
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 08/117/01. The contractual start date was in November 2009. The draft report began editorial review in June 2012 and was accepted for publication in January 2014. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
The Health Technology Assessment (HTA) programme commissioned this project following a bid by the authors, based at the Wessex Institute, University of Southampton. James Raftery is Professor of HTA at the Wessex Institute. He is a member of the HTA Editorial Board. Amanda Young has been employed by NETSCC since 2008. Louise Stanton was previously employed by NETSCC from 2008 to 2011. Ruairidh Milne is Director of the Wessex Institute and Head of NETSCC. He was employed by NETSCC from 2006 to 2012. Andrew Cook has been employed by NETSCC since 2006. David Turner was previously employed by the Wessex Institute from 2006 to 2011. Peter Davidson is a member of the HTA Editorial Board and has been Director of the HTA programme since 2006. As academics and professional researchers, the authors do not believe they have allowed bias to affect the design of the work, the analysis or the conclusions. Measures to prevent bias included an eminent advisory group and prospective specification of questions.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2015. This work was produced by Raftery et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
The Health Technology Assessment programme
The Health Technology Assessment (HTA) programme, established in 1993, recently celebrated its 20th birthday, part of which included an account of its history. 1 In brief, the programme funds assessments of health technologies with the aim of meeting the research needs of the NHS with scientifically robust evidence. These assessments take two forms: reviews of existing evidence and new research. The latter generally takes the form of clinical trials, most but not all of which are randomised. The overarching aim of the study is to assess the extent to which these trials contributed to meeting the needs of the NHS with scientifically robust evidence.
These randomised controlled trials (RCTs) are of interest for several reasons. First, although over 100 projects involving RCTs had been published by the end of 2011, no systematic compilation exists. Projects may include more than one trial. Some projects report on trials that either failed to recruit or had to depart from plans. As over 200 RCTs funded by the programme were in progress in 2011, a systematic list was required. Second, a small but growing literature studies RCTs. Reviewed below, this literature highlights the desirability of having standardised descriptions of key aspects of these trials.
The RCTs funded by the HTA programme are distinctive in being pragmatic, as opposed to explanatory or licensing trials. They aimed to evaluate the technology of interest in real-world conditions. Inclusion criteria were wide rather than narrow. Patient-related outcomes were preferred to intermediate or surrogate outcomes. Economic analysis was almost always included, sometimes along with qualitative studies. As most of the guidelines for the design, conduct and performance of clinical trials were designed for explanatory and licensing trials, their application to the pragmatic trials funded by the HTA programme may pose problems.
Studies of randomised controlled trials funded by the Health Technology Assessment programme
The Strategies for Trial Enrolment and Participation Study (STEPS),2 summarised in Chapter 6, reviewed recruitment in a cohort of trials funded by the HTA programme and the Medical Research Council (MRC) between 1994 and 2003, and showed that 80% failed to recruit 80% of planned patients.
A study of the impact of the HTA programme, published in 2007,3 reviewed all HTA-funded projects completed between 1993 and 2005, including many which did not include clinical trials, using Buxton and Haney’s payback method. 4 Data were drawn from HTA files and monograph reports supplemented by a survey of lead investigators and a random selection of case studies. It recommended collection of routine data on key headings from the payback approach (all peer-reviewed publications, data on other publications and presentations, capacity development linked to the project, etc.). The assessment of the impact of studies on policies was limited to the lead investigators’ views, which were explored in case studies.
One study considered how many trials funded by the programme showed a statistically significant difference in the primary outcome. 5 In the period 1993–2008 some two-thirds did not report such a difference. 5 This proportion was shown to be similar to other trial portfolios, notably that of National Institutes of Health (NIH) cancer trials. 6
Trials that fail to show such differences can make valuable contributions via their contributions to meta-analyses based on systematic reviews. Another study, which analysed one HTA trial and explored its contribution to the meta-analysis, showed that, although the question posed had been important at the time of commissioning the trial (large effect size, wide confidence interval), the reporting of six other trials in the interim meant that its eventual contribution to the meta-analyses was limited to narrowing the confidence intervals. 7 To follow up this work, one would need to know how many clinical trials funded by the HTA programme had both a relevant preceding and a subsequent meta-analysis.
Finally, a review of economic analyses in HTA trials showed that economic analyses were generally included in trials funded by the HTA programme. 8
Metadata
Metadata is a term commonly used with regard to digital equipment such as cameras to refer to the data routinely recorded about each item, such as time and date. Additional data headings can be added, such as place and persons. Any document prepared using a standard word processing package contains metadata indicating date, person, computer, etc. Non-digital indexes such as a traditional library indexing system can also be described as metadata.
Some databases already contain metadata on clinical trials, such as the International Standard Randomised Controlled Trial Number (ISRCTN) register (www.controlled-trials.com) and the US ClinGov register (https://clinicaltrials.gov). These registers, discussed more fully in Chapter 2, register trials under around 20 descriptive headings including title, start and planned completion dates, disease, intervention, primary outcomes, planned recruitment and contacts. No headings are specified for the reporting or analysis of results, or for the conduct or performance of the trial.
Aims
The aim of the project was to develop and pilot ‘metadata’ on clinical trials funded by the HTA programme. In exploring how to extend the metadata held in existing clinical trial registries, we considered two options. We could either aim to specify a comprehensive data set capable of answering all potential questions, or design a data set to answer particular questions. We pursued the latter option, starting with a set of themes and related questions that might plausibly be answered by such metadata.
We explored questions under six themes using classification systems in answering particular questions. Some classification systems were simple (yes/no) and some complex (16 headings for the European Medicines Agency guideline on handling missing data). Questions that had to do with whether or not analyses were as planned required not only classification of the planned and actual analyses, but also of their (dis)agreement. Data sources comprised both published and unpublished documents. Published sources were largely those in the HTA journal monograph series but also study protocols (most but not all published on the HTA website since 2006). Key unpublished sources included final application forms as well as vignettes, commissioning briefs and project protocol change forms.
The project explored the extent to which metadata could provide standardised data which would be useful not only in managing that portfolio but also in enabling assessment of the conduct, analysis and cost of those trials. Such assessment of the trials would require high-quality data, which had been subject to explicit quality assurance.
The four project objectives, as stated in the final application funded by the HTA programme, were:
-
to develop, pilot and validate metadata definitions and classification systems to answer specified questions within six themes
-
to extract data under these headings from published RCTs funded by the HTA programme
-
to analyse these data to answer specific questions grouped by theme
-
to consider further development and uses of the data set, including refinements of the metadata headings for their application to ongoing and future HTA trials.
The protocol stated that:
Metadata would provide standardised data about the portfolio of HTA trials. These data would enable assessment of questions such as how well the trials were conducted, and the extent to which their results were as expected. Some limited metadata are already publicly available; their extension as proposed here will require appropriate data headings (or classification systems), some of which would be developed in this project.
It also stated that:
The provision of such data would enable performance of the trial portfolio to be monitored over time. Such data would also indicate foci for improvement and help assess the contribution of the ‘needs-led’ and ‘value added’ scientific inputs. To the extent that similar data could be collated for other trials, these could be compared with the HTA trials.
Our research themes
The themes of most immediate interest was based around the composition and performance of the ‘portfolio’ of clinical trials funded by the HTA programme. The provision of such data was seen as enabling performance of the trial portfolio to be monitored over time. Such data would also indicate foci for improvement and help to assess the contribution of the ‘needs-led’ and ‘value-added’ elements of the programme.
The project proposal aimed to extend these trial registration metadata to include data required to answer questions under the following six broad themes:
-
How was the trial seen as meeting the needs of the NHS?
-
How well designed was the trial?
-
How well conducted was the trial?
-
Were the statistical analyses appropriate?
-
What, if any, kind of economic analysis was performed?
-
What was the cost of the trial?
Themes 1 (meeting the needs of the NHS) and 5 (economics) relate to the HTA programme’s overarching aim of meeting the needs of the NHS. Themes 2, 3 and 4 address the robustness of the scientific evidence. Theme 6, on the cost of trials, helps explore value for money.
The choice of the above themes was that of the authors, guided by the literature and aiming to update or replicate previous studies. Four of the authors (JR, RM, PD and AC), having worked for the National Institute for Health Research Evaluation, Trials and Studies Coordinating Centre (NETSCC) in a range of senior roles, identified these themes as of concern to the programme. Relevant published studies were updated where possible. As part of the first theme (the origin of the research question), an earlier paper by Chase et al. 9 was updated. For the second theme (design of trials), previous studies by Chan et al. 10 were drawn on. For performance (the third theme), STEPS,2 which examined the recruitment success of multicentre RCTs, was important. For the fourth theme (statistical analysis), besides Chan et al. 10 and Chan and Altman,11 the issues of concern were with the appropriate analysis of primary outcomes, including the congruence of planned and actual analyses. For economic analysis (the fifth theme), the widely used 1996 BMJ guideline12 provided a starting point. Few studies have been published on the costs of trials, mainly to do with commercial trials in the USA. Guidance from the UK Department of Health specified how non-commercial trials13 should be costed but its application had not been studied. This was the sixth theme. Each of these themes was operationalised into more specific questions and iterated against the data available (see Chapters 4–9).
Project team
Details of each authors' contributions are provided in Acknowledgements (p.113). An external advisory group, also detailed in Acknowledgements, provided valuable input for which we are grateful.
Structure of the report
The aims and objectives related to the development of the metadata were described in this chapter. Chapter 2 provides a review of the existing databases, Chapter 3 reports on methods and Chapters 4–9 report on each of the six themes. Chapters 10 and 11 provide a discussion of the main findings, draw conclusions for the overall project and discuss recommendations of the type of questions that are plausible for future use in the metadata database.
Chapter 2 Data quality and reporting in existing clinical trial registries: a review of existing databases
This chapter briefly reviews existing databases of clinical trials, including descriptions of the main databases and studies which have used them, based on a systematic literature search (detailed in Appendix 1).
Trial registries: the USA
The impetus for the US clinical trials database, ClinGov, came from legislation in 1997, against the background of the human immunodeficiency virus (HIV) epidemic, that mandated a registry of clinical trials for both federally and privately funded trials ‘of experimental treatments for serious or life-threatening diseases or conditions.’14 Patient groups had demanded ready access to information about clinical research studies so that they might be more fully informed about a range of potential treatment options, particularly for very serious diseases. The law emphasised that the information in such a registry must be easily accessible and available to patients, the public, health-care providers and researchers in a form that can be readily understood. 15
Previous attempts to establish clinical trials information systems had focused less on patient access than on clinician and researcher access and use. ClinGov had been concerned that if relevant data about trials were not published or are poorly reported, publication bias and, ultimately, poor care could result.
The design of ClinGov was guided by the following principles:
-
to ensure that design and implementation was guided by the needs of the primary intended audience, patients and other members of the public
-
to get broad agreement on a common set of data elements with a standard syntax and semantics
-
to acknowledge that requirements would evolve over time, implying a modular and extensible design.
A web-based system resulted, which aimed to be easy for novice users but which had extensive functionality. As all NIH-sponsored trials were to be included, ClinGov worked closely with the 21 NIH institutes, each of which had varying approaches to data management and collection and varying levels of technical expertise. The 21 institutes agreed on a common set of data elements for the clinical trials data. Just over a dozen required data elements and another dozen or so optional elements were agreed. The elements fell into several high-level categories: descriptive information such as titles and summaries; recruitment information to let patients know whether or not it is still possible to enrol in a trial; location and contact information to enable patients and their doctors to discuss with the persons who are actually conducting the trials; administrative data, such as trial sponsors and identification numbers; and optional supplementary information, such as literature references and keywords. Table 1 lists the 15 required and 12 optional data elements.
| Required data elements | Optional data elements |
|---|---|
| 1. Study identification number | 1. NIH grant or contract number |
| 2. Study sponsor | 2. Investigator |
| 3. Brief title | 3. Official title |
| 4. Brief summary | 4. Detailed description |
| 5. Location of trial | 5. Study start date |
| 6. Recruitment status | 6. Study completion date |
| 7. Contact information | 7. References for background citations |
| 8. Eligibility criteria | 8. References for completed studies |
| 9. Study type | 9. Results |
| 10. Study design | 10. Keywords |
| 11. Study phase | 11. Supplementary information |
| 12. Condition | 12. URL for trial information |
| 13. Intervention | |
| 14. Data provider | |
| 15. Date last modified |
The study sponsor was defined as the primary institute, agency or organisation responsible for conducting and funding the clinical study. Additional sponsors could be listed in the database. Investigator names were included at the discretion of the data provider. Data providers were asked to provide brief, readily understood titles and summaries, including why the study was being performed, what drugs or other interventions were being studied, which populations were being targeted, how participants were assigned to a treatment design and what primary and secondary outcomes were being examined for change (e.g. tumour size, weight gain, quality of life).
Location information included contact information and status of a clinical trial at specific locations. As many trials were being conducted at multiple locations, sometimes dozens of sites, contact information and recruitment status for all sites had to be accurate and current. Six categories of recruitment status applied: not yet recruiting (the investigators have designed the study but are not yet ready to recruit patients); recruiting (the study is ready to begin and is actively recruiting and enrolling subjects); no longer recruiting (the study is under way and has completed its recruiting and enrolment phase); completed (the study has ended and the results have been determined); suspended (the study has stopped recruiting or enrolling subjects, but may resume recruiting); and terminated (the study has stopped enrolling subjects and there is no potential to resume recruiting). Information about start and completion dates of the study were included, as was contact information including a name and a telephone number for further enquiries.
Eligibility criteria were defined as the conditions that an individual must meet to participate in a clinical study, based on inclusion and exclusion criteria and context.
Besides clinical trials designed to investigate new therapies, nine other study types were included: diagnostic, genetic, monitoring, natural history, prevention, screening, supportive care, training and treatment. Study design types included randomised and observational study designs as well as methods (e.g. double-blind method) and other descriptors (e.g. multicentre site).
ClinGov required certain items as separate data elements specifically to ensure optimal search capabilities. These included the study phase, the condition under study and the intervention being tested. The phase of the study was important information for patients who were considering enrolling in a particular trial. Data providers were requested to name the condition and intervention being studied using the medical subject headings (MeSH) of the Unified Medical Language System, if at all possible.
Optional information included references for publications that either led to the design of a study or reported on the study results. In these cases, data providers were asked to provide a MEDLINE unique identifier so that it could be linked directly to a MEDLINE citation record. A summary of the results could also be prepared specifically for inclusion in the database and the use of MeSH keywords was also encouraged. Supplementary information could include uniform resource locators (URLs) of websites related to the clinical trial.
The agreement of a common set of data elements was completed by the end of 1998. The next step was concerned with methods for receiving data for inclusion in a centralised database at the National Library of Medicine. Data were sent to ClinGov in Extensible Markup Language (XML) format according to a document type definition (DTD). Each clinical trial record was stored in a single XML document. A validator process performed checks on each record; each XML document was analysed and checked for adherence to the DTD. Adherence to the DTD helped identify structural errors in the document. Once the XML document was structurally correct, a Java object was created to facilitate content validation. Content validation could be performed on any data elements that did not contain free text.
Trial registries: World Health Organization clinical trials registry platform
Following the Declaration of Helsinki statement in 2000,16 the World Health Assembly vote to establish the International Clinical Trials Registry Platform (ICTRP)17 in 2004 and the International Committee of Medical Journal Editors (ICMJE) 2004 declaration,18 the World Health Organization (WHO) established the ICTRP to facilitate the prospective registration of clinical trials. Trials could not be registered with WHO but with either a primary registry in the WHO Registry Network or with an ICMJE-approved registry. As regulatory, legal, ethical, funding and other requirements differ from country to country, the approved registries vary to some extent. WHO specified a 20-item minimum data set. This list was very similar to that of ClinGov but differed in several ways:
-
The WHO list included sources of funding, which was not explicitly included in ClinGov.
-
Primary and secondary outcomes were included by WHO but not ClinGov.
-
ClinGov used eligibility whereas WHO used inclusion/exclusion criteria.
-
WHO distinguished between public and scientific in both titles and contacts.
-
Different identification numbers were used (ISRCTN and ClinGov).
The WHO Registry Network comprises primary registries, partner registries and registries working towards becoming primary registries. Any registry that enters clinical trials into its database prospectively (that is, before the first participant is recruited), and that meets the WHO Registry Criteria or is working with ICTRP towards becoming a primary registry, can be part of the WHO Registry Network.
Primary registries in the WHO Registry Network meet specific criteria for content, quality and validity, accessibility, unique identification, technical capacity and administration. Primary registries meet the requirements of the ICMJE. The nine primary registries as at December 2011 are shown in Box 1.
Australian New Zealand Clinical Trials Registry.
Brazilian Clinical Trials Registry.
Chinese Clinical Trial Registry.
Clinical Research Information Service, Republic of Korea.
Clinical Trials Registry, India.
Cuban Public Registry of Clinical Trials.
EU Clinical Trials Register.
German Clinical Trials Register.
Iranian Registry of Clinical Trials.
EU, European Union.
Partner registries meet the same criteria as primary registries in the WHO Registry Network (i.e. for content, quality and validity, etc.), except that they do not need to:
-
have a national or regional remit or the support of government
-
be managed by a not-for-profit agency
-
be open to all prospective registrants.
All partner registries must also be affiliated with either a primary registry in the WHO Registry Network or an ICMJE-approved registry. It is the responsibility of primary registries in the WHO Registry Network to ensure that their partner registries meet WHO Registry Criteria. Partner registries at the end of 2011 included the Clinical Trial Registry of the University Medical Center Freiburg, German Registry for Somatic Gene-Transfer Trials, the Centre for Clinical Trials, and Clinical Trials Registry – Chinese, University of Hong Kong.
The US registry, ClinGov, is not a partner of any kind in the WHO network. Two different realms thus exist in clinical registries: the USA and the rest of the world. Inevitably, the headings for trial registration, although broadly the same, differ. One striking difference is that the US register does not require data on the funding source of the trial, whereas this is required in the rest of the world. Another is that whereas the USA has moved towards requiring the registration of results of trials, the rest of the world has not.
Trial registries: the UK
The ISRCTN, a primary partner in the WHO platform, is run by Controlled Clinical Trials, which registers any clinical trial in the UK designed to assess the efficacy of a health-care intervention in humans. The ISRCTN collects the 20-point WHO list and makes this available on a trial-by-trial basis on the internet. The EU Clinical Trials Registry, a secondary partner in WHO, is confined to investigational drugs and includes the UK Medicines and Healthcare products Regulatory Agency (MHRA) as one of its data-providing agencies. It does not provide data on individual trials.
UK trials register mainly with ISRCTN but a proportion register with ClinGov. This appears to be partly for historical reasons (ClinGov came first), but also because registration is free in ClinGov but ISRCTN charges a small fee (£200 in 2012). Although this charge is met by the UK Department of Health for approved trials [those funded by the National Institute for Health Research (NIHR), research councils or UK charities], other trials which would have to pay may choose to register with ClinGov. A recent review of registration of UK non-commercial trials showed a rise in the proportion registering with ClinGov to around 30% in 2010. 19
Registries and reporting of results
An exploration of the issues raised by including reporting of the findings of clinical trials in databases under the aegis of WHO20–22 discussed issues to do with multiple outcomes and the importance of context in interpretation of results. It noted that, historically, access to the results of a trial had been achieved through publication in a peer-reviewed journal but that this publication model has its limitations, particularly in an environment where the end users of research information include health-care policy-makers, consumers, regulators and legislators who want rapid access to high-quality information in a ‘user-friendly’ format. It noted that in the future, researchers may be legally required to make their findings publicly available within a specific time frame (assuming any legislation created does not have escape clauses built in). In the USA, it noted that such legislation was already in place (available at www.fda.gov/oc/initiatives/HR3580.pdf).
Since the development of trial registration databases in 2000, research has been conducted to:
-
describe the characteristics of trials registered23
-
review the compliance and quality of entries in ClinGov,14 the WHO portal20–22 and several registers24
-
report on scientific leadership (ISRCTN and ClinGov)
-
compare planned and actual trial analyses, including analysis of primary outcomes in major journals27 and comparisons between protocols and registered entries to published reports. 28
Details of the literature searches on trial registration, uses and data quality are provided in Appendices 1–3. In summary, many trials registered were small, with 62% of interventional trials registered in ClinGov in 2007–10 enrolling fewer than 100 patients. The quality and compliance of registration was not good, with trials often registered late (whether defined as after recruitment had commenced or after the trial had been completed) and with missing registration data, specifically to do with contacts, primary outcomes and the processes of randomisation. Compliance was found to be improving, at least for the period 2005–7. 21 Around one-third of registered trials had not reported 24 months after completion, with worse performance for industry-funded trials.
Comparisons of planned and actual trial behaviour, summarised in Chapters 4–9, show that discrepancies between protocols or trial entries and trial reports were common.
Conclusions
This brief review of the literature indicates that:
-
Two main registry types have emerged – the US ClinGov and the WHO platform which provides an infrastructure for the rest of the world.
-
The data required differs between ClinGov and the WHO platform, with the latter including primary and secondary outcomes and source of financing. The former has more detail on patient eligibility and appears more patient oriented.
-
Prospective registration of planned RCTs has become common and mandatory in many countries.
-
In the UK, RCTs register mainly with the ISRCTN via Current Controlled Trials (CCT), but some register with ClinGov.
-
The quality of the data registered has been poor, with all studies indicating poor compliance.
-
Both the US and WHO registries are moving towards inclusion of results, with ClinGov further advanced owing to such reporting having become mandatory in the USA from 2006.
-
No studies were identified that went beyond the minimum data set for prospective registration to include conduct, performance, cost and results of trials.
Chapter 3 Methods
Introduction
This chapter outlines the target ‘population’, inclusion criteria, data sources and quality assurance methods used.
Population
The population of interest was completed RCTs funded by the HTA programme. The starting point was the published HTA monograph series which published the results of almost all funded projects. Projects were distinguished from trials, as projects can comprise several trials. Some trials were described as pilot or feasibility trials.
A RCT was defined for this study as:
An experiment in which two or more interventions, possibly including a control intervention or no intervention, are compared by being randomly allocated to participants. In most trials one intervention is assigned to each individual but sometimes assignment is to defined groups of individuals (for example, in a household) or interventions are assigned within individuals (for example, in different orders or to different parts of the body).
Reproduced with permission from www.nets.nihr.ac.uk/glossary?result_1655_result_page=R
Published HTA-funded projects which included a RCT were identified from the HTA monograph series (www.hta.ac.uk/). The title and the ISRCTN number for each published monograph were reviewed and cross-referenced with the HTA Management Information System (HTA MIS).
Inclusion criteria
The inclusion criteria were HTA-funded projects that had reported the results of at least one RCT and had been published as a HTA monograph by the end of February 2011. One project which included a clinical trial but failed to submit the draft final report and did not publish a HTA monograph was excluded on these grounds. One hundred and nine projects were included.
These criteria implied to the inclusion of pilot and feasibility studies. This mattered to varying degrees for the different themes. A full list of the RCTs included in the database is shown in Appendix 2.
Data sources
Data on each randomised clinical trial were extracted from seven sources:
-
the published HTA monograph (publicly available)
-
protocol changes form, if available (a confidential document submitted with the final report)
-
the final, most current version of the protocol (project protocols were not available for older HTA-funded trials)
-
the full proposal attached to the contract of agreement (confidential document)
-
the commissioning brief (publicly available)
-
the vignette (confidential document)
-
the HTA MIS (confidential).
As sources 2–6 above were mostly only available on paper, paper files were scanned to create electronic portable document format (PDF) copies, which were directly linked to the database.
As the HTA programme changed the format of these sources over time, a timeline was drawn within which each project was situated. These changes sometimes limited the data available for particular questions.
Quality control
Our approach to quality assurance was guided by Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA), which, although designed for systematic reviews, can be applied to the processes we used for data extraction. Out of the 27 PRISMA checklist items, 12 items are listed under ‘Methods’. Of these 12 statements, we used six items to help define the design of the project (eligibility criteria, information sources, search, study selection, data collection process and data items). PRISMA states: ‘Describe the method of data extraction from reports and any processes for obtaining and confirming data from investigators’ (Liberati et al. 200929).
Data extraction forms were developed and piloted on five trials, leading to refinement. This led to the identification of five types of data field, each with a different process of quality assurance, as shown in Table 2 along with their relevance by theme.
| Type of data field | Description | Application to theme | Quality check |
|---|---|---|---|
| 1. HTA MIS | Data fields obtained directly from the HTA MIS (read only) | 37 | Reasonableness, outliers |
| 2. Straightforward fields | Data fields relatively straightforward to extract and did not usually involve a judgement call (e.g. trial design, number of arms, details on project extensions, protocol changes) | 60 | Random sample checked against source documents |
| 3. Numeric | Data fields where the highest level of transcription errors (human error) were likely to occur (e.g. in conduct questions on number of patients recruited/randomised/followed up and number of centres) | 284 | All fields checked against source documents [with the exception of 58 fields for the health economics data, where a random sample was double-checked by the health economist (DT)]. The project statistician (LS) checked data extracted |
| 4. Judgement call fields | Data fields which involved a subjective judgement, for example whether or not the researchers had adequately specified the method of randomisation sequence generation, reported all CONSORT fields or the type of trial intervention | ||
| 5. Specialist data fields | Data fields which required specialised training or knowledge to understand and extract data accurately; these were likely to lead to the highest errors during data extraction (e.g. sample size calculation fields, planned method of statistical analysis, all health economics fields) |
Two members of the team went through discrepancies and queries relating to the complete extraction of data, came to an explicit agreement and amended the database accordingly. The level of checking for the 125 trials by the second team member varied; all data were checked for themes 1, 2, 3 and 4. A percentage of data checking was completed by DT for theme 5 [40% for the BMJ checklist (BCL)] and JR for theme 6 (40%).
Quality assurance by type of data (Table 2) was applied as follows. With a few exceptions, fields relating to the design of the trial, conduct of the trial, statistical analysis and Consolidated Standards of Reporting Trials (CONSORT) were classified as type 3, 4 or 5 in Table 2. All health economics fields were classified as type 5, the cost of trials fields were classified as type 4 and the NHS need fields were classified as type 2.
Errors noted in the data extracted were corrected and the data changed were recorded in a central Microsoft Office Excel 2010 (Microsoft Corporation, Redmond, WA, USA) spreadsheet. If the change needed further discussion it was noted in the spreadsheet and discussed with AY and/or other members of the steering group.
These quality assurance processes were carried out weekly to ensure issues with fields could be spotted quickly. Monthly reports were provided to the project steering group.
Most of the fields in the database were either numeric or categorical. For categorical fields, the possible categories for data entry were listed as a drop-down menu and locked to these codes to prevent errors.
Data extraction
Data extraction specification forms were developed for each question (see Appendix 3). Free-text entries were allowed only when no classification system could be employed. Classification systems were used to specify the forms, showing for each item the data to be extracted, the type of field and, if categorical, the classification we planned to use. Existing classifications from the published literature were used where possible. Where there were two competing classifications we used both, and if there was no published classification, we used either the HTA MIS (if applicable) or, in extremis, a simple hierarchical system (yes/no, if yes, then detail).
The forms were developed by the research fellow and statistician in conjunction with the research lead for each theme and reviewed by the steering group. The project advisory group was sent a full list of the data fields that were planned to be included in the metadata database for comment. The data items finally included in the metadata database were based on consensus.
Projects and trials included in the database
This section reports on the number of trials that met the eligibility criteria along with problems encountered.
From its start date in 1997 to the end of February 2011, the HTA programme published 574 projects in the HTA monograph series (in 15 annual volumes). The executive summary for each report was independently reviewed by two members of the steering group to assess whether or not the monograph included the results of a RCT. One hundred and twelve projects were identified as potentially including a RCT. From screening, three of the projects were excluded and full data were extracted from 109 monographs (Figure 1).
FIGURE 1.
Flow diagram of included and excluded RCTs and monographs.

The three excluded projects were:
-
one report originally funded as a RCT on paramedic training for serious trauma, which failed to randomise and went on as a non-randomised study (1998, Volume 2, Number 17)
-
one economic evaluation of pre-existing RCT data, which was not funded by the HTA programme (1999, Volume 3, Number 23)
-
one report of the long-term outcomes of patients in 10 RCTs of cognitive–behavioural therapy (CBT) conducted between 1985 and 2001 (2005, Volume 9, Number 42).
Narrowly included projects
One trial was narrowly included, in which participants were randomly assigned to be offered two different types of hearing aids. This study of a screening programme included a small RCT, the results of which were reported in the monograph.
Pilot and feasibility studies
Four monographs reported pilot or feasibility studies:
-
The feasibility of a RCT of treatments for localised prostate cancer (2003, Volume 7, Number 14) (trial ID15).
-
A two-centre, three-arm pilot conducted to assess the acceptability of a RCT for comparing arthroscopic lavage with a placebo surgical procedure (2010, Volume 14, Number 5) (trial ID97).
-
A pilot study conducted to assess the safety and efficacy of reducing blood pressure with two types of medication for patients with hypertension. The pressor phase of the trial was terminated owing to poor recruitment (2009, Volume 13, Number 9) (trial ID78 and ID110).
-
A pilot study on impact of early inhaled corticosteroids prophylaxis, conducted to assess recruitment rates and project protocol, pilot assessment tools and refine the sample size calculation for a definitive study (2000, Volume 4, Number 28) (trial ID121).
From the 109 included projects, 125 RCTs were identified and constituted the cohort of trials included in this study. Eleven monographs included the results of more than one RCT. Five of these included three RCTs and six included two RCTs.
Unique trial identification number
Each project funded by the HTA programme had a unique reference number (e.g. 10/07/99) given when the outline proposal was submitted. We used this to link records in the database to the HTA Management Information System. For the 11 projects that included more than a single RCT, we developed an additional identifier. A free-text field was included in the database to identify the specific clinical trial. Fields were also included in the database showing the number of trials in the monograph, listing the trial ID numbers for those trials reported in the same monograph. For example, the HTA-funded project 96/15/05, ‘Which anaesthetic agents and techniques are cost-effective in day surgery? Literature review, national survey of practice and randomised controlled trial’, contained two RCTs, a two-arm trial for the paediatric population and a four-arm trial for the adult population. This project has a single ISRCTN number (87609400). We created one unique ID for each RCT: ID13 for the adult study and ID14 for the paediatric study.
Completeness of data sources
Data extraction took place from 6 August 2010 to 8 November 2011. Table 3 shows the completeness of the sources of information used for data extraction.
| Document | Number available for data extraction (%) |
|---|---|
| Vignette | 99/109 (90.8) |
| Commissioning brief | 99/109 (90.8) |
| Application form (proposal) | 106/109 (97.2) |
| Protocol | 58/109 (53.2) |
| Monograph | 109/109 (100) |
| Protocol change form | 78/109 (71.6) |
| Progress reports/extension requests | Multiple documents per trial available; too many to count |
| Total number of monographs | 109 |
The least complete category of documents retrieved was the project protocols (65/125, or 53%). Only 30 out of the 65 were available in the public domain (via the HTA programme website, www.hta.ac.uk/). These were mainly linked to monographs published after 2009. Twenty-six protocols were extracted directly from the HTA project paper folders; these were not in the public domain and were treated as confidential. For the 60 trials lacking a protocol, we contacted the chief investigator (a) to see if a project protocol existed; (b) if one did exist, to request a copy of the document to include in the database; and (c) to seek permission to forward the protocol to the HTA programme. We succeeded in contacting 59 chief investigators and were unable to find current contact details for one. Of the 59 chief investigators contacted, 35 responded (59.3%), leading to retrieval of an additional nine project protocols.
Vignettes and commissioning briefs were located for 99 projects (90.8%). Out of 109 projects, 106 application forms were retrieved. Seventy-eight projects produced a protocol changes form which only applied to those that required such a change (for two trials, a non-standard form was used, which was included in these figures).
Database
Data for each trial were stored in a Microsoft Access 2003 (Microsoft Corporation, Redmond, WA, USA) database. Each clinical trial entered had a unique ID number which linked data to every table and form.
Relevant direct website links were included to assist in data entry. A hyperlink to the International Classification of Diseases, Tenth Edition (Theme 2) (http://apps.who.int/classifications/icd10/browse/2010/en) led to a drop-down menu for the disease chapters. Other hyperlinks included the UK Clinical Research Collaboration (UKCRC), Health Research Classification System (HRCS) for interventions (www.hrcsonline.net/rac/overview),30 CONSORT (www.consort-statement.org/), International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) (www.fda.gov/regulatoryinformation/guidances/ucm122049.htm) and BMJ health economics checklist (www.bmj.com/content/313/7052/275.full).
Classification systems
Data for each question were structured and entered into a classification system. The classification systems generated during the development of the data extraction specification forms were entered into the tables as value lists (source type). For example, trials type was classified as 1;‘Superiority’;2;‘Non-inferiority’;3;‘Equivalence’. Data fields sharing the same classification system were set up in an Access table. For example, data extraction for treatment of missing data was required for the clinical effectiveness and cost-effectiveness analysis of the trial, with 14 options to choose from. Instead of replicating these in each of the data fields, an Access table was generated.
Data collection and management
The data extraction specification forms defined the metadata database, using Access forms. The database was designed to directly link to the HTA programme’s MIS (also Access) so that relevant fields in the HTA MIS could be included in the metadata database and automatically updated. The metadata database was linked to the HTA MIS using either the project number (number given at the time of the outline proposal, which stays with the project until publication) or priority area number (number given at the time of commissioning).
The database was designed to be comprehensive, storing all data items and queries raised during data extraction and decisions made in relation to these. As it linked directly to the information sources for each trial, the database is a repository for all documents about included trials. Further, as suggested by a member of the advisory group, the source and page number of relevant data fields were incorporated into the database. These measures also mean that future researchers can use the links and page numbers to check on the accuracy of data previously extracted, indicate if there are any disagreements and include their own comments.
An escalation process was set up to deal with uncertainty and to resolve any disagreements. The research fellow extracted all data and recorded it in the database, with problems logged under the trial’s additional comments section. Regular meetings were held between the research fellow and statistician to resolve queries logged under each trial. If they could not resolve the issue, the research fellow was to discuss it with the relevant research lead, with escalation to the steering group if necessary. Most queries were resolved at early stages and none were escalated to the steering group.
The metadata database
The Access metadata database included 429 data fields on the 125 trials (Table 4).
| Theme | Actual number of data fields | Source of information |
|---|---|---|
| Core trial information | 48 | Monograph |
| Theme 1: meeting the needs of the NHS | 22 | Vignette, commissioning brief, monograph and HTA MIS |
| Theme 2: design and adherence to protocol | 72 | Protocol or proposal, monograph and HTA MIS |
| Theme 3: performance and delivery of the NIHR HTA programme-funded RCTs | 72 | Protocol or proposal, monograph, HTA MIS and protocol change form |
| Theme 4: statistical analyses appropriate and as planned | 97 | Protocol or proposal and monograph |
| Theme 5: economic analyses alongside clinical trials | 58 | Proposal, monograph and HTA MIS |
| Theme 6: cost of RCTs, trends and determinants | 60 | Protocol or proposal and monograph |
| Total | 429 |
Security, back up and confidentiality
The project had a dedicated research folder on the University of Southampton’s secure server. Access to the folder was only visible to the research team. Access to the folder was not possible without permission from AY.
Any documents containing sensitive data, such as named principal investigators or confidential information, were stored using a protected password; only two members of the project team had these. Other members of the research team were able to access these documents via these two people if deemed necessary.
The HTA programme policy for access to unpublished data requires signing a confidentiality agreement. That confidentiality agreement was signed by all members of the research team at the start of the project. We suggest below that the same rules, updated as appropriate, should govern future access to the database by other researchers.
Some data from older sources of information were deemed confidential, such as failed trials, the funding details of particular trials and problems with the conduct of the trials. Project protocols before 2007, which were often only in the form of proposals, might be seen as confidential as they are not in the public domain. Since 2007, the most current version of the project protocol has been published with researchers’ consent on the NETSCC HTA programme website.
This project assumed that protocols for trials funded before 2007 should be treated as confidential. All final proposals attached to the contract of agreement to the Department of Health were also treated as confidential, including the vignette.
For each trial record, a drop-down menu specified whether or not the source was confidential (not in the public domain). Data fields were positioned on the Access form based on where the information was extracted from and by theme. Page numbers and source of information boxes determined whether or not the data extracted were confidential.
Questions for which data should be extracted
This section outlines the method used to decide the questions for which data should be extracted.
Some questions under each theme were quickly shown as not feasible owing to lack of data or time required to extract data. These questions are discussed under each theme in the relevant chapters (see Chapters 4–9).
The questions deemed feasible were taken further by extracting data, entering it in the relevant classification systems and assessing it.
For each question, a judgement was made regarding whether it should be:
-
kept
-
amended
-
dropped.
These options were developed from those suggested by Thabane et al. 31 Three criteria were used to reach these judgements:
-
How complete were the data required?
-
Were changes recommended to the classification system?
-
What skills and resource (linked to data type and need for judgement) did the classification system require?
The completeness of the data was measured for each question. Whether or not changes were recommended to the classification system helped indicate if it should be amended. The final criterion, with regard to the skills and resources, was based on records kept by AY.
The criteria were applied hierarchically, with only those for which data were available being assessed against the other criteria. A threshold of 80% was set for data completeness on the basis of representativeness. However, instead of applying the criteria mechanically, the steering group retained the option to consider retaining any question that seemed particularly important.
Changes/deviations from the protocol for this study
Given that one of the questions explored in this project concerns protocol changes, this section discusses changes to the protocol for this study. We deviated from our protocol twice. These deviations were because of:
-
the number of trials included; we included 125 trials in the metadata database, more than the 120 maximum that we specified
-
planned quality assurance; we abandoned our plan to invite chief investigators to check our data extraction on the basis of the results of a small pilot study.
Further, the project was originally funded for 1 year to extract data on 63 RCTs. The piloting of data extraction showed that only around 40 RCTs, or around half the total published to mid-2009, could be done within that time scale. The steering and advisory groups agreed that the value of the database would increase with the number of trials included. The project steering group requested and received a 1-year extension.
None of these in our opinion was likely to have introduced bias.
Chapter 4 Theme 1: meeting the needs of the NHS
This chapter considers questions linked to the theme: ‘How was the trial seen as meeting the needs of the NHS by the HTA programme?’ After a brief review of the relevant literature, it summarises available data on how trials funded by the HTA programme could answer questions about meeting the needs of the NHS. It explores how topics of trials were generated and prioritised. It also explores the outcomes used and the time from prioritisation to publication of the findings. The methods used to answer each question are described and the results are followed by analysis and discussion.
