
Notes
Article history
This issue of Health Technology Assessment contains a project originally commissioned by the MRC but managed by the Efficacy and Mechanism Evaluation Programme. The EME programme was created as part of the National Institute for Health Research (NIHR) and the Medical Research Council (MRC) coordinated strategy for clinical trials. The EME programme is funded by the MRC and NIHR, with contributions from the CSO in Scotland and NISCHR in Wales and the HSC R&D, Public Health Agency in Northern Ireland. It is managed by the NIHR Evaluation, Trials and Studies Coordinating Centre (NETSCC) based at the University of Southampton.
The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from the material published in this report.
Declared competing interests of authors
John Zajicek reports grants and personal fees from the Medical Research Council, personal fees from Bayer Schering, personal fees from Institut für klinische Forschung, Berlin, grants from the Multiple Sclerosis Society and grants from the Multiple Sclerosis Trust outside the submitted work. David Miller reports grants from Multiple Sclerosis Society of Great Britain and Northern Ireland, grants from University College London/University College London Hospitals Biomedical Research Centre, during the conduct of the study; grants and other from Biogen Idec, grants and other from Novartis, grants and other from GlaxoSmithKline, grants from the National Institute for Health Research, grants from Genzyme, grants from the US National Multiple Sclerosis Society and the Multiple Sclerosis Society of Great Britain and Northern Ireland, other from Bayer Schering, other from Mitsubishi Pharma Ltd, other from Merck, other from Chugai and personal fees from McAlpines Multiple Sclerosis, 4th edition, outside the submitted work. David MacManus reports grants from Biogen Idec, grants from GlaxoSmithKline, grants from Apitope, grants from Novartis and grants from Richmond Pharma outside the submitted work.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2015. This work was produced by Ball et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
This study was based on a randomised controlled trial (RCT) of oral Δ9-tetrahydrocannabinol (Δ9-THC) compared with matching placebo, in patients with progressive multiple sclerosis (MS), incorporating a magnetic resonance imaging (MRI) substudy. 1 We aimed to test the hypothesis that oral Δ9-THC will slow progression of primary progressive MS (PPMS) and secondary progressive MS (SPMS) over 3 years.
Structure of this report
The report begins with a summary of the background to MS and treatments for the disease, findings from our previous study testing symptomatic benefits from cannabinoid use in MS and a review of trials focusing on alteration of disease course in progressive MS. This first chapter concludes with a description of the research aims.
The report then describes the methods from the main study and MRI substudy, including study design, recruitment and randomisation, outcome assessment, sample size and pre-specified statistical analyses. This is followed by results from pre-specified statistical analyses of the primary and secondary outcomes in the main study and MRI substudy. Subsequent chapters cover further results from post-hoc exploratory analyses, Rasch measurement theory (RMT) analysis of rating scale data and economic evaluation.
The final chapters summarise the findings from the study, provide interpretation in the light of previous studies and discuss the strengths and limitations of the research, implications for health care and recommendations for future research.
Background and objectives
Multiple sclerosis and cannabinoids
Multiple sclerosis is the commonest cause of neurological disability in young adults, affecting around 100,000 people in the UK. The disease is thought to be due to a complex interaction between genes (over 100 having been identified in recent association studies) and environment (with possible factors including low levels of vitamin D and exposure to environmental agents such as the Epstein–Barr virus), leading to an autoimmune attack on the myelin insulation around central nervous system neurons. Clinically it typically initially manifests as episodes of relapse and remission in young adults [relapsing–remitting MS (RRMS)]. Around 15% of people may present with an initial gradual deterioration in neurological function, mostly at a slightly later age (PPMS). The majority of people who start with RRMS will eventually go on to a more progressive clinical course (SPMS). MS is fundamentally unpredictable and it is not possible to be certain about the clinical disease course in any single individual.
Treatments for MS fall into one of four main categories: symptom treatments; treatments for relapse (corticosteroids); disease-modifying treatments; and other treatments (including physiotherapy). Although there are an increasing number of disease-modifying treatments, these are all targeted at reducing MS relapses and, therefore, are only effective in the earlier stages of MS. There are no treatments with proven efficacy for MS progression in the absence of relapses.
Our initial Medical Research Council (MRC)-funded Cannabinoids in MS (CAMS) study2 focused on testing symptomatic benefits from cannabinoids over a 15-week period. Participants were included on the basis of having relatively stable MS in the 6 months prior to study recruitment and, of the 630 who received treatment, 95% had progressive disease. Following the main 15-week trial period, patients were offered the opportunity to continue medication in a blinded fashion for up to 12 months, during which period both disability measures and symptomatic assessments were performed. 3 The primary outcome measure assessed spasticity using the best available measurement at the time – the Ashworth scale. No treatment effect on spasticity was found during the main study, although patients felt that active medication was much more helpful than placebo in alleviating some of their distressing symptoms. This may partly demonstrate the relative insensitivity of the Ashworth scale and certainly suggests that spasticity is a very complex phenomenon. During the course of the study, experimental evidence was emerging to suggest that cannabinoids might have a neuroprotective action, which led to particular interest in the results of the 12-month follow-up study. 3 The results of the follow-up study showed significant effects on spasticity scores in the Δ9-THC arm, but not in the cannabis extract arm. There was also some evidence for an effect on disability, measured by the Expanded Disability Status Scale (EDSS) and the Rivermead Mobility Index (RMI). It is worth stressing that although the effect size in the follow-up study may appear modest, we had not expected to see any effect over a relatively short follow-up period in this group. A degree of possible self-selection bias could be countered by the intention-to-treat (ITT) analysis (which may have diluted the effect size), but it must be acknowledged that data acquisition for the long-term phase was not as complete as it would have been if long-term follow-up had been the initial study aim. However, the CAMS follow-up results did provide the first clinical evidence to support increasing experimental data raising the possibility of a neuroprotective effect of cannabinoids, as well as confirming that these medicines continued to ameliorate patient symptoms.
Most cannabinoid effects appear to be mediated through cannabinoid receptors, two types of which have been isolated and cloned: CB1 and CB2. CB1 receptors are distributed widely in the nervous system and seem to have a general role in the inhibition of neurotransmitter release, whereas CB2 receptors are principally found on cells of the immune system. The discovery of a range of endogenous endocannabinoids, the most important of which are thought to be 2-arachidonoylglycerol and arachidonoylethanolamide (anandamide), has also provoked considerable interest. The experimental basis behind a neuroprotective action for cannabinoids is becoming more convincing, with neuroprotective effects having been demonstrated in animal models of head injury and MS. There is also in vitro evidence showing cannabinoids reduce glutamate release and calcium flux as well as being antioxidants, thereby reducing free radical damage. Excess excitatory neurotransmitter (especially glutamate), release increased calcium influx and free radical damage have all been implicated in neuronal death and treatment strategies in neurodegenerative conditions have focused on reducing the impact of some or all of these mechanisms. In addition, CB1 receptor activation has been shown to reduce oligodendrocyte apoptosis in vitro, which may be of significance to some progressive forms of MS.
Trials in progressive multiple sclerosis
We conducted a systematic review by searching MEDLINE (1950 to May 2013), EMBASE (1980 to May 2013) and all Cochrane databases, using the search terms ‘cannabinoid’, ‘tetrahydrocannabinol’, ‘THC’, ‘multiple sclerosis’, ‘clinical trial’, ‘progression’, ‘primary progressive’, ‘secondary progressive’ and ‘disease course’. We included all human clinical trials, focusing on alteration of disease course rather than symptomatic benefit. These searches confirmed that there has never been a clinical trial using cannabinoids to alter disease course in progressive MS. Although several other treatments have been tested in Phase II and III clinical trials in progressive disease, including beta-interferons, rituximab, intravenous pooled immunoglobulins, myelin basic protein peptide and glatiramer acetate, none has demonstrated convincing clinical benefit.
Despite the considerable interest in developing neuroprotective treatments, several methodological problems have been encountered in the successful delivery of RCTs in MS, which have affected recent trials of progressive disease. In particular, difficulties in choice of outcome measure, coupled with high drop-out rates make long-term studies difficult. Outcome measures used in MS research have been very labour-intensive, deterring investigators from entering patients in long-term studies. Poor sensitivity means effect sizes have needed to be large so that smaller effects, which may nevertheless be important from the patient perspective, may have been missed. Ideally very large studies, with easily measured outcomes, would provide more convincing evidence of effectiveness. At the moment, however, the field is limited by methodology and the reluctance to accept alternative ways to measure disease impact. We have been focusing on developing patient-orientated outcome measures in the context of RCTs in MS, in order to concentrate on the patient perspective, increase sensitivity (thereby intending to reduce length, size and cost of RCTs), improve ease of administration and also aiming to reduce drop-out rates. One of the aims in the Cannabinoid Use in Progressive Inflammatory brain Disease (CUPID) trial was, therefore, to build on previous trial methods, incorporate a patient-reported outcome (PRO) as a coprimary outcome measure and use the trial as a vehicle to develop improved methods for future trial design.
Research aims
-
To assess the efficacy of treatment with Δ9-THC in slowing progressive MS over a follow-up period of 3 years.
-
To assess the safety of Δ9-THC.
-
To assess the use of patient-orientated outcomes [in particular the MS Impact Scale-29 version 2 (MSIS-29v2) and 20-point physical subscale (MSIS-29phys)], in order to improve the methodology by which clinical trials are conducted in progressive MS.
Chapter 2 Methods: main study and magnetic resonance imaging substudy
Study design
The CUPID study was a double-blind, randomised, placebo-controlled, multicentre, parallel-group trial incorporating a MRI substudy. The study was designed to assess the safety and effectiveness of Δ9-THC in slowing MS disease progression over a 3-year period.
Adults with progressive MS4 and an EDSS score of 4.0–6.5 (walking affected by disease but still able to walk at least 20 metres without rest, with aids if necessary) were randomised individually to receive either oral Δ9-THC or matching placebo, in a 2 : 1 ratio.
Setting
The study was conducted in 27 NHS study sites in England, Wales and Scotland, comprising 25 hospital neurology departments and two rehabilitation departments (see Appendix 1). Principal investigators (PIs) were consultant neurologists or consultants in rehabilitation medicine. The majority of study sites had expressed an interest in the study and provided feasibility data before the trial started, but two sites were recruited once the study was under way. Despite early identification of study sites, there was wide variability in the time taken to obtain NHS research and development (R&D) approval at each site, putting early pressure on the rate of participant recruitment.
Study approvals
Applications for approval to conduct the study were submitted to the South and West Devon Research Ethics Committee (REC) and the Medicine and Healthcare products Regulatory Agency (MHRA) in December 2005.
Research Ethics Committee approval was confirmed on 4 April 2006 following some minor clarifications to the protocol. The REC reference was 06/Q2103/1. A clinical trial authorisation was granted by the MHRA on 2 May 2006 following revision of the protocol to include an annual assessment of depression and to strengthen the contraceptive advice for participants and partners. The trial was assigned European Union Drug Regulating Authorities Clinical Trials (EudraCT) Number 2005–002728–33 and the International Standard Randomised Controlled Trial Number (ISRCTN) ISRCTN62942668. NHS R&D approvals were obtained from all participating sites (see Appendix 2).
Training
All research site staff including neurologists, MS specialist nurses, research nurses, physiotherapists, pharmacists and MRI staff were invited to attend one of a series of study-specific training days held across the UK before the start of the study. Training was provided on all aspects of the study protocol including eligibility, recruitment, consent, prescription and titration of study treatment, study visit and assessment schedule, study assessments, blinding, pharmacovigilance, data collection documentation, and laboratory and MRI procedures. Individual training was provided for sites joining the study at a later stage.
A short EDSS training film was produced specifically for the study and provided to sites in DVD format. All neurologists undertaking EDSS assessments for the study were required to document that they had viewed the EDSS training material prior to assessing study participants. A training DVD was also provided for those site staff conducting the MS Functional Composite (MSFC) assessments and standardised equipment for conducting the MSFC components [9-hole peg test (9-HPT), timed 25-foot walk (T25-FW) and Paced Auditory Serial Addition Test (PASAT)] was provided to all sites.
All study team members involved in the recruitment and consent process, data collection, prescription of trial treatment, adverse event (AE) reporting and participant assessment were required to undertake Good Clinical Practice (GCP) training and to keep this up to date throughout their involvement in the trial.
Participant eligibility
The study eligibility criteria were as broad and pragmatic as possible to maximise both recruitment and generalisability of results. As the aim of the study was to investigate the effectiveness of Δ9-THC in slowing MS progression, patients whose disease had been stable for 12 months or more were excluded from participation. In the absence of recent documented evidence of disease activity or status, the assessment of disease stability relied on clinical judgement in discussion with the patient and/or family member.
Factors potentially influencing baseline EDSS assessment (e.g. recent relapse or steroid therapy) were grounds for exclusion from the trial, but patients in this category could be rescreened for inclusion in the trial at a later date. It was also deemed necessary to exclude patients with baseline EDSS scores of > 6.5 [an EDSS score of 6.5 was defined as constant bilateral support (canes, crutches or braces) required to walk about 20 metres without resting] because of the relative difficulties of identifying further progression in this group.
Inclusion criteria
Potential participants had to satisfy the following criteria to be enrolled in the study:
-
18–65 years old
-
diagnosis of PPMS or SPMS
-
evidence of disease progression in the preceding year
-
EDSS score of 4.0–6.5
-
willingness to abstain from other cannabis use during the trial.
Exclusion criteria
Potential participants who met any of the following criteria were excluded from study participation:
-
immunosuppressive or immunomodulatory therapy in the previous 12 months
-
corticosteroids in the previous 3 months
-
significant MS relapse in the previous 6 months
-
serious illness or medical condition likely to interfere with study assessment
-
previous history of psychotic illness
-
sesame seed allergy
-
pregnancy
-
cannabinoids (including nabilone) taken in the previous 4 weeks (positive urinary cannabinoid test prior to study entry).
Recruitment of participants
Patients were prospectively recruited to the study between May 2006 and July 2008. Potential participants were identified by a health-care professional at each study site, usually from an existing caseload of patients known to have a diagnosis of PPMS or SPMS. A small number of patients were referred to study site neurologists by colleagues at non-participating hospitals close to recruiting sites. A few patients self-referred as a result of publicity about the study in various local and national media or from information obtained via the internet.
Arrangements for inviting patients to participate in the study varied according to circumstances, local practice and available resources. As the study started before the inception of the Comprehensive Local Research Networks, it was adopted onto the UK Clinical Research Network Dementias and Neurodegenerative Diseases Research Network portfolio which allowed access to research support staff at a few study sites. Study participants were recruited either individually or in cohorts, depending on practical arrangements at each site. Some sites held dedicated research clinics and were thus able to follow-up several study participants in one session. In terms of grouping EDSS assessments and batching laboratory samples, this was generally an efficient model and was often well received by participants who had contact with one another as a result. Other sites were only able to recruit and follow-up participants on an individual basis. Monthly recruitment totals by site are shown in Appendix 3.
Initial identification of potential participants was predominantly by direct consultant referral to the study team or by screening of hospital records. Potentially eligible patients were then contacted individually – usually by a research nurse or MS specialist nurse on behalf of the PI – to discuss the study, ascertain interest and check major eligibility criteria. Group information sessions were held at some sites for interested patients and their families. Regardless of the mode of contact, all potentially eligible patients were provided with a participant information sheet (see Appendix 4) for the study. Apparently eligible patients who were interested in taking part in the study were subsequently invited to attend a screening visit (visit 1).
Each study site kept a record of the number of patients invited for screening and the number of ineligible, eligible, non-consenting, consenting, randomised and non-randomised patients was monitored by the co-ordinating team. Throughout the study, the number of participants who discontinued trial medication prematurely or were lost to follow-up was recorded, with reasons where known.
Study site personnel
Each study site was required to nominate a treating physician and a separate assessing physician for the study, with appropriate arrangements for cover in case of staff absence. Treating physicians were responsible for assessing patient eligibility, obtaining informed consent, prescription and titration of trial treatment, reviewing participant progress and monitoring and recording AEs and concomitant medications. Assessing physicians conducted the EDSS assessments. Other study assessments (i.e. the MSFC and RMI) could be conducted by the assessing physician or a non-clinician with appropriate training. To reduce bias from potential unblinding, clinicians were advised not to change their role during the study, particularly from treating physician to assessor; this was monitored by the study co-ordinating team.
Screening visit (consent and entry to trial)
Patients attending the screening visit were given the opportunity to discuss the study and have any questions answered. The main study inclusion and exclusion criteria were checked by the treating physician (including EDSS score, assessed by the assessing clinician). Patients who were eligible and willing to participate gave written consent for the trial, including consent for their name and address to be held by the study co-ordinating centre for central management of postal and web-based study questionnaires.
All patients were required to provide blood samples for pre-trial biochemical and haematological analysis and a urine sample for baseline cannabinoid testing. Demographic data, medical history, concomitant medications and vital signs were recorded at this visit. The RMI and a MSFC practice run were completed.
Inclusion in the study was confirmed within 2 weeks of the screening visit, following results of the laboratory tests. Participants’ general practitioners (GPs) were notified of patients’ involvement in the trial once enrolment had been confirmed.
Study diary
Each participant was given a study diary which provided space for the participant to record any side effects or other relevant information to aid recall at subsequent study visits. The diary also contained advice on when/how to take the trial medication, instructions on weekly dosage, research team contact details and space for study appointment details. The diary contents were not used as part of the study data set.
Randomisation and masking
Once eligibility had been confirmed following receipt of laboratory results, participants were assigned via a secure computer-generated randomisation system to receive oral Δ9-THC (dronabinol) or matched placebo capsules for 36 months in a 2 : 1 active-to-placebo ratio. The allocation ratio was chosen in order to increase the acceptability of the trial to patients, with the aim of maximising recruitment and retention rates. The randomisation allocation sequence was generated by an independent statistician using a stochastic minimisation algorithm and balanced according to EDSS score, study site and disease type (PPMS or SPMS) with a random component. During the study, the independent statistician made periodic checks of the allocation ratios according to the minimisation factors, to ensure that the randomisation schedule was performing as intended.
To ensure allocation concealment, treatment assignment was undertaken by the co-ordinating pharmacy based at the lead hospital site, independently of the research team. The randomisation list was stored securely within the co-ordinating pharmacy and written procedures were in place in the event of a request for emergency unblinding, either for clinical reasons or to facilitate monitoring of unexpected serious adverse reactions.
Participants and all other personnel directly involved in the study were blinded to treatment allocation. The discussion of symptoms and/or side effects between participants and assessing physicians was actively discouraged, although assessing physicians occasionally had to review study patients outside the context of the study, for routine clinical follow-up or during episodes of hospitalisation. On completion of the study, each participant and the treating and assessing physicians were asked which treatment they thought the participant had been allocated.
Trial interventions
Trial medication was supplied in bottles by Insys Therapeutics, Inc. (Chandler, AZ, USA), labelled in accordance with relevant clinical trial guidelines. Active treatment (oral Δ9-THC, dronabinol) consisted of hard gelatin capsules each containing 3.5 mg of Δ9-THC. Placebo treatment consisted of identically matched sesame oil capsules such that dronabinol and placebo capsules were indistinguishable.
Trial medication was originally supplied for storage at room temperature, with a 1-year expiry. Despite the logistics of scheduling resupply at regular intervals, there were clear advantages to both participants and study sites in providing trial medication with room temperature stability. However, economic pressures on the suppliers during the course of the trial led to the introduction of refrigerated medication in order to increase shelf life and reduce wastage.
Medication was distributed to sites on an individual participant basis by the co-ordinating pharmacy. Initial supplies were provided on a weight-related basis following randomisation. Once participants were established on a steady dose, subsequent resupplies were provided on a dose-related basis.
Outcome assessments
The primary clinician-based outcome was time to first EDSS score progression. This was defined as an increase of at least 1 point from a baseline EDSS score of 4.0–5.0, or at least 0.5 points from a baseline EDSS score of 5.5–6.5, confirmed at the next scheduled 6-monthly visit. The primary patient-based outcome measure was change in MSIS-29phys score.
Secondary outcomes included the number and nature of AEs, MS Walking Scale-12 (MSWS-12v2) score, MSFC score, RMI score, Short Form questionnaire-36 items version 2 (SF-36v2) score, European Quality of Life-5 Dimensions (EQ-5D) questionnaire score, MS Spasticity Scale-88 (MSSS-88) score and a category rating scale. In addition, for a subgroup of patients allocated to the MRI substudy, outcomes included brain atrophy, in terms of annual percentage brain volume change (PBVC) and occurrence of new or newly enlarging T2 and T1 lesions from annual cranial MRI.
The EDSS was assessed at each site by a neurologist, following study-specific training. The MSFC assessment was performed by a non-physician, as long as training had been given. The RMI was similarly assessed every 6 months.
Participant-completed questionnaires
Between the screening and baseline visits, participants were asked to complete a questionnaire booklet comprising the MSIS-29v2, MSWS-12v2, SF-36v2, EQ-5D, MSSS-88 and category rating scale. The questionnaire booklet was repeated in its entirety at 3, 12, 24 and 36 months and (excluding the MSSS-88 and category rating scale) at 6, 18 and 30 months.
The baseline questionnaire was sent to all participants by post from the co-ordinating centre with a pre-paid reply envelope. Thereafter, participants could choose to complete subsequent questionnaires by post or online via a secure web-based system. In the event of non-receipt of completed questionnaires, repeat copies were sent, with e-mail reminders to those participants opting for online completion.
Participants were also asked to complete the Beck Depression Inventory-II (BDI-II) at baseline and annually thereafter. As the BDI-II was included as a safety measure rather than an outcome assessment, total scores were reported directly to PIs as they were provided.
Baseline visit (provision of trial medication)
The baseline visit (visit 2) was held approximately 2–4 weeks after the screening visit, following confirmation of eligibility and randomisation. Vital signs and AEs were recorded at this visit and the baseline MSFC assessment was completed. A prescription for trial medication was provided, with a starting dose of one capsule (3.5 mg of Δ9-THC equivalent) twice daily for all participants.
Owing to the large interindividual dose variation with oral cannabinoids, there was a 4-week titration period in which participants in both study arms could increase their dose by one capsule twice daily at weekly intervals, to a maximum weight-related dose (Table 1). Dose progression depended on adverse effects. If unwanted side effects developed, participants were advised not to increase the dose. If these side effects were considered intolerable, the dose was reduced.
| Weight (kg) | Week 1 | Week 2 | Week 3 | Week 4 to study end |
|---|---|---|---|---|
| < 60 | One capsule twice a day | Two capsules twice a day | Two capsules twice a day | Two capsules twice a day |
| 60–80 | One capsule twice a day | Two capsules twice a day | Three capsules twice a day | Three capsules twice a day |
| > 80 | One capsule twice a day | Two capsules twice a day | Three capsules twice a day | Four capsules twice a day |
Subsequent participant follow-up
Participants were scheduled to attend follow-up visits with the treating physician at 2 and 4 weeks after the baseline visit, for AE monitoring and dose adjustment. Once participants had been settled on a suitable treatment dose, clinician-based outcome data were collected at assessment visits at 3 and 6 months, then 6-monthly up to 36 months. Participants who demonstrated new EDSS score progression according to the defined study end point at 36 months attended a further visit at 42 months to confirm EDSS score progression. Monitoring of AEs, recording of vital signs and collection of safety blood samples were undertaken at the clinic visits. Urine samples for cannabinoid analysis were requested from each participant approximately four times during the study according to a random schedule, with the aim of detecting illicit cannabis use in the placebo group. Study medication was gradually discontinued over a period of a few weeks from the final visit (36 or 42 months depending on EDSS status). Table 2 shows the CUPID study visit and questionnaire completion schedule.
| Visit | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | (12) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Start of week/month | Week –2 to –4 | Week 1 | Week 3 | Week 5 | Week 13 | Month 6 | Month 12 | Month 18 | Month 24 | Month 30 | Month 36 | (Month 42) | ||
| Treating physician | ||||||||||||||
| Screening, eligibility, consent | ✗ | |||||||||||||
| Physical examination, weight | ✗ | ✗ | ||||||||||||
| Vital signs (P and BP) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | ||
| Randomisation | ✗ | |||||||||||||
| Start trial treatment | ✗ | |||||||||||||
| Dose adjustment | ✗ | ✗ | ||||||||||||
| AE monitoring | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | |||
| Issue prescription | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | ||||||
| Assessor | ||||||||||||||
| EDSS | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | |||||
| T25-FW | } | MSFC | Practice | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | |||||
| 9-HPT | Practice | ✗ | ✗ | ✗ | ✗ | (✗) | ||||||||
| PASAT | Practice | ✗ | ✗ | ✗ | ✗ | (✗) | ||||||||
| RMI | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | |||||
| Other | ||||||||||||||
| MRI (selected study sites) | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Urine | ✗ | Plus random urine testing throughout study | ||||||||||||
| Blood (genetics and biomarkers) | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Blood (FBC, U&Es, LFTs) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | |||||
| Self-completion questionnaires (postal/web) | ||||||||||||||
| MSIS-29v2 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | |||||
| MSWS-12v2 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | |||||
| SF-36v2 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | |||||
| EQ-5D | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | |||||
| Category rating scale | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | ||||||||
| MSSS-88 | ✗ | ✗ | ✗ | ✗ | ✗ | (✗) | ||||||||
| BDI-II | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Health economics dataa | ✗ | ✗a | ✗a | ✗a | ✗a | ✗a | ✗a | |||||||
Various strategies were implemented during the course of the study to improve follow-up and data collection. It was identified that the completion rate for online questionnaires was lower than for postal questionnaires, despite e-mail reminders. Recognising that study participants may access their e-mail sporadically and, therefore, may miss electronic reminders, the co-ordinating centre added postal reminders, leading to an improvement in online completion rates.
The number and length of PROs appeared burdensome for a number of participants, including some who discontinued study medication prematurely but agreed to remain in follow-up. At the discretion of the co-ordinating centre, the questionnaire battery was reduced for some individual participants in order to secure at least some outcome data which otherwise were at risk of being lost completely. In these circumstances, priority was given to capturing the patient-reported primary outcome data (MSIS-29v2) at the expense of other measures, if necessary.
For participants in whom visual or motor/sensory impairment made questionnaire completion difficult, telephone support was provided by the co-ordinating centre to supply responses. Some participants with accumulating disability found it increasingly troublesome or tiring to attend clinic visits for EDSS assessment. Participants in this category who had also stopped medication prematurely were particularly at risk of being lost to follow-up since they had no need to attend in person for repeat supply of medication. To improve data completeness, the option of validated telephone EDSS assessment for participants unable or unwilling to attend clinic visits was introduced from June 2010. All telephone EDSS assessments were conducted by a trained clinician from the Plymouth co-ordinating team, following formal consent from participants.
Safety monitoring
In addition to the annual assessment of depression (BDI-II), safety monitoring included standard clinical laboratory assessments (chemistry, haematology, liver function) at 3 and 6 months and 6-monthly thereafter. Blood samples were analysed by a central laboratory and results reported to the PI by the co-ordinating centre.
Assessment of AEs was undertaken by the investigator at each study visit, with particular emphasis on the titration phase. If participants could not attend either the 2- or 4-week visit, site teams were encouraged to review AEs by telephone in order to optimise the individual dose of study treatment for each participant. Signs and symptoms were graded as mild, moderate or severe. Seriousness and causality were assessed by the reporting PI. AEs satisfying the criteria for serious AEs (SAEs) were subject to expedited reporting to the co-ordinating centre where a second assessment of causality was made. If either causality assessment indicated a definite, probable or possible relationship to study medication, an assessment of expectedness was made with reference to the current Investigator’s Brochure.
Magnetic resonance imaging substudy
Thirteen study sites participated in a MRI substudy. MRI site selection was on the basis of capacity and cost, with all MRI sites being required to perform a pre-enrolment qualifying scan prior to the start of participant recruitment. Non-contrast brain MRI was performed at baseline and annually in 273 participants (183 allocated to active treatment, 91 allocated to placebo). Images were analysed for PBVC and new and enlarging lesions at the Queen Square Multiple Sclerosis Centre, NMR Research Unit Trials Office, University College London’s Institute of Neurology, London, UK.
Data management and monitoring
Paper-based, two-part, no-carbon-required case report forms (CRFs) were used for both treating physician- and assessor-acquired data. Original data were sent by post to the co-ordinating centre in Plymouth and entered onto a central database held on a secure server. A system of double data entry was used, enabling generation of a single data set following a process of data comparison. Bespoke database reports were created to track participant status throughout the trial and ensure that data were requested and received from sites in a timely manner. A robust process was in place for clarification of queries in the case of missing or ambiguous data. The quality of the trial data were monitored using a combination of centralised data checking and site visits at which participant CRFs were compared with clinical case notes. Participants returned their self-completion questionnaires in Freepost™ envelopes directly to the co-ordinating office for double data entry.
Trial oversight
A Trial Steering Committee (TSC) (see Appendix 5 for details), including an independent chairperson, a neurologist, a statistician and a lay representative, was responsible for trial oversight and met on an annual basis. An independent Data Monitoring Committee (IDMC) (see Appendix 5 for details) met annually to review unblinded safety and efficacy data. Interim analyses of primary outcomes were produced by an independent statistician on request from the IDMC, using the pre-defined Haybittle–Peto boundary stopping rule. Four interim analyses were undertaken after 298 and 493 participants had been recruited and, then, annually during the follow-up period. The IDMC recommended continuation of the trial following all interim analyses. Appendix 6 gives details of patient and public involvement in the CUPID study.
Sample size and power
Previous data suggested a likely progression rate of approximately 70% in the placebo group. 5 Based on this and an expected 5% annual loss to follow-up rate, recruiting 492 patients provided 90% power, at a two-sided 5% significance level, to detect a one-third reduction in hazard of progression [i.e. hazard ratio (HR) 0.67], corresponding to a relative reduction in risk of progression over 3 years of 21% (from 70% to 55% progression in the Δ9-THC group).
Initial data from 210 patients with MS admitted in acute relapse for intravenous steroid treatment baseline and post treatment showed the standard deviation (SD) of the scores from MSIS-29v2 at baseline and follow-up were 21.8 and 24.3, respectively (Professor Jeremy Hobart, Institute of Neurology, London, 1999–2002, unpublished data). The SD of the difference in scores was estimated to be 20.6. With this SD, a difference in means of seven points (one-third of a SD) could be detected in a sample of 492 on a two-sided test carried out at the 5% level, with power in excess of 90%.
For the MRI substudy, allowing for losses to follow-up at a rate of 5% per year, it was estimated that a total of 261 patients, allocated to active treatment and placebo in a 2 : 1 ratio, would give 90% power to detect 40% slowing in rate of atrophy, with scans performed pre treatment and then at years 1, 2 and 3.
End Point Committee
An End Point Committee (EPC) was convened to adjudicate on EDSS score progression in participants for whom missing EDSS scores prevented confirmation of this end point according to protocol. The committee, comprising the chief investigator and two other consultant neurologists (one independent), met once on 27 February 2012. The EPC review was blinded to treatment allocation and included consideration of additional clinical details obtained from participants’ medical records. The outcomes determined by this committee were used in sensitivity analyses of EDSS score progression. Terms of reference are outlined in Appendix 7.
Statistical methods
The statistical analysis plan (SAP) was finalised and agreed by the TSC before unblinding. Data analysis, using the statistical software R version 2.14.2 (The R Foundation for Statistical Computing, Vienna, Austria), was undertaken on an ITT basis. All tests were carried out at the 5% significance level, with no adjustments for multiple testing.
In models for each outcome, main effects of study site, baseline EDSS score, disease type, age at registration, sex and weight were considered, as well as treatment allocation.
Pre-specified analyses of primary clinical outcomes
Primary analysis of time to first Expanded Disability Status Scale score progression
Kaplan–Meier estimates were used to show probability of EDSS score progression in the two treatment groups and in groups defined in terms of baseline EDSS score. Analysis of time to first EDSS score progression used a Cox proportional hazards (PH) model. Primary analysis was based on EDSS data obtained according to trial schedule; losses to follow-up before confirmed progression were considered as missing data and treated as censored observations at the time of the last visit for which EDSS measurements were taken.
Sensitivity analyses of time to first Expanded Disability Status Scale score progression
Sensitivity of conclusions from the main analysis of time to first EDSS score progression were assessed by repeating the analysis, considering all losses to follow-up as progression events at the time of the scheduled visit after the last visit for which EDSS measurements were taken.
Evidence for the sensitivity of conclusions to the effect of study sites with high losses to follow-up was assessed by repeating the analysis, under each way of dealing with losses to follow-up (censored observations or progression events), sequentially removing study sites with high rates of loss to follow-up.
Furthermore, sensitivity of conclusions to the decisions from the EPC review regarding EDSS score progressions was assessed by repeating the analysis, under each way of dealing with losses to follow-up, incorporating the findings from the EPC review.
Subgroup analyses of time to first Expanded Disability Status Scale score progression
Hazard ratios (active : placebo) for EDSS score progression, in subgroups defined by sex, baseline EDSS score, disease type, weight and age, were estimated.
Primary analysis of change in Multiple Sclerosis Impact Scale-29 20-point physical subscale
Total MSIS-29phys scores were calculated using previously published methods. 6 Repeated measures data on MSIS-29phys were analysed using multilevel models, which included time (visit) as well as the other pre-specified covariates. Individual differences in scores were incorporated using random coefficients. 7 Backward elimination was used to identify a final, reduced model, including effects significant at the 5% level, as well as effects of time and treatment allocation.
Pre-specified analyses of secondary clinical outcomes
Multiple Sclerosis Functional Composite, Multiple Sclerosis Walking Scale-12, Rivermead Mobility Index, Short Form questionnaire-36 items (physical health subscale), Multiple Sclerosis Spasticity Scale-88
Scores for each MSFC component were calculated using previously published methods. 8 MSFC component-wise z-scores9 were computed using results from all participants at visit 2 as the reference population. 8 MSFC composite scores were calculated from the mean of the component-wise z-scores. Total scores for MSWS-12v2,10 RMI,6 SF-36v2 physical health subscale [SF-36(PH)]11 and MSSS-8812 were calculated using an algorithm analogous to that used for MSIS-29phys for dealing with missing data.
Repeated measures of MSFC (composite and component-wise z-scores), MSWS-12v2 (total score), RMI (total score), SF-36(PH) (total score) and MSSS-88 [total score for each of three sections from the eight subscales, where MSSS-88 (1) combines subscales 1–3 and concerns muscle stiffness/spasms, pain and discomfort; MSSS-88 (2) combines subscales 4–6 and concerns activity, walking and body movements; and MSSS-88 (3) combines subscales 7 and 8 and concerns feelings and social functioning], were analysed using multilevel models, using the same covariates and variable selection procedure as for MSIS-29phys.
Investigation of adverse events and serious adverse events
Serious adverse events and the most common AEs (i.e. those which affected at least 10% of all participants) were summarised in terms of frequencies and relative frequencies.
Category rating scales
Category rating scales, included in patient-completed questionnaires sent at 3 months, 1, 2 and 3 years from baseline (visits 5, 7, 9 and 11), consisted of 16 questions: 1–8 related to how the patient felt over the past week and 9–16 related to how the patient felt at the time of completing the questionnaire compared with just before the start of the study. Analysis focused on questions 9–16, which were on a 1–11 scale, where 1 = very much better; 6 = no difference; and 11 = very much worse. Responses to these questions were grouped as 1–5 = better; 6 = no change; 7–11 = worse and summarised in terms of frequencies and relative frequencies in the two treatment groups, at each follow-up. Chi-squared tests for trend were used to test for an association between treatment allocation and response to question, allowing for the ordered nature of the grouped responses. No adjustments were made for multiple comparisons.
Analysis of premature discontinuations of trial medication and losses to follow-up
Kaplan–Meier estimates were used to show probability of discontinuation of trial medication in the two treatment groups. Analyses of time to discontinuation of trial medication or loss to follow-up used Cox PH models, in order to investigate the effect of treatment allocation and any potential pre-randomisation variables, on the risk of discontinuation. A forward selection procedure was used to identify a suitable model, including effects significant at the 5% level.
Pre-specified analyses of magnetic resonance imaging substudy
A multilevel model was used to analyse repeated measures data on PBVC13,14 from cranial MRI, transformed to cumulative, relative PBVC on the log10 scale.
Logistic regression models were used to examine the effect of treatment allocation and other pre-specified covariates on new T1 hypointense and new or enlarging T2 hyperintense lesions during follow-up. Participants were classified as having either no, or at least one, new or enlarging lesion(s) during follow-up. Final models were identified using a forward selection procedure and included main effects and interactions significant at the 5% level, as well as the main effect of treatment.
Further analyses
Details of post-hoc exploratory analyses performed on outcomes from the main study and MRI substudy are presented in Chapter 4.
One of the goals of the CUPID trial was to examine the contribution of advanced, but not widely used, methods, particularly rating scale psychometric methods. With this in mind we undertook a RMT-based analysis of data generation by the MSIS-29v2, MSWS-12v2 and MSSS-88. Specifically, when compared with traditional psychometric methods, RMT enables a more sophisticated evaluation of rating scale performance, the generation (contingent on appropriate findings) of interval-level measurements for analysis (rather than ordinal scores) and legitimate analyses of changes and differences at the individual person level. These methods are studied in detail in Chapter 5. An economic evaluation is detailed in Chapter 6.
Ethics and research governance approval
The study was approved by the South and West Devon REC and conducted in accordance with GCP guidelines. Eligible patients provided written informed consent before participation.
Trial registration
The trial was registered as Current Controlled Trials ISRCTN62942668.
Role of the funding source
The funders had no role in study design, data collection, analysis, interpretation or writing of the report.
Summary of changes to the study protocol
The protocol as approved by MHRA and REC at the start of the study underwent four amendments of significance to the conduct of the study. An additional exclusion criterion (recent use of any experimental therapies with potential disease-modifying actions) was added during the first few months of the study in order to reduce potential confounding of results. At the start of 2009, refrigerated storage conditions for the trial medication were introduced. In April 2009, an additional follow-up phase was added to the study – this used the fact that participants with new EDSS progression at 36 months were required to be reviewed at 42 months on treatment as an opportunity to follow up the remaining participants off treatment from months 36 and 42. The protocol was last amended in May 2010 to include the option of EDSS assessment by telephone as opposed to face to face.
Chapter 3 Results: main study and magnetic resonance imaging substudy
Patients were randomised between May 2006 and July 2008 and the final follow-up data collection took place in January 2012.
Unblinding of randomised treatments
The treatment allocation was unblinded six times during the course of the trial at the request of the sponsor organisation to assist in the management of suspected unexpected serious adverse reactions (SUSARs). In addition, unblinding of four participants at four separate sites was carried out at the request of the local investigator, on clinical grounds.
Telephone-based assessment of Expanded Disability Status Scale score
Of 3812 assessments of EDSS score over the study period, 42 (1.1%) were by telephone, rather than face to face. These telephone assessments were carried out on a total of 39 patients (25 assigned to active; 14 assigned to placebo).
Figure 1 shows the trial profile and Table 3 shows discontinuations of trial medication and losses to follow-up. A total of 498 patients were randomly assigned to active treatment (n = 332) or matching placebo (n = 166). The data from three patients (two randomised to active, one to placebo) were removed from the trial because they withdrew their consent after randomisation. A further two patients (one randomised to active, one to placebo) were found to be ineligible after randomisation. Four hundred and ninety-three (329 active, 164 placebo) received their allocated intervention and were therefore included in an ITT analysis. Of the 493 randomised and treated participants, 415 (84%) completed follow-up, of whom 119 (29%) had prematurely discontinued trial medication (Figure 1).
FIGURE 1.
The Consolidated Standards of Reporting Trials (CONSORT) flow diagram. a, Nine patients initially did not fulfil entry criteria, but did so on subsequent rescreening. DNA, did not attend; PEP, primary end point of EDSS score progression.

| Analysis population and scenarios for follow-up | All randomised patients (N = 498) | |||||
|---|---|---|---|---|---|---|
| Active (n = 329; 66.7%) | Placebo (n = 164; 33.3%) | All (n = 493) | ||||
| Analysis population, n (%) | ||||||
| Full analysis set | 329 | (99.1) | 164 | (98.8) | 493 | (99.0) |
| Scenarios for follow-up, n (%) of full analysis set | ||||||
| Completed follow-up on trial treatment | 178 | (54.1) | 118 | (72.0) | 296 | (60.0) |
| Completed follow-up having prematurely discontinued trial medication | 89 | (27.1) | 30 | (18.3) | 119 | (24.1) |
| Discontinued trial medication and subsequently lost to follow-up | 51 | (15.5) | 10 | (6.1) | 61 | (12.4) |
| Lost to follow-up without previous discontinuation of trial medication | 11 | (3.3) | 6 | (3.7) | 17 | (3.4) |
Baseline comparability of randomised groups
Baseline patient and disease characteristics were similar in both treatment groups (Table 4). At baseline, 59% of participants were women, 61% had SPMS and 78% had an EDSS score of 6.0 or 6.5. There were no important differences in outcome measures assessed at baseline (Table 5).
| Patient baseline characteristics | Randomised patients (N = 493;a 100%) | |||||
|---|---|---|---|---|---|---|
| Active (n = 329; 66.7%) | Placebo (n = 164; 33.3%) | All (n = 493) | ||||
| Age in years, mean (SD) | 52.29 | (7.6) | 51.97 | (8.2) | 52.19 | (7.8) |
| Weight in kg, mean (SD) | 74.54 | (16.5) | 75.93 | (16.5) | 75.00 | (16.5) |
| Men, n (%) | 133 | (40.4) | 68 | (41.5) | 201 | (40.8) |
| Women, n (%) | 196 | (59.6) | 96 | (58.5) | 292 | (59.2) |
| Disease type, n (%)b | ||||||
| PPMS | 126 | (38.3) | 65 | (39.6) | 191 | (38.7) |
| SPMS | 203 | (61.7) | 99 | (60.4) | 302 | (61.3) |
| EDSS score, n (%)b | ||||||
| 4.0 | 20 | (6.1) | 9 | (5.5) | 29 | (5.9) |
| 4.5 | 18 | (5.5) | 7 | (4.3) | 25 | (5.1) |
| 5.0 | 22 | (6.7) | 10 | (6.1) | 32 | (6.5) |
| 5.5 | 16 | (4.9) | 8 | (4.9) | 24 | (4.9) |
| 6.0 | 169 | (51.4) | 85 | (51.8) | 254 | (51.5) |
| 6.5 | 84 | (25.5) | 45 | (27.4) | 129 | (26.2) |
| Median (25th–75th percentiles) | 6.0 | (6.0–6.5) | 6.0 | (6.0–6.5) | 6.0 | (6.0–6.5) |
| Mean (SD) | 5.83 | (0.69) | 5.88 | (0.67) | 5.85 | (0.69) |
| Outcome measures at baseline | Randomised patients (N = 493;a 100%) | |||||
|---|---|---|---|---|---|---|
| Active (n = 329; 66.7%) | Placebo (n = 164; 33.3%) | All (n = 493) | ||||
| EDSS scoreb | ||||||
| Mean (SD) | 5.83 | (0.69) | 5.88 | (0.67) | 5.85 | (0.69) |
| Median (25th–75th percentiles) | 6.0 | (6.0–6.5) | 6.0 | (6.0–6.5) | 6.0 | (6.0–6.5) |
| MSIS-29phys scorec | ||||||
| Mean (SD) | 55.03 | (10.81) | 55.19 | (10.96) | 55.08 | (10.85) |
| Median (25th–75th percentiles) | 55 | (47.00–63.00) | 56 | (47.25–63.00) | 55.78 | (47.00–63.00) |
| Not reported, n (%) | 3 | (0.9) | 2 | (1.2) | 5 | (1.0) |
| MSFC componentsd | ||||||
| T25-FW | ||||||
| Time in seconds,e mean (SD) | 20.34 | 30.16 | 15.25 | 13.41 | 18.64 | 25.9 |
| Median (25th–75th percentiles) | 10.95 | (7.95–18.60) | 10.85 | (7.85–16.55) | 10.90 | (7.90–17.54) |
| Not reported, n (%) | 4 | (1.2) | 1 | (0.6) | 5 | (1.0) |
| 9-HPT (dominant hand) | ||||||
| Time in seconds, mean (SD) | 36.74 | (41.68) | 38.65 | (43.42) | 37.37 | (42.23) |
| Median (25th–75th percentiles) | 27.27 | (22.39–34.1) | 27.4 | (22.89–35.34) | 27.33 | (22.55–34.79) |
| Not reported, n (%) | 1 | (0.3) | 0 | (0.0) | 1 | (0.2) |
| 9-HPT (non-dominant hand) | ||||||
| Time in seconds, mean (SD) | 41.79 | (49.31) | 34.82 | (27.13) | 39.46 | (43.3) |
| Median (25th–75th percentiles) | 28.08 | (23.25–36.94) | 28.62 | (24.74–35.45) | 28.12 | (23.44–36.55) |
| Not reported, n (%) | 1 | (0.3) | 0 | (0.0) | 1 | (0.2) |
| 9-HPT (standard score)f | ||||||
| Mean (SD) | 0.04 | (0.01) | 0.03 | (0.01) | 0.04 | (0.01) |
| Median (25th–75th percentiles) | 0.04 | (0.03–0.04) | 0.04 | (0.03–0.04) | 0.04 | (0.03–0.04) |
| Not reported, n (%) | 1 | (0.3) | 0 | (0.0) | 1 | (0.2) |
| PASAT scoreg | ||||||
| Mean (SD) | 41.43 | (13.75) | 41.02 | (13.42) | 41.29 | (13.63) |
| Median (25th–75th percentiles) | 43 | (30–53) | 42 | (31–53) | 43 | (31–53) |
| Not reported, n (%) | 2 | (0.6) | 3 | (1.8) | 5 | (1.0) |
| MSWS-12v2h | ||||||
| Mean (SD) | 45.51 | (6.96) | 45.26 | (7.14) | 45.42 | (7.01) |
| Median (25th–75th percentiles) | 47 | (42–51) | 47 | (41–51) | 47 | (41–51) |
| Not reported, n (%) | 3 | (0.9) | 5 | (3.0) | 8 | (1.6) |
| RMIi | ||||||
| Mean (SD) | 11.40 | 2.51 | 11.64 | 2.20 | 11.48 | 2.41 |
| Median (25th–75th percentiles) | 12 | (10–13) | 12 | (10–13) | 12 | (10–13) |
| Not reported, n (%) | 1 | (0.3) | 0 | (0.0) | 1 | (0.2) |
| SF-36(PH)j | ||||||
| Mean (SD) | 44.31 | 6.08 | 44.18 | 5.76 | 44.26 | 5.97 |
| Median (25th–75th percentiles) | 44 | (40.5–48.0) | 44 | (40.25–47.00) | 44 | (40–48) |
| Not reported, n (%) | 2 | (0.6) | 2 | (1.2) | 4 | (0.8) |
| MSSS-88 | ||||||
| Section 1k | ||||||
| Mean (SD) | 71.97 | 21.14 | 73.60 | 22.10 | 72.51 | 21.45 |
| Median (25th–75th percentiles) | 70 | (55–84) | 71 | (56–88) | 70 | (55.26–85.00) |
| Not reported, n (%) | 2 | (0.61) | 1 | (0.61) | 3 | (0.61) |
| Section 2l | ||||||
| Mean (SD) | 77.96 | 20.78 | 80.30 | 20.90 | 78.73 | 20.83 |
| Median (25th–75th percentiles) | 77 | (63.5–93.0) | 81 | (66.00–94.97) | 78.23 | (64–94) |
| Not reported, n (%) | 2 | (0.61) | 3 | (1.83) | 5 | (1.00) |
| Section 3m | ||||||
| Mean (SD) | 44.29 | 15.13 | 45.19 | 15.67 | 44.59 | 15.30 |
| Median (25th–75th percentiles) | 42 | (32–55) | 44 | (32.25–56.75) | 43 | (32.0–55.5) |
| Not reported, n (%) | 8 | (2.43) | 2 | (1.22) | 10 | (2.00) |
Prescribed dose of trial medication
Prescribed daily doses of trial medication at each 6-monthly follow-up are summarised in Table 6 for those patients not discontinuing trial medication and for all patients. Among those patients not withdrawing from trial medication (n = 178 active, n = 118 placebo), median prescribed daily dose during final year of follow-up was four capsules (25th–75th percentiles 2–6 capsules) in the active group compared with six (25th–75th percentiles 4–8 capsules) in the placebo group. Final year medians among all patients were four capsules (25th–75th percentiles 2.0–5.5) for active and six capsules (25th–75th percentiles 4–8 capsules) for placebo. Percentiles of prescribed daily dose among non-withdrawals, by treatment group and weight group, are shown in Figure 2.
| Prescribed daily dose (number of capsules) of trial medication | Visit | ||||||
|---|---|---|---|---|---|---|---|
| Week 5 | Week 13 | Month 7 | Month 13 | Month 19 | Month 25 | Month 31 | |
| Non-withdrawals (n = 296) | |||||||
| Active (n = 178) | |||||||
| n | 177 | 176 | 177 | 178 | 178 | 177 | 177 |
| Missing | 1 | 2 | 1 | 0 | 0 | 1 | 1 |
| Mean | 5 | 4.48 | 4.26 | 4.06 | 4.05 | 3.99 | 3.91 |
| SD | 1.91 | 1.97 | 2.02 | 2 | 2.02 | 1.98 | 1.93 |
| Median | 5 | 4 | 4 | 4 | 4 | 4 | 4 |
| 25th percentile | 4 | 3 | 3 | 2 | 2 | 2 | 2 |
| 75th percentile | 6 | 6 | 6 | 6 | 6 | 6 | 5 |
| Minimum | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Maximum | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Placebo (n = 118) | |||||||
| n | 117 | 118 | 118 | 118 | 118 | 118 | 118 |
| Missing | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mean | 6.32 | 6.14 | 5.97 | 5.92 | 5.85 | 5.85 | 5.85 |
| SD | 1.57 | 1.67 | 1.81 | 1.91 | 1.92 | 1.92 | 1.92 |
| Median | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| 25th percentile | 6 | 5.25 | 4 | 4 | 4 | 4 | 4 |
| 75th percentile | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Minimum | 2 | 1 | 1 | 1 | 1 | 1 | 1 |
| Maximum | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| All patients (n = 493) | |||||||
| Active (n = 329) | |||||||
| n | 315 | 290 | 260 | 235 | 215 | 198 | 189 |
| Missing | 14 | 39 | 69 | 94 | 114 | 131 | 140 |
| Mean | 4.98 | 4.31 | 4.08 | 3.94 | 3.91 | 3.94 | 3.91 |
| SD | 1.96 | 2 | 2.03 | 2.02 | 2.02 | 1.97 | 1.95 |
| Median | 5 | 4 | 4 | 4 | 4 | 4 | 4 |
| 25th percentile | 4 | 3 | 2 | 2 | 2 | 2 | 2 |
| 75th percentile | 6 | 6 | 6 | 6 | 6 | 6 | 5 |
| Minimum | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Maximum | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Placebo (n = 164) | |||||||
| n | 161 | 157 | 146 | 138 | 131 | 125 | 123 |
| Missing | 3 | 7 | 18 | 26 | 33 | 39 | 41 |
| Mean | 6.2 | 6.05 | 5.93 | 5.9 | 5.91 | 5.92 | 5.92 |
| SD | 1.57 | 1.68 | 1.76 | 1.89 | 1.89 | 1.91 | 1.92 |
| Median | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| 25th percentile | 6 | 5 | 4 | 4 | 4 | 4 | 4 |
| 75th percentile | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Minimum | 2 | 1 | 1 | 1 | 1 | 1 | 1 |
| Maximum | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
FIGURE 2.
Percentiles of prescribed daily dose of trial medication among non-withdrawals at each visit, by treatment group and weight group. (a) Active, weight < 60 kg; (b) active, weight 60–80 kg; (c) active, weight > 80 kg; (d) placebo, weight < 60 kg; (e) placebo, weight 60–80 kg; and (f) active, weight > 80 kg. Heavy solid line, median; narrow solid lines, 25th and 75th percentiles; dashed lines, 5th and 90th percentiles. The maximum weight-related daily dose is superimposed in green.
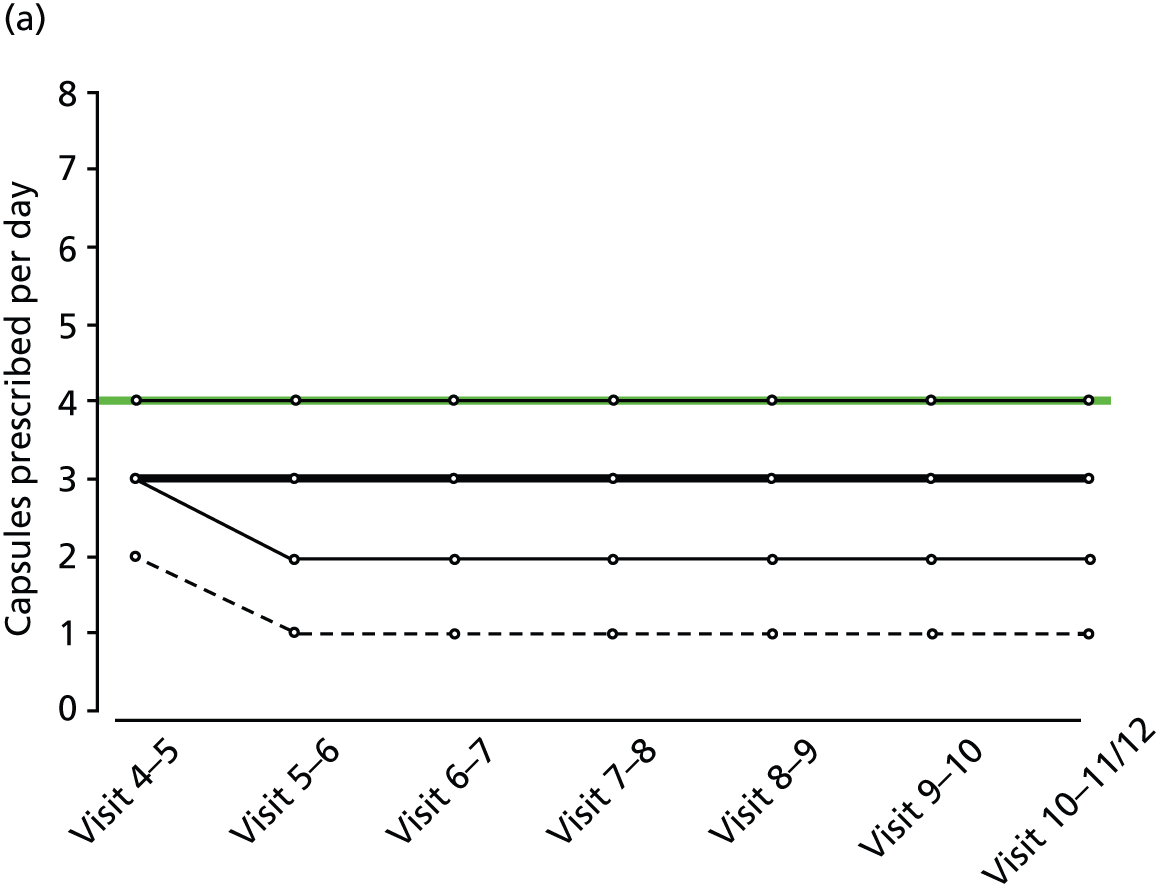
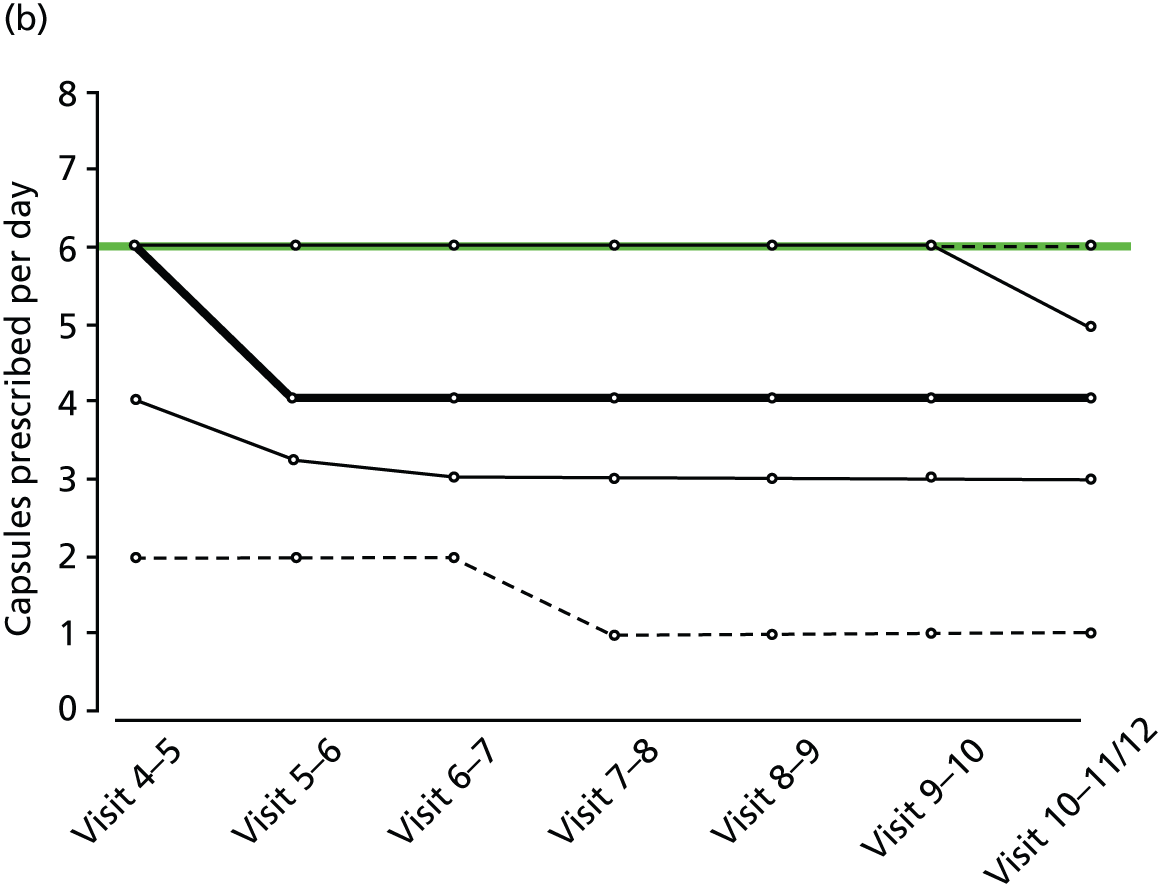


