaxel did not receive a dose of study drug and was therefore not included in the safety analysis. No deaths were categorised as treatment related. Non-haematological toxicity was similar between treatment groups, with the exception of hypersensitivity reactions, which occurred more frequently with combination treatment than with weekly paclitaxel alone (Table 81). A larger proportion of patients treated with weekly paclitaxel plus carboplatin experienced grade 3 and grade 4 leucopenia and neutropenia. Discontinuation rate because of adverse effects was also higher in the group receiving combination therapy (see Table 81). No patient in the weekly paclitaxel group discontinued treatment because of haematological toxicity, whereas 14% in the weekly paclitaxel plus carboplatin group discontinued treatment for this reason.
| Event | Weekly paclitaxel plus carboplatin (n = 51) | Weekly paclitaxel (n = 57) |
|---|---|---|
| Withdrawal for toxicity | 29% | 2% |
| Withdrawal for haematological toxicity | 14% | 0 |
| Grades 3 and 4 | ||
| Leucopenia | 31% | 7% |
| Neutropenia | 54% | 13% |
| Febrile neutropenia | 4% | 0 |
| Anaemia | 19% | 6% |
| Thrombocytopenia | 4% | 2% |
| Grade 2–4 | ||
| Hypersensitivity | 16% | 2% |
| Peripheral neuropathy | 20% | 32% |
| Vomiting | 20% | 17% |
| Fatigue | 61% | 59% |
| Mucositis (grade 2) | 6% | 7% |
| Alopecia (grade 2) | 46% | 33% |
Piccart et al. 63 reported safety analysis based on all 86 patients randomised: all patients had received at least one treatment cycle and were assessable for the safety analysis. Only grade 3 and grade 4 AEs were reported (presented in Table 82), with grade assigned according to NCI-CTC. Considering haematological toxicities, severe neutropenia (grades 3 and 4) occurred only in the paclitaxel group [9/41 (22%)], whereas grade 3 thrombocytopenia was reported only in the oxaliplatin group [2/45 (4%)]. Severe anaemia was rare, and no episodes of febrile neutropenia were observed. Of the non-haematological AEs reported, the number of patients experiencing an AE was low in each group. No episodes of grade 4 nausea and vomiting were reported. The most frequently reported non-haematological adverse effect was pain, with 12% (5/41) and 4% (2/45) of patients in the paclitaxel and oxaliplatin groups, respectively, experiencing a grade 3 pain event (see Table 82). The proportion of patients experiencing a grade 3 neurosensory AE was similar between the two treatment groups (7% with paclitaxel vs. 9% with oxaliplatin; see Table 82).
| Event | Paclitaxel (n = 41) | Oxaliplatin (n = 45) | ||
|---|---|---|---|---|
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | |
| Haematological | ||||
| Neutropenia | 6 (15%) | 3 (7%) | – | – |
| Anaemia | – | 1 (2%) | 1 (2%) | – |
| Thrombocytopenia | – | – | 2 (4%) | – |
| Liver function | ||||
| AST | – | – | – | – |
| ALT | 2 (5%) | – | – | – |
| Gastrointestinal | ||||
| Nausea | 1 (2%) | NA | 2 (4%) | NA |
| Vomiting | 1 (2%) | – | 3 (7%) | – |
| Diarrhoea | – | – | 2 (4%) | – |
| Neurosensory | 3 (7%) | NA | 4 (9%) | NA |
| Other | ||||
| Lethargy | 3 (7%) | NA | 3 (7%) | NA |
| Pain | 5 (12%) | – | 2 (4%) | – |
Gore et al. 24 reported that neutropenia and leucopenia were the most common haematological toxicities occurring in both treatment groups, although the rate of both AEs was higher in the group receiving topotecan intravenously rather than orally (Table 83). Seven deaths were attributed to haematological toxicity, two in the oral treatment group and five in the i.v. treatment group. A similar proportion of patients in each group developed grade 3 and grade 4 thrombocytopenia or anaemia. Gastrointestinal disturbances were the most common non-haematological toxicity, with most events reported as mild to moderate in severity. Incidence of gastrointestinal adverse effects was higher in the oral topotecan group (see Table 83). Grades 3 and 4 non-haematological toxicities generally occurred in <10% of patients. Incidence of Grades 3 and 4 nausea, diarrhoea, vomiting, and fever was marginally higher in patients treated with oral topotecan compared with i.v. topotecan (see Table 83).
| Event | Oral topotecan (n = 135) | Intravenous topotecan (n = 131) | ||
|---|---|---|---|---|
| Deaths due to haematological toxicity | 2 | 5 | ||
| Haematological | ||||
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | |
| Patients | ||||
| Neutropenia | 40 (30%) | 67 (50%) | 15 (11%) | 110 (84%) |
| Anaemia | 51 (38%) | 5 (4%) | 43 (33%) | 10 (8%) |
| Leucopenia | 59 (44%) | 28 (21%) | 78 (60%) | 40 (31%) |
| Thrombocytopenia | 30 (22%) | 27 (20%) | 27 (21%) | 23 (18%) |
| Courses | n = 729 | n = 778 | ||
| Neutropenia | 190 (26%) | 106 (15%) | 249 (32%) | 393 (51%) |
| Anaemia | 163 (22%) | 31 (4%) | 371 (48%) | 68 (9%) |
| Leucopenia | 70 (10%) | 42 (6%) | 90 (12%) | 29 (4%) |
| Thrombocytopenia | 85 (12%) | 7 (1%) | 78 (10%) | 10 (1%) |
| Non-haematological | ||||
| All grades | Grades 3 and 4 | All grades | Grades 3 and 4 | |
| Patients | ||||
| Nausea | 92 (68%) | 12 (9%) | 80 (61%) | 6 (5%) |
| Diarrhoea | 76 (56%) | 13 (10%) | 40 (31%) | 6 (5%) |
| Vomiting | 74 (55%) | 10 (7%) | 52 (40%) | 4 (3%) |
| Alopecia | 72 (53%) | 10 (7%) | 68 (52%) | 8 (6%) |
| Fatigue | 50 (37%) | 5 (4%) | 50 (38%) | 5 (4%) |
| Abdominal pain | 49 (36%) | 9 (7%) | 39 (30%) | 9 (7%) |
| Constipation | 47 (35%) | 4 (3%) | 42 (32%) | 7 (5%) |
| Fever | 38 (28%) | 14 (10%) | 31 (24%) | 7 (5%) |
Sehouli et al. 23 report that, of the 194 patients randomised, five patients did not receive any dose of study drug, which differs slightly from the number reported in the CONSORT diagram (two patients in each group). The methods state that all analyses are based on the ITT principle. However, it is unclear from the reporting of the adverse effects whether all patients have been analysed. It should be noted that, although the comparator is referred to as conventional topotecan, the dose administered in this group is 1.25 mg/m2 for five consecutive days compared with the licensed dose of 1.5 mg/m2.
Compared with the conventional dosing schedule, weekly topotecan was associated with significantly fewer episodes of severe (grades 3 and 4) haematological events (anaemia, leucopenia, neutropenia and thrombocytopenia; see Table 84). Incidence of severe non-haematological events was low in each group, with no difference between groups reaching statistical significance (Table 84).
| Event | Topotecan weekly (n = 97) | Topotecan conventional (n = 97) | p-value |
|---|---|---|---|
| Grades 3 and 4 | |||
| Anaemia | 7 (7.2%) | 20 (20.6%) | 0.007 |
| Leucopenia | 13 (13.4%) | 56 (57.7%) | < 0.001 |
| Neutropenia | 15 (15.5%) | 39 (40.2%) | < 0.001 |
| Lymphopenia | 1 (1.0%) | 5 (5.2%) | 0.097 |
| Thrombocytopenia | 5 (5.2%) | 22 (22.7%) | < 0.001 |
| Febrile neutropenia | 1 (1.0%) | 4 (4.1%) | 0.174 |
| Fever | 0 | 1 (1.0%) | 0.316 |
| Infection | 5 (5.1%) | 4 (4.1%) | 0.733 |
| Nausea | 1 (1.0%) | 5 (5.2%) | 0.097 |
| Vomiting | 4 (4.1%) | 3 (3.1%) | 0.700 |
| Diarrhoea | 1 (1.0%) | 1 (1.0%) | 1.000 |
| Constipation | 2 (2.1%) | 3 (3.1%) | 0.650 |
| Ileus | 7 (7.2%) | 7 (7.2%) | 1.000 |
| Fatigue | 10 (10.3%) | 6 (6.2%) | 0.296 |
| Motor neuropathy | 1 (1.0%) | 0 | 0.316 |
| Sensory neuropathy | 1 (1.0%) | 0 | 0.316 |
| Pain | 12 (12.4%) | 6 (6.2%) | 0.138 |
| Pleural effusion | 2 (2.1%) | 1 (1.0%) | 0.561 |
| Pneumonia | 1 (1.0%) | 1 (1.0%) | 1.000 |
| Dyspnoea | 5 (5.2%) | 2 (2.1%) | 0.248 |
Omura et al. 68 reported that febrile neutropenia was the most commonly observed severe toxicity. After the first cycle of therapy, the incidence of neutropenic fever did not differ significantly between:
-
patients receiving paclitaxel 175 mg/m2 (without filgrastim) and those assigned to paclitaxel 250 mg/m2 with filgrastim (22% paclitaxel 175 mg/m2 and no filgrastim vs. 19% with paclitaxel 250 mg/m2 and filgrastim; p-value not reported)
-
filgrastim 10 µg/kg and filgrastim 5 µg/kg among women receiving paclitaxel 250 mg/m2 (19% with 5 µg/kg filgrastim vs. 18% with 10 µg/kg filgrastim; 95% CI –11% to 13%, no point estimate reported).
Patients receiving the higher paclitaxel dose (250 mg/m2) reported a numerically greater incidence of anaemia, thrombocytopenia, nausea and vomiting, neuropathy and myalgia/arthralgia than those receiving paclitaxel 175 mg/m2. The difference between groups was statistically significant for thrombocytopenia (15% with 250 mg/m2 vs. 7% with 175 mg/m2; p = 0.009), neuropathy (16% with 250 mg/m2 vs. 7% with 175 mg/m2; p = 0.024) and myalgia/arthralgia (10% with 250 mg/m2 vs. 3% with 175 mg/m2; p = 0.022). Adverse effects as reported in Omura et al. 68 are summarised in Table 85.
| Adverse effect | Paclitaxel regimen | p-value | |
|---|---|---|---|
| 175 mg/m2 (%) | 250 mg/m2 plus filgrastim (%) | ||
| Anaemia | 7 | 15 | 0.102 |
| Thrombocytopenia | 5 | 15 | 0.009 |
| Nausea and vomiting | 5 | 10 | 0.211 |
| Neuropathy | 7 | 16 | 0.024 |
| Myalgia/arthralgia | 3 | 10 | 0.022 |
Of the 208 patients randomised in the trial reported by Rosenberg et al. ,60 205 received at least one dose of paclitaxel and were included in the safety analysis. No treatment-related deaths occurred in the trial. Considering haematological adverse effects, paclitaxel given every 3 weeks was associated with a significantly higher incidence of severe neutropenia (grades 3 and 4) compared with the once weekly regimen [19/104 (18%) with paclitaxel weekly vs. 45/101 (45%) with paclitaxel every 3 weeks; p < 0.001; Table 86]. Of the other haematological adverse effects assessed, number of episodes of severe anaemia, leucopenia and thrombocytopenia were similar between the two treatment groups, with none of the differences between groups reaching statistical significance. However, assessment of haematological toxicities of grades 1–4 identified a statistically significantly higher incidence of anaemia in patients treated with paclitaxel weekly compared with every 3 weeks [81/104 (78%) with paclitaxel weekly vs. 65/101 (64%) with paclitaxel every 3 weeks; p = 0.04; see Table 86]. The difference between groups in neutropenia remained significant and favoured paclitaxel weekly (i.e. smaller proportion of patients experienced an event; see Table 86).
No grade 4 non-haematological adverse effects were reported. Grade 1–3 non-haematological adverse effects were common, with high incidences of neuropathy, alopecia and arthralgia/myalgia (see Table 86). The difference between the two paclitaxel regimens in neuropathy and in alopecia was not statistically significant. However, paclitaxel every 3 weeks was associated with a significantly higher incidence of arthralgia/myalgia compared with the weekly regimen [61/104 (59%) with paclitaxel weekly vs. 85/101 (84%) with paclitaxel every 3 weeks; p = 0.04; see Table 86]. A larger proportion of patients treated with weekly paclitaxel experienced problems with their nails (discolouration and/or loosening from the nail bed) compared with patients treated every 3 weeks [37/104 (36%) with paclitaxel weekly vs. 2/101 (2%) with paclitaxel every 3 weeks; p < 0.001; see Table 86]. Considering only grade 3 non-haematological events, episodes of grade 3 neuropathy and grade 3 alopecia were significantly higher in the paclitaxel every 3 weeks regimen compared with the weekly regimen (see Table 86). Problems with nail changes remained significantly more common in the paclitaxel weekly group. Incidence of nausea/vomiting and of arthralgia/myalgia was similar in each group, with no statistically significant difference between the two treatment groups (see Table 86).
| Event | Paclitaxel weekly (n = 104) | Paclitaxel 3 weekly (n = 101) | p-value |
|---|---|---|---|
| Withdrawals due to toxicity | 1 | 4 | NR |
| Haematological toxicity | |||
| Grades 3 and 4 | |||
| Anaemia (haemoglobin) | 4 (4%) | 4 (4%) | 1.0 |
| Leucopenia (WBC) | 17 (16%) | 17 (17%) | 1.0 |
| Neutropenia (neutrophils) | 19 (18%) | 45 (45%) | < 0.001 |
| Thrombocytopenia (platelets) | 0 | 1 (1%) | 0.49 |
| Grades 1–4 | |||
| Anaemia | 81 (78%) | 65 (64%) | 0.04 |
| Leucopenia | 74 (71%) | 79 (78%) | 0.27 |
| Neutropenia | 63 (61%) | 80 (79%) | < 0.01 |
| Thrombocytopenia | 1 (1%) | 5 (5%) | 0.12 |
| Non-haematological | |||
| Grade 3 | |||
| Neuropathy | 11 (11%) | 29 (29%) | < 0.001 |
| Alopecia | 48 (46%) | 80 (79%) | < 0.001 |
| Arthralgia/myalgia | 5 (5%) | 8 (8%) | 0.40 |
| Nausea/vomiting | 4 (4%) | 3 (3%) | 1.0 |
| Nails | 9 (9%) | 0 | < 0.01 |
| Grades 1–3 | |||
| Neuropathy | 84 (81%) | 86 (85%) | 0.72 |
| Alopecia | 85 (82%) | 91 (90%) | 0.11 |
| Arthralgia/myalgia | 61 (59%) | 85 (84%) | < 0.001 |
| Nausea/vomiting | 48 (46%) | 42 (42%) | 0.57 |
| Nails | 37 (36%) | 2 (2%) | < 0.001 |
Network meta-analysis
For the NMA, studies that reported combined grades of AEs (e.g. grades 2–4, including grades 3 and 4) were excluded from the analysis. When data were reported separately for vomiting and nausea in the same study, this was combined for the purposes of the analysis, as were data on neurosensory events. It is acknowledged that this might have led to double-counting. For trials that specified they would record all AEs, events rates of zero were not imputed; only data reported in the papers were used to inform the analysis. Network diagrams for the AEs analysed in the NMA are presented in Appendix 4.
To give focus to the evaluation of AEs, the TAG consulted with its expert clinical advisors and identified the following severe AEs (grades 3 and 4) as those most problematic for patients or most likely to consume substantial health-care resource:
-
allergic reaction
-
alopecia
-
anaemia
-
fatigue
-
febrile neutropenia
-
nausea and vomiting
-
neuropathy.
The treatments evaluated for these serious AEs are as follows:
-
gemcitabine plus carboplatin
-
platinum monotherapy
-
PLDH monotherapy
-
PLDH plus carboplatin
-
paclitaxel monotherapy, i.e. 175 mg/m2 or 200 mg/m2 every 21 days
-
paclitaxel monotherapy (weekly), i.e. paclitaxel 67 mg/m2 every week for 21 days
-
topotecan monotherapy (i.v.), i.e. topotecan 1.25 mg/m2 or 1.5 mg/m2 daily for 5 days every 21 days
-
topotecan monotherapy (oral)
-
topotecan monotherapy (i.v., weekly), i.e. topotecan 4.0 mg/m2 (weekly) on days 1, 8 and 15 of a 28-day cycle.
Unlike the efficacy outcomes reported earlier, the evaluation of severe AEs is based on the total population regardless of PFI, i.e. it is not broken down by the various subgroups based on platinum sensitivity (or insensitivity). However, for consistency the baseline treatment for each network assessed are consistent with the efficacy analyses.
The absolute numbers for the RCTs included in the NMA evaluating allergic reaction in patients with recurrent ovarian cancer are reported in Adverse effects, above. Unfortunately, as described earlier, a single network could not be constructed out of the available trials. The two networks constructed for this outcome are depicted in Appendix 4.
The results from this NMA are presented in Table 87. Overall, only PLDH plus carboplatin was found to have significantly less risk of an allergic reaction (at the 5% level) than paclitaxel plus carboplatin. PLDH plus carboplatin is also associated with significantly less risk of allergic reaction than platinum as monotherapy. No other comparison of chemotherapies was found to have a statistically significant difference.
As only one trial62 provided data on this AE for network 2, it was not possible to conduct a NMA. Lortholary et al. 62 compared low-dose paclitaxel (80 mg/m2) with low-dose paclitaxel (80 mg/m2) plus carboplatin. Low-dose paclitaxel was found to have significantly less risk of causing an allergic reaction than paclitaxel plus carboplatin (OR 0.114, 95% CI 0.014 to 0.942).
| Comparison | OR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| Network 1 | |||
| vs. paclitaxel plus carboplatin (OR < 1 favours comparator, OR > 1 favours paclitaxel plus carboplatin) | |||
| Platinum monotherapy | 0.755 | 0.057 | 3.043 |
| PLDH plus carboplatin | 0.130 | 0.001 | 0.705 |
| Gemcitabine plus carboplatin | 0.757 | 0.030 | 3.798 |
| vs. platinum monotherapy (OR < 1 favours comparator, OR > 1 favours platinum monotherapy) | |||
| PLDH plus carboplatin | 0.213 | 0.004 | 0.965 |
| Gemcitabine plus carboplatin | 0.997 | 0.183 | 3.091 |
| vs. PLDH plus carboplatin (OR < 1 favours comparator, OR > 1 favours PLDH plus carboplatin) | |||
| Gemcitabine plus carboplatin | 6.680 | 0.495 | 242.200 |
The absolute numbers for the RCTs included in the NMA evaluating alopecia in patients with recurrent ovarian cancer are reported in Adverse effects, above. Unfortunately, as described earlier, a single network could not be constructed out of the available trials. The two networks constructed for this outcome are depicted in Appendix 4.
As only one trial29 provided data on this AE for network 1 it was not possible to conduct a NMA. Bafaloukos et al. 29 compared PLDH plus carboplatin to paclitaxel plus carboplatin. PLDH plus carboplatin was found to have significantly less risk of causing alopecia than paclitaxel plus carboplatin (OR 0.235, 95% CI 0.077 to 0.724).
The results for the NMA of network 2 are presented in Table 88. Overall, all chemotherapies assessed were found to have a significantly higher risk of alopecia (at the 5% level) than PLDH monotherapy. Paclitaxel monotherapy was also found to have a significantly higher risk of alopecia than paclitaxel monotherapy (weekly). No other comparison of chemotherapies was found to have a statistically significant difference.
| Comparison | OR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| Network 2 | |||
| vs. PLDH monotherapy (OR < 1 favours comparator, OR > 1 favours PLDH monotherapy) | |||
| Topotecan monotherapy (i.v.) | 6.099 | 1.578 | 18.780 |
| Topotecan monotherapy (oral) | 8.621 | 1.344 | 31.990 |
| Paclitaxel monotherapy (weekly) | 3.512 | 0.643 | 12.920 |
| Paclitaxel monotherapy | 15.160 | 3.444 | 52.790 |
| vs. topotecan monotherapy (i.v.) (OR < 1 favours comparator, OR > 1 favours topotecan monotherapy: i.v.) | |||
| Topotecan monotherapy (oral) | 1.415 | 0.467 | 3.390 |
| Paclitaxel monotherapy | 0.841 | 0.081 | 3.584 |
| Paclitaxel monotherapy (weekly) | 3.623 | 0.409 | 14.760 |
| vs. topotecan monotherapy (oral) (OR < 1 favours comparator, OR > 1 favours topotecan oral monotherapy: oral) | |||
| Paclitaxel monotherapy | 0.770 | 0.050 | 3.648 |
| Paclitaxel monotherapy (weekly) | 3.312 | 0.249 | 15.130 |
| vs. paclitaxel monotherapy (OR < 1 favours comparator, OR > 1 favours paclitaxel monotherapy) | |||
| Paclitaxel monotherapy (weekly) | 4.766 | 2.467 | 8.489 |
The absolute numbers for the RCTs included in the NMA evaluating anaemia in patients with recurrent ovarian cancer are reported in Adverse effects, above. Unfortunately, as described earlier, a single network could not be constructed out of the available trials. The two networks constructed for this outcome are depicted in Appendix 4.
The results of the NMA from network 1 are presented in Table 89. Overall, PLDH plus carboplatin and gemcitabine plus carboplatin were found to have significantly higher risk of anaemia (at the 5% level) than paclitaxel plus carboplatin. Gemcitabine plus carboplatin was also found to have a significantly higher risk of anaemia than platinum monotherapy. No other comparison of chemotherapies was found to have a statistically significant difference.
The results of the NMA from network 2 are also presented in Table 89. Overall, topotecan monotherapy (i.v.), topotecan monotherapy (oral) and PLDH plus trabectedin were found to have significantly higher risk of anaemia (at the 5% level) than PLDH monotherapy. PLDH plus trabectedin, paclitaxel monotherapy and topotecan monotherapy (i.v., weekly) were also found to have significantly higher risk of anaemia than topotecan monotherapy (i.v.). Paclitaxel monotherapy was found to have significantly less risk than topotecan monotherapy (oral) and topotecan monotherapy (i.v.). No other comparison of chemotherapies was found to have a statistically significant difference.
One additional trial62 provided data on this AE but it was not possible to include this in either network owing to the atypical doses of paclitaxel compared. Lortholary et al. 62 compared low-dose paclitaxel (80 mg/m2) with low-dose paclitaxel (80 mg/m2) plus carboplatin. No significant difference in risk of anaemia was identified (OR 0.273, 95% CI 0.071 to 1.048).
| Comparison | OR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| Network 1 | |||
| vs. paclitaxel plus carboplatin (OR < 1 favours comparator, OR > 1 favours paclitaxel plus carboplatin) | |||
| Platinum monotherapy | 1.255 | 0.305 | 3.479 |
| PLDH plus carboplatin | 1.926 | 1.164 | 3.039 |
| Gemcitabine plus carboplatin | 5.848 | 1.158 | 18.040 |
| vs. platinum monotherapy (OR < 1 favours comparator, OR > 1 favours platinum monotherapy) | |||
| PLDH plus carboplatin | 2.205 | 0.527 | 6.289 |
| Gemcitabine plus carboplatin | 4.664 | 2.366 | 8.600 |
| vs. PLDH plus carboplatin (OR < 1 favours comparator, OR > 1 favours PLDH plus carboplatin) | |||
| Gemcitabine plus carboplatin | 3.152 | 0.609 | 9.880 |
| Network 2 | |||
| vs. PLDH monotherapy (OR < 1 favours comparator, OR > 1 favours PLDH monotherapy) | |||
| Topotecan monotherapy (i.v.) | 7.374 | 3.775 | 13.590 |
| Topotecan monotherapy (oral) | 7.949 | 3.305 | 16.680 |
| PLDH plus trabectedin | 2.940 | 1.559 | 5.202 |
| Paclitaxel monotherapy | 0.742 | 0.209 | 1.848 |
| Paclitaxel monotherapy (weekly) | 2.551 | 0.407 | 9.425 |
| Topotecan monotherapy (i.v., weekly) | 2.346 | 0.625 | 6.118 |
| vs. topotecan monotherapy (i.v.) (OR < 1 favours comparator, OR > 1 favours topotecan monotherapy: i.v.) | |||
| Topotecan monotherapy (oral) | 1.078 | 0.640 | 1.714 |
| PLDH plus trabectedin | 0.443 | 0.166 | 0.958 |
| Paclitaxel monotherapy | 0.101 | 0.036 | 0.209 |
| Paclitaxel monotherapy (weekly) | 0.385 | 0.051 | 1.519 |
| Topotecan monotherapy (i.v., weekly) | 0.318 | 0.107 | 0.704 |
| vs. topotecan monotherapy (oral) (OR < 1 favours comparator, OR > 1 favours topotecan monotherapy: oral) | |||
| PLDH plus trabectedin | 0.438 | 0.140 | 1.044 |
| Paclitaxel monotherapy | 0.099 | 0.031 | 0.231 |
| Paclitaxel monotherapy (weekly) | 0.381 | 0.046 | 1.549 |
| Topotecan monotherapy (i.v., weekly) | 0.314 | 0.091 | 0.765 |
| vs. PLDH plus trabectedin (OR < 1 favours comparator, OR > 1 favours PLDH plus trabectedin) | |||
| Paclitaxel monotherapy | 0.277 | 0.064 | 0.766 |
| Paclitaxel monotherapy (weekly) | 0.951 | 0.128 | 3.676 |
| Topotecan monotherapy (i.v., weekly) | 0.876 | 0.192 | 2.531 |
| vs. paclitaxel monotherapy (OR < 1 favours comparator, OR > 1 favours paclitaxel monotherapy) | |||
| Paclitaxel monotherapy (weekly) | 4.701 | 0.445 | 20.380 |
| Topotecan monotherapy (i.v., weekly) | 3.869 | 0.866 | 11.400 |
| vs. paclitaxel monotherapy (weekly) (OR < 1 favours comparator, OR > 1 favours paclitaxel monotherapy: weekly) | |||
| Topotecan monotherapy (i.v., weekly) | 1.749 | 0.149 | 7.204 |
The absolute numbers for the RCTs included in the NMA evaluating fatigue in patients with recurrent ovarian cancer are reported in Adverse effects, above. Unfortunately, as described earlier, a single network could not be constructed out of the available trials. The two networks constructed for this outcome are depicted in Appendix 4.
A NMA of network 1 could not be performed owing to zero events in a link in the network29 and non-comparable doses and/or treatment regimen in the remaining available trials. Individual trial results are presented in Table 90.
The results of the NMA from network 2 are presented in Table 91. No comparison of chemotherapies was found to have a statistically significant difference (at the 5% level).
| Comparison | OR | 95% CI | Trial | |
|---|---|---|---|---|
| Lower limit | Upper limit | |||
| PLDH plus carboplatin vs. paclitaxel plus carboplatin | Infinitya | NA | NA | Bafaloukos et al.29 |
| PLDH monotherapy (every 3 weeks) vs. PLDH monotherapy (every 4 weeks) | 0.454 | 0.204 | 1.012 | Monk et al.30 |
| Gemcitabine plus carboplatin vs. platinum monotherapy | 1.326 | 0.292 | 6.011 | Pfisterer et al.50 |
| Paclitaxel monotherapy vs. paclitaxel plus carboplatin | 1.031 | 0.555 | 1.917 | Lortholary et al.62 |
| PLDH plus carboplatin vs. platinum monotherapy | 1.452 | 0.226 | 9.309 | Alberts et al.28 |
| Comparison | OR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| vs. paclitaxel monotherapy (OR < 1 favours comparator, OR > 1 favours paclitaxel monotherapy) | |||
| Topotecan monotherapy (i.v.) | 1.570 | 0.479 | 3.978 |
| Topotecan monotherapy (oral) | 1.896 | 0.242 | 7.042 |
| Topotecan monotherapy (i.v., weekly) | 3.334 | 0.548 | 11.390 |
| vs. topotecan monotherapy (i.v.) (OR < 1 favours comparator, OR > 1 favours topotecan monotherapy: i.v.) | |||
| Topotecan monotherapy (oral) | 1.213 | 0.256 | 3.645 |
| Topotecan monotherapy (i.v., weekly) | 2.123 | 0.627 | 5.573 |
| vs. topotecan monotherapy (oral) (OR < 1 favours comparator, OR > 1 favours topotecan oral monotherapy: oral) | |||
| Topotecan monotherapy (i.v., weekly) | 2.761 | 0.342 | 10.540 |
The absolute numbers for the RCTs included in the NMA evaluating febrile neutropenia in patients with recurrent ovarian cancer are reported in Adverse effects, above. Unfortunately, no NMA could be performed due to zero events in three of the available trials. 28,50,62 Individual trial results are presented in Table 92.
| Comparison | OR | 95% CI | Trial | |
|---|---|---|---|---|
| Lower limit | Upper limit | |||
| PLDH plus carboplatin vs. platinum monotherapy | Infinitya | NA | NA | Alberts et al.28 |
| PLDH plus carboplatin vs. paclitaxel plus carboplatin | 0.614 | 0.299 | 1.263 | Pujade-Lauraine et al.31 |
| PLDH plus trabectedin vs. PLDH monotherapy | 3.256 | 1.378 | 7.692 | Monk et al.30 |
| Gemcitabine plus carboplatin vs. platinum monotherapy | Infinitya | NA | NA | Pfisterer et al.50 |
| Paclitaxel plus carboplatin vs. paclitaxel monotherapy | Infinityb | NA | NA | Lortholary et al.62 |
| Topotecan monotherapy vs. topotecan monotherapy (weekly) | 4.000 | 0.439 | 36.439 | Sehouli et al.23 |
The absolute numbers for the RCTs included in the NMA evaluating nausea and vomiting in patients with recurrent ovarian cancer are reported in Adverse effects, above. Unfortunately, as described earlier, a single network could not be constructed out of the available trials. The two networks constructed for this outcome are depicted in Appendix 4.
The results of the NMA from network 1 are presented in Table 93. Overall, PLDH plus carboplatin was found to have significantly higher risk of nausea and vomiting (at the 5% level) than paclitaxel plus carboplatin. No other comparison of chemotherapies was found to have a statistically significant difference.
The results of the NMA from network 2 are also presented in Table 93. Overall, paclitaxel monotherapy was found to have significantly lower risk of nausea and vomiting (at the 5% level) than PLDH monotherapy. Topotecan monotherapy (oral) and PLDH plus trabectedin were found to have significantly higher risk of nausea and vomiting than PLDH monotherapy (and any of the other chemotherapies assessed). However, when compared with each other no significant difference was found. No other comparison of chemotherapies was found to have a statistically significant difference.
| Comparison | OR | 95% CrI | |
|---|---|---|---|
| Lower limit | Upper limit | ||
| Network 1 | |||
| vs. paclitaxel plus carboplatin (OR < 1 favours comparator, OR > 1 favours paclitaxel plus carboplatin) | |||
| Platinum monotherapy | 4.897 | 0.415 | 23.550 |
| PLDH plus carboplatin | 426.200 | 2.000 | 709.700 |
| vs. platinum monotherapy (OR < 1 favours comparator, OR > 1 favours platinum monotherapy) | |||
| PLDH plus carboplatin | 109.700 | 0.721 | 234.900 |
| Network 2 | |||
| vs. PLDH monotherapy (OR < 1 favours comparator, OR > 1 favours PLDH monotherapy) | |||
| Topotecan monotherapy (oral) | 3.849 | 1.377 | 8.921 |
| Topotecan monotherapy (i.v.) | 1.460 | 0.886 | 2.294 |
| PLDH plus trabectedin | 5.291 | 2.866 | 9.342 |
| Paclitaxel monotherapy (weekly) | 0.554 | 0.061 | 2.237 |
| Paclitaxel monotherapy | 0.279 | 0.120 | 0.535 |
| Topotecan monotherapy (i.v., weekly) | 1.023 | 0.219 | 2.915 |
| vs. topotecan monotherapy (oral) (OR < 1 favours comparator, OR > 1 favours topotecan oral monotherapy: oral) | |||
| Topotecan monotherapy (i.v.) | 0.449 | 0.180 | 0.904 |
| PLDH plus trabectedin | 1.724 | 0.486 | 4.403 |
| Paclitaxel monotherapy (weekly) | 0.176 | 0.015 | 0.765 |
| Paclitaxel monotherapy | 0.089 | 0.024 | 0.223 |
| Topotecan monotherapy (i.v., weekly) | 0.315 | 0.055 | 0.985 |
| vs. topotecan monotherapy (i.v.) (OR < 1 favours comparator, OR > 1 favours topotecan monotherapy: i.v.) | |||
| PLDH plus trabectedin | 3.840 | 1.698 | 7.673 |
| Paclitaxel monotherapy (weekly) | 0.392 | 0.043 | 1.596 |
| Paclitaxel monotherapy | 0.197 | 0.084 | 0.379 |
| Topotecan monotherapy (i.v., weekly) | 0.701 | 0.166 | 1.869 |
| vs. PLDH plus trabectedin (OR < 1 favours comparator, OR > 1 favours PLDH plus trabectedin) | |||
| Paclitaxel monotherapy (weekly) | 0.114 | 0.011 | 0.484 |
| Paclitaxel monotherapy | 0.058 | 0.019 | 0.130 |
| Topotecan monotherapy (i.v., weekly) | 0.211 | 0.038 | 0.655 |
| vs. paclitaxel monotherapy (weekly) (OR < 1 favours comparator, OR > 1 favours paclitaxel monotherapy: weekly) | |||
| Paclitaxel monotherapy | 1.029 | 0.134 | 3.613 |
| Topotecan monotherapy (i.v., weekly) | 4.260 | 0.257 | 19.750 |
| vs. paclitaxel monotherapy (every 3 weeks) (OR < 1 favours comparator, OR > 1 favours paclitaxel monotherapy: every 3 weeks) | |||
| Topotecan monotherapy (i.v., weekly) | 4.107 | 0.753 | 12.880 |
The absolute numbers for the RCTs included in the NMA evaluating neuropathy in patients with recurrent ovarian cancer are reported in Adverse effects, above. Unfortunately, no NMA could be performed due to zero events in four of the available trials. 21,23,29,48 Individual trial results are presented in Table 94.
| Comparison | OR | 95% CI | Trial | |
|---|---|---|---|---|
| Lower limit | Upper limit | |||
| PLDH plus carboplatin vs. paclitaxel plus carboplatin | Infinitya | NA | NA | Bafaloukos et al.29 |
| Platinum monotherapy vs. paclitaxel plus carboplatin | Infinityb | NA | NA | Gonzalez-Martin et al.48 |
| Gemcitabine plus carboplatin vs. platinum monotherapy | 0.994 | 0.198 | 4.994 | Pfisterer et al.50 |
| PLDH plus trabectedin vs. PLDH monotherapy | 1.404 | 0.815 | 2.419 | Monk et al.30 |
| Paclitaxel monotherapy (weekly) vs. paclitaxel monotherapy | 0.368 | 0.175 | 0.777 | Rosenberg et al.60 |
| Topotecan monotherapy (i.v.) vs. paclitaxel monotherapy | Infinityb | NA | NA | ten Bokkel Huinink et al.21 |
| Topotecan monotherapy (i.v.) vs. topotecan (i.v., weekly) monotherapy | Infinityc | NA | NA | Sehouli et al.23 |
| Paclitaxel monotherapy vs. paclitaxel plus carboplatin | 1.639 | 0.693 | 3.878 | Lortholary et al.62 |
Discussion
The population of ovarian cancer patients that is the focus of this MTA is those who have relapsed following first-line treatment with platinum-based therapy or have disease that is refractory to platinum-based chemotherapy. Diagnosis of recurrent disease varies in UK clinical practice, with diagnosis based on clinical examination, biochemical markers (CA125) or radiological confirmation – or any combination of these three. Clinical expert advice is that, typically, a patient is diagnosed as relapsed if they have a serial rise in CA125 level or have developed clinical signs, such as ascites. Diagnosis is typically confirmed with radiological scans. If a patient has no clinical symptoms but does have a rise in CA125 level, although possibly classified as relapse, the patient might not start a new chemotherapeutic regimen until they go on to develop symptoms. Date of relapse by CA125 level is likely to be about 4 months earlier than date of relapse based on radiological scans.
A patient’s response to first-line platinum-based therapy is indicative of their response to second and subsequent lines of platinum-based treatment, with the length of the PFI and the extent of relapse (site and number of tumours) particularly prognostic of response. However, most patients will develop resistance to platinum-based therapy over time, with decreasing length of PFI with increasing rounds of treatment. Platinum-resistant ovarian cancer has a particularly poor prognosis, with a reported median OS of < 12 months.
The systematic review of clinical effectiveness evidence carried out to address the decision problem that is the focus of this MTA identified 16 RCTs, evaluating 14 pairwise comparisons. Of the 16 RCTs identified, five evaluated the intervention and comparator within their licensed indication, and dose and route of administration. The remaining 11 RCTs evaluated the intervention or comparator outside the parameters specified in the licence. However, the scope of the evidence identified was insufficient to fully address the decision problem; therefore, where possible the TAG has carried out synthesis of the evidence within NMAs.
Based on clinical expert advice, the TAG has focused on the clinical effectiveness of interventions in populations defined by degree of platinum sensitivity [i.e. platinum sensitive (i.e. recurrence ≥ 6 months after last platinum-based treatment) and platinum resistant (i.e. recurrence < 6 months after last platinum-based treatment) or refractory (progression during platinum-based treatment)].
The identified RCTs facilitated the construction of three distinct networks for the outcomes of OS and PFS, two of which considered patients with platinum-sensitive disease; the remaining network considered patients with disease that is PRR. As the systematic review was conducted in such a way as to identify all trials with at least one intervention of interest, a wider selection of treatments were assessed, but, unfortunately, this did not uncover one or more trials that could link the disconnected networks in patients with platinum-sensitive disease. Furthermore, owing to time constraints, the decision was taken not to search for non-randomised trials.
The two networks, for OS and PFS, constructed in patients with platinum-sensitive disease were:
-
platinum sensitive network 1, which compared regimens containing platinum, in particular: platinum plus paclitaxel, PLDH plus platinum, gemcitabine plus carboplatin, and platinum alone
-
platinum sensitive network 2, which compared non-platinum-based therapies, in particular: PLDH, trabectedin plus PLDH, paclitaxel and topotecan.
Platinum-sensitive patients
Overall survival and PFS data were identified for eight and seven different head-to-head comparisons of interventions and comparators of interest, respectively. Of these, three reported a statistically significant difference in OS between the treatments considered. In particular, Parmar et al. 61 reported a statistically significant difference in OS between paclitaxel plus platinum vs. conventional platinum treatment (HR 0.82, 95% CI 0.69 to 0.97) observed in the ICON4/AGO-OVAR 2.261 trial. Gonzalez-Martin et al. 48 reported a statistically significant difference between paclitaxel plus carboplatin vs. carboplatin alone (HR 0.31, 95% CI 0.14 to 0.68) and Gordon et al. 54 present a statistically significant difference between PLDH and topotecan (HR 1.43, 95% CI 1.07 to 1.92). Six of the identified head-to-head comparisons identified a statistically significant difference in PFS. These were:
-
CALYPSO31 PLDH plus carboplatin vs. paclitaxel plus carboplatin (HR 0.82, 95% CI 0.72 to 0.94)
-
ICON4/AGO-OVAR 2.261 Paclitaxel plus platinum vs. conventional platinum treatment (HR 0.76, 95% CI 0.66 to 0.89)
-
Gonzalez-Martin et al. 48 Paclitaxel plus carboplatin vs. carboplatin alone (HR 0.54, 95% CI 0.32 to 0.92)
-
Alberts et al. 28 PLDH plus carboplatin vs. carboplatin alone (HR 0.54, 95% CI 0.32 to 0.93)
-
OVA-30130 Trabectedin plus PLDH vs. PLDH (HR 0.73, 95% CI 0.56 to 0.95)
-
Pfisterer et al. 50 Gemcitabine plus carboplatin vs. carboplatin alone (HR 0.72, 95% CI 0.58 to 0.90).
In the NMA evaluating platinum-based chemotherapies, PLDH plus carboplatin and paclitaxel plus carboplatin were found to significantly improve OS compared with platinum monotherapy. However, no statistically significant differences in OS were identified between the remaining treatments considered in the network. When compared with platinum monotherapy, PFS was estimated to significantly improve in patients treated with paclitaxel plus carboplatin, gemcitabine plus carboplatin or PLDH plus carboplatin. In addition, a statistically significant difference in PFS was estimated for paclitaxel plus carboplatin compared with PLDH plus carboplatin.
However, the TAG consider it important to note that examination of the baseline characteristics of trials included in NMAs of platinum-based therapies, revealed an imbalance in baseline performance score (ECOG) within one of the included trials. In particular, the trial carried out by Gonzalez-Martin et al. ,48 in which paclitaxel plus carboplatin is compared with platinum monotherapy; the proportion of patients with a baseline ECOG score of 2 that were randomised to treatment with platinum monotherapy was 17.9% vs. 5.6% of patients randomised to treatment with paclitaxel plus carboplatin. The TAG notes that this imbalance is likely to result in an overestimation of the relative treatment effect of paclitaxel plus carboplatin vs. platinum monotherapy.
Furthermore, the TAG notes the presence of clinical heterogeneity in the duration of PFI between trials. In particular, patients enrolled in the ICON-4/AGO-OVAR 2.261 trial had a comparably longer PFI than patients enrolled in the other trials included in NMA of OS and PFS data. Similarly, a comparatively high proportion of patients enrolled in the trial carried out by Gonzalez-Martin et al. 48 were diagnosed as recurrent based on assessment of CA125 levels; therefore these patients are likely to be more susceptible to platinum therapy than patients enrolled in the other included trials. However, the TAG notes that although patients in ICON-4/AGO-OVAR 2.261 and Gonzalez-Martin et al. 48 may be expected to experience greater benefit than patients enrolled in the other trials, the magnitude of this difference is unlikely to affect estimates of the relative effect of treatment.
Network meta-analysis of non-platinum-based therapies indicated that PLDH monotherapy and trabectedin plus PLDH are both significantly more effective at prolonging OS than topotecan monotherapy. No other significant OS differences were identified. Analysis of non-platinum-based regimens indicates that trabectedin plus PLDH statistically significantly improves PFS compared with PLDH, paclitaxel and topotecan when given as monotherapies. No statistically significant differences in PFS were identified among the monotherapies evaluated (PLDH, topotecan, and paclitaxel). However, as a result of the use of subgroup data to inform these analyses, assessment of the presence of clinical heterogeneity was not possible. In addition, the TAG considers it import to highlight that subgroup data from the included trials were not sufficiently powered to detect a difference in OS or PFS.
Overall response rate was reported for 11 different head-to-head comparisons of interventions and comparators of interest. Of these, only two were statistically significant: trabectedin plus PLDH vs. PLDH from OVA-30130 (OR 1.57, 95% CI 1.04 to 2.35); gemcitabine plus carboplatin vs. carboplatin alone from Pfisterer et al. 50 (OR 1.527, 95% CI 1.025 to 2.275).
Based on the trials identified, it was not possible to construct a complete network informing ORR. Akin to analyses of OS and PFS, two discrete networks were generated: one evaluating platinum-based therapies (paclitaxel plus carboplatin, gemcitabine plus carboplatin, PLDH plus carboplatin and platinum monotherapy) and the second comparing non-platinum-based regimens [PLDH, trabectedin plus PLDH, topotecan (i.v.), paclitaxel (every 3 weeks), topotecan (oral) and paclitaxel (weekly)].
In the NMA evaluating platinum-based chemotherapies, paclitaxel plus carboplatin and gemcitabine plus carboplatin were found to have a significantly higher ORR than platinum monotherapy. There was no significant difference between PLDH plus carboplatin and any of the chemotherapeutic treatments assessed. Analysis of non-platinum-based regimens indicates that trabectedin plus PLDH significantly improves ORR compared with PLDH, and oral topotecan. Compared with oral topotecan, i.v. topotecan was found to be associated with a significant increase in the proportion of patients achieving CR or PR. No other statistically significant differences were identified.
Platinum-resistant/-refractory patients
The OS and PFS data were reported for five and four different head-to-head comparisons in PRR patients, respectively. Two RCTs enrolled only patients with PRR, with the remaining RCTs reporting results from a subgroup of patients within the trial. None of the trials identified a significant difference in OS or PFS between the two treatment groups evaluated. Furthermore, no statistically significant differences in ORR were reported in the eight different head-to-head comparisons involving PRR patients. Similarly, no statistically significant differences in OS or PFS were identified in the NMA of treatment with paclitaxel, PLDH and topotecan. However, NMA of ORR estimated that PLDH significantly increased ORR compared with paclitaxel (175 mg/m2) every 21 days and with an alternative regimen in which paclitaxel was given weekly at a dose of 67 mg/m2. PLDH monotherapy was also significantly more effective than an unconventional regimen of topotecan in which topotecan was administered weekly at a dose of 4 mg/m2. As a result of the use of subgroup data to inform these analyses, the TAG notes that the individual trial data may have been underpowered to detect a difference in OS, PFS or ORR. Furthermore, as baseline characteristics were not reported for the subgroups, an assessment of the presence of clinical heterogeneity was not possible.
Health-related quality of life
Treatments for newly diagnosed ovarian cancer are given with curative intent; however, for women with advanced, recurrent disease, second- and subsequent-line therapies are typically given with palliative rather than curative intent, with the aim of alleviating symptoms and prolonging survival. Thus, key considerations in the choice of treatment at these stages in the pathway are maintaining the patient’s QoL. Of the 16 RCTs identified, 10 reported some level of data on QoL. However, reporting of results was generally limited, with few trials reporting scores generated from responses to the questionnaires. The most commonly used scale in the identified trials is the EORTC QLQ-C30 questionnaire,81 which was developed to assess the QoL of cancer patients and can be supplemented with disease-specific modules for individual cancers, including ovarian cancer. For many comparisons, scores on QoL scales were similar between treatments. Differences in QoL include:
-
For PLDH plus platinum versus paclitaxel plus platinum, at 3 months, PLDH plus platinum was associated with a significant improvement in global health compared with paclitaxel plus platinum. However, this benefit was not maintained at 6 months.
-
For paclitaxel plus platinum versus platinum-based chemotherapy patients receiving platinum monotherapy scored significantly worse on the nausea and vomiting symptom scale than did the paclitaxel plus platinum-based chemotherapy group. However, this difference seemed to be transient and was observed for only the first 15 weeks after randomisation.
-
For trabectedin plus PLDH versus PLDH in the subgroup of patients with PPS ovarian cancer, it is indicated that there is difference in global health status score among responding patients beyond cycle 5, with patients in the trabectedin plus PLDH group having a higher score than those receiving PLDH alone (higher score is favourable).
-
In comparison with PLDH, topotecan was associated with a significantly more favourable rating on the pain subscale of the EORTC QLQ-C30.
-
For paclitaxel plus platinum versus paclitaxel, patients receiving weekly paclitaxel plus platinum experienced improvements in constipation, abdominal/gastrointestinal symptoms, appetite loss, pain and emotional functioning. Patients treated with weekly paclitaxel alone experienced improvements in attitude to disease and insomnia, but worsening of dyspnoea and peripheral neuropathy.
-
For paclitaxel versus oxaliplatin, mean QoL score on the EORTC QLQ-C3081 increased by > 10 points between baseline and cycle 4 for patients in the paclitaxel group, irrespective of study withdrawal. By contrast, in the oxaliplatin group, the mean QoL score decreased through cycle 2, but by < 10 points, after which most patients’ mean scores returned to baseline levels.
Adverse events
An important consideration in the choice of second-line treatment is the adverse effect of neurotoxicity, which is commonly associated with paclitaxel and also with carboplatin. Neurotoxicity can persist for up to 2 years after the end of treatment. Patients who relapse after first-line treatment with paclitaxel–platinum combination therapy and are subsequently rechallenged with the same regimen within 12 months (i.e. those who are PPS) are at an increased risk of developing neurotoxicity. However, despite the associated increased risk of neurotoxicity, paclitaxel plus carboplatin is generally the preferred second-line treatment in UK practice in recurrent platinum-sensitive cancer, particularly for patients who relapse > 12 months after completion of first-line chemotherapy. Carboplatin is chosen over cisplatin because of its more favourable adverse effect profile.
Within each of the identified trials, the most frequently reported adverse effects were as expected for the individual treatments based on the SmPC. Commonly occurring adverse effects were alopecia, nausea and vomiting, haematological toxicities (neutropenia, anaemia, thrombocytopenia and leucopenia). Based on expert clinical advice the TAG restricted its comparison of AEs to those considered most problematic for patients or most likely to consume substantial health-care resource.
The potential for a NMA was, therefore, investigated for the following severe (grades 3 and 4) AEs: allergic reaction, alopecia, anaemia, fatigue, febrile neutropenia, nausea and vomiting, and neuropathy. In many cases a NMA was not possible owing to the lack of available data in the trials assessed. In these instances, the individual trial results are reported with the ORs and 95% CIs that were calculated. The majority of NMA results, supplemented by the individual trial results where a NMA was not possible, indicated that the likelihood of AEs were not statistically significantly different across treatment regimens. However, in some instances, chemotherapies were estimated as having significantly lower risks of one or more AEs but significantly higher risks of other AEs. For example, when compared with paclitaxel plus platinum, PLDH plus platinum is associated with significantly lower risks of allergic reaction and alopecia but significantly higher risks of anaemia and nausea and vomiting. Overall, no chemotherapy was consistently associated with either a lower risk or a higher risk of the severe AEs assessed.
Chapter 4 Assessment of cost-effectiveness
This chapter contains reference to confidential information provided as part of the NICE appraisal process. This information has been removed from the relevant sections and the results, discussions and conclusions do not include the confidential information. These sections are clearly marked in the report.
Review of existing cost-effectiveness evidence
This section provides a review of the existing cost-effectiveness evidence, both published and presented within MSs, for treatments in recurrent ovarian cancer covered in the scope of this MTA. 38 Review of TA91 and TA222 cost-effectiveness evidence summarises the cost-effectiveness evidence presented within TA91 and TA222. Technology Assessment Group systematic review of existing cost-effectiveness evidence presents findings from the TAG systematic review of cost-effectiveness evidence. Description and critique of manufacturer submitted evidence provides a description and critique of manufacturer submitted evidence. Summary and conclusion of available cost-effectiveness evidence summarises the available evidence and draws conclusions about the published and submitted assessments of cost-effectiveness.
Review of TA91 and TA222 cost-effectiveness evidence
This MTA is, in part, a review and update of TA9110 (Paclitaxel, pegylated liposomal doxorubicin hydrochloride and topotecan for second-line or subsequent treatment of advanced ovarian cancer) and TA22215 (Trabectedin for the treatment of relapsed ovarian cancer). The economic evidence presented within TA9110 and TA22215 was therefore considered to be a relevant source of information, and the cost-effectiveness analyses presented within both technology appraisals are summarised below.
Multiple Technology Appraisal no. 91: Paclitaxel, pegylated liposomal doxorubicin hydrochloride and topotecan for second-line or subsequent treatment of advanced ovarian cancer
Three manufacturers submitted cost-effectiveness evidence for consideration in TA91;10 GSK (topotecan), Schering-Plough (PLDH) and BMS (paclitaxel). GSK and Schering-Plough submitted cost-minimisation analyses comparing topotecan with PLDH. BMS submitted a cost-effectiveness analysis, which estimated the incremental cost per life-year gained (LYG) of paclitaxel, paclitaxel in combination with platinum, PLDH and topotecan. In addition to the submitted analyses, the TAG for TA91 identified four economic evaluations from the published literature: Smith et al. ,82 Ojeda et al. ,83 Capri and Cattaneo84 and Prasad et al. ,85 the first three of which were cost-minimisation analyses that compared topotecan with PLDH. Prasad et al. 85 reported the costs and effects associated with topotecan and gemcitabine, but did not carry out a formal economic evaluation. The TAG for TA91 concluded that the limitations of the submitted and published cost-effectiveness evidence were such that it was not possible to make a reliable comparison of the relative cost-effectiveness of the treatments considered in the scope of TA91. Therefore, to facilitate a comparison of the relative cost-effectiveness of the treatments considered, the TAG developed a new decision-analytic model. The model developed by the TAG was a semi-Markov cost–utility analysis, formed of three health states: SD, PD and death (Figure 13). The model evaluated OS in relation to the mean TTP, and the time from progression to death (estimated as mean OS minus mean TTP).
FIGURE 13.
Structure of the economic model developed for TA91 (reproduced from TA91 assessment report p. 17913).
Two analyses were carried out by the TAG for TA91;13 the main analysis considered a population of patients with refractory, resistant disease (PFI < 6 months) or platinum-sensitive disease (PFI ≥ 6 months) (full population), and the second analysis considered people with platinum-sensitive disease only.
For the main analysis, treatment effects in the form of HRs were extracted from two published RCTs, the first of which compared paclitaxel monotherapy with topotecan (ten Bokkel Huinink et al. 52) and the second of which compared topotecan with PLDH (data submitted for TA9113). Baseline estimates of PFS and OS were derived for the common comparator, topotecan, to which HRs from the two identified RCTs were applied to estimate PFS and OS for paclitaxel monotherapy and PLDH. In sensitivity analysis, of the main analysis, a third RCT was included, which compared paclitaxel with PLDH (Trial 30–5772). This RCT was excluded from the base-case analysis, as the trial was terminated early, and therefore the results were likely to be preliminary. In the sensitivity analysis, data from the three identified RCTs were combined via a NMA to estimate HRs for each treatment compared with topotecan. HRs were then applied to the baseline estimates of PFS and OS for patients treated with topotecan.
For the second analysis (people with platinum-sensitive disease) a further two RCTs were identified which were considered relevant; Cantu et al. 51 [paclitaxel vs. cyclophosphamide plus doxorubicin plus cisplatin (CAP)] and ICON4/AGO-OVAR 2.261 (paclitaxel plus platinum vs. platinum). ICON4/AGO-OVAR 2.2 could not be connected to the network owing to a lack of a common comparator; therefore, for the analysis, the TAG estimated the relative treatment effect associated with paclitaxel plus platinum using ‘an exponential approximation to estimate the absolute hazard associated with paclitaxel combination and topotecan respectively, and then take the ratio of these to provide the relative treatment effect’ (Assessment Report, p. 190). 13 This relative treatment effect was then included in a NMA, establishing a network of five RCTs in total. As before, HRs calculated from this NMA were applied to a baseline estimate of PFS and OS for patients treated with topotecan, resulting in estimates of PFS and OS for topotecan, paclitaxel monotherapy, PLDH, paclitaxel-plus-platinum combination therapy, platinum monotherapy and CAP.
The costs included in the analysis comprised the costs of study drugs, premedication, monitoring, drug administration and the cost of managing AEs. Long-term costs, including subsequent chemotherapy costs, were excluded from the model as a result of the lack of data. Sources of cost data included the British National Formulary (BNF) for drug costs (BNF 47,86 cost year 2004), data submitted by manufacturers (cost year 1999/2000) and national cost sources (Unit Costs of Health and Social Care,87 cost year 2000).
Quality-adjusted life-years (QALYs) were estimated by applying health-state utility values to the mean time spent in the SD and the PD health states. The utility associated with SD (0.63, applied to the mean time spent by patients in the health state of SD) was sourced from a study by Tengs and Wallace,88 identified in a systematic search of the literature carried out by the TAG for TA91. 13 However, no estimate of utility for PD was identified. Therefore, the TAG used a proxy measure of utility in PD from patients with breast cancer presented in a study by Brown and Hutton. 89 Although the TAG recognised the importance of the impact of treatment-related toxicity on QoL, no suitable or relevant QoL data were identified or submitted that could inform the disutility associated with the treatments considered.
Results were presented for the full population with recurrent ovarian cancer, and also separately for people with platinum-sensitive disease. For the full population, topotecan was extendedly dominated by PLDH, and a cost-effectiveness estimate of £24,606 per additional QALY was estimated for PLDH compared with paclitaxel. For the platinum-sensitive population: topotecan, paclitaxel and PLDH were dominated by platinum monotherapy, CAP was extendedly dominated, and a cost-effectiveness estimate of £3561 per additional QALY was estimated for paclitaxel plus platinum compared with platinum monotherapy (Table 95).
| Treatment | PFS (weeks) | OS (weeks) | Quality-adjusted survival (weeks) | Cost (£) | ICER (incremental cost per additional QALY) | Probability of being cost-effectiveness at a maximum WTP of £30,000 (%) |
|---|---|---|---|---|---|---|
| Full population | ||||||
| Topotecan | 24.5 | 86.0 | 34.2 | 8448 | Extendedly dominated | 2 |
| Paclitaxel | 20.1 | 79.7 | 30.9 | 4146 | – | 37 |
| PLDH | 27.5 | 104.8 | 40.9 | 8902 | £24,606 | 61 |
| Platinum-sensitive population (PFI ≥ 6 months) | ||||||
| Topotecan | 33.1 | 101.4 | 41.7 | 8330 | Dominated | 0 |
| Paclitaxel | 28.0 | 105.1 | 41.2 | 4066 | Dominated | 0 |
| PLDH | 43.0 | 145.8 | 58.5 | 8851 | Dominated | 1 |
| Paclitaxel plus platinum | 82.0 | 178.8 | 81.2 | 6828 | £3561 | 60 |
| Platinum monotherapy | 63.5 | 149.7 | 66.3 | 3383 | – | 1 |
| CAP | 47.9 | 176.7 | 69.5 | 3512 | Extendedly dominated | 38 |
The TAG also presented results for the full population in which data from an early terminated trial comparing paclitaxel with PLDH were incorporated (Trial 30–57). 72 For this analysis, topotecan was strictly dominated by paclitaxel, and PLDH compared with paclitaxel was associated with an incremental cost-effectiveness ratio (ICER) of £58,475. The results of this analysis are presented in Table 96.
| Treatment | PFS (weeks) | OS (weeks) | Quality-adjusted survival (weeks) | Cost (£) | ICER (incremental cost per additional QALY) | Probability of being cost-effectiveness at a maximum WTP of £30,000 (%) |
|---|---|---|---|---|---|---|
| Full population | ||||||
| Topotecan | 24.5 | 86.0 | 34.2 | 8448 | Dominated | 1 |
| Paclitaxel | 20.1 | 92.1 | 34.6 | 4146 | – | 81 |
| PLDH | 27.5 | 98.1 | 38.9 | 8902 | £58,475 | 18 |
Single Technology Appraisal no. 222: trabectedin for the treatment of relapsed ovarian cancer
Technology appraisal 22215 was a STA of trabectedin for the treatment of relapsed ovarian cancer. The manufacturer presented a cost–utility analysis based on the model developed by the TAG for TA91. 13 Results were presented separately for the platinum-sensitive population (PFI ≥ 6 months), the PPS population (PFI of 6–12 months) and the FPS population (PFI > 12 months).
Estimates of mean OS and mean TTP were derived by replicating the NMA used in TA9113 for the platinum-sensitive population for topotecan, paclitaxel and PLDH (i.e. excluding CAP, paclitaxel plus platinum, and platinum monotherapy) with the addition of data from OVA-301,30 a clinical trial for trabectedin plus PLDH compared with PLDH in the recurrent setting. It was not clear within the MS whether the early terminated trial (30–5772) was included within this analysis. The Evidence Review Group (ERG) for TA22290 commented that ‘the manufacturer stated that three trials were included in the mixed-treatment comparison (MTC) analysis: 039 (ten Bokkel Huinink et al. ;51 Gore et al. 24) 30–49 (Schering-Plough;13 Gordon et al. 54) and OVA-301 (Monk et al. 30,64). The ERG believes that Trial 30–5772 was also included in the MTC for OS in order to provide the paclitaxel and PLDH comparison in the network of evidence’ (ERG report for TA222, p. 8590).
For PLDH monotherapy, the manufacturer estimated mean PFS and OS, to which HRs estimated from the NMA were applied, thereby providing estimates of mean PFS and OS for topotecan, paclitaxel and trabectedin in combination with PLDH. Baseline estimates of PFS and OS for PLDH monotherapy were obtained by assuming that survival data for both interventions considered within OVA-30130 were represented by exponential distributions; however, the ERG considering the evidence submitted in TA22290 noted that exponential distributions were not the most appropriate fit to the patient-level data.
The costs included in the analysis comprised the costs of study drugs, premedication, monitoring, drug administration and the cost of managing AEs. Following a clarification request from the ERG for TA222,90 the manufacturer also included an estimate of the cost of palliative care. Sources of cost data included the BNF for drug costs (BNF 58,91 cost year 2009), and national cost databases (National Tariff 2010/11; NHS Reference Costs92 2007–8).
Quality-adjusted life-years were estimated by applying health-state utility values to the mean time spent in each health state (i.e. SD, PD and death). Utility values were estimated from EQ-5D data collected within OVA-30130 and were presented by health state: SD mean estimate, 0.718; PD mean estimate, 0.649; death, assumed to be 0.
The manufacturer presented results for the entire platinum-sensitive population (relapse at ≥ 6 months following previous platinum therapy) and the PPS patients (relapse within 6–12 months of previous platinum therapy) separately. Results from the manufacturer’s analyses are presented in Table 97.
| Population | Treatment | Total cost (£) | Total QALYs | ICER (incremental cost per additional QALY, deterministic) |
|---|---|---|---|---|
| Platinum sensitive (PFI ≥ 6 months) | Paclitaxel | 4738 | 1.17 | – |
| PLDH | 9355 | 1.54 | £12,680 | |
| Topotecan | 15,726 | 1.27 | Dominated | |
| Trabectedin plus PLDH | 26,389 | 1.81 | £62,619 | |
| PPS (PFI 6–12 months) | PLDH | 9350 | 1.34 | – |
| Trabectedin plus PLDH | 26,349 | 1.78 | £38,668 |
The ERG for TA22290 considered the comparison of trabectedin in combination with PLDH compared with PLDH in people with PPS disease to be the most pertinent decision problem. This was because PLDH was not listed as a comparator of interest in the NICE scope for people with FPS (PFI > 12 months) disease. The ERG therefore did not present any results for the FPS population within their report; instead, the ERG focused on results for the PPS population (PFI 6–12 months). The ERG for TA22290 investigated a number of changes to the model for PPS patients, including amending the parametric distribution used to calculate the mean PFS and OS time for PLDH. The ERG concluded that ‘the most plausible ICER for trabectedin in combination with PLDH compared with PLDH alone in women who relapse between 6 to 12 months after initial platinum-based chemotherapy ranges between £46,503 and £54,607’ (TA222 ERG report, p.127). 90
Technology Assessment Group’s systematic review of existing cost-effectiveness evidence
A systematic review was carried out in December 2012 to identify relevant published economic evaluations to support the development of this MTA. The following databases were searched:
-
MEDLINE (via Ovid)
-
EMBASE (via Ovid)
-
Health Technology Assessment (HTA) database
-
NHS Economic Evaluations Database (NHS EED).
The search strategy for MEDLINE and EMBASE combined terms capturing the interventions and comparators of interest (topotecan, PLDH, paclitaxel, trabectedin, gemcitabine, best supportive care, bevacizumab, carboplatin, cisplatin and etoposide); the target condition (ovarian cancer); and terms to capture economic evaluations. As this MTA is, in part, an update of TA9113 – in which a systematic review was carried out (search date of April 2004) to evaluate the cost-effectiveness of topotecan, PLDH and paclitaxel – searches for these interventions were carried out with a date limit of 2004. Databases were searched from inception for gemcitabine and trabectedin. The search strategy for HTA and NHS EED combined terms for the target condition (ovarian cancer) with no further limits. Full details of the search terms are presented in Appendix 5.
In addition to searches of the above databases, the following sources of potentially relevant publications were explored:
-
Experts in the field were contacted with a request for details of relevant published and unpublished studies of which they may have knowledge.
-
The NICE website was searched for any recently published technology appraisals in ovarian cancer that had not already been identified via the database searches.
-
Reference lists of key identified studies were reviewed for any potentially relevant studies.
No restrictions on language or setting were applied to any of the searches. The titles and abstracts of papers identified through the searches were independently assessed for inclusion by two health economists using the criteria outlined in Table 98.
| Inclusion criteria | Exclusion criteria |
|---|---|
|
|
The systematic review was updated in May 2013 while the report was under peer review. The search strategy remained the same as outlined above; however, results were limited from 4 December 2012 to 21 May 2013 in order to identify only additional relevant studies.
A total of 842 papers were identified from the December 2012 search (Figure 14). Of these papers, 740 were excluded on the basis of title and abstract. A total of 102 papers were therefore identified as potentially relevant and were ordered for full review. Of the 102 ordered papers, 59 were excluded following review of the full paper. For a description of the reason for exclusion of the ordered papers, see Appendix 6. A total of 43 papers were identified as economic evaluations from the December 2012 search.
FIGURE 14.
Identified economic evaluation studies: December 2012 search.
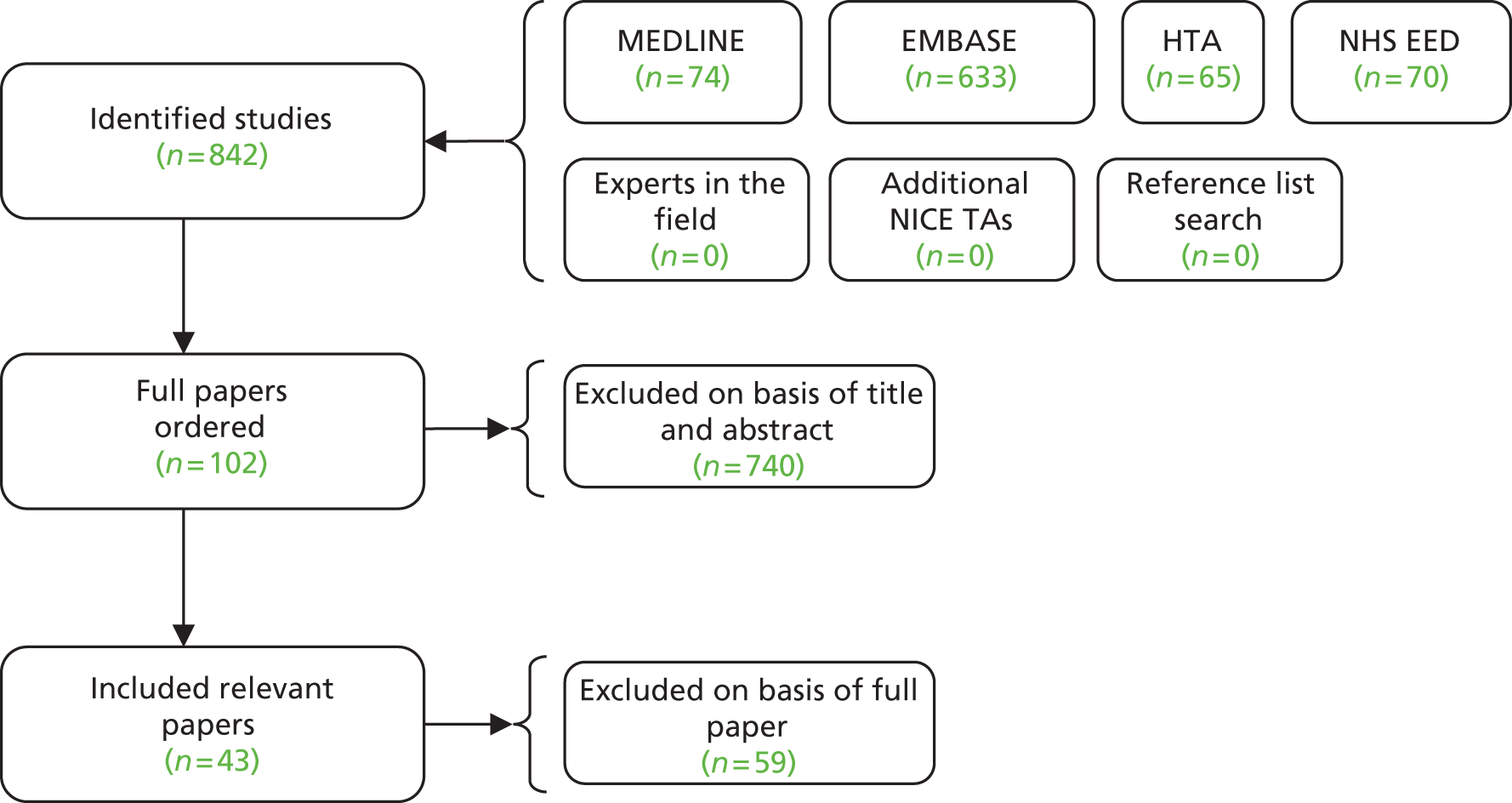
A further 91 papers were identified from the updated search in May 2013. Of these, 90 were excluded on the basis of title and abstract, with one paper identified as potentially relevant and ordered for full review. Additionally, two relevant NICE TAs were identified from the NICE website and were reviewed in full: TA28411 and TA285. 16
Of the 46 economic evaluation studies identified from the December 2012 (43 papers) and May 2013 (three papers) searches, 21 related specifically to recurrent ovarian cancer (Table 99). These 21 studies were considered by the TAG to be the most relevant to this MTA, and were extracted in full (see Appendix 7); the remaining included papers are presented as short summaries (see Appendix 7).
| Study | Additional studies | ||
|---|---|---|---|
| Identified in TA9113 | Related to TA9113 | Related to TA22217 | |
| Capri and Cattaneo et al.84 | Griffin et al.94 | NICE 201115 | Forbes et al.95 |
| TA28516 | |||
| Papaioannou et al.90 | Chan et al.96 | ||
| Ojeda et al.83 | Main et al.97 | Havrilesky et al.98 | |
| Papaioannou et al.99 | Lesnock et al.100 | ||
| Lesnock et al.101 | |||
| Smith et al.82 | NICE 200510 | Gore et al.102 | Case et al.103 |
| Havrilesky et al.104 | |||
| Montalar et al.105 | Rocconi et al.106 | ||
| Lee107 | |||
Of the 21 economic evaluations identified in patients with recurrent ovarian cancer,10,15,16,82–86,90,94–107 four studies82–84,95 were published prior to 2004 and describe cost-minimisation analyses comparing PLDH with topotecan. These studies were carried out from the perspective of Italy,84 Spain,83 the UK,82,95 and the USA. 82 Three of these studies82–84 were identified from the literature search for economic evaluations carried out in TA91, and are reviewed in detail within the TA91 Technology Assessment Report. 13
Three15,90,99 of the 21 identified studies10,15,16,82–86,90,94–107 were directly related to TA222,15 of which this MTA is, in part, a review and update (for a description of TA222,15 see Review of TA91 and TA222 cost-effectiveness evidence, above). A further two studies102,105 were published subsequent to TA222:15 Gore et al. 102 is a poster describing a cost–utility analysis of trabectedin in combination with PLDH compared with PLDH using more recent estimates of survival. Montalar et al. 105 is a cost–utility analysis carried out from the perspective of Spain comparing trabectedin in combination with PLDH compared with PLDH monotherapy; the analysis was based upon the model developed for TA91. 13 A further three identified studies10,94,97 were related to TA9110 of which this MTA is also, in part, a review and update. A description of the analysis carried out in TA9110 is presented above (see Review of TA91 and TA222 cost-effectiveness evidence, above).
Of the remaining nine economic evaluations identified,16,96,98,100,101,103,104,106,107 one was carried out from the perspective of the UK (TA28515) and was a STA considering the cost-effectiveness of bevacizumab in recurrent ovarian cancer. The model developed by the manufacturer for this STA was a semi-Markov model based upon the model structure outlined in TA9113 (i.e. SD, PD and death).
Of the remaining eight economic evaluations,96,98,100,101,104,106,107 seven were from the perspective of the USA96,98,100,101,103,104,106 and one was from the perspective of Korea. 107 Four98,100,101,107 of the eight economic evaluations were cost–utility analyses, i.e. assessed the incremental cost per additional QALY. Of these cost–utility analyses, Lesnock et al. 101 developed a Markov model with equivalent health states to those used in the TA91 TAG model: PFS, recurrence and death. Havrilesky et al. 98 developed a Markov model with health states including no evidence of disease and PD; in addition, AEs (specifically neurotoxicity) were accounted for within the model structure. Two studies, Lesnock100 and Lee,107 were presented as abstracts; Lee107 described the health states included within the model as responsive; progressive; clinical remission; and death. Lesnock et al. 100 did not describe the model structure in sufficient detail to enable reporting of the health states.
All studies identified within recurrent ovarian cancer with the exception of the three studies appraised within TA9182–84 were quality assessed against the NICE reference case, and Philips checklist (see Appendix 8). 108
Description and critique of manufacturer submitted evidence
Two manufacturers [Eli Lilly (gemcitabine); PharmaMar (trabectedin)] submitted evidence for consideration for this MTA. Of these, one manufacturer (PharmaMar) submitted cost-effectiveness evidence. PharmaMar did not carry out a systematic review of the existing cost-effectiveness evidence; instead, the manufacturer developed an economic analysis based upon the model developed for TA91. 13 The analysis and results are described below.
Trabectedin for the treatment of patients with relapsed platinum-sensitive ovarian cancer
The manufacturer developed an economic analysis based upon the model developed within TA91. 13 With this model, the manufacturer evaluated the cost-effectiveness of trabectedin (1.1 mg/m2) in combination with PLDH (30 mg/m2) administered every 3 weeks, compared with PLDH monotherapy (50 mg/m2) administered every 4 weeks, for the treatment of patients with relapsed platinum-sensitive ovarian cancer. The TAG’s appraisal of the manufacturer’s economic evaluation against the requirements set out in the NICE reference case checklist for a base-case analysis, and appraisal of the quality of the manufacturer’s economic evaluation using the Philips checklist,108 are summarised in Appendix 8.
Patient population
Trabectedin, in combination with PLDH, is indicated for the treatment of patients with relapsed platinum-sensitive (PFI of ≥ 6 months) ovarian cancer. The patient population for whom the manufacturer is requesting consideration within this MTA comprises a subset of this indication, specifically people who:
-
are not suitable for, or not best managed with, platinum-based chemotherapy because of an allergy or an intolerance owing to residual toxicities, and
-
have PPS disease (PFI of 6–12 months).
The manufacturer illustrated this group of patients diagrammatically within the MS (Figure 15).
FIGURE 15.
Patient population for which the manufacturer is positioning trabectedin (reproduced with permission from MS: figure 2.1, p. 7).
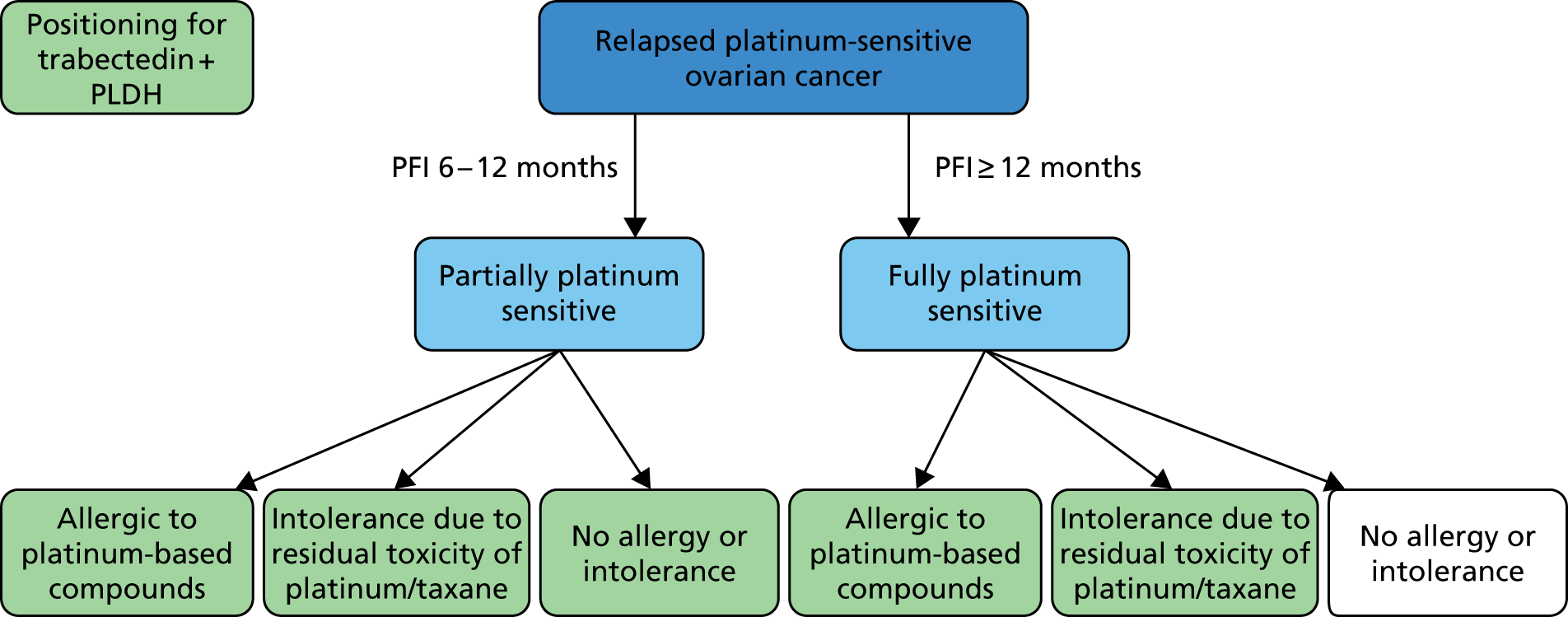
The manufacturer justified the choice of patient population by stating: ‘this patient population represents a restricted subgroup of the licensed platinum-sensitive population, and is chosen to align with the inclusion criteria of the OVA-301 trial and the clinical unmet need for non-platinum alternatives in these populations’ (MS, p. 30).
The TAG reviewed the inclusion criteria for OVA-301 supplied by the manufacturer within the submission and notes that OVA-301 included patients with platinum-resistant, platinum-refractory and platinum-sensitive disease; the licence for trabectedin in combination with PLDH is for patients with platinum-sensitive disease only. In addition, the TAG notes that the patients enrolled were those who ‘were not expected to benefit from or who were ineligible for or were not willing to receive retreatment with platinum-based chemotherapy’ (MS, p. 30 and MS, see Appendix 2). The TAG notes that it is unclear from the MS what proportion of patients included within OVA-301 were allergic or intolerant to platinum therapy compared with those who were not. However, following discussion with clinical experts, the TAG notes that the efficacy of non-platinum-based treatments (such as trabectedin and/or PLDH) is unlikely to differ between people with an allergy or intolerance to platinum therapy compared with people without. Therefore, the TAG considers that results are unlikely to differ between patients with or without the presence of allergy or intolerance.
Model structure
The model structure developed by the manufacturer was identical to the model developed within TA91; disease was classified into three distinct periods: SD, PD and death (Figure 16). The time spent within each health state was determined by the mean TTP and mean OS data from OVA-301. Costs and QALYs accumulated for each treatment were calculated based upon the mean time spent in each health state.
FIGURE 16.
Model structure used in the PharmaMar submission.

The TAG considers that the model structure used by the manufacturer was generally appropriate and in line with previous published model structures identified from the TAG systematic review of the cost-effectiveness literature (see Technology Assessment Group systematic review of existing cost-effectiveness evidence, above). However, the TAG notes a key critique of the same model structure provided by the ERG for TA222: ‘the ERG believes that there are potential limitations to this simplicity, which can impose constraints regarding the assignment of costs, utilities and discounting’ (ERG report for TA222, p. 93). 90
Comparators
The relevant comparators listed in the NICE scope for patients with platinum-sensitive ovarian cancer were:38
-
paclitaxel monotherapy
-
paclitaxel in combination with platinum therapy
-
PLDH monotherapy
-
PLDH in combination with platinum therapy
-
gemcitabine in combination with carboplatin
-
topotecan
-
platinum-based monotherapy.
Additionally, the relevant comparators listed in the scope for patients with an allergy to platinum-based compounds were:
-
paclitaxel monotherapy
-
PLDH monotherapy
-
topotecan
-
etoposide (Vepesid®, BMS)
-
best supportive care.
The comparator therapy assessed by the manufacturer was PLDH monotherapy. This represented one comparator listed within the NICE scope.
The manufacturer did not compare trabectedin in combination with PLDH with platinum-based regimens because data from the key clinical trial OVA-30130 were restricted to patients who, upon enrolment,‘were not expected to benefit from or who were ineligible for or were not willing to receive retreatment with platinum-based chemotherapy’ (MS, p. 30). However, the TAG notes that the patient group for which the manufacturer is seeking a recommendation includes PPS patients with no allergy or intolerance to platinum-based chemotherapy, but who were not expected to benefit from platinum therapy. Following discussion with clinical experts, the TAG considers that PPS patients may be treated with a platinum agent in clinical practice. The TAG acknowledges that patients with a PFI of close to 6 months would be less likely to receive platinum; however, platinum therapy remains an important treatment option for this group of patients.
Nonetheless, the TAG notes that although a comparison of trabectedin in combination with PLDH compared with a platinum agent is desirable, the clinical systematic review carried out by the TAG found no comparative clinical data linking (either directly or indirectly) trabectedin plus PLDH with a platinum agent administered either as monotherapy or in combination with another therapy. This issue and the importance of future research in this area are discussed in greater detail in Chapter 7 (see Suggested research priorities).
In addition, the manufacturer did not compare trabectedin in combination with PLDH with paclitaxel or topotecan. The TAG notes that the manufacturer’s rationale for omitting these comparisons was based on conclusions reached by the ERG responsible for assessing for TA222 (Box 1). 90
Non platinum-based regimens including paclitaxel, PLDH and topotecan were previously evaluated as comparators to trabectedin plus PLDH during the NICE STA. A MTC including paclitaxel, PLDH and topotecan as comparators was presented during the submission. However, it was concluded by both the Appraisal Committee and the NICE ERG (Sheffield University) that when compared with paclitaxel or topotecan monotherapy:
PLDH is the most clinically and cost-effective treatment within the platinum-sensitive population. As PLDH is the recommended second-line therapy, and trabectedin plus PLDH cannot be used where PLDH is contraindicated, the relative cost-effectiveness of trabectedin plus PLDH compared to paclitaxel or topotecan monotherapy is not needed, since there would never be a choice between these interventions. As such, a direct comparison of trabectedin plus PLDH is sufficient to address the decision problem.
Since no additional evidence has become available for PLDH, topotecan or paclitaxel since 2009 (see section 3), in line with NICE and ERG guidance (from TA222), we have not considered paclitaxel and topotecan as comparators for trabectedin plus PLDH.
The TAG acknowledges that based upon the NMA (referred to as a MTC within Box 1) and economic analysis carried out for TA91,13 for people with platinum-sensitive disease, PLDH extendedly dominated topotecan, and resulted in an incremental cost per additional QALY compared with paclitaxel at a value of < £20,000. The TAG also notes that the manufacturer updated the clinical systematic review undertaken, in 2009, as part of TA222,93 and found two additional studies, both of which were related to OVA-301. 30 The results from the clinical systematic review carried out by the TAG accorded with this. The TAG therefore acknowledges the rationale for restriction by the manufacturer; however, for completeness, we have carried out a full NMA, including paclitaxel and topotecan, and included these comparisons within the TAG economic model (see Independent economic assessment, below). This is to ensure that the most up-to-date information on clinical practice, costs, QoL and the most mature survival data from OVA-30130 have been used to inform the decision problem that is the focus of this MTA.
Finally, the manufacturer did not compare trabectedin in combination with PLDH with etoposide or best supportive care because no comparative clinical evidence was found by the manufacturer to enable such a comparison. The TAG acknowledges that, similarly to the manufacturer, no comparative data between trabectedin plus PLDH and etoposide or best supportive care were found during the TAG systematic review of the clinical literature. This lack of data makes a robust comparison with etoposide or best supportive care unfeasible. The TAG explores this issue further below (see Base-case results).
Effectiveness data (progression-free survival and overall survival)
Estimates of mean PFS and OS were calculated from Kaplan–Meier data obtained from the OVA-301 clinical trial; a RCT providing head-to-head data for trabectedin in combination with PLDH compared with PLDH monotherapy in patients with relapsed ovarian cancer. Specifically, the manufacturer fitted a variety of parametric curves (exponential, Weibull, Gompertz, log-logistic and log-normal) to OS and PFS Kaplan–Meier data for patients with platinum-sensitive disease. These curves were fitted separately by treatment arm, i.e. treatment was not included as a covariate; the manufacturer did not provide a rationale within the submission for this methodology. In addition, the manufacturer used explanatory variables to control for the following baseline characteristics:
-
age (continuous)
-
race (categorical)
-
PFI (continuous)
-
CA125 (categorical)
-
liver or lung involvement (binary)
-
prior taxane use (binary).
The manufacturer used the Akaike information criterion (AIC) and Bayesian information criterion associated with each survival distribution to select the preferred distribution for PFS and OS. The Weibull distribution was selected to inform mean PFS for both trabectedin in combination with PLDH and PLDH monotherapy. The log-logistic distribution was selected to inform mean OS for both trabectedin in combination with PLDH and PLDH monotherapy. The results are summarised in Figures 17 and 18 and Table 100.
FIGURE 17.
Survival distribution and Kaplan–Meier plots for PFS (reproduced with permission from MS, p. 35).

FIGURE 18.
Survival distribution and Kaplan–Meier plots for OS (reproduced with permission from MS, p. 35).

| Time point of interest | Trabectedin in combination with PLDH | PLDH monotherapy |
|---|---|---|
| Mean TTP (months) | 11.26 | 8.25 |
| Mean OS (months) | 44.69 | 34.97 |
The PFS and OS data used within the manufacturer’s economic model were obtained from the full platinum-sensitive patient population of OVA-301, i.e. including both patients with partially (PFI 6–12 months) or fully (PFI > 12 months) platinum-sensitive disease. The manufacturer’s rationale for using these data was that OVA-301 was not powered for post hoc analysis of subgroups within the platinum-sensitive stratum.
The TAG notes that within the analysis of PFS and OS, the manufacturer controlled for PFI (as a continuous variable). The TAG recognises that PFI is considered to be a prognostic factor for patients with relapsed ovarian cancer; patients with a longer PFI typically have an improved prognosis when compared with patients with a shorter PFI. The TAG also acknowledges that, when PFI is considered as a continuous rather than categorical variable, there exists a baseline imbalance in the PFI between patients in the PLDH plus trabectedin compared with PLDH arms of OVA-301 (Table 101). The manufacturer calculated that the mean PFI for the two treatment arms was [Commercial-in-confidence (CIC) information has been removed] [mean 13.3 months for PLDH monotherapy and mean 10.6 months for trabectedin plus PLDH, (CIC information has been removed) for the full population; mean 14.3 months for PLDH monotherapy, and mean 19.0 months for trabectedin plus PLDH, (CIC information has been removed) for the platinum-sensitive population]. Moreover, clinical opinion supports the manufacturer’s use of a continuous, rather than categorical variable to control for PFI. Consequently, the TAG considers the analysis carried out by the manufacturer to be appropriate, and recognises that the manufacturer explored the impact of controlling for PFI upon the ICER in the sensitivity analysis.
| Mean PFI | Trabectedin in combination with PLDH | PLDH monotherapy |
|---|---|---|
| For all patients within OVA-301 (months) | 10.6 | 13.3 |
| For platinum-sensitive patients within OVA-301 (months) | 14.3 | 19.0 |
However, the TAG has one key area of concern around the extrapolated PFS and OS data used within the manufacturer’s model; the degree of censoring observed within the PFS and OS data.
The PFS data used by the manufacturer within the economic model was subject to a high degree of censoring; for the full population (i.e. platinum sensitive and PRR) within OVA-301, 38.8% and 40.5% of people receiving treatment with PLDH alone and trabectedin in combination with PLDH were censored, respectively. The manufacturer did not include details around the reasons for censoring within the MS, nor did the manufacturer provide an explanation for the quantity of censoring encountered. Moreover, the manufacturer did not provide detail around censoring for the platinum-sensitive subgroup separately. The manufacturer reported that a total of 189 patients were censored in the final PFS analysis for platinum-sensitive patients (approximately 45%); however, it is unclear for what reasons, and in which arm, these patients were censored.
In addition, limited details around censoring within the OS analysis have been presented within the MS. The manufacturer reports that a total of 114 patients were censored in the final OS analysis for platinum-sensitive patients (approximately 27%); however, it is unclear for what reasons, and in which arm, these patients were censored.
The TAG requested the clinical study report (CSR) for OVA-301 to explore this issue of censoring in both the PFS and OS analyses in further detail; however, (CIC data removed). Nevertheless, the number of people with platinum-sensitive disease censored in the PFS and OS analyses by treatment arm was presented within the CSR. The TAG notes that the degree of censoring for PFS in people with platinum-sensitive disease was approximately (CIC data removed) in both arms, and the degree of censoring for OS in people with platinum-sensitive disease was approximately (CIC data removed).
As a result of the high degree of censoring, and the lack of information provided around the reasons for censoring, the TAG notes that censoring within this analysis may be informative. The TAG notes that the presence of informative censoring may reduce the validity of the Kaplan–Meier data presented and used within the model. The TAG does note, however, that censoring is (CIC data removed) (in terms of both number of patients and proportion of patients) between the two arms of the study on aggregate, although it is unclear at what time points censoring occurred.
Adverse event incidence
The manufacturer included AEs of grade 3 or 4 (or those associated with a notable cost) within the model. Table 102 summarises the AE incidence used in the economic model.
| Women with platinum-sensitive disease (n = 425) | Grade 3 AE (%) | Grade 4 AE (%) | |
|---|---|---|---|
| Treatment | AE | ||
| PLDH | Neutropenia | 20.0 | 11.2 |
| Neutropenia, febrile | 1.4 | 0.5 | |
| Neutropenic infection | – | – | |
| Neutropenic sepsis | – | – | |
| Plateletsa | 2.4 | 2.4 | |
| Haemoglobina | 4.9 | 2.0 | |
| Nausea/vomiting | 1.9 | – | |
| Diarrhoea | 1.9 | – | |
| PPE syndrome | 20.2 | 0.5 | |
| Stomatitis | 4.3 | – | |
| Trabectedin plus PLDH | Neutropenia | 31.8 | 41.0 |
| Neutropenia, febrile | 4.1 | 1.8 | |
| Neutropenic infection | 0.5 | – | |
| Neutropenic sepsis | 0.5 | – | |
| Plateletsa | 11.5 | 9.7 | |
| Haemoglobina | 13.8 | 4.1 | |
| Nausea/vomiting | 14.3 | 0.5 | |
| Diarrhoea | 2.8 | – | |
| PPE syndrome | 3.7 | – | |
| Stomatitis | 1.4 | – | |
Quality of life
The manufacturer did not carry out a systematic search of the utilities literature; instead, the manufacturer used EQ-5D data derived from the OVA-301 trial for the health states described within the economic model (SD and PD). Mean utilities in the stable and progressed health states were estimated to be 0.718 and 0.649, respectively. Although not reported, the TAG considers it likely that the estimates of HRQoL were derived from the full population included within the OVA-301 trial, i.e. both platinum-sensitive patients and platinum-resistant patients. The manufacturer undertook a number of sensitivity analyses using HRQoL data by platinum sensitivity and treatment arm. Disutilities associated with AEs were not included in the model as it was considered that the impact of AEs on QoL would be captured within the mean estimates obtained from trial data.
The TAG notes that the HRQoL data used by the manufacturer is in line with the NICE Guide to the Methods of Technology Appraisal,109 in which it is stated that EQ-5D is the preferred measure of HRQoL in adults.
Resource use and costs
The manufacturer included the following costs within the economic analysis: treatment, administration and preparation, management of disease, and treatment of AEs. Costs by treatment arm are summarised in Table 103.
| Cost | Trabectedin in combination with PLDH | PLDH monotherapy | Stated source |
|---|---|---|---|
| Drug cost per cycle (based upon a BSA of 1.72 m2) and assuming no vial sharing | £3167 | £1425 | BNF 2013110 |
| Drug administration costs | £334 per attendance £440 one-off cost of central venous line insertion |
£203 per attendance | NHS Reference Costs 2011–12111 |
| Medical management | Stable period: one outpatient visit per month (£121) and one CT scan every 2 months (£125) | NHS Reference Costs 2011–12111 | |
| Progressed period: estimated £6667 annual cost | Guest et al.112 | ||
| AEs, total cost per patient | £398 | £147 | NHS Reference Costs 2011–12111 |
Subsequent to initial submission, the manufacturer submitted a proposed patient access scheme (PAS) affecting the total chemotherapy costs associated with trabectedin in combination with PLDH. For the PAS, the manufacturer proposes that the NHS pays for the first five cycles of chemotherapy, after which acquisition costs would be met by the manufacturer. To reflect this within the economic model, the manufacturer assumed that patients would receive a lower number of cycles (and therefore a lower cost – efficacy is not affected by this assumption) of therapy with trabectedin plus PLDH; without the PAS, patients received 6.86 cycles of trabectedin on average and 4.28 cycles on average with the PAS. In addition, the manufacturer included implementation/administrative costs associated with the PAS and estimated that the total discounted implementation cost of the PAS would be £560.74.
Results
The manufacturer presented discounted, deterministic and probabilistic results from the analysis within the MS. The manufacturer presented results both without the PAS (Table 104), and, subsequently, following an updated submission, results including the PAS (Table 105).
Without the PAS, the manufacturer estimated an incremental cost per additional QALY for trabectedin in combination with PLDH compared with PLDH monotherapy to be £39,306 in the deterministic base case and £39,447 in the probabilistic base case. The TAG notes that in the base case, probabilistic and deterministic results are similar.
With the PAS, the manufacturer estimated an incremental cost per additional QALY for trabectedin in combination with PLDH compared with PLDH monotherapy to be £27,573 in the deterministic base case and £27,761 in the probabilistic base case.
| Treatment | Total costs (£) | Total LYG | Total QALYs | Incremental costs (£) | Incremental LYG | Incremental QALYs | ICER (£) |
|---|---|---|---|---|---|---|---|
| Deterministic results | |||||||
| Trabectedin plus PLDH | 43,907 | 3.72 | 2.33 | – | – | – | – |
| PLDH | 24,809 | 2.91 | 1.85 | 19,098 | 0.81 | 0.49 | 39,306 |
| Probabilistic results | |||||||
| Trabectedin plus PLDH | 44,203 | 3.724 | 2.35 | – | – | – | – |
| PLDH | 24,931 | 2.914 | 1.86 | 19,273 | 0.810 | 0.49 | 39,447 |
| Treatment | Total costs (£) | Total LYG | Total QALYs | Incremental costs (£) | Incremental LYG | Incremental QALYs | ICER (£) |
|---|---|---|---|---|---|---|---|
| Deterministic results | |||||||
| Trabectedin plus PLDH | 38,206 | 3.72 | 2.33 | – | – | – | – |
| PLDH | 24,809 | 2.91 | 1.85 | 13,397 | 0.81 | 0.49 | 27,573 |
| Probabilistic results | |||||||
| Trabectedin plus PLDH | 38,206 | 3.724 | 2.35 | – | – | – | – |
| PLDH | 24,931 | 2.914 | 1.86 | 13,563 | 0.810 | 0.49 | 27,761 |
Sensitivity analysis
The manufacturer carried out a number of sensitivity analyses both deterministic (one-way sensitivity analysis, scenario analyses) and probabilistic for results with and without the PAS.
In one-way sensitivity analysis, the 10 variables that the cost-effectiveness results were most sensitive to were presented in a tornado plot. Cost-effectiveness results were most sensitive to estimates of OS. The TAG notes that, although not reported, the manufacturer varied OS between an upper and lower 20% of the base-case figure. For the analyses without the PAS, the TAG notes that the x-axis on the tornado diagram was limited to £107,000 when the result using the low value for trabectedin in combination with PLDH for OS within the economic model was in fact £266,114 (without the PAS). The TAG updated the tornado diagram presented within the manufacturer’s model to reflect this (Figure 19; without the PAS). Figure 20 presents results of the one-way sensitivity analysis for the results with the PAS.
FIGURE 19.
Results from the manufacturer’s one-way sensitivity analysis updated by the TAG to reflect the full range of ICERs; without PAS. Tornado diagram: trabectedin plus PLDH vs. PLDH.
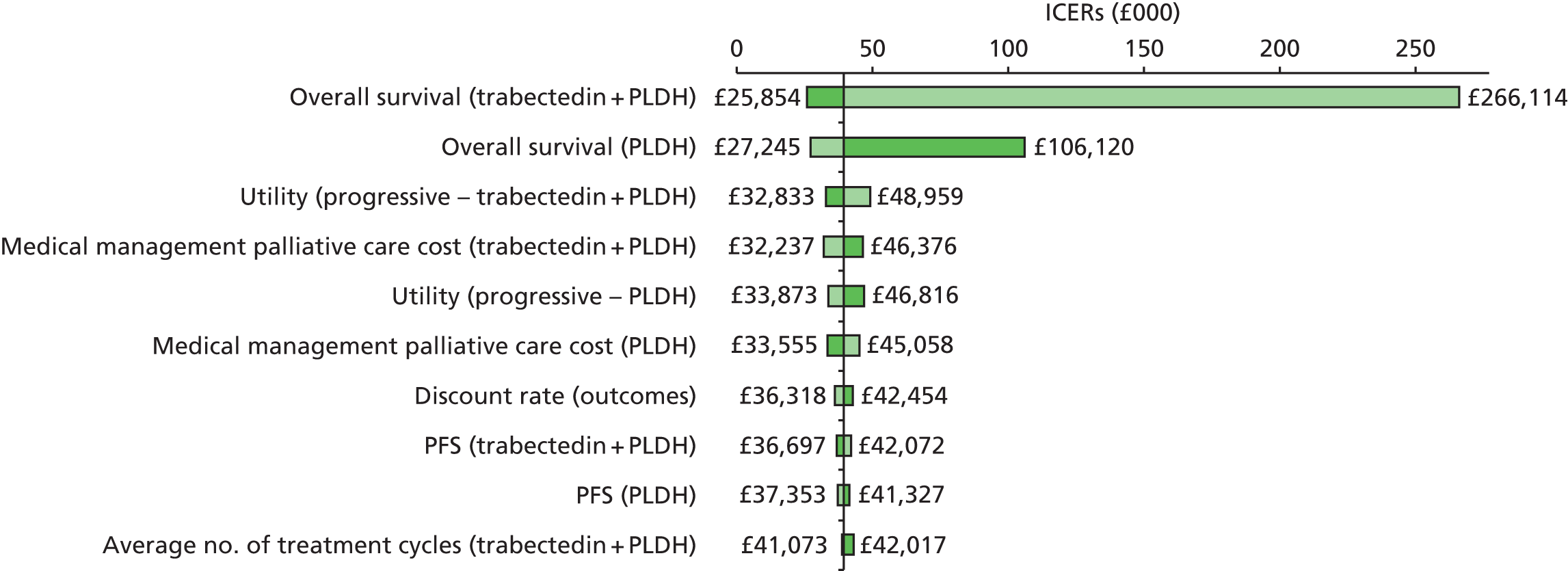
FIGURE 20.
Results from the manufacturer’s one-way sensitivity analysis; with PAS (reproduced with permission from PAS submission, p. 25). Tornado diagram: trabectedin plus PLDH vs. PLDH – with PAS.