Introduction
Several terms may usefully be defined. By commissioned research, we mean research where the topics to be researched are defined by the programme and not by the researchers who do the work. This implies that the programme is acting on behalf of the NHS and must have mechanisms for ‘knowing’ the needs of the NHS. This differs from both response-mode research (the traditional mode, with funders taking bids from expert teams of researchers) and researcher-led research (HTA’s term for the work stream introduced in 2006 where proposals are submitted by researchers but are rigorously assessed against NHS need). 1
To be relevant to decision-making in the NHS, any clinical trials would need to be pragmatic as opposed to explanatory. Pragmatic trials have been defined as those with broad inclusion criteria, carried out in many centres and with patient-relevant outcomes. 32
To employ a term given prominence in the Cooksey Report (2006),33 NHS-funded research had to be restricted to public interest or market failure research, that is, work that the private sector would not be interested in carrying out. This is often due to the inability to patent that which is being tested (difficult outside new drugs or, in particular, interventions made up of services rather than tightly defined products).
As the HTA programme is a commissioned programme, one might expect it to prioritise research focused on the needs of the NHS. A substantial literature discusses methods for research prioritisation but there is much less on how potential topics should be identified, or on assessments of whether or not prioritised research is indeed ‘needs-led’. 9,34–37
Since its inception, the NHS research and development (R&D) has focused on identifying gaps in research relevant to the NHS and prioritising them. Setting priorities is difficult and complex, partly because there is ‘no agreed upon definition for successful priority setting, so . . . no way of knowing if an organisation achieves it’. 38
Different methods have been suggested, such as multidisciplinary involvement, public and patient involvement, the use of scoring systems, the Delphi process and information specialist involvement. Economic impact approaches include the payback approach or expected value of information models. Priority setting means an allocation of limited resources, which can be highly political and controversial. Developing a structured topic prioritisation process helps address this challenge.
Chase et al. 9 described the different sources used by the HTA programme in 1998 to identify potential priorities. Overall, there were 1100 suggestions for the programme from four main sources: (1) a widespread consultation of health-care commissioners, providers and patients; (2) research recommendations from systematic reviews; (3) reconsideration of previous research priorities; and (4) horizon scanning. Nearly half (46%) of final programme priorities were from the widespread consultation, with 20% from systematic reviews and 10% from each of the other two areas. (The remainder came simultaneously from more than one source.) Chase et al. 9 concluded that there was value in having a mix of sources. One of the aims of this chapter was to apply the approach of Chase et al. 9 to all the RCTs published to mid-2011.
A small literature discussed the patient relevance of outcomes in publications, through surveys of trials published in a particular disease area. There are three notable examples:
-
Gandhi et al. 39 looked at diabetes trials and found that primary outcomes were patient important in only 78 of 436 RCTs (18%).
-
Montori et al. 40 also looked at diabetes trials and found that primary outcomes were patient important in only 42 of 199 RCTs (21%).
-
Rahimi et al. 41 looked at cardiovascular trials and found that primary outcomes were solely patient important (death, morbidity or patient-reported outcomes) in only 93 of 413 trials (23%).
Chalmers and Glasziou42 proposed a framework for considering avoidable waste in research, with four stages. The first concerned whether or not the questions addressed by research are relevant to clinicians and patients; if they are not, Chalmers and Glasziou42 argue that the research is wasted.
Although Chalmers and Glasziou42 give some examples of the ways in which research fails to address relevant questions, they provide no quantifiable measures of waste in this stage of the framework, unlike the other three stages (design, publication and useable report), for each of which empirical estimates of waste are provided.
The extent to which RCTs have been preceded by systematic reviews can indicate the source of the topic. A recent review of 48 trials funded by the HTA programme between 2006 and 2008 indicated that 80% had been preceded by a systematic review. 43
Questions addressed
The questions on which data were extracted are shown in Box 2.
-
T1.1. Type of commissioning work stream?
-
T1.2. Prior systematic review?
-
T1.3. The source for topic identification?
-
T1.4. Type of HTA advisory panel?
-
T1.5. What was the priority given by the programme to the research?
-
T1.6. Did the statement of need change?
-
T1.7. Frequency and accuracy of reporting the primary outcome?
-
T1.8. Adequate reporting of the proposed and published primary outcome?
-
T1.9. What was the time lag between prioritisation and publication of the monograph?
Methods
Nine questions were piloted in theme 1 (hereafter T1). One question (‘How was the relevance to the NHS assessed?’) was deemed not feasible owing to lack of data. However, data were available on the work stream (commissioned or researcher led) (T1.1), whether or not a prior systematic review existed (T1.2) and the source of the topic (T1.3). These are explored below.
Denominators
For questions T1.1, T1.3 and T1.4 the denominator was the number of priority areas (n = 100) which precede any call for a trial. (Note: ‘T’ refers to theme. Each of the six themes are numbered with additional numerals referring to questions within that theme.) One hundred research suggestions/priority areas made it through to the commissioning brief stage, which led to 107 projects being funded containing 123 RCTs. The denominator for questions T1.2 and T1.5–T1.9 was the total number of projects (n = 109) (107 projects via the commissioned work stream and two projects via direct commissioning).
Results
Question T1.1: type of commissioning work stream
Out of the 100 priority areas, 107 (98.2%) projects were funded through the commissioned work stream. The other two projects were ‘directly commissioned’ {09/94/01 [head-to-head comparison of two H1N1 swine influenza vaccines in children aged 6 months to 12 years] and 99/01/01 [conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR)]}.
Question T1.2: prior systematic review
Of the 109 projects, 56% reported a prior systematic review in the published monograph.
Question T1.3: the source for topic identification
Of the four main sources of identification, ‘widespread consultation’ contributed 64 (66.7%) topics followed by systematic reviews (25%, 24/100) and the Horizon Scanning Centre (3%, 3/100).
The balance of these sources shifted over time. When the number of trials increased in 2001–2, the proportion of topics from systematic reviews rose to 65% (Table 5).
| Source of topic identification | 1993–4, n (%) | 1995–6, n (%) | 1997–8, n (%) | 1999–2000, n (%) | 2001–2, n (%) | 2003–4, n (%) | 2005–6, n (%) | Total, n (%) |
|---|---|---|---|---|---|---|---|---|
| Widespread consultation | 17 (89.5) | 22 (84.6) | 11 (73.3) | 6 (66.7) | 3 (15.0) | 4 (66.7) | 1 (100) | 64 (66.7) |
| Systematic reviews | 2 (10.5) | 2 (7.7) | 3 (20.0) | 2 (22.2) | 13 (65.0) | 2 (33.3) | 0 | 24 (25.0) |
| Horizon Scanning Centre | 0 | 1 (3.8) | 1 (6.7) | 0 | 1 (5.0) | 0 | 0 | 3 (3.1) |
| Reconsidered topics | 0 | 1 (3.8) | 0 | 1 (11.1) | 3 (15.0) | 0 | 0 | 5 (5.2) |
| Total | 19 (19.8) | 26 (27.1) | 15 (15.6) | 9 (9.4) | 20 (20.8) | 6 (6.3) | 1 (1.0) | 96 (100.0) |
Question T1.4: type of Health Technology Assessment advisory panel
The source of topics varied by advisory panel (Table 6). Widespread consultation was the main commissioning source for two of the three panels (83.3%, 15/18 and 72.2%, 39/54, respectively). The exception was the pharmaceutical panel, where 50% (12/24) of the commissioned topics were from systematic reviews.
| Source of topic identification | Diagnostic and screening panel, n (%) | Pharmaceutical panel, n (%) | Therapeutic panel, n (%) | Total, n (%) |
|---|---|---|---|---|
| Widespread consultation | 15 (83.3) | 10 (41.7) | 39 (72.2) | 64 (66.7) |
| Systematic reviews | 2 (11.1) | 12 (50.0) | 10 (18.5) | 24 (25.0) |
| Horizon Scanning Centre | 0 | 1 (4.2) | 2 (3.7) | 3 (3.1) |
| Reconsidered topics | 1 (5.6) | 1 (4.2) | 3 (5.6) | 5 (5.2) |
| Total | 18 (18.8) | 24 (25.0) | 54 (56.3) | 96 (100.0) |
Question T1.5: what was the priority given by the programme to the research?
The programme prioritised 70% of projects in the top band. Of the 71 projects prioritised up to and including 1999, 50 (70.4%) were classified as A-list topics (‘recommended for commissioning – must commission’) and 18 (25.4%) were B-list topics (‘recommended for commissioning’ only). The HTA MIS database did not provide sufficient information for 4.2% of trials (3/71) (Table 7).
| Priority status and HTA advisory panel description | n (%) |
|---|---|
| Priority band (up to and including publication date 1999) | |
| Recommended for commissioning – must commission (A) | 50 (70.4) |
| Recommended for commissioning (B) | 18 (25.4) |
| Category unknown | 3 (4.2) |
| Total | 71 |
| HTA advisory panel | |
| Diagnostic and screening | 21 (19.3) |
| Pharmaceutical | 25 (22.9) |
| Therapeutic procedures | 61 (56.0) |
| Department of Health – Direct Project Commissioned | 2 (1.8) |
| Total | 109 |
Question T1.6: did the ‘statement of need’ change?
This question asked if researchers undertaking research drifted from the programme’s initial assessment of NHS need for the research. The statement of need did not change between the commissioning brief and the monograph in 101 trials (94.4%, 101/107). No data were available for the remaining six projects. For these six projects, we were unable to compare the information reported in the commissioning brief with that reported in the monograph for three trials (2.8%). The reasons were ‘No commissioning brief or vignette was available’ (trials ID121 and ID122) and ‘No commissioning brief or vignette was prepared. It was a fast track topic’ (trial ID106). For the final three projects, we were unclear about the reporting of the statement and whether or not it changed from the advertisement to the executive summary in the monograph (trials ID60, ID79 and ID86). Owing to the complexity of data extracted to answer this question, it was not possible to analyse the data further. In addition, it was agreed that all data fields related to the ‘statement of change’ question would be dropped from further analyses.
Question T1.7: frequency and accuracy of reporting the primary outcomes
The 109 funded projects included 125 clinical trials. The main primary outcome, defined as that used for sample size calculation, was reviewed independently by two researchers (RM and AY) for the 109 projects. Four projects lacked requisite information (the monograph did not clearly state what the main primary outcome was nor was it possible to determine what the main primary outcome was during the data extraction process). In this instance, consensus was reached by both researchers reviewing the monograph (specifically, the sample size calculation section reported in the methods chapter of the monograph) to determine the actual type of primary outcome. It was not possible to accurately identify what the main primary outcome was for one project (trial ID68).
Seventy-eight (73%) of the 107 projects in the commissioned work stream reported sufficiently on the proposed primary outcome. Twenty-one projects reported limited information. Eight commissioning briefs (7.5%) contained no information about what the primary outcome was.
Question T1.8: adequate reporting of the proposed and published primary outcome
All projects were analysed to compare the proposed primary outcomes with those published (n = 109). We were able to classify the proposed primary outcome for 97 projects (88.9%) and the published primary outcome for 108 projects (99.1%) (Table 8); little changed between these two stages. Patient-important outcomes were reported in more than half of the HTA-commissioned projects, at both the proposed and published stages of the project (67%, 73/109 and 73.4%, 80/109, respectively). A number of outcomes could not be classified using the Gandhi et al. 39 three main headings. Fourteen proposed primary outcomes and 18 published primary outcomes were categorised as ‘other’.
| Type of primary outcomes reported | n (%) |
|---|---|
| Type of primary outcome reported at the commissioning stagea | |
| Patient important (including others) | 75 (70.1) |
| Surrogate | 0 |
| Physiological/laboratory | 0 |
| Other | 3 (2.8) |
| Limited information reported in the commissioning brief | 21 (19.6) |
| No information available | 8 (7.5) |
| Total | 107 |
| Type of primary outcome reported in the proposal/protocol | |
| Patient important (including others) | 73 (67.0) |
| Surrogate | 8 (7.3) |
| Physiological/laboratory | 2 (1.8) |
| Other | 14 (12.8) |
| No information available | 12 (11.0) |
| Total | 109 |
| Type of primary outcome reported in the monograph | |
| Patient important (including others) | 80 (73.4) |
| Surrogate | 9 (8.3) |
| Physiological/laboratory | 1 (0.9) |
| Other | 18 (16.5) |
| No information available | 1 (0.9) |
| Total | 109 |
Thirteen projects (11.9%) had differences between the planned and actual type of primary outcome. These discrepancies were mainly due to a lack of information or having no information on the primary outcome in the planned documentation (proposal/protocol) (n = 12). The monograph was able to provide sufficient information for 10 of these projects to enable the primary outcome to be classified accordingly. Table 9 highlights where these discrepancies were between the planned and actual reporting of the primary outcome.
| Planned primary outcome as reported in the proposal/protocol | Actual primary outcome measure as reported in the monograph | |||||
|---|---|---|---|---|---|---|
| Patient important (including others), n (%) | Surrogate, n (%) | Physiological/laboratory, n (%) | Other, n (%) | No information available, n (%) | Total, n (%) | |
| Patient important (including others), n (%) | 72 (90.0) | 0 | 0 | 1 (5.6) | 0 | 73 (67.0) |
| Surrogate, n (%) | 0 | 8 (88.9) | 0 | 0 | 0 | 8 (7.3) |
| Physiological/laboratory, n (%) | 1 (1.3) | 0 | 1 (100.0) | 0 | 0 | 2 (1.8) |
| Other, n (%) | 0 | 0 | 0 | 14 (77.8) | 0 | 14 (12.8) |
| No information available, n (%) | 7 (8.7) | 1 (11.1) | 0 | 3 (16.7) | 1 (100.0) | 12 (11.0) |
| Total, n (%) | 80 (73.4) | 9 (8.3) | 1 (0.9) | 18 (16.5) | 1 (0.9) | 109 (100.0) |
When diagnostic and screening projects (n = 20) were excluded, patient-important outcomes increased from 67% (n = 73) to 73% (65/89).
Over the period 1993–2002, 82.7% (67/81) of reported primary outcomes were patient important (Table 10). The years 1993–2002 provide a more accurate report of the type of primary outcome reported in the monograph, as a number of projects funded during the period 2003–10 have not yet published.
| Type of primary outcome | 1993–4, n (%) | 1995–6, n (%) | 1997–8, n (%) | 1999–2000, n (%) | 2001–2, n (%) | 2003–4, n (%) | 2005–6, n (%) | 2007–8, n (%) | 2009–10, n (%) | Total, n (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient important (including others) | 16 (80.0) | 20 (83.4) | 12 (100.0) | 4 (66.7) | 15 (78.9) | 3 (75.0) | 1 (100.0) | 0 | 0 | 71 (80.6) |
| Surrogate | 1 (5.0) | 2 (8.3) | 0 | 1 (16.7) | 3 (15.8) | 0 | 0 | 0 | 1 (50.0) | 8 (9.1) |
| Physiological/laboratory | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (50.0) | 1 (1.1) |
| Other | 3 (15.0) | 2 (8.3) | 0 | 0 | 1 (5.3) | 1 (25.0) | 0 | 0 | 0 | 7 (8.0) |
| No information available | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 1 (1.1) |
| Total | 20 (100.0) | 24 (100.0) | 12 (100.0) | 6 (100.0) | 19 (100.0) | 4 (100.0) | 1 (100.0) | 0 | 2 (100.0) | 88 (100.0) |
Question T1.9: what was the time lag between prioritisation and publication of the monograph?
This question asked about the time taken between the programme prioritising a topic and the monograph publishing the results. The interval was 8–10 years (Table 11). The average was 8 years for trials prioritised in 1993 and 9 years for those prioritised in 1999.
| Year of publication | PAR year | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1993, n (%) | 1994, n (%) | 1995, n (%) | 1996, n (%) | 1997, n (%) | 1998, n (%) | 1999, n (%) | 2000, n (%) | 2001, n (%) | 2002, n (%) | 2003, n (%) | 2004, n (%) | 2005, n (%) | 2009, n (%) | Total, n (%) | |
| 1999 | 1 (8.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.9) |
| 2000 | 5 (41.7) | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 (5.5) |
| 2001 | 2 (16.7) | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (2.8) |
| 2002 | 1 (8.3) | 1 (7.1) | 1 (10.0) | 1 (5.0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 (3.7) |
| 2003 | 1 (8.3) | 1 (7.1) | 1 (10.0) | 2 (10.0) | 1 (8.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 (5.5) |
| 2004 | 1 (8.3) | 3 (21.4) | 3 (30.0) | 4 (20.0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 11 (10.1) |
| 2005 | 1 (8.3) | 5 (35.7) | 0 | 7 (35.0) | 1 (8.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 14 (12.8) |
| 2006 | 0 | 0 | 2 (20.0) | 4 (20.0) | 4 (33.3) | 0 | 1 (11.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 11 (10.1) |
| 2007 | 0 | 2 (14.3) | 2 (20.0) | 1 (5.0) | 1 (8.3) | 1 (33.3) | 3 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 10 (9.1) |
| 2008 | 0 | 0 | 1 (10.0) | 0 | 2 (16.7) | 1 (33.3) | 1 (11.1) | 0 | 2 (12.5) | 0 | 0 | 0 | 0 | 0 | 7 (6.4) |
| 2009 | 0 | 0 | 0 | 0 | 2 (16.7) | 1 (33.3) | 1 (11.1) | 0 | 10 (62.5) | 2 (50.0) | 2 (33.3) | 0 | 0 | 0 | 18 (16.5) |
| 2010 | 0 | 0 | 0 | 1 (5.0) | 1 (8.3) | 0 | 3 (33.3) | 0 | 4 (25.0) | 2 (50.0) | 3 (50.0) | 0 | 0 | 1 (50.0) | 15 (13.9) |
| 2011 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (100.0) | 1 (50.0) | 3 (2.7) |
| Range (years) | 6–12 | 6–13 | 7–13 | 6–14 | 6–13 | 9–11 | 7–11 | 0 | 7–9 | 7–8 | |||||
| Median (years) | 7.5 | 10.5 | 10.0 | 9.0 | 9.5 | 10.0 | 9.0 | 8.0 | 7.5 | ||||||
| Mean (years) | 8.25 | 10.07 | 10.1 | 9.0 | 9.92 | 10.0 | 9.22 | 8.13 | 7.5 | ||||||
| Total number of projects (%) | 12 (11.0) | 14 (12.8) | 10 (9.2) | 20 (18.3) | 12 (11.0) | 3 (2.8) | 9 (8.3) | 0 | 16 (14.7) | 4 (3.7) | 6 (5.5) | 0 | 1 (0.9) | 2 (1.8) | 109 |
Analysis
In Chase et al. ’s9 review, 46% of programme priorities in 1998 came from the widespread consultation and 20% from systematic reviews. Our data show greater reliance on consultation but with variation from year to year. The key question concerns what can be inferred about the importance of the HTA projects to the NHS. It would be a mistake to equate widespread consultation with NHS relevance and systematic reviews with academic interest. There is no reason why this should be so. The processes the HTA programme had in place between the identification of topics and their advertisement as commissioning briefs means that the initial topic only served as a starter for the real work on NHS relevance.
Unsurprisingly, most projects that were funded had been prioritised; 70% had been given the top band (A) by the programme’s prioritisation processes. Band A, ‘recommended for commissioning – must commission’, meant that the programme would ‘go the extra mile’ to ensure that research was funded in that area. What to make of this 70% figure? The priority banding was the end of a process that started with the source of the topic, addressed in the previous question. This process involved detailed consideration of potential research priorities by panels of NHS experts (patients, clinicians, managers) as well as an overarching standing group on health technologies, meeting annually for 2 days. The priority band was a summary score produced by the whole process. The process was producing research proposals of which 70% were thought to be of high relevance to the NHS and so of a high priority.
By contrast with the finding by Jones et al. 43 that 80% of RCTs funded by the HTA programme and published between 2006 and 2008 were preceded by a systematic review, we found that 56% of all trial published to 2011 were preceded by a systematic review.
The finding that the statement of need did not change between the commissioning brief and that reported in the monograph in 101 out of 107 trials (94%) suggests no evidence of ‘drift’. Unfortunately, the data available in the database were not detailed enough to allow us to make further informative assessments in this area.
Primary outcomes tended to be patient relevant. Excluding those projects relating to ‘diagnostic technologies and screening’ increased these figures from 67% to 73%, much higher than previous studies (18% in Gandhi et al. ,39 21% in Montori et al. 40 and 23% in Rahimi et al. 41).
The lag between the programme prioritising a topic and publishing the results in the monograph series was 8–10 years. As this measures the time to publication in the HTA journal and not to publication in any journal, it overestimates to some extent. The key question is choice of benchmark: what is the right length of time against which 8–10 years should be compared?
Discussion
Question T1.9 on the 8- to 10-year time lag from topic identification to monograph publication was striking. However, we were unable to find any comparable estimate in the literature.
Although the answers to most questions were largely as expected, these questions only relate to meeting NHS need in an oblique and indirect way. Data availability limited the questions that could be asked regarding the core aim of the HTA programme, that is, how well the research it funds aims to meet the needs of the NHS. This is something that the programme should consider how best to address.
Strengths and weaknesses of the study
Addressing this overall question based on NHS need was hampered more than other questions in this report by the limitations of the database before 2000. This is because so much of the needs-related information is captured at the very start of a project, rather than during or at its completion.
The strength of the work has been given a new focus by the work of Chalmers and Glasziou42 in highlighting avoidable waste in research. Their framework starts with posing questions that matter, something that is key to the HTA programme.
This project looked only at trials funded through the HTA programme’s commissioned work stream. Since 2006, the programme has developed a growing portfolio through its researcher-led work stream. Proposals for this work stream are also assessed in terms of NHS need.
Recommendations for future work
Any future work will need to take account of the data limitations on how the trials funded aimed to meet the needs of the NHS. Any future work should include seven of the questions explored in this chapter, five as is (T1.1, T1.3, T1.7, T1.8 and T1.9) and two to be amended (T1.2 and T1.4).
Unanswered questions and future research
We offer recommendations for any future similar analyses. We found it difficult to identify data that usefully, consistently or richly characterised the NHS need in these trials. This matters given the importance to the HTA programme of meeting (and being seen to meet) NHS need. We recommend that NETSCC should work with the HTA programme to develop trial metadata that more usefully, consistently and richly characterise NHS need (linked as appropriate to potential impact and reduced avoidable waste).
Chapter 5 Theme 2: design and adherence to protocol
This chapter considers questions regarding the reporting of HTA-funded trials. The relevant literature is noted before summarising the piloting of the questions. The degree to which trial reports met the CONSORT checklist is examined along with how well they reported on trial design, interventions and controls. Comparisons are made between what was planned in protocols and what was reported in the monographs.
Introduction
Well-conducted RCTs have become the ‘gold standard’ for evaluating interventions in health care. The WHO defines a clinical trial as ‘any research study that prospectively assigns human participants or groups of humans to one or more health-related interventions to evaluate the effects on health outcomes’ [reproduced, with the permission of the publisher, from Bulletin of the World Health Organization – Guidelines for Contributors. Geneva: WHO; 2006. URL: www.who.int/bulletin/volumes/84/current_guidelines/en/ (accessed 3 October 2014)].
An explanatory paper to the CONSORT 2010 statement summarises why particular design features help reduce bias and improve study power. 44 Randomisation should be rigorously done and allocation to groups should be adequately concealed from participants and researchers. Blinding (or masking) should be maintained when possible for participants and clinicians, and particularly for observers who measure outcomes. Participants lost to follow-up should be minimised, accounted for and analysed in their randomised groups. Properly designed trials reduce susceptibility to bias, whether due to selection, outcome reporting or attrition bias. The sample size should be sufficient to give adequate precision to estimate the effect of the intervention in the relevant wider population.
The design of the trial should be fully recorded in the study protocol. That protocol should be carefully followed. Failure to follow a protocol happens for many reasons, some beyond the control of the investigators. For example, the introduction or removal of outcome measures later in a trial (‘post hoc’) raises the possibility of outcome reporting bias and increases the play of chance in the trial through multiplicity of analysis.
For a reader to judge the quality (validity) of a completed trial, the design, methods, results and interpretation must be fully and fairly reported.
Good evidence in the literature shows that many trials are poorly planned, conducted or reported, or all of these. 44,45 This chapter describes our investigation of the design, conduct and reporting of HTA-funded trials. We provide a descriptive analysis of the design of the interventions tested. We compare the planned (in the protocol) and reported (published) methods to identify deviations from protocol and post-hoc analysis. We assess the quality of reporting of the trials against the CONSORT statement.
A tool developed to enhance the quality of reporting and reduce methodological flaws, CONSORT was established in 1996 in response to concern about the quality of reporting (www.consort-statement.org/about-consort/history). The CONSORT statement was developed by evidence and expert consensus. Widely used by authors and journals, it has been adopted by the HTA monograph series. Extensions to the main CONSORT statement include the reporting of special types of trials, such as pragmatic trials, non-inferiority or equivalence trials and cluster trials. For this study, we focus on the main CONSORT statement, which applies particularly to parallel arm trials.
In 2008, Enhancing the Quality and Transparency of Health Research (EQUATOR) was established to promote accurate reporting of health research and provide an international network to improve the quality of scientific publications. The EQUATOR website acts as a library of reporting guidelines in health research (www.equator-network.org/).
The CONSORT statement (and particularly the checklist), although useful, has limitations. It can only list the categories of information that should be reported in most trials; it cannot assess the completeness of that information. Selective reporting of whole trials46 or outcomes10 leads to biased estimates of effectiveness through ‘publication bias’ and (closely related) ‘outcomes reporting bias’, usually resulting in overestimates of effectiveness.
Absence of publication or partial publication can only be assessed by knowing what research has or should have been reported. Concerns over the lack of availability of trial results prompted the development of clinical trials registers such as the ICTRP (the Registry Platform) based at the WHO. Ghersi et al. 22 emphasised the need for greater accessibility of trial data such as protocols and final reports. Transparency is needed to overcome academic and/or commercial vested interests. We address transparency by comparing the planned research with the reported methods and results. This is usually from the study protocol which, for recent trials, is usually in the public domain through the HTA website. For the early trials in the cohort, conducted when submission of detailed protocols was not normally required, we used the full application forms for grant funding. These contain a detailed description of the planned study, but not changes that may legitimately occur before the trial starts, for instance at the request of the ethics committee.
Any assessment tool should need to include not only the completeness of its reporting, but also an assessment of the design or conduct of the study. The Cochrane Collaboration’s handbook47 suggests that the assessment of validity of trials is best done in a framework rather than using a specific tool (available from www.cochrane-handbook.org). A full assessment of the trials using such a framework was beyond the scope of this study. Instead, we provide a descriptive analysis of the design of the trials (including the study interventions). We also compared key features of the study design in the protocol and final report to assess adherence to protocol and completeness of reporting of planned primary outcomes and analyses. More detailed analysis and statistical methods are provided in Chapter 7.
Questions addressed
Box 3 shows the questions explored in this section.
-
T2.1. Was the trial adequately reported? (Using the revised 2010 CONSORT checklist44 for core trial information, methods and results).
-
T2.2. Trial design framework.
-
T2.3. Type of comparison.
-
T2.4. Type of care.
-
T2.5. Type of setting.
-
T2.6. Pilot and feasibility.
-
T2.7. Number of interventions.
-
T2.8. Whether the intervention was an ‘add-on’ or ‘substitute’.
-
T2.9. Type of intervention using the HRCS. 30
-
T2.10. Type of intervention using Chalmers’ classification.
-
T2.11. Type of control.
-
T2.12. Type of comparison.
-
T2.13. The number of proposed and reported arms.
-
T2.14. The number of proposed and reported trial centres.
-
T2.15. Number of primary outcomes.
-
T2.16. Primary time point.
-
T2.17. Specifying the primary time point.
Methods
Seventeen questions were piloted, as shown in Box 3. Two were considered not feasible: one on pragmatic–explanatory continuum indicator summary (PRECIS),48 the other on complex interventions. The PRECIS question has 10 headings with up to six subheadings requiring approximately 68 data fields, or around one-third of the total fields for this theme. Besides requiring considerable data extraction, matters of judgement were also likely to be required.
The issue of whether or not the intervention was complex as defined by the MRC involved four headings, each with three subheadings, thus requiring 12 questions as well as judgements on the interactions between them. Given the lack of data on basic aspects of the HTA RCTs, such as the number and types of interventions, more detailed work such as that required by PRECIS48 or complex interventions seemed a task for further work.
The main information sources were the final protocol (if this was not available, then the funding application form) and the published monograph.
Denominators
The unit of analysis for reporting was 123 trials, that is the 125 trials identified from the 109 monographs but excluding two pilot trials.
Results
Question T2.1: was the trial adequately reported? (Using the revised 2010 Consolidated Standards of Reporting Trials checklist for core trial information, methods and results)
The CONSORT checklist has six sections/topics and 37 items (the CONSORT statement checklist lists 25 items, but there are subsections listed as a or b for 12 items, making a total of 37 CONSORT items). We extracted data on four sections: ‘title and abstract’, ‘introduction’, ‘methods’ and ‘results’. No data were extracted for 16 of the 37 items. Out of these 16, six items were under ‘discussion’ and ‘other information’, four were under ‘results’, five were under ‘methods’ and one was under ‘introduction’. Data on sample size and primary outcome are further discussed in Chapter 7. The 21 items included in this chapter are listed in Table 12.
| CONSORT section/topic | CONSORT checklist item |
|---|---|
| Title and abstractb | Identification as a randomised trial in the title (1a) Structured summary of trial design, methods, results and conclusions (1b) |
| Introduction | Specific objectives or hypotheses (2) |
| Methods (trial design) | Description of trial design (such as parallel, factorial) including allocation ratio (3a) Important changes to methods after trial commencement (such as eligibility criteria), with reasons (3b) |
| Methods (participants) | Eligibility criteria for participants (4a)b Settings and locations where the data were collected (4b) |
| Methods (interventions) | The interventions for each group with sufficient details to allow replication, including how and when they were actually administered (5) |
| Methods (outcomes) | Completely defined prespecified primary and secondary outcome measures, including how and when they were assessed (6) |
| Methods (sample size) | How sample size was determined (7) |
| Methods (randomisation: sequence generation) | Methods used to generate the random allocation sequence (8a) Type of randomisation; details of any restriction (such as blocking and block size) (8b) |
| Methods (randomisation: allocation concealment mechanism)c | Mechanism used to implement the random allocation sequence (such as sequentially numbered containers), describing any steps taken to conceal the sequence until interventions were assigned (9) |
| Methods (randomisation: implementation)b | Who generated the random allocation sequence, who enrolled participants and who assigned participants to interventions (10) |
| Methods (blinding)d | If done, who was blinded after assignment to interventions (for example, participants, care providers, those assessing outcomes) and how (11a) |
| Results (participant flow)b | For each group, the numbers of participants who were randomly assigned, received intended treatment and were analysed for the primary outcome (13a) For each group, losses and exclusions after randomisation, together with reasons (13b) |
| Results (recruitment) | Dates defining the periods of recruitment and follow-up (14a) Why the trial ended or was stopped (14b) |
| Results (baseline data)b | A table showing baseline demographic and clinical characteristics for each group (15) |
| Results (harms)b | All important harms or unintended effects in each group (19) |
Consolidated Standards of Reporting Trials checklist items: 1 and 2
These two sections cover the title, abstract and introduction. Of the 123 trials, 100 reported that they were randomised clinical trials in their titles, 122 had a structured summary and all trials included their objectives and hypotheses in the introduction (Table 13).
| CONSORT description | Yes, n (%) | No, n (%) | Total |
|---|---|---|---|
| Title: identified as a randomised trial in the title (1a) | 100 (81.3) | 23 (18.7) | 123 |
| Abstract: structured summary of trial design, methods, results and conclusions (1b) | 122 (99.2) | 1 (0.8) | 123 |
| Introduction: objectives and hypotheses (2b) | 123 (100.0) | 0 | 123 |
Consolidated Standards of Reporting Trials checklist items: methods (items 3–12)
Trial design
Of the 123 included trials, the unit of analysis in 111 was individual patient. Twelve trials were cluster randomised.
Participants
One hundred and twenty-one trials reported the eligibility criteria and provided details about the setting and location where data were collected (98.4% for both CONSORT items).
Interventions and controls
All trials provided sufficient information about the intervention groups. For drug interventions, the drug name, dose and method of administration were provided. The reporting of the control group was, however, less complete.
Most trials compared the intervention with ‘standard care’ (52.8%, 65/123). Next most common were placebo (8.1%, 10/123), ‘next best service’ (2.4%, 3/123) and ‘no treatment’ (1.6%, 2/123). Forty-three trials (35%) were classified as ‘control undefined’ as they provided insufficient detail (see Table 14).
Outcomes
Forty trials reported more than one primary outcome. Of these, it was not possible to determine the ‘main’ primary outcome from the monograph for two trials. Reporting of the primary time point was a weakness in some trials. Eighty out of 123 trials covered this CONSORT item sufficiently (65%), the rest did not (see Table 14).
Sample size
For the 109 superiority trials, the four elements of the sample size required by the CONSORT guidelines (2010) were considered. Sixty per cent of superiority trials (66/109) reported all elements as detailed by the CONSORT guidelines (see Table 14). For each of the four elements:
-
94.5% (103/109) reported the statistical power.
-
90.8% (99/109) reported the alpha error level.
-
Out of the 109 trials, 45 had a binary outcome and 84.4% (38/45) sufficiently reported the estimated outcomes in each group.
-
Of the 109 trials, 53 had a continuous outcome and 56.6% (30/53) sufficiently reported the standard deviation (SD) of the measurements.
[The remaining trials were classified as ‘time to event’ (n = 3) or ‘effect size’ (n = 6). Two trials had missing data for the comparative analysis.]
Sequence generation
The method used to generate the random allocation sequence was adequately described in 94.3% of trials (116/123) (CONSORT statement 8a) and the type of randomisation including details about the restriction (CONSORT statement 8b) was adequately reported by 86.2% of trials (106/123) (Table 14). This item was not included in the 1996 version of CONSORT;50 however, 18 out of 21 trials using this version still reported the item.
| CONSORT description | Yes, n (%) | No, n (%) | Unable to report, n (%) |
|---|---|---|---|
| Trial design: Description of trial design (such as parallel, factorial), including allocation ratio (3a) | 123 (100.0) | 0 | 0 |
| Trial design: Important changes to methods after trial commencement (such as eligibility criteria), with reasons (3b) | 47 (38.2) | 22 (17.9) | 54 (43.9) |
| Participants: Eligibility criteria (4a) | 121 (98.4) | 2 (1.6) | 0 |
| Participants: Settings and locations where data were collected (4b) | 121 (98.4) | 2 (1.6) | 0 |
| Interventions: Interventions for each group with sufficient detail (5) | 80 (65.0) | 43 (35.0) | 0 |
| Outcomes: Completely defined pre-specified primary and secondary outcomes, including how and when they were assessed (6a) | 80 (65.0) | 43 (35.0) | 0 |
| Sample size: How the sample size was determined (7a)a | 66 (60.6) | 43 (39.4) | 0 |
| Sequence generation (8a): Method used to generate the random allocation sequence | 116 (94.3) | 3 (2.4) | 4 (3.2) |
| Sequence generation (8b): Type of randomisation; details of any restriction (such as blocking and block size) | 106 (86.2) | 10 (8.1) | 7 (5.7) |
| Allocation concealment: Mechanism used to implement the random allocation sequence (such as sequentially numbered containers) (9) | 97 (78.9) | 16 (13.0) | 10 (8.2) |
| Allocation concealment: Described any steps taken to conceal the sequence until interventions were assigned (9) | 105 (85.4) | 9 (7.3) | 9 (7.3) |
| Implementation: Described who generated the allocation sequences (10) | 90 (73.2) | 26 (21.1) | 7 (5.7) |
| Implementation: Described who enrolled the participants (10) | 111 (90.2) | 5 (4.1) | 7 (5.7) |
| Implementation: Described who assigned participants to interventions (10) | 105 (85.4) | 3 (2.4) | 15 (12.2) |
| Blinding: If done, who was blinded after assignment to interventions (for example, participants, care providers, those assessing outcomes) (11a) | 103 (83.7) | 0 | 20 (16.3) |
Consolidated Standards of Reporting Trials checklist items: results (items 13–19)
Most trials (97.6%, 120/123) included a flow diagram. Clear improvements were evident in the reporting of the losses and exclusions after randomisation for each intervention between the 199650 and 200151 CONSORT statements. Only 61.9% of trials (13/21) using the 1996 CONSORT statement reported group losses and exclusions after randomisation. By comparison, of those using the 2001 revised CONSORT statement, 85.1% of trials (86/101) reported such losses and exclusions in the participant flow diagram (one trial used the 2010 CONSORT statement) (see Table 15). The reasons were not explored here.
Baseline data
Most trials provided information about the demographic and clinical characteristics of the patient groups (91.9%, 113/123).
Harms
A notable improvement in the reporting of harms and unintended effects was found between the different CONSORT statements. Only 38.1% of trials (8/21) using CONSORT 1996 reported all important harms, compared with 64.4% of trials (65/101) reporting using the 2001 revised version (the one trial using the 2010 CONSORT statement did report all important harms or unintended effects). The results of 18 trials were unclear (Table 15).
| CONSORT description | Yes, n (%) | No, n (%) | Unable to report/N/A, n (%) |
|---|---|---|---|
| Participant flow: For each group, the number of participants who were randomly assigned (13a) | 120 (97.6) | 2 (1.6) | 1 (0.8) |
| Participant flow: For each group losses and exclusions after randomisation (13b) | 100 (81.3) | 18 (14.6) | 5 (4.0) |
| Recruitment: Dates defining the periods of recruitment (14a) | 105 (85.4) | 18 (14.6) | 0 |
| Recruitment: Why the trial ended or was stopped (14b)a | 5 (100.0) | 0 | 0 |
| Baseline data: A table showing baseline demographics (15) | 113 (91.9) | 6 (4.9) | 4 (3.2) |
| Harms: All important harms or unintended effects (19) | 74 (60.2) | 31 (25.2) | 18 (14.4) |
Questions T2.2–T2.11: what were the design characteristics of the included trials?
Question T2.2: trial design framework
All 123 trials reported the design of the trial in the published monograph, with none indicating a change in design from that planned. More than four-fifths were designed as parallel arm trials (87%, 107/123), 10 were factorial (8.1%, 10/123) and six were designed as crossovers (4.9%, 6/123). Thirteen trials reported having included a preference arm to the main clinical trial (10.6%, 13/123).