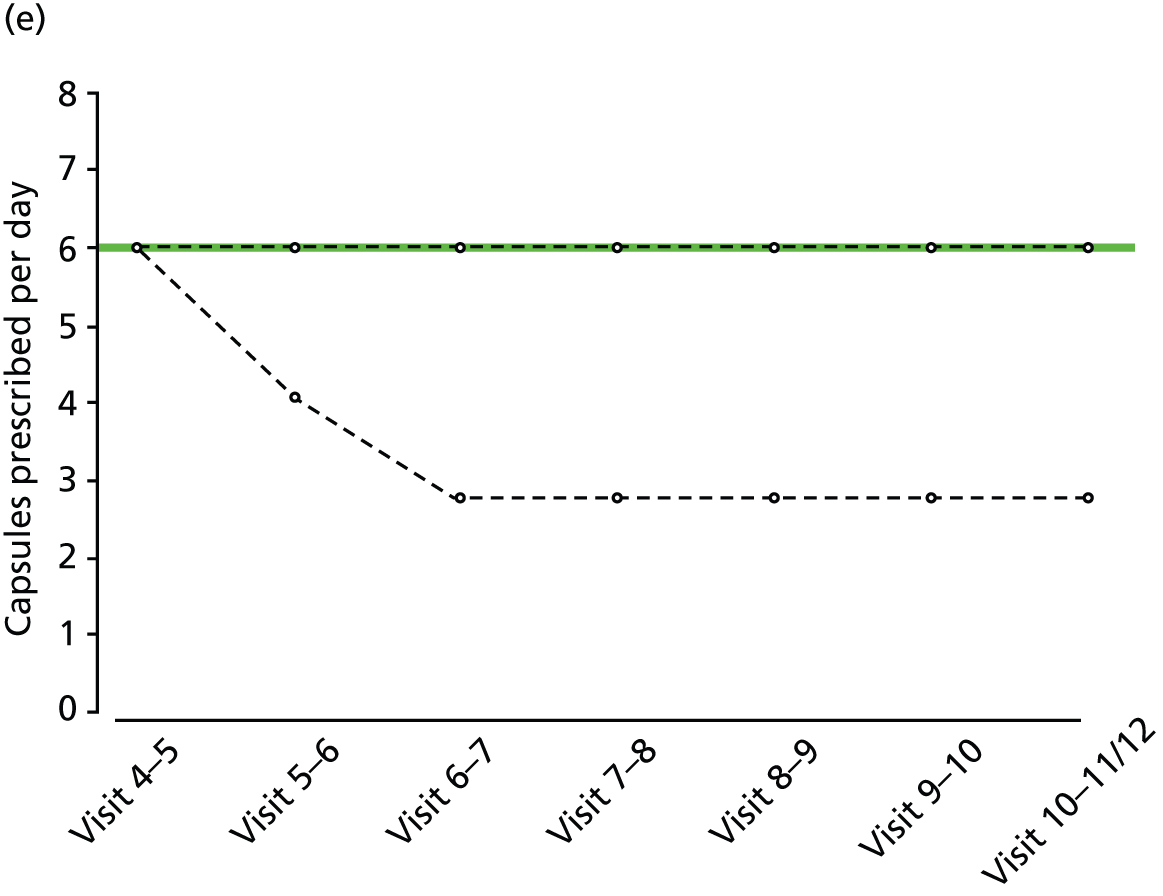

Random urine testing to determine any illicit cannabis use
Results from urinalyses throughout the study are given in Table 7. These results showed little illicit cannabis use in the placebo group and an increasing proportion of negative test results within the active group over time.
| Visit | 1 (N = 493) | 2 (N = 16) | 3 (N = 100) | 4 (N = 117) | 5 (N = 220) | 6 (N = 215) | 7 (N = 196) | 8 (N = 217) | 9 (N = 209) | 10 (N = 194) | 11 (N = 210) | AFU (N = 51) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment group | Active | Placebo | Active | Placebo | Active | Placebo | Active | Placebo | Active | Placebo | Active | Placebo | Active | Placebo | Active | Placebo | Active | Placebo | Active | Placebo | Active | Placebo | Active | Placebo |
| n | 329 | 164 | 10 | 6 | 60 | 40 | 71 | 46 | 155 | 65 | 142 | 73 | 133 | 63 | 136 | 81 | 136 | 73 | 119 | 75 | 131 | 79 | 34 | 17 |
| Positive | 1 (0.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 59 (98.3) | 1 (2.5) | 67 (94.4) | 1 (2.2) | 138 (89.0) | 0 (0.0) | 114 (80.3) | 0 (0.0) | 103 (77.4) | 0 (0.0) | 105 (77.2) | 4 (4.9) | 108 (79.4) | 3 (4.1) | 91 (76.5) | 2 (2.7) | 93 (71.0) | 1 (1.3) | 3 (8.8) | 3 (17.6) |
| Negative | 328 (99.7) | 164 (100.0) | 10 (100.0) | 6 (100.0) | 1 (1.7) | 38 (95.0) | 3 (4.2) | 45 (97.8) | 17 (11.0) | 65 (100.0) | 28(19.7) | 73 (100.0) | 29 (21.8) | 63 (100.0) | 31 (22.8) | 77 (95.1) | 28 (20.6) | 70 (95.9) | 26 (21.8) | 73 (97.3) | 38 (29.0) | 78 (98.7) | 31 (91.2) | 14 (82.4) |
| No result | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.5) | 1 (1.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (1.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
The main results are summarised in Table 8 and detailed below.
| Outcome measure | Summary statistics | Treatment group | Analysis | Estimated treatment effect (95% CI) | p-value | |
|---|---|---|---|---|---|---|
| Main study | Active (n = 329) | Placebo (n = 164) | ||||
| EDSS (all participants) | No. of first progression events | 145 | 73 | Primary: HR (active : placebo) from Cox regression analysis (losses to follow-up considered as censored observations) | 0.92 (0.68 to 1.23) | 0.57 |
| No. of first progression events per patient-yeara | 0.24 | 0.23 | ||||
| No. of first progression events | 201 | 88 | Sensitivity: HR (active : placebo) from Cox regression analysis (losses to follow-up considered as progression events) | 1.11 (0.86 to 1.44) | 0.41 | |
| No. of first progression events per patient-yeara | 0.34 | 0.27 | ||||
| No. of first progression events | 157 | 83 | Sensitivity: HR (active : placebo) from Cox regression analysis (EPC derived data, losses to follow-up considered as censored observations) | 0.88 (0.67 to 1.17) | 0.39 | |
| No. of first progression events per patient-yeara | 0.26 | 0.26 | ||||
| No. of first progression events | 204 | 90 | Sensitivity: HR (active : placebo) from Cox regression analysis (EPC-derived data, losses to follow-up considered as progression events) | 1.11 (0.86 to 1.43) | 0.42 | |
| No. of first progression events per patient-yeara | 0.34 | 0.28 | ||||
| Subgroup analyses: HR (active : placebo) from Cox regression analysis | Baseline EDSS score of 4.0–5.5: 0.52 (0.32 to 0.85) | 0.01 | ||||
| Baseline EDSS score of 6.0: 1.15 (0.76 to 1.73) | 0.51 | |||||
| Baseline EDSS score of 6.5: 1.63 (0.85 to 3.10) | 0.14 | |||||
| MSIS-29phys | Mean (SD) annual change | 0.62 (3.29) | 1.03 (3.74) | Multilevel model: estimated between-group difference (active–placebo) | –0.91 (–2.01 to 0.19) | 0.11 |
| MSFC composite (z-score) | Mean (SD) annual change | –0.17 (0.28) | –0.16 (0.30) | Multilevel model: estimated between-group difference (active–placebo) | –0.03 (–0.19 to 0.09) | 0.72 |
| T25-FW (z-score) | Mean (SD) annual change | –0.37 (0.73) | –0.41 (0.74) | Multilevel model: estimated between-group difference (active–placebo) | –0.08 (–0.25 to 0.09) | 0.37 |
| Main study | Active (n = 329) | Placebo (n = 164) | ||||
| 9-HPT (z-score) | Mean (SD) annual change | –0.13 (0.23) | –0.14 (0.27) | Multilevel model: estimated between-group difference (active–placebo) | 0.05 (–0.04 to 0.13) | 0.28 |
| PASAT (z-score) | Mean (SD) annual change | –0.025 (0.21) | –0.0074 (0.20) | Multilevel model: estimated between-group difference (active–placebo) | –0.01 (–0.10 to 0.09) | 0.92 |
| MSWS-12v2 | Mean (SD) annual change | 0.37 (2.33) | 0.52 (2.68) | Multilevel model: estimated between-group difference (active–placebo) | –0.19 (–0.97 to 0.60) | 0.74 |
| RMI | Mean (SD) annual change | –0.58 (0.96) | –0.72 (1.08) | Multilevel model: estimated between-group difference (active–placebo) | 0.04 (–0.24 to 0.32) | 0.76 |
| SF-36(PH) | Mean (SD) annual change | –0.58 (2.07) | –0.49 (2.06) | Multilevel model: estimated between-group difference (active–placebo) | –0.15 (–0.83 to 0.53) | 0.67 |
| MSSS-88 (1) | Mean (SD) annual change | 0.20 (6.25) | 0.54 (7.42) | Multilevel model: estimated between-group difference (active–placebo) | 0.26 (–1.99 to 2.52) | 0.82 |
| MSSS-88 (2) | Mean (SD) annual change | 1.27 (6.71) | 1.30 (6.50) | Multilevel model: estimated between-group difference (active–placebo) | –0.02 (–2.35 to 2.32) | 0.99 |
| MSSS-88 (3) | Mean (SD) annual change | –0.34 (4.88) | –0.97 (5.03) | Multilevel model: estimated between-group difference (active–placebo) | 1.00 (–0.70 to 2.70) | 0.25 |
| MRI substudy | Active (n = 182) | Placebo (n = 91) | ||||
| PBVC | Mean (SD) annual change | –0.68 (0.95) | –0.66 (0.98) | Multilevel model: estimated between-group difference (active–placebo) | –0.01 (–0.26 to 0.24) | 0.94 |
| Occurrence of new or enlarging T2 lesions | n (%) | 60 (37) | 34 (40) | Logistic regression model: estimated OR (active : placebo) | 0.89 (0.50 to 1.58) | 0.70 |
| Occurrence of new or enlarging T1 lesions | n (%) | 54 (34) | 28 (33) | Logistic regression model: estimated OR (active : placebo) | 1.05 (0.59 to 1.88) | 0.87 |
Pre-specified analyses of primary clinical outcomes
Primary analysis of time to first confirmed Expanded Disability Status Scale score progression
Primary analysis using a Cox regression model showed no evidence of an effect of age (p = 0.36), disease type (p = 0.12), sex (p = 0.56), weight (p = 0.11) or treatment (p = 0.57; see Table 8) on time to confirmed EDSS score progression. The HR for first EDSS score progression event in patients randomly assigned to dronabinol compared with those assigned to placebo was 0.92 [95% confidence interval (CI) 0.68 to 1.23; see Table 8]. At trial completion, Kaplan–Meier estimates of the probability of EDSS score progression were 0.55 (95% CI 0.46 to 0.63) in the dronabinol group compared with 0.60 (95% CI 0.44 to 0.71) in the placebo group (Figure 3).
FIGURE 3.
Kaplan–Meier estimates of the probability of progression on the EDSS in the two treatment groups, confirmed after 6 months within the study period. The numbers at risk (cumulative number of censored observations) are given. Those patients who were lost to follow-up during the trial are marked by +. Those who reached the end of the trial without progressing are marked by x.

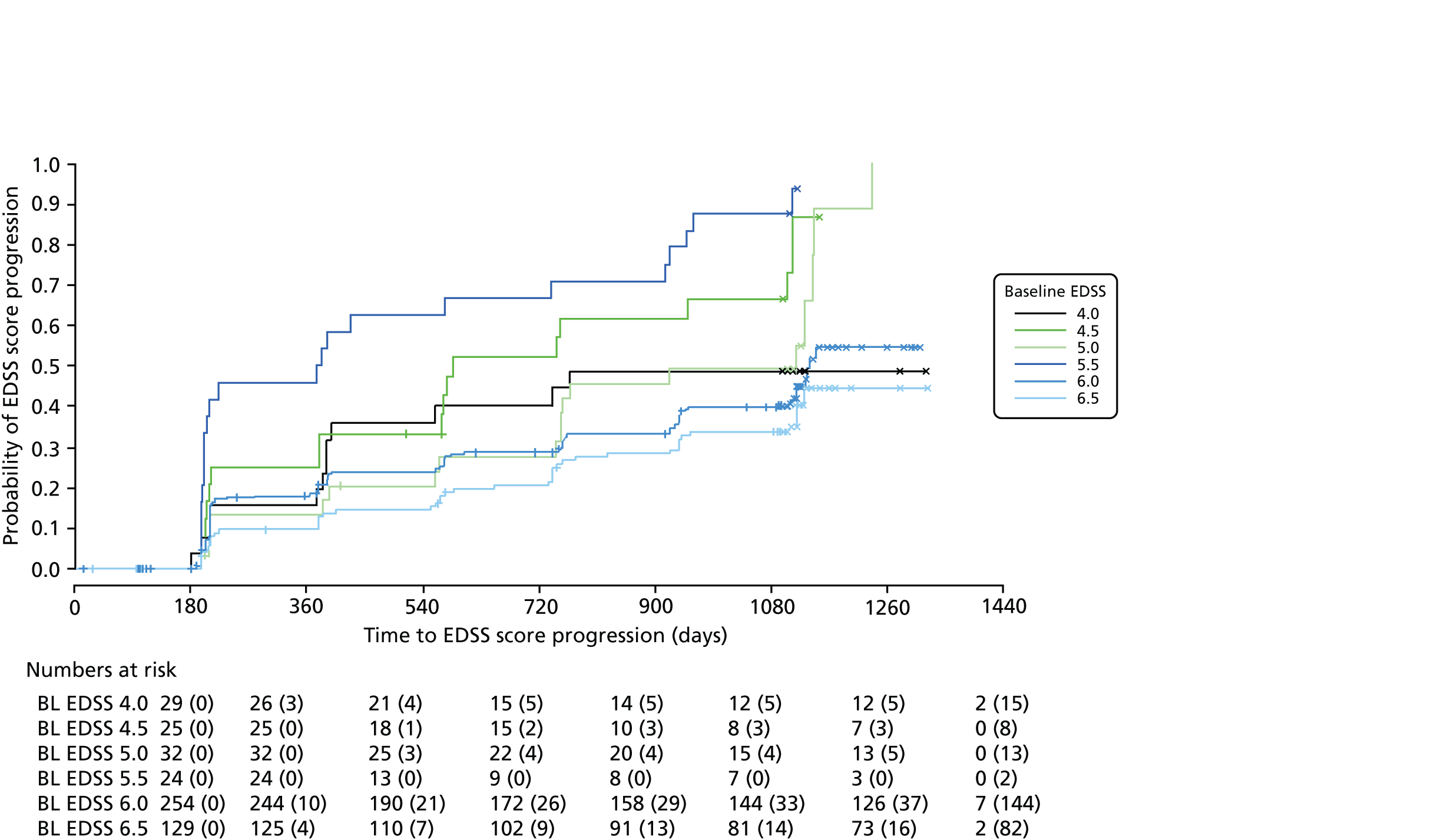
We noted evidence of some study site effects and of an effect of baseline EDSS score on time to confirmed progression (Figure 4). Most notably, relative to a baseline EDSS score of 4.0, there was an increased hazard of disease progression among those with a baseline EDSS score of 5.5 (HR 3.17, 95% CI 1.45 to 6.93; p = 0.004) and a reduced hazard among those with a baseline EDSS score of 6.5 (HR 0.49, 95% CI 0.24 to 0.98; p = 0.04). However, the numbers of participants in the individual EDSS groups are small (Figure 5), as are the numbers in some study sites (see Table 16 and Appendix 3).
FIGURE 4.
Estimated HRs and 95% CIs for EDSS score progression from a Cox regression model. HRs for study sites are compared with site 01; HR for SPMS is compared with PPMS; active compared with placebo; each baseline EDSS is compared with a baseline EDSS score of 4.0; and men are compared with women. BL, baseline.

FIGURE 5.
Kaplan–Meier estimates of the probability of progression on the EDSS, by baseline EDSS score, confirmed after 6 months within the study period. The numbers at risk (cumulative number of censored observations) are given. Those patients who were lost to follow-up during the trial are marked by +. Those who reached the end of the trial without progressing are marked by x. BL, baseline.
The global PH test gave no evidence that the PH assumption was violated (χ2 = 36, 36 degrees of freedom; p = 0.47).
Sensitivity analyses of time to first confirmed Expanded Disability Status Scale score progression
Results of sensitivity analysis showed that when losses to follow-up were treated as progression events rather than censored observations, the estimated HR (active : placebo) for EDSS score progression changed to 1.11 (95% CI 0.86 to 1.44; see Table 8), but the estimated effect of treatment remained non-significant (p = 0.41). This change in HR might be because the dronabinol group had a higher proportion of losses to follow-up for EDSS assessment [56 of 71 (79%)] than the placebo group [15 of 71 (21%)] and represents a worst-case scenario in terms of patient deterioration and hence the potential benefit of dronabinol.
The EPC reviewed data on 95 patients [71 active (74.7%); 24 placebo (25.3%)], for which there were ambiguities regarding EDSS scores. The EPC considered 22 (12 active; 10 placebo) of these patients to have progressed. These patients had no confirmed progression according to the data collected from the trial schedule. A further four patients (three active; one placebo) were considered to have progressed prior to the time of progression determined from the trial schedule. Clinical information on the remaining 69 patients reviewed by the EPC either confirmed non-progression or was insufficient to draw any further conclusions over those made on the primary data. As a result, data derived following EPC review consisted of a total of 240 first progression events compared with 218 in the primary data (with losses to follow-up considered as censored observations in both).
Conclusions from the main analyses of time to first EDSS score progression were robust to sensitivity analyses in terms of whether or not conclusions from the EPC were considered in defining EDSS progressions under both approaches to dealing with losses to follow-up, that is treated as censored observations or as progression events (see Table 8).
Furthermore, estimated HRs (active : placebo) for EDSS score progression remained similar after sequential removal of study sites with high loss to follow-up rates, under each of the two ways of treating losses to follow-up and each of the two data sets, that is according to trial schedule or following EPC review (Figure 6).
FIGURE 6.
Estimated HRs (active : placebo) for EDSS score progression, with 95% CIs and p-values from Cox regression models, sequentially removing study sites with high loss to follow-up for each of the two data sets and each of the two ways of dealing with losses to follow-up. (a) Data = according to trial schedule, losses to follow-up = censored observations; (b) data = according to trial schedule, losses to follow-up = progression events; (c) data = following EPC review, losses to follow-up = censored observations; and (d) data = following EPC review, losses to follow-up = progression events. The two rows of n (%) on horizontal axes show top row = number of sites removed (% losses to follow-up in remaining data); bottom row = site number removed (within-site % losses to follow-up). n, number of participants included in each fitted model.
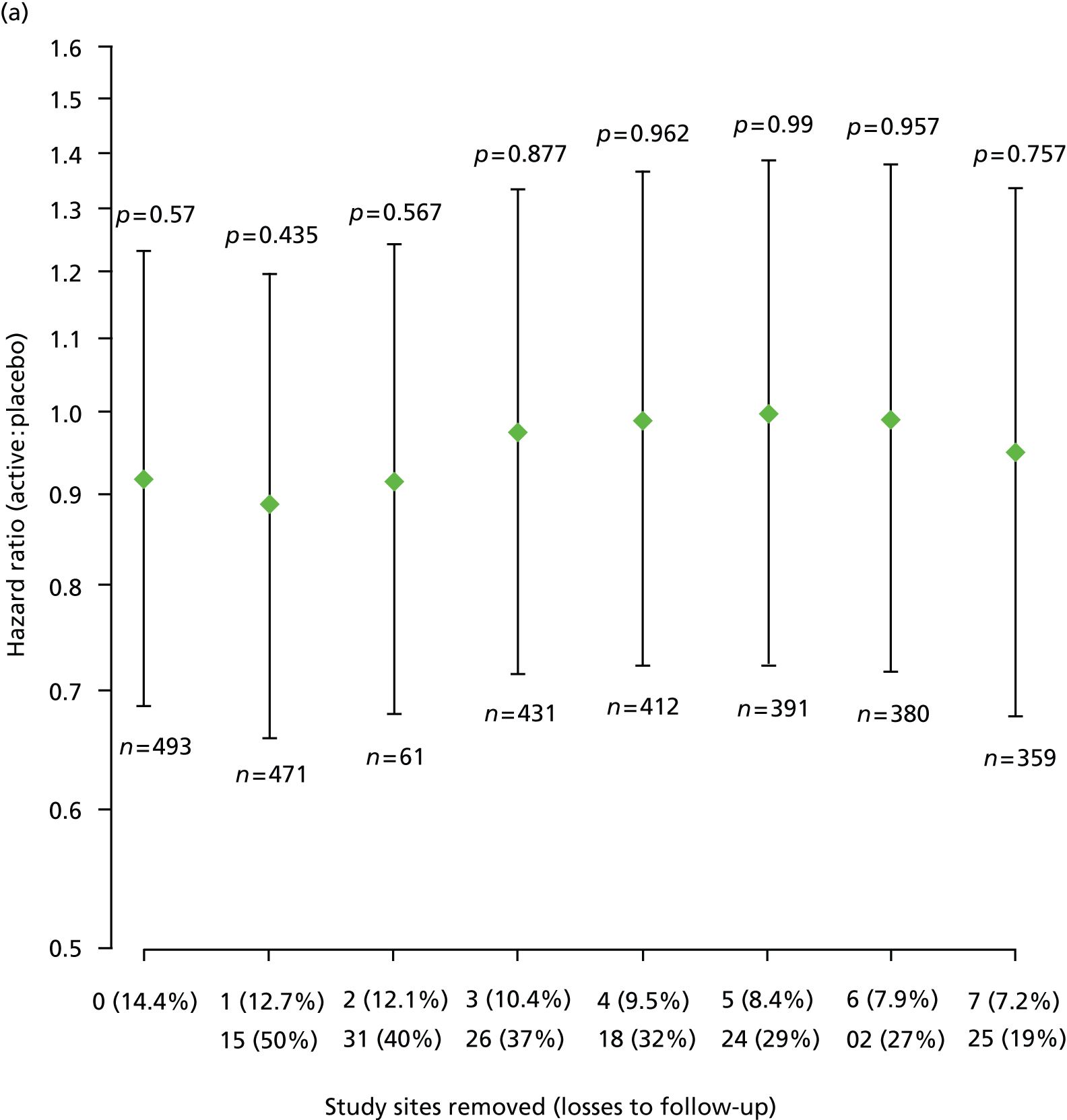
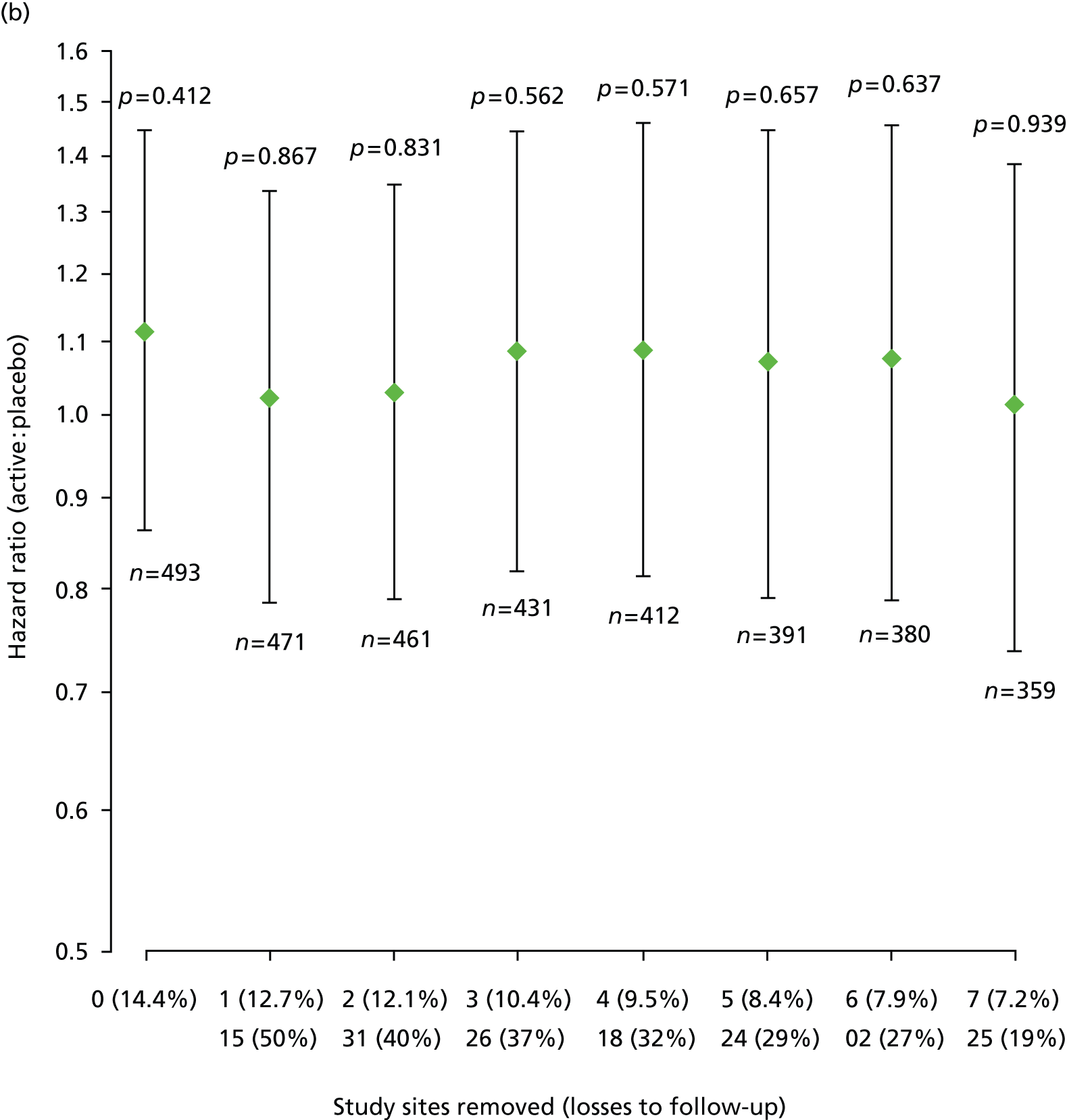
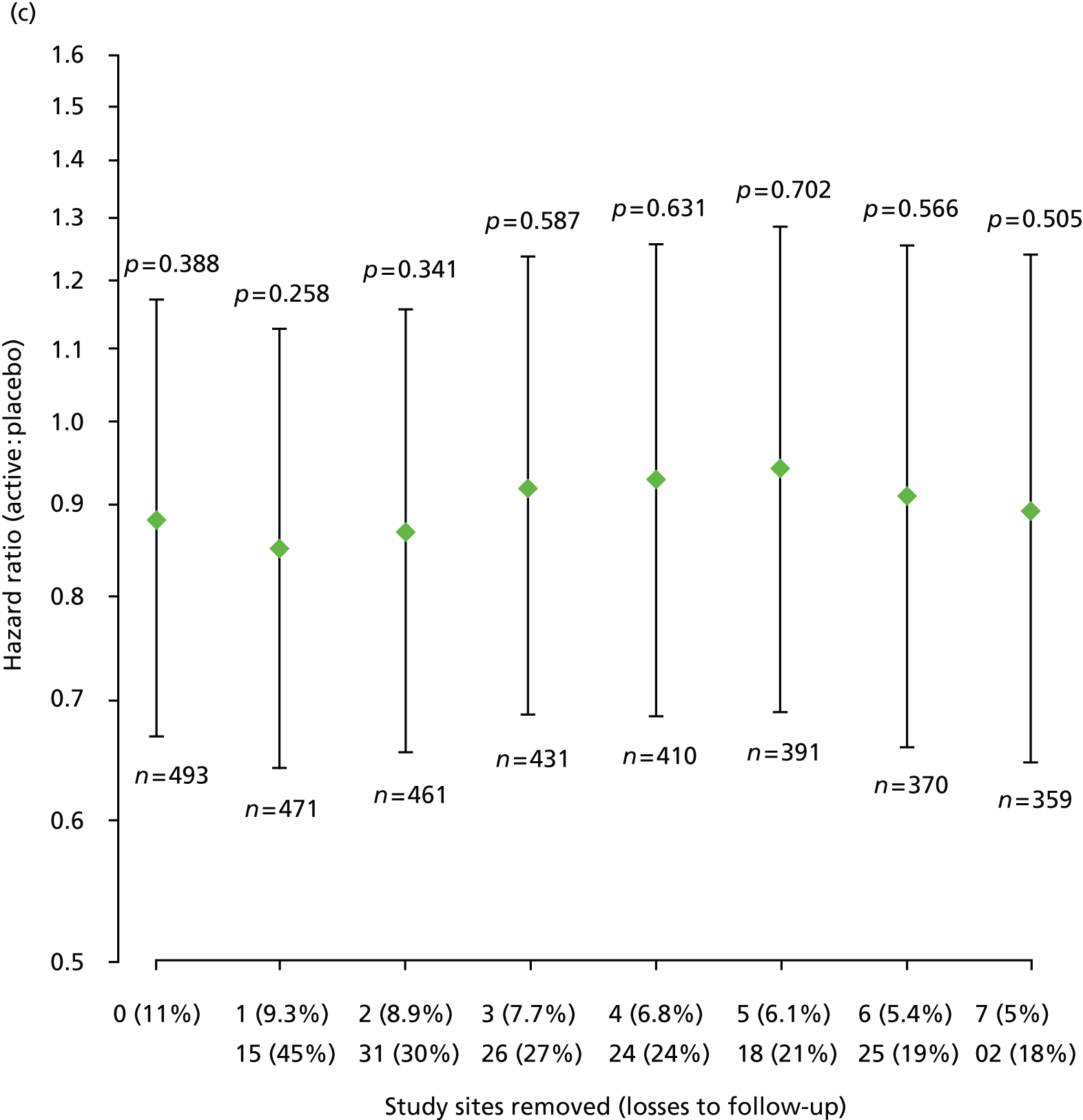

Pre-specified subgroup analyses of time to first confirmed Expanded Disability Status Scale score progression
Pre-specified subgroup analyses of time to first EDSS score progression suggested a differential effect of treatment between participants with lower (4.0–5.5) and higher (6.0–6.5) baseline EDSS scores (Figure 7). There was little evidence of differential effects of treatment among subgroups defined in terms of sex, disease type, or age or weight at registration.
FIGURE 7.
Estimated HRs (active : placebo) and 95% CIs for EDSS score progression by subgroup.

Primary analysis of change in Multiple Sclerosis Impact Scale-29 20-point physical subscale
A multilevel model fitted to repeated measures of MSIS-29phys score showed no evidence of an effect of treatment [estimated between-group difference (active–placebo) −0.91 points, 95% CI −2.01 to 0.19 points; p = 0.11; see Table 8], or of disease type, sex, weight or study site (data not shown; p > 0.05 for all).
It was estimated that MSIS-29phys score reduced by a mean of 1.4 points (95% CI 0.3 to 2.5 points; p = 0.02) for every 10-year increase in age. In both treatment groups, mean MSIS-29phys score decreased from baseline to month 3, after which it tended to increase (Figure 8).
FIGURE 8.
Estimated mean MSIS-29phys score, with 95% CIs, at each visit, separated by treatment group. The numbers of patients with total scores calculated at each visit, in each treatment group, are given directly above the CI for placebo and directly below the CI for active. Green points, active treatment; black points, placebo.
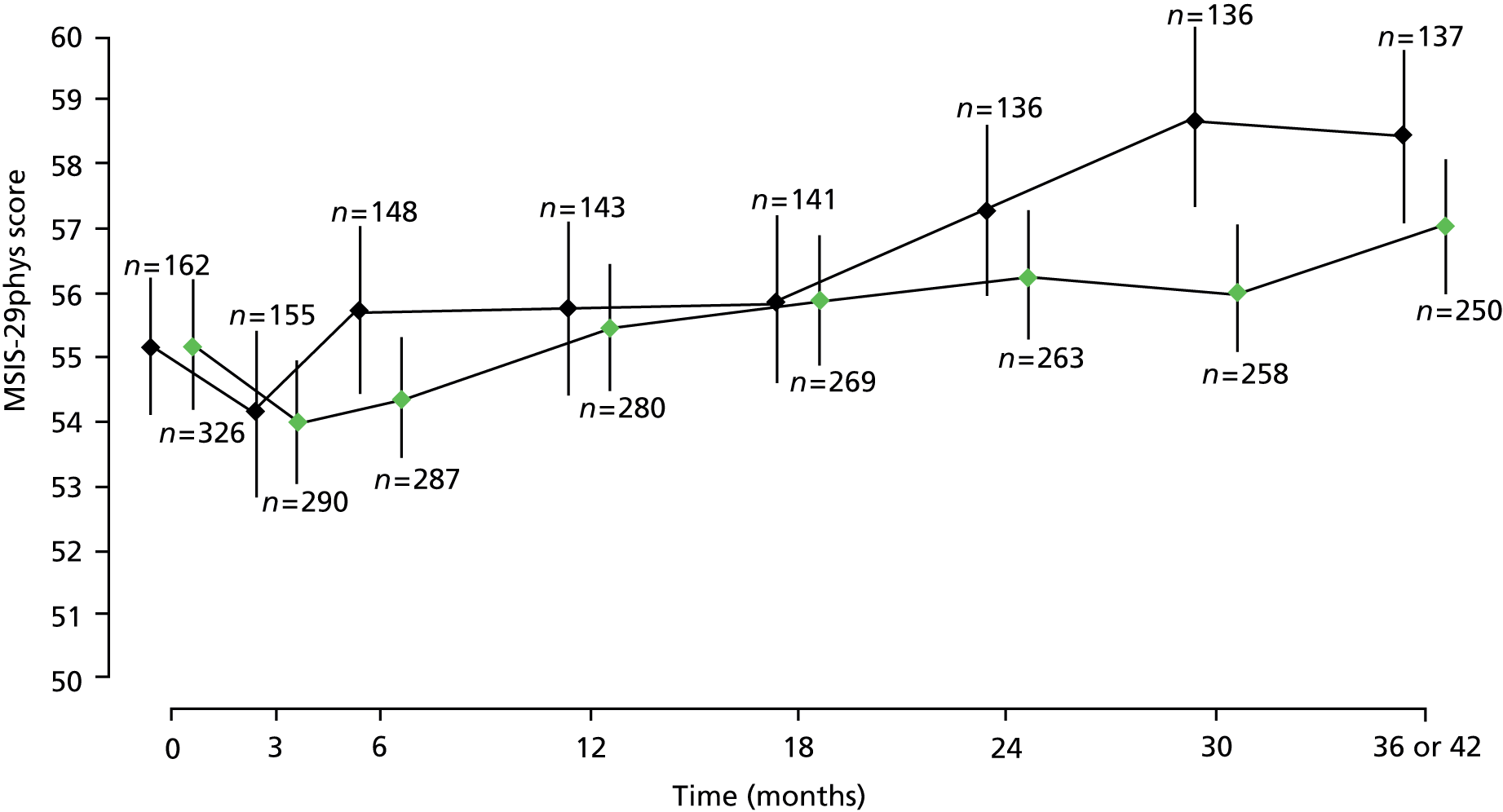
With the exception of a small reduction in MSIS-29phys score in patients with a baseline EDSS score of 5.0 compared with those with a score of 4.0, MSIS-29phys score tended to increase with increasing baseline EDSS score (data not shown).
Results from the primary analysis of repeated measures of MSIS-29phys remained unchanged after removal of non-significant terms from the fitted model and under an alternative analysis based on comparison of treatment groups in terms of change from baseline to last valid observation [estimated between-group difference (active–placebo) –1.4 points, 95% CI –3.3 to 0.4 points; p = 0.13].
Pre-specified analyses of secondary outcomes
Results of multilevel models fitted to data on the secondary outcomes MSWS-12v2, MSFC, RMI, SF-36(PH) and MSSS-88 are summarised in Table 8 and detailed below.
Multiple Sclerosis Walking Scale-12
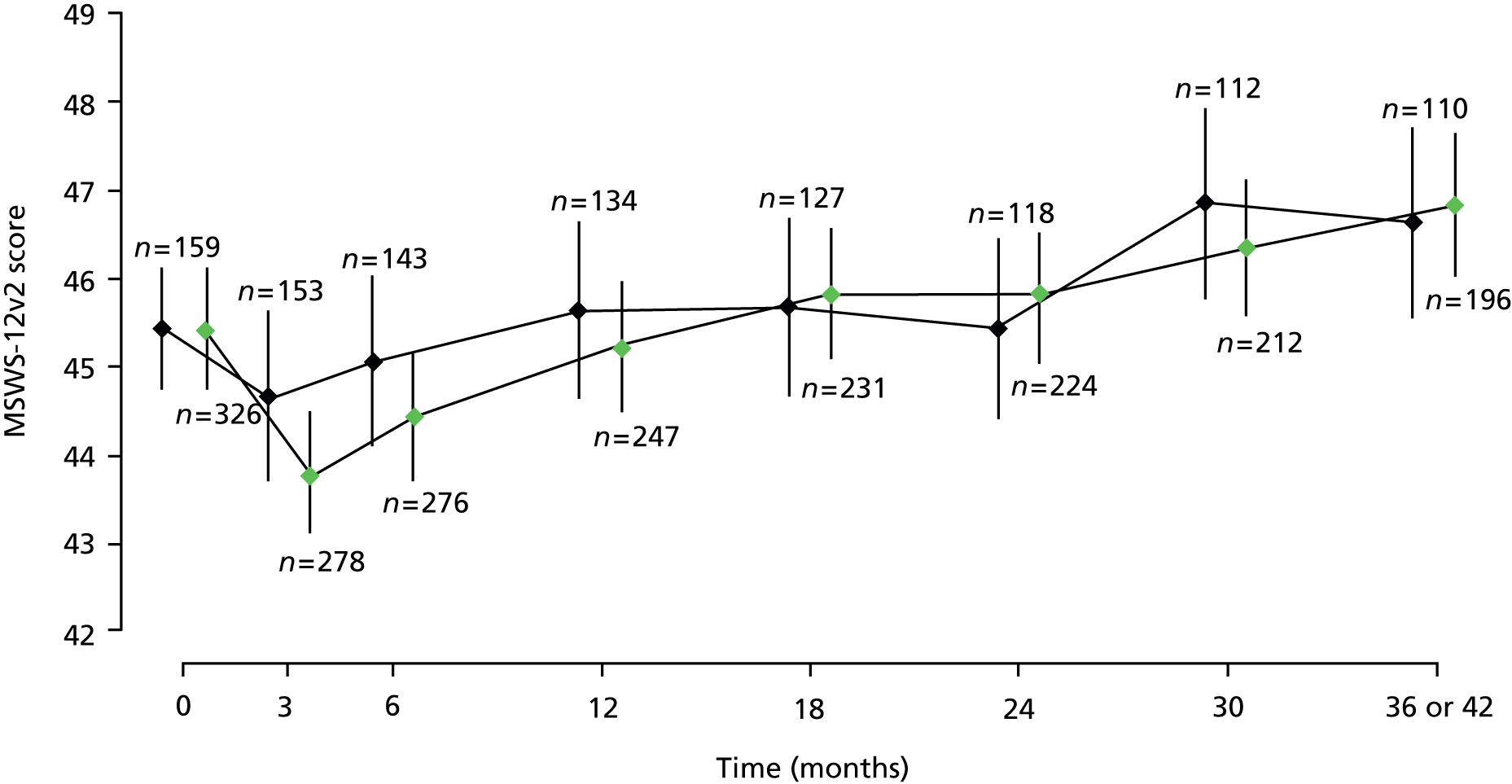
A multilevel model fitted to repeated measures of MSWS-12v2 score showed no evidence of an effect of treatment [estimated effect –0.19 (95% CI –0.97 to 0.60); p = 0.74; see Table 8], or of disease type, sex or weight (data not shown; p > 0.05 for all). There was some evidence of study site effects (data not shown) and of effects of baseline EDSS score. Compared with those with a baseline EDSS score of 4.0, MSWS-12v2 was estimated to be, on average, 5.7 (95% CI 2.3 to 9.0), 6.1 (95% CI 3.7 to 8.5) and 9.3 (95% CI 6.8 to 11.8) points higher in those with a baseline EDSS score 5.5, 6.0 and 6.5, respectively. In both treatment groups, mean MSWS-12v2 score decreased from baseline to month 3, after which it tended to increase (Figure 9).
FIGURE 9.
Estimated mean MSWS-12v2, with 95% CIs, at each visit, separated by treatment group. The numbers of patients with total scores calculated at each visit, in each treatment group, are given directly above the CI for placebo and directly below the CI for active. Green points, active treatment; black points, placebo.
Multiple Sclerosis Functional Composite
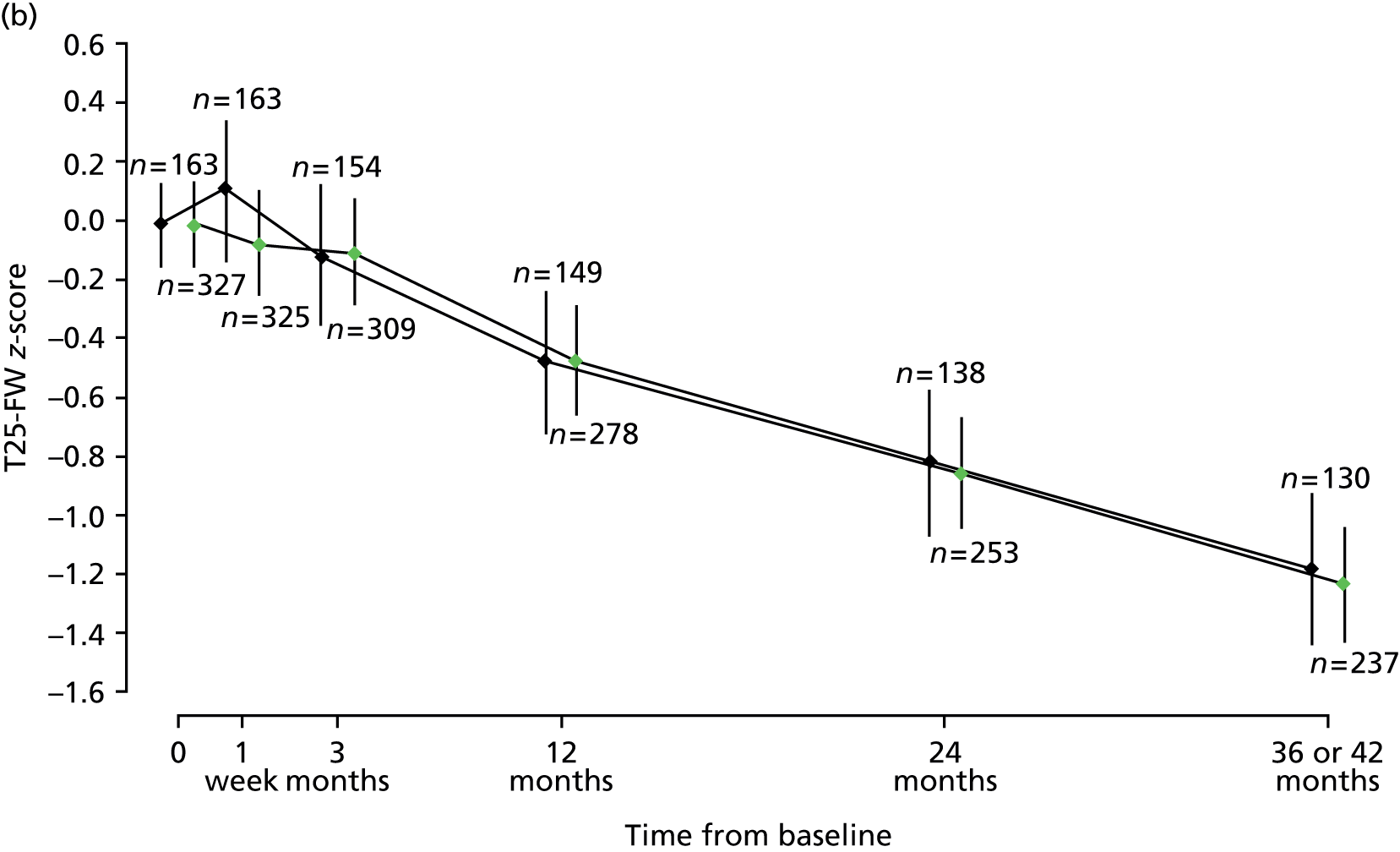
A multilevel model fitted to repeated measures of MSFC composite z-score showed no evidence of an effect of treatment; estimated between-group difference (active–placebo) –0.03 (95% CI –0.19 to 0.09; p = 0.72; see Table 8). Multilevel models fitted to the MSFC component-wise z-scores each showed no evidence of an effect of treatment. Estimated between-group differences (active–placebo) were: T25-FW –0.08 (95% CI –0.25 to 0.09; p = 0.37); 9-HPT 0.05 (95% CI –0.04 to 0.13; p = 0.28); and PASAT –0.01 (95% CI –0.10 to 0.09; p = 0.92). Across both treatment groups, mean T25-FW, 9-HPT and composite z-scores increased from baseline to week 1, after which they tended to decrease. After an initial increase at week 1, PASAT z-scores remained relatively constant over the 3-year study period (Figure 10).
FIGURE 10.
Estimated mean MSFC composite (a), T25-FW (b), 9-HPT (c) and PASAT (d) z-scores, with 95% CIs, at each visit, separated by treatment group. The numbers of patients with scores calculated at each visit, in each treatment group, are given directly above the CI for placebo and directly below the CI for active. Green points, active treatment; black points, placebo.



Rivermead Mobility Index
A multilevel model fitted to repeated measures of RMI showed no evidence of an effect of treatment [the estimated between-group difference (active–placebo) was 0.04 (95% CI –0.24 to 0.32; p = 0.76; see Table 8)] or of disease type, sex or weight (data not shown; p > 0.05 for all). There was some evidence of study site effects (data not shown) and of effects of baseline EDSS score. Compared with those with a baseline EDSS score of 4.0, RMI was estimated to be, on average, 1.57 points (95% CI 0.38 to 2.76 points), 2.44 points (95% CI 1.59 to 3.30 points) and 4.99 points (95% CI 4.10 to 5.88 points) lower in those with a baseline EDSS score of 5.5, 6.0 and 6.5, respectively. In both treatment groups, RMI decreased from baseline to 30 months, after which it remained fairly constant (Figure 11).
FIGURE 11.
Estimated mean RMI, with 95% CIs, at each visit, separated by treatment group. The numbers of patients with total scores calculated at each visit, in each treatment group, are given directly above the CI for placebo and directly below the CI for active. Green points, active treatment; black points, placebo.