A number of scenario analyses were presented within the MS; results with and without the PAS are summarised in Tables 106–108.
| Scenario | Trabectedin plus PLDH (PFS) | PLDH (PFS) | Trabectedin plus PLDH (OS) | PLDH (OS) | ICER without PAS (£) | ICER with PAS (£) |
|---|---|---|---|---|---|---|
| Base case | Weibull, AIC = 451.2 | Weibull, AIC = 380.9 | Log-logistic, AIC = 508.0 | Log-logistic, AIC = 472.7 | 39,306 | 27,573 |
| Distribution 1 | Gompertz, AIC = 457.7 | Gompertz, AIC = 384.0 | Log-logistic, AIC = 508.0 | Log-logistic, AIC = 472.7 | 39,320 | 27,572 |
| Distribution 2 | Weibull, AIC = 451.2 | Weibull, AIC = 380.9 | Weibull, AIC = 508.1 | Weibull, AIC = 478.6 | 52,589 | 35,485 |
| Distribution 3 | Gompertz, AIC = 457.7 | Gompertz, AIC = 384.0 | Weibull, AIC = 508.1 | Weibull, AIC = 478.6 | 52,611 | 35,485 |
| Base-case distribution (no PFI adjustment) | Weibull, AIC = 456.3 | Weibull, AIC = 385.8 | Log-logistic, AIC = 514.3 | Log-logistic, AIC = 504.7 | 109,892 | 70,222 |
| Scenario | Trabectedin plus PLDH | PLDH | Incremental analysis | |||
|---|---|---|---|---|---|---|
| Total cost (£) | Total QALYs | Total cost (£) | Total QALYs | ICER (£) | Difference (£) | |
| Base case | 43,907 | 2.33 | 24,809 | 1.85 | 39,306 | – |
| Treatment and platinum-sensitive specific utilities | 43,907 | 2.46 | 24,809 | 1.98 | 39,975 | –669 |
| Neutropenia Grade 3 (base case: £0, alternative scenario: £122.31) Grade 4 (base case: £0, alternative scenario: £2346.49) |
44,901 | 2.33 | 25,094 | 1.85 | 40,766 | –1460 |
| Neutropenic infection Grades 3 and 4 (base case: £2346, alternative scenario: £2108) |
43,906 | 2.33 | 24,809 | 1.85 | 39,304 | –2 |
| Neutropenic sepsis Grades 3 and 4 (base case: £2346, alternative scenario: £2108) |
43,906 | 2.33 | 24,809 | 1.85 | 39,304 | –2 |
| Scenario | Trabectedin plus PLDH | PLDH | Incremental analysis | |||
|---|---|---|---|---|---|---|
| Total cost (£) | Total QALYs | Total cost (£) | Total QALYs | ICER (£) | Difference (£) | |
| Base case | 38,206 | 2.33 | 24,809 | 1.85 | 27,573 | – |
| Treatment and platinum-sensitive specific utilities | 38,206 | 2.46 | 24,809 | 1.98 | 28,042 | 469 |
| Neutropenia Grade 3 (base case: £0, alternative scenario: £122.31) Grade 4 (base case: £0, alternative scenario: £2346.49) |
39,200 | 2.33 | 25,094 | 1.85 | 29,033 | 1,460 |
| Neutropenic infection Grades 3 and 4 (base case: £2,346, alternative scenario: £2108) |
38,205 | 2.33 | 24,809 | 1.85 | 27,571 | –2 |
| Neutropenic sepsis Grades 3 and 4 (base case: £2,346, alternative scenario: £2108) |
38,205 | 2.33 | 24,809 | 1.85 | 27,571 | –2 |
The manufacturer also presented results from probabilistic analysis, through both plots upon the cost-effectiveness plane (Figures 21 and 22) and cost-effectiveness acceptability curves (CEACs; Figures 23 and 24). According to the manufacturer’s analysis, at a willingness-to-pay (WTP) threshold of £20,000, the probability that trabectedin in combination with PLDH is cost-effective compared with PLDH monotherapy is 11% and 10% with and without the PAS, respectively. At a WTP threshold of £30,000, the probability of cost-effectiveness increases to 53% with the PAS and 20% without the PAS.
FIGURE 21.
Cost-effectiveness plane presented by the manufacturer summarising the results of probabilistic analysis (reproduced with permission from MS: figure 7.7, p. 43) without PAS.

FIGURE 22.
Cost-effectiveness plane presented by the manufacturer summarising the results of probabilistic analysis (reproduced with permission from PAS submission: figure 6, p. 28) with PAS.
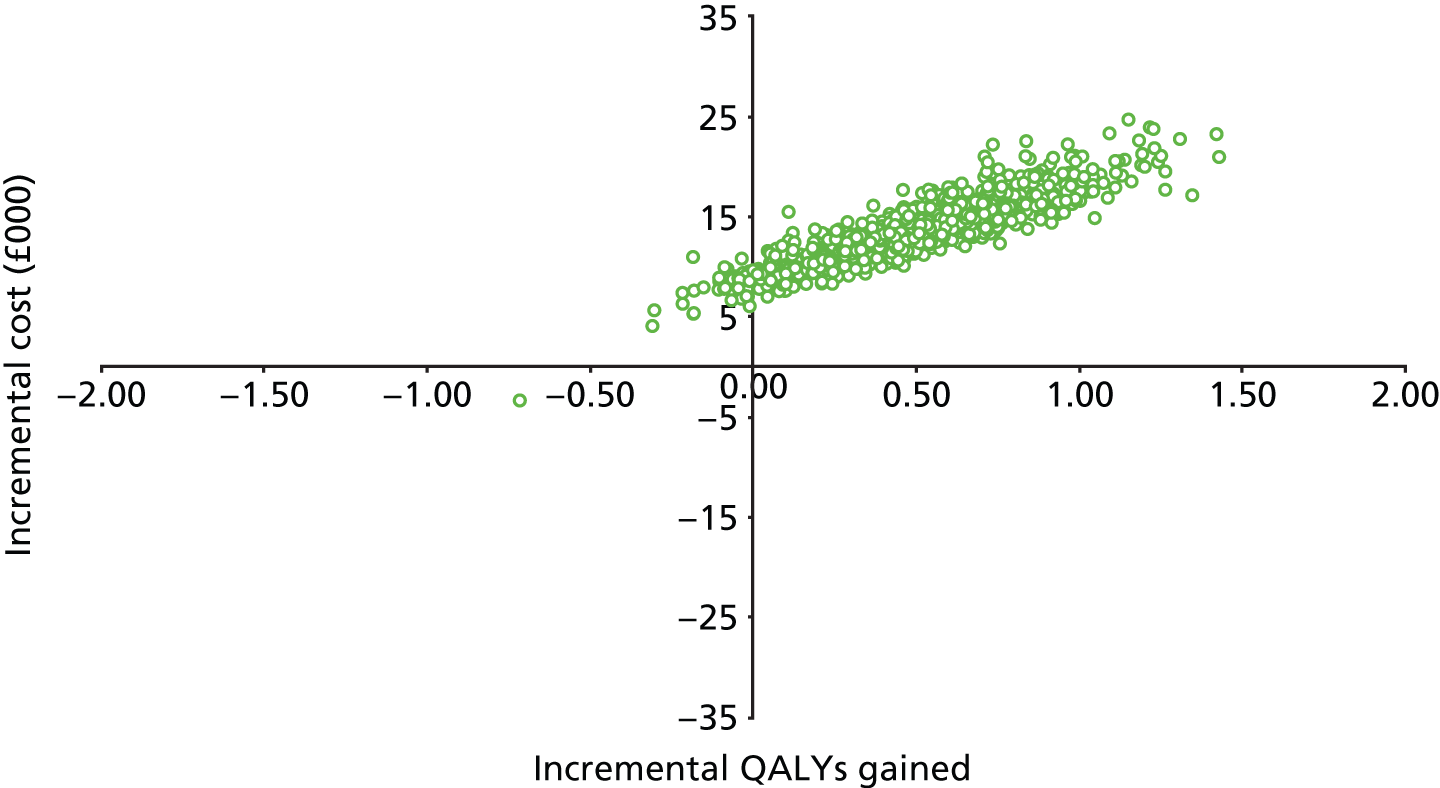
FIGURE 23.
Cost-effectiveness acceptability curve presented by the manufacturer summarising the results of probabilistic analysis (reproduced with permission from MS: figure 7.8, p. 43) without PAS.
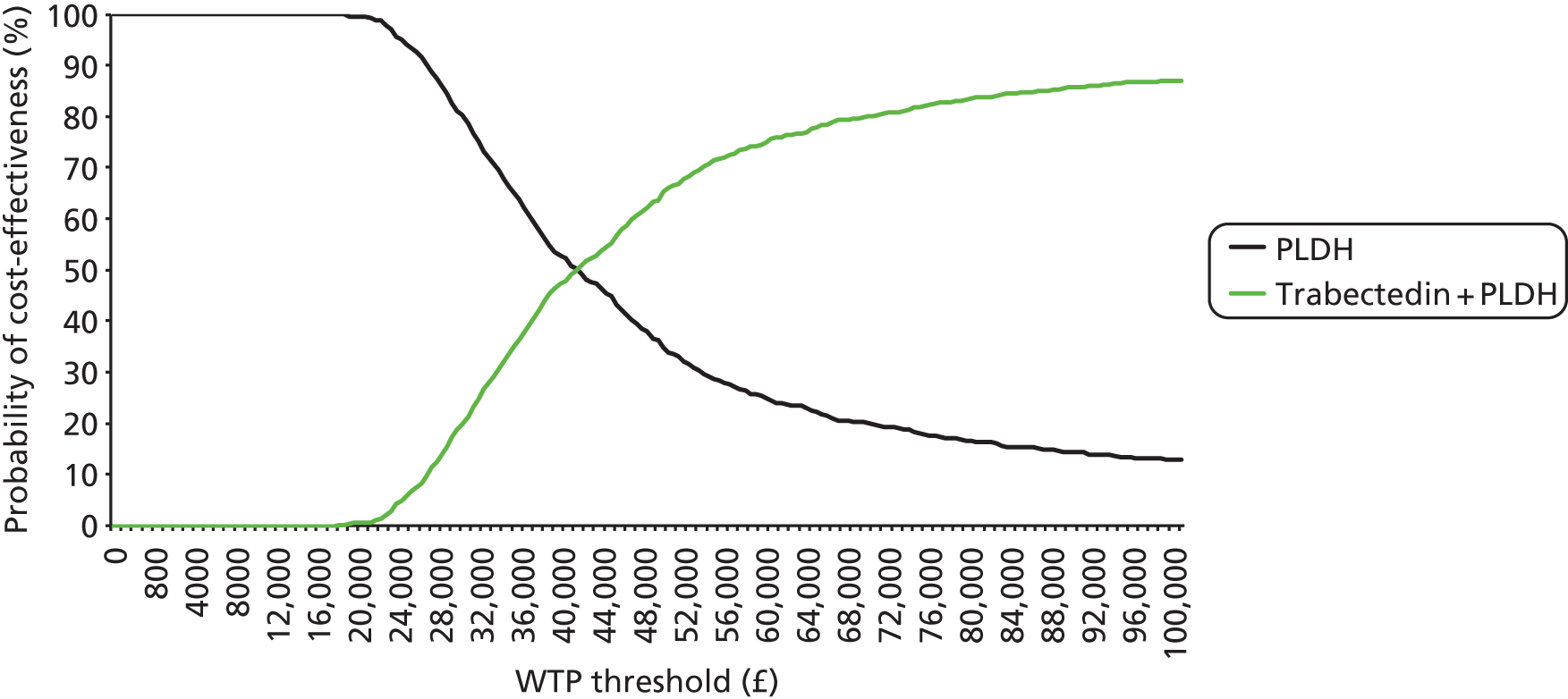
FIGURE 24.
Cost-effectiveness acceptability curve presented by the manufacturer summarising the results of probabilistic analysis (reproduced with permission from PAS submission: figure 7, p. 28) with PAS.
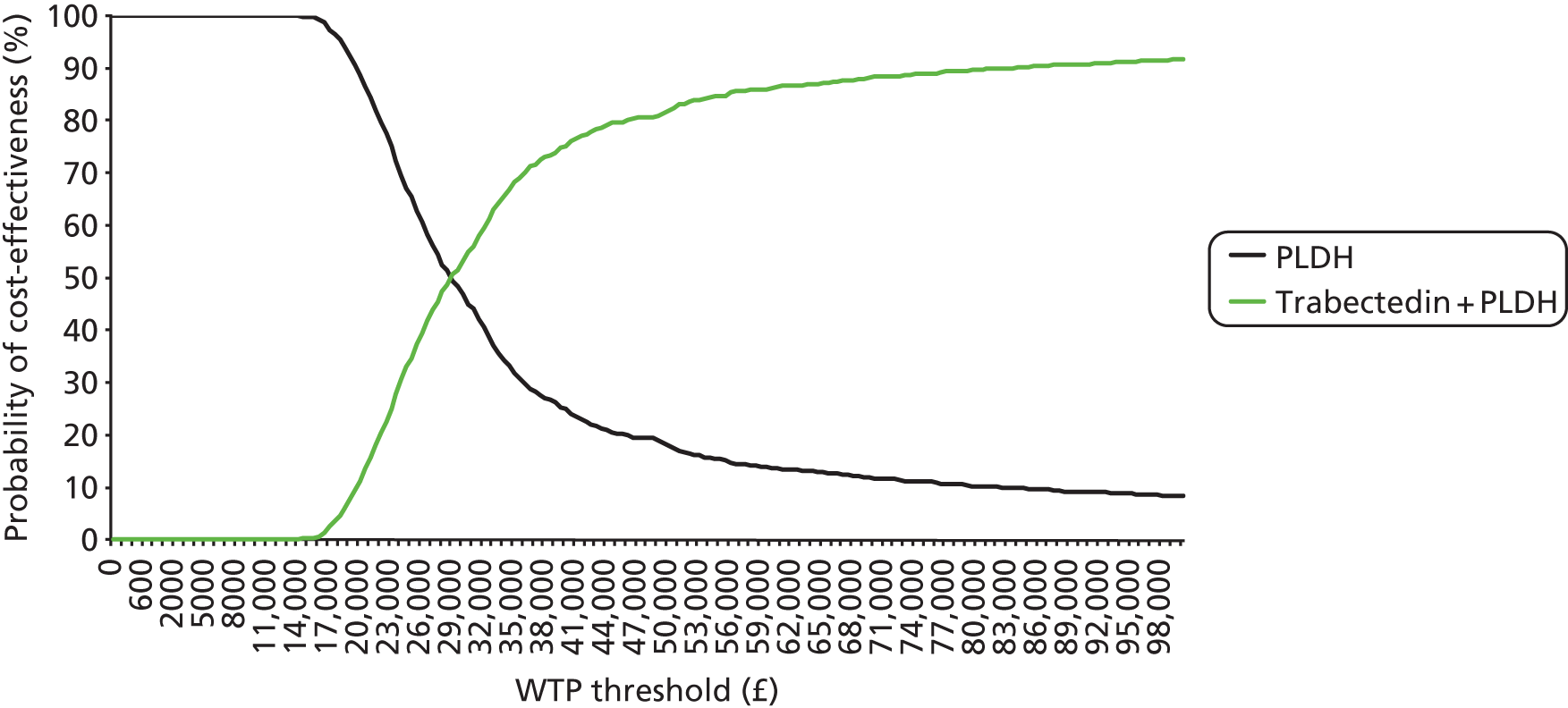
The TAG considers that the sensitivity analyses presented by the manufacturer identified estimates of OS as the key driver of model results and the main accumulator of QALYs, in particular through changes in the functional form and through controlling for PFI in the extrapolated estimates of OS.
Without the PAS, the manufacturer concluded that ‘the ICER of £39,306 per QALY could be considered cost-effective in the UK setting despite it being above the traditional NICE threshold values of £20,000 to £30,000 per QALY as a consequence of trabectedin plus PLDH being a candidate for end-of-life criteria’ (MS, p. 43). The manufacturer’s rationale for claiming that trabectedin in combination with PLDH is a candidate for end of life is outlined in Table 109. End of life is discussed in greater detail in Chapter 5 (see End-of-life criteria); however, the TAG considers it important to note that although median OS for PLDH was estimated by the manufacturer to be 19.4 months in the platinum-sensitive population (after controlling for PFI and other prognostic factors), mean OS for PLDH was estimated to be 35 months. Therefore, baseline life expectancy, as indicated by mean OS for patients treated with PLDH, is likely to be > 24 months.
| NICE end-of life criteria | Eligibility of trabectedin for consideration under end of life |
|---|---|
| The treatment is indicated for patients with a short life expectancy, normally less than 24 months | Trabectedin plus PLDH is indicated for patients with a life expectancy expected to be of less than 2 years without treatment The final analysis showed that median overall survival in the platinum-sensitive and partially platinum-sensitive populations were 24.1 months and 16.4 months for patients treated with PLDH. Accounting for the imbalance in PFI and other prognostic factors in the platinum-sensitive population reduced the median to 19.4 months |
| There is sufficient evidence to indicate that the treatment offers an extension to life, normally of at least an additional 3 months compared with current NHS treatment | For patients with platinum-sensitive and partially platinum-sensitive relapsed ovarian cancer, median survival (after correction of prognostic factors including PFI) shows an extension in life of 4 months, which is well in excess of the 3 months required. Estimated mean survival suggests that this extension of life could be in excess of 9 months |
| No alternative treatment with comparable benefits is available through the NHS | For the population considered (i.e. patients with relapsed ovarian cancer who are unsuitable to platinum-based compounds and who would otherwise be treated with PLDH) no alternative treatment has shown similar benefits |
| The treatment is licensed or otherwise indicated, for small patient populations | It is estimated that there are 2617 patients with relapsed platinum-sensitive ovarian cancer in England and Wales. In this submission, only relapsed platinum-sensitive ovarian cancer patients who are unsuitable for treatment with platinum-based chemotherapy because of allergy or intolerance due to residual toxicities or because they have partially platinum-sensitive disease will be considered for treatment with trabectedin plus PLDH. It is estimated that approximately 491 patients will fall into this group in 2014 |
| The estimates of the extension to life are robust and can be shown or reasonably inferred from either PFS or OS | Extension of life can be seen by the difference in both median and mean survival when considering OS adjusted for prognostic factors including PFI. Even when the PFI imbalance is not accounted for (which biases OS results in favour of PLDH), the platinum-sensitive population is associated with a 2.9 month survival gain and the partially platinum-sensitive population is associated with a 6 month survival gain |
| The assumptions used in the reference case economic modelling are plausible objective and robust | Adjusting for imbalances in pre-specified prognostic factors that significantly affect OS and PFS reduces bias and has been performed by ERGs previously (NICE TA222) |
Budget impact
The manufacturer submitted a budget impact analysis for trabectedin use. The manufacturer estimated that the total budget impact of introducing trabectedin in combination with PLDH would be £3,284,036 in 2014 (491 patients), increasing to £4,359,077 in 2018 (506 patients). The TAG notes that these costs were based upon the submission without the PAS. The manufacturer did not provide an updated budget impact analysis within the PAS submission. However, the TAG notes that within the submitted budget impact model, estimates with the PAS were presented. With the PAS, the manufacturer estimated a total budget impact of £1,439,204 in 2014 (491 patients), increasing to £1,910,333 in 2018 (506 patients).
The calculations used by the manufacturer to estimate the population are summarised in Table 110. The figures relate to the population for which the manufacturer has requested consideration; i.e. people who are not suitable for, or not best managed with, platinum-based chemotherapy because of an allergy or an intolerance due to residual toxicities; and people with PPS disease (PFI of 6–12 months).
| Population | 2014 | 2018 |
|---|---|---|
| Population England and Wales | 56,839,104 | 58,679,898 |
| Percentage women | 51 | 51 |
| Female population England and Wales | 28,419,552 | 29,339,949 |
| Incidence cases per 100,000 of the population per year | 20.9 | 20.9 |
| Total incident cases per year | 6058 | 6255 |
| Proportion of ovarian cancer that is epithelial (%) | 90 | 90 |
| No. with epithelial ovarian cancer | 5453 | 5629 |
| Proportion of ovarian cancer diagnosed at stages III/IV (%) | 75 | 75 |
| No. of patients with epithelial stage III/IV ovarian cancer | 4089 | 4222 |
| Proportion of ovarian cancer cases that are recurrent (%) | 80 | 80 |
| No. of patients with recurrent epithelial stage III/IV ovarian cancer | 3272 | 3378 |
| Proportion of patients with recurrent stage III/IV ovarian cancer that are platinum-sensitive (%) | 80 | 80 |
| No. of patients with recurrent stage III/IV ovarian cancer that are platinum sensitive | 2617 | 2702 |
| Proportion of platinum-sensitive patients who are PPS (6–12 months) (%) | 30 | 30 |
| Proportion of partially sensitive patients unsuitable for platinum-based therapy (%) | 50 | 50 |
| No. of PPS patients that are unsuitable for platinum-based therapy (6–12 months) | 393 | 405 |
| Proportion of FPS patients (> 12 months) with hypersensitive reactions (%) | 20 | 20 |
| Proportion of FPS patients with severe hypersensitivity reactions (%) | 47 | 47 |
| Proportion of FPS patients with severe allergies who abandon platinum treatment (%) | 25 | 25 |
| No. of FPS patients (> 12 months) unsuitable for treatment with platinum-based therapy due to allergies | 43 | 44 |
| Proportion of FPS patients with occurrence of neuropathy (%) | 20 | 20 |
| Persistent neurological toxicity among FPS patients and intolerant patients to be retreated with platinum at 1 year after the end of therapy (%) | 15 | 15 |
| No. of FPS patients unsuitable for treatment with platinum-based therapy due to intolerance | 55 | 57 |
| Total incidence patients eligible for treatment | 491 | 506 |
The TAG notes that the calculations used to estimate the eligible population were based solely around incident patients. The TAG considers that the budget impact would increase should prevalent patients who experience further relapses and have not previously been treated with trabectedin be included within the calculations. The TAG estimated that based upon an incidence of 6058 patients per year, and a death rate of 4295 (see Chapter 1, Incidence and prevalence), the remaining prevalent patients with ovarian cancer would be 1763. Using the manufacturer’s calculations for the year 2014 results in an estimate of the total number of patients eligible for treatment of 633.
Summary and conclusions of available cost-effectiveness evidence
No single cost-effectiveness analysis considering the full range of interventions and comparators relevant for this MTA was identified in the TAG systematic review (see Technology Assessment Group systematic review of existing cost-effectiveness evidence, above). The existing published UK cost-effectiveness evidence in recurrent ovarian cancer related largely to TA22215,90,99 and TA91. 10,94,97 In addition, three further studies16,82,95 considering the UK perspective were identified: the manufacturer for TA28516 built a model based upon the model used in TA9113 and TA222,15 and the remaining two studies were cost-minimisation analyses published prior to 2004.
The majority of the published UK evidence, therefore, evaluated the cost-effectiveness of treatments in recurrent ovarian cancer based upon the model developed for TA91. This model comprised three health states: the SD period, the PD period and death. Other recently published cost–utility models in recurrent ovarian cancer also considered similar health states98,101,107 from the perspective of the USA96,99 and Korea. 105
One MS was received that included economic evidence (PharmaMar). The manufacturer used the TA91 model structure, and used Weibull and log-logistic distributions to estimate the mean time spent in each health state (SD, PD and death). Clinical data from a single head-to-head comparison of trabectedin in combination with PLDH compared with PLDH monotherapy (OVA-301) was used to inform the parametric distributions used.
As such, although there exist studies that compare the cost-effectiveness of the treatments relevant to the scope of this MTA, there does not exist a simultaneous comparison of all of the interventions of interest. A de novo decision-analytic model was therefore developed to address this issue, and was based upon the model structure developed within TA91 (see Model structure, below). The model structure developed within TA91 was considered to be the most appropriate for this decision problem. This is because the structure has been widely used within recurrent ovarian cancer, and because the health states within this model capture clinically important aspects relating to the treatment of recurrent ovarian cancer, both extending survival, but also extending the stable, progression-free period.
Independent economic assessment
Overview
As no single published study, or MS, simultaneously compared the cost-effectiveness of treatments relevant to the scope of this MTA, the TAG carried out an independent assessment and developed a de novo economic analysis.
Comparison to scope
The summary of the final scope issued by NICE for this MTA is presented below in Table 111, alongside a commentary detailing to what extent the de novo analysis carried out by the TAG satisfies the scope.
| NICE scope | TAG de novo analysis | |
|---|---|---|
| Interventions | For people with platinum-sensitive ovarian cancer:
|
Yes For people with platinum-sensitive ovarian cancer all interventions of interest were considered; however, owing to the data available from the literature, two independent networks were constructed |
For people with platinum-resistant or platinum-refractory ovarian cancer:
|
Partially Data for paclitaxel in combination with platinum were not available from the literature. Therefore, this intervention was omitted from the base-case analysis |
|
For people who are allergic to platinum-based compounds:
|
Yes Based upon expert clinical advice it was considered that response to therapy was independent of presence or absence of platinum allergy. It was therefore assumed that results from non-platinum-based regimens for the platinum-sensitive and PRR populations were applicable to people allergic to platinum-based compounds |
|
| Population(s) | People with ovarian cancer that has recurred after first-line (or subsequent) platinum-based chemotherapy or is refractory to platinum-based chemotherapy | Yes |
| Comparators | For people with platinum-sensitive ovarian cancer:
|
Yes All comparators of interest were considered; however, owing to the data available from the literature, two independent networks were constructed Bevacizumab in platinum-containing chemotherapy was not recommended for use in TA28516 and therefore was not considered in this analysis |
For people with platinum-resistant or platinum-refractory ovarian cancer:
|
Partially Data for paclitaxel in combination with platinum was not available from the literature. Therefore, this intervention was omitted from the base-case analysis. Data for etoposide alone or in combination with platinum chemotherapy and data for best supportive care were not available. Therefore, these comparators were omitted from the base-case analysis |
|
For people who are allergic to platinum-based compounds:
|
Partially Based upon expert clinical advice, it was considered that response to therapy was independent of presence or absence of platinum allergy. It was therefore assumed that results from non-platinum-based regimens for the platinum-sensitive and PRR populations were applicable to people allergic to platinum-based compounds |
|
| Outcomes | The outcome measures to be considered include:
|
Partially Response rate was not utilised in the economic analysis; this outcome was considered in the clinical review |
| Economic analysis | The reference case stipulates that the cost-effectiveness of treatments should be expressed in terms of incremental cost per QALY The reference case stipulates that the time horizon for estimating clinical effectiveness and cost-effectiveness should be sufficiently long to reflect any differences in costs or outcomes between the technologies being compared Costs will be considered from a NHS and PSS perspective |
Yes |
| Other considerations | If the evidence allows the following subgroups will be considered:
|
Partially Data for people with PPS and FPS disease was not sufficient to carry out a full economic analysis |
Population
The population of interest for this MTA is people with ovarian cancer that has recurred after first-line (or subsequent) platinum-based chemotherapy or is refractory to platinum-based chemotherapy. Specifically, the following subgroups are described:
-
people with platinum-sensitive ovarian cancer (cancer that responds to initial chemotherapy but recurs 6 months or more after completion of the regimen), i.e. PFI ≥ 6 months
-
people with platinum-resistant (cancer that responds to initial chemotherapy but recurs within 6 months after completion of the regimen) and platinum-refractory cancer (cancer does not respond to initial therapy), i.e. PFI of < 6 months
-
people who are allergic to platinum-based compounds.
Following consultation with clinical experts, it was noted that the duration of the PFI is a key prognostic factor. Moreover, it was noted that platinum sensitivity (as indicated by the PFI), is a continuum, rather than a categorical variable. That is, patients’ response to treatment would be expected to gradually decline with decreasing PFI. Furthermore, clinical experts fed back that, in conjunction with factors such as neuropathy and patient preference, the duration of PFI would affect the treatment options considered (see Chapter 2).
Furthermore, the TAG notes that clinical effectiveness data (identified in the TAG’s clinical effectiveness review; see Chapter 3, Results), is presented by categories of platinum sensitivity, most frequently for patients with platinum-sensitive disease (PFI of ≥ 6 months) and patients with PRR disease (PFI of < 6 months).
Therefore, based on expert clinical opinion, and on the data available to inform the analysis, the TAG considers that disaggregation of the results by platinum sensitivity is more clinically relevant than presentation of the results in the full population (i.e. people with platinum-resistant, -refractory or -sensitive disease). Consequently, results from the PRR subgroup and the platinum-sensitive subgroup are presented separately, with no explicit analysis of the full population (see Base-case results, below).
The TAG notes that some data were available for patients with FPS (PFI > 12 months) and PPS (PFI 6–12 months) disease (see Chapter 3, Results). However, these data were insufficient to inform robust cost-effectiveness analysis. Therefore, consideration of the cost-effectiveness of treatments in patients with partially or FPS disease has been considered in sensitivity rather than base-case analysis.
Additionally, the TAG sought clinical advice around expected response to treatment for patients with an allergy to platinum-based compounds compared with those without an allergy. It was noted by clinical experts that response to non-platinum-based therapies would be expected to be consistent between patients with or without an allergy or intolerance to platinum-based therapy. The TAG therefore considers it appropriate to include platinum-allergic patients in the platinum-sensitive and platinum-refractory subgroups. Therefore, a separate analysis of platinum-allergic patients has not been carried out; however, treatment options for platinum-allergic patients are assumed to exclude platinum-based therapies.
Model structure
The model structure used by the TAG, to facilitate a comparison of the cost-effectiveness of the interventions and comparators outlined for this MTA, is derived from the cohort model developed in TA9113 (Figure 25).
The TAG elected to use a cohort model approach rather than individual patient modelling. This approach was considered to be the most appropriate because, with the exception of PFI, there is limited evidence of the effect of individual patient characteristics/history on disease course. Furthermore, data were not available at a sufficiently disaggregated level in order to model at the individual level.
FIGURE 25.
Model structure for the TAG’s de novo economic evaluation.
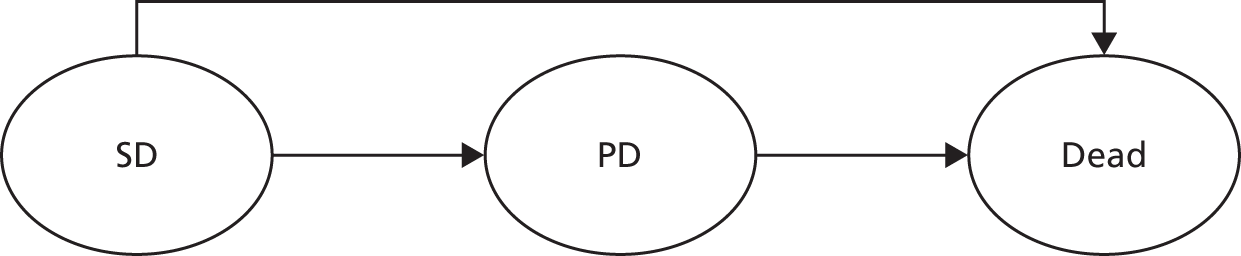
As evidenced from the systematic review of the cost-effectiveness literature carried out by the TAG (see Technology Assessment Group systematic review of existing cost-effectiveness evidence, above), this model structure has previously been used to assess interventions for recurrent ovarian cancer. Moreover, the health states within this model capture clinically important aspects relating to the treatment of recurrent ovarian cancer – both extending survival, but also extending the stable, progression-free period. In TA285, a recent STA considering bevacizumab use in recurrent advanced ovarian cancer, the importance of PFS was highlighted. 16 Specifically, patient experts stated that ‘increasing PFS gives additional time to deal with the physical, emotional and psychological effects of ovarian cancer and its treatment, and allows patients and their families to come to terms with the implications of relapse’ and that ‘this additional period of time is extremely important in helping them to recover from the shock of relapse, and enables them to use the period of wellbeing to make the most of their lives’. The model structure developed within TA91,13 which incorporated both overall and PFS, was therefore considered to be the most appropriate for this decision problem.
Within TA91,13 the MS for TA222,93 and the submission from PharmaMar for this MTA, the time spent in each health state is based upon the estimated mean TTP (time spent in the SD health state) and mean time to death (time spent in the PD health state, after subtracting time spent in the SD health state). For this MTA, a similar methodology (the partition method) has been used to estimate the proportion of patients in each health state; however, full survival curves, rather than mean estimates, have been derived from the clinical data for each therapy. This ensures that time is appropriately captured within the economic model, and facilitates the assignment of costs, utilities and discounting. As highlighted by the ERG for TA222, models constructed around mean time estimates may be constrained in the application of costs, utilities and discounting. 90
To capture the full costs and benefits associated with therapies for recurrent ovarian cancer, a lifetime time horizon was considered to be appropriate. In the base-case analysis this is set as 15 years, because at this time point, over 99.9% of patients within the model have died. Furthermore, as per the NICE reference case, costs and benefits are discounted at a rate of 3.5% per annum, and a NHS and Personal Social Services (PSS) perspective was considered. 109 The time horizon and discount rates used have been varied in sensitivity analysis (see Approach to uncertainty, below).
Interventions and comparators
The interventions and comparators of interest for this MTA, for both the platinum-sensitive and PRR subgroups outlined above (see Population), are presented in Table 112. In addition, the treatment options for patients considered platinum allergic are presented.
| Interventions/comparators | Patient population | ||
|---|---|---|---|
| Platinum sensitive | PRR | Platinum allergic | |
| Interventions | |||
| Paclitaxel plus platinum | ✓ | ✓ | NA |
| PLDH plus platinum | ✓ | NA | NA |
| Gemcitabine plus carboplatin | ✓ | NA | NA |
| PLDH | ✓ | ✓ | ✓ |
| Paclitaxel | ✓ | ✓ | ✓ |
| Trabectedin plus PLDH | ✓ | NA | ✓ |
| Topotecan | ✓ | ✓ | ✓ |
| Comparators | |||
| Platinum | ✓ | NA | NA |
| Etoposide | NA | ✓ | ✓ |
| Etoposide plus platinum | NA | ✓ | NA |
| Best supportive care | NA | ✓ | ✓ |
In order to assess the relative cost-effectiveness of these therapies, relative clinical effectiveness data, with respect to PFS and OS, were required. However, as reported in Chapter 3 a paucity of comparative clinical data was identified in the clinical systematic review, therefore, the full range of desirable comparisons, as outlined in Table 112, is not possible. Instead, Table 113 summarises, by population, the comparisons that are possible based on the availability of relative clinical data.
| Therapy | Paclitaxel plus platinum | PLDH plus platinum | Gemcitabine plus carboplatin | PLDH | Paclitaxel | Trabectedin plus PLDH | Topotecan | Platinum | Etoposide | Etoposide plus platinum | Best supportive care |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Platinum sensitive | |||||||||||
| Paclitaxel plus platinum | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✓ | ||||
| PLDH plus platinum | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✓ | ||||
| Gemcitabine plus carboplatin | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✓ | ||||
| PLDH | ✗ | ✗ | ✗ | ✓ | ✓ | ✓ | ✗ | ||||
| Paclitaxel | ✗ | ✗ | ✗ | ✓ | ✓ | ✓ | ✗ | ||||
| Trabectedin plus PLDH | ✗ | ✗ | ✗ | ✓ | ✓ | ✓ | ✗ | ||||
| Topotecan | ✗ | ✗ | ✗ | ✓ | ✓ | ✓ | ✗ | ||||
| PRR | |||||||||||
| Paclitaxel | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | |||||
| Paclitaxel plus platinum | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| PLDH | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | |||||
| Topotecan | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | |||||
| Platinum allergic | |||||||||||
| Paclitaxel | ✓ | ✓ | ✓ | ✗ | ✗ | ||||||
| PLDH | ✓ | ✓ | ✓ | ✗ | ✗ | ||||||
| Trabectedin plus PLDH | ✓ | ✓ | ✓ | ✗ | ✗ | ||||||
| Topotecan | ✓ | ✓ | ✓ | ✗ | ✗ | ||||||
For patients with platinum-sensitive disease, clinical data were retrieved for each of the interventions and comparators outlined within Table 112. However, as a result of the trials available (see Table 113), it was not possible to construct a single complete network comparing all interventions with all comparisons and with one another. Instead, two separate, disconnected networks formed the basis of the clinical analysis in the platinum-sensitive subgroup:
-
Network 1 comprises paclitaxel plus platinum, PLDH plus platinum, gemcitabine plus carboplatin, and platinum (hereafter referred to as ‘PS network 1’)
-
Network 2 comprises PLDH, paclitaxel, PLDH plus trabectedin and topotecan (hereafter referred to as ‘PS network 2’).
The use of two distinct networks to inform the relative clinical effectiveness of treatments for patients with platinum-sensitive disease necessitated the disaggregation of the economic analysis in this patient population. Therefore, the incremental cost-effectiveness of treatments in PS network 1 is considered separate to the incremental cost-effectiveness of treatments in PS network 2. The TAG notes that the ICERs estimated from these two networks are not comparable with each other and should be interpreted as independent analyses. This is discussed in more detail below (see Discussion).
For the PRR subgroup, clinical effectiveness data were available for PLDH, paclitaxel, paclitaxel plus platinum, and topotecan. However, the comparisons available (see Table 113) resulted in a network limited to PLDH, paclitaxel, and topotecan. No data were found for etoposide, either as monotherapy or in combination with a platinum agent, and no data regarding best supportive care were identified. However, following clinical advice that the prognosis of patients with PRR disease is often poor across available treatment options, a sensitivity analysis assuming equivalent efficacy between all treatments was carried out. The results of this sensitivity analysis are presented below (see Results of the sensitivity analysis).
For each of the interventions and comparators investigated within the de novo economic analysis, the treatment regimens modelled are those most representative of UK clinical practice (Table 114). Specification of the treatment regimens has been obtained through review of each relevant SmPC, followed by clinical expert verification to ensure accurate reflection of UK clinical practice. Etoposide is not licensed within ovarian cancer and therefore expert advice was sought to inform the doses used within the analysis. The TAG notes that the expert advice indicated that there is variation in clinical practice with regards to etoposide regimens used. However, as consideration of treatment with etoposide is limited to sensitivity analysis, the TAG does not expect this uncertainty to impact on the base-case cost-effectiveness results.
| Chemotherapy | Regimen description |
|---|---|
| Paclitaxel | For PRR disease: paclitaxel 80 mg/m2 weekly for 18 weeks or until progression For platinum-sensitive disease: paclitaxel 175 mg/m2 day 1 every 21-day cycle (maximum six cycles) |
| Paclitaxel plus platinum | For PRR disease:a paclitaxel 80 mg/m2 plus carboplatin AUC 3, weekly for 18 weeks or until progression For platinum-sensitive disease: paclitaxel 175 mg/m2 and carboplatin AUC 5, day 1 every 21-day cycle (maximum six cycles) |
| PLDH | 40 mg/m2 day 1 every 28-day cycle (maximum six cycles) |
| PLDH plus platinum | PLDH 30 mg/m2; carboplatin target AUC of 5, day 1 every 28-day cycle (maximum six cycles) |
| Gemcitabine plus carboplatin | Gemcitabine 1000 mg/m2, day 1 and 8, every 21-day cycle; carboplatin target AUC of 4, day 1 every 21-day cycle (maximum six cycles) |
| Trabectedin plus PLDH | Trabectedin 1.1 mg/m2; PLDH 30 mg/m2, day 1 every 21-day cycle (maximum six cycles) |
| Topotecan | 1.5 mg/m2, days 1–5 every 21-day cycle (maximum six cycles) |
| Platinum monotherapy | Carboplatin target AUC of 5, day 1 every 21-day cycle (maximum six cycles) |
| Etoposide a | 50 mg (oral) days 1–21 every 28 days (maximum six cycles) |
| Etoposide plus platinum a | Etoposide 50 mg (oral) days 1–21 every 28 days plus cisplatin i.v. 50 mg/m 2 day 1, 8 and 15 every 28 days (maximum six cycles) |
| Best supportive care a | No chemotherapeutic regimen modelled; interventions associated with supporting patients, for example in their control of pain, nausea, vomiting or constipation |
Overview of model parameters, sources and assumptions
See Tables 115 and 116.
| Parameter | Mean value | Variance | Source | Section | ||
|---|---|---|---|---|---|---|
| PFS (PS network 1) | ||||||
| PFS distribution used for the baseline treatment, paclitaxel plus platinum | Weibull distribution: | Cholesky matrix | (Intercept) | Log(scale) | Analysis of CALYPSO data31 using methods outlined in Hoyle and Henley113 | Treatment effectiveness |
| Intercept = 2.546 | (Intercept) | 0.026 | 0.000 | |||
| Log_scale = –0.656 | Log(scale) | −0.006 | 0.038 | |||
| HR for PLDH plus platinum vs. paclitaxel plus platinum | 0.817 | 95% CrI 0.717 to 0.927 | TAG NMA | |||
| HR for gemcitabine plus carboplatin vs. paclitaxel plus platinum | 0.985 | 95% CrI 0.748 to 1.273 | TAG NMA | |||
| HR for platinum vs. paclitaxel plus platinum | 1.361 | 95% CrI 1.182 to 1.559 | TAG NMA | |||
| PFS (PS network 2) | ||||||
| PFS distribution used for the baseline treatment, PLDH | Kaplan–Meier data | NA | MS | Treatment effectiveness | ||
| HR for paclitaxel vs. PLDH | 1.615 | 95% CrI 0.939 to 2.586 | TAG NMA | |||
| HR for trabectedin plus PLDH vs. PLDH | 0.736 | 95% CrI 0.560 to 0.949 | TAG NMA | |||
| HR for topotecan vs. PLDH | 1.298 | 95% CrI 0.979 to 1.688 | TAG NMA | |||
| PFS (PRR) | ||||||
| PFS distribution used for the baseline treatment, PLDH | Weibull distribution: | Cholesky matrix | (Intercept) | Log(scale) | Analysis of OVA-301 data30 using methods outlined in Hoyle and Henley113 | Treatment effectiveness |
| Intercept = 1.665 | (Intercept) | 0.081 | 0.000 | |||
| Log_scale = –0.345 | Log(scale) | −0.013 | 0.080 | |||
| HR for paclitaxel vs. PLDH | 1.360 | 95% CrI 0.817 to 2.123 | TAG NMA | |||
| HR for topotecan vs. PLDH | 0.998 | 95% CrI 0.767 to 1.277 | TAG NMA | |||
| OS (PS network 1) | ||||||
| OS distribution used for the baseline treatment, paclitaxel plus platinum | Weibull distribution: | Cholesky matrix | (Intercept) | Log(scale) | Analysis of CALYPSO data56 using methods outlined in Hoyle and Henley113 | Treatment effectiveness |
| Intercept = 3.750 | (Intercept) | 0.032 | 0.000 | |||
| Log_scale = –0.534 | Log(scale) | 0.004 | 0.046 | |||
| HR for PLDH plus platinum vs. paclitaxel plus platinum | 1.023 | 95% CrI 0.889 to 1.172 | TAG NMA | |||
| HR for gemcitabine plus carboplatin vs. paclitaxel plus platinum | 1.247 | 95% CrI 0.921 to 1.652 | TAG NMA | |||
| HR for platinum vs. paclitaxel plus platinum | 1.290 | 95% CrI 1.096 to 1.509 | TAG NMA | |||
| OS (PS network 2) | ||||||
| OS distribution used for the baseline treatment, PLDH | Weibull distribution: | Cholesky matrix | (Intercept) | Log(scale) | Analysis of manufacturer Kaplan–Meier data using methods outlined in Hoyle and Henley113 | Treatment effectiveness |
| Intercept = 3.449 | (Intercept) | 0.057 | 0.000 | |||
| Log_scale = –0.304 | Log(scale) | –0.008 | 0.066 | |||
| HR for paclitaxel vs. PLDH | 1.219 | 95% CrI 0.850 to 1.690 | TAG NMA | |||
| HR for trabectedin plus PLDH vs. PLDH | 0.835 | 95% CrI 0.667 to 1.032 | TAG NMA | |||
| HR for topotecan vs. PLDH | 1.367 | 95% CrI 1.035 to 1.770 | TAG NMA | |||
| OS (PRR) | ||||||
| OS distribution used for the baseline treatment, PLDH | Weibull distribution | (CIC data removed) | Analysis of manufacturer CSR data using methods outlined in Hoyle and Henley113 | Treatment effectiveness | ||
| HR for paclitaxel vs. PLDH | 1.053 | 95% CrI 0.783 to 1.382 | TAG NMA | |||
| HR for topotecan vs. PLDH | 0.973 | 95% CrI 0.764 to 1.221 | TAG NMA | |||
| Probability of allergic reaction (%) | ||||||
| Paclitaxel | 20.0 | Estimated 95% CI 11% to 31% | Clinical opinion | Adverse event incidence | ||
| Paclitaxel plus platinum | 3.9 | Estimated 95% CI 2.2% to 6.1% | Weighted average of Bafaloukos et al.29 (one event, 89 patients) and Gonzalez-Martin et al.48 (four events, 38 patients) | |||
| PLDH | 5.0 | Estimated 95% CI 3% to 8% | Clinical opinion | |||
| PLDH plus platinum | 0.5 | Estimate based upon the OR vs. paclitaxel plus platinum (OR 0.130, 95% CrI 0.001 to 0.705) | TAG NMA | |||
| Gemcitabine plus carboplatin | 3.9 | Set equal to paclitaxel plus platinum | TAG NMA | |||
| Trabectedin plus PLDH | 5.0 | Estimated 95% CI 3% to 8% | Clinical opinion | |||
| Topotecan | 0.0 | NA | Clinical opinion | |||
| Platinum | 3.9 | Set equal to paclitaxel plus platinum | TAG NMA | |||
| Probability of anaemia (%) | ||||||
| Paclitaxel | 4.7 | Set equal to PLDH | TAG NMA | Adverse event incidence | ||
| Paclitaxel plus platinum | 5.1 | Estimated 95% CI 2.9% to 7.9% | Weighted average of Bafaloukos et al.29 (three events, 89 patients), Gonzalez-Martin et al.48 (two events, 38 patients), and CALYPSO data31 (27 events, 501 patients) | |||
| PLDH | 4.7 | Estimated 95% CI 2.7% to 7.3% | Weighted average of Schering-Plough submission (30–57 trial) from TA9113 (three events, 108 patients), Gordon et al.49 (13 events, 239 patients) and OVA-301 data30 (16 events, 330 patients) | |||
| PLDH plus platinum | 9.4 | Estimate based upon the OR vs. paclitaxel plus platinum (OR 1.926, 95% CrI 1.164 to 3.039) | TAG NMA | |||
| Gemcitabine plus carboplatin | 23.9 | Estimate based upon the OR vs. paclitaxel plus platinum (OR 5.848, 95% CrI 1.158 to 18.040) | TAG NMA | |||
| Trabectedin plus PLDH | 12.7 | Estimate based upon the OR vs. PLDH (OR 2.940, 95% CrI 1.559 to 5.202) | TAG NMA | |||
| Topotecan | 26.8 | Estimate based upon the OR vs. PLDH (OR 7.374, 95% CrI 3.775 to 13.590) | TAG NMA | |||
| Platinum | 5.1 | Set equal to paclitaxel plus platinum | TAG NMA | |||
| Probability of febrile neutropenia (%) | ||||||
| Paclitaxel | 5.0 | Estimated 95% CI 2.8% to 7.7% | Clinical opinion | Adverse event incidence | ||
| Paclitaxel plus platinum | 4.2 | Estimated 95% CI 2.4% to 6.5% | CALYPSO data31 (21 events, 501 patients) | |||
| PLDH | 2.1 | Estimated 95% CI 1.2% to 3.3% | OVA-301 data30 (seven events, 330 patients) | |||
| PLDH plus platinum | 4.2 | Set equal to paclitaxel plus platinum | TAG NMA | |||
| Gemcitabine plus carboplatin | 4.2 | Set equal to paclitaxel plus platinum | Clinical opinion | |||
| Trabectedin plus PLDH | 6.6 | Estimate based upon the OR vs. PLDH (OR 3.256, 95% CrI 1.378 to 7.692) | TAG NMA | |||
| Topotecan | 5.0 | Estimated 95% CI 2.8% to 7.7% | Clinical opinion | |||
| Platinum | 0.0 | NA | Clinical opinion | |||
| Probability of nausea and vomiting (%) | ||||||
| Paclitaxel | 2.9 | Estimate based upon the OR vs. PLDH (OR 0.279, 95% CrI 0.120 to 0.535) | TAG NMA | Adverse event incidence | ||
| Paclitaxel plus platinum | 1.6 | Estimated 95% CI 0.9% to 2.4% | Weighted average of Bafaloukos et al.29 (one event, 89 patients) and Gonzalez-Martin et al.48 (one event, 38 patients) | |||
| PLDH | 9.8 | Estimated 95% CI 5.5% to 15.0% | Weighted average of OVA-301 data30 (15 events, 330 patients), Schering-Plough submission (30–57 trial) from TA9113 (19 events, 108 patients) and Gordon et al.49 (32 events, 239 patients) | |||
| PLDH plus platinum | 3.2 | Estimate based upon the OR vs. paclitaxel plus platinum (OR 2.055, 95% CrI 1.598, 2.608) | TAG NMA, based upon all grades AEs (see Adverse event incidence) | |||
| Gemcitabine plus carboplatin | 3.2 | Set equal to PLDH plus platinum | Clinical opinion | |||
| Trabectedin plus PLDH | 36.4 | Estimate based upon the OR vs. PLDH (OR 5.291, 95% CrI 2.866 to 9.342) | TAG NMA | |||
| Topotecan | 9.8 | Set equal to PLDH | TAG NMA | |||
| Platinum | 1.6 | Set equal to paclitaxel plus platinum | TAG NMA | |||
| Chemotherapy cost per cycle (£) | ||||||
| Paclitaxel 80mg/m2 weekly (cycle) for 18 weeks or until progression (plus dexamethasone pre-treatment) | 306 | Paclitaxel mean £302, se £2.03 | Standard error around the mean estimates for the IPD from Sacco et al.114 | Costs | ||
| Paclitaxel 175 mg/m2 day 1 every 21-day cycle (plus dexamethasone pre-treatment) | 638 | Paclitaxel mean £634, se £3.87 | ||||
| Paclitaxel 80mg/m2 plus carboplatin AUC 3, weekly for 18 weeks or until progression (plus dexamethasone pre-treatment) | 442 | Paclitaxel mean £302, se £2.03 Carboplatin mean £136, se £1.59 |
||||
| Paclitaxel 175 mg/m2 and carboplatin AUC 5, day 1 every 21-day cycle | 855 | Paclitaxel mean £634, se £3.87 Carboplatin mean £217, se £2.63 |
||||
| PLDH 40 mg/m2 day 1 every 28-day cycle | 1211 | se £9.62 | ||||
| PLDH 30 mg/m2; carboplatin target AUC 5, day 1 every 28-day cycle | 1137 | PLDH mean £920, se £9.95 Carboplatin mean £217, se £2.63 |
||||
| Gemcitabine 1000 mg/m2 day 1 and 8 every 21-day cycle, carboplatin target AUC 4 day 1 every 21-day cycle | 706 | Gemcitabine mean £265, se £1.68 Carboplatin mean £177, se £2.07 |
||||
| Trabectedin 1.1 mg/m2; PLDH 30 mg/m2, day 1 every 21-day cycle | 3679 | Trabectedin mean £2759, se £15.40 PLDH mean £920, se £9.95 |
||||
| Topotecan 1.5 mg/m2, day 1–5 every 21 days | 1305 | Topotecan mean £261, se £0.38 | ||||
| Carboplatin target AUC 5, day 1 every 21 days | 217 | Carboplatin mean £217, se £2.63 | ||||
| Administration cost | ||||||
| Minutes pharmacy preparation required per single chemotherapy agent | 20 minutes | Estimated 95% CI 10.2 to 29.8 minutes | Clinical opinion | Costs | ||
| Cost per hour of pharmacist time | £47 | Estimated 95% CI £26.86 to £72.67 | Unit Costs of Health and Social Care 2012115 | |||
| Deliver complex chemotherapy, including prolonged infusional treatment at first attendance (SB14Z) | £331 | Estimated 95% CI £230 to £388 | NHS Reference Costs 2011/12111 | |||
| Deliver more complex parenteral chemotherapy at first attendance (SB13Z) | £249 | Estimated 95% CI £177 to £301 | NHS Reference Costs 2011/12111 | |||
| Deliver simple parenteral chemotherapy at first attendance (SB12Z) | £200 | Estimated 95% CI £128 to £241 | NHS Reference Costs 2011/12111 | |||
| Deliver subsequent elements of a chemotherapy cycle (SB15Z) | £270 | Estimated 95% CI £192 to £326 | NHS Reference Costs 2011/12111 | |||
| Health-state cost | ||||||
| Cost of outpatient visit; gynaecologic oncology | £135 | Estimated 95% CI £91 to £172 | NHS Reference Costs 2011/12111 | Costs | ||
| One off cost of CT scan | £109 | Estimated 95% CI £93 to £127 | NHS Reference Costs 2011/12111 | |||
| 2000–1 estimate of palliative care cost, 399 days | £4789 | Estimated 95% CI £4277 to £5301 | Guest et al.112 | |||
| Proportion of single-agent carboplatin AUC5 for platinum-sensitive disease | 75% | sd 18.9% | Assumption | |||
| No. of months between outpatient visits | 3 months | Estimated 95% CI 1.53 to 4.47 months | Clinical opinion | |||
| AE cost (£) | ||||||
| SA01F plastic anaemia without CC | 1077 | Estimated 95% CI £661 to £1318 | NHS Reference Costs 2011/12111 | Costs | ||
| SA13A Single Plasma Exchange, Leucophoresis or Red Cell Exchange | 473 | Estimated 95% CI £300 to £586 | NHS Reference Costs 2011/12111 | |||
| SA14Z Plasma Exchanges 2 to 9 | 2479 | Estimated 95% CI £1184 to £3315 | NHS Reference Costs 2011/12111 | |||
| SA15Z Plasma Exchanges 10 to 19 | 5520 | Estimated 95% CI £2007 to £5959 | NHS Reference Costs 2011/12111 | |||
| SA16Z Plasma Exchanges 20 or more | 13,187 | Estimated 95% CI £4147 to £12,524 | NHS Reference Costs 2011/12111 | |||
| Utilities | ||||||
| SD | 0.718 | 95% CI 0.70 to 0.74 | TA22215 | Health-related quality-of-life data | ||
| PD | 0.649 | 95% CI 0.61 to 0.69 | TA22215 | |||
| Other | ||||||
| Time horizon | 15 years | NA | Assumption | Model structure | ||
| Discount rate (costs) | 3.5% | NA | Assumption | |||
| Discount rate (benefits) | 3.5% | NA | Assumption | |||
| Assumption | Rationale | Relevant section | Related sensitivity analyses |
|---|---|---|---|
| The time horizon was assumed to be 15 years in the base case | Fifteen years was considered to be of sufficient duration to capture the differences in costs and QALYs for the majority of women with recurrent ovarian cancer over their lifetime | Model structure | Time horizon varied in one-way sensitivity analysis |
| Carboplatin, rather than cisplatin, was assumed to constitute the platinum therapy of choice in UK clinical practice | The majority of the clinical data obtained for platinum was carboplatin (see Treatment effectiveness); in addition, carboplatin and cisplatin are considered in practice to have equivalent efficacy (see Treatment effectiveness),116 but carboplatin is associated with less toxicity and therefore may be considered the first choice of platinum therapy in clinical practice | Interventions and comparators; Treatment of effectiveness | NA |
| The efficacy of PLDH was assumed to be the same at a dose of 40 mg/m2 as at a dose of 50 mg/m2, as seen in clinical trials | The licensed indication for PLDH monotherapy is presented as 50 mg/m2 and clinical data used within the model for PLDH monotherapy was at this dose; however, clinical advice suggested that a dose of 40 mg/m2 was more likely in clinical practice for reasons of tolerability, and this was not anticipated to affect efficacy | Interventions and comparators; treatment of effectiveness | A scenario analysis was carried out in which the cost of PLDH was associated with a 50-mg/m2 dose rather than a 40-mg/m2 dose |
| For PRR patients, it was assumed that the efficacy estimates for 3-weekly paclitaxel were representative of efficacy from weekly paclitaxel | Clinical advice indicated that for PRR patients, paclitaxel was more likely to be administered via a weekly regimen rather than a 3-weekly regimen. No clinical data were found for weekly paclitaxel that could be included in the PFS and OS networks. This lack of data therefore necessitated an assumption of equivalent efficacy The TAG understands from clinical experts that this assumption is likely to result in an underestimate of the efficacy of weekly paclitaxel |
Interventions and comparators; treatment of effectiveness | OS and PFS was varied in one-way sensitivity analysis |
| For sensitivity analysis, etoposide was assumed to be administered as a flat dose of 50–75 mg days 1–21 out of 28 days, with oral etoposide for a further 7 weeks | Dose based upon clinical advice and used for costing in sensitivity analysis only. Etoposide does not have a licensed indication for ovarian cancer. For that reason, the SmPC did not provide sufficient information around dosing in recurrent ovarian cancer | Approach to uncertainty | This assumption is related to a specific sensitivity analysis |
| No vial sharing | It was assumed in the base case that chemotherapy vials were not shared in clinical practice | Costs | A scenario analysis was carried out whereby this assumption was relaxed and vial sharing was possible |
| It was assumed that every chemotherapy would require 20 minutes’ pharmacist preparation | Based upon clinical advice received for bevacizumab in recurrent ovarian cancer (TA28516) | Costs | Varied in one-way sensitivity analysis, and PSA |
| In the stable period, it was assumed that a patient would require a single outpatient visit every 3 months | Based upon clinical advice | Costs | Varied in one-way sensitivity analysis, and PSA |
| It was assumed that 100% of platinum-sensitive patients entering the model would receive one further line of therapy upon progression of their disease | This was a simplifying assumption designed to reflect the fact that although not all women will go on to receive another line of chemotherapy; some women will receive more than one line of chemotherapy | Costs | The cost within the PD health state was varied in one-way sensitivity analysis and PSA |
| It was assumed that for those women going on to receive a further line of therapy, 75% would receive single-agent carboplatin and 25% would receive PLDH monotherapy | This was a simplifying assumption based upon the proportions of patients receiving platinum-based and non-platinum-based therapy upon progression in Kaye et al.66 | Costs | This probability was varied in one-way sensitivity analysis and PSA |
| Assumption of proportional treatment hazards | The TAG did not have access to either a single clinical trial or patient-level data for the full range of interventions and comparators of interest for this MTA. For that reason, summary HRs were used to estimate the relative effects between treatments considered within the economic analysis and this necessitated the assumption of proportional hazards | Treatment of effectiveness | The appropriateness of the assumption of proportional hazards was investigated using log-cumulative hazard plots and is discussed below (see Discussion) |
| The likelihood of an adverse reaction was independent of the PFI | To increase the available data, the TAG analysed AEs without distinction between platinum-sensitive and platinum-resistant disease | Adverse event incidence | The probability of AEs was varied in sensitivity analysis |
| AEs occurred in the first month of the model | A simplifying assumption reflecting the likelihood that AEs would be experienced upon commencement of chemotherapy | Adverse event incidence | NA |
Treatment effectiveness
Throughout the 15-year time horizon of the TAG’s economic model, monthly estimates of PFS and OS are used to capture the effectiveness of treatments for recurrent ovarian cancer. PFS represents the length of time spent within the SD health state, and OS represents the length of time spent alive within the model in total. The length of time spent alive in the PD health state is calculated as OS minus PFS.
For each treatment, estimates of PFS and OS have been derived and applied in the model as follows (each step is described in more detail in the sections that follow):
-
Networks of treatments, by subgroup, for which PFS and OS data were available, were established via the clinical systematic review (see Chapter 3, Results).
-
For each network, a baseline treatment has been selected and monthly estimates of PFS and OS obtained from Kaplan–Meier data. Where required, parametric survival distributions have been fitted to Kaplan–Meier data, to allow extrapolation beyond the trial duration.
-
In each network, relative estimates of PFS and OS for each therapy have been synthesised in NMA, using HRs as the measure of relative effect compared with the baseline treatment (see Chapter 3, Results).
-
HRs obtained from the NMAs are applied to baseline estimates of PFS and OS. Thus providing, for every therapy in each network, monthly estimates of PFS and OS, and therefore the proportion of patients within each health state.
Establishing networks of treatments
No single trial comparing all relevant treatments, for either the platinum-sensitive or PRR subgroup, was identified from the clinical literature review. It was therefore necessary to assess which trials could be linked via a network, in order to establish the relative efficacy of treatments using NMA (see Chapter 3, Results).
For the platinum-sensitive subgroup, two independent networks have been constructed (Figures 26 and 27). Collectively, these two networks contain information on every intervention and comparator outlined within the NICE scope for platinum-sensitive disease;38 however, the absence of a common comparator between these two networks necessitated the separate analysis of these networks. For the PRR subgroup, a single network has been identified (Figure 28); however, this network contains no information for one of the interventions (paclitaxel plus platinum) and three of the comparators (etoposide monotherapy, etoposide plus platinum, best supportive care) specified in the NICE scope. 38
FIGURE 26.
Network diagram for the platinum-sensitive subgroup (network 1).

FIGURE 27.
Network diagram for the platinum-sensitive subgroup (network 2).
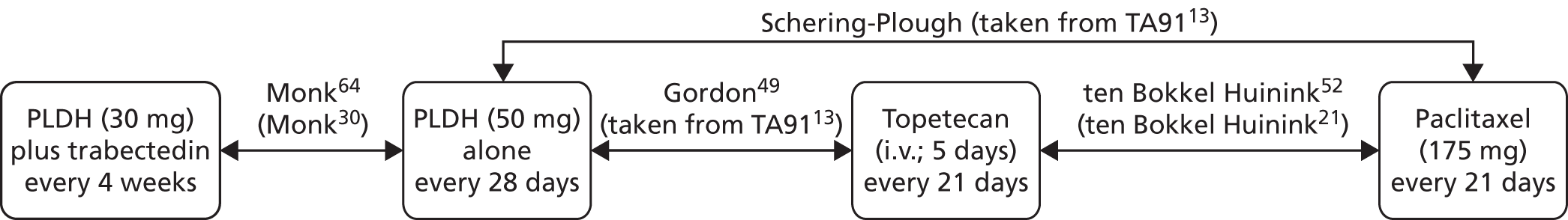
FIGURE 28.
Network diagram for the PRR subgroup.
Establishing baseline progression-free survival and overall survival for each network
For each network (PS network 1, PS network 2, and PRR), the proportions of patients with PFS and OS were estimated monthly, over a lifetime time horizon (15 years in the base case), for the baseline treatment.
To estimate baseline PFS and OS, the TAG used submitted Kaplan–Meier data or published Kaplan–Meier plots. Published Kaplan–Meier plots were digitised using an online digitising tool, WebPlotDigitzer117 (version 2; Ankit Rohatgi, Austin, TX, USA) and the underlying Kaplan–Meier data estimated using methods described in Hoyle and Henley. 113 Hoyle and Henley113 present an algorithm, informed by the Kaplan–Meier plot and the numbers of patients at risk at given time points, which can be used to estimate the underlying Kaplan–Meier data. Where required (e.g. where at the end of follow-up some patients remained at risk), parametric survival curves may then be fitted to the estimated Kaplan–Meier data using maximum likelihood estimation (MLE).
For each baseline treatment requiring extrapolation of estimated Kaplan–Meier data, Weibull, exponential, log-normal and log-logistic survival curves were fitted using methods of MLE described in Hoyle and Henley. 113 The fit of each survival distribution to the (estimated or actual) Kaplan–Meier data was assessed visually and using the AIC; the distribution chosen to inform the base-case analysis is varied in sensitivity analysis (see Results of the sensitivity analysis, below). Details of the distributions selected to inform baseline PFS and OS in each network are presented below.
Platinum-sensitive network 1 (baseline treatment: paclitaxel plus carboplatin)
For PS network 1, paclitaxel plus carboplatin has been selected as the baseline treatment as a result of the quality of information available for this intervention (Kaplan–Meier plots and numbers of patients at risk data available). Furthermore, paclitaxel plus carboplatin is connected to more than one of the therapies considered within PS network 1 (Figure 29; green square indicates baseline treatment). For paclitaxel plus platinum, three sources of published survival data are available for PFS (CALYPSO from Pujade-Lauraine et al. ,31 Gonzalez-Martin et al. ,48 ICON4/AGO-OVAR 2.261) and OS (CALYPSO from Wagner et al. ,56 Gonzalez-Martin et al. ,48 ICON4/AGO-OVAR 2.261). However, no complete PFS or OS data (i.e. no patients remaining at risk at the end-of-trial follow-up) exist for patients treated with paclitaxel plus carboplatin; therefore, parametric extrapolation has been used.
FIGURE 29.
Network diagram for the platinum-sensitive subgroup (network 1); green square indicates base-line treatment.
For PFS, data (Kaplan–Meier plots and numbers of patients at risk) presented for CALYPSO from Pujade-Lauraine et al. 31 are used to inform PFS for paclitaxel plus carboplatin in the base-case analysis. Pujade-Lauraine et al. 31 was chosen to inform the base-case analysis because of the quality of data presented, study date and purity of comparison made. That is, Pujade-Lauraine et al. 31 provides the number of patients at risk at different time points, required in order to use the methods described in Hoyle and Henley;113 this information is not presented in Gonzalez-Martin et al. 48 Furthermore, although Parmar et al. 61 present PFS data for ICON4/AGO-OVAR 2.2 with sufficient information to allow estimation of Kaplan–Meier data (as described by Hoyle and Henley113), the year of analysis for Pujade-Lauraine et al. 31 is more recent than that of Parmar et al. 61 (2010 vs. 2003, respectively) and therefore more likely to reflect current clinical practice. In addition, a proportion of patients considered in ICON4/AGO-OVAR 2.2 received paclitaxel in combination with cisplatin, rather than carboplatin. However, data from ICON4/AGO-OVAR 2.261 are used in sensitivity analysis (see Results of the sensitivity analysis, below).
Of the parametric survival distributions considered to extrapolate Kaplan–Meier PFS data estimated from Pujade-Lauraine et al. 31 (CALYPSO), the log-logistic distribution could be considered to be the best fit based upon the associated AIC value (Table 117). However, the TAG notes that model fit for each distribution is similar, and that while the log-logistic distribution results in the lowest AIC, the range of AIC values is not large. Moreover, the TAG notes that in a technical support document recently published by NICE’s DSU it is stated that the application of a HR to the entire modelled period ‘can be used within proportional hazards models such as the exponential, Gompertz or Weibull but log-logistic and log-normal models are accelerated failure time models and do not produce a single hazard ratio (HR) and thus the proportional hazards assumption does not hold with these models’. 118 In acknowledgement of this, and given the similarity of the AIC values, a Weibull distribution is used to inform the base-case analysis. However, to test the sensitivity of the cost-effectiveness results to the baseline curve selected, log-logistic, log-normal and exponential distributions are used in sensitivity analyses (see Results of the sensitivity analysis, below).
| Selected distribution | AIC |
|---|---|
| Weibull | 2404.564 |
| Exponential | 2618.638 |
| Log-normal | 2388.568 |
| Log-logistic | 2351.657 |
To estimate the monthly probability of PFS for patients receiving treatment with paclitaxel plus carboplatin, the TAG used the following formula (derived from that outlined in the DSU technical support document118):
where t is time in months and the intercept and scale parameters have been estimated using MLE methods described in Hoyle and Henley113 (intercept = 2.546, scale = –0.656).
Figure 30 presents the survival curve, for paclitaxel plus carboplatin, obtained from digitisation of the Kaplan–Meier plot presented in CALYPSO by Pujade-Lauraine et al. 31 compared with the Weibull extrapolation.

For OS, a similar methodology to that used for PFS was used to derive OS estimates for the baseline treatment; paclitaxel plus carboplatin. OS data (Kaplan–Meier plot and numbers of patients at risk) presented in CALYPSO by Wagner et al. 56 are used to inform the base-case analysis. Of the three studies presenting OS data for paclitaxel plus carboplatin, Wagner et al. 56 was chosen to inform the base-case OS distribution because of quality and maturity of data reported, the date of analysis and the purity of the comparison made. That is, the numbers of patients at risk, required for the methods described by Hoyle and Henley,113 although presented in Wagner et al. ,56 are not presented in Gonzalez-Martin et al. 48 Furthermore, data presented in Gonzalez-Martin et al. 48 were immature compared with data presented in Wagner et al. ;56 70% versus 20% of patients remained alive at the end of follow-up, respectively. As before, data from ICON4/AGO-OVAR 2.2 reported by Parmar et al. 61 are used in sensitivity analysis (see Results of the sensitivity analysis, below).
Based upon the AIC values (Table 118) of the distributions fitted to the Kaplan–Meier data estimated from CALYPSO reported by Wagner et al. ,56 the log-logistic distribution could be considered to provide the best fit. However, akin to PFS, the TAG notes that the fit of each considered OS distribution was similar, and that use of the log-logistic distribution may not be appropriate for the application of HRs. For these reasons, a Weibull distribution is used to inform the base-case analysis and the impact of using log-logistic, log-normal and exponential distributions are tested in sensitivity analyses (see Results of the sensitivity analysis, below).
| Selected distribution | AIC |
|---|---|
| Weibull | 2473.965 |
| Exponential | 2581.839 |
| Log-normal | 2475.084 |
| Log-logistic | 2463.795 |
As for PFS, to estimate the monthly probability of OS, for patients receiving treatment with carboplatin plus paclitaxel, the TAG used the following formula:
where t is time in months, and the intercept and scale parameters have been estimated using MLE methods described in Hoyle and Henley113 (intercept = 3.750, scale = –0.534).
Figure 31 presents the survival curve, for paclitaxel plus carboplatin, obtained from digitisation of the Kaplan–Meier plot for CALYPSO presented in Wagner et al. 56 compared with the Weibull extrapolation.
Platinum-sensitive network 2 (baseline treatment: pegylated liposomal doxorubicin hydrochloride)
For PS network 2, PLDH has been selected as the baseline treatment as a result of the quality of data available for this intervention (Figure 32; green square indicates baseline treatment). Furthermore, the TAG notes that relative efficacy (relative to other treatments of interest) data are available to a greater degree for PLDH than for other treatments included in the network. For PLDH, three sources of published survival data are available for PFS (OVA-301 from Monk et al. ,30 Gordon et al. 49 and Trial 30–57, Schering-Plough submitted data within the Assessment Report for TA9113) and OS (OVA-301 from Monk et al. ,64 Gordon et al. 54 and Trial 30–57, Schering-Plough submitted data within the Assessment Report for TA9113). In addition, the MS from PharmaMar and CSR for OVA-301, provide PFS and OS Kaplan–Meier data.
FIGURE 32.
Network diagram for the platinum-sensitive subgroup (network 2); green square indicates baseline treatment.

For PFS, Kaplan–Meier data provided in the MS from PharmaMar are used, in the base-case analysis, to provide monthly estimates of PFS for patients treated with PLDH. These data represent the most up-to-date information on PFS for patients treated with PLDH. In addition, rather than requiring digitisation, these data had the advantage of being presented within a Microsoft Excel (2010 version) worksheet (Microsoft Corporation, Redmond, WA, USA). Furthermore, the TAG notes that Kaplan–Meier PFS data for PLDH contained within the PharmaMar submission were complete, i.e. 0% of patients remained at risk at the end of follow-up (although the TAG notes that these data were subject to a large degree of censoring: see Description and critique of manufacturer-submitted evidence, above). Consequently, no extrapolation of these data was necessary.
However, for the purposes of sensitivity analysis, and to provide a smoothed survival curve, the TAG fitted a number of parametric survival distributions to the manufacturer’s Kaplan–Meier data. Based upon the AIC values associated with these distributions (Table 119), the TAG considers the Weibull distribution to provide the best fit of the Kaplan–Meier data.
| Selected distribution | AIC |
|---|---|
| Weibull | 734.896 |
| Exponential | 751.511 |
| Log-normal | 741.603 |
| Log-logistic | 746.192 |
For the sensitivity analysis, the TAG estimated monthly PFS using the following formula:
where t is time in months and the intercept (2.186) and scale (–0.320) parameters have been estimated using MLE methods described in Hoyle and Henley. 113
Figure 33 presents the manufacturer’s Kaplan–Meier data, for PLDH, compared with the extrapolated Weibull survival curve.
FIGURE 33.
Progression-free survival for PLDH as estimated from the PharmaMar MS Kaplan–Meier data compared with the extrapolated Weibull survival curve using methods from Hoyle and Henley113 (sensitivity analysis only).

For OS, Kaplan–Meier data presented in the model submitted by PharmaMar as part of this MTA, are used to inform the base-case OS distribution for PLDH. These data represent the most recent information, and did not require estimation as a result of being provided within an Excel worksheet.
Of the parametric distributions considered to extrapolate Kaplan–Meier OS data, the TAG considers the Weibull distribution to be the best fit based upon the AIC values (Table 120). However, the TAG notes that model fit was similar for each considered OS distribution, and that although the Weibull distribution resulted in the lowest AIC, the range of AIC values was not large. For this reason, the baseline distribution is varied in sensitivity analyses to test the sensitivity of the cost-effectiveness results to the baseline curve selected (see Results of the sensitivity analysis, below).
| Selected distribution | AIC |
|---|---|
| Weibull | 1116.346 |
| Exponential | 1134.797 |
| Log-normal | 1147.63 |
| Log-logistic | 1139.511 |
To estimate monthly OS for patients treated with PLDH, the TAG used the following formula:
where t is time in months and the intercept (3.449) and scale (–0.304) parameters have been estimated using MLE methods described in Hoyle and Henley. 113
Figure 34 presents the manufacturer’s Kaplan–Meier data, for PLDH, compared with the extrapolated Weibull survival curve.
FIGURE 34.
Overall survival for PLDH as estimated from the PharmaMar MS Kaplan–Meier data, vs. the extrapolated Weibull survival curve using methods from Hoyle and Henley. 113
Platinum resistant/refractory (baseline treatment: pegylated liposomal doxorubicin hydrochloride)
For the PRR network, PLDH has been selected as the baseline treatment (Figure 35; green square indicates baseline treatment). Three sources of published survival data are available for PFS (Gordon et al. ,49 data submitted by Schering-Plough presented within the Assessment Report for TA91 for Trial 30–57,13 and OVA-301 as reported in Monk et al. 30) and OS (data for Trial 30–57 submitted by Schering-Plough presented within the Assessment Report for TA91,13 Gordon et al. ,54 and OVA-301 from Monk et al. 64). In addition, the CSR for OVA-301 provided by PharmaMar contains PFS and OS Kaplan–Meier data. However, no complete PFS or OS data (i.e. no patients remaining at risk at the end-of-trial follow-up) exist for patients treated with PLDH; therefore, parametric extrapolation has been used.
FIGURE 35.
Network diagram for the PRR subgroup; green square indicates base-line treatment.
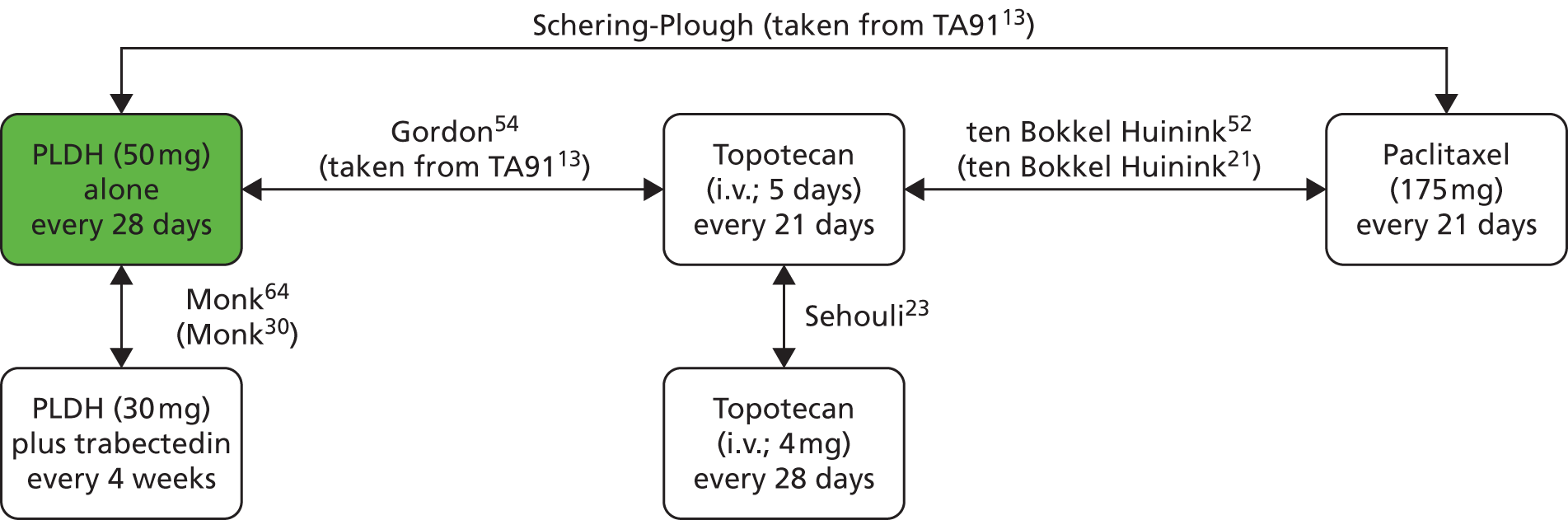
For PFS, data from OVA-301 in Monk et al. 30 are used to inform the distribution of PFS, used in the base case for patients treated with PLDH. This is because neither data presented within Gordon et al. 49 nor data contained within Trial 30–57 from TA9113 were sufficient to facilitate use of the methods described in Hoyle and Henley;113 i.e. no numbers of patients at risk were presented on Kaplan–Meier plots. The TAG notes that the comparison within OVA-301 from Monk et al. 30 is not relevant for the decision problem for this MTA (i.e. trabectedin plus PLDH is not an intervention or comparator of interest for the PRR subgroup); however, the TAG considers the information contained within Monk et al. 30 to be informative for the network, and notes that the trial represents the most recent data identified for PLDH in the PRR subgroup.
Therefore, Kaplan–Meier PFS data were estimated (from digitisation of the Kaplan–Meier plot and the reported numbers of patients at risk) and a number of parametric survival distributions fitted. Based on the AIC of the survival distributions considered (Table 121), the log-normal distribution could be considered to be the best fit of these data. However, as before, the TAG considers that given the similar AIC values, and acknowledging DSU guidance on the application of HRs, the Weibull distribution represents the most appropriate approximation of PFS. 118 Therefore, the Weibull distribution is used to inform the base-case analysis. However, log-normal, log-logistic and exponential distributions are used in sensitivity analyses (see Results of the sensitivity analysis, below).
| Selected distribution | AIC |
|---|---|
| Weibull | 514.0249 |
| Exponential | 528.9606 |
| Log-normal | 502.1343 |
| Log-logistic | 504.6734 |
The TAG calculated monthly PFS using the following formula:
where t is time in months, and the intercept (1.665) and scale (–0.345) parameters have been estimated using MLE methods described in Hoyle and Henley. 113
Figure 36 presents the Kaplan–Meier data, for PLDH, estimated from data presented in Monk et al. ,30 compared with the extrapolated Weibull curve.