Question T2.3: type of comparison
One hundred and nine trials (88.6%, 109/123) reported the type of comparison as superiority at the planning (as reported in the protocol or proposal) stage of the trial. The remaining 14 trials were either equivalence (6.5%, 8/123) or non-inferiority (4.1%, 5/123) and one trial did not report the planned type of comparison (0.8%, 1/123). The reported type of comparison was as planned for all non-inferiority trials (n = 5). There were three discrepant trials: two designed as superiority at the planning stage were actually reported as equivalence trials (ID36 and ID37) and one designed as an equivalence trial at the planned stage was actually reported as a superiority design (ID61).
Question T2.4: type of care
Eleven trials (8.9%) were reported to have been conducted in both primary and secondary care. More than half of the trials (56.1%, 69/123) were in secondary care and one-third (33.3%, 41/123) were in primary care. Two trials (1.6%, 2/123) were conducted in neither primary nor secondary care, one in a leisure centre (trial ID64) and the other in a school setting (trial ID66). These were classified as ‘other’ (see Table 16).
Question T2.5: type of setting
Almost half of the trials (44.7%, 55/123) were conducted only in a hospital setting, one-quarter (24.4%, 30/123) only in a general practitioner (GP) setting and eight (6.5%) in a community setting. Thirteen were categorised as ‘other type of setting/place’ (10.6%), which included settings such as non-NHS acupuncture clinics (trial ID39), community mental health services (trial ID42) and a health psychology department for chronic illness (trial ID60). In the remaining 17 trials, more than one type of setting was reported in the monograph (13.8%) (see Table 16).
Question T2.6: pilot and feasibility study
The NETSCC definitions of pilot and feasibility studies were used to determine whether the trial involved a pilot or feasibility study prior to conducting the main clinical trial (www.netscc.ac.uk/glossary). A pilot study is a rehearsal for the main study, whereas a feasibility study estimates important parameters for a main trial. Almost half of the trials (48%, 59/123) included a pilot study prior to conducting the main clinical trial. Six (4.9%, 6/123) conducted a feasibility study (Table 16).
| Description of data | n (%) |
|---|---|
| Type of care | |
| Primary care setting | 41 (33.3) |
| Secondary care setting | 69 (56.1) |
| Both | 11 (8.9) |
| Other | 2 (1.6) |
| Total | 123 |
| Type of place | |
| Hospital | 55 (44.7) |
| Community | 8 (6.5) |
| GP | 30 (24.4) |
| Other | 13 (10.6) |
| More than one type of place | 17 (13.8) |
| Total | 123 |
| Was a pilot or feasibility study conducted? | |
| Pilot study | |
| Yes | 59 (48.0) |
| No | 56 (45.5) |
| Not clear | 6 (4.9) |
| No information available | 1 (0.8) |
| Missing data | 1 (0.8) |
| Total | 123 |
| Feasibility study | |
| Yes | 6 (4.9) |
| No | 115 (93.5) |
| Not clear | 1 (0.8) |
| No information available | 1 (0.8) |
| Total | 123 |
Question T2.7: number of interventions
Three hundred and twenty-one interventions were reported from the 123 clinical trials (mean 2.6 interventions per trial).
Question T2.8: whether the intervention was an ‘add-on’ or ‘substitute’
Almost half (48%, 59/123) of the interventions described in the HTA monograph were reported as substitutions for another intervention and one-quarter (26%, 32/123) were reported as additional (Table 17).
| Description of data | n (%) |
|---|---|
| Intervention group type | |
| Add-on | 32 (26.0) |
| Substitute | 59 (48.0) |
| Neither | 31 (25.2) |
| Missing data | 1 (0.8) |
| Total | 123 |
Questions T2.9 and T2.10: type of intervention using the Health Research Classification System and Chalmers’ classification
Two classification systems were used to report the clinical trial interventions. Tables 18 and 19 illustrate the two classifications systems used (UKCRC HRCS30 and Chalmers’ classification52) by trial.
| Description of data | n (%) |
|---|---|
| 3. Prevention | |
| 3.1 Primary prevention interventions to modify behaviours or promote well-being | 0 |
| 3.2 Interventions to alter physical and biological environmental risks | 0 |
| 3.3 Nutrition and chemoprevention | 3 (2.4) |
| 3.4 Vaccines | 2 (1.6) |
| 3.5 Resources and infrastructure | 0 |
| 4. Detection and diagnosis | |
| 4.1 Discovery and preclinical testing of markers and technologies | 0 |
| 4.2 Evaluation of markers and technologies | 2 (1.6) |
| 4.3 Influences and impact | 0 |
| 4.4 Population screening | 3 (2.4) |
| 4.5 Resources and infrastructure | 0 |
| 5. Treatment development | |
| 5.1 Pharmaceuticals | 2 (1.6) |
| 5.2 Cellular and gene therapies | 0 |
| 5.3 Medical devices | 1 (0.8) |
| 5.4 Surgery | 4 (3.3) |
| 5.5 Radiotherapy | 0 |
| 5.6 Psychological and behavioural | 0 |
| 5.7 Physical | 0 |
| 5.8 Complementary | 0 |
| 5.9 Resources and infrastructure | 0 |
| 6. Treatment evaluation | |
| 6.1 Pharmaceuticals | 22 (17.9) |
| 6.2 Cellular and gene therapies | 0 |
| 6.3 Medical devices | 17 (13.8) |
| 6.4 Surgery | 12 (9.8) |
| 6.5 Radiotherapy | 2 (1.6) |
| 6.6 Psychological and behavioural | 11 (8.9) |
| 6.7 Physical | 9 (7.3) |
| 6.8 Complementary | 2 (1.6) |
| 6.9 Resources and infrastructure | 0 |
| 7. Disease management | |
| 7.1 Individual care needs | 8 (6.5) |
| 7.2 End-of-life care | 0 |
| 7.3 Management and decision-making | 4 (3.3) |
| 7.4 Resources and infrastructure | 0 |
| 8. Health services | |
| 8.1 Organisation and delivery of services | 18 (14.6) |
| 8.2 Health and welfare economics | 0 |
| 8.3 Policy, ethics and research governance | 0 |
| 8.4 Research design and methodologies | 0 |
| 8.5 Resources and infrastructure | 0 |
| Trial interventions not coded – missing data | 1 (0.8) |
| Total | 123 |
| Description of data | n (%) |
|---|---|
| Drug | 25 (20.3) |
| Radiotherapy | 2 (1.6) |
| Surgery | 12 (9.8) |
| Diagnostic | 9 (7.3) |
| Education and training | 4 (3.3) |
| Service delivery | 14 (11.4) |
| Psychological therapy | 9 (7.3) |
| Vaccines and biologicals | 1 (0.8) |
| Devices | 18 (14.7) |
| Physical therapies | 9 (7.3) |
| Contraception | 0 |
| Exercise | 1 (0.8) |
| Complementary therapies | 2 (1.6) |
| Social care | 1 (0.8) |
| Mixed or complex | 5 (4.1) |
| Diet | 0 |
| Perioperative | 0 |
| Other | 3 (2.4) |
| Interventions included more than one category | 8 (6.5) |
| Total | 123 |
The UKCRC HRCS30 was used to classify interventions in the included clinical trials. Twenty-two trials were classified as treatment evaluation of pharmaceuticals (17.9%), followed closely by organisation and delivery of services (‘health services’) (14.6%, 18/123) and treatment evaluation of medical devices (13.8%, 17/123).
The system of Chalmers et al. 52 was also used to classify interventions. For those trials in which the technologies compared fell into the same class, the two commonest interventions were drugs (21.7%) and devices (15.7%).
It was not possible using this classification to classify 8 of the 123 trials as the interventions included two or more categories, such as ‘drug’ and ‘mixed and complex’; ‘drug’ and ‘service delivery’; ‘drug’ and ‘psychological therapy’; ‘drug’ and ‘education and training’; ‘surgery’ and ‘mixed and complex’; and ‘surgery’ and ‘drug’.
The type of intervention for three trials was classified as ‘other’, which referred to ‘nutritional supplement in addition to the normal hospital diet’ (trial ID54), ‘self-monitoring intervention’ (trial ID80) and ‘intravenous fluids were to be administered following primary patient assessment/to be withheld for the first hour of pre-hospital care’ (trial ID122).
Question T2.11: type of control
More than one-third (35%, 43/123) of controls could not be defined as placebo, standard care, no treatment or next best (Table 20). There appeared to be no improvement over time. It was not possible, based on the monograph, to report or extract data with certainty about the type of control used. For the 80 clinical trials where the type of control could be defined, 65 (81.3%) reported the control as standard care and 10 (12.5%) as placebo.
| Description of data | n (%) |
|---|---|
| Placebo | 10 (8.1) |
| Standard care | 65 (52.8) |
| No treatment | 2 (1.6) |
| Next best | 3 (2.4) |
| Control undefined | 43 (35.0) |
| Total | 123 |
Questions T2.12–T2.17: did the trial conform to the protocol?
To assess whether or not the design of the trial, as described in the protocol (or the application form) differed from that published in the HTA monograph, we compared the type of comparison, the number of arms and the primary outcomes.
Question T2.12: type of comparison
As shown in Table 21, 119 of 122 trials reported the type of comparison as had been planned. Three trials changed: two designed as superiority at the planning stage reported as equivalence trials (trials ID36 and ID37), and one designed as an equivalence trial reported as a superiority design (trial ID61). This trial reported a protocol change relating to the design of the trial: ‘A protocol amendment to amalgamate the two arms of the study and to compare cost-effectiveness of endoscopies in general rather than by the site of endoscopy was approved by MREC [Multicentre Research Ethics Committee] and the HTA programme.’53
| Actual design framework | Planned design framework | |||
|---|---|---|---|---|
| Superiority | Non-inferiority | Equivalence | Total | |
| Superiority | 107 | 0 | 1 | 108 |
| Non-inferiority | 0 | 5 | 0 | 5 |
| Equivalence | 2 | 0 | 7 | 9 |
| Total | 109 | 5 | 8 | 122a |
The planned type of comparison was not reported in one trial. The importance of this change was not clear because the status of the change was not recorded in the monograph. The change of comparison could have been agreed as part of the analysis plan by the trial Data Monitoring Committee before data were examined.
Question T2.13: the number of proposed and reported arms
The mean number of planned arms was 2.67 (n = 328) and the number reported in the monograph was 2.63 (n = 323). One hundred and seventeen trials had the same number of arms as planned (Table 22). The six discrepant trials (4.9%, 6/123) (trials ID2, ID4, ID19, ID21, ID86 and ID95) are explored in Table 23.
| Published number of arms | Proposed number of arms | ||||||
|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | 7 | Total | |
| 2 | 73 | 3 | 1 | 0 | 0 | 0 | 77 |
| 3 | 0 | 24 | 0 | 0 | 0 | 0 | 24 |
| 4 | 0 | 0 | 14 | 0 | 0 | 0 | 14 |
| 5 | 0 | 1 | 0 | 5 | 0 | 1 | 7 |
| 6 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Total | 73 | 28 | 15 | 5 | 1 | 1 | 123 |
| Trial design characteristics | ID2 | ID4 | ID19 | ID21 | ID86 | ID95 |
|---|---|---|---|---|---|---|
| Intervention classification | Mixed or complex | Psychological therapy | Service delivery | Service delivery | Psychological therapy | Devices |
| Intervention types | New care/new therapy | Standard care/existing care | New care/new therapy | New care/new therapy | New care/new therapy | Standard care/existing care |
| Intervention add-on or substitute? | Neither | Substitute | Substitute | Add-on | Add-on | Substitute |
| Type of control | Standard care | Control undefined | Standard care | Standard care | Standard care | Standard care |
| Type of comparison | Superiority | Superiority | Superiority | Superiority | Superiority | Superiority |
| Type of framework | Factorial | Parallel | Parallel | Parallel | Parallel | Parallel |
| Preference arm | No | Yes | No | No | Yes | Yes |
| Type of place | Hospital | GP | GP | Hospital | GP | More than one type of place |
The number of arms for the 123 trials was 323. The number of interventions was 321. The difference was due to one trial having a factorial design in which two interventions were tested on two different groups.
The design characteristics of the six trials with a discrepant number of arms (see Table 23) shows that four were published before 2005 (trials ID2, ID4, ID19 and ID21) and the remaining two in 2009 (trials ID82 and ID95). Of the four trials published before 2005, none submitted a project protocol.
Question T2.14: the number of proposed and reported trial centres
One hundred and three trials reported the number of proposed centres (Table 24), with a mean of 17.05 and a median of 5. One hundred and nineteen trials with available data reported the actual mean number of centres as 26.82, median 11.
| Description of data | Proposed number (%) | Published number (%) |
|---|---|---|
| Number of arms | ||
| 2 | 73 (59.3) | 77 (62.6) |
| 3 | 28 (22.8) | 24 (19.5) |
| 4 | 15 (12.2) | 14 (11.4) |
| 5+ | 7 (5.7) | 8 (6.5) |
| Total | 123 | 123 |
| Total number of centres | ||
| 10 or fewer | 69 (56.1) | 59 (48.0) |
| 11–20 | 9 (7.3) | 15 (12.2) |
| 21–30 | 8 (6.5) | 13 (10.6) |
| 31–40 | 4 (3.3) | 4 (3.3) |
| 41–50 | 2 (1.6) | 5 (4.1) |
| 51–60 | 1 (0.8) | 5 (4.1) |
| 61–70 | 4 (3.3) | 3 (2.4) |
| 71–80 | 1 (0.8) | 2 (1.6) |
| 81–90 | 1 (0.8) | 5 (4.1) |
| 91–100 | 4 (3.3) | 2 (1.6) |
| 101+ | 0 | 6 (4.9) |
| No information reported | 20 (16.3) | 4 (3.3) |
| Total | 123 | 123 |
Thirty-nine trials (31.7%) had unchanged numbers of proposed centres. It was not possible to compare the planned and actual for 22 trials (17.9%). For the remaining trials (50.4%, 62/123), the number of planned and actual trial centres differed. Four-fifths of these (80.6%, 50/62) increased the number of centres from that planned, whereas the rest reduced it. Table 25 provides an overview of these discrepancies.
| Published number of centres reported | Proposed number of centres reported | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 or fewer | 11–20 | 21–30 | 31–40 | 41–50 | 51–60 | 61–70 | 71–80 | 81–90 | 91–100 | Total | |
| 10 or fewer | 53 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 53 |
| 11–20 | 9 | 3 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 13 |
| 21–30 | 1 | 4 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 12 |
| 31–40 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 4 |
| 41–50 | 0 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 3 |
| 51–60 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 4 |
| 61–70 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 2 |
| 71–80 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 2 |
| 81–90 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 3 |
| 91–100 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 2 |
| 101+ | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 2 |
| Total | 66 | 9 | 8 | 4 | 2 | 1 | 4 | 1 | 1 | 4 | 100 |
Change in the number of centres plausibly reflects difficulties with recruitment. For those trials using fewer centres than planned, this may reflect difficulties in obtaining the support hoped for from centres. Changes in the number of centres may reduce generalisability if the centres lost or gained were unrepresentative.
Question T2.15: number of primary outcomes
Two hundred and one planned primary outcomes were reported from 122 trials (no data for one trial) (mean 1.65 and median 1 planned primary outcome per trial). Two hundred and twenty-eight primary outcomes were reported in the monographs (mean 1.85 and median 1 primary outcome per trial) (Table 26). Ninety-five trials (78%) reported the planned number of primary outcomes. Eighty trials (65%) reported one primary outcome as planned. In 27 trials (22.4%), discrepancies were reported between the proposed number of primary outcomes and that reported in the published monograph. Fourteen trials (52%, 14/27) increased the number of primary outcomes in the published monograph from the original proposed number, the other 13 (48%) reduced it. [The number of primary outcomes differs to that reported in Chapter 7. This is because of the denominator of the analyses. This chapter reports on 123 trials whereas Chapter 7 reports on the full cohort (n = 125 trials).]
| Actual number of primary outcomes | Proposed number of primary outcomes | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 7 | Total | |
| 1 | 73 | 4 | 3 | 0 | 2 | 0 | 82 |
| 2 | 6 | 17 | 0 | 2 | 0 | 1 | 26 |
| 3 | 0 | 1 | 3 | 0 | 0 | 0 | 4 |
| 4 | 0 | 0 | 0 | 1 | 1 | 0 | 2 |
| 5 | 0 | 1 | 1 | 0 | 1 | 0 | 3 |
| 6 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| 7 | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| 15 | 0 | 0 | 0 | 3 | 0 | 0 | 3 |
| Total | 80 | 23 | 8 | 6 | 4 | 1 | 122 |
Three trials reported in one monograph (trials ID128, ID129 and ID134) had proposed four primary outcomes but actually reported 15. Another trial (ID79) planned seven primary outcomes but reported only two in the monograph.
Other changes in the primary outcomes are explored in Chapter 7 regarding statistical analysis.
Questions T2.16 and T2.17: reporting and specifying the time points of primary outcomes
Out of the 123 clinical trials, 68 (55.3%) did and 55 (44.7%) did not specify the planned primary time point in the protocol or application form. Of the 55 that did not specify the planned primary time point in the protocol or application form, 36 (65.5%) did not report the actual primary time point in the published monograph. Five trials (4.1%) reported discrepancies between the proposed and published reporting of the primary time point. In two, the proposed time point was 12 months, yet in the published monograph two primary time points (12 and 24 months, and 4 and 12 months, respectively) were reported (Table 27).
| Description of data | Proposed number (%) | Published number (%) |
|---|---|---|
| Was the primary time point provided? | ||
| Yes | 68 (55.3) | 74 (60.2) |
| No | 55 (44.7) | 49 (39.8) |
| Total | 123 | 123 |
| If yes, how were the data presented? | ||
| Less than 1 month | 7 (5.7) | 8 (6.5) |
| 1 month, up to and including 6 months | 29 (23.6) | 35 (28.5) |
| 7 months, up to and including 12 months | 23 (18.7) | 21 (17.1) |
| 13 months, up to and including 24 months | 8 (6.5) | 9 (7.3) |
| More than 25 months | 1 (0.8) | 1 (0.8) |
| No time frame given/stated | 5 (4.1) | 6 (4.9) |
| Unable to compare data | 50 (40.7) | 43 (35.0) |
| Total | 123 | 123 |
| Number of secondary outcomes published in the monograph | ||
| 1–5 | 0 | 41 (33.3) |
| 6–10 | 0 | 55 (44.7) |
| 11–20 | 0 | 25 (20.3) |
| Missing data | 0 | 2 (1.6) |
| Total | 0 | 123 |
Analysis
Adherence to those sections of the CONSORT checklist that were examined was fairly high, but with some exceptions, including lack of detail on interventions, prespecified outcomes and sample size calculation. About one-third of trials failed on each of these. This was a greater problem for older trials.
A high proportion (88.6%, 109/123) were designed as superiority trials with parallel arms. Almost all the rest were equivalence or non-inferiority trials. Almost half of all trials conducted pilots but few had feasibility studies.
Around half of all interventions were substitutes for standard care and about one-third were add-ons. More than half of controls (53%, 65/123) were standard care, but one-third of controls (35%, 43/123) could not be classified.
Both the Chalmers and HRCS classification systems could be applied. Although some categories in the latter were not relevant, the Chalmers system provided less detail. The more comprehensive HRCS system30 should probably be used in future.
Most trials were conducted in line with protocol and followed both the study framework and the planned type of comparison. In six trials, the number of arms changed but it could not be ascertained if those changes had been agreed with the programme. These trials were all in the early years of the programme.
The number of primary outcomes changed in 27 trials (half increased and half decreased). Changes were mainly in early trials. The outcome used to plan the sample size (the most important primary outcome) was unchanged in 106 (82%) trials.
The time point at which the primary outcome was measured was not specified in 45% (55/123) of proposals and 40% (49/123) of monograph reports.
On average, trials needed about twice as many centres as planned to complete the study, reflecting the difficulties in recruiting.
Discussion
Strengths and weaknesses of the study
Overall, sufficient data existed for the trials to be assessed against a selection of core CONSORT criteria. Comparison of planned and reported analyses showed the HTA trials reporting more faithfully to protocol than the cohort examined by Chan et al. 10
The HRCS classification of interventions of the trial proved slightly more comprehensive than Chalmers’ classification and should therefore be adopted. Further work is required on the classification of controls.
Recommendations for future work
Any such further work should include 14 questions, eight to remain as they are (T2.2, T2.3, T2.4, T2.5, T2.7, T2.8, T2.12 and T2.13) and six to be amended (T2.1, T2.6, T2.9, T2.11, T2.14 and T2.15).
Unanswered questions and future research
Should similar work be continued, the HTA programme might usefully clarify:
-
the extent to which the programme wishes to assess the extent of compliance with CONSORT
-
the importance it attaches to trials classifying the control group
-
how it wishes to classify cluster trials
-
the number of primary outcomes.
Chapter 6 Theme 3: performance and delivery of randomised controlled trials
This chapter considers questions under the theme of the performance of RCTs funded by the HTA programme. After a brief review of the relevant literature it considers data on recruitment and follow-up, changes in team composition, extensions and protocol changes and the time taken.
Introduction
As complex, highly regulated experiments involving humans, clinical trials often experience delays and changes in plan. The HTA programme operates a proactive monitoring system comprising welcome meetings, monitoring visits and regular progress reports in order to ensure that trials deliver their results, and to assist underperforming studies. 54 Nonetheless, Campbell et al. 2 found in 2007 that less than one-third of UK publicly funded (NIHR/MRC) studies recruited according to plan.
Performance of trials
Once commissioned, trials move through four main phases, at each of which problems may present:
-
start-up
-
recruitment of both centres and patients
-
delivery of the interventions, follow-up and data collection
-
analysis and reporting.
Two external barriers present themselves once funding has been agreed: ethics approval and research governance. Both can cause delays and alterations to protocol. For many years, ethics approval was the biggest challenge facing new trials. Permission from a NHS research ethics committee is legally required before research can take place in the NHS involving human subjects, or their tissue or data. 55 Issues with obtaining ethical approval for research have been addressed at the national level. When first introduced in 1991, NHS ethics committees dealt with research within limited geographical areas. Multicentre Research Ethics Comnittee followed in 1997. The major change which affected HTA trials was the provision of a single UK-wide ethics opinion in 2004. This relieved HTA trials of the necessity of obtaining ethics approval from multiple sites. 56–58 The National Research Ethics Advisory Service, established in 2007, further improved the process of obtaining ethics approval for trials. 59
The second major hurdle is local NHS R&D permissions, which must be obtained in each NHS organisation where patients will interact with the study. A recent review of the regulation and governance of health research highlighted that these permissions cause significant delays, duplication of checks, lack of consistent advice and interpretation, performance variation and process inconsistency, all of which are commonly experienced by researchers. 59
The HTA programme requires triallists to predict likely recruitment and to monitor actual against predicted recruitment. Five types of prediction models are commonly used. Most common are simple, straight line predictions, with or without conditions. Less used are models based on Poisson processes and Baysian approaches, and simulations based on underlying Markov models. These models can be used either before or during a trial to predict likely recruitment, in terms of both individuals and required recruitment duration. Those which can be adapted to developing information (the Baysian approach and to a lesser extent the Poisson) tend to be used to inform investigators about the adequacy of an ongoing recruitment process. 60,61 Those which produce a fixed or simulated answer lend themselves to pre-trial prediction. Of course, the second group can be used to re-estimate the point of recruitment completion midway through an accrual period, but they do not lend themselves well to dynamic re-estimation.
Historically, triallists funded by the HTA programme have used the unconditional model. 62 This has the advantage of simplicity and ease of use but often requires unsupportable assumptions. The major assumption required is that all the study centres start recruiting to their maximum capacity on the first day of a trial. No multicentre trial managed by the HTA programme has ever managed this.
Since around 2006, some applications to the HTA programme have adopted a conditional model approach to recruitment prediction. This modifies the unconditional by varying the recruitment rate conditional on other events,62,63 usually recruitment of centres. The investigator can apply what he or she knows or suspects about the likely times of start-up of his or her various centres and apply this to his or her prediction. Some groups have also added phenomena such as learning (centres learning the best ways of recruiting patients) and fatigue (centres getting bored with a trial and becoming less efficient at recruitment) to their conditional models.
Figure 2 illustrates the effect that adopting different prediction models can have on expectations. In the example, a further 3 months of recruitment is predicted by the conditional model, which assumes that the centres will gradually start recruiting over the first 6 months of the trial, rather than all on day 1.
FIGURE 2.
Comparison of the results of two prediction models.
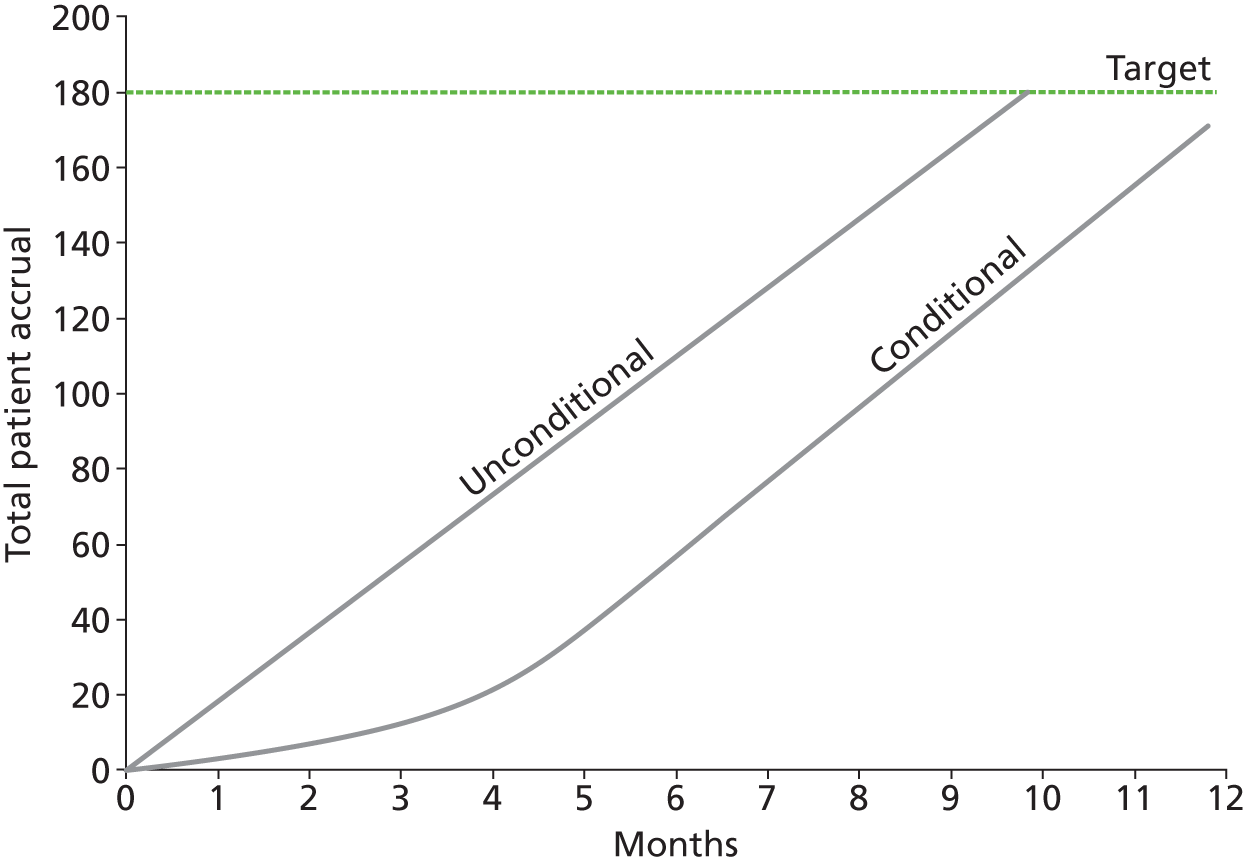
Failure of recruitment to match predictions is often taken as a flag for the HTA monitoring process to review and consider whether the project is at risk. More robust prediction modelling could help to prevent these ‘false positives’.
Having managed to recruit participants into a trial, a subsequent challenge is to collect data from them at the various study end points. Investigators normally strive to collect as many data as possible, as differential loss to follow-up in different arms of a study can introduce bias. 64
Extensions and protocol changes resulting from these challenges have different implications for interpretation of trial results. Extensions can be due to delays arising from researchers being unfamiliar with the particular topic and/or overconfident during the competitive bidding process. The HTA programme attempts to put in place robust assessment for extensions and protocol changes; almost all are reviewed by appropriate members of the secretariat with scientific expertise. Protocol changes are more important as they could undermine the scientific validity of a study. Changes in protocol should be agreed with the HTA programme and acknowledged in reports of findings.
Trials involve multidisciplinary teams, which often change over lengthy trials. It has been reported that the group which finishes a research project is often not the same as the group which starts it. 24,65 This could have a similar impact to protocol changes. Failure to report such changes is common. 24
Recruitment
The challenges and deficiencies of trial accrual prediction in the UK were highlighted by STEPS. 2 Campbell et al. 2 looked at a cohort of 114 multicentre trials funded by the MRC and the HTA programme, which started in or after 1994, and were due to end before 2003. Of the 114 studies, less than one-third recruited their target number of participants according to the original plan, and one-third required an extension to the duration of the study.
The STEPS identified as indicators of success:
-
having a dedicated trial manager
-
being a cancer or drug trial
-
having interventions which were only available inside the trial
-
addressing a clinically important question in a timely way.
Watson et al. 66 and Treweek et al. 67 reviewed the published literature around improving recruitment to trials. Watson et al. 66 included 14 papers describing 20 interventions, but excluding studies assessing recruitment to hypothetical studies. Treweek et al. 67 assessed 27 trials of interventions to improve recruitment to clinical trials, with a total of more than 26,000 participants but with wider inclusion criteria. The trials reviewed by Treweek were essentially a superset of those assessed by Watson et al. 66. Unsurprisingly, their findings were very similar, both suggesting the following strategies which may improve recruitment:
-
telephone reminders to non-responders
-
opt-out, rather than opt-in, procedures for contacting potential participants
-
open designs, where participants know which intervention they are receiving
-
monetary incentives
-
making trial materials culturally sensitive.
Both studies reflected on issues which these approaches may raise. For example, an open design is by necessity unmasked to the participant, with a subsequent risk of bias if the outcome measures are anything but objective and a potential difference of response between the arms due to a ‘placebo’ effect. They also identified remedies which have been used, but with little supporting evidence of effectiveness, such as providing trial details through information videos and recruiter training.
The UK Collaborative Trial of Ovarian Cancer Screening set out in 2000 to recruit 202,638 women, and met that target in 2005. 68 The authors highlighted meticulous planning as key to recruitment. Of the strategies recommended by Watson et al. 66 and Treweek et al. ,67 the only one adopted was an open design, but this was necessitated by the design of the study and was not driven by the need to recruit. They claimed that their information video was of great benefit, despite the lack of evidence identified in the earlier review. It seems likely that the major impacts on recruitment were personalised invitation letters and an intervention which offered a health benefit in a condition of particular concern to those invited.
Fletcher et al. ’s69 systematic review in 2011 of incentivising clinicians’ recruitment to clinical trials found that the available evidence was poor. The most promising method, they suggested, was to identify barriers to clinician recruitment through qualitative work.
Retention and follow-up
The literature on retention is less extensive than that on recruitment, investigating more tightly defined questions related to individual clinical areas or study methods.
Booker et al. 70 considered retention in long-term prospective cohort studies, which tend to be more vulnerable to attrition than most clinical trials. Their systematic review considered 28 studies, of which 11 were randomised trials of retention strategies. The strategies fell into three groups: incentives (including cash and other gifts), reminders (by letter, phone or, more recently, text message) and other (alternative data collection). They found that incentives were the best way to ensure retention, but it was unclear whether financial or non-financial greater-value incentives had a larger effect.
Meyers et al. 71 studied issues around follow-up of minors, especially in the context of substance abuse research, emphasising the need for intensive effort for follow-up.
Fisher et al. 72 built on some of the findings from Meyers et al. ’s71 work, incorporating a novel psychological and educational model to develop a programme aimed at increasing recruitment and retention in RCTs [anticipate, acknowledge, standardise, accept, plan (AASAP)]. Tested in a three-arm RCT of distress reduction techniques in diabetics, the scheme improved retention.
Prostate Testing for Cancer and Treatment (ProtecT) (ISRCTN 20141297) is a large HTA trial investigating treatments for men with localised prostate cancer. 73 Men registered at randomly selected GP practices were invited by letter to enrol. 74 A cohort of 89,000 men was recruited between 1999 and 2008, with 2600 developing prostate cancer and 1672 agreeing to randomisation. The team developed a framework, known as the Peer Review Intervention for Monitoring and Evaluating sites (PRIME), to assess site performance, training needs and good clinical practice adherence of the recruiting sites. 75 Each of the eight sites in the trial was assessed annually, looking at its recruitment processes but also its adherence to follow-up protocols. A review of PRIME found that it enhanced study conduct and consistency, manifested in a more complete follow-up for ProtecT.
Delivery of trials
Transparent reporting of extensions and protocol changes
Chan and Altman11 discussed outcome reporting bias in trials by reviewing publications and surveying authors. Incomplete reporting of trial results was common, with primary and reported outcomes often not in compliance with the predefined protocol, but instead driven by results which might be considered interesting. They recommended publication of all trial protocols to allow assessment of this bias by future reviewers and decision-makers, a recommendation followed up in the 2010 revision of CONSORT. 44,76 They noted that reporting bias may be promoted by the limited space available in traditional journals, a restriction the HTA journal, with its generous with space restrictions, might be expected to overcome.
Questions addressed
We reviewed how well the included HTA randomised trials performed, including recruitment patterns, frequency of protocol changes (and extension requests), team composition and delays to do with obtaining permissions.
Box 4 shows the questions explored in this section.
To what degree did actual recruitment match planned recruitment?:
-
T3.1. Expected and reported number of participants recruited.
-
T3.2. Number of centres (including multicentre).
-
T3.3. Date when recruitment took place.
-
T3.4. Sample size calculation changes.
-
T3.5. Follow-up (including how many participants followed up).
-
T3.6. Recruitment comparison with STEPS.
-
T3.7. What was the composition of the team and did it change?
Project protocol changes and extension approvals:
-
T3.8. Evidence of project protocol changes.
-
T3.9. The number of protocol changes reported.
-
T3.10. The type of protocol changes reported.
-
T3.11. Approved extension applications submitted to the HTA programme.
-
T3.12. Time and cost implications.
-
T3.13. Number of extension request approvals for the included projects.
-
T3.14. Reasons given for the submission of an extension request.
-
T3.15. What were the planned and actual contract start and end dates for those included projects?
Methods
Fifteen questions were piloted, as shown in Box 4. One question concerning delays in obtaining ethical committee and R&D approval from trusts was dropped, for which no data were available.
Denominators
The denominator for the questions regarding recruitment, centres and follow-up was the number of trials (n = 125). The denominator for questions regarding the team and extension/protocol changes was the number of projects (n = 109).
Of the 125 trials, five were reported to have been abandoned, stopped or closed down. Box 5 shows details of these five trials, including the planned and actual recruitment. One monograph reported two trials (ID110 and ID78), one of which (ID110) had been closed down owing to poor recruitment.
Trial ID62 was formally abandoned with agreement from the HTA programme and the Trial Steering Committee. The planned recruitment number was 1000 and the actual number of participants recruited was 208.
Trial ID75 was stopped and formal closure of the trial was initiated in May 2005 after 5 months of recruitment. The planned recruitment number was 1002 and the actual number of participants recruited was 19. Nine participants completed the trial.
Trial ID97 did not continue into a full trial; a pilot was conducted to see if it could continue as a full trial. A full trial did not commence owing to low recruitment numbers and placebo control group. The planned recruitment number was 70 and the actual number of participants recruited was nine.
Trial ID110 (linked to trial ID78) was stopped with agreement from the HTA programme 14 months after consultation. This arm of the trial was considered inappropriate and unfeasible. The planned recruitment number was 400 and the actual number of participants recruited was one.
Trial ID111 was stopped with agreement from the HTA programme and the Trial Steering Committee. The planned recruitment number was 1425 and the actual number of participants recruited was 154. Despite low recruitment, patient follow-up continued for these 154 participants, with analysis of response remaining descriptive.
Results
Questions T3.1–T3.6: to what degree did actual recruitment match planned recruitment?
Question T3.1: expected and reported numbers of participants recruited
The extent to which trials recruited as planned varied, as shown in Figure 3.
FIGURE 3.
Recruitment of trials compared with original protocol.

The x-axis measures the proportion of the original target recruitment achieved and the y-axis measures the proportion of trials in the cohort which achieved a certain recruitment or better. Seventy-two per cent of trials (y-axis) managed to achieve 50% or better (x-axis) of their original target recruitment. About 40% of studies managed to recruit to 80% or better of their original targets.
A common response to being unable to recruit to target is to revise the target. This may be justified by consciously sacrificing power (e.g. dropping from 90% to 80%), discovering that the event rate within the trial is different to that assumed at application or changing the hypothesised effect size sought within the trial.
Figure 4 shows the studies from Figure 3 divided into two groups. The centre line shows the success in recruitment of the studies which did not revise their targets. The two outer lines reflect the other group, comprising those studies which did change their recruitment target while under way. The top line shows these studies’ performance against their new targets and the bottom line performance against their original ones. Unsurprisingly, this group were doing far worse than the non-changers before target revision and substantially better afterwards. However, even in studies which revised their targets, just over 80% achieved 80% of their new goals.
FIGURE 4.
Recruitment of trials compared with revised targets.

We observed that not achieving these targets does not mean that the studies failed. They may have ended up being underpowered compared with what was commissioned (though the HTA process looks at the consequences for power when taking decisions about when to extend studies), or power may have been preserved because of unforeseen changes in event rate.
Question T3.2: number of centres
A major driver of recruitment is the recruitment of trial centres. Of the 120 trials, 112 (93.3%) were reported to be multicentre. Half of the trials reported a difference between the planned number of centres in the application form/protocol and that reported in the monograph (52.5%, 63/125).
About 90% of trials were able to recruit the number of centres that they set out to attain during the course of a trial (Figure 5) and about 40% over-recruited their centres. Twenty per cent recruited double the number of centres they initially planned.
FIGURE 5.
Recruitment of trial centres (planned and actual).

Question T3.3: date when recruitment took place
Of the 120 trials, 108 (90%) reported the planned start date of recruitment and 107 (89.2%) reported the actual end date of the recruitment period in the published monograph. We had planned to extract the start and end dates of recruitment from the protocol/application form but such data were lacking.