Short Form questionnaire-36 items (physical health subscale)
A multilevel model fitted to repeated measures of SF-36(PH) showed no evidence of an effect of treatment [the estimated between-group difference (active–placebo) was –0.15 (95% CI –0.83 to 0.53; p = 0.67; see Table 8)], or of disease type, sex or weight (data not shown; p > 0.05 for all). In both treatment groups, mean SF-36(PH) score increased from baseline to month 3, after which it tended to decrease (Figure 12).
FIGURE 12.
Estimated mean SF-36(PH), with 95% CIs, at each visit, separated by treatment group. The numbers of patients with total scores calculated at each visit, in each treatment group, are given directly above the CI for placebo and directly below the CI for active. Green points, active treatment; black points, placebo.
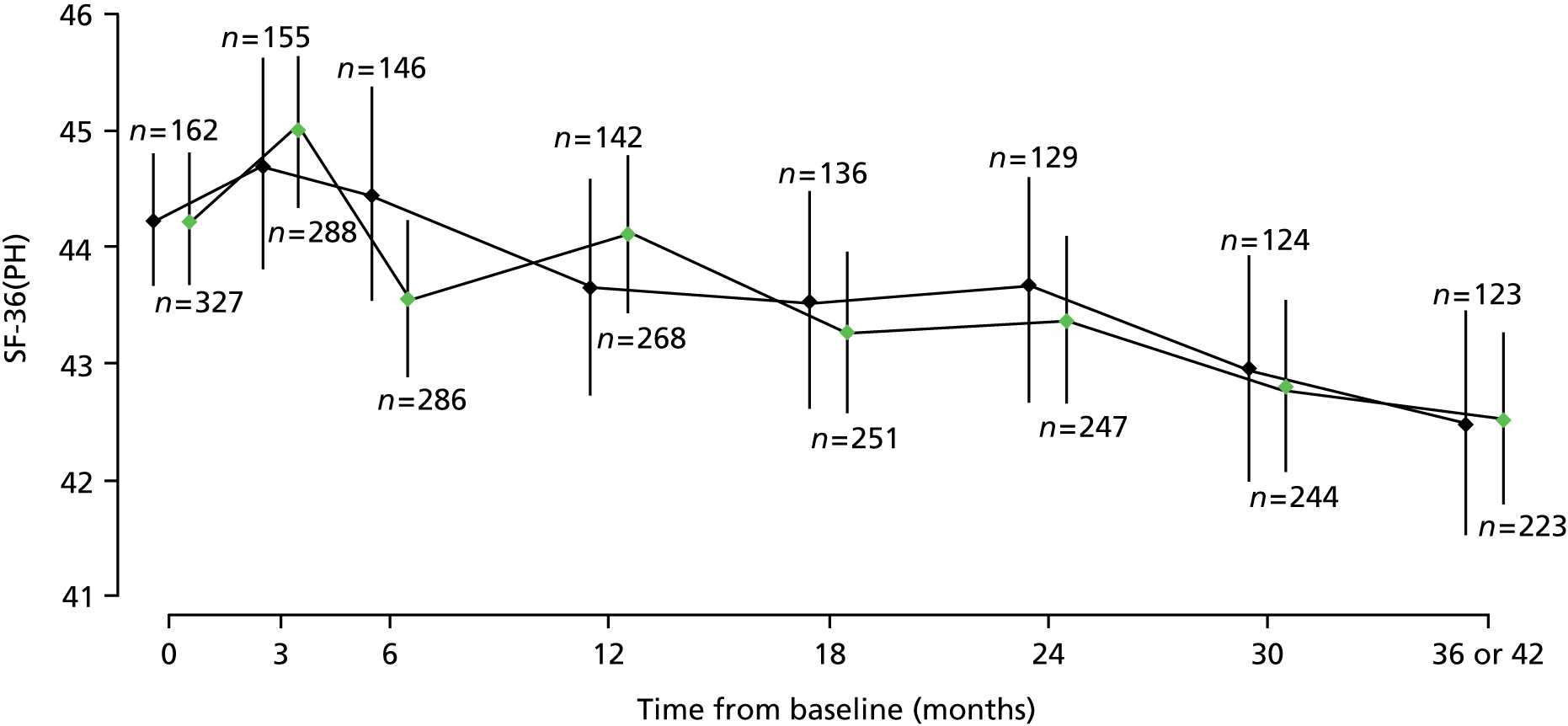
With the exception of a small increase in SF-36(PH) score in patients with a baseline EDSS score of 5.0 compared with those with a score of 4.0, SF-36(PH) score tended to decrease with increasing baseline EDSS score (data not shown).
Multiple Sclerosis Spasticity Scale-88
Figure 13 shows estimated mean MSSS-88 scores, with 95% CIs, by visit and treatment group, for each of the eight subscales.
FIGURE 13.
Estimated mean MSSS-88 subscales 1–8 scores, with 95% CIs, at each visit, separated by treatment group. The numbers of patients with total scores calculated at each visit, in each treatment group, are given directly above the CI for placebo and directly below the CI for active. Subscales relate to how bothered the patient has been by their spasticity in the past 2 weeks, where 1 = muscle stiffness; 2 = pain and discomfort; 3 = muscle spasms; 4 = effect of spasticity on daily activities; 5 = effect of spasticity on ability to walk; 6 = effect of spasticity on body movements; 7 = effect of spasticity on feelings; and 8 = effect of spasticity on social functioning. Green points, active treatment; black points, placebo.
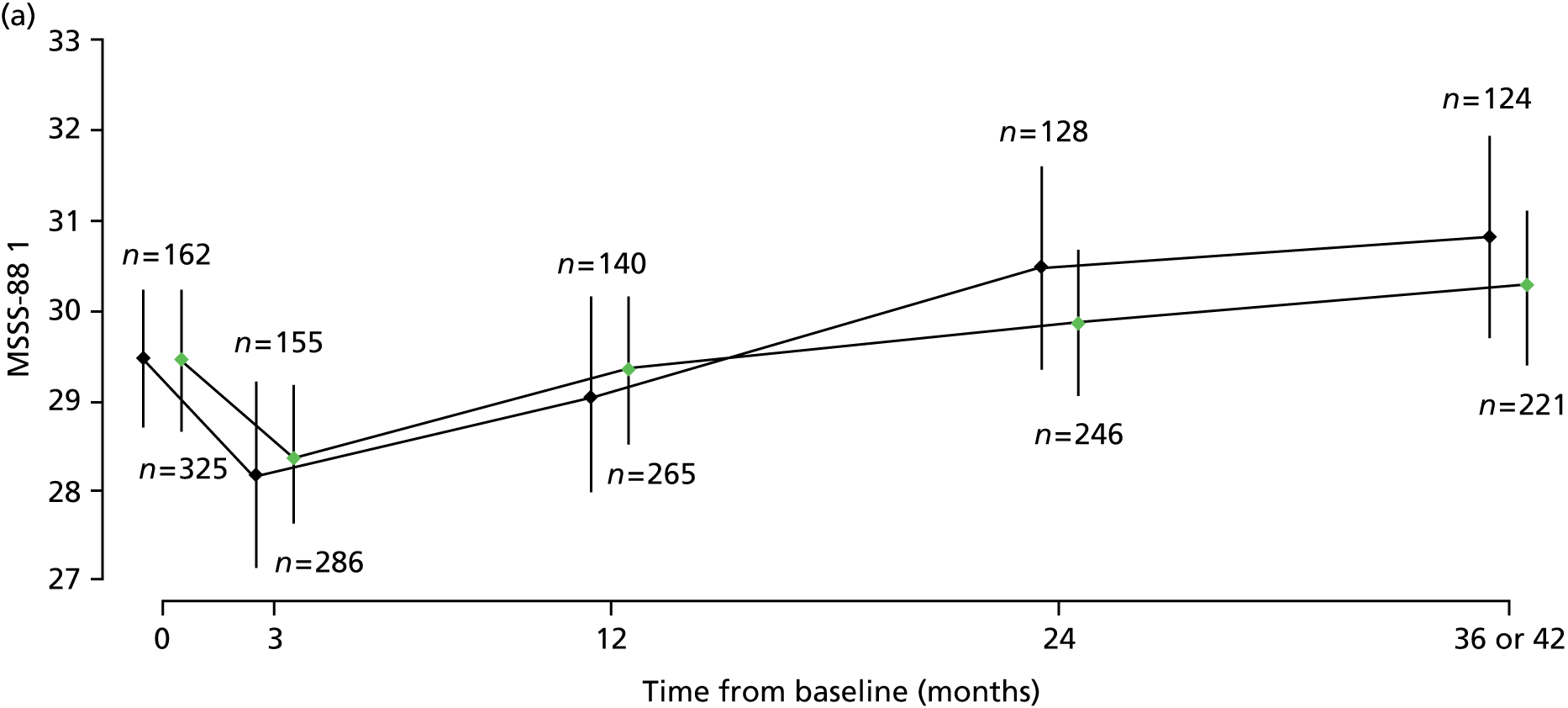

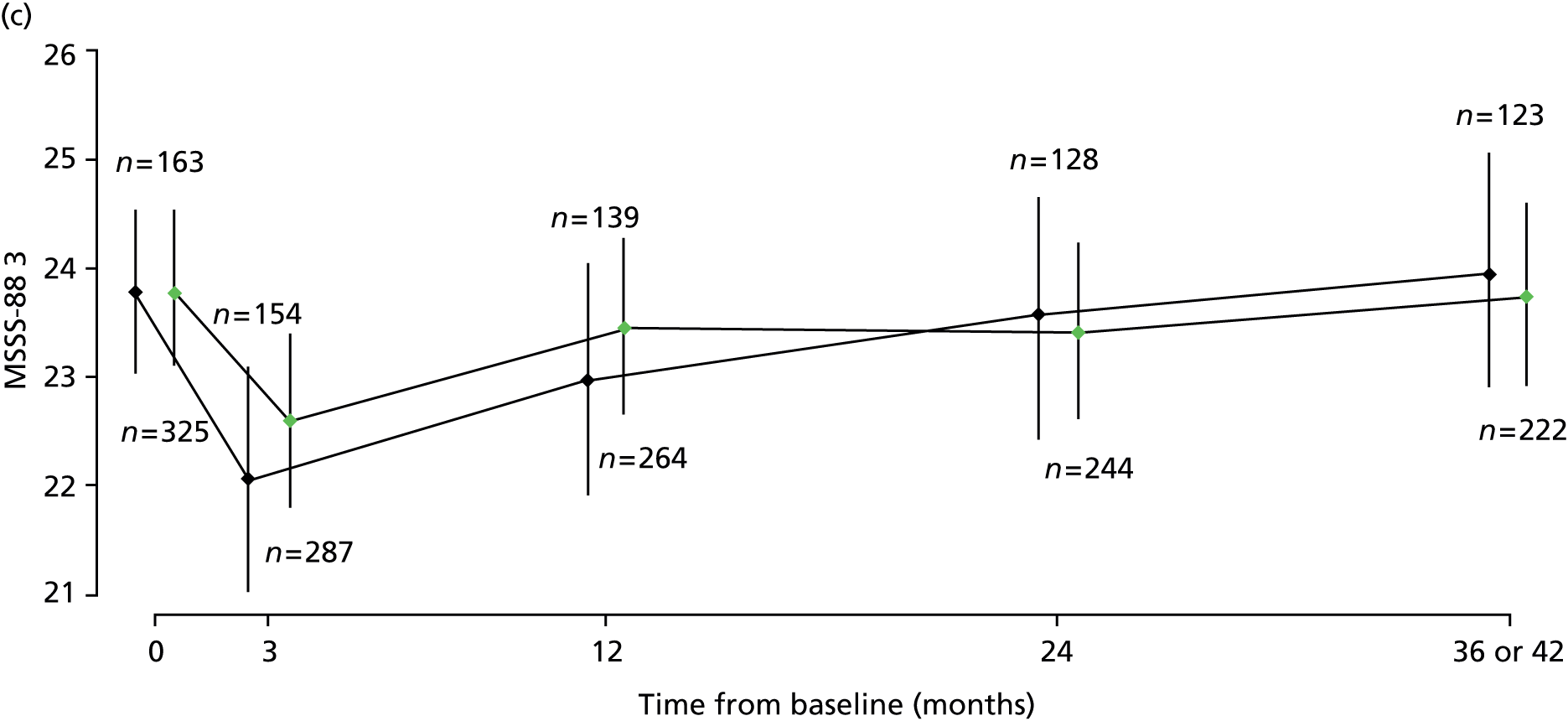




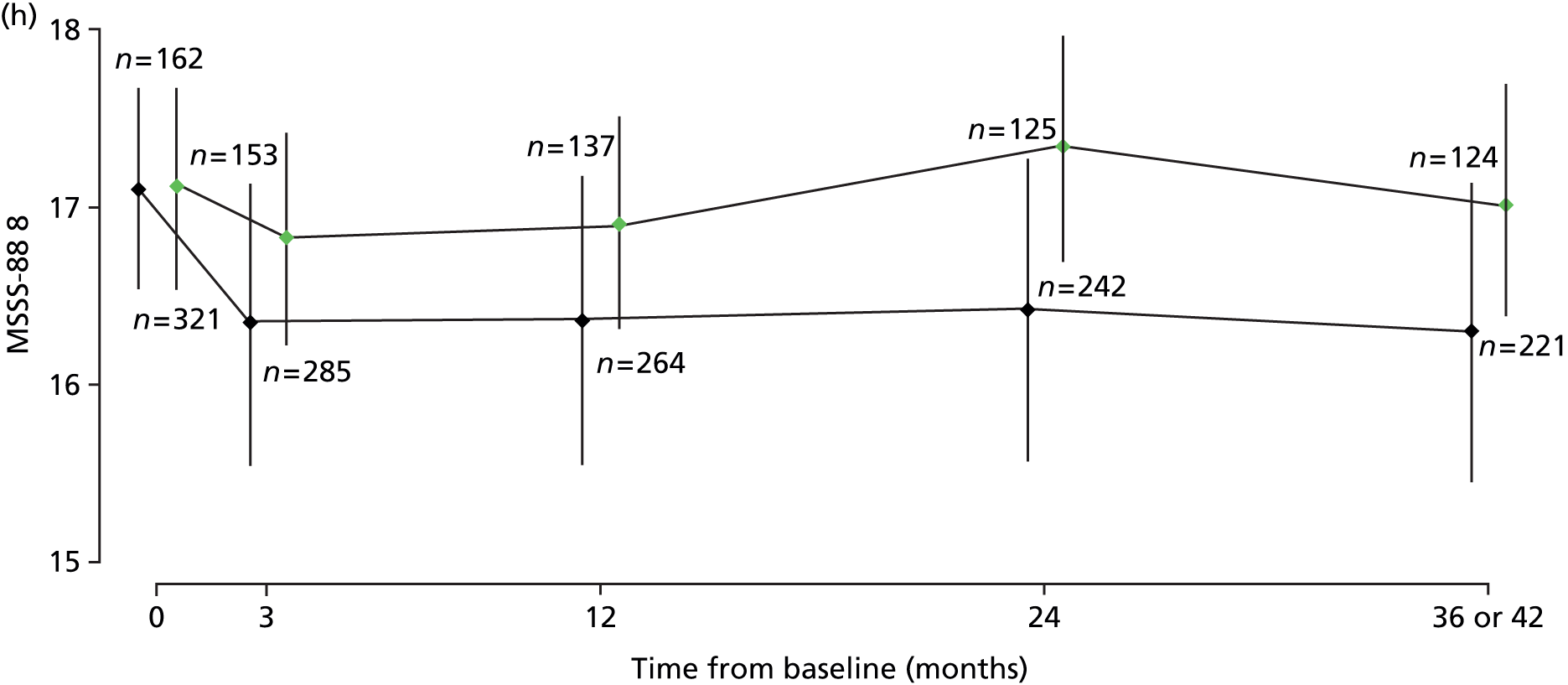
For each of the physical components of the MSSS-88, subscales 1 to 6 inclusive, after an initial decrease from baseline to month 3, mean scores tended to increase. Estimated means were consistent across treatment groups, as seen by the overlapping CIs. For the two psychological components, subscales 7 and 8, after an initial decrease from baseline to month 3, mean scores remained relatively constant over the study period. Estimated mean scores for these components tended to be higher in the active group than in placebo, but any differences failed to reach statistical significance.
Multilevel models fitted to three groups of the MSSS-88, where MSSS-88 (1) combines subscales 1–3; MSSS-88 (2) combines subscales 4–6 and MSSS-88 (3) combines subscales 7 and 8 (as described in Chapter 2), each showed no evidence of an effect of treatment. Estimated between-group difference (active–placebo) for MSSS-88 (1) was 0.26 (95% CI –1.99 to 2.52; p = 0.82; see Table 8); for MSSS-88 (2) was –0.02 (95% CI –2.35 to 2.32; p = 0.99; see Table 8); and for MSSS-88 (3) was 1.00 (95% CI –0.70 to 2.70; p = 0.25; see Table 8). In both treatment groups, mean MSSS-88 (1) and mean MSSS-88 (2) decreased from baseline to month 3, after which they tended to increase (Figure 14). After an initial decrease from baseline to month 3, mean MSSS-88 (3) remained relatively constant over the study period (see Figure 14).
FIGURE 14.
Estimated mean MSSS-88 (1) total score from subscales 1–3, concerning muscle stiffness/spasms, pain and discomfort; MSSS-88 (2) total score from subscales 4–6, concerning activity, walking and body movements; and MSSS-88 (3) total score from subscales 7 and 8, concerning feelings and social functioning, with 95% CIs, at each visit, separated by treatment group. The numbers of patients with total scores calculated at each visit, in each treatment group, are given directly above the CI for placebo and directly below the CI for active. Green points, active treatment; black points, placebo.

Investigation of adverse events and serious adverse events
The number of participants experiencing at least one SAE was 114 (35%) in the Δ9-THC group and 46 (28%) in the placebo group, the most common SAE being admission to hospital for MS-related events and infections. The number and nature of SAEs experienced was similar across treatment groups (Table 9).
| Classification or description of event | Number of patients (% of group) | ||
|---|---|---|---|
| Active (n = 329) | Placebo (n = 164) | All (n = 493) | |
| SAEs | |||
| Death | 6 (1.8) | 1 (0.6) | 7 (1.4) |
| Admission to hospital | 106 (32) | 44 (27) | 150 (30) |
| Life-threatening or important medical event | 10 (3.0) | 4 (2.4) | 14 (2.8) |
| At least one of the above | 114 (35) | 46 (28) | 160 (32) |
| Most common AEs | |||
| Falls and injuries | 101 (31) | 51 (31) | 152 (31) |
| Mobility, balance and co-ordination problems | 108 (33) | 43 (26) | 151 (31) |
| Infections (excluding urinary tract) | 95 (29) | 47 (29) | 142 (29) |
| Fatigue and tiredness | 81 (25) | 38 (23) | 119 (24) |
| Dizziness and light-headedness | 105 (32) | 12 (7) | 117 (24) |
| Muscle disorders (spasticity, stiffness, spasms or tremor) | 78 (24) | 38 (23) | 116 (24) |
| Muscle disorders (weakness) | 74 (22) | 32 (20) | 106 (22) |
| Dissociative and thinking or perception disorders | 98 (30) | 6 (4) | 104 (21) |
| Mood disorders (depression) | 66 (20) | 26 (16) | 92 (19) |
| Musculoskeletal pain and aches | 49 (15) | 41 (25) | 90 (18) |
| Constipation, diarrhoea, faecal incontinence | 56 (17) | 22 (13) | 78 (16) |
| Joint disorders | 47 (14) | 28 (17) | 75 (15) |
| Urinary tract infections | 44 (13) | 28 (17) | 72 (15) |
There were numerous non-serious AEs in both groups, consistent with the effects of MS and the known safety profile of cannabinoids. The median number of events per participant in the active group was 11 (25th–75th percentiles 7–17) compared with 10 (25th–75th percentiles 6–14) in the placebo group. Of those events judged to be either moderate or severe, the most frequent are documented in Table 9. Among these AEs, there was some suggestion that those participants on active treatment were more likely to experience dizziness or light-headedness and dissociative and thinking or perception disorders. On the other hand, a higher proportion of patients in the placebo group experienced musculoskeletal pain and aches than in the active group.
Six SAEs were classified as potential SUSARs in accordance with European clinical trials legislation. Three events occurred in each of the active and placebo groups. Trial treatment was discontinued in three participants as a result of the SAE. Three SAEs were classified as nervous system disorders: two were psychiatric events and one related to the gastrointestinal tract.
Category rating scales
Responses to questions 9–16 of the category rating scales, relating to how the patient felt at the time of completing the questionnaire, compared with just before the start of the study, have been grouped (as described in Chapter 2) and summarised, in terms of frequencies and relative frequencies in the two treatment groups, at each follow-up (Tables 10–13). Unadjusted p-values from chi-squared tests for trend are given.
| Visit 5 (3 months after baseline) | Better (scores 1–5) | No change (score 6) | Worse (scores 7–11) | p-valuea | |||
|---|---|---|---|---|---|---|---|
| Active | Placebo | Active | Placebo | Active | Placebo | ||
| Fatigue (n = 422; na = 276; np = 146) | 58 (21) | 27 (18) | 118 (43) | 67 (46) | 100 (36) | 52 (36) | 0.80 |
| Forgetfulness (n = 386; na = 254; np = 132) | 32 (13) | 18 (14) | 141 (56) | 94 (71) | 81 (32) | 20 (15) | 0.0067 |
| Sensory loss or numbness (n = 394; na = 254; np = 140) | 40 (16) | 21 (15) | 152 (60) | 74 (53) | 62 (24) | 45 (32) | 0.21 |
| Co-ordination (n = 445; na = 290; np = 155) | 188 (65) | 107 (69) | 35 (12) | 19 (12) | 67 (23) | 29 (19) | 0.29 |
| Irritability (n = 370; na = 238; np = 132) | 43 (18) | 22 (17) | 130 (55) | 80 (61) | 65 (27) | 30 (23) | 0.65 |
| Depression (n = 356; na = 238; np = 118) | 43 (18) | 27 (23) | 130 (55) | 67 (57) | 65 (27) | 24 (20) | 0.12 |
| Tremor (n = 438; na = 284; np = 154) | 34 (12) | 20 (13) | 103 (36) | 64 (42) | 147 (52) | 70 (45) | 0.29 |
| Bladder problems (n = 412; na = 270; np = 142) | 56 (21) | 30 (21) | 129 (48) | 72 (51) | 85 (31) | 40 (28) | 0.61 |
| Visit 7 (1 year after baseline) | Better (scores 1–5) | No change (score 6) | Worse (scores 7–11) | p-valuea | |||
|---|---|---|---|---|---|---|---|
| Active | Placebo | Active | Placebo | Active | Placebo | ||
| Fatigue (n = 385; na = 250; np = 135) | 32 (13) | 25 (19) | 80 (32) | 39 (29) | 138 (55) | 71 (53) | 0.29 |
| Forgetfulness (n = 340; na = 222; np = 118) | 18 (8) | 12 (10) | 96 (43) | 56 (47) | 108 (49) | 50 (42) | 0.25 |
| Sensory loss or numbness (n = 348; na = 222; np = 126) | 25 (11) | 15 (12) | 95 (43) | 58 (46) | 102 (46) | 53 (42) | 0.55 |
| Co-ordination (n = 368; na = 239; np = 129) | 23 (10) | 9 (7) | 98 (41) | 61 (47) | 118 (49) | 59 (46) | 0.89 |
| Irritability (n = 349; na = 225; np = 124) | 29 (13) | 16 (13) | 116 (52) | 58 (47) | 80 (36) | 50 (40) | 0.52 |
| Depression (n = 335; na=225; np =110) | 29 (13) | 19 (17) | 116 (52) | 45 (41) | 80 (36) | 46 (42) | 0.81 |
| Tremor (n = 274; na = 117; np = 97) | 22 (12) | 23 (24) | 78 (44) | 43 (44) | 77 (44) | 31 (32) | 0.011 |
| Bladder problems (n = 380; na = 246; np = 134) | 41 (17) | 30 (22) | 80 (33) | 53 (40) | 125 (51) | 51 (38) | 0.023 |
| Visit 9 (2 years after baseline) | Better (scores 1–5) | No change (score 6) | Worse (scores 7–11) | p-valuea | |||
|---|---|---|---|---|---|---|---|
| Active | Placebo | Active | Placebo | Active | Placebo | ||
| Fatigue (n = 358; na = 232; np = 126) | 30 (13) | 15 (12) | 51 (22) | 31 (25) | 151 (65) | 80 (63) | 0.94 |
| Forgetfulness (n = 327; na = 218; np = 109) | 18 (8) | 12 (11) | 75 (34) | 49 (45) | 125 (57) | 48 (44) | 0.037 |
| Sensory loss or numbness (n = 339; na = 220; np = 119) | 25 (11) | 9 (8) | 91 (41) | 48 (40) | 104 (47) | 62 (52) | 0.25 |
| Co-ordination (n = 344; na = 222; np = 122) | 18 (8) | 7 (6) | 70 (32) | 36 (30) | 134 (60) | 79 (65) | 0.34 |
| Irritability (n = 320; na = 207; np = 113) | 37 (18) | 15 (13) | 83 (40) | 45 (40) | 87 (42) | 53 (47) | 0.26 |
| Depression (n = 316; na = 207; np = 109) | 37 (18) | 19 (17) | 83 (40) | 43 (39) | 87 (42) | 47 (43) | 0.86 |
| Tremor (n = 276; na = 117; np = 99) | 27 (15) | 9 (9) | 76 (43) | 45 (45) | 74 (42) | 45 (45) | 0.25 |
| Bladder problems (n = 357; na = 233; np = 124) | 39 (17) | 18 (15) | 63 (27) | 32 (26) | 131 (56) | 74 (60) | 0.50 |
| Visit 11 (3 years after baseline) | Better (scores 1–5) | No change (score 6) | Worse (scores 7–11) | p-valuea | |||
|---|---|---|---|---|---|---|---|
| Active | Placebo | Active | Placebo | Active | Placebo | ||
| Fatigue (n = 332; na = 209; np = 123) | 33 (16) | 10 (8) | 44 (21) | 30 (24) | 132 (63) | 83 (67) | 0.14 |
| Forgetfulness (n = 301; na = 191; np = 110) | 22 (12) | 11 (10) | 54 (28) | 49 (45) | 115 (60) | 50 (45) | 0.11 |
| Sensory loss or numbness (n = 315; na = 201; np = 114) | 25 (12) | 8 (7) | 80 (40) | 46 (40) | 96 (48) | 60 (53) | 0.19 |
| Co-ordination (n = 321; na = 203; np = 118) | 21 (10) | 5 (4) | 65 (32) | 44 (37) | 117 (58) | 69 (58) | 0.35 |
| Irritability (n = 296; na = 185; np = 111) | 29 (16) | 10 (9) | 87 (47) | 58 (52) | 69 (37) | 43 (39) | 0.31 |
| Depression (n = 283; na = 185; np = 98) | 29 (16) | 18 (18) | 87 (47) | 42 (43) | 69 (37) | 38 (39) | 0.89 |
| Tremor (n = 252; na = 161; np = 91) | 24 (15) | 9 (10) | 54 (34) | 38 (42) | 83 (52) | 44 (48) | 0.84 |
| Bladder problems (n = 330; na = 209; np = 121) | 38 (18) | 21 (17) | 46 (22) | 32 (26) | 125 (60) | 68 (56) | 0.75 |
Generally, a higher proportion of patients on active treatment than on placebo reported being more forgetful at the time of follow-up compared with before the study. At 3 months from baseline, there was an approximately twofold increase in proportion of responses classified as ‘worse’ in the active group compared with placebo (32% active, 15% placebo; p = 0.0067; see Table 10). These proportions were similar across treatment groups at 1-year follow-up (49% active, 42% placebo; p = 0.25; see Table 11), at 2 years there was an approximately 30% increase in ‘worse’ responses in the active group compared with placebo (57% active, 44% placebo; p = 0.037; see Table 12) and similarly at 3 years, with a 33% increase (60% active, 45% placebo; p = 0.11; see Table 13). Responses to the remaining questions were similar across treatment groups.
Analysis of premature discontinuations of trial medication and losses to follow-up
Of the 493 patients included in the ITT analysis, 119 (24.1%; 89 active, 30 placebo) prematurely discontinued trial medication but remained in follow-up. Seventy-eight patients (15.8%; 62 active, 16 placebo) were lost to follow-up, meaning that 296 (60%) patients completed the study on trial treatment.
There was evidence of an increased risk of discontinuation of trial medication in the active group compared with placebo (p < 0.001, log-rank test) (Figure 15).
FIGURE 15.
Kaplan–Meier estimates of the probability of discontinuation of trial medication in the two treatment groups. Those patients who were lost to follow-up during the trial, without previous discontinuation of trial medication, are marked by +. Those patients who reached the end of the trial without discontinuing trial medication are marked by x.
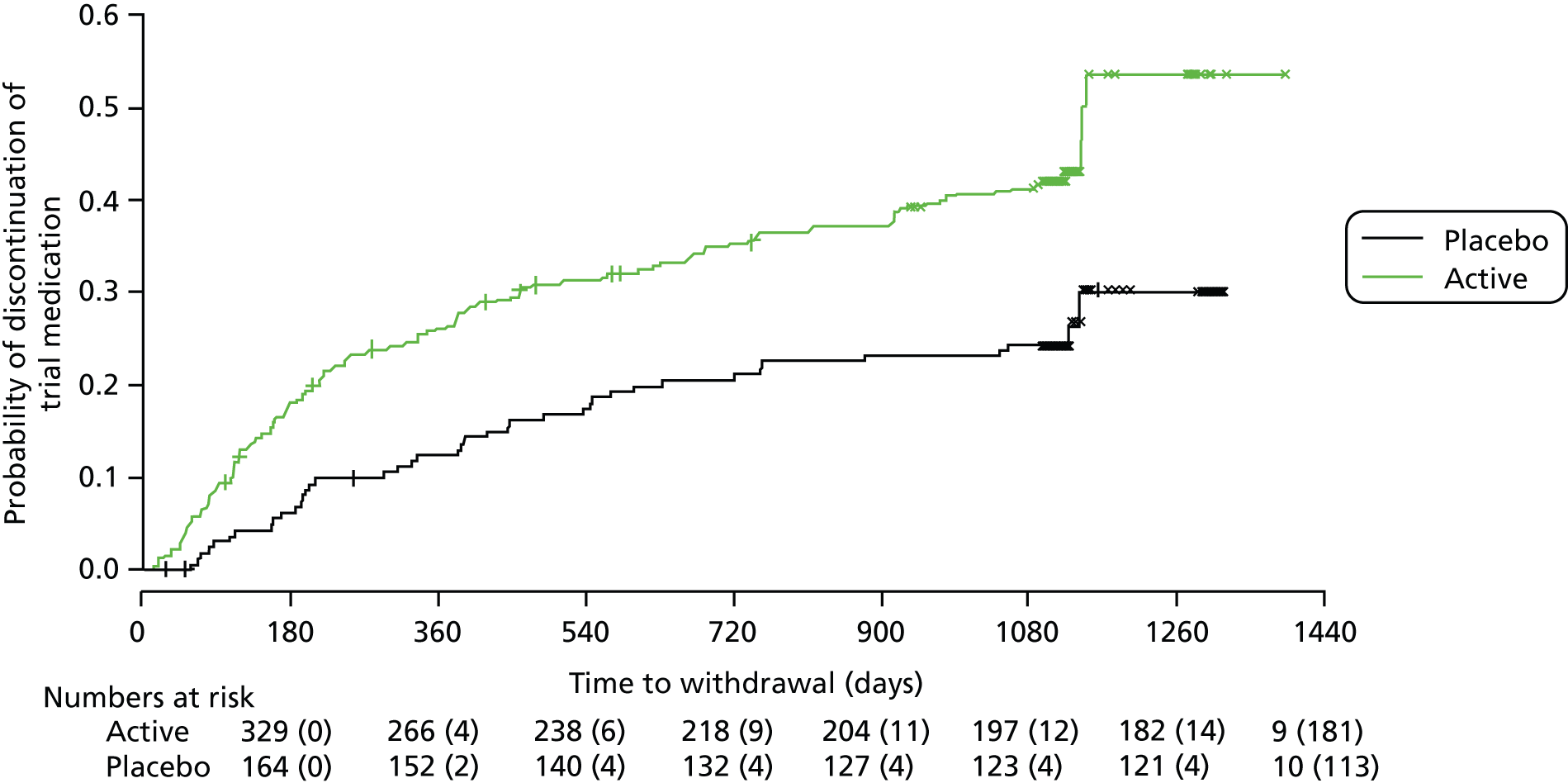
Reasons for discontinuation of trial medication or loss to follow-up were dominated by AEs, accounting for 65% of all early discontinuations (Table 14). Reasons for loss to follow-up are summarised in Table 15. The most common reasons were reported as ‘MS or other health issues’ and ‘other’, accounting for 50% (39 out of 78) of all losses to follow-up. ‘Travel or burden of the trial’ accounted for 22% (17 out of 78) of reasons for loss to follow-up and accounted for a larger proportion of losses in placebo patients [5 out of 16 (31%) compared with 12 out of 62 (19%) of the losses in the active group].
| Patient characteristics | Total | Reason for discontinuation of trial medication or loss to follow-up | |||
|---|---|---|---|---|---|
| AE | Death | Lack of efficacy | Other | ||
| Treatment group | |||||
| Active | 151 | 113 (74.8) | 3 (2.0) | 20 (13.2) | 15 (9.9) |
| Placebo | 46 | 14 (30.4) | 1 (2.2) | 22 (47.8) | 9 (19.6) |
| Disease type | |||||
| PPMS | 80 | 48 (60.0) | 4 (5.0) | 20 (25.0) | 8 (10.0) |
| SPMS | 117 | 79 (67.5) | 0 (0.0) | 22 (18.8) | 16 (13.7) |
| Baseline EDSS score | |||||
| 4.0–5.5 | 44 | 29 (65.9) | 1 (2.3) | 7 (15.9) | 7 (15.9) |
| 6.0 | 102 | 71 (69.6) | 1 (1.0) | 20 (19.6) | 10 (9.8) |
| 6.5 | 51 | 27 (52.9) | 2 (3.9) | 15 (29.4) | 7 (13.7) |
| Sex | |||||
| Women | 107 | 68 (63.6) | 2 (1.9) | 24 (22.4) | 13 (12.1) |
| Men | 90 | 59 (65.6) | 2 (2.2) | 18 (20.0) | 11 (12.2) |
| Overall | 197 | 127 (64.5) | 4 (2.0) | 42 (21.3) | 24 (12.2) |
| Patient characteristics | Total | Reason for loss to follow-up | ||||||
|---|---|---|---|---|---|---|---|---|
| Death | Moved out of area | AE | MS or other health issue | Travel or burden of the trial | Ineligible | Other | ||
| Treatment group | ||||||||
| Active | 62 | 7 (11.3) | 5 (8.1) | 6 (9.7) | 17 (27.4) | 12 (19.4) | 1 (1.6) | 14 (22.6) |
| Placebo | 16 | 1 (6.3) | 1 (6.3) | 0 (0.0) | 3 (18.8) | 5 (31.3) | 1 (6.3) | 5 (31.3) |
| Disease type | ||||||||
| PPMS | 32 | 4 (12.5) | 2 (6.3) | 3 (9.4) | 8 (25.0) | 4 (12.5) | 1 (3.1) | 10 (31.2) |
| SPMS | 46 | 4 (8.7) | 4 (8.7) | 3 (6.5) | 12 (26.1) | 13 (28.3) | 1 (2.2) | 9 (19.6) |
| Baseline EDSS score | ||||||||
| 4.0–5.5 | 15 | 2 (13.3) | 1 (6.7) | 2 (13.3) | 3 (20.0) | 1 (6.7) | 0 (0.0) | 6 (40.0) |
| 6.0 | 41 | 3 (7.3) | 3 (7.3) | 3 (7.3) | 10 (24.4) | 11 (26.8) | 1 (2.4) | 10 (24.4) |
| 6.5 | 22 | 3 (13.6) | 2 (9.1) | 1 (4.5) | 7 (31.8) | 5 (22.7) | 1 (4.5) | 3 (13.6) |
| Sex | ||||||||
| Women | 42 | 4 (9.5) | 2 (4.8) | 3 (7.1) | 7 (16.7) | 11 (26.2) | 1 (2.4) | 14 (33.3) |
| Men | 36 | 4 (11.1) | 4 (11.1) | 3 (8.3) | 13 (36.1) | 6 (16.7) | 1 (2.8) | 5 (13.9) |
| Overall | 78 | 8 (10.3) | 6 (7.7) | 6 (7.7) | 20 (25.6) | 17 (21.8) | 2 (2.6) | 19 (24.4) |
Rates of discontinuations from trial medication or loss to follow-up varied across study sites (Table 16).
| Participant group | Study site | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 01 | 02 | 04 | 05 | 06 | 07 | 08 | 10 | 11 | 12 | 13 | 14 | 15 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 31 | 32 | 33 | |
| Total recruited, n | 77 | 11 | 11 | 18 | 4 | 16 | 10 | 12 | 8 | 6 | 19 | 14 | 22 | 19 | 21 | 14 | 22 | 26 | 12 | 21 | 21 | 30 | 11 | 40 | 10 | 8 | 10 |
| Premature discontinuation of trial medication or loss to follow-up, n (%) | 23 (29.9) | 8 (72.7) | 5 (45.5) | 7 (38.9) | 1 (25.0) | 4 (25.0) | 3 (30.0) | 8 (66.7) | 5 (62.5) | 0 (0.0) | 8 (42.1) | 2 (14.3) | 12 (54.5) | 7 (36.8) | 8 (38.1) | 4 (28.6) | 6 (27.3) | 11 (42.3) | 1(8.3) | 12 (57.1) | 12 (57.1) | 17 (56.7) | 2 (18.2) | 17 (42.5) | 7 (70.0) | 3 (37.5) | 4 (40.0) |
Following a forward selection procedure, a Cox regression model fitted to data on time to discontinuation of trial medication or loss to follow-up showed evidence of effects of treatment allocation, sex and study site on the risk of withdrawal or loss to follow-up. The risk of withdrawal or loss to follow-up was estimated to be higher in men than in women [HR (men : women) 1.37, 95% CI 1.02 to 1.84] and higher in the active group than in the placebo group [HR (active : placebo) 1.97, 95% CI 1.41 to 2.76].
Pre-specified analyses of magnetic resonance imaging substudy
Two hundred and seventy-four patients from 13 study sites were allocated to the MRI substudy. Of these, one patient was excluded at baseline visit (as baseline scan was deemed problematic, due to patient tremor).
Baseline data on demographic and disease characteristics were similar across treatment groups (Table 17). Fifty-nine per cent of patients were women, 64% had SPMS and 76% had an EDSS score of 6.0 or 6.5.
| Patient baseline characteristics | Treatment group | All (N = 273) | |
|---|---|---|---|
| Active (N = 182; 66.7%) | Placebo (N = 91; 33.3%) | ||
| Age in years at registration, mean (SD) | 52.4 (7.3) | 52.2 (8.1) | 52.3 (7.6) |
| Weight in kg at registration, mean (SD) | 74.3 (16.1) | 75.7 (17.5) | 74.8 (16.6) |
| Men, n (%) | 80 (44.0) | 31 (34.1) | 111 (40.7) |
| Women, n (%) | 102 (56.0) | 60 (65.9) | 162 (59.3) |
| PPMS, n (%) | 60 (33.0) | 38 (41.8) | 98 (35.9) |
| SPMS, n (%) | 122 (67.0) | 53 (58.2) | 175 (64.1) |
| EDSS score at baseline, n (%) | |||
| 4.0 | 14 (7.7) | 6 (6.6) | 20 (7.3) |
| 4.5 | 12 (6.6) | 5 (5.5) | 17 (6.2) |
| 5.0 | 12 (6.6) | 6 (6.6) | 18 (6.6) |
| 5.5 | 8 (4.4) | 3 (3.3) | 11 (4.0) |
| 6.0 | 95 (52.2) | 47 (51.6) | 142 (52.0) |
| 6.5 | 41 (22.5) | 24 (26.4) | 65 (23.8) |
| Normalised brain volume, mean (SD) | 1422 (91.0) | 1417 (85.1) | 1420 (88.9) |
| Not reported, n (%) | 24 (13.2) | 8 (8.8) | 32 (11.7) |
Forty-seven of the 182 patients (25.8%) on active treatment and 17 of the 91 patients (18.7%) on placebo were lost to follow-up during the study period. Figure 16 shows the flow of patients over the 3-year follow-up period.
FIGURE 16.
Flow of participants through the MRI substudy.
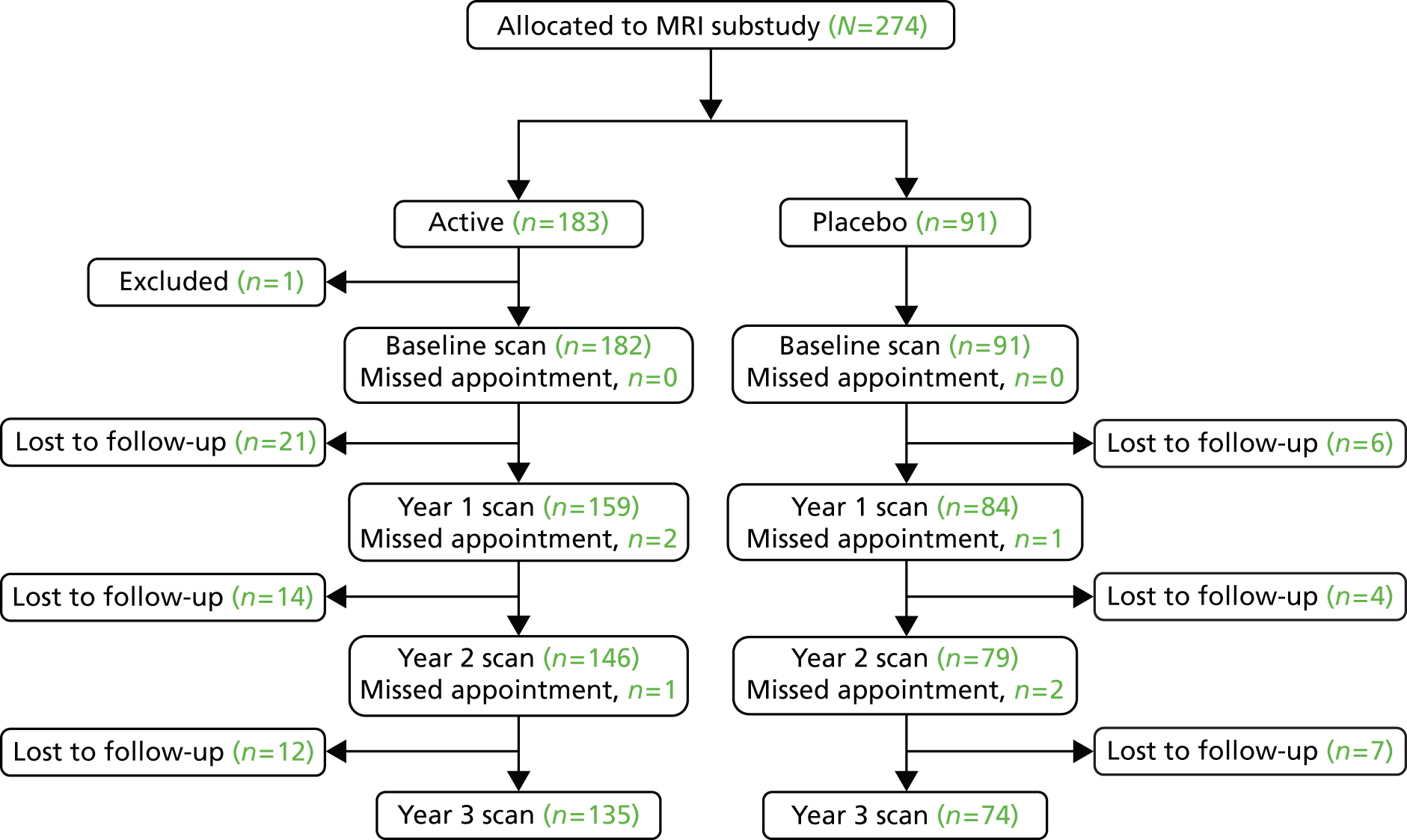
Between-treatment-group comparisons of PBVC and numbers of new or enlarging T2 and new T1 lesions at each annual follow-up showed little evidence of an association between treatment allocation and these outcomes (Table 18).
| Outcome measures | Year 1 | Year 2 | Year 3 | |||
|---|---|---|---|---|---|---|
| Active (n = 159) | Placebo (n = 84) | Active (n = 146) | Placebo (n = 79) | Active (n = 135) | Placebo (n = 74) | |
| PBVC | ||||||
| Mean (SD) | –0.60 (0.99) | –0.59 (0.95) | –0.58 (0.96) | –0.65 (0.95) | –0.88 (0.87) | –0.76 (1.04) |
| Median (25th–75th percentiles) | –0.60 (–1.32, –0.05) | –0.47 (–1.03, –0.08) | –0.52 (–1.21, –0.06) | –0.78 (–1.17, –0.10) | –0.78 (–1.43, –0.44) | –0.81 (–1.42, 0.20) |
| Not reported, n (%) | 3 (1.9) | 1 (1.2) | 5 (3.4) | 2 (2.5) | 7 (5.2) | 2 (2.7) |
| p-valuea | 0.9 | 0.6 | 0.4 | |||
| New or newly enlarging T2 lesions, n (%) | ||||||
| 0 | 118 (74.2) | 56 (66.7) | 111 (76.0) | 71 (89.9) | 113 (83.7) | 61 (82.4) |
| 1 | 20 (12.6) | 15 (17.9) | 23 (15.8) | 5 (6.3) | 16 (11.9) | 9 (12.2) |
| ≥ 2 | 21 (13.2) | 13 (15.5) | 12 (8.2) | 3 (3.8) | 6 (4.4) | 4 (5.4) |
| Not reported | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| p-valueb | 0.40 | 0.05 | 0.96 | |||
| New T1 lesions, n (%) | ||||||
| 0 | 123 (77.4) | 64 (76.2) | 122 (83.6) | 74 (93.7) | 118 (87.4) | 62 (83.8) |
| 1 | 20 (12.6) | 12 (14.3) | 16 (11.0) | 3 (3.8) | 13 (9.6) | 10 (13.5) |
| ≥ 2 | 16 (10.1) | 8 (9.5) | 8 (5.5) | 2 (2.5) | 4 (3.0) | 2 (2.7) |
| Not reported | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| p-valueb | 0.9 | 0.1 | 0.7 | |||
A multilevel model fitted to cumulative PBVC showed no evidence of an effect of active treatment on brain atrophy compared with placebo over the course of the study [estimated between-group difference in PBVC (active–placebo) was −0.01%, 95% CI −0.26% to 0.24%; p = 0.94; see Table 8]. However, brain atrophy did change significantly over time (p < 0.0001); using a fitted model, cumulative PBVC was estimated to be a mean of −0.58% at year 1, −1.20% at year 2 and −2.02% at year 3 (Figure 17).
FIGURE 17.
Estimated mean cumulative PBVC (%), and 95% CI, by treatment group, measured at yearly MRI visits. Green points, active treatment; black points, placebo. n, number of patients with cumulative PBVC calculated at each visit, given directly above the CI for placebo and directly below the CI for active.
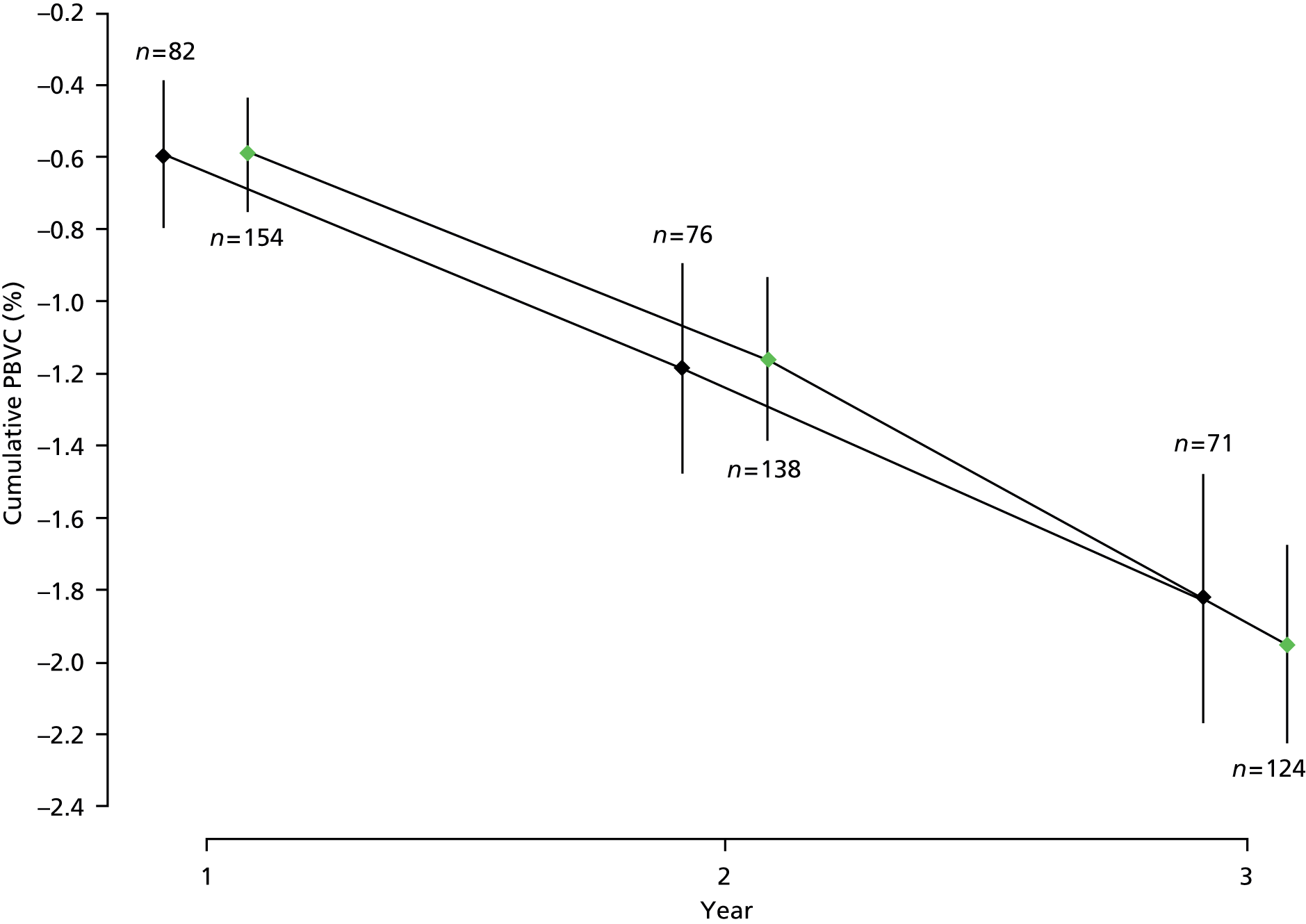
There was evidence of an effect of baseline normalised brain volume (NBV) on brain atrophy. Using a fitted model, it was estimated that, for a 100-unit reduction in baseline NBV, brain atrophy increased by a mean of 0.21% (95% CI 0.08% to 0.34%; p = 0.003).
Treatment did not significantly affect the occurrence of new or newly enlarging T2 lesions [estimated odds ratio (OR) (active : placebo) 0.89, 95% CI 0.50 to 1.58; p = 0.70; see Table 8] or new T1 lesions [estimated OR (active : placebo) 1.05, 95% CI 0.59 to 1.88; p = 0.87; see Table 8].
Chapter 4 Post-hoc exploratory analyses: main study and magnetic resonance imaging substudy
Introduction
Post-hoc exploratory analyses covered three main areas, as described below.
Firstly, the suggestion of a treatment effect from pre-specified subgroup analysis of time to first confirmed EDSS score progression led to a post-hoc analysis of time to EDSS score progression among patients in this EDSS group. Further analysis examined the effect of treatment allocation in the two participant groups with a baseline EDSS score of 4.0–5.5 and 6.0–6.5, on change in MSIS-29phys and PBVC.
Secondly, an on-treatment data set included follow-up data on all patients up to the time of withdrawal from trial treatment, loss to follow-up or end of study – whichever came first. Further analyses of time to first confirmed EDSS score progression, change in MSIS-29phys and PBVC were carried out using these data sets.
Thirdly, in contrast to pre-specified analyses, presented in the previous chapter, which were based on time to first progression event (or last follow-up) with any subsequent changes in EDSS score not considered, post-hoc exploratory analyses considered EDSS progression as an unconfirmed, recurrent event, using the following criteria:
The first progression event is when EDSS score increases by at least 1 point from a baseline score of 4.0, 4.5 or 5.0, or by at least 0.5 points from a baseline score of 5.5 or higher. Subsequent progression events are when the EDSS score increases by these amounts, from the score at the previous progression. Progressions do not need to be confirmed at the next scheduled 6-monthly visit.
Methods
Subgroup analyses
Subgroup analysis of time to EDSS score progression, based on a total of 110 patients with a baseline EDSS score of 4.0–5.5, included inspection of Kaplan–Meier estimates of the probability of EDSS score progression, by treatment group and comparison using a log-rank test.
The effect of treatment allocation in the two participant groups with a baseline EDSS score of 4.0–5.5 and 6.0–6.5, on change in MSIS-29phys and on PBVC, was examined. This was done by including separate effects of treatment in the groups of patients with a baseline EDSS score of 4.0–5.5 and 6.0–6.5 in the multilevel models fitted to the data sets used in the primary analyses of change in MSIS-29phys and of PBVC.
On-treatment analyses
Kaplan–Meier estimates were used to show probability of EDSS score progression in the on-treatment data set, by treatment group.
In the on-treatment analysis, in which there were fewer observed first progression events than in the primary analysis, there were one or more study sites for which no progression events were observed. Therefore, two Cox PH models were fitted; the first included individual study site effects and the second included individual effects for the larger sites (with 20 or more patients) and a single, combined effect for the study sites with low throughput (n < 20).
Multilevel models, analogous to those fitted in the primary analyses of change in MSIS-29phys and PBVC (including single main effects of treatment and other pre-specified covariates), were fitted to the on-treatment data sets, which included all available data on MSIS-29phys scores and MRI scans performed, respectively, up to time of withdrawal from trial treatment, loss to follow-up or end of study – whichever came first.
Expanded Disability Status Scale score transitions and recurrent progression events
Transitions in EDSS scores from one 6-monthly visit to the next were investigated. Probabilities of each possible transition from given starting EDSS scores and, hence, the probability of EDSS score progression (unconfirmed) were estimated. EDSS scores were also grouped into low (5.5 or lower), medium (6.0) and high (6.5 or higher) and transitions between these states and probabilities of progression from these states were estimated. In these analyses, patients were allowed to have more than one unconfirmed progression event during the study period.
Results
Results from post-hoc exploratory subgroup and on-treatment analyses are summarised in Table 19 and detailed below.
| Outcome measure | Summary statistics | Treatment group | Analysis | Estimated treatment effect (95% CI) | p-value | |
|---|---|---|---|---|---|---|
| Active | Placebo | |||||
| EDSS (subgroup of participants with a baseline EDSS score of 4.0–5.5) | No. of first progression events | 44 | 26 | Post-hoc subgroup analyses: log-rank test | – | 0.01 |
| No. of first progression events per patient-yeara | 0.35 | 0.64 | ||||
| MSIS-29phys | Mean (SD) annual change; subgroup, a baseline EDSS score of 4.0–5.5 | 0.50 (3.16) | 1.72 (3.46) | Post-hoc multilevel model with separate treatment effects for EDSS groups 4.0–5.5 and 6.0–6.5: estimated between-group differences (active–placebo) | Baseline EDSS score of 4.0–5.5: –3.26 (–4.89 to –1.63) | 0.0001 |
| Mean (SD) annual change; subgroup, baseline EDSS score of 6.0–6.5 | 0.64 (3.35) | 0.90 (3.79) | Baseline EDSS score of 6.0–6.5: –0.21 (–1.37 to 0.96) | 0.73 | ||
| MRI: PBVC | Mean (SD) annual change; subgroup, baseline EDSS score of 4.0–5.5 | –0.57 (0.49) | –0.36 (0.37) | Post-hoc multilevel model with separate treatment effects for EDSS groups 4.0–5.5 and 6.0–6.5: estimated between-group differences (active–placebo) | Baseline EDSS score of 4.0–5.5: –0.06% (–0.42% to 0.29%) | 0.73 |
| Mean (SD) annual change; subgroup, baseline EDSS score of 6.0–6.5 | –0.70 (0.52) | –0.70 (0.50) | Baseline EDSS score of 6.0–6.5: 0.01% (–0.26 to 0.28%) | 0.95 | ||
| EDSS (on-treatment data set; participants on trial treatment at end of study or last follow-up) | No. of first progression events | 114 | 63 | HR (active : placebo) from post-hoc Cox regression analysis (losses to follow-up considered as censored observations), individual study site effects | 0.96 (0.69 to 1.34) | 0.83 |
| No. of first progression events per patient-yeara | 0.23 | 0.22 | ||||
| EDSS (on-treatment data set; participants on trial treatment at end of study or last follow-up) | No. of first progression events | 114 | 63 | HR (active : placebo) from post-hoc Cox regression analysis (losses to follow-up considered as censored observations), grouped study site effects | 0.95 (0.69 to 1.31) | 0.76 |
| No. of first progression events per patient-yeara | 0.23 | 0.22 | ||||
| MSIS-29phys (on-treatment data set; participants on trial treatment at end of study or last follow-up) | Mean (SD) annual change | 0.59 (3.37) | 0.87 (3.85) | Post-hoc multilevel model: estimated between-group difference (active–placebo) | –0.77 (–1.92 to 0.38) | 0.19 |
| MRI: PBVC (on-treatment data set; participants on trial treatment at end of study or last follow-up) | Mean (SD) annual change | –0.64 (0.53) | –0.62 (0.40) | Post-hoc multilevel model: estimated between-group difference (active–placebo) | 0.03% (–0.24% to 0.31%) | 0.80 |
Subgroup analyses
Figure 18 shows Kaplan–Meier estimates of the probability of EDSS score progression in the subgroup of patients with a baseline EDSS score of 4.0–5.5, separated by treatment group. There was some evidence of a potentially beneficial effect of active treatment, compared with placebo in this low-score EDSS subgroup (p = 0.01, log-rank test).
FIGURE 18.
Kaplan–Meier estimates of the probability of progression on the EDSS in the two treatment groups, confirmed after 6 months within the study period, among the subgroup of patients with a baseline EDSS score of 4.0–5.5. The numbers at risk (cumulative number of censored observations) are given. Those patients who were lost to follow-up during the trial are marked by +. Those who reached the end of the trial without progressing are marked by x.

There was some evidence of an effect of treatment on change in MSIS-29phys score in the group of patients with a baseline EDSS score of 4.0–5.5. On average, MSIS-29phys scores in the active treatment group were estimated to be 3.26 points lower (95% CI for reduction 1.63 to 4.89 points; p = 0.0001; see Table 19) than in the placebo group. There was no significant effect of treatment on change in MSIS-29phys score in the group of participants with a baseline EDSS score of 6.0–6.5 [estimated between-group difference (active–placebo) –0.21, 95% CI –1.37 to 0.96; p = 0.73; see Table 19].
There was no significant effect of treatment on brain atrophy in the two participant groups defined by baseline EDSS score; estimated between-group differences in PBVC (active–placebo) were –0.06% (95% CI –0.42% to 0.29%; p = 0.73; see Table 19) and 0.01% (95% CI –0.26% to 0.28%; p = 0.95; see Table 19) for baseline EDSS scores of 4.0–5.5 and 6.0–6.5, respectively.
On-treatment analyses
Analysis of time to first confirmed EDSS score progression was carried out using the on-treatment data set described above. Considering all withdrawals from trial treatment as losses to follow-up at the time of withdrawal, the on-treatment data set included 177 first progression events (114 in the active group, 63 in the placebo group), compared with 218 (145 active, 73 placebo) in the ITT data set used in the primary analysis. A Cox regression model provided no evidence of an effect of treatment on probability of progression [HR (active : placebo) 0.96, 95% CI 0.69 to 1.34; p = 0.83; see Table 19]. This estimated treatment effect was similar when study sites with low throughput (< 20 patients) were combined in a single effect in the fitted model [HR (active : placebo) 0.95, 95% CI 0.69 to 1.31; p = 0.76; see Table 19]. The global PH test gave no evidence that the PH assumption was violated under either fitted model (χ2 = 30.2, 36 degrees of freedom, p = 0.74 for a Cox model including individual study site effects; and χ2 = 13.3, 19 degrees of freedom, p = 0.82 for a Cox model including a single effect for sites with low throughput).
At trial completion, Kaplan–Meier estimates of the probability of EDSS score progression were 0.54 (95% CI 0.43 to 0.62) in the dronabinol group, compared with 0.58 (95% CI 0.41 to 0.71) in the placebo group (Figure 19).
FIGURE 19.
Kaplan–Meier estimates of the probability of progression on the EDSS in the two treatment groups, confirmed after 6 months within the study period, based on an on-treatment data set, in which withdrawals from trial treatment are considered as losses to follow-up at the time of withdrawal. The numbers at risk (cumulative number of censored observations) are given. Those patients who were lost to follow-up during the trial are marked by +. Those who reached the end of the trial without progressing are marked by x.
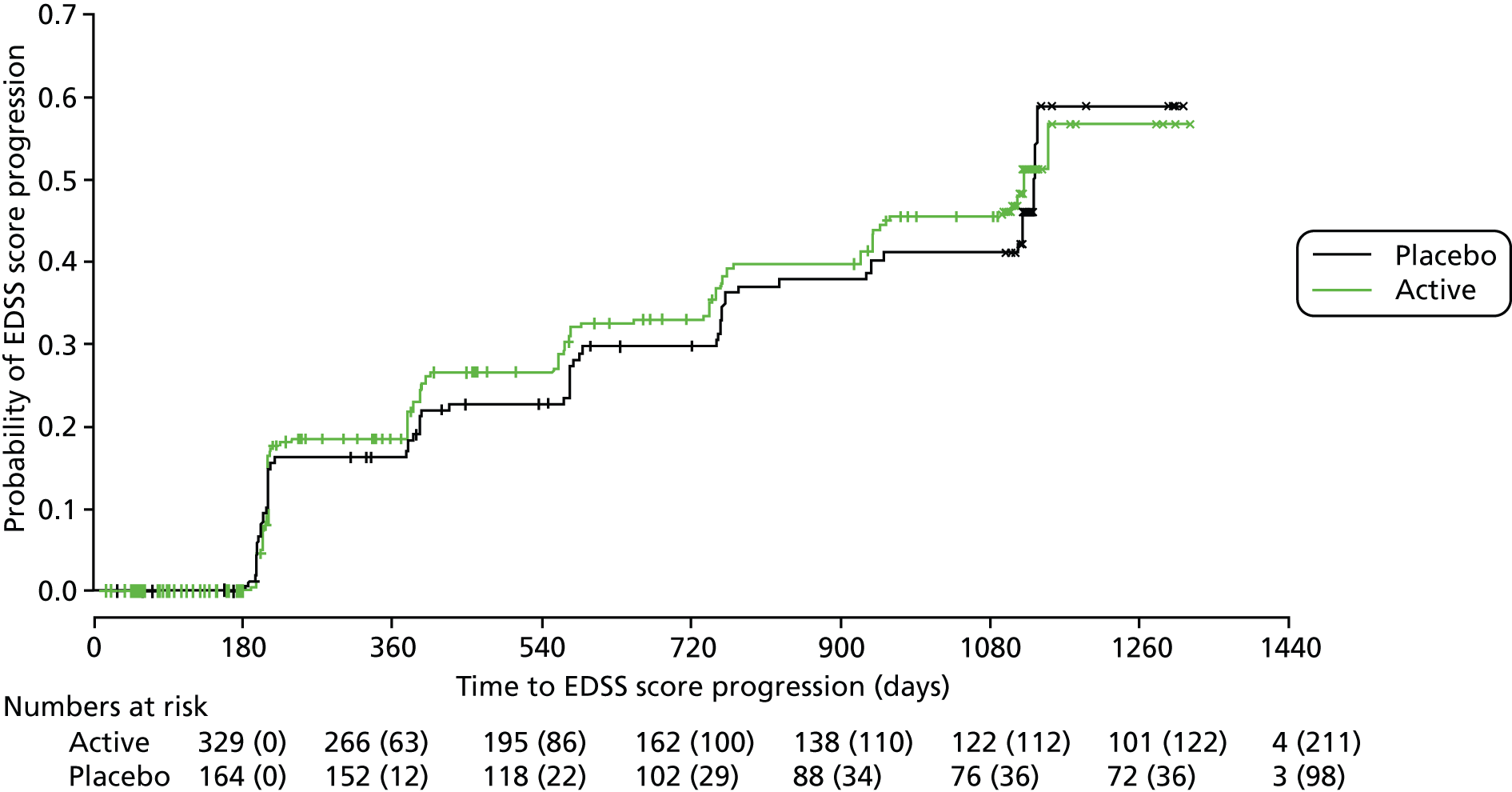
A multilevel model fitted to repeated measures of MSIS-29phys score in the on-treatment data set showed no evidence of an effect of treatment. The estimated between-group difference in MSIS-29phys (active–placebo) score was –0.77 (95% CI –1.92 to 0.38; p = 0.19; see Table 19).
Analysis of brain atrophy in the on-treatment data set on the MRI substudy was based on a total of 418 observations among 202 patients and included 200 measures of PBVC at year 1, 175 measures of cumulative PBVC at year 2 and 43 at year 3. There was no evidence of an effect of treatment on brain atrophy; the estimated between-group difference in PBVC (active–placebo) was 0.03% (95% CI –0.24% to 0.31%; p = 0.80; see Table 19). Using a fitted model, cumulative PBVC was estimated to be a mean of −0.60% at year 1, −1.17% at year 2 and −2.01% at year 3 (Figure 20).
FIGURE 20.
Estimated mean cumulative PBVC (%), and 95% CI, by treatment group, measured at yearly MRI visits, in the on-treatment data. Green points, active treatment; black points, placebo. n, number of patients with cumulative PBVC calculated at each visit, given directly above the CI for placebo and directly below the CI for active.