For OS, the Kaplan–Meier OS data available in the CSR for OVA-301 are used to inform the base-case OS distribution for PLDH. These data represent the only source of information around numbers of patients at risk at given time points.
The TAG fitted a number of parametric survival distributions to the estimated OS Kaplan–Meier data (estimated from digitisation of Kaplan–Meier plot and numbers of patients at risk). Based on the AIC values of the considered distributions (Table 122), the TAG notes that the log-normal distribution could be considered to be the best fit to the estimated data. However, as before, recognising that AIC values were similar, and that the log-logistic and log-normal distributions may not represent the most appropriate baseline curve from which to apply HRs,118 the Weibull distribution has been selected for use in the base case. However, to test the sensitivity of the cost-effectiveness results to the baseline curve selected, the choice of baseline distribution was varied in sensitivity analysis (see Results of the sensitivity analysis, below).
| Selected distribution | AIC |
|---|---|
| Weibull | 848.2284 |
| Exponential | 850.8514 |
| Log-normal | 837.5367 |
| Log-logistic | 841.1105 |
To estimate monthly OS for patients treated with PLDH, the TAG used the following formula:
where t is time in months, and the intercept and scale parameters have been estimated using MLE methods described in Hoyle and Henley. 113
Network meta-analysis of progression-free survival and overall survival
For each network constructed, HRs of the relative effect of treatment (vs. the baseline treatment) on PFS and OS have been obtained from NMA (see Chapter 3, Results). These are summarised in Table 123 below.
| Treatment regimen | PFS | OS | ||
|---|---|---|---|---|
| Mean HR vs. baseline | 95% CrI | Mean HR vs. baseline | 95% CrI | |
| PS network 1 (paclitaxel-plus-carboplatin baseline) | ||||
| Paclitaxel plus platinum (carboplatin) (baseline treatment) | 1.000 | – | 1.000 | – |
| PLDH plus platinum (carboplatin) | 0.817 | 0.717 to 0.927 | 1.023 | 0.889 to 1.172 |
| Gemcitabine plus carboplatin | 0.985 | 0.748 to 1.273 | 1.247 | 0.921 to 1.652 |
| Platinum (carboplatin) | 1.361 | 1.182 to 1.559 | 1.290 | 1.096 to 1.509 |
| PS network 2 (PLDH baseline) | ||||
| PLDH (baseline treatment) | 1.000 | – | 1.000 | – |
| PLDH plus trabectedin | 0.736 | 0.560 to 0.949 | 0.835 | 0.667 to 1.032 |
| Paclitaxel | 1.615 | 0.939 to 2.586 | 1.219 | 0.850 to 1.690 |
| Topotecan | 1.298 | 0.979 to 1.688 | 1.367 | 1.035 to 1.770 |
| PRR network (PLDH baseline) | ||||
| PLDH (baseline treatment) | 1.000 | – | 1.000 | – |
| Paclitaxel | 1.360 | 0.817 to 2.123 | 1.053 | 0.783 to 1.382 |
| Topotecan | 0.998 | 0.767 to 1.277 | 0.973 | 0.764 to1.221 |
Adjusted HRs (calculated from data adjusted for baseline characteristics) are available for some of the treatments considered (e.g. trabectedin plus PLDH, PLDH monotherapy, PLDH plus carboplatin, and paclitaxel plus carboplatin); however, unadjusted HRs were used in all TAG NMAs. This is because data used to inform the adjusted HRs, identified in the clinical systematic review, differed by trial; moreover, some trials reported only unadjusted HRs. Therefore, the TAG considered synthesis of unadjusted HRs to be the most comparable measure of relative effect across trials. Moreover, the TAG considers that the use of a consistent data set within the NMA to be the most appropriate methodology.
Estimating progression-free survival and overall survival for the remaining treatments in the network
For each network unadjusted HRs of treatment effect relative to the baseline treatment were obtained from the NMAs described in Chapter 3 (see Results). Within the TAG economic model, each HR is applied to the monthly PFS and OS estimates for the baseline treatment, thus providing monthly estimates of PFS and OS for all treatments in the network. To do this, the monthly probabilities of PFS and OS for the baseline treatment are converted into survival rates using the following formula: survival ratet = – ln(1 – pt), where pt is the proportion of patients surviving at month ‘t’.
The HR obtained from the NMA is then applied to the survival rate and the resultant rate converted back into a probability using the following formula: survival proportiont = 1 – exp (–HR × survival ratet), where t is the time in months and HR is the HR expressed as the relative hazard of survival (rather than the relative hazard of death).
For each network, the survival curves (PFS and OS) estimated using this method are presented in Appendix 9. A summary of the estimated mean PFS and mean OS from the TAG analysis are presented in Table 124 for each therapy.
| PS network 1 | ||
|---|---|---|
| Treatment | Mean PFS (months) | Mean OS (months) |
| Platinum | 10.3 | 33.9 |
| Gemcitabine plus carboplatin | 11.9 | 34.5 |
| Paclitaxel plus platinum | 11.8 | 38.4 |
| PLDH plus platinum | 12.8 | 38.0 |
| PS network 2 | ||
| Treatment | Mean PFS (months) | Mean OS (months) |
| Paclitaxel | 6.9 | 26.3 |
| PLDH | 8.9 | 29.3 |
| Topotecan | 7.8 | 24.6 |
| Trabectedin plus PLDH | 10.3 | 32.2 |
| PRR network | ||
| Treatment | Mean PFS (months) | Mean OS (months) |
| Paclitaxel | 4.6 | 18.0 |
| PLDH | 5.3 | 18.6 |
| Topotecan | 5.3 | 18.9 |
Issues considered by the Technology Assessment Group
The TAG notes that the effectiveness data used in the model was subject to a number of limitations. Therefore, the likely impact of these limitations has been explored in a variety of sensitivity analyses (see Approach to uncertainty, below); a summary of the keys issues and conclusions is provided below.
Appropriateness of clinical data used for the decision problem
Table 125 outlines the treatment regimens used in the clinical trials upon which the NMAs are based compared with the treatment regimens assumed to be used in the economic model. The following differences between the treatment regimens used to inform the effect of treatment (with respect to PFS and OS) within the model, and the modelled regimens have been identified:
-
No regimens used to inform estimates of treatment effectiveness were limited to six cycles, whereas the number of cycles of therapy modelled is limited to six.
-
The efficacy of platinum monotherapy and platinum in combination with paclitaxel has been estimated from, among other trials, a trial that included treatment with cisplatin, whereas only treatment with carboplatin (with or without paclitaxel) is modelled.
-
Clinical effectiveness data from paclitaxel administered at 3-weekly intervals were used to inform estimates of PFS and OS in the PRR population, whereas a weekly paclitaxel regimen is modelled.
-
Estimates of the treatment effectiveness of PLDH monotherapy was based upon a dose of 50 mg/m2, whereas PLDH monotherapy at 40 mg/m2 is modelled.
These differences are expected to have minimal impact upon the model results; however, for completeness, the potential impact of the differences is discussed in more detail below.
For all clinical data used to inform PFS and OS in the economic model, estimates are based on treatment regimens in which patients could receive more than six cycles of therapy. However, in the economic model, cycles are limited to a maximum of six to reflect UK clinical practice. The TAG considers that this difference is unlikely to materially impact upon the cost-effectiveness results. This is because it is generally considered that treatment beyond six cycles is unlikely to impact upon efficacy. 19
Progression-free survival and OS data from Parmar et al. 61 have been used to inform the effectiveness of treatment with platinum, and platinum in combination with paclitaxel, through the TAG NMA. These data include information from patients treated with either carboplatin or cisplatin; although, carboplatin was the agent used most commonly (71% of monotherapy patients, 80% of combination therapy patients). The TAG notes that in 2010 a Cochrane review was published in which a systematic review and meta-analysis comparing carboplatin and cisplatin in advanced ovarian cancer were carried out. The review estimated that the relative difference in survival, expressed as an OR, for patients treated with these two agents was 1.02 (95% CI 0.93 to 1.12, favours cisplatin). 116 The TAG considers that this result implies that the two agents may be considered similar. Moreover, clinical expert advice received by the TAG suggested that cisplatin and carboplatin have similar efficacy, with carboplatin preferred as a result of greater tolerability. For these reasons, the TAG considers that the assumption of equivalent efficacy between cisplatin and carboplatin is unlikely to impact upon the cost-effectiveness results.
For the PRR population, clinical advice suggested that paclitaxel monotherapy would be administered weekly rather than 3-weekly. This is because weekly administration is perceived to be more efficacious than administration every 3 weeks. However, for the PRR population, no PFS or OS data are available for paclitaxel administered weekly. Therefore, although a weekly paclitaxel regimen is modelled, 175 mg/m2 paclitaxel administered every 3 weeks has been used to inform PFS and OS. However, evidence from Rosenberg et al. 60 suggests that efficacy may not be affected by the use of weekly rather than 3-weekly administrations. Rosenberg et al. 60 presented evidence on the safety and efficacy, in patients with platinum-resistant or platinum refractory disease, of paclitaxel administered at a dose of 67 mg/m2 per week compared with paclitaxel administered at a dose of 200 mg/m2 every 3 weeks. The study concluded that paclitaxel administered weekly was better tolerated yet comparably efficacious to paclitaxel administered every 3 weeks. Therefore, the TAG considers it unlikely that the efficacy of paclitaxel will be understated to an extent likely to materially affect the cost-effectiveness results.
Finally, estimates of the clinical effectiveness of PLDH monotherapy were based upon a dose of 50 mg/m2, whereas PLDH monotherapy administered at a dose of 40 mg/m2 is modelled. This is because, as a result of tolerability issues, clinical advice highlighted that a 50 mg/m2 dose would not typically be used in clinical practice. Clinical opinion considered that efficacy would not be affected by this dose reduction; therefore the TAG considers that the assumption of equivalent efficacy between 50 mg/m2 and 40 mg/m2 PLDH is unlikely to impact upon model results. However, for completeness, the TAG investigated the impact of modelling 50 mg/m2 of PLDH in sensitivity analysis (see Approach to uncertainty, below).
| Chemotherapy | Regimen modelled, and typically used in clinical practice | Regimens from which data are used to inform the effectiveness estimates |
|---|---|---|
| Paclitaxel | For PRR disease: paclitaxel 80 mg/m2 weekly for 18 weeks or until progression For platinum-sensitive disease: paclitaxel 175 mg/m2 on day 1 of every 21-day cycle (maximum six cycles) |
For both platinum-sensitive and PRR disease: paclitaxel 175 mg/m2 day 1 of every 21-day cycle. For the Schering-Plough submission for TA91,13 minimum number of cycles was six. The number of cycles was not limited, although the median number of cycles was five (ten Bokkel et al.21) |
| Paclitaxel plus platinum | For PRR disease: paclitaxel 80 mg/m2 plus carboplatin AUC 3, weekly for 18 weeks or until progressiona For platinum-sensitive disease: paclitaxel 175 mg/m2 and carboplatin AUC 5, on day 1 of every 21-day cycle (maximum six cycles) |
No clinical data were found for the PRR population and this intervention was modelled only in sensitivity analysis For the platinum-sensitive population, the regimens on which the clinical data were based was a combination of:
|
| PLDH | 40 mg/m2 on day 1 of every 28-day cycle (maximum six cycles) | The regimen on which the clinical data were based was: |
| PLDH plus platinum | PLDH 30 mg/m2; carboplatin target AUC of 5, on day 1 of every 28-day cycle (maximum six cycles) | The regimens on which the clinical data were based were: |
| Gemcitabine plus carboplatin | Gemcitabine 1000 mg/m2 on days 1 and 8 of every 21-day cycle, carboplatin target AUC of 4 on day 1 of every 21-day cycle (maximum six cycles) | Gemcitabine 1000 mg/m2 on days 1 and 8 of every 21-day cycle, carboplatin target AUC of 4 on day 1 of every 21-day cycle; maximum of 10 cycles (median six cycles) (Pfisterer et al.50) |
| Trabectedin plus PLDH | Trabectedin 1.1 mg/m2; PLDH 30 mg/m2, on day 1 of every 21-day cycle (maximum six cycles) | Trabectedin 1.1 mg/m2; PLDH 30 mg/m2, on day 1 of every 21-day cycle, until progression (Monk et al.30) |
| Topotecan | 1.5 mg/m2, on days 1–5 of every 21-day cycle (maximum six cycles) | The regimen on which the clinical data were based was 1.5 mg/m2, days 1–5 of every 21-day cycle until progression (Gordon et al.,49 ten Bokkel Huinink et al.21) |
| Platinum monotherapy | Carboplatin target AUC of 5, on day 1 of every 21-day cycle (maximum six cycles) | The regimens on which the clinical data were based were: |
| Etoposidea | 50-mg flat dose on days 1–21 of every 28-day cycle (maximum six cycles) | No clinical data were identified and costs were included in sensitivity analysis only |
| Etoposide plus cisplatina | Etoposide 50-mg flat dose on days 1–21 of every 28-day cycle plus cisplatin i.v. 50 mg days 1, 8 and 15 every 28 days (maximum six cycles) | No clinical data were identified and costs were included in sensitivity analysis only |
| Best supportive carea | Costs associated with supportive care | No clinical data were identified and costs were included in sensitivity analysis only |
Appropriateness of hazard ratios obtained from the literature
The TAG considers that the HR is the most appropriate measure of relative treatment effect for survival (PFS and OS). This is because the HR is specifically designed to account for time-to-event data and allows for censoring frequently present in time-to-event data. Ideally, an IPD NMA would have been carried out to estimate HRs for all treatments, by subgroup; IPD NMA has the potential to account for differences in baseline characteristics within and between trials through the incorporation of covariates. However, the TAG did not have access to IPD that was sufficiently granular to facilitate such an analysis. Therefore, syntheses of published HRs within standard NMAs were carried out. Many (although not all) of the studies identified for inclusion within the networks provided HRs and, for those studies for which HRs were not available and sufficient information was provided, they were calculated using methods outlined in Tierney. 77
Additionally, although some of the clinical trials identified for inclusion in the NMA reported HRs adjusted for particular baseline characteristics, the TAG used unadjusted HRs within the NMAs and therefore economic analyses. The TAG recognises that imbalances in baseline characteristics between treatment arms may introduce bias into the HR; however, of those trials reporting adjusted HRs, each had adjusted for different factors. Moreover, for some comparisons only unadjusted HRs were reported. Therefore, the TAG considers the use of unadjusted HRs to be the most equitable way to compare therapies. Moreover, the TAG considers that the use of consistent data is appropriate for meta-analysis.
The TAG notes that within the DSU technical support document it is acknowledged that there are practical difficulties in modelling survival based upon summary data such as HRs rather than patient-level data, and notes that it is anticipated that this issue will be considered in a future technical support document. 116 Specifically, two key concerns are raised within this document about the use of summary HRs:
-
The assumption of proportional hazards (discussed below): ‘where one HR is applied to the entire modelled period, the proportional hazards assumption must be made – that is, the treatment effect is proportional over time and the survival curves fitted to each treatment group have a similar shape.’118
-
That HRs should be obtained from the same parametric model as used to estimate baseline survival: ‘care should be taken to ensure that only the HR obtained from the chosen parametric model is applied to the control group survival curve derived from the parametric model fitted with the treatment group as a covariate – it is theoretically incorrect to apply a HR derived from a different parametric model, or one derived from a Cox proportional hazards model’. 118
For the analyses carried out for this MTA, IPD were not available for all treatments considered; therefore, it was not possible to estimate HRs for each treatment using the same parametric model as fitted for the baseline treatment. Consequently, HRs were obtained from published or submitted literature. The TAG recognises that the use of published HRs is a weakness of the analysis and notes that it is unclear what impact this would have upon model results. However, to provide an indication of how sensitive model results were to the effect of treatment on PFS and OS, the survival curves estimated from application of the HR were tested in sensitivity analysis (see Approach to uncertainty, below).
Appropriateness of the proportional hazards assumption
The TAG did not have access to either a single clinical trial, or IPD for the full range of interventions and comparators of interest for this MTA. For that reason, as discussed, the TAG used summary HRs, synthesised from published or submitted literature, to estimate the relative effects of treatments considered within the economic analysis. Consequently, it is implicitly assumed that the relative treatment effects captured by the HRs holds true across all time points. In other words, use of HRs in the economic model assumes that the relative hazards between treatments are proportional.
The TAG explored whether the assumption of proportional hazards was appropriate for the data used within the analysis. This was explored, as per the DSU technical support document for survival analysis, with log-cumulative hazard (LCH) plots. 118 The LCH plots were created by digitising (where available) Kaplan–Meier plots for each of the treatments included within the analysis, ‘ln(time)’ was then plotted against ‘ln(-ln(survival probability)’. For each network, LCH plots based on Kaplan–Meier data used to inform PFS and OS are presented in Appendix 10; LCH plots are presented for the individual and total comparisons made.
Based on the LCH plots, the TAG considers that the assumption of proportional hazards may not be entirely appropriate, in particular, for PFS in platinum-sensitive patients, for whom in many cases the relative hazards of progression seem to decrease over time. The impact, on model results, of incorrectly assuming proportional hazards will depend on the nature of the true hazard function. In cases where the relative hazard (treatment A vs. treatment B) decreases over time (for both PFS and OS), the model is likely to overestimate the relative benefit of treatment A compared with treatment B. Conversely, when relative hazards increase over time (for PFS and OS), the model is likely to underestimate the benefit of treatment A over treatment B. In cases where the relative hazards are non-monotonic (i.e. increase and then decrease or vice versa) or differ between PFS and OS, it is more challenging to determine the possible direction of bias. With this in mind, when reporting the cost-effectiveness results the TAG has endeavoured to indicate the potential direction of bias resulting from inappropriate assumption of proportional hazards (see Base-case results, below).
Crossover bias
Crossover bias occurs when a patient switches from a control therapy to the treatment being evaluated during a clinical trial. Here, the switch of therapy results in a possibility that any clinical benefit associated with the experimental treatment will be underestimated. 119 In the clinical trials evaluated for this review, several allowed women to undergo further therapy following progression. This means that it is possible that crossover bias will have influenced OS results used within the analysis; indeed, confounding of OS data is a well-recognised complexity in clinical trials evaluating treatments for cancer.
A number of approaches have been suggested that attempt to quantify the degree of confounding; these are discussed in detail in Morden et al. 119 Within this paper it is suggested that the iterative parameter estimation algorithm put forward by Branson and Whitehead,120 may be considered when analysing the degree of bias.
The TAG was unable to investigate the degree of crossover bias within the estimates of OS for this MTA. This is because not all trials described the further treatments received by the women within the trial, and furthermore, application of the Branson and Whitehead method120 requires IPD in order to assess the degree of bias. As such, the degree to which crossover bias has influenced results is unclear. The TAG considers that underestimation of survival benefit may have affected all comparisons, although the degree to which comparisons are affected is unknown. It is possible, however, that the degree of bias may be balanced.
However, for completeness, and to address this uncertainty, the TAG carried out sensitivity analyses on the OS curves included in the economic analysis (see Approach to uncertainty, below).
Adverse event incidence
Following appraisal of the studies identified as part of the clinical effectiveness review and after discussion with clinical experts, a shortlist was drawn up of AEs considered to have a noteworthy impact on cost or patient QoL. These were allergic reaction, alopecia, anaemia, fatigue, febrile neutropenia, nausea and vomiting, and neuropathy (see Chapter 3, Adverse events). For the purposes of the economic model, only AEs of grade 3 and grade 4 were considered; this was consistent with the approach taken in TA91 and reflected the likelihood that grade 1 or 2 AEs are likely to impact little on cost or QoL.
In the base case, only the subset of AEs associated with a notable cost were included in the analysis; QoL decrements are included in sensitivity analysis only. This is because the reliability of the estimates identified for QoL decrements is uncertain. In addition, the impact of AEs on patient QoL associated with trabectedin plus PLDH and PLDH monotherapy are implicitly included within the health-state utility estimates from TA222;15 therefore, the addition of disutility values may result in double counting of the impact of AEs for these therapies (see Health-related quality-of-life data, below).
Of the AEs considered for inclusion in the model, four were deemed to result in a cost to the NHS (see Costs, below). These are allergic reaction, anaemia, febrile neutropenia, and nausea and vomiting. However, when data were available, the impact of AEs on patient QoL is considered in sensitivity analysis (see Approach to uncertainty, below).
The relative likelihood of an AE associated with each therapy was estimated from a series of NMAs carried out by the TAG (see Chapter 3, Adverse events). The outcome measure selected to assess the relative likelihood was the OR. As a result of data paucity, AEs were not analysed by population; instead, AE data from any population (platinum sensitive or PRR) were included in analysis. The TAG considered this approach to be appropriate in order to utilise all available data. However, the TAG notes that this approach necessitates the assumption that the likelihood of an adverse reaction is independent of the PFI.
Inconsistent reporting between trials led to differences in the networks of treatments available to assess the relative effect of treatment on each AE. Consequently, estimates of the impact of treatment on the rates of AEs were not available for all treatments for all AEs. Therefore, within the model, the following steps are taken:
-
For the baseline treatment in each network (PS network 1, PS network 2, and PRR) the probability of each AE has been estimated.
-
Where available, ORs for treatments within the same network are used to inform the probability of each AE.
-
ORs that are statistically significant (at the 5% level) are converted into a probability using the following formula: (7)oddsB=oddsBoddsA⋅pA(1−pA), and (8)pB=oddsB1+oddsB where pA is the probability of an AE for the baseline treatment, and where pB is the probability of an AE for all other treatments.
-
ORs that are not statistically significant (at the 5% level) are assumed to be equal to 1 (i.e. the baseline probability is used).
-
-
Where no OR was calculable, and therefore the relative effect is unknown, or where resultant probabilities were considered by clinical experts to represent unlikely values, expert opinion was sought in order to inform the rate of AEs.
The AE rates used in the base-case model are presented, by network (PS network 1, PS network 2, and PRR), in Tables 126–128.
| Chemotherapy | OR (95% CrI) | AE probability (%) | Comments |
|---|---|---|---|
| Allergic reaction | |||
| Paclitaxel plus platinum | Baseline treatment | 3.94 | Source of baseline probability: a weighted average of Bafaloukos et al.29 (one event, 89 patients) and Gonzalez-Martin et al.48 (four events, 38 patients) |
| PLDH plus platinum | 0.130 (0.001 to 0.705) | 0.53 | – |
| Gemcitabine plus carboplatin | 0.757 (0.030 to 3.798) | 3.94 | Non-statistically significant difference, therefore OR assumed to equal 1; probability was set to equal the baseline treatment |
| Platinum | 0.755 (0.057 to 3.043) | 3.94 | Non-statistically significant difference, therefore OR assumed to equal 1; probability was set to equal the baseline treatment |
| Anaemia | |||
| Paclitaxel plus platinum | Baseline treatment | 5.10 | Source of baseline probability: a weighted average of Bafaloukos et al.29 (3 events, 89 patients), Gonzalez-Martin et al.48 (2 events, 38 patients), and Pujade-Lauraine et al.31 (27 events, 501 patients) |
| PLDH plus platinum | 1.926 (1.164 to 3.039) | 9.38 | – |
| Gemcitabine plus carboplatin | 5.848 (1.158 to 18.040) | 23.91 | – |
| Platinum | 1.255 (0.305 to 3.479) | 5.10 | Non-statistically significant difference, therefore OR assumed to equal 1; probability was set to equal the baseline treatment |
| Febrile neutropenia | |||
| Paclitaxel plus platinum | Baseline treatment | 4.19 | Source of baseline probability: Pujade-Lauraine et al.31 (21 events, 501 patients) |
| PLDH plus platinum | 0.614 (0.299 to 1.263) | 4.19 | Non-statistically significant pairwise difference, therefore OR assumed to equal 1; probability was set to equal the baseline treatment |
| Gemcitabine plus carboplatin | NA | 4.19 | No OR calculable, therefore set equal to baseline treatment (4.19%) based upon clinical advice |
| Platinum | NA | 0 | No OR calculable, therefore set equal to 0% based upon clinical advice |
| Nausea and vomiting | |||
| Paclitaxel plus platinum | Baseline treatment | 1.57 | Source of baseline probability: weighted average of Bafaloukos et al.29 (1 event, 89 patients) and Gonzalez-Martin et al.48 (1 event, 38 patients). Clinical expert opinion implied that this rate appeared low; therefore, this was varied in a scenario analysis (see Approach to uncertainty, below) |
| PLDH plus platinum | 2.055 (1.598 to 2.608) | 3.17 | Given the uncertainty associated with this network, ORs estimated from analysis of all grades were used. OR for grade 3/4 provided extreme values; therefore, ORs estimated from analysis of all grades were used. Probabilities based upon clinical expert opinion were used in scenario analysis (see Approach to uncertainty, below) |
| Gemcitabine plus carboplatin | NA | 3.17 | No data; therefore, set equal to PLDH plus platinum in the base case based upon clinical advice |
| Platinum | 1.305 (0.981 to 1.706) | 1.57 | Given the uncertainty associated with this network, ORs estimated from analysis of all grades were used. This analysis provided a non-statistically significant difference between platinum and the baseline therapy; therefore, OR assumed to equal 1; probability was set to the same as the baseline treatment. Probabilities based upon clinical expert opinion were used in scenario analysis (see Approach to uncertainty, below) |
| Chemotherapy | OR (95% CrI) | AE rate (%) | Comments |
|---|---|---|---|
| Allergic reaction | |||
| Paclitaxel | NA | 20 | Set equal to 20% based upon clinical advice |
| PLDH | NA | 5 | Set equal to 5% based upon clinical advice |
| Trabectedin plus PLDH | NA | 5 | Set equal to 5% based upon clinical advice |
| Topotecan | NA | 0 | Set equal to 0% based upon clinical advice |
| Anaemia | |||
| Paclitaxel | 0.742 (0.209 to 1.848) | 4.73 | Non-statistically significant difference, therefore OR assumed to equal 1; probability was set to equal the baseline treatment |
| PLDH | Baseline | 4.73 | Source of baseline probability: weighted average of Schering-Plough submission from TA9113 (3 events, 108 patients), Gordon et al.49 (13 events, 239 patients) and Monk et al.30 (16 events, 330 patients) |
| Trabectedin plus PLDH | 2.940 (1.559 to 5.202) | 12.74 | – |
| Topotecan | 7.374 (3.775 to 13.590) | 26.80 | – |
| Febrile neutropenia | |||
| Paclitaxel | NA | 5 | Set equal to 5% based upon clinical advice |
| PLDH | Baseline | 2.12 | Source of baseline probability: Monk et al.30 (7 events, 330 patients) |
| Trabectedin plus PLDH | 3.256 (1.378 to 7.692) | 6.59 | – |
| Topotecan | NA | 5 | Set equal to 5% based upon clinical advice |
| Nausea and vomiting | |||
| Paclitaxel | 0.279 (0.120 to 0.535) | 2.93 | – |
| PLDH | Baseline | 9.75 | Source of baseline probability: a weighted average of Monk et al.30 (15 events, 330 patients), Schering-Plough submission from TA9113 (19 events, 108 patients), and Gordon et al.49 (32 events, 239 patients) |
| Trabectedin plus PLDH | 5.291 (2.866 to 9.342) | 36.37 | – |
| Topotecan | 1.460 (0.886 to 2.294) | 9.75 | Non-statistically significant difference, therefore OR assumed to equal 1; probability was set to equal the baseline treatment |
| Chemotherapy | OR (95% CrI) | AE rate (%) | Comments |
|---|---|---|---|
| Allergic reaction | |||
| Paclitaxel | NA | 20 | Set equal to 20% based upon clinical advice |
| PLDH | NA | 5 | Set equal to 5% based upon clinical advice |
| Trabectedin plus PLDH | NA | 5 | Set equal to 5% based upon clinical advice |
| Topotecan | NA | 0 | Set equal to 0% based upon clinical advice |
| Etoposidea | NA | 0 | Set equal to 0% based upon clinical advice |
| Etoposide plus carboplatina | NA | 10 | Set equal to 10% based upon clinical advice |
| Anaemia | |||
| Paclitaxel | 0.742 (0.209 to 1.848) | 4.73 | Non-statistically significant difference, therefore OR assumed to equal 1; probability was set to equal the baseline treatment |
| PLDH | Baseline | 4.73 | Source of baseline probability: weighted average of Schering-Plough submission from TA9113 (3 events, 108 patients), Gordon et al.49 (13 events, 239 patients) and Monk et al.30 (16 events, 330 patients) |
| Trabectedin plus PLDH | 2.940 (1.559 to 5.202) | 12.74 | – |
| Topotecan | 7.374 (3.775 to 13.590) | 26.80 | – |
| Etoposidea | NA | 4.73 | Set equal to paclitaxel (4.73%) based upon clinical advice |
| Etoposide plus carboplatina | NA | 4.73 | Set equal to paclitaxel (4.73%) based upon clinical advice |
| Febrile neutropenia | |||
| Paclitaxel | NA | 5 | Set equal to 5% based upon clinical advice |
| PLDH | Baseline | 2.12 | Source of baseline probability: Monk et al.30 (7 events, 330 patients) |
| Trabectedin plus PLDH | 3.256 (1.378 to 7.692) | 6.59 | – |
| Topotecan | NA | 5 | Set equal to 5% based upon clinical advice |
| Etoposidea | NA | 0 | No data |
| Etoposide plus carboplatina | NA | 0 | No data |
| Nausea and vomiting | |||
| Paclitaxel | 0.279 (0.120 to 0.535) | 2.93 | – |
| PLDH | Baseline | 9.75 | Weighted average of Monk et al.30 (15 events, 330 patients), Schering-Plough submission from TA9113 (19 events, 108 patients) and Gordon et al.49 (32 events, 239 patients) |
| Trabectedin plus PLDH | 5.291 (2.866 to 9.342) | 36.37 | – |
| Topotecan | 1.460 (0.886 to 2.294) | 9.75 | Non-statistically significant difference, therefore OR assumed to equal 1; probability was set to equal the baseline treatment |
| Etoposidea | NA | 9.75 | Set equal to PLDH (9.75%) based upon clinical advice |
| Etoposide plus carboplatina | NA | 9.75 | Set equal to PLDH (9.75%) based upon clinical advice |
Health-related quality-of-life data
Technology Assessment Group‘s systematic review of health-related quality-of-life data
A systematic review was carried out in December 2012 to identify relevant published HRQoL evidence to support the development of this MTA. The following databases were searched:
-
MEDLINE (Ovid)
-
EMBASE (Ovid)
-
HTA database
-
NHS EED.
The search strategy for all databases combined terms to capture the target condition (ovarian cancer) and terms to capture QoL. As this MTA is in part an update of TA91 in which a systematic review was carried out (search date of April 2004) to identify HRQoL studies, searches were limited from 2004. Full details of the search terms are presented in Appendix 5.
In addition to searches of the above databases, the following sources of potentially relevant publications were explored:
-
Experts in the field were contacted with a request for details of relevant published and unpublished studies of which they may have knowledge.
-
The NICE Technology Appraisal website was searched for any recently published Technology Appraisals in ovarian cancer that had not already been identified via the database searches or that may include additional HRQoL data.
-
Reference lists of key identified studies were reviewed for any potentially relevant studies.
No restrictions on language or setting were applied to any of the searches. Two health economists reviewed a sample of citations identified from the search and, upon confirming that the same inclusions and exclusions were applied for those papers, one health economist reviewed the remaining papers. Inclusion and exclusion criteria are presented in Table 129.
| Inclusion criteria | Exclusion criteria |
|---|---|
| Q1: possible generic, preference-based measure of HRQoL (e.g. EQ-5D, SF-6D, HUI) or standard gamble/TTO studies any setting (to be as inclusive as possible) Q2: possible generic, non-preference-based measure of HRQoL (e.g. SF-36) Q3: possible condition-specific measure of HRQoL |
Abstracts with insufficient methodological details, systematic reviews |
The systematic review was updated in May 2013 while the report was under peer review. The search strategy remained the same as outlined above; however, results were limited from 5 December 2012 to 23 May 2013 in order to identify only additional relevant studies.
A total of 3090 studies were identified from the December 2012 search of MEDLINE, EMBASE, HTA and NHS EED (Figure 37). Two health economists reviewed the first 100 citations identified from the search and, upon confirming consistency in the inclusions and exclusions made, one health economist reviewed the remaining 2990 papers. Of these, 722 were identified as duplicates and 2101 studies were excluded on the basis of title and abstract. A total of 267 papers were therefore identified as potentially relevant. Of these papers, 96 were identified, from the abstract, as either condition-specific measures of HRQoL or generic non-preference-based measures of HRQoL. Furthermore, 171 papers were identified as possible generic, preference-based measures of HRQoL (see Table 129, Q1). If it was unclear which type of HRQoL measure was included in the study, the reviewer was inclusive and labelled the study as a potential generic, preference-based measure of HRQoL.
The 96 studies identified as either condition-specific measures of HRQoL or generic non-preference-based measures of HRQoL during the December search were provisionally included, i.e. these studies were not ordered in full in the first instance. Instead, studies identified as reporting possible generic, preference-based measures of HRQoL were reviewed in full (171 papers). This is because a generic, preference-based measure of HRQoL, in particular the EQ-5D, is preferable for use within an economic evaluation. 109 It was therefore considered appropriate to assess the suitability of condition-specific or generic non-preference-based measures of HRQoL, if and only if, no suitable generic, preference-based measures of HRQoL were identified.
In addition to the studies identified through the database search, the ERG report for TA222,90 was identified through review of the NICE technology appraisal website. The ERG report for TA22290 was not detected in the database search; the TAG notes that this was because the date of the report was erroneously indexed within the search engine as the year 2000 rather than 2011, and was therefore excluded when date filters were applied to the search results. Additionally, through review of the reference lists of included studies, six studies were identified as possible preference-based measures of HRQoL. All six studies88,89,121–124 were published prior to 2004 and therefore were not detected in the database search; however, because these studies were referenced as the source of HRQoL data included within identified studies, they were included for completeness.
The studies identified from the database search and additional sources were reviewed in full. Of the 178 identified studies, a total of 22 studies included generic, preference-based HRQoL data. See Appendix 6 for an overview of reasons for exclusion of papers that were reviewed in full.
FIGURE 37.
Identified HRQoL studies: December 2012 search.
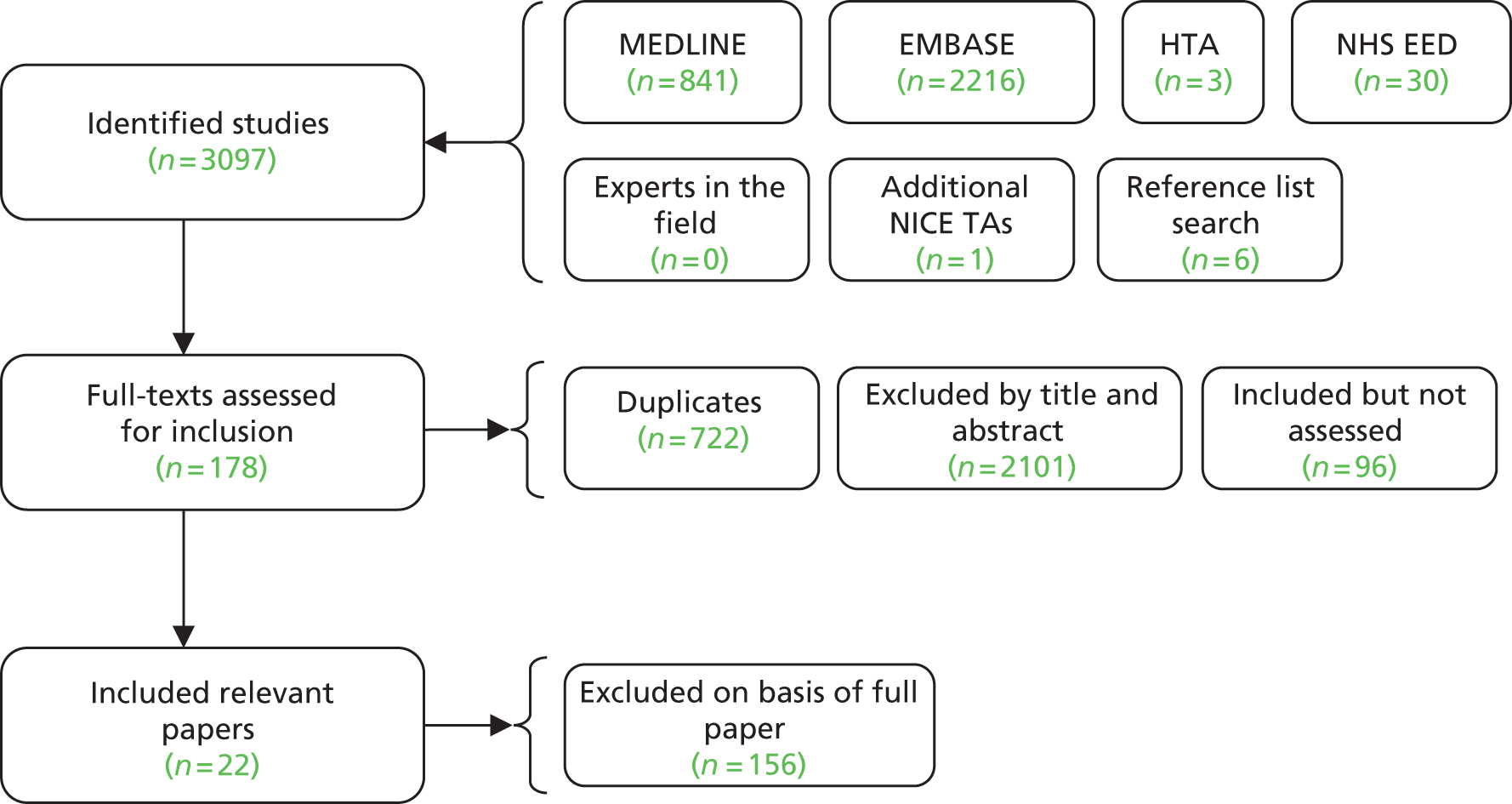
A further 239 papers were identified from the updated search in May 2013. Of these, a total of seven papers were identified as potentially relevant and ordered for full review. Of the seven ordered papers, four were excluded (see Appendix 6, May 2013 search) on the basis of the full paper, and three papers105,125,126 were identified as including generic, preference-based HRQoL data. For a detailed description of the reasons for exclusion, see Appendix 6. In addition to the included three papers, TA28411 and TA28516 were identified from an updated review of the NICE TA website.
Of the 187 papers included in the December 2012 and May 2013 searches, a total of 27 papers reported generic, preference-based HRQoL data. Information on the populations, health states, instruments and utility values reported in these studies are presented in Appendix 7; a summary of the HRQoL instrument used in each included study is presented in Table 130.
| Study | Instrument |
|---|---|
| Identified from the literature search and previous NICE TA | |
| Hess et al.125 | Valuation of the FACT questionnaire using methods described in Cheung et al.127 and Dobrez et al.128 |
| NICE 2013; TA285 (ERG report)129 | NA – utilities sourced from TA222 90 |
| NICE 2013; TA284 (MS)11 | EQ-5D |
| Bradford et al.126 | TTO |
| Montalar et al.105 | NA – utilities sourced from OVA-301 as reported in Krasner et al.67 |
| Havrilesky et al.98 | Valuation of the FACT questionnaire using TTO |
| Havrilesky et al.130 | NA – utilities stated as sourced from Leung et al.121 |
| Krasner et al.67 | EQ-5D |
| Pickard et al.131 | EQ-5D |
| Grann et al.132 | NA – utilities stated as sourced from Grann et al.133 |
| Lesnock et al.101 | NA – utilities stated as sourced from Greving et al.134 |
| NICE 2011; TA222 (ERG report)90 | EQ-5D |
| Gordon et al.135 | SF-6D |
| Grann et al.133 | TTO |
| Hess et al.136 | Standard gamble |
| Greving et al.134 | NA – utilities stated as sourced from Grann et al.122 and Grann et al.123 |
| Havrilesky et al.124 | TTO |
| Havrilesky et al.104 | NA – utilities stated as sourced from Sun et al.137 |
| Stein et al.138 | Standard gamble |
| Main et al.97 | NA – utilities stated as sourced from Tengs and Wallace88 and Brown and Hutton89 |
| Calhoun et al.139 | TTO |
| Identified from review of reference lists of the above identified studies | |
| Sun et al.137 | TTO |
| Tengs and Wallace88 | NA – utilities stated as sourced from Grann et al.122 |
| Grann et al.123 | TTO |
| Leung et al.121 | TTO |
| Brown and Hutton89 | Standard gamble |
| Grann et al.122 | TTO |
Of the included studies, four11,67,90,131 reported using EQ-5D questionnaires to collect QoL data. However, no EQ-5D scores for people with ovarian cancer were presented within the study by Pickard et al. ;131 therefore, this study could not be used to inform the economic model. In both Krasner et al. 67 and TA22290 EQ-5D data collected as part of OVA-301 were reported. OVA-301 was a Phase III clinical trial that recruited women with recurrent ovarian cancer after failure of first-line, platinum-based chemotherapy. Women were randomised to either PLDH or PLDH with trabectedin. For each treatment group, Krasner et al. 67 reported baseline EQ-5D scores and the change in EQ-5D from baseline to end of follow-up. By contrast, TA22290 reported EQ-5D data by health state (progression-free disease and PD) regardless of treatment received (Table 131).
| Study | Health state | Mean estimate of EQ-5D valuation | Measure of variance | n |
|---|---|---|---|---|
| Krasner et al.67 | PLDH (baseline) | 0.78 | 0.163 (sd) | 318 |
| PLDH (change from baseline) | –0.05 | 0.191 (sd) | 211 | |
| Trabectedin plus PLDH (baseline) | 0.78 | 0.171 (sd) | 323 | |
| Trabectedin plus PLDH (change from baseline) | –0.05 | 0.201 (sd) | 233 | |
| TA22290 | PFS | 0.718 | 0.010 (se) | NR |
| PD | 0.649 | 0.019 (se) | NR |
TA28411 reported EQ-5D data from ICON 7, a randomised, two arm, multicentre, Phase III trial considering the addition of bevacizumab to first-line treatment with carboplatin and paclitaxel (vs. carboplatin and paclitaxel) in patients with epithelial ovarian cancer. EQ-5D data were presented for SD and for PD, with utilities associated with SD dependent upon time (Table 132).
| Health state | Mean EQ-5D | se | n |
|---|---|---|---|
| SD weeks 0–2 | 0.6571 | 0.0133 | 335 |
| SD weeks 3–5 | 0.7153 | 0.0118 | 378 |
| SD weeks 6–8 | 0.7443 | 0.0110 | 375 |
| SD weeks 9–11 | 0.7683 | 0.0100 | 361 |
| SD weeks 12–14 | 0.7643 | 0.0112 | 363 |
| SD weeks 15–20 | 0.7444 | 0.0121 | 353 |
| SD weeks 21–26 | 0.7638 | 0.0131 | 303 |
| SD weeks 27–32 | 0.7718 | 0.0129 | 295 |
| SD weeks 33–38 | 0.7638 | 0.0136 | 282 |
| SD weeks 39–44 | 0.7785 | 0.0155 | 220 |
| SD weeks 45–50 | 0.7533 | 0.0165 | 202 |
| SD weeks 51–53 | 0.7760 | 0.0170 | 178 |
| SD weeks 54+ | 0.8129 | 0.0113 | 338 |
| PD | 0.7248 | – | – |
One study135 reported Short Form questionnaire-6 Dimensions (SF-6D) data. In this study, utility scores from 85 Australian women were reported by stage of disease (stage I/II; stage III; stage IV). For each disease stage a mix of drug therapies, platinum status and line of therapy were possible. No data by progression status were presented.
Ten studies valued health states using the time trade-off (TTO) method:
-
Hess et al. 125 used algorithms developed by Dobrez et al. 128 and Cheung et al. 127 to value responses to the Functional Assessment of Cancer Therapy (FACT) questionnaire from 746 people with ovarian cancer, through which Dobrez et al. 128 used TTO to value FACT questionnaire health states, and Cheung et al. 127 developed a mapping algorithm between FACT and EQ-5D.
-
Bradford et al. 126 used TTO to value sexual dysfunction and other hypothetical treatment-related side effects.
-
Havrilesky et al. 98 used estimates developed by Dobrez et al. 128 using TTO to value FACT questionnaire health states.
-
Grann et al. 133 estimated a single mean preference rating for ovarian cancer of between 0.83 and 0.84, based upon the responses from Canadian women with (n = 83) or without (n = 160) a personal or family history of breast or ovarian cancer.
-
Havrilesky et al. 124 valued 25 different health states based upon the responses of 37 female members of the public, and 13 women with a prior diagnosis of ovarian cancer. Health states valued included cancer states and AE states.
-
Calhoun et al. 139 valued six health states that reflected various levels of toxicity in women with ovarian cancer, based upon the responses of 39 ovarian cancer patients, 15 women at increased risk, 39 women in the general population and 11 gynaecological oncologists.
-
Sun et al. 137 valued AE health states based upon the responses from 34 women with ovarian cancer.
-
Grann et al. 123 estimated a mean preference rating for ovarian cancer, and metastatic cancer based upon the responses of 21 patients with breast cancer, 28 women with a personal history of multiple breast biopsies or a family history of breast cancer, and 135 women without these conditions.
-
Leung et al. 121 valued nine health states for breast cancer: toxicity from treatment, response to treatment, no response to treatment for each of treatment with paclitaxel, docetaxel and vinorelbine in patients with breast cancer. Values were estimated based upon the responses of 25 healthy volunteers and 25 women with breast cancer.
-
Grann et al. 122 estimated a mean preference rating for ovarian cancer, and metastatic cancer based upon the responses of 54 participants. The mean ovarian cancer utility was estimated to be 0.82, with metastatic disease estimated at 0.63.
In addition, three studies valued health states using the standard gamble technique. Hess et al. 136 valued six health states (with varying degrees of efficacy and AEs), based upon the responses of 51 women with ovarian cancer and 34 oncologists in the USA. Stein et al. 138 valued six clusters of patient characteristics (with varying proportions of performance status, disease stage and response after treatment), based upon the responses of 39 ‘Value of Health’ panel members. Brown and Hutton89 valued breast cancer health states based upon the responses from 29 US oncology nurses and 25–30 nurses from each of Germany, Italy, the Netherlands, Spain and the UK.
The remaining nine included studies88,97,101,104,105,129,130,132,134 were not the primary source of utility data. For example, four studies88,97,101,134 referenced (either directly or indirectly) Grann et al. ,122 although it is unclear how Greving et al. 134 used the data in Grann et al. 122 to estimate the utility values stated within the study.
Quality-of-life data included in the manufacturers’ submissions
One manufacturer (PharmaMar) submitted cost-effectiveness evidence, including estimates of HRQoL used in the economic model. The estimates used by the manufacturer were not obtained from a systematic review; instead, as described above (see Description and critique of manufacturer-submitted evidence), the manufacturer used EQ-5D data obtained from the OVA-301 clinical trial. The mean estimates of utility in the SD and PD health states were estimated to be 0.718 and 0.649, respectively. These estimates were used within TA222,90 and were therefore identical to the EQ-5D data identified by the TAG from the systematic review of the literature.
Quality-of-life data selected for the Technology Assessment Group economic analysis
In order to assess QALYs in the de novo economic analysis, it was necessary to identify health-state utility values for the SD (progression-free) and PD health states (see Model structure, above). In addition, given the importance of adverse treatment effects on QoL, it was desirable to identify disutilities associated with adverse treatment effects.
The health-state utility values selected for use within the TAG economic model are those used within TA22290 (Table 133). This is because TA22290 represents the only literature source identified that reports EQ-5D utility values in the recurrent ovarian cancer population by the health states required for the economic model. As described within the NICE Guide to the methods of technology appraisal, EQ-5D represents the preferred measure of HRQoL in adults. 109 In addition, the TAG notes that HRQoL data within TA22290 were based upon a sample of over 600 patients, the largest sample identified from the included HRQoL studies. EQ-5D data from TA28411 were not used in the economic analysis because these data were reflective of patients with ovarian cancer undergoing first-line treatment.
| Health state | Mean estimate | se |
|---|---|---|
| SD | 0.718 | 0.01 |
| PD | 0.649 | 0.02 |
With respect to disutilities associated with adverse treatment effects, four studies124,136,137,139 were identified that reported utilities associated with AEs in ovarian cancer. Havrilesky et al. 124 and Calhoun et al. 139 report mean values of health state valuations carried out by members of the public. By contrast, Sun et al. 137 and Hess et al. 136 report median values of patient and physician health-state valuation. Therefore, as mean versus median values and public versus patient preferences are recommended for use in economic evaluations,109 utility data from Havrilesky et al. 124 and Calhoun et al. 139 were selected over utility data from Sun et al. 137 and Hess et al. 136
The mean utility values reported for AEs in Calhoun et al. 139 are presented in Table 134. The mean utility values reported for AEs in Havrilesky et al. 125 are presented in Table 135.
| AE | Mean | n | sd |
|---|---|---|---|
| Mild ototoxicity | 0.88 | 39 | NR |
| Mild nephrotoxicity | 0.95 | 39 | |
| Mild neurotoxicity | 0.92 | 39 | |
| Severe ototoxicity | 0.38 | 39 | |
| Severe nephrotoxicity | 0.27 | 39 | |
| Severe neurotoxicity | 0.47 | 39 |
| AE | Mean | n | sd |
|---|---|---|---|
| Alopecia, grade 2 | 0.84 | 14 | 0.29 |
| Peripheral neuropathy, grades 1 and 2 | 0.81 | 15 | 0.29 |
| Stomatitis, grade 2 | 0.91 | 14 | 0.08 |
| Myalgia/pain, grades 1 and 2 | 0.89 | 15 | 0.12 |
| Nausea/vomiting, grades 1 and 2 | 0.76 | 15 | 0.28 |
| Myalgia/pain, grades 3 and 4 | 0.46 | 15 | 0.39 |
| Neutropenia, grade 4 | 0.64 | 16 | 0.36 |
| Peripheral neuropathy, grades 3 and 4 | 0.65 | 14 | 0.31 |
| Nausea/vomiting, grades 3 and 4 | 0.63 | 16 | 0.30 |
| Fatigue, grades 3 and 4 | 0.58 | 13 | 0.33 |
| Febrile neutropenia | 0.56 | 15 | 0.34 |
Havrilesky et al. 124 also describe health state valuations via TTO for recurrent ovarian cancer with and without grades 1 and 2 and grades 3 and 4 toxicity. These valuations were based upon both volunteers and women with ovarian cancer. These are presented within Table 136.
| Health state | Mean | n | sd |
|---|---|---|---|
| Recurrent ovarian cancer, responding to chemotherapy grades 1 and 2 toxicity | 0.50 | 15 | 0.34 |
| Recurrent ovarian cancer, responding to chemotherapy grades 3 and 4 toxicity | 0.61 | 14 | 0.24 |
| Recurrent ovarian cancer, progressive grades 1 and 2 toxicity | 0.40 | 16 | 0.33 |
| Recurrent ovarian cancer, progressive grades 3 and 4 toxicity | 0.47 | 15 | 0.34 |
| End-stage ovarian cancer | 0.16 | 15 | 0.25 |
There are a number of reliability issues with the incorporation, in the economic model, of disutility values calculated from either Calhoun et al. 139 or Havrilesky et al. 124
First, the sample size on which the estimates are based is small, ranging from 13 people to 16 people for Havrilesky et al. 124 and up to 39 people for Calhoun et al. 139 Second, for Havrilesky et al. 124 certain mean values (presented in Table 136) are counter intuitive. For example, the utility value for recurrent ovarian cancer with grades 1 and 2 AEs is lower than recurrent ovarian cancer with grades 3 and 4 AEs (whether progressive ovarian cancer or responding to therapy). Finally, the impact of AEs on patient QoL associated with trabectedin plus PLDH and PLDH monotherapy are already implicitly included within health-state EQ-5D estimates from TA222. 90 This means that addition of disutility values may result in double counting of the impact of AEs for these therapies. For these reasons, the impact of applying disutilities as a result of treatment-related AEs has been excluded from the base-case analysis and tested in sensitivity analysis. Therefore, the base-case analysis assumes that the impact of AEs on patient QoL is accounted for in the mean estimates of utility associated with the model health states; however, costs of grade 3 and grade 4 AEs were applied in the base case (see Costs, below).
Costs
The following costs are captured within the TAG’s economic model: chemotherapy; administration; health-state related; and AEs. A systematic search for UK-based cost studies to populate these parameters was carried out as part of the systematic review for economic evaluation studies. The TAG considered that UK-based costing studies would provide the most relevant information for the economic analysis, which was carried out from a UK perspective. The search strategy is described above (see Technology Assessment Group systematic review of existing cost-effectiveness evidence). A total of 18 studies were identified as purely costing studies; however, none of these studies was UK based and were therefore not considered relevant for this MTA. (The country for each excluded costing study is listed in Appendix 6 for information.) Consequently, where appropriate, costs included in the de novo analysis have been estimated from standard UK sources; these are described in further detail below.
Intervention/comparator chemotherapy costs
A summary of the chemotherapy regimens, and cost per administration applied within the economic model is presented in Table 137.
| Chemotherapy | Regimen description | Chemotherapy cost per cycle (£) |
|---|---|---|
| Paclitaxel | For PRR disease: paclitaxel 80 mg/m2 weekly for 18 weeks or until progression | 306 |
| For platinum-sensitive disease: paclitaxel 175 mg/m2 day 1 every 21-day cycle (maximum of six cycles) | 638 | |
| Paclitaxel plus platinum | For PRR disease: paclitaxel 80 mg/m2 plus carboplatin AUC 3,a weekly for 18 weeks or until progression | 442 |
| For platinum-sensitive disease: paclitaxel 175 mg/m2 and carboplatin AUC 5,a day 1 every 21-day cycle (maximum of six cycles) | 855 | |
| PLDH | 40 mg/m2 day 1 every 28-day cycle (maximum of six cycles) | 1211 |
| PLDH plus platinum | PLDH 30 mg/m2; carboplatin target AUC of 5,a day 1 every 28-day cycle (maximum of six cycles) | 1137 |
| Gemcitabine plus carboplatin | Gemcitabine 1000 mg/m2 day 1 and 8 every 21-day cycle, carboplatin target AUC of 4a day 1 every 21-day cycle (maximum of six cycles) | 706 |
| Trabectedin plus PLDH | Trabectedin 1.1 mg/m2; PLDH 30 mg/m2, day 1 every 21-day cycle (maximum of six cycles) | 3679 |
| Topotecan | 1.5 mg/m2, days 1–5 every 21-day cycle (maximum of six cycles) | 1305 |
| Platinum monotherapy | Carboplatin target AUC of 5,a day 1 every 21-day cycle (maximum of six cycles) | 217 |
| Etoposide (sensitivity analysis only) | 50 mg (oral) days 1–21 every 28 days (maximum of six cycles) | 200 |
| Etoposide plus platinum (sensitivity analysis only) | Etoposide 50 mg (oral) days 1–21 every 28-day cycle plus cisplatin i.v. 50 mg/m2 day 1, 8 and 15 every 28-day cycle (maximum of six cycles) | 340 |
The regimen descriptions presented in Table 137 were obtained through review of each relevant SmPC, with verification and amendment from clinical experts. The costs per cycle outlined in Table 137 are applied within the model to people within the SD health state, for up to the stated maximum number of cycles. The single exception to this is for patients treated with trabectedin plus PLDH. For this regimen, although the maximum number of cycles likely to be used in clinical practice is six cycles, the manufacturer for trabectedin has submitted a PAS. The manufacturer’s PAS limits the number of cycles for which the NHS would bear the cost to five cycles. Therefore, in the base-case analysis, for trabectedin plus PLDH, a maximum of five cycles were costed. Furthermore, as highlighted by PharmaMar, the implementation of such a PAS would result in an administration cost which would be borne by the NHS. Therefore, a PAS implementation cost was included within the TAG model. Within the manufacturer’s PAS submission, the total cost of PAS administration was estimated as £560.74 (Table 138). The TAG notes that this cost was subject to discounting and, given that treatment would occur in the first year of the model, the TAG included the non-discounted cost (£598.04) within the TAG economic analysis.
| PAS implementation costs | Annual cost (£) | Years | Discounted cost (£) |
|---|---|---|---|
| NHS trust costs of the PAS | 151.74 | 0.938 | 140.07 |
| NHS trust costs of claiming free of charge stock | 204.36 | 0.938 | 188.64 |
| NHS trust implementation and training | 66.50 | – | 66.50 |
| NHS trust scheme agreement | 46.80 | – | 46.80 |
| PCT costs of the PAS | 128.64 | 0.938 | 118.74 |
| Total | 598.04 | – | 560.74 |
For each regimen, the cost of treatment relies upon one or more patient characteristics, for example age, weight or BSA in square metres. To reflect the variation in such characteristics at a patient level, and the associated impact upon estimated cost, data from 321 patients with ovarian cancer described by Sacco et al. 114 have been used to estimate dose, and therefore cost, at an individual level. The cost reported in Table 137 is an average of the cost associated with each of the 321 patients for which treatment cost has been calculated. Therefore, uncertainty associated with patient characteristics has been accounted for in the base-case analysis. Full details of the calculations used to estimate the average cost per administration are presented below.
Patient-level characteristics
Patient-level characteristics of age and BSA were taken from Sacco et al. ,114 who report the results of a multicentre, retrospective study of the BSA of adult cancer patients in the UK. Sacco et al. 114 measured the BSA of 3613 patients receiving chemotherapy for various cancers, including 321 patients with ovarian cancer, for which the age and BSA of each patient is freely available online. The average age and BSA of the 321 patients with ovarian cancer as reported in Sacco et al. 114 are 61.4 years and 1.71 m2, respectively.
For the majority of the chemotherapy agents of interest for this MTA, doses are based upon BSA, for which IPD were available for analysis from Sacco et al. ;114 however, for estimates of carboplatin dose, individual weight data are also required. Therefore, a calculation was necessary to estimate, given their BSA and age, weight for each of the 321 patients assessed by Sacco et al. 114 The TAG used the commonly used Du Bois and Du Bois formula to estimate weight based upon BSA and height, noting that data on individual height were unknown and therefore required estimation,141 where if:
then:
Estimates of individual BSA were taken directly from the ovarian cancer data set in Sacco et al. 114 For each individual patient, height was estimated, based upon the age of the patients within Sacco et al. ,114 using the Health Survey for England (HSE),142 in which average height by age and gender is provided. The BSA and height information for each individual patient were then used to estimate weight for each individual patient; the average weight for the patient cohort estimated using this formula is 69.1 kg.
The TAG compared this estimate of weight with the estimate of weight determined using the HSE,142 in which average weight by age and gender is provided. For the patient cohort, the average weight, using the HSE data,142 is 72.1 kg. The TAG notes that the difference in average weight between the two estimates was 3 kg. The TAG considers that the estimates based upon BSA are more likely to reflect the weight associated with women with ovarian cancer; however, a scenario analysis using weight estimated from the HSE142 was tested in sensitivity analysis (see Approach to uncertainty, below).
Unit prices of chemotherapy agents
Unit prices were obtained from the BNF 65 (Table 139). 110 Where available, unit costs were obtained for the non-proprietary formulation. The impact of branded unit costs was explored in sensitivity analysis (see Approach to uncertainty, below).
| Chemotherapy (i.v.) | Vial size | mg/ml | mg | Price per vial (£) |
|---|---|---|---|---|
| Paclitaxel (non-proprietary) | 5 ml | 6 | 30 | 66.85 |
| 16.7 ml | 6 | 100.2 | 200.35 | |
| 25 ml | 6 | 150 | 300.52 | |
| 50 ml | 6 | 300 | 601.03 | |
| Carboplatin (non-proprietary) | 5 ml | 10 | 50 | 22.04 |
| 15 ml | 10 | 150 | 56.92 | |
| 45 ml | 10 | 450 | 168.85 | |
| 60 ml | 10 | 600 | 260.00 | |
| Cisplatin (non-proprietary)a | 10 ml | 1 | 10 | 5.85 |
| 50 ml | 1 | 50 | 24.50 | |
| 100 ml | 1 | 100 | 50.22 | |
| PLDH | 10 ml | 2 | 20 | 360.23 |
| 25 ml | 2 | 50 | 712.49 | |
| Gemcitabine (non-proprietary) | 200 mg | NA | 200 | 32.00 |
| 1000 mg | NA | 1000 | 162.00 | |
| 1500 mg | NA | 1500 | 213.93 | |
| 2000 mg | NA | 2000 | 324.00 | |
| Trabectedin | 0.25 mg | NA | 0.25 | 363.00 |
| 1 mg | NA | 1 | 1366.00 | |
| Topotecan (non-proprietary) | 1 ml | 1 | 1 | 87.88 |
| 4 ml | 1 | 4 | 261.55 | |
| Chemotherapy (tablets) | Tablets per pack | mg per tablet | Price per pack (£) | Price per tablet (£) |
| Etoposidea | 20 | 50 | 99.82 | 4.99 |
For the purposes of the base-case analysis, it was assumed that no vial sharing would occur. Therefore, for each therapy, a series of dosing ‘rules’ were established to indicate, for a given individual dose, which vial(s) would be used. It was assumed that for each individual, the selected vial or combination of vials would be those resulting in the lowest possible total cost. Vial sharing was included as a scenario analysis (see Approach to uncertainty, below).
Additional treatment costs: pre-, concomitant and maintenance treatment
In clinical practice, patients would typically be pretreated with a variety of anti-sickness therapies [e.g. ondansetron (Zofran®, GSK); granisetron (Kytril®, Roche)], with such treatment typically continuing throughout the course of treatment. For simplicity, given that these costs are applicable for all therapies, they have been excluded from the economic analysis.
For regimens including paclitaxel, in addition to the usual pre-treatment with anti-sickness therapies, pre-treatment with corticosteroids is required to prevent severe hypersensitivity reactions. This requirement is not necessary for other therapies; therefore, it was considered appropriate to include a cost associated with dexamethasone within the analysis (cost per cycle £4.15, based upon the cost of five 1-ml ampoules at 83p each142).
Finally, in the sensitivity analysis assessing the difference in cost of therapies used to treat patients with PRR disease, the two etoposide regimens are associated with the additional cost of maintenance therapy. Specifically, following completion of the etoposide regimen, 6–8 weeks of oral etoposide at 50 mg/day would typically be prescribed on an outpatient basis; this cost is included in the analysis with the assumption that packs of oral etoposide could not be shared. In addition, an average duration of 7 weeks of therapy is assumed (i.e. midway between 6 and 8 weeks), equating to 49 tablets or three packs of tablets. The cost for a single pack of oral etoposide tablets is £99.82, resulting in a total cost for the maintenance period of £299.46 (Table 140). 142
| Chemotherapy (tablets) | Tablets per pack | mg per tablet | Price per pack (£) | Cost per 7 weeks treatment (no pack sharing) (£) |
|---|---|---|---|---|
| Vepesid (etoposide) | 20 | 50 | 99.82 | 299.46 |
Administration costs
With the exception of oral etoposide, every chemotherapy regimen is assumed to be administered as an infusion within a hospital. To capture the costs associated with this, for each regimen, a cost of administration is applied within the economic model. This cost is assumed to comprise the cost of preparing the infusion(s) in the pharmacy and the cost associated with delivering the infusion in the hospital. A summary of the administration costs applied within the economic model is presented in Table 141. The calculation of these costs is described below.
| Regimen | Pharmacy preparation cost per cycle (£) | First cycle delivery cost (£)a | Subsequent cycle delivery costs (£)a | |
|---|---|---|---|---|
| Paclitaxel | Paclitaxel 80 mg/m2 weekly for 18 weeks or until progression | 16 | 200 | 270 |
| Paclitaxel 175 mg/m2 day 1 every 21-day cycle | 16 | 331 | 270 | |
| Paclitaxel plus platinum | Paclitaxel 80 mg/m2 plus carboplatin AUC 3, weekly for 18 weeks or until progression | 31 | 200 | 270 |
| Paclitaxel 175 mg/m2 and carboplatin AUC 5, day 1 every 21-day cycle | 31 | 331 | 270 | |
| PLDH 40 mg/m2 day 1 every 28-day cycle | 16 | 249 | 270 | |
| PLDH 30 mg/m2; carboplatin target AUC of 5, day 1 every 28-day cycle | 31 | 331 | 270 | |
| Gemcitabine 1000 mg/m2 days 1 and 8 every 21-day cycle, carboplatin target AUC of 4, day 1, every 21-day cycle | 47 | 520 | 541 | |
| Trabectedin 1.1 mg/m2; PLDH 30 mg/m2, day 1 every 21-day cycle | 31 | 331 | 270 | |
| Topotecan 1.5 mg/m2, days 1–5 every 21-day cycle | 78 | 1281 | 1351 | |
| Carboplatin target AUC5, day 1 every 21-day cycle | 16 | 200 | 270 | |
| Etoposideb 50 mg (oral) days 1–21 every 28 days | 0 | 0 | 0 | |
| Etoposideb 50 mg (oral) days 1–21 and cisplatin 50 mg/m2, days 1, 8 and 15, every 28-day cycle | 47 | 872 | 811 | |
For the cost of preparing the infusion(s) it was assumed, based upon clinical expert opinion described in a recently published STA, that preparation of one infusion would in practice take approximately 20 minutes. 16 Therefore, for all single agents, the cost of preparation of each infusion was estimated as the cost per minute associated with a hospital pharmacist, multiplied by 20 minutes. For combination therapies, the cost of preparation of each infusion was estimated as the cost per minute associated with a hospital pharmacist multiplied by 40 minutes (two agents at 20 minutes each). The cost associated with a hospital pharmacist was taken from the Unit Costs of Health and Social Care 2012 (cost per hour, £47; cost per minute, £0.78). 115
For the cost of delivering the infusion, the following NHS Reference Costs were selected. 111
To deliver:
-
complex chemotherapy, including prolonged infusional treatment at first attendance (SB14Z), £331
-
more complex parenteral chemotherapy at first attendance (SB13Z), £249
-
simple parenteral chemotherapy at first attendance (SB12Z), £200
-
subsequent elements of a chemotherapy cycle (SB15Z), £270.
The selection of the relevant first attendance code was based upon the maximum infusion time recommended within the associated SmPC:
-
For chemotherapy agents considered to require up to 60 minutes’ infusion time (weekly paclitaxel, carboplatin monotherapy, topotecan), the cost of SB12Z: Deliver simple parenteral chemotherapy at first attendance (£200) is applied.
-
For agents considered to require up to 120 minutes’ infusion time (PLDH, gemcitabine and carboplatin combination, etoposide and carboplatin combination), the cost of SB13Z: Deliver more complex parenteral chemotherapy at first attendance (£249) is applied.
-
For agents considered to require > 120 minutes’ infusion (cisplatin, paclitaxel monotherapy, paclitaxel combination therapy, PLDH and carboplatin combination, PLDH and trabectedin combination), the cost of SB14Z: Deliver complex chemotherapy, including prolonged infusional treatment at first attendance (£331) is applied.
-
For combination therapies, based upon clinical advice it was considered that infusions would occur sequentially, and therefore the combined duration of infusion was used to infer the relevant Healthcare Resource Group (HRG) code at first attendance.
-
For subsequent cycles, for all therapies, the cost of SB15Z: Deliver subsequent elements of a chemotherapy cycle (£270) is applied.
It is noted within the trabectedin SmPC that insertion of a central line is required for administration of trabectedin. The manufacturer for trabectedin, PharmaMar, accounted for this within the submission for this MTA by including a one-off cost associated with insertion of a central line. Following consultation with clinical experts, the TAG notes that, in clinical practice, many women eventually require insertion of a central line owing to increasing difficulties gaining venous access. For this reason, the TAG considers that the cost of insertion of a central line would be similar across treatment regimens and has therefore omitted this cost from the economic analysis.
Health-state costs
Costs attributable to the SD period and the PD period are included in the economic analysis. A summary of these costs is presented in Table 142. These costs are applied monthly to the number of patients residing in each health state. The calculation of these costs is discussed in detail below.
| Cost | SD health state | PD health state (platinum-sensitive patients) | PD health state (PRR patients) |
|---|---|---|---|
| One-off initial cost (£) | NA | 109 | 109 |
| Cost (£) per month (first 6 months) | 45 | 796 | 531 |
| Cost (£) per month (subsequent months) | 45 | 531 | 531 |
Stable disease health-state costs
The cost associated with the SD health state comprises the cost of monitoring for patients with SD. In the base-case analysis it is assumed that, based upon discussions with clinical experts, patients with SD require one outpatient visit every 3 months. The cost of an outpatient visit was estimated based upon NHS Reference Costs (2011–12) outpatient attendance data, service code 503, gynaecologic oncology, to be £135. 111 This equates to £45 per month for a patient within the SD health state.
Progressed disease health-state costs
For patients with recurrent ovarian cancer whose disease progresses after treatment for recurrent disease, treatment can vary. A proportion of women will receive subsequent lines of chemotherapy and may respond to these agents; many women may complete a further five or more lines of chemotherapy. However, for other patients with recurrent disease, treatment following progression can be considered palliative in intent, and may focus on ameliorating symptoms of disease. 4
To better understand these differences in treatment following progression, the TAG consulted with clinical experts, following which, the TAG considers that for women who subsequently progress following treatment for PRR disease, prognosis would indicate that subsequent treatment may more typically be palliative in intent. For these women, a cost associated with palliative care is applied for each month spent in the PD health state. For women who subsequently progress following treatment for platinum-sensitive disease, a cost associated with a further line of chemotherapy is applied. This cost is applied for 6 months, following progression. After this point, a cost associated with palliative care is applied for each month spent in the PD health state. The TAG acknowledges that the approach taken is a simplification of the reality of treatment following progression, which can and does vary for every woman; however, the TAG considers that by applying costs in this way, some key aspects of the cost of PD may be captured within the model. These costs were tested in sensitivity analysis (see Approach to uncertainty, below).
The cost associated with a further line of therapy (for patients progressing following treatment for platinum-sensitive disease) included the cost of chemotherapy, administration, and further monitoring for a 6-month period. A study by Kaye et al. ,66 reporting the use of chemotherapy agents following progression in patients treated for recurrent ovarian cancer, was used to inform the cost of chemotherapy. They reported that ∼80% of platinum-sensitive women went on to receive at least one subsequent therapy. Of these, the majority of women received chemotherapy (≈75%). For women who received chemotherapy, ∼75% received platinum-based chemotherapy and ≈25% received non-platinum-based therapy. 66
Therefore, in the economic analysis it was assumed that 100% of women who progressed following treatment for platinum-sensitive disease went on to receive a further line of therapy. This simplifying assumption was designed to reflect the fact that although not all women will go on to receive another line of chemotherapy, some women will receive more than one line of chemotherapy. Following discussion with clinical experts, who advised that PLDH monotherapy and platinum monotherapy would be the most likely treatment options, the cost applied within the economic model was estimated as 75% of the cost of carboplatin AUC 5 (to reflect the ∼75% of women receiving platinum based therapy), and 25% of the cost of PLDH (to reflect the ∼25% of women receiving non-platinum-based therapy) (Table 143).
| Treatment | Cost of chemotherapy agent per cycle (£) | Cost of administration/pharmacy infusion per cycle (£) | Total cost per cycle (£) | Weight (%) |
|---|---|---|---|---|
| Carboplatin AUC 5 | 216 | 286 | 539 | 75 |
| PLDH | 1211 | 286 | 1497 | 25 |
| Total | 751 |
Given that, in clinical practice, both carboplatin and PLDH monotherapy would typically be limited to six cycles, the average cost per month over a 6-month period was estimated to be £751 for chemotherapy and administration. Including the cost of monitoring, as estimated for patients in the SD period (£45 per month), the cost per month applied to platinum-sensitive patients for the first 6 months following progression is £796. The TAG recognises that this estimate is a simplification of the true value and therefore tested this figure in sensitivity analysis (see Approach to uncertainty, below).
To establish a published source of palliative care, the TAG carried out a rapid review of the literature in PubMed in February 2013. The TAG used broad disease terms [(ovarian or ovary) and (cancer)] alongside terms for palliative care [(palliative care) or (end of life)], cost (cost), and country (UK OR united kingdom OR britain OR england OR scotland OR wales OR ireland). A total of three studies were identified from this search, of which one study112 was considered relevant. This study, by Guest et al. ,112 has previously been identified by the ERG responsible for considering evidence submitted for TA222. 90
Guest et al. 112 investigated the resource use and cost associated with patients with a malignant neoplasm from the time they started strong opioid treatment until death. The study estimated the cost associated with a total of 547 patients, of which 21 patients (4% of the sample) were diagnosed with ovarian cancer. The palliative cost associated with ovarian cancer was estimated by Guest et al. 112 to be £4789 (at 2000–1 prices) for an average time period of 399 days. This cost predominantly consisted of hospitalisation costs (71% of costs), updating the estimate of palliative care for patients with ovarian cancer from Guest et al. 112 to current prices using the Hospital & Community Health Services index results in a cost of £6963, equating to £531 per month. This cost is applied monthly to all PRR patients following entry into the PD health state, and all platinum-sensitive patients following 6 months of residence in the PD health state.
The TAG notes that the analysis carried out by Guest et al. 112 has several weaknesses. In particular, ovarian cancer estimates are based upon a small sample size (n = 21) and does not consider costs for patients not requiring a strong opioid. In addition, the analysis was carried out in 2000–1 and may no longer reflect clinical practice. Therefore, in recognition of the uncertainty associated with the cost of palliative care, the TAG has tested this parameter in sensitivity analysis (see Approach to uncertainty, below).
Finally, in addition to the cost of further treatment and care, a one-off cost associated with a CT scan is applied at progression. This is to reflect that, during routine outpatient appointments in the stable period, a CA125 test is typically carried out. If a CA125 test indicates possible disease progression, a CT scan is then undertaken. The TAG acknowledges that some CT scans following raised CA125 levels would not necessarily indicate disease progression; however, for simplicity, these additional scans have been excluded from the cost. The TAG considers it likely that such additional scans would be equally likely across treatments and therefore the variation is unlikely to materially impact upon results. The cost of a CT scan was estimated to be £109, based upon NHS Reference Costs 2012. 111 It was estimated as the weighted average of outpatient CT scans (RA08A, RA09A, RA10Z-RA14Z; weighted by activity).
Adverse event costs
As described in Adverse event incidence, following discussion with clinical experts, the key AEs identified from the clinical review are allergic reaction, alopecia, anaemia, fatigue, febrile neutropenia, nausea and vomiting, and neuropathy. The costs ascribed to each of these AEs within the economic model are presented in Table 144.
| AE | Mean cost (£) | Source |
|---|---|---|
| Allergic reaction | 145 |
|
| Alopecia | NA | No cost ascribed to alopecia |
| Anaemia | 488 | Weighted (by activity) average of NHS Reference Costs,111 SA13A Single Plasma Exchange, Leucophoresis or Red Cell Exchange, with length of stay ≤ 2 days, ≥ 19 years, SA14Z Plasma Exchanges 2 to 9, SA15Z Plasma Exchanges 10 to 19, SA16Z Plasma Exchanges 20 or more |
| Fatigue | NA | No cost ascribed to fatigue |
| Febrile neutropenia | 1077 | NHS Reference Costs111 weighted mean HRG cost SA01F: A plastic Anaemia without CC |
| Nausea and vomiting | 160 |
|
| Neuropathy | NA | No cost ascribed to neuropathy |
No costs were ascribed to alopecia, neuropathy or fatigue within the economic analysis. This is because, in practice, these AEs are not easily treated and a cost to the NHS is not, in general, incurred. For alopecia and neuropathy, alternative therapies or a reduction in dose of chemotherapy would be more likely to be considered. The TAG recognises that, in particular, alopecia and fatigue can be distressing and problematic conditions for both patients and the clinicians treating them. The TAG attempted to capture the impact of these conditions through a sensitivity analysis, which included a QoL decrement (see Approach to uncertainty, below).
The AE costs detailed in Table 144 are applied to the AE incidence (see Adverse event incidence) to estimate a total cost of treating AEs for each treatment regimen. For simplicity, it was assumed that these costs were incurred at the start of treatment within the economic model.
Cost summary
A summary of the costs, by treatment regimen, included within the TAG’s de novo economic analysis is presented in Table 145.
| Chemotherapy regimen | Chemotherapy cost per cycle (£) | Administration cost cycle 1 (£) | Administration cost cycle 2 onwards (£) | Cost of AEs (during treatment) (£) | Health-state costs (per month) (£) | ||
|---|---|---|---|---|---|---|---|
| Stable period | Progressed period, PS patients, months 1–6 | Progressed period, PS patients months 6+ or PRR patients from progression | |||||
| Paclitaxel 80 mg/m2 weekly (cycle) for 18 weeks or until progression | 306 | 215 | 286 | 111 | 45 | 796 | 531 |
| Paclitaxel 175 mg/m2 day 1 every 21-day cycle | 638 | 347 | 286 | 111 | 45 | 796 | 531 |
| Paclitaxel 80 mg/m2 plus carboplatin AUC 3, weekly for 18 weeks or until progression | 442 | 231 | 302 | 78 | 45 | 796 | 531 |
| Paclitaxel 175 mg/m2 and carboplatin AUC 5, day 1 every 21-day cycle | 855 | 363 | 302 | 78 | 45 | 796 | 531 |
| PLDH 40 mg/m2 day 1 every 28-day cycle | 1211 | 265 | 286 | 69 | 45 | 796 | 531 |
| PLDH 30 mg/m2; carboplatin target AUC of 5, day 1 every 28-day cycle | 1137 | 363 | 302 | 97 | 45 | 796 | 531 |
| Gemcitabine 1000 mg/m2 days 1 and 8 every 21-day cycle, carboplatin target AUC of 4, day 1 every 21-day cycle | 706 | 567 | 588 | 172 | 45 | 796 | 531 |
| Trabectedin 1.1 mg/m2; PLDH 30 mg/m2, day 1 every 21-day cycle | 3679 | 363 | 302 | 198 | 45 | 796 | 531 |
| Topotecan 1.5 mg/m2, days 1–5 every 21-day cycle | 1305 | 1359 | 1430 | 200 | 45 | 796 | 531 |
| Carboplatin target AUC 5, day 1 every 21-day cycle | 217 | 215 | 286 | 33 | 45 | 796 | 531 |
| Etoposide 50 mg (oral) days 1–21 every 28-day cyclea | 200 | 0 | 0 | 39 | 45 | 796 | 531 |
| Etoposide 50 mg (oral) days 1–21 and cisplatin 50 mg/m2 days 1, 8 and 15 every 28-day cyclea | 340 | 919 | 858 | 53 | 45 | 796 | 531 |
Approach to uncertainty
The impact of parameter uncertainty upon model results has been investigated in both probabilistic sensitivity analyses (PSAs) and deterministic (one-way) sensitivity analyses. In addition, (where possible) structural assumptions have been varied in deterministic scenario analyses. As a result of time constraints and the volume of sensitivity analysis carried out, deterministic rather than probabilistic analysis was selected to inform one-way sensitivity and scenario analysis. However, based on the consistency observed between probabilistic and deterministic base-case results, the TAG considers that deterministic assessment of model sensitivity is reasonable.
Probabilistic sensitivity analyses
Within the TAG’s economic model, PSA has been used to investigate the simultaneous impact of parameter uncertainty on the cost-effectiveness results. Probability distributions were assigned to each parameter (except drug acquisition costs) used within the model, from which values have been simultaneously sampled 1000 times. Based on assessment of the stability of model results, ‘1000’ was chosen as the sample size for probabilistic analysis, assessed by comparing deterministic and probabilistic results obtained for sample sizes of 1000, 2000 and 5000. There was assumed to be zero uncertainty associated with drug acquisition costs. Table 146 summarises the type of distribution, and rationale for selection of the distribution, used to inform each group of parameters; full details of distributional specifications are provided in Table 115.
| Parameter type | Parameter description | Distribution(s) used | Rationale |
|---|---|---|---|
| Probability of PFS and OS associated with baseline curve | Parameters associated with selected distribution (Weibull in the base case) | Multivariate normal | Each parameter is sampled from a multivariate normal distribution using the Cholesky decomposition method143 |
| HRs | HRs estimated from TAG’s NMA | NA | The CODA output, from WinBUGS, provides a list of all values generated from the full posterior distribution. Therefore, rather than resampling from the posterior distribution, the output itself has been used in PSA144 |
| Costs | Unit costs of drug administration and delivery, unit costs of patient follow-up and care, cost of palliative care, unit costs associated with AEs | Gamma or log-normal | Either the gamma or log-normal distribution may be considered suitable for the sampling of cost data.143 Therefore, the distribution selected to inform each individual cost was dependent on the ability of that distribution to reproduce the inputted 95% CI or standard error. Note: where 95% CIs or standard errors were not available from the literature a standard error of 0.25 was assumed |
| OR | AEs | Log-normal | The CODA output, from WinBUGS provides a list of all values generated from the full posterior distribution. Therefore, rather than resampling from the posterior distribution, the output itself has been used in PSA144 |
| Probability of: | Treatment selected for further lines of therapy. Baseline probability of AEs. Probabilities of AEs based on clinical opinion | Beta | Probabilities that are based on the proportion of observed outcomes (i.e. probability of event is 1-probability of non-event) may be assumed to follow a binomial distribution. Therefore, the beta distribution was used as it is the conjugate of the binomial distribution and is bounded by 0 and 1143 Note: where 95% CIs or standard errors were not available from the literature a standard error of 0.25 was assumed |
| Utilities/disutilities | SD, PD utilities | Beta | The beta distribution was chosen based on the (0,1) boundary imposed by this distribution143 |
One-way sensitivity analysis
For each therapy, by subgroup, all model parameters with the exception of drug costs were varied in one-way sensitivity analysis. Parameters were assigned low and high values according to the 95% CI used in the PSA. The deterministic cost-effectiveness result was recorded for each one-way change in each parameter estimate. The variables associated with the greatest impact upon cost-effectiveness results are presented in tornado diagram format below (see Results of the sensitivity analysis, below).
Scenario analyses
A variety of structural assumptions have been made in the construction of the TAG’s base-case model. Where possible. these have been tested in scenario analysis. Table 147 lists the scenario analyses carried out by the TAG, the parameters used to inform these scenarios, and the rationale for each analysis.
| Scenario analysis | Parameter definition | Rationale |
|---|---|---|
| Cost scenarios | ||
| Costs associated with a 50-mg rather than 40-mg dose of PLDH | Cost per cycle for a 50-mg dose estimated to be £1443 using the methods described above (see Costs) | To establish the impact of using the cost likely to be incurred in clinical practice in the base case |
| Patient weight (used to inform drug costs) estimated from the HSE 2011142 | Estimating individual patient weight from Sacco et al.114 using HSE 2011,142 based upon the patient’s age | To assess the potential impact of patient-level data used to inform drug cost calculations |
| Branded costs of drugs |
|
To assess the potential impact of the use of branded drugs |
| Calculating cost based upon the selection of vials that resulted in the least number of vials used | For each chemotherapy, the combination of vials which resulted in the fewest number of vials used was investigated | To assess the robustness of the cost-effectiveness results of the calculation of drug costs |
| Vial sharing | For each chemotherapy, an average cost per mg was estimated and applied to the dose (mg) required per patient | To assess the potential impact of wastage on the cost-effectiveness results |
| Efficacy scenarios | ||
| Equivalent efficacy assumed for all therapies outlined within the NICE scope for patients with resistant/refractory disease with differences | Efficacy for all pharmacotherapies set to the baseline PFS and OS for PLDH in resistant/refractory patients
|
To reflect clinical advice that prognosis is often similar, and to investigate the cost impact associated with each therapy outlined in the NICE scope |
| Baseline PS PFS survival curve network 1 using alternative functional forms |
|
To assess the impact of the data and functional form of the baseline PFS and OS estimates |
| Baseline PS PFS survival curve network 1 using Parmar et al.61 | Weibull curve fitted to the ICON4/AGO-OVAR 2.2 data from Parmar et al.61 using methods outlined in Hoyle and Henley,113 rather than to the CALYPSO data from Pujade-Lauraine et al.31 data | |
| Baseline PS OS survival curve network 1 using alternative functional forms for Wagner et al.56 |
|
|
| Baseline PS OS survival curve network 1 using Parmar et al.61 | Weibull curve fitted to the ICON4/AGO-OVAR 2.2 data from Parmar et al.61 using methods outlined in Hoyle and Henley,113 rather than to the CALYPSO data from Wagner et al.56 | |
| Baseline PS PFS survival curve network 2 using extrapolated estimates rather than Kaplan–Meier data |
|
|
| Baseline PS OS survival curve network 2 using alternative function forms for the Kaplan–Meier data |
|
|
| Baseline PRR PFS survival curve using alternative functional forms for Monk et al.30 | ||
| Baseline PRR OS survival curve using alternative functional forms for CSR data | ||
| Head-to-head comparison of trabectedin plus PLDH with PLDH in platinum-sensitive patients, using adjusted PFS and OS estimates from the PharmaMar submission | The manufacturer base-case PFS and OS extrapolations were used within the TAG economic model | To assess the impact of using adjusted survival estimates within the TAG economic model, and to assess the face validity of the TAG and manufacturer ICERs for PLDH vs. trabectedin when using the same efficacy data |
| Patient subgroups | ||
| Analysis of the results considering the PPS subgroup alone (PFI 6–12 months) | Exploratory analysis using the OS NMA results for the PPS subgroup. Baseline survival for PLDH for the platinum-sensitive population was used; this was because no numbers of patients at risk were available on published Kaplan–Meier graphs. In addition, no PFS data were inputted owing to no possible network | To provide exploratory results for this patient subgroup; sufficient data were not available from the FPS subgroup in order to assess this comparison in addition |
| Other | ||
| Alternative discount rates for costs and benefits | Discount rate for costs and benefits assumed to be 1% or 6% | As per NICE guidelines |
| Disutilities for AEs applied | Disutilities from Havrilesky et al.124 (see Health-related quality-of-life data, above) for nausea and vomiting, fatigue, and febrile neutropenia; applied assuming:
|
To assess the potential impact of the different AE profiles associated with the treatments of interest |
| Nausea and vomiting probabilities for PS network 1 estimated from clinical expert opinion |
|
To assess the potential impact of alternative sources of AE probabilities on model results |
| Half-cycle correction | Half-cycle correction was applied to the estimates of PFS and OS | To assess the potential impact of half-cycle correction on model results |
Base-case results
Fully incremental probabilistic and deterministic results for each of the subgroups analysed are presented below (see Tables 148–150, below). For each set of results, interventions are ordered with respect to their total cost. Interventions with higher incremental costs and lower incremental QALYs than their predecessor are considered to be strictly dominated, by their predecessor, and are therefore removed from consideration in the final ICER calculations. Similarly, interventions with higher incremental costs and lower incremental QALYs (vs. the baseline treatment) than their predecessor are considered to be extendedly dominated, by their predecessor, and are removed from consideration in the final ICER calculations.
People with platinum-sensitive disease
As described above (see Interventions and comparators), no single network comprising the interventions and comparators of interest as outlined in the NICE scope was possible from the data identified. Instead, two separate networks were constructed. Network 1 comprised platinum; paclitaxel plus platinum; PLDH plus platinum; and gemcitabine plus carboplatin. Network 2 comprised paclitaxel; PLDH; PLDH plus trabectedin; and topotecan.
For network 1, base-case deterministic results indicated that PLDH plus platinum is strictly dominated (is more costly and less effective than) by paclitaxel plus platinum. Similarly, gemcitabine plus carboplatin was estimated to be extendedly dominated by paclitaxel plus platinum. Therefore, PLDH plus platinum and gemcitabine plus carboplatin are removed from consideration in the final ICER calculation, leaving paclitaxel plus platinum compared with platinum monotherapy as the only relevant comparison for this network. For this comparison, the ICER is estimated as £24,361; paclitaxel plus platinum was associated with an estimated incremental cost of £5694 and an additional 0.23 QALYs when compared with platinum monotherapy (Table 148).
Probabilistic results were largely consistent with deterministic results. That is, PLDH plus platinum and gemcitabine plus carboplatin are estimated to be strictly dominated and extendedly dominated by paclitaxel plus platinum, respectively. Similar to the deterministic base-case result, the ICER of paclitaxel plus platinum compared with platinum monotherapy has been estimated as £24,539 per QALY gained.
However, the TAG considers it important to note that the costs and QALYs associated with PLDH plus platinum and paclitaxel plus platinum are similar. Consequently, small changes in total costs or QALYs associated with either treatment, may alter the results (see Results of the sensitivity analysis, below).
For network 2, base-case results (deterministic and probabilistic) indicate that topotecan is strictly dominated by PLDH. Topotecan was therefore removed from the analysis, leaving the relevant comparisons of PLDH versus paclitaxel, and trabectedin plus PLDH versus PLDH monotherapy. PLDH compared with paclitaxel results in estimated ICERs of £23,733 and £25,931 in deterministic and probabilistic analyses, respectively. When compared with paclitaxel, PLDH was associated with incremental costs of approximately £3,900 and approximately 0.16 additional QALYs. The ICERs for trabectedin plus PLDH compared with PLDH alone are estimated to be £85,212 and £81,353, deterministically and probabilistically, respectively. When compared with PLDH monotherapy, trabectedin plus PLDH is associated with approximately £13,000 incremental costs and 0.16 additional QALYs (Table 149).
| Treatment | Modelled regimen | Total cost (discounted) (£) | Total QALYs (discounted) | Incremental cost (discounted) (£) | Incremental QALYs (discounted) | Incremental ICER (cost/QALY) (£) | Incremental ICER (cost/QALY (excluding dominated options) (£) |
|---|---|---|---|---|---|---|---|
| Probabilistic results | |||||||
| Platinum | Carboplatin target AUC of 5, on day 1 of every 21-day cycle | 15,935 | 1.805 | – | – | – | – |
| Gemcitabine plus carboplatin | Gemcitabine 1000 mg/m2 on days 1 and 8 of every 21-day cycle, carboplatin target AUC of 4 on day 1 of every 21-day cycle | 20,426 | 1.852 | 4491 | 0.047 | 94,984 | Extendedly dominated |
| Paclitaxel plus platinum | Paclitaxel 175 mg/m2 and carboplatin AUC 5, on day 1 of every 21-day cycle | 21,604 | 2.036 | 1178 | 0.184 | 6411 | 24,539 |
| PLDH plus platinum | PLDH 30 mg/m2; carboplatin target AUC of 5, on day 1 of every 28-day cycle | 22,625 | 2.027 | 1021 | –0.009 | Strictly dominated | |
| Deterministic results | |||||||
| Platinum | Carboplatin target AUC of 5, on day 1 of every 21-day cycle | 15,949 | 1.799 | – | – | – | – |
| Gemcitabine plus carboplatin | Gemcitabine 1000 mg/m2 on days 1 and 8 of every 21-day cycle, carboplatin target AUC of 4 on day 1 of every 21-day cycle | 20,381 | 1.837 | 4432 | 0.039 | 114,410 | Extendedly dominated |
| Paclitaxel plus platinum | Paclitaxel 175 mg/m2 and carboplatin AUC 5, on day 1 of every 21-day cycle | 21,643 | 2.032 | 1262 | 0.195 | 6472 | 24,361 |
| PLDH plus platinum | PLDH 30 mg/m2; carboplatin target AUC of 5, on day 1 of every 28-day cycle | 22,620 | 2.018 | 977 | –0.015 | Strictly dominated | |
| Treatment | Modelled regimen | Total cost (discounted) (£) | Total QALYs (discounted) (£) | Incremental cost (discounted) (£) | Incremental QALYs (discounted) | Incremental ICER (cost/QALY) | Incremental ICER (cost/QALY) (excluding dominated options) (£) |
|---|---|---|---|---|---|---|---|
| Probabilistic results | |||||||
| Paclitaxel | 175 mg/m2 on day 1 of every 21-day cycle | 15,777 | 1.421 | – | – | ||
| PLDH | 40 mg/m2 on day 1 of every 28-day cycle | 19,591 | 1.568 | 3814 | 0.147 | 25,931 | 25,931 |
| Topotecan | 1.5 mg/m2, on days 1–5 of every 21-day cycle | 23,889 | 1.330 | 4298 | –0.238 | Strictly dominated | |
| Trabectedin plus PLDH | Trabectedin 1.1 mg/m2; PLDH 30 mg/m2, on day 1 of every 21-day cycle | 32,687 | 1.729 | 8798 | 0.399 | 54,893 | 81,353 |
| Deterministic results | |||||||
| Paclitaxel | 175 mg/m2 on day 1 of every 21-day cycle | 15,668 | 1.398 | – | – | – | – |
| PLDH | 40 mg/m2 on day 1 of every 28-day cycle | 19,599 | 1.564 | 3931 | 0.166 | 23,733 | 23,733 |
| Topotecan | 1.5 mg/m2, on days 1–5 of every 21-day cycle | 23,793 | 1.317 | 4194 | –0.247 | Strictly dominated | |
| Trabectedin plus PLDH | Trabectedin 1.1 mg/m2; PLDH 30 mg/m2, on day 1 of every 21-day cycle | 32,640 | 1.717 | 8847 | 0.400 | 22,131 | 85,212 |
People with platinum-resistant/-refractory disease
The network of interventions and comparators identified for the PRR subgroup was limited by the availability of data to three of the therapies outlined in the scope: paclitaxel, PLDH and topotecan (see Interventions and comparators, above).
Base-case deterministic and probabilistic results indicate that paclitaxel is strictly dominated by PLDH, resulting in topotecan versus PLDH being the only comparison considered in the final cost-effectiveness results. The ICER for this comparison was estimated to be £449,553 and £324,188, deterministically and probabilistically, respectively. When compared with PLDH, topotecan was associated with approximately £7000 incremental costs and 0.02 incremental QALYs (Table 150).
However, the TAG considers it important to note that the costs and QALYs associated with paclitaxel are similar to those associated with PLDH. Consequently, small changes in total costs or QALYs associated with either treatment may alter the results.
People with platinum-allergic disease
Clinical advice indicated that response to therapy for patients with or without a platinum allergy was unlikely to differ for the same non-platinum containing therapy (see Population, above). Moreover, given that the PS network 1 contained only platinum-based therapies, the TAG considers that the results for PS network 2, and the network identified in PRR patients are applicable for the platinum-allergic population (see Tables 149 and 150).
| Treatment | Modelled regimen | Total cost (discounted) (£) | Total QALYs (discounted) | Incremental cost (discounted) (£) | Incremental QALYs (discounted) | Incremental ICER (cost/QALY) (£) | Incremental ICER (cost/QALY) (excluding dominated options) (£) |
|---|---|---|---|---|---|---|---|
| Probabilistic results | |||||||
| PLDH | 40 mg/m2 on day 1 of every 28-day cycle | 14,232 | 1.004 | – | – | – | – |
| Paclitaxel | 80 mg/m2 weekly for 18 weeks or until progression | 15,132 | 0.981 | 901 | –0.022 | Strictly dominated | |
| Topotecan | 1.5 mg/m2, on days 1–5 of every 21-day cycle | 21,232 | 1.025 | 6100 | 0.044 | 139,697 | 324,188 |
| Deterministic results | |||||||
| PLDH | 40 mg/m2 on day 1 of every 28-day cycle | 14,320 | 1.004 | – | – | – | – |
| Paclitaxel | 80 mg/m2 weekly for 18 weeks or until progression | 15,095 | 0.971 | 775 | –0.033 | Strictly dominated | |
| Topotecan | 1.5 mg/m2, on days 1–5 of every 21-day cycle | 21,271 | 1.020 | 6176 | 0.049 | 127,117 | 449,553 |
Results of the sensitivity analysis
Probabilistic sensitivity analyses
Following consideration of the probabilistic base-case results, some interventions have been excluded from final ICER calculations; based on strict or extended dominance by other interventions (see Base-case results, above). The remaining comparisons by subgroup are as follows:
-
PS network 1:
-
paclitaxel plus platinum compared with platinum monotherapy.
-
-
PS network 2:
-
PLDH compared with paclitaxel
-
trabectedin plus PLDH compared with paclitaxel
-
trabectedin plus PLDH compared with PLDH.
-
-
PRR patients:
-
topotecan compared with PLDH.
-
For each of these comparisons, probabilistic results have been summarised in scatterplots on the cost-effectiveness plane and CEACs (see Figures 38–53).
However, as highlighted in Base-case results (above), in PS network 1 and PRR there exist comparisons with highly similar total costs and total QALYs. In particular, in PS network 1, the comparison of PLDH plus platinum versus paclitaxel plus platinum. Also, in PRR, the comparison of paclitaxel versus PLDH. These similarities result in unstable estimates of mean cost-effectiveness. Therefore, to enable decision-makers to assess the likelihood that the interventions considered in these unstable comparisons are cost-effective, probabilistic cost-effectiveness results (vs. each other and vs. the baseline treatment) have been summarised in scatterplots and CEACs (see Figures 38–53).
Platinum-sensitive network 1
For the subgroup of patients with platinum-sensitive disease, probabilistic analysis of PS network 1 revealed that, for the majority of simulations, the addition of paclitaxel or PLDH to platinum therapy results in greater costs and greater QALYs than treatment with platinum alone. In particular, for a WTP threshold of £20,000 per additional QALY, the probabilities of paclitaxel plus platinum or PLDH plus platinum being considered cost-effective versus platinum monotherapy are 13% and 3%, respectively. At a WTP threshold of £30,000, the probabilities of being cost-effective versus platinum therapy increase to 78% and 48% for paclitaxel plus platinum and PLDH plus platinum, respectively.
Furthermore, the addition of PLDH to platinum therapy was estimated to be almost as likely to result in greater costs and QALYs as to be dominated by the addition of paclitaxel to platinum therapy. However, as discussed above (see Base-case results), the costs and QALYs accumulated by the addition of paclitaxel or PLDH to platinum therapy are similar, producing cost-effectiveness estimates that are sensitive to minor changes in parameter estimates. (Figures 38–43.)
FIGURE 38.
Scatterplot of cost-effectiveness results for paclitaxel plus platinum vs. platinum monotherapy (dark green line indicates threshold of £30,000 per additional QALY, light green line indicates threshold of £20,000 per additional QALY).
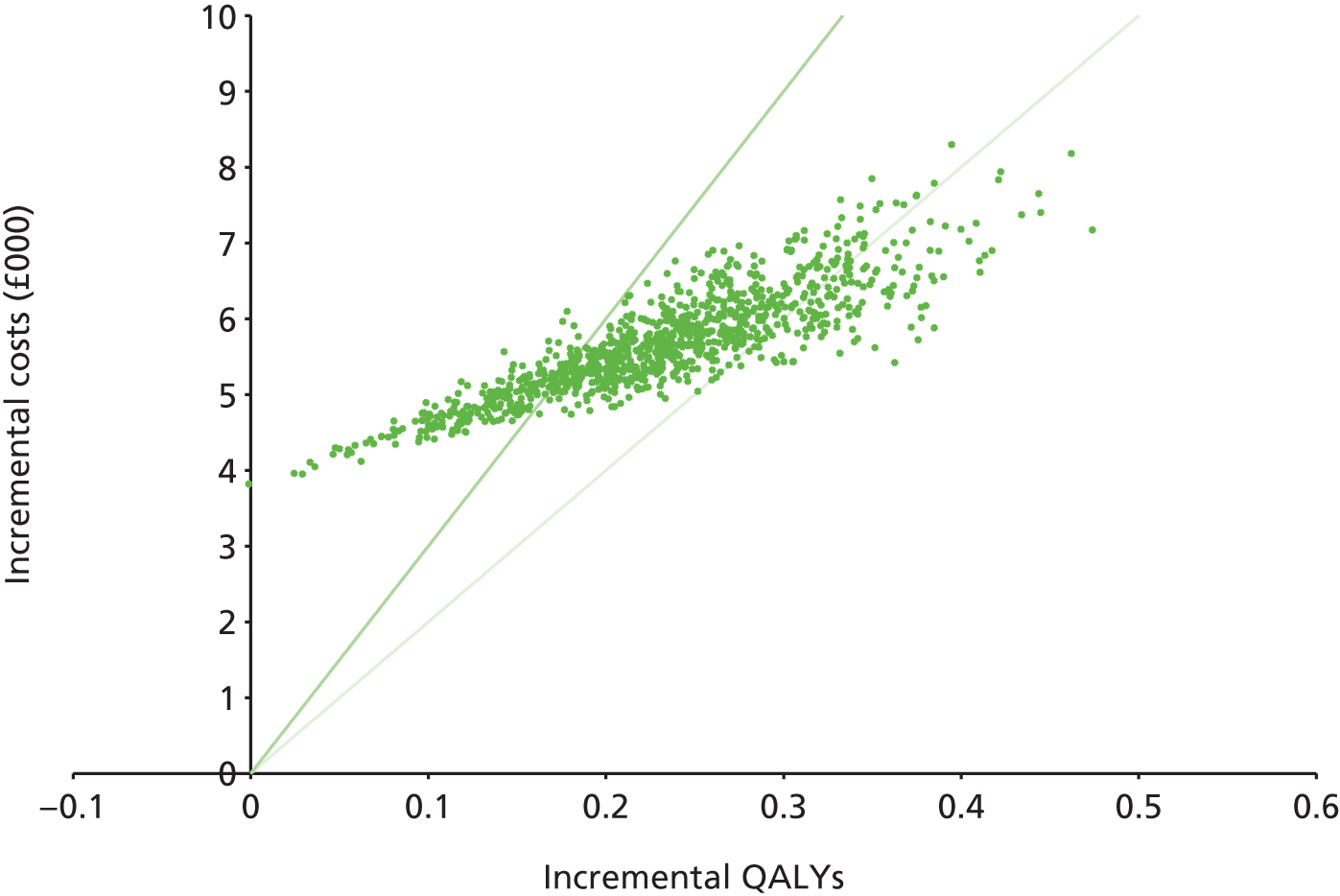
FIGURE 39.
Cost-effectiveness acceptability curve for paclitaxel plus platinum vs. platinum monotherapy.
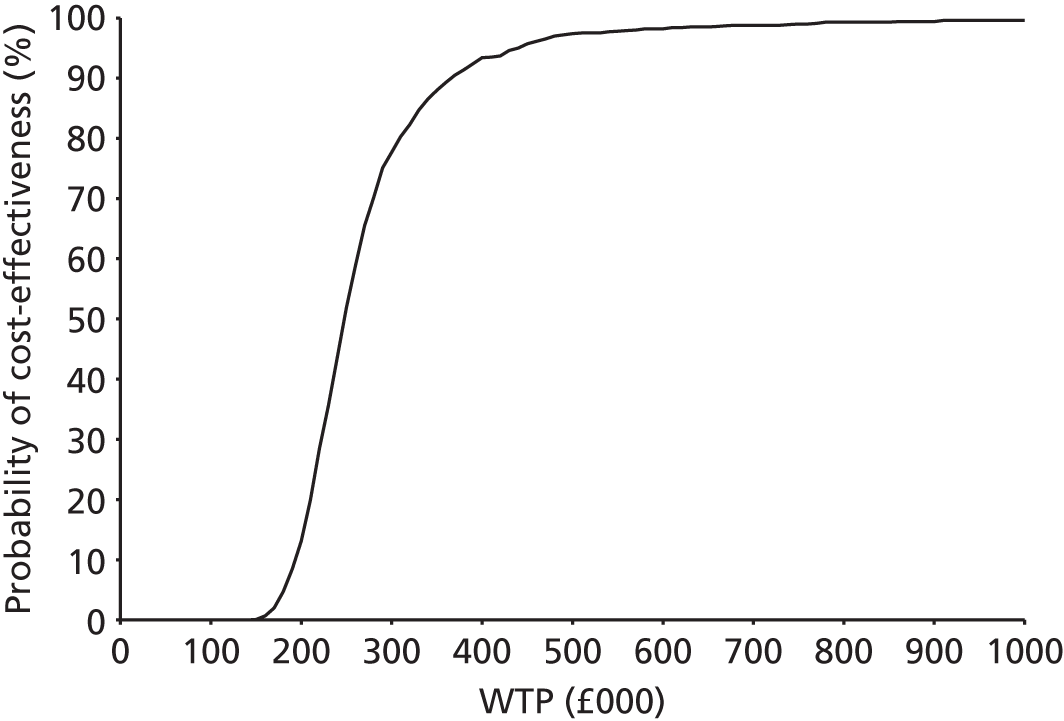
FIGURE 40.
Scatterplot of cost-effectiveness results for PLDH plus platinum vs. platinum monotherapy (dark green line indicates threshold of £30,000 per additional QALY, light green indicates threshold of £20,000 per additional QALY).
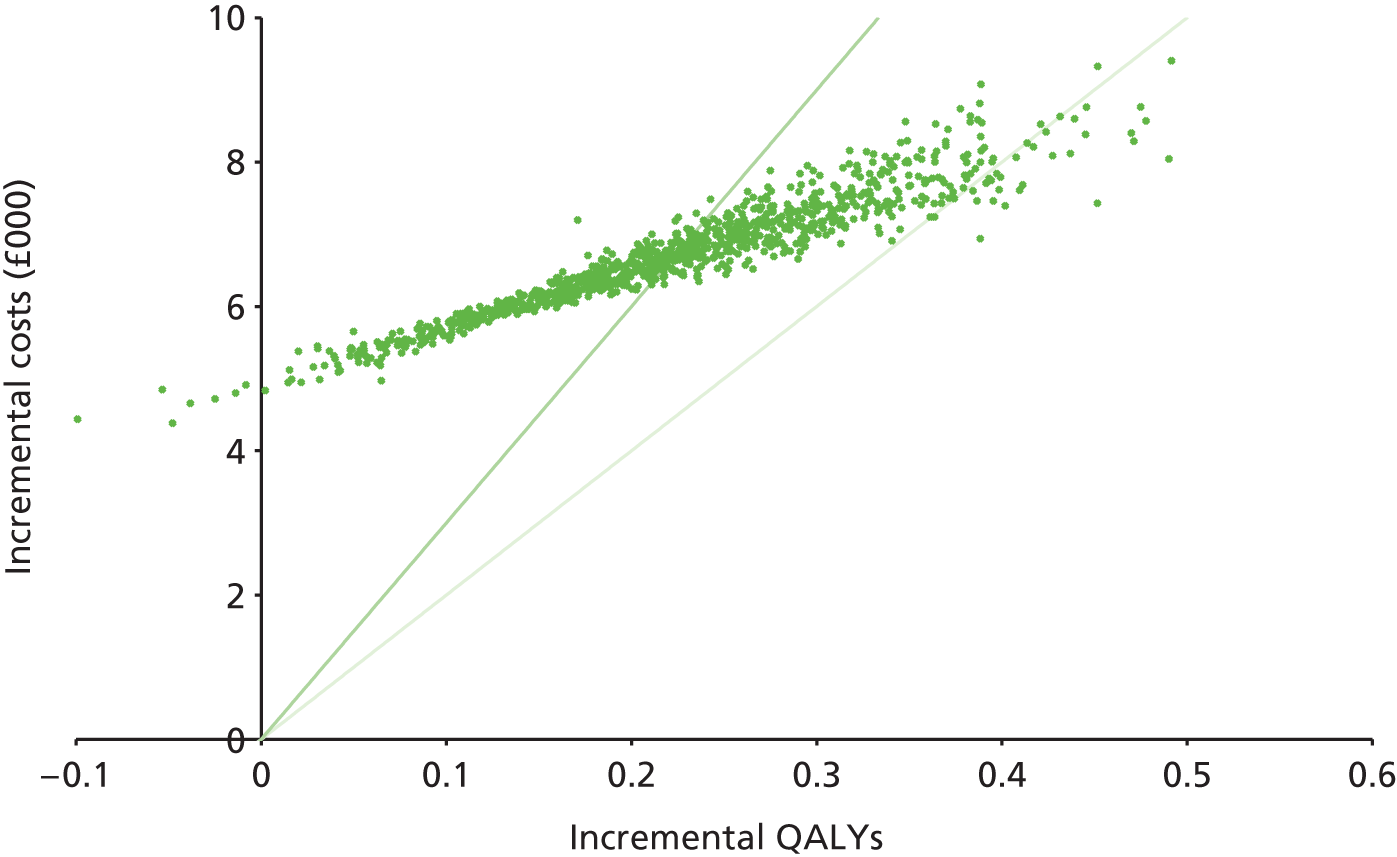
FIGURE 41.
Cost-effectiveness acceptability curve for PLDH plus platinum vs. platinum monotherapy.
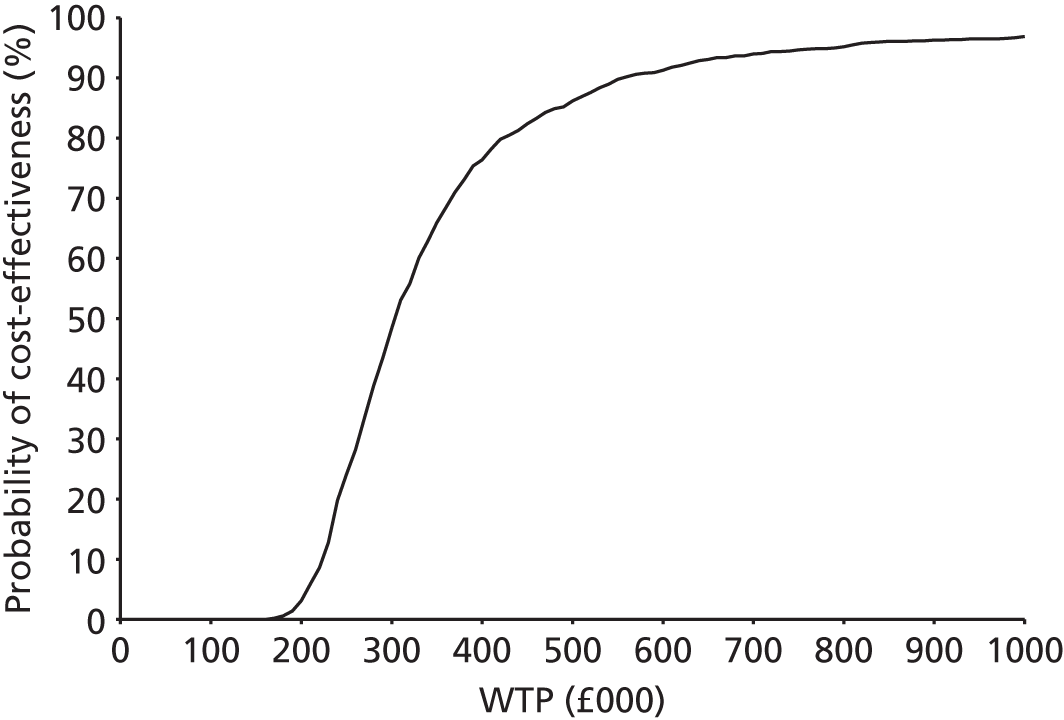
FIGURE 42.
Scatterplot of cost-effectiveness results for PLDH plus platinum vs. paclitaxel plus platinum (dark green line indicates threshold of £30,000 per additional QALY, light green line indicates threshold of £20,000 per additional QALY).
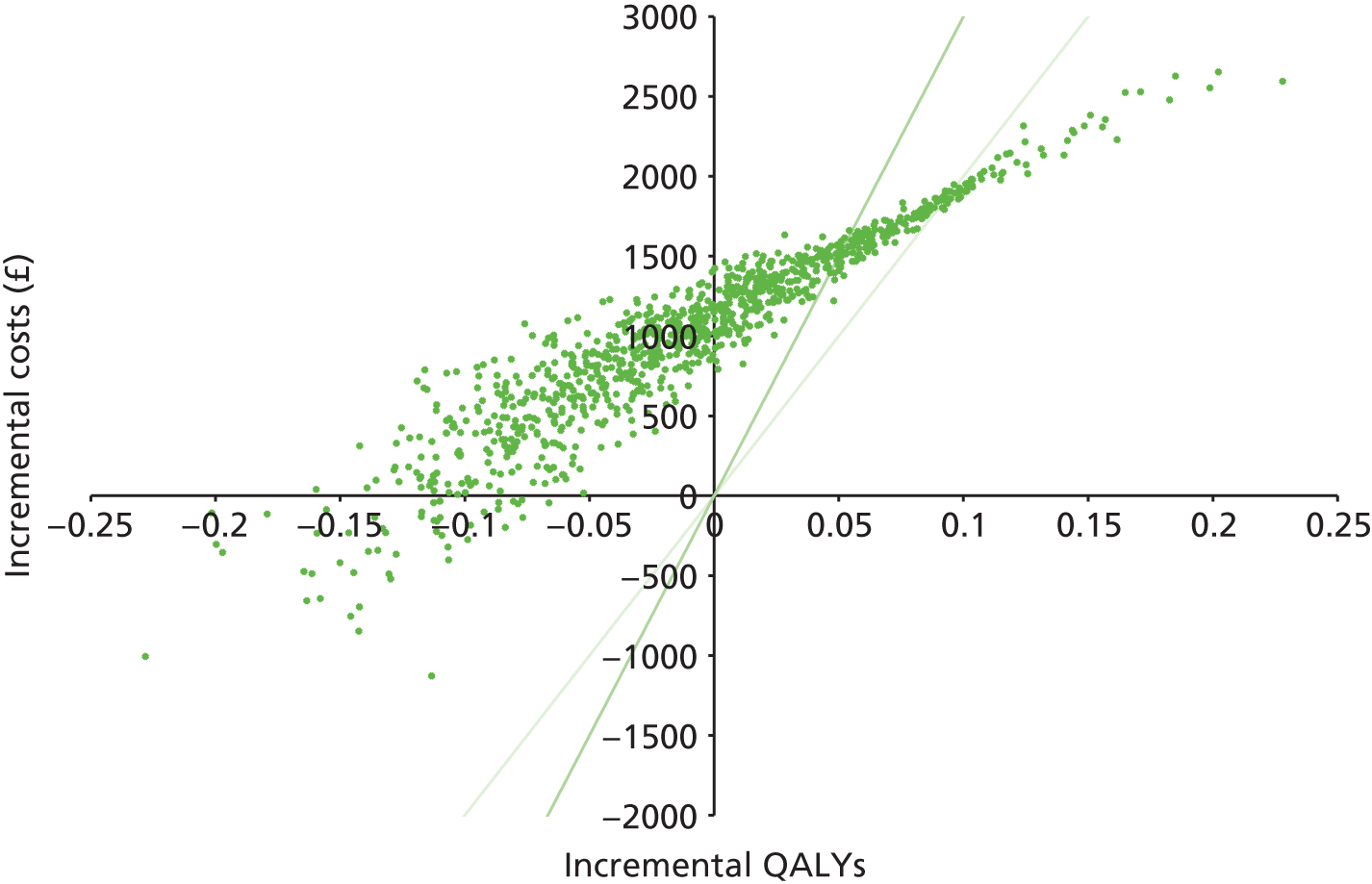
FIGURE 43.
Cost-effectiveness acceptability curve for PLDH plus platinum vs. paclitaxel plus platinum.