Question T3.4: sample size calculation changes
Of the 120 trials, 36 (30%) changed the sample size calculation after the trial had commenced (these are a subset of those represented in the two outer lines in Figure 4). More than 80% of these trials (88.9%, 32/36) decreased the number of participants needed for the trial in comparison with 11.1% of trials (4/36) which increased the numbers required. The remaining 89 trials did not report any changes to the sample size calculation once the trial had commenced.
The reasons why the target sample size was changed cannot be derived from the available data. Staff involved with the HTA monitoring programme suggest that the most common reason is that the triallists revisit target recruitment. Less commonly, this process is initiated by a Data Monitoring Ethics Committee because it has noticed that underlying assumptions, such as the event rate, do not hold up. Five of these 125 trials were abandoned before completion (see Box 5).
Of the 36 trials that changed the sample size calculation, 34 (94.4%) completed full follow-up, one trial did not complete full follow-up and no data were available for one trial. More than three-quarters of these trials (77.8%, 28/36) recruited 80% or more of the total number of participants required.
Question T3.5: follow-up
Of the 120 trials, 97.5% (117/120) completed full follow-up (five trials are excluded owing to the reasons explained in Box 5). Follow-up was not applicable to two trials as one was a feasibility study (trial ID15) and the other had no follow-up period (trial ID113). It was not possible to determine full follow-up for one trial (trial ID16).
Comparison between the planned and actual follow-up periods found that in 22.2% of trials (26/117) the follow-up period had changed from that reported in the proposal/protocol. One-quarter of trials (6/26) reported an increase in the follow-up period compared with that reported in the proposal document.
Question T3.6: recruitment comparison with the Strategies for Trial Enrolment and Participation Study
Previous work by STEPS2 was compared with those trials included in the metadata database. The recruitment eligibility criteria for inclusion in the STEPS study were:
-
trials funded by the MRC or HTA programme
-
recruitment start date from 1 January 1994
-
recruitment end date, as stated in the application form or first protocol report, before 31 December 2002.
Cluster randomised design trials and single-centre trials were excluded from STEPS. Thirty-two HTA-funded clinical trials were included in STEPS. A comparison of these with 42 later HTA-funded trials meeting the STEPS criteria showed that of those 42 projects, 69% (29/42) recruited more than 80% of the original target number and 92.9% (39/42) completed full follow-up (Table 28). By analysing the recruitment start and end dates from the metadata database, we found 48 eligible projects since the STEPS. Six projects were excluded (five were cluster randomised design trials and one was a single-centre trial).
| Item | STEPS cohort | Post-STEPS cohort |
|---|---|---|
| Recruitment to ≥ 80% of target | 54.8% | 69.0% |
Question T3.7: what was the composition of the team and did it change?
A comparison of applicants named on the final application form with authors of the monograph showed changes in all projects. Out of the 107 projects legible for analysis (two had no application form), 19.6% (21/107) were reported to have included the original proposal team applicants as well as other people as co-authors of the monograph. The most common change from the application form to the monograph was ‘a subset of the original team and additional team members’ (78%, 84/107). Table 29 shows the three different compositions of the research team.
| Composition of the team from proposal to monograph | n (%) |
|---|---|
| Original proposal team and additional others | 21 (19.6) |
| Subset of original team | 2 (1.9) |
| Subset of original team and additional others | 84 (78.5) |
| No change | 0 (0) |
Some changes in the trial team are to be expected but not changes in all. Retirement and death are unavoidable, and occasionally job changes, especially moving abroad, can lead to investigators leaving the team. We were unable to examine whether additional investigators or authors were used to replace expertise which was no longer available, or to supplement the team with expertise which was missing from the original application. Difficult to assess, although important to look at, is guest authorship and guest applicant status.
Questions T3.8–T3.14: project protocol changes and extension approvals
Question T3.8: evidence of project protocol changes
Since 2001, all projects have had to complete a protocol change form. These were located for 69.7% of projects (76/109). (Two trials submitted a non-standard protocol change form. These were included in subsequent analyses.) The protocol change form changed several times (three versions were identified). Only version 3 asked if any of the project protocol changes were reported in the final report. Of the 41 projects which submitted version 3, only half (51.2%, 21/41) reported the changes in the published monograph, one-third of projects (34.1%, 14/41) were not consistent in reporting the project protocol changes in the monograph and 14.6% (6/41) did not report any amendments or no information was available to report. By ‘not consistent’ in their reporting we mean that these projects may not have reported all protocol changes in the monograph. They only reported a select number of protocol changes.
Only projects that submitted a protocol change form were included in the two subsequent analyses (76 projects provided a protocol change form and two projects used a non-standardised form).
Question T3.9: the number of protocol changes reported
Seventy-four projects out of 78 (94.9%) reported more than one protocol change and the mean number of changes was six (exact mean 5.97) (out of those that submitted a protocol change form; n = 78 projects). A total of 466 protocol changes were reported from the 78 projects.
Question T3.10: the type of protocol changes reported
‘Clinical assessment’ was the most reported protocol change, with 122 mentions (26.2% of all reported changes). Examples included changes such as laboratory processes or specific diagnostic tests used. Occasionally the staff undertaking assessments changed, for example from a doctor to a specially trained nurse. Issues related to recruitment were reported 66 times (14.2% of changes) and patient eligibility criteria were reported 64 times (13.7% of changes). Three project protocol changes were reported as ‘other’ and these consisted of adverse event reporting (n = 1), GPs to flag records of participants who defaulted at 12 months (n = 1) and reduction in the amount of data verification (n = 1) (Table 30).
| Description of data | n (%) |
|---|---|
| Were there any protocol amendments? | |
| Yes | 78 (71.6) |
| Not applicable | 1 (0.9) |
| No information available | 23 (21.1) |
| Protocol changes were reported in the monographa | 7 (6.4) |
| Total | 109 |
| Number of protocol changes | |
| 1 | 4 (5.1) |
| 2 | 4 (5.1) |
| 3 | 9 (11.5) |
| 4 | 9 (11.5) |
| 5 | 11 (14.1) |
| 6 | 9 (11.5) |
| 7 | 9 (11.5) |
| 8 | 8 (10.3) |
| 9 | 7 (9.0) |
| 10+ | 8 (10.3) |
| Total | 78 |
| Project protocol changes record form submitted | |
| Yes | 76 (69.7) |
| No information submitted | 31 (28.4) |
| Non-standard forms were used | 2 (1.8) |
| Total | 109 |
| Type of protocol change reportedb,c | |
| Patient eligibility criteria | 64 (13.7) |
| Outcome measures | 15 (3.2) |
| Intervention | 13 (2.8) |
| Sample size | 18 (3.9) |
| Alternations to analyses | 31 (6.7) |
| Clinical assessment | 122 (26.2) |
| Study design | 47 (10.1) |
| All issues related to recruitment | 66 (14.2) |
| Randomisation process | 13 (2.8) |
| Trial management | 52 (11.2) |
| Number of centres | 22 (4.7) |
| Other | 3 (0.6) |
| Total | 466 |
Question T3.11: approved extension applications submitted to the Health Technology Assessment programme
Out of the 109 projects, 94 submitted extension request(s), which were approved by the HTA programme.
Question T3.12: time and cost implications
Seventy-seven projects (70.6%) submitted extensions for time and additional research cost compared with eight (7.3%) which requested an extension for time only and nine (8.3%) which requested an extension for additional research cost only.
Question T3.13: number of extension request approvals for the included projects
Of the 94 projects, 72 (76.6%) had more than one approved extension request, with a mean of 3.03. Two hundred and eighty-five approved extensions were reported from the 94 projects.
Question T3.14: reasons given for the submission of an extension request
Of these 285 approvals, 36 (12.6%, 36/285) referred to salary inflation and one (0.4%, 1/285) referred to start date change. As these did not have a direct relevance to the trial itself they were excluded from the descriptive statistics shown in Table 31 (n = 248) and any further analyses.
Adjusting for the total number of extension requests reported by each project, the most reported extension request was based on ‘recruitment issues’, reported 107 times (43.1% of requests). All other categories were below 10%, representing a small number of approved extension requests (Table 31).
| Description of data | n (%) |
|---|---|
| Did the trial receive an extension request? | |
| Yes | 94 (86.2) |
| No | 15 (13.8) |
| Number of extension requests | |
| 0 | 15 (13.8) |
| 1 | 22 (20.2) |
| 2 | 18 (16.5) |
| 3 | 21 (19.3) |
| 4 | 10 (9.2) |
| 5 | 14 (12.8) |
| 6 | 8 (7.3) |
| 7 | 1 (0.9) |
| Total | 109 |
| Type of approved extension requesta,b | |
| Recruitment | 107 (43.1) |
| Staffing issues | 18 (7.3) |
| Extended follow-up period | 7 (2.8) |
| Extra drug or equipment costs | 8 (3.2) |
| Additional work/work greater than expected | 12 (4.8) |
| Revised project costs | 12 (4.8) |
| Report writing | 12 (4.8) |
| Data issues | 6 (2.4) |
| Research governance | 7 (2.8) |
| EU directive work | 13 (5.2) |
| New host institution | 4 (1.6) |
| Additional support | 5 (2.0) |
| External factors | 8 (3.2) |
| Funding reduced | 9 (3.6) |
| Other | 20 (8.1) |
| Total | 248 |
Seventy-eight per cent of projects (85/109) submitted a time extension request to the HTA programme. Between these 85 projects, 141 requests for additional time were submitted and approved by the HTA programme (49.4%, 141/285 approved extensions). The mean length of time extensions was 4.06 months.
Question T3.15: what were the planned and actual contract start and end dates for the included projects?
The mean duration of the projects (n = 109) increased from a planned 39.7 months to the actual mean duration of 50.4 months. Almost half of the projects (49.5%, 54/109) had a different contract start date from the original date proposed in the Department of Health contract of agreement and this was particularly marked from 1997 to 2002.
Eighty-seven per cent of projects (95/109) had a different end date to that originally proposed and 89.5% (85/95) had a change to the total duration of the project.
Analysis
Hardly any trials recruited as planned. Around three-quarters recruited more than 50% of their planned sample; one-quarter recruited less than 50%. Most trials had to agree revised targets with the programme. More, but not all, of those that revised their targets came close to meeting them, but some did not.
Considering only studies which did not change their target sample size, 50% obtained more than 80% of their initial target sample, and over 90% obtained half their target sample.
Unsurprisingly, studies which revisited their sample size during the course of the trial tended to perform poorly on their ability to recruit to the target in their original protocol but did much better when compared against their revised target. The prompt to revisit the sample size, sometimes in response to a non-related factor such as an updated external estimate of effect size, is commonly poor recruitment performance.
Recruitment of centres is a major driver for overall recruitment in multicentre studies. 77 Ninety per cent of trials in this cohort recruited their target number of centres and 50% recruited more than the initial number that they thought they needed. Most surprisingly, 20% of studies recruited more than twice the number of centres initially planned.
We have not investigated how efficient these centres were, for example to asses if this was a good use of resources. The bulk of patients in a multicentre trial often come from a very small subset of centres. The excess centres were perhaps needed because the bulk of the originally planned centres did not recruit as well as expected. Alternatively, clinicians can be keen to join in with a trial that they perceive as successful, and this may be the reason for centre over-recruitment. Either way, the efficiency of centres could be worthy of further investigation.
A notable finding is that of all the studies in the cohort, none had the same team at the end as at the beginning. Further work may be required to investigate the process by which teams changed, whether or not those changes were agreed with the programme (either in approving the change, or assessing if the changed team could still deliver the contracted work) and whether new team members replaced outgoing ones or provided new skills missing in the original bid. The HTA programme should consider requiring teams to explain any changes in their final reports.
Protocol changes were common and not always well reported. Thirty projects in this cohort did not indicate using the appropriate form that there had been protocol changes at the time of submission of their report. Despite this, seven reported changes in their monographs.
Of those projects which notified the programme of changes to protocol, it is concerning that less than half acknowledged this in the monograph. This may have improved recently as the editorial process has matured. It would be useful to reassess this issue with more recent projects.
The extent of a protocol change can vary from minor to major. It should be possible to categorise proposed changes to allow a proportionate response, but consideration of this was outside the scope of this project.
The mean duration of projects increased from a planned duration of just under 40 months to an actual duration of just over 50 months. This delay varied over time, with a peak just after 2000, mostly resolved by 2003.
Far more studies were affected by delays in the planned start and end dates. Important causes of delay may be delays in obtaining ethical and governance approvals. Further, applicants tend to underestimate the time required to recruit participating centres. Investigators often underestimate the effort required to write the project’s final report, which will, after an editorial process, be published in Health Technology Assessment, and many projects therefore request short time-only extensions to give more time for report preparation. More recently, but probably not affecting this cohort of trials, studies have run into problems obtaining excess treatment costs, that is, the clinical costs of undertaking a trial which are in excess of those that a primary care trust (PCT) might usually expect to pay for the routine care of its population.
Discussion
Applicants need to be more realistic about trial set-up times and the programme can help by pointing out to applicants where it feels insufficient time has been allowed; however, the fundamental problems with the NHS’s current research approvals process can only be addressed at a national level, in much the same way that the previous problems with ethics committees were resolved.
The issue of excess treatment costs is one which can only be solved at a national level; the programme has no power to compel health service funders to pay excess treatment costs, despite the existence of instructions from the Department of Health which say that they should. 58,78,79
Strengths and weaknesses of the study
The main weakness in addressing these questions derives from data limitations. The HTA MIS did not store monthly recruitment in a machine-readable form. Even the programme’s paper records usually only noted recruitment every 6 months. It was not possible to examine how trials’ recruitment performance varies across time and by centre.
All trials in this cohort began before the introduction of the NIHR research networks. Newer trials will have been able to use these networks to speed up their start-up processes.
We identified several changes which could make the routine data more relevant, mainly reconfiguration of data collected regarding changes to projects. We have also identified some data which were less useful and probably not worth the effort of prospective collection.
Recommendations for future work
Should similar work continue, we consider that data should be extracted for 13 questions: five to be kept as they are (T3.3, T3.4, T3.9, T3.13 and T3.15) and eight to be amended (T3.1, T3.2, T3.5, T3.6, T3.7, T3.10, T3.12 and T3.14).
The finding that hardly any of the included trials recruited as planned merits consideration by the HTA programme. Although trials funded more recently may have recruited closer to planned targets, recruitment problems are likely to remain. At the very least, the data collected routinely should take fuller account of the knock-on effects, including on protocol changes and revised timescales.
Unanswered questions and future research
There is scope for further investigation of specific questions:
-
What is the influence of study under-recruitment on power? Following on from that, what power should the programme expect a study to deliver?
-
Why do studies systematically overestimate their ability to recruit?
-
How can the differential effectiveness of recruiting centres be addressed, preferably by increasing the effectiveness of the poorest performers?
-
Why do study teams change? How can that process of change best be managed?
-
How can protocol changes best be categorised, to allow the programme to respond (and assess the changes) in an appropriate way?
Chapter 7 Theme 4: were the statistical analyses appropriate and as planned?
This chapter considers questions surrounding the appropriateness of the statistical analyses. After a brief review of the relevant literature, 19 questions were explored. The results are summarised and discussed.
Introduction
Outcome reporting bias has been widely reported. 10,11,27,80–87 However, only a few papers have reported on whether or not researchers adequately specify planned analyses in the protocol and, subsequently, whether or not they follow the prespecified analysis. 88,89 This matters because failure to follow the prespecified analysis can result in bias. One study suggested that protocols were not sufficiently precise to identify deviations from planned analyses. 89 Two reviewed whether or not sample size calculations were adequately specified. 88–90 Another recently questioned whether or not the current method of sample size calculations was appropriate. 91 These are summarised below.
The primary outcomes in protocols were compared with those in published reports for 102 trials approved by the scientific ethics committees for Copenhagen and Frederiksberg, Denmark, between 1994 and 1995. 10 Selective reporting was revealed, with 62% of trials reviewed having at least one primary outcome added, omitted or changed.
A similar review of 48 trials funded by the Canadian Institutes for Health Research81 found that in 33% of trials, the outcome listed as primary in the publication differed from that in the protocol. They also found that outcome results were incompletely reported.
A pilot study conducted in 2000 reviewed 15 applications received by a single local research ethics committee in the 1990s and compared the outcomes, analysis and sample size in the protocol with that presented in the final study report. 89 The authors found that six protocols (40%) stated the primary outcome and, of these, four (67%) matched that in the published report. Eight mentioned an analysis plan but only one (12%) followed its prescribed plan. The study concluded that selective reporting may be substantial but that bias could only be broadly identified as protocols were not sufficiently precise.
In 2008, Chan et al. 88 compared the statistical analysis and sample size calculations specified in the protocol with those specified in the published paper. They found evidence of discrepancies in the sample size calculations (18/34 trials), the methods of handling protocol deviations (18/34 trials), methods of handling missing data (39/49 trials), primary outcome analyses (25/42 trials), subgroup analyses (25/25 trials) and adjusted analyses (23/28 trials). These discrepancies could affect the reliability of results, introduce bias and indicate selective reporting. They concluded that the reliability of trial reports cannot be assessed without access to the protocol.
A 2008 comparison of the sample size calculation specified in the protocol with that in the publication found that only 11 of the 62 trials reviewed adequately described the sample size calculation in both the protocol and published report. 88
Charles et al. ,90 in a review of the reporting of sample size calculations in 215 trials published between 2005 and 2006, found that 43% did not report all the required sample size calculation parameters.
A study of 18 trials that reported on traumatic brain injury reviewed the covariates adjusted for and subgroup analyses performed. 92 Protocols could be obtained for 6 of the 18 trials and it found that all six trials reported subgroup effects which differed from those specified in their protocols.
In collaboration with journal editors, triallists, methodologists and ethicists, Chan et al. 93,94 have launched the Standard Protocol Items for Randomised Trials (SPIRIT) initiative to establish evidence-based recommendations for the key content of trial protocols.
The above studies may not reflect current practice because either the number of trials reviewed was small or the studies reviewed were relatively old (1994–5 for Chan88 and similar for Hahn89). Practice may have improved since, following the introduction of CONSORT and other guidelines.
Our objective was to repeat these analyses on the cohort of all HTA published RCTs, assessing the extent of these discrepancies and whether or not they improved over time.
Questions addressed
The aim was to review the appropriateness of the statistical analyses for all published HTA clinical trials, including the sufficiency of the proposed statistical plan, handling of missing data and whether or not there were discrepancies between what was proposed and what was actually reported in the published monograph.
The questions posed (Box 6) fall under the following six subheadings:
-
Did the protocol specify the planned method of analysis for the primary outcome in sufficient detail?
-
Was the analysis planned in the proposal/protocol for the primary outcome carried out?
-
How was the sample size estimated?
-
How adequate was the reporting of planned and actual subgroup analysis?
-
Other information: what graphical presentation of data was reported in HTA trials?
-
Were conclusions justified given the analysis results?
Did the protocol specify the planned method of analysis for the primary outcome in sufficient detail? In relation to:
-
T4.1. how many specified a method of analysis for the primary outcome.
-
T4.2. whether or not this improved over time.
-
T4.3. statistical test applied.
-
T4.4. significance level.
-
T4.5. hypothesis testing.
-
T4.6. adjustment for covariates.
-
T4.7. analysis population.
-
T4.8. adjustment for multiple testing.
-
T4.9. missing data.
-
T4.10. sufficient detail including all of the above seven elements recorded in the protocol.
Was the analysis planned in the proposal/protocol for the primary outcome carried out? In relation to:
-
T4.11. statistical test/model used.
-
T4.12. significance level.
-
T4.13. analysis population.
-
T4.14. missing data.
-
T4.15. covariates adjusted for in the analysis.
How was the sample size estimated (power, confidence intervals, etc.)?
-
T4.16. Was sufficient information on the sample size calculation provided?
-
T4.17. Does the sample size calculation in the protocol match the sample size calculation shown in the monograph? What discrepancies were found?
-
T4.18. What values of alpha, power and drop out were used in the sample size calculation?
-
T4.19. Other information: what graphical presentation of data was reported in HTA trials?
Methods
Nineteen questions were piloted as shown in Box 6. Four questions were considered but not proceeded with, regarding:
-
the number of statistical tests and number of primary statistical tests
-
whether or not authors measured more outcomes than they reported
-
adequate reporting of subgroup analyses
-
whether or not the conclusions were justified given the analysis results.
Difficulties arose with each of these questions. Firstly, results were not presented in a standard format in the monographs. Secondly, as the monographs were lengthy, data extraction meant searching and reading through many pages. Thirdly, as the HTA trials are pragmatic, they include a large number of outcomes measured at multiple time points, which increased the number of tables/amount of text to be reviewed. Fourthly, extracting information on subgroup analyses planned and carried out was difficult because authors seldomly labelled analyses as subgroup analyses. Lastly, we found it difficult to specify data that could answer the question regarding the conclusions being justified by the analyses.
For the 19 questions explored, the methods used in the literature reviewed above were used as a framework to detail the questions. For example, the paper by Chan et al. 88 provided the key components of data that needed to be extracted on the sample size calculation. Data on these components were expanded to include other types of outcome measures and study designs (e.g. time-to-event data, non-inferiority and cluster randomised trials). We extracted these data from the protocol or project proposal (if a protocol was not available) and monograph, and analysed the data in a similar way.
Denominators
All trials were included (n = 125). The unit of analysis for questions T4.1–T4.15 was each trial’s primary outcome with complete analysis (n = 164 planned and n = 161 reported). The unit of analysis for T4.16–T4.18 was the individual trial.
Results
Questions T4.1–T4.10: did the protocol specify the planned method of analysis for the primary outcome in sufficient detail?
Question T4.1: how many specified a method of analysis for the primary outcome?
The 125 trials included 206 planned primary outcomes and reported on 232 primary outcomes. Of these, 164 and 161, respectively, were ‘complete for analysis’ (these are the denominators for questions T4.1–T4.10).
The method of analysis was prespecified for 111 out of 164 planned primary outcomes (67.7%), with little difference between those that did and did not have protocols (65.9%, 54/82 from the proposal and 69.5%, 57/82 from the protocol) (Table 32).
| Planned primary analysis | Protocol available or not? | Total, n (%) | |
|---|---|---|---|
| Yes, n (%) | No, n (%) | ||
| Yes | 57 (69.5) | 54 (65.9) | 111 (67.7) |
| No | 14 (17.1) | 13 (15.9) | 27 (16.5) |
| Not clear | 1 (1.2) | 1 (1.2) | 2 (1.2) |
| Not applicable | 1 (1.2) | 0 | 1 (0.6) |
| No information available | 9 (11.0) | 14 (17.0) | 23 (14.0) |
| Total number of primary outcomes | 82 (100.0) | 82 (100.0) | 164 (100.0) |
| Total number of trials | 65 (52.0) | 60 (48.0) | 125 (100.0) |
Question T4.2: has this improved over time?
There is a slight indication that the specification of the primary outcome analyses has improved over time. This could be due to the increasing number of protocols available (Table 33 and Figure 6) but the low numbers preclude strong conclusions.
| Year of commissioning brief | Yes, n (%) | No,a n (%) | Not clear, n (%) | Total, n (%) |
|---|---|---|---|---|
| 1993 | 12 (70.6) | 5 (29.4) | 0 | 17 (100.0) |
| 1994 | 13 (52.0) | 11 (44.0) | 1 (4.0) | 25 (100.0) |
| 1995 | 19 (73.0) | 7 (27.0) | 0 | 26 (100.0) |
| 1996 | 16 (55.2) | 13 (44.8) | 0 | 29 (100.0) |
| 1997 | 6 (50.0) | 5 (42.0) | 1 (8.0) | 12 (100.0) |
| 1998 | 3 (75.0) | 1 (25.0) | 0 | 4 (100.0) |
| 1999 | 10 (83.3) | 2 (16.7) | 0 | 12 (100.0) |
| 2001 | 20 (87.0) | 3 (13.0) | 0 | 23 (100.0) |
| 2002 | 3 (75.0) | 1 (25.0) | 0 | 4 (100.0) |
| 2003 | 6 (75.0) | 2 (25.0) | 0 | 8 (100.0) |
| 2005b | 1 (100.0) | 0 | 0 | 1 (100.0) |
| 2009b | 2 (66.7) | 1 (33.3) | 0 | 3 (100.0) |
| Total | 111 (67.8) | 51 (33.5) | 2 (1.2) | 164 (100.0) |
FIGURE 6.
Proportion of trials with a protocol available by year of commissioning brief.
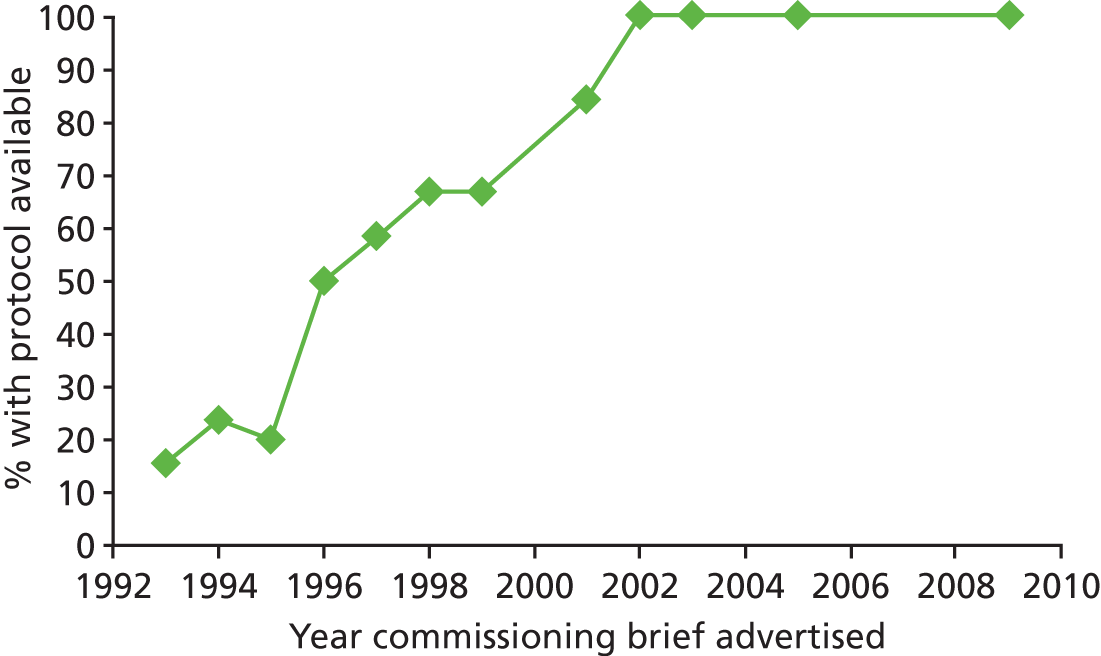
Question T4.3: statistical test applied
Of the 111 planned primary outcomes with a prespecified method, the proposed statistical test/choice of model was described in 108 (96.4%). The most frequently reported planned methods of analysis were logistic regression (23.4%, 26/111) and analysis of covariance (ANCOVA)/linear regression (17.1%, 19/111), followed by t-test (14.4%, 16/111) (Table 34).
| Description of planned statistical analyses | Planned from protocol/proposal, n (%) | Reported in the monograph, n (%) |
|---|---|---|
| Planned statistical test | ||
| t-test | 16 (14.4) | 14 (9.4) |
| Chi-squared test | 8 (7.2) | 20 (13.4) |
| ANOVA | 6 (5.4) | 0 |
| ANCOVA/linear regression | 19 (17.1) | 48 (32.2) |
| Logistic regression | 26 (23.4) | 21 (14.1) |
| Mixed model | 5 (4.5) | 18 (12.1) |
| Poisson regression | 3 (2.7) | 2 (1.3) |
| Cox proportional hazards | 7 (6.3) | 8 (5.4) |
| Log-rank test | 1 (0.9) | 4 (2.7) |
| Mann–Whitney | 1 (0.9) | 1 (0.7) |
| Non-parametric analyses | 1 (0.9) | 0 |
| Confidence interval | 11 (9.9) | 9 (6.0) |
| Other | 3 (2.7) | 4 (2.7) |
| Not specified | 4 (3.6) | 0 |
| Total | 111 (100.0) | 149 (100.0) |
| Significance level | ||
| 1% | 0 | 13 (8.7) |
| 2.5% | 2 (1.8) | 1 (0.7) |
| 5% | 17 (15.3) | 22 (14.8) |
| 95% confidence interval specified | 20 (18.0) | 47 (31.5) |
| Not specified | 72 (64.9) | 66 (44.3) |
| Total | 111 (100.0) | 149 (100.0) |
| Hypothesis testing | ||
| One-sided | 3 (2.7) | 3 (2.0) |
| Two-sided | 11 (9.9) | 28 (18.8) |
| Not specified | 97 (87.4) | 118 (79.2) |
| Total | 111 (100.0) | 149 (100.0) |
| Planned covariates to adjust for | ||
| Yes | 68 (61.3) | 0 |
| No | 9 (8.1) | 0 |
| Not clear | 3 (2.7) | 0 |
| No information available | 31 (27.9) | 0 |
| Total | 111 (100.0) | 0 |
| Analysis population | ||
| ITT analysis | 60 (55.0) | 117 (78.5) |
| PP analysis | 0 | 3 (2.0) |
| AT analysis | 0 | 0 |
| ITT and PP analysis | 5 (3.6) | 14 (9.4) |
| ITT and AT analysis | 0 | 0 |
| PP and AT analysis | 0 | 0 |
| No available information | 46 (41.4) | 15 (10.1) |
| Total | 111 (100.0) | 149 (100.0) |
| Adjustment for multiple comparisons | ||
| Bonferroni correction | 4 (3.6) | 8 (5.4) |
| Bonferroni–Dunn | 1 (0.9) | 3 (2.0) |
| Other | 2 (1.8) | 5 (3.4) |
| None specified | 104 (93.7) | 133 (89.3) |
| Total | 111 (100.0) | 149 (100.0) |
| Method of handling missing data | ||
| Complete case analysis | 3 (2.7) | 20 (13.4) |
| LOCF – single imputation method | 4 (3.6) | 14 (9.4) |
| WCI – single imputation method | 0 | 2 (1.3) |
| HDI – single imputation method | 0 | 1 (0.7) |
| RM – single imputation method | 0 | 5 (3.4) |
| Multiple imputation | 0 | 7 (4.7) |
| Mixed model | 3 (2.7) | 6 (4.0) |
| Generalised estimating equation | 1 (0.9) | 1 (0.7) |
| Survival analysis | 7 (6.3) | 11 (7.4) |
| Mean – single imputation method | 1 (0.9) | 3 (2.0) |
| More than one method was used to deal with missing data | 2 (1.8) | 8 (5.4) |
| Sensitivity analysis | 7 (6.3) | 9 (6.0) |
| None/no available information | 83 (74.8) | 62 (41.6) |
| Total | 111 (100.0) | 149 (100.0) |
Question T4.4: significance level
Of the 111 primary outcomes with a specified method of analysis, the significance level/confidence interval level to be used was specified in 39 (35.1%). Table 34 shows that the most commonly used level of statistical significance was 5%.
Question T4.5: hypothesis testing
The majority did not specify whether one-sided or two-sided analysis would be performed (87.4%, 97/111) (see Table 34).
Question T4.6: adjustment for covariates
Sixty-eight of the 111 (61.3%) planned primary outcomes specified the covariates that they planned to adjust for in the final analysis.
Question T4.7: analysis population
The planned population for the primary analysis was not specified by 41.4% (46/111). This appears to have improved over time (apart from anomalies in 1998 and 2003), with a big increase in 1996, the year in which CONSORT was published.
Question T4.8: adjustment for multiple testing
Almost all studies failed to specify a method of adjustment for multiple testing (93.7%, 104/111). As HTA trials are pragmatic as opposed to licensing trials, looking at a range of outcomes over short- and long-term periods, adjustment for multiple testing may matter less than transparency.
Question T4.9: missing data
Most studies did not specify a method for handling missing data (74.8%, 83/111). Of those that did, the methods used varied (see Table 34).
Question T4.10: is sufficient detail including all of the above seven elements recorded in the protocol?
The number of protocols meeting all seven criteria was low, at 1.8% (2/111). When we limited the criteria to three (model/test, significance level and analysis population), of the 111 primary outcomes for which a method of analysis was specified in the protocol/proposal, 30 primary outcomes qualified (27%, 30/111). This increased slightly over time, from 22.7% before 1998 to 35.6% after.
Questions T4.11–T4.15: was the analysis planned in the protocol/proposal for the primary outcome carried out?
Question T4.11: statistical test/model used
Of the 82 trials whose primary outcome was as planned, the authors changed the planned method of statistical analysis (model/test) in 20 (24.4%). Some changed to more complex methods (t-test changed to linear regression in five instances) and others to simpler methods (in three, a chi-squared test was carried out instead of logistic regression, linear regression was used instead of a mixed model and Fisher’s exact test was used instead of Cox proportional hazards). These could be legitimate changes or selective reporting depending on the results, something we did not explore (examples are given in Box 7).
-
In three trials (ID131, ID132 and ID133) reported in one monograph, the authors stated in the protocol that they would analyse the primary outcome score data using logistic regression. They actually analysed the continuous score data using ordinal regression (which they classified as linear regression).
-
Trial ID65 planned in the proposal to analyse the second primary outcome as follows: ‘Six month follow up data, relapse rates will be analysed by comparing relapse rates between the groups by survival analysis using cox’s regression controlling for baseline depression, age, sex and centre.’ They actually compared the percentage relapsing in each group at the end of treatment using a Fisher’s exact test and yielding a significant result (p < 0.005).
Question T4.12: significance level
All but six trials used the 5% significance level. Of the six discrepancies between the significance stated in the protocol/proposal and that used in the monograph, one led to an increase in the significance level used but this seems to be an error (trial ID42 stated in the protocol that ‘Differences will be judged significant at the 2.5% level to take account of two primary comparisons being drawn’; the monograph stated that 95% confidence intervals would be calculated but a 2.5% significance level was stated in the sample size calculation in the protocol.
Question T4.13: analysis population
Of those trials that stated the planned analysis population for the primary outcome analysis in the protocol/proposal, 90% (56/62) followed the plan. Most carried out what they described as an ‘intention-to-treat’ analysis. In two trials, the triallists stated in the protocol/proposal that they would carry out both an intention-to-treat and per-protocol analysis but reported only on the per-protocol analysis. Both of these were from trial ID109, where ‘The data were analysed per protocol. As planned, no intention-to-treat analyses were conducted, as < 10% of subjects would have been classified differently in such an analysis.’ Therefore, this change of analysis population was justified because the authors had specified in the protocol a rule which was used to decide which population to use.
Question T4.14: missing data
Of the 28 trials for which a method of handling missing data was specified in the protocol, the method used was different in 12 (42.9%).
Question T4.15: covariates adjusted for in the analysis
Sixty-eight of the 111 trials (61.3%) outlined their planned analysis of covariates and for 31 (27.9%) it was unclear (Table 35). Some trials did not specify which covariates they would adjust for in the protocol or, if they did, they failed to specify exactly which covariates would be adjusted for, for example ‘adjusting for baseline variables’ or ‘taking into account any statistically important imbalances’. This made it difficult to compare planned covariates with actual covariates adjusted for, in many trials.
| Trial ID | Covariates which trial planned to adjust for | Actual covariates adjusted for |
|---|---|---|
| 65 | Controlling for baseline HRSD, treatment centre, age and sex. Duration of index depressive episode, degree of treatment resistance, psychosis, antidepressant medication equivalents and cognitive impairment | Prerandomisation baseline HRSD scores were included as a covariate, as were NHS trusts to adjust for centre effects |
| 59 | No information | Adjusted for age, sex, surgical status, major presumptive clinical syndrome, SOFA score at time of randomisation and APACHE II score at ICU admission |
| 74 | Adjusting for group differences at baseline if necessary | With baseline HADS depression score and stratification categories (urban/rural location; horizontal/vertical kinship) as covariates |
| 78 | Age, sex, time to treatment and stroke type. Presence or absence of dysphagia | Time to treatment |
| 86 | Individual-level covariates, e.g. age of mother, parity, and health visitor confounders such as age | After adjusting for covariates such as 6-week EPDS score, living alone, previous history of PND and any life events experienced |
| 90 | Severity at initial presentation, age and sex | None specified |
| 94 | Two stratification variables – centre and size of ulcer – were to be adjusted for in the analyses, as were ulcer type, duration of episodes, weight of patient, ankle mobility and a binary variable for the presence/absence of infection at baseline. Authors were to present an unadjusted analysis, but the adjusted analysis would have primacy | A Cox proportional hazards model was used to adjust the analysis for the randomisation stratification factors (centre, baseline ulcer area), as well as duration and ulcer type. Actual baseline area (as measured from the tracings) and duration of ulcer were used |
| 103 | Group, time, group by time, model using a linear trend over time and a quadratic trend if necessary (group by time interaction) | Adjusted for baseline HbA1c based on those who completed their 12-month HbA1c measurement |
In summary, the analyses planned in the proposal/protocol for the primary outcome were carried out in 68 of the 82 trials (76%) and changed in 20 (24%) (considering statistical test/model only). The method of handling missing data specified in the protocol/proposal did not match what was carried out 43% of the time. The analysis population and significance level changed 10% of the time in trials. More detailed examination suggests that some of the changes were legitimate. Without knowing if a statistical analysis plan was drawn up before the analysis and subsequently followed, one cannot conclude departures from proper practice.
Questions T4.16–T4.18: how was the sample size estimated (power, confidence interval, etc.)?
We followed the methods and tables used by Chan et al. ,88 expanded to incorporate the different types of sample size calculation observed in the HTA trials (e.g. width of confidence interval calculations, time-to-event data, standardised effect size, non-inferiority, equivalence).
Question T4.16: was sufficient information on the sample size calculation provided?