There was evidence of an effect of baseline NBV on brain atrophy. Using a fitted model, it was estimated that, for a 100-unit reduction in baseline NBV, atrophy increased by a mean of 0.20% (95% CI 0.06% to 0.35%; p = 0.008).
Expanded Disability Status Scale score transitions and recurrent progression events
Based on the definition of unconfirmed, recurrent progression events introduced earlier in this chapter, there were a total of 380 (245 among 182 patients on active treatment, 135 among 97 patients on placebo) progression events observed over the course of the trial. Frequencies and relative frequencies of patients having different numbers of events during follow-up are given in Table 20.
| Patient characteristics | Number of progression events | |||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 or 4 | |
| Treatment group | ||||
| Active (n = 329) | 147 (44.7) | 128 (38.9) | 45 (13.7) | 9 (2.7) |
| Placebo (n = 164) | 67 (40.9) | 68 (41.5) | 21 (12.8) | 8 (4.9) |
| Baseline EDSS score | ||||
| 4.0–5.5 (n = 110) | 33 (30.0) | 58 (52.7) | 17 (15.5) | 2 (1.8) |
| 6.0 (n = 254) | 111 (43.7) | 107 (42.1) | 25 (9.8) | 11 (4.3) |
| 6.5 (n = 129) | 70 (54.3) | 31 (24.0) | 24 (18.6) | 4 (3.1) |
| Total (N = 493) | 214 (43.4) | 196 (39.8) | 66 (13.4) | 17 (3.4) |
The probability of unconfirmed EDSS score progression appeared to depend on the starting EDSS score (Figures 21 and 22). For example, the probability of progression from a score of 5.5 tended to be higher than probabilities of progression from other starting scores. It is notable that this is the starting score for which the definition of progression changes from a 1-point increase to a 0.5-point increase. As starting EDSS score increased from 5.5, the probability of progression tended to decrease.
FIGURE 21.
Estimated probabilities of EDSS score progression (unconfirmed), with 95% CI, by starting EDSS score, in each 6-monthly interval from baseline. (a) 0–6 months (0–182 days from baseline); (b) 6–12 months (183–365 days from baseline); (c) 12–18 months (366–548 days from baseline); (d) 18–24 months (549–731 days from baseline); (e) 24–30 months (732–914 days from baseline); and (f) 30–36/42 months (915 or more days from baseline). n, number of patients with given starting EDSS score who have an EDSS score recorded at the end of the 6-monthly interval.

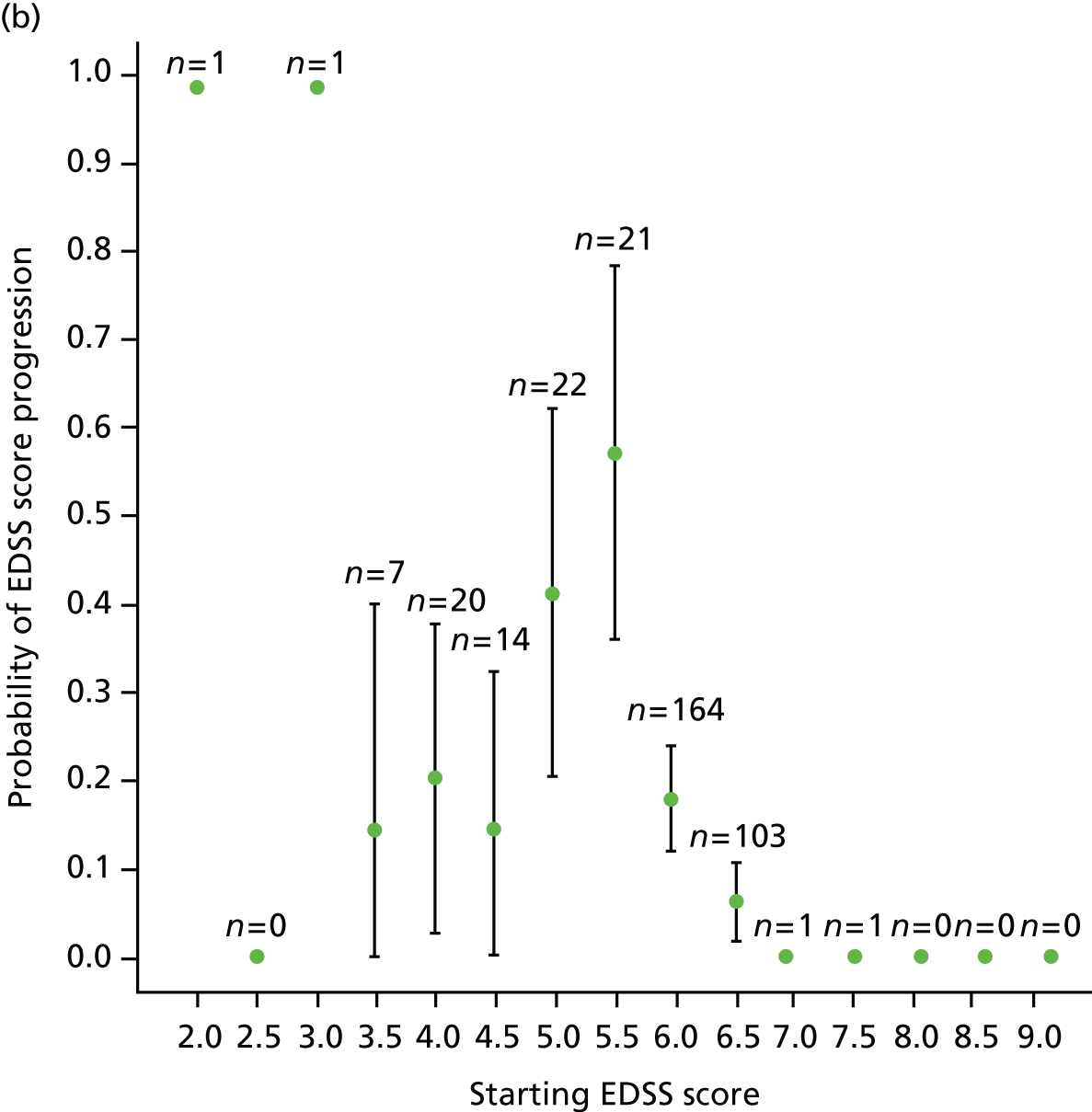

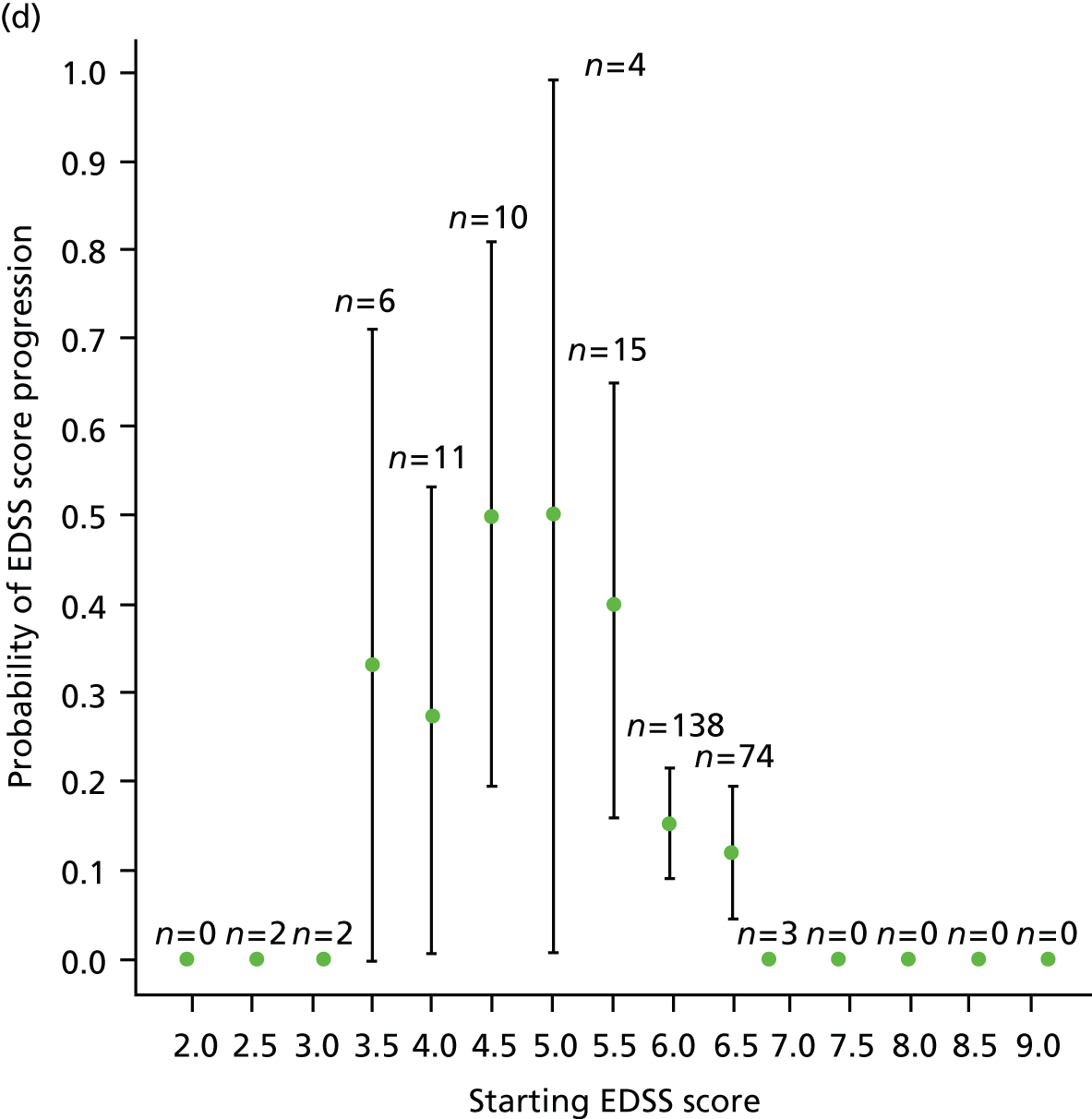
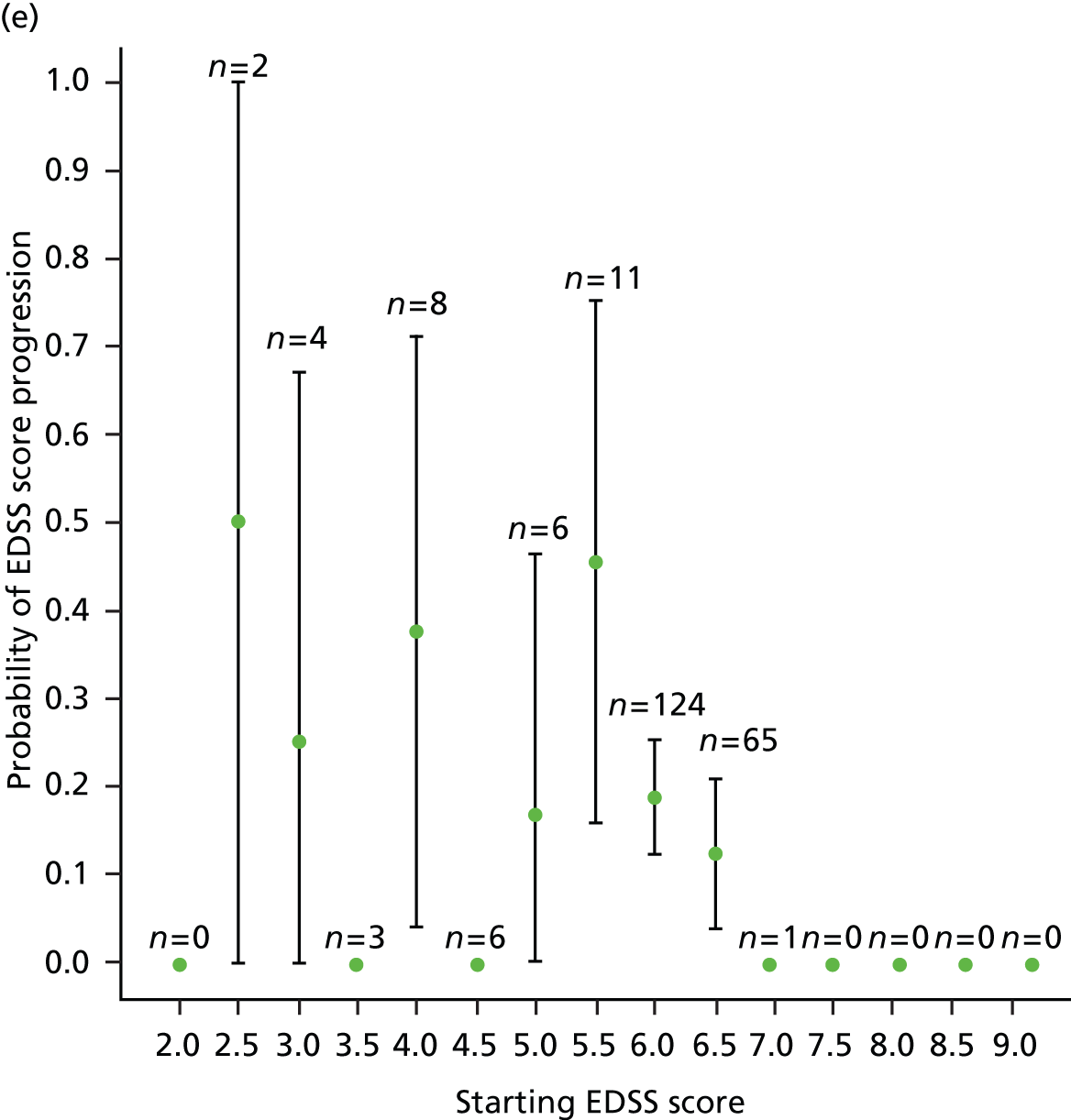
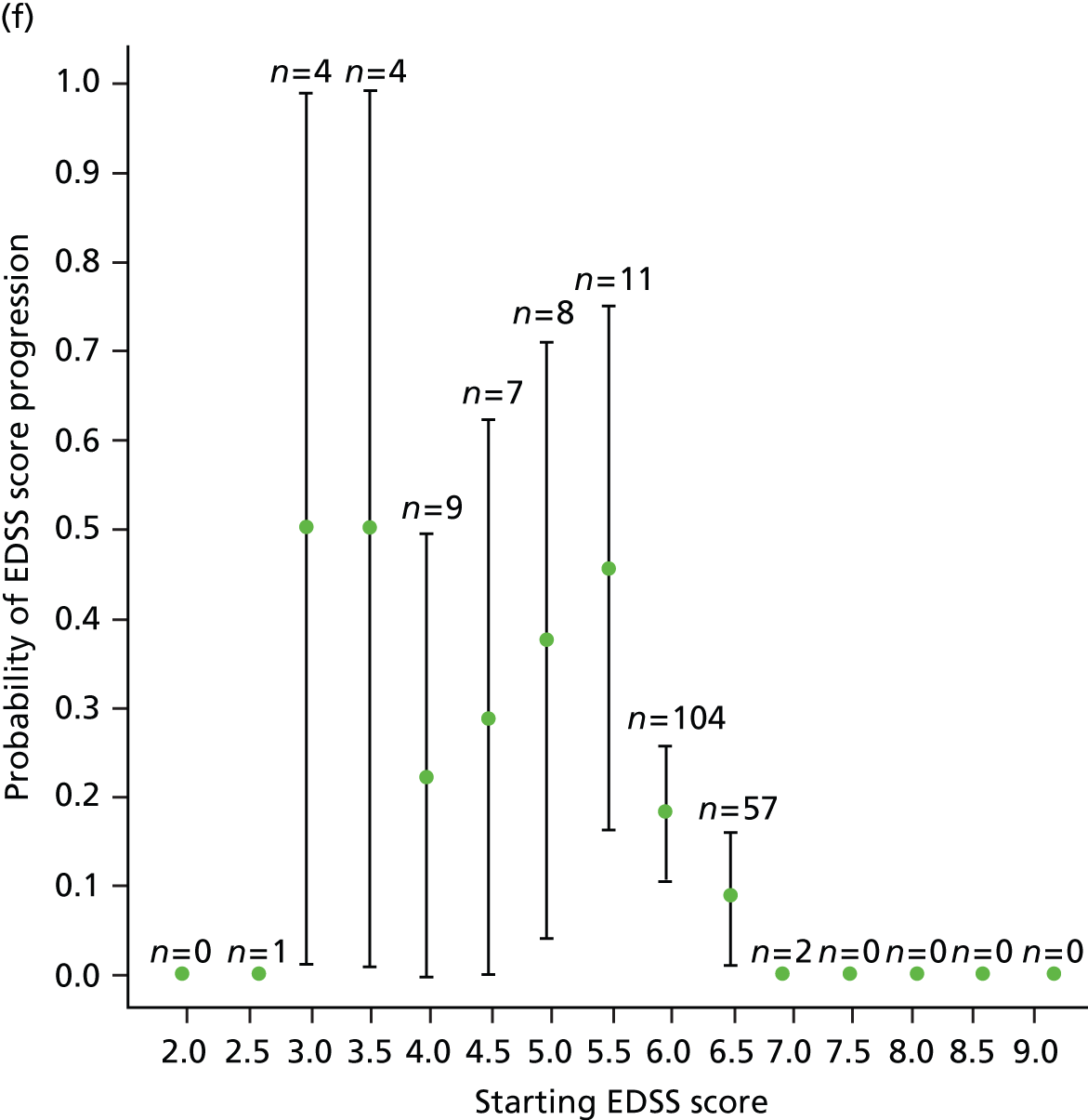
FIGURE 22.
Estimated probabilities of EDSS score progression (unconfirmed), with 95% CI, by starting EDSS score, in all 6-monthly intervals from baseline. n, number of patients with given starting EDSS score who have an EDSS score recorded at the end of the 6-monthly interval.

Overall, at a starting EDSS score of < 5.5, the probability of progression was slightly lower than for a starting EDSS score of 5.5 and slightly higher than for a starting EDSS score > 5.5 (see Figure 22). However, the number of observations at this lower end of the EDSS is relatively small, as reflected in the wide CIs.
Starting EDSS scores at baseline and at each 6-monthly follow-up were grouped into L, M and H, as described earlier in this chapter. Transition matrices were found for moving between states (Table 21). When considering progression from these three states, with the exception of the time period from baseline to 6 months, the probability of progression decreased with increasing starting EDSS score, for each 6-monthly period and overall (Figure 23).
| Time interval (number of observations, % of total) | Counts | Row proportions | ||||||
|---|---|---|---|---|---|---|---|---|
| From | To | From | To | |||||
| L | M | H | L | M | H | |||
| 0–6 months (0–182 days from baseline) (n = 457, 92.7%) | L | 79 | 20 | 4 | L | 0.7670 | 0.1942 | 0.0388 |
| M | 16 | 149 | 69 | M | 0.0684 | 0.6368 | 0.2949 | |
| H | 2 | 19 | 99 | H | 0.0167 | 0.1583 | 0.8250 | |
| 6–12 months (183–365 days from baseline) (n = 355, 72.0%) | L | 60 | 23 | 3 | L | 0.6977 | 0.2674 | 0.0349 |
| M | 6 | 129 | 29 | M | 0.0366 | 0.7866 | 0.1768 | |
| H | 0 | 25 | 80 | H | 0.0000 | 0.2381 | 0.7619 | |
| 12–18 months (366–548 days from baseline) (n = 300, 60.9%) | L | 47 | 9 | 0 | L | 0.8393 | 0.1607 | 0.0000 |
| M | 7 | 126 | 26 | M | 0.0440 | 0.7925 | 0.1635 | |
| H | 0 | 13 | 72 | H | 0.0000 | 0.1529 | 0.8471 | |
| 18–24 months (549–731 days from baseline) (n = 265, 53.8%) | L | 38 | 11 | 1 | L | 0.7600 | 0.2200 | 0.0200 |
| M | 5 | 112 | 21 | M | 0.0362 | 0.8116 | 0.1522 | |
| H | 0 | 12 | 65 | H | 0.0000 | 0.1558 | 0.8442 | |
| 24–30 months (732–914 days from baseline) (n = 230, 46.7%) | L | 34 | 6 | 0 | L | 0.8500 | 0.1500 | 0.0000 |
| M | 9 | 92 | 23 | M | 0.0726 | 0.7419 | 0.1855 | |
| H | 0 | 11 | 55 | H | 0.0000 | 0.1667 | 0.8333 | |
| 30–36/42 months (915 or more days from baseline) (n = 207, 42.0%) | L | 35 | 9 | 0 | L | 0.7955 | 0.2045 | 0.0000 |
| M | 0 | 85 | 19 | M | 0.0000 | 0.8173 | 0.1827 | |
| H | 0 | 8 | 51 | H | 0.0000 | 0.1356 | 0.8644 | |
FIGURE 23.
Estimated probabilities of unconfirmed EDSS score progression, with 95% CI, in each 6-monthly interval from baseline and overall. 0–6 months (0–182 days from baseline); 6–12 months (183–365 days from baseline); 12–18 months (366–548 days from baseline); 18–24 months (549–731 days from baseline); 24–30 months (732–914 days from baseline); 30–36/42 months (915 or more days from baseline). In each case, the three estimates and intervals correspond to different starting EDSS score groups: left = L (starting EDSS score of 5.5 or less); centre = M (starting EDSS score of 6.0); right = H (starting EDSS score of 6.5 or higher).
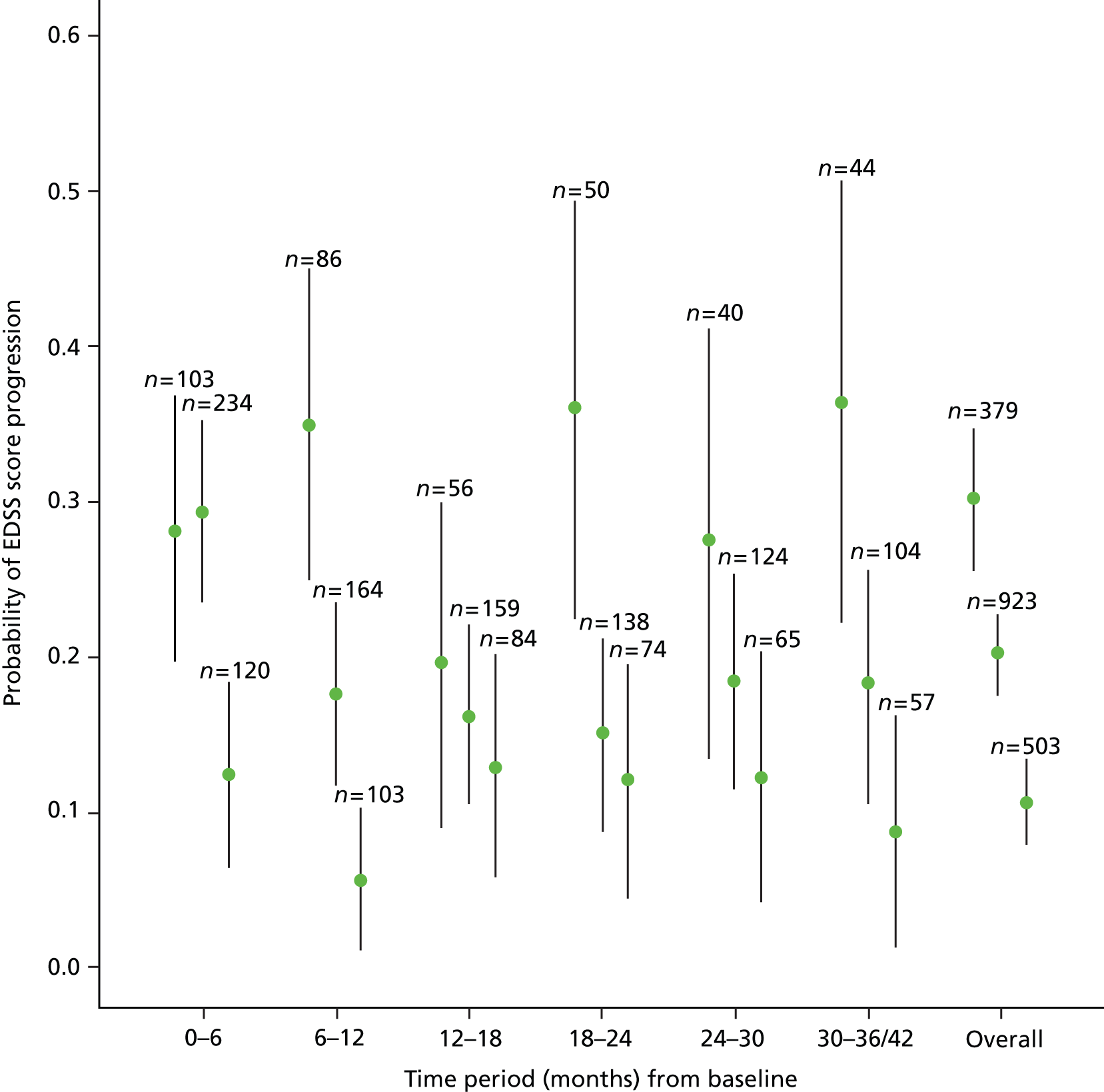
Conclusions
Post-hoc exploratory analyses showed some evidence of a potentially beneficial effect of active treatment in terms of time to first confirmed EDSS score progression and change in MSIS-29phys score among those patients with a baseline EDSS score of 4.0–5.5. However, this subgroup of participants consists of just 110 individuals and so findings should be interpreted with caution. This beneficial effect was not seen in the MRI outcome, PBVC.
Results from analysis of time to first EDSS score progression, change in MSIS-29phys score and PBVC based on an on-treatment data set showed no evidence of a treatment effect on these outcomes and supported the conclusions from the primary analyses.
Detailed inspection of transition between EDSS scores highlighted the relatively low probability of progression on the EDSS when starting from the more disabled end of this scale. It also suggested an increased probability of progression from an EDSS score of 5.5; the lowest score at which a 0.5-point increase is deemed a progression. There was also an indication of an increased probability of progression from a baseline EDSS score of 5.5 in the primary analysis of time to first confirmed EDSS score progression (see Figures 4 and 5) but, once again, these findings must be interpreted with caution because of the small numbers of patients in the individual groups defined by baseline EDSS score.
Chapter 5 Rasch measurement theory analysis of multiple sclerosis rating scale data
Introduction
This chapter concerns the application in the CUPID study of RMT analyses. RMT is a modern psychometric method for constructing and evaluating rating scales. It has a number of advantages for clinical trials over traditional methods of analysing rating scale data. These advantages include the ability to derive interval-level measurement estimates from necessarily ordinal rating scale scores and the ability to examine change legitimately at the individual person level. In addition, RMT enables a very sophisticated evaluation of rating scale performance. This chapter, which has five sections, capitalises on those advantages of RMT.
Section 1 gives a brief introduction to RMT.
Section 2 reports the RMT-based evaluation of the performance as measurement instruments of MS-specific PROs used in the CUPID study: MSIS-29v2, MSWS-12v2 and MSSS-88 scores. Specifically, for each subscale within each instrument (e.g. MSIS-29v2 has two subscales; MSSS-88 has eight subscales; MSWS-12v2 has one scale), three performance-related issues were examined: scale-to-sample targeting (relative range of person and item locations, shape of person distribution); aspects of scale’s item performance [response category working, mapped continuum mapped by items, item fit statistics, item bias, differential item functioning (DIF)]; and aspects of derived person measurements [person separation index (PSI), person fit residuals, extreme scores]. It was concluded that all 11 scales/subscales performed well. This enabled interval-level measurement estimates for individual patients, with individual person standard error (SE) estimates, to be derived and taken forward to sections 3 and 4. Naturally, there were some issues of scale performance that could have been better.
Section 3 reports and compares the changes associated with active and placebo for those people in each treatment group, who remained on trial medication and had paired measurements at the two appropriate time points. These analyses used the interval-level measurement estimates and individual person SE estimates derived from section 2. Specifically, two potential benefits of dronabinol were examined: a symptomatic benefit between baseline and visit 5, and a disease-modifying benefit between baseline and the end of the study. For each benefit analysis, relative changes in the active and placebo groups at the group and individual person level were examined. At the group level, for each scale/subscale, the statistical significance of change scores [analysis of variance (ANOVA) paired sample t-tests] and the clinical significance of change scores [Cohen’s effect size; standardised response means (SRMs)] was examined in paired samples. At the individual person level, the proportions of people in each treatment group who achieved five levels of change (significantly better, non-significantly better, no change, non-significantly worse, significantly worse) were examined. In essence, analyses showed no significant differences between people treated with dronabinol or placebo. A notable finding was limited progression in the placebo group implying less progression over the study period than might have been expected in a cohort of people with progressive MS.
Section 4 reports post-hoc exploratory analyses. These analyses capitalise on the finding of a suggestion of a treatment effect in people with less EDSS-measured disability at baseline. Here the analyses of section 3 were repeated in two subgroups of people: those with a baseline EDSS score of 4.0–5.5; and those with a baseline EDSS score of 6.0–6.5. These post-hoc exploratory analyses alluded to the possibility of a clinically significant treatment effect in people with lower disability at baseline. It was also notable that placebo-treated people with higher levels of disability at baseline had surprisingly little progression during the CUPID study. This implies a limited pathophysiological substrate for examining the hypothesis that any treatment may have a disease-modifying effect.
Section 5 reflects on the findings and the lessons learned from the RMT analyses of the CUPID data.
Section 1: rating scales, rating scale data analysis and the added value of Rasch measurement theory
This section aims to give a very brief account of how rating scales work as measurement instruments, the scientific methods that underpin them and the case for choosing RMT as the most appropriate method for analysing rating scales data in the CUPID study. Fuller and heavily referenced accounts are given elsewhere. 6,15–17
Rating scales attempt to measure variables that cannot be easily quantified using other methods. For example, the MSWS-12v2 attempts to measure the walking ability of people with MS. Rating scales achieve measurement through a set of questions, each of which has two or more response options. The response options are allotted sequential integer scores. People answer the questions, choosing the most relevant response option. Measurements are derived from these data. This method of measurement stems from a body of research beginning in the early 1900s in education and psychology and with it developed the methods of testing the quality of the measurement process known as reliability and validity testing. The methods for developing rating scales and examining their reliability and validity have become known as psychometric methods.
Any rating scale can be considered a hypothesis of how a variable might be measured. A number of reasons underpin this statement. First, the aspects of people who rating scales are seeking to measure are complex socially constructed variables; here, aspects of the impact of MS. As such, their measurement cannot be easy. Second, there is uncertainty concerning the definitions of these variables. This hampers the construction of rating scales and opens the door for a range of potential measurement methods. Third, socially constructed variables are measured through their manifestations. For example, the walking ability of an individual is estimated from their performance on a finite set of tasks. It follows then, that the extent to which performance on a set of tasks can be combined is an empirical question. Finally, there is uncertainty of the extent to which the numbers generated by any rating scales satisfy criteria as reliable and valid measurements. For these reasons, any scale is a hypothesis of how a complex clinical variable might be measured and as such this hypothesis requires careful testing. For example, the MSWS-12v2 should be viewed as a hypothesis of how walking ability might be measured that requires careful testing. RMT provides a criterion for part of that hypothesis test.
Most rating scales assign successive integer scores to two or more ordered-item response categories that imply increasing problems. For example, in the MSIS-29v2 the four response options are: not at all = 1; slightly = 2; moderately = 3; and extremely = 4. Then, scores for groups of items that form subscales (e.g. the 20 physical impact items of the MSIS-29v2) are generally summed to produce a subscale score, which is used as a numerical index for a person on the physical scale and likewise for the other scales.
The general goal of psychometric evaluations of rating scales is to test whether or not this process of item scoring and summation satisfies the criteria for deriving measurements. More specifically, the goal is to establish whether or not it is legitimate to sum the integer-scored responses of a group of items and to determine the extent to which these summed scores are free from random error (whether or not they are reliable) in accounting for differences among people and measuring the attributes they purport to measure (whether or not they are valid). There are three main paradigms for developing, analysing and modifying rating scales: classical test theory (CTT), item response theory and RMT.
An evaluation of rating scales is well suited to the RMT paradigm because the Rasch model, a mathematical equation (model), provides a hypothesis test. This is because it articulates, a priori, the requirements of rating scale data for rating scales to satisfy criteria as measurement instruments. The model was derived from theory and is independent of any data set. Therefore, discrepancies detected by the analysis, that is between the hypothesis (scale data) and the hypothesis test (Rasch model requirements), indicate anomalies in the hypothesis (scale) as a measurement instrument. In this way, a RMT analysis provides diagnostic information-informing measurement instrument development by exposing anomalies to be understood and improved empirically.
Rasch measurement theory analyses use a mathematical model to test scale performance and generate measurements of people. The Rasch model, a probabilistic measurement model, was developed to express the requirement of invariant comparisons. By this we mean that the performance of the measurement method (here, a rating scale’s items) should, within reason, be independent of the people it is tested on and the measurements of people should be independent of the measurement method (here, a set of scale items) used to measure them. The Rasch model does not arise from the need to model (explain or summarise) any particular data set, and is compatible with the requirements of measurement methods used in physics, called fundamental or additive conjoint measurement.
One important property of the Rasch model is that for items with ordered response categories, as they are in the MS-specific rating scales using in the CUPID study, the successive categories should be scored with successive integers. This is a consequence of the requirement of invariance and not an assumption of the Rasch model. The use of both successive integer scores for scoring successive categories within items, and then the total sum score across items to characterise a person, is also a feature of the theory that underpins traditional psychometric methods (called traditional true score theory). However, in traditional psychometric methods (CTT), these are essentially starting points of the theory and do not arise from a priori requirements as they do in RMT.
A Rasch analysis of data, therefore, examines the extent to which the observed data accord with the requirements of the model. Therefore, at a general level it assesses the degree to which the responses of the persons to the items can be summed to provide a single score for each person which summarises each person’s location (measurement) on a scale. In other words, it tests whether or not the scale is able to place people in order on a scale, such that those who are ‘worse’ (in this case, suffer more from the effects of fatigue) produce higher scores than those who are ‘better’ (suffer less fatigue). This test of accord between the observed data and the expectations of the model is generally referred to as a ‘test of fit’. Tests of fit can be constructed to assess various specific hypotheses, such as that of multidimensionality and of unexpected dependence between pairs of items within subscales.
The procedure for testing the fit of the responses to the Rasch model involves first estimating the parameters of the model, which include a location parameter for each person and location parameters of the thresholds of the items which define the categories of a rating scale. A threshold is the transition point between adjacent item response categories. Specifically, it is the point on the scale at which the probability of scoring in adjacent categories is 50%. For example, each MSIS-29v2 item has four response categories: 1, 2, 3 and 4. Therefore, each item has three thresholds, 1–2, 2–3 and 3–4, which mark the three points on the scale at which the probability is 50% of scoring 1 and 2, 2 and 3, and 3 and 4, respectively. These parameter estimates are effectively the relevant summaries of the data. Given these estimates and the model, evidence is found of the extent to which the actual responses of people to the scale’s items can be recovered. This evidence is examined to assess the fit of the responses of each item to the Rasch model.
There is no necessary and sufficient test of fit between the data and the model. Therefore, multiple pieces of evidence of fit are required. Each focuses on different but related aspects as to where the responses might diverge from the model’s expectations. Typically, when using RUMM2030 (Rasch Unidimensional Measurement Models, Perth, WA, Australia), three statistical tests are used: chi-squared, fit residual and the residual correlation.
The chi-squared test of fit operates at the level of class intervals formed on the basis of the total scores of persons and then provides an estimate of the magnitude of departure of the mean score for an item in each class interval from the mean value expected according to the model. This is the most general test of fit and provides a graphical counterpart [item characteristic curves (ICCs)], which is also examined.
The fit residual test of fit operates at the level of the response of each person to each item and provides evidence that an item discriminates either more or less than is expected. Although there are multiple reasons why an item might discriminate more or less than expected, a smaller discrimination than expected might imply that the item is assessing a somewhat different construct from that assessed by the majority of items, whereas one greater than expected might imply that it is part of a subset of items that are overly dependent and in some sense are redundant.
The residual correlations test of fit provides evidence regarding which subsets of items might either be assessing some aspect that is common but different from the majority of the items, or which items might be redundant in their assessment relative to the rest. Which of these hypotheses is believed to be the case depends on a qualitative understanding of the construct, the items and the response formats.
Evidence not directly involving statistical tests of fit is also relevant. First, an important aspect of validity is examining the evidence that the scoring of successive categories by successive integers is justified. If it is not justified, it implies that the endorsement of a higher score does not imply more of the construct being assessed (impact of MS) than a lower score. In principle, it reflects some operational or conceptual problem with the ordering of the categories, such that respondents might consistently interpret the response categories in a different way than intended. Second, for the same reasons that we examine the empirical ordering of the item categories, we also consider whether or not the item locations represent a conceptually meaningful order, such that they constitute a measurement continuum. In other words, responses to items in a scale should follow a consistent order that represents the level of ‘severity’ of the construct. Finally, reliability was quantified using the PSI, which is analogous to Cronbach’s alpha. This index can be interpreted in terms of the spread of the persons generated by the items, and is used in two ways. First, a low PSI means that the power of the test of fit to detect misfit between the responses and the model is weak. Second, if an item shows marginal fit and removing the item reduces the PSI, it suggests that the item is adding random error and may not be consistent with the majority of the items.
The information provided by a RMT analysis is both sophisticated and extensive. Information from multiple tests is integrated. These are considered simultaneously and interactively, rather than individually and sequentially. Test result interpretation requires professional judgement, rather than adherence to rigid criteria, because the information needs to be contextualised and most statistical tests are sample size dependent. Additionally, as the analyses compare observed rating scale data against a stringent mathematical model, anomalies are expected. To facilitate interpretation, analyses are grouped under three broad, clinically relevant, simple (but not simplistic) questions: (1) is the scale to sample targeting adequate for making judgements about the performance of the scale and the measurement of people?; (2) has a measurement ruler been constructed successfully (scale/item analysis)?; and (3) how have the people been measured by the ruler (person measurement)? Data analyses reported here used RUMM2030.
Section 2: within-study measurement performance of the Multiple Sclerosis Impact Scale-29, Multiple Sclerosis Walking Scale-12 and Multiple Sclerosis Spasticity Scale-88 using Rasch measurement theory
Background
Very few studies report a performance evaluation of the rating scales they use as clinical outcome assessments (COAs). This appears to be because there is a belief that once scales are ‘validated’ further evaluations are not necessary. This is somewhat misguided as there is no such thing as a ‘validated’ scale because rating scale performance is based on an evaluation of the observed data generated by their use. Theoretically, every time a scale is used its performance should be examined and the implications considered. Here, a RMT analysis of the data generated by three MS-specific PROs used in the CUPID study is reported: MSIS-29v2, MSWS-12v2 and MSSS-88.
Methods
The information provided by a RMT analysis is both sophisticated and extensive. Information from multiple tests is integrated. These are considered simultaneously and interactively, rather than individually and sequentially. Test result interpretation requires professional judgement, rather than adherence to rigid criteria, because the information needs to be contextualised and most statistical tests are sample size dependent. In addition, as the analyses compare observed rating scale data against a stringent mathematical model, anomalies are expected. To facilitate interpretation, analyses are grouped under three broad, clinically relevant, simple (but not simplistic) headings: scale-to-sample targeting, scale performance (item analysis) and person measurement (person analysis). Data analysis used RUMM2030 and SPSS (SPSS Inc., Chicago, IL, USA). Analyses included all data for the three scales generated during the CUPID study.
Scale-to-sample targeting
Scale-to-sample targeting analyses seek to determine if the match between the range of the construct measured by the scale and the range of the construct measured in the sample, is adequate enough to enable judgements about the performance of the scale and the measurement of people. There are no binary criteria to assess scale-to-sample targeting.
Scale-to-sample targeting concerns the match between the range of the target variable (e.g. walking ability) measured by the scale (e.g. MSWS-12v2) and the range of the target variable measured in the CUPID sample by the scale. A simple examination of histograms of two relative distributions – person locations from the sample and item location from the scale – provides a frame of reference for interpreting the other results, informs about the suitability of the sample for evaluating the scale and the suitability of the scale for measuring the sample. Not surprisingly, the better the targeting, the better the information. Scale-to-sample targeting was examined for MSIS-29v2, MSWS-12v2 and MSSS-88 across all time points.
Scale performance (item analysis)
A set of analyses were undertaken to determine if a measurement ruler had been constructed successfully. The MSWS-12v2 is used for illustrative purposes when needed, but the concepts apply to all of the scales examined (MSWS-29v2, MSWS-12v2 and MSSS-88).
Item response categories: to what extent do the item response categories work as intended?
Each scale item has multiple response categories labelled to imply an ordered continuum from less to more. This continuum is implied further by assigning sequential integer scores to the response categories. For example, consider item 6 of the MSWS-12v2 ‘limited your balance when standing and walking’: 1 = not at all, 2 = a little, 3 = moderately, 4 = quite a bit and 5 = extremely.
While this rank ordering is intuitively sound and clinically sensible at the individual item level, it must also work when an item is part of a set. By this we mean that the item’s response categories must have the same logical sequence when a person moves up and down the variable measured by the whole MSWS-12v2 set (here, walking ability). For example, as a person’s walking ability worsens, their scores on all the 12 items components should progress sequentially, that is 1, 2, 3, 4, 5 for items 4–12 and 1, 2, 3 for items 1–3.
Rasch measurement theory analyses test this requirement empirically by estimating the location, on the walking ability scale, of the points of transition (thresholds) between adjacent categories. A threshold is the location, on the cognitive performance variable, at which the probability of responding in adjacent categories is 50%. Thus, the MSWS-12v2 item 6, which has five response categories, has four transition points: 1–2, 2–3, 3–4 and 4–5. When the categories are working as intended the thresholds are ordered sequentially along the continuum: threshold 0–1 < threshold 1–2 < threshold 2–3 < threshold 3–4 < threshold 4–5.
When the thresholds are not correctly ordered (i.e. they are disordered), the implication is that the response categories for that component are not working as intended. Clinically, for the MSWS-12v2, this means that a higher score does not necessarily mean more walking disability. This has huge implications for clinical trials. Visually, thresholds are displayed as category probability curves which provide potential diagnostic information.
The extent to which the item response categories worked as intended was examined for each of the three scales. Data from all time points were pooled to maximise the power of the analysis to detect anomalies.
Mapped continuum: to what extent do the items map out a continuum on which people might be measured?
Before anything can be measured, the variable (or continuum) along which measurements are to be made needs to be marked out. Rating scales, such as the MSWS-12v2, use a set of items to define the variable they intend to measure. Therefore, for the MSWS-12v2 to define a walking ability variable along which measures can be interpreted, the items must be located at different points so that the direction and meaning of the variable can be identified. This question is addressed by examining the MSWS-12v2 threshold locations, their range, how they are spread, their proximity to each other and the precision of the estimates (SE). An item threshold location estimate is the point on the continuum at which the probability of scoring adjacent responses is 50%. For example, item 6 of the MSWS-12v2 has four thresholds, which mark the points on the walking ability continuum mapped out by the set of 12 MSWS-12v2 items at which there is a 50% probability of scoring: 1 and 2, 2 and 3, 3 and 4, and 4 and 5. The item location estimate is the mean of all the threshold location estimates for an item.
The extent to which the items of each instrument (scale and/or subscale) mapped out a continuum for measurement was examined. Data from all time points were pooled to maximise the power of the analysis to detect anomalies.
Item fit: to what extent do the items of a scale work together?
The components of a scale should work together as a conformable set both clinically and statistically. Otherwise it does not make sense conceptually, logically, clinically or empirically to sum component responses to get a total score and consider using that total score as a measurement of a person. If the components spread out and work together to define a single continuum then the responses to items should be predictable. Thus, examining the responses to each item for their consistency is important to determine if the components define a cohesive continuum. Specifically, the responses to components should be in general agreement with the ordering of persons implied by the majority of components. When this is not the case, the validity of the components and the higher-order construct they seek to measure is questioned.
These ideas are examined formally using indicators of goodness of fit of the observed rating scale data to the requirements of the Rasch mathematical model. No one indicator is sufficient to describe fit. We examined two statistical (fit residuals and chi-squared statistics) and one graphical (ICCs) indicator of fit.
Two item fit statistics (fit residuals and chi-squared values) and ICCs were examined for each instrument (scale and/or subscale). Data from all time points were pooled to maximise the power of the analysis to detect anomalies.
Item bias: do responses to one component bias responses to others?
The response to one scale item is expected, in general, to be related to another. For example, people who are less walking disabled are likely to perform better on all MSWS-12v2 items than people who are more walking disabled. However, the response to one scale item should not directly influence (or be dependent on) the response to another scale item. When this happens measurement estimates are artificially inflated or deflated (biased) and reliability is artificially elevated. Therefore, it is important to look actively for dependence among scale items. This is done by examining three indicators: correlations among the residuals, fit residuals and, when necessary, subtest analyses.
A residual is the difference between a person’s observed score on an item and their expected value for that item derived from the RMT analysis. Correlations among residuals, derived from the whole sample, reflect the degree of the inter-relationships between the residuals of the scale’s items. When measurement error is random, residuals are randomly distributed and correlations among residuals of components are low (rule of thumb range –0.30 to +0.30). However, when people’s responses to one component are biased by (dependent on) their responses to another component, the resulting residuals are not randomly distributed and higher correlations among residuals result (–0.30 < r > +0.30).
Residuals also provide a statistical indication of the observed data’s ’fit’ to the requirements of the Rasch measurement model. For each item, residuals are combined across individuals and standardised to produce the fit residual summary statistic. When there is dependency among components, a high score on one component results in an unexpectedly high score on another component. Likewise, a low score on one component results in an unexpectedly low score on another component. When viewed across the range of the measurement continuum and shown on the ICC, this pattern of dependency leads to the curve of observed scores being steeper than the curve of expected scores. This is reflected in the fit residual statistic as a high negative value. As a rule of thumb fit residual values are recommended to lie in the range –2.50 to +2.50 and values < –2.50 points to potential dependency. Naturally, as fit residuals are sample size dependent they need to be interpreted with this in mind.
In a subtest analysis, potentially dependent items are combined together to form a single ‘super item’ or subtest. This neutralises the dependency between the components. Dependency is determined by examining the impact of subtesting on the PSI, a reliability indicator. The magnitude of the drop in PSI, when the subtest analysis is compared with the ‘non-subtest’ analysis, indicates the extent to which the reliability of the latter is falsely elevated and the degree of dependency between components.
For each instrument (scale and/or subscale) the correlations among the residuals were examined to determine if there was evidence to suggest item score dependency. Fit residual values were also examined and where appropriate subtest analyses were undertaken. Data from all time points were pooled to maximise the power of the analysis to detect anomalies.
Item stability: is item performance stable across important groups?
When the ruler mapped out by a rating scale’s items is stable, the measurements generated by them can be used to make meaningful comparisons. Thus, we need the scale items to perform similarly across important groups that we might wish to study and compare (e.g. men and women, different age groups, different time points, different treatments). When item performance is not stable across important groups and display DIF, the measurement ruler is not stable across circumstances and measurement is affected to an unknown degree. The three MS PROs (MSIS-29v2, MSWS-12v2 and MSSS-88) were examined for DIF across randomisation groups (active and placebo) and time points.
Person measurement (person analysis)
When targeting is reasonable and scale performance is adequate it is possible to go on and examine the measurements derived for individuals. A range of analyses are possible and two specific questions were examined.
Person separation: to what extent are people in the sample separated by the items of the scale?
The aim of measurement is to locate people on a line (continuum) and detect differences between people and changes over time. It is, therefore, valuable to examine the extent to which a scale can detect differences between people in any study sample. In RMT analyses this is quantified as the PSI, computed as the ratio of error-corrected person variance to the total person variance. In addition, the distribution of person measurements and the percentage of extremes also provide information on the success of the scale to separate the sample. It is important to note that the PSI is sample specific.
The PSI of RMT is analogous to a Cronbach’s alpha coefficient in CTT, i.e. it is a reliability statistic that can range from 0 to 1, with greater values indicating greater separation of the people in this specific sample by this specific scale. Values do not generalise directly from sample to sample. Although CTT posits recommended values for alphas, this is somewhat misleading as it is a finding about the data. However, the PSI has implications for the power of the tests of fit. The greater the separation index the greater the power of the tests of fit to detect fit when it is present.
The PSI was examined for each instrument (scale and/or subscale) in the pooled data from all samples.
Person fit statistics: how valid are person measurements?
When a person is measured using a scale it is important to know that the scale has been used in the expected way. That is, consistent with the idea that the items map out a variable along which the items have a unique order. This can be determined by examining the extent to which the responses for an individual person are in general agreement with the ordering of items implied by the majority of persons. If not, the validity of that person’s measurement is questionable. This is determined by examining the person fit residual, which is analogous to the item fit residual.
Person fit residuals were examined for each instrument (scale and/or subscale) in the pooled data from all samples.
Results
Table 22 summarises the results from the RMT analysis of the MSIS-29v2, MSWS-12v2 and MSSS-88. These three COAs contain 11 different measurement scales/subscales that differ in their content and number of items (from 8 to 20).
| Scale | MSIS-29v2 | MSWS-12v2 | MSSS-88 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Symptoms (q1–12; 13–21; 22–35) | Physical (q36–46; 47–56; 57–67) | Psychological (q68–80; 81–88) | |||||||||
| Subscale | Physical | Psychological | Walking | Stiffness | Pain | Spasms | ADL | Walking | Body Mt | Feelings | Social function |
| Items | 20 | 9 | 12 | 12 | 9 | 14 | 11 | 10 | 11 | 13 | 8 |
| Samples | |||||||||||
| Entered into project | 3686 | 3667 | 3319 | 2343 | 2338 | 2341 | 2335 | 2223 | 2331 | 2328 | 2325 |
| Invalid | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Extremes | 16 | 83 | 429 | 94 | 155 | 391 | 165 | 269 | 160 | 204 | 255 |
| Item analysis | 3670 | 3584 | 2850 | 2249 | 2183 | 1950 | 2170 | 1954 | 2171 | 2124 | 2070 |
| Scale sample targeting | |||||||||||
| Item threshold range | –2.9/+2.6 | –2.5/+2.1 | –3.2/+4.1 | –3.9/+4.2 | –3.5/+2.6 | –3.0/+2.3 | –4.0/+4.0 | –3.4/+2.7 | –4.2/+3.3 | –4.4/+3.0 | –3.1/+2.7 |
| Person measure range | –3.0/+5.2 | –4.4/+4.2 | –5.4/+5.9 | –6.0/+5.8 | –5.1/+4.6 | –5.0/+4.7 | –5.8/+5.8 | –5.3/+4.8 | –5.8/+5.3 | –5.7/+5.0 | –4.7/+4.3 |
| Distribution shape | No skew | L skew+ | R skew+++ | No skew | L skew++ | L skew | L skew++ | R skew++ | L skew+ | R skew+ | L skew++ |
| Scale performance | |||||||||||
| Reversed thresholds | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Continuum | –2.9/+2.6 | –2.5/+2.1 | –3.2/+4.1 | –3.9/+4.2 | –3.5/+2.6 | –3.0/+2.3 | –4.0/+4.0 | –3.4/+2.7 | –4.2/+3.3 | –4.4/+3.0 | –3.1/+2.7 |
| Item fit residuals (total n) | –9.7/+ 19.2 | –12.7/+17.5 | –10.3/+8.5 | –8.1/+ 4.5 | –5.4/+5.1 | –6.7/+5.9 | –7.2/+9.7 | –10.9/+12.5 | –6.9/+6.8 | –9.7/+8.8 | –6.5/+3.3 |
| Sign chi-squared (n = 500) | Item 20 | Items 2, 5 | Item 3 | 0 | 0 | 0 | Item 2 | Items 4, 10 | 0 | 0 | 0 |
| Item bias (residual r > 0.30) | 5 (max. + 0.41) | 0 | 1 (–0.37) | 5 (max. +0.57) | 4 (max. –0.35) | 3 (max. +0.47) | 1 (max. –0.31) | 9 (max. –0.35) | 4 (max. +0.61) | 4 (max. +0.45) | 3 (max. –0.31) |
| DIF (treatment) | 1 (item 15) | 2 (items 26, 27) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (item 6) | 0 | 0 |
| Person measurement | |||||||||||
| PSI | 0.92 | 0.85 | 0.88 | 0.93 | 0.88 | 0.88 | 0.93 | 0.89 | 0.93 | 0.92 | 0.86 |
| Person fit range | –5.7/+5.1 | –5.9/+2.9 | –4.3/+3.9 | –5.2/+4.0 | –5.1/+3.2 | –6.3/+3.6 | –4.5/+3.7 | –5.3/+3.8 | –4.2/+3.6 | –6.1/+4.0 | –4.6/+3.6 |
| < –2.5 | 214 | 236 | 46 | 137 | 165 | 131 | 203 | 148 | 216 | 130 | 247 |
| > +2.5 | 92 | 21 | 6 | 32 | 11 | 39 | 25 | 23 | 32 | 38 | 17 |
| Extremes | 16 | 83 | 469 | 94 | 155 | 391 | 165 | 269 | 160 | 204 | 255 |
| Person measures | –3.0/+5.2 | –4.4/+4.2 | –5.4/+5.9 | –6.0/+5.8 | –5.1/+ 4.6 | –5.0/+4.7 | –5.8/+5.8 | –5.3/+4.8 | –5.8/+5.3 | –5.7/+5.0 | –4.7/+4.3 |
All available scale completions were included in the analysis. The total number of measurements entered into the RMT analyses varied across the scales (2223–3686). These represent the person’s x time points and are influenced by the number of people dropping out over time, the frequency with which the scale is administered and whether or not scale completion was appropriate. For example, people who were unable to walk did not complete the walking scales.
The number of extremes varied across scales from a low of 16 (4.3%, MSIS-29phys) to a high of 429 (12.9%, MSWS-12v2). However, the highest percentage of extremes was 16.7% (391/2341, MSSS-88 spasms subscale). Extreme scores are similar to floor and ceiling effects. An extreme is person scale completion in which either all items have the minimum possible score or all items have the maximum possible score. As such, extremes are dependent on the number of items answered. However, extremes differ from floor and ceiling effects which are the percentage of person scale completions in which the maximum possible and minimum possible scale scores are achieved. Extremes are important because people at the extremes may have true changes in the target variables underestimated or not detected by the scale.
Scale-to-sample targeting
Scale-to-sample targeting was generally adequate. For all scales the spread of person measures exceeded the spread of item thresholds. This is a common finding because thresholds are the points on the continuum at which a person is likely to have a 50% chance of responding in adjacent response categories. There are two implications. Firstly, the CUPID sample was adequate for examining the performance of all 11 scales/subscales of the three MS-specific COAs. Secondly, this raises the possibility that all 11 scales may have the potential to underestimate the true change in target variables.
The shapes of the person measurement distributions of the individual scales/subscale were examined. Although all person measure distributions were generally Gaussian in nature, most distributions (9 out of 11) were skewed to some extent. The two non-skewed distributions were for the MSIS-29phys (Figure 24) and MSSS-88 muscle stiffness subscales. Six distributions were left skewed (towards less problems), three mildly (MSIS-29v2 psychological; MSSS-88 spasms and body movement), three moderately [MSSS-88 pain/discomfort, activities of daily living (ADL) and social function]. These scales have the potential to underestimate some improvements if they occur.
FIGURE 24.
Person-item threshold distribution for MSIS-29phys (grouping set to interval length of 0.20 making 60 groups).
Three person measure distributions were right skewed (towards more problems): one mildly (MSSS-88 feelings), one moderately (MSSS-88 walking) and one notably (MSWS-12v2; Figure 25). Two distributions were not skewed (MSIS-29phys and MSSS-88 stiffness). These scales have the potential to underestimate some worsening if they occur.
FIGURE 25.
Person-item threshold distribution for MSWS-12v2 (grouping set to interval length of 0.20 making 60 groups).