Platinum-sensitive network 2
For the subgroup of patients with platinum-sensitive disease, probabilistic analysis of PS network 2 revealed that, at WTP thresholds of £20,000 and £30,000, treatment with PLDH (vs. paclitaxel) is more likely to be cost-effective than treatment with trabectedin plus PLDH (vs. paclitaxel). That is, treatment with PLDH has a 30% and 59% chance of being cost-effective at WTP thresholds of £20,000 and £30,000, respectively. Whereas, trabectedin plus PLDH has a 0.1% and 1.4% chance of being cost-effective at WTP thresholds of £20,000 and £30,000, respectively.
However, the TAG considers it important to note that 15% of PLDH compared with paclitaxel simulations fall in the north-west quadrant (i.e. dominance by paclitaxel), whereas 3% of simulations for trabectedin plus PLDH compared with paclitaxel fall into this quadrant. This suggests that there is a greater degree of uncertainty associated with the benefit of PLDH over paclitaxel compared with the benefit of trabectedin plus PLDH over paclitaxel. This is emphasised further by considering the comparison of trabectedin plus PLDH compared with PLDH alone in which 95% of simulations fall in the north-east quadrant, suggesting that the addition of trabectedin to treatment with PLDH is likely to improve outcomes as well as increasing cost. However, according to the TAG analysis trabectedin plus PLDH has a 0% probability of being cost-effective over PLDH at WTP thresholds of £20,000 or £30,000. (Figures 44–49.)
FIGURE 44.
Scatterplot of cost-effectiveness results for PLDH vs. paclitaxel (dark green line indicates threshold of £30,000 per additional QALY, light green line indicates threshold of £20,000 per additional QALY).

FIGURE 45.
Cost-effectiveness acceptability curve for PLDH vs. paclitaxel.
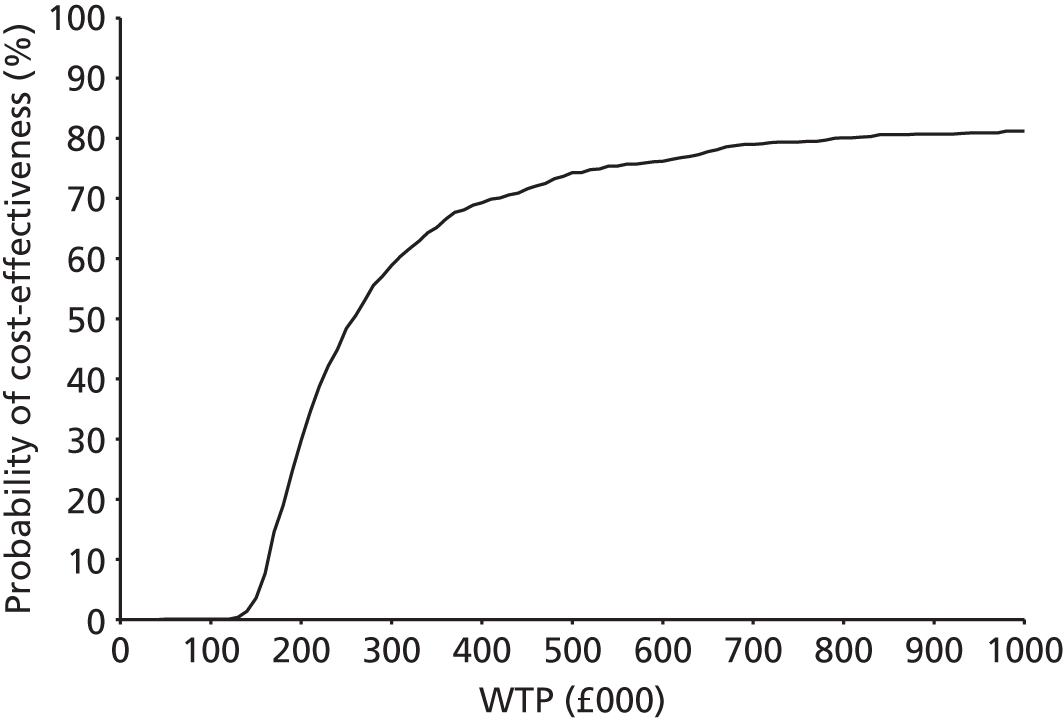
FIGURE 46.
Scatterplot of cost-effectiveness results for trabectedin plus PLDH vs. paclitaxel (dark green line indicates threshold of £30,000 per additional QALY, light green line indicates threshold of £20,000 per additional QALY).

FIGURE 47.
Cost-effectiveness acceptability curve for trabectedin plus PLDH vs. paclitaxel.
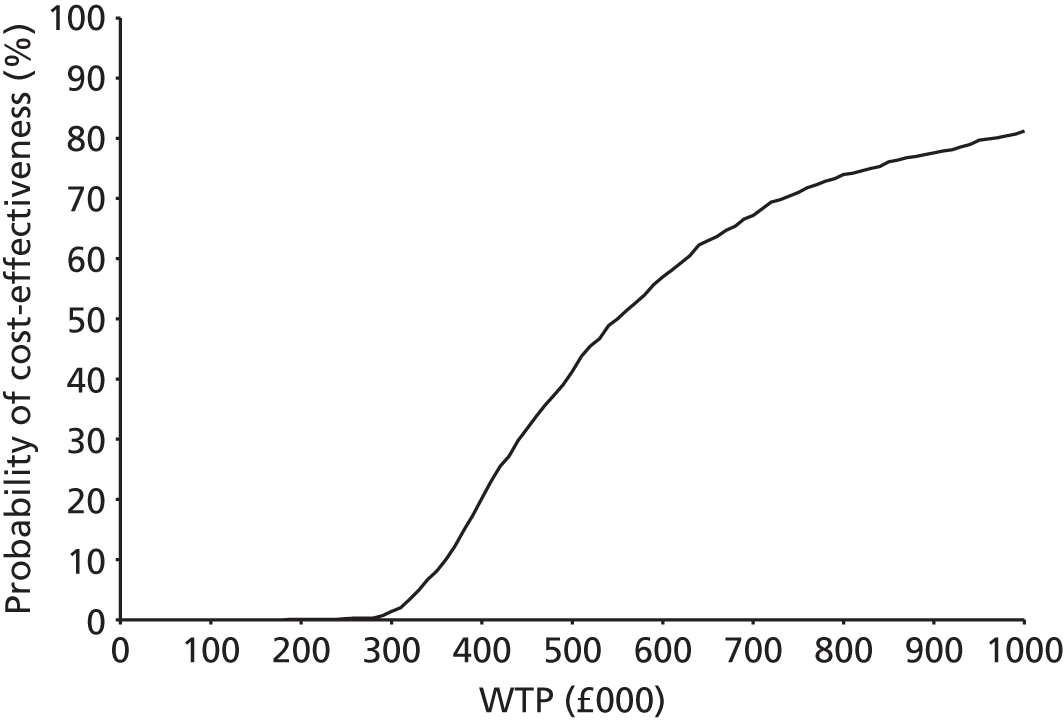
FIGURE 48.
Scatterplot of cost-effectiveness results for trabectedin plus PLDH vs. PLDH (dark green line indicates threshold of £30,000 per additional QALY, light green line indicates threshold of £20,000 per additional QALY).

FIGURE 49.
Cost-effectiveness acceptability curve for trabectedin plus PLDH vs. PLDH.

Platinum-resistant/-refractory network
For the subgroup of patients with PRR disease, probabilistic analysis revealed that, on average, treatment with paclitaxel is dominated by treatment with PLDH. Therefore, based on mean estimates, the key comparison in PRR patients is topotecan compared with PLDH. However, at WTP thresholds of £20,000 and £30,000, topotecan has a 0% chance of being cost-effective, whereas in 39% of simulations paclitaxel provides greater QALYs at a higher cost (compared with PLDH), with probabilities of being cost-effective of 3% and 14%, at WTP thresholds of £20,000 and £30,000, respectively. In addition, the TAG considers it important to note that in 23% of simulations paclitaxel was less expensive and less effective than PLDH (Figures 50–53).
FIGURE 50.
Scatterplot of cost-effectiveness results for topotecan vs. PLDH (dark green line indicates threshold of £30,000 per additional QALY, light green line indicates threshold of £20,000 per additional QALY).
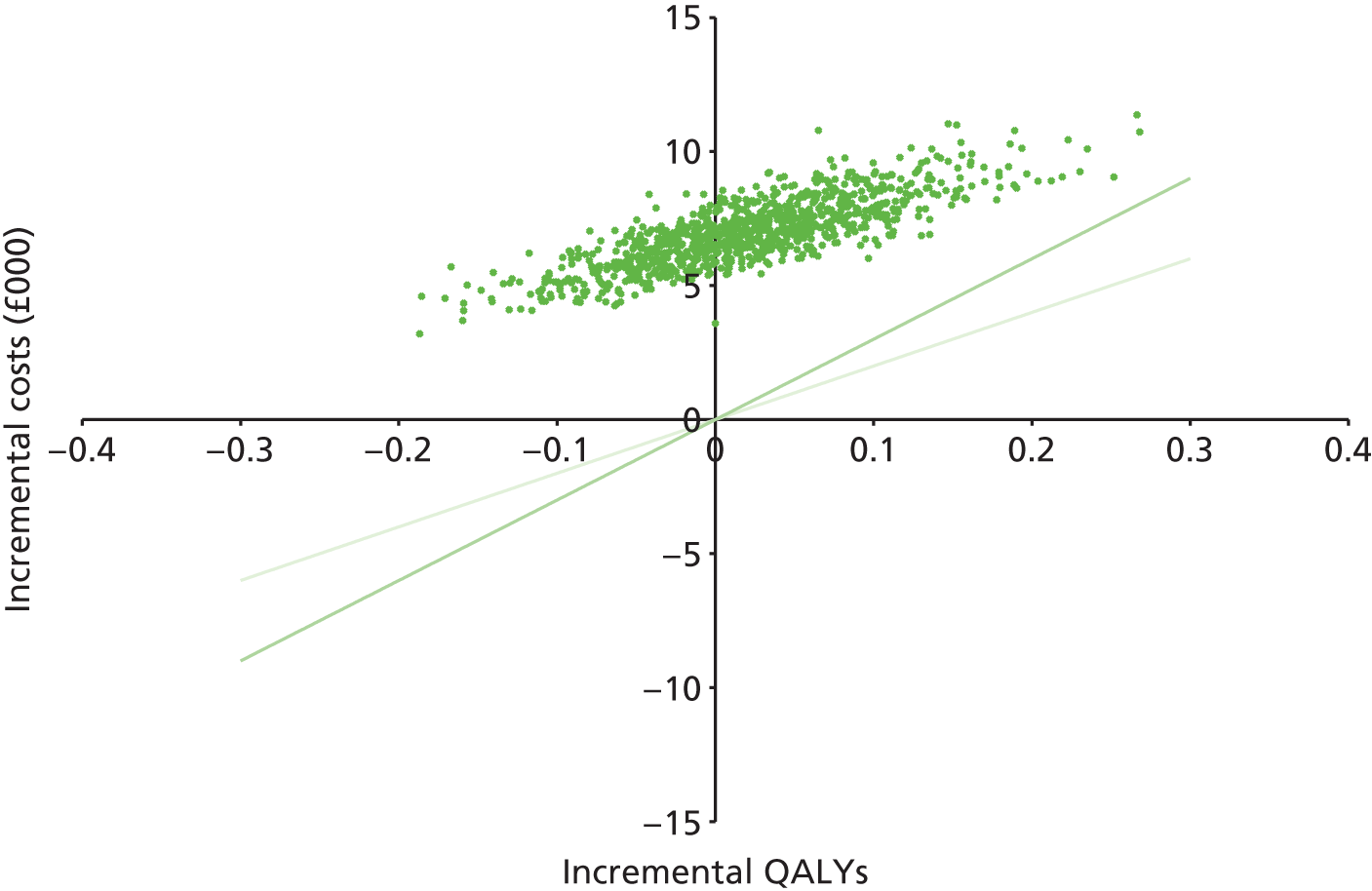
FIGURE 51.
Cost-effectiveness acceptability curve for topotecan vs. PLDH.

FIGURE 52.
Scatterplot of cost-effectiveness results for paclitaxel vs. PLDH (dark green line indicates threshold of £30,000 per additional QALY, light green line indicates threshold of £20,000 per additional QALY).

FIGURE 53.
Cost-effectiveness acceptability curve for paclitaxel vs. PLDH.