The results of classifying the trials by the five components suggested by Chan et al. 88 are shown in Table 36. Of the 125 trials, 75 proposals/protocols (60%) and 66 monographs (52.8%) reported all the required sample size components. Individual components were reported in 60.7–100% of proposals/protocols and 49.6–100% of monographs. The required sample size was reported in the proposal/protocol in 93% of trials (116/125), in the monograph in 90% (112/125) and in both in 89%. The result from the sample size calculation was presented in the proposal/protocol in 57% of trials (111/125), in the monograph in 46% (58/125) and in both in 42% (e.g. the sample size calculation showed that the trial will have to recruit 326 participants. Taking account of the participant dropout rate, this will increase the number needed per arm to 350). Forty-two per cent of trials (52/125) reported all the required components of the sample size calculation in both the proposal/protocol and monograph.
| Component of sample size calculation | Number of trials reporting each component (n = 117)a | ||
|---|---|---|---|
| Protocol, n/N (%) | Monograph, n/N (%) | Both,b n/N (%) | |
| 1. Name of outcome measure | 113/117 (96.6) | 113/117 (96.6) | 111/117 (94.9) |
| 2. Alpha (type 1 error rate) | 108/117 (92.3) | 109/117 (93.2) | 104/117 (88.9) |
| 3. (a) Method of calculation: powerc | 113/116 (97.4) | 113/116 (97.4) | 110/115 (95.7) |
| Continuous outcome | |||
| Minimum clinically important effect size (delta)d and | 43/49 (87.8) | 46/58 (79.3) | 39/47 (83.0) |
| SD for deltad or | 33/49 (67.3) | 32/58 (55.2) | 29/47 (61.7) |
| Standardised effect size | 12/12 (100.0) | 7/7 (100.0) | 7/7 (100.0) |
| Binary outcome | |||
| Estimated event rate in each arme | 41/53 (77.4) | 37/48 (77.1) | 36/48 (75.0) |
| Time-to-event outcome | |||
| Time-to-event dataf | 2/2 (100.0) | 2/2 (100.0) | 2/2 (100.0) |
| Type of outcome not specified | |||
| No components for sample size calculation specified | N/A | 1/1 (100.0) | 1/1 (100.0) |
| 3. (b) Method of calculation: width of confidence interval | 1/1 (100.0) | 1/1 (100.0) | 1/1 (100.0) |
| Binary outcome: event rate in each arm and precision/width of confidence interval required | 1/1 (100.0) | 1/1 (100.0) | 1/1 (100.0) |
| Continuous outcome: SD and precision/width of confidence interval required | 0 | 0 | 0 |
| 4. Calculated sample size | |||
| 4. (a) Included result from sample size calculation on number required to recruit | 71/117 (60.7) | 58/117 (49.6) | 53/117 (45.3) |
| 4. (b) Presented total number of participants required to recruit | 116/117 (99.1) | 112/117 (95.7) | 111/117 (94.9) |
| 5. All components required | 75/117 (64.1) | 66/117 (56.4) | 52/117 (44.4) |
Question T4.17: does the sample size calculation in the protocol match the sample size calculation shown in the monograph? What discrepancies were found?
Of the 117 trials reporting a sample size calculation in both the proposal/protocol and the monograph, we observed discrepancies between that planned and that reported in 45 trials (38.5%). A component of the sample size calculation was reported in the monograph but not reported in the protocol/proposal in 18 trials. In 39 trials, there was a discrepancy in at least one component reported in both the protocol/proposal and the monograph. These discrepancies were not acknowledged in the monograph. Where a discrepancy was observed between the number of patients, the trial planned to recruit and the number actually recruited, this was twice as likely to be because the number specified in the monograph was smaller than that in the protocol/proposal than vice versa (19 trials vs. 10 trials). Where a discrepancy existed in the minimum clinically important effect size, this was also almost twice as likely to be due to the effect size being reported as larger in the monograph than in the protocol (Table 37). These discrepancies could be due to reductions in the planned sample size after the study started which were not reported in the monograph, or attempts to justify the fewer patients actually recruited.
| Component of sample size calculation | Number of trials reporting each component (n = 117) | ||
|---|---|---|---|
| Total, n/N | Not prespecified,a n | Different from protocol description, n | |
| 1. Name of outcome measure | 18/113 | 2 | 16 |
| 2. Alpha (type 1 error rate) | 7/109 | 5 | 2 |
| 3. (a) Method of calculation: power | 18/113 | 3 | 15: nine larger in monograph; six larger in protocol/proposal |
| Continuous outcome | |||
| Minimum clinically important effect size (delta) and | 19/46 | 6/46 | 13: five larger in monograph, three larger in protocol/proposal and five not comparable as primary outcomes in protocol and monograph are different |
| SD for delta or | 5/32 | 3/32 | 2: one larger in protocol and one not comparable as primary outcomes in protocol and monograph are different |
| Standardised effect size | 1/7 | 0 | 1: one larger in protocol |
| Binary outcome | |||
| Estimated event rate in each arm | 4/37 | 1 | 3: in one values reported were higher in the monograph and one not comparable as primary outcomes in protocol and monograph are different |
| Time-to-event outcome | |||
| Time-to-event data | 0/2 | 0 | 0 |
| Type of outcome not specified | |||
| No components for sample size calculation specified in publication | 1 | 0 | 1: values specified in protocol for minimum difference aim to detect (delta), SD for delta, alpha and power |
| 3. (b) Method of calculation: width of confidence interval | 1 | ||
| Binary outcome: event rate in each arm and precision/width of confidence interval required | 0/1 | 0 | 0 |
| Continuous outcome: SD and precision/width of confidence interval required | 1/1 | 0 | Not comparable as primary outcome in protocol and monograph are different |
| 4. Calculated sample size | |||
| 4. (a) Included result from sample size calculation on number required to recruit | 20/58 | 5 | 15: 10 larger in protocol and five larger in monograph (note these figures include six trials where the primary outcome used for sample size calculation is different in protocol/proposal and monograph) |
| 4. (b) Presented total number of participants required to recruit | 30/112 | 1 | 29: 10 larger in monograph and 19 larger in protocol/proposal (note this includes five trials where the primary outcome used is different so not comparable) |
| 5. Any component | 45 | 18 | 39 |
Question T4.18: what values of alpha, power and dropout were used in the sample size calculation?
In the proposal/protocol, a 5% significance level was used in the sample size calculation 94.4% of the time (102/108). Eighty per cent power was specified in half of the protocols (52.2%, 59/113) and 90% power was specified in over one-third (37.1%, 42/113). The triallists inflated the sample size for participant loss to follow-up 61.5% of the time (72/122) in the protocol/proposal and 48.3% of the time (58/120) in the monograph (Table 38).
| Component of sample size calculation | Protocol, n (%) | Publication, n (%) |
|---|---|---|
| Alpha | ||
| 5% | 102 (94.4) | 100 (91.7) |
| 1% | 3 (2.8) | 3 (2.8) |
| Other | 3 (2.8) | 6 (5.5) |
| Total | 108 | 109 |
| Power | ||
| < 80% | 1 (0.9) | 1 (0.9) |
| 80% | 59 (52.2) | 63 (55.8) |
| 81–84% | 3 (2.7) | 1 (0.9) |
| 85% | 1 (0.9) | 4 (3.5) |
| 86–89% | 2 (1.8) | 1 (0.9) |
| 90% | 42 (37.1) | 40 (35.3) |
| > 90% | 5 (4.4) | 3 (2.7) |
| Total | 113 | 113 |
| Did they consider dropout? | ||
| Yes | 72 (61.5) | 58 (48.3) |
| No | 12 (10.3) | 27 (22.5) |
| Not clear | 1 (0.8) | 0 |
| No information available | 37 (31.6) | 35 (29.2) |
| Total | 122 | 120 |
Question T4.19: other information – what graphical presentation of data was reported in Health Technology Assessment trials?
We reviewed each HTA monograph and assessed whether it included a repeated measures plot, a Kaplan–Meier plot or a forest plot, as these were the top reported figures in Pocock et al. 95 (accounting for 92% of figures published in the 77 RCT reports that they reviewed in five general medical journals). A repeated measures plot was presented in the HTA monograph for 38.4% of the trials (48/125), followed in frequency by a Kaplan–Meier plot (20%, 25/125) and a forest plot (16.8%, 21/125) (Table 39). A repeated measures plot was observed more frequently in the HTA monographs than in Pocock et al. ’s95 sample, and a Kaplan–Meier plot less often. This could be due to differences in the types of trials reviewed, with HTA trials more likely to involve a longer follow-up at multiple time points and less likely to include survival outcomes.
| Description of data | n (%) | n (%) from Pocock et al.95 |
|---|---|---|
| What type of figures was used to illustrate results? | ||
| Kaplan–Meier plot | 25 (20.0) | 32 (41.6) |
| Repeated measures plot | 48 (38.4) | 20 (26.0) |
| Forest plot | 21 (16.8) | 21 (27.3) |
| None of the above | 31 (24.9) | N/A |
| Total | 125 | 73 |
Analysis
The planned method of analysis for the primary outcome was not specified in the protocol/proposal in one-third of the 125 trials. Of those that specified a method of analysis, only two (1.8%) fully specified the method of analysis using the six core criteria. Twenty-seven per cent met three criteria (statistical test/model, significance level and analysis population). Improvements occurred over time from 22.7% before 1998 to 35.6% thereafter. There did not appear to be differences in the level of detail reported in the protocol compared with the proposal, but this could be due to small numbers or confounding (with the year the commissioning brief was advertised).
Out of the 125 trials reviewed, only 52 (41.6%) reported all the required components of the sample size calculation in both the proposal/protocol and monograph. Of these, the information in the proposal/protocol matched the information in the monograph in only 43 trials (34%) (see Tables 36 and 37). Where discrepancies were observed, they were twice as likely to indicate a smaller sample size planned in the monograph than stated as planned in the protocol.
Discussion
We were able to extract data to answer a number of questions on the planned and actual method of statistical analysis and sample size calculation. The degree to which this study was successful varied by the three broad sets of questions:
-
Questions T4.1–T4.10: did the protocol specify the planned method of analysis for the primary outcome in sufficient detail? The study indicated that this set of questions could be answered and indicated some cause for concern as around one-third of trials provided insufficient detail, particularly on planned statistical analysis.
-
Questions T4.11–T4.15: was the analysis planned in the proposal/protocol for the primary outcome carried out? We showed that it was difficult to complete this set of questions owing to lack of data.
-
Questions T4.16 and T4.17: was sufficient information on the sample size calculation provided? And does the sample size calculation in the protocol match the sample size calculation shown in the monograph? What discrepancies were found? The study showed that it was difficult to complete this set of questions owing to lack of data.
One general finding from this study relates to the limitation of retrospective analysis. Standards changed over time. We were unable to discuss details with those responsible for the analyses in the trials. In particular, we had no way of knowing if statistical analysis plans had been drawn up separately from the protocol. We understand that such plans are common practice but often not until the trial is close to completion. The key issue is that such plans are specified in advance before the data are examined. We have no way of knowing if this happened.
This is the first study we are aware of that has reviewed whether or not the method of statistical analysis was recorded in sufficient detail in the protocol, as defined by a minimum set of criteria.
Sample size calculation is a vitally important aspect of any clinical trial to ensure that the number of patients included is large enough to answer the question reliably and as few patients as possible are exposed to a potentially inferior treatment. It is important that all parameters used in the sample size calculation(s) are clearly and accurately reported in both the grant proposal/protocol and final trial publication. The level of detail reported should enable another statistician to replicate the sample size calculation if necessary. The sample size calculation reported in the final trial protocol and final publication should match and any changes to the sample size that were made after the trial had started should be reported.
We found that sample size calculation information was not being recorded in sufficient detail in both the protocol and publication for RCTs. Where the information was recorded, the level of unexplained discrepancies was surprisingly high. Changes to the sample size calculation after a trial has started are allowed for much the same reasons as listed in relation to changes to the statistical analysis plan [e.g. advances in knowledge, change in trial design or better understanding of the trial data (SD or control group event rate)], but should be minimised as much as possible.
We observed fewer discrepancies than other studies with regard to the method of statistical analyses and whether or not the authors followed the protocol or the sample size calculations. The discrepancies observed could be legitimate changes not reported in the monograph or could be hiding unacknowledged reductions in the sample size carried out after the trial started due to recruitment problems (reported in Chapter 6), or they could be evidence of selective reporting bias indicating the results to be more clinically meaningful than they were (e.g. by increasing clinically meaningful difference specified) or typographical mistakes. Of these, given the large number of trials which failed to recruit the original planned sample size as reported in Chapter 6, we think the most likely explanations are the first two listed above.
Questions T4.11–T4.15 explored potential selective reporting. We found that potential selective reporting bias in sample size calculation information and in methods of analysis exists. This is perhaps not so serious, as a previous review of a subset of the RCTs in this cohort found that only 24% of primary outcome results were statistically significant. 5 If there was selective reporting bias we might expect this percentage to be higher.
Chan et al. 88 found that the statistical test for primary outcome measures differed between the protocol and publication in 60% of trials; we found a smaller percentage in our cohort of 25%. This could be because we had access to the final version of the protocol whereas Chan et al. 88 had access to the protocol submitted to an ethics committee. In addition, Chan et al. 88 studied protocols from the 1994–5 period, before CONSORT had been developed (in 1996). Chan et al. 88 observed that 32.6% of protocols described the planned method of handling missing data, higher than our finding of 25.2%.
Chan et al. 88 found that 11 out of 62 trials (17.7%) fully and consistently reported all of the requisite components of the sample size calculation in both the protocol and publication. The corresponding figure in our sample was 34%; this is twice as large as in Chan et al. 88 but is still much lower than expected.
We found a similar proportion of trials reporting all the required sample size calculation parameters as Charles et al. . 90 Charles found that 57% of 206 trials reported all the required sample size calculation parameters; we found that 56.4% of our trials did so.
The figures in the paper by Hahn et al. 89 are similar to ours, although their studies were few and dated.
Strengths and weaknesses of the study
The biggest strength of this study was that we had access to a protocol/proposal for all the trials. This is the largest cohort study that we are aware of to have compared the method of analysis and sample size calculation planned in the protocol with that reported in a publication. This is also the first such study of UK-funded RCTs. Further, previous studies comparing protocols with publications may not reflect current practice because either the number of trials reviewed was small or the studies reviewed were relatively old (1994–5 for Chan et al. 88 and similar for Hahn et al. 89).
A limitation of our work was that we only analysed the first sample size calculation reported and compared that with the monograph.
We were surprised at the lack of detail in statistical analysis plans reported in the protocol/proposal and how few met our criteria. However, as statisticians often create statistical analysis plans separate from the protocol prior to final analysis, these may well provide more detail.
Key questions for the HTA programme concern whether or not it requires audit of planned analyses and, if so, how and at what level of detail. Our study shows the limits of retrospective audit based on the protocol/application form and the monograph. More generally, the HTA programme should consider requiring information to be recorded on the statistical test/model planned for use in the analysis, the significance level/confidence interval level to be used and the analysis population.
Recommendations for future work
Should the database be continued, we recommend that the questions on statistical analysis are reviewed alongside the SPIRIT checklist. 94 Any further data extraction should include 13 questions: four should remain as they are (T4.1, T4.2, T4.18 and T4.19) and nine should be amended (T4.3, T4.4, T4.7, T4.10, T4.11, T4.12, T4.13, T4.16 and T4.17).
We observed that if trials funded by the HTA programme are to continue to qualify as one of the four cohorts of trials included in Djulbegovic et al. ,96 then data will have to be extracted on the relevant fields. 5 Dent and Raftery5 assessed treatment success and whether or not the results were compatible with equipoise using six categories: (1) statistically significant in favour of the new treatment; (2) statistically significant in favour of the control treatment; (3) true negative; (4) truly inconclusive; (5) inconclusive in favour of the new treatment; or (6) inconclusive in favour of the control treatment. Trials were classified by comparing the 95% confidence interval for the difference in primary outcome with the difference specified in the sample size calculation. The recent Cochrane Review used data extracted for this project and combined them with the only three other similar cohorts. 96
Unanswered questions and future research
We analysed whether or not the planned analyses were carried out. We did not attempt to investigate whether or not the planned analyses were appropriate.
We compared individual components of the planned method of analysis with individual components reported in the monograph but did not calculate how often all of the components of the analysis plan matched those presented in the monograph. Again, this could be the subject of further work.
Small numbers constrained our analysis of trends in time. If the work continues, time trend analyses should be repeated and extended.
Further work could explore whether or not the amount of detail provided in the protocol on planned analyses is affected by the seniority of the statistician involved, including if he/she was a co-applicant.
Chapter 8 Theme 5: economic analysis alongside clinical trials
This chapter describes the economic evaluations carried out alongside the clinical trials using available data. The questions posed include establishing if an economic evaluation was included and, if so, what type and whether or not the type planned was the type reported. The usefulness of the BCL for economic submissions was explored along with more detailed questions regarding particular methods. A final question considered the relationships between differences in costs and benefits.
Introduction
An economic evaluation considers the costs of providing a health-care intervention relative to its benefits. It always considers at least two options, for example a new intervention versus standard care. By including both costs and benefits it addresses issues of efficiency, that is, whether or not the intervention is a good use of health-care resources. Economic evaluations calculate incremental costs and benefits, that is, the extra costs and benefits generated by the intervention compared with the comparator.
The results of economic analysis can be shown graphically on the cost-effectiveness plane (Figure 7). 97,98 This compares the incremental costs (y-axis) and incremental effects (x-axis) of an intervention with those of a comparator. The position on the cost-effectiveness plane has implications for decision-making. An intervention located in the top-left quadrant is less effective and more costly than the comparator, which should consequently be the preferred option. An intervention that lies in the bottom-right quadrant is more effective and cheaper and will be preferred. An intervention in the bottom-left quadrant is cheaper and less effective. If an intervention lies in the top-right quadrant, it is more costly and more effective. In the last two cases, the question is whether or not the intervention is cost-effective, that is, are extra benefits gained worth the extra cost (top right) or are cost savings worth the benefits forgone (bottom left). The dashed line in Figure 7 shows a series of points that all have the same incremental cost-effectiveness ratio (ICER). If the ICER represented by the dashed line is an accepted value per unit of effectiveness, then all points on or under this dashed line can be considered cost-effective.
FIGURE 7.
Cost-effectiveness acceptability plane.
Different types of economic analyses, widely described in the literature (e.g. by Drummond et al. 99), are distinguished by the approach that they take to measuring outcomes. Cost–benefit analysis assigns a monetary value to benefits. Cost–utility analysis (CUA) uses a single utility measure encompassing both duration of life and quality of life, usually quality-adjusted life-years (QALYs). Cost-effectiveness analysis uses some ‘natural’ unit of health as the outcome measure, for example life-years saved or cases detected. Cost-minimisation analysis applies if two interventions have equivalent effectiveness enabling them to be compared solely on cost. Some studies do not conduct a formal economic analysis but collect data on the resource implications of interventions. These costing-only studies do not constitute an economic evaluation but can provide useful information for decision-making.
Economic analyses are widely used in informing health-care decision-making. Examples include the National Institute for Health and Care Excellence (NICE) (www.nice.org.uk), the Scottish Intercollegiate Guidelines Network100 and the Australian Pharmaceutical Benefit Scheme. 101 A cost–utility study (cost/QALY) is favoured by NICE as it can be generalised across a range of health-care settings and interventions. 102 However, NICE also considers other factors before reaching a decision. 103,104
Little has been published on the extent and types of economic evaluations included in trials funded by the HTA programme. Further, as with statistical analyses, the intended and reported economic evaluations may differ. For example, if the health-related quality of life measure was poorly completed, a cost–utility study may not have been feasible. Alternatively, if the investigators found no difference in clinical outcomes they might not have pursued the economic evaluation. It is not clear how often such changes occurred, or for what reasons. 105,106
A 2006 study107 showed that, since 1994, approximately 30% of the economic evaluations included in the NHS Economic Evaluation Database have used data obtained from a single RCT. It is likely that more trials include economics. A review by Barber and Thompson108 found that although 45 studies published in 1995 reported patient-level cost data, only half provided results on the comparison of costs between the interventions. A later, similar review found 115 studies using patient-level data in 2003. 109
Advantages of designing economic analyses alongside clinical trials were highlighted by O’Sullivan et al. 110 First, the economic analysis benefits from the elements of trial design, such as blinding and randomisation, which reduce the potential for bias. Second, trials provide an opportunity to collect patient-level data on costs and outcomes. Third, the evaluation alongside the trial is likely to be less costly than funding a stand-alone economic evaluation.
Attention has also been drawn to problems associated with carrying out economic analyses alongside clinical trials,107,110 notably by Sculpher et al. 107 Few trials directly compare all relevant options. The maximum follow-up in the trial is often shorter than the appropriate time frame for an economic analysis. The trial setting may not be relevant to the decision being made, for instance if a trial is run in a different country or health-care setting where costs are likely to be different. Finally, trials are unlikely to use all available evidence, such as the results of other trials and epidemiological data. For these reasons, the authors suggested that the role of trials should be to provide a means for the collection of relevant economic data. Conversely, it has been argued that although problems exist, many of these can be addressed with careful design. A role remains for economic analyses alongside trials:
In sum, when the various challenges posed by piggyback evaluations are acknowledged and addressed with rigor, clinical trials can indeed be an efficient and appropriate means through which to measure the economic impact of medical interventions.
O’Sullivan et al., p. 77110
A successful economic evaluation alongside a clinical trial requires close collaboration between health economists and clinical researchers at all stages of the design and implementation process. 105,111 Since it began, the HTA programme has funded economic evaluations alongside most trials. This has helped to ensure that health economists are involved in the trial design phase, contributions are properly planned and resourced, and trial processes are designed to include relevant health economics components.
There is no established ‘gold standard’ of what constitutes a good economic study. Different organisations have produced a range of guidelines on methods. This has led to a number of checklists for economic evaluations, including the BCL of economic quality,12 the Consensus Health Economic Criteria112 Quality of Health Economic Studies113 and the Drummond checklist. 114 Of these, the BCL12 is widely used, not least as it is intended to be accessible to non-economists. Published in 1996, it was the most likely to be used.
The conclusions of the clinical study and the economic study can differ. The measure of interest in the economic evaluation, the ICER is a composite of two variables (cost and health economics outcome), and hence this may differ from the results based on clinical outcomes alone. Further, the outcome measure used in the economic analysis (e.g. QALY) is unlikely to be the primary outcome measure used in the trial. Trials are likely to be powered on the clinical primary outcome measure, so the economic evaluation may not be powered to show differences. Finally, the statistical methods differ, with clinical trials generally being assessed on the basis of statistical significance and economic evaluations on the probability that an intervention is cost-effective. We do not know how often the clinical and economic results differ, what factors this is associated with and what to do if this occurs. An examination of the congruence between clinical and economic results in the HTA programme would help to answer some of these questions.
Questions addressed
The questions for which data were extracted are listed in Box 8.
-
T5.1. What is the methodological quality of HTA economic evaluations and do they adhere to good practice guidelines for economic analysis (BCL12)?
What, if any, type of economic evaluation was included at the planning and at the reporting stages?
-
T5.2. Was an economic analysis performed?
-
T5.3. What type of economic analysis was reported at each stage?
-
T5.4. Did the planned economic evaluation match the actual evaluation?
Is the extraction of metadata on a small number of study characteristics useful in describing the HTA programme of economic analyses?
-
T5.5. Perspective.
-
T5.6. Cost year.
-
T5.7. Data analysis and interpretation (bootstrapping and CEAC).
-
T5.8. Missing data.
-
T5.9. Utility measure.
-
T5.10. QALYs.
-
T5.11. Reporting and interpreting the ICER.
-
T5.12. Can the economic results be usefully shown on the cost-effectiveness plane?
CEAC, cost-effectiveness acceptability curve.
Methods
Twelve questions were piloted as shown in Box 8. A question posed in the bid for this theme asked if the clinical and economic results agreed. We approached this by locating the results of trials on the cost-effectiveness plane.
To answer the questions set out in Box 8, data were extracted from the 109 published monographs, with the exception of the planned economic analyses, which were taken from the project protocol or, if necessary, the final application bid. For the question concerned with the cost-effectiveness plane, data were extracted by DT and checked by AY.
Quality assurance of the question regarding the BCL involved two states: identification of questions requiring judgements and review of these by a health economist (DT).
Denominators
Two different denominators were used: trials and comparisons. Questions relating to the conduct of the economic evaluation focused on eligible trials, whereas those relating to the cost-effectiveness plane looked at comparisons. Of the 125 trials, 117 (93.6%) were eligible, eight were excluded, five trials did not include an economic analysis (trials ID2, ID63, ID97, ID109 and ID112), two trials closed owing to poor recruitment (trials ID75 and ID110) and one trial reported the cost data poorly (trial ID62).
For the cost-effectiveness plane, 95 trials were included, with others excluded owing to lack of sufficient detail on both costs and clinical outcomes. Of the 95 trials included, 70 trials reported one comparison, 16 trials reported two comparisons, seven trials reported three comparisons and two trials reported four comparisons, yielding 131 comparisons.
Operationalising the BCL12 (Table 40) led to omitting one question, with regard to the congruence between clinical and economic conclusions.
| Checklist number | Description |
|---|---|
| Study design | |
| 1 | The research question is stated |
| 2 | The economic importance of the research question is stated |
| 3 | The viewpoint(s) of the analysis are clearly stated and justified |
| 4 | The rationale for choosing the alternative programmes or interventions compared is stated |
| 5 | The alternatives being compared are clearly described |
| 6 | The form of economic evaluation used is stated |
| 7 | The choice of form of economic evaluation is justified in relation to the questions addressed |
| Data collection | |
| 11 | The primary outcome measure(s) for the economic evaluation are clearly stated |
| 12 | Methods to value health states and other benefits are stated |
| 13 | Details of the subjects from whom valuations were obtained are given |
| 14 | Productivity changes (if included) are reported separately |
| 15 | The relevance of productivity changes to the study question is discussed |
| 16 | Quantities of resources are reported separately from their costs |
| 17 | Methods for the estimation of quantities and unit costs are described |
| 18 | Currency and price data are recorded |
| 19 | Details of currency of price adjustments for inflation or currency conversion are given |
| 20 | Details of any model used are given |
| 21 | Choice of model used and the key parameters on which it was based are justified |
| Analysis and interpretation of results | |
| 22 | Time horizon of costs and benefits is stated |
| 23 | The discount rate(s) is stated |
| 24 | The choice of rate(s) is justified |
| 25 | An explanation is given if the costs and benefits are not discounted |
| 26 | Details of statistical tests and confidence intervals are given for stochastic data |
| 27 | The approach to sensitivity is given |
| 28 | The choice of variables for sensitivity analysis is justified |
| 29 | The ranges over which the variables were varied are stated |
| 30 | Relevant alternatives are compared |
| 31 | Incremental analysis is reported |
| 32 | Major outcomes are presented in a disaggregated as well as aggregated form |
| 33 | The answer to the study question is given |
| 34 | Conclusions follow from the data reported |
| 35 | Conclusions are accompanied by the appropriate caveats |
Analysis of a summary of the clinical and economic conclusions showed that results were often presented in an arbitrary and incomplete way. We omitted three items from the BCL as uninformative: number 8, ‘the sources of effectiveness estimates used are stated’; number 9, ‘details of the design and results of effectiveness study are given’; and number 10, ‘details of the method of synthesis or meta-analysis of estimates are given (if based on an overview of a number of effectiveness studies)’. 12 This was because all our studies were randomised trials.
The remaining BCL questions were completed with a four-item checklist: ‘yes’, ‘no’, ‘NA [not applicable]’ or ‘not clear’. To compare the quality of different studies we compiled a numerical index of quality {[yes/(yes + no + cannot tell)] × 100}. A study with a score of 100 met all relevant criteria; a study with a score of 0 met none.
Data were extracted on costs, primary outcome and QALYs including point estimates of differences and statistical significance. Each comparison was located on two cost-effectiveness planes, one using the primary outcome, the other using QALYs, using the framework in Figure 8. Although this enables comparisons to be placed on the plane, it does not enable a distinction to be made between two comparisons placed in the same box (see Figure 8). Ranking comparisons within any one box would require ICERs for both comparisons. Furthermore, where the effectiveness measure differs between two ICERs, ranking would also require a valuation of both these effectiveness measures.
FIGURE 8.
Framework for locating results from HTA-funded clinical trials on to cost-effectiveness planes.

Results
Question T5.1: what is the methodological quality of Health Technology Assessment economic evaluations and do they adhere to good practice guidelines for economic analysis (BMJ checklist)?
Out of the 117 trials reporting an economic analysis in the published monograph, seven (6%) met/adhered to all relevant checklist items (n = 32) in the BMJ guidelines for reporting an economic analysis. 12 The interquartile range was 8 and the mean quality score was 84. The majority of trials (76%, 89/117) reported a quality score of more than 80; few trials (4%, 5/117) reported a score of less than 49.
Figure 9 shows results for the BCL for each of the 32 questions assessed here. The figure shows how many of the 117 trials were categorised as ‘yes’, ‘no’ or ‘unclear’ for each of the BCL questions. For the majority of categories, the HTA trials had a high degree of reporting completeness (i.e. most questions had a high number of ‘yes’ responses). Exceptions were in relation to the discussion of the relevance of productivity changes (discussed in around one-third of studies), details of price adjustments for inflation or currency conversions (only discussed in approximately 40%) and reporting of discounting (included in around two-thirds).
FIGURE 9.
Overall BCL quality score for trials involving an economic analysis (n = 117).
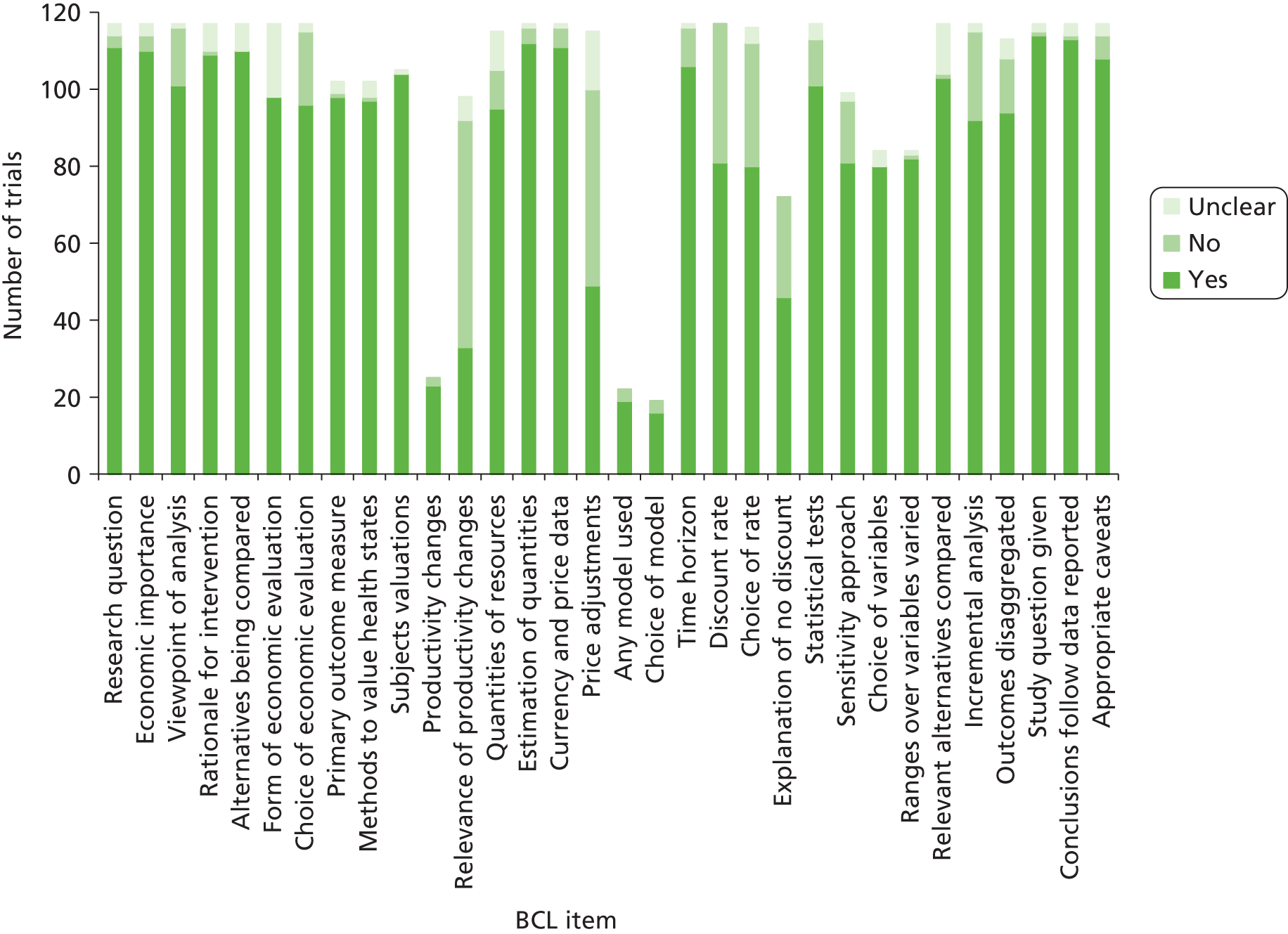
Questions T5.2–T5.4: what, if any, type of economic evaluation was included at the planning stage and at the reporting stage?
Question T5.2: was an economic analysis performed?
One hundred and seventeen of the 125 trials (93.6%) reported results of an economic analysis alongside the clinical trial. Five trials did not include an economic analysis, two were closed due to poor recruitment and the cost data were poorly reported in one.
Question T5.3: what type of economic analysis was reported at each stage?
The types of analysis planned and carried out are given in Table 41. Cost–utility and cost-effectiveness studies were planned in 70% of trials (82/117). Thirteen per cent contained insufficient information to determine study type. In some cases this arose because the type of analysis planned was stated in advance to depend on the results found. For example, a number of trials stated that a CUA would be performed depending on the outcomes.
| Type of economic analysis | Planned type, n (%) | Reported type, n (%) |
|---|---|---|
| CUA | 45 (38.5) | 60 (51.3) |
| Cost-effectiveness analysis | 37 (31.6) | 32 (27.4) |
| Cost–benefit analysis | 10 (8.5) | 1 (0.9) |
| Cost minimisation analysis | 3 (2.6) | 8 (6.8) |
| Costing only | 7 (6.0) | 15 (12.8) |
| Unclear | 15 (12.9) | 1 (0.9) |
| Total | 117 | 117 |
Three trials (ID75, ID97 and ID110) did not report that an economic evaluation had been planned.
Sixty trials (51.3%, 60/117) reported a CUA. Fewer (27.4%, 32/117) conducted a cost-effectiveness analysis than planned. Cost–benefit analysis was rare, with only one reported. Out of the 117 trials reported to have an economic analysis, 104 trials were superiority (88.9%), nine were equivalence (7.7%) and four were non-inferiority (3.4%).
The type of economic evaluation actually performed is given in Table 42, broken down by publication year. No CUAs were reported for 1999–2002, after which the proportion of these rose to around 60%.
| Utility measure | Time period (monograph publication date) | Total, N (%) | |||
|---|---|---|---|---|---|
| 1999–2002, n (%) | 2003–5, n (%) | 2006–8, n (%) | 2009–11, n (%) | ||
| CUA | 0 | 20 (57.1) | 21 (63.7) | 19 (57.5) | 60 (51.7) |
| Cost-effectiveness analysis | 7 (46.7) | 5 (14.3) | 10 (30.3) | 10 (30.3) | 32 (27.6) |
| Cost–benefit analysis | 0 | 0 | 1 (3.0) | 0 | 1 (0.9) |
| Cost minimisation analysis | 2 (13.3) | 4 (11.5) | 0 | 2 (6.1) | 8 (6.9) |
| Cost only | 6 (40.0) | 6 (17.1) | 1 (3.0) | 2 (6.1) | 15 (12.9) |
| Total | 15 (100.0) | 35 (100.0) | 33 (100.0) | 33 (100.0) | 116 (100.0) |
Question T5.4: did the planned economic evaluation match the actual evaluation?
Sixty-two trials had a discrepancy between the planned and reported type of economic analysis. Of the 15 trials where evaluation type was unclear at the planning stage, only one did not report some form of economic study. Some analyses shifted from cost-effectiveness to cost–utility. Of the 37 trials that planned a cost-effectiveness study, 15 (41%) reported a cost–utility study. Planned economic analysis was a poor predictor of the type of economic analysis carried out.
Questions T5.5–T5.11: is the extraction of metadata on a small number of study characteristics useful in describing the Health Technology Assessment programme of economic analyses?
Question T5.5: perspective
Of the 117 trials that conducted an economic analysis, 46 (39.3%) took a societal perspective, 42 (35.9%) a NHS-only perspective and 22 (18.8%) a NHS and social services perspective, with the remainder categorised as ‘other’ (2.6%, 3/117) or no perspective reported (3.4%, 4/117). Analysis of perspective over time showed no clear patterns.
Question T5.6: cost year
Data on the year that cost was incurred (needed to adjust for inflation) was well reported.
Question T5.7: data analysis and interpretation (bootstrapping and cost-effectiveness acceptability curve)
Eighty-one (69.2%) of the 117 trials reported using bootstrapping in the economic analysis and 62 (53%) presented cost-effectiveness acceptability curves (CEACs) in the reporting of the economic analysis. Over time, the reporting of CEACs became more prominent. From 2004 onwards, more than half of HTA clinical trials published presented CEACs in the reporting of the economic analysis. This increased to over 70% of published HTA clinical trials in 2009 (13/18).
Question T5.8: missing data
Almost one-third of trials (31.6%, 37/117) did not report methods for handling missing data and 19 (16.2%) did not provide sufficient information to determine whether or not they considered missing data. The remaining 61 trials (52.1%) reported how missing data were handled during the economic analysis, including multiple imputation (23%, 14/61) and complete case analysis (only including cases with complete data in the analysis) (18%, 11/61). Thirteen trials (21.3%) reported more than one method for handling missing data.
Question T5.9: utility measure
Seventy-seven trials reported the utility measure and, of these, 16 reported using two utility measures, with 93 responses from these 77 trials (Table 43).
| Utility measure | Time period (monograph publication date) | Total | |||
|---|---|---|---|---|---|
| 1999–2002 | 2003–5 | 2006–8 | 2009–11 | ||
| EQ-5D | 4 | 17 | 20 | 19 | 60 |
| SF-6D (SF-36) | 3 | 8 | 7 | 8 | 26 |
| SF-6D (SF-12) | 0 | 3 | 0 | 1 | 4 |
| HUI3 | 0 | 0 | 1 | 2 | 3 |
| Total number of utility measures | 7 | 28 | 28 | 30 | 93 |
| Total number of trials | 6 | 24 | 23 | 24 | 77 |
The European Quality of Life-5 Dimensions (EQ-5D) was by far the most common utility measure, used in 60 trials. Thirty trials used the Short Form questionnaire-6 Dimensions (SF-6D), the majority of which were derived from the Short Form questionnaire-36 items (SF-36) instrument and not the Short Form questionnaire-12 items (SF-12). No trials used direct elicitation methods (standard gamble or time trade-off). Where the EQ-5D was used as the primary instrument, it tended to be used on its own, with only one study reporting the use of the SF-6D. Ten studies using the SF-6D as the primary measure also used the EQ-5D. Two out of the three studies using the Health Utilities Index – Mark 3 (HUI3) also used the EQ-5D. The EQ-5D was often used secondary to non-EQ-5D measures, raising a question of possible redundancy.