Scale performance (item analysis)
Response categories: to what extent did the item response categories work as intended?
Only one of the 11 scales/subscales (the MSWS-12v2) had any reversed thresholds and only one of the 12 items was affected (running). A look at the response category endorsement frequencies indicate a bimodal distribution with very few people responding to the middle category (sometimes limited). This may imply that people with MS can either run or they cannot and that further gradations may be empirically supported. This required further evaluation.
Mapped continuum: to what extent did the items map out a continuum on which people might be measured?
Item threshold location estimates for all 11 scales/subscales spread over good ranges (minimum 4.6 logits to maximum 8.1 logits) indicating that each item set (i.e. scale/subscale) mapped out a continuum on which people might be measured.
Item fit: to what extent do the items of a scale work together?
Item fit statistics indicated misfit for all scales. However, misfit was expected as the CUPID sample was large (range for item analysis, from 1950 to 3670) and fit statistics are sample size dependent. Therefore, we examined the implications of smaller samples. At adjusted sample sizes of n = 500 there was very little item misfit. Also, the ICCs showed adequate coherence between observed scores and predicted values. These findings implied that each of the 11 scales/subscales was made up of a statistically cohesive item set.
Item scoring bias: did responses to one item bias responses to others?
Across the 11 scales/subscales examined, very few correlations among residuals exceeded the recommended range of ± 0.30. This implied little item scoring bias.
Item stability: is item performance stable across important groups?
Item stability was examined across randomisation groups. There was good item performance stability (10 class intervals) and very little evidence of statistically significant instability despite the large sample sizes. Visual examination of the DIF plots implied that the statistically significant DIF that was detected was unlikely to be clinically meaningful.
Person measurement (person analysis)
The results of both the targeting analyses and the scale performance analyses implied that an examination of person measures was necessary.
Person separation: to what extent were people separated by the scales?
Person separation index for the 11 scales/subscales ranged from 0.85 to 0.93. This indicated that all 11 scales/subscales separated the people well within the sample in terms of the target constructs being measured. This provides a good basis for measurement in clinical trials.
Person fit statistics: how valid are person measurements?
For each scale, a number of individual person measurements had associated fit residuals outside the rule of thumb range of –2.5 to +2.5. This number was typically < 10% except for the MSSS-88 feelings subscale (12.7% of the samples). This indicated that for these scale completions the patterns of responses across the items was out of keeping with expectation. Fit residuals cannot be computed for people at the extremes.
For all 11 scales/subscales there were typically many more out of range fit residuals that were < –2.5 than were > +2.5. Person fit residuals < –2.5 imply response patterns that are more consistent than expected and tend to occur for people who have given the same response to all, or most, of the items in the set. In contrast, person fit residuals exceeding +2.5 can indicate response patterns that are clinically inconsistent.
Summary
Rasch measurement theory analyses of CUPID data for MSIS-29v2, MSWS-12v2 and MSSS-88 implied that performance was generally good. This means that it is legitimate to use the interval estimates for people and their SEs in subsequent analyses. The targeting plots raise some questions about whether or not some of the scales might underestimate changes and differences occurring in the study.
Section 3: evaluation of treatment effect per protocol
Methods
In the CUPID study, 493 people with MS were randomised in a ratio of 2 : 1 to receive active treatment (n = 329) or placebo (n = 164). As has been outlined before, there were multiple visits. The change between baseline and visit 5 was chosen as an evaluation of symptomatic treatment effect of dronabinol and the change from baseline to end of study as an indicator of disease-modifying effect. This leads to two possible hypotheses. First, if dronabinol had a symptomatic benefit there would be an improvement relative to placebo in visit 5 PROs compared with baseline. Second, if dronabinol had a disease-modifying effect there would be less deterioration by the end of the study compared with placebo.
There were three main reasons why it was thought reasonable to consider the change between baseline and visit 5 as an evaluation of the symptomatic treatment effect of dronabinol, despite fully recognising that the choice of time points was arbitrary. First, visit 5, week 13 after randomisation to treatment group saw the end of the dose titration period. Second, from a clinic perspective, 3 months represents a reasonable recall period for an individual to judge a symptomatic benefit, whereas 6 months was more likely to be associated with recall bias. Third, 13 weeks on treatment was a similar time duration to the CAMS study,2 which studied the symptomatic effect of cannabinoids on MS-related spasticity.
The dropout rates have been discussed earlier. Therefore, an accurate and fair comparison would be of the paired measurements of the subgroups of people within the randomised groups who had paired measurements at the two relevant time points (baseline and visit 5; baseline and end).
The interval-level person measurement estimates (person locations) derived from the RMT analysis of rating scale data were analysed, rather than the raw scores generated by summing item scores. Analyses of change, for both the symptomatic effect and disease-modifying effect, were conducted at the group and individual person level.
Group-level analyses consisted of assessments of statistical and clinical significance of changes. Statistical significance of change was assessed using a one-way ANOVA on the change in person locations using the randomisation treatment as the grouping variable and paired samples t-tests for each treatment. Clinical significance of change was determined by computing two effect sizes from the person location estimates: Cohen’s effect size (mean change/SD baseline) and SRMs (mean change/SD change). Effect sizes were interpreted using Cohen’s widely used and cited criteria: 0.2 as the threshold for a small change; 0.5 the threshold for a moderate change; and 0.8 the threshold for a large change.
Rasch measurement theory also enables a legitimate assessment of change at the individual person level. This is because RMT provides an estimate of the SE associated with every person location estimate. This enables the significance of each person’s change in location to be examined as follows:
where
Significance of change values were categorised as:
Sig Change ≥ +1.96 = significant improvement;
0 < Sig Change ≤ +1.95 = non-significant improvement;
Sig Change = 0 = no change;
–1.95 ≤ Sig Change < 0 = non-significant worsening; and
Sig Change ≤ –1.96 = significant worsening.
The distribution of people across the significance of change categories for active and placebo can be determined and compared using a chi-squared test for contingency tables.
Results
Symptomatic effect (baseline to visit 5)
Tables 23–33 show the group and individual person change for each of the 11 scales/subscales. Changes in scores are computed as baseline minus visit 5 so that an improvement is a positive change and a worsening is a negative change. Results are very consistent across scales.
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 149) | Active (n = 266) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | 0.543 (1.155) | 0.539 (1.131) |
| On treatment (visit 5), mean (SD) | 0.451 (1.110) | 0.427 (1.180) |
| Change (baseline to visit 5), mean (SD) | 0.0916 (0.899) | 0.112 (0.881) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.052 (0.820) | |
| Paired samples t-test, t-statistic (p-value) | 1.243 (0.216) | 2.078 (0.039) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.079 | 0.099 |
| SRM (mean change/SD change) | 0.102 | 0.127 |
| Magnitude of change – individual person level | ||
| Better – significantly | 16.8% (n = 25) | 17.7% (n = 47) |
| Better – not significantly | 27.5% (n = 41) | 32.3% (n = 86) |
| No change | 10.7% (n = 16) | 7.5% (n = 20) |
| Worse – not significantly | 36.2% (n = 54) | 33.1% (n = 88) |
| Worse – significantly | 8.7% (n = 13) | 9.4% (n = 25) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 147) | Active (n = 265) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.442 (1.458) | –0.410 (1.400) |
| On treatment (visit 5), mean (SD) | –0.461 (1.477) | –0.567 (1.400) |
| Change (baseline to visit 5), mean (SD) | 0.0193 (1.1830) | 0.157 (1.155) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 1.319 (0.251) | |
| Paired samples t-test, t-statistic (p-value) | 0.198 (0.843) | 2.211 (0.028) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.013 | 0.112 |
| SRM (mean change/SD change) | 0.016 | 0.136 |
| Magnitude of change – individual person level | ||
| Better – significantly | 9.5% (n = 14) | 9.8% (n = 26) |
| Better – not significantly | 32.7% (n = 48) | 44.5% (n = 118) |
| No change | 10.9% (n = 16) | 6.4% (n = 17) |
| Worse – not significantly | 38.8% (n = 57) | 31.7% (n = 84) |
| Worse – significantly | 8.2% (n = 12) | 7.5% (n = 20) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 146) | Active (n = 257) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | 2.573 (1.792) | 2.626 (1.786) |
| On treatment (visit 5), mean (SD) | 2.455 (1.901) | 2.351 (2.078) |
| Change (baseline to visit 5), mean (SD) | 0.118 (1.459) | 0.275 (1.690) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.883 (0.348) | |
| Paired samples t-test, t-statistic (p-value) | 0.981 (0.328) | 2.611 (0.010) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.066 | 0.154 |
| SRM (mean change/SD change) | 0.081 | 0.163 |
| Magnitude of change – individual person level | ||
| Better – significantly | 13.0% (n = 19) | 21.8% (n = 56) |
| Better – not significantly | 39.7% (n = 58) | 29.2% (n = 75) |
| No change | 6.8% (n = 10) | 13.6% (n = 35) |
| Worse – not significantly | 30.8% (n = 45) | 25.7% (n = 66) |
| Worse – significantly | 9.6% (n = 14) | 9.7% (n = 25) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 148) | Active (n = 262) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.024 (2.115) | –0.125 (2.088) |
| On treatment (visit 5), mean (SD) | –0.384 (2.108) | –0.447 (2.220) |
| Change (baseline to visit 5), mean (SD) | 0.360 (1.811) | 0.321 (1.785) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.044 (0.835) | |
| Paired samples t-test, t-statistic (p-value) | 2.417 (0.170) | 2.913 (0.004) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.170 | 0.154 |
| SRM (mean change/SD change) | 0.199 | 0.180 |
| Magnitude of change – individual person level | ||
| Better – significantly | 22.3% (n = 33) | 20.2% (n = 53) |
| Better – not significantly | 27.0% (n = 40) | 34.0% (n = 89) |
| No change | 6.8% (n = 10) | 5.3% (n = 14) |
| Worse – not significantly | 31.8% (n = 47) | 26.0% (n = 68) |
| Worse – significantly | 12.2% (n = 18) | 14.5% (n = 38) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n=148) | Active (n=264) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.745 (1.934) | –0.906 (1.983) |
| On treatment (visit 5), mean (SD) | –1.137 (1.768) | –1.175 (1.899) |
| Change (baseline to visit 5), mean (SD) | 0.393 (1.528) | 0.269 (1.528) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.625 (0.430) | |
| Paired samples t-test, t-statistic (p-value) | 3.126 (0.002) | 2.855 (0.005) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.203 | 0.135 |
| SRM (mean change/SD change) | 0.257 | 0.176 |
| Magnitude of change – individual person level | ||
| Better – significantly | 18.2% (n = 27) | 15.5% (n = 41) |
| Better – not significantly | 29.7% (n = 44) | 36.7% (n = 97) |
| No change | 10.1% (n = 15) | 11.4% (n = 30) |
| Worse – not significantly | 32.4% (n = 48) | 26.9% (n = 71) |
| Worse – significantly | 9.5% (n = 14) | 9.5% (n = 25) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 148) | Active (n = 262) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –2.053 (2.064) | –2.081 (1.905) |
| On treatment (visit 5), mean (SD) | –2.419 (1.803) | –2.331 (1.865) |
| Change (baseline to visit 5), mean (SD) | 0.366 (1.713) | 0.250 (1.388) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.555 (0.457) | |
| Paired samples t-test, t-statistic (p-value) | 2.599 (0.010) | 2.916 (0.004) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.177 | 0.131 |
| SRM (mean change/SD change) | 0.214 | 0.180 |
| Magnitude of change – individual person level | ||
| Better – significantly | 15.5% (n = 23) | 17.2% (n = 45) |
| Better – not significantly | 30.4% (n = 45) | 30.5% (n = 80) |
| No change | 20.3% (n = 30) | 16.8% (n = 44) |
| Worse – not significantly | 24.3% (n = 36) | 25.6% (n = 67) |
| Worse – significantly | 9.5% (n = 14) | 9.9% (n = 26) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 148) | Active (n = 262) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –1.111 (2.142) | –1.275 (2.108) |
| On treatment (visit 5), mean (SD) | –1.185 (2.227) | –1.474 (2.113) |
| Change (baseline to visit 5), mean (SD) | 0.0739 (1.4350) | 0.199 (1.624) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.614 (0.434) | |
| Paired samples t-test, t-statistic (p-value) | 0.626 (0.532) | 1.988 (0.048) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.034 | 0.095 |
| SRM (mean change/SD change) | 0.051 | 0.123 |
| Magnitude of change – individual person level | ||
| Better – significantly | 14.9% (n = 22) | 15.6% (n = 41) |
| Better – not significantly | 33.8% (n = 50) | 36.3% (n = 95) |
| No change | 8.1% (n = 12) | 10.7% (n = 28) |
| Worse – not significantly | 32.4% (n = 48) | 24.0% (n = 63) |
| Worse – significantly | 10.8% (n = 16) | 13.4% (n = 35) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 145) | Active (n = 255) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | 1.385 (2.234) | 1.196 (2.048) |
| On treatment (visit 5), mean (SD) | 1.011 (2.098) | 0.910 (2.304) |
| Change (baseline to visit 5), mean (SD) | 0.374 (1.833) | 0.286 (1.905) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.200 (0.655) | |
| Paired samples t-test, t-statistic (p-value) | 2.454 (0.015) | 2.399 (0.017) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.167 | 0.140 |
| SRM (mean change/SD change) | 0.204 | 0.150 |
| Magnitude of change – individual person level | ||
| Better – significantly | 19.3% (n = 28) | 19.2% (n = 49) |
| Better – not significantly | 28.3% (n = 41) | 30.2% (n = 77) |
| No change | 20.0% (n = 29) | 14.1% (n = 36) |
| Worse – not significantly | 21.4% (n = 31) | 27.1% (n = 69) |
| Worse – significantly | 11.0% (n = 16) | 9.4% (n = 24) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 147) | Active (n = 263) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.143 (2.436) | –0.376 (2.262) |
| On treatment (visit 5), mean (SD) | –0.609 (2.260) | –0.629 (2.446) |
| Change (baseline to visit 5), mean (SD) | 0.466 (1.885) | 0.253 (2.045) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 1.079 (0.299) | |
| Paired samples t-test, t-statistic (p-value) | 2.995 (0.003) | 2.006 (0.046) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.191 | 0.112 |
| SRM (mean change/SD change) | 0.247 | 0.124 |
| Magnitude of change – individual person level | ||
| Better – significantly | 25.2% (n = 37) | 22.4% (n = 59) |
| Better – not significantly | 25.2% (n = 37) | 23.6% (n = 62) |
| No change | 8.8% (n = 13) | 9.5% (n = 25) |
| Worse – not significantly | 28.6% (n = 42) | 30.4% (n = 80) |
| Worse – significantly | 12.2% (n = 18) | 14.1% (n = 37) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 147) | Active (n = 261) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.997 (2.200) | –1.023 (2.154) |
| On treatment (visit 5), mean (SD) | –1.270 (2.262) | –1.257 (2.343) |
| Change (baseline to visit 5), mean (SD) | 0.273 (1.614) | 0.234 (1.856) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.045 (0.832) | |
| Paired samples t-test, t-statistic (p-value) | 2.050 (0.042) | 2.037 (0.043) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.124 | 0.109 |
| SRM (mean change/SD change) | 0.169 | 0.126 |
| Magnitude of change – individual person level | ||
| Better – significantly | 20.4% (n = 30) | 19.9% (n = 52) |
| Better – not significantly | 32.0% (n = 47) | 30.3% (n = 79) |
| No change | 14.3% (n = 21) | 9.2% (n = 24) |
| Worse – not significantly | 22.4% (n = 33) | 23.4% (n = 61) |
| Worse – significantly | 10.9% (n = 16) | 17.2% (n = 45) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 146) | Active (n = 260) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.792 (1.988) | –1.004 (1.865) |
| On treatment (visit 5), mean (SD) | –1.112 (1.993) | –1.061 (2.012) |
| Change (baseline to visit 5), mean (SD) | 0.320 (1.700) | 0.0568 (1.6470) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 2.328 (0.128) | |
| Paired samples t-test, t-statistic (p-value) | 2.272 (0.025) | 0.556 (0.579) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.161 | 0.030 |
| SRM (mean change/SD change) | 0.188 | 0.034 |
| Magnitude of change – individual person level | ||
| Better – significantly | 19.9% (n = 29) | 11.5% (n = 30) |
| Better – not significantly | 26.7% (n = 39) | 36.5% (n = 95) |
| No change | 15.8% (n = 23) | 11.9% (n = 31) |
| Worse – not significantly | 28.8% (n = 42) | 28.5% (n = 74) |
| Worse – significantly | 8.9% (n = 13) | 11.5% (n = 30) |
Group-level changes
For all 11 scales, both active and placebo groups had positive mean change scores indicating an average improvement in all 11 target variables. However, the mean change scores were small and none of the changes for each group was statistically significant. There were no significant differences between the change scores for active and placebo.
The corresponding effect sizes were small, typically but not always < 0.20 (none exceeding 0.25) and generally similar for both active and placebo.
There were no clear trends at group level to imply that dronabinol, or placebo, may be superior.
There was no treatment effect on aspects of psychosocial function (MSIS-29v2 psychological impact subscale; MSSS-88 feelings subscale; and MSSS-88 psychosocial function subscale).
Individual person level change
The tables show that the proportions of people undergoing different degrees of change were relatively similar between active and placebo. None of the chi-squared values were significant.
Disease-modifying effect
Tables 34–44 show the group and individual person change for each of the 11 scales/subscales between baseline and the end of the CUPID study. Change scores are computed as baseline minus end so that an improvement is a positive change and a worsening is a negative change. As CUPID was a study of people with progressive MS we would expect there to be deterioration over time, on average, particularly in terms of motor-related functions and symptoms. Psychological functions can be affected by other factors, so it is hard to anticipate what would happen to these variables over time. However, if dronabinol has a mood enhancing effect, or simply makes people ‘feel better’, we might expect to see this reflected in the results.
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 112) | Active (n = 173) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | 0.475 (1.202) | 0.472 (1.143) |
| On treatment (end), mean (SD) | 0.861 (1.509) | 0.642 (1.189) |
| Change (baseline to end), mean (SD) | –0.3864 (1.3080) | –0.170 (1.070) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 2.324 (0.129) | |
| Paired samples t-test, t-statistic (p-value) | –3.127 (0.002) | –2.091 (0.038) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | –0.321 | –0.149 |
| SRM (mean change/SD change) | –0.295 | –0.159 |
| Magnitude of change – individual person level | ||
| Better – significantly | 10.7% (n = 12) | 16.2% (n = 28) |
| Better – not significantly | 23.2% (n = 26) | 23.1% (n = 40) |
| No change | 2.7% (n = 3) | 3.5% (n = 6) |
| Worse – not significantly | 33.9% (n = 38) | 33.5% (n = 58) |
| Worse – significantly | 29.5% (n = 33) | 23.7% (n = 41) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 109) | Active (n = 171) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.539 (1.481) | –0.520 (1.397) |
| On treatment (end), mean (SD) | –0.488 (1.542) | –0.574 (1.383) |
| Change (baseline to end), mean (SD) | –0.050 (1.385) | 0.055 (1.375) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.388 (0.534) | |
| Paired samples t-test, t-statistic (p-value) | –0.379 (0.705) | 0.522 (0.602) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | –0.034 | 0.039 |
| SRM (mean change/SD change) | –0.036 | 0.040 |
| Magnitude of change – individual person level | ||
| Better – significantly | 9.2% (n = 10) | 12.9% (n = 22) |
| Better – not significantly | 33.9% (n = 37) | 34.5% (n = 59) |
| No change | 11.0% (n = 12) | 8.8% (n = 15) |
| Worse – not significantly | 35.8% (n = 39) | 31.6% (n = 54) |
| Worse – significantly | 10.1% (n = 11) | 12.3% (n = 21) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 92) | Active (n = 147) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | 2.391 (1.768) | 2.452 (1.709) |
| On treatment (end), mean (SD) | 2.891 (2.083) | 2.831 (1.931) |
| Change (baseline to end), mean (SD) | –0.500 (2.052) | –0.378 (1.792) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.235 (0.629) | |
| Paired samples t-test, t-statistic (p-value) | –2.338 (0.022) | –2.559 (0.012) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | –0.283 | –0.221 |
| SRM (mean change/SD change) | –0.244 | –0.211 |
| Magnitude of change – individual person level | ||
| Better – significantly | 12.0% (n = 11) | 10.9% (n = 16) |
| Better – not significantly | 25.0% (n = 23) | 27.9% (n = 41) |
| No change | 8.7% (n = 8) | 8.2% (n = 12) |
| Worse – not significantly | 30.4% (n = 28) | 35.4% (n = 52) |
| Worse – significantly | 23.9% (n = 22) | 17.7% (n = 26) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 104) | Active (n = 158) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.174 (2.025) | –0.276 (2.120) |
| On treatment (end), mean (SD) | 0.213 (2.179) | –0.154 (2.244) |
| Change (baseline to end), mean (SD) | –0.387 (2.254) | –0.122 (2.070) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.956 (0.329) | |
| Paired samples t-test, t-statistic (p-value) | –1.751 (0.083) | –0.742 (0.459) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | –0.191 | –0.058 |
| SRM (mean change/SD change) | –0.172 | –0.059 |
| Magnitude of change – individual person level | ||
| Better – significantly | 21.2% (n = 22) | 20.9% (n = 33) |
| Better – not significantly | 13.5% (n = 14) | 22.8% (n = 36) |
| No change | 2.9% (n = 3) | 6.3% (n = 10) |
| Worse – not significantly | 30.8% (n = 32) | 32.3% (n = 51) |
| Worse – significantly | 31.7% (n = 33) | 17.7% (n = 28) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 104) | Active (n = 156) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.745 (1.791) | –1.041 (1.992) |
| On treatment (end), mean (SD) | –0.805 (2.039) | –1.082 (1.925) |
| Change (baseline to end), mean (SD) | 0.061 (1.907) | 0.0418 (1.730) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.007 (0.935) | |
| Paired samples t-test, t-statistic (p-value) | 0.324 (0.747) | 0.302 (0.763) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.034 | 0.021 |
| SRM (mean change/SD change) | 0.032 | 0.024 |
| Magnitude of change – individual person level | ||
| Better – significantly | 16.3% (n = 17) | 14.7% (n = 23) |
| Better – not significantly | 30.8% (n = 32) | 26.3% (n = 41) |
| No change | 6.7% (n = 7) | 13.5% (n = 21) |
| Worse – not significantly | 26.9% (n = 28) | 31.4% (n = 49) |
| Worse – significantly | 19.2% (n = 20) | 14.1% (n = 22) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 103) | Active (n = 156) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –2.129 (2.086) | –2.108 (1.916) |
| On treatment (end), mean (SD) | –2.099 (1.894) | –2.105 (1.855) |
| Change (baseline to end), mean (SD) | –0.030 (2.071) | –0.004 (1.657) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.013 (0.911) | |
| Paired samples t-test, t-statistic (p-value) | –0.146 (0.884) | –0.028 (0.978) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | –0.014 | –0.002 |
| SRM (mean change/SD change) | –0.014 | –0.002 |
| Magnitude of change – individual person level | ||
| Better – significantly | 17.5% (n = 18) | 20.5% (n = 32) |
| Better – not significantly | 21.4% (n = 22) | 22.4% (n = 35) |
| No change | 11.7% (n = 12) | 9.6% (n = 15) |
| Worse – not significantly | 28.2% (n = 29) | 33.3% (n = 52) |
| Worse – significantly | 21.4% (n = 22) | 14.1% (n = 22) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 103) | Active (n = 154) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.976 (2.091) | –1.446 (2.046) |
| On treatment (end), mean (SD) | –0.417 (2.865) | –1.213 (2.443) |
| Change (baseline to end), mean (SD) | –0.559 (2.228) | –0.233 (2.220) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 1.329 (0.250) | |
| Paired samples t-test, t-statistic (p-value) | –2.547 (0.012) | –1.302 (0.195) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | –0.267 | –0.114 |
| SRM (mean change/SD change) | –0.251 | –0.105 |
| Magnitude of change – individual person level | ||
| Better – significantly | 15.5% (n = 16) | 18.2% (n = 28) |
| Better – not significantly | 15.5% (n = 16) | 25.3% (n = 39) |
| No change | 5.8% (n = 6) | 3.9% (n = 6) |
| Worse – not significantly | 35.0% (n = 36) | 26.6% (n = 41) |
| Worse – significantly | 28.2% (n = 29) | 26.0% (n = 40) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 98) | Active (n = 145) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | 1.352 (2.281) | 0.790 (1.998) |
| On treatment (end), mean (SD) | 1.335 (2.191) | 1.001 (2.064) |
| Change (baseline to end), mean (SD) | 0.017 (1.915) | –0.211 (2.220) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.685 (0.409) | |
| Paired samples t-test, t-statistic (p-value) | 0.085 (0.932) | –1.145 (0.254) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.007 | –0.106 |
| SRM (mean change/SD change) | 0.009 | –0.095 |
| Magnitude of change – individual person level | ||
| Better – significantly | 13.3% (n = 13) | 19.3% (n = 28) |
| Better – not significantly | 34.7% (n = 34) | 21.4% (n = 31) |
| No change | 9.2% (n = 9) | 7.6% (n = 11) |
| Worse – not significantly | 23.5% (n = 23) | 31.7% (n = 46) |
| Worse – significantly | 19.4% (n = 19) | 20.0% (n = 29) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 100) | Active (n = 157) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.058 (2.422) | –0.750 (2.190) |
| On treatment (end), mean (SD) | 0.063 (2.299) | –0.507 (2.378) |
| Change (baseline to end), mean (SD) | –0.121 (2.181) | –0.242 (2.097) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 0.197 (0.657) | |
| Paired samples t-test, t-statistic (p-value) | –0.557 (0.579) | –1.449 (0.149) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | –0.050 | –0.111 |
| SRM (mean change/SD change) | –0.056 | –0.116 |
| Magnitude of change – individual person level | ||
| Better – significantly | 19.0% (n = 19) | 17.8% (n = 28) |
| Better – not significantly | 26.0% (n = 26) | 25.5% (n = 40) |
| No change | 5.0% (n = 5) | 7.6% (n = 12) |
| Worse – not significantly | 25.0% (n = 25) | 28.0% (n = 44) |
| Worse – significantly | 25.0% (n = 25) | 21.0% (n = 33) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 102) | Active (n = 156) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.858 (2.106) | –1.182 (2.141) |
| On treatment (end), mean (SD) | –1.341 (2.215) | –1.349 (2.164) |
| Change (baseline to end), mean (SD) | 0.482 (2.002) | 0.167 (2.181) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 1.372 (0.243) | |
| Paired samples t-test, t-statistic (p-value) | 2.434 (0.017) | 0.959 (0.339) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.229 | 0.078 |
| SRM (mean change/SD change) | 0.241 | 0.077 |
| Magnitude of change – individual person level | ||
| Better – significantly | 25.5% (n = 26) | 25.6% (n = 40) |
| Better – not significantly | 37.3% (n = 38) | 26.3% (n = 41) |
| No change | 3.9% (n = 4) | 7.1% (n = 11) |
| Worse – not significantly | 18.6% (n = 19) | 19.2% (n = 30) |
| Worse – significantly | 14.7% (n = 15) | 21.8% (n = 34) |
| Analyses | Treatment group | |
|---|---|---|
| Placebo (n = 104) | Active (n = 155) | |
| Descriptive statistics | ||
| Pre treatment (baseline), mean (SD) | –0.783 (1.963) | –1.286 (1.816) |
| On treatment (end), mean (SD) | –1.228 (2.123) | –1.271 (1.995) |
| Change (baseline to end), mean (SD) | 0.445 (2.066) | –0.014 (1.953) |
| Magnitude of change – group level | ||
| One-way ANOVA, F-statistic (p-value) | 3.287 (0.071) | |
| Paired samples t-test, t-statistic (p-value) | 2.197 (0.030) | –0.091 (0.928) |
| Effect sizes | ||
| Cohen (mean change/SD pre treatment) | 0.227 | –0.008 |
| SRM (mean change/SD change) | 0.215 | –0.007 |
| Magnitude of change – individual person level | ||
| Better – significantly | 24.0% (n = 25) | 18.1% (n = 28) |
| Better – not significantly | 26.0% (n = 27) | 29.7% (n = 46) |
| No change | 15.4% (n = 16) | 6.5% (n = 10) |
| Worse – not significantly | 20.2% (n = 21) | 26.5% (n = 41) |
| Worse – significantly | 14.4% (n = 15) | 19.4% (n = 30) |
Group-level changes
For eight of the 11 scales/subscales there is no real difference between active- and placebo-treated people.
For three of 11 scales/subscales (MSIS-29phys; MSSS-88 muscle stiffness and ADL subscales), there was a suggestion that people treated with dronabinol have deteriorated less than people treated with placebo. These changes within the placebo group and within the active group, and between placebo and active, were not significant statistically.
Effect size calculations implied that the deterioration in placebo-treated people exceeded the threshold for small clinical worsening (< –0.20) for two subscales (MSIS-29phys and MSSS-88 ADL subscales) and is borderline small for the third (MSSS-88 muscle stiffness). The corresponding effect size calculations for dronabinol-treated people are approximately half the magnitude. This hinted at the possibility of a disease-modifying treatment effect.
There was no treatment effect on aspects of psychosocial function (MSIS-29v2 psychological impact subscale; MSSS-88 feelings subscale; or MSSS-88 psychosocial function subscale).
There are two notable findings. First, the mean changes over a considerable period of time are very small. This implies only clinically small progression during the study in a large cohort of people with a diagnosis of progressive MS. Second was the high dropout rate. The CUPID study recruited 493 people, randomising 329 to active and 164 to placebo. By the end of the study the maximum number of people completing a PRO was 173 in the active group, 53% of the original dronabinol-treated cohort and 112 in the placebo group, 68% of the original placebo-treated cohort.
Individual person-level change
The proportion of people in each of the five change groups is very similar for placebo and active although, as expected, this changes across scales. The similarity between placebo and active is notable even for the three scales/subscales where the group analyses hint at a disease-modifying treatment effect.
Perhaps the most striking findings from the individual person-level analysis are the proportions of people who appear to have improved by the end of the study. In the placebo group, this proportion ranged from 31% (MSSS-88 ADL subscale) to 63% (MSSS-88 feelings subscale). The proportion of people categorised as having a significant improvement ranged from 9% (MSIS-29v2 psychological impact scale) to 26% (MSSS-88 feeling subscale), with a mean of 17%. This warrants further examination.
For illustration, Figures 26 and 27 show the SRMs for the two MSIS-29v2 subscales and the individual person change for the MSIS-29phys.
FIGURE 26.
Standardised response means for the two MSIS-29v2 subscales, by treatment group. (a) MSIS-29v2 physical impact subscale; and (b) MSIS-29v2 psychological subscale.

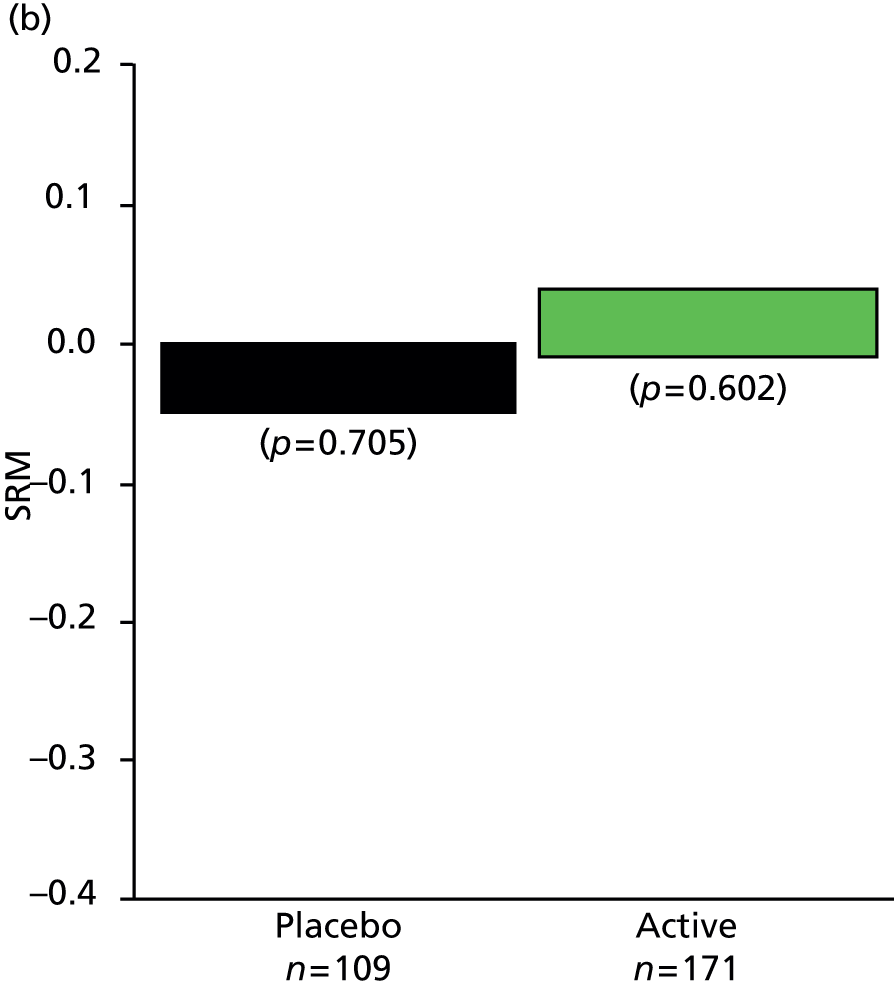
FIGURE 27.
Individual-person change for the MSIS-29phys, by treatment group.
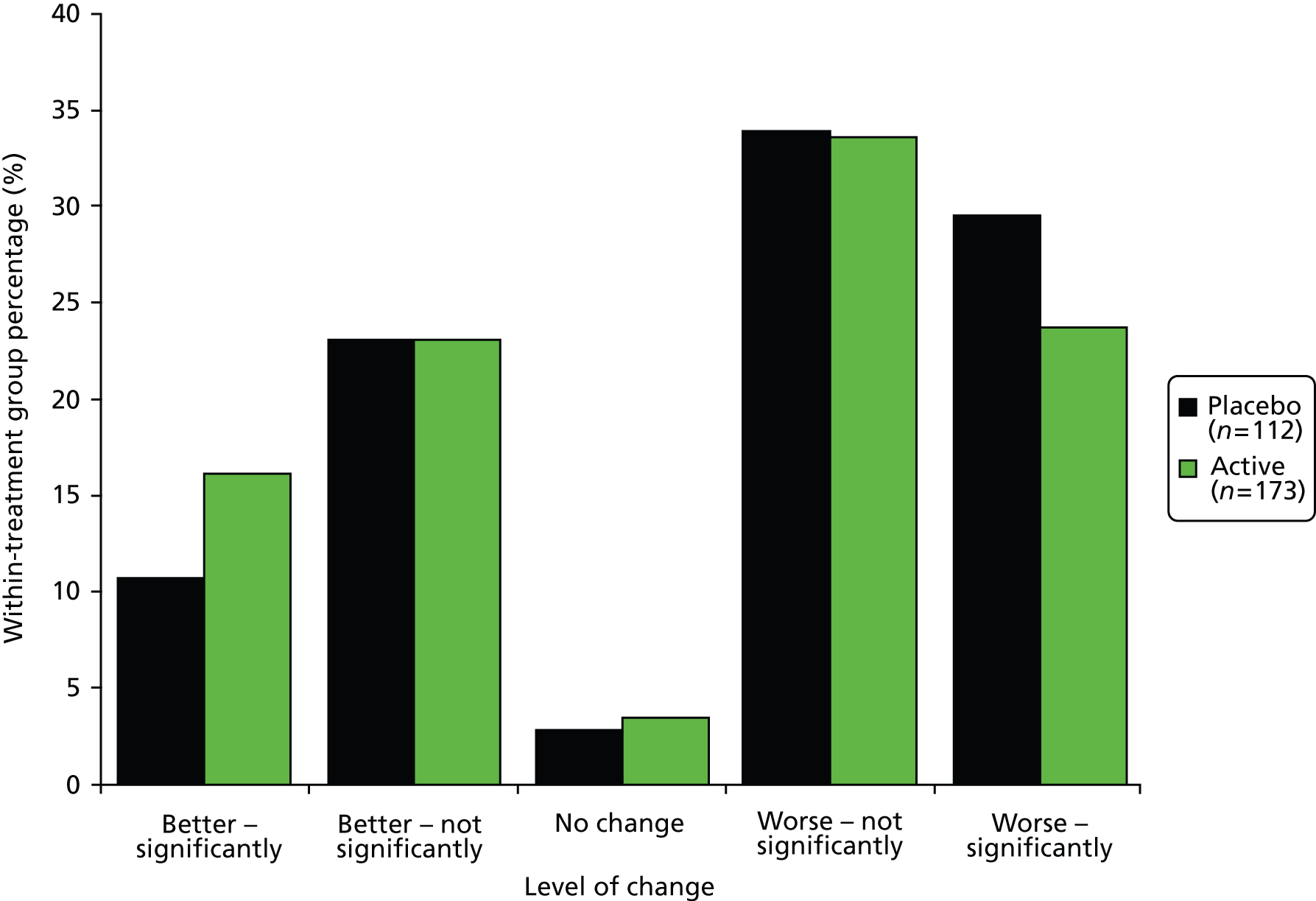
Summary
The results imply that dronabinol was not associated with either a symptomatic or a disease-modifying benefit. There was also no evidence that dronabinol improved people’s psychosocial functioning in either the short or the long term.
There were, however, some particularly notable findings. First, the degree of progression at a group and individual person level in the placebo group was smaller than might be expected for a cohort of people with a diagnosis of progressive MS. The largest deterioration in any one scale only just exceeded clinically a small change according to Cohen’s criteria for interpreting effect sizes. This makes it difficult to show a disease-modifying effect even if it were present. Second, and related to this, notable numbers of people appeared to have improved (up to 63% on one subscale), some significantly (up to 26% on one subscale), by the end of the study. Third, the dropout rate from both treatment groups, particularly the active group, was very high (nearly 50%).
Section 4: exploratory evaluation of treatment effect by baseline disability level
Analyses presented earlier in this report hinted towards a treatment effect in people with lower EDSS scores at baseline and limited progression in participants with high EDSS scores. For this reason we explored the changes in the MS-specific PROs of people who were less disabled (baseline EDSS score of 4.0–5.5) and those who were more disabled (baseline EDSS score of 6.0–6.5). These analyses were undertaken in the full knowledge of being exploratory post-hoc analyses in non-randomised groups. As such, any results should be interpreted with caution.
Methods
Patient-reported outcomes were compared in active and placebo for the two disability-defined subgroups. Specifically, these were examined for evidence of a symptomatic effect (change from visit 1 to visit 5) and a disease-modifying effect (change from visit 1 to end of study). The statistical significance of change scores in people with paired data using paired samples t-tests and clinical significance was determined using two effect size calculations: Cohen’s effect size (mean change/SD baseline) and the SRM (mean change/SD change). Effect sizes were interpreted using Cohen’s criteria (0.2 is the threshold for a clinically small change; 0.5 is the threshold for a clinically moderate change; and 0.8 is the threshold for a clinically large change).
Results
Symptomatic effect
Table 45 shows the results of analyses to determine evidence of a symptomatic effect. Sample sizes of the comparison groups varied notably, with the more disabled subgroup being far larger that the less disabled subgroup.
| Scale | Subscale | Treatment | Baseline EDSS subgroup | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EDSS score of 4.0–5.5 | EDSS score of 6.0–6.5 | |||||||||
| n | t-statistic (p-value) | Cohen’s ES | SRM | n | t-statistic (p-value) | Cohen’s ES | SRM | |||
| MSIS-29v2 | Physical | Active | 68 | 1.887 (0.063) | +0.181 | +0.229 | 222 | 1.215 (0.226) | +0.067 | +0.082 |
| Placebo | 33 | 1.650 (0.190) | +0.245 | +0.287 | 121 | 0.663 (0.509) | +0.046 | +0.060 | ||
| Psychological | Active | 68 | 2.365 (0.021) | +0.246 | +0.287 | 221 | 0.738 (0.462) | +0.041 | +0.050 | |
| Placebo | 33 | 1.597 (0.120) | +0.291 | +0.278 | 118 | –0.388 (0.700) | –0.028 | –0.036 | ||
| MSWS-12v2 | Active | 68 | 3.331 (0.001) | +0.357 | +0.404 | 212 | 1.249 (0.213) | +0.088 | +0.086 | |
| Placebo | 33 | 2.894 (0.007) | +0.310 | +0.504 | 11 | 0.0375 (0.708) | +0.031 | +0.035 | ||
| MSSS-88 | Stiffness | Active | 67 | 1.159 (0.251) | +0.114 | +0.142 | 217 | 2.309 (0.022) | +0.147 | +0.157 |
| Placebo | 33 | 1.930 (0.062) | +0.239 | +0.336 | 120 | 1.656 (0.100) | +0.135 | +0.151 | ||
| Pain and discomfort | Active | 54 | 0.955 (0.343) | +0.085 | +0.116 | 219 | 2.326 (0.021) | +0.129 | +0.157 | |
| Placebo | 33 | 2.345 (0.025) | +0.321 | +0.408 | 120 | 2.504 (0.014) | +0.176 | +0.229 | ||
| Spasms | Active | 67 | 1.602 (0.292) | +0.097 | +0.130 | 218 | 2.621 (0.009) | +0.135 | +0.178 | |
| Placebo | 33 | 0.213 (0.833) | +0.034 | +0.037 | 120 | 2.937 (0.004) | +0.212 | +0.268 | ||
| ADL | Active | 67 | 0.057 (0.955) | +0.004 | +0.007 | 218 | 1.543 (0.124) | +0.091 | +0.105 | |
| Placebo | 33 | 0.641 (0.526) | +0.066 | +0.112 | 120 | 0.084 (0.933) | +0.005 | +0.008 | ||
| Walking | Active | 67 | 0.6330 (0.0529) | +0.056 | +0.077 | 211 | 1.814 (0.071) | +0.126 | +0.125 | |
| Placebo | 33 | 2.391 (0.023) | +0.347 | +0.416 | 117 | 1.178 (0.241) | +0.087 | +0.109 | ||
| Body movements | Active | 67 | 0.394 (0.695) | +0.035 | +0.048 | 219 | 1.603 (0.110) | +0.110 | +0.108 | |
| Placebo | 33 | 2.044 (0.049) | +0.177 | +0.356 | 119 | 2.422 (0.017) | +0.186 | +0.222 | ||
| Feelings | Active | 67 | 1.757 (0.083) | +0.166 | +0.215 | 217 | 1.219 (0.224) | +0.071 | +0.083 | |
| Placebo | 32 | 1.604 (0.119) | +0.172 | +0.284 | 120 | 1.097 (0.275) | +0.078 | +0.100 | ||
| Social function | Active | 67 | 0.392 (0.696) | +0.045 | +0.048 | 216 | 0.510 (0.611) | +0.030 | +0.035 | |
| Placebo | 32 | 1.694 (0.100) | +0.250 | +0.300 | 119 | 1.751 (0.083) | +0.139 | +0.161 | ||
Results for all 11 scales/subscales implied an improvement at visit 5 relative to visit 1. No t-statistics were significant at the p < 0.001 level (no Bonferroni correction). Effect sizes did not suggest any patterns in the data in terms of the comparison between active treatment in the less and more disabled subgroups. Some scales had larger effect sizes in the less disabled subgroup, some had larger effect sizes in the more disabled subgroup, and some were equal.
The comparisons of active with placebo were similar for most scales. Five MSSS-88 subscales (stiffness, pain, walking, body movements and social function) recorded notably larger benefits in the placebo group than in the active group among the less disabled cohort.
The two effect sizes (Cohen’s effect size and SRM) produced different results, as we have noted in our previous work.
Disease-modifying effect
Table 46 shows the results of analyses to determine evidence of a disease-modifying effect. Again, sample sizes of the comparison groups varied notably, with the more disabled subgroup being far larger that the less disabled subgroup. The sample size for the less disabled people on placebo was particularly small (between 23 and 29).
| Scale | Subscale | Treatment | Baseline EDSS subgroup | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EDSS score of 4.0–5.5 | EDSS score of 6.0–6.5 | |||||||||
| n | t-statistic (p-value) | Cohen’s ES | SRM | n | t-statistic (p-value) | Cohen’s ES | SRM | |||
| MSIS-29v2 | Physical | Active | 61 | –1.191 (0.238) | –0.142 | –0.153 | 191 | –2.940 (0.004) | –0.202 | –0.213 |
| Placebo | 29 | –2.326 (0.027) | –0.431 | –0.432 | 111 | –2.770 (0.007) | –0.297 | –0.263 | ||
| Psychological | Active | 60 | 0.856 (0.395) | +0.095 | +0.111 | 189 | 0.390 (0.697) | +0.028 | +0.029 | |
| Placebo | 29 | 0.665 (0.512) | +0.187 | +0.123 | 108 | –0.187 (0.852) | –0.016 | –0.018 | ||
| MSWS-12v2 | Walking | Active | 54 | –1.622 (0.111) | –0.212 | –0.221 | 147 | –2.870 (0.005) | –0.274 | –0.237 |
| Placebo | 23 | –2.978 (0.007) | –0.880 | –0.621 | 88 | –1.683 (0.096) | –0.210 | –0.179 | ||
| MSSS-88 | Stiffness | Active | 54 | –1.884 (0.065) | –0.260 | –0.256 | 170 | –1.160 (0.248) | –0.086 | –0.089 |
| Placebo | 24 | –0.677 (0.505) | –0.123 | –0.138 | 102 | –1.640 (0.140) | –0.191 | –0.162 | ||
| Pain and discomfort | Active | 54 | –0.927 (0.358) | –0.108 | –0.126 | 169 | 0.780 (0.437) | +0.051 | +0.060 | |
| Placebo | 24 | –0.097 (0.923) | –0.019 | –0.020 | 102 | 0.401 (0.690) | +0.043 | +0.040 | ||
| Spasms | Active | 53 | –1.111(0.272) | –0.147 | –0.153 | 171 | 0.446 (0.656) | +0.031 | +0.034 | |
| Placebo | 24 | –0.786 (0.440) | –0.139 | –0.160 | 101 | 0.118 (0.906) | +0.012 | +0.012 | ||
| ADL | Active | 53 | –2.613 (0.012) | –0.372 | –0.359 | 169 | –2.500 (0.013) | –0.238 | –0.192 | |
| Placebo | 24 | –1.376 (0.182) | –0.254 | –0.281 | 101 | –2.981 (0.004) | –0.344 | –0.297 | ||
| Walking | Active | 53 | –1.917 (0.061) | –0.244 | –0.263 | 148 | –1.035 (0.302) | –0.093 | –0.085 | |
| Placebo | 24 | 0.002 (0.998) | 0.0004 | 0.0004 | 93 | 0.370 (0.712) | +0.033 | +0.038 | ||
| Body movements | Active | 54 | –2.565 (0.013) | –0.281 | –0.349 | 171 | –1.924 (0.056) | –0.147 | –0.152 | |
| Placebo | 24 | –1.752 (0.093) | –0.285 | –0.358 | 98 | –0.335 (0.738) | –0.032 | –0.034 | ||
| Feelings | Active | 54 | 1.100 (0.277) | +0.132 | +0.150 | 170 | 1.077 (0.283) | +0.084 | +0.083 | |
| Placebo | 23 | –0.200 (0.843) | –0.052 | –0.042 | 100 | 2.612 (0.010) | +0.253 | +0.261 | ||
| Social function | Active | 54 | 0.593 (0.555) | +0.084 | +0.081 | 169 | –0.292 (0.770) | –0.022 | –0.022 | |
| Placebo | 23 | –0.348 (0.731) | –0.089 | –0.073 | 102 | 2.473 (0.015) | +0.258 | +0.245 | ||
In the less disabled subsample, change scores were mostly negative, implying a worsening in these outcomes between the beginning and end of the study. Effect sizes for placebo and active were similar for eight scales. For two scales/subscales (MSWS-12v2; MSIS-29phys) there was notably less worsening in the active than the placebo group. In contrast, there was greater worsening in the active group for the MSSS-88 walking subscale.
In the more disabled group, effect sizes for both active and placebo were generally similar across the scales. It is notable that the group-based deterioration in physical function in the placebo group of the more disabled group was surprisingly small over the 3 years of the study, with the largest change being considered small by Cohen’s criteria.
Figures 28 and 29 show the SRM plots for the MSIS-29phys and MSWS-12v2, in the two subgroups defined by baseline EDSS score.
FIGURE 28.
Standardised response means for the MSIS-29phys, by treatment group and baseline EDSS score subgroup. (a) Baseline EDSS score of 4.0–5.5; and (b) baseline EDSS score of 6.0–6.5. Effect size difference (SRM) = 0.28; effect size difference (Cohen’s) = 0.29.

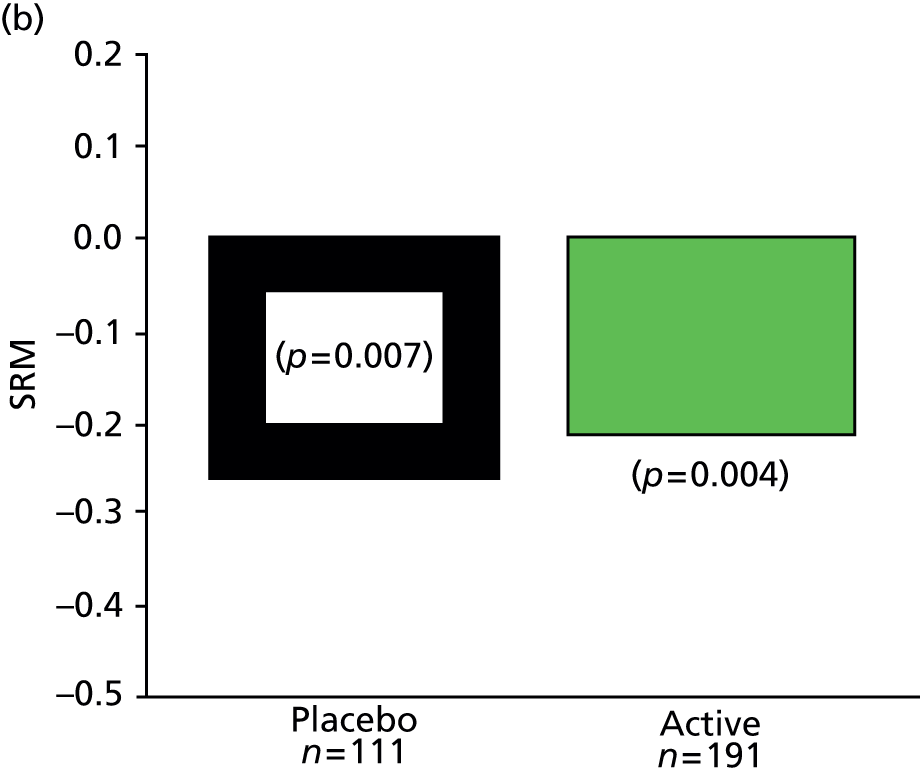
FIGURE 29.
Standardised response means for the MSWS-12v2, by treatment group and baseline EDSS score subgroup. (a) Baseline EDSS score of 4.0–5.5; and (b) baseline EDSS score of 6.0–6.5. Effect size difference (SRM) = 0.40; effect size difference (Cohen’s) = 0.67.


Finally, individual person changes were examined (computed as described previously), for the MSIS-29v2 and MSWS-12v2. These results are shown in Figures 30 and 31. For the MSIS-29phys, the plot shows less difference between the two disability groups than is implied by the effect sizes. For the MSWS-12v2, the differences between the two disability groups are notable.
FIGURE 30.
Individual-person change for the MSIS-29v2phys, in each treatment group, by disability subgroup. (a) Baseline EDSS score of 4.0–5.5; and (b) baseline EDSS score of 6.0–6.5.