One-way sensitivity analyses
As discussed in Base-case results (above), in addition to probabilistic analysis, one-way sensitivity analysis has been carried out on all model parameters. The TAG notes that many of the parameters tested in sensitivity analysis had minimal impact on the deterministic cost-effectiveness results. Therefore, the TAG have produced tornado diagrams depicting only the impact of the variables which the cost-effectiveness results were most sensitive to (these are presented in Appendix 15). For each subgroup, the key findings from one-way sensitivity analyses are discussed in the following sections.
Platinum-sensitive network 1
In patients considered to have platinum-sensitive disease, one-way sensitivity analysis of PS network 1 revealed that the comparisons of paclitaxel plus platinum versus platinum and PLDH plus platinum versus platinum are most sensitive to the relative effect of treatment on OS. For example, use of the lower bound of the 95% credible interval (CrI) (estimated from TAG NMA) for the HR of OS (platinum monotherapy vs. paclitaxel plus platinum) increases the deterministic base-case ICERs by > £20,000. This is because the base-case value of the OS HR (platinum vs. paclitaxel plus platinum) is 1.29, indicating that relative to paclitaxel plus platinum, platinum monotherapy increases the risk of death. Therefore assuming a lower value for this parameter directly results in a lower relative treatment effect for paclitaxel plus platinum and indirectly results in a lower relative treatment effect for PLDH plus platinum. The impact of other parameters, such as the relative effect of treatment on PFS and the utility value associated with each health state, are relatively minimal.
Similarly, when considering the comparison of PLDH plus platinum with paclitaxel plus platinum, the relative effect of treatment on OS has the largest impact of all variables tested on the cost-effectiveness results. That is, when the lower bound of the OS HR (PLDH plus platinum vs. paclitaxel plus platinum) is used to inform the cost-effectiveness analysis, the ICER moves from the dominance of PLDH plus platinum by paclitaxel plus platinum to an ICER of approximately £20,000 for PLDH plus platinum compared with paclitaxel plus platinum. This is because lowering the HR (1.023 in the base case) reduces the relative benefit of paclitaxel plus platinum over PLDH plus platinum; however, the magnitude of change observed in this sensitivity analysis reflects the instability of the mean cost-effectiveness estimate for this comparison.
Platinum-sensitive network 2
Similar to the network of treatments for patients with platinum-sensitive disease considered in PS network 1, the cost-effectiveness of treatments considered in PS network 2 appears to be driven by the relative effect of treatment on OS. For example, in the comparison of PLDH to paclitaxel, use of the lower bound of the 95% CrI for the HR of OS (paclitaxel vs. PLDH) results in a move from an ICER of approximately £25,000 (PLDH vs. paclitaxel) to dominance of PLDH by paclitaxel. This is because the base-case HR used to inform this comparison is 1.22 (paclitaxel vs. PLDH), indicating that, relative to PLDH, paclitaxel results in a higher risk of death. Therefore, assuming a lower value (0.80) for this parameter (i.e. representing a situation where, relative to PLDH, paclitaxel decreases the risk of death) results in a dramatic reversal of the cost-effectiveness results. As is the case in the comparison of paclitaxel plus platinum with PLDH plus platinum in PS network 1, the magnitude of change observed in this sensitivity analysis reflects the instability of the mean cost-effectiveness estimate for the comparison of PLDH with paclitaxel.
The relative effect of treatment on OS has a similar impact on the cost-effectiveness results of trabectedin plus PLDH compared with PLDH. In this comparison, use of the lower bound of the OS HR (trabectedin plus PLDH vs. PLDH) results in a £40,000 reduction in the ICER, whereas use of the upper bound of the OS HR (trabectedin plus PLDH vs. PLDH) results in dominance of trabectedin plus PLDH by PLDH.
With respect to the comparison of trabectedin plus PLDH compared with paclitaxel, the impact of treatment effect on OS remains high, although it is not as influential as in the comparison of PLDH to paclitaxel. In particular, use of the lower bound of the OS HR (paclitaxel vs. PLDH) increases the ICER from approximately £55,000 to £400,000, whereas use of the upper bound of the OS HR (paclitaxel vs. PLDH) decreases the ICER to approximately £35,000. This is because, in the base case, the OS HR (paclitaxel vs. PLDH) is 1.22, and the OS HR (trabectedin plus PLDH vs. PLDH) is 0.84, suggesting that, compared with PLDH, paclitaxel increases the risk of death and trabectedin plus PLDH decreases the risk of death. Use of the lower 95% CrI (0.85) of OS HR (paclitaxel vs. PLDH) effectively removes the difference in OS benefit between trabectedin plus PLDH and paclitaxel and therefore increases the ICER. Conversely, a larger relative difference in the effect of treatment on OS (between trabectedin plus PLDH and paclitaxel), through use of the upper bound (1.69) of the OS HR (paclitaxel vs. PLDH), decreases the ICER. However, unlike the comparison of PLDH vs. paclitaxel, sensitivity analysis around the relative effect of treatment on OS does not alter the quadrant of the cost-effectiveness plane in which the result falls.
Platinum-resistant/-refractory network
In patients with resistant or refractory disease, the relative effect of treatment on OS continues to be a key driver of cost-effectiveness results. Moreover, one-way sensitivity analysis revealed that the comparisons of topotecan with PLDH and paclitaxel with PLDH are unstable. That is, for both comparisons, sensitivity analysis around the relative effect of treatment on OS altered the quadrant in which the cost-effectiveness result falls. In particular, when the lower bound of the OS HRs (topotecan vs. PLDH and paclitaxel vs. PLDH) was used, the ICERs of topotecan compared with PLDH and paclitaxel compared with PLDH were £53,288 and £17,903, respectively.
Scenario analyses
In addition to probabilistic and one-way sensitivity analyses, several scenario analyses have been carried out to assess the sensitivity of the cost-effectiveness results to structural assumptions made. Full results of these analyses are presented in Appendix 11, with a summary of the key results, for each network, presented below.
Platinum-sensitive network 1
For PS network 1 (platinum; gemcitabine plus carboplatin; PLDH plus platinum; paclitaxel plus platinum) two scenarios materially impacted the results and conclusions of the base-case analysis. These scenarios were those in which branded (Abraxane and Taxol) rather than non-proprietary drug acquisition costs of paclitaxel were used.
Using the cost associated with Abraxane, the total discounted cost associated with paclitaxel plus platinum increases from £21,643 to £22,940. Increasing the ICER associated with paclitaxel plus platinum compared with platinum from £24,361 to £29,912. This increase results in a shift from strict dominance of PLDH plus platinum by paclitaxel plus platinum, to extended dominance of PLDH plus platinum by paclitaxel plus platinum. That is, when Abraxane is used, treatment with paclitaxel plus platinum results in higher costs than treatment with PLDH plus platinum. However, the additional benefit provided by using paclitaxel rather than PLDH in combination with platinum therapy, may be considered to provide better value for money (i.e. results in a lower ICER vs. platinum).
Use of Taxol rather than non-proprietary paclitaxel produces very similar results and conclusions to the use of Abraxane; PLDH plus paclitaxel switches from being strictly dominated by paclitaxel plus platinum to being extendedly dominated by paclitaxel plus platinum. The ICER associated with paclitaxel plus platinum vs. platinum increases from £24,361 to £36,092.
For all other scenarios, results are robust to the changes made; gemcitabine plus carboplatin remains extendedly dominated, PLDH plus carboplatin remains strictly dominated, and the ICER for paclitaxel plus platinum compared with platinum ranges between £19,113 and £30,084.
Platinum-sensitive network 2
For PS network 2 (paclitaxel; PLDH; PLDH plus trabectedin; topotecan) base-case incremental results were robust to the majority of scenarios modelled. In particular, topotecan continued to be dominated by trabectedin plus PLDH, in every modelled scenario. In addition, with the exception of one scenario, the ICER associated with PLDH compared with paclitaxel remained at < £30,000; increasing the dose of PLDH (from 40 mg/m2 to 50 mg/m2) used in drug acquisition calculations, increased the ICER from £23,733 to £31,222. Furthermore, the ICER associated with trabectedin plus PLDH compared with PLDH remained at > £60,000 in all scenarios assessing incremental base-case results.
In addition to scenario analyses of incremental base-case results, a further two scenario analyses, examining the cost-effectiveness of a subset of comparisons of interest, were carried out. These were:
-
exploratory analysis of the cost-effectiveness of PLDH, trabectedin plus PLDH and topotecan in patients with PPS (PFI 6–12 months) disease
-
head-to-head comparison of trabectedin plus PLDH compared with PLDH using clinical effectiveness data from the PharmaMar submission (i.e. adjusted for baseline characteristics) within the TAG economic model.
Scenario analysis in the PPS patient population was carried out using OS HRs (trabectedin plus PLDH vs. PLDH and topotecan, vs. PLDH) estimated from TAG NMA (see Chapter 3, Overall survival). For the following reasons, the TAG considers this analysis as highly uncertain and therefore exploratory. First, as a result of data paucity, it was not possible to estimate baseline survival for the PPS (PFI 6–12 months) population; instead estimates of baseline survival from platinum-sensitive (PFI ≥ 6 months) patients treated with PLDH were used. Second, HRs were available only for OS and not PFS; estimates of PFS from platinum-sensitive patients were used as proxies. Results of this exploratory scenario analysis were dominance of topotecan by PLDH and an ICER of £37,691 for trabectedin plus PLDH compared with PLDH.
Head-to-head comparison, in the platinum-sensitive (PFI > 6 months) population, of trabectedin plus PLDH compared with PLDH, based on PFS and OS from the PharmaMar submission (i.e. adjusted for baseline characteristics) within the TAG economic model, resulted in an ICER of £35,646. By contrast, the ICERs, of trabectedin plus PLDH compared with PLDH in the platinum-sensitive (PFI > 6) population, estimated by the TAG and PharmaMar base-case analyses were £85,212 and £27,573, respectively. The deterministic incremental costs and QALYs associated with the TAG’s base-case and scenario analyses and the manufacturer’s base-case analysis are presented in Table 151.
| Treatment | Total (discounted) costs (£) | Total (discounted) QALYs | Incremental costs (£) | Incremental QALYs | ICER (£) |
|---|---|---|---|---|---|
| TAG‘s base-case estimates | |||||
| Trabectedin plus PLDH | 32,640 | 1.717 | – | – | – |
| PLDH | 19,599 | 1.564 | 13,041 | 0.15 | 85,212 |
| PharmaMar‘s estimates | |||||
| Trabectedin plus PLDH | 38,206 | 2.33 | – | – | – |
| PLDH | 24,809 | 1.85 | 13,397 | 0.49 | 27,573 |
| TAG‘s scenario analysis estimates | |||||
| Trabectedin plus PLDH | 34,569 | 2.08 | – | – | – |
| PLDH | 21,063 | 1.70 | 13,506 | 0.38 | 35,646 |
The TAG notes that the differences between the TAG (ICER of £85,212) and PharmaMar (ICER of £27,573) base-case estimates of the cost-effectiveness of trabectedin plus PLDH compared with PLDH alone is largely driven by the use of adjusted data to inform the manufacturer’s base case; when used in the TAG model the ICER fell to £35,646.
However, the TAG notes that, given both the TAG scenario analysis (ICER of £35,646) and the PharmaMar model (ICER of £27,573) utilise the same PFS, OS and utility data, it would be expected that the difference in the estimated ICERs would be explained through a difference in estimated incremental cost. However, incremental costs were similar between the TAG and PharmaMar analyses (£13,506 vs. £13,397, respectively), with the difference in incremental QALYs (0.38 vs. 0.49, respectively) being the main driver of the difference in the ICER estimates. Therefore, the TAG investigated potential causes of this discrepancy and considers that it likely to be a result of:
-
difference in model structure and therefore discounting methodology and time horizon used
-
minor differences in costs.
The manufacturer’s model was based upon the model developed in TA9113 (used in TA22215), whereby the mean TTP and mean time to death, to which costs and QALYs were applied, were estimated from survival data (see Description and critique of manufacturer-submitted evidence, above). In order to apply discounting to costs and QALYs in this model structure, the manufacturer stated: ‘The exponential discounting method was used whereby costs and QALYs were discounted continuously based on the time spent in the model health states. The instantaneous rate of 3.44% (Ln[1.035)] was therefore considered’ (MS, p. 31). Within TA222, a key critique of the manufacturer’s model, by the ERG responsible for reviewing this STA, related to the method of discounting used as a result of the model structure: ‘Discounting cannot be easily implemented in such a model structure. Ideally a state transition-type Markov trace element should be constructed to facilitate the implementation of discounting’. 90 The TAG economic analysis did not rely upon mean estimates of PFS or OS. Instead, costs and QALYs were estimated monthly for each health state; these costs and QALYs were then discounted depending upon the year in which they fell.
The TAG considers that, as a result of the discounting methodology used, the manufacturer may have overestimated the QALY gain. This is because, application of discounting to average estimates is unlikely to be as accurate as discounting based on monthly estimates, as the granularity of patient proportions, by health state, over time, is not captured.
However, the TAG considers that the difference in the ICER between the TAG’s and manufacturer’s base-case analyses is predominantly a consequence of the use of adjusted clinical effectiveness data; adjusted for baseline characteristics such as PFI (as a continuous variable). The TAG notes that adjustment of clinical effectiveness data for key prognostic factors, such as PFI, is likely to result in more accurate estimates of PFS and OS.
For these reasons, the TAG considers that the ICER estimated in the TAG’s scenario analysis is likely to be the most accurate reflection of the cost-effectiveness of trabectedin plus PLDH compared with PLDH.
Platinum-resistant/-refractory network
For the PRR network (paclitaxel; PLDH; topotecan) results are robust to the majority of scenarios modelled. The ICER for topotecan compared with PLDH ranges between £374,963 and £503,885. Paclitaxel is dominated in all but one scenario, where costs associated with a 50-mg dose of PLDH, rather than costs associated with a dose of 40 mg are used. In this scenario, paclitaxel becomes the least costly treatment option and therefore represents the baseline for incremental assessment of cost-effectiveness results. In this scenario, the ICER associated with PLDH compared with paclitaxel is estimated to be £10,480.
The TAG modelled a scenario in which only the costs associated with treatment for PRR patients differed between chemotherapy regimens. In this scenario, PFS and OS were set equal to the baseline treatment (PLDH). The purpose of this scenario was twofold; first, to provide a comparison including all interventions and comparators of interest as listed in the NICE scope and second, to reflect clinical advice that the prognosis of patients with PRR disease is often poor across available treatment options. In this scenario, the cost of etoposide 50 mg (oral) days 1–21 every 28 days for a maximum of six cycles followed by maintenance with oral etoposide was estimated to be the cheapest treatment option (£8194), followed by best supportive care (£12,622). The TAG considers that the cost associated with best supportive care may have been overestimated. The palliative cost associated with ovarian cancer was estimated by Guest et al. 112 to be £4789 (at 2000–1 prices) for an average time period of 399 days. This cost predominantly consisted of hospitalisation costs (71% of costs). Updating the estimate of palliative care for ovarian cancer patients from Guest et al. 112 to current prices using the Hospital & Community Health Services index results in a cost of £6963;115 equating to £531 per month. This cost is applied monthly to all PRR patients following entry into the PD health state, and all platinum-sensitive patients following 6 months of residence in the PD health state.
The TAG notes that the analysis carried out by Guest et al. 112 has several weaknesses. In particular, ovarian cancer estimates are based upon a small sample size (n = 21) and does not consider costs for patients not requiring a strong opioid. In addition, the analysis was carried out in 2000–1 and may no longer reflect clinical practice. To establish the impact of this uncertainty, the TAG varied this cost in sensitivity analysis for the base case; however, the TAG considers that future research into the cost of best supportive care for women with ovarian cancer may be warranted (see Chapter 7, Suggested research priorities).
Summary of the Technology Assessment Group de novo economic evaluation
Following review of the economic literature and MSs, the TAG developed a de novo economic model to address the decision problem outlined for this MTA. The economic model was based upon the model structure for TA91, in which three health states were modelled: SD, PD and death. Within the TA91 model, the proportions of patients within each health state were calculated from estimates of mean TTP and mean time to death, available from the literature. Utilities and costs were then applied to mean estimates of time spent within each health state. The ERG responsible for appraisal of a subsequent STA (TA22290), in which the same model structure was applied, commented that the use of mean estimates resulted in difficulties in the application of discounting; this is because the proportion of patients in each health state over time is not explicitly modelled. Therefore, in order to address this concern, the model used in TAG analyses incorporates monthly estimates of PFS and OS over time (see Treatment effectiveness, above).
Furthermore, based on the data identified in the clinical systematic review and consultation with clinical experts, the TAG carried out separate analyses of patients with platinum-sensitive disease (PFI of ≥ 6 months) and PRR disease (PFI of < 6 months). Moreover, as no single trial assessing the full range of interventions and comparators was identified in the platinum-sensitive or PRR patient populations, NMAs were used to synthesise the available clinical effectiveness data (see Chapter 3, Results). However, as a result of the trials available for patients with platinum-sensitive disease it was not possible to construct a single complete network comparing all interventions with all comparisons and with one another. Instead, two separate, disconnected networks form the basis of analyses in the platinum-sensitive subgroup. For patients with PRR disease, the trials available enabled the TAG to analyse a subset of the interventions and comparators listed within the scope. Finally, following consultation with clinical experts, the TAG considers that patients who are platinum allergic are likely to respond to non-platinum therapies in a similar way to patients without a platinum allergy. Therefore, a separate analysis of platinum-allergic patients has not been carried out; however, treatment options for platinum-allergic patients are assumed to exclude platinum-based therapies (see Population, above).
Within the TAG’s economic model, costs associated with drug acquisition and administration, patient care (health-state costs) and AEs are accounted for. QALYs are used to assess the benefit of each treatment to patients. QALYs are calculated by the application of health-state utility values, identified from the published literature, to the proportion of patients in each health state over time. AEs which, following consultation with clinical experts, are considered to be associated with a noteworthy cost are included in the base-case analyses. However, the impact of treatment-related toxicity on QoL is not explicitly assessed in the TAG’s base-case analysis. The rationale for exclusion of utility decrements associated with AEs is twofold. In particular, the TAG notes that the impact of AEs on patient QoL associated with trabectedin plus PLDH and PLDH monotherapy is implicitly included within the health-state utility estimates used (health-state utility estimates are sourced from TA22290). Furthermore, the reliability of the estimates identified for QoL decrements is uncertain. Finally, in line with the NICE reference case, analysis is carried out from the perspective of the NHS and PSS, costs and benefits are discounted at a rate of 3.5% per annum over a 15-year time horizon.
A summary of the results of the TAG’s base-case analyses is presented in Table 152.
| PS network 1 | PS network 2 (including platinum-allergic patients) | PRR (including platinum-allergic patients) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | Incremental ICER probabilistic (deterministic) | Prob. cost-effective at threshold of:a | Treatment | Incremental ICER probabilistic (deterministic) | Prob. cost-effective at threshold of:a | Treatment | Incremental ICER probabilistic (deterministic) | Prob. cost-effective at threshold of:a | |||||
| £20,000 | £30,000 | £20,000 | £30,000 | £20,000 | £30,000 | ||||||||
| Platinum | – | – | – | Paclitaxel | – | – | – | PLDH | – | – | – | ||
| Gemcitabine plus carboplatin | Extendedly dominatedb | PLDH | £25,931 (£23,733) | 30% | 59% | ||||||||
| Paclitaxel plus platinum | £24,539 (£24,361) | 13% | 78% | Topotecan | Strictly dominatedc | Paclitaxel | Strictly dominatedc | ||||||
| PLDH plus platinum | Strictly dominatedc | Trabectedin plus PLDH | £81,353 (£85,212) | 0% | 0% | Topotecan | £324,188 (£449,553) | 0% | 0% | ||||
Discussion
As highlighted above (see Summary of the Technology Assessment Group de novo economic evaluation), economic analysis has been carried out separately for patients with platinum-sensitive disease (PFI ≥ 6 months) and PRR disease (PFI < 6 months). In addition, as a result of the limited number of trials identified, two separate networks, of interventions and comparators outlined in the scope of this MTA, have been constructed in patients with platinum-sensitive disease. Consequently, cost-effectiveness is assessed for three networks of treatment, of which two consider a population of patients with platinum-sensitive disease and one considers a population of patients with PRR disease.
For each network, OS and PFS data, synthesised in NMA, are used to inform the economic model. In the absence of IPD of sufficient granularity to allow IPD NMA, these data were synthesised from summary measures, available in the literature, of relative treatment effect in the form of HRs. Furthermore, although some of the clinical trials identified for inclusion in the NMAs reported HRs adjusted for particular baseline characteristics, unadjusted HRs are used within the NMAs and therefore economic analyses. This is because adjusted HRs were not available for all included trials and, of those trials reporting adjusted HRs, adjustments for different factors had been carried out. Therefore, the TAG considers the synthesis of unadjusted HRs to be the most equitable way to compare therapies.
Within each network, the TAG selected a baseline treatment for which monthly estimates of PFS and OS could be obtained from submitted or published Kaplan–Meier data. Where Kaplan–Meier data were incomplete (i.e. when a proportion of patients remained at risk at the end-of-trial follow-up), parametric survival distributions were fitted to allow extrapolation beyond the trial duration. HRs obtained from the TAG’s NMAs are applied to baseline estimates of PFS and OS.
However, by using this methodology, the TAG implicitly makes three key assumptions. First, it assumes that data combined within the NMAs were homogeneous or that any differences between the trials included in the analysis would not bias estimates of relative treatment effect. Second, it assumes that the relative effect of treatment (relative to the baseline treatment) is constant over time, namely the assumption of proportional hazards. Third, and perhaps most importantly, as a result of using a consistent data set (i.e. unadjusted HRs rather than a combination of adjusted and unadjusted HRs), the methodology used assumes that estimates of relative treatment effect based on unadjusted data would not meaningfully differ from estimates of relative treatment effect based on adjusted data.
The homogeneity, or otherwise, of the trials included in the TAG’s NMAs was assessed from a clinical perspective. That is, baseline characteristics of key prognostic indicators were compared both within and across included trials. Where differences were identified, expert clinical advice was sought to determine the potential magnitude of impact (on estimates of relative treatment effect) that imbalances in these characteristics were likely to have. However, statistical assessment of heterogeneity was not possible as a result of the low number of trials identified and the predominantly linear nature of the networks constructed.
Furthermore, for each network, the pertinence of assuming that the relative effect of treatment (relative to the baseline treatment) is constant over time was investigated through assessment of the hazards (of progression or death) associated with each treatment. In particular, LCH plots based on submitted or published Kaplan–Meier data were constructed and visually examined to determine the presence or absence of hazards that were proportional between treatments.
Finally, the potential impact of adjustments for baseline characteristics on estimates of relative treatment effect was assessed by considering individual trial comparisons for which HRs calculated from adjusted and unadjusted data were presented. For example, in the evidence submitted by PharmaMar as part of this MTA, OS HRs (trabectedin plus PLDH vs. PLDH) calculated from unadjusted Kaplan–Meier data and from Kaplan–Meier data that adjusted for PFI (as a continuous variable), ECOG performance score, race, baseline CA125 level, age, baseline liver/lung involvement and prior taxane therapy were presented; the HRs obtained from these analyses were 0.83 (95% CI 0.67 to 1.04; p = 0.106) and 0.78 (95% CI 0.62 to 0.98; p = 0.032), respectively. This suggests that use of unadjusted data in the NMAs and therefore economic analyses may introduce bias into estimates of relative treatment effect. However, in the absence of consistently adjusted data for all treatments of interest the TAG is unable to account for the magnitude or direction of any bias introduced from the use of unadjusted data.
In the sections that follow, the results of the TAG’s base-case and sensitivity analyses are discussed. In addition, the potential impact, with respect to the magnitude and direction of bias, that may have been introduced as a result of non-proportional hazards or potential clinical heterogeneity within the network of trials informing the TAG’s NMAs, is discussed.
Patients with platinum-sensitive disease
For patients with platinum-sensitive disease, a single network linking all the interventions and comparators of interest was not identified from the literature; instead, two independent networks were constructed:
-
PS network 1, which compared regimens containing platinum, in particular platinum plus paclitaxel, PLDH plus platinum, gemcitabine plus carboplatin, and platinum.
-
PS network 2, which compared therapies not containing platinum, in particular: PLDH, trabectedin plus PLDH, paclitaxel and topotecan.
The TAG notes that the ICERs estimated from these two networks are not comparable with each other and should be interpreted as independent analyses. Furthermore, the TAG acknowledges that the use of two independent analyses to inform this aspect of the decision problem (i.e. the comparative cost-effectiveness of treatments in patients with platinum-sensitive disease) is a limitation. However, following consultation with clinical experts, the TAG considers that the use of separate analyses for platinum and non-platinum therapies may not be unreasonable. This is because it is generally accepted that, in clinical practice, patients who are platinum sensitive and able to (and willing to) tolerate further platinum treatment would be treated with platinum. Therefore, for these patients, PS network 1 may be considered to provide information on the network of therapies most likely to be considered in clinical practice. Similarly, PS network 2 (PLDH, trabectedin plus PLDH, paclitaxel and topotecan) may be considered to provide information on the network of treatments suitable for platinum-sensitive patients who are unable or unwilling to tolerate further platinum-based therapy.
As a result of limited data, in particular PFS data, available for patients with PPS (PFI 6–12 months) and FPS (PFI > 12 months) disease, base-case analyses were not carried out for these subgroups. Furthermore, the TAG notes that identified trials which reported subgroup analyses in patients with partially and FPS disease were not sufficiently powered.
Platinum-sensitive network 1
Of the treatments considered in PS network 1 (platinum, gemcitabine plus carboplatin, paclitaxel plus platinum and PLDH plus platinum), base-case probabilistic and deterministic analysis estimated that treatment with gemcitabine plus carboplatin was extendedly dominated by treatment with paclitaxel plus platinum. That is, for the additional costs associated with paclitaxel plus platinum, the additional benefit was such that paclitaxel plus platinum may be considered better value for money than treatment with gemcitabine plus carboplatin.
Probabilistic analysis of the addition of paclitaxel or PLDH to platinum therapy resulted in similar estimates of mean total costs and QALYs. However, on average, treatment with paclitaxel plus platinum appeared to offer greater benefit than treatment with PLDH plus platinum. In addition, on average, treatment with PLDH plus platinum incurred higher costs than treatment with paclitaxel plus platinum, resulting in the dominance of PLDH plus platinum by paclitaxel plus platinum in probabilistic and deterministic analysis. The ICER associated with paclitaxel plus platinum compared with platinum was estimated from probabilistic analysis as £24,539.
However, the TAG considers it important to note that expert clinical advice highlighted that increased risk of neurotoxicity as a result of prior taxane therapy means that not all patients may tolerate further treatment with paclitaxel. With this in mind, the TAG consider it important to highlight that, at a WTP threshold of £30,000, the addition of PLDH to platinum therapy was associated with a 48% likelihood of being cost-effective compared with platinum monotherapy (probabilistic ICER vs. platinum was estimated to be £30,188).
Furthermore, one-way sensitivity analysis revealed that the relative effective of treatment on OS was the key driver of the cost-effectiveness results. However, visual inspection of the LCH plots for the outcome of OS (see Appendix 10) indicated that relative to the hazard of death associated with platinum therapy, the hazard of death associated with paclitaxel plus platinum may not be proportional. In fact, the relative hazard between these treatments appears to non-monotonically decrease over time. A similar relative hazard is observed between PLDH plus platinum compared with platinum monotherapy. With regards to the cost-effectiveness analysis, hazards that initially increase and then decrease over time are likely to lead to an initial underestimation of treatment effect, followed by an overestimation of treatment effect. However, it is unclear whether estimation of treatment effect will balance out over the time horizon of the economic model.
Furthermore, as discussed in Chapter 3 (see Comparability of baseline characteristics), there exists an imbalance in baseline performance score (ECOG) within one of the trials included in the OS NMA. In particular, the trial carried out by Gonzalez-Martin et al. ,48 in which paclitaxel plus carboplatin is compared with platinum monotherapy; the proportion of patients with a baseline ECOG score of 2, who were randomised to treatment with platinum monotherapy, was 17.9% compared with 5.6% of patients randomised to treatment with paclitaxel plus carboplatin. The TAG notes that this imbalance is likely to result in an overestimation of the relative treatment effect of paclitaxel plus carboplatin compared with platinum monotherapy.
In addition, the TAG notes the presence of clinical heterogeneity in the duration of PFI between trials. In particular, patients enrolled in the ICON-4/AGO-OVAR 2.2 trial61 had a comparably longer PFI than patients enrolled in the other trials included in NMA of OS and PFS data. Similarly, a comparatively high proportion of patients enrolled in the trial carried out by Gonzalez-Martin et al. 48 were diagnosed as recurrent based on assessment of CA125 levels; therefore these patients are likely to be more susceptible to platinum therapy than patients enrolled in the other included trials. However, the TAG notes that although patients in ICON-4/AGO-OVAR 2.261 and Gonzalez-Martin et al. 48 may be expected to experience greater benefit than patients enrolled in the other trials, the magnitude of this difference is unlikely to affect estimates of the relative effect of treatment.
For these reasons (non-proportional hazards and within trial heterogeneity) the TAG considers that it is unclear whether the relative effect of treatment with platinum monotherapy is overestimated or underestimated, particularly when compared with treatment with paclitaxel plus platinum.
Platinum-sensitive network 2
For PS network 2 (PLDH, trabectedin plus PLDH, paclitaxel and topotecan), base-case probabilistic and deterministic analysis estimated that treatment with topotecan was strictly dominated by (more expensive and less effective than) treatment with PLDH. Treatment with PLDH and treatment with trabectedin plus PLDH were estimated to provide benefit over treatment with paclitaxel. The existence of this benefit is more certain for trabectedin plus PLDH, than for PLDH. However, based on the TAG’s probabilistic analysis the cost per QALY of trabectedin plus PLDH compared with paclitaxel is £54,893 and the ICER associated with trabectedin compared with PLDH is £81,353, whereas the ICER associated with PLDH compared with paclitaxel is £25,931.
The key driver of the cost-effectiveness results in PS network 2 was identified in one-way sensitivity analysis as the relative effect of treatment on OS. As discussed above, the relative effect of treatment on OS has been estimated in NMA under the assumptions of proportional hazards and homogeneity of included trials. However, as a result of the absence of Kaplan–Meier data, it was not possible to construct LCH plots examining the hazards of OS associated with PLDH compared with paclitaxel; therefore, the proportionality or otherwise of these hazards is unknown. Furthermore, as a result of insufficient reporting, the TAG was not able to assess the baseline characteristics of included trials; trials were generally carried out in a mixed population of patients with platinum-resistant or -sensitive disease, therefore, baseline characteristics were not disaggregated by the subgroups of platinum sensitivity.
The TAG consider it important to highlight that the manufacturer of trabectedin, PharmaMar, submitted an analysis considering the head-to-head comparison of trabectedin plus PLDH compared with PLDH based on clinical effectiveness data that had been adjusted for baseline characteristics. Of particular importance within this analysis was the adjustment of PFS and OS data using PFI as a continuous variable. Following consultation with clinical experts, the TAG considers the use of adjusted data, in particular data adjusting for PFI as a continuous variable, to be appropriate. This is because, platinum sensitivity, as indicated by PFI, is a continuum related to the prognosis of the patient. That is, the longer the PFI, the more favourable the patient’s prognosis. The ICER of trabectedin plus PLDH compared with PLDH, estimated by the manufacturer, is £27,573 [including PAS (see Description and critique of manufacturer submitted evidence)].
However, as discussed above (see Description and critique of manufacturer submitted evidence), previous appraisal of the manufacturer’s model, by the ERG responsible for critical appraisal of the evidence submitted as part of TA222,90 highlighted limitations associated with the model used; in particular, the difficulty in applying discounting. Therefore, in order to assess the impact of using adjusted survival estimates within the TAG’s economic model, and to assess the validity of the manufacturer’s ICER, the TAG carried out a head-to-head comparison of trabectedin plus PLDH compared with PLDH. That is, adjusted PFS and OS data presented within the manufacturer’s model were used in the TAG’s model; costs, utilities and discounting applied within the TAG’s model were not altered. The ICER of trabectedin plus PLDH compared with PLDH, estimated by the TAG’s scenario analysis, is £35,646. Following inspection of the manufacturer’s model, the TAG notes that the difference in ICERs between the TAG’s scenario and manufacturer’s base-case analyses is likely to be a result of the method with which discounting is applied. The TAG considers that the method used in the TAG analysis is likely to be more accurate as a result of a model structured around monthly rather than mean estimates of PFS and OS. However, as efficacy data used in the TAG’s base-case model was unadjusted (to provide a consistent data set), the TAG notes that the head-to-head ICER generated from using adjusted efficacy data is not comparable with ICERs estimated for other treatments in the TAG’s base-case analyses.
Patients with platinum-resistant/-refractory disease
The network of interventions and comparators considered for the PRR subgroup was limited by the availability of data to three of the therapies, paclitaxel, PLDH and topotecan, outlined in the scope. However, based on expert clinical opinion that the prognosis of patients with PRR disease is often poor across available treatment options, a sensitivity analysis assuming equivalent efficacy between all treatments was carried out. Sensitivity analysis estimated that treatment with etoposide resulted in the lowest overall cost. However, the TAG notes that the cost associated with best supportive care may have been overestimated as only patients requiring strong opioid treatment were accounted for in the cost calculations.
Platinum-resistant/-refractory network
Of the treatments considered in the PRR network base-case probabilistic and deterministic analysis estimated that treatment with paclitaxel is strictly dominated by treatment with PLDH. However, probabilistic analysis estimated the ICER of topotecan compared with PLDH as £324,188, with 0% probability of being cost-effective at a WTP threshold of £30,000. Furthermore, the costs and QALYs associated with paclitaxel are similar to those associated with PLDH, with paclitaxel being dominated by PLDH in 39% of probabilistic simulations. As highlighted for patients with platinum-sensitive disease, increased risk of neurotoxicity following prior taxane therapy means that not all patients may tolerate further treatment with paclitaxel.
One-way sensitivity analysis revealed that the relative effect of treatment on OS was the key driver of the cost-effectiveness results. Assessment of the LCH plots for the outcome of OS (see Appendix 10) indicated that the hazard of death associated with topotecan is generally proportional to the hazard of death associated with PLDH. However, as a result of the absence of Kaplan–Meier data, it was not possible to construct LCH plots examining the hazards of OS associated with paclitaxel compared with PLDH; therefore, the proportionality or otherwise of these hazards is unknown. Furthermore, as a result of insufficient reporting, the TAG was not able to assess the baseline characteristics of included trials; trials were generally carried out in a mixed population of patients with platinum-resistant or platinum-sensitive disease, therefore, baseline characteristics were not disaggregated by the subgroups of platinum sensitivity.
Chapter 5 Assessment of factors relevant to the NHS and other parties
End-of-life criteria
Tables 153–155 assess the treatments against the NICE end-of-life criteria,145 by network. The TAG considers that it is likely that the criteria for end of life have not been met by any treatment. For the platinum-sensitive networks (PS network 1 and PS network 2) life expectancy for the baseline treatments are estimated by the TAG to be > 24 months. For the platinum-resistant population, no evaluable treatment offers a survival gain of greater than three months.
| NICE end-of-life criterion | Gemcitabine plus carboplatin | Paclitaxel plus platinum | PLDH plus platinum |
|---|---|---|---|
| Life expectancy on current standard care < 24 months | Mean OS for platinum monotherapy estimated from the TAG de novo analysis to be approximately 34 months Median OS for platinum monotherapy estimated from the TAG de novo analysis to be approximately 30 months |
||
| Treatment provides extension to life expectancy compared with current standard care of > 3 months | Mean OS estimated by TAG to be 35 months; gain in estimated mean OS < 1 month | Mean OS estimated by TAG to be 38 months; gain in estimated mean OS > 4 months | Mean OS estimated by TAG to be 38 months; gain in estimated mean > 4 months |
| Median OS estimated by TAG to be 30 months; no gain in estimated median OS | Median OS estimated by TAG to be 35 months; gain in estimated median OS of approximately 5 months | Median OS estimated by TAG to be 34 months; gain in estimated median OS of approximately 4 months | |
| The treatment is licensed or otherwise indicated for small populations | The incident population with platinum-sensitive disease was estimated by the manufacturer for trabectedin to be 2617 patients (see Table 121); however, this population does not include prevalent patients who relapse or take into account multiple relapses that may increase the number of treatable patients. The TAG estimates that including prevalent patients who may relapse and require treatment, would result in approximately 3379 patients | ||
| The estimates of the extension to life are robust | The HR for OS vs. platinum monotherapy was estimated by the TAG to be non-statistically significant. Therefore, the extension to life may not be considered to be robust | The HR for OS vs. platinum monotherapy was estimated by the TAG to be statistically significant. Therefore, the extension to life may be considered to be robust | The HR for OS vs. platinum monotherapy was estimated by the TAG to be statistically significant. Therefore, the extension to life may be considered to be robust |
| Overall assessment | All criteria not met Current life expectancy > 24 months; gain in OS < 3 months; gain in OS not statistically significant |
All criteria not met Current life expectancy > 24 months |
All criteria not met Current life expectancy > 24 months |
| NICE end-of-life criterion | PLDH | Topotecan | Trabectedin plus PLDH |
|---|---|---|---|
| Life expectancy on current standard care < 24 months | Mean OS for paclitaxel estimated from the TAG de novo analysis to be approximately 26 months Median OS for paclitaxel estimated from the TAG de novo analysis to be approximately 21 months |
||
| Treatment provides extension to life expectancy compared with current standard care of > 3 months | Mean OS estimated by TAG to be 29 months; gain in estimated mean OS approximately 3 months Median OS estimated by TAG to be 25 months; gain in estimated median OS of approximately 4 months |
Mean OS estimated by TAG to be 25 months; reduction in estimated mean OS Median OS estimated by TAG to be 19 months; reduction in estimated median OS |
Mean OS estimated by TAG to be 32 months; gain in estimated mean > 6 months vs. paclitaxel (approximately 3 months vs. PLDH) Median OS estimated by TAG to be 28 months; gain in estimated median OS of approximately 7 months (approximately 3 months vs. PLDH) |
| The treatment is licensed or otherwise indicated for small populations | The incident population with platinum-sensitive disease was estimated by the manufacturer for trabectedin to be 2617 patients (see Table 121); however, this population does not include prevalent patients who relapse or take into account multiple relapses that may increase the number of treatable patients. The TAG estimates that including prevalent patients who may relapse and require treatment, would result in approximately 3379 patients. The number of eligible patients may be greater than this if multiple relapses are taken into account The TAG notes that the manufacturer for trabectedin is requesting consideration for a subset of this population, and the manufacturer estimates the patient population to be 491 patients in 2014. The TAG considers that this number is likely to be an underestimate if prevalent and multiple relapses were taken into consideration |
||
| The estimates of the extension to life are robust | The HR for OS vs. paclitaxel monotherapy was estimated by the TAG to be non-statistically significant. Therefore, the extension to life may not be considered to be robust | NA | The HR for OS vs. platinum monotherapy or PLDH monotherapy was estimated by the TAG to be non-statistically significant. Therefore, the extension to life may not be considered to be robust |
| Overall assessment | All criteria not met Current life expectancy > 24 months; gain in OS not statistically significant |
All criteria not met Current life expectancy > 24 months; no gain in OS |
All criteria not met Current life expectancy > 24 months; gain in OS not statistically significant |
| NICE end-of-life criterion | Paclitaxel | Topotecan |
|---|---|---|
| Life expectancy on current standard care < 24 months | Mean OS for PLDH estimated from the TAG de novo analysis to be approximately 18.5 months Median OS for PLDH estimated from the TAG de novo analysis to be approximately 14 months |
|
| Treatment provides extension to life expectancy compared with current standard care of > 3 months | Mean OS estimated by TAG to be 18 months; reduction in estimated mean OS Median OS estimated by TAG to be 14 months; no gain in estimated median OS |
Mean OS estimated by TAG to be 19 months; gain in mean OS < 1 months Median OS estimated by TAG to be 15 months; gain in estimated median OS of approximately 1 month |
| The treatment is licensed or otherwise indicated for small populations | The incident population with recurrent advanced ovarian cancer was estimated by the manufacturer for trabectedin to be 3272 patients (see Table 121); given that the manufacturer estimated that 80% of these patients would be platinum-sensitive disease, this implies that 20% patients would have PRR disease approximately 654 patients However, this population does not include prevalent patients who relapse or take into account multiple relapses that may increase the number of treatable patients. The TAG estimates that including prevalent patients who may relapse and require treatment, would result in approximately 845 patients |
|
| The estimates of the extension to life are robust | NA | The HR for OS vs. PLDH monotherapy was estimated by the TAG to be non-statistically significant. Therefore, the extension to life may not be considered to be robust |
| Overall assessment | All criteria not met No gain in OS |
All criteria not met Gain in OS < 3 months; gain in OS not statistically significant |
Chapter 6 Discussion
The systematic review of clinical effectiveness evidence carried out to address the decision problem that is the focus of this MTA identified 16 RCTs, evaluating 14 pairwise comparisons. Furthermore, 21 economic evaluations considering patients with recurrent ovarian cancer were identified in the TAG’s review of the economic literature. However, the scope of the evidence identified was insufficient to fully address the decision problem; therefore, where possible the TAG has carried out synthesis of the evidence within network meta-analyses and de novo economic analyses.
Following consideration of the data identified and consultation with clinical experts, separate analyses have been carried out for patients with platinum-sensitive disease (PFI of ≥ 6 months) and PRR disease (PFI of < 6 months). The identified RCTs facilitated the construction of three distinct networks for the outcomes of OS and PFS, two of which considered patients with platinum-sensitive disease; the remaining network considered patients with disease that is PRR. As the systematic review was conducted in such a way as to identify all trials with at least one intervention of interest, a wider selection of treatments were assessed but, unfortunately, this did not uncover trials that could link the disconnected networks, in patients with platinum-sensitive disease, together. Furthermore, owing to time constraints, the decision was taken not to search for non-randomised trials.
The two networks constructed in patients with platinum-sensitive disease were:
-
PS network 1, which compared regimens containing platinum, in particular platinum plus paclitaxel, PLDH plus platinum, gemcitabine plus carboplatin, and platinum alone.
-
PS network 2, which compared non-platinum-based therapies, in particular PLDH, trabectedin plus PLDH, paclitaxel and topotecan.
Statement of main findings
Patients with platinum-sensitive disease
Overall survival and PFS data were identified for eight and seven different head-to-head comparisons of interventions and comparators of interest, respectively. Of these, three reported a statistically significant difference in OS between the treatments considered. In particular, Parmar et al. 61 reported a statistically significant difference in OS between paclitaxel plus platinum compared with conventional platinum treatment (HR 0.82, 95% CI 0.69 to 0.97), observed in the ICON4/AGO-OVAR 2.2 trial. Gonzalez-Martin et al. 48 reported a statistically significant difference between paclitaxel plus carboplatin compared with carboplatin alone (HR 0.31, 95% CI 0.14 to 0.68) and Gordon et al. 54 present a statistically significant difference between PLDH and topotecan (HR 1.43, 95% CI 1.07 to 1.92). Six of the identified head-to-head comparisons identified a statistically significant difference in PFS. These were:
-
CALYPSO31 PLDH plus carboplatin compared with paclitaxel plus carboplatin (HR 0.82, 95 CI 0.72 to 0.94)
-
ICON4/AGO-OVAR 2.261 Paclitaxel plus platinum compared with conventional platinum treatment (HR 0.76, 95% CI 0.66 to 0.89)
-
Gonzalez-Martin et al. 48 Paclitaxel plus carboplatin compared with carboplatin alone (HR 0.54, 95% CI 0.32 to 0.92)
-
Alberts et al. 28 PLDH plus carboplatin compared with carboplatin alone (HR 0.54, 95% CI 0.32 to 0.93)
-
OVA-30130 Trabectedin plus PLDH compared with PLDH alone (HR 0.73, 95% CI 0.56 to 0.95)
-
Pfisterer et al. 50 Gemcitabine plus carboplatin compared with carboplatin alone (HR 0.72, 95% CI 0.58 to 0.90).
In the NMA evaluating platinum-based chemotherapies, PLDH plus carboplatin and paclitaxel plus carboplatin were found to significantly improve OS compared with platinum monotherapy. However, no statistically significant differences in OS were identified between the remaining treatments considered in the network. When compared with platinum monotherapy, PFS was estimated to significantly improve in patients treated with paclitaxel plus carboplatin, gemcitabine plus carboplatin or PLDH plus carboplatin. In addition, a statistically significant difference in PFS was estimated for paclitaxel plus carboplatin compared with PLDH plus carboplatin.
Network meta-analysis of non-platinum-based therapies indicated that PLDH monotherapy and trabectedin plus PLDH are both significantly more effective at prolonging OS than topotecan monotherapy. No other significant OS differences were identified. Analysis of non-platinum-based regimens indicates that trabectedin plus PLDH statistically significantly improves PFS compared with PLDH, paclitaxel and topotecan when given as monotherapies. No statistically significant differences were identified among the monotherapies evaluated (PLDH, topotecan and paclitaxel).
Overall response rate was reported for 11 different head-to-head comparisons of interventions and comparators of interest. Of these, only two were statistically significant: trabectedin plus PLDH compared with PLDH from OVA-301 (OR 1.57, 95% CI 1.04 to 2.35); gemcitabine plus carboplatin compared with carboplatin alone from Pfisterer et al. (OR 1.527, 95% CI 1.025 to 2.275).
Based on the trials identified, it was not possible to construct a complete network informing relative ORR. Akin to analyses of OS and PFS, two discrete networks were generated: one evaluating platinum-based therapies (paclitaxel plus carboplatin, gemcitabine plus carboplatin, PLDH plus carboplatin and platinum monotherapy) and the second comparing non-platinum-based regimens [PLDH, trabectedin plus PLDH, topotecan (i.v.), paclitaxel (every 3 weeks), topotecan (oral) and paclitaxel weekly].
In the network evaluating platinum-based chemotherapies, paclitaxel plus carboplatin and gemcitabine plus carboplatin were found to have a significantly higher ORR than platinum monotherapy. There was no significant difference between PLDH plus carboplatin compared with any of the chemotherapeutic treatments assessed. Analysis of non-platinum-based regimens indicates that trabectedin plus PLDH significantly improves ORR compared with PLDH, and oral topotecan. Compared with oral topotecan, i.v. topotecan was found to be associated with a significant increase in the proportion of patients achieving CR or PR. No other statistically significant differences were identified.
Probabilistic economic analyses of PS network 1 indicated that treatment with gemcitabine plus platinum was extendedly dominated by treatment with paclitaxel plus platinum. That is, for the additional costs associated with paclitaxel plus platinum, the additional benefit was such that paclitaxel plus platinum may be considered better value for money than treatment with gemcitabine plus platinum.
Furthermore, the addition of paclitaxel or PLDH to platinum therapy resulted in similar estimates of mean total costs and QALYs. However, on average PLDH plus platinum was strictly dominated by (more expensive and less effective than) paclitaxel plus platinum. However, the increased risk of neurotoxicity as a result of prior taxane therapy means that not all patients may tolerate further treatment with paclitaxel. The ICERs associated with paclitaxel plus platinum compared with platinum, and PLDH plus platinum compared with platinum are £24,539 and £30,188, respectively.
In PS network 2, probabilistic economic analysis estimated that topotecan was strictly dominated by treatment with PLDH. In addition, treatment with PLDH and treatment with trabectedin plus PLDH were estimated to provide benefit over treatment with paclitaxel. However, based on the TAG’s probabilistic analysis, the ICER associated with trabectedin plus PLDH compared with paclitaxel is £54,893, and the ICER associated with trabectedin compared with PLDH is £81,353, whereas the ICER associated with PLDH compared with paclitaxel is £25,931.
However, the TAG considers it important to note that head-to-head comparison of trabectedin plus PLDH compared with PLDH, submitted by PharmaMar, estimated the ICER of trabectedin plus PLDH compared with PLDH as £27,573 (including PAS). This analysis was based on adjusted efficacy data, adjusted for, among other factors, PFI as a continuous variable. When efficacy data from the manufacturer’s model were used in the TAG’s model, the head-to-head ICER became £35,646. The TAG notes that the discrepancy in ICERs (between the manufacturer’s and the TAG’s analyses) is likely to be a result of the different methodologies used in the application of discounting. Furthermore, the TAG considers that the method used in TAG analysis is likely to be more accurate as a result of a model structured around monthly rather than mean estimates of PFS and OS. Moreover, the TAG considers that the ICER of £35,646 estimated using adjusted data is more likely to represent the cost-effectiveness of trabectedin plus PLDH compared with PLDH. However, as efficacy data used in the TAG’s base-case model was unadjusted (to provide a consistent data set), the TAG notes that the head-to-head ICER generated from using adjusted efficacy data is not comparable with ICERs estimated for other treatments in the TAG’s base-case analyses.
Patients with platinum-resistant/-refractory disease
The OS and PFS data were reported for five and four different head-to-head comparisons in PRR patients, respectively. Two RCTs enrolled only patients with PRR, with the remaining RCTs reporting results from a subgroup of patients within the trial. None of the trials identified a significant difference in OS or PFS between the two treatment groups evaluated. Furthermore, no statistically significant differences in ORR were reported in the eight different head-to-head comparisons involving PRR patients. Similarly, no statistically significant differences in OS or PFS were identified in NMA of treatment with paclitaxel, PLDH and topotecan. However, NMA of ORR estimated that PLDH significantly increased ORR compared with paclitaxel (175 mg/m2) every 21 days and with an alternative regimen in which paclitaxel was given weekly at a dose of 67 mg/m2. PLDH monotherapy was also significantly more effective than an unconventional regimen of topotecan in which topotecan was administered weekly at a dose of 4 mg/m2.
Probabilistic economic analysis estimated that similar costs and QALYs were accrued from treatment with PLDH and treatment with paclitaxel; however, on average, treatment with paclitaxel was dominated by treatment with PLDH. As highlighted for patients with platinum-sensitive disease, increased risk of neurotoxicity following prior taxane therapy means that not all patients may tolerate further treatment with paclitaxel.
Strengths and limitations of the assessment
Strengths
-
The evidence used to inform the decision problem that is the focus of this MTA has been identified following the general principles published by the CRD.
-
The methods used for the NMA followed the guidance described in the NICE DSU’s TSDs for evidence synthesis.
-
Economic analyses have been carried out in accordance with the NICE guide to methods of technology appraisal, International Society for Pharmacoeconomics and Outcomes Research (ISPOR) guidance, and, where possible, in adherence to recommendations made by the NICE DSU.
-
The economic model used to provide a framework for analysis has been widely used in the indication that is the focus of this MTA. In addition, amendments to the structure based on previous critiques have been made.
-
Expert clinical input has been sought and received throughout the project, in particular with respect to assumptions made in clinical and economic analyses and the face validity of final results and conclusions.
Weaknesses
The key weaknesses of the evidence synthesis used to address the decision problem are related to the limitations of the data available from the literature.
-
The absence of data linking the networks of treatment identified in patients with platinum-sensitive disease prevented consistent appraisal of the clinical effectiveness and cost-effectiveness of therapies of interest to patients with platinum-sensitive disease.
-
Limited data available for treatments of interest to patients with PRR disease led to assessment of the clinical effectiveness and cost-effectiveness of a subset of therapies of interest.
-
Clinical heterogeneity identified within the Gonzalez-Martin et al. trial48 included in NMA of PS may have introduced bias into the estimates of relative treatment effect.
-
The use of clinical effectiveness data unadjusted for key prognostic indicators, such as the PFI (measured continuously), may have introduced bias into the relative estimates of treatment effectiveness estimated from NMAs. Confounding from the use of postprogression therapy may have introduced bias into relative estimates of OS benefit, particularly in trials in which all patients cross over to the alternative group after progression or in trials in which the ‘new’ therapy is available as a postprogression treatment in the control group. The assumption of proportional hazards may have introduced bias into clinical and economic analyses.
Uncertainties
The magnitude and direction of potential bias introduced from use of unadjusted clinical effectiveness data, the assumption of proportional hazards and the potential clinical heterogeneity among trials included within the NMAs is uncertain. However, based on expert clinical opinion the TAG considers that the trials included in NMA were sufficiently homogeneous to facilitate the comparison of the clinical effectiveness of treatments. Furthermore, the TAG considers that the identified heterogeneity is unlikely to significantly impact estimates of relative treatment effect.
As a result of the absence of Kaplan–Meier data, the validity or otherwise of the assumption of proportional hazards is unknown for all comparisons considered in clinical and economic analysis. However, for the treatments identified in PS network 1, the TAG think it is likely that underestimates and overestimates of the relative effect of treatment may balance out over the time horizon of the economic model.
Other relevant factors
Based on criteria outlined by NICE,145 the TAG considers that none of the treatments identified within the scope of this MTA are eligible for consideration as end-of-life treatments.
Chapter 7 Conclusions
Suggested research priorities
Provided that this was thought to be of interest to the wider clinical community, RCT evidence comparing platinum-containing regimens with non-platinum-containing regimens should be sought. Furthermore, RCT evidence of the efficacy of etoposide and best supportive care in patients with resistant/refractory disease may be desirable.
Given the palliative nature of second-line or later treatment for recurrent ovarian cancer, and the limited data available on QoL, particularly for patients with PRR disease, research to determine reliable estimates of QoL in recurrent advanced ovarian cancer might be warranted.
Future trials in recurrent ovarian cancer should endeavour to carry out analysis on patient-level data which has been adjusted for a consistent array of variables; of particular importance is the adjustment of clinical effectiveness data for PFI (measured as a continuous rather than categorical variable).
Limited information on best supportive care was identified. Some people may choose to not receive further treatment, and research into what constitutes best supportive care, and impact of best supportive care on QoL, might help to inform the decision-making process from the perspective of both the clinician and the person with advanced ovarian cancer. In addition, further research into the cost of best supportive care may also be warranted.
Acknowledgements
The Assessment Group would like to thank Professor Nicholas Reed (Consultant Clinical Oncologist) and Professor Gordon Rustin (Consultant Medical Oncologist) for providing clinical advice throughout the project. Thanks also to Dr Timothy Perren for advising on the protocol and to Mr Khalil Razvi (Gynaecological Oncologist) for providing comments on the background section of the technology assessment report. The Assessment Group would also like to thank Dr Susan Griffin (Senior Research Fellow) and Dr Laura Bojke (Senior Research Fellow) for providing feedback on the proposed economic analysis, and the economic sections of the report. Thanks also to Taryn Krause and Ashwini Sreekanta for their contributions to appraisal of the abstracts identified from the literature search and the validation of the included studies.
Contribution of authors
Steve Edwards Project lead: supervised the production of the final report; carried out the network meta-analyses and critical appraisal of the economic evidence and company submissions.
Samantha Barton Devised and carried out the clinical literature searches; study selection; data extraction; report writing; and critical appraisal of the company submissions.
Elizabeth Thurgar Devised and carried out the economic literature searches; study selection; data extraction; development of the economic model; report writing; and critical appraisal of the company submissions.
Nicola Trevor Devised and carried out the economic literature searches; study selection; data extraction; development of the economic model; report writing; and critical appraisal of the company submissions.
All authors read and commented on draft versions of the TAG report.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Cancer Research UK (CRUK) . Ovarian Cancer Statistics 2013. www.cancerresearchuk.org/cancer-info/cancerstats/types/ovary (accessed July 2013).
- Benedet JL, Bender H, Jones H III, Ngan HY, Pecorelli S. FIGO staging classifications and clinical practice guidelines in the management of gynecologic cancers. FIGO Committee on Gynecologic Oncology. Int J Gynaecol Obstet 2000;70:209-62. http://dx.doi.org/10.1016/S0020-7292(00)90001-8.
- National Institute for Health and Care Excellence (NICE) . Ovarian Cancer: The Recognition and Initial Management of Ovarian Cancer 2011. www.nice.org.uk/nicemedia/live/13464/54194/54194.pdf (accessed July 2013).
- Scottish Intercollegiate Guidelines Network (SIGN) . Epithelial Ovarian Cancer: A National Clinical Guideline. 2003. www.sign.ac.uk/pdf/sign75.pdf (accessed July 2013).
- National Institute for Health and Care Excellence (NICE) . Guidance on the Use of Paclitaxel in the Treatment of Ovarian Cancer 2003. www.nice.org.uk/nicemedia/live/11486/32539/32539.pdf (accessed July 2013).
- Nossov V, Amneus M, Su F, Lang J, Janco JM, Reddy ST, et al. The early detection of ovarian cancer: from traditional methods to proteomics. Can we really do better than serum CA-125?. Am J Obstet Gynecol 2008;199:215-23. http://dx.doi.org/10.1016/j.ajog.2008.04.009.
- Sorensen SS, Mosgaard BJ. Combination of cancer antigen 125 and carcinoembryonic antigen can improve ovarian cancer diagnosis. Dan Med Bull 2011;58.
- Hess LM, Stehman FB. State of the science in ovarian cancer quality of life research: a systematic review. Int J Gynecol Cancer 2012;22:1273-80. http://dx.doi.org/10.1097/IGC.0b013e318263f02e.
- Health and Social Care Information Centre (HSCIC) . Hospital Episode Statistics 2011–2012 2012. www.hscic.gov.uk/catalogue/PUB08288/hosp-epis-stat-admi-prim-diag-3cha-11-12-tab.xls (accessed July 2013).
- National Institute for Health and Care Excellence (NICE) . Paclitaxel, Pegylated Liposomal Doxorubicin Hydrochloride and Topotecan for Second-Line or Subsequent Treatment of Advanced Ovarian Cancer. Systematic Review 2005. www.nice.org.uk/nicemedia/pdf/TA091guidance.pdf (accessed July 2013).
- National Institute for Health and Care Excellence (NICE) . Bevacizumab in Combination With Paclitaxel and Carboplatin for First-Line Treatment of Advanced Ovarian Cancer 2013. http://publications.nice.org.uk/bevacizumab-in-combination-with-paclitaxel-and-carboplatin-for-first-line-treatment-of-advanced-ta284 (accessed July 2013).
- National Institute for Health and Care Excellence (NICE) . NICE Pathway for Management of Advanced (stage II-IV) Ovarian Cancer 2013. http://pathways.nice.org.uk/pathways/ovarian-cancer/management-of-advanced-stage-ii-iv-ovarian-cancer (accessed July 2013).
- Main C, Bojke L, Griffin S, Norman G, Barbieri M, Mather L, et al. Topotecan, Pegylated Liposomal Doxorubicin Hydrochloride and Paclitaxel for Second-Line or Subsequent Treatment of Advanced Ovarian Cancer: A Systematic Review and Economic Evaluation 2004. www.nice.org.uk/nicemedia/live/11553/33023/33023.pdf (accessed July 2013).
- Naumann RW, Coleman RL. Management strategies for recurrent platinum-resistant ovarian cancer. Drugs 2011;71:1397-412. http://dx.doi.org/10.2165/11591720-000000000-00000.
- Trabectedin for the Treatment of Relapsed Ovarian Cancer. Report. London: NICE; 2011.
- National Institute for Health and Care Excellence (NICE) . Bevacizumab in Combination With Gemcitabine and Carboplatin for Treating the First Recurrence of Platinum-Sensitive Advanced Ovarian Cancer 2013. http://publications.nice.org.uk/bevacizumab-in-combination-with-gemcitabine-and-carboplatin-for-treating-the-first-recurrence-of-ta285 (accessed July 2013).
- National Institute for Health and Care Excellence (NICE) . Trabectedin for the Treatment of Relapsed Ovarian Cancer. 2013. http://publications.nice.org.uk/trabectedin-for-the-treatment-of-relapsed-ovarian-cancer-ta222 (accessed July 2013).
- Pfisterer J, Ledermann JA. Management of platinum-sensitive recurrent ovarian cancer. Semin Oncol 2006;33:S12-16. http://dx.doi.org/10.1053/j.seminoncol.2006.03.012.
- Colombo N, Peiretti M, Parma G, Lapresa M, Mancari R, Carinelli S, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010;21:v23-30. http://dx.doi.org/10.1093/annonc/mdq244.
- Hounsome L, Gillatt D, Persad R, Verne J. Hospital Care for Cancer Patients in the Last Year of Life 2012. www.swpho.nhs.uk/resource/item.aspx?RID=97136 (accessed July 2013).
- ten Bokkel Huinink W, Gore M, Carmichael J, Gordon A, Malfetano J, Hudson I, et al. Topotecan versus paclitaxel for the treatment of recurrent epithelial ovarian cancer. J Clin Oncol 1997;15:2183-93.
- Electronic Medicines Compendium . Topotecan Hospira 4 mg 4 ml Concentrate for Solution for Infusion. Summary of Product Characteristics 2012. www.medicines.org.uk/emc/medicine/25101/SPC (accessed July 2013).
- Sehouli J, Stengel D, Harter P, Kurzeder C, Belau A, Bogenrieder T, et al. Topotecan weekly versus conventional 5-day schedule in patients with platinum-resistant ovarian cancer: a randomized multicenter phase II trial of the North-Eastern German Society of Gynecological Oncology Ovarian Cancer Study Group. J Clin Oncol 2011;10:242-8. http://dx.doi.org/10.1200/JCO.2009.27.8911.
- Gore M, Oza A, Rustin G, Malfetano J, Calvert H, Clarke-Pearson D, et al. A randomised trial of oral versus intravenous topotecan in patients with relapsed epithelial ovarian cancer. Eur J Cancer 2002;38:57-63. http://dx.doi.org/10.1016/S0959-8049(01)00188-5.
- Horenstein MS, Vander Heide RS, L’Ecuyer TJ. Molecular basis of anthracycline-induced cardiotoxicity and its prevention. Mol Genet Metab 2000;71:436-44. http://dx.doi.org/10.1006/mgme.2000.3043.
- Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet 2003;42:419-36. http://dx.doi.org/10.2165/00003088-200342050-00002.
- Electronic Medicines Compendium . Caelyx 2 mg Ml Concentrate for Solution for Infusion 2010. www.medicines.org.uk/emc/medicine/7017/SPC/Caelyx+2mg+ml+concentrate+for+solution+for+infusion (accessed July 2013).
- Alberts DS, Liu PY, Wilczynski SP, Clouser MC, Lopez AM, Michelin DP, et al. Randomized trial of pegylated liposomal doxorubicin (PLD) plus carboplatin versus carboplatin in platinum-sensitive (PS) patients with recurrent epithelial ovarian or peritoneal carcinoma after failure of initial platinum-based chemotherapy (Southwest Oncology Group Protocol S0200). Gynecol Oncol 2008;108:90-4. http://dx.doi.org/10.1016/j.ygyno.2007.08.075.
- Bafaloukos D, Linardou H, Aravantinos G, Papadimitriou C, Bamias A, Fountzilas G, et al. A randomized phase II study of carboplatin plus pegylated liposomal doxorubicin versus carboplatin plus paclitaxel in platinum sensitive ovarian cancer patients: a Hellenic Cooperative Oncology Group study. BMC Med 2010;8. http://dx.doi.org/10.1186/1741-7015-8-3.
- Monk BJ, Herzog TJ, Kaye SB, Krasner CN, Vermorken JB, Muggia FM, et al. Trabectedin plus pegylated liposomal doxorubicin in recurrent ovarian cancer. J Clin Oncol 2010;28:3107-14. http://dx.doi.org/10.1200/JCO.2009.25.4037.
- Pujade-Lauraine E, Wagner U, Avall-Lundqvist E, Gebski V, Heywood M, Vasey PA, et al. Pegylated liposomal doxorubicin and carboplatin compared with paclitaxel and carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol 2010;28:3323-9. http://dx.doi.org/10.1200/JCO.2009.25.7519.
- Jennewein S, Croteau R. Taxol: biosynthesis, molecular genetics, and biotechnological applications. Appl Microbiol Biotechnol 2001;57:13-9. http://dx.doi.org/10.1007/s002530100757.
- Electronic Medicines Compendium . Paclitaxel 6 mg Ml Concentrate for Solution for Infusion 2012. www.medicines.org.uk/emc/medicine/25881/SPC/Paclitaxel+6+mg+ml+Concentrate+for+Solution+for+Infusion (accessed July 2013).
- van Kesteren C, de Vooght MM, Lopez-Lazaro L, Mathot RA, Schellens JH, Jimeno JM, et al. Yondelis (trabectedin, ET-743): the development of an anticancer agent of marine origin. Anticancer Drugs 2003;14:487-502. http://dx.doi.org/10.1097/00001813-200308000-00001.
- Electronic Medicines Compendium . Yondelis 0.25 mg Powder for Concentrate for Solution for Infusion Yondelis 1 mg Powder for Concentrate for Solution for Infusion 2012. www.medicines.org.uk/emc/medicine/20457/SPC/Yondelis+0.25+mg+powder+for+concentrate+for+solution+for+infusion+Yondelis+1+mg+powder+for+concentrate+for+solution+for+infusion (accessed July 2013).
- Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol 1995;22:3-10.
- Electronic Medicines Compendium . Gemcitabine 100 mg Ml Concentrate for Solution for Infusion 2012. www.medicines.org.uk/emc/medicine/27136/SPC/Gemcitabine+100+mg+ml+Concentrate+for+Solution+for+Infusion (accessed July 2013).
- National Institute for Health and Care Excellence (NICE) . Ovarian Cancer: Topotecan, Pegylated Liposomal Doxorubicin Hydrochloride, Paclitaxel, Trabectedin and Gemcitabine for Advanced Recurrent Disease Only (Review of TA 91): Final Protocol 2013. www.nice.org.uk/nicemedia/live/13843/62152/62152.pdf (accessed July 2013).
- Centre for Reviews and Dissemination (CRD) . CRD’s Guidance for Undertaking Reviews in Healthcare 2011. www.york.ac.uk/inst/crd/SysRev/!SSL!/WebHelp/SysRev3.htm (accessed July 2013).
- Scottish Intercollegiate Guidelines Network (SIGN) . Search Filters 2013. www.sign.ac.uk/methodology/filters.html#random.
- Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration; 2011.
- Davis S, Tappenden P, Cantrell A. A Review of Studies Examining the Relationship Between Progression-Free Survival and Overall Survival in Advanced or Metastatic Cancer 2012. www.nicedsu.org.uk/PFSOS%20Report.FINAL.06.08.12.pdf (accessed July 2013).
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials 2011. www.nicedsu.org.uk/TSD2%20General%20meta%20analysis%20corrected%20Mar2013.pdf (accessed July 2013).
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 1: Introduction to Evidence Synthesis for Decision-Making 2013. www.nicedsu.org.uk/TSD1%20Introduction.final.08.05.12.pdf (accessed July 2013).
- US Department of Health and Human Services . Common Terminology Criteria for Adverse Events (CTCAE) 2009. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf (accessed July 2013).
- Cancer Therapy Evaluation Program . Common Toxicity Criteria Manual 1999. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcmanual_v4_10-4-99.pdf.
- O’Byrne KJ, Bliss P, Graham JD, Gerber J, Vasey PA, Khanna S, et al. A phase III study of Doxil/Caelyx versus paclitaxel in platinum-treated taxane-naive relapsed ovarian cancer. Proc Am Soc Clin Oncol 2002;21.
- Gonzalez-Martin AJ, Calvo E, Bover I, Rubio MJ, Arcusa A, Casado A, et al. Randomized phase II trial of carboplatin versus paclitaxel and carboplatin in platinum-sensitive recurrent advanced ovarian carcinoma: a GEICO (Grupo Espanol de Investigacion en Cancer de Ovario) study. Ann Oncol 2005;16:749-55. http://dx.doi.org/10.1093/annonc/mdi147.
- Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ. Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol 2001;19:3312-22.
- Pfisterer J, Plante M, Vergote I, du Bois A, Hirte H, Lacave AJ, et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol 2006;24:4699-707. http://dx.doi.org/10.1200/JCO.2006.06.0913.
- Cantu MG, Buda A, Parma G, Rossi R, Floriani I, Bonazzi C, et al. Randomized controlled trial of single-agent paclitaxel versus cyclophosphamide, doxorubicin, and cisplatin in patients with recurrent ovarian cancer who responded to first-line platinum-based regimens. J Clin Oncol 2002;20:1232-7. http://dx.doi.org/10.1200/JCO.20.5.1232.
- ten Bokkel Huinink W, Lane SR, Ross GA. Long-term survival in a phase III, randomised study of topotecan versus paclitaxel in advanced epithelial ovarian carcinoma. Ann Oncol 2004;15:100-3. http://dx.doi.org/10.1093/annonc/mdh025.
- Gore M, ten Bokkel Huinink W, Carmichael J, Gordon A, Davidson N, Coleman R, et al. Clinical evidence for topotecan-paclitaxel non-cross-resistance in ovarian cancer. J Clin Oncol 2001;19:1893-900.
- Gordon AN, Tonda M, Sun S, Rackoff W. Long-term survival advantage for women treated with pegylated liposomal doxorubicin compared with topotecan in a phase 3 randomized study of recurrent and refractory epithelial ovarian cancer. Gynecol Oncol 2004;95:1-8. http://dx.doi.org/10.1016/j.ygyno.2004.07.011.
- Markman M, Moon J, Wilczynski S, Lopez AM, Rowland J, Michelin DP, et al. Single agent carboplatin versus carboplatin plus pegylated liposomal doxorubicin in recurrent ovarian cancer: final survival results of a SWOG (S0200) phase 3 randomized trial. Gynecol Oncol 2010;116:323-5. http://dx.doi.org/10.1016/j.ygyno.2009.11.026.
- Wagner U, Marth C, Largillier R, Kaern J, Brown C, Heywood M, et al. Final overall survival results of phase III GCIG CALYPSO trial of pegylated liposomal doxorubicin and carboplatin vs paclitaxel and carboplatin in platinum-sensitive ovarian cancer patients. Br J Cancer 2012;107:588-91. http://dx.doi.org/10.1038/bjc.2012.307.
- Gladieff L, Ferrero A, De Rauglaudre G, Brown C, Vasey P, Reinthaller A, et al. Carboplatin and pegylated liposomal doxorubicin versus carboplatin and paclitaxel in partially platinum-sensitive ovarian cancer patients: results from a subset analysis of the CALYPSO phase III trial. Ann Oncol 2012;23:1185-9. http://dx.doi.org/10.1093/annonc/mdr441.
- Kurtz JE, Kaminsky MC, Floquet A, Veillard AS, Kimmig R, Dorum A, et al. Ovarian cancer in elderly patients: carboplatin and pegylated liposomal doxorubicin versus carboplatin and paclitaxel in late relapse: a gynecologic cancer intergroup (GCIG) CALYPSO sub-study. Ann Oncol 2011;22:2417-23. http://dx.doi.org/10.1093/annonc/mdr001.
- Brundage M, Gropp M, Mefti F, Mann K, Lund B, Gebski V, et al. Health-related quality of life in recurrent platinum-sensitive ovarian cancer: results from the CALYPSO trial. Ann Oncol 2012;23:2020-7. http://dx.doi.org/10.1093/annonc/mdr583.
- Rosenberg P, Andersson H, Boman K, Ridderheim M, Sorbe B, Puistola U, et al. Randomized trial of single agent paclitaxel given weekly versus every three weeks and with peroral versus intravenous steroid premedication to patients with ovarian cancer previously treated with platinum. Acta Oncol 2002;41:418-24. http://dx.doi.org/10.1080/028418602320404998.
- Parmar MK, Ledermann JA, Colombo N, du Bois A, Delaloye JF, Kristensen GB, et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet 2003;361:2099-106. http://dx.doi.org/10.1016/S0140-6736(03)13718-X.
- Lortholary A, Largillier R, Weber B, Gladieff L, Alexandre J, Durando X, et al. Weekly paclitaxel as a single agent or in combination with carboplatin or weekly topotecan in patients with resistant ovarian cancer: the CARTAXHY randomized phase II trial from Groupe d’Investigateurs Nationaux pour l’Etude des Cancers Ovariens (GINECO). Ann Oncol 2012;23:346-52. http://dx.doi.org/10.1093/annonc/mdr149.
- Piccart MJ, Green JA, Lacave AJ, Reed N, Vergote I, Benedetti-Panici P, et al. Oxaliplatin or paclitaxel in patients with platinum-pretreated advanced ovarian cancer: a randomized phase II study of the European Organization for Research and Treatment of Cancer Gynecology Group. J Clin Oncol 2000;18:1193-202.
- Monk BJ, Herzog TJ, Kaye SB, Krasner CN, Vermorken JB, Muggia FM, et al. Trabectedin plus pegylated liposomal doxorubicin (PLD) versus PLD in recurrent ovarian cancer: overall survival analysis. Eur J Cancer 2012;48:2361-8. http://dx.doi.org/10.1016/j.ejca.2012.04.001.
- Poveda A, Vergote I, Tjulandin S, Kong B, Roy M, Chan S, et al. Trabectedin plus pegylated liposomal doxorubicin in relapsed ovarian cancer: outcomes in the partially platinum-sensitive (platinum-free interval 6-12 months) subpopulation of OVA-301 phase III randomized trial. Ann Oncol 2011;22:39-48. http://dx.doi.org/10.1093/annonc/mdq352.
- Kaye SB, Colombo N, Monk BJ, Tjulandin S, Kong B, Roy M, et al. Trabectedin plus pegylated liposomal doxorubicin in relapsed ovarian cancer delays third-line chemotherapy and prolongs the platinum-free interval. Ann Oncol 2011;22:49-58. http://dx.doi.org/10.1093/annonc/mdq353.
- Krasner CN, Poveda A, Herzog TJ, Vermorken JB, Kaye SB, Nieto A, et al. Patient-reported outcomes in relapsed ovarian cancer: results from a randomized Phase III study of trabectedin with pegylated liposomal doxorubicin (PLD) versus PLD alone. Gynecol Oncol 2012;127:161-7. http://dx.doi.org/10.1016/j.ygyno.2012.06.034.
- Omura GA, Brady MF, Look KY, Averette HE, Delmore JE, Long HJ, et al. Phase III trial of paclitaxel at two dose levels, the higher dose accompanied by filgrastim at two dose levels in platinum-pretreated epithelial ovarian cancer: an intergroup study. J Clin Oncol 2003;21:2843-8. http://dx.doi.org/10.1200/JCO.2003.10.082.
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92:205-16. http://dx.doi.org/10.1093/jnci/92.3.205.
- Gynecologic Cancer InterGroup . CA125 Definitions Agreed by GCIG November 2005 2005. www.gcig.igcs.org/CA125/respdef_nov2005.pdf (accessed 27 August 2014).
- European Organisation for Research in the Treatment of Cancer . Quality of Life n.d. http://groups.eortc.be/qol.
- Clinical Study Abbreviated Report. A Phase III, Randomized, Open-label, Comparative Study of Caelyx Versus Paclitaxel HCI in Patients with Epithelial Ovarian Carcinoma Following Failure of First-line, Platinum-based Chemotherapy, Protocol 30-57. Saunderton, Bucks: Johnson & Johnson Pharmaceutical Research and Development; 2004.
- National Institute for Health and Care Excellence (NICE) . Trabectedin for the Treatment of Relapsed Ovarian Cancer. Final Appraisal Determination 2013. www.nice.org.uk/nicemedia/live/12094/50814/50814.pdf (accessed July 2013).
- Mandrekar SJ, Sargent DJ. Pick the winner designs in phase II cancer clinical trials. J Thorac Oncol 2006;1:5-6. http://dx.doi.org/10.1097/01243894-200601000-00003.
- Sorensen JB, Klee M, Palshof T, Hansen HH. Performance status assessment in cancer patients. An inter-observer variability study. Br J Cancer 1993;67:773-5. http://dx.doi.org/10.1038/bjc.1993.140.
- Rustin GJ, Timmers P, Nelstrop A, Shreeves G, Bentzen SM, Baron B, et al. Comparison of CA-125 and standard definitions of progression of ovarian cancer in the intergroup trial of cisplatin and paclitaxel versus cisplatin and cyclophosphamide. J Clin Oncol 2006;24:45-51. http://dx.doi.org/10.1200/JCO.2005.01.2757.
- Food and Drug Administration (FDA) . Guidance for Industry. Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics 2007. www.fda.gov/downloads/Drugs/. /Guidances/ucm071590.pdf (accessed 27 August 2014).
- Booth CM, Eisenhauer EA. Progression-free survival: meaningful or simply measurable?. J Clin Oncol 2012;30:1030-3. http://dx.doi.org/10.1200/JCO.2011.38.7571.
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical Methods for Incorporating Summary Time-to-event Data into Meta-analysis. Trials 2007;8. http://dx.doi.org/10.1186/1745-6215-8-16.
- Rustin GJ, Nelstrop AE, Bentzen SM, Piccart MJ, Bertelsen K. Use of tumour markers in monitoring the course of ovarian cancer. Ann Oncol 1999;10:S21-7. http://dx.doi.org/10.1023/A:1008351216605.
- Aaronson NK, Ahmedzai S, Bergman B, Bullinger M, Cull A, Duez NJ, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst 1993;85:365-76. http://dx.doi.org/10.1093/jnci/85.5.365.
- Smith DH, Adams JR, Johnston SR, Gordon A, Drummond MF, Bennett CL. A comparative economic analysis of pegylated liposomal doxorubicin versus topotecan in ovarian cancer in the USA and the UK. Ann Oncol 2002;13:1590-7. http://dx.doi.org/10.1093/annonc/mdf275.
- Ojeda B, de Sande LM, Casado A, Merino P, Casado M. Cost-minimisation analysis of pegylated liposomal doxorubicin hydrochloride versus topotecan in the treatment of patients with recurrent epithelial ovarian cancer in Spain. Br J Cancer 2003;89:1002-7. http://dx.doi.org/10.1038/sj.bjc.6601228.
- Capri S, Cattaneo G. Cost-minimization analysis of pegylated liposomal doxorubicin versus topotecan for the treatment of ovarian cancer in Italy. Clin Ther 2003;25:1826-45. http://dx.doi.org/10.1016/S0149-2918(03)80172-8.
- Prasad M, Ben-Porat L, Hoppe B, Aghajanian C, Sabbatini P, Chi DS, et al. Costs of treatment and outcomes associated with second-line therapy and greater for relapsed ovarian cancer. Gynecol Oncol 2004;93:223-8. http://dx.doi.org/10.1016/j.ygyno.2004.01.014.
- British National Formulary. London: BMA and RPS; 2004.
- Netten AP, Curtis LA. Unit Costs of Health and Social Care 2000. Canterbury: PSSRU, University of Kent; 2000.
- Tengs TO, Wallace A. One thousand health-related quality-of-life estimates. Med Care 2000;38:583-637. http://dx.doi.org/10.1097/00005650-200006000-00004.
- Brown RE, Hutton J. Cost-utility model comparing docetaxel and paclitaxel in advanced breast cancer patients. Anticancer Drugs 1998;9:899-907. http://dx.doi.org/10.1097/00001813-199811000-00009.
- Papaioannou D, Rafia R, Stevenson MD, Stevens JW, Evans P, Papaioannou D, et al. Trabectedin for the Treatment of Relapsed Ovarian Cancer: A Single Technology Appraisal 2011. www.nice.org.uk/guidance/index.jsp?action=download&o=49230 (accessed July 2013).
- British National Formulary. London: BMA and RPS; 2009.
- NHS Trusts and PCTs Combined Reference Cost Schedules 2007–08. London: DoH; 2009.
- National Institute for Health and Care Excellence (NICE) . Trabectedin (Yondelis®) for Treatment of Patients With Ovarian Cancer (relapsed). 2010. www.nice.org.uk/guidance/index.jsp?action=download&o=49233 (accessed July 2013).
- Griffin S, Bojke L, Main C, Palmer S. Incorporating direct and indirect evidence using Bayesian methods: an applied case study in ovarian cancer. Value Health 2006;9:123-31. http://dx.doi.org/10.1111/j.1524-4733.2006.00090.x.
- Forbes C, Wilby J, Richardson G, Sculpher M, Mather L, Riemsma R. A systematic review and economic evaluation of pegylated liposomal doxorubicin hydrochloride for ovarian cancer. Health Technol Assess 2002;6.
- Chan JM. An economic analysis of bevacizumab in recurrent treatment of ovarian cancer. Gynecol Oncol 2011;125:S15-16. http://dx.doi.org/10.1016/j.ygyno.2011.12.037.
- Main C, Bojke L, Griffin S, Norman G, Barbieri M, Mather L, et al. Topotecan, pegylated liposomal doxorubicin hydrochloride and paclitaxel for second-line or subsequent treatment of advanced ovarian cancer: a systematic review and economic evaluation. Health Technol Assess 2006;10.
- Havrilesky LJ, Pokrzywinski R, Revicki D, Higgins RV, Nycum LR, Kohler MF, et al. Cost-effectiveness of combination versus sequential docetaxel and carboplatin for the treatment of platinum-sensitive, recurrent ovarian cancer. Cancer 2012;118:386-91. http://dx.doi.org/10.1002/cncr.26199.
- Papaioannou D, Rafia R, Stevenson MD, Stevens JW, Evans P, Papaioannou D, et al. Trabectedin for the treatment of relapsed ovarian cancer. Health Technol Assess 2011;15.
- Lesnock JF. Consolidation paclitaxel is more cost-effective than bevacizumab following upfront treatment of advanced ovarian cancer. Gynecol Oncol 2011;120:S66-7. http://dx.doi.org/10.1016/j.ygyno.2010.12.159.
- Lesnock JL, Farris C, Krivak TC, Smith KJ, Markman M. Consolidation paclitaxel is more cost-effective than bevacizumab following upfront treatment of advanced epithelial ovarian cancer. Gynecol Oncol 2011;122:473-8. http://dx.doi.org/10.1016/j.ygyno.2011.05.014.
- Gore M, Vergote I, Vasanthan S, Chan S, Arranz JM, Colombo N, et al. Cost-effectiveness of trabectedin in combination with pegylated liposomal doxorubicin hydrochloride for the treatment of women with relapsed platinum-sensitive ovarian cancer in the UK: analysis based on the final survival data. Eur J Cancer 2011;47. http://dx.doi.org/10.1016/S0959-8049(11)72138-4.
- Case AS, Rocconi RP, Partridge EE, Straughn JM. A cost-effectiveness analysis of chemotherapy for patients with recurrent platinum-sensitive epithelial ovarian cancer. Gynecol Oncol 2007;105:223-7. http://dx.doi.org/10.1016/j.ygyno.2006.11.018.
- Havrilesky LJ, Secord AA, Kulasingam S, Myers E. Management of platinum-sensitive recurrent ovarian cancer: a cost-effectiveness analysis. Gynecol Oncol 2007;107:211-18. http://dx.doi.org/10.1016/j.ygyno.2007.06.029.
- Montalar JC. Trabectedin plus PLD versus PLD monotherapy in patients with platinum-sensitive relapsed ovarian cancer: a cost-effectiveness analysis in Spain. Eur J of Hosp Pharm Sci Pract 2012;19:364-9.
- Rocconi RP, Case AS, Straughn JM, Estes JM, Partridge EE. Role of chemotherapy for patients with recurrent platinum-resistant advanced epithelial ovarian cancer: a cost-effectiveness analysis. Cancer 2006;107:536-43. http://dx.doi.org/10.1002/cncr.22045.
- Lee HYH. Cost-utility analysis of combination therapy of pegylated liposomal doxorubicin (PLD) and carboplatin for Korean women with platinum-sensitive ovarian cancer. Value in Health Conference 2011 n.d.
- Philips Z, Ginnelly L, Sculpher M, Claxton K, Golder S, Riemsma R, et al. Review of guidelines for good practice in decision-analytic modelling in health technology assessment. Health Technol Assess 2004;8.
- National Institute for Health and Care Excellence (NICE) . Guide to the Methods of Technology Appraisal 2008. www.nice.org.uk/media/B52/A7/TAMethodsGuideUpdatedJune2008.pdf (accessed July 2013).
- British National Formulary. London: BMA and RPS; 2013.
- Department of Health (DH) . National Schedule of Reference Costs 2011-12 for NHS Trusts and NHS Foundation Trusts 2012. www.gov.uk/government/publications/nhs-reference-costs-financial-year-2011-to-2012 (accessed July 2013).
- Guest JF, Ruiz FJ, Greener MJ, Trotman IF. Palliative care treatment patterns and associated costs of healthcare resource use for specific advanced cancer patients in the UK. Eur J Cancer Care(Engl) 2006;15:65-73. http://dx.doi.org/10.1111/j.1365-2354.2005.00623.x.
- Hoyle MW, Henley W. Improved curve fits to summary survival data: application to economic evaluation of health technologies. BMC Med Res Methodol 2011;11. http://dx.doi.org/10.1186/1471-2288-11-139.
- Sacco JJ, Botten J, Macbeth F, Bagust A, Clark P. The average body surface area of adult cancer patients in the UK: a multicentre retrospective study. PLOS One 2010;5. http://dx.doi.org/10.1371/journal.pone.0008933.
- Curtis L. Unit Costs of Health and Social Care 2012 2012. www.pssru.ac.uk/archive/pdf/uc/uc2012/full-with-covers.pdf (accessed July 2013).
- Stewart L. Chemotherapy for Advanced Ovarian Cancer. The Cochrane Library; 2011.
- Rohatgi A. WebPlotDigitizer 2012. http://arohatgi.info/WebPlotDigitizer (accessed July 2013).
- Latimer N. Nice DSU Technical Support Document 14: Survival Analysis for Economic Evaluations Alongside Clinical Trials – Extrapolation With Patient-Level Data 2011. www.nicedsu.org.uk/NICE%20DSU%20TSD%20Survival%20analysis.updated%20March%202013.pdf (accessed July 2013).
- Morden JP, Lambert PC, Latimer N, Abrams KR, Wailoo AJ. Assessing methods for dealing with treatment switching in randomised controlled trials: a simulation study. BMC Med Res Methodol 2011;11. http://dx.doi.org/10.1186/1471-2288-11-4.
- Branson M, Whitehead J. Estimating a treatment effect in survival studies in which patients switch treatment. Stat Med 2002;21:2449-63. http://dx.doi.org/10.1002/sim.1219.
- Leung PP, Tannock IF, Oza AM, Puodziunas A, Dranitsaris G. Cost-utility analysis of chemotherapy using paclitaxel, docetaxel, or vinorelbine for patients with anthracycline-resistant breast cancer. J Clin Oncol 1999;17:3082-90.
- Grann VR, Panageas KS, Whang W, Antman KH, Neugut AI. Decision analysis of prophylactic mastectomy and oophorectomy in BRCA1-positive or BRCA2-positive patients. J Clin Oncol 1998;16:979-85.
- Grann VR, Jacobson JS, Sundararajan V, Albert SM, Troxel AB, Neugut AI. The quality of life associated with prophylactic treatments for women with BRCA1/2 mutations. Cancer J Sci Am 1999;5:283-92.
- Havrilesky LJ, Broadwater G, Davis DM, Nolte KC, Barnett JC, Myers ER, et al. Determination of quality of life-related utilities for health states relevant to ovarian cancer diagnosis and treatment. Gynecol Oncol 2009;113:216-20. http://dx.doi.org/10.1016/j.ygyno.2008.12.026.
- Hess LM, Brady WE, Havrilesky LJ, Cohn DE, Monk BJ, Wenzel L, et al. Comparison of methods to estimate health state utilities for ovarian cancer using quality of life data: a Gynecologic Oncology Group study. Gynecol Oncol 2013;128:175-80. http://dx.doi.org/10.1016/j.ygyno.2012.10.024.
- Bradford AS. Preferences for Treatment-Related Sexual Dysfunction in Ovarian Cancer Patients and Physicians n.d.
- Cheung YB, Thumboo J, Gao F, Ng GY, Pang G, Koo WH, et al. Mapping the English and Chinese versions of the Functional Assessment of Cancer Therapy-General to the EQ-5D utility index. Value Health 2009;12:371-6. http://dx.doi.org/10.1111/j.1524-4733.2008.00448.x.
- Dobrez D, Cella D, Pickard AS, Lai JS, Nickolov A. Estimation of patient preference-based utility weights from the functional assessment of cancer therapy: general. Value Health 2007;10:266-72. http://dx.doi.org/10.1111/j.1524-4733.2007.00181.x.
- Edwards SJ, Barton S, Thurgar E, Nherera L, Hamilton V, Karner C, et al. Bevacizumab for the Treatment of Recurrent Advanced Ovarian Cancer: A Single Technology Appraisal 2012. http://guidance.nice.org.uk/TA/Wave28/1/Consultation/EvaluationReport/ERGReport/pdf/English (accessed July 2013).
- Havrilesky LJ, Garfield CF, Barnett JC, Cohn DE. Economic impact of paclitaxel shortage in patients with newly diagnosed ovarian cancer. Gynecol Oncol 2012;125:631-4. http://dx.doi.org/10.1016/j.ygyno.2012.03.028.
- Pickard S, Ray S, Ganguli A. Preference scores for 6 types of cancer using fact and EQ-5D. Value Health 2012;15. http://dx.doi.org/10.1016/j.jval.2012.03.1214.
- Grann VR, Patel PR, Jacobson JS, Warner E, Heitjan DF, Ashby-Thompson M, et al. Comparative effectiveness of screening and prevention strategies among BRCA1/2-affected mutation carriers. Breast Cancer Res Treat 2011;125:837-47. http://dx.doi.org/10.1007/s10549-010-1043-4.
- Grann VR, Patel PR, Jacobson JS, Warner E, Heitjan DF, Ashby-Thompson M, et al. Comparative Effectiveness of Screening, Surgery, and Chemo Prevention Among BRCA1 2 Mutation Carriers n.d.
- Greving JP, Vernooij F, Heintz AP, van der GY, Buskens E, Greving JP, et al. Is centralization of ovarian cancer care warranted? A cost-effectiveness analysis. Gynecol Oncol 2009;113:68-74. http://dx.doi.org/10.1016/j.ygyno.2008.12.008.
- Gordon LG, Scuffham PA, Beesley VL, Green AC, DeFazio A, Wyld DK, et al. Medical costs and outcomes for Australian women with ovarian cancer: a patient-level analysis over 2.5 years. Int J Gynecol Cancer 2010;20:757-65. http://dx.doi.org/10.1111/IGC.0b013e3181dbd13f.
- Hess LM, Malone DC, Skrepnek GH, Reed PG, Armstrong EP, Weihs K. Preferences of patients and oncologists for advanced ovarian cancer treatment-related health states. Health Outcomes Res Med 2010;1:e51-9. http://dx.doi.org/10.1016/j.ehrm.2010.02.001.
- Sun CC, Bodurka DC, Donato ML, Rubenstein EB, Borden CL, Basen-Engquist K, et al. Patient preferences regarding side effects of chemotherapy for ovarian cancer: do they change over time?. Gynecol Oncol 2002;87:118-28. http://dx.doi.org/10.1006/gyno.2002.6807.
- Stein K, Sugar C, Velikova G, Stark D, Stein K, Sugar C, et al. Putting the ‘Q’ in quality adjusted life years (QALYs) for advanced ovarian cancer: an approach using data clustering methods and the internet. Eur J Cancer 2007;43:104-13. http://dx.doi.org/10.1016/j.ejca.2006.09.007.
- Calhoun EA, Fishman DA, Lurain JR, Welshman EE, Bennett CL, Calhoun EA, et al. A comparison of ovarian cancer treatments: analysis of utility assessments of ovarian cancer patients, at-risk population, general population, and physicians. Gynecol Oncol 2004;93:164-9. http://dx.doi.org/10.1016/j.ygyno.2004.01.017.
- Medicines Complete . Creatinine Clearance 2013. www.medicinescomplete.com/mc/bnf/current/PHP18586-creatinine-clearance.htm (accessed July 2013).
- Du Bois D, Du Bois EF. A formula to estimate the approximate surface area if height and weight be known. Nutrition 1989;5:303-11.
- Health and Social Care Information Centre (HSCIC) . Health Survey for England 2011, Trend Tables 2012. www.hscic.gov.uk/catalogue/PUB09302 (accessed July 2013).
- Briggs A, Sculpher M, Claxton K. Decision Modelling for Health Economic Evaluation. Oxford: Oxford University Press; 2007.
- Dias S, Sutton AJ, Welton NJ, Ades AE. NICE DSU Technical Support Document 6: Embedding Evidence Synthesis in Probabilistic Cost-Effectiveness Analysis: Software Choices. Report by the Decision Support Unit 2011. www.nicedsu.org.uk/TSD6%20Software.final.08.05.12.pdf (accessed July 2013).
- National Institute of Health and Clinical excellence . Appraising Life-Extending, End of Life Treatments 2009. www.nice.org.uk/guidance/gid-tag387/resources/appraising-life-extending-end-of-life-treatments-paper2.
- Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels. Boston, MA: Little, Brown & Co; 1994.
- Cancer Therapy Evaluation Program 1 Revised March 23, 1998, Common Toxicity Criteria, Version 2.0 n.d.
- 2006 update of ASCO practice guideline recommendations for the use of white blood cell growth factors: guideline summary. J Oncol Pract 2006;2:196-201. http://jop.ascopubs.org/content/2/4/196.full.
- Calvert AH, Newell DR, Gumbrell LA, O’Reilly S, Burnell M, Boxall FE, et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol 1989;7:1748-56.
- Cockcroft DW, Gault MH. Cockcroft formula: [rediction of creatinine clearance from serum creatinine]. Nephron 1976;16:31-4.
- National Institute for Health and Care Excellence (NICE) . Ovarian Cancer (Advanced) – Paclitaxel, Pegylated Liposomal Doxorubicin Hydrochloride and Topotecan 2002. www.nice.org.uk/guidance/ta45/resources/guidance-ovarian-cancer-advanced-paclitaxel-pegylated-liposomal-doxorubicin-hydrochloride-and-topotecan-review-pdf (accessed July 2013).
- Barnett J, Alvarez-Secord A, Cohn D, Leath C, Peterson B, Myers E, et al. Cost-effectiveness of a predictive biomarker for bevacizumab responsiveness in the primary treatment of ovarian cancer. Gynecol Oncol 2012;125. http://dx.doi.org/10.1016/j.ygyno.2011.12.157.
- Chan JH. A cost effective strategy of bevacizumab in treatment of primary ovarian cancer – a subset analysis of ICON 7 trial. Gynecol Oncol 2012;125:S15-16. http://dx.doi.org/10.1016/j.ygyno.2011.12.037.
- Dalton HJ, Yu X, Hu L, Kapp DS, Benjamin I, Monk BJ, et al. An economic analysis of dose dense weekly paclitaxel plus carboplatin versus every-3-week paclitaxel plus carboplatin in the treatment of advanced ovarian cancer. Gynecol Oncol 2012;124:199-204. http://dx.doi.org/10.1016/j.ygyno.2011.09.028.
- Geisler JL. Chemotherapeutic regimens for early high risk ovarian cancer. Gynecol Oncol 2012;125. http://dx.doi.org/10.1016/j.ygyno.2011.12.216.
- Havrilesky LG. Economic Impact of Paclitaxel Shortage in Patients With Newly Diagnosed Ovarian Cancer n.d.
- Lechuga DA. Economic evaluation of bevacizumab for the treatment of advanced ovarian cancer in Mexico. Value Health 2012;15.
- Neymark N, Gorlia T, Adriaenssen I, Baron B, Piccart M. Cost effectiveness of paclitaxel/cisplatin compared with cyclophosphamide/cisplatin in the treatment of advanced ovarian cancer in Belgium. Pharmacoeconomics 2002;20:485-97. http://dx.doi.org/10.2165/00019053-200220070-00006.
- Cohn DE, Kim KH, Resnick KE, O’Malley DM, Straughn JM. At what cost does a potential survival advantage of bevacizumab make sense for the primary treatment of ovarian cancer? A cost-effectiveness analysis. J Clin Oncol 2011;29:1247-51. http://dx.doi.org/10.1200/JCO.2010.32.1075.
- Dalton H, Yu X. An economic analysis of intravenous carboplatin plus dose-dense weekly paclitaxel versus intravenous carboplatin plus every three-weeks paclitaxel in the upfront treatment of ovarian cancer. Gynecol Oncol 2011;120:S2-S133.
- Fuh KC. Is It More Cost-Effective to Use Bevacizumab in the Primary Treatment Setting or at Recurrence? An Economic Analysis n.d.
- Krysinski JP. Treatment of advanced ovarian cancer: cost-effectiveness analysis. Curr Gynecol Oncol 2011;9:147-57.
- Cohn DK. At what cost does a potential survival advantage of bevacizumab make sense for the primary treatment of ovarian cancer? A cost-effectiveness analysis. Gynecol Oncol 2010;116:S12-13.
- Havrilesky LJ, Secord AA, Darcy KM, Armstrong DK, Kulasingam S. Gynecologic Oncology Group . Cost effectiveness of intraperitoneal compared with intravenous chemotherapy for women with optimally resected stage III ovarian cancer: a gynecologic oncology group study. J Clin Oncol 2008;26:4144-50. http://dx.doi.org/10.1200/JCO.2007.13.1961.
- Bristow RE, Santillan A, Salani R, Diaz-Montes TP, Giuntoli RL, Meisner BC, et al. Intraperitoneal cisplatin and paclitaxel versus intravenous carboplatin and paclitaxel chemotherapy for Stage III ovarian cancer: a cost-effectiveness analysis. Gynecol Oncol 2007;106:476-81. http://dx.doi.org/10.1016/j.ygyno.2007.05.043.
- Fedders M, Hartmann M, Schneider A, Kath R, Camara O, Oelschlager H, et al. Markov-modeling for the administration of platinum analogues and paclitaxel as first-line chemotherapy as well as topotecan and liposomal doxorubicin as second-line chemotherapy with epithelial ovarian carcinoma. J Cancer Res Clinical Oncol 2007;133:619-25. http://dx.doi.org/10.1007/s00432-007-0313-y.
- Dranitsaris G, Elia-Pacitti J, Cottrell W, Dranitsaris G, Elia-Pacitti J, Cottrell W. Measuring treatment preferences and willingness to pay for docetaxel in advanced ovarian cancer. Pharmacoeconomics 2004;22:375-87. http://dx.doi.org/10.2165/00019053-200422060-00004.
- Limat S, Woronoff-Lemsi MC, Menat C, Madroszyk-Flandin A, Merrouche Y. From randomised clinical trials to clinical practice: a pragmatic cost-effectiveness analysis of paclitaxel in first-line therapy for advanced ovarian cancer. Pharmacoeconomics 2004;22:633-41. http://dx.doi.org/10.2165/00019053-200422100-00002.
- Bennett CL, Golub RM, Calhoun EA, Weinstein J, Fishman D, Lurain J, et al. Cost-utility assessment of amifostine as first-line therapy for ovarian cancer. Int J Gynecol Cancer 1998;8:64-72.
- Berger K, Fischer T, Szucs TD. Cost-effectiveness analysis of paclitaxel and cisplatin versus cyclophosphamide and cisplatin as first-line therapy in advanced ovarian cancer: a European perspective. Eur J Cancer 1998;34:1894-901. http://dx.doi.org/10.1016/S0959-8049(98)00260-3.
- Messori A, Trippoli S, Becagli P, Tendi E. Treatments for newly diagnosed advanced ovarian cancer: analysis of survival data and cost-effectiveness evaluation. Anticancer Drugs 1998;9:491-502.
- Elit LM, Gafni A, Levine MN. Economic and policy implications of adopting paclitaxel as first-line therapy for advanced ovarian cancer: an Ontario perspective. J Clin Oncol 1997;15:632-9.
- McGuire W, Neugut A, Arikian S, Doyle JL, Dezii CM. Analysis of the cost-effectiveness of paclitaxel as alternative combination therapy for advanced ovarian cancer. J Clin Oncol 1997;15:640-5.
- Papaioannou D, Rafia R, Stevens JW, Stevenson M, Evans P. Trabectedin for the Treatment of Relapsed Ovarian Cancer: A Single Technology Appraisal. Sheffield: ScHARR, University of Sheffield; 2010.
Appendix 1 Literature search strategies
Clinical searches
Database searched: Ovid MEDLINE® In-Process & Other Non-Indexed Citations and Ovid MEDLINE
Date range searched: 1946 to present.
Date searched: initially searched 18 January 2013 and updated 23 May 2013.
| # | Term |
|---|---|
| 1 | exp ovarian neoplasms/ |
| 2 | (ovar$ adj4 (cancer$ or tumo?r$ or malignan$ or oncolog$ or carcinoma$ or neoplas$ or mass$ or growth$ or cyst$)).mp. |
| 3 | (adenexa$ adj4 mass$).mp. |
| 4 | 1 or 2 or 3 |
| 5 | exp Topotecan/ |
| 6 | topotecan.mp. |
| 7 | (hycam$ or potactasol).mp. |
| 8 | exp Doxorubicin/ |
| 9 | (doxorubicin hydrochloride or doxorubicin hcl).mp. |
| 10 | liposomal doxorubicin.mp. |
| 11 | liposome encapsulated doxorubicin.mp. |
| 12 | doxil.mp. |
| 13 | caelyx.mp. |
| 14 | exp Paclitaxel/ |
| 15 | paclitaxel.mp. |
| 16 | taxol.mp. |
| 17 | trabectedin.mp. |
| 18 | yondelis.mp. |
| 19 | gemcitabine.mp. |
| 20 | gemzar.mp. |
| 21 | or/5–20 |
| 22 | 4 and 21 |
| 23 | Randomized Controlled Trials as Topic |
| 24 | randomized controlled trial |
| 25 | Random Allocation |
| 26 | Double Blind Method/ |
| 27 | Single Blind Method/ |
| 28 | clinical trial/ |
| 29 | clinical trial, phase i.pt. |
| 30 | clinical trial, phase ii.pt. |
| 31 | clinical trial, phase iii.pt. |
| 32 | clinical trial, phase iv.pt. |
| 33 | controlled clinical trial.pt. |
| 34 | randomized controlled trial.pt. |
| 35 | multicenter study.pt. |
| 36 | clinical trial.pt. |
| 37 | exp Clinical Trials as topic/ |
| 38 | (clinical adj trial$).tw. |
| 39 | ((singl$ or doubl$ or treb$ or tripl$) adj (blind$3 or mask$3)).tw. |
| 40 | PLACEBOS/ |
| 41 | placebo$.tw. |
| 42 | randomly allocated.tw. |
| 43 | (allocated adj2 random$).tw. |
| 44 | or/23–43 |
| 45 | case report.tw. |
| 46 | letter/ (795094) |
| 47 | historical article |
| 48 | 45 or 46 or 47 |
| 49 | 44 not 48 |
| 50 | 22 and 49 |
Database: Ovid EMBASE
Date range searched: inception to present.
Date searched: 18 January 2013 and updated 23 May 2013.
| # | Term |
|---|---|
| 1 | exp ovary cancer |
| 2 | (ovar$ adj4 (cancer$ or tumo?r$ or malignan$ or oncolog$ or carcinoma$ or neoplas$ or mass$ or growth$ or cyst$)).mp. |
| 3 | (adenexa$ adj4 mass$).mp. |
| 4 | 1 or 2 or 3 |
| 5 | exp topotecan/ |
| 6 | topotecan.mp. |
| 7 | (hycam$ or potactasol).mp. |
| 8 | exp doxorubicin/ |
| 9 | (doxorubicin hydrochloride or doxorubicin hcl).mp. |
| 10 | liposomal doxorubicin.mp. |
| 11 | liposome encapsulated doxorubicin.mp. |
| 12 | doxil.mp. |
| 13 | caelyx.mp. |
| 14 | exp paclitaxel/ |
| 15 | paclitaxel.mp. |
| 16 | taxol.mp. |
| 17 | exp trabectedin/ |
| 18 | trabectedin.mp. |
| 19 | yondelis.mp. |
| 20 | exp gemcitabine/ |
| 21 | gemcitabine.mp. |
| 22 | gemzar.mp. |
| 23 | or/5–22 |
| 24 | 4 and 23 |
| 25 | Clinical trial/ |
| 26 | Randomized controlled trial/ |
| 27 | Randomization/ |
| 28 | Single blind procedure/ |
| 29 | Double blind procedure/ |
| 30 | Crossover procedure/ |
| 31 | Placebo/ |
| 32 | Randomi?ed controlled trial$.tw. |
| 33 | Rct.tw. |
| 34 | Random allocation.tw. |
| 35 | Randomly allocated.tw. |
| 36 | Allocated randomly.tw. |
| 37 | (allocated adj2 random).tw. |
| 38 | Single blind$.tw. |
| 39 | Double blind$.tw. |
| 40 | ((treble or triple) adj blind$).tw. |
| 41 | Placebo$.tw. |
| 42 | Prospective study/ |
| 43 | or/25–42 |
| 44 | Case study/ |
| 45 | Case report.tw. |
| 46 | Abstract report/ or letter/ |
| 47 | 44 or 45 or 46 |
| 48 | 43 not 47 |
| 49 | 24 and 48 |
Database searched: Cochrane Central Register of Controlled Trials
Date searched: initially searched from inception 18 January 2013 and updated 23 May 2013.
| # | Term |
|---|---|
| 1 | OVARIAN NEOPLASMS explode all trees (MeSH) |
| 2 | (ovar* near cancer*) |
| 3 | (ovar* near tumor*) |
| 4 | (ovar* near tumour*) |
| 5 | (ovar* near malignan*) |
| 6 | (ovar* near oncolog*) |
| 7 | (ovar* near carcinoma) |
| 8 | (ovar* near neoplas*) |
| 9 | (ovar* near mass*) |
| 10 | (ovar* near growth*) |
| 11 | (ovar* near cyst*) |
| 12 | (adenexa* near mass*) |
| 13 | (#1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12) |
| 14 | TOPOTECAN explode all trees (MeSH) |
| 15 | (topotecan or hycamtin or hycamptamine or potactasol) |
| 16 | (#14 or #15) |
| 17 | DOXORUBICIN explode all trees (MeSH) |
| 18 | (doxil or (doxorubicin next hydrochloride) or (doxorubicin next hcl)) |
| 19 | (liposomal next doxorubicin) |
| 20 | (caelyx or adriamycin or rubex) |
| 21 | (liposome next encapsulated next doxorubicin) |
| 22 | (#17 or #18 or #19 or #20 or #21) |
| 23 | PACLITAXEL explode all trees (MeSH) |
| 24 | (paclitaxel or taxol or taxotere or abraxane) |
| 25 | (#23 or #24) |
| 26 | (trabectedin or yondelis or ecteinascidin or ET-743 or ecteinascidin 743) |
| 27 | (gemcitabine or gemzar) |
| 28 | (#16 or #22 or #25 or #26 or #27) |
| 29 | (#13 and #28) |
Database searched: European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (ENCePP®)
URL: www.encepp.eu/
Date searched: initially searched from inception 18 January 2013 and updated 23 May 2013.
| Field | Term |
|---|---|
| Substance: | Topotecan or paclitaxel or pegylated liposomal doxorubicin hydrochloride, trabectedin or gemcitabine |
| Medical condition: | Ovarian cancer |
| Status of study: | Planned; ongoing; finalised |
| No limits placed on: | Study type |
| Coordinating entity of study | |
| Research network | |
| Population age | |
| Scope of study |
Appendix 2 Data abstraction
Data abstraction of clinically relevant details from included studies
Alberts et al.28
| Item | Details | |
|---|---|---|
| Study | Alberts et al.28 | |
| Location | USA (no. of institutions not reported) | |
| Trial sponsor | Grant awarded by the NCI and supported in part by Ortho Biotech | |
| Patient enrolment | Between August 2002 and December 2004 | |
| Trial design | Phase II (initially designed as Phase III but deemed to be Phase II due to low patient accrual), RCT with an active control Level of masking is unclear |
|
| Line of therapy | Second line (all) | |
| Inclusion criteria |
|
|
| Exclusion criteria | None | |
| Outcomes reported | OS, PFS, tumour response and toxicity | |
| Subgroups | None | |
| Stratification | Disease measurability, no. of disease sites and serous histology | |
| Measure of disease response or progression | Objective response and disease progression were defined according to standard RECIST criteria.69 GCIG CA125 progression criteria70 were also implemented in defining disease progression | |
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | All platinum sensitive (PFI 6–24 months) | |
| Other definitions | OS, PFS and confirmed response rate not defined | |
| Treatment | PLDH plus carboplatin | Carboplatin alone |
| Randomised, n | 31 | 30 |
| Withdrawals, n (%) | NR | NR |
| Treatment | Intravenous infusion: PLDH 30 mg/m2 as a 1-hour i.v. infusion plus carboplatin (AUC 5 mg/ml/minute) administered over a minimum of 15 minutes every 4 weeks | Carboplatin alone (AUC 5 mg/ml/minute) administered over a minimum of 15 minutes every 4 weeks |
| Treatment duration | Median number of cycles: 7 (range 1–18) | Median number of cycles: 6 (range 2–16) |
| Treatment discontinuation | Treatment was given until progression, intolerable toxicity or physician/patient desire for removal from study. The maximum cumulative dose allowed for PLDH was 600 mg/m2. Any patient with a compromised LVEF (< 45% or decreases by a relative 20% from baseline) was removed from PLDH and continued on the carboplatin treatment. For PPE or stomatitis and bilirubin toxicity, a dose-reduction schedule was created, based on grade and previous history in order to minimise this side effect. For all other grade 3 and grade 4 events, PLDH was withheld for up to 4 weeks until the toxicity resolved to ≤ grade 2, after which treatment resumed at a one-level dose reduction (level 1 = 25 mg/m2, level 2 = 20 mg/m2). If treatment was delayed for >4 weeks PLDH was permanently discontinued. Patients with persistently ≥ grade 2 peripheral neuropathy, despite dose reduction, were permanently taken off carboplatin treatment | Treatment was given until progression, intolerable toxicity or physician/patient desire for removal from study. Patients with persistently ≥ grade 2 peripheral neuropathy, despite dose reduction, were permanently taken off carboplatin treatment |
| Concomitant medications | Prophylactic use of G-CSF or GM-CSF was not allowed but was allowed to treat neutropenia according to ASCO guidelines168 | |
| Duration of follow-up | Median 22.4 months | |
| Baseline patient characteristics | ||
| Age, years (range) | Median 66.9 (range 43–87) | Median 62.5 (range 31–80) |
| Previous treatment | NR | NR |
| Duration of PFI | Median 430 (range 253–774) days Proportion with PFI > 365 days: 57% |
Median 382 (range 192–790) days Proportion with PFI > 365 days: 57% |
| Prior chemotherapy, n (%) | ||
| One regimen | 31 (100) | 30 (100) |
| Primary site of disease | NR | NR |
| No. of sites of lesions, n (%) | ||
| ≤ 2 | 24 (77) | 22 (73) |
| ≥ 3 | 7 (23) | 8 (27) |
| Histological type, n (%) | ||
| Serous | 25 (81) | 25 (83) |
| Non-serous (not broken down further) | 6 (19) | 5 (17) |
| Histological grade | NR | NR |
| Tumour size, cm | NR | NR |
| Disease measurability, n (%) | ||
| Measurable disease | 19 (61) | 20 (67) |
| Elevated CA125 level | 4 (13) | 2 (7) |
| Other non-measurable disease | 8 (26) | 8 (27) |
| FIGO stage at diagnosis | NR | NR |
| Performance status | Zubrod performance status at study entry | |
| 0 | 20 (65%) | 16 (53%) |
| 1 | 11 (35%) | 14 (47%) |
| Comments | Study closed early because of slow patient accrual | |
Bafaloukos et al.29
| Item | Details | |
|---|---|---|
| Study | Bafaloukos et al.29 | |
| Location | Greece; number of institutions not reported | |
| Trial sponsor | NR | |
| Patient enrolment | Between October 1999 and December 2005 | |
| Trial design | Phase II RCT with an active control Level of masking unclear |
|
| Line of therapy | Predominantly second line | |
| Inclusion criteria |
|
|
| Exclusion criteria |
|
|
| Outcomes reported | Primary end points: RR and toxicity of the two treatment regimens Secondary end points: TTP and OS |
|
| Subgroups | None | |
| Stratification | No stratification criteria applied at randomisation | |
| Measure of disease response or progression | WHO criteria for those with measurable disease and CA125 level according to Rustin’s criteria80 for those without measurable disease | |
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | Platinum sensitive: patients with ovarian cancer relapsing ≥ 6 months after first-line platinum-based therapy | |
| Other definitions | OS was estimated from the initiation of treatment to the date of last follow-up or until the patient’s death TTP was calculated from the initiation of treatment to the first disease progression |
|
| Treatment | PLDH plus carboplatin | Paclitaxel plus carboplatin |
| Randomised, n | 93 | 96 |
| Withdrawals, n (%) | 20 (21.5) | 24 (25) |
| Treatment | Intravenous infusion: PLDH 45 mg/m2 as a 90-minute i.v. infusion followed by carboplatin AUC 5 | Intravenous infusion: Paclitaxel 175 mg/m2 as a 3-hour i.v. infusion followed by carboplatin at an AUC 5 on day 1 |
| Treatment duration | Median number of cycles: 6 (range 1–8) | Median number of cycles: 6 (range 1–9) |
| Median length per cycle: 28 days | Median length per cycle: 21 days | |
| Treatment discontinuation | Maximum of 2 weeks’ delay was allowed for toxicity and treatment was discontinued if longer toxicity-related delays occurred. In cases of prolonged neutropenia (> 7 days with ANC of < 0.5 × 109/l) despite G-CSF use or febrile neutropenia, a 25% dose reduction for all drugs was applied additionally to G-CSF. For grades 3 and 4 thrombocytopenia, a 25% and a 50% dose reduction, respectively, was recommended for all drugs. If creatinine clearance was calculated as < 30 ml/minute, treatment was delayed for a maximum of 2 weeks until recovery; otherwise the patient was withdrawn from the study. For cardiac arrhythmia, grade 3 hypersensitivity reactions and any non-haematological toxicity grade > 2, treatment was discontinued. Specifically, for grade 2 PPE, treatment was delayed for a maximum of 2 weeks until recovery to grade 0 or 1 | |
| Concomitant medications | All patients received standard premedication of dexamethasone, diphenhydramine and ranitidine prior to PLDH infusion | All patients received standard premedication of dexamethasone, diphenhydramine and ranitidine prior to paclitaxel, orally 12 hours prior to and again intravenously 30-minutes prior to paclitaxel infusion |
| Duration of follow-up | Median 43.6 (range 0.1–74.8) months | Median 43.6 (range 0.1–74.8) months |
| Baseline patient characteristics | ||
| Age, years (range) | Median 62 (range 38–89) | Median 63 (range 37–81) |
| Previous treatment | Surgery: 76 (82%) | Surgery: 85 (89%) |
| Taxane-containing therapy: 86/93 (92%) | Taxane-containing therapy: 84/96 (88%) | |
| PFI from last therapy | ||
| Median | 17.3 (range 6–119) months | 14.8 (range 6–96) months |
| 6–12 months | 22 (23%) | 32 (33%) |
| 12.1–24 months | 38 (41%) | 32 (33%) |
| > 24 months | 29 (31% | 23 (24%) |
| Unknown | 4 (4%) | 9 (9%) |
| Previous chemotherapy, n (%): | ||
| One regimen | 89/93 (96) | 92/96 (96) |
| Two or more regimens | 4/93 (4) | 4/96 (4) |
| Primary site of disease | Not broken down by affected site | |
| No. of sites of lesions, n (%) | NR | NR |
| Histological type, n (%): | ||
| Serous | 72 (77) | 71 (74) |
| Mucinous | 3 (3) | 0 (0) |
| Endometrioid | 7 (8) | 6 (6) |
| Clear cell | 3 (3) | 3 (3) |
| Other | 5 (5) | 9 (9) |
| Unknown | 3 (3) | 7 (7) |
| Histological grade, n (%): | ||
| I | 5 (5) | 8 (8) |
| II | 30 (32) | 27 (28) |
| III | 44 (47) | 48 (50) |
| IV | 2 (2) | 1 (1) |
| Unknown | 12 (13) | 12 (13) |
| Tumour size, cm | NR | NR |
| Disease measurability, n (%): | ||
| Elevated CA125 level only | 9 (10) | 7 (7) |
| FIGO stage at diagnosis, n (%) | ||
| I | 5 (5) | 9 (9) |
| II | 7 (8) | 9 (9) |
| III | 62 (67) | 56 (58) |
| IV | 13 (14) | 15 (16) |
| Unknown | 6 (7) | 7 (7) |
| Performance status, ECOG score, n (%): | ||
| 0 | 55 (59) | 62 (65) |
| 1 | 30 (32) | 27 (28) |
| 2 | 1 (1) | 0 (0) |
| Unknown | 7 (8) | 7 (7) |
| Comments | None | |
Gonzalez-Martin et al.48
| Item | Details | |
|---|---|---|
| Study | Gonzalez-Martin et al.48 | |
| Location | Spain; no. of institutions not reported | |
| Trial sponsor | Not specified | |
| Patient enrolment | Between May 2000 and December 2002 | |
| Trial design | Phase II RCT, ‘pick the winner’ design (no formal statistical analysis between the treatment arms was planned) | |
| Line of therapy | Second and third | |
| Inclusion criteria |
|
|
| Exclusion criteria | No additional criteria listed | |
| Outcomes reported | Response rate, OS, TTP, tolerability and QoL | |
| Subgroups | None specified | |
| Stratification | Stratification by PFI (6–12 months vs. > 12months) and no. of previous lines of therapy (one vs. two) | |
| Measure of disease response or progression | WHO criteria for those with measurable disease and CA125 level according to Rustin’s criteria for those without measurable disease | |
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | Platinum sensitive | |
| Other definitions | OS: date of randomisation to death TTP: date of randomisation to date of documentation of tumour progression |
|
| Treatment | Paclitaxel plus carboplatin | Carboplatin |
| Randomised, n | 41 | 40 |
| Withdrawals, n (%) | NR | NR |
| Treatment | Paclitaxel 175 mg/m2 over 3 hours plus carboplatin (AUC 5) every 3 weeks for a minimum of six cycles unless there was progression, unacceptable toxicity or patient refusal | Carboplatin AUC 5 every 3 weeks for a minimum of six cycles unless there was progression, unacceptable toxicity or patient refusal |
| In both groups, therapy was continued after six cycles if, in the opinion of the attending physician, further clinical benefit could be expected | ||
| Treatment duration | Median number of cycles 6 (range 1–8) | Median number of cycles 6 (range 2–9) |
| Treatment discontinuation | Progression, unacceptable toxicity or patient refusal | Progression, unacceptable toxicity or patient refusal |
| Concomitant medications | Premedication of dexamethasone, diphenhydramine and ranitidine approximately 30 minutes before infusion of paclitaxel | None |
| Duration of follow-up | Median 67.7 weeks | |
| Baseline patient characteristics | ||
| Age, years (range) | Median 59 (range 40–77) | Median 61 (range 35–77) |
| Previous treatment | Previous paclitaxel: | Previous paclitaxel: |
| In any regimen: 35/38 (92.1%) | In any regimen: 33/40 (82.5%) | |
| In last regimen: 32/38 (84.2%) | In last regimen: 33/40 (82.5%) | |
| TFI, months: | ||
| Median (range) | 13.5 (7–147) | 14 (6–60) |
| 6–12 months | 17 (45%) | 16 (40%) |
| > 12 months | 21 (55%) | 24 (60%) |
| Previous chemotherapy, n (%): | ||
| One regimen | 31 (81.6) | 35 (87.5) |
| Two regimens | 7 (18.4) | 5 (12.5) |
| Primary site of disease | NR | NR |
| No. of involved sites, n (%): | ||
| 1–2 | 25 (65.8) | 33 (82.5) |
| > 2 | 13 (34.2) | 7 (17.5) |
| Histological type, n (%): | ||
| Serous | 29 (76.3) | 27 (67.5) |
| Mucinous | 2 (5.3) | – |
| Endometrioid | 2 (5.3) | 2 (5.0) |
| Clear cell | 2 (5.3) | 5 (12.5) |
| Undifferentiated | 1 (2.6) | 5 (12.5) |
| Other | 2 (5.3) | 1 (2.5) |
| Histological grade: | ||
| Poorly differentiated grade | 16 (48.5%) | 20 (54.1%) |
| Tumour size, cm | > 5 cm: 8 (21.1%) | > 5 cm: 5 (12.5%) |
| Disease measurability: | ||
| WHO criteria | 27 (71%) | 25 (62.5%) |
| CA125 criteria | 11 (28.9%) | 15 (37.5%) |
| FIGO stage at diagnosis | NR | NR |
| Performance status, ECOG: | ||
| 0 | 17 (47.2%) | 14 (35.9%) |
| 1 | 17 (47.2%) | 18 (46.2%) |
| 2 | 2 (5.6%) | 7 (17.9%) |
| NR | 2 | 1 |
| Comments | None | |
Gordon et al.49,54
| Item | Details | |
|---|---|---|
| Study | Gordon et al.49,54 | |
| Location | USA, Canada and Europe (104 sites) | |
| Trial sponsor | Alza Corporation, Mountain View, CA, USA; Johnson & Johnson Pharmaceutical, Raritan, NJ, USA; and Tibotec Therapeutics, a division of Biotech Products | |
| Patient enrolment | Between May 1997 and March 1999 | |
| Trial design | Multicentre | |
| Line of therapy | Second line | |
| Inclusion criteria |
|
|
| Exclusion criteria |
|
|
| Outcomes reported | OS, PFS and ORR | |
| Subgroups | None specified | |
| Stratification | Stratified by platinum sensitivity (platinum refractory vs. platinum sensitive); presence or absence of bulky disease (tumour mass > 5 cm) | |
| Measure of disease response or progression | CR: complete disappearance of all measurable and assessable disease, no new lesions and no disease-related symptoms PR: ≥ 50% decrease in the sum of the products of bidimensional perpendicular diameters of all measurable lesions progression of assessable disease and new lesions were not allowed PD: ≥ 50% increase in the sum of the products of bidimensionally measured lesions over the smallest sum obtained at best response or reappearance of any lesion that had disappeared, or clear worsening of any assessable disease, or failure to return for evaluation because of death or deteriorating condition, or the appearance of any new lesion or site SD: If the patient did not qualify for CR, PR or PD Objective tumour assessments CR and PR were confirmed by radiological assessment at least 4 weeks later |
|
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | Platinum refractory: progressed during initial platinum-based chemotherapy, demonstrated SD, or relapsed within 6 months after completing platinum-based chemotherapy Platinum sensitive: PFS > 6 months after first-line platinum therapy |
|
| Other definitions | Measurable disease was defined as bidimensionally measurable lesion(s) with clearly defined margins by plain radiograph, with at least one lesion of diameter of ≥ 0.5 cm (excluding bone lesions), or CT, MRI or another imaging scan with both diameters greater than the distance between cuts of imaging study or palpation with both diameters ≥ 2 cm Assessable disease included unidimensionally measurable lesion(s), mass(es) with margins not clearly defined, lesion(s) with both diameters of ≤ 2 cm, and malignant ascites or pleural effusion in conjunction with serum CA125 levels > 100 U/ml in absence of cirrhosis |
|
| Treatment | PLDH | Topotecan |
| Randomised, n | 239 | 235 |
| Withdrawals, n (%) | Seven patients did not receive study drug but number not given by arm | |
| Treatment | PLDH 50 mg/m2 via 1-hour infusion every 28 days | Topotecan administered at 1.5 mg/m2/day as 30-minute infusion daily for five consecutive days every 21 days, beginning on day 1 of a 21-day cycle |
| Treatment duration | Median number of cycles: 6 Median cycle length: 30 (range 27–56) days |
Median number of cycles: 8 Median cycle length: 24 (range 20–38) days |
| Treatment discontinuation | Treatment was temporarily suspended or discontinued if a person had disease progression, developed serious or intolerable AEs precluding further treatment, was unable to tolerate study drug despite dose modification, had LVEF of < 45% or 20% decrease from baseline, or decided to withdraw participation. Patients requiring radiation were removed from treatment | Treatment was temporarily suspended or discontinued if a person had disease progression, developed serious or intolerable AEs precluding further treatment, was unable to tolerate study drug despite dose modification, had LVEF of < 45% or 20% decrease from baseline, or decided to withdraw participation. Patients requiring radiation were removed from treatment |
| Concomitant medications | Prophylactic cytokine administration was not recommended during the first cycle of either study drug. However, growth factor support was allowed in subsequent cycles for any patient with grade 4 neutropenia lasting > 7 days or failure of absolute neutrophil count to recover within 22 days. All patients who developed febrile neutropenia were also eligible for prophylactic growth factor administration in the next cycles | |
| Duration of follow-up | NR | NR |
| Baseline patient characteristics | ||
| Age, years (range) | Median 60 (range 27–87) | Median 60 (range 25–85) |
| Previous treatment: | ||
| Prior platinum and taxane | 74% | 72% |
| TFI: | ||
| Median, months | 7.0 (range 0.9–82.1) | 6.7 (range 0.5–109.6) |
| Platinum sensitive | 109 (45.6%) | 110 (46.8%) |
| Platinum refractory | 130 (54.4%) | 125 (53.2%) |
| Previous chemotherapy, n (%): | ||
| One regimen | 100% | 100% |
| Primary site of disease | NR | NR |
| No. of lesions, median (range) | Median of 20 (1–441) | Median of 20 (1–296) |
| Histological type, n (%) | NR | NR |
| Histological grade | NR | NR |
| Tumour size, bulky disease: | ||
| Present | 111 (46%) | 111 (47%) |
| Absent | 128 (54%) | 124 (53%) |
| Disease measurability | Breakdown of measurable disease vs. assessable disease at baseline not reported | |
| FIGO stage at diagnosis: | ||
| I | 11 (5%) | 15 (6%) |
| II | 13 (5%) | 8 (3%) |
| III | 175 (73%) | 164 (70%) |
| IV | 40 (17%) | 48 (20%) |
| Performance status: baseline KPS, n (%) | ||
| < 80 | 39 (16.3%) | 37 (15.7%) |
| ≥ 80 | 199 (83.3%) | 195 (83.0%) |
| Unknown | 1 (0.4%) | 3 (1.3%) |
| Comments | None | |
Gore et al.24
| Item | Details | |
|---|---|---|
| Study | Gore et al.24 | |
| Location | Europe, South Africa and North America; multicentre | |
| Trial sponsor | SmithKline Beecham | |
| Patient enrolment | NR | |
| Trial design | Open-label, multicentre | |
| Line of therapy | Second line | |
| Inclusion criteria |
|
|
| Exclusion criteria |
|
|
| Outcomes reported | ORR, OS, AEs | |
| Subgroups | None specified | |
| Stratification | Stratification by response to previous platinum chemotherapy, tumour size (< or ≥ 5 cm in diameter) and whether or not the previous regimen had included a taxane | |
| Measure of disease response or progression | Based on WHO criteria, confirmed by independent, blinded radiological review. Time to response, TTP and OS measured from time of first dose and response duration measured from time of first documented CR or PR Response also measured by serial CA125 values. Response defined as 50% decrease in two samples, confirmed by third, or serial decrease over three samples of > 75%. Final sample at least 25 days after previous sample |
|
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | Platinum refractory: progressive or SD during initial chemotherapy Platinum resistant: responded and subsequently relapsed within 6 months of discontinuing initial chemotherapy Platinum sensitive: responded to initial therapy but subsequently relapsed at > 6 months |
|
| Other definitions | Measurable disease was defined as one lesion ≥ 2 cm in diameter (or ≥ 1 cm for skin lesions) CR: complete disappearance of all known measurable and evaluable disease determined by two measurements not less than 4 weeks apart PR: > 50% decrease in measurable lesion size for at least 4 weeks with no simultaneous increase in a known lesion or appearance of new lesions or increase in evaluable disease |
|
| Treatment | Oral topotecan | Topotecan |
| Randomised, n | 135 | 131 |
| Withdrawals, n | 0 | 0 |
| Treatment | Oral topotecan 2.3 mg/m2/day. Duration of therapy depended on response to treatment and at discretion of investigator | Intravenous topotecan 1.5 mg/m2/day for 5 days every 21 days dependent on response to treatment and at discretion of investigator |
| Treatment duration | Median number of cycles 4 (range 1–23) | Median number of cycles 6 (range 1–26) |
| Treatment discontinuation | NR | NR |
| Concomitant medications | NR | NR |
| Duration of follow-up | NR | NR |
| Baseline patient characteristics | ||
| Age, years (range) | Median 60 (23–80) | Median 60 (27–80) |
| Previous treatment: | ||
| First-line platinum/paclitaxel | 53 | 54 |
| TFI: | ||
| Median | NR | NR |
| Platinum sensitive | 58 (43%) | 56 (43%) |
| Platinum resistant | 37 (27%) | 36 (27%) |
| Platinum refractory | 40 (30%) | 39 (30%) |
| Previous chemotherapy, %: | ||
| One regimen | 100 | 100 |
| Primary site of disease | NR | NR |
| No. of sites of lesions | NR | NR |
| Histological type | NR | NR |
| Histological grade: | NR | NR |
| Tumour size, cm: | ||
| < 5 | 66 (49%) | 65 (50%) |
| 5–10 | 58 (43%) | 50 (38%) |
| > 10 | 10 (7%) | 11 (8%) |
| Missing data | 1 (1%) | 5 (4%) |
| Disease measurability | Patients all had measurable disease at baseline | |
| FIGO stage at diagnosis: | ||
| III | 84 (62%) | 82 (63%) |
| IV | 43 (32%) | 42 (32%) |
| Missing | 8 (6%) | 7 (5%) |
| Performance status, ECOG score: | ||
| 0 | 59 (45%) | 47 (35%) |
| 1 | 60 (46%) | 77 (57%) |
| 2 | 12 (9%) | 11 (8%) |
| Comments | None | |
CARTAXHY (Lortholary et al.62)
| Item | Details | ||
|---|---|---|---|
| Study | CARTAXHY62 | ||
| Location | NR; patients randomised at the ‘GINECO’ data centre | ||
| Trial sponsor | NR | ||
| Patient enrolment | Between April 2004 and August 2008 | ||
| Trial design | Phase II, multicentre, open label Three-armed trial; third arm evaluated weekly paclitaxel plus weekly topotecan, which is outside of the scope of the review |
||
| Line of therapy | Second/third line | ||
| Inclusion criteria |
|
||
| Exclusion criteria |
|
||
| Outcomes reported | Primary end point: comparison of PFS Secondary end points: ORR, OS, QoL and safety |
||
| Subgroups | None specified | ||
| Stratification | NR | ||
| Measure of disease response or progression | Response determined according to RECIST69 for measurable disease and Rustin’s criteria for non-measurable disease. Progression determined according to the definition of the GCIG. Objective response: radiologically confirmed at least 4 weeks after baseline assessments | ||
| Ethnicity | NR | ||
| Disease classifications according to platinum sensitivity | Progression during or within 6 months of platinum-containing therapy: progression during treatment, relapse at between 0 and 3 months, or relapse > 3 months and ≤ 6 months | ||
| Other definitions | No other definitions | ||
| Treatment | Weekly paclitaxel plus carboplatin | Weekly paclitaxel | |
| Randomised, n | 51 | 57 | |
| Withdrawals, n (%) | |||
| PD | 20 (39.2) | 29 (50.8) | |
| Toxicity | 15 (29.4) | 1 (1.8) | |
| Other | 2 (3.9) | 3 (5.3) | |
| Treatment | Weekly paclitaxel plus carboplatin dosed to an AUC 5 mg/ml/minute on day 1 of a 4-week cycle given for six to nine cycles or until progression | Paclitaxel 80 mg/m2 on days 1, 8 and 15 of a 4-week cycle given for six to nine cycles or until progression | |
| Treatment duration | Median number of cycles 3 (range 1–23) | Median number of cycles 3 (range 1–23) | |
| Treatment discontinuation | On progression, patients treated with weekly paclitaxel plus carboplatin received treatment as per the investigator | On progression, patients treated with weekly paclitaxel received carboplatin (AUC 5) | |
| Concomitant medications | NR | NR | |
| Duration of follow-up | Median 15 months | Median 15 months | |
| Baseline patient characteristics | |||
| Age, years (range) | Median 60 (43–77) | Median 60 (30–80) | |
| Previous treatment | NR | NR | |
| Disease-free interval: | |||
| Progression during treatment | 0% | 4% | |
| < 3 months | 47% | 42% | |
| > 3months | 53% | 54% | |
| Previous chemotherapy | |||
| One regimen | 71% | 74% | |
| Two regimens | 29% | 19% | |
| More than two regimens | 0% | 7% | |
| Primary site of disease | NR | NR | |
| No. of sites of lesions, n (%) | NR | NR | |
| Histological type, n (%): | |||
| Serous | 76% | 79% | |
| Clear cell | 2% | 2% | |
| Other | 18% | 18% | |
| Unknown | 4% | 2% | |
| Histological grade | NR | NR | |
| Tumour size, cm | NR | NR | |
| Disease measurability: | |||
| Measurable (RECIST) | 68% | 57% | |
| Elevated CA125 level only (GCIG) | 28% | 37% | |
| FIGO stage at diagnosis | NR | NR | |
| Performance status, ECOG score | |||
| 0–1 | 92% | 95% | |
| 2 | 8% | 5% | |
| Comments | None | ||
OVA-301 (Monk et al.30)
| Item | Details | |
|---|---|---|
| Study | OVA-30130 | |
| Location | 124 centres in 21 countries | |
| Trial sponsor | Johnson & Johnson | |
| Patient enrolment | Between April 2005 and May 2007 | |
| Trial design | Phase III, open label, international, multicentre | |
| Line of therapy | Second line | |
| Inclusion criteria |
|
|
| Exclusion criteria |
|
|
| Outcomes reported | OS, PFS, ORR duration of response | |
| Subgroups | None specified | |
| Stratification | Stratified by ECOG performance status (0 to 1 vs. 2) and platinum sensitivity (sensitive vs. resistant) | |
| Measure of disease response or progression | PFS by independent radiology assessment based on only RECIST criteria.69 Secondary analyses of PFS based on independent oncologist (radiological evaluation in conjunction with prespecified clinical data) and investigator’s assessments | |
| Ethnicity: | ||
| PLDH plus trabectedin | PLDH alone | |
| White | 265 (79%) | 259 (77%) |
| Asian | 66 (20%) | 71 (21%) |
| Black | 2 (1%) | 3 (1%) |
| Other | 4 (1%) | 2 (1%) |
| Disease classifications according to platinum sensitivity | Platinum sensitive or resistant but not refractory | |
| Other definitions | ORR: response maintained for ≥ 4 weeks by the RECIST criteria69 Duration of response: date of first documentation of response to date of PD or death due to PD |
|
| Treatment | PLDH plus trabectedin | PLDH |
| Randomised, n | 337 | 335 |
| Withdrawals, n | ||
| Did not receive allocated treatment | 3 | 6 |
| Lost to follow-up | 2 | 0 |
| Discontinued trabectedin/PLDH | 325 | 322 |
| Disease progression | 139 | 178 |
| Patient choice | 57 | 50 |
| AE | 69 | 39 |
| Other | 28 | 33 |
| CR (confirmed) | 24 | 14 |
| Treatment | PLDH 30 mg/m2 followed immediately by trabectedin 1.1 mg/m2 (3-hour infusion) through a central venous catheter every 3 weeks | PLDH 50 mg/m2 every 4 weeks |
| Treatment duration | NR | NR |
| Treatment discontinuation | Treatment continued until disease progression or confirmation of CR and could be continued for two or more cycles beyond confirmed CR | Treatment continued until disease progression or confirmation of CR and could be continued for two or more cycles beyond confirmed CR |
| Concomitant medications | Before treatment with PLDH, patients were given i.v. dexamethasone 20 mg (or equivalent) followed by treatment regimen after 30 minutes. Colony-stimulating factors were permitted after cycle 1 as per ASCO guidelines; additional antiemetics were permitted at investigator’s discretion | Colony-stimulating factors were permitted after cycle 1 as per ASCO guidelines;148 additional antiemetics were permitted at investigator’s discretion |
| Duration of follow-up | NR | NR |
| Baseline patient characteristics | ||
| Age, years (range) | Median 56 (26–82) | Median 58 (27–87) |
| Previous treatment: | ||
| Prior taxane use | 269 (80%) | 271 (81%) |
| Prior consolidation chemotherapy | 27 (8%) | 32 (10%) |
| PFI, months: | ||
| < 6 | 115/333 (35%) | 117/330 (35%) |
| 6 to < 12 | 123/333 (37%) | 91/330 (28%) |
| ≥ 12 | 95/333 (28%) | 122/330 (37%) |
| Previous chemotherapy: | ||
| One regimen | 100% | 100% |
| Primary site of disease | NR | NR |
| No. of sites of lesions: | ||
| 0 | 6 (2%) | 3 (1%) |
| 1–3 | 278 (82%) | 295 (88%) |
| > 3 | 53 (16%) | 37 (11%) |
| Histological type: | ||
| Papillary/serous | 225 (67%) | 230 (69%) |
| Endometrioid | 23 (7%) | 17 (5%) |
| Clear cell carcinoma | 13 (4%) | 16 (5%) |
| Mucinous | 5 (1%) | 4 (1%) |
| Transitional cell carcinoma | 2 (1%) | 2 (1%) |
| Mixed epithelial tumour | 4 (1%) | 5 (1%) |
| Peritoneal carcinoma | 11 (3%) | 9 (3%) |
| Fallopian tube carcinoma | 3 (1%) | 3 (1%) |
| Other | 50 (15%) | 49 (15%) |
| Histological grade: | ||
| 1 | 18 (5%) | 10 (3%) |
| 2 | 58 (17%) | 59 (18%) |
| 3 | 175 (52%) | 174 (52) |
| Unknown | 85 (25%) | 91 (27%) |
| Tumour size, cm | NR | NR |
| Disease measurability | All patients had measurable disease at baseline | |
| FIGO stage at diagnosis | NR | NR |
| Performance status, ECOG score: | ||
| 0 | 230 (68%) | 192 (57%) |
| 1 | 98 (29%) | 132 (39%) |
| 2 | 9 (3%) | 11 (3%) |
| Comments | None | |
ICON4/AGO-OVAR2.2 (Parmar et al.61)
| Item | Details | ||
|---|---|---|---|
| Study | ICON4/AGO-OVAR 2.261 | ||
| Location | 199 hospitals in UK, Norway, Switzerland, Italy | ||
| Trial sponsor | BMS | ||
| Patient enrolment | ICON4 MRC CTU: between May 1996 and March 2002 ICON4 Italy: between January 1996 and March 2002 AGO: between October 1996 and September 1999 |
||
| Trial design | Parallel RCTs, three different protocols:
|
||
| Line of therapy | MRC CTU: second line and greater AGO: second line ICON4: second line |
||
| Inclusion criteria |
|
||
| Exclusion criteria | None apart from above | ||
| Outcomes reported | OS, PFS | ||
| Subgroups | OS and PFS results were reported by trial and by prespecified subgroups | ||
| Stratification | Stratification by centre, age, last chemotherapy received, time since completion of last chemotherapy, and intended platinum treatment | ||
| Measure of disease response or progression | PD defined by clinical or radiological evidence Raised CA125 concentrations alone (in the absence of clinical or radiological evidence of PD) were not deemed to show disease progression |
||
| Ethnicity | NR | ||
| Disease classifications according to platinum sensitivity | Platinum sensitivity: treatment free (following platinum therapy) for > 6 months | ||
| Other definitions | OS: time from randomisation to death from any cause; patients known to be alive at the time of analysis were censored at the time of their last follow-up PFS: time from randomisation to first appearance of PD or death from any cause; patients known to be alive and without PD at the time of analysis were censored at their last follow-up |
||
| Treatment | Paclitaxel plus platinum chemotherapy | Conventional platinum-based chemotherapy | |
| Randomised, n | 392 | 410 | |
| Withdrawals, n | 1 (treatment never began); 7 (missing details) | 2 (treatment did not begin); 16 (missing details) | |
| Treatment | Paclitaxel AGO: 185 mg/m2 paclitaxel (3-hour infusion) followed by carboplatin ICON4: 175 mg/m2 paclitaxel (3-hour infusion) plus platinum followed by carboplatin or cisplatin Carboplatin If determined by method of Calvert et al.,149 AUC was a minimum of 5 If the dose was assessed by the Cockcroft formula,150 the AUC was 6 The planned minimum dose of cisplatin, in ICON4 patients only, was 50 mg/m2 if given in combination |
Carboplatin If determined by method of Calvert and colleagues, AUC was a minimum of 5 If the dose was assessed by Cockcroft formula, the AUC was a minimum of 6 Cisplatin The planned minimum dose of cisplatin, in ICON4 patients only, was 75 mg/m2 if given as a single agent |
|
| Treatment duration | 309 received ≥ 6 cycles, 75 received < 6 cycles | 271 received ≥ 6 cycles, 121 received < 6 cycles | |
| Treatment discontinuation | Reasons for not completing six cycles were: disease progression or death 109 (56%); toxicity 77 (39%); patient preference 9 (5%) (not shown separately by group) | ||
| Concomitant medications | NR | NR | |
| Duration of follow-up | Median 42 months | Median 42 months | |
| Baseline patient characteristics | |||
| Age, years: | |||
| Median | 60 | 59.2 | |
| < 55 | 127/392 (32%) | 123/410 (30%) | |
| 55–65 | 151 (39%) | 162 (40%) | |
| > 65 | 114 (29%) | 125 (30%) | |
| Previous treatment – last chemotherapy received: | |||
| Paclitaxel and carboplatin | 133/392 (34%) | 141/410 (34%) | |
| Carboplatin | 119 (30%) | 128 (31%) | |
| CAP | 62 (16%) | 72 (18%) | |
| Paclitaxel and cisplatin | 27 (7%) | 20 (5%) | |
| Docetaxel and carboplatin | 7 (2%) | 14 (3%) | |
| Other platinum based | 34 (9%) | 30 (7%) | |
| Other non-platinum | 10 (3%) | 5 (1%) | |
| TFI (months): | |||
| ≤ 12 | 92 (23%) | 111 (27%) | |
| > 12 | 300 (77%) | 299 (73%) | |
| Previous chemotherapy, n (%): | |||
| One regimen | 354 (90) | 380 (93) | |
| Two regimens (MRC CTU patients only) | 22 (6) | 22 (5) | |
| More than Two regimens (MRC CTU patients only) | 15 (4) | 15 (4) | |
| Not yet known | 1 (0.2) | 1 (0.2) | |
| Primary site of disease | NR | NR | |
| No. of sites of lesions | NR | NR | |
| Histological type, n (%) | NR | NR | |
| Histological grade | NR | NR | |
| Tumour size, cm | NR | NR | |
| Disease measurability | NR | NR | |
| FIGO stage at diagnosis | NR | NR | |
| Performance status, WHO score: | |||
| 0 | 246 (63%) | 262 (64%) | |
| 1 | 121 (31%) | 122 (30%) | |
| 2–3 | 25 (6%) | 26 (6%) | |
| Comments | None | ||
Pfisterer et al.50
| Item | Details | ||
|---|---|---|---|
| Study | Pfisterer et al.50 | ||
| Location | Germany | ||
| Trial sponsor | AGO-OVAR, National Cancer Institute of Canada Clinical Trials Group, and EORTC Gynecologic Cancer Group; supported by Lilly Deutschland GmbH | ||
| Patient enrolment | Between September 1999 and April 2002 | ||
| Trial design | Phase III, RCT, active controlled | ||
| Line of therapy | Second line | ||
| Inclusion criteria |
|
||
| Exclusion criteria | No others reported | ||
| Outcomes reported | PFS (primary), ORR, duration of response, OS, QoL and toxicity | ||
| Subgroups | Age (> 60 vs. ≤ 60) Performance status (0 vs. 1–2) Prior platinum therapy (platinum plus non-paclitaxel vs. platinum plus paclitaxel) Disease status (bidimensionally measurable vs. assessable) Duration of PFI (6–12 months vs. > 12 months) |
||
| Stratification | Stratified according to PFI (6–12 months vs. ≥ 12 months), first-line therapy (platinum paclitaxel vs. other platinum-based therapy) and bidimensionally measurable disease (yes vs. no) | ||
| Measure of disease response or progression | PD was based on clinical and/or radiological evaluation according to SWOG criteria. Categorisation of PD was not based on CA125 elevation without other clinical or radiological evidence of disease progression | ||
| Ethnicity | NR | ||
| Disease classifications according to platinum sensitivity | Platinum-sensitive recurrent ovarian cancer: recurrent ovarian cancer at least 6 months after completion of first-line, platinum-based therapy | ||
| Other definitions | PFS was defined as the time from the date of random assignment to the date of disease progression or death from any cause OS was measured from the date of random assignment to the date of death from any cause. OS was assessed when 71% of the study population had died |
||
| Treatment | Gemcitabine plus carboplatin | Carboplatin alone | |
| Randomised, n | 178 | 178 | |
| Withdrawals, n | One ineligible after randomisation, two withdrew consent | Three withdrew consent, one thrombocytopenia | |
| Treatment | Gemcitabine (1000 mg/m2) on days 1 and 8 plus carboplatin AUC 4 on day 1 every 21 days | Carboplatin AUC 5, based on the Calvert formula, on day 1 every 21 days | |
| Treatments were given for six cycles in the absence of PD or unacceptable toxicity. Patients showing benefit could receive up to 10 cycles, based on the discretion of the investigator | |||
| Treatment duration | Median number of cycles: 6 | Median number of cycles: 6 | |
| Treatment discontinuation | Day 8 gemcitabine was reduced by 50% if ANC ≥ 1.0 to 1.4 × 109/l and/or platelets 75 to 99 × 109/l, and it was omitted if below these values. For grade 3 non-haematological toxicities (excluding nausea/vomiting), dose modifications and/or study discontinuation were at the investigator’s discretion. Successive reductions by one dose level were required for treatment delays 1 week or longer due to toxicity, ANC < 0.5 × 109/l for > 5 days (or < 0.1 × 109/l for > 3 days), febrile neutropenia, platelets < 25 × 109/l, and grade 3/4 non-haematological toxicities (except nausea/vomiting). Dose level −1 of gemcitabine was 800 mg/m2, and dose level −2 was omission of day 8 gemcitabine; carboplatin was not reduced | Dose level −1 was a reduction to AUC 4; if additional dose reductions were required, patients were discontinued | |
| Cycles could be postponed up to 2 weeks owing to toxicity, and longer toxicity-related delays led to treatment discontinuation. Treatment resumed after recovery from non-haematological and haematological toxicities (ANC ≥ 1.5 × 109/l and platelets ≥ 100 × 109/l) | |||
| Concomitant medications | NR | NR | |
| Duration of follow-up | 17 months | 17 months | |
| Baseline patient characteristics | |||
| Age, years (range) | Median 59 (36–78) | Median 58 (21–81) | |
| Previous treatment: | |||
| Surgery | 178 (100%) | 178 (100%) | |
| Radiotherapy | 4 (2%) | 3 (2%) | |
| Prior taxane use | 125 (70.2%) | 127 (71.3%) | |
| Immunotherapy | 4 (2.2%) | 4 (2.2%) | |
| Hormonal therapy | 6 (3.4%) | 2 (1.1%) | |
| PFI: | |||
| < 6 months | 1 (0.6%) | 0 | |
| 6–12 months | 71 (39.9%) | 71 (39.9%) | |
| > 12 months | 106 (59.6%) | 107 (60.1%) | |
| Previous chemotherapy, %: | |||
| One regimen | 100 | 100 | |
| Primary site of disease | NR | NR | |
| No. of sites of lesions, n (%) | NR | NR | |
| Histological type, n (%): | |||
| Well differentiated | 15 (8.4) | 13 (7.3) | |
| Moderately differentiated | 51 (28.7) | 49 (27.5) | |
| Poorly differentiated | 78 (43.8) | 88 (49.4) | |
| Undifferentiated | 10 (5.6) | 7 (3.9) | |
| Unknown | 24 (13.5) | 21 (11.8) | |
| Histological grade: | NR | NR | |
| Tumour size, cm | NR | NR | |
| Disease measurability | NR | NR | |
| FIGO stage at diagnosis: | |||
| IA–IIA | 16 (9.0%) | 14 (7.9%) | |
| IIB–IIIA | 22 (12.4%) | 12 (6.7%) | |
| IIIB | 16 (9.0%) | 22 (12.4%) | |
| IIIC | 97 (54.5%) | 107 (60.1%) | |
| IV | 27 (15.2%) | 22 (12.4%) | |
| Unspecified | 0 | 1 (0.6%) | |
| Performance status, ECOG score: | |||
| ND | 5 (2.8%) | 4 (2.2%) | |
| 0 | 83 (46.6%) | 93 (52.2%) | |
| 1 | 79 (44.4%) | 72 (40.4%) | |
| 2 | 11 (6.2%) | 9 (5.1%) | |
| Comments | None | ||
Piccart et al.63
| Item | Details | |
|---|---|---|
| Study | Piccart et al.63 | |
| Location | 17 European centres | |
| Trial sponsor | Debiopharm SA, Lausanne, Switzerland | |
| Patient enrolment | Between January 1996 and December 1997 | |
| Trial design | Multicentre, open-label trial | |
| Line of therapy | Second and third | |
| Inclusion criteria |
|
|
| Exclusion criteria |
|
|
| Outcomes reported | Response rate, OS, PFS | |
| Subgroups | Potentially platinum-sensitive vs. platinum-refractory | |
| Stratification | Stratification by centre, performance status (0 or 1 vs. 2), PFI (0–6 months vs. 6–12 months), no. of prior platinum-based regimens (1 vs. 2) | |
| Measure of disease response or progression | Target lesions measured by CT scan or MRI every two cycles CR, PR and disease progression as defined:
|
|
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | Platinum sensitive: relapse at > 6 months but < 12 months after their last platinum-based chemotherapy regimen Platinum refractory: disease progression after a minimum of two cycles of chemotherapy, no change under chemotherapy for at least four cycles, or a relapse that occurred < 6 months after the end of prior chemotherapy |
|
| Other definitions | OS: day 1 of treatment to date of first observation of disease progression, treatment failure [tumour progression or change of treatment, including crossover (which was allowed), or death] Confirmed response: verified by two independent radiologists and defined as PR or CR observed in at least two consecutive evaluations at least 4 weeks apart |
|
| Treatment | Paclitaxel | Oxaliplatin |
| Randomised, n | 41 | 45 |
| Withdrawals, n (%) | NR | NR |
| Treatment | Paclitaxel (175 mg/m2) administered as a 3-hour i.v. infusion every 21 days | Oxaliplatin (130 mg/m2) administered as a 3-hour i.v. infusion every 21 days |
| Treatment duration | Median number of cycles: 6 (range 1–8) | Median number of cycles: 4 (range 1–8) |
| Treatment discontinuation | Continued until time of disease progression, unacceptable toxicity, or patient refusal. Doses could not fall below established minimum doses per cycle (90 mg/m2) | Continued until time of disease progression, unacceptable toxicity, or patient refusal. Doses could not fall below established minimum doses per cycle (75 mg/m2) |
| Concomitant medications | Premedication with oral dexamethasone 20 mg 12 and 6 hours before paclitaxel infusion, diphenhydramine 50 mg i.v. and cimetidine 300 mg or ranitidine 50 mg i.v. 30 minutes before paclitaxel | Premedication of antiemetic with serotonin antagonist (5-HT3) with a single dose of corticosteroid (e.g. dexamethasone 20 mg) |
| Duration of follow-up | NR | NR |
| Baseline patient characteristics | ||
| Age, years (range) | Median 62 (37–81) | Median 59 (28–71) |
| Previous treatment – regimens containing: | ||
| Cisplatin | 16 (39%) | 19 (42%) |
| Median dose, mg | 442 | 440 |
| Range, mg | 223–456 | 223–478 |
| Carboplatin | 21 (51%) | 21 (47%) |
| Median dose, mg | 1970 | 1888 |
| Range, mg | 652–3600 | 1402–3568 |
| Both cisplatin and carboplatin | 4 (10%) | 5 (11%) |
| TFI, months: | ||
| 0–6 | 31 (76%) | 32 (71%) |
| 6–12 | 10 (24%) | 13 (29%) |
| Previous chemotherapy, n (%): | ||
| One regimen | 30 (73) | 29 (64) |
| Two regimens | 11 (27) | 16 (36) |
| Sites involved (not primary): | ||
| Pelviperineum | 30 (73%) | 25 (56%) |
| Lymph nodes | 15 (37%) | 13 (29%) |
| Lung | 2 (5%) | 2 (4%) |
| Liver | 7 (17%) | 15 (33%) |
| Other | 7 (17%) | 7 (16%) |
| No. of sites of lesions: | ||
| 0 | 0 | 1 (2%) |
| 1 | 22 (54%) | 30 (67%) |
| 2 | 17 (41%) | 11 (25%) |
| 3 | 2 (5%) | 2 (4%) |
| > 3 | 0 | 1 (2%) |
| Histological type, n (%): | ||
| Serous | 17 (41) | 33 (73) |
| Other | 24 (59) | 12 (27) |
| Histological grade | NR | NR |
| Tumour size, cm | NR | NR |
| Disease measurability | NR | NR |
| FIGO stage at diagnosis: | ||
| I | 5 (12%) | 7 (16%) |
| II | 2 (5%) | 1 (2%) |
| III | 26 (63%) | 29 (64%) |
| IV | 8 (20%) | 8 (18%) |
| Performance status, WHO score: | ||
| 0–1 | 35 (85%) | 38 (84%) |
| 2 | 6 (15%) | 7 (16%) |
| Comments | None | |
CALYPSO (Pujade-Lauraine et al.31,56,57)
| Item | Details | |
|---|---|---|
| Study | CALYPSO31,56,57 | |
| Location | Multicentre, multinational | |
| Trial sponsor | Schering–Plough | |
| Patient enrolment | Between April 2005 and September 2007 | |
| Trial design | Phase III, non-inferiority, multicentre, multinational trial | |
| Line of therapy | Second or third line | |
| Inclusion criteria |
|
|
| Exclusion criteria | Pre-existing neuropathy (NCI-CTCAE grade > 1) | |
| Outcomes reported | PFS, AEs | |
| Subgroups | ‘Exploratory analyses’ only | |
| Stratification | Stratified by therapy-free interval from last chemotherapy (6 to 12 vs. > 12 months), measurable disease (yes vs. no) and centre | |
| Measure of disease response or progression | Disease progression based on RECIST69 and GCIG70 criteria or histologically proven diagnosis of relapse RECIST and GCIG modifications may have included: occurrence (clinically or imaging signs) of any new lesion; increase in measurable and/or non-measurable tumour defined by RECIST; CA125 elevation defined by GCIG criteria; health status deterioration attributable to disease; and death of any cause before progression is diagnosed Evaluation assessments were independently reviewed |
|
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | Platinum sensitive: disease recurrence > 6 months after first or second-line platinum therapy | |
| Other definitions | None | |
| Treatment | PLDH plus carboplatin | Paclitaxel plus carboplatin |
| Randomised, n | 467 | 509 |
| Withdrawals, n | 1 (ineligible) | 2 (missing data) |
| Treatment | PLDH (30 mg/m2 intravenously on day 1) and carboplatin (AUC 5 on day 1) every 4 weeks | Paclitaxel (175 mg/m2 intravenously on day 1) and carboplatin (AUC 5 intravenously on day 1) every 3 weeks |
| Treatment duration | Median no. of cycles: 6 (range 1–14) | Median no. of cycles: 6 (range 1–12) |
| Treatment discontinuation | Treatment continued until disease progression or unacceptable toxicity | |
| Concomitant medications | All patients received antiemetics, including serotonin antagonist and corticosteroid. Patients assigned to carboplatin received premedication to prevent hypersensitivity reactions | |
| Duration of follow-up | 5 years | 5 years |
| Baseline patient characteristics | ||
| Age, years (range) | Median 60.5 (24–82) | Median 61 (27–82) |
| Previous treatment: | ||
| Prior taxane use, n (%) | 396 (85%) | 407 (80%) |
| Surgery for this relapse | 87 | 100 |
| TFI, months: | ||
| Median | 15.2 | 15.0 |
| 6–12 | 161/466 (35%) | 183/507 (36%) |
| > 12 | 305 (65%) | 324 (64%) |
| Prior chemotherapy: | ||
| One regimen | 408/466 (88%) | 419/507 (83%) |
| Two regimens | 58 (12%) | 88 (17%) |
| Primary site: | ||
| Ovarian | 416 (89.2%) | 452 (89.2%) |
| Fallopian | 18 (3.9%) | 19 (3.7%) |
| Peritoneal | 32 (6.9%) | 36 (7.1%) |
| No. of sites of lesions: | ||
| 1 | 217 (46.6%) | 243 (47.9%) |
| > 1 | 249 (53.4%) | 264 (52.0%) |
| Histological type: | ||
| Serous | 334 (71.4%) | 366 (72.2%) |
| Endometrioid | 38 (8.2%) | 35 (6.9%) |
| Clear cell | 14 (3%) | 13 (2.6%) |
| Mixed epithelial | 8 (1.7%) | 17 (3.3%) |
| Mucinous | 9 (1.9%) | 8 (1.6%) |
| Other | 37 (7.9%) | 42 (8.3%) |
| Unspecified | 26 (5.6%) | 26 (5.1%) |
| Histological grade: | ||
| 1 | 29 (6.2%) | 23 (4.5%) |
| 2 | 100 (21.5%) | 128 (25.2%) |
| 3 | 257 (55.1%) | 270 (53.3%) |
| Unknown | 80 (17.2%) | 89 (17.0%) |
| Tumour size, cm: | ||
| < 5 | 377 (80.9%) | 417 (82.3%) |
| ≥ 5 | 89 (19.1%) | 90 (17.7%) |
| Measurable disease: | ||
| Yes | 281 (60.3%) | 321 (63.3%) |
| No | 185 (39.7%) | 186 (36.7%) |
| FIGO stage at diagnosis: | ||
| I/II | 57 (12.3%) | 66 (13.0%) |
| III/IV | 400 (85.8%) | 427 (84.2%) |
| Missing | 9 (1.9%) | 14 (2.8%) |
| Performance status, ECOG score: | ||
| 0 | 286 (61.4%) | 317 (62.5%) |
| 1 | 158 (33.9%) | 164 (32.3%) |
| 2 | 13 (2.8%) | 15 (3.0%) |
| Missing | 9 (1.9%) | 11 (2.2%) |
| Comments | Crossover: 43% PLDH and carboplatin group; 68% paclitaxel and carboplatin group | |
Rosenberg et al.60
| Item | Details | |
|---|---|---|
| Study | Rosenberg et al.60 | |
| Location | Sweden | |
| Trial sponsor | NR | |
| Patient enrolment | Between February 1995 and June 1998 | |
| Trial design | Bifactorial, stratified, multicentre trial, phase not reported | |
| Line of therapy | Second line only | |
| Inclusion criteria |
|
|
| Exclusion criteria |
|
|
| All outcomes reported | OS, TTP, response | |
| Subgroups | None | |
| Stratification | Platinum resistance (i.e. relapse at ≤ 6 months vs. > 6 months after primary platinum-based therapy) | |
| Measure of disease response or progression | Progression and response assessed according to WHO tumour response criteria | |
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | Platinum resistant/sensitive: relapse ≤ 6 months and > 6 months after primary platinum-based therapy | |
| Other definitions | OS: from the day of randomisation to the day of death or censored observation TTP: from the first day of study treatment to the day of documented progression or censored observation Response duration for patients with CR: from the day of first observation of CR to the day of documented progression or censored observation Response duration for patients with PR: from the first day of study treatment to the day of documented progression or censored observation |
|
| Treatment | Paclitaxel weekly | Paclitaxel 3 weekly |
| Randomised, n | 105 | 103 |
| Withdrawals, n (%) | 32 (30) | 16 (15.5) |
| Treatment | Paclitaxel 67 mg/m2 weekly | Paclitaxel 200 mg/m2 every 3 weeks |
| Patients within paclitaxel groups also randomised to oral steroids 12 and 6 hours before paclitaxel or parenteral steroids 30 minutes before paclitaxel | ||
| Treatment duration | Median no. of courses: 5.7 (range 1–16) | Median no. of courses: 7 (range 1–17) |
| Treatment discontinuation | Protocol allowed indefinite treatment: if no haematological toxicity occurred the dose was escalated maximally by two steps. Dose reduction was performed in case of severe cytopenia. Patients who could not tolerate the lowest dose level were taken off the study treatment. No dose escalation was allowed once a dose reduction had been made. If infusion was interrupted due to a hypersensitivity reaction patients could be re-treated at the investigator’s discretion. Decision on whether or not to continue treatment was made on basis of tumour assessments every 6 weeks. Patients with PD were taken off the study. Patients with SD received treatment until either progression or unacceptable toxicity occurred. Patients who achieved a CR or a PR continued study treatment for a minimum of 6 weeks and thereafter at the investigator’s discretion to tumour progression/relapse or unacceptable toxicity whichever came first. Cycles were to be given as planned – not permissible to prolong TFI | |
| Concomitant medications | Oral dexamethasone 20 mg or its equivalent 12 and 6 hours before paclitaxel (group A1) or dexamethasone 20 mg i.v. 30 minutes before paclitaxel (group A2). All patients received clemastine 2 mg and cimetidine 300 mg or ranitidine 50 mg 30-minutes before paclitaxel | Oral dexamethasone 20 mg or its equivalent 12 and 6 hours before paclitaxel (group B1) or dexamethasone 20 mg i.v. 30 minutes before paclitaxel (group B2). All patients received clemastine 2 mg and cimetidine 300 mg or ranitidine 50 mg 30-minutes before paclitaxel |
| Duration of follow-up | Median 27 months (range 7–47+) | Median 27 months (range 7–47+) |
| Baseline patient characteristics | ||
| Age, years (range) | Median 59 (37–74) | Median 60 (40–76) |
| Previous treatment | NR | NR |
| PFI – defined as platinum-resistant tumour (relapse at ± 6 months after primary chemotherapy): | ||
| Yes | 57 | 51 |
| No | 48 | 52 |
| Prior chemotherapy: | One prior platinum-containing regimen of chemotherapy not containing a taxane | One prior platinum-containing regimen of chemotherapy not containing a taxane |
| One regimen | 100% | 100% |
| Primary site of disease | Epithelial (all patients) | |
| No. of sites of lesions, n (%) | NR | NR |
| Histological type, n (%) | NR | NR |
| Histological grade | NR | NR |
| Tumour size, cm: | ||
| ≤ 2 cm | 7 | 11 |
| 2–5 cm | 34 | 26 |
| 5–10 cm | 30 | 26 |
| ≥ 10 cm | 33 | 40 |
| Unknown | 1 | 0 |
| Measurable disease | All patients had measurable disease at baseline | |
| FIGO stage at diagnosis | NR | NR |
| Performance status: | WHO criteria | |
| 0 | 57 | 56 |
| 1 | 40 | 33 |
| 2 | 8 | 14 |
| Comments | None | |
Sehouli et al.23
| Item | Details | |
|---|---|---|
| Study | Sehouli et al.23 | |
| Location | Germany: 54 German institutions | |
| Trial sponsor | North Eastern Germany Society of Gynaecologic Oncology | |
| Patient enrolment | Between September 2005 and February 2008 | |
| Trial design | Phase II, RCT, active control | |
| Line of therapy | Second line | |
| Inclusion criteria |
|
|
| Exclusion criteria |
|
|
| Outcomes reported | Response rate, PFS, OS, toxicity, tolerability, QoL, symptom control with both regimens | |
| Subgroups | Response rate stratified by best response, CA125 response and tumour response | |
| Stratification | None stated | |
| Measure of disease response or progression | CR and PR were defined according to RECIST criteria69 for measurable disease or GCIG criteria70 for serum CA125 levels | |
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | Platinum resistance was defined as clinical disease progression after a TFI of < 6 months after a platinum-based regimen Platinum refractory patients had stable or PD while receiving platinum |
|
| Other definitions | OS was measured from random assignment to the date of death resulting from any cause or, for living patients, the date of last contact CR was defined as complete disappearance of all measurable and assessable disease by physical examination, imaging and normalisation of CA125 as determined before the study began PR was assumed in case of a 50% reduction in the sum of the product of two perpendicular diameters of the tumour SD was considered for all patients who had less than PR, but no evidence of PD PD was defined as an increase of least 25% in the sum of the product of the dimensions of the lesion or evidence of new tumour |
|
| Treatment | Topotecan weekly | Topotecan conventional |
| Randomised, n | 97 | 97 |
| Withdrawals, n (%) | NR | NR |
| Treatment | Topotecan 4.0 mg/m2 once each week every 21 days | Topotecan 1.25 mg/m2 daily for five consecutive days every 28 days |
| Treatment duration | Mean no. of cycles (sd): 3.5 (2.5) | Mean no. of cycles (sd): 4.8 (3.3) |
| Treatment discontinuation | Treatment was continued until intolerable toxicity or disease progression or until the patient refused further therapy. The protocol mandated a maximum treatment duration of 12 months after random assignment | |
| Concomitant medications | All patients received 5-HT3 antagonists intravenously for prophylaxis of nausea and emesis | |
| Duration of follow-up | 23.4 months (range 12.7–41.4 months) | |
| Baseline patient characteristics | ||
| Age, years (range) | Median 65 (41–82) | Median 61 (36–85) |
| Previous treatment | NR (all patients received prior paclitaxel) | |
| PFI, months | NR | NR |
| Previous chemotherapy, n (%): | ||
| One regimen | 69 (71) | 66 (68) |
| Two regimens | 28 (29) | 31 (32) |
| Primary site of disease | NR | NR |
| No. of sites of lesions, n (%) | NR | NR |
| Histological type, n (%): | ||
| Serous papillary adenocarcinoma | 78 (80) | 73 (75) |
| Mucinous carcinoma | 1 (1) | 2 (2) |
| Endometrioid carcinoma | 0 (0) | 3 (3) |
| Other | 15 (15) | 15 (15) |
| Undifferentiated | 1 (1) | 2 (2) |
| Peritoneal carcinoma | 0 (0) | 1 (1) |
| Unknown | 1 (1) | 2 (2) |
| Histological grade: | ||
| 1 | 5 (5%) | 6 (6%) |
| 2 | 5 (5%) | 2 (2%) |
| 3 | 22 (23%) | 30 (31%) |
| 4 | 63 (65%) | 55 (57%) |
| Unclear | 2 (2%) | 3 (3%) |
| Tumour size, cm | NR | NR |
| Measurable disease: | ||
| Yes | 86 (89%) | 90 (93%) |
| FIGO stage at diagnosis | ||
| I | 2 (2%) | 0 (0%) |
| II | 2 (2%) | 2 (2%) |
| III | 73 (75%) | 76 (78%) |
| IV | 16 (16%) | 17 (18%) |
| Unclear | 4 (4%) | 2 (2%) |
| Performance status, ECOG score: | ||
| 0 | 33 (34%) | 34 (35%) |
| 1 | 48 (49%) | 50 (52%) |
| 2 | 12 (12%) | 11 (11%) |
| Unknown | 4 (4%) | 2 (2%) |
| Comments | None | |
ten Bokkel Huinink et al.52
| Item | Details | |
|---|---|---|
| Study | ten Bokkel Huinink et al.52 | |
| Location | International; countries not reported | |
| Trial sponsor | SmithKline Beecham | |
| Patient enrolment | NR | |
| Trial design | Phase III, multicentre, stratified open-label RCT | |
| Line of therapy | Second line | |
| Inclusion criteria |
|
|
| Exclusion criteria |
|
|
| All outcomes reported | Response rate, duration of response, TTP, OS | |
| Subgroups | Age (≥ 65 years vs. < 65 years), platinum sensitivity, and presence or absence of ascites | |
| Stratification | Patients stratified by age (≥ 65 years vs. < 65 years), ascites (present vs. absent) and platinum sensitivity (resistant, early, interim or late) | |
| Measure of disease response or progression | Response and progression assessed according to WHO criteria | |
| Ethnicity | NR | |
| Disease classifications according to platinum sensitivity | Refractory: progression during chemotherapy Disease relapse was categorised as early (within 3 months), interim (between 3 and 6 months) or late (> 6 months) |
|
| Other definitions | CR defined as the complete disappearance of all known measurable and assessable disease on two separate measurements at least 4 weeks apart PR defined as a 50% reduction in the sum of products of the perpendicular diameters of all measurable lesions for at least 4 weeks and with no new lesion or progression of assessable disease PD defined as a 25% increase in a single measurable lesion, reappearance of measurable disease, clear worsening of assessable disease, or the development of new metastatic disease SD defined as any measurement not fulfilling the criteria for response or progression, and lasting > 8 weeks TTP measured from the time of first study drug administration to documented PD or initiation of third-line therapy Duration of response measured from the time of initial documented response to the first sign of disease progression |
|
| Treatment | Topotecan | Paclitaxel |
| Randomised, n | 117 (112 received intervention) | 118 (114 received intervention) |
| Withdrawals, n (%) | 11/112 (10) | 4/114 (3.5) |
| Treatment | Topotecan 1.5 mg/m2 as a 30-minute infusion on five consecutive days every 21 days | Paclitaxel 175 mg/m2 as a 3-hour infusion every 21 days |
| Treatment duration | Median no. of cycles: 5 (range 1–17) | Median no. of cycles: 5 (range 1–12) |
| Treatment discontinuation | Patients were withdrawn from treatment if there was a > 2-week delay in treatment at the minimum dose of either medication because of toxicity. The number of cycles of both the topotecan and paclitaxel interventions were determined by the patients’ response. Patients with a CR/PR continued until progression or for 6 months after the maximal response. Patients who progressed during treatment were removed from the study. Those whose best response was SD after six courses were removed or switched to the other treatment | |
| Concomitant medications | Premedication was not given to the topotecan group unless nausea or vomiting occurred, in which case it was permitted in subsequent cycles. Prophylactic recombinant G-CSF was allowed after the first course of therapy to maintain dose intensity, on day 6 of the topotecan group, if participants had experienced any of: grade 4 neutropenia with fever or infection, grade 4 neutropenia lasting > 7 days, or grade 3 neutropenia that required a delay in treatment. Dependent on toxicity the dose could vary from 1.0 to 2.0 mg/m2/day | Patients received premedication with dexamethasone, and both H1- and H2-receptor antagonists to prevent hypersensitivity reactions. Prophylactic recombinant G-CSF was allowed after the first course of therapy to maintain dose intensity, on day 2 of the paclitaxel group, if patients had experienced any of: grade 4 neutropenia with fever or infection, grade 4 neutropenia lasting > 7 days, or grade 3 neutropenia that required a delay in treatment. Dependent on toxicity, the dose could vary from 135 to 175 mg/m2 |
| Duration of follow-up | Long-term follow-up was 4 years | |
| Baseline patient characteristics | ||
| Age, years (range) | Mean 59.2 (29–85) | Mean 58.3 (29–79) |
| Previous treatment: | ||
| Cyclophosphamide | 66.0% | 69.0% |
| Carboplatin | 55.0% | 61.0% |
| Cisplatin | 54.0% | 51.0% |
| Epirubicin | 8.0% | 5.3% |
| Doxorubicin hydrochloride | 4.5% | 6.1% |
| Doxorubicin | 3.6% | 3.5% |
| Etoposide | 1.8% | 0.9% |
| Mitoxantrone | 1.8% | 0.9% |
| Ifosfamide | 1.8% | 0.0% |
| Epirubicin hydrochloride | 0.9% | 1.8% |
| Chlorambucil | 0.9% | 0.9% |
| Prednimustine | 0.9% | 0.0% |
| Fluorouracil | 0.0% | 0.9% |
| Pirarubicin | 0.0% | 0.9% |
| TFI | ||
| Platinum refractory | 52/112 (46.4%) | 55/114 (48.4%) |
| Platinum sensitive | 60/112 (53.6%) | 59/114 (51.8%) |
| Previous chemotherapy, n (%): | ||
| One regimen | 100% | 100% |
| Primary site of disease | NR | NR |
| No. of sites of lesions, n (%) | NR | NR |
| Histological type, n (%): | ||
| Malignant serous | 58 (51.8) | 59 (51.8%) |
| Malignant mucinous | 6 (5.4) | 6 (5.3) |
| Malignant endometrial | 10 (8.9) | 15 (13.2) |
| Undifferentiated carcinoma | 18 (16.1) | 8 (7.0) |
| Other | 20 (17.9) | 26 (22.8) |
| Histological grade: | ||
| 0–1 | 6 (5.0%) | 8 (7.0%) |
| 2 | 23 (20.5%) | 29 (25.4%) |
| 3 | 56 (50.0%) | 50 (43.9%) |
| 4 | 10 (8.9%) | 12 (10.5%) |
| Not determined | 17 (15.2%) | 15 (13.2%) |
| Tumour size, cm: | ||
| < 5 cm | 54 (48.2%) | 53 (46.5%) |
| ≥ 5 cm | 56 (50.0%) | 59 (51.8%) |
| Not determined | 2 (1.8%) | 2 (1.8%) |
| Measurable disease | All patients had measurable disease at baseline | |
| FIGO stage at diagnosis | NR | NR |
| Performance status, ECOG score: | ||
| 0 | 41 (36.6%) | 42 (36.8%) |
| 1 | 51 (45.5%) | 53 (46.5%) |
| 2 | 20 (17.9%) | 17 (14.9%) |
| 3 | 0 | 2 (1.8%) |
| Comments | The methods section of the report states that HRs with 95% CI were calculated. Survival curves were presented for the duration of response, TTP and survival, but HRs were not reported. It was also not clear from the data presented whether the median times quoted were based on Kaplan–Meier estimates | |
Trial 30–57 (details taken from TA9113)
| Item | Details | ||
|---|---|---|---|
| Study | 30–57; Johnson & Johnson Pharmaceutical Research & Development | ||
| Location | NR | ||
| Trial sponsor | NR | ||
| Patient enrolment | NR | ||
| Trial design | Phase III, randomised, open label, non-inferiority trial | ||
| Line of therapy | Second line | ||
| Inclusion criteria |
|
||
| Exclusion criteria |
|
||
| Outcomes reported | OS | ||
| Subgroups | Platinum-sensitive disease; platinum-refractory disease | ||
| Stratification | By platinum-sensitivity [platinum sensitive (PFI > 6 months), platinum refractory (PFI < 6 months)] and bulky disease (presence or absence of a tumour mass > 5 cm in size) | ||
| Measure of disease response or progression | NR | ||
| Ethnicity | White: 210/216 (97.2%); black: 1/216 (0.5%); Hispanic: 2/216 (0.9%); Asian: 3/216 (1.4%) | ||
| Disease classifications according to platinum sensitivity | Participants who had initially responded to platinum-based therapy and who had a PFI of > 6 months off treatment were classified as platinum sensitive Participants who progressed during treatment, or who had SD in response to initial platinum-based therapy, or whose disease relapsed within 6 months of cessation of therapy, were classified as having platinum-refractory disease |
||
| Other definitions | CR: complete disappearance of all measurable and evaluable disease. No new lesions and no disease-related symptoms PR: > 50% decrease in the sum of the products of biodimensional perpendicular diameters of all measurable lesions. No progression of evaluable disease. No new lesions |
||
| Treatment | PLDH | Paclitaxel | |
| Randomised, n | 108 | 108 | |
| Withdrawals, n (%) | |||
| Disease progression | 42/108 (38.9) | 30/108 (27.8) | |
| Death | 12/108 (11.1) | 5/108 (4.6) | |
| AE | 18/108 (16.7) | 7/108 (6.5) | |
| Lost to follow-up | 0 (0) | 1/108 (0.1) | |
| Other/unknown | 10/108 (9.3) | 12/108 (11.1) | |
| Completed protocol treatment | 26/108 (24.1) | 53/108 (49.1) | |
| Treatment | PLDH 50 mg/m2 (1-hour infusion) every 28 days | Paclitaxel 175 mg/m2 (3-hour infusion) every 21 days | |
| Treatment duration | Mean (sd): 98.7 (77.05) days Median (range): 85.0 (1–448) days |
Mean (sd): 106.2 (50.13) days Median (range): 106.0 (1–260) days |
|
| Treatment discontinuation | NR | NR | |
| Concomitant medications | None; the prophylactic use of haematopoietic cytokines was discouraged in conjunction with the first dose of study drug. Their use was recommended in subsequent cycles under specific circumstances: in participants with prolonged neutropenia (grade 4 neutropenia lasting > 7 days or failure of ANC to recover within 22 days), or the occurrence of febrile neutropenia in a prior cycle of treatment. Pyridoxine (vitamin B6) was recommended for the treatment of hand–foot syndrome symptoms | All paclitaxel-treated participants were to be premedicated with corticosteroids, antihistamines and H2 antagonists prior to paclitaxel administration. The prophylactic use of haematopoietic cytokines was discouraged in conjunction with the first dose of study drug. Their use was recommended in subsequent cycles under specific circumstances: in participants with prolonged neutropenia (grade 4 neutropenia lasting > 7 days or failure of ANC to recover within 22 days), or the occurrence of febrile neutropenia in a prior cycle of treatment. Pyridoxine was recommended for the treatment of hand–foot syndrome symptoms | |
| Duration of follow-up | NR | NR | |
| Baseline patient characteristics | |||
| Age, years (range) | Mean 58.4 (27–80) | Mean 59.5 (20–78) | |
| Previous treatment | Platinum-based first-line monotherapy regimen | Platinum-based first-line monotherapy regimen | |
| Prior anthracycline therapy: 10/108 (9.3%) | Prior anthracycline therapy: 15/108 (13.9%) | ||
| Duration of PFI | Mean (sd): 9.0 (9.98) months | Mean (sd): 11.1 (17.34) months | |
| Median (range): 6.6 (1.0–69.4) months | Median (range): 6.7 (0.9–109.1) months | ||
| Prior chemotherapy, n (%): | |||
| One regimen | 108/108 (100) | 108/108 (100) | |
| Primary site of disease | NR | NR | |
| No. of sites of lesions, n (%) | NR | NR | |
| Histological type, n (%): | |||
| Serous papillary | 29/105 (26.9) | 24/102 (22.2) | |
| Mucinous | 0 (0) | 1/102 (0.9) | |
| Unspecified adenocarcinoma | 12/105 (11.1) | 18/102 (16.7) | |
| Not specified | 67/105 (62) | 65/102 (60.2) | |
| Histological grade, n (%) | |||
| Moderately differentiated | 1/108 (0.9) | 6.108 (5.6) | |
| Poorly differentiated | 12/108 (11.1) | 13/108 (12.0) | |
| Unspecified differentiated | 28/108 (25.9) | 24/108 (22.2) | |
| Not specified | 67/108 (62) | 65/108 (60.2) | |
| Tumour size, cm | NR | NR | |
| Measurable disease | NR | NR | |
| FIGO stage at diagnosis: | |||
| I | 10/108 (9.3%) | 10/108 (9.3%) | |
| II | 11/108 (10.2%) | 8/108 (7.4%) | |
| III | 64/108 (59.3%) | 77/108 (71.3%) | |
| IV | 22/108 (20.4%) | 13/108 (12.0%) | |
| Performance status, KPS at study entry: | |||
| 0 | < 80: 11/108 (10.2%) | < 80: 12/108 (11.1%) | |
| 1 | > 80: 95/108 (88%) | > 80: 90/108 (83.3%) | |
| Comments | None | ||
Omura et al.68
| Item | Details | |
|---|---|---|
| Study | Omura et al.68 | |
| Location | USA (intergroup/multicentre: no. of institutions not reported) | |
| Trial sponsor | Study supported by NCI grants of the Gynecologic Oncology Group Administrative Office (grant no. CA 27469), the Gynecologic Oncology Group Statistical Office (grant no. CA 37517), the SWOG, the ECOG and the North Central Cancer Treatment Group | |
| Patient enrolment | Between August 1992 and February 1995 | |
| Trial design | Phase III, RCT with active control. Treatment regimen sequentially assigned from permuted blocks. Masking unclear | |
| Line of therapy | Second line | |
| Inclusion criteria |
|
|
| Exclusion criteria |
|
|
| Outcomes reported | PFS, OS, tumour response in patients with measurable disease (pleural effusion or elevated CA125 level were not regarded as measurable disease), toxicity | |
| Subgroups | None prespecified | |
| Stratification | Clinically measurable disease, platinum sensitivity, co-operative group (see Trial sponsor) | |
| Measurement of disease response or progression | CR: disappearance of all gross evidence of disease for at least 4 weeks PR: ≥ 50% reduction in the product of perpendicular measurements of each lesion for at least 4 weeks |
|
| Ethnicity: | ||
| Paclitaxel 250 mg/m2 (n = 166 evaluated) | Paclitaxel 175 mg/m2 (n = 164 evaluated) | |
| Black | 7 (4%) | 4 (2%) |
| Hispanic | 6 (4%) | 5 (3%) |
| White | 146 (88%) | 149 (91%) |
| Other/NS | 7 (4%) | 6 (4%) |
| Disease classifications according to platinum sensitivity | Platinum-resistant: progression during first-line platinum treatment or within 6 months of completing therapy, a best response of SD after six courses of platinum, or SD with rising CA125 level while on platinum Platinum-sensitive: initial response to platinum therapy lasting at least 6 months, followed by progression or recurrence |
|
| Other definitions | PFS: date of first progression or death from any cause OS: death or last contact if the date of death was unknown |
|
| Treatment | Paclitaxel 250 mg/m 2 (plus filgrastim 5 or 10 µg/kg) | Paclitaxel 175 mg/m 2 |
| Randomised, n | 188 | 184 |
| Withdrawals, n | Seven women randomised to this group were not assessed for response because of death, toxicity or withdrawal. They were classified as not responding for an ITT analysis among eligible patients | Three women randomised to this group were not assessed for response because of death, toxicity or withdrawal. They were classified as not responding for an ITT analysis among eligible patients |
| Reasons for ineligibility in the two treatment groups included inappropriate disease site (n = 34), improper prior treatment (n = 7), inadequately documented histology (n = 3), second primary cancer (n = 3), inadequate documentation of recurrence (n = 2), borderline tumour histology (n = 1) and wrong disease stage (n = 1) | ||
| Treatment | Paclitaxel 250 mg/m2 by 24-hour i.v. infusion every 3 weeks (patients in this group also randomised to filgrastim 5 or 10 µg/kg/day subcutaneously) | Paclitaxel 175 mg/m2 by 24-hour i.v. infusion every 3 weeks |
| Treatment duration | Six or more cycles (55% of patients) | Six or more cycles (58% of patients) |
| Treatment discontinuation | Patients who did not exhibit clinical progression or excessive toxicity after six cycles of therapy could continue treatment indefinitely. Paclitaxel dose could be reduced for some grade 3 or greater toxicities Over the initial six cycles, approximately 70% of patients received their planned ideal dose |
Patients who did not exhibit clinical progression or excessive toxicity after six cycles of therapy could continue treatment indefinitely. Paclitaxel dose could be reduced for some grade 3 or greater toxicities Over the initial six cycles, approximately 76% of patients received their planned ideal dose |
| Concomitant medications | Filgrastim 5 or 10 µg/kg/day subcutaneously | Patients experiencing neutropenic fever were permitted to receive filgrastim during subsequent therapy cycles |
| Duration of follow-up | NR | |
| Baseline patient characteristics (eligible patients) | ||
| Age, years (range) | Median 62 (24–80) | Median 60 (23–88) |
| Previous treatment | NR (no more than one prior platinum-based regimen and no prior taxane) | NR (no more than one prior platinum-based regimen and no prior taxane) |
| Duration of PFI | NR | NR |
| Platinum resistant: 132 (79%) | Platinum resistant: 125 (76%) | |
| Platinum sensitive: 34 (21%) | Platinum sensitive: 39 (24%) | |
| Prior chemotherapy, n (%): | ||
| One regimen | 166 (100) | 164 (100) |
| Primary site of disease | Histologically confirmed epithelial ovarian cancer | |
| No. of sites of lesions, n (%) | NR | NR |
| Histological type, n (%): | ||
| Serous | 100 (60) | 105 (63) |
| Endometrioid Mucinous Clear cell Other |
22 (13) 7 (4) 11 (7) 26 (16) |
17 (10) 0 (0) 8 (5) 34 (21) |
| Histological grade | NR | NR |
| Tumour size, cm | NR | NR |
| Measurable disease | 134 (81%) | 131 (80%) |
| FIGO stage at diagnosis | NR | NR |
| Performance status, GOG performance status at study entry: | ||
| 0 | 88 (53%) | 89 (54%) |
| 1 2 |
63 (38%) 15 (9%) |
65 (40%) 10 (6%) |
| Comments | At initiation, the study included a paclitaxel 135 mg/m2 treatment arm. Accrual to this low-dose arm decreased when paclitaxel became commercially available and enrolment ceased in October 1993 Patients treated with paclitaxel 250 mg/m2 were randomly assigned to receive filgrastim (5 or 10 µg/kg/day subcutaneously) to assess its effect on the incidence of febrile neutropenia |
|
Appendix 3 Table of excluded studies with rationale
| Paper excluded | Full reference details | Reason for exclusion |
|---|---|---|
| Aghajanian 2011 | Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. Int J Gynecol Cancer 2011;21:S11 | Comparator (bevacizumab plus gemcitabine plus carboplatin not approved by NICE) |
| Aghajanian 2012a | Aghajanian C, Blank SV, Goff BA, Judson PL, Nycum LR, Sovak MA. An updated safety analysis of OCEANS, a randomized, double-blind, phase III trial of gemcitabine (G) and carboplatin (C) with bevacizumab (BV) or placebo (PL) followed by BV or PL to disease progression (PD) in patients with platinum-sensitive (Plat-S) recurrent ovarian cancer. J Clin Oncol: ASCO annual meeting proceedings 2012;30 | Comparator (bevacizumab plus gemcitabine plus carboplatin not approved by NICE) |
| Aghajanian 2012b | Aghajanian C, Blank SV, Goff BA. An updated safety analysis of OCEANS, a randomized, double-blind, phase III trial of gemcitabine (G) and carboplatin (C) with bevacizumab (BV) or placebo (PL) followed by BV or PL to disease progression (PD) in patients with platinum-sensitive (PS) recurrent ovarian cancer [abstract]. J Clin Oncol: ASCO annual meeting proceedings 2012;30 | Comparator (bevacizumab plus gemcitabine plus carboplatin not approved by NICE) |
| Aghajanian 2012c | Aghajanian C, Makhija S, Rutherford T, Sharma S, Nycum L, Sovak M, et al. Independent radiologic review of OCEANS, a phase III trial of carboplatin, gemcitabine, and bevacizumab or placebo for the treatment of platinum-sensitive, recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. Gynecol Oncol 2012;125:S30–1 | Comparator (bevacizumab plus gemcitabine plus carboplatin not approved by NICE) |
| Aghajanian 2012d | Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol 2012;30:2039–45 | Comparator (bevacizumab plus gemcitabine plus carboplatin not approved by NICE) |
| Alberts 2007 | Alberts DS, Liu PY, Wilczynski S, Clouser M, Lopez A, Lange M, et al. Phase III randomized trial of pegylated liposomal doxorubicin plus carboplatin versus carboplatin in platinum-sensitive patients with recurrent epithelial ovarian or peritoneal carcinoma after failure of initial platinum-based chemotherapy: Southwest Oncology Group Protocol S0200. J Clin Oncol: ASCO annual meeting proceedings 2007;25(1) | Conference abstract of an already identified full publication |
| Alexandre 2012 | Alexandre J, Brown C, Coeffic D, Raban N, Pfisterer J, Maenpaa J, et al. CA-125 can be part of the tumour evaluation criteria in ovarian cancer trials: experience of the GCIG CALYPSO trial. Br J Cancer 2012;106:633–7 | Not RCT |
| Alvarez 2009 | Alvarez RD, Mannel R, Garcia AA, Gallion HH, Lucci J III, Kilgore LC, et al. Fixed-dose rate gemcitabine plus carboplatin in relapsed, platinum-sensitive ovarian cancer patients: results of a three-arm Phase I study. Gynecol Oncol 2009;115:389–95 | Not RCT |
| Andersson 2000 | Andersson H, Boman K, Ridderheim M, Rosenberg P, Sorbe B, Puistola U, et al. An updated analysis of a randomized study of single agent paclitaxel (P) given weekly vs. every 3 weeks to patients (PTS) with ovarian cancer (OV) treated with prior platinum therapy. Proc Am Soc Clin Oncol 2000;19:380a | Conference abstract of an already identified full publication |
| Armstrong 2006 | Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med 2006;354:34–43 | First-line therapy |
| Bamias 2012 | Bamias A, Timotheadou E, Aravantinos G. Randomized, phase III study of carboplatin plus paclitaxel for 8 cycles (CP8) versus carboplatin × 8 cycles plus paclitaxel × 4 cycles (C8P4) in advanced ovarian, fallopian, or primary peritoneal carcinoma. J Clin Oncol: ASCO annual meeting proceedings 2012;30 | First-line therapy |
| Basu 2011 | Basu C. Second line chemotherapy in platinum potentially resistant recurrent epithelial ovarian cancer: experience from Eastern India. Int J Gynecol Cancer 2011;21:99 | Author contacted with a request for additional information; insufficient information to include |
| Bidzinski 2009 | Bidzinski M, Poveda A, Vermorken J, Kaye S, Makhson A, Jagiello-Gruszfeld A, et al. Influence of an independent review on PFS and response assessments in a phase III clinical trial in relapsed ovarian cancer. Eur J Cancer 2009; 7(Suppl.):468 | Not RCT |
| Bokkel Huinink 1996 | Bokkel Huinink W, Gore M, Spaczynski M, Carmichael J, Davison N, Hudson I, et al. Topotecan, a new active drug vs. paclitaxel in advanced epithelial ovarian carcinoma: International Topotecan Study Group Trial. Proc Eur Soc Med Oncol 1996:15:282 | Conference abstract of an already identified full publication |
| Bolis 2004 | Bolis G, Scarfone G, Polverino G, Raspagliesi F, Tateo S, Richiardi G, et al. Paclitaxel 175 or 225 mg per meters squared with carboplatin in advanced ovarian cancer: a randomized trial. J Clin Oncol 2004;22:686–90 | First-line therapy |
| Boman 2010 | Boman K, Colombo N, Runnebaum IB, Vergote I, Gore M, Oaknin A, et al. Tolerability of trabectedin (TR) plus pegylated liposomal doxorubicin (PLD) in platinum sensitive (p-s) vs. platinum resistant (P-R) patients (PTS) with relapsed ovarian cancer. Ann Oncol 2010;21:viii 306 | Not RCT |
| Coleman 2007 | Coleman RL, Gordon A, Barter J, Sun S, Rackoff W, Herzog TJ. Early changes in CA125 after treatment with pegylated liposomal doxorubicin or topotecan do not always reflect best response in recurrent ovarian cancer patients. Oncologist 2007;12:72–8 | Not RCT |
| Colombo 2011 | Colombo N. Efficacy of trabectedin in platinum-sensitive-relapsed ovarian cancer: new data from the randomized OVA-301 study. Int J Gynecol Cancer 2011;21:S12–16 | Not RCT |
| de Jongh 2002 | de Jongh FE, de Wit R, Verweij J, Sparreboom A, van den Bent MJ, Stoter G, et al. Dose-dense cisplatin/paclitaxel: a well-tolerated and highly effective chemotherapeutic regimen in patients with advanced ovarian cancer. Eur J Cancer 2002;38:2005–13 | First-line therapy |
| Diebolder 2010 | Diebolder H, Runnebaum I, Poveda A, Monk BJ, Zintl P, Lehmann-Willenbrock E, et al. Extending platinum-free interval (PFI) in partially platinum-sensitive (PPS) patients (pts) with recurrent ovarian cancer (ROC) treated with trabectedin (Yondelis) plus pegylated liposomal doxorubicin (Caelyx [PLD]) combination versus PLD alone: results from a PPS cohort of the OVA-301 phase III study. Arch Gynecol Obstet 2010;282:S50 | Conference abstract of an already identified full publication |
| Eisenhauer 1997 | Eisenhauer E, Hoskins P, Beare S, Roy M, Drouin P, Stuart G, et al. Randomized phase II study of two schedules of topotecan in previously treated epithelial ovarian cancer. Proc Am Soc Clin Oncol 1997;16:349a | Not in TA9113 |
| Eisenhauer 1997 | Eisenhauer EA, ten Bokkel Huinink WW, Swenerton KD, Gianni L, Myles J, van der Burg ME, et al. European-Canadian randomized trial of paclitaxel in relapsed ovarian cancer: high-dose versus low-dose and long versus short infusion. J Clin Oncol 1994;12:2654–66 | Not in TA9113 |
| Gladieff 2009 | Gladieff L, Lortholary A, Largillier R, Weber B, Alexandre J, Durando X, et al. Weekly paclitaxel (wP) as single agent or in combination with weekly topotecan (wT) or carboplatin (C) in patients with resistant ovarian cancer (ROC): the phase II CARTAXHY randomized trial from GINECO. J Clin Oncol 2009;27:291 | Conference abstract of an already identified full publication |
| Gladieff 2009 | Gladieff L, Lortholary A, Largillier R, Weber B, Alexandre J, Durando X, et al. Weekly paclitaxel (wP) as single agent or in combination with weekly topotecan (wT) or carboplatin (C) in patients with resistant ovarian cancer (ROC): the phase II CARTAXHY randomized trial from GINECO. 45th Annual Meeting of the American Society of Clinical Oncology; Orlando, FL, USA, 29 May to 2 June 2009 | Conference abstract of an already identified full publication |
| Gonzalez-Martin 2003 | Gonzalez-Martin AA, Calvo E, Bover I, Rubio MJ, Arcusa A, Casado A, et al. Randomised phase II study of carboplatin (C) versus paclitaxel-carboplatin (PC) in platinum-sensitive (PS) recurrent advanced ovarian carcinoma (AOC) with assessment of QoL (QoL): a GEICO study (Spanish Group for Investigation on Ovarian Carcinoma). Proc Am Soc Clin Oncol 2003;22:451 | Conference abstract of an already identified full publication |
| Gordon 1998 | Gordon A, Carmichael J, Malfetano J, Gore M, Spaczynski M, Clarke D, et al. Final analysis of a phase III, randomized study of topotecan (T) vs. paclitaxel (P) in advanced epithelial ovarian carcinoma (Oc): International Topotecan Study Group. Proc Ann Meet Am Soc Clin Oncol 1998 | Conference abstract of an already identified full publication |
| Gordon 2002 | Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore M, Lacave AJ, Mutch D. Interim analysis of a phase III randomized trial of Doxil/Caelyx (D) versus topotecan (T) in the treatment of patients with relapsed ovarian cancer. Proc Am Soc Clin Oncol 2000;19:380a | Conference abstract of an already identified full publication |
| Gordon 2003 | Gordon A, Teitelbaum A. Overall survival advantage for pegylated liposomal doxorubicin compared to topotecan in recurrent epithelial ovarian cancer. Eur J Cancer 2003;1:S51 | Unobtainable |
| Gordon 2006 | Gordon A, Sun S, Rackoff W. Incidence of adverse events in women (≤ 65 or > 65 years) with recurrent ovarian cancer receiving pegylated liposomal doxorubicin or topotecan. Gynecol Oncol 2006;101:S59–60 | Conference abstract of an already identified full publication |
| Gore 1998 | Gore M, Rustin G, Calvert H, Bezwoda W, Carmichael J, Oza A, et al. A multicentre, randomised, phase III study of topotecan (T) administered intravenously or orally for advanced epithelial ovarian carcinoma. Proc Ann Meet Am Soc Clin Oncol 1998;17:349a | Conference abstract of an already identified full publication |
| Greimel 2006 | Greimel ER, Bjelic-Radisic V, Pfisterer J, Hilpert F, Daghofer F, du Bois A. Randomized study of the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group comparing quality of life in patients with ovarian cancer treated with cisplatin/paclitaxel versus carboplatin/paclitaxel. J Clin Oncol 2006;24:579–86 | First-line therapy |
| Herzog 2011 | Herzog TJ, Sill MW, Walker JL, O’Malley D, Shahin M, Degeest K, et al. A phase II study of two topotecan regimens evaluated in recurrent platinum-sensitive ovarian, fallopian tube or primary peritoneal cancer: a Gynecologic Oncology Group study (GOG 146Q). Gynecol Oncol 2011;120:454–8 | Not RCT |
| Hoskins 1998 | Hoskins P, Eisenhauer E, Beare S, Roy M, Drouin P, Stuart G, et al. Randomized phase II study of two schedules of topotecan in previously treated patients with ovarian cancer: a National Cancer Institute of Canada Clinical Trials Group study. J Clin Oncol 1998;16:2233–7 | Not in TA9113 |
| Isonishi 2008 | Isonishi S, Yasuda M, Takahashi F, Katsumata N, Kimura E, Aoki D, et al. Randomized phase III trial of conventional paclitaxel and carboplatin (c-TC) versus dose dense weekly paclitaxel and carboplatin (dd-TC) in women with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer: Japanese Gynecologic Oncology. J Clin Oncol: ASCO annual meeting proceedings 2008;26:29 | Unobtainable |
| Isonishi 2008 | Isonishi S, Yasuda M, Takahashi F, Katsumata N, Kimura E, Aoki D, et al. Randomized phase III trial of conventional paclitaxel and carboplatin (c-TC) versus dose dense weekly paclitaxel and carboplatin (dd-TC) in women with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer: Japanese Gynecologic Oncology. J Clin Oncol: ASCO annual meeting proceedings 2008;26:294. 44th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, 30 May to 3 June 2008 | Unobtainable |
| Katsumata 2009 | Katsumata N, Yasuda M, Takahashi F, Isonishi S, Jobo T, Aoki D, et al. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: a phase 3, open-label, randomised controlled trial. Lancet 2009;374:1331–8 | First-line therapy |
| Katsumata 2012 | Katsumata N, Yasuda M, Isonishi S. Long-term follow-up of a randomized trial comparing conventional paclitaxel and carboplatin with dose-dense weekly paclitaxel and carboplatin in women with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer: JGOG 3016 trial. J Clin Oncol: ASCO annual meeting proceedings 2012;30 | First-line therapy |
| Krasner 2009 | Krasner CN, Poveda A, Herzog T, Vermorken J, Monk B, Zintl P, et al. Health-related quality of life/patient-reported outcomes in relapsed ovarian cancer: results from a randomized phase III study of trabectedin with pegylated doxorubicin (PLD) versus PLD alone. J Clin Oncol: ASCO annual meeting proceedings 2009;27 | Conference abstract of an already identified full publication |
| Ledermann 2003a | Ledermann JA. Randomised trial of paclitaxel in combination with platinum chemotherapy versus platinum-based chemotherapy in the treatment of relapsed ovarian cancer (ICON4/OVAR 2.2). Br J Cancer 2003;88:S9CT2 | Not RCT |
| Ledermann 2003b | Ledermann JA. Randomized trial of paclitaxel in combination with platinum chemotherapy versus platinum/based chemotherapy in the treatment of relapsed ovarian cancer (ICON4/OVAR 2.2). Proc Am Soc Clin Oncol 2003;22:603 | Not RCT |
| Lehmann-Willenbrock 2010 | Lehmann-Willenbrock E, Runnebaum I, Nieto A, Poveda A, Monk BJ, De La Riba MI, et al. Extending platinum-free interval (PFI) in partially platinum-sensitive (PPS) patients (pts) with recurrent ovarian cancer (ROC) treated with trabectedin (Yondelis) plus pegylated liposomal doxorubicin (Caelyx[PLD]) combination versus PLD alone: results from a PPS cohort of the OVA-301 phase III study. Onkologie 2010;33:190 | Conference abstract of an already identified full publication |
| Luck 2010 | Luck HJ, Jackisch C, Schmalfeldt B, Stahle A, Burges A, Kurzeder C, et al. Ovar 2.9: a phase III study comparing PLD-doxorubicine-carboplatin (CD) with carboplatin-paclitaxel (CP) in recurrent platin-sensible ovarian cancer. A GCIG study. Arch Gynecol Obstet 2010;282:S129–30 | Conference abstract of an already identified full publication |
| Mahner 2011 | Mahner S, Meier W, du Bois A. Carboplatin and pegylated liposomal doxorubicin versus carboplatin and paclitaxel in very platinum-sensitive ovarian cancer patients: result from a subset analysis of the CALYPSO phase III GCIG trial. J Clin Oncol: ASCO annual meeting proceedings 2011;29 | Conference abstract of an already identified full publication |
| Markman 2007 | Markman M. Re: ‘Randomized phase III trial of three versus six cycles of adjuvant carboplatin and paclitaxel in early stage epithelial ovarian carcinoma: a Gynecologic Oncology Group study [letter]. Gynecol Oncol 2007;105:279–80 | Not RCT |
| Marth 2011 | Marth C, Alexandre J, Hanker LC. Pegylated liposomal doxorubicin and carboplatin (C-PLD) versus paclitaxel and carboplatin (C-P) in platinum-sensitive ovarian cancer (OC) patients (pts): treatment at recurrence and overall survival (OS) final analysis from CALYPSO Phase III GCIG trial. J Clin Oncol: ASCO annual meeting proceedings 2011;29 | Conference abstract of an already identified full publication |
| Meden 2000 | Meden H. Cisplatin/Paclitaxel vs. Carboplatin/Paclitaxel in ovarian cancer FIGO IIB-IV: update of an AGO (Arbeitsgemeinschaft Gynaenkologischer Onkologie) Study Group trial (OVAR-3). J Cancer Res Clin Oncol 2000;126(Suppl. 1):R55 | First-line therapy |
| Meier 1999 | Meier W, du Bois A, Olbricht S, Nitz U, Jackisch C, Richter B, et al. Cisplatin/paclitaxel vs. carboplatin/paclitaxel in ovarian cancer: results of a prospective randomized phase III study. Int J Gynecol Cancer 1999;9:48A146 | First-line therapy |
| Monk 2011 | Monk BJ, Herzog TJ, Kaye SB. Final survival results of the randomized phase III study of trabectedin with pegylated liposomal doxorubicin (PLD) versus PLD in recurrent ovarian cancer. Clin J Oncol: ASCO annual meeting proceedings 2011;29 | Conference abstract of an already identified full publication |
| Muggia 1997 | Muggia FM, Braly PS, Brady MF, Sutton G, Copeland LJ, Lentz SL, et al. Phase III of cisplatin or paclitaxel versus their combination in suboptimal stage III and IV epithelial ovarian cancer: Gynecologic Oncology Group study #132. Proc Am Soc Clin Oncol 1997;16:352a | First-line therapy |
| Muggia 2000 | Muggia FM, Braly PS, Brady MF, Sutton G, Niemann TH, Lentz, SL, et al. Phase III randomized study of cisplatin versus paclitaxel versus cisplatin and paclitaxel in patients with suboptimal stage III or IV ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 2000;18:106–15 | First-line therapy |
| Pfisterer 2004a | Pfisterer J, Plante M, Vergote I, du Bois A, Wagner U, Hirte H, et al. Gemcitabine/carboplatin vs. carboplatin in platinum sensitive recurrent ovarian cancer. Results of a Gynecologic Cancer Intergroup randomized phase III trial of the AGO OVAR, the NCIC CTG and the EORTC GCG. J Clin Oncol 2004;22(Suppl. 14) | Conference abstract of an already identified full publication |
| Pfisterer 2004b | Pfisterer J, Plante M, Vergote I, du Bois A, Wagner U, Hirte H, et al. Gemcitabine/carboplatin (GC) vs. carboplatin (C) in platinum sensitive recurrent ovarian cancer (OVCA). Results of a Gynaecologic Cancer Intergroup randomized phase III trial of the AGO OVAR, the NCIC CTG and the EORTC GCG. Ann Meet Proc Am Soc Clin Oncol 2004;449 | Conference abstract of an already identified full publication |
| Piccart 1998a | Piccart-Gebhart M, Green J, Lacave A, Benedetti-Panici P, Reed N, Vergote I, et al. A randomized phase II study of taxol or oxaliplatin in platinum-pretreated epithelial ovarian cancer patients. Proc Am Soc Clin Oncol 1998;17:349a | Conference abstract of an already identified full publication |
| Piccart 1998b | Piccart-Gebhart M, Green J, Lacave A, Benedetti-Panici P, Reed, N, Vergote I, et al. A randomized phase II study of taxol or oxaliplatin in platinum-pretreated epithelial ovarian cancer patients. Proc Ann Meet Am Soc Clin Oncol 1998;17 | Conference abstract of an already identified full publication |
| Poveda 2010a | Poveda A, Tjulandin S, Kong B, Roy M, Chan S, Filipczyk-Cisarz E, et al. Extending platinum-free interval (PFI) in partially platinum-sensitive (PPS) patients (pts) with recurrent ovarian cancer (ROC) treated with trabectedin (Tr) plus pegylated liposomal doxorubicin (Tr+PLD) versus PLD alone: results from a PPS cohort of a phase III study. J Clin Oncol 2010;28 | Conference abstract of an already identified full publication |
| Poveda 2010b | Poveda A, Tjulandin S, Kong B, Roy M, Chan S. Extending platinum-free interval (PFI) in partially platinum-sensitive (PPS) patients (pts) with recurrent ovarian cancer (ROC) treated with trabectedin (Tr) plus pegylated liposomal doxorubicin (Tr + PLD) versus PLD alone: results from a PPS cohort of a phase III study. J Clin Oncol: ASCO annual meeting proceedings 2010;28 | Conference abstract of an already identified full publication |
| Pujade-Lauraine 2009 | Pujade-Lauraine E, Mahner S, Kaern J, Gebski V, Heywood M, Vasey P, et al. A randomized phase III study of carboplatin and pegylated liposomal doxorubicin versus carboplatin and paclitaxel in relapsed platinum-sensitive ovarian cancer (OC): CALYPSO study of the Gynecologic Cancer Intergroup (GCIG). J Clin Oncol: ASCO annual meeting proceedings 2009;27 | Conference abstract of an already identified full publication |
| Rosenberg 1999 | Rosenberg P, Andersson H, Boman K, Ridderheim M, Sorbe B, Puistola U, et al. A randomized multicenter study of single agent paclitaxel (TAXOL©) given weekly versus every three weeks to patients (PTS) with ovarian cancer (OC) previously treated with platinum therapy. Proc Am Soc Clin Oncol 1999;18:368a | Conference abstract of an already identified full publication |
| Ross 2001 | Ross G, Lane S, Dane G. Long term survival in a phase III randomised study of topotecan (T) vs. paclitaxel (P) in advanced epithelial ovarian carcinoma. Eur J Cancer 2001;37(Suppl. 6):S326 | Unobtainable |
| Runnebaum 2010 | Runnebaum IB, Poveda A, Hagberg H, Lebedinsky C, Zintl P, Hossfeld M, et al. Extending platinum-free interval (PFI) in partially platinum-sensitive (PPS) patients (pts) with recurrent ovarian cancer (ROC) treated with trabectedin (Tr) plus pegylated liposomal doxorubicin (Tr + PLD) versus PLD alone: results from a PPS cohort of a phase III study. Arch Gynecol Obstet 2010;282:S116O | Conference abstract of an already identified full publication |
| Runnebaum 2011 | Runnebaum I, Sehouli J, Gebauer G, Lehmann-Willenbrock E, Schutte J, Zieger W, et al. Trabectedin + PLD significantly prolongs survival in platinum sensitive + partially platinum sensitive relapsed ovarian cancer (ROC) patients in comparison to PLD alone. Onkologie 2011;34:222 | Conference abstract of an already identified full publication |
| Scarfone 2001 | Scarfone G, Parazzini F, Sciatta C, Rabaiotti E, Richiardi G, Tateo S, et al. A multicenter randomized trial comparing two different doses of TAXOL (T) plus a fixed dose of carboplatin (C) in advanced ovarian cancer (AOC). Proc Am Soc Clin Oncol 2001;20(1):205a | First-line therapy |
| Scarfone 2006 | Scarfone G, Presti M, Scarabelli C, Polverino GP, Polonio N, Bertoglio S. Pegylated liposomal doxorubicin alone or in combination with platinum compounds in recurrent ovarian cancer after first line chemotherapy containing paclitaxel and carboplatin. Int J Gynecol Cancer 2006;16:668 | Abstract only; insufficient information |
| Sehouli 2007 | Sehouli J, Oskay-Oezcelik G, Stengel D, du Bois A, Markmann S, Loibl S, et al. Topotecan weekly versus routine 5-day schedule in patients with platinum-resistant ovarian cancer (TOWER): a randomized, two-stage phase II study of the North-Eastern German Society of Gynaecological Oncology (NOGGO). J Clin Oncol: ASCO annual meeting proceedings 2007;25(1) | Conference abstract of an already identified full publication |
| Sehouli 2009a | Sehouli J, Oskay-Oezcelik G, Stengel D, Harter D, Kurzeder C, Belau A, et al. Topotecan weekly versus routine 5-day schedule in patients with platinum-resistant ovarian cancer (TOWER): a randomized, multicenter trial of the North-Eastern German Society of Gynaecological Oncology (NOGGO). J Clin Oncol 2009;27:290 | Conference abstract of an already identified full publication |
| Sehouli 2009b | Sehouli J, Oskay-Oezcelik G, Stengel D, Harter P, Kurzeder C, Belau A, et al. Topotecan weekly versus routine 5-day schedule in patients with platinum-resistant ovarian cancer (TOWER): a randomised multicenter trial of the North-Eastern German Society of Gynecological Oncology (NOGGO). J Clin Oncol: ASCO annual meeting proceedings 2009;27: Abstract. 45th Annual Meeting of the American Society of Clinical Oncology, Orlando, Florida, USA, 29 May to 2 June 2009 | Conference abstract of an already identified full publication |
| Spriggs 2004 | Spriggs DR, Brady M, Rubin S, Hanley M, Copeland LJ, Clarke-Pearson D, et al. A phase III randomised trial of cisplatin and paclitaxel administered by either 24 hour or 96 hour infusion in patients with selected stage III or stage IV epithelial ovarian cancer (GOG162). Proc Am Soc Clin Oncol 2004;23:449 | First-line therapy |
| Spriggs 2007 | Spriggs DR, Brady MF, Vaccarello L, Clarke-Pearson DL, Burger RA, Mannel R, et al. Phase III randomised trial of intravenous cisplatin plus a 24- or 96-hour infusion of paclitaxel in epithelial ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 2007;25:4466–71 | First-line therapy |
| Swenerton 1993 | Swenerton K, Eisenhauer E, Bokkel Huinink W, Myles J, Mangioni C, Burg M, et al. Taxol in relapsed ovarian cancer: high vs. low dose and short vs. long infusion: a European-Canadian study coordinated by the NCI Canada Clinical Trials Group. Proc Am Soc Clin Oncol 1993;12:256 | Unobtainable |
| ten Bokkel Huinink 1993 | ten Bokkel Huinink WW, Eisenhauer E, Swenerton K. Preliminary evaluation of a multicenter, randomized comparative study of TAXOL (paclitaxel) dose and infusion length in platinum-treated ovarian cancer. Canadian-European Taxol Cooperative Trial Group. Cancer Treat Rev 1993;19:79–86 | Not in TA9113 |
| Avall-Lundqvist 2008 | Avall-Lundqvist E, Wimberger P, Gladieff L, Gebski V, Huober JB, Floquet A, et al. Pegylated liposomal doxorubicin (PLD)-carboplatin (C) (C-D) in relapsing sensitive ovarian cancer (OC): a 500-patient interim safety analysis of the CALYPSO GCIG Intergroup phase III study. J Clin Oncol: ASCO annual meeting proceedings 2008;26 | Conference abstract of an already identified full publication |
| Vasey 2009 | Vasey P, Largillier R, Gropp M, Gebski V, Sandvei R, Elit L, et al. A GCIG randomized phase III study of carboplatin (C) & pegylated liposomal doxorubicin (PLD) (C-D) vs. carboplatin (C) & paclitaxel (P) (C-P): CALYPSO results in partially platinum-sensitive ovarian cancer (OC) patients. Eur J Cancer 2009;7(Suppl. 1):11 | Conference abstract of an already identified full publication |
| Vergote 2004 | Vergote I, Plante M, Richter B, Emmerich J, Hirte H, Costa S, et al. Improved progression-free survival (PFS) and quality of life (QOL) in a randomized study comparing gemcitabine/carboplatinum (GC) vs. carboplatin (C) in platinum sensitive ovarian cancer (OVCA). Int J Gynecol Cancer 2004;14(Suppl. 1):45–6 | Abstract only; insufficient information to include |
| Vergote 2007 | Vergote I, Finkler N, Campo J, Lohr A, Hunter J, Matei D, et al. Single agent, canfosfamide (C, TLK286) vs. pegylated liposomal doxorubicin or topotecan in 3rd-line treatment of platinum refractory or resistant ovarian cancer: phase III study results. J Clin Oncol: ASCO annual meeting proceedings 2007;25(1) | Incorrect comparator; results for PLDH and topotecan not reported separately |
| Vergote 2009 | Vergote I, Finkler N, del Campo J, Lohr A, Hunter J, Matei D, et al. Study Group Phase 3 randomised study of canfosfamide (Telcyta, TLK286) versus pegylated liposomal doxorubicin or topotecan as third-line therapy in patients with platinum-refractory or -resistant ovarian cancer. Eur J Cancer 2009;45:2324–32 | Incorrect comparator; results for PLDH and topotecan not reported separately |
| Vermorken 2001 | Vermorken J, Gore M, Perren T, Vergote I, Colombo N, Harper P, et al. Multicenter randomized phase II study of oxaliplatin (OXA) or topotecan (TOPO) in platinum-pretreated epithelial ovarian cancer (EOC) patients (pts). Proc Am Soc Clin Oncol 2001;20(1):212a | Abstract only; insufficient information to include |
Appendix 4 Networks for the adverse effects network meta-analysis
All potential links are displayed in the networks. In some cases, zero events may have precluded analysis.
FIGURE 54.
Allergic reaction.
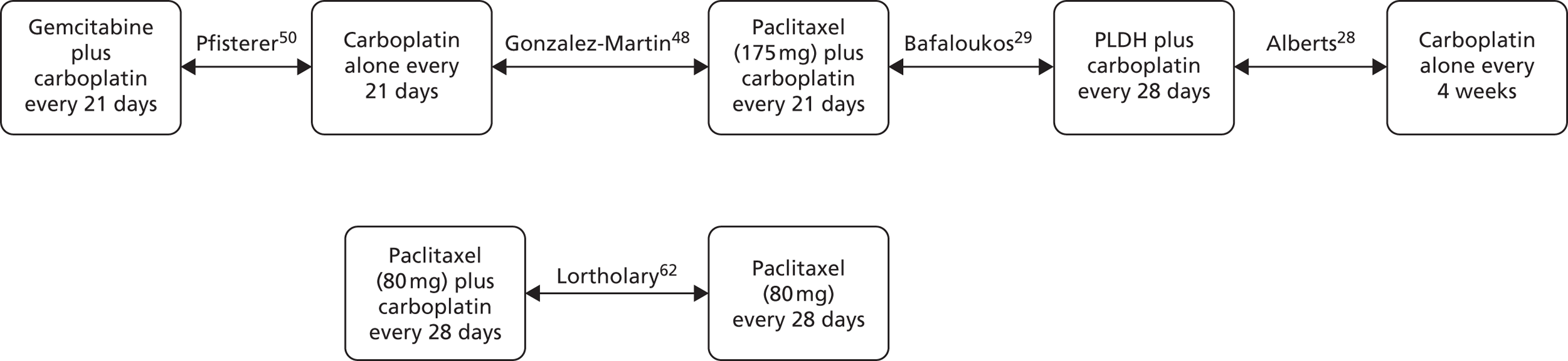
FIGURE 55.
Alopecia.