Question T5.10: quality-adjusted life-years
More than half of the trials (56.4%, 66/117) reported an incremental QALY difference and of these 66 trials, 60 (90.9%) specified the QALY time frame, which ranged from 5 days to lifetime. The most common time frames were 12 months (28.8%, 19/66) and 24 months (15.2%, 10/66).
Question T5.11: reporting and interpreting the incremental cost-effectiveness ratio
Ninety-five of the 117 trials reported an incremental analysis in the published monograph (BCL item number 32). Additional data were extracted on the two interventions being compared and the within-trial estimate of the ICER for each comparison was reported.
Seventy trials named the two interventions being compared. Twenty-five trials reported more than one comparison, leading to 131 cost-effectiveness comparisons.
Question T5.12: can the economic results be usefully shown on the cost-effectiveness plane?
The results for clinical outcomes and cost are shown in Figure 10. A similar framework has previously been used by Briggs and O’Brien. 115 Most comparisons fell into the centre box (66/131 comparisons), that is neither costs nor effects were significantly different. Twenty comparisons fell in the top-middle box, with statistically significant differences in outcomes but statistically significantly more expensive. Sixteen fell in the middle-right box, with statistically significant improvements in effectiveness but not in costs. Comparisons in the top-right box showed statistically significant differences in both effectiveness and costs. Only four comparisons showed an unambiguous preference for one of the comparators, that is comparisons that fell in either the top-left (one comparison) or the bottom-right (three comparisons) boxes. Forty comparisons showed a statistically significant increase in costs between intervention and comparator, but only five showed a statistically significant decrease in costs. Comparisons were concentrated towards the top right of the cost-effectiveness plane. Many interventions were likely to be both more effective and more costly than comparators, even if many of these differences may be small and non-significant. The appropriate use of frequentist and Bayesian approaches might usefully be clarified by the HTA programme.
FIGURE 10.
Results of trial comparisons: incremental costs and effects (n = 131 comparisons).

A similar approach was taken to the 65 comparisons of incremental cost per QALY. Table 44 classifies studies into the boxes shown in Figure 10, dividing comparisons by ICER into those with a cost per QALY greater or less than £30,000 (the upper NICE threshold). 102 Table 44 also indicates whether the intervention dominates or was dominated by the comparator and when the intervention is cheaper but less effective.
| Cost | Effectiveness | Number | CUA comparisons | < £30,000/QALY | > £30,000/QALY | Intervention is dominated | Intervention cheaper and less effective | Intervention dominates comparator |
|---|---|---|---|---|---|---|---|---|
| Not statistically different | Not statistically different | 66 | 29 | 10 | 9 | 7 | 3 | 0 |
| Not statistically different | Statistically significantly better | 16 | 7 | 2 | 0 | 0 | 1 | 4 |
| Not statistically different | Statistically significantly worse | 3 | 3 | 2 | 0 | 1 | 0 | 0 |
| Statistically significantly more expensive | Not statistically different | 20 | 13 | 3 | 5 | 5 | 0 | 0 |
| Statistically significantly more expensive | Statistically significantly better | 20 | 9 | 7 | 2 | 0 | 0 | 0 |
| Statistically significantly more expensive | Statistically significantly worse | 1 | 1 | 0 | 0 | 1 | 0 | 0 |
| Statistically significantly cheaper | Not statistically different | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| Statistically significantly cheaper | Statistically significantly better | 3 | 2 | 0 | 0 | 0 | 0 | 2 |
| Statistically significantly cheaper | Statistically significantly worse | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 131 | 65 | 24 | 16 | 14 | 4 | 7 |
The three boxes on the right of Figure 10 are of particular interest as they indicate statistically significant improvements in outcome. We wanted to see whether or not the economic studies reached the same conclusions.
The top right box of Figure 10 had 20 comparisons with statistically significant differences for both costs and effects (line 5 in Table 44). Of these 20 comparisons, nine had cost–utility data. Seven comparisons showed ICERs less than £30,000 per QALY (trials ID29, ID77, ID107, ID31, ID39, ID69 and ID99) and two studies (ID103 and ID106) showed higher ICERs. One comparison (trial ID103) compared motivational enhancement therapy and CBT with usual care in diabetes. This found the cost per QALY at 12 months to be £312,000. However, as this study showed a reduction in glycated haemoglobin at 12 months, it is possible that a longer-term study may have shown a lower ICER. A second study (trial ID106) compared conventional ventilator support with extracorporeal membrane oxygenation for severe adult respiratory failure. This trial found an extremely high ICER of over £1.6M per QALY at 6 months but a long-term model showed an ICER of less than £20,000 per QALY.
The majority of economic analyses presented here support and reinforce the clinical conclusions, that is not only is there evidence that these interventions may be effective, there is also evidence that they may represent reasonable value for money, that is ICERs of less than £30,000 per QALY.
The second line in Table 44 shows 16 studies with no statistically significant difference in costs but in which the clinical study indicated a statistically significant difference. Cost-per-QALY results for seven of these comparisons indicated that for four out of seven comparisons (trials ID28, ID78, ID86 and ID90) the intervention dominated the control. In two comparisons (trials ID125 and ID23) the intervention generated QALYs at less than £30,000 per QALY. One study (trial ID17) showed non-significant reductions in estimated QALYs and so the point estimate for the cost–utility study lay in the south-west quadrant. As in the last example, the health economic results reinforce the clinical ones.
The most common category in Figure 10 (see Table 44) was the central box composed of those comparisons where neither cost nor outcomes of interventions were statistically significantly different from comparators. Sixty-six comparisons fell into this category. Of these, 29 also involved a cost–utility study. The CUA in 16 of these studies indicated that the intervention would not be cost-effective; of these, seven showed the intervention to be dominated and nine showed ICERs greater than £30,000 per QALY. For one-third of cost–utility studies (34.5%, 10/29), the ICER indicated that the intervention could be cost-effective even though neither cost nor clinical outcome was statistically significantly different. For studies falling into this category, the health economic component adds useful information. Not only was this the most common category, it is also one where one-third of the cost–utility studies were contradicting (to some extent) the clinical results. That is, these were cases where the clinical study had not shown a statistically significant difference and yet the cost–utility study was indicating that these were potentially cost-effective. This is because CUA can be based on differences that were not statistically significant.
The fourth line in Table 44 shows studies that were statistically significantly more expensive but also not statistically significantly more clinically effective. A priori, we would expect these to have a low probability of being cost-effective and 10 out of 13 showed CUA results that indicate either that the intervention was dominated or that it generated QALYs at greater than £30,000 per QALY. However, even in this case, there were three comparisons where the cost per QALY indicated potential cost-effectiveness (trials ID50, ID71 and ID115).
Analysis
Although the BCL12 was easy to use, many questions required some skills in health economics. It was not satisfactory as a test of methodological quality for a number of reasons. For instance, it indicated whether or not something was done rather than whether or not it was done well. Take, for example, BCL item 17: ‘Methods for the estimation of the quantities and unit costs are described’. 12 A ‘yes’ response is obtained if the methods are described. However, this does not access the underlying strength of the methodology, i.e. whether resource use was taken from patient self-report or notes, how outliers were handled, whether or not the methods of costing were robust, etc.
For the BCL to be used as a measure of quality, it would need to be completed by a health economist to assess whether or not issues were addressed well, rather than whether or not they were addressed at all. A better way to quality assure economic evaluations might be through the current peer review process. HTA trials are assessed by peer review at the proposal stage by external health economic peer reviewers as well as health economists on HTA funding boards. They are also peer reviewed as part of the monograph publication process. Either a checklist tool for health economics could be incorporated into this process, or referees could be asked to complete a more structured report.
Out of the 125 trials considered in this chapter, 117 (94%) included an economic evaluation or a costing study. Reasons for not including health economics included the trial being stopped or having an equivalence or non-inferiority design. Cost-per-QALY analysis was used in almost 60% of HTA trials after 2002. The type of economic evaluation planned and that reported sometimes differed, usually due to a switch from cost-effectiveness to cost–utility. The proportion of economic evaluations that were cost–utility studies was constant over time (after 1999–2002).
Putting the results of trial comparisons on the cost-effectiveness plane was more successful when using clinical outcomes than QALY outcomes. Most results were located in the top right quadrant (more effective, more costly), the section where health economics is most needed. These results are not useful in evaluating the results of one comparison against another, as it is not possible to make comparisons between them. For making comparisons between different trials and comparisons, the cost–utility approach is required. We were able to carry this out for 66 out of 131 comparisons (50.4%), roughly the proportion that might be expected given that about 60% of trials perform CUA. The results indicated that some interventions which failed to show differences that were statistically significant could be cost-effective within a Bayesian perspective. However, more robust results would require meta-analytic data rather than those from a single trial.
Discussion
Although we were able to apply the BCL, we query its value as it is a check of reporting completeness rather than methodological quality. It is questionable if the information obtained would be worth the effort required to extract it in future. If a formal assessment of the quality of any economic analysis is required, then the HTA programme should investigate whether or not this is achievable as part of the existing peer review process.
Only around 40% of HTA trials took a societal perspective. It is possible that important cost issues are being missed in some trials but we did not explore the reasons given for the perspective adopted.
Advanced analytic techniques such as bootstrapping and CEACs are widely and increasingly used in HTA trials. However, the issue of missing data is not handled well in most. The EQ-5D was the most widely used utility measure in HTA trials. Most studies used it as the sole preference-based utility measure. When other utility measures were used, it was usually in combination with the EQ-5D. Most studies calculated QALYs, although the time frame varied greatly between studies.
This study demonstrated that study characteristics, such as perspective, can be successfully extracted by a non-health economist. These are potentially a check on quality. We suggest that any future database should continue to extract information on the characteristics of economic evaluations.
We showed that data could be extracted on incremental costs and clinical outcomes for the majority of trials (see Question T5.12: can the economic results be usefully shown on the cost-effectiveness plane?) and inserted on to a cost-effectiveness plane. This could be done for 131 clinical outcomes, with around half of these also having cost-per-QALY estimates. This enabled identification of cases where economic and clinical studies agreed or disagreed. It would be feasible to collect simple data on results of both clinical and economic studies in future. We recommend that simple data on results be collected and used in any ongoing work.
Strengths and weaknesses of the study
Weaknesses included the use of a cohort funded over a long period of time, which may not be representative, and an inability to probe much beyond the BCL in relation to how well particular aspects were performed. Strengths included access to planned and actual analyses, and a large cohort.
Other studies have attempted to quality assess some parts of the economic analysis component of the HTA programme of trials,8 but only in relation to resource data collection, albeit in more detail than we have attempted.
Others have classified the results of the programme of HTA-funded clinical trials. Dent and Raftery5 compared 85 comparisons from 51 superiority studies from the HTA programme based around the confidence interval and included six different categories. This was a more sophisticated classification than the one used here although the current work covers more comparisons. The aim of the current approach was limited to placing results on the cost-effectiveness plane rather than analysing the pattern of HTA programme clinical results.
This study has shown the high importance placed on economic evaluations and costing alongside trials funded by the HTA programme. Almost all planned and reported on an economic analysis. Most were well reported according to the BMJ economic analysis checklist. 12 The current work supports the HTA policy of funding economic analyses alongside clinical trials. Many of the interventions evaluated in HTA trials occurred in the top right corner of the cost-effectiveness plane, i.e. they were more effective and more costly than comparators.
The current work also emphasises the importance of incorporating a cost–utility study alongside the economic evaluation. Just over 51% of trials reported a cost-per-QALY analysis. This slightly improved with later studies, but even for the period 2006–11 only around 60% of published monographs featured a CUA. Although there may be good reasons why some trials do not carry out CUA, we favour as many studies as possible doing so.
Recommendations for future work
Should the database be continued, we recommend that data should be extracted for 10 questions, nine unchanged (T5.2, T5.3, T5.4, T5.6, T5.7, T5.9, T5.10, T5.11 and T5.12) and one to be amended (T5.5).
Unanswered questions and future research
If economic evaluations are included as part of any future database, methodological quality should be considered, perhaps linked to the current refereeing process rather than a quality checklist. Data on planned and actual economic analyses should also be retained for the database and expanded where required (planned and actual perspective). Data on key methodological issues should be collected.
Chapter 9 Theme 6: the cost of randomised trials, trends and determinants
This chapter considers questions regarding the costs of RCTs funded by the HTA programme. After a brief review of the relevant literature and guidance on costing, it explores questions that might be answered using available data. It distinguishes the roles of different types of costs, particularly ‘NHS support costs’ and ‘NHS excess treatment costs’. Actual and planned costs are compared. The trend in costs over time is graphed and multivariate analysis used to identify factors associated with differences in projects’ costs.
Introduction
The sparse literature on the costs of clinical trials focuses mainly on the cost to the pharmaceutical industry of bringing a drug to market. A series of studies by DiMasi et al. 116,117 have used data supplied by pharmaceutical companies, putting the drug-to-market cost at $800M in 2000. An update put the figure at $1.2B. 118 This approach has been strongly criticised by Light and Warburton,119 who query the many assumptions involved. About half of the total cost estimate was due to the cost of clinical trials.
A literature search focused on the costs of clinical trials located 15 studies, only two of which concerned UK trials. One was a case study of a lung cancer trial in 84 centres which was delayed by major differences in approach in different centres, but did not explicitly address costs. 120 The other was a case study of the NIHR-funded Support and Assessment for Fall Emergency Referrals121 trial, evaluating different protocols for response to 999 calls involving falls by elderly persons. Factors which delayed the trial were outlined, leading to the recommendation that the NIHR be given sole responsibility for allocating research, excess treatment and service support costs. A qualitative study indicated the complexity imposed by the UK system of separately funding research, NHS support and treatment costs. This required the trial team to negotiate with multiple funders. The divisions were ‘somewhat malleable and the funding system was used differently in each trial.’122 Research governance has been blamed for delays leading to increased costs of trials. 123
An example from the USA shows that these complexities are not confined to the UK. The Comparison of Age-related Macular Degeneration Treatments Trial (CATT), funded by NIH, was delayed because of funding issues. 124,125 It is generally accepted that costs which would have been incurred in the absence of the trial should not be set against the cost of the trial. The most obvious such cost is that of treatment that would have been provided in the absence of the trial. The comparisons in CATT were between two similar drugs, bevacizumab (Avastin®, Roche) and ranibizumab (Lucentis®, Novartis), for age-related macular degeneration. As ranibizumab, unlike bevacizumab, was licensed for the condition, it was deemed the standard treatment, that is, the treatment that would have been provided in the absence of the trial. However, CATT was delayed for a year because of difficulties in getting the Center for Medical Services, which funds Medicare, to agree that the cost of ranibizumab in CATT should be seen as standard treatment and funded accordingly. Although CATT and a similar UK trial, Inhibit VEGF in Age-related choroidal Neovascularisation (IVAN), may be extreme examples (costly drugs in head-to-head trials), they illustrate both the principle of excluding the costs of normal treatment from those of trials and the difficulty of applying that principle in practice. 124,126
In both CATT and IVAN trials, the cost of both drugs had to be funded before the trials could proceed. Although companies often donate drugs free for use in trials, this did not apply in these examples as the relevant companies did not wish to support the trials.
The principle that costs which would have been incurred in the absence of the trial should not be attributed to the trial was formally stated in the UK in 1997 NHS guidance, Attributing Revenue Costs of Externally Funded Non-commercial Research in the NHS (ARCO),13 and reiterated in Attributing the Cost of Health and Social Care Research and Development (AcoRD) in 2012. 127 These distinguished research costs (costs arising from the research) from treatment costs (those costs that would have been incurred in the absence of the trial) and excess treatment costs (defined as those additional costs due to the trial). In the CATT/IVAN example, ranibizumab would be a treatment cost (as it would be used in the absence of the trial) and bevacizumab would be an excess treatment cost (as it was only used because of the trial).
The ARCO/AcoRD13,127 cost categories are defined in Table 45. Although the distinction between research and treatment costs is universal, the subdivision of research costs into those directly incurred by research and those arising from its support appears to be unique to England, and can be explained by how research funding has evolved there. Research costs relate to the costs of the research itself, such as data collection and analysis. NHS support costs cover extra patient tests and stays, as well as recruitment. These also include costs concerned with governance, obtaining consent and any additional clinic visits. Support costs since 2006 are met by the NIHR Clinical Research Network.
| Type of cost | Definitiona | Examples | Funder in non-commercial trials |
|---|---|---|---|
| Research cost | The costs of the R&D itself | Costs of data collection, analysis and other activities. Often include pay and indirect costs of staff employed to carry out the R&D, registration of trials and publication costs | NIHR research funder |
| Support cost | Additional patient-related costs associated with the research, which would end once the R&D activity in question had stopped | Might cover extra patient tests, extra inpatient days and extra nursing attention. Might also include costs of informed consent, managing and undertaking a portfolio of projects and research-active professionals employed in the NHS | NIHR Clinical Research Network |
| Treatment cost | Patient care costs which would continue to be incurred if the patient care service in question continued to be provided after the R&D activity had stopped | Cover all types of patient care services, including diagnostic, preventative, continuing care and rehabilitative care services and health promotion | NHS |
| Excess treatment costs | Where patient care is provided that is either an experimental treatment or in a different location from normal, and differs from the normal, standard treatment for that condition, the difference between the total treatment costs and the costs of the standard treatment (if any) is called excess treatment costs. These costs are part of the treatment costs | Might include the cost of a new treatment | PCTs to 2013, Clinical Commissioning Group from 2013 |
The ARCO/AcoRD guidance means that the HTA programme should fund only direct research costs, leaving support costs to be met by the NHS, mainly the hospitals or other NHS organisations within which the research occurred. The rationale for this was that a large part of the NHS R&D budget has historically been held by NHS trusts for research support activities. Since 1996, these have gone under various titles (Culyer, Budget 2, Support for Science). 128 Following the establishment of NIHR in 2006, these costs were to be met by the new NIHR Clinical Research Network.
The definition of excess treatment costs set out by ARCO13 meant that these had to be met by local NHS funders, various health authorities (until their abolition in 1998), PCTs (until their abolition in 2013) and Clinical Commissioning Groups from 2013. Excess treatment costs, according to ARCO, had to be met by the NHS (although given that these were extra costs resulting from research, the rationale for having the NHS meet them was unclear). In those instances where the excess treatment costs were lower than those of the intervention it was being compared with (i.e. the control), this might not pose a problem as the trial would reduce costs. However, problems might well arise if excess treatment costs exceeded treatment costs, with the increased cost falling on the local NHS.
In summary, the full cost of a clinical trial in the NHS should be the sum of the research cost, the service support cost and the excess treatment cost. Any treatment costs which would have been incurred in the absence of the trial should be met by the NHS and not included in the cost of the trial.
As the HTA programme preceded ARCO, it had to devise its own solutions to ensure that trials had the necessary funding. These might have sometimes involved funding what later came to be known as NHS support costs and excess treatment costs. NHS support costs should pay for research nurses to identify and recruit patients, but these might have sometimes been funded by the research grant. Excess treatment costs might have sometimes been funded, notably in trials of services not routinely provided by the NHS, such as CBT (trials ID3, ID4, ID53, ID60, ID76 and ID103), acupuncture (trials ID29 and ID39) or other novel therapies such as larval therapy (trial ID94). This was less of a problem for low-cost interventions, but could pose serious problems when the cost of the intervention was large and had to be met by the NHS.
Although the HTA programme was responsible only for the research cost, its application forms required data on other costs from 2000. This was the result of one particular trial experiencing problems due to the magnitude of support costs and excess treatment costs. Progress depended on agreement on who should fund them. From 1998, the HTA programme required applicants to estimate the service support and excess treatment costs incurred by their trial. It also required that bids which included either NHS support costs or excess treatment costs should be itemised and signed off by a clinical director, R&D manager or chief executive of the NHS trust.
However, these estimates were planned, not actual, costs. One of the surprising early findings of the current work was that no estimates were available of the actual support costs incurred by the Clinical Research Networks in supporting HTA programme trials. We had intended to cross-check the data on support costs in the application form with data from NHS trusts (or later, the Clinical Research Networks), but this was not possible as neither hospitals nor the networks collect or compile such data. The same applies to excess treatment costs, although estimates of those actually incurred could be estimated on a trial-by-trial basis, based on the numbers actually recruited.
Questions addressed
The aim was to review available cost data for published HTA-funded clinical trials, by attempting to answer specific questions concerning what the data show on the different types of cost (research, research support and excess treatment costs); how the planned and actual costs relate; the cost of additional elements in the projects; whether or not costs are increasing; and what factors determine differences in costs. Box 9 shows the questions explored.
-
T6.1. What do available data show regarding research, research support and excess treatment costs?
-
T6.2. What is the relationship between planned and actual costs?
-
T6.3. What was the cost of additional elements, such as economic and statistical analysis, within clinical trials?
-
T6.4. What is the trend in time for the costs of HTA-funded clinical trials?
-
T6.5. What factors help explain variations in the cost of individual trials?
-
T6.6. What is the cost per patient per year?
Methods
Six questions were piloted, as shown in Box 9. An attempt was made to record cost data under the five headings on the application form – staff, travel, consumables, exceptional items and equipment – and the 40% overhead allowed on staff costs. Data extracted on a pilot basis under these headings proved unhelpful in distinguishing types of costs for the early projects. The alternative approach was adopted of searching under each heading for support costs, excess treatment costs, health economics or statistical consultancy. Putative support costs were reviewed against the ARCO definition of support costs. 13 Any bid for research nurses to carry out such tasks was deemed a service support cost. Similarly, with excess treatment costs, each application for funding intervention(s) was assessed against the ARCO definition.
From 1998, data were required in applications on support and treatment costs. Appendix A in the HTA programme grant application form required data on two items: ‘service support costs’ and ‘excess treatment cost’. (The form changed slightly over time. Appendix A in the HTA programme grant application form from 1998 required data on two items: ‘service support costs’ and ‘treatment costs’, retitled in 1999 as ‘excess costs’ and again in 2000 as ‘excess treatment costs’.) These headings were extracted in addition to any such costs already identified in the HTA application form.
The exclusion criteria (Table 46) led to a total of 14 projects being excluded. Two were not trials (ID35) or were marginally concerned with trials (ID67), four were pilot studies (ID15, ID78, ID97 and ID121) and one trial was abandoned (ID75).
| Exclusion criteria | Trial IDs of those excluded | Number of projects |
|---|---|---|
| Monographs | 109 | |
| Not a RCT, or included small RCT | ID135, ID67 | 107 |
| Pilot studies | ID15, ID121, ID97, ID78 | 103 |
| Abandoned trials | ID75 | 102 |
| Overhead on staff not 40% | ID12, ID29, ID64, ID71, ID109, ID112, ID115 | 95 |
Seven further projects were excluded on the basis of non-standard overheads; either a lower than usual overhead was charged (trials ID12, ID29, ID64 and ID71) or the overhead was replaced by full economic costing (FEC) introduced in 2006 (trials ID109, ID112 and ID115). The total number of projects finally included for costing was 95. For analysis of trends and variances, a further 10 multitrial projects were excluded (ID3 and ID4; ID13 and ID14; ID24 and ID25; ID128, ID129 and ID134; ID35, ID36 and ID37; ID54, ID55 and ID56; ID48, ID49 and ID50; ID51 and ID52; ID69 and ID70; ID131, ID132 and ID133), reducing the total to 85 trials.
The planned cost referred to the research cost agreed at the time when the trial officially started. Many trials changed their start dates owing to delays in ethical approval or staff recruitment. Contract variation applied if the change of start date was more than 1 month. We took the research cost at the time of commencement of the trial and extracted data automatically from the HTA MIS. As many trials apply successfully for extensions with additional research costs, actual costs often exceed those planned. The cost of these extensions is included in the final cost. The data fields used to extract data from the HTA MIS for the planned and actual cost of the trial were verified by the HTA finance department.
The HTA application form evolved over time with variations in the questions asked about costs.
Data on costs were extracted under the following headings:
-
research cost, planned as outlined in the application form, with any amendments, along with any items that could be termed support costs (specifying whether funded by the HTA programme or separately by the NHS)
-
excess treatment costs (with any appropriate detail on type), again specifying whether funded by the HTA programme or the NHS
-
health economics, statistics, surveys, qualitative research or other additional items, noting any costs attributed to them in the application form
-
actual cost of the project from NETSCC records.
We reported whether or not the application form requested the following information:
-
service support costs
-
treatment costs
-
excess treatment costs.
For final application forms submitted from October 1994 up to and including February 1998, no information about treatment cost or service support costs was requested. For these trials, the ‘no information available’ code entry was used from the standardised ‘yes’/‘no’ table. However, during the period from March 1998 to January 1999 there were two versions of the application form being submitted. Although version 2 included support costs, errors were found in the application form section for applicants to provide cost data on support costs, treatment costs and excess treatment costs. Version 3 was therefore implemented and in use from June 2000 onwards.
For those application forms submitted post 1998, the ‘no information available’ code entry was used to refer to projects where we were unable to retrieve the full application form.
More than half of these projects (56.3%, 9/16) were based in a primary care setting using primary care networks, made up of ‘research practices’. The remaining projects were divided, with 5 out of 16 (31.3%) in secondary care and 2 out of 16 (12.4%) in both primary and secondary care.
Costs in different years were expressed in 2010 prices using the Higher Education Institutions pay expenditure index. 129 This was chosen as most of the funding awards were to universities. A comparison with the NHS price index showed little difference between the two indices.
Data were reviewed on an Excel spreadsheet and examined for gross outliers. Two very low planned/actual cost estimates were investigated (trials ID87 and ID100), shown to be incorrect from the application form and corrected.
Denominators
The unit of analysis depended on the question. For some it was the project, for others the trial. The 109 projects included 125 trials. For analysis of trend and variance (including two questions: cost factors and patient cost), the analysis was single-trial projects. Examples of multitrial projects include one that randomised adults and children separately to different dosages (trial ID13). Other trials randomised mild, moderate and severe patients separately to different interventions (trials ID128, ID129 and ID130). Had the interventions not been different for each of these groups, the trials would have been single trials with stratification by adults/children or by severity.
Results
Question T6.1: what do the available data show regarding research, research support and excess treatment costs?
The mean total cost per included project, along with its breakdown by support and treatment cost, is shown in Figure 11. This varied between £0.5M and £2.4M at current prices between 1995 and 2005. In all years but 2000, which had a peak of £2.4M, the mean total was below £1.6M. The peak in 2000 was due to one project with particularly high treatment costs. Leaving 2000 aside, the trend in mean total cost was upwards, with 4 out of 5 years after 2000 above £1M, and none above that figure before 2000.
FIGURE 11.
Health Technology Assessment-funded projects 1995–2005, by cost component (HTA research cost, HTA-funded support cost, HTA-funded excess treatment cost, externally funded support or externally funded excess treatment cost). For abbreviations see Table 47 (current prices).
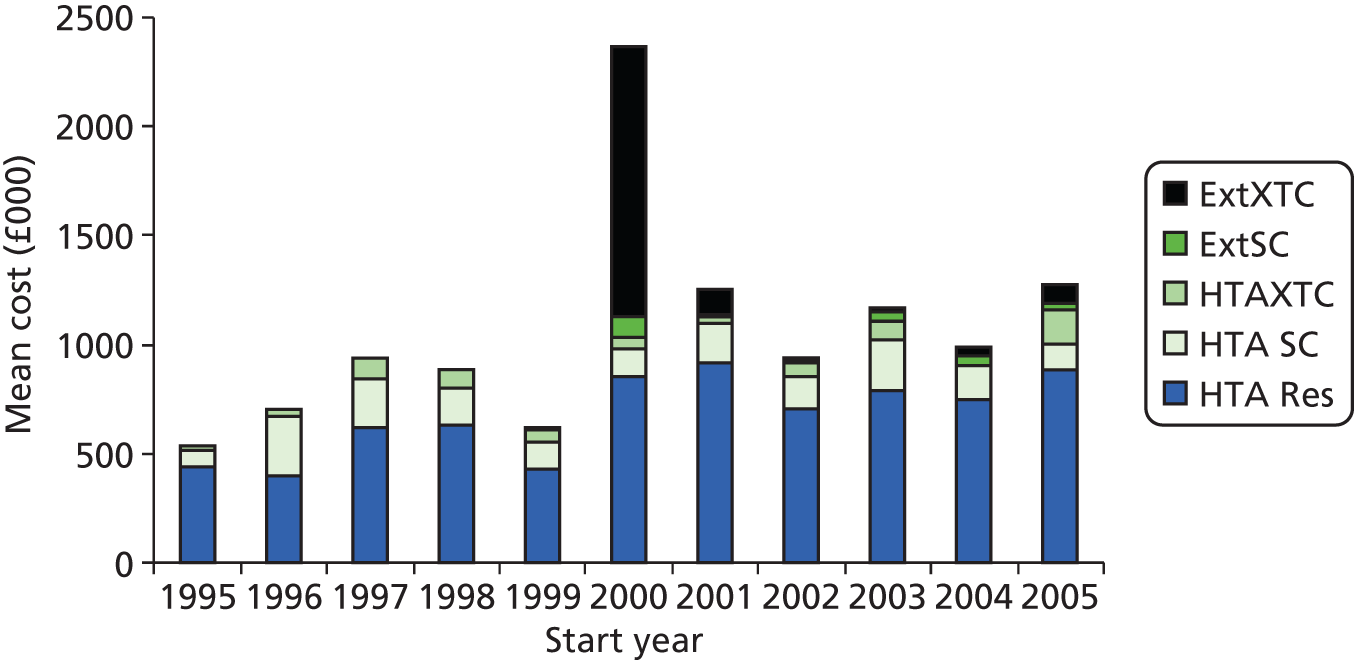
Research cost, as funded by the HTA programme, accounted for some 70% of the total cost in every year but 2000. Support costs were higher than treatment costs in all years but 2000 (Table 47).
| Cost category | Mean cost by year (£) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1995 | 1996 | 1997 | 1998 | 1999 | 2000 | 2001 | 2002 | 2003 | 2004 | 2005 | Total | |
| HTA research cost | 444,374 | 392,363 | 617,124 | 633,267 | 433,273 | 855,248 | 914,004 | 704,555 | 792,995 | 750,972 | 884,308 | 7,422,483 |
| HTA support costa | 68,914 | 282,801 | 226,038 | 168,146 | 122,790 | 119,187 | 186,132 | 148,441 | 228,108 | 158,832 | 118,151 | 1,827,539 |
| HTA excess treatment costb | 23,681 | 28,701 | 93,282 | 79,021 | 49,681 | 52,948 | 29,476 | 61,096 | 84,215 | 0 | 161,172 | 663,273 |
| Extra support costc | 0 | 0 | 0 | 0 | 0 | 105,001 | 9711 | 8566 | 44,220 | 34,202 | 22,427 | 224,128 |
| Extra excess treatment costd | 0 | 0 | 0 | 0 | 2171 | 1,234,253 | 116,521 | 6581 | 23,310 | 49,417 | 90,707 | 1,522,960 |
| Total | 536,968 | 703,866 | 936,444 | 880,435 | 607,915 | 2,366,637 | 1,255,844 | 929,240 | 1,172,847 | 993,424 | 1,276,765 | |
The HTA programme funded what appear to be support costs in 65 out of 95 projects, or roughly two-thirds. These costs amounted to almost half (45.4%) of the planned cost on average for those projects (mean cost £560,000). Some of these were concerned with trials based in primary care, which included payment to GP practices for their involvement, which could be seen as a form of NHS support cost. Out of the 95 projects, 16 (16.8%) included funding to GPs.
Excess treatment costs were also funded nearly as often by the programme (40/95 projects, or just over 40%) and accounted for a similar share of planned cost: £144,033 against a mean of £346,664 (or 41.5%).
When support and excess treatment costs were funded by the NHS, the following applied.
Support costs were noted in 13 projects, with a mean planned cost of £125,808 and externally funded support costs of £155,236, or 123% of the direct research cost funded by the HTA programme. NHS-funded excess treatment costs were noted for 19 projects, with a mean cost of £165,426 and a mean excess treatment cost of £786,977, or 476%. This included one outlier with excess treatment costs of £12M. After its exclusion, the mean project cost was £150,261, with a mean excess treatment cost of £157,966, or 123%.
The outlier in 2000 was trial ID106 [Conventional ventilator support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR)]. This project was commissioned on request by the HTA programme for the National Specialist Commissioning Advisory Group, which funded the excess treatment costs.
Question T6.2: what is the relationship between planned and actual costs?
The planned and actual mean project research cost, as funded by the HTA programme, is shown in Figure 12 for each project by year of funding. The mean cost in 2010 prices rose from around £0.5M in 1994 to just over £1M in 2000, after which it fell back to between £0.7M and £0.8M. A wide dispersion existed around these means (see Figure 12).
FIGURE 12.
Planned and actual mean project cost, by start year, 1995–2005.
The actual cost exceeded the planned cost in almost all instances, with a mean difference of 21% (range 0–74%). Of the 95 projects, 74 (78%) had actual costs that exceeded the planned cost. Of these, 54 exceeded by 10% or more, 11 exceeded by 50% or more but none exceeded by 100%. The relationship between planned and actual mean cost of HTA-funded clinical trial projects, by year, shows the actual higher than the planned each year, with the difference widening up to 2002 and then declining slightly (see Figure 12).
Question T6.3: what was the cost of additional elements, such as economic and statistical analysis within clinical trials?
The contribution of health economics was separately identified in 65 projects. It had a mean cost of £47,618 compared with the mean HTA programme-funded cost per project of £577,798, or 8.2%. The projects that did not itemise health economics costs provided that input by other means, often by having a health economist funded as a co-applicant.
Similarly, the contribution of statistician input, which was identified in 43 projects, had a mean cost of £34,251 compared with the mean HTA programme-funded cost per project of £377,699, or 9.1%.
Question T6.4: what is the trend in time for the costs of Health Technology Assessment-funded clinical trials?
The actual cost to the HTA programme of its funded projects in constant 2010 prices (Figure 13) varied widely by project but with an upwards, if uneven, trend. The range of project costs was from £0.25M to almost £2.5M, or 10-fold. The mean cost rose each year from around £0.5M in 1993 to £1.2M in 1998, after which it fell slightly and levelled off at just over £1M.
FIGURE 13.
Health Technology Assessment trial mean and individual project costs, by start year, 1995–2005.

The effect of including externally funded support and excess treatment costs in the HTA programme costed annual averages is shown in Table 48 to have relatively little impact on the mean cost of HTA-funded projects from 1997, with the exception of 2000, when the ratio of total to research costs jumped. This, however, was due almost entirely to the impact of a single project which had very large excess treatment costs (trial ID106). For the other years after 1997, the ratio ranged between 106% and 135%.
| Year | HTA cost (£) | Total cost (£) | Ratio of total to HTA (%) |
|---|---|---|---|
| 1995 | 536,968 | 536,968 | 100 |
| 1996 | 703,866 | 703,866 | 100 |
| 1997 | 936,444 | 936,444 | 100 |
| 1998 | 880,435 | 880,435 | 100 |
| 1999 | 605,744 | 607,915 | 100 |
| 2000 | 1,027,382 | 2,366,637 | 230 |
| 2001 | 1,129,612 | 1,255,844 | 111 |
| 2002 | 914,092 | 929,240 | 102 |
| 2003 | 1,105,317 | 1,172,847 | 106 |
| 2004 | 909,804 | 993,424 | 109 |
| 2005 | 1,163,631 | 1,276,765 | 110 |
The mean cost per year is shown in Figure 14, with the dots representing mean values weighted by the number of project trials starting in each year. The progressively increasing costs after 2000 look now more like a series of continuously increasing total project costs over time.
FIGURE 14.
Health Technology Assessment-funded project costs, 1995–2005. Mean cost in 2010 prices is shown, with the size of each dot proportional to the number of projects started by year.

Question T6.5: what factors help explain variations in the cost of individual trials?
The three different definitions of cost led to three analyses: planned project costs to the HTA programme, actual costs to the HTA programme and a wider definition made up of actual costs to the HTA programme plus externally funded planned support and excess treatment costs. As this wider definition of costs was the most comprehensive, and the findings were similar for the other cost definitions, only the results of the widest definition are reported here.
Univariate analysis of costs (wide definition) indicated correlations with recruitment, number of centres and duration of trial. Multivariate regression, based on transforming the skewed distribution of costs, used generalised linear models (GLMs) to explore how six different models fitted the data (combination of identity, square root or log link functions, normal or gamma distributions).
Fourteen variables were included after three had been excluded because of collinearity (care in both primary and secondary settings, GP practice and UKCRC Health Services category).
A square root transformation of cost data produced the distribution closest to normal (lowest chi-squared) compared with other common transformations, yet not achieving normality. For the GLMs estimated, the Akaike information criterion (AIC) was used as a measure of goodness of fit (based on information loss after model fit; the lower, the better). The square root link function under a normal distribution seemed to fit the data well (AIC=28.76); however, the identity link function under the normal distribution (equivalent to common ordinary least squares) produced a slightly lower loss of data than the rest (AIC=28.70) (Table 49).
| Link distribution | Square root (normal) | Identity (normal) | Log (normal) | Square root (gamma) | Identity (gamma) | Log (gamma) |
|---|---|---|---|---|---|---|
| AIC | 28.758 | 28.704 | 28.758 | 29.414 | 29.400 | 29.464 |
The coefficients and their statistical significance identified two parameters – the actual number of centres and trial duration – as statistically significant in all six models. A few additional explanatory variables were also significant in some but not all models. The proportion of the variance explained in the best models was 40% (R2 = 0.40). The relation between the cost predicted by the best model and actual costs (wide definition) is shown in Figure 15.
FIGURE 15.
Actual and predicted HTA project costs (wide definition).

Question T6.6: what is the cost per patient per year?
As the cost per patient recruited is sometimes considered for benchmarking, these data are analysed here. As a comparison of cost per patient recruited was very little different using HTA actual costs or using the wider definition (HTA actual plus planned service support and excess treatment costs), only the latter is reported.