FIGURE 31.
Individual-person change for the MSWS-12v2, in each treatment group, by disability subgroup. (a) Baseline EDSS score of 4.0–5.5; and (b) baseline EDSS score of 6.0–6.5.
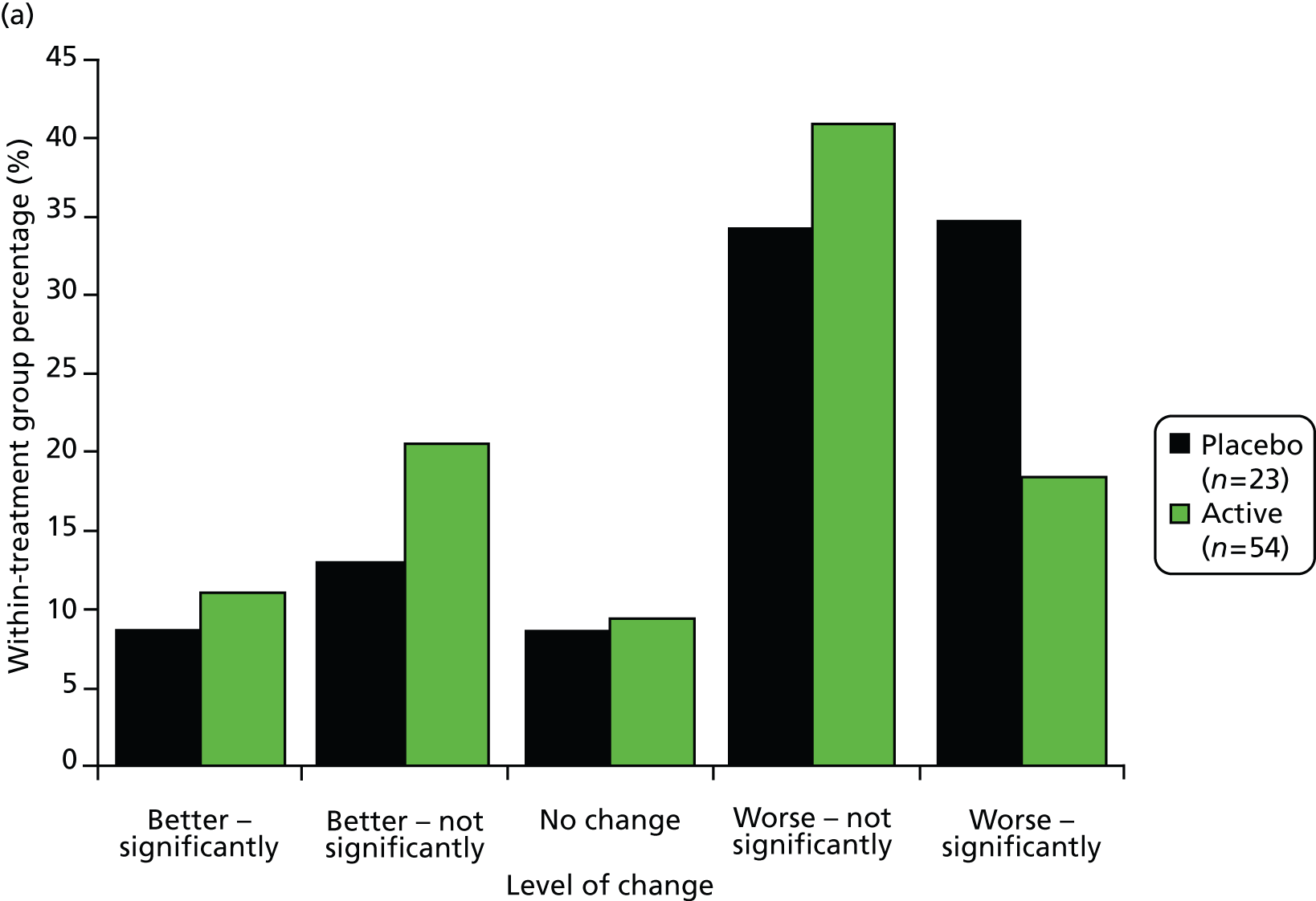

Summary
Exploratory post-hoc analyses in generally small, non-randomised samples of disability subgroups did not suggest a clear symptomatic or a disease-modifying treatment effect on the MS-specific PROs used in the CUPID study. There were some hints of a potential disease-modifying effect, with reduced progression measured by the MSIS-29phys and MSWS-12v2. Indeed, the effect size differences between active and placebo for these two scales/subscales was striking, regardless of calculation method. However, these effects were not supported by a benefit on MSSS-88 physical function subscales (ADL, walking, body movements). Indeed, the MSSS-88 walking subscale came to the opposite conclusion, favouring placebo. Perhaps the most notable finding from these analyses was the size of the worsening over time in the placebo group. In the more disabled subgroup, which was reasonably large (n = 88–111), the effect size-based worsening on the five self-report physical function scales/subscales ranged from –0.01 to –0.34, implying very little deterioration. Deterioration in the (very small) placebo-treated lower disability subgroup (n = 23–29) was larger, with effects sizes for four of the five scale/subscales ranging from clinically small to clinically large.
Section 5: reflections and lessons learned from Rasch measurement theory analysis of Cannabinoid Use in Progressive Inflammatory brain Disease data
This chapter has concerned the application of the modern, sophisticated psychometric method RMT to data generated by MS-specific PRO measures used in a large multicentre Phase III pivotal clinical trial. Historically, despite its availability since the 1960s, RMT has been used frugally before. There may be two main reasons for this: the inaccessibility of RMT to clinicians, and the lack of emphasis on COAs as primary and secondary end points. The first reason is changing as RMT is becoming more widely known, understood and used. The second reason is changing, as there is increasing recognition of the importance of measuring patient-focused outcomes. In particular, the US Food and Drug Administration (FDA) has emphasised the importance of patient-focused COA and the use of PROs. Indeed, recently, two important figures related to COAs in general – the road map to patient-focused outcome assessment (Figure 32) and the wheel and spokes diagram for the qualification of COAs (Figure 33) – have appeared on their website.
FIGURE 32.
Roadmap to patient-focused outcome measurement in clinical trials. US FDA. COA Roadmap Diagram. URL: www.fda.gov/downloads/drugs/developmentapprovalprocess/drugdevelopmenttoolsqualificationprogram/ucm370174.pdf (accessed 29 April 2014).

FIGURE 33.
Qualification of COAs. US FDA. Qualification of Clinical Outcome Assessments (COAs). URL: www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/UCM370175.pdf (accessed 29 April 2014).

The evaluation of scale performance was informative. Analyses show the scales performed adequately, although the suboptimal targeting for some scales/subscales means there is room for improvement. It is difficult to quantify the implication of limitations in scale performance. However, our findings mean that we can be relatively confident in the interpretation of subsequent analyses.
The main finding of the study was that Δ9-THC does not appear to have either a symptomatic or a disease-modifying effect as measured using these MS-specific PROs. There were some suggestions of improvement but no consistent findings. It seems that the chance of detecting a disease-modifying effect, if present, was slim as the progression in the sample, on average, was statistically and clinically small. Also, a reasonably large proportion of people appeared to improve their function over time, which is surprising for people diagnosed as having a chronic progressive disabling incurable neurological disease.
These findings raised questions of the completeness of our understanding of progressive MS and the need for more basic research to understand exactly what is progressing in progressive MS.
Chapter 6 Economic evaluation
Introduction
The aim of the economic evaluation was to compare the costs and consequences of cannabinoids (Δ9-THC) with those of usual care in progressive MS in the UK outpatient setting. The primary analytical perspective was the NHS and Personal and Social Services (PSS), with the patient perspective considered in secondary analyses. The primary health economic outcome was the quality-adjusted life-year (QALY) estimated using the EQ-5D18 and the primary economic end point was the 36-month follow-up. Costs and QALYs were discounted after the first year at the UK treasury rate of 3.5%.
Methods
Resource use
Resource use data were collected at participant level using a combination of patient self-report questionnaires and clinical records. Table 47 details the resource use data collected in the trial and the time points at which it was collected.
| Resource use item | Sources | Time period (figures in brackets indicate follow-up intervals) |
|---|---|---|
| Medication (see Appendix 8) | CRF | 36-month trial period |
| Other intervention costs (additional neurology consultations, management of AEs) | Expert opinion | 36-month trial period |
| CRF | ||
| SAEs | ||
| Hospital admissions | CRF | 36-month trial period |
| SAEs | ||
| Primary and acute care services (GP, community nurse, MS specialist nurse, physiotherapist, rehabilitation clinic visit, occupational therapist, speech therapist, neurologist, psychologist, chiropodist, optician, continence advisor, social worker) | Patient questionnaire | Baseline, 4.5 months (1, 2) and 6 months (3, 4, 5, 6) |
| Alternative practitioners (reflexologist, osteopath, homeopath, herbalist, masseuse, chiropractor, acupuncturist, hypnotist) | Patient questionnaire | Baseline, 4.5 months (1, 2) and 6 months (3, 4, 5, 6) |
| Informal care from friends/family | Patient questionnaire | Baseline, 4.5 months (1, 2) and 6 months (3, 4, 5, 6) |
| Formal personal care services | Patient questionnaire | Baseline, 4.5 months (1, 2) and 6 months (3, 4, 5, 6) |
| Home adaptations and equipment | Patient questionnaire | Baseline, 4.5 months (1, 2) and 6 months (3, 4, 5, 6) |
| Day care | Patient questionnaire | Baseline, 4.5 months (1, 2) and 6 months (3, 4, 5, 6) |
| Respite care | Patient questionnaire | Baseline, 4.5 months (1, 2) and 6 months (3, 4, 5, 6) |
| Treatment-related travel | Patient questionnaire | Baseline, 4.5 months (1, 2) and 6 months (3, 4, 5, 6) |
Intervention resource use, for delivery of Δ9-THC (other than medication which was based on patient-level data) was estimated based on the clinical protocol for delivery of the intervention.
Unit costs for resource use
Resource use data (i.e. number of visits, hours of care, respite episodes, hospital admissions) were combined with unit costs to estimate resource costs. Unit costs attached to health service resource use were based as far as possible on those faced by the NHS. Unit costs used were for 2010/11. Resource use was valued using available national unit costs for the NHS, usually the Personal and Social Services Research Unit (PSSRU)19 or NHS reference costs (www.gov.uk/government/publications/2010–11-reference-costs-publication) (Table 48). Future costs were discounted at the UK treasury rate of 3.5% per year, in line with National Institute for Health and Care Excellence (NICE) methods guidance. 20 The value of adaptations and equipment were annualised over a 10-year time period. This is a pragmatic and simplifying assumption, used to reflect the costs over the longer term use of these items of expenditure (e.g. bathroom adaptations, stair lifts), but this assumption does not impact on the results presented here. Hospitalisation data were limited to episodes reported as MS- or medication-related AEs or SAEs in trial CRFs, consistent with the trial protocol. Inpatient episodes were valued using relevant Healthcare Resource Group codes for MS patients (see Table 48). Informal (unpaid) care provided by friends and family was valued using the equivalent NHS/PSS hourly home care rate (as a shadow price) based on the replacement valuation method. Travel mileage was valued using national Automobile Association rates including standing and running costs. Sources and assumptions for all unit costs are reported.
| Resource use item | Unit | Unit cost (£) | Source | Notes |
|---|---|---|---|---|
| Cost to NHS/PSS | ||||
| GP (at practice) | Per visit | 36 | PSSRU19 | 11.7-minute consultation |
| Nurse (at practice) | Per visit | 15 | PSSRU19 | 15.5-minute surgery visit |
| Nurse home visit | Per visit | 30 | PSSRU19 | 25-minute home visit |
| MS nurse clinic visit | Per visit | 25 | PSSRU19 | Clinical nurse specialist |
| MS nurse home visit | Per visit | 38 | PSSRU19 | 25-minute home visit |
| Physiotherapist clinic visit | Per visit | 34 | PSSRU19 | 1-hour visit |
| Physiotherapist home visit | Per visit | 47 | PSSRU19 | Per visit |
| Occupational therapist clinic visit | Per visit | 34 | PSSRU19 | 1-hour visit |
| Rehabilitation clinic visit | Per visit | 34 | Assumed equivalent to occupational therapy clinic visit | |
| Speech therapist clinic visit | Per visit | 34 | PSSRU19 | 1-hour visit |
| Speech therapist home visit | Per visit | 47 | PSSRU19 | Per visit |
| Neurologist consultation | Per visit | 145 | NHS reference costs | Per consultation |
| Psychologist consultation | Per visit | 135 | PSSRU19 | 1-hour visit |
| Chiropodist clinic visit | Per visit | 31 | PSSRU19 | 1-hour visit |
| Chiropodist home visit | Per visit | 47 | Assumed equivalent to physiotherapist home visit | |
| Optician visit | Per visit | 20 | Assumed equivalent to private visit | |
| Continence advisor clinic visit | Per visit | 25 | PSSRU19 | Clinical nurse specialist |
| Continence advisor home visit | Per visit | 38 | PSSRU19 | 25-minute visit |
| Social worker home visit | Per visit | 212 | PSSRU19 | 1-hour visit |
| Acupuncturist clinic visit | Per visit | 34 | Assumed equivalent to physiotherapist clinic visit | |
| Personal care | Per hour | 18 | PSSRU19 | 1-hour visit |
| Day care | Per session | 36 | PSSRU19 | 3-hour session |
| Respite care | Per session | 1005 | PSSRU19 | 1-week stay |
| Elective inpatient HRG for MS | Per admission | 1511 | NHS reference costs | Average length of stay 2.66 days |
| Non-elective inpatient HRG for MS | Per admission | 2263 | NHS reference costs | Average length of stay 4.97 days |
| Non-elective inpatient HRG for MS short stay | Per admission | 501 | NHS reference costs | Overnight admission for drug complications |
| MS-related outpatient procedures (urinary tract infections, catheters, Botox to bladder) | Per procedure | 128–206 | NHS reference costs | |
| Private patient costs | ||||
| Physiotherapist clinic visit | Per visit | 34 | Assumed equivalent to NHS cost | |
| Chiropodist clinic visit | Per visit | 31 | Assumed equivalent to NHS cost | |
| Alternative practitioners clinic visits | Per visit | 34 | Assumed equivalent to a physiotherapist clinic visit | |
| Home care | Per hour | 18 | Assumed equivalent to NHS hourly rate for home care workers | |
| Travel costs | Per mile | 0.6862 | Automobile Association | Automobile running and standing costs |
Outcomes
The primary health economic outcome is the QALY calculated using the EuroQol EQ-5D. QALYs are a generic measure of health outcome which simultaneously capture changes in health-related quality of life (HRQoL) and survival combined into a single measure of treatment effect. Patient HRQoL was assessed by responses to the EQ-5D, a generic measure of health status with five domains (mobility, self-care, usual activity, pain/discomfort and anxiety/depression). Health states described by the EQ-5D have been valued on a scale of 0 (dead) to 1 (full health), with some states at worse than dead, based on the preferences of a community sample of people in the UK for time spent in each health state. 18 The EQ-5D is the measure preferred by NICE in health technology assessments. 20 Patient-level QALYs are calculated by applying an area under the curve method,21 which assumes linear change between discrete follow-up points in time. As with future costs, future QALYs after the first year are discounted using the UK treasury rate of 3.5% per annum.
Statistical methods
Descriptive analyses are undertaken and the means and distributions of resource use and costs, by type, and QALYs at each time point are presented, using complete case data, with no discounting applied. Discounted aggregated (category subgroup) costs are then presented with means, measures of variability and 95% CIs at each time point. Between-group comparisons are presented using multivariable regression based on a generalised linear model (GLM) used to estimate the treatment effect on costs and outcomes controlling for baseline costs and six other baseline covariates specified a priori in the trial SAP (study site, sex, age, weight, MS type and EDSS score). Because cost data are skewed, a GLM with an identity link and gamma distribution was used to estimate the regression coefficients. 22 The goodness of fit of the selected link and family function was confirmed using the modified Park test. Statistical analyses were performed on Stata version 12.0 (StataCorp LP, College Station, TX, USA). Primary analyses of between-group differences are based on data with missing values replaced using multiple imputation (see Methods, below) and where future costs and QALYs are discounted.
Post-hoc subgroup analysis
Consistent with the analyses of effectiveness data (Chapter 3), post-hoc economic analyses were undertaken among those patients with a baseline EDSS score of 4.0–5.5. Accordingly, the relationship between costs and outcomes with treatment allocation and covariates pre-specified in the trial SAP were examined in a regression analysis on the subgroup of patients with a baseline EDSS score of 4.0–5.5.
Missing data
As a consequence of participants not returning or submitting postal and internet questionnaires or dropping out of the study, there was a large number of missing data. Additionally, levels of missing cost and QALY data were different because of differences in the timing of questionnaires. The number of missing questionnaires in each treatment group for resource use and the EQ-5D are shown in Tables 49 and 50. Half of the cost data are missing, although most of this (74% or 179/241 missing questionnaires) was intermittent missing data (i.e. these patients missed one or more questionnaires, but provided subsequent data and did not drop out of the study entirely). This suggests that these data were missing at random. There were no between-group differences in missing cost (χ2 = 0.122; p = 0.73) or EQ-5D data (χ2 = 1.805; p = 0.18).
| Time point | Active n (%) | Placebo n (%) |
|---|---|---|
| N | 329 | 164 |
| Baseline | 5 (2) | 1 (1) |
| 4.5-month follow-up | 71 (22) | 28 (17) |
| 9-month follow-up | 73 (22) | 33 (20) |
| 15-month follow-up | 84 (26) | 38 (23) |
| 21-month follow-up | 95 (29) | 48 (29) |
| 27-month follow-up | 109 (33) | 44 (27) |
| 33-month follow-up | 107 (33) | 46 (28) |
| Completed all questionnaires | 159 (48) | 82 (50) |
| Time point | Active n (%) | Placebo n (%) |
|---|---|---|
| N | 329 | 164 |
| Baseline | 2 (1) | 0 (0) |
| 3-month follow-up | 38 (12) | 9 (5) |
| 6-month follow-up | 40 (12) | 16 (10) |
| 12-month follow-up | 44 (13) | 17 (10) |
| 18-month follow-up | 58 (18) | 23 (14) |
| 24-month follow-up | 62 (19) | 24 (15) |
| 30-month follow-up | 68 (21) | 30 (18) |
| 36-month follow-up | 79 (24) | 25 (15) |
| Completed all questionnaires | 217 (66) | 118 (72) |
Missing cost and QALY data were associated with study site (p = 0.004 and p < 0.001, respectively), age (p = 0.05 and p = 0.003, respectively) (younger patients more likely to have missing data); and, for QALY outcomes, hospitalisation costs (p = 0.02) (patients with missing data had significantly higher hospitalisation costs). No other baseline or cost variables had a significant effect on missing cost or QALY data.
Imputation of missing data
Data analysis confined to complete cases would ignore data from most patients departing from an ITT analysis. Missing data are handled using the method of multiple imputation using chained equations (ICE)23 on costs and EQ-5D scores. This method imputes values based on the available data. Missing data were not imputed for patients who died or did not have baseline cost or utility data. The sets of predictor variables were based on the dependent variable at all other time points and on the six baseline covariates selected a priori in the multivariable regression. The incomplete response variables were aggregated health-care costs and social care costs. The data set was imputed 50 times and the ICE program uses the 50 data sets simultaneously for statistical analysis, thereby accounting for both within- and between-data set variability.
Results
Intervention costs (delivery of Δ9-tetrahydrocannabinol)
Intervention costs reflect the costs of resources required to deliver a new intervention (Δ9-THC) over and above usual care. This required an understanding of the resources typically involved in the usual care of progressive MS patients and the additional resources expected to be required to deliver the CUPID intervention in current UK outpatient settings. This information was obtained from the clinical protocol and advice provided from the clinical teams delivering the intervention. Frequency of appointments with consultant neurologists in this patient group is usually one per year, more if patients are experiencing problems, for example with walking or bladder control. The additional resources that would be needed to add the CUPID intervention (Δ9-THC) to current UK practice comprise drug acquisition, an additional hospital consultation to monitor induction and management of AEs, as well as additional patient costs to collect monthly prescriptions.
Table 51 presents the mean cost of the 3-year CUPID Δ9-THC intervention, estimated at £27,433 per patient enrolled in the active treatment group. Medication use was based on observed patient-level data. Dose was estimated using the median dose achieved by patients in the active treatment group who were compliant for the duration of the trial (n = 178). This was two capsules twice per day. The cost of the medication was based on a quote of £561.50 from a pharmaceutical importer (Pharmarama) provided on 6 March 2012 for a pack of 60 2.5-mg capsules of Marinol® dispensed against a private prescription (excluding any VAT charge). This unit cost was established with agreement from the trial team for use as the base case cost for intervention medication, in the absence of any other published informative unit cost (at that time). On this basis the price per capsule was £9.36. Noting that 3.5-mg capsules were used in the CUPID study, the daily cost associated with the median dose in the CUPID study was £37.43 or, alternatively, £13,663.17 per year for fully compliant patients. As a long-term treatment, it is important to factor in compliance otherwise the cost of the treatment will be overestimated. Thus, the median dose was multiplied by the number of days patients were medication compliant (i.e. days between induction date and date of medication discontinuation). On this basis the average cost per treated patient per year was £10,780. Pharmacy and drug wastage costs are assumed to be included in these estimates.
| Cost category | Cost | Source | Level cost incurred | Total per compliant patient first year | Mean cost, all treated patients first year | Mean discounted cost, all treated patients 3 years |
|---|---|---|---|---|---|---|
| Marinol® (3.5-mg capsule) | £9.36 | Quote for 2.5-mg capsule (Pharmarama/distributor price) | Median daily dose (four capsules) | £13,663 | £10,780 | £27,303 |
| Additional Induction consultation | £140 | PSSRU hospital consultant | One visit | £140 | £140 | £140 |
| Managing AEs | £501 | NHS reference costs (per admission) | Five overnight inpatient admissions | N/A | £8 | Included in hospital cost data |
| Total | £13,803 | £10,928 | £27,443 |
Health and social care resource use and costs, NHS/personal and social services perspective
Table 52 summarises the key NHS-/PSS-funded health and social care cost data collected in the trial. Detailed resource use and cost data measured for the 6-month period preceding baseline and 4.5, 9, 15, 21, 27 and 33 months thereafter are presented in Appendix 8, Tables 67 and 68. These summary results are based on the non-imputed and undiscounted data set. It can be seen that the resource use cost data for all categories in both groups are positively skewed (i.e. the mean is greater than the median). Mean costs for the treatment group (Δ9-THC) were almost £5900 over 36 months, compared with a mean estimate in placebo participants of £8849; however, median costs were similar, highlighting the non-normal distribution of data. To highlight the reason for differences in mean total cost estimates, the mean total costs were higher in the placebo group due to an outlying value in NHS/PSS homecare (where we see a mean cost of £5641 compared with £1871; see Figure 35), otherwise, costs were generally higher in the active group in most resource categories. CIs indicate wide variation in the data and that there were no statistically significant differences between the treatment groups.
| Item | Active (n = 159) | Placebo (n = 82) | ||||||
|---|---|---|---|---|---|---|---|---|
| Mean (£) | SD (£) | Median (£) (range) | 95% CI (£) | Mean (£) | SD (£) | Median (£) (range) | 95% CI (£) | |
| GP (at practice or clinic) | 386.04 | 491.91 | 288.00 (0.00–5220.00) | 309.14 to 462.93 | 324.00 | 303.10 | 252.00 (0.00–1980.00) | 257.40 to 390.60 |
| Practice or clinic nurse (at GP practice) | 68.58 | 165.76 | 0.00 (0.00–1095.00) | 42.62 to 94.55 | 45.37 | 112.22 | 15.00 (0.00–735.00) | 20.71 to 70.02 |
| MS nurse | 82.97 | 85.75 | 63.00 (0.00–354.00) | 69.54 to 96.40 | 72.74 | 69.50 | 50.00 (0.00–325.00) | 57.47 to 88.01 |
| Physiotherapist | 368.16 | 492.90 | 188.00 (0.00–2346.00) | 290.95 to 445.36 | 300.00 | 397.08 | 172.50 (0.00–1972.00) | 212.75 to 387.25 |
| Rehabilitation clinic visit | 31.22 | 84.24 | 0.00 (0.00–612.00) | 18.02 to 44.41 | 22.80 | 55.02 | 0.00 (0.00–374.00) | 10.71 to 34.89 |
| Occupational therapist | 85.53 | 142.10 | 34.00 (0.00–1054.00) | 63.28 to 107.98 | 77.12 | 134.21 | 34.00 (0.00–986.00) | 47.63 to 106.61 |
| Speech therapist | 18.71 | 67.50 | 0.00 (0.00–544.00) | 8.14 to 29.28 | 13.51 | 48.22 | 0.00 (0.00–269.00) | 2.92 to 24.11 |
| Neurologist | 477.86 | 582.64 | 290.00 (0.00–3480.00) | 386.60 to 569.12 | 479.21 | 468.99 | 290.00 (0.00–2175.00) | 376.16 to 582.25 |
| Psychologist | 60.28 | 230.67 | 0.00 (0.00–2025.00) | 24.15 to 96.41 | 52.68 | 162.60 | 0.00 (0.00–1080.00) | 16.95 to 88.41 |
| Chiropodist | 41.10 | 130.67 | 0.00 (0.00–992.00) | 20.63 to 61.57 | 33.27 | 76.83 | 0.00 (0.00–372.00) | 16.39 to 50.15 |
| Optician | 20.13 | 27.33 | 20.00 (0.00–140.00) | 15.84 to 24.41 | 18.29 | 26.33 | 0.00 (0.00–100.00) | 12.51 to 24.08 |
| Continence advisor | 50.15 | 93.94 | 0.00 (0.00–760.00) | 35.44 to 64.68 | 39.07 | 64.26 | 0.00 (0.00–342.00) | 24.55 to 53.19 |
| Social worker | 210.67 | 986.47 | 0.00 (0.00–11,872.00) | 56.15 to 365.18 | 155.12 | 372.52 | 0.00 (0.00–2120.00) | 73.27 to 236.97 |
| Acupuncture | 8.77 | 44.15 | 0.00 (0.00–442.00) | 1.85 to 15.68 | 21.56 | 132.44 | 0.00 (0.00–1156.00) | 0.00 to 50.66 |
| Home care | 1871.71 | 9,721.10 | 0.00 (0.00–83,444.40 | 349.04 to 3394.37 | 5641.11 | 25,545.98 | 0.00 (0.00–211,770.00) | 28.05 to 11,254.18 |
| Day care | 69.06 | 596.22 | 0.00 (0.00–7272.00) | 0.00 to 162.44 | 73.76 | 341.43 | 0.00 (0.00–2196.00) | 0.00 to 148.78 |
| Respite care | 303.40 | 1590.07 | 0.00 (0.00–16,080.00) | 54.33 to 552.46 | 147.07 | 526.66 | 0.00 (0.00–3015.00) | 31.35 to 262.79 |
| Hospital episodes | 592.81 | 1427.75 | 0.00 (0.00–10,057.00) | 437.96 to 747.66 | 422.65 | 1140.30 | 0.00 (0.00–7060.00) | 246.83 to 598.48 |
| Adaptations/equipment | 149.73 | 397.22 | 3.00 (0.00–2765.75) | 87.51 to 211.95 | 194.87 | 541.35 | 16.50 (0.00–4206.13) | 75.92 to 313.82 |
| Concomitant medications | 978.71 | 1174.36 | 593.58 (0.00–5995.76) | 794.76 to 1162.65 | 848.98 | 958.94 | 523.14 (0.00–4843.92) | 638.28 to 1059.68 |
| Total undiscounted NHS costs | 5897.89 | 12,477.72 | 2799.40 (67.71–110,327.72) | 3943.45 to 7852.34 | 8949.96 | 26,621.87 | 2491.96 (413.00–222,161.54) | 3100.49 to 14,799.42 |
NHS-/personal and social services-funded other health-care costs
The proportion of costs attributable to each type of NHS-/PSS-funded primary or acute care service provider in each treatment group is illustrated in Figure 34. Resource use was higher in the active treatment group for almost all categories, based on the complete-case data set. Patients attended an average of three or more consultations with neurologists during the trial accounting for 25–29% of total NHS health-care costs. GP visits accounted for approximately 20% of NHS primary/acute sector health-care costs in both groups. Physiotherapy was the third largest cost category, accounting for almost 20% of NHS primary/acute sector health-care costs.
FIGURE 34.
Proportion (%) of costs attributable to each type of NHS-/PSS-funded primary or acute care service provider in each treatment group. The estimated cost (percentage of total within-treatment group cost) is given for each type of service provider. CN, practice or clinic nurse (at GP practice); MSN, MS nurse; OT, occupational therapist; physio, physiotherapist; rehab, rehabilitation clinic visit.

NHS-/personal and social services-funded social care costs
Social care costs are included in Figure 35. Most variations between treatment groups were due to outliers (i.e. individual patients reporting very high use of home care, day care or respite care). This can particularly skew results where unit costs are high, such as in respite care, or where the use of resources is high, such as in home care use in some instances. One participant in the placebo group reported over 12,000 hours of home care, at an estimated cost in excess of £200,000 compared with a mean cost of home care in other placebo participants of £5641 (see Table 52).
FIGURE 35.
Proportion (%) of costs attributable to each type of NHS-/PSS-funded social care provider in each treatment group. The estimated cost (percentage of total within-treatment group cost) is given for each type of care provider. Note: percentages may not sum to 100 due to rounding.

NHS-/personal and social services-funded multiple sclerosis-related hospital episodes
Unplanned admissions accounted for approximately 70% of hospital costs in both treatment groups (Figure 36). These mainly involved urinary tract infections (30%), relapses (18%) and MS-related fractures and falls (14%). The average length of stay for an unplanned admission was 10.7 days. Each hospital-based rehabilitation episode averaged 7 days. Planned MS-related admission involved mostly treatment with medications such as Botox (63%), MS reviews (21%) or insertion/removal of catheters (16%). Planned admission lasted, on average, 3.4 days. Most day cases involved insertion/removal of catheters (52%) or administration of medications such as Botox or intravenous steroids (26%). Admissions for suspected side effects were mainly for overnight observation for mood disturbances (average length of stay, 1.33 days). Most were in the active group (five out of six cases).
FIGURE 36.
Proportion (%) of costs attributable to each type of MS-related hospital episode in each treatment group. The estimated cost (percentage of total within-treatment group cost) is given for each type of episode. Note: percentages may not sum to 100 due to rounding.

Total costs by treatment group and follow-up (complete cases and imputed data)
Estimated total discounted NHS/PSS other health and social care cost data are presented in Appendix 8, Tables 69 and 70, to illustrate the evolution of costs within each treatment group over each study follow-up. Overlapping 95% CIs at every time point suggest no statistically significant between-treatment-group differences in costs.
Comparison of NHS/personal and social services resource use costs by treatment group
To assess differences in NHS/PSS resource use costs between treatment groups, regression analyses were applied to consider the relationship between total NHS costs and treatment allocation, adjusting for covariates pre-specified in the trial SAP (Table 53). As the cost data were skewed, a GLM with an identity link and gamma distribution was used to estimate the regression coefficients. 22 Total costs for health and social care were £165.22 higher in the placebo group, although this was not significant after adjusting for other factors (p = 0.75). Only baseline EDSS score and baseline costs had a statistically significant effect on total other costs in the study.
| Variable | Coefficient | SE | p-value | Direction |
|---|---|---|---|---|
| Treatment group | –165.22 | 524.09 | 0.753 | Placebo > active |
| Study site | –0.293 | 2.462 | 0.905 | |
| Sex | –266.49 | 579.36 | 0.646 | Men > women |
| Age | –41.93 | 35.88 | 0.243 | Younger > older |
| Weight | –1.96 | 19.91 | 0.921 | |
| MS type | –174.70 | 532.19 | 0.743 | PPMS > SPMS |
| Baseline EDSS score | 847.81 | 293.98 | 0.004 | Higher > lower |
| Baseline cost | 4.21 | 0.81 | < 0.001 | Higher > lower |
| Constant | 803.21 | 3232.16 | 0.804 |
Confidence intervals, SEs and associated p-values for each of the main components of total aggregated discounted costs were also estimated using a GLM including covariates for treatment group, study site, sex, age, weight, MS type, baseline EDSS score and baseline costs (Table 54). Once the costs of the CUPID intervention are included (mainly medication costs), the additional (adjusted) cost per treated patient over 3 years was £27,794.31.
| Resource use item | Active, mean (SE); n | Placebo, mean (SE); n | Adjusted mean difference, mean (SE) | 95% CI | p-value |
|---|---|---|---|---|---|
| Total other costs | 6111.58 (896.77); n = 325 | 7217.84 (1520.78); n = 163 | –165.22 (524.09) | –1193.50 to 863.07 | 0.753 |
| Intervention costs | 27,442.30 (857.37); n = 329 | 0 | 27,442.30 | 25,755.66 to 29,128.95 | – |
| Total costs | 33,553.88 (1837.75); n = 325 | 7217.84 (1520.78); n = 163 | 27,529.14 (3363.06) | 20,957.66 to 34,120.61 | < 0.001 |
Post-hoc subgroup analysis of total NHS costs by treatment group
From the main analysis of time to EDSS score progression, there was a suggestion of a differential effect of treatment between those patients with a baseline EDSS score of 4.0–5.5 and those with a baseline EDSS score of 6.0–6.5 (see Chapter 3). Accordingly, the relationship between total NHS costs with treatment allocation and covariates pre-specified in the trial SAP were examined in a regression based on the subgroup of patients with a baseline EDSS score of 4.0–5.5 (Tables 55 and 56). Intervention costs were higher for treated patients in this subgroup, suggesting enhanced medication compliance. Including intervention costs (mainly the cost of medication), the additional adjusted cost per treated patient over 3 years was £30,130.37. Study site also emerged as significantly related to overall NHS costs, possibly as a result of poorer treatment retention in some sites.
| Variable | Coefficient | SE | p-value | Direction |
|---|---|---|---|---|
| Treatment group | 30,130.37 | 2767.07 | < 0.001 | Active > placebo |
| Study site | –39.57 | 12.52 | 0.002 | Later < earlier |
| Sex | –3082.87 | 2877.98 | 0.284 | Women > men |
| Age | –152.78 | 177.88 | 0.390 | Older < younger |
| Weight | 123.50 | 93.07 | 0.185 | Heavier > lighter |
| MS type | –1180.88 | 2464.73 | 0.632 | SPMS > PPMS |
| Baseline EDSS score | 1820.88 | 2187.51 | 0.405 | Higher > lower |
| Baseline cost | –1.49 | 3.77 | 0.693 | Lower > higher |
| Constant | 54,009.09 | 15,629.37 | 0.001 |
| Resource use item | Active, mean (SE); n | Placebo, mean (SE); n | Adjusted mean difference, mean (SE) | 95% CI | p-value |
|---|---|---|---|---|---|
| Total other costs | 3304.69 (678.21); n = 75 | 2391.40 (369.29); n = 34 | 436.67 (428.55) | –404.28 to 1275.62 | 0.309 |
| Intervention costs | 29,948.03 (1697.11); n = 75 | 0 | 29,948.03 | 26,567.20 to 33,328.86 | – |
| Total costs | 33,252.72 (1792.98); n = 75 | 2391.40 (369.29); n = 34 | 30,130.37 (2767.07) | 35,554.43 to 24,706.31 | < 0.001 |
Private-/patient-funded other health and social care resource use
Table 57 summarises the key private-/patient-funded costs for other health and social care, based on data collected in the trial. Detailed private/patient resource use and cost data measured for the 6-month period preceding baseline and 4.5, 9, 15, 21, 27 and 33 months thereafter are presented in Appendix 8, Tables 71 and 72. These summary results are based on complete case data and are not discounted. It can be seen that the resource cost data for all categories in both groups are positively skewed (i.e. the mean is greater than the median). The table also shows that for all observed resource data there were no statistically significant differences between the treatment groups although, unlike the NHS-/PSS-funded resource use, mean total costs were higher in the active group. In the informal/home care costs there was an outlier, in the active treatment group, reporting in excess of £1.3M of estimated informal home care use, over the follow-up time frame (see Table 57). This highlights the wide variation in the burden on informal care. Although these extreme estimates are rare, such impacts are likely to fall on some providers. The estimated costs for home care (informal care), estimated using a shadow price for unpaid care, represents almost 95% of the total estimated private and patient costs (see further detail below).
| Item | Treatment group | |||||||
|---|---|---|---|---|---|---|---|---|
| Active (n = 159) | Placebo (n = 82) | |||||||
| Mean | SD | Median (range) | 95% CI | Mean | SD | Median (range) | 95% CI | |
| Physiotherapist | 227.74 | 717.45 | 0.00 (0.00–4386.00) | 115.36 to 340.11 | 288.17 | 690.34 | 0.00 (0.00–3026.00) | 136.49 to 439.85 |
| Chiropodist | 82.80 | 233.78 | 0.00 (0.00–1269.00) | 46.19 to 119.42 | 98.37 | 219.45 | 0.00 (0.00–893.00) | 50.15 to 146.58 |
| Optician | 20.75 | 26.99 | 20.00 (0.00–140.00) | 16.53 to 24.98 | 21.46 | 25.10 | 20.00 (0.00–100.00) | 15.95 to 26.98 |
| Acupuncture | 69.28 | 368.18 | 0.00 (0.00–3740.00) | 11.61 to 126.95 | 64.27 | 261.79 | 0.00 (0.00–1530.00) | 6.75 to 121.79 |
| Alternative practitioners | 309.85 | 1029.58 | 0.00 (0.00–9350.00) | 148.58 to 471.12 | 461.07 | 977.42 | 0.00 (0.00–5236.00) | 246.31 to 675.83 |
| Travel | 159.06 | 193.20 | 89.89 (0.00–1169.89) | 128.80 to 189.32 | 161.01 | 178.51 | 106.02 (0.00–891.11) | 121.79 to 200.24 |
| Home care | 1642.42 | 3390.93 | 0.00 (0.00–19,656.00) | 1111.28 to 2173.55 | 2637.35 | 7253.95 | 0.00 (0.00–49842.00) | 1043.4 to 4231.22 |
| Informal care (unpaid) | 56,697.24 | 119,349.22 | 35,568.00 (0.00–1,359,072.00) | 38,002.98 to 75,391.51 | 52,794.85 | 50,782.44 | 36,621.00 (0.00–201,708.00) | 41,636.73 to 63,952.98 |
| Day care | 27.85 | 137.71 | 0.00 (0.00–972.00) | 6.28 to 49.42 | No private day care episodes reported | |||
| Respite care | 6.32 | 79.70 | 0.00 (0.00–1005.00) | 0.00 to 18.80 | 49.02 | 269.04 | 0.00 (0.00–2010.00) | 0.00 to 108.14 |
| Adaptations/equipment | 707.97 | 1261.98 | 171.50 (0.00–7123.75) | 510.30 to 905.64 | 610.57 | 1007.70 | 166.25 (0.00–5397.50) | 389.15 to 831.99 |
| Total undiscounted private costs | 59,951.29 | 119,255.98 | 37,574.57 (36.48–1,362,452.54) | 41,271.63 to 78,630.95 | 57,186.15 | 51,332.35 | 37,388.13 (0.00–202,322.57) | 45,907.20 to 68,465.11 |
Private-/patient-funded other health-care costs
The proportion of costs attributable to each type of private-/patient-funded primary or acute care service provider in each treatment group is illustrated in Figure 37. The relative importance of each cost component was the same in both treatment groups. Resource use was higher in the placebo group for all major categories, based on the complete case data set.
FIGURE 37.
Proportion (%) of costs attributable to each type of private-/patient-funded primary or acute care service provider in each treatment group. The estimated cost (percentage of total within-treatment group cost for primary or acute care services) is given for each type of service provider. Physio, physiotherapist.

Private-/patient-funded social care costs
Privately provided social care costs are included in Figure 38. Note that access to unpaid informal care and adaptations/equipment supplied by the NHS/PSS may be influenced by other factors including living arrangements and income.
FIGURE 38.
Proportion (%) of costs attributable to each type of private-/patient-funded social care provider in each treatment group. The estimated cost (percentage of total within-treatment group cost) is given for each type of care provider. Note: percentages may not sum to 100 due to rounding.