FIGURE 56.
Anaemia.
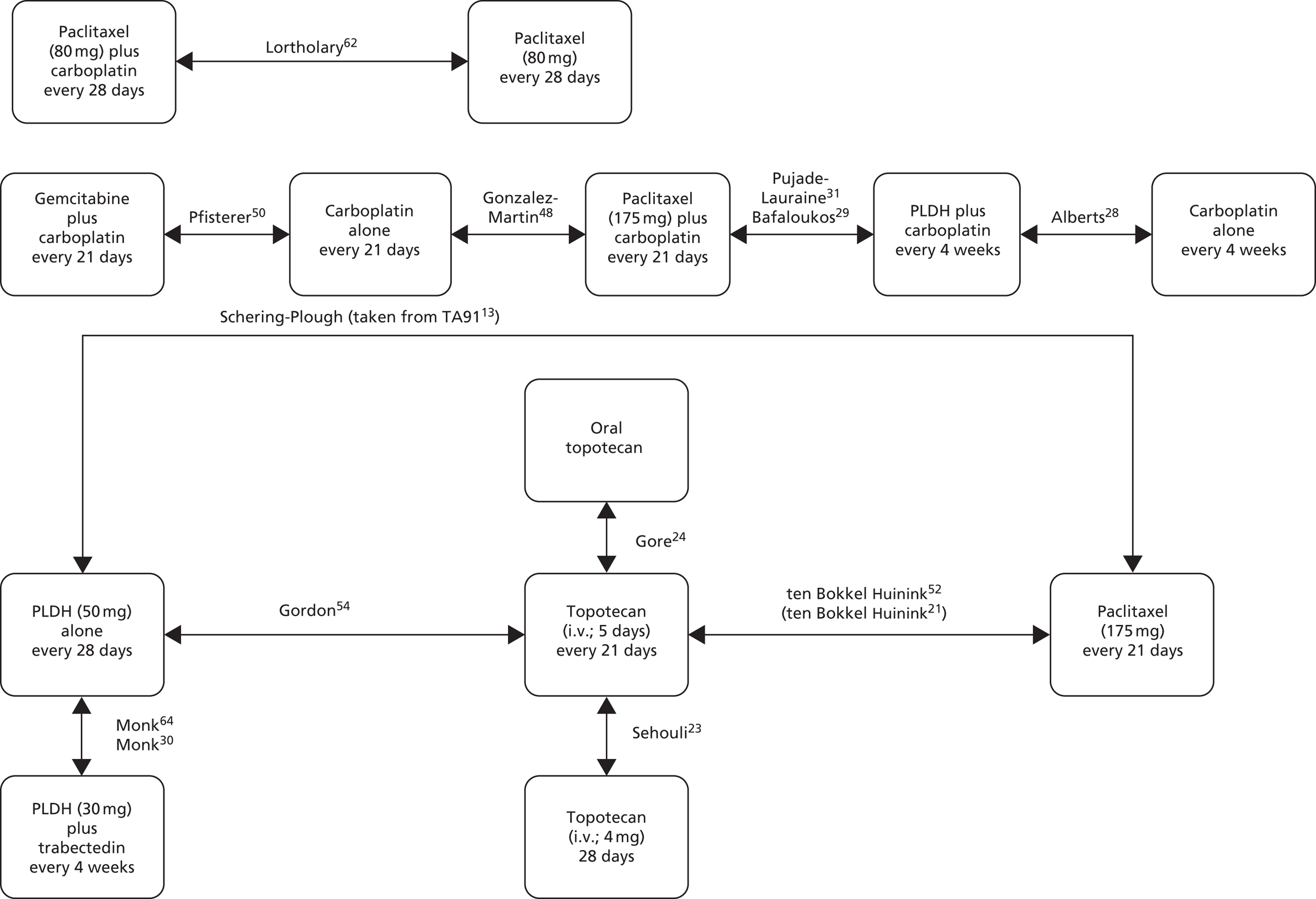
FIGURE 57.
Fatigue.

FIGURE 58.
Febrile neutropenia.
FIGURE 59.
Nausea and vomiting.
FIGURE 60.
Neuropathy.