The mean costs, along with minimum and maximum values, are shown in Table 50 for the same 84 trials analysed above (excluding one outlier, trial ID106). The same data are shown in Figure 16 along with outliers and one SD from the mean. The mean cost per patient rose in constant 2010 prices from under £1500 in 1995 and 1996 to over £2500 in each of 1997 and 1998, with a fall to just under £2000 in 1999. From 2000, it exceeded £3000 in all years except 2005, when it was just over £2000. The range of the mean cost per patient was wide, as shown in Figure 16.
| Actual start year | Number of trial | Mean cost per patient (£) | Minimum (£) | Maximum (£) |
|---|---|---|---|---|
| 1995 | 7 | 1220 | 124 | 1900 |
| 1996 | 6 | 1490 | 309 | 2472 |
| 1997 | 4 | 2755 | 539 | 6457 |
| 1998 | 9 | 2971 | 826 | 9199 |
| 1999 | 13 | 1853 | 207 | 4909 |
| 2000 | 10 | 3907 | 43 | 15,295 |
| 2001 | 8 | 3451 | 97 | 16,510 |
| 2002 | 9 | 4830 | 995 | 10,457 |
| 2003 | 10 | 3228 | 1475 | 5552 |
| 2004 | 4 | 3741 | 568 | 5618 |
| 2005 | 4 | 2206 | 24 | 4476 |
FIGURE 16.
Mean cost (wide definition) per patient recruited to HTA-funded projects, 1995–2005 (n = 84), with one SD and outliers.
Analysis
The feasibility study was helpful in refining one question and dropping the question regarding payments to centres. Consideration might be given, should the database continue, to exploring inclusion of data on how centres are remunerated for recruitment to clinical trials funded later than those reviewed here.
The data on costing of projects were complex owing to the HTA programme only funding one element, the research cost, with the support costs and treatment costs each funded by different bodies. Projects varied, with around half incurring neither support nor excess treatment costs. For those that did, these costs accounted for around 40% of the HTA grant. Although the HTA programme funding was confined to research costs, it sometimes funded what appears to be support and/or excess treatment costs according to the ARCO definitions. 13 From 1998, the HTA application form began to collect data on externally funded support and excess treatment costs, but only on planned costs. The HTA programme continued to fund what appear to be support and excess treatment costs, but interpretation of these depends on the nuances of the ARCO rules.
Data on actual and planned costs were available only for research costs, with no data on actual costs available for support or excess treatment costs. The actual research cost exceeded the planned cost in 78% of projects, with a mean excess of 25% for those projects and 21% for all projects. All cost increases were agreed with the HTA programme secretariat.
Data on the cost of health economics and statistical advice, although not required on the application form, are often bid for on a consultancy basis. The data for those projects that itemised these costs put the cost of health economics at 8% of research costs and the equivalent figure for statistics was 9%.
The cost of HTA-funded projects varied widely, as shown by a wide spread around the mean value each year, but with an increase each year from 1993 to 1998, after which the mean value fell slightly and levelled off. Inclusion of externally funded planned support and treatment costs increased the totals for years after 1998, but, with the exception of one project with very high cost in 2000, made little difference to the trend over time.
In all multivariate analyses, two factors were shown to be statistically significant: the number of centres and the duration of the trial. The proportion of variance explained – at best 42% – was similar but slightly lower for the narrowed cost definitions. Further consideration of the use of the model for benchmarking the cost of bids to the HTA programme seems worthwhile.
The mean cost per patient rose most years to a peak of just under £5000 in 2002, after which it fell to just over £2200 in 2005. The dispersion around the mean was wide. The number of patients was shown not to be a statistically significant predictor of the total cost of the trial in the multivariate analysis. This may be due to the heterogeneity of the trials analysed. Cost per patient may be more useful for comparing trial costs within more homogeneous trials.
Discussion
If further work is to continue to collect data on HTA programme-funded trial-related projects, it should collect data on all the relevant costs, including NHS support and excess treatment costs. Although the externally funded support and excess treatment costs in the projects analysed were as planned in the HTA application forms, rather than actual costs, at some stage actual support costs (and perhaps excess treatment costs) may become available. Similarly, the analyses presented above predated the change from overheads to FEC, with three projects being excluded on account of having used FEC. The most appropriate way to extract FEC data should be piloted on a selection of projects and the results integrated into the database.
The collection of data on the cost of health economics and statistics could be dropped as these elements are relatively small and difficult to extract. The finding that each of health economics and statistics cost, on average, around 10% of the HTA grant might be used to benchmark costs.
More generally, the findings that the main predictors of the cost of the HTA grant were number of centres and duration of project might be used to provide predicted costs for use in benchmarking bids.
Strengths and weaknesses of the study
Weaknesses surrounding this theme relate mainly to the complexity of the cost data. Projects were very heterogeneous in terms of their costs, with some involving large and others no support/excess treatment costs. The interplay between ARCO guidance and practice was unclear. Only planned, not actual, costs were available for both externally funded support costs and excess treatment costs.
We know of no other such study.
Recommendations for future work
Any further work should extract data on the cost of trials on four questions: two as they currently are (T6.3 and T6.4) and two to be amended (T6.1 and T6.2).
Full costing data were not requested to 1998 and remained planned rather than actual. The lack of any facility to monitor and manage support costs inevitably reduces the power of both the HTA programme and researchers to manage the projects. Provision of actual support costs seems a priority.
Unanswered questions and future research
The heterogeneity of trials and the implication of this for cost analysis remains a topic to be explored in future research.
Chapter 10 Discussion of main findings
Introduction
This chapter discusses the main findings in relation to the six themes and identifies which questions can readily be answered should the database continue. It considers the strengths and limitations of the study before considering how it might be used by other funders of randomised trials.
Before discussing the implications of the pilot, one general point should be made. Neither the ISRCTN nor the titles of the projects proved reliable in identifying RCTs. Although titles may have improved owing to the CONSORT requirements44 for titles, our initial reliance on titles led to considerable delays. The HTA programme and NETSCC should consider its policy on how the trials it funds can best be identified. Enforcement of the CONSORT requirement that the title should include the words ‘randomised trial’ would go some of the way, but not sufficiently unless an indication is also given when the project includes more than one RCT.
The review of the extent to which the trials could be seen as meeting the needs of the NHS (see Chapter 4) showed that basic data were lacking. Improved ways are required of recording the source of commissioned topics, how they are prioritised and how they have an impact on the NHS. This requires clarification by the programme as to how this might best be recorded. One way of assessing the importance of the trial to the NHS might be through assessing its impact on guidelines such as those issued by NICE, and on meta-analyses such as those of the Cochrane Collaboration. Work is ongoing assessing the impact of particular trials on relevant meta-analyses,130 and their impact on guidelines.
Trials were shown to comply with the CONSORT reporting requirements,44 at least in general. Adherence to those sections of the CONSORT checklist examined was fairly high, but with some exceptions, including a lack of detail on interventions, prespecified outcomes and the basis for the sample size calculation. About one-third of trials failed on each of these. The number of primary outcomes changed in 27 trials (in half the number increased and in half it decreased). The time point at which the primary outcome was measured was not specified in 45% of proposals and 40% of monograph reports. Although these weaknesses were mainly in early trials, they continued to appear in those that were funded later.
Almost half of trials conducted pilots but few had had feasibility studies. Around half of all interventions were substitutes for standard care and about one-third were add-ons. Almost two-thirds (64%) of the controls were standard care, but one-third of controls could not be classified.
Most trials were conducted in line with the protocol and followed both the study framework and the planned type of comparison. For those variations from the protocol that could cause bias, the results compare well with those reported for other cohorts of trials, such as in the study by Chan et al. 10,11,88 This may reflect the monitoring programme and publication requirements of the HTA programme.
On average, trials needed about twice as many centres as planned to complete the study, but this may reflect the practical difficulty of running large trials.
Chapter 5 showed that although reporting checklists such as CONSORT44 and ICH131 can be operationalised, assessing compliance is difficult. Compliance in terms of ‘yes/no’ often required closer examination, expertise and interpretation. Although a ‘yes/no’ response in an author’s checklist may provide a useful reminder, it is difficult for an outside assessor to interpret compliance without further detail. Comparing planned and reported analyses showed that a higher proportion of the HTA programme’s trials reported as planned than in the cohort examined by Chan et al. 10,88 Another key point was that many ‘audit’-type questions could not be answered owing to incomplete data. If audit is required, this study shows that it cannot be achieved retrospectively from published reports. The HTA programme needs to decide if it wants to assess compliance with CONSORT and other relevant checklists, such as the PRECIS tool,48 as well as the extent to which it wishes to compare planned and actual reported analyses.
The performance of trials centres on recruitment (see Chapter 6), is almost always underachieved. Modelling of the factors linked to recruitment was shown to be rare. Estimates of recruitment were almost always overoptimistic. However, the development of the NIHR Clinical Research Network (www.crncc.nihr.ac.uk/), with responsibility for recruitment, happened after most of the trials included in this study had started. Any future questions to do with recruitment need to be framed with the role of those networks in mind.
The statistical analysis of primary outcomes (see Chapter 7) had to be limited as many trials had more than one primary outcome. This was surprising and may be something the HTA programme should review. Comparing planned and actual statistical analyses of the primary outcomes proved difficult and required statistical expertise. More importantly, a key document, the statistical plan, was not available. Such plans are not included in either the bid or the protocol, but are often prepared close to the end of the trial but before unblinding. If analysis of the statistical analysis is required, then such plans will have to be collected.
The review of economic evaluation (see Chapter 8) showed that it was included in almost all trials funded by the HTA programme. The BCL12 was silent on many of the more recent developments in cost-effectiveness analysis. Further, it could only indicate if something had been done, not how well it was done. Much more detail would be required to establish the latter. This chapter showed the value of separate presentation of differences in costs and outcomes. A previous analysis (by LS and JR) of the first 65 HTA-published trials found no statistically significant difference in their primary outcomes in two-thirds. 5 This work led to inclusion of those trials as one of four cohorts in an international review of old versus new interventions in trials. 96 (This Cochrane Review showed that it identified four such cohorts globally, one based on US NIH cancer trials, one on 30 UK MRC trials and another on a cohort of 30 Canadian trials. The fourth cohort was made up of some 60 HTA-funded trials based on the previous work by JR and LS.) One reason for continuing the metadata database would be to enable that analysis to be updated and for ongoing comparisons to be made.
The analysis of the cost of trials (see Chapter 9) showed that basic data were absent, notably on NHS support and excess treatment costs. Without such data, the total cost of RCTs cannot be determined. Only estimates of planned support and treatment costs are available. A key issue for the HTA programme involves collection of actual as opposed to planned NHS support and excess treatment cost data. As with data on recruitment, closer links with the NIHR Clinical Research Network seem appropriate.
The overarching aim of this study was to assess the extent to which the HTA programme trials were helping to meet the needs of the NHS with scientifically robust evidence. The results indicate more difficulties in answering questions concerned with meeting the needs of the NHS than with scientific evidence. This is to some extent inevitable. Although data could be improved on how trials aimed to meet the needs of the NHS, answering this question properly requires the trial to have completed. Assessing the scientific quality of the trials is, by contrast, an easier task.
Continuation of the database?
We recommend that the answers to the recommended questions be updated as more trials are completed and that the included trials be extended to all those funded by NIHR research programmes.
A key question relates to whether or not the metadata database should be maintained and, if so, by who and with what terms of reference. We recommend that it continue to be maintained on the grounds of the relevance to the programme of the questions that were shown to be answerable. If it were to be continued, this study has shown which questions should be included in any further database.
Limitations of the study
Limitations include:
-
this review being limited to a cohort of HTA programme projects funded between 1993 and 2003, which may be atypical of trials funded later, mainly as these included a new researcher-initiated work stream
-
reliance, for around half of the included trials, on data from application forms rather than protocols
-
reliance on ‘insiders’ (several of the research team are or were NETSCC employees) who may have biases (offset to some extent by the advisory group and by quality assurance)
-
reliance on a fairly limited number of questions determined largely by data availability.
The strengths largely mirror the weaknesses:
-
Given that some 220 similar trials were funded by the HTA programme by the end of 2011 using similar criteria, the trials included in this database are likely to be typical of the programme, if not more generally.
-
Although the application form was the source for around half the included trials, protocols were increasingly common from 2000. Although the application form changed over time, those changes were confined to costs and involved the addition of questions rather than changes to existing questions.
-
The authors were well informed because they were ‘insiders’ with good access to documents and detailed working knowledge of many of the trials. This enabled a focus on the data required to answer particular questions, which might have delayed a less experienced team.
-
Finally, the database is unique in going well beyond the metadata in trial registries to include source of topic, adherence to protocol, planned and actual recruitment, quality of statistical and economic analyses and costs.
Implications for other funders
The findings of this study on the kinds of metadata that can be collected has implications for other research funders (research councils, medical charities), most obviously other NIHR research programmes but also other UK funders as well as funders in other countries.
The NIHR funds RCTs through programmes other than the HTA programme, including the Public Health Research (PHR), Efficacy and Mechanism Evaluation (EME) and Health Services and Delivery Research (HS&DR) programmes (all managed by NETSCC), as well as the Programme Grants for Applied Research (PGfAR). As the NIHR Journals Library will publish these, data will be available. A strong case exists for the database covering all NIHR-funded trials. However, given the different types of trial (EME funding early-stage RCTs, and PGfAR funding pilots and feasibility studies as well as RCTs), the types of questions may need to be revisited.
Other UK funders include the medical charities and the MRC; trials funded by each of these tend to be earlier stage than those of the HTA programme. As with such trials funded by NIHR, some revision may be required if the database is to cover these.
International funders, particularly in the USA, given the requirement for the results of trials to be registered, may be able to benefit from the work reported here. This may apply to large pragmatic trials of the sort likely to be funded by the Patient-Centered Outcomes Research Institute (www.pcori.org/).
Further research/implementation
Further research might usefully:
-
Compare this portfolio of HTA-funded trials with trials funded by others. Extension of the database to include all NIHR-funded trials would be an obvious step, as would extending it to include those trials funded by charities, the second biggest funder of non-commercial trials.
-
Consider extending the CCT registration to include results on the headings specified here. Given that ClinGov has moved towards requiring inclusion of results, CCT may move in a similar direction, in which case some of the headings in the database could be used.
-
Make comparisons with other research, such as the cost casemix study of RCTs in the USA132 and the analysis of cohorts of RCTs (as in the Cochrane Review by Djulbegovic et al. 96).
-
Explore the implications of adding questions to the database, such as regarding the role and type of patient and public involvement, and similarly with regard to peer review.
-
Research the effectiveness of tick box checklists, such as CONSORT and the BMJ economic analysis checklist, for quality assurance versus more detailed analysis.
Concluding remarks
The overall finding on RCTs funded by the HTA programme might be ‘good, but could do better’. The main topics for improvement concerned data on the ways in which the programme met the needs of the NHS and on the overall cost of the trials it part funds.
The main finding on the database is that the set of metadata headings presently used in trial registries could be expanded to include aspects of design, performance, results, analyses and costs. This study showed that much of the data required could be extracted on a routine basis from administrative systems, but mainly from the application forms, protocols and monographs.
Chapter 11 Conclusions
Introduction
This project has shown that the ‘metadata’ collected in trial registries can be expanded to include aspects of performance, conduct, results and costs. It has also indicated the limits of available data.
Recommendations for the future metadata database
The main recommendation is that the metadata should continue to be extracted in order to update the answers to the questions we have posed. Such data can make a substantial contribution to understanding each of the six themes. If NIHR is to maximise the added value of funding of trials to meet the needs of the NHS with high-quality science, then metadata on those trials is likely to play a key role.
We suggest that the cost of maintaining the database could be minimised by integrated data collection into NETSCC’s MIS and publication processes. However, this would need some dedicated resource as well as commitment from the HTA and other NIHR research programmes.
We also recommend that any future data extraction should include all NIHR-funded RCTs. We included 125 completed HTA trials. Any future data extraction should include data on the 220 other HTA-funded trials that were active at the end of December 2012. As other NIHR research programmes also fund RCTs, notably the PHR, EME and HS&DR programmes, but also the PGfAR, inclusion of all RCTs by these funders would greatly enhance the value of the database.
Recommendations have been offered on how the database might be refined to learn from the experiences reported here. If the database is to be continued, we recommend that the changes suggested for particular classification systems should be piloted using the 20 or so trials published since the cut-off date for this project. We have also recommended that the database be extended in relation to its inclusion of sufficient data on differences in results between new and old interventions, to enable it to remain part of the portfolio of cohorts of trials included in the ongoing Cochrane Review. 96
We recommend that the database be made available to other researchers, subject to similar confidentiality agreements that governed the present project (similar because the rules governing data access have changed since this project began). We also recommend that the database include aspects aimed at enabling the quality of its data to be improved by such use. Given the inclusion of all source documents within it, future users could have the facility and the requirement to note any differences of data extraction and/or interpretation. This would need careful piloting but could provide a unique feature that would enhance the value of the database to NETSCC as a scientific secretariat.
Other relevant research noted previously includes work on the factors associated with differences in the cost of trials. 132 This work, being carried out for NIH in the USA using a portfolio of commercially funded trials, should be monitored and compared.
The metadata database is unique in extending well beyond trial registration databases and in including source documentation. We believe it addresses key issues for any research funder and we recommend that it continue in the ways discussed above.
This project also indicated the limits of available data. For each theme, several important questions were deemed not feasible owing to lack of data or difficulties in their extraction. The most important of these concerned how the programme was meeting the needs of the NHS. Data on the factors leading to a proposed trial being prioritised and funded will inevitably be limited in relation to the eventual impact of that trial. However, Chapter 4 suggests that the programme could improve the data it collects on these factors.
Reflections by the team on the project
Classification systems proved key to the database as they provided the options within boxes. Relatively few well-established, relevant classification systems could be identified as we had to sometimes improvise. This contributed to the finding that around one-third of our questions required amendment.
Our methods, in retrospect, could have been better specified. We aimed to construct a database, use it to answer questions and, based on that experience, make recommendations for how the database might be further developed. Doing this with six lead researchers, each with different degrees of detailed knowledge and commitments, proved challenging. In retrospect, each of the unfunded research leads would have benefited from having had specific funded time. This would also have facilitated project management. Further, the process of iteration and piloting might have been better specified and ‘protocolised’.
We carried out two small micro-pilot studies, one concerned with extracting data on trial results from academic journals rather than the monographs, and the other relating to having principal investigators quality assure the data extracted. Neither of these proved a success and neither was proceeded with. Many of the academic articles could not be readily accessed. The process of having a principal investigator quality assure the data extracted proved laborious. However, the time required for this might be reduced if it could be done contemporaneously. To do so with trials, some of which had been started up to 20 years ago, was never likely to be feasible.
In retrospect, the database should have included questions on results similar to those in the Cochrane Review of new versus old interventions. 96 As two of us (LS, JR) had extracted and classified data on 65 published HTA programme trials, this would have been a considerable but not major challenge. Given that those 65 trials made up one of only four such cohorts globally, it is unfortunate that all 125 trials in the database cannot constitute an update on that earlier work. We recommend that the database is expanded to include the relevant questions.
Finally, we underestimated the work involved in piloting the database in ways that were rigorous and transparent. We should have developed earlier criteria linked to the aims of the project which discriminated success and failure.
On the positive side, we were able to identify weaknesses in the data and how they might be classified. One of the surprises was that such databases are very uncommon, with the research referred to above indicating that the HTA cohort of studies was not only one of four existing globally, but also that it was the second largest. Interest in assessing the costs and benefits of publicly funded RCTs is likely to grow. We hope that the work reported here will facilitate the inclusion of trials funded by the HTA programme in those assessments.
Acknowledgements
We would like to thank members of the advisory group (Professor Marion Campbell, Professor Doug Altman, Professor Mike Clarke, Professor Jon Nicholl, Professor Janet Peacock, Professor Ken Stein and Professor Hywel Williams) for their valuable advice and support throughout the duration of the project.
The authors also wish to thank Rafael Pinedo Villanueva from the University of Southampton’s Wessex Institute for his contribution to the cost of trial theme (Chapter 9), and Antonia Perez from NETSCC’s finance department, who provided advice on financial payments to clinical trials and the HTA MIS.
We are grateful to the HTA editor and referees for their valuable comments during the editorial stage of the report.
The research team also acknowledges the funding supplied by the HTA programme.
Contributions of authors
James Raftery (Professor of HTA) was the chief investigator, originated the proposal, led on theme 6 and prepared the report for publication.
Amanda Young (Senior Research Fellow) was a member of the research team and led on the design of the database, extracted data, ran analyses, worked closely with James Raftery to keep the project running and prepared the report for publication.
Louise Stanton (Senior Medical Statistician) was a member of the research team, provided statistical input, quality assured data, led on theme 4, contributed to the early drafts of the theme-related sections of the report and reviewed a draft of the report.
Ruairidh Milne (head of NETSCC) was a member of the research team, led on theme 1, contributed to the early drafts of the theme-related sections of the report and reviewed a draft of the report.
Andrew Cook (Consultant in Public Health Medicine and Fellow in HTA) was a member of the research team, led on theme 3, contributed to the early drafts of the theme-related sections of the report and reviewed a draft of the report.
David Turner (Senior Health Economist) led on theme 5, extracted cost-effectiveness plane data, quality assured health economics data, contributed to the early drafts of the theme-related sections of the report and reviewed a draft of the report.
Peter Davidson (NETSCC, Director of the HTA programme) was a member of the research team, led on theme 2, contributed to the early drafts of the theme-related sections of the report and reviewed a draft of the report.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Raftery J, Powell J. Health Technology Assessment in the UK. Lancet 2013;382:1278-85. http://dx.doi.org/10.1016/S0140-6736(13)61724-9.
- Campbell MK, Snowdon C, Francis D, Elbourne D, McDonald AM, Knight R, et al. Recruitment to randomised trials: strategies for trial enrolment and participation study. The STEPS study. Health Technol Assess 2007;11. http://dx.doi.org/10.3310/hta11480.
- Hanney S, Buxton M, Green C, Coulson D, Raftery J. An assessment of the impact of the NHS Health Technology Assessment Programme. Health Technol Assess 2007;11. http://dx.doi.org/10.3310/hta11530.
- Buxton M, Hanney S. How can payback from health services research be assessed?. J Health Serv Res Policy 1996;1:35-43.
- Dent L, Raftery J. Treatment success in pragmatic randomised controlled trials: a review of trials funded by the UK Health Technology Assessment programme. Trials 2011;12. http://dx.doi.org/10.1186/1745-6215-12-109.
- Djulbegovic B, Kumar A, Soares HP, Hozo I, Bepler G, Clarke M, et al. Treatment success in cancer: new cancer treatment successes identified in phase 3 randomized controlled trials conducted by the National Cancer Institute-sponsored cooperative oncology groups, 1955 to 2006. Arch Intern Med 2008;168:632-42. http://dx.doi.org/10.1001/archinte.168.6.632.
- Jolly K, Taylor R, Lip G, Greenfield S, Raftery J, Mant J, et al. The Birmingham Rehabilitation Uptake Maximisation Study (BRUM). Home-based compared with hospital-based cardiac rehabilitation in a multi-ethnic population: cost effectiveness and patient adherence. Health Technol Assess 2007;11. http://dx.doi.org/10.3310/hta11350.
- Ridyard C, Hughes D. Methods for the collection of resource use data within clinical trials: a systematic review of studies funded by the UK Health Technology Assessment Program. Value Health 2010;13:867-72. http://dx.doi.org/10.1111/j.1524-4733.2010.00788.x.
- Chase D, Milne R, Stein K, Stevens A. What are the relative merits of the sources used to identify potential research priorities for the NHSHTA programme?. Int J Technol Assess Health Care 2000;16:743-50. http://dx.doi.org/10.1017/S0266462300102028.
- Chan A, Hrobjartsson A, Haahr M, Gotzsche P, Altman D. Empirical evidence for selective reporting of outcomes in randomized trials: comparison of protocols to published articles. JAMA 2004;291:2457-65. http://dx.doi.org/10.1001/jama.291.20.2457.
- Chan AW, Altman DG. Identifying outcome reporting bias in randomised trials on PubMed: review of publications and survey of authors. BMJ 2005;330. http://dx.doi.org/10.1136/bmj.38356.424606.8F.
- Drummond MF, Jefferson TO. Guidelines for authors and peer reviewers of economic submissions to the BMJ. The BMJ Economic Evaluation Working Party. BMJ 1996;313:275-83. http://dx.doi.org/10.1136/bmj.313.7052.275.
- Attributing Revenue Costs of Externally-Funded Non-Commercial Research in the NHS (ARCO). London: Department of Health; 2005.
- Viergever RF, Ghersi D. The quality of registration of clinical trials. PLOS ONE 2011;6. http://dx.doi.org/10.1371/journal.pone.0014701.
- ClinicalTrials.gov . FDAAA 801 Requirements n.d. https://clinicaltrials.gov/ct2/manage-recs/fdaaa (accessed 29 October 2014).
- World Medical Association . Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects 2008. www.wma.net/en/30publications/10policies/b3/17c.pdf (accessed 29 October 2014).
- World Health Organization . International Clinical Trials Registry Platform (ICTRP) n.d. www.who.int/ictrp/en/ (accessed 29 October 2014).
- International Committee of Medical Journal Editors (ICMJE) . ICMJE Recommendations (‘The Uniform Requirements’) 2004. www.icmje.org/about-icmje/faqs/icmje-recommendations/ (accessed 29 October 2014).
- Raftery J, Fairbank E, Douet L, Dent L, Price A, Milne R, et al. Registration of noncommercial randomised clinical trials: the feasibility of using trial registries to monitor the number of trials. Trials 2012;13. http://dx.doi.org/10.1186/1745-6215-13-140.
- Reveiz L, Cortés-Jofré M, Asenjo Lobos C, Nicita G, Ciapponi A, Garcìa-Dieguez M, et al. Influence of trial registration on reporting quality of randomized trials: study from highest ranked journals. J Clin Epidemiol 2010;63:1216-22. http://dx.doi.org/10.1016/j.jclinepi.2010.01.013.
- Moja L, Moschetti I, Nurbhai M, Compagnoni A, Liberati A, Grimshaw J, et al. Compliance of clinical trial registries with the World Health Organization minimum data set: a survey. Trials 2009;10. http://dx.doi.org/10.1186/1745-6215-10-56.
- Ghersi D, Clarke M, Berlin J, Gulmezoglu AM, Kush R, Lumbiganon P, et al. Reporting the findings of clinical trials: a discussion paper. Bull World Health Organ 2008;86:492-93. http://dx.doi.org/10.2471/BLT.08.053769.
- Califf R, Zarin D, Kramer J, Sherman R, Aberle L, Tasneem A. Characteristics of clinical trials registered in ClinicalTrials.gov, 2007–2010. JAMA 2012;307:1838-47. http://dx.doi.org/10.1001/jama.2012.3424.
- Sekeres M, Gold JL, Chan AW, Lexchin J, Moher D, Van Laethem MLP, et al. Poor reporting of scientific leadership information in clinical trial registers. PLOS ONE 2008;3. http://dx.doi.org/10.1371/journal.pone.0001610.
- Ross JS, Mulvey GK, Hines EM, Nissen SE, Krumholz HM. Trial publication after registration in ClinicalTrials.Gov: a cross-sectional analysis. PLOS Med 2009;6. http://dx.doi.org/10.1371/journal.pmed.1000144.
- Bourgeois FT, Murthy S, Mandl KD. Outcome reporting among drug trials registered in ClinicalTrials.gov. Ann Intern Med 2010;153:158-66. http://dx.doi.org/10.7326/0003-4819-153-3-201008030-00006.
- Mathieu S, Boutron I, Moher D, Altman DG, Ravaud P. Comparison of registered and published primary outcomes in randomized controlled trials. JAMA 2009;302:977-84. http://dx.doi.org/10.1001/jama.2009.1242.
- Dwan K, Altman DG, Cresswell L, Blundell M, Gamble CL, Williamson PR. Comparison of protocols and registry entries to published reports for randomised controlled trials. Cochrane Database Syst Rev 2011;1. http://dx.doi.org/10.1002/14651858.MR000031.pub2.
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLOS Med 2009;6. http://dx.doi.org/10.1371/journal.pmed.1000100.
- UK Clinical Research Collaboration Health Research Classification System (UKCRC HRCS) Online . List of Research Activity Codes n.d. www.hrcsonline.net/rac/overview (accessed 3 October 2014).
- Thabane L, Ma J, Chu R, Cheng J, Ismaila A, Rios L, et al. A tutorial on pilot studies: the what, why and how. BMC Med Res Methodol 2010;10. http://dx.doi.org/10.1186/1471-2288-10-1.
- Schwartz D, Lellouch J. Explanatory and pragmatic attitudes in therapeutical trials. J Clin Epidemiol 2009;62:499-505. http://dx.doi.org/10.1016/j.jclinepi.2009.01.012.
- Cooksey D. A Review of UK Health Research Funding. Norwich: The Stationery Office; 2006.
- Chalkidou K, Whicher D, Kary W, Tunis S. Comparative effectiveness research priorities: identifying critical gaps in evidence for clinical and health policy decision making. Int J Technol Assess Health Care 2009;25:241-48. http://dx.doi.org/10.1017/S0266462309990225.
- Jones R, Lamont T, Haines A. Setting priorities for research and development in the NHS: a case study on the interface between primary and secondary care. BMJ 1995;311:1076-80. http://dx.doi.org/10.1136/bmj.311.7012.1076.
- Noorani HZ, Husereau DR, Boudreau R, Skidmore B. Priority setting for health technology assessments: a systematic review of current practical approaches. Int J Technol Assess Health Care 2007;23:310-15. http://dx.doi.org/10.1017/S026646230707050X.
- Tomlinson M, Swartz L, Officer A, Chan KY, Rudan I, Saxena S. Research priorities for health of people with disabilities: an expert opinion exercise. Lancet 2009;374:1857-62. http://dx.doi.org/10.1016/S0140-6736(09)61910-3.
- Sibbald SL, Singer PA, Upshur R, Martin DK. Priority setting: what constitutes success? A conceptual framework for successful priority setting. BMC Health Serv Res 2009;9. http://dx.doi.org/10.1186/1472-6963-9-43.
- Gandhi GY, Murad MH, Fujiyoshi A, Mullan RJ, Flynn DN, Elamin MB, et al. Patient-important outcomes in registered diabetes trials. JAMA 2008;299:2543-9. http://dx.doi.org/10.1001/jama.299.21.2543.
- Montori VM, Wang YG, Alonso-Coello P, Bhagra S. Systematic evaluation of the quality of randomized controlled trials in diabetes. Diabetes Care 2006;29:1833-8. http://dx.doi.org/10.2337/dc06-0077.
- Rahimi K, Malhotra A, Banning A, Jenkinson C. Outcome selection and role of patient reported outcomes in contemporary cardiovascular trials: systematic review. BMJ 2010;341. http://dx.doi.org/10.1136/bmj.c5707.
- Chalmers I, Glasziou P. Avoidable waste in the production and reporting of research evidence. Lancet 2009;374:86-9. http://dx.doi.org/10.1016/S0140-6736(09)60329-9.
- Jones A, Conroy E, Williamson P, Clarke M, Gamble C. The use of systematic reviews in the planning, design and conduct of randomised trials: a retrospective cohort of NIHR HTA funded trials. BMC Med Res Methodol 2013;13. http://dx.doi.org/10.1186/1471-2288-13-50.
- Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. J Clin Epidemiol 2010;63:e1-37. http://dx.doi.org/10.1016/j.jclinepi.2010.03.004.
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias – dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408-12. http://dx.doi.org/10.1001/jama.1995.03520290060030.
- Dickersin K, Chan S, Chalmers TC, Sacks HS, Smith J. Publication bias and clinical trials. Control Clin Trials 1987;8:343-53. http://dx.doi.org/10.1016/0197-2456(87)90155-3.
- Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions 2011. www.cochrane-handbook.org (accessed 25 November 2014).
- Thorpe KE, Zwarenstein M, Oxman AD, Treweek S, Furberg CD, Altman DG, et al. A pragmatic-explanatory continuum indicator summary (PRECIS): a tool to help trial designers. J Clin Epidemiol 2009;62:464-75. http://dx.doi.org/10.1016/j.jclinepi.2008.12.011.
- Devereaux PJ, Manns BJ, Ghali WA, Quan H, Lacchetti C, Montori VM, et al. Physician interpretations and textbook definitions of blinding terminology in randomized controlled trials. JAMA 2001;285:2000-3. http://dx.doi.org/10.1001/jama.285.15.2000.
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials: the CONSORT statement. JAMA 1996;276:637-9. http://dx.doi.org/10.1001/jama.1996.03540080059030.
- Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel group randomized trials. BMC Med Res Methodol 2001;1. http://dx.doi.org/10.1186/1471-2288-1-2.
- Chalmers I, Rounding C, Lock K. Descriptive survey of non-commercial randomised controlled trials in the United Kingdom, 1980–2002. BMJ 2003;327. http://dx.doi.org/10.1136/bmj.327.7422.1017.
- Williams J, Russell I, Dural D, Cheung W-Y, Farrin A, Bloor K, et al. What are the clinical outcome and cost effectiveness of endoscopy undertaken by nurses when compared with doctors? A Multi-Institution Nurse Endoscopy Trial (MINuET). Health Technol Assess 2006;10. http://dx.doi.org/10.3310/hta10400.
- Walley T. Health technology assessment in England: assessment and appraisal. Med J Aust 2007;187:283-5.
- Medicines and Healthcare products Regulatory Agency (MHRA) . Description of the Medicines for Human Use (Clinical Trials) Regulations; 2004 n.d. www.mhra.gov.uk/home/groups/l-unit1/documents/websiteresources/con2022633.pdf (accessed 25 November 2014).
- NHS Executive . Ethics Committee Review of Multicentre Research. HSG (97) 1997. http://webarchive.nationalarchives.gov.uk/+/www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_4009191 (accessed 25 November 2014).
- Department of Health (DH) . Local Research Ethics Committees 1991. http://webarchive.nationalarchives.gov.uk/+/www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_4002874 (accessed 25 November 2014).
- Department of Health (DH) . Requirements to Support Research in the NHS 2009. www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/documents/digitalasset/dh_102098.pdf (accessed 25 November 2014).
- A New Pathway for the Regulation and Governance of Health Research. London: Academy of Medical Sciences; 2011.
- Gajewski B, Simon S, Carlson S. Predicting accrual in clinical trials with Bayesian posterior predictive distributions. Stat Med 2008;27:2328-40. http://dx.doi.org/10.1002/sim.3128.
- Williford W, Bingham S, Weiss D, Collins JF, Rains K, Krol WF. The ‘constant intake rate’ assumption in interim recruitment goal methodology for multicenter clinical trials. J Chronic Dis 1987;40:297-30. http://dx.doi.org/10.1016/0021-9681(87)90045-2.
- Carter R, Sonne S, Brady K. Practical considerations for estimating clinical trial accrual periods: application to a multi-center effectiveness study. BMC Med Res Methodol 2005;5. http://dx.doi.org/10.1186/1471-2288-5-11.
- Moussa MAA. Planning a clinical trial with allowance for cost and patient recruitment rate. Comput Programs Biomed 1984;18:173-9. http://dx.doi.org/10.1016/0010-468X(84)90049-7.
- Guyatt GH, Oxman AD, Vist G, Kunz R, Brozek J, Alonso-Coello P, et al. GRADE guidelines: 4. Rating the quality of evidence: cost study limitations (risk of bias). J Clin Epidemiol 2011;64:407-15. http://dx.doi.org/10.1016/j.jclinepi.2010.07.017.
- Buchan JC, Spokes DM. Do recorded abstracts from scientific meetings concur with the research presented?. Eye 2010;24:695-8. http://dx.doi.org/10.1038/eye.2009.133.
- Watson J, Torgerson D. Increasing recruitment to randomised trials: a review of randomised controlled trials. BMC Med Res Methodol 2006;6. http://dx.doi.org/10.1186/1471-2288-6-34.
- Treweek S, Mitchell E, Pitkethly M, Cook J, Kjeldstrom M, Taskila T, et al. Strategies to improve recruitment to randomised controlled trials. Cochrane Database Syst Rev 2010;1. http://dx.doi.org/10.1002/14651858.MR000013.pub4.
- Menon U, Gentry-Meharaj A, Ryan A, Sharma A, Burnell M, Hallett R, et al. Recruitment to multicentre trials – lessons from UKCTOCS: descriptive study. BMJ 2008;337. http://dx.doi.org/10.1136/bmj.a2079.
- Fletcher B, Gheorghe A, Moore D, Wilson S, Damery S. Improving the recruitment activity of clinicians in randomised controlled trials: a systematic review. BMJ Open 2012;2. http://dx.doi.org/10.1136/bmjopen-2011-000496.
- Booker C, Harding S, Benzeval M. A systematic review of the effect of retention methods in population-based cohort studies. BMC Public Health 2011;11. http://dx.doi.org/10.1186/1471-2458-11-249.
- Meyers K, Webb A, Frantz J, Randall M. What does it take to retain substance-abusing adolescents in research protocols? Delineation of effort required, strategies undertaken, costs incurred, and 6-month post-treatment differences by retention difficulty. Drug Alcohol Depend 2003;69:73-85. http://dx.doi.org/10.1016/S0376-8716(02)00252-1.
- Fisher L, Hessler D, Naranjo D, Polonsky W. AASAP: A program to increase recruitment and retention in clinical trials. Patient Educ Couns 2012;86:372-7. http://dx.doi.org/10.1016/j.pec.2011.07.002.
- Hamdy F. Evaluating the Effectiveness of Treatment for Clinically Localised Prostate Cancer n.d. www.isrctn.com/ISRCTN20141297?q=20141297&filters=&sort=&offset=1&totalResults=1&page=1&pageSize=10&searchType=basic-search (accessed 25 November 2014).
- Donovan J, Hamdy F, Neal D, Peters T, Oliver S, Brindle L, et al. Prostate Testing for Cancer and Treatment (ProtecT) feasibility study. Health Technol Assess 2003;7. http://dx.doi.org/10.3310/hta7140.
- Lane JA, Wade J, Down L, Bonnington S, Holding PN, Lennon T, et al. A Peer Review Intervention for Monitoring and Evaluating sites (PRIME) that improved randomized controlled trial conduct and performance. J Clin Epidemiol 2011;64:628-36. http://dx.doi.org/10.1016/j.jclinepi.2010.10.003.
- Schulz KF, Altman DG, Moher D. CONSORT 2010 Statement: Updated guidelines for reporting parallel group randomised trials. J Clin Epidemiol 2010;63:834-40. http://dx.doi.org/10.1016/j.jclinepi.2010.02.005.