Aggregated resource use costs by treatment group and follow-up (complete cases and imputed data)
Aggregated and discounted private/patient other health and social care cost data are presented in Appendix 8, Table 73 and illustrate the evolution of costs within each treatment group over each study follow-up. There were no statistically significant differences between groups at any time point.
Comparison of private/patient resource use costs by treatment group
Regression analyses were used to assess the relationship between total other private costs and treatment allocation, adjusting for covariates pre-specified in the trial SAP (Table 58). As the cost data were skewed, a GLM regression model with an identity link and gamma distribution was used to estimate the regression coefficients. 22 Once other factors were included, total other private patient costs were higher in the treatment group by an estimated £5637.19, although this difference was not statistically significant (p = 0.65).
| Variable | Coefficient | SE | p-value | Direction |
|---|---|---|---|---|
| Treatment group | 5637.19 | 12,561.61 | 0.654 | Active > placebo |
| Study site | 132.05 | 60.79 | 0.032 | Later > earlier |
| Sex | 24,773.05 | 14,276.24 | 0.085 | Women > men |
| Age | –411.49 | 827.17 | 0.621 | Younger > older |
| Weight | 975.74 | 458.85 | 0.036 | Heavier > lighter |
| MS type | 1279.35 | 11,090.55 | 0.908 | SPMS > PPMS |
| Baseline EDSS score | 12,372.36 | 8184.30 | 0.132 | Higher > lower |
| Baseline cost | 0.79 | 0.37 | 0.038 | Higher > lower |
| Constant | –129,423.40 | 71,718.17 | 0.072 |
Post-hoc subgroup analysis of total private costs by treatment group
A subgroup analysis previously described was performed regressing costs and treatment allocation to patients in earlier EDSS stages (baseline EDSS score of 4.0–5.5). There were no significant differences between treatment groups on total private/patient costs based on the subgroup analysis (Table 59). Once other factors were included, total other private patient costs were higher in the placebo group by an estimated £8784.21, although this difference was not statistically significant (p = 0.33).
| Variable | Coefficient | SE | p-value | Direction |
|---|---|---|---|---|
| Treatment group | –8784.21 | 9062.82 | 0.333 | Placebo > active |
| Study site | 28.14 | 47.71 | 0.557 | Later > earlier |
| Sex | 6748.30 | 11,394.18 | 0.555 | Men > women |
| Age | 700.13 | 644.04 | 0.279 | Older > younger |
| Weight | 45.91 | 347.14 | 0.895 | Heavier > lighter |
| MS type | 16,016.74 | 8486.58 | 0.061 | Secondary > primary |
| Baseline EDSS score | 13,433.89 | 8440.93 | 0.114 | Higher > lower |
| Baseline cost | 1.38 | 0.50 | 0.007 | Higher > lower |
| Constant | –101,605.00 | 59,700.67 | 0.091 |
Health outcomes: quality-adjusted life-years
The primary health economic outcome was the QALY, over 36 months, estimated using the EQ-5D. The EQ-5D questionnaire is based on five dimensions (mobility, self-care, usual activity, pain/discomfort and anxiety/depression), with each dimension having three levels (no problem, some problem or extreme problems, i.e. unable to perform the task in question). The percentages of the different responses to each of the five dimensions, at each follow-up, are presented in Figure 39. There appeared to be very little change in the domains of mobility, usual activities and pain/discomfort regardless of treatment group or time. Most patients reported some problem with mobility (over 90%), usual activities (over 70%) and pain/discomfort (over 60%) at every time point. Self-care appeared to be the only domain where changes were seen over time. At baseline the majority of patients reported no problems with self-care and almost none reported severe problems. By the final follow-up the majority reported some problems with self-care and a small proportion in both treatment groups reported severe problems. Placebo patients consistently reported fewer problems with anxiety/depression over the study period, although this might have been due to baseline differences.
FIGURE 39.
Percentages of the different responses to each of the five dimensions of the EQ-5D, at each follow-up, separated by treatment group (complete cases).
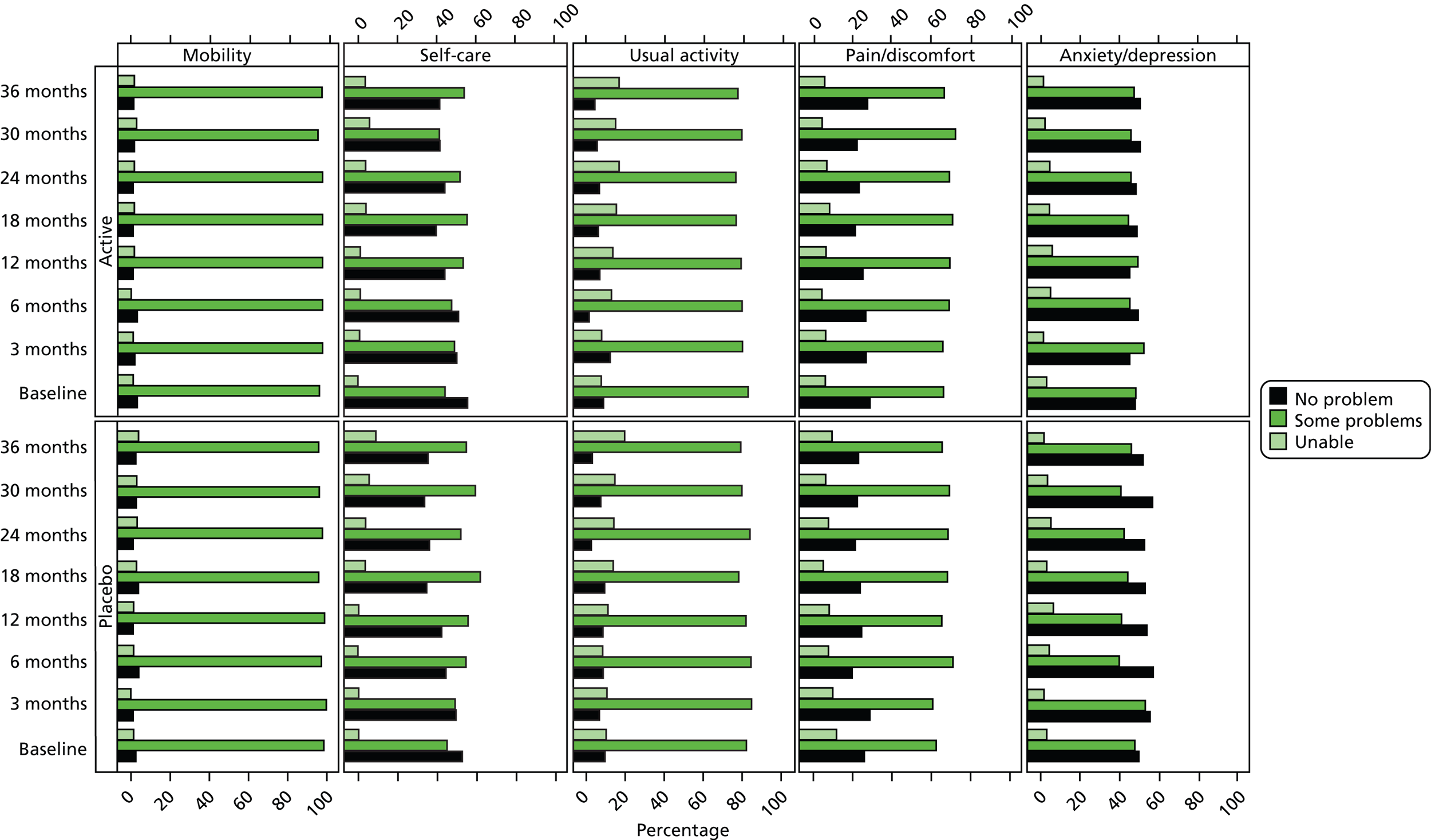
European Quality of Life-5 Dimensions scores by treatment group and follow-up
Mean EQ-5D scores in each treatment group at each follow-up are presented in Figure 40. There were no statistically significant differences at any time point.
FIGURE 40.
Mean EQ-5D scores, with 95% CIs, by treatment group and follow-up. The number of patients with an EQ-5D score reported at each follow-up is given directly above the CI for placebo and directly below the CI for active.
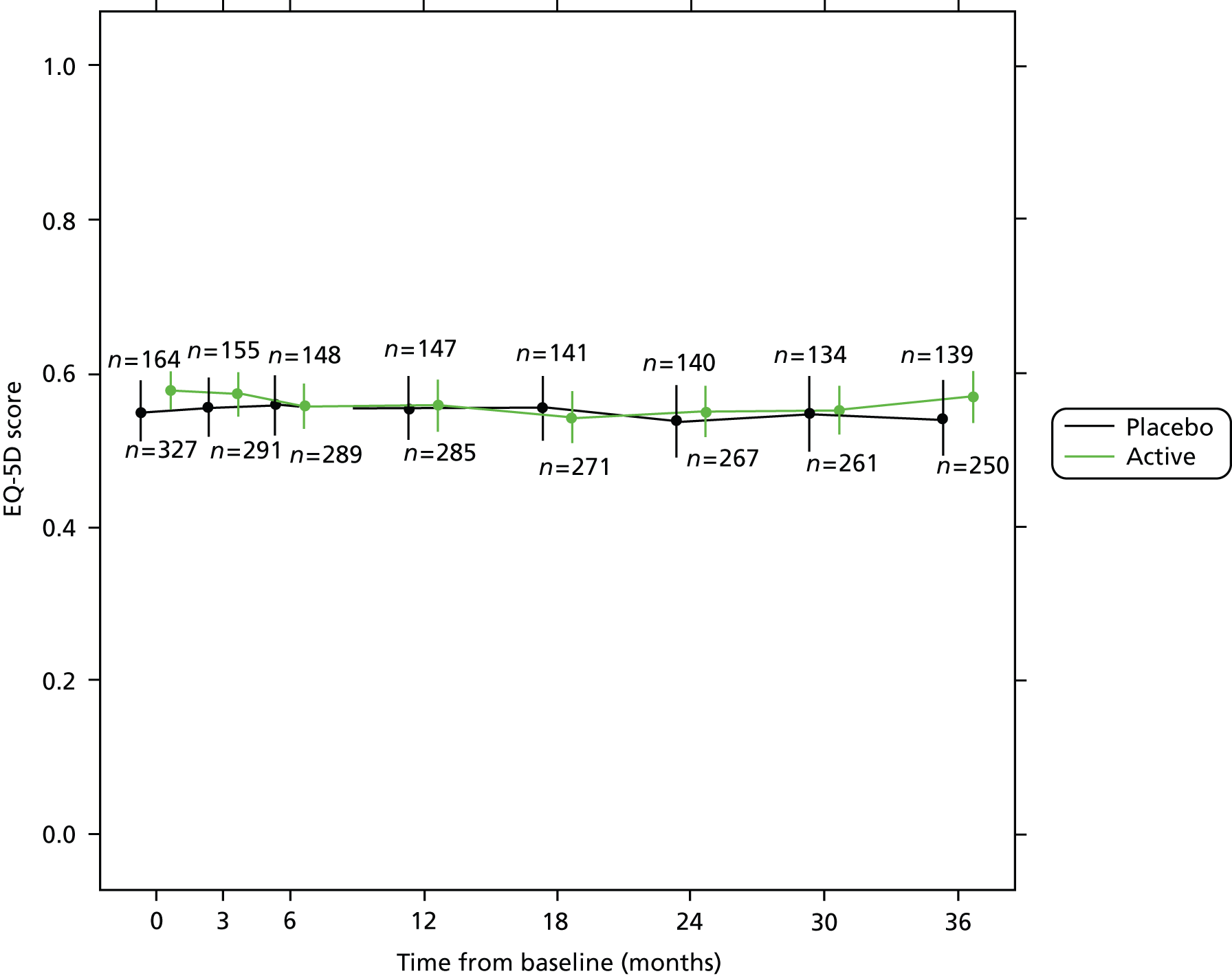
Comparison of quality-adjusted life-year outcomes between treatment groups
For the final regression analyses, the relationship between total QALYs with treatment allocation and covariates pre-specified in the trial SAP were examined (Tables 60 and 61). Over the 36-month follow-up there was no difference between the treatment groups. Baseline EDSS and EQ-5D scores were significant predictors of QALY outcomes at the 0.001 level.
| Variable | Coefficient | SE | p-value | Direction |
|---|---|---|---|---|
| Treatment group | –0.0001 | 0.04 | 0.998 | No direction |
| Study site | 0.0001 | <0.001 | 0.675 | No direction |
| Sex | –0.0758 | 0.05 | 0.121 | Women > men |
| Age | –0.0019 | 0.002 | 0.507 | No direction |
| Weight | –0.0010 | 0.002 | 0.494 | No direction |
| MS type | 0.0087 | 0.04 | 0.844 | No direction |
| Baseline EDSS score | –0.1094 | 0.03 | < 0.001 | Higher < lower |
| Baseline EQ-5D score | 1.5102 | 0.09 | < 0.001 | Higher |
| Constant | 1.4666 | 0.31 |
| Time point | Treatment group | Adjusted mean difference, mean (SE) | 95% CI | p-value | |
|---|---|---|---|---|---|
| Active, mean (SE); n | Placebo, mean (SE); n | ||||
| Year 1 | 0.5633 (0.01); n = 321 | 0.5553 (0.02); n = 163 | –0.0098 (0.01) | –0.0380 to 0.0183 | 0.491 |
| Year 2 | 0.5310 (0.01); n = 321 | 0.5193 (0.02); n = 163 | –0.0040 (0.02) | –0.0399 to 0.0319 | 0.826 |
| Year 3 | 0.5214 (0.02); n = 321 | 0.4933 (0.02); n = 163 | 0.0135 (0.02) | –0.0239 to 0.0510 | 0.478 |
| Total QALYs | 1.6152 (0.03); n = 321 | 1.5669 (0.05); n = 163 | –0.0001 (0.04) | –0.0846 to 0.0848 | 0.998 |
Post-hoc subgroup analysis of quality-adjusted life-year outcomes by treatment group
A subgroup analysis was performed regressing QALYs and treatment allocation limited to patients in earlier EDSS stages (baseline EDSS score of 4.0–5.5). There were no statistically significant between-group differences in the subgroup analysis although this was based on a diminished sample size (Tables 62 and 63). In contrast to the full sample, QALYs among active patients were higher in this subgroup compared with placebo where QALYs were little changed from those in the full sample. The overall QALY-adjusted difference in favour of treatment in the subgroup (0.0658), while sufficient to support a cost-effectiveness analysis, remains a relatively small benefit compared with the additional treatment costs outlined earlier. Differences may also have been due to random chance because of the small number of placebo patients (n = 34) in this subgroup.
| Variable | Coefficient | SE | p-value | Direction |
|---|---|---|---|---|
| Treatment group | 0.0658 | 0.08 | 0.436 | Active > placebo |
| Study site | 0.0001 | < 0.001 | 0.778 | No direction |
| Sex | –0.1004 | 0.09 | 0.278 | Women > men |
| Age | –0.0043 | 0.006 | 0.447 | Older < younger |
| Weight | –0.0047 | 0.003 | 0.153 | Heavier < lighter |
| MS type | –0.0035 | 0.08 | 0.965 | No direction |
| Baseline EDSS score | –0.0419 | 0.07 | 0.547 | Higher < lower |
| Baseline EQ-5D score | 1.4565 | 0.18 | < 0.001 | Higher |
| Constant | 1.4666 | 0.31 |
| Time point | Treatment group | Adjusted mean difference, mean (SE) | 95% CI | p-value | |
|---|---|---|---|---|---|
| Active, mean (SE); n | Placebo, mean (SE); n | ||||
| Year 1 | 0.6208 (0.02); n = 75 | 0.5448 (0.03); n = 34 | 0.0109 (0.03) | –0.0552 to 0.0669 | 0.704 |
| Year 2 | 0.5959 (0.02); n = 75 | 0.5173 (0.04); n = 34 | 0.0131 (0.04) | –0.0610 to 0.0873 | 0.728 |
| Year 3 | 0.5877 (0.02); n = 75 | 0.4978 (0.04); n = 34 | 0.0419 (0.04) | –0.0337 to 0.1175 | 0.277 |
| Total QALYs | 1.8041 (0.06); n = 75 | 1.5599 (0.10); n = 34 | 0.0658 (0.08) | –0.1000 to 0.2316 | 0.436 |
Subgroup EQ-5D scores over the study period are illustrated in Figure 41. These appear to show a consistent trend for higher EQ-5D scores in patients in the active group who had a baseline EDSS score of 4.0–5.5, compared with placebo. Adjusting for baseline, the difference at the final follow-up was 0.047 based on a complete case analysis. In patients with higher baseline EDSS score (6.0–6.5), the mean EQ-5D score appears to have the same trajectory in both active and placebo groups (adjusted difference at final follow-up 0.012). Neither finding was statistically significant.
FIGURE 41.
European Quality of Life-5 Dimensions weighted index score by follow-up and EDSS subgroup. Top panel = baseline EDSS score of 4.0–5.5; bottom panel = baseline EDSS score of 6.0–6.5.
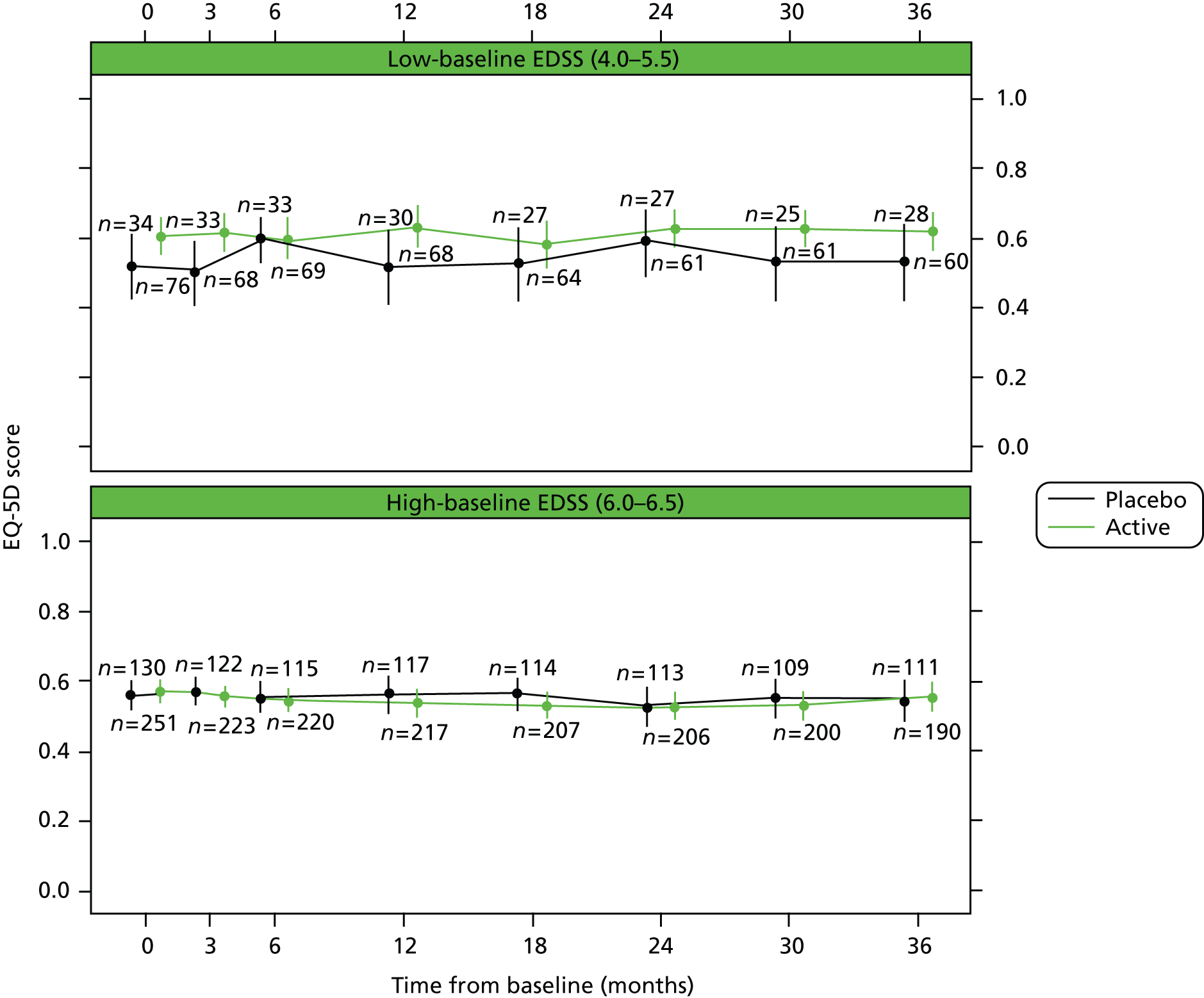
Conclusions
The CUPID trial did not show any consistent or marked between-group differences either in mean costs (other than the treatment cost) or mean QALYs. As no evidence was found for the clinical effectiveness of the CUPID intervention, a cost-effectiveness analysis combining group differences in costs and effects was not undertaken.
In terms of point estimates, after controlling for baseline covariates and imputation of missing data, costs from the NHS/PSS perspective were significantly lower in the placebo group with no between-group difference in QALYs. The mean additional cost of the 3-year CUPID intervention was estimated to be £27,529.14 per patient (see Table 54) with a negligible difference in QALYs (–0.0001) (see Table 60). This indicates that placebo dominates the CUPID intervention in this treatment population achieving comparable health outcomes at lower cost.
The clinical effectiveness analysis of the CUPID study suggested that patients in earlier EDSS stages (score of 4.0–5.5) at baseline may have represented a subgroup of positive-treatment responders. A post-hoc subgroup analysis, restricted to this subgroup, estimated the mean additional cost of Δ9-THC in patients with less severe disease at baseline to be £30,130.37 (see Table 55), with an additional 0.0658 QALYs over 3 years (see Table 62). This indicates that the CUPID intervention may provide modest health benefits for progressive MS patients in earlier disease stages but at a high cost. These data indicate a cost per QALY gained for Δ9-THC in this subgroup in excess of £400,000, which is well above the cost-per-QALY threshold values used by NICE in their guidance on value for money and use of interventions in NHS in England. Owing to the small sample size, particularly in the placebo group, findings from the subgroup analysis should be treated with caution.
Chapter 7 Discussion
Primary outcome measures
No treatments have as yet shown clinical efficacy in the modification of progressive MS in the absence of relapses. Results of this study did not show an overall treatment effect of oral dronabinol on the clinical disease course in progressive MS, as assessed by either sustained EDSS progression or using the MSIS-29phys. Several factors might have reduced the ability to detect any potential treatment effect, in particular treatment discontinuation, loss to follow-up and less overall disease progression than expected. Long-term studies in progressive neurological diseases are notoriously difficult to undertake: loss to follow-up might hinder interpretation of results and low event rates and discontinuation of study medication also decrease the power to detect a treatment effect. Overall, loss to follow-up in the CUPID study was around the level expected, but more attrition occurred in the dronabinol group. This coincided with a higher number of AEs than in the placebo group, which also largely accounted for premature treatment discontinuation, although no major safety concerns were reported. As in previous studies of cannabinoids, most AEs occurred during the dose titration period, but the high lipid solubility of cannabinoids means that long-term build-up can occur some months after treatment initiation. Any future long-term studies to investigate disease-modifying effects of cannabinoids should use lower doses to reduce the risk of AEs, which in turn should increase compliance and reduce potential error in any ITT analysis.
Low progression rates make the identification of a treatment effect less likely and further work is necessary to establish optimum inclusion criteria for trials of progressive MS. Although recent studies have tried to be more specific about monitoring pre-trial progression to fulfil inclusion criteria, the CUPID study was designed to be a pragmatic study for people with progressive MS, testing some of the findings from the CAMS extension phase, which suggested that dronabinol might have an effect on MS progression. Future studies should ensure that the population recruited has a high chance of progression. Our study population was skewed towards the higher end of the EDSS score range (52% had a baseline score of 6.0; 26% had a baseline score of 6.5). Mean EDSS score at baseline was 5.9 (SD 0.69), higher than most other recent studies of treatments in progressive disease. Some studies have taken account of the slower progression rates in patients with higher EDSS scores by adjusting recruitment to ensure a lower overall mean EDSS score. The PROMiSe study24 was adjusted to 40% recruitment of patients with an EDSS score of 3.0–5.0, producing a mean EDSS score of 4.9 and yearly progression rate of 16% in patients with primary progressive MS, compared with 24% estimated in the CUPID study.
Secondary outcome measures
When the overall population was considered, none of the secondary outcome measures demonstrated a treatment effect. Once again, mean change over time was generally less than expected, reducing the opportunity to identify any potential treatment effect. The same arguments can be applied to secondary measures as for primary outcomes: premature discontinuation, selective drop-out and lack of progression may have affected our ability to detect treatment effects.
Although brain atrophy, as assessed using longitudinal comparison of MRI, is being used as a surrogate for clinical disability progression (particularly in Phase II studies), there remains controversy over its validity in this regard. Although a recent meta-analysis demonstrated reasonable correlation between treatment effects on brain atrophy and disability progression over 2 years in RRMS (R2 = 0.48),25 fewer studies exist in progressive MS and treatment effects on both MRI and clinical end points have not been demonstrated. Our current results showed that overall rate of brain atrophy was as expected, whereas clinical change was less than expected. Even in the less disabled subgroup (baseline EDSS score of 4.0–5.5), where disability did alter significantly over time and there was an apparent treatment effect, this was not mirrored in a treatment effect on brain atrophy from MRI.
Further analyses
Pre-specified subgroup analyses suggested that dronabinol might have a slight potentially beneficial effect in terms of EDSS score progression and other outcome measures in less disabled patients (baseline EDSS score of < 6.0). Conversely, dronabinol might have a slight potentially negative effect in patients with higher EDSS scores. One possible explanation for these findings is that cannabinoids have been shown to reduce muscle stiffness2,26,27 and, although an antispasticity effect might improve function at lower levels of disability, if walking is compromised by weakness, dronabinol might reduce muscle tone to the point where muscle power becomes affected and walking is more difficult. This side effect is well known with agents such as baclofen. Distinguishing a symptomatic effect from a disease-modifying effect can be a difficult task. In Parkinson’s disease, for example, it is generally accepted that L-dopa has a symptomatic effect that can have a profound long-term impact on disability, although it is generally regarded as not having a neuroprotective role.
Treatment effects shown in pre-specified analyses at each EDSS level below 6.0 led us to combine EDSS score levels of 4.0 to 5.5 in a post-hoc fashion, which confirmed treatment effects in both primary outcomes. The suggestion of a potential effect in lower disability groups might have relevance for inclusion criteria in future clinical trials in progressive MS. It is well known that the EDSS is not a linear scale and people affected by MS tend to spend considerably more time at EDSS scores of 6.0 and 6.5 (needing one or two sticks for mobility), yet progress relatively rapidly through scores of 4.0 to 5.5. It is, therefore, not surprising that this subpopulation progressed faster than more disabled individuals and that a treatment effect was detected in this subgroup.
Economic evaluation
Dronabinol is associated with high additional costs and has no evidence of incremental health gains and is, therefore, not cost-effective compared with standard care, which dominates dronabinol in this patient group. Exploratory analyses in the subgroup of patients with lower EDSS scores, suggests potential health gains, but at a high incremental cost relative to potential incremental QALY gains, with estimates of cost per QALY likely to be in excess of £400,000 per QALY gained.
Rasch analysis
All MS-specific scales performed well as measurement instruments. Some scales were less optimally targeted to the study sample. This may have influenced the interpretation of changes and differences. Typically suboptimal targeting reduces the ability to detect changes and differences. There was no clear and consistent evidence for a symptomatic or disease-modifying treatment effect. There was some suggestion, from exploratory post-hoc analyses, that less disabled people might derive a disease-modifying benefit. However, this was not a consistent effect across scales measuring related constructs and at best provides a potential hypothesis for formal testing.
Symptomatic evaluation
This study was not designed to demonstrate symptomatic benefits and the high level of treatment discontinuation is likely to have had an effect on our ability to show this at the end of the 3-year study.
Safety
Serious adverse events (including death or hospitalisation because of MS-related problems) were expected to occur relatively frequently in this patient population and about one-third of all participants experienced at least one SAE. The number and nature of SAEs were broadly similar in the two treatment groups. Two SUSARs were reported in the active treatment group. Of these, one participant with a previous history of seizure was hospitalised following a grand mal seizure and trial treatment was discontinued. Seizures are known to be more prevalent in people with MS than in the general population and may also occur following overdose of dronabinol in patients with existing seizure disorders. The second SUSAR was reported following the non-urgent hospitalisation of a participant for colonoscopic investigation of altered bowel habit. In this case, the SUSAR classification was largely as a result of the interpretation of reporting guidelines and trial treatment was continued.
The number and nature of minor AEs was also similar across the two groups with the exception of known cannabinoid-related effects (dizziness and light-headedness; dissociative and thinking or perception disorders) which occurred more frequently in the active group. These treatment-related AEs contributed to the higher incidence of premature discontinuation of trial medication in participants in the active group than in the placebo group.
Further work
This independent study provides the largest data set on cannabinoid exposure over time currently available. Most outcomes, both primary and secondary, did not provide any evidence for a treatment effect with dronabinol. However, the results do suggest that further study of less disabled people with progressive MS may be warranted. In addition, results suggest that studies of antiprogressive treatments in progressive MS should pay careful attention to the pre-study progression of potential participants. A reasonable degree of ongoing progression is a prerequisite for antiprogressive treatments to have a substrate to effect. Our results therefore elude to an incomplete understanding of progressive MS and further advancing our understanding of progression in people with progressive MS would be very valuable.
Chapter 8 Conclusions
Implications for health care
The CUPID study has a few important implications for health care. First, it will come as a disappointment to people with progressive MS who have witnessed, over some 40 years, multiple negative clinical trials for progressive MS.
Another implication for health care concerns the potential medicinal use of cannabinoids in MS. This will strengthen the case against their use. While this is a reasonable interpretation if one takes the negative clinical trials at face value, it is known that many people with MS find benefit from cannabinoids. This anomaly warrants further investigation. One option is that the measures used to determine effectiveness are missing important benefits.
Recommendations for research
The results from the CUPID study point to three major themes for research.
First, research is required to better understand exactly what is progressing in progressive MS so that clinical trials are better informed. A concerted effort is required to carefully and systematically document and monitor over time cohorts of people with a diagnosis of progressive disease. This will lead to disease profiles and trajectories. In addition, the CUPID study implied that a substantial proportion of people improved in terms of aspects of their physical function. This would seem surprising for a disease whose hallmark is purported to be progressive physical disability. One explanation for this is that people adapt to their worsening function – the so-called response shift. If this were the explanation, we would expect to see RMT analyses to show DIF by time point; this was not a finding in the CUPID study.
A second major theme for research builds on the hypothesis that cannabinoids may have an effect in less disabled people. This may be because people with higher levels of disability did not progress in the outcomes measured or that there is a treatment effect in only the less disabled.
A third area for research is further development of COAs. Although the measures were shown to be robust, limitations were identified. The role of the measurement instruments in clinical trials should not be underestimated and their quality must not be compromised.
Acknowledgements
We thank the patients who took part in this study and their families, and we acknowledge the contribution of all principal investigators and their study site teams. Particular thanks go to the co-ordinating team in Plymouth (especially Jacqui Mathews, Chris Hayward, Margie Berrow, Brian Wainman, Mark Warner, Linda Sutcliffe, Chris Rollinson, Lorraine Underwood, Caroline Snelgrove, Corinna Phillips and Liz Ford) and also to Mike Marner, Suzi Reilly, Nick Pilkington, Adrian Pace, Richard Hosking and Claire Fox.
Contributions of authors
Susan Ball (Research Fellow in Statistics, Plymouth University Peninsula Schools of Medicine and Dentistry): analysis and interpretation of the data and writing of the report.
Jane Vickery (Senior Trial Manager, Peninsula Clinical Trials Unit, Plymouth University Peninsula Schools of Medicine and Dentistry): co-ordination of the study and writing of the report.
Jeremy Hobart (Professor of Clinical Neurology and Health Measurement, Plymouth University Peninsula Schools of Medicine and Dentistry): study design, conduct, patient enrolment, data analysis and interpretation and writing of the report.
Dave Wright (Professor of Applied Statistics, Plymouth University Peninsula Schools of Medicine and Dentistry): study design, analysis and interpretation of the data and writing of the report.
Colin Green (Professor of Health Economics, University of Exeter Medical School): analysis and interpretation of the economic evaluation and writing of the report.
James Shearer (Lecturer in Health Economics, King’s College London, formerly at University of Exeter): analysis and interpretation of the economic evaluation and writing of the report.
Andrew Nunn (Associate Director and Chair of Infections Research Theme, MRC Clinical Trials Unit, London): study design and conduct.
Mayam Gomez Cano (Research Assistant in Statistics, Plymouth University Peninsula Schools of Medicine and Dentistry): data analysis and writing of the report.
David MacManus (Principal Research Associate, Queen Square Multiple Sclerosis Centre, University College London’s Institute of Neurology): supervision of MRI data collection and quality assurance.
David Miller (Professor of Clinical Neurology, Queen Square Multiple Sclerosis Centre, University College London’s Institute of Neurology): design of the MRI substudy and supervision of the analysis of imaging outcomes.
Shahrukh Mallik (Clinical Research Associate, Queen Square Multiple Sclerosis Centre, University College London’s Institute of Neurology): MRI analysis.
John Zajicek (Professor of Clinical Neuroscience, Peninsula Clinical Trials Unit, Plymouth University Peninsula Schools of Medicine and Dentistry): study concept, design and conduct, patient enrolment, interpretation of data and writing of the report.
All authors have approved the final version of the report to be published.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the HTA programme, the EME programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme, the EME programme or the Department of Health.
Publications
Zajicek J, Ball S, Wright D, Vickery J, Nunn A, Miller D, et al. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol 2013;51:857–65.
References
- Zajicek J, Ball S, Wright D, Vickery J, Nunn A, Miller D, et al. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol 2013;51:857-65. http://dx.doi.org/10.1016/S1474-4422(13)70159-5.
- Zajicek J, Fox P, Sanders H, Wright D, Vickery J, Nunn A. UK MS Research Group . Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo-controlled trial. Lancet 2003;362:1517-26. http://dx.doi.org/10.1016/S0140-6736(03)14738-1.
- Zajicek J, Fox P, Teare L. Cannabinoids in multiple sclerosis (CAMS) study: safety and efficacy data for 12-months follow up. J Neurol Neurosurg Psychiatry 2005;76:1664-9. http://dx.doi.org/10.1136/jnnp.2005.070136.
- McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol 2001;50:121-7. http://dx.doi.org/10.1002/ana.1032.
- Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon-Beta-1a in MS (SPECTRIMS) Study Group . Randomised controlled trial of interferon-beta-1a in secondary progressive MS: clinical results. Neurology 2001;56:1496-504. http://dx.doi.org/10.1212/WNL.56.11.1496.
- Hobart J, Cano S. Improving the evaluation of therapeutic interventions in multiple sclerosis: the role of new psychometric methods. Health Technol Assess 2009;13.
- Pinheiro JC, Bates DM. Mixed-Effects Models in S and S-PLUS. New York, NY: Springer Verlag; 2000.
- Fischer JS, Jak AJ, Kniker JE, Rudick RA, Cutter GR. Multiple Sclerosis Functional Composite (MSFC) Administration and Scoring Manual. New York, NY: National Multiple Sclerosis Society; 2001.
- Cohen JA, Cutter GR, Fischer JS, Goodman AD, Heidenreich FR, Jak AJ, et al. Use of the Multiple Sclerosis Functional Composite as an outcome measure in a phase 3 clinical trial. Arch Neurol 2001;58:961-7. http://dx.doi.org/10.1001/archneur.58.6.961.
- Molt RW, McAuley E, Mullen S. Longitudinal measurement invariance of the Multiple Sclerosis Walking Scale-12. J Neurol Sci 2011;305:75-9. http://dx.doi.org/10.1016/j.jns.2011.03.008.
- Ware JE. SF-36 health survey update. Spine 2000;25:3130-9. http://dx.doi.org/10.1097/00007632-200012150-00008.
- Hobart JC, Riazi A, Thompson AJ, Styles IM, Ingram W, Vickery PJ, et al. Getting the measure of spasticity in multiple sclerosis: the Multiple Sclerosis Spasticity Scale (MSSS-88). Brain 2006;129:224-34. http://dx.doi.org/10.1093/brain/awh675.
- Smith SM, De Stefano N, Jenkinson M, Matthews PM. Normalized accurate measurement of longitudinal brain change. J Comput Assist Tomogr 2001;25:466-75. http://dx.doi.org/10.1097/00004728-200105000-00022.
- Smith SM, Zhang Y, Jenkinson M, Chen J, Matthews PM, Federico A, et al. Accurate, robust and automated longitudinal and cross-sectional brain change analysis. Neuroimage 2002;17:479-89. http://dx.doi.org/10.1006/nimg.2002.1040.
- Andrich D. A rating formulation for ordered response categories. Psychometrika 1978;43:561-73. http://dx.doi.org/10.1007/BF02293814.
- Andrich D. Rasch Models for Measurement. Beverley Hills, CA: Sage Publications; 1988.
- Hobart J, Cano S, Zajicek J, Thompson AJ. Rating scales as outcome measures for clinical trials in neurology: problems, solutions, and recommendations. Lancet Neurol 2007;6:1094-105. http://dx.doi.org/10.1016/S1474-4422(07)70290-9.
- Dolan P. Modeling valuations for EuroQol health states. Med Care 1997;35:1095-108. http://dx.doi.org/10.1097/00005650-199711000-00002.
- Curtis L. Unit Costs of Health and Social Care 2011. Canterbury: PSSRU, University of Kent; 2011.
- Guide to the Methods of Technology Appraisal. London: NICE; 2008.
- Brazier J, Ratcliffe J, Salomon J, Tsuchiya A. Measuring and Valuing Health Benefits for Economic Evaluation. Oxford: Oxford University Press; 2007.
- Barber JA, Thompson SG. Multiple regression of cost data: use of generalised linear models. J Health Serv Res Policy 2004;9:197-204. http://dx.doi.org/10.1258/1355819042250249.
- White I, Royston P, Wood A. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 2011;30:377-99. http://dx.doi.org/10.1002/sim.4067.
- Wolinsky JS, Narayana PA, O’Connor P, Coyle PK, Ford C, Johnson K, et al. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double-blind, placebo controlled trial. Ann Neurol 2007;61:14-2. http://dx.doi.org/10.1002/ana.21079.
- Sormani MP, Bruzzi P. MRI lesions as a surrogate for relapses in multiple sclerosis: a meta-analysis of randomised trials. Lancet Neurol 2013;12:669-76. http://dx.doi.org/10.1016/S1474-4422(13)70103-0.
- Novotna A, Mares J, Ratcliffe S, Novakova I, Vachova M, Zapletalova O, et al. A randomized, double-blind placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex®), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol 2011;18:1122-31. http://dx.doi.org/10.1111/j.1468-1331.2010.03328.x.
- Zajicek J, Hobart J, Slade A, Barnes D, Mattison P. MUSEC Research Group . Multiple Sclerosis and Extract of Cannabis: results of the MUSEC trial. J Neurol Neurosurg Psychiatry 2012;83:1125-32. http://dx.doi.org/10.1136/jnnp-2012-302468.
Appendix 1 Participating sites and principal investigators
| Site | PI |
|---|---|
| Plymouth, Derriford Hospital | Professor J Zajicek |
| Aberdeen Royal Infirmary | Dr R Coleman |
| Birmingham, West Midlands Rehabilitation Centre | Dr C Ko-Ko |
| Bristol, Frenchay Hospital | Professor N Scolding |
| Cambridge, Addenbrookes Hospital | Rev Dr A Coles |
| Cardiff, University Hospital of Wales | Professor N Robertson |
| Coventry, University Hospitals Coventry & Warwickshire | Dr A Shehu |
| Edinburgh, Western General Hospital | Dr B Weller |
| Gloucestershire Royal Hospital | Dr R Martin |
| Hertford County Hospital | Dr D Kidd |
| Irvine, Ayrshire Central Hospital | Dr P Mattison |
| Leicester General Hospital | Dr B Kendall |
| London, Charing Cross Hospital | Dr R Nicholas |
| Manchester, Hope Hospital | Dr P Talbot |
| Newcastle, Royal Victoria Infirmary | Dr M Duddy |
| Norfolk and Norwich University Hospital | Dr M Lee |
| Nottingham University Hospital | Professor C Constantinescu |
| Oxford, John Radcliffe Hospital | Dr J Palace |
| Poole General Hospital | Dr C Hillier |
| Preston, Royal Preston Hospital | Dr P Tidswell |
| Sheffield, Royal Hallamshire Hospital | Dr S Howell |
| Stoke on Trent, University Hospital of North Staffordshire | Professor C Hawkins |
| Taunton & Somerset Hospital | Dr E Fathers |
| Truro, Royal Cornwall Hospital | Dr B McLean |
| London, Barts and London NHS Trust | Professor G Giovannoni |
| University Hospital of Birmingham | Dr G Mazibrada |
| Reading, Royal Berkshire Hospital | Dr A Weir |
Appendix 2 NHS research and development approval dates
| Site | Date approval received |
|---|---|
| Plymouth, Derriford Hospital | 19 May 2006 |
| Birmingham, West Midlands Rehabilitation Centre | 13 June 2006 |
| Nottingham University Hospital | 13 June 2006 |
| Truro, Royal Cornwall Hospital | 20 June 2006 |
| Aberdeen Royal Infirmary | 23 June 2006 |
| Manchester, Hope Hospital | 28 June 2006 |
| Bristol, Frenchay Hospital | 28 June 2006 |
| Irvine, Ayrshire Central Hospital | 29 June 2006 |
| Preston, Royal Preston Hospital | 29 June 2006 |
| Norfolk and Norwich University Hospital | 4 July 2006 |
| Edinburgh, Western General Hospital | 7 July 2006 |
| Stoke on Trent, University Hospital of North Staff | 14 July 2006 |
| Taunton & Somerset Hospital | 24 July 2006 |
| Newcastle, Royal Victoria Infirmary | 30 August 2006 |
| Sheffield, Royal Hallamshire Hospital | 31 August 2006 |
| Cardiff, University Hospital of Wales | 31 August 2006 |
| London, Charing Cross Hospital | 14 September 2006 |
| Oxford, John Radcliffe Hospital | 26 September 2006 |
| Poole General Hospital | 11 October 2006 |
| Cambridge, Addenbrookes Hospital | 27 October 2006 |
| Coventry, University Hospitals Coventry & Warwickshire | 23 January 2007 |
| Leicester General Hospital | 23 March 2007 |
| The Barts and London NHS Trust | 4 April 2007 |
| Gloucestershire Royal Hospital | 20 April 2007 |
| Reading, Royal Berkshire Hospital | 14 August 2007 |
| University Hospital of Birmingham | 24 September 2007 |
| Hertford County Hospital | 6 November 2007 |
Appendix 3 Study recruitment May 2006–July 2008 by site
| Site | 2006 | 2007 | 2008 | Total | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| May | June | July | August | September | October | November | December | January | February | March | April | May | June | July | August | September | October | November | December | January | February | March | April | May | June | July | ||
| Plymouth | 18 | 18 | 16 | 11 | 14 | 77 | ||||||||||||||||||||||
| Aberdeen | 2 | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 11 | |||||||||||||||||||
| Birmingham | 2 | 3 | 3 | 3 | 11 | |||||||||||||||||||||||
| Bristol | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 18 | ||||||||||||
| Cambridge | 1 | 3 | 4 | |||||||||||||||||||||||||
| Cardiff | 15 | 1 | 16 | |||||||||||||||||||||||||
| Coventry | 6 | 4 | 10 | |||||||||||||||||||||||||
| Edinburgh | 5 | 1 | 4 | 2 | 12 | |||||||||||||||||||||||
| Gloucester | 2 | 6 | 8 | |||||||||||||||||||||||||
| Hertford | 4 | 2 | 6 | |||||||||||||||||||||||||
| Irvine | 19 | 19 | ||||||||||||||||||||||||||
| Leicester | 2 | 2 | 1 | 2 | 1 | 2 | 2 | 2 | 14 | |||||||||||||||||||
| Charing Cross | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 4 | 5 | 2 | 1 | 1 | 22 | |||||||||||||||
| Manchester | 1 | 1 | 1 | 1 | 1 | 3 | 4 | 4 | 1 | 1 | 1 | 19 | ||||||||||||||||
| Newcastle | 4 | 3 | 3 | 3 | 1 | 3 | 1 | 2 | 1 | 21 | ||||||||||||||||||
| Norwich | 4 | 5 | 3 | 2 | 14 | |||||||||||||||||||||||
| Nottingham | 2 | 1 | 2 | 3 | 5 | 4 | 3 | 1 | 1 | 22 | ||||||||||||||||||
| Oxford | 7 | 2 | 4 | 4 | 3 | 3 | 1 | 2 | 26 | |||||||||||||||||||
| Poole | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 12 | ||||||||||||||||||||
| Preston | 3 | 5 | 2 | 2 | 5 | 4 | 21 | |||||||||||||||||||||
| Sheffield | 1 | 4 | 3 | 3 | 5 | 1 | 1 | 1 | 1 | 1 | 21 | |||||||||||||||||
| Stoke on Trent | 10 | 5 | 1 | 3 | 3 | 5 | 3 | 30 | ||||||||||||||||||||
| Taunton | 3 | 2 | 3 | 3 | 11 | |||||||||||||||||||||||
| Truro | 6 | 4 | 7 | 5 | 3 | 5 | 4 | 6 | 40 | |||||||||||||||||||
| Royal London | 7 | 2 | 1 | 10 | ||||||||||||||||||||||||
| Birmingham QE | 6 | 2 | 8 | |||||||||||||||||||||||||
| Reading | 2 | 1 | 2 | 1 | 4 | 10 | ||||||||||||||||||||||
| Totals | 18 | 0 | 2 | 20 | 4 | 38 | 15 | 6 | 28 | 31 | 22 | 21 | 29 | 38 | 19 | 13 | 18 | 26 | 18 | 9 | 23 | 15 | 19 | 22 | 15 | 16 | 8 | 493 |
Appendix 4 Participant information sheet
Cannabinoid Use in Progressive Inflammatory brain Disease (CUPID study)
PARTICIPANT INFORMATION SHEET
Invitation to participate
We would like to invite you to take part in a research study. Before you decide whether or not to participate, it is important for you to understand why the research is being done and what it will involve. Please take time to read this information carefully and discuss it with family and friends or your own doctor if you wish.
-
Part 1 explains the purpose of this study and what will happen to you if you take part.
-
Part 2 gives more detailed information about the conduct of the study.
If anything is not clear or you would like more information, please ask us. Remember, your participation is entirely voluntary – it’s up to you.
PART 1
What is the purpose of this study?
Current treatments for primary or secondary progressive multiple sclerosis (MS) are aimed at relieving specific symptoms. Few treatments actually help the disease itself, so the next step is to find treatments that can slow down the disease in the long term.
Results from a previous study have shown that tetrahydrocannabinol (THC) – found in the cannabis plant – appears to have an effect on slowing disability in progressive MS, when taken for a year. To find out if THC really does slow progression of disability, we need to conduct a new study over a longer period of time. In this study, we will assess the effects of THC on progressive MS by comparing it with an inactive substance, called a placebo (or dummy).
Why might I be suitable for the study?
People who have primary or secondary progressive MS and are between 18–65 years old are being invited to take part in this study. To be included in the study, your MS must have become worse over the last year and you must be able to walk at least 20 metres (with or without a walking aid). You must not have taken any cannabis in any form in the month prior to joining the study.
If there is a possibility that you may be pregnant or you are breastfeeding or planning a pregnancy, you may not take part in this study. More details are included in Part 2. If you have had a significant relapse in the last six months or have taken steroids in the last three months, you will not be able to join the study immediately but may be considered for entry at a later date. Your study doctor will confirm all these details with you if you would like to take part and will also check other aspects of your health to ensure your suitability to participate.
If you decide to take part in the study, please note:
-
Cannabis may cause side effects that may affect your ability to drive or operate dangerous machinery, so you should take extra care and consult your GP or study doctor if you do get any side effects. If your work or daily life involves driving or operating machinery, it may be better not to take part in this study. You should avoid driving or operating machinery if your concentration is affected by medication.
-
If you drive, you should inform your insurance company that you are participating in the study and may have been prescribed cannabis medication. Some insurers will require a letter from your doctor stating that you are fit to drive whilst taking medication.
-
Although we are authorised to use cannabis for this trial, it is an illegal drug. You must not under any circumstances give your study medication to anyone else.
-
You must not take your medication out of the United Kingdom before checking with your study doctor or nurse. Some countries will not allow cannabis-based medicines and others will need to see official documentation, which we will be able to provide.
-
It is important that you do not take any extra cannabis preparations during the study period, since this would interfere with the results of the study. Such preparations include nabilone, smoked cannabis and cannabis taken in food or drink. We will check your urine for cannabis at intervals throughout the study.
-
This is a long-term study so you should be prepared to take part for up to three and a half years.
Do I have to take part?
No. Participation in this study is entirely voluntary and it is up to you to decide whether or not to take part. If you do decide to take part you will be asked to sign a consent form, but you are still free to withdraw at any time in the future and without giving a reason. You will be given a copy of this information sheet and a copy of your signed consent form to keep. If you decide not to take part, or you withdraw from the study at any point, your usual medical care will not be affected in any way.
Who decides which study medication I will receive?
This type of study is known as a ‘randomised trial’ which means that your treatment will be chosen randomly (by chance) by a computer at the beginning of the trial. If you decide to take part, you will be allocated to receive either the active cannabis treatment or the placebo (dummy) treatment. Two thirds of the people in the study will be allocated to active treatment and one third to placebo. Both treatments are in the form of capsules which are taken by mouth and all capsules look identical. You will not be given anything to smoke. This is a ‘double-blind’ study which means that neither you nor the study team will know which treatment you are taking until the end of the study, in order that study assessments are not biased. However, your study doctor would be able to find out what you are taking very quickly, should it be necessary. It is important to note that once you are allocated to your treatment, you will remain on the same treatment throughout the study.
How many capsules do I have to take and for how long?
You will be asked to take the capsules twice a day for 3 years (possibly three and a half years). During the first four weeks of the study, your study doctor will adjust the number of capsules you are taking in order to find the amount that suits you best. If you have any side effects from the study medication which you find unacceptable, then you may be advised to reduce your dose. When the correct dosage has been found for you, you will continue to take the same amount each day, usually between two and four capsules twice a day.
What about my usual medications?
You should continue as normal with all your usual medicines and any other therapy prescribed by your family doctor or specialist. We will ask you about any medicines that you are currently taking (include supplements, vitamins and alternative remedies) at your first visit and will check whether these have changed at each subsequent visit.
What else is involved if I take part?
Clinic visits
All participants will need to attend the study clinic 11 times (six times during the first six months and then once every six months for the remainder of the trial). Most people will end the study at this point (after 3 years). Some participants (depending on their final clinic assessment) will be asked to continue in the study for an extra six months and to attend one extra clinic.
At each visit your study doctor will ask about your general health and whether you have had any side effects from your medication. You will then be given a new prescription for your study medication. A different doctor will make an assessment of disability (the EDSS score) at every visit. This may take up to 20 minutes. About half of the study visits also include a series of assessments to measure your progress. These include a timed 25 feet walk, a peg test (placing small pegs in holes and removing them by hand) and a number counting exercise. These assessments take about 15–20 minutes to complete and will be carried out by a nurse, a doctor or a therapist.
Questionnaires
If you take part in the study, the co-ordinating centre will send you a questionnaire booklet to complete approximately every three months throughout the trial. This booklet will take about half an hour to complete. If you have any difficulties in completing the questionnaires, you can telephone the trial co-ordinating centre on [telephone number supplied] (Freephone) during normal office hours and a member of the research team will be able to help you. Freepost envelopes will be provided for you to return the questionnaires.
Some of the questionnaires ask about your general health and well-being and about how MS is affecting your day to day life. Other questionnaires will ask specifically about services you have used (e.g. visiting your GP) or equipment and adaptations that you have needed for your home because of your MS. During the first and last six months of the study we will also send you a very short questionnaire (six times in total), asking specifically about services that you have used in the previous 2 days. One questionnaire specifically asks about symptoms relating to depression. The co-ordinating centre will send the results of this questionnaire to your study doctor. Only your trial number will appear on these results and no-one at the co-ordinating centre will be able to identify you from them. Your study doctor will be able to identify you from your trial number and if your study doctor thinks that you may be depressed, he/she may contact your MS Specialist Nurse or GP so that you can receive support and treatment as appropriate.
If you have access to a computer and the internet there will be an option to complete the questionnaire booklets online. All questionnaires will be anonymous and identified only by a study number and initials.
Blood samples
There are two reasons for taking blood samples during the study. Unfortunately, if you are unwilling to provide blood samples then you will not be able to participate in the study.
i) To assess the safety of taking study medication: At each visit you will be asked to provide a small blood sample (equivalent to two teaspoons). This sample will be tested for factors that indicate your general health, including liver function and blood count. By regularly checking your blood, your study doctor can see the effect of medication on your general well-being. Should the results of these blood tests change significantly during the study, your study doctor will inform both you and your GP.
ii) To try to identify genes and markers that may be linked to MS and cannabinoids: This sample (approximately four teaspoons) will be collected from you at the start of the study and at the end of each year (four samples in all). We hope that identifying genes and markers linked to MS may lead to a better understanding of why the condition develops and possibly lead to improved treatments. There are more details about this research in Part 2.
MRI scans
At your study centre, all participants will undergo regular MRI (Magnetic Resonance Imaging) brain scans at the start of the trial, and then at the end of each study year (four scans in total). The scans will usually be performed at the hospital where your study clinics are held and each scan takes approximately 20 minutes. If you are claustrophobic (have a fear of enclosed spaces), or have had any metallic implants (such as a cardiac pacemaker) you will not be able to have a scan and will unfortunately not be able to join the study. Orthopaedic metal is usually safe to scan but it is important to tell your study doctor if you have ever had any metal implanted. You may have had an MRI scan in the past, but please ask your study doctor or nurse for further information about the procedure if necessary.
Study diary and identity card
Each participant will be given a study diary. This contains useful information about storing and taking study medication and also serves as an appointment card. The blank pages can be used to write down any changes in your MS or other symptoms, or to record visits to your GP, specialist nurse etc. This will help when filling in your study questionnaires or talking to your study doctor.
You will also be given an identity card (the size of a credit card) which should be carried at all times. The card states that the holder is taking part in a clinical trial and provides a contact telephone number at the trial co-ordinating centre in case of query or emergency.
What are the possible risks or disadvantages of taking part?
The study involves attending a study clinic every six months, usually in addition to any other hospital appointments you may have. If you live a long way from your study centre, or if you can foresee any other difficulties in taking part in the study, please consider these carefully before agreeing to take part.
Cannabis can cause an increase in heart rate and a reduction in blood pressure. For this reason, people with a history of heart disease will not be permitted to take part in the study. There may be other illnesses that might exclude you from participation in the study, which your study doctor will check before you are accepted into the study.
The House of Lords Select Committee assessed the safety of cannabis in 1998 and found that no-one has ever died as a direct result of recreational or medicinal use of cannabis. It is generally thought that cannabis may possibly be mildly addictive but in our previous study we found no evidence of withdrawal syndrome in participants after twelve months of treatment. There is no convincing evidence for any long term toxic effects of cannabis.
During the collection of blood samples, you may experience pain and/or bruising where the blood has been taken. Infection or blood clots are a very rare complication. Some people feel faint or light headed when they have blood taken. If this happens to you, you should lie down prior to having the blood taken to avoid falling and causing injury to yourself.
What side effects may I experience?
Possible side effects of cannabis may include a “high” feeling, although in previous studies some patients taking the placebo drug have also experienced this. Other possible symptoms include increased appetite, weakness, dry mouth, dizziness and poor concentration.
Any new symptoms should be discussed with the study doctor at your clinic visits. If you have any queries between visits you should also contact your study doctor, or you can telephone the Freephone study helpline [telephone number supplied] for advice. If necessary, the dose of your treatment can be adjusted to reduce unwanted side effects. Please note, though, that you may not notice any side effects at all.
What are the benefits of taking part?
You have two chances in three of receiving an active cannabis treatment that may help to slow down progression of your disability, although improvement cannot be guaranteed. By taking part, you will be helping in an important study to decide the usefulness of THC as a treatment for multiple sclerosis in the future.
Will I have to pay for travel?
No. We will pay any travel or parking expenses incurred as a result of attending study visits at a fixed rate per mile for use of your own vehicle or the full amount if using public transport (bus, taxi or train), provided that receipts or tickets are produced.
Will the information collected during the study be kept confidential?
Yes. All the information collected about you during the study will remain strictly confidential. The details are included in Part 2.
Contact for further information
If you have any questions, please do not hesitate to contact your local study team (Local Contact Details). Alternatively, you may contact the co-ordinating centre in Plymouth on [telephone number supplied] (Freephone) or visit the study website at [web address supplied].
This completes Part 1 of the Information Sheet. If you are considering taking part in the study, please read the information in Part 2 before making any decision.
PART 2
What if new information about cannabis-based medicines becomes available?
If new and relevant information becomes available during the course of the study, we will tell you and give you the opportunity to discuss whether you want to continue in the study.
What happens if I don’t want to carry on with the study?
If you decide to withdraw from the study at any time we will still need to use the information collected up to this time.
What if something goes wrong?
The study organisers do not believe that you will suffer any injury by participating in this study so there are no special compensation arrangements. If you have any concerns about the way that you have been approached or treated during this study, you are free to follow the usual NHS complaints procedure. If you are harmed due to someone’s negligence then you may have grounds for legal action but you may have to pay for this yourself. Your right to claim for compensation for injury where you can prove negligence is not affected.
Will the information collected during the study be kept confidential?
The study will be conducted in accordance with the Data Protection Act (1998). All information collected about you during the study will remain strictly confidential.