Appendix 5 Literature search strategies for Technology Assessment Group economic evaluation
Economic evaluation searches
Databases searched: Ovid MEDLINE In-Process & Other Non-Indexed Citations and Ovid MEDLINE
Date range searched: 1946 to present.
Date of searched: initially searched 4 December 2012 and updated 23 May 2013.
| # | Terms | Hits (4 December 2012) | Hits (23 May 2013) |
|---|---|---|---|
| 1 | ovarian neoplasms/ | 57,969 | 58,587 |
| 2 | exp ovarian neoplasms/ | 60,059 | 60,739 |
| 3 | (ovar$ adj4 (cancer$ or tumo?r$ or malignan$)).ti. | 27,883 | 28,450 |
| 4 | (ovar$ adj4 (cancer$ or tumo?r$ or malignan$)).ab. | 41,322 | 42,450 |
| 5 | (ovar$ adj4 (oncolog$ or carcinoma$)).ab. | 12,353 | 12,521 |
| 6 | or/1-5 | 74,774 | 75,993 |
| 7 | Topotecan/ | 1704 | 1725 |
| 8 | topotecan.mp. | 2446 | 2482 |
| 9 | hycamtin.mp. | 70 | 69 |
| 10 | or/7-9 | 2447 | 2483 |
| 11 | exp Doxorubicin/ | 40,241 | 41,211 |
| 12 | doxil.mp. | 281 | 290 |
| 13 | (doxorubicin hydrochloride or doxorubicin hcl).mp. | 562 | 585 |
| 14 | liposomal doxorubicin.mp. | 1287 | 1354 |
| 15 | (caelyx or adriamycin or rubex).mp. | 13,911 | 14,054 |
| 16 | liposome encapsulated doxorubicin.mp. | 88 | 88 |
| 17 | (PLDH or pegylated liposomal doxorubicin hydrochloride).mp | 143 | 163 |
| 18 | or/11-17 | 44,840 | 45,884 |
| 19 | paclitaxel/ | 17,785 | 18,248 |
| 20 | paclitaxel.mp. | 22,571 | 23,229 |
| 21 | taxol.mp. or abraxane.mp | 6031 | 6155 |
| 22 | or/19-21 | 24,178 | 24,878 |
| 23 | carboplatin/ | 8360 | 8580 |
| 24 | (carboplatin or paraplatin).mp. | 11,703 | 12,003 |
| 25 | or/23-24 | 11,703 | 12,003 |
| 26 | cisplatin/ | 37,783 | 38,561 |
| 27 | cisplatin.mp. | 50,320 | 51,546 |
| 28 | or/26-27 | 50,320 | 51,546 |
| 29 | 10 or 18 or 22 or 25 or 28 | 112,024 | 114,904 |
| 30 | Limit 29 to yr = 2004-2012 (2013) | 46,970 | 49,826 |
| 31 | gemcitabine.mp. | 9065 | 9504 |
| 32 | gemzar.mp | 212 | 216 |
| 33 | or/31-32 | 9078 | 9520 |
| 34 | Trabectedin.mp | 388 | 396 |
| 35 | ecteinascidin 743.mp. | 131 | 131 |
| 36 | ET-743.mp. | 171 | 174 |
| 37 | yondelis.mp | 96 | 93 |
| 38 | or/34-37 | 432 | 442 |
| 39 | bevacizumab.mp | 7354 | 7994 |
| 40 | avastin.mp | 927 | 947 |
| 41 | or/39-40 | 7430 | 8069 |
| 42 | etoposide.mp | 19,804 | 20,237 |
| 43 | Eposin.mp | 0 | 0 |
| 44 | or/42-43 | 19,804 | 20,237 |
| 45 | (best supportive care).mp | 974 | 1003 |
| 46 | 33 or 38 or 41 or 44 or 45 | 36,770 | 38,290 |
| 47 | 30 or 46 | 76,853 | 80,860 |
| 48 | economics/ | 26,664 | 26,636 |
| 49 | exp costs/ and cost analysis/ | 40,385 | 40,679 |
| 50 | exp economics, hospital/ | 18,425 | 18,679 |
| 51 | economics, medical/ | 8511 | 8501 |
| 52 | economics, pharmaceutical/ | 2387 | 2442 |
| 53 | (economic$ or pharmaeconomic$ or pharmacoeconomic$ or pharmaco-economic$).tw. | 138,434 | 144,495 |
| 54 | (cost or costs or costly or costing or costed).tw. | 294,675 | 306,620 |
| 55 | value for money.tw. | 857 | 869 |
| 56 | cost utility.mp. | 2172 | 2212 |
| 57 | cost effectiveness/ | 56,140 | 56,826 |
| 58 | cost benefit/ | 56,140 | 56,826 |
| 59 | cost consequence.mp. | 107 | 108 |
| 60 | cost minimi*ation.mp. | 781 | 803 |
| 61 | economic evaluation.mp. | 4598 | 4683 |
| 62 | Or/48-61 | 465,315 | 482,157 |
| 63 | 6 and 47 and 62 | 74 | 71 |
| 64 | limit 63 to ed = 20121201-20130523 | NA | 2 |
Database searched: EMBASE
Date range searched: 1974 to present.
Date of searched: initially searched 4 December 2012 and updated 23 May 2013.
| # | Terms | Hits (4 December 2012) | Hits (23 May 2013) |
|---|---|---|---|
| 1 | exp Ovary Cancer/ | 65,122 | 67,668 |
| 2 | (ovar$ adj4 (cancer$ or tumo?r$ or malignan$)).ti. | 34,811 | 35,910 |
| 3 | (ovar$ adj4 (cancer$ or tumo?r$ or malignan$)).ab. | 51,886 | 53,822 |
| 4 | (ovar$ adj4 (oncolog$ or carcinoma$)).ab. | 15,047 | 15,442 |
| 5 | or/1-4 | 90,905 | 94,505 |
| 6 | Topotecan/ | 7884 | 8187 |
| 7 | topotecan.mp. | 8124 | 8437 |
| 8 | hycamtin.mp. | 581 | 591 |
| 9 | or/6-8 | 8124 | 8437 |
| 10 | exp Doxorubicin/ | 125,207 | 129,170 |
| 11 | doxil.mp. | 1521 | 1619 |
| 12 | (doxorubicin hydrochloride or doxorubicin hcl).mp. | 645 | 674 |
| 13 | liposomal doxorubicin.mp. | 1867 | 1983 |
| 14 | (caelyx or adriamycin or rubex).mp. | 24,081 | 24,468 |
| 15 | liposome encapsulated doxorubicin.mp. | 107 | 112 |
| 16 | (PLDH or pegylated liposomal doxorubicin hydrochloride).mp. | 220 | 247 |
| 17 | or/10-16 | 127,421 | 131,439 |
| 18 | paclitaxel/ | 57,308 | 60,283 |
| 19 | paclitaxel.mp. | 59,414 | 62,489 |
| 20 | (taxol or abraxane).mp. | 11,748 | 12,030 |
| 21 | or/18-20 | 60,485 | 63,583 |
| 22 | carboplatin/ | 38,672 | 40,505 |
| 23 | (carboplatin or paraplatin).mp. | 39,961 | 41,870 |
| 24 | or/22-23 | 39,961 | 41,870 |
| 25 | cisplatin/ | 112,665 | 116,858 |
| 26 | cisplatin.mp. | 117,604 | 121,966 |
| 27 | or/25-26 | 117,604 | 121,966 |
| 28 | 9 or 17 or 21 or 24 or 27 | 255,147 | 264,864 |
| 29 | limit 28 to yr = 2004-2012 (2013) | 130,702 | 140,463 |
| 30 | gemcitabine.mp. | 28,137 | 29,972 |
| 31 | gemzar.mp. | 1706 | 1751 |
| 32 | or/30-31 | 28,148 | 29,985 |
| 33 | Trabectedin.mp. | 1198 | 1267 |
| 34 | ecteinascidin 743.mp. | 178 | 181 |
| 35 | ET-743.mp. | 477 | 490 |
| 36 | yondelis.mp. | 329 | 344 |
| 37 | or/33-36 | 1224 | 1293 |
| 38 | bevacizumab.mp. | 24,620 | 27,022 |
| 39 | avastin.mp. | 6598 | 6921 |
| 40 | or/38-39 | 24,651 | 27,054 |
| 41 | etoposide.mp. | 62,570 | 64,348 |
| 42 | Eposin.mp. | 20 | 20 |
| 43 | or/41-42 | 62,576 | 64,354 |
| 44 | best supportive care.mp. | 1624 | 1786 |
| 45 | 32 or 37 or 40 or 43 or 44 | 107,900 | 113,504 |
| 46 | 29 or 45 | 200,564 | 213,305 |
| 47 | economics/ | 207,721 | 209,851 |
| 48 | exp costs/ and cost analysis/ | 16,393 | 16,842 |
| 49 | exp economics, hospital/ | 567,261 | 584,236 |
| 50 | economics, medical/ | 32,131 | 32,624 |
| 51 | economics, pharmaceutical/ | 5762 | 5828 |
| 52 | (economic$ or pharmaeconomic$ or pharmacoeconomic$ or pharmaco-economic$).tw. | 176,979 | 184,758 |
| 53 | (cost or costs or costly or costing or costed).tw. | 378,322 | 395,583 |
| 54 | value for money.tw. | 1152 | 1213 |
| 55 | cost utility.mp. | 5779 | 6082 |
| 56 | cost effectiveness/ | 84,693 | 88,469 |
| 57 | cost benefit/ | 62,729 | 64,078 |
| 58 | cost consequence.mp. | 166 | 173 |
| 59 | cost minimi*ation.mp. | 2723 | 2824 |
| 60 | economic evaluation.mp. | 11,827 | 12,399 |
| 61 | or/47-60 | 981,563 | 1,014,510 |
| 62 | 4 and 46 and 61 | 633 | 712 |
| 65 | limit 62 to em = 201247-201321 | NA | 77 |
Database searched: HTA database
URL: www.crd.york.ac.uk/CRDWeb/
Initially searched 4 December 2012 and updated 23 May 2013.
| Date of search | 4 December 2012 | 21 May 2013 |
|---|---|---|
| Search terms (and fields searched) | Ovarian neoplasm (all fields) Ovarian cancer (all fields) Ovary cancer (all fields) |
Ovarian neoplasm (all fields) Ovarian cancer (all fields) Ovary cancer (all fields) Limit 4 December 2012 to 21 May 2013 |
| No. of hits | 65 | 5 |
Database searched: NHS Economic Evaluation Database
URL: www.crd.york.ac.uk/CRDWeb/
Initially searched 4 December 2012 and updated 23 May 2013.
| Date of search | 4 December 2012 | 21 May 2013 |
|---|---|---|
| Search terms (and fields searched) | Ovarian neoplasm (all fields) Ovarian cancer (all fields) Ovary cancer (all fields) |
Ovarian neoplasm (all fields) Ovarian cancer (all fields) Ovary cancer (all fields) Limit 4 December 2012 to 21 May 2013 |
| No. of hits | 70 | 7 |
Health-related quality-of-life searches
Databases searched: Ovid MEDLINE In-Process & Other Non-Indexed Citations and Ovid MEDLINE
Date range searched: 1946 to present.
Date of searched: initially searched 4 December 2012 and updated 23 May 2013.
| # | Terms | Hits (4 December 2012) | Hits (23 May 2013) |
|---|---|---|---|
| 1 | exp Ovarian Neoplasms/ | 60,059 | 60,739 |
| 2 | (ovar$ adj4 (cancer$ or tumo?r$ or malignan$ or oncolog$ or carcinoma$ or neoplas$ or mass$ or growth$ or cyst$)).mp. | 81,853 | 83,124 |
| 3 | (adenexa$ adj4 mass$).mp. | 7 | 7 |
| 4 | or/1-3 | 83,643 | 84,962 |
| 5 | animal/ not (animal/ and human/) | 3,720,388 | 3,757,872 |
| 6 | 4 not 5 | 78,554 | 79,809 |
| 7 | exp Life Tables/ | 12,317 | 12,163 |
| 8 | exp “Quality of Life”/ | 104,747 | 108,376 |
| 9 | Health Status/ | 54,169 | 55,687 |
| 10 | exp Health Status Indicators/ | 177,713 | 182,827 |
| 11 | (utilit$ approach$ or health gain or hui or hui2 or hui 2 or hui3 or hui 3).ti,ab. | 1287 | 1344 |
| 12 | (health measurement$ scale$ or health measurement$ questionnaire$).ti,ab. | 39 | 39 |
| 13 | (standard gamble$ or categor$ scal$ or linear scal$ or linear analog$ or visual scal$ or magnitude estmat$).ti,ab. | 3223 | 3312 |
| 14 | (time trade off$ or rosser$ classif$ or rosser$ matrix or rosser$ distress$ or hrqol).ti,ab. | 6670 | 7134 |
| 15 | (index of wellbeing or quality of wellbeing or qwb).ti,ab. | 165 | 166 |
| 16 | (rating scale$ or multiattribute$ health ind$ or multi attribute$ health ind$).ti,ab. | 31,065 | 32,283 |
| 17 | (health utilit$ index or health utilit$ indices).ti,ab. | 582 | 620 |
| 18 | (multiattribute$ theor$ or multi attribute$ theor$ or multiattribute$ analys$ or multi attribute$ analys$).ti,ab. | 9 | 9 |
| 19 | (health utilit$ scale$ or classification of illness state$ or 15d or 15 d or 15 dimension).ti,ab. | 3303 | 3393 |
| 20 | (health state$ utilit$ or 12d or 12 d or 12 dimension).ti,ab. | 2279 | 2350 |
| 21 | well year$.ti,ab. | 22 | 21 |
| 22 | (multiattribute$ utilit$ or multi attribute$ utilit$).ti,ab. | 173 | 179 |
| 23 | health utilit$ scale$.ti,ab. | 8 | 9 |
| 24 | (qol or 5d or 5-d or 5 dimension or quality of life or eq-5d or eq5d or eq 5d or euroqol).ti,ab. | 139,143 | 145,531 |
| 25 | (qualy or qaly or qualys or qalys or quality adjusted life year$).ti,ab. | 6089 | 6240 |
| 26 | life year$ gain$.ti,ab. | 1573 | 1613 |
| 27 | willingness to pay.ti,ab. | 1978 | 2039 |
| 28 | (hye or hyes or health$ year$ equivalent$).ti,ab. | 62 | 62 |
| 29 | (person trade off$ or person tradeoff$ or time tradeoff$ or time trade off$).ti,ab. | 915 | 942 |
| 30 | theory utilit$.ti,ab. | 7 | 7 |
| 31 | life table$.ti,ab. | 7420 | 7166 |
| 32 | health state$.ti,ab. | 3326 | 3467 |
| 33 | (sf36 or sf 36).ti,ab. | 11,840 | 12,389 |
| 34 | (short form 36 or shortform 36 or sf thirtysix or sf thirty six or shortform thirtysix or shortform thirty six or short form thirtysix or short form thirty six).ti,ab. | 5504 | 5736 |
| 35 | (6d or 6-d or 6 dimension).ti,ab. | 5207 | 5553 |
| 36 | or/7-35 | 430,827 | 444,856 |
| 37 | 6 and 36 | 1518 | 1539 |
| 38 | letter.pt. | 785,671 | 794,959 |
| 39 | editorial.pt. | 322,998 | 330,055 |
| 40 | comment.pt. | 527,227 | 538,874 |
| 41 | or/38-40 | 1,223,799 | 1,246,592 |
| 42 | 37 not 41 | 1474 | 1496 |
| 43 | limit 42 to yr = 2004-2012 (2013) | 841 | 865 |
| 44 | limit 43 to ed = 20121201-20130523 | NA | 54 |
Database searched: EMBASE
Date range searched: 1974 to present.
Date of searched: initially searched 4 December 2012 and updated 23 May 2013.
| # | Terms | Hits (4 December 2012) | Hits (23 May 2013) |
|---|---|---|---|
| 1 | exp Ovarian Cancer/ | 90,833 | 93,629 |
| 2 | (ovar$ adj4 (cancer$ or tumo?r$ or malignan$ or oncolog$ or carcinoma$ or neoplas$ or mass$ or growth$ or cyst$)).mp. | 123,449 | 127,280 |
| 3 | (adenexa$ adj4 mass$).mp. | 13 | 13 |
| 4 | or/1-3 | 126,934 | 130,811 |
| 5 | animal/ not (animal/ and human/) | 1,354,956 | 1,367,021 |
| 6 | 4 not 5 | 122,457 | 126,280 |
| 7 | exp Life Tables/ | 3392 | 3446 |
| 8 | exp “Quality of Life”/ | 221,902 | 234,293 |
| 9 | Health Status/ | 75,649 | 78,135 |
| 10 | exp Health Status Indicators/ | 141,853 | 1113 |
| 11 | (utilit$ approach$ or health gain or hui or hui2 or hui 2 or hui3 or hui 3).ti,ab. | 1625 | 1702 |
| 12 | (health measurement$ scale$ or health measurement$ questionnaire$).ti,ab. | 50 | 53 |
| 13 | (standard gamble$ or categor$ scal$ or linear scal$ or linear analog$ or visual scal$ or magnitude estmat$).ti,ab. | 3738 | 3847 |
| 14 | (time trade off$ or rosser$ classif$ or rosser$ matrix or rosser$ distress$ or hrqol).ti,ab. | 9305 | 9977 |
| 15 | (index of wellbeing or quality of wellbeing or qwb).ti,ab. | 188 | 194 |
| 16 | (rating scale$ or multiattribute$ health ind$ or multi attribute$ health ind$).ti,ab. | 41,599 | 43,458 |
| 17 | (health utilit$ index or health utilit$ indices).ti,ab. | 713 | 739 |
| 18 | (multiattribute$ theor$ or multi attribute$ theor$ or multiattribute$ analys$ or multi attribute$ analys$).ti,ab. | 14 | 14 |
| 19 | (health utilit$ scale$ or classification of illness state$ or 15d or 15 d or 15 dimension).ti,ab. | 3968 | 4109 |
| 20 | (health state$ utilit$ or 12d or 12 d or 12 dimension).ti,ab. | 2682 | 2785 |
| 21 | well year$.ti,ab. | 24 | 24 |
| 22 | (multiattribute$ utilit$ or multi attribute$ utilit$).ti,ab. | 232 | 234 |
| 23 | health utilit$ scale$.ti,ab. | 10 | 11 |
| 24 | (qol or 5d or 5-d or 5 dimension or quality of life or eq-5d or eq5d or eq 5d or euroqol).ti,ab. | 192,239 | 202,999 |
| 25 | (qualy or qaly or qualys or qalys or quality adjusted life year$).ti,ab. | 8724 | 9257 |
| 26 | life year$ gain$.ti,ab. | 2118 | 2202 |
| 27 | willingness to pay.ti,ab. | 2720 | 2874 |
| 28 | (hye or hyes or health$ year$ equivalent$).ti,ab. | 83 | 90 |
| 29 | (person trade off$ or person tradeoff$ or time tradeoff$ or time trade off$).ti,ab. | 1115 | 1145 |
| 30 | theory utilit$.ti,ab. | 8 | 8 |
| 31 | life table$.ti,ab. | 7641 | 7769 |
| 32 | health state$.ti,ab. | 4797 | 4995 |
| 33 | (sf36 or sf 36).ti,ab. | 16,506 | 17,526 |
| 34 | (short form 36 or shortform 36 or sf thirtysix or sf thirty six or shortform thirtysix or shortform thirty six or short form thirtysix or short form thirty six).ti,ab. | 6610 | 7022 |
| 35 | (6d or 6-d or 6 dimension).ti,ab. | 5644 | 5831 |
| 36 | or/7-35 | 525,702 | 420,454 |
| 37 | 6 and 36 | 3356 | 3221 |
| 38 | letter.pt. | 806,544 | 823,694 |
| 39 | editorial.pt. | 421,004 | 431,762 |
| 40 | comment.pt. | 0 | 0 |
| 41 | or/38-40 | 1,227,548 | 1,255,456 |
| 42 | 37 not 41 | 3155 | 3026 |
| 43 | limit 42 to yr = 2004-2012 (2013) | 2216 | 2179 |
| 44 | limit 43 to em = 201247-201321 | NA | 184 |
Health Technology Assessment database
| Date of search | 5 December 2012 | 23 May 2013 |
|---|---|---|
| Search terms (and fields searched) | Ovarian neoplasm (all fields) or Ovarian cancer (all fields) or Ovary cancer (all fields) or and quality of life (all fields) or qol (all fields) or qaly (all fields) or |
Ovarian neoplasm (all fields) or Ovarian cancer (all fields) or Ovary cancer (all fields) or and quality of life (all fields) or qol (all fields) or qaly (all fields) or |
| Date restriction | 2004 to 2012 | 4 December 2012 to 21 May 2013 |
| No. of hits | 3 | 0 |
NHS Economic Evaluation Database
| Date of search | 5 December 2012 | 23 May 2013 |
|---|---|---|
| Search terms (and fields searched) | Ovarian neoplasm (all fields) or Ovarian cancer (all fields) or Ovary cancer (all fields) or and quality of life (all fields) or qol (all fields) or qaly (all fields) or |
Ovarian neoplasm (all fields) or Ovarian cancer (all fields) or Ovary cancer (all fields) or and quality of life (all fields) or qol (all fields) or qaly (all fields) or |
| Date restriction | 2004 to 2012 | 4 December 2012 to 21 May 2013 |
| No. of hits | 30 | 1 |
Appendix 6 Excluded studies for Technology Assessment Group economic evaluation
Summary of reasons for excluding economic evaluation studies
| Reference | Primary reason for exclusion |
|---|---|
| December 2012 search | |
| Havrilesky LJ, Pokrzywinski R, Revicki D, Higgins RV, Nycum LR, Kohler MF, et al. Cost-effectiveness of combination versus sequential docetaxel and carboplatin for the treatment of platinum-sensitive, recurrent ovarian cancer. Cancer 2012;118:386–91 | Duplicate paper |
| Dranitsaris Kim T. The lifecycle value of oncology medicines. Value in Health Conference 2012;15: var. pagings, abstract no. 24 | Review paper |
| Koczorek M. Angiogenesis inhibition: bevacizumab in ovarian carcinoma is approved. Arzneimitteltherapie 2012;30:320–1 | Not an economic evaluation |
| NHSC. Farletuzumab for ovarian cancer: relapsed, platinum-sensitive – in combination with carboplatin and a taxane. Birmingham: National Horizon Scanning Centre (NHSC). Horizon Scanning Review; 2012 | Not an economic evaluation |
| Pike CT, Birbaum HG, Muehlenbein CE, Pohl GM, Natale RB. Healthcare costs and workloss burden of patients with chemotherapy- associated peripheral neuropathy in breast, ovarian, head and neck, and nonsmall cell lung cancer. Chemother Res Pract 2012; article no. 913848 | Not an economic evaluation |
| Basu C. Second line chemotherapy in epithelial ovarian cancer: Experience from a cancer institute of Eastern India. International Journal of Gynecological Cancer Conference 2011;21: var. pagings, 98 | Not an economic evaluation |
| Basu C. Second line chemotherapy in platinum potentially resistant recurrent epithelial ovarian cancer: Experience from Eastern India. International Journal of Gynecological Cancer Conference 2011; var. pagings, 99 | Not an economic evaluation |
| Comitè d’Avaluació de Medicaments d’Utilització Hospitalaria (CAMUH). [Trabectedin (Yondelis) for the treatment of ovarian cancer.] Anonymous. 2011 | Review paper |
| Hensley ML. Big costs for little gain in ovarian cancer. J Clin Oncol 2011;29:1230–2 | Review paper |
| Manahan Wood K. The cost effectiveness of bevacizimab in the primary treatment of ovarian cancer. International Journal of Gynecological Cancer Conference 2011;21(Suppl. 3): var. pagings, 674 | Not an economic evaluation |
| NHSC. Paclitaxel (Paclical) for epithelial ovarian cancer, fallopian tube cancer or peritoneal cancer: second or third line. Anonymous. 2011 | Not an economic evaluation |
| Kazazi-Hyseni F, Beijhen JH, Schellens JH. Bevacizumab. Oncologist 2010;15:819–25 | Not an economic evaluation |
| Benard J. Enhance the cancer cell in platinum, at all costs. B Cancer 2010;97:1029 | Not an economic evaluation |
| Faure S. Cytotoxic antineoplastics. Actual Pharm 2010;497:51–4 | Not an economic evaluation |
| Gordon LG, Scuffham PA, Beesley VL, Green AC, DeFazio A, Wyld DK, et al. Medical costs and outcomes for Australian women with ovarian cancer: a patient-level analysis over 2.5 years. Int J Gynecol Cancer 2010;20:757–65 | Not an economic evaluation |
| Hintringer K. Trabectedin (Yondelis) for second-line recurrent platinum-sensitive ovarian cancer. Anonymous. 2010 | Not an economic evaluation |
| Jungmayr P. The 29th German Cancer Congress –Trabectedin: approval for soft tissue and ovary carcinoma. Deutsche Apotheker Zeitung 2010;150:49–50 | Not an economic evaluation |
| Mkele G. Rational selection of cancer chemotherapy. SAfr Pharm J 2010;77:32–4 | Not an economic evaluation |
| Anonymous. Gemcitabine: new indication. Relapsed ovarian cancer: simply more toxic. Increases haematologic toxicity but not overall survival. Prescrire Int 2009;18:156 | Not an economic evaluation |
| Murphy M, Cunningham J. Intraperitoneal chemotherapy for ovarian cancer patients: a review of the clinical and cost-effectiveness. Anonymous. 2009 | Review paper |
| National Horizon Scanning Centre. Bevacizumab (Avastin) for advanced metastatic ovarian cancer. Anonymous. 2009 | Not an economic evaluation |
| Anonymous. Avastin (bevacizumab) for the treatment of ovarian cancer. Anonymous. 2008 | Not an economic evaluation |
| Petit T. Gynecological cancers. Oncologie 2008;10:463–5 | Not an economic evaluation |
| Marosi Preusser M. Topotecan (Hycamtin). Gynakologische Praxis 2008;32:337–40 | Not an economic evaluation |
| Marosi Preusser M. Topotecan (Hycamtin). Internistische Praxis 2008;48:401–4 | Not an economic evaluation |
| Szucs TD, Dedes KJ. Balancing costs and benefits in cancer therapy and prevention. Ann Oncol 2008;19(Suppl. 7):vii313–19 | Not an economic evaluation |
| Weiss J. Which treatment is cost-effective in recurrent ovarian cancer? Geburtsh Frauenheilk 2008;68:466–7 | Review paper |
| Fedders M, Hartmann MM, Schneider A, Kath R, Camara O, Oelschläger H. Markov-modeling for the administration of platinum analogues and paclitaxel as first-line chemotherapy as well as topotecan and liposomal doxorubicin as second-line chemotherapy with epithelial ovarian carcinoma. J Cancer Res Clin Oncol 2007;133:619–25 | Duplicate paper |
| Purins A, Mundy L, Hiller, JE. Ovarian cancer symptom index. Anonymous. 2007 | Did not include interventions or comparators of interest |
| Anonymous. Off-label uses of bevacizumab: renal cell carcinoma and other miscellaneous non-colorectal cancer indications. Technol Eval Cent Asses Prog Exec Summ 2006;21:1–4 | Not an economic evaluation |
| Anonymous. Trading places. Lancet Oncol 2006;7:275 | Not an economic evaluation |
| Campos SM. Phase II study of CT-2103 in patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Womens Oncol Rev 2005;5:105–7 | Not an economic evaluation |
| Gradishar WJ. Albumin-bound nanoparticle paclitaxel. Clin Adv Hematol Oncol 2005;3:348–9 | Not an economic evaluation |
| Herzog TJ. The challenge of paying for our targeted future. Womens Oncol Rev 2005;5:1 | Not an economic evaluation |
| Lidouren G. Anticancers. Actual Pharm 2005;443:58–63 | Not retrievable |
| Possinger Schmid P. Gemcitabin (Gemzar). Chirurgische Praxis 2005;64:351–8 | Not an economic evaluation |
| Possinger Schmid P. Gemcitabin (Gemzar). Gynakologische Praxis 2005;29:351–8 | Not an economic evaluation |
| Possinger Schmid P. Gemcitabine (Gemzar). Tagliche Praxis 2005;46:415–22 | Not retrievable |
| Jungmayr P, Muller-Bohn I. Tumor disease: Prevention, treatment, health economics. Deutsche Apotheker Zeitung 2004;144:56–69 | Not retrievable |
| Prasad M, Ben-Porat L, Hoppe B, Aghajanian C, Sabbatini P, Chi DS, et al . Costs of treatment and outcomes associated with second-line therapy and greater for relapsed ovarian cancer. Gynecol Oncol 2004;93:223–8 | Did not include interventions or comparators of interest |
| Anonymous. Trabectedin: ET 743, Ecteinascidin 743, Yondelis. Drugs R&D 2003;4:75–81 | Not an economic evaluation |
| Exposito J, Hernández J, Fernández Feijóo A, Nieto T, Briones E. New chemotherapy treatments in advanced cancer patients: an easily applicable evaluation of clinical efficacy and cost-effectiveness. Acta Oncol 2003;42:895–902 | Not an economic evaluation |
| NHSC. Gemcitabine for Recurrent Ovarian Cancer: Horizon Scanning Review. Birmingham: National Horizon Scanning Centre (NHSC); 2003 | Paper not retrievable; archived |
| National Institute for Clinical Excellence (NICE). Guidance on the Use of Pegylated Liposomal Doxorubicin Hydrochloride (PLDH) for the Treatment of Advanced Ovarian Cancer. Anonymous. 2002 | Paper not retrievable; archived |
| Anonymous. Clinical and pharmacoeconomic aspects both play an important role in the treatment of ovarian cancer. Drugs Ther Perspect 2001;17:12–15 | Review paper |
| Anonymous. Taxanes (ovarian cancer): update. Health Technol Assess; 2001 | Paper not retrievable; archived |
| National Institute for Clinical Excellence (NICE). Guidance on the use of topotecan for the treatment of advanced ovarian cancer. Anonymous. 2001 | Paper not retrievable; replaced |
| Anonymous. Is top-level care for ovarian cancer patients more cost-effective than regular care? The Netherlands Organisation for Health Research and Development (ZonMw); 2000 | Not an economic evaluation |
| National Horizon Scanning Centre. Trabectedin (Yondelis) for Ovarian Cancer – Relapsed, Second Line: Horizon Scanning Technology Briefing. Birmingham: National Horizon Scanning Centre (NHSC); 2000 | Paper not retrievable; archived |
| National Institute for Clinical Excellence (NICE). Guidance on the Use of Taxanes for Ovarian Cancer. Anonymous. 2000 | Paper not retrievable; replaced |
| Greenspan EM. New chemoimmunotherapy: courtesy of a more flexible Food and Drug Administration. Cancer Invest 1999;17:371–3 | Review paper |
| NHS Centre for Reviews and Dissemination. Management of Gynaecological Cancers. Anonymous. 1999 | Review paper |
| Orr JW, Orr P, Kern DH. Cost-effective treatment of women with advanced ovarian cancer by cytoreductive surgery and chemotherapy directed by an in vitro assay for drug resistance. Cancer J Sci Am 1999;5:174–8 | Not an economic evaluation |
| Stinson TJ, Calhoun E, Yang T, Lurain JR, Bennett CL, Stinson TJ, et al. Cost analysis of second-line therapies for platinum-refractory ovarian cancer: reimbursement dilemmas for Medicare patients. Cancer Invest 1999;17:559–65 | Not an economic evaluation |
| Bishop JF, Arounas-Kirchman K, Bishop JF, Arounas-Kirchman K. The pharmacoeconomics of cancer therapies. Semin Oncol 1997;24:S19 | Review paper |
| Best L. Paclitaxel as a First Line Chemotherapy Agent in the Treatment of Ovarian Cancer. Southampton: Wessex Institute for Health Research and Development (WIHRD); 1996 | Paper not retrievable; archived |
| Lynch T. Topotecan today. J Clin Oncol 1996;14:3053–5 | Not an economic evaluation |
| Bertelsen K, Kruhoffer A. What have we achieved in ovarian cancer: a comparison of survivals and resources in two different periods. Int J Gynecol Cancer 1995;5:148–55 | Did not include interventions or comparators of interest |
| Chica Marchal AML. Pharmacoeconomic study of intravenous antineoplastic therapy in a centralized cytostatics unit. Farmacie Clin 1995;12:202–9 | Not an economic evaluation |
Summary of reasons for excluding health-related quality-of-life papers reviewed in full
| Study | Reason for exclusion |
|---|---|
| December 2012 search | |
| Andersen MR, Sweet E, Lowe KA, Standish LJ, Drescher CW, Goff BA, et al. Involvement in decision-making about treatment and ovarian cancer survivor quality of life. Gynecol Oncol 2012;124:465–70 | Generic non-preference-based QoL |
| Cui S, Ba M, Tang Y, Liu J, Wu Y, Zhang X, et al. B ultrasound-guided hyperthermic intraperitoneal perfusion chemotherapy for the treatment of malignant ascites. Oncol Rep 2012;28:1325–31 | Review paper |
| Dhillon S. Bevacizumab combination therapy: For the first-line treatment of advanced epithelial ovarian, fallopian tube or primary peritoneal cancer. Drugs 2012;72:917–30 | Review paper |
| Farghaly S. Long term survival of female patients with peritoneal carcinomatosis utilizing robot assisted laparoscopic ultra radical cytoreductive surgery and hyperthermic intraperitoneal chemotherapy (HIPEC). International Journal of Gynecology & Obstetrics Conference, 2012, var. pagings | No QoL data |
| Frampton JE. Catumaxomab: in malignant ascites. Drugs 2012;72:1399–410 | Review paper |
| Gilbertson-White S, Aouizerat BE, Jahan T, Paul SM, West C, Schemacher K, et al. Determination of cutpoints for low and high number of symptoms in patients with advanced cancer. J Palliat Med 2012;15:1027–36 | Condition-specific QoL |
| Hilpert F, Wimberger P, du Bois A, Pfisterer J, Harter P. Treatment of elderly ovarian cancer patients in the context of controlled clinical trials: a joint analysis of the AGO Germany experience. Onkologie 2012;35:76–81 | Condition-specific QoL |
| Izzo AA. Limited value of traditional Chinese medicine in improving quality of life in cancer patients. Focus Alt Complement Ther 2012;17:228–9 | Generic non-preference-based QoL |
| Jayatilleke N, Pashayan N, Powles JW. Burden of disease due to cancer in England and Wales. J Pub Health 2012;34:287–95 | Generic non-preference-based QoL |
| Lindemann K, Christensen RD, Vergote I, Stuart G, Izquierdo MA, Kaern J, et al. First-line treatment of advanced ovarian cancer with paclitaxel/carboplatin with or without epirubicin (TEC versus TC): a gynecologic cancer intergroup study of the NSGO, EORTC GCG and NCIC CTG. Ann Oncol 2012;23:2613–9 | Condition-specific QoL |
| Maccio A, Madeddu C, Gramignano G, Mulas C, Floris C, Sanna E, et al. A randomized phase III clinical trial of a combined treatment for cachexia in patients with gynaecologic cancers: Evaluating the impact on metabolic and inflammatory profiles and quality of life. Gynecol Oncol 2012;124:417–25 | Condition-specific QoL |
| Nagel C, Street J, Kehoe S, Richardson D, Miller D, Lea J. Clinical course of ovarian cancer after two salvage regimens. Gynecol Oncol 2012;127(Suppl.1):S27–8 | No QoL data |
| Perwitasari DA, Atthobari J, Mustofa M, Dwiprahasto I, Hakimi M, Gelderblom H, et al. Impact of chemotherapy-induced nausea and vomiting on quality of life in Indonesian patients with gynecologic cancer. Int J Gynecol Cancer 2012;22:139–45 | Generic non-preference-based QoL |
| Pilger A, Richter R, Fotopoulou C, Beteta C, Klapp C, Sehouli J, et al. Quality of life and sexuality of patients after treatment for gynaecological malignancies: results of a prospective study in 55 patients. Anticancer Res 2012;32:5045–9 | Generic non-preference-based QoL |
| Richter R, Oskay-Oezcelik G, Chekerov R, Pilger A, Hindenburg HJ, Sommer H, et al. Health-related quality of life during sequential chemotherapy with carboplatin followed by weekly paclitaxel in advanced ovarian cancer: a multicenter phase ii study of the North Eastern German Society of Gynecological Oncology. Anticancer Res 2012;3:3969–76 | Condition-specific QoL |
| Robinson KM, Christensen KB, Ottesen B, Krasnik A. Diagnostic delay, quality of life and patient satisfaction among women diagnosed with endometrial or ovarian cancer: a nationwide Danish study. Qual Life Res 2012;21:1519–25 | Condition-specific QoL |
| Alvarez Secord A, Berchuck A, Higgins RV, Nycum LR, Kohler MF, Puls LE, et al. A multicenter, randomized, phase 2 clinical trial to evaluate the efficacy and safety of combination docetaxel and carboplatin and sequential therapy with docetaxel then carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Cancer 2012;118:3283–93 | Condition-specific QoL |
| Sorbe B, Graflund M, Nygren L, Horvath G, Swahn M, Boman K, et al. A phase II study of docetaxel weekly in combination with carboplatin every three weeks as first line chemotherapy in stage IIB-IV epithelial ovarian cancer: neurological toxicity and quality-of-life evaluation. Int J Oncol 2012;40:773–81 | Condition-specific QoL |
| Sommeijer DWP. Quality of life and coping in ovarian cancer: the last year of life. Asia-Pacific Journal of Clinical Oncology Conference 2012, var. pagings | No QoL data |
| Stavraka C, Ford A, Ghaem-Maghami S, Crook T, Agarwal R, Gabra H, et al. A study of symptoms described by ovarian cancer survivors. Gynecol Oncol 2012;125:59–64 | Condition-specific QoL |
| Patidar S, Telepak L, Lipe M, Sannes T, Dodd S, Bishop M, et al. A ‘snapshot’ of photovoice as a psychosocial intervention for individuals affected by ovarian cancer. Psycho-Oncology Conference, February 2012, var. pagings | Condition-specific QoL |
| Basu C. Second line chemotherapy in epithelial ovarian cancer: Experience from a cancer institute of Eastern India. International Journal of Gynecological Cancer Conference 2011, var. pagings, 98 | No QoL data |
| Basu C. Second line chemotherapy in platinum potentially resistant recurrent epithelial ovarian cancer: Experience from Eastern India. International Journal of Gynecological Cancer Conference 2011, var. pagings, 99 | No QoL data |
| Beesley VL, Price MA, Butow PN, Green AC, Olsen CM, Australian Ovarian Cancer Study Group, Australian Ovarian Cancer Study – Quality of Life Study Investigators, et al. Physical activity in women with ovarian cancer and its association with decreased distress and improved quality of life. Psychooncology 2011;20:1161–9 | Condition-specific QoL |
| Donnelly CM, Blaney JM, Lowe-Strong A, Rankin JP, Campbell A, Crum-Gardner E, et al. A randomised controlled trial testing the feasibility and efficacy of a physical activity behavioural change intervention in managing fatigue with gynaecological cancer survivors. Gynecol Oncol 2011;122:618–24 | Condition-specific QoL |
| Faul LA, Jim HS, Minton S, Fishman M, Tanvetyanon T, Jacobsen PB. Relationship of exercise to quality of life in cancer patients beginning chemotherapy. J Pain Symptom Manage 2011;41:859–69 | Generic non-preference-based QoL |
| Gorasia TKK. Phase II study of intraperitoneal chemotherapy in inoperable epithelial ovarian and primary peritoneal cancers. International Journal of Gynecological Cancer Conference 2011, var. pagings, 115 | No QoL data |
| Guimaraes GC, Baiocchi G, Ferreira FO, Kumagai LY, Fallopa CC, Aguiar S, et al. Palliative pelvic exenteration for patients with gynecological malignancies. Arch Gynecol Obstet 2011;283:1107–12 | No QoL data |
| Judson PL, Dickson EL, Argenta PA, Xiong Y, Geller MA, Carson LF. A prospective, randomized trial of integrative medicine for women with ovarian cancer. Gynecol Oncol 201;123:346–50 | Condition-specific QoL |
| Krishnappa S. Pattern of care by primary surgery vs neoadjuvant chemotherapy followed by interval debulking surgery in advanced epithelial ovarian cancer. International Journal of Gynecological Cancer Conference 2011, var. pagings, 127 | No QoL data |
| Lee HY, Hong JM, Yang BM, Lee TJ, Kim BG, Kang SB, et al. Cost-utility analysis of combination therapy of pegylated liposomal doxorubicin (PLD) and carboplatin for Korean women with platinum-sensitive ovarian cancer. Value in Health Conference 2011, var. pagings, A455 | No QoL data |
| Harter Ledermann J. Phase 2 randomized placebo-controlled study of olaparib (AZD2281) in patients with platinum-sensitive relapsed serous ovarian cancer (PSR SOC). International Journal of Gynecological Cancer Conference 2011, var. pagings, S13 | No QoL data |
| Lesnock JL, Farris C, Krivak TC, Smith KJ, Markman M. Consolidation paclitaxel is more cost-effective than bevacizumab following upfront treatment of advanced ovarian cancer. Gynecol Oncol 2011;122:473–8 | No QoL data |
| Pace Lugini A. The combination of weekly carboplatin and paclitaxel is active and tolerated for the treatment of advanced ovarian cancer in elderly patients. European Journal of Cancer Conference, September 2011, var. pagings | No QoL data |
| Catarina R, Pimenta F, Leal I, Maroco J. Menopause-specific quality of life: a comparison between menopausal women with and without a diagnosis of cancer. Climacteric Conference, June 2011, var. pagings | Generic non-preference-based QoL |
| Netzer IML. Reduced weekly docetaxel regimen in combination with carboplatin for treatment of ovarian cancer. International Journal of Gynecological Cancer Conference 2011, var. pagings, S574 | No QoL data |
| Pace Lugini A. Aprepitant in the prevention of acute and delayed chemotherapy-induced nausea and vomiting (CINV) in elderly patients with advanced ovarian cancer. International Journal of Gynecological Cancer Conference 2011, var. pagings, S1272 | No QoL data |
| Papaioannou D, Rafia R, Stevenson MD, Stevens JW, Evans P. Trabectedin for the treatment of relapsed ovarian cancer. Health Technol Assess 2011;15(1) | No QoL data |
| Fujisaka Y, Sugiyama T, Saito H, Nagase S, Kudoh S, Endo M, et al. Randomised, phase III trial of epoetin-B to treat chemotherapy-induced anaemia according to the EU regulation. Br J Cancer 2011;105:1267–72 | Condition-specific QoL |
| Wilailak S, Lertkhachonsuk A, Lohachroenvanich N, Luengsukcharoen SC, Jirajaras M, Likitanasombat P, et al. Quality of life in gynecologic cancer survivors compared to healthy check-up women. J Gynecol Oncol 2011;22:103–9 | Condition-specific QoL |
| Jurczyk Polocka-Molinska M. Quality of life of women with inoperable ovarian cancer. Current Gynecol Oncol 2011;9:82–94 | Condition-specific QoL |
| Rochet N, Kieser M, Sterzing F, Krause S, Lindel K, Harms W, et al. Phase II study evaluating consolidation whole abdominal intensity-modulated radiotherapy (IMRT) in patients with advanced ovarian cancer stage FIGO III: the OVAR-IMRT-02 Study. BMC Cancer 2011;11:41 | Condition-specific QoL |
| von Gruenigen VE, Frasure HE, Kavanagh MB, Lerner E, Waggoner SE, Courneya KS. Feasibility of a lifestyle intervention for ovarian cancer patients receiving adjuvant chemotherapy. Gynecol Oncol 2011;122:328–33 | Condition-specific QoL |
| Pokrzywinski R, Secord AA, Havrilesky LJ, Puls LE, Holloway RW, Lewandowski GS, et al. Health-related quality of life outcomes of docetaxel/carboplatin combination therapy vs. sequential therapy with docetaxel then carboplatin in patients with relapsed, platinum-sensitive ovarian cancer: results from a randomized clinical trial. Gynecol Oncol 2011;123:505–10 | Condition-specific QoL |
| van de Poll-Franse LV, Nicolaije KA, Vos MC, Pijnenborg JM, Boll D, Husson O, et al. The impact of a cancer Survivorship Care Plan on gynecological cancer patient and health care provider reported outcomes (ROGY Care): study protocol for a pragmatic cluster randomized controlled trial. Trials 2011;12:256 | Condition-specific QoL |
| Bidzinski Vergote I. Health-related quality of life (HRQOL)/patient reported outcomes (PRO) of patients (pts) with partially platinum sensitive (PPS) recurrent ovarian cancer (ROC) treated in a randomised phase III trial of trabectedin and pegylated liposomal doxorubicin (PLD) vs PLD alone (OVA-301): an exploratory analysis. European Journal of Cancer Conference, September 2011, var. pagings | Condition-specific QoL |
| Nankivell Stark D. Quality of life in the ICON7 GCIG phase III randomised clinical trial. European Journal of Cancer Conference, September 2011, var. pagings | Condition-specific QoL |
| M Vergote Gore I. Cost-effectiveness of trabectedin in combination with pegylated liposomal doxorubicin hydrochloride for the treatment of women with relapsed platinum-sensitive ovarian cancer in the UK: analysis based on the final survival data. European Journal of Cancer, September 2011, var. pagings | No QoL data |
| Dean-Clower E, Doherty-Gilman AM, Keshaviah A, Baker F, Kaw C, Lu W, et al. Acupuncture as palliative therapy for physical symptoms and quality of life for advanced cancer patients. Integr Cancer Ther 2010;9:158–67 | Condition-specific QoL |
| Hisanaga T, Shinjo T, Morita T, Nakajima N, Ikenaga M, Tanimizu M, et al. Multicenter prospective study on efficacy and safety of octreotide for inoperable malignant bowel obstruction. Jpn J Clin Oncol 2010;40:739–45 | Condition-specific QoL |
| Dean-Clower E, Doherty-Gilman AM, Keshaviah A, Baker F, Kaw C, Lu W, et al. Acupuncture as palliative therapy for physical symptoms and quality of life for advanced cancer patients. Integr Cancer Ther 2010;9:158–67 | Duplicate paper |
| Henry M, Cohen SR, Lee V, Sauthier P, Provencher D, Drouin P, et al. The Meaning-Making intervention (MMi) appears to increase meaning in life in advanced ovarian cancer: a randomized controlled pilot study. Psychooncology 2010;19:1340–7 | Generic non-preference-based QoL |
| Johns S. Dignity therapy for women with metastatic cancer: effects and lessons learned. Psycho-Oncology Conference, February 2010, var. pagings | No QoL data |
| Grann VR, Patel PR, Jacobson JS, Warner E, Heitjan DF, Ashby-Thompson M, et al. Comparative effectiveness of screening, surgery, and chemoprevention among BRCA1/2 mutation carriers. Journal of Clinical Oncology Conference, 2010, var. pagings. J Clin Oncol ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2010;28(Suppl. 20 May), 6011. URL: http://meeting.ascopubs.org/cgi/content/abstract/28/15_suppl/6011?sid = a784435c-fe71–4deb-bf50-ffaf39b981c7 | No QoL data |
| Burger RAB. Phase III trial of bevacizumab (BEV) in the primary treatment of advanced epithelial ovarian cancer (EOC), primary peritoneal cancer (PPC), or fallopian tube cancer (FTC): a Gynecologic Oncology Group study. Journal of Clinical Oncology Conference 2010, var. pagings | No QoL data |
| Burger RAB. Safety and subgroup efficacy analyses in GOG218, a phase III trial of bevacizumab (BEV) in the primary treatment of advanced epithelial ovarian cancer (EOC), primary peritoneal cancer (PPC) or fallopian tube cancer (FTC): a gynecologic oncology group study. Annals of Oncology Conference, October 2010, var. pagings | No QoL data |
| Basu S, Mukhopadhyay S, Bose CK, Pandey R, Basak J, Mukhopadhyay A. Do adult cancer survivors require psychotherapy? An experience from Eastern India. Annals of Oncology Conference ,October 2010, var. pagings | No QoL data |
| Friedlander MV. Symptom burden among patients with platinum resistant/refractory recurrent ovarian cancer (PRR ROC): stage 1 of the GCIG symptom benefit study. Asia-Pacific Journal of Clinical Oncology Conference, November 2010, var. pagings | No QoL data |
| Hay Ding Y. Cost-effectiveness analysis of multimodal screening for ovarian cancer. Value in Health Conference 2010, var. pagings, A37 | No QoL data |
| von Gruenigen VE, Huang HQ, Gil KM, Gibbons HE, Monk BJ, Rose PG, et al. A comparison of quality-of-life domains and clinical factors in ovarian cancer patients: a gynecologic oncology group study. J Pain Symptom Manage 2010;39:839–46 | Condition-specific QoL |
| Lluch Palli C. Sexuality, communication and emotions: a situational study in women affected by gynecologic cancer. Psicooncologia 2010;7:153–73 | Condition-specific QoL |
| Sawada NOZ. The outcomes of visualization and acupuncture on the quality of life of adult cancer patients receiving chemotherapy. Cancer Nurs 2010;33:E21–8 | Condition-specific QoL |
| Pujade-Lauraine E, Wagner U, Avall-Lundqvist E, Gebski V, Heywood M, Vasey PA, et al. Pegylated liposomal doxorubicin and carboplatin compared with paclitaxel and carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol 2010;28:3323–9 | Condition-specific QoL |
| Zamurovic M, Mitrovic-Jovanovic A, Jurisic A. Ovarian carcinoma patients: life quality analysis in the postoperative period – how to improve it? Eur J Gynaecol Oncol 2010;31:672–4 | Condition-specific QoL |
| von Gruenigen V. The association between quality of life and overall survival in ovarian cancer patients during adjuvant chemotherapy: a Gynecologic Oncology Group study. Journal of Clinical Oncology Conference 2010, var. pagings. URL: http://meeting.ascopubs.org/cgi/content/abstract/28/15_suppl/5075?sid = 663fdb93–2fe5–47e0–89bb-5fa625839dc6 | No QoL data |
| Wright AAP. Associations between age and quality of life in advanced ovarian cancer. Journal of Clinical Oncology Conference 2010, var. pagings | Condition-specific QoL |
| Pignata S, Scambia. Carboplatin (C) plus paclitaxel (P) versus carboplatin plus pegylated liposomal doxorubicin (PLD) in patients with advanced ovarian cancer (AOC): Final analysis of the MITO-2 randomized multicenter trial. Journal of Clinical Oncology Conference 2010, var. pagings | No QoL data |
| Zhukovsky Ramondetta L. Factors contributing to anxiety, depression in newly diagnosed ovarian cancer patients. Support Care in Cancer Conference, June 2010, var. pagings | Condition-specific QoL |
| Sandadi S, Frasure HE, Broderick MJ, Waggoner SE, Miller JA, von Grunigen VE. The Effect of Sleep Disturbance on Quality of Life in Women with Ovarian Cancer. Gynecologic Oncology Conference 2010, var. pagings: S140–1 | Condition-specific QoL |
| Mikkelsen EM, Sunde L, Johansen C, Johnsen SP. Psychosocial consequences of genetic counseling: a population-based follow-up study. Breast J 2009;15:61–8 | Generic non-preference-based QoL |
| Averbeck Arriagada E. OECI Workshop on late side-effects of cancer treatments. Eur J Cancer 2009;45:354–9 | No QoL data |
| Bielawska Leszek A. Quality of life among patients with ovarian cancer. Ginek Prakt 2009;17:3–6 | Condition-specific QoL |
| Fischer A. Better quality of life: a new combination with advantages for ovarian cancer. Klinikarzt 2009;38:362 | No QoL data |
| Ozanne EMC. Cost-effectiveness of surgical interventions for BRCA gene mutation carriers: Impact of delaying decision-making. Cancer Reseach Conference, 2009, var. pagings | No QoL data |
| Ozanne EMC. Cost-effectiveness of genetic testing for BRCA1 and BRCA2 mutations. Cancer Research Conference 2009, var. pagings | No QoL data |
| Lortholary Gladieff L. Weekly paclitaxel (wP) as single agent or in combination with weekly topotecan (wT) or carboplatin (C) in patients with resistant ovarian cancer (ROC): The phase II CARTAXHY randomized trial from GINECO. Journal of Clinical Oncology Conference 2009, var. pagings, 5557 | No QoL data |
| Ferguson Helpman Bek L. Use of complementary medicine (CAM) among women receiving chemotherapy for ovarian cancer: a comparison of attitudes between two patient populations. Journal of Clinical Oncology Conference 2009, var. pagings: e20545. URL: http://meeting.ascopubs.org/cgi/content/abstract/27/15S/e20545?sid = 2ba30803-e476–47d3-af1a-2afb134588ab | No QoL data |
| Cohen Henry M. Randomized control trial of the Meaning-Making intervention (MMi) for people newly diagnosed with advanced ovarian cancer: A pilot study. Psychooncology Conference June 2009, var. pagings | Condition-specific QoL |
| Lisyanskya AST. Restoration of ovarian function after cryopreserved ovarian tissue transplantation in women exposed to complex treatment for gynecological cancer: Feasibility of this option in pediatric cancer patients. Cellular Therapy Transplantation Conference 2009, var. pagings | No QoL data |
| Steppan I, Reimer D, Sevelda U, Ulmer H, Marth C, Zeimet AG. Treatment of recurrent platinum-resistant ovarian cancer with pegylated liposomal doxorubicin: an evaluation of the therapeutic index with special emphasis on cardiac toxicity. Chemotherapy 2009;55:391–8 | Condition-specific QoL |
| Morita Yamagishi A. Symptom prevalence and longitudinal follow-up in cancer outpatients receiving chemotherapy. J Pain Symptom Manage 2009;37:823–30 | Condition-specific QoL |
| von Gruenigen V. Assessment of factors that contribute to decreased quality of life in gynecologic oncology group ovarian cancer trials. Cancer 2009;115:4857–64 | Condition-specific QoL |
| Thomas SGJ. Prospective phase II trial of fulvestrant in the treatment of recurrent ovarian carcinoma. Gynecological Oncology Conference 2009, var. pagings: S32 | Condition-specific QoL |
| Wakabayashi MTO. Integration of palliative care during the administration of intraperitoneal chemotherapy for ovarian cancer. Gynecologic Oncology Conference 2009, var. pagings, S164 | Condition-specific QoL |
| Van Der Burg MEL. Randomized MRC OV05/EORTC 55955 trial in recurrent ovarian cancer: Early treatment based on increased serum CA125 alone versus delayed treatment based on conventional clinical indicators. European Journal Cancer Supplement Conference 2009, var. pagings, 3 | No QoL data |
| von Gruenigen V. A double-blind randomized trial of pyridoxine versus placebo for the prevention of pegylated liposomal doxorubicin hydrochloride-related palmar-plantar erythrodysesthesia. Journal of Clinical Oncology Conference 2009, var. pagings, 5594 | No QoL data |
| Pignata S, Scambia G. Carboplatin plus paclitaxel (CP) versus carboplatin plus stealth liposomal doxorubicin (CLD) in patients with advanced ovarian cancer (AOC): Activity and safety results of the MITO-2 randomized multicenter trial. Journal of Clinical Oncology Conference 2009, var. pagings, LBA5508 | No QoL data |
| Pujade-Lauraine E, Mahner A. A randomized, phase III study of carboplatin and pegylated liposomal doxorubicin versus carboplatin and paclitaxel in relapsed platinum-sensitive ovarian cancer (OC): CALYPSO study of the Gynecologic Cancer Intergroup (GCIG). Journal of Clinical Oncology Conference 2009, var. pagings, LBA5509 | No QoL data |
| Krasner CNP. Health-related quality of life/patient-reported outcomes in relapsed ovarian cancer: Results from a randomized phase III study of trabectedin with pegylated liposomal doxorubicin (PLD) versus PLD alone. Journal of Clinical Oncology Conference, 2009, var. pagings, 5526 | No QoL data |
| Hackbarth M, Haas N, Fotopoulou C, Lichtenegger W, Sehouli J. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. Results of a prospective study. Support Care Cancer 2008;16:267–73 | Condition-specific QoL |
| Liavaag AH, Dorum A, Bjoro T, Oksefjell H, Fossa SD, Trope C, et al. A controlled study of sexual activity and functioning in epithelial ovarian cancer survivors. A therapeutic approach. Gynecol Oncol 2008;108:348–54 | Condition-specific QoL |
| Matulonis UA, Kornblith A, Lee H, Bryan J, Gibson C, Wells C, et al. Long-term adjustment of early-stage ovarian cancer survivors. Int J Gynecol Cancer 2008;18:1183–93 | Condition-specific QoL |
| Ferrandina G, Ludovisi M, Lorusso D, Pignata S, Breda E, Savarese A, et al. Phase III trial of gemcitabine compared with pegylated liposomal doxorubicin in progressive or recurrent ovarian cancer. J Clin Oncol 2008;26:890–6 | Condition-specific QoL |
| Danhauer SC, Tooze JA, Farmer DF, Campbell CR, McQuellon RP, Barrett R, et al. Restorative yoga for women with ovarian or breast cancer: findings from a pilot study. J Soc Integrat Oncol 2008;6:47–58 | Generic non-preference-based QoL |
| Absolom K, Eiser C, Turner L, Ledger W, Ross R, Davies H, et al. Ovarian failure following cancer treatment: current management and quality of life. Human Reproduct 2008;23:2506–12 | Generic non-preference-based QoL |
| Liavaag AHD. A controlled study of sexual activity and functioning in epithelial ovarian cancer survivors. A therapeutic approach. Gynecol Oncol 2008;108:348–54 | Duplicate paper |
| Litterini AJF. The change in fatigue, strength, and quality of life following a physical therapist prescribed exercise program for cancer survivors. Rehabil Oncol 2008;26:11–17 | Generic non-preference-based QoL |
| Meropol NJE. Cancer patient preferences for quality and length of life. Cancer 2008;113:3459–66 | Generic non-preference-based QoL |
| Caruso A, Vigna C, Maggi G, Sega FM, Cognetti F, Savarese A. The withdrawal from oncogenetic counselling and testing for hereditary and familial breast and ovarian cancer. A descriptive study of an Italian sample. J Exp Clin Cancer Res 2008;27:1 | No QoL data |
| Shinjyo Hisanaga T. Efficacy of octreotide acetate for malignant gastrointestinal obstruction. Annals of Oncology Conference (ESMO), Stockholm, 2008, viii 255 | Condition-specific QoL |
| Havrilesky LJ, Secord AA, Darcy KM, Armstrong DK, Kulasingam S, Gynecologic Oncology Group. Cost effectiveness of intraperitoneal compared with intravenous chemotherapy for women with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 2008;26:4144–50 | Condition-specific QoL |
| Wagner LI, Beaumont JL, Ding B, Malin J, Peterman A, Calhoun E, et al. Measuring health-related quality of life and neutropenia-specific concerns among older adults undergoing chemotherapy: validation of the Functional Assessment of Cancer Therapy-Neutropenia (FACT-N). Support Care Cancer 2008;16:47–56 | Condition-specific QoL |
| Sehouli J, Stengel D, Oskay-Oezcelik G, Zeimet AG, Sommer H, Klare P, et al. Nonplatinum topotecan combinations versus topotecan alone for recurrent ovarian cancer: results of a phase III study of the North-Eastern German Society of Gynecological Oncology Ovarian Cancer Study Group. J Clin Oncol 2008;26:3176–82 | Condition-specific QoL |
| Schulman-Green D, Ercolano E, Dowd M, Schwartz P, McCorkle R. Quality of life among women after surgery for ovarian cancer. Palliat Support Care 2008;6:239–47 | Generic non-preference-based QoL |
| Huang Wenzel L. Validation of FACT/GOG-AD subscale for ovarian cancer-related abdominal discomfort: A Gynecologic Oncology Group study. Gynecol Oncol 2008;110:60–4 | Condition-specific QoL |
| Stefanie S, Zahasky KM. Psychological aspect of chemotherapy. CME J Gynecol Oncol 2008;13:7–20 | No QoL data |
| Champion V, Williams SD, Miller A, Reuille KM, Wagler-Ziner K, Monahan PO, et al. Quality of life in long-term survivors of ovarian germ cell tumors: A Gynecologic Oncology Group study. Gynecol Oncol 2007;105:687–94 | Generic non-preference-based QoL |
| Liavaag AH, Dorum A, Fossa SD, Trope C, Dahl AA, Astrid H. Controlled study of fatigue, quality of life, and somatic and mental morbidity in epithelial ovarian cancer survivors: how lucky are the lucky ones? J Clin Oncol 2007;25:2049–56 | Condition-specific QoL |
| Mori T, Hosokawa K, Kinoshita Y, Watanabe A, Yamaguchi T, Kuroboshi H, et al. A pilot study of docetaxel-carboplatin versus paclitaxel-carboplatin in Japanese patients with epithelial ovarian cancer. Int J Clin Oncol 2007;12:205–11 | Condition-specific QoL |
| Bristow RE, Santillan A, Salani R, Diaz-Montes TP, Giuntoli RL, Meisner BC, et al. Intraperitoneal cisplatin and paclitaxel versus intravenous carboplatin and paclitaxel chemotherapy for Stage III ovarian cancer: a cost-effectiveness analysis. Gynecol Oncol 2007;106:476–81 | Condition-specific QoL |
| Fox SW, Lyon D. Symptom clusters and quality of life in survivors of ovarian cancer. Cancer Nurs 2007;30:354–61 | Generic non-preference-based QoL |
| Hopkins ML, Coyle D, Le T, Fung MF, Wells G. Cancer antigen 125 in ovarian cancer surveillance: a decision analysis model. Current Oncol 2007;14:167–72 | Generic non-preference-based QoL |
| Levine EG, Silver B. A pilot study: evaluation of a psychosocial program for women with gynecological cancers. J Psychosoc Oncol 2007;25:75–98 | Condition-specific QoL |
| Cohen L, de Moor CA, Eisenberg P, Ming EE, Hu H. Chemotherapy-induced nausea and vomiting: incidence and impact on patient quality of life at community oncology settings. Support Care Cancer 2007;15:497–503 | Condition-specific QoL |
| Liavaag AHD. Controlled study of fatigue, quality of life, and somatic and mental morbidity in epithelial ovarian cancer survivors: how lucky are the lucky ones? J Clin Oncol 2007;25:2049–56 | Duplicate paper |
| Fasching PAT. Association of complementary methods with quality of life and life satisfaction in patients with gynecologic and breast malignancies. Support Care Cancer 2007;15:1277–84 | Condition-specific QoL |
| Zhang MM, Chan JK, Husain A, Guo HY, Teng NN. Safety and efficacy of lenalidomide (Revlimid) in recurrent ovarian and primary peritoneal carcinoma. Gynecol Oncol 2007;105:194–8 | Condition-specific QoL |
| Wenzel LB, Huang HQ, Armstrong DK, Walker JL, Cella D, Gynecologic Oncology Group. Health-related quality of life during and after intraperitoneal versus intravenous chemotherapy for optimally debulked ovarian cancer: a Gynecologic Oncology Group Study. J Clin Oncol 2007;25:437–43 | Condition-specific QoL |
| Stevinson C, Faught W, Steed H, Tonkin K, Ladha AB, Vallance JK, et al. Associations between physical activity and quality of life in ovarian cancer survivors. Gynecol Oncol 2007;106:244–250 | Condition-specific QoL |
| Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med 2006;354:34–43 | Condition-specific QoL |
| Hirte H, Vergote IB, Jeffrey JR, Grimshaw RN, Coppieters S, Schwartz B, et al. A phase III randomized trial of BAY 12–9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: a National Cancer Institute of Canada Clinical Trials Group Study. Gynecol Oncol 2006;102:300–8 | Condition-specific QoL |
| Apte SM, Vadhan-Raj S, Cohen L, Bassett RL, Gordon IO, Levenback CF, et al. Cytokines, GM-CSF and IFNgamma administered by priming and post-chemotherapy cycling in recurrent ovarian cancer patients receiving carboplatin. J Transl Med 2006;4:16 | Condition-specific QoL |
| de Moor JS, de Moor CA, Basen-Engquist K, Kudelka A, Bevers MW, Cohen L, et al. Optimism, distress, health-related quality of life, and change in cancer antigen 125 among patients with ovarian cancer undergoing chemotherapy. Psychosom Med 2006;68:555–62 | Condition-specific QoL |
| Livartowski Buron C. Considering simultaneously quality of life and quantity of life in oncology. Oncologie 2006;8:483–8 | No QoL data |
| Griffin S, Bojke L, Main C, Palmer S. Incorporating direct and indirect evidence using Bayesian methods: an applied case study in ovarian cancer. Value Health 2006;9:123–31 | No QoL data |
| Markman M. Intraperitoneal chemotherapy as primary treatment of advanced ovarian cancer. Comm Oncol 2006;3:352–3 | No QoL data |
| Wolf JK, Bodurka DC, Verschraegen C, Sun CC, Branham D, Jenkins AD, et al. A phase II trial of oral capecitabine in patients with platinum- and taxane-refractory ovarian, fallopian tube, or peritoneal cancer. Gynecol Oncol 2006;102:468–74 | Condition-specific QoL |
| von Gruenigen V, Frasure HE, Jenison EL, Hopkins MP, Gil KM. Longitudinal assessment of quality of life and lifestyle in newly diagnosed ovarian cancer patients: the roles of surgery and chemotherapy. Gynecologic Oncol 2006;103:120–6 | Condition-specific QoL |
| Wilkinson PM, Antonopoulos M, Lahousen M, Lind M, Kosmidis P, EPO-INT-45 Study Group. Epoetin alfa in platinum-treated ovarian cancer patients: results of a multinational, multicentre, randomised trial. Br J Cancer 2006;94:947–54 | Condition-specific QoL |
| Pfisterer J, Plante M, Vergote I, du Bois A, Hirte H, Lacave AJ, et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol 2006;24:4699–707 | Condition-specific QoL |
| Wasta V. Intraperitoneal chemotherapy improves survival in ovarian cancer patients when combined with intravenous chemotherapy. Cancer Biol Ther 2006;5:130–1 | Review paper |
| Yan Zhu Y-P. Effect of polysaccharide-peptide plus chemotherapy on the immune function and quality of life in patients with ovarian or endometrial cancer. Chin J Clin Rehab 2006;10:212–4 | Condition-specific QoL |
| De Vos FY, Bos AM, Schaapveld M, de Swart CA, de Graaf H, van der Zee AG, et al. A randomized phase II study of paclitaxel with carboplatin +/– amifostine as first line treatment in advanced ovarian carcinoma. Gynecol Oncol 2005;97:60–7 | Condition-specific QoL |
| Fushiki H, Yoshimoto H, Ikoma T, Ota S. [A trial of biweekly paclitaxel administration in consideration of QOL for advanced or recurrent gynecologic cancer.] Gan to Kagaku Ryoho 2005;32:691–3 | Not retrievable |
| Costanzo ES, Lutgendorf SK, Sood AK, Anderson B, Sorosky J, Lubaroff DM. Psychosocial factors and interleukin-6 among women with advanced ovarian cancer. Cancer 2005;104:305–13 | Condition-specific QoL |
| Alvarez Secord A, Jones EL, Hahn CA, Petros WP, Yu D, Havrilesky LJ, et al. Phase I/II trial of intravenous Doxil and whole abdomen hyperthermia in patients with refractory ovarian cancer. Int J Hypertherm 2005;21:333–47 | Condition-specific QoL |
| Dedes KJ, Bramkamp M, Szucs TD. Paclitaxel: cost-effectiveness in ovarian cancer. Expert Rev Pharmacoecon Outcomes Res 2005;5:235–43 | Review paper |
| Gonzalez-Martin AJC. Randomized phase II trial of carboplatin versus paclitaxel and carboplatin in platinum-sensitive recurrent advanced ovarian carcinoma: A GEICO (Grupo Espanol de Investigacion en Cancer de Ovario) study. Ann Oncol 2005;16:749–55 | Condition-specific QoL |
| Stauch Oberhoff C. Prevention and therapy of anemia in tumor patients with Epoetin beta (NeoRecormon). Tumordiagn Ther 2005;26:166–71 | Condition-specific QoL |
| Denniston Baker F. Adult cancer survivors: how are they faring? Cancer 2005;104(Suppl. 11):2565–76 | Condition-specific QoL |
| Miller B. Spiritual journey during and after cancer treatment. Gynecol Oncol 2005;99(Suppl. 3):129–30 | Condition-specific QoL |
| Sun CC, Bodurka DC, Weaver CB, Rasu R, Wolf JK, Bevers MW, et al. Rankings and symptom assessments of side effects from chemotherapy: insights from experienced patients with ovarian cancer. Support Care Cancer 2005;13:219–27 | Generic non-preference-based QoL |
| Wenzel L, Huang HQ, Monk BJ, Rose PG, Cella D. Quality-of-life comparisons in a randomized trial of interval secondary cytoreduction in advanced ovarian carcinoma: a Gynecologic Oncology Group study. J Clin Oncol 2005;23:5605–12 | Condition-specific QoL |
| Passik SDK. A pilot examination of the impact of cancer patients’ fatigue on their spousal caregivers. Palliat Support Care 2005;3:273–9 | No QoL data |
| Presant CAT. Effects of weekly paclitaxel or paclitaxel plus carboplatin on functionality and symptoms of geriatric patients with cancer as measured by a brief geriatric oncology module: A pilot experience. Cancer 2005;103:2623–8 | Generic non-preference-based QoL |
| Secord AAJ. Phase I/II trial of intravenous Doxil and whole abdomen hyperthermia in patients with refractory ovarian cancer. Int J Hypertherm 2005;21:333–47 | No QoL data |
| Pujade-Lauraine E, du Bois A, Goupil A, Rochon H, Möbus V, Weber B, et al. Epirubicin/paclitaxel/carboplatin (TEC) vs paclitaxel/carboplatin (TC) in first-line treatment of ovarian cancer FIGO stages IIB-IV. Results of a randomized AGO-GINECO GCIG Intergroup phase III trial. International Journal of Gynecological Cancer 2005;15(6 Suppl. 3):222–3 | No QoL data |
| Advani R, Peethambaram P, Lum BL, Fisher GA, Hartmann L, Long HJ, et al. A Phase II trial of aprinocarsen, an antisense oligonucleotide inhibitor of protein kinase C alpha, administered as a 21-day infusion to patients with advanced ovarian carcinoma. Cancer 2004;100:321–6 | Condition-specific QoL |
| Butler L, Bacon M, Carey M, Zee B, Tu D, Bezjak A. Determining the relationship between toxicity and quality of life in an ovarian cancer chemotherapy clinical trial. J Clin Oncol 2004;22:2461–8 | Condition-specific QoL |
| Berek JS, Taylor PT, Gordon A, Cunningham MJ, Finkler N, Orr J Jr, et al. Randomized, placebo-controlled study of oregovomab for consolidation of clinical remission in patients with advanced ovarian cancer. J Clin Oncol 2004;22:3507–16 | Condition-specific QoL |
| Limat S, Woronoff-Lemsi MC, Menat C, Madroszyk-Flandin A, Merrouche Y. From randomised clinical trials to clinical practice: a pragmatic cost-effectiveness analysis of paclitaxel in first-line therapy for advanced ovarian cancer. Pharmacoecon 2004;22:633–41 | Generic non-preference-based QoL |
| Piao BK, Wang YX, Xie GR, Mansmann U, Matthes H, Beuth J, et al. Impact of complementary mistletoe extract treatment on quality of life in breast, ovarian and non-small cell lung cancer patients. A prospective randomized controlled clinical trial. Anticancer Res 24:303–9 | Condition-specific QoL |
| Solov’ev VI, Semkina EN. [Impact of special treatment methods on life quality and lifespan of patients with widespread forms of ovarian cancer.]. Antibiot Khimioter 2004;49:14–18 | Condition-specific QoL |
| Vasey PA, Jayson GC, Gordon A, Gabra H, Coleman R, Atkinson R, et al. Phase III randomized trial of docetaxel-carboplatin versus paclitaxel-carboplatin as first-line chemotherapy for ovarian carcinoma. J Nat Cancer Inst 2004;96:1682–91 | Condition-specific QoL |
| Rothenberg ML, Liu PY, Wilczynski S, Nahhas WA, Winakur GL, Jiang CS, et al. Phase II trial of vinorelbine for relapsed ovarian cancer: a Southwest Oncology Group study. Gynecol Oncol 2004;95:506–12 | Generic non-preference-based QoL |
| ten Bokkel Huinink W, Lane SR, Ross GA, International Topotecan Study Group. Long-term survival in a phase III, randomised study of topotecan versus paclitaxel in advanced epithelial ovarian carcinoma. Ann Oncol 2004;15:100–3 | Condition-specific QoL |
| May 2013 search | |
| Hilpert E. AURELIA: A randomized phase III trial evaluating bevacizumab (BEV) plus chemotherapy (CT) for platinum (PT)-resistant recurrent ovarian cancer (OC). Journal of Clinical Oncology Conference 2012, var. pagings | No QoL data |
| Uppal S, Hernandez E, Dutta M, Dandolu V, Rose S, Hartenbach E. Prolonged postoperative venous thrombo-embolism prophylaxis is cost-effective in advanced ovarian cancer patients. Gynecol Oncol 2012;127:631–7 | No QoL data |
| Sidhu Kiss N. Quality of life and patient preferences in platinum sensitive ovarian cancer. Value in Health Conference 2012, var. pagings, A429 | No QoL data |
| Nankivell Stark D. Standard chemotherapy with or without bevacizumab in advanced ovarian cancer: Quality-of-life outcomes from the International Collaboration on Ovarian Neoplasms (ICON7) phase 3 randomised trial. Lancet Oncol 2013;14:236–43 | Condition-specific QoL |
Summary of reasons for excluding costing studies
| Reference | Primary reason for exclusion |
|---|---|
| December 2012 search | |
| Geisler Walter A. Annual cost of bevacizumab in the adjuvant treatment of ovarian cancer to the U.S. Medicare system. Gynecol Oncol Conference, March 2012, var. pagings | US study |
| Cajaraville Oyaguez I. Budget impact of trabectedin and pegylated liposomal doxorubicin (PLD) for the treatment of partially platinum-sensitive ovarian cancer. Pharmacoecon – Spanish Research Articles 2012;9:83–94 | Spanish study |
| Pike CTB. Healthcare costs and workloss burden of patients with chemotherapy- associated peripheral neuropathy in breast, ovarian, head and neck, and nonsmall cell lung cancer. Chemother Res Prac 2012. Article no. 913848 | US study |
| Jacobs VRM. Financial quality control of in-patient chemotherapy in Germany: Are additional payments cost-covering for pharmaco-oncological expenses? Breast Care 2011;6:120–5 | German study |
| Frederick Barnes M. Evaluating the true cost of a major phase III GOG clinical trial: a cause for concern. Gynecol Oncol Conference 2010 (var. pagings): S13 | US study |
| Gordon LGS. Medical costs and outcomes for Australian women with ovarian cancer: a patient-level analysis over 2.5 years. Int J Gynecol Cancer 2010;20:757–65 | Australian study |
| Havrilesky LJK. Impact of a chemoresponse assay on treatment costs for recurrent ovarian cancer. Am J Obstet Gynecol 2010;203:160 | US study |
| Gao Parthan A. Health care resource utilization (HRU) in advanced ovarian cancer-findings from linked SEER-Medicare data. Value in Health Conference 2010, var. pagings, A31 | US study |
| Gao Parthan A. Health care resource use (HRU) with nonplatinum chemotherapy for previously treated advanced ovarian cancer (aOC): Findings from SEER-Medicare data. Journal of Clinical Oncology Conference 2010, var. pagings | US study |
| Cooper ALN. Long-term survival and cost of treatment in patients with stage IIIC epithelial ovarian cancer. Curr Womens Health Rev 2009;5:44–50 | US study |
| Havrilesky LJK. Cost analysis of ovarian cancer chemotherapy based on the use of a chemoresponse assay. Gynecologic Oncol Conference, 2009, var. pagings: S22 | US study |
| Nagai Nomura H. [Compared medical costs of treating ovarian cancer patients with weekly paclitaxel, carboplatin (TC) chemotherapy.] Gan To Kagaku Ryoho 2007;34:1091–4 | Japanese study |
| Prasad M, Ben-Porat L, Hoppe B, Aghajanian C, Sabbatini P, Chi DS, et al. Costs of treatment and outcomes associated with second-line therapy and greater for relapsed ovarian cancer. Gynecol Oncol 2004;93:223–8 | US study |
| Evans WKN. Cancer care Ontario’s new drug funding program: Controlled introduction of expensive anticancer drugs. Chron Dis Canada 2002;23:152–6 | Canadian study |
| Bennett CL, Stinson TJ, Yang T, Lurain JR. The effect of reimbursement policies on the management of Medicare patients with refractory ovarian cancer. Seminars Oncol 1999;26(Suppl. 1):40–5 | US study |
| Stinson TJ, Calhoun E, Yang T, Lurain, JR. Cost analysis of second-line therapies for platinum-refractory ovarian cancer: reimbursement dilemmas for Medicare patients. Cancer Invest 1999;17:559–65 | US study |
| Rozek RPB. The costs to the U.S. health care system of extending marketing exclusivity for Taxol. J Res Pharmaceut Econ 1998:21–41 | US study |
| Chica Marchal AML. Pharmacoeconomic study of intravenous antineoplastic therapy in a centralized cytostatics unit. Farmacie Clinica 1995;12:202–9 | Spanish study |
Appendix 7 Data abstraction for Technology Assessment Group economic evaluation
Identified economic evaluations in people with recurrent ovarian cancer
| Author; year; country | Overview | Patient population | Intervention/comparator | Costs and source | Outcomes and source | ICER | Uncertainty |
|---|---|---|---|---|---|---|---|
| NICE; 2013; UK16 | MS, ERG comments and appraisal committee conclusions for NICE TA285 Cost–utility analysis from the perspective of the UK NHS Manufacturer developed a semi-Markov economic model with three health states (PFS, PD, death) based upon NICE TA9113 |
Recurrent platinum-sensitive ovarian cancer | Bevacizumab plus carboplatin and gemcitabine vs. carboplatin and gemcitabine | Costs captured included costs relating to treatment, costs of managing SD, cost of further therapies, cost of AEs, cost of palliative care. Costs discounted at 3.5% | OCEANS provided PFS and OS data on which survival distributions were fitted to extrapolate beyond the trial duration QoL data were taken from TA222.15 Outcomes discounted at 3.5% |
Incremental cost per additional QALY Bevacizumab in addition to gemcitabine and carboplatin vs. gemcitabine and carboplatin was estimated by the manufacturer to be £149,050 (deterministic) in the base case |
Model uncertainty was tested in one-way sensitivity analysis and via Monte Carlo simulations. The Appraisal Committee considered that the uncertainty in estimates of OS in particular meant that the true ICER was likely to be much higher than £149,050 |
| Montalar; 2012; Spain105 | Cost–utility analysis Semi-Markov model with lifetime time horizon. Model based upon TA9113 |
Recurrent platinum-sensitive ovarian cancer | Trabectedin plus PLDH vs. PLDH monotherapy | Costs discounted at 3% Costs captured included drug costs, medical management costs, AE management cost |
Outcomes discounted at 3% PFS and OS taken from OVA-301 Utility taken from EQ-5D data collected as part of OVA-301 |
Incremental cost per QALY Addition of trabectedin vs. PLDH alone resulted in an estimated ICER of 45,592 Euros (2011) |
Addressed through deterministic sensitivity analysis and PSA |
| Havrilesky; 2012; USA98 | Cost–utility analysis Markov model with 24-month time horizon Health states include probability of completed treatment (no disease); PD; and active treatment with or without neurotoxicity |
Recurrent, platinum-sensitive ovarian cancer | Docetaxel and carboplatin combination; docetaxel and carboplatin sequentially | 2010 cost year Costs included: costs associated with AEs with a significant difference in incidence between treatment arms; treatment cost; infusion treatment charges; costs of standard pre-treatment medications Costs were estimated using national 2010 Medicare reimbursement data |
PFS was taken from the published literature and modelled for 24 months at which time > 95% patients had experienced recurrence or had died in each arm Rates of grade 2 and above AEs with a significant difference was documented between treatment arms and modelled QoL was obtained as FACTG and converted to a utility using Dobrez et al.128 QoL estimates were not estimated for health states |
Incremental cost per additional QALY Combination vs. sequential: US$25,239 |
Model uncertainty was tested in one-way sensitivity analysis and via Monte Carlo simulations. At a threshold of US$50,000 the combination was estimated to be cost-effective in 72% of simulations |
| Chan; 2011; USA96 | Cost-effectiveness analysis In-trial analysis |
Recurrent ovarian cancer | Gemcitabine and carboplatin; gemcitabine, carboplatin and bevacizumab | Details of costs included and source of data was not reported | PFS was taken from the OCEANS clinical trial Data on bowel perforation was also taken from OCEANS |
Incremental cost per life-year saved for the addition of bevacizumab to gemcitabine and carboplatin combination therapy was US$253,968 | A series of threshold analyses were carried out on the cost of bevacizumab, PFS and rate of bowel perforation |
| Gore; 2011; UK102 | Cost–utility analysis from the perspective of the UK NHS Decision-analytic model |
Patients with relapsed platinum-sensitive ovarian cancer | Trabectedin plus PLDH; PLDH | Costs were discounted at a rate of 3.5% Drug, administration, medical management and AE costs were based on BNF prices and UK HRG codes |
Outcomes were discounted at a rate of 3.5% Effectiveness data for PFS and OS was based on the Phase III randomised trial OVA301 in 672 patients with relapsed ovarian cancer; parametric survival distributions were fitted to the data from the platinum-sensitive subgroup to calculate mean PFS and OS for each treatment. QoL was measured by EQ-5D data collected in the OVA-301 trial |
Incremental cost per additional QALY Trabectedin plus PLDH vs. PLDH: £37,206 (deterministic) and £39,505 (probabilistic) |
Uncertainty was explored through univariate and PSAs |
| Lee; 2011; Korea107 | Cost–utility analysis Markov model with four health states: responsive; progressive; clinical remission; death. The model time horizon was 10 years, with 9-week cycle length | Korean women with platinum-sensitive ovarian cancer at second line | PLDH and carboplatin; paclitaxel and carboplatin | Both direct and indirect costs were included in the model: drug acquisition costs; test costs; monitoring costs; best supportive care costs; out of pocket costs; transportation related expenses | Median TTP and OS was either estimated from a literature review and meta-analysis or from an expert panel Utilities were obtained from existing literature (reference not reported) |
Incremental cost per QALY PLDH and carboplatin vs. paclitaxel and carboplatin: 19,712,349 Korean won (equivalent to US$18,093) |
Uncertainty was explored though deterministic and PSAs. In deterministic analyses the model was robust to all changes except median TTP. In the probabilistic analysis the probability of cost-effectiveness for PLDH and carboplatin combination was 50.6% at a WTP threshold of 22,000,000 Korean won (US$20,202), the Korean GDP per capita |
| Lesnock; 2011; USA100 | Cost–utility analysis Decision model with three health states: PFS, recurrence, and death |
Women with relapsed ovarian cancer | Carboplatin and paclitaxel; carboplatin and paclitaxel followed by paclitaxel; carboplatin and paclitaxel followed by carboplatin, paclitaxel and bevacizumab | 2009 cost year Costs captured included reimbursement costs of medication and administration, major complications and surveillance With the exception of bevacizumab cost, all costs were estimated based on hospital costs, Medicare reimbursement rates, the Agency for Healthcare Research and Quality database, the American Medical Association database, the Centers for Medicare & Medicaid Services Physician Payment database or Red Book medication costs Bevacizumab cost was included at the cost to the authors’ home institution |
OS, PFS, complications of treatment all taken from the published data QoL adjustments were estimated using a panel of three gynaecological oncology experts |
Incremental cost per additional QALY Carboplatin, paclitaxel and paclitaxel following initial treatment vs. carboplatin and paclitaxel: US$13,402 Carboplatin, paclitaxel and bevacizumab was dominated |
Uncertainty was explored in two-way sensitivity analysis and threshold analyses Sensitivity analyses demonstrated that model conclusions were robust to variation across parameters |
| Lesnock; 2011; USA101 | Cost–utility analysis | Women with relapsed ovarian cancer | Carboplatin and paclitaxel; carboplatin and paclitaxel followed by paclitaxel; carboplatin and paclitaxel followed by carboplatin, paclitaxel and bevacizumab | Reimbursement costs of chemotherapy, administration, complications and surveillance Key data based upon Medicare reimbursement rates, and the Agency for Healthcare Research and Quality database |
OS, PFS, complications of treatment all taken from the published data QoL adjustments were estimated using a panel of three gynaecological oncology experts |
Incremental cost per additional QALY Carboplatin, paclitaxel and paclitaxel following initial treatment vs. carboplatin and paclitaxel: US$12,888 Carboplatin, paclitaxel and bevacizumab dominated when compared with carboplatin, paclitaxel and paclitaxel following initial treatment |
Sensitivity analyses were performed to account for uncertainty and demonstrated that results were robust to PFS variation |
| NICE; 2011; UK15 | MS,93 ERG comments90 and appraisal committee conclusions for NICE TA22215 Cost–utility analysis from the perspective of the UK NHS Manufacturer developed a semi-Markov economic model with three health states (PFS, PD, death) based upon NICE TA9113 |
Women with relapsed platinum-sensitive ovarian cancer | Trabectedin plus PLDH; topotecan; paclitaxel; PLDH | Costs captured included costs relating to treatment, costs of managing SD, cost of PD, cost of AEs | Outcomes were discounted at 3.5% Manufacturer used PFS from OVA-301 and a meta-analysis and presented results for the entire platinum-sensitive population, the PPS population; and the FPS population Interim analyses of OS were taken from OVA-301 AE rates were taken from OVA-301, utilities were derived from the OVA-301 trial that collected EQ-5D |
Incremental cost per QALY gained PLDH vs. paclitaxel: £15,234 Topotecan was dominated by PLDH Trabectedin plus PLDH compared with PLDH alone: £70,076 Alternative results using differing assessments of efficacy through the OVA-301 trial were also presented |
Uncertainty was explored by the manufacturer using one-way and PSA Results showed that key drivers of cost-effectiveness were OS, average number of treatment cycles, drug costs and utility weights. The probability of trabectedin plus PLDH being cost-effective compared with PLDH was approximately 23% at a threshold of £30,000 |
| Papaioannou; 2011; UK99 | ERG assessment and additional analysis associated with a cost–utility analysis submitted by a manufacturer from the perspective of the UK NHS. Manufacturer developed a semi-Markov model with three health states: SD, PD, death Model derived from NICE TA9113 |
Women with relapsed platinum-sensitive ovarian cancer | Trabectedin plus PLDH; paclitaxel; topotecan; PLDH | NR | Evidence on mean TTP and death provided by the manufacturer for NICE TA22215 was derived from a Phase III RCT (OVA-301). The manufacturer extrapolated estimates of survival using the exponential function. The ERG did not agree that this was appropriate and used alternative distributions to represent the data Utilities were taken from OVA-301 |
Incremental cost per QALY gained ERG estimates:Trabectedin plus PLDH vs. PLDH: £46,503 to £54,607 in the PPS population Manufacturer estimates: in the entire population trabectedin plus PLDH vs. PLDH: £94,832; in the PPS population, trabectedin plus PLDH vs. PLDH: £43,996; in the FPS population trabectedin plus PLDH vs. PLDH: £31,092 |
Uncertainty was explored in univariate sensitive analyses for the main analysis and PSA |
| Papaioannou; 2011; UK90 | ERG report for NICE TA222: cost–utility analysis from the perspective of the UK NHS. ERG review and amends to the MS in which the manufacturer developed a semi-Markov economic model with three health states (PFS, PD, death) based upon NICE TA9113 | Women with relapsed platinum-sensitive ovarian cancer | Trabectedin plus PLDH; topotecan; paclitaxel; PLDH | Cost captured included drug and administration costs from the BNF and national reference costs; management costs from assumptions around management requirement and reference costs for costs; costs associated with AEs | Outcomes were discounted at 3.5% Manufacturer estimated efficacy using OVA-301 trial and a meta-analysis with extrapolation using an exponential function. The ERG did not believe that an exponential distribution was appropriate to extrapolate survival. HRQoL was taken from patients within the OVA-301 trial and the values across treatment arms were used. Data by platinum sensitivity was not used by the manufacturer, and the ERG deemed this to be appropriate because the estimated values were counterintuitive |
Incremental cost per QALY gained The ERG reviewed the comparison of PLDH in combination with trabectedin vs. PLDH monotherapy. The ERG considered that the oncologist assessment of progression was most appropriate and noted that the manufacturer’s ICER with this was £39,262 |
The ERG changed a number of parameters and believed that the most plausible ICER for trabectedin in combination with PLDH vs. PLDH in women with PPS disease to be within the range of £46,503 to £54,607 |
| Case; 2007; USA103 | Trial-based cost-effectiveness analysis Perspective of third-party payer |
Hypothetical cohort of 10,000 platinum-sensitive patients with advanced, recurrent, epithelial ovarian cancer | BSC; second-line monotherapy; second–line combination therapy; third-line chemotherapy after disease progression on second-line chemotherapy; fourth-line chemotherapy after disease progression on third-line chemotherapy | 2004 cost year Drug costs and costs associated with chemotherapy administration were included in the economic evaluation Costs were estimated by adjusting local charges using a cost–charge ratio of 60% The University of Alabama was used for all laboratory and procedure cost estimates Pharmacy costs were calculated using average wholesale drug costs |
PFS was used to estimate OS (average PFS plus time in hospice care). PFS data were estimated from the literature, with the exception of BSC, where PFS was estimated based upon clinical experience | Incremental cost per life-year saved: Second-line monotherapy vs. BSC: US$24,228 Second–line combination (vs. second-line monotherapy): US$46,068 Third-line previous combination (vs. second-line combination): US$66,012 Fourth-line previous combination (vs. third-line chemotherapy): US$162,552. Third- and fourth-line previous monotherapy strategies were dominated |
One-way sensitivity analysis was carried out on survival and total costs. No rationale was provided for the selected ranges |
| Havrilesky; 2007; USA104 | Cost-effectiveness analysis with some adjustment for QoL in a sensitivity analysis. A Markov model with 42-month time horizon was developed from the payer perspective | Patients with ovarian cancer recurring at > 6 months following completion of first-line platinum based therapy | Carboplatin; gemcitabine and carboplatin; paclitaxel and carboplatin | 2006 cost year Costs were not discounted Costs of chemotherapy were calculated for a hypothetical 58-year-old woman Costs of AEs were applied to treatment of AEs whose rates differed significantly between treatment groups. All costs were inflated to 2006 US$ using the medical component of the Consumer Price Index |
Survival data were taken from published sources Data on toxicity was taken from published sources. AEs were included if direct medical costs would be incurred and whose rates differed significantly between arms in the published trials |
Incremental cost per progression-free life-year Paclitaxel and carboplatin vs. carboplatin: US$15,564 Gemcitabine and carboplatin vs. paclitaxel and carboplatin: US$278,388 |
PFS was varied using the 95% CIs; one-way sensitivity analysis was undertaken on AE rates and cost of thrombocytopenia; costs of chemotherapy were varied; QoL was included for neurotoxicity |
| Griffin; 2006; UK94 | A publication reporting on the meta-analysis carried out, and the model developed by the assessment group for TA91 Cost–utility analysis from the UK NHS perspective with three health states: SD; PD; death |
Second-line ovarian cancer | PLDH; topotecan; paclitaxel | NR | A systematic review identified RCTs reporting PFS and OS. Data were combined via a MTC meta-analysis | Incremental cost per QALY gained Topotecan: dominated by paclitaxel; PLDH vs. paclitaxel £16,714 |
NR |
| Main; 2006; UK97 | Cost–utility analysis from the perspective of the UK NHS Model with three health states: SD, PD, death |
Advanced second-line ovarian cancer | PLDH; topotecan; paclitaxel | 2003/4 cost year. Costs were not discounted as they were assumed to be incurred in year 1 The costs captured were drug acquisition cost; costs of monitoring; costs of administration; costs of managing AEs Costs were sourced from the literature (AEs), BNF (drug costs), and via data from MSs for NICE TA9113 |
Outcomes were discounted at 1.5% Efficacy was estimated from data obtained from a literature search and manufacturers. Data were meta-analysed using MTC techniques Data were available by subgroup (platinum sensitive vs. platinum refractory and whole population), such that two separate analyses were carried out Utility values were obtained through a systematic review for SD; however, no value for PD was obtained. A proxy was therefore used in breast cancer |
Incremental cost per QALY ERG estimates (analysis 1): PLDH vs. paclitaxel: £7033 in the overall patient population; £5777 in the platinum-sensitive population; and £9555 in the PRR population ERG estimates (analysis 2): cyclophosphamide, doxorubicin and cisplatin vs. platinum monotherapy:£16,421 in the platinum-sensitive population;paclitaxel–platinum combination therapy compared with cyclophosphamide, doxorubicin and cisplatin: £20,950 for the platinum-sensitive population |
Uncertainty was explored through deterministic and PSAs In sensitivity analysis subgroup-specific treatment estimates were applied; results remained similar Cost assumptions were also varied and results remain similar An additional trial was also included; this reduced the effectiveness of PLDH |
| Rocconi; 2006; USA106 | Cost-effectiveness analysis from the perspective of a third-party payer decision–analytic model | A hypothetical cohort of 4000 platinum-resistant patients with recurrent ovarian cancer | BSC; second-line chemotherapy (monotherapy); second-line chemotherapy (combination); third-line chemotherapy after disease progression on second-line monotherapy; third-line chemotherapy after disease progression on second-line combination | 2004 cost year Direct costs were calculated for each strategy Costs were estimated by adjusting local charges using a cost to charge ratio of 60% Laboratory and procedure estimates were taken from the University of Alabama at Birmingham Pharmacy costs were calculated using average wholesale drug costs. AE costs were not included. Cost of BSC was US$135.50 per day |
Clinical estimates were obtained from a review of published literature and included both Phase II and Phase III trials | Incremental cost per life-year saved Second-line monotherapy vs. BSC: US$64,104 Second-line combination vs. second-line monotherapy: US$302,316 Third-line previous combination vs. second-line combination: US$303,984 |
Uncertainty was tested in sensitivity analysis (one way) |
| NICE; 2005; UK10 | MSs for NICE TA91: GSK, Schering-Plough: cost-minimisation analysis BMS: cost-effectiveness analysis Assessment group: summarised in Main et al.97 |
Women with second-line or subsequent advanced ovarian cancer | GSK: topotecan; PLDH Schering-Plough: topotecan; PLDH BMS: paclitaxel and platinum; paclitaxel; topotecan; PLDH |
GSK: costs taken from a published analysis with inclusion of additional costs associated with toxicity monitoring Schering-Plough: similar to published cost-minimisation analysis except expert opinion was used to estimate number and types of resources used to treat all AEs BMS: costs included drug acquisition costs and costs of administration No costs of AEs were included |
BMS: three trials were used to estimate effectiveness in the model. No adjustments were made for differences in baseline characteristics. Survival was estimated up to 3 years | BMS: incremental cost per LYG from paclitaxel/platinum relative to single-agent paclitaxel was £12,120 | Sensitivity analyses carried out by the assessment group were discussed |
| Capri and Cattaneo; 2003; Italy84 | Cost-minimisation analysis from the perspective of Italy’s National Health Service Trial-based estimation of cost based on rationale that PLDH and topotecan efficacy data were similar |
Women with second-line advanced ovarian cancer | PLDH; topotecan | 2002 cost year Direct medical costs including cost of drug, medical visits, laboratory tests, AEs and hospital stays Dosages quantified according to Gordon et al. 200149 A panel of experts determined the resource consumption related to AEs (five oncologists) |
Efficacy data were considered to be similar for PLDH vs. topotecan based upon findings from a Phase III RCT with 474 patients The following AEs were included: anaemia, thrombocytopenia, neutropenia, sepsis, fever, stomatitis/pharyngitis, nausea/vomiting, diarrhoea, PPE |
Mean total cost of treatment with PLDH per patient was €8812 vs. topotecan at €15,788 | Sensitivity analysis tested AE cost and found variability; however, the authors concluded that PLDH was the most efficient choice of treatment |
| Ojeda; 2003; Spain83 | Cost minimisation analysis with trial-based estimation of cost based on rationale that PLDH and topotecan efficacy data were similar | A total of 474 patients with ovarian cancer, all of whom had failed or relapsed after first-line chemotherapy with a platinum-based regimen | PLDH; topotecan | 2001 cost year Direct medical costs (study drug, drug administration, cost of managing AEs) were included in the economic evaluation Cost of study drug was taken from the Spanish Catalogue of Medicinal Products 2001. Unit costs of procedures were taken from the Spanish Data Base of Sanitary Costs and the published literature. Costs were converted from pesetas to euros at the rate of 166.386 pesetas per euro Estimates of resource utilisation associated with treatments when managing AEs was made through an expert panel |
Efficacy data were considered to be similar for PLDH vs. topotecan based upon findings from a Phase III RCT with 474 patients Incidence of the following AEs were included: anaemia, thrombocytopenia, neutropenia, sepsis, fever, stomatitis/pharyngitis, nausea/vomiting, diarrhoea, PPE |
Total cost of PLDH was €9614.72 vs. topotecan where total cost was €11,824.69 The estimated difference in cost in the base case was €2209.97 |
Uncertainty was tested in one-way sensitivity analysis via changes in a number of key variables Results remained favourable to PLDH |
| Forbes; 2002; UK95 | Cost-effectiveness analysis from the perspective of the UK NHS | A total of 474 patients with ovarian cancer, all of whom had failed or relapsed after first-line chemotherapy with a platinum-based regimen | PLDH; topotecan | Costs were taken from Smith 200282 | OS was extrapolated from median survival presented from a MS for NICE TA45.151 Extrapolation was based upon an exponential distribution. HRQoL was not derived from the literature – instead, a sensitivity analysis was conducted to explore what relative magnitude of HRQoL might cause the conclusions of the cost-effectiveness analysis based on life-years to alter | Incremental cost per incremental survival PLDH was dominant compared with topotecan (PLDH was cost saving and improved mean survival duration vs. topotecan) |
Uncertainty was explored in scenario analyses and Monte Carlo simulation The authors found that 80% simulations were dominant for PLDH, and 20% of Monte Carlo simulations resulted in estimates of lower cost and a reduction in survival for PLDH vs. topotecan |
| Smith; 2002; USA and UK82 | Cost minimisation analysis from the payer perspective Trial-based estimation of cost based on rationale that PLDH and topotecan efficacy data were similar |
A total of 474 patients with ovarian cancer, all of whom had failed or relapsed after first-line chemotherapy with platinum-based regimen | PLDH; topotecan | Costs included cost of study drug, cost of drug administration, and management of AEs UK costs were presented as US dollars using the conversion rate of US$1.4 = £1 Cost data were taken from a clinical trial for study drug volume and BNF for estimates of drug cost; clinical trial data for quantities of resource use estimated were used to estimate cost of AE treatment as well as estimates from a panel of oncologists from the USA and UK; costs of blood products come from the National Blood Authority, 2000 tariff; cost of inpatient stay came from a national costing database on literature from a UK Trust that studied patients in ICU; costs of an outpatient clinic visit and a chemotherapy administration come from tariffs at a UK cancer centre and were similar to costs at two other major cancer centres in England |
NA | Total UK cost per patient, US$1.4 = £1: Topotecan, US$16,906 (95% CI US$15,617 to US$18,847); PLDH US$13,997 (95% CI US$12,863 to US$15,392) Incremental cost (P–T): –US$2909 (95% CI –US$3415 to –US$779) |
Uncertainty: in an extreme analysis to favour topotecan, 89% of the replicates showed PLDH to be cost saving |
Additional identified economic evaluations
| Author; year; country | Overview |
|---|---|
| NICE; 2013; UK11 | UK cost–utility analysis modelled using a semi-Markov model comparing bevacizumab plus paclitaxel and carboplatin vs. paclitaxel and carboplatin. Patients were those with first-line ovarian cancer |
| Barnett; 2012; USA152 | US cost-effectiveness analysis modelled using a Markov model comparing bevacizumab incorporated into standard platinum–taxane chemotherapy for all with bevacizumab incorporated into treatment and maintenance for suboptimally debulked stage IV disease, and a predictive biomarker test that would identify a subset of women who derive survival advantage from the addition of bevacizumab. Patients were those with first-line ovarian cancer |
| Chan; 2012; USA153 | US cost-effectiveness analysis comparing the addition of bevacizumab and maintenance bevacizumab to paclitaxel and carboplatin for stage IIIC and stage IV ovarian cancer after primary surgery |
| Dalton; 2012; USA154 | US cost-effectiveness analysis comparing dose dense weekly paclitaxel plus carboplatin vs. paclitaxel plus carboplatin in patients with first-line ovarian cancer |
| Geisler; 2012; USA155 | US cost-effectiveness analysis comparing carboplatin and paclitaxel at four alternative dosages in patients with first-line, high-risk, ovarian cancer |
| Havrilesky; 2012; USA130 | US cost–utility analysis modelled using a Markov model comparing standard treatment; paclitaxel and carboplatin; paclitaxel drug shortage; docetaxel and carboplatin. Patients had newly diagnosed, untreated ovarian cancer |
| Havrilesky; 2012; USA156 | US cost–utility analysis modelled using a Markov model comparing standard treatment; paclitaxel and carboplatin; paclitaxel drug shortage; docetaxel and carboplatin. Patients had newly diagnosed, untreated ovarian cancer |
| Lechuga; 2012; Mexico157 | Mexican cost-effectiveness analysis modelled using a Markov model with three health states (PFS, PD, death), comparing carboplatin plus paclitaxel with bevacizumab plus carboplatin plus paclitaxel, in patients with first-line ovarian cancer |
| Neymark; 2012; Belgium158 | Belgian within-trial cost-effectiveness analysis comparing cisplatin and cyclophosphamide with cisplatin and paclitaxel in women with first-line ovarian cancer stage IIB–IV |
| Cohn; 2011; USA159 | US cost-effectiveness analysis modelled using a decision tree, comparing paclitaxel plus carboplatin (PC) vs. PC plus bevacizumab (PCB) vs. PCB plus bevacizumab maintenance therapy (PCB+B) in patients with first-line ovarian cancer |
| Dalton; 2011; USA160 | US cost-effectiveness analysis modelled using a Markov model comparing dose dense paclitaxel plus carboplatin vs. standard paclitaxel plus carboplatin in women with first-line advanced ovarian cancer |
| Fuh; 2011; USA161 | US cost-effectiveness analysis comparing paclitaxel, carboplatin and bevacizumab and maintenance bevacizumab with gemcitabine, carboplatin and bevacizumab and maintenance bevacizumab. The study investigated cost-effectiveness in the recurrent setting with first-line data |
| Krysinski; 2011; Poland162 | Polish retrospective cost-effectiveness analysis comparing cisplatin plus paclitaxel vs. cisplatin plus cyclophosphamide in women with ovarian cancer stage III and IV |
| Cohn; 2010; USA163 | US cost-effectiveness analysis comparing paclitaxel plus carboplatin (PC) vs. PC plus bevacizumab in patients with advanced first-line ovarian cancer |
| Havrilesky; 2008; USA164 | US cost–utility analysis modelled using a decision analysis and comparing cisplatin plus paclitaxel with carboplatin plus paclitaxel in women with first line stage III optimally resected ovarian cancer |
| Bristow; 2007; USA165 | US cost–utility analysis comparing paclitaxel and cisplatin in patients with first-line ovarian cancer with stage III disease |
| Fedders; 2007; Germany166 | German cost-effectiveness analysis modelled using a Markov model comparing paclitaxel and platinum vs. carboplatin in women with first-line ovarian cancer, as well as topotecan and liposomal doxorubicin as second-line chemotherapy |
| Dranitsaris; 2004; Canada167 | Canadian cost–benefit analysis comparing docetaxel and paclitaxel in patients with first-line advanced ovarian cancer |
| Limat; 2004; France168 | French retrospective cost-effectiveness analysis comparing cyclophosphamide and cisplatin with paclitaxel and cisplatin in patients with first-line advanced ovarian cancer |
| NICE; 2003; UK5 | Guidance on the use of first-line paclitaxel in the treatment of ovarian cancer and summary of submitted manufacturer models for TA55; second-line recommendations were superceded by TA9113 |
| Bennett; 1998; USA169 | US cost–utility analysis comparing the addition of amifostine as an adjunctive supportive therapy to cyclophosphamide plus cisplatinum in patients with newly diagnosed advanced ovarian cancer |
| Berger; 1998; Germany, Spain, France, Italy, the Netherlands and the UK170 | Cost-effectiveness analysis comparing cisplatin plus cyclophosphamide or paclitaxel in women with first-line advanced ovarian cancer |
| Messori; 1998; USA171 | US cost–utility analysis comparing cisplatin based chemotherapy with or without paclitaxel at either a conventional or high dose in patients with newly diagnosed ovarian cancer |
| Elit; 1997; Canada172 | Canadian cost-effectiveness analysis comparing cisplatin and cyclophosphamide with cisplatin and paclitaxel in women with first line stage III/IV ovarian cancer |
| McGuire; 1997; USA173 | US cost-effectiveness analysis comparing paclitaxel plus cisplatin vs. cyclophosphamide plus cisplatin in patients with first-line advanced ovarian cancer |
Identified studies including utility data
| Author; year; country | Population | Health states | Instrument (valuation) | Utility results | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Studies identified from the literature search | ||||||||||||||||||||||||||||||||||
| Hess; 2013; USA125 | People with ovarian cancer enrolled within GOG-0152 or GOG-0172 | No specific health states; instead mean utility at different time points, and overall, were reported | Valuation of FACT scores using two methods: | GOG-0152GOG-0172Mean utilitynMean utilitynCheung et al.1270.8113620.761323Dobrez et al.1280.8413420.801294 | GOG-0152 | GOG-0172 | Mean utility | n | Mean utility | n | Cheung et al.127 | 0.81 | 1362 | 0.76 | 1323 | Dobrez et al.128 | 0.84 | 1342 | 0.80 | 1294 | ||||||||||||||
| GOG-0152 | GOG-0172 | |||||||||||||||||||||||||||||||||
| Mean utility | n | Mean utility | n | |||||||||||||||||||||||||||||||
| Cheung et al.127 | 0.81 | 1362 | 0.76 | 1323 | ||||||||||||||||||||||||||||||
| Dobrez et al.128 | 0.84 | 1342 | 0.80 | 1294 | ||||||||||||||||||||||||||||||
| NICE; 2013; UK (TA285)16 | People with recurrent, platinum-sensitive ovarian cancer enrolled on OVA-301 | SD, PD | EQ-5D | Mean SD 0.718 (se 0.01); mean PD 0.649 (se 0.019) | ||||||||||||||||||||||||||||||
| NICE; 2013; UK (TA284)11 | People with first-line ovarian cancer enrolled on ICON7 | SD, PD | EQ-5D | Mean utilityPFS weeks 0–20.6571PFS weeks 3–50.7153PFS weeks 6–80.7443PFS weeks 9–110.7683PFS weeks 12–140.7643PFS weeks 15–200.7444PFS weeks 21–260.7638PFS weeks 27–320.7718PFS weeks 33–380.7638PFS weeks 39–440.7785PFS weeks 45–500.7533PFS weeks 51–530.7760PFS weeks 54+0.8129PD0.7248 | Mean utility | PFS weeks 0–2 | 0.6571 | PFS weeks 3–5 | 0.7153 | PFS weeks 6–8 | 0.7443 | PFS weeks 9–11 | 0.7683 | PFS weeks 12–14 | 0.7643 | PFS weeks 15–20 | 0.7444 | PFS weeks 21–26 | 0.7638 | PFS weeks 27–32 | 0.7718 | PFS weeks 33–38 | 0.7638 | PFS weeks 39–44 | 0.7785 | PFS weeks 45–50 | 0.7533 | PFS weeks 51–53 | 0.7760 | PFS weeks 54+ | 0.8129 | PD | 0.7248 | |
| Mean utility | ||||||||||||||||||||||||||||||||||
| PFS weeks 0–2 | 0.6571 | |||||||||||||||||||||||||||||||||
| PFS weeks 3–5 | 0.7153 | |||||||||||||||||||||||||||||||||
| PFS weeks 6–8 | 0.7443 | |||||||||||||||||||||||||||||||||
| PFS weeks 9–11 | 0.7683 | |||||||||||||||||||||||||||||||||
| PFS weeks 12–14 | 0.7643 | |||||||||||||||||||||||||||||||||
| PFS weeks 15–20 | 0.7444 | |||||||||||||||||||||||||||||||||
| PFS weeks 21–26 | 0.7638 | |||||||||||||||||||||||||||||||||
| PFS weeks 27–32 | 0.7718 | |||||||||||||||||||||||||||||||||
| PFS weeks 33–38 | 0.7638 | |||||||||||||||||||||||||||||||||
| PFS weeks 39–44 | 0.7785 | |||||||||||||||||||||||||||||||||
| PFS weeks 45–50 | 0.7533 | |||||||||||||||||||||||||||||||||
| PFS weeks 51–53 | 0.7760 | |||||||||||||||||||||||||||||||||
| PFS weeks 54+ | 0.8129 | |||||||||||||||||||||||||||||||||
| PD | 0.7248 | |||||||||||||||||||||||||||||||||
| Montalar; 2012; Spain105 | People with recurrent, platinum-sensitive ovarian cancer enrolled on OVA-301 | SD, PD | EQ-5D | SD 0.72, PD 0.65 | ||||||||||||||||||||||||||||||
| Havrilesky; 2012; USA98 | People with recurrent, platinum-sensitive ovarian cancer who had completed the FACT questionnaire as part of a Phase II RCT. Sample size was not reported, however, the text indicates that participants who completed the FACT questionnaire were enrolled in a Phase II clinical trial with 150 participants | No specific health states were described (e.g. PFS), instead the study reported utility by study arm at different time points | Dobrez et al.129 valued the FACT questionnaire using the TTO method with 1433 patients with cancer who had one of 10 different cancer diagnoses and was 53% male | Utility at randomisation was mean 0.87 for both arms, and 0.83 to 0.84 at the end of the study | ||||||||||||||||||||||||||||||
| Havrilesky; 2012; USA130 | QoL data were sources from a previous study, Leung et al.121 | Utility on treatment with carboplatin and paclitaxel; utility on treatment with carboplatin and docetaxel | Utilities were derived from Leung et al.121 | Utility on treatment with carboplatin and paclitaxel (0.62); utility on treatment with carboplatin and docetaxel (0.51) | ||||||||||||||||||||||||||||||
| Krasner; 2012; UK67 | Six-hundred and seventy-two patients treated with PLDH (n = 335) and trabectedin plus PLDH (n = 337) in a Phase III clinical trial. Although not reported within the paper, the trial recruited women with recurrent ovarian cancer after failure of first-line, platinum-based chemotherapy | Health states were not described, instead QoL was assessed at baseline, and at end of study by treatment group | EQ-5D, valuation was not described | PLDH: 0.78 (sd 0.163) at baseline (n = 318), with –0.05 (sd 0.191) change from baseline (n = 211) Trabectedin plus PLDH; 0.78 (sd 0.171) at baseline (n = 323) with –0.05 (sd 0.201) change from baseline (n = 233) |
||||||||||||||||||||||||||||||
| Pickard; 2012; USA/UK131 | People with advanced breast, brain, colorectal, hepatobiliary system, lung and ovarian cancer. n = 41–49 for each subgroup | The aim of the study was to compare preference based scores between the EQ-5D and FACT, by cancer type. No health states within each cancer were described | EQ-5D and FACT | No utility results for ovarian cancer were presented within the abstract | ||||||||||||||||||||||||||||||
| Grann; 2011; NA132 | QoL data were sourced from a previous study, Grann et al.132 | A QoL value was reported for ovarian cancer | Grann et al.132 valued health states using TTO in two groups of women: women without ovarian cancer, and women with BRCA1/2 mutations | The utility estimated for ovarian cancer was 0.83 (women without ovarian cancer n = 160) and 0.84 (women with BRCA1/2 mutations n = 83) | ||||||||||||||||||||||||||||||
| Lesnock; 2011; USA101 | QoL data were sourced from a previous study, Greving et al.134 | PFS | Utilities were derived from Greving et al.,134 who derived utilities from Grann et al.122 and Grann et al.123 where TTO was used to value health states | The utility for PFS was 0.85 | ||||||||||||||||||||||||||||||
| TA222; 2011; UK90 | IPD within the OVA-301 trial. The OVA-301 trial included 6 72 patients treated with PLDH (n = 335) and trabectedin plus PLDH (n = 337) | PFS; PD | EQ-5D, valuation was not described | As there was no evidence of an interaction between experimental group and health utility, and also no evidence of a systematic difference in health–utility within a health state (stable or progressive) over time, health–utility was based upon the first health–utility estimate for each subject in that health state. Mean SD 0.718 (se 0.01); mean PD 0.649 (se 0.019) | ||||||||||||||||||||||||||||||
| Gordon; 2010; Australia135 | Eighty-five Australian women aged 18–79 years referred for chemotherapy for ovarian cancer but newly presenting or recurrent completed the SF-6D questionnaire; 60 women had recurrent disease of which 55% were platinum sensitive and 37% were platinum resistant (remainder unknown) | Mean SF-6D score by stage of disease (I/II; III; IV) and as a whole group. These groups were a mix of drug therapies, platinum status, first line/subsequent line, etc. | SF-6D, valuation was not described | Stage I/II (n = 13) 0.74 (sd 0.11) Stage III 0.68 (n = 63) (sd 0.09) Stage IV 0.69 (n = 9) (sd 0.08) Total population 0.69 (sd 0.10) |
||||||||||||||||||||||||||||||
| Grann; 2010; Canada133 | One hundred and sixty Canadian women without a personal or family history of breast or ovarian cancer and without known high risk for either breast or ovarian cancer, and 83 women with known BRCA1/2 mutation carrier status were recruited to value health states using the TTO method | Breast and ovarian cancer health states; gene positive health states; prophylactic surgery health states; chemopreventative health states; screening methods | TTO | Mean preference rating for ovarian cancer was 0.84 for mutation carriers and 0.83 for control subjects | ||||||||||||||||||||||||||||||
| Hess; 2010; USA136 | Thirty-four US oncologists who prescribed treatment for women with ovarian cancer. 51 US women with ovarian cancer | Six health states:
|
Standard gamble | Utility scores presented graphically. Reading values from the graph:
|
||||||||||||||||||||||||||||||
| Greving; 2009; the Netherlands134 | Utilities were derived from the literature from studies Grann et al.122 and Grann et al.123 | PFS; relapsed disease | Utilities were derived from the literature from studies Grann et al.122 and Grann et al.123 Both studies used TTO to value health states | Utility for PFS (0.85) and relapsed disease (0.65), although unclear how utilities from Grann et al.122,123 related to these numbers | ||||||||||||||||||||||||||||||
| Havrilesky; 2009; USA124 | Thirty-seven female members of the public without a personal history of ovarian cancer and 13 women with a prior diagnosis of ovarian cancer were recruited to evaluate the 25 health states; the average age of patients was 58, the average age of the volunteers was 41 | 25 descriptive health states | TTO | Diagnosis health states:
|
||||||||||||||||||||||||||||||
| Author; year; country | Population | Health states | Instrument (valuation) | Utility results |
|---|---|---|---|---|
| Havrilesky; 2007; USA104 | QoL for neurotoxicity taken from Sun et al.137 | Neurotoxicity | Utility was derived from Sun et al.137 in which utilities were estimated via TTO | Utility score for neurotoxicity was varied from 0.28 to 1.00. In Sun et al.137 the median utility weight was 0.90 to 1.00 |
| Stein; 2007; UK138 | Sixty-six people with advanced ovarian cancer on chemotherapy who had participated in a RCT of routine QoL measurement, completed the EORTC QLQ-C30 | No specific health states were described (e.g. PFS), instead six clusters of patients were described as health states and included varying proportions of performance status, disease stage and response after treatment | Each health state was valued by the Value of Health Panel, a panel that contains members of the public recruited from the electoral registers of four UK cities. The panel included 39 panel members. Health states were valued using the standard gamble technique | The mean utility for each cluster ranged from 0.694 (cluster 6, high levels of physical, role, and social impairment, poor emotional and cognitive function, older than average age, and highest proportion of metastatic disease) to 0.977 (cluster 1, good performance status, few limitations) |
| Main; 2006; UK97 | Utility for SD taken from Tengs and Wallace,88 a US review of QoL weights in the literature. Utility for PD was taken from Brown and Hutton89 for breast cancer | Health states were SD and PD | Not reported in the study. SD taken from Tengs and Wallace88 from ‘ovarian cancer, metastatic’ with 54 participants via TTO, which, in itself, was taken from Grann et al.122 In Grann et al.,122 the utility of 0.63 was for ‘metastatic disease’ For progressed disease, a utility value was estimated from Brown and Hutton89 by subtracting the utility for progressed disease from the utility for SD |
SD 0.63, PD 0.34 |
| Calhoun; 2004; USA139 | Thirty-nine ovarian cancer patients, 15 women at increased risk, 39 women at baseline risk, and 11 gynaecological oncologists completed utility assessment surveys | Fifteen specific health states reflecting varying levels of toxicity severity, patient functioning and progressive cancer disease for neurotoxicity, nephrotoxicity and ototoxicity | Modified TTO | Mean utility scores were presented for those with disease, those at risk, the general population and physicians. The mean utility scores for the general population (n = 39) were estimated to be: Mild ototoxicity: 0.88 Mild nephrotoxicity: 0.95 Mild neurotoxicity: 0.92 Severe ototoxicity: 0.38 Severe nephrotoxicity: 0.27 Severe neurotoxicity: 0.47 |
| Studies identified from review of reference lists | ||||
| Sun; 2002; USA137 | Forty patients with ovarian cancer enrolled in Phase II trials of high-dose chemotherapy with peripheral stem cell support were asked to participate. 34 completed two surveys. These patients were either second line (n = 27) or third line (n = 7). All women had prior platinum/paclitaxel therapy | Side effects associated with chemotherapy: alopecia, pancytopenia, fatigue, neuropathy, ototoxicity, dysuria, mucositis, nausea/vomiting, hepatotoxicity, ‘ideal’ chemotherapy, ‘worst’ chemotherapy | TTO | Median values, where T1 refers to survey 1, and T2 refers to survey 2:
|
| Tengs; 2000; USA88 | Study was a review of HRQoL weights from the literature. The weight for ‘ovarian cancer, metastatic’ used within Main et al.97 for TA91 was taken from Grann et al.122 from 54 participants via TTO | Ovarian cancer, metastatic and other ovarian cancer health states that were treatment related and captured from the literature | The weight for ‘ovarian cancer, metastatic’ used within Main et al.97 for TA91 was taken from Grann et al.122 from 54 participants via TTO | The weight for ‘ovarian cancer, metastatic’ used within Main et al.97 for TA91 was 0.63 |
| Grann; 1999; USA123 | Twenty-one breast cancer patients, 28 women with a personal history of multiple breast biopsies or a family history of breast cancer, and 135 women without these conditions | Ovarian cancer, metastatic cancer and other cancer states, preventative measures and genetic risk | TTO | Valued by reference group aged 20–32 years (n = 92) Ovarian cancer mean 0.84 (sd 0.22) Metastatic mean 0.73 (sd 0.27) Valued by reference group aged 33–50 years (n = 42) Ovarian cancer mean 0.58 (sd 0.36) Metastatic cancer 0.52 (sd 0.35) |
| Leung; 1999; Canada121 | Twenty-five healthy volunteers and 25 women with breast cancer | Health states by treatment: paclitaxel, docetaxel and vinorelbine; toxicity from treatment, response to treatment and no response to treatment | TTO | No ovarian cancer health states reported |
| Brown; 1998; USA89 | Twenty-nine US oncology nurses at two large oncology centres provided one estimate of average utility; in addition, 25–30 nurses – from each of Germany, Italy, the Netherlands, Spain and the UK – also estimated patient preferences | All breast cancer:
|
Standard gamble | No ovarian cancer health states reported |
| Grann; 1998; USA123 | A sample of 54 participants (unclear whether these participants had or did not have the condition) | Well post oophorectomy; well post mastectomy and oophorectomy; breast cancer; ovarian cancer; metastatic disease | TTO | Ovarian cancer mean = 0.82 (IQR 0.750–1.00); metastatic disease mean = 0.63 (IQR 0.50–0.83) |
Appendix 8 Quality assessment of cost-effectiveness evidence
Quality assessment of the PharmaMar submission compared with the NICE reference case
| Attribute | Reference case | Does the de novo economic evaluation match the reference case? |
|---|---|---|
| Decision problem | The scope developed by NICE | Partially; within the scope, trabectedin was listed as a comparator in people with platinum-sensitive ovarian cancer, and people who are allergic to platinum-based compounds. Within the economic evaluation, the manufacturer considered trabectedin in combination with PLDH in people who are PPS (i.e. a subset of the full platinum-sensitive population) and people who are allergic to platinum-based compounds. The manufacturer’s rationale for this was to ‘align with the inclusion criteria of the OVA-301 trial and the clinical unmet need for non-platinum alternatives in these populations’ (MS, p. 29) |
| Comparator(s) | Alternative therapies routinely used in the NHS | Partially; the manufacturer compared trabectedin in combination with PLDH vs. PLDH monotherapy. However, within the scope, a number of additional therapies were listed as comparator treatments.38 The manufacturer provided rationale for not including these comparators within the submission; however, the TAG considers that consideration of platinum-based therapies as comparators for the group of patients with no allergy or intolerance to platinum would have been appropriate. |
| Perspective costs | NHS and PSS | Yes |
| Perspective benefits | All health effects on individuals | Yes |
| Form of economic evaluation | Cost–utility analysis | Yes |
| Time horizon | Sufficient to capture differences in costs and outcomes | Yes, lifetime time horizon |
| Synthesis of evidence on outcomes | Systematic review | No; utilities were obtained from head-to-head trial data (OVA-301) |
| Outcome measure | QALYs | Yes |
| Health states for QALY | Described using a standardised and validated instrument | Yes, EQ-5D |
| Benefit valuation | Time-trade off or standard gamble | Yes; EQ-5D |
| Source of preference data for valuation of changes in HRQoL | Representative sample of the public | Yes; EQ-5D |
| Discount rate | An annual rate of 3.5% on both costs and health effects | Yes |
| Equity | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | Yes; however, the manufacturer requested consideration under end-of-life criteria |
| Sensitivity analysis | PSA | Yes |
Quality assessment of the PharmaMar submission using the Philips checklist108
| Attribute | Assessment | Comment |
|---|---|---|
| Structure | ||
| S1: Statement of decision problem/objective | Yes | Stated |
| S2: Statement of scope/perspective | Yes | Stated |
| S3: Rationale for structure | Yes | Stated; based upon TA91,13 a previous technology appraisal in recurrent ovarian cancer |
| S4: Structural assumptions | Yes | Stated |
| S5: Strategies/comparators | ? | Partial; the manufacturer compared trabectedin in combination with PLDH vs. PLDH monotherapy. However, within the scope, a number of additional therapies were listed as comparator treatments.38 The manufacturer provided rationale for not including these comparators within the submission; however, the TAG considers that the exclusion of platinum-based therapies as comparators for the group of patients with no allergy or intolerance to platinum was inappropriate |
| S6: Model type | Yes | Stated, semi-Markov model |
| S7: Time horizon | Yes | Stated, lifetime |
| S8: Disease states/pathways | Yes | Stated, PFS and OS |
| S9: Cycle length | – | NA |
| Data | ||
| D1: Data identification | Yes | Stated |
| D2: Pre-model data analysis | ? | Partial; pre-analysis of PFS and OS extrapolation is discussed, but it is not possible to validate the regression analysis controlling for baseline characteristics |
| D2a: Baseline data | ? | Partial, with the exception of the extrapolated curves for PFS and OS all data sources are described |
| D2b: Treatment effects | Yes | Stated |
| D2d: QoL weights (utilities) | Yes | Stated |
| D3: Data incorporation | Yes | Stated |
| D4: Assessment of uncertainty | Yes | Deterministic and probabilistic analysis |
| D4a: Methodological | No | NR |
| D4b: Structural | Yes | Assessed through alternative functional forms for the extrapolated PFS and OS curves |
| D4c: Heterogeneity | Yes | Assessed through consideration of the PPS and FPS populations |
| D4d: Parameter | Yes | Assessed through deterministic and probabilistic analysis |
| Consistency | ||
| C1: Internal consistency | Yes | Discussed |
| C2: External consistency | No | Not assessed; in particular the long tail for OS established via use of the log-logistic extrapolation is not discussed |
Quality assessment of the included economic evaluations against the NICE reference case
| Attribute | Reference case | Comments | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NICE; 2013; UK (TA285)16 | Montalar; 2012; Spain105 | Chan; 2011; USA96 | Havrilesky; 2012; USA98 | Gore; 2011; UK102 | Lee; 2011; Korea107 | Lesnock; 2011; USA100 | Lesnock; 2011; USA101 | NICE; 2011; UK15 | ||
| Decision problem | The scope developed by NICE | Partial | Partial | Partial | Partial | Partial | Partial | Partial | Partial | Partial |
| Comparator(s) | Alternative therapies routinely used in the NHS | Partial | Partial | Partial | Partial | Partial | Partial | Partial | Partial | Partial |
| Perspective costs | NHS and PSS | Yes | No | No | No | Yes | No | No | No | Yes |
| Perspective benefits | All health effects on individuals | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Form of economic evaluation | Cost–utility analysis | Yes | Yes | No, cost per life-year saved | Yes | Yes | Yes | Yes | Yes | Yes |
| Time horizon | Sufficient to capture differences in costs and outcomes | Yes, 10 years | Yes, lifetime | No, trial duration | No, 24 months | Yes | Yes | No, not reported | No, not reported | Yes |
| Synthesis of evidence on outcomes | Systematic review | No, head-to-head clinical trial | No, head-to-head clinical trial | No, head-to-head clinical trial | No, head-to-head clinical trial | No, head-to-head clinical trial | Yes | No, not reported | Yes | No, head-to-head clinical trial |
| Outcome measure | QALYs | Yes | Yes | No, life-years saved | Yes | Yes | Yes | Yes | Yes | Yes |
| Health states for QALY | Described using a standardised and validated instrument | Yes, EQ-5D | Yes, EQ-5D | NA | Yes, FACT mapped to utility | Yes, EQ-5D | No, not reported | No, not reported | No, expert opinion | Yes, EQ-5D |
| Benefit valuation | Time trade-off or standard gamble | Yes, TTO via EQ-5D | Yes, TTO via EQ-5D | NA | No, not reported | Yes, TTO via EQ-5D | No, not reported | No, not reported | No, expert opinion | Yes, TTO via EQ-5D |
| Source of preference data for valuation of changes in HRQoL | Representative sample of the public | Yes, the public via EQ-5D | Yes, the public via EQ-5D | NA | No, not reported | Yes, the public via EQ-5D | No, not reported | No, not reported | No, expert opinion | Yes, the public via EQ-5D |
| Discount rate | An annual rate of 3.5% on both costs and health effects | Yes | No, 3% on both costs and benefits | No, not reported | No | Yes | No, not reported | No, not reported | No, not reported | Yes |
| Equity | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | Yes | Yes | NA | Yes | Yes | Yes | Yes | Yes | Yes |
| Sensitivity analysis | PSA | Yes | Yes | No | Yes | Yes | No | No | No | Yes |
| Attribute | Reference case | Comments | ||||||||
| Papaioannou; 2011; UK99 | Papaioannou; 2010; UK174 | Case; 2007; USA103 | Havrilesky; 2007; USA104 | Griffin; 2006; UK94 | Main; 2006; UK97 | Rocconi; 2006; USA106 | NICE; 2005; UK10 | Forbes; 2002; UK95 | ||
| Decision problem | The scope developed by NICE | Partial | Partial | Partial | Partial | Partial | Partial | Partial | Partial | Partial |
| Comparator(s) | Alternative therapies routinely used in the NHS | Partial | Partial | Partial | Partial | Partial | Partial | Partial | Partial | Partial |
| Perspective costs | NHS and PSS | Yes | Yes | No | No | Yes | Yes | No | Yes | Yes |
| Perspective benefits | All health effects on individuals | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Form of economic evaluation | Cost–utility analysis | Yes | Yes | No | No | Yes | Yes | No | Yes | No |
| Time horizon | Sufficient to capture differences in costs and outcomes | Yes | Yes | Yes, implicitly lifetime | No, 42 months | Yes | Yes | Yes | Yes | Yes |
| Synthesis of evidence on outcomes | Systematic review | No, head-to-head clinical trial | No, head-to-head clinical trial | Partial; assumptions made for best supportive care | Partial | Yes | Yes | Partial | Yes | Yes |
| Outcome measure | QALYs | Yes | Yes | No | No | Yes | Yes | No | Yes | No |
| Health states for QALY | Described using a standardised and validated instrument | Yes, EQ-5D | Yes, EQ-5D | NA | NA | ? | ? | NA | ? | NA |
| Benefit valuation | Time trade-off or standard gamble | Yes, time trade-off via EQ-5D | Yes, time trade-off via EQ-5D | NA | NA | Yes | Yes | NA | Yes | NA |
| Source of preference data for valuation of changes in HRQoL | Representative sample of the public | Yes, the public via EQ-5D | Yes | NA | NA | ? | ? | NA | ? | NA |
| Discount rate | An annual rate of 3.5% on both costs and health effects | Yes | Yes | No | No | Yes | Yes | No | Yes | Partial, discounting at 6% |
| Equity | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | Yes | Yes | NA | NA | Yes | Yes | Yes | Yes | NA |
| Sensitivity analysis | PSA | Yes | Yes | No | No | No | Yes | No | Yes | Yes |
Quality assessment of the included economic evaluations using the Philips checklist108
| Dimension of quality | Comments | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study | Chan; 2011; USA96 | Havrilesky; 2012; USA98 | Gore; 2011; UK102 | Lee; 2011; Korea107 | Lesnock; 2011; USA100 | Lesnock; 2011; USA101 | ||||||
| Structure | ||||||||||||
| S1: Statement of decision problem/objective | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| S2: Statement of scope/perspective | ✗ | Not stated | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✗ | Not stated | ✓ | Stated |
| S3: Rationale for structure | ✗ | Not stated | ✗ | Not stated | ✗ | Not stated | ✗ | Not stated | ✗ | Not stated | ✗ | Not stated |
| S4: Structural assumptions | ✗ | Not stated; the analysis was a within-trial evaluation | ✓ | Stated | ? | Partially | ✓ | Stated | ✗ | Not stated | ✓ | Stated |
| S5: Strategies/comparators | ? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
| S6: Model type | ✓ | Within-trial economic evaluation | ✓ | Markov model based upon RCT | ✓ | Decision-analytic model based upon RCT | ✓ | Markov model | ✓ | Decision-analytic model based upon RCT | ✓ | Markov model |
| S7: Time horizon | ✗ | Trial duration, < 1 year; unlikely to reflect the lifetime horizon for these patients | ✗ | 24 months; unlikely to reflect the lifetime horizon for these patients | ✓ | Lifetime time horizon | ✓ | Ten years; likely to reflect the lifetime time horizon for these patients | ✗ | NR | ✗ | NR |
| S8: Disease states/pathways | ? | Partially, PFS and bowel perforation was captured, but OS was not considered | ? | Partially, PFS and neurotoxicity was captured, but OS was not considered | ✓ | PFS, OS | ✓ | Responsive, progressive, clinical remission, death | ✓ | PFS, OS | ✓ | PFS, OS, complications |
| S9: Cycle length | – | NA | ✓ | 21 days, equivalent to one chemotherapy cycle | – | NA | ? | 9 weeks; no rationale for this duration provided | ✗ | NR | ✗ | NR |
| Data | ||||||||||||
| D1: Data identification | ? | Partially | ✓ | Stated | ? | Partially | ? | Partially | ? | Partially | ✓ | Stated |
| D2: Pre-model data analysis | ? | NR | ✓ | Stated | ? | NR | ✓ | Stated | ? | NR | ? | NR |
| D2a: Baseline data | ? | NR | ✓ | Stated | ? | NR | ? | NR | ? | NR | ✓ | Stated |
| D2b: Treatment effects | ? | PFS and bowel perforation, although not reported in sufficient detail to obtain estimates | ✓ | Stated | ? | Relative treatment effects were reported for PFS and OS | ✗ | NR | ✗ | NR | ✓ | Stated |
| D2d: QoL weights (utilities) | ✗ | NA; no QoL weights were used | ✓ | Condition-specific weights were mapped to utilities | ✓ | EQ-5D data were used | ✗ | NA; no QoL weights were used | ? | Utilities, but with no description of how these have been obtained | ? | Utilities, but limited description of methods |
| D3: Data incorporation | ✗ | It is not possible to validate the incorporation of data due to a lack of reporting | ✓ | Stated | ✗ | It is not possible to validate the incorporation of data due to a lack of reporting | ✗ | It is not possible to validate the incorporation of data due to a lack of reporting | ✗ | It is not possible to validate the incorporation of data due to a lack of reporting | ✓ | Stated |
| D4: Assessment of uncertainty | ? | Some scenario analyses were carried out | ? | A number of sensitivity analyses were carried out | ? | A number of sensitivity analyses were carried out | ? | A number of sensitivity analyses were carried out | ? | A number of sensitivity analyses were carried out | ? | A number of sensitivity analyses were carried out |
| D4a: Methodological | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR |
| D4b: Structural | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR |
| D4c: Heterogeneity | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR |
| D4d: Parameter | ✓ | Some scenario analyses on PFS benefit and bowel perforation | ✓ | A number of one-way sensitivity analyses were carried out, as were Monte Carlo simulations, accounting for simultaneous uncertainty | ✓ | PSA | ✓ | One-way sensitivity analysis | ✓ | Some scenario analyses on PFS and OS | ✓ | A number of deterministic sensitivity analyses were carried out, as were Monte Carlo simulations, accounting for simultaneous uncertainty |
| Consistency | ||||||||||||
| C1: Internal consistency | ✗ | NR | ✓ | Discussed | ✗ | NR | ✗ | NR | ✗ | NR | ✓ | Discussed |
| C2: External consistency | ✗ | NR | ✓ | Comparison with results from previous cost-effectiveness analyses was considered | ✗ | NR | ✗ | NR | ✗ | NR | ✗ | NR |
| Dimension of quality | Comments | |||||||||||
| Study | NICE; 2011; UK15 | Papaioannou; 2011; UK99 | Papaioannou; 2011; UK90 | Case; 2007; USA103 | Havrilesky; 2007; USA104 | Griffin; 2006; UK94 | ||||||
| Structure | ||||||||||||
| S1: Statement of decision problem/objective | ✓ | Discussion of MS | ✓ | Discussion of manufacturer submission | ✓ | Discussion of manufacturer submission | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| S2: Statement of scope/perspective | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| S3: Rationale for structure | – | NA; discussion of manufacturer’s structure only | – | NA; discussion of manufacturer’s structure only | – | NA; discussion of manufacturer’s structure only | ✗ | NR | ✗ | NR | ✗ | NR |
| S4: Structural assumptions | – | NA; discussion of manufacturer’s structure only | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| S5: Strategies/comparators | ? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
| S6: Model type | ✓ | Stated, semi-Markov model | ✓ | Stated, semi-Markov model | ✓ | Stated, semi-Markov model | ✓ | Stated, decision-analytic model | ✓ | Stated, Markov model | ✓ | Stated, semi-Markov model |
| S7: Time horizon | ✓ | Lifetime | ✓ | Lifetime | ✓ | Lifetime | ✓ | Although not explicitly stated, implicitly lifetime | ✗ | 42 months; unlikely to reflect the lifetime horizon for these patients | ✓ | Lifetime |
| S8: Disease states/pathways | ✓ | PFS, OS | ✓ | PFS, OS | ✓ | PFS, OS | ✓ | PFS, OS | ? | PFS and neurotoxicity | ✓ | PFS, OS |
| S9: Cycle length | – | NA | – | NA | – | NA | – | NA | ✓ | 3 months | – | NA |
| Data | ||||||||||||
| D1: Data identification | ✓ | Reported | ✓ | Reported | ✓ | Reported | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| D2: Pre-model data analysis | ✓ | Reported | ✓ | Reported | ✓ | Reported | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| D2a: Baseline data | ✓ | Reported | ✓ | Reported | ✓ | Reported | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| D2b: Treatment effects | ✓ | Reported | ✓ | Reported | ✓ | Reported | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| D2d: QoL weights (utilities) | ✓ | Reported | ✓ | Reported | ✓ | Reported | ✗ | QoL not considered | ✗ | QoL not considered in the main analysis | ✓ | Not reported in detail |
| D3: Data incorporation | ✓ | Reported | ✓ | Reported | ✓ | Reported | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| D4: Assessment of uncertainty | ✓ | Reported | ✓ | Reported | ✓ | Reported | ✓ | Stated | ✓ | Stated | ? | Not reported in detail |
| D4a: Methodological | – | NA | ✓ | Reported | ✓ | Reported | ✗ | NR | ✗ | NR | ✓ | Stated; related to the NMA |
| D4b: Structural | ✓ | Reported | ✓ | Reported | ✓ | Reported | ✗ | NR | ✓ | Stated | ✗ | NR |
| D4c: Heterogeneity | ✓ | Subgroups reported | ✓ | Subgroups reported | ✓ | Subgroups reported | ✗ | NR | ✓ | Stated | ✗ | NR |
| D4d: Parameter | ✓ | Reported | ✓ | Reported | ✓ | Reported | ✓ | Stated | ✓ | Stated | ✗ | NR |
| Consistency | ||||||||||||
| C1: Internal consistency | ✓ | Discussed | ✓ | Discussed | ✓ | Discussed | ✗ | NR | ✗ | NR | ✓ | Stated |
| C2: External consistency | ✓ | Discussed | ✓ | Discussed | ✓ | Discussed | ✗ | NR | ✓ | Stated | ✗ | NR |
| Dimension of quality | Comments | |||||||||||
| Study | Main; 2006; UK97 | Rocconi; 2006; USA106 | NICE; 2005; UK10 | Forbes; 2002; UK95 | NICE; 2013; UK (TA285)16 | Montalar; 2012; Spain105 | ||||||
| Structure | ||||||||||||
| S1: Statement of decision problem/objective | ✓ | Stated | ✓ | Stated | ✓ | Discussion of manufacturers’ submissions and TAG report | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| S2: Statement of scope/perspective | ✓ | Stated | ? | Partially | ✓ | Stated | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| S3: Rationale for structure | ✓ | Stated | ✗ | NR | – | NA | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| S4: Structural assumptions | ✓ | Stated | ✓ | Stated | – | NA | ✓ | Stated | ✓ | Stated | ✓ | Stated |
| S5: Strategies/comparators | ? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
? | Did not include the full range of comparators, but considered:
|
| S6: Model type | ✓ | Stated, semi-Markov | ✓ | Stated, decision-analytic model | ✓ | Stated, semi-Markov model | ✓ | Stated, cost-minimisation analysis | ✓ | Stated, semi-Markov model | ✓ | Stated, semi-Markov model |
| S7: Time horizon | ✓ | Lifetime | ✓ | Implicitly lifetime | ✓ | Lifetime | ✓ | Lifetime | ✓ | 10 years | ✓ | Lifetime |
| S8: Disease states/pathways | ✓ | PFS, OS | ✓ | PFS, OS | ✓ | PFS, OS | ✓ | OS | ✓ | PFS, OS | ✓ | PFS, OS |
| S9: Cycle length | – | NA | – | NA | – | NA | – | NA | ✓ | 1 week | – | NA |
| Data | ||||||||||||
| D1: Data identification | ✓ | Stated | ✓ | Stated | ✓ | Reported | ✓ | Stated | ✓ | Reported | ✓ | Stated |
| D2: Pre-model data analysis | ✓ | Stated | ✓ | Stated | ✓ | Reported | ✓ | Stated | ✓ | Reported | ✓ | Stated |
| D2a: Baseline data | ✓ | Stated | ✓ | Stated | ✓ | Reported | ✓ | Stated | ✓ | Reported | ✓ | Stated |
| D2b: Treatment effects | ✓ | Stated | ✓ | Stated | ✓ | Reported | ✓ | Stated | ✓ | Reported | ✓ | Stated |
| D2d: QoL weights (utilities) | ✓ | Stated | ✗ | QoL not considered | ✓ | Reported | ✗ | QoL not considered | ✓ | Reported | ✓ | Stated |
| D3: Data incorporation | ✓ | Stated | ✓ | Stated | ✓ | Reported | ✓ | Stated | ✓ | Reported | ✓ | Stated |
| D4: Assessment of uncertainty | ✓ | Stated | ✓ | Stated | ✓ | Reported | ✓ | Stated | ✓ | Reported | ✓ | Stated |
| D4a: Methodological | ✓ | Stated | ✗ | NR | – | NA | ✓ | Stated | ✓ | Reported | ✓ | Stated |
| D4b: Structural | ✗ | NR | ✗ | NR | – | NA | ✓ | Stated | ✓ | Reported | ✓ | Stated |
| D4c: Heterogeneity | ✓ | Stated; subgroups | ✗ | NR | ✓ | Subgroups reported | ✗ | NR | ✓ | Reported | ✗ | NR |
| D4d: Parameter | ✓ | Stated | ✓ | Stated | ✓ | Reported | ✓ | Stated | ✓ | Reported | ✓ | Stated |
| Consistency | ||||||||||||
| C1: Internal consistency | ✓ | Discussed | ✗ | NR | ✓ | Discussed | ✓ | Discussed | ✓ | Discussed | ✓ | Discussed |
| C2: External consistency | ✓ | Discussed | ✓ | Discussed | ✓ | Discussed | ✓ | Discussed | ✓ | Discussed | ✗ | NR |
Appendix 9 Survival curves for the Technology Assessment Group economic model
Platinum-sensitive network 1
FIGURE 61.
Progression-free survival proportions for PS network 1.

FIGURE 62.
Overall survival proportions for PS network 1.
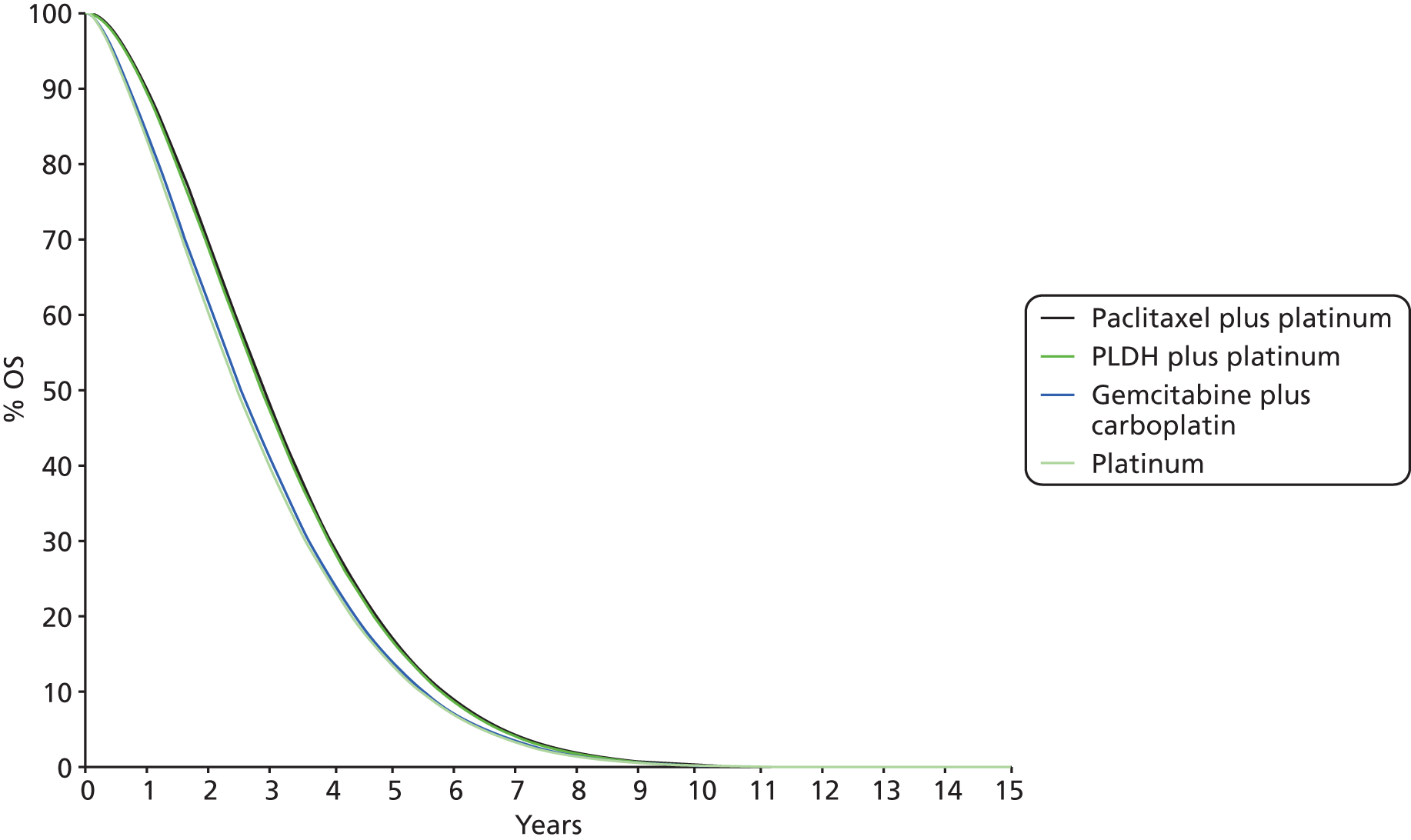
FIGURE 63.
Progressed disease proportions for PS network 1.
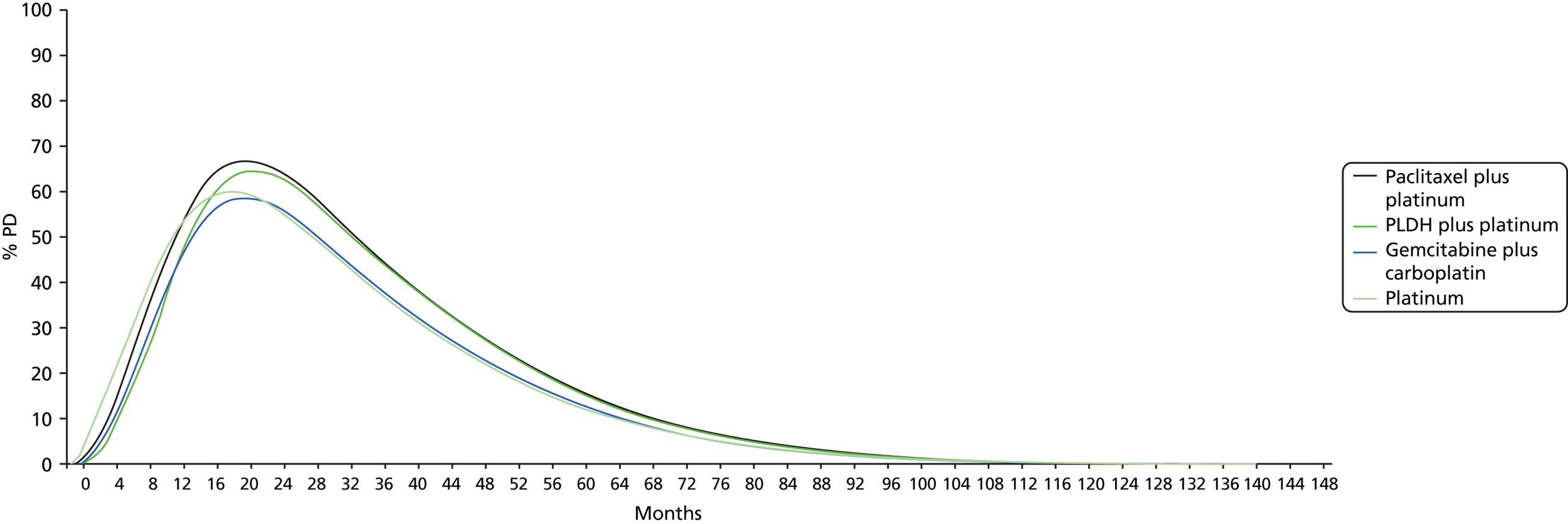
Platinum-sensitive network 2
FIGURE 64.
Progression-free survival proportions for PS network 2.
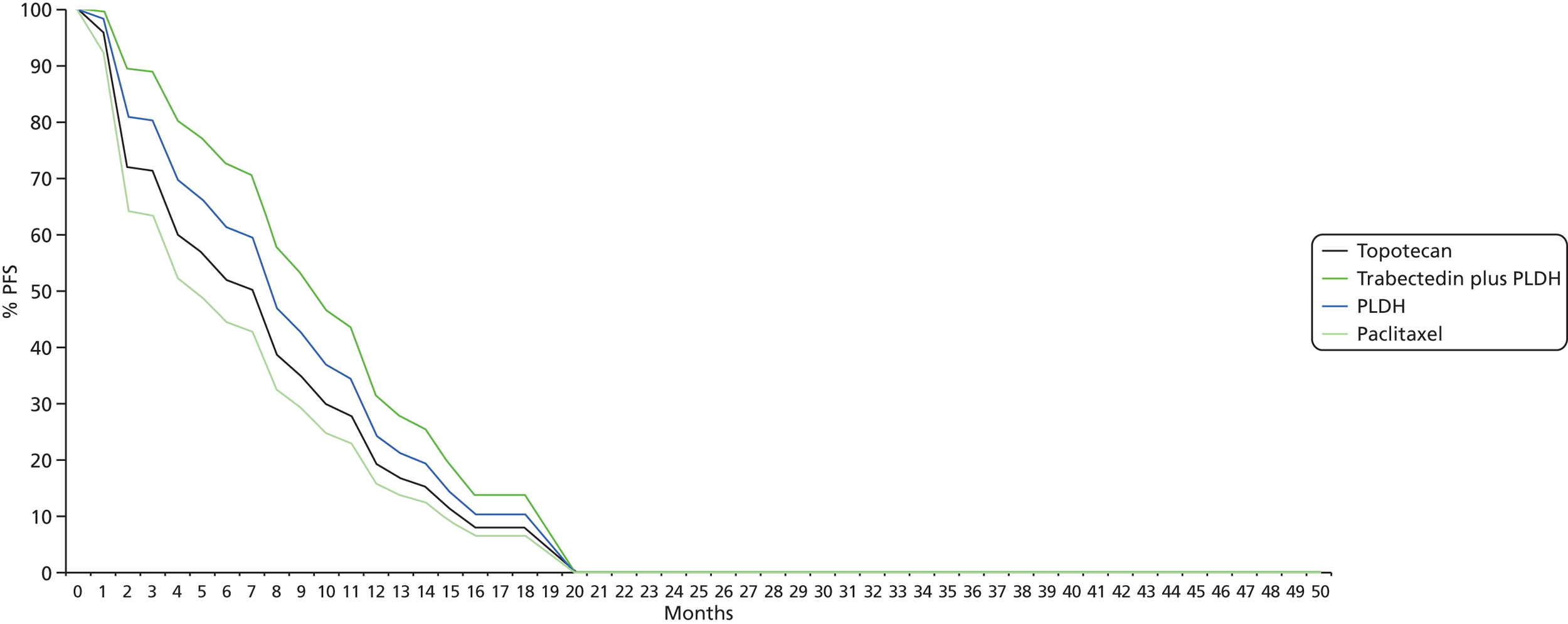
FIGURE 65.
Overall survival proportions for PS network 2.
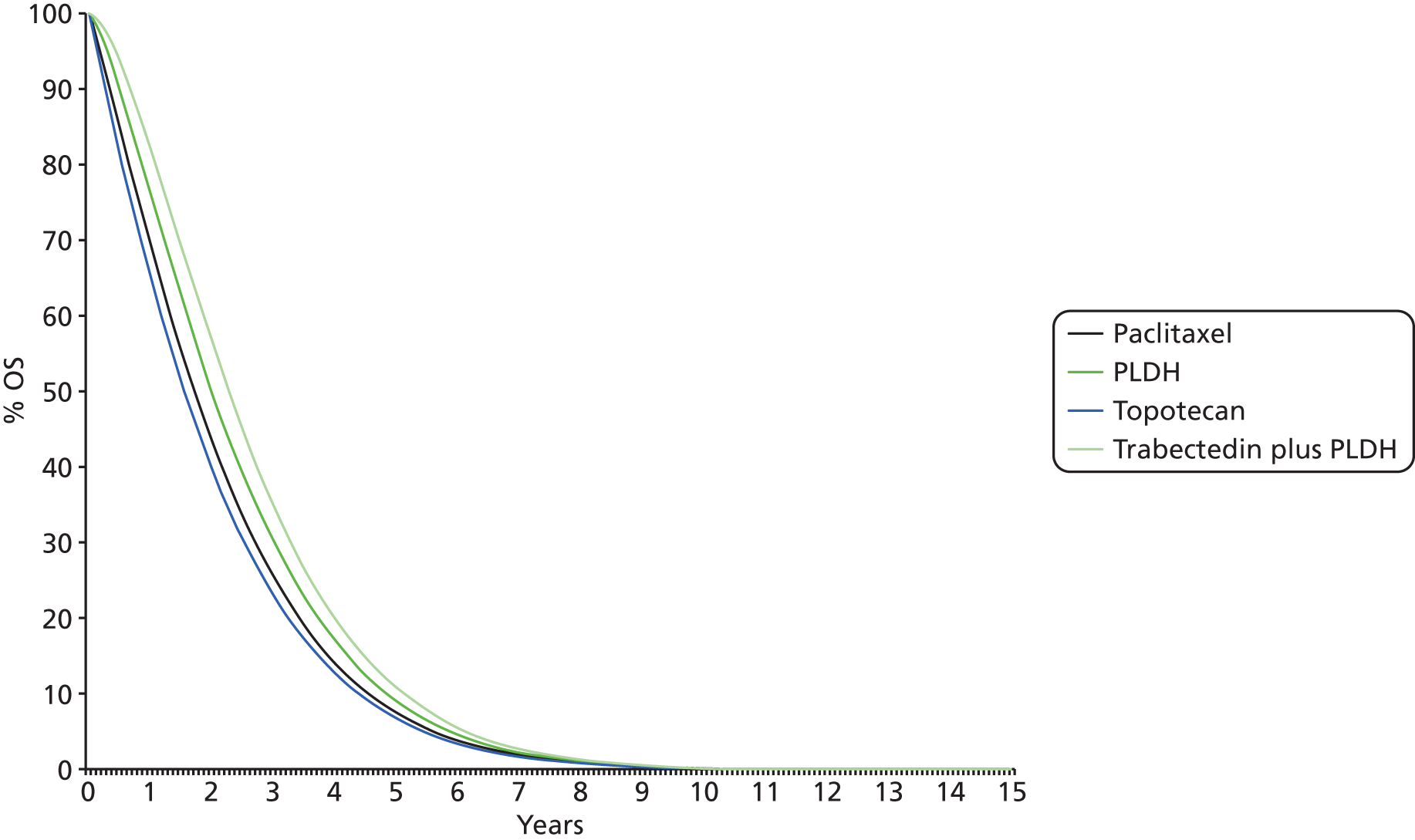
FIGURE 66.
Progressed disease proportions for PS network 2.
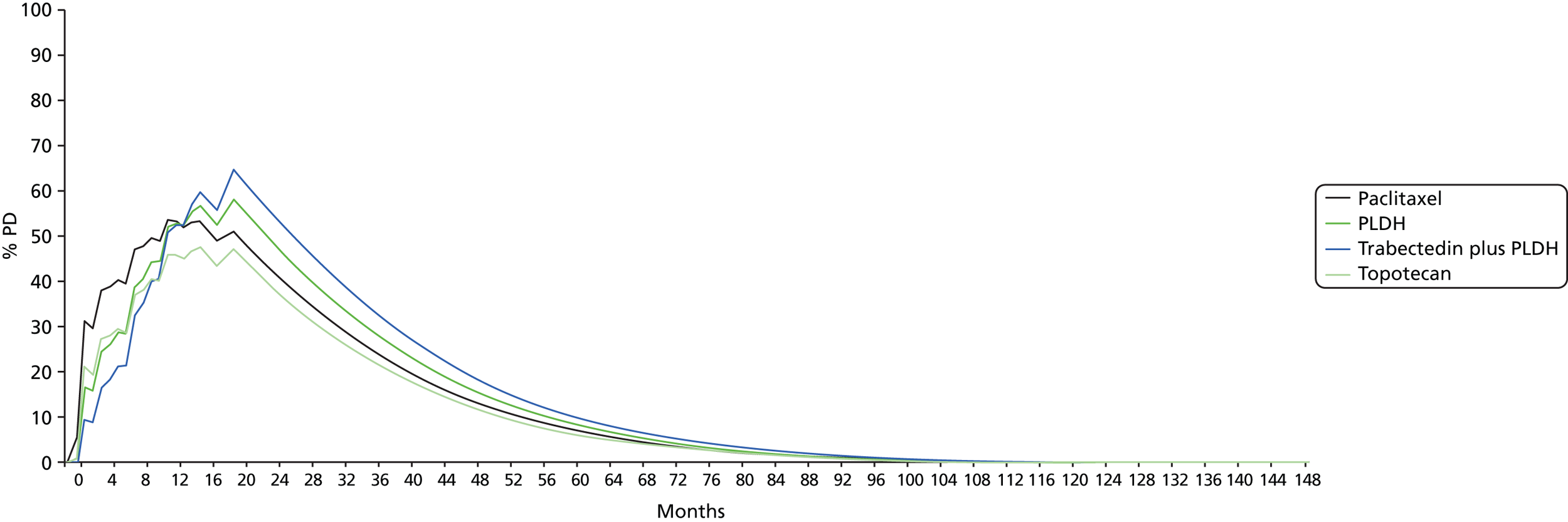
Platinum-resistant/-refractory network
FIGURE 67.
Progression-free survival proportions for PRR network.