- Barnard K, Dent L, Cook A. A systematic review of models to predict recruitment to multicentre clinical trials. BMC Med Res Methodol 2010;10. http://dx.doi.org/10.1186/1471-2288-10-63.
- Department of Health (DH) . Guidance on Funding Excess Treatment Costs Related to Non-Commercial Research Studies and Applying for a Subvention 2009. http://webarchive.nationalarchives.gov.uk/20130107105354/http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/documents/digitalasset/dh_097627.pdf (accessed 25 November 2014).
- Responsibilities for Meeting Patient Care Costs Associated with Research and Development in the NHS. HSG(97)32. London: Department of Health; 1997.
- Al-Marzouki S, Roberts I, Evans S, Marshall T. Selective reporting in clinical trials: analysis of trial protocols accepted by The Lancet. Lancet 2008;372. http://dx.doi.org/10.1016/S0140-6736(08)61060-0.
- Chan AW, Krleza-Jeric K, Schmid I, Altman DG. Outcome reporting bias in randomized trials funded by the Canadian Institutes of Health Research. CMAJ 2004;171:735-40. http://dx.doi.org/10.1503/cmaj.1041086.
- Ewart R, Lausen H, Millian N. Undisclosed changes in outcomes in randomized controlled trials: an observational study. Ann Fam Med 2009;7:542-6. http://dx.doi.org/10.1370/afm.1017.
- Kavvoura FK, McQueen MB, Khoury MJ, Tanzi RE, Bertram L, Ioannidis JPA. Evaluation of the potential excess of statistically significant findings in published genetic association studies: application to Alzheimer’s disease. Am J Epidemiol 2008;168:855-65. http://dx.doi.org/10.1093/aje/kwn206.
- Pildal J, Chan AW, Hrobjartsson A, Forfang E, Altman DG, Gotzsche PC. Comparison of descriptions of allocation concealment in trial protocols and the published reports: cohort study. BMJ 2005;330. http://dx.doi.org/10.1136/bmj.38414.422650.8F.
- Scharf O, Colevas AD. Adverse event reporting in publications compared with sponsor database for cancer clinical trials. J Clin Oncol 2006;24:3933-8. http://dx.doi.org/10.1200/JCO.2005.05.3959.
- Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med 2008;358:252-60. http://dx.doi.org/10.1056/NEJMsa065779.
- Vedula SS, Bero L, Scherer RW, Dickersin K. Outcome reporting in industry-sponsored trials of gabapentin for off-label use. N Engl J Med 2009;361:1963-71. http://dx.doi.org/10.1056/NEJMsa0906126.
- Chan AW, Hrobjartsson A, Jorgensen KJ, Gotzsche PC, Altman DG. Discrepancies in sample size calculations and data analyses reported in randomised trials: comparison of publications with protocols. BMJ 2008;337. http://dx.doi.org/10.1136/bmj.a2299.
- Hahn S, Williamson PR, Hutton JL. Investigation of within-study selective reporting in clinical research: follow-up of applications submitted to a local research ethics committee. J Eval Clin Pract 2002;8:353-9. http://dx.doi.org/10.1046/j.1365-2753.2002.00314.x.
- Charles P, Giraudeau B, Dechartres A, Baron G, Ravaud P. Reporting of sample size calculation in randomised controlled trials: review. BMJ 2009;338. http://dx.doi.org/10.1136/bmj.b1732.
- Bland JM. The tyranny of power: is there a better way to calculate sample size?. BMJ 2009;339. http://dx.doi.org/10.1136/bmj.b3985.
- Hernández A, Steyerberg E, Taylor G, Marmarou A, Habbema J, Maas I. Subgroup analysis and covariate adjustment in randomized clinical trials of traumatic brain injury: a systematic review. Neurosurgery 2005;57:1244-53. http://dx.doi.org/10.1227/01.NEU.0000186039.57548.96.
- Chan AW. Bias, spin, and misreporting: time for full access to trial protocols and results. PLOS Med 2008;5:1533-5. http://dx.doi.org/10.1371/journal.pmed.0050230.
- Chan A-W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jeric K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med 2013;158:200-7. http://dx.doi.org/10.7326/0003-4819-158-3-201302050-00583.
- Pocock S, Travison T, Wruck L. Figures in clinical trial reports: current practice and scope for improvement. Trials 2007;8. http://dx.doi.org/10.1186/1745-6215-8-36.
- Djulbegovic B, Kumar A, Glasziou PP, Perera R, Reljic T, Dent L, et al. New treatments compared to established treatments in randomized trials (review). Cochrane Database Syst Rev 2012;10.
- Anderson JP, Bush JW, Chen M, Dolen D. Policy space areas and properties of benefit cost/utility analysis. JAMA 1986;255:794-5. http://dx.doi.org/10.1001/jama.1986.03370060108029.
- Black WC. The CE plane: a graphic representation of cost-effectiveness. Med Decis Mak 1990;10:212-14. http://dx.doi.org/10.1177/0272989X9001000308.
- Drummond MF, Sculpher MJ, Torrance GW, O’Brien BJ, Stoddart GL. Methods for the Economic Evaluation of Health Care Programmes. Oxford: Oxford University Press; 2005.
- Scottish Intercollegiate Guidelines Network (SIGN) . Healthcare Improvement Scotland 2012. www.sign.ac.uk./guidelines/index.html (accessed 25 November 2014).
- Australian Pharmaceutical Benefits Scheme (PBS) . Australian Government Department of Health and Ageing 2012. www.health.gov.au/pbs (accessed 25 November 2014).
- National Institute for Health and Care Excellence (NICE) . Guide to the Methods of Technology Appraisal n.d. www.nice.org.uk/article/PMG9/chapter/Foreword (accessed 3 May 2012).
- Raftery J. Should NICE’s threshold range for cost per QALY be raised? No. BMJ 2009;338. http://dx.doi.org/10.1136/bmj.b185.
- Towse A. Should NICE’s threshold range for cost per QALY be raised? Yes. BMJ 2009;338. http://dx.doi.org/10.1136/bmj.b181.
- Petrou S, Gray A. Economic evaluation alongside randomised controlled trials: design, conduct, analysis, and reporting. BMJ 2011;342. http://dx.doi.org/10.1136/bmj.d1548.
- Petrou S, Gray A. Economic evaluation using decision analytical modelling: design, conduct, analysis, and reporting. BMJ 2011;342. http://dx.doi.org/10.1136/bmj.d1766.
- Sculpher M, Claxton K, Drummond M. Whither trial-based economic evaluation for health care decision making?. Health Econ 2006;15:677-87. http://dx.doi.org/10.1002/hec.1093.
- Barber JA, Thompson SG. Analysis and interpretation of cost data in randomised controlled trial: review of published studies. BMJ 1998;317:1195-200. http://dx.doi.org/10.1136/bmj.317.7167.1195.
- Doshi JA, Glick HA, Polsky D. Analyses of cost data in economic evaluations conducted alongside randomized controlled trials. Value Health 2006;9:334-40. http://dx.doi.org/10.1111/j.1524-4733.2006.00122.x.
- O’Sullivan AK, Hompson D, Rummond MF. Collection of health economic data alongside clinical trials: is there a future for piggyback evaluations?. Value Health 2005;8:67-79. http://dx.doi.org/10.1111/j.1524-4733.2005.03065.x.
- Glick HA, Doshi JA, Sonnad SS, Polsky D. Economic Evaluation in Clinical Trials. Oxford: Oxford University Press; 2007.
- Evers S, Goossens M, de Vet H, van Tulder M, Ament A. Criteria list for assessment of methodological quality of economic evaluations: consensus on health economic criteria. Int J Technol Assess Health Care 2005;21:240-5.
- Chiou CF, Hay JW, Wallace JF, Bloom BS, Neumann PJ, Sullivan SD, et al. Development and validation of a grading system for the quality of cost-effectiveness studies. Med Care 2003;41:32-44. http://dx.doi.org/10.1097/00005650-200301000-00007.
- Drummond MF, Sculpher MJ, Torrance GW, O’Brien BJ, Stoddart GL. Methods for the Economic Evaluation of Health Care Programmes. Oxford: Oxford University Press; 2005.
- Briggs A, O’Brien BJ. The death of cost-minimization analysis?. Health Econ 2001;10:179-84. http://dx.doi.org/10.1002/hec.584.
- Kaitin KI, DiMasi JA. Pharmaceutical innovation in the 21st century: new drug approvals in the first decade, 2000–2009. Clin Pharmacol Ther 2011;89:183-8. http://dx.doi.org/10.1038/clpt.2010.286.
- DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ 2003;22:151-85. http://dx.doi.org/10.1016/S0167-6296(02)00126-1.
- Adams C, Brantner VV. Spending on new drug development. Health Econ 2010;19:130-41. http://dx.doi.org/10.1002/hec.1454.
- Light D, Warburton R. Demythologizing the high costs of pharmaceutical research. BioSocieties 2011;6:34-50. http://dx.doi.org/10.1057/biosoc.2010.40.
- Hackshaw A, Farrant H, Bulley S, Seckl M, Ledermann J. Setting up non-commercial clinical trials takes too long in the UK: findings from a perspective study. J R Soc Med 2008;101:299-304. http://dx.doi.org/10.1258/jrsm.2008.070373.
- Hutchings H, Lloyd G, Snooks H, Russell I. A1 financial and time costs of R&D governance and regulation in England and Wales: evidence from the SAFER 2 trial. Emerg Med J 2011;28. http://dx.doi.org/10.1136/emermed-2011-200645.1.
- Snowdon C, Elbourne D, Garcia J, Campbell M, Entwistle V, Francis D, et al. Financial considerations in the conduct of multi-centre randomised controlled trials: evidence from a qualitative study. Trials 2006;7. http://dx.doi.org/10.1186/1745-6215-7-34.
- Al-Shahi S, Brock T, Dennis M, Sandercock P, White P, Warlow C. Research governance impediments to clinical trials: a retrospective survey. J R Soc Med 2007;100:101-4. http://dx.doi.org/10.1258/jrsm.100.2.101.
- Martin D, Maguire M, Fine S. Identifying and eliminating the roadblocks to comparative-effectiveness research. N Engl J Med 2010;363:105-7. http://dx.doi.org/10.1056/NEJMp1001201.
- Abernethy A, Lapointe N, Wheeler J, Irvine R, Patwardhanm M, Matchar D. Horizon Scan: To What Extent Do Changes in Third Party Payment Affect Clinical Trials and the Evidence Base? 2009. www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/id67ata.pdf (accessed 25 November 2014).
- Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Culliford LA, et al. on behalf of the IVAN study investigators. Alternative treatments to inhibit VEGF in age related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet 2013;382:1258-67. http://dx.doi.org/10.1016/S0140-6736(13)61501-9.
- Attributing the costs of health and social care Research & Development (AcoRD). London: DH; 2012.
- Bailey C, Baker R, Kirk S. Research and Development for the NHS. 3rd edn. Oxford: Radcliffe Medical Press; 1998.
- Phillips C, Moustaki I. Higher Education Pay and Prices Index: July 2009 2009. www.universitiesuk.ac.uk/Publications/Pages/HEPPI1July2009.aspx (accessed 24 November 2014).
- Dent L, Taylor R, Jolly K, Raftery J. ‘Flogging dead horses’: evaluating when have clinical trials achieved sufficiency and stability? A case study in cardiac rehabilitation. Trials 2011;12. http://dx.doi.org/10.1186/1745-6215-12-83.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) . Statistical Principles For Clinical Trials 1998. www.ich.org (accessed 24 November 2014).
- Getz KA, Zuckerman R, Cropp AB, Hindle AL, Krauss R, Kaitin KI. Measuring the incidence, causes, and repercussions of protocol amendments. Drug Info J 2011;45:265-75. http://dx.doi.org/10.1177/009286151104500307.
Appendix 1 Literature search strategy
Uses and limitations of trial registration databases
Date searched: 9 February 2012.
Sources searched:
Ovid MEDLINE(R) without Revisions (1996 to February week 1, 2012) Ovid MEDLINE(R) In-Process & Other Non-Indexed Citations Ovid EMBASE (1996 to week 5, 2012) Cochrane Methodology Register.
Database: Ovid MEDLINE(R) without Revisions (1996 to February week 1, 2012)
Search strategy
-
Randomized Controlled Trials as Topic/ or Clinical Trials as Topic/ (141,375)
-
Database Management Systems/ (5285)
-
Registries/ (32,826)
-
(trial* adj3 regist*).ti,ab. (14,862)
-
clinicaltrials.ti,ab. (46)
-
2 and 4 (3)
-
(trial registries or trial registers).ti,ab. (318)
-
evaluation studies.pt. (157,963)
-
7 and 8 (5)
-
from 9 keep 3,5 (2)
-
1 and 3 (1292)
-
International Committee of Medical Journal Editors.mp. (160)
-
7 and 12 (3)
-
from 13 keep 1-3 (3)
-
(evaluation or usability).ti,ab. (398,106)
-
7 and 15 (20)
-
3 and 15 (1724)
-
from 16 keep 13 (1)
-
1 and 3 (1292)
-
usability.ti,ab. (2559)
-
19 and 20 (1)
-
"Databases, Factual"/ (29,221)
-
limitation*.ti,ab. (101,377)
-
1 and 3 and 22 (103)
-
23 and 24 (4)
-
from 21 keep 1 (1)
-
from 25 keep 3 (1)
-
current controlled trials.ti,ab. (707)
-
clinicaltrials gov.ti,ab. (13)
-
28 or 29 (720)
-
7 and 28 (2)
-
10 or 14 or 18 or 26 or 27 (8)
-
8 and 11 (13)
-
from 33 keep 10 (1)
-
comparative study.pt. (830,359)
-
11 and 35 (144)
-
from 36 keep 8-9,11,26,72 (5)
Appendix 2 Full list of randomised controlled trials included in the metadata database
The following is a list of all published HTA-funded RCTs included in the metadata database project (Table 51). The year, volume and issue of the HTA report is included along with description details where funded projects reported more than one trial. The list is in chronological order by HTA publication.
| HTA publication (year, volume, issue) | Project title | Trial ID number | Journal series ID number | Description of trial |
|---|---|---|---|---|
| HTA 1999, 3, 4 | A randomised controlled trial of different approaches to universal antenatal HIV testing: acceptability, costs and benefits | 2 | 1 | |
| HTA 2000, 4, 6 | The costs and benefits of post-natal midwifery support – a randomised controlled trial | 7 | 2 | |
| HTA 2000, 4, 19 | Effectiveness of counselling, cognitive behavioural therapy and GP care for depression in general practice | 3 | 3 | Randomised three-way (GP, CBT and non-directive counselling) |
| HTA 2000, 4, 19 | 4 | 3 | Randomised two-way (CBT and non-directive counselling) | |
| HTA 2000, 4, 20 | Is the outcome for patients with low back pain influenced by GP’s referral for plain radiography? | 5 | 4 | |
| HTA 2000, 4, 28 | Early asthma prophylaxis, natural history, skeletal development and economy (EASE) | 121 | 5 | |
| HTA 2000, 4, 31 | A randomised controlled trial of infusion protocols in adult pre-hospital care | 122 | 6 | |
| HTA 2000, 4, 36 | A randomised controlled trial to evaluate the efficacy and cost-effectiveness of counselling with patients with chronic depression and anxiety | 6 | 7 | |
| HTA 2001, 5, 20 | Multi-centre randomised controlled trial of nurse practitioners and pre-registration house officers in pre-operative workup | 8 | 8 | |
| HTA 2001, 5, 27 | The cost-effectiveness of MRI for investigation of the knee joint | 9 | 9 | |
| HTA 2001, 5, 30 | A randomised controlled trial to assess the effectiveness, cost-effectiveness and cost benefit of routine referral for lumbar spine radiography in patients with low back pain | 10 | 10 | |
| HTA 2002, 6, 20 | Clinical medication review by a pharmacist of patients on repeat prescriptions in general practice | 11 | 11 | |
| HTA 2002, 6, 27 | A randomised crossover trial of nurse versus doctor-led outpatient care in a bronchiectasis clinic | 12 | 12 | |
| HTA 2002, 6, 30 | Which anaesthetic agents and techniques are most cost-effective in day surgery? | 13 | 13 | Adult population |
| HTA 2002, 6, 30 | 14 | 13 | Paediatric population | |
| HTA 2002, 6, 34 | A comparative study of hypertonic saline, daily and alternate day rhDNase in cystic fibrosis | 123 | 14 | |
| HTA 2003, 7, 8 | A multi-centre randomised controlled trial assessing the costs and benefits of using structured information and analysis of women’s preferences in the management of menorrhagia | 20 | 15 | |
| HTA 2003, 7, 14 | The feasibility of conducting a multicentre randomised trial of treatment for localised prostate cancer: early detection, recruitment strategies and a pilot study | 15 | 16 | |
| HTA 2003, 7, 24 | Cost–benefit evaluation of routine influenza immunisation in subjects 65–74 years of age | 16 | 17 | |
| HTA 2003, 7, 28 | Randomised controlled trial to assess the impact of a package comprising a patient-orientated, evidence-based self-help guidebook and patient-centred consultations on disease management and satisfaction in inflammatory bowel disease | 17 | 18 | |
| HTA 2003, 7, 36 | Central line insertion project (CLIP) | 18 | 19 | |
| HTA 2003, 7, 37 | Redesigning postnatal care: a randomised controlled trial of protocol-based, midwifery-led care | 19 | 20 | |
| HTA 2004, 8, 8 | Psychological treatment in the regulation of long-term hypnotic drug use | 31 | 21 | |
| HTA 2004, 8, 14 | Extending midwife/nurse roles in the routine examination of the newborn: randomised controlled evaluation and cost-effectiveness (EMREN trial) | 21 | 22 | |
| HTA 2004, 8, 16 | A multi-centre randomised controlled trial of minimally invasive bypass grafting vs angioplasty with stenting for single vessel disease of the left anterior descending coronary artery | 22 | 23 | |
| HTA 2004, 8, 17 | Does early imaging influence management and improve outcome in patients with low back pain? | 23 | 24 | |
| HTA 2004, 8, 26 | A randomised trial to assess the effectiveness, costs and cost-effectiveness of laparoscopic, vaginal and abdominal hysterectomy | 24 | 25 | Laparoscopic hysterectomy and abdominal hysterectomy |
| HTA 2004, 8, 26 | 25 | 25 | Laparoscopic hysterectomy and vaginal hysterectomy | |
| HTA 2004, 8, 29 | A randomised controlled trial of two bandages for treating venous leg ulcers | 26 | 26 | |
| HTA 2004, 8, 32 | Randomised controlled trial and economic evaluation of two alternative strategies of providing support for socially disadvantaged inner city families with infants | 27 | 27 | |
| HTA 2004, 8, 34 | Diagnosis of endometrial abnormality: comparison of outpatient procedures within cohorts defined by age and menopausal status | 128 | 28 | Moderate-risk group |
| HTA 2004, 8, 34 | 129 | 28 | High-risk group | |
| HTA 2004, 8, 34 | 134 | 28 | Low-risk group | |
| HTA 2004, 8, 46 | A randomised controlled trial of intensive physiotherapy vs a home-based exercise treatment programme in knee osteoarthritis | 28 | 29 | |
| HTA 2004, 8, 48 | Acupuncture for migraine and headache in primary care: a pragmatic, randomised trial | 29 | 30 | |
| HTA 2004, 8, 50 | Virtual outreach: a randomised controlled trial and economic appraisal | 30 | 31 | |
| HTA 2005, 9, 1 | Identification of the most cost-effective, microbiologically safe antimicrobial treatments for acne | 32 | 32 | |
| HTA 2005, 9, 3 | Improving the referral process for familial breast cancer genetic counselling: an evaluation of complementary interventions | 35 | 33 | Primary care trial – Aberdeen only |
| HTA 2005, 9, 3 | 36 | 33 | Nurse counsellor trial in Aberdeen (trial 1) | |
| HTA 2005, 9, 3 | 37 | 33 | Nurse counsellor trial in Cardiff (trial 2) | |
| HTA 2005, 9, 4 | Randomised evaluation of alternative electrosurgical modalities to treat bladder outflow obstruction in men with benign prostatic hyperplasia (BPH) | 44 | 34 | |
| HTA 2005, 9, 5 | A pragmatic randomised controlled trial of the cost-effectiveness of palliative therapies for patients with oesophageal cancer | 47 | 35 | |
| HTA 2005, 9, 16 | A randomised controlled trial to compare the cost-effectiveness of tricyclic antidepressants, selective serotonin re-uptake inhibitors and lofepramine (AHEAD) | 33 | 36 | |
| HTA 2005, 9, 18 | A controlled comparison of alternative strategies in stroke rehabilitation | 34 | 37 | |
| HTA 2005, 9, 31 | Randomised controlled trial of the cost-effectiveness of water-based therapy for lower limb osteoarthritis (ROAR) | 38 | 38 | |
| HTA 2005, 9, 32 | Longer term clinical and economic benefits of offering acupuncture to patients with chronic low back pain | 39 | 39 | |
| HTA 2005, 9, 33 | Wessex epidural steroids trial (WEST) | 40 | 40 | |
| HTA 2005, 9, 34 | The British Rheumatoid Outcome Study Group (BROSG) trial of symptomatic versus aggressive therapy in established rheumatoid arthritis | 41 | 41 | |
| HTA 2005, 9, 37 | Trial of problem-solving by community psychiatric nurses (CPNs) for anxiety, depression and life difficulties among general practice patients | 42 | 42 | |
| HTA 2005, 9, 39 | Is hydrotherapy cost-effective? The costs and outcome measures of hydrotherapy programmes compared with physiotherapy land techniques in children with rheumatoid conditions | 43 | 43 | |
| HTA 2005, 9, 40 | Randomised controlled trial and cost-effectiveness study of targeted screening versus systematic population screening for atrial fibrillation in the over 65s: the SAFE study | 45 | 44 | |
| HTA 2005, 9, 41 | Scottish trial of arthroplasty or reduction for subcapital fractures (STARS) | 46 | 45 | |
| HTA 2006, 10, 2 | FOOD: a multicentre international randomised trial to evaluate percutaneous endoscopic gastrostomy and nasogastric tube feeding in patients admitted to hospital with a recent stroke | 54 | 46 | Trial 1: normal hospital diet vs. normal hospital diet plus oral supplements |
| HTA 2006, 10, 2 | 55 | 46 | Trial 2: early enteral tube feeding vs. avoid enteral tube feeding | |
| HTA 2006, 10, 2 | 56 | 46 | Trial 3: nasogastric tube feeding vs. percutaneous endoscopic gastrostomy tube feeding | |
| HTA 2006, 10, 13 | Assessment of cost-effectiveness of the treatment of varicose veins | 48 | 47 | Clinical group 1 |
| HTA 2006, 10, 13 | 49 | 47 | Clinical group 2 | |
| HTA 2006, 10, 13 | 50 | 47 | Clinical group 3 | |
| HTA 2006, 10, 17 | Cost utility of the latest antipsychotics in severe schizophrenia (CUtLASS): a multi-centre, randomised, controlled trial | 51 | 48 | Band 1 compared older, inexpensive conventional drugs with new atypical drugs (broadly defined) |
| HTA 2006, 10, 17 | 52 | 48 | Band 2 compared the new (non-clozapine) atypical drugs with clozapine (narrowly defined) | |
| HTA 2006, 10, 19 | Cognitive behavioural therapy versus antispasmodic therapy for irritable bowel syndrome in primary care | 53 | 49 | |
| HTA 2006, 10, 21 | Health benefits from anti-viral therapy for mild chronic hepatitis C | 57 | 50 | |
| HTA 2006, 10, 22 | Randomised controlled trial comparing alternating pressure overlays with alternating pressure mattresses for pressure sore prevention and treatment | 58 | 51 | |
| HTA 2006, 10, 29 | An evaluation of the clinical and cost-effectiveness of pulmonary artery flotation catheters (PAC-Man) in intensive care | 59 | 52 | |
| HTA 2006, 10, 37 | Cognitive behavioural therapy in chronic fatigue syndrome: a randomised controlled trial of an outpatient group programme | 60 | 53 | |
| HTA 2006, 10, 40 | What is the cost-effectiveness of endoscopy undertaken by nurses? A multi-institution nurse endoscopy trial (MINUET) | 61 | 54 | |
| HTA 2006, 10, 43 | Randomised controlled trial of asynchronous and synchronous telemedicine in dermatology – RCT-ASTID | 62 | 55 | |
| HTA 2006, 10, 50 | Amniocentesis results: investigation of anxiety (ARIA) | 63 | 56 | |
| HTA 2007, 11, 8 | A study to evaluate the most cost-effective way to screen for chlamydia trachomatis genital tract infection and reduce its prevalence and associated burden of disease (ClaSS) | 126 | 57 | |
| HTA 2007, 11, 10 | EXERT (exercise evaluation randomised trial) – randomised trial comparing leisure centre-based exercise on prescription, home-based walking and usual advice in primary care | 64 | 58 | |
| HTA 2007, 11, 24 | Clinical effectiveness and cost of repetitive transcranial magnetic stimulation versus ECT in severe depression: a multi-centre randomised controlled trial and economic analysis | 65 | 59 | |
| HTA 2007, 11, 25 | An RCT and economic evaluation of direct versus indirect and individual versus group modes of speech and language therapy for children with primary language impairment | 66 | 60 | |
| HTA 2007, 11, 31 | The PRIME breast cancer trial (postoperative radiotherapy in minimum-risk elderly) | 67 | 61 | |
| HTA 2007, 11, 35 | Birmingham rehabilitation uptake maximisation study (BRUM). Home-based versus hospital-based cardiac rehabilitation in a multi-ethnic population: cost-effectiveness and patient adherence | 68 | 62 | |
| HTA 2007, 11, 37 | A randomised controlled trial of longer-term clinical outcomes and cost-effectiveness of standard and new antiepileptic drugs (SANAD) | 69 | 63 | Arm A: carbamazepine as standard drug |
| HTA 2007, 11, 37 | 70 | 63 | Arm B: valproate as standard drug | |
| HTA 2007, 11, 49 | The cost-effectiveness of functional cardiac testing in the diagnosis and management of coronary heart disease | 71 | 64 | |
| HTA 2007, 11, 16 | Efficacy and cost-effectiveness of physiotherapy for children less than four years old with cerebral palsy | 127 | 65 | |
| HTA 2007, 11, 42 | Acceptability, benefit and costs of early screening for hearing disability | 135 | 66 | |
| HTA 2008, 12, 4 | Does befriending by trained lay workers improve psychological well-being and quality of life for carers of people with dementia, and at what cost? A randomised controlled trial | 74 | 67 | |
| HTA 2008, 12, 13 | STOOL – Stepped Treatment of Older adults On Laxatives | 75 | 68 | |
| HTA 2008, 12, 14 | Randomised trial of fluoxetine and cognitive behavioural therapy versus fluoxetine alone in adolescents with persistent major depression | 76 | 69 | |
| HTA 2008, 12, 22 | Are topical or oral Ibuprofen equally effective for the treatment of chronic knee pain in older people (Topical or Oral Ibuprofen) | 72 | 70 | |
| HTA 2008, 12, 23 | A prospective randomised comparison of minor surgery in primary and secondary care | 73 | 71 | |
| HTA 2008, 12, 29 | Absorbent products for urinary/faecal incontinence: A comparative evaluation of key product categories | 131 | 72 | Clinical trial 1, module 3: a comparison of the performance and cost-effectiveness of disposable and washable designs for light incontinence when used by women living in the community |
| HTA 2008, 12, 29 | 132 | 72 | Clinical trial 2a, module 2: a comparison of the performance and cost-effectiveness of disposable and washable designs for moderate/heavy incontinence when used by men and women living in the community | |
| HTA 2008, 12, 29 | 133 | 72 | Clinical trial 2b, module 1: a comparison of the performance and cost-effectiveness of disposable designs for moderate/heavy incontinence when used by men and women living in nursing homes | |
| HTA 2008, 12, 31 | The place of minimal access surgery amongst people with gastro-oesophageal reflux disease (GORD) – a UK collaborative study | 77 | 73 | |
| HTA 2009, 13, 9 | Controlling hypertension and hypotension immediately post stroke (CHHIPS) trial | 110 | 74 | Pressor arm of the trial |
| HTA 2009, 13, 9 | 78 | 74 | This describes the depressor limb only | |
| HTA 2009, 13, 13 | A randomised controlled trial to estimate the clinical and cost-effectiveness of four different methods of mechanical support in severe ankle sprains (CAST) | 79 | 75 | |
| HTA 2009, 13, 15 | A randomised controlled trial to determine the effect of blood glucose self-monitoring in people with type 2 diabetes (DiGEM) | 80 | 76 | |
| HTA 2009, 13, 19 | Development and randomised controlled trial of dipsticks and diagnostic algorithms for the management of UTI | 81 | 77 | |
| HTA 2009, 13, 21 | Neuroleptics in adults with aggressive challenging behaviour and intellectual disability (NACHBID) | 82 | 78 | |
| HTA 2009, 13, 22 | Randomised controlled trial to determine the cost-effectiveness of fluoxetine for mild to moderate depression with somatic symptoms in primary care – THREshold for AntiDepressant treatment (THREAD) | 125 | 79 | |
| HTA 2009, 13, 27 | Ibuprofen and paracetamol in combination and separately for fever in pre-school children presenting to primary care: a randomised controlled trial (PITCH) | 84 | 80 | |
| HTA 2009, 13, 28 | A randomised controlled trial to compare minimally invasive glucose monitoring devices to conventional monitoring in the management of insulin-treated diabetes mellitus (MITRE) | 85 | 81 | |
| HTA 2009, 13, 30 | Psychological interventions for postnatal depression – randomised controlled trial and economic evaluation (PONDER) | 86 | 82 | |
| HTA 2009, 13, 33 | Randomised controlled trial of continuous positive airways pressure and non-invasive positive pressure ventilation in the management of patients presenting with acute cardiogenic pulmonary oedema (3CPO) | 87 | 83 | |
| HTA 2009, 13, 37 | A double-blind randomised placebo-controlled trial of topical nasal steroids in 4- to 11-year-old children with persistent bilateral otitis media with effusion (OME) in primary care | 88 | 84 | |
| HTA 2009, 13, 39 | Rehabilitation of older patients: day hospital compared to rehabilitation at home | 89 | 85 | |
| HTA 2009, 13, 47 | Use of aciclovir and/or prednisolone for the early treatment of Bel’s palsy: the Bells study | 90 | 86 | |
| HTA 2009, 13, 51 | A randomised trial of human papilloma virus testing in primary cervical screening (ARTISTIC) | 91 | 87 | |
| HTA 2009, 13, 53 | A randomised preference trial of medical versus surgical termination of pregnancies less than 14 weeks’ gestation (TOPS) | 92 | 88 | |
| HTA 2009, 13, 54 | Randomised controlled trial of the use of three dressing regimens in the management of chronic ulcers of the foot in diabetes | 93 | 89 | |
| HTA 2009, 13, 55 | VenUS II: larval therapy venous ulcer study | 94 | 90 | |
| HTA 2009, 13, 56 | Randomised controlled trial and economic modelling to evaluate the place of antimicrobial agents in the management of venous leg ulcers (VULCAN) | 95 | 91 | |
| HTA 2010, 14, 1 | Multi-centre randomised controlled trial examining the cost-effectiveness of contrast-enhanced high field magnetic resonance imaging in women scheduled for wide local excision (COMICE) | 96 | 92 | |
| HTA 2010, 14, 5 | Effectiveness and cost-effectiveness of arthroscopic lavage in the treatment of osteoarthritis of the knee (the KORAL study) | 97 | 93 | |
| HTA 2010, 14, 6 | A randomised 2 × 2 trial of community versus hospital rehabilitation, followed by telephone or conventional follow-up: impact on quality of life, exercise capacity and use of health care resources | 98 | 94 | |
| HTA 2010, 14, 13 | North of England study of tonsillectomy and adeno-tonsillectomy in children (NESSTAC) | 99 | 95 | |
| HTA 2010, 14, 14 | Multi-centre randomised controlled trial of the cost-effectiveness of infra-inguinal percutaneous transluminal angioplasty (PTA) versus reconstructive surgery for severe limb ischaemia (BASIL) | 100 | 96 | |
| HTA 2010, 14, 15 | A randomised controlled multicentre trial of treatments for adolescent anorexia nervosa including assessment of cost-effectiveness and patient acceptability – the TOuCAN trial | 101 | 97 | |
| HTA 2010, 14, 20 | Antenatal screening for haemoglobinopathies in primary care: a cluster randomised trial to inform a simulation model (SHIFT) | 102 | 98 | |
| HTA 2010, 14, 22 | A randomised controlled trial of cognitive behaviour therapy and motivational interviewing for people with type 1 diabetes mellitus and suboptimal glycaemic control (ADaPT) | 103 | 99 | |
| HTA 2010, 14, 23 | A single blind randomised controlled trial to determine the effectiveness and cost utility of manual chest physiotherapy techniques in the management of exacerbations of chronic obstructive pulmonary disease (MATREX) | 104 | 100 | |
| HTA 2010, 14, 26 | What is the clinical effect and cost-effectiveness of treating upper limb spasticity due to stroke with botulinum toxin? | 105 | 101 | |
| HTA 2010, 14, 35 | Conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR) | 106 | 102 | |
| HTA 2010, 14, 41 | A multicentred randomised controlled trial of a primary care-based cognitive behavioural programme for low back pain (UK-Best) | 107 | 103 | |
| HTA 2010, 14, 43 | Antidepressant drug therapy vs a community-based psychosocial intervention for the treatment of moderate postnatal depression: a pragmatic randomised controlled trial (RESPOND) | 108 | 104 | |
| HTA 2010, 14, 46 (1–130) | Head-to-head comparison of two H1N1 swine influenza vaccines in children aged 6 months to 12 years | 109 | 105 | |
| HTA 2010, 14, 52 | LIFELAX – Diet and lifestyle vs laxatives in the management of constipation in older people | 111 | 106 | |
| HTA 2011, 14, 55 | A randomised, partially observer-blind, multicentre, head-to-head comparison of a two-dose regimen of Baxter and GSK H1N1 pandemic vaccines, administered 21 days apart | 112 | 107 | |
| HTA 2011, 15, 3 | A comparison of automated technology and manual cervical screening (MAVARIC) | 113 | 108 | |
| HTA 2011, 15, 8 | Randomised controlled trial of ion-exchange water softeners for the treatment of atopic eczema in children (SWET) | 115 | 109 |
Appendix 3 Data extraction specification form
| Number | What is the subquestion this field is used to answer? | Field name | Description | Field format | Any additional links to the field format | Reference to classification | Source | Anticipated problems | Is information in this field publicly available? | Can this field be filled in for ongoing trials? |
|---|---|---|---|---|---|---|---|---|---|---|
| e.g. 1 | Xxxxx | Include in here field name used in the database | Include in here exactly what information needs to be included in this field | Include in here whether this will be a numerical value or text, and what, if any, classifications will be used | If yes, more information is inputted | Is there a reference for the classification? | Where is this information likely be found, e.g. monograph, NETSCC Information Management System, project files? | Any information about foreseeable problems | Yes/no | Yes/no |
| 1 | What type of design? | Planned_design_framework | What was the planned design framework of the trial (primary hypothesis)? | Drop-down menu: superiority, non-inferiority, equivalence | Chan AW, Hrobjartsson A, Jorgensen KJ, Gotzsche PC, Altman DG. Discrepancies in sample size calculations and data analyses reported in randomised trials: comparison of publications with protocols. BMJ 2008;337:a2299 | Protocol | Authors may not clearly distinguish between non-inferiority and equivalence; suggested solution to use the sample size calculation to help identify | Yes for future trials as protocol published on the HTA website | Yes | |
| 2 | What type of design? | Actual_design_framework | What was the actual design framework of the trial (primary hypothesis)? | Drop-down menu: superiority, non-inferiority, equivalence | As above | As above | Monograph | Authors may not clearly distinguish between non-inferiority and equivalence; suggested solution to use the sample size calculation to help identify | Yes | No |
List of abbreviations
- AcoRD
- Attributing the costs of health and social care Research and Development
- AIC
- Akaike information criterion
- ANCOVA
- analysis of covariance
- ARCO
- Attributing revenue costs of externally funded non-commercial Research in the NHS
- BCL
- BMJ checklist
- CATT
- Comparison of Age-related Macular Degeneration Treatments Trial
- CBT
- cognitive–behavioural therapy
- CCT
- Current Controlled Trials
- CEAC
- cost-effectiveness acceptability curve
- CONSORT
- Consolidated Standards of Reporting Trials
- CUA
- cost–utility analysis
- DTD
- document type definition
- EME
- Efficacy and Mechanism Evaluation
- EQ-5D
- European Quality of Life-5 Dimensions
- EQUATOR
- Enhancing the Quality and Transparency of Health Research
- EU
- European Union
- FEC
- full economic costing
- GLM
- generalised linear model
- GP
- general practitioner
- HIV
- human immunodeficiency virus
- HRCS
- Health Research Classification System
- HS&DR
- Health Services and Delivery Research
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- ICH
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
- ICMJE
- International Committee of Medical Journal Editors
- ICTRP
- International Clinical Trials Registry Platform
- ID
- identification
- ISRCTN
- International Standard Randomised Controlled Trial Number
- IVAN
- Inhibit VEGF in Age-related Choroidal Neovascularisation
- MeSH
- medical subject heading
- MIS
- Management Information System
- MRC
- Medical Research Council
- NETSCC
- NIHR Evaluation, Trials and Studies Coordinating Centre
- NICE
- National Institute for Health and Care Excellence
- NIH
- National Institutes of Health
- NIHR
- National Institute for Health Research
- PCT
- primary care trust
- PGfAR
- Programme Grants for Applied Research
- PHR
- Public Health Research
- PRECIS
- pragmatic–explanatory continuum indicator summary
- PRIME
- Peer Review Intervention for Monitoring and Evaluating sites
- PRISMA
- Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- ProtecT
- Prostate Testing for Cancer and Treatment
- QALY
- quality-adjusted life-year
- R&D
- research and development
- RCT
- randomised controlled trial
- SD
- standard deviation
- SF-6D
- Short Form questionnaire-6 Dimensions
- SPIRIT
- Standard Protocol Items for Randomised Trials
- SPSS
- Statistical Product and Service Solutions
- STEPS
- Strategies for Trial Enrolment and Participation Study
- UKCRC
- UK Clinical Research Collaboration
- URL
- uniform resource locator
- WHO
- World Health Organization
- XML
- Extensible Markup Language