Your name and address will be held by the co-ordinating office in Plymouth so that study questionnaires can be mailed directly to you. Your personal details will be stored securely on a computer, accessible only by members of the co-ordinating team. Your name and address will not appear on any study forms or questionnaires so that you cannot be recognised from them. All other information collected about you during this study will be entered onto a separate, secure database at the co-ordinating centre and will only be identifiable by a study number and initials. Only members of the co-ordinating team will have direct access to these data.
If you consent to take part in the study, your medical records may be inspected by the doctors looking after you and by members of the co-ordinating team responsible for monitoring the safe conduct of the study. During the study an audit may be carried out by responsible members of either the Medical Research Council, the study sponsor (Plymouth Hospitals NHS Trust) or the Medicines and Healthcare Products Regulatory Agency (MHRA) who would also have access to your medical records for this purpose.
Anonymised data collected during the study may be transferred to the supplier of study medication outside the European Economic Area. This information may be used in applications to gain a marketing authorisation for study medication. Some countries outside Europe may not have laws which protect your privacy to the same extent as the Data Protection Act in the UK or European Law, however no-one will be able to identify you from the data transferred.
If you agree to take part we will inform your general practitioner, unless you specifically ask us not to.
What about pregnancy?
Cannabis may disrupt reproductive hormones. If you or your partner usually rely on a hormonal form of contraception (the contraceptive pill or an injectable or implanted contraceptive), you must also use a barrier method of contraception (condom or diaphragm with spermicidal jelly) to prevent pregnancy occurring to you or your partner during the study period. No extra precautions are required if you are post-menopausal (women) or have been surgically sterilised (men or women), if you have an intra-uterine contraceptive device (coil) in place or you abstain from intercourse for the study period. If you or your partner becomes pregnant during the study, please inform your study doctor as soon as possible so that appropriate advice can be given.
What will happen to my blood samples?
-
Your samples will be stored by Professor John Zajicek at the Peninsula Medical School in Exeter.
-
Your name will not appear anywhere on the samples. Only your initials and a study number will be used.
-
Your blood will not be tested for any inherited diseases.
-
Your samples will only be used for research about MS and cannabinoids. No other research can be performed on your samples without your permission.
-
Samples will be stored indefinitely. This is because new scientific techniques related to MS and the relationship of cannabinoids to MS may emerge in the future, allowing further research that was not possible at the time of collection.
-
Future work may involve collaborations with other academic groups outside the Peninsula Medical School. If this occurs, no personal details or information that could identify you would be shared with these groups.
-
You will not derive any direct benefit from this part of our research nor will you be informed of any results.
-
If at any time you decide to withdraw your consent for participation in the CUPID study, you may also request that any samples already collected are destroyed.
Will I be able to find out how other participants are getting on?
About 500 people will be recruited to the study from all over UK. At your study visits, you may meet other people who are taking part in the study. We encourage you not to discuss your progress with each other because there is a chance that this could affect the outcome of the study. For the same reason, we cannot give you any specific details about how people are getting on in other parts of UK. However, we can give you some general information about the study, in the form of a newsletter, including updates about recruitment.
What happens at the end of the study?
At the end of the study, you will have to stop taking your study medication and return any unused medication to your study centre. We will plan a programme to reduce your study medication gradually after your last visit. We hope that it may be possible to offer active trial medication for a fixed period to all participants who have completed the study, whilst we are preparing the results, but this cannot be guaranteed. Whether or not the study medication then becomes available on prescription after the study will depend upon the study results and the decision of the relevant authorities.
What will happen to the results of the study?
We intend to publish the study results in a medical journal approximately 9 months after the last participant completes the trial. Each participant will receive a summary of the results at the time of publication and this summary will also be posted on the website (www.pms.ac.uk/cnrg/cupid). If you want to find out what treatment you were taking during the study, you will be able to do so, but only after the last participant completes the trial. It may take up to five years for all participants to complete the study, so you may have to wait a while.
Who is organising and funding the study?
The study is being conducted by the Clinical Neurology Research Group at the Peninsula Medical School (Universities of Exeter and Plymouth) in conjunction with the Department of Mathematics and Statistics, University of Plymouth. The study is funded by the Medical Research Council with support from the MS Society and the MS Trust. The study has been approved by the South and West Devon Research Ethics Committee.
Thank you for considering taking part in this study. You may also find it helpful to read the enclosed leaflet “Medical Research and You”, published by Consumers for Ethics in Research (CERES). The leaflet gives more information about medical research and considers some questions you may want to ask.
Appendix 5 Cannabinoid Use in Progressive Inflammatory brain Disease trial organisation
Trial Steering Committee
The TSC members were Professor Nigel Leigh, Professor of Neurology, Brighton and Sussex Medical School, University of Sussex (chairperson); Dr Carl Counsell, Reader in Neurology, University of Aberdeen; Professor David Jones, Professor of Medical Statistics, University of Leicester; Professor Andrew Nunn, Associate Director & Chair of Infections Research Theme, MRC Clinical Trials Unit, London; Ms Nicola Russell, Director of Special Projects, Multiple Sclerosis Trust; and Professor Alan Thompson, Professor of Clinical Neurology and Neurorehabilitation, University College London.
Independent Data Monitoring Committee
The IDMC members were Professor Richard Grey, Professor of Medical Statistics, Clinical Trial Service Unit, University of Oxford (chairperson); Professor Ian Bone, Formerly Consultant Neurologist, Institute of Neurological Sciences, University of Glasgow; and Professor Michael Hutchinson, Consultant Neurologist, University College Dublin.
Appendix 6 Patient and public involvement in the Cannabinoid Use in Progressive Inflammatory brain Disease study
The CUPID study was developed as a consequence of findings from our previous CAMS study2,3 which supported the theory that cannabis-based medicines might have a neuroprotective effect in progressive MS. On completion of the CAMS study, feedback about the experience of clinical trial participation was obtained from semistructured interviews with 10 study participants and an end-of-trial postal survey with more than 500 respondents. As the target population (people with MS) of the two studies were similar, findings from the post-CAMS survey were used to inform certain aspects of the design and conduct of the CUPID study.
Specifically, previous study participants reported that physical limitations led to practical difficulties in completing study questionnaires, with a significant proportion signifying a willingness or preference to have completed web-based questionnaires instead. As one of the joint primary outcomes in the CUPID study was a patient-reported measure, both postal and web-based options for self-report instruments were included in this study, with additional telephone support for questionnaire completion from the co-ordinating centre.
The treatment and follow-up period for the CAMS study was only 1 year and from participant feedback it was clear that one of the challenges of the CUPID study would be to keep participants with accumulating disability engaged and involved for 3 years. Various strategies were therefore introduced from the start of the CUPID study to optimise participant retention and data completeness. These included regular participant newsletters, practical and financial support for travel and parking for study visits and a 24-hour access Freephone helpline. In participants for whom the study became burdensome or who developed ‘trial fatigue’, a policy of reducing the number or frequency of questionnaires and/or visits was adopted, with an emphasis on maximising completeness of primary outcome data.
During the set-up and development of the CUPID study, members of the MRC consumer group reviewed and commented upon the draft participant information sheet. Local people with MS and lay volunteers also commented on the readability and acceptability of the study information and self-completion questionnaires. Members of a local MS support group undertook the specific task of reviewing the resource use questionnaire for the health economic component of the study. Lay membership of the TSC comprised representatives from the two major UK MS charities (the MS Trust and the MS Society), one as a full member and one as an observer. Apart from supporting and publicising the study, these representatives made an important contribution to TSC meetings on behalf of the MS community and provided advice on the interpretation and dissemination of the study results.
Patient and public involvement in the CUPID study contributed to various aspects of study conduct including the development of accessible study information and high-quality documentation, the introduction of a number of strategies to promote participant retention and data completeness and representation of the interests of people with MS by inclusion of relevant TSC members.
Appendix 7 End Point Committee terms of reference
Cannabinoid Use in Progressive Inflammatory brain Disease study End Point Committee
Terms of reference
Background
Primary outcomes
As described in the study protocol, the CUPID study has two joint primary outcome measures:
-
Physician-based EDSS: time to EDSS progression of at least 1 point from a baseline EDSS score of 4.0, 4.5 or 5.0 or at least 0.5 points from a baseline EDSS score ≥ 5.5. Once identified, deterioration must be confirmed at the next scheduled 6-monthly visit.
-
Patient-based MSIS-29 physical impact scale version 2: overall mean change from baseline to end of study.
For the purpose of any licensing application for treatment effects on progression, the EDSS score will be considered as the single primary outcome measure. The MSIS-29v2 will be treated as secondary.
Missing outcome data
In a proportion of study participants, some of the eight or nine scheduled EDSS scores are missing as a result of non-attendance at individual visits (e.g. through illness), study site organisational problems (e.g. lack of assessor) or complete loss to follow-up. As a result, in these participants it may not be possible to judge whether or not their MS has progressed according to the protocol definition.
Collection of additional clinical details
For some participants in whom EDSS scores are missing, supplementary clinical details have been collected from hospital records in order to help estimate EDSS score for secondary analysis purposes. All such supplementary information has been obtained within the limits of REC approval and the informed consent process. Hospital notes of participants with missing EDSS scores were examined during routine site monitoring visits. Where these records contained either direct entries relating to inpatient or outpatient episodes or documentary evidence in the form of clinical correspondence relating to the period(s) for which EDSS data were missing, relevant information was transcribed (verbatim where possible) on to purpose-designed forms. Information acquired in this way was largely, but not exclusively, related to the participant’s mobility in order to facilitate estimation of EDSS. Dates of original entries or correspondence were recorded and all data collection forms were signed and dated by the person responsible for recording the data.
End Point Committee
Composition
The EPC will comprise Professor John Zajicek (chief investigator), Professor Jeremy Hobart (Professor of Clinical Neurology and Health Measurement, Peninsula College of Medicine & Dentistry) and Dr Timothy Harrower (independent chairperson, Consultant Neurologist, Royal Devon & Exeter NHS Foundation Trust).
Aim
The EPC has been set up with the aim of adjudicating on MS progression in participants for whom confirmation of this end point is not clear due to missing EDSS scores. In accordance with the wishes of the TSC, decisions made by the EPC will be included in a secondary analysis, rather than the primary analysis of study data.
Scope
It is not necessary or expected that the EPC will review EDSS end points for every trial participant. The committee will only be responsible for judging EDSS progression (and/or the estimated timing of any such progression) in participants for whom missing data renders this unclear.
At the study end, 95 participants have missing EDSS scores such that either (i) disease progression cannot be confirmed or (ii) the onset date of confirmed progression is unclear. The committee will not be required to adjudicate in the case of 57 of these participants who have been irretrievably lost to follow-up (e.g. moved house or died) and/or for whom no additional clinical information is available (these cases have been scrutinised by the Trial Co-ordinator and decisions verified by the Assistant Trial Co-ordinator). In X of the remaining 38 cases, the committee will use the available additional clinical information to consider whether or not progression can be confirmed. In Y cases where progression is confirmed but the onset date is uncertain, the committee will review the available information in an effort to make a realistic estimate of the date of onset of progression.
Process
It is anticipated that the committee will meet once only. Members will be provided with a summary sheet for each participant being reviewed and, blinded to the treatment allocation, will consider all available study EDSS scores and any supplementary clinical information available from the notes review process. Any information which might unblind (e.g. trial/centre number or details of adverse events) will be removed before data are presented to the committee. Where possible, dates of recorded clinical information will be provided for comparison with scheduled study time points and existing EDSS score dates. An audit trail providing a record of the additional information source, date sourced and person responsible for acquiring/providing the information is available separately.
On the basis of information provided, the committee may decide that a participant should be:
-
Treated as having progressed at a particular time within the 3-year follow-up period.
-
Treated as if there was no MS progression during the 3 years of follow-up.
-
Treated as progression unknown/lost to follow-up.
In view of the complexity of the disease area and the features of the EDSS scale it is not possible to stipulate what level of evidence would be sufficient to be convincing of disease progression or to give specific guidance regarding the criteria for decision-making. Members of the EPC will use their clinical judgement and, in each case, the final decision of the EPC will be based on agreement of all three members. Decisions will be documented on the individual summary sheet provided, signed and dated by the committee Chair. These data will then be incorporated into the final study database.
Appendix 8 Economic evaluation
Detailed NHS/personal and social services resource use by treatment group and follow-up
| Resource use item | Active | Placebo | ||||
|---|---|---|---|---|---|---|
| Range | Mean | SD | Range | Mean | SD | |
| GP visits | ||||||
| Baseline | 0–15 | 2.18 | 1.98 | 0–20 | 2.39 | 2.82 |
| 4.5 months | 0–15 | 1.52 | 2.00 | 0–8 | 1.38 | 1.61 |
| 9 months | 0–12 | 1.34 | 1.74 | 0–9 | 1.31 | 1.64 |
| 15 months | 0–25 | 1.73 | 2.39 | 0–6 | 1.54 | 1.50 |
| 21 months | 0–36 | 1.89 | 3.07 | 0–18 | 1.68 | 2.41 |
| 27 months | 0–35 | 1.94 | 3.06 | 0–16 | 2.05 | 2.42 |
| 33 months | 0–28 | 1.81 | 2.71 | 0–15 | 1.64 | 2.15 |
| Total | 0–145 | 10.72 | 13.64 | 0–55 | 9.00 | 8.42 |
| Community nurse | ||||||
| Baseline | 0–10 | 0.35 | 1.13 | 0–24 | 0.66 | 2.35 |
| 4.5 months | 0–6 | 0.23 | 0.77 | 0–7 | 0.31 | 0.87 |
| 9 months | 0–8 | 0.18 | 0.79 | 0–4 | 0.16 | 0.56 |
| 15 months | 0–78 | 0.84 | 5.42 | 0–15 | 0.35 | 1.48 |
| 21 months | 0–12 | 0.43 | 1.46 | 0–20 | 0.54 | 2.02 |
| 27 months | 0–10 | 0.47 | 1.30 | 0–24 | 0.60 | 2.53 |
| 33 months | 0–160 | 1.70 | 11.33 | 0–21 | 0.51 | 2.07 |
| Total | 0–37 | 2.87 | 6.05 | 0–49 | 0.51 | 6.87 |
| MS nurse | ||||||
| Baseline | 0–8 | 0.81 | 1.09 | 0–6 | 0.81 | 1.11 |
| 4.5 months | 0–8 | 0.68 | 1.49 | 0–5 | 0.57 | 1.09 |
| 9 months | 0–4 | 0.34 | 0.67 | 0–11 | 0.39 | 1.17 |
| 15 months | 0–4 | 0.42 | 0.66 | 0–3 | 0.41 | 0.71 |
| 21 months | 0–6 | 0.48 | 0.83 | 0–3 | 0.32 | 0.57 |
| 27 months | 0–7 | 0.51 | 0.89 | 0–7 | 0.51 | 0.93 |
| 33 months | 0–8 | 0.51 | 0.95 | 0–6 | 0.54 | 1.03 |
| Total | 0–12 | 3.13 | 3.20 | 0–13 | 2.72 | 2.66 |
| Physiotherapist | ||||||
| Baseline | 0–40 | 2.19 | 4.34 | 0–24 | 2.34 | 3.99 |
| 4.5 months | 0–24 | 1.50 | 3.21 | 0–12 | 1.35 | 2.54 |
| 9 months | 0–26 | 1.32 | 2.94 | 0–45 | 1.44 | 4.77 |
| 15 months | 0–52 | 2.14 | 5.19 | 0–26 | 1.62 | 3.57 |
| 21 months | 0–30 | 1.98 | 4.17 | 0–15 | 1.30 | 2.64 |
| 27 months | 0–50 | 1.62 | 4.75 | 0–12 | 1.18 | 2.34 |
| 33 months | 0–40 | 2.19 | 5.23 | 0–19 | 1.39 | 3.15 |
| Total | 0–69 | 9.99 | 13.86 | 0–58 | 8.33 | 11.31 |
| Rehabilitation | ||||||
| Baseline | 0–5 | 0.15 | 0.62 | 0–2 | 0.11 | 0.37 |
| 4.5 months | 0–14 | 0.18 | 1.15 | 0–6 | 0.13 | 0.61 |
| 9 months | 0–2 | 0.09 | 0.32 | 0–8 | 0.13 | 0.75 |
| 15 months | 0–4 | 0.15 | 0.51 | 0–8 | 0.15 | 0.77 |
| 21 months | 0–3 | 0.15 | 0.49 | 0–4 | 0.18 | 0.64 |
| 27 months | 0–3 | 0.12 | 0.42 | 0–3 | 0.08 | 0.38 |
| 33 months | 0–12 | 0.23 | 1.08 | 0–6 | 0.19 | 0.79 |
| Total | 0–18 | 0.92 | 2.48 | 0–11 | 0.67 | 1.62 |
| Occupational therapist | ||||||
| Baseline | 0–8 | 0.43 | 0.94 | 0–6 | 0.58 | 1.19 |
| 4.5 months | 0–12 | 0.41 | 1.19 | 0–4 | 0.24 | 0.61 |
| 9 months | 0–6 | 0.37 | 0.90 | 0–21 | 0.45 | 1.99 |
| 15 months | 0–6 | 0.46 | 0.99 | 0–6 | 0.51 | 1.17 |
| 21 months | 0–7 | 0.43 | 0.98 | 0–6 | 0.53 | 1.00 |
| 27 months | 0–20 | 0.47 | 1.63 | 0–12 | 0.43 | 1.42 |
| 33 months | 0–10 | 0.53 | 1.51 | 0–16 | 0.45 | 1.64 |
| Total | 0–31 | 2.51 | 4.18 | 0–29 | 2.27 | 3.95 |
| Speech therapy | ||||||
| Baseline | 0–4 | 0.09 | 0.48 | 0–4 | 0.08 | 0.46 |
| 4.5 months | 0–4 | 0.06 | 0.35 | 0–3 | 0.04 | 0.28 |
| 9 months | 0–2 | 0.03 | 0.20 | 0–2 | 0.05 | 0.29 |
| 15 months | 0–4 | 0.06 | 0.35 | 0–3 | 0.07 | 0.36 |
| 21 months | 0–4 | 0.09 | 0.44 | 0–2 | 0.04 | 0.24 |
| 27 months | 0–7 | 0.06 | 0.50 | 0–3 | 0.07 | 0.36 |
| 33 months | 0–8 | 0.51 | 1.94 | 0–2 | 0.34 | 1.21 |
| Total | 0–16 | 0.51 | 1.94 | 0–6 | 0.34 | 1.21 |
| Neurologist | ||||||
| Baseline | 0–6 | 0.83 | 0.93 | 0–5 | 0.94 | 1.01 |
| 4.5 months | 0–8 | 0.96 | 1.73 | 0–7 | 0.98 | 1.52 |
| 9 months | 0–3 | 0.34 | 0.55 | 0–4 | 0.43 | 0.70 |
| 15 months | 0–6 | 0.50 | 0.77 | 0–2 | 0.44 | 0.56 |
| 21 months | 0–11 | 0.47 | 0.93 | 0–3 | 0.47 | 0.60 |
| 27 months | 0–9 | 0.43 | 0.81 | 0–3 | 0.49 | 0.64 |
| 33 months | 0–6 | 0.47 | 0.77 | 0–2 | 0.43 | 0.56 |
| Total | 0–24 | 3.29 | 4.02 | 0–15 | 3.30 | 3.23 |
| Psychologist | ||||||
| Baseline | 0–3 | 0.04 | 0.28 | 0–10 | 0.08 | 0.82 |
| 4.5 months | 0–3 | 0.02 | 0.21 | 0–3 | 0.05 | 0.33 |
| 9 months | 0–3 | 0.06 | 0.34 | 0–3 | 0.07 | 0.40 |
| 15 months | 0–7 | 0.07 | 0.52 | 0–4 | 0.05 | 0.38 |
| 21 months | 0–14 | 0.19 | 1.22 | 0–3 | 0.09 | 0.41 |
| 27 months | 0–6 | 0.06 | 0.45 | 0–1 | 0.03 | 0.18 |
| 33 months | 0–4 | 0.06 | 0.42 | 0–8 | 0.09 | 0.78 |
| Total | 0–15 | 0.45 | 1.71 | 0–8 | 0.39 | 1.20 |
| Chiropodist | ||||||
| Baseline | 0–7 | 0.17 | 0.69 | 0–4 | 0.17 | 0.60 |
| 4.5 months | 0–5 | 0.16 | 0.60 | 0–2 | 0.08 | 0.32 |
| 9 months | 0–6 | 0.14 | 0.57 | 0–5 | 0.21 | 0.71 |
| 15 months | 0–5 | 0.17 | 0.67 | 0–5 | 0.21 | 0.71 |
| 21 months | 0–6 | 0.17 | 0.69 | 0–6 | 0.28 | 0.84 |
| 27 months | 0–7 | 0.22 | 0.87 | 0–4 | 0.23 | 0.63 |
| 33 months | 0–5 | 0.21 | 0.72 | 0–7 | 0.36 | 1.05 |
| Total | 0–32 | 1.26 | 4.10 | 0–12 | 1.07 | 2.48 |
| Optician | ||||||
| Baseline | 0–3 | 0.18 | 0.45 | 0–4 | 0.24 | 0.56 |
| 4.5 months | 0–3 | 0.18 | 0.44 | 0–4 | 0.18 | 0.54 |
| 9 months | 0–6 | 0.16 | 0.54 | 0–3 | 0.12 | 0.39 |
| 15 months | 0–4 | 0.14 | 0.46 | 0–1 | 0.11 | 0.32 |
| 21 months | 0–4 | 0.17 | 0.52 | 0–2 | 0.19 | 0.44 |
| 27 months | 0–2 | 0.20 | 0.44 | 0–2 | 0.17 | 0.42 |
| 33 months | 0–2 | 0.14 | 0.38 | 0–2 | 0.16 | 0.43 |
| Total | 0–7 | 1.01 | 1.37 | 0–5 | 0.91 | 1.32 |
| Continence advice | ||||||
| Baseline | 0–6 | 0.32 | 0.84 | 0–6 | 0.47 | 1.08 |
| 4.5 months | 0–4 | 0.29 | 0.70 | 0–3 | 0.27 | 0.66 |
| 9 months | 0–17 | 0.24 | 1.16 | 0–5 | 0.27 | 0.79 |
| 15 months | 0–8 | 0.26 | 0.79 | 0–3 | 0.29 | 0.67 |
| 21 months | 0–4 | 0.23 | 0.66 | 0–3 | 0.25 | 0.63 |
| 27 months | 0–5 | 0.35 | 0.88 | 0–4 | 0.30 | 0.74 |
| 33 months | 0–3 | 0.24 | 0.62 | 0–3 | 0.29 | 0.66 |
| Total | 0–20 | 1.77 | 3.08 | 0–9 | 1.35 | 2.13 |
| Social worker | ||||||
| Baseline | 0–4 | 0.15 | 0.59 | 0–4 | 0.08 | 0.40 |
| 4.5 months | 0–4 | 0.11 | 0.54 | 0–4 | 0.17 | 0.62 |
| 9 months | 0–4 | 0.09 | 0.45 | 0–1 | 0.04 | 0.19 |
| 15 months | 0–3 | 0.12 | 0.46 | 0–5 | 0.17 | 0.73 |
| 21 months | 0–26 | 0.25 | 1.79 | 0–2 | 0.07 | 0.29 |
| 27 months | 0–26 | 0.24 | 1.80 | 0–4 | 0.13 | 0.56 |
| 33 months | 0–2 | 0.14 | 0.47 | 0–3 | 0.14 | 0.53 |
| Total | 0–56 | 0.99 | 4.65 | 0–10 | 0.73 | 1.76 |
| Acupuncturist | ||||||
| Baseline | 0–7 | 0.06 | 0.52 | 0–8 | 0.13 | 0.91 |
| 4.5 months | 0–2 | 0.02 | 0.15 | 0–6 | 0.08 | 0.58 |
| 9 months | 0–1 | 0.01 | 0.09 | 0–5 | 0.10 | 0.59 |
| 15 months | 0–3 | 0.01 | 0.19 | 0–6 | 0.11 | 0.72 |
| 21 months | 0–6 | 0.06 | 0.51 | 0–1 | 0.01 | 0.09 |
| 27 months | 0–8 | 0.05 | 0.56 | 0–15 | 0.13 | 1.37 |
| 33 months | 0–6 | 0.04 | 0.42 | 0–9 | 0.08 | 0.83 |
| Total | 0–13 | 0.26 | 1.30 | 0–34 | 0.63 | 3.89 |
| Home care hours | ||||||
| Baseline | 0–546 | 8.06 | 58.76 | 0–1014 | 19.82 | 119.73 |
| 4.5 months | 0–377 | 6.22 | 42.60 | 0–2028 | 34.40 | 228.75 |
| 9 months | 0–1456 | 29.78 | 176.00 | 0–2496 | 40.90 | 278.66 |
| 15 months | 0–676 | 12.34 | 77.28 | 0–2340 | 45.02 | 266.21 |
| 21 months | 0–611 | 13.30 | 70.56 | 0–2288 | 53.84 | 274.30 |
| 27 months | 0–897 | 16.70 | 98.24 | 0–2119 | 61.99 | 295.61 |
| 33 months | 0–1378 | 26.30 | 146.20 | 0–1664 | 77.24 | 287.32 |
| Total | 0–4635.80 | 103.98 | 540.06 | 0–11,765 | 313.40 | 1419.23 |
| Day-care episodes | ||||||
| Baseline | 0–40 | 0.16 | 2.33 | 0–26 | 0.25 | 2.34 |
| 4.5 months | 0–14 | 0.10 | 1.15 | 0–18 | 0.13 | 1.54 |
| 9 months | 0–32 | 0.14 | 2.01 | 0–135 | 1.03 | 11.80 |
| 15 months | 0–40 | 0.30 | 2.86 | 0–48 | 0.38 | 4.28 |
| 21 months | 0–52 | 0.31 | 3.47 | 0–26 | 0.48 | 3.10 |
| 27 months | 0–78 | 0.48 | 5.54 | 0–10 | 0.12 | 0.95 |
| 33 months | 0–26 | 0.12 | 1.75 | 0–52 | 0.59 | 5.05 |
| Total | 0–202 | 1.92 | 16.56 | 0–61 | 2.05 | 9.48 |
| Respite episodes | ||||||
| Baseline | 0–20 | 0.07 | 1.11 | 0–2 | 0.02 | 0.17 |
| 4.5 months | 0–1 | 0.02 | 0.15 | 0–1 | 0.01 | 0.09 |
| 9 months | 0–8 | 0.04 | 0.51 | 0–1 | 0.02 | 0.12 |
| 15 months | 0–8 | 0.05 | 0.55 | 0–2 | 0.03 | 0.22 |
| 21 months | 0–5 | 0.03 | 0.36 | 0–2 | 0.03 | 0.21 |
| 27 months | 0–1 | 0.02 | 0.13 | 0–2 | 0.02 | 0.18 |
| 33 months | 0–2 | 0.05 | 0.25 | 0–2 | 0.03 | 0.22 |
| Total | 0–16 | 0.30 | 1.58 | 0–3 | 0.15 | 0.52 |
| Hospital episodes | ||||||
| Baseline | Not collected | |||||
| 4.5 months | 0–2 | 0.05 | 0.22 | 0–1 | 0.02 | 0.15 |
| 9 months | 0–2 | 0.07 | 0.28 | 0–1 | 0.02 | 0.13 |
| 15 months | 0–2 | 0.05 | 0.22 | 0–2 | 0.05 | 0.24 |
| 21 months | 0–3 | 0.07 | 0.29 | 0–3 | 0.05 | 0.30 |
| 27 months | 0–2 | 0.07 | 0.27 | 0–2 | 0.08 | 0.29 |
| 33 months | 0–2 | 0.07 | 0.26 | 0–2 | 0.05 | 0.24 |
| Total | 0–5 | 0.37 | 0.84 | 0–10 | 0.27 | 0.93 |
| Resource use item | Active | Placebo | ||||
|---|---|---|---|---|---|---|
| Range | Mean | SD | Range | Mean | SD | |
| GP | ||||||
| Baseline | 0–540 | 78.44 | 71.32 | 0–720 | 86.13 | 101.46 |
| 4.5 months | 0–540 | 54.56 | 71.88 | 0–288 | 49.76 | 57.98 |
| 9 months | 0–432 | 48.09 | 62.52 | 0–324 | 46.99 | 58.88 |
| 15 months | 0–900 | 62.45 | 85.92 | 0–216 | 55.43 | 54.00 |
| 21 months | 0–1296 | 68.00 | 110.35 | 0–648 | 60.52 | 86.84 |
| 27 months | 0–1260 | 69.87 | 110.10 | 0–576 | 73.80 | 87.17 |
| 33 months | 0–1008 | 65.19 | 97.71 | 0–540 | 58.88 | 77.58 |
| Total | 0–5220 | 386.04 | 490.91 | 0–1980 | 324.00 | 303.10 |
| Clinic nurse | ||||||
| Baseline | 0–210 | 7.22 | 25.33 | 0–360 | 13.90 | 49.68 |
| 4.5 months | 0–120 | 4.53 | 16.43 | 0–120 | 5.51 | 16.51 |
| 9 months | 0–180 | 3.93 | 16.89 | 0–60 | 3.32 | 10.57 |
| 15 months | 0–2340 | 22.35 | 161.46 | 0–450 | 7.74 | 41.47 |
| 21 months | 0–360 | 9.68 | 37.28 | 0–300 | 9.83 | 32.21 |
| 27 months | 0–180 | 10.16 | 29.76 | 0–360 | 10.75 | 40.00 |
| 33 months | 0–4800 | 48.11 | 339.81 | 0–315 | 9.28 | 34.53 |
| Total | 0–1095 | 68.58 | 165.76 | 0–735 | 45.37 | 112.22 |
| MS nurse | ||||||
| Baseline | 0–228 | 21.82 | 30.96 | 0–228 | 21.76 | 31.34 |
| 4.5 months | 0–200 | 17.31 | 37.48 | 0–152 | 15.01 | 29.02 |
| 9 months | 0–114 | 8.80 | 18.07 | 0–275 | 10.43 | 30.48 |
| 15 months | 0–100 | 11.04 | 17.76 | 0–114 | 11.66 | 21.75 |
| 21 months | 0–150 | 12.96 | 22.78 | 0–76 | 8.76 | 15.91 |
| 27 months | 0–266 | 14.67 | 28.52 | 0–175 | 14.66 | 27.21 |
| 33 months | 0–200 | 14.24 | 27.35 | 0–150 | 14.55 | 27.32 |
| Total | 0–354 | 82.97 | 85.75 | 0–325 | 72.74 | 69.50 |
| Physiotherapist | ||||||
| Baseline | 0–1360 | 79.46 | 157.63 | 0–816 | 82.95 | 141.31 |
| 4.5 months | 0–940 | 55.25 | 120.82 | 0–564 | 49.19 | 96.82 |
| 9 months | 0–884 | 47.55 | 102.63 | 0–1530 | 51.53 | 164.28 |
| 15 months | 0–1768 | 78.53 | 184.07 | 0–940 | 59.28 | 134.72 |
| 21 months | 0–1410 | 74.75 | 162.13 | 0–564 | 47.97 | 99.21 |
| 27 months | 0–1700 | 60.51 | 176.92 | 0–470 | 41.68 | 84.59 |
| 33 months | 0–1360 | 81.05 | 194.45 | 0–893 | 51.00 | 121.80 |
| Total | 0–2346 | 368.16 | 492.90 | 0–1972 | 300.00 | 397.08 |
| Rehabilitation | ||||||
| Baseline | 0–170 | 5.25 | 20.92 | 0–68 | 3.75 | 12.53 |
| 4.5 months | 0–476 | 6.19 | 39.13 | 0–204 | 4.53 | 20.69 |
| 9 months | 0–68 | 2.92 | 10.88 | 0–272 | 4.41 | 25.44 |
| 15 months | 0–136 | 5.13 | 17.33 | 0–272 | 5.17 | 26.29 |
| 21 months | 0–102 | 4.94 | 16.53 | 0–136 | 6.16 | 21.78 |
| 27 months | 0–102 | 4.02 | 14.33 | 0–102 | 2.83 | 12.91 |
| 33 months | 0–408 | 7.66 | 36.79 | 0–204 | 6.63 | 26.77 |
| Total | 0–612 | 31.22 | 84.24 | 0–374 | 22.80 | 55.02 |
| Occupational therapist | ||||||
| Baseline | 0–272 | 14.59 | 31.85 | 0–204 | 19.61 | 40.32 |
| 4.5 months | 0–408 | 14.10 | 40.61 | 0–136 | 8.00 | 20.79 |
| 9 months | 0–204 | 12.75 | 30.56 | 0–714 | 15.31 | 67.50 |
| 15 months | 0–204 | 15.54 | 33.49 | 0–204 | 17.27 | 39.82 |
| 21 months | 0–238 | 14.68 | 33.44 | 0–204 | 17.88 | 33.99 |
| 27 months | 0–680 | 16.07 | 55.37 | 0–408 | 14.45 | 48.42 |
| 33 months | 0–340 | 18.07 | 51.21 | 0–544 | 15.27 | 55.81 |
| Total | 0–1054 | 85.53 | 142.10 | 0–986 | 77.12 | 134.21 |
| Speech therapy | ||||||
| Baseline | 0–141 | 3.67 | 18.26 | 0–136 | 2.95 | 16.54 |
| 4.5 months | 0–136 | 2.46 | 13.53 | 0–102 | 1.35 | 10.01 |
| 9 months | 0–68 | 1.11 | 6.94 | 0–68 | 2.02 | 10.48 |
| 15 months | 0–136 | 2.24 | 12.78 | 0–141 | 3.15 | 16.54 |
| 21 months | 0–136 | 3.20 | 15.83 | 0–94 | 1.91 | 11.03 |
| 27 months | 0–238 | 2.22 | 17.19 | 0–102 | 2.48 | 13.59 |
| 33 months | 0–272 | 3.60 | 23.21 | 0–68 | 2.35 | 11.41 |
| Total | 0–544 | 18.71 | 67.50 | 0–269 | 13.51 | 48.22 |
| Neurologist | ||||||
| Baseline | 0–870 | 119.94 | 135.08 | 0–725 | 136.99 | 146.12 |
| 4.5 months | 0–1160 | 139.38 | 250.27 | 0–1015 | 141.80 | 220.76 |
| 9 months | 0–435 | 49.84 | 80.07 | 0–580 | 61.98 | 101.79 |
| 15 months | 0–870 | 72.80 | 111.87 | 0–290 | 63.29 | 80.98 |
| 21 months | 0–1595 | 67.54 | 135.09 | 0–435 | 68.75 | 86.50 |
| 27 months | 0–1305 | 62.61 | 117.57 | 0–435 | 71.29 | 92.08 |
| 33 months | 0–870 | 67.93 | 111.76 | 0–290 | 62.67 | 81.49 |
| Total | 0–3480 | 477.86 | 582.64 | 0–2175 | 479.21 | 468.99 |
| Psychologist | ||||||
| Baseline | 0–405 | 5.83 | 37.85 | 0–1350 | 10.77 | 110.21 |
| 4.5 months | 0–405 | 2.62 | 27.81 | 0–405 | 6.95 | 44.46 |
| 9 months | 0–405 | 7.91 | 46.40 | 0–405 | 9.27 | 53.45 |
| 15 months | 0–945 | 9.37 | 70.12 | 0–540 | 6.43 | 50.82 |
| 21 months | 0–1890 | 25.38 | 164.92 | 0–405 | 11.64 | 55.07 |
| 27 months | 0–810 | 8.59 | 61.27 | 0–135 | 4.50 | 24.33 |
| 33 months | 0–540 | 8.51 | 56.80 | 0–1080 | 12.58 | 105.88 |
| Total | 0–2025 | 60.28 | 230.67 | 0–1080 | 52.68 | 162.60 |
| Chiropodist | ||||||
| Baseline | 0–217 | 5.46 | 22.63 | 0–141 | 5.62 | 20.43 |
| 4.5 months | 0–188 | 5.67 | 21.86 | 0–62 | 2.51 | 10.02 |
| 9 months | 0–186 | 4.48 | 18.19 | 0–155 | 6.63 | 22.09 |
| 15 months | 0–155 | 5.25 | 20.93 | 0–155 | 6.64 | 22.05 |
| 21 months | 0–186 | 5.64 | 22.90 | 0–186 | 8.55 | 26.04 |
| 27 months | 0–217 | 7.27 | 27.96 | 0–124 | 6.97 | 19.47 |
| 33 months | 0–155 | 6.85 | 22.84 | 0–217 | 11.43 | 32.71 |
| Total | 0–992 | 41.10 | 130.68 | 0–372 | 33.27 | 76.83 |
| Optician | ||||||
| Baseline | 0–60 | 3.64 | 8.92 | 0–80 | 4.79 | 11.30 |
| 4.5 months | 0–60 | 3.50 | 8.76 | 0–80 | 3.53 | 10.85 |
| 9 months | 0–120 | 3.28 | 10.85 | 0–60 | 2.44 | 7.85 |
| 15 months | 0–80 | 2.78 | 9.17 | 0–20 | 2.22 | 6.31 |
| 21 months | 0–80 | 3.42 | 10.41 | 0–40 | 3.79 | 8.71 |
| 27 months | 0–40 | 4.00 | 8.88 | 0–40 | 3.33 | 8.33 |
| 33 months | 0–40 | 2.79 | 7.69 | 0–40 | 3.22 | 8.66 |
| Total | 0–140 | 20.13 | 27.33 | 0–100 | 18.29 | 26.33 |
| Continence advisor | ||||||
| Baseline | 0–228 | 9.51 | 26.66 | 0–228 | 14.20 | 34.66 |
| 4.5 months | 0–152 | 8.48 | 21.50 | 0–114 | 8.04 | 20.74 |
| 9 months | 0–646 | 7.28 | 42.50 | 0–190 | 7.87 | 25.04 |
| 15 months | 0–304 | 7.59 | 25.81 | 0–114 | 8.17 | 20.17 |
| 21 months | 0–100 | 6.38 | 18.21 | 0–76 | 6.87 | 17.54 |
| 27 months | 0–152 | 9.75 | 24.99 | 0–100 | 7.93 | 19.42 |
| 33 months | 0–114 | 6.61 | 17.43 | 0–114 | 8.53 | 20.03 |
| Total | 0–760 | 50.15 | 93.94 | 0–342 | 39.07 | 64.26 |
| Social worker | ||||||
| Baseline | 0–848 | 32.72 | 125.00 | 0–848 | 16.91 | 84.87 |
| 4.5 months | 0–848 | 23.83 | 115.09 | 0–848 | 35.85 | 130.44 |
| 9 months | 0–848 | 18.22 | 95.83 | 0–212 | 8.09 | 40.78 |
| 15 months | 0–636 | 25.09 | 97.46 | 0–1060 | 37.02 | 154.23 |
| 21 months | 0–5512 | 52.55 | 379.75 | 0–424 | 14.62 | 60.77 |
| 27 months | 0–5512 | 50.11 | 381.10 | 0–848 | 28.27 | 119.59 |
| 33 months | 0–424 | 28.65 | 98.75 | 0–636 | 30.54 | 111.82 |
| Total | 0–11,872 | 210.67 | 986.47 | 0–2120 | 155.12 | 372.52 |
| Acupuncturist | ||||||
| Baseline | 0–238 | 2.20 | 17.71 | 0–272 | 4.59 | 31.04 |
| 4.5 months | 0–68 | 0.53 | 5.17 | 0–204 | 2.75 | 19.87 |
| 9 months | 0–34 | 0.27 | 3.00 | 0–170 | 3.37 | 20.16 |
| 15 months | 0–102 | 0.42 | 6.52 | 0–204 | 3.78 | 24.41 |
| 21 months | 0–204 | 1.89 | 17.29 | 0–34 | .29 | 3.16 |
| 27 months | 0–272 | 1.70 | 19.01 | 0–510 | 4.25 | 46.56 |
| 33 months | 0–204 | 1.38 | 14.22 | 0–306 | 2.88 | 28.32 |
| Total | 0–442 | 8.77 | 44.15 | 0–1156 | 21.56 | 132.44 |
| Home care | ||||||
| Baseline | 0–9828 | 145.14 | 1057.62 | 0–18,252 | 356.71 | 2155.09 |
| 4.5 months | 0–6786 | 111.96 | 766.82 | 0–36,504 | 619.24 | 4117.43 |
| 9 months | 0–26,208 | 536.13 | 3185.79 | 0–44,928 | 736.24 | 5015.79 |
| 15 months | 0–12,168 | 222.15 | 1390.97 | 0–42,120 | 810.44 | 4791.79 |
| 21 months | 0–10,998 | 239.33 | 1269.98 | 0–41,184 | 969.10 | 4937.43 |
| 27 months | 0–16,146 | 300.65 | 1768.36 | 0–38,142 | 1115.78 | 5320.96 |
| 33 months | 0–24,804 | 473.33 | 2523.55 | 0–29,952 | 1390.30 | 5171.76 |
| Total | 0–83,444 | 1871.71 | 9721.10 | 0–211,770 | 5641.11 | 25,545.98 |
| Day care | ||||||
| Baseline | 0–1440 | 5.89 | 84.04 | 0–936 | 9.06 | 84.41 |
| 4.5 months | 0–504 | 3.63 | 41.25 | 0–648 | 4.76 | 55.57 |
| 9 months | 0–1152 | 4.92 | 72.29 | 0–4860 | 37.10 | 424.62 |
| 15 months | 0–1440 | 10.87 | 102.80 | 0–1728 | 13.71 | 153.94 |
| 21 months | 0–1872 | 11.23 | 125.07 | 0–936 | 17.38 | 111.69 |
| 27 months | 0–2808 | 17.18 | 199.28 | 0–360 | 4.20 | 34.35 |
| 33 months | 0–936 | 4.22 | 62.82 | 0–1872 | 21.36 | 181.88 |
| Total | 0–7272 | 69.06 | 596.22 | 0–2196 | 73.76 | 341.43 |
| Respite care | ||||||
| Baseline | 0–20,100 | 68.24 | 1119.10 | 0–2010 | 18.50 | 175.58 |
| 4.5 months | 0–1005 | 23.37 | 151.76 | 0–1005 | 7.39 | 86.18 |
| 9 months | 0–8040 | 39.26 | 509.78 | 0–1005 | 15.34 | 123.70 |
| 15 months | 0–8040 | 53.33 | 554.62 | 0–2010 | 31.90 | 217.84 |
| 21 months | 0–5025 | 34.36 | 358.97 | 0–2010 | 25.99 | 207.92 |
| 27 months | 0–1005 | 18.27 | 134.58 | 0–2010 | 16.75 | 183.49 |
| 33 months | 0–2010 | 45.27 | 248.85 | 0–2010 | 34.07 | 225.00 |
| Total | 0–16080 | 303.40 | 1590.07 | 0–3015 | 147.07 | 526.66 |
| Hospital | ||||||
| Baseline | Not collected | |||||
| 4.5 months | 0–4526 | 57.19 | 361.84 | 0–2263 | 34.62 | 260.69 |
| 9 months | 0–3268 | 128.19 | 522.81 | 0–2263 | 26.05 | 207.67 |
| 15 months | 0–4526 | 81.78 | 434.70 | 0–2263 | 74.84 | 376.07 |
| 21 months | 0–6789 | 116.79 | 553.21 | 0–2263 | 76.38 | 380.67 |
| 27 months | 0–3268 | 103.75 | 451.35 | 0–2263 | 134.67 | 504.22 |
| 33 months | 0–3268 | 105.11 | 442.53 | 0–2263 | 76.09 | 380.21 |
| Total | 0–10057 | 592.81 | 1427.75 | 0–7060 | 422.65 | 1140.30 |
| Adaptations and equipment | ||||||
| Baseline | 0–921.50 | 17.26 | 83.11 | 0–991.00 | 36.53 | 136.68 |
| 4.5 months | 0–1292.50 | 27.41 | 132.71 | 0–4870.25 | 61.77 | 426.59 |
| 9 months | 0–4671.63 | 34.95 | 302.77 | 0–4206.13 | 55.15 | 383.37 |
| 15 months | 0–988.00 | 33.29 | 135.14 | 0–1202.00 | 37.49 | 138.04 |
| 21 months | 0–2656.50 | 30.49 | 205.41 | 0–772.50 | 41.70 | 129.02 |
| 27 months | 0–1817.00 | 42.24 | 242.68 | 0–861.00 | 20.55 | 98.62 |
| 33 months | 0–1274.00 | 17.24 | 107.33 | 0–198.00 | 7.75 | 31.04 |
| Total | 0–2765.75 | 149.73 | 397.22 | 0–4206.13 | 194.87 | 541.35 |
| Concomitant medications | ||||||
| Baseline | Not collected | |||||
| 4.5 months | 0–830.34 | 133.05 | 163.10 | 0–658.66 | 112.26 | 129.61 |
| 9 months | 0–816.52 | 131.80 | 159.38 | 0–659.66 | 115.66 | 130.49 |
| 15 months | 0–1090.68 | 177.56 | 214.31 | 0–891.22 | 155.17 | 178.64 |
| 21 months | 0–1090.68 | 180.65 | 216.93 | 0–881.15 | 154.02 | 174.15 |
| 27 months | 0–1090.68 | 178.74 | 213.63 | 0–881.15 | 155.51 | 176.14 |
| 33 months | 0–1090.68 | 181.70 | 215.93 | 0–881.15 | 156.36 | 175.61 |
| Total | 0–5995.76 | 983.50 | 1176.54 | 0–4843.92 | 848.98 | 958.94 |
| Resource use type | Active | Placebo | ||||
|---|---|---|---|---|---|---|
| Mean | SD | 95% CI | Mean | SD | 95% CI | |
| Total other costs | ||||||
| Baseline | 750.11 | 2819.92 | 306.99 to 1193.22 | 957.92 | 2642.96 | 377.20 to 1538.64 |
| 4.5 months | 672.58 | 1085.26 | 502.85 to 843.12 | 1105.80 | 4216.66 | 179.30 to 2032.30 |
| 9 months | 1038.15 | 3615.01 | 470.10 to 1606.21 | 1122.36 | 5028.73 | 17.42 to 2227.29 |
| 15 months | 839.90 | 1964.29 | 532.22 to 1147.58 | 1364.94 | 4994.17 | 267.60 to 2462.28 |
| 21 months | 987.31 | 2094.31 | 659.27 to 1315.35 | 1372.94 | 5018.67 | 270.22 to 2475.66 |
| 27 months | 913.21 | 2185.58 | 570.88 to 1255.55 | 1658.66 | 5279.56 | 498.61 to 2818.71 |
| 33 months | 1032.92 | 2563.26 | 631.42 to 1434.41 | 1801.33 | 5114.86 | 677.47 to 2925.19 |
| Total | 5473.78 | 11,980.30 | 3597.25 to 7350.32 | 8426.03 | 25,922.73 | 2730.18 to 14,121.88 |
| Resource use type | Active | Placebo | ||||
|---|---|---|---|---|---|---|
| Mean | SD | 95% CI | Mean | SD | 95% CI | |
| Total other costs | ||||||
| Baseline | 750.11 | 2819.92 | 306.99 to 1193.22 | 957.92 | 2642.96 | 377.20 to 1538.64 |
| 4.5 months | 885.12 | 232.97 | 426.74 to 1343.50 | 905.28 | 242.88 | 425.58 to 1384.99 |
| 9 months | 1106.70 | 238.89 | 636.26 to 1577.13 | 1079.20 | 336.36 | 414.08 to 1744.33 |
| 15 months | 968.53 | 167.81 | 638.21 to 1298.84 | 1078.76 | 287.13 | 511.62 to 1645.91 |
| 21 months | 1017.91 | 128.26 | 765.25 to 1270.56 | 1276.82 | 307.79 | 668.38 to 1885.26 |
| 27 months | 919.30 | 138.64 | 645.80 to 1192.80 | 1365.83 | 328.06 | 717.23 to 2014.42 |
| 33 months | 1126.78 | 169.21 | 793.04 to 1460.52 | 1526.62 | 347.05 | 839.82 to 2213.41 |
| Total | 6111.58 | 896.77 | 4346.67 to 7876.49 | 7217.84 | 1520.78 | 4213.67 to 10,222.01 |
Detailed private/patient resource use by treatment group and follow-up
| Resource use item | Active | Placebo | ||||
|---|---|---|---|---|---|---|
| Range | Mean | SD | Range | Mean | SD | |
| Physiotherapist | ||||||
| Baseline | 0–52 | 0.77 | 4.10 | 0–26 | 1.21 | 5.05 |
| 4.5 months | 0–75 | 0.86 | 5.31 | 0–28 | 0.96 | 4.07 |
| 9 months | 0–26 | 0.64 | 2.97 | 0–32 | 1.21 | 4.33 |
| 15 months | 0–52 | 1.01 | 4.93 | 0–26 | 1.68 | 5.17 |
| 21 months | 0–26 | 1.09 | 4.41 | 0–25 | 1.37 | 4.60 |
| 27 months | 0–29 | 1.12 | 4.39 | 0–26 | 1.98 | 5.90 |
| 33 months | 0–52 | 1.30 | 5.32 | 0–40 | 1.92 | 6.07 |
| Total | 0–129 | 6.70 | 21.10 | 0–89 | 8.48 | 20.30 |
| Chiropodist | ||||||
| Baseline | 0–6 | 0.26 | 0.89 | 0–5 | 0.21 | 0.80 |
| 4.5 months | 0–6 | 0.27 | 0.96 | 0–5 | 0.18 | 0.66 |
| 9 months | 0–4 | 0.27 | 0.80 | 0–3 | 0.15 | 0.48 |
| 15 months | 0–6 | 0.33 | 1.02 | 0–6 | 0.39 | 1.14 |
| 21 months | 0–8 | 0.36 | 1.12 | 0–5 | 0.40 | 1.15 |
| 27 months | 0–6 | 0.39 | 1.18 | 0–8 | 0.46 | 1.30 |
| 33 months | 0–12 | 0.42 | 1.33 | 0–6 | 0.45 | 1.26 |
| Total | 0–27 | 2.03 | 5.50 | 0–21 | 2.51 | 5.41 |
| Optician | ||||||
| Baseline | 0–2 | 0.23 | 0.51 | 0–4 | 0.25 | 0.59 |
| 4.5 months | 0–2 | 0.14 | 0.39 | 0–2 | 0.10 | 0.32 |
| 9 months | 0–3 | 0.16 | 0.46 | 0–2 | 0.15 | 0.42 |
| 15 months | 0–4 | 0.17 | 0.47 | 0–4 | 0.29 | 0.63 |
| 21 months | 0–4 | 0.17 | 0.47 | 0–2 | 0.16 | 0.44 |
| 27 months | 0–2 | 0.19 | 0.45 | 0–2 | 0.20 | 0.51 |
| 33 months | 0–2 | 0.17 | 0.45 | 0–2 | 0.15 | 0.38 |
| Total | 0–7 | 1.04 | 1.35 | 0–5 | 1.07 | 1.25 |
| Acupuncturist | ||||||
| Baseline | 0–14 | 0.29 | 1.60 | 0–24 | 0.22 | 1.99 |
| 4.5 months | 0–12 | 0.28 | 1.49 | 0–12 | 0.26 | 1.47 |
| 9 months | 0–14 | 0.19 | 1.21 | 0–10 | 0.33 | 1.61 |
| 15 months | 0–24 | 0.39 | 2.14 | 0–25 | 0.63 | 3.36 |
| 21 months | 0–24 | 0.30 | 1.97 | 0–12 | 0.16 | 1.20 |
| 27 months | 0–24 | 0.27 | 1.96 | 0–12 | 0.30 | 1.67 |
| 33 months | 0–24 | 0.23 | 1.85 | 0–7 | 0.06 | 0.64 |
| Total | 0–110 | 2.04 | 10.83 | 0–45 | 1.89 | 7.70 |
| Alternative practice | ||||||
| Baseline | 0–58 | 1.78 | 5.42 | 0–32 | 2.01 | 5.63 |
| 4.5 months | 0–136 | 1.77 | 9.13 | 0–24 | 1.32 | 3.93 |
| 9 months | 0–60 | 1.39 | 5.69 | 0–25 | 1.52 | 4.18 |
| 15 months | 0–51 | 1.51 | 4.93 | 0–41 | 2.01 | 5.78 |
| 21 months | 0–52 | 1.56 | 5.47 | 0–32 | 1.76 | 4.87 |
| 27 months | 0–52 | 1.58 | 5.47 | 0–32 | 2.08 | 5.07 |
| 33 months | 0–64 | 1.58 | 6.23 | 0–25 | 2.16 | 4.95 |
| Total | 0–275 | 9.11 | 30.28 | 0–154 | 13.52 | 28.75 |
| Home care hours | ||||||
| Baseline | 0–130 | 8.89 | 24.69 | 0–780 | 32.37 | 123.29 |
| 4.5 months | 0–156 | 12.84 | 31.88 | 0–338 | 12.05 | 46.08 |
| 9 months | 0–143 | 10.04 | 27.17 | 0–273 | 16.96 | 51.71 |
| 15 months | 0–260 | 15.22 | 40.69 | 0–676 | 35.61 | 121.95 |
| 21 months | 0–468 | 18.84 | 51.60 | 0–840 | 36.24 | 140.93 |
| 27 months | 0–429 | 15.96 | 47.07 | 0–468 | 17.50 | 63.57 |
| 33 months | 0–728 | 18.92 | 67.69 | 0–819 | 28.16 | 118.12 |
| Total | 0–1092 | 91.25 | 188.38 | 0–2769 | 146.52 | 402.99 |
| Unpaid care hours | ||||||
| Baseline | 0–16,640 | 542.76 | 1410.61 | 0–2951 | 471.24 | 569.92 |
| 4.5 months | 0–2834 | 407.37 | 533.29 | 0–5954 | 555.85 | 905.82 |
| 9 months | 0–2730 | 370.38 | 487.03 | 0–3302 | 546.00 | 720.25 |
| 15 months | 0–26,208 | 571.86 | 2135.95 | 0–2678 | 437.68 | 522.19 |
| 21 months | 0–39,312 | 746.68 | 3170.85 | 0–2158 | 457.64 | 485.93 |
| 27 months | 0–34,632 | 636.34 | 2767.60 | 0–1846 | 472.87 | 484.96 |
| 33 months | 0–3562 | 437.15 | 606.98 | 0–2106 | 463.01 | 515.38 |
| Total | 0–75,504 | 3149.85 | 6630.51 | 0–11,206 | 2933.05 | 2821.25 |
| Resource use item | Active | Placebo | ||||
|---|---|---|---|---|---|---|
| Range | Mean | SD | Range | Mean | SD | |
| Physiotherapist | ||||||
| Baseline | 0–1768 | 26.23 | 139.55 | 0–884 | 41.30 | 171.54 |
| 4.5 months | 0–2550 | 29.24 | 180.65 | 0–952 | 32.75 | 138.27 |
| 9 months | 0–884 | 21.91 | 100.87 | 0–1088 | 41.01 | 147.28 |
| 15 months | 0–1768 | 34.28 | 167.50 | 0–884 | 57.21 | 175.94 |
| 21 months | 0–884 | 36.91 | 149.92 | 0–850 | 46.60 | 156.45 |
| 27 months | 0–986 | 38.02 | 149.12 | 0–884 | 67.43 | 200.69 |
| 33 months | 0–1768 | 44.11 | 180.76 | 0–1360 | 65.41 | 206.32 |
| Total | 0–4386 | 227.74 | 717.45 | 0–3026 | 288.17 | 690.34 |
| Chiropodist | ||||||
| Baseline | 0–235 | 8.14 | 32.26 | 0–235 | 10.16 | 36.35 |
| 4.5 months | 0–155 | 7.11 | 24.89 | 0–188 | 10.09 | 35.37 |
| 9 months | 0–94 | 5.23 | 17.12 | 0–188 | 10.67 | 43.26 |
| 15 months | 0–282 | 15.23 | 47.60 | 0–282 | 13.51 | 43.65 |
| 21 months | 0–235 | 15.60 | 47.08 | 0–248 | 14.34 | 45.28 |
| 27 months | 0–248 | 18.61 | 52.31 | 0–282 | 15.97 | 50.34 |
| 33 months | 0–282 | 19.08 | 55.24 | 0–564 | 18.10 | 60.43 |
| Total | 0–893 | 98.37 | 219.45 | 0–1269 | 82.81 | 233.78 |
| Optician | ||||||
| Baseline | 0–40 | 4.57 | 10.14 | 0–80 | 4.91 | 11.78 |
| 4.5 months | 0–40 | 2.71 | 7.72 | 0–40 | 1.91 | 6.38 |
| 9 months | 0–60 | 3.28 | 9.13 | 0–40 | 3.05 | 8.40 |
| 15 months | 0–80 | 3.35 | 9.42 | 0–80 | 5.71 | 12.61 |
| 21 months | 0–80 | 3.33 | 9.49 | 0–40 | 3.28 | 8.73 |
| 27 months | 0–40 | 3.82 | 8.96 | 0–40 | 4.00 | 10.24 |
| 33 months | 0–40 | 3.33 | 9.01 | 0–40 | 3.05 | 7.68 |
| Total | 0–140 | 20.75 | 26.99 | 0–100 | 21.46 | 25.10 |
| Acupuncturist | ||||||
| Baseline | 0–476 | 9.76 | 54.25 | 0–816 | 7.51 | 67.69 |
| 4.5 months | 0–408 | 9.62 | 50.83 | 0–408 | 8.75 | 50.00 |
| 9 months | 0–476 | 6.37 | 41.01 | 0–340 | 11.16 | 54.73 |
| 15 months | 0–816 | 13.18 | 72.78 | 0–850 | 21.59 | 114.40 |
| 21 months | 0–816 | 10.32 | 67.02 | 0–408 | 5.57 | 40.84 |
| 27 months | 0–816 | 9.12 | 66.63 | 0–408 | 10.20 | 56.89 |
| 33 months | 0–3740 | 7.66 | 62.75 | 0–238 | 2.02 | 21.91 |
| Total | 0–3740 | 69.28 | 368.18 | 0–1530 | 64.27 | 261.79 |
| Alternative practitioners | ||||||
| Baseline | 0–1972 | 60.65 | 184.24 | 0–1088 | 68.42 | 191.51 |
| 4.5 months | 0–4624 | 60.09 | 310.43 | 0–816 | 44.75 | 133.76 |
| 9 months | 0–2040 | 47.41 | 193.54 | 0–850 | 51.65 | 142.22 |
| 15 months | 0–1734 | 51.35 | 167.72 | 0–1394 | 68.27 | 196.40 |
| 21 months | 0–1768 | 53.03 | 185.82 | 0–1088 | 59.79 | 165.70 |
| 27 months | 0–1768 | 53.78 | 185.82 | 0–1088 | 70.83 | 172.39 |
| 33 months | 0–2176 | 53.76 | 211.97 | 0–850 | 73.47 | 168.22 |
| Total | 0–9350 | 309.85 | 1029.58 | 0–5236 | 461.07 | 977.42 |
| Home care | ||||||
| Baseline | 0–2340 | 159.95 | 444.37 | 0–14,040 | 582.72 | 2219.18 |
| 4.5 months | 0–2808 | 231.04 | 573.78 | 0–6084 | 216.88 | 829.38 |
| 9 months | 0–2574 | 180.68 | 489.06 | 0–4914 | 305.34 | 930.70 |
| 15 months | 0–4680 | 273.99 | 732.48 | 0–12,168 | 640.93 | 2195.17 |
| 21 months | 0–8424 | 339.15 | 928.72 | 0–15,116 | 652.35 | 2536.82 |
| 27 months | 0–7722 | 287.32 | 847.16 | 0–8424 | 315.04 | 1144.33 |
| 33 months | 0–13,104 | 340.63 | 1218.51 | 0–14,742 | 506.81 | 2126.14 |
| Total | 0–19,656 | 1642.42 | 3390.93 | 0–49,842 | 2637.35 | 7253.95 |
| Informal care | ||||||
| Baseline | 0–299,520 | 9769.74 | 25,390.93 | 0–53,118 | 8482.39 | 10,258.59 |
| 4.5 months | 0–51,012 | 7332.73 | 9599.26 | 0–107,172 | 10,005.38 | 16,304.73 |
| 9 months | 0–49,140 | 6666.81 | 8766.52 | 0–59,436 | 9828.00 | 12,964.54 |
| 15 months | 0–471,744 | 10,293.51 | 38,447.11 | 0–48,204 | 7878.21 | 9399.37 |
| 21 months | 0–707,616 | 13,440.26 | 57,074.99 | 0–38,844 | 8237.54 | 8746.79 |
| 27 months | 0–623,376 | 11,454.15 | 49,816.83 | 0–33,228 | 8511.61 | 8729.32 |
| 33 months | 0–64,116 | 7868.62 | 10,925.71 | 0–37,908 | 8334.11 | 9276.78 |
| Total | 0–1,359,072 | 56,697.24 | 119,349.22 | 0–201,708 | 52,794.85 | 50,782.44 |
| Adaptations and equipment | ||||||
| Baseline | 0–1036 | 62.32 | 145.15 | 0–894 | 74.05 | 143.36 |
| 4.5 months | 0–5714 | 203.33 | 670.79 | 0–4903 | 196.48 | 594.72 |
| 9 months | 0–9158 | 151.10 | 735.89 | 0–969 | 84.59 | 218.65 |
| 15 months | 0–3542 | 139.35 | 360.34 | 0–1972 | 170.16 | 346.81 |
| 21 months | 0–3099 | 100.81 | 320.03 | 0–1180 | 77.48 | 188.99 |
| 27 months | 0–2463 | 67.83 | 210.03 | 0–2126 | 80.41 | 279.33 |
| 33 months | 0–1224 | 32.34 | 105.36 | 0–885 | 27.39 | 93.36 |
| Total | 0–7123 | 707.97 | 1261.98 | 0–5397 | 610.57 | 1007.70 |
| Travel | ||||||
| Baseline | 0–643 | 32.85 | 55.74 | 0–238 | 33.51 | 42.89 |
| 4.5 months | 0–388 | 24.32 | 44.52 | 0–186 | 25.52 | 33.80 |
| 9 months | 0–736 | 23.80 | 62.11 | 0–125 | 20.62 | 29.51 |
| 15 months | 0–507 | 22.58 | 45.40 | 0–304 | 29.80 | 44.99 |
| 21 months | 0–400 | 23.31 | 43.86 | 0–191 | 24.12 | 36.69 |
| 27 months | 0–219 | 24.37 | 38.51 | 0–219 | 24.63 | 38.09 |
| 33 months | 0–249 | 22.05 | 33.38 | 0–175 | 23.28 | 31.36 |
| Total | 0–1169 | 159.06 | 193.20 | 0–891 | 161.01 | 178.51 |
| Respite care | ||||||
| Baseline | 0–6030 | 18.61 | 335.00 | 0–936 | 5.74 | 73.31 |
| 4.5 months | 0–2010 | 8.91 | 126.35 | 0–2736 | 20.12 | 234.61 |
| 9 months | 0–10230 | 42.91 | 640.16 | 0 | 0.00 | 0.00 |
| 15 months | 0 | 0 | 0.00 | 0 | 0.00 | 0.00 |
| 21 months | 0–720 | 3.08 | 47.07 | 0–1872 | 33.47 | 216.58 |
| 27 months | 0–936 | 4.25 | 63.11 | 0–1005 | 8.38 | 91.74 |
| 33 months | 0–1005 | 12.31 | 98.05 | 0–1872 | 24.38 | 194.89 |
| Total | 0–1005 | 34.17 | 157.99 | 0–2010 | 49.02 | 269.04 |
| Resource use item | Active | Placebo | ||||
|---|---|---|---|---|---|---|
| Mean | SD | 95% CI | Mean | SD | 95% CI | |
| Health care | ||||||
| Baseline | 108.40 | 240.51 | 70.61 to 146.19 | 124.23 | 284.46 | 61.73 to 186.73 |
| 4.5 months | 123.19 | 226.77 | 87.56 to 158.83 | 123.64 | 210.36 | 77.42 to 169.86 |
| 9 months | 119.61 | 277.35 | 76.17 to 163.05 | 164.22 | 332.97 | 91.06 to 237.38 |
| 15 months | 147.63 | 312.20 | 98.73 to 196.53 | 216.42 | 432.10 | 121.48 to 311.36 |
| 21 months | 144.11 | 304.13 | 96.47 to 191.74 | 161.57 | 310.46 | 93.36 to 229.79 |
| 27 months | 152.82 | 312.08 | 103.94 to 201.70 | 206.88 | 328.68 | 134.66 to 279.10 |
| 33 months | 156.13 | 358.64 | 99.95 to 212.31 | 187.94 | 298.05 | 122.45 to 253.43 |
| Total | 842.72 | 1542.18 | 601.16 to 1084.28 | 1060.67 | 1605.21 | 707.96 to 1413.37 |
| Social care | ||||||
| Baseline | 10,038.87 | 25,358.73 | 6054.07 to 14,023.69 | 9169.28 | 10,261.99 | 6914.47 to 11,424.08 |
| 4.5 months | 7760.10 | 9499.35 | 6272.16 to 6599.69 | 10,452.99 | 16,039.83 | 6869.33 to 14,036.66 |
| 9 months | 6949.40 | 8819.82 | 5567.90 to 8330.89 | 10,204.19 | 13,184.49 | 7307.24 to 13,101.14 |
| 15 months | 10,444.95 | 37,631.73 | 4550.51 to 16,339.40 | 8501.89 | 9485.25 | 6417.75 to 10,586.03 |
| 21 months | 13,343.61 | 54,982.61 | 4731.41 to 21,955.81 | 8681.22 | 8521.63 | 6808.81 to 10,553.63 |
| 27 months | 11,140.18 | 47,135.25 | 3757.15 to 18,523.21 | 8471.07 | 8393.07 | 6626.91 to 10,315.23 |
| 33 months | 766.89 | 10,310.85 | 6047.85 to 9277.93 | 8292.46 | 8640.36 | 6393.96 to 10,190.95 |
| Total | 57,301.13 | 114,682.41 | 39,337.85 to 75,264.41 | 54,603.82 | 50,018.62 | 43613.52 to 65,594.11 |
| Total private costs | ||||||
| Baseline | 10,147.28 | 25,352.84 | 6163.39 to 14,131.16 | 9293.51 | 10,232.74 | 7045.13 to 11,541.89 |
| 4.5 months | 7932.40 | 9497.69 | 6439.96 to 9424.85 | 10,576.63 | 16,324.61 | 6989.72 to 14,163.54 |
| 9 months | 7113.42 | 8813.43 | 5782.50 to 8498.34 | 10,368.41 | 13,176.49 | 7473.21 to 13,263.60 |
| 15 months | 10,592.58 | 37,684.42 | 4689.89 to 16,495.28 | 8718.31 | 9499.53 | 6631.04 to 10,805.59 |
| 21 months | 13,487.72 | 54,988.66 | 4874.57 to 22,100.87 | 8842.79 | 8523.04 | 6970.08 to 10,715.51 |
| 27 months | 11,293.01 | 47,142.36 | 3908.86 to 18,677.15 | 8677.94 | 8427.20 | 6826.28 to 10,529.60 |
| 33 months | 7819.02 | 10,334.86 | 6200.22 to 9437.82 | 8480.40 | 8682.45 | 6572.65 to 10,388.14 |
| Total | 58,143.84 | 114,838.26 | 40,156.16 to 76,131.53 | 55,664.49 | 50,115.70 | 44,652.86 to 66,676.12 |
Method for estimating concomitant medication costs
Costs were calculated for MS-related medications as per clinical guidance. This list was further refined to exclude low-cost or short-course treatments (e.g. antibiotics). On this basis, costs were estimated for 35 drugs prescribed during the trial (Table 74). The British National Formulary online was consulted on 20 August 2012 for unit costs. Dosages were estimated on typical doses observed in the trial assuming 100% adherence. Short courses of normally longer-term medication were excluded.
| Drug class | Medication | Cost (£) | % of total |
|---|---|---|---|
| Antidepressants | Citalopram | 15,336.51 | 3.7 |
| Nortriptyline | 2123.02 | 0.5 | |
| Escitalopram | 1550.85 | 0.3 | |
| Mirtazapine | 858.32 | 0.2 | |
| Imipramine | 566.91 | 0.1 | |
| Lofepramine | 555.96 | 0.1 | |
| Subtotal | 20,991.57 | 5.0 | |
| Antiepileptic | Pregabalin | 37,054.35 | 8.9 |
| Gabapentin | 32,297.42 | 7.8 | |
| Clonazepam | 8202.94 | 2.0 | |
| Carbamazepine | 5025.58 | 1.2 | |
| Levetiracetam | 2692.02 | 0.6 | |
| Oxcarbazepine | 1716.66 | 0.4 | |
| Primidone | 1853.20 | 0.4 | |
| Sodium valproate | 741.28 | 0.2 | |
| Subtotal | 89,583.45 | 21.5 | |
| Antispasticity | Baclofen | 20,496.24 | 4.9 |
| Botulinum toxin | 4490.79 | 1.1 | |
| Dantrolene | 3979.56 | 0.9 | |
| Subtotal | 28,966.59 | 6.9 | |
| Disease-modifying drugs | Mitoxantrone | 1392.98 | 0.3 |
| Alemtuzumab | 756.91 | 0.2 | |
| Azathioprine | 419.40 | 0.1 | |
| Methotrexate | 453.38 | 0.1 | |
| Subtotal | 3022.67 | 0.7 | |
| Fatigue | Modafinil | 80,955.70 | 19.5 |
| Amantadine | 15,473.02 | 3.7 | |
| Subtotal | 96,428.72 | 23.2 | |
| Laxatives | Macrogol | 13,562.9 | 3.3 |
| Sodium picosulphate | 214.58 | 0.1 | |
| Subtotal | 13,777.48 | 3.4 | |
| Steroids | Methylprednisolone | 6758.90 | 1.6 |
| Urine/bladder | Tolterodine | 57,429.76 | 13.8 |
| Solifenacin | 33,266.58 | 8.0 | |
| Oxybutynin | 31,953.48 | 7.7 | |
| Trospium | 15,342.60 | 3.7 | |
| Desmopressin | 11,333.84 | 2.7 | |
| Duloxetine | 2574.98 | 0.6 | |
| Propiverine | 2442.29 | 0.6 | |
| Tamsulosin | 1316.79 | 0.3 | |
| Alfuzosin | 1014.38 | 0.2 | |
| Subtotal | 156,674.70 | 37.6 | |
| Total | 416,204.10 | 100.00 | |
List of abbreviations
- Δ9-THC
- Δ9-tetrahydrocannabinol
- 9-HPT
- 9-hole peg test
- ADL
- activities of daily living
- AE
- adverse event
- ANOVA
- analysis of variance
- BDI-II
- Beck Depression Inventory-II
- CAMS
- Cannabinoids in Multiple Sclerosis
- CI
- confidence interval
- COA
- clinical outcome assessment
- CRF
- case report form
- CTT
- classical test theory
- CUPID
- Cannabinoid Use in Progressive Inflammatory brain Disease
- DIF
- differential item functioning
- EDSS
- Expanded Disability Status Scale
- EPC
- End Point Committee
- EQ-5D
- European Quality of Life-5 Dimensions
- FDA
- Food and Drug Administration
- GCP
- Good Clinical Practice
- GLM
- generalised linear model
- GP
- general practitioner
- HR
- hazard ratio
- HRQoL
- health-related quality of life
- ICC
- item characteristic curve
- ICE
- imputation using chained equations
- IDMC
- Independent Data Monitoring Committee
- ISRCTN
- International Standard Randomised Controlled Trial Number
- ITT
- intention to treat
- MHRA
- Medicine and Healthcare products Regulatory Agency
- MRC
- Medical Research Council
- MRI
- magnetic resonance imaging
- MS
- multiple sclerosis
- MSFC
- Multiple Sclerosis Functional Composite
- MSIS-29phys
- Multiple Sclerosis Impact Scale-29 version 2 20-point physical subscale
- MSIS-29v2
- Multiple Sclerosis Impact Scale-29 version 2
- MSSS-88
- Multiple Sclerosis Spasticity Scale-88
- MSWS-12v2
- Multiple Sclerosis Walking Scale-12 version 2
- NBV
- normalised brain volume
- NICE
- National Institute For Health and Care Excellence
- OR
- odds ratio
- PASAT
- Paced Auditory Serial Addition Test
- PBVC
- percentage brain volume change
- PH
- proportional hazard
- PI
- principal investigator
- PPMS
- primary progressive multiple sclerosis
- PRO
- patient-reported outcome
- PSI
- person separation index
- PSS
- Personal and Social Services
- PSSRU
- Personal and Social Services Research Unit
- QALY
- quality-adjusted life-year
- R&D
- research and development
- RCT
- randomised controlled trial
- REC
- research ethics committee
- RMI
- Rivermead Mobility Index
- RMT
- Rasch measurement theory
- RRMS
- relapsing–remitting multiple sclerosis
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SE
- standard error
- SF-36v2
- Short Form questionnaire-36 items version 2
- SF-36(PH)
- Short Form questionnaire-36 items version 2 (physical health subscale)
- SPMS
- secondary progressive multiple sclerosis
- SRM
- standardised response mean
- SUSAR
- suspected unexpected serious adverse reaction
- T25-FW
- timed 25-foot walk
- THC
- tetrahydrocannabinol
- TSC
- Trial Steering Committee