Appendix 10 Cumulative log-hazard plots
Platinum-sensitive network 1
FIGURE 68.
Cumulative log-hazards associated with Kaplan–Meier PFS data for carboplatin plus paclitaxel vs. carboplatin plus PLDH.
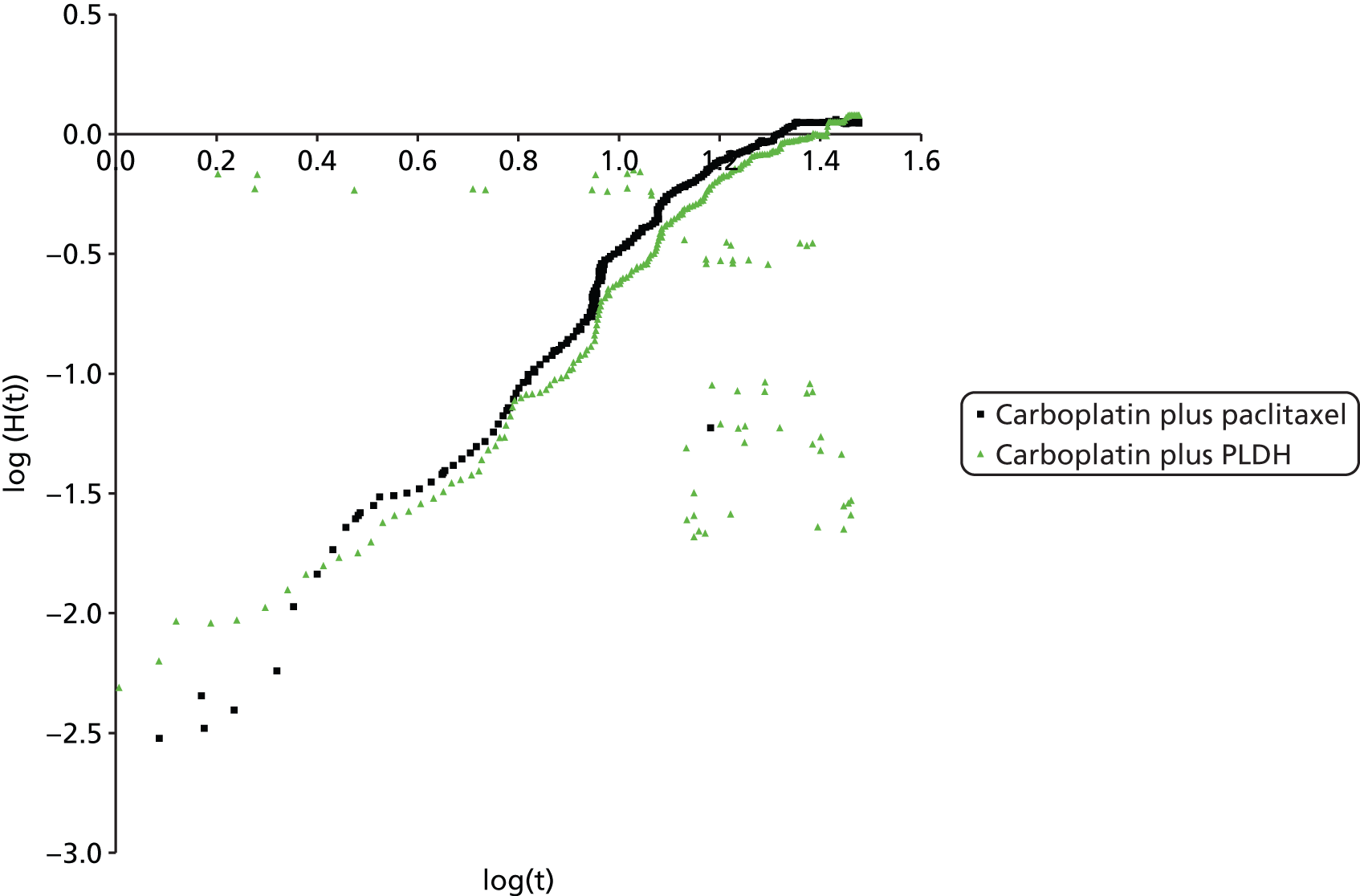
FIGURE 69.
Cumulative log-hazards associated with Kaplan–Meier PFS data for carboplatin vs. carboplatin plus paclitaxel.

FIGURE 70.
Cumulative log-hazards associated with Kaplan–Meier PFS data for platinum vs. paclitaxel plus platinum.

FIGURE 71.
Cumulative log-hazards associated with Kaplan–Meier PFS data for carboplatin vs. gemcitabine plus carboplatin.

FIGURE 72.
Cumulative log-hazards associated with Kaplan–Meier PFS data for carboplatin plus PLDH vs. carboplatin.
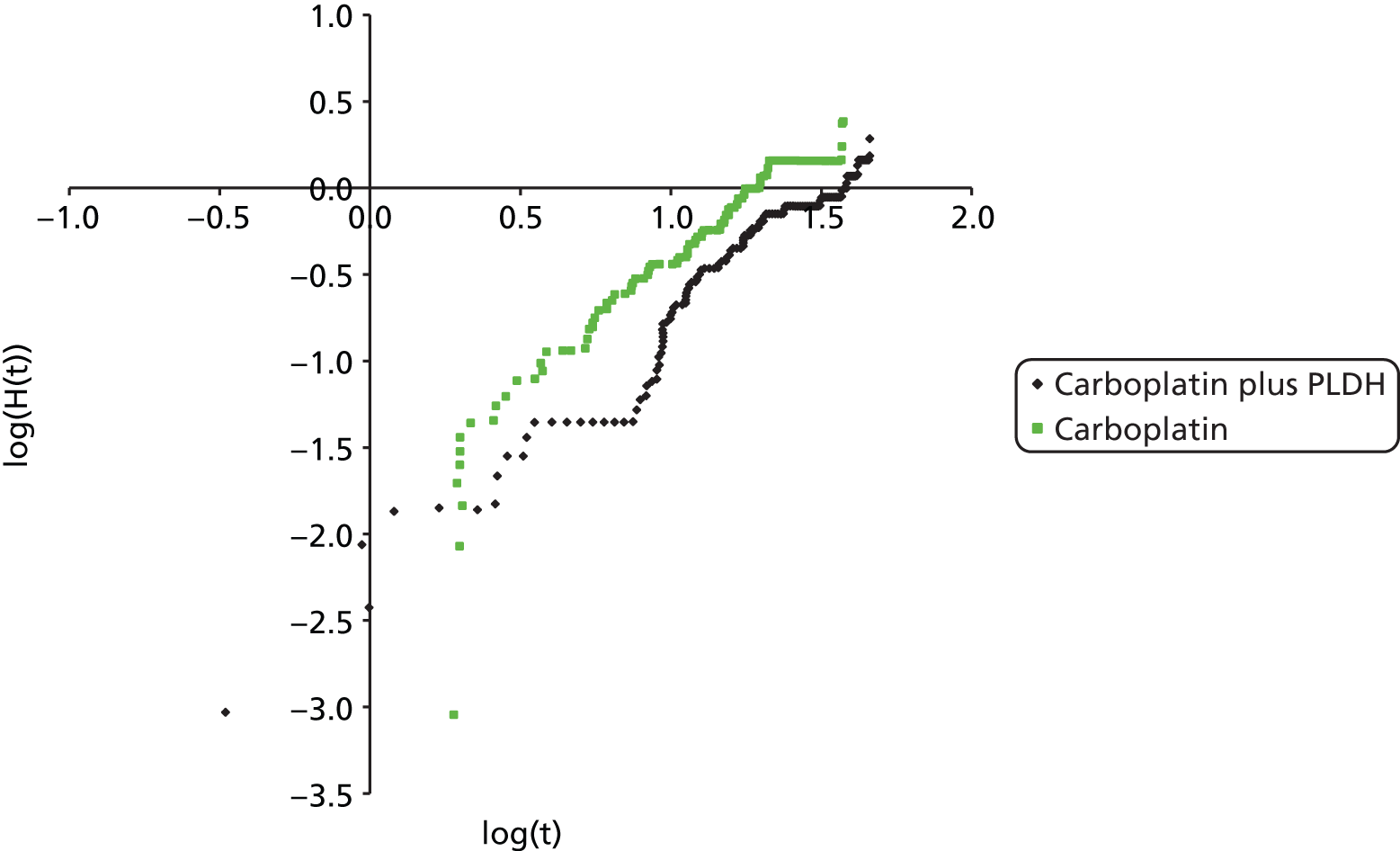
FIGURE 73.
Cumulative log-hazards associated with Kaplan–Meier PFS data for all treatments considered.

FIGURE 74.
Cumulative log-hazards associated with Kaplan–Meier OS data for carboplatin plus paclitaxel vs. carboplatin plus PLDH.

FIGURE 75.
Cumulative log-hazards associated with Kaplan–Meier OS data for carboplatin vs. carboplatin plus paclitaxel.

FIGURE 76.
Cumulative log-hazards associated with Kaplan–Meier OS data for platinum vs. paclitaxel plus platinum.

FIGURE 77.
Cumulative log-hazards associated with Kaplan–Meier OS data for carboplatin vs. gemcitabine plus carboplatin.

FIGURE 78.
Cumulative log-hazards associated with Kaplan–Meier OS data for carboplatin plus PLDH vs. carboplatin.
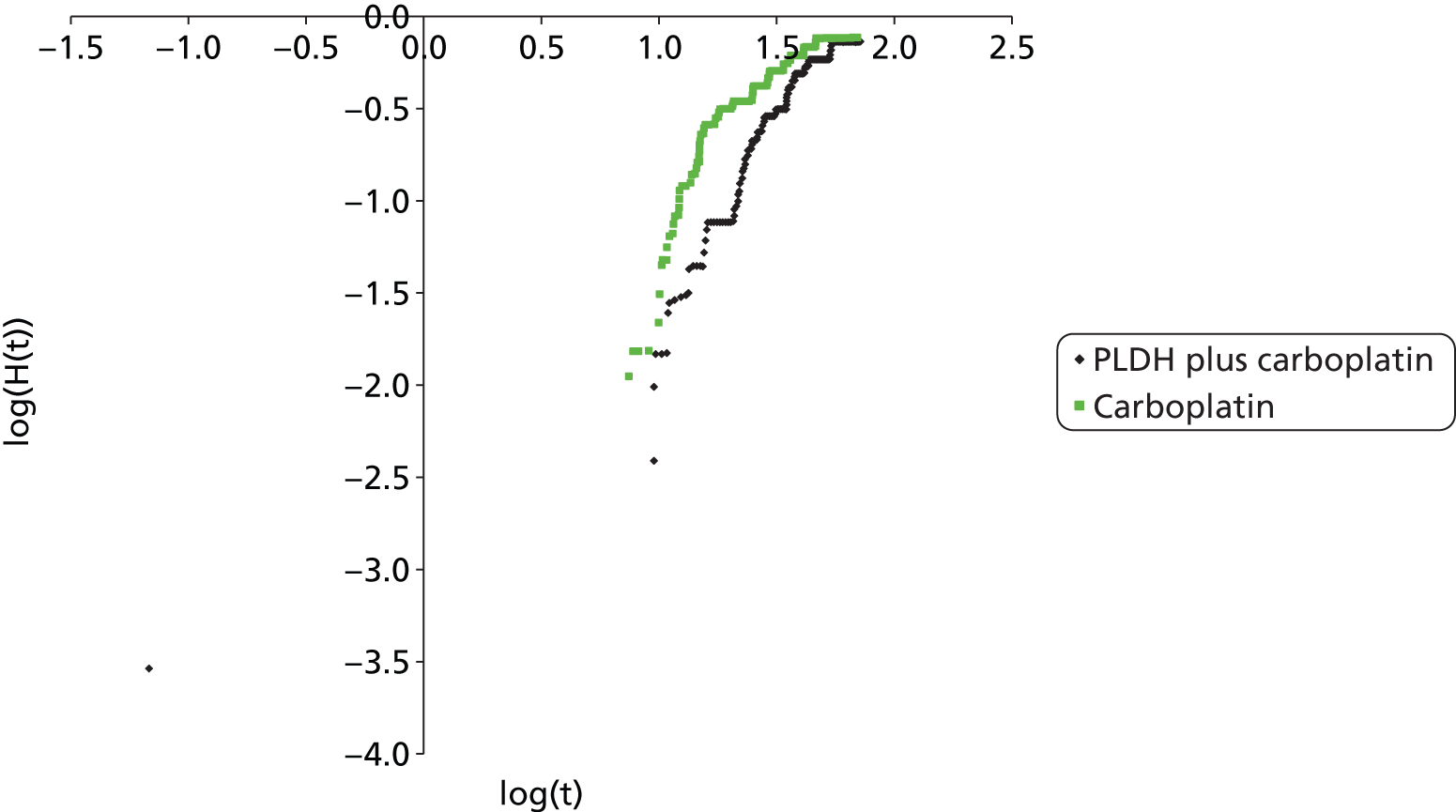
FIGURE 79.
Cumulative log-hazards associated with Kaplan–Meier OS data for all treatments considered.

Platinum-sensitive network 2
FIGURE 80.
Cumulative log-hazards associated with Kaplan–Meier PFS data for trabectedin plus PLDH vs. PLDH.
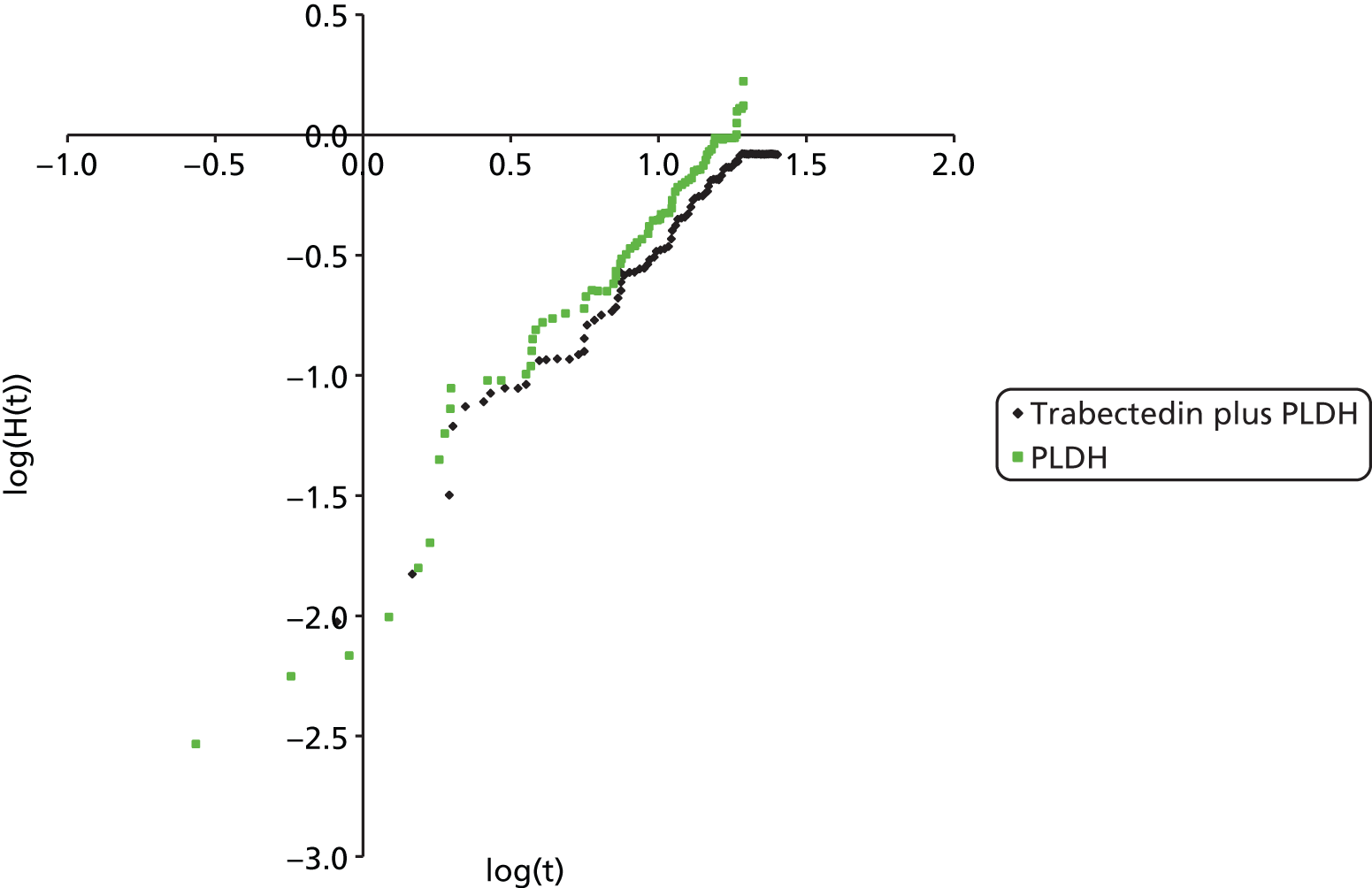
FIGURE 81.
Cumulative log-hazards associated with Kaplan–Meier PFS data for topotecan vs. PLDH.
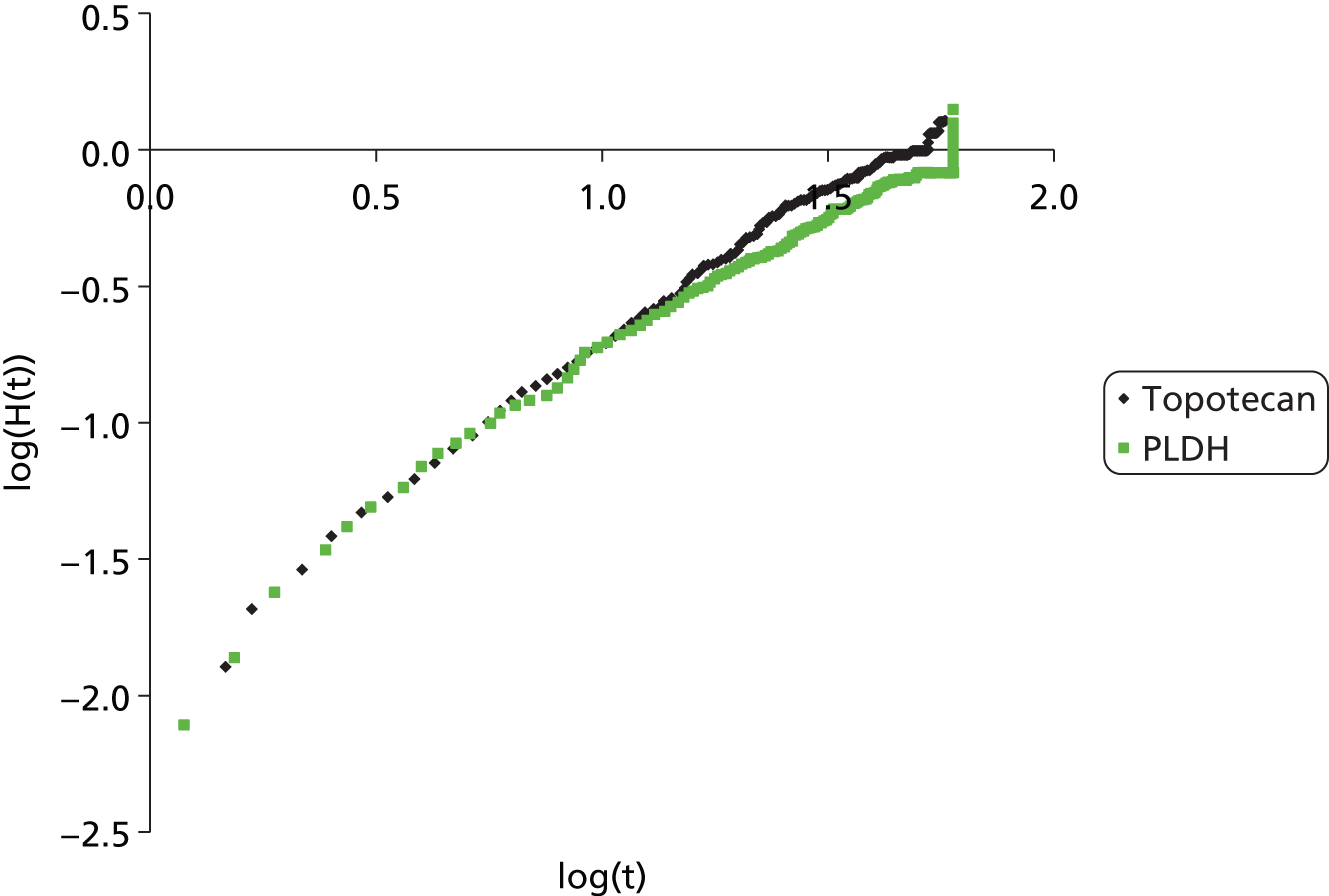
FIGURE 82.
Cumulative log-hazards associated with Kaplan–Meier PFS data for all treatments considered.

FIGURE 83.
Cumulative log-hazards associated with Kaplan–Meier OS data for trabectedin plus PLDH vs. PLDH.

FIGURE 84.
Cumulative log-hazards associated with Kaplan–Meier OS data for topotecan vs. PLDH.

FIGURE 85.
Cumulative log-hazards associated with Kaplan–Meier OS data for topotecan vs. paclitaxel.

FIGURE 86.
Cumulative log-hazards associated with Kaplan–Meier OS data for all treatments considered.

Platinum-resistant/-refractory network
FIGURE 87.
Cumulative log-hazards associated with Kaplan–Meier PFS data for trabectedin plus PLDH vs. PLDH.
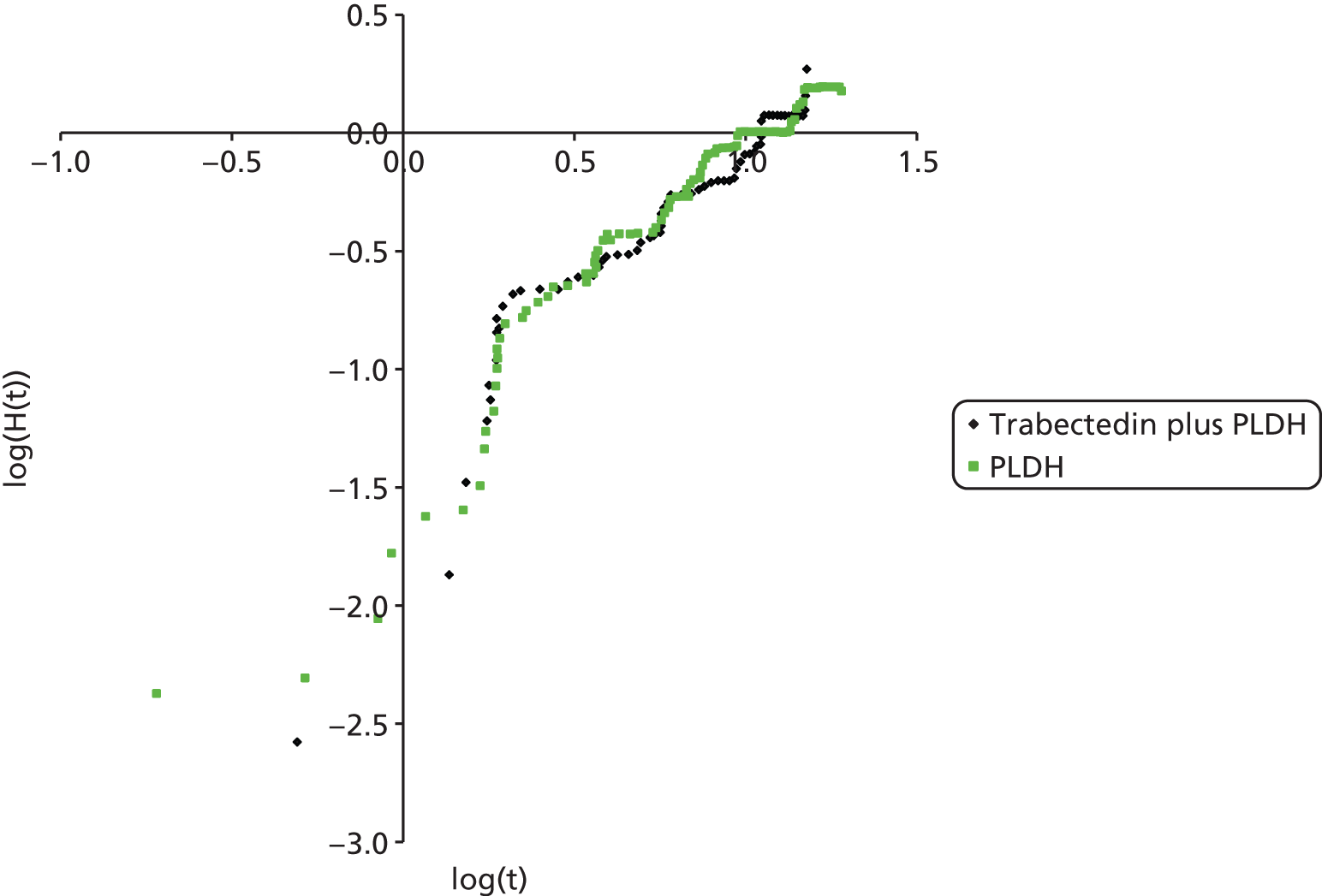
FIGURE 88.
Cumulative log-hazards associated with Kaplan–Meier PFS data for topotecan vs. PLDH.

FIGURE 89.
Cumulative log-hazards associated with Kaplan–Meier PFS data for all treatments considered.
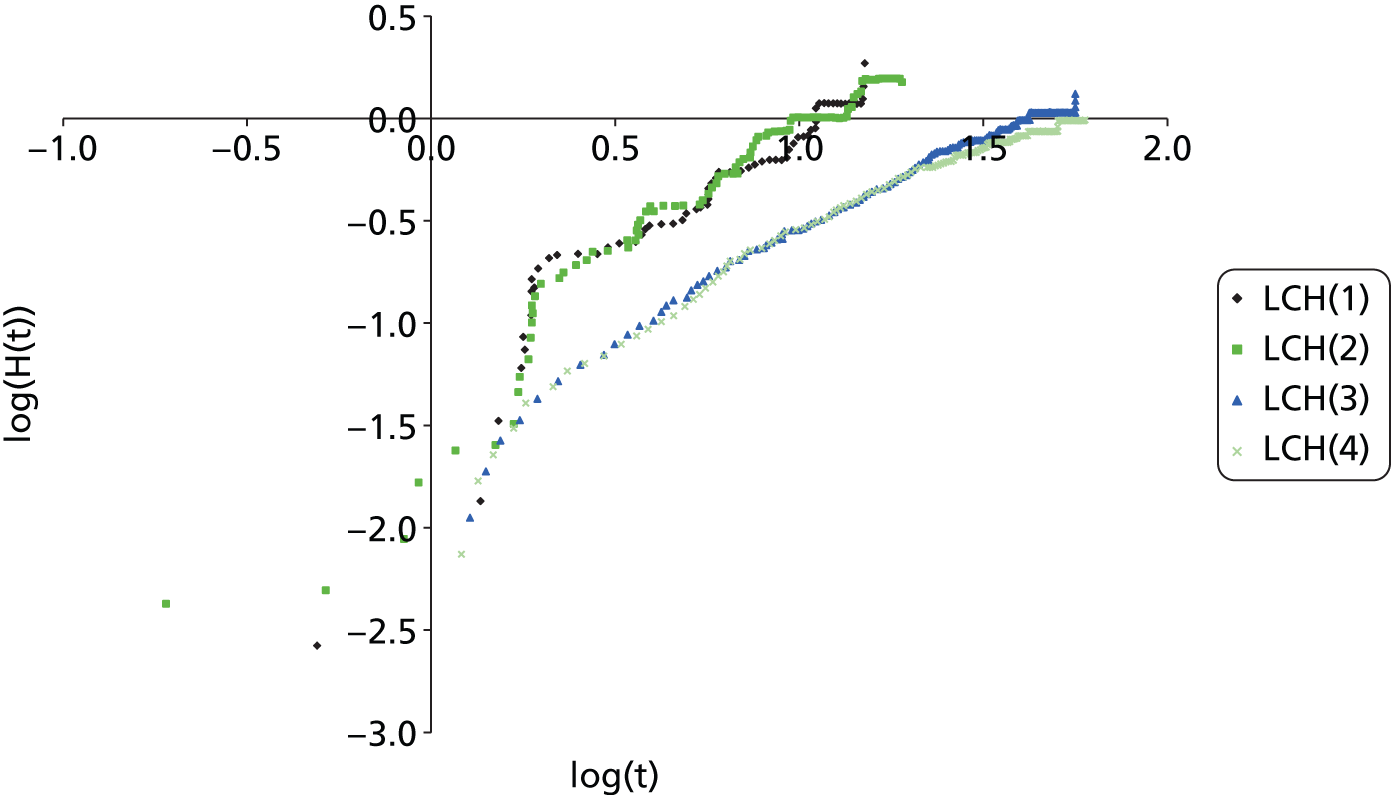
FIGURE 90.
Cumulative log-hazards associated with Kaplan–Meier OS data for trabectedin plus PLDH vs. PLDH.

FIGURE 91.
Cumulative log-hazards associated with Kaplan–Meier OS data for topotecan vs. PLDH.

FIGURE 92.
Cumulative log-hazards associated with Kaplan–Meier OS data for all treatments considered.

Appendix 11 Scenario analysis results
Deterministic scenario analyses, results for platinum-sensitive network 1
| Scenario | Outcomes | Platinum | Gemcitabine plus carboplatin | Paclitaxel plus platinum | PLDH plus platinum |
|---|---|---|---|---|---|
| Base case | Total discounted cost (£) | 15,949 | 20,381 | 21,643 | 22,620 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICER a | – | Extendedly dominated | £24,361 | Strictly dominated | |
| Costs associated with a 50-mg dose rather than 40-mg dose of PLDH | Total discounted cost (£) | 16,155 | 20,581 | 21,871 | 22,839 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £24,455 | Strictly dominated | |
| Patient weight (used to inform drug costs) estimated from the HSE 2011142 | Total discounted cost (£) | 16,015 | 20,432 | 21,713 | 22,689 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £24,377 | Strictly dominated | |
| Branded costs of drugs (Abraxane) | Total discounted cost (£) | 15,949 | 20,381 | 22,940 | 22,620 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £29,912 | Extendedly dominated | |
| Branded costs of drugs (Taxol) | Total discounted cost (£) | 15,949 | 20,381 | 24,384 | 22,620 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £36,092 | Extendedly dominated | |
| Branded costs of drugs (Gemzar) | Total discounted cost (£) | 15,949 | 20,555 | 21,643 | 22,620 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £24,361 | Strictly dominated | |
| Calculating cost based upon the selection of vials that resulted in the least number of vials used | Total discounted cost (£) | 16,293 | 21,329 | 22,128 | 22,979 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £24,961 | Strictly dominated | |
| Vial sharing | Total discounted cost (£) | 15,896 | 20,348 | 21,484 | 22,025 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £23,908 | Strictly dominated | |
| Baseline PFS survival curve network 1 using alternative functional forms (log-logistic) | Total discounted cost (£) | 15,768 | 20,184 | 21,430 | 22,361 |
| Total discounted QALYs | 1.80 | 1.84 | 2.04 | 2.02 | |
| ICERa | – | Extendedly dominated | £24,213 | Strictly dominated | |
| Baseline PFS survival curve network 1 using alternative functional forms (exponential) | Total discounted cost (£) | 15,379 | 19,104 | 20,436 | 21,036 |
| Total discounted QALYs | 1.80 | 1.84 | 2.04 | 2.03 | |
| ICERa | – | Extendedly dominated | £21,239 | Strictly dominated | |
| Baseline PFS survival curve network 1 using alternative functional forms (log-normal) | Total discounted cost (£) | 15,806 | 20,209 | 21,455 | 22,360 |
| Total discounted QALYs | 1.80 | 1.84 | 2.04 | 2.02 | |
| ICERa | – | Extendedly dominated | £24,177 | Strictly dominated | |
| Baseline PS PFS survival curve network 1 using Parmar et al.60 (rather than Pujade-Lauraine et al.31) fitted with a Weibull extrapolation | Total discounted cost (£) | 11,861 | 15,213 | 16,536 | 16,817 |
| Total discounted QALYs | 1.84 | 1.89 | 2.09 | 2.08 | |
| ICERa | – | Extendedly dominated | £19,113 | Strictly dominated | |
| Baseline OS survival curve network 1 using alternative functional forms (log-logistic) | Total discounted cost (£) | 17,672 | 22,144 | 23,779 | 24,709 |
| Total discounted QALYs | 1.97 | 2.02 | 2.25 | 2.23 | |
| ICERa | – | Extendedly dominated | £22,064 | Strictly dominated | |
| Baseline OS survival curve network 1 using alternative functional forms (exponential) | Total discounted cost (£) | 17,329 | 21,974 | 24,063 | 24,994 |
| Total discounted QALYs | 1.96 | 2.02 | 2.30 | 2.28 | |
| ICERa | – | Extendedly dominated | £19,927 | Strictly dominated | |
| Baseline OS survival curve network 1 using alternative functional forms (log-normal) | Total discounted cost (£) | 16,165 | 20,483 | 21,125 | 22,158 |
| Total discounted QALYs | 1.80 | 1.83 | 1.96 | 1.95 | |
| ICERa | – | Extendedly dominated | £30,084 | Strictly dominated | |
| Baseline OS survival curve network 1 using Parmar et al.60 (rather than Wagner et al.55) fitted with a Weibull extrapolation | Total discounted cost (£) | 15,544 | 19,984 | 21,296 | 22,267 |
| Total discounted QALYs | 1.76 | 1.80 | 2.00 | 1.99 | |
| ICERa | – | Extendedly dominated | £24,030 | Strictly dominated | |
| Alternative discount rates for costs and benefits (costs at 1%) | Total discounted cost (£) | 16,584 | 21,030 | 22,414 | 23,375 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £24,944 | Strictly dominated | |
| Alternative discount rates for costs and benefits (costs at 6%) | Total discounted cost (£) | 15,376 | 19,796 | 20,948 | 21,940 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £23,841 | Strictly dominated | |
| Alternative discount rates for costs and benefits (benefits at 1%) | Total discounted cost (£) | 15,949 | 20,381 | 21,643 | 22,620 |
| Total discounted QALYs | 1.86 | 1.91 | 2.11 | 2.10 | |
| ICERa | – | Extendedly dominated | £22,970 | Strictly dominated | |
| Alternative discount rates for costs and benefits (benefits at 6%) | Total discounted cost (£) | 15,949 | 20,381 | 21,643 | 22,620 |
| Total discounted QALYs | 1.74 | 1.78 | 1.96 | 1.95 | |
| ICERa | – | Extendedly dominated | £25,755 | Strictly dominated | |
| Disutilities for AEs applied | Total discounted cost (£) | 15,949 | 20,381 | 21,643 | 22,620 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £24,446 | Strictly dominated | |
| Nausea and vomiting probabilities estimated from clinical expert opinion for network 1 | Total discounted cost (£) | 15,962 | 20,399 | 21,672 | 22,638 |
| Total discounted QALYs | 1.80 | 1.84 | 2.03 | 2.02 | |
| ICERa | – | Extendedly dominated | £24,429 | Strictly dominated | |
| Half-cycle correction | Total discounted cost (£) | 15,859 | 20,286 | 21,553 | 22,542 |
| Total discounted QALYs | 1.77 | 1.81 | 2.00 | 1.99 | |
| ICERa | – | Extendedly dominated | £24,326 | Strictly dominated |
Deterministic scenario analyses, results for platinum-sensitive network 2
| Scenario | Outcomes | Paclitaxel | PLDH | Topotecan | Trabectedin plus PLDH |
|---|---|---|---|---|---|
| Base case | Total discounted cost (£) | 15,668 | 19,599 | 23,793 | 32,640 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICER a | – | £23,733 | Strictly dominated | £85,212 | |
| Costs associated with a 50-mg dose rather than 40-mg dose of PLDH | Total discounted cost (£) | 15,878 | 21,049 | 23,987 | 32,878 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £31,222 | Strictly dominated | £77,290 | |
| Patient weight (used to inform drug costs) estimated from the HSE 2011142 | Total discounted cost (£) | 15,689 | 19,621 | 23,813 | 32,665 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £23,740 | Strictly dominated | £85,223 | |
| Branded costs of drugs (Abraxane) | Total discounted cost (£) | 16,736 | 19,599 | 23,793 | 32,640 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £17,285 | Strictly dominated | £85,212 | |
| Branded costs of drugs (Taxol) | Total discounted cost (£) | 17,925 | 19,599 | 23,793 | 32,640 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £10,106 | Strictly dominated | £85,212 | |
| Branded costs of drugs (Hycamtin) | Total discounted cost (£) | 15,668 | 19,599 | 24,534 | 32,640 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £23,733 | Strictly dominated | £85,212 | |
| Calculating cost based upon the selection of vials that resulted in the least number of vials used | Total discounted cost (£) | 15,880 | 19,717 | 23,910 | 33,277 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £23,167 | Strictly dominated | £88,605 | |
| Vial sharing | Total discounted cost (£) | 15,505 | 18,951 | 22,343 | 31,612 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £20,810 | Strictly dominated | £82,723 | |
| Baseline PFS survival curve network 2 using alternative functional forms (Weibull) | Total discounted cost (£) | 15,791 | 19,690 | 24,086 | 32,959 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £23,565 | Strictly dominated | £86,700 | |
| Baseline PFS survival curve network 2 using alternative functional forms (log-logistic) | Total discounted cost (£) | 15,044 | 18,366 | 23,144 | 31,234 |
| Total discounted QALYs | 1.42 | 1.59 | 1.34 | 1.75 | |
| ICERa | – | £19,188 | Strictly dominated | £78,954 | |
| Baseline PFS survival curve network 2 using alternative functional forms (exponential) | Total discounted cost (£) | 15,148 | 18,640 | 22,737 | 31,478 |
| Total discounted QALYs | 1.40 | 1.57 | 1.32 | 1.72 | |
| ICERa | – | £20,694 | Strictly dominated | £82,280 | |
| Baseline PFS survival curve network 2 using alternative functional forms (log-normal) | Total discounted cost (£) | 15,313 | 18,772 | 23,448 | 31,805 |
| Total discounted QALYs | 1.40 | 1.57 | 1.32 | 1.73 | |
| ICERa | – | £20,465 | Strictly dominated | £83,213 | |
| Baseline OS survival curve network 2 using alternative functional forms (log-logistic) | Total discounted cost (£) | 17,965 | 22,333 | 25,868 | 35,859 |
| Total discounted QALYs | 1.63 | 1.84 | 1.53 | 2.05 | |
| ICERa | – | £20,660 | Strictly dominated | £66,604 | |
| Baseline OS survival curve network 1 using alternative functional forms (exponential) | Total discounted cost (£) | 15,939 | 20,191 | 23,922 | 33,584 |
| Total discounted QALYs | 1.44 | 1.63 | 1.34 | 1.82 | |
| ICERa | – | £21,550 | Strictly dominated | £71,009 | |
| Baseline OS survival curve network 1 using alternative functional forms (log-normal) | Total discounted cost (£) | 17,242 | 21,536 | 25,193 | 34,998 |
| Total discounted QALYs | 1.56 | 1.77 | 1.46 | 1.96 | |
| ICERa | – | £20,974 | Strictly dominated | £68,262 | |
| Alternative discount rates for costs and benefits (costs at 1%) | Total discounted cost (£) | 16,090 | 20,097 | 24,175 | 33,217 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £24,196 | Strictly dominated | £85,726 | |
| Alternative discount rates for costs and benefits (costs at 6%) | Total discounted cost (£) | 15,286 | 19,148 | 23,446 | 32,118 |
| Total discounted QALYs | 1.40 | 1.56 | 1.32 | 1.72 | |
| ICERa | – | £23,316 | Strictly dominated | £84,750 | |
| Alternative discount rates for costs and benefits (benefits at 1%) | Total discounted cost (£) | 15,668 | 19,599 | 23,793 | 32,640 |
| Total discounted QALYs | 1.44 | 1.62 | 1.36 | 1.78 | |
| ICERa | – | £22,669 | Strictly dominated | £80,986 | |
| Alternative discount rates for costs and benefits (benefits at 6%) | Total discounted cost (£) | 15,668 | 19,599 | 23,793 | 32,640 |
| Total discounted QALYs | 1.36 | 1.52 | 1.28 | 1.66 | |
| ICERa | – | £24,779 | Strictly dominated | £89,400 | |
| Disutilities for AEs applied | Total discounted cost (£) | 15,668 | 19,599 | 23,793 | 32,640 |
| Total discounted QALYs | 1.40 | 1.56 | 1.31 | 1.71 | |
| ICERa | – | £23,635 | Strictly dominated | £87,916 | |
| Head-to-head comparison of trabected in plus PLDH vs. PLDH using adjusted PFS and OS estimates directly from the PharmaMar submission | Total discounted cost (£) | NA | 21,063 | NA | 34,569 |
| Total discounted QALYs | NA | 1.70 | NA | 2.08 | |
| ICERa | NA | – | NA | £35,646 | |
| Analysis of the results considering the PPS HRs for OS | Total discounted cost (£) | NA | 19,599 | 22,705 | 34,610 |
| Total discounted QALYs | NA | 1.56 | 1.20 | 1.96 | |
| ICERa | NA | – | Strictly dominated | £37,691 | |
| Half-cycle correction | Total discounted cost (£) | 15,250 | 19,238 | 23,044 | 32,323 |
| Total discounted QALYs | 1.37 | 1.54 | 1.29 | 1.69 | |
| ICERa | – | £24,050 | Strictly dominated | £85,377 |
Deterministic scenario analyses, results for the platinum-resistant/-refractory network
| Scenario | Outcomes | PLDH | Paclitaxel | Topotecan | ||||
|---|---|---|---|---|---|---|---|---|
| Base case | Total discounted cost (£) | 14,320 | 15,095 | 21,271 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £449,553 | |||||
| Costs associated with a 50-mg dose rather than a 40-mg dose of PLDH | Total discounted cost (£) | 15,442 | 15,095 | 21,271 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | £10,480 (vs. paclitaxel) | – | £376,985 (vs. PLDH) | |||||
| Patient weight (used to inform drug costs) estimated from the HSE 2011142 | Total discounted cost (£) | 14,320 | 15,095 | 21,271 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £449,553 | |||||
| Branded costs of drugs (Abraxane) | Total discounted cost (£) | 14,320 | 17,635 | 21,271 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £449,553 | |||||
| Branded costs of drugs (Taxol) | Total discounted cost (£) | 14,320 | 18,074 | 21,271 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £449,553 | |||||
| Branded costs of drugs (Hycamtin) | Total discounted cost (£) | 14,320 | 15,095 | 22,011 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £497,418 | |||||
| Calculating cost based upon the selection of vials that resulted in the least number of vials used | Total discounted cost (£) | 14,320 | 15,794 | 21,284 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £450,435 | |||||
| Vial sharing | Total discounted cost (£) | 13,808 | 14,824 | 19,901 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £394,035 | |||||
| Baseline PFS survival curve using alternative functional forms (log-logistic) | Total discounted cost (£) | 14,101 | 15,055 | 21,195 | ||||
| Total discounted QALYs | 1.01 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £458,313 | |||||
| Baseline PFS survival curve using alternative functional forms (exponential) | Total discounted cost (£) | 13,681 | 14,267 | 20,115 | ||||
| Total discounted QALYs | 1.01 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £415,929 | |||||
| Baseline PFS survival curve using alternative functional forms (log-normal) | Total discounted cost (£) | 14,227 | 15,156 | 21,304 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £457,727 | |||||
| Baseline OS survival curve using alternative functional forms (log-logistic) | Total discounted cost (£) | 15,394 | 16,114 | 22,375 | ||||
| Total discounted QALYs | 1.11 | 1.07 | 1.13 | |||||
| ICER | – | Strictly dominated | £374,963 | |||||
| Baseline OS survival curve using alternative functional forms (exponential) | Total discounted cost (£) | 14,459 | 15,210 | 21,422 | ||||
| Total discounted QALYs | 1.02 | 0.98 | 1.04 | |||||
| ICER | – | Strictly dominated | £414,866 | |||||
| Baseline OS survival curve using alternative functional forms (log-normal) | Total discounted cost (£) | 14,927 | 15,670 | 21,896 | ||||
| Total discounted QALYs | 1.07 | 1.03 | 1.08 | |||||
| ICER | – | Strictly dominated | £402,379 | |||||
| Alternative discount rates for costs and benefits (costs at 1%) | Total discounted cost (£) | 14,522 | 15,288 | 21,478 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £449,873 | |||||
| Alternative discount rates for costs and benefits (costs at 6%) | Total discounted cost (£) | 14,135 | 14,918 | 21,081 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £449,261 | |||||
| Alternative discount rates for costs and benefits (benefits at 1%) | Total discounted cost (£) | 14,320 | 15,095 | 21,271 | ||||
| Total discounted QALYs | 1.02 | 0.99 | 1.04 | |||||
| ICER | – | Strictly dominated | £435,381 | |||||
| Alternative discount rates for costs and benefits (benefits at 6%) | Total discounted cost (£) | 14,320 | 15,095 | 21,271 | ||||
| Total discounted QALYs | 0.99 | 0.95 | 1.00 | |||||
| ICER | – | Strictly dominated | £463,366 | |||||
| Disutilities for AEs applied | Total discounted cost (£) | 14,320 | 15,095 | 21,271 | ||||
| Total discounted QALYs | 1.00 | 0.97 | 1.02 | |||||
| ICER | – | Strictly dominated | £503,885 | |||||
| Half-cycle correction | Total discounted cost (£) | 13,782 | 14,290 | 20,266 | ||||
| Total discounted QALYs | 0.98 | 0.94 | 0.99 | |||||
| ICER | – | Strictly dominated | £418,861 | |||||
| Scenario | Outcomes | Etoposide | Best supportive care | Etoposide plus platinum | PLDH | Paclitaxel | Paclitaxel plus platinum | Topotecan |
| Equivalent efficacy assumed for all therapies outlined within the NICE scope for patients with PRR disease (cost analysis only) | Total discounted cost (£) | 8194 | 12,622 | 13,095 | 14,320 | 15,822 | 18,023 | 21,114 |
Appendix 12 Quality assessment
Quality assessment of the clinical evidence
Alberts et al.28
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | No details reported | ||
| Allocation concealment | ✓ | No details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data | ✓ | Modified ITT analysis | |||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
OVA-301 (Monk et al.30)
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Permuted block | ||
| Allocation concealment | ✓ | No details given | |||
| Selective reporting | ✓ | All outcomes mentioned are reported | |||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias as an outcome measure | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Blinded independent radiology and oncology review | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Blinded independent radiology and oncology review | |||
| Incomplete outcome data | ✓ | ITT analysis | |||
| QoL | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Independent data monitoring committee | |||
| Incomplete outcome data | ✓ | ||||
CARTAXHY (Lortholary et al.62)
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | No details reported | ||
| Allocation concealment | ✓ | No details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | No details reported as to level of masking of assessor | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | No details reported as to level of masking of assessor | |||
| Incomplete outcome data | ✓ | ||||
| QoL | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | Limited details reported on proportion of patients returning questionnaire and scores on completed questionnaires | |||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
Pfisterer et al.50
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Random assignment through central office, using ‘block size of 10’; no additional details reported | ||
| Allocation concealment | ✓ | No details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | No details reported as to level of masking of assessor | |||
| Incomplete outcome data | ✓ | ITT analysis | |||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | No details reported as to level of masking of assessor | |||
| Incomplete outcome data | ✓ | ||||
| QoL | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | No details reported as to level of masking of assessor | |||
| Incomplete outcome data | ✓ | Limited details reported on proportion of patients returning questionnaire and scores on completed questionnaires | |||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
Piccart et al.63
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Not reported | ||
| Allocation concealment | ✓ | Assigned by European Organisation for Research and Treatment of Cancer Data Centre. Method not reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Verified by two independent radiologists; level of masking unclear | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Verified by two independent radiologists. Level of masking unclear | |||
| Incomplete outcome data | ✓ | ||||
| QoL | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | Limited details reported on proportion of patients returning questionnaire and scores on completed questionnaires | |||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
Bafaloukos et al.29
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Performed at central HeCOG Data Office; no further details reported | ||
| Allocation concealment | ✓ | No details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| TTP | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
Gonzalez-Martin et al.48
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Reported to have been carried out at a central data centre; no details reported | ||
| Allocation concealment | ✓ | No details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| TTP | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
| QoL | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | Limited details reported on proportion of patients returning questionnaire and scores on completed questionnaires | |||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
Rosenberg et al.60
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Reported to have been carried out at BMS office in Stockholm; no further details reported | ||
| Allocation concealment | ✓ | No details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| TTP | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
CALYPSO (Pujade-Lauraine et al.31)
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Permuted blocks of six; centrally randomised | ||
| Allocation concealment | ✓ | ||||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Responses reviewed by an independent assessor masked to treatment | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Responses reviewed by an independent assessor masked to treatment | |||
| Incomplete outcome data | ✓ | ||||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
Gordon et al.49,54
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | No details reported in full publication; TA91 indicated that method of randomisation was robust13 | ||
| Allocation concealment | ✓ | No details reported in full publication; TA91 indicated that allocation of treatment was concealed13 | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | Open-label design | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | Open-label design | ||
| Blinding of outcome assessment | ✓ | Radiological scans underwent independent radiological review, but level of masking of assessor is unclear | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | Open-label design | ||
| Blinding of outcome assessment | ✓ | Radiological scans underwent independent radiological review, but level of masking of assessor is unclear | |||
| Incomplete outcome data | ✓ | ||||
| QoL | Blinding [who (participants, personnel) and method] | ✓ | Open-label design | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | Limited details reported on proportion of patients returning questionnaire and scores on completed questionnaires | |||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | Open-label design | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
ICON4/AGO-OVAR 2.2 (Parmar et al.61)
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Minimisation by computer | ||
| Allocation concealment | ✓ | ‘Telephone or facsimile’ – no extra details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
| QoL | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | Limited details reported on proportion of patients returning questionnaire and scores on completed questionnaires | |||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
Gore et al.24
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | No others details reported | ||
| Allocation concealment | ✓ | Reported to be ‘by telephone’, but no additional details given | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| TTP | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Independent, blinded radiological review | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Independent, blinded radiological review | |||
| Incomplete outcome data | ✓ | ||||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
ten Bokkel Huinink et al.21,52
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Paper states ‘telephone randomisation system’; no further details given | ||
| Allocation concealment | ✓ | No details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | OS associated with low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ||||
| TTP | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Independent, blinded review of response | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | Independent, blinded review of response | |||
| Incomplete outcome data | ✓ | ||||
| QoL | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | Open label | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | ||||
Sehouli et al.23
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Central randomisation with permutated blocks | ||
| Allocation concealment | ✓ | Telephone or facsimile. No additional details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | No details reported | |||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | Confirmed by second evaluation; no details reported for blinding | |||
| Incomplete outcome data | ✓ | ||||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | Confirmed by second evaluation; no details reported for blinding | |||
| Incomplete outcome data | ✓ | ||||
| QoL | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
Omura et al.68
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | Treatment regimens sequentially assigned from stratified, permuted blocks | ||
| Allocation concealment | ✓ | No details reported | |||
| Selective reporting | ✓ | ||||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | Not all patients randomised were included in analysis. Reasons for ineligibility not clearly reported | |||
| PFS | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | Not all patients randomised were included in analysis. Reasons for ineligibility not clearly reported | |||
| Response rate | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | Not all patients randomised were included in analysis. Reasons for ineligibility not clearly reported | |||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | No details reported | ||
| Blinding of outcome assessment | ✓ | No details reported | |||
| Incomplete outcome data | ✓ | Not all patients randomised were included in analysis. Reasons for ineligibility not clearly reported | |||
Trial 30–57 (taken from TA91)13
| Outcome | Risk of bias | Comments | |||
|---|---|---|---|---|---|
| Low | Unclear | High | |||
| Trial methodology | Random sequence generation | ✓ | |||
| Allocation concealment | ✓ | ||||
| Selective reporting | ✓ | Unclear if all outcomes reported | |||
| Other bias | ✓ | ||||
| OS | Blinding [who (participants, personnel) and method] | ✓ | Personnel and patients not masked | ||
| Blinding of outcome assessment | ✓ | OS is associated with a low risk of bias | |||
| Incomplete outcome data (patients who discontinued/changed treatment, patients lost to follow-up) | ✓ | ITT | |||
| AEs | Blinding [who (participants, personnel) and method] | ✓ | |||
| Blinding of outcome assessment | ✓ | ||||
| Incomplete outcome data | ✓ | ||||
Appendix 13 Completed and ongoing clinical trials of interest
| Trial title | Sponsor | ID | Intervention | Comparator | Status |
|---|---|---|---|---|---|
| Phase III International Multicentre Randomized Study Testing the Effect on Survival of Prolonging Platinum-free Interval in Patients With Ovarian Cancer Recurring Between 6 and 12 Months After Previous Platinum Based Chemotherapy | NCI, Naples | EudraCT no. 2008-001755-22 ClinicalTrials.gov identifier NCT00657878 |
A non-platinum-based therapy (corresponding to stealth liposomal doxorubicin, or topotecan, or gemcitabine, or any other drug approved in clinical practice for the treatment of patients with ovarian cancer after previous platinum-based chemotherapy) followed by a platinum-based chemotherapy at disease progression | Platinum-based chemotherapy (corresponding to the combination of carboplatin plus paclitaxel, or carboplatin plus gemcitabine for patients with significant but lower than grade 3 neuropathy at baseline) followed by a non-platinum-based chemotherapy at disease progression | Recruiting |
| An Open, Randomized, Multicenter Study in Patients With Recurrent Epithelian Cancer, Primary Peritoneal Cancer or Fallopian Tube Cancer to Compare the Efficacy and Safety of Paclitaxel (Micellar) Nanoparticles and Paclitaxel (Cremophor® EL) | Oasmia Pharmaceutical AB | EudraCT no. 2008-002668-32 ClinicalTrials.gov Identifier: NCT00989131 |
Paclitaxel (Paclical®) plus carboplatin | Paclitaxel (Taxol®) plus carboplatin | Ongoing |
| Phase III International, Randomised Study of Trabectedin plus Pegylated Liposomal Doxorubicin (PLD) versus Carboplatin plus PLD in Patients With Ovarian Cancer Progressing Within 6–12 Months of Last Platinum | IRFMN | EudraCT no. 2010-022949-17 ClinicalTrials.gov identifier: NCT01379989 |
Trabectedin plus PLDH | Carboplatin plus PLDH | Suspended due to limited availability of PLDH |
| An Open-Label Multicenter Randomized Phase 3 Study Comparing the Combination of DOXIL/CAELYX and YONDELIS With DOXIL/CAELYX Alone in Subjects With Advanced Relapsed Ovarian Cancer | Johnson & Johnson Pharmaceutical Research & Development, LLC | NCT00113607 | Doxil plus trabectedin | Doxil | Completed |
| National, Randomized, Phase II Study Comparing Efficacy of Weekly Administration of Paclitaxel in Monotherapy or in Combination With Topotecan or Carboplatin in Patients With Epithelial Ovarian Cancer in Early Relapse | ARCAGY/GINECO Group | NCT00189566 | Paclitaxel monotherapy | Paclitaxel combination with topotecan or carboplatin | Completed |
| A Randomized Phase III Study Comparing Gemcitabine Plus Carboplatin versus Carboplatin Monotherapy in Patients With Advanced Epithelial Ovarian Carcinoma who Failed First-Line Platinum-Based Therapy | AGO Study Group | NCT00102414 | Gemcitabine plus carboplatin | Carboplatin monotherapy | Completed |
| A Randomized, Open-Label Study Comparing the Combination of YONDELIS and DOXIL/CAELYX With DOXIL/CAELYX Monotherapy for the Treatment of Advanced-Relapsed Epithelial Ovarian, Primary Peritoneal, or Fallopian Tube Cancer | Janssen Research & Development, LLC | NCT01846611 | Trabectedin plus PLDH | PLDH | Not yet recruiting |
| A Phase II Randomized Controlled Clinical Trial of Carboplatin and Paclitaxel or Carboplatin and Gemcitabine in Platinum-sensitive, Recurrent, Ovarian, Fallopian Tube, and Primary Peritoneal Cancer | Korean Gynecologic Oncology Group | NCT01570582 | Carboplatin and paclitaxel | Carboplatin and gemcitabine | Active, not recruiting |
| A Randomized Phase II Evaluation of Topotecan Administered Daily × 5 Every 3 Weeks vs. Weekly Topotecan in the Treatment of Recurrent Platinum-Sensitive Ovarian, Fallopian Tube, or Primary Peritoneal Cancer | Gynecologic Oncology Group | NCT00114166 | Topotecan administered daily × 5 every 3 weeks | vs. weekly | Completed |
Appendix 14 WinBUGS code
Overall survival and progression-free survival
Overall response rate and all safety outcomes
Appendix 15 Tornado diagrams
Tornado diagram of parameters to which the cost-effectiveness of paclitaxel plus platinum versus platinum monotherapy is most sensitive (PS network 1). pac, paclitaxel; plat, platinum; pts, patients
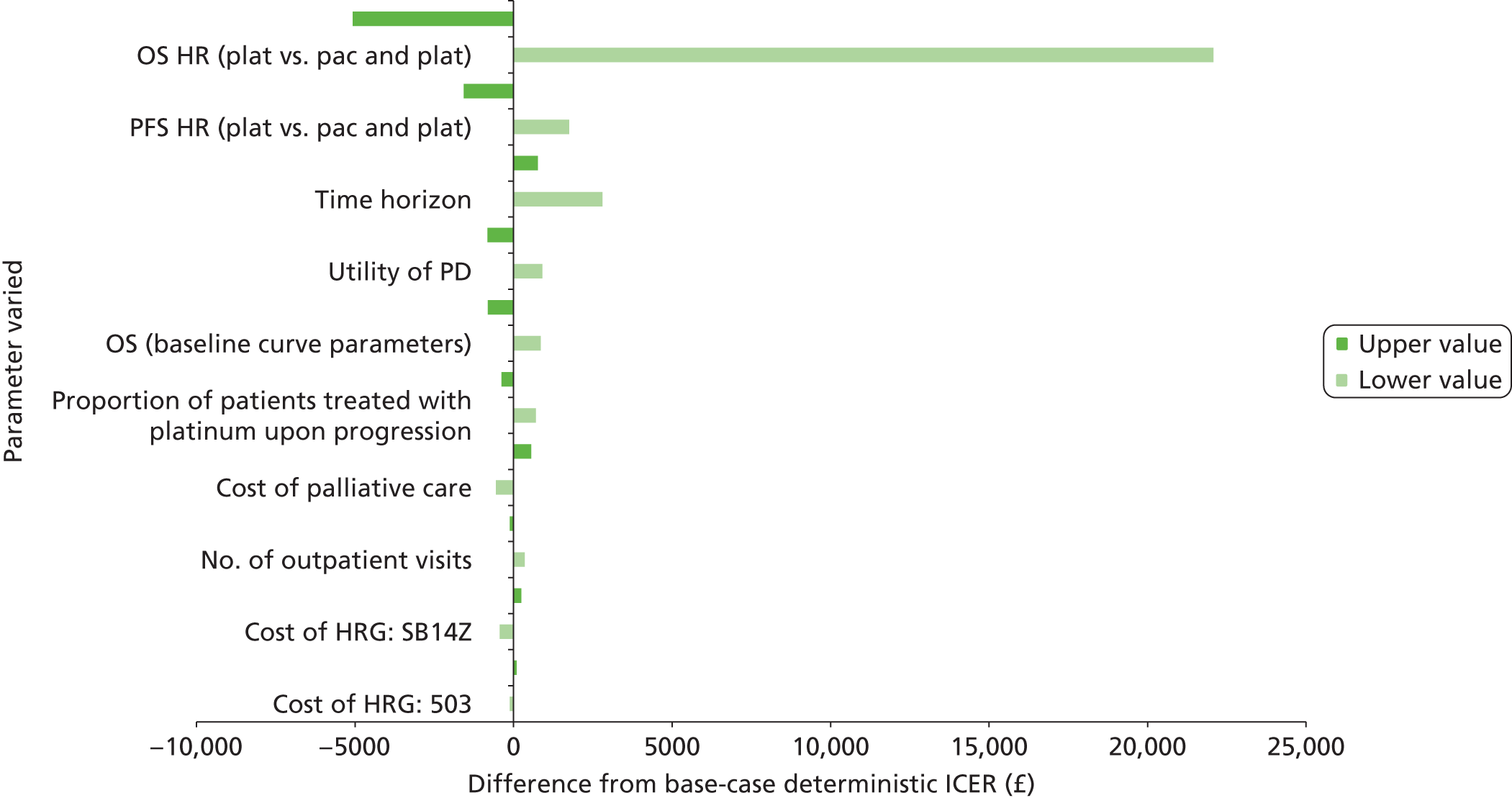
Tornado diagram of parameters to which the cost-effectiveness of PLDH plus platinum versus platinum monotherapy is most sensitive (PS network 1). pac, paclitaxel; plat, platinum
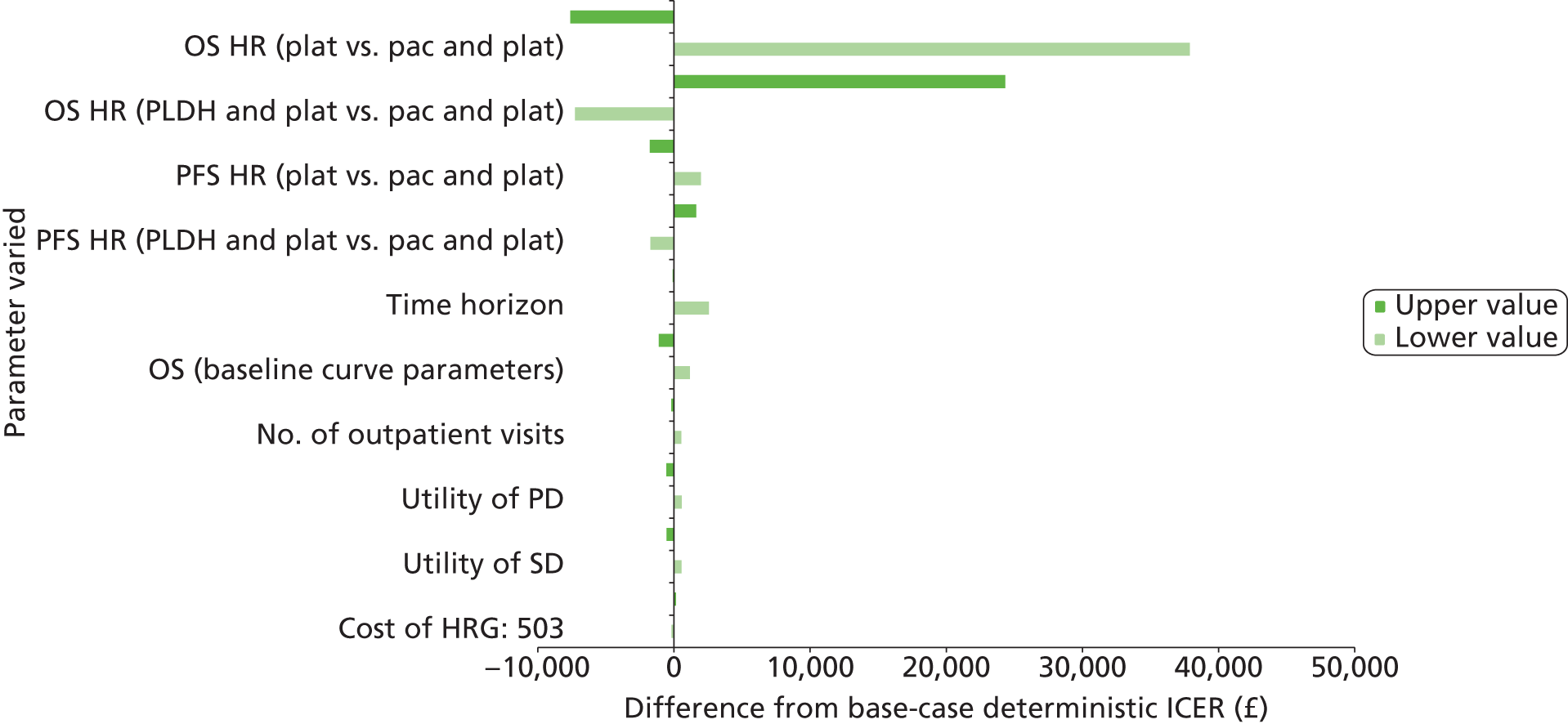
Tornado diagram of parameters to which the cost-effectiveness of PLDH plus platinum versus paclitaxel plus platinum is most sensitive (PS network 1). pac, paclitaxel; plat, platinum
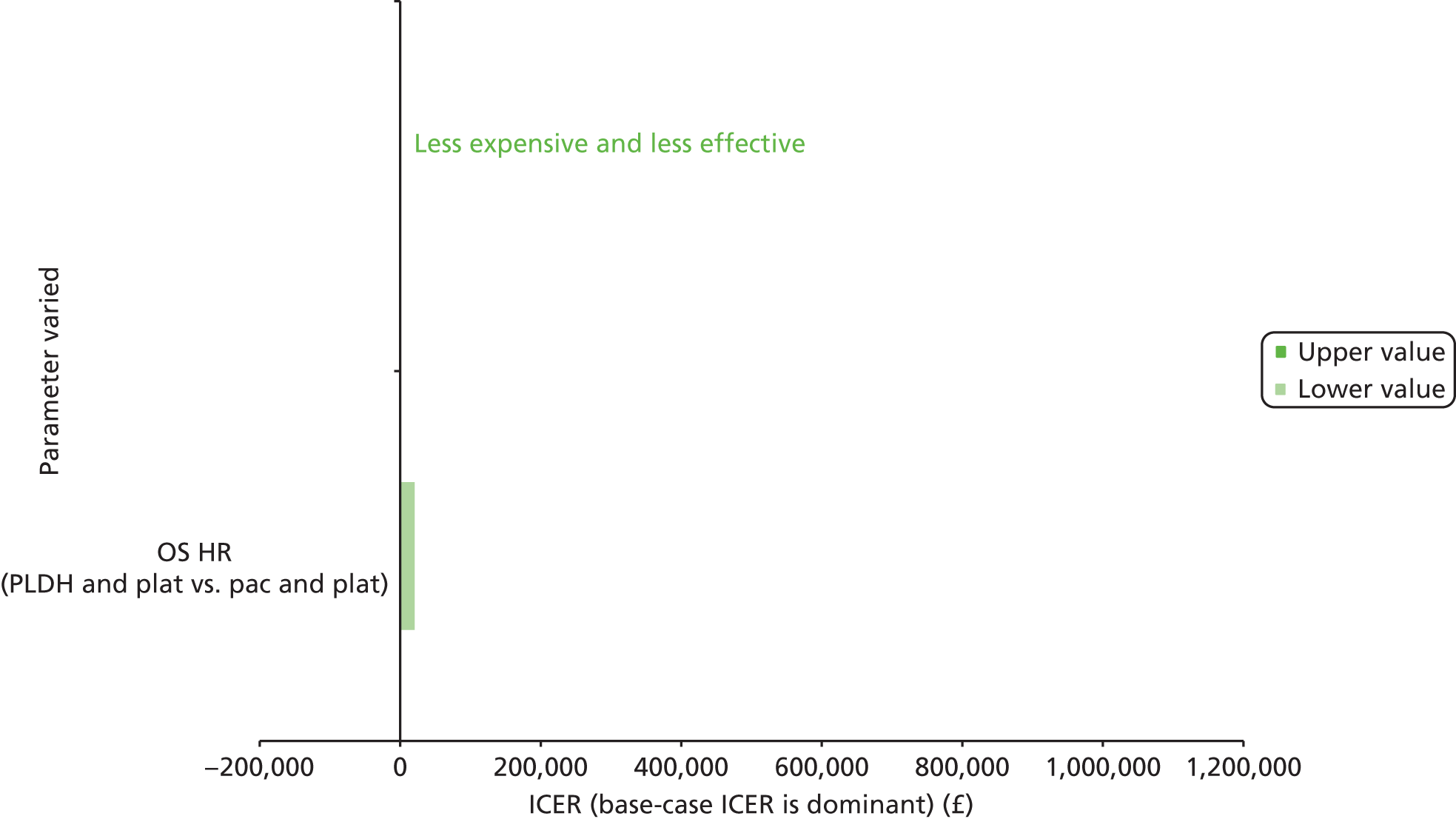
Tornado diagram of parameters to which the cost-effectiveness of PLDH versus paclitaxel is most sensitive (PS network 2). pac, paclitaxel
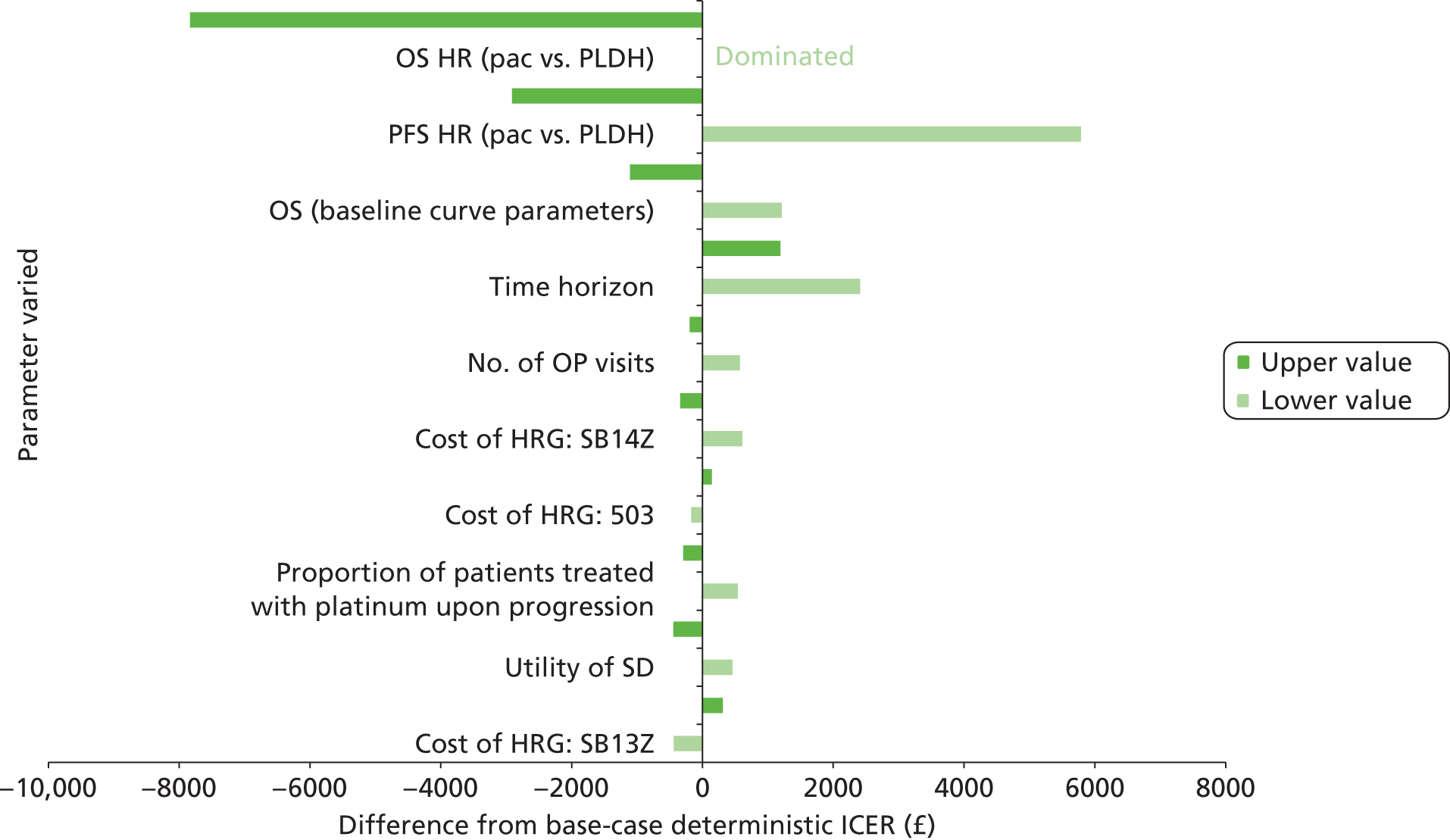
Tornado diagram of parameters to which the cost-effectiveness of trabectedin plus PLDH versus paclitaxel is most sensitive (PS network 2). pac, paclitaxel; Trab, trabectedin

Tornado diagram of parameters to which the cost-effectiveness of trabectedin plus PLDH versus PLDH is most sensitive (PS network 2). Trab, trabectedin

Tornado diagram of parameters to which the cost-effectiveness of topotecan versus PLDH is most sensitive (platinum resistant/refractory). Top, topotecan
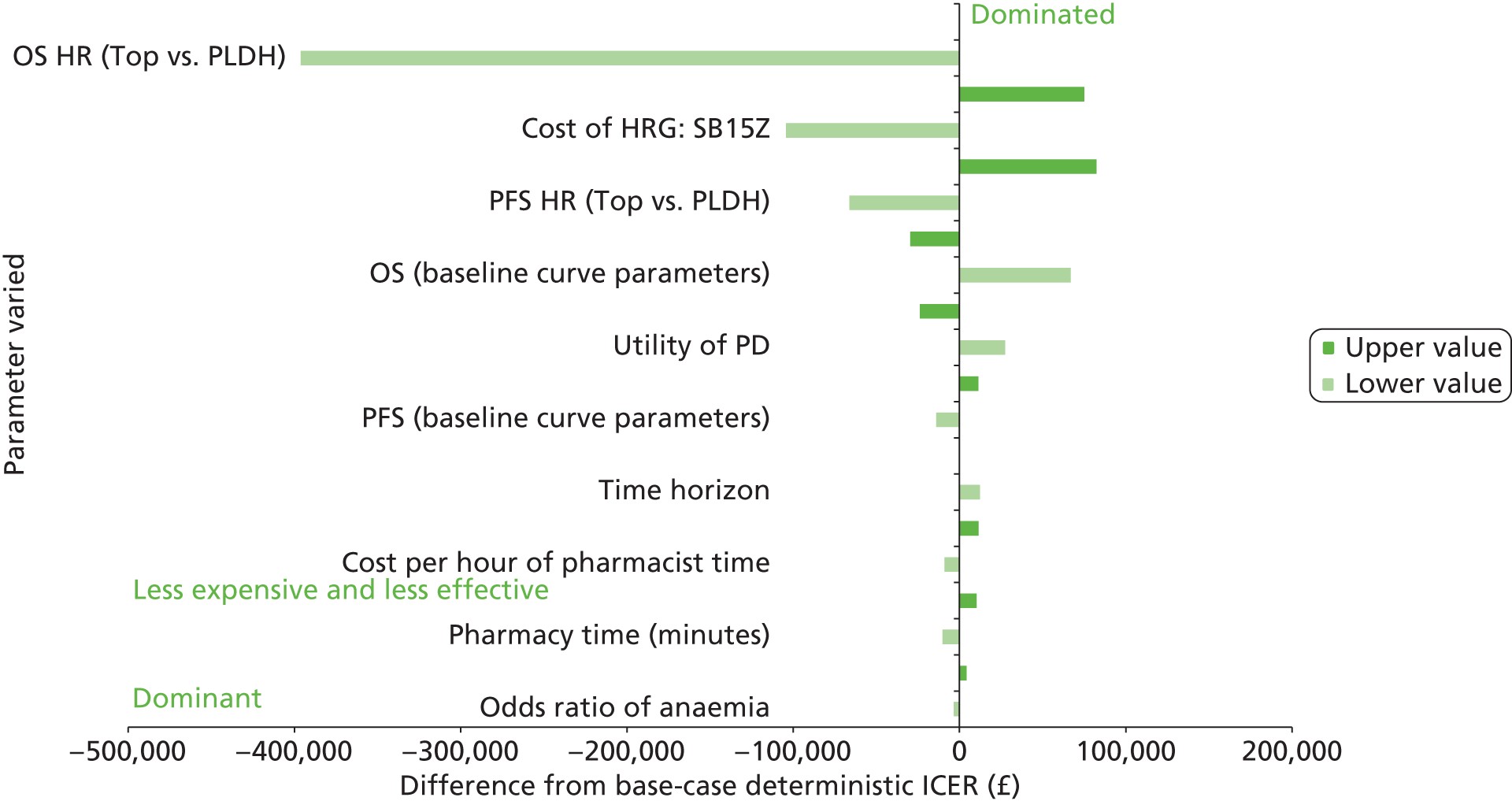
Tornado diagram of parameters to which the cost-effectiveness of paclitaxel versus PLDH is most sensitive (platinum resistant/refractory). pac, paclitaxel
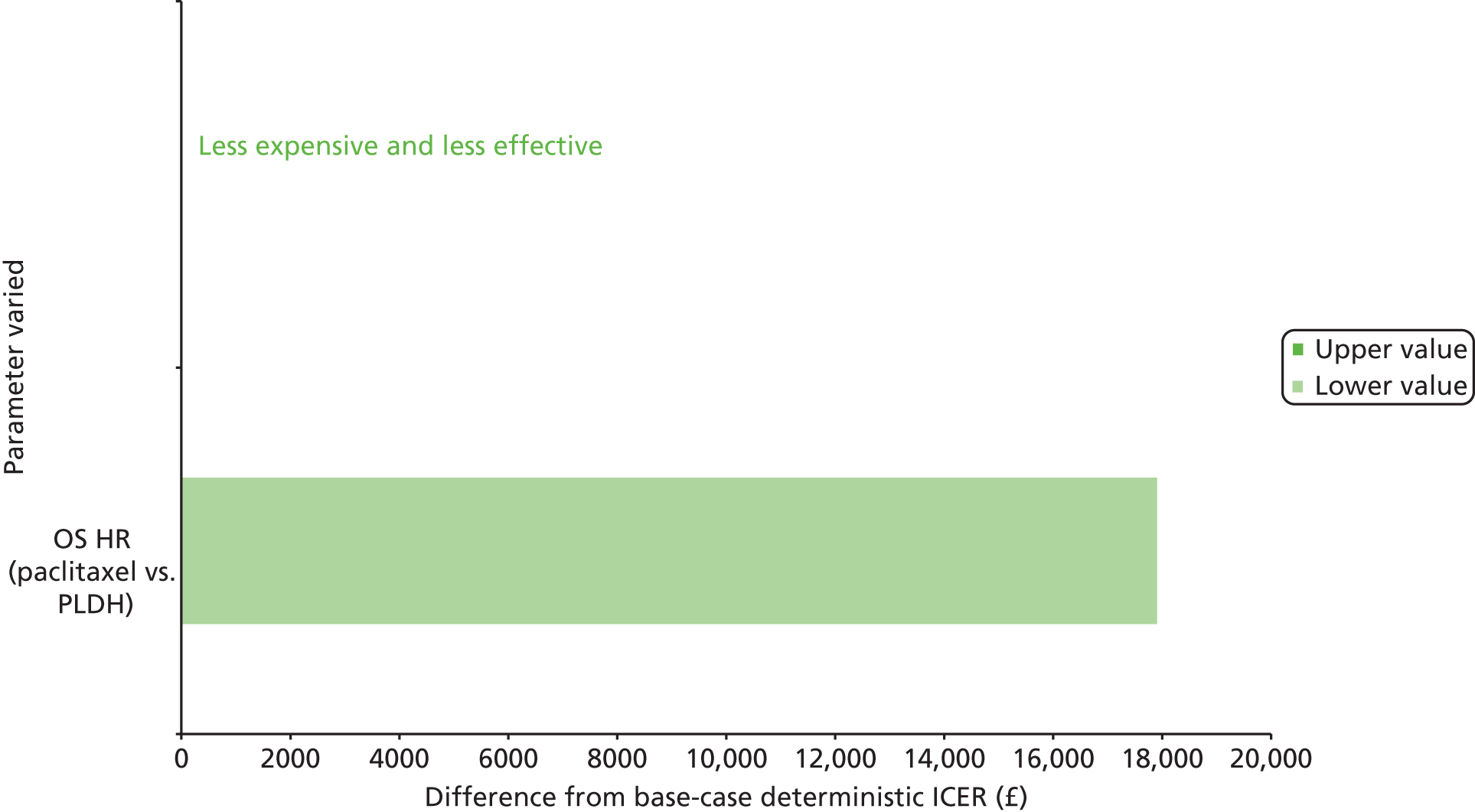
Glossary
- Advanced ovarian cancer
- Disease classified as International Federation of Gynecology and Obstetrics stages III and IV.
- BRAC1/2
- BRCA1 and BRCA2 are two genes associated with hereditary breast cancer (BRCA stands for BReast CAncer). A mutation in either BRCA1 or BRCA2 increases a woman’s lifetime risk of developing breast cancer. Mutations to BRCA genes are also linked with an increased risk of developing ovarian cancer.
- CA125
- A cell surface marker found in serum. A response according to CA125 has occurred if there is at least a 50% reduction in CA125 levels from a pre-treatment sample. The response must be confirmed and maintained for at least 28 days.
- Chemotherapy
- The use of drugs that are capable of killing cancer cells or preventing/slowing their growth.
- Complete response
- The total disappearance of all detectable malignant disease for at least 4 weeks.
- Cost-effectiveness acceptability curve
- A graphical representation of the probability of an intervention being cost-effective over a range of monetary values for society’s willingness to pay for an additional unit of health gain.
- Debulking
- Surgical removal of a substantial proportion of cancer tissue. Optimal debulking refers to the removal of the largest possible amount of tumour while limiting the damage to the surrounding normal tissue; interval debulking refers to the surgical removal of a tumour after chemotherapy, aimed at further reducing its bulk.
- Eastern Cooperative Oncology Group performance status
- Scores are as follows:
- First-line therapy
- The first chemotherapy regimen (usually administered with curative intent) given to patients who have been newly diagnosed with ovarian cancer, or who had an early stage of the disease that has been previously treated with surgery alone but has since relapsed and requires chemotherapy.
- Histological grade
- The degree of malignancy of a tumour as judged by histology.
- Histological type
- The type of tissue found in a tumour as determined by histology.
- Incremental cost-effectiveness ratio
- An expression of the additional cost of health gain associated with an intervention relative to an appropriate comparator. Expressed as the difference in mean costs (relative to the comparator) divided by the difference in mean effects. Sometimes expressed with confidence intervals.
- Kaplan–Meier curves
- Also called product limit method. A non-parametric method of compiling life or survival tables, developed by Kaplan and Meier in 1958. This combines calculated probabilities of survival and estimates to allow for censored observations, which are assumed to occur randomly. The intervals are defined as ending each time an event (e.g. death, withdrawal) occurs and are therefore unequal.
- Karnofsky performance status scale
- A performance measure for rating the ability of a person to perform usual activities, evaluating a patient’s progress after a therapeutic procedure, and determining a patient’s suitability for therapy. It is used most commonly in the prognosis of cancer therapy, usually after chemotherapy and customarily administered before and after therapy. A measure is given by a physician to a patient’s ability to perform certain ordinary tasks:
- Partial response
- At least a 50% decrease in tumour size for > 4 weeks without an increase in the size of any area of known malignant disease or the appearance of new lesions.
- Phase II trial
- A study with a small number of patients diagnosed with the disease for which the drug is being studied. In this study, the safety of the new drug is tested. Early effectiveness data are also collected for varying doses of the drug.
- Phase III trial
- A study with a large number of patients diagnosed with the disease for which the drug is being studied and is unlicensed for the indication. In this study, the drug is tested against a placebo or alternative treatment.
- Proportional hazards model
- Regression method for modelling survival times. The outcome variable is whether or not the event of interest has occurred and, if so, after what period; if not, the duration of follow-up. The model predicts that hazard or risk of the event in question at any given time.
- Quality-adjusted life-year
- A term originally developed in cancer studies to balance poor quality of life (possibly with long life expectancy) with good quality of life (possibly with short life expectancy).
- Quality of life
- A concept incorporating all of the factors that might impact on an individual’s life, including factors such as the absence of disease or infirmity, as well as other factors that might affect their physical, mental and social well-being.
- Response Evaluation Criteria for Solid Tumors
- Complete response – disappearance of all target lesions and confirmed at 4 weeks; partial response – at least a 30% decrease in the sum of longest diameters of target lesions (taking as reference the baseline sum of longest diameters) and confirmed at 4 weeks; disease progression – at least a 20% increase in the sum of longest diameters of target lesions (taking as reference the smallest sum of longest diameter recorded since treatment started) with no documentation of complete response, partial response or stable disease before disease progression; stable disease – neither sufficient decrease in sum of longest diameters to meet criteria for partial response nor sufficient increase in sum of longest diameters to meet criteria for disease progression.
- Staging
- The allocation of categories (e.g. for ovarian cancer International Federation of Gynecology and Obstetrics stages I–IV) to tumours, defined by internationally agreed criteria. Tumour stage is an important determinant of treatment and prognosis.
List of abbreviations
- AE
- adverse event
- AGO
- Arbeitsgemeinschaft Gynaekologische Onkologie
- AIC
- Akaike information criterion
- AUC
- area under the curve
- BMI
- body mass index
- BMS
- Bristol-Myers Squibb
- BNF
- British National Formulary
- BSA
- body surface area
- CAP
- cyclophosphamide plus doxorubicin plus cisplatin
- CEA
- carcinoembryonic antigen
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CIC
- commercial-in-confidence
- CR
- complete response
- CRD
- Centre for Reviews and Dissemination
- CrI
- credible interval
- CSR
- clinical study report
- CT
- computed tomography
- CTU
- Clinical Trials Unit
- DSU
- Decision Support Unit
- ECOG
- Eastern Cooperative Oncology Group
- EORTC
- European Organisation for Research in the Treatment of Cancer
- EQ-5D
- European Quality of Life-5 Dimensions
- ERG
- Evidence Review Group
- FACT
- Functional Assessment of Cancer Therapy
- FAD
- Final Appraisal Determination
- FIGO
- International Federation of Gynecology and Obstetrics
- FPS
- fully platinum sensitive
- GCIG
- Gynecologic Cancer Intergroup
- GINECO
- Group d‘Investigateurs Nationaux pour l’Etude des Cancers Ovariens
- GOG
- Gynecologic Oncology Group
- GSK
- GlaxoSmithKline
- HeCOG
- Hellenic Cooperative Oncology Group
- HR
- hazard ratio
- HRG
- Healthcare Resource Group
- HRQoL
- health-related quality of life
- HRT
- hormone replacement therapy
- HSE
- Health Survey for England
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- ICON
- International Collaborative Ovarian Neoplasm
- IPD
- individual patient data
- IRFMN
- Istituto di Ricerche Farmacologiche Mario Negri
- ITT
- intention to treat
- i.v.
- intravenous
- KPS
- Karnofsky performance status
- LCH
- log-cumulative hazard
- LVEF
- left ventricular ejection fraction
- LYG
- life-year gained
- MCAR
- missing completely at random
- MeSH
- medical subject heading
- MLE
- maximum likelihood estimation
- MRC
- Medical Research Council
- MRI
- magnetic resonance imaging
- MS
- manufacturer submission
- MTA
- multiple technology appraisal
- MTC
- mixed-treatment comparison
- NCI
- National Cancer Institute
- NCI-CTC
- National Cancer Institute Common Toxicity Criteria
- NHS EED
- NHS Economic Evaluation Database
- NICE
- National Institute for Health and Care Excellence
- NMA
- network meta-analysis
- OR
- odds ratio
- ORR
- overall response rate
- OS
- overall survival
- OVAR
- Ovarian Cancer Study Group
- PAS
- patient access scheme
- PD
- progressive disease
- PFI
- platinum-free interval
- PFS
- progression-free survival
- PLDH
- pegylated liposomal doxorubicin hydrochloride
- PPE
- palmar–plantar erythrodysaesthesia
- PPS
- partially platinum sensitive
- PR
- partial response
- PRR
- platinum resistant/refractory
- PSA
- probability sensitivity analysis
- PSS
- Personal Social Services
- QALY
- quality-adjusted life-year
- QLQ-C30
- quality of life questionnaire C30
- QoL
- quality of life
- RCT
- randomised controlled trial
- RECIST
- Response Evaluation Criteria in Solid Tumours
- RR
- relative risk
- SD
- stable disease
- sd
- standard deviation
- SF-6D
- Short Form questionnaire-6 Dimensions
- SmPC
- Summary of Product Characteristics
- STA
- single technology appraisal
- SWOG
- Southwest Oncology Group
- TA
- technology appraisal
- TAG
- Technology Assessment Group
- TFI
- treatment-free interval
- TSD
- Technical Support Document
- TTO
- time trade-off
- TTP
- time to progression
- ULN
- upper limit of normal
- WHO
- World Health Organization
- WTP
- willingness to pay
This monograph is based on the Technology Assessment Report produced for NICE. The full report contained a considerable number of data that were deemed commercial-in-confidence. The full report was used by the Appraisal Committee at NICE in their deliberations. The full report with each piece of commercial-in-confidence data removed and replaced by the statement ‘commercial-in-confidence data removed’ is available on the NICE website: www.nice.org.uk.
The present monograph presents as full a version of the report as is possible while retaining readability, but some sections, sentences, tables and figures have been removed. Readers should bear in mind that the discussion, conclusions and implications for practice and research are based on all the data considered in the original full NICE report.